EP0487103B1 - Verfahren zur Herstellung von Arylensulfidpolymer - Google Patents
Verfahren zur Herstellung von Arylensulfidpolymer Download PDFInfo
- Publication number
- EP0487103B1 EP0487103B1 EP91119960A EP91119960A EP0487103B1 EP 0487103 B1 EP0487103 B1 EP 0487103B1 EP 91119960 A EP91119960 A EP 91119960A EP 91119960 A EP91119960 A EP 91119960A EP 0487103 B1 EP0487103 B1 EP 0487103B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- polymerization
- polymer
- carbon atoms
- arylene sulfide
- dichlorobenzene
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 229920000642 polymer Polymers 0.000 title claims abstract description 113
- -1 arylene sulfide Chemical compound 0.000 title claims abstract description 95
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 12
- 238000006116 polymerization reaction Methods 0.000 claims abstract description 73
- 239000000203 mixture Substances 0.000 claims abstract description 63
- 238000000034 method Methods 0.000 claims abstract description 40
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 claims abstract description 40
- 150000001336 alkenes Chemical class 0.000 claims abstract description 37
- 150000001875 compounds Chemical class 0.000 claims abstract description 26
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims abstract description 20
- 229910052717 sulfur Inorganic materials 0.000 claims abstract description 20
- 239000011593 sulfur Substances 0.000 claims abstract description 20
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 42
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 claims description 32
- 125000004432 carbon atom Chemical group C* 0.000 claims description 30
- 229920000069 polyphenylene sulfide Polymers 0.000 claims description 25
- ZGEGCLOFRBLKSE-UHFFFAOYSA-N 1-Heptene Chemical compound CCCCCC=C ZGEGCLOFRBLKSE-UHFFFAOYSA-N 0.000 claims description 24
- OCJBOOLMMGQPQU-UHFFFAOYSA-N 1,4-dichlorobenzene Chemical compound ClC1=CC=C(Cl)C=C1 OCJBOOLMMGQPQU-UHFFFAOYSA-N 0.000 claims description 20
- 125000000217 alkyl group Chemical group 0.000 claims description 17
- 229910052783 alkali metal Inorganic materials 0.000 claims description 16
- 150000001993 dienes Chemical class 0.000 claims description 11
- 150000008044 alkali metal hydroxides Chemical class 0.000 claims description 9
- PVEOYINWKBTPIZ-UHFFFAOYSA-N but-3-enoic acid Chemical compound OC(=O)CC=C PVEOYINWKBTPIZ-UHFFFAOYSA-N 0.000 claims description 8
- 229910052739 hydrogen Inorganic materials 0.000 claims description 7
- 239000001257 hydrogen Substances 0.000 claims description 7
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 7
- RFFLAFLAYFXFSW-UHFFFAOYSA-N 1,2-dichlorobenzene Chemical compound ClC1=CC=CC=C1Cl RFFLAFLAYFXFSW-UHFFFAOYSA-N 0.000 claims description 6
- 229910052977 alkali metal sulfide Inorganic materials 0.000 claims description 6
- IHYNKGRWCDKNEG-UHFFFAOYSA-N n-(4-bromophenyl)-2,6-dihydroxybenzamide Chemical compound OC1=CC=CC(O)=C1C(=O)NC1=CC=C(Br)C=C1 IHYNKGRWCDKNEG-UHFFFAOYSA-N 0.000 claims description 6
- ZPQOPVIELGIULI-UHFFFAOYSA-N 1,3-dichlorobenzene Chemical compound ClC1=CC=CC(Cl)=C1 ZPQOPVIELGIULI-UHFFFAOYSA-N 0.000 claims description 4
- PRBHEGAFLDMLAL-UHFFFAOYSA-N 1,5-Hexadiene Natural products CC=CCC=C PRBHEGAFLDMLAL-UHFFFAOYSA-N 0.000 claims description 4
- OQILOJRSIWGQSM-UHFFFAOYSA-N 1-methylpyrrolidine-2-thione Chemical compound CN1CCCC1=S OQILOJRSIWGQSM-UHFFFAOYSA-N 0.000 claims description 4
- RWSOTUBLDIXVET-UHFFFAOYSA-N Dihydrogen sulfide Chemical compound S RWSOTUBLDIXVET-UHFFFAOYSA-N 0.000 claims description 4
- PYGSKMBEVAICCR-UHFFFAOYSA-N hexa-1,5-diene Chemical compound C=CCCC=C PYGSKMBEVAICCR-UHFFFAOYSA-N 0.000 claims description 4
- 229910000037 hydrogen sulfide Inorganic materials 0.000 claims description 4
- 229910018830 PO3H Inorganic materials 0.000 claims description 3
- 229910006069 SO3H Inorganic materials 0.000 claims description 3
- 229940117389 dichlorobenzene Drugs 0.000 claims description 2
- 150000004816 dichlorobenzenes Chemical class 0.000 claims description 2
- 230000000379 polymerizing effect Effects 0.000 abstract 1
- 239000004734 Polyphenylene sulfide Substances 0.000 description 22
- 238000001125 extrusion Methods 0.000 description 16
- 150000001491 aromatic compounds Chemical class 0.000 description 9
- 238000006243 chemical reaction Methods 0.000 description 9
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 8
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 7
- 230000018044 dehydration Effects 0.000 description 7
- 238000006297 dehydration reaction Methods 0.000 description 7
- 239000000243 solution Substances 0.000 description 7
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 5
- 239000000460 chlorine Substances 0.000 description 5
- 229910052801 chlorine Inorganic materials 0.000 description 5
- 229920001577 copolymer Polymers 0.000 description 5
- UHOVQNZJYSORNB-UHFFFAOYSA-N monobenzene Natural products C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 5
- 239000001632 sodium acetate Substances 0.000 description 5
- 235000017281 sodium acetate Nutrition 0.000 description 5
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 4
- 125000003342 alkenyl group Chemical group 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 239000000155 melt Substances 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 239000000376 reactant Substances 0.000 description 4
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 238000005538 encapsulation Methods 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 229920000098 polyolefin Polymers 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- 238000011084 recovery Methods 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 239000010936 titanium Substances 0.000 description 3
- 229910052719 titanium Inorganic materials 0.000 description 3
- VXNZUUAINFGPBY-UHFFFAOYSA-N 1-Butene Chemical compound CCC=C VXNZUUAINFGPBY-UHFFFAOYSA-N 0.000 description 2
- JTPNRXUCIXHOKM-UHFFFAOYSA-N 1-chloronaphthalene Chemical compound C1=CC=C2C(Cl)=CC=CC2=C1 JTPNRXUCIXHOKM-UHFFFAOYSA-N 0.000 description 2
- LIKMAJRDDDTEIG-UHFFFAOYSA-N 1-hexene Chemical compound CCCCC=C LIKMAJRDDDTEIG-UHFFFAOYSA-N 0.000 description 2
- 229910000619 316 stainless steel Inorganic materials 0.000 description 2
- SHOJXDKTYKFBRD-UHFFFAOYSA-N 4-Methyl-3-penten-2-one, 9CI Chemical compound CC(C)=CC(C)=O SHOJXDKTYKFBRD-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- KAKZBPTYRLMSJV-UHFFFAOYSA-N Butadiene Chemical compound C=CC=C KAKZBPTYRLMSJV-UHFFFAOYSA-N 0.000 description 2
- 238000005033 Fourier transform infrared spectroscopy Methods 0.000 description 2
- 238000001157 Fourier transform infrared spectrum Methods 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- RRHGJUQNOFWUDK-UHFFFAOYSA-N Isoprene Chemical compound CC(=C)C=C RRHGJUQNOFWUDK-UHFFFAOYSA-N 0.000 description 2
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- IAQRGUVFOMOMEM-UHFFFAOYSA-N but-2-ene Chemical compound CC=CC IAQRGUVFOMOMEM-UHFFFAOYSA-N 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 150000001734 carboxylic acid salts Chemical class 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- JBKVHLHDHHXQEQ-UHFFFAOYSA-N epsilon-caprolactam Chemical compound O=C1CCCCCN1 JBKVHLHDHHXQEQ-UHFFFAOYSA-N 0.000 description 2
- 239000000835 fiber Substances 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- 238000002329 infrared spectrum Methods 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 238000005272 metallurgy Methods 0.000 description 2
- CFEYBLWMNFZOPB-UHFFFAOYSA-N pent-4-enenitrile Chemical compound C=CCCC#N CFEYBLWMNFZOPB-UHFFFAOYSA-N 0.000 description 2
- HVAMZGADVCBITI-UHFFFAOYSA-N pent-4-enoic acid Chemical compound OC(=O)CCC=C HVAMZGADVCBITI-UHFFFAOYSA-N 0.000 description 2
- YWAKXRMUMFPDSH-UHFFFAOYSA-N pentene Chemical compound CCCC=C YWAKXRMUMFPDSH-UHFFFAOYSA-N 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- 230000002787 reinforcement Effects 0.000 description 2
- CPRMKOQKXYSDML-UHFFFAOYSA-M rubidium hydroxide Chemical compound [OH-].[Rb+] CPRMKOQKXYSDML-UHFFFAOYSA-M 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229910052979 sodium sulfide Inorganic materials 0.000 description 2
- GRVFOGOEDUUMBP-UHFFFAOYSA-N sodium sulfide (anhydrous) Chemical compound [Na+].[Na+].[S-2] GRVFOGOEDUUMBP-UHFFFAOYSA-N 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- UHEPJGULSIKKTP-UHFFFAOYSA-N sulcatone Chemical compound CC(C)=CCCC(C)=O UHEPJGULSIKKTP-UHFFFAOYSA-N 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 230000010512 thermal transition Effects 0.000 description 2
- PMJHHCWVYXUKFD-SNAWJCMRSA-N (E)-1,3-pentadiene Chemical compound C\C=C\C=C PMJHHCWVYXUKFD-SNAWJCMRSA-N 0.000 description 1
- UNKZCXQBGMGLDG-UHFFFAOYSA-N 1,2,4,5-tetrabutyl-3,6-dichlorobenzene Chemical compound CCCCC1=C(Cl)C(CCCC)=C(CCCC)C(Cl)=C1CCCC UNKZCXQBGMGLDG-UHFFFAOYSA-N 0.000 description 1
- ASWQRHRFJXTXBJ-UHFFFAOYSA-N 1,4-dibromo-2-butyl-5-ethylbenzene Chemical compound CCCCC1=CC(Br)=C(CC)C=C1Br ASWQRHRFJXTXBJ-UHFFFAOYSA-N 0.000 description 1
- KZHZSALFKRDIBQ-UHFFFAOYSA-N 1,4-dibromo-2-ethyl-5-propan-2-ylbenzene Chemical compound CCC1=CC(Br)=C(C(C)C)C=C1Br KZHZSALFKRDIBQ-UHFFFAOYSA-N 0.000 description 1
- SWJPEBQEEAHIGZ-UHFFFAOYSA-N 1,4-dibromobenzene Chemical compound BrC1=CC=C(Br)C=C1 SWJPEBQEEAHIGZ-UHFFFAOYSA-N 0.000 description 1
- PTIQIITULDNGAF-UHFFFAOYSA-N 1,4-dichloro-2,3,5,6-tetramethylbenzene Chemical compound CC1=C(C)C(Cl)=C(C)C(C)=C1Cl PTIQIITULDNGAF-UHFFFAOYSA-N 0.000 description 1
- UTGSRNVBAFCOEU-UHFFFAOYSA-N 1,4-dichloro-2,5-dimethylbenzene Chemical group CC1=CC(Cl)=C(C)C=C1Cl UTGSRNVBAFCOEU-UHFFFAOYSA-N 0.000 description 1
- KFAKZJUYBOYVKA-UHFFFAOYSA-N 1,4-dichloro-2-methylbenzene Chemical compound CC1=CC(Cl)=CC=C1Cl KFAKZJUYBOYVKA-UHFFFAOYSA-N 0.000 description 1
- LFMWZTSOMGDDJU-UHFFFAOYSA-N 1,4-diiodobenzene Chemical compound IC1=CC=C(I)C=C1 LFMWZTSOMGDDJU-UHFFFAOYSA-N 0.000 description 1
- AZAMPMUEKXNYFW-UHFFFAOYSA-N 1-[2-(2-oxopyrrolidin-1-yl)ethyl]pyrrolidin-2-one Chemical compound O=C1CCCN1CCN1C(=O)CCC1 AZAMPMUEKXNYFW-UHFFFAOYSA-N 0.000 description 1
- NHDODQWIKUYWMW-UHFFFAOYSA-N 1-bromo-4-chlorobenzene Chemical compound ClC1=CC=C(Br)C=C1 NHDODQWIKUYWMW-UHFFFAOYSA-N 0.000 description 1
- UCCUXODGPMAHRL-UHFFFAOYSA-N 1-bromo-4-iodobenzene Chemical compound BrC1=CC=C(I)C=C1 UCCUXODGPMAHRL-UHFFFAOYSA-N 0.000 description 1
- YZJKRCHJFHUTTK-UHFFFAOYSA-N 1-butyl-2,5-dichloro-3-ethylbenzene Chemical compound CCCCC1=CC(Cl)=CC(CC)=C1Cl YZJKRCHJFHUTTK-UHFFFAOYSA-N 0.000 description 1
- GWQSENYKCGJTRI-UHFFFAOYSA-N 1-chloro-4-iodobenzene Chemical compound ClC1=CC=C(I)C=C1 GWQSENYKCGJTRI-UHFFFAOYSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- MFGOFGRYDNHJTA-UHFFFAOYSA-N 2-amino-1-(2-fluorophenyl)ethanol Chemical compound NCC(O)C1=CC=CC=C1F MFGOFGRYDNHJTA-UHFFFAOYSA-N 0.000 description 1
- AKWXOKCDAFZPCM-UHFFFAOYSA-N 2-butyl-1,4-dichlorobenzene Chemical compound CCCCC1=CC(Cl)=CC=C1Cl AKWXOKCDAFZPCM-UHFFFAOYSA-N 0.000 description 1
- WBTOYDCQRYRSAH-UHFFFAOYSA-N 2-ethyl-1,4-diiodobenzene Chemical compound CCC1=CC(I)=CC=C1I WBTOYDCQRYRSAH-UHFFFAOYSA-N 0.000 description 1
- YHQXBTXEYZIYOV-UHFFFAOYSA-N 3-methylbut-1-ene Chemical compound CC(C)C=C YHQXBTXEYZIYOV-UHFFFAOYSA-N 0.000 description 1
- MVQHVFFNSQIDLB-UHFFFAOYSA-N 3-methylhex-5-en-2-amine Chemical compound CC(N)C(C)CC=C MVQHVFFNSQIDLB-UHFFFAOYSA-N 0.000 description 1
- OIRBZGZDHXBOFW-UHFFFAOYSA-M 4-ethyltetradecanoate;rubidium(1+) Chemical compound [Rb+].CCCCCCCCCCC(CC)CCC([O-])=O OIRBZGZDHXBOFW-UHFFFAOYSA-M 0.000 description 1
- ODPYDILFQYARBK-UHFFFAOYSA-N 7-thiabicyclo[4.1.0]hepta-1,3,5-triene Chemical compound C1=CC=C2SC2=C1 ODPYDILFQYARBK-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 239000005641 Methyl octanoate Substances 0.000 description 1
- ZWXPDGCFMMFNRW-UHFFFAOYSA-N N-methylcaprolactam Chemical compound CN1CCCCCC1=O ZWXPDGCFMMFNRW-UHFFFAOYSA-N 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 229910001508 alkali metal halide Inorganic materials 0.000 description 1
- 150000008045 alkali metal halides Chemical class 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 125000002877 alkyl aryl group Chemical group 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 150000001555 benzenes Chemical class 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- HYWCISCKYCCFMI-UHFFFAOYSA-M butanoate;rubidium(1+) Chemical compound [Rb+].CCCC([O-])=O HYWCISCKYCCFMI-UHFFFAOYSA-M 0.000 description 1
- 229910052792 caesium Inorganic materials 0.000 description 1
- TVFDJXOCXUVLDH-UHFFFAOYSA-N caesium atom Chemical compound [Cs] TVFDJXOCXUVLDH-UHFFFAOYSA-N 0.000 description 1
- HUCVOHYBFXVBRW-UHFFFAOYSA-M caesium hydroxide Inorganic materials [OH-].[Cs+] HUCVOHYBFXVBRW-UHFFFAOYSA-M 0.000 description 1
- 238000001460 carbon-13 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 239000003575 carbonaceous material Substances 0.000 description 1
- IBSGAWQJFSDRBJ-UHFFFAOYSA-M cesium sulfanide Chemical compound [SH-].[Cs+] IBSGAWQJFSDRBJ-UHFFFAOYSA-M 0.000 description 1
- WDPREJLILODKKB-UHFFFAOYSA-M cesium;cyclododecanecarboxylate Chemical compound [Cs+].[O-]C(=O)C1CCCCCCCCCCC1 WDPREJLILODKKB-UHFFFAOYSA-M 0.000 description 1
- NMKVMUXQLXZRNQ-UHFFFAOYSA-M cesium;hexanoate Chemical compound [Cs+].CCCCCC([O-])=O NMKVMUXQLXZRNQ-UHFFFAOYSA-M 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 239000008199 coating composition Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- QTNDMWXOEPGHBT-UHFFFAOYSA-N dicesium;sulfide Chemical compound [S-2].[Cs+].[Cs+] QTNDMWXOEPGHBT-UHFFFAOYSA-N 0.000 description 1
- 238000000113 differential scanning calorimetry Methods 0.000 description 1
- XNMQEEKYCVKGBD-UHFFFAOYSA-N dimethylacetylene Natural products CC#CC XNMQEEKYCVKGBD-UHFFFAOYSA-N 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 230000009477 glass transition Effects 0.000 description 1
- 150000002367 halogens Chemical group 0.000 description 1
- CKDDRHZIAZRDBW-UHFFFAOYSA-N henicosanoic acid Chemical compound CCCCCCCCCCCCCCCCCCCCC(O)=O CKDDRHZIAZRDBW-UHFFFAOYSA-N 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- XVQUOJBERHHONY-UHFFFAOYSA-N isometheptene Chemical compound CNC(C)CCC=C(C)C XVQUOJBERHHONY-UHFFFAOYSA-N 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- XIXADJRWDQXREU-UHFFFAOYSA-M lithium acetate Chemical compound [Li+].CC([O-])=O XIXADJRWDQXREU-UHFFFAOYSA-M 0.000 description 1
- 229940031993 lithium benzoate Drugs 0.000 description 1
- GLNWILHOFOBOFD-UHFFFAOYSA-N lithium sulfide Chemical compound [Li+].[Li+].[S-2] GLNWILHOFOBOFD-UHFFFAOYSA-N 0.000 description 1
- IFXWSHQRYWTGOH-UHFFFAOYSA-M lithium;2-(4-ethylcyclohexyl)acetate Chemical compound [Li+].CCC1CCC(CC([O-])=O)CC1 IFXWSHQRYWTGOH-UHFFFAOYSA-M 0.000 description 1
- IIDVGIFOWJJSIJ-UHFFFAOYSA-M lithium;2-methylpropanoate Chemical compound [Li+].CC(C)C([O-])=O IIDVGIFOWJJSIJ-UHFFFAOYSA-M 0.000 description 1
- ZSICDRPAYOOLQB-UHFFFAOYSA-M lithium;2-phenylacetate Chemical compound [Li+].[O-]C(=O)CC1=CC=CC=C1 ZSICDRPAYOOLQB-UHFFFAOYSA-M 0.000 description 1
- LDJNSLOKTFFLSL-UHFFFAOYSA-M lithium;benzoate Chemical compound [Li+].[O-]C(=O)C1=CC=CC=C1 LDJNSLOKTFFLSL-UHFFFAOYSA-M 0.000 description 1
- OYTJIZNGQNUSAK-UHFFFAOYSA-M lithium;cyclohexanecarboxylate Chemical compound [Li+].[O-]C(=O)C1CCCCC1 OYTJIZNGQNUSAK-UHFFFAOYSA-M 0.000 description 1
- RQZHWDLISAJCLK-UHFFFAOYSA-M lithium;heptanoate Chemical compound [Li+].CCCCCCC([O-])=O RQZHWDLISAJCLK-UHFFFAOYSA-M 0.000 description 1
- KDDRURKXNGXKGE-UHFFFAOYSA-M lithium;pentanoate Chemical compound [Li+].CCCCC([O-])=O KDDRURKXNGXKGE-UHFFFAOYSA-M 0.000 description 1
- AXMOZGKEVIBBCF-UHFFFAOYSA-M lithium;propanoate Chemical compound [Li+].CCC([O-])=O AXMOZGKEVIBBCF-UHFFFAOYSA-M 0.000 description 1
- HXQGSILMFTUKHI-UHFFFAOYSA-M lithium;sulfanide Chemical compound S[Li] HXQGSILMFTUKHI-UHFFFAOYSA-M 0.000 description 1
- GPSDUZXPYCFOSQ-UHFFFAOYSA-M m-toluate Chemical compound CC1=CC=CC(C([O-])=O)=C1 GPSDUZXPYCFOSQ-UHFFFAOYSA-M 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- LQAVWYMTUMSFBE-UHFFFAOYSA-N pent-4-en-1-ol Chemical compound OCCCC=C LQAVWYMTUMSFBE-UHFFFAOYSA-N 0.000 description 1
- QYZLKGVUSQXAMU-UHFFFAOYSA-N penta-1,4-diene Chemical compound C=CCC=C QYZLKGVUSQXAMU-UHFFFAOYSA-N 0.000 description 1
- 238000005191 phase separation Methods 0.000 description 1
- PMJHHCWVYXUKFD-UHFFFAOYSA-N piperylene Natural products CC=CC=C PMJHHCWVYXUKFD-UHFFFAOYSA-N 0.000 description 1
- 229920002959 polymer blend Polymers 0.000 description 1
- 229960003975 potassium Drugs 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 239000004300 potassium benzoate Substances 0.000 description 1
- 235000010235 potassium benzoate Nutrition 0.000 description 1
- 229940103091 potassium benzoate Drugs 0.000 description 1
- ZOCLAPYLSUCOGI-UHFFFAOYSA-M potassium hydrosulfide Chemical compound [SH-].[K+] ZOCLAPYLSUCOGI-UHFFFAOYSA-M 0.000 description 1
- DPLVEEXVKBWGHE-UHFFFAOYSA-N potassium sulfide Chemical compound [S-2].[K+].[K+] DPLVEEXVKBWGHE-UHFFFAOYSA-N 0.000 description 1
- FDJJMQRIEWTAQS-UHFFFAOYSA-M potassium;2-(4-methylphenyl)acetate Chemical compound [K+].CC1=CC=C(CC([O-])=O)C=C1 FDJJMQRIEWTAQS-UHFFFAOYSA-M 0.000 description 1
- HLCXPTFJMZUPPP-UHFFFAOYSA-M potassium;2-cyclohexylacetate Chemical compound [K+].[O-]C(=O)CC1CCCCC1 HLCXPTFJMZUPPP-UHFFFAOYSA-M 0.000 description 1
- HIDKSOTTZRMUML-UHFFFAOYSA-M potassium;dodecanoate Chemical compound [K+].CCCCCCCCCCCC([O-])=O HIDKSOTTZRMUML-UHFFFAOYSA-M 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000004064 recycling Methods 0.000 description 1
- 229910052701 rubidium Inorganic materials 0.000 description 1
- IGLNJRXAVVLDKE-UHFFFAOYSA-N rubidium atom Chemical compound [Rb] IGLNJRXAVVLDKE-UHFFFAOYSA-N 0.000 description 1
- LXOXXUIVMOYGST-UHFFFAOYSA-M rubidium(1+);sulfanide Chemical compound [SH-].[Rb+] LXOXXUIVMOYGST-UHFFFAOYSA-M 0.000 description 1
- AHKSSQDILPRNLA-UHFFFAOYSA-N rubidium(1+);sulfide Chemical compound [S-2].[Rb+].[Rb+] AHKSSQDILPRNLA-UHFFFAOYSA-N 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 229960003885 sodium benzoate Drugs 0.000 description 1
- HYHCSLBZRBJJCH-UHFFFAOYSA-M sodium hydrosulfide Chemical compound [Na+].[SH-] HYHCSLBZRBJJCH-UHFFFAOYSA-M 0.000 description 1
- RYYKJJJTJZKILX-UHFFFAOYSA-M sodium octadecanoate Chemical compound [Na+].CCCCCCCCCCCCCCCCCC([O-])=O RYYKJJJTJZKILX-UHFFFAOYSA-M 0.000 description 1
- JXKPEJDQGNYQSM-UHFFFAOYSA-M sodium propionate Chemical compound [Na+].CCC([O-])=O JXKPEJDQGNYQSM-UHFFFAOYSA-M 0.000 description 1
- 239000004324 sodium propionate Substances 0.000 description 1
- 235000010334 sodium propionate Nutrition 0.000 description 1
- 229960003212 sodium propionate Drugs 0.000 description 1
- ZZYMZNLESRCHHY-UHFFFAOYSA-M sodium;3-methylcyclopentane-1-carboxylate Chemical compound [Na+].CC1CCC(C([O-])=O)C1 ZZYMZNLESRCHHY-UHFFFAOYSA-M 0.000 description 1
- KDGFSUSPVCYLFX-UHFFFAOYSA-M sodium;4-phenylcyclohexane-1-carboxylate Chemical compound [Na+].C1CC(C(=O)[O-])CCC1C1=CC=CC=C1 KDGFSUSPVCYLFX-UHFFFAOYSA-M 0.000 description 1
- LHYPLJGBYPAQAK-UHFFFAOYSA-M sodium;pentanoate Chemical compound [Na+].CCCCC([O-])=O LHYPLJGBYPAQAK-UHFFFAOYSA-M 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- OHEFFKYYKJVVOX-UHFFFAOYSA-N sulcatol Natural products CC(O)CCC=C(C)C OHEFFKYYKJVVOX-UHFFFAOYSA-N 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G75/00—Macromolecular compounds obtained by reactions forming a linkage containing sulfur with or without nitrogen, oxygen, or carbon in the main chain of the macromolecule
- C08G75/02—Polythioethers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G75/00—Macromolecular compounds obtained by reactions forming a linkage containing sulfur with or without nitrogen, oxygen, or carbon in the main chain of the macromolecule
- C08G75/02—Polythioethers
- C08G75/0204—Polyarylenethioethers
- C08G75/025—Preparatory processes
- C08G75/0259—Preparatory processes metal hydrogensulfides
Definitions
- This invention relates to the production of arylene sulfide polymers. In one aspect, this invention relates to the production of phenylene sulfide polymers. In another aspect, this invention relates to the production of arylene sulfide polymers having a high extrusion rate or a low inherent viscosity. In a further aspect, this invention relates to the production of arylene sulfide polymers in which the functionality of the polymer end groups is controllable. In a still further aspect, this invention relates to arylene sulfide polymers having alkyl, alkenyl, functionalized alkyl or functionalized alkenyl end groups.
- Arylene sulfide polymers can be characterized at least in part in terms of a melt flow rate. It is generally considered that a melt flow rate is inversely related to molecular weight for polymeric materials in general and for arylene sulfide polymers in particular. Extrusion rate, which is more specifically defined hereinafter, is a specific type of melt flow rate which is particularly useful for characterizing arylene sulfide polymers in the lower molecular weight range.
- Arylene sulfide polymers can also be characterized at least in part in terms of inherent viscosity. It is generally considered that inherent viscosity, which is more specifically defined hereinafter, is directly related to molecular weight for polymeric materials in general and for arylene sulfide polymers in particular.
- Arylene sulfide polymers having a relatively high extrusion rate or relatively low inherent viscosity are desirable for a variety of applications such as encapsulation of electronic components and coating formulations.
- US-A-4,437,182 and 4,482,665 provide exemplary disclosures of compositions comprising arylene sulfide polymers which are employed in the encapsulation of electronic components.
- Arylene sulfide polymers in which the functionality of the polymer end groups is controllable are desirable for improving adhesion of the polymer to metal substrates and/or fiber reinforcement. Such polymers are also desirable for use in the preparation of copolymers by reaction at the polymer end groups having the desired functionality. The copolymers would be particularly useful as compatabilizers in polymer blends.
- a process for preparing arylene sulfide polymers comprises contacting at least one sulfur source, at least one cyclic organic amide, and at least one dihaloaromatic compound to form a polymerization mixture, subjecting the polymerization mixture to polymerization conditions of temperature and time sufficient to form the arylene sulfide polymer, and recovering the arylene sulfide polymer, wherein the process is conducted in the presence of a suitable olefin.
- the olefin may be added to the polymerization mixture prior to subjecting the polymerization mixture to polymerization conditions, or during the time the polymerization mixture is being subjected to polymerization conditions.
- an arylene sulfide polymer composition comprising end groups resulting from polymerization in the presence of suitable olefins selected from the group consisting of alkyl radicals, alkenyl radicals, functionalized alkyl radicals, functionalized alkenyl radicals and combinations thereof.
- This invention relates to arylene sulfide polymer compositions and a process for preparing arylene sulfide polymers having a readily controllable molecular weight as measured by extrusion rate or inherent viscosity and/or polymer end group functionality which is readily controllable comprising the steps of: (a) dehydrating an aqueous mixture comprising at least one sulfur source and at least one cyclic organic amide thereby forming a dehydrated mixture, (b) contacting at least one dihaloaromatic compound with the dehydrated mixture to produce a polymerization mixture, (c) subjecting the polymerization mixture to polymerization conditions of temperature and time sufficient to form the arylene sulfide polymer, and (d) recovering the arylene sulfide polymer, wherein the process is conducted in the presence of an olefin having 2 to about 20 carbon atoms.
- the arylene sulfide polymers having a relatively high extrusion rate or low inherent viscosity made according to this invention are readily recoverable and well suited for use in applications where such relatively high extrusion rate or low inherent viscosity arylene sulfide polymers are desired, e.g. encapsulation of electronic components and coatings.
- the arylene sulfide polymers having polymer end groups of desired functionality made according to this invention are readily recoverable and well suited for use in applications where such polymer end group functionality is desired, e.g. improved adhesion to metal substrates and/or fiber reinforcement and in the preparation of copolymers for use as blend compatabilizers.
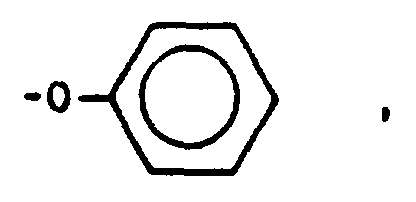
- arylene sulfide polymer is defined by the formula wherein -Ar-S- is the repeating unit, i is the number of repeating units in the polymer molecule, and Ar is selected from the group consisting of wherein each R is selected from the group consisting of hydrogen and alkyl radicals having 1 to about 4 carbon atoms, the total number of carbon atoms in all of the R groups in the repeat unit being 0 to about 12.
- extrusion rate refers to a flow rate measurement on molten polymer based on ASTM D 1238-86, Condition 315/0.345, modified to use an orifice having a length of 31.8 mm and a 5 minute preheat time.
- extrusion rate is a specific type of melt flow measurement which is particularly useful in characterizing arylene sulfide polymers in the lower molecular weight range.
- inherent viscosity refers to dilute solution viscosity which is the ratio of the natural logarithm of the relative viscosity to the polymer solution concentration in grams per deciliter.
- the relative viscosity is the ratio of the flow time of a specific solution of the polymer to the flow time of the pure solvent.
- inherent viscosities for arylene sulfide polymers are measured generally according to the method described in ASTM D 1243-79 wherein samples of dried polymer are dissolved in 1-chloronaphthalene at 210°C at a polymer concentration of 0.4 grams per deciliter (g/dL) utilizing a No. 50 Cannon-Fenske viscometer.
- inherent viscosity is a measurement which is a function of molecular weight and which is particularly useful in characterizing arylene sulfide polymers in the lower molecular weight range.
- Suitable olefins which are utilized according to the invention are selected from the group consisting of alkenes having 2 to about 20 carbon atoms, dienes having 2 to about 20 carbon atoms, functionalized alkenes represented by the formulas and and functionalized dienes represented by the formula wherein R' is selected from the group consisting of hydrogen and alkyl groups having 1 to about 10 carbon atoms, R" is selected from the group consisting of
- R"' is selected from the group consisting of -H and -CH3
- Y is selected from the group consisting of -H and -COO ⁇
- m is 0 or 1
- n is 0 to 10
- p is 0 or 1
- Y, n and R" in any of the above formulas may be the same or different and wherein the total number of carbon atoms in the functionalized alkene of formula (II) and the functionalized diene is from 2 to about 20 and the total number of carbon atoms in the functionalized alkene of formula (I) is 3 or 4.
- a Y group of COO ⁇ can be obtained by addition of the corresponding carboxylate salt or it can be prepared in-situ by reaction of the free carboxylic acid with a stoichiometric amount of a base, e.g. an alkali metal hydroxide.
- a base e.g. an alkali metal hydroxide.
- the alkenes, dienes, functionalized alkenes of formula (II) and functionalized dienes which are employed according to the invention are compounds having 2 to about 20 carbon atoms per molecule, preferably about 4 to about 9 carbon atoms per molecule, and most preferably about 5 to about 7 carbon atoms per molecule.
- Suitable olefins which can be employed in the process of the invention include 1-butene, 1-pentene, 1-hexene, 1-heptene, 1-nonene, 2-butene, 3-methyl-1-butene, 1,3-butadiene, 1,3-pentadiene, 1,4-pentadiene, 1,5-hexadiene, 2-methyl-1,3-butadiene, 3-butenoic acid, 4-pentenenitrile, 4-penten-1-ol, 4-methyl-3-penten-2-one, 5-amino-4-methyl-1-hexene, 2-methyl-6-methylamino-2-heptene and 6-methyl-5-hepten-2-one, and the like, and mixtures of any two or more thereof.
- the presently preferred compounds are 1-heptene, 1,5-hexadiene, 1-nonene, and 3-butenoic acid because of their effectiveness.
- the amount of olefin employed according to the process of the invention can be conveniently expressed in terms of a molar ratio based on the sulfur source.
- the molar ratio of olefin to sulfur source will be about 0.02:1 to about 0.3:1, preferably about 0.05:1 to about 0.25:1, and most preferably about 0.1:1 to about 0.2:1.
- the olefin will be effective in the process of the invention when added to the polymerization mixture prior to subjecting the polymerization mixture to polymerization conditions, or during the time the polymerization mixture is being subjected to polymerization conditions. If an aqueous sulfur-containing compound is used and a dehydration step is required, the olefin may only be added prior to dehydration if the olefin is not volatile under dehydration conditions.
- the dihaloaromatic compounds which are employed according to the invention are compounds having 6 to about 22 carbon atoms per molecule.
- the halogen substituent on the dihaloaromatic compound can be selected from the group consisting of chlorine, bromine and iodine.
- the dihaloaromatic compound will be dihalo-substituted benzene and more preferably dichloro-substituted benzene.
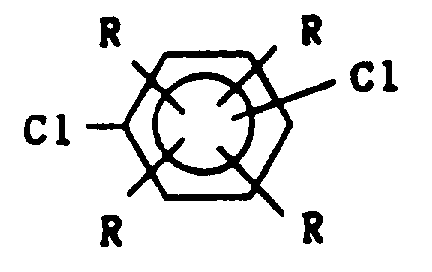
- the dihaloaromatic compound is selected from the group consisting of p-dichlorobenzene and mixtures of p-dichlorobenzene with a total of about 0 to about 10 mole percent of at least one of m-dichlorobenzene, o-dichlorobenzene and alkyl-substituted dichlorobenzene having the formula wherein R is as defined herein and at least one R is not hydrogen.
- dihaloaromatic compounds examples include p-dichlorobenzene, p-dibromobenzene, p-diiodobenzene, 1-chloro-4-bromobenzene, 1-chloro-4-iodobenzene, 1-bromo-4-iodobenzene, 2,5-dichlorotoluene, 2,5-dichloro-p-xylene, 1-ethyl-4-isopropyl-2,5-dibromobenzene, 1,2,4,5-tetramethyl-3,6-dichlorobenzene, 1,2,4,5-tetrabutyl-3,6-dichlorobenzene, 1-ethyl-3-butyl-2,5-dichlorobenzene, 1-ethyl-2,5-diiodobenzene, 1-butyl-2,5-dichlorobenzene, 1-butyl-4-ethyl-2,5-ddi
- the cyclic organic amide used in the process of the invention should be substantially liquid at the reaction temperatures and pressures employed.
- the cyclic organic amides can have 5 to about 12 carbon atoms per molecule.
- suitable cyclic organic amides include N,N'-ethylenedipyrrolidone, N-methyl-2-pyrrolidone, pyrrolidone, caprolactam, N-ethyl caprolactam, N-methyl caprolactam and mixtures thereof.
- the presently preferred cyclic organic amide is N-methyl-2-pyrrolidone because of its effectiveness and commercial availability.
- the amount of cyclic organic amide employed according to the process of the invention can be expressed in terms of a molar ratio of cyclic organic amide to sulfur source compounds. Broadly, the molar ratio of cyclic organic amide to sulfur source compound will be about 1.5:1 to about 25:1, preferably about 2:1 to about 8:1.
- suitable sulfur sources which can be employed in the production of the arylene sulfide polymers include alkali metal sulfides, alkali metal bisulfides, N-methyl-2-pyrrolidinethione, and hydrogen sulfide.
- the alkali metal sulfides can be employed with good results in the absence of any added alkali metal hydroxide whereas the other suitable sulfur sources are preferably employed in the process of the invention in the presence of an added alkali metal hydroxide.
- the amount of added alkali metal hydroxide will generally be in the range of from about 0.3:1 to about 4:1, preferably about 0.4:1 to about 2:1 moles per mole of alkali metal bisulfides or N-methyl-2-pyrrolidinethione.
- the amount of added alkali metal hydroxide is generally within the range of about 1.3:1 to about 5:1 preferably about 1.4:1 to about 3:1 moles per mole of hydrogen sulfide employed.
- Alkali metal bisulfides that can be employed according to the invention include sodium bisulfide, lithium bisulfide, potassium bisulfide, rubidium bisulfide, cesium bisulfide, and mixtures thereof.
- Sodium bisulfide is preferred because of ready availability and good results obtained therewith.
- the alkali metal bisulfide can conveniently be utilized in the process of the invention as an aqueous solution. For example, an aqueous solution of sodium bisulfide having about 60 weight percent sodium bisulfide is convenient to use.
- Alkali metal sulfides which can be employed in the process of the invention include lithium sulfide, sodium sulfide, potassium sulfide, rubidium sulfide, cesium sulfide, and mixtures thereof.
- the alkali metal sulfide can be used in anhydrous form, as a hydrate, or as an aqueous mixture.
- Sodium sulfide is preferred because of ready availability and good results obtained therewith.
- the ratio of reactants for the preparation of arylene sulfide polymers according to the invention can vary considerably, the ratio of moles of dihaloaromatic compound to atoms of divalent sulfur in the sulfur source should be within the range of about 0.8:1 to about 2:1, preferably about 0.95:1 to about 1.3:1.
- a polymerization modifier such as an alkali metal carboxylate can be employed in the process of the invention particularly when the olefin utilized is a functionalized alkene or a functionalized diene.
- the alkali metal carboxylates that can be employed in the process of the invention can be represented by the formula R''''CO2M where R'''' is a hydrocarbyl radical selected from alkyl, cycloalkyl, and aryl and combinations thereof such as alkaryl, aralkyl, and the like, the number of carbon atoms in said R'''' being within the range of 1 to about 20, and M is an alkali metal selected from lithium, sodium, potassium, rubidium, and cesium.
- alkali metal carboxylates examples include lithium acetate, sodium acetate, potassium acetate, lithium propionate, sodium propionate, lithium 2-methylpropionate, rubidium butyrate, lithium valerate, sodium valerate, cesium hexanoate, lithium heptanoate, lithium 2-methyl octanoate, potassium dodecanoate, rubidium 4-ethyltetradecanoate, sodium octadecanoate, sodium heneicosanoate, lithium cyclohexane carboxylate, cesium cyclododecane carboxylate, sodium 3-methylcyclopentane carboxylate, potassium cyclohexylacetate, potassium benzoate, lithium benzoate, sodium benzoate, potassium m-toluate, lithium phenylacetate, sodium 4-phenylcyclohexane carboxylate, potassium p-tolylacetate, lithium 4-ethylcyclohexyla
- the amount of alkali metal carboxylate employed according to the invention can be expressed in terms of molar ratio based on the sulfur source compound employed. Broadly, the molar ratio of alkali metal carboxylate to sulfur source compound will be from about 0.002:1 to about 4:1, preferably about 0.1:1 to about 2:1.
- the alkali metal carboxylate can be used in anhydrous form, as a hydrate, or as an aqueous mixture.
- Suitable polymerization conditions include a reaction temperature which can vary over a wide range but will generally be within the range of about 200°C to about 450°C, preferably from about 210°C to about 350°C.
- the reaction time will be within the range of about 10 minutes to about 72 hours and preferably about 1 hour to about 8 hours.
- the pressure need only be sufficient to maintain the dihaloaromatic compound and the cyclic organic amide substantially in the liquid phase, and to substantially retain the sulfur source therein.
- the flashed reaction mixture residue can be slurried with a liquid diluent such as water in which the alkali metal halides and other impurities are soluble.
- a liquid diluent such as water in which the alkali metal halides and other impurities are soluble.
- the liquid diluent is removed with dissolved impurities such as by filtration leaving a particular arylene sulfide polymer. This washing process can be repeated until the desired level of arylene sulfide polymer purity is attained.
- Another known method that can be employed is the "water quench" process described in US-A-4,415,729, wherein the polymerization mixture is contacted at a temperature above that at which the arylene sulfide polymer is soluble in the polymerization mixture with a sufficient amount of a separation agent that is soluble in the cyclic organic amide and is a non-solvent for the arylene sulfide polymer, e.g. water, to cause or enhance a phase separation.
- a separation agent that is soluble in the cyclic organic amide and is a non-solvent for the arylene sulfide polymer, e.g. water
- phase-separated mixture produces a slurry of particulate arylene sulfide polymer in the cyclic organic amide which can be filtered to recover the particulate arylene sulfide polymer.
- the separated polymer can be washed as described above.
- the arylene sulfide polymer will comprise end groups selected from the group consisting of alkyl radicals having 2 to about 20 carbon atoms, alkenyl radicals having 2 to about 20 carbon atoms, functionalized alkyl radicals derived from the formula (I) having 3 or 4 carbon atoms, functionalized alkyl radicals derived from the formula (II) having 2 to about 20 carbon atoms, functionalized alkenyl radicals having 2 to about 20 carbon atoms and combinations thereof wherein the end groups are a result of the use of olefins as defined herein according to the process of the invention.
- arylene sulfide polymer composition end groups will comprise structures selected from the group consisting of and combinations thereof wherein m, n, p, R', R", R"' and Y are as defined herein and Y, n and R" in any of end groups (III) - (X) may be the same or different, and wherein the total number of carbon atoms in the end groups of formulas (III) and (IV) is 3 or 4, and the total number of carbon atoms in the end groups of formulas (V), (VI), (VII), (VIII), (IX) and (X) is 2 to about 20.
- Polymer thermal transitions were determined by differential scanning calorimetry (DSC) on a Perkin-Elmer DSC, Model DSC-2, with a nitrogen atmosphere at a sample heating rate of 20°C/min.
- the polymer glass transition (Tg) and melting point (Tm) are reported as °C.
- Polymer ash levels were determined by burning a weighed sample of the polymer in a platimum dish. Residual carbonaceous material was removed by heating the residue at 540°C in a muffle furnace. The weight of the residue (ash) is expressed as a percentage of the original weight of the polymer.
- the polymer chlorine content was determined by neutron activation, with the results reported as weight percent chlorine based on the original sample weight.
- Fourier transform infrared (FTIR) spectra were determined using film pressed from the polymer without heat. The presence of alkyl groups was indicated by a peak at approximately 2925 cm-1.
- This example is a control run in which PPS is made without the addition of an olefin.
- a one-liter, stainless steel reactor was charged with 1.00 g-mol of sodium hydrosulfide (NaSH) as an aqueous mixture containing about 60 weight percent NaSH, 1.03 g-mol sodium hydroxide (NaOH) as a solid, and 2.5 g-mol N-methyl-2-pyrrolidone (NMP).
- NaSH sodium hydrosulfide
- NaOH sodium hydroxide
- NMP N-methyl-2-pyrrolidone
- PPS polymerization run 3 was carried out in a manner similar to that described in Example 2 except that the dehydrated mixture was charged with 0.515 g-mol meta-DCB and 0.515 g-mol para-DCB with the 0.25 g-mol 1-nonene and NMP. In this polymerization, the polymerization mixture was held at 235°C for one hour and at 265°C for two hours.
- the product of this polymerization was a copolymer recovered in a yield of 56 mole percent with an IV of 0.06 dL/g.
- the polymer Tg was about 25° C.
- Carbon-13 NMR spectra of the copolymer in benzene and tetrahydrofuran (THF) showed that the polymer contained alkyl groups.
- Example 2 After the reaction had been exposed to conditions noted in Example 1, the addition of a large quantity of water to the mixture revealed that no polyolefin was formed as evidenced by the lack of a precipitate forming upon addition of the water. The conditions used for the PPS polymerization do not produce any significant amounts of polyolefins from the added olefins.
- the extrusion rates of polymers 6 and 7 are higher than for control run 5. IR spectra of polymers 6 and 7 show that alkyl groups are present in the polymer. The presence of sodium acetate in run 7 does not significantly effect the polymer molecular weight. 1-Heptene added to a PPS polymerization is incorporated into the polymer and reduces the PPS melt viscosity.
- An invention run was carried out with an unsaturated carboxylic acid added to the polymerization to determine the effect of unsaturated carboxylic acid salts on PPS polymerizations.
- control run 8 0.25 g-mol of 4-pentenoic acid was charged with the NaSH. NaOH and NMP before dehydration in a polymerization procedure similar to that described for run 5 in Example 5.
- invention run 9 0.29 g-mol 3-butenoic acid was charged with the NaSH, NaOH and NMP before dehydration. Both polymerizations were carried out for one hour at 235°C and three hours at 265°C. Additional NaOH was used in these polymerization runs for neutralization of the acid groups. The chemicals used and the results of these two runs are summarized in Table II. Run 5 from Example 5 is included in the table for comparison.
- Control run 8 with 4-pentenoic acid did not produce a decrease in the polymer melt viscosity.
- An FTIR spectrum of the polymer from run 8 showed no alkyl groups to be present while an FTIR spectrum of the polymer from run 9 did show the presence of alkyl groups.
- Run 10 is a control run made in a titanium reactor without any added 1-heptene and with polymerization conditions of one hour at 235°C and two hours at 265°C.
- Run 11 is an invention run made in a 316 stainless steel reactor with 0.25 g-mol 1-heptene and polymerization conditions of three hours at 245°C.
- Runs 12 through 16 all used a titanium reactor and 0.25 g-mol 1-heptene. 0.30 g-mol of NaOAc was also present in run 16.
- the polymerization conditions for run 12 were three hours at 245°C; run 13, 2.5 hours at 255°C; run 14, 4 hours at 235°C; run 15, two hours at 265°C; and run 16, three hours at 265°C.
- Table III The chemicals used and the results of these runs are summarized in Table III.
- PPS polymerization run 17 was carried out in a manner similar to that described in run 11 of Example 7 except that 0.10 g-mol of 1,5-hexadiene was added to the dehydrated mixture instead of 1-heptene.
- a control PPS polymerization was carried out to produce a truly low molecular weight PPS for comparison with the products of the invention runs above.
- Run 18 was carried out in a procedure similar to that described in Example 1 except that the reactor was charged with 0.5 g-mol NaSH, 0.5 g-mol NaOH, and 1.25 g-mol NMP. The dehydrated mixture was charged with 1.03 g-mol DCB and 1.0 g-mol NMP. Due to the large excess of DCB over the sulfur source, a very low molecular weight PPS was produced. Polymer 18 was recovered in 94 mole percent yield and had a degree of polymerization of about 13 as determined by 13C NMR.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Polymers With Sulfur, Phosphorus Or Metals In The Main Chain (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
Claims (12)
- Verfahren zur Herstellung von Arylensulfidpolymeren, das folgende Stufen umfaßt:(a) Kontaktieren mindestens einer Schwefelquelle, mindestens eines cyclischen organischen Amids und mindestens einer aromatischen Dihalogenverbindung unter Bildung eines Polymerisationsgemisches;(b) Unterwerfen des Polymerisationsgemisches unter eine Polymerisation; und(c) Gewinnung des Arylensulfidpolymeren;wobei das Verfahren in Gegenwart eines Olefins durchgeführt wird, das unter Alkenen mit 2 bis 20 Kohlenstoffatomen, Dienen mit 2 bis 20 Kohlenstoffatomen, funktionalisierten Alkenen mit der Formel:



-H, -CN, -NR'₂, -NO₂, -Aryl,
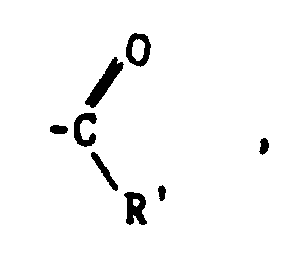

ausgewählt ist, R"' unter -H und -CH₃ ausgewählt ist, Y unter -H und -COO⁻ ausgewählt ist, m den Wert 0 oder 1 hat, n einen Wert von 0 bis 10 hat, p den Wert 0 oder 1 hat und Y, n und R" in jeder der vorstehenden Formeln gleich oder verschieden sein können und wobei die Gesamtzahl der Kohlenstoffatome in dem funktionalisierten Alken der Formel (II) und dem funktionalisierten Dien 2 bis 20 beträgt und die Gesamtzahl der Kohlenstoffatome in dem funktionalisierten Alken der Formel (I) 3 oder 4 beträgt. - Verfahren nach Anspruch 1, wobei das molare Verhältnis des Olefins zur Schwefelquelle 0,02:1 bis 0,3:1 beträgt.
- Verfahren nach Anspruch 1 oder 2, wobei das Olefin zu dem Polymerisationsgemisch gegeben wird, bevor das Polymerisationsgemisch der Polymerisation unterworfen wird.
- Verfahren nach Anspruch 1 oder 2, wobei das Olefin während der Zeit, in der das Polymerisationsgemisch der Polymerisation unterworfen wird, zugegeben wird.
- Verfahren nach Anspruch 3 oder 4, wobei die Schwefelquelle unter Alkalimetallsulfiden, Alkalimetallbisulfiden, N-Methyl-2-pyrrolidinthion und Schwefelwasserstoff ausgewählt ist.
- Verfahren nach Anspruch 5, wobei das Polymerisationsgemisch ferner ein Alkalimetallhydroxid umfaßt.
- Verfahren nach Anspruch 6, wobei das cyclische organische Amid N-Methyl-2-pyrrolidon umfaßt und die aromatische Dihalogenverbindung unter p-Dichlorbenzol und Gemischen von p-Dichlorbenzol mit insgesamt bis zu etwa 10 Mol-% mindestens eines der Bestandteile m-Dichlorbenzol, o-Dichlorbenzol und alkylsubstituierten Dichlorbenzolen mit der Formel
- Verfahren nach Anspruch 7, wobei das Alkalimetallbisulfid Natriumbisulfid umfaßt, das Alkalimetallhydroxid Natriumhydroxid umfaßt und die aromatische Dihalogenverbindung p-Dichlorbenzol umfaßt.
- Verfahren nach einem der vorstehenden Ansprüche, wobei das Polymerisationsgemisch ferner ein Alkalimetallcarboxylat umfaßt.
- Verfahren zur Herstellung von Poly-(phenylensulfid), das folgende Stufen umfaßt:(a) Dehydratisieren eines wäßrigen Gemisches, das Natriumbisulfid, Natriumhydroxid und N-Methyl-2-pyrrolidon umfaßt, unter Bildung eines dehydratisierten Gemisches;(b) Kontaktieren von p-Dichlorbenzol mit dem dehydratisierten Gemisch unter Bildung eines Polymerisationsgemisches;(c) Unterwerfen des Polymerisationsgemisches einer Polymerisation; und(d) Gewinnung des Poly-(phenylensulfids);
wobei das Verfahren in Gegenwart eines Olefins durchgeführt wird, das unter 1-Hepten, 1-Nonen, 1,5-Hexadien und 3-Butensäure ausgewählt ist. - Arylensulfidpolymer, umfassend Endgruppen, die unter



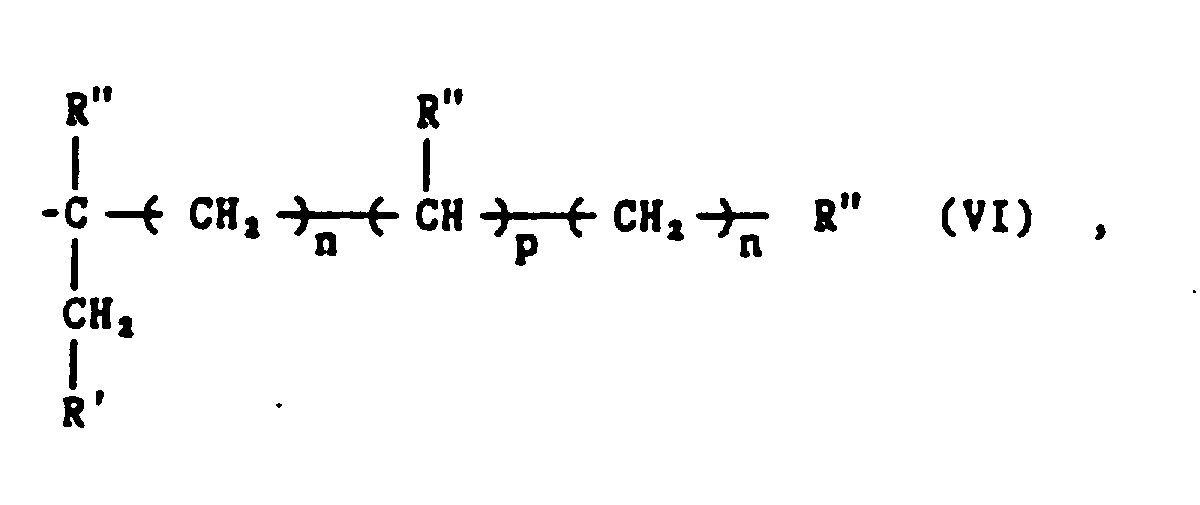




-H, -CN, -NR'₂, -NO₂, -Aryl,
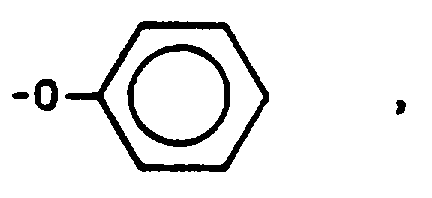


ausgewählt ist, R"' unter -H und -CH₃ ausgewählt ist, Y unter -H und -COO⁻ ausgewählt ist, m den Wert 0 oder 1 hat, n einen Wert von 0 bis 10 hat, p den Wert 0 oder 1 hat und Y, n und R" in jeder der vorstehenden Endgruppen gleich oder verschieden sein können und wobei die Gesamtzahl der Kohlenstoffatome in den Endgruppen der Formeln (III) und (IV) 3 oder 4 beträgt und die Gesamtzahl der Kohlenstoffatome in den Endgruppen der Formeln (V), (VI), (VII), (VIII), (IX) und (X) 2 bis 20 beträgt. - Polymer nach Anspruch 11, wobei es sich bei dem Arylensulfidpolymeren um Poly-(phenylensulfid) handelt.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US617228 | 1990-11-23 | ||
| US07/617,228 US5115093A (en) | 1990-11-23 | 1990-11-23 | Process for preparing arylene sulfide polymers in the presence of functionalized olefin |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP0487103A1 EP0487103A1 (de) | 1992-05-27 |
| EP0487103B1 true EP0487103B1 (de) | 1996-01-10 |
Family
ID=24472788
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP91119960A Expired - Lifetime EP0487103B1 (de) | 1990-11-23 | 1991-11-22 | Verfahren zur Herstellung von Arylensulfidpolymer |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US5115093A (de) |
| EP (1) | EP0487103B1 (de) |
| JP (1) | JP2690228B2 (de) |
| KR (1) | KR920009876A (de) |
| AT (1) | ATE132880T1 (de) |
| CA (1) | CA2052015A1 (de) |
| DE (1) | DE69116320T2 (de) |
| DK (1) | DK0487103T3 (de) |
| ES (1) | ES2082102T3 (de) |
| GR (1) | GR3018917T3 (de) |
| TW (1) | TW215096B (de) |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3354129A (en) * | 1963-11-27 | 1967-11-21 | Phillips Petroleum Co | Production of polymers from aromatic compounds |
| US3800845A (en) * | 1971-12-14 | 1974-04-02 | Phillips Petroleum Co | Solvent removal from poly(arylene sulfide) reaction slurry |
| US4337182A (en) * | 1981-03-26 | 1982-06-29 | Phillips Petroleum Company | Poly (arylene sulfide) composition suitable for use in semi-conductor encapsulation |
| US4415729A (en) * | 1982-06-04 | 1983-11-15 | Phillips Petroleum Company | Recovering granular poly(arylene sulfide) particles from a poly(arylene sulfide) reaction mixture |
| US4482665A (en) * | 1982-09-14 | 1984-11-13 | Phillips Petroleum Company | Encapsulation of electronic components with calcium silicate-containing poly(arylene sulfide) compositions |
| DE3421608A1 (de) * | 1984-06-09 | 1985-12-12 | Bayer Ag, 5090 Leverkusen | Verfahren zur herstellung von polyarylensulfiden mit funktionellen endgruppen |
| EP0228268B1 (de) * | 1985-12-27 | 1990-05-23 | Toray Industries, Inc. | Polyphenylensulfidharzzusammensetzung und Verfahren zu ihrer Herstellung |
| US4786711A (en) * | 1987-01-23 | 1988-11-22 | Phillips Petroleum Company | P-phenylene sulfide polymer preparation with dehydrated mixture of alkali metal hydroxide and excess alkali metal bisulfide |
| US4767841A (en) * | 1987-02-24 | 1988-08-30 | Phillips Petroleum Company | Arylene sulfide polymer preparation from dehydrated admixture comprising sulfur source, cyclic amide solvent and water |
| US4820800A (en) * | 1988-02-25 | 1989-04-11 | Phillips Petroleum Company | Arylene sulfide polymers |
| EP0384191A3 (de) * | 1989-02-07 | 1991-09-18 | Mitsubishi Rayon Company, Ltd. | Poly(arylensulfid)-Harzzusammensetzung |
-
1990
- 1990-11-23 US US07/617,228 patent/US5115093A/en not_active Expired - Fee Related
-
1991
- 1991-09-23 CA CA002052015A patent/CA2052015A1/en not_active Abandoned
- 1991-10-28 JP JP3281209A patent/JP2690228B2/ja not_active Expired - Fee Related
- 1991-11-05 TW TW080108724A patent/TW215096B/zh active
- 1991-11-07 KR KR1019910019725A patent/KR920009876A/ko not_active Abandoned
- 1991-11-22 DK DK91119960.2T patent/DK0487103T3/da active
- 1991-11-22 EP EP91119960A patent/EP0487103B1/de not_active Expired - Lifetime
- 1991-11-22 ES ES91119960T patent/ES2082102T3/es not_active Expired - Lifetime
- 1991-11-22 DE DE69116320T patent/DE69116320T2/de not_active Expired - Fee Related
- 1991-11-22 AT AT91119960T patent/ATE132880T1/de active
-
1996
- 1996-02-07 GR GR960400323T patent/GR3018917T3/el unknown
Also Published As
| Publication number | Publication date |
|---|---|
| ATE132880T1 (de) | 1996-01-15 |
| GR3018917T3 (en) | 1996-05-31 |
| TW215096B (de) | 1993-10-21 |
| JP2690228B2 (ja) | 1997-12-10 |
| CA2052015A1 (en) | 1992-05-24 |
| KR920009876A (ko) | 1992-06-25 |
| DE69116320D1 (de) | 1996-02-22 |
| DE69116320T2 (de) | 1996-05-23 |
| JPH04292626A (ja) | 1992-10-16 |
| ES2082102T3 (es) | 1996-03-16 |
| US5115093A (en) | 1992-05-19 |
| DK0487103T3 (da) | 1996-02-19 |
| EP0487103A1 (de) | 1992-05-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US4038259A (en) | Production of p-phenylene sulfide polymers | |
| EP0280274B1 (de) | Polyarylensulfide | |
| US4096132A (en) | Production of p-phenylene sulfide polymers | |
| US4748231A (en) | Reprecipitation of poly(arylene sulfide) to increase molecular weight thereof | |
| US4089847A (en) | Temperature programming in the production of arylene sulfide polymers | |
| EP0513730B1 (de) | Verfahren zur Herstellung von Arylensulfidpolymeren | |
| CA2096652C (en) | Process for the preparation of poly(arylene sulfide) with low metal contamination and polymer produced | |
| US5175243A (en) | Process for preparing arylene sulfide polymers with halo benzene containing deactivating group | |
| EP0086487B1 (de) | Verfahren zur Herstellung von Arylensulfidpolymeren | |
| US5235032A (en) | Process for the preparation of poly(arylene sulfide) with low metal contamination | |
| US5328980A (en) | Method of preparing poly(arylene sulfide) polymers, polymers and polymer blends | |
| US5053486A (en) | Process for preparing poly(arylene sulfide sulfone) with controlled ratio of alkali metal carboxylate to sulfur source | |
| JPS63243133A (ja) | 硫化アリーレン重合体の製法 | |
| EP0487103B1 (de) | Verfahren zur Herstellung von Arylensulfidpolymer | |
| US4988796A (en) | Process for preparing poly(arylene sulfide sulfone) | |
| US5144004A (en) | Process for preparing arylene sulfide sulfone/sulfoxide polymers | |
| KR0182327B1 (ko) | 폴리(페닐렌설파이드) 중합체의 제조 방법 | |
| US5003033A (en) | Process for preparing branched poly(arylene sulfide ketone) | |
| JPH04275334A (ja) | フェニレンスルフィドポリマーの製造法 | |
| US5064936A (en) | Process for preparing high extrusion rate arylene sulfide polymers | |
| US5331069A (en) | Method of treating poly(arylene sulfide sulfone) polymers and polymers | |
| US4882416A (en) | Preparation of poly arylene sulfide with specified ratio of sulfur source to cyclic organic amide | |
| EP0534359A1 (de) | Herstellungsverfahren für Polyarylensulfide | |
| US5177174A (en) | Branched aromatic sulfide/sulfone polymer production | |
| EP0431613A2 (de) | Verfahren zur Herstellung von Polyarylensulfidsulfonen |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IT LI LU NL SE |
|
| 17P | Request for examination filed |
Effective date: 19920929 |
|
| 17Q | First examination report despatched |
Effective date: 19950216 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AT BE CH DE DK ES FR GB GR IT LI LU NL SE |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 19960110 |
|
| REF | Corresponds to: |
Ref document number: 132880 Country of ref document: AT Date of ref document: 19960115 Kind code of ref document: T |
|
| ITF | It: translation for a ep patent filed | ||
| REG | Reference to a national code |
Ref country code: DK Ref legal event code: T3 |
|
| REF | Corresponds to: |
Ref document number: 69116320 Country of ref document: DE Date of ref document: 19960222 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: NV Representative=s name: KIRKER & CIE SA |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FG2A Ref document number: 2082102 Country of ref document: ES Kind code of ref document: T3 |
|
| ET | Fr: translation filed | ||
| REG | Reference to a national code |
Ref country code: GR Ref legal event code: FG4A Free format text: 3018917 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Effective date: 19961122 Ref country code: DK Effective date: 19961122 Ref country code: AT Effective date: 19961122 |
|
| REG | Reference to a national code |
Ref country code: DK Ref legal event code: EBP |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SE Effective date: 19961123 Ref country code: ES Free format text: LAPSE BECAUSE OF THE APPLICANT RENOUNCES Effective date: 19961123 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19961130 Ref country code: LI Effective date: 19961130 Ref country code: CH Effective date: 19961130 Ref country code: BE Effective date: 19961130 |
|
| 26N | No opposition filed | ||
| BERE | Be: lapsed |
Owner name: PHILLIPS PETROLEUM CY Effective date: 19961130 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Effective date: 19970601 |
|
| REG | Reference to a national code |
Ref country code: GR Ref legal event code: MM2A Free format text: 3018917 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 19961122 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Effective date: 19970731 |
|
| NLV4 | Nl: lapsed or anulled due to non-payment of the annual fee |
Effective date: 19970601 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Effective date: 19970801 |
|
| EUG | Se: european patent has lapsed |
Ref document number: 91119960.2 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FD2A Effective date: 20010402 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES;WARNING: LAPSES OF ITALIAN PATENTS WITH EFFECTIVE DATE BEFORE 2007 MAY HAVE OCCURRED AT ANY TIME BEFORE 2007. THE CORRECT EFFECTIVE DATE MAY BE DIFFERENT FROM THE ONE RECORDED. Effective date: 20051122 |