EP4043476B1 - Verfahren zur herstellung einer glycosidverbindung - Google Patents
Verfahren zur herstellung einer glycosidverbindung Download PDFInfo
- Publication number
- EP4043476B1 EP4043476B1 EP20874517.4A EP20874517A EP4043476B1 EP 4043476 B1 EP4043476 B1 EP 4043476B1 EP 20874517 A EP20874517 A EP 20874517A EP 4043476 B1 EP4043476 B1 EP 4043476B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- group
- producing
- glycoside compound
- formula
- producing according
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H11/00—Compounds containing saccharide radicals esterified by inorganic acids; Metal salts thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H23/00—Compounds containing boron, silicon or a metal, e.g. chelates or vitamin B12
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the present invention relates to a method for producing glycoside compound.
- RNA which is a nucleic acid oligomer comprising ribose
- RNA probe an antisense RNA, a ribozyme, a siRNA, or an aptamer and so on, which is a useful material.
- a phosphoramidite compound of nucleoside in which a 3'-position hydroxy group is phosphoramidited.
- a glycoside compound in which a 3'-position hydroxy group and a 5'-position hydroxy group of the nucleoside are substituted with a protecting group, and a 2'-position hydroxy group is substituted with a protecting group that is capable of leaving under mild condition.
- Non-Patent Literature 1 relates to the synthesis of a long RNA oligomer using a CEM protecting group.
- Non-Patent Literature 1 Shiba et al., Nucleic Acids Res. 2007, 35, 3287-3296 .
- An object of the present invention is to provide a method for producing a glycoside compound having high purity.
- the present invention provides a method for producing the below-mentioned glycoside compounds.
- the present invention encompasses aspects described in the below-mentioned Items, but are not limited thereto.
- a formation of impurities of a glycoside compound which is a starting material for oligonucleic acid synthesis can be suppressed.
- the glycoside compound is suitable for a synthesis of oligonucleic acid.
- a method for producing a glycoside compound represented by formula (3) comprises reacting a glycoside compound represented by formula (1) with an ether compound represented by formula (2) in 4-methyltetrahydropyran in the presence of one or more halogenating agents selected from halogen, N-halogenated succinimide, and N-halogenated hydantoin.
- the adenine group which may be optionally substituted with an acyl group indicated as B a has the following structure. wherein R 2 represents a hydrogen atom or an acyl group.
- An acyl group represents a straight chain or branched chain of aliphatic acyl group, or an aromatic acyl group, in which the total number of carbon atom including the carbon atom contained in carbonyl group is 2 to 12, and preferably is 2 to 7.
- acyl group examples include aliphatic acyl groups (such as an acetyl group, a propionyl group, a butanoyl group (a butyryl group), an isobutanoyl group (an isobutyryl group), a pentanoyl group, a hexanoyl group, a heptanoyl group, an octanoyl group, a nonanoyl group, a decanoyl group, and an undecanoyl group, and so on); and an aromatic acyl groups (such as a benzoyl group, a 1-naphtoyl group, and a 2-naphtoyl group), and preferably include an acetyl group or a benzoyl group.
- aliphatic acyl groups such as an acetyl group, a propionyl group, a butanoyl group (a butyryl group), an isobutanoyl
- halogenated agent examples include halogens (such as iodine and bromine); N-halogenated succinimides (such as N-chloro succinimide, N-bromo succinimide (NBS), and N-iodo succinimide (NIS) and so on); N-halogenated hydantoins (such as 1,3-diiodo-5,5-dimethyl hydantoin, 1,3-dibromo-5,5-dimethyl hydantoin, and 1-bromo-3-chloro-5,5-dimethyl hydantoin, and so on).
- halogens are preferably used, and iodine is further preferably used.
- the reaction of the present invention may be conducted by adding an acid in addition to the halogenated agents.
- the acid include perfluoroalkyl carboxylic acids and salts thereof, alkyl sulfonic acids and salts thereof, aryl sulfonic acids and salts thereof, perfluoroalkyl sulfonic acid and its salt, and alkyl sulfonic acids and salts thereof, as well as any combinations of two or more of these acids.
- the salt include metal salts (such as copper salt and silver salt).
- the acid include methanesulfonic acid, p-toluenesulfonic acid, camphor sulfonic acid, trifluoromethanesulfonic acid, and silver trifluoromethanesulfonate, as well as any combinations of two or more of these acids.
- the alkyl group of alkylsulfonic acid may be a straight chain or branched chain thereof, and preferably is an alkyl group having 1 to 6 of carbon atoms.
- alkyl group methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, tert-butyl, n-pentyl, isopentyl, and hexyl are exemplified.
- aryl group of aryl sulfonic acid include phenyl group and tolyl group.
- the alkyl group indicated as R 1 may be a straight chain or branched chain thereof, and preferably is an alkyl group having 1 to 6 of carbon atoms.
- the alkyl group include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, tert-butyl, n-pentyl, isopentyl, and hexyl.
- Examples of the alkyl group include preferably a methyl group.
- the solvent for the reaction of the present invention includes 4-methyltetrahydropyran (MTHP).
- the amount of solvent is within a range of usually 0.5 v/w to 20 v/w %, and preferably 1 v/w to 10 v/w %, as opposed to the glycoside compound represented by formula (1).
- the amount of the ether compound represented by formula (2) is within a range of usually 0.8 to 5 moles, preferably 1 to 3 mole(s), and more preferably 1 to 1.5 mole(s), as opposed to 1 mole of the glycoside compound represented by formula (1).
- the amount of the halogenated agents is within a range of usually 0.8 to 10 moles, preferably 3 to 10 moles, and more preferably 3 to 6 moles, as opposed to 1 mole of the glycoside compound represented by formula (1).
- the amount of the acid is within a range of usually 0 to 5 moles, preferably 0 to 1.5 moles, and more preferably 0 to 0.1 moles, as opposed to 1 mole of the glycoside compounds represented by formula (1).
- the reaction temperature of the present invention is usually within a range of -30 to 30°C, preferably -20 to 25°C, and more preferably -15 to 5°C.
- the reaction period of the present reaction is within a range of usually 0.5 to 10 hours, and preferably 0.5 to 6 hours.
- the glycoside compound represented by formula (1) can be prepared by a publicly known method, or can be obtained as a commercially available product.
- the ether compound represented by formula (2) can be prepared according to a publicly known method (see the above-mentioned patent literatures 1, 2 and JP 2016-50203 A ).
- the glycoside compound obtained by the present invention may be used as a crude product in a next reaction, and as needed, may be purified by a silica gel column chromatography.
- the purity of the synthesized glycoside compound was measured by HPLC.
- the glycoside compound was separated to each ingredient by HPLC, and for the amount of by-products, the total of area percentages of the by-products in the obtained chromatograms were calculated.
- the used HPLC apparatus was set such that any peaks having peak area of 1,000 or more can be detected, and the peak having minimum peak area which was detectable under this condition was 0.03 % as area percentage.
- Table 1 Column XBridge BEH C18 XP Column.
- CEM represents 2-cyanoethoxymethyl
- EMM represents 2-cyanoethoxymethoxymethyl
- N 6 -Acetyl-3',5'-O-(tetraisopropyldisiloxan-1,3-diyl)adenosine (1.0 g, 1.8 mmol) was dissolved in 4-methyltetrahydropyran (10 mL), and the mixture was cooled to 0°C. Thereto were added methanesulfonic acid (17 mg, 0.18 mmol), iodine (2.7 g, 10.8 mmol), and 2-cyanoethoxymethyl methylthiomethyl ether (0.44 g, 2.7 mmol), and the mixture was stirred at 0°C under nitrogen atmosphere for 3 hours.
- reaction solutions were added to a mixed solution of saturated aqueous sodium thiosulfate solution and saturated aqueous sodium hydrocarbonate solution, and the mixture was extracted with ethyl acetate. The solvents were evaporated to obtain a crude product containing a desired product.
- N 6 -Acetyl-3',5'-O-(tetraisopropyldisiloxan-1,3-diyl)adenosine (1.0 g, 1.8 mmol) was dissolved in 4-methyltetrahydropyran (5 mL), and the mixture was cooled to 0°C. Thereto were added methanesulfonic acid (17 mg, 0.18 mmol), iodine (2.7 g, 10.8 mmol), and methylthiomethyl 2-cyanoethyl ether (0.35 g, 2.7 mmol), and the mixture was stirred at 0°C under nitrogen atmosphere for 3 hours.
- reaction solutions were added to a mixed solution of saturated aqueous sodium thiosulfate solution and saturated aqueous sodium hydrocarbonate solution, and the mixture was extracted with ethyl acetate. The solvents were evaporated to obtain a crude product containing a desired product.
- N 6 -Acetyl-3',5'-O-(tetraisopropyldisiloxan-1,3-diyl)adenosine (5.0 g, 9.0 mmol) was dissolved in toluene (25 mL), and the mixture was concentrated to 15 mL as a solution volume. To this solution was added 4-methyltetrahydropyran (10 mL), and the mixture was cooled to -10°C. Thereto were added iodine (13.8 g, 55.2 mmol), and 2-cyanoethoxymethyl methylthiomethyl ether (2.18 g, 13.4 mmol), and the mixture was stirred at 0°C under nitrogen atmosphere for 1 hour.
- reaction solution was added to a mixed solution of saturated aqueous sodium thiosulfate solution and saturated aqueous sodium hydrocarbonate solution, and the mixture was extracted with toluene. The solvents were evaporated to obtain a crude product containing a desired product.
- N 6 -Acetyl-3',5'-O-(tetraisopropyldisiloxan-1,3-diyl)adenosine (1.0 g, 1.8 mmol) was dissolved in tetrahydrofuran (10 mL), and the mixture was cooled to 0°C. Thereto were added methanesulfonic acid (17 mg, 0.18 mmol), iodine (2.7 g, 10.8 mmol), and 2-cyanoethoxymethyl methylthiomethyl ether (0.44 g, 2.7 mmol), and the mixture was stirred at 0°C under nitrogen atmosphere for 3 hours.
- reaction solution was added to a mixed solution of saturated aqueous sodium thiosulfate solution and saturated aqueous sodium hydrocarbonate solution, and the mixture was extracted with ethyl acetate. The solvents were evaporated to obtain a crude product containing a desired product.
- N 6 -Acetyl-3',5'-O-(tetraisopropyldisiloxan-1,3-diyl)adenosine (1.0 g, 1.8 mmol) was dissolved in tetrahydrofuran (5 mL), and the mixture was cooled to 0°C. Thereto were added methanesulfonic acid (17 mg, 0.18 mmol), iodine (2.7 g, 10.8 mmol), and methylthiomethyl 2-cyanoethyl ether (0.35 g, 2.7 mmol), and the mixture was stirred at 0°C under nitrogen atmosphere for 3 hours.
- reaction solutions were added to a mixed solution of saturated aqueous sodium thiosulfate solution and saturated aqueous sodium hydrocarbonate solution, and the mixture was extracted with ethyl acetate. The solvents were evaporated to obtain a crude product containing a desired product.
- N 6 -Acetyl-3',5'-O-(tetraisopropyldisiloxan-1,3-diyl)adenosine (5.0 g, 9.0 mmol) was dissolved in toluene (25 mL), and the mixture was concentrated to 15 mL as a solution volume. To this solution was added tetrahydrofuran (10 mL), and the mixture was cooled to -10°C. Thereto were added iodine (13.8 g, 55.2 mmol) and 2-cyanoethoxymethyl methylthiomethyl ether (2.18 g, 13.4 mmol), and the mixture was stirred at 0°C under nitrogen atmosphere for 1 hour.
- reaction solution was added to a mixed solution of saturated aqueous sodium thiosulfate solution and saturated aqueous sodium hydrocarbonate solution, and the mixture was extracted with toluene. The solvents were evaporated to obtain a crude product containing a desired product.
- a method for producing a glycoside compound is provided.
- the glycoside compound that is obtained according to the process has a high purity, and is suitable for synthesis of oligonucleotide with high purity.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Saccharide Compounds (AREA)
Claims (8)
- Verfahren zur Herstellung einer Glykosidverbindung, die dargestellt ist durch Formel (3):
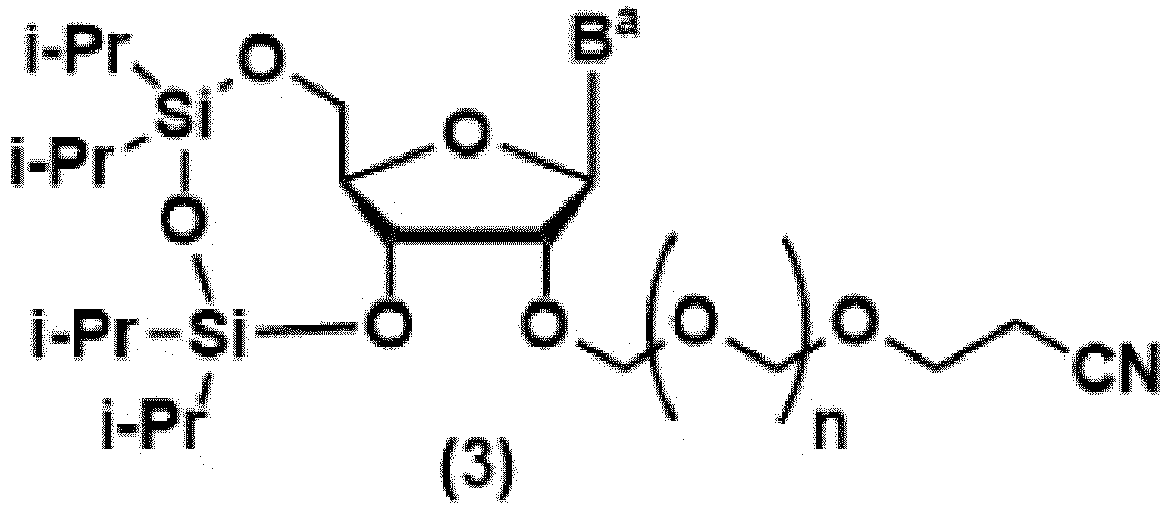
 wobei Ba eine Adenin-Gruppe darstellt, die optional mit einer Acyl-Gruppe substituiert sein kann, und die i-Pr-Gruppe eine Isopropyl-Gruppe darstellt,mit einer Etherverbindung, die dargestellt ist durch Formel (2):
wobei Ba eine Adenin-Gruppe darstellt, die optional mit einer Acyl-Gruppe substituiert sein kann, und die i-Pr-Gruppe eine Isopropyl-Gruppe darstellt,mit einer Etherverbindung, die dargestellt ist durch Formel (2): wobei R1 eine C1-C6-Alkylgruppe oder eine Phenylgruppe darstellt, und n 0 oder 1 ist,in 4-Methyltetrahydropyran in Gegenwart von einem oder mehreren Halogenierungsmitteln, ausgewählt aus Halogen, N-halogeniertem Succinimid und N-halogeniertem Hydantoin.
wobei R1 eine C1-C6-Alkylgruppe oder eine Phenylgruppe darstellt, und n 0 oder 1 ist,in 4-Methyltetrahydropyran in Gegenwart von einem oder mehreren Halogenierungsmitteln, ausgewählt aus Halogen, N-halogeniertem Succinimid und N-halogeniertem Hydantoin. - Verfahren zur Herstellung nach Anspruch 1, wobei n 1 ist.
- Verfahren zur Herstellung nach Anspruch 1, wobei n 0 ist.
- Verfahren zur Herstellung nach einem der Ansprüche 1 bis 3, wobei das Halogenierungsmittel Halogen ist.
- Verfahren zur Herstellung nach einem der Ansprüche 1 bis 4, wobei das Halogenierungsmittel Iod ist.
- Verfahren zur Herstellung nach einem der Ansprüche 1 bis 5, wobei R1 eine Methylgruppe darstellt.
- Verfahren zur Herstellung nach einem der Ansprüche 1 bis 6, wobei Ba eine Adenin-Gruppe darstellt, die mit einer Acetyl-Gruppe oder einer Benzoyl-Gruppe substituiert ist.
- Verfahren zur Herstellung nach einem der Ansprüche 1 bis 6, wobei Ba eine Adenin-Gruppe darstellt, die mit einer Acetyl-Gruppe substituiert ist.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019185109 | 2019-10-08 | ||
| PCT/JP2020/032609 WO2021070507A1 (ja) | 2019-10-08 | 2020-08-28 | 配糖体化合物の製造方法 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| EP4043476A1 EP4043476A1 (de) | 2022-08-17 |
| EP4043476A4 EP4043476A4 (de) | 2023-11-01 |
| EP4043476B1 true EP4043476B1 (de) | 2024-12-11 |
Family
ID=75437155
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP20874517.4A Active EP4043476B1 (de) | 2019-10-08 | 2020-08-28 | Verfahren zur herstellung einer glycosidverbindung |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US12559512B2 (de) |
| EP (1) | EP4043476B1 (de) |
| JP (1) | JP7522753B2 (de) |
| KR (1) | KR102938084B1 (de) |
| CN (1) | CN114502567B (de) |
| DK (1) | DK4043476T3 (de) |
| ES (1) | ES2998864T3 (de) |
| HU (1) | HUE070788T2 (de) |
| WO (1) | WO2021070507A1 (de) |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003089684A (ja) * | 2001-09-20 | 2003-03-28 | Sankyo Co Ltd | ゾフィマリン誘導体を含有する抗真菌剤 |
| US8158775B2 (en) * | 2006-02-27 | 2012-04-17 | Nippon Shinyaku Co., Ltd. | Method for detaching protecting group on nucleic acid |
| US8158774B2 (en) | 2006-08-02 | 2012-04-17 | Nippon Shinyaku Co., Ltd. | Method for introducing a nucleic-acid protecting group |
| US20100099640A1 (en) | 2007-05-04 | 2010-04-22 | Joannes Geuns | Tissue degeneration protection |
| BR122020014853B8 (pt) | 2011-08-25 | 2023-02-28 | Bonac Corp | Éter, e, método de produção de um éter |
| JP6459852B2 (ja) | 2014-08-29 | 2019-01-30 | 住友化学株式会社 | エーテル化合物の製造方法 |
| KR20170129264A (ko) * | 2015-04-02 | 2017-11-24 | 가부시키가이샤 보낙 | 글리코시드 화합물의 제조 방법 |
| JP2019185109A (ja) | 2018-04-02 | 2019-10-24 | パイオニア株式会社 | 検知装置、検知方法及びプログラム |
| WO2019208571A1 (ja) * | 2018-04-24 | 2019-10-31 | 住友化学株式会社 | アミダイト化合物及び該化合物を用いたポリヌクレオチドの製造方法 |
-
2020
- 2020-08-28 DK DK20874517.4T patent/DK4043476T3/da active
- 2020-08-28 EP EP20874517.4A patent/EP4043476B1/de active Active
- 2020-08-28 KR KR1020227010695A patent/KR102938084B1/ko active Active
- 2020-08-28 HU HUE20874517A patent/HUE070788T2/hu unknown
- 2020-08-28 CN CN202080070122.1A patent/CN114502567B/zh active Active
- 2020-08-28 WO PCT/JP2020/032609 patent/WO2021070507A1/ja not_active Ceased
- 2020-08-28 US US17/766,984 patent/US12559512B2/en active Active
- 2020-08-28 ES ES20874517T patent/ES2998864T3/es active Active
- 2020-08-28 JP JP2021550439A patent/JP7522753B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| DK4043476T3 (da) | 2025-02-24 |
| EP4043476A1 (de) | 2022-08-17 |
| EP4043476A4 (de) | 2023-11-01 |
| KR20220079540A (ko) | 2022-06-13 |
| KR102938084B1 (ko) | 2026-03-11 |
| ES2998864T3 (en) | 2025-02-21 |
| HUE070788T2 (hu) | 2025-07-28 |
| US20230022212A1 (en) | 2023-01-26 |
| WO2021070507A1 (ja) | 2021-04-15 |
| CN114502567B (zh) | 2024-11-19 |
| US12559512B2 (en) | 2026-02-24 |
| JP7522753B2 (ja) | 2024-07-25 |
| CN114502567A (zh) | 2022-05-13 |
| JPWO2021070507A1 (de) | 2021-04-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12473321B2 (en) | Compositions and methods for phosphoramidite and oligonucleotide synthesis | |
| EP3172218B1 (de) | Verfahren zur herstellung von gemcitabin-[phenyl(benzoxy-l-alaninyl)]phosphat | |
| US20090209754A1 (en) | Process for the preparation of capecitabine | |
| EP4047004B1 (de) | Phosphoramidit-aktivator | |
| EP4043476B1 (de) | Verfahren zur herstellung einer glycosidverbindung | |
| EP2878604B1 (de) | Monomer zur synthese von rna, herstellungsverfahren dafür und verfahren zur herstellung von rna | |
| JP2025532629A (ja) | 改善されたオリゴヌクレオチド合成 | |
| JP7584410B2 (ja) | 二分岐脂質結合オリゴヌクレオチドの製造方法及び中間体 | |
| JPH06135988A (ja) | ヌクレオシド誘導体 | |
| KR102935864B1 (ko) | 배당체 화합물의 제조 방법 | |
| EP4049996A1 (de) | Glycosidverbindung, amiditverbindung und herstellungsverfahren für polynukleotid unter verwendung dieser verbindungen | |
| US20100267940A1 (en) | Method for Producing 4-Deoxy-4-Fluoro-D-Glucose Derivative | |
| EP2860172A1 (de) | Verfahren zur Herstellung von Acetamidophenylderivaten | |
| KR101241321B1 (ko) | 수율 및 순도가 개선된 데시타빈의 제조방법 | |
| JP2011148738A (ja) | リボヌクレオシド誘導体またはその塩の製造方法 | |
| KR20260003278A (ko) | 올리고뉴클레오타이드 합성용 고지용성 뉴클레오타이드 빌딩 블록 및 이를 이용한 올리고뉴클레오타이드의 합성 | |
| JP4627625B2 (ja) | N−アセチルシチジン類の製造方法 | |
| WO2004104017A2 (en) | Methods for preparing 2-alkynyladenosine derivatives | |
| FR2766189A1 (fr) | Synthese regiospecifique de 5-aminoimidazol et 5-aminotriazol-1-yl desoxyglucofuranose et de leurs derives 1,5'-cyclo-5-(5'-desoxy-b-d-glucofuranosylamino), produits obtenus et leurs applications | |
| HK1232546A1 (en) | Process for the preparation of gemcitabine-[phenyl(benzoxy-l-alaninyl)] phosphate | |
| HK1232546B (en) | Process for the preparation of gemcitabine-[phenyl(benzoxy-l-alaninyl)] phosphate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: THE INTERNATIONAL PUBLICATION HAS BEEN MADE |
|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: REQUEST FOR EXAMINATION WAS MADE |
|
| 17P | Request for examination filed |
Effective date: 20220401 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AL AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR |
|
| DAV | Request for validation of the european patent (deleted) | ||
| DAX | Request for extension of the european patent (deleted) | ||
| REG | Reference to a national code |
Ref country code: DE Free format text: PREVIOUS MAIN CLASS: C07H0023000000 Ipc: C07H0001000000 Ref country code: DE Ref legal event code: R079 Ref document number: 602020043130 Country of ref document: DE Free format text: PREVIOUS MAIN CLASS: C07H0023000000 Ipc: C07H0001000000 |
|
| A4 | Supplementary search report drawn up and despatched |
Effective date: 20230928 |
|
| RIC1 | Information provided on ipc code assigned before grant |
Ipc: C07H 23/00 20060101ALI20230922BHEP Ipc: C07H 1/00 20060101AFI20230922BHEP |
|
| GRAP | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOSNIGR1 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: GRANT OF PATENT IS INTENDED |
|
| INTG | Intention to grant announced |
Effective date: 20240711 |
|
| GRAS | Grant fee paid |
Free format text: ORIGINAL CODE: EPIDOSNIGR3 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: THE PATENT HAS BEEN GRANTED |
|
| P01 | Opt-out of the competence of the unified patent court (upc) registered |
Free format text: CASE NUMBER: APP_55516/2024 Effective date: 20241009 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AL AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: EP |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R096 Ref document number: 602020043130 Country of ref document: DE |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FG2A Ref document number: 2998864 Country of ref document: ES Kind code of ref document: T3 Effective date: 20250221 |
|
| REG | Reference to a national code |
Ref country code: DK Ref legal event code: T3 Effective date: 20250221 |
|
| REG | Reference to a national code |
Ref country code: LT Ref legal event code: MG9D |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: HR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241211 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FI Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241211 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: BG Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241211 |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: MP Effective date: 20241211 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NO Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20250311 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LV Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241211 Ref country code: GR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20250312 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: RS Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20250311 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241211 |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: MK05 Ref document number: 1750327 Country of ref document: AT Kind code of ref document: T Effective date: 20241211 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SM Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241211 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: PL Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241211 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IS Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20250411 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: PT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20250411 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: EE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241211 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: AT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241211 Ref country code: RO Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241211 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241211 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CZ Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241211 |
|
| REG | Reference to a national code |
Ref country code: HU Ref legal event code: AG4A Ref document number: E070788 Country of ref document: HU |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241211 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20241211 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R097 Ref document number: 602020043130 Country of ref document: DE |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: HU Payment date: 20250815 Year of fee payment: 6 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: ES Payment date: 20250901 Year of fee payment: 6 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 20250724 Year of fee payment: 6 Ref country code: DK Payment date: 20250723 Year of fee payment: 6 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: GB Payment date: 20250725 Year of fee payment: 6 Ref country code: BE Payment date: 20250723 Year of fee payment: 6 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 20250725 Year of fee payment: 6 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: CH Payment date: 20250901 Year of fee payment: 6 |
|
| 26N | No opposition filed |
Effective date: 20250912 |