EP0010891B1 - Heterocyclisch substituierte Triazolylphosphorverbindungen, sie enthaltende insektizide Zusammensetzungen und Verfahren zur Bekämpfung von Insekten - Google Patents
Heterocyclisch substituierte Triazolylphosphorverbindungen, sie enthaltende insektizide Zusammensetzungen und Verfahren zur Bekämpfung von Insekten Download PDFInfo
- Publication number
- EP0010891B1 EP0010891B1 EP79302206A EP79302206A EP0010891B1 EP 0010891 B1 EP0010891 B1 EP 0010891B1 EP 79302206 A EP79302206 A EP 79302206A EP 79302206 A EP79302206 A EP 79302206A EP 0010891 B1 EP0010891 B1 EP 0010891B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- triazol
- carbon atoms
- pyridinyl
- phosphorothioate
- diethyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired
Links
- 239000000203 mixture Substances 0.000 title claims description 85
- -1 triazolyl phosphorous compounds Chemical class 0.000 title claims description 47
- 238000000034 method Methods 0.000 title claims description 28
- 241000238631 Hexapoda Species 0.000 title claims description 15
- 230000000749 insecticidal effect Effects 0.000 title claims description 10
- 125000000623 heterocyclic group Chemical group 0.000 title description 7
- 150000001875 compounds Chemical class 0.000 claims description 103
- 125000004432 carbon atom Chemical group C* 0.000 claims description 34
- 229910052739 hydrogen Inorganic materials 0.000 claims description 19
- 239000001257 hydrogen Substances 0.000 claims description 19
- 125000004414 alkyl thio group Chemical group 0.000 claims description 15
- 125000000217 alkyl group Chemical group 0.000 claims description 14
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 14
- 125000001246 bromo group Chemical group Br* 0.000 claims description 13
- 125000001153 fluoro group Chemical group F* 0.000 claims description 12
- 125000003545 alkoxy group Chemical group 0.000 claims description 9
- 125000004644 alkyl sulfinyl group Chemical group 0.000 claims description 9
- 125000004663 dialkyl amino group Chemical group 0.000 claims description 7
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 claims description 7
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 6
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 claims description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 5
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 claims description 5
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 5
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 5
- 125000004390 alkyl sulfonyl group Chemical group 0.000 claims description 4
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 4
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 4
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 4
- 125000003356 phenylsulfanyl group Chemical group [*]SC1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 claims description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 3
- 125000000858 thiocyanato group Chemical group *SC#N 0.000 claims description 3
- 239000004480 active ingredient Substances 0.000 claims description 2
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 claims description 2
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 claims 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims 1
- 238000006243 chemical reaction Methods 0.000 description 56
- 239000000047 product Substances 0.000 description 53
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 48
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 44
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 34
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 33
- 239000002904 solvent Substances 0.000 description 31
- 239000000243 solution Substances 0.000 description 24
- 239000006185 dispersion Substances 0.000 description 23
- 238000001914 filtration Methods 0.000 description 23
- 239000011541 reaction mixture Substances 0.000 description 21
- 239000007787 solid Substances 0.000 description 19
- 229910052757 nitrogen Inorganic materials 0.000 description 18
- 241000196324 Embryophyta Species 0.000 description 17
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 15
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 15
- 239000000376 reactant Substances 0.000 description 15
- 239000004094 surface-active agent Substances 0.000 description 15
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 14
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 14
- 229910052799 carbon Inorganic materials 0.000 description 14
- 229910052783 alkali metal Inorganic materials 0.000 description 13
- 235000013601 eggs Nutrition 0.000 description 13
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 12
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 12
- 238000010992 reflux Methods 0.000 description 12
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 11
- 238000001704 evaporation Methods 0.000 description 11
- 230000008020 evaporation Effects 0.000 description 11
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 10
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- 239000007800 oxidant agent Substances 0.000 description 9
- 231100000167 toxic agent Toxicity 0.000 description 9
- 239000003440 toxic substance Substances 0.000 description 9
- FQKFPGMGQXQHLP-UHFFFAOYSA-N 1-hydroxytriazole Chemical class ON1C=CN=N1 FQKFPGMGQXQHLP-UHFFFAOYSA-N 0.000 description 8
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 7
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- 239000003054 catalyst Substances 0.000 description 7
- 238000004821 distillation Methods 0.000 description 7
- 239000012442 inert solvent Substances 0.000 description 7
- 239000007788 liquid Substances 0.000 description 7
- 238000007254 oxidation reaction Methods 0.000 description 7
- 239000002689 soil Substances 0.000 description 7
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 7
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 6
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 6
- 239000012141 concentrate Substances 0.000 description 6
- 239000002270 dispersing agent Substances 0.000 description 6
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 6
- 239000003960 organic solvent Substances 0.000 description 6
- 230000003647 oxidation Effects 0.000 description 6
- 238000002360 preparation method Methods 0.000 description 6
- 239000012265 solid product Substances 0.000 description 6
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 6
- 241001674044 Blattodea Species 0.000 description 5
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 5
- 241000220225 Malus Species 0.000 description 5
- 241000721621 Myzus persicae Species 0.000 description 5
- 235000021016 apples Nutrition 0.000 description 5
- 239000000428 dust Substances 0.000 description 5
- 229910000027 potassium carbonate Inorganic materials 0.000 description 5
- 239000002244 precipitate Substances 0.000 description 5
- 239000012429 reaction media Substances 0.000 description 5
- 150000003839 salts Chemical class 0.000 description 5
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 description 5
- 150000003512 tertiary amines Chemical class 0.000 description 5
- KDAFHWYEIJPHPC-UHFFFAOYSA-N 2-pyridin-2-yl-1h-1,2,4-triazol-5-one Chemical compound N1=C(O)N=CN1C1=CC=CC=N1 KDAFHWYEIJPHPC-UHFFFAOYSA-N 0.000 description 4
- HXRPGJSKTFBJHN-UHFFFAOYSA-N 3-chloro-2-pyridin-2-yl-1h-1,2,4-triazol-5-one Chemical compound N1=C(O)N=C(Cl)N1C1=CC=CC=N1 HXRPGJSKTFBJHN-UHFFFAOYSA-N 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- 0 CN1N(*)C(*)=NC1OP(*)(O*)=S Chemical compound CN1N(*)C(*)=NC1OP(*)(O*)=S 0.000 description 4
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 4
- 241000256244 Heliothis virescens Species 0.000 description 4
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 4
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 4
- 241000256247 Spodoptera exigua Species 0.000 description 4
- LBQXBGCPOXMFGY-UHFFFAOYSA-N [(6-fluoropyridin-2-yl)amino]urea Chemical compound NC(=O)NNC1=CC=CC(F)=N1 LBQXBGCPOXMFGY-UHFFFAOYSA-N 0.000 description 4
- IRZJMYBVNZUYDQ-UHFFFAOYSA-N [benzoyl(pyridin-2-yl)amino]urea Chemical compound C=1C=CC=NC=1N(NC(=O)N)C(=O)C1=CC=CC=C1 IRZJMYBVNZUYDQ-UHFFFAOYSA-N 0.000 description 4
- 239000000370 acceptor Substances 0.000 description 4
- WFDIJRYMOXRFFG-UHFFFAOYSA-N acetic acid anhydride Natural products CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 4
- 150000001340 alkali metals Chemical class 0.000 description 4
- 239000006227 byproduct Substances 0.000 description 4
- 239000007795 chemical reaction product Substances 0.000 description 4
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 4
- 239000003995 emulsifying agent Substances 0.000 description 4
- 239000012433 hydrogen halide Substances 0.000 description 4
- 229910000039 hydrogen halide Inorganic materials 0.000 description 4
- 229960002523 mercuric chloride Drugs 0.000 description 4
- LWJROJCJINYWOX-UHFFFAOYSA-L mercury dichloride Chemical compound Cl[Hg]Cl LWJROJCJINYWOX-UHFFFAOYSA-L 0.000 description 4
- 239000008096 xylene Substances 0.000 description 4
- APOYQJOPORRGDX-UHFFFAOYSA-N (pyridin-2-ylamino)urea Chemical compound NC(=O)NNC1=CC=CC=N1 APOYQJOPORRGDX-UHFFFAOYSA-N 0.000 description 3
- NDQXKKFRNOPRDW-UHFFFAOYSA-N 1,1,1-triethoxyethane Chemical compound CCOC(C)(OCC)OCC NDQXKKFRNOPRDW-UHFFFAOYSA-N 0.000 description 3
- KLNTUXYXMQMTEG-UHFFFAOYSA-N 2-(6-fluoropyridin-2-yl)-1h-1,2,4-triazol-5-one Chemical compound FC1=CC=CC(N2NC(=O)N=C2)=N1 KLNTUXYXMQMTEG-UHFFFAOYSA-N 0.000 description 3
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 3
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 3
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 241000238657 Blattella germanica Species 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- 241000257226 Muscidae Species 0.000 description 3
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 3
- 241001454293 Tetranychus urticae Species 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 241000607479 Yersinia pestis Species 0.000 description 3
- 229960000583 acetic acid Drugs 0.000 description 3
- 229910001508 alkali metal halide Inorganic materials 0.000 description 3
- 150000008045 alkali metal halides Chemical class 0.000 description 3
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 3
- 125000004429 atom Chemical group 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- KMJJJTCKNZYTEY-UHFFFAOYSA-N chloro-diethoxy-sulfanylidene-$l^{5}-phosphane Chemical compound CCOP(Cl)(=S)OCC KMJJJTCKNZYTEY-UHFFFAOYSA-N 0.000 description 3
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 3
- 235000019253 formic acid Nutrition 0.000 description 3
- 229910052736 halogen Inorganic materials 0.000 description 3
- 150000002367 halogens Chemical group 0.000 description 3
- 125000000717 hydrazino group Chemical group [H]N([*])N([H])[H] 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 229910052500 inorganic mineral Inorganic materials 0.000 description 3
- 239000011707 mineral Substances 0.000 description 3
- 229910017604 nitric acid Inorganic materials 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- JLTDJTHDQAWBAV-UHFFFAOYSA-N phenyldimethylamine Natural products CN(C)C1=CC=CC=C1 JLTDJTHDQAWBAV-UHFFFAOYSA-N 0.000 description 3
- 150000004714 phosphonium salts Chemical class 0.000 description 3
- 150000003015 phosphoric acid halides Chemical class 0.000 description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 3
- DUIOPKIIICUYRZ-UHFFFAOYSA-N semicarbazide Chemical class NNC(N)=O DUIOPKIIICUYRZ-UHFFFAOYSA-N 0.000 description 3
- RMVRSNDYEFQCLF-UHFFFAOYSA-N thiophenol Chemical compound SC1=CC=CC=C1 RMVRSNDYEFQCLF-UHFFFAOYSA-N 0.000 description 3
- 231100000331 toxic Toxicity 0.000 description 3
- 230000002588 toxic effect Effects 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- TXUICONDJPYNPY-UHFFFAOYSA-N (1,10,13-trimethyl-3-oxo-4,5,6,7,8,9,11,12,14,15,16,17-dodecahydrocyclopenta[a]phenanthren-17-yl) heptanoate Chemical compound C1CC2CC(=O)C=C(C)C2(C)C2C1C1CCC(OC(=O)CCCCCC)C1(C)CC2 TXUICONDJPYNPY-UHFFFAOYSA-N 0.000 description 2
- SZSBOOZVDCBSAE-UHFFFAOYSA-N 2-(6-fluoropyridin-2-yl)-3-methyl-1h-1,2,4-triazol-5-one Chemical compound CC1=NC(O)=NN1C1=CC=CC(F)=N1 SZSBOOZVDCBSAE-UHFFFAOYSA-N 0.000 description 2
- YKGHOFYAPOSLEZ-UHFFFAOYSA-N 3-amino-2-pyridin-2-yl-1h-1,2,4-triazol-5-one Chemical compound NC1=NC(O)=NN1C1=CC=CC=N1 YKGHOFYAPOSLEZ-UHFFFAOYSA-N 0.000 description 2
- NVFJYGVMHBGDCZ-UHFFFAOYSA-N 3-methyl-2-pyridin-2-yl-1h-1,2,4-triazol-5-one Chemical compound CC1=NC(O)=NN1C1=CC=CC=N1 NVFJYGVMHBGDCZ-UHFFFAOYSA-N 0.000 description 2
- SHOOGROXMSZHEL-UHFFFAOYSA-N 3-phenyl-2-pyridin-2-yl-1h-1,2,4-triazol-5-one Chemical compound C=1C=CC=NC=1N1N=C(O)N=C1C1=CC=CC=C1 SHOOGROXMSZHEL-UHFFFAOYSA-N 0.000 description 2
- 241001124076 Aphididae Species 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 241001635274 Cydia pomonella Species 0.000 description 2
- 241000489973 Diabrotica undecimpunctata Species 0.000 description 2
- 240000002024 Gossypium herbaceum Species 0.000 description 2
- 235000004341 Gossypium herbaceum Nutrition 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 241000258916 Leptinotarsa decemlineata Species 0.000 description 2
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 2
- 241000254022 Locusta migratoria Species 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 241000691880 Planococcus citri Species 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- 229910021626 Tin(II) chloride Inorganic materials 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- QIHKGSVZVCDJIW-UHFFFAOYSA-N [(2,3,5-trichloropyridin-4-yl)amino]urea Chemical compound NC(=O)NNC1=C(Cl)C=NC(Cl)=C1Cl QIHKGSVZVCDJIW-UHFFFAOYSA-N 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- 229910001963 alkali metal nitrate Inorganic materials 0.000 description 2
- 150000008055 alkyl aryl sulfonates Chemical class 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- 150000001649 bromium compounds Chemical class 0.000 description 2
- QARVLSVVCXYDNA-UHFFFAOYSA-N bromobenzene Chemical compound BrC1=CC=CC=C1 QARVLSVVCXYDNA-UHFFFAOYSA-N 0.000 description 2
- XJHCXCQVJFPJIK-UHFFFAOYSA-M caesium fluoride Chemical compound [F-].[Cs+] XJHCXCQVJFPJIK-UHFFFAOYSA-M 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 238000005660 chlorination reaction Methods 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 238000007598 dipping method Methods 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 239000010440 gypsum Substances 0.000 description 2
- 229910052602 gypsum Inorganic materials 0.000 description 2
- 230000012447 hatching Effects 0.000 description 2
- 229910001385 heavy metal Inorganic materials 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- 238000011065 in-situ storage Methods 0.000 description 2
- 239000002917 insecticide Substances 0.000 description 2
- 229960004592 isopropanol Drugs 0.000 description 2
- 230000001418 larval effect Effects 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- UAEPNZWRGJTJPN-UHFFFAOYSA-N methylcyclohexane Chemical compound CC1CCCCC1 UAEPNZWRGJTJPN-UHFFFAOYSA-N 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- IOLCXVTUBQKXJR-UHFFFAOYSA-M potassium bromide Chemical compound [K+].[Br-] IOLCXVTUBQKXJR-UHFFFAOYSA-M 0.000 description 2
- GKKCIDNWFBPDBW-UHFFFAOYSA-M potassium cyanate Chemical compound [K]OC#N GKKCIDNWFBPDBW-UHFFFAOYSA-M 0.000 description 2
- 238000010791 quenching Methods 0.000 description 2
- 230000000171 quenching effect Effects 0.000 description 2
- 238000011084 recovery Methods 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 239000001119 stannous chloride Substances 0.000 description 2
- 235000011150 stannous chloride Nutrition 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 125000004434 sulfur atom Chemical group 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- GKASDNZWUGIAMG-UHFFFAOYSA-N triethyl orthoformate Chemical compound CCOC(OCC)OCC GKASDNZWUGIAMG-UHFFFAOYSA-N 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical class OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 1
- GESCDDOSISTEKA-UHFFFAOYSA-N (6-fluoropyridin-2-yl)hydrazine Chemical compound NNC1=CC=CC(F)=N1 GESCDDOSISTEKA-UHFFFAOYSA-N 0.000 description 1
- KOPMZTKUZCNGFY-UHFFFAOYSA-N 1,1,1-triethoxybutane Chemical compound CCCC(OCC)(OCC)OCC KOPMZTKUZCNGFY-UHFFFAOYSA-N 0.000 description 1
- FGWYWKIOMUZSQF-UHFFFAOYSA-N 1,1,1-triethoxypropane Chemical compound CCOC(CC)(OCC)OCC FGWYWKIOMUZSQF-UHFFFAOYSA-N 0.000 description 1
- PAAZPARNPHGIKF-UHFFFAOYSA-N 1,2-dibromoethane Chemical compound BrCCBr PAAZPARNPHGIKF-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- ODNBVEIAQAZNNM-UHFFFAOYSA-N 1-(6-chloroimidazo[1,2-b]pyridazin-3-yl)ethanone Chemical compound C1=CC(Cl)=NN2C(C(=O)C)=CN=C21 ODNBVEIAQAZNNM-UHFFFAOYSA-N 0.000 description 1
- GSOZVENRSFXLPV-UHFFFAOYSA-N 1-butyl-1h-pyrrol-1-ium;chloride Chemical class [Cl-].CCCC[NH+]1C=CC=C1 GSOZVENRSFXLPV-UHFFFAOYSA-N 0.000 description 1
- FMPJPCHSXGNEMD-UHFFFAOYSA-N 1-butylpyrazole Chemical compound CCCCN1C=CC=N1 FMPJPCHSXGNEMD-UHFFFAOYSA-N 0.000 description 1
- IWDFHWZHHOSSGR-UHFFFAOYSA-N 1-ethylimidazole Chemical compound CCN1C=CN=C1 IWDFHWZHHOSSGR-UHFFFAOYSA-N 0.000 description 1
- FLNMQGISZVYIIK-UHFFFAOYSA-N 1-ethylpyrazole Chemical compound CCN1C=CC=N1 FLNMQGISZVYIIK-UHFFFAOYSA-N 0.000 description 1
- WAUUWCXTWPHKOU-UHFFFAOYSA-N 1-hexylimidazole;1h-pyrazole Chemical compound C=1C=NNC=1.CCCCCCN1C=CN=C1 WAUUWCXTWPHKOU-UHFFFAOYSA-N 0.000 description 1
- SDZFVDDMTMLQGG-UHFFFAOYSA-N 1-hexylpyrazole Chemical compound CCCCCCN1C=CC=N1 SDZFVDDMTMLQGG-UHFFFAOYSA-N 0.000 description 1
- INJKWASQTISCEJ-UHFFFAOYSA-M 1-hexylpyridin-1-ium;iodide Chemical compound [I-].CCCCCC[N+]1=CC=CC=C1 INJKWASQTISCEJ-UHFFFAOYSA-M 0.000 description 1
- MCTWTZJPVLRJOU-UHFFFAOYSA-N 1-methyl-1H-imidazole Chemical compound CN1C=CN=C1 MCTWTZJPVLRJOU-UHFFFAOYSA-N 0.000 description 1
- ANFXTILBDGTSEG-UHFFFAOYSA-N 1-methyl-4,5-dihydroimidazole Chemical compound CN1CCN=C1 ANFXTILBDGTSEG-UHFFFAOYSA-N 0.000 description 1
- QAIGYXWRIHZZAA-UHFFFAOYSA-M 1-methylpyridin-1-ium;chloride Chemical compound [Cl-].C[N+]1=CC=CC=C1 QAIGYXWRIHZZAA-UHFFFAOYSA-M 0.000 description 1
- AVFZOVWCLRSYKC-UHFFFAOYSA-N 1-methylpyrrolidine Chemical compound CN1CCCC1 AVFZOVWCLRSYKC-UHFFFAOYSA-N 0.000 description 1
- GEPBNSNQBGDCTO-UHFFFAOYSA-N 1-pentylpyrazole Chemical compound CCCCCN1C=CC=N1 GEPBNSNQBGDCTO-UHFFFAOYSA-N 0.000 description 1
- IYVYLVCVXXCYRI-UHFFFAOYSA-N 1-propylimidazole Chemical compound CCCN1C=CN=C1 IYVYLVCVXXCYRI-UHFFFAOYSA-N 0.000 description 1
- CAHSGICMADEGCU-UHFFFAOYSA-N 1h-1,2,4-triazol-5-yl dihydrogen phosphate Chemical class OP(O)(=O)OC=1N=CNN=1 CAHSGICMADEGCU-UHFFFAOYSA-N 0.000 description 1
- GNVSDQAIXSNCFI-UHFFFAOYSA-N 2-(6-chloropyrazin-2-yl)-1h-1,2,4-triazol-5-one Chemical compound N1=C(O)N=CN1C1=CN=CC(Cl)=N1 GNVSDQAIXSNCFI-UHFFFAOYSA-N 0.000 description 1
- QZYRANVXDPTSFK-UHFFFAOYSA-N 2-(6-chloropyridazin-3-yl)-1h-1,2,4-triazol-5-one Chemical compound N1=C(O)N=CN1C1=CC=C(Cl)N=N1 QZYRANVXDPTSFK-UHFFFAOYSA-N 0.000 description 1
- YNJSNEKCXVFDKW-UHFFFAOYSA-N 3-(5-amino-1h-indol-3-yl)-2-azaniumylpropanoate Chemical compound C1=C(N)C=C2C(CC(N)C(O)=O)=CNC2=C1 YNJSNEKCXVFDKW-UHFFFAOYSA-N 0.000 description 1
- GSVVUPOBHIQNOJ-UHFFFAOYSA-M 3-ethyl-1,3-thiazol-3-ium;chloride Chemical compound [Cl-].CC[N+]=1C=CSC=1 GSVVUPOBHIQNOJ-UHFFFAOYSA-M 0.000 description 1
- LGYGPXRSSPCFFI-UHFFFAOYSA-N 3-phenylsulfanyl-2-pyridin-2-yl-1h-1,2,4-triazol-5-one Chemical compound C=1C=CC=NC=1N1N=C(O)N=C1SC1=CC=CC=C1 LGYGPXRSSPCFFI-UHFFFAOYSA-N 0.000 description 1
- QXFJJCJBDOXWDA-UHFFFAOYSA-M 4,4-dibutylmorpholin-4-ium;chloride Chemical compound [Cl-].CCCC[N+]1(CCCC)CCOCC1 QXFJJCJBDOXWDA-UHFFFAOYSA-M 0.000 description 1
- 150000000565 5-membered heterocyclic compounds Chemical class 0.000 description 1
- 150000000644 6-membered heterocyclic compounds Chemical class 0.000 description 1
- 241000238876 Acari Species 0.000 description 1
- 241000132121 Acaridae Species 0.000 description 1
- 241000256118 Aedes aegypti Species 0.000 description 1
- 241000256186 Anopheles <genus> Species 0.000 description 1
- GUNJVIDCYZYFGV-UHFFFAOYSA-K Antimony trifluoride Inorganic materials F[Sb](F)F GUNJVIDCYZYFGV-UHFFFAOYSA-K 0.000 description 1
- 241000239223 Arachnida Species 0.000 description 1
- 241000238888 Argasidae Species 0.000 description 1
- 241000238421 Arthropoda Species 0.000 description 1
- 241000238662 Blatta orientalis Species 0.000 description 1
- 241000238660 Blattidae Species 0.000 description 1
- 241000907223 Bruchinae Species 0.000 description 1
- 241001388466 Bruchus rufimanus Species 0.000 description 1
- DKPFZGUDAPQIHT-UHFFFAOYSA-N Butyl acetate Natural products CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 235000008534 Capsicum annuum var annuum Nutrition 0.000 description 1
- 235000002568 Capsicum frutescens Nutrition 0.000 description 1
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 241001124134 Chrysomelidae Species 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 241000256057 Culex quinquefasciatus Species 0.000 description 1
- 241000256113 Culicidae Species 0.000 description 1
- 241001641896 Dermestes lardarius Species 0.000 description 1
- 241001513837 Dermestes maculatus Species 0.000 description 1
- 241000131287 Dermestidae Species 0.000 description 1
- 241000255925 Diptera Species 0.000 description 1
- 241000630736 Ephestia Species 0.000 description 1
- 241000122098 Ephestia kuehniella Species 0.000 description 1
- 241000238889 Ixodidae Species 0.000 description 1
- 239000005909 Kieselgur Substances 0.000 description 1
- LSDPWZHWYPCBBB-UHFFFAOYSA-N Methanethiol Chemical compound SC LSDPWZHWYPCBBB-UHFFFAOYSA-N 0.000 description 1
- 241000257159 Musca domestica Species 0.000 description 1
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 description 1
- DJEQZVQFEPKLOY-UHFFFAOYSA-N N,N-dimethylbutylamine Chemical compound CCCCN(C)C DJEQZVQFEPKLOY-UHFFFAOYSA-N 0.000 description 1
- AHVYPIQETPWLSZ-UHFFFAOYSA-N N-methyl-pyrrolidine Natural products CN1CC=CC1 AHVYPIQETPWLSZ-UHFFFAOYSA-N 0.000 description 1
- UQFQONCQIQEYPJ-UHFFFAOYSA-N N-methylpyrazole Chemical compound CN1C=CC=N1 UQFQONCQIQEYPJ-UHFFFAOYSA-N 0.000 description 1
- 244000061176 Nicotiana tabacum Species 0.000 description 1
- 235000002637 Nicotiana tabacum Nutrition 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 241000238675 Periplaneta americana Species 0.000 description 1
- 229920003171 Poly (ethylene oxide) Chemical class 0.000 description 1
- 241001415279 Pseudococcidae Species 0.000 description 1
- 241000255893 Pyralidae Species 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical class C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- 241001247145 Sebastes goodei Species 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 241000254179 Sitophilus granarius Species 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- 241001494115 Stomoxys calcitrans Species 0.000 description 1
- 244000186561 Swietenia macrophylla Species 0.000 description 1
- 241000254109 Tenebrio molitor Species 0.000 description 1
- 241000254107 Tenebrionidae Species 0.000 description 1
- 241001454295 Tetranychidae Species 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- 230000000895 acaricidal effect Effects 0.000 description 1
- 150000008065 acid anhydrides Chemical class 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 150000003973 alkyl amines Chemical class 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 239000008365 aqueous carrier Substances 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 150000004982 aromatic amines Chemical class 0.000 description 1
- 229960000892 attapulgite Drugs 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M bisulphate group Chemical group S([O-])(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 229910052792 caesium Inorganic materials 0.000 description 1
- TVFDJXOCXUVLDH-UHFFFAOYSA-N caesium atom Chemical compound [Cs] TVFDJXOCXUVLDH-UHFFFAOYSA-N 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 150000003857 carboxamides Chemical class 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- RLGQACBPNDBWTB-UHFFFAOYSA-N cetyltrimethylammonium ion Chemical group CCCCCCCCCCCCCCCC[N+](C)(C)C RLGQACBPNDBWTB-UHFFFAOYSA-N 0.000 description 1
- 230000001055 chewing effect Effects 0.000 description 1
- XFBJRFNXPUCPKU-UHFFFAOYSA-N chloro-dimethoxy-sulfanylidene-$l^{5}-phosphane Chemical compound COP(Cl)(=S)OC XFBJRFNXPUCPKU-UHFFFAOYSA-N 0.000 description 1
- WBLIXGSTEMXDSM-UHFFFAOYSA-N chloromethane Chemical compound Cl[CH2] WBLIXGSTEMXDSM-UHFFFAOYSA-N 0.000 description 1
- 239000003426 co-catalyst Substances 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 239000007859 condensation product Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- OEBMLQSJSSJIRO-UHFFFAOYSA-M diethyl-methyl-phenylazanium;hydrogen sulfate Chemical compound OS([O-])(=O)=O.CC[N+](C)(CC)C1=CC=CC=C1 OEBMLQSJSSJIRO-UHFFFAOYSA-M 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 244000078703 ectoparasite Species 0.000 description 1
- 235000013399 edible fruits Nutrition 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 239000004495 emulsifiable concentrate Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 244000079386 endoparasite Species 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 150000002429 hydrazines Chemical class 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical class [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- 150000002431 hydrogen Chemical group 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N hydrogen thiocyanate Natural products SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 238000005470 impregnation Methods 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 239000003350 kerosene Substances 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-M methyl sulfate(1-) Chemical compound COS([O-])(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-M 0.000 description 1
- YQRNTVUSJHYLNZ-UHFFFAOYSA-N methyl(tridecyl)azanium;chloride Chemical class [Cl-].CCCCCCCCCCCCC[NH2+]C YQRNTVUSJHYLNZ-UHFFFAOYSA-N 0.000 description 1
- GYNNXHKOJHMOHS-UHFFFAOYSA-N methyl-cycloheptane Natural products CC1CCCCCC1 GYNNXHKOJHMOHS-UHFFFAOYSA-N 0.000 description 1
- 125000004092 methylthiomethyl group Chemical group [H]C([H])([H])SC([H])([H])* 0.000 description 1
- ZUZLIXGTXQBUDC-UHFFFAOYSA-N methyltrioctylammonium Chemical group CCCCCCCC[N+](C)(CCCCCCCC)CCCCCCCC ZUZLIXGTXQBUDC-UHFFFAOYSA-N 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- DAZXVJBJRMWXJP-UHFFFAOYSA-N n,n-dimethylethylamine Chemical compound CCN(C)C DAZXVJBJRMWXJP-UHFFFAOYSA-N 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 229910052625 palygorskite Inorganic materials 0.000 description 1
- 230000000149 penetrating effect Effects 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 230000000361 pesticidal effect Effects 0.000 description 1
- 239000003209 petroleum derivative Substances 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- 235000021317 phosphate Nutrition 0.000 description 1
- 150000003018 phosphorus compounds Chemical class 0.000 description 1
- FAIAAWCVCHQXDN-UHFFFAOYSA-N phosphorus trichloride Chemical compound ClP(Cl)Cl FAIAAWCVCHQXDN-UHFFFAOYSA-N 0.000 description 1
- 239000002574 poison Substances 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 238000011027 product recovery Methods 0.000 description 1
- 239000012264 purified product Substances 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- SBYHFKPVCBCYGV-UHFFFAOYSA-N quinuclidine Chemical compound C1CC2CCN1CC2 SBYHFKPVCBCYGV-UHFFFAOYSA-N 0.000 description 1
- 239000000344 soap Substances 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 238000007711 solidification Methods 0.000 description 1
- 230000008023 solidification Effects 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 238000005507 spraying Methods 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 150000005621 tetraalkylammonium salts Chemical class 0.000 description 1
- CCBCJOPAGXYIQL-UHFFFAOYSA-M tetrabenzylazanium;chloride Chemical compound [Cl-].C=1C=CC=CC=1C[N+](CC=1C=CC=CC=1)(CC=1C=CC=CC=1)CC1=CC=CC=C1 CCBCJOPAGXYIQL-UHFFFAOYSA-M 0.000 description 1
- DZLFLBLQUQXARW-UHFFFAOYSA-N tetrabutylammonium Chemical group CCCC[N+](CCCC)(CCCC)CCCC DZLFLBLQUQXARW-UHFFFAOYSA-N 0.000 description 1
- DTIFFPXSSXFQCJ-UHFFFAOYSA-N tetrahexylazanium Chemical group CCCCCC[N+](CCCCCC)(CCCCCC)CCCCCC DTIFFPXSSXFQCJ-UHFFFAOYSA-N 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 125000005490 tosylate group Chemical group 0.000 description 1
- 150000003852 triazoles Chemical class 0.000 description 1
- IMNIMPAHZVJRPE-UHFFFAOYSA-N triethylenediamine Chemical compound C1CN2CCN1CC2 IMNIMPAHZVJRPE-UHFFFAOYSA-N 0.000 description 1
- KUSDSMLUXMFMCB-UHFFFAOYSA-M trimethyl(2-phenylethyl)azanium;chloride Chemical class [Cl-].C[N+](C)(C)CCC1=CC=CC=C1 KUSDSMLUXMFMCB-UHFFFAOYSA-M 0.000 description 1
- YCYOWIYHCRGAAH-UHFFFAOYSA-M trimethyl(naphthalen-1-yl)azanium;chloride Chemical compound [Cl-].C1=CC=C2C([N+](C)(C)C)=CC=CC2=C1 YCYOWIYHCRGAAH-UHFFFAOYSA-M 0.000 description 1
- GNMJFQWRASXXMS-UHFFFAOYSA-M trimethyl(phenyl)azanium;bromide Chemical compound [Br-].C[N+](C)(C)C1=CC=CC=C1 GNMJFQWRASXXMS-UHFFFAOYSA-M 0.000 description 1
- CLPINXBPSKSFNU-UHFFFAOYSA-M trimethyl(pyridin-4-yl)azanium;chloride Chemical compound [Cl-].C[N+](C)(C)C1=CC=NC=C1 CLPINXBPSKSFNU-UHFFFAOYSA-M 0.000 description 1
- DGOVSCVARCXHRS-UHFFFAOYSA-M trimethyl-(4-methylphenyl)azanium;chloride Chemical compound [Cl-].CC1=CC=C([N+](C)(C)C)C=C1 DGOVSCVARCXHRS-UHFFFAOYSA-M 0.000 description 1
- VFJMZEKQTOYWLP-UHFFFAOYSA-N tritylazanium;fluoride Chemical compound [F-].C=1C=CC=CC=1C(C=1C=CC=CC=1)([NH3+])C1=CC=CC=C1 VFJMZEKQTOYWLP-UHFFFAOYSA-N 0.000 description 1
- 239000004563 wettable powder Substances 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/76—Nitrogen atoms to which a second hetero atom is attached
- C07D213/77—Hydrazine radicals
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01N—PRESERVATION OF BODIES OF HUMANS OR ANIMALS OR PLANTS OR PARTS THEREOF; BIOCIDES, e.g. AS DISINFECTANTS, AS PESTICIDES OR AS HERBICIDES; PEST REPELLANTS OR ATTRACTANTS; PLANT GROWTH REGULATORS
- A01N57/00—Biocides, pest repellants or attractants, or plant growth regulators containing organic phosphorus compounds
- A01N57/02—Biocides, pest repellants or attractants, or plant growth regulators containing organic phosphorus compounds having alternatively specified atoms bound to the phosphorus atom and not covered by a single one of groups A01N57/10, A01N57/18, A01N57/26, A01N57/34
- A01N57/08—Biocides, pest repellants or attractants, or plant growth regulators containing organic phosphorus compounds having alternatively specified atoms bound to the phosphorus atom and not covered by a single one of groups A01N57/10, A01N57/18, A01N57/26, A01N57/34 containing heterocyclic radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6515—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having three nitrogen atoms as the only ring hetero atoms
- C07F9/6518—Five-membered rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65583—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system each of the hetero rings containing nitrogen as ring hetero atom
Definitions
- This invention is directed to heterocyclic substituted triazolyl phosphorous compounds corresponding to the formula wherein R represents a nitrogen containing heterocyclic radical corresponding to one of the formulae each X independently represents chloro, fluoro, bromo, nitro, alkyl of 1 to 4 carbon atoms, amino, mono- or dialkylamino wherein each alkyl group independently contains from 1 to 4 carbon atoms, alkoxy of 1 to 4 carbon atoms, alkylthio of 1 to 4 carbon atoms, alkylsulfinyl of 1 to 4 carbon atoms, alkylsulfonyl of 1 to 4 carbon atoms, cyano, trifluoromethyl, trichloromethyl, phenoxy or substituted phenoxy of the formula wherein each Z independently represents chloro, fluoro, bromo, nitro, cyano, alkoxy of 1 to 4 carbon atoms or alkylthio of 1 to 4 carbon atoms, with the proviso that when either n
- sterically compatible is employed to designate X and Z substituent groups which are not affected by steric hindrance as defined in "The Condensed Chemical Dictionary," 7th edition, Reinhold Publishing Co. N.Y., page 893 (1966) which definition is as follows:
- German Offenlegungsschrifts Nos. 2360631 and 2547971 describe certain 1,2,4-triazolylphosphoric acid esters having pesticidal activity.
- the compounds of the present invention differ from these prior art compounds mainly having regard to the nature of the substituent group R.
- the compounds of the present invention possess a superior foliar and systemic insecticidal activity and a broader spectrum of activity than the prior art compounds.
- the triazolyl phosphorous compounds of the present invention possess excellent insecticidal properties and are very useful for the control of insects and for the protection of plants and stored goods from destruction by insects.
- the phosphorous compounds of the present invention are crystalline solids or liquids which are sparingly soluble in water and which are soluble in most organic solvents.
- R 2 is C 1 to C 3 alkyl
- R 3 is C 1 to C 3 alkoxy
- X is chloro, fluoro, bromo or phenoxy
- n is 0 or 1
- R 1 is C, to C 3 alkyl, C 1 to C 2 alkoxy or C 1 to C 2 dialkylamino.
- the triazolyl phosphorous compounds of the present invention can be prepared by the reaction of an appropriate substituted 3-hydroxytriazole or an alkali metal salt thereof which corresponds to the formula with a phosphoric acid halide corresponding to the formula in the presence of an acid binding agent and a solvent in the above formulae, R, R 1 , R and R 3 are as hereinbefore defined; Me represents sodium, potassium, lithium or cesium and X represents chloro or bromo.
- the 3-hydroxytriazole reactant or the alkali metal salt thereof, in a solvent is mixed with the phosphoric acid halide and the mixture heated at a temperature of from 20°C up to the boiling point of the specific solvent employed.
- the reaction can be carried out if desired in the presence of a catalyst.
- the reaction is usually complete in from 0.1 to 24 hours.
- the reaction consumes the reactants in stoichiometric proportions, i.e. one equivalent of the 3-hydroxytriazole reactant per equivalent of the phosphoric acid halide and for the most part, these amounts can be employed. It should be noted however, that the actual amount of the reactants to be employed is not critical as some of the desired product is formed when employing any proportions.
- the alkali metal salt of 3-hydroxytriazole reactant can be prepared by the reaction of molecular equivalent amounts of the 3-hydroxytriazole and an alkali metal hydroxide or carbonate in the presence of a solvent at temperatures of from 20°C up to the reflux temperature of the solvent employed for from 10 minutes to 4 hours. From a practical standpoint, the salt conversion is usually carried out in situ. In addition, it is preferred to employ the alkali metal salt since during the subsequent reaction with the phosphorous acid halide, an insoluble alkali metal halide by-product is formed rather than an acid and no acid binding agent is necessary. Additionally, the insoluble salt can be easily removed allowing for a more convenient product recovery.
- catalysts useful in carrying out this process include tertiary amines having a pKa of at least 9.5, co-catalysts which are mixtures of quaternary ammonium or phosphonium salts and organic tertiary amines which include quaternary ammonium compounds such as tetraalkylammonium salts, such as tetra-n-butyl-, tetrahexyl-, tri-n-butylmethyl-, cetyltrimethyl-, trioctylmethyl- and tridecylmethyl ammonium chlorides, bromides, bisulfates, tosylates, etc.; aralkylammonium salts, such as tetrabenzylammonium chloride, benzyltrimethyl-, benzyltriethyl-, benzyltributyl-, and phenethyl- trimethylammonium chlorides, bromides, etc.; arylammoni
- ammonium salts are currently preferred over the phosphonium salts due to cost and commercial availability.
- the most preferred catalysts are benzyltrimethyl-, benzyltriethyl- tetra-n-butyi and tri-n-butylmethyl ammonium-salts.
- Suitable tertiary amines include aliphatic trihydrocarbyl amines (e.g. trimethylamine, ethyldimethylamine, butyldimethylamine, N,N,N',N'-tetramethylethylenediamine, and the like); aliphatic heterocyclic amines (e.g. 1,4-diazabicyclo[2.2.2]octane, 1-azabicyclo[2.2.2]octane, 1-methyl-2-imidazoline, 1-methylpyrrolidine, and the like); mixed aliphatic/aromatic amines (e.g. 4-dimethylaminopyridine, 4-(N-pyrrolidion)pyridine phenyldimethylamine, and other like organic, sterically unhindered, nucleophilic, tertiary amines.
- aliphatic trihydrocarbyl amines e.g. trimethylamine, ethyldimethylamine, butyld
- suitable diazoles include imidazole, 1-methylimidazole, 1-ethylimidazole, 1-propylimidazole, 1-hexylimidazole pyrazole, 1-methylpyrazole, 1-ethylpyrazole, 1-butylpyrazole, 1-amylpyrazole and 1-hexylpyrazole, and the like.
- catalysts include a heavy metal or heavy metal salt such as, for example, metallic copper or mercuric chloride.
- a catalyst it can be added at any stage of the process. In order to achieve the best yield, it is preferred that the catalyst be added at the beginning of the reaction. If, however, the catalyst is a tertiary amine, it is preferred that it be added just prior to the addition of the phosphorous acid chloride.
- Suitable acid-binding agents are, for example, organic amines such as triethylamine, dimethylaniline, pyridine, inorganic bases such as the hydroxides and carbonates of alkali metals and alkaline earth metals, such as sodium, potassium, calcium, or lithium. These agents are employed in from about about an equimolar amount up to a 10 percent excess based on the triazole reactant.
- Suitable solvents which can be employed are all the usual organic liquids which are inert under the reaction conditions, for example, acetone, methylethyl ketone, acetonitrile, ethyl acetate, butyl acetate, tetrahydrofuran, dioxane, methylene chloride, carbon tetrachloride, benzene, chlorobenzene, polychlorobenzenes, bromobenzene, dimethylformamide, and xylene.
- the reaction mixture was cooled and filtered to remove the insoluble by-product and the solvent then removed by evaporation at 50°C under reduced pressure.
- the oil product which remained was taken up in 250 mls of methylene chloride and then washed twice with 200 mls of water, separated, dried over anhydrous sodium sulfate, filtered and the solvent removed by evaporation under reduced pressure.
- the crude 0,0 - diethyl - 0 - (1 - (6 - chloro - 2 - pyrazinyl) - 1 H - 1,2,4 - triazol - 3 - yl) phosphorothioate product thus obtained was purified by recrystallization from hexane.
- Those compounds wherein R 1 is hydrogen can be prepared by the reaction of an appropriate substituted hydrazine carboxamide with excess formic acid or triethyl orthoformate under reflux conditions with or without a solvent being present.
- Representative solvents include dimethyl sulfoxide, xylene and dimethylformamide.
- the product is thereafter recovered by cooling the reaction mixture and pouring it over ice and filtering off the solid product which precipitates. Alternatively the reaction mixture is concentrated by distillation and the solid product which precipitates is recovered by filtration.
- the product regardless of the method of recovery can be further purified by being either washed with a solvent and then dried or washed with water and dried. When water washing is employed it is often advantageous to neutralize the filtrate to recover any product remaining therein.
- Those compounds where R 1 is chloro are usually prepared by chlorinating the appropriate compound wherein R 1 is hydrogen. This chlorinating step employs a convention chlorinating procedure wherein chlorine gas is bubbled through the triazol-3-ol compound at 100-200C. After the chlorination is complete, the desired product is recovered by filtration.
- Those compounds wherein R 1 is bromo or fluoro can be prepared by conventional halogen exchange procedures wherein the appropriate triazol-3-ol compound, wherein R 1 is chloro, is treated with potassium bromide or cesium fluoride, respectively. The desired product is then recovered by quenching the reaction mixture with ice and filtering of the solid product.
- Those compounds wherein R 1 is amino or alkylamino can be prepared by reacting the appropriate triazol-3-ol compound wherein R 1 is chloro, fluoro or bromo with ammonia or an alkylamine in the presence of a solvent such as, for example, dimethyl sulfoxide or isopropylalcohol and at temperatures in the range of about 45° to about 150°C for from 1 to about 3 hours.
- a solvent such as, for example, dimethyl sulfoxide or isopropylalcohol
- Those compounds wherein R 1 is alkoxy, alkylthio or phenylthio can be prepared by the reaction of an appropriate triazol-3-ol compound wherein R 1 is chloro, bromo, or fluoro and an appropriate alkali metal alkoxide, alkali metal alkyl- or phenylmercaptide, in the presence of an organic solvent of the type conventionally employed for such reactions.
- the alkali metal is dissolved in an alcohol corresponding to the alkyl or phenyl group being added.
- the alkali metal oxy compounds is inherently formed and is employed as such in the following reactions.
- the mercaptide is formed by adding the appropriate mercaptan to the alcohol solution.
- To the appropriate above mixture is added the triazol-3-ol reactant and the resulting mixture is refluxed for a time sufficient to complete the reaction, usually from about 1 to about 3 hours.
- the reaction products are filtered to remove any alkali metal halide, followed by solvent removal by evaporation, distillation or other conventional separatory procedures, leaving the desired product.
- R 1 is alkylsulfinyl or sulfonyl
- Those compounds wherein R 1 is alkylsulfinyl or sulfonyl can be prepared by reacting an appropriate triazol-3-ol with an oxidizing agent.
- the oxidation of any of the alkylthio substituted compounds results, at least partially, in the formation of the corresponding alkylsulfinyl substituted compound.
- the oxidation of one molecular of the alkylthio substituted compound to the corresponding alkylsulfinyl substituted compound or the oxidation of one molecule of a alkylsulfinyl compound to the corresponding alkylsulfonyl compound requires one atom of oxygen for each sulfur atom oxidized.
- the oxidation of the alkylthio compound directly to the corresponding alkylsulfonyl compound consumes two atoms of oxygen for each sulfur atom so oxidized.
- the alkylsulfinyl compounds can be prepared and subjected to continuing oxidative conditions so as to be further oxidized in situ to the corresponding sulfonyl compound. In some instances, depending on the oxidizing agent and process conditions the oxidation proceeds to the sulfonyl compound so rapidly that it is not practical to isolate the sulfinyl compound.
- Representative oxidizing agents for the production of the sulfinyl compounds include nitric acid and hydrogen peroxide and representative oxidizing agents to be employed in the preparation of the sulfonyl compounds include hydrogen peroxide, and perbenzoic acid.
- Hydrogen peroxide and conveniently an aqueous solution thereof, can be employed as the oxidizing agent in the production of the sulfinyl and sulfonyl containing derivatives of the present invention.
- the reaction is carried out in the presence of a liquid reaction medium, such as trifluoroacetic acid, glacial acetic acid or a mixture of acetic acid and acetic anhydride.
- a liquid reaction medium such as trifluoroacetic acid, glacial acetic acid or a mixture of acetic acid and acetic anhydride.
- the acid-anhydride mixture is employed as the liquid reaction medium.
- the reaction takes place at temperatures of from about 75° to about 120°C. In a convenient method, the reaction is carried out at the boiling temperature of the reaction mixture and under reflux.
- the reactants are contracted in any order or fashion, and preferably in amounts stoichiometric for the preparation of the desired product.
- the reaction mixture is then maintained at a temperature within the reaction temperature range until the desired degree of conversion is achieved.
- the sulfinyl or sulfonyl product can be separated by conventional procedures such as evaporation of the reaction medium to obtain the product as a solid residue.
- the reaction mixture is washed with cold water and is thereafter filtered, centrifuged or the like to obtain the crystalline product.
- Nitric acid is conveniently employed to oxidize the alkylthio starting material to the corresponding sulfinyl derivative.
- the reaction can be carried out in the presence of a halocarbon reaction medium such as carbon tetrachloride, methylene dichloride, ethylene dibromide, etc. In a preferred procedure, excess nitric acid is employed as the reaction medium.
- the reaction proceeds at temperatures between about 15° and about 120°C.
- the reaction is carried out under reflux conditions at temperatures of from 80° to 120°C and requires only a short period of time for completion, i.e., about 2 to about 7 minutes.
- the reactants are mixed and the temperature is allowed to rise to the desired temperature and maintained at or about this temperature during the refluxing.
- chlorine water can also be employed as the oxidizing agent in the preparation of alkylsulfinyl or sulfonyl derivatives from the corresponding alkylthio derivatives.
- a slurry of the alkylthio containing compound to be oxidized is prepared in water and the slurry agitated while chlorine gas is bubbled in. The mixture is maintained at room temperature until no starting alkylthio material is left unoxidized. If it is desired to convert the sulfinyl compound to the sulfonyl state, the temperature is raised to about 90°C and the mixture maintained at this temperature until oxidation is complete.
- Those compounds wherein R 1 is alkyl are prepared by the reaction of an appropriate substituted hydrazine carboxamide with an excess of an appropriate triethylorthoester under reflux conditions in the presence or absence of a solvent.
- Representative solvents include dimethylsulfoxide and xylene. The product is thereafter recovered as set forth hereinbefore for compounds wherein R is hydrogen.
- Representative ester reactants include compounds corresponding to the formula wherein R 4 is methyl, ethyl or propyl. Specific compounds include triethyl ortho acetate, triethyl ortho propionate and triethyl ortho butyrate.
- Those compounds wherein R 1 is phenyl can be prepared by the reaction of an appropriate 2-benzoyl-2-(2-pyridinyl)hydrazine carboxamide with an alkali metal hydroxide, at a temperature of from 25° to 75°C for from one minute to one hour, followed by acidifying the reaction product.
- Those compounds wherein R 1 is methylthiomethyl can be prepared by the reaction of an appropriate triazol-3-ol compound wherein R 1 is chloromethyl with an alkali metal methylmercaptide in the presence of an organic solvent of the type conventionally employed for such reactions under reflux conditions.
- Those compounds wherein R 1 is hydroxymethyl can be prepared by the reaction of an appropriate triazol-3-ol compound wherein R 1 is chloromethyl with an alkali metal hydroxide in the presence of an organic solvent of the type conventionally employed for such reactions.
- Those compounds wherein R 1 is thiocyanato can be prepared by the reaction of an appropriate triazol-3-ol compound wherein R 1 is chloro, bromo or fluoro and an appropriate alkali metal thiocyanate under reflux conditions for a time sufficient to complete the reaction, usually from about 1 to about 3 hours.
- the reaction products are filtered to remove any alkali metal halide, followed by solvent removal by evaporation, distillation or other conventional separatory procedures, leaving the desired product.
- Those compounds wherein R 1 is chloromethyl or trichloromethyl can be prepared by the selective chlorination of the appropriate compounds wherein R 1 is methyl.
- chlorine gas is passed into the appropriate triazol-3-ol compound at temperatures of from 20° to 30°C in the presence of a solvent such as carbon tetrachloride for a period of from 30 minutes to 5 hours in the presence of UV light.
- Those compounds wherein R 1 is trifluoromethyl can be prepared by halogen exchange whereby the appropriate trichloromethyl substituted compound is treated with antimony trifluoride under conventional halogen exchange conditions.
- a solution was prepared by admixing 17 g (0.10 m) of 2-(6-fluoro-2-pyridinyl)hydrazinecarboxamide and 60 mls of 98 percent formic acid. This mixture was stirred and refluxed for 2 hours and allowed to cool to room temperature. The insolubles were filtered off and dried to produce 3.5 g of the desired 1 - (6 - fluoro - 2 - pyridinyl) - 1 H - 1,2,4 - triazol - 3 - ol which melted at 290°C with decomposition. The excess formic acid in the remaining liquid phase was distilled off under reduced pressure and the solids which precipitated were washed and dried.
- This product was a second crop of the desired product which also melted at 290°C.
- the product was recovered in a yield of 60 percent of theoretical and upon analysis, was found to have carbon, hydrogen and nitrogen contents of 46.80, 2.89 and 31.17 percent, respectively, as compared with the theoretical contents of 46.47,2.80 and 31.10 percent, respectively, as calculated for the above named compound.
- the product melted at 260 0- 262°C and upon analysis was found to have carbon, hydrogen and nitrogen contents of 52.26, 3.90 and 34.79 percent, respectively, as compared with the theoretical contents of 51.89, 3.73 and 34.55 percent, respectively, calculated for the above named compound.
- a solution was prepared by admixing 17.02 g (0.10 m) of 2-(6-fluoro-2-pyridinyl)hydrazinecarboxamide with 100 ml of triethyl orthoacetate. The mixture was refluxed overnight and the reaction mixture cooled to room temperature and poured over ice water. The solids which precipitated out were recovered by filtration. The solids were washed with methylene chloride and dried under vacuum to yield 11.4 g (58.7 percent of theoretical) of the desired 1 - (6 - fluoro - 2 - pyridinyl) - 5 - methyl - 1H - 1,2,4 - triazol - 3 - ol.
- a reaction mixture was prepared by suspending 457.6 g (3 m) of 2-pyridinyl-hydrazine carboxamide in 2600 ml of triethylorthoacetate and the mixture refluxed for about 9 hours.
- the ethanol by-product formed was continuously removed by distillation allowing the reaction temperature to stay at 125°-130°C.
- the 1 - (2 - pyridinyl) - 5 - methyl - 1H - 1,2,4 - triazol - 3 - ol product was recovered by filtration in a yield of 116 g (66 percent of theoretical) and was washed with chloroform.
- the white solid which precipitated was recovered by filtration and dried under vacuum to yield 9.5 g (- 70 percent of theoretical) of the desired 1 - (2 - pyridinyl) - 5 - (phenylthio) - 1 H - 1,2,4 - triazol - 3 - ol product.
- the product melted at 235°-237°C and upon analysis was found to have carbon, hydrogen and nitrogen contents of 55.87,3.79 and 19.90 percent, respectively, as compared with the theoretical contents of 55.91, 3.93 and 20.06 percent, respectively, calculated for the above named compound.
- a mixture was prepared by admixing 1.84 g (0.00718 m) of 2-benzoyl-2-(2-pyridinyl)hydrazinecarboxamide with 30 ml of 10 percent sodium hydroxide. The mixture was warmed to 50°C to dissolve the carboxamide. The solution was held at 50°C for 10 minutes and the reaction mixture was cooled to room temperature. The reaction product was acidified with 30 ml or 50 percent acetic acid and the white solid which precipitated was recovered by filtration, washed with water and air dried.
- the 5 - phenyl - 1 - (2 - pyridinyl) - 1 H - 1,2,4 - triazol - 3 - ol product was recovered in a yield of 1.12 g (66 percent of theoretical).
- the product melted at 240 0- 241 °C and upon analysis was found to have carbon, hydrogen and nitrogen contents of 64.83, 4.35 and 22.46 percent, respectively, as compared with the theoretical contents of 65.53, 4.23 and 23.52.
- a one liter flask equipped with a dry ice condenser, gas inlet tube, thermometer and a magnetic stirrer was charged with 32.43 g (0.20 m) of 1-(2-pyridinyl)-1 H-1,2,4-triazol-3-ol and 300 ml of water. To this mixture was bubbled 16.3 g (0.23 m) of condensed chlorine gas at 10°-20°C. Thereafter, the reaction mixture was stirred at room temperature for one hour.
- the substituted hydrazine carboxamide corresponding to the formula wherein R is as hereinabove defined can be prepared by the reaction of an appropriate substituted hydrazino compound of the formula in the presence of a solvent and/or water and a mineral acid with an alkali metal cyanate.
- the appropriate hydrazino compound in the solvent and/or water is mixed with concentrated mineral acid and the mixture stirred at a temperature of from 20° to 40°C until a clear solution is formed.
- the alkali metal cyanate is added as an aqueous solution and the mixture stirred at room temperature until the reaction is complete, usually from 1 to 4 hours.
- the product is thereafter recovered by cooling the reaction mixture and filtering off the solid product which precipitates.
- the product can be further purified, if desired, by washing with a solvent such as methylene chlorine and/or water and drying.
- the compounds can be prepared by the reaction of an appropriately substituted haloheterocyclic compound with hydrazine or its hydrate.
- This reaction can be characterized as follows: wherein X, R and n are as hereinbefore defined, q is chloro, fluoro or bromo and no attempt is made to present a balanced equation.
- This procedure is especially preferred when preparing compounds wherein the hydrazino group is in a ring position ortho or para to the heterocyclic nitrogen.
- a solvent such as for example ethanol or isopropanol and a hydrogen halide acceptor
- Representative hydrogen halide acceptors include, triethylamine, pyridine, or other such conventional material.
- a large excess of hydrazine can be employed which then acts as a reactant and as the hydrogen halide acceptor.
- the mixture is maintained under reflux conditions for from 1 to 8 hours.
- about two-thirds of the solvent is distilled off and the remaining mixture cooled, diluted with water, extracted with a solvent such as methylene chloride and the extracts dried.
- the insolubles are filtered off and the solvent removed by evaporation under reduced pressure leaving the desired product.
- the above reactants can be placed into sealed reaction vessel and stirred for from 2 to 16 hours or more at a temperature of from 100° to 150°C, or more, depending upon the reactants.
- the reaction mixture is cooled and the solid product which precipitates are recovered by filtration and washed with water and methylene chloride, filtered off and dried.
- one molar equivalent of the appropriately substituted meta-amino compound is mixed with concentrated hydrochloric acid and cooled to 0 to -10°C while a slight (",10 to 2096) excess of the alkali metal nitrate reactant in water is added at a rate to maintain the reaction temperature at below 0°C.
- This mixture is thereafter slowly added to a fresh solution of stannous chloride in concentrated hydrochloric acid at a rate to maintain the reaction temperature below 0°C.
- the mixture is stirred for from 1 to 3 hours and the solution made basic.
- the mixture is extracted with a solvent such as methylene chloride and the extract dried filtered and distilled to recover the crude product.
- the crude product is purified by dissolving in dilute hydrochloric acid and extracting with methylene chloride.
- the aqueous phase is made basic and the desired product which precipitates is recovered by filtration, water washed and dried.
- a solution was prepared by admixing 55 g (0.43 m) of 2-hydrazino-6-fluoropyridine in 240 ml of water with 40 g of concentrated hydrochloric acid in 60 ml of water. The mixture was stirred at 35°C until a clear solution formed. To this reaction mixture was added 37 g (0.46 m) of potassium cyanate in 100 ml of water. After the addition was completed, the reaction mixture was stirred for 2 1/2 hours at room temperature and then cooled to 10°C on an ice bath. The 2-(6-fluoro-2-pyridinyl)hydrazinecarboxamide product was isolated by filtration and washed with methylene chloride in quantitative yield.
- a solution was prepared by admixing 43 g (0.20 m) of 2-hydrazino-2,3,5-trichlo ' ropyridine in 100 ml of dimethylformamide with 20 g of concentrated hydrochloric acid in 250 ml of water. The mixture was stirred at 35°C until a clear solution formed. To this reaction mixture was added 17 g (0.20 m) of potassium cyanate in 80 ml of water. After the addition was completed, the reaction mixture was stirred for 3 hours at room temperature and then cooled to 10°C on an ice bath.
- a mixture was prepared by admixing 15.22 g (0.1 mole) of 2-(2-pyridinyl)hydrazinecarboxamide with a solution of 16.2 g (0.16 m) of triethylamine in 200 ml of acetonitrile. The mixture was stirred while a solution of 21.08 g (0.15 m) of benzoyl chloride in 50 ml of acetonitrile was added. After an induction period of N 5 minutes, the mixture exothermed reaching a peak temperature of 52 °C after 10 minutes. The reaction mixture was then stirred, without heating, for 5 1/2 hours and then heated under reflux for 2 hours.
- the new triazolyl phosphates of the present invention possess excellent insecticidal and acaracidal properties and are very suitable for the control of chewing and sucking insects for the protection of plants and stored goods.
- the compounds penetrate into the tissues of plants and are highly effective as contact and stomach poison insecticides. Owing to their low toxicity to warm- blooded animals, they are also suitable for the control of ecto and endo-parasites on and in animals.
- the compounds of the present invention can be employed for the control of one or more of the following arthropods, among others, and their various stages of development (larvae and eggs): insects of the families Muscidae and Culicidae, house flies (Musca domestica), stable flies (Stomoxys calcitrans) and mosquitoes (e.g. Aedes Aegypti, Culex fatigans, Anopheles stephensi7; against insects of the families Curculonidae, Bruchidae, Dermestidae, Tenebrionidae and Chrysomelidae, e.g.
- green peach aphids Myzus persicae
- Pseudococcidae e.g. citrus mealybugs (Planococcus citri)
- Locustidae e.g. migratory locusts (Locusta migratoria)
- arachnids including the families Acaridae, Ixodidae, Tetranychidae and Argasidae.
- an insecticidal amount of the phosphorous compound per se or a composition incorporating an insecticidal amount of the compound is used as the toxicant for contact with the pest insect or its habitat.
- the insecticidal amount is that quantity which elicits toxic mortality among the treated pests.
- such insecticidal response results by contacting the target pests or their habitat with a composition containing from 0.00001 to 99 or more percent of the active compound in the total composition. Good results are achieved upon contact with a composition containing about 1000 parts of the active compound per million by weight.
- Suitable compositions include those which are in the form of liquid solutions, liquid emulsifiable concentrates, and dust or granular preparations. Such can be further diluted as and where appropriate with convention diluents.
- Liquid compositions containing the active compound are prepared by dissolving the active compound in a suitable inert organic solvent such as acetone, toluene, xylene, methylene chloride, chlorobenzene, ethyl ether or petroleum distillates or by dispersing the active compound in water with or without the aid of a suitable surface acting dispersing agent such as can be provided by ionic or nonionic dispersing and emulsifying agents.
- a suitable inert organic solvent such as acetone, toluene, xylene, methylene chloride, chlorobenzene, ethyl ether or petroleum distillates
- a suitable surface acting dispersing agent such as can be provided by ionic or nonionic dispersing and emulsifying agents.
- the aqueous compositions may contain one or more water-immiscible solvents for the toxicants.
- the carrier comprises an aqueous emulsion, that is, a mixture of water-immiscible solvent, emulsifying agent and water.
- aqueous emulsion that is, a mixture of water-immiscible solvent, emulsifying agent and water.
- Dispersing and emulsifying agents which may be employed in the compositions include the condensation products of alkylene oxides with phenols and organic acids, alkylarylsulfonates, polyoxyethylene derivatives or sorbitan esters, complex ether alcohols, mahogany soaps, and the like.
- the surface active agents are usually employed in the amount of from 1 to 20 percent by weight of the combined weight of the surface active agent and the active compound.
- the active compound is dispersed in and on a finely divided inert solid such as talcum, chalk, gypsum, and the like.
- the carriers are mechanically ground with the compounds or wet with a volatile organic solvent solution thereof.
- dust compositions containing the compound may be prepared from bentonite, fuller's earth, attapulgite, and other clays.
- these dust compositions may be employed as concentrates and subsequently diluted with additional solid surface acting dispersing agent or with talc, chalk, or gypsum and the like to obtain a desired amount of active agent in a composition adapted to be applied for insect control.
- such concentrate dust compositions may be dispersed in water with or without the aid of a dispersing agent to form spray mixtures.
- Granular formulations are conveniently prepared by impregnations, such as through simple mechanical mixing, of the active compound in a presized carrier, usually of the type hereinbefore set forth.
- the active compound is distributed so as to provide contact of the target insect with toxic amounts of the active compound.
- Such contact can be achieved through direct contact of the active compound with the target insect or by more indirect means such as by application to its food and/or habitat.
- the active compound hereof or a composition thereof can be spread throughout the environs of the target host so as to both provide direct and indirect contact thereof or bait compositions incorporating a toxic amount of the active compound or composition thereof can be readily prepared and strategically located so as to provide ultimate contact of the host species therewith.
- 1 Part of one of the compounds numbered 8, 1 or 20 is mixed with 99 parts of purified kerosene to obtain an oil preparation having an active ingredient concentration of 1 percent.
- the composition can be atomized or sprayed as is.
- compositions may be further diluted in their concentrate state and/or dispersed in water to prepare aqueous compositions which have desirable wetting and penetrating properties.
- These compositions are adapted to be employed to treat target insect life and thus distribute the active compounds to provide contact of such insect life in insecticidal concentrations.
- Paper cylindrical cartons about 3-5/8 inches (9.2 cms) in diameter by 3-1/4 inches (8.26 cms) high were fitted with wire screen on the top and bottom.
- Into each cage were placed 6 German cockroaches (Blattella germanica).
- additional cockroaches were sprayed with a solvent-water-surfactant mixture containing no active toxicant to serve as controls.
- An aqueous dispersion was prepared by dispersing a predetermined amount of one of the test compounds, which had been dissolved in a suitable inert solvent, and a predetermined amount of a surfactant in a predetermined amount of water.
- a water-surfactant-solvent mixture containing none of the compounds was also prepared to serve as a control.
- Sheets containing egg masses of codling moths were pinned to apples and the egg sheets and apples were drenched with an aqueous dispersion of one of the hereinafter set forth compounds. Separate egg masses and apples were also treated with the control mixture. The egg masses/apples were incubated under conditions conducive to the hatching of the eggs and the growth of the larvae therefrom. Ten days after treatment, the apples were examined for the presence of egg hatch and larvae. Counts of the number of larval penetration in the treated fruit were compared to the number present in the untreated control to determine the percent control obtained with the test compounds.
- This examination determined the minimum concentration in parts of the active compound per million parts of the ultimate dispersion necessary to give at least a 70 percent kill and control of codling moth larvae and/or eggs and the results of this examination are set forth in Table III.
- aqueous dispersions were prepared by admixing one of the hereinafter set forth compounds, dissolved in a suitable inert solvent, with a predetermined quantity of water and a predetermined amount of a surfactant to give aqueous dispersions containing varying predetermined amounts of one of the compounds as the sole active toxicant.
- Separate cotton plant leaves were thoroughly wetted by briefly dipping into one of the dispersions and the wetted leaves placed in an open petri dish and permitted to dry. After the leaves were dry, 5 live beet armyworm larvae, approximately late 2nd instar were placed in each petri dish.
- aqueous dispersions were prepared by admixing one of the hereinafter set forth compounds, dissolved in a suitable inert solvent, with a predetermined quantity of water and a predetermined amount of a surfactant to give aqueous dispersions of varying predetermined amounts of one of the compounds as the sole active toxicant.
- Separate 3 inch discs cut from tobacco plant leaves were thoroughly wetted by briefly dipping into one of the dispersions and the wetted leaves placed in an open petri dish and permitted to dry. After the leaves were dry, 5 live tobacco budworm larvae, approximately late 2nd instar were placed in each petri dish.
- Aqueous dispersions were prepared by admixing one of the hereinafter set forth compounds, dissolved in a suitable inert solvent with a predetermined quantity of water and a predetermined amount of a surfactant to give aqueous dispersions containing varying predetermined amounts of one of the compounds as the sole toxicant.
- Separate chili pepper plants were infested with 20 green peach aphids and the plants sprayed with one of the dispersions to run off.
- 20 green peach aphids were placed on control plants and the,plants sprayed to run off with a solution containing only water and surfactant. The plants were maintained under conditions conducive to the growth of the plants and aphids.
- Aqueous dispersions were prepared by admixing one of the hereinafter set forth compounds, dissolved in a suitable inert solvent, with a predetermined quantity of water and a predetermined amount of a surfactant to give aqueous dispersions containing varying predetermined amounts of one of the compounds as the sole toxicant.
- Separate cotton plants were infested with 20 two spotted spider mites and the plants sprayed with one of the dispersions to run off.
- 20 two spotted spider mites were placed on control plants and the plants sprayed to run off with a solution containing only water and surfactant. The plants were maintained under conditions conducive to the growth of the plants and mites.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Pest Control & Pesticides (AREA)
- Agronomy & Crop Science (AREA)
- Plant Pathology (AREA)
- Engineering & Computer Science (AREA)
- Dentistry (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Environmental Sciences (AREA)
- Plural Heterocyclic Compounds (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
Claims (19)



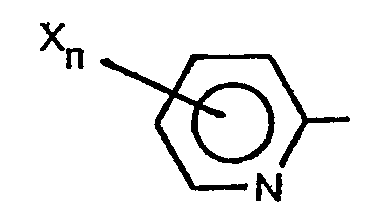
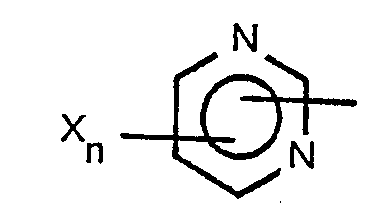
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US95192378A | 1978-10-13 | 1978-10-13 | |
| US951923 | 1978-10-13 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP0010891A1 EP0010891A1 (de) | 1980-05-14 |
| EP0010891B1 true EP0010891B1 (de) | 1982-09-01 |
Family
ID=25492339
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP79302206A Expired EP0010891B1 (de) | 1978-10-13 | 1979-10-12 | Heterocyclisch substituierte Triazolylphosphorverbindungen, sie enthaltende insektizide Zusammensetzungen und Verfahren zur Bekämpfung von Insekten |
Country Status (15)
| Country | Link |
|---|---|
| EP (1) | EP0010891B1 (de) |
| JP (1) | JPS5553297A (de) |
| AR (1) | AR226690A1 (de) |
| AU (1) | AU529628B2 (de) |
| BR (1) | BR7906567A (de) |
| CA (1) | CA1100967A (de) |
| DE (1) | DE2963622D1 (de) |
| DK (1) | DK426979A (de) |
| GB (1) | GB2035325B (de) |
| HU (1) | HU182946B (de) |
| IL (1) | IL58427A (de) |
| IN (1) | IN151525B (de) |
| MY (1) | MY8500034A (de) |
| NZ (1) | NZ191781A (de) |
| ZA (1) | ZA795431B (de) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4562801A (en) * | 1983-07-28 | 1986-01-07 | Sanshin Kogyo Kabushiki Kaisha | Engine control system for marine propulsion device |
| WO2024208143A1 (zh) * | 2023-04-05 | 2024-10-10 | 青岛清原化合物有限公司 | 一种三氮唑类化合物及其制备方法、除草组合物和应用 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NL7316468A (de) * | 1972-12-08 | 1974-06-11 | ||
| DE2424571A1 (de) * | 1974-05-21 | 1975-12-04 | Bayer Ag | S-triazolopyridinmethyl-thiolo(thion)phosphor(phosphon)-saeureester bzw. -esteramide, verfahren zu ihrer herstellung und ihre verwendung als insektizide und akarizide |
| DE2515794A1 (de) * | 1975-04-11 | 1976-10-21 | Bayer Ag | S-triazolobenzopyrazinmethyl(thiono) thiolphosphor(phosphon)saeureester, verfahren zu ihrer herstellung und ihre verwendung als insektizide |
| DE2547971A1 (de) * | 1975-10-27 | 1977-05-12 | Bayer Ag | O-triazolylthiono(thiol)-phosphor (phosphon)-saeureester bzw. -esteramide, verfahren zu ihrer herstellung und ihre verwendung als insektizide, akarizide und nematizide |
-
1979
- 1979-10-08 AU AU51573/79A patent/AU529628B2/en not_active Ceased
- 1979-10-08 NZ NZ191781A patent/NZ191781A/xx unknown
- 1979-10-10 IL IL58427A patent/IL58427A/xx unknown
- 1979-10-10 CA CA337,298A patent/CA1100967A/en not_active Expired
- 1979-10-10 DK DK426979A patent/DK426979A/da not_active Application Discontinuation
- 1979-10-11 ZA ZA00795431A patent/ZA795431B/xx unknown
- 1979-10-11 HU HU79DO437A patent/HU182946B/hu unknown
- 1979-10-12 GB GB7935448A patent/GB2035325B/en not_active Expired
- 1979-10-12 EP EP79302206A patent/EP0010891B1/de not_active Expired
- 1979-10-12 IN IN1069/CAL/79A patent/IN151525B/en unknown
- 1979-10-12 JP JP13173179A patent/JPS5553297A/ja active Pending
- 1979-10-12 AR AR278478A patent/AR226690A1/es active
- 1979-10-12 BR BR7906567A patent/BR7906567A/pt unknown
- 1979-10-12 DE DE7979302206T patent/DE2963622D1/de not_active Expired
-
1985
- 1985-12-30 MY MY34/85A patent/MY8500034A/xx unknown
Also Published As
| Publication number | Publication date |
|---|---|
| IL58427A (en) | 1983-10-31 |
| HU182946B (en) | 1984-03-28 |
| EP0010891A1 (de) | 1980-05-14 |
| BR7906567A (pt) | 1980-07-15 |
| ZA795431B (en) | 1980-09-24 |
| AU529628B2 (en) | 1983-06-16 |
| IN151525B (de) | 1983-05-14 |
| GB2035325B (en) | 1983-03-02 |
| DK426979A (da) | 1980-04-14 |
| GB2035325A (en) | 1980-06-18 |
| NZ191781A (en) | 1982-03-16 |
| AR226690A1 (es) | 1982-08-13 |
| IL58427A0 (en) | 1980-01-31 |
| AU5157379A (en) | 1980-04-17 |
| CA1100967A (en) | 1981-05-12 |
| MY8500034A (en) | 1985-12-31 |
| JPS5553297A (en) | 1980-04-18 |
| DE2963622D1 (en) | 1982-10-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US4080443A (en) | (Substituted pyridyl)phenoxy phosphorous compounds and their use as insecticides | |
| US4298602A (en) | Heterocyclic substituted triazolyl phosphorous compounds and their use as insecticides | |
| US4429125A (en) | Phosphorus esters of 5-pyrimidinols | |
| US4132783A (en) | (Substituted pyridyl)phenoxy phosphorus compounds and their use as insecticides | |
| EP0010891B1 (de) | Heterocyclisch substituierte Triazolylphosphorverbindungen, sie enthaltende insektizide Zusammensetzungen und Verfahren zur Bekämpfung von Insekten | |
| US4028377A (en) | O,S-dialkyl- and O-alkyl-S-alkoxyalkyl-S-1,2,4-oxa-diazolyl-3-methylene dithiophosphates | |
| US4212861A (en) | Isoxazole-5-alkyl dithiophosphoric acid derivatives | |
| US4558039A (en) | Phosphorus derivatives of 2-fluoroalkyl-5-pyrimidinols useful as insecticides | |
| US4172080A (en) | Phosphorus esters of 1-cyanoethyl-1,2,4-triazol-3-ols | |
| EP0023841B1 (de) | Phosphorige Ester von 5-Pyrimidinolen und Verfahren zu ihrer Herstellung, diese Ester enthaltende Zusammensetzungen und ihre Verwendung als Pestizide | |
| JPS598277B2 (ja) | 置換されたエチル−りん酸(ホスホン酸)エステル、それらの製造方法並びに殺昆虫剤、殺ダニ剤及び殺線虫剤としてのそれらの使用 | |
| KR830000616B1 (ko) | 살충제로 유용한 헤테로 고리치환 트리아졸릴 포스포러스 화합물의 제조방법 | |
| US4223025A (en) | 6-Fluoro-3,5-dichloro-2-pyridinyl phosphorus compounds | |
| US4729987A (en) | Insecticidal O,O-diethyl-O-(2-(1,1-dimethylethyl)-5-pyrimidinyl)-phosphorothioate | |
| PL88487B1 (de) | ||
| US3475452A (en) | Phosphates and phosphonates of cyclic sulfones | |
| US4357328A (en) | Heterocyclic substituted triazolyl phosphorous compounds and their use as insecticides | |
| US4465675A (en) | Insecticidal and fungicidal 1-alkyl-5-alkylsulfonyl-4-chloropyrazole-3-yl-(thio)phosphates and (thio)phosphonates | |
| US4654329A (en) | Insecticidal, miticidal or nematocidal phosphorus esters of 5-pyrimidinols | |
| US3551562A (en) | Insecticidal use of s-((4-oxo-1,2,3-benzotriazin - 3(4h)-yl) - methyl) phosphorothioates and phosphorodithioates | |
| US4424217A (en) | Substituted phenyl phosphorothioates and their use as pesticides | |
| US3320261A (en) | Pyridyl and quinolyl mercapto methyl phosphorus esters | |
| EP0011363B1 (de) | Pyrazolphosphate und -phosphonate, ihre Herstellung und ihre Verwendung als Insektizide | |
| US4400516A (en) | Heterocyclic substituted triazole-3-ol compounds | |
| US4670425A (en) | Oximinophosphoric acid derivatives and their use for controlling pests |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Designated state(s): BE CH DE FR IT LU NL SE |
|
| 17P | Request for examination filed | ||
| ITF | It: translation for a ep patent filed | ||
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Designated state(s): BE CH DE FR IT LU NL SE |
|
| REF | Corresponds to: |
Ref document number: 2963622 Country of ref document: DE Date of ref document: 19821028 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19821031 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: SE Payment date: 19830831 Year of fee payment: 5 Ref country code: DE Payment date: 19830831 Year of fee payment: 5 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 19830906 Year of fee payment: 5 Ref country code: CH Payment date: 19830906 Year of fee payment: 5 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: BE Payment date: 19830930 Year of fee payment: 5 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: LU Payment date: 19831005 Year of fee payment: 5 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: NL Payment date: 19831031 Year of fee payment: 5 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SE Effective date: 19841013 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CH Effective date: 19841031 Ref country code: BE Effective date: 19841031 |
|
| BERE | Be: lapsed |
Owner name: THE DOW CHEMICAL CY Effective date: 19841012 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Effective date: 19850501 |
|
| NLV4 | Nl: lapsed or anulled due to non-payment of the annual fee | ||
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19850628 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Effective date: 19850702 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST |
|
| EUG | Se: european patent has lapsed |
Ref document number: 79302206.2 Effective date: 19851007 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |