CN110914456A - 检测胎儿染色体异常的方法 - Google Patents
检测胎儿染色体异常的方法 Download PDFInfo
- Publication number
- CN110914456A CN110914456A CN201880035942.XA CN201880035942A CN110914456A CN 110914456 A CN110914456 A CN 110914456A CN 201880035942 A CN201880035942 A CN 201880035942A CN 110914456 A CN110914456 A CN 110914456A
- Authority
- CN
- China
- Prior art keywords
- chromosome
- size
- fetal
- fragments
- target
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 238000000034 method Methods 0.000 title claims abstract description 154
- 208000031404 Chromosome Aberrations Diseases 0.000 title claims abstract description 33
- 210000003754 fetus Anatomy 0.000 title claims abstract description 33
- 206010008805 Chromosomal abnormalities Diseases 0.000 title abstract description 17
- 239000012634 fragment Substances 0.000 claims abstract description 188
- 230000001605 fetal effect Effects 0.000 claims abstract description 162
- 201000010374 Down Syndrome Diseases 0.000 claims abstract description 15
- 210000000349 chromosome Anatomy 0.000 claims description 147
- 239000000523 sample Substances 0.000 claims description 128
- 238000012163 sequencing technique Methods 0.000 claims description 57
- 230000008774 maternal effect Effects 0.000 claims description 41
- 210000002381 plasma Anatomy 0.000 claims description 36
- 150000007523 nucleic acids Chemical class 0.000 claims description 34
- 238000002360 preparation method Methods 0.000 claims description 22
- 238000005516 engineering process Methods 0.000 claims description 20
- 210000001766 X chromosome Anatomy 0.000 claims description 19
- 230000002759 chromosomal effect Effects 0.000 claims description 18
- 238000007481 next generation sequencing Methods 0.000 claims description 16
- 239000012472 biological sample Substances 0.000 claims description 15
- 238000007847 digital PCR Methods 0.000 claims description 15
- 230000008439 repair process Effects 0.000 claims description 13
- 210000002593 Y chromosome Anatomy 0.000 claims description 12
- 239000002773 nucleotide Substances 0.000 claims description 12
- 125000003729 nucleotide group Chemical group 0.000 claims description 12
- 238000000926 separation method Methods 0.000 claims description 12
- 230000005856 abnormality Effects 0.000 claims description 10
- 208000036878 aneuploidy Diseases 0.000 claims description 10
- 231100001075 aneuploidy Toxicity 0.000 claims description 10
- 108020004707 nucleic acids Proteins 0.000 claims description 10
- 102000039446 nucleic acids Human genes 0.000 claims description 10
- 206010044688 Trisomy 21 Diseases 0.000 claims description 9
- 210000003765 sex chromosome Anatomy 0.000 claims description 9
- 230000035772 mutation Effects 0.000 claims description 8
- 201000009928 Patau syndrome Diseases 0.000 claims description 5
- 206010044686 Trisomy 13 Diseases 0.000 claims description 5
- 208000006284 Trisomy 13 Syndrome Diseases 0.000 claims description 5
- 238000012217 deletion Methods 0.000 claims description 5
- 230000037430 deletion Effects 0.000 claims description 5
- 206010053884 trisomy 18 Diseases 0.000 claims description 5
- 201000006360 Edwards syndrome Diseases 0.000 claims description 4
- 208000007159 Trisomy 18 Syndrome Diseases 0.000 claims description 4
- 210000004369 blood Anatomy 0.000 claims description 4
- 239000008280 blood Substances 0.000 claims description 4
- 230000007614 genetic variation Effects 0.000 claims description 4
- 230000008995 epigenetic change Effects 0.000 claims description 2
- 238000003780 insertion Methods 0.000 claims description 2
- 230000037431 insertion Effects 0.000 claims description 2
- 210000002966 serum Anatomy 0.000 claims description 2
- 239000007787 solid Substances 0.000 claims description 2
- 238000006467 substitution reaction Methods 0.000 claims description 2
- 230000005945 translocation Effects 0.000 claims description 2
- 210000002700 urine Anatomy 0.000 claims description 2
- 238000001514 detection method Methods 0.000 abstract description 13
- 208000037280 Trisomy Diseases 0.000 description 51
- 238000004458 analytical method Methods 0.000 description 45
- 239000003153 chemical reaction reagent Substances 0.000 description 36
- 150000002500 ions Chemical class 0.000 description 36
- 238000006243 chemical reaction Methods 0.000 description 34
- 238000009826 distribution Methods 0.000 description 30
- 238000012360 testing method Methods 0.000 description 27
- 230000000694 effects Effects 0.000 description 23
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 22
- 239000011324 bead Substances 0.000 description 21
- 238000013459 approach Methods 0.000 description 16
- 230000035945 sensitivity Effects 0.000 description 14
- 239000000203 mixture Substances 0.000 description 12
- 230000035935 pregnancy Effects 0.000 description 10
- 239000000499 gel Substances 0.000 description 9
- 208000011580 syndromic disease Diseases 0.000 description 9
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 9
- 239000000463 material Substances 0.000 description 8
- 238000002156 mixing Methods 0.000 description 8
- 230000002829 reductive effect Effects 0.000 description 8
- 238000004364 calculation method Methods 0.000 description 7
- 230000008859 change Effects 0.000 description 7
- 210000002826 placenta Anatomy 0.000 description 7
- 230000008901 benefit Effects 0.000 description 6
- 238000010790 dilution Methods 0.000 description 6
- 239000012895 dilution Substances 0.000 description 6
- 238000000605 extraction Methods 0.000 description 6
- 239000008188 pellet Substances 0.000 description 6
- 239000000243 solution Substances 0.000 description 6
- 239000006228 supernatant Substances 0.000 description 6
- 230000008569 process Effects 0.000 description 5
- 239000000376 reactant Substances 0.000 description 5
- 238000013179 statistical model Methods 0.000 description 5
- 229920000936 Agarose Polymers 0.000 description 4
- 108700028369 Alleles Proteins 0.000 description 4
- 238000007400 DNA extraction Methods 0.000 description 4
- 208000017924 Klinefelter Syndrome Diseases 0.000 description 4
- 102000003960 Ligases Human genes 0.000 description 4
- 108090000364 Ligases Proteins 0.000 description 4
- 230000001351 cycling effect Effects 0.000 description 4
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 4
- 238000010348 incorporation Methods 0.000 description 4
- 238000002955 isolation Methods 0.000 description 4
- 239000012160 loading buffer Substances 0.000 description 4
- 238000011068 loading method Methods 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 238000001556 precipitation Methods 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- 238000012546 transfer Methods 0.000 description 4
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 208000026928 Turner syndrome Diseases 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 210000004027 cell Anatomy 0.000 description 3
- 230000001186 cumulative effect Effects 0.000 description 3
- 230000001627 detrimental effect Effects 0.000 description 3
- 238000002405 diagnostic procedure Methods 0.000 description 3
- 208000035475 disorder Diseases 0.000 description 3
- 239000000975 dye Substances 0.000 description 3
- 230000007774 longterm Effects 0.000 description 3
- 238000013507 mapping Methods 0.000 description 3
- 239000003550 marker Substances 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 238000010606 normalization Methods 0.000 description 3
- 229920003023 plastic Polymers 0.000 description 3
- 239000004033 plastic Substances 0.000 description 3
- 239000004065 semiconductor Substances 0.000 description 3
- 238000007619 statistical method Methods 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 235000018185 Betula X alpestris Nutrition 0.000 description 2
- 235000018212 Betula X uliginosa Nutrition 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 108091028043 Nucleic acid sequence Proteins 0.000 description 2
- 208000007254 Tetrasomy X Diseases 0.000 description 2
- 239000011543 agarose gel Substances 0.000 description 2
- 238000007622 bioinformatic analysis Methods 0.000 description 2
- 238000003766 bioinformatics method Methods 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 238000012937 correction Methods 0.000 description 2
- 238000001962 electrophoresis Methods 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 230000008014 freezing Effects 0.000 description 2
- 238000007710 freezing Methods 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 230000003278 mimic effect Effects 0.000 description 2
- 230000008775 paternal effect Effects 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 238000011002 quantification Methods 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 238000010187 selection method Methods 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 230000008685 targeting Effects 0.000 description 2
- 238000010998 test method Methods 0.000 description 2
- 208000010543 22q11.2 deletion syndrome Diseases 0.000 description 1
- 208000026817 47,XYY syndrome Diseases 0.000 description 1
- 208000024143 48,XXYY syndrome Diseases 0.000 description 1
- 208000009575 Angelman syndrome Diseases 0.000 description 1
- 238000012935 Averaging Methods 0.000 description 1
- 208000014392 Cat-eye syndrome Diseases 0.000 description 1
- 108091061744 Cell-free fetal DNA Proteins 0.000 description 1
- 208000003449 Classical Lissencephalies and Subcortical Band Heterotopias Diseases 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- 206010011385 Cri-du-chat syndrome Diseases 0.000 description 1
- 201000003883 Cystic fibrosis Diseases 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- 102000053602 DNA Human genes 0.000 description 1
- 238000001712 DNA sequencing Methods 0.000 description 1
- 230000004568 DNA-binding Effects 0.000 description 1
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 1
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 1
- 208000000398 DiGeorge Syndrome Diseases 0.000 description 1
- 108700024394 Exon Proteins 0.000 description 1
- 208000022471 Fetal disease Diseases 0.000 description 1
- 208000018478 Foetal disease Diseases 0.000 description 1
- 208000001914 Fragile X syndrome Diseases 0.000 description 1
- 208000026350 Inborn Genetic disease Diseases 0.000 description 1
- 108091092195 Intron Proteins 0.000 description 1
- 208000004706 Jacobsen Distal 11q Deletion Syndrome Diseases 0.000 description 1
- 208000029279 Jacobsen Syndrome Diseases 0.000 description 1
- 201000004246 Miller-Dieker lissencephaly syndrome Diseases 0.000 description 1
- 208000035022 Miller-Dieker syndrome Diseases 0.000 description 1
- 208000001804 Monosomy 5p Diseases 0.000 description 1
- 208000016679 Monosomy X Diseases 0.000 description 1
- 208000010610 Mosaic trisomy 8 Diseases 0.000 description 1
- 239000012807 PCR reagent Substances 0.000 description 1
- 241000286209 Phasianidae Species 0.000 description 1
- 201000010769 Prader-Willi syndrome Diseases 0.000 description 1
- 201000001388 Smith-Magenis syndrome Diseases 0.000 description 1
- 208000002903 Thalassemia Diseases 0.000 description 1
- 206010053871 Trisomy 8 Diseases 0.000 description 1
- 206010071547 Trisomy 9 Diseases 0.000 description 1
- 208000006254 Wolf-Hirschhorn Syndrome Diseases 0.000 description 1
- 206010056894 XYY syndrome Diseases 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 238000002669 amniocentesis Methods 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 238000010876 biochemical test Methods 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 238000004422 calculation algorithm Methods 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 210000004252 chorionic villi Anatomy 0.000 description 1
- 239000013611 chromosomal DNA Substances 0.000 description 1
- 238000010835 comparative analysis Methods 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000005520 cutting process Methods 0.000 description 1
- 238000005202 decontamination Methods 0.000 description 1
- 230000003588 decontaminative effect Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 239000012470 diluted sample Substances 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 238000005315 distribution function Methods 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 230000006862 enzymatic digestion Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 230000005669 field effect Effects 0.000 description 1
- 238000013100 final test Methods 0.000 description 1
- 230000004907 flux Effects 0.000 description 1
- 230000014509 gene expression Effects 0.000 description 1
- 208000016361 genetic disease Diseases 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 229920001519 homopolymer Polymers 0.000 description 1
- 238000009396 hybridization Methods 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- -1 hydrogen ions Chemical class 0.000 description 1
- GPRLSGONYQIRFK-UHFFFAOYSA-N hydron Chemical compound [H+] GPRLSGONYQIRFK-UHFFFAOYSA-N 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 210000005259 peripheral blood Anatomy 0.000 description 1
- 239000011886 peripheral blood Substances 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 238000003752 polymerase chain reaction Methods 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 238000003793 prenatal diagnosis Methods 0.000 description 1
- 230000037452 priming Effects 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 238000010926 purge Methods 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 238000012175 pyrosequencing Methods 0.000 description 1
- 239000013643 reference control Substances 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000009933 reproductive health Effects 0.000 description 1
- 210000003296 saliva Anatomy 0.000 description 1
- 239000012898 sample dilution Substances 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 238000005204 segregation Methods 0.000 description 1
- 238000002864 sequence alignment Methods 0.000 description 1
- 238000000060 site-specific infrared dichroism spectroscopy Methods 0.000 description 1
- 238000004513 sizing Methods 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000009987 spinning Methods 0.000 description 1
- 230000003068 static effect Effects 0.000 description 1
- 238000000551 statistical hypothesis test Methods 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 230000002992 thymic effect Effects 0.000 description 1
- 206010044689 trisomy 22 Diseases 0.000 description 1
- 208000026485 trisomy X Diseases 0.000 description 1
- 208000034298 trisomy chromosome 8 Diseases 0.000 description 1
- 230000007306 turnover Effects 0.000 description 1
- 238000002604 ultrasonography Methods 0.000 description 1
- 238000012795 verification Methods 0.000 description 1
- 238000011179 visual inspection Methods 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 238000012070 whole genome sequencing analysis Methods 0.000 description 1
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1034—Isolating an individual clone by screening libraries
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6879—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for sex determination
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B20/00—ICT specially adapted for functional genomics or proteomics, e.g. genotype-phenotype associations
- G16B20/20—Allele or variant detection, e.g. single nucleotide polymorphism [SNP] detection
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B30/00—ICT specially adapted for sequence analysis involving nucleotides or amino acids
- G16B30/10—Sequence alignment; Homology search
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/156—Polymorphic or mutational markers
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Biotechnology (AREA)
- Biophysics (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- General Engineering & Computer Science (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- Immunology (AREA)
- Theoretical Computer Science (AREA)
- Medical Informatics (AREA)
- Evolutionary Biology (AREA)
- Bioinformatics & Computational Biology (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Biomedical Technology (AREA)
- Pathology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Plant Pathology (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
本发明涉及检测胎儿染色体异常的新方法,特别是,本发明涉及三体性21(唐氏综合征)的检测,其包括约100 bp‑约150 bp的所分析片段大小的富集。本发明还涉及用于实施所述方法的试剂盒。本发明还涉及预测怀孕女性受试者体内的胎儿性别的方法。
Description
发明领域
本发明涉及一种检测胎儿染色体异常的新方法,特别是,本发明涉及三体性21(唐氏综合征)的检测,其包括所分析片段大小约100 bp-约150 bp的富集。本发明还涉及用于实施所述方法的试剂盒。本发明还涉及预测怀孕女性受试者体内胎儿性别的方法。
发明背景
唐氏综合征为一种相对常见的遗传障碍,每800名活产婴儿中约有一名受到影响。该综合征是由存在额外的全染色体21 (三体性21,T21)引起,或不太常见地,由存在额外的该染色体的大部分引起。在活产婴儿也发生涉及其他常染色体的三体性(即T13或T18),但比T21更罕见。
通常,其中存在由额外染色体或由染色体缺陷引起的胎儿非整倍性的病症,在母体无细胞血浆DNA中的胎儿DNA分子群体产生失衡,这是可检测的。
开发可靠的用于产前诊断胎儿染色体异常的方法已成为生殖保健的长期目标(Puszyk等人, 2008, Prenat Diagn 28, 1-6)。基于通过羊膜穿刺术或绒毛膜绒毛取样获取胎儿材料的方法为侵入性的,并且甚至是在熟练的临床医生手中也对妊娠带来不可忽视的风险。在目前的实践中,这种侵入性的诊断方法通常用于由于母体年龄或通过事先使用生化测试或超声波检查进行筛查而指征唐氏妊娠的可能性增加时。需要可靠的、适用于头3个月妊娠期的、快速返回结果的和廉价的非侵入性产前诊断(NIPD)方法。
通过利用发现孕妇血浆中的无细胞DNA包含胎儿来源的成分,已经朝着实现这一目标取得了进展(Lo等人, 1997, Lancet 350, 485-487)。无细胞血浆DNA (下文称为“血浆DNA”)主要由短DNA分子(80-200 bp)组成,其中一般地5%-20%属于胎儿来源的,其余为母体的(Birch等人, 2005, Clin Chem 51, 312-320; Fan等人, 2010, Clin Chem 56,1279-1286)。对于血浆DNA分子的细胞来源及其进入血液并随后从循环中清除的机制了解甚少。然而,普遍认为胎儿成分很大程度上是胎盘内的凋亡细胞死亡的结果(Bianchi,2004, Placenta 25, S93-S101)。胎儿来源的血浆DNA分子的分数因情况而异,存在很大的个体差异。叠加于个体差异上的为胎儿成分随着孕龄的增加而增加的总体趋势(Birch等人, 2005, 同上; Galbiati等人, 2005, Hum Genet 117, 243-248)。胎儿成分可易于在妊娠早期,一般地早在第8周就可检测到。
原则上,如果血浆中的无细胞胎儿DNA没有被母体成分稀释,那么通过与正常妊娠相比较,表征T21的额外染色体预期会导致源自该染色体的DNA分子过量50%。然而,取胎儿来源的无细胞血浆DNA成分的典型值为10%,则结果的失衡预期仅为5%,或者相对于正常妊娠为1.00,21号染色体来源的片段数量相对增加至1.05的值。在其中血浆DNA的胎儿成分小于或大于10%的值的情况下,母体血浆中的分子群体中21号染色体来源的分子数量失衡将相应地更小或更大。
因此,对于T21进行诊断测试的基础为从母体血浆中获取DNA分子的核苷酸序列数据(“DNA测序”)。一旦从单个DNA分子获取了部分或完整的核苷酸序列信息,就必须应用生物信息学技术,以最简单地通过与一个或多个参考人类基因组进行比较,将单个分子指定给其来源的染色体。在涉及具有T21的胎儿的妊娠的情况下,可检测到分子群体的轻微失衡,因为21号染色体来源的分子数量超过正常妊娠所预期的数量。
考虑到21号染色体仅占人类基因组的一小部分(少于2%)这一事实,为了从该染色体收集足够大量进行可靠诊断,必须对来自母体血浆的大量DNA分子进行随机采样、测序并根据生物信息学将其指定给特定染色体。以下两项所需的血浆DNA分子的总数小于对全部或大多数胎儿基因组采样所需的量,但其至少是数十万个分子:(1) 特征在于从其来源的核苷酸序列信息,和然后(2) 可靠地指定给染色体位置。所需的最小数量为构成母体无细胞血浆DNA分子群体的胎儿成分的血浆DNA分数的函数。一般地,数量介于一百万或几百万个分子之间。
由于对来自特定染色体位置的DNA分子进行计数需要很高的定量准确性,因此应用该方法的挑战相当大。此外,来自母体血浆的DNA为基因组的混合物,在其中胎儿成分占小部分。这种定量技术问题性质上不同于鉴定DNA样品中特定基因座处的突变。
鉴于对于足够大量的血浆DNA可获取一些核苷酸序列数据,和鉴于可以可靠地应用生物信息学方法将足够大量指定给其染色体来源,可应用统计方法来以统计置信度确定血浆DNA分子群体中是否存在染色体失衡。
这种对来自母体血浆的DNA片段的随机样品(但该样品仅构成完整基因组的一小部分)进行测序的想法,为Fan等人, 2008, Proc Natl Acad Sci U S A 105, 16266-16271和Chiu等人, 2008, Proc Natl Acad Sci U S A 105, 20458-20463所述的NIPD方法学的基础。
利用测序数据的一种明显方法为排除指定范围之外的所有片段,从而在计算机中增加胎儿分数。然而,这种方法将使大多数测序数据无用,并且需要显著增加所处理和测序样品的量。因此,可认为数字富集对于在常规实验室环境中使用是昂贵、低效且不切实际的。
一种备选解决方案为在测序之前富集源自胎儿的DNA的比例。一般地已经由大小选择方法(去除约200 bp或更大的片段)利用这种富集。这种方法具有有限的灵敏度和富集胎儿分数的能力。迄今为止,还没有描述使得能够从生物样品高度准确和精确地富集胎儿DNA的方法。
因此,需要一种用于从生物样品富集胎儿DNA以检测胎儿染色体异常的高度准确、非侵入性且简化的方法。这种方法可提高针对常见染色体异常(比如三体性13、18和21)的非侵入性产前测试的性能。其也将显著提高检测小得多的染色体异常(比如微缺失)的能力,其中相对于更常见的染色体异常,目前的性能非常差。
发明概述
本发明的第一方面提供一种检测胎儿染色体异常的方法,其包括以下步骤:
(a) 从获自怀孕女性受试者的生物样品中分离核酸;
(b1) 选择120 bp-135 bp之间的核酸片段大小值用于最佳胎儿分数;
(b2) 分离大小在步骤(b1)中选择的片段大小值的20 bp以内的核酸片段;
(c) 确定与目标染色体的目标区域对齐的所述片段的第一数量和确定与参考染色体内的一个或多个目标区域对齐的所述片段的第二数量;
(d) 计算第一和第二数量之间的比率或差异;
(e) 基于所述比率或差异确定所述目标染色体的胎儿异常的存在。
本发明的第二方面提供一种预测怀孕女性受试者体内的胎儿性别的方法,所述方法包括以下步骤:
(a) 从获自怀孕女性受试者的生物样品中分离核酸;
(b1) 选择120 bp-135 bp之间的核酸片段大小值用于最佳胎儿分数;
(b2) 分离大小在步骤(b1)中选择的片段大小值的20 bp之内的核酸片段;
(c) 确定与性染色体对齐的所述片段的第一数量,和确定与一个或多个参考染色体对齐的所述片段的第二数量;
(d) 计算第一和第二数量之间的比率或差异;
(e) 基于与所述参考染色体相比较是过多还是等同的片段与X染色体对齐或者是否存在Y染色体来确定所述胎儿的性别。
附图简述
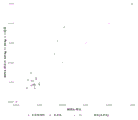
图1:135 bp目标片段大小(±10 bp)处的21号染色体比率。
图2: 135 bp目标片段大小(±10 bp)处的胎儿分数估计。
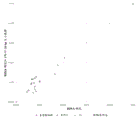
图3:135 bp目标片段大小(±10 bp)处的X染色体比率。
图4:135 bp目标片段大小(±10 bp)处的片段大小分布,和相对较小的母体峰。
图5:135 bp目标片段大小(±10 bp)处的重复性数据。
图6:135 bp目标片段大小(±5 bp)处的21号染色体比率。
图7:135 bp目标片段大小(±20 bp)处的21号染色体比率。
图8:120 bp目标片段大小(±10 bp)处的21号染色体比率。
图9:170 bp目标片段大小(±10 bp)处的21号染色体比率。
图10和图11:几个目标片段大小和范围处的胎儿分数估计。
图12:给定大小的片段为胎儿来源的建模概率和典型的母体片段大小分布(10%胎儿分数)。
图13:描绘给定大小的片段为胎儿的概率的图示。
图14:常染色体比率比较,大小加权相对于未加权(21号染色体)。
图15:常染色体比率比较,大小加权相对于未加权(18号染色体)。
图16:常染色体比率比较,大小加权相对于未加权(13号染色体)。
图17:未加权和加权分析方法的T21受影响和未受影响的样品组的分布。
图18:未加权和加权分析方法的T18受影响和未受影响的样品组的分布。
图19:未加权和加权分析方法的T13受影响和未受影响的样品组的分布。
图20:未加权和大小加权分析方法两者分析时的有效胎儿分数。
图21:在未加权和大小加权分析方法之间,分析时受三体性影响的样品的有效胎儿分数的比较。
发明详述
本发明的第一方面提供一种检测胎儿染色体异常的方法,其包括以下步骤:
(a) 从获自怀孕女性受试者的生物样品中分离核酸;
(b1) 选择120 bp-135 bp之间的核酸片段大小值用于最佳胎儿分数;
(b2) 分离大小在步骤(b1)中选择的片段大小值的20 bp之内的核酸片段;
(c) 确定与目标染色体的目标区域对齐的所述片段的第一数量,和确定与参考染色体内的一个或多个目标区域对齐的所述片段的第二数量;
(d) 计算第一和第二数量之间的比率或差异;
(e) 基于所述比率或差异确定所述目标染色体的胎儿异常的存在。
可提及的本公开的一个方面提供一种检测胎儿染色体异常的方法,所述方法包括以下步骤:
(a) 从获自怀孕女性受试者的生物样品中分离核酸;
(b) 分离大小为约80 bp-约150 bp的核酸片段;
(c) 确定与目标染色体的目标区域对齐的所述片段的第一数量,和确定与参考染色体内的一个或多个目标区域对齐的所述片段的第二数量;
(d) 计算第一和第二数量之间的比率或差异;
(e) 基于所述比率或差异确定所述目标染色体的胎儿异常的存在。
在检测胎儿染色体异常中,重要的是确保尽可能不会确定假结果。特别是,特别期望减少确定假阴性结果的概率。然而,同样重要的是确保有效地使用数据,并且在可接受数量的情况或测试下产生阳性且准确的结果。理想地,测试结果应在可能的情况下声明,而不是测试表明由于与测试相关的一个或多个参数而导致结果不可靠。
源自胎儿的无细胞DNA的比例为用于检测胎儿样品中染色体异常的关键参数。准确检测需要最小比例的DNA结合其他因素。特别是,较小的染色体异常(比如微缺失)需要较大比例的源自胎儿的DNA以便可检测到。本发明的发明人已经令人惊讶地鉴定出,将胎儿材料富集成约80 bp-约150 bp的片段大小已显著改善这种测试的准确性和性能,如本文呈现的数据所支持。另外,本发明的方法产生显著更低量的测序数据,从而导致更省时和更经济的胎儿染色体异常检测方法。
自2004年以来的许多出版物(即EP2728014)均指出,母体循环中的母体和胎儿无细胞DNA片段具有不同的大小分布。另外,在文献中已经注意到,有可能在分析中利用这种差异进行非侵入性产前检查(NIPT)。然而,迄今为止,在文献中尚未公布从混合的母体/胎儿cfDNA样品提取的DNA片段的大小与所述片段为胎儿或母体来源的概率之间直接的确定性理论关系。需要这种模型才能确定最佳大小范围,以用于基于大小选择的方法,从而有效富集样品中胎儿DNA的分数以经受NIPT分析,并且尤其可用于在关于允许的大小范围的与仪器相关的实际考虑因素起作用时告知范围选择。
为了支持本发明,图12显示发明人采用先前公布的大小分布数据构建的这种模型。实线代表给定大小的片段为胎儿来源的建模概率,和作为参考,虚线代表总胎儿分数为10%的样品按大小的总片段分布(即独立于大小,任何给定片段来自胎儿的总概率为0.1)。
通过选择尽可能使大概率值最大化的大小范围,可将图12中实线所示的模型直接用于告知大小分数的选择,以最佳地富集胎儿DNA的NIPT样品。独立于关于大小范围的任何其他考虑因素,典型的最佳范围可为例如120±10 bp (以仅包括概率的峰),然而出于实际实施的目的,可选择仍有效富集胎儿DNA的备选范围,比如:
实际上,已针对总体母体和胎儿片段(即不考虑大小)同样可能在样品中出现的情况构建图12的概率模型。当然,这种总体平衡在实际样品中会有所不同,胎儿分数在约3%-25%之间,然而母体和胎儿片段对于大小的相对浓度仍然会遵循相同的分布,独立于总体胎儿分数。
此外,本发明的方法使得能够显著提高所得测序的效率。例如,分析之前胎儿分数百分比的增加使得能够显著减少准确检测胎儿染色体异常所需的数据量。
应当意识到,术语“在20 bp内”是指+/- 20 bp,即核酸片段的总体范围为40 bp。在一个实施方案中,步骤(b2)中的分离为分离10 bp内的核酸片段(即片段总体范围为20bp)。在另一个实施方案中,步骤(b2)中的分离为分离5 bp内的核酸片段(即片段总体范围为10 bp)。
发明人已经确定100 bp-155 bp的任何40 bp“窗口”均提供最佳结果,如本文呈现和讨论的数据所示。因此,在分析之前,从120 bp-135 bp选择任意值(如本文所述的步骤(b1))。发明人已经令人惊讶地发现,在这些范围之间的任何值均提供最佳的胎儿分数,因为大多数胎儿染色体片段具有该大小。在一个实施方案中,在步骤(b1)中所选的值为120bp、或121 bp、或122 bp、或123 bp、或124 bp、或125 bp、或126 bp、或127 bp、或128 bp、或129 bp、或130 bp、或131 bp、或132 bp、或133 bp、或134 bp、或135 bp。
本发明的一个关键方面为认识到,用户不仅必须在步骤(b1)中选择上述任意值,而且还必须然后确保然后分析与该大小非常接近的大小范围。这很重要,因为如果在步骤(b1)中选择125 bp作为任意值,并且仅识别出具有该大小的片段,则读取次数将不足以产生显著且最关键的足够准确的结果。因此,分析在步骤(b1)中选择的大小的20 bp或10 bp或5 bp内(即总核酸片段范围为40 bp或20 bp或10 bp)的所有片段将提供更大量的大多数胎儿染色体片段,以显著提高结果的灵敏度和准确性。因此,总而言之,在步骤(b1)和(b2)之间存在协同作用,使得步骤(b1)为最大胎儿浓度提供最佳大小值,并且步骤(b2)中的范围使胎儿片段的总数最大化。
胎儿染色体异常
应当意识到,本文提及的“胎儿染色体异常”是指胎儿染色体内的任何遗传变异,并且包括所述胎儿的天然、非突变或野生型遗传密码的任何变异。这种遗传变异的实例包括:非整倍性、重复、易位、突变(例如点突变)、取代、缺失、单核苷酸多态性(SNP)、染色体异常、拷贝数变异(CNV)、表观遗传变化和DNA倒置。
本文提及的术语“单核苷酸多态性(SNP)”旨在指当给定基因中的单个核苷酸在物种的成员之间或个体的成对染色体之间不同时发生的DNA序列变异。
在一个实施方案中,遗传变异为一种功能突变,即一种为临床相关的胎儿疾病或障碍的起因的突变。除了片段长度障碍(比如脆性X综合征)之外,这种疾病或障碍的实例还包括地中海贫血和囊性纤维化。突变可为功能性的,因为其影响氨基酸编码或通过破坏调控元件(例如其可调控基因表达,或通过破坏参与剪接调控的序列(其可为外显子或内含子))。
本发明可用于检测的合适的胎儿染色体异常的实例包括:唐氏综合征(三体性21)、Edward综合征(三体性18)、Patau综合征(三体性13)、三体性9、Warkany综合征(三体性8)、猫眼综合征(22号染色体的4个拷贝)、三体性22和三体性16。
另外或者备选地,对基因、染色体或染色体的一部分、拷贝数异常的检测可包括检测和/或诊断选自以下的病症:Wolf-Hirschhorn综合征(4p-)、Cri du chat综合征(5p-)、Williams-Beuren综合征(7-)、Jacobsen综合征(11-)、Miller-Dieker综合征(17-)、Smith-Magenis综合征(17-)、22ql l.2缺失综合征(也称为Velocardiofacial综合征、DiGeorge综合征、椎干异常面容综合征、先天性胸腺发育不全和Strong综合征)、Angelman综合征(15-)和Prader-Willi综合征(15-)。
另外或者备选地,染色体拷贝数异常的检测可包括检测和/或诊断选自以下的病症:Turner综合征(Ullrich-Turner综合征或单体性X)、Klinefelter综合征、47,XXY或XXY综合征、48,XXYY综合征、49,XXXXY综合征、三X综合征、XXXX综合征(也称为四体性X、四倍X、或48,XXXX)、XXXXX综合征(也称为五体性X或49,XXXXX)和XYY综合征。
在一个实施方案中,目标染色体为13号染色体、18号染色体、21号染色体、X染色体或Y染色体。
在一个实施方案中,胎儿染色体异常为胎儿染色体非整倍性。在另一个实施方案中,胎儿染色体非整倍性为三体性13、三体性18或三体性21。在仍然另一个实施方案中,胎儿染色体非整倍性为三体性21 (唐氏综合征)。在该实施方案中,本领域的技术人员将易于理解,本发明的方法学可应用于诊断其中胎儿携带21号染色体的大部分而不是整个染色体的情况。
在一个实施方案中,胎儿染色体异常为例如高达1 Mb、高达5 Mb、高达10 Mb或高达20 Mb或大于20 Mb的染色体插入或缺失。
样品提取
应当意识到,样品可根据常规程序从怀孕的女性受试者获取。在一实施方案中,生物样品为母体血液、血浆、血清、尿液或唾液。 在另一个实施方案中,生物样品为母体血浆。
获取母体血浆的步骤一般地包括从怀孕的女性受试者抽取(一般地通过静脉穿刺) 5-20 ml血液样品(一般地为外周血液样品)。因此,获取这种样品的特征在于对胎儿空间的非侵入性和对母亲的侵入性最小。在通过离心去除细胞物质之后,通过常规方法制备血浆(Maron等人, 2007, Methods Mol Med 132, 51-63)。
通过对于血浆DNA的核苷酸序列无偏倚的常规方法学从母体血浆提取DNA (Maron等人, 2007, 同上)。血浆DNA分子的群体一般地将包含胎儿来源的分数和母体来源的分数。
从生物样品中分离核酸
在一个实施方案中,步骤(a)中的分离步骤包括制备核酸片段的文库。应当意识到,分离、片段化和文库制备的步骤可根据技术人员众所周知的常规程序进行。在另一个实施方案中,文库制备包括DNA末端修复、衔接子连接、净化和PCR的依序步骤。有关可如何制备合适的核酸文库的全部实验细节描述于本文的方法部分,特别是步骤1-49。
富集
在一个实施方案中,分离步骤(b2)包括富集大小为步骤(b1)中选择的片段大小值的10bp以内,比如为步骤(b1)中选择的片段大小值的5 bp以内的核酸片段。
在本公开的一个实施方案中,分离步骤(b2)包括富集大小为115±35 bp (即80-150 bp),比如115±30 bp、115±25 bp、115±20 bp、115±15 bp、115±10 bp、120±10bp、110±10 bp、135±10 bp、140±10 bp、115±5 bp或115 bp的核酸片段。
在另一个实施方案中,分离步骤(b2)包括富集大小为120±10 bp、110±10 bp、135±10 bp、140±10 bp、115±5 bp或115 bp的核酸片段。
应当意识到,这种富集步骤可根据技术人员众所周知的常规程序进行。在一个实施方案中,分离步骤(b2)包括使用大小选择进行富集。在另一个实施方案中,分离步骤(b2)包括使用基于凝胶的大小选择进行富集。在另一个实施方案中,分离步骤(b2)包括使用自动化的基于凝胶的大小选择进行富集。
自动化的基于凝胶的大小选择的一个这种实例包括来自Coastal Genomics的Ranger Technology™。
Ranger Technology™利用隔离的盒子,其创建黑暗环境,以防止光对分析的影响。目前,盒具有专有大小而不是SSID,以匹配其他自动化足迹。盒含有形成的带有12个通道的琼脂糖凝胶以供使用。
样品按照标准电泳进行处理,藉此盒末端产生的电荷导致DNA片段依大小(以及因此电荷)而移动和分离。没有使用梯度,但提供上下标志物的混合物以确保可在样品内进行大小确定。输出可以电泳图或凝胶图像形式显示。
所需大小的样品将被处理到确定要取出的孔内含有的溶液中,在此,整个体积将被取出并按照Ranger软件的提示进行多次补充。
Ranger Technology™在蓝光和红光下拍摄整个迁移过程的凝胶图像,所述光基于在存在该光的情况下变为激发的相关染料(每种具有其自己的荧光,这可减少与不正确的标志识别相关的不正确结果)为样品和标志物提供可见性。Ranger Technology™的全部细节可见于http://coastalgenomics.com/。
在一个可提及的备选实施方案中,本发明的方法可包括基于低熔点琼脂糖的方法。该实施方案需要将来自样品的DNA片段在合适的琼脂糖凝胶上运行,然后使用手动方法从凝胶上切下(例如使用一次性刀切下的凝胶的细带)。
在一个可提及的备选实施方案中,本发明的方法可包括基于珠粒的大小选择方法而不是基于凝胶的大小选择。该实施方案需要基于珠粒的方法,该方法基于碱基对中的其大小以很高程度的准确性和精确度来选择DNA片段。
在一个可提及的备选实施方案中,本发明的方法可包括基于PCR的方法。该实施方案需要建立PCR,藉此长于指定碱基对长度的片段不能扩增(或以大大降低的效率扩增)。
在一个可提及的备选实施方案中,本发明的方法可包括基于酶消化的方法。该实施方案需要使用酶来消化(或优先地消化)超过指定长度的DNA片段。
上文的一些实施方案利用基于其大小的DNA片段的物理分离,这需要后续步骤来从不需要的片段纯化出期望大小的片段。这可通过基于已知大小的DNA片段的行为构建标准曲线,并然后使用该标准曲线分离含有目标片段的大小分离的片段区域来实现。
或者,可构建一种或多种标记的分子,当经受大小分离方法时,其具有与目标DNA片段类似的行为。可将这些标记的分子与DNA混合以进行大小分离,并然后在分离过程后,含有标记的片段的区域也将含有目标DNA片段。
分离后,有必要分离大小分离的目标DNA片段组。
有许多不同的方法可分离所需大小范围的DNA片段。在一种方法中,使用已知大小的DNA片段构建标准曲线,然后将其用于分离目标DNA片段。
片段对齐
本发明方法的步骤(c)进行对齐或匹配分析。这种分析最初将需要测量在步骤(b2)中分离的片段内一个或多个目标序列的存在或者对所述片段进行测序。因此,在一个实施方案中,步骤(c)最初包括在对齐之前对在步骤(b2)中分离的片段进行测序或使所述片段经受基于数字PCR或SNP的方法学。
测序
在另一个实施方案中,步骤(c)最初包括对在步骤(b2)中分离的片段进行测序。技术人员将意识到,本发明不限于用于对富集的片段进行测序并获取序列数据的任何特定技术。在一个实施方案中,通过包括使用聚合酶链反应的测序平台获取序列数据。在另一个实施方案中,使用下一代测序平台获取序列数据。这种测序平台已在以下文献中进行了广泛讨论和综述:Loman等人(2012) Nature Biotechnology 30(5), 434-439; Quail等人(2012)BMC Genomics 13, 341; Liu等人(2012) Journal of Biomedicine and Biotechnology2012, 1-11; 和Meldrum等人(2011) Clin Biochem Rev.32(4): 177-195,其测序平台通过参考结合至本文中。
合适的下一代测序平台的实例包括:Roche 454 (即Roche 454 GS FLX)、AppliedBiosystems的SOLiD系统(即SOLiDv4)、Illumina的GAIIx、HiSeq 2000和MiSeq测序仪、LifeTechnologies的Ion Torrent基于半导体的测序仪、Pacific Biosciences的PacBio RS和Sanger的3730xl。
Roche的454平台的每一个均采用焦磷酸测序,藉此化学发光信号表明碱基掺入,并且通过均聚物读取,信号强度与掺入的碱基数量相关。
在一个实施方案中,通过包括使用基于半导体的测序方法学的测序平台对富集的片段进行测序。基于半导体的测序方法学的优点为仪器、芯片和试剂的制造非常廉价,测序过程快速(尽管被emPCR偏置(off-set))和系统可按比例缩放,尽管这可能会有点受到用于emPCR的珠粒大小的限制。
在一个实施方案中,通过包括使用通过合成测序的测序平台对富集的片段进行测序。Illumina的通过合成测序(SBS)技术为目前成功的和全球范围内广泛采用的下一代测序平台。TruSeq技术支持使用专有的基于可逆终止子的方法进行大规模平行测序,该方法使得能够在将单个碱基掺入到生长中的DNA链时对其进行检测。随着添加每个dNTP对荧光标记的终止子进行成像并然后切割以使得能够掺入下一个碱基。由于在每个测序循环期间均存在全部4种可逆终止子结合的dNTP,因此天然竞争使得掺入偏倚最小化。
在一个实施方案中,通过包括使用基于纳米孔的测序方法学的测序平台对富集的片段进行测序。在另一个实施方案中,基于纳米孔的方法学包括使用模拟活细胞中的细胞膜和蛋白通道的情况的有机型纳米孔,比如在Oxford Nanopore Technologies使用的技术中的(例如Branton D, Bayley H,等人(2008). Nature Biotechnology 26 (10), 1146-1153)。在仍然另一个实施方案中,基于纳米孔的方法学包括使用由金属、聚合物或塑料材料构建的纳米孔。
在一个实施方案中,下一代测序平台选自Life Technologies的Ion Torrent平台或Illumina的MiSeq。该实施方案的下一代测序平台在大小上均小和特征在于周转率快,但是提供的数据通量有限。
在另一个实施方案中,下一代测序平台为个人基因组仪(Personal GenomeMachine, PGM),其为Life Technologies的Ion Torrent个人基因组仪(Ion TorrentPGM)。Ion Torrent装置使用类似于通过合成测序(SBS)的策略,但通过核苷酸掺入期间因DNA聚合酶活性导致的氢离子释放来检测信号。本质上,Ion Torrent芯片为非常灵敏的pH计。每个离子芯片均含有数百万个离子灵敏场效应晶体管(ISFET)传感器,其使得能够平行检测多个测序反应。ISFET装置的使用为本领域的技术人员众所周知的,并且完全处于可用于获取本发明方法所需的序列数据的技术范围内(Prodromakis等人(2010) IEEEElectron Device Letters 31(9), 1053-1055; Purushothaman等人(2006) Sensors andActuators B 114, 964-968; Toumazou和Cass (2007) Phil. Trans. R. Soc. B, 362,1321-1328; WO 2008/107014 (DNA Electronics Ltd); WO 2003/073088 (Toumazou);US 2010/0159461 (DNA Electronics Ltd); 每一个的测序方法学通过参考结合至本文中)。
在一个实施方案中,通过包括使用离子(比如氢离子)的释放的测序平台对富集的片段进行测序。该实施方案提供许多关键优势。例如,Ion Torrent PGM 在Quail等人(2012; 同上)中描述为市场上最便宜的个人基因组仪(即约80000美元)。此外,Loman等人(2012; 同上)将Ion Torrent PGM描述为产生最快通量(80-100 Mb/h)和最短运行时间(~3h)。
应当意识到,后代Ion Torrent装置也可在本发明中发现实用性,例如在一个实施方案中,序列数据通过基于Life Technologies的Ion Torrent平台(比如带有PI或PII芯片的Ion Proton)及其进一步的衍生装置和组件的能够多路复用的迭代获取。
数字PCR
在另一个实施方案中,步骤(c)最初包括使步骤(b2)中分离的片段经受数字PCR。技术人员应当意识到,本发明不限于用于所富集的片段的数字PCR和获取数据的任何特定技术。本发明本身特别适合于使用数字PCR作为片段分析方法,因为当胎儿分数为至少20%时,数字PCR工作最佳,并且本发明提供能够提供这种胎儿分数水平的方法学。EP 1981995中描述了可如何对母体血浆样品进行数字PCR的合适方法学。合适的数字PCR系统的实例包括选自以下的数字PCR系统:Quant studio数字PCR系统(ThermoFisher)和RainDrop Plus数字PCR系统(RainDance technologies)。
对齐分析
这种匹配分析一般地包括生物信息学分析,其使用合适的软件来进行,并且基于所述片段是否与所述染色体对齐或认为源自所述染色体来为给定染色体(即目标或参考染色体)的每个片段分配命中。
在一个实施方案中,使用IONA®软件(Premaitha Helath plc)、Bowtie2或BWA-SW(Li和Durbin (2010) Bioinformatics, Epub)比对软件或采用最大精确匹配技术的比对软件比如BWA-MEM (lh3lh3.users.sourceforge.net/download/mem-poster.pdf)或CUSHAW2 (http://cushaw2.sourceforge.net/)软件进行对齐。在另一个实施方案中,使用Bowtie2软件进行对齐。在仍然另一个实施方案中,Bowtie2软件为Bowtie2 2.0.0-beta7。
在一个备选的实施方案中,使用采用最大精确匹配(MEM)技术的比对软件比如BWA-MEM (lh3lh3.users.sourceforge.net/download/mem-poster.pdf)或CUSHAW2(http://cushaw2.sourceforge.net/)软件进行对齐。认为MEM算法具有提供更高准确性的优势。
应当意识到,对于最大准确性,应将序列映射或对齐到唯一的染色体位置。例如,如果片段既映射到目标染色体的目标区域又映射到另一个染色体,则应将其从分析中删除,因为不能认为其与目标染色体的目标区域唯一对齐。在一个实施方案中,所述方法另外包括从对齐步骤(c)之前获取的序列数据塌缩(collapsing)重复读取的步骤。
因此,在一个实施方案中,步骤(c)包括确定与目标染色体的目标区域唯一对齐的所述片段的第一数量和确定与参考染色体内的一个或多个目标区域唯一对齐的所述片段的第二数量。
应当意识到,本文提及“目标区域”是指所述目标和/或参考染色体的一部分或全部。
在一个实施方案中,目标染色体为染色体内的区域和参考染色体为与目标染色体相同的染色体内的区域。
在一个实施方案中,所述方法另外包括针对怀疑含有胎儿染色体异常的基因组区域富集样品。这种实施方案一般地利用通过基于杂交的技术进行选择的方法,并且将使得能够预选择以在测序之前保留或去除预选择的目标序列。
为了将序列映射到唯一的染色体位置,插入缺失/错配成本加权必须参数化以在该分析中降低。用这些前提条件确定非严格性片段长度匹配。使用这种生物信息学方法,一般地约有95%的样品读取被映射到基因组。如果读取与基因组中的唯一位置匹配,才将读取计数为指定给染色体位置,一般地使唯一匹配和随后对染色体指定计数的样品读取的比例为约50%。
在一个实施方案中,对于全染色体进行对齐,例如,因此分析将包括检测过多的给定染色体。在一个备选的实施方案中,对于所述染色体的一部分进行对齐,例如,将仅对于染色体的特定预定区域来分析匹配。据信本发明的该实施方案通过针对染色体的特定区域而提供更灵敏的匹配技术。
大小加权
在本文所述的任何方法(即异常检测和性别预测方法)的一个实施方案中,可进行另外的大小加权步骤。因此,在一个实施方案中,所述方法另外包括以下步骤:
(i) 通过计算每个片段大小(s)为胎儿来源的概率(w),将与目标染色体的目标区域对齐的每个片段进行大小加权;
(ii) 通过计算每个片段大小(s)为胎儿来源的概率(w),将与一个或多个参考染色体内的一个或多个目标区域对齐的每个片段进行大小加权;
(iii) 通过将步骤(i)中获取的值求和,计算总目标加权计数(Nc目标);
(iv) 通过将步骤(ii)中获取的值求和,计算总参考加权计数(Nc);
(v) 计算在步骤(iii)和(iv)中获取的Nc目标和Nc值之间的比率或差异;和
(vi) 基于所述比率或差异确定所述目标染色体的胎儿异常的存在。
已知源自孕妇胎盘的无细胞DNA片段的大小分布(因此在大多数情况下反映一个或多个胎儿的核型)遵循与源自孕妇自身(即来自除了胎盘以外的组织)的片段明显不同的分布。从根本上说,发现胎儿DNA片段平均短于母体DNA片段。
在专注于胎儿基因组的分析中,可通过优先选择更有可能为胎儿的测序片段,利用大小分布的差异来提高灵敏度。先前已知的生物信息学方法包括仅传递给定(较短)大小范围内的片段用于进一步分析。然而,尽管这种幼稚的方法可导致分析中胎儿DNA的相对富集,但其也导致大多数被计数的片段丢失,从而导致分析的显著的更加不确定性,然后这会抵消(offsets)富集胎儿DNA的许多灵敏度益处。
发明人已经开发出一种备选方法,其更好地利用所分析的所有片段。这利用了胎儿和母体DNA分子之间的片段大小分布的已知差异来在分析中优先将片段加权(即给予其优先权)(如果相比于母亲核型,这些具有更高的反映胎儿核型的概率),反之,则不予强调来自具有更高的源自胎盘以外的母体组织的概率的片段的贡献。
重要的是,这项新的分析并没有丢弃来自目标大小范围之外的计数的片段,而是继续考虑计数过程中的所有片段。因此,其保留了通过计数大部分DNA片段提供的统计能力,同时降低源自整倍体母亲的片段的稀释作用。
图13和表A中提供的数据表示从母体血浆提取的无细胞DNA片段为胎儿来源的概率,其为片段大小的函数。已从表示已知为胎儿或母体来源的片段的相对频率的数据计算出这种关系(Chandrananda等人(2015) BMC medical genomics 8.1: 29)。
根据本发明采取的生物信息学方法的概述如下。先前的方法学会为每个染色体c生成片段计数值Nc,其中每个片段的贡献值为1。因此,Nc仅为发现映射到染色体c的片段数量。
在利用每个片段的大小加权的本发明的改进方法中,每个片段代之以贡献值为w[s],其中w为加权函数和s为核苷酸片段的大小,其作为测序方法的一部分测定。此处使用的加权函数w为片段为胎儿来源的概率,如图13所绘制的并以表A所示的特定概率(w)值外推的。然后,通过将发现与该染色体对齐的所有片段的所有w[s]值求和得出指定给染色体c的总加权计数Nc。
通过将发现与目标染色体c目标对齐的所有片段的所有w[s]值求和得出的总加权计数Nc本文称为 。因此,在一个实施方案中,在步骤(i)和(ii)中计算每个片段大小(s)为胎儿来源的概率(w)的步骤包括识别每个对齐的片段的大小(s)并基于表A所示的值为所述片段分配w值。
。因此,在一个实施方案中,在步骤(i)和(ii)中计算每个片段大小(s)为胎儿来源的概率(w)的步骤包括识别每个对齐的片段的大小(s)并基于表A所示的值为所述片段分配w值。
在一个实施方案中,使 值经受GC校正步骤(如在先前方法学中),并计算样品中来自该染色体的片段的存在的标准化量度;对于目标染色体c目标而言这通过形成针对所有常染色体计数的片段比例(该比例相对于针对所有常染色体计算的Nc值的总和;这些Nc值也都经受了GC校正步骤)来进行。
值经受GC校正步骤(如在先前方法学中),并计算样品中来自该染色体的片段的存在的标准化量度;对于目标染色体c目标而言这通过形成针对所有常染色体计数的片段比例(该比例相对于针对所有常染色体计算的Nc值的总和;这些Nc值也都经受了GC校正步骤)来进行。
比率(即步骤(v)中的“常染色体比率”)的计算称为计算 值:
值:

然后将该常染色体比率用作统计模型的输入,该统计模型估计三体性的概率以产生步骤(vi)中计算出的最终测试结果。
表A:每个片段的大小加权值
| 以碱基对表示的片段大小(s) | 概率(胎儿/大小) (w) |
| 75 | 0.742 |
| 76 | 0.740 |
| 77 | 0.710 |
| 78 | 0.688 |
| 79 | 0.679 |
| 80 | 0.682 |
| 81 | 0.686 |
| 82 | 0.694 |
| 83 | 0.705 |
| 84 | 0.712 |
| 85 | 0.717 |
| 86 | 0.724 |
| 87 | 0.731 |
| 88 | 0.737 |
| 89 | 0.733 |
| 90 | 0.728 |
| 91 | 0.732 |
| 92 | 0.734 |
| 93 | 0.742 |
| 94 | 0.750 |
| 95 | 0.757 |
| 96 | 0.756 |
| 97 | 0.749 |
| 98 | 0.744 |
| 99 | 0.741 |
| 100 | 0.738 |
| 101 | 0.739 |
| 102 | 0.744 |
| 103 | 0.748 |
| 104 | 0.755 |
| 105 | 0.761 |
| 106 | 0.766 |
| 107 | 0.767 |
| 108 | 0.768 |
| 109 | 0.765 |
| 110 | 0.758 |
| 111 | 0.756 |
| 112 | 0.758 |
| 113 | 0.761 |
| 114 | 0.764 |
| 115 | 0.766 |
| 116 | 0.770 |
| 117 | 0.772 |
| 118 | 0.769 |
| 119 | 0.760 |
| 120 | 0.751 |
| 121 | 0.744 |
| 122 | 0.742 |
| 123 | 0.747 |
| 124 | 0.761 |
| 125 | 0.776 |
| 126 | 0.786 |
| 127 | 0.788 |
| 128 | 0.790 |
| 129 | 0.783 |
| 130 | 0.773 |
| 131 | 0.760 |
| 132 | 0.745 |
| 133 | 0.735 |
| 134 | 0.734 |
| 135 | 0.738 |
| 136 | 0.743 |
| 137 | 0.740 |
| 138 | 0.728 |
| 139 | 0.713 |
| 140 | 0.695 |
| 141 | 0.683 |
| 142 | 0.675 |
| 143 | 0.670 |
| 144 | 0.664 |
| 145 | 0.661 |
| 146 | 0.655 |
| 147 | 0.647 |
| 148 | 0.637 |
| 149 | 0.627 |
| 150 | 0.617 |
| 151 | 0.605 |
| 152 | 0.598 |
| 153 | 0.594 |
| 154 | 0.591 |
| 155 | 0.582 |
| 156 | 0.573 |
| 157 | 0.559 |
| 158 | 0.542 |
| 159 | 0.524 |
| 160 | 0.513 |
| 161 | 0.502 |
| 162 | 0.489 |
| 163 | 0.475 |
| 164 | 0.460 |
| 165 | 0.447 |
| 166 | 0.436 |
| 167 | 0.434 |
| 168 | 0.431 |
| 169 | 0.427 |
| 170 | 0.422 |
| 171 | 0.421 |
| 172 | 0.416 |
| 173 | 0.411 |
| 174 | 0.405 |
| 175 | 0.399 |
| 176 | 0.393 |
| 177 | 0.388 |
| 178 | 0.390 |
| 179 | 0.391 |
| 180 | 0.391 |
| 181 | 0.390 |
| 182 | 0.391 |
| 183 | 0.389 |
| 184 | 0.385 |
| 185 | 0.382 |
| 186 | 0.378 |
| 187 | 0.374 |
| 188 | 0.369 |
| 189 | 0.369 |
| 190 | 0.369 |
| 191 | 0.367 |
| 192 | 0.364 |
| 193 | 0.362 |
| 194 | 0.357 |
| 195 | 0.353 |
| 196 | 0.351 |
| 197 | 0.348 |
| 198 | 0.344 |
| 199 | 0.340 |
| 200 | 0.336 |
| 201 | 0.334 |
| 202 | 0.335 |
| 203 | 0.337 |
| 204 | 0.338 |
| 205 | 0.336 |
| 206 | 0.333 |
| 207 | 0.331 |
| 208 | 0.329 |
| 209 | 0.326 |
| 210 | 0.324 |
| 211 | 0.323 |
| 212 | 0.323 |
| 213 | 0.323 |
| 214 | 0.324 |
| 215 | 0.324 |
| 216 | 0.323 |
| 217 | 0.322 |
| 218 | 0.321 |
| 219 | 0.318 |
| 220 | 0.319 |
| 221 | 0.321 |
| 222 | 0.328 |
| 223 | 0.339 |
| 224 | 0.344 |
| 225 | 0.346 |
| 226 | 0.340 |
| 227 | 0.335 |
| 228 | 0.326 |
| 229 | 0.317 |
| 230 | 0.313 |
| 231 | 0.313 |
| 232 | 0.324 |
| 233 | 0.323 |
| 234 | 0.327 |
| 235 | 0.330 |
| 236 | 0.332 |
| 237 | 0.335 |
| 238 | 0.342 |
| 239 | 0.334 |
| 240 | 0.333 |
| 241 | 0.331 |
| 242 | 0.346 |
| 243 | 0.331 |
| 244 | 0.341 |
| 245 | 0.353 |
| 246 | 0.353 |
| 247 | 0.355 |
| 248 | 0.357 |
| 249 | 0.357 |
| 250 | 0.363 |
已知源自一个或多个孕妇胎盘的无细胞DNA片段的大小分布(因此在大多数情况下反映一个或多个胎儿的核型)遵循与源自孕妇自身(即来自胎盘以外的组织)的片段明显不同的分布。本发明的方法利用这种现象。
如果相比于母亲核型,这些片段具有更高的反映胎儿核型的概率,还可利用片段大小分布的这些已知差异来优先将片段加权以进行染色体比率计算,反之,则不予强调来自具有更高的源自胎盘以外的母体组织的概率的片段的贡献。
一种特定的非限制性的大小加权方法描述如下:
方法
需要在软件的配置数据中包含以下静态数据项:
对于任何唯一的片段对齐事件,方法如下进行,生成计数增量u.
如果CountWeightFragSizeEnable为假,则不应进行任何加权操作,即
u=1。
如果CountWeightFragSizeEnable为真,则如下参考片段大小和加权映射生成计数增量。
首先,每计数累计值被初始化:
W ← 0
Ncontrib ← 0
然后,对于塌缩为单个计数事件(即在对齐后视为重复项)的读取组中的每个读取,
1. 尝试获取片段大小测量值(s)。获取该测量值的确切方式取决于所使用的特定测序平台,但例如可为:在采用单端测序方案的Thermo Fisher Ion Torrent系列测序系统的情况下,可与读取一起存储于Basecaller BAM文件中的ZA标签的含量,或者如果已使用双端测序方案,则从序列对齐步骤提取的数值。技术人员将意识到,可存在许多其他可能的机制来获取大小值,所述方法根据所使用的测序平台和方案定制。如果可获取s的值,则尝试在权重映射中查找它:如果该值s在权重映射边界之内(即smin≤s≤smax),则如下更新每计数累计值:
W ← W + w[s]
Ncontrib ← Ncontrib + 1。
或者,如果s<smin或s> smax,则不会更新累计值。
2. 如果没有ZA标签与读取相关,则采取的操作取决于CountWeightFragSizeMissingAction的值,如下所示:
a. 如果CountWeightFragSizeMissingAction的值为集成(Integrate),则片段(l)的测序读取长度用作大小范围的下限,范围的上限为CountWeightFragSizeMax。然后,通过对大小范围进行平均来计算综合权重,并更新累计值:

b. 如果CountWeightFragSizeMissingAction的值为忽略(Ignore),则丢弃该片段,并且不会更新累计值。
当考虑了所有有助于计数事件的读取,然后如下生成计数增量:

将u的最终确定的值最终添加到发现与其对齐的染色体的累计对齐片段计数(Nc)中。如先前的方法学一样,以这种方式确定的累计的加权对齐片段计数将根据GC含量进行校正,并然后将经校正的值用于计算常染色体和其他染色体比率,以输入三体性似然模型(Rin值)、胎儿分数估计(Rx)和性别确定(Rx以及任选地Ry)。
对此有一个例外:用作运行控制有效性检查的一部分的染色体比率不应根据片段大小进行加权(但仍应进行GC校正)。
当将常染色体或染色体片段的计数用于计算常染色体或其他染色体比率以外的任何目的,则该计数不应根据片段大小进行加权。
比率计算
一旦根据本文定义的片段对齐分析将命中总数指定给给定染色体,则一般地然后将命中标准化为普通数值(common number)。然后根据简单的数学计算目标染色体的目标区域的每个命中与一个或多个参考染色体上的命中相比较的比率。
除了标准化为如上文所述的普通数值之外,一般地有用的是能够估计母体血浆DNA中胎儿来源的分数;这将证实母体血浆DNA样品中有足够的胎儿DNA用于检测胎儿染色体异常。例如,在一个实施方案中,本发明的方法另外包括基于样品内胎儿DNA的量来标准化或调整匹配命中的数量的步骤。
统计显著性
为了将本发明的诊断测试置于统计学基础上,本发明的方法另外包括计算目标染色体的目标区域的每个命中与其他染色体上的命中相比较的比率的统计显著性的步骤。在一个实施方案中,统计显著性检验包括根据减少的计数数据的常规统计分析来计算z分数。然而,应当意识到,本领域的技术人员可应用其他统计方法。
在假定计数比率“目标染色体/一个或多个参考染色体”的误差分布近似正态的情况下,z分数表示要素与平均值之间的标准偏差为多少。
z-分数可根据下式计算:
z = (X - μ)/σ
其中z为z分数,X为要素的值,μ为群体平均值,和σ为群体值的标准偏差。当根据本发明测试三体性21的存在时,计数比率的z-分数值为2.0或更高表明该计数比率值指示三体性21妊娠的概率为约98%。
在一个实施方案中,步骤(e)包括计算似然比,其指示目标染色体的胎儿染色体异常,并且一般地基于许多因素,比如胎儿分数、上述z分数等。WO2014/033455中描述了可如何计算似然比的全部细节。
预测性别的方法
从胎儿的父系遗传获得的染色体Y DNA的存在为男性胎儿的诊断标志物。本发明的另一方面为检测胎儿的性别,如染色体Y序列的存在所示。
当胎儿为女性,则排除Y染色体成分的使用,然而代替父系遗传获得的Y染色体,可检测父系来源的基因等位基因。这其中为胎儿SNP (单核苷酸多态性),其作为等位基因是明显的,所述等位基因作为母体血浆DNA中DNA序列的次要成分而存在(Dhallan等人,Lancet 369, 474-481)。当如在本发明中仅对胎儿基因组的一小部分进行测序时,从胎儿父亲遗传获得并作为与相对更丰富的母体等位基因不同的变体检测的这种等位基因的数量,为血浆胎儿DNA分数的函数。这提供一种备选的独立于性别的用于估计胎儿来源的母体血浆DNA分数的方法。
本发明的第二方面提供一种预测怀孕女性受试者体内的胎儿性别的方法,所述方法包括以下步骤:
(a) 从获自怀孕女性受试者的生物样品中分离核酸;
(b1) 选择120 bp-135 bp之间的核酸片段大小值用于最佳胎儿分数;
(b2) 分离大小在步骤(b1)中选择的片段大小值的20 bp以内的核酸片段;
(c) 确定与性染色体对齐的所述片段的第一数量和确定与一个或多个参考染色体对齐的所述片段的第二数量;
(d) 计算第一和第二数量之间的比率或差异;
(e) 基于与所述参考染色体相比较是过多还是等同的片段与X染色体对齐或者是否存在Y染色体来确定所述胎儿的性别。
可提及的本公开的另一方面提供一种预测怀孕女性受试者体内的胎儿性别的方法,所述方法包括以下步骤:
(a) 从获自怀孕女性受试者的生物样品中分离核酸;
(b) 分离大小为约80 bp-约150 bp的核酸片段;
(c) 确定与性染色体对齐的所述片段的第一数量,和确定与一个或多个参考染色体对齐的所述片段的第二数量;
(d) 计算第一和第二数量之间的比率或差异;
(e) 基于与所述参考染色体相比较是过多还是等同的片段与X染色体对齐或者是否存在Y染色体来确定所述胎儿的性别。
在一个实施方案中,所述方法另外包括以下步骤:
(i) 通过计算每个片段大小(s)为胎儿来源的概率(w),将与性染色体对齐的每个片段进行大小加权;
(ii) 通过计算每个片段大小(s)为胎儿来源的概率(w),将与一个或多个参考染色体对齐的每个片段进行大小加权;
(iii) 通过将步骤(i)中获取的值求和,计算总目标加权计数(Nc目标);
(iv) 通过将步骤(ii)中获取的值求和,计算总参考加权计数(Nc);
(v) 计算在步骤(iii)和(iv)中获得的Nc目标和Nc值之间的比率或差异;和
(vi) 基于与所述一个或多个参考染色体相比较是过多还是等同的片段与X染色体对齐或者是否存在Y染色体来确定所述胎儿的性别。
本文提及的性染色体,即异染色体,包括X或Y染色体。
在实施方案中,参考染色体选自常染色体(即非性染色体)。
应当意识到,与所述参考染色体相比较,与X染色体对齐的片段过多指示女性性别预测(即XX)。
应当意识到,与所述参考染色体相比较,与X染色体对齐的片段等同指示男性性别预测(即XY)。
应当意识到,存在与Y染色体对齐的片段指示男性性别预测(即XY)。
应当意识到,不存在与Y染色体对齐的片段指示女性性别预测(即XX)。
应当意识到,适用于本发明第一方面的每个实施方案均同样适用于本发明第二方面的性别预测方法。
试剂盒
本发明的另一方面提供一种用于实施本文定义的任何方法的试剂盒,其包含用于根据本文定义的任何方法使用试剂盒的说明书。
在一个实施方案中,试剂盒另外包含一种或多种本文定义的试剂和/或一种或多种消耗品。
本发明的另一方面提供如本文定义的试剂盒在检测怀孕女性受试者体内的胎儿染色体异常的方法或预测怀孕女性受试者体内的胎儿性别的方法中的用途。
以下研究说明本发明。
材料和方法
应当意识到,本文所述的方法可使用可见于http://www.premaitha.com/the-iona-test的IONA®测试来进行。然而,以下详细方案提供有关可如何实施本发明方法的指南。
材料
除了IONA®文库制备试剂盒中提供的试剂之外,使用手动方案的DNA文库制备还需要以下材料:















注释:如果将96孔板磁铁用于手动文库制备方案,则可使用IONA®塑料消耗品试剂盒(Plastics Consumables Kit)中提供的消耗品。
DNA提取
IONA®测试利用源自全血血浆分数的无细胞DNA (cfDNA)作为输入样品进行分析。当进行用于IONA®测试工作流程的手动DNA提取方案时,必须使用经验证可用于从血浆提取cfDNA的DNA提取试剂盒。
样品处理应根据DNA提取试剂盒制造商提供的说明书或根据本领域的技术人员已知的确立程序进行。
DNA文库制备
IONA®测试中用于DNA文库制备的手动方案利用IONA®文库制备试剂盒(LibraryPreparation Kit)中提供的试剂。与自动化方案相比较,当对IONA®文库制备试剂盒使用手动方案时,建议分批处理样品,以避免样品通量减小。
1. 从冷冻柜中取出DNA (如果需要),并在环境温度(15-25℃)下解冻30分钟。
2. 从冷冻柜中取出IONA®文库制备试剂盒板1,并在环境温度(15-25℃)下解冻30分钟。
3. 从冰箱中取出IONA®文库制备试剂盒板2,并平衡至环境温度(15-25℃)达最少30分钟。
4. 在平板离心机中脉冲旋转IONA®文库制备试剂盒板1和2持续5秒,以在平板底部收集试剂。
注释:IONA®文库制备试剂盒板2在环境温度下保持在工作台上,直至后续步骤需要。
5. IONA®文库制备试剂盒板1和2的试剂布局如以下表1和2所示。


8. 将以下体积的末端修复反应的每种试剂添加到每个样品中以制备反应物:
表3:末端修复反应体积
| 试剂 | 板1的试剂列 | 体积 |
| 样品 | - | 51 µL |
| 末端修复缓冲液I | 1 | 6 µL |
| 末端修复酶I | A2 | 3 µL |
| 总计 | 60 µL |
注释:如果要测试多个DNA样品,则可为该步骤制备末端修复试剂的主混合物。建议过量的至少一种反应物。如果使用这种方法,可减少可使用IONA®文库制备试剂盒测试的样品数量。
9. 对于每个样品,将末端修复反应物用移液器上下吸取10次以混合和使用适当的台式离心机脉冲旋转5秒。
10. 将IONA®文库制备试剂盒板1储存于冰箱中,直至需要进行衔接子连接反应。
11. 将每个样品管(或96孔反应板)转移至热循环仪上以进行末端修复反应。将热循环仪设定为以下循环条件并运行末端修复反应:
表4:末端修复温度循环

12. 在末端修复反应完成时,将样品从热循环仪转移至工作台上以准备衔接子连接反应。
13. 从冰箱中取出IONA®文库制备试剂盒板1。用移液器吸取IONA®文库制备试剂盒板1 (表2)的第3列的衔接子连接缓冲液I (ALB I)、第4列的衔接子连接酶I (ALE I)和位置A5的衔接子连接酶II (ALE II)上下10次以进行混合。
注释:对将要使用的任何试剂孔均进行该操作。
14. 将以下体积的衔接子连接反应的每种试剂直接添加到来自末端修复反应的每个样品中:
表5:末端修复温度循环
| 试剂 | 板1的试剂列 | 体积 |
| 样品 (来自末端修复) | - | 60 µL |
| 无核酸酶水 | - | 15 µL |
| 衔接子连接缓冲液I | 3 | 10 µL |
| 衔接子连接酶I | 4 | 6 µL |
| 衔接子连接酶II | A5 | 1 µL |
| 92 µL |
注释:如果要测试多个DNA样品,则可为该步骤制备末端修复试剂的主混合物。建议过量的至少一种反应物。如果使用这种方法,可减少可使用IONA®文库制备试剂盒测试的样品数量。
15. 用移液器吸取IONA®文库制备试剂盒板1 (表2)第6-9列中的带条形码的衔接子上下10次以进行混合。
注释:对制备的适当数量的样品进行该操作。每个样品使用一种带条形码的衔接子。
16. 将8 µL指定给适当样品的带条形码的衔接子添加到样品的衔接子连接反应中。衔接子连接反应体积为100 µL。
注释:必须记录每个样品使用的带条形码的衔接子编号(表2,第6-9列),因为每个样品将根据其各自的条形码在后续步骤中进行分析。
17. 对于每个样品,用移液器吸取衔接子连接反应物上下10次以进行混合,并使用适当的台式离心机脉冲旋转5秒。
18. 将IONA®文库制备试剂盒板1储存于冰箱中直至文库PCR反应需要。
19. 将每个样品转移至经验证的热循环仪上以进行衔接子连接反应。将热循环仪设定为以下循环条件并运行衔接子连接反应:
表6:衔接子连接循环

注释:确保反应体积设定为100 µL。
20. 在衔接子连接反应完成时,将样品从热循环仪转移至工作台上以进行衔接子连接反应的净化。
21. (如果需要)将每个样品转移至适当的容器中,用于与可用于样品净化的磁力板/架一起使用。
22. 通过将40 mL 100%乙醇与10 mL无核酸酶水混合,制备80%乙醇的溶液(总体积50 mL)。
23. 用移液器吸取储存于环境温度(15-25℃)下的IONA®文库制备试剂盒板2(表3)中的珠粒上下25次以进行混合。
注释:对要自其取出珠粒的任何孔均进行该操作。珠粒的充分混合是重要的。
24. 将100 µL珠粒直接加入到来自衔接子连接反应的每个样品中,并用移液器吸取上下10次以进行混合。
25. 将每个样品在环境温度(15-25℃)下温育5分钟。
26. 将样品在适当的台式离心机中脉冲旋转5秒。
27. 将样品(在1.5 mL管或96孔平板中)转移至磁力板/架2分钟。
28. 在将样品保持在磁力板/架上的同时,取出并丢弃上清液。注意不要扰动沉淀。
29. 向每个样品添加80%乙醇。确保浸没全部珠粒沉淀。
注释:如果使用96孔板,建议200 µL 80%乙醇。如果使用1.5 mL管,建议500 µL80%乙醇。
30. 将每个样品在80%乙醇中温育30秒。
31. 将样品保持在磁力板/架上。取出并丢弃上清液。注意不要扰动沉淀。
32. 重复步骤29-31。
33. 将每个样品在环境温度(15-25℃)下,于磁力板/架上风干5分钟。
注释:如果使用1.5 mL管,请确保试管盖打开。
34. 从磁力板/架上取下每个样品,并将珠粒沉淀重悬于43 µL无核酸酶水中。
注释:确保所有珠粒处于悬浮状态。用吸移器沿着在其上珠粒靠着磁铁保持的板孔/管的侧面吸取,以回收全部珠粒沉淀。
35. 将每个样品在环境温度(15-25℃)下温育3分钟。
36. 将样品在适当的台式离心机中脉冲旋转5秒。
37. 将样品转移至磁力板/架2分钟。
38. 将40 µL上清液转移至新的板孔/管中以进行文库PCR。
39. 从冰箱取出IONA®文库制备试剂盒板1。用移液器吸取IONA®文库制备试剂盒板1的位置10A/B的PCR引物混合物I和第11和12列的PCR主混合物I。
40. 用移液器吸取上下10次以进行混合。
注释:对于要使用的任何试剂孔均进行该操作。
41. 将以下体积的文库PCR反应的每种试剂直接添加到来自衔接子连接反应的净化的每个样品中:
表7

注释:如果要测试多个DNA样品,则可为该步骤制备文库PCR试剂的主混合物。建议过量的至少一种反应物。如果使用这种方法,可减少可使用IONA®文库制备试剂盒测试的样品数量。
42. 对于每个样品,用移液器吸取文库PCR反应物上下10次以进行混合,并使用适当的台式离心机脉冲旋转5秒。
注释:将IONA®文库制备试剂盒板1放回冷冻柜中进行储存。
43. 将每个样品转移至热循环仪以进行文库PCR反应,将热循环仪设定为以下循环条件并运行文库PCR反应:
表8

注释:确保反应体积设定为100 µL。
44. 在文库PCR反应完成时,将样品从热循环仪转移至工作台以进行文库量化。
注释:PCR扩增的文库可储存于冷冻柜(-15至-25℃)中,并且工作流程在20个工作日内完成。将IONA®文库制备试剂盒板2放回冰箱中储存直至需要。
注释:如果从将PCR扩增的文库存储于冷冻柜(-15至-25℃)之后这一时间点开始工作流程,则在后续步骤之前将文库解冻30分钟。确保将IONA®文库制备试剂盒板2从冰箱储存中取出30分钟以进行后续步骤。
45. 根据制造商的说明书,使用DNA分析仪平台(例如Perkin Elmer LabChip®GX, Agilent 2100 Bioanalyser®)对每个样品文库进行量化。
注释:PCR扩增的文库可能过于浓缩而无法在某些DNA分析仪平台上未经稀释直接运行。建议为要量化的每个文库制备1/5稀释液。
注释:在测序之前,必须以摩尔浓度记录每个文库的浓度用于随后进行标准化和多路复用。根据需要针对稀释因子校正浓度。
注释:确保要量化的任何文库的浓度处于所用的DNA分析仪平台的检测限以内。
46. 在使用以下步骤的大小选择之前进行样品的标准化和多路复用。单次运行可多路复用多达8个样品进行测序。确保没有将带相同条形码衔接子编号的样品文库添加到同一多路复用池中。
47. 从量化中选择多达8个样品及其相应浓度(摩尔浓度;nM)。鉴定具有最低浓度的样品–该值为所有样品将对其进行标准化的目标浓度。
48. 对要合并的每个样品使用以下计算来确定文库的多路复用所需的体积:
样品文库体积:
目标浓度(nM) x 20 µL样品文库浓度(nM)
水体积:
20 µL - 样品文库体积
例如样品5=最低浓度=目标浓度
实例中带下划线的体积表示要添加的最终体积。
表9

49. 在1.5 mL管中,将针对所有要多路复用的样品的标准化计算出的水和样品文库体积合并在一起。
注释:后续步骤需要最小体积为100 µL的合并样品。如果少于5个样品进行多路复用,则以上计算所示的20 µL体积可根据需要增加。
使用大小选择进行富集(使用Ranger Technology™)
设置
- 为样品选择适当的双染料上样缓冲液(取决于上样体积)和琼脂糖盒。
- 将双染料上样缓冲液和样品在单个0.2 ml微量离心管中合并。
- 大小选择(体积28.5 µl):3.5 µl上样缓冲液+ 25 µl样品
- 大小选择(体积50 µl):6 µl上样缓冲液+ 44 µl样品
- 用移液器吸取混合物并使管旋下。
- 使用以下引发和校准盒:
- 28.5 µl体积盒= 15 µl (上样孔) + 75 µl (提取孔)的[TBE]
- 50 µl体积盒= 25 µl (上样孔) + 75 µl (提取孔)的[TBE]
- 将盒置于平台上
- 启动软件
- 一旦提示,跳过净化步骤
- 不管上样体积多大,4 x 50 µl提取提示从提取孔中提取。
软件
- 打开Ranger软件
- 选择适当的平台布局(文件>新建>运行>)
- 定义源板的内容(点击源板图像)
- 定义每个盒的琼脂糖类型(完全套索每个盒,然后右键点击以选择凝胶类型)
- 样品类型管理器;100-300 bp标志物,指定目标碱基对范围(205-227 bp),100%速度。
- 定义盒的琼脂糖百分比类型,例如2%、3%
- 对平台进行目视检查并关闭仪器门。
- 点击软件的“开始”按钮开始运行。
- 在整个运行持续期间遵循屏幕上的提示并完成适当的洗涤和提取
Ranger Technology™运行完成,取出样品
从Ranger Technology™转移200 µL经大小选择的样品文库。分成2 x 100 µL反应物,每个与700 ul珠粒混合以净化样品。
用移液器吸取在环境温度(15-25℃)下储存的IONA®文库制备试剂盒板2中的珠粒上下25次以进行混合。
注释:对将要使用的任何试剂孔均进行该操作。
表10:IONA®测试板2的板布局
表2:IONA®测试板2的板布局

将样品在环境温度(15-25℃)下温育5分钟。
将样品在适当的台式离心机中脉冲旋转5秒。
将样品转移至磁力板/架上2分钟。
在将样品保持在磁力板/架上的同时,取出并丢弃上清液。注意不要扰动珠粒沉淀。
向样品添加80%乙醇。确保浸没全部珠粒沉淀。
注释:如果使用96孔板,建议200 µL 80%乙醇。如果使用1.5 mL管,建议500 µL80%乙醇。
将样品在80%乙醇中温育30秒。
在将样品保持在磁力板/架上的同时,取出并丢弃上清液。注意不要扰动珠粒沉淀。
重复步骤56-58。
将样品在环境温度(15-25℃)下,于磁力板/架上风干5分钟。
注释:如果使用1.5 mL管,请确保试管盖打开。
从磁力板/架上取下样品,并将珠粒沉淀重悬于18 µL无核酸酶水中。
注释:确保所有珠粒处于悬浮状态。用吸移器沿着在其上珠粒靠着磁铁保持的板孔/管的侧面吸取,以回收全部珠粒沉淀。
将样品在环境温度(15-25℃)下温育3分钟。
将样品在适当的台式离心机中脉冲旋转5秒。
将样品转移至磁力板/架2分钟。
将2x 15 µL上清液转移至新的板孔/管中。
根据制造商的说明书,使用DNA分析仪平台(例如Perkin Elmer LabChip® GX,Agilent 2100 Bioanalyser®)对经大小选择的多路复用样品进行量化。
注释:经大小选择的多路复用样品可在DNA分析仪平台上未经稀释而运行。确保样品的浓度处于所用的DNA分析仪平台的检测限以内。样品可根据需要进行稀释和重新量化。
注释:必须以摩尔浓度记录每个文库的浓度用于测序的随后稀释。根据需要针对稀释因子校正浓度。
使用对来自DNA分析仪的经大小选择的多路复用样品池确定的浓度进行稀释至所用的下一代测序平台所需的输入浓度。
对最终大小选择的多路复用样品池使用40 pM 的输入浓度(如果使用Ion PI V2芯片则为50 pM),已使用Ion Chef™仪器和Ion Proton™下一代测序平台(ThermoFisher)验证IONA®测试。使用以下计算来确定样品稀释所需的体积:
样品文库体积:
目标浓度= 样品文库需要的40 pM (或50 pM) x (120 µL) x µL
样品文库浓度
运行对照1/25制备:
2 µL储备液运行对照+ 48 µL水= 50 µL的1/25运行对照
用于测序的样品制备:
样品文库需要的x µL + 22.2 µL 1/25运行对照
用水使最终体积达到120 µL总体积
注释:建议在待使用的下一代测序平台的反应设置之前立即进行该稀释。
注释:避免将稀释的多路复用样品冷冻,因为来自冷冻/解冻循环的可能影响对低浓度的DNA更为不利。在用于测序的反应设置之前,可将稀释的样品在冰箱中储存长达24小时。
下一代测序反应设置
下一代测序反应可使用半自动或全自动化方案进行。
IONA®测试已使用Ion Chef™仪器和Ion Proton™下一代测序平台(ThermoFisher)进行了验证。该自动化DNA文库方案的工作流程如下所述。
注释:对于备选方案,请根据制造商的方案准备样品。
使用Ion Chef™和Ion Proton™仪器进行下一代测序需要以下消耗品:



异丙醇,如果使用Ion PI V2 BC芯片则为分子生物学等级(例如目录号11388461;Fisher Scientific)
按照IC 200试剂盒用户指南(IC 200 Kit User Guide)的Ion PI™ Hi-Q Chef概述的方案,使用针对Ion Chef™和Ion Proton™平台的自动化方案进行下一代测序的运行。
如果尚未制备好,则将要测试的大小选择的多路复用样品稀释至手动文库制备方案的步骤88所述的所需输入浓度。
将Ion Chef™/ Ion Proton™运行计划成根据制造商的说明书进行。
注释:在运行设置期间扫描测序芯片上的条形码,以将正确的多路复用文库样品池与正确的芯片配对。将样品ID指定给适当的衔接子条形码由IONA®分析软件进行。
注释:如果使用备选的下一代测序方案或平台,请确保将正确的样品ID指定给正确的衔接子条形码。
注释:针对Ion Chef™和Ion Proton™仪器的每种设置准备两个测序反应和运行。工作流程在两天内发生。可预先选择Ion Chef™反应的完成时间,以使反应过夜进行。
在Ion Proton™测序运行完成时,将数据直接传输以由IONA®软件进行分析,以确定对所有测试的样品所研究的三体性的似然状态。
注释:测序后的数据传输和分析可能需要长达6小时。在此期间,请勿关闭IonProton™仪器或IONA®软件工作站PC。
数据分析 - IONA软件
总之,进行以下步骤:
- 将样品解复用;
- 将读取对齐并分箱(bin)到染色体中;
- 将读取GC校正;和
- 计算染色体比率,计算FF%估计。
通过使用IONA®软件,主要生物信息学流水线的实施如下进行:对于8个样品的每次测序运行,均以未映射的BAM文件的形式从测序平台检索多路复用的序列读取。读取的多路复用组装最初要经受条形码分类步骤,在该步骤中识别带条形码的5'衔接子并针对预定义的集合进行匹配,以将多路复用拆分成针对单个样品的读取以进行进一步处理。
在早期过滤步骤以去除小量非常短的读取后,使用耐空位的读取对齐模块将片段映射到“hg19”人类基因组参考。然后进行对齐结果的后过滤以去除在测试工作流程的PCR阶段产生的重复读取,其经确定为其5'末端与任何其他读取映射到参考的同一位置的那些。
然后,将被确定为在基因组参考中唯一对齐的片段根据常染色体进行分箱,使所得计数经受校准步骤,以校正与GC含量相关的测序覆盖度偏倚;这通过首先根据片段在整个基因组参考中分箱时的平均GC含量来表征片段的代表性过度或不足的水平,并然后反转和作为校正加权应用于片段计数/染色体来实现。
最后,将所得的片段计数数据用作一组混合模型的输入,所述混合模型掺入在针对三体性13、18和21测试的受三体性影响和未受影响的两种假设下的预期值分布。每个模型生成测试似然比,然后在考虑年龄和相应的DNA测试结果的同时,将其与母体年龄相关的三体性先验概率一起用于量化每个三体性的概率。
IONA®软件还进行内部有效性检查。进行工作流程数据质量检查,该检查利用测序和对齐指标来确保序列数据具有足够的质量以进行进一步分析。另外,在生成每个常染色体片段计数后,进行运行有效性检查。该步骤首先分离源自测序In-Run Control (其被设计用于模拟胎儿分数为约10%的三体性21阳性样品)的片段,并然后使用在软件配置中先前设置的参考范围比较来自针对21号染色体对齐的这些片段的计数比例。如果比例符合参考标准,则运行有效性检查通过。
对每个样品也进行单独的有效性检查。这些确保对齐的片段计数足以用于要使用的似然混合模型,并且确保样品中胎儿来源的cfDNA的分数足以报告结果。对于该最后的检查,首先使用X染色体表示的测量(在可能的情况下,即在男性胎儿情形中)和评价其中X染色体表示不能说明胎儿分数的胎儿富集大小区域中物质的相对量的方法的组合,对胎儿分数进行独立地量化。
1. 富集结果
该文件所述的结果表明,相对于母体背景DNA含量,胎儿DNA大量富集。
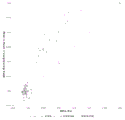
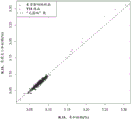
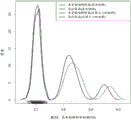
图1说明使用片段大小富集方法进行的富集对21号染色体比率的影响。未受影响的(整倍体)样品(正方形)的比率围绕预期值簇集;即相对于参考结果没有变化,而三体性21样品(三角形)的21号染色体比率相对于参考结果显著增加。富集方法显著增加了比率最高的整倍体样品与比率最低的T21样品之间21号染色体比率的差异。这大大提高区分整倍体和三体性样品的能力。图1中生成的数据表明,只有通过富集胎儿成分才能发生21号染色体DNA的富集。在该数据集中,21个样品中的20个以这种方式富集,而1个T21样品没有富集。在测序步骤期间使用32个样品多路复用生成了富集的数据,而参考数据为8-16个样品多路复用。预计32plex数据应显示T21与未受影响的样品之间的区分度较差(由于每个样品的数据量减少),然而结果表明由于富集而使其得到改善。
图2说明使用片段大小富集方法进行的富集对男性样品中胎儿分数估计的影响。数据表明,相对于参考结果,通过本文所述的方法,源自胎儿的DNA比例被大量富集。图2中生成的数据表明,除一个样品之外的所有样品的胎儿分数均由于富集而增加,平均增加至约2-2.5倍。这种富集使得能够进行更高的多路复用和/或改善性能(灵敏度/特异性),同时预计还可降低失败率。当胎儿分数较低时,富集还可使得NIPT能够在妊娠的早期阶段进行。不受理论的束缚,据信一个没有富集的样品可能实际上没有富集,或者仅仅是由于偶然因素而高估了参考结果和略微低估了富集的胎儿分数%。这种富集将可能具有显著改善微缺失(Mdel)测试性能的作用。迄今为止,Mdel的NIPT中阳性预测值(PPV)相对差。富集胎儿分数可校正这种性能不佳并改善PPV。
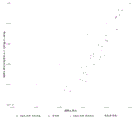
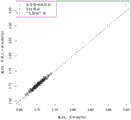
图3说明使用片段大小富集方法进行的富集对X染色体比率的影响。女性胎儿样品的X染色体比率围绕预期值聚集,即相对于参考结果没有变化,而X染色体的比率相对于参考结果显著降低。图3中对男性样品所示的结果表明,使用富集方法,除一个样品之外的所有样品均显示出X染色体比率降低。正如预期的那样,男性胎儿样品中胎儿分数的增加将导致Y染色体比率的相应增加,从而样品的X染色体比率降低。一个样品显示X染色体比率小幅增加(即因此没有富集)。图3中对女性样品所示的结果表明,6个中有5个位于图右上方的参考线上,即富集方法对女性胎儿样品的X比率没有影响(正如预期的那样)。一个样品显示出稍微富集。这可能这个样品实际上在一开始就是低胎儿分数男性样品,被错误地标记为女性样品。因此,数据还表明,可利用富集方法来提高性别确定测试的准确性。
孤立地,图1、2或3中的数据可能是由于将样品的大小选择到非常窄的期望的范围所导致的假象结果。结合起来,通过增加T21样品中21号染色体的比率(对整倍体样品没有影响),男性样品中X染色体的比率降低和对女性样品中的X染色体比率的影响最小以及相应的胎儿分数估计的增加,表明这些样品中胎儿DNA的大量富集。
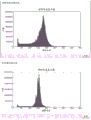
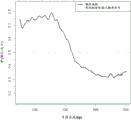
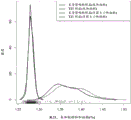
图4说明典型的全基因组测序运行的片段大小分布(上图)和使用RangerTechnology™处理的样品的分布。在使用富集方法之后,所测序样品的片段大小分布明显更窄,大多数片段落在20-30 bp范围内,并以135 bp DNA样品片段大小的目标为中心。注释:片段大小还包括13 bp的衔接子序列。
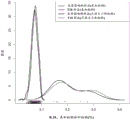
图5显示富集方法的可重复性。使用相同的方法学将同一组样品处理3次至135 bp+/- 10 bp的目标范围(图中左手的3个分布)。在全部3种情况下,胎儿分数值在整个实验中可比,并且高于参考对照(图中右手的分布)。
图6说明利用围绕135 bp目标的较窄大小选择范围(+/-5 bp),使用片段大小富集方法进行的富集对21号染色体比率的影响。未受影响的(整倍体)样品的比率围绕预期值聚集;即相对于参考结果没有变化,而21号染色体的比率相对于参考结果显著增加。
图7说明利用较宽的135 bp +/- 20 bp大小选择范围,使用片段大小富集方法进行的富集对21号染色体比率的影响。未受影响的(整倍体)样品的比率围绕预期值聚集;即相对于参考结果没有变化,而21号染色体的比率相对于参考结果显著增加。然而,尽管未受影响的(整倍体)与三体性样品之间染色体比率的差异相对于参考结果得以增加,但比使用+/- 5或+/- 10 bp目标捕获范围时该差异似乎略较不明显,尽管相对于参考测试方法富集仍然很明显。
图8说明利用备选大小选择范围(120 bp +/-10 bp),使用片段大小富集方法进行的富集对21号染色体比率的影响。未受影响的(整倍体)样品的比率围绕预期值聚集;即相对于参考结果没有变化,而21号染色体的比率相对于参考结果,以与135 bp +/- 10 bp目标区域可比的方式显著增加。这些结果表明,合理地预期靶向低至80 bp和高至150 bp的片段大小提供等同的结果。
图9说明利用较高碱基对目标值(170 bp +/-10 bp),使用片段大小富集方法进行的富集对21号染色体比率的影响。与参考结果相比较,三体性样品的21号染色体比率相对没有变化。然而,整倍体样品的结果显示出更大可变性,这具有减小整倍体与三体性样品之间的染色体比率差异的作用。因此,以该片段大小进行富集似乎对区分整倍体和三体性样品的能力具有不利影响。
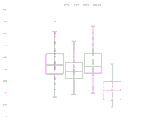
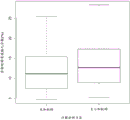
图10显示相对于参考结果在几个片段大小目标和范围处的胎儿分数估计。120 bp和135 bp目标均显示出大量的胎儿分数富集。170 bp目标显示对胎儿分数的影响的宽可变性,几个样品显示胎儿分数增加,而其他样品显示胎儿分数减少。该图中的数据支持了对图6-9中观察到的染色体比率的影响。
图11为显示图10中呈现的数据的不同方式,其显示盒须图中几个片段大小目标和范围处的胎儿分数估计。相对于参考结果,120 bp和135 bp目标均显示出大量的胎儿部分富集。170 bp目标显示与参考结果可比的数据。该图中的数据支持了对图6-9中观察到的染色体比率的影响。
2. 片段大小方法实施方案的评估
先前在IONA®临床性能评估研究中采用的样品用于通过比较在更新的大小加权分析方法下13、18和21号染色体的常染色体比率值与来自先前软件版本中采用的基线未加权方法的那些值来评估该方法。这些样品取自IONA研究样品收集和其他Premaitha Health样品收集。
总共405个样品通过了IONA®软件有效性检查(关于序列数据的质量和一致性、片段计数密度和胎儿分数),并将其用于比较分析。就三体性状态而言,这些样品的分布如表11所示。
表11. 研究中样品的三体性状态
| 三体性状态 | 样品计数 |
| 未受影响的(整倍体) | 351 |
| 受三体性21影响的(‘T21’) | 40 |
| 受三体性18影响的(‘T18’) | 9 |
| 受三体性13影响的(‘T13’) | 5 |
与这些样品对应的测序数据集提取自数据档案,并使用以下两个生物信息学分析流水线进行分析:
1. 如经验证的IONA®软件版本1.6中使用的流水线(具有未加权计数);
2. 如更新的IONA®软件版本1.7.0中实施的更新的流水线(具有片段大小加权计数)。
比较每种情况下对13、18和21号染色体生成的常染色体比率,以测量未受影响和受三体性影响的组之间的分离增加,以及评价在分析时有效胎儿分数的任何变化。
2.1 结果
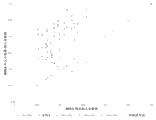
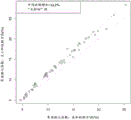
图14、15和16分别为21、18和13号3个染色体的每一个的散点图,其涉及通过每个样品的未加权和大小加权分析两者生成的常染色体比率。这些分别对应于针对三体性21、18和13的测试。每个绘图还通过未加权和加权分析方法之间相等的常染色体比率含有虚线。
可以看出,在每种情况下,尽管未受三体性影响的样品的常染色体比率保持围绕“无影响”线聚集,这表明未受影响的样品的分布没有变化,但与基线未加权方法相比较,对于大小加权方法而言对受三体性影响的样品计算的常染色体比率得以增加。另外,可以看出,较大的常染色体比率的增加大于较小的常染色体比率的增加,表明大小加权方法对受三体性影响的常染色体比率赋予按比例缩放(放大)作用。
图17、18和19给出了数据的一种备选表示;这些显示分别对于未受三体性影响和受三体性影响组的经验分布函数(核密度估计)以及起作用的绘制的常染色体比率值。可以清楚地看出,在片段大小加权方法下,相对于未加权方法的情况,受影响的样品组分布向上移动并缩大,而未受影响的样品组则保持在其初始位置。
2.1.1对性能的影响
使用统计模型在IONA®软件中确定三体性,所述模型被拟合至群体的预期未受影响和三体性组分布。
系统比如IONA®测试的灵敏度和特异性性能测量为以下的函数:正确分类的未受影响和受影响的真实病例数以及用于确定结果的统计模型或截断值。因此,增加未受影响与受影响的数据之间的分离将具有改善整体性能的作用。反之,减少分离将对整体性能产生不利影响。
在这项研究中,在片段大小加权方法下,当与基线未加权方法相比较时(即在新方法和目前方法之间),在未受影响和受影响组的常染色体比率值之间观察到分离始终增加。在三体性13、18和21情况中发生这种增加,因此,在重新拟合用于三体性测定的统计模型之后,从长远来看,预计提高针对三体性13、18和21的测试的灵敏度和特异性。特别是,通过将样品结果的似然比从<1调整为似然比>1,这将使得能够改善对胎儿分数低的三体性13、18和21样品的检测,否则胎儿分数低可能导致测试失败或假阴性结果。还预计在检测其他异常(比如性染色体非整倍性和微缺失)方面提高灵敏度和特异性。
2.1.2在有效胎儿分数上的增加的评价
有可能直接从其常染色体比率计算出由三体性分析程序对三体性样品看到的有效胎儿分数。在给定的三体性样品中分析时的胎儿分数与该样品的常染色体比率与对未受影响的样品看到的预期值(平均值)常染色体比率之间的差异成正比,因此:

在此,


计算了研究数据集中54个受三体性影响的样品的Feff值,其中使用了现有的未加权和新的大小加权两种分析方法来生成常染色体比率。图20含有计算出的胎儿分数值分布的盒须图,如分析所示。可以看到中值胎儿分数(FF)的增加(未加权中位数FF:11.1%;大小加权中位数FF:12.6%)。另外,在大小加权的情况下数值的分布比在未加权的情况下更宽,表明通过改进的大小加权分析方法实现的有效胎儿分数的按比例缩放。图21进一步证明由于包含每个片段的大小加权所致的胎儿分数按比例缩放效应。该图涉及对于初始的未加权和新的大小加权分析情况,如根据其常染色体比率计算的单个受三体性影响的样品在分析时的胎儿分数值。由于改进的计数方案,对于所包括的所有受三体性影响的样品在三体性分析阶段看到的胎儿分数的平均比例增加为13.2%。
2.2 结论
已经开发出一种方法并入IONA®软件,以提高三体性测定的性能,或等同地降低给定性能水平所需的测序密度。本文的文本描述了验证练习,进行其以评估更新的软件程序并确认其可满足这些要求。
这项研究检查了通过利用现有的(未加权)计数分析方法和新的计数分析方法(其并入通过片段大小的加权)两者,使用IONA®测试方法分析受三体性影响的和未受三体性影响的样品生成的常染色体比率分布之间的分离。
该研究考虑了来自405个临床样品的数据,其由未受三体性影响和受三体性13、18和21影响的情况的混合组成。在片段大小加权方法下,当与基线未加权方法相比较时(即新的和目前方法之间),在未受影响和受影响组的常染色体比率值之间观察到的分离始终增加。在三体性13、18和21情况中发生这种增加。因此,在重新拟合用于三体性测定的统计模型之后,对于给定的测序密度水平,从长远来看,预计提高针对三体性13、18和21的测试的灵敏度和特异性。
进一步的研究已表明,与现有方法相比较,由于使用新的大小加权方法,未受影响和受三体性影响的常染色体比率之间的分离增加对应于在三体性分析时13.2%的三体性样品胎儿分数的有效扩增。
Claims (26)
1.一种检测胎儿染色体异常的方法,其包括以下步骤:
(a) 从获自怀孕女性受试者的生物样品中分离核酸;
(b1) 选择120 bp-135 bp之间的核酸片段大小值用于最佳胎儿分数;
(b2) 分离大小在步骤(b1)中选择的片段大小值的20 bp以内的核酸片段;
(c) 确定与目标染色体的目标区域对齐的所述片段的第一数量和确定与参考染色体内的一个或多个目标区域对齐的所述片段的第二数量;
(d) 计算所述第一和第二数量之间的比率或差异;
(e) 基于所述比率或差异确定所述目标染色体的胎儿异常的存在。
2.如权利要求1定义的方法,其中所述胎儿染色体异常为选自以下的遗传变异:非整倍性、重复、易位、突变(例如点突变)、取代、缺失、单核苷酸多态性(SNP)、染色体异常、拷贝数变异(CNV)、表观遗传变化和DNA倒置。
3.如权利要求1定义的方法,其中所述目标染色体为13号染色体、18号染色体、21号染色体、X染色体或Y染色体。
4.如权利要求1-3的任何一项定义的方法,其中所述胎儿染色体异常为胎儿染色体非整倍性。
5.如权利要求4定义的方法,其中所述胎儿染色体非整倍性为三体性13、三体性18或三体性21。
6.如权利要求5定义的方法,其中所述胎儿染色体非整倍性为三体性21 (唐氏综合征)。
7.如权利要求1-6的任何一项定义的方法,其中所述胎儿染色体异常为例如高达1 Mb、高达5 Mb、高达10 Mb或高达20 Mb或大于20 Mb的染色体插入或缺失。
8.如权利要求1-7的任何一项定义的方法,其中所述目标染色体为染色体内的区域和所述参考染色体为与所述目标染色体相同的染色体内的区域。
9.如权利要求1-8的任何一项定义的方法,其另外包括针对怀疑含有所述胎儿染色体异常的基因组区域富集样品。
10.如权利要求1-9的任何一项定义的方法,其另外包括以下步骤:
(i) 通过计算每个片段大小(s)为胎儿来源的概率(w),将与目标染色体的目标区域对齐的每个片段进行大小加权;
(ii) 通过计算每个片段大小(s)为胎儿来源的概率(w),将与一个或多个参考染色体内的一个或多个目标区域对齐的每个片段进行大小加权;
(iii) 通过将步骤(i)中获取的值求和,计算总目标加权计数(Nc目标);
(iv) 通过将步骤(ii)中获得的值求和,计算总参考加权计数(Nc);
(v) 计算在步骤(iii)和(iv)中获得的Nc目标和Nc值之间的比率或差异;和
(vi) 基于所述比率或差异确定所述目标染色体的胎儿异常的存在。
11.一种预测怀孕女性受试者体内的胎儿性别的方法,所述方法包括以下步骤:
(a) 从获自怀孕女性受试者的生物样品中分离核酸;
(b1) 选择120 bp-135 bp之间的核酸片段大小值用于最佳胎儿分数;
(b2) 分离大小在步骤(b1)中选择的片段大小值的20 bp以内的核酸片段;
(c) 确定与性染色体对齐的所述片段的第一数量和确定与一个或多个参考染色体对齐的所述片段的第二数量;
(d) 计算所述第一和第二数量之间的比率或差异;
(e) 基于与所述参考染色体相比较是过多还是等同的片段与X染色体对齐或者是否存在Y染色体来确定所述胎儿的性别。
12.如权利要求11定义的方法,其另外包括以下步骤:
(i) 通过计算每个片段大小(s)为胎儿来源的概率(w),将与性染色体对齐的每个片段进行大小加权;
(ii) 通过计算每个片段大小(s)为胎儿来源的概率(w),将与一个或多个参考染色体对齐的每个片段进行大小加权;
(iii) 通过将步骤(i)中获得的值求和,计算总目标加权计数(Nc目标);
(iv) 通过将步骤(ii)中获得的值求和,计算总参考加权计数(Nc);
(v) 计算在步骤(iii)和(iv)中获得的Nc目标和Nc值之间的比率或差异;和
(vi) 基于与所述一个或多个参考染色体相比较是过多还是等同的片段与X染色体对齐或者是否存在Y染色体来确定所述胎儿的性别。
13.如权利要求1-12的任何一项定义的方法,其中所述生物样品为母体血液、血浆、血清或尿液。
14.如权利要求13定义的方法,其中所述生物样品为母体血浆。
15.如权利要求1-14的任何一项定义的方法,其中在步骤(a)中的分离步骤包括制备核酸片段的文库。
16.如权利要求15定义的方法,其中所述文库制备包括DNA末端修复、衔接子连接、净化和PCR的依序步骤。
17.如权利要求1-16的任何一项定义的方法,其中分离步骤(b2)包括富集大小为步骤(b1)中选择的片段大小值的10 bp以内,比如为步骤(b1)中选择的片段大小值的5 bp以内的核酸片段。
18.如权利要求1-17的任何一项定义的方法,其中分离步骤(b2)包括使用大小选择,比如基于凝胶的大小选择,特别是自动化的基于凝胶的大小选择进行富集。
19.如权利要求1-17的任何一项定义的方法,其中分离步骤(b2)包括使用计算机大小选择进行富集。
20.如权利要求1-19的任何一项定义的方法,其中步骤(c)最初包括在对齐之前对在步骤(b2)中分离的片段进行测序或使所述片段经受数字PCR。
21.如权利要求20定义的方法,其中所述测序包括选自以下的下一代测序系统:LifeTechnologies的Ion Torrent个人基因组仪(Ion Torrent PGM)或带有PI或PII芯片的IonProton及其进一步的衍生装置和组件;或Roche 454 (即Roche 454 GS FLX),AppliedBiosystems的SOLiD系统(即SOLiDv4),Illumina的NextSeq、GAIIx、HiSeq 2000和MiSeq测序仪,Pacific Biosciences的PacBio RS和Sanger的3730xl和QIAGEN的GeneReader。
22.如权利要求20定义的方法,其中所述测序包括选自以下的数字PCR系统:Quantstudio数字PCR系统(ThermoFisher)和RainDrop Plus数字PCR系统(RainDancetechnologies)。
23.如权利要求1-22的任何一项定义的方法,其另外包括从对齐步骤(c)之前获取的序列数据塌缩重复读取的步骤。
24.如权利要求23定义的方法,其中步骤(c)包括确定与目标染色体的区域唯一对齐的所述片段的第一数量和确定与参考染色体内的一个或多个目标区域唯一对齐的所述片段的第二数量。
25.如权利要求1-24的任何一项定义的方法,其中所述对齐步骤(c)通过IONA®、Bowtie2或BWA-SW软件或采用最大精确匹配技术的软件比如BWA-MEM或CUSHAW2软件进行。
26.如权利要求1-25的任何一项定义的方法,其另外包括基于所述样品内胎儿DNA的量来标准化或调整匹配命中的数量的步骤。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB1705281.2 | 2017-03-31 | ||
| GBGB1705281.2A GB201705281D0 (en) | 2017-03-31 | 2017-03-31 | Method of detecting a fetal chromosomal abnormality |
| GBGB1718623.0A GB201718623D0 (en) | 2017-11-10 | 2017-11-10 | Method of detecting a fetal chromosomal abnormality |
| GB1718623.0 | 2017-11-10 | ||
| PCT/GB2018/050855 WO2018178700A1 (en) | 2017-03-31 | 2018-03-29 | Method of detecting a fetal chromosomal abnormality |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN110914456A true CN110914456A (zh) | 2020-03-24 |
Family
ID=61906775
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201880035942.XA Pending CN110914456A (zh) | 2017-03-31 | 2018-03-29 | 检测胎儿染色体异常的方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20200109452A1 (zh) |
| EP (1) | EP3601591A1 (zh) |
| JP (1) | JP2020512000A (zh) |
| CN (1) | CN110914456A (zh) |
| AU (1) | AU2018244815A1 (zh) |
| CA (1) | CA3058551A1 (zh) |
| WO (1) | WO2018178700A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112037174A (zh) * | 2020-08-05 | 2020-12-04 | 湖南自兴智慧医疗科技有限公司 | 染色体异常检测方法、装置、设备及计算机可读存储介质 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102532991B1 (ko) * | 2019-12-23 | 2023-05-18 | 주식회사 랩 지노믹스 | 태아의 염색체 이수성 검출방법 |
| WO2024049915A1 (en) * | 2022-08-30 | 2024-03-07 | The General Hospital Corporation | High-resolution and non-invasive fetal sequencing |
| CN115798578A (zh) * | 2022-12-06 | 2023-03-14 | 中国人民解放军军事科学院军事医学研究院 | 一种分析与检测病毒新流行变异株的装置及方法 |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103108960A (zh) * | 2010-02-19 | 2013-05-15 | 西昆诺姆有限公司 | 用于检测胎儿核酸和诊断胎儿异常的方法 |
| CN103403183A (zh) * | 2011-06-29 | 2013-11-20 | 深圳华大基因健康科技有限公司 | 胎儿遗传异常的无创性检测 |
| CN103923987A (zh) * | 2014-04-01 | 2014-07-16 | 中山大学达安基因股份有限公司 | 一种基于高通量测序检测13、18、21三体综合征的方法 |
| CN104232778A (zh) * | 2014-09-19 | 2014-12-24 | 天津华大基因科技有限公司 | 同时确定胎儿单体型及染色体非整倍性的方法及装置 |
| CN104968800A (zh) * | 2012-08-30 | 2015-10-07 | 普莱梅沙有限公司 | 检测染色体异常的方法 |
| CN105296606A (zh) * | 2014-07-25 | 2016-02-03 | 深圳华大基因股份有限公司 | 确定生物样本中游离核酸比例的方法、装置及其用途 |
| WO2016071369A1 (en) * | 2014-11-04 | 2016-05-12 | Genesupport Sa | Method for determining the presence of a biological condition by determining total and relative amounts of two different nucleic acids |
| WO2016094853A1 (en) * | 2014-12-12 | 2016-06-16 | Verinata Health, Inc. | Using cell-free dna fragment size to determine copy number variations |
| CN105695567A (zh) * | 2015-11-30 | 2016-06-22 | 北京昱晟达医疗科技有限公司 | 一种用于检测胎儿染色体非整倍体的试剂盒、引物和探针序列及检测方法 |
| CN105926043A (zh) * | 2016-04-19 | 2016-09-07 | 苏州贝康医疗器械有限公司 | 一种提高孕妇血浆游离dna测序文库中胎儿游离dna占比的方法 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2496713B1 (en) * | 2009-11-06 | 2018-07-18 | The Chinese University of Hong Kong | Size-based genomic analysis |
| EP2768978B1 (en) * | 2011-10-18 | 2017-11-22 | Multiplicom NV | Fetal chromosomal aneuploidy diagnosis |
-
2018
- 2018-03-29 AU AU2018244815A patent/AU2018244815A1/en not_active Abandoned
- 2018-03-29 CA CA3058551A patent/CA3058551A1/en active Pending
- 2018-03-29 CN CN201880035942.XA patent/CN110914456A/zh active Pending
- 2018-03-29 US US16/499,849 patent/US20200109452A1/en not_active Abandoned
- 2018-03-29 WO PCT/GB2018/050855 patent/WO2018178700A1/en unknown
- 2018-03-29 EP EP18715932.2A patent/EP3601591A1/en not_active Withdrawn
- 2018-03-29 JP JP2019553832A patent/JP2020512000A/ja active Pending
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103108960A (zh) * | 2010-02-19 | 2013-05-15 | 西昆诺姆有限公司 | 用于检测胎儿核酸和诊断胎儿异常的方法 |
| CN103403183A (zh) * | 2011-06-29 | 2013-11-20 | 深圳华大基因健康科技有限公司 | 胎儿遗传异常的无创性检测 |
| CN104968800A (zh) * | 2012-08-30 | 2015-10-07 | 普莱梅沙有限公司 | 检测染色体异常的方法 |
| CN103923987A (zh) * | 2014-04-01 | 2014-07-16 | 中山大学达安基因股份有限公司 | 一种基于高通量测序检测13、18、21三体综合征的方法 |
| CN105296606A (zh) * | 2014-07-25 | 2016-02-03 | 深圳华大基因股份有限公司 | 确定生物样本中游离核酸比例的方法、装置及其用途 |
| CN104232778A (zh) * | 2014-09-19 | 2014-12-24 | 天津华大基因科技有限公司 | 同时确定胎儿单体型及染色体非整倍性的方法及装置 |
| WO2016071369A1 (en) * | 2014-11-04 | 2016-05-12 | Genesupport Sa | Method for determining the presence of a biological condition by determining total and relative amounts of two different nucleic acids |
| WO2016094853A1 (en) * | 2014-12-12 | 2016-06-16 | Verinata Health, Inc. | Using cell-free dna fragment size to determine copy number variations |
| CN105695567A (zh) * | 2015-11-30 | 2016-06-22 | 北京昱晟达医疗科技有限公司 | 一种用于检测胎儿染色体非整倍体的试剂盒、引物和探针序列及检测方法 |
| CN105926043A (zh) * | 2016-04-19 | 2016-09-07 | 苏州贝康医疗器械有限公司 | 一种提高孕妇血浆游离dna测序文库中胎儿游离dna占比的方法 |
Non-Patent Citations (2)
| Title |
|---|
| STEPHANIE C Y YU 等: "Size-based molecular diagnostics using plasma DNA for noninvasive prenatal testing" * |
| 殷旭阳 等: "孕妇血浆游离核酸高通量测序检测胎儿遗传异常" * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112037174A (zh) * | 2020-08-05 | 2020-12-04 | 湖南自兴智慧医疗科技有限公司 | 染色体异常检测方法、装置、设备及计算机可读存储介质 |
| CN112037174B (zh) * | 2020-08-05 | 2024-03-01 | 湖南自兴智慧医疗科技有限公司 | 染色体异常检测方法、装置、设备及计算机可读存储介质 |
Also Published As
| Publication number | Publication date |
|---|---|
| CA3058551A1 (en) | 2018-10-04 |
| WO2018178700A1 (en) | 2018-10-04 |
| US20200109452A1 (en) | 2020-04-09 |
| AU2018244815A1 (en) | 2019-10-31 |
| EP3601591A1 (en) | 2020-02-05 |
| JP2020512000A (ja) | 2020-04-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10619214B2 (en) | Detecting genetic aberrations associated with cancer using genomic sequencing | |
| KR102339760B1 (ko) | 대규모 병렬 게놈 서열분석을 이용한 태아 염색체 이수성의 진단 방법 | |
| KR20150070111A (ko) | 염색체 이상의 검출 방법 | |
| CN110914456A (zh) | 检测胎儿染色体异常的方法 | |
| CN112662754B (zh) | 用于预测小耳畸形发生概率的组合物的应用方法 | |
| CN111763742A (zh) | 甲基化标志物及确定个体年龄的方法和应用 | |
| WO2019092438A1 (en) | Method of detecting a fetal chromosomal abnormality | |
| AU2013203077B2 (en) | Diagnosing fetal chromosomal aneuploidy using genomic sequencing | |
| Gómez-Manjón et al. | Noninvasive Prenatal Testing: Comparison of Two Mappers and Influence in the Diagnostic Yield | |
| AU2013200581B2 (en) | Diagnosing cancer using genomic sequencing | |
| CN113969310A (zh) | 胎儿dna浓度的评估方法及应用 | |
| AU2008278843B2 (en) | Diagnosing fetal chromosomal aneuploidy using genomic sequencing | |
| CN113999900A (zh) | 以孕妇游离dna评估胎儿dna浓度的方法及应用 | |
| WO2020226528A1 (ru) | Способ определения кариотипа плода беременной женщины | |
| KR20200137875A (ko) | 2단계 Z-score에 기반한 비침습적 산전 검사 방법 및 장치 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Application publication date: 20200324 |
|
| WD01 | Invention patent application deemed withdrawn after publication |