CN1511192A - 聚酮化合物的制备 - Google Patents
聚酮化合物的制备 Download PDFInfo
- Publication number
- CN1511192A CN1511192A CNA018087116A CN01808711A CN1511192A CN 1511192 A CN1511192 A CN 1511192A CN A018087116 A CNA018087116 A CN A018087116A CN 01808711 A CN01808711 A CN 01808711A CN 1511192 A CN1511192 A CN 1511192A
- Authority
- CN
- China
- Prior art keywords
- epothilone
- pks
- host cell
- module
- gene
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 229930001119 polyketide Natural products 0.000 title claims abstract description 94
- 125000000830 polyketide group Chemical group 0.000 title abstract description 54
- 238000004519 manufacturing process Methods 0.000 title description 108
- 229930013356 epothilone Natural products 0.000 claims abstract description 392
- 150000003883 epothilone derivatives Chemical class 0.000 claims abstract description 319
- 238000000855 fermentation Methods 0.000 claims abstract description 71
- 230000004151 fermentation Effects 0.000 claims abstract description 71
- 241000863420 Myxococcus Species 0.000 claims abstract description 41
- 108010030975 Polyketide Synthases Proteins 0.000 claims description 393
- 150000001875 compounds Chemical class 0.000 claims description 198
- HESCAJZNRMSMJG-KKQRBIROSA-N epothilone A Chemical class C/C([C@@H]1C[C@@H]2O[C@@H]2CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 HESCAJZNRMSMJG-KKQRBIROSA-N 0.000 claims description 178
- 108090000623 proteins and genes Proteins 0.000 claims description 161
- 238000000034 method Methods 0.000 claims description 132
- 101001110310 Lentilactobacillus kefiri NADP-dependent (R)-specific alcohol dehydrogenase Proteins 0.000 claims description 119
- XOZIUKBZLSUILX-UHFFFAOYSA-N desoxyepothilone B Natural products O1C(=O)CC(O)C(C)(C)C(=O)C(C)C(O)C(C)CCCC(C)=CCC1C(C)=CC1=CSC(C)=N1 XOZIUKBZLSUILX-UHFFFAOYSA-N 0.000 claims description 102
- 102000004867 Hydro-Lyases Human genes 0.000 claims description 83
- 108090001042 Hydro-Lyases Proteins 0.000 claims description 83
- 241000863422 Myxococcus xanthus Species 0.000 claims description 79
- 108091026890 Coding region Proteins 0.000 claims description 68
- XOZIUKBZLSUILX-GIQCAXHBSA-N epothilone D Chemical compound O1C(=O)C[C@H](O)C(C)(C)C(=O)[C@H](C)[C@@H](O)[C@@H](C)CCC\C(C)=C/C[C@H]1C(\C)=C\C1=CSC(C)=N1 XOZIUKBZLSUILX-GIQCAXHBSA-N 0.000 claims description 67
- XOZIUKBZLSUILX-SDMHVBBESA-N Epothilone D Natural products O=C1[C@H](C)[C@@H](O)[C@@H](C)CCC/C(/C)=C/C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C XOZIUKBZLSUILX-SDMHVBBESA-N 0.000 claims description 63
- 230000015572 biosynthetic process Effects 0.000 claims description 63
- 239000001257 hydrogen Substances 0.000 claims description 59
- 229910052739 hydrogen Inorganic materials 0.000 claims description 59
- 238000011068 loading method Methods 0.000 claims description 49
- 150000003881 polyketide derivatives Chemical class 0.000 claims description 49
- 239000013598 vector Substances 0.000 claims description 47
- 101100382105 Sorangium cellulosum cyp167A1 gene Proteins 0.000 claims description 44
- 108090000790 Enzymes Proteins 0.000 claims description 41
- 102000004190 Enzymes Human genes 0.000 claims description 37
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 36
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 34
- 239000013604 expression vector Substances 0.000 claims description 32
- 241000862997 Sorangium cellulosum Species 0.000 claims description 28
- 125000003118 aryl group Chemical group 0.000 claims description 28
- 239000011347 resin Substances 0.000 claims description 27
- 229920005989 resin Polymers 0.000 claims description 27
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 26
- 238000012217 deletion Methods 0.000 claims description 23
- 230000037430 deletion Effects 0.000 claims description 23
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 20
- 239000011203 carbon fibre reinforced carbon Substances 0.000 claims description 20
- 229910052799 carbon Inorganic materials 0.000 claims description 19
- 229910052760 oxygen Inorganic materials 0.000 claims description 18
- 230000035772 mutation Effects 0.000 claims description 16
- 239000004606 Fillers/Extenders Substances 0.000 claims description 15
- 235000018417 cysteine Nutrition 0.000 claims description 15
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 claims description 15
- 238000006467 substitution reaction Methods 0.000 claims description 13
- 229940024606 amino acid Drugs 0.000 claims description 12
- 235000001014 amino acid Nutrition 0.000 claims description 12
- 150000001413 amino acids Chemical class 0.000 claims description 12
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 12
- 239000001301 oxygen Substances 0.000 claims description 12
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 claims description 10
- 230000008859 change Effects 0.000 claims description 10
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 10
- 229930027917 kanamycin Natural products 0.000 claims description 10
- 229960000318 kanamycin Drugs 0.000 claims description 10
- SBUJHOSQTJFQJX-NOAMYHISSA-N kanamycin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N SBUJHOSQTJFQJX-NOAMYHISSA-N 0.000 claims description 10
- 229930182823 kanamycin A Natural products 0.000 claims description 10
- 229960001153 serine Drugs 0.000 claims description 10
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 claims description 8
- 235000014113 dietary fatty acids Nutrition 0.000 claims description 8
- 229930195729 fatty acid Natural products 0.000 claims description 8
- 239000000194 fatty acid Substances 0.000 claims description 8
- 150000004665 fatty acids Chemical class 0.000 claims description 8
- 229910052736 halogen Inorganic materials 0.000 claims description 8
- 150000002367 halogens Chemical group 0.000 claims description 8
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 claims description 7
- 238000012258 culturing Methods 0.000 claims description 7
- 241001377010 Pila Species 0.000 claims description 6
- 239000001963 growth medium Substances 0.000 claims description 6
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 5
- 238000003259 recombinant expression Methods 0.000 claims description 5
- 241001646999 Corallococcus Species 0.000 claims description 4
- 125000002877 alkyl aryl group Chemical group 0.000 claims description 4
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 claims description 4
- 230000032258 transport Effects 0.000 claims description 4
- 241001600177 Cystobacterineae Species 0.000 claims description 3
- 241000863421 Myxococcaceae Species 0.000 claims description 3
- 125000004103 aminoalkyl group Chemical group 0.000 claims description 3
- 238000009630 liquid culture Methods 0.000 claims description 3
- 238000002414 normal-phase solid-phase extraction Methods 0.000 claims description 3
- 238000004587 chromatography analysis Methods 0.000 claims description 2
- 238000002425 crystallisation Methods 0.000 claims description 2
- 230000008025 crystallization Effects 0.000 claims description 2
- 150000002431 hydrogen Chemical group 0.000 claims 5
- 125000005594 diketone group Chemical group 0.000 claims 2
- JDMUPRLRUUMCTL-VIFPVBQESA-N D-pantetheine 4'-phosphate Chemical compound OP(=O)(O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCS JDMUPRLRUUMCTL-VIFPVBQESA-N 0.000 claims 1
- 241000219000 Populus Species 0.000 claims 1
- 241000329138 Protocystis Species 0.000 claims 1
- 150000002118 epoxides Chemical class 0.000 claims 1
- 239000010685 fatty oil Substances 0.000 claims 1
- 210000004027 cell Anatomy 0.000 description 221
- 108700016155 Acyl transferases Proteins 0.000 description 104
- 102000057234 Acyl transferases Human genes 0.000 description 104
- QYDYPVFESGNLHU-KHPPLWFESA-N methyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC QYDYPVFESGNLHU-KHPPLWFESA-N 0.000 description 79
- QYDYPVFESGNLHU-UHFFFAOYSA-N elaidic acid methyl ester Natural products CCCCCCCCC=CCCCCCCCC(=O)OC QYDYPVFESGNLHU-UHFFFAOYSA-N 0.000 description 78
- 229940073769 methyl oleate Drugs 0.000 description 77
- 239000000047 product Substances 0.000 description 70
- -1 but not limited to Natural products 0.000 description 66
- HESCAJZNRMSMJG-HGYUPSKWSA-N epothilone A Natural products O=C1[C@H](C)[C@H](O)[C@H](C)CCC[C@H]2O[C@H]2C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C HESCAJZNRMSMJG-HGYUPSKWSA-N 0.000 description 61
- 101001014220 Monascus pilosus Dehydrogenase mokE Proteins 0.000 description 56
- 101000573542 Penicillium citrinum Compactin nonaketide synthase, enoyl reductase component Proteins 0.000 description 56
- 239000002609 medium Substances 0.000 description 55
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 50
- 235000018102 proteins Nutrition 0.000 description 47
- 102000004169 proteins and genes Human genes 0.000 description 47
- 239000000203 mixture Substances 0.000 description 42
- 108010000785 non-ribosomal peptide synthase Proteins 0.000 description 41
- ZIYVHBGGAOATLY-UHFFFAOYSA-N methylmalonic acid Chemical compound OC(=O)C(C)C(O)=O ZIYVHBGGAOATLY-UHFFFAOYSA-N 0.000 description 39
- 239000013612 plasmid Substances 0.000 description 38
- 108020004414 DNA Proteins 0.000 description 34
- 108091008053 gene clusters Proteins 0.000 description 34
- 239000000306 component Substances 0.000 description 33
- 101710146995 Acyl carrier protein Proteins 0.000 description 32
- 230000012010 growth Effects 0.000 description 30
- 229940088598 enzyme Drugs 0.000 description 29
- 230000000694 effects Effects 0.000 description 28
- 235000013350 formula milk Nutrition 0.000 description 28
- 239000003921 oil Substances 0.000 description 28
- 235000019198 oils Nutrition 0.000 description 28
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 27
- LTYOQGRJFJAKNA-KKIMTKSISA-N Malonyl CoA Natural products S(C(=O)CC(=O)O)CCNC(=O)CCNC(=O)[C@@H](O)C(CO[P@](=O)(O[P@](=O)(OC[C@H]1[C@@H](OP(=O)(O)O)[C@@H](O)[C@@H](n2c3ncnc(N)c3nc2)O1)O)O)(C)C LTYOQGRJFJAKNA-KKIMTKSISA-N 0.000 description 27
- LTYOQGRJFJAKNA-DVVLENMVSA-N malonyl-CoA Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCSC(=O)CC(O)=O)O[C@H]1N1C2=NC=NC(N)=C2N=C1 LTYOQGRJFJAKNA-DVVLENMVSA-N 0.000 description 27
- QXRSDHAAWVKZLJ-PVYNADRNSA-N epothilone B Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 QXRSDHAAWVKZLJ-PVYNADRNSA-N 0.000 description 26
- QXRSDHAAWVKZLJ-OXZHEXMSSA-N Epothilone B Natural products O=C1[C@H](C)[C@H](O)[C@@H](C)CCC[C@@]2(C)O[C@H]2C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C QXRSDHAAWVKZLJ-OXZHEXMSSA-N 0.000 description 25
- 229910021529 ammonia Inorganic materials 0.000 description 25
- 108020004511 Recombinant DNA Proteins 0.000 description 24
- 230000014509 gene expression Effects 0.000 description 24
- 238000003786 synthesis reaction Methods 0.000 description 24
- 230000006870 function Effects 0.000 description 23
- 239000002243 precursor Substances 0.000 description 23
- VZCCETWTMQHEPK-QNEBEIHSSA-N gamma-linolenic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/CCCCC(O)=O VZCCETWTMQHEPK-QNEBEIHSSA-N 0.000 description 22
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 22
- 238000010923 batch production Methods 0.000 description 20
- 238000011282 treatment Methods 0.000 description 20
- 239000001888 Peptone Substances 0.000 description 19
- 108010080698 Peptones Proteins 0.000 description 19
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 19
- 235000019319 peptone Nutrition 0.000 description 19
- 239000013587 production medium Substances 0.000 description 19
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 19
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 19
- 229960002930 sirolimus Drugs 0.000 description 19
- 101150035879 epoA gene Proteins 0.000 description 18
- 239000012634 fragment Substances 0.000 description 18
- 239000011573 trace mineral Substances 0.000 description 17
- 235000013619 trace mineral Nutrition 0.000 description 17
- ULGZDMOVFRHVEP-RWJQBGPGSA-N Erythromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 ULGZDMOVFRHVEP-RWJQBGPGSA-N 0.000 description 16
- 206010028980 Neoplasm Diseases 0.000 description 16
- 239000003814 drug Substances 0.000 description 16
- BEFZAMRWPCMWFJ-QJKGZULSSA-N epothilone C Chemical compound O1C(=O)C[C@H](O)C(C)(C)C(=O)[C@H](C)[C@@H](O)[C@@H](C)CCC\C=C/C[C@H]1C(\C)=C\C1=CSC(C)=N1 BEFZAMRWPCMWFJ-QJKGZULSSA-N 0.000 description 16
- 229910001868 water Inorganic materials 0.000 description 16
- BEFZAMRWPCMWFJ-JRBBLYSQSA-N Epothilone C Natural products O=C1[C@H](C)[C@@H](O)[C@@H](C)CCC/C=C\C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C BEFZAMRWPCMWFJ-JRBBLYSQSA-N 0.000 description 15
- 101000691656 Streptomyces venezuelae Narbonolide/10-deoxymethynolide synthase PikA1, modules 1 and 2 Proteins 0.000 description 15
- 101000691655 Streptomyces venezuelae Narbonolide/10-deoxymethynolide synthase PikA2, modules 3 and 4 Proteins 0.000 description 15
- 101000691658 Streptomyces venezuelae Narbonolide/10-deoxymethynolide synthase PikA3, module 5 Proteins 0.000 description 15
- 101001125873 Streptomyces venezuelae Narbonolide/10-deoxymethynolide synthase PikA4, module 6 Proteins 0.000 description 15
- ZDQSOHOQTUFQEM-NURRSENYSA-N ascomycin Chemical compound C/C([C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@]2(O)O[C@@H]([C@H](C[C@H]2C)OC)[C@@H](OC)C[C@@H](C)C/C(C)=C/[C@H](C(C[C@H](O)[C@H]1C)=O)CC)=C\[C@@H]1CC[C@@H](O)[C@H](OC)C1 ZDQSOHOQTUFQEM-NURRSENYSA-N 0.000 description 15
- BEFZAMRWPCMWFJ-UHFFFAOYSA-N desoxyepothilone A Natural products O1C(=O)CC(O)C(C)(C)C(=O)C(C)C(O)C(C)CCCC=CCC1C(C)=CC1=CSC(C)=N1 BEFZAMRWPCMWFJ-UHFFFAOYSA-N 0.000 description 15
- 235000019867 fractionated palm kernal oil Nutrition 0.000 description 15
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 14
- 241001465754 Metazoa Species 0.000 description 13
- 102000002932 Thiolase Human genes 0.000 description 13
- 108060008225 Thiolase Proteins 0.000 description 13
- 125000001721 carboxyacetyl group Chemical group 0.000 description 13
- 230000010261 cell growth Effects 0.000 description 13
- 201000010099 disease Diseases 0.000 description 13
- 230000010354 integration Effects 0.000 description 13
- 239000000463 material Substances 0.000 description 13
- 239000002246 antineoplastic agent Substances 0.000 description 12
- 201000011510 cancer Diseases 0.000 description 12
- 230000008569 process Effects 0.000 description 12
- 239000000243 solution Substances 0.000 description 12
- 238000001727 in vivo Methods 0.000 description 11
- 230000002401 inhibitory effect Effects 0.000 description 11
- 108020004707 nucleic acids Proteins 0.000 description 11
- 150000007523 nucleic acids Chemical class 0.000 description 11
- 102000039446 nucleic acids Human genes 0.000 description 11
- 238000002360 preparation method Methods 0.000 description 11
- 108090000765 processed proteins & peptides Proteins 0.000 description 11
- 238000006722 reduction reaction Methods 0.000 description 11
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 10
- 108060004795 Methyltransferase Proteins 0.000 description 10
- 102000016397 Methyltransferase Human genes 0.000 description 10
- 125000001931 aliphatic group Chemical group 0.000 description 10
- 125000000217 alkyl group Chemical group 0.000 description 10
- 238000003780 insertion Methods 0.000 description 10
- 230000037431 insertion Effects 0.000 description 10
- 230000004048 modification Effects 0.000 description 10
- 238000012986 modification Methods 0.000 description 10
- 229920001184 polypeptide Polymers 0.000 description 10
- 102000004196 processed proteins & peptides Human genes 0.000 description 10
- 150000003839 salts Chemical class 0.000 description 10
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 10
- 239000000126 substance Substances 0.000 description 10
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 9
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 9
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 9
- 229960003276 erythromycin Drugs 0.000 description 9
- ROBFUDYVXSDBQM-UHFFFAOYSA-N hydroxymalonic acid Chemical compound OC(=O)C(O)C(O)=O ROBFUDYVXSDBQM-UHFFFAOYSA-N 0.000 description 9
- 238000011081 inoculation Methods 0.000 description 9
- 239000003120 macrolide antibiotic agent Substances 0.000 description 9
- 239000003550 marker Substances 0.000 description 9
- 230000009467 reduction Effects 0.000 description 9
- 238000011218 seed culture Methods 0.000 description 9
- 235000004400 serine Nutrition 0.000 description 9
- 125000001424 substituent group Chemical group 0.000 description 9
- 239000000758 substrate Substances 0.000 description 9
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 8
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 8
- 210000004436 artificial bacterial chromosome Anatomy 0.000 description 8
- 230000015556 catabolic process Effects 0.000 description 8
- 238000006731 degradation reaction Methods 0.000 description 8
- 229940079593 drug Drugs 0.000 description 8
- 230000006872 improvement Effects 0.000 description 8
- 101150014100 pilA gene Proteins 0.000 description 8
- 239000008389 polyethoxylated castor oil Substances 0.000 description 8
- 230000006798 recombination Effects 0.000 description 8
- 238000005215 recombination Methods 0.000 description 8
- 230000002829 reductive effect Effects 0.000 description 8
- 239000004593 Epoxy Substances 0.000 description 7
- 241000588724 Escherichia coli Species 0.000 description 7
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 7
- YFFOFFWSBYZSOI-UHFFFAOYSA-N Narbonolid Natural products CCC1OC(=O)C(C)C(=O)C(C)C(O)C(C)CC(C)C(=O)C=CC1C YFFOFFWSBYZSOI-UHFFFAOYSA-N 0.000 description 7
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 7
- 125000002252 acyl group Chemical group 0.000 description 7
- 238000004458 analytical method Methods 0.000 description 7
- 210000000349 chromosome Anatomy 0.000 description 7
- 150000002148 esters Chemical class 0.000 description 7
- 238000009472 formulation Methods 0.000 description 7
- YFFOFFWSBYZSOI-HQWJGCFGSA-N narbonolide Chemical compound CC[C@H]1OC(=O)[C@H](C)C(=O)[C@H](C)[C@@H](O)[C@@H](C)C[C@@H](C)C(=O)\C=C\[C@H]1C YFFOFFWSBYZSOI-HQWJGCFGSA-N 0.000 description 7
- 108010001814 phosphopantetheinyl transferase Proteins 0.000 description 7
- 238000012216 screening Methods 0.000 description 7
- GVJHHUAWPYXKBD-UHFFFAOYSA-N (±)-α-Tocopherol Chemical compound OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- 201000001320 Atherosclerosis Diseases 0.000 description 6
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 6
- 241000863434 Myxococcales Species 0.000 description 6
- 108700026244 Open Reading Frames Proteins 0.000 description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- 239000004098 Tetracycline Substances 0.000 description 6
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 6
- 230000006154 adenylylation Effects 0.000 description 6
- 239000003242 anti bacterial agent Substances 0.000 description 6
- 229940088710 antibiotic agent Drugs 0.000 description 6
- 125000003710 aryl alkyl group Chemical group 0.000 description 6
- 230000001684 chronic effect Effects 0.000 description 6
- 230000006378 damage Effects 0.000 description 6
- 238000011161 development Methods 0.000 description 6
- 230000018109 developmental process Effects 0.000 description 6
- 239000002552 dosage form Substances 0.000 description 6
- UKFXDFUAPNAMPJ-UHFFFAOYSA-N ethylmalonic acid Chemical compound CCC(C(O)=O)C(O)=O UKFXDFUAPNAMPJ-UHFFFAOYSA-N 0.000 description 6
- 229930182830 galactose Natural products 0.000 description 6
- 239000003018 immunosuppressive agent Substances 0.000 description 6
- 230000001404 mediated effect Effects 0.000 description 6
- 229910052751 metal Inorganic materials 0.000 description 6
- 239000002184 metal Substances 0.000 description 6
- 150000002739 metals Chemical class 0.000 description 6
- IJXHLVMUNBOGRR-UHFFFAOYSA-N methyl nonanoate Chemical compound CCCCCCCCC(=O)OC IJXHLVMUNBOGRR-UHFFFAOYSA-N 0.000 description 6
- 150000003254 radicals Chemical class 0.000 description 6
- 239000007787 solid Substances 0.000 description 6
- 239000007858 starting material Substances 0.000 description 6
- 208000024891 symptom Diseases 0.000 description 6
- 229960002180 tetracycline Drugs 0.000 description 6
- 229930101283 tetracycline Natural products 0.000 description 6
- 235000019364 tetracycline Nutrition 0.000 description 6
- 150000003522 tetracyclines Chemical class 0.000 description 6
- 238000009966 trimming Methods 0.000 description 6
- FCCNKYGSMOSYPV-DEDISHTHSA-N (-)-Epothilone E Natural products O=C1[C@H](C)[C@H](O)[C@@H](C)CCC[C@H]2O[C@H]2C[C@@H](/C(=C\c2nc(CO)sc2)/C)OC(=O)C[C@H](O)C1(C)C FCCNKYGSMOSYPV-DEDISHTHSA-N 0.000 description 5
- VFLSVROIWGPRPN-BFVWCIFVSA-N (4s,7r,8s,9r,13z,16s)-4,8-dihydroxy-5,5,7,9,13-pentamethyl-16-[(e)-1-(2-methyl-1,3-thiazol-4-yl)prop-1-en-2-yl]-1-oxacyclohexadec-13-ene-2,6,10-trione Chemical compound C1\C=C(C)/CCC(=O)[C@H](C)[C@H](O)[C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O[C@@H]1C(\C)=C\C1=CSC(C)=N1 VFLSVROIWGPRPN-BFVWCIFVSA-N 0.000 description 5
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 5
- 229920001817 Agar Polymers 0.000 description 5
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical compound NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 5
- 101150002621 EPO gene Proteins 0.000 description 5
- 239000007995 HEPES buffer Substances 0.000 description 5
- 101150062179 II gene Proteins 0.000 description 5
- 108700011325 Modifier Genes Proteins 0.000 description 5
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 5
- 201000004681 Psoriasis Diseases 0.000 description 5
- 241000187747 Streptomyces Species 0.000 description 5
- 102000005488 Thioesterase Human genes 0.000 description 5
- 238000009825 accumulation Methods 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 239000003463 adsorbent Substances 0.000 description 5
- 235000010419 agar Nutrition 0.000 description 5
- 239000008272 agar Substances 0.000 description 5
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 5
- 230000003115 biocidal effect Effects 0.000 description 5
- 239000013611 chromosomal DNA Substances 0.000 description 5
- 238000009833 condensation Methods 0.000 description 5
- 230000005494 condensation Effects 0.000 description 5
- 238000010276 construction Methods 0.000 description 5
- 230000001472 cytotoxic effect Effects 0.000 description 5
- 238000006114 decarboxylation reaction Methods 0.000 description 5
- 239000008367 deionised water Substances 0.000 description 5
- 229910021641 deionized water Inorganic materials 0.000 description 5
- FCCNKYGSMOSYPV-UHFFFAOYSA-N epothilone E Natural products O1C(=O)CC(O)C(C)(C)C(=O)C(C)C(O)C(C)CCCC2OC2CC1C(C)=CC1=CSC(CO)=N1 FCCNKYGSMOSYPV-UHFFFAOYSA-N 0.000 description 5
- FCCNKYGSMOSYPV-OKOHHBBGSA-N epothilone e Chemical compound C/C([C@@H]1C[C@@H]2O[C@@H]2CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(CO)=N1 FCCNKYGSMOSYPV-OKOHHBBGSA-N 0.000 description 5
- 101150045500 galK gene Proteins 0.000 description 5
- 125000000623 heterocyclic group Chemical group 0.000 description 5
- 230000002779 inactivation Effects 0.000 description 5
- 239000002054 inoculum Substances 0.000 description 5
- 238000001990 intravenous administration Methods 0.000 description 5
- 239000002502 liposome Substances 0.000 description 5
- 235000015097 nutrients Nutrition 0.000 description 5
- 229920000642 polymer Polymers 0.000 description 5
- 239000000651 prodrug Substances 0.000 description 5
- 229940002612 prodrug Drugs 0.000 description 5
- 125000006239 protecting group Chemical group 0.000 description 5
- 238000011160 research Methods 0.000 description 5
- 229960005322 streptomycin Drugs 0.000 description 5
- 239000006228 supernatant Substances 0.000 description 5
- 150000007970 thio esters Chemical class 0.000 description 5
- 108020002982 thioesterase Proteins 0.000 description 5
- 210000001519 tissue Anatomy 0.000 description 5
- 210000004509 vascular smooth muscle cell Anatomy 0.000 description 5
- 239000005660 Abamectin Substances 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 239000002028 Biomass Substances 0.000 description 4
- 108010076119 Caseins Proteins 0.000 description 4
- 108020004705 Codon Proteins 0.000 description 4
- 101100334784 Escherichia coli (strain K12) fimA gene Proteins 0.000 description 4
- VUGZQVCBBBEZQE-VRQRJWBYSA-N Ethylmalonyl-CoA Chemical group O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)C(O)C(=O)NCCC(=O)NCCSC(=O)C(C(O)=O)CC)O[C@H]1N1C2=NC=NC(N)=C2N=C1 VUGZQVCBBBEZQE-VRQRJWBYSA-N 0.000 description 4
- 102000002274 Matrix Metalloproteinases Human genes 0.000 description 4
- 108010000684 Matrix Metalloproteinases Proteins 0.000 description 4
- 108010074633 Mixed Function Oxygenases Proteins 0.000 description 4
- 108010083674 Myelin Proteins Proteins 0.000 description 4
- 102000006386 Myelin Proteins Human genes 0.000 description 4
- 241000246398 Myxococcus phage Mx8 Species 0.000 description 4
- 241001111421 Pannus Species 0.000 description 4
- 241000531819 Streptomyces venezuelae Species 0.000 description 4
- 125000003342 alkenyl group Chemical group 0.000 description 4
- 125000003545 alkoxy group Chemical group 0.000 description 4
- 230000008901 benefit Effects 0.000 description 4
- 239000000969 carrier Substances 0.000 description 4
- 239000005018 casein Substances 0.000 description 4
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 4
- 235000021240 caseins Nutrition 0.000 description 4
- 230000006037 cell lysis Effects 0.000 description 4
- 230000004663 cell proliferation Effects 0.000 description 4
- 238000012512 characterization method Methods 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 231100000433 cytotoxic Toxicity 0.000 description 4
- 230000001419 dependent effect Effects 0.000 description 4
- 239000003937 drug carrier Substances 0.000 description 4
- 238000004520 electroporation Methods 0.000 description 4
- 239000000839 emulsion Substances 0.000 description 4
- 108010017796 epoxidase Proteins 0.000 description 4
- 238000006735 epoxidation reaction Methods 0.000 description 4
- 101150049376 ftsY gene Proteins 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- 229960003444 immunosuppressant agent Drugs 0.000 description 4
- 230000001976 improved effect Effects 0.000 description 4
- 238000000338 in vitro Methods 0.000 description 4
- 230000000415 inactivating effect Effects 0.000 description 4
- 238000010348 incorporation Methods 0.000 description 4
- 230000004054 inflammatory process Effects 0.000 description 4
- 125000000468 ketone group Chemical group 0.000 description 4
- 150000002576 ketones Chemical class 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 4
- 235000019341 magnesium sulphate Nutrition 0.000 description 4
- HUEBIMLTDXKIPR-UHFFFAOYSA-N methyl heptadecanoate Chemical compound CCCCCCCCCCCCCCCCC(=O)OC HUEBIMLTDXKIPR-UHFFFAOYSA-N 0.000 description 4
- BDXAHSJUDUZLDU-UHFFFAOYSA-N methyl nonadecanoate Chemical compound CCCCCCCCCCCCCCCCCCC(=O)OC BDXAHSJUDUZLDU-UHFFFAOYSA-N 0.000 description 4
- MZFOKIKEPGUZEN-FBMOWMAESA-N methylmalonyl-CoA Chemical group O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCSC(=O)C(C(O)=O)C)O[C@H]1N1C2=NC=NC(N)=C2N=C1 MZFOKIKEPGUZEN-FBMOWMAESA-N 0.000 description 4
- 210000005012 myelin Anatomy 0.000 description 4
- 150000002924 oxiranes Chemical class 0.000 description 4
- 238000011084 recovery Methods 0.000 description 4
- 108091008146 restriction endonucleases Proteins 0.000 description 4
- 229930000044 secondary metabolite Natural products 0.000 description 4
- 210000003491 skin Anatomy 0.000 description 4
- 239000002904 solvent Substances 0.000 description 4
- 239000003381 stabilizer Substances 0.000 description 4
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 4
- 238000006257 total synthesis reaction Methods 0.000 description 4
- 231100000331 toxic Toxicity 0.000 description 4
- 230000002588 toxic effect Effects 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- 238000012546 transfer Methods 0.000 description 4
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 4
- UKIMCRYGLFQEOE-RLHMMOOASA-N (-)-Epothilone F Natural products O=C1[C@H](C)[C@H](O)[C@@H](C)CCC[C@@]2(C)O[C@H]2C[C@@H](/C(=C\c2nc(CO)sc2)/C)OC(=O)C[C@H](O)C1(C)C UKIMCRYGLFQEOE-RLHMMOOASA-N 0.000 description 3
- HQZOLNNEQAKEHT-UHFFFAOYSA-N (3R,4S,5R,6S,7S,9R,11R,12S,13R,14R)-14-ethyl-4,6,12-trihydroxy-3,5,7,9,11,13-hexamethyloxacyclotetradecane-2,10-dione Natural products CCC1OC(=O)C(C)C(O)C(C)C(O)C(C)CC(C)C(=O)C(C)C(O)C1C HQZOLNNEQAKEHT-UHFFFAOYSA-N 0.000 description 3
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 3
- 108020004638 Circular DNA Proteins 0.000 description 3
- 206010009944 Colon cancer Diseases 0.000 description 3
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 3
- 206010012735 Diarrhoea Diseases 0.000 description 3
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 3
- 241000196324 Embryophyta Species 0.000 description 3
- UKIMCRYGLFQEOE-UHFFFAOYSA-N Epothilone F Natural products O1C(=O)CC(O)C(C)(C)C(=O)C(C)C(O)C(C)CCCC2(C)OC2CC1C(C)=CC1=CSC(CO)=N1 UKIMCRYGLFQEOE-UHFFFAOYSA-N 0.000 description 3
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 3
- 229930182566 Gentamicin Natural products 0.000 description 3
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 3
- 206010061218 Inflammation Diseases 0.000 description 3
- 108010061833 Integrases Proteins 0.000 description 3
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 3
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 3
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- 102000029749 Microtubule Human genes 0.000 description 3
- 108091022875 Microtubule Proteins 0.000 description 3
- 102000008109 Mixed Function Oxygenases Human genes 0.000 description 3
- 241001647006 Myxococcus virescens Species 0.000 description 3
- 108091028043 Nucleic acid sequence Proteins 0.000 description 3
- 239000004100 Oxytetracycline Substances 0.000 description 3
- 101150053185 P450 gene Proteins 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical class CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 3
- 241000187559 Saccharopolyspora erythraea Species 0.000 description 3
- 241000194017 Streptococcus Species 0.000 description 3
- 241000187438 Streptomyces fradiae Species 0.000 description 3
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 3
- 239000004473 Threonine Substances 0.000 description 3
- PHYFQTYBJUILEZ-UHFFFAOYSA-N Trioleoylglycerol Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC(OC(=O)CCCCCCCC=CCCCCCCCC)COC(=O)CCCCCCCC=CCCCCCCCC PHYFQTYBJUILEZ-UHFFFAOYSA-N 0.000 description 3
- 239000004182 Tylosin Substances 0.000 description 3
- 229930194936 Tylosin Natural products 0.000 description 3
- 229930003427 Vitamin E Natural products 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 229960003767 alanine Drugs 0.000 description 3
- 235000004279 alanine Nutrition 0.000 description 3
- 150000001298 alcohols Chemical class 0.000 description 3
- 150000001299 aldehydes Chemical class 0.000 description 3
- 125000000304 alkynyl group Chemical group 0.000 description 3
- 230000004075 alteration Effects 0.000 description 3
- 125000003277 amino group Chemical group 0.000 description 3
- 230000033115 angiogenesis Effects 0.000 description 3
- 229910052786 argon Inorganic materials 0.000 description 3
- 230000033228 biological regulation Effects 0.000 description 3
- 230000001851 biosynthetic effect Effects 0.000 description 3
- CRFNGMNYKDXRTN-CITAKDKDSA-N butyryl-CoA Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCSC(=O)CCC)O[C@H]1N1C2=NC=NC(N)=C2N=C1 CRFNGMNYKDXRTN-CITAKDKDSA-N 0.000 description 3
- 229940041514 candida albicans extract Drugs 0.000 description 3
- 108010079058 casein hydrolysate Proteins 0.000 description 3
- 230000002759 chromosomal effect Effects 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000010367 cloning Methods 0.000 description 3
- 238000006482 condensation reaction Methods 0.000 description 3
- 239000013078 crystal Substances 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- FFHWGQQFANVOHV-UHFFFAOYSA-N dimethyldioxirane Chemical compound CC1(C)OO1 FFHWGQQFANVOHV-UHFFFAOYSA-N 0.000 description 3
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 3
- 229940043264 dodecyl sulfate Drugs 0.000 description 3
- 238000012377 drug delivery Methods 0.000 description 3
- AEAGZTSEFUFUKN-UHFFFAOYSA-N epothilone 490 Natural products O1C(=O)CC(O)C(C)(C)C(=O)C(C)C(O)C(C)CC=CC(C)=CCC1C(C)=CC1=CSC(C)=N1 AEAGZTSEFUFUKN-UHFFFAOYSA-N 0.000 description 3
- HESCAJZNRMSMJG-XOVLCIRJSA-N epothilone a Chemical compound C([C@@H]1O[C@@H]1CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)C1C(\C)=C\C1=CSC(C)=N1 HESCAJZNRMSMJG-XOVLCIRJSA-N 0.000 description 3
- UKIMCRYGLFQEOE-RGJAOAFDSA-N epothilone f Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(CO)=N1 UKIMCRYGLFQEOE-RGJAOAFDSA-N 0.000 description 3
- 239000012530 fluid Substances 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 3
- BRZYSWJRSDMWLG-CAXSIQPQSA-N geneticin Chemical compound O1C[C@@](O)(C)[C@H](NC)[C@@H](O)[C@H]1O[C@@H]1[C@@H](O)[C@H](O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](C(C)O)O2)N)[C@@H](N)C[C@H]1N BRZYSWJRSDMWLG-CAXSIQPQSA-N 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 3
- 238000002744 homologous recombination Methods 0.000 description 3
- 230000006801 homologous recombination Effects 0.000 description 3
- 150000002430 hydrocarbons Chemical group 0.000 description 3
- 230000033444 hydroxylation Effects 0.000 description 3
- 238000005805 hydroxylation reaction Methods 0.000 description 3
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 3
- 230000003463 hyperproliferative effect Effects 0.000 description 3
- 150000002632 lipids Chemical class 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 230000000813 microbial effect Effects 0.000 description 3
- 239000003094 microcapsule Substances 0.000 description 3
- 210000004688 microtubule Anatomy 0.000 description 3
- 201000006417 multiple sclerosis Diseases 0.000 description 3
- 239000002105 nanoparticle Substances 0.000 description 3
- 230000008520 organization Effects 0.000 description 3
- 230000003647 oxidation Effects 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- 229960000625 oxytetracycline Drugs 0.000 description 3
- IWVCMVBTMGNXQD-PXOLEDIWSA-N oxytetracycline Chemical compound C1=CC=C2[C@](O)(C)[C@H]3[C@H](O)[C@H]4[C@H](N(C)C)C(O)=C(C(N)=O)C(=O)[C@@]4(O)C(O)=C3C(=O)C2=C1O IWVCMVBTMGNXQD-PXOLEDIWSA-N 0.000 description 3
- 235000019366 oxytetracycline Nutrition 0.000 description 3
- 229960001592 paclitaxel Drugs 0.000 description 3
- 239000000546 pharmaceutical excipient Substances 0.000 description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 3
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 230000003389 potentiating effect Effects 0.000 description 3
- 230000035755 proliferation Effects 0.000 description 3
- 108010009004 proteose-peptone Proteins 0.000 description 3
- 238000010188 recombinant method Methods 0.000 description 3
- 230000010076 replication Effects 0.000 description 3
- 208000037803 restenosis Diseases 0.000 description 3
- 206010039073 rheumatoid arthritis Diseases 0.000 description 3
- 239000000523 sample Substances 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 238000002741 site-directed mutagenesis Methods 0.000 description 3
- 239000003549 soybean oil Substances 0.000 description 3
- 235000012424 soybean oil Nutrition 0.000 description 3
- 230000009469 supplementation Effects 0.000 description 3
- 210000001258 synovial membrane Anatomy 0.000 description 3
- 210000002437 synoviocyte Anatomy 0.000 description 3
- 201000004595 synovitis Diseases 0.000 description 3
- IWVCMVBTMGNXQD-UHFFFAOYSA-N terramycin dehydrate Natural products C1=CC=C2C(O)(C)C3C(O)C4C(N(C)C)C(O)=C(C(N)=O)C(=O)C4(O)C(O)=C3C(=O)C2=C1O IWVCMVBTMGNXQD-UHFFFAOYSA-N 0.000 description 3
- 230000008719 thickening Effects 0.000 description 3
- 125000001544 thienyl group Chemical group 0.000 description 3
- 210000004881 tumor cell Anatomy 0.000 description 3
- 229960004059 tylosin Drugs 0.000 description 3
- WBPYTXDJUQJLPQ-VMXQISHHSA-N tylosin Chemical compound O([C@@H]1[C@@H](C)O[C@H]([C@@H]([C@H]1N(C)C)O)O[C@@H]1[C@@H](C)[C@H](O)CC(=O)O[C@@H]([C@H](/C=C(\C)/C=C/C(=O)[C@H](C)C[C@@H]1CC=O)CO[C@H]1[C@@H]([C@H](OC)[C@H](O)[C@@H](C)O1)OC)CC)[C@H]1C[C@@](C)(O)[C@@H](O)[C@H](C)O1 WBPYTXDJUQJLPQ-VMXQISHHSA-N 0.000 description 3
- 235000019375 tylosin Nutrition 0.000 description 3
- 238000000825 ultraviolet detection Methods 0.000 description 3
- 235000019165 vitamin E Nutrition 0.000 description 3
- 229940046009 vitamin E Drugs 0.000 description 3
- 239000011709 vitamin E Substances 0.000 description 3
- 239000012138 yeast extract Substances 0.000 description 3
- JTPNRXUCIXHOKM-UHFFFAOYSA-N 1-chloronaphthalene Chemical compound C1=CC=C2C(Cl)=CC=CC2=C1 JTPNRXUCIXHOKM-UHFFFAOYSA-N 0.000 description 2
- RCQKLWAPRHHRNN-UHFFFAOYSA-N 2-Methyl-4-pentenal Chemical compound O=CC(C)CC=C RCQKLWAPRHHRNN-UHFFFAOYSA-N 0.000 description 2
- VZWOXDYRBDIHMA-UHFFFAOYSA-N 2-methyl-1,3-thiazole Chemical group CC1=NC=CS1 VZWOXDYRBDIHMA-UHFFFAOYSA-N 0.000 description 2
- 229920000936 Agarose Polymers 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 206010006187 Breast cancer Diseases 0.000 description 2
- 208000026310 Breast neoplasm Diseases 0.000 description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 2
- 238000003512 Claisen condensation reaction Methods 0.000 description 2
- 101710095468 Cyclase Proteins 0.000 description 2
- 241000863004 Cystobacter Species 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- 229930006677 Erythromycin A Natural products 0.000 description 2
- 230000005526 G1 to G0 transition Effects 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 235000010469 Glycine max Nutrition 0.000 description 2
- 241000701044 Human gammaherpesvirus 4 Species 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- 206010020751 Hypersensitivity Diseases 0.000 description 2
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 2
- 240000008415 Lactuca sativa Species 0.000 description 2
- HPEUJPJOZXNMSJ-UHFFFAOYSA-N Methyl stearate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC HPEUJPJOZXNMSJ-UHFFFAOYSA-N 0.000 description 2
- 108010085220 Multiprotein Complexes Proteins 0.000 description 2
- 102000007474 Multiprotein Complexes Human genes 0.000 description 2
- 208000008238 Muscle Spasticity Diseases 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 2
- 239000004104 Oleandomycin Substances 0.000 description 2
- RZPAKFUAFGMUPI-UHFFFAOYSA-N Oleandomycin Natural products O1C(C)C(O)C(OC)CC1OC1C(C)C(=O)OC(C)C(C)C(O)C(C)C(=O)C2(OC2)CC(C)C(OC2C(C(CC(C)O2)N(C)C)O)C1C RZPAKFUAFGMUPI-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 229930012538 Paclitaxel Natural products 0.000 description 2
- 208000002193 Pain Diseases 0.000 description 2
- 208000031481 Pathologic Constriction Diseases 0.000 description 2
- 102000035195 Peptidases Human genes 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 239000004365 Protease Substances 0.000 description 2
- 108010001267 Protein Subunits Proteins 0.000 description 2
- 102000002067 Protein Subunits Human genes 0.000 description 2
- 235000019484 Rapeseed oil Nutrition 0.000 description 2
- 229930189077 Rifamycin Natural products 0.000 description 2
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 2
- 235000014680 Saccharomyces cerevisiae Nutrition 0.000 description 2
- 206010039491 Sarcoma Diseases 0.000 description 2
- 206010040030 Sensory loss Diseases 0.000 description 2
- 238000012300 Sequence Analysis Methods 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- 241001532577 Sorangium Species 0.000 description 2
- 241000186988 Streptomyces antibioticus Species 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 235000019486 Sunflower oil Nutrition 0.000 description 2
- 241000656145 Thyrsites atun Species 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 206010047700 Vomiting Diseases 0.000 description 2
- ZSLZBFCDCINBPY-ZSJPKINUSA-N acetyl-CoA Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCSC(=O)C)O[C@H]1N1C2=NC=NC(N)=C2N=C1 ZSLZBFCDCINBPY-ZSJPKINUSA-N 0.000 description 2
- 230000006978 adaptation Effects 0.000 description 2
- 125000005236 alkanoylamino group Chemical group 0.000 description 2
- 150000001336 alkenes Chemical class 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 230000003698 anagen phase Effects 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- 230000000843 anti-fungal effect Effects 0.000 description 2
- 229940121375 antifungal agent Drugs 0.000 description 2
- 229940041181 antineoplastic drug Drugs 0.000 description 2
- YZXBAPSDXZZRGB-DOFZRALJSA-N arachidonic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O YZXBAPSDXZZRGB-DOFZRALJSA-N 0.000 description 2
- 210000001367 artery Anatomy 0.000 description 2
- 125000001769 aryl amino group Chemical group 0.000 description 2
- 210000001130 astrocyte Anatomy 0.000 description 2
- RRZXIRBKKLTSOM-XPNPUAGNSA-N avermectin B1a Chemical compound C1=C[C@H](C)[C@@H]([C@@H](C)CC)O[C@]11O[C@H](C\C=C(C)\[C@@H](O[C@@H]2O[C@@H](C)[C@H](O[C@@H]3O[C@@H](C)[C@H](O)[C@@H](OC)C3)[C@@H](OC)C2)[C@@H](C)\C=C\C=C/2[C@]3([C@H](C(=O)O4)C=C(C)[C@@H](O)[C@H]3OC\2)O)C[C@H]4C1 RRZXIRBKKLTSOM-XPNPUAGNSA-N 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- 229920000249 biocompatible polymer Polymers 0.000 description 2
- 230000017531 blood circulation Effects 0.000 description 2
- 239000006227 byproduct Substances 0.000 description 2
- FATUQANACHZLRT-KMRXSBRUSA-L calcium glucoheptonate Chemical compound [Ca+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O FATUQANACHZLRT-KMRXSBRUSA-L 0.000 description 2
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 2
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 101150108672 ccr gene Proteins 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- JQXXHWHPUNPDRT-BQVAUQFYSA-N chembl1523493 Chemical compound O([C@](C1=O)(C)O\C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)/C=C\C=C(C)/C(=O)NC=2C(O)=C3C(O)=C4C)C)OC)C4=C1C3=C(O)C=2C=NN1CCN(C)CC1 JQXXHWHPUNPDRT-BQVAUQFYSA-N 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- 229960005091 chloramphenicol Drugs 0.000 description 2
- WIIZWVCIJKGZOK-RKDXNWHRSA-N chloramphenicol Chemical compound ClC(Cl)C(=O)N[C@H](CO)[C@H](O)C1=CC=C([N+]([O-])=O)C=C1 WIIZWVCIJKGZOK-RKDXNWHRSA-N 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 238000002648 combination therapy Methods 0.000 description 2
- 230000000295 complement effect Effects 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 229920001577 copolymer Polymers 0.000 description 2
- 150000001944 cysteine derivatives Chemical class 0.000 description 2
- 230000009089 cytolysis Effects 0.000 description 2
- 229940127089 cytotoxic agent Drugs 0.000 description 2
- 239000002254 cytotoxic agent Substances 0.000 description 2
- 231100000599 cytotoxic agent Toxicity 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 238000006297 dehydration reaction Methods 0.000 description 2
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 2
- 229960003957 dexamethasone Drugs 0.000 description 2
- 229960003668 docetaxel Drugs 0.000 description 2
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- 150000003889 epothilone F derivatives Chemical class 0.000 description 2
- 229910052564 epsomite Inorganic materials 0.000 description 2
- 125000004185 ester group Chemical group 0.000 description 2
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 2
- 210000003527 eukaryotic cell Anatomy 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 235000019197 fats Nutrition 0.000 description 2
- 239000000417 fungicide Substances 0.000 description 2
- 125000002541 furyl group Chemical group 0.000 description 2
- 108020001507 fusion proteins Proteins 0.000 description 2
- 102000037865 fusion proteins Human genes 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- 238000010353 genetic engineering Methods 0.000 description 2
- 229960002518 gentamicin Drugs 0.000 description 2
- 150000004676 glycans Chemical class 0.000 description 2
- 230000005484 gravity Effects 0.000 description 2
- 239000003102 growth factor Substances 0.000 description 2
- 150000004820 halides Chemical class 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- 230000002209 hydrophobic effect Effects 0.000 description 2
- 125000002883 imidazolyl group Chemical group 0.000 description 2
- 229940125721 immunosuppressive agent Drugs 0.000 description 2
- 238000011065 in-situ storage Methods 0.000 description 2
- 125000001041 indolyl group Chemical group 0.000 description 2
- 230000008595 infiltration Effects 0.000 description 2
- 238000001764 infiltration Methods 0.000 description 2
- 230000002757 inflammatory effect Effects 0.000 description 2
- 230000028709 inflammatory response Effects 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 210000002510 keratinocyte Anatomy 0.000 description 2
- 238000010930 lactamization Methods 0.000 description 2
- 229940070765 laurate Drugs 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 230000033001 locomotion Effects 0.000 description 2
- 229940041033 macrolides Drugs 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 239000000401 methanolic extract Substances 0.000 description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 2
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 2
- 208000024191 minimally invasive lung adenocarcinoma Diseases 0.000 description 2
- YNFMRVVYUVPIAN-AQUURSMBSA-N nemadectin Chemical compound C1[C@H](O)[C@H](C)[C@@H](C(/C)=C/C(C)C)O[C@]11O[C@H](C\C=C(C)\C[C@@H](C)\C=C\C=C/2[C@]3([C@H](C(=O)O4)C=C(C)[C@@H](O)[C@H]3OC\2)O)C[C@H]4C1 YNFMRVVYUVPIAN-AQUURSMBSA-N 0.000 description 2
- 229950009729 nemadectin Drugs 0.000 description 2
- YNFMRVVYUVPIAN-UHFFFAOYSA-N nemadectin alpha Natural products C1C(O)C(C)C(C(C)=CC(C)C)OC11OC(CC=C(C)CC(C)C=CC=C2C3(C(C(=O)O4)C=C(C)C(O)C3OC2)O)CC4C1 YNFMRVVYUVPIAN-UHFFFAOYSA-N 0.000 description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- RZPAKFUAFGMUPI-KGIGTXTPSA-N oleandomycin Chemical compound O1[C@@H](C)[C@H](O)[C@@H](OC)C[C@@H]1O[C@@H]1[C@@H](C)C(=O)O[C@H](C)[C@H](C)[C@H](O)[C@@H](C)C(=O)[C@]2(OC2)C[C@H](C)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C RZPAKFUAFGMUPI-KGIGTXTPSA-N 0.000 description 2
- 229960002351 oleandomycin Drugs 0.000 description 2
- 235000019367 oleandomycin Nutrition 0.000 description 2
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 2
- 229940127084 other anti-cancer agent Drugs 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- 210000000496 pancreas Anatomy 0.000 description 2
- 229940124531 pharmaceutical excipient Drugs 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 235000021317 phosphate Nutrition 0.000 description 2
- 229920002643 polyglutamic acid Polymers 0.000 description 2
- 229920001282 polysaccharide Polymers 0.000 description 2
- 239000005017 polysaccharide Substances 0.000 description 2
- 230000002028 premature Effects 0.000 description 2
- 230000000750 progressive effect Effects 0.000 description 2
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 125000004076 pyridyl group Chemical group 0.000 description 2
- 125000000714 pyrimidinyl group Chemical group 0.000 description 2
- 238000002708 random mutagenesis Methods 0.000 description 2
- ATEBXHFBFRCZMA-VXTBVIBXSA-N rifabutin Chemical compound O([C@](C1=O)(C)O/C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)\C=C\C=C(C)/C(=O)NC(=C2N3)C(=O)C=4C(O)=C5C)C)OC)C5=C1C=4C2=NC13CCN(CC(C)C)CC1 ATEBXHFBFRCZMA-VXTBVIBXSA-N 0.000 description 2
- 229960000885 rifabutin Drugs 0.000 description 2
- 229960001225 rifampicin Drugs 0.000 description 2
- 229960003292 rifamycin Drugs 0.000 description 2
- 238000007363 ring formation reaction Methods 0.000 description 2
- 235000012045 salad Nutrition 0.000 description 2
- 229960001860 salicylate Drugs 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 2
- 230000028327 secretion Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 2
- DAEPDZWVDSPTHF-UHFFFAOYSA-M sodium pyruvate Chemical compound [Na+].CC(=O)C([O-])=O DAEPDZWVDSPTHF-UHFFFAOYSA-M 0.000 description 2
- YVOFSHPIJOYKSH-NLYBMVFSSA-M sodium rifomycin sv Chemical compound [Na+].OC1=C(C(O)=C2C)C3=C([O-])C=C1NC(=O)\C(C)=C/C=C/[C@H](C)[C@H](O)[C@@H](C)[C@@H](O)[C@@H](C)[C@H](OC(C)=O)[C@H](C)[C@@H](OC)\C=C\O[C@@]1(C)OC2=C3C1=O YVOFSHPIJOYKSH-NLYBMVFSSA-M 0.000 description 2
- 210000004872 soft tissue Anatomy 0.000 description 2
- 239000012453 solvate Substances 0.000 description 2
- 208000018198 spasticity Diseases 0.000 description 2
- 241000894007 species Species 0.000 description 2
- UNFWWIHTNXNPBV-WXKVUWSESA-N spectinomycin Chemical compound O([C@@H]1[C@@H](NC)[C@@H](O)[C@H]([C@@H]([C@H]1O1)O)NC)[C@]2(O)[C@H]1O[C@H](C)CC2=O UNFWWIHTNXNPBV-WXKVUWSESA-N 0.000 description 2
- 229960000268 spectinomycin Drugs 0.000 description 2
- 230000036262 stenosis Effects 0.000 description 2
- 208000037804 stenosis Diseases 0.000 description 2
- 230000001954 sterilising effect Effects 0.000 description 2
- 238000004659 sterilization and disinfection Methods 0.000 description 2
- 238000005556 structure-activity relationship Methods 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 2
- 125000005420 sulfonamido group Chemical group S(=O)(=O)(N*)* 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 239000002600 sunflower oil Substances 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 230000008961 swelling Effects 0.000 description 2
- 238000010189 synthetic method Methods 0.000 description 2
- 229940095064 tartrate Drugs 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 125000000335 thiazolyl group Chemical group 0.000 description 2
- 239000002562 thickening agent Substances 0.000 description 2
- 150000003573 thiols Chemical class 0.000 description 2
- YWWDBCBWQNCYNR-UHFFFAOYSA-N trimethylphosphine Chemical compound CP(C)C YWWDBCBWQNCYNR-UHFFFAOYSA-N 0.000 description 2
- PHYFQTYBJUILEZ-IUPFWZBJSA-N triolein Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC(OC(=O)CCCCCCC\C=C/CCCCCCCC)COC(=O)CCCCCCC\C=C/CCCCCCCC PHYFQTYBJUILEZ-IUPFWZBJSA-N 0.000 description 2
- 229940117972 triolein Drugs 0.000 description 2
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 2
- 238000011144 upstream manufacturing Methods 0.000 description 2
- 235000013311 vegetables Nutrition 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- AADVCYNFEREWOS-UHFFFAOYSA-N (+)-DDM Natural products C=CC=CC(C)C(OC(N)=O)C(C)C(O)C(C)CC(C)=CC(C)C(O)C(C)C=CC(O)CC1OC(=O)C(C)C(O)C1C AADVCYNFEREWOS-UHFFFAOYSA-N 0.000 description 1
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical compound CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 description 1
- VEEGZPWAAPPXRB-BJMVGYQFSA-N (3e)-3-(1h-imidazol-5-ylmethylidene)-1h-indol-2-one Chemical compound O=C1NC2=CC=CC=C2\C1=C/C1=CN=CN1 VEEGZPWAAPPXRB-BJMVGYQFSA-N 0.000 description 1
- FELGMEQIXOGIFQ-CYBMUJFWSA-N (3r)-9-methyl-3-[(2-methylimidazol-1-yl)methyl]-2,3-dihydro-1h-carbazol-4-one Chemical compound CC1=NC=CN1C[C@@H]1C(=O)C(C=2C(=CC=CC=2)N2C)=C2CC1 FELGMEQIXOGIFQ-CYBMUJFWSA-N 0.000 description 1
- WRRSFOZOETZUPG-FFHNEAJVSA-N (4r,4ar,7s,7ar,12bs)-9-methoxy-3-methyl-2,4,4a,7,7a,13-hexahydro-1h-4,12-methanobenzofuro[3,2-e]isoquinoline-7-ol;hydrate Chemical compound O.C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC WRRSFOZOETZUPG-FFHNEAJVSA-N 0.000 description 1
- DEQANNDTNATYII-OULOTJBUSA-N (4r,7s,10s,13r,16s,19r)-10-(4-aminobutyl)-19-[[(2r)-2-amino-3-phenylpropanoyl]amino]-16-benzyl-n-[(2r,3r)-1,3-dihydroxybutan-2-yl]-7-[(1r)-1-hydroxyethyl]-13-(1h-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentazacycloicosane-4-carboxa Chemical compound C([C@@H](N)C(=O)N[C@H]1CSSC[C@H](NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](CC=2C3=CC=CC=C3NC=2)NC(=O)[C@H](CC=2C=CC=CC=2)NC1=O)C(=O)N[C@H](CO)[C@H](O)C)C1=CC=CC=C1 DEQANNDTNATYII-OULOTJBUSA-N 0.000 description 1
- 239000001149 (9Z,12Z)-octadeca-9,12-dienoate Substances 0.000 description 1
- WTTJVINHCBCLGX-UHFFFAOYSA-N (9trans,12cis)-methyl linoleate Natural products CCCCCC=CCC=CCCCCCCCC(=O)OC WTTJVINHCBCLGX-UHFFFAOYSA-N 0.000 description 1
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- QBWLKDFBINPHFT-UHFFFAOYSA-L 1,3,2$l^{2}-benzodioxabismin-4-one;hydrate Chemical compound O.C1=CC=C2C(=O)O[Bi]OC2=C1 QBWLKDFBINPHFT-UHFFFAOYSA-L 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- RZRNAYUHWVFMIP-KTKRTIGZSA-N 1-oleoylglycerol Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC(O)CO RZRNAYUHWVFMIP-KTKRTIGZSA-N 0.000 description 1
- MQLACMBJVPINKE-UHFFFAOYSA-N 10-[(3-hydroxy-4-methoxyphenyl)methylidene]anthracen-9-one Chemical compound C1=C(O)C(OC)=CC=C1C=C1C2=CC=CC=C2C(=O)C2=CC=CC=C21 MQLACMBJVPINKE-UHFFFAOYSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- KAESVJOAVNADME-UHFFFAOYSA-N 1H-pyrrole Natural products C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 1
- 101150028074 2 gene Proteins 0.000 description 1
- YQTCQNIPQMJNTI-UHFFFAOYSA-N 2,2-dimethylpropan-1-one Chemical group CC(C)(C)[C]=O YQTCQNIPQMJNTI-UHFFFAOYSA-N 0.000 description 1
- 239000000263 2,3-dihydroxypropyl (Z)-octadec-9-enoate Substances 0.000 description 1
- NFJFUDGBWGXTHS-UHFFFAOYSA-N 2-benzhydryloxyethanamine Chemical compound C=1C=CC=CC=1C(OCCN)C1=CC=CC=C1 NFJFUDGBWGXTHS-UHFFFAOYSA-N 0.000 description 1
- VLDSMCHWNWDAKQ-UHFFFAOYSA-N 2-methoxypropanedioic acid Chemical compound COC(C(O)=O)C(O)=O VLDSMCHWNWDAKQ-UHFFFAOYSA-N 0.000 description 1
- 229940080296 2-naphthalenesulfonate Drugs 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- FDVCQFAKOKLXGE-UHFFFAOYSA-N 216978-79-9 Chemical compound C1CC(C)(C)C2=CC(C=O)=CC3=C2N1CCC3(C)C FDVCQFAKOKLXGE-UHFFFAOYSA-N 0.000 description 1
- LNJCGNRKWOHFFV-UHFFFAOYSA-N 3-(2-hydroxyethylsulfanyl)propanenitrile Chemical compound OCCSCCC#N LNJCGNRKWOHFFV-UHFFFAOYSA-N 0.000 description 1
- LOZMZPIMDDLWRS-KOQRGSBKSA-N 3-[2-[3-[[(2r)-4-[[[(2r,3s,4r,5r)-5-(6-aminopurin-9-yl)-4-hydroxy-3-phosphonooxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl]oxy-2-hydroxy-3,3-dimethylbutanoyl]amino]propanoylamino]ethylsulfanyl]-2-hydroxy-3-oxopropanoic acid Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCSC(=O)C(O)C(O)=O)O[C@H]1N1C2=NC=NC(N)=C2N=C1 LOZMZPIMDDLWRS-KOQRGSBKSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- ALKYHXVLJMQRLQ-UHFFFAOYSA-M 3-carboxynaphthalen-2-olate Chemical compound C1=CC=C2C=C(C([O-])=O)C(O)=CC2=C1 ALKYHXVLJMQRLQ-UHFFFAOYSA-M 0.000 description 1
- ZRPLANDPDWYOMZ-UHFFFAOYSA-N 3-cyclopentylpropionic acid Chemical compound OC(=O)CCC1CCCC1 ZRPLANDPDWYOMZ-UHFFFAOYSA-N 0.000 description 1
- RZRNAYUHWVFMIP-GDCKJWNLSA-N 3-oleoyl-sn-glycerol Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@H](O)CO RZRNAYUHWVFMIP-GDCKJWNLSA-N 0.000 description 1
- XMIIGOLPHOKFCH-UHFFFAOYSA-M 3-phenylpropionate Chemical compound [O-]C(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-M 0.000 description 1
- LOLKAJARZKDJTD-UHFFFAOYSA-N 4-Ethoxy-4-oxobutanoic acid Chemical class CCOC(=O)CCC(O)=O LOLKAJARZKDJTD-UHFFFAOYSA-N 0.000 description 1
- 125000001999 4-Methoxybenzoyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C(*)=O 0.000 description 1
- WFJIVOKAWHGMBH-UHFFFAOYSA-N 4-hexylbenzene-1,3-diol Chemical compound CCCCCCC1=CC=C(O)C=C1O WFJIVOKAWHGMBH-UHFFFAOYSA-N 0.000 description 1
- ZYMHCFYHVYGFMS-UHFFFAOYSA-N 5-methyl-1,3-oxazole Chemical group CC1=CN=CO1 ZYMHCFYHVYGFMS-UHFFFAOYSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 102100033350 ATP-dependent translocase ABCB1 Human genes 0.000 description 1
- 108091006112 ATPases Proteins 0.000 description 1
- 208000004998 Abdominal Pain Diseases 0.000 description 1
- 208000000187 Abnormal Reflex Diseases 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 235000006491 Acacia senegal Nutrition 0.000 description 1
- 241000186361 Actinobacteria <class> Species 0.000 description 1
- 108700037654 Acyl carrier protein (ACP) Proteins 0.000 description 1
- 102000048456 Acyl carrier protein (ACP) Human genes 0.000 description 1
- 102100022089 Acyl-[acyl-carrier-protein] hydrolase Human genes 0.000 description 1
- 102000057290 Adenosine Triphosphatases Human genes 0.000 description 1
- 102100027211 Albumin Human genes 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 108010063905 Ampligase Proteins 0.000 description 1
- 241001468213 Amycolatopsis mediterranei Species 0.000 description 1
- 241000094551 Amyloporia xantha Species 0.000 description 1
- 206010073478 Anaplastic large-cell lymphoma Diseases 0.000 description 1
- 208000008822 Ankylosis Diseases 0.000 description 1
- 241000203069 Archaea Species 0.000 description 1
- 241000863009 Archangium gephyra Species 0.000 description 1
- 241001647019 Archangium violaceum Species 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 208000036170 B-Cell Marginal Zone Lymphoma Diseases 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 206010069632 Bladder dysfunction Diseases 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- 241000222455 Boletus Species 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- 240000002791 Brassica napus Species 0.000 description 1
- 235000004977 Brassica sinapistrum Nutrition 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 208000011691 Burkitt lymphomas Diseases 0.000 description 1
- VUHSKSHHOLONGT-UHFFFAOYSA-N C1(=CC=CC=C1)N(O)C1=CC=CC=C1.Cl Chemical compound C1(=CC=CC=C1)N(O)C1=CC=CC=C1.Cl VUHSKSHHOLONGT-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 241000589876 Campylobacter Species 0.000 description 1
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 1
- 206010007882 Cellulitis Diseases 0.000 description 1
- 241000186321 Cellulomonas Species 0.000 description 1
- 206010008531 Chills Diseases 0.000 description 1
- 240000007706 Chionanthus macrocarpus Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- HZZVJAQRINQKSD-UHFFFAOYSA-N Clavulanic acid Natural products OC(=O)C1C(=CCO)OC2CC(=O)N21 HZZVJAQRINQKSD-UHFFFAOYSA-N 0.000 description 1
- 244000060011 Cocos nucifera Species 0.000 description 1
- 235000013162 Cocos nucifera Nutrition 0.000 description 1
- 208000028698 Cognitive impairment Diseases 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 108060005980 Collagenase Proteins 0.000 description 1
- 102000029816 Collagenase Human genes 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- 108091035707 Consensus sequence Proteins 0.000 description 1
- 229910021592 Copper(II) chloride Inorganic materials 0.000 description 1
- 101001030456 Coprinopsis cinerea (strain Okayama-7 / 130 / ATCC MYA-4618 / FGSC 9003) Adenylate-forming reductase 03009 Proteins 0.000 description 1
- 101001000081 Coprinopsis cinerea (strain Okayama-7 / 130 / ATCC MYA-4618 / FGSC 9003) Adenylate-forming reductase 06235 Proteins 0.000 description 1
- 241001647000 Corallococcus exiguus Species 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 101000979117 Curvularia clavata Nonribosomal peptide synthetase Proteins 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- 241000863003 Cystobacter fuscus Species 0.000 description 1
- 241000884527 Cystobacter velatus Species 0.000 description 1
- 241000968335 Cystococcus Species 0.000 description 1
- 108010015742 Cytochrome P-450 Enzyme System Proteins 0.000 description 1
- 102000003849 Cytochrome P450 Human genes 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- CKLJMWTZIZZHCS-UWTATZPHSA-N D-aspartic acid Chemical compound OC(=O)[C@H](N)CC(O)=O CKLJMWTZIZZHCS-UWTATZPHSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- 230000006820 DNA synthesis Effects 0.000 description 1
- 241001495437 Dactylosporangium Species 0.000 description 1
- 208000016192 Demyelinating disease Diseases 0.000 description 1
- 206010012305 Demyelination Diseases 0.000 description 1
- 201000004624 Dermatitis Diseases 0.000 description 1
- 208000036828 Device occlusion Diseases 0.000 description 1
- 240000001879 Digitalis lutea Species 0.000 description 1
- 208000003164 Diplopia Diseases 0.000 description 1
- AADVCYNFEREWOS-OBRABYBLSA-N Discodermolide Chemical compound C=C\C=C/[C@H](C)[C@H](OC(N)=O)[C@@H](C)[C@H](O)[C@@H](C)C\C(C)=C/[C@H](C)[C@@H](O)[C@@H](C)\C=C/[C@@H](O)C[C@@H]1OC(=O)[C@H](C)[C@@H](O)[C@H]1C AADVCYNFEREWOS-OBRABYBLSA-N 0.000 description 1
- CYQFCXCEBYINGO-DLBZAZTESA-N Dronabinol Natural products C1=C(C)CC[C@H]2C(C)(C)OC3=CC(CCCCC)=CC(O)=C3[C@H]21 CYQFCXCEBYINGO-DLBZAZTESA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- NVTRPRFAWJGJAJ-UHFFFAOYSA-L EDTA monocalcium salt Chemical compound [Ca+2].OC(=O)CN(CC([O-])=O)CCN(CC(O)=O)CC([O-])=O NVTRPRFAWJGJAJ-UHFFFAOYSA-L 0.000 description 1
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 1
- 206010015084 Episcleritis Diseases 0.000 description 1
- 241000702191 Escherichia virus P1 Species 0.000 description 1
- 101150034814 F gene Proteins 0.000 description 1
- 101150021185 FGF gene Proteins 0.000 description 1
- 108010039731 Fatty Acid Synthases Proteins 0.000 description 1
- 208000028387 Felty syndrome Diseases 0.000 description 1
- 102000009123 Fibrin Human genes 0.000 description 1
- 108010073385 Fibrin Proteins 0.000 description 1
- BWGVNKXGVNDBDI-UHFFFAOYSA-N Fibrin monomer Chemical group CNC(=O)CNC(=O)CN BWGVNKXGVNDBDI-UHFFFAOYSA-N 0.000 description 1
- 201000008808 Fibrosarcoma Diseases 0.000 description 1
- 206010016654 Fibrosis Diseases 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 206010017577 Gait disturbance Diseases 0.000 description 1
- 206010018341 Gliosis Diseases 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 244000068988 Glycine max Species 0.000 description 1
- 229930186217 Glycolipid Natural products 0.000 description 1
- 102000051366 Glycosyltransferases Human genes 0.000 description 1
- 108700023372 Glycosyltransferases Proteins 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 244000020551 Helianthus annuus Species 0.000 description 1
- 235000003222 Helianthus annuus Nutrition 0.000 description 1
- 208000032843 Hemorrhage Diseases 0.000 description 1
- 101001124319 Heterobasidion annosum Adenylate-forming reductase Nps10 Proteins 0.000 description 1
- 208000017604 Hodgkin disease Diseases 0.000 description 1
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 1
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 1
- 241001354531 Holozonia Species 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000833492 Homo sapiens Jouberin Proteins 0.000 description 1
- 101000651236 Homo sapiens NCK-interacting protein with SH3 domain Proteins 0.000 description 1
- 101001065541 Homo sapiens Protein LYRIC Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 241001562081 Ikeda Species 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102100034343 Integrase Human genes 0.000 description 1
- 208000005016 Intestinal Neoplasms Diseases 0.000 description 1
- 229910021578 Iron(III) chloride Inorganic materials 0.000 description 1
- 206010023198 Joint ankylosis Diseases 0.000 description 1
- 206010023215 Joint effusion Diseases 0.000 description 1
- 206010023230 Joint stiffness Diseases 0.000 description 1
- 102100024407 Jouberin Human genes 0.000 description 1
- 108010076876 Keratins Proteins 0.000 description 1
- 102000011782 Keratins Human genes 0.000 description 1
- 229930194542 Keto Natural products 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 208000032004 Large-Cell Anaplastic Lymphoma Diseases 0.000 description 1
- ZHTRILQJTPJGNK-FYBAATNNSA-N Leinamycin Chemical compound N([C@@H](C=1SC=C(N=1)\C=C/C=C/C(=O)[C@H](O)/C=C(C)/CC1)C)C(=O)C[C@@]21S(=O)SC(=O)[C@]2(C)O ZHTRILQJTPJGNK-FYBAATNNSA-N 0.000 description 1
- ZHTRILQJTPJGNK-UHFFFAOYSA-N Leinamycin Natural products C1CC(C)=CC(O)C(=O)C=CC=CC(N=2)=CSC=2C(C)NC(=O)CC21S(=O)SC(=O)C2(C)O ZHTRILQJTPJGNK-UHFFFAOYSA-N 0.000 description 1
- 108090000364 Ligases Proteins 0.000 description 1
- 102000003960 Ligases Human genes 0.000 description 1
- OJMMVQQUTAEWLP-UHFFFAOYSA-N Lincomycin Natural products CN1CC(CCC)CC1C(=O)NC(C(C)O)C1C(O)C(O)C(O)C(SC)O1 OJMMVQQUTAEWLP-UHFFFAOYSA-N 0.000 description 1
- 102000004882 Lipase Human genes 0.000 description 1
- 108090001060 Lipase Proteins 0.000 description 1
- 239000004367 Lipase Substances 0.000 description 1
- 206010025323 Lymphomas Diseases 0.000 description 1
- 201000003791 MALT lymphoma Diseases 0.000 description 1
- 208000006644 Malignant Fibrous Histiocytoma Diseases 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 229910021380 Manganese Chloride Inorganic materials 0.000 description 1
- GLFNIEUTAYBVOC-UHFFFAOYSA-L Manganese chloride Chemical compound Cl[Mn]Cl GLFNIEUTAYBVOC-UHFFFAOYSA-L 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 208000025205 Mantle-Cell Lymphoma Diseases 0.000 description 1
- 102000000422 Matrix Metalloproteinase 3 Human genes 0.000 description 1
- 241001647015 Melittangium boletus Species 0.000 description 1
- 108010047230 Member 1 Subfamily B ATP Binding Cassette Transporter Proteins 0.000 description 1
- 102000005741 Metalloproteases Human genes 0.000 description 1
- 108010006035 Metalloproteases Proteins 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- PKIXXJPMNDDDOS-UHFFFAOYSA-N Methyl linoleate Natural products CCCCC=CCCC=CCCCCCCCC(=O)OC PKIXXJPMNDDDOS-UHFFFAOYSA-N 0.000 description 1
- 241000192041 Micrococcus Species 0.000 description 1
- 108010011756 Milk Proteins Proteins 0.000 description 1
- 102000014171 Milk Proteins Human genes 0.000 description 1
- 229930192392 Mitomycin Natural products 0.000 description 1
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 1
- 101100174763 Mus musculus Galk1 gene Proteins 0.000 description 1
- 208000023178 Musculoskeletal disease Diseases 0.000 description 1
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 1
- 241000863423 Myxococcus fulvus Species 0.000 description 1
- 241000863424 Myxococcus macrosporus Species 0.000 description 1
- 241001647011 Myxococcus stipitatus Species 0.000 description 1
- 108010045510 NADPH-Ferrihemoprotein Reductase Proteins 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- 229930193140 Neomycin Natural products 0.000 description 1
- 244000187664 Nerium oleander Species 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 108010016076 Octreotide Proteins 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 241000315040 Omura Species 0.000 description 1
- 208000001132 Osteoporosis Diseases 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 102000004316 Oxidoreductases Human genes 0.000 description 1
- 108090000854 Oxidoreductases Proteins 0.000 description 1
- 238000012408 PCR amplification Methods 0.000 description 1
- 240000005373 Panax quinquefolius Species 0.000 description 1
- 235000003140 Panax quinquefolius Nutrition 0.000 description 1
- 101001053399 Paxillus involutus Atromentin synthetase invA1 Proteins 0.000 description 1
- 101001053402 Paxillus involutus Atromentin synthetase invA2 Proteins 0.000 description 1
- 101000599707 Paxillus involutus Atromentin synthetase invA5 Proteins 0.000 description 1
- 101001053396 Paxillus involutus Inactive atromentin synthetase invA3 Proteins 0.000 description 1
- 101000599706 Paxillus involutus Inactive atromentin synthetase invA4 Proteins 0.000 description 1
- 101000599710 Paxillus involutus Inactive atromentin synthetase invA6 Proteins 0.000 description 1
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical class OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 1
- UZQBOFAUUTZOQE-UHFFFAOYSA-N Pikromycin Natural products CC1CC(C)C(=O)C=CC(O)(C)C(CC)OC(=O)C(C)C(=O)C(C)C1OC1C(O)C(N(C)C)CC(C)O1 UZQBOFAUUTZOQE-UHFFFAOYSA-N 0.000 description 1
- 241000896242 Podosphaera Species 0.000 description 1
- 239000004952 Polyamide Substances 0.000 description 1
- 229920002732 Polyanhydride Polymers 0.000 description 1
- 229920001273 Polyhydroxy acid Polymers 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 108010076039 Polyproteins Proteins 0.000 description 1
- 102000029797 Prion Human genes 0.000 description 1
- 108091000054 Prion Proteins 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 108010009736 Protein Hydrolysates Proteins 0.000 description 1
- 102100032133 Protein LYRIC Human genes 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 1
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 1
- 108700005075 Regulator Genes Proteins 0.000 description 1
- 208000002392 Rheumatic Nodule Diseases 0.000 description 1
- 240000000296 Sabal minor Species 0.000 description 1
- 241001326564 Saccharomycotina Species 0.000 description 1
- 101100214703 Salmonella sp aacC4 gene Proteins 0.000 description 1
- 240000000984 Sauvagesia erecta Species 0.000 description 1
- 206010039705 Scleritis Diseases 0.000 description 1
- 101001124317 Serpula lacrymans var. lacrymans (strain S7.9) Adenylate-forming reductase Nps11 Proteins 0.000 description 1
- 101000577223 Serpula lacrymans var. lacrymans (strain S7.9) Adenylate-forming reductase Nps9 Proteins 0.000 description 1
- 101001124346 Serpula lacrymans var. lacrymans (strain S7.9) Atromentin synthetase nps3 Proteins 0.000 description 1
- 235000003434 Sesamum indicum Nutrition 0.000 description 1
- 244000040738 Sesamum orientale Species 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 206010041067 Small cell lung cancer Diseases 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 238000002105 Southern blotting Methods 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 241000863002 Stigmatella Species 0.000 description 1
- 241000863001 Stigmatella aurantiaca Species 0.000 description 1
- 241001468227 Streptomyces avermitilis Species 0.000 description 1
- 241001509463 Streptomyces caelestis Species 0.000 description 1
- 241000187391 Streptomyces hygroscopicus Species 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 101001034059 Suillus grevillei Atromentin synthetase greA Proteins 0.000 description 1
- OUUQCZGPVNCOIJ-UHFFFAOYSA-M Superoxide Chemical compound [O-][O] OUUQCZGPVNCOIJ-UHFFFAOYSA-M 0.000 description 1
- 238000006069 Suzuki reaction reaction Methods 0.000 description 1
- 206010042674 Swelling Diseases 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- CYQFCXCEBYINGO-UHFFFAOYSA-N THC Natural products C1=C(C)CCC2C(C)(C)OC3=CC(CCCCC)=CC(O)=C3C21 CYQFCXCEBYINGO-UHFFFAOYSA-N 0.000 description 1
- 108700012920 TNF Proteins 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 102000012463 Thioesterase domains Human genes 0.000 description 1
- 108050002018 Thioesterase domains Proteins 0.000 description 1
- NSFFHOGKXHRQEW-UHFFFAOYSA-N Thiostrepton B Natural products N1C(=O)C(C)NC(=O)C(=C)NC(=O)C(C)NC(=O)C(C(C)CC)NC(C(C2=N3)O)C=CC2=C(C(C)O)C=C3C(=O)OC(C)C(C=2SC=C(N=2)C2N=3)NC(=O)C(N=4)=CSC=4C(C(C)(O)C(C)O)NC(=O)C(N=4)CSC=4C(=CC)NC(=O)C(C(C)O)NC(=O)C(N=4)=CSC=4C21CCC=3C1=NC(C(=O)NC(=C)C(=O)NC(=C)C(N)=O)=CS1 NSFFHOGKXHRQEW-UHFFFAOYSA-N 0.000 description 1
- 208000007536 Thrombosis Diseases 0.000 description 1
- 108700009124 Transcription Initiation Site Proteins 0.000 description 1
- 102000004357 Transferases Human genes 0.000 description 1
- 108090000992 Transferases Proteins 0.000 description 1
- BAECOWNUKCLBPZ-HIUWNOOHSA-N Triolein Natural products O([C@H](OCC(=O)CCCCCCC/C=C\CCCCCCCC)COC(=O)CCCCCCC/C=C\CCCCCCCC)C(=O)CCCCCCC/C=C\CCCCCCCC BAECOWNUKCLBPZ-HIUWNOOHSA-N 0.000 description 1
- 102000004243 Tubulin Human genes 0.000 description 1
- 108090000704 Tubulin Proteins 0.000 description 1
- 208000015778 Undifferentiated pleomorphic sarcoma Diseases 0.000 description 1
- 206010046542 Urinary hesitation Diseases 0.000 description 1
- 206010046543 Urinary incontinence Diseases 0.000 description 1
- 206010046555 Urinary retention Diseases 0.000 description 1
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 1
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 1
- 208000024248 Vascular System injury Diseases 0.000 description 1
- 208000012339 Vascular injury Diseases 0.000 description 1
- 206010047115 Vasculitis Diseases 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 208000033559 Waldenström macroglobulinemia Diseases 0.000 description 1
- 108010084455 Zeocin Proteins 0.000 description 1
- 101150067314 aadA gene Proteins 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 235000011054 acetic acid Nutrition 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000001252 acrylic acid derivatives Chemical class 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 108010011384 acyl-CoA dehydrogenase (NADP+) Proteins 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 238000005273 aeration Methods 0.000 description 1
- 108010045649 agarase Proteins 0.000 description 1
- 238000005882 aldol condensation reaction Methods 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 150000007933 aliphatic carboxylic acids Chemical class 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 150000008055 alkyl aryl sulfonates Chemical class 0.000 description 1
- 229940045714 alkyl sulfonate alkylating agent Drugs 0.000 description 1
- 150000008052 alkyl sulfonates Chemical class 0.000 description 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 230000002152 alkylating effect Effects 0.000 description 1
- AEMOLEFTQBMNLQ-BKBMJHBISA-N alpha-D-galacturonic acid Chemical compound O[C@H]1O[C@H](C(O)=O)[C@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-BKBMJHBISA-N 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- BIGPRXCJEDHCLP-UHFFFAOYSA-N ammonium bisulfate Chemical compound [NH4+].OS([O-])(=O)=O BIGPRXCJEDHCLP-UHFFFAOYSA-N 0.000 description 1
- 239000004037 angiogenesis inhibitor Substances 0.000 description 1
- 229940121369 angiogenesis inhibitor Drugs 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 208000022531 anorexia Diseases 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 230000003474 anti-emetic effect Effects 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 238000011230 antibody-based therapy Methods 0.000 description 1
- 239000003793 antidiarrheal agent Substances 0.000 description 1
- 229940125683 antiemetic agent Drugs 0.000 description 1
- 239000002111 antiemetic agent Substances 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 230000030741 antigen processing and presentation Effects 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 239000003972 antineoplastic antibiotic Substances 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- XZNUGFQTQHRASN-XQENGBIVSA-N apramycin Chemical compound O([C@H]1O[C@@H]2[C@H](O)[C@@H]([C@H](O[C@H]2C[C@H]1N)O[C@@H]1[C@@H]([C@@H](O)[C@H](N)[C@@H](CO)O1)O)NC)[C@@H]1[C@@H](N)C[C@@H](N)[C@H](O)[C@H]1O XZNUGFQTQHRASN-XQENGBIVSA-N 0.000 description 1
- 229950006334 apramycin Drugs 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 229940114079 arachidonic acid Drugs 0.000 description 1
- 235000021342 arachidonic acid Nutrition 0.000 description 1
- 125000005140 aralkylsulfonyl group Chemical group 0.000 description 1
- 125000003435 aroyl group Chemical group 0.000 description 1
- 125000005239 aroylamino group Chemical group 0.000 description 1
- 230000008321 arterial blood flow Effects 0.000 description 1
- 125000001691 aryl alkyl amino group Chemical group 0.000 description 1
- 125000004659 aryl alkyl thio group Chemical group 0.000 description 1
- 125000004391 aryl sulfonyl group Chemical group 0.000 description 1
- 125000005110 aryl thio group Chemical group 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 238000003149 assay kit Methods 0.000 description 1
- 208000037875 astrocytosis Diseases 0.000 description 1
- 230000007341 astrogliosis Effects 0.000 description 1
- 101150063145 atoA gene Proteins 0.000 description 1
- 239000005667 attractant Substances 0.000 description 1
- OISFUZRUIGGTSD-LJTMIZJLSA-N azane;(2r,3r,4r,5s)-6-(methylamino)hexane-1,2,3,4,5-pentol Chemical compound N.CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO OISFUZRUIGGTSD-LJTMIZJLSA-N 0.000 description 1
- 150000001540 azides Chemical class 0.000 description 1
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 210000003578 bacterial chromosome Anatomy 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- XMIIGOLPHOKFCH-UHFFFAOYSA-N beta-phenylpropanoic acid Natural products OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 1
- 210000003445 biliary tract Anatomy 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000000975 bioactive effect Effects 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 239000004621 biodegradable polymer Substances 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 238000010170 biological method Methods 0.000 description 1
- 229920001222 biopolymer Polymers 0.000 description 1
- 230000006696 biosynthetic metabolic pathway Effects 0.000 description 1
- 229960000782 bismuth subsalicylate Drugs 0.000 description 1
- 230000000740 bleeding effect Effects 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 230000008499 blood brain barrier function Effects 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 210000001218 blood-brain barrier Anatomy 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 244000082778 bunchy flat sedge Species 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 150000004648 butanoic acid derivatives Chemical class 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 230000009702 cancer cell proliferation Effects 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 150000001718 carbodiimides Chemical class 0.000 description 1
- 125000000837 carbohydrate group Chemical group 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 230000006860 carbon metabolism Effects 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- 210000000845 cartilage Anatomy 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 238000002701 cell growth assay Methods 0.000 description 1
- 230000019522 cellular metabolic process Effects 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 235000013339 cereals Nutrition 0.000 description 1
- 239000002962 chemical mutagen Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- ZPEIMTDSQAKGNT-UHFFFAOYSA-N chlorpromazine Chemical compound C1=C(Cl)C=C2N(CCCN(C)C)C3=CC=CC=C3SC2=C1 ZPEIMTDSQAKGNT-UHFFFAOYSA-N 0.000 description 1
- 229960001076 chlorpromazine Drugs 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 229940090805 clavulanate Drugs 0.000 description 1
- HZZVJAQRINQKSD-PBFISZAISA-N clavulanic acid Chemical compound OC(=O)[C@H]1C(=C/CO)/O[C@@H]2CC(=O)N21 HZZVJAQRINQKSD-PBFISZAISA-N 0.000 description 1
- 238000011259 co-electroporation Methods 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 229960004126 codeine Drugs 0.000 description 1
- OROGSEYTTFOCAN-DNJOTXNNSA-N codeine Natural products C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC OROGSEYTTFOCAN-DNJOTXNNSA-N 0.000 description 1
- 208000010877 cognitive disease Diseases 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 229960002424 collagenase Drugs 0.000 description 1
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 239000000356 contaminant Substances 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- 208000006111 contracture Diseases 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- ORTQZVOHEJQUHG-UHFFFAOYSA-L copper(II) chloride Chemical compound Cl[Cu]Cl ORTQZVOHEJQUHG-UHFFFAOYSA-L 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 239000003246 corticosteroid Substances 0.000 description 1
- 229960001334 corticosteroids Drugs 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- 125000004093 cyano group Chemical group *C#N 0.000 description 1
- 229940095074 cyclic amp Drugs 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000000392 cycloalkenyl group Chemical group 0.000 description 1
- 125000000000 cycloalkoxy group Chemical group 0.000 description 1
- 125000006310 cycloalkyl amino group Chemical group 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 125000005366 cycloalkylthio group Chemical group 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 206010061428 decreased appetite Diseases 0.000 description 1
- 230000032459 dedifferentiation Effects 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 239000007857 degradation product Substances 0.000 description 1
- 230000000593 degrading effect Effects 0.000 description 1
- CYQFCXCEBYINGO-IAGOWNOFSA-N delta1-THC Chemical compound C1=C(C)CC[C@H]2C(C)(C)OC3=CC(CCCCC)=CC(O)=C3[C@@H]21 CYQFCXCEBYINGO-IAGOWNOFSA-N 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 238000001212 derivatisation Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 101150116498 devS gene Proteins 0.000 description 1
- AAOVKJBEBIDNHE-UHFFFAOYSA-N diazepam Chemical compound N=1CC(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 AAOVKJBEBIDNHE-UHFFFAOYSA-N 0.000 description 1
- 229960003529 diazepam Drugs 0.000 description 1
- 150000001993 dienes Chemical class 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 238000009792 diffusion process Methods 0.000 description 1
- 102000038379 digestive enzymes Human genes 0.000 description 1
- 108091007734 digestive enzymes Proteins 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- FSBVERYRVPGNGG-UHFFFAOYSA-N dimagnesium dioxido-bis[[oxido(oxo)silyl]oxy]silane hydrate Chemical compound O.[Mg+2].[Mg+2].[O-][Si](=O)O[Si]([O-])([O-])O[Si]([O-])=O FSBVERYRVPGNGG-UHFFFAOYSA-N 0.000 description 1
- SPCNPOWOBZQWJK-UHFFFAOYSA-N dimethoxy-(2-propan-2-ylsulfanylethylsulfanyl)-sulfanylidene-$l^{5}-phosphane Chemical compound COP(=S)(OC)SCCSC(C)C SPCNPOWOBZQWJK-UHFFFAOYSA-N 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- HYPPXZBJBPSRLK-UHFFFAOYSA-N diphenoxylate Chemical compound C1CC(C(=O)OCC)(C=2C=CC=CC=2)CCN1CCC(C#N)(C=1C=CC=CC=1)C1=CC=CC=C1 HYPPXZBJBPSRLK-UHFFFAOYSA-N 0.000 description 1
- 229960004192 diphenoxylate Drugs 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 208000037765 diseases and disorders Diseases 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- 150000002019 disulfides Chemical class 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 229960004242 dronabinol Drugs 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 210000002969 egg yolk Anatomy 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 201000009409 embryonal rhabdomyosarcoma Diseases 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 150000002081 enamines Chemical class 0.000 description 1
- 101150016100 entF gene Proteins 0.000 description 1
- 150000003885 epothilone B derivatives Chemical class 0.000 description 1
- 150000003887 epothilone D derivatives Chemical class 0.000 description 1
- CAMHHLOGFDZBBG-UHFFFAOYSA-N epoxidized methyl oleate Natural products CCCCCCCCC1OC1CCCCCCCC(=O)OC CAMHHLOGFDZBBG-UHFFFAOYSA-N 0.000 description 1
- 235000020774 essential nutrients Nutrition 0.000 description 1
- AFAXGSQYZLGZPG-UHFFFAOYSA-L ethane-1,2-disulfonate Chemical compound [O-]S(=O)(=O)CCS([O-])(=O)=O AFAXGSQYZLGZPG-UHFFFAOYSA-L 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 239000002024 ethyl acetate extract Substances 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 229940071106 ethylenediaminetetraacetate Drugs 0.000 description 1
- 125000006351 ethylthiomethyl group Chemical group [H]C([H])([H])C([H])([H])SC([H])([H])* 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 229950003499 fibrin Drugs 0.000 description 1
- 230000004761 fibrosis Effects 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 201000003444 follicular lymphoma Diseases 0.000 description 1
- 150000004675 formic acid derivatives Chemical class 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 238000012215 gene cloning Methods 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 230000004077 genetic alteration Effects 0.000 description 1
- 231100000118 genetic alteration Toxicity 0.000 description 1
- 230000002518 glial effect Effects 0.000 description 1
- 229960001731 gluceptate Drugs 0.000 description 1
- KWMLJOLKUYYJFJ-VFUOTHLCSA-N glucoheptonic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)C(O)=O KWMLJOLKUYYJFJ-VFUOTHLCSA-N 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 229940049906 glutamate Drugs 0.000 description 1
- 229960002989 glutamic acid Drugs 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 229960002449 glycine Drugs 0.000 description 1
- 230000013595 glycosylation Effects 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- 108700014210 glycosyltransferase activity proteins Proteins 0.000 description 1
- 125000002795 guanidino group Chemical group C(N)(=N)N* 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 125000001475 halogen functional group Chemical group 0.000 description 1
- 238000003306 harvesting Methods 0.000 description 1
- 210000003128 head Anatomy 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- XLYOFNOQVPJJNP-ZSJDYOACSA-N heavy water Substances [2H]O[2H] XLYOFNOQVPJJNP-ZSJDYOACSA-N 0.000 description 1
- 230000002440 hepatic effect Effects 0.000 description 1
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 1
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- AILKHAQXUAOOFU-UHFFFAOYSA-N hexanenitrile Chemical compound CCCCCC#N AILKHAQXUAOOFU-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 229960003258 hexylresorcinol Drugs 0.000 description 1
- 239000012510 hollow fiber Substances 0.000 description 1
- 235000003642 hunger Nutrition 0.000 description 1
- 229920002674 hyaluronan Polymers 0.000 description 1
- 229960003160 hyaluronic acid Drugs 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 150000002429 hydrazines Chemical class 0.000 description 1
- 150000007857 hydrazones Chemical class 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- OROGSEYTTFOCAN-UHFFFAOYSA-N hydrocodone Natural products C1C(N(CCC234)C)C2C=CC(O)C3OC2=C4C1=CC=C2OC OROGSEYTTFOCAN-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 239000000413 hydrolysate Substances 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 1
- 206010020718 hyperplasia Diseases 0.000 description 1
- 206010020745 hyperreflexia Diseases 0.000 description 1
- 230000035859 hyperreflexia Effects 0.000 description 1
- 210000003026 hypopharynx Anatomy 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 125000001841 imino group Chemical group [H]N=* 0.000 description 1
- 230000009390 immune abnormality Effects 0.000 description 1
- 230000003832 immune regulation Effects 0.000 description 1
- 230000003053 immunization Effects 0.000 description 1
- 238000002649 immunization Methods 0.000 description 1
- 239000002955 immunomodulating agent Substances 0.000 description 1
- 229940121354 immunomodulator Drugs 0.000 description 1
- 230000002584 immunomodulator Effects 0.000 description 1
- 230000001506 immunosuppresive effect Effects 0.000 description 1
- 230000001861 immunosuppressant effect Effects 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 230000008676 import Effects 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 239000012678 infectious agent Substances 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 201000002313 intestinal cancer Diseases 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 230000009545 invasion Effects 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- MGFYSGNNHQQTJW-UHFFFAOYSA-N iodonium Chemical compound [IH2+] MGFYSGNNHQQTJW-UHFFFAOYSA-N 0.000 description 1
- 201000004614 iritis Diseases 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- RBTARNINKXHZNM-UHFFFAOYSA-K iron trichloride Chemical compound Cl[Fe](Cl)Cl RBTARNINKXHZNM-UHFFFAOYSA-K 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 210000000281 joint capsule Anatomy 0.000 description 1
- 150000003951 lactams Chemical group 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-N lactobionic acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-N 0.000 description 1
- 150000002596 lactones Chemical class 0.000 description 1
- 238000007273 lactonization reaction Methods 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 238000011031 large-scale manufacturing process Methods 0.000 description 1
- 210000000867 larynx Anatomy 0.000 description 1
- 201000010260 leiomyoma Diseases 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- OJMMVQQUTAEWLP-KIDUDLJLSA-N lincomycin Chemical compound CN1C[C@H](CCC)C[C@H]1C(=O)N[C@H]([C@@H](C)O)[C@@H]1[C@H](O)[C@H](O)[C@@H](O)[C@@H](SC)O1 OJMMVQQUTAEWLP-KIDUDLJLSA-N 0.000 description 1
- 229960005287 lincomycin Drugs 0.000 description 1
- 125000005647 linker group Chemical group 0.000 description 1
- 235000019421 lipase Nutrition 0.000 description 1
- 206010024627 liposarcoma Diseases 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 1
- 229960004844 lovastatin Drugs 0.000 description 1
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000002751 lymph Anatomy 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 201000007919 lymphoplasmacytic lymphoma Diseases 0.000 description 1
- 230000002132 lysosomal effect Effects 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 229940099273 magnesium trisilicate Drugs 0.000 description 1
- 229910000386 magnesium trisilicate Inorganic materials 0.000 description 1
- 235000019793 magnesium trisilicate Nutrition 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 201000009020 malignant peripheral nerve sheath tumor Diseases 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- 239000011565 manganese chloride Substances 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 229960004961 mechlorethamine Drugs 0.000 description 1
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical class ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 1
- 239000013028 medium composition Substances 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-M methyl sulfate(1-) Chemical compound COS([O-])(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-M 0.000 description 1
- 230000011987 methylation Effects 0.000 description 1
- 238000007069 methylation reaction Methods 0.000 description 1
- 125000004092 methylthiomethyl group Chemical group [H]C([H])([H])SC([H])([H])* 0.000 description 1
- HUKYPYXOBINMND-UHFFFAOYSA-N methymycin Natural products CC1CC(C)C(=O)C=CC(O)(C)C(CC)OC(=O)C(C)C1OC1C(O)C(N(C)C)CC(C)O1 HUKYPYXOBINMND-UHFFFAOYSA-N 0.000 description 1
- TTWJBBZEZQICBI-UHFFFAOYSA-N metoclopramide Chemical compound CCN(CC)CCNC(=O)C1=CC(Cl)=C(N)C=C1OC TTWJBBZEZQICBI-UHFFFAOYSA-N 0.000 description 1
- 229960004503 metoclopramide Drugs 0.000 description 1
- 230000025090 microtubule depolymerization Effects 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 235000021239 milk protein Nutrition 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 238000007479 molecular analysis Methods 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- RZRNAYUHWVFMIP-UHFFFAOYSA-N monoelaidin Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC(O)CO RZRNAYUHWVFMIP-UHFFFAOYSA-N 0.000 description 1
- 210000005087 mononuclear cell Anatomy 0.000 description 1
- 230000036651 mood Effects 0.000 description 1
- 230000007659 motor function Effects 0.000 description 1
- 210000000214 mouth Anatomy 0.000 description 1
- 238000002703 mutagenesis Methods 0.000 description 1
- 231100000350 mutagenesis Toxicity 0.000 description 1
- 239000003471 mutagenic agent Substances 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-M naphthalene-2-sulfonate Chemical compound C1=CC=CC2=CC(S(=O)(=O)[O-])=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-M 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 210000003928 nasal cavity Anatomy 0.000 description 1
- 210000001989 nasopharynx Anatomy 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- 210000003739 neck Anatomy 0.000 description 1
- 230000001338 necrotic effect Effects 0.000 description 1
- 229960004927 neomycin Drugs 0.000 description 1
- 208000029974 neurofibrosarcoma Diseases 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 235000021049 nutrient content Nutrition 0.000 description 1
- 229960002700 octreotide Drugs 0.000 description 1
- PFDLUBNRHMFBGI-HRVFELILSA-N oleandolide Chemical compound O=C1[C@H](C)[C@@H](O)[C@@H](C)[C@@H](C)OC(=O)[C@H](C)[C@@H](O)[C@H](C)[C@@H](O)[C@@H](C)C[C@]11OC1 PFDLUBNRHMFBGI-HRVFELILSA-N 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 229960005343 ondansetron Drugs 0.000 description 1
- 229940005483 opioid analgesics Drugs 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000013110 organic ligand Substances 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 210000003300 oropharynx Anatomy 0.000 description 1
- 201000008968 osteosarcoma Diseases 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 150000002923 oximes Chemical class 0.000 description 1
- 125000001820 oxy group Chemical group [*:1]O[*:2] 0.000 description 1
- 125000003232 p-nitrobenzoyl group Chemical group [N+](=O)([O-])C1=CC=C(C(=O)*)C=C1 0.000 description 1
- 229940014662 pantothenate Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- 208000007312 paraganglioma Diseases 0.000 description 1
- 210000003695 paranasal sinus Anatomy 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 208000008494 pericarditis Diseases 0.000 description 1
- 230000002572 peristaltic effect Effects 0.000 description 1
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical compound [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 239000008194 pharmaceutical composition Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- WXVUCMFEGJUVTN-UHFFFAOYSA-N phenyl methanesulfonate Chemical compound CS(=O)(=O)OC1=CC=CC=C1 WXVUCMFEGJUVTN-UHFFFAOYSA-N 0.000 description 1
- 125000003170 phenylsulfonyl group Chemical group C1(=CC=CC=C1)S(=O)(=O)* 0.000 description 1
- CWCMIVBLVUHDHK-ZSNHEYEWSA-N phleomycin D1 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC[C@@H](N=1)C=1SC=C(N=1)C(=O)NCCCCNC(N)=N)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C CWCMIVBLVUHDHK-ZSNHEYEWSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 150000003003 phosphines Chemical class 0.000 description 1
- 150000003009 phosphonic acids Chemical class 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 238000002428 photodynamic therapy Methods 0.000 description 1
- 229940075930 picrate Drugs 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-M picrate anion Chemical compound [O-]C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-M 0.000 description 1
- 101150090633 pikC gene Proteins 0.000 description 1
- UZQBOFAUUTZOQE-VSLWXVDYSA-N pikromycin Chemical compound C[C@H]1C[C@@H](C)C(=O)\C=C\[C@@](O)(C)[C@@H](CC)OC(=O)[C@H](C)C(=O)[C@H](C)[C@H]1O[C@H]1[C@H](O)[C@@H](N(C)C)C[C@@H](C)O1 UZQBOFAUUTZOQE-VSLWXVDYSA-N 0.000 description 1
- 235000015047 pilsener Nutrition 0.000 description 1
- 101150045760 pimA gene Proteins 0.000 description 1
- NVPICXQHSYQKGM-UHFFFAOYSA-N piperidine-1-carbonitrile Chemical compound N#CN1CCCCC1 NVPICXQHSYQKGM-UHFFFAOYSA-N 0.000 description 1
- 229950010765 pivalate Drugs 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 210000004180 plasmocyte Anatomy 0.000 description 1
- 239000005014 poly(hydroxyalkanoate) Substances 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 229920002627 poly(phosphazenes) Polymers 0.000 description 1
- 229920002647 polyamide Polymers 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920000903 polyhydroxyalkanoate Polymers 0.000 description 1
- 229930001118 polyketide hybrid Natural products 0.000 description 1
- 125000003308 polyketide hybrid group Chemical group 0.000 description 1
- 229920001470 polyketone Polymers 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- 239000002952 polymeric resin Substances 0.000 description 1
- 238000012809 post-inoculation Methods 0.000 description 1
- 230000004481 post-translational protein modification Effects 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 238000011027 product recovery Methods 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- QAQREVBBADEHPA-IEXPHMLFSA-N propionyl-CoA Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCSC(=O)CC)O[C@H]1N1C2=NC=NC(N)=C2N=C1 QAQREVBBADEHPA-IEXPHMLFSA-N 0.000 description 1
- BVWMJLIIGRDFEI-QXMHVHEDSA-N propyl (z)-octadec-9-enoate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCCC BVWMJLIIGRDFEI-QXMHVHEDSA-N 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000003531 protein hydrolysate Substances 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 239000003642 reactive oxygen metabolite Substances 0.000 description 1
- 230000003362 replicative effect Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 201000006845 reticulosarcoma Diseases 0.000 description 1
- 208000029922 reticulum cell sarcoma Diseases 0.000 description 1
- 210000003705 ribosome Anatomy 0.000 description 1
- 210000003079 salivary gland Anatomy 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 230000037390 scarring Effects 0.000 description 1
- 230000024053 secondary metabolic process Effects 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 125000003607 serino group Chemical group [H]N([H])[C@]([H])(C(=O)[*])C(O[H])([H])[H] 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 1
- 206010040882 skin lesion Diseases 0.000 description 1
- 231100000444 skin lesion Toxicity 0.000 description 1
- 208000000587 small cell lung carcinoma Diseases 0.000 description 1
- 210000000329 smooth muscle myocyte Anatomy 0.000 description 1
- AWUCVROLDVIAJX-GSVOUGTGSA-N sn-glycerol 3-phosphate Chemical compound OC[C@@H](O)COP(O)(O)=O AWUCVROLDVIAJX-GSVOUGTGSA-N 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 229940054269 sodium pyruvate Drugs 0.000 description 1
- WPMGNXPRKGXGBO-OFQQMTDKSA-N soraphen A Chemical compound C1([C@H]2OC(=O)[C@@H](C)[C@@]3(O)O[C@@H]([C@H](/C=C/[C@@H](OC)[C@@H](OC)CCCC2)C)[C@@H](C)[C@H](O)[C@H]3OC)=CC=CC=C1 WPMGNXPRKGXGBO-OFQQMTDKSA-N 0.000 description 1
- WPMGNXPRKGXGBO-UHFFFAOYSA-N soraphen A1alpha Natural products COC1C(O)C(C)C(C(C=CC(OC)C(OC)CCCC2)C)OC1(O)C(C)C(=O)OC2C1=CC=CC=C1 WPMGNXPRKGXGBO-UHFFFAOYSA-N 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 230000037351 starvation Effects 0.000 description 1
- 229940114926 stearate Drugs 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 108091007196 stromelysin Proteins 0.000 description 1
- 125000003107 substituted aryl group Chemical group 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 150000003457 sulfones Chemical group 0.000 description 1
- 150000003460 sulfonic acids Chemical group 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 230000000153 supplemental effect Effects 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 206010042863 synovial sarcoma Diseases 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 229920003002 synthetic resin Polymers 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229940127072 targeted antineoplastic agent Drugs 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 1
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000000037 tert-butyldiphenylsilyl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1[Si]([H])([*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 101150061166 tetR gene Proteins 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- XCTYLCDETUVOIP-UHFFFAOYSA-N thiethylperazine Chemical compound C12=CC(SCC)=CC=C2SC2=CC=CC=C2N1CCCN1CCN(C)CC1 XCTYLCDETUVOIP-UHFFFAOYSA-N 0.000 description 1
- 229960004869 thiethylperazine Drugs 0.000 description 1
- 125000004149 thio group Chemical group *S* 0.000 description 1
- 125000002813 thiocarbonyl group Chemical group *C(*)=S 0.000 description 1
- 150000003568 thioethers Chemical group 0.000 description 1
- 238000006177 thiolation reaction Methods 0.000 description 1
- NSFFHOGKXHRQEW-AIHSUZKVSA-N thiostrepton Chemical compound C([C@]12C=3SC=C(N=3)C(=O)N[C@H](C(=O)NC(/C=3SC[C@@H](N=3)C(=O)N[C@H](C=3SC=C(N=3)C(=O)N[C@H](C=3SC=C(N=3)[C@H]1N=1)[C@@H](C)OC(=O)C3=CC(=C4C=C[C@H]([C@@H](C4=N3)O)N[C@H](C(N[C@@H](C)C(=O)NC(=C)C(=O)N[C@@H](C)C(=O)N2)=O)[C@@H](C)CC)[C@H](C)O)[C@](C)(O)[C@@H](C)O)=C\C)[C@@H](C)O)CC=1C1=NC(C(=O)NC(=C)C(=O)NC(=C)C(N)=O)=CS1 NSFFHOGKXHRQEW-AIHSUZKVSA-N 0.000 description 1
- 229940063214 thiostrepton Drugs 0.000 description 1
- 229930188070 thiostrepton Natural products 0.000 description 1
- NSFFHOGKXHRQEW-OFMUQYBVSA-N thiostrepton A Natural products CC[C@H](C)[C@@H]1N[C@@H]2C=Cc3c(cc(nc3[C@H]2O)C(=O)O[C@H](C)[C@@H]4NC(=O)c5csc(n5)[C@@H](NC(=O)[C@H]6CSC(=N6)C(=CC)NC(=O)[C@@H](NC(=O)c7csc(n7)[C@]8(CCC(=N[C@@H]8c9csc4n9)c%10nc(cs%10)C(=O)NC(=C)C(=O)NC(=C)C(=O)N)NC(=O)[C@H](C)NC(=O)C(=C)NC(=O)[C@H](C)NC1=O)[C@@H](C)O)[C@](C)(O)[C@@H](C)O)[C@H](C)O NSFFHOGKXHRQEW-OFMUQYBVSA-N 0.000 description 1
- 125000000464 thioxo group Chemical group S=* 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 229910021654 trace metal Inorganic materials 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 238000010361 transduction Methods 0.000 description 1
- 230000026683 transduction Effects 0.000 description 1
- 238000005809 transesterification reaction Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 230000001131 transforming effect Effects 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 125000002306 tributylsilyl group Chemical group C(CCC)[Si](CCCC)(CCCC)* 0.000 description 1
- 230000004102 tricarboxylic acid cycle Effects 0.000 description 1
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- 125000000025 triisopropylsilyl group Chemical group C(C)(C)[Si](C(C)C)(C(C)C)* 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 230000005747 tumor angiogenesis Effects 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- KPGXUAIFQMJJFB-UHFFFAOYSA-H tungsten hexachloride Chemical compound Cl[W](Cl)(Cl)(Cl)(Cl)Cl KPGXUAIFQMJJFB-UHFFFAOYSA-H 0.000 description 1
- 238000010396 two-hybrid screening Methods 0.000 description 1
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical compound CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 1
- 229940070710 valerate Drugs 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 230000006442 vascular tone Effects 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 230000004393 visual impairment Effects 0.000 description 1
- 230000008673 vomiting Effects 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
Images








Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/10—Cells modified by introduction of foreign genetic material
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P17/00—Preparation of heterocyclic carbon compounds with only O, N, S, Se or Te as ring hetero atoms
- C12P17/18—Preparation of heterocyclic carbon compounds with only O, N, S, Se or Te as ring hetero atoms containing at least two hetero rings condensed among themselves or condensed with a common carbocyclic ring system, e.g. rifamycin
- C12P17/181—Heterocyclic compounds containing oxygen atoms as the only ring heteroatoms in the condensed system, e.g. Salinomycin, Septamycin
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D313/00—Heterocyclic compounds containing rings of more than six members having one oxygen atom as the only ring hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/06—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D407/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00
- C07D407/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings
- C07D407/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N1/00—Microorganisms, e.g. protozoa; Compositions thereof; Processes of propagating, maintaining or preserving microorganisms or compositions thereof; Processes of preparing or isolating a composition containing a microorganism; Culture media therefor
- C12N1/20—Bacteria; Culture media therefor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N1/00—Microorganisms, e.g. protozoa; Compositions thereof; Processes of propagating, maintaining or preserving microorganisms or compositions thereof; Processes of preparing or isolating a composition containing a microorganism; Culture media therefor
- C12N1/20—Bacteria; Culture media therefor
- C12N1/205—Bacterial isolates
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/52—Genes encoding for enzymes or proenzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/74—Vectors or expression systems specially adapted for prokaryotic hosts other than E. coli, e.g. Lactobacillus, Micromonospora
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P17/00—Preparation of heterocyclic carbon compounds with only O, N, S, Se or Te as ring hetero atoms
- C12P17/16—Preparation of heterocyclic carbon compounds with only O, N, S, Se or Te as ring hetero atoms containing two or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12R—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES C12C - C12Q, RELATING TO MICROORGANISMS
- C12R2001/00—Microorganisms ; Processes using microorganisms
- C12R2001/01—Bacteria or Actinomycetales ; using bacteria or Actinomycetales
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Medicinal Chemistry (AREA)
- Biophysics (AREA)
- Tropical Medicine & Parasitology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Physics & Mathematics (AREA)
- Virology (AREA)
- Plant Pathology (AREA)
- Cell Biology (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Macromolecular Compounds Obtained By Forming Nitrogen-Containing Linkages In General (AREA)
- Polyesters Or Polycarbonates (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
重组粘球菌属宿主细胞可用于产生聚酮化合物,所述聚酮化合物包括可从发酵肉汤纯化并结晶的埃坡霉素和埃坡霉素类似物。
Description
发明领域
本发明提供了用于在重组宿主细胞内制备聚酮化合物的重组方法和材料;提供了产生聚酮化合物的重组宿主细胞;提供了结构与埃坡霉素类(epothilones)相关的新聚酮化合物;提供了用于纯化埃坡霉素类的方法;以及提供了埃坡霉素D的晶体形式。在一个优选的实施方案中,本发明的重组宿主细胞来自孢囊杆菌亚目(Suborder Cystobacterineae),优选地来自粘球菌属(粘球菌)和标桩菌属(Stigmatella),这些菌已用编码组件式(modular)或迭代式(iterative)聚酮化合物合酶(PKS)基因的本发明的重组DNA表达载体转化。重组宿主细胞产生包括但不限于埃坡霉素及埃坡霉素衍生物的已知和新聚酮化合物。本发明涉及农业、化学、药用化学、医学、分子生物学和药学领域。
发明背景
聚酮化合物构成了一类结构各异的化合物,它们至少部分是由以二碳单位为结构单元的化合物经一系列克莱森缩合和随后的修饰而合成。聚酮化合物包括抗生素(如四环素和红霉素)、抗癌剂(如埃坡霉素和道诺霉素)和免疫抑制剂(如FK506、FK520和雷帕霉素)。聚酮化合物在多种生物体中天然存在,其中所述生物体包括真菌和丝状细菌。在体内聚酮化合物经聚酮化合物合酶合成,这类酶通常被称为PKS酶。已知两种主要类型的PKS,它们的结构和合成聚酮化合物的方式不同。这两类酶通常被称为I型或组件式PKS酶和II型或迭代式(芳香)PKS酶。
本发明提供了用于产生组件式和迭代式PKS酶的方法、重组表达载体和宿主细胞,以及由这些酶产生的聚酮化合物。组件式PKS酶一般为多蛋白复合体,其中每种蛋白质包含多个活性位点,每个位点在碳链装配和修饰过程中只用一次。迭代式PKS酶一般为多蛋白复合体,其中每种蛋白仅含有一个或最多两个活性位点,每个位点在碳链装配和修饰过程中多次使用。如下文更详细描述的那样,已克隆了大量的组件式和芳香PKS酶的基因。
组件式PKS基因由编码一个装载组件、若干延伸组件和一个释放结构域的编码序列组成。如下文更详细描述的那样,这些结构域和组件中的每一个结构域和组件都与一条具有一种或多种特定功能的多肽相对应。通常,装载组件负责与用于合成聚酮化合物的第一个结构单元结合,然后将其转移至第一个延伸组件。用于形成复杂聚酮化合物的结构单元一般为酰基硫酯类,最通常地为乙酰、丙酰、丙二酸单酰、甲基丙二酸单酰、羟基丙二酸单酰、甲氧基丙二酸单酰和乙基丙二酸单酰CoA。其它结构单元包括氨基酸和氨基酸样酰基硫酯类。PKSs在酰基硫酯结构单元之间通过反复的脱羧克莱森缩合而催化聚酮化合物的生物合成。每一组件负责与一个结构单元结合,在该结构单元上行使一种或多种功能,并将所得的化合物转移至下一组件。下一个组件接下来负责结合下一个结构单元,并将生长的化合物转移至下一个组件,直至合成结束。此时,通常是具有硫脂酶(TE)活性的释放结构域将聚酮化合物从PKS上切割下来。
被称为6-脱氧赤酮酸环内酯(6-deoxyerythronolide;B6-dEB)的聚酮化合物由原型的组件式PKS合成。称为eryAI、eryAII和eryAIII的基因编码用以合成6-dEB的称为脱氧赤酮酸环内酯B合酶或DEBS的多亚单位蛋白质(每个亚单位称作DEBS1、DEBS2和DEBS3),这些基因在美国专利号5,672,491、5,712,146和5,824,513中已有描述,它们在此引用作为参考文献。
DEBS PKS的装载组件由一个酰基转移酶(AT)和一个酰基载体蛋白(ACP)组成。DEBS装载组件的AT识别丙酰CoA(其它装载组件的ATs能识别其它酰基CoAs,例如乙酰、丙二酸单酰、甲基丙二酸单酰或丁酰CoA),并将其作为硫酯转移至装载组件的ACP。与此同时,DEBS六个延伸组件上的每个ATs识别甲基丙二酸单酰CoA(其它延伸组件的ATs能够识别其它CoA,例如丙二酸单酰或α-取代的丙二酸单酰CoAs,即丙二酸单酰、乙基丙二酸单酰和2-羟基丙二酸单酰CoA),并将其转移至该组件的ACP以形成硫酯。一旦DEBS上装填了丙酰和甲基丙二酸单酰-ACPs,运载组件的酰基基团就会移动,以在第一个延伸组件的KS处形成硫酯(酯交换);在此阶段,组件1具有一个靠近甲基丙二酸单酰ACP的酰基-KS。来源于DEBS装载组件上的酰基基团然后在伴随的脱羧反应驱动下与延伸基团的α-碳共价结合形成碳-碳键,并产生主链比加载单位多出两个碳(延长或延伸)的酰基-ACP。生长的聚酮化合物链由ACP转移至DEBS的下一个组件的KS,并且反应继续进行。
由DEBS的每一个组件增加两个碳而生长的聚酮化合物链以共价结合的硫酯形式顺序地从一个组件至另一个组件以流水线样的方式传递。仅通过这种方法产生的碳链在每隔一个碳原子上拥有一个酮,形成了一种聚酮,聚酮化合物的名字由此而来。但一般而言,在已添加了二碳单元的聚酮化合物链转移至下一组件前,由其它的酶活性修饰该聚酮化合物链的β酮基。这样,除了含有形成碳-碳键所必须的KS、AT和ACP的最小组件外,组件还可含有一个使β酮基还原成醇的酮还原酶(KR)。组件也可含有一个KR外加一个使醇脱水形成双键的脱水酶(DH)。组件也可含有一个KR、一个DH和一个用β碳原子行使亚甲基功能以使双键转化为饱和单键的烯酰还原酶(ER)。DEBS组件包括那些仅有一个KR结构域、仅有一个失活KR结构域以及含全部三个KR、DH和ER结构域的组件。
一旦聚酮化合物链经过PKS的最后一个组件,它将遇到释放结构域,该结构域一般为硫酯酶,存在于大多数组件式PKS酶的羧基端。在此,聚酮化合物从酶上被切割下来,并且许多(但并非全部的)聚酮化合物被环化。聚酮化合物可被修整酶或修饰酶进一步修饰;这些酶在聚酮化合物的核心分子和/或其取代基上加上糖类基团或甲基基团,或进行如氧化、还原反应的其它修饰。例如,6-dEB为羟基化和糖基化的,其中的一个糖基取代基被甲基化后,就在天然产生抗生素红霉素A的Saccharopolysporaerythraea细胞中形成了人们熟知的该抗生素。
虽然以上描述通常适用于组件式PKS酶,且特别适用于DEBS,自然界存在许多变异。例如,许多PKS酶包含的装载组件与DEBS的装载组件不同,前者含一个具有脱羧酶功能的“失活”KS结构域。多数情况下,该失活的KS被称为KSQ,其中上标Q是谷氨酰胺的单字母缩写,表示酮合酶活性所必需的半胱氨酸活性位点的取代。埃坡霉素PKS的装载组件含一个半胱氨酸活性位点被酪氨酸取代的KSY结构域。此外,其它聚酮化合物的合成起始于与DEBS或埃坡霉素装载组件所结合的起始单位不同的起始单位。与这些起始单位结合的酶例如可包括诸如用于FK520、FK506和雷帕霉素生物合成的AMP连接酶、诸如用于leinamycin生物合成中的非核糖体肽合酶(NRPS)或可溶性CoA连接酶。
PKS酶中的其它重要变异涉及作为延伸单元掺入的结构单元的类型。关于起始单元,一些PKS酶用一个或多个NRPS组件作为延伸组件来掺入氨基酸样酰基硫酯结构单元。埃坡霉素PKS例如包含一个NRPS组件。另一此类变异发现存在于FK506、FK520和雷帕霉素的PKS酶中,它们含有一个掺入2-哌啶酸基(pipecolate)并也可用作PKS释放结构域的NRPS。而另一变异涉及到延伸组件的其它活性。例如,埃坡霉素PKS的一个组件含甲基转移酶(MT)结构域,该结构域向聚酮化合物中掺入一个甲基基团。
操作组件式和迭代式PKS基因的重组方法在美国专利号5,962,290、5,672,491、5,712,146、5,830,750和5,843,718以及在PCT专利公开号98/49315和97/02358中已有描述,它们在此引用作为参考。这些和其它专利描述了异源产生聚酮化合物的重组表达载体以及组合产生新聚酮化合物的两种或多种不同PKS基因或基因簇的部分而装配的重组PKS基因。目前,此类方法已用来在生物体中产生已知或新聚酮化合物,所述生物体例如可天然产生聚酮化合物的链霉菌(Streptomyces)及不天然产生聚酮化合物的大肠杆菌(E.coli)和酵母(见美国专利号6,033,883,在此引用作为参考)。在后两种宿主中,聚酮化合物的产生依赖于磷酸泛酰巯基乙胺转移酶的异源表达,该酶激活PKS的ACP结构域(见PCT公开号97/13845,在此引用作为参考)。
尽管这些方法重要且非常有用,但某些聚酮化合物在所用的异源宿主细胞中仅以非常低的水平表达,或者对所用的异源宿主细胞具有毒性。举一个例子,抗癌剂埃坡霉素A、B、C和D通过埃坡霉素PKS基因的异源表达而得以在链霉菌中产生(Tang等,2000年1月28日,埃坡霉素基因簇的克隆和异源表达,科学(Science),287:640-642,以及PCT公开号号00/031247,它们在此引用作为参考)。埃坡霉素A和B的产量低于约50-100μg/L,并且似乎对生产细胞产生了毒性效应。
埃坡霉素A和B首先被鉴定为具有抗真菌活性,它们提取自粘球菌的纤维堆囊菌(Sorangium cellulosum)(见Gerth等,1996,抗生素杂志(J.Antibiotics)49:560-563和德国专利号DE 4138 042,它们在此引用作为参考),随后在微管蛋白聚合试验中发现具有活性(见Bollag等,1995,癌研究(Cancer Res)55:2325-2333,在此引用作为参考)。埃坡霉素A、B和某些天然存在的和合成的衍生物从此作为癌症治疗中潜在的抗肿瘤剂被广泛研究。由纤维堆囊菌菌株So ce 90产生的埃坡霉素的化学结构已被描述,见文献:Hofle等,1996,埃坡霉素A和B-具有细胞毒性的新16元大环内酯:分离、晶体结构和溶液构象,Angew.Chem.Int.Ed.Engl.35(13/14):1567-1569,在此引用作为参考。埃坡霉素A(R=H)和B(R=CH3)具有如下所示的结构,并显示广谱的抗真核细胞的细胞毒活性,以及对乳腺癌和结肠癌细胞系有明显的活性和选择性。

埃坡霉素A和B的脱氧对应物,也被称为埃坡霉素C(R=H)和D(R=CH3),已从头化学合成,但它们在纤维堆囊菌(S.cellulosum)的发酵中也作为次要产物存在。埃坡霉素C和D的细胞毒性要低于埃坡霉素A和B;它们的结构为如下所示。

已描述了其它天然存在的埃坡霉素。它们包括埃坡霉素E和F以及许多其它埃坡霉素化合物(见PCT公开号99/65913,在此引用作为参考),其中埃坡霉素A和B的噻唑部分的甲基侧链已分别被羟基化,以产生埃坡霉素E和F。
由于埃坡霉素潜在的作为抗癌剂的用途,且由于天然的So ce 90菌株低水平产生埃坡霉素,许多研究小组致力于合成埃坡霉素的研究。如前所述,这种尝试已获成功(见Balog等,1996,(-)-埃坡霉素A的全合成,Angew.Chem.Int.Ed.Engl.35(23/24):2801-2803;Su等,1997,(-)-埃坡霉素B的全合成:铃木偶合法的扩展以及对埃坡霉素结构-活性关系的了解,Angew.Chem.Int.Ed.Engl.36(7):757-759;Meng等,1997,埃坡霉素A和B的全合成,JACS 119(42):10073-10092;以及Balog等,1998,与2-甲基-4-戊烯醛的一种新羟醇缩合以及其在改进的埃坡霉素B全合成中的应用,Angew.Chem.Int.Ed.Engl.37(19):2675-2678,它们在此引用作为参考)。尽管这些尝试获得成功,但埃坡霉素的化学合成是费力、费时而且昂贵的。实际上,认为这些方法对于用作抗癌剂的任何埃坡霉素的大规模药物研制都是不切实际的。
已在体内和体外对一些埃坡霉素衍生物以及埃坡霉素A-D进行了研究(见Su等,1997,埃坡霉素的结构-活性关系研究以及与紫杉醇的首次体内比较,Angew.Chem.Int.Ed.Engl.36(19):2093-2096;和Chou等,1998年8月,脱氧埃坡霉素B:一种与埃坡霉素B有关的、具有所期望体内特性的有效的微管靶向的抗肿瘤剂,美国国家科学院院刊(Proc.Natl.Acad.Sci.USA)95:9642-9647,它们于此处引用作为参考)。其它的埃坡霉素衍生物以及合成埃坡霉素类和埃坡霉素衍生物的方法在PCT公开号00/23452、00/00485、99/67253、99/67252、99/65913、99/54330、99/54319、99/54318、99/43653、99/43320、99/42602、99/40047、99/27890、99/07692、99/02514、99/01124、98/25929、98/22461、98/08849和97/19086;美国专利号5,969,145和德国专利公开号DE 4138 042中已有描述,它们在此引用作为参考。
仍需要用实用的方法来产生不仅是天然存在的埃坡霉素类,而且还有它们的衍生物或前体,以及具有改进特性的新埃坡霉素衍生物。仍需要较天然生成者纤维堆囊菌更易操作和发酵的、产生埃坡霉素类或其衍生物的宿主细胞,并且所述宿主细胞产生更多的所需聚酮化合物产物。本发明通过提供高水平产生聚酮化合物的宿主细胞满足这些要求,且所述宿主细胞不仅用于产生埃坡霉素类(包括本文描述的新埃坡霉素衍生物)而且用于产生其它聚酮化合物。
发明概述
在一个实施方案中,本发明提供了孢囊杆菌亚目的重组宿主细胞,其中所述重组宿主细胞包含编码异源PKS基因的重组表达载体,并产生由这些载体上的基因编码的PKS酶所合成的聚酮化合物。在一个优选的实施方案中,宿主细胞来自粘球菌属或标桩菌属。在特别优选的实施方案中,宿主细胞选自叶柄粘球菌(M.stipitatus)、橙色粘球菌(M.fulvus)、黄色粘球菌(M.xanthus)、变绿粘球菌(M.virescens)、直立标桩菌(S.erecta)和橙色标桩囊菌(S.aurantiaca)。
在另一个实施方案中,本发明提供了可在本发明的宿主细胞内进行染色体整合或染色体外复制的重组DNA载体。本发明的载体至少包含部分PKS编码序列,并可在本发明的宿主细胞中指导功能PKS酶的表达。在一个相关的实施方案中,本发明提供了以下的载体和宿主细胞,其中包含产生用于聚酮化合物生物合成的底物所需的基因及基因产物,所述底物在本发明的宿主细胞不产生或以低丰度产生。在一个实施方案中,基因及基因产物催化乙基丙二酸单酰CoA的合成。在另一个实施方案中,基因及基因产物催化丁酰CoA的合成。
在另外的实施方案中,本发明提供了在孢囊杆菌亚目的宿主细胞中产生聚酮化合物的方法,该聚酮化合物在所述宿主细胞中不能天然产生,所述方法包括转染了重组DNA载体的宿主细胞在一定条件下的培养,这样载体上编码的PKS基因得以表达并产生该聚酮化合物。在一相关的实施方案中,本发明提供了发酵本发明的导致聚酮化合物以高产量产生的宿主细胞的方法。
在一个优选的实施方案中,本发明的重组宿主细胞产生埃坡霉素或埃坡霉素衍生物。这样,本发明提供了产生所需埃坡霉素或埃坡霉素衍生物的重组宿主细胞。在一个优选的实施方案中,宿主细胞以等于或大于10mg/L的量产生一种或多种埃坡霉素。在一个实施方案中,本发明提供了这样的宿主细胞,其产生一种或几种埃坡霉素的水平高于天然存在的产生埃坡霉素的生物体所产生的水平。在另一实施方案中,本发明提供了产生埃坡霉素类混合物的宿主细胞,其中所述混合物较天然存在的宿主细胞所产生的埃坡霉素类混合物的复杂性要低。本发明的重组宿主细胞也包括仅产生以一种所需埃坡霉素或埃坡霉素衍生物作为主要产物的宿主细胞。
在一个相关的优选实施方案中,本发明提供了编码全部或部分埃坡霉素PKS的重组DNA表达载体。这样,本发明提供了在本发明的宿主细胞中编码产生埃坡霉素A、B、C和D所需的蛋白质的重组DNA表达载体。本发明还提供了编码这些蛋白质的一部分的重组DNA表达载体。本发明也提供了编码杂合蛋白质的重组DNA化合物,该杂合蛋白质包括参与埃坡霉素生物合成的一种蛋白质的全部或一部分和参与另一种聚酮化合物或非核糖体来源肽生物合成的一种蛋白质的全部或一部分。
在另一实施方案中,本发明以相当纯的形式提供了用于农业、兽医和医学的新埃坡霉素衍生物化合物。这些化合物包括埃坡霉素A、B、C和D的16-去甲基、14-甲基、13-氧代、13-氧代-11,12-脱氢、12-乙基、13-羟基-10,11-脱氢、11-氧代、11-羟基、10-甲基、10,11-脱氢、9-氧代、9-羟基、8-去甲基、6-去甲基和2-甲基类似物,以及天然存在的埃坡霉素类的甲基噻唑部分被替换为其它部分的一系列类似物。在一个实施方案中,化合物被用作杀真菌剂。在另一实施方案中,化合物在癌症化学治疗中用作抗癌剂。在一个优选的实施方案中,化合物是一种埃坡霉素衍生物,其在抗肿瘤细胞方面至少象埃坡霉素B或D一样有效。在另一实施方案中,化合物用作免疫抑制剂。在另一实施方案中,化合物用于制备另一化合物。在一个优选的实施方案中,化合物以混合物或溶液方式制剂,用来施与人或动物。
在另一实施方案中,本发明提供了纯化埃坡霉素的方法。在一个优选的实施方案中,埃坡霉素被从发酵培养基中纯化。
在另一实施方案中,本发明提供了高纯度形式的埃坡霉素化合物。在一个优选的实施方案中,埃坡霉素的纯度高于95%。在一个更为优选的实施方案中,埃坡霉素的纯度高于99%。在一个特别优选的实施方案中,本发明提供了晶体形式的埃坡霉素。在一个特别优选的实施方案中,本发明提供了结晶的埃坡霉素D。
在另一实施方案中,本发明提供了治疗癌症的方法,该方法包括施用治疗有效量的本发明的新埃坡霉素化合物。本发明的化合物和组合物也用于治疗其它过度增生性疾病和病症,它们包括但不限于银屑病和炎症。
本发明的这些和其它实施方案在以下的描述、实施例和权利要求中将有更为详细的描述。
附图简述
图1显示了N-乙酰基半胱胺硫酯衍生物的一系列前体化合物,这些化合物可被提供给NRPS-样组件1或组件2的KS结构域已被失活的本发明的埃坡霉素PKS,以产生新的埃坡霉素衍生物。还示出了产生此类化合物的通用合成步骤。
图2显示的是质粒pKOS35-82.1和pKOS35-82.2的限制性位点和功能图谱。
图3显示的是质粒pKOS35-154和pKOS90-22的限制性位点和功能图谱。
图4显示了如实施例2中描述的将埃坡霉素PKS和修饰酶基因导入黄色粘球菌宿主细胞染色体的方法示意图。
图5示出了在实施例2中描述的pBeloBACII的图谱。
图6显示了在仅含5g/L酪胨(一种胰的酪蛋白消化物)和2g/L硫酸镁的简单生产培养基中的黄色粘球菌K111-40-1株的基线特性。图例:分批法中,在基本CTS培养基中的生长(●)、产量(■)和氨产生(▲)曲线;培养条件如在实施例3的材料和方法中所描述。
图7显示了XAD-16树脂对黄色粘球菌K111-40-1株在CTS生产培养基中的发酵特性的影响。图例:在分批法中将20g/L XAD-16树脂加入到CTS生产培养基中的生长(●)和产量(■)曲线。
图8显示的是酪胨对生长和产物产量的影响。图例:酪胨浓度对生长(●)、产量(■)和比生产率(▲)的影响。
图9显示了痕量元素和较高的油酸甲酯浓度对生长和产物产量的影响。图例:油酸甲酯(●)和痕量元素(■)对产量的影响。
图10A示出了在分批发酵法中黄色粘球菌菌株在最佳浓度的油酸甲酯和痕量元素存在下的生长和产量。接种后的头两天发生了指数级生长。埃坡霉素D的产生始于稳定期的开始,并在第5天油酸甲酯耗尽、细胞裂解时停止。图10B示出了在分批发酵法中当存在最佳浓度的油酸甲酯和痕量元素时,黄色粘球菌菌株的生长和产生期间,对应于油酸甲酯的消耗和氨生成的时程。图例:A.)分批法中在CTS生产培养基中加入最佳浓度的油酸甲酯(7mL/L)和痕量元素(4mL/L)的生长(●)和产量(■)曲线。B.)对应于油酸甲酯的消耗(●)和氨生成(■)的时程。
图11A示出了间歇补料分批法对黄色粘球菌菌株的生长和产量的影响。图例:在摇瓶中,间歇补料分批法的生长(●)和产量(■)曲线。酪胨和油酸甲酯的补料速率分别为2g/L/天和3mL/L/天。图11B示出了在发酵过程中油酸甲酯的恒定消耗速率和氨的恒定生成速率。图例:对应于加入到培养基中的油酸甲酯的总添加量(●)、油酸甲酯的总消耗量(■)和氨生成(▲)的时程。
图12示出了在5-L的生物反应器中,间歇补料分批法的生产曲线。酪胨和油酸甲酯的补料速率分别为2g/L/天和3mL/L/天。
图13A显示了连续补料对生长和产量的影响。图例:在5-L生物反应器中,连续补料分批法的生长(●)和产量(■)曲线。酪胨和油酸甲酯的补料速率分别为2g/L/天和3mL/L/天。图13B显示了在连续补料分批法中油酸甲酯的加入、消耗和氨生成的时程。图例:对应于加入到培养基中的油酸甲酯的总添加量(●)、油酸甲酯的总消耗量(■)和氨生成(▲)的时程;培养条件如在材料和方法中所描述的。
发明详述
有关本发明范围的陈述和此处所用术语的定义列出如下。这些定义适用于本说明书中单独用到的或作为一个较大组的一部分用到的术语,除非限定在某一特定的情况下。
本发明化合物的所有立体异构体,不管是纯化合物还是它的混合物,均包括在本发明的范围之内。单个的对映异构体、非对映立体异构体、几何异构体,以及它们的组合和混合物均包含在本发明中。此外,化合物的一些晶体形式可以多晶型物存在,且它们同样包括在本发明中。另外,一些化合物可以与水或普通有机溶剂形成溶剂合物(例如与水形成水合物),此类溶剂合物也包括在本发明的范围内。
所发明化合物的被保护形式包括在本发明的范围内。大量的保护基已经公开,例如见T.H.Greene和P.G.M.Wuts,有机合成中的保护基(Protective Groups in Organic Synthesis),第三版,John Wiley & Sons,纽约(1999),在此全部并入作为参考。例如,本发明化合物的羟基被保护形式为其羟基中至少有一个被羟基保护基保护的形式。羟基保护基的示例包括但并不仅限于四氢吡喃基;苯甲基;甲硫基甲基;乙硫基甲基;三甲基乙酰;苯磺酰基;三苯甲基;三取代甲硅烷基,例如三甲代甲硅烷基、三乙基甲硅烷基、三丁基甲硅烷基、三异丙基甲硅烷基、叔丁基二甲基甲硅烷基、三叔丁基甲硅烷基、甲基二苯基甲硅烷基、乙基二苯基甲硅烷基、叔丁基二苯基甲硅烷基等;酰基和芳酰基,例如乙酰基、三甲基乙酰苯甲酰基、4-甲氧苯甲酰基、4-硝基苯甲酰基和脂族酰芳基等。本发明化合物中的酮基基团可同样地被保护。
本发明化合物的前体药物也包括在本发明范围之内。一般而言,这些前体药物是化合物的功能衍生物,在体内易于转化为所需的化合物。这样在本发明的治疗方法中,术语“施用”包含用明确公开的化合物或用可能未明确公开的化合物对多种所述疾病的治疗,其中所述未明确公开的公合物在施用于需要此类治疗的受试者后,在体内转化为所说明的化合物。例如在“前体药物的设计”(“Design of Prodrugs”)H.Bundgaard编辑,Elsevier,1985一书中描述了筛选和制备适宜前体药物衍生物的常规方法。
此处所用术语“脂族”是指可在一个或多个位置处任意取代的饱和和不饱和的直链、支链、环状或多环烃。脂族基的示例包括烷基、链烯基、炔基、环烷基、环链烯基和环炔基部分。术语“烷基”是指直链或支链饱和烃取代基。“链烯基”是指具有至少一个碳-碳双键的直链或支链烃取代基。“炔基”是指具有至少一个碳-碳三键的直链或支链烃取代基。
术语“芳基”是指具有至少一个芳香环结构的单环或多环基团,其中所述芳香环结构任选地含有一个或多个杂原子,且优选地包括3到14个碳原子。芳基取代基可任选地在一个或多个位置进行取代。芳基的示例包括但不仅限于:呋喃基、咪唑基、2,3-二氢化茚基、茚基、吲哚基、异噁唑基、异喹啉基、萘基、噁唑基、噁二唑基、苯基、吡嗪基、吡啶基、嘧啶基、吡咯基、吡唑基、喹啉基、喹握啉基、四氢萘基、四唑基、噻唑基、噻吩基、苯硫基等。
脂族(即烷基、链烯基等)和芳族部分可任选地用一个或多个取代基取代,取代基优选地为1个到5个,更为优选地为1个到3个,最为优选地为1个到2个取代基。对分子内某特定部位的任一取代基或变量的定义不受该分子内其它部位处对其定义的限制。应当理解可以用一种本领域中常用技术选择本发明化合物的取代基和取代方式,以提供化学稳定的、且可用本领域公知技术及在此所示的方法易于合成的化合物。合适取代基的实例包括但不限于:烷基、链烯基、炔基、芳基、卤素;三氟甲基;三氟甲氧基;羟基;烷氧基;环烷氧基;杂环氧基;氧代;烷酰基(-C(=O)-烷基也被称为“酰基”));芳氧基;烷酰氧基;氨基;烷基氨基;芳基氨基;芳烷基氨基;环烷基氨基;杂环氨基;双取代胺,其中两个氨基取代基选自烷基、芳基或芳烷基;烷酰基氨基;芳酰基氨基;芳烷酰基氨基;取代烷酰基氨基;取代芳基氨基;取代芳烷酰基氨基;硫醇;烷硫基;芳硫基;芳烷硫基;环烷硫基;杂环硫基;烷基硫羰;芳基硫羰;芳烷基硫羰;烷基磺酰基;芳基磺酰基;芳烷基磺酰基;磺酰氨基(如SO2NH2);取代磺酰氨基;硝基;氰基;羧基;氨甲酰基(如CONH2);取代氨甲酰基(例如-C(=O)NRR′,其中R和R′每一个为独立的氢、烷基、芳基、芳烷基等);烷氧羰基、芳基、取代芳基、胍基以及杂环,例如吲哚基、咪唑基、呋喃基、噻吩基、噻唑基、吡咯烷基、吡啶基、嘧啶基等。根据需要,取代基可进一步被诸如烷基、烷氧基、芳基、芳烷基、卤素、羟基等取代。
术语“烷基芳基”或“芳基烷基”是指带有脂族取代基的芳基,其通过脂族基结合到化合物上。烷基芳基或芳基烷基的示例是苯甲基,为具有一个甲基的苯基,通过甲基结合到化合物上(-CH2Ph,其中Ph为苯基)。
术语“酰基”是指-C(=O)R,其中R为脂族基,优选地为C1-C6基。
术语“烷氧基”是指-OR,其中O是氧,且R为脂族基。
术语“氨基烷基”是指-RNH2,其中R为脂族基。
术语“卤素”、“卤”或“卤化物”是指氟、氯、溴和碘。
术语“卤烷基”是指-RX,其中R为脂族基,且X为一种或多种卤素。
术语“羟烷基”是指-ROH,其中R为脂族基。
术语“氧代”是指一个羰基氧(=O)。
除了对以上描述的基团进行明确取代外,本发明的化合物根据需要可包括其它取代。例如,内酯或内酰胺主链或主链取代基可另外地(例如通过取代一个氢或通过衍生一个非氢基团)被一个或多个以下取代基取代,诸如C1-C5脂族基、C1-C5烷氧基、芳基或功能基团。合适的功能基团的示例包括但不限于:缩醛、醇、醛、酰胺、胺、硼化物、氨基甲酸酯、烷氧羰基、碳酸盐、碳二亚胺、羧酸、羟腈、二硫化物、烯胺、酯、醚、卤素、肼、腙、二酰亚胺、亚氨基、亚胺、异氰酸盐、酮缩醇、酮、硝基、肟、膦、膦酸酯、膦酸、季铵、氧硫基、硫化物、砜、磺酸、硫醇等。
此处在提到本发明的化合物时所用的术语“分离的”是指通过人为干预使其从天然状态发生“改变”。例如,如果化合物是天然存在的,则它已经发生改变或从其最初环境分开,或两种情况均存在。换言之,在活生物体中天然存在的化合物不是“分离的”,如在此应用的术语所示,从其天然状态的共存材料中分离的同一化合物却是“分离的”。术语“分离的”也可指在一种基本无污染或无不需要物质的制品中的化合物。至于在自然界中发现的化合物,“分离的”是指基本不存在与该化合物或组合物在天然状态下相关的物质。
在提到化合物时所用术语“纯化的”是指在这样一种制品中的化合物,其中该化合物组成了该制品的主要成分,例如化合物在制品组成中的重量占到约50%、约60%、约70%、约80%、约90%、约95%或更多。
此处所用术语“受试者”是指成为治疗、观察或实验对象的一种动物,该动物优选地为哺乳动物,且最优选地为作为治疗和/或观察对象的人。
此处所用术语“治疗有效量”意指在研究者、兽医、内科医生或其它临床医生所应用的组织系统、动物或人中引起生物学或医学反应的活性化合物或药物制剂的剂量,其中所述反应包括受治疗疾病或紊乱症状的缓解。
术语“组合物”指含有特定剂量的特定组分的产品,以及由特定剂量的特定组分组合直接或间接产生的任一产品。
术语“可药用盐”是指一种或多种本发明化合物的盐。化合物的合适可药用盐包括酸加成盐,其例如可通过将化合物的溶液和可药用酸的溶液混合而形成,所述可药用酸诸如盐酸、硫酸、富马酸、顺丁烯二酸、琥珀酸、乙酸、苯甲酸、柠檬酸、酒石酸、碳酸或磷酸。此外,当本发明的化合物带有一个酸性部分时,其合适的可药用盐可包括碱金属盐(例如钠盐或钾盐);碱土金属盐(例如钙盐或镁盐);以及与合适的有机配体所形成的盐(例如用如卤化物、氢氧化物、羧酸盐、硫酸盐、磷酸盐、硝酸盐、烷基磺酸盐和芳基磺酸盐的反荷阴离子形成的铵、季铵和胺阳离子)。可药用盐的示例包括但不限于:醋酸盐、己二酸盐、藻酸盐、抗坏血酸盐、天冬氨酸盐、苯磺酸盐、苯甲酸盐、碳酸氢盐、硫酸氢盐、酒石酸氢盐、硼酸盐、溴化物、丁酸盐、乙二胺四乙酸钙、樟脑酸盐、樟脑磺酸盐、右旋樟脑磺酸、碳酸盐、氯化物、柠檬酸盐、克拉维酸盐、环戊烷丙酸盐、二葡萄糖酸盐(digluconate)、二氢氯化物、十二烷基硫酸盐、乙二胺四乙酸盐、乙二磺酸盐、丙烯酯十二烷基硫酸盐、乙磺酸盐、乙烷磺酸盐、甲酸盐、富马酸盐、葡庚糖酸盐(gluceptate)、葡庚糖酸盐、葡糖酸盐、谷氨酸盐、甘油磷酸盐、乙醇酰阿散酸盐、半硫酸盐、庚酸盐、己酸盐、间苯二酚己酯、海巴明、氢溴酸盐、氢氯化物、氢碘化物、2-羟基-乙烷磺酸盐、羟基萘甲酸盐、碘化物、异硫代硫酸盐、乳酸盐、乳糖醛酸盐、月桂酸盐、十二烷基硫酸盐、苹果酸盐、马来酸盐、丙二酸盐、扁桃酸盐、甲磺酰盐、甲磺酸盐、甲基硫酸盐、粘酸盐、2-萘磺酸盐、napsylate、烟酸盐、硝酸盐、N-甲基葡糖胺铵盐、油酸盐、草酸盐、双羟萘酸盐、棕榈酸盐、泛酸盐、pectinate、过硫酸盐、丙酸3-苯酯、磷酸盐/二磷酸盐、苦味酸盐、三甲基乙酸盐、聚半乳糖醛酸盐、丙酸盐、水杨酸盐、硬脂酸盐、硫酸盐、碱式乙酸盐、琥珀酸盐、鞣酸盐、酒石酸盐、8-氯荼碱、苯甲磺酸盐、三乙基碘、十一酸酯、戊酸盐等。
术语“可药用载体”是用于制备所需要的本发明化合物剂型的介质。可药用载体包括溶剂、稀释剂或其它液体载体;分散助剂或悬浮助剂;表面活性剂;等渗剂;增稠或乳化剂;防腐剂;固体粘合剂;润滑剂等。Remington药物科学(Remington′s Pharmaceutical Sciences),第15版,E.W.Martin(Mack出版公司,Easton,Pa.,1975)和药物赋形剂手册(Handbook of Pharmaceutical Excipients),第3版,A.H.Kibbe,编辑(Amer.Pharmaceutical Assoc.2000),公开了多种用于制备药学组合物的载体以及制备它们的公知技术,二者在此全部并入作为参考。
术语“可药用酯”是指可在体内水解成本发明的化合物或其盐的酯。合适的酯基团的示例包括例如衍生自可药用的脂族羧酸的酯基团,诸如甲酸酯、乙酸酯、丙酸酯盐、丁酸酯、丙烯酸酯和琥珀酸乙酯。
本发明提供了用于在重组宿主细胞中产生聚酮化合物的重组方法和材料;提供了产生聚酮化合物的重组宿主细胞;提供了结构上与埃坡霉素类相关的新聚酮化合物;提供了纯化埃坡霉素类的方法;以及提供了埃坡霉素D的晶体形式。
在一个实施方案中,本发明提供了来自孢囊杆菌亚目的重组宿主细胞,该细胞含有编码异源PKS基因的重组表达载体,并可由这些载体编码的PKS酶产生聚酮化合物。如此处所用,术语重组指通过人类干预产生的细胞、化合物或组合物,这种干预一般是通过对基因或部分基因进行特异且定向的操作来实现。孢囊杆菌亚目是粘球菌目中的两个成员之一(另一成员是堆囊菌亚目,其包括埃坡霉素的生成者纤维堆囊菌属)。孢囊杆菌亚目包括粘球菌科、孢囊杆菌科。粘球菌科包括囊球菌属(Angiococcus)(即碟形囊球菌(A.disciformis))、粘球菌属和软骨球菌属(Corallococcus)(即大孢软骨球菌(C.macrosporus)、珊瑚软骨球菌(C.corralloides)和C.exiguus)。孢囊杆菌科包括孢囊杆菌属(Cystobacter)(即深褐孢囊杆菌(C.fuscus),锈色孢囊杆菌(C.ferrugineus),小孢囊杆菌(C.minor),C.velatus和C.violaceus)、蜂窝囊菌属(Melittangium)(即牛肝菌蜂窝囊菌(M.boletus)和栖地衣蜂窝囊菌(M.lichenicola)、标桩菌属(即直立标桩菌和橙色标桩囊菌)和原囊菌属(Archangium)(即过渡原囊菌(A.gephyra))。本发明特别优选的宿主细胞为这样的宿主细胞,其产生聚酮化合物的产量等于或大于10至20mg/L,更优选地等于或大于100至200mg/L,且最优选地等于或大于1至2g/L。
在一个优选的实施方案中,本发明的宿主细胞来自粘球菌属或标桩菌属。在特别优选的实施方案中,宿主细胞选自叶柄粘球菌、橙色粘球菌、黄色粘球菌、变绿粘球菌、直立标桩菌和橙色标桩囊菌。特别优选的本发明的粘球菌宿主细胞为这样的宿主细胞,其产生聚酮化合物的产量等于或大于10至20mg/L,更优选地等于或大于100至200mg/L,最优选地等于或大于1至2g/L。特别优选黄色粘球菌宿主细胞,其在此水平产生。可用于本发明的黄色粘球菌宿主细胞包括但不限于DZ1细胞系(Campos等,1978,分子生物学杂志(J.Mol.Biol.)119:167-178,在此引用作为参考),产生TA的细胞系ATCC 31046,DK1219细胞系(Hodgkin和Kaiser,1979,分子普通遗传学(Mol.Gen.Genet.)171:177-191,在此引用作为参考),以及DK1622细胞系(Kaiser,1979,美国国家科学院院刊(Proc.Natl.Acad.Sci.USA)76:5952-5956,在此引用作为参考)。
本发明的宿主细胞包含重组DNA表达载体,且在另一个实施方案中,本发明提供的重组DNA载体能够在这些宿主细胞内进行染色体整合和染色体外复制。本发明的载体至少包含部分的PKS编码序列,并能够在本发明的宿主细胞内指导功能PKS酶的表达。如此处所用,术语表达载体指任一可导入宿主细胞内的核酸。表达载体能够在细胞中稳定地或瞬时存在,不管是作为染色体的一部分还是作为细胞内或任一细胞腔区室内的其它DNA,例如细胞质中的复制载体。表达载体也包含用于指导RNA合成的基因,该RNA在细胞或细胞提取物中被翻译成多肽。这样,载体或者含有启动子以促进基因表达,或是整合到染色体的某一位点以实现基因表达。此外,表达载体一般包含额外的功能元件,如用作选择标记的抗性赋予基因和增强启动子活性的调节基因。
一般而言,表达载体含一个或多个标记基因,含该载体的宿主细胞可通过这些基因进行鉴定和/或筛选。用于本发明载体的抗生素抗性赋予基因的示例包括ermE(赋予红霉素和林可霉素抗性)、tsr(赋予硫链丝菌肽抗性)、aadA(赋予壮观霉素和链霉素抗性)、aacC4(赋予阿泊拉霉素、卡那霉素、庆大霉素、遗传霉素(G418)和新霉素抗性)、hyg(赋予潮霉素抗性),以及vph(赋予紫霉素抗性)抗性赋予基因。用于黄色粘球菌的选择标记包括卡那霉素、四环素、氯霉素、zeocin、壮观霉素以及链霉素抗性赋予基因。
表达载体的各种组分差异很大,这取决于载体的特定用途。特别是组分取决于载体将要用到的宿主细胞,以及它发挥作用的方式。例如,本发明的某些优选的载体是整合载体:载体整合到宿主细胞的染色体DNA中。这些载体可包含一个噬菌体附着位点或与宿主细胞染色体DNA片段互补的DNA片段以指导整合。此外,如此处所示,一系列的此类载体可用于在宿主细胞内构建PKS基因簇,而每个载体仅包含完整PKS基因簇的一部分。这样,本发明的重组DNA表达载体可仅含有部分的PKS基因或基因簇。也可用同源重组方法来缺失、破坏或改变基因,其中所述基因包括先期导入宿主细胞的异源PKS基因。
在一个优选的实施方案中,本发明提供了表达载体和重组粘球菌宿主细胞,优选地黄色粘球菌宿主细胞,这些宿主细胞含有产生聚酮化合物所需的那些表达载体。目前,在黄色粘球菌中染色体外进行复制的载体还未见发表,尽管已有一篇未发表的报道涉及到一种基于Mx4噬菌体复制子的人工质粒。但是,已知有很多噬菌体整合到黄色粘球菌的染色体DNA,所述噬菌体包括Mx8、Mx9、Mx81和Mx82。可将这些噬菌体的整合和附着功能导入质粒,以构建出可整合到黄色粘球菌染色体DNA的基于噬菌体的表达载体。其中,噬菌体Mx9和Mx8优选地用于本发明。质粒pPLH343是在大肠杆菌中复制的质粒,且包含编码附着和整合功能的噬菌体Mx8基因,在Salmi等,1998年2月,温和的黄色粘球菌噬菌体Mx8的免疫及整合的遗传决定部位,细菌学杂志(J.Bact.)180(3):614-621中已有描述。
大量的启动子可用于本发明的优选的粘球菌表达载体。见以下的实施例8。例如,纤维堆囊菌埃坡霉素PKS基因的启动子(见PCT公开号00/031247,在此引用作为参考)在黄色粘球菌宿主细胞中行使功能。埃坡霉素PKS基因启动子可在重组宿主细胞内驱动一个或多个埃坡霉素PKS基因或其它PKS基因产物的表达。另一个用于在黄色粘球菌宿主细胞内表达本发明的重组PKS的优选启动子是黄色粘球菌的pilA基因启动子。该启动子以及两株黄色粘球菌在Wu和Kaiser,1997年12月,pilA基因在黄色粘球菌中的表达调控,细菌学杂志179(24):7748-7758一文中已有有描述,在此引用作为参考,上述两株黄色粘球菌为pilA缺失株和pilS缺失株,可由被pilA启动子控制的基因表达高水平的基因产物。本发明也提供了同时含有pilA和pilS缺失的重组粘球菌宿主细胞。另一优选的启动子是sdeK基因的饥饿依赖启动子。
本发明提供了用于制备重组的黄色粘球菌表达载体的优选表达载体,以及提供了本发明的宿主细胞。命名为质粒pKOS35-82.1和pKOS35-82.2(图2)的这些载体能够在大肠杆菌宿主细胞内复制并整合到黄色粘球菌的染色体DNA中。这些载体包含Mx8附着和整合基因以及PilA启动子,且在下游方便地带有限制性酶识别位点。两种载体相互间的不同仅为载体上pilA启动子的方向不同,并可容易地改变为包含本发明的埃坡霉素PKS和修饰酶基因,或包含其它PKS和修饰酶基因。载体的构建在实施例1中描述。
在另一个实施方案中,本发明提供了在不能天然产生聚酮化合物的孢囊杆菌亚目宿主细胞中制备聚酮化合物的方法,该方法包括转染了本发明的重组DNA载体的宿主细胞的培养,培养条件为使载体上编码的PKS基因得以表达,且产生该聚酮化合物。用此方法可制备出由组件式或迭代式PKS酶所产生的任何一种聚酮化合物的多样化成员。此外,用此方法也可制备由杂合或其它重组PKS基因所产生的新聚酮化合物。在一个优选的实施方案中,PKS基因编码一个杂合组件式PKS。
已克隆了大量的组件式PKS基因,并可立即应用于本发明的载体和方法。由PKS酶产生的聚酮化合物经常由聚酮化合物修饰酶(该酶也称修整酶)进一步修饰,以使PKS的聚酮化合物产物发生羟化、环氧化、甲基化和/或糖基化。根据本发明的方法,这些基因也可导入宿主细胞以制备出经过修饰的目标聚酮化合物。下列表格列出了阐述PKS基因和相应的PKS酶的参考文献,这些基因和酶可用于构建本发明的重组PKSs和编码它们的相应DNA化合物。也列出了描述聚酮化合物修整酶和修饰酶,以及可用于制备本发明的重组DNA化合物的相应基因的多个参考文献。
PKS和聚酮化合物修整酶基因
除虫菌素
美国专利号5,252,474;美国专利号4,703,009;和EP公开号118,367(Merck)。
MacNeil等,1993,工业微生物学:基础和应用分子遗传学(IndustrialMicroorganisms:Basic and Applied Molecular Genetics),Baltz,Hegeman,和Skatrud编辑(ASM),245-256页,除虫菌素、红霉素和Nemadectin的聚酮化合物合酶编码基因的比较。
MacNeil等,1992,基因(Gene),115:119-125,Streptomycesavermitilis编码除虫菌素聚酮化合物合酶的基因的复杂组织。
Ikeda和Omura,1997,化学研究(Chem.Res.)97:2599-2609,除虫菌素的生物合成。
杀假丝菌素(FR008)
Hu等,1994,分子微生物学(Mol.Microbiol.)14:163-172.
埃坡霉素
PCT公开号99/66028(Novartis)。
PCT公开号00/031247(Kosan)。
红霉素
PCT公开号93/13663;美国专利号6,004,787;和美国专利号5,824,513(Abbott)。
Donadio等,1991,科学(Science)252:675-9。
Cortes等,1990年11月8日,自然(Nature)348:176-8,红色糖多孢菌(Saccharopolyspora erythraea),产生红霉素的聚酮化合物合酶中一类异常巨大的多功能多肽。
糖基化酶
PCT公开号97/23630和美国专利号5,998,194(Abbott)。
FK-506
Motamedi等,1998,免疫抑制剂FK-506大环内酯环的生物合成基因簇,欧洲生物化学杂志(Eur.J.biochem.)256:528-534。
Motamedi等,1997,参与大环内酯类免疫抑制剂FK-506生物合成的多功能聚酮化合物合酶的结构组织,欧洲生物化学杂志244:74-80。
甲基转移酶
美国专利号5,264,355和美国专利号5,622,866(Merck)。
Motamedi等,1996,参与免疫抑制剂FK506和FK-520生物合成的甲基转移酶和羟化酶的特征,细菌学杂志178:5243-5248。
FK-520
PCT公开号00/20601 to Kosan。
Nielsen等,1991,生物化学(Biochem.)30:5789-96。
洛伐他汀
美国专利号5,744,350(Merck)。
Nemadectin
MacNeil等,1993,同上。
尼达霉素
PCT公开号98/51695(Abbott)。
Kakavas等,1997,来自天青链霉菌(Streptomyces caelestis)的尼达霉素聚酮化合物合酶基因的鉴定及特征,细菌学杂志179:7515-7522。
夹竹桃霉素
Swan等,1994,具有罕见编码序列的编码I型聚酮化合物合酶的一种抗生链霉菌(Streptomyces antibioticus)基因的特征,分子普通遗传法242:358-362。
PCT公开号00/026349(Kosan)。
Olano等,1998,参与夹竹桃霉素生物合成的抗生链球菌染色体区域的分析,该区域编码负责大环内酯环糖基化的两种糖基转移酶,分子普通遗传学259(3):299-308。
PCT公开号99/05283(Hoechst)。
苦霉素
PCT公开号99/61599(Kosan)。
PCT公开号00/00620(明尼苏达大学)。
Xue等,1998,在委内瑞接链霉菌(Streptomyces venezuelae)中大环内酯YC-17和冥菌素的羟化由编码pikC的细胞色素P450介导,化学和生物学(Chemistry & Biology)5(11):661-667。
Xue等,1998年10月,委内瑞拉链霉菌中用于大环内酯类抗生素生物合成的基因簇:代谢多样性的体系结构,美国国家科学院院刊95:12111-12116。
Platenolide
欧洲公开号791,656;和美国专利号5,945,320 to Lilly。
雷帕霉素
Schwecke等,1995年8月,用于聚酮化合物雷帕霉素生物合成的基因簇,美国国家科学院院刊92:7839-7843。
Aparicio等,1996,吸水链球菌(Streptomyces hygroscopicus)中雷帕霉素生物合成基因簇的组织:组件式聚酮化合物合酶的酶活性结构域分析,基因(Gene)169:9-16。
利福霉素
PCT公开号98/07868(Novartis)。
August等,1998年2月13日,袢霉素(ansamycin)抗生素利福霉素的生物合成:推导自Amycolatopsis mediterranei S669的rif生物合成基因簇的分子分析,化学和生物学,5(2):69-79。
堆囊菌属PKS
美国专利号6,090,601(Kosan)。
Soraphen
美国专利号5,716,849(Novartis)。
Schupp等,1995,细菌学杂志(J.Bacteriology)177:3673-3679.用于大环内酯类抗生素Soraphen A生物合成的纤维堆囊菌(粘球菌)基因簇:克隆、特征及与来自放线菌纲的聚酮化合物合酶基因的同源性。
Spinocyn
PCT公开号99/46387(DowElanco)。
螺旋霉素
美国专利号5,098,837(Lilly)。
激活基因
美国专利号5,514,544(Lilly)。
泰乐菌素
美国专利号5,876,991;美国专利号5,672,497;美国专利号5,149,638;欧洲公开号791,655;和欧洲公开号238,323(Lilly)。
Kuhstoss等,1996,基因(Gene)183:231-6,通过构建杂合聚酮化合物合酶产生新聚酮化合物。
修整酶
Merson-Davies和Cundliffe,1994,分子微生物学,13:349-355。来自弗氏链霉菌(Streptomyces fradiae)基因组tyIBA区域中5种泰乐菌素生物合成基因的分析。
带有或不带有用于聚酮化合物修饰的基因的以上任何基因可用于本发明的重组DNA表达载体。此外,可通过用多种载体转化来构建本发明的宿主细胞,其中所述每种载体含所需PKS和修饰酶基因簇的一部分;见美国专利号6,033,883,在此引用作为参考。
为提高聚酮化合物在本发明的宿主细胞(包括粘球菌宿主细胞)中的产量,可转化细胞,以表达异源磷酸泛酰巯基乙胺基转移酶。PKS蛋白质需要装载和延伸组件的ACP结构域以及任何NRPS的PCP结构域进行磷酸泛酰巯基乙胺化作用。磷酸泛酰巯基乙胺化作用由称为磷酸泛酰巯基乙胺基转移酶(PPTases)的酶介导。为了在不天然表达可作用于所需PKS酶的PPTase的宿主细胞中产生功能PKS酶,或增加PPTase有限存在的宿主细胞中功能PKS酶的量,可导入异源PPTase,所述PPTase包括但不限于Sfp,如在PCT公开号97/13845和98/27203,以及美国专利号6,033,883中所述,它们在此引用作为参考。另一个可用于此目的合适的PPTase为来自橙色标桩囊菌的MtaA。
本发明提供的在任一生物体中提高聚酮化合物产量的另一方法是筛选链霉素、利福平和/或庆大霉素的抗性细胞,其中所述的任一生物体包括但不限于粘球菌属、链霉菌属和堆囊菌属宿主细胞。在一个优选的实施方案中,生成聚酮化合物的宿主依次用这些化合物(或结构与其类似的化合物)中的每一个进行筛选,分离聚酮化合物产生能力提高的抗性细胞,并用于下一轮的筛选。以这种方式可获得例如(但不限于)以高水平产生埃坡霉素或埃坡霉素衍生物并对链霉素、利福平和庆大霉素具有抗性的黄色粘球菌宿主细胞。
本发明的宿主细胞不仅可用于产生天然存在的聚酮化合物,也可用于产生由重组PKS基因产物和修饰酶所产生的聚酮化合物。在一个重要的实施方案中,本发明提供了包含杂合PKS的重组DNA表达载体。对本发明而言,杂合PKS为重组PKS,其包含第一种PKS的一个或多个延伸组件、装载组件和硫酯酶/环化酶结构域的全部或一部分,以及第二种PKS的一个或多个延伸组件、装载组件和硫酯酶/环化酶结构域的全部或一部分。
本领域技术人员将认识到:无需从天然存在源中分离本发明的杂合PKS中的第一种或第二种PKS的全部或部分。例如,AT结构域中仅一小部分决定它的特异性。见PCT公开号00/001838,在此引用作为参考。DNA合成技术的陈述使得技术人员可从头合成大小足以构建PKS组件或结构域的有用部分的DNA化合物。对于本发明,这样的合成DNA化合物被视为PKS的一部分。
如上表所示,存在有大量的作为易于获得的DNA源的PKS基因和序列信息用于构建本发明的编码杂合PKS的DNA化合物。构建编码杂合PKS的DNA化合物的方法在美国专利号6,022,731;5,962,290;5,672,491;和5,712,146以及PCT公开号98/49315;99/61599;和00/047724中已有描述,它们均在此引用作为参考。本发明的编码杂合PKS的DNA化合物可为两种以上PKS基因的杂合体。即使仅应用两种基因时,在该杂合基因中往往有两个或更多组件,其中组件的全部或一部分来自第二种PKS基因。本领域的技术人员将会理解,本发明的杂合PKS包括但不限于任一下列类型的PKS:(i)PKS包含一个这样的组件,其中至少有一个结构域来自异源组件;(ii)PKS包含一个来自异源PKS的组件;(iii)PKS含有一种来自异源PKS的蛋白质;以及(iv)以上的组合。
本发明的杂合PKS酶往往这样构建:通过将第一种PKS的一个组件的一个或多个结构域的编码序列替换为来自第二种PKS的一个组件的一个或多个结构域的编码序列而构建重组编码序列。一般而言,此处谈到的插入或替换KR、DH和/或ER结构域,包括对组件进行相关KR、DH或ER结构域的替换,通常是该组件的相对应结构域替换,从而获得插入或替换结构域。如果需要或有利的话,任何组件的KS和/或ACP也可用另一KS和/或ACP进行替换。例如,如果由于组件中的变化而导致埃坡霉素衍生物化合物的产量降低,则可通过改变随后组件的KS和/或ACP结构域来提高产量。在这些替换或插入的每一种替换或插入中,异源KS、AT、DH、KR、ER或ACP编码序列可来自相同或不同PKS的另一组件的编码序列或来自化学合成,以获得杂合PKS编码序列。
虽然本发明的一个重要实施方案涉及到杂合PKS基因,但本发明也提供了不存在第二种PKS基因序列却与天然存在的PKS基因存在一处或多处突变和/或缺失的不同的重组PKS基因。缺失可包括一个或多个组件或结构域的缺失,和/或限于一个或多个组件或结构域内的缺失。当缺失包含了一个完整的延伸组件(而不是NRPS组件),所得的聚酮化合物衍生物比衍生缺失变体的PKS所产生的化合物至少少两个碳。缺失也可包含NRPS组件和/或装载组件。缺失限于一个组件内时,缺失可仅包含一个单一结构域,一般为KR、DH或ER结构域的缺失,或包含多个结构域,如DH和ER结构域,或KR和DH结构域,或全部三个KR、DH和ER结构域的缺失。PKS的结构域也可通过突变如随机或定点诱变发生功能“缺失”。因此,如此处所示的那样,KR结构域通过突变呈现出无功能或低于其全部功能的状态。此外,AT结构域的特异性也可通过突变如随机或定点诱变而发生改变。
为构建本发明的任何PKS,可应用在PCT公开号98/27203和美国专利号6,033,883(它们在此引用作为参考)中所描述的技术,这些文献中,PKS的多种基因和任选地一种或多种聚酮化合物修饰酶的基因被分作两个或多个片段(一般是三个),并且每个片段都置于单独的表达载体(也见PCT公开号00/063361,二者在此引用作为参考)。以这种方式,可组装整套基因并更易于异源表达的操作,且可以改变基因的每一个片段,并且可将多种改变片段组合入单一的宿主细胞以提供本发明的重组PKS基因。此项技术使得构建重组PKS基因的大文库、构建表达这些基因的载体以及构建包含这些载体的宿主细胞变得更为有效。在所有上下文中,编码所需PKS的基因不仅可存在于两个或更多的载体上,也可与基因所来自的天然生产者生物体中存在不同地进行安排或排列。
在优选的和阐述的实施方案中,本发明的重组宿主细胞产生埃坡霉素或埃坡霉素衍生物。天然存在的埃坡霉素(包括埃坡霉素A、B、C、D、E和F)和与它们结构相关的非天然存在的化合物(埃坡霉素衍生物或类似物)是高效的对真核细胞特异的细胞毒性剂。这些化合物已用作抗真菌剂、癌症化学治疗剂和免疫抑制剂,并通常用于治疗炎症或任何过度增生性疾病,如银屑病、多发性硬化症、动脉硬化症和斯坦特固定模堵塞。埃坡霉素在其得以鉴定的天然存在的纤维堆囊菌细胞中以很低的水平产生。此外,纤维堆囊菌的生长非常缓慢,且发酵纤维堆囊菌菌株是困难和费时的。本发明赋予的一个重要的优点是在非纤维堆囊菌宿主细胞中以简单的方式产生埃坡霉素或埃坡霉素衍生物的能力。本发明的另外一个优点是在本发明提供的重组宿主细胞中以较天然存在的埃坡霉素生产者细胞相比具有更高水平和更大量产生埃坡霉素的能力。而另一个优点是在重组宿主细胞中产生埃坡霉素衍生物的能力。这样,本发明提供了产生所需埃坡霉素或埃坡霉素衍生物的重组宿主细胞。在一个优选的实施方案中,宿主细胞产生埃坡霉素或埃坡霉素衍生物的量等于或大于10mg/L。在一个实施方案中,本发明提供了较天然存在的产生埃坡霉素的生物体相比,以更高水平产生一种或多种埃坡霉素或埃坡霉素衍生物的宿主细胞。在另一实施方案中,本发明提供了产生埃坡霉素混合物的宿主细胞,其中所述混合物的复杂性要低于天然存在的产生埃坡霉素的宿主细胞所生成的混合物。
在一个特别优选的实施方案中,本发明的宿主细胞产生埃坡霉素混合物的复杂性低于天然存在的产生埃坡霉素的细胞所产生的混合物。作为一个实例,本发明的某些宿主细胞产生的埃坡霉素D在一种较由天然存在的纤维堆囊菌所产生的混合物的复杂性要低的混合物中,因为埃坡霉素D在前者是主产物,而在后者是副产物。天然存在的产生埃坡霉素的纤维堆囊菌一般产生埃坡霉素A、B、C、D、E、F和极少量的其它产物的混合物,其中只有埃坡霉素A和B作为主要产物存在。下列表1概述了本发明的不同的示例宿主细胞所产生的埃坡霉素。
表1
| 细胞类型 | 所产生的埃坡霉素 | 不能产生的埃坡霉素* |
| 1 | A,B,C,D | E,F |
| 2 | A,C | B,D,E,F |
| 3 | B,D | A,C,E,F |
| 4 | A,B | C,D |
| 5 | C,D | A,B |
| 6 | B | A,C,D,E,F |
| 7 | D | A,B,C,E,F |
*或仅作为副产物产生
这样,本发明的重组宿主细胞也包括仅将一种所需埃坡霉素或埃坡霉素衍生物作为主产物产生的宿主细胞。
仅基于对埃坡霉素PKS结构域的分析,可预测出PKS酶催化产生随意命名为“G”和“H”的埃坡霉素,它们的结构如下所示:
埃坡霉素G(R=H)和H(R=CH3).
这些化合物相互间的区别之处在于,埃坡霉素G在C-12处为氢,而埃坡霉素H在该部位是甲基。经预测C-12位的差异是源于对应的PKS的AT结构域(延伸组件4)结合能力的不同,AT结构域或者结合丙二酸单酰CoA导致氢,或者结合甲基丙二酸单酰CoA导致甲基。但是,并未在自然界或本发明的重组宿主细胞中观察到埃坡霉素G和H。相反,认为PKS的产物是埃坡霉素C和D,它们具有一个C-12到C-13的双键和缺少一个C-13羟基取代基而分别区别于埃坡霉素G和H。如下文更详细描述的那样,基于埃坡霉素PKS基因在异源宿主细胞中的表达,以及这些基因经遗传改造后所产生的产物,认为在埃坡霉素C和D中形成C12-C13双键的脱水反应是由埃坡霉素PKS本身完成的。埃坡霉素A和B分别由埃坡霉素C和D通过epoK基因产物进行C-12到C-13双键的环氧化作用得以形成。埃坡霉素E和F可分别由埃坡霉素A和B通过C-21甲基的羟基化作用或通过埃坡霉素PKS装载组件掺入羟基丙二酸单酰CoA而不是丙二酸单酰CoA得以形成,这一点在下面进一步讨论。
这样,埃坡霉素PKS基因和epoK基因在本发明宿主细胞中的表达导致了埃坡霉素A、B、C和D的产生。如果epoK基因不存在,或通过突变使其失活或部分失活,那么埃坡霉素C和D作为主要产物得以生成。如果延伸组件4的AT结构域被丙二酸单酰CoA特异的AT结构域替换,那么埃坡霉素A和C得以产生,并且如果没有功能epoK基因,那么埃坡霉素C就作为主产物得以产生。如果延伸组件4的AT结构域被甲基丙二酸单酰CoA特异的AT结构域替换,那么埃坡霉素B和D就作为主产物得以产生,并且如果没有功能epoK基因,那么埃坡霉素D就作为主产物得以产生。
埃坡霉素PKS和修饰酶基因克隆自产生埃坡霉素的纤维堆囊菌SMP44菌株。用Jaoua等,1992,质粒(Plasmid)28:157-165所描述的方法(在此引用作为参考)从该菌株中制备总DNA。粘粒文库制备自pSupercos(Stratagene)中的纤维堆囊菌基因组DNA。完整的PKS和修饰酶基因簇分离自4个重叠的粘粒克隆(根据布达佩斯条约于1999年2月17日保藏于美国典型培养物保藏中心(ATCC),10801 University Blvd.,马纳萨斯,VA,20110-2209美国,且授予的ATCC保藏号如下:pKOS35-70.1A2(ATCC 203782),pKOS35-70.4(ATCC 203781),pKOS35-70.8A3(ATCC 203783)和pKOS35-79.85(ATCC 203780)),且测定的DNA序列如在PCT公开号00/031237中所描述的,在此引用作为参考。DNA序列分析表明PKS基因簇带有一个装载组件和九个延伸组件。PKS序列的下游是一个开放读码框(ORF),命名为epoK,该读码框显示出与细胞色素P450氧化酶基因有强同源性,且编码埃坡霉素环氧酶(修饰酶)。
PKS基因被组织在6个ORF中。在多肽水平上,装载组件、延伸组件1(一种NRPS)、2和9出现在单独的多肽上;它们相应的基因分别命名为epoA、epoB、epoC和epoF。组件3、4、5和6包含在一条单独的多肽上,其基因命名为epoD,且组件7和8在另外一条多肽上,其基因命名为epoE。ORFs之间的间隔提示epoC、epoD、epoE和epoF构成了一个操纵子。epoA、epoB和epok基因也可为该巨大操纵子的一部分,但在epoB和epoC之间有约100bp的间隔,在epoF和epoK之间有约115bp的间隔,这些间隔可能含有启动子。埃坡霉素PKS基因簇在图解1中概要显示。
图解1
紧靠epoK(P450环氧酶基因)的下游是ORF1,其编码的多肽似乎包括跨膜结构域,并可参与转运埃坡霉素。该ORF之后是一系列ORFs,它们包含编码参与转运和调控的蛋白质的基因。对组件的详细检查显示出与埃坡霉素生物合成相一致的组织和组成。
随后的描述是在多肽水平上进行的。装载组件和延伸组件3、4、5和9中AT结构域的序列显示出与丙二酸单酰装载组件的共有序列相似,这与分别在C-14、C-12(埃坡霉素A和C)、C-10和C-2位存在H侧链一致。组件2、6、7和8的AT结构域与甲基丙二酸单酰特异的AT结构域类似,同样与分别在C-16、C-8、C-6和C-4位存在甲基侧链一致。
装载组件包含这样的KS结构域,通常在其活性部位存在的半胱氨酸残基由酪氨酸取代。该结构域命名为KSy,并行使脱羧酶功能(为其正常功能的一部分),但不具有缩合酶功能。这样,预计装载组件可装载丙二酸单酰CoA,将其移动到ACP,并使其脱羧以产生与半胱氨酸缩合所需的乙酰基残基。延伸组件1是非核糖体肽合酶,可激活半胱氨酸并催化与装载组件上的乙酸酯缩合。序列包含活化氨基酸所需的、与ATP-结合和ATPase结构域高度类似的片段,磷酸泛酰巯基乙胺化作用位点,氧化结构域,环化结构域和延伸结构域。延伸组件2决定埃坡霉素C-15-C-17的结构。组件2中存在的DH结构域在分子中产生C-16-17脱氢部分。组件3中的结构域与埃坡霉素C-14和C-15的结构一致;KR反应产生的OH用于分子的内酯化。延伸组件4控制C-12和C-13的结构,其中埃坡霉素C和D在此处为一双键。尽管AT结构域的序列似乎与特异装载丙二酸酯的序列类似,它也可装载丙二酸甲酯,因此部分导致了在天然产生埃坡霉素的生物体的发酵培养基中发现有埃坡霉素类混合物。
发现延伸组件4存在有与预期明显偏离的功能。该结构域预期含有一个DH结构域,从而指导埃坡霉素C和D作为PKS的产物而合成。分析显示组件4中AT和KR结构域间的间距并没有大到可容纳下一个功能DH结构域。这样,组件4长度的减少似乎并非发生在β-酮的酮还原之后,上述β-酮形成于由延伸组件4指导的缩合反应之后。如此处所示,埃坡霉素PKS基因本身足以使本发明的宿主细胞具备产生埃坡霉毒C和D的能力。埃坡霉素C和D的异源产生表明,一定存在有可引入双键的脱水酶功能。基于埃坡霉素PKS基因的异源表达和由改变的埃坡霉素基因所产生的产物,认为形成此双键的脱水反应是由埃坡霉素PKS延伸组件5的DH结构域介导,且共轭二烯前体的生成早于组件5ER结构域引起的还原反应。
延伸组件5和6各有一整套还原结构域(KR、DH和ER)以在C-11和C-9位产生亚甲基功能。延伸组件7和9具有KR结构域,以在C-7和C-3位产生羟基,且延伸组件8不具有功能性KR结构域,这与C-5位存在酮基一致。延伸组件8也包含甲基转移酶(MT)结构域,该结构域导致了在C-4位成对的二甲基功能的存在。延伸组件9也有一个终止聚酮化合物合成并催化环闭合的硫酯酶结构域。
下表2概述了埃坡霉素PKS的基因、蛋白质、组件和结构域。
表2
| 基因 | 蛋白质 | 组件 | 存在的结构域 |
| epoA | EpoA | 装载 | KSY mAT ER ACP |
| epoB | EpoB | 1 | NRPS,缩合结构域,杂环化结构域, |
| 腺苷酸化结构域,硫醇化结构域,PCP | |||
| epoC | EpoC | 2 | KS mmAT DH KR ACP |
| epoD | EpoD | 3-6 | KS mAT KR ACP;KS mAT KR ACP; |
| KS | |||
| mAT DH ER KR ACP;KS mmAT DHER | |||
| KR ACP | |||
| epoE | EpoE | 7-8 | KS mmAT KR ACP;KS mmAT MTDH*KR*ACP |
| epoF | EpoF | 9 | KS mAT KR DH* ER* ACP TE |
NRPS-非核糖体肽合酶;KS-酮合酶;mAT-丙二酸单酰CoA特异的酰基转移酶;mmAT-甲基丙二酸单酰CoA特异的酰基转移酶;DH-脱水酶;ER-烯酰基还原酶;KR-酮还原酶;MT-甲基转移酶;TE硫酯酶;*-失活结构域。
序列分析显示,epoA和epoB(装载组件和延伸组件1NRPS)之间以及epoC和epoD之间存在翻译偶联。发现epoD和epoE以及epoE和epoF之间的间隔非常小,而epoB和epoC以及epoF和epoK之间的间隔超过100bp。这些基因间的区域可能含有启动子。
这样,埃坡霉素PKS是由epoA、epoB、epoC、epoD、epoE和epoF基因的基因产物组成的多蛋白质复合体。为赋予宿主细胞产生埃坡霉素类的能力,提供了带有本发明的重组epoA、epoB、epoC、epoD、epoE和epoF基因以及任选地其它可在该宿主细胞内表达的基因(例如epoK基因)的宿主细胞。本领域技术人将会理解:尽管埃坡霉素以及其它PKS酶此处可作为单一实体被提及,但这些酶一般为多亚单位蛋白质。这样,可通过改变一个或多个编码构成PKS的一种或多种多蛋白质的基因来制备衍生PKS(通过缺失或突变形成的与天然存在的PKS不同的PKS)或杂合PKS(由两种不同的PKS酶的部分组成的PKS)。
埃坡霉素的后PKS(post-PKS)修饰或修整包括由多种酶介导的多个步骤。这些酶此处称为修整酶或修饰酶。埃坡霉素PKS基因epoA、epoB、epoC、epoD、epoE和epoF在本发明的不表达epoK的宿主细胞内的表达导致埃坡霉素C和D作为主产物得以产生,埃坡霉素C和D缺少埃坡霉素A和B的C-12-C-13环氧键,代之以C-12-C-13双键。这样,埃坡霉素C和D由epoK基因编码的环氧酶转化为埃坡霉素A和B。埃坡霉素A和B可由羟化酶基因转化为埃坡霉素E和F,其中所述羟化酶基因可由与埃坡霉素PKS基因簇相关联的基因编码或另一纤维堆囊菌的内源基因编码。另外,这些化合物可由结合了非丙二酸单酰CoA的起始单位(如羟基丙二酸单酰CoA)的装载组件形成。这样,可通过提供带有一种或多种本发明的重组修饰酶基因的宿主细胞,或使用可天然表达(或不表达)修饰酶的宿主细胞,和/或提供非丙二酸单酰CoA的起始单位,从而在宿主细胞中产生如所希望进行了修饰的埃坡霉素或埃坡霉素衍生物。
这样,本发明提供了大量用于表达天然存在的埃坡霉素A、B、C和D及其衍生物的重组DNA化合物和宿主细胞。本发明也提供了重组宿主细胞,该细胞以类似于埃坡霉素E和F的方式产生进行了修饰的埃坡霉素衍生物。此外,本发明的任一埃坡霉素或埃坡霉素衍生物可按照PCT专利公开号00/039276中所描述的方法转化为对应的埃坡霉素E或F衍生物,其中所述专利在此引用作为参考。
本发明也提供了制备埃坡霉素衍生物的大量重组DNA化合物和宿主细胞。如此处所用的,短语埃坡霉素衍生物是指由重组埃坡霉素PKS所产生的化合物,其中所述重组埃坡霉素PKS中至少已插入了一个结构域,或它的一个结构域通过缺失或突变已失活,或结构域已被突变而改变了它的催化功能,或结构域被具有不同功能的结构域替换。在任何情况下,因此产生的埃坡霉素衍生物PKS的功能在于产生与天然存在的选自埃坡霉素A、B、C、D、E和F的结构不同的化合物。为有助于更好地理解本发明提供的重组DNA化合物和宿主细胞,以下提供了有关埃坡霉素PKS及其新重组衍生物的装载组件和每一组件的详细讨论。
埃坡霉素PKS的装载组件包含一个“失活”KS结构域(该结构域由于在“活性”KS结构域的半胱氨酸残基被酪氨酸(Y)残基取代而被命名为KSY),该结构域不能进行由活性KS结构域介导的缩合反应。该KSY结构域确可进行由KS结构域介导的脱羧反应。此类“失活”KS结构域也存在于其它PKS酶中,通常其活性位点半胱氨酸被谷氨酰胺(Q)残基取代,且被命名为KSQ结构域。已显示大鼠脂肪酸合酶的KSQ结构域不能进行缩合反应,但在脱羧反应中呈现出2个数量级的增加。见Witkowski等,1999年9月7日,生物化学(Biochem.)38(36):11643-11650,在此引用作为参考。KSQ结构域在脱羧反应中比KSY结构域更为有效,因此埃坡霉素PKS的KSY结构域被KSQ结构域取代可增加在某些宿主细胞内或在某特定培养条件下的埃坡霉素的生物合成效率。这可仅仅通过将酪氨酸密码子改变为谷氨酰胺密码子而得以实现,如以下实施例6中所描述的。这也可通过将KSY结构域替换为另一PKS的KSQ结构域而得以实现,其中所述另一PKS例如oleandolide PKS或narbonolide PKS(见以上表中引用的与夹竹桃霉素、冥霉素和苦霉素PKS及修饰酶有关的参考文献)。
埃坡霉素装载组件也包含丙二酸单酰CoA特异的AT结构域(认为其被KSY结构域脱羧基而产生乙酰基),以及一个ACP结构域。本发明提供了重组埃坡霉素衍生物的装载组件或它们的编码DNA序列,其中丙二酸单酰基特异的AT结构域或其编码序列已被改变为另一特异性,诸如对甲基丙二酸单酰CoA、乙基丙二酸单酰CoA和2-羟基丙二酸单酰CoA特异。当与埃坡霉素PKS的其它蛋白质表达时,此类装载组件产生这样的埃坡霉素,其中埃坡霉素的噻唑环的甲基取代基分别用乙基、丙基和羟甲基取代。本发明提供了包含此类装载组件的重组PKS酶和用于产生此类酶及由此产生聚酮化合物的宿主细胞。当AT结构域改变为对2-羟基丙二酸单酰CoA特异的结构域时,相应的埃坡霉素PKS衍生物将产生埃坡霉素E和F衍生物。2-羟基丙二酸单酰CoA特异的AT结构域将在所产生的聚酮化合物的相应部位具有一个羟基的聚酮化合物;羟基可被聚酮化合物修饰酶甲基化而产生甲氧基。见例如在上表中与FK-520 PKS有关的引用文献。结果,此处谈到的具有2-羟基丙二酸单酰特异的AT结构域同样是指由该PKS产生的聚酮化合物在其相应部位具有羟基或甲氧基。
埃坡霉素PKS的装载组件也含有ER结构域。该结构域可参与埃坡霉素中噻唑部分的双键之一的形成(与其正常反应相反),它也可为无功能的。在两种情况中的任何一种情况下,本发明提供了这样的重组DNA化合物,该DNA化合物编码带有或不带有ER结构域的埃坡霉素PKS装载组件,以及编码杂合装裁组件,其中所述杂合装载组件包含用来自另一PKS的ER结构域(活性或失活的,带有或不带有伴随的KR和DH结构域)替代埃坡霉素装载组件的ER结构域。本发明也提供了包含此类装载组件的重组PKS酶和用于产生此类酶并由此产生聚酮化合物的宿主细胞。
埃坡霉素PKS的装载组件也可用来自异源PKS的装载组件替换,以形成可产生埃坡霉素衍生物的杂合PKS。在一个实施方案中,用NRPS替换埃坡霉素PKS的装载组件,这一点在下列实施例中描述。
在埃坡霉素PKS装载组件之后是PKS的第一延伸组件,该组件为对半胱氨酸特异的NRPS延伸组件。该NRPS组件本身表达为独立蛋白质,为epoB基因产物。在一个实施方案中,NRPS组件编码序列的一部分用于连接异源编码序列。在此实施方案中,本发明提供了例如将埃坡霉素PKS的NRPS组件的特异性由半胱氨酸变为另一氨基酸。也可通过构建编码序列来获得改变,编码序列中埃坡霉素PKS NRPS组件编码序列的全部或一部分被替换为编码具有不同特异性的NRPS组件的编码序列。
在一个示例的实施方案中,埃坡霉素NRPS组件的特异性由半胱氨酸变为丝氨酸或苏氨酸。当如此改变的NRPS结构域与埃坡霉素PKS的其它蛋白质表达时,该重组PKS产生了这样的埃坡霉素衍生物,其中埃坡霉素(或埃坡霉素衍生物)的噻唑部分分别变成唑或5-甲基唑部分。这样,在一个示例的实施方案中,本发明提供了宿主细胞、载体和重组埃坡霉素PKS酶,其中NRPS结构域由埃坡霉素NRPS的腺苷酸化结构域替换为entF基因(对于丝氨酸而言)编码的NRPS的腺苷酸化结构域而改变。在另一示例的实施方案中,本发明提供了宿主细胞、载体和重组埃坡霉素PKS酶,其中NRPS结构域已经通过将埃坡霉素NRPS的腺苷酸化结构域替换为vib F基因(对于苏氨酸而言)编码的NRPS的腺苷酸化结构域而改变。在一个实施方案中,在一种埃坡霉素PKS中进行这些NRPS的替换,其中所述埃坡霉素PKS中也包含结合丙二酸单酰CoA而非甲基丙二酸单酰CoA的延伸组件2,以产生唑和甲基唑埃坡霉素衍生物的16-去甲基衍生物。
另外,本发明提供了由不带有NRPS组件或带有来自异源PKS的NRPS组件的epoA、epoC、epoD、epoE和epoF基因(或其修饰变体)产物组成的重组PKS酶。异源NRPS组件可以独立的蛋白质形式表达或与epoA基因或epoC基因以融合蛋白质的形式表达。将PKS的一个组件替换为另一组件时,重要的是确保保留或另外应用了相容的模件间连接子序列。见PCT公开号00/047724,在此引用作为参考。
在另一实施方案中,本发明提供了重组埃坡霉素PKS酶、相应的重组DNA化合物和载体,它们中的NRPS组件已被失活或缺失。本发明提供的失活NRPS组件的蛋白质和编码序列包括那些将PCP结构域全部或部分缺失,或将活性位点丝氨酸用于(磷酸泛酰巯基乙胺化的位点)变换为其它氨基酸(如丙氨酸)而使PCP结构域失活,或将腺苷酸化结构域缺失或使之失活。在一个实施方案中,装载组件和NRPS均被缺失或失活。在任何情况下,所产生的埃坡霉素PKS只要提供结合埃坡霉素PKS或埃坡霉素衍生物的PKS延伸组件2(或一个随后的组件)的KS结构域的底物,然后就能够行使功能。在本发明提供的方法中,这样的修饰细胞然后用活化的酰基硫酯饲养,该酰基硫酯优选地与第二个延伸组件结合,但可能与随后的任何组件结合,并加工成新埃坡霉素衍生物。宿主细胞被饲以活化的酰基硫酯以产生本发明的新埃坡霉素衍生物。表达PKS的宿主细胞或含有PKS的无细胞提取物可被饲以或提供新前体分子N-酰基半胱胺硫酯(NACS)来制备埃坡霉素衍生物。见PCT公开号US99/03986和00/044717,二者在此引用作为参考,还见以下的实施例9和10。
埃坡霉素PKS的第二个(第一个非NRPS)延伸组件包括KS、甲基丙二酸单酰CoA特异的AT、DH、KR和ACP。埃坡霉素PKS的第二个延伸组件为epoC基因产生的独立蛋白质。全部或仅部分的第二延伸组件的编码序列可连接其它PKS编码序列用于产生杂合组件。在此实施方案中,本发明提供了例如用丙二酸单酰CoA、乙基丙二酸单酰CoA或2-羟基丙二酸单酰CoA特异的AT替换甲基丙二酸单酰CoA;缺失DH或KR或二者都缺失;将DH、KR或二者替换为具有不同立体化学特异性的DH、KR或二者;和/或插入ER。所产生的异源第二延伸组件的编码序列可与其它构成PKS的蛋白质共表达,其中所述PKS合成埃坡霉素、埃坡霉素衍生物或其它聚酮化合物。另外,可将埃坡霉素PKS的第二延伸组件缺失或替换为来自异源PKS的组件,上述来自异源PKS的组件可表达为独立蛋白质或与表达为与epoB或epoD基因产物融合的融合蛋白质。
示例的本发明的重组PKS基因包括以下重组PKS基因:其中埃坡霉素PKS第二延伸组件的AT结构域编码序列已被改变或替换,从而将其编码的AT结构域由甲基丙二酸单酰基特异的AT替换为丙二酸单酰基特异的AT。可分离此类编码丙二酸单酰基特异的AT结构域的核酸,例如且不限于,从编码narbonolide PKS、soraphen PKS、雷帕霉素PKS(即延伸组件2和12)和FK520 PKS(即延伸组件3、7和8)的PKS基因分离。当此杂合第二延伸组件与其它构成埃坡霉素PKS的蛋白质共表达时,最终生成的埃坡霉素衍生物为16-去甲基埃坡霉素。在一个实施方案中,杂合PKS也在延伸组件4包含一个甲基丙二酸单酰CoA特异的AT结构域,且在缺乏功能性epoK基因的宿主细胞中表达从而所产生的化合物为16-去甲基埃坡霉素D。在另一实施方案中,杂合PKS也包含已改变的苏氨酸特异NRPS,导致了5-甲基唑-16-去甲基埃坡霉素的生成。
此外,本发明提供了编码重组埃坡霉素PKS酶及相应重组蛋白质的DNA化合物、载体,其中如以下的实施例9中所述,第二(或随后的)延伸组件已被失活或缺失。在一个优选的实施方案中,失活是通过将活性半胱氨酸位点的密码子变换为丙氨酸密码子得以实现。如以上所描述的NRPS组件的相应变体,所产生的重组埃坡霉素PKS酶不能产生埃坡霉素或埃坡霉素衍生物,除非提供可与重组PKS酶的其余结构域和组件结合并延伸的前体。示例的前体化合物在以下实施例10中描述。另外,可简单地将此类前体提供给仅表达epoD、epoE和epoF基因的宿主细胞。
埃坡霉素PKS的第三延伸组件包括KS、丙二酸单酰CoA特异的AT、KR和ACP。埃坡霉素第三延伸组件以一种蛋白质的形式表达,为epoD基因产物,该基因也包含组件4、5和6。为制备由于延伸组件3到6中任一延伸组件的改变而产生埃坡霉素衍生物的重组埃坡霉素PKS,一般表达包含所有四个延伸组件的一种蛋白质。在一个实施方案中,全部或部分的第三延伸组件编码序列用于与其它PKS编码序列连接用以产生杂合组件。在此实施方案中,本发明提供了例如用甲基丙二酸单酰CoA、乙基丙二酸单酰CoA或2-羟基丙二酸单酰CoA特异的AT替换丙二酸单酰CoA特异的AT;缺失KR;用具有不同立体化学特异性的KR替换KR;和/或插入DH或DH和ER。所产生的异源第三延伸组件编码序列可用于与合成埃坡霉素、埃坡霉素衍生物或另一聚酮化合物的PKS编码序列连接。
本发明示例的重组PKS包括以下重组PKS基因:其中埃坡霉素PKS第三延伸组件的AT结构域编码序列已被改变或替换,从而将其编码的AT结构域由丙二酸单酰基特异的AT变为甲基丙二酸单酰特异的AT。可分离此类编码甲基丙二酸单酰基特异的AT结构域的核酸,例如且不限于,分离自编码DEBS、narbonolide PKS、雷帕霉素PKS和FK-520 PKS的PKS基因。当与埃坡霉素PKS或埃坡霉素PKS衍生物的剩余组件和蛋白质共表达时,重组PKS产生本发明的14-甲基埃坡霉素衍生物。
本领域技术人员将认识到:PKS第三延伸组件的KR结构域负责形成参与埃坡霉素环化的羟基。结果,去除第三延伸组件的KR结构域或添加DH或DH和ER结构域将妨碍环化,导致产生线性分子或产生在不同于埃坡霉素A、B、C、D、E和F的环化部位环化的分子。
埃坡霉素PKS的第四延伸组件包括KS、可结合丙二酸单酰CoA或甲基丙二酸单酰CoA的AT、KR和ACP。在一个实施方案中,全部或部分的第四延伸组件编码序列用于连接其它的PKS编码序列,以产生杂合组件。在此实施方案中,本发明提供了,例如用丙二酸单酰CoA、甲基丙二酸单酰CoA、乙基丙二酸单酰CoA或2-羟基丙二酸单酰CoA特异的AT替换丙二酸单酰CoA和甲基丙二酸单酰CoA特异的AT;缺失ER;和/或替换ER,其包括任选地替换为具不同立体化学特异性的ER;和/或插入DH或DH和ER。产生的异源第四延伸组件编码序列掺入到合成埃坡霉素、埃坡霉素衍生物或另一聚酮化合物的重组PKS的蛋白质亚单位。另外,本发明提供了埃坡霉素和埃坡霉素衍生物的重组PKS酶,该重组PKS酶的全部第四延伸组件已被缺失或被异源PKS组件替换。
在一个优选的实施方案中,本发明提供了包含埃坡霉素PKS第四延伸组件编码序列的重组DNA化合物,其中所述第四延伸组件编码序列已改变为编码结合甲基丙二酸单酰CoA而非丙二酸单酰CoA(或结合丙二酸单酰CoA而非甲基丙二酸单酰CoA)的AT。在一个实施方案中,该特异性的变化是通过突变延伸组件4的AT结构域的编码序列得以实现。此类突变可应用诱变因子(如紫外线),或通过定点诱变实现。在另一实施方案中,该特异性的变化是通过将全部或部分的延伸组件4 AT结构域的编码序列替换为异源AT结构域的编码序列而实现的。这样,本发明提供了重组DNA化合物、表达载体和相应的重组PKS,其中掺入了具有甲基丙二酸单酰基特异AT的杂合第四延伸组件。甲基丙二酸单酰基特异的AT编码序列可来自(例如且不限于):包含甲基丙二酸单酰基特异的AT结构域的oleandolide PKS、DEBS、narbonolide PKS、雷帕霉素PKS或任一其它PKS的编码序列。
按照本发明,此编码序列表达的杂合第四延伸组件可掺入到埃坡霉素PKS(或埃坡霉素衍生物的PKS)中,一般作为衍生的epoD基因产物,其包含已改变的第四延伸组件以及延伸组件3、5和6,其中的任一组件或多个组件可任选地为埃坡霉素PKS的衍生形式。本发明提供的甲基丙二酸单酰基特异的重组埃坡霉素第四延伸组件编码序列因此提供了在宿主细胞内产生所需埃坡霉素的其它方法。特别是,此类化合物将为埃坡霉素D、B和F或其衍生物,其中埃坡霉素B的产生依赖于功能epoK基因的存在。
本发明也提供了包含埃坡霉素PKS第四延伸组件编码序列的重组DNA化合物,其中所述第四延伸组件编码序列已改变为编码结合丙二酸单酰CoA而非甲基丙二酸单酰CoA的AT。本发明提供了重组DNA化合物、载体和相应的重组PKS,其中该杂合第四延伸组件已被掺入到衍生的epoD基因产物中。当掺入到埃坡霉素PKS(或埃坡霉素衍生物的PKS)时,所产生的重组埃坡霉素PKS产生埃坡霉素C、A和E,其中埃坡霉素A的产生依赖于是否存在功能epoK基因。
在另一实施方案中,本发明提供了用于产生12-去甲基-12-乙基-埃坡霉素D的重组宿主细胞。在该实施方案中,本发明提供了表达重组埃坡霉素PKS衍生物的宿主细胞,其中所述的PKS衍生物延伸组件4的AT结构域已替换为例如来自FK520或尼达霉素PKS酶的乙基丙二酸单酰CoA特异的延伸组件。在一个实施方案中,宿主细胞为表达巴豆酰基CoA还原酶的宿主,所述还原酶由来自异源宿主细胞的基因(ccr基因)编码或在异源启动子的控制下,以增加乙基丙二酸单酰CoA的产量。在一个实施方案中,宿主细胞为表达分离自链霉菌宿主细胞的ccr基因的粘球菌宿主细胞。在另一实施方案中,宿主细胞已被修饰,可表达或过表达大肠杆菌atoA、D和E基因,这些基因产物可转运丁酸酯并将其转化为丁酰CoA,然后丁酰CoA转化为乙基丙二酸单酰CoA。
除了将内源AT编码序列替换为甲基丙二酸单酰CoA特异的AT编码序列,也可将KR结构域编码序列替换为另一KR、DH和KR(例如但不限于来自雷帕霉素PKS的组件10或FK-520 PKS的组件1或5)或DH、KR和ER的编码序列。如果将KR替换为另一KR,或替换成KR和DH,且在延伸组件5(或PKS的其它部位)中没有变化,则重组埃坡霉素PKS产生埃坡霉素C和D,因为延伸组件5的DH结构域介导埃坡霉素C和D中C-12-C-13双键的形成。如果将KR替换为KR、DH和ER,且延伸组件5(或PKS的其它部位)没有变化,则重组埃坡霉素产生12,13-二氢-埃坡霉素C和D。如果将KR用失活KR替换或使KR失活,则重组埃坡霉素PKS产生13-氧代-11,12-脱氢-埃坡霉素C和D。
这样,本发明提供了这样的重组埃坡霉素PKS,其延伸组件4的KR结构域已通过突变、缺失或用来自另一PKS的无功能KR结构域替换而失活。此重组PKS主要产生13-氧代-11,12-脱氢埃坡霉素B;该生物体产生的化合物中观察到的C-11-C-12双键被认为是来自新生聚酮化合物链中由延伸组件5的DH结构域所形成的双键在延伸组件5的ER结构域还原之前双键的迁移。本发明也提供了产生该新聚酮化合物的宿主细胞。例如,黄色粘球菌菌株K122-56(该菌株根据布达佩斯条约于2000年11月21日保藏于美国典型培养物保藏中心(ATCC),10801 University Blvd.,马纳萨斯,VA,20110-2209美国,且可在保藏号PTA 2714下得到)包含埃坡霉素PKS基因,其中组件4的KR结构域已通过缺失而失活,且其产生13-氧代埃坡霉素A和B以及它们的脱氢衍生物(主要为13-氧代-11,12脱氢埃坡霉素B)。本发明也提供了由该菌株产生的新埃坡霉素衍生物。
埃坡霉素PKS的第五延伸组件包括KS、结合丙二酸单酰CoA的AT、DH、ER、KR和ACP结构域。在一个实施方案中,将包含编码埃坡霉素PKS第五延伸组件的序列的DNA化合物插入到包含编码埃坡霉素PKS和产生埃坡霉素衍生物的重组埃坡霉素PKS的序列的DNA化合物中。在另一实施方案中,第五延伸组件编码序列的一部分用于连接其它PKS编码序列,以产生杂合组件编码序列及由其编码的杂合组件。在此实施方案中,本发明提供了例如用甲基丙二酸单酰CoA、乙基丙二酸单酰CoA或2-羟基丙二酸单酰CoA特异的AT替换丙二酸单酰CoA特异的AT;缺失ER、DH和KR中的任何一个、两个或全部的三个;和/或用KR或DH和KR、KR、DH和ER(任选地包括具有不同立体化学特异性)替换该ER、DH和KR中的任何一个、两个或全部的三个。所产生的杂合第五延伸组件编码序列可用于连接合成埃坡霉素、埃坡霉素衍生物或另一聚酮化合物的PKS的编码序列。另外,埃坡霉素PKS的第五延伸组件可被全部缺失或用异源PKS组件替换,以产生这样的蛋白质,其与埃坡霉素PKS或其衍生物的其它蛋白质一起构成产生埃坡霉素衍生物的PKS。
本发明示例的重组PKS基因包括重组epoD基因衍生物,其中埃坡霉素PKS的第五延伸组件的AT结构域编码序列已被改变或替换,以将其编码的AT结构域由丙二酸单酰特异的AT改变为甲基丙二酸单酰基特异的AT。此甲基丙二酸单酰基特异的基AT结构域编码核酸可分离自,例如且不限于编码DEBS、narbonolide PKS、雷帕霉素PKS和FK-520 PKS的PKS基因。当此重组epoD基因衍生物与epoA、epoB、epoC、epoE、epoF和/或epoK基因(或它们的衍生物)共表达时,由它们组成的PKS产生10-甲基埃坡霉素或其衍生物。本发明提供的另一重组epoD基因衍生物不仅包括这种改变的组件5编码序列,还包括编码仅结合甲基丙二酸单酰CoA的AT结构域的组件4编码序列。当重组epoD基因衍生物与epoA、epoB、epoC、epoE、epoF和/或epoK基因一同掺入到PKS时,重组epoD基因衍生物产物导致10-甲基埃坡霉素B和/或D衍生物的产生。
本发明的其它示例的重组epoD基因衍生物包括:埃坡霉素PKS的第五延伸组件的一个或多个ER、DH和KR结构域编码序列已被替换或突变以提供:(i)无功能的ER、DH或KR结构域;(ii)仅一个功能的KR结构域;(iii)仅KR和DH结构域有功能;或(iv)来自另一PKS的功能ER、DH或KR结构域。发现延伸组件5的DH结构域负责形成埃坡霉素C和D中的C-12-C-13双键,此发现提供了一种用于在任一生物体中制备埃坡霉素和埃坡霉素衍生物的本发明的新方法,其中所述生物体包括含埃坡霉素PKS基因的纤维堆囊菌和重组宿主细胞。此外,目前已发现延伸组件6的DH结构域也可对结合在前述组件上的新生聚酮化合物的β-羰基起作用,可根据本发明的方法利用该发现以制备新埃坡霉素衍生物。
这样,当延伸组件5的所有三个结构域KR、DH和ER均缺失或失活时,重组埃坡霉素PKS产生埃坡霉素A和B的13-羟基-11-氧代类似物。当DH和ER结构域缺失或失活时,重组埃坡霉素PKS产生13-羟基-10,11-脱氢-埃坡霉素类,主要为13-羟基-10,11-脱氢-埃坡霉素D。本发明也提供了产生此新聚酮化合物的宿主细胞。例如,黄色粘球菌菌株K122-148(该菌株根据布达佩斯条约于2000年11月21日保藏于美国典型培养物保藏中心(ATCC),10801 University Blvd.,马纳萨斯,VA,20110-2209美国,且可在保藏号PTA-2711下获得)包含这样的埃坡霉素PKS基因,其中的延伸组件5的DH、KR和ER结构域被替换为仅一个KR结构域,且其产生13-羟基-10,11-脱氢-埃坡霉素D。本发明也提供了由该菌株产生的新埃坡霉素衍生物。只有当ER结构域缺失或失活时,重组埃坡霉素PKS产生埃坡霉素C和D的10,11-脱氢类似物,主要为10,11-脱氢埃坡霉素。这样,一方面本发明提供了这样的重组埃坡霉素PKS,其中延伸组件5的ER结构域通过突变缺失或失活,且其产生10,11-脱氢-埃坡霉素D。在另一实施方案中,本发明提供了产生10,11-脱氢-埃坡霉素D的纤维堆囊菌宿主细胞,其中由于埃坡霉素PKS延伸组件5的ER结构域的编码序列突变而产生10,11-脱氢-埃坡霉素D。
本发明的这些重组epoD基因衍生物与epoA、epoB、epoC、epoE和epoF基因或与含其它改变(且可本身含有其它变化)的重组epo基因共表达,以产生制备相应埃坡霉素衍生物的PKS。例如,本发明提供的一种重组epoD基因衍生物也包括编码仅结合甲基丙二酸单酰CoA的AT结构域的组件4编码序列。如前所示,用于产生埃坡霉素C-11衍生物的功能类似的epoD基因也可通过将埃坡霉素第五延伸组件的ER、DH和KR结构域中的一个、两个或全部三个失活而得以产生。用于改变任一组件中此类结构域的另一方式是通过与取自同一或异源PKS编码序列的另一组件的一整套所需结构域进行替换。此方法中,PKS的天然结构得以保留。同样,如果存在,在天然PKS中共同起作用的KR和DH或者KR、DH和ER结构域优选地用于重组PKS。以上描述用于替换的示例替换结构域包括:例如且不限于,来自雷帕霉素PKS延伸组件3的失活KR结构域,来自雷帕霉素PKS延伸组件5的KR结构域;以及来自雷帕霉素PKS延伸组件4的KR和DH结构域。编码其它此类失活KR、活性KR以及活性KR和DH结构域的核酸可分离自:例如且不限于,编码DEBS、narbonolide PKS和FK-520 PKS的PKS基因。每一种产生的PKS酶产生一种聚酮化合物,后者可经标准的化学方法在体外进一步衍生,以产生本发明的半合成埃坡霉素衍生物。
埃坡霉素PKS的第六延伸组件包括KS、结合甲基丙二酸单酰CoA的AT、DH、ER、KR和ACP。在一个实施方案中,第六延伸组件编码序列的一部分用于连接其它PKS编码序列,以产生杂合组件。在此实施方案中,本发明提供了例如将甲基丙二酸单酰CoA特异的AT替换为丙二酸单酰CoA、乙基丙二酸单酰CoA或2-羟基丙二酸单酰CoA特异的AT;缺失ER、DH和KR中的一个、两个或全部三个;和/或将ER、DH和KR中的一个、两个或全部三个替换为KR或DH和KR或KR、DH和ER(任选地包括具有不同立体化学特异性)。所产生的异源第六延伸组件编码序列可用于与制备埃坡霉素、埃坡霉素衍生物或另一聚酮化合物的PKS的蛋白质亚单位的编码序列连接。另外,埃坡霉素PKS的第六延伸组件可全部被缺失或用异源PKS组件全部替换,以产生埃坡霉素衍生物的PKS。
本发明的示例的重组PKS基因包括:埃坡霉素PKS的第六延伸组件的AT结构域编码序列已被改变或替换,以将其编码的AT结构域由甲基丙二酸单酰基特异的AT替换为丙二酸单酰基特异的AT。编码此丙二酸单酰基特异的AT结构域的核酸可分离自例如且不限于编码narbonolide PKS、雷帕霉素PKS和FK-520 PKS的PKS基因。当编码此杂合组件6的本发明的重组epoD基因与其它埃坡霉素PKS基因共表达时,该重组PKS产生8-去甲基埃坡霉素衍生物。该重组epoD基因衍生物也可与含其它变化的重组epo基因衍生物共表达或可自身进一步改变,以生成制备相应8-去甲基埃坡霉素衍生物的PKS。例如,本发明提供的一种重组也epoD基因包括编码仅结合甲基丙二酸单酰CoA的AT结构域的组件4编码序列。当与epoA、epoB、epoC、epoE和epoF基因掺入到PKS时,重组epoD基因产物导致埃坡霉素B(如果存在功能epoK基因)和D的8-去甲基衍生物的产生。
本发明的其它示例的重组epoD基因衍生物包括:埃坡霉素PKS第六延伸组件的ER、DH和KR结构域编码序列被替换为编码(i)KR和DH结构域;(ii)KR结构域;和(iii)失活KR结构域的编码序列。当本发明的这些重组epoD基因衍生物与其它埃坡霉素PKS基因共表达时,生成了相应的(i)C-9烯烃、(ii)C-9羟基(两种差向异构体,仅其中之一可经下游组件处理,除非在下一个组件中进行了额外的KS和/或ACP替换)和(iii)C-9酮(C-9-氧代)埃坡霉素衍生物。也可通过失活埃坡霉素第六延伸组件的ER、DH和ER结构域中的一个、两个或全部三个来制备功能等效的第六延伸组件。例如,本发明提供了黄色粘球菌菌株K39-164(该菌株根据布达佩斯条约于2000年11月21日保藏于美国典型培养物保藏中心(ATCC),10801 University Blvd.,马纳萨斯,VA,20110-2209美国,且可在保藏号PTA-2716下获得),该菌株含有这样的埃坡霉素PKS基因,其中延伸组件6的KR结构域已通过突变而失活,且其产生9-酮-埃坡霉素D。本发明也提供了由该菌株产生的新埃坡霉素衍生物。
这样,重组epoD基因衍生物也可与其它含其它变化的epo基因衍生物共表达或它们自身可进一步改变,以产生可制备相应C-9埃坡霉素衍生物的PKS。例如,本发明提供的一种重组epoD基因衍生物也包括编码仅结合甲基丙二酸单酰CoA的AT结构域的组件4编码序列。当与epoA、epoB、epoC、epoE和epoF基因掺入到PKS时,重组epoD基因产物导致了埃坡霉素B和D的C-9衍生物的产生,这取决于是否存在功能epoK基因。
以上描述的用于替换的示例性替换结构域包括但不限于,来自雷帕霉素PKS组件3的失活KR结构域以形成酮,来自雷帕霉素PKS组件5的KR结构域以形成醇,以及来自雷帕霉素PKS组件4的KR和DH结构域形成了烯。其它此类失活KR、活性KR及活性KR和DH结构域编码核酸可分离自,例如且不限于,编码DEBS、narbonolide PKS和FK-520 PKS的PKS基因。每一种产生的PKS酶产生一种聚酮化合物,后者在其C-9位包含功能基团,且可经标准的化学方法在体外进一步衍生,以产生本发明的半合成埃坡霉素衍生物。
埃坡霉素PKS的第七延伸组件包括KS、甲基丙二酸单酰CoA特异的AT、KR和ACP。埃坡霉素PKS的第七延伸组件包含在epoE基因的基因产物内,该epoE基因也包含第八延伸组件。在一个实施方案中,表达包含编码埃坡霉素PKS第七延伸组件的序列的DNA化合物并形成这样的蛋白质,其与其它蛋白质一起构成了埃坡霉素PKS或产生埃坡霉素衍生物的PKS。在这些及相关的实施方案中,埃坡霉素PKS或其衍生物的第七和第八延伸组件一般表达为单个蛋白质,并与epoA、epoB、epoC、epoD和epoF基因或其衍生物共表达以构成PKS。在另一实施方案中,第七延伸组件编码序列的部分或全部用于与其它PKS编码序列连接,以产生杂合组件。在此实施方案中,本发明提供了例如用丙二酸单酰CoA、乙基丙二酸单酰CoA或2-羟基丙二酸单酰CoA特异的AT替换甲基丙二酸单酰CoA特异的AT;缺失KR;替换KR为对不同立体化学特异的KR;和/或插入DH或DH和ER。所产生的异源第七延伸组件编码序列用于任选地与其它编码序列连接,以表达蛋白质,表达蛋白质与其它蛋白质一起构成合成埃坡霉素、埃坡霉素衍生物或另一聚酮化合物的PKS。另外,epoE基因中第七延伸组件的编码序列可被缺失或被异源组件的编码序列替换,以制备重组epoE基因衍生物,后者与epoA、epoB、epoC、epoD和epoF基因一起进行表达以制备埃坡霉素衍生物的PKS。
本发明示例的重组epoE基因衍生物包括,其中埃坡霉素PKS第七延伸组件的AT结构域编码序列被改变或替换,以将其编码的AT结构域由甲基丙二酸单酰特异的AT替换为丙二酸单酰特异的AT。此类丙二酸单酰基特异的AT结构域编码核酸可分离自,例如且不限于,编码narbonolidePKS、雷帕霉素PKS和FK-520 PKS的PKS基因。当与其它埃坡霉素PKS基因(epoA、epoB、epoC、epoD和epoF或它们的衍生物)共表达时,产生用于具有C-6氢而非C-6甲基的埃坡霉素衍生物的PKS。这样,如果基因不含其它变化,产生的化合物为6-去甲基埃坡霉素。
埃坡霉素PKS第八延伸组件包括KS、甲基丙二酸单酰CoA特异的AT、失活KR和DH结构域、甲基转移酶(MT)结构域和ACP。在一个实施方案中,包含编码埃坡霉素PKS第八延伸组件序列的DNA化合物与其它构成埃坡霉素PKS或产生埃坡霉素衍生物的PKS的蛋白质共表达。在另一实施方案中,第八延伸组件编码序列的部分或全部用于与其它PKS编码序列连接,以产生杂合组件。在此实施方案中,本发明提供了例如将甲基丙二酸单酰CoA特异的AT替换为丙二酸单酰CoA、乙基丙二酸单酰CoA或2-羟基丙二酸单酰CoA特异的AT;缺失失活KR和/或失活DH;将失活KR和/或DH替换为活性KR和/或DH;和/或插入ER。所产生的异源第八延伸组件编码序列作为一个蛋白质进行表达,表达的蛋白质用于与其它构成合成埃坡霉素、埃坡霉素衍生物或另一聚酮化合物的PKS的蛋白质连接。另外,epoE基因中第八延伸组件的编码序列可被缺失或被异源组件的编码序列取代以制备重组epoE基因,该epoE基因与epoA、epoB、epoC、epoD和epoF基因一同表达以制备产生埃坡霉素衍生物的PKS。
埃坡霉素PKS第八延伸组件也包含具有活性的甲基化或甲基转移酶(MT)结构域,该结构域可使埃坡霉素的前体甲基化。该功能可被缺失以产生本发明的重组epoD基因衍生物,后者可与其它埃坡霉素PKS基因或其衍生物一同表达,这些基因产生的埃坡霉素衍生物在相应的埃坡霉素分子的C-4位缺少一个或两个甲基,这取决于第八延伸组件的AT结构域是否被改变为丙二酸单酰基特异的AT结构域。
埃坡霉素PKS第九延伸组件包括KS、丙二酸单酰CoA特异的AT、KR、失活的DH和ACP。埃坡霉素PKS第九延伸组件作为一个蛋白质表达,为epoF基因产物,其也含有埃坡霉素PKS的TE结构域。在一个实施方案中,包含编码埃坡霉素PKS第九延伸组件序列的DNA化合物被以一个蛋白质表达,该表达的蛋白质与其它蛋白质一起构成埃坡霉素PKS或产生埃坡霉素衍生物的PKS。在这些实施方案中,第九延伸组件一般表达为也包含埃坡霉素PKS或异源PKS TE结构域的蛋白质。在另一实施方案中,第九延伸组件编码序列的部分或全部编码序列用于与其它PKS编码序列连接以产生杂合组件。在此实施方案中,本发明提供了例如用甲基丙二酸单酰CoA、乙基丙二酸单酰CoA或2-羟基丙二酸单酰CoA特异的AT替换丙二酸单酰CoA特异的AT;缺失KR;替换KR为对不同立体化学特异的KR;和/或插入DH或DH和ER。例如,将延伸组件9的AT结构域替换为对甲基丙二酸单酰CoA特异的AT结构域导致生成了这样的重组埃坡霉素PKS,其在本发明的重组粘球菌宿主细胞中产生2-甲基-埃坡霉素A、B、C和D。所产生的异源第九延伸组件编码序列与其它蛋白质共表达,构成了合成埃坡霉素、埃坡霉素衍生物或另一聚酮化合物的PKS。另外,本发明提供了这样的用于合成埃坡霉素或埃坡霉素衍生物PKS,其第九延伸组件被异源PKS的组件替换或全部缺失。在后一实施方案中,TE结构域作为独立蛋白质表达或与第八延伸组件融合表达。
在另一实施方案中,本发明提供了这样的本发明宿主,其中包含异源PKS基因簇(该PKS基因簇不存在于相同类型的未加修饰、天然存在的宿主细胞)以及编码硫酯酶II型蛋白质(“TE II”)的基因。在一个优选的实施方案中,TE II基因异源于PKS基因簇-TE II基因并非来源于同一PKS的基因簇。作为一个实例,在一个实施方案中的本发明重组宿主细胞包含编码表达埃坡霉素PKS或埃坡霉素PKS衍生物的基因。根据本发明的此方面,宿主细胞被改变成含有这样的TE II基因,基分离自除埃坡霉素PKS基因簇外的PKS基因簇。示例的实施方案包括例如来自委内瑞拉链霉菌苦霉素PKS基因簇的TE II基因和来自纤维堆囊菌tmbA PKS基因簇(该PKS基因簇在美国专利号6,090,601;美国专利申请系列号144,085,于1998年8月31日申请;和美国临时专利申请系列号60/271,245,2001年2月15日申请,均在此引用作为参考)的TE II基因。
本发明的重组埃坡霉素衍生物的PKS基因通过列出杂合组件的改变的特异性(其它组件具有与埃坡霉素PKS相同的特异性)得以鉴定,这些PKS基因的示例包括:
(a)组件4带有甲基丙二酸单酰特异的AT(mmAT)和KR,且组件2带有丙二酸单酰特异的AT(mAT)和KR;
(b)组件4带有mmAT且组件3带有mmAT;
(c)组件4带有mmAT且组件5带有mmAT;
(d)组件4带有mmAT,且组件5带有mmAT并仅含一个DH和KR;
(e)组件4带有mmAT,且组件5带有mmAT并仅含一个KR;
(f)组件4带有mmAT,且组件5带有mmAT并仅含失活KR;
(g)组件4带有mmAT且组件6带有mAT;
(h)组件4带有mmAT,且组件6带有mAT并仅含一DH和KR;
(i)组件4带有mmAT,且组件6带有mAT且仅含一个KR;
(j)组件4带有mmAT,且组件6带有mAT并且仅含一失活KR;
(k)组件4带有mmAT,且组件7带有mAT;
(l)除了组件5含mAT外的(d)至(f)的杂合体;
(m)除了组件6含mmAT外的(h)至(j)的杂合体;以及
(n)除了组件4含mAT外的(a)至(m)的杂合体。
上面所列出的仅为例证性的,并不构成对本发明的限制,本发明包括其它重组埃坡霉素PKS基因和酶,它们不仅为非所示的那些带有两个杂合组件,还可带有三个或更多的杂合组件。
本发明的宿主细胞可在本领域公知的用于其它目的的条件下生长和发酵,以用来产生本发明的化合物。本发明也提供了发酵本发明的宿主细胞的新方法。本发明的化合物可分离自这些培养细胞的发酵培养基,并通过如以下实施例3中的方法纯化。
本发明提供了有关发酵用于产生聚酮化合物和其它产物的粘球菌的一系列方法。在本发明之前,对粘球菌的发酵并不用于生产除TA和番红霉素(二者由某些粘球菌菌株天然产生)之外的其它任何聚酮化合物的生产目的。这样,一方面,本发明使得可应用粘球菌作为生产宿主用于通过发酵产生有用的生物活性化合物,包括但不限于,聚酮化合物、非核糖体肽类、埃坡霉素类、脂肪酶、蛋白酶、其它蛋白质、脂类、糖脂、鼠李糖脂和聚羟基链烷酸酯。
本发明提供的方法中包括制备和储存细胞库的方法和使粘球菌菌株适应发酵培养基的方法。这些方法是重要的,因为在本发明之前,尚没有制备出适应在基于油的发酵培养基中产生的冷冻粘球菌菌株细胞库,且缺乏适应性时,粘球菌菌株往往会死亡,特别是在基于油的发酵培养基中。
本发明也提供了用于生长粘球菌的发酵方法及该方法中有用的发酵培养基。令人惊讶的是,黄色粘球菌和其它粘球菌菌株不能将碳水化合物、甘油、醇或TCA循环中间体作为碳源加以利用。在本发明之前,黄色粘球菌的发酵在基于蛋白质的培养基中进行。但是,在基于蛋白质的培养基中NH4累积到对生长有毒性的水平,且因此限制发酵。根据本发明,粘球菌菌株在含有作为碳源的油和/或脂肪酸的培养基中进行发酵。
在方法中有用的油和脂肪酸的示例包括且不限于,油酸甲酯;来自椰子、猪油、油菜籽、芝麻、大豆和向日葵的油类;色拉油;自身乳化的油类(如Agrimul CoS2、R505和R503);甘油油酸酯,包括甘油单油酸酯和甘油三油酸酯;奇数链的酯如十七烷酸甲酯、十九烷酸甲酯和壬酸甲酯;酯链如油酸丙酯和油酸乙酯;植物油酸甲酯;硬脂酸甲酯;亚油酸甲酯;油酸;以及纯的或来自大豆和卵黄的磷脂酰胆碱。这样,任何植物或谷类来源的油(如向日葵油或大豆油)、任何动物来源的油脂(如猪油)、任意链长的饱和或不饱和游离或酯化脂肪酸、天然和合成的脂肪酸混合物(分别如磷脂酰胆碱或壬酸甲酯)以及工业发酵的油类,如Cognis公司的Agrimul系列,均可用于方法中。在一个优选的实施方案中,发酵培养基用油酸甲酯作为碳源。一般而言,室温下为流体的油类比固体油类更为优选,主要是由于易于分散。发酵培养基的其它重要组分包括痕量金属如Fe和Cu,二者在复合和特定的的培养基中以及在分批和补料分批法中提高生长和产量。包含油酸甲酯和痕量金属的培养基优选地用于埃坡霉素类的生产。
在一个实施方案中,本发明提供了用于本发明宿主细胞的发酵培养基,该培养基含有减量的或不含有动物来源的材料。由于潜在的被感染因子(诸如病毒和朊病毒)污染的可能,在发酵过程中应用动物副产物用于产生欲施与人或动物的化合物时,可优选使用含减量或不含动物来源材料的发酵培养基。提供了用于本发明方法的此类培养基。发酵培养基中含有的油类和脂肪酸可来自非动物源,例如植物。例如植物来源的油酸甲酯可由商业途径获得。此外,可将动物来源的材料替换为等效的但来自非动物源的不同材料。例如,酪胨,为酪蛋白(一种乳蛋白)的胰消化物,可用来自非动物源的蛋白质的水解产物替代,包括且不限于植物,如植物来源的蛋白质水解物。
一般而言,补料分批发酵优选地用于发酵。补料促使细胞有效地应用营养物(例如,细胞将碳代谢为CO2和H2O而不是产生有毒的有机酸)。高营养水平可抑制次级代谢,且如果发酵以低于抑制阈值的速率馈与营养物,产量可更高。
本发明的发酵方法也包括具体地与埃坡霉素产生相关的方法以及用于这些方法的发酵培养基。作为一个实例,丙酸酯和乙酸酯可用于影响埃坡霉素D∶C(或B∶A)的比率以及所获埃坡霉素的滴度。此效应在优选的油酸甲酯/痕量金属发酵培养基中是很小的,但在其它培养基如CTS培养基中的效应却相当显著。增加在发酵培养基中乙酸酯的含量可增加粘球菌的生长和埃坡霉素的生成。乙酸酯单独可显著提高埃坡霉素C(或埃坡霉素A)的滴度,且降低埃坡霉素D(或埃坡霉素A)的滴度。丙酸酯单独不能提高埃坡霉素的滴度,且在高浓度时可降低滴度。但是,丙酸酯和乙酸酯一起可使产物由埃坡霉素C(或埃坡霉素A)变为埃坡霉素D(或埃坡霉素B)。用于产生埃坡霉素D的一个优选的培养基包含酪胨、10mM乙酸酯和30mM丙酸酯。用含奇数链脂肪酸的培养基发酵产生埃坡霉素C和D的黄色粘球菌细胞时,可减少埃坡霉素C的产量。痕量金属也可在存在乙酸酯而不存在任何油类的发酵培养基中增加埃坡霉素D的产量且提高埃坡霉素D∶C的比率。
本发明也包含用于从发酵培养基中纯化埃坡霉素类的方法以及用于制备晶体形式埃坡霉素的方法。一般而言,纯化方法包括在发酵中将埃坡霉素捕获到XAD树脂、从树脂洗脱、固相提取、层析和结晶。方法在实施例3中详述,虽然该方法优选地且作为示例用于埃坡霉素D,该方法通常也可用于制备结晶的埃坡霉素类,后者包括但不限于其它天然存在的埃坡霉素类和由本发明宿主细胞产生的埃坡霉素类似物。
这样,在另一实施方案中,本发明以分离的或纯化的形式提供了新埃坡霉素衍生物化合物,用于农业、兽医和医学。在一个实施方案中,化合物用作杀真菌剂。在另一实施方案中,化合物用于癌症的化学治疗。在另一实施方案中,化合物用于阻止不需要的细胞生长,包括但不限于治疗过度增生性疾病如炎症、自身免疫病和银屑病,以及防止细胞在斯坦特固定模上的生长。在一个优选的实施方案中,化合物为在抗肿瘤细胞方面至少与埃坡霉素B和D一样高效的埃坡霉素衍生物。在另一实施方案中,化合物用作免疫抑制剂。在另一实施方案中,化合物用于制造另一化合物。在一个优选的实施方案中,化合物以被制剂为混合物或溶液,用于施与人或动物。
本发明的新埃坡霉素类似物,以及由本发明宿主细胞产生的埃坡霉素类,可如在PCT专利公开号93/10121,97/19086,98/08849,98/22461,98/25929,99/01 124,99/02514,99/07692,99/27890,99/39694,99/40047,99/42602,99/43320,99/43653,99/54318,99/54319,99/54330,99/65913,99/67252,99/67253和00/00485,以及在美国专利号5,969,145中所述进行衍生和制剂,它们在此引用作为参考。
本发明的化合物包括14-甲基埃坡霉素衍生物(通过利用本发明的杂合组件3而得以生成,其中所述杂合组件3具有结合甲基丙二酸单酰CoA而不是丙二酸单酰CoA的AT);8,9-脱氢埃坡霉素衍生物(通过利用本发明的具有DH和KR而不是ER、DH和KR的杂合组件6得以生成);10-甲基埃坡霉素衍生物(通过利用本发明具有结合甲基丙二酸单酰CoA而不是丙二酸单酰CoA的杂合组件5得以生成);9-羟基埃坡霉素衍生物(通过利用本发明具有KR而不是ER、DH和KR的杂合组件6得以生成);8-去甲基-14-甲基-埃坡霉素衍生物(通过利用本发明的具有结合甲基丙二酸单酰CoA而非丙二酸单酰CoA的杂合组件3以及结合丙二酸单酰CoA而非甲基丙二酸单酰CoA的杂合组件6得以生成);8-去甲基-8,9-脱氢埃坡霉素衍生物(通过利用本发明具有DH和KR而非ER、DH和KR且具有对丙二酸单酰CoA特异而非对甲基丙二酸单酰CoA特异的AT的杂合组件6得以生成);以及9-氧代-埃坡霉素D。本发明的其它优选的新埃坡霉素包括以下及在实施例11中所描述的。
本发明的一方面,提供了下式的化合物:
其中:
R1、R2、R3、R5、R11和R12每一个独立地为氢、甲基或乙基;
R4、R6和R9每一个独立地为氢、羟基或氧;或者,
R5和R6一起形成碳碳双键;
R7为氢、甲基或乙基;
R8和R10均为氢或一起形成碳碳双键或环氧化物;
Ar是芳基;且,
W是O或NR13,其中R13为氢、C1-C10脂族、芳基或烷基芳基。在另一实施方案中,提供了式I的化合物,其中R1、R2、R3、R4、R5、R6、R7、R8、R9、R10、R11、R12、R13、Ar和W为如前所述的,条件是R1、R4、R5、R6、R9和R11中的至少一个不为氢。
在另一实施方案中,提供了式I的化合物,其中R1、R2、R3和R11每一个独立地为氢或甲基;R4和R9每一个独立地为氢、羟基或氧;R5和R6均为氢或一起形成碳碳双键;R7和R12均为甲基;R8和R10均为氢或一起形成碳碳双键;Ar为杂芳基;且,W为O或NR13,其中R13为氢或C1-C5烷基,条件是R1、R4、R5、R6、R9和R11中的至少一个不为氢。
在本发明的另一方面中,提供了式II的化合物
R4、R6和R9每一个独立地为氢、羟基或氧;
R5、R11、R12每一个独立地为氢、甲基或乙基;或者
R5和R6一起形成碳碳双键;
R7为氢、甲基或乙基;
R8和R10均为氢或一起形成碳碳双键或环氧化物;
Ar为芳基;且,
W为O或NR13,其中R13为氢或C1-C5烷基。在另一实施方案中,提供了式II的化合物,其中R4、R5、R6、R7、R8、R9、R10、R12、R13、Ar和W为前面所描述的,条件是R4、R5、R6和R9中的至少一个不为氢。
在另一实施方案中,提供了式II的化合物,其中
R4和R9每一个独立地为氢、羟基或氧;
R5和R6每一个为氢或一起形成碳碳双键;
R7和R12均为甲基;
R8和R10均为氢或一起形成碳碳双键;
Ar为2-甲基-1,3-噻唑啉基、2-甲基-1,3-噁唑啉基、2-羟甲基-1,3-噻唑啉基或2-羟甲基-1,3-噁唑啉基;且,
W为O或NH,条件是R4、R5、R6和R9中的至少一个不是氢。
在本发明的另一方面中,提供了式III的化合物
其中R1、R2、R3、R5、R11、R12每一个独立地为氢、甲基或乙基;
R6为氢;或者,
R5和R6一起形成碳碳双键;
R7为氢、甲基或乙基;
Ar为芳基;且,
W为O或NR13,其中R13为氢或C1-C5烷基。在另一实施方案中,提供了式III的化合物,其中R1、R2、R3、R5、R6、R7、R11、R12、R13、Ar和W为如先前描述的,条件是R1、R5、R6和R11中的至少一个不是氢。
在另一实施方案中,提供了式III的化合物,其中
R1、R2、R3、R11每一个独立地为氢、甲基或乙基;
R5和R6均为氢或一起形成碳碳双键;
R7和R12均为甲基;
Ar为2-甲基-1,3-噻唑啉基、2-甲基-1,3-噁唑啉基、2-羟甲基-1,3-噻唑啉基或2-羟甲基-1,3-噁唑啉基;且,W为O或NH,条件是R1、R5、R6和R11中的至少一个不为氢。
在本发明的另一方面中,提供了式IV的化合物

其中
R4为氢或氧;
R5和R6均为氢或一起形成碳碳双键;
R7为氢或甲基;R9为氢或羟基;
R8和R10均为氢或一起形成碳碳双键或环氧化物;
W为O或NH;
X为O或S;且
R14为甲基或羟甲基。
在本发明的另一方面中,化合物属于式IV的化合物

其中
R4为氢或氧;
R5和R7每一个独立地为氢或甲基;
R6为氢;
R8和R10均为氢或一起形成碳碳双键或环氧化物;或者
R6和R8一起形成双键;
R9为氢、羟基或氧;
W为O或NH;
X为O或S;且
R14为甲基或羟甲基。
在本发明的另一方面,提供了下列化合物:
和

本发明的化合物是细胞毒性剂,可以任何适合的方式应用,包括但不限于作为抗癌剂。用于评估细胞毒性程度和微管蛋白聚合的阐述性测试在实施例12中描述。
可用一系列方法制造本发明的化合物。在本发明的一方面中,化合物由表达埃坡霉素PKS的重组宿主细胞产生。在一个实施方案中,式V的化合物

(其中R1到R12如先前在式I中所述)通过在一个或多个组件改变AT的特异性和/或在一个或多个组件改变酶活性结构域而制备。实施例11描述了改变的种类以及可由此方法生成的特定化合物。
在本发明另一实施方案中,式V的噁唑对应物可通过调整宿主细胞的发酵条件得以生成,其中所述宿主细胞正常条件下产生式V的化合物。式V化合物的噻唑部分源自在NRPS上结合半胱氨酸。埃坡霉素H1和H2为埃坡霉素C和D的噁唑对应物,由宿主细胞以痕量生成,且被认为是由于偶尔的丝氨酸而非半胱氨酸结合到埃坡霉素NRPS得以生成。
本方法利用了在埃坡霉素PKS的NRPS结合部位丝氨酸与半胱氨酸的明显竞争,且应用利于在埃坡霉素NRPS上结合丝氨酸而不是半胱氨酸的发酵条件。已发现在补充丝氨酸(例如高于基础水平50倍)的培养基中培养宿主细胞导致主要生成了含噁唑的化合物,而不是通常产生的含噻唑的化合物。结果,设计为制备特定埃坡霉素化合物或式V化合物的重组宿主细胞可在补充丝氨酸的培养基中生长,以使这些同样的细胞现在有利于产生噁唑对应物即式VI的化合物:

换言之,本方法是一个简单和巧妙的付出一次代价获得两种化合物的途径,所述化合物一种对应于式V,且它的对应物对应于式VI。补充丝氨酸用于制备含噁唑化合物的方法在实施例13中更为详细地进行了描述。该实施例描述了用于降低由K111-40-1菌株正常产生的埃坡霉素D和C的水平以利于分别生成埃坡霉素H2和H1以及埃坡霉素D和C的噁唑对应物的条件,产生本发明的其它式V化合物的其它重组构建体可应用制备式VI化合物的类似条件进行培养。
在本发明的另一方面,可用称为化学生物合成法的方法生产化合物。该方法应用的埃坡霉素PKS已经以某种方式发生了改变,使得PKS在指定部位接受并结合一种合成的前体。该合成前体然后由PKS以常规方式从该结合点向前加工。
化学生物合成法所需的改变类型的示例在实施例9中描述,该实施例描述了一株黄色粘球菌菌株的KS2敲除变体的构建,其中所述黄色粘球菌菌株通常以埃坡霉素A、B、C和D作为主产物生成。KS2敲除是指失活延伸组件2的KS结构域,以使产生的PKS不能装载和处理先前组件、装载结构域和NRPS(认为是延伸组件1)的产物。结果,PKS指导的合成在延伸组件2的ACP处停止,且该菌株在缺少合成前体时不能生成埃坡霉素产物。但是,当提供给菌株合成前体时,该前体物与装载结构域和延伸组件1的产物类似,这样延伸组件2的ACP结合前体且PKS从该点向前加工前体。例如,提供给实施例9中的敲除菌株以合成前体

导致生成了埃坡霉素B和D(埃坡霉素A和B也生成但以痕量生成),如在实施例10中更详细描述的。也见图1。在另外的实施例中,提供给实施例9中的敲除菌株以合成前体,例如,
其中R为氢、羟基、卤素、氨基、C1-C5烷基、C1-C5羟基烷基、C1-C5烷氧基以及C1-C5氨基烷基,更优选地为氢或甲基,得到了下列埃坡霉素化
合物

和

以及它们分别的12,13-环氧对应物。
这样,通过改变合成前体,单一的KS2敲除菌株可用于制备大量的化合物。实际上,在实施例9中描述的菌株可通过提供下式的合成前体
来制备下式的化合物
和

其中Ar为芳基且R7为氢或甲基。合适的芳基的示例包括但不限于
其中R为氢、羟基、卤素、氨基、C1-C5烷基、C1-C5羟基烷基、C1-C5烷氧基、C1-C5氨基烷基。在更优选的实施方案中,R为氢或甲基。实施例10描述了个多种前体的合成以及它们在化学生物合成中的用途,及它们分别的12,13-环氧对应物。
在另一实施方案中,装载结构域组件敲除用于生成本发明的某些化合物。例如,用于实施例9中的起始材料的装载结构域敲除也可通过提供下式的合成前体用于制备式VII和VIII的化合物。
在其它实施方案中,制备了本发明的其它菌株的KS2或装载结构域敲除体,其中所述敲除体包括但不限于那些在实施例11中描述的菌株,且用于产生带有除2-甲基噻唑外的芳基部分的化合物。例如,将式IX的合成前体馈与一种主要产生9-氧代-埃坡霉素D构建体的KS2敲除体将产生下式的化合物
其中Ar为芳基。
在本发明的另一方面,从表达埃坡霉素PKS的宿主细胞产生的化合物可用生物法和/或合成法进一步修饰。在一个实施方案中,式I的化合物中Ar为

可用微生物来源的羟化酶在C-21碳处进行羟化。用于完成此转变的方法例如在PCT公开号WO 00/39276中描述(其全部并入本文作为参考)以及此处的实施例14中描述。
在另一实施方案中,在相应于埃坡霉素A-D的C-12和C-13位具有碳碳双键的本发明化合物可用EpoK或另一P450环氧酶环氧化。用EpoK进行环氧化作用的通用方法在PCT公开WO 00/31247的实施例5描述(其在此引用作为参考),以及此处的实施例15中描述。另外,环氧化反应可通过将含有双键的埃坡霉素化合物与表达功能Epo K的细胞培养物接触而进行,其中所述双键的位置对应于C-12和C-13之间的键。此类细胞包括粘球菌目纤维堆囊菌。在特别优选的实施方案中,纤维堆囊菌表达Epo K但不含有功能埃坡霉素聚酮化合物合酶(“PKS”)基因。此类菌株可通过诱变获得,其中在埃坡霉素PKS基因中的一处或多处突变使得菌株无效力。此类突变体可天然存在(可通过筛选发现)或可通过应用如化学诱变剂或射线或通过遗传操作来实现。用于制备带有无效埃坡霉素PKS的菌株的一个尤其有效的策略是同源重组,如在PCT公开WO00/31247中所描述的。
在另一实施方案中,环氧化反应可用合成进行法。例如,如在图解2中所示,本发明的脱氧化合物可通过将脱氧化合物与二甲基二氧杂环丙烷(dimethyldioxirane)反应而转变成环氧对应物。
图解2
实施例16更为详细地描述了该合成方法。
在另一实施方案中,本发明的大环内酯可转变为本发明的大环内酰胺。如在图解3中所述的,本发明的脱氧大环内酯如先前图解2中所描述的用二甲基二氧杂环丙烷环氧化以提供含氧对应物。
图解3
含氧大环内酯用叠氮化钠和四(三苯基膦)钯处理以打开环并形成叠氮酸。然后叠氮化物用三甲基膦还原以形成氨基羧酸。
本发明的W位置为NH的环氧化合物可由氨基羧酸的大环内酰胺化得以生成。
图解4
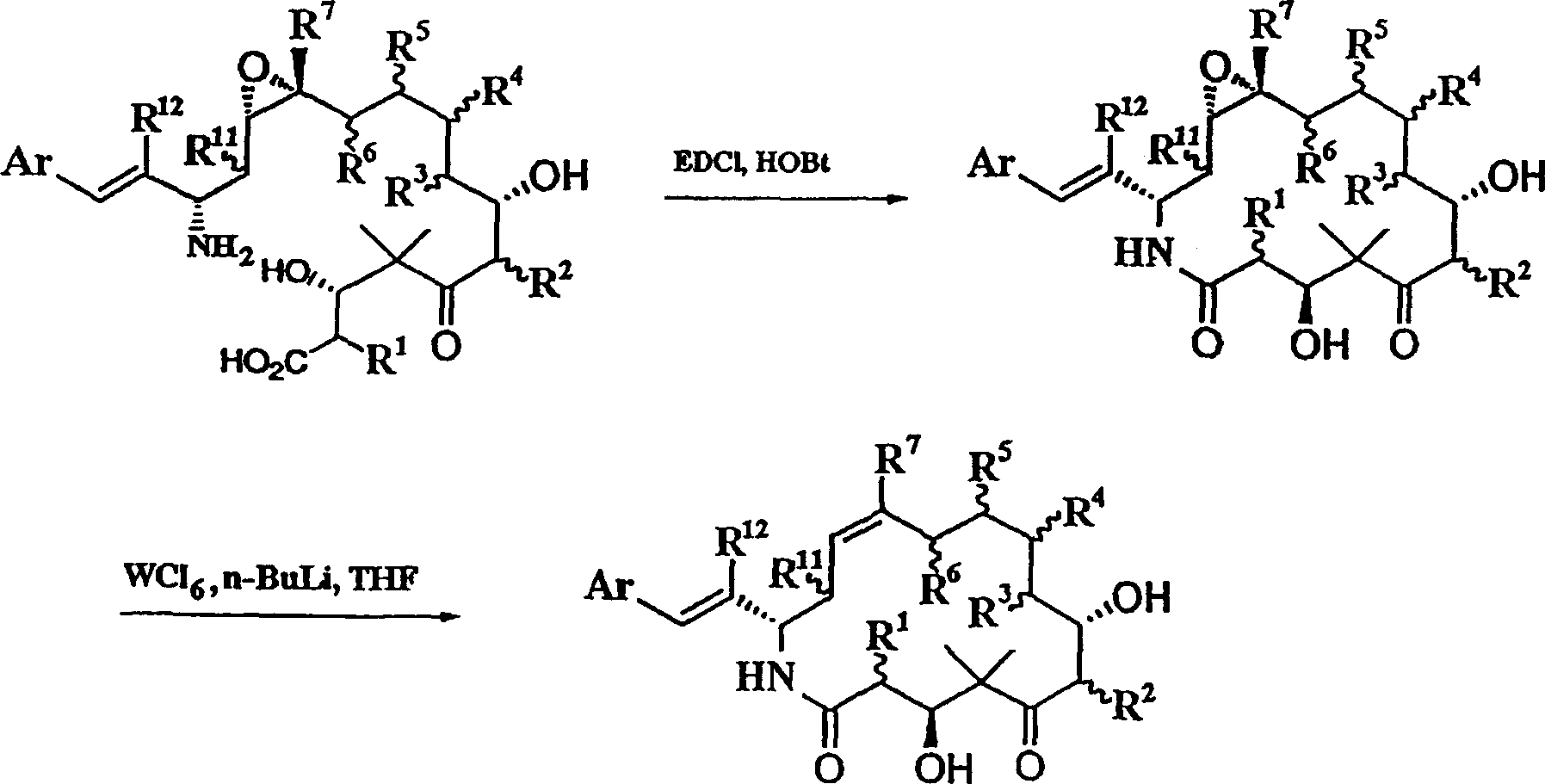
如图解4所示,氨基羧酸用1-(3-二甲氨基丙基)-3-乙基-碳二亚胺和1-羟基苯并三唑处理以形成环氧-大环内酰胺。脱氧-大环内酰胺可通过用六氯化钨和丁基锂处理环氧-大环内酰胺得以生成。
本发明的W位置为NR13且R13非氢的环氧-化合物可通过在大环内酰胺化之前用醛和氰基氢硼化钠处理氨基羧酸得以生成。
图解5


如图解5所示,氨基羧酸用醛、R13HO和氰基氢硼化钠处理以形成取代氨基酸,然后该氨基酸被大环内酰胺化且任选地如图解4中所述去氧,以提供R13不为氢的环氧化和脱氧大环内酰胺。
制造本发明大环内酰胺的合成方法也在实施例17-19中更为详细地描述。实施例17描述了应用9-氧代-埃坡霉素作为示例的起始材料来形成氨基酸。实施例18和19分别描述了9-氧代-埃坡霉素D环氧大环内酰胺变体和脱氧大环内酰胺变体的形成。实施例20和21分别描述了9-氧代-埃坡霉素D环氧大环内酰胺变体和脱氧大环内酰胺变体的形成。
本发明的组合物往往包含本发明的化合物和可药用载体。本发明化合物可为游离形式,或根据需要,可为所发明化合物的可药用衍生物,如其前体药物、其盐和酯。组合物可为任何适宜的形式,如固体、半固体或流体。见药物剂型和药物释放体系(Pharmaceutical Dosage Forms and DrugDelivery Systems),第5版,Lippicott Williams & Wilkins(1991),在此引用作为参考。一般而言,药物制品包含作为活性成分的一种或多种本发明的化合物,其与适于外部、肠道或非肠道施用的有机或无机载体或赋形剂混合。活性成分可与例如通常的无毒、药学可接受载体化合,用于制备片剂、小丸剂、胶囊剂、栓剂、阴道栓剂、溶液、乳剂、悬浮剂以及任一其它适用剂型。可用的载体包括水、葡萄糖、乳糖、阿拉伯树胶、明胶、甘露醇、淀粉糊、三硅酸镁、滑石、玉米淀粉、角蛋白、胶态二氧化硅、马铃薯淀粉、尿素,以及其它适用于生产固体、半固体或流体形式制剂的载体。此外,也可应用辅助的稳定剂、增稠剂和着色剂。
在一个实施方案中,包含本发明化合物的组合物不含Cremophor)的发明化合物。Cremophor(BASF Aktiengesellschaft)为聚乙氧基化的蓖麻油,其在制剂低溶解度药物时用一般用作表面活性剂。但是,因为Cremophor可对受试者引起过敏反应,因此减少或消除Cremophor的组合物为优选的。消除Cremophor的埃坡霉素A或B的制剂在例如PCT公开WO 99/39694(其在此引用作为参考)中有所描述,并可适宜用于本发明的化合物。
根据需要,本发明的化合物可制剂成微胶囊和纳米级颗粒。例如于医学和制药中的微胶囊和纳米级颗粒(Microcapsules and Nanoparticles inMedicine and Pharmacy),Max Donbrow编辑,CRC Press(1992)和美国专利号5,510,118;5,534,270和5,662,883中描述了常规方法,均在此引用作为参考。通过增加表面积对体积的比率,这些剂型使得不适合经口递送的化合物可经口递送。
本发明的化合物也可用先前描述用于低溶解度药物的其它方法来进行制剂。例如,化合物可与维生素E或其PEG化的衍生物形成乳剂,如在WO 98/30205和00/71163中所描述的,它们在此引用作为参考。一般将来发明的化合物溶解在含乙醇(优选的为低于1%w/v)的水溶液中。加入维生素E或PEG化的维生素E。然后移去乙醇以形成可制剂成用于静脉或经口途径施用的前乳剂。另一策略涉及将本发明化合物包裹入脂质体中。用于形成作为药物递送载体的脂质体的方法在本领域已为人熟知。合适的方法包括在美国专利号5,683,715;5,415,869和5,424,073中所描述的涉及另一相对低溶解度的癌症药物紫杉醇的方法,在此引用作为参考,以及在PCT公开WO 01/10412中所描述的涉及埃坡霉素B的方法,在此引用作为参考。可应用的多种脂类中,特别优选地用于制备胶囊化埃坡霉素脂质体的脂类,其包括磷脂酰胆碱和聚乙二醇衍生的二硬脂基磷脂酰乙醇胺。实施例22提供了用于制备含9-氧代-埃坡霉素D的脂质体的示例性方法,该通用方法易于改变为制备含本发明其它化合物的脂质体的方法。
然而另一方法涉及用聚合物制剂本发明的化合物,其中聚合物如生物聚合物或生物相容的聚合物(合成的或天然存在的)。生物相容的聚合物可分类为可生物降解的和非生物降解的。可生物降解的聚合物的体内降解与化学组合物、生产方法和内植体的结构相关。合成的聚合物的示例包括聚酐类、多羟基酸(如聚乳酸、聚乙二醇酸及其共聚体)、聚酯类、聚酰胺类、聚原酸酯以及一些多磷腈。天然存在的聚合物的示例包括蛋白质和多糖如胶原、透明质酸、白蛋白和明胶。
另一方法涉及将本发明的化合物结合到可增加水溶解度的聚合物。适宜的聚合物的示例包括聚乙二醇、聚-(d-谷氨酸)、聚-(1-谷氨酸)、聚-(1-谷氨酸)、聚-(d-天冬氨酸)、聚-(1-天冬氨酸)、聚-(1-天冬氨酸)及其共聚物。具有分子量在约5,000到约100,000之间的聚谷氨酸是优选的,更为优选的是具有分子量在约20,000到80,000之间,且最为优选的是具有分子量在约30,000到60,000之间。聚合物通过酯键共轭连接到本发明的埃坡霉素的一个或多个羟基,所用方法为如在美国专利号5,977,163(在此引用作为参考)以及实施例23中所描述。优选的结合部位包括:在为本发明的21-羟基衍生物的情况下,结合部位为21-碳的羟基。其它结合部位包括3-碳上的羟基和7-碳上的羟基。
在另一方法中,本发明的化合物连接到单克隆抗体。该策略使得本发明的化合物靶向特定靶标。用于设计和应用结合抗体的常用方法在基于单克隆抗体的癌症治疗(Monoclonal Antibody-Based Therapy of Cancer),Michael L.Grossbard,编辑(1998)一书中描述,在此引用作为参考。
可与载体材料结合以产生单一剂型的活性成分的量随受治疗的受试者以及具体的施用模式而有所变化。例如,用于静脉内施用的制剂含本发明化合物的剂量为从约1mg/mL到约25mg/mL,优选地从约5mg/mL到约15mg/mL,且更优选地为约10mg/mL。静脉内制剂一般用普通生理盐水或5%葡萄糖溶液在使用前稀释约2倍到30倍之间。
在本发明的一个方面,本发明的化合物用于治疗癌症。在一个实施方案中,本发明的化合物用于治疗头、颈部的癌症,其包括头、颈、鼻腔、鼻旁窦、鼻咽、口腔、口咽、喉、下咽部、唾液腺和副神经节瘤的肿瘤。在另一实施方案中,本发明的化合物用于治疗肝和胆管树的癌症,特别是肝细胞癌。在另一实施方案中,本发明的化合物用于治疗肠道癌症,特别是结肠直肠癌。在另一实施方案中,本发明的化合物用于治疗卵巢癌。在另一实施方案中,本发明的化合物用于治疗小细胞和非小细胞肺癌。在另一实施方案中,本发明的化合物用于治疗乳腺癌。在另一实施方案中,本发明的化合物用于治疗肉瘤,其包括纤维肉瘤、恶性纤维组织细胞瘤、胚胎横纹肌肉瘤、平滑肌瘤、神经纤维肉瘤、骨肉瘤、滑膜肉瘤、脂肪肉瘤和软组织腺泡状肉瘤。在另一实施方案中,本发明的化合物用于治疗中枢神经系统肿瘤,特别是脑癌。在另一实施方案中,本发明的化合物用于治疗淋巴瘤,其包括何杰金氏淋巴瘤、淋巴浆细胞样淋巴瘤、滤泡淋巴瘤、粘膜相关淋巴组织淋巴瘤、外膜细胞淋巴瘤、B-细胞系大细胞淋巴瘤、伯基特淋巴瘤和T细胞退行发育的大细胞淋巴瘤。
本方法包括给患癌症的受试者施用治疗有效量的本发明化合物。必要时可重复该方法以限制(即防止进一步的生长)或消除癌症。临床上,该方法的应用会导致癌性肿物大小或数量的减少和/或相关症状的减轻(如果适用的话)。病理方面,该方法的应用将产生至少以下情况之一:抑制癌细胞增殖、减小癌或肿瘤的大小、防止进一步转移以及抑制肿瘤血管生成。
本发明的化合物和组合物可用于联合治疗。换言之,本发明的化合物和组合物可在一个或多个其它所需的治疗方法或医学方法的同时、之前或之后施用。在联合疗法中治疗和方法的具体联合应考虑到治疗和/或方法以及所要达到的治疗效果的相容性。
在一个实施方案中,本发明的化合物和组合物用于与其它抗癌剂和方法联合。其它抗癌剂的示例包括但不限于:(i)烷基化药物如氮芥、苯丁酸氮芥、环磷酰胺、苯丙氨酸氮芥、异环磷酰胺;(ii)抗代谢物如氨甲蝶呤;(iii)微管稳定剂如长春灭瘟碱、紫杉醇、多西他赛(docetaxel)和discodermolide;(iv)血管发生抑制剂;以及(v)细胞毒抗生素如阿霉素、博来霉素和丝裂霉素。其它抗癌方法的示例包括:(i)外科;(ii)放射疗法和(iii)光动力学疗法。
在另一实施方案中,本发明的化合物和组合物用于与试剂和方法联合应用,以减轻本发明的化合物或组合物的潜在副作用,如腹泻、恶心和呕吐。腹泻可用抗腹泻剂治疗,如阿片样物质(例如可待因、苯乙哌啶、氰苯哌啶和loeramide)、碱式水杨酸铋和奥曲肽。恶心和呕吐可用止吐剂治疗,如地塞米松、甲氧氯普胺、二苯甲氧乙胺、安定、奥丹西隆、氯丙嗪、硫乙拉嗪和屈大麻酚。对那些包括聚乙氧基化的蓖麻油如Cremophor,用皮质类固醇如地塞米松和甲基脱氧皮质醇和/或H1拮抗剂(如盐酸二苯羟基胺)和/或H2拮抗剂进行预处理可用于缓解过敏反应。用于静脉使用的制剂和预处理治疗方案的示例分别在实施例24和25中描述。
在本发明的另一方面,本发明的化合物用于治疗以细胞过度增生为特征的非癌病症。在一个实施方案中,本发明的化合物用于治疗银屑病,该病以角化细胞的细胞性过度增生并在皮肤表面形成隆起的有磷屑的损害为特征。方法包括给患银屑病的受试者施用治疗有效量的本发明的化合物。如果需要可重复本方法以减少损害的数目或严重程度或消除损害。在临床上,该方法的应用导致了皮肤病灶大小和数量的减少,皮肤症状(受损皮肤的疼痛、烧灼感和出血)的减轻和/或相关症状的减轻(例如,关节红、热、肿、腹泻、腹痛)。在病理上,该方法的应用将导致至少以下之一:抑制角化细胞增生、减少皮肤炎症(例如,通过影响吸引因子和生长因子、抗原呈递、产生活性氧类和基质金属蛋白酶)以及抑制皮肤的血管发生。
在另一实施方案中,本发明的化合物用于治疗多发性硬化症,该疾病以脑的渐进性脱髓鞘为特点。尽管参与髓磷脂丧失的确切机制还不清楚,但存在星形胶质细胞增生的增加和髓磷脂损害区域的积聚。在这些部位,有巨噬细胞样活性和增强的蛋白酶活性,这些至少部分对髓鞘的降解负有责任。方法包括给患多发性硬化症的受试者施用治疗有效量的本发明化合物。如果需要,该方法可以重复以抑制星形胶质细胞增生和/或减轻运动功能丧失的程度和/或防止或减轻疾病的慢性进程。临床上,应用该方法可导致改善视觉症状(视力丧失、复视)、步态障碍(虚弱、轴向不稳、感觉缺失、痉挛状态、反射亢进、灵巧性丧失)、上肢极度功能障碍(虚弱、痉挛状态、感觉缺失)、膀胱功能障碍(尿急、尿失禁、尿迟疑、排尿不完全)、抑郁症、情绪不稳定和认知缺损。病理上,该方法的应用导致一种或多种以下情况的减轻,如髓磷脂损失、血脑屏障的破坏、血管周围单核细胞浸润、免疫异常、胶质瘢痕形成和星形胶质细胞增生、金属蛋白酶生成以及传导速率受损。
在另一实施方案中,本发明的化合物用于治疗类风湿性关节炎,该疾病为多系统的慢性复发性炎性疾病,有时导致受损关节的损坏和关节强硬。类风湿性关节炎以滑膜的显著增厚(增厚的滑膜形成放射状延伸到关节腔)、滑膜细胞排列为多层(滑膜增生)、白细胞(巨噬细胞、淋巴细胞、浆细胞和淋巴小结;称为“炎性滑膜炎”)浸润滑膜以及纤维蛋白与坏死细胞在滑膜的沉积为特点。此过程最终形成的组织称为关节翳,且最终关节翳将生长至充满关节腔。关节翳在血管发生过程中形成了广泛的新血管网络,其中所述血管发生是滑膜炎进程中所必须的。来自关节翳组织细胞的消化酶的释放(基质金属蛋白酶(例如胶原酶、溶基质素)和炎症过程中的其它介质(例如过氧化氢、超氧化物、溶酶体酶和花生四烯酸代谢产物)导致软骨组织的渐进性破坏。关节翳侵袭关节软骨导致软骨组织的腐蚀和断裂。最终在所涉及的关节发生软骨下骨的腐蚀,发生纤维性关节强直并最终骨性关节强直。
方法包括给患类风湿性关节炎的受试者施用治疗有效量的本发明化合物。如果需要可重复该方法以抑制滑膜细胞增生和/或减轻受影响关节运动损失的程度和/或预防或减轻疾病的慢性进程。临床上,应用该方法可导致一种或多种以下情况:(i)降低症状(受损关节的疼痛、肿胀和触痛;早晨强直、虚弱、疲劳、厌食和体重减轻)的严重性;(ii)减轻疾病临床指征(关节囊的增厚、滑膜肥大、关节渗出、软组织挛缩、运动幅度减少、关节强硬和刚性连接畸形)的严重性;(iii)减少疾病关节外的表现(风湿性小结、脉管炎、肺小结、间质纤维化、心包炎、巩膜外层炎、虹膜炎、Felty综合征、骨质疏松征);(iv)增加疾病缓解/无症状时期的频率和持续时间;(v)预防固定的损害和失能;和/或(vi)预防/减轻疾病的慢性进程。病理上,应用本发明将产生至少一种的如下情况:(i)减轻炎性反应;(ii)破坏炎性细胞因子(如IL-I、TNFa、FGF、VEGF)的活性;(iii)抑制滑膜细胞增生;(iv)抑制基质金属蛋白酶的活性,和/或(v)抑制血管发生。
在另一实施方案中,本发明的化合物用于治疗动脉粥样硬化和/或再狭窄,特别是用于可能会应用血管内斯坦特固定模治疗阻塞的患者。动脉粥样硬化为慢性的血管损伤,其中动脉壁的一些正常血管平滑肌细胞(“VSMC”)(通常控制血管紧张度以调控血流)改变它们的特性,并显现出“癌样”行为。这些VSMC变得非正常增生,分泌物质(生长因子、组织降解酶和其它蛋白质)以使其能够侵入和扩散到血管内层、阻塞血流且使该血管异常敏感易于被局部血凝块完全堵塞。再狭窄为矫正操作后再发生的狭窄或动脉狭窄,是动脉粥样硬化的加重形式。
本方法包含在斯坦特固定模上包被治疗有效量的本发明的化合物,并将斯坦特固定模递送到患动脉粥样硬化受试者的发病动脉。用化合物包被斯坦特固定模的方法在例如美国专利号6,156,373和6,120,847中描述。临床上,应用本发明可导致一种或多种以下情况:(i)增加动脉血流;(ii)降低疾病临床指征的严重性;(iii)降低再狭窄的速率;或(iv)防止/减轻动脉粥样硬化的慢性进程。病理上,应用本发明可在斯坦特固定模植入的部位产生至少一种以下情况:(i)降低炎症反应;(ii)抑制VSMC分泌基质金属蛋白酶;(iii)抑制平滑肌细胞的积聚;和(iv)抑制VSMC表型的去分化。
在一个实施方案中,对患癌症或以细胞增生为特点的非癌病症的受试者的用药剂量水平从约1mg/m2到约200mg/m2的等级,药物可以大丸剂施用(以任一适宜的施用途径)或按需要每周、每两周或每三周连续静脉输液(例如1小时、3小时、6小时、24小时、48小时或72小时)。但应当理解,任何特定患者的特定剂量水平依赖于一系列的因素。这些因素包括应用的特定化合物的活性;受试者的年龄、体重、总体健康情况、性别和饮食;药物的施用时间和途径及药物排泄的速率;是否在治疗中联合应用药物;以及受治疗疾病的严重程度。
在另一实施方案中,剂量水平根据需要和可忍受的程度每三周一次,从约10mg/m2到约150mg/m2,优选地从约10到约75mg/m2,且更优选地从约15mg/m2到约50mg/m2。在另一实施方案中,剂量水平根据需要和可忍受的程度每两周一次,从约1mg/m2到约150mg/m2,优选地从约10mg/m2到约75mg/m2且更优选地从约25mg/m2到约50mg/m2。在另一实施方案中,剂量水平根据所需或可忍受的程度每周一次,从约1mg/m2到约100mg/m2,优选地从约5mg/m2到约50mg/m2,且更优选地从约10mg/m2到约25mg/m2。在另一实施方案中,剂量水平根据需要或可忍受的程度每天一次,从约0.1到约25mg/m2,优选地从约0.5到约15mg/m2,且更优选地从约1mg/m2到约10mg/m2。
以上提供了本发明的详细描述,给出以下的实施例用于阐述本发明,且不应构成对本发明或权利要求范围的限制。
实施例1
黄色粘球菌表达载体的构建
提供整合和附着功能的噬菌体Mx8的DNA插入到市售的pACYC184(New England Biolabs)。从质粒pPLH343(见Salmi等,1998年2月,细菌学杂志180(3):614-621)分离~2360 bp MfeI-SmaI片段,并连接到质粒pACYC184的大EcoRI-XmnI限制性片段。这样形成的环状DNA大小~6kb,并被命名为质粒pKOS35-77。
质粒pKOS35-77作为方便的载体,用于在埃坡霉素PKS基因启动子控制下表达本发明的重组PKS基因。在一个实施方案示例中,带有其同源启动子的完整埃坡霉素PKS基因以一个或多个片段插入到质粒,以产生本发明的表达载体。
本发明也提供了这样的表达载体,其中本发明的重组PKS基因位于黄色粘球菌启动子控制之下。为构建示例的载体,以PCR扩增产物分离黄色粘球菌pilA基因的启动子。将包含pilA基因启动子的质粒pSWU357(在Wu和Kaiser,1997年12月,细菌学杂志179(24):7748-7758中描述)与PCR引物Seq 1和Mxpi 11混合:
Seq1:5′-AGCGGATAACAATTTCACACAGGAAACAGC-3′和
Mxpil1:5′-TTAATTAAGAGAAGGTTGCAACGGGGGGC-3′
并且用标准PCR条件扩增以产生~800bp的片段。该片段用限制性酶KpnI切割并连接入市售质粒pLitmus 28(New England Biolabs)的大KpnI-EcoRV限制性片段。所产生的环状DNA命名为质粒pKOS35-71B。
来自质粒pKOS35-71B的pilA基因启动子以~800bp EcoRV-SnaBI限制性片段分离,并连接到质粒pKOS35-77的大MscI限制性片段以产生6.8kb大小的环状DNA。由于~800bp片段可以两个方向中的任何一个方向被插入,连接产生了大小相等的两个质粒,命名为质粒pKOS35-82.1和pKOS35-82.2。这些质粒的限制性位点和功能图谱在图2中描述。
质粒pKOS35-82.1和pKOS35-82.2作为方便的起始材料用作本发明的载体,载体中重组PKS基因被置于黄色粘球菌pilA基因启动子控制之下。这些质粒包含置于紧靠启动子转录起始位点下游的单一PacI限制性酶识别序列。在一个示例的实施方案中,不带有同源启动子的完整埃坡霉素PKS基因以一个或多个片段在PacI位点插入到质粒,以产生本发明的表达载体。
这些载体上的pilA启动子序列如下所示:
CGACGCAGGTGAAGCTGCTTCGTGTGCTCCAGGAGCGGAAGGTGAAGCCG
GTCGGCAGCGCCGCGGAGATTCCCTTCCAGGCGCGTGTCATCGCGGCAAC
GAACCGGCGGCTCGAAGCCGAAGTAAAGGCCGGACGCTTTCGTGAGGACC
TCTTCTACCGGCTCAACGTCATCACGTTGGAGCTGCCTCCACTGCGCGAGC
GTTCCGGCGACGTGTCGTTGCTGGCGAACTACTTCCTGTCCAGACTGTCGG
AGGAGTTGGGGCGACCCGGTCTGCGTTTCTCCCCCGAGACACTGGGGCTAT
TGGAGCGCTATCCCTTCCCAGGCAACGTGCGGCAGCTGCAGAACATGGTG
GAGCGGGCCGCGACCCTGTCGGATTCAGACCTCCTGGGGCCCTCCACGCTT
CCACCCGCAGTGCGGGGCGATACAGACCCCGCCGTGCGTCCCGTGGAGGG
CAGTGAGCCAGGGCTGGTGGCGGGCTTCAACCTGGAGCGGCATCTCGACG
ACAGCGAGCGGCGCTATCTCGTCGCGGCGATGAAGCAGGCCGGGGGCGTG
AAGACCCGTGCTGCGGAGTTGCTGGGCCTTTCGTTCCGTTCATTCCGCTACC
GGTTGGCCAAGCATGGGCTGACGGATGACTTGGAGCCCGGGAGCGCTTCG
GATGCGTAGGCTG ATCG ACAGTTATCGTCAGCGTCACTGCCGAATTTTGTC
AGCCCTGGACCCATCCTCGCCGAGGGGATTGTTCCAAGCCTTGAGAATTGG
GGGGCTTGGAGTGCGCACCTGGGTTGGCATGCGTAGTGCTAATCCCATCCG
CGGGCGCAGTGCCCCCCGTTGCAACCTTCTCTTAATTAA
为产生本发明的重组黄色粘球菌宿主细胞,黄色粘球菌细胞在CYE培养基中(Campos和Zusman,1975,黄色粘球菌发育的调节:3′:5′-环AMP、ADP和营养物的影响,美国国家科学院院报72:518-522)于30℃、300转/分钟生长到100克莱特。方法的其余部分除非特别指出均在25℃操作。然后离心沉淀细胞(用SS34或SA600转头,8000转/分钟,离心10分钟)并重悬于去离子水中。细胞再次沉淀并用其1/100原始体积重悬。
DNA(1至2μL)用0.1cm电击杯在室温下电穿孔进入细胞,条件为400欧姆、25μFD、0.65V、时间常数在8.8-9.4之间。DNA不含盐,并重悬在蒸馏且去离子水中,或经0.025μm型VS膜(Millipore)透析。电穿孔效率低时,DNA被点透析,且增生物在CYE中。电穿孔后立即加入1mLCYE,电击杯中的细胞加到50mL锥形瓶中,该瓶已预先加入了另外的1.5mL CYE(总体积为2.5ml)。细胞在30到32℃以300转/分钟生长4到8小时(或过夜)以使得选择标记表达。然后,将细胞铺在平板的选择性CYE软琼脂中。用卡那霉素作为选择标志时,每μg DNA的产量通常为103到105。用链霉素作选择标记时,其包含在上层琼脂中,因为它可结合琼脂。
用此步骤,本发明的重组DNA表达载体被电穿孔入粘球菌宿主细胞,该细胞表达本发明的重组PKSs,并产生其编码的埃坡霉素、埃坡霉素衍生物和其它新聚酮化合物。
实施例2
染色体整合以及用于在黄色粘球菌中表达埃坡霉素的
一种细菌人工染色体(BAC)
为在异源宿主中表达埃坡霉素PKS和修饰酶基因,以通过发酵产生埃坡霉素,根据本发明的方法,使用了黄色粘球菌,其与纤维堆囊菌密切相关,并可得到其一系列克隆载体。黄色粘球菌和纤维堆囊菌为粘细菌,并可能具有基因表达、免疫调控和翻译后修饰的通用元件。黄色粘球菌已被开发为用于基因克隆和表达:DNA可通过电穿孔导入,且一系列的载体和遗传标志可用于导入外源DNA(包括那些允许DNA稳定插入到染色体的载体和遗传标志)。黄色粘球菌可在发酵罐的复杂培养基中相对易于生长,并在需要时可进行操作以提高基因表达。
为将埃坡霉素基因簇导入黄色粘球菌,可利用同源重组的方法将埃坡霉素簇构建到染色体中,以组装完整的基因簇。另外,完整的埃坡霉素基因簇可克隆入细菌人工染色体(BAC),然后转移入黄色粘球菌用于整合到染色体。
为从粘粒pKOS35-70.1A2和pKOS35-79.85组装基因簇,将来自这些粘粒的同源小区域(2kb或更大)导入黄色粘球菌以提供用于基因簇的较大片段重组位点。如在图3中所示,构建出质粒pKOS35-154和pKOS90-22以导入这些重组位点。在黄色粘球菌染色体中组装埃坡霉素基因簇的策略如图4所示。起初,在细菌染色体上选择一个并不破坏任何基因或转录单位的中立位点。一个此类区域在devS基因的下游,已显示此区域不影响黄色粘球菌的生长和发育。第一个质粒pKOS35-154用DraI线性化,并电穿孔入黄色粘球菌。该质粒包含在埃坡霉素基因簇两个片段侧翼的dev基因座的两个区域。插入到epo基因区域之间的是包含卡那霉素抗性标志和大肠杆菌galk基因的盒子。见Ueki等,1996,基因(Gene)183:153-157,在此引用作为参考。如果DNA应用dev序列作为同源区域通过二次重组重组到dev区域,菌落出现卡那霉素抗性。
K35-159菌株包含允许pKOS35-79重组的埃坡霉素基因簇的小区域(2.5kb)。由于pKOS35-79.85上的抗性标志与K35-159上的相同,四环素转座子被转位到粘粒,筛选包含插入卡那霉素标志的转座子的粘粒。该粘粒pKOS90-23经电穿孔导入K35-159,并选择土霉素抗性菌落,以产生K35-174株。为从粘粒上移去不需要的区域并仅留下埃坡霉素基因,将细胞被铺在含1%半乳糖的CYE平板上。galK基因的存在使得细胞对1%的半乳糖敏感。K35-174的半乳糖抗性菌落代表细胞通过重组或通过galK基因的突变而丧失了galK标志。如果重组发生,则半乳糖抗性菌株对卡那霉素和土霉素敏感。对这两种抗生素都敏感的菌株通过Southern印迹分析得以证实。确定正确的菌株并命名为K35-175,且含有从组件7到epoK终止密码下游4680bp的埃坡霉素基因簇。
为导入组件1到组件7,再次重复上述步骤。质粒pKOS90-22用DraI线性化,并电穿孔导入K35-175以产生K111-13.2。该菌株用pKOS35-70.1A2,pKOS90-38四环素抗性变体电穿孔,并选择土霉素抗性菌落。此过程产生了菌株K111-13.23。含有整个埃坡霉素基因簇的重组体通过对1%半乳糖的抗性进行筛选。此过程导致了K111-32.25、K111-32.26和K111-32.35克隆。菌株K111-32.25根据布达佩斯条约于2000年4月14日保藏于美国典型培养物保藏中心(ATCC),10801 University Blvd.,马纳萨斯,VA,20110-2209美国,且可在保藏号PTA-1700下获得。该菌株含所有埃坡霉素基因和它们的启动子。
通过在50mL烧瓶中将菌株接种到5mL CYE(每升中含10g酪胨、5g酵母提取物和1g MgSO4·7H2O)中进行发酵,并过夜生长直到培养基处于中间对数生长期。取100μL等份涂到CTS平板,且平板在32℃孵育4-5天。为提取埃坡霉素,将来自平板的琼脂和细胞浸软,放入50mL圆锥管中,并将管中加满丙酮。该溶液摇动孵育4-5小时,蒸发丙酮,且残留液体用等体积的乙酸乙酯提取两次。通过加入硫酸镁从乙酸乙酯提取物中去除水。硫酸镁被滤除,且液体蒸发至干。将埃坡霉素重悬在200μL乙腈中,并用LC/MS进行分析。分析显示菌株产生了埃坡霉素A和B,培养物中存在的埃坡霉素B约0.1mg/L,且埃坡霉素A的水平要低5到10倍。
该菌株也可用于在液体培养基中产生埃坡霉素。将埃坡霉素产生菌株接种到含CYE的烧瓶。第二天,当细胞处于中间对数生长期,将5%的接种物加入到含0.5%CMM(0.5%酪胨、0.2%MgSO4·7H2O、10mMMOPS pH7.6)以及1mg/mL丝氨酸、丙氨酸和甘氨酸以及0.1%丙酮酸钠的烧瓶中。可加到0.5%以增加埃坡霉素B的产量,但引起埃坡霉素B对埃坡霉素A的比率降低。培养物于30℃生长60-72小时。更长的孵育时间会导致埃坡霉素的滴度降低。为收获埃坡霉素,培养物在SS34转头中以10,000转/分钟离心10分钟。上清用乙酸乙酯提取两次并旋转蒸发(“rotavaped”)至干。液体培养物产生2到3mg/L的埃坡霉毒A和B,比率与平板培养观察到的类似。如果向培养物中加入XAD(0.5-2%),将观察到埃坡霉素C和D,存在的埃坡霉素D为0.1mg/L,而存在的埃坡霉素C的水平低5到10倍。
为将所有的基因簇作为一个片段克隆,构建了细菌人工染色体(BAC)文库。首先,将SMP44细胞埋入琼脂糖,按照BIO-RAD基因组DNA填料试剂盒将其裂解。含DNA的填料用限制酶如Sau3AI或HindIII部分消化后,在FIGE或CHEF凝胶上电泳。通过从琼脂上电洗脱DNA或用琼脂酶将琼脂糖降解而分离DNA片段。选择分离片段的方法是电洗脱,如Strong等,1997,核酸研究(Nucleic Acids Res),19:3959-3961中所述,在此引用作为参考。将DNA连接入用适当酶消化的BAC(pBeloBACII)。PBeloBACII的图谱如图5所示。
将DNA电穿孔导入DH10B细胞以产生纤维堆囊菌基因组文库,所用方法为Sheng等,1995,核酸研究,23:1990-1996,在此引用作为参考。用来自埃坡霉素簇NRPS区域的探针筛选克隆。挑取阳性克隆,并分离DNA进行限制性分析,以证明完整基因簇的存在。阳性克隆命名为pKOS35-178。
为产生可用于导入pKOS35-178的菌株,构建了含埃坡霉素基因簇上游和下游同源区的质粒pKOS35-164,埃坡霉素基因簇上游和下游两侧是dev基因座和包含卡那霉素抗性的galK盒,类似于质粒pKOS90-22和pKOS35-154。该质粒以DraI线性化,电穿孔导入黄色粘球菌,以产生K35-183,方法参见Kafeshi等,1995,分子微生物学15:483-494。质粒pKOS35-178可通过电穿孔或者用噬菌体P1转导的方法导入K35-183,挑选氯霉素抗性克隆。另外,可构建含来自pRP4结合转移起点的pKOS35-178变体,用于将DNA从大肠杆菌转移至K35-183。该质粒的制备为:首先,构建含来自RP4 oriT区域和来自pACYC184的四环素抗性标记的转座子,然后在体外或体内将该转座子转位至pKOS35-178上。该质粒转化入S17-1,并结合进黄色粘球菌。菌株K35-190在存在1%半乳糖时生长,以对第2次重组事件进行选择。该菌株含有所有的埃坡霉素基因和所有潜在的启动子。发酵该菌株,并测试埃坡霉素类A和B的产量。
另外,使用温度敏感质粒pMAK705或pK03通过将该转座子转位至pMAK705或pK03上,并筛选tetR和camS质粒可将转座子重组入BAC;重组按Hamilton等,1989年9月,细菌学杂志,171(9):4617-4622和Link等1997年10月,细菌学杂志,179(20):6228-6237所述进行,二者在此处引用作为参考。
除了将pKOS35-178整合入dev基因座外,它也可利用真菌噬菌体Mx8或Mx9的整合功能整合入噬菌体附着位点。构建包含来自Mx8或Mx9的整合基因和att位点以及来自pACYC184的四环素基因的转座子。该转座子的其它变体可只有附着位点。在此变体中,整合基因为接下来由共电穿孔含整合酶基因的质粒反式提供或由任一组成型启动子如mgl启动子使得整合酶蛋白质在电穿孔菌株中表达(参见Magrini等,1999年7月,细菌学杂志,181(13):4062-4070,此处引用作为参考)。一旦构建了转座子,它就被转位至pKOS35-178上,以产生pKOS35-191。将该质粒导入如上所述的黄色粘球菌。该菌株含有所有埃坡霉素基因及所有潜在的启动子。发酵该菌株,并测试埃坡霉素类A和B的产生。另外,含有att位点和oriT区的菌株可被转位至BAC上,并将所得到的BAC接合入黄色粘球菌。
一旦埃坡霉素基因已在黄色粘球菌的一株菌株中建立后,操作该基因簇的任一部分,如改变启动子或交换组件,可利用卡那霉素抗性和galK盒进行,如下所述。含epo基因的黄色粘球菌培养物在多种培养基中生长,并检测埃坡霉素类的产生。如果埃坡霉素类(尤其是B或D)的产生水平低,则如下面的实施例所述对产生黄色粘球菌的克隆进行培养基的开发和基于突变的菌株改良。
实施例3
埃坡霉素类的产生和纯化方法
A、优化埃坡霉素D在黄色粘球菌中的异源产生
埃坡霉素D在黄色粘球菌中的异源产生通过掺入吸附树脂、确定适当的碳源。并执行补料分批方法,可从起始滴度0.16毫克/升提高140倍。
为减少埃坡霉素D在基本培养基中的降解,加入XAD-16(20克/升)以稳定细胞外产物。这极大的促进了产物的回收,并使产量增大了3倍。也评估了使用油类作为细胞生长和产物形成的碳源。通过筛选多种油类,显示出油酸甲酯具有最大的效果。在分批法中,当最佳浓度为7毫升/升时,最大细胞密度可从0.4克干细胞重(DCW)/升增加到2克干细胞重/升。产物产量依赖于生产培养基中痕量元素的存在。对基本培养基外源补充痕量金属时,峰值埃坡霉素D的滴度增加8倍,显示了金属离子在细胞代谢和埃坡霉素生物合成中的重要作用。为进一步增加产物产量,采用连续补料分批法以引起更高的细胞密度和维持延长的生产期。优化的补料分批培养物始终产生7克干细胞重/升的细胞密度,且平均产生滴度为23毫克/升。
埃坡霉素类为由粘球菌目纤维堆囊菌的多株菌株天然产生的次级代谢物(Gerth等,1996;Gerth等,2001;在本实施例中引用的参考文献列于本部分的最后,并于此处引用作为参考)。它们是微管解聚的有效抑制剂,作用机制类似于抗癌药物紫杉醇(Taxol)(Bollag等,1995)。它们对具多种药物抗性、表达P-糖蛋白的肿瘤细胞系的细胞毒效果使它们成为潜在的具有大的商业价值的治疗化合物(Su等,1997;Kowalski等,1997)。它们在水中相对高的溶解度也有利于它们用于临床评估的制剂。
埃坡霉素类A和B是天然宿主的主要发酵产物(Gerth等,1996)。这些多肽分子的大环核心是通过连续脱羧缩合乙酸基和丙酸基单元而形成的(Gerth等,2000)。埃坡霉素类A和B的不同为在其碳骨架的C-12位一个甲基的不同。此结构差异源自在组装埃坡霉素A时掺入了一个乙酸基,而在组装埃坡霉素B时掺入了一个丙酸基。埃坡霉素类C和D分别是埃坡霉素类A和B生物合成途径中的中间体(Tang等,2000;Molnar等,2000)。它们在发酵过程中作为次要产物分泌出来,合成产量约为0.4毫克/升。因为初步的体内研究显示,埃坡霉素D在4种化合物中是最有希望发展为抗肿瘤药物的(Chou等,1998),所以人们有相当大的兴趣来大规模产生这种分子。
负责埃坡霉素类生物合成的基因簇已测序(Tang等,2000;Molnár等,2000),并已用于在与纤维堆囊菌紧密相关、且更易于遗传操作的微生物宿主黄色粘球菌中制备这些复合物。为提高埃坡霉素D的产生,已构建了该重组菌株的缺失突变体,以灭活可分别催化埃坡霉素C和D转化为埃坡霉素A和B的P450环氧酶(Tang等,2000)(如下面的实施例4所述)。这种遗传改变有效促进了作为黄色粘球菌发酵的唯一产物的埃坡霉素C和D的分泌,其中埃坡霉素D比C的比率是4∶1。得到的突变体与天然宿主相比,在所需产物的回收和纯化方面具有明显的优势。在本实施例中,描述了培养基成分和发酵策略的改进,这种改进使得埃坡霉素D在黄色粘球菌中的产量增加了140倍。
为连续获得低量产生的生物活性分子,吸附树脂已被用于粘球菌的发酵(Reichenbach和Hfle,1993)。为便于分离埃坡霉素D,将疏水树脂XAD-16加入培养基中。因为用适当的溶剂易于将结合产物从树脂上洗脱下来,因此回收大大简化了。而且,XAD-16的使用通过稳定埃坡霉素使产物的降解最小化。
黄色粘球菌通常在主要由含酪蛋白的酶水解物组成的培养基中培养,如蛋白胨和酪胨,依赖氨基酸作为其唯一的碳源和氮源(Reichenbach和Dworkin,1991)。结果,由于氨基酸降解使得氨在发酵肉汤中聚集。Gerth等(1986)显示在变绿粘球菌培养物中,其胞外氨浓度为35-42mM时,与之对应的是令人惊讶的细胞内高氨浓度80-140mM。更重要的是,已表明使用原位膜方法通过连续移去多余的氨,使其浓度在8mM以下,则细胞块和二级代谢产物的产生都急剧增加(Hecht等,1990)。因为推测高水平氨的产生抑制黄色粘球菌的生长和埃坡霉素D的产生,需要另一种可降低氨基酸消耗的碳源。
虽然需要适合的方法,但通过广泛筛选不同油类,确定油酸甲酯为可被黄色粘球菌代谢的底物。往生长培养基中加入外源的痕量元素溶液,埃坡霉素D在简单分批发酵中,产生增加8倍,达到3.3毫克/升的产量。为进一步对方法进行优化,采用了使用间歇或连续馈与酪胨和油酸甲酯的补料分批法延长细胞的生产期。在本实施例中,报道了由两种不同补料策略所获得结果的比较。
材料和方法
接种物的制备
为在无油酸甲酯的培养基中生产埃坡霉素D,在50毫升的玻璃培养管中,用1毫升含20%(V/V)甘油的冷冻黄色粘球菌菌株K111-40.1细胞接种3毫升含10克/升酪胨(Difco)、5克/升酵母提取物(Difco)、1克/升MgSO4·7H2O和50mM HEPES,pH7.6的CYE培养基。用氢氧化钾将HEPES缓冲液的pH滴定至7.6。细胞在旋转摇床上于30℃、175转/分孵育3天。然后转到含50毫升CYE培养基的250毫升锥形烧瓶中,在同样的条件下生长2天。得到的种子培养物按5%(v/v)的接种大小接种50毫升的生产瓶。
为在含油酸甲酯的培养基中培养黄色粘球菌,细胞不得不适应在含油的环境中培养。将一瓶冷冻细胞的种子瓶接种到3毫升补加有3μl油酸甲酯(Emerest 2301)(Cognis Corp.)的CYE培养基中。细胞在玻璃培养管于30℃、175转/分生长2-6天,直到培养物在显微镜下足够密集为止。然后将它们转移到装有50毫升CYE-MOM培养基的250毫升锥形瓶,其中所述50毫升CYE-MOM培养基含10克/升酪胨(Difco)、5克/升酵母提取物(Difco)、1克/升MgSO4·7H2O、2毫升/升油酸甲酯和50mM HEPES,pH7.6。生长2天后,冷冻细胞,并按含20%(v/v)甘油的1毫升小份贮存于-80℃。
为在含油酸甲酯的培养基中制备埃坡霉素D,将1毫升已适应油的冷冻细胞接种到加有3毫升CYE-MOM培养基的玻璃培养管中。细胞于30℃、175转/分孵育2天,并转到含50毫升CYE-MOM培养基的250毫升锥形烧瓶中。得到的种子培养物在同样的条件下生长2天,并用于接种50毫升生产瓶,接种大小为5%(v/v)。
在准备5升发酵的接种物时,将25毫升已适应油的种子培养物转移到含475毫升CYE-MOM培养基的2.8升费氏烧瓶中。细胞于30℃、175转/分生长2天。随后,将250毫升的二级种子培养物接种到含4.75升生产培养基的5升发酵罐中,最终的接种物浓度为5%(v/v)。
摇瓶生产
在不含油酸甲酯的情况下,黄色粘球菌K111-40-1的分批培养物如下制备。1克XAD-16树脂(Rohm和Haas)和5毫升去离子水在250毫升锥形烧瓶中,于121℃高压灭菌30分钟。然后去掉烧瓶中多余的水,并加入50毫升含5克/升酪胨、2克/升MgSO4·7H2O和50mM HEPES,pH7.6的CTS培养基。因为和在存在生产培养基时高压灭菌吸附树脂会使细胞所需要的基本营养成分结合到树脂上,所以树脂和培养基要分开灭菌。向生产烧瓶中接种2.5毫升种子培养物,并于30℃、175转/分孵育6天。
在存在油酸甲酯时,分批培养物的制备按如上所述。另外,向生产培养基中补加7毫升/升油酸甲酯和4毫升/升过滤除菌的痕量元素溶液,其中所述痕量元素溶液包含10毫升/升浓缩H2SO4、14.6克/升FeCl3·6H2O、2.0克/升ZnCl3、1.0克/升MnCl2·4H2O、0.43克/升CuCl2·2H2O、0.31克/升H3BO3、0.24克/升CaCl2·6H2O和0.24克/升Na2MO4·2H2O。然后,向生产烧瓶接种2.5毫升已适应油的种子培养物,并于30℃、175转/分孵育5天。
在间歇供给酪胨和油酸甲酯时,如下准备补料分批培养物。1克XAD-16树脂和5毫升去离子水在250毫升锥形烧瓶中于121℃高压灭菌30分钟。灭菌后,去掉烧瓶中多余的水,并加入50毫升补加有2毫升/升油酸甲酯、4毫升/升痕量元素溶液和50mM HEPES,pH7.6的CTS培养基。向生产烧瓶中接种2.5毫升已适应油的种子培养物,并于30℃、175转/分孵育。接种后2天,每间隔24小时向培养基中加入2克/升酪胨和3毫升/升油酸甲酯。酪胨补料制备为浓缩的100克/升溶液。培养物生长10-12天,直至观察到相当多的细胞裂解。
在同样的生长条件下,所有的生产培养物可在2.8升的费氏烧瓶中按500毫升的规模生长。
发酵生产
在间歇或连续供给酪胨和油酸甲酯的5升规模的补料分批发酵按如下制备。20克/升XAD-16、2克/升MgSO4·7H2O和4.75升去离子水在5升发酵罐(B.Braun)中于121℃高压灭菌30分钟。灭菌后,将浓缩的酪胨溶液(150克/升)、油酸甲酯和痕量元素无菌添加到生物反应器,使酪胨、油酸甲酯和痕量元素的最终浓度分别为5克/升、2毫升/升和4毫升/升。然后向该培养基中接种250毫升已适应油的种子培养物。发酵在30℃进行,通气速率为0.4-0.5 v/v/m,初始搅拌速率为400转分。通过在400-700转/分之间的阶式搅拌,使溶解氧控制在50%饱和。通过自动加入2.5N KOH和2.5M H2SO4,使培养pH维持在7.4。接种后24小时,将酪胨(150克/升)和油酸甲酯加入生产培养基,供给速率为酪胨2克/升/天,油酸甲酯3毫升/升/天。补料每24小时给予一块,或者以蠕动泵(W.Marlow)连续给予。使细胞生长10-12天,直至观察到相当多的细胞裂解。
埃坡霉素的定量
发酵中,在使用XAD-16树脂之前,从生产烧瓶或生物反映器中取1毫升培养肉汤,并于13,000g离心10分钟。上清中埃坡霉素产物的定量用Hewlett Packard 1090 HPLC进行,UV检测的波长为250nm。将500微升上清注入通过4.6×10mm保护柱(Inertsil,ODS-3、5um)。然后进行在线提取,流速为1毫升/分钟。用100%水洗0.5分钟,接着用至50%乙腈的梯度洗1.5分钟。开始2分钟的洗脱剂转变为废物,之后通过一个更长的分离柱(4.6×150mm,Inertsil,ODS-3,5μm)。埃坡霉素类C和D的分离在50%-100%乙腈梯度下进行8分钟,然后用100%乙腈洗3分钟。在这些条件下,埃坡霉素C在9.4分钟时洗脱,埃坡霉素D在9.8分钟时洗脱。
随着XAD-16树脂的使用,从生产烧瓶或生物反应器中取5-50毫升混合很好的培养肉汤和树脂。树脂在重力作用下沉降后,倾倒培养肉汤。树脂用5-50毫升的水洗涤,并在重力作用下再次沉降。完全移弃含水混合物,并用100%甲醇从树脂提取埃坡霉素产物。溶剂的使用量是样品体积的50%。埃坡霉素类C和D的定量通过HPLC分析进行,UV检测波长为250nm。将50微升甲醇提取物注入通过2个4.6×10mm保护柱(Inertsil,ODS-3,5μm)和一个更长的4.6×150mm柱(Inertsil,ODS-3,5μm)。测定方法是用60%乙腈和40%水恒溶剂洗脱18分钟,流速为1毫升/分钟。在这些条件下,埃坡霉素C在10.3分钟时检测到,埃坡霉素D在13.0分钟时可检测到。用纯化的埃坡霉素D制备标准品。
细胞生长测定
在无油酸甲酯时,通过测定600nm处的光学密度(OD)来监测细胞生长。样品用水稀释至最终OD值低于0.4。因为将油酸甲酯加到培养基中会导致在600nm有强吸收的乳液形成,存在油酸甲酯时的细胞生长通过干细胞重(DCW)测定。40毫升培养肉汤在已称重的试管中于4200g离心20分钟。然后沉淀用40毫升水洗涤,并在称重前于80℃干燥2天。
氨的测定
1毫升发酵肉汤通过在13,000g离心5分钟而澄清。然后上清用氨测定试剂盒(Sigma)进行氨的分析。样品用水稀释20-100倍,直至最终浓度低于880μM。
油酸甲酯的测定
1毫升发酵肉汤中残留的油酸甲酯用5毫升乙腈提取。将混合物涡旋,并于4200g离心20分钟使其澄清。油酸甲酯的定量通过HPLC分析进行,UV检测波长为210nm。将50微升上清注入通过2个4.6×10mm保护柱(Inertsil,ODS-3、5μm)和一个更长的4.6×150mm柱(Inertsil,ODS-3、5μm)。柱子用乙腈∶水(1∶1)洗2分钟,流速为1毫升/分钟。然后,用50%-100%的乙腈梯度洗脱24分钟以上,接着用100%乙腈洗5分钟。因为商品化油酸甲酯碳链长度的异质性,该化合物在洗脱时有2个在25.3分钟和27.1分钟检测到的主峰。结合在XAD-16树脂上的来自甲醇提取物的油酸甲酯用同样的HPLC方法进行定量。油酸甲酯标准品于83.3%已腈中制备。细胞消耗的油酸甲酯如下计算:加入的总油酸甲酯—培养基中残留的油酸甲酯—结合于树脂上的油酸甲酯。
结果
在分批发酵法中,黄色粘球菌株K111-40-1最初使用简单的只含5克/升酪胨(一种胰的酪蛋白消化物)和2克/升硫酸镁的生产培养基培养。细胞的基线特性如图6所示。
接种后3天,获得最大细胞密度和最大埃坡霉素D的产生量,OD600为1.6,且相应滴度为0.16mg/L。其后细胞密度和产物产量均明显降低。随着细胞消耗酪胨,在生产培养基中也可检测到铵的逐渐积累。在5天发酵的最后,最终的氨浓度接近20mM。
XAD-16对产物稳定性的影响
为防止埃坡霉素D的快速降解,将疏水吸附树脂XAD-16加入到生产培养基中,以结合和稳定分泌产物。XAD-16是芳族聚合物树脂,之前Gerth等(1996)用该树脂从微生物生产者纤维堆囊菌So ce90的发酵产物中分离埃坡霉素类A和B。如图7所示,吸附树脂的存在对细胞的生长没有影响。然而,它有效减少发酵肉汤中埃坡霉素D的损失,其可使该产物的回收增加3倍。
培养基的研发
在致力于开发可支持更高细胞密度和埃坡霉素产量的培养基时,通过使生产培养基中酪胨的浓度在1克/升-40克/升之间变化,评价酪胨对生长和产物产量的影响。虽然酪胨浓度的增加可刺激细胞生长,但细胞的比生产力率显著降低,如图8所示。产生埃坡霉素D的最佳酪胨浓度为5克/升,而更高的酪胨浓度导致滴度降低。
由于酪胨的使用,培养基的改进受限,所以评估了作为基本生产培养基添加成分的其它底物。根据详细筛选的不同油类,确定油酸甲酯为可促进埃坡霉素D产量增加最大的碳源,如表3所概述的。
表3
| 油(7毫升/升) | 相对于无油添加剂的对照的埃坡霉素D产量(%) |
| 油酸甲酯 | 780 |
| 油酸乙酯 | 740 |
| 椰子油 | 610 |
| 豚脂 | 470 |
| 油酸丙酯 | 420 |
| 芝麻油 | 380 |
| 三油酸甘油酯 | 370 |
| 色拉油 | 360 |
| 向日葵油 | 330 |
| 大豆油 | 290 |
| 十七烷酸甲酯 | 190 |
| 无油(对照) | 100 |
| 十九烷酸甲酯 | 96 |
| 壬酸甲酯 | 40 |
| 菜籽油 | 40 |
然而,直接将油酸甲酯加入生产培养基中会导致细胞过早裂解。因此,种子培养物在进行生产发酵之前生长于含油酸甲酯的培养基。有趣的是,这种适应方法使细胞对裂解具有较低的易感性。如表4所示(与分批发酵中CTS培养基的基线特性相比,对生长和产量的改善),在油酸甲酯浓度为7毫升/升,痕量元素浓度为4毫升/升时,达到2.1克/升的峰高生物量浓度和3.3毫克/升的埃坡霉素D滴度。
表4
| 发酵条件 | 最大细胞密度(gDCW/L) | 最大埃坡霉素D产量(毫克/升) |
| 在分批法中,无XAD-16的CTS培养基 | 0.44±0.04 | 0.16±0.03 |
| 在分批法中,有XAD-16的CTS培养基 | 0.44±0.04 | 0.45±0.09 |
| 在分批法中,具7毫升/升油酸甲酯的CTS培养基 | 1.2±0.1 | 0.12±0.02 |
| 在分批法中,具7毫升/升油酸甲酯和4毫升/升痕量元素的CTS培养基 | 2.1±0.2 | 3.3±0.7 |
| 间歇补料分批法 | 6.3±0.6 | 9.8±2.0 |
| 连续补料分批法 | 7.3±0.7 | 23±4.6 |
在更高浓度的油酸甲酯,没有观察到滴度的进一步提高,如图9所示。
除显示了油酸甲酯对细胞生长和产量的重要性外,上述图表也强调了生长培养基中痕量元素的重要性。预测酪胨提供的营养物对旺盛的细胞生长可能不够,进行了与油酸甲酯一同加入的外源痕量金属的添加。令人吃惊的是,发现这种补充对细胞生长和产量提高是必需的。缺少这种补充时,最大生物量浓度和埃坡霉素D的滴度分别只有1.2克/升和0.12毫克/升。这种低滴度与由基本培养基所获得的滴度相当。
补料分批法的开发
在分批发酵法中,当存在最佳浓度的油酸甲酯和痕量元素时,黄色粘球菌菌株在接种后的开始2天以指数增长。埃坡霉素D的产生开始于稳定期的初期,并随着油酸甲酯耗尽、当细胞裂解出现的时候于第5天停止,如图10A和10B所示。也显示了油酸甲酯消耗和氨产生的时程。生产培养基中的氨浓度在整个发酵过程中低于4mM。
为延长烧瓶培养物中细胞的生产期间,向培养基中加入酪胨和油酸甲酯,每天1次,加入速率分别为2克/升/天和3毫升/升/天。接种后48小时启动底物补料,且其后细胞以指数生长3天。如图11A所示,生物量浓度在第5天开始为平高线,并在第10天达到最大值6.3克/升。此外,埃坡霉素D的产生与稳定生长期一致,在第12天结束后,终产量达到9.8毫克/升。如图11B所示,在发酵过程中,油酸甲酯的消耗和氨的产生都以恒定的速率增加。
油酸甲酯的耗尽速率与其加入生物反映器的速率一样,且氨以3.2mM/天的速率累积。较低的酪胨或油酸甲酯供给速率会极大减少埃坡霉素D的滴度,而较高的供给速率会使细胞在达到显著产量之前过早裂解。
为测试补料分批法在更大规模中的有效性,每间隔24小时向生物反应器中的5升发酵罐间隔加入酪胨和油酸甲酯。如图12所示,所得的生产曲线与烧瓶培养物的生产曲线极为相似。底物补料在接种后24小时启动,且埃坡霉素的产生在第4天开始。在接种后第10天获得埃坡霉素D的峰滴度9.2毫克/升。
为评估更为明确定义的供给策略对生长和产量的影响,不断地往生物反应器中加入加倍的补料。如图13A所示,进行连续供给不影响细胞的生长,但是使细胞的产率增加了几乎3倍。接种后10天,埃坡霉素D的最终滴度达到27毫克/升。如图13B所示,在发酵过程中油酸甲酯消耗速率与其加入生产培养基中的速率一样,且氨以6.4mM/天的稳定速率释放。讨论
虽然最近可实现埃坡霉素D的化学合成(Harris等,1999;Meng等,1998;Sinha等,1998),但是,对该化合物的大规模生产而言,这种复杂的20步方法并非经济可行。虽然埃坡霉素D在黄色粘球菌菌株中的最初生产产量为0.16毫克/升,但是本发明改良的发酵方法基本上使产量水平增加至23毫克/升。
获得更高埃坡霉素D滴度的一个主要障碍是产物在发酵肉汤中的迅速降解。由于往培养基中加入吸附树脂,该问题得到缓解。加入XAD-16不影响细胞的生长,但是将分泌产物的损失减到最小,同时使其回收增加了3倍。
由于使用酪胨作为基本碳源和氮源,增加产物产量的另一个障碍是滴度的有限提高。虽然增加酪胨浓度可刺激黄色粘球菌菌株的生长,但是浓度超过5克/升导致滴度显著降低。在高浓度胨或酪胨存在时,次生代谢产物的抑制作用以前已在一些其它粘细菌发酵中得到证实,并且把它归因于培养基中氨的积累。
因为黄色粘球菌不能代谢多糖和糖(Reichenbach和Dworkin,1991),所以测试了它利用油作为碳源的能力。因为脂肪酸的氧化作用不但可作为细胞的能量来源,而且作为降解产物的乙酰CoA的形成也可提供埃坡霉素生物合成的前体,所以油类是具有吸引力的碳源。红色糖多孢菌、弗氏链霉菌和吸水链球菌发酵时加入油类,显示可分别增加聚酮化合物分子的产量,其中所述聚酮化合物如红霉素泰乐菌素和免疫调节剂L-683590(Mirjalili等,1999;Choi等,1998;Junker等,1998)。
根据对不同油类的筛选,确定油酸甲酯为促进细胞生长和埃坡霉素D产生的主要候选者。然而,这些改善只有在同时往生产培养基中加入痕量金属才可观察到。单独加入油酸甲酯至7毫升/升,可将最大细胞密度从0.4克DCW/升增至1.2克DCW/升,但产量保持在基线水平。随着外源补加4毫升/升痕量元素,最大生物量浓度增加至2.1克DCW/升,同时埃坡霉素D的滴度从0.45毫克/升增加至3.3毫克/升。这些发现显示,酪胨中营养成分缺陷可能对黄色粘球菌的生长和埃坡霉素类的形成至关重要。
随着分批法中油酸甲酯和痕量元素最佳浓度的确立,正努力开发可保持细胞旺盛生长和延长生产周期的补料策略。间歇和连续地以恒定速率供给酪胨和油酸甲酯得到评估,且两种方法在生长曲线上均有类似的改善。由于这两种底物的最佳供给,得到了为大约6.8克DCW/升的最大细胞密度。在这两种情况下,油酸甲酯随着加入发酵培养基而耗尽。
与细胞生长和油酸甲酯消耗不同,埃坡霉素和氨的产生受所选择的供给策略影响极大。在连续补料分批法培养中,埃坡霉素D最高滴度可达23毫克/升。这大约是间歇补料分批法培养所获滴度的2.5倍。随着连续供给,细胞释放氨的速率也高达2倍,表明酪胨的消耗速率更高。这些结果共同显示,与间歇补料分批法相关的较低滴度可能是由分解代谢产物阻抑所导致的,而不是氨的积累。此外,这些表明,黄色粘球菌菌株的产率对培养基中底物的量敏感,并且在限制底物的条件下,产率可最大。这也和增加生产培养基中的酪胨浓度会导致更高的细胞密度,但滴度更低的观察一致。
与用基本培养基进行分批法培养相比,连续补料分批培养使细胞密度增加17倍,滴度增加140倍。这种方法已从5升扩大到1000升,并且显示可同等执行。以生产规模产生埃坡霉素D所显示的结果表明,黄色粘球菌可用作从粘细菌生产其它生物活性分子的宿主。
参考文献
Bollag等,1995,埃坡霉素类,一类具有紫杉醇样作用机制的新微管稳定剂,癌研究(Cancer Res),55:2325-2333。
Choi等,1998,在弗氏链霉菌培养中,菜籽油对甲基丙二酸单酰-CoA羧基转移酶活性的影响,Biosci Biotechnol Biochem 62:902-906。
Chou等,1998,脱氧埃坡霉素B:一种有效的微管靶向抗肿瘤剂,其具有与埃坡霉素B相关的有应用前景的体内特性,美国国家科学院院报95:9642-9647。
Gerth等,1996,埃坡霉素A和B:来自纤维堆囊菌(粘细菌)抗真菌和细胞毒化合物一产生、物理化学特性和生物学特性,抗生素杂志(JAntibiot)(东京)49:560-563。
Gerth等,2000,埃坡霉素类的生物合成研究:碳骨架生物合成的起始,抗生素杂志(东京)53:1373-1377。
Gerth等,2001,埃坡霉素类的生物合成研究:PKS和埃坡霉素C/D单加氧酶,抗生素杂志(东京)54:144-148。
Harris等,1999,有前景的癌症化疗剂12,13-脱氧埃坡霉素B的新化学合成:对羟醛缩合的非对映选择性令人吃惊的长期效果的发现,美国化学会杂志(J Am Chem Soc)12:7050-7062。
Hecht等,1990,在抗生素发酵中,中空纤维支持的气膜用于原位去除铵,生物技术与生物工程(Biotechnol Bioeng)35:1042-1050。
Junker等,1998,使用大豆油和硫酸氨添加剂优化次生代谢物的产生,生物技术与生物工程60:580-588。
Kowalski等,1997,微管稳定剂埃坡霉素A和B与纯化的微管蛋白的活性,以及对紫杉醇抗性细胞的活性,生物化学杂志(J Biol Chem)272:2534-41。
Meng等,1997,通过成环烯烃的取代形成大环内酯的远期效应:应用于合成具完全活性的埃坡霉素同类物,美国化学会杂志119:2733-2734。
Mirjalili等,1999,红色糖多孢菌中,菜籽油的吸收对红霉素和三酮内酯(triketide lactone)产生的影响。生物科学进展(Biotechnol Prog)15:911-918。
Molnar等,2000。来自纤维堆囊菌So ce90的微管稳定剂埃坡霉素类A和B的生物合成基因簇,化学与生物学(Chem Biol)7:97-109。
Reichenbach等。原核生物(The Prokaryotes),第2版,纽约:Springer-Verlag.3417-3487页。
Reichenbach等,1993,生物活性次生代谢物的生产。Dworkin M、Kaiser D编辑。粘细菌II(Mycobacteria II)。华盛顿DC:美国微生物学会,347-397页。
Sinha等,1998,全合成埃坡霉素类的抗体催化途径,美国国家科学院院报95:14603-14608。
Su等,1997,埃坡霉素类的结构与活性关系以及首次在体内与紫杉醇的比较,Angew Chem Int Ed Engl 36:2093-2096。
Tang等,2000,埃坡霉素基因簇的克隆和异源表达,科学(Science)287:640-642。
B、埃坡霉素B的生产
烧瓶
将一支1毫升小瓶K111-32-25菌株融化,并将内含物转移到装有3毫升CYE种子培养基的玻璃管中。培养物在30℃孵育72±12小时,接着在250毫升折流锥形瓶中,将3毫升该试管培养物在50毫升CYE培养基中传代培养。该CYE瓶在30℃孵育24±8小时,同时,用2.5毫升这种种子(5%v/v)接种埃坡霉素生产瓶(250毫升折流锥形瓶中装有50毫升CTS-TA培养基)。然后,这些锥形瓶在30℃孵育48±12小时,培养基初始pH为7.4。
发酵罐
如上所述,对K111-32-25进行类似的接种物扩大,添加的步骤是将50毫升CYE种子中的25毫升在500毫升CYE中传代培养。该次级种子用于接种含9.5升CTS-TA和1克/升丙酮酸钠的10升发酵罐。该发酵方法的参数设定值为:pH-7.4;搅拌-400转/分;喷射速度-0.15vvm。这些参数足以保持DO在80%饱和以上。通过往培养基中加入2.5N硫酸和氢氧化钠来控制pH。埃坡霉素的最高滴度在48±8小时获得。
C、埃坡霉素D的生产
烧瓶
将一支1毫升小瓶的K111-40-1菌株(实施例4中所述)融化,同时将内含物转移到装有3毫升CYE种子培养基的玻璃管中。培养物在30℃孵育72±12小时,接着将3毫升这种试管培养物在装有50毫升CYE培养基的250毫升折流锥形瓶中传代培养。CYE瓶在30℃孵育24±8小时,同时用2.5毫升该种子(5%v/v)接种埃坡霉素生产瓶(250毫升折流锥形瓶中装有50毫升1X小麦谷蛋白培养基)。然后,将这些瓶在30℃孵育48±12小时,培养基初始pH为7.4。
发酵罐
如上所述,对K111-40-1进行类似的接种物扩大,添加的步骤是将50毫升CYE种子中的25毫升在500毫升CYE中传代培养。250毫升次级种子用于接种含4.5升CTS-TA的5升发酵罐,每天供给1克/升丙酮酸钠。该发酵方法的参数设定值为:pH-7.4;搅拌-400转/分;喷射速度-0.15vvm。这些参数足以保持DO在80%饱和以上。通过往培养基中加入2.5N硫酸和氢氧化钠来控制pH。埃坡霉素的最高滴度在36±8小时获得,埃坡霉素C的最高滴度为0.5毫克/升,埃坡霉素D的最高滴度为1.6毫克/升。
表5是所用培养基和它们各自组分的概述。
表5
| CYE种子培养基 | 组分 | 浓度 |
| 酪胨(Difco) | 10克/升 |
| 酵母提取物(Difco) | 5克/升 | |
| MgSO4·7H2O(EM Science) | 1克/升 | |
| HEPES缓冲液 | 50mM | |
| CTS-TA生产培养基 | 组分 | 浓度 |
| 酪胨(Difco) | 5克/升 | |
| MgSO4·7H2O(EM Science) | 2克/升 | |
| L-丙氨酸、L-丝氨酸、甘氨酸 | 1毫克/升 | |
| HEPES缓冲液 | 50mM | |
| 1X小麦谷蛋白生产培养基 | 组分 | 浓度 |
| 小麦谷蛋白(Sigma) | 5克/升 | |
| MgSO4·7H2O(EM Science) | 2克/升 | |
| HEPES缓冲液 | 50mM |
CYE种子培养基和CTS-TA生产培养基通过自动灭菌的高压灭菌法于121℃灭菌30分钟。小麦谷蛋白生产培养基通过高压灭菌法于121℃灭菌45分钟。
D、从黄色粘球菌中生产埃坡霉素C和D
一方面,本发明为粘球菌菌株提供了一种改良的发酵方法,其中所述粘细菌包括但不限于黄色粘球菌K111-40-1,其中发酵培养基提供可以利用而不产生氨的碳源。在一个优选的实施方案中,碳源是油,如油酸甲酯或类似的油。在用多种供给比例进行摇瓶测定时,这些方法会导致埃坡霉素类C和D的产生,其中大部分为埃坡霉素D,产量为15-25毫克/升,如下所述。
种子培养
将已在50毫升含2毫升/升油酸甲酯的CYE培养基中生长过的一支1毫升小瓶黄色粘球菌K111-40-1冻存细胞接种于含1毫升/升油酸甲酯的3毫升新鲜CYE培养基的无菌玻璃管中。试管在摇床中于30℃、250转/分钟孵育24小时。然后,将玻璃管中的接种物转移到250毫升非折流瓶中,该瓶装有50毫升含有2毫升/升油酸甲酯的新鲜CYE培养基。该瓶在摇床中于30℃、250转/分钟孵育48小时。
生产瓶
通过高压灭菌法,1克安伯来特(Amberlite)XAD-16在250毫升非折流瓶中于121℃灭菌30分钟。然后,将50毫升灭菌的生产培养基加入瓶中。瓶用5%(v/v)种子培养物接种,同时放置在孵育摇床中,转速为250转/分钟,温度为30℃。接种后2天,开始供给油酸甲酯,速率为3毫升/升/天,同时,接种后1天,开始供给酪胨,速率为2克/升/天。
产物提取
14天后,将生产瓶中的XAD树脂转移到50毫升离心管中。弃掉管中多余的培养基,而不移去树脂。然后用25毫升水洗涤树脂,并让树脂沉淀。弃掉管中的水,而不移去任何的树脂,同时往管中加入20毫升甲醇。为将产物埃坡霉素从树脂上提取出来,将离心管放置在摇床中,于175转/分摇动20-30分钟。将甲醇提取物转移到新离心管中,以备存储和LC/MS分析。表6是所用培养基和它们各自组分的概述。
表6
| CYE培养基 | 组分 | 浓度 |
| 酪胨(Difco) | 10克/升 | |
| 酵母提取物(Difco) | 5克/升 | |
| MgSO4·7H2O(EM Science) | 1克/升 | |
| 生产培养基 | 组分 | 浓度 |
| 酪胨(Difco) | 5克/升 | |
| MgSO4·7H2O(EM Science) | 2克/升 | |
| 高压灭菌后加入生 |
| 产培养基 | ||
| 1000x痕量元素溶液 | 4毫升/升 | |
| 油酸甲酯 | 2毫升/升 |
CYE培养基和生产培养基通过自动灭菌的高压灭菌法于121℃灭菌30分钟。痕量元素溶液过滤除菌;油酸甲酯单独高压灭菌。
将表7中的所有成分混合、往溶液中加入10毫升/升的浓缩H2SO4、并使最终体积为1升,依此制备痕量元素溶液。
表7
| 1000x痕量元素溶液 | 组分 | 浓度 |
| FeCl3 | 8.6克/升 | |
| ZnCl2 | 2.0克/升 | |
| MnCl2·4H2O | 1.0克/升 | |
| CuCl2·2H2O | 0.43克/升 | |
| H3BO3 | 0.31克/升 | |
| CaCl2·6H2O | 0.24克/升 | |
| Na2MoO4·2H2O | 0.24克/升 |
将得到的溶液过滤除菌。
E、发酵、产生和纯化来自黄色粘球菌的埃坡霉素类
黄色粘球菌菌株的描述
菌株K111-25-1是埃坡霉素B的产生菌株,它也可产生埃坡霉素A。菌株K111-40-1是埃坡霉素D的产生菌株,它也可产生埃坡霉素C。
平板上黄色粘球菌的保存
黄色粘球菌保存在CYE琼脂平板(平板组分见表8)上。在平板上划线分离后大约3天出现菌落。为获得预期的生长水平,平板在32℃孵育,然后存储于室温,可达3周(平板存储于4℃会杀死细胞)。
表8
| CYE琼脂平板 | 组分 | 浓度 |
| 水解的酪蛋白(胰消化物) | 10克/升 | |
| 酵母提取物 | 5克/升 | |
| 琼脂 | 15克/升 | |
| MgSO4 | 1克/升 | |
| 1M MOPS缓冲液(pH7.6) | 10毫升/升 |
*1升分批的培养基高压灭菌45分钟,然后倒于培养皿中。
用于细胞建库的黄色粘球菌对油的适应
将CYE平板上未适应油的菌落或冷冻的小瓶细胞转移到50毫升玻璃培养管中,该培养管中含3毫升CYE种子培养基和1滴来自100微升移液管的油酸甲酯。让细胞生长2-6天(30℃,175转/分钟),直至培养物在显微镜下显得致密为止。因为这些细胞不总是很好地适应油,启动几个(5-7)平行管。
细胞建库的方法(主细胞库)
启动如上所述的适应油的试管培养物。当试管培养物足够致密(OD=5+/-1)时,将所有试管内含物转移到含50毫升CYE-MOM种子培养基(见下表的培养基组分)的250毫升无菌摇瓶中。在摇床培养箱(30℃,175转/分钟)中生长48±12小时后,将5毫升该种子培养物转移到装有100毫升CYE-MOM的500毫升摇瓶中。让培养物在摇床培养箱(30℃,175转/分钟)中生长1天。在显微镜下检查培养物,以使细胞生长适当且无污染。
在无菌的250毫升摇瓶中,将80毫升种子培养物和24毫升无菌的90%甘油混合。使漩涡至彻底混匀,同时将该混合物按每小份1毫升加入100个无菌的、预先标记的低温小瓶。将它们放置于-80℃冰箱,慢慢冷冻。
细胞建库步骤(工作细胞库)
通过将如上所述制备的一小瓶主细胞库在室温融化,从而启动试管培育,然后将小瓶中的全部内含物置于装有3毫升CYE-MOM种子培养基的玻璃管中。当试管培育物足够致密(OD=5+/-1)时,将试管的全部内含物转移到含50毫升CYE-MOM种子培养基的无菌250毫升摇瓶中。生长48±12小时(30℃,175转/分钟)后,将5毫升该种子培养物转移到装有100毫升CYE-MOM的500毫升摇瓶中。让它生长1天(30℃,175转/分钟)。在显微镜下检查生长和污染。
在无菌的250毫升摇瓶中,将80毫升种子培养物和24毫升无菌的90%甘油混合。使漩涡至彻底混匀,同时将该混合物按每小份1毫升加入100个无菌的、预先标记的低温小瓶。将它们放置于-80℃冰箱,慢慢冷冻。种子培养基的组分
如表9所述的同样的种子培养基用于细胞建库并扩大细胞库小瓶至需要的容量。
表9
| CYE-MOM种子培养基* | 组分 | 浓度 |
| 水解的酪蛋白(胰消化物)-Difco | 10克/升 | |
| 酵母提取物-Difco | 5克/升 | |
| MgSO4·7H2O-EM Science | 1克/升 | |
| 油酸甲酯-Cognis | 2毫升/升 |
*注意:因为油酸甲酯在酪胨中形成乳剂,并且与其它组分不能完全混匀,所以在其它组分之后加入。
摇瓶、5升和1000升发酵的接种物放大
融化冷冻的工作细胞库小瓶的油酸甲酯适应细胞。将小瓶的全部内含物转移到含3毫升CYE-MOM种子培养基的50毫升玻璃培养管中。管放置在摇床(30℃,175转/分钟)中,生长48±24小时。将培养管的全部内含物转移到含50毫升CYE-MOM种子培养基的250毫升摇瓶中。摇瓶放置在摇床(30℃,175转/分钟)中,生长48±24小时。为了用于摇瓶实验,通过将10毫升该培养物在含40毫升新鲜CYE-MOM的5个新种子瓶中传代培养,以扩大种子。将种子瓶在摇床中孵育24±12小时,用作瓶容量(30-100毫升)生产培养的接种物。按4.5%混合(种子和生产培养基)的初始体积接种生产瓶。
为准备小规模(5-10升)发酵的种子,将1个50毫升种子瓶的全部内含物在含500毫升CYE-MOM的2.8升无菌费氏烧瓶中传代培养。该费氏烧瓶在摇床(30℃,175转/分钟)中孵育48±24小时,用作发酵罐的接种物。按5%混合的初始体积接种生产发酵。
大规模(1000升)发酵需要种子的进一步扩大。此时,将1升费氏烧瓶中的种子用来接种(以5%体积)含9升CYE-MOM的10升种子发酵罐。通过加入2.5N氢氧化钾和2.5N硫酸,将发酵罐的pH控制在7.4。温度设定在30℃。通过在400-700转/分钟之间的阶式搅拌速率,使溶解氧维持在50%饱和或者50%以上饱和。起始搅拌速率设定为400转/分钟,且喷射速率维持在0.1v/v/m。在10升发酵罐中生长24±12小时后,将全部培养物转移到含90升CYE-MOM的150升发酵罐中。PH再次用2.5N氢氧化钾和2.5N硫酸控制在7.4。温度设定在30℃。通过在400-700转/分钟之间的阶式搅拌速率,使溶解氧维持在50%饱和或者50%以上饱和。起始搅拌速率设定为400转/分钟,且喷射速率维持在0.1v/v/m。
准备用于发酵的XAD-16树脂
将所需量的XAD-16树脂(Rohm & Haas)转移到安全的甲醇容器中,该容器的最小体积为XAD-16树脂重量的3倍(即1.2千克树脂需要最少为3.6升的容器)。用100%甲醇充分洗涤树脂,以去除存在于未用过的树脂上的单体。加入甲醇,甲醇的量按升计,为树脂按千克计的2倍(即3千克XAD-16需要6升甲醇)。将甲醇和XAD悬液混合5分钟,以去除存在于XAD-16上的单体。为将树脂的碎裂减至最小,在混合时轻轻搅拌浆液。停止搅拌,让树脂在重力下至少沉降15分钟。排出容器中的甲醇,在XAD层上留下0.5-1英寸的甲醇层。将XAD和甲醇从搅拌容器中转移至Amicon VA250柱。将上层床支撑物加到柱中,同时,通过顺时针转动密封调节旋钮密封床支撑物。用至少5倍柱体积的甲醇洗涤柱中的XAD,速度为300±50厘米/小时。用溶剂废物容器收集流出的甲醇。用至少10倍柱体积的去离子水洗涤柱中的XAD,速度为300±50厘米/小时。
埃坡霉素生产培养基的组分如表10所述
表10
| CTS-MOM生产培养基 | 组分 | 浓度 |
| 酪胨(Difco) | 5克/升 | |
| MgSO4·7H2O(EM Science) | 2克/升 | |
| XAD-16 | 20克/升 | |
| 高压灭菌后加入 | ||
| 1000x痕量元素溶液 | 4毫升/升 | |
| 油酸甲酯 | 2毫升/升 |
*注意:因为油酸甲酯在酪胨中形成乳剂,并且与其它组分不能完全混匀,所以在其它组分之后加入。痕量元素溶液如表7所述。
准备和瓶规模(50毫升)的埃坡霉素生产发酵
在250毫升摇瓶中,1克XAD-16用足够的去离子水(~3毫升)覆盖,进行高压灭菌。通过高压灭菌法,瓶于121℃灭菌30分钟。往瓶中无菌加入如下培养基组分:50毫升CTS-MOM生产培养基和2.5毫升1M HEPES缓冲液(用氢氧化钾将pH滴定为7.6)。用2.5毫升CYE种子瓶(4.5%体积/接种物体积)接种培养物。生产瓶在摇床上于30℃和175转/分钟孵育。
接种后24±6小时,开始供给酪胨和油酸甲酯。在这一时间点和以后每24±6小时,供给1毫升100克/升酪胨溶液和150微升油酸甲酯。继续这种供给方案,直至初次供给后13天,或者直至观察到细胞开始裂解(11-14天)。为确定产物埃坡霉素的动力学,取5毫升有代表性的发酵肉汤和XAD的充分混合物作为样品。此外,每天取小份(0.25-0.5毫升)无XAD的肉汤,在视觉上检查培养物的生长状况。当观察到大量细胞裂解时,应收获剩余体积的培养物。
准备和5升规模的埃坡霉素生产发酵
100克XAD和8克MgSO4·7H2O与3.9升去离子水混合,并且在5升的B-Braun生物反应器中灭菌(90分钟,121℃)。将足够(133毫升)的预先灭菌的酪胨/去离子水溶液(150克/升)无菌泵进,使发酵罐中酪胨的最终浓度达到5克/升。通过加入10毫升油酸甲酯,使油的最初浓度达到2毫升/升。最后,在孵育之前无菌加入16毫升预先灭菌的痕量元素溶液。然后发酵罐用200毫升CYE种子培养物接种(4.8%体积/体积),并且允许生长24±6小时。在这一时间点,开始供给酪胨(2克/升/天,连续供给)和油酸甲酯(共3毫升/升/天,每90分钟进行一次半连续供给)。生物反应器中的气流保持恒定,速度为0.4-0.5vvm(如供给程序一样,增加发酵体积导致这种变化)。通过阶式搅拌(400-700转/分钟),将溶解氧浓度控制在50%饱和。通过设定初始搅拌速度为400转/分钟和初始气流为0.5vvm,建立100%饱和溶解氧校准点。通过自动加入2.5N H2SO4和2.5NKOH,保持pH为7.4。接种后埃坡霉素可连续生产11-14天,当肉汤样品中的细胞裂解明显和对氧的需求(如搅拌速率所示)急剧下降时,收集生物反应器。在本发酵方法中,埃坡霉素D的滴度一般可达到18-25毫克/升。
准备和1000升规模的埃坡霉素生产发酵
用作埃坡霉素生产的1000升发酵罐按如下准备。600升水和18升(11.574千克)XAD-16在发酵罐中灭菌(45分钟,121℃)。痕量金属和MgSO4过滤除菌(通过预先灭菌的0.2微米聚醚砜膜囊状过滤器),直接导入发酵容器。用同样的囊状过滤器,加入2.9升痕量元素溶液和足够量的浓缩MgSO4溶液(发酵罐中的最终浓度达到2克/升)。约200升117克/升酪胨和175毫升/升油酸甲酯的混合物在260升的供给容器中灭菌。将大约32升灭菌混合物加入1000升发酵罐中。将水过滤(通过同样的囊状过滤器)入容器中,使最终体积为710升。搅拌速度为100转/分钟。反压保持在100-300毫巴。当接种后溶解氧(DO)达到50%时,搅拌速度增加到150转/分钟。当DO再次达到50%时,搅拌速度增加到200转/分钟。通过阶式气流(0Lpm-240Lpm),控制DO在50%饱和。通过自动加入2.5MKOH和2.5N H2SO4,使pH设定值保持在7.4。用38升来自150升发酵罐的种子接种发酵罐(5%体积/体积)。
DO达到50%后(第2次,大约接种后10±5小时),开始加入0.570升/小时的酪胨-油酸甲酯供给溶液,并且继续直至收获发酵罐。接种后10天收获生物反应器。埃坡霉素D的最后滴度测定为大约20±5毫克/升。
发酵采样方法
为进行瓶中的动力学实验,用25毫升移液管采集5-50毫升充分混合的肉汤和XAD树脂,并置于10或50毫升的圆锥形试管中。对生物反应器样品,将50毫升混合的肉汤和树脂样品置于50毫升圆锥形试管中。然后让圆锥形试管静置10分钟,使XAD沉降于试管底部。此时,从XAD树脂中弃掉肉汤。如果XAD不沉淀,则用10-25毫升吸量管移去肉汤。
为定量埃坡霉素滴度,用甲醇提取XAD树脂
XAD树脂已在重力作用下沉降于样品管底部后(如前述采样方法),将所有上清转移到一个新50毫升圆锥形管中。加水至50毫升标记洗涤XAD树脂1次,通过颠倒充分混合以及在重力作用下让XAD树脂再次沉降。轻轻倒掉试管中的水样混合物,而不倒掉XAD。用1毫升移液器把枪尖推入试管的底部移去最后几毫升水。往试管中加入甲醇至25毫升,并且盖上试管的盖子。将圆锥形试管水平放在摇床上30分钟(温度20-30℃),以从XAD树脂中充分提取所有埃坡霉素。
埃坡霉素定量的HPLC方法
利用Hewlett Packard 1090 HPLC,进行埃坡霉素类C和D的分析,UV检测波长为250nm。来自XAD树脂(50微升)的甲醇提取溶液通过2个4.6×10mm保护柱(Inertsil,C18 OD 53,5μm)和一个更长的用于层析法分离相同材料的柱(4.6×150mm)注入。该方法为用60%乙腈和40%水恒溶剂运行18分钟。用这种方法,埃坡霉素D在13分钟洗脱,埃坡霉素C在10.3分钟洗脱。用从发酵肉汤中纯化的埃坡霉素D制备标准品。
用于生长曲线中的干细胞称重方法
将Sorvall RC5B离心机(用SH-3000斗式转子)的温度设定为20℃。称重至少已在80℃烤箱烘烤至少1天的50毫升圆锥形试管。在试管侧面记录皮重和发酵样品的身份。将40毫升肉汤(不含XAD)倒进或用吸量管吸进称量管。圆锥形管于4700转/分钟(4200g)离心30分钟。沉淀后,倒掉上清,同时用40毫升去离子水重悬细胞沉淀,以洗涤沉淀。管再次离心(4200g,30分钟)。弃掉上清,并将管在80℃干燥箱中至少放置2天。对管称重,并在管上记录最后重量。然后干细胞重(DCW)按下述等式计算:DCW(克/升)=(管最后重量(克)-管的皮重(克))/.04升。
铵离子浓度的测定
在埃坡霉素发酵中,例行测定发酵肉汤中的氨浓度。在微量离心机中离心(5分钟,12000转/分钟),使1毫升发酵肉汤澄清。采用Sigma(Catalog#171-UV)的氨测定试剂盒进行定量,其中用澄清的发酵肉汤取代试剂盒操作程序中的血浆。因为该比色测定的线性响应范围只为0.01176-0.882mM/升,所以澄清的发酵样品一般在去离子水中稀释20-100倍,以在此范围内测定铵浓度。
测定残存的油酸甲酯浓度
通过用甲醇提取发酵肉汤样品和在HPLC上运行这些提取肉汤样品,可测定存在于发酵肉汤中残存油酸甲酯的量。油酸甲酯浓度的定量用Hewlett Packard 1090 HPLC进行,UV检测波长为210nm。全部肉汤样品(1-4毫升)用等体积甲醇提取,并在12,000g离心,以沉淀不溶成分。澄清上清(50微升)注入4.6×10mm提取柱(Inertsil,C18 OD 53,5μm)、用50%乙腈洗2分钟、然后在主柱(4.6×150mm,同样的固定相和流速)上洗脱24分钟,乙腈梯度从50%开始,100%结束。100%乙腈在柱中的流动保持5分钟。由于油酸甲酯不均一的特性,它洗脱时有许多不同的峰,而不是单一的纯化合物。然而,在全部提取出的油酸甲酯中,大约64-67%出现在2个主峰,分别在25.3±0.2和27.1±0.2分钟洗脱。甲醇提取的发酵样品中的油酸甲酯可通过测定这2个主峰的总面积进行估计,然后用50%水/甲醇配制的油酸甲酯标准物的这2个主峰的总面积进行校准。
埃坡霉素D的纯化和结晶
本发明提供了埃坡霉素类和埃坡霉素D纯化方法,并提供了高纯度埃坡霉素D制剂,包括结晶形态的埃坡霉素D。本方法的优点包括最初的纯化步骤只需要乙醇(如甲醇)和水,这使得可高效利用产物合并液,并对费时和费力的蒸发步骤的需求减至最少。本方法只需要单一的蒸发步骤,该方法地每10-15克埃坡霉素需要蒸发1升乙醇。本方法中,用合成的聚苯乙烯-二乙烯基苯树脂如HP20SS填充柱子来去除极性和亲脂的杂质。该柱产生含10%埃坡霉素类的中间产物,并且无需使用高度可燃或者有毒溶剂的液体/液体提取。
另一个改进涉及具分布有40-60微米颗粒的C18树脂的使用,如Bakerbond C18,它使得可用低压柱和泵(低于50psi),这极大降低成本。C18层析步骤的起始材料是溶于稀释装填溶剂的溶液。溶剂非常弱,以便埃坡霉素类以高度浓缩、政协委员密带位于柱的顶部,这使柱子在大装载量(2-5克埃坡霉素/升树脂)的情况下良好运行。因为一般的柱装载量是1克/升或者更少,且层析通常是纯化中最为昂贵的步骤,所以这种改进使成本大量节约。此外,本方法允许使用醇,如甲醇,而不是层析步骤中的乙腈。含埃坡霉素的收集物从二相溶剂系统中结晶,在该溶剂系统中,水是主要溶剂,以提供结晶形式的埃坡霉素。
在一个实施方案中,纯化方法由下述步骤和材料组成。发酵肉汤中的XAD树脂(1)收集于过滤蓝,(2)并洗脱出来,提供XAD提取物,(3)该提取物用水稀释,(4)并通过HP20SS柱以提供HP20SS收集物。(5)HP20SS收集物用水稀释,(6)经过C18层析以提供埃坡霉素收集物,(7)该埃坡霉素收集物用水稀释,(8)经过溶剂交换以提供浓缩的埃坡霉素收集物。浓缩的埃坡霉素收集物经过(9)活性炭过滤、(10)蒸发和(11)结晶,提供高度纯化的材料。
从2个1000升黄色粘球菌发酵中(1031001K和1117001K),总共分离到11克埃坡霉素D,并纯化成白色晶体粉末。最终产物的纯度大于95%,埃坡霉素D的回收效率为71%。
表11概述了纯化中使用的HPLC方法
表11
| Epol法 | ||
| 柱 | Inertsil ODS3,5μm,4.6×150mm | |
| 流速 | 1毫升/分钟 | |
| 柱加热炉 | 50℃ | |
| 运行时间 | 15分钟 | |
| 检测 | UV 250nm处 | |
| 梯度 | 0分钟;60∶40 ACN/水12分钟;100∶0 ACN/水12.1分钟;60∶40 ACN/水 | |
| Epo78法 |
| 柱 | Inertsil ODS3,5μm,4.6×150mm | |
| 流速 | 1毫升/分钟 | |
| 柱加热炉 | 50℃ | |
| 运行时间 | 5分钟 | |
| 检测 | UV 250nm处 | |
| 梯度 | 0分钟;78∶22 ACN/水 |
本部分使用的材料如下。HP20SS树脂购自Mitsubishi。C18购自Bakerbond C18 40g,甲醇购自Fisher Bulk(55gal)。使用去离子水。
发酵运行1031001K
步骤1 XAD洗脱(K125-173)
使用带有13升、150μm捕获蓝的主流过滤装置,从发酵培养物中过滤17升XAD-16树脂。用捕获的XAD树脂装填Amicon VA250柱,并用65升(3.8倍柱体积)水洗涤,流速1.0升/分钟。然后用230升溶于水的80%甲醇将产物埃坡霉素D从树脂上洗脱。
步骤2 固相提取(K125-175)
将77升水加入步骤1的产物合并液(230升)中,以将装载溶剂稀释成溶于水的60%甲醇。将得到的悬液(307升)混合,并装载用5升HP20SS树脂填充的Amicon VA180柱,该树脂之前已用5倍柱体积的60%甲醇平衡。装载流速为1升/分钟。装载后,柱用13升60%甲醇洗涤,并用77升75%甲醇洗脱,流速为325毫升/分钟。收集31个2.5升洗脱级分。发现级分10-26(42.5升)含有埃坡霉素D,将这些部分合并在一起。
步骤3 层析(K125-179)
用2个20升的旋转蒸发器将步骤2中的产物合并液蒸发成油。在蒸发过程中,为将泡沫减至最小,有必要加入乙醇。干燥材料重悬于1.0升甲醇中,并用0.67升水稀释,制成1.67升60%甲醇溶液。将得到的溶液装载1升C18层析柱(55×4.8厘米),该柱之前已用3倍柱体积的60%甲醇平衡。装载流速平均为64毫升/分钟。装载柱用1升60%甲醇洗涤,并用等梯度70%甲醇洗脱产物埃坡霉素D,流速为33毫升/分钟。其收集了27个级分,第1级分含3.8升。接着是3个500毫升级分和23个250毫升级分。认为级分5-20为最好的收集物(K125-179-D),含有4.8克埃坡霉素D。级分3-4(K125-179-C)含有1.4克埃坡霉素D。因为该收集物中也含有高浓度埃坡霉素C,所以将它放到一边以便重做(步骤3b)。
步骤4 层析(K119-153)
也含有高浓度类似物埃坡霉素C的埃坡霉素D级分在C18树脂上重新进行如下层析。用C18树脂填充2.5×50厘米柱,以1升100%甲醇洗涤,并用1升溶于水的55%甲醇平衡,流速为20毫升/分钟。压力差为125psi。起始材料(K125-179-C,1040毫升)用260毫升水稀释,以便装载溶液含有溶于水的55%甲醇。得到的溶液(1300毫升)装载在树脂上,并将另外250毫升55%甲醇通过该柱。柱开始用5升65%甲醇洗脱,接着用3升溶于水的70%甲醇。在65%甲醇洗脱期间,共收集了48个100毫升级分。当转为用70%甲醇后,共收集了10个250毫升的级分。级分50-58是最好的埃坡霉素D收集物(K119-153-D),含有1.0克目标产物。
步骤5a的结晶(K119-158)
本步骤的起始材料是步骤3和4层析产物的合并。开始,将120毫升乙醇加入7.9克含5.5克埃坡霉素D的固体颗粒中。随着轻轻混合,固体颗粒完全溶解,并将溶液转移到放置在通风橱搅拌平台上的400毫升烧杯中。加入1″搅拌棒,同时溶液得到迅速搅拌。同时在大约5分钟的时间内慢慢加入100毫升水。当观察到白色小晶体形成时,将溶液再搅拌15分钟,直至溶液变稠,并有白色固体。然后将烧杯从搅拌平台上移开,用铝箔覆盖,并在冰箱(2℃)中放置12小时。白色固体用Whatman #50滤纸过滤,并且在头次收获时,不进行另外的洗涤。固体放置在结晶盘中,并且在真空烘箱(40℃,15毫巴)中干燥1小时。随后,材料从烘箱中移去,制成更细的颗粒,并在真空烘箱中再干燥4小时。本结晶方法产生3.41克灰白色固体。Epol HPLC方法用于测定最终产物的层析纯度。HPLC结果和相应的1H和13C NMR数据一起,都证实干燥材料含有95%以上的埃坡霉素D。头次收获物的回收率为58%。
步骤5b结晶化(K119-167)
本步骤的起始材料是来自步骤5a的蒸发母液。开始,70毫升乙醇和30毫升水加入3.4克含有2.1克埃坡霉素D的固体颗粒。将清澈溶液转移到烧杯中,并往杯中加入1克脱色的活性炭。混合物在中速设置上搅拌10分钟,然后用Whatman #50滤纸过滤。活性炭用2个10毫升乙醇洗涤,并再次过滤。用旋转蒸发器使合并的滤出液干燥,并将固体重悬于50毫升乙醇中。得到的溶液置于250毫升烧杯中,随着充分搅拌,慢慢加入50毫升水。为促进晶体的形成,往混合物中加入少量的种子晶体(1毫克)。搅拌几分钟后,观察到有另外的白色固体形成。当搅拌继续时,使一阵氮气轻轻吹过混合物表面。15分钟后,将烧杯在冰箱2℃放置36小时。用Whatman #50滤纸过滤混合物,以捕获晶体,同时用另外的7毫升50∶50的乙醇∶水洗涤固体。随后,晶体在真空烘箱中干燥4小时。该结晶步骤产生1.46克含95%以上埃坡霉素D的白色晶体。
发酵运行1117001K
步骤1XAD的洗脱(K125-182)
使用带有13升、150μm捕获蓝的主流过滤装置,从发酵培养物中过滤17升XAD-16树脂。用捕获的XAD树脂装填Amicon VA250柱,并用58升(3.4倍柱体积)水洗涤,流速1.0升/分钟。然后用170升溶于水的80%甲醇将产物埃坡霉素D从树脂上洗脱。在用水洗涤和首次柱体积洗脱期间,柱的反压稳定地增至3巴以上,伴随着最终流速在300毫升/分钟以下。因此,将XAD树脂从柱中移去,并重新填充另一个Amicon VA250柱。交换后,反压下降至1巴以下,并且流速保持在1.0升/分钟。将单一的170升洗脱级分收集在600升的不锈钢容器中。根据HPLC分析,发现
步骤1中的产物收集物中含8.4克埃坡霉素D。
步骤2固相提取(K145-150)
将57升水加入步骤1的产物合并液(170升)中,以将装载溶剂稀释成溶于水的60%甲醇。得到的悬液(227升)用高架闪电搅拌器(overheadlightning mixer)搅拌,并装载用6.5升HP20SS树脂填充的Amicon VA180柱,该树脂之前已用5倍柱体积的60%甲醇平衡。装载流速为1升/分钟。装载后,柱用16升60%甲醇洗涤,并用84升75%甲醇洗脱,流速为300毫升/分钟。收集的7个级分的体积分别是18升、6升、6升、6升、36升、6升和6升。级分4和5含埃坡霉素D总计8.8克,将它们合并在一起。
步骤3层析(K145-160)
用2个20升的旋转蒸发器,将步骤2的产物合并液蒸发成油。在蒸发过程中,为将泡沫减至最小,往混合物中加入10升乙醇。干燥材料重悬于2.8升甲醇中,并用3.4升水稀释,制成6.2升45%的甲醇溶液。得到的溶液泵进1升C18层析柱(55×4.8厘米),该柱之前已用5倍柱体积的45%甲醇平衡。装载流速平均为100毫升/分钟。用1升60%甲醇洗涤装载柱,并用分级梯度将产物埃坡霉素D从树脂上洗脱下来,流速为100毫升/分钟。柱用5升55%甲醇、11.5升60%甲醇和13.5升65%甲醇洗脱。在用55%甲醇洗脱过程中,共收集了10个500毫升的洗脱级分。转为用60%甲醇后,共收集了23个500毫升的洗脱级分。在用最后的65%甲醇洗脱过程中收集了11个500毫升的洗脱级分,接着是8个1升的洗脱级分。最好的埃坡霉素D收集液(K145-160-D)由级分28-50组成,含有8.3克目标产物。污染有0.4克埃坡霉素C的级分26-27(K145-160-C)含有0.2克埃坡霉素D。将所有的25个级分混合在一起。
为将产物合并液稀释成溶于水的40%甲醇,将9.5升水加至15.8升装载溶液中。然后将得到的溶液(25.3升)泵进700毫升C18层析柱(9×10厘米),该柱之前已用4倍柱体积的40%甲醇平衡。装载流速平均为360毫升/分钟。用1升40%甲醇洗涤装载柱,用3.75升100%乙醇将产物埃坡霉素D从树脂上洗脱。洗脱物用旋转蒸发器蒸发干。将该固体颗粒重悬于100毫升丙酮,不溶物质用Whatman #2滤纸从溶液中滤出。将过滤颗粒用另外115毫升丙酮洗涤,并再次过滤。丙酮提取之后,往合并滤液中加入2克脱色活性炭。混合物在中速设置上搅拌1小时,并用Whatman #50滤纸过滤。活性炭用180毫升乙醇洗涤,并再次过滤。合并滤液,并旋转蒸发至干。
步骤4层析(K119-174)
来自步骤3的干燥材料重悬于5.0升溶于水的50%甲醇,并装载1升C18层析柱(55×4.8厘米),该柱之前已用3倍柱体积50%甲醇平衡。装载流速平均为80毫升/分钟。随后,用1升50%甲醇洗涤柱子,并且产物埃坡霉素D用70%甲醇、按相同的流速从树脂上等梯度洗脱下来。共收集了48个洗脱级分,其中开始47个级分含有240毫升,最后一个级分含有1升。级分25-48认为是最好的合并液(K119-174-D),含有7.4克埃坡霉素D。级分21-24(K119-174-C)含有1.1克埃坡霉素D。因为该合并液也含有高浓度的埃坡霉素C,所以将它放到一边,以便重做。
步骤5结晶(K119-177)
为在结晶步骤之前进行溶剂的交换,将3.9升水加入来自步骤5的6.4升最好的埃坡霉素D合并液(K119-174-D),以将装载溶液稀释成溶于水的40%甲醇。然后将所得溶液装载200毫升C18层析柱(2.5×10厘米),该柱之前已用3倍柱体积的40%甲醇平衡。用200毫升40%甲醇洗涤装载柱,并用1升100%乙醇将产物埃坡霉素D从树脂上洗脱下来。洗脱物用旋转蒸发器蒸发至干,并将固体重悬于150毫升100%乙醇。将清澈溶液转移到烧杯中,并伴随着良好的搅拌,慢慢加入175毫升水。溶液中也加入小块种子晶体(1毫克),以促进晶体形成。当可观察到白色小晶体形成时,溶液再搅拌15分钟,直至溶液变稠,并伴随有白色固体。然后将烧杯从搅拌平台上移开,覆盖铝箔,并在冰箱(2℃)中放置12小时。白色固体用Whatman #50滤纸过滤,在此首次收获时,没有进行另外的洗涤。将固体置于结晶盘,并在真空烘箱(40℃,15毫巴)中干燥6小时。本结晶方法产生6.2克含95%以上埃坡霉素D的白色固体。首次收获的回收率是74%。
结果
运行1031001K的埃坡霉素D回收率是纯度约为97.5-98.8%的4.8克晶体物质。运行1117001K的埃坡霉素D回收率是纯度约为97.7%的6.2克晶体物质。这些运行方法的杂质特性如表12所示。
表12
| 1031001K运行 | 步骤 | 产物 | Epo C | Epo490 | Epo D |
| 2 | SPE | 23 | 4 | 74 | |
| 3-4 | 总色谱 | 0.7 | 0.7 | 90.6 | |
| 5a | 结晶 | 1.0 | 1.0 | 97.5 | |
| 5b | 结晶 | 0.7 | 0.5 | 98.8 | |
| 1117001K运行 | 步骤 | 产物 | Epo C | Epo490 | Epo D |
| 2 | SPE | 18 | 2 | 60 | |
| 3 | C18色谱 | 5.2 | 1.6 | 81.4 | |
| 4 | C18色谱 | 1.6 | 1.8 | 96.6 | |
| 5 | 结晶 | 0.8 | 1.4 | 97.7 |
“Epo490”是本发明的一种新埃坡霉素化合物,为10,11-脱氢埃坡霉素D,该化合物由粘球菌宿主细胞生产。
本纯化方法起于对埃坡霉素D纯化方法而努力做的大规模改进,为在发酵培养基中适应于油酸甲酯的使用。埃坡霉素D产物从XAD树脂上的洗脱以直接的方式进行。不使用100%甲醇,而用10倍柱体积的80%甲醇从柱中的珠上洗脱产物。在XAD洗脱步骤中,注意到发酵肉汤中裂解细胞的存在可能会使纯化柱堵塞。103100-1K发酵运行在明显的细胞裂解发生之前收获,而111700-1K发酵运行只在大量细胞裂解发生后才收获。然而,在洗脱过程时,只在后一运行中观察到高的反压和低流速。因此,可能是此运行中的裂解细胞聚集,并随后堵塞了柱的过滤器。
这些纯化运行表明,埃坡霉素D在80%甲醇中于室温至少稳定存在1天。根据HPLC分析,在这些条件下,未检测到产物的降解。该发现允许将来自XAD洗脱步骤的170升产物合并液在600升不锈钢容器中过夜,而不需冷冻。为进一步改善该方法,使用溶剂交换柱,这比用旋转蒸发来浓缩产物合并液的体积费时和费力更少。因此,可用溶剂交换步骤代替大规模旋转蒸发。
虽然在XAD洗脱步骤中,有显著量的油依然结合在树脂上,但在洗脱液中仍存在相当量的油。甚至在HP20SS固相提取后,在产物合并液中可清楚看到油滴,且在C18层析中是个问题。为得到最佳层析特性,装载溶液中埃坡霉素D的浓度应维持在2克/升以下。在更高浓度时,起始材料在柱子上出油的倾向。
当供给物质含有3%以上埃坡霉素C或epo490时,结晶是不可能的。在1117001K纯化就是如此。第1次层析步骤产生的产物含有5%埃坡霉素C。多次尝试后,没有完成该物质的结晶。然而,将该物质通过第2次层析步骤可将埃坡霉素C降低至1%,并产生易于结晶的供给物质。
实施例4
构建具无功能epoK基因的粘球菌菌株
通过插入灭活epoK基因,从K111-32.25菌株构建K111-40-1菌株。为构建该epoK突变体,将卡那霉素抗性盒插入epoK基因。从pKOS35-79.85分离含epoK基因的4879bp片段,并将其连入pBluescript SKII+的NotI位点,从而完成该构建。该质粒pKOS35-83.5用ScaI部分切割,并且将7.4kb片段和1.5kb含pBJ180-2的卡那霉素抗性基因的EcoRI-BamHI片段连接,产生质粒pKOS90-55,其中后者的1.5kb片段具有用DNA聚合酶I的克列诺片段补平的DNA平端。最后,将来自pBJ183的约400bpRP4 oriT片段连入XbaI和EcoRI位点,以产生pKOS90-63。该质粒用DraI线性化,并电穿孔入黄色粘球菌菌株K111-32.25,挑选转化子,以提供黄色粘球菌菌株K111-40.1。菌株K111-40.1根据布达佩斯条约于2000年11月21日保藏于美国典型培养物保藏中心(ATCC),10801University Blvd.,马纳萨斯,VA,20110-2209美国,且在保藏号PTA-2712下得到。
为产生无标记的epoK突变,pKOS35-83.5用ScaI切割,并将2.9kb和4.3kb的片段连接起来。质粒pKOS90-101在epoK上有一框架内缺失。接着,将KG2中的3kb BamHI和NdeI片段连入pKOS90-101的DraI位点,以构建pKOS90-105,其中前者的3kb片段有用DNA聚合酶I的克列诺片段补平的DNA平端,并含有卡那霉素抗性和galK基因。将该质粒电穿孔入K111-32.25,同时挑选卡那霉素抗性电穿孔子。为用缺失体替换野生型epoK拷贝,通过在半乳糖平板上生长来挑选第2次重组事件。检测半乳糖抗性菌落的埃坡霉素C和D的产生,并将生产菌株命名为K111-72.4.4,并且根据布达佩斯条约于2000年11月21日保藏于美国典型培养物保藏中心(ATCC),10801 University Blvd.,马纳萨斯,VA,20110-2209美国,且在保藏号PTA-2713下可获得。
实施例5
matBC的添加
matBC基因分别编码丙二酸单酰CoA合成酶和二羧酸酯载体蛋白。见An和Kim,1998,欧洲生物化学杂志(Eur.J.Biochem.)257:395-402,此处引用作为参考。这2种蛋白质在细胞内负责将外源丙二酸酯转换为丙二酸单酰CoA。这2种基因产物可运输二羧酸,并将它们转换成CoA衍生物(见PCT专利申请号US00/28573,此处引用作为参考)。这2种基因可插入黄色粘球菌染色体,以增加丙二酸单酰CoA和甲基丙二酸单酰CoA的细胞浓度,从而增加聚酮化合物的产量。这可通过如下途径实现,用BglII和SpeI切割pMATOP-2,并将其连入pKOS35-82.1的BgIII和SpeI位点,其中pKOS35-82.1含有四环素抗性赋予基因、Mx8 att位点和驱使matBC表达的黄色粘球菌pilA启动子。该质粒可电穿孔导入黄色粘球菌。因为pilA启动子被高效转录,所以在本事件中有必要插入较弱的启动子,因为过多的MatB和MatC影响细胞生长。其它启动子包括卡那霉素抗性赋予基因的启动子。
实施例6
装载组件中的KSY突变
启动埃坡霉素生物合成的假设机制是丙二酸酯与装载结构域的ACP结合,以及随后通过装载KS结构域的脱羧作用。装载KS结构域在活性位点的半胱氨酸处含有一个酪氨酸(KSY),这使得该活性位点不能进行缩合反应。然而,认为它仍可进行脱羧反应。用鼠脂肪酸合酶进行的实验显示,在活性中心半胱氨酸处含有一个谷氨酰胺(KSQ)的KS结构域,它与野生型相比可使脱羧作用上升2个数量级,但将该氨基酸改变成丝氨酸、丙氨酸、天冬酰胺、甘氨酸或苏氨酸却不会提高脱羧作用。因此,将KSY改变成KSQ会增加埃坡霉素的起动,从而使埃坡霉素的产量增加。为在菌株K111-32.25中产生该变化,构建具有约850bp埃坡霉素KS装载组件编码序列的质粒pKOS39-148。通过定点诱变在该质粒中产生KSQ突变。为在K111-32.25中进行基因替换,将来自KG2的卡那霉素抗性和galK基因插入pKOS39-148的DraI位点,以产生质粒pKOS111-56.2A和pKOS111-56.2B。这些质粒中的卡那霉素-galK盒的方向不同。这些质粒电穿孔入K111-32.25,并挑选卡那霉素抗性菌落,从而产生菌株K111-63。为取代野生型装载组件KS,将K111-63铺于CYE半乳糖平板,并通过PCR和测序来筛选KSQ突变。
实施例7
mtaA的添加
为增加磷酸泛酰巯基乙基转移酶(PPTase)蛋白质的水平,将来自橙色标桩囊菌菌株DW4的PPTase加入K111-32.25。这通过如下方法实现:用以下引物
111-44.1(AAAAGCTTCGGGGCACCTCCTGGCTGTCGGC)
和111-44.4(GGTTAATTAATCACCCTCCTCCCACCCCGGGCAT).
从DW4的染色体DNA通过PCR扩增matA。见Silakowski等,1999,生物化学杂志274(52):37391-37399,此处引用作为参考。该约800bp片段用NcoI切割,并连入用PstI切割后,将其DNA末端用DNA聚合酶I的克列诺片段补平、并用NcoI切割的pUHE24-2B。该质粒命名为pKOS111-54。将mtaA基因转移到含有四环素抗性赋予基因、Mx8 att位点和驱动mtaA表达的黄色粘球菌pilA启动子的质粒pKOS35-82.1中。将该质粒导入黄色粘球菌,并整合Mx8噬菌体附着位点。
实施例8
启动子置换质粒的构建
为提高埃坡霉素的产量水平,并阐明在本发明的宿主细胞中可用于表达PKS基因的多种启动子,用其它适当启动子置换纤维堆囊菌埃坡霉素PKS基因启动子,构建了一系列载体和宿主细胞,如本实施例中所述。
A、带有下游侧翼区质粒的构建
粘粒pKoS35-70.8A3用NsiI和AvrII切割。将9.5kb片段与用PstI和AvrII切割的pSL1190连接,产生pKOS90-13。质粒pKOS90-13约为12.9kb。将它用EcoRI/AvrII切割。将5.1kb片段与用EcoRI/SpeI消化的pBluescript连接,以产生pKOS90-64(~8.1kb)。该质粒含有启动子(epoA和起始密码子上游的一些序列)的下游侧翼区。EcoRI位点在epoA基因的起始密码子上游约220bp。AvrII位点在EcoRI位点下游5100bp。
B、上游侧翼区的克隆
用引物90-66.1和90-67(如下所示)克隆上游侧翼区。引物90-67在PCR片段的5’末端,且引物90-66.1在PCR片段的3’末端,该片段终止于epoA基因起始密码子之前的2481bp。将~2.2kb片段用HindIII切割。加入克列诺聚合酶补平HindIII位点。将该片段连入pNEB193的HincII位点。挑选那些具有正确插入方向,也就是EcoRI位点在插入片段下游末端、并且HindIII在插入片段上游末端的克隆,并命名为pKOS90-90。
90-66.1:5′GCGGG AAGCTT TCACGGCGCAGGCCCTCGTGGG 3′
| |
接头 HindIII 引物
90-67:5′GC GGTACC TTCAACAGGCAGGCCGTCTCATG 3′
| |
接头 KpnI 引物
C、最终质粒的构建
质粒pKOS90-90用EcoRI和HindIII切割。将2.2kb片段与用EcoRI/HindIII消化的pKOS90-64连接,构建pKOS 90-91(10.3kb)。质粒pKOS90-91含有启动子的上游侧翼区,接着是pBluescript的下游侧翼区。在2个侧翼区中间,有一个可克隆目标启动子的PacI位点。然后将galK/kanr盒插入,使其能够重组进入黄色粘球菌。质粒pKOS90-91用DraI切割。DraI在amp基因内切割一次,在载体内(amp基因附近)切割2次。质粒KG-2用BamHI/NdeI切割,并加入克列诺聚合酶将片段补平。将3kb片段(含galK/kanr基因)与pKOS90-91的~9.8kb DraI片段连接,以产生pKOS90-102(12.8kb)。
D、构建带有其它前导序列的质粒
埃坡霉素PKS基因的天然前导区可用具不同核糖体结合位点的前导序列替换。质粒pKOS39-136(如美国专利申请系列号09/443,501所述,申请日1999年11月19日)用PacI/AscI切割。分离含有前导序列和部分epoA序列的3 kb片段,并与pKOS90-102的9.6kb PacI/AscI片段连接,以产生pKOS90-106(~12.7kb)。
E、构建启动子置换质粒
I、MTA(粘噻唑菌醇)启动子
用引物111-44.3和111-44.5(如下所示)从橙色标桩囊菌染色体DNA(菌株DW4)中PCR扩增粘噻唑菌醇启动子。将~454bp条带克隆入pNEB193的HincH位点,以产生pKOS90-107。质粒pKOS90-107用PstI和XbaI切割,并用克列诺补平。将560bp条带克隆入用PacI切割、并用克列诺补平的pKOS90-102和pKOS90-106(PacI在pKOS90-102和pKOS90-106中只切割1次)。筛选正确插入方向的质粒。MTA启动子/pKOS90102质粒命名为pKOS90-114(13.36kb),同时,MTA启动子/pKOS90-106质粒命名为pKOS90-113(13.26kb)。
111-443 5′AA AAGCTT AGGCGGTATTGCTTTCGTTGCACT 3′
| |
接头 HindIII 引物
11144.5 5′GG TTAATTAA GGTCAGCACACGGTCCGTGTGCAT 3′
| |
接头 PacI 引物
将这些质粒电穿孔入含有埃坡霉素PKS基因的粘球菌宿主细胞,挑选卡那霉素抗性重组予以鉴定单一交换体重组子。对这些重组子筛选半乳糖抗性,以确定双交换重组子,这些重组子通过Southern分析和PCR进行筛选,从而鉴定那些含有目标重组事件的重组子。将目标重组子进行培养,并检测埃坡霉素的产生。
II、TA启动子
用下列引物:
111-44.8(AAAGATCTCTCCCGATGCGGGAAGGC)和11144.9
(GGGGATCCAATGGAAGGGGATGTCCGCGGAA).
从菌株TA中PCR扩增推定的TA启动子和taA,taA编码推定的转录抗终止子。该大约1.1kb片段用BamHI和BglII切割,并连入用BamHI切割的pNEB193。该质粒命名为pKOS111-56.1。质粒pKOS111-56.1用EcoRI和HindIII切割,并用克列诺补平。该~1.1kb条带克隆入用PacI切割、并用克列诺补平的pKOS90-102和pKOS90-106(PacI在pKOS90-102和pKOS90-106中只切割1次)。筛选具有正确插入方向的质粒。TA启动子/90-102质粒命名为pKOS90-115(13.9Kb),TA启动子/pKOS90-106质粒命名为pKOS90-111(13.8kb)。
这些质粒电穿孔入含有埃坡霉素PKS基因的粘球菌宿主细胞,同时挑选卡那霉素抗性重组子以鉴定单一交换重组子。对这些重组子挑选半乳糖抗性,以鉴定双交换重组子,这些重组子通过Southern分析和PCR进行筛选,从而鉴定那些含有目标重组事件的重组子。将目标重组子进行培养,并检测埃坡霉素的产生。
III、pilA启动子
质粒pKOS35-71B用EcoRI切割,并用克列诺补平。将800bp片段克隆入用PacI切割、并用克列诺补平的pKOS90-102和pKOS90-106。筛选具有正确插入方向的质粒。pilA启动子/pKOS90-102质粒命名为pKOS90-120(13.6kb),pilA启动子/pKOS90-106命名为pKOS90-121(13.5kb)。
这些质粒电穿孔入含有埃坡霉素PKS基因的粘球菌宿主细胞,同时挑选卡那霉素抗性重组子以鉴定单一交换重组子。对这些重组子挑选半乳糖抗性,以鉴定双交换重组子,这些重组子通过Southern分析和PCR进行筛选,从而鉴定那些含有目标重组事件的重组子。将目标重组子进行培养,并检测埃坡霉素的产生。
IV、kan启动子
质粒pBJ180-2用BamHI/BglII切割,并用克列诺补平。将350bp片段克隆入用PacI切割、并用克列诺补平的pKOS90-102和pKOS90-106。筛选具有正确插入方向的质粒。kan启动子/pKOS90-102质粒命名为pKOS90-126(13.15kb),kan启动子pKOS/90-106质粒命名为pKOS90-122(13.05kb)。
这些质粒电穿孔导入含有埃坡霉素PKS基因的粘球菌宿主细胞,同时挑选卡那霉素抗性重组子以鉴定单一交换具有重组子。对这些重组子挑选半乳糖抗性,以鉴定双交换重组子,这些重组子通过Southern分析和PCR进行筛选,从而鉴定那些含有目标重组事件的重组子。将目标重组子进行培养,并检测埃坡霉素的产生。
V、So ce90启动子
用引物111-44.6和111-44.7(如下所示),从So ce90染色体DNA扩增So ce90启动子。该~900bp条带用Pacl切割,并克隆入用PacI切割的pNEB193,以产生pKOS90-125。质粒pKOS90-125用PacI切割。将924bp条带克隆入用PacI切割的pKOS90-102和pKOS90-106。筛选具有正确插入方向的质粒。Soce90启动子/pKOS90-102质粒命名为pKOS90-127(13.6kb),Soce90启动子/pKOS90-106质粒命名为pKOS90-128(13.7kb)。
这些质粒电穿孔入含有埃坡霉素PKS基因的粘球菌宿主细胞,同时挑选卡那霉素抗性重组子以鉴定单一交换重组子。对这些重组子挑选半乳糖抗性,以鉴定双交换重组子,这些重组子通过Southern分析和PCR进行筛选,从而鉴定那些含有目标重组事件的重组子。将目标重组子进行培养,并检测埃坡霉素的产生。
111-44.6 5′GG TTAATTAA CATCGCGCTATCAGCAGCGCTGAG 3′
| |
接头 PacI 引物
111-44.7 5′GG TTAATTAA TCCTCAGCGGCTGACCCGCTCGCG 3′
| |
接头 PacI 引物
实施例9
KS2敲除菌株的构建
本实施例描述了以下埃坡霉素PKS衍生物的构建,其中通过突变将活性位点半胱氨酸密码子改变为丙氨酸密码子,使延伸组件2的KS结构域失活。对所得的衍生物PKS提供合成的前体(如下面的实施例所述),以制备本发明的埃坡霉素衍生物。
用下列引物:90-103(5′-
AAAAAATGCATCTACCTCGCTCGTGGCGGTT-3′)和90-107.1
(5′-CCCCC TCTAGA ATAGGTCGGCAGCGGTACCCG-3′)
从质粒pKOS35-78.2中扩增埃坡霉素PKS基因的下游侧翼区。~2kb的PCR产物用NsiI/XbaI切割,并与用NsiI和SpeI消化的pSL1190连接,以产生pKOS90-123(~5.4kb)。用下列引物:
90-105(5′-
TTTTTATGCATGCGGCAGTTTGAACGG-AGATGCT-3′)和
90-106(5′-CCCCCGAATTCTCCCGGAAGGCACACGGAGAC-3′)
从pKOS35-78.2 DNA扩增~2kb的PCR片段,将该片段用NsiI切割,并与用NsiI/EcoRV消化的pKOS90-123连接,以产生pKOS90-130(~7.5kb)。当该质粒用NsiI切割、DNA末端用DNA聚合酶I的克列诺片段补平,并重新连接时,产生了质粒pKOS90-131。为将galK/kanr盒克隆入该质粒,用BamHI/NdeI切割质粒KG-2,并用DNA聚合酶的克列诺片段补平。将3kb的片段克隆入pKOS90-131的DraI位点(DraI在该载体中切割3次),构建质粒pKOS90-132(10.5kb)。为产生从半胱氨酸到丙氨酸的预期改变,使用NsiI位点以实现KS2敲除。当pKOS90-130用NsiI切割、用DNA聚合酶I的克列诺片段补平,并重新连接时,丙氨酸密码子替换了半胱氨酸密码子。根据上面所述产生目标菌株的方法,将得到的质粒导入本发明的黄色粘球菌菌株,以产生目标菌株。
通过本方法(使用埃坡霉素A、B、C和D生产株K111-32.25),构建黄色粘球菌菌株K90-132.1.1.2,并根据布达佩斯条约于2000年11月21日保藏于美国典型培养物保藏中心(ATCC),10801 University Blvd.,马纳萨斯,VA,20110-2209美国,且在保藏号PTA-2715下可得到。为表明当提供适当的二酮起始单位时,由菌株生产的所得到的PKS可合成埃坡霉素类,使菌株K90-132.1.1.2在50毫升CTS加20%XAD中于30℃生长3天,然后提供200毫克/毫升如下所示的噻唑二酮:

菌株再培养另一个5天,收集XAD,并用10%甲醇提取埃坡霉素类。将提取物干燥,重悬于0.2毫升乙腈,并用LC/MS分析0.05毫升样品,结果如预期一样,显示存在埃坡霉素类B和D。如下列实施例所讨论的一样,本系统可用于生产多种埃坡霉素类似物。
实施例10
来自化学生物合成的修饰埃坡霉素
本实施例描述了用于通过化学生物合成产生埃坡霉素衍生物的一系列硫酯。来自纤维堆囊菌的埃坡霉素生物合成基因簇的DNA序列显示,PKS的起动涉及到聚酮化合物和氨基酸组分的混合物。起动涉及用丙二酸单酰CoA装载装载组件的PKS样部分,接着是脱羧作用和用半胱氨酸装载延伸组件的NRPS,然后进行缩合作用形成酶结合的N-乙酰半胱氨酸。环化作用形成二氢噻唑之后是氧化作用,以形成酶结合的2-甲基噻唑-4-羧酸酯,它是装载组件和NRPS的产物。随后通过组件2的酮合酶与甲基丙二酸单酰CoA的缩合作用提供二酮的等同物,如图解6所示。
图解6

本发明以一种类似于PCT公开号99/03986和97/02358中描述的制备6-dEB和红霉素类似物的方式提供了用于化学生物合成产生埃坡霉素衍生物的方法和试剂。提供了2类供给底物:NRPS产物的类似物和二酮等同物的类似物。NRPS产物类似物和带突变NRPS样结构域的PKS酶一起使用,且二酮等同物和在组件2内带突变KS结构域(如实施例9中所述)的PKS酶一起使用。在下面方案7和8的结构中,R、R1和R2可独立地选自甲基、乙基、低碳烷基(C1-C6)和取代的低碳烷基。
图解7显示的是装载组件类似物的例子。
图解7
通过活化相应的羧酸,并用N-乙酰半胱胺处理来制备装载组件类似物。活化方法包括形成酰基氯、混合酸酐,或与缩合试剂如碳化二亚胺反应。
图解8显示的是二酮等同物的例子:
二酮等同物用三步法制备。首先,将相应的醛用维悌希试剂或等同物处理,形成取代的丙烯酸酯。将该酯皂化成酸,然后活化,并用N-乙酰半胱胺处理。
接着是制备装载组件产物类似物和二酮等同物的反应图解的示例。其它适合制备二酮等同物的化合物如图1所示的羧酸(或者可转换成羧酸的醛),它们可转化成N-乙酰半胱胺,用于提供本发明的宿主细胞。
A、噻吩-3-羧酸-N-乙酰半胱胺-硫酯
噻吩-3-羧酸(128毫克)在2毫升无水四氢呋喃中的溶液在惰性环境下,用三乙胺(0.25毫升)和二苯基磷酰基氮化物(0.50毫升)处理。1小时后,加入N-乙酰半胱胺(0.25毫升),并让反应进行12小时。将混合物倒入水中,并用等体积乙酸乙酯提取3次。合并有机提取物,接下来用水、1N HCl、饱和CuSO4和盐水洗涤,然后在MgSO4上干燥、过滤,并于真空浓缩。用乙醚,接着用乙酸乙酯在SiO2上层析,可提供在静止状态下可结晶的纯产物。
B、呋喃-3-羧酸-N-乙酰半胱胺-硫酯
呋喃-3-羧酸(112毫克)在2毫升无水四氢呋喃中的溶液在惰性环境下,用三乙胺(0.25毫升)和二苯基磷酰基氮化物(0.50毫升)处理。1小时后,加入N-乙酰半胱胺(0.25毫升)并让反应进行12小时。将混合物倒入水中,并用等体积乙酸乙酯提取3次。合并有机提取物,接下来用水、1N HCl、饱和CuSO4和盐水洗涤,然后在MgSO4上干燥、过滤,并在真空中浓缩。用乙醚,接着用乙酸乙酯在SiO2上层析,可提供在静止状态下可结晶的纯产物。
C、吡咯-2-羧酸-N-乙酰半胱胺-硫酯
吡咯-2-羧酸(112毫克)在2毫升无水四氢呋喃中的溶液在惰性环境下,用三乙胺(0.25毫升)和二苯基磷酰基氮化物(0.50毫升)处理。1小时后,加入N-乙酰半胱胺(0.25毫升)并让反应进行12小时。将混合物倒入水中,并用等体积乙酸乙酯提取3次。合并有机提取物,接下来用水、1N HCl、饱和CuSO4和盐水洗涤,然后在MgSO4上干燥、过滤,并在真空中浓缩。用乙醚,接着用乙酸乙酯在SiO2上层析,可提供在静止状态下可结晶的纯产物。
D、2-甲基-3-(3-噻吩基)丙烯酸-N-乙酰半胱胺-硫酯
(1)乙基-2-甲基-3-(3-噻吩基)丙烯酸酯∶噻吩-3-羧醛(1.12克)和(乙氧甲酰基亚乙基)三苯基膦(4.3克)在无水四氢呋喃(20毫升)中的混合物加热回流16小时。将混合物冷至周围环境温度,并在真空下浓缩至干。将固体残留物悬于1∶1的乙醚/己烷中,并过滤除去三苯基膦氧化物。滤出物用1∶1的乙醚/已烷,使滤出物通过SiO2填料过滤,以提供浅黄色油形式的产物(1.78克,91%)。
(2)2-甲基-3-(3-噻吩基)丙烯酸:来自(1)的酯溶于甲醇(5毫升)和8N KOH(5毫升)的混合物中,并加热回流30分钟。将混合物冷至周围环境温度,用水稀释,并用乙醚洗2次。水相用1N HCl酸化,然后用等体积乙醚提取3次。合并有机提取物,用MgSO4干燥,过滤,并在真空下浓缩至干。从2∶1己烷/乙醚中结晶可提供无色针状形式的产物。
(3)2-甲基-3-(3-噻吩基)丙烯酸N-乙酰半胱胺硫酯:2-甲基-3-(3-噻吩基)丙烯酸(168毫克)在2毫升无水四氢呋喃中的溶液,在惰性环境下用三乙胺(0.56毫升)和二苯基磷酰基氮化物(0.45毫升)处理。15分钟后,加入N-乙酰半胱胺(0.15毫升),并让反应进行4小时。将混合物倒入水中,并用等体积乙酸乙酯提取3次。合并有机抽体物,接下来用水、1N HCl、饱和CuSO4和盐水洗涤,然后用MgSO4干燥,过滤,并在真空下浓缩。用乙酸乙酯在SiO2上层析,可提供在静止状态下可结晶的纯产物。
将上述化合物提供给含有本发明的重组埃坡霉素PKS的宿主细胞培养物,以制备相应的本发明的埃坡霉素衍生物,其中所述本发明的重组埃坡霉素PKS中的延伸组件2的NRPS或KS结构域已通过突变而失活。
实施例11
埃坡霉素类似物的的生产
A、13-酮-埃坡霉素类似物的产生
失活埃坡霉素PKS中延伸组件4的KR结构域,导致产生了用于产生13-酮-埃坡霉素的本发明的杂合PKS。通过用多种如下所述的缺失变体替换野生型基因来改变延伸组件4的KR结构域。首先,用质粒pKOS39-118B(来自粘粒pKOS35-70.4 epoD基因的亚克隆)作为模板,扩增片段。形成该缺失左侧的寡核苷酸引物是TL3和TL4,如下所示:
TL3:5′-ATGAATTCATGATGGCCCGAGCAGCG;和
TL4:5′-ATCTGCAGCCAGTACCGCTGCCGCTGCCA.
形成该缺失右侧的寡核苷酸引物是TL5和TL6,如下所示:
TL5:5′-GCTCTAGAACCCGGAACTGGCGTGGCCTGT;和
TL6:5-GCAGATCTACCGCGTGAGGACACGGCCTT将PCR片段克隆入载体Litmus 39,并测序以证实已获得目标片段。然后,用限制性酶PstI和BamHI消化含有TL3/TL4片段的克隆,并分离~4.6kb片段。将用引物TL5/TL6获得的2.0kb PCR片段用限制酶BgIII和XbaI处理,然后连入(i)短KR接头TL23和TL24(它们一起退火,形成带有单链突出的双链接头),产生pKOS122-29;或(ii)用引物TL33+TL34通过PCR获得、然后用限制酶NsiI和SpeI处理的长(epoDH3*)接头,产生质粒pKOS122-30。这些寡核苷酸接头和引物的序列如下所示:
TL23:5′-GGCGCCCGGCCAAGAGCGCCGCGCCGGTCGGCGGGCCAGCCGGGGACGGGT;
TL24:5′-CTAGACCCGTCCCCGGCTGGCCCGCCGACCGGCGCGGCGCTCTTGGCCG-GCGCCTGCA;
TL33:5′-GGATGCATGCGCCGGCCGAAGGGCTCGGA;和
TL34:5′-TCACTAGTCAGCGACACCGGCGCTGCGTTT.
通过测序确认含有预期置换的质粒,然后用限制酶DraI消化。然后,将每个克隆的大片段与卡那霉素抗性和gaIK基因(KG或kan-gal)盒连接,以提供递送质粒。通过电穿孔将递送质粒转化入埃坡霉素B生产菌株黄色粘球菌K111-32.25。筛选转化子,并挑选卡那霉素抗性、半乳糖抗性存活株,以鉴定除去KG基因的克隆。通过PCR证明重组菌株中已除去KG,并且已替换预期基因。重组菌株在装有50毫升CTS培养基和2%XAD-16的瓶中发酵5天,埃坡霉素类似物用10毫升甲醇从XAD上洗脱。根据LC/MS谱和NMR测定其结构。命名为K122-56的菌株根据布达佩斯条约于2000年11月21日保藏于美国典型培养物保藏中心(ATCC),10801 University Blvd.,马纳萨斯,VA,20110-2209美国,且在保藏号PTA-2714下可得到。K122-56菌株(来自质粒pKOS122-29)产生的产物以13-酮-11,12-脱氢-埃坡霉素D为主,其结构如下所示。
K122-56菌株也产生作为次要产物的13-酮-埃坡霉素C和D,它们各自的结构如下。

从来源于质粒pKOS122-30的菌株K122-30得到类似的结果。这些化合物、菌株和产生它们的PKS酶是本发明的新化合物、新菌株和新PKS酶。
可生产13-酮-11,12脱氢埃坡霉素类的其它本发明的菌株,包括那些通过一个或多个点突变而使KR结构域失活的菌株。例如,将KR结构域的组成型酪氨酸残基突变成苯丙氨酸,使KR活性下降大约10%,并导致某些13-酮-埃坡霉素的产生。KR结构域的其它突变可使KR活性下降更多,或者消除所有的KR活性,但也使埃坡霉素产量下降。
B、13-羟基-埃坡霉素类似物的产生
用异源KR结构域,如雷帕霉素PKS的延伸组件2或FK520 PKS的延伸组件3的KR结构域,替换埃坡霉素PKS的延伸组件5的KR、DH和ER区,将导致用于产生13-羟基-埃坡霉素的杂合PKS的形成。按与本实施例的A部分所述类似的方法完成构建。用质粒pKOS39-118B作为模板来扩增epoD基因目标部分的寡核苷酸引物是:
TL7:5′-GCGCTCGAGAGCGCGGGTATCGCT;
TL8:5′-GAGATGCATCCAATGGCGCTCACGCT;
TL9:5′-GCTCTAGAGCCGCGCGCCTTGGGGCGCT;和
TL10:5-GCAGATCTTGGGGCGCTGCCTGTGGAA.
将由引物TL7/TL8产生的PCR片段克隆入载体LITMUS 28,得到的克隆用限制酶NsiI和BglII消化,分离5.1kb片段,与用TL9/TL10产生的、用限制酶BgIII和XbaI处理、并连接到KR盒的2.2kb片段连接。用引物TL31和TL32,通过PCR制备FK520 PKS的KR盒,然后用限制酶XbaI和PstI消化。这些引物如下所示:
TL31:5′-GGCTGCAGACCCAGACCGCGGGGGACGC;和
TL32:5′-GCTCAGAGGTGGCGCCGGCCGCCCGGCG
除了黄色粘球菌KIII-72.4.4用作受体菌外,菌株构建的其余部分类似于本实施例的A部分所述进行。用FK520 PKS延伸组件3的KR结构域替换埃坡霉素PKS延伸组件5的KR、DH和ER结构域的菌株命名为K122-148,其根据布达佩斯条约于2000年11月21日保藏于美国典型培养物保藏中心(ATCC),10801 University Blvd.,马纳萨斯,VA,20110-2209美国,且在保藏号PTA-2711下可得到。菌株K122-148产生的主要产物为13-羟基-10,11-脱氢-埃坡霉素D,C衍生物为次要产物,其结构如下所示。
命名为K122-52的类似菌株产生同样的化合物,在该菌株内,用雷帕霉素PKS延伸组件2的KR结构域作为替换物。这些化合物、菌株和产生它们的PKS酶是本发明中的新化合物、新菌株和新PKS酶。
C、9-酮-埃坡霉素类似物的产生
失活埃坡霉素PKS延伸组件6的KR结构域,导致本发明的能产生9-酮-埃坡霉素类的新PKS产生。KR结构域可通过位点专一诱变改变一个或多个保守残基而失活。埃坡霉素PKS延伸组件6的KR结构域的DNA和氨基酸序列如下所示:
36710 36720 36730 36740 36750
GACGGCACCTACCTCGTGACCGGCGGTCTGGGTGGGCTCGGTCTGA
D G T Y L V T G G L G G L G L>
36760 36770 36780 36790 36800
GCGTGGCTGGATGGCTGGCCGAGCAGGGGGCTGGGCATCTGGTGCTGGTG
S V A G W L A E Q G A G H L V L V>
36810 36820 36830 36840 36850
GGCCGCTCCGGTGCGGTGAGCGCGGAGCAGCAGACGGCTGTCGCCGCGCT
G R S G A V S A E Q Q T A V A A L>
36860 36870 36880 36890 36900
CGAGGCGCACGGCGCGCGTGTCACGGTAGCGAGGGCAGACGTCGCCGATC
E A H G A R V T V A R A D V A D>
36910 36920 36930 36940 36950
GGGCGCAGATCGAGCGGATCCTCCGCGAGGTTACCGCGTCGGGGATGCCG
R A Q I E R I L R E V T A S G M P>
36960 36970 36980 36990 37000
CTCCGCGGCGTCGTTCATGCGGCCGGTATCCTGGACGACGGGCTGCTGAT
L R G V V H A A G I L D D G L L M>
37010 37020 37030 37040 37050
GCAGCAAACCCCCGCGCGGTTCCGCGCGGTCATGGCGCCCAAGGTCCGAG
Q Q T P A R F R A V M A P K V R>
37060 37070 37080 37090 37100
GGGCCTTGCACCTGCATGCGTTGACACGCGAAGCGCCGCTCTCCTTCTTC
G A L H L H A L T R E A P L S F F>
37110 37120 37130 37140 37150
GTGCTGTACGCTTCGGGAGCAGGGCTCTTGGGCTCGCCGGGCCAGGGCAA
V L Y A S G A G L L G S P G Q G N>
37160 37170 37180 37190 37200
CTACGCCGCGGCCAACACGTTCCTCGACGCTCTGGCACACCACCGGAGGG
Y A A A N T F L D A L A H H R R>
37210 37220 37230 37240 37250
CGCAGGGGCTGCCAGCATTGAGCATCGACTGGGGCCTGTTCGCGGACGTG
A Q G L P A L S I D W G L F A D V>
GGTTTG
G L>
本发明提供的新9-酮-埃坡霉素PKS延伸组件6的突变和失活KR结构域的DNA和氨基酸序列如下所示:
36710 36720 36730 36740 36750
GACGGCACCTACCTCGTGACCGGCGCTCTGGGTGGGCTCGGTCTGA
D G T Y L V T G A L G G L G L>
36760 36770 36780 36790 36800
GCGTGGCTGGATGGCTGGCCGAGCAGGGGGCTGGGCATCTGGTGCTGGTG
S V A G W L A E Q G A G H L V L V>
36810 36820 36830 36840 36850
GGCCGCTCCGGTGCGGTGAGCGCGGAGCAGCAGACGGCTGTCGCCGCGCT
G R S G A V S A E Q Q T A V A A L>
36860 36870 36880 36890 36900
CGAGGCGCACGGCGCGCGTGTCACGGTAGCGAGGGCAGACGTCGCCGATC
E A H G A R V T V A R A D V A D>
36910 36920 36930 36940 36950
GGGCGCAGATCGAGCGGATCCTCCGCGAGGTTACCGCGTCGGGGATGCCG
R A Q I E R I L R E V T A S G M P>
36960 36970 36980 36990 37000
CTCCGCGGCGTCGTTCATGCGCCCGGTATCCTGGACGACGGGCTGCTGAT
L R G V V H A A G I L D D G L L M>
37010 37020 37030 37040 37050
GCAGCAAACCCCCGCGCGGTTCCGCGCGGTCATGGCGCCCAAGGTCCGAG
Q Q T P A R F R A V M A P K V R>
37060 37070 37080 37090 37100
GGGCCTTGCACCTGCATGCGTTGACACGCGAAGCGCCGCTCTCCTTCTTC
G A L H L H A L T R E A P L S F F>
37110 37120 37130 37140 37150
GTGCTGTACGCTTCGGGAGCAGGGCTCTTGGGCTCGCCGGGCCAGGGCAA
V L Y A S G A G L L G S P G Q G N>
37160 37170 37180 37190 37200
CTTCGCCACGGCCAACACGTTCCTCGACGCTCTGGCACACCACCGGAGGG
F A T A N T F L D A L A H H R R>
37210 37220 37230 37240 37250
CGCAGGGGCTGCCAGCATTGAGCATCGACTGGGGCCTGTTCGCGGACGTG
A Q G L P A L S I D W G L F A D V>
GGTTTG
G L>
除了将黄色粘球菌K111-72.4.4作为受体菌外,包括该突变KR结构域编码序列的菌株一般按本实施例的A部分所述进行构建。延伸组件6的KR结构域已失活的菌株命名为K39-164,并且根据布达佩斯条约于2000年11月21日保藏于美国典型培养物保藏中心(ATCC),10801 UniversityBlvd.,马纳萨斯,VA,20110-2209美国,且在保藏号PTA-2716下可得到。菌株K39-164产生作为主产物的9-酮-埃坡霉素D,C衍生物为次要产物,其结构如下所示:

这些化合物、菌株和产生它们的PKS酶是本发明的新化合物、新菌株和新PKS酶。
D、2-甲基-埃坡霉素类似物的产生
通过将延伸组件9的AT结构域("epoAT9")编码序列用甲基丙二酸单酰CoA特异的AT结构域的编码序列替换,构建埃坡霉素A、B、C和D的2-甲基-埃坡霉素类似物。合适的置换体AT结构域编码序列可例如从编码FK520 PKS的延伸组件2(“FKAT2”;见PCT公开号00/020601,此处引用作为参考)、埃坡霉素PKS的延伸组件2(“epoAT2”)和tmbA基因编码的PKS的延伸组件3(“tmbAT3”;见美国专利号6,090,601和美国专利申请系列号60/271,245,申请日2001年2月15日,此处引用作为参考)的基因获得。置换一般按如上所述进行,产生的具体埃坡霉素仅仅取决于已转导置换体的粘球菌宿主产生何种埃坡霉素。
这样,用带有设计的BglII(AGATCT)和NsiI(ATGCAT)限制酶交接处识别序列的epoAT2(核苷酸12251至核苷酸13287)、或FKAT2、或tmbAT3编码序列替换epoAT9编码序列(从核苷酸50979至核苷酸52026)。
以pKOS39-125 DNA作为模板,第1轮PCR用来产生~1.6kb片段。将该PCR片段亚克隆入载体LITMUS28的HindIII和BgIII位点,并测序;带有目标序列的质粒命名为P1。该PCR使用的寡核苷酸是:
TLII-1:5′-ACAAGCTTGCGAAAAAGAACGCGTCT;和
TLII-2:5′-CGAGATCTGCCGGGCGAGGAAGCGGCCCTG.
以pKOS39-125 DNA作为模板,第2轮PCR用来产生~1.9kb片段。将该PCR片段亚克隆入载体LITMUS28的NsiI和SpeI位点,并测序;带有目标序列的质粒命名为P2。该PCR使用的寡核苷酸是:
TLII-3B:5′-GCATGCATGCGCCGGTCGATGGTGAG;和
TLII-4:5′-AGACTAGTCACCGGCTGGCCCACCACAAGG.
然后用限制酶BglII和SpeI消化质粒P1,分离4.5kb片段,与来自质粒P2的~1.9kb NsiI-SpeI限制性片段连接,并与作为NsiI-BglII限制性片段分离的3个置换体AT片段(FKAT2,epoAT2,tmbAT3)中的1个连接,以获得质粒P3.1、P3.2和P3.3。通过PCR产生置换体AT片段,使用了下面的寡核苷酸引物:
对FKAT2:
TLII-20:5′-GCATGCATCCAGTAGCGGTCACGGCGGA;和
TLII-21:5′-CGAGATCTGTGTTCGCGTTCCCCGGGCAG;
对tmbAT3:
TLII-13:5′-GCATGCATCCAGTAGCGCTGCCGCTGGAAT;和
TLII-14:5′-GCAGATCTGTGTTCGTGTTCCCCGGCCA;以及
对epoAT2:
TLII-17:5′-GCATGCATCCAGTACCGCTCGCGCTG;和
TLII-18:5′-CGAGATCTGTCTTCGTCTTTCCCGGCCAG.
然后通过在kan-gal盒的DraI位点的插入,对质粒P3.1、P3.2和P3.3进行改变。得到的质粒转化本发明的埃坡霉素生产菌黄色粘球菌宿主细胞(即KIII-72.4.4),培养细胞,并如上所述挑选双交换重组事件的细胞。通过PCR筛选挑选的克隆。显示有预期重组事件的克隆在50毫升培养基中培养,并通过LC/MS筛选产生目标化合物的克隆。预期产物是2-甲基-埃坡霉素D和2-甲基-埃坡霉素C,其结构如下所示。
E、6-去甲基-埃坡霉素类似物的产生
通过用丙二酸单酰CoA特异的AT结构域的编码序列替换延伸组件7AT结构域(“epoAT7”)的编码序列,构建埃坡霉素A、B、C和D的6-去甲基-埃坡霉素类似物。合适的置换体AT结构域编码序列例如可从编码FK520 PKS的延伸组件3、埃坡霉素PKS的延伸组件5(“epoAT5”)和tmbA基因编码的PKS的延伸组件4的基因获得,此处引用作为参考。置换一般按如上所述进行,产生的具体埃坡霉素仅仅取决于已转导置换体的粘球菌宿主产生何种埃坡霉素。
这样,用带有设计的BalII(AGATCT)和NsiI(ATGCA7)限制酶交接处识别序列的epoAT5(核苷酸26793至核苷酸27833)、或FKAT3、或tmbAT4编码序列替换epoAT7编码序列(从核苷酸39585至核苷酸40626)。
以pKOS39-125 DNA作为模板,第1轮PCR用来产生~1.8kb片段。将该PCR片段亚克隆入载体LITMUS28的NsiI和SpeI位点,并测序;带有目标序列的质粒命名为P4。该PCR使用的寡核苷酸是:
TLII-S:5′-GGATGCATGTCGAGCCTGACGCCCGCCG;和
TLII-6:5′-GCACTAGTGATGGCGATCTCGTCATCCGCCGCCAC.
以pKOS039-118B DNA作为模板,第2轮PCR用来产生~2.1kb片段。该PCR使用的寡核苷酸是:
TL16:ACAGATCTCGGCGCGCTGCCGCCGGAG;和
TL15:GGTCTAGACTCGAACGGCTCGCCACCGC.
将PCR片段亚克隆入LITMUS 28的EcoRV限制位点,通过测序鉴定带有目标序列的质粒,并命名为质粒pKOS122-4。然后,用限制酶BglII和SpeI消化质粒pKOS122-4,分离4.8kb片段,与来自质粒P4的~1.8kbNsiI-SpeI限制片段连接,并与作为NsiI-BglII限制片段分离的3个置换体AT片段(FKAT3,epoAT5,tmbAT4)中的1个连接,获得质粒P5.1、P5.2和P5.3。通过PCR产生置换体AT片段,使用了下面的寡核苷酸引物:对FKAT3:
TLII-11:5′-GTATGCATCCAGTAGCGGACCCGCTCGA;和
TLII-12:5′-GCAGATCTGTGTGGCTCTTCTCCGGACA;
对tmbAT4:
TLII-15;5′-GCATGCATCCAGTAGCGCTGCCGCTGGAAC;和
TLII-16;5′-GGAGATCTGCGGTGCTGTTCACGGGGCA;以及
对epoAT5的PCR:
TLII-19; 5′-GTAGATCTGCTTTCCTGTTCACCGGACA;和TL8(见本实施例的B部分)。
然后,通过在kan-gal盒DraI位点的插入,对质粒P5.1、P5.2和P5.3进行改变。得到的质粒转化本发明的埃坡霉素生产菌黄色粘球菌宿主细胞(即K111-72.4.4),培养细胞,并如上所述挑选双交换重组事件的细胞。通过PCR筛选挑选的克隆。显示有预期重组事件的克隆在50毫升培养基中培养,并通过LC/MS筛选产生目标化合物的克隆。预期产物是6-去甲基-埃坡霉素D和6-去甲基-埃坡霉素C,其结构如下所示。

F、10-甲基-埃坡霉素类似物的产生
通过用甲基丙二酸单酰CoA特异的AT结构域的编码序列替换延伸组件5 AT结构域(“epoAT5”)的编码序列,构建埃坡霉素类A、B、C和D的10-甲基-埃坡霉素类似物。合适的置换体AT结构域编码序列可例如从编码FK520 PKS的延伸组件2(此处引用作为参考)、埃坡霉素PKS的延伸组件2(“epoAT2”)和tmbA基因编码的PKS的延伸组件3的基因获得。置换一般按如上所述进行,产生的具体埃坡霉素类仅仅取决于已转导置换体的粘球菌宿主产生何种埃坡霉素。
这样,用交接处带有设计的BglII(AGATCT)和NsiI(ATGCAT)限制酶识别序列的epoAT2(核苷酸12251至核苷酸13287)、或FKAT2、或tmbAT3编码序列替换epoAT5编码序列(从核苷酸26793至核苷酸27833)。
将用质粒pKOS39-118B为模板、根据引物TL11和TL12产生的PCR片段克隆入载体LITMUS 28。使用的PCR引物是:
TL11:5′-GGATGCATCTCACCCCGCGAAGCG;和
TT12:5′-GTACTAGTCAAGGGCGCTGCGGAGG.
通过DNA测序鉴定含有目标插入片段的质粒。然后用限制酶NsiI和XbaI消化质粒,并分离4.6kb片段。该片段与从引物TL5和TL6(如本实施例的A部分所述)获得的、已用限制酶BglII和XbaI消化的2.0kb PCR片段连接,并与作为NsiI-BglII限制片段分离的3个置换AT片段(FKAT2,epoAT2,tmbAT3)中的1个连接,以获得质粒P6.1、P6.2和P6.3。然后,通过在kan-gal盒DraI位点的插入,对后面的3个质粒进行改变。得到的质粒转化本发明的埃坡霉素生产菌黄色粘球菌宿主细胞(即K111-72.4.4),培养细胞,并如上所述挑选双交换重组事件的细胞。通过PCR筛选挑选的克隆。显示有目标重组事件的克隆在50毫升培养基中培养,并通过LC/MS筛选产生预期化合物的克隆。预期产物是10-甲基-埃坡霉素D和10-甲基-埃坡霉素C,其结构如下所示。

G、14-甲基-埃坡霉素类似物的生产
通过用甲基丙二酸单酰CoA特异的AT结构域的编码序列替换延伸组件3 AT结构域(“epoAT3”)的编码序列,构建埃坡霉素类A、B、C和D的14-甲基-埃坡霉素类似物。合适的置换体AT结构域编码序列可例如从编码FK520 PKS延伸组件2、埃坡霉素PKS的延伸组件2(“epoAT2”)和tmbA基因编码的PKS的延伸组件3的基因获得。置换一般按如上所述进行,产生的具体埃坡霉素仅仅取决于已转导置换体的粘球菌宿主产生何种埃坡霉素。
这样,用交接处带有设计的BglII(AGATCT)和NsiI(ATGCAT)限制酶识别序列的epoAT2(核苷酸12251至核苷酸13287)、或FKAT2、或tmbAT3编码序列替换epoAT3编码序列(从核苷酸17817至核苷酸18858)。
以pKOS39-124 DNA作为模板,第1轮PCR用来产生~1.8kb片段。将该PCR片段亚克隆入载体LITMUS28的XbaI和BglII位点,并测序;带有目标序列的质粒命名为P9。该PCR使用的寡核苷酸是:
TLII-7:5′-GCAGATCTGCCGCGCGAGGAGCTCGCGAT;和
TLII-8:5′-CATCTAGAGCCGCTCCTGTGGAGTCAC.
以pKOS39-124 DNA作为模板,第2轮PCR用来产生~1.9kb片段。将该PCR片段亚克隆至载体LITMUS28的NsiI和SpeI,并测序;带有目标序列的质粒命名为P10。该PCR使用的寡核苷酸是:
TLII-9B:5′-GGATGCATGCGCCGGCCGAAGGGCTCGGAG;
和
TLII-10:5′-GCACTAGTGATGGCGATCGGGTCCTCTGTCGC.
然后,质粒P9用限制酶BglII和SpeI消化,分离4.5kb片段,与来自质粒P4的~1.9kb NsiI-SpeI限制片段,并与作为NsiI-BglII限制片段分离的3个置换体AT片段(FKAT2,epoAT2,tmbAT3)中的1个连接,以获得质粒P11.1、P11.2和P11.3。然后,通过在kan-gal盒DraI位点的插入,对后面的3个质粒进行改变。得到的质粒转化本发明的埃坡霉素生产菌黄色粘球菌宿主细胞(即K111-72.4.4),培养细胞,并如上所述挑选双交换重组事件的细胞。通过PCR筛选挑选的克隆。显示有目标重组事件的克隆在50毫升培养基中培养,并通过LC/MS筛选产生预期化合物的克隆。预期产物是14-甲基-埃坡霉素D和14-甲基-埃坡霉素C,其结构如下所示。

在一个实施方案中,本发明提供了一种新埃坡霉素,即10,11-脱氢-埃坡霉素D,以及产生该化合物的重组宿主细胞。10,11-脱氢-埃坡霉素D的结构如下所示。
在另一个实施方案中,本发明提供了通过灭活产生相应埃坡霉素的埃坡霉素PKS延伸组件5的ER区而制备任一10,11-脱氢-埃坡霉素类似物的方法。
在一个实施方案中,通过灭活延伸组件5的烯酰还原酶(ER)区,构建产生10,11-脱氢埃坡霉素D的菌株。在一个实施方案中,通过将NADPH结合区的2个甘氨酸(-Gly-Gly-)改变成丙氨酸和丝氨酸(-Ala-Ser-),实现ER的失活。来自质粒pKOS39-118B(来自粘粒pKOS35-70.4的epoD基因的亚克隆)的2.5kb BbvCI-HindIII片段已克隆入pLitmus28,得到的pTL7作为定点诱变的模板。在NADPH结合区引入-Gly-Gly-至-Ala-Ser-突变的寡核苷酸引物是:
TLII-22,5′-TGATCCATGCTGCGGCCGCTAGCGTGGGCATGGCCGC.
TLII-23,5′-GCGGCCATGCCCACGCTAGCGGCCGCAGCATGGATCA.
通过测序确定含有置换的PCR克隆,并用限制酶DraI消化和用虾碱性磷酸酶处理。然后,将每个克隆的大片段与卡那霉素抗性和galK基因(KG或kan-gal)盒连接,以提供递送质粒。通过电穿孔,将递送质粒转化埃坡霉素D生产菌黄色粘球菌K111-72-4.4或K111-40-1。筛选转化子,挑选卡那霉素敏感、半乳糖抗性的存活者以鉴定除去KG基因的克隆。重组菌株中KG的除去和目的基因的置换的确认通过PCR进行。重组菌株在装有50毫升CTS培养基(酪胨,5克/升;MgSO4,2克/升;L-丙氨酸,1毫克/升;L-丝氨酸,1毫克/升;甘氨酸,1毫克/升和HEPES缓冲液,50mM)和2%XAD-16的瓶中发酵7天,并且用10ml甲醇将10,11-脱氢-埃坡霉素D从XAD树脂上洗脱下来。
I、通过发酵生产含噁唑的埃坡霉素类
在一个实施方案中,通过在补加L-丝氨酸的培养基中发酵产生埃坡霉素的菌株,如本发明提供的纤维堆囊菌菌株或粘球菌菌株,本发明提供了获得含噁唑的埃坡霉素类(相应埃坡霉素的噻唑部分用噁唑取代)的方法。
为说明本发明的该方面,除L-丝氨酸以分批培养基中基本丝氨酸浓度(2.3mM)的11、51x、101x和201x出现外,黄色粘球菌菌株K111-40.1或K111-72.4.4的培养根据实施例3的方法发酵。这样,含50毫升培养物的分批培养基含有:20克/升XAD-16;5克/升酪胨;2克/升MgSO4;7毫升/升油酸甲酯和4毫升/升痕量金属溶液,并加入适当浓度过滤除菌的1.25M L-丝氨酸溶液。在基本培养基中观察到的分批滴度是:Epo C:0.4毫克/升,Epo D:2毫克/升,Epo H1(C的噁唑类似物):未检测到,以及Epo H2(D的噁唑类似物):0.02毫克/升。增加丝氨酸浓度会降低epoC和epoD的浓度(在51X补加时几乎达到检测不到的水平)。这样,培养基中补加51X L-丝氨酸的基本滴度是:Epo C:0.03毫克/升,Epo D:0.05毫克/升,Epo H1:0.12毫克/升和Epo H2:0.13毫克/升。补料分批法可使观察到的滴度增加大约10倍。
J、埃坡霉素类似物的构建
在一个实施方案中,本发明提供了由本发明的重组埃坡霉素PKS酶产生的埃坡霉素和埃坡霉素衍生物,其中(i)延伸组件1 NRPS的特异性已由半胱氨酸改变成另一个氨基酸;(ii)装载结构域已改变成NRPS或CoA连接酶);或(iii)(i)和(ii)都有。本实施例描述了怎样构建本发明中这样的重组埃坡霉素PKS酶;本实施例引用的参考文献列于本实施例的末尾,通过引用号查阅文中的参考文献,此处引用作为参考。
埃坡霉素类含有氨基酸半胱氨酸,该氨基酸已被环化和氧化形成了噻唑。其它2个氨基酸,丝氨酸和苏氨酸,可经过相似的环化和氧化,分别产生噁唑和甲基噁唑。例如,埃坡霉素D的噁唑和甲基噁唑衍生物如下所示。

为构建带有噁唑或甲基噁唑的埃坡霉素类似物,进行延伸组件1的NRPS结构域的设计。延长正在生长分子的NRPS结构域最少包括激活氨基酸区、腺苷酰化区、将氨基酸限制在NRPS的PCP或肽基载体蛋白质区和将氨基酸缩合至正在生长分子的羧基以形成肽键的缩合区(5、7)。发现决定氨基酸特异性的识别序列位于腺苷酰化区的内部,具体地说在A4和A5共有序列之间(4)。分析该区域显示,该蛋白质区域的主要氨基酸可预测NRPS利用哪个氨基酸(2、8)。将该完整的NRPS腺苷酰化区与另一个完整的NRPS腺苷酰化区进行交换的实验已进行,该实验导致具有新腺苷酰化区的氨基酸特异性的杂合NRPS的形成(6、9)。使用腺苷酰化区的较小区域,如A4和A5共有序列之间的区域的实验还未见报道。在一个实施方案中,通过用利用苏氨酸、来自vibF和blmVII的腺苷酰化区的A4和A5共有序列之间的区域和利用丝氨酸、来自博来霉素基因簇blmVI的NRPS-4区域替换来自epoB的腺苷酰化区的A4和A5共有序列之间的区域,构建本发明的杂合PKS(3)。
最近的实验显示,如果不正确氢基酸已接合至PCP上,可以检测到缩合区(1)。一旦检测到不正确氨基酸,缩合反应的效率降低。为避免这种现象,除交换腺苷酰化区外,也可带有同源缩合结构域,以便改变腺苷酰化区的特异性和设计具有完全活性的NRPS。
本发明也提供了产生埃坡霉素的噁唑和甲基噁唑形式的16去甲基衍生物的重组埃坡霉素PKS酶。通过将延伸组件2的AT结构域epoC从甲基丙二酸单酰特异改变成丙二酸单酰特异来构建这样的酶。可用于制备构建体的AT结构域包括,来自埃坡霉素簇的延伸组件5和9以及来自soraphen基因簇的延伸组件2和4的AT结构域。
本发明也提供了已用NRPS置换EpoA蛋白质的重组PKS酶。本发明也提供这些酶产生的新埃坡霉素类。通过将丙二酸酯装载至装载组件的ACP,即EpoA,起始埃坡霉素的生物合成。随后,该丙二酸酯通过KS区脱羧,然后以乙酰基转移至NRPS组件EpoB。该分子已通过EpoB起作用后,得到的化合物是2-甲基噻唑。
为制备氨基酸附着在噻唑2位上的类似物,需要对epoA进行缺失,并插 NRPS组件。可使用任何NRPS组件;然而,为制备最保守的变化,可用NRPS组件置换可天然与下游NRPS组件交流的epoA。而且,因为置换epoA的NRPS是装载组件,所以不需要缩合区。这可通过获得延伸NRPS组件并移去缩合区,或者使用本身为装载组件且因此缺少缩合区的NRPS而实现。一个示例的NRPS装载组件来自safB的NRPS装载组件,其利用丙氨酸且来源于黄色粘球菌。
在构建epoA位置含有safB装载组件的黄色粘球菌菌株时,可确定新装载组件和epoB的最佳边界。PKS蛋白质之间的接头区域对那些蛋白质之间的“交流”常常很关键。可构建3个不同的菌株来检验最佳接头。首先,将EpoA的ACP区融合到safB装载组件的腺苷酰化区。该构建需要EpoA的ACP行使PCP的功能。虽然PCP和ACP区在功能上类似,但是它们没有显示出高度的序列同源性,并且这样可对它们的识别和结合位点有限制。第2个构建体融合了SafB装载组件的PCP区的EpoA下游的最后几个氨基酸,这样提供用于与EpoB“交流”的杂合装载组件的必要接头区域。最后,SafB装载组件和EpoB的融合将得到构建。因为SafB包含2个组件,所以可取出SafB的所有装载组件,并直接将它融合至EpoB,从而产生融合蛋白质,该蛋白质应优化SafB装载组件和EpoB之间的交流。
一旦使用SafB或任何其它装载NRPS区用氨基酸替换埃坡霉素噻唑上的2-甲基,则可在新装载NRPS组件中发生变化,从而便任何氨基酸可用于起始埃坡霉素类似物的合成。可用于这些交换的潜在氨基酸和它们相应的NRPS组件的综合列表由Challis等提供(2)。
可在含有埃坡霉素基因的黄色粘球菌菌株K111-32.25、或者在含有埃坡霉素类基因而其中的epoK基因不生产功能性产物的黄色粘球菌菌株K111-40-1,或本发明的任一其它埃坡霉素生产菌株、或在纤维堆囊菌中进行所有这些置换。在粘球菌中,使用galK和卡那霉素选择,在质粒上可产生适当的构建体,并可应用于用工程基因置换野生型基因。例如,用丝氨酸特异的NRPS置换K111-40-1中的NRPS,预期会产生埃坡霉素D的2-甲基-噁唑衍生物和埃坡霉素C的2-甲基-噁唑衍生物,它们分别为主要和次要产物。这些化合物的结构如下所示:
用苏氨酸特异的NRPS置换K111-40-1中的NRPS,预期会产生下面的化合物,且分别是主要和次要产物。
用甘氨酸、丙氨酸、谷氨酸、天冬氨酸、苯丙氨酸、组氨酸、异亮氨酸、亮氨酸、甲硫氨酸、天冬酰胺、谷氨酰胺、精氨酸、缬氨酸和酪氨酸特异的NRPS置换K111-40-1中的NRPS,每个预期产生下面的化合物,它们分别作为主要和次要产物。
其中R′对应于氨基酸的特异侧链(例如,在氨基酸通式NH2-CHR′COOH中对甘氨酸而言R′是H,对丙氨酸而言是甲基,等等)。
用脯氨酸特异的NRPS置换KIII-40-1的NRPS,预期会产生下面的化合物,它们分别作为主要和次要产物。
本小节中引用的参考文献如下:
1、Belshaw等,1999,氨酰基-CoAs在非核糖体肽合成中作为缩合结构域选择性的探测剂,科学,284:486-9。
2、Challis等,2000,预测的、基于结构的非核糖体肽合成酶腺苷酰化结构域氨基酸识别模型,化学与生物学(Chem Biol),7:211-24。
3、Du等,2000,来自支持非核糖体肽合成酶和聚酮化合物合酶功能性相互作用的轮枝链霉菌(Streptomyces verticillus)ATCC15003抗肿瘤药物博来霉素的生物合成基因簇,化学与生物学,7:623-42。
4、Konz等,1999,肽合成酶怎样产生结构多样性?化学与生物学,6:R39-48。
5、Marahiel等,1997,组件式肽合成酶参与非核糖体肽合成,化学评论(Chem.Rev),97:2651-2673。
6、Schneider等,1998,通过合理的组件交换,靶向性改变肽合成酶的底物特异性,分子普通遗传学(Mol Gen Genet),257:308-18。
7、Stachelhaus等,1995,通过分解多功能酶GrsA揭示肽合成酶的组件式结构,生物化学杂志,270:616-39。
8、Stachelhaus等,1999,在非核糖体肽合成酶中腺苷酰化结构域的特异性赋予密码,化学与生物学,6:493-505。
9、Stachelhaus等,1995,通过靶向性置换细菌和真菌结构域合理设计肽抗生素,科学,269:69-72。
实施例12
生物活性
利用硫氰酸胺B(sulforhodamine B;SRB)实验,在4种不同的人肿瘤细胞系中筛选10,11-脱氢-埃坡霉素D的抗癌活性。10,11-脱氢-埃坡霉素D在所有4种细胞系中显示生长抑制效果,IC50S为28nM-40nM。通过基于细胞的微管蛋白聚合分析确定作用机制,该分析揭示该化合物促进微管蛋白的聚合。从国立癌症研究所获得人癌细胞系MCF-7(乳腺)、NCI/ADR-Res(乳腺,MDR)、SF-268(胶质瘤)和NCI-H460(肺)。细胞维持在补加10%胎牛血清(Hyclone)和2mM L-谷氨酰胺的RPMI1640培养基(Life Technology)中,置于5%CO2和潮湿环境以及37℃维持。
10,11-脱氢-埃坡霉素D的细胞毒性通过SRB实验确定(Skehan等,国立癌症研究所杂志(J.Natl.Cancer Inst),82:1107-1112(1990),此处引用作为参考)。培养细胞用胰蛋白酶消化、记数,并将每100微升用生长培养基稀释至下面的浓度:MCF-7,5000;NCI/ADR-Res,7500;NCI-H460,5000和SF-268,7500。将细胞按100微升/孔种于96孔微滴定板。24小时后,将100微升10,11脱氢-埃坡霉素D(用生长培养基稀释,浓度为1000nM-0.001nM)加至每孔中。与该化合物孵育3天后,细胞用100微升10%三氯酸(“TCA”)于4℃固定1小时,并用0.2%SRB/1%乙酸于室温染色20分钟。未结合的染料用1%乙酸漂洗掉,然后用200微升10mM Tris碱提取结合的SRB。与总细胞蛋白含量相关的结合染料的量通过OD 515nm确定。然后,用Kaleida Graph程序分析数据,并计算IC50’S。平行测试化学合成的埃坡霉素D作为比较。
对于微管蛋白聚合实验,MCF-7细胞在35mm培养皿中生长至汇合,并用1μM 10,11-脱氢-埃坡霉素D或埃坡霉素D于37℃处理0、1或2小时(Giannakakou等,生物化学杂志,271:17118-17125(1997);国际癌杂志(Int.J.Cancer),75:57-63(1998),此处引用作为参考)。用2毫升不含钙或镁的PBS洗涤细胞2次后,细胞用300微升裂解缓冲液(20mMTris,PH6.8,1mM MgCl2,2mM EGTA,1%Triton X-100和蛋白酶抑制剂)于室温裂解5-10分钟。将细胞刮下,并将裂解物转移至1.5毫升微离心管。然后,在室温于18000g离心裂解物12分钟。从含不溶的或聚合的(细胞骨架)微管蛋白的沉淀中分离含可溶或未聚合(胞液)微管蛋白的上清,并转移至新管中。然后将沉淀重悬于300微升裂解缓冲液。通过用SDS-PAGE、接着使用抗微管蛋白抗体(Sigma)的免疫印迹法,分析每一样品的等体积小份,确定细胞中微管蛋白聚合的变化。
几个实验的结果显示,10,11-脱氢-埃坡霉素D(命名为"Epo490")针对4种不同人肿瘤细胞系的IC50范围为28nM-40nM。
表13
细胞系 EpoD(nM) Epo490(nM)
N=3 N=2
MCF-7 21±10 28±8
NCI/ADR 40±12 35±9
SF-268 34±8 40±5
NCI-H460 30±2 34±1微管蛋白聚合实验揭示,10,11-脱氢埃坡霉素D具有与埃坡霉素D同样的作用机制。在MCF-7细胞中,10,11-脱氢-埃坡霉素D在测定条件下强烈促进微管蛋白的聚合,具有与埃坡霉素D类似的动力学和效果。通过替换10,11-脱氢-埃坡霉素D的目的化合物,可用类似方法测定本发明的其它化合物。
实施例13
噁唑衍生物
本实施例描述了应用发酵条件调节宿主细胞产生埃坡霉素化合物的类型。通过给宿主细胞补加过量的丝氨酸,由宿主细胞正常产生的化合物以一种利于噁唑对应物产生的方式而被调节。例如,可使主要产生式V的一种化合物或多种化合物的细胞有利于产生相应于分子式VI的化合物的噁唑对应物。
在一个实施方案中,主要产生埃坡霉素C和D的黄色粘球菌菌株K111-40-1,用于显著增加埃坡霉素C和D的噁唑对应物埃坡霉素H1和H2的产生。菌株K111-40-1(PTA-2712)于2000年11月21日保藏于美国典型培养物保藏中心(ATCC),10801 University Blvd.,马纳萨斯,VA,20110-2209美国。菌株K111-40-1在补加或不补加额外丝氨酸的培养基中生长。未补加的培养基中成分的最终浓度是:水解酪蛋白(胰消化物,购自Difco,商标名酪胨),5克/升;MgSO4·7H2O,2克/升;XAD-16,20克/升;痕量元素溶液,4毫升/升;油酸甲酯,7毫升/升和Hepes缓冲液,40mM(用KOH将pH滴定至7.6)。痕量元素溶液包括:浓缩H2SO4,10毫升/升;FeCl3·6H2O,14.6克/升;ZnCl2,2.0克/升;MnCl2·4H2O,1.0克/升;CuCl2·2H2O,0.42克/升;H3BO3,0.31克/升;CaCl2·6H2O,0.24克/升和Na2MoO4·2H2O,0.24克/升。丝氨酸的基本水平认为是4.82%w/w,该值通过对酪胨具体批次的Difco氨基酸分析而确定。因此,丝氨酸的基本浓度是2.3mM,该值从培养基中5克/升酪胨的终浓度计算而来。丝氨酸补加的培养基含有50倍的更高丝氨酸浓度:117mM。
细胞于匣式摇床上的瓶中,在30℃、250转/分钟生长120小时。通过捕获树脂、在水中洗涤树脂1次,并在20毫升甲醇中提取树脂中的化合物30分钟,从而提取菌株在发酵过程中产生的化合物。通过HPLC和质谱对样品进行分析。
分析细胞生产的化合物显示,与在未补加丝氨酸的培养基中生长的细胞产生的产物相比,丝氨酸水平增加50倍导致埃坡霉素H1的产量增加30倍(0.12mg/L),同时埃坡霉素H2的产量增加5倍(0.12mg/L)。值得注意的是,细胞产生的埃坡霉素C和D几乎检测不到(<50微克/升)。
由于丝氨酸的供给,含噁唑的化合物增加同时含噻唑的化合物减少,这提供了从正常情况下产生含噻唑对应物的宿主细胞中获得噁唑化合物的方法。例如,制备9-氧代-埃坡霉素D的重组构建体(如实施例11的子部分C所述)可在如上所述的类似条件下生长,以制备9-氧代-埃坡霉素D的噁唑对应物17-去(2-甲基-4-噻唑基)-17-(2-甲基-4-噁唑基)-9-氧代-埃坡霉素D。同样,本发明的其它重组构建体,包括实施例11所描述的那些,可在过量丝氨酸中生长,以提供相应的噁唑化合物。
实施例14
从C-21甲基到C-21羟甲基的微生物转化
本实施例描述了从式I化合物的C-21甲基到C-21羟甲基的微生物转化,其中Ar是
或

一瓶冷冻(约2毫升)的如PCT公开号WO 00/39276中所述的Amycolataautotrophica ATCC 35203或放线菌sp.菌株PTA-XXX用来接种含有100毫升培养基的1500毫升瓶。1升去离子水中的植物培养基包含1克葡萄糖、10麦芽膏提取物、10克酵母提取物和1克蛋白胨。植物培养物在旋转振荡器上于28℃、250转/分钟孵育3天。将得到的1毫升培养物加入62个500毫升瓶中的每一瓶,这些瓶中含有与植物培养基同样组分的转化培养基。培养物在28℃、250转/分钟孵育24小时。将本发明中的适当化合物溶于155毫升乙醇,并将溶液分送至该62瓶。然后,将瓶放回振荡器,并于28℃、250转/分钟再孵育43小时。然后,处理反应培养物,以回收起始化合物的21-羟基对应物。
实施例15
利用EpoK的环化作用
本实施例描述了式I化合物的酶环化作用,其中R8和R10一起形成碳碳双键(本发明的脱氧化合物)。epoK基因产物在大肠杆菌中表达为带有多聚组氨酸标签(his标签)的融合蛋白质,并按PCT公开WO 00/31247所述进行纯化,此处引用作为参考。100μL体积中反应由50mM Tris(pH7.5)、21μM菠菜铁氧化还原蛋白、0.132单位菠菜铁氧化还原蛋白:NADP+氧化还原酶、0.8单位葡萄糖-6-磷酸脱氢酶、1.4mM NADP和7.1mM葡萄糖-6-磷酸、100μM或200μM本发明的脱氧化合物和1.7μM氨基端组氨酸标记的EpoK或1.6μM羧基端组氨酸标记的EpoK。反应于30℃孵育67分钟,并于90℃加热2分钟终止。不溶物质通过离心移去,并将50微升含预期产物的上清在LC/MS上分析。
实施例16
化学环化
本实施例描述了式I化合物的化学环化,其中R8和R10一起形成碳碳双键(本发明的脱氧化合物)。将二甲基二氧杂环丙烷(0.1M,溶于丙酮,17毫升)溶液于-78℃逐滴加入本发明的脱氧化合物(505毫克)在10毫升CH2Cl2中的溶液。混合物升温至-50℃,维持1小时,然后加入另一部分二甲基二氧杂环丙烷溶液(5毫升),反应于-50℃继续另外1.5小时。然后反应在氮气流中于-50℃干燥。通过在SiO2上快速层析对产物进行纯化。
实施例17
(3S,6R,7S,8R,12R,13S,15S,16E)-15-氨基-3,7-二羟基-5,9-二氧代-12,13-环氧-4,4,6,8,12,16-六甲基-17-(2-甲基噻唑-4-基-16-十七碳烯酸

步骤1 9-氧代-埃坡霉素B。二甲基二氧杂环丙烷溶液(0.1M,溶于丙酮,17毫升)于-78℃逐滴加入9-氧代-埃坡霉素D(505毫克)在10毫升CH2Cl2中的溶液。混合物升温至-50℃,维持1小时,然后加入另一部分的二甲基二氧杂环丙烷溶液(5毫升),反应于-50℃继续另外1.5小时。然后反应在氮气流中于-50℃干燥。通过在SiO2上快速层析对产物进行纯化。
步骤2(3S,6R,7S,8R,12R,13S,15S,16E)-15-叠氮基-3,7-二羟基-5,9-二氧代-12,13-环氧-4,4,6,8,12,16-六甲基-17-(2-甲基噻唑-4-基)-16-十七碳烯酸。在氩气下,用四(三苯基膦)钯(0.58克)处理9-氧代-埃坡霉素B(2.62克)和叠氮化钠(0.49克)在55毫升脱气的四氢呋喃/水(10∶1 v/v)中的溶液。混合物在45℃维持1小时,然后用50毫升水稀释,并用乙酸乙酯提取。提取物用盐水洗涤,在Na2SO4上干燥,过滤,并蒸发。通过在SiO2上快速层析,对产物进行纯化。
步骤3(3S,6R,7S,8R,12R,13S,15S,16E)-15-氨基-3,7-二羟基-5,9-二氧代12,13-环氧-4,4,6,8,12,16-六甲基-17-(2-甲基噻唑-4-基)-16-十七碳烯酸。在氩气下,于环境温度,用1.0M三甲基膦在甲苯中的溶液(3毫升)处理(3S,6R,7S,8R,12R,13S,15S,16E)-15-叠氮基-3,7-二羟基-5,9-二氧代-12,13-环氧-4,4,6,8,12,16-六甲基-17-(2-甲基噻唑-4-基)-16-十七碳烯酸(565毫克)在15毫升THF/水(10∶1 v/v)中的溶液2小时。浓缩混合物,通过在SiO2上快速层析,对产物进行纯化。
实施例18
(4S,7R,8S,9R,13R,14S,16S)-13,14-环氧-4,8-二羟基-2,6,10-三氧代-5,5,7,9,13-五甲基-16-(1-(2-甲基噻唑-4-基)丙烯-2-基)-1-氮杂-11-环十六碳烯。

将(3S,6R,7S,8R,12R,13S,15S,16E)-15-氨基-3,7-二羟基-5,9-二氧代-12,13-环氧-4,4,6,8,12,16-六甲基-17-(2-甲基噻唑-4-基)-16-十七碳烯酸(540毫克)在乙腈/二甲基甲酰胺(20∶1 v/v,150毫升)中的溶液冷至0℃,并接下来用1-羟基苯并三唑(0.135克)和1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸(0.5克)处理。将混合物保温至环境温度,并维持12小时,然后用水稀释,并用乙酸乙酯提取。提取物接下来用水、饱和NaHCO3和盐水洗涤,然后在Na2SO4上干燥,过滤,并蒸发。通过在SiO2上快速层析,对产物进行纯化。
实施例19
(4S,7R,8S,9R,13Z,16S)-4,8-二羟基-2,6,10-三氧代-5,5,7,9,13-五甲基-16-(1-(2-甲基噻唑-4-基)丙烯-2-基)-1-氮杂-11-环十六碳烯
在-78℃的六氯化钨(0.76克)在四氢呋喃中的溶液(20ml)用1.6M正丁基锂在己烷中的溶液(2.5毫升)处理。让混合物保温至环境温度20分钟。在环境温度下,将得到的绿色溶液中的13.8毫升室温下加入(4S,7R,8S,9R,13R,14S,16S)-4,8-二羟基-13,14-环氧-2,6,10-三氧代-5,5,7,9,13-五甲基-1 6-(1-(2-甲基噻唑-4-基)丙烯-2-基)-1-氮杂-11-环十六碳烯(360毫克)在2毫升四氢呋喃中的溶液。30分钟后,将反应冷却至0℃,并用饱和NaHCO3(10毫升)处理。混合物用水稀释,并用CH2Cl2提取。提取物在Na2SO4上干燥,过滤,并蒸发。通过在SiO2上快速层析,对产物进行纯化。
实施例20
(4S,7R,8S,9R,13R,14S,1 6S)-13,14-环氧-4,8-二羟基-2,6,10-三氧代-1,5,5,7,9,13-六甲基-16-(1-(2-甲基噻唑-4-基)丙烯-2-基)-1-氮杂-11-环十六碳烯。

步骤1(3S,6R,7S,8R,12R,13S,15S,16E)-3,7-二羟基-5,9-二氧代-12,13-环氧-4,4,6,8,12,16-六甲基-15-(甲基氨基)-17-(2-甲基噻唑-4-基)-16-十七碳烯酸。用37%含水甲醛(1毫升)、乙酸(25微升)和氰基氢硼化钠(100毫克)处理(3S,6R,7S,8R,12R,13S,15S,16E)-15-氨基-3,7-二羟基-5,9-二氧代-12,13-环氧-4,4,6,8,12,16-六甲基-17-(2-甲基噻唑-4-基)-16-十七碳烯酸(540毫克)在10毫升甲醇中的溶液。1小时后,用1NHCL处理混合物,然后用乙酸乙酯和水稀释。水相用乙酸乙酯提取,并合并有机相,在Na2SO4上干燥,过滤,并蒸发。通过在SiO2上快速层析,对产物进行纯化。
步骤2(4S,7R,8S,9R,13R,14S,16S)-13,14-环氧-4,8-二羟基-2,6,10-三氧代-1,5,5,7,9,13-六甲基-6-(1-(2-甲基噻唑-4-基)丙烯-2-基)-1-氮杂-11-环十六碳烯。将(3S,6R,7S,8R,12R,13S,15S,16E)-3,7-二羟基-5,9-二氧代-12,13-环氧-4,4,6,8,12,16-六甲基-15-(甲基氨基)-17-(2-甲基噻唑-4-基)-16-十七碳烯酸(554毫克)在乙腈/二甲基甲酰胺(20∶1 v/v,150毫升)中的溶液冷却至0℃,接下来1-羟基苯并三唑(0.135克)和1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸(0.5克)处理。混合物保温至环境温度,并维持12小时,然后用水稀释,并用乙酸乙酯提取。提取物继续用水、饱和NaHCO3、盐水洗涤,然后在Na2SO4上干燥,过滤,并蒸发。通过在SiO2上快速层析,对产物进行纯化。
实施例21
(4S,7R,8S,9R,13Z,16S)-4,8-二羟基-2,6,10-三氧代-1,5,5,7,9,13-六甲基-16-(1-(2-甲基噻唑-4-基)丙烯-2-基)-1-氮杂-11-环十六碳烯。

用正丁基锂在己烷中的1.6M溶液(2.5毫升)处理-78℃的六氯化钨(0.76克)在四氢呋喃中的溶液(20毫升)。让混合物保温至环境温度20分钟。将得到的绿色溶液中的13.8毫升于环境温度下加入(4S,7R,8S,9R,13R,14S,16S)-13,14-环氧-4,8-二羟基-2,6,10-三氧-1,5,5,7,9,13-六甲基-1 6-(1-(2-甲基噻唑-4-基)丙烯-2-基)-1-氮杂-11-环十六碳烯(370毫克)在2毫升四氢呋喃中的溶液。30分钟后,将反应冷却至0℃,并用饱和NaHCO3(10毫升)处理。用水稀释混合物,并用CH2Cl2提取。提取物在Na2SO4上干燥,过滤,蒸发。通过在SiO2上快速层析,对产物进行纯化。
实施例22
脂质体组合物
本实施例描述了含9-氧代埃坡霉素的脂质体组合物。脂类和9-氧代-埃坡霉素D的混合物溶解于乙醇中,并在减压下通过旋转将该溶液干燥成薄膜。通过加入水相,使得到的脂膜被水合,并将该含脂质体的埃坡霉素衍生物的粒度调至预期范围。优选地,平均颗粒直径小于10微米,优选地从约0.5微米至约4微米。粒度可减少至预期水平,例如,通过使用研磨机(如空气喷射磨机、球磨机或振动磨机)、微沉淀、喷雾干燥、冷冻干燥、高压均化、从超临界介质中重结晶,或通过使脂质体的含水悬液挤压穿过具有所选选的统一孔径的一系列膜(如聚碳酸脂膜)。在一个实施方案中,脂质体组合物包含:发明的化合物(1.00毫克);磷脂酰胆碱(16.25毫克);胆固醇(3.75毫克);聚乙二醇衍生的二硬脂基磷脂酰乙醇胺(5.00毫克);乳糖(80.00毫克);柠檬酸(4.20毫克);酒石酸(6.00毫克);NaOH(5.44毫克);水(至1毫升)。在另一实施方案中,脂质体组合物包含:发明的化合物(1.00毫克);磷脂酰胆碱(19.80毫克);胆固醇(3.75毫克);二硬脂基磷脂酰胆碱(1.45毫克);乳糖(80.00毫克);柠檬酸(4.20毫克);酒石酸(6.00毫克);NaOH(5.44毫克);水(至1毫升)。又在另一个实施方案中,脂质体组合物包含:本发明的化合物(1.00毫克);1-棕榈酰-2-油基-sn-甘油基-3-胆碱磷酸(17.50毫克);1-棕榈酰-2-油基-sn-甘油基-3-甘油磷酸,Na(7.50毫克);乳糖(80.毫克);柠檬酸(4.20毫克);酒石酸(6.00毫克);NaOH(5.44mg);水(至1毫升)。包含本发明的其它化合物的脂质体组合物用类似于如上所述的条件制备。
实施例23
聚谷氨酸共轭物
本实施例描述了聚谷氨酸-21-羟基-9-氧代-埃坡霉素D共轭物的制造。用水溶解聚(l-谷氨酸)(″PG″)钠盐(MW 34 K,Sigma,0.35克)。用0.2M HCl将水溶液的pH调至2。收集沉淀,用蒸馏水透析,并冷冻干燥,产生0.29克PG。将20毫克21-羟基-9-氧代-埃坡霉素D、15毫克二环己基碳二亚胺(“DCC”)和痕量的二甲基氨基吡啶加入PG(75毫克,重复单元FW 170,0.44mM)在无水DMF的溶液中(1.5毫升)。反应在室温进行4小时,或者直至薄层层析指示结束。将反应混合物倒入氯仿,收集得到的沉淀,并在真空中干燥,产生大约65毫克PG-21-羟基-9-氧代-埃坡霉素D共轭物。改变起始材料中本发明化合物与PG的重量比将导致21-羟基-10,11-脱氢埃坡霉素D的多种浓度的聚合共轭物。本发明的其它化合物的共轭物用类似于如上所述的条件制备。
实施例24
静脉内制剂
本实施例描述了9-氧代-埃坡霉素D的静脉内制剂。制剂含有10毫克/毫升9-氧代-埃坡霉素D,甚至含30%丙二醇、20%Creomophor EL和50%乙醇的载体中。将称量的乙醇(591.8克)放入装有搅拌棒的烧杯中,往溶液中加入Creomophor EL(315.0克),并混合10分钟;然后往溶液中加入丙二醇(466.2克),并再混合10分钟,依此制备载体。将9-氧代-埃坡霉素D(1克)加入装有400-600毫升载体的1升容量瓶,并混合5分钟。10,11-脱氢-埃坡霉素D加入溶液后,将体积补至1升;再混合10分钟;并用0.22μm Millipore Millipak滤器过滤。用测量蠕动泵将得到的溶液无菌注入无菌的5毫升小瓶,目标注入体积为5.15毫升/瓶。注满的小瓶立即塞紧,并折缝。
用生理盐水或5%葡萄糖溶液稀释含10毫克/毫升9-氧代-埃坡霉素D的小瓶以施与患者,并用非PVC、非-DEHP袋和施用装置施与。产品在1-6小时期间注入,并递送预期的剂量。
在一个实施方案中,在静脉注入之前,用无菌生理盐水将制剂稀释20倍。最终注入浓度是0.5毫克/毫升本发明的化合物、1.5%丙二醇、1%Chremophor EL和2.5%乙醇,在1-6小时期间注入,并递送预期的剂量。
含本发明的其它化合物的静脉内制剂可用类似的方法制备和使用。
实施例25
Cremophor毒性的预处理
本实施例描述了Cremophor毒性的预处理方案。包含Cremophort的本发明化合物的制剂对患者可导致毒性。用类固醇预处理可用于预防过敏反应。可使用任何适当的皮质类固醇或将皮质类固醇与H1拮抗剂和/或H2拮抗剂使用。在一个实施方案中,受试者在用本发明化合物在含有Cremophor制剂处理前1小时,口服50毫克二苯基羟基胺和300毫克甲氰咪胍。在另一实施方案中,受试者在用本发明化合物在含有Cremophor制剂处理前至少1小时,静脉内施用20毫克地塞米松。在另一个实施方案中,受试者在用本发明化合物在含有Cremophor制剂处理前至少1小时,静脉内施用50毫克二苯基羟基胺、300毫克甲氰咪胍和20毫克地塞米松。又在另一个实施方案中,将受试者的体重考虑进去,且受试者在用本发明化合物在含有Cremophor制剂处理前至少1小时,预先施用二苯基羟基胺(5毫克/千克,i.v.)、甲氰咪胍(5毫克/千克,i.v)和地塞米松(1毫克/千克,i.m.)。
此处的所有科学公开和专利公开在此处引用作为参考。本发明现在已通过书面描述和实施例进行了描述,本领域技术人员将认识到,本发明可用于多种实施方案,前面的描述和实施例是用于阐述的目的,并不用于限制下面的权利要求。
Claims (47)
1、孢囊杆菌亚目的包含重组表达载体的重组宿主细胞,该重组表达载体编码异源聚酮化合物合酶(PKS)基因并产生由所述载体编码的PKS酶合成的聚酮化合物。
2、权利要求1的宿主细胞,其选自粘球菌科。
3、权利要求1的宿主细胞,其选自孢囊杆菌科。
4、权利要求2的宿主细胞,其选自囊球菌属、粘球菌属和软骨球菌属。
5、权利要求3的宿主细胞,其选自包括孢囊杆菌属、蜂窝囊菌属、标桩菌属和原囊菌属。
6、权利要求4的宿主细胞,其选自粘球菌属。
7、权利要求5的宿主细胞,其选自标桩菌属。
8、权利要求6的宿主细胞,其选自叶柄粘球菌、橙色粘球菌、黄色粘球菌、变绿粘球菌。
9、权利要求7的宿主细胞,其选自直立标桩菌和橙色标桩囊菌。
10、权利要求8的宿主细胞,其为黄色粘球菌。
11、在孢囊杆菌亚目的宿主细胞中产生聚酮化合物的方法,其中聚酮化合物不在所述宿主细胞中天然产生,所述方法包括在表达载体上编码的PKS基因且产生所述聚酮化合物的条件下培养权利要求1的宿主细胞。
12、权利要求1的重组宿主细胞,其产生埃坡霉素或埃坡霉素衍生物。
13、权利要求10的宿主细胞,其产生埃坡霉素或埃坡霉素衍生物。
14、权利要求13的宿主细胞,其产生埃坡霉素A、B、C和D。
15、权利要求14的宿主细胞,其为黄色粘球菌K111-32.25。
16、权利要求14的宿主细胞,其产生作为主要产物的埃坡霉素A和B,及作为次要产物的埃坡霉素C和D。
17、权利要求13的宿主细胞,其产生作为主要产物的埃坡霉素C和D。
18、权利要求17的宿主细胞,其不含epoK基因或不表达完全功能的epoK基因产物。
19、权利要求18的宿主细胞,其为黄色粘球菌K111-40.1。
20、权利要求18的宿主细胞,其为黄色粘球菌K111-72.4.4。
21、权利要求13的宿主细胞,其包含埃坡霉素PKS基因,其中所述PKS的一个组件的编码序列已通过突变、缺失或替换而改变。
22、权利要求21的宿主细胞,其中所述组件是延伸组件6。
23、权利要求22的宿主细胞,其中所述组件缺少功能性酮还原酶结构域,并产生9-酮-埃坡霉素。
24、权利要求21的宿主细胞,其中所述组件是延伸组件5。
25、权利要求24的宿主细胞,其中所述组件5缺少功能性脱水酶结构域,并产生13-羟基-埃坡霉素。
26、权利要求21的宿主细胞,其中所述组件是延伸组件4。
27、权利要求26的宿主细胞,其中所述组件缺少功能性酮还原酶结构域,并产生13-酮-埃坡霉素。
28、权利要求21的宿主细胞,其中所述组件是延伸组件2,已通过突变将活性位点半胱氨酸更换成另一个氨基酸而改变酮合酶结构域编码序列,且其中必须给宿主细胞提供二酮等同化合物,以产生埃坡霉素或埃坡霉素衍生物。
29、权利要求28的宿主细胞,其为黄色粘球菌菌株K90-132.1.1.2。
30、权利要求21的宿主细胞,其中所述组件是延伸组件1。
31、权利要求30的宿主细胞,其中所述组件已被改变,以致于它结合除半胱氨酸外的氨基酸。
32、权利要求21的宿主细胞,其中所述组件是装载组件。
33、权利要求32的宿主细胞,其中所述组件已用结合氨基酸的组件替换。
34、下式的埃坡霉素衍生物:
其通过用下式的二酮等同化合物培养权利要求28所述的宿主细胞而产生,

其中R7为氢或甲基,且Ar是选自以下的芳基:
其中R为氢、羟基、卤素、氨基、C1-C5烷基、C1-C5羟基烷基、C1-C5烷氧基和C1-C5氨基烷基。
35、权利要求1的宿主细胞,其还包含编码一种酶的异源基因,其中所述酶选自:将一种用于聚酮化合物生物合成的化合物转运入所述细胞的酶、合成用于聚酮化合物生物合成的化合物的酶,以及使PKS磷酸泛酰巯基乙胺化的酶。
36、权利要求35的宿主细胞,其中所述酶是MatB。
37、权利要求35的宿主细胞,其中所述酶是MatC。
38、权利要求35的宿主细胞,其中所述酶是MtaA。
39、权利要求13的宿主细胞,其中所述埃坡霉素或埃坡霉素衍生物由在选自以下的启动子控制下的PKS基因产生,其中所述启动子选自:来自纤维堆囊菌埃坡霉素PKS基因的启动子、来自粘噻唑菌醇生物合成基因的启动子、来自TA生物合成基因的启动子、pilA启动子、来自卡那霉素抗性赋予基因的启动子以及So ce90启动子。
40、从产生埃坡霉素的细胞中纯化埃坡霉素的方法,所述方法包括在XAD树脂存在下培养所述细胞、从所述树脂洗脱埃坡霉素、对从所述树脂上洗脱的埃坡霉素进行固相提取,以及从所述固相提取得到的埃坡霉素进行层析。
41、权利要求36的方法,其还包括结晶步骤。
42、结晶的埃坡霉素D。
43、发酵粘球菌宿主细胞的方法,该方法包括在含有脂肪酸或油作为碳源的液体培养基中培养所述细胞。
44、权利要求43的方法,其上述发酵为补料分批发酵。
45、权利要求43的方法,其中所述粘球菌宿主细胞产生埃坡霉素或埃坡霉素衍生物。
46、权利要求45的方法,其中所述宿主细胞产生含有噁唑、而不是噻唑的埃坡霉素衍生物,且所述液体培养基含有L-丝氨酸。
47、下式的分离化合物:

其中:
R1、R2、R3、R5、R11和R12每一个独立地为氢、甲基或乙基;
R4、R6和R9每一个独立地为氢、羟基或氧;或者,
R5和R6一起形成碳碳双键;
R7为氢、甲基或乙基;
R8和R10均为氢或一起形成碳碳双键或环氧化物;
Ar是芳基;且,
W是O或NR13,其中R13为氢、C1-C10脂族、芳基或烷基芳基。
Applications Claiming Priority (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US09/560,367 | 2000-04-28 | ||
| US09/560,367 US6410301B1 (en) | 1998-11-20 | 2000-04-28 | Myxococcus host cells for the production of epothilones |
| US23269600P | 2000-09-14 | 2000-09-14 | |
| US60/232,696 | 2000-09-14 | ||
| US25751700P | 2000-12-21 | 2000-12-21 | |
| US60/257,517 | 2000-12-21 | ||
| US26902001P | 2001-02-13 | 2001-02-13 | |
| US09/825,856 | 2001-04-03 | ||
| US09/825,876 US6589968B2 (en) | 2001-02-13 | 2001-04-03 | Epothilone compounds and methods for making and using the same |
| US09/825,856 US6489314B1 (en) | 2001-04-03 | 2001-04-03 | Epothilone derivatives and methods for making and using the same |
| US09/825,876 | 2001-04-03 | ||
| US60/269,020 | 2001-04-13 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1511192A true CN1511192A (zh) | 2004-07-07 |
| CN100480389C CN100480389C (zh) | 2009-04-22 |
Family
ID=27559233
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB018087116A Expired - Fee Related CN100480389C (zh) | 2000-04-28 | 2001-04-26 | 聚酮化合物的制备 |
Country Status (15)
| Country | Link |
|---|---|
| EP (2) | EP1652926B1 (zh) |
| KR (2) | KR100832145B1 (zh) |
| CN (1) | CN100480389C (zh) |
| AT (2) | ATE444368T1 (zh) |
| AU (2) | AU9519501A (zh) |
| CA (1) | CA2404938C (zh) |
| CY (1) | CY1109698T1 (zh) |
| DE (2) | DE60140087D1 (zh) |
| DK (1) | DK1652926T3 (zh) |
| ES (2) | ES2254493T3 (zh) |
| IL (1) | IL151960A0 (zh) |
| MX (1) | MXPA02010565A (zh) |
| NZ (2) | NZ521788A (zh) |
| PT (1) | PT1652926E (zh) |
| WO (1) | WO2001083800A2 (zh) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102015756A (zh) * | 2008-03-20 | 2011-04-13 | 辛文特公司 | Nrps-pks基因簇及其操纵和应用 |
| CN105793424A (zh) * | 2013-07-12 | 2016-07-20 | 亥姆霍兹感染研究中心有限公司 | 孢囊菌酰胺 |
| WO2017133706A1 (zh) * | 2016-02-06 | 2017-08-10 | 北京华昊中天生物技术有限公司 | 脱环氧埃坡霉素衍生物制剂、制备及其治疗肿瘤的应用 |
| WO2018149282A1 (zh) * | 2017-02-20 | 2018-08-23 | 复旦大学 | 一种异源表达埃博霉素的方法 |
Families Citing this family (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2273083C (en) | 1996-12-03 | 2012-09-18 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto, analogues and uses thereof |
| FR2775187B1 (fr) | 1998-02-25 | 2003-02-21 | Novartis Ag | Utilisation de l'epothilone b pour la fabrication d'une preparation pharmaceutique antiproliferative et d'une composition comprenant l'epothilone b comme agent antiproliferatif in vivo |
| US6410301B1 (en) | 1998-11-20 | 2002-06-25 | Kosan Biosciences, Inc. | Myxococcus host cells for the production of epothilones |
| EP1135470A2 (en) | 1998-11-20 | 2001-09-26 | Kosan Biosciences, Inc. | Recombinant methods and materials for producing epothilone and epothilone derivatives |
| US6998256B2 (en) | 2000-04-28 | 2006-02-14 | Kosan Biosciences, Inc. | Methods of obtaining epothilone D using crystallization and /or by the culture of cells in the presence of methyl oleate |
| GB0029895D0 (en) * | 2000-12-07 | 2001-01-24 | Novartis Ag | Organic compounds |
| US6893859B2 (en) | 2001-02-13 | 2005-05-17 | Kosan Biosciences, Inc. | Epothilone derivatives and methods for making and using the same |
| WO2003022844A2 (en) * | 2001-09-06 | 2003-03-20 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones intermediates thereto and analogues thereof |
| WO2003042217A2 (en) * | 2001-11-15 | 2003-05-22 | Kosan Biosciences, Inc. | Method for making epothilone compounds by bioconversion with microorganisms |
| WO2003045324A2 (en) * | 2001-11-26 | 2003-06-05 | Kosan Biosciences, Inc. | 14-methyl-epothilones |
| US7220560B2 (en) * | 2002-02-25 | 2007-05-22 | Kosan Biosciences Incorporated | Secondary metabolite congener distribution modulation |
| AU2003212457A1 (en) * | 2002-03-01 | 2003-09-16 | University Of Notre Dame | Derivatives of epothilone b and d and synthesis thereof |
| US20040072882A1 (en) * | 2002-05-20 | 2004-04-15 | Kosan Biosciences, Inc., A Delaware Corporation | Methods to administer epothilone D |
| WO2004014295A2 (en) * | 2002-07-29 | 2004-02-19 | Optimer Pharmaceuticals, Inc. | Tiacumicin production |
| AR040956A1 (es) | 2002-07-31 | 2005-04-27 | Schering Ag | Nuevos conjugados de efectores, procedimientos para su preparacion y su uso farmaceutico |
| JP2006505627A (ja) * | 2002-07-31 | 2006-02-16 | シエーリング アクチエンゲゼルシャフト | 新規エフェクター接合体、それらの生成方法及びそれらの医薬使用 |
| EP2135867B1 (en) | 2002-11-07 | 2013-09-25 | Kosan Biosciences Incorporated | Trans-9, 10-dehydroepothilone C and trans-9, 10-dehydroepothilone D, analogs thereof and methods of making the same |
| BR0317150A (pt) * | 2002-12-09 | 2005-11-01 | Novartis Ag | Estabilizadores de microtúbulos em stents para o tratamento de estenose |
| US7378508B2 (en) | 2007-01-22 | 2008-05-27 | Optimer Pharmaceuticals, Inc. | Polymorphic crystalline forms of tiacumicin B |
| EP2141242A1 (en) * | 2008-07-04 | 2010-01-06 | Universität des Saarlandes | Synthetic pathway enzymes for the production of Argyrins |
| JO3464B1 (ar) | 2013-01-15 | 2020-07-05 | Astellas Pharma Europe Ltd | التركيبات الخاصة بمركبات التياكوميسين |
| KR101575295B1 (ko) | 2014-01-21 | 2015-12-09 | 선문대학교 산학협력단 | 신규한 에포틸론 글리코시드 및 이의 제조방법 |
| US20200231562A1 (en) * | 2014-07-07 | 2020-07-23 | Helmholtz-Zentrum für Infektionsforschung GmbH | Novel macrolide antibiotics |
| CN107629994B (zh) * | 2017-10-25 | 2021-11-02 | 中国科学院上海有机化学研究所 | 一种构建fk520高产工程菌株的方法和吸水链霉菌高产菌株 |
| CN113969255A (zh) * | 2020-07-24 | 2022-01-25 | 北京华昊中天生物医药股份有限公司 | 生产脱环氧埃坡霉素b的重组菌及其用途 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK1367057T3 (da) * | 1996-11-18 | 2009-01-19 | Biotechnolog Forschung Gmbh | Epothiloner E og F |
| US6194181B1 (en) * | 1998-02-19 | 2001-02-27 | Novartis Ag | Fermentative preparation process for and crystal forms of cytostatics |
| NZ508326A (en) * | 1998-06-18 | 2003-10-31 | Novartis Ag | A polyketide synthase and non ribosomal peptide synthase genes, isolated from a myxobacterium, necessary for synthesis of epothiones A and B |
| DE19846493A1 (de) * | 1998-10-09 | 2000-04-13 | Biotechnolog Forschung Gmbh | DNA-Sequenzen für die enzymatische Synthese von Polyketid- oder Heteropolyketidverbindungen |
| EP1135470A2 (en) * | 1998-11-20 | 2001-09-26 | Kosan Biosciences, Inc. | Recombinant methods and materials for producing epothilone and epothilone derivatives |
| AU2174102A (en) | 2000-10-31 | 2002-05-15 | Boehringer Ingelheim Pharma | Inhalative solution formulation containing a tiotropium salt |
-
2001
- 2001-04-26 MX MXPA02010565A patent/MXPA02010565A/es active IP Right Grant
- 2001-04-26 DE DE60140087T patent/DE60140087D1/de not_active Expired - Lifetime
- 2001-04-26 AT AT05023081T patent/ATE444368T1/de active
- 2001-04-26 NZ NZ521788A patent/NZ521788A/en not_active IP Right Cessation
- 2001-04-26 PT PT05023081T patent/PT1652926E/pt unknown
- 2001-04-26 AT AT01973782T patent/ATE309369T1/de not_active IP Right Cessation
- 2001-04-26 AU AU9519501A patent/AU9519501A/xx active Pending
- 2001-04-26 WO PCT/US2001/013793 patent/WO2001083800A2/en active IP Right Grant
- 2001-04-26 NZ NZ546497A patent/NZ546497A/en not_active IP Right Cessation
- 2001-04-26 KR KR1020027014529A patent/KR100832145B1/ko not_active IP Right Cessation
- 2001-04-26 IL IL15196001A patent/IL151960A0/xx unknown
- 2001-04-26 KR KR1020077019766A patent/KR20070092334A/ko not_active Application Discontinuation
- 2001-04-26 DK DK05023081.2T patent/DK1652926T3/da active
- 2001-04-26 DE DE60114865T patent/DE60114865T2/de not_active Expired - Lifetime
- 2001-04-26 EP EP05023081A patent/EP1652926B1/en not_active Expired - Lifetime
- 2001-04-26 CN CNB018087116A patent/CN100480389C/zh not_active Expired - Fee Related
- 2001-04-26 CA CA2404938A patent/CA2404938C/en not_active Expired - Fee Related
- 2001-04-26 EP EP01973782A patent/EP1320611B1/en not_active Expired - Lifetime
- 2001-04-26 ES ES01973782T patent/ES2254493T3/es not_active Expired - Lifetime
- 2001-04-26 AU AU2001295195A patent/AU2001295195B2/en not_active Ceased
- 2001-04-26 ES ES05023081T patent/ES2332727T3/es not_active Expired - Lifetime
-
2009
- 2009-12-30 CY CY20091101352T patent/CY1109698T1/el unknown
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102015756A (zh) * | 2008-03-20 | 2011-04-13 | 辛文特公司 | Nrps-pks基因簇及其操纵和应用 |
| CN102015756B (zh) * | 2008-03-20 | 2014-07-30 | 辛文特公司 | Nrps-pks基因簇及其操纵和应用 |
| CN105793424A (zh) * | 2013-07-12 | 2016-07-20 | 亥姆霍兹感染研究中心有限公司 | 孢囊菌酰胺 |
| WO2017133706A1 (zh) * | 2016-02-06 | 2017-08-10 | 北京华昊中天生物技术有限公司 | 脱环氧埃坡霉素衍生物制剂、制备及其治疗肿瘤的应用 |
| CN108778335A (zh) * | 2016-02-06 | 2018-11-09 | 北京华昊中天生物技术有限公司 | 脱环氧埃坡霉素衍生物制剂、制备及其治疗肿瘤的应用 |
| US10980782B2 (en) | 2016-02-06 | 2021-04-20 | Beijing Biostar Technologies, Ltd. | De-epoxidized epothilone derivative preparation, preparation of same and use thereof in the treatment of tumor |
| CN108778335B (zh) * | 2016-02-06 | 2022-03-11 | 北京华昊中天生物医药股份有限公司 | 脱环氧埃坡霉素衍生物制剂、制备及其治疗肿瘤的应用 |
| CN114288290A (zh) * | 2016-02-06 | 2022-04-08 | 北京华昊中天生物医药股份有限公司 | 脱环氧埃坡霉素衍生物制剂、制备及其治疗肿瘤的应用 |
| CN114288290B (zh) * | 2016-02-06 | 2023-11-03 | 北京华昊中天生物医药股份有限公司 | 脱环氧埃坡霉素衍生物制剂、制备及其治疗肿瘤的应用 |
| WO2018149282A1 (zh) * | 2017-02-20 | 2018-08-23 | 复旦大学 | 一种异源表达埃博霉素的方法 |
| CN108456703A (zh) * | 2017-02-20 | 2018-08-28 | 复旦大学 | 一种异源表达埃博霉素的方法 |
| CN108456703B (zh) * | 2017-02-20 | 2022-01-14 | 复旦大学 | 一种异源表达埃博霉素的方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2001083800A2 (en) | 2001-11-08 |
| WO2001083800A9 (en) | 2003-09-12 |
| DE60114865T2 (de) | 2006-07-27 |
| EP1652926B1 (en) | 2009-09-30 |
| EP1652926A2 (en) | 2006-05-03 |
| KR20030032942A (ko) | 2003-04-26 |
| KR100832145B1 (ko) | 2008-05-27 |
| ATE309369T1 (de) | 2005-11-15 |
| CY1109698T1 (el) | 2014-08-13 |
| ATE444368T1 (de) | 2009-10-15 |
| PT1652926E (pt) | 2009-12-03 |
| ES2254493T3 (es) | 2006-06-16 |
| EP1652926A3 (en) | 2006-08-09 |
| ES2332727T3 (es) | 2010-02-11 |
| CA2404938C (en) | 2011-11-01 |
| IL151960A0 (en) | 2003-04-10 |
| AU2001295195B2 (en) | 2007-02-01 |
| DE60114865D1 (de) | 2005-12-15 |
| CA2404938A1 (en) | 2001-11-08 |
| NZ521788A (en) | 2007-04-27 |
| KR20070092334A (ko) | 2007-09-12 |
| NZ546497A (en) | 2008-04-30 |
| DK1652926T3 (da) | 2010-02-15 |
| WO2001083800A3 (en) | 2003-04-10 |
| EP1320611A2 (en) | 2003-06-25 |
| CN100480389C (zh) | 2009-04-22 |
| MXPA02010565A (es) | 2004-05-17 |
| DE60140087D1 (de) | 2009-11-12 |
| WO2001083800A8 (en) | 2003-01-03 |
| AU9519501A (en) | 2001-11-12 |
| EP1320611B1 (en) | 2005-11-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1511192A (zh) | 聚酮化合物的制备 | |
| US7323573B2 (en) | Production of polyketides | |
| US6410301B1 (en) | Myxococcus host cells for the production of epothilones | |
| AU2001295195A1 (en) | Myxococcus host cells for the production of epothilones | |
| CN1333820A (zh) | 产生环氧噻酮及其衍生物的重组方法和材料 | |
| CN1179046C (zh) | 新型红霉素、其制备方法、用途和含其的药物组合物 | |
| Lau et al. | Optimizing the heterologous production of epothilone D in Myxococcus xanthus | |
| CN101056880A (zh) | 17-去甲基雷帕霉素及其类似物、它们的制备方法和它们作为免疫抑制剂、抗癌剂、抗真菌剂等的用途 | |
| EP1303615A2 (en) | Fermentation process for epothilones | |
| JP2008092958A (ja) | エポチロン生合成用遺伝子 | |
| AU2007200160B2 (en) | Heterologous production of polyketides | |
| JP4280810B2 (ja) | 二次代謝物同属種分布の調節 | |
| JP2004508810A (ja) | ポリケチドの生成 | |
| ZA200207688B (en) | Heterologous production of polyketides. | |
| CN1521258A (zh) | 一类新型埃坡霉素化合物及其制备方法和用途 | |
| AU2001279025B2 (en) | Fermentation process for epothilones | |
| AU2001279025A1 (en) | Fermentation process for epothilones |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20090422 Termination date: 20190426 |