EP1523025B1 - Field emission-type electron source - Google Patents
Field emission-type electron source Download PDFInfo
- Publication number
- EP1523025B1 EP1523025B1 EP02022014A EP02022014A EP1523025B1 EP 1523025 B1 EP1523025 B1 EP 1523025B1 EP 02022014 A EP02022014 A EP 02022014A EP 02022014 A EP02022014 A EP 02022014A EP 1523025 B1 EP1523025 B1 EP 1523025B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- layer
- electron
- conductive
- carbide
- surface electrode
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 150000004767 nitrides Chemical class 0.000 claims description 85
- 229910000510 noble metal Inorganic materials 0.000 claims description 49
- 229910026551 ZrC Inorganic materials 0.000 claims description 11
- OTCHGXYCWNXDOA-UHFFFAOYSA-N [C].[Zr] Chemical compound [C].[Zr] OTCHGXYCWNXDOA-UHFFFAOYSA-N 0.000 claims description 11
- -1 hafnium nitride Chemical class 0.000 claims description 8
- WHJFNYXPKGDKBB-UHFFFAOYSA-N hafnium;methane Chemical compound C.[Hf] WHJFNYXPKGDKBB-UHFFFAOYSA-N 0.000 claims description 7
- MTPVUVINMAGMJL-UHFFFAOYSA-N trimethyl(1,1,2,2,2-pentafluoroethyl)silane Chemical group C[Si](C)(C)C(F)(F)C(F)(F)F MTPVUVINMAGMJL-UHFFFAOYSA-N 0.000 claims description 7
- NRTOMJZYCJJWKI-UHFFFAOYSA-N Titanium nitride Chemical compound [Ti]#N NRTOMJZYCJJWKI-UHFFFAOYSA-N 0.000 claims description 5
- 229910052735 hafnium Inorganic materials 0.000 claims description 5
- MZLGASXMSKOWSE-UHFFFAOYSA-N tantalum nitride Chemical group [Ta]#N MZLGASXMSKOWSE-UHFFFAOYSA-N 0.000 claims description 4
- ZVWKZXLXHLZXLS-UHFFFAOYSA-N zirconium nitride Chemical compound [Zr]#N ZVWKZXLXHLZXLS-UHFFFAOYSA-N 0.000 claims description 4
- 239000010410 layer Substances 0.000 description 401
- 238000000034 method Methods 0.000 description 182
- 239000000758 substrate Substances 0.000 description 102
- 239000010408 film Substances 0.000 description 90
- 229910021420 polycrystalline silicon Inorganic materials 0.000 description 89
- 229910052751 metal Inorganic materials 0.000 description 86
- 239000002184 metal Substances 0.000 description 86
- 230000008569 process Effects 0.000 description 73
- 238000004544 sputter deposition Methods 0.000 description 40
- 239000007789 gas Substances 0.000 description 39
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Substances [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 35
- 239000004065 semiconductor Substances 0.000 description 34
- 230000003647 oxidation Effects 0.000 description 33
- 238000007254 oxidation reaction Methods 0.000 description 33
- 230000006866 deterioration Effects 0.000 description 28
- 238000010438 heat treatment Methods 0.000 description 27
- 238000004519 manufacturing process Methods 0.000 description 26
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 25
- 229910052710 silicon Inorganic materials 0.000 description 25
- 239000010703 silicon Substances 0.000 description 25
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 22
- 238000001771 vacuum deposition Methods 0.000 description 22
- 239000002159 nanocrystal Substances 0.000 description 21
- 239000010409 thin film Substances 0.000 description 21
- 229910052799 carbon Inorganic materials 0.000 description 20
- 229910052697 platinum Inorganic materials 0.000 description 20
- 238000005546 reactive sputtering Methods 0.000 description 20
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 18
- 229910052814 silicon oxide Inorganic materials 0.000 description 17
- 239000011521 glass Substances 0.000 description 16
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 15
- 238000007743 anodising Methods 0.000 description 15
- 229910052737 gold Inorganic materials 0.000 description 15
- 239000010931 gold Substances 0.000 description 15
- 229910052721 tungsten Inorganic materials 0.000 description 15
- 229910052782 aluminium Inorganic materials 0.000 description 14
- 239000000463 material Substances 0.000 description 14
- 238000005229 chemical vapour deposition Methods 0.000 description 13
- 238000001755 magnetron sputter deposition Methods 0.000 description 13
- 238000000137 annealing Methods 0.000 description 11
- 238000009792 diffusion process Methods 0.000 description 11
- 238000000151 deposition Methods 0.000 description 10
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 10
- 239000012535 impurity Substances 0.000 description 10
- 229910052719 titanium Inorganic materials 0.000 description 10
- 239000010936 titanium Substances 0.000 description 10
- 239000010937 tungsten Substances 0.000 description 10
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 9
- 230000008018 melting Effects 0.000 description 9
- 238000002844 melting Methods 0.000 description 9
- 229910052757 nitrogen Inorganic materials 0.000 description 9
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 description 9
- 239000012298 atmosphere Substances 0.000 description 8
- 238000005054 agglomeration Methods 0.000 description 7
- 230000002776 aggregation Effects 0.000 description 7
- 125000004429 atom Chemical group 0.000 description 7
- 230000004888 barrier function Effects 0.000 description 7
- 239000000919 ceramic Substances 0.000 description 7
- 230000005684 electric field Effects 0.000 description 7
- 229910052750 molybdenum Inorganic materials 0.000 description 7
- 230000003746 surface roughness Effects 0.000 description 7
- 229910021417 amorphous silicon Inorganic materials 0.000 description 6
- 238000007789 sealing Methods 0.000 description 6
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 5
- 229910045601 alloy Inorganic materials 0.000 description 5
- 239000000956 alloy Substances 0.000 description 5
- 229910052804 chromium Inorganic materials 0.000 description 5
- 239000011651 chromium Substances 0.000 description 5
- 229910052802 copper Inorganic materials 0.000 description 5
- 239000010949 copper Substances 0.000 description 5
- 230000008021 deposition Effects 0.000 description 5
- 238000010894 electron beam technology Methods 0.000 description 5
- 238000005121 nitriding Methods 0.000 description 5
- 238000001552 radio frequency sputter deposition Methods 0.000 description 5
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 4
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 4
- 239000003513 alkali Substances 0.000 description 4
- 239000002131 composite material Substances 0.000 description 4
- 239000011261 inert gas Substances 0.000 description 4
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 4
- 239000000203 mixture Substances 0.000 description 4
- 238000005268 plasma chemical vapour deposition Methods 0.000 description 4
- 238000012545 processing Methods 0.000 description 4
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 4
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 3
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 3
- CFJRGWXELQQLSA-UHFFFAOYSA-N azanylidyneniobium Chemical compound [Nb]#N CFJRGWXELQQLSA-UHFFFAOYSA-N 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 238000001803 electron scattering Methods 0.000 description 3
- 229910052741 iridium Inorganic materials 0.000 description 3
- GKOZUEZYRPOHIO-UHFFFAOYSA-N iridium atom Chemical compound [Ir] GKOZUEZYRPOHIO-UHFFFAOYSA-N 0.000 description 3
- 230000005641 tunneling Effects 0.000 description 3
- 238000007738 vacuum evaporation Methods 0.000 description 3
- INZDTEICWPZYJM-UHFFFAOYSA-N 1-(chloromethyl)-4-[4-(chloromethyl)phenyl]benzene Chemical compound C1=CC(CCl)=CC=C1C1=CC=C(CCl)C=C1 INZDTEICWPZYJM-UHFFFAOYSA-N 0.000 description 2
- QIJNJJZPYXGIQM-UHFFFAOYSA-N 1lambda4,2lambda4-dimolybdacyclopropa-1,2,3-triene Chemical compound [Mo]=C=[Mo] QIJNJJZPYXGIQM-UHFFFAOYSA-N 0.000 description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- 229910039444 MoC Inorganic materials 0.000 description 2
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 description 2
- 230000004075 alteration Effects 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- UFGZSIPAQKLCGR-UHFFFAOYSA-N chromium carbide Chemical compound [Cr]#C[Cr]C#[Cr] UFGZSIPAQKLCGR-UHFFFAOYSA-N 0.000 description 2
- 239000004020 conductor Substances 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 238000010586 diagram Methods 0.000 description 2
- 239000008151 electrolyte solution Substances 0.000 description 2
- 239000002784 hot electron Substances 0.000 description 2
- 238000004050 hot filament vapor deposition Methods 0.000 description 2
- 238000005468 ion implantation Methods 0.000 description 2
- 238000005224 laser annealing Methods 0.000 description 2
- 238000004518 low pressure chemical vapour deposition Methods 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- UNASZPQZIFZUSI-UHFFFAOYSA-N methylidyneniobium Chemical compound [Nb]#C UNASZPQZIFZUSI-UHFFFAOYSA-N 0.000 description 2
- NFFIWVVINABMKP-UHFFFAOYSA-N methylidynetantalum Chemical compound [Ta]#C NFFIWVVINABMKP-UHFFFAOYSA-N 0.000 description 2
- 239000011733 molybdenum Substances 0.000 description 2
- 239000012299 nitrogen atmosphere Substances 0.000 description 2
- 230000001590 oxidative effect Effects 0.000 description 2
- 238000000059 patterning Methods 0.000 description 2
- 239000005361 soda-lime glass Substances 0.000 description 2
- 229910003468 tantalcarbide Inorganic materials 0.000 description 2
- 238000002230 thermal chemical vapour deposition Methods 0.000 description 2
- XOLBLPGZBRYERU-UHFFFAOYSA-N tin dioxide Chemical compound O=[Sn]=O XOLBLPGZBRYERU-UHFFFAOYSA-N 0.000 description 2
- 229910003470 tongbaite Inorganic materials 0.000 description 2
- UONOETXJSWQNOL-UHFFFAOYSA-N tungsten carbide Chemical compound [W+]#[C-] UONOETXJSWQNOL-UHFFFAOYSA-N 0.000 description 2
- HUVAFPHSQFFCID-UHFFFAOYSA-N 5-chlorocyclopenta-1,3-diene titanium(2+) Chemical compound [Ti++].Cl[c-]1cccc1.Cl[c-]1cccc1 HUVAFPHSQFFCID-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 1
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- QZPSXPBJTPJTSZ-UHFFFAOYSA-N aqua regia Chemical compound Cl.O[N+]([O-])=O QZPSXPBJTPJTSZ-UHFFFAOYSA-N 0.000 description 1
- SJKRCWUQJZIWQB-UHFFFAOYSA-N azane;chromium Chemical compound N.[Cr] SJKRCWUQJZIWQB-UHFFFAOYSA-N 0.000 description 1
- GPBUGPUPKAGMDK-UHFFFAOYSA-N azanylidynemolybdenum Chemical compound [Mo]#N GPBUGPUPKAGMDK-UHFFFAOYSA-N 0.000 description 1
- SKKMWRVAJNPLFY-UHFFFAOYSA-N azanylidynevanadium Chemical compound [V]#N SKKMWRVAJNPLFY-UHFFFAOYSA-N 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 150000001722 carbon compounds Chemical class 0.000 description 1
- 229910052681 coesite Inorganic materials 0.000 description 1
- 229910052593 corundum Inorganic materials 0.000 description 1
- 229910052906 cristobalite Inorganic materials 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- VBJZVLUMGGDVMO-UHFFFAOYSA-N hafnium atom Chemical compound [Hf] VBJZVLUMGGDVMO-UHFFFAOYSA-N 0.000 description 1
- PDPJQWYGJJBYLF-UHFFFAOYSA-J hafnium tetrachloride Chemical compound Cl[Hf](Cl)(Cl)Cl PDPJQWYGJJBYLF-UHFFFAOYSA-J 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 229910000040 hydrogen fluoride Inorganic materials 0.000 description 1
- 229910000765 intermetallic Inorganic materials 0.000 description 1
- 230000001678 irradiating effect Effects 0.000 description 1
- 150000002736 metal compounds Chemical class 0.000 description 1
- 229910001092 metal group alloy Inorganic materials 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- 239000012495 reaction gas Substances 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 239000004054 semiconductor nanocrystal Substances 0.000 description 1
- 229910021332 silicide Inorganic materials 0.000 description 1
- FVBUAEGBCNSCDD-UHFFFAOYSA-N silicide(4-) Chemical compound [Si-4] FVBUAEGBCNSCDD-UHFFFAOYSA-N 0.000 description 1
- 239000002356 single layer Substances 0.000 description 1
- 238000009751 slip forming Methods 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 229910052682 stishovite Inorganic materials 0.000 description 1
- 229910052715 tantalum Inorganic materials 0.000 description 1
- XJDNKRIXUMDJCW-UHFFFAOYSA-J titanium tetrachloride Chemical compound Cl[Ti](Cl)(Cl)Cl XJDNKRIXUMDJCW-UHFFFAOYSA-J 0.000 description 1
- 229910052905 tridymite Inorganic materials 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 1
- 229910001845 yogo sapphire Inorganic materials 0.000 description 1
- 229910052726 zirconium Inorganic materials 0.000 description 1
- DUNKXUFBGCUVQW-UHFFFAOYSA-J zirconium tetrachloride Chemical compound Cl[Zr](Cl)(Cl)Cl DUNKXUFBGCUVQW-UHFFFAOYSA-J 0.000 description 1
Images






Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01J—ELECTRIC DISCHARGE TUBES OR DISCHARGE LAMPS
- H01J1/00—Details of electrodes, of magnetic control means, of screens, or of the mounting or spacing thereof, common to two or more basic types of discharge tubes or lamps
- H01J1/02—Main electrodes
- H01J1/30—Cold cathodes, e.g. field-emissive cathode
- H01J1/312—Cold cathodes, e.g. field-emissive cathode having an electric field perpendicular to the surface, e.g. tunnel-effect cathodes of metal-insulator-metal [MIM] type
Definitions
- an oxidized or nitrided porous polycrystalline silicon layer is used as the electron transit layer.
- a collector is positioned opposed to the surface electrode while exposing the surface electrode to a vacuum space. Then, a DC current is applied between the surface and lower electrodes such that the surface electrode has a higher potential than that of the lower electrode, and simultaneously another DC current is applied between the collector and surface electrodes such that the collector has a higher potential than that of the surface electrode.
- electrons are injected from the lower electrode to the electron transit layer, and then emitted through the surface electrode after drifting within the electron transit layer.
- the surface electrode is typically made of chemically stable metal (e.g. noble metal such as gold) .
- the thickness of the surface electrode is set at about 10 nm.
- the terms "diode current Ips" and “emission current (emission electron current) Ie” generally mean a current flowing between the surface and lower electrodes and a current flowing between the collector and surface electrodes, respectively. Greater ratio of the emission current Ie to the diode current (Ie / Ips) provides higher electrode emitting efficiency [(Ie / Ips) x 100 (%)].
- the above electron source can emit electrons even if the DC voltage to be applied between the surface and lower electrodes is in a low range of about 10 to 20 V.
- the electron source can exhibit enhanced electron emission characteristics having a low dependence on vacuum degree, and emit electrons stably without occurrence of the so-called popping phenomenon during electron emission.
- the electron source disclosed in Japanese Patent Laid-Open Publication No. 2001-126610 comprises a surface electrode partially including a carbon region made of carbon or carbon compounds. This electron source can advantageously prevent excessive diode current Ips to provide enhanced electron emitting efficiency.
- the surface electrode is composed of a metal thin-film.
- the thickness of the surface electrode is set at 10 nm as in the foregoing electron source.
- a gold thin-film commonly used as the surface electrode is involved with agglomeration at a temperature of 400 °C or more.
- the resulting agglomeration deteriorates the thickness uniformity and continuity of the thin film, and thereby the electrical resistance of the surface electrode is increased, resulting in deteriorated electron emission characteristics.
- Tungsten and aluminum are known as metal having high resistibility to such agglomeration.
- the surface electrode is made of tungsten or aluminum, the surface of the electrode will be subject to oxidation, which leads to deteriorated electron emitting efficiency.
- the lower electrode has an increased electrical resistance. The following factors are assumed as the reason for the increased electrical resistance.
- a surface electrode includes a metal thin-film portion having a flat surface, and a plurality of island-shaped raised portions protruding from the surface of the metal thin-film portion, the raised portions being continuously and integrally formed with the thin-film portion.
- the respective metal raised portions of this electron sources are connected with each other by the metal thin-film portion, and thereby the film thickness of the metal thin-film portion will impose a restriction on a lower limit of the electrical resistance of the surface electrode.
- the thickness of the metal thin-film portion cannot be sufficiently reduced or the electron emitting efficiency cannot be sufficiently enhanced.
- WO0074098 discloses a conductive material with a wider forbidden band than silicon is used as a material of an upper electrode of a thin-film electron source.
- preferable materials include a conductive oxide such as SnO2 and ITO film, and semiconductor with a wide forbidden band such as GaN and SiC.
- the electron source of the present invention can provide enhanced thermal resistance of the surface electrode and/or the lower electrode while suppressing the deterioration of electron emission characteristics. This makes it possible to prevent electron emission characteristics from being deteriorated due to a thermal process such as a vacuum-sealing process.
- an electron source 10 includes a substrate 1 composed of an insulative substrate (e.g. glass substrate or ceramic substrate), and a lower electrode 2 made of layer-structured conductive carbide and formed on one (diffusing surface) of the principal surfaces of the substrate 1.
- a semiconductor layer or a non-doped polycrystalline silicon layer 3 is formed on the lower electrode 2.
- An electron transit layer 6 strong field drift layer
- a surface electrode 7 made of layer-structured conductive carbide is formed on the electron transit layer 6.
- the surface electrode 7 and the lower electrode 2 are disposed opposed to one another, and the electron transit layer 6 is interposed between the surface electrode 7 and the lower electrode 2.
- the thickness of the surface electrode 7 is set at 10 nm or less.
- the first embodiment employs the insulative substrate as the substrate 1
- any other suitable semiconductor substrate such as a silicon substrate may be used as the substrate 1.
- the polycrystalline silicon layer 3 is interposed between the electron transit layer 6 and the lower electrode 2 in the first embodiment, the electron transit layer 6 may be formed directly on the lower electrode 2 without interposing the polycrystalline silicon layer 3.
- a collector 21 may be positioned opposed to the surface electrode 7, as shown in Fig. 2 . Then, after evacuating the space between the surface electrode 7 and the collector 21 to provide a vacuum space, a DC voltage Vps is applied between the surface electrode 7 and the lower electrode 2 such that the surface electrode 7 has a higher potential than that of the lower electrode 2, and simultaneously a DC voltage Vc is applied between the collector 21 and the surface electrode 7 such that the collector 21 has a higher potential than that of the surface electrode 7.
- Vps DC voltage
- Vc DC voltage
- greater ratio (Ie / Ips) of the emission current Ie flowing between the collector 21 and the surface electrode 7 to the diode current Ips flowing between the surface electrode 7 and the lower electrode 2 provides higher electron emitting efficiency.
- the lower electrode 2 and the surface electrode 7 is made of layer-structured conductive carbide.
- the electron source according to the first embodiment can provide enhanced thermal resistance of the lower electrode 2 and the surface electrode 7 while suppressing the deterioration of electron emission characteristics. This makes it possible to prevent not only the increase of electrical resistance in the lower electrode 2 and the surface electrode 7 due to a thermal process such as a vacuum-sealing process but also agglomeration in the surface electrode 7, and thereby to avoid undesirable deterioration of electron emission characteristics.
- the lower electrode 2 made of the conductive carbide can prevent respective atoms constituting the substrate 1 and the semiconductor layer 3 (the electron transit layer 6 in case of omitting the semiconductor layer 3) from being diffused therebetween.
- the materials of the substrate 1 and the semiconductor layer 3 (the electron transit layer 6 in case of omitting the semiconductor layer 3) can be selected from various alternatives.
- the surface electrode 7 made of the conductive carbide can prevent respective atoms constituting the surface electrode 7 and the electron transit layer 6 from being diffused therebetween.
- the materials of the surface electrode 7 and the electron transit layer 6 can be selected from various alternatives.
- each of the lower electrode 2 and the surface electrode 7 is entirely made of layer-structured conductive carbide.
- at least a portion of each of the lower electrode 2 and the surface electrode 7 may be made of layer-structured conductive carbide, and the remaining potion may be made of metal such as Cr, W, Ti, Al, Cu, Au, Pt or Mo; alloy containing such metal; or semiconductor material such as impurity-doped polycrystalline silicon.
- either one of the lower electrode 2 and the surface electrode 7 may be devoid of the conductive carbide.
- a porous region is provided up to a given depth of the polycrystalline silicon layer 3 by means of an anodizing process.
- an anodizing process a processing bath is used which contains an electrolytic solution consisting of a mixture prepared by mixing a water solution containing 55 wt% of hydrogen fluoride with ethanol at the ratio of about 1 : 1.
- a lamp annealing apparatus is used in the rapid heating method for the oxidation process.
- a furnace is kept in O 2 gas atmosphere.
- the substrate is heated from room temperature up to a given oxidation temperature (e.g. 900 °C) at a specific programming rate (e.g. 80 °C/sec).
- the substrate is maintained at the oxidation temperature only for a given oxidation time-period (e.g. one hour) to perform a rapid thermal oxidation (RTO) processing.
- RTO rapid thermal oxidation
- the oxidation process is not limited to the rapid heating method.
- the electron transit layer 6 including the grains 51, the silicon nanocrystals 63 and the silicon oxide films 52, 64 may be formed by using an oxidation-processing bath containing an electrolytic solution (e.g. 1 mole/L of H 2 SO 4 , 1 mole/L of HNO 3 , or aqua regia), and applying a constant current between a platinum electrode (not shown) and the lower electrode 2 to electrochemically oxidize the porous polycrystalline silicon layer 4.
- an electrolytic solution e.g. 1 mole/L of H 2 SO 4 , 1 mole/L of HNO 3 , or aqua regia
- an existing sputtering apparatus can be readily diverted to an apparatus for forming the conductive carbide layer, which provides a downsized equipment investment and a reduced equipment cost.
- the electron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics can be provided at a lower cost.
- the conductive carbide layer can be formed at a relatively low process temperature.
- the film forming method for the conductive carbide layer may further include a method of annealing a metal film (e.g. titanium film, zirconium film or hafnium film) under gas atmosphere including carbon. This method is advantageously to improve the purity and film quality of the conductive carbide layer.
- the film forming method for the metal film may include a CVD method, vacuum evaporation method and sputtering method. In view of productivity and film quality, it is preferable to use a DC magnetron sputtering method using Ar gas. For example, when CH 4 gas is used as the gas including carbon, the conductive carbide layer can be formed by annealing the metal film at a temperature of 1000 °C .
- chloride such as titanium chloride, zirconium chloride or hafnium chloride
- a mixed gas of hydrogen and gas including carbon atom such as CH 4 (methane), C 3 H 8 (propane), C 6 H 5 CH 3 (toluene), CCl 4 (carbon tetrachloride) or CH 3 OH (methanol) may be used as the reactive gas.
- a thermal CVD method involves a process temperature of 600 °C or more, it is preferable to use the plasma CVD method capable of lowering the process temperature.
- the conductive carbide layer may be formed by annealing a multilayer consisting of a metal film (e.g. titanium film) and a carbon film.
- the conductive carbide layer may be formed by annealing a film made of a mixture of metal (e.g. titanium) and carbon.
- the electron transit layer 6 is composed of the oxidized porous polycrystalline silicon layer.
- the electron transit layer 6 may be composed of a nitrided porous polycrystalline silicon layer or an oxynitrided porous polycrystalline silicon layer.
- the electron transit layer 6 may be composed of any other suitable oxidized, nitrided or oxynitrided porous semiconductor layer.
- a nitriding process of nitriding the porous polycrystalline silicon layer 4 through a rapid heating method using NH 3 gas may be employed, as a substitute for the oxidation process of oxidizing the layer 4 through the rapid heating method using O 2 gas.
- an oxynitriding process of oxynitriding the porous polycrystalline silicon layer 4 through a rapid heating method using a mixed gas of O2 gas and NH3 gas may be employed, as a substitute for the oxidation process of oxidizing the layer 4 through the rapid heating method using 02 gas.
- a silicon oxynitride film will be formed instead of the silicon oxide films 52, 64 in Fig. 3 .
- an electron source 10 comprises a substrate 1 composed of an insulative substrate (e.g. insulative glass substrate or insulative ceramic substrate), and an electron source device 10a formed on one of the principal surfaces of the substrate 1.
- the electron source device 10a comprises a lower electrode 2 formed on the selected principal surface of the substrate 1, a semiconductor layer or a non-doped polycrystalline silicon layer 3 formed on the lower electrode 2, an after-mentioned electron transit layer 6 formed on the polycrystalline silicon layer 3, and a surface electrode 7 formed on the electron transit layer 6.
- the surface electrode 7 and the lower electrode 2 are disposed opposed to one another, and the electron transit layer 6 is interposed between the surface and lower electrodes.
- the thickness of the lower electrode 2 is set at about 300 nm, and the thickness of the surface electrode 7 is set such that it does not exceed 10 nm.
- the second embodiment employs the insulative substrate as the substrate 1
- any other suitable semiconductor substrate such as a silicon substrate may be used as the substrate 1
- the lower electrode 2 may be comprised of the semiconductor substrate and a conductive layer (e.g. ohmic electrode) deposited on the semiconductor substrate.
- the polycrystalline silicon layer 3 is interposed between the electron transit layer 6 and the lower electrode 2 in the first embodiment, the electron transit layer 6 may be formed directly on the lower electrode 2 without interposing the polycrystalline silicon layer 3.
- the lower electrode 2 is composed of a single layer thin-film made of a metal (e.g. Mo, Cr, W, Ti, Ta, Ni, Al, Cu, Au or Pt; alloy containing such metal; or intermetallic compound such as silicide).
- a metal e.g. Mo, Cr, W, Ti, Ta, Ni, Al, Cu, Au or Pt; alloy containing such metal; or intermetallic compound such as silicide.
- the lower electrode 2 may be composed of a multilayer thin-film made of such a metal.
- the lower electrode 2 may be made of a semiconductor material such as impurity-doped polycrystalline silicon.
- the electron transit layer 6 is formed by subjecting the polycrystalline silicon layer to a anodizing process and an oxidation process as described later.
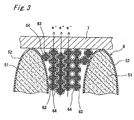
- the electron transit layer 6 in the second embodiment includes grains 51, silicon oxide films 52, silicon nanocrystals 63, and silicon oxide films 64 (see Fig. 3 ).
- the remaining region other than the grains 51, silicon nanocrystals 63 and silicon oxide films 52, 64 is formed as an amorphous region made of amorphous silicon or partially oxidized amorphous silicon.
- the polycrystalline silicon and the number of silicon nanocrystals 63 residing adjacent to the grain boundary of the polycrystalline silicon are mixed together within the electron transit layer 6.
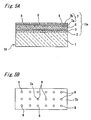
- the surface electrode 7 is composed of a multilayer film having a conductive carbide or conductive nitride layer 7a deposited on the electron transit layer 6 and a noble metal layer 7b deposited on the conductive carbide or conductive nitride layer 7a.
- the surface of the multilayer film is formed with a number of concave portions 8 allowing the multilayer film to have a locally reduced thickness.
- the thickness of the conductive carbide or conductive nitride layer 7a is set such that it does not exceed 4 nm
- the total thickness of the conductive carbide or conductive nitride layer 7a and the noble metal layer 7b is set such that it does not exceed 10 nm.
- the surface of the surface electrode 7 is formed with the number of concave portions 8. In the region having the concave portions 8, the surface of the conductive carbide or conductive nitride layer 7a is exposed to outside. That is, the depth of the concave portion 8 is approximately equal to the thickness of the noble metal layer 7b. However, the depth of the concave portion 8 may be less than, for example about one-half of, the thickness of the noble metal layer 7b.
- the glass substrate When a glass substrate is used as the substrate 1, the glass substrate may be appropriately selected from a silica glass substrate, no-alkali glass substrate, low-alkali glass substrate or soda-lime glass substrate, depending on a process temperature.
- the substrate 1 In case of employing a ceramic substrate, the substrate 1 may be composed, for example, of an alumina substrate.
- the electron source 10 according to the first embodiment may be used as an electron source for display devices. In this case, a suitable patterning of the lower electrode 2, the surface electrode 7 and the electron transit layer 6 may be appropriately selected.
- a lower electrode 2 composed of a metal film (e.g. molybdenum film) having a given film thickness (e.g. about 300 nm) is first formed on one of the principal surfaces of a substrate 1 composed of a silica glass substrate. Then, a non-doped polycrystalline silicon layer 3 having a given film thickness (e.g. 1.5 ⁇ m) is formed on the lower electrode 2 to provide the structure as shown in Fig. 6A .
- the lower electrode 2 may be formed, for example, through a sputtering method or a CVD method.
- the lower electrode 2 may be formed through a method of forming a non-doped polycrystalline silicon layer and then doping an n-type impurity into the polycrystalline silicon layer by means of a thermal diffusion method, or another method of forming a non-doped polycrystalline silicon layer and simultaneously doping an n-type impurity into the polycrystalline silicon layer (i.e. forming an conductive polycrystalline silicon layer directly on the substrate 1 without using an ion-implantation method or thermal diffusion method) .
- an anodizing process is performed to form a porous polycrystalline silicon layer 4 in which polycrystalline silicon grains 51 (see Fig. 3 ), silicon nanocrystals 63 (see Fig. 3 ) and amorphous silicons are mixed together.
- the structure as shown in Fig. 6B is obtained.
- the procedure for performing the anodizing process is as same as that of the first embodiment.
- an oxidation process is performed to form an electron transit layer 6 composed of the composite nanocrystal layer having the structure as shown in Fig. 3 .
- an oxidation process is performed to form an electron transit layer 6 composed of the composite nanocrystal layer having the structure as shown in Fig. 3 .
- the structure as shown in Fig. 6C is obtained.
- the porous polycrystalline silicon layer 4 is oxidized, for example, through a rapid heating method to form the electron transit layer 6 (see Fig. 3 ) including the grains 51, the silicon nanocrystals 63, and the silicon oxide films 52, 64.
- the procedure for performing the oxidation process is as same as that of the first embodiment.
- a conductive carbide or conductive nitride layer 7a having a given film thickness (appropriately selected in the range of 1 nm to 4 nm) and a noble metal layer 7b having a given film thickness (e.g. 3 nm) are sequentially formed on the electron transit layer 6, for example, through a sputtering method.
- a multi layer composed of the conductive carbide or conductive nitride layer 7a and the noble metal layer 7b is formed, and the structure as shown in Fig. 6D is obtained.
- a method for forming the conductive carbide or conductive nitride layer 7a may be selected from various thin-film forming methods such as a sputtering method (e.g. RF sputtering method, RF magnetron sputtering method, DC sputtering method, DC magnetron sputtering method, or reactive sputtering method), a vacuum evaporation method, and a CVD method.
- a sputtering method e.g. RF sputtering method, RF magnetron sputtering method, DC sputtering method, DC magnetron sputtering method, or reactive sputtering method
- a vacuum evaporation method e.g. a sputtering method
- a vacuum evaporation method e.g. a vacuum evaporation method, and a CVD method.
- the conductive carbide or conductive nitride layer 7a may be formed through a method of depositing a metal film on the electron transit layer 6 by means of the sputtering method or vacuum evaporation method and then annealing the metal film under gas atmosphere including carbon or nitrogen, or through another method of depositing a metal film on the electron transit layer 6 by means of the sputtering method or vacuum evaporation method and then implanting carbon ion or nitrogen ion into the metal film.
- a method for forming the noble metal layer 7b may be selected from various thin-film forming methods such as a sputtering method (e.g. RF sputtering method, RF magnetron sputtering method, DC sputtering method or DC magnetron sputtering method), a vacuum evaporation method, and a CVD method.
- a sputtering method e.g. RF sputtering method, RF magnetron sputtering method, DC
- a number of concave portions are formed on the multilayer composed of the conductive carbide or conductive nitride layer 7a and the noble metal layer 7b, and the electron source 10 having the structure as shown in Fig. 6E is obtained.
- the multilayer is subjected to a heat treatment at a temperature in the range of 300 °C to 450 °C for a given time-period (e.g. 15 minutes to 120 minutes) under nitrogen atmosphere.
- the concave portion can be formed at a relatively low process temperature.
- the heat treatment is performed under nitrogen atmosphere in this heat treatment process, it may be performed under any other suitable inert gas atmosphere or in vacuum.
- a usable heat treatment may include an annealing using an electric furnace, an annealing based on light irradiation (e.g. lamp annealing), and a laser annealing.
- the manufacturing process of the electron source 10 according to the second embodiment can facilitate manufacturing the electron source 10 having enhanced electron emitting efficiency with suppressing the increase of electrical resistance in the surface electrode 7.
- the oxidation process may be substituted with a nitriding process or an oxynitriding process as same as the case of the first embodiment.
- the electron transit layer 6 is composed of the composite nanocrystal layer as shown in Fig. 3 .
- the electron transit layer 6 may be composed of an insulative layer made, for example, of Al2O3, Si02 or the like.
- the electron source provided with a semiconductor layer can operate as with an electron source having the MIS structure.
- the electron source provided with no semiconductor layer can operate as with an electron source having the MIM structure.
- electron emission characteristics can be improved by appropriately setting the thickness of the electron transit layer.
- the forming process of the electron transit layer can be simplified.
- an electron source 10 comprises a substrate 1 composed of an insulative substrate (e.g. insulative glass substrate or insulative ceramic substrate), and an electron source device 10a formed on one of the principal surfaces of the substrate 1.
- the electron source device 10b comprises a lower electrode 2 formed on the selected principal surface of the substrate 1, a semiconductor layer or a non-doped polycrystalline silicon layer 3 formed on the lower electrode 2, an after-mentioned electron transit layer 6 formed on the polycrystalline silicon layer 3, and a surface electrode 7 formed on the electron transit layer 6.
- the surface electrode 7 and the lower electrode 2 are disposed opposed to one another, and the electron transit layer 6 is interposed between the surface and lower electrodes.
- the thickness of the lower electrode 2 is set at about 300 nm, and the thickness of the surface electrode 7 is set such that it does not exceed 10 nm.
- the third embodiment employs the insulative substrate as the substrate 1
- any other suitable semiconductor substrate such as a silicon substrate may be used as the substrate 1
- the lower electrode 2 may be comprised of the semiconductor substrate and a conductive layer (e.g. ohmic electrode) deposited on the semiconductor substrate.
- the polycrystalline silicon layer 3 is interposed between the electron transit layer 6 and the lower electrode 2, the electron transit layer 6 may be formed directly on the lower electrode 2 without interposing the polycrystalline silicon layer 3.
- the lower electrode 2 is composed of a thin film made of the same metal as that of the lower electrode 2 of the electron source according to the second embodiment.
- the electron transit layer 6 is formed by subjecting the polycrystalline silicon layer to a anodizing process and an oxidation process as described later.
- the electron transit layer 6 in the third embodiment includes grains 51, silicon oxide films 52, silicon nanocrystals 63, and silicon oxide films 64 as with the electron transit layer 6 in the first embodiment (see Fig. 3 ), and the remaining region other than the grains 51, silicon nanocrystals 63 and silicon oxide films 52, 64 is formed as an amorphous region made of amorphous silicon or partially oxidized amorphous silicon as with the electron transit layer 6 in the second embodiment.
- the surface electrode 7 is composed of a conductive nitride layer 7c deposited on the electron transit layer 6 and a metal layer 7d deposited on the conductive nitride layer 7c.
- the conductive nitride layer 7c is made of conductive nitride consisting of titanium nitride.
- the conductive nitride has a relatively high conductivity, a melting point higher than that of noble metal such as gold, and an excellent diffusion barrier performance. In addtion, the conductive nitride has an oxidation resistance superior to that of metal such as tungsten or aluminum.
- the conductive nitride having these properties usable in the conductive nitride layer 7c is not limited to titanium nitride, and it may be chromium nitride, molybdenum nitride, tungsten nitride, vanadium nitride, niobium nitride, tantalum nitride, zirconium nitride, or hafnium nitride. In view of thermal stability, work function and reproducibility, it is preferable to use either one of titanium nitride, zirconium nitride, hafnium nitride, niobium nitride and tantalum nitride.
- the metal layer 7d is made of platinum which is one of noble metals.
- the material of the metal layer 7d is not limited to platinum, and the metal layer 7d may be made of any other suitable noble metal such as gold or iridium. However, platinum is particularly effective to assure thermal stability.
- the thickness of the conductive nitride layer 7c is set such that it does not exceed 4 nm, and the total thickness of the conductive nitride layer 7c and the metal layer 7d is set such that it does not exceed 10 nm, in order to suppress the deterioration of electron emitting efficiency or provide other advantages.
- the electron source 10 or the electron source device 10b as a ballistic electron surface-emitting type electron source according to the third embodiment steps of emitting electrons or an electron emission model is the same as that of the electron source 10 according to the first embodiment (see Figs. 2 and 3 ).
- the electron source 10 according to the third embodiment exhibits enhanced electron emission characteristics having a low dependence on vacuum degree and free from the popping phenomenon, and thereby can emit electrons stably.
- the glass substrate may be substituted with any other suitable substrate in the same manner as that in the second embodiment.
- the electron source 10 according to the third embodiment may also be used as an electron source for display devices in the same manner as that in the second embodiment.
- a lower electrode 2 composed of a metal film (e.g. molybdenum film) having a given film thickness (e.g. about 300 nm) is first formed on one of the principal surfaces of a substrate 1 composed of a silica glass substrate. Then, a non-doped polycrystalline silicon layer 3 having a given film thickness (e.g. 1.5 ⁇ m) is formed on the lower electrode 2 to provide the structure as shown in Fig. 8A .
- the procedure for forming the lower electrode 2 and the procedure for forming the non-doped polycrystalline silicon layer 3 are as same as those of the second embodiment.
- an anodizing process is performed to form a porous polycrystalline silicon layer 4 in which polycrystalline silicon grains 51 (see Fig. 3 ), silicon nanocrystals 63 (see Fig. 3 ) and amorphous silicons are mixed together.
- the structure as shown in Fig. 8B is obtained.
- the procedure for performing the anodizing process is as same as that of the first embodiment.
- an oxidation process is performed to form an electron transit layer 6 composed of the composite nanocrystal layer having the structure as shown in Fig. 3 .
- the structure as shown in Fig. 8C is obtained.
- the composite nanocrystal layer 4 is oxidized, for example, through a rapid heating method to form the electron transit layer 6 (see Fig. 3 ) including the grains 51, the silicon nanocrystals 63, and the silicon oxide films 52, 64.
- the procedure for performing the oxidation process is as same as that of the first embodiment.
- a conductive nitride layer 7c having a given film thickness (e.g. 1 nm) and a metal layer 7d having a given film thickness (e.g. 3 nm) are sequentially formed on the electron transit layer 6, for example, through a sputtering method.
- a surface electrode 7 composed of the conductive nitride layer 7c and the metal layer 7d is formed, and the electron source 10 having the structure as shown in Fig. 8D is obtained.
- the manufacturing process of the electron source 10 according to the third embodiment can provide the electron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics.
- the conductive nitride layer 7c of the surface electrode 7 is formed through a sputtering method.
- a usable sputtering method may be selected from various types such as an RF sputtering method, RF magnetron sputtering method, DC sputtering method and DC magnetron sputtering method.
- the conductive nitride layer 7c is formed through a sputtering method using a target consisting of conductive nitride. This method can provide the conductive nitride layer 7c having a low surface roughness with excellent reproducibility and stability as well as a high throughput, which achieves a reduced manufacturing cost and an increased electron emitting area of the electron source 10.
- an existing sputtering apparatus can be readily diverted to an apparatus for forming the conductive nitride layer 7c, which provides a downsized equipment investment and a reduced equipment cost.
- the electron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics can be provided at a lower cost.
- the conductive nitride 7c layer can be formed at a relatively low process temperature.
- the method for forming the conductive nitride layer 7c is not limited to the aforementioned sputtering methods.
- the conductive nitride layer may be formed through a vacuum evaporation method using a deposition source consisting of conductive nitride, or a reactive sputtering method using a target consisting of metal (pure metal) and gas (reactive gas) including nitrogen.
- the vacuum evaporation method or the reactive sputtering method can also provide the conductive nitride layer 7c having a low surface roughness with excellent reproducibility and stability as well as a high throughput, which achieves a reduced manufacturing cost and an increased electron emitting area of the electron source 10.
- the electron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics can be provided at a lower cost.
- the conductive nitride layer can be formed at a relatively low process temperature.
- an existing vacuum evaporation apparatus can be readily diverted to an apparatus for forming the conductive nitride layer 7c, which provides a downsized equipment investment and a reduced equipment cost.
- the reactive sputtering method using a target consisting of metal (pure metal) and gas (reactive gas) including nitrogen can facilitate the control of nitrogen concentration in the conductive nitride layer 7c. That is, in the reactive sputtering method, a type of the reaction gas and a mixing ratio of inert gas such as Ar to the reactive gas including nitrogen atoms can be selected to control the composition of nitrogen in the conductive nitride layer 7c. Thus, the reactive sputtering method can provide the conductive nitride layer 7c having a nitrogen composition controlled at a desired value.
- the conductive nitride layer 7c may be formed through a CVD method.
- the CVD method can also provide the conductive nitride layer 7c having a low surface roughness with excellent reproducibility and stability as well as a high throughput, which achieves a reduced manufacturing cost and an increased electron emitting area of the electron source 10.
- the electron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics can be provided at a lower cost.
- the CVD method is excellent in a step-coverage performance. Since a thermal CVD method typically involves a process temperature of 600 °C or more, it is preferable to use the plasma CVD method capable of lowering the process temperature.
- the conductive nitride layer 7c may be formed by depositing a metal film (e.g. titanium film) on the electron transit layer 6 through a vacuum evaporation method or a sputtering method and then implanting nitrogen ions into the metal film.
- a metal film e.g. titanium film
- This method can also form the conductive nitride layer 7c having a low surface roughness with excellent reproducibility and stability.
- this method can reduce the roughness of the interface between the conductive nitride layer 7c and the metal layer 7b, which achieves a reduced manufacturing cost and an increased electron emitting area of the electron source 10.
- the electron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics can be provided at a lower cost. Further, this method can form the conductive nitride layer 7c at a relatively low process temperature, and facilitate the control of conductivity in the conductive nitride layer 7c.
- the lower electrode 2 may be formed to have a multilayer structure, and at least one of the layers may be formed as a conductive nitride layer.
- the conductive nitride has a relatively high conductivity, a melting point higher than that of noble metal such as gold, an excellent diffusion barrier performance, and an oxidation resistance superior to that of metal such as tungsten or aluminum.
- the lower electrode 2 having the conductive nitride layer can have enhanced thermal resistance with suppressing the deterioration of electron emission characteristics to prevent the deterioration of electron emission characteristics due to a thermal process such as the vacuum-sealing process.
- the conductive nitride layer can provide enhanced thermal resistance of the lower electrode 2.
- another layer deposited on the conductive nitride layer can be arranged to provide reduced electric resistance of the lower electrode 2, and enhanced adhesiveness between the lower electrode 2 and a layer forming an interface together with the lower electrode 2. It is to be understood that the entire lower electrode 2 may be composed of a conductive nitride layer.
- the conductive nitride layer used in the lower electrode 2 may be made of the same conductive nitride as the material used in the conductive nitride layer 7c of the surface electrode 7. Further, the conductive nitride layer of the lower electrode 2 may be formed through the same method as that for forming the conductive nitride layer 7c of the surface electrode 7.
- the electron source 10 as a ballistic electron surface-emitting type electron source steps of emitting electrons or an electron emission model is the same as that of the electron source 10 according to the first embodiment (see Figs. 2 and 3 ).
- the electron source 10 according to the fourth embodiment exhibits enhanced electron emission characteristics having a low dependence on vacuum degree and free from the popping phenomenon, and thereby can emit electrons stably.
- the surface electrode 7 is composed of a conductive carbide layer 7e deposited on the electron transit layer 6 and a noble metal layer 7f deposited on the conductive carbide layer 7e.
- the conductive carbide layer 7e is made of conductive carbide consisting of zirconium carbide.
- the conductive carbide has a relatively high conductivity, a relatively low work function, a melting point higher than that of noble metal such as gold, and an excellent diffusion barrier performance.
- the conductive carbide has an oxidation resistance superior to that of metal such as tungsten or aluminum.
- the conductive carbide having these properties usable in the conductive carbide layer 7e is not limited to zirconium carbide, and it may be chromium carbide, molybdenum carbide, tungsten carbide, vanadium carbide, niobium carbide, tantalum carbide, titanium carbide, or hafnium carbide. In view of thermal stability, work function and reproducibility, it is preferable to use either one of zirconium carbide, titanium carbide and hafnium carbide.
- the noble metal layer 7f is made of platinum which is one of noble metals (That is, the noble metal layer 7f is composed of a layer-structured platinum layer).
- the noble metal to be used is not limited to platinum, and the noble metal layer 7f may be made of any other suitable noble metal such as gold or iridium.
- a portion of the noble metal layer 7f may be composed of a layer-structured platinum layer, and the remaining portion may be made of different noble metal other than platinum.
- the surface electrode 7 composed of the conductive carbide layer 7e and the noble metal layer 7f can prevent respective atoms constituting the surface electrode 7 and the electron transit layer 6 from being diffused therebetween.
- the materials of the surface electrode 7 and the electron transit layer 6 can be selected from various alternatives.
- a non-doped polycrystalline silicon layer having a given film thickness (e.g. 1 ⁇ m) is first formed on one of the principal surfaces of the substrate 1, and an n-type impurity is doped into the polycrystalline silicon layer through an ion-implantation method or heat treatment to form a lower electrode 2 composed of a conductive polycrystalline silicon layer (n-type polycrystalline silicon layer). Then, an additional non-doped polycrystalline silicon layer 3 having a given film thickness (e.g. 1.5 ⁇ m) is formed on the side of the selected principal surface of the substrate 1 (on the lower electrode 2) to provide the structure as shown in Fig. 10A .
- the procedure for forming the non-doped polycrystalline silicon layer 3 is as same as that of the second embodiment.
- the lower electrode 2 is made of metal such as Cr, W, Ti, Al, Cu, Au, Pt or Mo, or alloy containing such meta, the lower electrode 2 may be formed, for example, through a sputtering method or vacuum evaporation method.
- a porous region is provided up to a given depth of the polycrystalline silicon layer 3 by means of an anodizing process.
- a porous semiconductor layer or a porous polycrystalline silicon layer 4 is formed, and thus the structure as shown in Fig. 10B is obtained.
- the procedure for performing the anodizing process is as same as that of the first embodiment.
- the porous polycrystalline silicon layer 4 is oxidized through an oxidation process. Through this step, an electron transit layer 6 composed of the oxidized porous polycrystalline silicon layer is formed, and thus the structure as shown in Fig. 10C is obtained.
- the porous polycrystalline silicon layer 4 is oxidized, for example, through a rapid heating method to form the electron transit layer 6 including the grains 51, the silicon nanocrystals 63, and the silicon oxide films 52, 64 (see Fig. 3 ).
- the procedure for performing the oxidation process is as same as that of the first embodiment.
- a conductive carbide layer 7e and a noble metal layer 7f are sequentially formed, for example, through a sputtering method. Through this step, a surface electrode 7 composed of the conductive carbide layer 7e and the noble metal layer 7f is formed on the electron transit layer 6, and thus the electron source 10 having the structure as shown in Fig. 10D is obtained.
- the manufacturing process of the electron source 10 according to the fourth embodiment can provide the electron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics.
- the conductive carbide layer 7e of the surface electrode 7 is formed through a sputtering method.
- a usable sputtering method may be selected from various types such as an RF sputtering method, RF magnetron sputtering method, DC sputtering method and DC magnetron sputtering method.
- the conductive carbide layer 7e is formed through a sputtering method using a target consisting of conductive carbide. These methods can provide the conductive carbide layer 7e having a low surface roughness with excellent reproducibility and stability as well as a high throughput, which achieves a reduced manufacturing cost and an increased electron emitting area of the electron source 10.
- an existing sputtering apparatus can be readily diverted to an apparatus for forming the conductive carbide layer 7e, which provides a downsized equipment investment and a reduced cost.
- the electron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics can be provided at a lower cost.
- the conductive carbide layer 7e can be formed at a relatively low process temperature.
- the electron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics can be provided at a lower cost.
- the conductive carbide layer 7e can be formed at a relatively low process temperature. In addtion, when the conductive carbide layer 7e is formed through the vacuum evaporation method, an existing vacuum evaporation apparatus can be readily diverted to an apparatus for forming the conductive carbide layer 7e, which provides a downsized equipment investment and a reduced equipment cost.
- An EB (Electron Beam) vacuum evaporation method is one of appropriate vacuum evaporation methods to be used, because conductive carbide has a high melting point.
- the reactive sputtering method using a target consisting of metal (pure metal) and gas (reactive gas) including carbon can facilitate the control of carbon concentration in the conductive carbide layer 7e. That is, in the reactive sputtering method, a type of the reactive gas or a mixing ratio of inert gas such as Ar to the reactive gas including carbon atoms can be appropriately selected to control a carbon concentration in the conductive carbide layer 7e so as to provide the conductive carbide layer 7e having a desired carbon concentration. Further, when using the reactive sputtering method, a desired film of titanium carbide, zirconium carbide or hafnium carbide can be formed at a relatively low substrate temperature of 150 to 500 °C.
- the reactive sputtering method can provide the conductive carbide layer 7e including a less amount of metal impurities as compared to the sputtering method using conductive carbide as a target, and thereby performance decrement due to impurities can be suppressed.
- the sputtering method, vacuum evaporation method or reactive sputtering method is more advantageous than other film forming methods to form such a conductive carbide layer.
- a silicon substrate was used as the substrate 1, an n-type polycrystalline silicon layer being used as the lower electrode 2, an oxidized porous polycrystalline silicon layer being used as the electron transit layer 6 (strong field drift layer), a zirconium carbide layer being used as the conductive carbide layer 7e of the surface electrode 7, and a platinum layer being used as the noble metal layer 7f of the surface electrode 7.
- the thickness of the conductive carbide layer 7e was 2 nm, and the thickness of the noble metal layer 7f was 4 nm.
- the conductive carbide layer 7e was formed through an RF magnetron sputtering method using a target consisting of zirconium carbide under Ar gas atmosphere.
- the electron source 10 was introduced into a vacuum chamber (not shown) to measure the electron emission characteristics of the electron source 10 in the state as shown in Fig. 2 . That is, the collector 21 was positioned opposed to the surface electrode 7. Then, a DC voltage Vps was applied between the surface and lower electrodes such that the surface electrode 7 had a higher potential than that of the lower electrode 7, while another DC voltage Vc was applied between the collector and surface and electrodes such that the collector 21 had a higher potential than that of the surface electrode 7.
- Fig. 11 shows the measurement result of electron emission characteristics in the state when vacuum degree in the vacuum chamber was set at 5 x 10 -5 Pa, and the DC voltage Vc was set at a constant value of 1000 V.
- the horizontal axis indicates the DC voltage Vps
- the vertical axis indicates the current density.
- the marks "A” indicate the current density of diode current Ips in the state before the heat treatment.
- the marks “B” indicate the current density of emission current Ie in the state before the heat treatment.
- the marks “C” indicate the current density of diode current Ips in the state after the heat treatment.
- the marks “D” indicate the current density of emission current Ie in the state after the heat treatment.
- the heat treatment was performed under N 2 gas atmosphere at 400 °C for 1 hour.
Landscapes
- Cold Cathode And The Manufacture (AREA)
Description
- The present invention relates to a field emission-type electron source for emitting electron beams by means of the field emission phenomenon.
- Heretofore, there has been well known in the art a field emission-type electron source (hereinafter referred to as "electron source" for brevity), as disclosed, for example, in Japanese Patent Publication No.
2987140 2001-126610 - The electron source disclosed in Japanese Patent Publication No.
2987140 - For example, an oxidized or nitrided porous polycrystalline silicon layer is used as the electron transit layer. In activating the electron source, a collector is positioned opposed to the surface electrode while exposing the surface electrode to a vacuum space. Then, a DC current is applied between the surface and lower electrodes such that the surface electrode has a higher potential than that of the lower electrode, and simultaneously another DC current is applied between the collector and surface electrodes such that the collector has a higher potential than that of the surface electrode. As a result of the above operation, electrons are injected from the lower electrode to the electron transit layer, and then emitted through the surface electrode after drifting within the electron transit layer. Alteration such as oxidation of the surface of the surface electrode causes deterioration in the efficiency of emitting electrons or the electron emitting efficiency. Thus, the surface electrode is typically made of chemically stable metal (e.g. noble metal such as gold) . In this case, the thickness of the surface electrode is set at about 10 nm.
- In electron sources, the terms "diode current Ips" and "emission current (emission electron current) Ie" generally mean a current flowing between the surface and lower electrodes and a current flowing between the collector and surface electrodes, respectively. Greater ratio of the emission current Ie to the diode current (Ie / Ips) provides higher electrode emitting efficiency [(Ie / Ips) x 100 (%)]. The above electron source can emit electrons even if the DC voltage to be applied between the surface and lower electrodes is in a low range of about 10 to 20 V. Thus, the electron source can exhibit enhanced electron emission characteristics having a low dependence on vacuum degree, and emit electrons stably without occurrence of the so-called popping phenomenon during electron emission.
- The lower electrode of the electron source comprises a semiconductor substrate having a resistivity relatively close to that of a conductor, and an ohmic electrode formed on the back surface of the semiconductor substrate. Alternatively, the lower electrode comprises an insulative substrate and a metal conductive layer formed on the insulative substrate.
- On the other hand, the electron source disclosed in Japanese Patent Laid-Open Publication No.
2001-126610 - The conventional electron sources as described above are used in a vacuum-sealed state. In this connection, an assembling process of such electron sources involves a relatively high-temperature thermal process including a vacuum-sealing process (which is performed at about 500 °C), which inevitably increases the electrical resistance of the surface electrode and/or the lower electrode. The increased electrical resistance makes it difficult for voltage to be adequately applied between the surface and lower electrodes and/or between the collector and surface electrodes, resulting in deteriorated electron emission characteristics (such as lowered emission current or electron emitting efficiency).
- The surface electrode is composed of a metal thin-film. The thickness of the surface electrode is set at 10 nm as in the foregoing electron source. A gold thin-film commonly used as the surface electrode is involved with agglomeration at a temperature of 400 °C or more. The resulting agglomeration deteriorates the thickness uniformity and continuity of the thin film, and thereby the electrical resistance of the surface electrode is increased, resulting in deteriorated electron emission characteristics. Tungsten and aluminum are known as metal having high resistibility to such agglomeration. However, if the surface electrode is made of tungsten or aluminum, the surface of the electrode will be subject to oxidation, which leads to deteriorated electron emitting efficiency.
- Similarly, the lower electrode has an increased electrical resistance. The following factors are assumed as the reason for the increased electrical resistance.
- (1) Agglomeration of metal
- (2) Decrease in the film thickness of the lower electrode due to thermal diffusion of atoms (metal atoms) constituting the lower electrode toward a layer (e.g. electron transit layer) deposited on the lower electrode
- (3) Decrease in the film thickness of the lower electrode due to thermal diffusion of atoms constituting a layer (e.g. electron transit layer) deposited on the lower electrode toward the lower electrode
- (4) Increase in resistivity of the lower electrode
- In addition to the aforementioned electron sources, various modified electron sources for emitting electrons by means of the field emission phenomenon have been proposed. For example, one of the proposed electron sources has a MIM (Metal-Insulator-Metal) structure including an insulating layer serving as the electron transit layer. Another electron source has a MIS (Metal-Insular-Semiconductor) including a semiconductor layer provided between the electron transit layer and the lower electrode.
- In view of industrial availability of the various electron sources, it is desired to provide increased emission current and enhanced electron emitting efficiency contributing to reduction in power consumption. In all of the above electron sources, electrons are emitted through the surface electrode. Thus, the electron emitting efficiency can be increased by reducing an energy loss due to electron scattering within the surface electrode. From this point of view, it is contemplable to reduce the thickness of the surface electrode insofar as device characteristics are not adversely affected. For example, Japanese Patent Laid-Open Publication No.
2001-243901 -
EP1117118 discloses an electron-emitting device includes an electron source layer made of a metal, a metal alloy or a semiconductor, an insulating layer formed on the electron source layer and a metal thin film electrode formed on the insulating layer. Electrons are emitted upon application of an electric field between the electron source layer and the metal thin film electrode. The insulating layer has at least one island region which constitutes an electron-emitting section in which the film thickness of the insulating layer is gradually reduced. -
WO0074098 - It is the object of the present invention to provide an electron source capable of suppressing the increase of electrical resistance in a surface electrode and achieving enhanced electron emission characteristics, whereby an electron source capable of suppressing deterioration of electron emission characteristics and achieving enhanced thermal resistance is provided.
- According to the present invention, there is provided an electron source comprising a lower electrode, a surface electrode, and an electron transit layer. The electron transit layer is interposed between the lower electrode and the surface electrode to allow an electron to pass therethrough. An electron passed through the electron transit layer is emitted through the surface electrode. The surface electrode is composed of a multilayer film including a conductive carbide or conductive nitride layer and a noble metal layer, the surface electrode having a surface formed with a number of concave portions allowing the multilayer film to have a locally reduced thickness, wherein the conductive carbide layer or conductive nitride layer of the surface electrode has a thickness of 4nm or less, while the total thickness of the conductive carbide or conductive nitride layer and the noble metal layer of the surface electrode does not exceed 10nm.
- In this electron source, an energy loss due to electron scattering will be reduced in the region of the surface electrode formed with the concave portions as comparted to other region. Further, electron emission characteristics can be enhanced while suppressing the increase of electrical resistance in the surface electrode because the electrical resistance of the surface electrode is substantially determined by the film thickness of the multilayer film composed of a conductive carbide layer or conductive nitride layer and a noble metal layer.
- The conductive carbide or conductive nitride of the surface electrode has a relatively high conductivity, a relatively low work function, a melting point higher than noble metal, and an excellent diffusion barrier performance. In addition, the conductive carbide or conductive nitride has an oxidation resistance superior to that of metal such as tungsten or aluminum. In the electron source of the present invention, at least a portion of the surface electrode and/or at least a portion of the lower electrode are made of layer-structured conductive carbide or layer-structured conductive nitride. Thus, as compared to an electron source having a surface electrode composed of a metal thin-film, the electron source of the present invention can provide enhanced thermal resistance of the surface electrode and/or the lower electrode while suppressing the deterioration of electron emission characteristics. This makes it possible to prevent electron emission characteristics from being deteriorated due to a thermal process such as a vacuum-sealing process.
- A more complete understanding of the invention will be apparent from the following detailed description when taken in conjunction with the accompanied drawings in which same elements have the same reference numerals, wherein:
-
Fig. 1 is a sectional view of an electron source according to a first embodiment which does not fall under the scope of the present invention; -
Fig. 2 is an explanatory diagram of a general electron emission operation of electron sources according to first to fourth embodiments; -
Fig. 3 is an explanatory diagram of a microscopic electron emission operation of the electron sources according to the first to fourth embodiments; -
Figs. 4A to 4D are sectional views of in-process and final products provided in major steps of a manufacturing process of the electron source according to the first embodiment, which does not fall under the scope of the present invention; -
Figs. 5A and 5B are sectional and top plan views of the electron source according to the second embodiment, which is an embodiment of - the present invention,
-
Figs. 6A to 6E are sectional views of in-process and final products provided in major steps of a manufacturing process of the electron source according to the second embodiment of the present invention; -
Fig. 7 is a sectional view of the electron source according to the third embodiment, which does not fall under the scope of the present invention; -
Figs. 8A to 8D are sectional views of in-process and final products provided in major steps of a manufacturing process of the electron source according to the third embodiment, which does not fall under the scope of the present invention ; -
Fig. 9 is a sectional view of the electron source according to the fourth embodiment, which does not fall under the scope of the present invention; -
Figs. 10A to 10D are sectional views of in-process and final products provided in major steps of a manufacturing process of the electron source according to the fourth embodiment, which does not fall under the scope of the present invention; and -
Fig. 11 is a graph showing the relationship between current density and voltage Vps before and after a heat treatment in the electron source according to the fourth embodiment, which does not fall under the scope of the present invention. - A first embodiment, which does not fall under the scope of the present invention, will now be described.
- As shown in
Fig. 1 , anelectron source 10 according to the first embodiment includes asubstrate 1 composed of an insulative substrate (e.g. glass substrate or ceramic substrate), and alower electrode 2 made of layer-structured conductive carbide and formed on one (diffusing surface) of the principal surfaces of thesubstrate 1. A semiconductor layer or a non-dopedpolycrystalline silicon layer 3 is formed on thelower electrode 2. An electron transit layer 6 (strong field drift layer) composed of an oxidized porous polycrystalline silicon layer is formed on thepolycrystalline silicon layer 3. Further, asurface electrode 7 made of layer-structured conductive carbide is formed on theelectron transit layer 6. In theelectron source 10, thesurface electrode 7 and thelower electrode 2 are disposed opposed to one another, and theelectron transit layer 6 is interposed between thesurface electrode 7 and thelower electrode 2. The thickness of thesurface electrode 7 is set at 10 nm or less. - While the first embodiment employs the insulative substrate as the
substrate 1, any other suitable semiconductor substrate such as a silicon substrate may be used as thesubstrate 1. Further, while thepolycrystalline silicon layer 3 is interposed between theelectron transit layer 6 and thelower electrode 2 in the first embodiment, theelectron transit layer 6 may be formed directly on thelower electrode 2 without interposing thepolycrystalline silicon layer 3. - For the purpose of allowing electrons to be emitted from the
electron source 10, acollector 21 may be positioned opposed to thesurface electrode 7, as shown inFig. 2 . Then, after evacuating the space between thesurface electrode 7 and thecollector 21 to provide a vacuum space, a DC voltage Vps is applied between thesurface electrode 7 and thelower electrode 2 such that thesurface electrode 7 has a higher potential than that of thelower electrode 2, and simultaneously a DC voltage Vc is applied between thecollector 21 and thesurface electrode 7 such that thecollector 21 has a higher potential than that of thesurface electrode 7. By appropriately setting the DC voltages Vps and Vc, electrons are injected from thelower electrode 2 to theelectron transit layer 6, and then emitted through thesurface electrode 7 after drifting within theelectron transit layer 6. The one-dot chain lines inFig. 2 indicate the flow of the electrons e- emitted through thesurface electrode 7. The electron reaching the surface of theelectron transit layer 6 can be considered as a hot electron. Thus, after readily tunneling through thesurface electrode 7, the electron will be emitted into the vacuum space. In the first embodiment, electrons pass through theelectron transit layer 6 under an electric field between thelower electrode 2 and thesurface electrode 7 which is induced when the voltage is applied between the lower and surface electrodes. The electrons are moved from thelower electrode 2 to thesurface electrode 7 through theelectron transit layer 6 by the electric field caused when thesurface electrode 7 has a higher potential than that of thelower electrode 2. - In the
electron source 10 according to the first embodiment, greater ratio (Ie / Ips) of the emission current Ie flowing between thecollector 21 and thesurface electrode 7 to the diode current Ips flowing between thesurface electrode 7 and thelower electrode 2 provides higher electron emitting efficiency. - As shown in
Fig. 3 , it can be assumed that theelectron transit layer 6, at least, includes a plurality of columnar polycrystalline silicon grains 51 (semiconductor crystals) disposed along with each other on the side of the selected principal surface of thesubstrate 1, a thinsilicon oxide film 52 formed on each surface of thegrains 51, a number of nano-order silicon nanocrystals 63 (semiconductor nanocrystals) interposed between theadjacent grains 51, and a number ofsilicon oxide films 64 each of which is formed on each surface of thesilicon nanocrystals 63 and is composed of an insulating film having a film thickness less than each crystal grain size of thesilicon nanocrystals 63. That is, in theelectron transit layer 6, each of the grains has a porous surface, and a crystalline state is maintained at each central region of the grains. Each of thegrains 51 extends in the direction perpendicular to thelower electrode 2 or in the thickness direction of thelower electrode 2. - In the
electron source 10 according to the first embodiment, the electron emission would be caused based on the following model. - A DC voltage Vps is applied between the
surface electrode 7 and thelower electrode 2 such that thesurface electrode 7 has a higher potential than that of thelower electrode 2, and simultaneously a DC voltage Vc is applied between thecollector 21 and thesurface electrode 7 such that thecollector 21 has a higher potential than that of thesurface electrode 7. When the DC voltage Vps reaches a given value (critical value), electrons e- are injected from thelower electrode 2 into theelectron transit layer 6 under thermal excitation. At the same time, the electric field applied to theelectron transit layer 6 mostly acts to thesilicon oxide films 64. Thus, the injected electrons e- are accelerated by the strong electric field acting to thesilicon oxide films 64. Then, the electrons drift toward the surface electrode or in the direction of the arrows inFig. 3 through the region between theadjacent grains 51 in theelectron transit layer 6. After tunneling through thesurface electrode 7, the electrons are emitted from thesurface electrode 7 to the vacuum space. The electrons injected from thelower electrode 2 into theelectron transit layer 6 are accelerated by the electric field acting tosilicon oxide films 64, and then emitted through thesurface electrode 7 after drifting approximately without scattering due to the silicon nanocrystals 63 (ballistic electron emission phenomenon). Further, heat generated at theelectron transit layer 6 can be released through thegrains 51. Thus, during electron emission, the electrons can be stably emitted without occurrence of the popping phenomenon. The electrons reaching the surface of the electron transit layer 6 (hot electrons) are emitted into the vacuum space after readily tunneling through thesurface electrode 7. The electron source based on such an operational principle is referred to as Ballistic electron Surface-emitting Device. - As described above, the
lower electrode 2 and thesurface electrode 7 is made of conductive carbide. Generally, the conductive carbide has a relatively high conductivity and a relatively low work function. The conductive carbide also has a melting point higher than that of noble metal such as gold, and an excellent diffusion barrier performance. In addition, the conductive carbide has an oxidation resistance superior to that of metal such as tungsten or aluminum. The conductive carbide having these properties includes chromium carbide, molybdenum carbide, tungsten carbide, vanadium carbide, niobium carbide, tantalum carbide, titanium carbide, zirconium carbide, and hafnium carbide. In view of thermal stability, work function and reproducibility, it is preferable to use titanium carbide, zirconium carbide or hafnium carbide. - In the
electron source 10 according to the first embodiment, thelower electrode 2 and thesurface electrode 7 is made of layer-structured conductive carbide. Thus, as compared to an electron source having lower and surface electrodes made of a metal, the electron source according to the first embodiment can provide enhanced thermal resistance of thelower electrode 2 and thesurface electrode 7 while suppressing the deterioration of electron emission characteristics. This makes it possible to prevent not only the increase of electrical resistance in thelower electrode 2 and thesurface electrode 7 due to a thermal process such as a vacuum-sealing process but also agglomeration in thesurface electrode 7, and thereby to avoid undesirable deterioration of electron emission characteristics. Further, thelower electrode 2 made of the conductive carbide can prevent respective atoms constituting thesubstrate 1 and the semiconductor layer 3 (theelectron transit layer 6 in case of omitting the semiconductor layer 3) from being diffused therebetween. Thus, the materials of thesubstrate 1 and the semiconductor layer 3 (theelectron transit layer 6 in case of omitting the semiconductor layer 3) can be selected from various alternatives. Similarly, thesurface electrode 7 made of the conductive carbide can prevent respective atoms constituting thesurface electrode 7 and theelectron transit layer 6 from being diffused therebetween. Thus, the materials of thesurface electrode 7 and theelectron transit layer 6 can be selected from various alternatives. - In the first embodiment, each of the
lower electrode 2 and thesurface electrode 7 is entirely made of layer-structured conductive carbide. Alternatively, at least a portion of each of thelower electrode 2 and thesurface electrode 7 may be made of layer-structured conductive carbide, and the remaining potion may be made of metal such as Cr, W, Ti, Al, Cu, Au, Pt or Mo; alloy containing such metal; or semiconductor material such as impurity-doped polycrystalline silicon. Further, either one of thelower electrode 2 and thesurface electrode 7 may be devoid of the conductive carbide. For example, thelower electrode 2 or thesurface electrode 7 may be made by using metal such as Cr, W, Ti, Al, Cu, Au, Pt or Mo; alloy containing such metal; or semiconductor material such as impurity-doped polycrystalline silicon. In this case, the deterioration of electron emission characteristics can also be suppressed as compared to the conventional electron sources. - When a glass substrate is used as the
substrate 1, the glass substrate may be appropriately selected from a silica glass substrate, no-alkali glass substrate, low-alkali glass substrate or soda-lime glass substrate, depending on a process temperature. In case of employing a ceramic substrate, thesubstrate 1 may be composed, for example, of an alumina substrate. Theelectron source 10 according to the first embodiment may be used as an electron source for display devices. In this case, a suitable patterning of thelower electrode 2, thesurface electrode 7 and theelectron transit layer 6 may be appropriately selected. - With reference to
Figs. 4A to 4D , a manufacturing process of theelectron source 10 according to the first embodiment will be described below. - A
lower electrode 2 composed of a conductive carbide layer is first formed on one of the principal surfaces of asubstrate 1 through a sputtering method or another suitable method. Then, a semiconductor layer or a non-dopedpolycrystalline silicon layer 3 is formed on the side of the selected principal surface of the substrate 1 (on thelower electrode 2 herein). Through the above step, the structure as shown inFig. 4A is obtained. A film forming method for a conductive carbide layer serving as thelower electrode 2 will be described later. Thepolycrystalline silicon layer 3 may be formed through various film forming methods such as a CVD method (e.g. LPCVD method, plasma CVD method or catalytic-CVD method), a sputtering method or a CGS (Continuous Grain Silicon) method. - After the non-doped
polycrystalline silicon layer 3 has been formed, a porous region is provided up to a given depth of thepolycrystalline silicon layer 3 by means of an anodizing process. Through this step, a porous semiconductor layer or a porouspolycrystalline silicon layer 4 is formed, and thus the structure as shown inFig. 4B is obtained. In the anodizing process, a processing bath is used which contains an electrolytic solution consisting of a mixture prepared by mixing a water solution containing 55 wt% of hydrogen fluoride with ethanol at the ratio of about 1 : 1. The anodizing process is performed by immersing a platinum electrode (not shown) and thelower electrode 2 into the processing bath, and applying a certain voltage between the platinum electrode and thelower electrode 2 at a constant current while irradiating thepolycrystalline silicon layer 3 with light, to form the porouspolycrystalline silicon layer 4. The obtained porouspolycrystalline silicon layer 4 includes a polycrystalline silicon grain and a silicon nanocrystal. While the porous region is formed in a portion (up to a given depth) of thepolycrystalline silicon layer 3 in the first embodiment, the porous region may be formed in the entire polycrystalline silicon layer 3 (up to a depth reaching the substrate 1). - After the completion of the anodizing process, the porous
polycrystalline silicon layer 4 is oxidized through an oxidation process. Through this step, anelectron transit layer 6 composed of the oxidized porous polycrystalline silicon layer is formed, and thus the structure as shown inFig. 4C is obtained. In the oxidation process, the porouspolycrystalline silicon layer 4 is oxidized, for example, through a rapid heating method to form theelectron transit layer 6 including thegrains 51, thesilicon nanocrystals 63, and thesilicon oxide films 52, 64 (seeFig. 3 ). - A lamp annealing apparatus is used in the rapid heating method for the oxidation process. A furnace is kept in O2 gas atmosphere. The substrate is heated from room temperature up to a given oxidation temperature (e.g. 900 °C) at a specific programming rate (e.g. 80 °C/sec). The substrate is maintained at the oxidation temperature only for a given oxidation time-period (e.g. one hour) to perform a rapid thermal oxidation (RTO) processing. Then, the substrate is cooled down to room temperature. The oxidation process is not limited to the rapid heating method. For example, the
electron transit layer 6 including thegrains 51, thesilicon nanocrystals 63 and thesilicon oxide films lower electrode 2 to electrochemically oxidize the porouspolycrystalline silicon layer 4. - After the
electron transit layer 6 has been formed, asurface electrode 7 composed of a conductive carbide layer is formed on theelectron transit layer 6, for example, through a sputtering method. Through this step, theelectron source 10 as shown inFig. 4D is obtained. A film forming method for the conductive carbide layer serving as thesurface electrode 7 will be described later in conjunction with the film forming method for thelower electrode 2. - The manufacturing process of the
electron source 10 according to the first embodiment can provide theelectron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics. - In the first embodiment, the
lower electrode 2 and thesurface electrode 7 each composed of the conductive carbide layer are formed through a sputtering method. A usable sputtering method may be selected from various types such as an RF sputtering method, RF magnetron sputtering method, DC sputtering method and DC magnetron sputtering method. In this case, the conductive carbide layer is formed through a sputtering method using a target consisting of conductive carbide. These methods can provide the conductive carbide layer with excellent stability and reproducibility as well as a high throughput, which achieves a reduced manufacturing cost and an increased electron emitting area of theelectron source 10. In addtion, an existing sputtering apparatus can be readily diverted to an apparatus for forming the conductive carbide layer, which provides a downsized equipment investment and a reduced equipment cost. Thus, theelectron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics can be provided at a lower cost. Further, the conductive carbide layer can be formed at a relatively low process temperature. - The film forming method for the conductive carbide layer is not limited to the aforementioned sputtering methods. For example, the conductive carbide layer may be 'formed through a vacuum evaporation method using a deposition source consisting of conductive carbide, or a reactive sputtering method using a target consisting of metal (pure metal) and gas (reactive gas) including carbon. The vacuum evaporation method or the reactive sputtering method can also provide the conductive carbide layer with excellent stability and reproducibility as well as a high throughput, which achieves a reduced manufacturing cost and an increased electron emitting area of the
electron source 10. Thus, theelectron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics can be provided at a lower cost. Further, the conductive carbide layer can be formed at a relatively low process temperature. In addtion, when the conductive carbide layer is formed through the vacuum evaporation method, an existing vacuum evaporation apparatus can be readily diverted to an apparatus for forming the conductive carbide layer, which provides a downsized equipment investment and a reduced equipment cost. - An EB (Electron Beam) vacuum evaporation method is one of appropriate vacuum evaporation methods to be used, because conductive carbide has a high melting point. On the other hand, the reactive sputtering method using a target consisting of metal (pure metal) and gas (reactive gas) including carbon can facilitate the control of carbon concentration in the conductive carbide layer. That is, in the reactive sputtering method, a type of the reactive gas or a mixing ratio of inert gas such as Ar to the reactive gas including carbon atoms can be appropriately selected to control a carbon concentration in the conductive carbide layer so as to provide the conductive carbide layer having a desired carbon concentration. Further, as compared to the carbide target, the pure metal target having a higher purity is widely placed on the market. Thus, the reactive sputtering method can provide the conductive carbide layer including a less amount of metal impurities as compared to the sputtering method using conductive carbide as a target, and thereby performance decrement due to impurities can be suppressed.
- The film forming method for the conductive carbide layer may further include a method of annealing a metal film (e.g. titanium film, zirconium film or hafnium film) under gas atmosphere including carbon. This method is advantageously to improve the purity and film quality of the conductive carbide layer. The film forming method for the metal film may include a CVD method, vacuum evaporation method and sputtering method. In view of productivity and film quality, it is preferable to use a DC magnetron sputtering method using Ar gas. For example, when CH4 gas is used as the gas including carbon, the conductive carbide layer can be formed by annealing the metal film at a temperature of 1000 °C . Thus, as with the aforementioned film forming methods for the conductive carbide layer, the method of annealing a metal film under gas atmosphere including carbon can provide the conductive carbide layer with excellent reproducibility and stability. This makes it possible to achieve a reduced manufacturing cost and an increased electron emitting area of the
electron source 10. In addtion, an existing apparatus (DC magnetron sputtering apparatus or appealing apparatus) can be readily diverted to an apparatus for forming the conductive carbide layer, which provides a downsized equipment investment and a reduced equipment cost. Thus, theelectron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics can be provided at a lower cost. Further, as compared to the reactive sputtering method, defects due to plasma damages will be reduced to form the conductive carbide layer having a higher purity and film quality. From this point of view, the above annealing method is suitable for forming thelower electrode 2, particularly, in a ceramic substrate used as thesubstrate 1. - Alternatively, the conductive carbide layer may be formed through a CVD method. The CVD method can provide the conductive carbide layer with excellent reproducibility and stability as well as a high throughput, which achieves a reduced manufacturing cost and an increased electron emitting area of the
electron source 10. Thus, theelectron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics can be provided at a lower cost. Further, the CVD method is excellent in a step-coverage performance. In the CVD method, an organic metal compound such as (C5H5)2 Ti Cl2 (Dichlorotitanocene) may be used as a deposition source. However, it is desired to use chloride such as titanium chloride, zirconium chloride or hafnium chloride, in view of a problem of stability of the deposition source itself. A mixed gas of hydrogen and gas including carbon atom such as CH4 (methane), C3H8 (propane), C6H5CH3 (toluene), CCl4 (carbon tetrachloride) or CH3OH (methanol) may be used as the reactive gas. Since a thermal CVD method involves a process temperature of 600 °C or more, it is preferable to use the plasma CVD method capable of lowering the process temperature. - The conductive carbide layer may be formed by annealing a multilayer consisting of a metal film (e.g. titanium film) and a carbon film. Alternatively, the conductive carbide layer may be formed by annealing a film made of a mixture of metal (e.g. titanium) and carbon. Theses methods can also provide the conductive carbide layer with excellent reproducibility and stability, which achieves a reduced manufacturing cost and an increased electron emitting area of the
electron source 10. Thus, theelectron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics can be provided at a lower cost. - In the first embodiment, the
electron transit layer 6 is composed of the oxidized porous polycrystalline silicon layer. Alternatively, theelectron transit layer 6 may be composed of a nitrided porous polycrystalline silicon layer or an oxynitrided porous polycrystalline silicon layer. Further, theelectron transit layer 6 may be composed of any other suitable oxidized, nitrided or oxynitrided porous semiconductor layer. When theelectron transit layer 6 is composed of the nitrided porous polycrystalline silicon layer, a nitriding process of nitriding the porouspolycrystalline silicon layer 4 through a rapid heating method using NH3 gas may be employed, as a substitute for the oxidation process of oxidizing thelayer 4 through the rapid heating method using O2 gas. Further, when theelectron transit layer 6 is composed of the oxynitrided porous polycrystalline silicon layer, an oxynitriding process of oxynitriding the porouspolycrystalline silicon layer 4 through a rapid heating method using a mixed gas of O2 gas and NH3 gas may be employed, as a substitute for the oxidation process of oxidizing thelayer 4 through the rapid heating method using 02 gas. In this case, a silicon oxynitride film will be formed instead of thesilicon oxide films Fig. 3 . - A second embodiment, which is an embodiment of the present invention, will be described below.
- As shown in
Figs. 5A and 5B , anelectron source 10 according to the second embodiment comprises asubstrate 1 composed of an insulative substrate (e.g. insulative glass substrate or insulative ceramic substrate), and anelectron source device 10a formed on one of the principal surfaces of thesubstrate 1. Theelectron source device 10a comprises alower electrode 2 formed on the selected principal surface of thesubstrate 1, a semiconductor layer or a non-dopedpolycrystalline silicon layer 3 formed on thelower electrode 2, an after-mentionedelectron transit layer 6 formed on thepolycrystalline silicon layer 3, and asurface electrode 7 formed on theelectron transit layer 6. Specifically, in theelectron source device 10a, thesurface electrode 7 and thelower electrode 2 are disposed opposed to one another, and theelectron transit layer 6 is interposed between the surface and lower electrodes. The thickness of thelower electrode 2 is set at about 300 nm, and the thickness of thesurface electrode 7 is set such that it does not exceed 10 nm. While the second embodiment employs the insulative substrate as thesubstrate 1, any other suitable semiconductor substrate such as a silicon substrate may be used as thesubstrate 1, and thelower electrode 2 may be comprised of the semiconductor substrate and a conductive layer (e.g. ohmic electrode) deposited on the semiconductor substrate. Further, while thepolycrystalline silicon layer 3 is interposed between theelectron transit layer 6 and thelower electrode 2 in the first embodiment, theelectron transit layer 6 may be formed directly on thelower electrode 2 without interposing thepolycrystalline silicon layer 3. - The
lower electrode 2 is composed of a single layer thin-film made of a metal (e.g. Mo, Cr, W, Ti, Ta, Ni, Al, Cu, Au or Pt; alloy containing such metal; or intermetallic compound such as silicide). Alternatively, thelower electrode 2 may be composed of a multilayer thin-film made of such a metal. Further, thelower electrode 2 may be made of a semiconductor material such as impurity-doped polycrystalline silicon. - The
electron transit layer 6 is formed by subjecting the polycrystalline silicon layer to a anodizing process and an oxidation process as described later. As with theelectron transit layer 6 in the first embodiment, theelectron transit layer 6 in the second embodiment includesgrains 51,silicon oxide films 52, silicon nanocrystals 63, and silicon oxide films 64 (seeFig. 3 ). In the second embodiment, the remaining region other than thegrains 51, silicon nanocrystals 63 andsilicon oxide films silicon nanocrystals 63 residing adjacent to the grain boundary of the polycrystalline silicon are mixed together within theelectron transit layer 6. - The
surface electrode 7 is composed of a multilayer film having a conductive carbide orconductive nitride layer 7a deposited on theelectron transit layer 6 and anoble metal layer 7b deposited on the conductive carbide orconductive nitride layer 7a. The surface of the multilayer film is formed with a number ofconcave portions 8 allowing the multilayer film to have a locally reduced thickness. In view of thermal stability and reproducibility in a film forming process, it is preferable to use titanium carbide, zirconium carbide, hafnium carbide, titanium nitride, zirconium nitride, hafnium nitride, niobium nitride, tantalum nitride or the like. - The
noble metal layer 7b is made of platinum as a noble metal. While the noble metal is not limited to platinum and thenoble metal layer 7b may be made, for example, of gold or iridium, it is preferable to use platinum in view of reproducibility in a film forming process. - In order to provide increased emission current and enhanced emitting efficiency, it is required to reduce the thickness of the
surface electrode 7. For this purpose, the thickness of the conductive carbide orconductive nitride layer 7a is set such that it does not exceed 4 nm, and the total thickness of the conductive carbide orconductive nitride layer 7a and thenoble metal layer 7b is set such that it does not exceed 10 nm. As described above, the surface of thesurface electrode 7 is formed with the number ofconcave portions 8. In the region having theconcave portions 8, the surface of the conductive carbide orconductive nitride layer 7a is exposed to outside. That is, the depth of theconcave portion 8 is approximately equal to the thickness of thenoble metal layer 7b. However, the depth of theconcave portion 8 may be less than, for example about one-half of, the thickness of thenoble metal layer 7b. - In the
electron source 10 or theelectron source device 10a as a ballistic electron surface-emitting type electron source according to the second embodiment, steps of emitting electrons or an electron emission model is the same as that of theelectron source 10 according to the first embodiment (seeFigs. 2 and3 ). Thus, as with theelectron source 10 according to the first embodiment, theelectron source 10 according to the second embodiment exhibits enhanced electron emission characteristics having a low dependence on vacuum degree and free from the popping phenomenon, and thereby can emit electrons stably. - As above, in the
electron source 10 according to the second embodiment, thesurface electrode 7 is composed of the conductive carbide orconductive nitride layer 7a and thenoble metal layer 7b, and the surface of the multilayer film is formed with the number ofconcave portions 8 allowing the multilayer film to have a locally reduced thickness. Thus, an energy loss due to electron scattering caused in thesurface electrode 7 will be reduced in the region of thesurface electrode 7 formed with theconcave portions 8 as comparted to other region. Further, electron emission characteristics can be enhanced while suppressing the increase of electrical resistance in thesurface electrode 7, because the electrical resistance of thesurface electrode 7 is substantially determined by the film thickness of the multilayer film composed of the conductive carbide orconductive nitride layer 7a and thenoble metal layer 7b. - When a glass substrate is used as the
substrate 1, the glass substrate may be appropriately selected from a silica glass substrate, no-alkali glass substrate, low-alkali glass substrate or soda-lime glass substrate, depending on a process temperature. In case of employing a ceramic substrate, thesubstrate 1 may be composed, for example, of an alumina substrate. Theelectron source 10 according to the first embodiment may be used as an electron source for display devices. In this case, a suitable patterning of thelower electrode 2, thesurface electrode 7 and theelectron transit layer 6 may be appropriately selected. - With reference to
Figs. 6A to 6E , a manufacturing process of theelectron source 10 according to the second embodiment will be described below. - A
lower electrode 2 composed of a metal film (e.g. molybdenum film) having a given film thickness (e.g. about 300 nm) is first formed on one of the principal surfaces of asubstrate 1 composed of a silica glass substrate. Then, a non-dopedpolycrystalline silicon layer 3 having a given film thickness (e.g. 1.5 µm) is formed on thelower electrode 2 to provide the structure as shown inFig. 6A . Thelower electrode 2 may be formed, for example, through a sputtering method or a CVD method. Alternatively, thelower electrode 2 may be formed through a method of forming a non-doped polycrystalline silicon layer and then doping an n-type impurity into the polycrystalline silicon layer by means of a thermal diffusion method, or another method of forming a non-doped polycrystalline silicon layer and simultaneously doping an n-type impurity into the polycrystalline silicon layer (i.e. forming an conductive polycrystalline silicon layer directly on thesubstrate 1 without using an ion-implantation method or thermal diffusion method) . - By using the co-doping method of doping simultaneously with film formation as the method for forming the
lower electrode 2, thelower electrode 2 and the non-dopedpolycrystalline silicon layer 3 can be continuously formed by the same film forming apparatus without any carry-in-and-out operation. Thelower electrode 2 is not limited to the n-type polycrystalline silicon layer, and it may be composed of a p-type polycrystalline silicon layer. In this case, a p-type impurity may be doped in the non-dopedpolycrystalline silicon layer 3. The non-dopedpolycrystalline silicon layer 3 may be formed through various film forming methods such as a CVD method (e.g. LPCVD method, plasma CVD method or catalytic-CVD method), a sputtering method, a CGS method, or a method of depositing amorphous silicon and then laser-annealing the amorphous silicon. - After the non-doped
polycrystalline silicon layer 3 has been formed, an anodizing process is performed to form a porouspolycrystalline silicon layer 4 in which polycrystalline silicon grains 51 (seeFig. 3 ), silicon nanocrystals 63 (seeFig. 3 ) and amorphous silicons are mixed together. Through this step, the structure as shown inFig. 6B is obtained. Hereupon, the procedure for performing the anodizing process is as same as that of the first embodiment. - After the completion of the anodizing process, an oxidation process is performed to form an
electron transit layer 6 composed of the composite nanocrystal layer having the structure as shown inFig. 3 . Through this step, the structure as shown inFig. 6C is obtained. - In the oxidation process, the porous
polycrystalline silicon layer 4 is oxidized, for example, through a rapid heating method to form the electron transit layer 6 (seeFig. 3 ) including thegrains 51, thesilicon nanocrystals 63, and thesilicon oxide films - After the
electron transit layer 6 has been formed, a conductive carbide orconductive nitride layer 7a having a given film thickness (appropriately selected in the range of 1 nm to 4 nm) and anoble metal layer 7b having a given film thickness (e.g. 3 nm) are sequentially formed on theelectron transit layer 6, for example, through a sputtering method. Through this step, a multi layer composed of the conductive carbide orconductive nitride layer 7a and thenoble metal layer 7b is formed, and the structure as shown inFig. 6D is obtained. A method for forming the conductive carbide orconductive nitride layer 7a may be selected from various thin-film forming methods such as a sputtering method (e.g. RF sputtering method, RF magnetron sputtering method, DC sputtering method, DC magnetron sputtering method, or reactive sputtering method), a vacuum evaporation method, and a CVD method. Alternatively, the conductive carbide orconductive nitride layer 7a may be formed through a method of depositing a metal film on theelectron transit layer 6 by means of the sputtering method or vacuum evaporation method and then annealing the metal film under gas atmosphere including carbon or nitrogen, or through another method of depositing a metal film on theelectron transit layer 6 by means of the sputtering method or vacuum evaporation method and then implanting carbon ion or nitrogen ion into the metal film. A method for forming thenoble metal layer 7b may be selected from various thin-film forming methods such as a sputtering method (e.g. RF sputtering method, RF magnetron sputtering method, DC sputtering method or DC magnetron sputtering method), a vacuum evaporation method, and a CVD method. - Then, through a heat treatment process, a number of concave portions are formed on the multilayer composed of the conductive carbide or
conductive nitride layer 7a and thenoble metal layer 7b, and theelectron source 10 having the structure as shown inFig. 6E is obtained. In the heat treatment process, the multilayer is subjected to a heat treatment at a temperature in the range of 300 °C to 450 °C for a given time-period (e.g. 15 minutes to 120 minutes) under nitrogen atmosphere. Thus, the concave portion can be formed at a relatively low process temperature. While the heat treatment is performed under nitrogen atmosphere in this heat treatment process, it may be performed under any other suitable inert gas atmosphere or in vacuum. A usable heat treatment may include an annealing using an electric furnace, an annealing based on light irradiation (e.g. lamp annealing), and a laser annealing. - The manufacturing process of the
electron source 10 according to the second embodiment can facilitate manufacturing theelectron source 10 having enhanced electron emitting efficiency with suppressing the increase of electrical resistance in thesurface electrode 7. - In the second embodiment, also, the oxidation process may be substituted with a nitriding process or an oxynitriding process as same as the case of the first embodiment.
- In the second embodiment, the
electron transit layer 6 is composed of the composite nanocrystal layer as shown inFig. 3 . Alternatively, theelectron transit layer 6 may be composed of an insulative layer made, for example, of Al2O3, Si02 or the like. In this case, the electron source provided with a semiconductor layer can operate as with an electron source having the MIS structure. On the other hand, the electron source provided with no semiconductor layer can operate as with an electron source having the MIM structure. In either electron source, electron emission characteristics can be improved by appropriately setting the thickness of the electron transit layer. In addition, the forming process of the electron transit layer can be simplified. - A third embodiment, which does not fall under the scope of the present invention, will be described below.
- As shown in
Fig. 7 , anelectron source 10 according to the third embodiment comprises asubstrate 1 composed of an insulative substrate (e.g. insulative glass substrate or insulative ceramic substrate), and anelectron source device 10a formed on one of the principal surfaces of thesubstrate 1. Theelectron source device 10b comprises alower electrode 2 formed on the selected principal surface of thesubstrate 1, a semiconductor layer or a non-dopedpolycrystalline silicon layer 3 formed on thelower electrode 2, an after-mentionedelectron transit layer 6 formed on thepolycrystalline silicon layer 3, and asurface electrode 7 formed on theelectron transit layer 6. In theelectron source device 10b, thesurface electrode 7 and thelower electrode 2 are disposed opposed to one another, and theelectron transit layer 6 is interposed between the surface and lower electrodes. The thickness of thelower electrode 2 is set at about 300 nm, and the thickness of thesurface electrode 7 is set such that it does not exceed 10 nm. - While the third embodiment employs the insulative substrate as the
substrate 1, any other suitable semiconductor substrate such as a silicon substrate may be used as thesubstrate 1, and thelower electrode 2 may be comprised of the semiconductor substrate and a conductive layer (e.g. ohmic electrode) deposited on the semiconductor substrate. Further, while thepolycrystalline silicon layer 3 is interposed between theelectron transit layer 6 and thelower electrode 2, theelectron transit layer 6 may be formed directly on thelower electrode 2 without interposing thepolycrystalline silicon layer 3. - The
lower electrode 2 is composed of a thin film made of the same metal as that of thelower electrode 2 of the electron source according to the second embodiment. - The
electron transit layer 6 is formed by subjecting the polycrystalline silicon layer to a anodizing process and an oxidation process as described later. Theelectron transit layer 6 in the third embodiment includesgrains 51,silicon oxide films 52, silicon nanocrystals 63, andsilicon oxide films 64 as with theelectron transit layer 6 in the first embodiment (seeFig. 3 ), and the remaining region other than thegrains 51, silicon nanocrystals 63 andsilicon oxide films electron transit layer 6 in the second embodiment. - The
surface electrode 7 is composed of aconductive nitride layer 7c deposited on theelectron transit layer 6 and ametal layer 7d deposited on theconductive nitride layer 7c. Theconductive nitride layer 7c is made of conductive nitride consisting of titanium nitride. Generally, the conductive nitride has a relatively high conductivity, a melting point higher than that of noble metal such as gold, and an excellent diffusion barrier performance. In addtion, the conductive nitride has an oxidation resistance superior to that of metal such as tungsten or aluminum. The conductive nitride having these properties usable in theconductive nitride layer 7c is not limited to titanium nitride, and it may be chromium nitride, molybdenum nitride, tungsten nitride, vanadium nitride, niobium nitride, tantalum nitride, zirconium nitride, or hafnium nitride. In view of thermal stability, work function and reproducibility, it is preferable to use either one of titanium nitride, zirconium nitride, hafnium nitride, niobium nitride and tantalum nitride. - The
metal layer 7d is made of platinum which is one of noble metals. The material of themetal layer 7d is not limited to platinum, and themetal layer 7d may be made of any other suitable noble metal such as gold or iridium. However, platinum is particularly effective to assure thermal stability. - In the
surface electrode 7, the thickness of theconductive nitride layer 7c is set such that it does not exceed 4 nm, and the total thickness of theconductive nitride layer 7c and themetal layer 7d is set such that it does not exceed 10 nm, in order to suppress the deterioration of electron emitting efficiency or provide other advantages. - In the
electron source 10 or theelectron source device 10b as a ballistic electron surface-emitting type electron source according to the third embodiment, steps of emitting electrons or an electron emission model is the same as that of theelectron source 10 according to the first embodiment (seeFigs. 2 and3 ). Thus, as with theelectron source 10 according to the first embodiment, theelectron source 10 according to the third embodiment exhibits enhanced electron emission characteristics having a low dependence on vacuum degree and free from the popping phenomenon, and thereby can emit electrons stably. - In the
electron source 10 according to the third embodiment, thesurface electrode 7 is composed of theconductive nitride layer 7c made of conductive nitride and deposited on theelectron transit layer 6, and themetal layer 7d deposited on theconductive nitride layer 7c. As described above, the conductive nitride has a relatively high conductivity, a melting point higher than that of noble metal such as gold, an excellent diffusion barrier performance, and an oxidation resistance superior to that of metal such as tungsten or aluminum. In addtion, theconductive nitride layer 7c has an excellent adhesiveness to themetal layer 7d. Thus, as compared to an electron source having a surface electrode composed of a metal thin-film, theelectron source 10 according to the third embodiment can provide enhanced thermal resistance of thesurface electrode 7 while suppressing the deterioration of electron emission characteristics. That is, theelectron source 10 according to the third embodiment can suppress undesirable agglomeration otherwise caused in the material of thesurface electrode 7 to prevent the deterioration of electron emission characteristics due to a thermal process such as the vacuum-sealing process. Further, thesurface electrode 7 composed of theconductive nitride layer 7c and themetal layer 7d can prevent respective atoms constituting thesurface electrode 7 and theelectron transit layer 6 from being diffused therebetween. Thus, the materials of thesurface electrode 7 and theelectron transit layer 6 can be selected from various alternatives. Thesurface electrode 7 may be formed of only the conductive nitride layer. - While the third embodiment employs the glass substrate as the
substrate 1, the glass substrate may be substituted with any other suitable substrate in the same manner as that in the second embodiment. Theelectron source 10 according to the third embodiment may also be used as an electron source for display devices in the same manner as that in the second embodiment. - With reference to
Figs. 8A to 8D , a manufacturing process of theelectron source 10 according to the third embodiment will be described below. - A
lower electrode 2 composed of a metal film (e.g. molybdenum film) having a given film thickness (e.g. about 300 nm) is first formed on one of the principal surfaces of asubstrate 1 composed of a silica glass substrate. Then, a non-dopedpolycrystalline silicon layer 3 having a given film thickness (e.g. 1.5 µm) is formed on thelower electrode 2 to provide the structure as shown inFig. 8A . The procedure for forming thelower electrode 2 and the procedure for forming the non-dopedpolycrystalline silicon layer 3 are as same as those of the second embodiment. - After the non-doped
polycrystalline silicon layer 3 has been formed, an anodizing process is performed to form a porouspolycrystalline silicon layer 4 in which polycrystalline silicon grains 51 (seeFig. 3 ), silicon nanocrystals 63 (seeFig. 3 ) and amorphous silicons are mixed together. Through this step, the structure as shown inFig. 8B is obtained. The procedure for performing the anodizing process is as same as that of the first embodiment. - After the completion of the anodizing process, an oxidation process is performed to form an
electron transit layer 6 composed of the composite nanocrystal layer having the structure as shown inFig. 3 . Through this step, the structure as shown inFig. 8C is obtained. In the oxidation process, thecomposite nanocrystal layer 4 is oxidized, for example, through a rapid heating method to form the electron transit layer 6 (seeFig. 3 ) including thegrains 51, thesilicon nanocrystals 63, and thesilicon oxide films - After the
electron transit layer 6 has been formed, aconductive nitride layer 7c having a given film thickness (e.g. 1 nm) and ametal layer 7d having a given film thickness (e.g. 3 nm) are sequentially formed on theelectron transit layer 6, for example, through a sputtering method. Through this step, asurface electrode 7 composed of theconductive nitride layer 7c and themetal layer 7d is formed, and theelectron source 10 having the structure as shown inFig. 8D is obtained. - The manufacturing process of the
electron source 10 according to the third embodiment can provide theelectron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics. - In the third embodiment, the
conductive nitride layer 7c of thesurface electrode 7 is formed through a sputtering method. A usable sputtering method may be selected from various types such as an RF sputtering method, RF magnetron sputtering method, DC sputtering method and DC magnetron sputtering method. Theconductive nitride layer 7c is formed through a sputtering method using a target consisting of conductive nitride. This method can provide theconductive nitride layer 7c having a low surface roughness with excellent reproducibility and stability as well as a high throughput, which achieves a reduced manufacturing cost and an increased electron emitting area of theelectron source 10. In addtion, an existing sputtering apparatus can be readily diverted to an apparatus for forming theconductive nitride layer 7c, which provides a downsized equipment investment and a reduced equipment cost. Thus, theelectron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics can be provided at a lower cost. Further, theconductive nitride 7c layer can be formed at a relatively low process temperature. - The method for forming the
conductive nitride layer 7c is not limited to the aforementioned sputtering methods. For example, the conductive nitride layer may be formed through a vacuum evaporation method using a deposition source consisting of conductive nitride, or a reactive sputtering method using a target consisting of metal (pure metal) and gas (reactive gas) including nitrogen. The vacuum evaporation method or the reactive sputtering method can also provide theconductive nitride layer 7c having a low surface roughness with excellent reproducibility and stability as well as a high throughput, which achieves a reduced manufacturing cost and an increased electron emitting area of theelectron source 10. Thus, theelectron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics can be provided at a lower cost. Further, the conductive nitride layer can be formed at a relatively low process temperature. In addtion, when theconductive nitride layer 7c is formed through the vacuum evaporation method, an existing vacuum evaporation apparatus can be readily diverted to an apparatus for forming theconductive nitride layer 7c, which provides a downsized equipment investment and a reduced equipment cost. - The reactive sputtering method using a target consisting of metal (pure metal) and gas (reactive gas) including nitrogen can facilitate the control of nitrogen concentration in the
conductive nitride layer 7c. That is, in the reactive sputtering method, a type of the reaction gas and a mixing ratio of inert gas such as Ar to the reactive gas including nitrogen atoms can be selected to control the composition of nitrogen in theconductive nitride layer 7c. Thus, the reactive sputtering method can provide theconductive nitride layer 7c having a nitrogen composition controlled at a desired value. - Generally, as compared to the nitride target, the pure metal target having a higher purity is widely placed on the market. Thus, the reactive sputtering method can provide the
conductive nitride layer 7c including a less amount of metal impurities as compared to the sputtering method using conductive nitride as a target, and thereby performance decrement due to impurities can be suppressed. - Alternatively, the
conductive nitride layer 7c may be formed through a CVD method. The CVD method can also provide theconductive nitride layer 7c having a low surface roughness with excellent reproducibility and stability as well as a high throughput, which achieves a reduced manufacturing cost and an increased electron emitting area of theelectron source 10. Thus, theelectron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics can be provided at a lower cost. Further, the CVD method is excellent in a step-coverage performance. Since a thermal CVD method typically involves a process temperature of 600 °C or more, it is preferable to use the plasma CVD method capable of lowering the process temperature. - In the step of forming the
conductive nitride layer 7c, theconductive nitride layer 7c may be formed by depositing a metal film (e.g. titanium film) on theelectron transit layer 6 through a vacuum evaporation method or a sputtering method and then implanting nitrogen ions into the metal film. This method can also form theconductive nitride layer 7c having a low surface roughness with excellent reproducibility and stability. In addition, this method can reduce the roughness of the interface between theconductive nitride layer 7c and themetal layer 7b, which achieves a reduced manufacturing cost and an increased electron emitting area of theelectron source 10. Thus, theelectron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics can be provided at a lower cost. Further, this method can form theconductive nitride layer 7c at a relatively low process temperature, and facilitate the control of conductivity in theconductive nitride layer 7c. - Alternatively, in the step of forming the
conductive nitride layer 7c, theconductive nitride layer 7c may be formed by depositing a metal film (e.g. titanium film) on theelectron transit layer 6 through a vacuum evaporation method or a sputtering method and then subjecting the metal film to a heat treatment under gas atmosphere including nitrogen. This method can also form theconductive nitride layer 7c having a low surface roughness with excellent reproducibility and stability. In addition, this method can reduce the roughness of the interface between theconductive nitride layer 7c and themetal layer 7b, which achieves a reduced manufacturing cost and an increased electron emitting area of theelectron source 10. Thus, theelectron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics can be provided at a lower cost. - In the third embodiment, also, the oxidation process may be substituted with a nitriding process or an oxynitriding process as same as the case of the first embodiment.
- The
lower electrode 2 may be formed to have a multilayer structure, and at least one of the layers may be formed as a conductive nitride layer. The conductive nitride has a relatively high conductivity, a melting point higher than that of noble metal such as gold, an excellent diffusion barrier performance, and an oxidation resistance superior to that of metal such as tungsten or aluminum. Thus, as compared to an electron source having alower electrode 2 made of a metal, thelower electrode 2 having the conductive nitride layer can have enhanced thermal resistance with suppressing the deterioration of electron emission characteristics to prevent the deterioration of electron emission characteristics due to a thermal process such as the vacuum-sealing process. Further, since thelower electrode 2 is composed of the multilayer structure having at least one of conductive nitride layer, the conductive nitride layer can provide enhanced thermal resistance of thelower electrode 2. Further, another layer deposited on the conductive nitride layer can be arranged to provide reduced electric resistance of thelower electrode 2, and enhanced adhesiveness between thelower electrode 2 and a layer forming an interface together with thelower electrode 2. It is to be understood that the entirelower electrode 2 may be composed of a conductive nitride layer. - The conductive nitride layer used in the
lower electrode 2 may be made of the same conductive nitride as the material used in theconductive nitride layer 7c of thesurface electrode 7. Further, the conductive nitride layer of thelower electrode 2 may be formed through the same method as that for forming theconductive nitride layer 7c of thesurface electrode 7. - A fourth embodiment, which does not fall under the scope of the present invention, will be described below.
- As shown in
Fig. 9 , anelectron source 10 according to the fourth embodiment comprises asubstrate 1 composed of an insulative substrate (e.g. insulative glass substrate or insulative ceramic substrate), and alower electrode 2 formed on one of the principal surfaces of thesubstrate 1. A semiconductor layer or a non-dopedpolycrystalline silicon layer 3 is formed on thelower electrode 2. An electron transit layer 6 (strong-field drift layer) composed of an oxidized porous polycrystalline silicon layer is formed on thepolycrystalline silicon layer 3. Further, asurface electrode 7 is formed on theelectron transit layer 6. Specifically, in theelectron source 10, thesurface electrode 7 and thelower electrode 2 are disposed opposed to one another, and theelectron transit layer 6 is interposed between the surface and lower electrodes. The thickness of thesurface electrode 7 is set at 12 nm or less. While the fourth embodiment employs the insulative substrate as thesubstrate 1, any other suitable semiconductor substrate such as a silicon substrate may be used as thesubstrate 1. Further, while thepolycrystalline silicon layer 3 is interposed between theelectron transit layer 6 and thelower electrode 2, theelectron transit layer 6 may be formed directly on thelower electrode 2 without interposing thepolycrystalline silicon layer 3. - In the
electron source 10 as a ballistic electron surface-emitting type electron source according to the fourth embodiment, steps of emitting electrons or an electron emission model is the same as that of theelectron source 10 according to the first embodiment (seeFigs. 2 and3 ). Thus, as with theelectron source 10 according to the first embodiment, theelectron source 10 according to the fourth embodiment exhibits enhanced electron emission characteristics having a low dependence on vacuum degree and free from the popping phenomenon, and thereby can emit electrons stably. - While the fourth embodiment employs the glass substrate as the
substrate 1, the glass substrate may be substituted with any other suitable substrate in the same manner as that in the second embodiment. Theelectron source 10 according to the fourth embodiment may also be used as an electron source for display devices in the same manner as that in the second embodiment. - The
lower electrode 2 is made of highly doped n-type polycrystalline silicon. However, the material of thelower electrode 2 is not limited to the n-type polycrystalline silicon, and thelower electrode 2 may be made of metal such as Cr, W, Ti, Al, Cu, Au, Pt or Mo; alloy containing such metal; or impurity-doped semiconductor other than polycrystalline silicon. - The
surface electrode 7 is composed of aconductive carbide layer 7e deposited on theelectron transit layer 6 and anoble metal layer 7f deposited on theconductive carbide layer 7e. Theconductive carbide layer 7e is made of conductive carbide consisting of zirconium carbide. Generally, the conductive carbide has a relatively high conductivity, a relatively low work function, a melting point higher than that of noble metal such as gold, and an excellent diffusion barrier performance. In addtion, the conductive carbide has an oxidation resistance superior to that of metal such as tungsten or aluminum. The conductive carbide having these properties usable in theconductive carbide layer 7e is not limited to zirconium carbide, and it may be chromium carbide, molybdenum carbide, tungsten carbide, vanadium carbide, niobium carbide, tantalum carbide, titanium carbide, or hafnium carbide. In view of thermal stability, work function and reproducibility, it is preferable to use either one of zirconium carbide, titanium carbide and hafnium carbide. - The
noble metal layer 7f is made of platinum which is one of noble metals (That is, thenoble metal layer 7f is composed of a layer-structured platinum layer). However, the noble metal to be used is not limited to platinum, and thenoble metal layer 7f may be made of any other suitable noble metal such as gold or iridium. Further, a portion of thenoble metal layer 7f may be composed of a layer-structured platinum layer, and the remaining portion may be made of different noble metal other than platinum. - In the
surface electrode 7, the thickness of theconductive carbide layer 7e is preferably set at 4 nm or less, and the thickness of thenoble metal layer 7f is preferably set at 8 nm or less. More preferably, the thickness of theconductive carbide layer 7e is set at 2 nm or less, and the thickness of thenoble metal layer 7f is set at 4 nm or less to provide enhanced electron emitting efficiency. - In the
electron source 10 according to the fourth embodiment, thesurface electrode 7 is composed of theconductive carbide layer 7e made of conductive carbide and thenoble metal layer 7f made of noble metal. As described above, the conductive carbide has a relatively high conductivity, a relatively low work function, a melting point higher than that of noble metal, an excellent diffusion barrier performance, and an oxidation resistance superior to that of metal such as tungsten or aluminum. In addtion, theconductive carbide layer 7e has an excellent adhesiveness to thenoble metal layer 7f. Thus, as compared to an electron source having a surface electrode composed of a metal thin-film, theelectron source 10 according to the fourth embodiment can provide enhanced thermal resistance of thesurface electrode 7 while suppressing the deterioration of electron emission characteristics. That is, theelectron source 10 according to the fourth embodiment can suppress undesirable agglomeration otherwise caused in the material of thesurface electrode 7 to prevent the deterioration of electron emission characteristics due to a thermal process such as the vacuum-sealing process. - Further, the
surface electrode 7 composed of theconductive carbide layer 7e and thenoble metal layer 7f can prevent respective atoms constituting thesurface electrode 7 and theelectron transit layer 6 from being diffused therebetween. Thus, the materials of thesurface electrode 7 and theelectron transit layer 6 can be selected from various alternatives. - With reference to
Figs. 10A to 10D , a manufacturing process of theelectron source 10 according to the fourth embodiment will be described below. - A non-doped polycrystalline silicon layer having a given film thickness (e.g. 1 µm) is first formed on one of the principal surfaces of the
substrate 1, and an n-type impurity is doped into the polycrystalline silicon layer through an ion-implantation method or heat treatment to form alower electrode 2 composed of a conductive polycrystalline silicon layer (n-type polycrystalline silicon layer). Then, an additional non-dopedpolycrystalline silicon layer 3 having a given film thickness (e.g. 1.5 µm) is formed on the side of the selected principal surface of the substrate 1 (on the lower electrode 2) to provide the structure as shown inFig. 10A . The procedure for forming the non-dopedpolycrystalline silicon layer 3 is as same as that of the second embodiment. When thelower electrode 2 is made of metal such as Cr, W, Ti, Al, Cu, Au, Pt or Mo, or alloy containing such meta, thelower electrode 2 may be formed, for example, through a sputtering method or vacuum evaporation method. - After the non-doped
polycrystalline silicon layer 3 has been formed, a porous region is provided up to a given depth of thepolycrystalline silicon layer 3 by means of an anodizing process. Through this step, a porous semiconductor layer or a porouspolycrystalline silicon layer 4 is formed, and thus the structure as shown inFig. 10B is obtained. The procedure for performing the anodizing process is as same as that of the first embodiment. - After the completion of the anodizing process, the porous
polycrystalline silicon layer 4 is oxidized through an oxidation process. Through this step, anelectron transit layer 6 composed of the oxidized porous polycrystalline silicon layer is formed, and thus the structure as shown inFig. 10C is obtained. In the oxidation process, the porouspolycrystalline silicon layer 4 is oxidized, for example, through a rapid heating method to form theelectron transit layer 6 including thegrains 51, thesilicon nanocrystals 63, and thesilicon oxide films 52, 64 (seeFig. 3 ). The procedure for performing the oxidation process is as same as that of the first embodiment. - After the
electron transit layer 6 has been formed, aconductive carbide layer 7e and anoble metal layer 7f are sequentially formed, for example, through a sputtering method. Through this step, asurface electrode 7 composed of theconductive carbide layer 7e and thenoble metal layer 7f is formed on theelectron transit layer 6, and thus theelectron source 10 having the structure as shown inFig. 10D is obtained. - The manufacturing process of the
electron source 10 according to the fourth embodiment can provide theelectron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics. - In the fourth embodiment, the
conductive carbide layer 7e of thesurface electrode 7 is formed through a sputtering method. A usable sputtering method may be selected from various types such as an RF sputtering method, RF magnetron sputtering method, DC sputtering method and DC magnetron sputtering method. In this case, theconductive carbide layer 7e is formed through a sputtering method using a target consisting of conductive carbide. These methods can provide theconductive carbide layer 7e having a low surface roughness with excellent reproducibility and stability as well as a high throughput, which achieves a reduced manufacturing cost and an increased electron emitting area of theelectron source 10. In addtion, an existing sputtering apparatus can be readily diverted to an apparatus for forming theconductive carbide layer 7e, which provides a downsized equipment investment and a reduced cost. Thus, theelectron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics can be provided at a lower cost. Further, theconductive carbide layer 7e can be formed at a relatively low process temperature. - The film forming method for the
conductive carbide layer 7e is not limited to the aforementioned sputtering methods. For example, theconductive carbide layer 7e may be formed through a vacuum evaporation method using a deposition source consisting of conductive carbide, or a reactive sputtering method using a target consisting of metal (pure metal) and gas (reactive gas) including carbon. The vacuum evaporation method or the reactive sputtering method can also provide theconductive carbide layer 7e having a low surface roughness with excellent reproducibility and stability as well as a high throughput, which achieves a reduced manufacturing cost and an increased electron emitting area of theelectron source 10. Thus, theelectron source 10 having enhanced thermal resistance with suppressing the deterioration of electron emission characteristics can be provided at a lower cost. Further, theconductive carbide layer 7e can be formed at a relatively low process temperature. In addtion, when theconductive carbide layer 7e is formed through the vacuum evaporation method, an existing vacuum evaporation apparatus can be readily diverted to an apparatus for forming theconductive carbide layer 7e, which provides a downsized equipment investment and a reduced equipment cost. - An EB (Electron Beam) vacuum evaporation method is one of appropriate vacuum evaporation methods to be used, because conductive carbide has a high melting point. On the other hand, the reactive sputtering method using a target consisting of metal (pure metal) and gas (reactive gas) including carbon can facilitate the control of carbon concentration in the
conductive carbide layer 7e. That is, in the reactive sputtering method, a type of the reactive gas or a mixing ratio of inert gas such as Ar to the reactive gas including carbon atoms can be appropriately selected to control a carbon concentration in theconductive carbide layer 7e so as to provide theconductive carbide layer 7e having a desired carbon concentration. Further, when using the reactive sputtering method, a desired film of titanium carbide, zirconium carbide or hafnium carbide can be formed at a relatively low substrate temperature of 150 to 500 °C. - Generally, as compared to the carbide target, the pure metal target having a higher purity is widely placed on the market. Thus, the reactive sputtering method can provide the
conductive carbide layer 7e including a less amount of metal impurities as compared to the sputtering method using conductive carbide as a target, and thereby performance decrement due to impurities can be suppressed. In view of film-thickness uniformity and coverage performance of the surface of aconductive carbide layer 7e having a thickness of 4 nm or less as in the fourth embodiment, the sputtering method, vacuum evaporation method or reactive sputtering method is more advantageous than other film forming methods to form such a conductive carbide layer. - In the fourth embodiment, the
electron transit layer 6 is composed of the oxidized porous polycrystalline silicon layer. Alternatively, theelectron transit layer 6 may be composed of a nitrided porous polycrystalline silicon layer or an oxynitrided porous polycrystalline silicon layer. Further, theelectron transit layer 6 may be composed of any other suitable oxidized, nitrided or oxynitrided porous semiconductor layer. The oxidation, nitriding or oxynitriding method and the characteristics or advantages in these cases are as described in the first embodiment. (Example) - With reference to
Fig. 11 , a measurement result of electron emission characteristics in both states before and after an actually producedelectron source 10 through the manufacturing process according to the fourth embodiment was subjected to a heat treatment will be described below. - In this example, a silicon substrate was used as the
substrate 1, an n-type polycrystalline silicon layer being used as thelower electrode 2, an oxidized porous polycrystalline silicon layer being used as the electron transit layer 6 (strong field drift layer), a zirconium carbide layer being used as theconductive carbide layer 7e of thesurface electrode 7, and a platinum layer being used as thenoble metal layer 7f of thesurface electrode 7. The thickness of theconductive carbide layer 7e was 2 nm, and the thickness of thenoble metal layer 7f was 4 nm. Theconductive carbide layer 7e was formed through an RF magnetron sputtering method using a target consisting of zirconium carbide under Ar gas atmosphere. - Set out below were formation conditions of the
conductive carbide layer 7e;
Pressure of Ar gas: 0.5 - 7 Pa
RF power density: 0.5 - 3.0 W/cm2
Substrate temperature: 30 - 150 °C
in which the data shown inFig. 11 was obtained from the electron source having theconductive carbide layer 7e of zirconium carbide formed under the following conditions.
Pressure of Ar gas: about 1.3 Pa
RF power density: about 1.7 W/cm2
Substrate temperature: about 50 °C - Set out below were formation conditions of the
noble metal layer 7f;
Pressure of Ar gas: about 0.7 Pa
RF power density: about 0.6 W/cm2
Substrate temperature: about 50 °C - The
electron source 10 was introduced into a vacuum chamber (not shown) to measure the electron emission characteristics of theelectron source 10 in the state as shown inFig. 2 . That is, thecollector 21 was positioned opposed to thesurface electrode 7. Then, a DC voltage Vps was applied between the surface and lower electrodes such that thesurface electrode 7 had a higher potential than that of thelower electrode 7, while another DC voltage Vc was applied between the collector and surface and electrodes such that thecollector 21 had a higher potential than that of thesurface electrode 7. -
Fig. 11 shows the measurement result of electron emission characteristics in the state when vacuum degree in the vacuum chamber was set at 5 x 10-5 Pa, and the DC voltage Vc was set at a constant value of 1000 V. InFig. 11 , the horizontal axis indicates the DC voltage Vps, and the vertical axis indicates the current density. The marks "A" indicate the current density of diode current Ips in the state before the heat treatment. The marks "B" indicate the current density of emission current Ie in the state before the heat treatment. The marks "C" indicate the current density of diode current Ips in the state after the heat treatment. The marks "D" indicate the current density of emission current Ie in the state after the heat treatment. The heat treatment was performed under N2 gas atmosphere at 400 °C for 1 hour. - As can been seen from
Fig. 11 , in theelectron source 10 of this example, neither emission current Ie nor emission emitting efficiency are degraded by the heat treatment. Further, in theelectron source 10 of this example, whereas the diode current Ips is reduced by the heat treatment, the emission current Ie is increased. This proves that electron emitting efficiency is improved.
While the invention has been described by reference to specific embodiments, various modifications and alterations will become apparent to those skilled in the art. Therefore, it is not intended that the invention be limited to the illustrative embodiments herein, but only by the appended claims.
Claims (2)
- A field emission-type electron source comprising:a lower electrode (2),a surface electrode (7), andan electron transit layer (6) interposed between said lower electrode (2) and said surface electrode (7) to allow an electron to pass therethrough,whereinthe field emission-type electron source is adapted to allow an electron passed through said electron transit layer (6) to be emitted through said surface electrode (7),whereinsaid surface electrode (7) is composed of a multilayer film including a conductive carbide layer or conductive nitride layer (7a) and a noble metal layer (7b), said surface electrode (7) having a surface formed with a number of concave portions (8) allowing the multilayer film to have a locally reduced thickness, characterized in that the conductive carbide layer or conductive nitride layer (7a) of the surface electrode (7) has a thickness of 4 nm or less, while the total thickness of the conductive carbide or conductive nitride layer (7a) and the noble metal layer (7b) of the surface electrode (7) does not exceed 10 nm.
- The field emission-type electron source according to claim 1, wherein the conductive carbide is titanium carbide, zirconium carbide or hafnium carbide, and the conductive nitride is tantalum nitride, titanium nitride, zirconium nitride or hafnium nitride.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002083928 | 2002-03-25 | ||
| JP2002083927A JP3755474B2 (en) | 2002-03-25 | 2002-03-25 | Electron source and manufacturing method thereof |
| JP2002083928A JP2003281993A (en) | 2002-03-25 | 2002-03-25 | Electron source and manufacturing method therefor |
| JP2002083927 | 2002-03-25 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| EP1523025A2 EP1523025A2 (en) | 2005-04-13 |
| EP1523025A3 EP1523025A3 (en) | 2005-04-20 |
| EP1523025B1 true EP1523025B1 (en) | 2012-12-19 |
Family
ID=34315543
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP02022014A Expired - Lifetime EP1523025B1 (en) | 2002-03-25 | 2002-10-01 | Field emission-type electron source |
Country Status (1)
| Country | Link |
|---|---|
| EP (1) | EP1523025B1 (en) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0367195A2 (en) * | 1988-10-31 | 1990-05-09 | Matsushita Electric Industrial Co., Ltd. | Mim cold-cathode electron emission elements and methods of manufacture thereof |
| WO2000074098A1 (en) * | 1999-05-28 | 2000-12-07 | Hitachi, Ltd. | Thin-film electron source, display and device |
| EP1117118A1 (en) * | 2000-01-13 | 2001-07-18 | Pioneer Corporation | Electron-emitting device and method of manufacturing the same and display apparatus using the same |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2987140B2 (en) | 1997-10-29 | 1999-12-06 | 松下電工株式会社 | Field emission electron source, method of manufacturing the same, flat light emitting device, display device, and solid-state vacuum device |
| DE69941811D1 (en) * | 1998-02-09 | 2010-01-28 | Panasonic Corp | ELECTRON EMISSIONING DEVICE, METHOD FOR HEELS; PICTURE DISPLAY WITH SUCH ELECTRON EMISSIONS DEVICE AND METHOD FOR THE PRODUCTION THEREOF |
| KR100374782B1 (en) * | 1999-10-18 | 2003-03-04 | 마츠시다 덴코 가부시키가이샤 | Field emision-type electron source and manufacturing method thereof |
| JP3789064B2 (en) * | 1999-10-27 | 2006-06-21 | パイオニア株式会社 | Electron emitter |
| JP3688970B2 (en) * | 2000-02-29 | 2005-08-31 | 株式会社日立製作所 | Display device using thin film type electron source and manufacturing method thereof |
-
2002
- 2002-10-01 EP EP02022014A patent/EP1523025B1/en not_active Expired - Lifetime
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0367195A2 (en) * | 1988-10-31 | 1990-05-09 | Matsushita Electric Industrial Co., Ltd. | Mim cold-cathode electron emission elements and methods of manufacture thereof |
| WO2000074098A1 (en) * | 1999-05-28 | 2000-12-07 | Hitachi, Ltd. | Thin-film electron source, display and device |
| US6818941B1 (en) * | 1999-05-28 | 2004-11-16 | Hitachi, Ltd. | Thin film electron emitter, display device using the same and applied machine |
| EP1117118A1 (en) * | 2000-01-13 | 2001-07-18 | Pioneer Corporation | Electron-emitting device and method of manufacturing the same and display apparatus using the same |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1523025A2 (en) | 2005-04-13 |
| EP1523025A3 (en) | 2005-04-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6720717B2 (en) | Field emission-type electron source | |
| US6940087B2 (en) | Quantum device | |
| WO2002089166A1 (en) | Field emission electron source and production method thereof | |
| US6765342B1 (en) | Field emission-type electron source and manufacturing method thereof | |
| US5436505A (en) | Heat-resisting ohmic contact on semiconductor diamond layer | |
| EP1523025B1 (en) | Field emission-type electron source | |
| EP1328003B1 (en) | Field emission-type electron source | |
| JP2003100201A (en) | Field emission electron source and manufacturing method thereof | |
| JP2003187688A (en) | Field emission type electron source and manufacturing method of the same | |
| JP4415922B2 (en) | Method for forming silicon oxide film | |
| JP3465657B2 (en) | Field emission type electron source and manufacturing method thereof | |
| JP3755474B2 (en) | Electron source and manufacturing method thereof | |
| JP3508652B2 (en) | Field emission type electron source and method of manufacturing the same | |
| JP3589172B2 (en) | Field emission electron source | |
| JP3363429B2 (en) | Field emission type electron source and manufacturing method thereof | |
| JP2003281993A (en) | Electron source and manufacturing method therefor | |
| TWI222653B (en) | Filed emission-type electron source | |
| JP3480464B2 (en) | Method for manufacturing field emission electron source | |
| JP3508651B2 (en) | Field emission type electron source and method of manufacturing the same | |
| JP3809808B2 (en) | Manufacturing method of field emission electron source | |
| JP3687520B2 (en) | Field emission electron source and manufacturing method thereof | |
| JP4616538B2 (en) | Manufacturing method of field emission electron source | |
| JP3852564B2 (en) | Field emission electron source and manufacturing method thereof | |
| JP3478279B2 (en) | Method for manufacturing field emission electron source | |
| JP3478206B2 (en) | Method for manufacturing field emission electron source |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| PUAL | Search report despatched |
Free format text: ORIGINAL CODE: 0009013 |
|
| 17P | Request for examination filed |
Effective date: 20021001 |
|
| AK | Designated contracting states |
Kind code of ref document: A2 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR IE IT LI LU MC NL PT SE SK TR |
|
| AX | Request for extension of the european patent |
Extension state: AL LT LV MK RO SI |
|
| AK | Designated contracting states |
Kind code of ref document: A3 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR IE IT LI LU MC NL PT SE SK TR |
|
| AX | Request for extension of the european patent |
Extension state: AL LT LV MK RO SI |
|
| APAF | Appeal reference modified |
Free format text: ORIGINAL CODE: EPIDOSCREFNE |
|
| APAF | Appeal reference modified |
Free format text: ORIGINAL CODE: EPIDOSCREFNE |
|
| AKX | Designation fees paid |
Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR IE IT LI LU MC NL PT SE SK TR |
|
| RAP1 | Party data changed (applicant data changed or rights of an application transferred) |
Owner name: PANASONIC ELECTRIC WORKS CO., LTD. |
|
| 17Q | First examination report despatched |
Effective date: 20110919 |
|
| RAP1 | Party data changed (applicant data changed or rights of an application transferred) |
Owner name: PANASONIC CORPORATION |
|
| GRAP | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOSNIGR1 |
|
| RIN1 | Information on inventor provided before grant (corrected) |
Inventor name: HONDA, YOSHIAKI Inventor name: TAKEGAWA, YOSHIYUKI Inventor name: ICHIHARA, TSUTOMU Inventor name: WATABE, YOSHIFUMI Inventor name: AIZAWA, KOICHI Inventor name: HATAI, TAKASHI Inventor name: BABA, TORU Inventor name: KOMODA, TAKUYA |
|
| GRAS | Grant fee paid |
Free format text: ORIGINAL CODE: EPIDOSNIGR3 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR IE IT LI LU MC NL PT SE SK TR |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: EP |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: REF Ref document number: 589774 Country of ref document: AT Kind code of ref document: T Effective date: 20130115 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R096 Ref document number: 60244238 Country of ref document: DE Effective date: 20130214 |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: T3 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20121219 Ref country code: ES Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130330 Ref country code: FI Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20121219 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130320 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: EE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20121219 Ref country code: CY Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20121219 Ref country code: BG Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130319 Ref country code: SK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20121219 Ref country code: BE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20121219 Ref country code: CZ Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20121219 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: PT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20130419 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20121219 |
|
| 26N | No opposition filed |
Effective date: 20130920 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R097 Ref document number: 60244238 Country of ref document: DE Effective date: 20130920 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: MC Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20121219 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: MM4A |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20131031 Ref country code: CH Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20131031 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20131001 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: NL Payment date: 20140910 Year of fee payment: 13 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 20141022 Year of fee payment: 13 Ref country code: DE Payment date: 20141022 Year of fee payment: 13 Ref country code: GB Payment date: 20141021 Year of fee payment: 13 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: AT Payment date: 20140925 Year of fee payment: 13 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: IT Payment date: 20141015 Year of fee payment: 13 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: TR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20121219 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20131001 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R119 Ref document number: 60244238 Country of ref document: DE |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: MM01 Ref document number: 589774 Country of ref document: AT Kind code of ref document: T Effective date: 20151001 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 20151001 |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: MM Effective date: 20151101 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20151001 Ref country code: GB Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20151001 Ref country code: DE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20160503 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST Effective date: 20160630 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20151101 Ref country code: AT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20151001 Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20151102 |