-
Die
vorliegende Erfindung betrifft substituierte Piperazinderivate (hier
als Verbindungen oder Verbindungen der Formel (1) bezeichnet) oder
Stereoisomere oder pharmazeutisch verträgliche Salze davon und deren
Verwendung als Tachykininrezeptor-Antagonisten. Diese Antagonisten
sind bei der Behandlung von Erkrankungen und Zuständen, die
durch Tachykinin vermittelt und hier offenbart werden, einschließlich Asthma, Husten
und Bronchitis, verwendbar.
-
ZUSAMMENFASSUNG
DER ERFINDUNG
-
Die
vorliegende Erfindung betrifft Verbindungen der Formel (1):
Formel
(1) wobei
G
1 -CH
2- oder -C(O)- ist;
G
2 -CH
2- oder -C(O)- ist;
G
3 -CH
2- oder -C(O)- ist;
m 0 oder 1 ist;
Ar
1 ein Rest ist, ausgewählt aus:
wobei
Z
1 1 bis 3 Substituenten bedeutet, welche
jeweils unabhängig
ausgewählt
sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF
3,
C
1-C
4-Alkyl und
C
1-C
4-Alkoxy;
Ar
2 ein Rest ist, ausgewählt aus:
wobei
Z
2 1
bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind
aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF
3,
C
1-C
4-Alkyl und
C
1-C
4-Alkoxy;
Ar
3 ein Rest ist, ausgewählt aus:
wobei
Z
3 1 bis 3 Substituenten bedeutet, welche
jeweils unabhängig
ausgewäht
sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF
3,
C
1-C
4-Alkyl und
C
1-C
4-Alkoxy; R
6 Wasserstoff, C
1-C
4-Alkyl, oder -CHO ist;
R
1 Wasserstoff,
C
1-C
4-Alkyl, -(CH
2)
qAr
4 oder
-CH
2C(O)Ar
4 ist,
wobei q eine ganze Zahl von 1 bis 4 und Ar
4 ein Rest
der Formel
ist, wobei
Z
4 1 bis 3 Substituenten bedeutet, welche
jeweils unabhängig
ausgewählt
sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF
3,
C
1-C
4-Alkyl und
C
1-C
4-Alkoxy;
oder
Stereoisomere oder pharmazeutisch verträgliche Salze davon;
mit
der Maßgabe,
dass, falls G
1 -C(O)- ist, dann G
2 nicht -C(O)- ist;
und mit der weiteren
Maßgabe,
dass, falls G
3 -CH
2-
ist, dann G
1 und G
2 -CH
2- sind.
-
Wie
Durchschnittsfachleuten klar ist, liegen die Verbindungen der Formel
(1) als Stereoisomere vor. Die Cahn-Ingold-Prelog-Bezeichnung (R)-
und (S)- für
die Stereochemie der Verbindungen der Formel (1) hängt von
der Art der vorhandenen Substituenten ab. In dieser Anmeldung soll
jede Bezugnahme auf eine der Verbindungen der Formel (1) entweder
spezielle Stereoisomere oder ein Gemisch von Stereoisomeren umfassen.
Die speziellen Stereoisomere können
durch stereospezifische Synthese hergestellt werden oder können mit
im Fachgebiet bekannten Verfahren getrennt und gewonnen werden,
wie Chromatographie an chiralen stationären Phasen, Amidbildung mit
einer chiralen Säure,
gefolgt von Trennung der resultierenden diastereomeren Amide und
Hydrolyse zum gewünschten
Stereoisomer, oder fraktionierte Umkristallisation von Additionssalzen,
die mit zu diesem Zweck verwendeten Reagenzien erzeugt werden, wie
in „Enantiomers,
Racemates, and Resolutions",
J. Jacques, A. Collet und S. H. Wilen, Wiley (1981), beschrieben.
-
Wie
in dieser Anmeldung verwendet:
- a) bezieht sich
der Begriff „Halogen" auf ein Fluoratom,
Chloratom, Bromatom oder Iodatom;
- b) bezieht sich der Begriff „C1-C4-Alkyl" auf
einen verzweigten oder geradkettigen Alkylrest mit 1 bis 4 Kohlenstoffatomen,
wie Methyl, Ethyl, n-Propyl, Isopropyl, n-Butyl, Isobutyl, t-Butyl
etc;
- c) bezieht sich der Begriff „C1-C4-Alkoxy" auf
einen geraden oder verzweigten Alkoxyrest mit 1 bis 4 Kohlenstoffatomen,
wie Methoxy, Ethoxy, n-Propoxy, Isopropoxy, n-Butoxy, Isobutoxy, t-Butoxy etc;
- d) bezieht sich die Bezeichnung -C(O)- oder C(O) auf eine Carbonylgruppe
der Formel:
- e) bezieht sich die Bezeichnung „" auf eine Bindung,
deren Stereochemie nicht gekennzeichnet ist.
- f) wie in den Herstellungen und Beispielen verwendet: bezieht
sich der Begriff „mg" auf Milligramm;
bezieht sich der Begriff „g" auf Gramm; bezieht
sich der Begriff „kg" auf Kilogramm; bezieht
sich der Begriff „mmol" auf Millimol; bezieht
sich der Begriff „ml" auf Milliliter;
bezieht sich der Begriff „°C" auf Grad Celsius;
bezieht sich der Begriff „Rf" auf
den Retentionsfaktor; bezieht sich der Begriff „mp" auf den Schmelzpunkt; bezieht sich
der Begriff „dec" auf die Zersetzung;
bezieht sich der Begriff „THF" auf Tetrahydrofuran;
bezieht sich der Begriff „DMF" auf Dimethylformamid;
bezieht sich der Begriff „[α] 20 / D" auf die spezifische
Drehung der Natrium-D-Linie bei 20°C, die in einer 1 dezimeter-Zelle
erhalten wurde; bezieht sich der Begriff „c" auf die Konzentration in g/ml; bezieht
sich der Begriff „DMSO" auf Dimethylsulfoxid;
bezieht sich der Begriff „M" auf molar; bezieht
sich der Begriff „HPLC" auf Hochleistungsflüssigchromatographie;
bezieht sich der Begriff „HRMS" auf hochauflösendes Massenspektrum;
- g) unter der Bezeichnung ist klar, dass der Rest an
der 1-Position verknüpft
ist und der Substituent oder die Substituenten Z an jeder der Positionen
2, 3, 4, 5 oder 6 verknüpft
sein können;
- h) unter der Bezeichnung ist klar, dass der Rest entweder
an der 1-Position oder der 2-Position verknüpft sein kann, es ist ferner
klar, dass wenn der Rest an der 1-Position verknüpft ist, der Substituent oder
die Substituenten Z an jeder der Positionen 2, 3, 4, 5, 6, 7 oder
8 verknüpft
sein können,
und dass wenn der Rest an der 2-Position verknüpft ist, der Substituent oder
die Substituenten Z an jeder der Positionen 1, 3, 4, 5, 6, 7 oder
8 verknüpft
sein können;
- i) bezieht sich der Begriff „pharmazeutisch verträgliche Salze
davon" auf entweder
ein Säureadditionssalz oder
ein Basenadditionssalz.
-
Der
Ausdruck „pharmazeutisch
verträgliche
Säureadditionssalze" soll auf alle nicht
toxischen organischen oder anorganischen Säureadditionssalze der Grundverbindungen
der Formel (1) oder aller ihrer Zwischenprodukte Anwendung finden.
Veranschaulichende anorganische Säuren, die geeignete Salze bilden, schließen die
Chlorwasserstoff-, Bromwasserstoff-, Schwefel- und Phosphorsäure und
saure Metallsalze ein, wie Natriummonohydrogenorthophosphat und
Kaliumhydrogensulfat. Veranschaulichende organische Säuren, die
geeignete Salze bilden, schließen
die Mono-, Di- und Tricarbonsäuren
ein. Veranschaulichend für
solche Säuren
sind beispielsweise Essig-, Glykol-, Milch-, Brenztrauben-, Malon-,
Bernstein-, Glutar-, Fumar-, Äpfel-,
Wein-, Zitronen-, Ascorbin-, Malein-, Hydroxymalein-, Benzoe-, Hydroxybenzoe-,
Phenylessig-, Zimt-, Salicyl-, 2-Phenoxybenzoe-,
p-Toluolsulfonsäure
und Sulfonsäuren,
wie Methansulfonsäure
und 2-Hydroxyethansulfonsäure. Diese
Salze können
entweder in einer hydratisierten oder einer im Wesentlichen wasserfreien Form
vorliegen. Im Allgemeinen sind die Säureadditionssalze dieser Verbindungen
in Wasser und verschiedenen hydrophilen organischen Lösungsmitteln
löslich
und zeigen im Vergleich zu ihren freien Grundformen im Allgemeinen
höhere
Schmelzpunkte.
-
Der
Ausdruck „pharmazeutisch
verträgliche
Basenadditionssalze" soll
auf alle nicht toxischen organischen oder anorganischen basischen
Additionssalze der Grundverbindungen der Formel (1) oder aller ihrer Zwischenprodukte
Anwendung finden. Veranschaulichende Basen, die geeignete Salze
bilden, schließen
Alkalimetall- oder Erdalkalimetallhydroxide, wie Natrium-, Kalium-,
Calcium-, Magnesium- oder Bariumhydroxide; Ammoniak, und aliphatische,
alicyclische oder aromatische organische Amine ein, wie Methylamin,
Dimethylamin, Trimethylamin und Picolin. Entweder ein- oder zweibasige
Salze können
mit diesen Verbindungen gebildet werden.
-
Wie
bei allen Gruppen von strukturell verwandten Verbindungen, die eine
spezielle generische Verwendbarkeit besitzen, werden bestimmte Reste
und Konfigurationen für
die Verbindungen der Formel (1) in ihrer Endanwendung bevorzugt.
-
Bevorzugte
Ausführungsformen
der Formel (1) werden nachstehend angegeben:
- 1)
Verbindungen, in denen G3 -C(O)- ist, werden
bevorzugt;
- 2) Verbindungen, in denen G1 -C(O)-
und G2 -CH2- ist,
werden bevorzugt und Verbindungen, in denen G1 -CH2- und G2 -C(O)-
ist, werden stärker
bevorzugt;
- 3) Verbindungen, in denen R1 Wasserstoff
ist, werden bevorzugt.
- 4) Verbindungen, in denen Ar3 der Rest ist, werden bevorzugt.
-
Es
versteht sich, dass weitere bevorzugte Ausführungsformen der Formel (1)
gewählt
werden können, indem
eine oder mehrere der bevorzugten Ausführungsformen 1 bis 4 der Formel
(1) gefordert werden, oder durch Bezugnahme auf die hier angegebenen
Beispiele.
-
Veranschaulichende
Verbindungen, die in der vorliegenden Erfindung eingeschlossen sind,
schließen die
folgenden ein. Diese Liste soll lediglich repräsentativ sein und soll den
Umfang der Erfindung in keiner Weise begrenzen:
(S)-2-[(S)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid];
(S)-2-[(S)-3-(1-Methylindol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid];
(S)-2-[(R)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid];
(S)-2-[(S)-3-Phenylmethyl-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid];
(S)-2-[(S)-3-Phenyl-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid];
(S)-2-[(S)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(naphth-2-yl)propionamid];
(S)-2-[(S)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-(2-methoxybenzyl)-3-(naphth-2-yl)propionamid];
(S)-2-[(S)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-(3,4-dichlorbenzyl)-3-(naphth-2-yl)propionamid];
(S)-2-[(S)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-(2-methoxybenzyl)-3-(phenyl)propionamid];
(S)-2-[3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-2-[[(S)-N-methyl-N-(3-phenylpropyl)]benzamid];
(S)-2-[3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-[(S)-N-methyl-N-benzyl-3-(phenyl)propylamin];
(R)-2-[(S)-3-(1H-Indol-3-ylmethyl)piperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid];
(R)-2-[(S)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid];
(R)-2-[(S)-3-(1-Methylindol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid];
(R)-2-[(S)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid];
(R)-2-[(S)-3-Phenylmethyl-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid];
(R)-2-[(S)-3-Phenyl-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid];
(R)-2-[(S)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(naphth-2-yl)propionamid];
(R)-2-[(S)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-(2-methoxybenzyl)-3-(naphth-2-yl)propionamid];
(R)-2-[(S)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-(3,4-dichlorbenzyl)-3-(naphth-2-yl)propionamid];
(R)-2-[(S)-3-Phenyl-2-oxopiperazin-1-yl]-1-[N-methyl-N-(2-methoxybenzyl)-3-(phenyl)propionamid];
(R)-2-[3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-2-[[(S)-N-methyl-N-(3-phenylpropyl)]benzamid];
(R)-2-[3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-[(S)-N-methyl-N-benzyl-3-(phenyl)propylamin];
(R)-2-[(S)-3-(1H-Indol-3-ylmethyl)piperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid];
(S)-2-[(R)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid];
(S)-2-[(R)-3-(1-Methylindol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid];
(S)-2-[(R)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3- (phenyl)propionamid];
(S)-2-[(R)-3-Phenylmethyl-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid];
(S)-2-[(R)-3-Phenyl-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid];
(S)-2-[(R)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(naphth-2-yl)propionamid];
(S)-2-[(R)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-(2-methoxybenzyl)-3-(naphth-2-yl)propionamid];
(S)-2-[(R)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-(3,4-dichlorbenzyl)-3-(naphth-2-yl)propionamid];
(S)-2-[(R)-3-Phenyl-2-oxopiperazin-1-yl]-1-[N-methyl-N-(2-methoxybenzyl)-3-(phenyl)propionamid];
(S)-2-[3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-2-[[(R)-N-methyl-N-(3-phenylpropyl)]benzamid];
(S)-2-[3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-[(R)-N-methyl-N-benzyl-3-(phenyl)propylamin];
(S)-2-[(R)-3-(1H-Indol-3-ylmethyl)piperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid];
(R)-2-[(R)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid];
(R)-2-[(R)-3-(1-Methylindol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid];
(R)-2-[(R)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid];
(R)-2-[(R)-3-Phenylmethyl-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid];
(R)-2-[(R)-3-Phenyl-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid];
(R)-2-[(R)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(naphth-2-yl)propionamid];
(R)-2-[(R)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-(2-methoxybenzyl)-3-(naphth-2-yl)propionamid];
(R)-2-[(R)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-(3,4-dichlorbenzyl)-3-(naphth-2-yl)propionamid];
(R)-2-[(R)-3-Phenyl-2-oxopiperazin-1-yl]-1-[N-methyl-N-(2-methoxybenzyl)-3-(phenyl)propionamid];
(R)-2-[3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-2-[[(R)-N-methyl-N-(3-phenylpropyl)]benzamid];
(R)-2-[3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-[(R)-N-methyl-N-benzyl-3- (phenyl)propylamin];
(R)-2-[(R)-3-(1H-Indol-3-ylmethyl)piperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid].
-
Die
Verbindungen der Formel (1) können
mittels der folgenden Syntheseverfahren hergestellt werden, wodurch
erfindungsgemäße Zwischenprodukte
oder Endverbindungen hergestellt werden:
- • Schema
A.1 betrifft die Synthese von Verbindungen der Formel (1), in der
G3 -C(O)- ist.
- • Schema
A.2 betrifft die Synthese von Verbindungen der Formel (1), in der
G3 -CH2- ist.
- • Schema
B betrifft die Synthese des Aldehyds der Struktur (3), in der G1 -CH2- und G2 -C(O)- ist, der als Ausgangsmaterial in
Schema A.1 eingesetzt wird.
- • Schema
C betrifft die Synthese des Aldehyds der Struktur (3), in der G1 -C(O)- und G2 -CH2- ist, der als Ausgangsmaterial in Schema
A.1 eingesetzt wird.
- • Schema
D betrifft die Synthese des Aldehyds der Struktur (3), in der G1 -CH2- und G2 -CH2- ist, der
als Ausgangsmaterial in Schema A.1 eingesetzt wird.
-
In
Schema A.1 wird ein allgemeines Syntheseverfahren zur Herstellung
dieser Verbindungen der Formel (1), in der G3 -C(O)-
ist, angegeben. Die Reagenzien und Ausgangsmaterialien sind für den Durchschnittsfachmann
leicht erhältlich.
In Schema A.1 sind alle Substituenten, sofern nicht anders angegeben,
wie zuvor definiert.
-
-
-
In
Schema A.1, Schritt a, wird ein geeigneter Aldehyd der Struktur
(3) mit einem geeigneten Amin der Struktur (2) oder einem Salz davon
in einer reduktiven Aminierung in Kontakt gebracht, so dass sich
eine Verbindung der Struktur (4) ergibt.
-
Ein
geeigneter Aldehyd der Struktur (3) ist einer, bei dem Stereochemie,
G1, G2, Ar1 und Ar2 so sind, wie
im Produkt der Formel (1) erwünscht,
oder kann auch einer sein, bei dem die Stereochene, Ar1 und
Ar2 nach Antipodentrennung, Schutzgruppenabspaltung
oder Modifikation die Stereochemie, Ar1 und
Ar2 ergeben, wie im Endprodukt der Formel
(1) erwünscht.
-
Ein
geeignetes Amin der Struktur (2) ist eines, bei dem R10 ein
C1-C4-Alkyl ist,
wobei Verbindungen der Struktur (2), bei denen R10 Methyl
ist, bevorzugt werden, die Stereochemie, m und Ar3 so
sind, wie im Endprodukt der Formel (1) erwünscht, oder kann auch eines
sein, bei dem die Stereochemie und Ar3 nach
Antipodentrennung, Schutzgruppenabspaltung oder Modifikation die
Stereochemie und Ar3 ergeben, wie im Endprodukt der
Formel (1) erwünscht.
-
Beispielsweise
wird ein geeigneter Aldehyd der Struktur (3) mit einem geeigneten
Amin der Struktur (2) oder einem Salz eines geeigneten Amins der
Struktur (2) in einer reduktiven Aminierung in Kontakt gebracht.
Die Reaktion wird unter Verwendung eines molaren Überschusses
eines geeigneten Reduktionsmittels durchgeführt, wie Natriumborhydrid oder
Natriumcyanoborhydrid, wobei Natriumcyanoborhydrid bevorzugt wird.
Die Reaktion wird in einem geeigneten Lösungsmittel, wie Methanol,
durchgeführt.
Die Reaktion wird bei Temperaturen von 0°C bis 50°C durchgeführt. Die Reaktion erfordert
im Allgemeinen 1 bis 72 Stunden. Das Produkt kann mit im Fachgebiet
bekannten Verfahren, wie Extraktion, Verdampfen, Chromatographie
und Umkristallisation, isoliert und gereinigt werden.
-
In
Schema A.1, Schritt b, wird an der Verbindung der Struktur (4) die
Schutzgruppe entfernt, so dass sich ein Diaminoester der Struktur
(5) ergibt.
-
Beispielsweise
wird eine Verbindung der Struktur (4) mit einer Protonensäure, wie
Salzsäure
oder Trifluoressigsäure,
umgesetzt. Die Reaktion wird in einem Lösungsmittel, wie Ethylacetat,
Dioxan, Methanol oder Ethanol, durchgeführt. Die Reaktion erfordert
im Allgemeinen 1 bis 48 Stunden und wird bei Zimmertemperatur durchgeführt. Das
Produkt kann mit im Fachgebiet bekannten Verfahren, wie Extraktion,
Verdampfen, Chromatographie und Umkristallisation, isoliert und
gereinigt werden.
-
In
Schema A.1, Schritt c, wird ein Diaminoester der Struktur (5), bei
dem R10 C1-C4-Alkyl ist, zu einer Diaminosäure der
Struktur (5a) hydrolysiert. Die Hydrolyse von Estern, wie die in
Protecting Groups in Organic Synthesis von T. Greene beschriebenen,
ist bekannt und im Fachgebiet anerkannt. Für Durchschnittsfachleute ist
klar, dass die Schritte b und c in beliebiger Reihenfolge durchgeführt werden
können.
-
Beispielsweise
wird ein Diaminoester der Struktur (5), bei dem R10 C1-C4-Alkyl ist, mit
einem geeigneten Hydrolysemittel, wie Natriumhydroxid, Kaliumhydroxid,
Lithiumhydroxid oder Natriumcarbonat, umgesetzt. Die Reaktion wird
in einem geeigneten Lösungsmittel,
wie Wasser oder Wasser/Methanol-Gemische, Wasser/Ethanol-Gemische,
Wasser/Tetrahydrofuran-Gemische, durchgeführt. Die Reaktionen werden
bei Temperaturen von 0°C
bis zur Rückflusstemperatur
des Lösungsmittels
durchgeführt
und erfordern im Allgemeinen 30 Minuten bis 48 Stunden. Das Produkt
kann mit im Fachgebiet bekannten Verfahren, wie Ansäuern, Extraktion,
Verdampfen, Chromatographie und Umkristallisation, isoliert und
gereinigt werden.
-
In
Schema A.1, Schritt d, wird eine Diaminosäure der Struktur (5a) oder
ein Salz davon zyklisiert, wodurch sich eine Verbindung der Formel
(1) ergibt, in der G3 -C(O)- und R1 Wasserstoff ist. Diese Zyklisierungsreaktion
kann in Gegenwart von Kupplungsmitteln, wie 2-Ethoxy-1-ethoxycarbonyl-1,2-dihydrochinolin
oder 1,3-Dicyclohexylcarbodiimid, durchgeführt werden oder kann über eine
aktivierte Zwischenstufe, wie (O)-Hydroxybenzotriazol, ablaufen, die vor
der Zyklisierung hergestellt werden kann, aber nicht notwendigerweise isoliert
wird.
-
Beispielsweise
wird eine Diaminosäure
der Struktur (5a) oder ein Salz davon mit 1-Hydroxybenzotriazol-Hydrat in Gegenwart
eines geringen molaren Überschusses
eines Kupplungsmittels, wie Dicyclohexylcarbodiimid oder 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid,
in Kontakt gebracht. Die Reaktion wird in Gegenwart einer geeigneten
Base, wie Diisopropylethylamin, durchgeführt. Die Reaktion wird in einem
geeigneten Lösungsmittel,
wie Dichlormethan oder Chloroform, durchgeführt. Die Reaktion wird bei
Temperaturen von –50°C bis zur
Rückflusstemperatur
des Lösungsmittels
durchgeführt.
Die Reaktion erfordert im Allgemeinen 1 Stunde bis 48 Stunden. Das
Produkt kann mit im Fachgebiet bekannten Verfahren, wie Extraktion,
Verdampfen, Chromatographie und Umkristallisation, isoliert und
gereinigt werden.
-
In
Schema A.1, optionaler Schritt e, kann von einer geschützten Verbindung
der Formel (1) die Schutzgruppe entfernt werden oder die Verbindung
so modifiziert werden, dass sich eine Verbindung der Formel (1) ergibt.
-
Eine
Reaktion zur Schutzgruppenabspaltung umfasst die Entfernung einer
Hydroxyschutzgruppe. Die Auswahl, Verwendung und Entfernung von
Schutzgruppen, die geeignete Schutzgruppen, wie die in Protecting Groups
in Organic Synthesis von T. Greene beschriebenen, einsetzen, ist
im Fachgebiet bekannt und anerkannt.
-
Eine
Modizierungsreaktion umfasst die Alkylierung eines Amins, eine Additionsreaktion
an einen Indolstickstoff oder die Bildung eines Amidats. Eine Verbindung
der Formel (1), in der G3 -C(O)- und R1 Wasserstoff ist, wird mit einem geeigneten
Alkylierungsmittel zu einer Verbindung der Formel (1) alkyliert,
in der R1 C1-C4-Alkyl, -(CH2)qAr4 oder -CH2C(O)Ar4 ist. Ein
geeignetes Alkylierungsmittel ist eines, das C1-C4-Alkyl, -(CH2)qAr4 oder -CH2C(O)Ar4 überträgt, wie
Iodmethan, Brommethan, Bromethan, Brompropan, Brombutan, Benzylbromid,
Benzylchlorid, Phenethylbromid, 3-Chlor-1-phenylpropan, 4-Chlor-1-phenylbutan, α-Chloracetophenon, α-Bromacetophenon,
3-[(Chlor)acetyl]indol usw. Modifizierungsreaktionen, die die Alkylierung
eines Indolstickstoffs beinhalten und an Verbindungen der Formel
(1), in denen R1 Wasserstoff ist, durchgeführt werden,
können
die Verwendung einer Schutzgruppe erfordern. In diesem Fall kann
eine t-BOC-Schutzgruppe verwendet werden. Die Verwendung und Entfernung
der t-BOC-Schutzgruppe, wie in Protecting Groups in Orgnic Synthesis
von T. Greene beschrieben, ist im Fachgebiet bekannt und anerkannt.
-
Beispielsweise
kann eine Modifizierung eine Verbindung der Formel (1), in der R1 Wasserstoff ist, betreffen, die mit einer
geringen molaren Menge eines geeigneten Alkylierungsmittels in Kontakt
gebracht wird. Die Reaktion wird in Gegenwart eines geringen molaren Überschusses
einer geeigneten Base, wie Natriumbicarbonat, Kaliumbicarbonat,
Diisopropylethylamin oder Triethylamin, durchgeführt. Die Reaktion wird in einem
geeigneten Lösungsmittel,
wie Acetonitril, Dimethylformamid, Ethanol oder Dimethylsulfoxid,
durchgeführt.
Die Reaktion kann in Gegenwart eines geeigneten Katalysators, wie
Kaliumiodid, Natriumiodid, Tetrabutylammoniumiodid, Trimethylbenzylammoniumiodid,
Tetraethylammoniumiodid, Tetrabutylammoniumbromid, Trimethylbenzylammoniumbromid,
Tetraethylammoniumbromid, Tetrabutylammoniumhydrogensulfat, Trimethylbenzylammoniumhydrogensulfat,
Tetraethylammoniumhydrogensulfat usw., durchgeführt werden. Die Reaktion wird
bei Temperaturen von 50°C
bis zur Rückflusstemperatur
des Lösungsmittels
durchgeführt. Die
Reaktion erfordert im Allgemeinen 1 bis 48 Stunden. Das Produkt
kann mit im Fachgebiet bekannten Verfahren isoliert und gereinigt
werden, wie Extraktion, Verdampfen, Chromatographie und Umkristallisation.
-
Wie
im Fachgebiet bekannt und anerkannt, können eine Anzahl von Schutz-,
Schutzgruppenabspaltungs- und Modifizierungsschritten, die in Schema
A.1, optionaler Schritt e, eingeschlossen sind, erforderlich sein
und in jeder Reihenfolge durchgeführt werden, die das passende
Einführen
von Resten ermöglicht,
wie sie im Endprodukt der Formel (1) erwünscht sind.
-
Die
folgenden Beispiele stellen typische Synthesen dar, wie sie in Schema
A.1 beschrieben sind. Diese Beispiele sind als lediglich veranschaulichend
zu verstehen und sollen den Umfang der Erfindung in keiner Weise
begrenzen.
-
BEISPIEL
1
(S)-2-[(S)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid]
-
Schema A.1, Schritt a: (S)-N-Benzyl-N-methyl-2-[[(S)-2-[(1H-indol-3-yl)-1-carboxymethyl]ethylamino]-N'-(t-butoxycarbonyl)ethylamino]-3-phenylpropionamid
-
Man
vereinigt (S)-N-Benzyl-N-methyl-2-[N'-(t-butoxycarbonyl)-2-oxoethylamino]-3-phenylpropionamid (4,87
g, 11,87 mmol) und (S)-2-(1H-Indol-3-yl)-1-carboxymethylethylamin-Hydrochloridsalz
((S)-Tryptophanmethylester-Hydrochloridsalz) (3,02 g, 11,86 mmol)
in Methanol (120 ml). Man fügt
Natriumcyanoborhydrid in Lösung
(9,5 ml, 1 M in THF, 9,5 mmol) zu und rührt 48 Stunden unter einer
Inertatmosphäre.
Man engt im Vakuum zu einem Rückstand
ein. Man verdünnt
den Rückstand
mit Ethylacetat und extrahiert mit Wasser. Man trennt die Phasen,
trocknet die organische Phase über
MgSO4, filtriert und verdampft im Vakuum.
Man chromatographiert an Silicagel, wobei mit 3% Methanol/Dichlormethan
eluiert wird, wodurch sich die Titelverbindung ergibt: DC Rf = 0,57 (Silicagel, 10% Methanol/Dichlormethan).
-
Schema A.1, Schritt b:
(S)-N-Benzyl-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]ethylamino]-3-phenylpropionamid
-
Man
vereinigt (S)-N-Benzyl-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]-N'-(t-butoxycarbonyl)ethylamino]-3-phenylpropionamid
(0,28 g, 0,58 mmol) und 4 M Dioxan (10 ml) und rührt 1 Stunde. Man verdampft
im Vakuum. Man reinigt durch Chromatographie an Silicagel, wobei
nacheinander mit Dichlormethan und dann 3% Methanol/Dichlormethan
eluiert wird, wodurch sich die Titelverbindung ergibt: DC Rf = 0,51 (Silicagel, 10% Methanol/Dichlormethan).
HRMS berechnet für
C29H36N3O3 474,2757. Gefunden 474,2755.
-
Schema A.1, Schritt c:
(S)-N-Benzyl-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxyethylamino]ethylamino]-3-phenylpropionamid
-
Man
vereinigt (S)-N-Benzyl-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]ethylamino]-3-phenylpropionamid
(0,48 g, 0,93 mmol) und 1 M Natriumhydroxid (10 ml, 10 mmol) in
Ethanol (20 ml) und rührt
18 Stunden unter einer Inertatmosphäre. Man verdünnt mit
Wasser und extrahiert mit Ethylacetat. Man säuert die wässrige Phase mit 1 N Salzsäure an und
extrahiert mit Ethylacetat. Man trocknet die organische Phase über MgSO4, filtriert und verdampft im Vakuum zur
Titelverbindung als Feststoff DC Rf = 0,43
(Silicagel, 85% Chloroform, 10% Methanol, 5% Essigsäure). HRMS
berechnet für
C30H35N4O3 499,2709. Gefunden 499,2696.
-
Schema A.1, Schritt d:
(S)-2-[(S)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid]
-
Man
vereinigt (S)-N-Benzyl-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxyethylamino]ethylamino]-3-phenylpropionamid
(0,10 g, 0,20 mmol), 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid-Hydrochlorid (0,043
g, 0,22 mmol), 1-Hydroxybenzotriazol-Hydrat
(0,033 g, 0,22 mmol), Diisopropylethylamin (0,053 ml, 0,22 mmol)
und Dimethylformamid (2 ml). Man rührt 18 Stunden unter einer
Inertatmosphäre.
Man verdünnt mit
Ethylacetat und extrahiert mit Wasser. Man trocknet die organische
Phase über
MgSO4, filtriert und verdampft im Vakuum.
Man chromatographiert, wobei nacheinander mit 50% Ethylacetat/Hexan,
Ethylacetat und 5% Methanol/Dichlormethan eluiert wird, wodurch
sich die Titelverbindung ergibt: DC Rf =
0,51 (Silicagel, 10% Methanol/Dichlormethan). HRMS berechnet für C30H33N4O2 481,2604. Gefunden 481,2582.
-
BEISPIEL
2
(S)-2-[(S)-3-(1-Methylindol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid]
-
Schema A.1, Schritt a: (S)-N-Benzyl-N-methyl-[[(S)-2-[(1-methylindol-3-yl)-1-carboxymethyl]ethylamino]-N'-(t-butoxycarbonyl)ethylamino]-3-phenylpropionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt a, wobei (S)-2-(1-Methylindol-3-yl)-1-carboxymethylethylamin-Hydrochloridsalz
(Lin-Hua Zhang und James M. Cook, Heterocycles 27, 2795–2802 (1988))
(1,0 mmol) und (S)-N-Benzyl-N-methyl-2-[N'-(t-butoxycarbonyl)-2-oxoethylamino]-3-phenylpropionamid
(1,0 mmol) verwendet werden, wodurch sich die Titelverbindung ergibt.
-
Schema A.1, Schritt b:
(S)-N-Benzyl-N-methyl-2-[[(S)-2-[(1-methylindol-3-yl)-1-carboxymethyl]ethylamino]ethylamino]-3-phenylpropionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt b, wobei (S)-N-Benzyl-N-methyl-2-[[(S)-2-[(1-methylindol-3-yl)-1-carboxymethyl]ethylamino]-N'-(t-butoxycarbonyl)ethylamino]-3-phenylpropionamid
verwendet wird, wodurch sich die Titelverbindung ergibt.
-
Schema A.1, Schritt c:
(S)-N-Benzyl-N-methyl-2-[[(S)-2-[(1-methylindol-3-yl)-1-carboxy]ethylamino]ethylamino]-3-phenylpropionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt c, wobei (S)-N-Benzyl-N-methyl-2-[[(S)-2-[(1-methylindol-3-yl)-1-carboxymethyl]ethylamino]ethylamino]-3-phenylpropionamid
verwendet wird, wodurch sich die Titelverbindung ergibt.
-
Schema A.1, Schritt d:
(S)-2-[3-(1-Methylindol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid]
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt d, wobei (S)-N-Benzyl-N-methyl-2-[[(S)-2-[(1-methylindol-3-yl)-1-carboxy]ethylamino]ethylamino]-3-phenylpropionamid
verwendet wird, wodurch sich die Titelverbindung ergibt.
-
BEISPIEL
3
(S)-2-[(R)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid]
-
Schema A.1, Schritt a: (S)-N-Benzyl-N-methyl-2-[[(R)-2-[(1H-indol-3-yl]-1-carboxymethyl]ethylamino]-N'-(t-butoxycarbonyl)ethylamino]-3-phenylpropionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt a, wobei (R)-2-(1H-Indol-3-yl)-1-carboxymethylethylamin-Hydrochloridsalz
((R)-Tryptophanmethylester-Hydrochloridsalz)
(0,25 g, 1,0 mmol) und (S)-N-Benzyl-N-methyl-2-[N'-(t-butoxycarbonyl)-2-oxoethylamino]-3-phenylpropionamid
(0,41 g, 1,0 mmol) verwendet werden, wodurch sich die Titelverbindung
ergibt: DC Rf = 0,54 (Silicagel, 10% Methanol/Dichlormethan).
-
Schema A.1, Schritt b:
(S)-N-Benzyl-N-methyl-2-[[(R)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]ethylamino]-3-phenylpropionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt b, wobei (S)-N-Benzyl-N-methyl-2-[[(R)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]-N'-(t-butoxycarbonyl)ethylamino]-3-phenylpropionamid
(0,31 g, 0,5 mmol) verwendet wird, wodurch sich die Titelverbindung
ergibt: DC Rf = 0,50 (Silicagel, 10% Methanol/Dichlormethan).
HRMS berechnet für
C31H37N4O3 513,2865. Gefunden 513,2839.
-
Schema A.1, Schritt c:
(S)-N-Benzyl-N-methyl-2-[[(R)-2-(1H-indol-3-yl)-1-carboxyethylamino]ethylamino]-3-phenylpropionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt c, wobei (S)-N-Benzyl-N-methyl-2-[[(R)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]ethylamino]-3-phenylpropionamid
verwendet wird, wodurch sich die Titelverbindung ergibt.
-
Schema A.1, Schritt d:
(S)-2-[(R)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid]
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt d, wobei (S)-N-Benzyl-N-methyl-2-[[(R)-2-(1H-indol-3-yl)-1-carboxyethylamino]ethylamino]-3-phenylpropionamid
verwendet wird, wodurch sich die Titelverbindung ergibt.
-
BEISPIEL
4
(S)-2-[(S)-3-Phenylmethyl-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid]
-
Schema A.1, Schritt a:
(S)-N-Benzyl-N-methyl-2-[[(S)-2-phenyl-1-carboxymethyl]ethylamino]-N'-(t-butoxycarbonyl)ethylamino-3-phenylpropionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, wobei (S)-2-Amino-3-phenylpropionsäuremethylester-Hydrochloridsalz
((S)-Phenylalaninmethylester-Hydrochloridsalz)
(0,26 g, 1,0 mmol) und (S)-N-Benzyl-N-methyl-2-[N'-(t-butoxycarbonyl)-2-oxoethylamino]-3-phenylpropionamid
(0,41 g, 1,0 mmol) verwendet werden, wodurch sich die Titelverbindung
ergibt: DC Rf = 0,51 (Silicagel, 10% Methanol/Dichlormethan).
-
Schema A.1, Schritt b:
(S)-N-Benzyl-N-methyl-2-[[(S)-2-phenyl-1-carboxymethyl]ethylamino]ethylamino-3-phenylpropionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt b, wobei (S)-N-Benzyl- N-methyl-2-[[(S)-2-phenyl-1-carboxymethyl]ethylamino]-N'-(t-butoxycarbonyl)ethylamino-3-phenylpropionamid
verwendet wird, wodurch sich die Titelverbindung ergibt: DC Rf = 0,51 (Silicagel, 10% Methanol/Dichlormethan).
-
Schema A.1, Schritt c:
(S)-N-Benzyl-N-methyl-2-[[(S)-2-phenyl-1-carboxy]ethylamino]ethylamino-3-phenylpropionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt c, wobei (S)-N-Benzyl-N-methyl-2-[[(S)-2-phenyl-1-carboxymethyl]ethylamino]ethylamino-3-phenylpropionamid
verwendet wird, wodurch sich die Titelverbindung ergibt.
-
Schema A.1, Schritt d:
(S)-2-[(S)-3-Phenylmethyl-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt d, wobei (S)-N-Benzyl-N-methyl-2-[[(S)-2-phenyl-1-carboxy]ethylamino]ethylamino-3-phenylpropionamid
verwendet wird, wodurch sich die Titelverbindung ergibt.
-
BEISPIEL
5
(S)-2-[(S)-3-Phenyl-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid]
-
Schema A.1, Schritt a:
(S)-N-Benzyl-N-methyl-2-[[(S)-1-phenyl-1-carboxymethylmethylamino]-N'-(t-butoxycarbonyl)ethylamino]-3-phenylpropionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt a, wobei (S)-1-Amino-1-phenylessigsäuremethylester-Hydrochloridsalz
((S)-Phenylglycinmethylester-Hydrochloridsalz)
(1,0 g, 4,96 mmol) und (S)-N-Benzyl-N-methyl-2-[N'-(t-butoxycarbonyl)-2-oxoethylamino]-3-phenylpropionamid
(2,04 g, 4,96 mmol) verwendet werden, wodurch sich die Titelverbindung
ergibt: DC Rf = 0,80 (Silicagel, 10% Methanol/Dichlormethan).
-
Schema A.1, Schritt b:
(S)-N-Benzyl-N-methyl-2-[[(S)-1-phenyl-1-carboxymethylmethylamino]ethylamino]-3-phenylpropionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt b, wobei (S)-N-Benzyl-N-methyl-2-[[(S)-1-phenyl-1-carboxymethylamino]ethylamino]-3-phenylpropionamid
verwendet wird, und Reinigung mit Chromatographie, wobei mit 5%
Methanol/Dichlormethan eluiert wird, wodurch sich die Titelverbindung
als farbloses Öl
ergibt: DC Rf = 0,76 (Silicagel, 10% Methanol/Dichlormethan).
Elementaranal. berechnet für C28H33N3O3·0,25H2O: C 72,47; H 7,23; N 9,05. Gefunden: C
72,47; H 7,53; N 9,10.
-
Schema A.1, Schritt c:
(S)-N-Benzyl-N-methyl-2-[[(S)-1-phenyl-1-carboxymethylamino]ethylamino]-3-phenylpropionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt c, wobei (S)-N-Benzyl-N-methyl-2-[[(S)-1-phenyl-1-carboxymethylmethylamino]ethylamino]-3-phenylpropionamid
verwendet wird, wodurch sich die Titelverbindung ergibt.
-
Schema A.1, Schritt d:
(S)-2-[(S)-3-Phenyl-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt d, wobei (S)-N-Benzyl-N-methyl-2-[[(S)-1-phenyl-1-carboxymethylamino]ethylamino]-3-phenylpropionamid
verwendet wird, wodurch sich die Titelverbindung ergibt.
-
BEISPIEL
6
(S)-2-[(S)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(naphth-2-yl)propionamid]
-
Schema A.1, Schritt a: (S)-N-Benzyl-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]-N'-(t-butoxycarbonyl)ethylamino]-3-(naphth-2-yl)propionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt a, wobei (S)-N-Benzyl-N-methyl-2-[N'-(t-butoxycarbonyl)-2-oxoethylamino]-3-(2-napthyl)propionamid
(0,46 g, 1,0 mmol) und (S)-2-(1H-Indol-3-yl)-1-carboxymethylethylamin-Hydrochloridsalz
((S)-Tryptophanmethylester-Hydrochloridsalz)
(0,26 g, 1,0 mmol) verwendet werden. Reinigung mit Chromatographie,
wobei mit 5% Methanol/Dichlormethan eluiert wird, wodurch sich die
Titelverbindung ergibt: DC Rf = 0,34 (Silicagel,
50% Ethylacetat/Hexan).
-
Schema A.1, Schritt b:
(S)-N-Benzyl-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]ethylamino]-3-(naphth-2-yl)propionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt b, wobei (S)-N-Benzyl-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]-N'-(t-butoxycarbonyl)ethylamino]-3-(naphth-2-yl)propionamid
verwendet wird, wodurch sich die Titelverbindung ergibt: DC Rf = 0,75 (Silicagel, 85% Chloroform, 10%
Methanol, 5% Essigsäure).
HRMS berechnet für
C35H39N4O3 563,3022. Gefunden 563,2996.
-
Schema A.1, Schritt c:
(S)-N-Benzyl-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxyethylamino]ethylamino]-3-(naphth-2-propionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt c, wobei (S)-N-Benzyl-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]ethylamino]-3-(naphth-2- yl)propionamid verwendet wird,
und Reinigung mit Chromatographie, wobei mit 5% Methanol/Dichlormethan
eluiert wird, wodurch sich die Titelverbindung ergibt.
-
Schema A.1, Schritt d:
(S)-2-[(S)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(naphth-2-yl)propionamid]
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt d, wobei (S)-N-Benzyl-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxyethylamino]ethylamino]-3-(naphth-2-yl)propionamid verwendet
wird, wodurch sich die Titelverbindung ergibt.
-
BEISPIEL
7
(S)-2-[(S)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl)-1-[N-methyl-N-(2-methoxybenzyl)-3-(naphth-2-yl)propionamid]
-
Schema A.1, Schritt a: (S)-N-(2-Methoxybenzyl)-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]-N'-(t-butoxycarbonyl)ethylamino]-3-(naphth-2-yl)propionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt a, wobei (S)-N-(2-Methoxybenzyl)-N-methyl-2-[N'-(t-butoxycarbonyl)-2-oxoethylamino]-3-(naphth-2-yl)propionamid (0,47
g, 0,96 mmol) und (S)-2-(1H-Indol-3-yl)-1-carboxymethylethylamin-Hydrochloridsalz
((S)-Tryptophanmethylester-Hydrochloridsalz) (0,28 g, 1,1 mmol)
verwendet werden. Reinigung mit Chromatographie, wodurch sich die
Titelverbindung ergibt.
-
Schema A.1, Schritt b:
(S)-N-(2-Methoxybenzyl)-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]ethylamino]-3-(naphth-2-yl)propionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt b, wobei (S)-N-(2- Methoxybenzyl)-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]-N'-(t-butoxycarbonyl)ethylamino]-3-(naphth-2-yl)propionamid
verwendet wird, wodurch sich die Titelverbindung ergibt. HRMS berechnet
für C32H39N4O4 543,2971. Gefunden 543,2980.
-
Schema A.1, Schritt c:
(S)-N-(2-Methoxybenzyl)-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxyethylamino]ethylamino]-3-(naphth-2-yl)propionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt c, wobei (S)-N-(2-Methoxybenzyl)-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]ethylamino]-3-(naphth-2-yl)propionamid verwendet
wird, wodurch sich die Titelverbindung ergibt.
-
Schema A.1, Schritt d:
(S)-2-[(S)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-(2-methoxybenzyl)-3-(naphth-2-yl)propionamid]
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt d, wobei (S)-N-(2-Methoxybenzyl)-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxyethylamino]ethylamino]-3-(naphth-2-yl)propionamid
verwendet wird, wodurch sich die Titelverbindung ergibt.
-
BEISPIEL
8
(S)-2-[(S)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-(3,4-dichlorbenzyl)-3-(naphth-2-yl)propionamid]
-
Schema A.1, Schritt a: (S)-N-(3,4-Dichlorbenzyl)-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]-N'-(t-butoxycarbonyl)ethylamino]-3-(naphth-2-yl)propionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt a, wobei (S)-N-(3,4-Dichlorbenzyl)-N-methyl-2-[N'-(t-butoxycarbonyl)-2-oxoethylamino]-3-(naphth-2- yl)propionamid (0,29
g, 0,55 mmol) und (S)-2-(1H-Indol-3-yl)-1-carboxymethylethylamin-Hydrochloridsalz
((S)-Tryptophanmethylester-Hydrochloridsalz) (0,14 g, 0,55 mmol)
verwendet werden. Reinigung mit Chromatographie, wobei mit 5% Methanol/Dichlormethan
eluiert wird, wodurch sich die Titelverbindung ergibt: DC Rf = 0,58 (Silicagel, 50% Ethylacetat/Hexan).
-
Schema A.1, Schritt b:
(S)-N-(3,4-Dichlorbenzyl)-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]ethylamino]-3-(naphth-2-yl)propionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt b, wobei (S)-N-(3,4-Dichlorbenzyl)-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]-N'-(t-butoxycarbonyl)ethylamino]-3-(naphth-2-yl)propionamid
verwendet wird, wodurch sich die Titelverbindung ergibt: DC Rf = 0,57 (Silicagel, 10% Methanol/Dichlormethan).
-
Schema A.1, Schritt c:
(S)-N-(3,4-Dichlorbenzyl-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxyethylamino]ethylamino]-3-(naphth-2-yl)propionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt c, wobei (S)-N-(3,4-Dichlorbenzyl)-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]ethylamino]-3-(naphth-2-yl)propionamid verwendet
wird, wodurch sich die Titelverbindung ergibt.
-
Schema A.1, Schritt d:
(S)-2-[(S)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-(3,4-dichlorbenzyl)-3-(naphth-2-yl)propionamid]
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt d, wobei (S)-N-(3,4-Dichlorbenzyl)-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxyethylamino]ethylamino]-3-(naphth-2-yl)propionamid
verwendet wird, wodurch sich die Titelverbindung ergibt.
-
BEISPIEL
9
(S)-2-[(S)-3-(1H-Indol-3-yl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-(2-methoxybenzyl)-3-(phenyl)propionamid]
-
Schema A, Schritt a: (S)-N-(2-Methoxybenzyl)-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]-N'-(t-butoxycarbonyl)ethylamino]-3-phenylpropionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt a, wobei (S)-N-(2-Methoxybenzyl)-N-methyl-2-[N'-(t-butoxycarbonyl)-2-oxoethylamino]-3-phenylpropionamid
(0,71 g, 1,6 mmol) und (S)-2-(1H-Imidol-3-yl)-1-carboxymethylethylamin-Hydrochloridsalz
((S)-Tryptophanmethylester-Hydrochloridsalz) (0,45 g, 1,76 mmol)
verwendet werden. Reinigung mit Chromatographie, wobei mit 50% Ethylacetat/Hexan
eluiert wird, wodurch sich die Titelverbindung ergibt: DC Rf = 0,56 (Silicagel, 50% Ethylacetat/Hexan).
-
Schema A.1, Schritt b:
(S)-N-(2-Methoxybenzyl)-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]ethylamino]-3-phenylpropionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt b, wobei (S)-N-(2-Methoxybenzyl)-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]-N'-(t-butoxycarbonyl)ethylamino]-3-phenylpropionamid
verwendet wird, wodurch sich die Titelverbindung ergibt.
-
Schema A.1, Schritt c:
(S)-N-(2-Methoxybenzyl)-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxyethylamino]ethylamino]-3-phenylpropionamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt c, wobei (S)-N-(2-Methoxybenzyl)-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]ethylamino]-3-phenylpropionamid
verwendet wird, wodurch sich die Titelverbindung ergibt.
-
Schema A.1, Schritt d:
(S)-2-[(S)-3-(1H-Indol-3)-2-oxopiperazin-1-yl]-1-[N-methyl-N-(2-methoxybenzyl)-3-(phenyl)propionamid]
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt d, wobei (S)-N-(2-Methoxybenzyl)-N-methyl-2-[[(S)-2-(1H-indol-3-yl)-1-carboxyethylamino]ethylamino]-3-phenylpropionamid
verwendet wird, wodurch sich die Titelverbindung ergibt.
-
BEISPIEL
10
(S)-2-[3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-2-[[(S)-N-methyl-N-(3-phenylpropyl)]benzamid]
Schema
A.1, Schritt a: N-Methyl-N-[(S)-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]-N'-(t-butoxycarbonyl)ethylamino]-3-phenylpropyl]benzamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt a, wobei (S)-N-Methyl-N-[2-[N'-(t-butoxycarbonyl)-2-oxoethylamino]-3-phenylpropyl]benzamid
(0,11 g, 0,27 mmol) und (S)-2-(1H-Indol-3-yl)-1-carboxymethylethylamin-Hydrochloridsalz
((S)-Tryptophanmethylester-Hydrochloridsalz)
(0,076 g, 0,3 mmol) verwendet werden. Reinigung mit Chromatographie,
wobei mit 30% Ethylacetat/Hexan eluiert wird, wodurch sich die Titelverbindung
ergibt.
-
Schema A.1, Schritt b:
N-Methyl-N-[(S)-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]ethylamino]-3-phenylpropyl]benzamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt b, wobei N-Methyl-N-[(S)-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]-N'-(t-butoxycarbonyl)ethylamino]-3-phenylpropyl]benzamid
verwendet wird, wodurch sich die Titelverbindung ergibt. HRMS berechnet
für C31H37N4O3 513,2865. Gefunden 513,2872.
-
Schema A.1, Schritt c:
N-Methyl-N-[(S)-2-[[(S)-2-(1H-Indol-3-yl)-1-carboxyethylamino]ethylamino]-3-phenylpropyl]benzamid
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt c, wobei N-Methyl-N-[(S)-2-[[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]ethylamino]-3-phenylpropyl]benzamid
verwendet wird, wodurch sich die Titelverbindung ergibt.
-
Schema A.1, Schritt d:
(S)-2-[3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-2-[[(S)-N-methyl-N-(3-phenylpropyl)]benzamid]
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt d, wobei N-Methyl-N-[(S)-2-[[(S)-2-(1H-indol-3-yl)-1-carboxyethylamino]ethylamino]-3-phenylpropyl]benzamid
verwendet wird, wodurch sich die Titelverbindung ergibt.
-
Beispiel
12
(S)-2-[3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-[(S)-N-methyl-N-benzyl-3-(phenyl)propylamin]
-
Schema A.1, Schritt a:
N-Methyl-N-benzyl-N-[(S)-2-[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino-N'-(t-butoxycarbonyl)ethylamino]-3-phenylpropyl
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt a, wobei (S)-N-Methyl-N-benzyl-N-[2-[N'-(t-butoxycarbonyl)-2-oxoethylamino]-3-phenylpropylamin
(0,5 mmol) und (S)-2-(1H-Indol-3-yl)-1-carboxymethylethylamin-Hydrochloridsalz
((S)-Tryptophanmethylester-Hydrochloridsalz)
(0,55 mmol) verwendet werden. Reinigung mit Chromatographie, wodurch
sich die Titelverbindung ergibt.
-
Schema A.1, Schritt b:
N-Methyl-N-benzyl-N-[(S)-2-[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]ethylamino]-3-phenylpropylamin
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt b, wobei N-Methyl-N-benzyl-N-[(S)-2-[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]-N'-(t-butoxycarbonyl)ethylamino]-3-phenylpropylamin
verwendet wird, wodurch sich die Titelverbindung ergibt.
-
Schema A.1, Schritt c:
N-Methyl-N-benzyl-N-[(S)-2-[(S)-2-(1H-indol-3-yl)-1-carboxyethylamino]ethylamino]-3-phenylpropylamin
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt c, wobei N-Methyl-N-benzyl-N-[(S)-2-[(S)-2-(1H-indol-3-yl)-1-carboxymethylethylamino]ethylamino]-3-phenylpropylamin
verwendet wird, wodurch sich die Titelverbindung ergibt.
-
Schema A.1, Schritt d:
(S)-2-[3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-[(S)-N-methyl-N-benzyl-3-(phenyl)propylamin]
-
Herstellung
mit dem Verfahren aus Beispiel 1, Schema A.1, Schritt d, wobei N-Methyl-N-benzyl-N-[(S)-2-[(S)-2-(1H-indol-3-yl)-1-carboxyethylamino]ethylamino]-3-phenylpropylamin
verwendet wird, wodurch sich die Titelverbindung ergibt.
-
In
Schema A.2 wird ein allgemeines Syntheseverfahren zur Herstellung
dieser Verbindungen der Formel (1), in der G3 -CH2- ist, angegeben. Die Reagenzien und Ausgangsmaterialien
sind für
den Durchschnittsfachmann leicht erhältlich. In Schema A.2 sind
alle Substituenten, sofern nicht anders angegeben, wie zuvor definiert.
-
-
In
Schema A.2, Schritt a, wird eine geeignete Verbindung der Formel
(1), in der G3 -C(O)- und R1 Wasserstoff
ist, zu einer Verbindung der Formel (1) reduziert, in der G3 -CH2- und R1 Wasserstoff ist.
-
Für Schema
A.2 ist eine geeignete Verbindung der Formel (1) eine, in der G1 und G2 entweder
-C(O)- oder -CH2- sind, G3 -C(O)-
ist und R1 Wasserstoff ist; und die Stereochemie,
m, Ar1, Ar2 und
Ar3 so sind, wie im Endprodukt der Formel
(1) erwünscht,
oder kann auch eine sein, bei der die Stereochemie und Ar1, Ar2 und Ar3 nach Antipodentrennung, Schutzgruppenabspaltung
oder Modifikation die Stereochemie und Ar1,
Ar2 und Ar3 ergeben,
wie im Endprodukt der Formel (1) erwünscht.
-
Beispielsweise
wird eine geeignete Verbindung der Formel (1) mit 1 bis 10 Äquivalenten
eines geeigneten Reduktionsmittels für Amide in Kontakt gebracht,
wie Lithiumaluminumhydrid, Diisobutylaluminumhydrid oder Boran-Dimethylsulfid-Komplex.
Die verwendete Menge an geeignetem Reduktionsmittel für Amide
hängt von
der Anzahl der Amide ab, die in Schema A.2, Schritt a, reduziert
werden, wenn beispielsweise entweder G1 oder
G2 -C(O)- sind, wird die verwendete Menge
an Reduktionsmittel für
Amide erhöht,
wie im Fachgebiet bekannt und anerkannt. Die Reaktion wird in einem
geeigneten Lösungsmittel,
wie Tetrahydrofuran, Toluol oder Diethylether, durchgeführt. Im
Allgemeinen wird die Reaktion bei Temperaturen von 0°C bis zur
Rückflusstemperatur
des Lösungsmittels
durchgeführt.
Die Reaktionen erfordern im Allgemeinen 1 bis 72 Stunden. Das Produkt
kann mit im Fachgebiet bekannten Verfahren, wie Quenchen, Extraktion,
Verdampfen, Chromatographie und Umkristallisation, isoliert und
gereinigt werden.
-
In
Schema A.2, optionaler Schritt b, kann von einer geschützten Verbindung
der Formel (1) die Schutzgruppe entfernt werden oder die Verbindung
so modifiziert werden, dass sich eine Verbindung der Formel (1) ergibt,
wie allgemein vorstehend in Schema A.1, optionaler Schritt f, gelehrt.
-
Die
folgenden Beispiele stellen typische Synthesen dar, wie sie in Schema
A.2 beschrieben sind. Diese Beispiele sind als lediglich veranschaulichend
zu verstehen und sollen den Umfang der Erfindung in keiner Weise
begrenzen.
-
BEISPIEL
13
(S)-2-[(S)-3-(1H-Indol-3-ylmethyl)piperazin-1-yl]-[N-methyl-N-benzyl-3-(phenyl)propylamin]
-
Schema A.2, Schritt a:
(S)-2-[(S)-3-(1H-Indol-3-ylmethyl)piperazin-1-yl]-[N-methyl-N-benzyl-3-(phenyl)propylamin]
-
Man
vereinigt (S)-2-[(S)-3-(1H-Indol-3-ylmethyl)-2-oxopiperazin-1-yl]-1-[N-methyl-N-benzyl-3-(phenyl)propionamid)
(5 mmol) und Tetrahydrofuran (25 ml). Man gibt langsam portionsweise
Lithiumaluminiumhydrid (16 mmol) hinzu. Man erhitzt 48 Stunden unter
Rückfluss.
Man kühlt
auf Zimmertemperatur ab, gibt langsam Wasser (0,6 ml), 15%ige Natriumhydroxidlösung (0,6
ml) und Wasser (1,8 ml) hinzu. Man rührt, bis alles Reduktionsmittel
gequencht ist. Man filtriert und verdampft im Vakuum zu einem Rückstand.
Man verteilt den Rückstand
zwischen Ethylacetat und Wasser. Man trennt die organische Phase
ab und trocknet über
MgSO4, filtriert und verdampft im Vakuum,
wodurch sich die Titelverbindung ergibt.
-
In
Schema B wird ein allgemeines Syntheseverfahren zur Herstellung
des Aldehyds der Struktur (3) angegeben, in der G1 -CH2- und G2 -C(O)-
ist, der als Ausgangsmaterial in Schema A.1 eingesetzt wird. Die Reagenzien
und Ausgangsmaterialien sind für
den Durchschnittsfachmann leicht erhältlich. In Schema B sind alle
Substituenten, sofern nicht anders angegeben, wie zuvor definiert.
-
-
-
In
Schema B, optionaler Schritt a, wird ein geeigneter Aminoester der
Struktur (11) oder ein Salz davon zu einem Allylaminoester der Struktur
(12) allyliert.
-
Ein
geeigneter Aminoester der Struktur (11) oder ein Salz davon ist
einer, bei dem die Stereochemie und Ar2 so
sind, wie im Produkt der Formel (1) gewünscht, oder kann einer sein,
der nach Antipodentrennung oder Schutzgruppenabspaltung die Stereochemie
oder Ar2 ergibt, wie im Endprodukt der Formel
(1) erwünscht.
-
Beispielsweise
wird ein geeigneter Aminoester der Struktur (11) oder ein Salz eines
geeigneten Aminoesters der Struktur (11) mit 1 bis 3 molaren Äquivalenten
Allylbromid oder Allylchlorid in Kontakt gebracht. Das Allylbromid
oder Allylchlorid wird vorzugsweise portionsweise im Verlauf der
Reaktion zugegeben. Wenn ein Salz eines geeigneten Aminoesters der
Struktur (11) verwendet wird, wird die Reaktion in Gegenwart einer äquimolaren
Menge einer geeigneten Base, wie Triethylamin oder Diisopropylethylamin,
durchgeführt.
Die Reaktion wird in einem geeigneten Lösungsmittel, wie Tetrahydrofuran,
durchgeführt.
Die Reaktion wird im Allgemeinen bei Temperaturen von 0°C bis 60°C durchgeführt. Die
Reaktionen erfordern im Allgemeinen 1 bis 72 Stunden. Das Produkt
kann mit im Fachgebiet bekannten Verfahren, wie Extraktion, Verdampfen,
Chromatographie und Umkristallisation, isoliert und gereinigt werden.
-
In
Schema B, optionaler Schritt b, wird die Allylaminogruppe eines
Allylaminoesters der Struktur (12) oder eines Salzes davon mit einem
geeigneten Reagens zur Bildung eines Carbamats in ein Carbamat überführt, wodurch
sich eine Verbindung der Struktur (14) ergibt.
-
Ein
geeignetes Reagens zur Bildung eines Carbamats ist eines, das den
Rest -CO2R5 auf
ein Amin überträgt, wie
Methylchlorformiat, Ethylchlorformiat, Propylchlorformiat, iso-Butylchlorformiat
und Di-t-butyldicarbonat usw.
-
Beispielsweise
wird ein Allylaminoester der Struktur (12) oder ein Salz davon mit
einem Reagens in Kontakt gebracht, das den Rest -CO2R5 auf ein Amin überträgt. Wenn ein Salz eines Allylaminoesters
der Struktur (12) verwendte wird, wird die Reaktion in Gegenwart
einer äquimolaren
Menge einer geeigneten Base, wie Triethylamin oder Diisopropylethylamin,
durchgeführt.
Wenn die Reaktion unter Verwendung eines Reagens zur Bildung eines
Carbamats, das Säure
freisetzt, wenn das Carbamat gebildet wird, wie Methylchlorformiat,
Ethylchlorformiat, Propylchlorformiat, iso-Butylchlorformiat etc.,
durchgeführt
wird, wird eine äquimolare Menge
einer geeigneten Base, wie Triethylamin oder Diisopropylethylamin,
verwendet, um die Säure
zu neutralisieren, die freigesetzt wird.
-
Die
Reaktion wird in einem geeigneten Lösungsmittel, wie Tetrahydrofuran,
Dimethylformamid, Ethylacetat oder Dimethylformamid/Ethylacetat-Gemische,
durchgeführt.
Im Allgemeinen werden die Reaktionen bei Zimmertemperatur durchgeführt. Das
Produkt kann mit im Fachgebiet bekannten Verfahren, wie Extraktion, Verdampfen,
Chromatographie und Umkristallisation, isoliert und gereinigt werden.
-
Alternativ
kann ein Carbamat der Struktur (14), in der R5 t-Butyl
ist, nach Schema B, optionale Schritte c und d, hergestellt werden.
-
In
Schema B, optionaler Schritt c, wird ein geeigneter Aminoester der
Struktur (11) oder ein Salz davon mit einem geeigneten Reagens zur
Bildung eines Carbamats in Kontakt gebracht, wodurch sich eine Verbindung
der Struktur (13) ergibt.
-
Ein
geeignetes Reagens zur Bildung eines Carbamats zur Verwendung auf
diesem alternativen Weg zur Herstellung von Verbindungen der Struktur
(14) ist eines, das ein t-Butylcarbamat überträgt, wie
Di-t-butyldicarbonat.
-
Ein
geeigneter Aminoester der Struktur (11) oder ein Salz davon ist
einer, bei dem die Stereochemie und Ar2 so
sind, wie im Produkt der Formel (1) gewünscht, oder kann einer sein,
der nach Antipodentrennung oder Schutzgruppenabspaltung die Stereochemie
oder Ar2 ergibt, wie im Endprodukt der Formel
(1) erwünscht.
-
Beispielsweise
wird ein Aminoester der Struktur (11) oder ein Salz davon mit einem
Reagens in Kontakt gebracht, das eine t-Butoxycarbonylgruppe überträgt, wie
Di-t-butyldicarbonat.
Wenn ein Salz eines Aminoesters der Struktur (11) verwendet wird,
wird die Reaktion in Gegenwart einer äquimolaren Menge einer geeigneten
Base, wie Triethylamin oder Diisopropylethylamin, durchgeführt. Die
Reaktion wird in einem geeigneten Lösungsmittel, wie Dimethylformamid,
Ethylacetat oder Dimethylformamid/Ethylacetat-Gemische, durchgeführt. Im
Allgemeinen werden die Reaktionen bei Zimmertemperatur durchgeführt. Das
Produkt kann mit im Fachgebiet bekannten Verfahren, wie Extraktion,
Verdampfen, Chromatographie und Umkristallisation, isoliert und
gereinigt werden.
-
In
Schema B, optionaler Schritt d, wird ein Carbamat-Ester der Struktur
(13), in der R5 t-Butyl ist, zu einem Allyl-Carbamat-Ester
der Struktur (14) allyliert, in der R5 t-Butyl
ist.
-
Beispielsweise
wird ein Carbamat-Ester der Struktur (13) mit Allylbromid oder Allylchlorid
in Kontakt gebracht. Die Reaktion wird in Gegenwart einer geeigneten
Base, wie Natriumhydrid, durchgeführt. Die Reaktion wird in einem
geeigneten Lösungsmittel,
wie Tetrahydrofuran, Dimethylformamid oder Tetrahydrofuran/Dimethylformamid-Gemische,
durchgeführt.
Die Reaktion wird bei Temperaturen von 0°C bis zur Rückflusstemperatur des Lösungsmittels
durchgeführt.
Das Produkt kann mit im Fachgebiet bekannten Verfahren, wie Extraktion,
Verdampfen, Chromatographie und Umkristallisation, isoliert und
gereinigt werden.
-
In
Schema B, Schritt e, wird ein Allyl-Carbamat-Ester der Struktur
(14) zu einer Allyl-Carbamat-Säure der
Struktur (15) hydrolysiert.
-
Beispielsweise
wird ein Allyl-Carbamat-Ester der Struktur (14) mit einer geeigneten
Base, wie Natriumhydroxid, Lithiumhydroxid oder Kaliumhydroxid,
in Kontakt gebracht. Die Reaktion wird in einem geeigneten Lösungsmittel,
wie Methanol, Ethanol, Wasser, Methanol/Wasser-Gemische, Ethanol/Wasser-Gemische
oder Tetrahydrofuran/Wasser-Gemische,
durchgeführt.
Im Allgemeinen wird die Reaktion bei Zimmertemperatur durchgeführt. Die
Reaktion erfordert im Allgemeinen 2 bis 72 Stunden. Das Produkt
kann mit im Fachgebiet bekannten Verfahren, wie Ansäuern, Filtration,
Extraktion, Verdampfen und Umkristallisation, isoliert und gereinigt
werden.
-
In
Schema B, Schritt f, durchläuft
eine Allyl-Carbamat-Säure
der Struktur (15) mit einem geeigneten Amin eine Amidierungsreaktion
zu einem Allyl-Carbamat-Amid der Struktur (16).
-
Ein
geeignetes Amin der Struktur HN(CH3)CH2Ar1 ist eines, bei
dem der Rest Ar1 so ist, wie im Produkt der
Formel (1) erwünscht,
oder nach Schutzgruppenabspaltung Ar1 ergibt,
wie im Endprodukt der Formel (1) erwünscht.
-
Eine
Amidierungsreaktion kann über
eine aktivierte Zwischenstufe verlaufen, wie ein gemischtes Anhydrid
oder ein (O)-Hydroxybenzotriazol, das hergestellt werden kann, aber
nicht notwendigerweise vor der Zugabe eines geeigneten Amins HN(CH3)CH2Ar1 isoliert
wird.
-
Beispielsweise
wird eine Allyl-Carbamat-Säure
der Struktur (15) mit 1,2 bis 1,7 Äquivalenten einer geeigneten
Base, wie N-Methylmorpholin, in einem geeigneten Lösungsmittel,
wie Tetrahydrofuran, in Kontakt gebracht. Im Allgemeinen wird das
Reaktionsgemisch vor der Zugabe von 1,2 bis 1,7 Äquivalenten Isobutylchlorformiat
auf eine Temperatur zwischen –50°C und 0°C abgekühlt, wobei –25°C bis –20°C bevorzugt
werden. Die Reaktion wird etwa 30 Minuten bis 3 Stunden rühren gelassen,
so dass sich das gemischte Anhydrid, eine aktivierte Zwischenstufe,
bilden kann. Während
die Temperatur zwischen –50°C und 0°C gehalten
wird, wird ein geeignetes Amin der Struktur HN(CH3)CH2Ar1 zugegeben. Die
Reaktion kann, nachdem die Zugabe des Amins beendet ist, auf Zimmertemperatur
erwärmt
werden. Die Reaktion erfordert 2 bis 48 Stunden. Das Produkt kann
mit im Fachgebiet bekannten Verfahren, wie Extraktion, Verdampfen,
Chromatographie und Umkristallisation, isoliert und gereinigt werden.
-
In
einer anderen Ausführungsform
beispielsweise wird eine Allyl-Carbamat-Säure der Struktur (15) mit einem
geringen molaren Überschuss
eines geeigneten Amins HN(CH3)CH2Ar1 und 1-Hydroxybenzotriazol-Hydrat
in Gegenwart eines geringen molaren Überschusses an Kupplungsmittel,
wie Dicyclohexylcarbodiimid oder 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid,
in Kontakt gebracht. Die Reaktion wird in Gegenwart einer geeigneten
Base, wie Diisopropylethylamin, durchgeführt. Die Reaktion wird in einem
geeigneten Lösungsmittel,
wie Dimethylformamid, Dichlormethan oder Chloroform, durchgeführt. Das
Produkt kann mit im Fachgebiet bekannten Verfahren, wie Extraktion,
Verdampfen, Chromatographie und Umkristallisation, isoliert und
gereinigt werden.
-
Alternativ
kann ein Allyl-Carbamat-Amid der Struktur (16) aus einer Aminosäure gemäß Schema
B, optionale Schritte j, k, und l, hergestellt werden.
-
In
Schema B, optionaler Schritt j, wird eine geeignete Aminosäure der
Struktur (11) oder ein Salz davon mit einem geeigneten Reagens zur
Bildung eines Carbamats in Kontakt gebracht, wodurch sich eine Verbindung
der Struktur (3) ergibt.
-
Ein
geeignetes Reagens zur Bildung eines Carbamats zur Verwendung auf
diesem alternativen Weg zur Herstellung von Verbindungen der Struktur
(13) ist eines, das ein t-Butylcarbamat überträgt, wie
Di-t-butyldicarbonat.
-
Eine
geeignete Aminosäure
der Struktur (11) oder ein Salz davon ist eine, bei der die Stereochemie und
Ar2 so sind, wie im Produkt der Formel (1)
gewünscht,
oder ist eine, die nach Antipodentrennung oder Schutzgruppenabspaltung
die Stereochemie oder Ar2 ergibt, wie im
Endprodukt der Formel (1) erwünscht.
-
Beispielsweise
wird eine geeignete Aminosäure
der Struktur (11) oder ein Salz davon mit einem Reagens in Kontakt
gebracht, das eine t-Butoxycarbonylgruppe überträgt, wie Di-t-butyldicarbonat.
Die Reaktion wird in Gegenwart einer äquimolaren Menge einer geeigneten
Base, wie Triethylamin oder Diisopropylethylamin, durchgeführt. Wenn
ein Salz einer geeigneten Aminosäure
der Struktur (11) verwendet wird, wird eine zusätzliche äquimolare Menge einer geeigneten
Base verwendet. Die Reaktion wird in einem geeigneten Lösungsmittel,
wie Dimethylformamid, Ethylacetat oder Dimethylformamid/Ethylacetat-Gemische, durchgeführt. Im
Allgemeinen werden die Reaktionen bei Zimmertemperatur durchgeführt. Das
Produkt kann mit im Fachgebiet bekannten Verfahren, wie Extraktion,
Verdampfen, Chromatographie und Umkristallisation, isoliert und gereinigt
werden.
-
In
Schema B, Schritt k, durchläuft
eine t-BOC-geschützte
Aminosäure
der Struktur (13) eine Amidierungsreaktion mit einem geeigneten
Amin, wie allgemein in Schema B, Schritt f, gelehrt, wodurch sich
ein t-BOC-geschütztes
Amino-Amid der Struktur (17) ergibt.
-
Ein
geeignetes Amin der Struktur HN(CH3)CH2Ar1 ist eines, bei
dem Ar1 so ist, wie im Produkt der Formel
(1) erwünscht,
oder nach Schutzgruppenabspaltung Ar1 ergibt,
wie im Endprodukt der Formel (1) erwünscht.
-
In
Schema B, optionaler Schritt 1, wird ein t-BOC-geschütztes Amino-Amid
der Struktur (17) allyliert, wie allgemein in Schema B, optionaler
Schritt d, gelehrt, wodurch sich ein Allyl-Carbamat-Amid der Struktur (16) ergibt,
in der R5 t-Butyl ist.
-
In
Schema B, Schritt g, wird ein Allyl-Carbamat-Amid der Struktur (16)
in einen Aldehyd der Struktur (3) überführt. Ein Allyl-Carbamat-Amid
der Struktur (16) kann durch entweder Ozonolyse in Gegenwart von Methanol,
gefolgt von reduktiver Aufarbeitung, oder eine über Osmiumtetroxid vermittelte
Bildung eines intermediären
Diols, gefolgt von oxidativer Spaltung mit Bleitetraacetat oder
Natriummetaperiodat, in einen Aldehyd der Struktur (3) überführt werden.
-
Die
folgenden Beispiele stellen typische Synthesen dar, wie sie in Schema
B beschrieben sind. Diese Beispiele und Herstellungen sind als lediglich
veranschaulichend zu verstehen und sollen den Umfang der Erfindung
in keiner Weise begrenzen.
-
BEISPIEL 44
-
2-(Methoxy)benzylmethylamin
-
Ausgangsmaterial für Beispiele
50 und 55:
-
Man
vereinigt o-Amisoylchlorid (2-Methoxybenzoylchlorid) (2,9 g, 17,0
mmol) und Tetrahydrofuran (170 ml) und kühlt auf 0°C ab. Man gibt Diisopropylethylamin
(5,92 ml, 34 mmol) hinzu. Man gibt Methylamin-Hydrochlorid (1,26
g, 18,7 mmol) hinzu. Man lässt
1 Stunde rühren
und engt im Vakuum ein. Man chromatographiert an Silicagel, wobei
nacheinander mit 50% Ethylacetat/Hexan eluiert wird, wodurch sich
N-Methyl-2- methoxybenzamid
ergibt: DC Rf = 0,45 (Silicagel, 50% Ethylacetat/Hexan).
-
Man
vereinigt N-Methyl-2-methoxybenzamid (1,55 g, 9,36 mmol) und Tetrahydrofuran
(100 ml) und erhitzt zum Rückfluss.
Man tropft langsam eine Lösung
des Boran-Dimethylsulfid-Komplexes
(28,1 ml, 2,0 M in Tetrahydrofuran, 56,2 mmol) zu. Man erhitzt 1
Stunde zum Rückfluss,
nachdem die Zugabe beendet ist. Man kühlt auf Zimmertemperatur ab
und engt im Vakuum ein, wodurch ein Rückstand erhalten wird. Man
kühlt den Rückstand
auf 0°C.
Man gibt langsam 6 M Salzsäurelösung zu.
Nach dem Ende der Zugabe wird das Gemisch 1 Stunde zum Rückfluss
erhitzt. Man kühlt
auf 0°C,
gibt 6 M Natriumhydroxidlösung
zu, bis der pH-Wert 7 beträgt.
Man extrahiert das Reaktionsgemisch mit Ethylacetat. Man trocknet
die organische Phase über
MgSO4, filtriert und verdampft im Vakuum,
wodurch sich die Titelverbindung ergibt.
-
BEISPIEL 45
-
3,4,5-(Trimethoxy)benzylmethylamin
-
Ausgangsmaterial für Beispiel
51:
-
Man
vereinigt 3,4,5-Trimethoxybenzoylchlorid (2,9 g, 17,0 mmol) und
Tetrahydrofuran (170 ml) und kühlt
auf 0°C
ab. Man gibt Diisopropylethylamin (5,92 ml, 34 mmol) hinzu. Man
gibt Methylamin-Hydrochlorid (1,26 g, 18,7 mmol) hinzu. Man lässt 1 Stunde
rühren
und engt im Vakuum ein. Man chromatographiert an Silicagel, wobei
nacheinander mit 50% Ethylacetat/Hexan eluiert wird, wodurch sich
N-Methyl-3,4,5-trimethoxybenzamid ergibt: DC Rf =
0,45 (Silicagel, 50% Ethylacetat/Hexan).
-
Man
vereinigt N-Methyl-3,4,5-trimethoxybenzamid (1,55 g, 9,36 mmol)
und Tetrahydrofuran (100 ml) und erhitzt zum Rückfluss. Man tropft langsam
eine Lösung
des Boran-Dimethylsulfid-Komplexes (28,1 ml, 2,0 M in Tetrahydrofuran,
56,2 mmol) zu. Man erhitzt 1 Stunde zum Rückfluss, nachdem die Zugabe
beendet ist. Man kühlt
auf Zimmertemperatur ab und engt im Vakuum ein, wodurch ein Rückstand
erhalten wird. Man kühlt den
Rückstand
auf 0°C.
Man gibt langsam 6 M Salzsäurelösung zu.
Nach dem Ende der Zugabe wird das Gemisch 1 Stunde zum Rückfluss
erhitzt. Man kühlt
auf 0°C,
gibt 6 M Natriumhydroxidlösung
zu, bis der pH-Wert 7 beträgt.
Man extrahiert das Reaktionsgemisch mit Ethylacetat. Man trocknet
die organische Phase über MgSO4, filtriert und verdampft im Vakuum, wodurch
sich die Titelverbindung ergibt.
-
BEISPIEL 46
-
(S)-2-Allylamino-3-phenylpropionsäuremethyl
-
Schema B, optionaler Schritt
a:
-
Man
vereinigt (S)-2-Amino-3-phenylpropionsäuremethylester-Hydrochloridsalz
((S)- Phenylalaninmethylester-Hydrochloridsalz)
(8,63 g, 40,0 mmol), Diisopropylethylamin (6,8 ml, 40,0 mmol) und
Allylbromid (1,8 ml, 20,0 mmol) in THF (200 ml). Man rührt 16 Stunden
unter einer Inertatmosphäre.
Man gibt Allylbromid (1,8 ml, 20,0 mmol) hinzu und rührt weitere
24 Stunden. Man engt im Vakuum zu einem Rückstand ein. Man verdünnt den
Rückstand
mit Ethylacetat und extrahiert mit Wasser. Man trennt die Phasen,
trocknet die organische Phase über
MgSO4, filtriert und verdampft im Vakuum.
Man chromatographiert an Silicagel, wobei mit 30% Ethylacetat/Hexan
eluiert wird, wodurch sich die Titelverbindung ergibt: DC Rf = 0,43 (Silicagel, 30% Ethylacetat/Hexan).
-
BEISPIEL 47
-
(S)-2-[N-(t-Butoxycarbonyl)allylamino]-3-phenylpropionsäuremethylester
-
Schema B, optionaler Schritt
b:
-
Man
vereinigt (S)-2-Allylamino-3-phenylpropionsäuremethylester (6,62 g, 30,4
mmol) und Di-t-butyldicarbonat (7,29 g, 33,5 mmol) in DMF/Ethylacetat
(30 ml/30 ml). Man rührt
16 Stunden unter einer Inertatmosphäre. Man verdünnt das
Reaktionsgemisch mit Ethylacetat und extrahiert mit Wasser. Man
trennt die Phasen, trocknet die organische Phase über MgSO4, filtriert und verdampft im Vakuum. Man
chromatographiert an Silicagel, wobei mit 10% Ethylacetat/Hexan
eluiert wird, wodurch sich die Titelverbindung ergibt.
-
BEISPIEL 48
-
(S)-2-[N-(t-Butoxycarbonyl)allylamino]-3-phenylpropionsäure
-
Schema B, Schritt e:
-
Man
vereinigt (S)-2-[N-(t-Butoxycarbonyl)allylamino-3-phenylpropionsäuremethylester
(0,32 g, 1,0 mmol) und 1 M Natriumhydroxid (10 ml, 10 mmol) in Ethanol
(10 ml). Man rührt
4 Stunden. Man säuert
das Reaktionsgemisch mit 1 M Salzsäure an und extrahiert mit Ethylacetat.
Man trennt die Phasen, trocknet die organische Phase über MgSO4, filtriert und verdampft im Vakuum. Man
chromatographiert an Silicagel, wobei mit 3% Methanol/Dichlormethan
eluiert wird, wodurch sich die Titelverbindung ergibt: DC Rf = 0,40 (Silicagel, 5% Methanol/Dichlormethan).
-
BEISPIEL 49
-
(S)-N-Benzyl-N-methyl-2-[N-(t-butoxycarbonyl)allylamino]-3-phenylpropionamid
-
Schema B, Schritt f:
-
Man
vereinigt (S)-2-[N-(t-Butoxycarbonyl)allylamino]-3-phenylpropionsäure (11,1
g, 36,35 mmol) und THF (360 ml). Man kühlt auf –22°C ab. Man gibt N-Methylmorpholin
(7,09 ml, 54,53 mmol) hinzu und rührt 10 Minuten. Man gibt Isobutylchlorformiat
(7,09 ml, 54,53 mmol) hinzu und rührt 30 Minuten bei –22°C. Man gibt N-Methyl-N-benzylamin
(7,09 ml, 54,53 mmol) hinzu. Man lässt auf Zimmertemperatur erwärmen und
rührt 2 Stunden.
Man verdünnt
das Reaktionsgemisch mit Ethylacetat und extrahiert mit Wasser.
Man trennt die Phasen, trocknet die organische Phase über MgSO4, filtriert und verdampft im Vakuum. Man
chromatographiert an Silicagel, wobei mit 10% Ethylacetat/Hexan
eluiert wird, wodurch sich die Titelverbindung ergibt.
-
BEISPIEL 50
-
(S)-N-(2-Methoxybenzyl)-N-methyl-2-[N'-(t-butoxycarbonyl)allylamino]-3-phenylpropionamid
-
Schema B, Schritt g:
-
Man
vereinigt (S)-2-[N-(t-Butoxycarbonyl)allylamino]-3-phenylpropionsäure (1,59
g, 5,20 mmol), (2-Methoxybenzyl)methylamin (0,79 g, 5,20 mmol),
1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid-Hydrochlorid
(1,12 g, 5,72 mmol), 1-Hydroxybenzotriazol (0,38 g, 2,52 mmol) und
Diisopropylethylamin (1,34 ml, 6,5 mmol) in Dichlormethan (50 ml)
und rührt
18 Stunden. Man verdünnt
mit Ethylacetat und extrahiert mit 1 M Salzsäure, einer gesättigten
wässrigen
Lösung
von Natriumbicarbonat und einer gesättigten wässrigen Lösung von Natriumchlorid. Man
trocknet die abgetrennte organische Phase über MgSO4,
filtriert und verdampft im Vakuum. Man chromatographiert an Silicagel,
wobei nacheinander mit 5% Ethylacetat/Hexan und 10% Ethylacetat/Hexan
eluiert wird, wodurch sich die Titelverbindung ergibt: DC Rf = 0,55 (Silicagel, 30% Ethylacetat/Hexan).
-
BEISPIEL 51
-
(S)-N-(3,4,5-Trimethoxybenzyl)-N-methyl-2-[N'-(t-butoxycarbonyl)allylamino]-3-phenylpropionamid
-
Schema B, Schritt g:
-
Man
vereinigt (S)-2-[N-(t-Butoxycarbonyl)allylamino]-3-phenylpropionsäure (0,91
g, 2,97 mmol), (3,4,5-Trimethoxy)benzylmethylamin (0,63 g, 2,97
mmol), 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid-Hydrochlorid
(0,63 g, 3,27 mmol), 1-Hydroxybenzotriazol
(0,349 g, 3,27 mmol) und Diisopropylethylamin (0,77 ml, 6,27 mmol)
in Dichlormethan (30 ml) und rührt
18 Stunden. Man verdünnt
mit Ethylacetat und extrahiert mit 1 M Salzsäure, einer gesättigten
wässrigen
Lösung
von Natriumbicarbonat und einer gesättigten wässrigen Lösung von Natriumchlorid. Man
trocknet die abgetrennte organische Phase über MgSO4,
filtriert und verdampft im Vakuum. Man chromatographiert an Silicagel,
wobei nacheinander mit 10% Methanol/Dichlormethan und 30% Methanol/Dichlormethan
eluiert wird, wodurch sich die Titelverbindung ergibt: DC Rf = 0,30 (Silicagel, 30% Ethylacetat/Hexan).
-
BEISPIEL 52
-
(S)-2-[N-(t-Butoxycarbonyl)amino[3-(naphth-2-yl)propionsäure
-
Schema B, optionaler Schritt
j:
-
Man
vereinigt (S)-2-Amino-3-(naphth-2-yl)propionsäure ((S)-(2-Napthyl)alanin)
(2,0 g, 9,29 mmol) und Di-t-butyldicarbonat (2,23 g, 10,22 mmol)
in 1/1 DMF/Ethylacetat (200 ml). Man gibt Diisopropylethylamin (2,0 ml)
hinzu, um die (S)-2-Amino-3-(2-napthyl)-propionsäure löslich zu machen, und rührt 18 Stunden.
Man verdünnt
mit Ethylacetat und extrahiert mit 1 M Salzsäurelösung. Man trocknet die abgetrennte
organische Phase über
MgSO4, filtriert und verdampft im Vakuum,
wodurch sich die Titelverbindung ergibt. DC Rf =
0,47 (Silicagel, 10% Methanol/Dichlormethan).
-
BEISPIEL 53
-
(S)-N-Benzyl-N-methyl-2-[N'-(t-butoxycarbonyl)amino]-3-(naphth-2-yl)propionamid
-
Schema B, optionaler Schritt
k:
-
Man
vereinigt (S)-2-[N-(t-Butoxycarbonyl)amino]-3-(naphth-2-yl)propionsäure (2,92
g, 9,3 mmol), 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid-Hydrochlorid
(1,96 g, 10,22 mmol), 1-Hydroxybenzotriazol (1,38 g, 10,22 mmol),
N-Methyl-N-benzylamin (9,3 mmol) und Diisopropylethylamin (1,78
ml, 10,22 mmol) in Dichlormethan (100 ml) und rührt 18 Stunden. Man verdünnt mit
Ethylacetat und extrahiert mit 1 M Salzsäure, einer gesättigten
wässrigen
Lösung
von Natriumbicarbonat und einer gesättigten wässrigen Lösung von Natriumchlorid. Man
trocknet die abgetrennte organische Phase über MgSO4,
filtriert und verdampft im Vakuum. Man chromatographiert an Silicagel,
wobei mit 20% Ethylacetat/Hexan eluiert wird, wodurch sich die Titelverbindung
ergibt: DC Rf = 0,29 (Silicagel, 20% Ethylacetat/Hexan).
-
BEISPIEL 54
-
(S)-N-(3,4-Dichlorbenzyl)-N-methyl-2-[N'-t-butoxycarbonyl)amino]-3-(naphth-2-yl)propionamid
-
Schema B, optionaler Schritt
k:
-
Man
vereinigt (S)-2-[N-(t-Butoxycarbonyl)amino]-3-(naphth-2-yl)propionsäure (1,23
g, 3,93 mmol), 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid-Hydrochlorid
(0,435 g, 2,2 mmol), 1-Hydroxybenzotriazol (0,297 g, 2,2 mmol),
N-Methyl-N-(3,4-dichlorbenzyl)amin (0,382, 2,0 mmol) und Diisopropylethylamin
(0,53 ml, 2,2 mmol) in Dichlormethan (20 ml) und rührt 72 Stunden.
Man verdünnt
mit Ethylacetat und extrahiert mit 1 M Salzsäure, einer gesättigten
wässrigen
Lösung
von Natriumbicarbonat und einer gesättigten wässrigen Lösung von Natriumchlorid. Man
trocknet die abgetrennte organische Phase über MgSO4,
filtriert und verdampft im Vakuum. Man chromatographiert an Silicagel,
wobei mit 10% Ethylacetat/Hexan eluiert wird, wodurch sich die Titelverbindung
ergibt.
-
BEISPIEL 55
-
(S)-N-(2-Methoxybenzyl)-N-methyl-2-[N'-(t-butoxycarbonyl)amino]-3-(naphth-2-yl)propionamid
-
Schema B, optionaler Schritt
k:
-
Man
vereinigt (S)-2-[N-(t-Butoxycarbonyl)amino]-3-(naphth-2-yl)propionsäure (1,89
g, 6,0 mmol), (2-Methoxybenzyl)methylamin (1,67 g, 11,1 mmol), 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid-Hydrochlorid
(1,30 g, 6,6 mmol), 1-Hydroxybenzotriazol (0,89 g, 6,6 mmol) und
Diisopropylethylamin (1,59 ml, 6,6 mmol) in Dichlormethan (60 ml)
und rührt
18 Stunden. Man verdünnt
mit Ethylacetat und extrahiert mit 1 M Salzsäure, einer gesättigten
wässrigen
Lösung
von Natriumbicarbonat und einer gesättigten wässrigen Lösung von Natriumchlorid. Man
trocknet die abgetrennte organische Phase über MgSO4,
filtriert und verdampft im Vakuum zur Titelverbindung als farblosem Öl.
-
BEISPIEL 56
-
(S)-N-Benzyl-N-methyl-2-[N'-(t-butoxycarbonyl)allylamino-3-(naphth-2-yl)propionamid
-
Schema B, optionaler Schritt
l:
-
Man
vereinigt (S)-N-Benzyl-N-methyl-2-[N'-(t-butoxycarbonyl)amino]-3-(naphth-2-yl)propionamid (3,3 g,
7,9 mmol) und THF/DMF (70 ml/ml) und kühlt in einem Eisbad auf 0°C ab. Man
gibt Natriumhydrid (0,7 g, 60% in Öl, 17,38 mmol) und Allylbromid
(4,1 ml, 47,4 mmol) hinzu. Man lässt
die Reaktion auf Zimmertemperatur erwärmen und erhitzt dann 18 Stunden
zum Rückfluss.
Man gießt
das Reaktionsgemisch in eine gesättigte
wässrige
Lösung
von Ammoniumchlorid. Man trennt die Phasen und extrahiert die wässrige Phase
mit Dichlormethan. Man vereinigt die organischen Phasen. Man trocknet über MgSO4, filtriert und verdampft im Vakuum. Man
chromatographiert an Silicagel, wobei nacheinander mit 10% Ethylacetat/Hexan
und 20% Ethylacetat/Hexan eluiert wird, wodurch sich die Titelverbindung
ergibt: DC Rf = 0,59 (Silicagel, 20% Ethylacetat/Hexan).
-
BEISPIEL 57
-
(S)-N-(3,4-Dichlorbenzyl)-N-methyl-2-[N'-(t-butoxycarbonyl)allylamino]-3-(naphth-2-yl)propionamid
-
Schema B, optionaler Schritt
l:
-
Man
vereinigt (S)-N-(3,4-Dichlorbenzyl)-N-methyl-2-[N'-(t-butoxycarbonyl)amino]-3-(naphth-2-yl)propionamid
(0,48 g, 7,9 mmol) und THF/DMF (9 ml/1 ml) und kühlt in einem Eisbad auf 0°C ab. Man
gibt Natriumhydrid (0,048 g, 60% in Öl, 2,0 mmol) und Allylbromid
(0,52 ml) hinzu. Man lässt
die Reaktion auf Zimmertemperatur erwärmen und erhitzt dann 18 Stunden
zum Rückfluss.
Man gießt
das Reaktionsgemisch in eine gesättigte
wässrige
Lösung
von Ammoniumchlorid. Man trennt die Phasen und extrahiert die wässrige Phase mit
Dichlormethan. Man vereinigt die organischen Phasen. Man trocknet über MgSO4, filtriert und verdampft im Vakuum. Man
chromatographiert an Silicagel, wobei nacheinander mit 10% Ethylacetat/Hexan
und 20% Ethylacetat/Hexan eluiert wird, wodurch sich die Titelverbindung
als Feststoff ergibt.
-
BEISPIEL 58
-
(S)-N-(2-Methoxybenzyl)-N-methyl-2-[N'-(t-butoxycarbonyl)allylamino]-3-(naphth-2-yl)propionamid
-
Schema B, optionaler Schritt
l:
-
Man
vereinigt (S)-N-(2-Methoxybenzyl)-N-methyl-2-[N'-(t-butoxycarbonyl)amino]-3-(naphth-2-yl)propionamid
(0,63 g, 1,41 mmol) und THF/DMF (15 ml/5 ml) und kühlt in einem
Eisbad auf 0°C
ab. Man gibt Natriumhydrid (0,067 g, 60% in Öl, 2,82 mmol) und Allylbromid
(0,73 ml, 8,46 mmol) hinzu. Man lässt die Reaktion auf Zimmertemperatur
erwärmen
und erhitzt dann 18 Stunden zum Rückfluss. Man gießt das Reaktionsgemisch
in eine gesättigte
wässrige
Lösung
von Ammoniumchlorid. Man trennt die Phasen und extrahiert die wässrige Phase
mit Dichlormethan. Man vereinigt die organischen Phasen. Man trocknet über MgSO4, filtriert und verdampft im Vakuum. Man
chromatographiert an Silicagel, wobei mit 10% Ethylacetat/Hexan
eluiert wird, wodurch sich die Titelverbindung ergibt: DC Rf = 0,55 (Silicagel, 20% Ethylacetat/Hexan).
-
BEISPIEL 59
-
(S)-N-Benzyl-N-methyl-2-[N'-(t-butoxycarbonyl)-2-oxoethylamino]-3-phenylpropionamid
-
Schema B, Schritt g:
-
Man
vereinigt (S)-N-Benzyl-N-methyl-2-[N'-(t-butoxycarbonyl)allylamino]-3-phenylpropionamid
(10,04 g, 24,5 mmol) und Pyridin (0,13 ml) in Dichlormethan/Methanol
(300 ml/30 ml). Man kühlt
auf –78°C ab. Man leitet
ozonierten Sauerstoff durch die Lösung, bis eine bleibende hellblaue
Färbung
erhalten wird. Man leitet Stickstoff durch die Lösung, bis die blaue Färbung verschwindet.
Man gibt Dimethylsulfid (55 ml) hinzu. Man lässt das Reaktionsgemisch auf
Zimmertemperatur erwärmen
und rührt
16 Stunden. Man engt im Vakuum zu einem Rückstand ein. Man verdünnt den
Rückstand
mit Ethylacetat und extrahiert mit Wasser. Man trennt die Phasen,
trocknet die organische Phase über
MgSO4, filtriert und verdampft im Vakuum.
Man chromatographiert an Silicagel, wobei mit 10% Ethylacetat/Hexan
eluiert wird, wodurch sich die Titelverbindung ergibt: DC Rf = 0,53 (Silicagel, 10% Ethylacetat/Hexan).
-
BEISPIEL 60
-
(S)-N-(2-Methoxybenzyl)-N-methyl-2-[N'-(t-butoxycarbonyl)-2-oxoethylamino]-3-phenylpropionamid
-
Schema D, Schritt g:
-
Man
vereinigt (S)-N-(2-Methoxybenzyl)-N-methyl-2-[N'-(t-butoxycarbonyl)allylamino]-3-phenylpropionamid
(0,66 g, 1,52 mmol), N-Methylmorpholin-N-oxid (0,20 g, 1,67 mmol),
Aceton (5 ml) und Wasser (5 ml). Man gibt Osmiumtetroxid (0,78 ml,
0,04 M in THF, 0,032 mmol) hinzu und rührt 18 Stunden unter einer
Inertatmosphäre.
Man gießt
das Reaktionsgemisch in eine gesättigte
Lösung
von Natriumbisulfit und extrahiert das intermediäre Diol in Ethylacetat. Man
trocknet die abgetrennte organische Phase über MgSO4,
filtriert und verdampft im Vakuum, wodurch sich das rohe Diol ergibt,
das ohne weitere Reinigung im nächsten
Schritt eingesetzt wird. Man löst
das rohe Diol in Chloroform (10 ml). Man gibt eine Lösung von
Bleitetraacetat (0,74 g, 1,67 mmol) in Chloroform (10 ml) hinzu.
Man rührt
30 Minuten und gießt
dann das Reaktionsgemisch in eine gesättigte Lösung von Natriumbicarbonat.
Man extrahiert mit Dichlormethan und trennt die organische Phase
ab. Man trocknet die abgetrennte organische Phase über MgSO4, filtriert und verdampft im Vakuum, wodurch
sich die Titelverbindung ergibt, die ohne weitere Reinigung verwendet
werden kann.
-
BEISPIEL 61
-
(S)-N-(3,4,5-Trimethoxybenzyl)-N-methyl-2-[N'-(t-butoxycarbonyl)-2-oxoethylamino]-3-phenylpropionamid
-
Schema B, Schritt g:
-
Man
vereinigt (S)-N-(3,4,5-Trimethoxybenzyl)-N-methyl-2-[N'-(t-butoxycarbonyl)-2-allylamino]-3-phenylpropionamid
(0,50 g, 1,0 mmol), N-Methylmorpholin-N-oxid (0,13 g, 1,1 mmol),
Aceton (15 ml) und Wasser (20 ml). Man gibt Osmiumtetroxid (0,51
ml, 0,04 M in THF, 0,021 mmol) hinzu und rührt 18 Stunden unter einer Inertatmosphäre. Man
gießt
das Reaktionsgemisch in eine gesättigte
Lösung
von Natriumbisulfit und extrahiert das intermediäre Diol in Ethylacetat. Man
trocknet die abgetrennte organische Phase über MgSO4,
filtriert und verdampft im Vakuum. Man nimmt das rohe Diol im nächsten Schritt
ohne weitere Reinigung. Man löst
das rohe Diol in Chloroform (10 ml). Man gibt Bleitetraacetat (0,48
g, 1,1 mmol) als Lösung
in Chloroform (10 ml) hinzu. Man rührt 30 Minuten. Man gießt die Reaktion
in eine gesättigte
wässrige
Lösung
von Natriumbicarbonat und extrahiert mit Dichlormethan. Man trocknet
die abgetrennte organische Phase über MgSO4,
filtriert und verdampft im Vakuum, wodurch sich die Titelverbindung
ergibt. Die Titelverbindung kann ohne weitere Reinigung verwendet
werden.
-
BEISPIEL 62
-
(S)-N-Benzyl-N-methyl-2-[N'-(t-butoxycarbonyl]-2-oxoethylamino)-3-(naphth-2-yl)propionamid
-
Schema B, Schritt g:
-
Man
vereinigt (S)-N-Benzyl-N-methyl-2-[N'-(t-butoxycarbonyl)allylamino]-3-(naphth-2-yl)-propionamid (3,34
g, 7,29 mmol) und Pyridin (0,03 ml) in Dichlormethan/Methanol (66
ml/7 ml). Man kühlt
auf –78°C ab. Man
leitet ozonierten Sauerstoff durch die Lösung, bis eine bleibende hellblaue
Färbung
erhalten wird. Man leitet Stickstoff durch die Lösung, bis die blaue Färbung verschwindet.
Man gibt Dimethylsulfid (12 ml) hinzu. Man lässt das Reaktionsgemisch auf
Zimmertemperatur erwärmen
und rührt
16 Stunden. Man engt im Vakuum zu einem Rückstand ein. Man verdünnt den
Rückstand
mit Ethylacetat und extrahiert mit Wasser. Man trennt die Phasen,
trocknet die organische Phase über
MgSO4, filtriert und verdampft im Vakuum.
Man chromatographiert an Silicagel, wobei mit 20% Ethylacetat/Hexan
eluiert wird, wodurch sich die Titelverbindung ergibt: DC Rf = 0,70 (Silicagel, 50% Ethylacetat/Hexan).
-
BEISPIEL 63
-
(S)-N-(3,4-Dichlorbenzyl)-N-methyl-2-[N'-(t-butoxycarbonyl)-2-oxoethylamino]-3-(naphth-2-yl)propionamid
-
Schema B, Schritt g:
-
Man
vereinigt (S)-N-(3,4-Dichlorbenzyl)-N-methyl-2-[N'-(t-butoxycarbonyl)allylamino]-3-(naphth-2-yl)propionamid
(0,26 g, 0,50 mmol), N-Methylmorpholin-N-oxid (0,065 g, 0,55 mmol),
Aceton (10 ml), Tetrahydrofuran (5 ml) und Wasser (5 ml). Man gibt
Osmiumtetroxid (0,26 ml, 0,04 M in THF, 0,042 mmol) hinzu und rührt 18 Stunden
unter einer Inertatmosphäre.
Man gießt
das Reaktionsgemisch in eine gesättigte
Lösung
von Natriumbisulfit und extrahiert das intermediäre Diol in Ethylacetat. Man
trocknet die abgetrennte organische Phase über MgSO4,
filtriert und verdampft im Vakuum, wodurch sich das rohe Diol ergibt, das
ohne weitere Reinigung im nächsten
Schritt eingesetzt wird. Man löst
das rohe Diol in Chloroform (10 ml). Man gibt eine Lösung von
Bleitetraacetat (0,24 g, 0,55 mmol) in Chloroform (10 ml) hinzu.
Man rührt
30 Minuten und gießt
dann das Reaktionsgemisch in eine gesättigte Lösung von Natriumbicarbonat.
Man extrahiert mit Dichlormethan und trennt die organische Phase
ab. Man trocknet die abgetrennte organische Phase über MgSO4, filtriert und verdampft im Vakuum, wodurch
sich die Titelverbindung ergibt, die ohne weitere Reinigung verwendet
werden kann.
-
BEISPIEL 64
-
(S)-N-(2-Methoxybenzyl)-N-methyl-2-[N'-(t-butoxycarbonyl)-2-oxoethylamino]-3-(naphth-2-yl)propionamid
-
Schema B, Schritt g:
-
Man
vereinigt (S)-N-(2-Methoxybenzyl)-N-methyl-2-[N'-(t-butoxycarbonyl)allylamino]-3-(naphth-2-yl)propionamid
(0,46 g, 0,96 mmol), N-Methylmorpholin-N-oxid (0,12 g, 1,06 mmol),
Aceton (20 ml) und Wasser (10 ml). Man gibt Osmiumtetroxid (0,50
ml, 0,04 M in THF, 0,02 mmol) hinzu und rührt 18 Stunden unter einer
Inertatmosphäre.
Man gießt
das Reaktionsgemisch in eine gesättigte
Lösung
von Natriumbisulfit und extrahiert das intermediäre Diol in Ethylacetat. Man
trocknet die abgetrennte organische Phase über MgSO4,
filtriert und verdampft im Vakuum, wodurch sich das rohe Diol ergibt,
das ohne weitere Reinigung im nächsten
Schritt eingesetzt wird. Man löst
das rohe Diol in Chloroform (10 ml). Man gibt eine Lösung von Bleitetraacetat
(0,46 g, 1,06 mmol) in Chloroform (10 ml) hinzu. Man rührt 30 Minuten
und gießt
dann das Reaktionsgemisch in eine gesättigte Lösung von Natriumbicarbonat.
Man extrahiert mit Dichlormethan und trennt die organische Phase
ab. Man trocknet die abgetrennte organische Phase über MgSO4, filtriert und verdampft im Vakuum, wodurch
sich die Titelverbindung ergibt, die ohne weitere Reinigung verwendet
werden kann.
-
BEISPIEL 65
-
(S)-N-Benzyl-N-methyl-2-[N'-(t-butoxycarbonyl)-3-hydroxypropylamino]-3-phenylpropionamid
-
Schema B, optionaler Schritt
h:
-
Man
kühlt eine
Lösung
von Boran (1,5 ml, 1 M in THF, 1,5 mmol) in einem Eisbad unter einer
Inertatmosphäre
auf 0°C
ab. Man gibt Cyclohexen (0,31 ml, 3,1 mmol) hinzu und rührt 15 Minuten
bei fortgesetzter Kühlung.
Man gibt die vorstehend hergestellte Suspension von Dicyclohexylboran
in THF zu (S)-N-Benzyl-N-methyl-2-[N'-(t-butoxycarbonyl)allylamino]-3-phenylpropionamid
(2 mmol) und rührt
15 Minuten in einem Eisbad. Man erwärmt auf Zimmertemperatur und
rührt 2
Stunden. Man verdünnt
das Gemisch mit Phosphatpuffer mit pH-Wert 7 (40 ml) und Ethanol
(20 ml). Man gibt 30%iges Wasserstoffperoxid (8 ml) hinzu. Man rührt 20 Stunden
bei Zimmertemperatur. Man engt im Vakuum zu einem Rückstand
ein. Man verdünnt
das Reaktionsgemisch mit Ethylacetat und extrahiert mit Wasser.
Man trennt die Phasen, trocknet die organische Phase über MgSO4, filtriert und verdampft im Vakuum. Man
chromatographiert an Silicagel, wodurch sich die Titelverbindung
ergibt.
-
BEISPIEL 66
-
(S)-N-Benzyl-N-methyl-2-[N'-(t-butoxycarbonyl)-3-oxopropylamino]-3-phenylpropionamid
-
Schema B, optionaler Schritt
i:
-
Man
vereinigt (S)-N-Benzyl-N-methyl-2-[N'-(t-butoxycarbonyl)-3-hydroxypropylamino]-3-phenylpropionamid
(20 mmol), Triethylamin (10 mmol) und Dimethylsulfoxid (4 ml). Man
gibt die vorstehend hergestellte Lösung zu einer Lösung von
Pyridin/Schwefeltrioxid-Komplex
(6,4 mmol) in Dimethylsulfoxid (12 ml). Man rührt 1 Stunde. Man verdünnt das
Reaktionsgemisch mit Ethylacetat und extrahiert mit Wasser. Man
trennt die Phasen, trocknet die organische Phase über MgSO4, filtriert und verdampft im Vakuum, wodurch
sich die Titelverbindung ergibt, die ohne weitere Reinigung im nächsten Schritt
eingesetzt wird.
-
In
Schema C wird ein allgemeines Syntheseverfahren zur Herstellung
des Aldehyds der Struktur (3) angegeben, in der G1 -C(O)-
und G2 -CH2- ist,
der als Ausgangsmaterial in Schema A.1 eingesetzt wird. Die Reagenzien
und Ausgangsmaterialien sind für
den Durchschnittsfachmann leicht erhältlich. In Schema C sind alle
Substituenten, sofern nicht anders angegeben, wie zuvor definiert.
-
-
In
Schema C, Schritt a, durchläuft
eine geeignete Allyl-Carbamat-Säure
der Struktur (15), die unter Verwendung der Verfahren aus Schema
B hergestellt wurde, eine Amidierungsreaktion mit Methylamin oder einem
Salz von Methylamin zu einem Allyl-Carbamat-Säure-N-methylamid der Struktur
(19).
-
Eine
geeignete Allyl-Carbamat-Säure
der Struktur (15) ist eine, bei der die Stereochemie, R5 und
Ar2 so sind, wie im Produkt der Formel (1)
gewünscht,
oder ist eine, die nach Antipodentrennung oder Schutzgruppenabspaltung
die Stereochemie oder Ar2 und R5 ergibt,
wie im Endprodukt der Formel (1) erwünscht.
-
Eine
Amidierungsreaktion kann über
eine aktivierte Zwischenstufe verlaufen, wie ein gemischtes Anhydrid
oder ein (O)-Hydroxybenzotriazol, das hergestellt werden kann, aber
nicht notwendigerweise vor der Zugabe von Methylamin oder einem
Salz von Methylamin isoliert wird.
-
Beispielsweise
wird eine geeignete Allyl-Carbamat-Säure der Struktur (15) mit 1,2
bis 1,7 Äquivalenten
einer geeigneten Base, wie N-Methylmorpholin, in einem geeigneten
Lösungsmittel,
wie Tetrahydrofuran, in Kontakt gebracht. Im Allgemeinen wird das
Reaktionsgemisch vor der Zugabe von 1,2 bis 1,7 Äquivalenten Isobutylchlorformiat
auf eine Temperatur zwischen –50°C und 0°C abgekühlt, wobei –25°C bis –20°C bevorzugt
werden. Die Reaktion wird im Allgemeinen 30 Minuten bis 3 Stunden
rühren
gelassen, so dass sich das gemischte Anhydrid, eine aktivierte Zwischenstufe,
bilden kann. Während
die Temperatur zwischen –50°C und 0°C gehalten
wird, wird Methylamin oder ein Salz von Methylamin zugegeben. Die
Reaktion kann, nachdem die Zugabe des Amins beendet ist, auf Zimmertemperatur
erwärmt
werden. Die Reaktion erfordert 2 bis 48 Stunden. Das Produkt kann
mit im Fachgebiet bekannten Verfahren, wie Extraktion, Verdampfen,
Chromatographie und Umkristallisation, isoliert und gereinigt werden.
-
In
einer anderen Ausführungsform
beispielsweise wird eine geeignete Allyl-Carbamat-Säure der Struktur
(15) mit einem geringen molaren Überschuss
an Methylamin oder einem Salz von Methylamin und 1-Hydroxybenzotriazol-Hydrat
in Gegenwart eines geringen molaren Überschusses an Kupplungsmittel,
wie Dicyclohexylcarbodiimid oder 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid,
in Kontakt gebracht. Die Reaktion wird in Gegenwart einer geeigneten
Base, wie Diisopropylethylamin, durchgeführt. Die Reaktion wird in einem
geeigneten Lösungsmittel,
wie Dichlormethan oder Chloroform, durchgeführt. Das Produkt kann mit im Fachgebiet
bekannten Verfahren, wie Extraktion, Verdampfen, Chromatographie
und Umkristallisation, isoliert und gereinigt werden.
-
In
Schema C, Schritt b, wird ein Allyl-Carbamat-Säure-N-methylamid der Struktur
(19) zu einer N-Methylaminoverbindung der Struktur (20) reduziert.
-
Beispielsweise
wird ein Allyl-Carbamat-Säure-N-methylamid
der Struktur (19) mit einem geeigneten Reduktionsmittel, wie Diisobutylaluminiumhydrid
oder Lithiumaluminiumhydrid, in Kontakt gebracht, wobei Diisobutylaluminiumhydrid
bevorzugt wird. Die Reaktion wird in einem geeigneten Lösungsmittel,
wie Tetrahydrofuran oder Toluol, durchgeführt. Im Allgemeinen wird die
Reaktion bei Temperaturen von –20°C bis zur Rückflusstemperatur
des Lösungsmittels
durchgeführt.
Nach geeigneter Aufarbeitung, wie im Fachgebiet bekannt, hängt die
eingesetzte Aufarbeitung von den hergestellten Produkten und dem
verwendeten Reduktionsmittel ab, kann das Produkt im Fachgebiet
bekannten Verfahren, wie Extraktion, Verdampfen, Chromatographie
und Umkristallisation, isoliert und gereinigt werden.
-
In
Schema C, Schritt c, wird eine N-Methylaminoverbindung der Struktur
(20) mit einem geeigneten Aroylsäurechlorid
zu einem N-Methylaroylamid der Struktur (21) aroyliert.
-
Ein
geeignetes Aroylsäurechlorid
Ar1C(O)Cl ist eines, bei dem Ar1 so
ist, wie im Produkt der Formel (1) erwünscht, oder nach Schutzgruppenabspaltung
Ar1 ergibt, wie im Endprodukt der Formel
(1) erwünscht.
-
Beispielsweise
wird eine N-Methylaminoverbindung der Struktur (20) mit einem geeigneten
Aroylsäurechlorid
Ar1C(O)Cl in Kontakt gebracht. Die Reaktion
wird in Gegenwart einer geeigneten Base, wie Triethylamin, Diisopropylethylamin
oder Pyridin, durchgeführt.
Die Reaktion wird in einem geeigneten Lösungsmittel, wie Dichlormethan,
Chloroform, Pyridin, Dioxan, Tetrahydrofuran oder Wasser, durchgeführt. Im
Allgemeinen wird die Reaktion bei Temperaturen von –20°C bis zur
Rückflusstemperatur
des Lösungsmittels
durchgeführt. Das
Produkt kann mit im Fachgebiet bekannten Verfahren, wie Extraktion,
Verdampfen, Chromatographie und Umkristallisation, isoliert und
gereinigt werden.
-
In
Schema C, Schritt d, wird ein N-Methylaroylamid der Struktur (21)
in einen Aldehyd der Struktur (3) überführt, in der G1 -C(O)-
und G2 -CH2- ist.
Ein N-Methylaroylamid der Struktur (21) kann durch entweder Ozonolyse
in Gegenwart von Methanol, gefolgt von reduktiver Aufarbeitung,
oder eine über
Osmiumtetroxid vermittelte Bildung eines intermediären Diols,
gefolgt von oxidativer Spaltung mit Bleitetraacetat oder Natriummetaperiodat,
in einen Aldehyd der Struktur (3), in der G1 -C(O)-
und G2 -CH2- ist, überführt werden.
-
Beispielsweise
wird ein N-Methylaroylamid der Struktur (21) mit Ozon in Gegenwart
von Methanol in Kontakt gebracht. Die Reaktion wird in einem geeigneten
Lösungsmittel,
wie Dichlormethan, durchgeführt.
Im Allgemeinen wird die Reaktion bei einer Temperatur von –100°C bis –60°C durchgeführt, wobei –70°C bevorzugt
wird. Die Reaktion wird reduktiv durch Zugabe eines geeigneten Reduktionsmittels,
wie Tributylphosphin oder Dimethylsulfid, aufgearbeitet. Das Produkt
kann durch Verdampfen aus der Reaktionszone isoliert werden und
kann ohne weitere Reinigung verwendet werden. Das Produkt kann mit
im Fachgebiet bekannten Verfahren, wie Chromatographie und Umkristallisation,
gereinigt werden.
-
In
einer anderen Ausführungsform
beispielsweise wird ein N-Methylaroylamid der Struktur (21) mit
Osmiumtetroxid in Kontakt gebracht, wodurch sich ein intermediäres Diol
ergibt. Die Reaktion kann unter Verwendung von 0,01 bis 0,05 molaren Äquivalenten
Osmiumtetroxid und eines geringen molaren Überschusses an Oxidationsmittel,
wie N-Methylmorpholin-N-oxid,
durchgeführt
werden. Die Reaktion wird in einem Lösungsmittel, wie Aceton/Wasser-Gemische,
durchgeführt.
Die Reaktion wird bei Zimmertemperatur durchgeführt und erfordert 12 bis 48
Stunden. Das Reaktionsgemisch wird zu einer gesättigten Lösung von Natriumbisulfit gegebem
und das intermediäre
Diol wird durch Extraktion und Verdampfen isoliert und ohne weitere
Reinigung verwendet. Das intermediäre Diol wird mit einem geringen
molaren Überschuss
Bleitetraacetat oder Natriummetaperiodat in Kontakt gebracht. Im
Allgemeinen wird die Reaktion in einem Lösungsmittel, wie Chloroform, durchgeführt. Die
Reaktion wird bei Zimmertemperatur durchgeführt und erfordert 30 Minuten
bis 8 Stunden. Das Produkt kann durch Extraktion und Verdampfen
aus der Reaktionszone isoliert werden und kann ohne weitere Reinigung
verwendet werden. Das Produkt kann mit im Fachgebiet bekannten Verfahren,
wie Chromatographie und Umkristallisation, gereinigt werden.
-
BEISPIEL 67
-
(S)-N-Methyl-2-[N'-(t-butoxycarbonyl)allylamino]-3-phenylpropionamid
-
Schema C, Schritt a:
-
Man
vereinigt (S)-2-[N-(t-Butoxycarbonyl)allylamino]-3-phenylpropionsäure (0,7
g, 2,29 mmol), 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid-Hydrochlorid
(0,50 g, 2,52 mmol), 1-Hydroxybenzotriazol (0,38 g, 2,52 mmol),
Methylamin-Hydrochlorid (0,17 g, 2,52 mmol) und Diisopropylethylamin
(0,59 ml, 2,52 mmol) in Dichlormethan (23 ml) und rührt 18 Stunden.
Man verdünnt
mit Ethylacetat und extrahiert mit 1 M Salzsäure, einer gesättigten
wässrigen
Lösung
von Natriumbicarbonat und einer gesättigten wässrigen Lösung von Natriumchlorid. Man
trocknet die abgetrennte organische Phase über MgSO4,
filtriert und verdampft im Vakuum. Man chromatographiert an Silicagel,
wobei nacheinander mit 5% Methanol/Dichlormethan und 10% Methanol/Dichlormethan
eluiert wird, wodurch sich die Titelverbindung ergibt: DC Rf = 0,44 (Silicagel, 30% Ethylacetat/Hexan).
-
BEISPIEL 68
-
(S)-N-Methyl-2-[N'-(t-butoxycarbonyl)allylamino]-3-phenylpropyl
-
Schema C, Schritt b:
-
Man
löst (S)-N-Methyl-2-[N'-(t-butoxycarbonyl)allylamino]-3-phenylpropionamid
(0,31 g, 0,98 mmol) in Dichlormethan (10 ml) und kühlt in einem
Trockeneis/Aceton-Bad auf –78°C ab. Man
gibt Diisobutylaluminiumhydrid (1,96 ml, 1,5 M in Toluol, 2,94 mmol)
hinzu. Man lässt
langsam auf Zimmertemperatur erwärmen
und rührt
16 Stunden. Man gibt langsam eine 15%ige wässrige Lösung von Natriumhydroxid (3,0
ml) hinzu. Man extrahiert mit Dichlormethan, trocknet die organische
Phase über
MgSO4, filtriert und verdampft im Vakuum, wodurch
sich die Titelverbindung als ein Gemisch ergibt, das ohne weitere
Reinigung im nächsten
Schritt eingesetzt wird.
-
BEISPIEL 69
-
(S)-N-Methyl-N-[2-[N'-(t-butoxycarbonyl)allylamino]-3-phenylpropyl]benzamid
-
Schema C, Schritt c:
-
Man
vereinigt (S)-N-Methyl-2-[N'-(t-butoxycarbonyl)allylamino]-3-phenylpropylamin
(1,23 g, 4,25 mmol) und Diisopropylethylamin (0,36 ml, 2,0 mmol)
in Dichlormethan (20 ml). Man kühlt
in einem Eisbad auf 0°C
ab. Man gibt Benzoylchlorid (0,24 ml, 2,0 mmol) hinzu und rührt die
Reaktion 2 Stunden bei 0°C.
Man extrahiert das Reaktionsgemisch mit Wasser, trocknet die organische
Phase über
MgSO4, filtriert und verdampft im Vakuum.
Man chromatographiert an Silicagel, wobei mit 20% Ethylacetat/Hexan
eluiert wird, wodurch sich die Titelverbindung ergibt: DC Rf = 0,59 (Silicagel, 20% Ethylacetat/Hexan).
-
BEISPIEL 70
-
(S)-N-Methyl-N-[2-[N'-(t-butoxycarbonyl)allylamino]-3-phenylpropyl]-(3,4,5-trimethoxy)benzamid
-
Schema C, Schritt c:
-
Man
vereinigt (S)-N-Methyl-2-[N'-(t-butoxycarbonyl)allylamino]-3-phenylpropylamin
(0,57 g, 1,87 mmol) und Diisopropylethylamin (0,65 ml, 3,74 mmol)
in Dichlormethan (40 ml). Man kühlt
in einem Eisbad auf 0°C
ab. Man gibt 3,4,5-Trimethoxybenzoylchlorid (0,43 g, 1,87 mmol)
hinzu und rührt
die Reaktion 4 Stunden bei 0°C.
Man extrahiert das Reaktionsgemisch mit Wasser, trocknet die organische
Phase über
MgSO4, filtriert und verdampft im Vakuum.
Man chromatographiert an Silicagel, wobei nacheinander mit 5% Ethylacetat/Hexan,
20% Ethylacetat/Hexan und 35% Ethylacetat/Hexan eluiert wird, wodurch
sich die Titelverbindung ergibt.
-
BEISPIEL 71
-
(S)-N-Methyl-N-[2-[N'-(t-butoxycarbonyl)-2-oxoethylamino]-3-phenylpropyl]-(3,4,5-trimethoxy)benzamid
-
Schema C, optionaler Schritt
d:
-
Man
vereinigt (S)-N-Methyl-N-[2-[N'-(t-butoxycarbonyl)allylamino]-3-phenylpropyl]-(3,4,5-trimethoxy)benzamid
(0,145 g, 0,29 mmol), N-Methylmorpholin-N-oxid (0,037 g, 0,32 mmol),
Aceton (5 ml) und Wasser (5 ml). Man gibt Osmiumtetroxid (0,15 ml,
0,04 M in THF, 0,006 mmol) hinzu und rührt 18 Stunden unter einer Inertatmosphäre. Man
gießt
das Reaktionsgemisch in eine gesättigte
Lösung
von Natriumbisulfit und extrahiert das intermediäre Diol in Ethylacetat. Man
trocknet die abgetrennte organische Phase über MgSO4,
filtriert und verdampft im Vakuum, wodurch das rohe Diol erhalten
wird, das ohne weitere Reinigung im nächsten Schritt eingesetzt wird.
Man löst
das rohe Diol in Chloroform (10 ml). Man gibt Bleitetraacetat (0,32
g, 0,32 mmol) als Lösung
in Chloroform (10 ml) hinzu. Man rührt 30 Minuten. Man gießt die Reaktion
in eine gesättigte
wässrige Lösung von
Natriumbicarbonat und extrahiert mit Dichlormethan. Man trocknet
die abgetrennte organische Phase über MgSO4,
filtriert und verdampft im Vakuum, wodurch sich die Titelverbindung
ergibt. DC Rf = 0,79 (Silicagel, 10% Methanol/Dichlormethan).
Die Titelverbindung kann ohne weitere Reinigung verwendet werden.
-
BEISPIEL 72
-
(S)-N-Methyl-N-[2-[N'-(t-butoxycarbonyl)-2-oxoethylamino]-3-phenylpropyl]benzamid
-
Schema C, optionaler Schritt
d:
-
Man
vereinigt (S)-N-Methyl-N-[2-[N'-(t-butoxycarbonyl)allylamino]-3-phenylpropyl]benzamid
(0,14 g, 0,35 mmol), N-Methylmorpholin-N-oxid (0,044 g, 0,38 mmol),
Aceton (5 ml) und Wasser (5 ml). Man gibt Osmiumtetroxid (0,18 ml,
0,04 M in THF, 0,0074 mmol) hinzu und rührt 18 Stunden unter einer
Inertatmosphäre. Man
gießt
das Reaktionsgemisch in eine gesättigte
Lösung
von Natriumbisulfit und extrahiert das intermediäre Diol in Ethylacetat. Man
trocknet die abgetrennte organische Phase über MgSO4,
filtriert und verdampft im Vakuum, wodurch das rohe Diol erhalten
wird, das ohne weitere Reinigung im nächsten Schritt eingesetzt wird. Man
löst das
rohe Diol in Chloroform (5 ml). Man gibt Bleitetraacetat (0,16 g,
0,38 mmol) als Lösung
in Chloroform (5 ml) hinzu. Man rührt 30 Minuten. Man gießt die Reaktion
in eine gesättigte
wässrige
Lösung
von Natriumbicarbonat und extrahiert mit Dichlormethan. Man trocknet
die abgetrennte organische Phase über MgSO4,
filtriert und verdampft im Vakuum, wodurch sich die Titelverbindung
ergibt. DC Rf = 0,76 (Silicagel, 50% Ethylacetat/Hexan).
Die Titelverbindung kann ohne weitere Reinigung verwendet werden.
-
In
Schema D wird ein allgemeines Syntheseverfahren zur Herstellung
des Aldehyds der Struktur (3) angegeben, in der G1 -CH2- und G2 -CH2- ist, der als Ausgangsmaterial in Schema
A.1 eingesetzt wird. Die Reagenzien und Ausgangsmaterialien sind
für den
Durchschnittsfachmann leicht erhältlich.
In Schema D sind alle Substituenten, sofern nicht anders angegeben,
wie zuvor definiert.
-
-
In
Schema D, optionaler Schritt a, wird ein geeigneter Aldehyd der
Struktur (3) unter Verwendung eines geeigneten Reduktionsmittels
zu einem Alkohol-Amid der Struktur (25) reduziert.
-
Ein
geeigneter Aldehyd der Struktur (3) ist einer, bei dem G1 -CH2- ist, G2 -C(O)- ist, Stereochemie, R5, Ar1, Ar2 so sind, wie
im Produkt der Formel (1) erwünscht,
oder kann auch einer sein, bei dem die Stereochemie und die Reste
Ar1 und Ar2 und
R5 nach Antipodentrennung und Schutzgruppenabspaltung
oder Modifikation die Stereochemie und die Reste Ar1 und
Ar2 und R5 ergeben,
wie im Endprodukt der Formel (1) erwünscht. Zur Herstellung von
Aldehyden der Struktur (3), in der G1 -CH2- und G2 -CH2- ist, wird die Verwendung von Aldehyden
(3), in denen G1 -CH2-,
G2 -C(O)- und R5 t-Butyl
ist, bevorzugt.
-
Beispielsweise
wird ein geeigneter Aldehyd der Struktur (3) mit 1 bis 4 Äquivalenten
eines geeigneten Reduktionsmittels, wie Natriumborhydrid, in Kontakt
gebracht. Ein geeignetes Reduktionsmittel im optionalen Schritt
a reduziert einen Aldehyd und wirkt nicht auf ein Amid irgendeiner
Schutzgruppe, die vorhanden sein kann. Die Reaktion wird in einem
geeigneten Lösungsmittel,
wie Methanol oder Ethanol, durchgeführt. Im Allgemeinen wird die
Reaktion bei Temperaturen von 0°C
bis zur Rückflusstemperatur
des Lösungsmittels
durchgeführt.
Die Reaktionen erfordern im Allgemeinen 1 bis 72 Stunden. Das Produkt
kann mit im Fachgebiet bekannten Verfahren, wie Quenchen, Extraktion,
Verdampfen, Chromatographie und Umkristallisation, isoliert und
gereinigt werden.
-
In
Schema D, optionaler Schritt b, wird ein Alkohol-Amid der Struktur
(25) unter Verwendung einer geeigneten Hydroxyschutzgruppe Pg1 geschützt,
wodurch sich eine geschützte
Hydroxy-Amid-Verbindung der Struktur (26) ergibt. Ein Alkohol-Amid
der Struktur (25) kann hergestellt werden, wie in Schema D, optionaler Schritt
a, gelehrt.
-
Eine
geeignete Hydroxyschutzgruppe ist eine, die die Reduktion eines
Amids ermöglicht,
solche Schutzgruppen schließen
Tetrahydropyran-2-yl, t-Butyldimethylsilyl oder t-Butyldiphenylsilyl
ein, sind aber nicht darauf begrenzt. Die Auswahl und Verwendung
geeigneter Hydroxyschutzgruppen ist im Fachgebiet bekannt und anerkannt
und wird in Protecting Groups in Organic Synthesis von T. Greene,
Wiley-Interscience (1981), beschrieben.
-
In
Schema D, optionaler Schritt c, wird eine geschützte Hydroxy-Amid-Verbindung
der Struktur (26) unter Verwendung eines geeigneten Reduktionsmittels
für Amide
reduziert, wodurch sich eine geschützte Hydroxy-Amin-Verbindung
der Struktur (27) ergibt.
-
Beispielsweise
wird eine geschützte
Hydroxy-Amid-Verbindung der Struktur (26) mit 1 bis 5 Äquivalenten
eines geeigneten Reduktionsmittels für Amide, wie Lithiumaluminumhydrid,
Diisobutylaluminumhydrid oder Boran-Dimethylsulfid-Komplex, in Kontakt
gebracht. Die Reaktion wird in einem geeigneten Lösungsmittel,
wie Tetrahydrofuran, Toluol oder Diethylether, durchgeführt. Im
Allgemeinen wird die Reaktion bei Temperaturen von 0°C bis zur
Rückflusstemperatur
des Lösungsmittels
durchgeführt.
Die Reaktionen erfordern im Allgemeinen 1 bis 72 Stunden. Das Produkt
kann mit im Fachgebiet bekannten Verfahren, wie Quenchen, Extraktion,
Verdampfen, Chromatographie und Umkristallisation, isoliert und
gereinigt werden.
-
In
Schema D, optionaler Schritt d, wird von einer geschützten Hydroxy-Amin-Verbindung
der Struktur (27) die Schutzgruppe entfernt, wodurch sich eine Hydroxy-Amin-Verbindung
der Struktur (28) ergibt.
-
Die
Verwendung und Entfernung geeigneter Hydroxyschutzgruppen ist im
Fachgebiet bekannt und anerkannt und wird in Protecting Groups in
Organic Synthesis von T. Greene, Wiley-Interscience (1981), beschrieben.
-
Wie
für Durchschnittsfachleute
klar ist, kann unter Verwendung eines geeigneten Reduktionsmittels
für Amide,
wie Lithiumaluminiumhydrid, Diisobutylaluminiumhydrid oder Boran-Dimethylsulfid-Komplex,
entweder ein Aldehyd der Struktur (3), in der G1 -CH2- und G2 -C(O)-
ist, oder ein Alkohol der Struktur (25) direkt zu einer Hydroxy-Amin-Verbindung der Struktur
(28) reduziert werden, wie in Schema D, optionaler Schritt d, gelehrt.
-
In
Schema D, Schritt e, wird eine Hydroxy-Amin-Verbindung der Struktur
(28) zu einem Aldehyd der Struktur (3) oxidiert, in der G1 -CH2- und G2 -CH2- ist.
-
Oxidationen
von Alkoholen in Verbindungen, die tertiäre Amine enthalten, sind im
Fachgebiet bekannt und anerkant und werden in T. P. Burkholder und
P. L. Fuchs, J. Am. Chem. Soc. 112, 9601 (1990) und M. P. Kotick
et al., J. Med. Chem. 26, 1050 (1983), beschrieben.
-
Beispielsweise
werden zwei molare Äquivalente
Dimethylsulfoxid zu einer Lösung
von Trifluoressigsäureanhydrid
in Dichlormethan bei ungefähr –60°C zugetropft.
Nach dem Ende der Zugabe wird die Reaktion ungefähr zwei Minuten gerührt. Ein
molares Äquivalent
einer Hydroxy-Amin-Verbindung der Struktur (28) als Lösung in
Dichlormethan wird zugetropft. Nach dem Ende der Zugabe wird das
Reaktionsgemisch ungefähr 40
Minuten gerührt,
dann wird ein 3- bis 5facher Überschuss
an Triethylamin zugegeben. Das Reaktionsgemisch wird rühren gelassen,
wobei es sich im Verlauf von 1 Stunde bis 5 Stunden auf Zimmertemperatur
erwärmt.
Das Produkt kann mit im Fachgebiet bekannten Verfahren, wie Extraktion,
Verdampfen, Chromatographie und Umkristallisation, isoliert und
gereinigt werden.
-
BEISPIEL 75
-
(S)-N-Benzyl-N-methyl-2-[N-(t-butoxycarbonyl)-2-hydroxyethylamino-3-phenylpropionamid
-
Schema D, optionaler Schritt
a:
-
Man
vereinigt (S)-N-Benzyl-N-methyl-2-[N-(t-butoxycarbonyl)-2-oxoethylamino]-3-phenylpropionamid (5,0
mmol) und Natriumborhydrid (5,0 mmol) in Ethanol (20 ml). Man rührt 16 Stunden.
Man engt im Vakuum zu einem Rückstand
ein. Man verdünnt
den Rückstand
mit Ethylacetat und extrahiert mit 0,5 M Salzsäurelösung und Wasser. Man trennt
die Phasen, trocknet die organische Phase über MgSO4,
filtriert und verdampft im Vakuum, wodurch sich die Titelverbindung
ergibt.
-
BEISPIEL 76
-
(S)-N-Benzyl-N-methyl-2-[N-(t-butoxycarbonyl)-2-tetrahydropyran-2-yloxyethylamino]-3-phenylpropionamid
-
Schema D, optionaler Schritt
b:
-
Man
vereinigt (S)-N-Benzyl-N-methyl-2-[N-(t-butoxycarbonyl)-2-hydroxyethylamino]-3-phenylpropionamid
(4 mmol), p-Toluolsulfonsäure
(50 mg) und Dihydropyran (4 mmol) in wasserfreiem Dichlormethan.
Nach 8 Stunden verteilt man das Reaktionsgemisch zwischen Dichlormethan
und 0,5 M Natriumhydroxidlösung. Man
trennt die organische Phase ab und trocknet über MgSO4,
filtriert und verdampft im Vakuum, wodurch sich die Titelverbindung
ergibt.
-
BEISPIEL 77
-
(S)-N-Benzyl-N-methyl-2-[N-(t-butoxycarbonyl)-2-tetrahydropyran-2-ylpropylamino]-3-phenylpropionamid
-
Schema D, optionaler Schritt
b:
-
Man
vereinigt (S)-N-Benzyl-N-methyl-2-[N'-(t-butoxycarbonyl)-3-hydroxypropylamino]-3-phenylpropionamid
(4 mmol), p-Toluolsulfonsäure
(50 mg) und Dihydropyran (4 mmol) in wasserfreiem Dichlormethan. Nach
8 Stunden verteilt man das Reaktionsgemisch zwischen Dichlormethan
und 0,5 M Natriumhydroxidlösung.
Man trennt die organische Phase ab und trocknet über MgSO4,
filtriert und verdampft im Vakuum, wodurch sich die Titelverbindung
ergibt.
-
BEISPIEL 78
-
(S)-N-Benzyl-N-methyl-N-[N-(t-butoxycarbonyl)-2-tetrahydropyran-2-yloxyethylamino]-3-phenylpropylamin
-
Schema D, optionaler Schritt
c:
-
Man
vereinigt (S)-N-Benzyl-N-methyl-2-[N-(t-butoxycarbonyl)-2-tetrahydropyran-2-yloxyethylamino]-3-phenylpropionamid
(4 mmol) und Lithiumaluminiumhydrid (8 mmol) in Tetrahydrofuran
(20 mmol). Man erhitzt 48 Stunden unter Rückfluss. Man kühlt auf
Zimmertemperatur ab, gibt langsam Wasser (0,3 ml), 15%ige Natriumhydroxidlösung (0,3
ml) und Wasser (0,9 ml) hinzu. Man rührt, bis alles Reagens gequencht
ist. Man filtriert und verdampft im Vakuum zu einem Rückstand.
Man verteilt den Rückstand
zwischen Ethylacetat und Wasser. Man trennt die organische Phase
ab und trocknet über
MgSO4, filtriert und verdampft im Vakuum,
wodurch sich die Titelverbindung ergibt.
-
BEISPIEL 79
-
(S)-N-Benzyl-N-methyl-N-[N-(t-butoxycarbonyl)-2-tetrahydropyran-2-yloxypropylamino]-3-phenylpropylamin
-
Schema D, optionaler Schritt
c:
-
Man
vereinigt (S)-N-Benzyl-N-methyl-2-[N-(t-butoxycarbonyl)-2-tetrahydropyran-2-yloxypropylamino]-3-phenylpropionamid
(4 mmol) und Lithiumaluminiumhydrid (8 mmol) in Tetrahydrofuran
(20 mmol). Man erhitzt 48 Stunden unter Rückfluss. Man kühlt auf
Zimmertemperatur ab, gibt langsam Wasser (0,3 ml), 15%ige Natriumhydroxidlösung (0,3
ml) und Wasser (0,9 ml) hinzu. Man rührt, bis alles Reagens gequencht
ist. Man filtriert und verdampft im Vakuum zu einem Rückstand.
Man verteilt den Rückstand
zwischen Ethylacetat und Wasser. Man trennt die organische Phase
ab und trocknet über
MgSO4, filtriert und verdampft im Vakuum,
wodurch sich die Titelverbindung ergibt.
-
BEISPIEL 80
-
(S)-N-Benzyl-N-methyl-N-[N-(t-butoxycarbonyl)-2-hydroxyethylamino]-3-phenylpropylamin
-
Schema D, optionaler Schritt
d:
-
Man
vereinigt (S)-N-Benzyl-N-methyl-N-[N-(t-butoxycarbonyl)-2-tetrahydropyran-2-yloxyethylamino)-3-phenylpropylamin
(2,0 mmol) und p-Toluolsulfonsäure
(3 mmol) in Methanol (20 ml). Nach 8 Stunden verdampft man im Vakuum.
Man verteilt den Rückstand
zwischen Dichlormethan und 0,5 M Natriumhydroxidlösung. Man
trennt die organische Phase ab und trocknet über MgSO4,
filtriert und verdampft im Vakuum, wodurch sich die Titelverbindung
ergibt.
-
BEISPIEL 81
-
(S)-N-Benzyl-N-methyl-N-[N-(t-butoxycarbonyl)-2-hydroxypropylamino)-3-phenylpropylamin
-
Schema D, optionaler Schritt
d:
-
Man
vereinigt (S)-N-Benzyl-N-methyl-N-[N-(t-butoxycarbonyl)-2-tetrahydropyran-2-yloxypropylamino]-3-phenylpropylamin
(2,0 mmol) und p-Toluolsulfonsäure
(3 mmol) in Methanol (20 ml). Nach 8 Stunden verdampft man im Vakuum.
Man verteilt den Rückstand
zwischen Dichlormethan und 0,5 M Natriumhydroxidlösung. Man
trennt die organische Phase ab und trocknet über MgSO4,
filtriert und verdampft im Vakuum, wodurch sich die Titelverbindung
ergibt.
-
BEISPIEL 82
-
(S)-N-Methyl-N-benzyl-N-[2-[N'-(t-butoxycarbonyl)-2-oxoethylamino]-3-phenylpropylamin
-
Schema D, Schritt e:
-
Man
vereinigt Trifluoressigsäureanhydrid
(4,8 mmol) mit Dichlormethan (10 ml) und kühlt auf –60°C ab. Man tropft eine Lösung von
Dimethylsulfoxid (9,6 mmol) in Dichlormethan (1 ml) zu, während die
Temperatur unter –55°C gehalten
wird. Nach dem Ende der Zugabe rührt
man 2 Minuten. Man gibt eine Lösung
von (S)-N-Benzyl-N-methyl-N-[N-(t-butoxycarbonyl)-2-hydroxyethylamino]-3-phenylpropylamin
(2 mmol) in Dichlormethan hinzu und rührt 45 Minuten. Man kühlt die
Reaktion auf –78°C ab und
tropft Triethylamin (10 mmol) zu. Man lässt die Reaktion auf Zimmertemperatur
erwärmen
und rührt
45 Minuten. Man gießt
die Reaktion in Wasser. Man extrahiert dieses Gemisch mit Diethylether.
Man trennt die organische Phase ab und trocknet über MgSO4,
filtriert und verdampft im Vakuum, wodurch sich die Titelverbindung
ergibt.
-
BEISPIEL 83
-
(S)-N-Methyl-N-benzyl-N-[2-[N'-(t-butoxycarbonyl)-2-oxopropylamino]-3-phenylpropylamin
-
Man
vereinigt Trifluoressigsäureanhydrid
(4,8 mmol) mit Dichlormethan (10 ml) und kühlt auf –60°C ab. Man tropft eine Lösung von
Dimethylsulfoxid (9,6 mmol) in Dichlormethan (1 ml) zu, während die
Temperatur unter –55°C gehalten
wird. Nach dem Ende der Zugabe rührt
man 2 Minuten. Man gibt eine Lösung
von (S)-N-Benzyl-N-methyl-N-[N-(t-butoxycarbonyl)-2-hydroxypropylamino]-3-phenylpropylamin
(2 mmol) in Dichlormethan hinzu und rührt 45 Minuten. Man kühlt die
Reaktion auf –78°C ab und
tropft Triethylamin (10 mmol) zu. Man lässt die Reaktion auf Zimmertemperatur
erwärmen
und rührt
45 Minuten. Man gießt
die Reaktion in Wasser. Man extrahiert dieses Gemisch mit Diethylether.
Man trennt die organische Phase ab und trocknet über MgSO4,
filtriert und verdampft im Vakuum, wodurch sich die Titelverbindung
ergibt.
-
Die
Tachykinine sind eine Klasse von Neuropeptiden, die eine gemeinsame
C-terminale Sequenz Phe-Xaa-Gly-Leu-Met-NH2 aufweisen.
Die Tachykinine sind weit verbreitet im peripheren und zentralen
Nervensystem, wo sie an mindestens drei Rezeptortypen binden. Die
NK1-, NK2- bzw.
NK3-Rezeptoren werden über die bevorzugte Bindungsaffinität von Substanz
P, Neurokinin A (NKA) bzw. Neurokinin B (NKB) definiert.
-
Ein
Antagonismus der Wirkungen von Substanz P an ihrem bevorzugten Rezeptor,
d. h. NK1, behindert nicht die Wirkungen
von NKA an seinem bevorzugten Rezeptor, d. h. NK2.
Deshalb sind die potenziellen Vorteile eines Antagonisten mit Affinität für sowohl
die NK1- als
auch NK2-Rezeptoren die Verringerung oder
Verhinderung klinischer Manifestationen von Erkrankungen und Zuständen, die über beide
Rezeptoren vermittelt werden.
-
Die
Verwendung von Tachykininantagonisten ist als Therapie für eine Vielzahl
von Erkrankungen und Zuständen,
die durch Tachykinin vermittelt werden, indiziert, einschließlich: Zystitis;
Bronchokonstriktion; Überempfindlichkeitsreaktionen;
Behandlung von Schmerz; periphere Neuropathie; post-herpetische
Neuralgie; ungünstige
immunologische Reaktionen; Erkrankungen der Atemwege, wie Asthma,
Bronchitis, Husten, Rhinitis und Allergien und dergleichen; Augenerkrankungen,
wie Konjunktivitis und Conjunctivitis vernalis; Hauterkrankungen,
wie Kontaktdermatitis, atopische Dermatitis und Urtikaria; entzündliche
Erkrankungen, wie rheumatoide Arthritis und Osteoarthritis und dergleichen;
gastrointestinale Zustände,
wie Morbus Crohn, Emesis und Colitis ulcerosa; Zustände auf
Grund von Vasodilatation, wie Angina und Migräne; und Erkrankungen und Zustände des
zentralen Nervensystems, wie Angst, Depression, Psychose, Schizophrenie,
Demenz.
-
Es
versteht sich, dass Erkrankungen und Zustände, die durch Tachykinin vermittelt
werden, diejenigen Erkrankungen und Zustände sind, bei denen die Tachykinine
entweder als ganzes oder zum Teil an deren klinischen Manifestationen)
beteiligt sind. Darüber
hinaus ist die Beteiligung der Tachykinine nicht notwendigerweise
ursächlich
für eine
bestimmte Erkrankung und Zustand, die durch Tachykinin vermittelt
wird. Tachykininantagonisten sind zur Steuerung oder zur Bereitstellung
von therapeutischer Erleichterung dieser Erkrankungen und Zustände, die
durch Tachykinin vermittelt werden, verwendbar.
-
Die
vorliegende Erfindung stellt neue und nützliche Tachykininantagonisten
der Formel (1) oder Stereoisomere oder pharmazeutisch verträgliche Salze
davon bereit. Insbesondere stellt die vorliegende Erfindung Verbindungen
der Formel (1) bereit, die NK1-Rezeptorantagonisten,
NK2-Rezeptorantagonisten und sowohl NK1- als auch NK2- Rezeptorantagonisten
sind.
-
Die
vorliegende Erfindung ermöglicht
also ein Verfahren zur Behandlung von Erkrankungen und Zuständen, die
durch Tachykinin vermittelt werden, an einem Patienten, der es benötigt, umfassend
das Verabreichen einer therapeutisch wirksamen Menge einer Verbindung
der Formel (1) an den Patienten. Zahlreiche Erkrankungen und Zustände, die
als hier zu behandelnd beschrieben werden, sind Fachleuten bekannt
und anerkannt. Es wird auch anerkannt, dass Fachleute die betreffenden
Erkrankungen und Zustände
beeinflussen können,
indem sie einen Patienten, der zurzeit an diesen Erkrankungen oder
Zuständen
leidet, behandeln oder indem sie einen Patienten, der an diesen
Erkrankungen oder Zuständen
leidet, prophylaktisch mit einer therapeutisch wirksamen Menge der
Verbindungen der Formel (1) behandeln.
-
Wie
hier verwendet, bezieht sich der Begriff „Patient" auf ein warmblütiges Tier, wie ein Säugetier,
das an einer speziellen, durch Tachykinin vermittelten Erkrankung
oder Zustand leidet. Es versteht sich, dass Meerschweine, Hunde,
Katzen, Ratten, Mäuse,
Pferde, Rinder, Schafe und Menschen Beispiele für Tiere innerhalb des Umfangs
der Bedeutung des Begriffs sind.
-
Wie
hier verwendet, bezieht sich der Begriff „therapeutisch wirksame Menge" einer Verbindung
der Formel (1) auf eine Menge, die bei der Kontrolle der durch Tachykinin
vermittelten Erkrankungen und Zustände wirksam ist. Der Begriff „Kontrollieren" soll sich auf alle
Verfahren beziehen, bei denen es eine Verlangsamung, Unterbrechung,
Hemmung oder Anhalten der Progression der hier beschriebenen Erkrankungen
und Zustände
geben kann, zeigt aber nicht notwendigerweise eine totale Beseitigung
aller Erkrankungs- und Zustandssymptome an und soll die prophylaktische
Behandlung der durch Tachykinin vermittelten Erkrankungen und Zustände einschließen.
-
Eine
therapeutisch wirksame Menge kann einfach durch den behandelnden
Arzt als Fachmann durch Anwendung herkömmlicher Verfahren und durch
Beachten von unter analogen Umständen
erhaltenen Ergebnissen bestimmt werden. Bei der Bestimmung der therapeutisch
wirksamen Menge, der Dosis, werden vom behandelnden Arzt eine Anzahl
von Faktoren in Betracht gezogen, einschließlich, aber nicht begrenzt
auf die Säugetierspezies;
seine Größe, Alter
und allgemeiner Gesundheitszustand; die spezielle, betreffende Erkrankung;
der Grad der oder die Beteiligung an oder die Schwere der Erkrankung;
die Antwort des einzelnen Patienten; die jeweilige verabreichte
Verbindung; der Modus der Verabreichung; die Bioverfügbarkeitseigenschaften
des verabreichten Präparats;
das gewählte
Dosierungsschema; die Verwendung von Begleitmedikation; und andere
relevante Umstände.
-
Von
einer therapeutisch wirksamen Menge einer Verbindung der Formel
(1) wird erwartet, dass sie von etwa 0,1 Milligramm pro Kilogramm
Körpergewicht
pro Tag (mg/kg/Tag) bis etwa 100 mg/kg/Tag variiert. Bevorzugte
Mengen können
von Fachleuten bestimmt werden.
-
Bei
der Behandlung eines Patienten, der an den vorstehend beschriebenen,
durch Tachykinin vermittelten Erkrankungen und Zuständen leidet,
kann eine Verbindung der Formel (1) in jeder Form oder Weise verabreicht
werden, die die Verbindung in einer wirksamen Menge bioverfügbar macht,
einschließlich
orale, Inhalations- und parenterale Wege. Beispielsweise können Verbindungen
der Formel (1) oral, durch Inhalation von Aerosol oder trockenem
Pulver, subkutan, intramuskulär,
intravenös,
transdermal, intranasal, rektal, topisch und dergleichen verabreicht
werden. Orale oder Inhalationsverabreichung wird im Allgemeinen
zur Behandlung von Erkrankungen und Zuständen der Atemwege, z. B. Asthma,
bevorzugt. Fachleute für
die Herstellung von Formulierungen können in Abhängigkeit von den speziellen
Eigenschaften der gewählten
Verbindung, der zu behandelnden Erkrankung oder Zustand, dem Stadium
der Erkrankung oder Zustand und anderen relevanten Umständen einfach
die passende Form und Weise der Verabreichung wählen. (Remington's Pharmaceutical
Sciences, 18. Auflage, Mack Publishing Co. (1990)).
-
Die
erfindungsgemäßen Verbindungen
können
allein oder in Form eines Arzneimittels in Kombination mit pharmazeutisch
verträglichen
Trägern
oder Excipienten verabreicht werden, deren Anteil und Art durch
die Löslichkeit
und chemischen Eigenschaften der gewählten Verbindung, den gewählten Verabreichungsweg
und pharmazeutische Standardverfahren bestimmt werden. Die erfindungsgemäßen Verbindungen
können,
auch wenn sie als solche wirksam sind, zu Zwecken von Stabilität, Einfachheit
der Kristallisation, erhöhter
Löslichkeit
und dergleichen in Form ihrer pharmazeutisch verträglichen
Salze formuliert und verabreicht werden, wie Säureadditionssalze oder Basenadditionssalze.
-
In
einer weiteren Ausführungsform
stellt die vorliegende Erfindung Arzneimittel bereit, umfassend
eine therapeutisch wirksame Menge einer Verbindung der Formel (1)
im Gemisch oder andersartig in Verbindung mit einem oder mehreren
pharmazeutisch verträglichen
Trägern
oder Excipienten.
-
Die
Arzneimittel werden auf eine Weise hergestellt, die im Fachgebiet
der Pharmazie bekannt ist. Der Träger oder Excipient kenn ein
festes, halbfestes oder flüssiges
Material sein, das als ein Vehikel oder Medium für den Wirkstoff dienen kann.
Geeignete Träger
oder Excipienten sind im Fachgebiet bekannt. Das Arzneimittel kann
für orale,
Inhalations-, parenterale oder topische Verwendung angepasst sein
und kann dem Patienten in Form von Tabletten, Kapseln, Aerosolen,
Inhalanda, Suppositorien, Lösung,
Suspensionen oder dergleichen verabreicht werden.
-
Die
erfindungsgemäßen Verbindungen
können
oral beispielsweise mit einem inerten Verdünnungsmittel oder mit einem
essbaren Träger
verabreicht werden. Sie können
in Gelatinekapseln eingeschlossen oder zu Tabletten verpresst werden.
Zum Zwecke der oralen therapeutischen Verabreichung können die
Verbindungen mit Excipienten vermischt und in Form von Tabletten,
Pastillen, Kapseln, Elixiren, Suspensionen, Sirupen, Waffeln, Kaugummis
und dergleichen verwendet werden. Diese Präparate sollten mindestens 4%
der erfindungsgemäßen Verbindung
als den Wirkstoff enthalten. Dies kann aber in Abhängigkeit
von der speziellen Form variiert werden und kann günstigerweise
zwischen 4% bis etwa 70% des Gewichts der Einheit betragen. Die
Menge an der Verbindung, die in Zusammensetzungen vorliegt, ist
so, dass eine geeignete Dosierung erhalten wird. Bevorzugte Zusammensetzungen
und Präparate
gemäß der vorliegenden
Erfindung können
von einem Fachmann bestimmt werden.
-
Die
Tabletten, Pillen, Kapseln, Pastillen und dergleichen können auch
einen oder mehrere der folgenden Hilfsstoffe enthalten: Bindemittel,
wie mikrokristalline Cellulose, Tragantgummi oder Gelatine; Excipienten, wie
Stärke
oder Lactose, Sprengmittel, wie Alginsäure, Primogel, Maisstärke und
dergleichen; Schmiermittel, wie Magnesiumstearat oder Sterotex;
Fließregulierungsmittel,
wie kolloidales Siliziumdioxid; und Süßstoffe, wie Saccharose oder
Saccharin, können
zugegeben wereden oder ein Geschmackstoff, wie Pfefferminz, Methylsalicylat
oder Orangengeschmack. Wenn die Dosierungseinheit eine Kapsel ist,
kann sie außer
den Materialien des vorstehenden Typs einen flüssigen Träger, wie Polyethylenglykol
oder ein fettes Öl,
enthalten. Andere Darreichungseinheitsformen können andere verschiedene Materialien
enthalten, die die physikalische Form der Dosierungseinheit modifizieren,
beispielsweise als Beschichtungen. So können Tabletten oder Pillen mit
Zucker, Schellack oder anderen magensaftresistenten Beschichtungsmitteln
beschichtet sein. Ein Sirup kann außer den erfindungsgemäßen Verbindungen
Saccharose als Süßstoff und
bestimmte Konservierungsmittel, Farbstoffe und Färbemittel und Geschmacksstoffe
enthalten. Materialien, die bei der Herstellung dieser verschiedenen
Zusammensetzungen verwendet werden, sollten pharmazeutisch rein
und in den verwendeten Mengen nicht toxisch sein.
-
Zum
Zwecke der parenteralen therapeutischen Verabreichung können die
erfindungsgemäßen Verbindungen
in eine Lösung
oder Suspension eingebracht werden. Diese Präparate sollten 0,1% einer erfindungsgemäßen Verbindung
enthalten. Dies kann aber zwischen 0,1 und etwa 50% ihres Gewichts
variiert werden. Die Menge an der Verbindung der Formel (1), die
in Zusammensetzungen vorliegt, ist so, dass eine geeignete Dosierung
erhalten wird. Bevorzugte Zusammensetzungen und Präparate können vom
Fachmann bestimmt werden.
-
Die
erfindungsgemäßen Verbindungen
können
auch durch Inhalation verabreicht werden, wie durch Aerosol oder
als trockenes Pulver. Die Abgabe kann durch ein verflüssigtes
oder verdichtetes Gas oder durch ein geeignetes Pumpensystem erfolgen,
das die erfindungsgemäßen Verbindungen
oder eine Formulierung davon abgibt. Formulierungen zur Verabreichung
durch Inhalation von Verbindungen der Formel (1) können in Einzelphasen-,
zweiphasigen oder dreiphasigen Systemen abgeliefert werden. Ein
Vielzahl von Systemen ist zur Verabreichung durch Aerosol der Verbindungen
der Formel (1) verfügbar.
Trockenpulverformulierungen werden entweder durch Pelletieren oder
Vermahlen der Verbindung der Formel (1) auf eine geeignete Teilchengröße oder
durch Vermischen der pelletierten oder vermahlenen Verbindung der
Formel (1) mit einem geeigneten Trägermaterial, wie Lactose und
dergleichen, hergestellt. Die Abgabe durch Inhalation schließt den notwendigen
Behälter,
Aktivatoren, Ventile, Unterbehälter
und dergleichen ein. Bevorzugte Aerosol- und Trockenpulverformulierungen
zur Verabreichung durch Inhalation können vom Fachmann bestimmt
werden.
-
Die
erfindungsgemäßen Verbindungen
können
auch topisch verabreicht werden und in diesem Fall kann der Träger geeigneterweise
eine Lösung,
Salbe oder Gelgrundlage umfassen. Die Grundlage kann beispielsweise
eines oder mehrere der folgenden umfassen: Vaselin, Lanolin, Polyethylenglykole,
Bienenwachs, Mineralöl
und dergleichen; Verdünnungsmittel,
wie Wasser und Alkohole; Emulgatoren; und Stabilisatoren. Topische
Formulierungen können
eine Konzentration der Formel (1) oder ihres pharmazeutisch verträglichen Salzes
von etwa 0,1 bis etwa 10% Gew./Vol. (Gewicht pro Volumeneinheit)
enthalten.
-
Die
Lösungen
oder Suspensionen können
auch einen oder mehrere der folgenden Hilfsstoffe einschließen: sterile
Verdünnungsmittel,
wie Wasser zur Injektion, Kochsalzlösung, nicht flüchtige Öle, Polyethylenglykole,
Glycerin, Propylenglykol oder andere synthetische Lösungsmittel;
antibakterielle Mittel, wie Benzylalkohol oder Methylparaben; Antioxidantien,
wie Ascorbinsäure
oder Natriumbisulfit; Chelatbildner, wie Ethylendiamintetraessigsäure; Puffer,
wie Acetate, Citrate oder Phosphate und Mittel zur Einstellung der
Tonizität, wie
Natriumchlorid oder Dextrose. Das parenterale Präparat kann in Ampullen, Einwegspritzen
oder Mehrfachdosisflaschen aus Glas oder Kunststoff eingeschlossen
sein.
-
Der
Fachmann kann die NK1-Rezeptor- und NK2-Rezeptoraffinität wie folgt in vitro bestimmen.
Die NK1-Rezeptoraffinität von Tachykininantagonisten
wird an Meerschweinchenlungen (Keystone Biologicals, Cleveland,
OH) bewertet und die Affinität
für den
NK2-Rezeptor wird an HSKR-1-Zellen bewertet
(die Maus-3T3-Fibroblasten sind, die den menschlichen jejunalen
NK2-Rezeptor exprimieren). Gewebe oder Zellen werden
mit einem Polytron in 15 Volumina 50 mM Tris-HCl-Puffer (pH-Wert
7,4, 4°C)
homogenisiert und zentrifugiert. Das Pellet wird erneut in Tris-HCl-Puffer
suspendiert und wird zentrifugiert; das Pellet wird zweimal durch
Resuspendieren gewaschen. Das endgültige Pellet wird in einer
Konzentration von 40 mg/ml bei Geweben und 20 mg/ml bei Zellen in
Inkubationspuffer resuspendiert und verbleibt vor der Verwendung
mindestens 15 min bei Zimmertemperatur. Die Rezeptorbindung wird
durch Zugabe von 250 μl
Membranpräparat
in doppelter Ausfertigung zu 0,1 nM der folgenden Radioliganden
gestartet: Substanz P, markiert mit 125I-Bolton
Hunter Lys-3, und 125Iodohistidyl-1-neurokinin
A; in einem Endvolumen von 500 μl
Puffer, der 50 mM Tris-HCl (pH-Wert 7,4 bei Zimmertemperatur), 0,1%
Rinderserumalbumin, 2 mM MnCl2, 40 μg/ml Bacitracin,
4 μg/ml Leupeptin
und Chymostatin, 10 μM
Thiorphan und verschiedene Dosen der mutmaßlichen Tachykininantagonisten
enthält.
Die Inkubationen werden 90 min (NK1-Rezeptorassays)
oder 2 Std. (NK2-Rezeptorassay) bei Zimmertemperatur
durchgeführt;
die Bindung wird durch Zugabe von 50 mM Tris-HCl-Puffer (pH-Wert
7,4, 4°C)
und Filtration im Vakuum durch GF/B-Filter, die mit 0,1% Polyethylenimin
(NK1-Rezeptorassays) oder 0,5% Rinderserumalbumin
(NK2-Rezeptorassay) vorgetränkt wurden,
beendet. Die an den Filter gebundene Radioaktivität wird in
einem Gammazähler
quantifiziert. Die nicht spezifische Bindung ist als Bindung in
Gegenwart von 1 μM
Substanz P oder Neurokinin A definiert. Die spezifische Bindung
wird durch Subtraktion der nicht spezifischen Bindung von der Gesamtbindung
berechnet. Die Konkurrenz um die Bindung von iodiertem SP oder NKA
durch Testverbindungen oder Standards wird als Prozentsatz dieser
maximalen Konkurrenz ausgedrückt.
IC50-Werte (Konzentration, die zur Hemmung
von 50% der Rezeptorbindung erforderlich sind) werden für jede der
Testverbindungen durch nicht lineare Regression mittels eines iterativen
Kurvenanpassungsprogramms erzeugt (GraphPAD Inplot, San Diego, CA).
-
Der
Fachmann kann auch den NK1-Rezeptor- und
NK2-Rezeptorantagonismus wie folgt in vitro
bestimmen. Durch Tachykinin vermittelte Phosphatidylinositol-(PI,
Inositolphosphat)akkumulation wird in UC11- oder SKLKB82#3-Zellen
in Gegenwart und Abwesenheit von NK1- bzw.
NK2-Rezeptorantagonisten gemessen. Die Zellen
werden zwei oder drei Tage vor dem Assay auf Platten mit 24 Vertiefungen
mit 125.000 Zellen/Vertiefung gesät. Die Zellen werden 20–24 Stunden
vor dem Assay mit 0,5 ml 0,2 μM
Myo[2-3H(N)]inositol beladen. Die kultivierten
Zellen werden bei 37°C
in einer Umgebung mit 95% O2 und 5% CO2 gehalten. Am Tag des Assays wird das Medium
abgesaugt und die Zellen werden in RPMI-1640-Medium (bei UC11-Zellen) oder D-MEM/F12-Medium
(bei SKLKB82#3-Zellen) (das 40 μg/ml
Bacitracin, jeweils 4 μg/ml
Leupeptin und Chymostatin, 0,1% Rinderserumalbumin und 10 μM Thiorphan
und 10 mM LiCl enthält)
inkubiert und die Testverbindung wird zugegeben. Nach 15 min wird
mit verschiedenen Konzentrationen SP zu den UC11-Zellen oder NKA zu
den SKLKB82#3-Zellen gegeben, um die Reaktion zu starten. Nach 60
min Inkubation bei Zimmertemperatur wird die Reaktion durch Entfernen
des Mediums und Zugabe von 0,1 ml Methanol in alle Vertiefungen beendet.
Zwei Aliquote Methanol (0,5 ml) werden zu den Vertiefungen gegeben,
um die Zellen zu ernten, gefolgt von Chloroform (1 ml), dann doppelt
destilliertes Wasser (0,5 ml). Die Proben werden verwirbelt, zentrifugiert
und 0,9 ml wässrige
(obere) Phase wird entfernt und zu 2 ml doppelt destilliertem H2O gegeben. Das Gemisch wird verwirbelt und
auf eine 50% Bio-Rad AG 1-X8 (Formiatform, 100–200 mesh) Austauschersäule (Bio-Rad
Laboratories, Hercules, CA) geladen. Die Säulen werden der Reihenfolge
nach gewaschen mit: 1) 10 ml doppelt destilliertem Wasser, 2) 5
ml 5 mM Dinatriumtetraborat/60 mM Natriumformiat und 3) 5 ml 1 M Ammoniumformiat/0,1
M Ameisensäure.
Die dritte Elution wird gesammelt und 1 ml in 7 ml Szintillationsflüssigkeit
gezählt.
Ein 50 μl
Aliquot der organischen (unteren) Phase wird entfernt, in einem
Szintillationsvial getrocknet und in 7 ml Szintillationsflüssigkeit
gezählt.
-
Das
Verhältnis
von DPM im Aliquot der wässrigen
Phase (gesamte Inositolphosphate) zum DPM im 50 μl Aliquot der organischen Phase
(gesamtes eingebautes [3H]Inositol) wird
für jede
Probe berechnet. Die Daten werden als Prozent durch Agonist induzierte
Akkumulation von [3H]-Inositolphosphaten über Grundniveaus
ausgedrückt.
Die Verhältnisse
in Gegenwart von Testverbindung und/oder Standards werden mit den Verhältnissen
für Kontrollproben
(d. h. ohne stimulierenden Agonist) verglichen. Dosis-Wirkungs-Graphen
werden ausgearbeitet und die Fähigkeit
der Testverbindungen, den durch Tachykinin induzierten Phosphatidyinositolumsatz
zu hemmen, wird mit Hilfe eines Computerprogramms bestimmt. Die
Daten werden als Prozent Stimulation der gesamten Inositolphosphatakkumulation über Grundniveaus
ausgedrückt
und auf die maximale Antwort, die von SP hervorgerufen wird, normalisiert.
Die Schild-Analyse wird unter Verwendung der Dosis-Wirkungs-Kurven
durchgeführt,
wodurch ein Wert erhalten wird, der die Stärke eines kompetitiven Antagonisten
angibt und als pA2 ausgedrückt wird,
was der negative Logarithmus der molaren Konzentration des Antagonisten
ist, die die Wirkung einer Dosis des Agonisten auf die Hälfte der
Wirkung verringert, die bei der Dosis des Agonisten erwartet wird.
-
Der
Fachmann kann bestimmen, dass die erfindungsgemäßen Verbindungen in vivo NK1-Rezeptorantagonisten
sind, indem die Fähigkeit
der Verbindung bewertet wird, die durch SP induzierte Plasmaproteinextravasation
an der Meerschweinchentrachea zu hemmen. Die durch SP induzierte
Proteinleckage durch postkapillare Venolen wird bewertet, indem
die durch Agonisten induzierte Evans Blue-Farbstoffakkumulation
in der Meerschweinchentrachea gemessen wird. Die Tiere werden mit
Pentobarbital anästhesiert,
dann wird ihnen Evans Blue-Farbstoff injiziert (20 mg/kg, i.v.,
bereitet in 0,9% NaCl-Lösung). Eine
Minute nach der Verabreichung des Farbstoffs wird der Antagonist
verabreicht (i.v.), gefolgt von SP (0,3 nmol/kg, i.v.), und nach
5 Min. wird überschüssiger Farbstoff
durch transkardiale Perfusion mit 50 ml 0,9% NaCl-Lösung aus
dem Kreislauf entfernt. Die Trachea und Bronchi principales werden
entfernt, trocken getupft und gewogen. Die Quantifizierung des Farbstoffs
wird spektrophotometrisch (620 nm) durchgeführt, nachdem die Gewebe 24
Std. bei 50°C in
Formamid extrahiert wurden. Die Werte werden vom Hintergrund subtrahiert
(lediglich Farbstoff, kein Agonist). Die ED50 (Dosis
der Verbindung, die die durch SP induzierte Plasmaproteinextravasation
um 50% hemmt) wird durch lineare Regressionsanalyse berechnet.
-
Der
Fachmann kann bestimmen, dass die erfindungsgemäßen Verbindungen in vivo NK2-Rezeptorantagonisten
sind, indem die Fähigkeit
der Verbindungen bewertet wird, die durch NKA induzierten Auswirkungen auf
die Atmung zu hemmen. Außerdem
kann sowohl NK1- als auch NK2-Antagonismus
nach der Verabreichung von Capsaicin bewertet werden, von dem bekannt
ist, dass es sowohl SP als auch NKA aus sensorischen Nerven des
Luftweges freisetzt. Der Antagonismus von durch NKA und Capsaicin
induzierten Auswirkungen auf die Atmung bei Meerschweinchen, die
bei Bewusstsein sind, wird wie folgt durchgefüht. Die in vivo-Experimente
werden unter Verwendung männlicher
Duncan Hartley-Meerschweinchen
(250–350
g) durchgeführt.
Veränderungen
im bewussten Atemmuster werden bei vier Tieren gleichzeitig überwacht,
wobei eine modifizierte Ganzkörperplethysmographie
eingesetzt wird, die aus vier kleinen Plexiglaskästen besteht, die jeweils über Validyne
DP 45-16 Druckdifferenzwandler mit einem Bezugskasten verbunden
sind. Die 4 Kästen
sind mit einem Luftzufuhrrohr (wird auch zur Aerosolabgabe verwendet)
und einem Abluftrohr ausgerüstet.
Zufuhr- und Abluftrohre haben dieselbe Länge und enge Bohrung und gehen
aus einer gemeinsamen Zufuhrkammer hervor und werden in eine gemeinsame
Abluftkammer entlüftet.
Dieses System wird verwendet, um zu gewährleisten, dass Fluktuationen
der Zufuhrluft und des Atmosphärendrucks
in Phase bleiben und von den Druckdifferenzwandlern aus dem Nettosignal
entfernt werden. Die analogen Drucksignale werden über einen
Data Translation DT2821 A/D-Wandler digitalisiert. Die Daten werden
mit einer Rate von 100 Proben/Sekunde/Tier gesammelt. Jeder Zyklus
der Druckänderung
wird unter Verwendung der folgenden Parameter analysiert: Steigung
von Anstieg und Abfall, die zwischen minimalen und maximalen Drücken bestimmt
wird, das Verhältnis der
Steigung des Anstiegs zu der des Abfalls und die Größe der Änderung
zwischen dem anfänglichen
Minimaldruck und Zyklusspitzendruck. Unter Verwendung dieser Werte
(und Beobachtung der Tiere) werden die Druckzyklen mit einem PCAT
286, auf dem ein System V UNIX Betriebssystem läuft, als normale Atemzüge, forcierte
Ausatmung (erkennbar durch Weitung des Abdomens), bemerkenswerte
Vorkommnisse bei der Atmung (SRE: significant respiratory events; üblicherweise
Husten, weniger oft Niesen oder Keuchen, die durch vorübergehende,
extrem große
Druckanstiege gekennzeichnet sind, die vom Rauschen unterscheidbar
sind) und Bewegung/Rauschen gekennzeichnet. Dyspnoe wird als signifikanter,
anhaltender Anstieg des Plethysmographendrucks definiert, der mit
einer erkennbaren Verlagerung zu angestrengter Atmung des Tieres
verbunden ist.
-
Im
Verlauf eines typischen Experiments, bei dem das Antwortverhalten
der Luftwege auf verschiedene Bronchokonstriktoren untersucht wird,
werden die Aerosole 19 Min. (0,33 ml/min) unter Verwendung eines
DeVilbiss Ultraneb 99 Ultraschallverneblers abgegeben und der Plethysmographendruck
während
dieser Zeit überwacht.
Vor dem Vernebeln werden 1 Min. lang Atemdruckdaten im Ruhezustand
gesammelt, wodurch ein Grundlininedruck ermittelt wird. In vorbereitenden
Experimenten werden verschiedene Konzentrationen der Bronchokonstriktoren
bewertet und die Konzentration gewählt, die die Anzahl der Tiere,
die Dyspnoe zeigten, maximierte, aber die Schwere der Antwort minimierte.
Deshalb wird Neurokinin A mit einer Endkonzentration von 0,05% und
Capsaicin mit 0,001% abgegeben. Das Vehikel zum Verneblen aller
Bronchokonstriktoren ist mit Phosphat gepufferte Kochsalzlösung (pH-Wert
7,4), die selbst keine Wirkung auf die Atemwege hervorruft. Mutmaßliche Tachykininrezeptorantagonisten
werden 20 Min. vor dem Beginn der Aerosolexposition verabreicht
(i.v.) oder oral 1 Stunde vor dem Beginn der Aerosolexposition.






















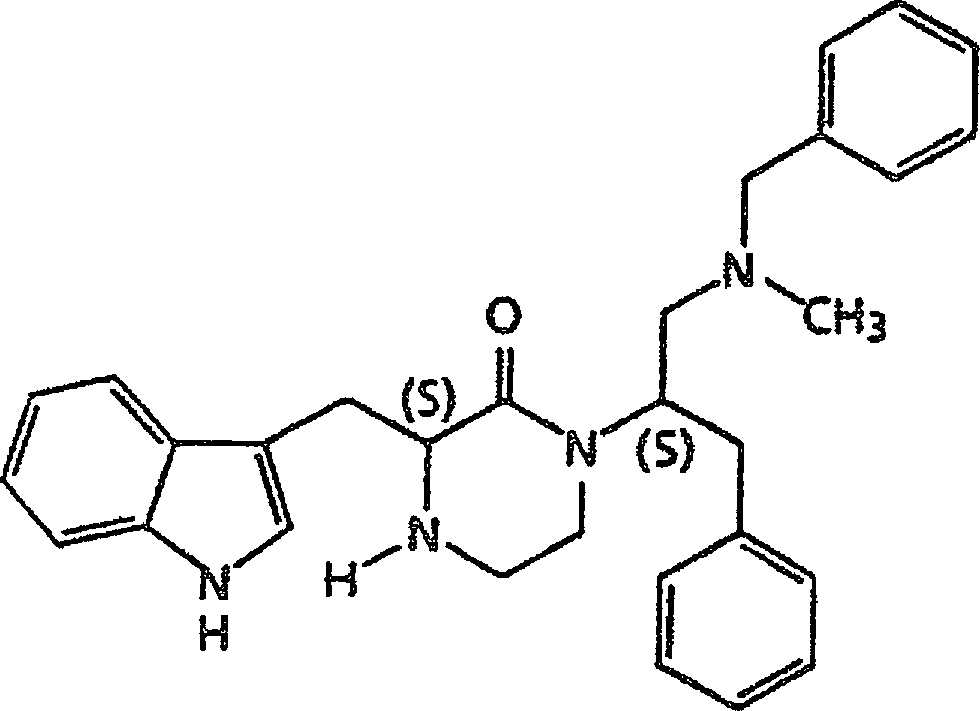






 wobei Z1 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; Ar2 ein Rest ist, ausgewählt aus:
wobei Z1 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; Ar2 ein Rest ist, ausgewählt aus: wobei Z2 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; Ar3 ein Rest ist, ausgewählt aus:
wobei Z2 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; Ar3 ein Rest ist, ausgewählt aus: wobei Z3 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; R6 Wasserstoff, C1-C4-Alkyl oder -CHO ist; R1 Wasserstoff, C1-C4-Alkyl, -(CH2)qAr4 oder -CH2C(O)Ar4 ist, wobei q eine ganze Zahl von 1 bis 4 ist und Ar4 ein Rest der Formel
wobei Z3 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; R6 Wasserstoff, C1-C4-Alkyl oder -CHO ist; R1 Wasserstoff, C1-C4-Alkyl, -(CH2)qAr4 oder -CH2C(O)Ar4 ist, wobei q eine ganze Zahl von 1 bis 4 ist und Ar4 ein Rest der Formel ist, wobei Z4 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; oder Stereoisomere oder pharmazeutisch verträgliche Salze davon; mit der Maßgabe, daß, falls G1 -C(O)- ist, dann G2 nicht -C(O)- ist; und mit der weiteren Maßgabe, daß, falls G3 -CH2- ist, dann G1 und G2 -CH2- sind.
ist, wobei Z4 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; oder Stereoisomere oder pharmazeutisch verträgliche Salze davon; mit der Maßgabe, daß, falls G1 -C(O)- ist, dann G2 nicht -C(O)- ist; und mit der weiteren Maßgabe, daß, falls G3 -CH2- ist, dann G1 und G2 -CH2- sind.
 wobei Z1 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; Ar2 ein Rest ist, ausgewählt aus:
wobei Z1 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; Ar2 ein Rest ist, ausgewählt aus: wobei Z2 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; Ar3 ein Rest ist, ausgewählt aus:
wobei Z2 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; Ar3 ein Rest ist, ausgewählt aus: wobei Z3 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; R6 Wasserstoff, C1-C4-Alkyl oder -CHO ist; R1 Wasserstoff, C1-C4-Alkyl, -(CH2)qAr4 oder -CH2C(O)Ar4 ist, wobei q eine ganze Zahl von 1 bis 4 ist und Ar4 ein Rest der Formel
wobei Z3 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; R6 Wasserstoff, C1-C4-Alkyl oder -CHO ist; R1 Wasserstoff, C1-C4-Alkyl, -(CH2)qAr4 oder -CH2C(O)Ar4 ist, wobei q eine ganze Zahl von 1 bis 4 ist und Ar4 ein Rest der Formel ist, wobei Z4 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; oder Stereoisomere oder pharmazeutisch verträgliche Salze davon; mit der Maßgabe, daß falls G1 -C(O)- ist, dann G2 nicht -C(O)- ist, umfassend das Cyclisieren einer Verbindung der Formel
ist, wobei Z4 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; oder Stereoisomere oder pharmazeutisch verträgliche Salze davon; mit der Maßgabe, daß falls G1 -C(O)- ist, dann G2 nicht -C(O)- ist, umfassend das Cyclisieren einer Verbindung der Formel wobei m, G1, G2, Ar1 und Ar3 wie vorstehend definiert sind, unter Verwendung eines Kupplungsmittels und gegebenenfalls Modifizieren durch Alkylieren eines Amins, einer Additionsreaktion an ein Indolstickstoffatom oder der Bildung eines Amidats und gegebenenfalls Entfernen von Schutzgruppen und gegebenenfalls Herstellen eines pharmazeutisch verträglichen Salzes durch weiteres Umsetzen mit einer verträglichen Säure oder einer verträglichen Base.
wobei m, G1, G2, Ar1 und Ar3 wie vorstehend definiert sind, unter Verwendung eines Kupplungsmittels und gegebenenfalls Modifizieren durch Alkylieren eines Amins, einer Additionsreaktion an ein Indolstickstoffatom oder der Bildung eines Amidats und gegebenenfalls Entfernen von Schutzgruppen und gegebenenfalls Herstellen eines pharmazeutisch verträglichen Salzes durch weiteres Umsetzen mit einer verträglichen Säure oder einer verträglichen Base.
 wobei Z1 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; Ar2 ein Rest ist, ausgewählt aus:
wobei Z1 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; Ar2 ein Rest ist, ausgewählt aus: wobei Z2 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; Ar3 ein Rest ist, ausgewählt aus:
wobei Z2 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; Ar3 ein Rest ist, ausgewählt aus: wobei Z3 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; R6 Wasserstoff, C1-C4-Alkyl oder -CHO ist; R1 Wasserstoff, C1-C4-Alkyl, -(CH2)qAr4 oder -CH2C(O)Ar4 ist, wobei q eine ganze Zahl von 1 bis 4 ist und Ar4 ein Rest der Formel
wobei Z3 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; R6 Wasserstoff, C1-C4-Alkyl oder -CHO ist; R1 Wasserstoff, C1-C4-Alkyl, -(CH2)qAr4 oder -CH2C(O)Ar4 ist, wobei q eine ganze Zahl von 1 bis 4 ist und Ar4 ein Rest der Formel ist, wobei Z4 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; oder Stereoisomere oder pharmazeutisch verträgliche Salze davon; mit der Maßgabe, daß falls G1 -C(O)- ist, dann G2 nicht -C(O)- ist, umfassend das Reduzieren einer Verbindung der Formel
ist, wobei Z4 1 bis 3 Substituenten bedeutet, welche jeweils unabhängig ausgewählt sind aus Wasserstoff, Halogen, Benzyloxy, Hydroxy, CF3, C1-C4-Alkyl und C1-C4-Alkoxy; oder Stereoisomere oder pharmazeutisch verträgliche Salze davon; mit der Maßgabe, daß falls G1 -C(O)- ist, dann G2 nicht -C(O)- ist, umfassend das Reduzieren einer Verbindung der Formel wobei m, G1, G2, Ar1, Ar2 und Ar3 wie vorstehend definiert sind, unter Verwendung eines Amid-Reduktionsmittels, ausgewählt aus Lithiumaluminiumhydrid, Diisobutylaluminiumhydrid und Borandimethylsulfidkomplex, und gegebenenfalls Modifizieren durch Alkylieren eines Amins, einer Additionsreaktion an ein Indolstickstoffatom oder der Bildung eines Amidats und gegebenenfalls Entfernen von Schutzgruppen und gegebenenfalls Herstellen eines pharmazeutisch verträglichen Salzes durch weiteres Umsetzen mit einer verträglichen Säure oder einer verträglichen Base.
wobei m, G1, G2, Ar1, Ar2 und Ar3 wie vorstehend definiert sind, unter Verwendung eines Amid-Reduktionsmittels, ausgewählt aus Lithiumaluminiumhydrid, Diisobutylaluminiumhydrid und Borandimethylsulfidkomplex, und gegebenenfalls Modifizieren durch Alkylieren eines Amins, einer Additionsreaktion an ein Indolstickstoffatom oder der Bildung eines Amidats und gegebenenfalls Entfernen von Schutzgruppen und gegebenenfalls Herstellen eines pharmazeutisch verträglichen Salzes durch weiteres Umsetzen mit einer verträglichen Säure oder einer verträglichen Base.