-
Gebiet der Erfindung
-
Die vorliegende Erfindung gehört dem Gebiet der In-vitro-Diagnostik an. Innerhalb von diesem Gebiet betrifft sie insbesondere die Probenpräparation von Nucleinsäuren zu diagnostischen Zwecken. Genauer stellt die Erfindung ein Verfahren zur gleichzeitigen Isolierung von mindestens einer ersten und einer zweiten Zielnucleinsäure aus einer Vielzahl von verschiedenen Typen von Fluidproben bereit.
-
Hintergrund der Erfindung
-
Die Isolierung von biologischen Materialien wie Nucleinsäuren oder Proteinen aus komplexen biologischen Gemischen wie z. B. klinischen Proben ist insbesondere für diagnostische Zwecke bereits von erheblicher Bedeutung.
-
Beispiele für diagnostische Anwendungen von Nucleinsäure-Probenpräparation umfassen die Präparation und den anschließenden Nachweis von Viren, wie Humanes Papilloma-Virus (HPV), West-Nil-Virus (WNV), oder das routinemäßige Screening von Blutspendeproben auf das Vorliegen des Humanen Immundefizienz-Virus (HIV), des Hepatitis B- (HBV) und/oder C-Virus (HCV). Weiterhin sind diese Amplifikationstechniken für bakteriellen Ziele, wie Mykobacteria oder Chlamydia trachomatis und Neisseria gonorrhoeae, oder für die Analyse von onkologischen Markern geeignet.
-
Zahlreiche verschiedene Verfahren wurden bereits auf dem Fachgebiet entwickelt, z. B. Denaturieren, Präzipitieren und Entfernen unerwünschter Komponenten in einer Probe mit anschließender Präzipitation und Isolierung des in Frage kommenden Analyten (beispielsweise alkoholbasierte Präzipitation von Nucleinsäuren). Ein weiterer Ansatz besteht im Binden des jeweiligen biologischen Materials an ein festes Trägermaterial, das z. B. in der Form von Chromatographiesäulen bereitgestellt sein kann. Für diagnostische Zwecke und insbesondere für die automatisierte Isolierung von biologischen Materialien, die einer anschließenden Mittel- bis Hochdurchsatzanalyse unterliegen, werden oft bindende Teilchen verwendet. Solche Teilchen können funktionalisierte Oberflächen aufweisen, d. h. sie sind oft mit Antikörpern, Nucleinsäure-Fangsonden oder dergleichen überzogen, um den gewünschten Analyten zu binden. Alternativ können sie unmodifizierte Oberflächen, wie Glasoberflächen, insbesondere zur Isolierung von Nucleinsäuren, aufweisen.
-
Allerdings können die zu analysierenden Zielnucleinsäuren für diagnostische Zwecke in einer Vielzahl von verschiedenen Quellen vorhanden sein. In der Praxis wird das Probenpräparationsverfahren für Nucleinsäuren in verschiedenen Quellen in der Regel angepasst an:
- 1. den Typ von Fluidprobe
- 2. den Typ von Nucleinsäure.
-
Auch andere Kriterien können beim Isolieren verschiedener Nucleinsäuren aus verschiedenen Quellen in Betracht gezogen werden. Die bisherige Technik hat dieser Diversität durch die Bereitstellung verschiedener Verfahren zur Präparation für die verschiedenen Typen von Proben Rechnung getragen.
-
Durch die vorliegende Erfindung wird ein verbessertes Verfahren zur Isolierung von mindestens einer ersten und einer zweiten Zielnucleinsäure aus einer Vielzahl von verschiedenen Typen von Fluidproben bereitgestellt.
-
Beschreibung der Erfindung
-
Die vorliegende Erfindung stellt ein Verfahren zur gleichzeitigen Isolierung von mindestens einer ersten und einer zweiten Zielnucleinsäure aus einer Vielzahl von verschiedenen Typen von Fluidproben bereit.
-
In einem ersten Aspekt betrifft die Erfindung ein Verfahren zur gleichzeitigen Isolierung von mindestens einer ersten und einer zweiten Zielnucleinsäure aus einer Vielzahl von verschiedenen Typen von Fluidproben, wobei das Verfahren die folgenden automatisierten Schritte umfasst:
- a. Gegenseitiges Kombinieren von einem festen Trägermaterial und der Vielzahl von verschiedenen Typen von Fluidproben in einer der Anzahl von Fluidproben entsprechenden Anzahl von Gefäßen für einen Zeitraum und unter Bedingungen, die ausreichen, um zu ermöglichen, dass Nucleinsäuren, die die Zielnucleinsäuren einschließen, auf dem festen Trägermaterial immobilisiert werden,
- b. Isolieren des festen Trägermaterials aus dem anderen in den Fluidproben vorhandenen Material in einer Trennstation,
- c. Reinigen der Nucleinsäuren in einer Trennstation durch Abtrennen der Fluidprobe aus dem festen Trägermaterial und ein- oder mehrmaliges Waschen des festen Trägermaterials mit einem Waschpuffer,
wobei die physikalischen Bedingungen und der Zeitraum für die Elemente aus der Vielzahl von verschiedenen Typen von Fluidproben identisch sind.
-
Es ist insbesondere, jedoch nicht nur für klinische Laboratorien mit hohem Probendurchsatz, äußerst günstig, mit einem solchen verbesserten Verfahren zur schnellen, leichten und zuverlässigen gleichzeitigen Isolierung von mehreren Zielnucleinsäuren aus einer Vielzahl von verschiedenen Typen von Fluidproben auserlaubt zu sein.
-
Das Verfahren, das die oben erwähnten automatisierten Schritte umfasst, zeigt verschiedene Vorteile.
-
Erstens setzt die automatisierte Kombination des Probenpräparationsverfahrens gemäß der vorliegenden Erfindung mit z. B. reverser Transkription von RNA und Amplifikation der Zielnucleinsäuren den Bedarf nach einem manuellen Eingriff und dadurch das potentielle Risiko von einer Verunreinigung deutlich herab.
-
Weiterhin trägt die Möglichkeit der Bereitstellung eines einzigen Verfahrens mit einer Vielzahl von verschiedenen Proben, d. h. verschiedene Quellen von Nucleinsäuren, deutlich zur Herabsetzung der Gesamtkomplexität der Nucleinsäurediagnostik bei. Wenn beispielsweise verschiedene Verfahren je Typ von Fluidprobe angewendet werden müssen, wie es im Stand der Technik bisher der Fall war, ist die Probenpräparation viel komplexer, zeitraubender und ressourcenintensiver. Meistens müssen verschiedene Reagenzien in Anspruch genommen werden, was zu erhöhten Kosten und zur Behinderung der Entwicklung von schnellen und unkomplizierten automatisierten Lösungen führt.
-
Die Probenpräparation gemäß der Erfindung zeigt die entsprechende Vielseitigkeit und den Arbeitsablauf, um mehrere verschiedene Probentypen, die verschiedene Typen von Nucleinsäuren enthalten, wie beispielsweise DNA und RNA, zu handhaben.
-
Verschiedene Quellen, d. h. Typen von Proben, umfassen unter anderem alle Arten von menschlichen Körperfluiden, wie beispielsweise Blut, Sputum, Nasenabstrich, Urin, Schweiß oder andere.
-
Das Verfahren gemäß der Erfindung erfordert viel weniger Zeit zur Durchführung und die Testung ist viel einfacher durchzuführen als Probenpräparationsverfahren, die in der bisherigen Technik verwendet werden. Das Verfahren gemäß der Erfindung bietet z. B. auf dem Gebiet der klinischen Virologie einen wesentlichen Vorteil, da es parallele Probenpräparation und nachgeschaltet vorzugsweise die Amplifikation von mehreren Viren in Parallelexperimenten erlaubt. Das Verfahren ist besonders bei der Handhabung von Posttransplantatpatienten geeignet, in denen häufige Virusüberwachung erforderlich ist. Dadurch erleichtert das Verfahren gemäß der Erfindung eine kostengünstige Diagnose und trägt zu einer geringeren Verwendung von antiviralen Mitteln und zu weniger Virus-Komplikationen und Krankenhausaufenthalten bei. Dies trifft auch auf das Gebiet der klinischen Mikrobiologie zu. Im Allgemeinen werden Wirkungen in einer schnelleren Umsatzzeit und verbesserten Testungsvielseitigkeit erzielt. In der Folge führt dies, um eine Diagnose zu erstellen, zu einer Abnahme in der Anzahl von an einem Patienten erforderlichen Testläufen und zu potentiell kürzeren Krankenhausaufenthalten (z. B. erhalten, wenn eine Diagnose schneller bereitgestellt werden kann, die Patienten, bei denen antimikrobielle Therapie erforderlich ist, sie schneller und genesen somit früher). Zusätzlich zeigen Patienten eine geringere Morbidität und verursachen darum im Zusammenhang mit Unterstützungstherapie (z. B. Intensivpflege im Zusammenhang mit einer verzögerten Diagnose von Sepsis) geringere Kosten. Das frühzeitigere Bereitstellen eines negativen Ergebnisses kann für die Überverschreibung von Antibiotika bedeutende Auswirkungen haben. Wenn beispielsweise ein unter Verwendung des Verfahrens gemäß der Erfindung erhaltenes Testergebnis das Pathogen schneller auszuschließen vermag als eine Standard-Probenpräparationsmethode mit z. B. anschließender Realzeit-PCR, wird der Kliniker nicht gezwungen sein, empirische Antibiotika zu verwenden. Wenn andererseits empirische Antibiotika verwendet werden, kann die Dauer der jeweiligen Behandlung verkürzt werden.
-
Mit Hinblick auf die Konzeption eines Tests einschließlich Probenpräparation mit dem Verfahren gemäß der Erfindung profitiert der Fachmann insbesondere, jedoch nicht nur, von den folgenden Vorteilen:
- • eine Reduktion in der Softwarekomplexität (was zu einem verminderten Risiko von Programmierfehlern führt),
- • Fokussieren der Testentwicklungsanstrengungen auf die Optimierung der Chemie anstelle Chemie plus Instrumentensteuerungsparameter,
- • ein viel zuverlässigeres System, da ein einziges Verfahren immer verwendet wird und die Hardware optimal zur Durchführung dieses Protokolls konzipiert werden kann,
- • der Fachmann, der das Verfahren gemäß der Erfindung durchführt, wird mit der Vielseitigkeit auserlaubt, mehrere verschiedene Isolierungen parallel als Teil des gleichen Verfahrens laufen zu lassen,
- • Kostenreduktion.
-
Im Sinne der Erfindung beziehen sich „Reinigung”, „Isolierung” oder „Extraktion” von Nucleinsäuren auf das Folgende: Bevor Nucleinsäuren in einem diagnostischen Test analysiert werden können, z. B. durch Amplifikation, müssen sie typischerweise gereinigt, isoliert oder aus biologischen Proben, die komplexe Gemische von verschiedenen Komponenten enthalten, extrahiert werden. Für die ersten Schritte können Verfahren verwendet werden, durch die sich die Nucleinsäuren anreichern lassen.
-
Oft sind die zu analysierenden Nucleinsäuren innerhalb der in Frage kommenden Fluidprobe nicht in der Lösung frei, sondern sind innerhalb von geschlossenen Strukturen, wie zum Beispiel Zellen oder Viren, lokalisiert. In diagnostischen Tests ist es oft das Ziel, insbesondere pathogene Zellen oder Viren in Fluidproben, wie klinische Proben, zu identifizieren. Solche Pathogene können z. B. RNA-Viren, wie beispielsweise das Humane Immundefizienz-Virus (HIV), das Hepatitis C-Virus (HCV), das West-Nil-Virus (WNV), das Humane Papilloma-Virus (HPV), das Japanische-Enzephalitis-Virus (JEV), St.-Louis-Enzephalitis-Virus (SLEV) und andere, oder DNA-Viren, wie das Hepatitis B-Virus (HBC), Cytomegalo-Virus (CMV) und andere, oder Bakterien, wie z. B. Chlamydia trachomatis (CT), Neisseria gonorrhoeae (NG) und andere umfassen. Das Verfahren gemäß der Erfindung ist zur Extraktion von Nucleinsäuren aus den oben genannten sowie anderen Organismen geeignet.
-
Darum ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei Schritt a. weiterhin die Freisetzung von Nucleinsäuren aus ihrer zellulären und/oder viralen Umgebung durch Lyse von Zellen und/oder viralen Kapsiden, die potentiell in der Vielzahl von verschiedenen Fluidproben vorhanden sind, umfasst.
-
Zur Freisetzung des Inhalts von Zellen oder Viruspartikeln können sie mit Enzymen oder mit Chemikalien zur Auflösung, zum Abbau oder zur Denaturierung der Zellwände oder der Viruspartikel behandelt werden. Dieses Verfahren wird im Allgemeinen als Lyse bezeichnet. Die resultierende Lösung, die solches lysiertes Material enthält, wird als Lysat bezeichnet.
-
Mittel, die zur Lyse von Zellen und/oder viralen Kapsiden oder vergleichbaren Strukturen geeignet sind, werden im Allgemeinen in einen Lysepuffer bereitgestellt. Daher umfasst das vorstehend beschriebene Verfahren in einer bevorzugten Ausführungsform der Erfindung weiterhin in Schritt a. die Zugabe von einem Lysepuffer zu einer Vielzahl von verschiedenen Fluidproben.
-
Da das Verfahren gemäß der Erfindung bezüglich hohem Durchsatz, Wirkungsgrad und Parallelisierung besonders vorteilhaft ist, besteht ein bevorzugter Aspekt der Erfindung in dem vorstehend beschriebenen Verfahren, wobei der Lysepuffer für die Mitglieder der Vielzahl von verschiedenen Typen von Fluidproben identisch ist.
-
Auf diesem Weg wird die Komplexität der Probenpräparationsverfahrens weiter vermindert, da keine verschiedenen Lysereagenzien einzeln für die verschiedenen zu behandelnden Proben bereitgestellt werden müssen. Weiterhin kann das Verfahren leichter kontrolliert werden, wenn mit einem einzigen Lysepuffer gearbeitet wird. Der Lysepuffer kann z. B. mit einem Multipipettierer aus einem einzigen Behälter aufgezogen und anschließend gleichzeitig in die verschiedenen Proben abgegeben werden.
-
In einer bevorzugten Ausführungsform der Erfindung umfasst der Lysepuffer in dem vorstehend beschriebenen Verfahren eine oder mehrere, aus der folgenden Gruppe ausgewählte Komponenten:
- • einem chaotropen Mittel
- • einer Puffersubstanz
- • einem Alkohol
- • einem Reduktionsmittel.
-
Chaotrope Mittel, die im Allgemeinen die geordnete Struktur von Wassermolekülen in Lösung und nicht-kovalente Bindungskräfte in und zwischen den Molekülen stören, können an dem Verfahren der Probenpräparation mehrfachen Anteil nehmen. Insbesondere, jedoch nicht nur, können sie durch Beeinträchtigen der Nuclease-Tertiärstruktur als RNase-Inhibitoren eingesetzt werden. In der Regel muss kein weiterer RNase-Inhibitor auf den Lysepuffer angewandt werden. Daneben tragen chaotrope Mittel dazu bei, biologische Membranen, wie Plasmamembranen oder die Membranen von Zellorganellen, sofern vorhanden, aufzuschließen. Auch können sie beim adhäsiven Binden von Nucleinsäuren an Oberflächen wie Glas eine signifikante Rolle spielen (siehe infra). Bevorzugte chaotrope Mittel im Zusammenhang mit der Erfindung sind Guanidiniumsalze, wie Guanidiniumthiocyanat oder Guanidinhydrochlorid oder Guanidiniumchlorid oder Guanidiniumisothiocyanat, Harnstoff, Perchlorat, wie z. B. Kaliumperchlorat, weitere Thiocyanate oder Kaliumiodid. Besonders bevorzugt ist Guanidiniumthiocyanat. Allerdings können im Umfang der Erfindung auch andere chaotrope Mittel verwendet werden.
-
Puffersubstanzen sind im Allgemeinen zum Aufrechterhalten eines bestimmten pH-Wertes oder eines bestimmten pH-Bereiches in einer Lösung von Bedeutung. Dies ist die Voraussetzung für die meisten biologischen Systeme und meistens auch für In-vitro-Reaktionen erwünscht. Es kann auch für das Verfahren der Erfindung von Vorteil sein. Bevorzugte Puffer im Zusammenhang mit der Erfindung sind Citratpuffer, wie Natriumcitrat, aber auch Tris(Tris(hydroxymethyl)-aminomethan)-Puffer, wie Tris-HCl, Phosphat, N-(2-Hydroxyethyl)-piperazin-N'-(2-ethansulfonsäure) (HEPES), Acetatpuffer, aber auch andere Puffer können im Zusammenhang mit der Erfindung verwendet werden.
-
Die Verwendung von Alkohol in einem Lysepuffer zur Nucleinsäurepräparation kann ebenfalls von Vorteil sein, wie es dem Fachmann bekannt ist. Besonders bevorzugt im Zusammenhang mit der Erfindung ist die Verwendung von Polidocanol, während im Lysepuffer, vorstehend beschrieben, auch ein anderer Alkohol verwendet werden kann. Die Verwendung von Polidocanol zur Präparation von Nucleinsäuren wurde z. B. bereits in der
EP 1 932 913 beschrieben.
-
Reduktionsmittel können ebenfalls zur Denaturierung von unerwünschten Komponenten, wie RNase A, vorstehend erwähnt, beitragen. Insbesondere spalten Reduktionsmittel, wie sie auf dem Fachgebiet weithin bekannt sind, inter- und intramolekulare Disulfidbindungen, die besonders für die Tertiärstruktur von vielen Proteinen von Bedeutung sind. Bevorzugt im Zusammenhang mit der Erfindung sind Reduktionsmittel, wie Dithiothreitol (DTT), aber andere, auf dem Fachgebiet bekannte Reduktionsmittel, wie z. B. 2-Mercaptoethanol, können im Zusammenhang mit der Erfindung ebenfalls in vorteilhafter Weise eingesetzt werden.
-
Im Hinblick auf das zuvor Genannte ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei der Lysepuffer die folgenden Komponenten umfasst:
- • Guanidiniumthiocyanat,
- • Na-Citrat,
- • Polydocanol,
- • DTT.
-
In einer stärker bevorzugten Ausführungsform der Erfindung sind die Konzentrationen der vorstehend erwähnten Komponenten des Lysepuffers wie folgt:
- • Guanidiniumthiocyanat: 4 M
- • Na-Citrat: 50 mM
- • Polydocanol: 5% Gew./Vol.
- • DTT: 2% Gew./Vol.
-
Der pH-Wert des vorstehend beschriebenen Lysepuffers ist nicht auf spezielle pH-Werte begrenzt. Allerdings hat der Lysepuffer in einer bevorzugten Ausführungsform einen sauren pH-Wert, stärker bevorzugt einen pH-Wert zwischen 5,5 und 6,5, am stärksten bevorzugt etwa 5,8. Ein während der Lyse oft angetroffenes Problem besteht darin, dass andere Enzyme, die die Komponente von Interesse abbauen, z. B. Desoxyribonucleasen oder Ribonucleasen, die Nucleinsäuren abbauen, wie RNase, vorstehend erwähnt, während des Lysevorgangs mit der Komponente von Interesse in Kontakt geraten. Diese abbauenden Enzyme können auch außerhalb der Zellen vorhanden sein oder können vor der Lyse räumlich in verschiedene Zellkompartimente getrennt worden sein. Wenn die Lyse stattfindet, wird die Komponente von Interesse gegenüber den abbauenden Enzymen exponiert. Weitere Komponenten, die während dieses Verfahrens freigesetzt werden, können z. B. Endotoxine sein, die der Familie der Lipopolysaccharide angehören, die für Zellen toxisch sind, und können Probleme für Produkte hervorrufen, die zur Verwendung in der Human- oder Tiertherapie beabsichtigt sind.
-
Es gibt eine Vielzahl von Mitteln, um das oben genannte Problem zu beheben. Es ist üblich, chaotrope Mittel (wie vorstehend beschrieben) oder anionische, kationische, zwitterionische oder nicht-ionische Detergentien, wenn es beabsichtigt ist, Nucleinsäuren freizusetzen, zu verwenden.
-
Es ist auch ein Vorteil, Proteasen zu verwenden, die schnell die zuvor beschriebenen Enzymen oder unerwünschte Proteine abbauen. Allerdings kann dies andere Probleme hervorrufen, da die Substanzen oder Enzyme Reagenzien oder Komponenten in den anschließenden Schritten stören können.
-
Enzyme, die bevorzugt bei solchen Lyse- oder Probenpräparationsverfahren verwendet werden, die vorstehend erwähnt sind, sind Enzyme, die die Amidverknüpfungen in Proteinsubstraten spalten, und die als Proteasen oder (synonym) als Peptidasen klassifiziert sind (siehe
Walsh, 1979, Enzymatic Reaction Mechanisms, W. H. Freeman and Company, San Francisco, Kapitel 3). Proteasen, die in der bisherigen Technik verwendet werden, umfassen alkalische Proteasen (
WO 98/04730 ) oder saure Proteasen (
US 5,386,024 ). Eine Protease, die für die Probenpräparation bei der Isolierung von Nucleinsäuren in der bisherigen Technik breit eingesetzt wird, ist Proteinase K aus Tritirachium album (siehe z. B.
Sambrook J. et al., Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 1989), die um den neutralen pH aktiv ist und einer Familie von Proteasen angehört, die dem Fachmann als Subtilisine bekannt ist. Besonders bevorzugt für die Verwendung bei Lyse- oder Probenpräparationsverfahren, die vorstehend erwähnt sind, ist das Enzym Esperase, eine robuste Protease, die ihre Aktivität sowohl bei hoher Alkalinität als auch bei hohen Temperaturen beibehält (
EP 1 201 753 ).
-
In den auf den Lyseschritt folgenden Probenpräparationsschritten wird die Komponente von Interesse weiter angereichert. Wenn die nicht-proteinartigen Komponenten von Interesse z. B. Nucleinsäuren sind, werden sie normalerweise aus den komplexen Lysegemischen extrahiert, bevor sie in einem Sonden-basierten Test verwendet werden.
-
Es gibt verschiedene Verfahren der Reinigung von Nucleinsäuren:
- – Sequenzabhängige oder biospezifische Verfahren, wie z. B.
• Affinitätschromatographie
• Hybridisierung mit immobilisierten Sonden
- – Sequenzunabhängige oder physikochemische Verfahren, wie z. B.
• Flüssig-Flüssig-Extraktion z. B. mit Phenol-Chloroform
• Präzipitation z. B. mit reinem Ethanol
• Extraktion mit Filterpapier
• Extraktion mit Mizellen-bildenden Mitteln, wie Cetyltrimethylammoniumbromid
• Binden an immobilisierte, interkalierende Farbstoffe, z. B. Acridinderivate
• Adsorption an Silicagel oder Diatomeenerden
• Adsorption an magnetische Glasteilchen (MGP) oder Organosilanpartikel unter chaotropen Bedingungen.
-
Besonders interessant zu Reinigungszwecken ist die Adsorption von Nucleinsäuren an eine Glasoberfläche, obwohl andere Oberflächen möglich sind. Viele Verfahren zur Isolierung von Nucleinsäuren aus ihrer natürlichen Umgebung wurden bereits in den letzten Jahren durch die Verwendung von ihrem Bindungsverhalten an Glasflächen vorgeschlagen. Wenn unmodifizierte Nucleinsäuren das Ziel sind, ist ein direktes Binden der Nucleinsäuren an ein Material mit einer Silicaoberfläche bevorzugt, da, unter anderen Gründen, die Nucleinsäuren nicht modifiziert werden müssen und sogar native Nucleinsäuren gebunden werden können. Diese Verfahren sind in verschiedenen Dokumenten ausführlich beschrieben. In
Vogelstein B. et al., Proc. Natl. Acad. USA 76 (1979) 615–9 beispielsweise wird ein Verfahren zum Binden von Nucleinsäuren aus Agarosegelen in Gegenwart von Natriumjodid auf gemahlenes Flintglas vorgeschlagen. Die Reinigung von Plasmid-DNA aus Bakterien auf Glasstaub in Gegenwart von Natriumperchlorat ist bei
Marko M. A. et al., Anal. Biochem. 121 (1982) 382–387 beschrieben. In
DE-A 37 34 442 ist die Isolierung von einzelsträngiger M13 Phagen-DNA auf Glasfaserfiltern durch Präzipitieren von Phagenpartikeln unter Verwendung von Essigsäure und Lyse der Phagenpartikel mit Perchlorat beschrieben. Die an die Glasfaserfilter gebundenen Nucleinsäuren werden gewaschen und dann mit einem Methanol enthaltenden Tris/EDTA-Puffer eluiert. Ein vergleichbares Verfahren zur Reinigung von DNA aus
Lambda-Phagen ist in Jakobi R. et al., Anal. Biochem. 175 (1988) 196–201 beschrieben. Das Verfahren hat das selektive Binden von Nucleinsäuren an Glasflächen in chaotropen Salzlösungen und das Abtrennen der Nucleinsäuren von Verunreinigungen wie Agarose, Proteine oder Zellrückstand zur Folge. Zur Abtrennung der Glaspartikel von Verunreinigungen können die Partikel entweder zentrifugiert werden oder es werden Fluide durch die Glasfaserfilter gezogen. Dies ist ein limitierender Schritt, der allerdings verhindert, dass das Verfahren dazu verwendet wird, große Mengen von Proben zu verarbeiten. Die Verwendung von magnetischen Partikeln zur Immobilisierung von Nucleinsäuren nach der Präzipitation durch Zugabe von Salz und Ethanol ist zweckmäßiger und z. B. in
Alderton R. P. et al., S. Anal. Biochem. 201 (1992) 166–169 und PCT
GB 91/00212 beschrieben. Bei diesem Verfahren werden die Nucleinsäuren zusammen mit den Magnetpartikeln agglutiniert. Das Agglutinat wird von dem Original-Lösungsmittel durch Anlegen eines Magnetfelds und Durchführen eines Waschschritts abgetrennt. Nach einem Waschschritt werden die Nucleinsäuren in einem Tris-Puffer gelöst. Dieses Verfahren hat allerdings insofern einen Nachteil, als die Präzipitation für Nucleinsäuren nicht selektiv ist. Stattdessen wird ebenso eine Vielzahl von festen und gelösten Substanzen agglutiniert. Als Ergebnis kann dieses Verfahren nicht zur Entfernung signifikanter Mengen von Inhibitoren für spezifische enzymatische Reaktionen eingesetzt werden, die vorhanden sein können. Magnetisches poröses Glas, das Magnetpartikel in einer porösen, bestimmten Glasmatrix enthält und mit einer Schicht, die Streptavidin enthält, bedeckt ist, ist ebenfalls im Handel erhältlich. Dieses Produkt kann zur Isolierung biologischer Materialien verwendet werden, z. B. Proteine oder Nucleinsäuren, wenn sie in einem komplexen Präparationsschritt modifiziert werden, so dass sie kovalent an Biotin binden. Magnetisierbare teilchenförmige Adsorbentien, haben sich zur automatischen Probenpräparation als sehr wirksam und geeignet erwiesen. Für diesen Zweck werden fernmagnetische und ferromagnetische sowie superparamagnetische Pigmente verwendet. Die am stärksten bevorzugten magnetischen Glaspartikel und die Verfahren, die sie verwenden, sind diejenigen, die in
WO 01/37291 beschrieben sind. Besonders geeignet für die Nucleinsäureisolierung im Zusammenhang mit der Erfindung ist das Verfahren gemäß
R. Boom et al. (J Clin Microbiol. 28 (1990), 495–503).
-
Die Vielseitigkeit des Verfahrens gemäß der Erfindung kann durch Anpassen des Volumens der jeweiligen in dem Verfahren verwendeten Fluidprobe weiter verbessert werden. Diese Ausführungsform konzentriert sich auf die Diversität der verschiedenen Typen von Fluidproben und möglicherweise die Typen von Organismen und Nucleinsäuren, die innerhalb von ihnen vorhanden sind. Z. B. können bestimmte Viren in einer Vollblutprobe mehr Ausgangsmaterial als andere Proben erfordern, wenn es bekannt ist, dass im Allgemeinen nur geringe Kopienanzahlen in diesen spezifischen Fällen vorhanden sind.
-
Somit ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei mindestens eine Fluidprobe der Vielzahl von verschiedenen Fluidproben ein verschiedenes Volumen als die anderen Fluidproben aufweist.
-
Es ist ebenfalls bevorzugt, dass, alternativ oder zusätzlich, verschiedene Volumina von Lysepuffer der Vielzahl von verschiedenen Fluidproben zugesetzt werden.
-
Bei einer weiteren bevorzugten Ausführungsform, wenn mindestens eine Fluidprobe der Vielzahl von verschiedenen Fluidproben ein verschiedenes Volumen als die anderen Fluidproben aufweist, wird den Proben Lysepuffer zugesetzt, dass alle Proben nach der Zugabe das gleiche Volumen aufweisen.
-
In dieser Ausführungsform ist es sogar noch zweckmäßiger, ein automatisiertes Verfahren mit verschiedenen Proben gleichzeitig durchzuführen. Die Vorteile, in der Lage zu sein, ein entsprechendes Ausgangsvolumen in Abhängigkeit von dem Probentyp zu wählen, und davon, dass identische Volumina zur Durchführung der Isolierung und gegebenenfalls z. B. der Amplifikation und Detektion vorhanden sind, werden in diesem Ansatz kombiniert.
-
Der Begriff „festes Trägermaterial” schließt sämtliche festen Materialien ein, die vorstehend in Verbindung mit der Immobilisierung von Nucleinsäuren erwähnt wurden, z. B. magnetische Glaspartikel, Glasfasern, Glasfaserfilter, Filterpapier etc., obgleich das feste Trägermaterial nicht auf diese Materialien beschränkt ist.
-
Ein bevorzugter Aspekt der Erfindung ist das vorstehend beschriebene Verfahren, wobei das feste Trägermaterial Nucleinsäure-bindende Partikel umfasst, vorzugsweise ein oder mehrere der Materialien, ausgewählt aus Silica, Metall, Metalloxiden, Kunststoff, Polymeren und Nucleinsäuren. In einer sehr bevorzugten Ausführungsform der Erfindung ist das feste Trägermaterial magnetische Glaspartikel.
-
„Immobilisieren” im Zusammenhang mit der Erfindung bedeutet Einfangen von Gegenständen, wie z. B. Nucleinsäuren, auf reversible oder irreversible Weise. Insbesondere bedeutet „immobilisiert auf dem festen Trägermaterial”, dass der Gegenstand oder die Gegenstände mit dem festen Trägermaterial für den Zweck ihrer Abtrennung aus umgebenden Medien assoziiert sind und z. B. durch Abtrennen von dem festen Trägermaterial zu einem späteren Zeitpunkt gewonnen werden können. In diesem Zusammenhang kann „Immobilisierung” z. B. die Adsorption von Nucleinsäuren an Glas oder an andere geeignete Oberflächen von festen Materialien, wie vorstehend beschrieben, einschließen. Ferner können Nucleinsäuren spezifisch durch Binden an Fängersonden „immobilisiert” werden, wobei Nucleinsäuren an im Wesentlichen komplementäre Nucleinsäuren gebunden werden, die an einen festen Träger durch Basenpaarung gebunden sind. Im letzteren Fall kann eine solche spezifische Immobilisierung zum überwiegenden Binden von Zielnucleinsäuren führen.
-
„Gleichzeitig” im Sinne der Erfindung bedeutet, dass zwei Vorgänge, wie Amplifizieren einer ersten und einer zweiten oder mehrerer Nucleinsäuren gleichzeitig unter den gleichen physikalischen Bedingungen durchgeführt werden. In einer Ausführungsform wird die gleichzeitige Amplifikation der mindestens ersten und zweiten Zielnucleinsäure in einem Gefäß durchgeführt. In einer anderen Ausführungsform wird die gleichzeitige Amplifikation mit mindestens einer Nucleinsäure in einem Gefäß und mindestens einer zweiten Nucleinsäure in einem zweiten Gefäß gleichzeitig und unter den gleichen physikalischen Bedingungen, insbesondere bezüglich Temperatur und Inkubationszeit, durchgeführt.
-
Die „erste Zielnucleinsäure” und die „zweite Zielnucleinsäure” sind verschiedene Nucleinsäuren.
-
Eine „Fluidprobe” ist jedes fluide Material, das einem diagnostischen Test, der auf Nucleinsäuren ausgerichtet ist, unterzogen werden kann und vorzugsweise von einer biologischen Quelle abgeleitet ist. Auch ist die Fluidprobe bevorzugt von einem Menschen abgeleitet und ist ein Körperfluid. In einer bevorzugten Ausführungsform der Erfindung ist die Fluidprobe Menschenblut, Urin, Sputum, Schweiß, Abstrich, pipettierbarer Stuhl oder Spinalfluid. Am stärksten bevorzugt ist die Fluidprobe Menschenblut.
-
Der Begriff „Reaktionsgefäß” umfasst, ist jedoch nicht beschränkt auf, Röhrchen oder die Vertiefungen von Platten, wie Mikroplatten, tiefe Platten oder andere Typen von Multiwellplatten, wobei eine Reaktion zur Analyse der Fluidprobe, wie z. B. reverse Transkription oder eine Polymerase-Kettenreaktion stattfindet. Die äußeren Grenzen oder Wände von solchen Gefäßen sind chemisch inert, derart dass sie nicht die analytische Reaktion, die darin stattfindet, beeinträchtigen. Vorzugsweise wird die Isolierung der Nucleinsäuren, wie vorstehend beschrieben, ebenfalls in einer Multiwellplatte durchgeführt.
-
In diesem Zusammenhang erlauben Multiwellplatten in analytischen Systemen die parallele Trennung und Analyse oder Lagerung von mehreren Proben. Multiwellplatten können zur maximalen Flüssigkeitsaufnahme oder zum maximalen Wärmeaustausch optimiert werden. Eine bevorzugte Multiwellplatte zur Verwendung in dem Zusammenhang der vorliegenden Erfindung wird zur Inkubation oder Trennung eines Analyten in einem automatisierten Analysator optimiert. Vorzugsweise ist die Multiwellplatte konstruiert und angeordnet, um eine magnetische Vorrichtung und/oder eine Heizvorrichtung zu kontaktieren.
-
Die bevorzugte Multiwellplatte, die im Zusammenhang mit der Erfindung synonym „Prozessplatte” genannt wird, umfasst:
- – eine obere Fläche, die mehrere in Reihen angeordnete Gefäße oben mit Öffnungen umfasst. Die Gefäße umfassen einen oberen Teil, einen mittleren Teil und einen unteren Teil. Der obere Teil ist mit der oberen Fläche der Multiwellplatte gekoppelt und umfasst zwei längere und zwei kürzere Seiten. Der mittlere Teil besitzt einen rechteckigen Querschnitt mit zwei längeren Seiten und zwei kürzeren Seiten;
- – zwei gegenüberliegende kürzere und zwei gegenüberliegende längere Seitenwände und
- – eine Grundfläche, wobei die Grundfläche eine zur Anordnung der Multiwellplatte in Kontakt mit der magnetischen Vorrichtung und/oder einer Heizvorrichtung konstruierte und angeordnete Öffnung umfasst.
-
In einer bevorzugten Ausführungsform der Multiwellplatte sind nebeneinander liegende Gefäße in einer Reihe an der längeren Seite der fast rechteckigen Form zusammengefügt.
-
Vorzugsweise umfasst die Multiwellplatte einen zusammenhängenden Raum, der zwischen nebeneinander liegenden Reihen von Gefäßen angeordnet ist. Der zusammenhängende Raum ist zur Aufnahme einer plattenförmigen magnetischen Vorrichtung konstruiert und angeordnet. In einer bevorzugten Ausführungsform umfasst der untere Teil der Gefäße einen kugeligen Boden.
-
In einer stärker bevorzugten Ausführungsform umfasst der untere Teil des Gefäßes einen konischen Teil, der zwischen dem mittleren Teil und dem kugeligen Boden angeordnet ist.
-
In einer bevorzugten Ausführungsform umfasst die obere Fläche Rippen, wobei die Rippen die Öffnungen der Gefäße umgeben. Vorzugsweise umfasst eine kürzere Seite des oberen Teils der Gefäße eine Aussparung, wobei die Aussparung eine gekrümmte Fläche einschließt, die sich von der Rippe zur Innenseite des Gefäßes erstreckt.
-
Weiterhin umfassen in einer bevorzugten Ausführungsform die Gefäße eine gerundete innere Form.
-
Zur Befestigung an der Arbeits- oder Inkubationsstation umfasst die Basis vorzugsweise einen Rand, der Aussparungen einschließt. Schnappbügel an der Station eines Analysators können in die Aussparung eingreifen, um die Platte an einer Station zu befestigen.
-
In einer bevorzugten Ausführungsform umfassen die Gefäße eine im Wesentlichen konstante Wanddicke.
-
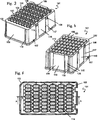
Die bevorzugte Arbeitsplatte (101) im Zusammenhang mit der vorliegenden Erfindung ist eine Einkomponentenplatte. Ihre obere Fläche (110) schließt mehrere Gefäße (103) ein (5, 6). Jedes Gefäß weist oben eine Öffnung (108) auf und ist am unteren Ende (112) geschlossen. Die obere Fläche (110) umfasst Rippen (104), die vorzugsweise relativ zur oberen Fläche (110) erhöht sind und die Öffnungen (108) der Gefäße (103) umgeben. Dies verhindert Verunreinigung des Inhalts der Gefäße (103) mit Tröpfchen von Flüssigkeit, die auf die obere Fläche (110) der Platte (101) fallen können. Ansichten einer bevorzugten Arbeitsplatte sind in den 3 bis 8 gezeigt.
-
Die Standfläche der Arbeitsplatte (101) umfasst vorzugsweise eine Länge und eine Breite der Basis entsprechend einem ANSI SBS Standplattenformat. Stärker bevorzugt beträgt die Länge 127,76 mm +/– 0,25 mm, und die Breite beträgt 85,48 mm +/– 0,25 mm. Somit weist die Platte (101) zwei nebeneinander liegende kürzere Seitenwände (109) und zwei nebeneinander liegende längere Seitenwände (118) auf. Die Arbeitsplatte (101) umfasst Formarretierelemente (106) zur Wechselwirkung mit einem Steuerungsprogramm (500, 12). Die Arbeitsplatte (101) kann gegriffen, befördert und schnell und sicher mit hoher Geschwindigkeit positioniert werden, während die korrekte Orientierung und Position beibehalten werden. Vorzugsweise sind die Formarretierelemente (106) zum Greifen innerhalb des oberen zentralen Teils angeordnet, vorzugsweise in dem oberen zentralen Drittel der Arbeitsplatte (101). Dies hat den Vorteil, dass eine potentielle Verformung der Arbeitsplatte (101) nur eine kleinere Auswirkung auf die Formarretierelemente (106) aufweist und dass die Handhabung der Platte (101) robuster ist.
-
Die Arbeitsplatte (101) umfasst vorzugsweise die Hardware-Kennzeichen (102) und (115). Die Hardware-Kennzeichen (102) und (115) sind für die Arbeitsplatte (101) einmalig und von den Hardware-Kennzeichen von anderen, in dem System verwendeten Verbrauchsmaterialien verschieden. Die Hardware-Kennzeichen (102, 115) umfassen vorzugsweise Rillen (119) und/oder Aussparungen (125) auf den Seitenwänden der Verbrauchsmaterialien, wobei das Muster von Rillen (119) und/oder Aussparungen (125) für einen spezifischen Typ von Verbrauchsmaterial einmalig ist, vorzugsweise für die Arbeitsplatte (101). Dieses einmalige Muster wird hierin auch als eine einmalige „Oberflächengeometrie” bezeichnet. Die Hardware-Kennzeichen (102, 115) gewährleisten, dass der Anwender nur die Arbeitsplatte (101) in die entsprechende Stapelposition von einem analytischen Instrument in der ordnungsgemäßen Orientierung laden kann. An den Seiten der Arbeitsplatte (101) sind Führungselemente (116) und (117) eigeschlossen (3, 4). Sie verhindern das Verkanten der Arbeitsplatte (101). Die Führungselemente (116, 117) erlauben es dem Anwender, die Arbeitsplatten (101) mit Führungselementen (116, 117) als ein Stapel in ein analytisches Instrument zu laden, der anschließend innerhalb des Instruments vertikal in eine Ablage ohne Verkanten der Platten übergeführt wird.
-
Der mittlere Teil (120) der Gefäße (103) weist einen fast rechteckigen Querschnitt auf (6, 7). Sie sind entlang der längeren Seite (118) der fast rechteckigen Form durch eine gemeinsame Wand (113) (3) getrennt. Die dadurch gebildete Reihe von Gefäßen (103) besitzt den Vorteil, dass sie trotz des verfügbaren Raums ein großes Volumen, vorzugsweise von 4 ml aufweisen. Ein weiterer Vorteil besteht darin, dass aufgrund der im Wesentlichen konstanten Wanddicke die Herstellung sehr wirtschaftlich ist. Ein weiterer Vorteil besteht darin, dass die Gefäße (103) sich gegenseitig stärken und somit eine hohe Stabilität der Form erhalten werden kann.
-
Zwischen den Reihen von Gefäßen (103) ist ein zusammenhängender Raum (121) angeordnet (6, 7). Der Raum (121) kann Magnete (202, 203) oder Heizvorrichtungen (128) (11) aufnehmen. Diese Magnete (202, 203) und Heizvorrichtungen (128) sind vorzugsweise massive Vorrichtungen. Somit können magnetische Partikel (216), die in Flüssigkeiten (215) eingeschlossen sind, die in den Gefäßen (103) gehalten werden können, von der Flüssigkeit (215) durch Anlegen eines Magnetfelds an die Gefäße (103) getrennt werden, wenn die Magnete (202, 203) in die Nähe der Gefäße (103) gebracht werden. Oder der Inhalt der Gefäße (103) kann bei einer erhöhten, kontrollierten Temperatur inkubiert werden, wenn die Arbeitsplatte (101) auf der Heizvorrichtung (128) angeordnet wird. Da die Magnete (202, 203) oder Heizvorrichtungen (128) massiv sein können, kann eine hohe Energiedichte erreicht werden. Die fast rechteckige Form des mittleren Teils (120) der Gefäße (103) (10) optimiert auch den Kontakt zwischen der Gefäßwand (109) und einem flach geformten Magnet (202) oder einer Heizvorrichtung (128) durch Optimieren der Kontaktfläche zwischen Gefäß (103) und Magnet (202) oder Heizvorrichtung (128) und verstärkt somit den Energieaustausch in das Gefäß (103).
-
Im Bereich des konischen Bodens (111) der Gefäße ist der Raum (121) sogar noch ausgeprägter und kann weitere Magnete (203) aufnehmen. Die Kombination der großen Magnete (202) im oberen Bereich und der kleineren Magnete (203) im konischen Bereich der Gefäße erlaubt die Trennung von magnetischen Partikeln (216) in größere oder kleine Volumina von Flüssigkeit (215). Die kleinen Magnete (203) machen es somit leichter, die magnetischen Teilchen (216) während des Eluat-Pipettierens zu sequestrieren. Dies ermöglicht das Pipettieren des Eluats mit minimalem Verlust durch Reduzieren des Totvolumens des magnetischen Partikelpellets (216). Weiterhin wird die Gegenwart von magnetischen Teilchen (216) in dem transferierten Eluat minimiert.
-
Am oberen Ende der Gefäße (103) umfasst eine der kürzeren Seitenwände (109) des Gefäßes (103) einen Reagenzeinlasskanal (105), der sich bis zu der Umfangsrippe (104) (3, 4, 7) erstreckt. Die Reagenzien werden auf den Reagenzeinlasskanal (105) pipettiert und laufen von dem Kanal (105) in das Gefäß (103) ab. Somit wird der Kontakt zwischen der Pipettennadel oder -spitze (3, 4) und der in dem Gefäß enthaltenen Flüssigkeit verhindert. Weiterhin werden Spritzer, die von der Flüssigkeit herrühren, die direkt in andere Flüssigkeit (215), die in den Gefäßen (103) enthalten ist, abgegeben wird, was Verunreinigung der Pipettennadel oder -spitze (3, 4) oder der benachbarten Gefäße (103) bewirken kann, verhindert. Sequentielles Pipettieren von kleinen Volumina von Reagenzien und anschließend von dem großen Volumen von einem anderen Reagenz auf den Reagenzeinlasskanal (105) stellt sicher, dass die Reagenzien, die nur in kleinen Mengen zugegeben werden, vollständig in das Gefäß (103) ablaufen. Somit ist das Pipettieren von kleinen Volumina von Reagenzien ohne Verlust von Genauigkeit des durchzuführenden Tests möglich.
-
Auf der Innenseite, auf dem Boden der Gefäße (111, 112) wird die Form konisch (111) und endet in einem kugeligen Boden (112) (6, 7). Die Innenform des Gefäßes (114), einschließlich des rechteckigen mittleren Teils (120), ist gerundet. Die Kombination von kugeligem Boden (112), gerundeter Innenform (114), konischem Teil (111) und verfeinerter Oberfläche der Gefäße (103) führt zu günstigen Fluiditäten, was eine wirksame Trennung und Reinigung von Analyten in der Arbeitsplatte (101) erleichtert. Die kugelige Boden (112) erlaubt eine im Wesentlichen vollständige Verwendung des abgetrennten Eluats und eine Reduktion des Totvolumens, welches das Mitschleppen von Reagenzien oder Proben-Kreuzverunreinigung vermindert.
-
Der Rand auf der Grundfläche (129) der Arbeitsplatte (101) umfasst Aussparungen (107) zum Eingriff mit Schnappbügeln (124) auf der Arbeitsstation (201) oder Heizvorrichtung (128) oder dem analytischen Instrument (126) (5, 9). Durch das Eingreifen der Schnappbügel (124) in die Aussparungen (107) lässt sich die Arbeitsplatte (101) auf der Arbeitsstation (201) anordnen und befestigen. Das Vorliegen der Aussparungen (107) erlaubt es, dass die Einrastkraft auf die Arbeitsplatte (101) fast vertikal zu der Grundfläche (129) wird. Somit können nur kleine Kräfte, die seitwärts wirken, auftreten. Dies vermindert das Auftreten von Spannung und somit die Verformung der Arbeitsplatte (101). Die vertikalen Einrastkräfte können auch alle Verformungen der Arbeitsplatte (101) neutralisieren, was zu einer exakteren Anordnung der kugeligen Böden (111) in der Arbeitsstation (201) führt. Im Allgemeinen vermindert die exakte Grenzfläche zwischen der Arbeitsplatte (101) und der Arbeitsstation (201) oder der Heizvorrichtung (128) in einem Analysator Totvolumina und vermindert auch das Risiko von Proben-Kreuzverunreinigung.
-
Eine „Trennstation” ist eine Vorrichtung oder eine Komponente eines analytischen Systems, das die Isolierung des festen Trägermaterials von dem anderen Material, das in der Fluidprobe vorhanden ist, erlaubt. Eine solche Trennstation kann z. B. eine Zentrifuge, ein Ständer mit Filterröhrchen, ein Magnet oder andere geeignete Komponenten umfassen, ist jedoch nicht darauf beschränkt. In einer bevorzugten Ausführungsform der Erfindung umfasst die Trennstation einen oder mehrere Magnete. Vorzugsweise werden ein oder mehrere Magnete zur Abtrennung von magnetischen Partikeln, vorzugsweise magnetischen Glaspartikeln, als ein fester Träger verwendet. Wenn beispielsweise die Fluidprobe und das feste Trägermaterial miteinander in den Vertiefungen einer Multiwellplatte kombiniert werden, kann ein oder mehrere Magnete, die in der Trennstation mit eingeschlossen sind, z. B. mit der Fluidprobe selbst durch Einbringen der Magnete in die Vertiefungen kontaktiert werden, oder der eine oder die mehreren Magnete können eng an die Außenwände der Wände gebracht werden, um die magnetischen Partikel anzuziehen und um sie anschließend aus der umgebenden Flüssigkeit abzutrennen.
-
In einer bevorzugten Ausführungsform ist die Trennstation eine Vorrichtung, die eine Multiwellplatte umfasst, die Gefäße mit einer Öffnung in der Oberfläche der Multiwellplatte und einem geschlossenen Boden umfasst. Die Gefäße umfassen einen oberen Teil, einen mittleren Teil und einen unteren Teil, wobei der obere Teil mit der Oberfläche der Multiwellplatte gekoppelt ist und vorzugsweise zwei längere und zwei kürzere Seiten einschließt. Der mittlere Teil weist einen im Wesentlichen rechteckigen Querschnitt mit zwei längeren Seiten auf, wobei die Gefäße in Reihen ausgerichtet sind. Zwischen zwei nebeneinander liegenden Reihen ist ein zusammenhängender Raum zum selektiven Kontaktieren von mindestens einem Magneten angeordnet, der auf einer Befestigung mit den Seitenwänden in mindestens zwei Z-Positionen montiert ist. Die Vorrichtung umfasst ferner eine magnetische Trennstation, die mindestens eine Haltevorrichtung umfasst. Die Haltevorrichtung umfasst mindestens einen Magneten, der ein Magnetfeld erzeugt. Ein beweglicher Mechanismus, der die mindestens eine Haltevorrichtung, die mindestens einen Magneten mindestens zwischen ersten und zweiten Positionen bezüglich der Gefäße der Multiwellplatte umfasst, vertikal bewegt, ist vorhanden. Vorzugsweise umfassen die mindestens zwei Z-Positionen der Gefäße die Seitenwände und den unteren Teil der Gefäße. Das Magnetfeld des mindestens einen Magneten zieht vorzugsweise die magnetischen Partikel zu einer inneren Fläche des Gefäßes, die an dem mindestens einen Magneten anliegt, wenn sich der mindestens eine Magnet in der ersten Position befindet. Die Auswirkung des Magnetfelds ist geringer, wenn sich der mindestens eine Magnet in der zweiten Position befindet, als wenn sich der mindestens eine Magnet in der ersten Position befindet. Vorzugsweise umfasst die Haltevorrichtung, die den mindestens einen Magneten umfasst, einen Rahmen. Die Gefäße weisen bevorzugte Merkmale auf, wie vorstehend im Zusammenhang mit der Multiwellplatte/Arbeitsplatte beschrieben. Ein solches Merkmal besteht darin, dass mindestens ein Teil der Gefäße einen im Wesentlichen rechteckigen Querschnitt orthogonal zu der Achse der Gefäße aufweist.
-
In der ersten Position liegt der mindestens eine Magnet an dem Teil der Gefäße an. Anliegend wird so verstanden, dass entweder in enger Nachbarschaft, derart, dass ein Magnetfeld auf den Inhalt des Gefäßes ausgeübt wird, oder in physikalischem Kontakt mit dem Gefäß gemeint ist.
-
Die Trennstation umfasst einen Rahmen, um die Multiwellplatte aufzunehmen, und Schnappbügel, um die Multiwellplatte zu befestigen. Vorzugsweise umfasst die Trennstation zwei Typen von Magneten. Diese bevorzugte Ausführungsform wird nachstehend weiter beschrieben.
-
Eine zweite bevorzugte Ausführungsform wird nachstehend beschrieben, die eine Feder einschließt, die einen Druck auf den Rahmen ausübt, der die Magnete umfasst, derart, dass die Magnete gegen die Gefäße der Multiwellplatte gepresst werden.
-
Die ersten Magnete sind vorzugsweise so konstruiert und angeordnet, um mit Gefäßen von einer Multiwellplatte zum Ausüben eines Magnetfelds auf ein großes Volumen von Flüssigkeit, das magnetische Partikel, die in den Gefäßen gehalten werden, zu Wechselwirken. Die zweiten Magnete sind vorzugsweise konstruiert und angeordnet, um mit Gefäßen von einer Multiwellplatte zum Ausüben eines Magnetfelds auf ein kleines Volumen von Flüssigkeit, das magnetische Partikel, die in dem Gefäß gehalten werden, umfasst, zu Wechselwirken. Die ersten und zweiten Magnete können zu verschiedenen Z-Positionen bewegt werden.
-
Geeignet im Zusammenhang mit der vorliegenden Erfindung und der Trennstation ist weiterhin ein Verfahren der Isolierung und Reinigung einer Nucleinsäure. Das Verfahren umfasst die Schritte des Bindens einer Nucleinsäure an magnetische Partikel in einem Gefäß von einer Multiwellplatte. Das Gefäß umfasst eine obere Öffnung, einen mittleren Teil und einen unteren Teil. Das gebundene Material wird anschließend von dem ungebundenen Material, das in einer Flüssigkeit enthalten ist, abgetrennt, wenn der Hauptteil der Flüssigkeit oberhalb des Abschnitts angeordnet ist, wo der konische Teil des Gefäßes durch den mittleren Teil mit der rechteckigen Form ersetzt wird, durch Bewegen eines Magnets von einer zweiten Position in eine erste Position und, in der ersten Position, Anlegen eines Magnetfelds an den mittleren Teil und gegebenenfalls zusätzlich Anlegen eines Magnetfelds an den unteren Teil des Gefäßes. Die magnetischen Partikel können gegebenenfalls mit einer Waschlösung gewaschen werden. Ein kleines Volumen von Flüssigkeit, wobei der Hauptteil der Flüssigkeit unterhalb des Abschnitts angeordnet ist, wo der konische Teil des Gefäßes durch den mittleren Teil mit der rechteckigen Form ersetzt wird, wird von den magnetischen Partikeln durch selektives Anlegen eines Magnetfelds an den Bodenteil des Gefäßes abgetrennt.
-
Geeignet im Zusammenhang mit der vorliegenden Erfindung ist auch eine magnetische Trennstation zum Abtrennen einer an magnetische Partikel gebundenen Nucleinsäure, wobei die Trennstation erste Magnete umfasst, die zur Wechselwirkung mit Gefäßen von einer Multiwellplatte zum Ausüben eines Magnetfelds auf ein großes Volumen von Flüssigkeit, umfassend magnetische Partikel, die in dem Gefäß gehalten werden, konstruiert und angeordnet sind, und zweite Magnete, die zur Wechselwirkung mit Gefäßen von einer Multiwellplatte zum Anlegen eines Magnetfelds an ein kleines Volumen von Flüssigkeit, das magnetische Partikel umfasst, die in den Gefäßen gehalten werden, konstruiert und angeordnet sind, und wobei die ersten und zweiten Magnete zu verschiedenen Z-Positionen bewegt werden können. Bevorzugte Ausführungsformen der magnetischen Trennstation sind hierin beschrieben.
-
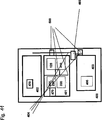
Eine erste bevorzugte Ausführungsform für eine Trennstation (201), die für die vorliegende Erfindung geeignet ist, ist nachstehend beschrieben. Die erste bevorzugte Ausführungsform der Trennstation (201) umfasst mindestens zwei Typen von Magneten (202, 203). Der erste, lange Typ von Magnet (202) ist konstruiert und angeordnet, um in den Raum (121) der Arbeitsplatte (101) zu passen. Der Magnet (202) übt somit ein Magnetfeld auf die Flüssigkeit (215) in dem Gefäß (103) aus, um magnetische Partikel (216) auf der Innenseite der Gefäßwand zu sequestrieren. Dies erlaubt die Abtrennung der magnetischen Partikel (216) und von jedem Material, das daran und die Flüssigkeit (215) im Inneren des Gefäßes (103) gebunden ist, wenn ein großes Volumen von Flüssigkeit (215) vorhanden ist. Der Magnet (202) besitzt eine längliche Struktur und ist zur Wechselwirkung mit dem im Wesentlichen rechteckigen mittleren Teil (120) des Gefäßes konstruiert und angeordnet. Somit wird der Magnet (202) verwendet, wenn der Hauptteil der Flüssigkeit (215) oberhalb des Abschnitts angeordnet ist, wo der konische Teil (111) des Gefäßes (103) durch den mittleren Teil (120) mit der rechteckigen Form ersetzt wird. Wie in 40 gezeigt, umfasst die bevorzugte Konstruktion der Magnete (202) die Haltevorrichtungen (204, 204a), die Magnete (202) umfasst, die in die Form (121) zwischen den Reihen von Gefäßen (103) in der Arbeitsplatte (101) passen. Eine weitere bevorzugte Ausführungsform der Magnete (202) umfasst Magnete (202), die auf Haltevorrichtungen (204, 204a) angeordnet sind. Die Magnete (203) der bevorzugten Trennstation (201) sind kleiner und können mit dem konischen Teil (111) des Gefäßes (103) wechselwirken. Dies ist in 10 gezeigt. Die Magnete (203) sind vorzugsweise auf einer Grundfläche (205) angeordnet, die in dem Raum (121) der Arbeitsplatte (101) bewegt werden kann. Jeder Magnet (202, 203) ist vorzugsweise zur Wechselwirkung mit zwei Gefäßen (103) in zwei nebeneinander liegenden Reihen konstruiert. In einer bevorzugten Ausführungsform besitzt die Arbeitsplatte (101) 6 Reihen von 8 Gefäßen (103). Eine Trennstation (201), die mit der bevorzugten Arbeitsplatte (101) Wechselwirken kann, besitzt drei Haltevorrichtungen (204, 204a), die Magnete (202) und vier Grundflächen (205), die Magnete (203) umfasst, umfassen. Eine Ausführungsform, wobei die Trennstation vier magnetische Haltevorrichtungen (204, 204a) aufweist, die Magnete (202), und drei magnetische Grundflächen (205), die Magnete (203) umfasst, umfassen, ist auch eingeschlossen.
-
Die Magnete (202, 203) sind beweglich. Die Trennstation (201) umfasst einen Mechanismus zur Bewegung der Haltevorrichtungen (204, 204a) und der Grundflächen (205). Sämtliche Haltevorrichtungen (204, 204a) sind durch eine Grundfläche (217) miteinander gekoppelt und werden somit koordinativ bewegt. Sämtliche Magnete (203) sind mit der Grundfläche (218) gekoppelt und werden somit koordinativ bewegt. Der Mechanismus zum Bewegen der magnetischen Platten (202) und (203) ist zur Bewegung der beiden Typen von magnetischen Platten (202, 203) zu insgesamt vier Endpositionen konstruiert und angeordnet:
In 40a–c sind die Magnete (203) in enger Nachbarschaft zu dem konischen Teil der Gefäße (103) der Arbeitsplatte (101) angeordnet. Dies ist die oberste Position von Magneten (203) und ist die Trennposition. In dieser Figur sind die Magnete (202) in der untersten Position angeordnet. Sie sind nicht an der Trennung beteiligt, wenn sie sich in dieser Position befinden.
-
In der in 10 gezeigten bevorzugten Ausführungsform ist die Grundfläche (217) der Magnete (202) mit einer Positionierscheibe (206) gekoppelt. Die Grundfläche (217) umfasst ein unteres Ende (207), das mit einem Verbindungselement (208) durch ein bewegliches Element (209) flexibel in Kontakt ist. Das bewegliche Element ist zur Bewegung des Verbindungselementes (208) entlang einer Schiene (212) von einer Seite zu der anderen konstruiert und angeordnet. Das bewegliche Element (209) ist an dem Verbindungselement (208) mit einem Stift (220) befestigt. Das Verbindungselement (208) ist an der Positionierscheibe (206) durch Schraube (210) befestigt. Das Verbindungselement (208) ist ebenfalls an der Achse (211) befestigt. Das Verbindungselement (208) ist vorzugsweise eine rechteckige Platte. Wenn sich die Positionierscheibe (206) exzentrisch um eine Achse (211) bewegt, derart dass sich die Schraube (210) von einem Punkt oberhalb der exzentrischen Achse zu einem Punkt unterhalb der exzentrischen Achse bewegt, werden das bewegliche Element (209) und das untere Ende (207) der Grundfläche (204) mit den daran angebrachten Magneten (202) von der obersten Position zu der untersten Position bewegt. Die Basis (218) ist an einem unteren Teil (219) befestigt und ist an ihrem unteren Ende mit einem Stift (213) an einem beweglichen Element (214) befestigt, welches vorzugsweise ein Rad ist, welches mit der Positionierscheibe (206) wechselwirkt. Wenn sich die Positionierscheibe (214) um die Achse (211) dreht, bewegt sich das Rad (214) entlang der Positionierscheibes (206). Wenn das Rad (214) an einem Abschnitt der Positionierscheibe (206) angeordnet ist, wo der Abstand von der Achse (211) kurz ist, sind die Magnete (203) in ihrer untersten Position. Wenn das Rad (214) an einem Abschnitt der Positionierscheibe (206) angeordnet ist, wo der Abstand von der Achse (211) maximal ist, sind die Magnete (203) in ihrer obersten Position. Somit wird in der bevorzugten Ausführungsform der ersten Ausführungsform der Trennstation die Anordnung der Magnete (203) durch die Form der Positionierscheibe (206) kontrolliert. Wenn sich das bewegliche Element (209) entlang des mittleren, gerundeten oberen oder unteren Teils (212a) der Schiene (212) bewegt, werden die kleinen Typen von Magneten (203) auf und ab bewegt. Wenn das bewegliche Element (209) auf der Seite (212b) des unteren Endes (207) angeordnet ist und sich auf und ab bewegt, werden die Magnete (202) nach oben oder nach unten bewegt. Die Positionierscheibe kann durch einen Motor (224) gedreht werden.
-
In einer bevorzugten Ausführungsform ist eine Feder (225) an der Grundfläche (222) der Trennstation und der Grundfläche (218) der Magnete (203) gekoppelt, um zu gewährleisten, dass die Magnete (203) in die unterste Position bewegt werden, wenn sie nach unten bewegt werden.
-
Der Begriff „Stift” wie hierin verwendet bezieht sich auf jedes Befestigungselement, darunter Schrauben oder Stifte.
-
In einer zweiten bevorzugten Ausführungsform umfasst die Trennstation (230) mindestens eine Haltevorrichtung (231), die mindestens einen Magneten (232), vorzugsweise eine einer Anzahl von Gefäßen (103) in einer Reihe (123) entsprechende Anzahl von Magneten umfasst. Vorzugsweise umfasst die Trennstation (230) eine Anzahl von Haltevorrichtungen (231), die der Anzahl von Reihen (123) der Multiwellplatte (101), die hierin zuvor beschrieben wurde, entspricht. Stärker bevorzugt sind sechs Haltevorrichtungen (231) auf der Trennstation (230) befestigt. Mindestens ein Magnet (232) ist an der Haltevorrichtung (231) befestigt. Vorzugsweise entspricht die Anzahl von Magneten (232) der Anzahl von Gefäßen (103) in einer Reihe (123). Am stärksten bevorzugt sind acht Magneten (232) an einer Haltevorrichtung (231) befestigt. Vorzugsweise ist ein Typ von Magnet (232) an der Haltevorrichtung (231) mit umfasst. Stärker bevorzugt ist der Magnet (232) auf einer Seite orientiert in Richtung der Gefäße, mit welchen der Magnet wechselwirkt.
-
Die Haltevorrichtung (231) ist auf einer Grundfläche (233) befestigt. Vorzugsweise ist diese Befestigung flexibel. Die Grundfläche (233) umfasst Federn (234), die daran befestigt sind. Die Anzahl von Federn (234) beträgt mindestens eine Feder pro Haltevorrichtung (231), die auf der Grundfläche (233) befestigt ist. Die Basis umfasst weiterhin eine Abschrägung (236), die die Bewegung der Feder und folglich der Haltevorrichtung (231), die die Magnete (232) einschließt, begrenzt. Vorzugsweise ist eine der Federn (234) zur Wechselwirkung mit einer Haltevorrichtung (231) konstruiert und angeordnet. Stärker bevorzugt ist die Feder (234) eine Winkelhebelfeder. Die Wechselwirkung kontrolliert die horizontale Bewegung der Haltevorrichtungen (231). Weiterhin umfasst die Trennstation (230) einen Rahmen (235). Die Basis (233) mit Haltevorrichtungen (231) ist an den Rahmen (235) über einen beweglichen Mechanismus, wie hierin zuvor für die Magnete (232) der ersten Ausführungsform beschrieben, gekoppelt.
-
Vorzugsweise sind Basis (233) und Haltevorrichtung (231) zur Bewegung vertikal (in Z-Richtung) konstruiert und angeordnet.
-
Die Multiwellplatte (101), die hierin zuvor beschrieben wurde, wird in die Trennstation (230) eingeführt. Die Haltevorrichtung (231), die die Magnete (232) einschließt, wird vertikal bewegt. Jede einzelne Haltevorrichtung (232) wird somit in einen Raum (121) zwischen zwei Reihen (123) von Gefäßen (103) hineinbewegt. Die vertikale Bewegung bringt die auf einer Haltevorrichtung (231) befestigten Magnete (232) mit den Gefäßen (103) in Kontakt. Die Z-Position wird in Abhängigkeit von dem Volumen von Flüssigkeit (215) im Inneren der Gefäße (103) gewählt. Für große Volumina kontaktieren die Magnete (232) die Gefäße (103) in einer zentralen Position (120), wobei die Gefäße (103) von fast rechteckiger Form sind. Für kleine Volumina von Flüssigkeit (215), wobei der Hauptteil der Flüssigkeit (215) unterhalb des mittleren Teils (120) der Gefäße (103) angeordnet ist, kontaktieren die Magnete (232) vorzugsweise den konischen Teil (111) der Gefäße (103).
-
An der Grundfläche (233) von jedem einzelnen Rahmen (231) ist eine Feder angebracht (9a), b)). Die Feder presst die Magnete (232) gegen die Gefäße (103). Dies gewährleistet einen Kontakt zwischen den Magneten (232) und den Gefäßen (103) während der magnetischen Trennung. Vorzugsweise kontaktiert der Magnet (232) das Gefäß (103) an der Seitenwand (109), die unterhalb des Einlasses (105) angeordnet ist. Dies hat den Vorteil, dass Flüssigkeit, die durch Pipettieren zugesetzt wird, über die sequestrierten magnetischen Partikel fließt und sicherstellt, dass Partikel resuspendiert werden, und dass alle Proben in allen Gefäßen identisch behandelt werden.
-
Diese Ausführungsform ist besonders zur Abtrennung einer Flüssigkeit (215), die in einer Multiwellplatte (101), wie hierein zuvor beschrieben, enthalten ist, von magnetischen Partikeln (216), geeignet, wenn verschiedene Niveaus von Flüssigkeit (215) in den Gefäßen (103) der Multiwellplatte (101) enthalten sind.
-
Ein „Waschpuffer” ist ein Fluid, das zur Entfernung unerwünschter Komponenten, insbesondere bei einem Reinigungsvorgang, ausgelegt ist. Solche Puffer sind auf dem Fachgebiet wohlbekannt. Im Zusammenhang mit der Reinigung von Nucleinsäuren ist der Waschpuffer zum Waschen des festen Trägermaterials geeignet, um die immobilisierte Nucleinsäure von sämtlichen unerwünschten Komponenten abzutrennen. Der Waschpuffer kann beispielsweise Ethanol und/oder chaotrope Mittel in einer gepufferten Lösung oder in Lösungen mit einem sauren pH-Wert ohne Ethanol und/oder chaotrope Mittel, wie vorstehend beschrieben, enthalten. Oft werden die Waschlösung oder andere Lösungen als Stammlösungen bereitgestellt, die vor der Verwendung verdünnt werden müssen.
-
Das Waschen in dem Verfahren gemäß der Erfindung erfordert einen mehr oder weniger intensiven Kontakt des festen Trägermaterials und der darauf immobilisierten Nucleinsäuren mit dem Waschpuffer. Verschiedene Verfahren sind möglich, um dies zu bewerkstelligen, z. B. Schütteln des Waschpuffers mit dem festen Trägermaterial in oder entlang des jeweiligen Gefäßes oder der jeweiligen Gefäße. Ein weiteres vorteilhaftes Verfahren ist ein- oder mehrmaliges Ansaugen und Abgeben der Suspension, die Waschpuffer und festes Trägermaterial umfasst. Dieses Verfahren wird vorzugsweise unter Verwendung einer Pipette durchgeführt, wobei die Pipette vorzugsweise eine Wegwerf-Pipettenspitze umfasst, in die die Suspension eingesaugt wird und aus der sie wieder abgegeben wird. Eine solche Pipettenspitze kann mehrmals verwendet werden, bevor sie weggeworfen und ersetzt wird. Wegwerf-Pipettenspitzen, die für die Erfindung geeignet sind, besitzen vorzugsweise ein Volumen von mindestens 10 μl, stärker bevorzugt von mindestens 15 μl, stärker bevorzugt von mindestens 100 μl, stärker bevorzugt von mindestens 500 μl, stärker bevorzugt von mindestens 1 ml, noch stärker bevorzugt von etwa 1 ml. Pipetten, die im Zusammenhang mit der Erfindung verwendet werden, können auch Pipettiernadeln sein.
-
Somit ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, bei das Waschen in Schritt c. das Ansaugen und Abgeben des Waschpuffers, der das feste Trägermaterial umfasst, umfasst.
-
Zur Leichtigkeit der Handhabung und zur Erleichterung der Automation ist es bevorzugt, die Gefäße, die vorstehend erwähnt sind, in einer integralen Anordnung zu kombinieren, so dass sie miteinander gehandhabt werden können.
-
Folglich ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei die Gefäße in einer integralen Anordnung kombiniert sind.
-
Integrale Anordnungen können z. B. Ampullen oder Röhrchen sein, die miteinander oder in einem Reagenzglasständer reversibel oder irreversibel gekoppelt sind. Vorzugsweise ist die integrale Anordnung eine Multiwellplatte. Stärker bevorzugt ist die Multiwellplatte eine Deepwellplatte.
-
Das Verfahren gemäß der Erfindung ist besonders geeignet, wenn verschiedene Typen von Nucleinsäuren herzustellen sind, da das Bereitstellen von einem einzigen Arbeitsablauf und den gleichen Reagenzien das Erfordernis ausschaltet, verschiedene Typen von Nucleinsäuren, wie DNA und RNA, aufgrund ihrer verschiedenen Eigenschaften einzeln zu isolieren.
-
Somit ist ein bevorzugter Aspekt der Erfindung das vorstehend erwähnte Verfahren, wobei die erste Zielnucleinsäure RNA und die zweite Zielnucleinsäure DNA umfasst.
-
Weiterhin können mehrere verschiedene Fluidproben verschiedene Organismen umfassen oder können aus verschiedenen Organismen abgeleitet sein. Auch dann ist es vorteilhaft, die jeweilige Nucleinsäure gleichzeitig mit dem gleichen Arbeitsablauf und den gleichen Reagenzien zu gewinnen. Die vorliegende Erfindung erlaubt eine solche gleichzeitige Präparation von Nucleinsäuren z. B. aus Bakterien, DNA-Viren und RNA-Viren, trotz ihrer unterschiedlichen Struktur und Eigenschaften.
-
Daher ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei die erste Zielnucleinsäure und die zweite Zielnucleinsäure aus verschiedenen Organismen stammen.
-
Ein weiterer bevorzugter Aspekt der Erfindung ist das vorstehend beschriebene Verfahren, wobei die erste und/oder die zweite Nucleinsäure eine nicht-virale Nucleinsäure ist.
-
Ebenfalls ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei die erste und/oder die zweite Zielnucleinsäure eine bakterielle Nucleinsäure ist.
-
Ein „Organismus”, wie hierin verwendet, bedeutet jede lebende einzellige oder mehrzellige Lebensform. Im Zusammenhang mit der Erfindung ist ein Virus ein Organismus.
-
Die vorliegende Erfindung ist auch geeignet, wenn verschiedene Nucleinsäuren aus einer Vielzahl von verschiedenen Typen von Fluidproben stammen sollen. Somit können verschiedene Nucleinsäuren in parallelen gleichzeitigen Extraktionen unter den gleichen physikalischen Bedingungen isoliert werden, und können dann z. B. analytisch in verschiedenen Gefäßen weiter bearbeitet werden.
-
Somit ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei die erste Nucleinsäure in einer ersten Fluidprobe vorhanden ist und die zweite Nucleinsäure in einer zweiten Fluidprobe vorhanden ist.
-
Eine solche Ausführungsform ist besonders geeignet, wenn die verschiedene Nucleinsäuren nicht in Kontakt miteinander sind und getrennt bearbeitet werden können. Darum ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei die zweite Zielnucleinsäure in der ersten Fluidprobe fehlt.
-
Allerdings können auch verschiedene Nucleinsäuren innerhalb derselben Probe vorhanden sein, aber es müssen nicht notwendigerweise alle von ihnen nach der Isolierung weiter bearbeitet werden. Die vorliegende Erfindung ist auch in diesen Fällen geeignet.
-
Daher ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei die zweite Nucleinsäure ebenfalls in der ersten Fluidprobe vorhanden ist.
-
Im Falle des nachgeschalteten Bearbeitens, insbesondere wenn diagnostische Techniken eingesetzt werden, wie Nucleinsäureamplifikationsverfahren, ist es oft wünschenswert oder sogar erforderlich, eine oder mehrere Kontrollnucleinsäuren einzuschließen. Auf diese Weise kann entweder die analytische Reaktion kontrolliert werden, wenn die Kontrolle der gereinigten Nucleinsäure zugesetzt wird, oder auch die Probenpräparation kann überwacht werden, wenn die Kontrolle vor oder während Nucleinsäureextraktion zugesetzt wird. Es ist auch üblich und bevorzugt, beide Typen von Kontrollen einzuschließen.
-
In dieser Hinsicht ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei eine Kontrollnucleinsäure der Fluidprobe und/oder der gereinigten Nucleinsäure bei einem der Schritte zugesetzt wird.
-
Zum Binden der Nucleinsäuren an das feste Trägermaterial und, sofern anwendbar, zur Lyse von Zellen und Viren, hat es sich als vorteilhaft erwiesen, bei Temperaturen bis 50°C zu inkubieren.
-
Somit ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei Schritt a. bei einer Temperatur von bis zu 50°C, vorzugsweise bei einer Temperatur zwischen 35°C und 45°C, stärker bevorzugt bei einer Temperatur von 40°C durchgeführt wird.
-
Zum nachgeschalteten Bearbeiten der isolierten Nucleinsäuren kann es zweckmäßig sein, sie aus dem festen Träger abzutrennen, bevor sie z. B. der Amplifikation unterzogen werden.
-
Darum ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei das Verfahren weiterhin nach Schritt c. den folgenden Schritt umfasst:
- d. Eluieren der Nucleinsäuren aus dem festen Trägermaterial mit einem Elutionspuffer.
-
Ein „Elutionspuffer” im Zusammenhang mit der Erfindung ist eine geeignete Flüssigkeit zum Abtrennen der Nucleinsäuren von dem festen Träger. Eine solche Flüssigkeit kann z. B. destilliertes Wasser oder wässrige Salzlösungen sein, wie z. B. Tris-Puffer wie Tris-HCl oder HEPES, oder andere geeignete, dem Fachmann bekannte Puffer. Der pH-Wert eines solchen Elutionspuffers ist vorzugsweise alkalisch oder neutral. Der Elutionspuffer kann weitere Komponenten enthalten, wie z. B. Chelatbildner, wie EDTA, die die isolierten Nucleinsäuren durch Inaktivierung von Abbauenzymen stabilisieren.
-
Die Elution wird vorzugsweise bei erhöhten Temperaturen durchgeführt, derart, dass eine bevorzugte Ausführungsform der Erfindung das vorstehend beschriebene Verfahren ist, wobei Schritt d. bei einer Temperatur zwischen 70°C und 90°C, stärker bevorzugt bei einer Temperatur von 80°C durchgeführt wird.
-
Wie vorstehend erwähnt, ist es oft wünschenswert, die durch das vorstehend beschriebene Verfahren isolierten Nucleinsäuren zu analysieren. Hierzu kann es zweckmäßig sein, die Menge an Ausgangsmaterial für die Analyse zu erhöhen.
-
Darum ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei das Verfahren weiterhin nach Schritt c. oder nach Schritt d. die folgenden Schritte umfasst:
- e. Überführen der gereinigten Nucleinsäuren und gegebenenfalls des festen Trägermaterials in eine Vielzahl von Reaktionsgefäßen,
- f. Amplifizieren der Zielnucleinsäuren.
-
In diesem Zusammenhang ist es besonders zweckmäßig, Amplifikations- und Nachweisverfahren einzusetzen, die die gleichzeitige Amplifikation und den Nachweis von mehreren verschiedenen Nucleinsäuren in zwei oder mehreren Reaktionsgefäßen unter den gleichen physikalischen Bedingungen und unter Verwendung der gleichen Reagenzien erlauben. Eine Kombination einer solchen Technik mit der schnellen und wirksamen Probenpräparation, die vorstehend beschrieben ist, kann zum Bereitstellen z. B. von integrierten automatisierten Lösungen sehr zweckmäßig sein, wobei der gleiche Arbeitsablauf an einer Vielzahl von verschiedenen Typen von Proben, die verschiedene Nucleinsäuren enthalten, durchgeführt wird. Diese Proben können parallel bearbeitet werden, um gleichzeitig die verschiedenen Nucleinsäuren, die sie enthalten, zu isolieren, und die Analyse der isolierten verschiedenen Nucleinsäuren kann anschließend ebenfalls gleichzeitig durchgeführt werden. Die Kombination von diesen Ansätzen vermindert signifikant die Komplexität und die Wartezeit auf Ergebnisse für solche Experimente, was insbesondere für diagnostische Laboratorien in einem klinischen Umfeld von beträchtlichem Vorteil ist.
-
Somit ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei Schritt f. die folgenden Schritte umfasst:
- i. Kontaktieren der gereinigten Nucleinsäuren mit einem oder mehreren Amplifikationsreagenzien, umfassend eine Polymerase mit reverser Transkriptaseaktivität in mindestens zwei Reaktionsgefäßen, wobei mindestens ein erstes Reaktionsgefäß mindestens die erste Zielnucleinsäure umfasst und mindestens ein zweite Reaktionsgefäß mindestens die zweite Zielnucleinsäure umfasst, und wobei die zweite Zielnucleinsäure in dem ersten Reaktionsgefäß fehlt;
- ii. Inkubieren in den Reaktionsgefäßen der gereinigten Nucleinsäuren mit dem einen oder den mehreren Amplifikationsreagenzien für einen Zeitraum und unter Bedingungen, die geeignet sind, dass die Transkription von RNA durch die Polymerase mit reverser Transkriptaseaktivität eintritt;
- iii. Inkubieren in den Reaktionsgefäßen der gereinigten Nucleinsäuren mit dem einen oder den mehreren Amplifikationsreagenzien für einen Zeitraum und unter Bedingungen, die ausreichen, dass eine Amplifikationsreaktion, die auf die Gegenwart oder Abwesenheit der ersten und zweiten Zielnucleinsäure hinweist, eintritt,
wobei die Bedingungen für Transkription und Amplifikation in den Schritten i. bis iii. für die mindestens erste und zweite Zielnucleinsäure identisch sind.
-
Im Hinblick auf den Amplifizierungsvorgang bestand eine Herausforderung an die bisherige Technik darin, dass die Anzahl von verschiedenen Zielnucleinsäuren in einem Multiplex-Test, der in einem einzigen Reaktionsgefäß ausgeführt wird, durch die Anzahl an entsprechenden Markern begrenzt ist. In einem PCR-Realzeit-Test hat beispielsweise die potentielle Überlappung von fluorochromen Spektren einen großen Einfluss auf die Testleistung (Risiko von falsch positiven Ergebnissen, geringere Genauigkeit, etc.) Darum müssen die jeweiligen Fluorophore mit Bedacht gewählt werden und spektral gut voneinander getrennt sein, um die gewünschte Leistung eines diagnostischen Tests sicherzustellen. Typischerweise entspricht die Anzahl von verschiedenen geeigneten Fluorophoren einer einstelligen Anzahl von PCR-Instrument-Fluoreszenzkanälen.
-
Im Gegensatz dazu erfolgt bei dem vorstehend beschriebenen Verfahren die Amplifikation von mindestens einer ersten und einer zweiten Zielnucleinsäure in mindestens zwei verschiedenen Reaktionsgefäßen, was die gleichzeitige Amplifikation von einer höheren Anzahl von verschiedenen Zielnucleinsäuren erlaubt, da Signale in verschiedenen Reaktionsgefäßen unabhängig voneinander nachgewiesen werden können. Immer noch innerhalb des Umfangs der vorliegenden Erfindung sind Ausführungsformen, wobei in einem oder in mehr als einem der mehreren Reaktionsgefäße Multiplex-Reaktionen durchgeführt werden, wodurch die Anzahl von Zielen, die gleichzeitig und unter den gleichen Bedingungen amplifiziert werden können, vervielfältigt wird.
-
„Amplifikationsreagenzien” im Zusammenhang mit der Erfindung sind chemische oder biochemische Komponenten, die die Amplifikation von Nucleinsäuren ermöglichen. Solche Reagenzien umfassen, sind aber nicht beschränkt auf, Nucleinsäurepolymerasen, Puffer, Mononucleotide, wie Nucleosidtriphosphate, Oligonucleotide, z. B. Oligonucleotidprimer, Salze und ihre jeweiligen Lösungen, Nachweissonden, Farbstoffe und mehr.
-
Wie es auf dem Fachgebiet bekannt ist, ist ein „Nucleosid” eine Base-Zucker-Kombination. Der Baseteil des Nucleosids ist normalerweise eine heterocyclische Base. Die zwei häufigsten Klassen von solchen heterocyclischen Basen sind Purine und Pyrimidine.
-
„Nucleotide” sind Nucleoside, die weiterhin eine kovalent an den Zuckerteil des Nucleosids gebundene Phosphatgruppe einschließen. Für diese Nucleoside, die Pentofuranosyl-Zucker einschließen, kann die Phosphatgruppe mit entweder der 2'-, 3'- oder 5'-Hydroxyleinheit des Zuckers verknüpft sein. Ein Nucleotid ist die monomere Einheit von einem „Oligonucleotid”, das allgemeiner als eine „oligomere Verbindung” oder ein „Polynucleotid” bezeichnet werden kann, das allgemeiner als eine „polymere Verbindung” bezeichnet wird. Ein weiterer allgemeiner Ausdruck für das zuvor Genannte ist Desoxyribonucleinsäure (DNA) und Ribonucleinsäure (RNA).
-
Gemäß der Erfindung ist eine „oligomere Verbindung” eine Verbindung, die aus „monomeren Einheiten” besteht, die Nucleotide allein oder nicht-natürliche Verbindungen (siehe nachstehend), spezieller modifizierte Nucleotide (oder Nucleotidanaloge) oder Nicht-Nucleotid-Verbindungen, allein oder Kombinationen davon, sein können.
-
„Oligonucleotide” und „modifizierte Oligonucleotide” (oder „Oligonucleotid-Analoge”) sind Untergruppen von oligomeren Verbindungen. Im Zusammenhang mit dieser Erfindung bezieht sich der Begriff „Oligonucleotid” auf Komponenten, die aus einer Vielzahl von Nucleotiden als ihre monomeren Einheiten gebildet sind. Die Phosphatgruppen werden allgemein als das Internucleosid-Rückgrat des Oligonucleotids bildend bezeichnet. Die normale Verknüpfung oder das Rückgrat von RNA und DNA ist eine 3'- zu 5'-Phosphodiesterverknüpfung. Oligonucleotide und modifizierte Oligonucleotide (siehe nachstehend), die für die Erfindung geeignet sind, können synthetisiert werden, wie es im Prinzip auf dem Fachgebiet beschrieben und dem Fachmann auf dem Gebiet bekannt. Verfahren zur Herstellung von oligomeren Verbindungen von speziellen Sequenzen sind auf dem Fachgebiet bekannt und umfassen beispielsweise Klonieren und Restriktion von entsprechenden Sequenzen und die direkte chemische Synthese. Chemische Syntheseverfahren können beispielsweise das Phosphotriester-Verfahren einschließen, das bei
Narang S. A. et al., Methods in Enzymology 68 (1979) 90–98 beschrieben ist, das Phosphodiester-Verfahren, das bei
Brown E. L., et al. Methods in Enzymology 68 (1979) 109–151 offenbart ist, das Phosphoramidit-Verfahren, das bei
Beaucage et al., Tetrahedron Letters 22 (1981) 1859 offenbart ist, das H-Phosphonat-Verfahren, das bei
Garegg et al., Chem. Scr. 25 (1985) 280–282 offenbart ist, und das Verfahren des festen Trägers, das in der
US 4,458,066 offenbart ist.
-
Bei dem Verfahren gemäß der Erfindung können die Oligonucleotide chemisch modifiziert werden, d. h. der Primer und/oder die Sonde umfassen ein modifiziertes Nucleotid oder eine Nicht-Nucleotid-Verbindung. Die Sonde oder der Primer ist dann ein modifiziertes Oligonucleotid.
-
„Modifizierte Nucleotide” (oder „Nucleotidanaloge”) unterscheiden sich von einem natürlichen Nucleotid durch eine Modifikation, bestehen aber immer noch aus einer Base, einem Pentofuranosyl-Zucker, einem Phosphatteil, einem basenartigen Teil, Pentofuranosyl-Zucker-artigen Teil, und einem Phosphat-artigem Teil, oder Kombinationen davon. Beispielsweise kann eine Markierung an dem Basenteil von einem Nucleotid angebracht sein, wodurch ein modifiziertes Nucleotid erhalten wird. Eine natürliche Base in einem Nucleotid kann auch durch z. B. ein 7-Desazapurin ersetzt werden, wodurch ebenso ein modifiziertes Nucleotid erhalten wird.
-
Ein „modifiziertes Oligonucleotid” (oder „Oligonucleotidanalog”), das einer anderen spezifischen Untergruppe von oligomeren Verbindungen angehört, besitzt ein oder mehrere Nucleotide und ein oder mehrere modifizierte Nucleotide als monomere Einheiten. Somit bezieht sich der Begriff „modifiziertes Oligonucleotid” (oder „Oligonucleotidanalog”) auf Strukturen, die in einer Weise funktionieren, die im Wesentlichen den Oligonucleotiden ähnlich ist, und kann im Zusammenhang mit der vorliegenden Erfindung synonym verwendet werden. Aus synthetischer Sicht kann ein modifiziertes Oligonucleotid (oder ein Oligonucleotidanalog) beispielsweise durch chemische Modifikation von Oligonucleotiden durch entsprechende Modifikation des Phosphatgerüstes, Riboseeinheit oder der Nucleotidbasen hergestellt werden (
Uhlmann und Peyman, Chemical Reviews 90 (1990) 543;
Verma S., und Eckstein F., Annu. Rev. Biochem. 67 (1998) 99–134). Repräsentative Modifikationen umfassen Phosphorothioat-, Phosphorodithioat-, Methylphosphonat-, Phosphotriester- oder Phosphoramidat-Internucleosid-Verknüpfungen anstelle der Phosphodiester-Internucleosidverknüpfungen; Deaza- oder Azapurine und -pyrimidine anstelle von natürlichen Purin- und Pyrimidinbasen, Pyrimidinbasen mit Substituentengruppen an der 5- oder 6-Position; Purinbasen mit veränderten Substituentengruppen an der 2-, 6- oder 8-Position oder 7-Position als 7-Desazapurine; Basen, die Alkyl-, Alkenyl-, Alkinyl- oder Aryleinheiten tragen, z. B. Niederalkylgruppen, wie Methyl-, Ethyl-, Propyl-, Butyl-, tert.-Butyl-, Pentyl-, Hexyl-, Heptyl-, Octyl-, Nonyl-, Decyl- oder Arylgruppen, wie Phenyl, Benzyl, Naphthyl; Zucker mit Substituentengruppen an beispielsweise ihrer 2'-Position; oder carbocyclische oder acyclische Zuckeranaloge. Weitere Modifikationen, die mit dem Geist dieser Erfindung im Einklang stehen, sind der Fachwelt bekannt. Derartige modifizierte Oligonucleotide (oder Oligonucleotidanaloge) werden am besten als funktionell äquivalent mit, jedoch strukturell verschieden von natürlichen Oligonucleotiden beschrieben. Im Einzelnen sind beispielhafte Modifikationen bei
Verma S. und Eckstein F., Annu. Rev. Biochem. 67 (1998) 99–134 oder
WO 02/12263 offenbart. Zusätzlich kann eine Modifikation vorgenommen werden, wobei Nucleosideinheiten über Gruppen verknüpft werden, die die Internucleosid-Phosphat- oder Zucker-Phosphat-Verknüpfungen ersetzen. Solche Verknüpfungen umfassen, diejenigen, die bei
Verma S. und Eckstein F. Annu. Rev. Biochem. 67 (1998) 99–134 offenbart sind. Wenn andere als Phosphatverknüpfungen zur Verknüpfung der Nucleosideinheiten verwendet werden, werden solche Strukturen ebenfalls als „Oligonucleoside” beschrieben.
-
Eine „Nucleinsäure” sowie die „Zielnucleinsäure” ist eine polymere Verbindung von Nucleotiden, wie sie dem Fachmann bekannt ist. „Zielnucleinsäure” wird hierin zur Bezeichnung einer Nucleinsäure in einer Probe verwendet, d. h. die Gegenwart, die Nicht-Gegenwart und/oder die Menge davon in einer Probe sollten bestimmt werden.
-
Der Begriff „Primer” wird hierin wie dem Fachmann bekannt verwendet und bezieht sich auf oligomere Verbindungen, hauptsächlich auf Oligonucleotide, aber auch auf modifizierte Oligonucleotide, die in der Lage sind, die DNA-Synthese durch eine templatabhängige DNA-Polymerase zu initiieren, d. h. das 3'-Ende von z. B. dem Primer stellt eine freie 3'-OH-Gruppe bereit, woran weitere Nucleotide von einer templatabhängigen DNA-Polymerase angeknüpft werden können, die eine 3'- zu 5'-Phosphodiesterverknüpfung entwickeln, wobei Desoxynucleosidtriphosphate verwendet werden und wobei Pyrophosphat freigesetzt wird.
-
Eine „Sonde” bezeichnet ebenfalls ein natürliches oder modifiziertes Oligonucleotid. Wie auf dem Fachgebiet bekannt, dient eine Sonde dem Zweck des Nachweises eines Analyten oder Amplifikats. Im Falle des Verfahrens gemäß der Erfindung können Sonden zum Nachweis der Amplifikate der Zielnucleinsäuren verwendet werden. Für diesen Zweck tragen die Sonden typischerweise Markierungen.
-
„Markierungen”, die oft als „Reportergruppen” bezeichnet werden, sind allgemein Gruppen, die eine Nucleinsäure, insbesondere Oligonucleotide oder modifizierte Oligonucleotide, sowie alle Nucleinsäuren, die daran gebunden sind, von dem Rest der Probe unterscheidbar machen (Nucleinsäuren mit einer angeknüpften Markierung können ebenfalls als markierte Nucleinsäure-bindende Verbindungen, markierte Sonden oder einfach als Sonden bezeichnet werden). Bevorzugte Markierungen gemäß der Erfindung sind fluoreszierende Markierungen, die z. B. fluoreszierende Farbstoffe sind, wie ein Fluoresceinfarbstoff, ein Rhodaminfarbstoff, ein Cyaninfarbstoff oder ein Cumarinfarbstoff. Bevorzugte fluoreszierende Farbstoffe gemäß der Erfindung sind FAM, HEX, JA270, CAL635, Coumarin343, Quasar705, Cyan500, Cy5.5, LC-Red 640, LC-Red 705.
-
Im Zusammenhang mit der Erfindung kann ein Primer und/oder eine Sonde chemisch modifiziert sein, d. h. der Primer und/oder die Sonde umfassen ein modifiziertes Nucleotid oder eine Nicht-Nucleotid-Verbindung. Die Sonde oder der Primer ist dann ein modifiziertes Oligonucleotid.
-
Ein bevorzugtes Verfahren der Nucleinsäureamplifikation ist die Polymerse-Kettenreaktion (PCR), die, unter anderen Druckschriften, in den
US-Patentschriften Nrn. 4,683,202 ,
4,683,195 ,
4,800,159 und
4,965,188 offenbart ist. Die PCR setzt typischerweise zwei oder mehr Oligonucleotid-Primer ein, die an ein ausgewähltes Nucleinsäuretemplat (z. B. DNA oder RNA) binden. Primer, die für die Nucleinsäureanalyse geeignet sind, umfassen Oligonucleotide, die in der Lage sind, als ein Initiationspunkt für die Nucleinsäuresynthese innerhalb der Nucleinsäuresequenzen der Zielnucleinsäuren zu wirken. Ein Primer kann aus einem Restriktionsverdau durch herkömmliche Verfahren aufgereinigt werden, oder er kann synthetisch hergestellt werden. Der Primer ist vorzugsweise zur maximalen Wirkung bei der Amplifikation einzelsträngig, aber der Primer kann doppelsträngig sein. Doppelsträngige Primer werden zuerst denaturiert, d. h. behandelt, um die Stränge zu trennen. Ein Verfahren der Denaturierung von doppelsträngiger Nucleinsäuren erfolgt durch Erhitzen. Eine „wärmestabile Polymerase” ist ein Polymeraseenzym, das wärmestabil ist, d. h. es ist ein Enzym, das die Bildung von Primerextensionsprodukten komplementär zu einem Templat katalysiert und nicht irreversibel denaturiert, wenn es den erhöhten Temperaturen für die Zeit unterworfen wird, die zur Herbeiführung der Denaturierung von doppelsträngigen Templat-Nucleinsäuren notwendig ist. Im Allgemeinen wird die Synthese jeweils am 3'-Ende des Primers initiiert und verläuft in der 5'- zu 3'-Richtung entlang des Templatstrangs. Wärmestabile Polymerasen wurden z. B. bereits aus Thermus flavus, T. ruber, T. thermophilus, T. aquaticus, T. lacteus, T. rubens, Bacillus stearothermophilus und Methanothermus fervidus isoliert. Dennoch können auch Polymerasen, die nicht wärmestabil sind, in den PCR-Tests eingesetzt werden, mit der Maßgabe, das Enzym wird wieder ergänzt.
-
Wenn die Templat-Nucleinsäure doppelsträngig ist, ist es notwendig, die beiden Stränge zu trennen, bevor sie als ein Templat bei der PCR verwendet werden kann. Die Strangtrennung kann durch jedes geeignete Denaturierungsverfahren, darunter physikalische, chemische oder enzymatische Mittel, bewerkstelligt werden. Ein Verfahren der Trennung der Nucleinsäurestränge umfasst das Erwärmen der Nucleinsäure, bis sie überwiegend denaturiert ist (z. B. mehr als 50%, 60%, 70%, 80%, 90% oder 95% denaturiert). Die zur Denaturierung von Templat-Nucleinsäure notwendigen Erwärmungsbedingungen hängen z. B. von der Puffersalzkonzentration und der Länge und der Nucleotidzusammensetzung der Nucleinsäuren, die denaturiert werden, ab, aber reichen typischerweise von etwa 90°C bis etwa 105°C für eine Dauer in Abhängigkeit von den Merkmalen der Reaktion, wie Temperatur und Nucleinsäurelänge. Die Denaturierung wird typischerweise etwa 5 Sekunden bis 9 Minuten durchgeführt. Um die jeweilige Polymerase, wie z. B. die Z05 DNA-Polymerase, gegenüber solchen hohen Temperaturen nicht zu lange zu exponieren und somit einen Verlust von funktionellem Enzym zu riskieren, ist es bevorzugt, kurze Denaturierungsschritte zu verwenden.
-
In einer bevorzugten Ausführungsform der Erfindung ist der Denaturierungsschritt bis zu 30 Sekunden, weiterhin bevorzugt bis zu 20 Sekunden, weiterhin bevorzugt bis zu 10 Sekunden, weiterhin bevorzugt bis 5 Sekunden, am stärksten bevorzugt bis etwa 5 Sekunden lang.
-
Wenn die doppelsträngige Templat-Nucleinsäure durch Wärme denaturiert wird, wird das Reaktionsgemisch auf eine Temperatur abkühlen gelassen, die das Anellieren von jedem Primer an seine Zielsequenz auf den Zielnucleinsäuren beschleunigt.
-
Die Temperatur zum Anellieren beträgt vorzugsweise von etwa 35°C bis etwa 70°C, stärker bevorzugt etwa 45°C bis etwa 65°C; stärker bevorzugt etwa 50°C bis etwa 60°C, stärker bevorzugt etwa 55°C bis etwa 58°C. Anellierungszeiten können von etwa 10 Sekunden bis etwa 1 Minute (z. B. etwa 20 Sekunden bis etwa 50 Sekunden; etwa 30 Sekunden bis etwa 40 Sekunden) betragen. In diesem Zusammenhang kann es zweckmäßig sein, verschiedene Anellierungstemperaturen zu verwenden, um die Einschließlichkeit des jeweiligen Tests zu verstärken. Kurz gesagt bedeutet dies, dass bei relativ niedrigen Anellierungstemperaturen auch Primer, mit einzelnen Fehlpaarungen, an Ziele binden können und so auch Varianten von bestimmten Sequenzen amplifiziert werden können. Dies kann wünschenswert sein, wenn z. B. ein bestimmter Organismus bekannte oder unbekannte genetische Varianten besitzt, die ebenfalls nachgewiesen werden sollten. Andererseits bergen relativ hohe Anellierungstemperaturen den Vorteil der Bereitstellung höherer Spezifität, da die Wahrscheinlichkeit des Primerbindens an nicht exakt passende Zielsequenzen in Richtung höherer Temperaturen stetig abnimmt. Um von beiden Phänomenen zu profitieren, ist es in einigen Ausführungsformen der Erfindung bevorzugt, dass das vorstehend beschriebene Verfahren das Anellieren bei unterschiedlichen Temperaturen einschließt, vorzugsweise zuerst bei einer niedrigeren, dann bei einer höheren Temperatur. Wenn z. B. eine erste Inkubation bei 55°C für etwa 5 Zyklen stattfindet, können nicht exakt passende Zielsequenzen(prä-)amplifiziert werden. Hierauf können z. B. etwa 45 Zyklen bei 58°C folgen, die für den Hauptteil des Experiments durchwegs eine höhere Spezifität bereitstellen. Auf diese Weise werden potenziell wichtige genetische Varianten nicht verfehlt, während die Spezifität relativ hoch bleibt.
-
Das Reaktionsgemisch wird dann auf eine Temperatur eingestellt, bei der die Aktivität der Polymerase beschleunigt oder optimiert wird, d. h. eine Temperatur, die ausreicht, dass von dem anellierten Primer aus Extension eintritt, um Produkte zu erzeugen, die zu der analysierenden Nucleinsäure komplementär sind. Die Temperatur sollte ausreichen, um ein Extensionsprodukt von jedem Primer, der an ein Nucleinsäure-Templat anelliert ist, zu synthetisieren, sollte aber nicht so hoch sein, dass ein Extensionsprodukt von seinem komplementären Templat denaturiert wird, z. B. reicht die Temperatur zur Extension im Allgemeinen von etwa 40°C bis etwa 80°C (z. B. etwa 50°C bis etwa 70°C; etwa 60°C). Die Extensionszeiten können von etwa 10 Sekunden bis etwa 5 Minuten, vorzugsweise etwa 15 Sekunden bis etwa 2 Minuten, stärker bevorzugt etwa 20 Sekunden bis etwa 1 Minute, stärker bevorzugt etwa 25 Sekunden bis etwa 35 Sekunden betragen. Die neu synthetisierten Stränge bilden ein doppelsträngiges Molekül, das in den folgenden Schritten der Reaktion verwendet werden kann. Die Schritte der Strangtrennung, des Anellierens und der Verlängerung können so oft wie nötig, um die gewünschte Menge an Amplifikationsprodukten entsprechend den Zielnucleinsäuren zu erzeugen, wiederholt werden. Die begrenzenden Faktoren bei der Reaktion sind die Mengen von Primern, wärmestabiles Enzym und Nucleosidtriphosphate, die in der Reaktion vorhanden sind. Die Zyklusschritte (d. h. Denaturierung, Anellierung und Extension) werden vorzugsweise mindestens einmal wiederholt. Zur Verwendung beim Nachweis hängt die Anzahl der Zyklusschritte z. B. von der Natur der Probe ab. Wenn die Probe ein komplexes Gemisch von Nucleinsäuren ist, sind mehr Zyklusschritte zur Amplifikation der Zielsequenz erforderlich, die zum Nachweis ausreichen. Im Allgemeinen werden die Zyklusschritte mindestens etwa 20 Mal wiederholt, können allerdings so viele Male wie 40-, 60- oder sogar 100 Mal wiederholt werden.
-
Innerhalb des Umfangs der Erfindung kann eine PCR durchgeführt werden, wobei die Schritte des Anellierens und der Extension in dem gleichen Schritt (einstufige PCR) oder wie oben beschrieben in getrennten Schritten (zweistufige PCR) durchgeführt werden. Das Durchführen von Anellierung und Extension zusammen und somit unter den gleichen physikalischen und chemischen Bedingungen mit einem geeigneten Enzym, wie beispielsweise die Z05 DNA-Polymerase, trägt den Vorteil des Einsparens von Zeit für einen zusätzlichen Schritt in jedem Zyklus, und auch des Beseitigens des Erfordernisses einer zusätzlichen Temperatureinstellung zwischen dem Anellieren und der Extension. Somit vermindert die einstufige PCR die Gesamtkomplexität des jeweiligen Tests.
-
Im Allgemeinen sind kürzere Zeiten für die Gesamtamplifikation bevorzugt, da die Zeit bis zum Ergebnis vermindert wird und zu einer möglichen früheren Diagnose führt.
-
Weitere bevorzugte Nucleinsäureamplifikationsverfahren, die im Zusammenhang mit der Erfindung zu verwenden sind, umfassen die Ligasekettenreaktion (
LCR; Wu D. Y. und Wallace R. B., Genomics 4 (1989) 560–69; und
Barany F., Proc. Genomics 4 (1989) 560–69; and
Barany F., Natl. Acad. Sci. USA 88 (1991) 189–193); die Polymerase-Ligase-Kettenreaktion (
Barany F., PCR Methods and Applic. 1 (1991) 5–16); die Gap-LCR (
WO 90/01069 ); die Reparatur-Kettenraktion (
EP 0439182 A2 ), 3SR (
Kwoh D. Y. et al., Proc. Natl. Acad. Sci. USA 86 (1989) 1173–1177;
Guatelli J. C., et al., Proc. Natl. Acad. Sci. USA 87 (1990) 1874–1878;
WO 92/08808 ) und NASBA (
US 5,130,238 ). Weiterhin existieren die Strangverschiebungsamplifikation (SDA), die Transkriptions-vermittelte Amplifikation (TMA) und die Qb-Amplifikation (für eine Übersicht siehe z. B.
Whelen A. C. und Persing D. H., Annu. Rev. Microbiol. 50 (1996) 349–373;
Abramson R. D. und Myers T. W., Curr Opin Biotechnol 4 (1993) 41–47).
-
Eine „Polymerase mit reverser Transkriptaseaktivität” ist eine Nucleinsäure-Polymerase, die in der Lage ist, DNA auf der Grundlage eines RNA-Templats zu synthetisieren. Sie ist auch zur Bildung einer doppelsträngigen DNA in der Lage, sobald die RNA in eine einzelsträngige cDNA revers transkribiert wurde. In einer bevorzugten Ausführungsform der Erfindung ist die Polymerase mit reverser Transkriptaseaktivität wärmestabil.
-
In einer bevorzugten Ausführungsform umfasst das Verfahren gemäß der Erfindung das Inkubieren einer Probe, die RNA-Templat enthält, mit einem Oligonucleotid-Primer, der ist zu dem RNA-Templat ausreichend komplementär, um mit letzterem zu hybridisieren, und eine vorzugsweise wärmestabile DNA-Polymerase in Gegenwart von mindestens allen vier natürlichen oder modifizierten Desoxyribonucleosidtriphosphaten, in einem entsprechenden Puffer, umfassend einen Metallionenpuffer, der, in einer bevorzugten Ausführungsform, sowohl den pH als auch die Metallionenkonzentration puffert. Diese Inkubation wird bei einer Temperatur durchgeführt, die ausreicht, dass der Primer an das RNA-Templat hybridisiert und dass die DNA-Polymerase die Polymerisation der Desoxyribonucleosidtriphosphate unter Bildung einer zu der Sequenz des RNA-Templats komplementären cDNA-Sequenz katalysiert.
-
Wie hierin verwendet, bezieht sich der Begriff „cDNA” auf ein komplementäres DNA-Molekül, das unter Verwendung eines Ribonucleinsäure-Strangs (RNA) als ein Templat synthetisiert wird. Die RNA kann z. B. mRNA, tRNA, rRNA oder eine andere Form von RNA sein, wie z. B. Virus-RNA. Die cDNA kann einzelsträngig, doppelsträngig oder an ein komplementäres RNA-Molekül wasserstoffgebunden, wie in einem RNA/cDNA-Hybrid, sein.
-
Ein Primer, der zum Anellieren an ein RNA-Templat geeignet ist, kann auch zur Amplifikation durch PCR geeignet sein. Für die PCR stellt ein zweiter, zu dem revers transkribierten cDNA-Strang komplementärer Primer eine Initiationsstelle für die Synthese eines Extensionsproduktes bereit.
-
Bei der Amplifikation von einem RNA-Molekül durch DNA-Polymerase ist die erste Extensionsreaktion die reverse Transkription unter Verwendung eines RNA-Templats, und ein DNA-Strang wird erzeugt. Die zweite Extensionsreaktion unter Verwendung des DNA-Templats erzeugt ein doppelsträngiges DNA-Molekül. Somit stellt die Synthese von einem komplementären DNA-Strang aus einem RNA-Templat durch eine DNA-Polymerase das Ausgangsmaterial zur Amplifikation bereit.
-
Wärmestabile DNA-Polymerasen können in einer gekoppelten reversen Ein-Enzym-Transkriptions/Amplifikationsreaktion verwendet werden. Der Begriff „homogen” bezieht sich in diesem Zusammenhang auf eine zweistufige Einzeladditionsreaktion für die reverse Transkription und Amplifikation von einem RNA-Ziel. Mit homogen ist gemeint, dass es nach dem reversen Transkriptions(RT)schritt nicht notwendig ist, das Reaktionsgefäß zu öffnen oder die Reaktionskomponenten anderweitig vor dem Amplifikationsschritt einzustellen. In einer nicht-homogenen RT/PCR-Reaktion nach der reversen Transkription und vor der Amplifikation werden eine oder mehrere der Reaktionskomponenten, wie die Amplifikationsreagenzien z. B. eingestellt, hinzugefügt oder verdünnt, wofür das Reaktionsgefäß geöffnet oder zumindest sein Inhalt manipuliert werden muss. Obgleich sowohl homogene als auch nicht-homogene Ausführungsformen im Umfang der Erfindung mit umfasst sind, ist das homogene Format für RT/PCR bevorzugt.
-
Die reverse Transkription ist in einer RT/PCR ein wichtiger Schritt. Es ist beispielsweise auf dem Fachgebiet bekannt, dass RNA-Template eine Neigung in Richtung der Bildung von Sekundärstrukturen zeigen, welche das Primerbinden und/oder die Verlängerung des cDNA-Strangs durch die jeweilige reverse Transkriptase beeinträchtigen können. Somit sind relativ hohe Temperaturen für eine RT-Reaktion bezüglich der Wirksamkeit der Transkription zweckmäßig. Andererseits impliziert das Erhöhen der Inkubationstemperatur auch höhere Spezifität, d. h. die RT-Primer anellieren nicht an Sequenzen, die Fehlpaarungen gegenüber der erwarteten Sequenz oder Sequenzen aufweisen. Besonders im Falle von mehreren verschiedenen Ziel-RNAs kann es wünschenswert sein, auch Sequenzen mit einzelnen Fehlpaarungen zu transkribieren und anschließend zu amplifizieren und nachzuweisen, z. B. im Falle des möglichen Vorliegens von unbekannten oder seltenen Untersträngen oder Unterspezies von Organismen in der Fluidprobe.
-
Um sowohl von den vorstehend beschriebenen Vorteilen, d. h. Reduktion der Sekundärstrukturen, als auch von der reversen Transkription von Templaten mit Fehlpaarungen, zu profitieren, ist es ein Aspekt der Erfindung, die RT-Inkubation bei mehr als einer verschiedenen Temperatur durchzuführen.
-
Darum ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei in Schritt ii. die Inkubation der Polymerase mit reverser Transkriptaseaktivität bei verschiedene Temperaturen von 30°C bis 75°C, vorzugsweise von 45°C bis 70°C, stärker bevorzugt von 55°C bis 65°C durchgeführt wird.
-
Als ein weiterer wichtiger Aspekt der reversen Transkription können lange RT-Schritte die DNA-Template, die in der Fluidprobe vorhanden sein können, beschädigen. Wenn die Fluidprobe sowohl RNA- als auch DNA-Spezies enthält, ist es somit von Vorteil, die Dauer der RT-Schritte so kurz wie möglich zu halten, aber gleichzeitig die Synthese von ausreichenden Mengen von cDNA für die anschließende Amplifikation und den fakultativen Nachweis von Amplifikaten sicherzustellen.
-
Somit ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei in Schritt ii. die Zeitdauer bis zu 30 Minuten, 20 Minuten, 15 Minuten, 12,5 Minuten, 10 Minuten, 5 Minuten oder 1 Minute beträgt.
-
Ein weiterer bevorzugter Aspekt der Erfindung ist das vorstehend beschriebene Verfahren, wobei die Polymerase mit reverser Transkriptaseaktivität und eine Mutation umfassend aus der Gruppe ausgewählt wird, die aus folgendem besteht:
- a. einer CS5 DNA-Polymerase
- b. einer CS6 DNA-Polymerase
- c. einer Thermotoga maritima DNA-Polymerase
- d. einer Thermus aquaticus-DNA-Polymerase
- e. einer Thermus thermophilus-DNA-Polymerase
- f. einer Thermus flavus DNA-Polymerase
- g. einer Thermus filiformis DNA-Polymerase
- h. einer Thermus sp. Sps17 DNA-Polymerase
- i. einer Thermus sp. Z05 DNA-Polymerase
- j. einer Thermotoga neapolitana DNA-Polymerase
- k. einer Termosipho africanus DNA-Polymerase
- l. einer Thermus caldophilus DNA-Polymerase.
-
Besonders geeignet für diese Anforderungen sind Enzyme, die eine Mutation in der Polymerasedomäne tragen, die ihre reverse Transkriptionseffizienz hinsichtlich einer schnelleren Extensionsrsate verstärkt.
-
Darum ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei die Polymerase mit reverser Transkriptaseaktivität eine Polymerase ist, die eine Mutation umfasst, die zu einer verbesserten Nucleinsäureextensionsrate und/oder zu einer verbesserten reversen Transkriptaseaktivität relativ zu der jeweiligen Wildtyp-Polymerase beiträgt.
-
In einer stärker bevorzugten Ausführungsform in dem vorstehend beschriebenen Verfahren ist die Polymerase mit reverser Transkriptaseaktivität eine Polymerase, die eine Mutation einschließt, die zu einer verbesserten reversen Transkriptaseaktivität relativ zu der jeweiligen Wildtyp-Polymerase beiträgt.
-
Polymerasen, die Punktmutationen tragen, die sie im Zusammenhang mit der Erfindung besonders geeignet machen, sind in der
WO 2008/046612 offenbart. Insbesondere sind bevorzugte Polymerasen, die im Zusammenhang mit der vorliegenden Erfindung zu verwenden sind, mutierte DNA-Polymerasen, die mindestens das folgende Motiv in der Polymerasedomäne umfassen:
T-G-R-L-S-S-X
b7-X
b8-P-N-L-Q-N; wobei X
b7 eine Aminosäure ist, die aus S oder T ausgewählt ist, und wobei X
b8 eine Aminosäure ist, die aus G, T, R, K oder L ausgewählt ist, wobei die Polymerase 3'-5'-Exonukleaseaktivität umfasst und eine verbesserte Nucleinsäureextensionsrate und/oder eine verbesserte reverse Transkriptionseffizienz relativ zu der Wildtyp-DNA-Polymerase aufweist, wobei in der Wildtyp-DNA-Polymerase X
b8 eine Aminosäure ist, die aus D, E oder N ausgewählt ist.
-
Ein besonders bevorzugtes Beispiel sind Mutanten der wärmestabilen DNA-Polymerase aus der Thermus-Spezies Z05 (z. B. in der
US 5,455,170 beschrieben), wobei die Variationen Mutationen in der Polymerasedomäne im Vergleich zu dem jeweiligen Wildtyp-Z05-Enzym umfassen. Besonders bevorzugt für das Verfahren gemäß der Erfindung ist eine mutante Z05 DNA-Polymerase, wobei die Aminosäure an Position 580 aus der aus G, T, R, K und L bestehenden Gruppe ausgewählt ist.
-
Zur reversen Transkription unter Verwendung einer wärmestabilen Polymerase ist Mn2+ als zweiwertiges Kation bevorzugt und ist typischerweise als Salz eingeschlossen, beispielsweise als Manganchlorid (MnCl2), Manganacetat (Mn(OAc)2) oder Mangansulfat (MnSO4). Wenn MnCl2 in eine Reaktion eingeschlossen ist, die 50 mM Tricinpuffer enthält, ist das MnCl2 beispielsweise im Allgemeinen in einer Konzentration von 0,5 bis 7,0 mM vorhanden; 0,8–1,4 mM sind bevorzugt, wenn 200 mM jeweils von dGTP, dATP, dUTP und dCTP verwendet werden; und 2,5 bis 3,5 mM MnCl2 sind am stärksten bevorzugt. Weiterhin ist auch die Verwendung von Mg2+ als ein zweiwertiges Kation zur reversen Transkription im Zusammenhang mit der vorliegenden Erfindung bevorzugt.
-
Da es im Umfang der Erfindung liegt, RNA-Zielnucleinsäuren in cDNA revers zu transkribieren, während die DNA-Zielnucleinsäuren konserviert werden, so dass sowohl die cDNA als auch DNA zur anschließenden Amplifikation verwendet werden können, ist das Verfahren gemäß der Erfindung besonders zur gleichzeitigen Amplifikation von Zielnucleinsäuren geeignet, die sich sowohl von Organismen mit einer RNA als auch von Organismen mit einem DNA-Genom ableiten. Dieser Vorteil erweitert das Spektrum der verschiedenen Organismen, insbesondere von Pathogenen, beträchtlich, die unter identischen physikalischen Bedingungen analysiert werden können.
-
Darum ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei die mindestens zwei Zielnucleinsäuren RNA und DNA umfassen.
-
Insbesondere aufgrund eines entsprechenden Temperaturoptimums sind Enzyme wie Tth-Polymerase oder vorzugsweise die vorstehend erwähnte mutante Z05 DNA-Polymerase zur Durchführung des anschließenden Schritts der Amplifikation der Zielnucleinsäuren geeignet. Die Ausnutzung des gleichen Enzyms sowohl für die reverse Transkription als auch Amplifikation trägt zur Leichtigkeit der Durchführung des Verfahrens bei und erleichtert seine Automation, da die Fluidprobe zwischen dem RT- und dem Amplifizierungsschritt nicht manipuliert werden muss.
-
In einer bevorzugten Ausführungsform wird darum bei dem vorstehend beschriebenen Verfahren die gleiche Polymerase mit reverser Transkriptaseaktivität in Schritt ii. und Schritt iii. verwendet. Vorzugsweise ist das Enzym die vorstehend beschriebene mutante Z05 DNA-Polymerase.
-
Um die Polymerase oder andere Komponenten des Reaktionsgemisches, das im Zusammenhang mit der Erfindung verwendet wird, nicht erhöhten Temperaturen für Zeiten auszusetzen, die länger sind als notwendig, sind in einer bevorzugten Ausführungsform Schritte über 90°C bis zu 20 Sekunden, vorzugsweise bis zu 15 Sekunden, stärker bevorzugt bis zu 10 Sekunden, stärker bevorzugt bis zu 5 Sekunden und am stärksten bevorzugt 5 Sekunden lang. Dies vermindert ebenfalls die Zeit bis zum Ergebnis und reduziert die erforderliche Gesamtzeit für den Test.
-
In einem solchen homogenen Umfeld kann es von beträchtlichem Vorteil sind, die Reaktionsgefäße vor Initiierung der RT und der Amplifikation zu verschließen, wodurch das Risiko von Verunreinigung vermindert wird. Das Verschließen kann z. B. durch Aufbringen einer Folie, die vorzugsweise transparent ist, einer Kappe oder durch Öl, das den Reaktionsgefäßen zugesetzt wird, und eine lipophile Phase als Versiegelungsschicht auf dem Fluid bildet, bewerkstelligt werden.
-
Somit ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, das weiterhin zwischen dem Schritt i. und dem Schritt ii. den Schritt des Verschließens der mindestens zwei Reaktionsgefäße umfasst.
-
Das Ziel des Amplifikationsschritts kann ein RNA/DNA-Hybridmolekül sein. Das Ziel kann eine einzelsträngige oder doppelsträngige Nucleinsäure sein. Obwohl die am breitesten eingesetzte PCR-Verfahrensweise ein doppelsträngiges Ziel verwendet, ist dies keine Notwendigkeit. Nach dem ersten Amplifikationszyklus eines einzelsträngigen DNA-Ziels enthält das Reaktionsgemisch ein doppelsträngiges DNA-Molekül, bestehend aus dem einzelsträngigen Ziel und einem neu synthetisierten Komplementärstrang. Gleichermaßen enthält das Reaktionsgemisch nach dem ersten Amplifikationszyklus eines RNA/cDNA-Ziels ein doppelsträngiges cDNA-Molekül. An diesem Punkt laufen nachfolgende Amplifikationszyklen ab, wie vorstehend beschrieben.
-
Da die Nucleinsäureamplifikation insbesondere, jedoch nicht nur, im Falle der PCR sehr wirksam ist, wenn sie als Zyklusreaktion durchgeführt wird, ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei die Amplifikationsreaktion in Schritt iii. aus mehreren Zyklusschritten besteht.
-
Geeignete Nucleinsäure-Nachweisverfahren sind dem Fachmann bekannt und in Standardtextbüchern beschrieben, wie Sambrook J. et al., Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 1989 und Ausubel F. et al.: Current Protocols in Molecular Biology 1987, J. Wiley and Sons, NY. Auch weitere Reinigungsschritte können vor Durchführen des Nucleinsäure-Nachweisschritts vorhanden sein, wie z. B. ein Präzipitationsschritt.
-
Die Nachweisverfahren können das Binden oder Interkalieren von spezifischen Farbstoffen als Ethidiumbromid, das in die doppelsträngige DNA interkaliert und ihre Fluoreszenz anschließend verändert, einschließen, sind jedoch nicht darauf beschränkt. Die gereinigte Nucleinsäure kann auch durch elektrophoretische Verfahren, gegebenenfalls nach einem Restriktionsverdau, abgetrennt und anschließend sichtbar gemacht werden. Es existieren auch Sonden-basierte Tests, die die Oligonucleotid-Hybridisierung an spezifische Sequenzen und den anschließenden Nachweis des Hybrids ausnutzen.
-
Es ist bevorzugt, die amplifizierten Zielnucleinsäuren während oder nach der Amplifikationsreaktion nachzuweisen, um das Ergebnis der Analyse zu bewerten. Insbesondere zum Nachweis in Realzeit ist es von Vorteil, Nucleinsäuresonden zu verwenden.
-
Somit ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei ein Zyklusschritt einen Amplifizierungsschritt und einen Hybridisierungsschritt umfasst, wobei der Hybridisierungsschritt das Hybridisieren der amplifizierten Nucleinsäuren mit Sonden umfasst.
-
Es kann günstig sein, die Amplifikationsreaktion in Realzeit zu überwachen, d. h. die Zielnucleinsäuren und/oder ihre Amplifikate während der Amplifikation selbst nachzuweisen.
-
Darum ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei die Sonden mit einer Donor-Fluoreszenzeinheit und einer entsprechenden Akzeptor-Fluoreszenzeinheit markiert werden.
-
Die vorstehend aufgeführten Verfahren basieren vorzugsweise auf Fluoreszenzresonanz-Energietransfer (FREI) zwischen einer Donor-Fluoreszenzeinheit und einer Akzeptor-Fluoreszenzeinheit. Eine repräsentative Donor-Fluoreszenzeinheit ist Fluoreszin, und repräsentative entsprechende Akzeptor-Fluoreszenzeinheiten umfassen LC-Red 640, LC-Red 705, Cy5 und Cy5.5. Typischerweise umfasst der Nachweis das Anregen der Probe bei einer Wellenlänge, die von der Donor-Fluoreszenzeinheit absorbiert wird, und das Visualisieren und/oder Messen der Wellenlänge, die von der entsprechenden Akzeptor-Fluoreszenzeinheit emittiert wird. Bei dem Verfahren gemäß der Erfindung folgt auf den Nachweis vorzugsweise die quantitative Bestimmung des FREI. Vorzugsweise wird der Nachweis nach jedem Zyklusschritt durchgeführt. Am stärksten bevorzugt wird der Nachweis in Realzeit durchgeführt. Durch Verwendung von im Handel erhältlichen Realzeit-PCR-Instrumenten (z. B. LightCyclerTM oder TagMan®) können die PCR-Amplifikation und der Nachweis des Amplifikationsprodukts in einer einzigen geschlossenen Küvette mit extrem verminderter Zykluszeit kombiniert werden. Da der Nachweis gleichzeitig mit der Amplifikation erfolgt, umgehen die Realzeit-PCR-Methoden das Erfordernis der Manipulation des Amplifikationsprodukts und vermindern das Risiko von Kreuzverunreinigung zwischen Amplifikationsprodukten. Die Realzeit-PCR setzt die Verweildauer sehr stark herab und ist eine attraktive Alternative zu herkömmlichen PCR-Techniken im klinischen Labor.
-
Die folgenden Patentanmeldungen beschreiben die Realzeit-PCR, wie sie in der LightCycler
TM-Technik verwendet wird:
WO 97/46707 ,
WO 97/46714 und
WO 97/46712 . Das LightCycler
TM-Instrument ist ein schneller Thermocycler in Kombination mit einem Mikrovolumenfluorometer unter Verwendung hochqualitativer Optik. Diese schnelle Thermocyclingtechnik verwendet dünne Glasküvetten als Reaktionsgefäße. Erwärmen und Abkühlen der Reaktionskammer werden durch abwechselnde erwärmte und Umgebungsluft kontrolliert. Aufgrund der geringen Luftmasse und des hohen Verhältnisses von spezifischer Oberfläche zu Volumen der Küvetten können in der Wärmekammer schnelle Temperaturaustauschraten erreicht werden.
-
Die TagMan®-Technologie verwendet eine einzelsträngige Hybridisierungssonde, die mit zwei fluoreszierenden Einheiten markiert ist. Wenn eine erste fluoreszierende Einheit mit Licht einer geeigneten Wellenlänge angeregt ist, wird die absorbierte Energie gemäß den Prinzipien von FREI auf eine zweite fluoreszierende Einheit übertragen. Die zweite fluoreszierende Einheit ist im Allgemeinen ein Quencher-Molekül. Typische fluoreszierende Farbstoffe, die in diesem Format verwendet werden, sind beispielsweise unter anderem FAM, HEX, CY5, JA270, Cyan und CY5.5. Während des Anellierungsschritts der PCR-Reaktion bindet die markierte Hybridisierungssonde an die Zielnucleinsäure (d. h. das Amplifikationsprodukt) und wird durch die 5'-zu-3'-Exonucleaseaktivität der Taq- oder einer anderen geeigneten Polymerase, wie sie dem Fachmann bekannt ist, wie die bevorzugte mutante Z05-Polymerase, während der anschließenden Verlängerungsphase abgebaut. Als Ergebnis werden die angeregte Fluoreszenzeinheit und die Quenchereinheit räumlich voneinander getrennt. Als Folge kann beim Anregen der ersten Fluoreszenzeinheit in Abwesenheit des Quenchers die Fluoreszenzemission aus der ersten Fluoreszenzeinheit nachgewiesen werden.
-
In beiden vorstehend beschriebenen Nachweisformaten kann die Intensität des emittierten Signals mit der Anzahl von ursprünglichen Zielnucleinsäuremolekülen korreliert werden.
-
Als Alternative zu FREI kann ein Amplifikationsprodukt unter Verwendung eines doppelsträngige DNA bindenden Farbstoffs, wie eines DNA-bindenden Fluoreszenzfarbstoffs (z. B. SYBRGREEN I® oder SYBRGOLD® (Molecular Probes)) nachgewiesen werden. Bei Wechselwirkung mit der doppelsträngigen Nucleinsäure emittieren solche DNA-bindenden Fluoreszenzfarbstoffe nach Anregung mit Licht bei einer geeigneten Wellenlänge ein Fluoreszenzsignal. Ein doppelsträngige DNA bindender Farbstoff, wie ein Nukleinsäure-Interkalationsfarbstoff, kann ebenfalls verwendet werden. Wenn doppelsträngige DNA bindende Farbstoffe verwendet werden, wird in der Regel eine Schmelzkurvenanalyse zur Bestätigung des Vorliegens des Amplifikationsprodukts durchgeführt.
-
Molecular Beacons in Verbindung mit FREI können ebenfalls zum Nachweis des Vorliegens eines Amplifikationsprodukts unter Verwendung der Realzeit-PCR-Verfahren der Erfindung verwendet werden. Die Molecular-Beacon-Technologie verwendet eine Hybridisierungssonde, die mit einer ersten fluoreszierenden Einheit und einer zweiten fluoreszierenden Einheit markiert ist. Die zweite fluoreszierende Einheit ist im Allgemeinen ein Quencher, und die fluoreszierenden Markierungen sind typischerweise an jedem Ende der Sonde angeordnet. Die Molecular-Beacon-Technologie verwendet ein Sonden-Oligonucleotid mit Sequenzen, die die Sekundärstrukturbildung (z. B. eine Haarnadel) erlauben. Als Ergebnis der Sekundärstrukturbildung in der Sonde befinden beide fluoreszierende Einheiten in räumlicher Nähe, wenn die Sonde in Lösung ist. Nach Hybridisierung mit den Amplifikationsprodukten wird die Sekundärstruktur der Sonde unterbrochen, und die fluoreszierenden Einheiten werden voneinander getrennt, derart dass nach der Anregung mit Licht von einer geeigneten Wellenlänge die Emission der ersten fluoreszierenden Einheit nachgewiesen werden kann.
-
Somit ist in einem bevorzugten Verfahren gemäß der Erfindung das vorstehend beschriebene Verfahren unter Verwendung von FREI, wobei die Sonden eine Nucleinsäuresequenz umfassen, die die Sekundärstrukturbildung erlaubt, wobei die Sekundärstrukturbildung die räumliche Nähe zwischen der ersten und der zweiten fluoreszierenden Einheit bewirkt.
-
Eine wirksame FREI kann nur stattfinden, wenn sich die fluoreszierenden Einheiten in direkter lokaler Nähe befinden und wenn sich das Emissionsspektrum der Donor-Fluoreszenzeinheit mit dem Absorptionsspektrum der Akzeptor-Fluoreszenzeinheit überlappt.
-
Somit sind in einer bevorzugten Ausführungsform der Erfindung die Donor- und Akzeptor-Fluoreszenzeinheiten innerhalb von nicht mehr als 5 Nucleotiden voneinander auf der Sonde vorhanden.
-
Bei einer weiteren bevorzugten Ausführungsform ist die Akzeptor-Fluoreszenzeinheit ein Quencher.
-
Wie vorstehend beschrieben bindet die markierte Hybridisierungssonde in dem TagMan-Format während des Anellierungsschritts der PCR-Reaktion an die Zielnucleinsäure (d. h. das Amplifikationsprodukt) und wird durch die 5'-zu-3'-Exonucleaseaktivität der Taq- oder einer anderen geeigneten Polymerase, wie sie dem Fachmann bekannt ist, wie die bevorzugte mutante Z05-Polymerase während der anschließenden Verlängerungsphase abgebaut.
-
Somit verwendet die Amplifikation in einer bevorzugten Ausführungsform in dem Verfahren gemäß der Erfindung eine Polymeraseenzym mit einer 5'-zu-3'-Exonucleaseaktivität.
-
Es ist weiterhin vorteilhaft, die Länge des Amplikons, das als ein Ergebnis des vorstehend beschriebenen Verfahrens gewonnen wird, mit Bedacht zu wählen. Im Allgemeinen erhöhen relativ kurze Amplikons die Wirksamkeit der Amplifikationsreaktion. Somit ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei die amplifizierten Fragmente bis zu 450 Basen, vorzugsweise bis zu 300 Basen, stärker bevorzugt bis zu 200 Basen, und stärker bevorzugt bis zu 150 Basen umfassen.
-
Gemäß der Erfindung kann es weiterhin von Vorteil sein, Kontrollnucleinsäuren zu verwenden. Es ist auf dem Fachgebiet bekannt, dass sowohl qualitative als auch quantitative Kontrollen insbesondere in einer diagnostischen Umgebung, von beträchtlicher Bedeutung sind.
-
In diesem Zusammenhang ist eine Kontrollnucleinsäure, die als eine „quantitative Standardnucleinsäure” dient, dazu geeignet, eine Referenz darzustellen und verwendet zu werden, um die Menge der Zielnucleinsäuren zu quantifizieren, d. h. zu bestimmen. Zu diesem Zweck durchlaufen eine oder mehrere quantitative Standardnucleinsäuren zusammen mit den Zielnucleinsäuren alle möglichen Probenpräparationsschritte. Ferner wird eine quantitative Standardnucleinsäure bei dem Verfahren durchwegs innerhalb des gleichen Reaktionsgemisches bearbeitet. Sie muss direkt oder indirekt ein nachweisbares Signal sowohl in Gegenwart als auch Abwesenheit der Zielnucleinsäure erzeugen. Zu diesem Zweck muss die Konzentration der quantitativen Standardnucleinsäure in jedem Test mit Bedacht optimiert werden, damit die Empfindlichkeit nicht beeinträchtigt, sondern ein nachweisbares Signal erzeugt wird, auch z. B. bei sehr hohen Zielkonzentrationen. Hinsichtlich der Nachweisgrenze (LOD, siehe nachstehend) des jeweiligen Tests beträgt der Konzentrationsbereich für die „quantitative Standardnucleinsäure” vorzugsweise 20–5000x LOD, stärker bevorzugt 20–1000x LOD, am stärksten bevorzugt 20–5000x LOD. Die Endkonzentration der quantitativen Standardnucleinsäure in dem Reaktionsgemisch ist von dem quantitativen Messbereich, der erreicht wird, abhängig. Die quantitative Standardnucleinsäure kann beispielsweise DNA, RNA oder PNA, armierte DNA oder RNA und modifizierte Formen davon sein.
-
„Nachweisgrenze” oder „LOD” bedeutet die geringste nachweisbare Menge oder Konzentration von einer Nucleinsäure in einer Probe. Eine niedrige „LOD” entspricht einer hohen Empfindlichkeit und umgekehrt. Die „LOD” wird in der Regel entweder mit der Einheit „cp/ml”, insbesondere wenn die Nucleinsäure eine Virus-Nukleinsäure ist, oder mit IU/ml ausgedrückt. „cp/ml” bedeutet „Kopien pro Milliliter”, wobei eine „Kopie” eine Kopie der jeweiligen Nucleinsäure ist. IU/ml steht für „International Units/ml” und bezeichnet den WHO-Standard.
-
Ein breit eingesetztes Verfahren zur Berechnung einer LOD ist die „Probit-Analyse”, die ein Verfahren der Analyse der Beziehung zwischen einem Stimulus (Dosis) und der Quanten(alles oder nichts)-Antwort ist. In einem typischen Quanten-Antwortexperiment werden an Gruppen von Tieren verschiedene Dosen eines Arzneimittels verabreicht. Der Prozentsatz, der jeweils bei einem Dosisniveau stirbt, wird dokumentiert. Diese Daten können unter Verwendung der Probit-Analyse ausgewertet werden. Das Probit-Modell nimmt an, dass die prozentuale Antwort mit der log-Dosis als die kumulative Normalverteilung zusammenhängt. D. h., die log-Dosen können als Variablen verwendet werden, um aus der kumulativen Normalverteilung den Prozentsatz abzulesen, der gestorben ist. Die Verwendung der Normalverteilung statt anderer Wahrscheinlichkeitsverteilungen beeinflusst die vorausberechnete Antwortreaktionsrate am unteren und oberen Ende der möglichen Dosen, aber hat wenig Einfluss in der Nähe der Mitte.
-
Die Probit-Analyse kann bei distinkten „Trefferraten” angewendet werden. Wie es auf dem Fachgebiet bekannt ist, wird eine „Trefferrate” im Allgemeinen in Prozent [%] ausgedrückt und gibt den Prozentsatz an positiven Ergebnissen bei einer spezifischen Konzentration von einem Analyten an. Somit kann beispielsweise eine LOD mit 95% Trefferrate bestimmt werden, was bedeutet, dass die LOD für ein Umfeld berechnet wird, in dem 95% der gültigen Ergebnis positiv sind.
-
In einer bevorzugten Ausführungsform stellt das vorstehend beschriebene Verfahren eine LOD von 1 bis 100 cp/ml oder 0,5 bis 50 IU/ml, stärker bevorzugt von 1 bis 75 cp/ml oder 0,5 bis 30 IU/ml, stärker bevorzugt von 1 bis 25 cp/ml bzw. 1 bis 20 IU/ml bereit.
-
Bezüglich einiger Beispiele für mögliche Zielnucleinsäuren aus bestimmten Viren stellt das Verfahren gemäß der Erfindung vorzugsweise die folgenden LODs bereit:
- • HIV: bis zu 60 cp/ml, stärker bevorzugt bis zu 50 cp/ml, stärker bevorzugt bis zu 40 cp/ml, stärker bevorzugt bis zu 30 cp/ml, stärker bevorzugt bis zu 20 cp/ml, stärker bevorzugt bis zu 15 cp/ml
- • HBV: bis zu 10 IU/ml, stärker bevorzugt bis zu 7,5 IU/ml, stärker bevorzugt bis 5 IU/ml
- • HCV: bis zu 10 IU/ml, stärker bevorzugt bis zu 7,5 IU/ml, stärker bevorzugt bis 5 IU/ml
- • WNV I: bis zu 20 cp/ml, stärker bevorzugt bis zu 15 cp/ml, stärker bevorzugt bis zu 10 cp/ml
- • WNV II: bis zu 20 cp/ml, stärker bevorzugt bis zu 15 cp/ml, stärker bevorzugt bis zu 10 cp/ml, stärker bevorzugt bis zu 5 cp/ml.
- • JEV: bis 100 cp/ml, stärker bevorzugt bis zu 75 cp/ml, stärker bevorzugt bis zu 50 cp/ml, stärker bevorzugt bis zu 30 cp/ml
- • SLEV: bis 100 cp/ml, stärker bevorzugt bis zu 75 cp/ml, stärker bevorzugt bis zu 50 cp/ml, stärker bevorzugt bis zu 25 cp/ml, stärker bevorzugt bis zu 10 cp/ml.
-
Ein Beispiel dafür, wie die Berechnung von quantitativen Ergebnissen in dem TagMan-Format auf der Grundlage einer quantitativen Standardnucleinsäure durchgeführt wird, ist im Folgenden beschrieben: Aus den Eingabedaten von Instrumenten-korrigierten Fluoreszenzwerten aus einem gesamten PCR-Lauf wird eine Titer berechnet. Eine Reihe von Proben, die eine Zielnucleinsäure und eine Kontrollnucleinsäure enthalten, die als eine quantitative Standardnucleinsäure dient, durchlaufen auf einem Thermocycler unter Verwendung eines spezifizierten Temperaturprofils eine PCR. Bei gewählten Temperaturen und Zeiten während des PCR-Profils werden die Proben mit gefiltertem Licht bestrahlt und die gefilterten Fluoreszenzdaten werden für jede Probe für die Zielnucleinsäure und die quantitative Standardnucleinsäure gesammelt. Nach Abschluss eines PCR-Laufes werden die Fluoreszenzablesungen verarbeitet, um eine Reihe von Farbstoffkonzentrationsdaten für die quantitative Standardnucleinsäure und eine Reihe von Farbstoffkonzentrationsdaten für die Zielnucleinsäure zu ergeben. Jede Reihe von Farbstoffkonzentrationsdaten wird auf die gleiche Weise verarbeitet. Nach mehreren Plausibilitätsprüfungen werden die Ellbogenwerte (CT) für die quantitative Standardnucleinsäure und die Zielnucleinsäure berechnet. Der Ellbogenwert ist als der Punkt definiert, an dem die Fluoreszenz der Zielnucleinsäure oder der quantitativen Standardnucleinsäure eine vorbestimmte Schwelle (Fluoreszenzkonzentration) kreuzt. Die Titerbestimmung beruht auf den Annahmen, dass die Zielnucleinsäure und die quantitative Standardnucleinsäure mit der gleichen Effizienz amplifiziert werden, und dass bei dem berechneten Ellbogenwert gleiche Mengen von Amplikonkopien von Zielnucleinsäure und quantitativer Standardnucleinsäure amplifiziert und nachgewiesen werden. Darum ist (CTQS-CT-Ziel) linear zu log(Zielkonz/QS-Konz), wobei „QS” für die interne quantitative Standardnucleinsäure steht. Der Titer T kann dann beispielsweise unter Verwendung einer Polynominalberechnungsformel wie in der folgenden Gleichung berechnet werden: T' = 10(a(CTQS – CTZiel)2 + b(CTQS – CTZiel) + c)
-
Die Polynominalkonstanten und die Konzentration der quantitativen Standardnucleinsäure sind bekannt, darum ist in der Gleichung die einzige Variable die Differenz (CTQS – CTZiel). Zusätzlich zu dem bloßen Nachweis der Gegenwart oder Abwesenheit von einer Zielnucleinsäure in einer Fluidprobe ist es oft wichtig, die Menge der Nucleinsäure zu bestimmen. Als ein Beispiel können das Stadium und eine Schwere von einer Viruserkrankung auf der Grundlage der Virusbelastung beurteilt werden. Weiterhin erfordert die Überwachung einer Therapie Informationen über die Menge von einem in einem Individuum vorhandenen Pathogen, um den Therapieerfolg zu beurteilen.
-
Hinsichtlich des oben Genannten ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, das weiterhin den Schritt der Bestimmung der Menge der Zielnucleinsäuren nach Schritt iii. umfasst.
-
Weiterhin können im Sinne der Erfindung eine oder mehrere Kontrollnucleinsäuren als eine „qualitative interne Kontrollnucleinsäure” dienen. Der qualitative Nachweis von einer Nucleinsäure in einer biologischen Probe ist unerlässlich z. B. zum Erkennen einer Infektion von einem Individuum. Dadurch ist eine bedeutende Anforderung für einen Test zum Nachweis einer mikrobiellen Infektion, dass falsch-negative oder falsch-positive Ergebnisse vermieden werden, da solche Ergebnisse hinsichtlich der Behandlung des jeweiligen Patienten fast unweigerlich zu schweren Folgen führen würden. Somit wird, insbesondere bei PCR-basierten Verfahren, dem Nachweisgemisch eine qualitative interne Kontrollnucleinsäure zugesetzt. Die Kontrolle ist besonders für die Bestätigung der Validität eines Testergebnisses von Bedeutung: Mindestens im Falle von einem negativen Ergebnis hinsichtlich der jeweiligen Zielnucleinsäure muss sich die qualitative interne Kontrollreaktion innerhalb gegebener Umfelder reaktiv verhalten, d. h. die qualitative interne Kontrolle muss nachgewiesen werden, ansonsten wird der Test an sich als inoperativ betrachtet. Allerdings muss in einem qualitativen Umfeld die qualitative interne Kontrolle nicht notwendigerweise im Falle eines positiven Ergebnisses nachgewiesen werden. Für qualitative Tests ist es von besonderer Bedeutung, dass die Empfindlichkeit der Reaktion sichergestellt ist und darum streng kontrolliert wird. Als eine Folge muss die Konzentration der qualitativen internen Kontrolle relativ niedrig sein, so dass sogar in einer Situation, z. B. von leichter Hemmung, die qualitative interne Kontrolle nicht nachgewiesen wird und darum der Test ungültig ist. Die muss dem jeweiligen Test und seiner Empfindlichkeit mit Bedacht angepasst werden. Vorzugsweise umfasst der Konzentrationsbereich für die qualitative interne Nucleinsäure, d. h. die zweite Kontrollnucleinsäure, einen Bereich von 1 Kopie pro Reaktion bis 1000 Kopien pro Reaktion. In Relation zu der jeweiligen Test-Nachweisgrenze (LOD) liegt ihre Konzentration vorzugsweise zwischen der LOD von einem Test und dem 25fachen Wert der LOD, stärker bevorzugt zwischen LOD und 10× LOD.
-
Stärker bevorzugt liegt sie zwischen 2× und 10× LOD. Noch stärker bevorzugt liegt sie zwischen 5× und 10× LOD. Am stärksten bevorzugt beträgt sie 5× oder 10× LOD.
-
Somit ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei die Gegenwart von einem Amplifikationsprodukt der internen Kontrollnucleinsäure ein Hinweis für eine Amplifikation ist, die in dem Reaktionsgemisch auch in Abwesenheit von Amplifikationsprodukten für eine oder mehrere der Zielnucleinsäuren stattfindet.
-
Die vorliegende Erfindung ist besonders für die Entwicklung von Simultantests auf eine Vielzahl von Parametern und/oder Nucleinsäuretypen, während die gleiche interne Kontrollnucleinsäuresequenz für die verschiedenen Parameter und/oder Nucleinsäuretypen verwendet wird, geeignet. Darum trägt sie zur Verminderung der Gesamtkomplexität der entsprechenden Experimente auf verschiedenen Niveaus bei: Beispielsweise muss nur eine interne Kontrollnucleinsäuresequenz konstruiert werden und den jeweiligen Amplifikationsgemischen zugesetzt werden, was somit Zeit und Kosten für Konstruktion und Synthese oder zum Kauf mehrerer Kontrollnucleinsäuresequenzen spart. Der Test oder die Tests können stromlinienförmig sein, und das Risiko von Handhabungsirrtümern wird vermindert. Zusätzlich, je mehr verschiedene Kontrollnucleinsäuresequenzen in einem Test oder in parallelen gleichzeitig unter den gleichen Bedingungen durchgeführten Tests eingesetzt werden, desto komplexer kann sich die Einstellung der jeweiligen Bedingungen ergeben. Ferner kann mit einer einzigen Kontrolle, die für eine Vielzahl von Nucleinsäuren geeignet ist, die Kontrolle aus einer einzigen Quelle, z. B. in verschiedene Gefäße, die die Zielnucleinsäuren enthält, abgegeben werden. Innerhalb des Umfangs der Erfindung kann die einzige Kontrollnucleinsäuresequenz auch als eine qualitative und als eine quantitative Kontrolle dienen.
-
Somit ist ein bevorzugter Aspekt der Erfindung ein Verfahren zur Isolierung und gleichzeitigen Amplifizierung von mindestens einer ersten und einer zweiten Zielnucleinsäure, die in einer oder mehreren Fluidproben vorhanden sein können, wobei das Verfahren die folgenden automatisierten Schritte umfasst:
- a. Zugeben einer internen Kontrollnucleinsäure von jeder der Fluidproben,
- b. Gegenseitiges Kombinieren eines festen Trägermaterials und der einen oder mehreren Fluidproben in einem oder mehreren Gefäßen für eine Zeit und unter Bedingungen, die ausreichen, um zu ermöglichen, dass die Nucleinsäuren, die die Zielnucleinsäure und die interne Kontrollnucleinsäure umfassen, auf dem festen Trägermaterial immobilisiert werden,
- c. Isolieren des festen Trägermaterials von dem anderen in der Fluidprobe vorhandenen Material in einer Trennstation,
- d. Reinigen der Nucleinsäuren in der Trennstation und ein- oder mehrmaliges Waschen des festen Trägermaterials mit einem Waschpuffer,
- e. Kontaktieren der gereinigten Zielnucleinsäuren und der gereinigten internen Kontrollnucleinsäure mit einem oder mehreren Amplifikationsreagenzien, die mindestens eine distinkte Reihe von Primern für jede der Zielnucleinsäuren und für die interne Kontrollnucleinsäure einschließen, in mindestens zwei Reaktionsgefäßen, wobei mindestens ein erstes Reaktionsgefäß mindestens die erste Zielnucleinsäure umfasst und mindestens ein zweites Reaktionsgefäß mindestens die zweite Zielnucleinsäure umfasst und wobei die zweite Zielnucleinsäure in dem ersten Reaktionsgefäß fehlt,
- f. Inkubieren in dem Reaktionsgefäß der gereinigten Zielnucleinsäuren und der gereinigten internen Kontrollnucleinsäure mit dem einen oder den mehreren Amplifikationsreagenzien für einen Zeitraum und unter Bedingungen, die ausreichen, dass eine Amplifikationsreaktion eintritt, die ein Hinweis auf die Gegenwart oder Abwesenheit der Zielnucleinsäuren ist,
- g. Nachweisen und Messen der Signale, die durch die Amplifikationsprodukte der Zielnucleinsäuren erzeugt wurden und die proportional zu der Konzentration der Zielnucleinsäuren sind, und Nachweisen und Messen eines von der internen Kontrollnucleinsäure erzeugten Signals,
wobei die Bedingungen für die Amplifikation und den Nachweis in den Schritten d. bis g. für die mindestens erste und zweite gereinigte Zielnucleinsäure und die interne Kontrollnucleinsäure identisch sind, und wobei die Sequenz der internen Kontrollnucleinsäure für die mindestens erste und zweite gereinigte Zielnucleinsäure identisch ist.
-
Als ein weiterer Vorteil des vorstehend beschriebenen Verfahrens muss die Testung von einer bestimmten biologischen Probe auf andere Nucleinsäuren in möglichen Folgeexperimenten kein weiteres Probenpräparationsverfahren umfassen, mit der Zugabe von einer unterschiedlichen internen Kontrollnucleinsäure, da die bei der Erfindung eingesetzte Kontrolle zur Kontrolle der Amplifikation von verschiedenen Nucleinsäuren verwendet werden kann. Somit können, sobald eine interne Kontrollnucleinsäure zugesetzt worden ist, andere Parameter in der gleichen Probe unter den gleichen Bedingungen getestet werden.
-
Die interne Kontrollnucleinsäure kann kompetitiv, nicht-kompetitiv oder teilweise kompetitiv sein.
-
Eine kompetitive interne Kontrollnucleinsäure trägt im Wesentlichen die gleichen Primerbindungsstellen wie das Ziel und konkurriert somit mit dem Ziel um die gleichen Primer. Obgleich dieses Prinzips eine gute Nachahmung der jeweiligen Zielnucleinsäure aufgrund ihrer ähnlichen Struktur gestattet, kann es die Amplifikationseffizienz hinsichtlich der Zielnucleinsäure oder -säuren erniedrigen und somit zu einem weniger empfindlichen Test führen.
-
Eine nicht-kompetitive interne Kontrollnucleinsäure hat unterschiedliche Primerbindungsstellen als das Ziel und bindet somit an unterschiedliche Primer. Vorteile eines solchen Umfelds umfassen unter anderem die Tatsache, dass Einzel-Amplifikationsereignisse der verschiedenen Nucleinsäuren in dem Reaktionsgemisch unabhängig voneinander ohne kompetitive Effekte erfolgen können. Somit treten keine nachteiligen Auswirkungen im Hinblick auf die Nachweisgrenze des Tests auf, wie es in einem kompetitiven Umfeld der Fall sein kann.
-
Schließlich konkurrieren bei einer Amplifikation unter Verwendung eines teilweise kompetitiven Umfelds die jeweilige Kontrollnucleinsäure und mindestens eine der Zielnucleinsäuren um die gleichen Primer, obgleich mindestens eine andere Zielnucleinsäure an verschiedene Primer bindet.
-
Die Tatsache, dass das vorstehend beschriebene Verfahren eine distinkte Reihe von Primern für jede der Zielnucleinsäuren und für die interne Kontrollnucleinsäure umfasst, macht das Verfahren beträchtlich flexibel. In diesem nicht-kompetitiven Umfeld ist es nicht notwendig, zielspezifische Bindungsstellen in die Kontrollnucleinsäure einzuführen, wie es für ein kompetitives Umfeld der Fall ist, und die Nachteile eines kompetitiven Umfelds, wie vorstehend erwähnt, werden vermieden. In einem nicht-kompetitiven Umfeld hat die interne Kontrollnucleinsäure eine von allen Zielsequenzen unterschiedliche Sequenz, so dass sie nicht um ihre Primer und/oder Sonden konkurriert. Vorzugsweise ist die Sequenz der internen Kontrollnucleinsäure von den anderen Nucleinsäuresequenzen in der Fluidprobe verschieden. Als ein Beispiel muss, wenn sich die Fluidprobe von einem Menschen ableitet, die interne Kontrollnucleinsäure nicht bevorzugt eine Sequenz aufweisen, die ebenfalls endogen in Menschen auftritt. Der Unterschied in der Sequenz sollte somit mindestens signifikant genug sein, um kein Binden von Primern und/oder Sonden an die jeweilige endogene Nucleinsäure oder Säuren unter stringenten Bedingungen zu erlauben und somit das Umfeld kompetitiv machen. Um eine solche Beeinträchtigung zu vermeiden, ist die Sequenz der internen Kontrollnucleinsäure, die bei der Erfindung verwendet wird, vorzugsweise von einer Quelle abgeleitet, die von dem Ursprung der Fluidprobe verschieden ist. Vorzugsweise ist sie von einem natürlich vorkommenden Genom, vorzugsweise einem Pflanzengenom, noch stärker bevorzugt von einem Traubengenom abgeleitet. In einer sehr bevorzugten Ausführungsform ist eine Nucleinsäure, die sich von einem natürlich vorkommenden Genom abgeleitet, gescrambelt. Wie auf dem Fachgebiet bekannt, bedeutet „Scrambling” das Einbringen von Basemutationen in eine Sequenz bis zu einem gewissen Ausmaß. Vorzugsweise ist die Sequenz der internen Kontrollnucleinsäure, die bei der Erfindung verwendet wird, im Wesentlichen bezüglich des natürlich vorkommenden Gens, von dem sie sich ableitet, geändert.
-
Im Zusammenhang mit der Erfindung ist eine „Sequenz” die Primärstruktur einer Nucleinsäure, d. h. die spezifische Anordnung der einzelnen Nucleobasen, aus der die jeweiligen Nucleinsäuren bestehen. Es ist selbstverständlich, dass der Begriff „Sequenz” keinen spezifischen Typ von Nucleinsäure bezeichnet, wie RNA oder DNA, sondern auf beide sowie auf andere Typen von Nucleinsäuren zutrifft, wie z. B. PNA oder andere. Wo Nucleobasen einander entsprechen, insbesondere im Fall von Uracil (in RNA vorhanden) und Thymin (in DNA vorhanden), können diese Basen als äquivalent zwischen RNA- und DNA-Sequenzen betrachtet werden, wie es auf dem einschlägigen Fachgebiet wohlbekannt ist.
-
Klinisch relevante Nucleinsäuren sind oft DNA, die z. B. von DNA-Viren wie Hepatitis-B-Virus (HBV), Cytomegalovirus (CMV) und anderen, oder von Bakterien, wie Chlamydia trachomatis (CT), Neisseria gonorrhoeae (NG) und anderen abgeleitet werden kann. In solchen Fällen kann es zweckmäßig sein, eine interne Kontrollnucleinsäure zu verwenden, die aus DNA besteht, um die Zielnucleinsäure-Eigenschaften wiederzugeben.
-
Darum ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei die interne Kontrollnucleinsäure DNA ist.
-
Andererseits sind zahlreiche Nucleinsäuren, die für die klinische Diagnostik relevant sind, Ribonucleinsäuren, wie z. B. Nucleinsäuren aus RNA-Viren, wie beispielsweise Humanes Immundefizienz-Virus (HIV), (HIV), Hepatitis-C-Virus (HCV), West-Nil-Virus (WNV), Humanes Papilloma-Virus (HPV), Japanisches-Enzephalitis-Virus (JEV), St.-Louis-Enzephalitis-Virus (SLEV) und andere. Die vorliegende Erfindung kann unschwer auf solche Nucleinsäuren angewendet werden. In diesem Fall kann es von Vorteil sein, eine interne, aus RNA bestehende Kontrollnucleinsäure zu verwenden, um die Zielnucleinsäuren-Eigenschaften wiederzugeben. Wenn sowohl RNA als auch DNA in dem vorstehend beschriebenen Verfahren analysiert werden sollen, ist es bevorzugt, dass die interne Kontrollnucleinsäure RNA ist, da die interne Kontrollnucleinsäure vorzugsweise das empfindlichste Ziel eines Tests, der mehrere Ziele umfasst, nachstellt und RNA-Ziele in der Regel exakter kontrolliert werden müssen.
-
Somit ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei die interne Kontrollnucleinsäure RNA ist.
-
Da RNA aufgrund von Einflüssen, wie alkalischer pH, Ribonucleasen, etc. eine größere Neigung zum Abbau besitzt als DNA, werden vorzugweise interne Kontrollnucleinsäuren, die aus RNA hergestellt sind, als armierte Partikel bereitgestellt. Armierte Partikel, wie insbesondere armierte RNA, werden z. B. in der
EP910643 beschrieben. Kurz gesagt ist die RNA, die chemisch oder vorzugsweise heterolog, z. B. durch Bakterien, wie E. coli, produziert werden kann, mindestens teilweise in einem viralen Mantelprotein verkapselt. Letzteres trägt zur Resistenz der RNA gegenüber externen Einflüssen, insbesondere Ribonucleasen, bei. Es muss verstanden werden, dass auch interne Kontroll-DNA als ein armiertes Partikel bereitgestellt werden kann. Sowohl armierte RNA als auch DNA sind als interne Kontrollnucleinsäuren im Zusammenhang mit der Erfindung geeignet. In einer bevorzugten Ausführungsform sind RNA-Kontrollnucleinsäuren mit dem MS2-Mantelprotein in E. coli armiert. In einer weiteren bevorzugten Ausführungsform sind DNA-Kontrollnucleinsäuren unter Verwendung des Lambda-Phagen GT11 armiert.
-
Darum ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, wobei die interne Kontrollnucleinsäure eine armierte Nucleinsäure ist.
-
Die Ergebnisse der vorstehend beschriebenen Tests können verfälscht werden und beispielsweise Falsch-Positive umfassen, in dem Fall einer Kreuzverunreinigung mit Nucleinsäuren aus Quellen, die anders sind als die Fluidprobe. Insbesondere können Amplifikate aus vorherigen Experimenten zu solchen unerwünschten Effekten beitragen. Ein besonderes Verfahren zur Minimierung der Auswirkungen von Kreuzverunreinigung von Nucleinsäureamplifikation ist in der
US-Patentschrift Nr. 5,035,996 beschrieben. Das Verfahren umfasst das Einbringen von unkonventionellen Nucleotidbasen, wie dUTP, in das amplifizierte Produkt und die Exposition von mitgeschleppten Produkten gegenüber enzymatischer und/oder physikochemischer Behandlung, um die Produkt-DNA unfähig zu machen, als ein Templat für anschließende Amplifikationen zu dienen. Enzyme für solche Behandlungen sind auf dem Fachgebiet bekannt. Beispielsweise entfernt Uracil-DNA-Glycosylase, auch als Uracil-N-Glycosylase oder UNG bekannt, Uracil-Reste aus PCR-Produkten, die die Base enthalten. Die Enzymbehandlung führt zum Abbau des verunreinigenden mitgeschleppten PCR-Produkts und dient der „Sterilisierung” der Amplifikationsreaktion.
-
Somit ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene Verfahren, das zwischen Schritt i. und Schritt ii. weiterhin die folgenden Schritte umfasst:
- • Behandeln der Fluidprobe mit einem Enzym unter Bedingungen, wobei Produkte aus Amplifikationen von kreuzverunreinigenden Nucleinsäuren aus anderen Proben enzymatisch abgebaut werden,
- • Inaktivierung des Enzyms.
-
Das Enzym ist vorzugsweise Uracil-N-Glycosylase.
-
Bei dem vorstehend beschriebenen Verfahren ist es bevorzugt, dass alle Schritte automatisiert sind. „Automatisiert” bedeutet, dass die Schritte eines Verfahrens dazu geeignet sind, mit einem Gerät oder einer Maschine durchgeführt zu werden, die in der Lage ist, mit wenig oder keiner externen Kontrolle oder Einfluss durch ein humanes Wesen zu arbeiten. Nur die Präparationsschritte für das Verfahren müssten per Hand vorgenommen werden, z. B. Lagerbehälter müssen gefüllt und vor Ort gebracht werden, die Wahl der Proben, und weitere, dem Fachmann bekannte Schritte, z. B. der Prozess einer Computerkontrolle, müssen von einem menschlichen Wesen durchgeführt werden. Das Gerät oder die Maschine können z. B. automatisch Flüssigkeiten zugeben, die Proben mischen und Inkubationsschritte bei spezifischen Temperaturen durchführen. Typischerweise ist eine solche Maschine oder ein solches Gerät ein Roboter, der von einem Computer gesteuert wird, der ein Programm ausführt, in dem die einzelnen Schritte und Befehle spezifiziert sind.
-
Ein weiterer Aspekt der Erfindung ist ein analytisches System (440) zur Isolierung und gleichzeitigen Amplifikation von mindestens zwei Zielnucleinsäuren, die in einer Fluidprobe vorhanden sein können, wobei das analytische System die folgenden Module umfasst:
- • eine Trennstation (230), umfassend ein festes Trägermaterial, wobei die Trennstation konstruiert und angeordnet ist, um eine Zielnucleinsäure, die in einer Fluidprobe mit eingeschlossen ist, abzutrennen und zu reinigen,
- • eine Amplifikationstation (405), die mindestens zwei Reaktionsgefäße umfasst, wobei die Reaktionsgefäße Amplifikationsreagenzien, mindestens eine erste gereinigte Zielnucleinsäure in mindestens einem ersten Reaktionsgefäß, und mindestens eine zweite gereinigte Nucleinsäure in mindestens einem zweiten Reaktionsgefäß, wobei die zweite Nucleinsäure in dem ersten Reaktionsgefäß fehlt, und eine Polymerase mit reverser Transkriptaseaktivität, wobei die Polymerase weiterhin eine Mutation einschließt, die zu einer verbesserten Nucleinsäure-Extensionsrate und/oder einer verbesserten reversen Transkriptaseaktivität relativ zu der jeweiligen Wildtyp-Polymerase beiträgt, umfassen.
-
Ein „analytisches System” ist eine Anordnung von Komponenten, wie Instrumente, die miteinander mit dem Endziel, eine gegebene Probe zu analysieren, Wechselwirken.
-
Das analytische System (440, 11) der vorliegenden Erfindung ist ein System (440), das ein Modul (401) zur Isolierung und/oder Reinigung eines Analyten umfasst. Weiterhin umfasst das System (440) zusätzlich ein Modul (403) zur Analyse des Analyten, um ein nachweisbares Signal zu erhalten. Das nachweisbare Signal kann in dem gleichen Modul (401, 402, 403) oder alternativ in einem getrennten Modul nachgewiesen werden. Der Begriff „Modul”, wie hierin verwendet, bezieht sich auf jeden räumlich definierten Ort innerhalb des Analysators (400). Zwei Module (401, 403) können durch Wände getrennt oder können in offener Beziehung sein. Jedes einzelne Modul (401, 402, 403) kann entweder autonom kontrolliert werden, oder die Kontrolle des Moduls (401, 402, 403) kann mit anderen Modulen geteilt werden. Vorzugsweise werden alle Module zentral gesteuert. Der Transfer zwischen den Modulen (401, 402, 403) kann manuell sein, aber ist vorzugsweise automatisiert. Somit sind eine Anzahl von verschiedenen Ausführungsformen von automatisierten Analysatoren (400) in der vorliegenden Erfindung mit umfasst.
-
Die „Trennstation” ist vorstehend beschrieben.
-
Eine „Amplifikationsstation” umfasst einen temperaturkontrollierten Inkubator zur Inkubation des Inhalts von mindestens zwei Reaktionsgefäßen. Sie umfasst weiterhin eine Vielzahl von Reaktionsgefäßen, wie Röhrchen oder Platten, in denen eine Reaktion für die Analyse der Probe, wie PCR, erfolgt. Die äußere Grenze oder die Wände von solchen Gefäßen sind chemisch inert, so dass sie die Amplifikationsreaktion, die darin stattfindet, nicht beeinträchtigt. Für die Leichtigkeit der Handhabung und zur leichteren Automation ist es bevorzugt, die mindestens zwei Reaktionsgefäße in einer integralen Anordnung zu kombinieren, so dass sie zusammen gehandhabt werden können.
-
Folglich ist ein bevorzugter Aspekt der Erfindung das vorstehend beschriebene analytische System, wobei die mindestens zwei Reaktionsgefäße in einer integralen Anordnung kombiniert sind.
-
Integrale Anordnungen können z. B. Ampullen oder Röhrchen sein, die reversibel oder irreversibel miteinander verknüpft oder in einem Ständer angeordnet sind. Vorzugsweise ist die integrale Anordnung eine Multiwellplatte.
-
Die Multiwellplatte wird vorzugsweise in einer Haltestation gehalten. In einer bevorzugteren Ausführungsform befördert ein Steuerungsprogramm ein Multiwellgefäß von einer Haltestation in eine Luftschleuse (460), und ein zweites Steuerungsprogramm befördert die Multiwellplatte von der Luftschleuse zur Amplifikationsstation, wobei beide Steuerungsprogramme mit der Multiwellplatte über eine formschlüssige Wechselwirkung Wechselwirken.
-
In einer bevorzugten Ausführungsform ist das analytische System voll automatisiert.
-
In einer Ausführungsform werden mindestens zwei in einer integralen Anordnung kombinierte Reaktionsgefäße zwischen Stationen des Systems befördert.
-
In einer zweiten Ausführungsform wird die gereinigte Zielnucleinsäure aus der Trennstation zu der Amplifikationsstation übergeführt. Vorzugsweise überführt ein Pipettiergerät, das Pipetten umfasst, mit angehefteten Pipettenspitzen die Flüssigkeit, die die gereinigte Nucleinsäure umfasst.
-
In einer dritten Ausführungsform wird die gereinigte Nucleinsäure von der Trennstation zu einem Reaktionsgefäß in einer integralen, in einer Haltestation gehaltenen Anordnung übergeführt. Vorzugsweise wird das Reaktionsgefäß in einer integralen Anordnung dann aus der Haltestation zur Amplifikationsstation übergeführt.
-
Das Analysesystem gemäß der Erfindung umfasst vorzugsweise weiterhin eine Pipettiereinheit. Die Pipettiereinheit umfasst mindestens eine Pipette, vorzugsweise mehrere Pipetten. In einer bevorzugten Ausführungsform sind die mehreren Pipetten in einer oder mehreren integralen Anordnungen kombiniert, innerhalb derer die Pipetten vorzugsweise einzeln manipuliert werden können. Pipetten, die im Zusammenhang mit der Erfindung verwendet werden, sind vorzugsweise Pipetten, die Pipettenspitzen umfassen, wie vorstehend beschrieben. In einer anderen bevorzugten Ausführungsform sind die Pipetten Pipettiernadeln.
-
Alternativ kann ein Reaktionsgefäß oder eine Anordnung von Reaktionsgefäßen, die für die Probenpräparation in der Trennstation verwendet wird, und die das Fluid, das die gereinigten Zielnucleinsäuren einschließt, aus der Trennstation in die Amplifikationsstation übergeführt werden.
-
Zu diesem Zweck umfasst das Analysesystem gemäß der Erfindung vorzugsweise weiterhin eine Überführungseinheit, wobei die Überführungseinheit vorzugsweise eine Robotervorrichtung umfasst, wobei die Vorrichtung vorzugsweise ein Steuerungsprogramm umfasst.
-
Aus den vorstehend ausgeführten Gründen im Zusammenhang mit dem Verfahren gemäß der Erfindung sind das Folgende weitere bevorzugte Aspekte der Erfindung:
- • Das vorstehend beschriebene Analysesystem (440), wobei mindestens ein Reaktionsgefäß eine RNA-Zielnucleinsäure und eine DNA-Zielnucleinsäure umfasst.
- • Das vorstehend beschriebene Analysesystem (440), wobei mindestens ein Reaktionsgefäß eine RNA-Zielnucleinsäure umfasst und mindestens ein anderes Reaktionsgefäß eine DNA-Zielnucleinsäure umfasst.
-
Vorzugsweise umfasst das vorstehend beschriebene Analysesystem (440) ein oder mehrere Elemente, die aus der Gruppe ausgewählt sind, bestehend aus:
- • einem Nachweismodul (403) zum Nachweis von Signalen, die von einem Analyten provoziert werden,
- • einem Verschluss (410),
- • einem Lagermodul (1008), für Reagenzien und/oder Abfall,
- • einer Steuereinheit (1006) zur Steuerung von Systemkomponenten.
-
Ein „Nachweismodul” (403) kann z. B. eine optische Nachweiseinheit zum Nachweis des Ergebnisses oder der Auswirkung des Amplifikationsvorgangs sein. Eine optische Nachweiseinheit kann eine Lichtquelle umfassen, z. B. eine Xenonlampe, optische Vorrichtungen, wie Spiegel, Linsen, optische Filter, Faseroptik zum Führen und Filtern des Lichts, einen oder mehrere Referenzkanäle, oder einer CCD-Kamera oder eine unterschiedliche Kamera.
-
Ein „Verschluss” (410) ist konstruiert und angeordnet, um alle im Zusammenhang mit dem Analysesystem gemäß der Erfindung verwendeten Gefäße zu verschließen. Ein solcher Verschluss kann beispielsweise Röhrchen mit passenden Kappen oder Multiwellplatten mit Folie oder anderen geeigneten Versiegelungsmaterialien verschließen.
-
Ein „Lagermodul” (1008) lagert die notwendigen Reagenzien, um eine chemische oder biologische Reaktion, die zur Analyse der Fluidprobe wichtig ist, zustande zu bringen. Es kann auch weitere Komponenten einschließen, die für das Verfahren der Erfindung geeignet sind, z. B. Wegwerfartikel, wie Pipettenspitzen oder Gefäße, die als Reaktionsgefäße in der Trennstation und/oder der Amplifikationsstation verwendet werden sollen.
-
Vorzugsweise umfasst das Analysesystem gemäß der Erfindung weiterhin eine Steuereinheit zur Steuerung der Systemkomponenten.
-
Eine solche „Steuereinheit” (1006) kann Software zur Sicherstellung einschließen, dass verschiedene Komponenten des Analysesystems korrekt und mit der korrekten Zeitgebung, wie auf koordinierte Weise, arbeiten und Wechselwirken, z. B. Bewegen und Manipulieren von Komponenten, wie Pipetten. Die Steuereinheit kann auch einen Prozessor einschließen, der ein Realzeitbetriebssystem (RTOS) betreibt, welches ein Multitasking-Betriebssystem ist, das für Realzeitanwendungen beabsichtigt ist. Mit anderen Worten ist der Systemprozessor in der Lage, Realzeiteinschränkungen zu verwalten, d. h. betriebsbedingte Fristen aus der Ereignis-zu-System-Reaktion ohne Rücksicht auf die Systemlast. Es kontrolliert in Realzeit, dass verschiedene Einheiten innerhalb des Systems arbeiten und korrekt nach gegebenen Anweisungen reagieren.
-
In einer bevorzugten Ausführungsform betrifft die vorliegende Erfindung ein Analysesystem (440) zum Verarbeiten eines Analyten, umfassend
- a. eine erste Position, umfassend erste Aufnahmegefäße (1001) in linearer Anordnung, umfassend flüssige Proben (1010), eine Arbeitsplatte (101), umfassend Aufnahmegefäße (103) in einer nxm-Anordnung zum Halten einer flüssigen Probe (1011), eine erste Pipettiervorrichtung (700), umfassend mindestens zwei Pipettiereinheiten (702) in linearer Anordnung, wobei die Pipettiereinheiten (702) an Pipettenspitzen (3, 4) gekoppelt sind, und einen Spitzenständer (70), umfassend Pipettierspitzen (3, 4) in einer ax(nxm)-Anordnung;
- b. eine zweite Position, umfassend eine Halterung (201, 128) für die Arbeitsplatte (101), eine Halterung (330) für eine Multiwellplatte, eine Halterung (470) für den Spitzenständer (70), und eine zweite Pipettiervorrichtung (35), wobei die zweite Pipettiervorrichtung (35) Pipettiereinheiten (702) in einer nxm-Anordnung zum Koppeln an Pipettenspitzen (3, 4) (12) umfasst. Der Begriff „Halterung”, wie hierin verwendet, bezieht sich auf jede Anordnung, die in der Lage ist, einen Ständer oder eine Arbeitsplatte aufzunehmen.
-
Die Vorteile des Analysesystems (440) der vorliegenden Erfindung sind wie vorstehend für das Verfahren der vorliegenden Erfindung beschrieben.
-
Vorzugsweise ist die Position der ersten Pipetteneinheiten (702) der ersten Pipettiervorrichtung (700) variabel. Bevorzugte Ausführungsformen der ersten Pipettiervorrichtung (700) sind hierein im Folgenden beschrieben.
-
In einer Ausführungsform umfasst der Spitzenständer (70) Pipettenspitzen (3, 4) in einer ax(nxm)-Anordnung. Vorzugsweise sind ein erster Typ (4) und ein zweiter Typ (3) von Pipettenspitzen in dem Spitzenständer (70) mit umfasst. In dieser Ausführungsform ist der erste Typ von Pipettenspitzen (4) in einer nxm-Anordnung angeordnet, und der zweite Typ von Pipettenspitzen (3) ist in einer nxm-Anordnung angeordnet. In diesem Zusammenhang bezeichnet „n” die Anzahl von Reihen und m die Anzahl von Spalten, wobei n vorzugsweise 6 und m vorzugsweise 8 ist. Stärker bevorzugt besitzt der erste Typ von Pipettenspitzen (4) ein unterschiedliches Volumen zum zweiten Typ von Pipettenspitzen (3), am stärksten bevorzugt ist das Volumen des ersten Typs von Pipettenspitzen (4) mehr als 500 μl, und das Volumen des zweiten Typs von Pipettenspitzen (3) beträgt weniger als 500 μl. In dieser Ausführungsform gilt a = 2. Allerdings sind Ausführungsformen der Erfindung mit mehr als zwei Typen von Pipettenspitzen und somit a > 2 ebenfalls in der vorliegenden Erfindung mit umfasst.
-
In einem Aspekt umfasst das Analysesystem (440) der vorliegenden Erfindung eine Steuereinheit (1006) zum Zuordnen von Probentypen und einzelnen Tests zu einzelnen Positionen der Arbeitsplatte (101). Vorzugsweise sind die Positionen getrennte Zellen (401, 402).
-
In einem Aspekt der Erfindung umfasst das System zusätzlich ein Übertragungssystem (480) zum Übertragen der Arbeitsplatte (101) und des Ständers (70) zwischen ersten (402) und zweiten (401) Positionen. Bevorzugte Ausführungsformen des Übertragungssystems (480) sind Förderbänder oder stärker bevorzugt ein oder mehrere Steuerungsprogramme.
-
Außerdem greifen die Pipetteneinheiten der zweiten Pipettiervorrichtung (35) vorzugsweise in die Pipettenspitzen (3, 4), die in der ersten Position (402) verwendet wurden, ein.
-
Eine bevorzugte Ausführungsform des Systems (440) der vorliegenden Erfindung umfasst zusätzlich eine dritte Station (403), umfassend einen temperaturgesteuerten Inkubator zum Inkubieren des Analyten mit Reagenzien, die notwendig sind, um ein nachweisbares Signal zu erhalten. Weitere bevorzugte Ausführungsformen dieses Systems sind im Folgenden beschrieben.
-
Eine optimalere Kontrolle der Zuordnung von Proben und Tests zu der nxm-Anordnung wird mit einem ersten Prozessor (1004), der in der ersten Position (402) eingeschlossen ist, zu der die Steuereinheit (1006) Anweisungen zum Zuordnen der Probentypen und einzelnen Tests auf spezifische Positionen in der nxm-Anordnung der Gefäße (103) der Arbeitsplatte (101) überträgt, und einen zweiten Prozessor (1005), der in der zweiten Position (401) eingeschlossen ist, zu der die Steuereinheit (1006) Anweisungen zum Zuordnen von Probentypen und einzelnen Tests zu spezifischen Positionen in der nxm-Anordnung von Gefäßen (103) der Arbeitsplatte überträgt, erreicht.
-
Vorzugsweise umfasst das System zusätzlich einen ersten in der ersten Position angeordneten Prozessor und einen zweiten in der zweiten Position angeordneten Prozessor.
-
Stärker bevorzugt steuert der erste Prozessor (1004) die erste Pipettiervorrichtung (700) und der zweite Prozessor (1005) steuert die zweite Pipettiervorrichtung (35).
-
Alle anderen bevorzugten Ausführungsformen und speziellen Beschreibungen von Ausführungsformen des Analysesystems gemäß der Erfindung sind diejenigen, die für das Verfahren gemäß der Erfindung erwähnt wurden.
-
Kurze Beschreibung der Figuren
-
Fig. 1
-
Eine schematische Abbildung des Probenpräparationsablaufs, wie in einer Ausführungsform der Erfindung verwendet.
-
Pfeile, die nach unten zeigen, bezeichnen die Zugabe von einer Komponente oder von einem Reagenz zu der jeweiligen Vertiefung der Deepwellplatte, die vorstehend erwähnt wurde, Pfeile, die nach oben zeigen, ihre jeweilige Entnahme. Diese Vorgänge wurden manuell in den Schritten 2, 3, 4, 21 und 22 durch den Arbeitskopf des Gerätes in den Schritten 10, 14, 16, 18 und 24, und durch den Reagenzkopf des Gerätes in den Schritten 5, 6, 7, 11, 15 und 19 durchgeführt.
-
Es ist selbstverständlich, dass die verwendeten Volumina im Geiste der Erfindung, vorzugsweise mindestens bis zu etwa 30% der offenbarten Werte, flexibel eingestellt werden können. Insbesondere in dem Falle von Schritt 2 ist das Probenvolumen vorzugsweise variabel, um die unterschiedlichen Typen von Fluidproben zu berücksichtigen, die mehr oder weniger Ausgangsmaterial zum Erhalt entsprechender Ergebnisse erfordern, wie es einem Fachmann bekannt ist. Vorzugsweise beträgt der Bereich von etwa 100 μl bis etwa 850 μl. Stärker bevorzugt beträgt er etwa 100 μl, etwa 500 μl oder etwa 850 μl. Vorzugsweise wird das Volumen in den jeweiligen Gefäßen mit dem Verdünnungsmittel in Schritt 3 auf ein identisches Gesamtvolumen eingestellt. Vorzugsweise, wie in dem Schema in 1 gezeigt, macht das Gesamtvolumen bis zu etwa 850 μl aus.
-
Fig. 2
-
Wachstumskurven der auf einem LightCycler480 (Roche Diagnostics GmbH, Mannheim, DE), wie in Beispiel 1 beschrieben, durchgeführten Amplifikationen der Zielnucleinsäuren, die sich von HIV, HBV und CT ableiten. Das das auf der y-Achse angegebene „Signal” ist ein normalisiertes Fluoreszenzsignal. Die x-Achse zeigt die Anzahl des jeweiligen PCR-Zyklus.
-
Die Wachstumskurven von HIV und HBV sind zusammen mit den Wachstumskurven der gezeigten entsprechenden internen Kontrollnucleinsäure gezeigt. Die jeweiligen Zielnukleinsäure-Kurven sind durch gerade Linien dargestellt, die Kontrollnucleinsäure-Kurven durch gepunktete Linien.
-
2a: Qualitativer HIV-Test, gemessen in dem Kanal zum Nachweis der Zielsonde.
-
2b: Qualitativer HIV-Test, gemessen in dem Kanal zum Nachweis der Kontrollsonde.
-
2c: Quantitativer HIV-Test, gemessen in dem Kanal zum Nachweis der Zielsonde.
-
2d: Quantitativer HIV-Test, gemessen in dem Kanal zum Nachweis der Kontrollsonde.
-
2e: Quantitativer HBV-Test, gemessen in dem Kanal zum Nachweis der Zielsonde.
-
2f: Quantitativer HBV-Test, gemessen in dem Kanal zum Nachweis der Kontrollsonde.
-
2g: CT-Test, gemessen in dem Kanal zum Nachweis der Zielsonde.
-
Fig. 3:
-
Perspektivische Ansicht der Arbeitslatte.
-
Fig. 4:
-
Perspektivische Ansicht der Arbeitsplatte aus dem Gegenwinkel.
-
Fig. 5:
-
Draufsicht auf die Arbeitsplatte.
-
Fig. 6:
-
Querschnittsansicht entlang der längeren Seite der Arbeitsplatte.
-
Fig. 7:
-
Eine Teilansicht der Querschnittsansicht.
-
Fig. 8:
-
Perspektivische Ansicht der längeren Seite der Arbeitsplatte.
-
Fig. 9:
-
a) bis d) zeigen verschiedene Ansichten der zweiten Ausführungsform der magnetischen Trennstation.
-
Fig. 10:
-
(a) bis (c) zeigen eine Ansicht der ersten Ausführungsform der magnetischen Trennstation, die die Arbeitsplatte hält, mit dem ersten Typ von Magneten in der obersten Z-Position und dem zweiten Typ von Magneten in der untersten Z-Position.
-
Fig. 11:
-
Schematische Zeichnungen von einem Analysator, umfassend verschiedene Stationen, Module oder Zellen.
-
Fig. 12:
-
zeigt ein Analysesystem der vorliegenden Erfindung.
-
Fig. 13:
-
Linearität des quantitativen HBV-Tests in EDTA-Plasma gemäß den Daten in Beispiel 2.
-
Fig. 14:
-
Linearität des quantitativen HBV-Tests in Serum gemäß den Daten in Beispiel 2.
-
Fig. 15:
-
Linearität des quantitativen HCV-Tests in EDTA-Plasma gemäß den Daten in Beispiel 2.
-
Fig. 16:
-
Linearität des quantitativen HCV-Tests in Serum gemäß den Daten in Beispiel 2.
-
Fig. 17:
-
Linearität des quantitativen HIV-Tests in EDTA-Plasma gemäß den Daten in Beispiel 2.
-
Beispiele
-
Die folgenden Beispiele beschreiben eine Ausführungsform, in der die Erfindung ausgeführt werden kann. Der Fachmann erkennt, dass diese Beispiele nicht einschränkend sind und ohne den Geist der Erfindung zu verlassen, modifiziert werden können.
-
Beispiel 1:
-
Dieses Beispiel beschreibt ein Verfahren zur Isolierung und gleichzeitigen Amplifizierung von mindestens einer ersten und einer zweiten Zielnucleinsäure unter Verwendung einer einzigen typischen internen Kontrollnucleinsäure.
-
Kurz gesagt wird in der beschriebenen Ausführungsform eine Realzeit-PCR gleichzeitig und unter identischen Bedingungen an einem Panel aus mehreren verschiedenen Zielen durchgeführt, das Bakterien (Chlamydia trachomatis, CT) sowie ein DNA-Virus (HBV) und ein RNA-Virus (HIV) einschloss. Sämtliche Proben wurden im gleichen Experiment verarbeitet und ausgewertet, d. h. auf der gleichen Deepwell- (zur Probenpräparation) bzw. Multiwellplatte (zur Amplifikation und zum Nachweis).
-
Die folgenden Proben wurden hergestellt und anschließend ausgewertet:
| Reagenz | Hersteller |
| HIV-1M Sekundärstandard, 50.000 cp/ML | Roche |
| HBV Sekundärstandard, 400 IU/ml | Roche |
| CT (DNA POS CTL pCHL-1) | Roche |
-
Geeignete Standards oder andere Typen von Zielen stehen dem Fachmann zur Verfügung.
-
Die in der folgenden Tabelle aufgeführten Instrumente wurden nach den Anweisungen des jeweiligen Herstellers verwendet:
| Instrument | Hersteller |
| Hamilton Star | Hamilton Medical AG (Bonaduz, CH) |
| Light Cycler 480 | Roche Diagnostics GmbH (Mannheim, DE) |
| Chameleon Sealer | K biosystems (Essex, UK) |
| Compressor | K biosystems (Essex, UK) |
-
Zur Probenpräparation wurden die folgenden Reagenzien als Verdünnungsmittel verwendet:
| Reagenz | Hersteller |
| PreservCyt | Thin Prep |
| K3 EDTA Plasma, PCR neg. | Roche |
-
Die folgenden Verdünnungen wurden zuvor hergestellt und über Nacht gelagert
(Plasmaverdünnungen bei –60 bis –90°C, PreservCyt-Verdünnungen bei 2–8°C):
| Ziel | Konzentration | Matrix |
| HBV | 50 | IU/ml | K3 EDTA-Plasma |
| HIV-1M | 100 | cp/ml | K3 EDTA-Plasma |
| CT | 2,5 | fg/ml | PreservCyt |
-
Die jeweilige Probe (500 μl) und das jeweilige Probenverdünnungsmittel (350 μl) wurden jeweils per Hand in eine Deepwellplatte pipettiert, wobei jede Probe für Dreifach-Auswertungen zu drei verschiedenen Vertiefungen zugesetzt wurde. Jeder Vertiefung, die eine HIV- oder HBV-Probe enthielt, wurden per Hand 50 μl der internen Kontrollnucleinsäure zugesetzt. Für den qualitativen HIV-Test wurde eine RNA, die als eine qualitative Kontrolle diente, zugesetzt (100 armierte Partikel/Probe). Für den quantitativen HIV-Test wurde eine RNA, die als ein quantitativer Standard diente, zugesetzt (500 armierte Partikel/Probe). Für den quantitativen HBV-Test wurde eine DNA, die als quantitativer Standard diente, zugesetzt (1E4 Kopien/Probe). Die Sequenz der Kontrollnucleinsäuren war in allen Fällen identisch und wurde aus der Gruppe der SEQ ID NOs 45–48 ausgewählt.
-
Die jeweilige Kontrollnucleinsäure wurde in dem folgenden Puffer aufbewahrt:
| IC/IQS-Lagerpuffer | Konz. oder pH |
| Tris (mM) | 10 |
| EDTA (mM) | 0,1 |
| Natriumazid (Gew./Vol., %) | 0,05 |
| Poly rA RNA (mg/l) | 20 |
| pH | 8 |
-
Die Probenpräparation wurde auf einem Hamilton Star (Hamilton, Bonaduz, CH) unter Befolgung des Arbeitsablaufs nach dem in
1 dargestellten Schema und unter Verwendung der folgenden Reagenzien durchgeführt:
| Proteasereagenz | Konz. oder pH |
| Tris (mM) | 10 |
| EDTA (mM) | 1 |
| Calciumchlorid (mM) | 5 |
| Calciumacetat (mg/ml) | 80 |
| Glycerin (Gew./Vol., %) | 50 |
| pH | 5,5 |
| MGP Reagenz | Konz. oder pH |
| MPG Pulver (mg/ml) | 60 |
| Tris (mM) | 30 |
| Methylparaben (Gew./Vol., %) | 0,1 |
| Natriumazid (Gew./Vol., %) | 0,095 |
| pH | 8,5 |
| Lysereagenz | Konz. oder pH |
| Guanidinthiocyanat (M) | 4 |
| Natriumcitrat (mM) | 50 |
| Polydocanol | 5 |
| Dithiotreitol (Gew./Vol., %) | 2 |
| pH | 5,8 |
| Waschpuffer | Konz. oder pH |
| Natriumcitrat (mM) | 7,5 |
| Methylparaben (Gew./Vol., %) | 0,1 |
| pH | 4,1 |
| Elutionspuffer | Konz. oder pH |
| Tris (mM) | 30 |
| Methylparaben (Gew./Vol., %) | 0,2 |
| pH | 8,5 |
-
Nach dem letzten Schritt setzte der Arbeitskopf des Hamilton-Star-Geräts jeder Vertiefung die jeweiligen Amplifikationsreagenzien enthaltenden Mastergemische (Mmxs) zu, mischte die Fluide, die die isolierten Nucleinsäuren enthielt, mit dem Mmx und überführte das resultierende Gemisch jeweils in eine entsprechende Vertiefung einer Mikrowellplatte, in der die Amplifikation durchgeführt wurde.
-
Die folgenden Mastergemische (bestehend jeweils aus den beiden Reagenzien R1 und R2) wurden verwendet: Für HIV:
| R1 Reagenz | Konzentration/50 μl-PCR (μM) |
| Wasser (PCR-Grad) | |
| Mn(Ac)2·4H2O (pH 6,1 eingestellt mit Essigsäure) | 3.000 |
| NaN3/Ri, gepuffert mit 10 mM Tris bei pH 7 [%] | 0,018 |
| | |
| R2 Reagenz | Konzentration/50 μl-PCR (μM) |
| DMSO [%] | 5,000% |
| NaN3/Ri, gepuffert mit 10 mM Tris bei pH 7 [%] | 0,027% |
| Kaliumacetat pH 7,0 | 110.000 |
| Glycerin [%] | 3,000% |
| Tricin pH 8,0 | 50.000% |
| Igepal [%] | 0,024% |
| dGTP | 337,5 |
| dATP | 337,5 |
| dCTP | 337,5 |
| dUTP | 675 |
| Primer/Sonden ausgewählt aus SEQ ID NOs 1–35 | 0,1–0,15 |
| SEQ ID NO 42 | 0,1 |
| SEQ ID NO 43 | 0,1 |
| SEQ ID NO 44 | 0,1 |
| Uracil-N-Glycosylase | 10 (U/Reaktion) |
| Z05-D Polymerase | 40 (U/Reaktion) |
| NTQ21-46A Aptamer | 0,222 |
| Wasser | |
Für HBV:
| R2 Reagenz | Konzentration/50 μl-PCR |
| H2O | 100 | % |
| Tricin 7,7 | 40 | mM |
| Tween | 0,03 | %(Vol./Vol.) |
| Glycerin | 5 | %(Vol./Vol.) |
| KOH | 25,2 | mM |
| KOAc | 121,8 | mM |
| NTQ21-46A (Aptamer) | 0,2625 | μM |
| dGTP | 0,42 | μM |
| dATP | 0,42 | μM |
| dCTP | 0,42 | μM |
| dUTP | 0,84 | μM |
| SEQ ID NO 36 | 1,2 | μM |
| SEQ ID NO 37 | 0,1 | μM |
| SEQ ID NO 38 | 1,2 | μM |
| SEQ ID NO 42 | 0,6 | μM |
| SEQ ID NO 43 | 0,6 | μM |
| SEQ ID NO 44 | 0,15 | μM |
| Z05D Polymerase | 35 | (U/Reaktion) |
| Uracil-N-Glycosylase | 2 | (U/Reaktion) |
| Natriumazid | 0,027 | %(m/v) |
| | | |
| R1 Reagenz | Konzentration/50 μl-PCR |
| H2O | 100 | % |
| MgOAc | 2,5 | mM |
| MnOAc pH 6,1 | 2,5 | mM |
| Natriumazid | 0,018 | % (m/v) |
Für CT:
| R1 Reagenz | Konzentration/50 μl-PCR (μM) |
| Wasser (PCR-Grad) | |
| Mn(Ac)2 (pH 6,5 in 0,002% (Vol./Vol.) Eisessigsäure | 2,7 mM |
| NaN3 | 0,0135% (Gew./Vol.) |
| | |
| R2 Reagenz | Konzentration/50 μl-PCR (μM) |
| NaN3/Ri, gepuffert mit 10 mM Tris bei pH 7 [%] | 0,0315% |
| Kaliumacetat | 112,4 mM |
| Glycerin [%] | 3,5% |
| Tricin | 61 mM |
| Kaliumhydroxid | 28,4 mM |
| dGTP | 525 μM |
| dATP | 525 μM |
| dCTP | 525 μM |
| dUTP | 1,05 mM |
| SEQ ID NO 39 | 750 nM |
| SEQ ID NO 40 | 600 nM |
| SEQ ID NO 41 | 116 nM |
| Aptamer NTQ21-46A | 175 nM |
| Uracil-N-Glycosylase | 5 U/Reaktion |
| Z05-D-Polymerase | 31 U/Reaktion |
-
Zur Amplifikation und zum Nachweis wurde die Mikrowellplatte mit einem automatisierten Plattenverschlussgerät (siehe vorstehend) verschlossen, und die Platte wurde in einen LightCycler 480 (siehe vorstehend) übergeführt.
-
Das folgende PCR-Profil wurde verwendet: Thermocyclingprofil:
| Programmname | Ziel (°C) | Aquisitionsmodus | Halten (hh:mm:s) | Anstiegsgeschwindigkeit [°C/s] | Zyklen | Analysemodus |
| Pre-PCR | 50 | ohne | 00:02:00 | 4,4 | 1 | ohne |
| 94 | ohne | 00:00:05 | 4,4 |
| 55 | ohne | 00:02:00 | 2,2 |
| 60 | ohne | 00:06:00 | 4,4 |
| 65 | ohne | 00:04:00 | 4,4 |
| 1. Messung | 95 | ohne | 00:00:05 | 4,4 | 5 | quantitativ |
| 55 | einfach | 00:00:30 | 2,2 |
| 2. Messung | 91 | ohne | 00:00:05 | 4,4 | 45 | quantitativ |
| 58 | einfach | 00:00:25 | 2,2 |
| Kühlen | 40 | ohne | 00:02:00 | 2,2 | 1 | ohne |
Nachweisformat (manuell)
| Filterkombination | Integrationszeit (Sekunden) |
| 435–470 | 1 |
| 495–525 | 0,5 |
| 540–580 | 0,5 |
| 610–645 | 0,5 |
| 680–700 | 1 |
-
Das Pre-PCR-Programm umfasst anfängliches Denaturieren und Inkubation bei 55, 60 und 65°C für die reverse Transkription von RNA-Templaten. Das Inkubieren bei drei Temperaturen vereinigte die vorteilhaften Wirkungen, dass bei niedrigeren Temperaturen auch geringfügig fehlgepaarte Zielsequenzen (wie genetische Varianten von einem Organismus) transkribiert werden, während bei höheren Temperaturen die Bildung von RNA-Sekundärstrukturen unterdrückt wird, was somit zu einer wirksameren Transkription führt.
-
PCR-Cycling wird in zwei Messungen unterteilt, wobei beide Messungen eine Ein-Schritt-Anordnung (wobei Anellieren und Extension kombiniert wird) anwenden. Die ersten 5 Zyklen bei 55°C gestatten eine erhöhte Inklusivität durch Preamplifikation von geringfügig fehlgepaarten Zielsequenzen, wohingegen die 45 Zyklen der zweiten Messung eine erhöhte Spezifität unter Verwendung einer Anellierungs-/Extensionstemperatur von 58°C bereitstellen.
-
Unter Verwendung dieses Profils bei allen Proben, die auf der vorstehend erwähnten Mikrowellplatte mit umfasst waren, wurden in allen Proben Amplifikation und Nachweis erreicht, wie in 2 dargestellt. Dies zeigt, dass auch die Probenpräparation vor der Amplifikation erfolgreich durchgeführt wurde.
-
Die Ergebnisse für die qualitativen und quantitativen internen HIV-Kontrollen und die quantitativen internen HBV-Kontrollen sind in 2 aus Gründen der Klarheit getrennt dargestellt. Es kann gesehen werden, dass auch die Kontrollen in allen Fällen erfolgreich amplifiziert wurden. Die Quantifizierung der HIV- und HBV-Ziele in der quantitativen Anordnung wurde durch Vergleich mit der internen Kontrollnucleinsäure, die als ein quantitativer Standard diente, berechnet.
-
Beispiel 2:
-
Das hierin zuvor beschriebene typische Amplifikationsverfahren wurde in getrennten Experimenten, aber unter identischen Bedingungen an einer Vielzahl von verschiedenen Zielnucleinsäuren durchgeführt. Die Isolierung der jeweiligen Nucleinsäure wurde wie unter Beispiel 1 beschrieben durchgeführt.
-
Die jeweilige generische interne Kontrollnucleinsäure wurde aus den SEQ ID NOs 45–49 ausgewählt und war für RNA-Ziele armierte RNA und für DNA-Ziele Lambda-gepackte DNA. Für qualitative RNA-Tests wurden 300 Partikel pro Probe zugesetzt, für quantitative RNA-Tests 3000 und für alle DNA-Tests 500. Das folgende PCR-Profil wurde bei allen Zielen verwendet:
| | | Ziel [°C] | Aquisitionsmodus | Plateau [hh:mm:ss] | Messung [hh:mm:ss] | Anstiegsgeschwindigkeit [°C/s] |
| Pre-PCR | UNG-Schritt | 50 | ohne | 00:02:00 | 00:00:00 | 2,2 |
| UNG/Templatdenaturierung | 94 | ohne | 00:00:05 | 00:00:00 | 4,4 |
| | RT-Schritt | 55 | ohne | 00:02:00 | 00:00:00 | 2,2 |
| 60 | ohne | 00:06:00 | 00:00:00 | 4,4 |
| 65 | ohne | 00:04:00 | 00:00:00 | 4,4 |
| erste Messung | 95 | ohne | 00:00:05 | 00:00:00 | 4,4 |
| 55 | einfach | 00:00:30 | 00:00:08 | 2,2 |
| zweite Messung | 91 | ohne | 00:00:05 | 00:00:00 | 4,4 |
| 58 | einfach | 00:00:25 | 00:00:08 | 2,2 |
| Kühlen | 40 | ohne | 00:02:00 | 00:00:00 | 2,2 |
| Name | Zyklen |
| Pre-PCR | 1 |
| erste Messung | 5 |
| zweite Messung | 45 |
| Kühlen | 1 |
-
Im Einzelnen wurden die folgenden Experimente durchgeführt: 1. Qualitative Multiplex-Analyse von HBV, HCV und HIV a. Mastergemisch R1:
| | Konzentration/50 μl-PCR (μM) |
| Mn(Ac)2·4H2O (pH 6,1 eingestellt mit Essigsäure) | 3.300 |
| NaN3/Ri, gepuffert mit 10 mM Tris bei pH 7 [%] | 0,018 |
| | pH: 6,41 |
R2:
| Reagenz | Konzentration/50 μl-PCR (μM) |
| DMSO [%] | 5,4 |
| NaN3/Ri, gepuffert mit 10 mM Tris bei pH 7 | 0,027 |
| KOAc (pH 7,0) | 120.000 |
| Glycerin (%) | 3 |
| Tween 20 (%) | 0,015 |
| Tricin pH 8,0 | 60.000 |
| NTQ21-46A-Aptamer | 0,2222 |
| Uracil-N-Glycosylase (U/uL) | 0,2 |
| dGTP | 400,0 |
| dATP | 400,0 |
| dCTP | 400,0 |
| dUTP | 800,0 |
| Z05-D Polymerase (U/uL)* | 0,9 |
| Primer/Sonden ausgewählt aus SEQ ID NOs 1–35 | 0,125–0,3 |
| SEQ ID NO 36 | 0,100 |
| SEQ ID NO 37 | 0,100 |
| SEQ ID NO 38 | 0,150 |
| Primer/Sonden ausgewählt aus SEQ ID NOs 60–76 | 0,050–0,250 |
| SEQ ID NO 42 | 0,200 |
| SEQ ID NO 43 | 0,200 |
| SEQ ID NO 44 | 0,100 |
-
Analytische Empfindlichkeit/LOD
-
Für jedes nachgewiesene Virus (HIV-1 Gruppe M und HIV-1 Gruppe O, HIV-2, HBV und HCV) bei mehreren Konzentrationen/Niveaus bei und in der Nähe der antizipierten LOD für EDTA-Plasma. Pro Virus und Konzentration wurde ein Panel mit mindestens 20 gültigen Replikaten pro Konzentration getestet. Die LOD wurde durch Probit-Analyse (siehe Tabelle 1–5) bestimmt. HIV Tabelle 1: HIV-1 Gruppe M-Trefferraten und Probit-LOD aus dem Einzelpanel
| Konzentration | Anzahl von Replikaten | Anzahl von Positiven | Trefferrate |
| 32 cp/mL | 21 | 21 | 100% |
| 16 cp/mL | 21 | 21 | 100% |
| 8 cp/mL | 21 | 21 | 100% |
| 4 cp/mL | 21 | 20 | 95% |
| 2 cp/mL | 21 | 15 | 71% |
| 1 cp/mL | 21 | 9 | 43% |
| 0 cp/mL (neg. Kontrolle) | 12 | 0 | 0% |
| | | | |
| LOD durch Probit-Analyse (95% Trefferrate) | | 4,06 cp/mL |
| 95% Konfidenzintervall für LOD durch Probit-Analyse | | 2,85–9,24 cp/mL |
-
Der Titer des WHO-Standards für die HIV-1 Gruppe M wurde in IU/mL umgewandelt.
-
-
Die HIV-1 Gruppe M-LOD in IU/mL ist daher folgende:
| LOD gemäß Probit-Analyse (95% Trefferrate): | 6,77 IU/mL |
| 95% Konfidenzintervall für LOD durch Probit-Analyse: | 4,75–15,4 IU/mL |
Tabelle 2: HIV-1 Gruppe O-Trefferrate und Probit-LOD aus dem Einzelpanel
| Konzentration | Anzahl von Replikaten | Anzahl von Positiven | Trefferrate |
| 60 cp/mL | 21 | 21 | 100% |
| 30 cp/mL | 20 | 20 | 100% |
| 20 cp/mL | 21 | 21 | 100% |
| 14 cp/mL | 21 | 19 | 90% |
| 7 cp/m L | 21 | 15 | 71% |
| 4,5 cp/mL | 21 | 12 | 57% |
| 0 cp/mL (neg. Kontrolle) | 12 | 0 | 0% |
| 0 cp/mL (neg. Kontrolle) | 12 | 0 | 0% |
| | | | |
| LOD durch Probit-Analyse (95% Trefferrate) | | 14,9 cp/mL |
| 95% Konfidenzintervall für LOD durch Probit-Analyse | | 10,9–31,5 cp/mL |
-
Der Titer des Primärstandards für die HIV-1-Gruppe O wurde wieder dem CBER HIV-1 Gruppe-O-Panel zugeordnet; der Berechnungsfaktor ist 0,586.
-
Die HIV-1 Gruppe O LOD ist daher folgende:
| LOD gemäß Probit-Analyse (95% Trefferrate): | 8,8 cp/mL |
| 95% Konfidenzintervall für LOD durch Probit-Analyse: | 6,4–18,5 cp/mL |
Tabelle 3: HIV-2-Trefferraten und Probit-LOD aus dem Einzelpanel
| Konzentration | Anzahl von Replikaten | Anzahl von Positiven | Trefferrate |
| 4 cp/mL | 21 | 21 | 100% |
| 2 cp/mL | 21 | 21 | 100% |
| 1 cp/mL | 21 | 20 | 95% |
| 0,5 cp/mL | 21 | 13 | 62% |
| 0,25 cp/mL | 21 | 13 | 62% |
| 0,125 cp/mL | 21 | 7 | 33% |
| 0 cp/mL (neg. Kontrolle) | 12 | 0 | 0% |
| | | | |
| LOD durch Probit-Analyse (95% Trefferrate) | | 1,29 cp/mL |
| 95% Konfidenzintervall für LOD durch Probit-Analyse | | –3,11 cp/mL |
-
Der Titer des Primärstandards für HIV-2 wurde wieder dem CBER HIV-2-Panel zugeordnet; der Berechnungsfaktor ist 26,7.
-
Die HIV-2 LOD ist daher folgende:
| LOD gemäß Probit-Analyse (95% Trefferrate): | 34,44 cp/mL |
| 95% Konfidenzintervall für LOD durch Probit-Analyse: | 21,89–83,04 cp/mL |
HBV Tabelle 4: HBV Trefferraten und Probit-LOD aus dem Einzelpanel:
| Konzentration | Anzahl von Replikaten | Anzahl von Positiven | Trefferrate |
| 7,6 IU/mL | 21 | 21 | 100% |
| 3,8 IU/mL | 21 | 21 | 100% |
| 1,9 IU/mL | 21 | 20 | 95% |
| 0,95 IU/mL | 21 | 14 | 67% |
| 0,6 IU/mL | 19 | 12 | 63% |
| 0,4 N/mL | 21 | 12 | 57% |
| 0 IU/mL (neg. Kontrolle) | 12 | 0 | 0% |
| | | | |
| LOD durch Probit-Analyse (95% Trefferrate) | | 2,27 IU/mL |
| 95% Konfidenzintervall für LOD durch Probit-Analyse | | 1,48–6,54 IU/mL |
HCV Tabelle 5: HCV-Trefferraten und Probit-LOD aus dem Einzelpanel
| Konzentration | Anzahl von Replikaten | Anzahl von Positiven | Trefferrate |
| 24 IU/mL | 21 | 21 | 100% |
| 12 IU/mL | 21 | 21 | 100% |
| 6 IU/mL | 21 | 21 | 100% |
| 3 IU/mL | 21 | 17 | 81% |
| 1,5 IU/mL | 21 | 14 | 67% |
| 0,75 IU/mL | 21 | 9 | 43% |
| 0 IU/mL (neg. Kontrolle) | 18 | 0 | 0% |
| | | | |
| LOD durch Probit-Analyse (95% Trefferrate) | | 4,76 IU/mL |
| 95% Konfidenzintervall für LOD durch Probit-Analyse | | 3,14–11,61 IU/mL |
2. Qualitative Analyse von WNV Mastergemisch R1:
| Reagenz | Konzentration/50 μl-PCR (μM) |
| Mn(Ac)2·4H2O (pH 6,1 eingestellt mit Essigsäure | 3.300 |
| NaN3/Ri, gepuffert mit 10 mM Tris bei pH 7 [%] | 0,018 |
| | pH: 6,41 |
R2:
| Reagenz | Konzentration/50 μl-PCR (μM) |
| DMSO [%] | 5,4 |
| NaN3/Ri, gepuffert mit 10 mM Tris bei pH 7 | 0,027 |
| K Acetat pH 7,0 | 120.000 |
| Glycerin (%) | 3 |
| Tween 20 (%) | 0,015 |
| Tricin pH 8,0 | 60.000 |
| NTQ21-46A-Aptamer | 0,2222 |
| Uracil-N-Glycosylase (U/μL) | 0,2 |
| dGTP | 400,0 |
| dATP | 400,0 |
| dCTP | 400,0 |
| dUTP | 800,0 |
| Z05-D Polymerase (U/μL)* | 0,9 |
| Primer/Sonden ausgewählt aus SEQ ID NOs 53–59 | 0,08–0,4 |
| SEQ ID NO 42 | 0,150 |
| SEQ ID NO 43 | 0,150 |
| SEQ ID NO 44 | 0,100 |
-
Analytische Empfindlichkeit/LOD
-
Für die Viren (WNV, SLEV und JEV) wurde ein unabhängiges Panel als eine Verdünnungsreihe des jeweiligen Standards, darunter mehrere Konzentrationen/Niveaus bei und in der Nähe der vorausberechneten LOD hergestellt. Ein Panel pro Virus und Konzentration wurde mit mindestens 20 gültigen Replikaten pro Konzentration getestet. Die LCD wurde durch Probit-Analyse bestimmt. Tabelle 6: WNV-Trefferraten und Probit-LOD aus dem Einzelpanel
| Konzentration Kontrolle) | Anzahl von Replikaten | Anzahl von Positiven | Trefferrate |
| 20 cp/mL | 21 | 21 | 100% |
| 12 cp/mL | 21 | 21 | 100% |
| 8 cp/m L | 21 | 21 | 100% |
| 5 cp/mL | 21 | 17 | 81% |
| 2,5 cp/mL | 21 | 15 | 71,4% |
| 0,5 cp/mL | 21 | 1 | 4,8% |
| 0 cp/mL (neg. Kontrolle) | 12 | 0 | 0% |
| | | | |
| LCD durch Probit-Analyse (95% Trefferrate) | | 6,57 cp/mL |
| 95% Konfidenzintervall für LOD durch Probit-Analyse | | 4,74–11,03 cp/mL |
Tabelle 7: SLEV Trefferraten und Probit-LOD aus dem Einzelpanel
| Konzentration | Anzahl von Replikaten | Anzahl von Positiven | Trefferrate |
| 140 cp/mL | 21 | 21 | 100% |
| 100 cp/mL | 21 | 20 | 95,2% |
| 70 cp/mL | 21 | 20 | 95,2% |
| 40 cp/mL | 21 | 17 | 81,0% |
| 20 cp/mL | 21 | 11 | 52,4% |
| 10 cp/mL | 21 | 6 | 28,6% |
| 0 cP/mL (neg. Kontrolle) | 12 | 0 | 0% |
| LOD durch Probit-Analyse (95% Trefferrate) | | 78,9 cp/mL |
| 95% Konfidenzintervall für LOD durch Probit-Analyse | | 55,4–145,7 cp/mL |
Tabelle 8: JEV Trefferraten und Probit-LOD aus dem Einzelpanel
| Konzentration Kontrolle) | Anzahl von Replikaten | Anzahl von Positiven | Trefferrate |
| 20 cp/mL | 21 | 20 | 95,2% |
| 12 cp/mL | 21 | 20 | 95,2% |
| 8 cp/mL | 21 | 18 | 85,7% |
| 5 cp/mL | 21 | 17 | 81,0% |
| 2,5 cp/mL | 21 | 14 | 66,7% |
| 0,5 cp/mL | 21 | 2 | 9,52% |
| 0 cp/mL (neg. | 12 | 0 | 0% |
| | | | |
| LOD durch Probit-Analyse (95% Trefferrate) | | 13,55 cp/mL |
| 95% Konfidenzintervall für LOD durch Probit-Analyse | | 8,78–27,7 cp/mL |
3. Quantitative Analyse von HBV Mastergemisch R1:
| | Endkonzentration/50 μl-PCR (μM) |
| Mn(Ac)2·4H2O (pH 6,1 eingestellt mit Essigsäure) | 3.300 |
| NaN3/Ri, gepuffert mit 10 mM Tris bei pH 7 [%] | 0,018 |
| | pH: 6,41 |
R2:
| Reagenz | Endkonzentration/50 μl-PCR (μM) |
| Glycerin (%) | 3% |
| Tricin | 60 mM |
| DMSO (% Vol./Vol.) | 5,4% |
| KOAc | 120 mM |
| Tween 20 (Vol./Vol.) | 0,015% |
| Aptamer NTQ21-46A | 0,222 μM |
| Z05-D Polymerase | 0,9 U/μl (45 U/rxn) |
| Uracil-N-Glycosylase | 0,2 U/μl (10 U/rxn) |
| Natriumazid (Gew./Vol.) | 0,027% |
| dCTPs | 400 μM |
| dGTPs | 400 μM |
| dATPs | 400 μM |
| dUTPs | 800 μM |
| SEQ ID NO 36 | 1,2 μM |
| SEQ ID NO 37 | 0,6 μM |
| SEQ ID NO 50 | 0,6 μM |
| SEQ ID NO 51 | 0,6 μM |
| SEQ ID NO 38 | 0,1 μM |
| SEQ ID NO 52 | M |
-
Analytische Empfindlichkeit/LOD
-
Zur Verdünnung wurden Panels mit HBV-Sekundärstandard (welcher den Gentyp A darstellte), hergestellt, d. h. zwei in HBV-negativem Serum für Probeneingangsvolumina von 200 μL und 500 μL und zwei in HBV-negativem EDTA-Plasma für Probeneingangsvolumina von 200 μL und 500 μL. Jedes Panel umfasste 7 Konzentrationsniveaus bei und in der Nähe der vorausberechneten LOD. Ein Panel pro Matrix wurde mit ≥ 21 Replikaten pro Konzentrationsniveau getestet. Mindestens 20 Replikate mussten gültig sein. Die LOD wurde durch Probit-Analyse bei 95% Trefferrate und ≥ 95% Trefferrate-Analyse bestimmt. Tabelle 9: LOD-Analyse für 200 μL Eingangsvolumen in EDTA-Plasma.*
| Konzentration | Anzahl von Replikaten | Anzahl von Positiven | Trefferrate |
| 25 IU/mL | 41 | 41 | 100% |
| 15 IU/mL | 41 | 39 | 95,1% |
| 10 IU/mL | 41 | 40 | 97,6% |
| 7 IU/mL | 41 | 40 | 97,6% |
| 4 IU/mL | 24 | 20 | 83,3% |
| 1 IU/mL | 24 | 4 | 16,7% |
| 0 IU/mL (neg. Kontrolle) | 24 | 0 | 0% |
| | | | |
| LOD durch Probit-Analyse (95% Trefferrate) | | 8,2 IU/mL |
| 95% Konfidenzintervall für LOD durch Probit-Analyse | | 4,8–26,0 IU/mL |
* es wurden zusätzliche Replikate getestet, um das festgestellte 95% Konfidenzintervall zu verschmälern. Tabelle 10: LOD-Analyse für 500 μL Eingangsvolumen in EDTA-Plasma
| Konzentration | Anzahl von Replikaten | Anzahl von Positiven | Trefferrate |
| 10 IU/mL | 21 | 21 | 100% |
| 7 IU/mL | 21 | 21 | 100% |
| 4 IU/mL | 21 | 21 | 100% |
| 2,5 IU/mL | 21 | 20 | 95,2% |
| 1 IU/mL | 21 | 14 | 66,7% |
| 0,2 IU/mL | 21 | 1 | 4,8% |
| 0 IU/mL (neg. Kontrolle) | 21 | 0 | 0% |
| | | | |
| LOD durch Probit-Analyse (95% Trefferrate) | | 2,3 IU/mL |
| 95% Konfidenzintervall für LOD durch Probit-Analyse | | 1,6–4,2 IU/mL |
Tabelle 11: LOD-Analyse für 200 μL Eingangsvolumen in Serum
| Konzentration | Anzahl von Replikaten | Anzahl von Positiven | Trefferrate |
| 25 IU/mL | 21 | 21 | 100% |
| 15 IU/mL | 21 | 20 | 95,2% |
| 10 IU/mL | 21 | 21 | 100% |
| 7 IU/mL | 21 | 20 | 95,2% |
| 4 IU/mL | 21 | 15 | 71,4% |
| 1 IU/mL | 21 | 8 | 38,1% |
| 0 IU/mL (neg. Kontrolle) | 21 | 0 | 0% |
| | | | |
| LOD durch Probit-Analyse (95% Trefferrate) | | 9,4 IU/mL |
| 95% Konfidenzintervall für LOD durch Probit-Analyse | | 6,2–19,0 IU/mL |
Tabelle 12: LOD-Analyse für 500 μL Eingangsvolumen in Serum
| Konzentration | Anzahl von Replikaten | Anzahl von Positiven | Trefferrate |
| 10 IU/mL | 21 | 21 | 100% |
| 7 IU/mL | 21 | 21 | 100% |
| 4 IU/mL | 21 | 21 | 100% |
| 2,5 IU/mL | 21 | 16 | 76,2% |
| 1 IU/mL | 21 | 16 | 76,2% |
| 0,2 IU/mL | 21 | 7 | 33,3% |
| 0 IU/mL (neg. Kontrolle) | 21 | 0 | 0% |
| | | | |
| LOD durch Probit-Analyse (95% Trefferrate) | | 4,1 IU/mL |
| 95% Konfidenzintervall für LOD durch Probit-Analyse | | 2,4–10,0 IU/mL |
-
Zusammenfassung LOD:
-
EDTA-Plasma: Die Probit-Analyse bei 95% Trefferrate ergab eine LOD von 8,2 IU/mL für 200 μL Probeneingangsvolumen und 2,3 IU/mL für 500 μL Probeneingangsvolumen für EDTA-Plasma.
-
Der 95%-Konfidenzintervallbereich für diese Konzentrationen betrug 4,8 bis 26,0 IU/mL für 200 μL Probeneingangsvolumen und 1,6 bis 4,2 IU/mL für 500 μL Probeneingangsvolumen.
-
Serum: Die Probit-Analyse bei 95% Trefferrate führte zu einer LOD von 9,02 IU/mL für 200 μL Probeneingangsvolumen und 4,1 IU/mL für 500 μL Probeneingangsvolumen für Serum.
-
Der 95%-Konfidenzintervallbereich betrug für diese Konzentrationen 6,2 bis 19,0 IU/mL für 200 μL Probeneingangsvolumen und 2,4 bis 10,0 IU/mL für 500 μL Probeneingangsvolumen.
-
Linearität
-
Ein EDTA-Plasmapanel und ein Serumpanel wurden unter Verwendung von HBV-Gentyp A (bereitgestellt von der Firma RMD Research Pleasanton, linearisiertes Plasmid, pHBV-PC_ADW2) hergestellt. Die Panels wurden zur Bestimmung des erwarteten dynamischen Bereiches (4 – 2E + 09 IU/mL) des Tests jeweils in 12 Konzentrationsniveaus ausgewertet. Sämtliche Konzentrationsniveaus/Panelmitglieder (PM) wurden in 21 Replikaten getestet.
-
Diese Studie wurde mit einem Probeneingangsvolumen von 500 μL durchgeführt. Die Konzentrationsniveaus wurden wie folgt gewählt: Ein Niveau unter der erwarteten quantitativen Untergrenze (LLOQ), eines bei der erwarteten LLOQ, eines oberhalb der erwarteten LLOQ, mehrere Konzentrationen auf intermediären Niveaus, bei der erwarteten quantitativen Obergrenze (ULOQ) und eines oberhalb der erwarteten ULOQ:
PM 12 – 2.0E + 09 IU/mL – oberhalb der erwarteten ULOQ.
PM 11 – 1,0E + 09 IU/mL – bei der erwarteten ULOQ.
PM 10 – 1,0E + 08 IU/mL – unterhalb der erwarteten ULOQ.
PM 9 – 1,0E + 07 IU/mL – intermediäres Konzentrationsniveau
PM 8 – 1,0E + 06 IU/mL – intermediäres Konzentrationsniveau
PM 7 – 1,0E + 05 IU/mL – intermediäres Konzentrationsniveau
PM 6 – 1,0E + 04 IU/mL – intermediäres Konzentrationsniveau
PM 5 – 1,0E + 03 IU/mL – intermediäres Konzentrationsniveau
PM 6a – 2,0E + 02 IU/mL – intermediäres Konzentrationsniveau (PM 6 verdünnt auf 2,0E + 02 IU/mL, verwendet als Titerzuordnung des Serumpanels)
PM 4 – 1,0E + 02 IU/mL – intermediäres Konzentrationsniveau (auch als Titerzuordnung des Plasmapanel verwendet)
PM 3 – 5,0E + 01 IU/mL – oberhalb der erwarteten LLOQ
PM 2 – 1,0E + 01 IU/mL – bei der erwarteten LLOQ
PM 1 – 4,0E + 00 IU/mL – unterhalb der erwarteten LLOQ
-
Für jede gültige Probe des Linearitätspanels wurde der festgestellte HBV-DNA-Titer in den log10-Titer umgewandelt, und der mittlere log10-Titer wurde pro Konzentrationsniveau berechnet. Tabelle 13: Linearität in EDTA-Plasma
| Nominaler Titer (IU/mL) | Zugeordneter Titer (IU/mL) | Zugeordneter log10-Titer | Mittlerer log10-Titer, festgestellt | Replikate |
| 4,00E + 00 | 3,50E + 00 | 0,54 | 0,52 | 17 |
| 1,00E + 01 | 8,70E + 00 | 0,94 | 0,91 | 21 |
| 5,00E + 01 | 4,40E + 01 | 1,64 | 1,69 | 21 |
| 1,00E + 02 | 8,70E + 01 | 1,94 | 2,04 | 21 |
| 1,00E + 03 | 8,70E + 02 | 2,94 | 3,01 | 21 |
| 1,00E + 04 | 8,70E + 03 | 3,94 | 3,9 | 21 |
| 1,00E + 05 | 8,70E + 04 | 4,94 | 4,88 | 21 |
| 1,00E + 06 | 8,70E + 05 | 5,94 | 5,87 | 21 |
| 1,00E + 07 | 8,70E + 06 | 6,94 | 6,92 | 21 |
| 1,00E + 08 | 8,70E + 07 | 7,94 | 8,01 | 21 |
| 1,00E + 09 | 8,70E + 08 | 8,94 | 9,04 | 21 |
| 2,00E + 09 | 1,70E + 09 | 9,24 | 9,38 | 21 |
-
Eine graphische Darstellung dieses Ergebnisses ist in
13 gezeigt. Tabelle 14: Linearität in Serum
| Nominaler Titer (IU/mL) | Zugeordneter Titer (IU/mL) | Zugeordneter log10-Titer | Mittlerer log10-Titer festgestellt | Replikate |
| 4,00E + 00 | 3,30E + 00 | 0,52 | 0,7 | 21 |
| 1,00E + 01 | 8,30E + 00 | 0,92 | 0,99 | 21 |
| 5,00E + 01 | 4,10E + 01 | 1,62 | 1,73 | 21 |
| 1,00E + 02 | 8,30E + 01 | 1,92 | 2,03 | 21 |
| 1,00E + 03 | 8,30E + 02 | 2,92 | 2,93 | 21 |
| 1,00E + 04 | 8,30E + 03 | 3,92 | 3,8 | 21 |
| 1,00E + 05 | 8,30E + 04 | 4,92 | 4,78 | 21 |
| 1,00E + 06 | 8,30E + 05 | 5,92 | 5,75 | 21 |
| 1,00E + 07 | 8,30E + 06 | 6,92 | 6,73 | 21 |
| 1,00E + 08 | 8,30E + 07 | 7,92 | 7,78 | 21 |
| 1,00E + 09 | 8,30E + 08 | 8,92 | 8,92 | 21 |
| 2,00E + 09 | 1,70E + 09 | 9,22 | 9,22 | 21 |
-
Eine graphische Darstellung dieses Ergebnisses ist in 14 gezeigt. Zusammenfassung Linearität: Der lineare Bereich, der als der Konzentrationsbereich definiert ist, für den die log10-Abweichung der festgestellten mittleren log10-Titer innerhalb von ±0,3 des nominalen log10-Titers liegt, wurde bestimmt als: 3,5E + 00 IU/mL – 1,7E + 09 IU/ml für EDTA-Plasma und 3,3E + 00 IU/mL – 1,7E + 09 IU/mL für Serum.
-
Die quantitative Untergrenze wurde für EDTA-Plasma und Serum als 4,0E + 00 IU/mL festgestellt. 4. Quantitative Analyse von HCV Mastergemisch R1:
| Reagenz | Endkonzentration/50 μl-PCR (μM) |
| Mn(Ac)2·4H2O (pH 6,1 eingestellt mit Essigsäure) | 3.300 |
| NaN3/Ri, gepuffert mit 10 mM Tris bei pH 7 | 0,018 |
| | pH: 6,41 |
R2:
| Reagenz | Endkonzentration/50 μl-PCR (μM) |
| Glycerin (% Gew./Vol.) | 3% |
| Tricin | 60 mM |
| DMSO (% Vol./Vol.) | 5,4% |
| KOAc | 120 mM |
| Tween 20 (Vol./Vol.) | 0,015% |
| NTQ21-46A | 0,222 μM |
| Z05D | 0,9 U/μl (45 U/rxn) |
| UNG | 0,2 U/μl (10 U/rxn) |
| Natriumazid (Gew./Vol.) | 0,027% |
| dCTPs | 400 μM |
| dGTPs | 400 μM |
| dATPs | 400 μM |
| dUTPs | 800 μM |
| Primer/Sonden ausgewählt aus SEQ ID NOs 60–76 | 0,1 μM |
| SEQ ID NO 42 | 0,3 μM |
| SEQ ID NO 43 | 0,3 μM |
| SEQ ID NO | 44 μM |
-
Analytische Empfindlichkeit/LOD
-
Ein Verdünnungspanel wurde mit Roche HCV Sekundärstandard in HCV-negativem EDTA-Plasma und Serum unter Verwendung von Probeneingangsvolumina von 200 μL und 500 μL hergestellt. Jedes Konzentrationsniveau wurde mit 21 Replikaten getestet. Mindestens z 20 Replikate müssen gültig sein. Die LOD wurde durch Probit-Analyse bei 95% Trefferrate und durch ≥ 95% Trefferrate-Analyse bestimmt. Tabelle 15: Trefferraten und Probit-Analyse mit 200 μL Probeneingangsvolumen für EDTA-Plasma
| Konzentration Kontrolle) | Anzahl von Replikaten | Anzahl von Positiven | Trefferrate |
| 55 IU/mL | 21 | 21 | 100% |
| 38 IU/mL | 21 | 21 | 100% |
| 25 IU/mL | 21 | 20 | 95% |
| 12,5 IU/mL | 21 | 19 | 90% |
| 6 IU/mL | 21 | 15 | 71% |
| 3 IU/mL | 21 | 6 | 29% |
| 0 IU/mL (neg. Kontrolle) | 21 | 0 | 0% |
| | | | |
| LOD durch Probit-Analyse (95% Trefferrate) | | 17,4 IU/mL |
| 95% Konfidenzintervall für LOD durch Probit-Analyse | | 12,1–34,4 IU/mL |
Tabelle 16: Trefferraten und Probit-Analyse mit 500 μL Probeneingangsvolumen für EDTA-Plasma
| Konzentration | Anzahl von Replikaten | Anzahl von Positiven | Trefferrate |
| 22 IU/mL | 21 | 21 | 100% |
| 15 IU/mL | 21 | 21 | 100% |
| 10 IU/mL | 20 | 20 | 100% |
| 5 IU/mL | 21 | 19 | 76% |
| 2,5 IU/mL | 21 | 15 | 71% |
| 1 IU/mL | 21 | 6 | 57% |
| 0 IU/mL (neg. Kontrolle) | 21 | 0 | 0% |
| | | | |
| LOD durch Probit-Analyse (95% Trefferrate) | | 9,0 IU/mL |
| 95% Konfidenzintervall für LOD durch Probit-Analyse | | 5,5–25,4 IU/mL |
Tabelle 17: Trefferraten und Probit-Analyse mit 200 μL Probeneingangsvolumen für Serum
| Konzentration | Anzahl von Replikaten | Anzahl von Positiven | Trefferrate |
| 55 IU/mL | 21 | 21 | 100% |
| 38 IU/mL | 21 | 21 | 100% |
| 25 IU/mL | 21 | 20 | 95% |
| 12,5 IU/mL | 21 | 18 | 86% |
| 6 IU/mL | 21 | 13 | 62% |
| 3 IU/mL | 21 | 6 | 29% |
| 0 IU/mL (neg. Kontrolle) | 21 | 0 | 0% |
| | | | |
| LOD durch Probit-Analyse (95% Trefferrate) | | 20,2 IU/mL |
| 95% Konfidenzintervall für LOD durch Probit-Analyse | | 14,0–39,3 IU/mL |
Tabelle 18: Trefferraten und Probit-Analyse mit 500 μL Probeneingangsvolumen für Serum
| Konzentration | Anzahl von Replikaten | Anzahl von Positiven | Trefferrate |
| 22 IU/mL | 21 | 21 | 100% |
| 15 IU/mL | 21 | 21 | 100% |
| 10 IU/mL | 21 | 20 | 95% |
| 5 IU/mL | 21 | 18 | 86% |
| 2,5 IU/mL | 21 | 12 | 57% |
| 1 IU/mL | 21 | 4 | 19% |
| 0 IU/mL (neg. Kontrolle) | 21 | 0 | 0% |
| LOD durch Probit-Analyse (95% Trefferrate) | | 8,2 IU/mL |
| 95% Konfidenzintervall für LOD durch Probit-Analyse | | 5,8–15,0 IU/mL |
-
Zusammenfassung LOD:
-
- 1. Die Probit-Analyse bei 95% Trefferrate ergab für 200 μL Probeneingangsvolumen eine LOD von 17,4 IU/mL und 9,0 IU/mL für 500 μL Probeneingangsvolumen für EDTA-Plasma. Das 95%-Konfidenzintervall für diese Konzentrationen beträgt 12,1 bis 34,3 IU/mL für 200 μL Probeneingangsvolumen und 5,5 bis 25,4 IU/mL für 500 μL Probeneingangsvolumen.
- 2. Die Werte der Probit-Analyse bei 95% Trefferrate betragen 20,2 IU/mL für 200 μL Probeneingangsvolumen 8,2 IU/mL für 500 μL Probeneingangsvolumen für Serum. Das 95%-Konfidenzintervall für diese Konzentrationen beträgt 14,0 bis 39,3 IU/mL für 200 μL Probeneingangsvolumen und 5,8 bis 15,0 IU/mL für 500 μL Probeneingangsvolumen.
-
Linearität
-
Eine Präparation eines EDTA-Plasmapanels und eine Präparation eines Serumpanels von HCV-aRNA, die auf den HCV-WHO-Standard zurückführbar war, wurden analysiert. Die Linearitätspanels wurden durch serielle Verdünnung hergestellt und bei 10 verschiedenen Konzentrationen ausgewertet. Die Untersuchung wurde mit 500 μL Probeneingangsvolumen vorgenommen. Die Konzentrationen wurden wie folgt gewählt: ein Niveau unterhalb der unteren quantitativen Nachweisgrenze (LLOQ), eines bei LLOQ, eines oberhalb von LLOQ, mehrere Konzentrationen auf intermediären Niveaus, bei erwarteter quantitativen oberen Nachweisgrenze (ULOQ) und eines bei oder oberhalb der ULOQ. Für sämtliche Konzentrationen wurden 21 Replikate getestet.
PM 1 – 2,0E + 08 IU/mL – oberhalb der erwarteten ULOQ
PM 2 – 1,0E + 08 IU/mL – bei der erwarteten ULOQ
PM 3 – 1,0E + 07 IU/mL – unter der erwarteten ULOQ
PM 4 – 1,0E + 06 IU/mL – intermediäres Konzentrationsniveau
PM 5 – 1,0E + 05 IU/mL – intermediäres Konzentrationsniveau
PM 6 – 1,0E + 04 IU/mL – intermediäres Konzentrationsniveau für die Titerzuordnung
PM 7 – 1,0E + 03 IU/mL – intermediäres Konzentrationsniveau
PM 8 – 1,0E + 02 IU/mL – oberhalb der erwarteten LLOQ
PM 9 – 1,0E + 01 IU/mL – bei der erwarteten LLOQ
PM 10 – 8,0E + 00 IU/mL – unter der erwarteten LLOQ Tabelle 19: Linearität in EDTA-Plasma
| Nominaler Titer (IU/mL) | Zugeordnet er Titer (IU/mL) | Zugeordnet er log10-Titer | Mittlerer log10-Titer, festgestellt | Replikate |
| 8,00E + 00 | 4,87E + 00 | 0,7 | 0,6 | 15 |
| 1,00E + 01 | 6,09E + 00 | 0,8 | 0,8 | 17 |
| 1,00E + 02 | 6,09E + 01 | 1,8 | 1,7 | 21 |
| 1,00E + 03 | 6,09E + 02 | 2,8 | 2,8 | 21 |
| 1,00E + 04 | 6,09E + 03 | 3,8 | 3,8 | 21 |
| 1,00E + 05 | 6,09E + 04 | 4,8 | 4,7 | 21/20 |
| 1,00E + 06 | 6,09E + 05 | 5,8 | 5,6 | 21/20 |
| 1,00E + 07 | 6,09E + 06 | 6,8 | 6,7 | 21 |
| 1,00E + 08 | 6,09E + 07 | 7,8 | 7,8/7,7 | 21/18 |
| 2,00E + 08 | 1,22E + 08 | 8,1 | 8 | 21/20 |
-
Eine graphische Darstellung dieses Ergebnisses ist in
15 gezeigt. Tabelle 20: Linearität in Serum
| Nominaler Titer (IU/mL) | Zugeordnet er Titer (IU/mL) | Zugeordnet er log10-Titer | Mittlerer log10-Titer festgestellt | Replikate |
| 8,00E + 00 | 3,90E + 00 | 0,6 | 0,7 | 10 |
| 1,00E + 01 | 4,96E + 00 | 0,7 | 0,7 | 14 |
| 1,00E + 02 | 4,96E + 01 | 1,7 | 1,6 | 21 |
| 1,00E + 03 | 4,96E + 02 | 2,7 | 2,8 | 21 |
| 1,00E + 04 | 4,96E + 03 | 3,7 | 3,7 | 21 |
| 1,00E + 05 | 4,96E + 04 | 4,7 | 4,7 | 21 |
| 1,00E + 06 | 4,96E + 05 | 5,7 | 5,7 | 21 |
| 1,00E + 07 | 4,96E + 06 | 6,7 | 6,7 | 21 |
| 1,00E + 08 | 4,96E + 07 | 7,7 | 7,7 | 21 |
| 2,00E + 08 | 9,92E + 07 | 8 | 8,1 | 21 |
-
Eine graphische Darstellung dieses Ergebnisses ist in 16 gezeigt.
-
Zusammenfassung Linearität:
-
Der Linearitätsbereich, der als der Konzentrationsbereich definiert ist, für den die log10-Abweichung des festgestellten mittleren log10-Titers innerhalb von ±0,3 des nominalen log10-Titers liegt, wurde wie folgt bestimmt: 4,87E + 00 IU/mL – 1,22E + 08 IU/mL für EDTA-Plasma und 3,90E + 00 IU/mL – 9,92E + 07 IU/mL für Serum. 5. Quantitative Analyse von HIV Mastergemisch R1:
| Reagenz | Endkonzentration/50 μl-PCR (μM) |
| Mn(Ac)2·4H2O (pH 6,1 eingestellt mit Essigsäure) | 3.300 |
| NaN3/Ri, gepuffert mit 10 mM Tris bei pH 7 | 0,018 |
| | pH: 6,41 |
R2:
| Reagenz | Endkonzentration/50 μl-PCR (μM) |
| Glycerin (% Gew./Vol.) | 3% |
| Tricin | 60 mM |
| DMSO (% Vol./Vol.) | 5,4% |
| KOAc | 120 mM |
| Tween 20 (Vol./Vol.) | 0,02% |
| Aptamer NTQ21-46A | 0,222 μM |
| Z05 D Polymerase | 0,9 U/μl (45 U/rxn) |
| UNG | 0,2 U/μl (10 U/rxn) |
| Natriumazid (Gew./Vol.) | 0,027% |
| dCTPs | 400 μM |
| dGTPs | 400 μM |
| dATPs | 400 μM |
| dUTPs | 800 μM |
| Primer/Sonden ausgewählt aus SEQ ID NOs 1–35 | 0,1 μM–0,3 μM |
| SEQ ID NO 50 | 0,3 μM |
| SEQ ID NO 51 | 0,3 μM |
| SEQ ID NO 52 | μM |
-
Analytische Empfindlichkeit/LOD
-
Ein Verdünnungspanel wurde mit HIV-1M Sekundärstandard in HIV-1-negativem EDTA-Plasma für Probeneingangsvolumina von 200 μL und 500 μL hergestellt. Jedes Konzentrationsniveau wurde mit 21 Replikaten getestet. Mindestens ≥ 20 Replikate müssen gültig sein. Die LOD wurde durch Probit-Analyse bei 95% Trefferrate und durch ≥ 95% Trefferrate-Analyse bestimmt. Tabelle 21: LOD-Analyse für 200 μL Eingabevolumen in EDTA-Plasma
| Konzentration | Anzahl von Replikaten | Anzahl von Positiven | Trefferrate |
| 200 cp/mL | 21 | 21 | 100% |
| 100 cp/mL | 21 | 21 | 100% |
| 80 cp/mL | 21 | 21 | 100% |
| 50 cp/mL | 21 | 20 | 95,2% |
| 30 cp/mL | 21 | 18 | 85,7% |
| 20 cp/mL | 21 | 17 | 81,0% |
| 10 cp/mL Kontrolle) | 21 | 8 | 38,1% |
| 0 cp/mL (neg. Kontrolle) | 21 | 0 | 0% |
| | | | |
| LOD durch Probit-Analyse (95% Trefferrate) | | 41,8 cp/mL |
| 95% Konfidenzintervall für LOD durch Probit-Analyse | | 30,9–74,9 cp/mL |
Tabelle 22: LOD-Analyse für 500 μL Eingabevolumen
| Konzentration | Anzahl von Replikaten | Anzahl von Positiven | Trefferrate |
| 30 cp/mL | 21 | 21 | 100% |
| 25 cp/mL | 21 | 20 | 95,2% |
| 20 cp/mL | 21 | 21 | 100% |
| 13,5 cp/mL | 21 | 18 | 85,7% |
| 9 cp/mL | 21 | 13 | 61,9% |
| 6 cp/mL | 21 | 9 | 42,9% |
| 0 cp/mL (neg. Kontrolle) | 21 | 0 | 0% |
| | | | |
| LOD durch Probit-Analyse (95% Trefferrate) | | 18,9 cp/mL |
| 95% Konfidenzintervall für LOD durch Probit-Analyse | | 14,9–29,4 cp/mL |
-
Zusammenfassung LOD
-
- 1. Die Probit-Analyse bei 95% Trefferrate ergab eine LOD von 41,8 cp/mL für 200 μL Eingabevolumen und 18,9 cp/mL für 500 μL Eingabevolumen.
- 2. Der 95% Konfidenzintervallbereich für diese Konzentrationen betrug 30,9 bis 74,9 cp/mL für 200 μL Eingabevolumen und 14,9 bis 29,4 cp/mL für 500 μL Eingabevolumen.
-
Linearität
-
Die in der Linearitäts/dynamischer Bereich/Genauigkeitsstudie verwendeten Proben bestanden aus einem Verdünnungspanel von einem HIV- 1-Zellkulturüberstandsmaterial, HIV-1 Gruppe M, Untertyp B.
-
Das Linearitätspanel wurde durch serielle Verdünnung hergestellt. Dieses Panel wurde auf 10 Konzentrationsniveaus analysiert.
-
Die Konzentrationen wurden wie folgt gewählt: Ein Niveau unterhalb der erwarteten quantitativen Nachweisuntergrenze (LLOQ), eines bei LLOQ, eines oberhalb der LLOQ, mehrere Konzentrationen auf intermediären Niveaus, bei der erwarteten quantitativen Nachweisobergrenze (ULOQ) und eines oberhalb der ULOQ. Für sämtliche Konzentrationen wurden 21 Replikate getestet. Die Linearitätsstudie wurde mit 500 μL Eingabevolumen vorgenommen.
PM 1 – 2,0E + 07 cp/mL – oberhalb der erwarteten ULOQ
PM 2 – 1,0E + 07 cp/mL – bei der erwarteten ULOQ
PM 3 – 1,0E + 06 cp/mL – unter der erwarteten ULOQ
PM 4 – 1,0E + 05 cp/mL – intermediäres Konzentrationsniveau
PM 5 – 3,0E + 04 cp/mL – intermediäres Konzentrationsniveau für die Titerzuordnung
PM 6 – 1,0E + 04 cp/mL – intermediäres Konzentrationsniveau
PM 7 – 1,0E + 03 cp/mL – intermediäres Konzentrationsniveau
PM 8 – 1,0E + 02 cp/mL – intermediäres Konzentrationsniveau
PM 9 – 5,0E + 01 cp/mL – oberhalb der erwarteten LLOQ
PM 10 – 2,0E + 01 cp/mL – bei der erwarteten LLOQ
PM 11 – 1,5E + 00 cp/mL – unter der erwarteten LLOQ Tabelle 23: Linearität in EDTA-Plasma
| Nominaler Titer (IU/mL) | Zugeordneter Titer (IU/mL) | Zugeordneter log10-Titer | Mittlerer log10-Titer, festgestellt | Replikate |
| 1,50E + 01 | 1,50E + 01 | 1,2 | 1,3 | 21 |
| 2,00E + 01 | 2,00E + 01 | 1,3 | 1,5 | 21 |
| 5,00E + 01 | 5,10E + 01 | 1,7 | 1,8 | 21 |
| 1,00E + 02 | 1,00E + 02 | 2 | 2 | 21 |
| 1,00E + 03 | 1,00E + 03 | 3 | 3 | 21 |
| 1,00E + 04 | 1,00E + 04 | 4 | 4 | 21 |
| 1,00E + 05 | 1,00E + 05 | 5 | 5 | 21 |
| 1,00E + 06 | 1,00E + 06 | 6 | 6 | 21 |
| 1,00E + 07 | 1,00E + 07 | 7 | 7 | 21 |
| 2,00E + 07 | 2,00E + 07 | 7,3 | 7,4 | 21 |
-
Eine graphische Darstellung dieses Ergebnisses ist in 17 gezeigt.
-
Zusammenfassung Linearität
-
Der Linearitätsbereich, der als der Konzentrationsbereich definiert ist, für den die log10-Abweichung der festgestellten mittleren log10-Titer innerhalb von ±0,3 des nominalen log10-Titers liegt, wurde als 1,5 E + 01 cp/mL – 2,0 E + 07 cp/mL bestimmt.
-
Es folgt ein Sequenzprotokoll nach WIPO St. 25.
Dieses kann von der amtlichen Veröffentlichungsplattform des DPMA heruntergeladen werden.
-
-
ZITATE ENTHALTEN IN DER BESCHREIBUNG
-
Diese Liste der vom Anmelder aufgeführten Dokumente wurde automatisiert erzeugt und ist ausschließlich zur besseren Information des Lesers aufgenommen. Die Liste ist nicht Bestandteil der deutschen Patent- bzw. Gebrauchsmusteranmeldung. Das DPMA übernimmt keinerlei Haftung für etwaige Fehler oder Auslassungen.
-
Zitierte Patentliteratur
-
- EP 1932913 [0028]
- WO 98/04730 [0035]
- US 5386024 [0035]
- EP 1201753 [0035]
- DE 3734442 A [0038]
- GB 91/00212 [0038]
- WO 01/37291 [0038]
- US 4458066 [0124]
- WO 02/12263 [0127]
- US 4683202 [0133]
- US 4683195 [0133]
- US 4800159 [0133]
- US 4965188 [0133]
- WO 90/01069 [0141]
- EP 0439182 A2 [0141]
- WO 92/08808 [0141]
- US 5130238 [0141]
- WO 2008/046612 [0157]
- US 5455170 [0158]
- WO 97/46707 [0176]
- WO 97/46714 [0176]
- WO 97/46712 [0176]
- EP 910643 [0214]
- US 5035996 [0216]
-
Zitierte Nicht-Patentliteratur
-
- Walsh, 1979, Enzymatic Reaction Mechanisms, W. H. Freeman and Company, San Francisco, Kapitel 3 [0035]
- Sambrook J. et al., Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 1989 [0035]
- Vogelstein B. et al., Proc. Natl. Acad. USA 76 (1979) 615–9 [0038]
- Marko M. A. et al., Anal. Biochem. 121 (1982) 382–387 [0038]
- Lambda-Phagen ist in Jakobi R. et al., Anal. Biochem. 175 (1988) 196–201 [0038]
- Alderton R. P. et al., S. Anal. Biochem. 201 (1992) 166–169 [0038]
- R. Boom et al. (J Clin Microbiol. 28 (1990), 495–503) [0038]
- Narang S. A. et al., Methods in Enzymology 68 (1979) 90–98 [0124]
- Brown E. L., et al. Methods in Enzymology 68 (1979) 109–151 [0124]
- Beaucage et al., Tetrahedron Letters 22 (1981) 1859 [0124]
- Garegg et al., Chem. Scr. 25 (1985) 280–282 [0124]
- Uhlmann und Peyman, Chemical Reviews 90 (1990) 543 [0127]
- Verma S., und Eckstein F., Annu. Rev. Biochem. 67 (1998) 99–134 [0127]
- Verma S. und Eckstein F., Annu. Rev. Biochem. 67 (1998) 99–134 [0127]
- Verma S. und Eckstein F. Annu. Rev. Biochem. 67 (1998) 99–134 [0127]
- LCR; Wu D. Y. und Wallace R. B., Genomics 4 (1989) 560–69 [0141]
- Barany F., Proc. Genomics 4 (1989) 560–69 [0141]
- Barany F., Natl. Acad. Sci. USA 88 (1991) 189–193 [0141]
- Barany F., PCR Methods and Applic. 1 (1991) 5–16 [0141]
- Kwoh D. Y. et al., Proc. Natl. Acad. Sci. USA 86 (1989) 1173–1177 [0141]
- Guatelli J. C., et al., Proc. Natl. Acad. Sci. USA 87 (1990) 1874–1878 [0141]
- Whelen A. C. und Persing D. H., Annu. Rev. Microbiol. 50 (1996) 349–373 [0141]
- Abramson R. D. und Myers T. W., Curr Opin Biotechnol 4 (1993) 41–47 [0141]
- Sambrook J. et al., Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 1989 [0169]
- Ausubel F. et al.: Current Protocols in Molecular Biology 1987, J. Wiley and Sons, NY [0169]