CN105612176B - Hla g修饰的细胞及方法 - Google Patents
Hla g修饰的细胞及方法 Download PDFInfo
- Publication number
- CN105612176B CN105612176B CN201380051169.3A CN201380051169A CN105612176B CN 105612176 B CN105612176 B CN 105612176B CN 201380051169 A CN201380051169 A CN 201380051169A CN 105612176 B CN105612176 B CN 105612176B
- Authority
- CN
- China
- Prior art keywords
- genetically modified
- cell
- mammalian cell
- hla
- modified mammalian
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images





















Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/569—Immunoassay; Biospecific binding assay; Materials therefor for microorganisms, e.g. protozoa, bacteria, viruses
- G01N33/56966—Animal cells
- G01N33/56977—HLA or MHC typing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; CARE OF BIRDS, FISHES, INSECTS; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K67/00—Rearing or breeding animals, not otherwise provided for; New breeds of animals
- A01K67/027—New breeds of vertebrates
- A01K67/0271—Chimeric animals, e.g. comprising exogenous cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/28—Bone marrow; Haematopoietic stem cells; Mesenchymal stem cells of any origin, e.g. adipose-derived stem cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/33—Fibroblasts
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/37—Digestive system
- A61K35/407—Liver; Hepatocytes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/44—Vessels; Vascular smooth muscle cells; Endothelial cells; Endothelial progenitor cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/70503—Immunoglobulin superfamily
- C07K14/70539—MHC-molecules, e.g. HLA-molecules
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0603—Embryonic cells ; Embryoid bodies
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/566—Immunoassay; Biospecific binding assay; Materials therefor using specific carrier or receptor proteins as ligand binding reagents where possible specific carrier or receptor proteins are classified with their target compounds
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
- G01N33/6893—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids related to diseases not provided for elsewhere
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; CARE OF BIRDS, FISHES, INSECTS; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2207/00—Modified animals
- A01K2207/12—Animals modified by administration of exogenous cells
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; CARE OF BIRDS, FISHES, INSECTS; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2227/00—Animals characterised by species
- A01K2227/10—Mammal
- A01K2227/105—Murine
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; CARE OF BIRDS, FISHES, INSECTS; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2267/00—Animals characterised by purpose
- A01K2267/02—Animal zootechnically ameliorated
- A01K2267/025—Animal producing cells or organs for transplantation
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2510/00—Genetically modified cells
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/435—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans
- G01N2333/705—Assays involving receptors, cell surface antigens or cell surface determinants
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/435—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans
- G01N2333/705—Assays involving receptors, cell surface antigens or cell surface determinants
- G01N2333/71—Assays involving receptors, cell surface antigens or cell surface determinants for growth factors; for growth regulators
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2800/00—Detection or diagnosis of diseases
- G01N2800/24—Immunology or allergic disorders
- G01N2800/245—Transplantation related diseases, e.g. graft versus host disease
Abstract
本发明公开了可持续表达HLA‑G(例如细胞表面HLA‑G)的遗传修饰的细胞和用于产生此类遗传修饰的细胞的核酸组合物。还公开了利用可持续表达HLA‑G的遗传修饰细胞的细胞治疗方法。本发明的HLA‑G遗传修饰可提供具有减低的免疫原性和/或改进的免疫抑制特征的细胞,以使得这些细胞可作为通用或改进的供体细胞用于移植、细胞和组织再生或重建以及其他治疗。
Description
相关专利申请的交叉引用
本申请要求2012年7月31日提交的美国临时专利申请序列号61/677,739的优先权益,其以引用方式全文纳入本文。
背景技术
细胞移植形式的再生医学是可用于治疗疑难医学病况例如糖尿病、心脏病和神经退行性疾病的最有希望的治疗方法之一。但是,临床中实现细胞移植的主要障碍是供体细胞的免疫排斥,特别当其来自外源宿主时。虽然通过给予免疫抑制药物可部分地解决免疫排斥,然而其也会产生严重的副作用。因此,需要开发改进的技术用于细胞移植治疗。
发明内容
本发明公开了基于细胞的组合物和基于在将要被移植到需要供体细胞的受试者中的供体细胞中长期强制表达至少一种外源HLA-G蛋白进行细胞移植治疗的方法。本发明提供的数据显示,经本发明所述方式修饰可表达外源HLA-G的细胞(无论多能性的或分化型的)具有减低的免疫原性和/或增加的免疫抑制。通过HLA-G遗传修饰产生的减低的免疫原性和/或增加的免疫抑制能力是稳定的并且可持续长时间,包括贯穿分化过程。这意味着本发明的HLA-G修饰的细胞可作为通用的供体细胞或组织(即,减少或排除供体细胞和受体之间I类和II类经典人白细胞抗原(HLA)分子配型的需要)用于多种损伤、疾病或病症。
相应地,本发明一方面是遗传修饰的哺乳动物细胞(HLA-G修饰的细胞),其与不含所述遗传修饰的哺乳动物细胞相比在异体移植受体(an allogeneic recipient)中具有减低的免疫原性和/或能够提供增加的免疫抑制,其中(i)所述遗传修饰的哺乳动物细胞包含:(a)外源核酸(例如表达载体),其包含编码与人HLA-G具有至少85%氨基酸序列同一度的HLA-G蛋白并且含有可减低内质网-高尔基体循环途径中HLA-G存留的一种或多种氨基酸突变的核酸序列,和/或(b)不含microRNA结合位点的3’UTR(非翻译区)序列例如SEQ IDNO:3或不含SEQ ID NO:4的序列;并且(ii)编码的HLA-G蛋白是由遗传修饰的哺乳动物细胞表达至少7周(例如至少20周或至少50周)。
另外的实施方案中,本发明提供了遗传修饰的哺乳动物细胞,其与不含所述遗传修饰的哺乳动物细胞相比具有减低的免疫原性和/或改进的免疫抑制,其中(i)所述遗传修饰的哺乳动物细胞含有外源核酸,其包括:(a)核酸序列(例如SEQ ID NO:2),其编码与同义野生型人HLA-G(例如SEQ ID NO:1)具有至少95%氨基酸序列同一度的HLA-G蛋白,并且含有可减低内质网-高尔基体循环途径中HLA-G存留的一种或多种氨基酸突变;和(b)3’非翻译区(UTR)(例如SEQ ID NO:3),其与同义野生型人HLA-G基因的3’非翻译区序列具有至少85%的同一度,并且不含SEQ ID NO:4;并且(ii)编码的HLA-G蛋白是由遗传修饰的哺乳动物细胞表达至少7周。
在一些实施方案中,遗传修饰的细胞具有减低的免疫原性和/或改进的免疫抑制,如果其显示出:(1)与不含所述遗传修饰的哺乳动物细胞相比,遗传修饰的细胞的NK-92细胞毒性减低,(2)与不含所述遗传修饰的哺乳动物细胞相比,遗传修饰细胞的体外的外周血单核细胞增殖减低,和/或(3)在人源化NSG小鼠中,与不含所述遗传修饰的哺乳动物细胞相比,遗传修饰的细胞增加肿瘤形成的大小和重量。
在一些实施方案中,遗传修饰的哺乳动物细胞与异体移植受体相比在一种或多种HLA抗原中不具有匹配(即,相同的等位基因),其中所述HLA抗原选自HLA-A、HLA-B、HLA-C、HLA-DP、HLA-DQ和HLA-DR。在某些实施方案中,遗传修饰的哺乳动物细胞与异体移植受体相比在一种或多种HLA抗原中只有1、2、3、4或5种匹配,其中所述HLA抗原选自HLA-A、HLA-B、HLA-C、HLA-DP、HLA-DQ和HLA-DR。在一个实施方案中,遗传修饰的哺乳动物细胞与异体移植受体的HLA-A、HLA-B、HLA-C、HLA-DP、HLA-DQ和HLA-DR不匹配。
在一些实施方案中,遗传修饰的细胞包括不含可减低内质网-高尔基体循环途径中HLA-G存留的一种或多种氨基酸突变的HLA-G转基因(即HLA-G野生型同义序列,例如SEQID NO:1),但是具有包括不含microRNA结合位点的3’UTR(非翻译区)序列例如SEQ ID NO:3或不含SEQ ID NO:4的序列的HLA-G转基因。
在一些实施方案中,可减低内质网-高尔基体循环途径中HLA-G存留的一种或多种氨基酸突变包括双赖氨酸(KK)基序突变。在一些实施方案中,所述KK基序突变包括K334A突变、K335A突变或其二者。
在一些实施方案中,在遗传修饰的细胞中表达的外源核酸包括不含SEQ ID NO:4的3’UTR序列。在一个实施方案中,当所述外源核酸的3’UTR序列不包括SEQ ID NO:4,所述核酸序列含有SEQ ID NO:3。
在一些实施方案中,表达的HLA-G存在于遗传修饰的哺乳动物细胞的细胞表面上。
在一些实施方案中,所述遗传修饰的哺乳动物细胞是人细胞、小鼠细胞、大鼠细胞、猴细胞或猪细胞。
在一些实施方案中,所述遗传修饰的哺乳动物细胞是干细胞、祖细胞或者由干细胞或祖细胞直接分化产生的细胞。在一些实施方案中,所述遗传修饰的哺乳动物细胞是导入外源HLA-G转基因之前已经分化(天然地或体外)的细胞。在一些实施方案中,所述遗传修饰的哺乳动物细胞是干细胞(例如多能干细胞)。在一些实施方案中,当所述遗传修饰的哺乳动物细胞是干细胞时,所述干细胞是胚胎干细胞、诱导的多能干细胞或者全能干细胞。在一个实施方案中,所述遗传修饰的哺乳动物细胞是胚胎干细胞。另一个实施方案中,所述遗传修饰的哺乳动物细胞是诱导的多能干细胞。又一个实施方案中,所述遗传修饰的哺乳动物细胞不属于免疫系统细胞类型。另一个实施方案中,所述遗传修饰的哺乳动物细胞是通过干细胞或祖细胞体外分化获得的细胞,其中所述干细胞或祖细胞是经过遗传修饰然后体外分化的。
在另外的实施方案中,所述遗传修饰的细胞是完全分化的细胞、表皮祖细胞、胰腺祖细胞、造血干细胞、多能干细胞分化产生的细胞、角质形成细胞、成纤维细胞、间质干细胞、心肌细胞、神经干细胞、神经元、星形胶质细胞或胰腺β祖细胞。
在一些实施方案中,当遗传修饰的哺乳动物细胞中的外源核酸是表达载体时,所述表达载体是转座子载体或逆转录病毒载体。在一些实施方案中,当所述外源核酸是表达载体时,所述表达载体是打靶载体,并且所述遗传修饰的哺乳动物细胞是通过同源重组所述打靶载体获得的。在一些实施方案中,所述表达载体还可包括编码报告蛋白例如绿色荧光蛋白(GFP)的核酸序列。
在一些实施方案中,所述外源核酸还包括这样的核酸序列:(i)与人HLA-G基因的3’非翻译区序列具有至少85%同一度的核酸序列;和(ii)在其3’非翻译区含有至少1个突变,所述突变可抑制同源microRNA与含有突变结合位点的mRNA中的突变位点的结合。在一个实施方案中,所述核酸序列包含SEQ ID NO:3。
在一些实施方案中,提供了含有所述遗传修饰细胞的人工组织。
另一方面,本发明提供了分离的核酸,其包括(i)编码与人HLA-G具有至少85%同一度的氨基酸序列的第一核酸序列;和(ii)与人HLA-G基因的3’非翻译区序列具有至少85%同一度并且可操作地连接到所述第一核酸序列的第二核酸序列,其中所述氨基酸序列包含可减低内质网-高尔基体循环途径中HLA-G存留的突变并且所述第二核酸序列的3’非翻译区包含可抑制同源microRNA与含有突变结合位点的mRNA结合的至少1个突变。
在一些实施方案中,所述分离核酸3’非翻译区序列不含SEQ ID NO:4。在一个实施方案中,当所述3’非翻译区序列不含SEQ ID NO:4时,所述3’非翻译区序列包含SEQ ID NO:3。
在一些实施方案中,提供了包含分离的核酸和可操作地连接到所述第一核酸序列的启动子的哺乳动物表达载体,其中所述启动子在干细胞中不是沉默的。在一些实施方案中,所述启动子包含中国仓鼠EF-1α(CHEF-1α)启动子或人EF-1α启动子的核酸序列。在一个实施方案中,所述CHEF-1α启动子包含SEQ ID NO:7。另外的实施方案中,用于驱动HLA-G转基因表达的启动子是组织或细胞类型选择性启动子。在一些实施方案中,所述哺乳动物表达载体含有编码报告蛋白的第三核酸序列。在一些实施方案中,所述哺乳动物表达载体是转座子载体。在一些实施方案中,提供了含有哺乳动物表达载体的遗传修饰的哺乳动物细胞。
在一些实施方案中,提供了分离的核酸,其包括:(i)编码与人HLA-G具有至少95%同一度的氨基酸序列的第一核酸序列,其中所述氨基酸序列包含可减低内质网-高尔基体循环途径中HLA-G存留的突变;和(ii)与人HLA-G基因的3’非翻译区序列具有至少95%同一度并且可操作地连接到所述第一核酸序列的第二核酸序列,其中所述第二核酸序列的3’非翻译区包含可抑制同源microRNA与含有突变结合位点的mRNA结合的至少1个突变。在一个实施方案中,所述第一核酸序列编码SEQ ID NO:2的氨基酸序列。另一个实施方案中,所述第二核酸序列不含SEQ ID NO:4。在一个实施方案中,所述第二核酸序列包含SEQ ID NO:3。另一个实施方案中,提供的哺乳动物表达载体含有所述第一和第二核酸序列,并且还含有可操作地连接到所述第一核酸序列的启动子,其中所述启动子在干细胞或由干细胞分化产生的细胞中不是沉默的。所述启动子可含有中国仓鼠EF-1α启动子的核酸序列。另一个实施方案中,所述哺乳动物表达载体还含有编码报告蛋白的核酸序列。另一个实施方案中,所述哺乳动物表达载体是转座子载体。另一个实施方案中,所述哺乳动物表达载体含有图1所示的所有元件。
在一个实施方案中,提供了哺乳动物表达载体,其包括:(a)中国仓鼠EF-1α启动子,(b)可操作地连接到所述启动子并且编码具有氨基酸序列SEQ ID NO:2的人HLA-G的核酸序列,和(c)含有SEQ ID NO:3的3’UTR序列。在一些实施方案中,提供了遗传修饰的哺乳动物细胞,其包含这样的表达载体。
在多个实施方案中,将HLA-G修饰的哺乳动物细胞(例如人HLA-G修饰的细胞)施用至遭受多种病况中的任意病况的受试者,所述病况包括但不限于心血管疾病、眼病(例如黄斑变性)、耳病(例如耳聋)、糖尿病、神经退行性疾病、阿尔茨海默氏病、帕金森病、多发性硬化、骨质疏松症、肝病、肾病、自身免疫疾病、关节炎、牙龈疾病、牙齿病况或增生性病症(例如肿瘤)。其他实例中,受试者正遭受或处于遭受急性健康病况例如中风、脊髓损伤、烧伤或创伤的高风险。其他实例中,受试者正遭受组织损失例如脂肪萎缩(lipatrophy)或者年龄相关的胶原损失。其他实例中,受试者遭受非愈合性溃疡或者需要试剂以辅助缺陷例如尿道下裂和尿道上裂的闭合。其他实例中,受试者需要持久或暂时的皮肤移植用于伤口愈合或皮肤替代。
在一些实施方案中,本发明提供了细胞或组织修复或再生的通用方法用于有需要的受试者,所述方法包括将含eHLA-G修饰的细胞群的细胞或组织组合物注射或移植到受试者,其中与eHLA-G修饰的细胞群相比所述受试者具有至少1种不匹配的经典I类HLA或II类HLA分子,并且其中所述eHLA-G修饰的细胞群与不含所述eHLA-G修饰的相同类型细胞相比显示出减低的免疫原性和/或改进的免疫抑制。所述减低的免疫原性和/或改进的免疫抑制可以确定,例如通过在NK-92细胞毒性测定、人源化NSG肿瘤生长测定和/或PBMC增殖测定中比较eHLA-G修饰细胞与不含所述eHLA-G修饰的相同类型对照细胞。在一个实施方案中,所述遗传修饰的细胞群包括eHLA-G遗传修饰的人真皮成纤维细胞群。另一个实施方案中,遗传修饰的细胞群包括eHLA-G遗传修饰的人表皮祖细胞群。另一个实施方案中,遗传修饰的细胞群包括eHLA-G遗传修饰的人间质干细胞群。另一个实施方案中,遗传修饰的细胞群包括eHLA-G遗传修饰的人胚胎干细胞群。另一个实施方案中,遗传修饰的细胞群包括由eHLA-G遗传修饰的人胚胎干细胞体外分化产生的细胞群。另外的实施方案中,所述遗传修饰的细胞群未被受试者的免疫系统排斥至少2、4、6、8、10、12、14、16、18、20、24、36、48或52周。
另一个实施方案中,本发明提供了可再生皮肤的方法用于需要其的受试者,所述方法包括将eHLA-G修饰的皮肤成纤维细胞群和/或eHLA-G修饰的胚胎表皮祖细胞群注射至受试者皮肤损伤的位点,其中与eHLA-G修饰的皮肤成纤维细胞群和/或eHLA-G修饰的胚胎表皮祖细胞群相比所述受试者具有至少1种不匹配的经典I类HLA或II类HLA分子。
另一个实施方案中,提供了细胞治疗方法,其包括向需要其的受试者施用本发明的含外源人β2-微球蛋白(β2m)分子和eHLA-G转基因的遗传修饰的哺乳动物细胞群。
附图说明
图1示出了增强的HLA-G(eHLA-G)转基因表达转座子载体的非限制性实施方案的示意图,其含有:磷酸甘油酸酯激酶(PGK)启动子驱动的选择标记(新霉素磷酸转移酶);“INS”侧翼绝缘子(insulator)元件;中国仓鼠EF-1α启动子,驱动eHLA-G表达;PGK启动子,驱动EGFP表达;和5’末端重复(TR)转座子元件。eHLA-G转基因包含可增强HLA-G蛋白表达尤其是细胞表面表达的突变组合和/或非编码元件(例如启动子或修饰的3’UTR)。对于本发明的实验,所述eHLA-G转基因上述元件,并且更具体地包括具有HLA-G的ER滞留基序(K334A/K335A)突变和2)HLA-G的3’UTR microRNA结合位点突变的如SEQ ID NO:2所示的人HLA-G编码序列,其中所述修饰的3’UTR具有如SEQ ID NO:3所示的序列。具体描述见实施例3。
图2示出了用于驱动共转染的转座子表达载体基因组整合的转座酶表达辅助载体的非限制性实施方案的示意图。
图3示出了用可驱动eHLA-G和EGFP表达的转座子表达载体稳定转染后多个时间电的人ES细胞克隆的荧光显微图像。
图4(上图)示出了描述使用含EGFP的表达载体转染的hES细胞起始数与转染10天后在细胞群中检测的部分EGFP+细胞之间关系的柱状图。x轴的数值是每5×105个细胞中的GFP+克隆数。y轴数值是细胞数。(下图)示出了描述多种转座子载体(使用不同启动子)和所述细胞群中检测的部分EGFP+细胞之间关系的柱状图。x轴的数值是每5×105个细胞中的GFP+克隆数。下图中,自左至右为:对照(con)、GFP-puro(转染载体大小为7.3kb)、eHLA-G(MSCV)-GFP-puro(转染载体大小为8.6kb)和eHLA-G(EF-1α)-GFP-puro(转染载体大小为9.2kb)。另外还进行了eHLA-G(EF-1α)-GFP-puro转让效率实验(未示出),其显示在转染后10天且嘌呤霉素选择10天后以溶液V和程序B16获得最高的效率(每5×105个细胞中~500个GFP+克隆)。
图5示出了显示HLA-G修饰的人ES细胞中HLA-G表达和DAPI染色(上图)、Oct 3/4和DAPI(中图)以及SSEA-4和DAPI(下图)的系列免疫荧光显微图像。
图6(上图)的流式细胞术散点图示出了eHLA-G修饰的hES细胞群中SSEA-4+/GFP+双阳性细胞的分布。(下图)示出了eHLA-G修饰的hES细胞群中Oct 3/4+细胞的分布。SSEA-4和Oct 3/4是多能性标记。该数据以及图5显示,eHLA-G修饰的hES细胞保持了其独特的自我更新多能性标记。另外,eHLA-G(EF-1α)-GFP-hESC保持了其多能性和体内正常核型。将人源化的NSG小鼠皮下注射eHLA-G(EF-1α)-GFP-hESC并观测畸胎瘤形成,显示出注射的/移植的细胞未被排斥,由于hESC显示出减低的免疫原性和/或增加的免疫抑制。所述畸胎瘤细胞的核型是正常的。
图7(上图)示出了第15天野生型(左图)和eHLA-G修饰的hESC产生的(右图)胚状体(EB)的相差显微照片。(下图)上图所示EB的荧光显微照片。野生型hESC EB中未检测到GFP信号,而两个eHLA-G修饰的hESC EB中都检测到强的GFP表达。所述数据表明,eHLA-G+hESC维持了EB形成。
图8示出了eHLA-G+hESC是沉默抗性的。(上图)示出了eHLA-G修饰的hESC细胞系中GFP表达的流式细胞术分布直方图,其显示传代6次和16次后类似的GFP强表达。(下图)示出了相同的hESC细胞系中HLA-G表达的流式细胞术分布直方图,也显示传代6次和16次时eHLA-G转基因的持续表达。
图9A(上图)示出了野生型(左侧直方图)、GFP修饰的(中间直方图)和eHLA-G(EF-1α)-GFP修饰的hESC(右侧直方图)中总(细胞内)HLA-G表达的流式细胞术分布直方图。(下图)示出了野生型(左侧直方图)、GFP修饰的(中间直方图)和eHLA-G(EF-1α)-GFP修饰的hESC(右侧直方图)中表面HLA-G表达的流式细胞术分布直方图。图9B(上图)示出了eHLA-G(MSCV)-GFP修饰的hESC中总HLA-G表达的流式细胞术分布直方图。图9B(下图)示出了eHLA-G(MSCV)-GFP修饰的hESC中表面HLA-G表达。这些数据表明,当转基因可操作地连接到EF-1α启动子时HLA-G高表达,相反地当所述转基因在MSCV启动子控制下时表达最低。另外的数据也可见图15,其示出HLA-G转基因表达受启动子活性的显著影响。
图10示出了野生型和eHLA-G+hESC中I类HLA和II类HLA的表达类似。该表比较了野生型和HLA-G修饰的hESC中多种HLA变体和β2微球蛋白的表达水平。
图11示出了NK92细胞毒性效应受eHLA-G+hESC显著抑制。(更多描述见实施例6)该图示出了显示NK92细胞毒性测定结果的柱状图。1:10和1:30值代表效应子(NK92)与靶细胞(只含GFP转基因的对照野生型细胞或者eHLA-G-GFP转基因修饰的细胞)的比例。该图是体外测定,用于确定与野生型hESC(灰色柱;GFP)相比较的eHLA-G修饰的hESC(黑色柱;eHLA-G-GFP)的免疫原性。与野生型hESC相比,存在NK92细胞的条件下,eHLA-G修饰的hESC显示出细胞毒性的大幅度减低。所述数据表明,外源HLA-G表达可为所述遗传修饰的细胞提供改进的供体能力,由于存在NK92细胞条件下减低的细胞毒性表明所述遗传修饰的细胞具有减低的免疫原性和/或改进的免疫抑制。结果是4次实验的平均值。更多描述见实施例6。
图12是一系列柱状图,示出了hESC体外直接分化形成胚胎表皮祖细胞(EEP)过程中多种多能性标记物和表皮祖细胞标记物的基因表达水平的时间进程。y-轴的值是通过半定量RT-PCR确定的mRNA相对表达水平。x-轴的值是测定表达时的天数。更多描述见实施例2。图12数据表明,表皮分化标记物K14、Tap63和ΔNp63在分化过程中逐渐增强。本文未示出的数据中,进行了K14和另外的表皮标记物p63、CD29和CD49f的免疫荧光检测研究。免疫荧光数据显示,分化的eHLA-G(EF-1α)-GFP hEEP是K14、p63、CD29和CD49f蛋白表达阳性的。
图13(上图)示出了稳定转染EF-1α启动子驱动eHLA-G表达的hESC细胞系中eHLA-G转基因表达的时间进程。(下图)示出了GFP修饰的hESC细胞系(阴性对照)、MSCV启动子驱动的eHLA-G hESC细胞系和EF-1α驱动的eHLA-G hESC细胞系中HLA-G(mRNA)表达的时间进程比较(黑色柱代表第0天;水平条纹柱代表第7天;灰色柱代表第14天)。注意,由EF-1α启动子驱动的eHLA-G表达水平更高且更持久。
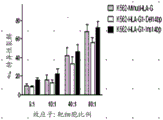
图14(K562-HLA-G1 14碱基对(bp)插入/缺失多态性对NK细胞的细胞毒活性的影响。)该图示出了比较NK细胞介导的对于野生型K562细胞(灰色柱)、表达3’UTR中含14bp插入的HLA-G变体的K562细胞(Ins14bp)(黑色柱)以及表达3’UTR中含14bp缺失的HLA-G变体的K562细胞(Del14bp)(灰色柱)的细胞毒性(%特异性裂解)的柱状图。共有4组数据,具有不同的效应子(NK细胞)与靶细胞(K562细胞)比例。
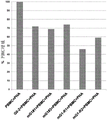
图15提供的数据显示HLA-G转基因表达受启动子活性的显著影响。RT-PCR显示,eHLA-G(EF-1α)-GFP-hESC细胞系中GFP和HLA-G转录本都高表达。免疫荧光检测还显示,HLA-G(EF-1α)-GFP-hESC细胞系中GFP和HLA-G蛋白质都高表达(本文中未示出)。但是,HLA-G(pMSCV)-GFP-hESC细胞系中HLA-G转录本或蛋白质很少检测到,无论通过RT-PCR或免疫荧光检测,虽然这些细胞中GFP高表达。(本文中数据未示出。)因此,在一些实施方案中,EF-1α启动子是HLA-G转基因表达优选的。
图16.纯化的hEEP显示出同源角质形成细胞的形态,如相差显微镜检测结果所示。eHLA-G-GFP-hEEP克隆18和21由修饰的eHLA-G(EF-1α)-GFP-hESC分化产生,如实施例2所述。
图17.通过流式细胞术验证了HLA-G转基因在分化的hEEP中的稳定性。与不含外源HLA-G的对照细胞(只含GFP的hEEP)和野生型hEEP相比,分化的eHLA-G(EF-1a)-GFP-hEEP(超过90%的细胞)中HLA-G总的表达(上图)和表面表达(下图)都是很强的。
图18.在另外的NK细胞毒性实验中重复和验证图11的结果。如图所示,eHLA-G(EF-1α)-GFP-hESC的杀死与只含GFP转基因(不含HLA-G转基因)的对照hESC相比减少超过100%。(注:本文中,“mHLA-G(EF-1α)-GFP”转基因是“eHLA-G(EF-1α)-GFP”的同义词。)所述数据表明,hESC中HLA-G转基因表达可产生免疫抑制和/或减低的免疫原性特征。更多描述见实施例6。
图19.在由hESC分化产生的hEEP中进行NK细胞毒性实验。如图所示,eHLA(EF-1α)-GFP-hEEP的杀死与对照hEEP相比显著减低超过100%(约3倍)。所述数据显示,HLA-G转基因表达可在分化的细胞中产生免疫抑制和/或减低的免疫原性特征,其还表明通过eHLA-G转基因由HLA-G表达产生的所述增强的功能性免疫逃逸特征存在于整个分化过程中。更多描述见实施例6。
图20示出了hESC异体移植入人源化小鼠的结果。“G0”hESC是对照野生型hESC,其不含eHLA-G转基因而只含GFP。“mG1(#1)”和“mG1(#2)”是两种不同的eHLA-G(EF-1α)-GFP核转染hESC克隆。对如图所示的G0、mG1(#1)和mG1(#2)肿瘤测量并称重。G0 hESC形成的肿瘤体积为126.9立方毫米,重32毫克。mG1(#1)hESC形成的肿瘤体积为748.4立方毫米,重318毫克。mG1(#2)hESC形成的肿瘤体积为1116.7立方毫米,重675毫克。更多描述见实施例7。
图21示出了将hESC异体移植入5只人源化NSG小鼠形成肿瘤的平均结果。上图示出了肿瘤重量(mg)结果。下图示出了肿瘤体积(立方毫米)结果。所述数据显示,HLA-G核转染的hESC(“mG1”)形成的肿瘤比野生型hESC(“G0”)移植入人源化NSG小鼠形成的肿瘤大得多(体积超过3倍)且重得多(重量超过2倍)。这表明,eHLA-G转基因表达可在异体移植的人环境(即NSG人源化小鼠)中产生减低的免疫原性和/或增加的免疫抑制。这些数据结合NK92细胞毒性研究,支持将本发明的eHLA-G转基因构建体普遍应用于将任意需要的细胞类型修饰成为通用或优良的异体供体用于治疗、移植、组织修复、细胞和组织替换等。更多描述见实施例7。
图22.测定了由eHLA-G(EF-1α)-GFP转基因(“HFD-m1-GFP”细胞)或单独的GFP对照构建体(“HFD-G0-GFP”细胞)稳定转染的人真皮成纤维细胞抑制PBMC增殖的能力。如图所示,HFD-mG1-GFP克隆“mG1-R1”抑制PBMC增殖大于对照和其他克隆,表明外源HLA-G表达可为分化的细胞例如成纤维细胞提供免疫抑制。更多描述见实施例9,包括使用HFD-m1-GFP和对照的NK-92细胞毒性研究总结,其显示出eHLA-G修饰人真皮成纤维细胞可减低其免疫原性。因此,这些数据还支持将本发明的eHLA-G转基因构建体应用于将任意需要的细胞类型无论是多能的、专能的(multipotent)或者完全分化的细胞类型修饰成为通用或优良的异体供体用于治疗、移植、组织修复、细胞和组织替换等。
具体实施方式
本发明的特征在于可持续表达外源HLA-G的遗传修饰的哺乳动物细胞(HLA-G修饰的细胞)以及可产生所述修饰的哺乳动物细胞的核酸组合物。本发明的eHLA-G遗传修饰可提供具有减低的免疫原性和/或改进的免疫抑制特征的细胞,以使得所述细胞可作为通用或改进的供体细胞用于移植、细胞和组织再生或重建以及其他治疗。
I.组合物:
A.可表达外源HLA-G的遗传修饰的哺乳动物细胞
本发明中,可产生能够表达外源HLA-G的大范围的哺乳动物细胞类型(HLA-G修饰的细胞)。所述细胞类型包括但不限于全能细胞、胚胎干细胞(例如人胚胎干细胞)、诱导的多能干细胞(例如诱导的人多能干细胞)、专能干细胞、表皮祖细胞、间质干细胞、胰腺β祖细胞、胰腺β细胞、心脏祖细胞、心肌细胞、肝脏祖细胞、肝细胞、肌细胞祖细胞、肌细胞、肾细胞、成骨细胞、造血祖细胞、牙囊细胞、毛囊细胞、视网膜色素上皮细胞、神经干细胞、神经元、星形胶质细胞、少突胶质细胞、内耳细胞和成纤维细胞(包括人真皮成纤维细胞(HFD))。在一些实施方案中,HLA-G修饰的细胞不是具有免疫系统细胞类型的细胞。所述哺乳动物细胞可来自包括例如人、小鼠、大鼠、猴或猪的数种中的一种。实质上,可用本发明的构建体转染任意细胞类型,然后测试HLA-G表达以及所述表达如何给予所述修饰细胞以减低的免疫原性和/或改进的免疫抑制。
在一些实施方案中,为获得大大富集的所需细胞类型的HLA-G修饰细胞群,可产生表达HLA-G的遗传修饰的多能干细胞系例如人胚胎干细胞系或诱导的人多能干细胞系或具有专能性特征的任意细胞系包括间质干细胞和免疫系统祖细胞,然后直接分化以获得表达HLA-G并且所需细胞类型大大富集的细胞群。在一些实施方案中,所述大大富集的细胞群包括至少约2%至约100%所需细胞类型,例如至少约3%、4%、5%、7%、8%、10%、20%、22%、25%、35%、40%、45%、50%、60%、70%、75%、80%、85%或者从至少约2%至约100%的其他百分率的所需细胞类型。用于富集所需细胞类型的细胞的方法是本领域公知的。见例如美国专利申请No.12/532,512。
用于获得人胚胎干细胞或诱导的多能干细胞的方法是本领域公知的,例如美国专利No.6,200,806和No.7,217,569(用于人胚胎干细胞诱导)以及No.8,048,999、No.8,058,065和No.8,048,675(用于产生诱导的人多能干细胞)中所述。
可稳定表达由本发明的核酸之一编码的HLA-G蛋白的遗传修饰的哺乳动物多能或专能干细胞系例如人胚胎干细胞或诱导的人多能干细胞等以及完全分化的遗传修饰的哺乳动物可通过本领域公知的任意数量的方法产生。在一些实施方案中,所述细胞系是通过使用含有用于表达本文所述HLA-G蛋白的表达盒、选择标记及任选报告蛋白的一种或多种核酸表达载体(例如质粒载体或微环载体)稳定转染进行遗传修饰的。由所述载体编码的适合的选择标记的实例包括提供对选择性试剂的抗性的蛋白。所述蛋白及其相应的选择性试剂包括但不限于嘌呤霉素N-乙酰转移酶(嘌呤霉素)、潮霉素磷酸转移酶(潮霉素)、杀稻瘟素-S-脱氨酶(杀稻瘟素)和新霉素磷酸转移酶(新霉素)。使用适合的选择性试剂进行选择可持续至少约3至约14天,例如约4、5、6、7、8、9、10、12、13天或从约3至约14天直至出现抗性克隆的另外的周期。
适合的荧光报告蛋白的实例包括但不限于EGFP及其变体例如YFP、Cyan和dEGFP;DS-红、单体橙、远红荧光蛋白“Katushka”(Shcherbo et al(2007),Nat Methods,4:741-746)或上述任意的变体。其他实施方案中,所述报告蛋白是酶,其可在荧光或发光底物存在下转化底物,其在过程中产生可检测信号例如荧光或发光信号。例如,在一些实施方案中,所述选择性标记酶包含萤光素酶例如萤火虫萤光素酶、叩头虫萤光素酶或海肾萤光素酶的氨基酸序列。萤光素酶活性可通过提供适合的激活发光底物检测,所述激活发光底物例如用于萤火虫萤光素酶的萤火虫荧光素或用于海肾萤光素酶的腔肠荧光素。存在适合底物时的萤光素酶活性的定量可通过发光法测定培养皿孔中全部细胞群的总萤光素酶活性,或者通过使用显微镜结合光子计数相机测定单个细胞或集落的萤光素酶活性。萤光素酶测定的细节包括高通量的方法公开于美国专利No.5,650,135、No.5,744,320和No.6,982,431中。其他实施方案中,所述报告酶包括修饰的β-内酰胺酶的氨基酸序列,其表达可通过分解荧光的β-内酰胺底物的比率荧光测定在活细胞中进行检测和定量,如美国专利No.5,741,657、No.6,031,094和美国专利公开No.20070184513中所述。也可见Qureshi(2007),Biotechniques,42(1):91-96综述。其他适合的报告酶包括但不限于 水解酶(Promega,Madison,Wis.,如美国专利No.7,238,842和专利公开No.20080026407和No.20080145882中所述)和β-半乳糖苷酶。
水解酶(Promega,Madison,Wis.,如美国专利No.7,238,842和专利公开No.20080026407和No.20080145882中所述)和β-半乳糖苷酶。
在一些实施方案中,所述遗传修饰的哺乳动物细胞还可经遗传修饰以表达人β2微球蛋白(GenBank登录号No.AY187687.1)。不希望囿于理论,可认为人β2微球蛋白的表达可增强遗传修饰的哺乳动物细胞中转基因HLA-G的表面表达。
在一些实施方案中,核酸表达载体是转座子载体,其包括在同源转座酶(例如piggyBAC转座酶)存在下导入宿主细胞时可促进转座子载体转座整合进入宿主基因组的转座元件,如美国专利申请No.12/728,943所述。转座子表达载体(例如PiggyBac载体)以及转座酶表达载体是可商购的,例如来自System Biosciences(Mountain View,CA)。在一些实施方案中,使用转座子载体时,没有选择性标记或报告蛋白表达盒是产生稳定转染的HLA-G修饰细胞系所必需的,由于所述载体的转染效率足够高以至不需要选择性标记。可选择地,表达载体可以是打靶载体,其可使eHLA-G转基因位点特异地整合进入宿主细胞基因组。打靶载体的设计和使用是本领域常规的,如美国专利No.5,464,764所示。
用于制备转染级核酸表达载体的方法和转染方法是公知的。见例如Sambrook andRussell(2001),"Molecular Cloning:A Laboratory Manual",第三版,(CSHL出版社)和Current Protocols in Molecular Biology,John Wiley&Sons,N.Y.(2005),9.1-9.14。高效转染方法的实例包括“核转染”,如Trompeter(2003),J Immunol.Methods,274(1-2):245-256及美国专利No.7,332,332、No.8,003,389、No.8,039,259和No.8,192,990 9中所述;使用基于脂质的转染试剂例如 (Roche)、DOTAP和LipofectamineTM(Invitrogen)的转染。
(Roche)、DOTAP和LipofectamineTM(Invitrogen)的转染。
其他实施方案中,遗传修饰的细胞例如分化的、专能的、多能的或全能干细胞系是通过重组病毒转导产生的。适合的重组病毒的实例包括但不限于反转录病毒(包括慢病毒)、腺病毒和腺相关病毒。通常,重组反转录病毒是莫洛尼鼠白血病病毒(MMLV),但是也可使用其他重组反转录病毒例如禽白血病病毒、牛白血病病毒、鼠白血病病毒(MLV)、水貂细胞病灶诱发病毒、鼠肉瘤病毒、网状内皮组织增殖病病毒、长臂猿白血病病毒、MasonPfizer猴病毒或劳斯肉瘤病毒,见例如美国专利No.6,333,195。
其他实例中,重组反转录病毒是慢病毒(例如人免疫缺陷病毒-1(HIV-1)、猴免疫缺陷病毒(SIV)或者猫免疫缺陷病毒(FIV)),见例如Johnston et al.,(1999),Journal ofVirology,73(6):4991-5000(FIV);Negre et al.,(2002),Current Topics inMicrobiology and Immunology,261:53-74(SIV);Naldini et al.,(1996),Science,272:263-267(HIV)。
重组反转录病毒可包含病毒多肽(例如逆转录病毒包膜蛋白)以辅助进入靶细胞。所述病毒多肽是本领域得到确认的,见例如美国专利No.5,449,614。所述病毒多肽可以是辅助进入来自多个物种的细胞包括原宿主物种以外的细胞的兼向性病毒多肽例如兼向性包膜蛋白。见例如id。所述病毒多肽可以是可辅助进入原宿主物种以外细胞的异嗜性病毒多肽。见例如id。在一些实施方案中,所述病毒多肽是可辅助进入原宿主物种细胞的亲嗜性病毒多肽例如亲嗜性包膜蛋白。见例如id。
病毒转导细胞可通过本领域已知的任意方法实现,例如Palsson,B.,et al.,(1995),WO95/10619;Morling,F.J.et al.,(1995),Gene Therapy,2:504-508;Gopp etal.,(2006),Methods Enzymol,420:64-81。例如,可通过旋转转染或“旋转接种”法进行转染,其包括将病毒加入细胞后尽快离心细胞。一些实例中,转染之前可通过例如超速离心将病毒浓缩。
用于转导将被遗传修饰的细胞的感染复数(m.o.i.)范围可为从约1m.o.i.到约50m.o.i.,例如约1m.o.i.、约5m.o.i.、约7.5m.o.i.、约10m.o.i.、约15m.o.i.、约20m.o.i.、约30m.o.i.、约40m.o.i.或约50m.o.i.。
可用于通过多能干细胞(例如人胚胎干细胞或诱导的人多能干细胞)直接分化产生多种细胞类型的方法是本领域公知的,如美国专利No.7,955,849、No.7,763,466、No.7,264,968及美国专利申请No.12/179,462和No.12/187,543所述。
一个示例性实施方案中,人胚胎表皮祖细胞(hEEP)来自于人多能干细胞系例如人胚胎干细胞或诱导的人多能干细胞,如下。
多能干细胞在含有补充20%敲除血清替代品的DMEM/F12(1:1)、0.1mM MEM非必需氨基酸、1mM GlutaMax、0.1mMβ-巯基乙醇(Sigma)的胚胎干细胞(ESC)生长介质中维持。ESC生长介质是通过将有丝分裂失活的小鼠胚胎成纤维细胞(MEF)(CF-1,ATCC)以5×104细胞/cm2的密度铺平板并孵育18–24小时预处理的。预处理后,加入4ng/ml bFGF并将完全预处理的介质无菌过滤。将hESC在 (BD Biosciences)涂覆的平板上每5-6天继代培养1次(1:3或1:4分流),使用1mg/ml分散酶移除细胞集落。hEEP分化多能干细胞成为K14+/p63+hEEP,通过先在6-孔板的ESC生长介质中培养多能干细胞4天,然后转移到2ml/孔的分化介质中,其包括含有1μM全反式维甲酸(Sigma)和25ng/ml BMP4的未经预处理的hESC生长介质。每日更换介质7天后,将细胞用分散酶处理、离心并重悬于以无血清介质(definedkeratinocyte-serum free medium,DSFM)中,并且以1:3的分流比种植于明胶涂覆的平板上。每隔一天更换DSFM持续3-4周。然后通过胰蛋白酶消化、离心、清洗并以10,000细胞/cm2铺板于明胶涂覆的组织培养平板(含DSFM)上继代培养细胞。为证实上皮细胞单层具有≥90%纯度并且表达K14,使用流式细胞仪测定细胞,方法依据Metallo et al(2010),Methods Mol Biol 585:83-92。为增加分离的hEEP的纯度,使用磁性活化细胞分选(MASC)与CD29抗体分选细胞。约92%的CD29 MASC分选的由eHLA-G(EF-1α)-GFP修饰的hESC分化产生的hEEP细胞培养细胞是K14(一种特异的角质形成细胞标记物)阳性的。
(BD Biosciences)涂覆的平板上每5-6天继代培养1次(1:3或1:4分流),使用1mg/ml分散酶移除细胞集落。hEEP分化多能干细胞成为K14+/p63+hEEP,通过先在6-孔板的ESC生长介质中培养多能干细胞4天,然后转移到2ml/孔的分化介质中,其包括含有1μM全反式维甲酸(Sigma)和25ng/ml BMP4的未经预处理的hESC生长介质。每日更换介质7天后,将细胞用分散酶处理、离心并重悬于以无血清介质(definedkeratinocyte-serum free medium,DSFM)中,并且以1:3的分流比种植于明胶涂覆的平板上。每隔一天更换DSFM持续3-4周。然后通过胰蛋白酶消化、离心、清洗并以10,000细胞/cm2铺板于明胶涂覆的组织培养平板(含DSFM)上继代培养细胞。为证实上皮细胞单层具有≥90%纯度并且表达K14,使用流式细胞仪测定细胞,方法依据Metallo et al(2010),Methods Mol Biol 585:83-92。为增加分离的hEEP的纯度,使用磁性活化细胞分选(MASC)与CD29抗体分选细胞。约92%的CD29 MASC分选的由eHLA-G(EF-1α)-GFP修饰的hESC分化产生的hEEP细胞培养细胞是K14(一种特异的角质形成细胞标记物)阳性的。
本发明表达转基因eHLA-G的遗传修饰的哺乳动物细胞与不表达转基因HLA-G的相应的哺乳动物细胞相比具有减低的免疫原性。例如,与不表达外源HLA-G的相应细胞类型相比免疫原性可减低至少约5%至约95%,例如与不表达外源HLA-G的相同细胞类型的细胞相比免疫原性减低约6%、7%、10%、12%、15%、20%、30%、40%、50%、65%、70%、80%、85%、90%或其他百分比例。
用于确定细胞的免疫原性的方法是本领域公知的。例如,在一些实施方案中,将HLA-G修饰的哺乳动物细胞(例如人胚胎干细胞,或由HLA-G修饰的胚胎干细胞分化的细胞,或者修饰之前已经完全分化的细胞等)或者未经修饰的哺乳动物细胞在存在异源自然杀伤细胞系(例如NK-92)条件下培养,然后通过多种标准细胞存活测定方法中的任意方法测定由NK-92诱导的对HLA-G修饰的细胞或未经修饰细胞的细胞毒性。
含有增强的HLA-G(eHLA-G)转基因的核酸
用于产生本发明HLA修饰的哺乳动物细胞的分离的核酸(例如哺乳动物质粒表达载体)含有增强的HLA-G“eHLA-G”转基因,与野生型HLA-G转基因驱动的细胞表面表达相比其可驱动HLA-G细胞表面表达和/或分泌增加。所述转基因典型地包括至少3种不同组分:启动子和5’非翻译区(5’UTR)序列、编码序列和3’非翻译区(3’UTR)序列。
在一些实施方案中,用于驱动eHLA-G转基因表达的启动子是能够驱动eHLA-G转基因在感兴趣的细胞类型中表达至少约7周至约50周,例如8周、9周、10周、12周、15周、20周、25周、30周、35周、40周、42周、45周、47周、48周或者至少约7周至约50周内的其他时间段的启动子。能够驱动所述时间段的表达的启动子对于避免多种细胞类型包括例如干细胞(例如胚胎干细胞、诱导的多能干细胞或间质干细胞)中产生的沉默是有效的。
适合的沉默抗性启动子包括但不限于中国仓鼠延伸因子-1α(CHEF-1α)启动子(见Running Deer et al(2004),Biotechnol.Prog.,20:880-889;和GenBank登录号No.AY188393.1)、鼠干细胞病毒(MSCV)启动子的M-U3/R-变体启动子(如Swindle et al(2004),J Biol Chem,279:34-41所述)、磷酸甘油酸酯激酶(PGK)启动子、人β-肌动蛋白启动子和泛素C启动子。
在一些实施方案中,用于驱动eHLA-G转基因表达的启动子是在一种或多种所需细胞类型中以高于其他细胞类型中的水平表达的。本领域技术人员应理解,例如当HLA-G修饰的干细胞分化为具体细胞类型时,可有利地选择在所述细胞类型中有活性甚至对该细胞类型是选择性的启动子。例如,对于给定的组织或细胞类型选择性启动子,所需细胞类型中的表达水平与其他细胞类型相比可以高约2倍到100倍,例如所需细胞类型中的表达水平与其他细胞类型相比可以高约3倍、4倍、5倍、10倍、20倍、25倍、30倍、40倍、50倍、70倍、80倍、90倍或其他倍数。组织和/或细胞类型选择性启动子的实例包括但不限于用于:神经元特异性烯醇酶(神经元的)、突触蛋白(神经元的)、CamKII(前脑神经元)、HB9(运动神经元)和多巴胺转运蛋白(多巴胺能神经元)、胶质细胞原纤维酸性蛋白(星形胶质细胞)、白蛋白(肝)、α-肌球蛋白重链(αMHC-心肌细胞)、神经元素3和胰腺-十二指肠同源盒1(胰腺)、角蛋白14(皮肤)和Bestrophin 1(视网膜色素上皮)的启动子。
典型地,所述eHLA-G转基因序列编码相对于人(GenBank No.NP_002118.1)或黑猩猩同义序列含有至少1个到约10个点突变的HLA-G蛋白,例如相对于上述HLA-G蛋白同义序列2、3、4、5、6、7、8、9或10个点突变。
所述人同义野生型序列(SEQ ID NO:1)如下所示:

在一些实施方案中,所述至少1个至约10个点突变可通过减低蛋白质加工和成熟过程中HLA-G在内质网中的存留来增加HLA-G蛋白在所述表达宿主细胞的细胞表面的表达水平。所述突变包括例如HLA-G“KK”基序突变。见例如Park et al(2001),Immunity,15:213-224。在一些实例中,KK基序突变包括K334A突变、K335A突变或两种置换突变。其他实例中,所述置换可使用不同的脂肪族氨基酸(例如亮氨酸)或其他类型的非碱性氨基酸实现。
在一个实例中,所编码的HLA-G蛋白具有氨基酸序列(SEQ ID NO:2),其中相对于野生型序列已导入K334A和K335A置换(下划线):

在一些实施方案中,eHLA-G转基因编码的HLA-G蛋白的氨基酸序列与SEQ ID NO:2具有至少75%到100%同一度,例如与SEQ ID NO:2的氨基酸序列具有77%、80%、82%、85%、87%、88%、90%、92%、95%、96%、97%、98%、99%或其他百分比的同一度。
本发明的eHLA-G转基因还包括含有多种可影响HLA-G转录本的表达/翻译效率的调节元件的3’非翻译区(3’UTR)。在一些实施方案中,eHLA-G转基因3’UTR序列为包括与SEQID NO:3序列具有至少75%同一度的核酸序列的核酸序列,例如与HLA-G 3’UTR序列(SEQID NO:3)具有至少77%、80%、82%、85%、87%、88%、90%、92%、94%、95%、96%、97%或其他百分比的同一度。

所述序列包含减低两个microRNA结合位点结合,并导致转基因编码的HLA-G表达增加的突变(下划线)。所述序列包括某些HLA-G等位基因的外显子8中14个碱基对序列的缺失。所述14个碱基对序列如下SEQ ID NO NO:4所示:
SEQ ID NO:4HLA3’UTR中14-bp插入序列
ATTTGTTCATGCCT
如下所示的SEQ ID NO:5对应于插入这14个碱基对序列(小写体)的SEQ ID NO:3:
SEQ ID NO:5(+14BP HLA-G 3’UTR变体)

已报道,具有包含上述14个碱基插入(见SEQ ID NO:4)的3’UTR的HLA-G等位基因可产生更加稳定的HLA-G mRNA转录本。见Rouseau et al(2003),Human Immunology,64:1005-1010。令人惊讶地,作为本公开的一些实施方案的基础,出乎意料地发现具有14个碱基对缺失(SEQ ID NO:3所示)的等位基因的表达显示出比含有这14个碱基对插入的等位基因具有更强的免疫保护能力。见图13。不希望囿于理论,据信该14bp的缺失对HLA-G表达水平的影响可以是细胞类型特异的,例如该14bp缺失可显著地增强人细胞(至少包括hESC、hEEP和人真皮成纤维细胞)中的eHLA-G表达。
在一些实施方案中,所述含eHLA-G转基因的核酸序列还包括eHLA-G表达盒侧翼的绝缘子序列。绝缘子序列能够缓解可假性影响整合的外源表达盒表达的基因组位置效应。在一些实施方案中,所用的绝缘子序列含有鸡β-球蛋白HS4核心绝缘子序列。
在一些实施方案中,所述含eHLA-G转基因的核酸还包括本文所述的选择性标记。可任选地,含eHLA-G转基因的分离的核酸还可包含本文所述的报告蛋白。
在一些实施方案中,eHLA-G修饰的细胞系是通过使用含eHLA-G表达盒但是不含选择性标记或报告蛋白表达盒的转座子载体产生的。将所述载体与转座酶表达载体一起导入需要修饰的细胞,然后进行有限稀释克隆。由于转染的细胞群被非常有效地修饰,避免了使用选择性标记或报告蛋白的需求。这是非常重要的因素,特别是对于将用于在人类患者中进行细胞治疗的细胞。通过基于转座子的稳定转染方法成功修饰细胞的百分率范围可从约0.5%到约50%,例如转染细胞的1%、2%、3%、5%、7%、8%、15%、20%、22%、30%、40%或者从约0.5%到约50%的其他比例。
在一些实施方案中,由编码2种或更多种蛋白的核酸编码的蛋白的表达是由独立的启动子驱动的。其他实施方案中,多顺反子表达盒中纳入的开放读码框之间可纳入一种或多种内部核糖体进入位点(IRES)序列。IRES序列及其使用是本领域已知的,如Martinez-Salas(1999),Curr Opin Biotechnol,10(5):458-464中所示。可选择地,多个开放读码框可通过插入口蹄疫病毒的2A肽序列(F2A)或来自其他病毒的2A-类似序列相互连接。见例如Hasegawa et al(2007),Stem Cells,25:1707-1712和Symczak et al(2004),NatBiotechnol,589-594。包含2A肽序列使得可翻译后剪切含eHLA-G和其他序列(例如报告蛋白序列或选择性标记蛋白序列)的连续多肽形成单独的蛋白。
虽然相对短的氨基酸或核酸序列之间的同一度可容易地通过目测确定,然而适合的算法分析,典型地通过计算机软件进行分析,通常用于确定较长的序列之间的同一度。当使用序列比较算法时,通常将测试序列和参照序列输入计算机,如果需要可指定子序列坐标,并且指定序列算法程序参数。然后序列比较算法可基于所指定的程序参数计算测试序列相对于参照序列的序列同一度百分数。可用于快速获得优选的比对并计算两个或更多个序列之间同一度的多种数学算法是已知的,并已被纳入多种可获得的软件程序中。所述程序的实例包括MATCH-BOX、MULTAIN、GCG、FASTA和ROBUST程序用于氨基酸序列分析,SIM、GAP、NAP、LAP2、GAP2和PIPMAKER程序用于核苷酸序列。可用于氨基酸和多核苷酸序列二者的分析的优选的软件分析程序包括ALIGN、CLUSTALW(例如version 1.6及其更新的版本)和BLAST程序(例如BLAST 2.1、BL2SEQ及其更新的版本)。
就氨基酸序列或核苷酸序列而言,“同一度”(有时称为“整体同一度”与下文的“局部同一度”相对)分别是指当两个氨基酸序列或核苷酸序列以最佳方式比对时所述两个氨基酸序列或核苷酸序列中相同氨基酸残基或核苷酸碱基的百分率。在最佳比对中,如果第一个序列中的位点在第二个对应的氨基酸或核苷酸序列中相应的位点被相同的氨基酸残基或核苷酸残基占据,则所述序列的所述残基位点是同一的。两个序列之间同一度水平(或“序列同一度百分率”)是序列共有的相同位点数相对于分析序列的大小的比率测定的(即序列同一度百分率=(相同位点数/位点总数)×100)。
本发明还包括可在低度、中度或高度严谨条件下可与来自编码SEQ ID NO:2氨基酸序列的核酸的至少100个核苷酸的探针或者SEQ ID NOS:3或SEQ ID NOS:5的核酸序列特异地杂交的核酸。
本文中,低度严谨杂交条件包括例如在2×SSC和0.1%SDS中在42℃与约40%到约70%GC含量的100个核苷酸的探针杂交。中度严谨条件包括例如在0.5×SSC和0.1%SDS中在50℃。高度严谨条件包括例如在0.2×SSC和0.1%SDS中在65℃与上述探针杂交。这些条件下,随着杂交温度的提高,可获得具有较高序列同源性的核酸。
含eHLA-G遗传修饰细胞的组合物
本发明还包括含有一种或多种HLA-G修饰哺乳动物细胞类型或使用其产生的药物组合物、局部用组合物、细胞移植物和人工组织。如文中所示,eHLA-G修饰的hESC显示出稳定且持续的HLA-G表达,甚至通过直接分化为hEEP。而且,稳定且持续的HLA-G表达可为所述遗传修饰的细胞提供减低的免疫原性和/或改进的免疫抑制。此外,还显示eHLA-G修饰的完全分化的人真皮成纤维细胞也具有可提供减低的免疫原性和/或改进的免疫抑制的稳定且持续的HLA-G表达。因此,本发明的eHLA-G构建体可用于产生通用的任意类型的供体细胞,无论来自遗传修饰的多能或专能细胞的直接分化或是完全分化的细胞的遗传修饰。
一方面,提供了用于皮肤再生或者修复的局部用组合物,其含有本发明的含eHLA-G转基因的遗传修饰的真皮成纤维细胞。另一方面,提供了用于注射的药物组合物,其含有本发明的含eHLA-G转基因的遗传修饰的真皮成纤维细胞。另一方面,提供了皮肤移植组合物,其含有本发明的含eHLA-G转基因的遗传修饰的真皮成纤维细胞。另一方面,提供了持久皮肤移植组合物,其含有本发明的含eHLA-G转基因的遗传修饰的胚胎表皮祖细胞。
另一方面,可用作人工组织的生物相容性合成支架及其产生方法是本领域已知的,并且可与本发明的HLA-G修饰细胞一起用于产生与含有不表达外源HLA-G的细胞的组织相比具有减低的免疫原性和/或改进的免疫抑制的人工组织。见例如美国专利No.7,960,166,名称为“Microfabricated compositions and processes for engineering tissuescontaining multiple cell types”。
II.方法
细胞治疗处理
由于经本发明的方法修饰可稳定表达外源HLA-G的细胞具有减低的免疫原性和/或增加的免疫抑制,这些特征使修饰的细胞可用作通用或改进的供体细胞或组织。这是由于提供给所述细胞的HLA-G介导的免疫原性减低和/或免疫抑制增加可减少或消除供体细胞和受体之间I类和II类经典人白细胞抗原(HLA)配型的需要,用于多种损伤、疾病或病症。
因此,本发明可稳定表达eHLA-G(以及可任选地表达外源人β2微球蛋白)的HLA-G修饰的细胞可用于治疗。所述治疗可以是直接处理病因,或者可选择地处理疾病或病况的影响。可将遗传修饰的细胞递送到或接近受试者的损伤位点,或者可以使细胞迁移或定位到损伤位点的方式将所述细胞导入受试者。所递送的细胞可有利地替换受伤害或损伤的细胞,并改善受试者的整体状况。在一些实例中,所递送的细胞可刺激组织再生或修复,包括皮肤再生或皮肤修复。
在多个实施方案中,将HLA-G修饰的哺乳动物细胞(例如人HLA-G修饰的细胞)给予遭受多种病况中的任意病况的受试者,所述病况包括但不限于心血管疾病、眼病(例如黄斑变性)、耳病(例如耳聋)、糖尿病、神经退行性疾病、阿尔茨海默氏病、帕金森病、多发性硬化、骨质疏松症、肝病、肾病、自身免疫疾病、关节炎、牙龈疾病、牙齿状况或增生性病症(例如肿瘤)。其他实例中,受试者正遭受或处于遭受急性健康病况例如中风、脊髓损伤、烧伤或创伤的高风险中。其他实例中,受试者正遭受组织损失例如脂肪萎缩或者年龄相关的胶原损失。其他实例中,受试者遭受非愈合性溃疡或者需要试剂以辅助愈合例如尿道下裂和尿道上裂的闭合。其他实例中,受试者需要持久或暂时的皮肤移植用于伤口愈合或皮肤替代。
一方面,本发明提供了对有需要的受试者进行细胞或组织移植的通用方法,所述方法包括将含eHLA-G修饰细胞群的细胞或组织组合物注射或移植至受试者,其中与eHLA-G修饰的细胞群相比所述受试者具有至少1种不匹配的经典I类HLA或II类HLA分子,并且其中所述eHLA-G修饰的细胞群与不含所述eHLA-G修饰的相同类型细胞相比显示出减低的免疫原性和/或改进的免疫抑制。所述减低的免疫原性和/或改进的免疫抑制可以确定,例如通过在NK-92细胞毒性测定、人源化NSG肿瘤生长测定和/或PBMC增殖测定中比较eHLA-G修饰细胞与不含所述eHLA-G修饰的相同类型对照细胞。
另一方面,本发明提供了用于有需要的受试者再生皮肤的方法,所述方法包括将eHLA-G修饰的真皮成纤维细胞群和/或eHLA-G修饰的胚胎表皮祖细胞群注射至受试者的皮肤损伤位点,其中与eHLA-G修饰的真皮成纤维细胞群和/或eHLA-G修饰的胚胎表皮祖细胞群相比所述受试者具有至少1种不匹配的经典I类HLA或II类HLA分子。
将施用至有需要的受试者的HLA-G修饰的细胞类型包括但不限于表皮祖细胞、间质干细胞、胰腺β祖细胞、胰腺β细胞、心脏祖细胞、心肌细胞、肝脏祖细胞、肝细胞、肌细胞祖细胞、肌细胞、肾细胞、成骨细胞、造血祖细胞、牙囊细胞、毛囊细胞、视网膜色素上皮细胞、神经干细胞、神经元、星形胶质细胞、少突胶质细胞或其任意组合。所述哺乳动物细胞可以是来自多个物种包括例如人、小鼠、大鼠、猴或猪中的一种。
所述治疗可以是直接处理病因,或者可选择地所述治疗可以是处理疾病或病况的影响。可将HLA-G修饰的细胞递送到或接近受试者的损伤位点,或者可以使细胞迁移或定位到损伤位点的方式将所述细胞导入受试者。所递送的细胞可有利地替换受伤害或损伤的细胞,并可改善受试者的整体状况。在一些实例中,所递送的细胞可刺激组织再生或修复。
所递送的细胞可以是由HLA-G修饰的多能(或全能)干细胞分化产生的细胞。所递送的细胞还可以是由多能的HLA-G修饰的细胞分化产生的专能干细胞。
施用至受试者治疗的数量可不同。将HLA-G修饰的和/或分化的细胞导入受试者可以是一次完成的,但在某些情况下,此类治疗可刺激有限时间段的改善并且需要持续重复的一系列治疗。其他情况下,可能需要多次施用所述细胞才能产生效果。本领域技术人员应理解,具体的治疗方案依赖于所治疗的疾病或病况以及疾病的阶段和具体受试者的参数。
HLA-G修饰的细胞可通过下述任意途径导入受试者:肠胃外、静脉内、动脉内、肌内、皮下、经皮、气管内、腹膜内或进入脊髓液。
HLA-G修饰的细胞可分化为细胞,然后递送到遭受多种疾病或病症的受试者。
胰岛细胞(或朗格罕氏岛的原代细胞)可移植到遭受糖尿病(例如1型糖尿病)的受试者,见例如Burns et al.,(2006)Curr.Stem Cell Res.Ther.,2:255-266。因此,在一些实施方案中,将来自HLA-G修饰细胞的胰腺β细胞移植入遭受糖尿病(例如1型糖尿病)的受试者。
其他实例中,将来自HLA-G修饰细胞的肝细胞或肝脏干细胞移植入遭受肝病例如肝炎、肝硬化或肝功能衰竭的受试者。
退行性心脏病如缺血性心肌病、传导疾病和先天性缺陷可受益于干细胞治疗。见例如Janssens et al.,(2006),Lancet,367:113-121。
来自HLA-G修饰细胞的造血细胞或造血干细胞(HSC)可移植入遭受血液癌症或者其他血液或免疫障碍的受试者。可能通过造血细胞或HSC处理的血液癌症的实例包括:急性淋巴细胞白血病、急性粒细胞白血病、慢性髓性白血病(CML)、霍奇金病、多发性骨髓瘤和非霍奇金淋巴瘤。通常,遭受所述疾病的受试者必须经受放射治疗和/或化学疗法以杀死快速分裂的血细胞。将来自HLA-G修饰细胞的HSC导入所述受试者中可帮助重新填充耗竭的细胞储库。
遭受神经系统疾病或病症的受试者可特别地受益于HLA-G修饰细胞的治疗,尤其是当血脑屏障可能已受损时。在一些方案中,可使HLA-G修饰的细胞分化为神经干细胞或神经元,然后将其移植到损伤位点用于治疗神经系统病况例如阿尔茨海默氏病、帕金森病、多发性硬化、脑梗死、脊髓损伤或其他中枢神经系统障碍,见例如Morizane et al.,(2008),Cell Tissue Res.,331(1):323-326;Coutts and Keirstead(2008),Exp.Neurol.,209(2):368-377;Goswami and Rao(2007),Drugs,10(10):713-719。
为治疗帕金森病,可使HLA-G修饰的细胞分化为多巴胺活性神经元(dopamine-acting neuron),然后植入帕金森病受试者的纹状体。为治疗多发性硬化,可使神经干细胞分化为少突胶质细胞或少突胶质细胞的祖细胞,然后将其递送到遭受MS的受试者。
为治疗任意神经性疾病或病症,一个成功的方案是将神经干细胞导入受试者。例如,为治疗阿尔茨海默氏病、脑梗死或脊髓损伤,可使HLA-G修饰的细胞分化为神经干细胞,然后将其移植入损伤位点。还可将HLA-G修饰的细胞加工以对可使其迁移入损伤位点的信号反应用于脑和脊髓修复,例如Chen et al.,(2007),Stem Cell Rev.,3(4):280-288。
可任选地,用于细胞治疗方法的HLA-G修饰的细胞还可表达本文所述的报告蛋白。在一些实施方案中,使用的报告蛋白可有利于体内检测(例如成像)所导入的细胞。例如,所述细胞可表达可发射远红光的荧光蛋白例如Katushka,其长的激发和发射波长非常适于在组织中成像。Katushka是可商购的,商品名为“TurboFP635”(Evrogen,Moscow,Russia)。
实施例
下述具体实施例应理解为仅用于说明,不以任何方式限定本发明其余部分的任何内容。除非另有说明,应理解本领域技术人员可基于本文所述利用本发明的全部内容。文中引用的所有出版物在此以引用方式全文纳入本文。当参考内容是以URL或其他类似的标识或地址时,应理解此类标识可改变并且网络信息可能会存在或删除,但是可通过网络搜索发现等价的信息。对其参考证明所述信息是可获得并可公开传播的。
实施例1与癌细胞的免疫耐受相关的基因表达模式的鉴定
首先筛选人软组织癌症阵列,搜索随着癌症演进为转移状态而显示出表达增加的候选免疫耐受基因,该表达增加可能是由于逃逸免疫监测机制。数据显示,成功转移和此前显示参与免疫耐受的多个基因的表达水平之间有很强的正相关。为评价MSC是否可诱导免疫耐受和异体移植接受,通过RT-PCR交叉筛选MSC以测定这些候选基因和多种HLA抗原的表达水平。表1显示,除表达CD200、CD47和吲哚胺2,3-二氧化酶(IDO)以外,第1次传代的MSC还可表达Ia类HLA、HLA-G和II类HLA标记。研究发现,MSC群可中等水平地表达HLA-G,虽然其低于具有积极转移能力的Jeg-3肿瘤系中的表达。
表1

由于MSC在体内的免疫抑制效应似乎是短暂的,将MSC连续传代并检测所选基因表达水平的变化。通过3次传代,MSC中的天然HLA-G表达缺失,虽然其他候选标记未发生改变。Jeg-3细胞中的HLA-G表达未改变。然后基于细胞表面的HLA-G表达通过FACS分离MSC,发现0.5-3%的MSC在细胞表面表达HLA-G。为评价HLA-G+MSC体内逃逸供体排斥的能力,将1-3×105个细胞注入具有免疫能力小鼠的尾静脉。从眶后静脉丛采取血液样本(200μl),并使用抗人I类HLA特异的mAb进行FACS分选。表2显示,移植1周后,HLA-G+MSC(和Jeg-3细胞)显示出分别超过HSC和Jurkat细胞(都是HLA-G-)24倍和270倍的生存优势。2周后,HLA-G+MSC的优势增加到27倍和311倍。HLA-G-MSC的存活率与HSC相同,表明MSC的HLA-G+亚群相对于未分选的MSC在体内环境中显示出增强的耐受效应。事实上,事后分析表明使用Jeg-3细胞移植12周后具有免疫能力小鼠的肺部有显著的肿瘤,但是使用G+MSC的没有(数据未示出)。
表2

然后,将修饰的HLA-G构建体在HF(人成纤维细胞)和K562细胞中过表达,并通过人NK细胞测试对裂解的防护(表3)。NK介导的细胞毒性在HLA-G+HF中下降75%,并且在HLA-G+K562细胞中几乎消失。这与检测到HLA-G+Jeg-3的防护相一致,其可通过与抗-HLA-G中和抗体(87G)共孵育被逆转,但同种型对照不能。这些数据表明,对NK杀伤的防护是HLA-G-依赖的。
表3

此前的研究显示,通过10周的培养,突变的MSCV启动子M-U3/R可避免沉默压力。本发明的研究表明,M-U3/R可在持续培养1年后抵御沉默,这优于野生型启动子,野生型启动子在培养4-6周后即被沉默。此外,3’UTR的突变或缺失可增加HLA-G表面表达,ER滞留基序(retrieval motif)的突变也如此。较高的HLA-G表面表达与细胞毒性裂解负相关,这增加了使用优化的基因递送构建体的重要性。
实施例2人胚胎干细胞的培养及分化为人表皮祖细胞(hEEP)
除非另有说明,否则所有组织培养试剂均来自Life Technologies。ESC生长介质含有补充20%敲除血清替代品的DMEM/F12(1:1)、0.1mM MEM非必需氨基酸、1mM GlutaMax、0.1mMβ-巯基乙醇(Sigma)。ESC生长介质是通过将有丝分裂失活的小鼠胚胎成纤维细胞(MEF)(CF-1,ATCC)以5×104细胞/cm2铺平板并孵育18–24小时预处理的。预处理后,加入4ng/ml bFGF并将完全预处理的介质无菌过滤。将hESC在Matrigel涂覆的平板上每5-6天继代培养1次(1:3或1:4分流),使用1mg/ml分散酶移除细胞集落。依据上述Metallo et al的方法产生K14+/p63+hEEP。简言之,将hESC在6-孔板中培养4天,然后用2ml/孔的分化介质处理,其包括含有1μM全反式维甲酸(Sigma)和25ng/ml BMP4的未经预处理的hESC生长介质。每日更换介质7天后,将细胞用分散酶处理、离心并重悬于以无血清介质(DSFM)中,并且以1:3的分流比接种于明胶涂覆的平板上。每隔一天更换DSFM,持续3-4周。然后通过胰蛋白酶消化、离心、清洗并以10,000细胞/cm2铺板于明胶涂覆的组织培养平板(含DSFM)上继代培养细胞。在以无血清介质中14天后,通过显微镜观察表皮分化的早期征兆,其特征在于形成表皮片层结构。培养4周后,表皮片层中的细胞显示出具有立方形态的典型的表皮分化表型。
从细胞中分离总RNA及反转录酶反应见Zhao et al(2010),Tissue Eng Part A,16(2):725-733中所述。具体的PCR扩增在Hybaid Omnigene thermal cycler(Bio-rad,Hercules,CA)中进行,使用图12所示的感兴趣基因的特异引物。PCR条件包括35个循环(94℃30s,65℃1min和72℃1min)和最后72℃延伸10min。每种PCR产物取10μl通过溴化乙锭凝胶电泳检测。图12数据表明,表皮分化标记物K14、Tap63和ΔNp63在分化过程中逐渐增强。本文未示出的数据中,进行了K14和另外的表皮标记物p63、CD29和CD49f的免疫荧光检测。免疫荧光数据显示,分化的eHLA-G(EF-1α)-GFP hEEP是K14、p63、CD29和CD49f蛋白表达阳性的。
为证实上皮细胞单层具有≥90%纯度并且表达K14,依据上述Metallo et al(2010)方法使用流式细胞仪测定细胞,并使用BD FACS Canto II分析。eHLA-G转基因表达(eHLA-G(EF1-α)-GFP-hESC)对hESC分化为EP(表皮祖细胞)的影响通过比较野生型、G--hEEP和G+-hEEP的K14阳性程度进行评估。为增加分离的hEEP的纯度,使用磁性活化细胞分选(MASC)通过CD29抗体分选细胞。约92%的CD29 MASC分选的由eHLA-G(EF-1α)-GFP修饰的hESC分化产生的hEEP细胞培养细胞是K14(一种特异的角质形成细胞标记物)阳性的。纯化的hEEP显示出同源角质形成细胞的形态,如相差显微镜检测结果所示(见图16)。
通过流式细胞术验证了HLA-G转基因在分化的hEEP中的稳定度。与不含外源HLA-G的对照细胞(只含GFP的hEEP)和野生型hEEP相比,分化的eHLA-G(EF-1a)-GFP-hEEP(超过90%的细胞)中HLA-G总的表达和表面表达都是很强的(见图17)。只选择可产生与野生型细胞类似分化能力的克隆用于进一步研究。
实施例3 eHLA-G构建设计及在hESC中的稳定表达
设计了一种新的HLA-G构建体,其组合了多种修饰:1)HLA-G的ER滞留基序突变(K334A/K335A);和2)HLA-G的3’UTR microRNA结合位点突变。由于病毒基因递送系统存在很大的调节困难,使用了基于转座子的PiggyBac系统,其是显示出在H1 hESC中可达到90%转染效率的非病毒方法(Lacoste et al(2009),Cell Stem Cell,5:332-342)。该系统需要含转座子的供体质粒(图1)和表达转座酶的辅助质粒(图2)。为产生辅助质粒,定制合成(GeneArt)了ePiggyBac密码子人源化的转座酶cDNA,然后将其克隆到pBluescript(Stratagene)中PGK启动子下游且SV40多腺苷酸化信号序列(pA)上游。为产生eHLA-G表达盒,比较了包括M-U3/R启动子、MSCV启动子和中国仓鼠EF1α(CHEF-1α)启动子的多种启动子,其序列如下文SEQ ID NO:6所示。
(SEQ ID NO:6)-CHEF-1α启动子的一个实施方案


为产生供体质粒,定制合成了T53C/C136T突变的长度为313bp的5’末端重复序列(TR)和长度为235bp的3’TR(如上文Lacoste所述),并将其分别克隆到下述表达盒的上游和下游:eHLA-G、250bp的鸡β-球蛋白HS4核心绝缘子序列(Recillas-Targa et al(2002),PNAS USA,99:6883-6888)、EGFP和pA。HS4用于阻止抑制性染色质蔓延至整合的构建体中。EGFP表达受磷酸甘油酸酯激酶(PGK)启动子驱动(见图1)。HS4元件的序列如下文SEQ IDNO:7所示。
(SEQ ID NO:7)HSF元件的一个实施方案

供体质粒的空载体(HLA-G-)除不含eHLA-G构建体外与eHLA-G供体质粒一致(图1)。基因转移之前,将hESC用10μM Y-27632处理1hr,其为可大大减低细胞分离诱导的凋亡并增加克隆效率的ROCK抑制剂(Watanabe et al(2007),Nat Biotechnol,25:681-686)。使hESC在0.25%胰蛋白酶-EDTA中37℃5分钟进行分离,在含Y-27632的经预处理的mTeSR介质中洗涤并重悬于核转染溶液L(Amaxa)中。每1.5×105个细胞中加入3μg辅助质粒和6μg转座子供体质粒,并将程序设定为B-016进行核转染。然后以每个6cm平皿2×105个细胞将hESC铺板于含Y-27632的CM中用于克隆选择。24小时后,将培养介质换为只有CM,此后每天更换一次。通过荧光显微镜选择同时表达tdT/eHLA-G最高的克隆,而非通过抗生素抗性或流式细胞术选择,由于hESC中常出现转基因沉默并且只有部分转单基因细胞可产生有标记的细胞系(Braam et al(2008),Nat Methods,5:389-392)。
实施例4 ePiggyBac基因递送系统评估
按每个6cm平皿2×103个细胞,将转染的细胞铺板于含Y-27632的CM中24小时,然后换为只有CM,用于测定eHLA-G转染效率。此后每天更换一次介质,共7天,并通过活细胞染色和免疫荧光显微镜检查评估细胞集落,如实施例5所述。对于每个克隆,计数3个高倍视野并计算报告蛋白+/eHLA-G+hESC的百分数。实验结果见图3-4。通过质粒拯救策略确定eHLA-G插入位点,如上文Lacoste et al中所述。简言之,从转基因hESC克隆中分离基因组DNA并使用BamHI/BglII/NotI消化,使用T4 DNA连接酶在低浓度16℃自连过夜,用100%异丙醇沉淀,70%乙醇洗涤,然后转化入DH10B E.coli中并用氨苄青霉素选择。使用SplinkTA PCR测定eHLA-G的拷贝数。每20次传代检测标准G带用于评估核型稳定度。每10次传代使用流式细胞仪评估eHLA-G和报告蛋白的基因沉默。分离及测定之前,使用10μM Y-27632处理hESC 1小时。在0.25%胰蛋白酶-EDTA中37℃5分钟使细胞分离,在含Y-27632的CM中洗涤,将细胞重悬于含0.1%BSA和0.5mM EDTA的冰冷PBS中,然后使用BD FACS Canto II流式细胞仪(Becton Dickinson)分析。如图8和图9所示,传代第16次时,几乎未检测到EGFP或eHLA-G表达沉默。每传代20次评估多能性,通过免疫细胞化学检测:1)多能性标记物Oct3/4、SSEA-4、Sox2和Nanog(图5和图6),2)报告蛋白+/eHLA-G+胚状体形成(图7)和3)分化的转基因hESC中的内胚层标记物Gata 6、中胚层标记物肌肉肌动蛋白和外胚层标记物神经丝重链(数据未示出),均使用Leica CTR6500荧光显微镜检测。除非另有说明,否则所有抗体均来自Abcam。
实施例5 eHLA-G及其他HLA蛋白的细胞表面定位
通过活细胞染色检测转基因及野生型hESC和hEEP中细胞表面的Ia类HLA、HLA-E、eHLA-G和II类HLA表达。简言之,收集细胞并在冷的PBS中洗涤,在含10%山羊血清和3%BSA的PBS中使用相应的1°mAb在4℃染色60min,洗涤,用1%多聚甲醛固定10min,然后用与FITC共轭的山羊抗小鼠IgG在4℃染色30min。对照等分w使用同种型配对IgG染色用于评估与靶细胞的非特异性结合。首先测试每种抗体(HLA-G的抗体MEMG/9、HLA-E的抗体MEM-E/08、Ia类HLA的抗体Bu8和II类HLA的抗体HKB1(Abbiotec,San Diego,CA))的多个稀释以确定可获得仅有特异性结合的最适条件。染色后,将细胞涂布于载玻片上,使其风干,然后使用含DAPI的抗淬灭介质封片(Vector Laboratories)。使用Leica CTR6500荧光显微镜立即观察载玻片。如图5和图10所示,野生型及eHLA-G修饰的人ES细胞中HLA-A、HLA-B、HLA-C、HLA-E、HLA-DP、DQ、DR和β-微球蛋白的表达类似,而eHLA-G修饰的人ES细胞中HLA-G的表达水平高出约7倍。
实施例6 eHLA-G表达对NK-92细胞诱导的细胞毒性影响评估
将表达eHLA-G的hESC培养并分化为hEEP,如实施例2中所述。将NK-92细胞(CRL-2407,美国典型培养物保藏中心,Manassas,VA)在补充12.5%FBS、12.5%马血清、0.2mM肌醇、0.1mMβ-巯基乙醇、0.02mM叶酸和100IU/ml重组IL-2(Sigma)的最低基础培养基α介质(α-MEM,Invitrogen)中,5%CO2加湿培养箱中37℃培养。
使用CytoTox96非放射性细胞毒性测定试剂盒(Promega,Madison,WI)依据说明书的方案测定细胞毒性。简言之,将效应细胞与5×103个靶细胞以NK-92(效应子或“E”)与hESC或hEEP(靶或“T”)E:T的多个细胞比例在U-底96孔板(Costar,Cambridge,MA)中混合。在5%CO2加湿培养箱中37℃培养4h后,收集50μl上清用于确定LDH释放。靶细胞自发释放和最大释放的LDH及效应细胞自发释放的LDH是通过将这些细胞在单独的介质中孵育测定的。每种测定三个重复,并将结果表述为裂解百分率%。特异性裂解百分率按如下方式确定:(实验的释放-效应子自发释放-靶自发释放/靶最大释放-靶自发释放)×100。所有实验中,自发释放<10%最大释放。
如图11所示,E:T比例为1:10时,杀死表达eHLA-G的hESC相对于只表达GFP的野生型细胞减少超过50%。E:T比例为1:30时,杀死表达eHLA-G的hESC减少约75%。两种E:T比例下,野生型hESC(只表达GFP)以适当比例被杀死,如图所示。此外,还显示K562细胞中3’UTR(Del 14bp)HLA-G等位基因的表达可导致NK细胞诱导的细胞毒性相对于表达(Ins 14bp)HLA-G等位基因的K562细胞和未修饰的K562细胞中的结果是减低的。见图13。
图11的结果已在另外的NK细胞毒性实验中进行重复和验证。如图18所示,杀死eHLA-G(EF-1a)-GFP-hESC与只含GFP转基因(不含HLA-G转基因)的对照hESC相比减少超过100%。所述数据表明,hESC中HLA-G转基因表达可产生免疫抑制和/或减低的免疫原性特征。
NK细胞毒性实验也在由hESC分化产生的hEEP中进行。如图19所示,杀死由eHLA(EF-1α)-GFP-hESC分化产生的eHLA(EF-1α)-GFP-hEEP与对照hEEP相比减低超过100%(约3倍)。所述数据显示,eHLA(EF-1α)-GFP转基因在整个分化过程中是稳定且持续的,并且HLA-G表达可在分化的细胞中产生免疫抑制和/或减低的免疫原性特征。
实施例7通过异体移植入人源化小鼠检测e-HLA-G+细胞在体内的免疫原性
具有人外周血淋巴细胞的人源化小鼠模型(Hu-PBL-NSG)显示出可在移植后1-2周内排斥不匹配的人胰岛(islets),但是野生型免疫缺陷NSG小鼠未显示(King et al(2008),Clin Immunol,126:303-314)。虽然在4-5周内可产生移植物抗宿主病(GVHD),但是直到符合安乐死标准才可检测移植物存活。仅存在低水平的GVHD使我们可扩展观察窗。NSG小鼠(雌性6周龄)购自Jackson实验室并且依据动物保护和利用委员会的指导以及实验动物保护和利用指南(实验动物资源研究所、国家研究理事会、国家科学院)的建议进行操作。功能性人源化NSG小鼠是通过静脉注射约20×106人PBMC进入NSG小鼠产生的,依据(Pearson et al(2008),Curr Protoc Immunol,Ch.15:Unit15.21和King et al见上文)。约第4周,通过使用EDTA-涂覆的毛细管(Drummond Scientific)和EDTA-处理的1.5ml管(Eppendorf)从麻醉小鼠的眶后静脉丛收集血液来验证移植物植入。然后将细胞处理后通过FACS分析检测人CD45阳性,参照上文中King et al。第4后,血液中的人CD45+细胞水平达到0.1%的即认为成功的移植物植入并可进行异体排斥研究。
通常使用hESC和hEEP培养系统将细胞暴露至免疫原性动物诱导药例如唾液酸Neu5Gc(Martin et al(2005),Nat Med,11:228-232)。因此,可加入去污步骤,近来的研究显示其可显著减低Neu5Gc水平(Heiskanen et al(2007),Stem Cells,25:197-202)。hESC可使用下述两种方法去除Neu5Gc污染:1)在人基质涂层板上在添加bFGF、LiCl、GABA和哌啶酸的TeSR1基质中培养,参照方法Ludwig et al(2006),Nat Biotechnol,24:185-187,和2)使用热失活AB Rh-血型的人血清替换KSR(Heiskanen et al(2007),Stem Cells,25:197-202)。可通过与抗-Neu5Gc mAb孵育然后流式细胞仪测定来评估两种方法培养的hESC。可采取第2种方法,由于Ludwig et al的方法可能需要hESC适应。hEEP按照生产商的说明在缺少动物诱导药的DFSM中培养。
使用HLA-G修饰的细胞进行异体移植的目的是评价是否HLA-G表达可减低免疫原性,如通过肿瘤大小增加显示。移植前,将预期的注射位点刮毛以方便临床观察。全程遵照动物准则,包括适当使用麻醉。将5×106个转基因eHLA-G+和HLA-G-hESC重悬于100μl含印度墨水以标记注射位点的适合的介质中。这有利于观察可能荧光不足的组织学测定。将细胞皮下注入5只3月龄的Hu-PBL-NSG小鼠的胸部乳腺脂肪垫。异体移植的结果见图20和图21。“G0”hESC是对照ESC,其不含eHLA-G转基因而只含GFP。“mG1(#1)”和“mG1(#2)”是两种不同的eHLA-G(EF-1α)-GFP核转染hESC克隆。G0、mG1(#1)和mG1(#2)肿瘤如图20所示,对其测量并称重。G0 hESC形成的肿瘤体积为126.9立方毫米,重32毫克。mG1(#1)hESC形成的肿瘤体积为748.4立方毫米,重318毫克。mG1(#2)hESC形成的肿瘤体积为1116.7立方毫米,重675毫克。
图21示出了将hESC异体移植至5只人源化NSG小鼠形成的肿瘤的平均结果。数据表明,HLA-G核转染的hESC(“mG1”)形成的肿瘤比野生型hESC(“G0”)移植入人源化NSG小鼠形成的肿瘤大得多(体积超过3倍)且重得多(重量超过2倍)。因此,所述数据表明,eHLA-G转基因表达可提供减低的免疫原性和/或增加的免疫抑制。这支持了将本发明的eHLA-G转基因构建体普遍应用于将任意需要的细胞类型修饰成为通用或优良的异源供体用于治疗、移植、组织修复、细胞和组织替换等。
上述使用人源化NSG小鼠的异体移植实验可使用可活跃地增殖或者可被诱导增殖的eHLA-G修饰的任意细胞类型实施。
此外,可使用光谱体内成像系统(Caliper,Mountain View,CA)使用620nm发射滤光片和2.5秒曝光时间检测人源化小鼠中的异体移植物。620nm或更大波长下,GFP的自发荧光消除,并且可检测到皮肤表面以下2.5cm深度的信号(Shaner et al(2004),NatBiotechnol,22:1567-1572)。每周对小鼠纵向成像并称重。如果在4周时间内未检测到信号,则将小鼠处死并使用荧光显微镜对所注射的胸部乳房脂肪垫进行组织学评估以检测所用的荧光报告蛋白。所有实例中,小鼠处死后,用苏木精和伊红将组织染色以测定畸胎瘤形成和免疫排斥。还通过免疫组织化学检测人β2微球蛋白(PBMC、hESC、hEEP)和CD45(PBMC),后者用作异体移植的免疫细胞浸润的补充测定。切片是由对所述实验条件不了解的病理学专家解读的。
实施例8 eHLA-G+-hEEP和HLA-G--hEEP的致肿瘤性测定
通过注射转基因G+和非转基因G--hEEP测定了eHLA-G对hEEP致肿瘤性的影响,除细胞是被移植入非人源化NSG小鼠以外所用方法与实施例8所述类似。使用纵向荧光活体动物成像检测了共14只小鼠,每月测定体重至9个月初或者符合安乐死标准。然后将小鼠处死,并收集注射的胸部乳腺至4%中性缓冲的多聚甲醛中。将固定的组织转移到70%乙醇中并用石蜡包埋。切片用苏木精和伊红染色,并经处理用于荧光报告蛋白显微镜检查。如果所述组织切片呈现为报告蛋白阴性的,则使用人β2微球蛋白和CD45特异的mAb将其染色,后者用于确定所述注射位点非PBMC的存在。
独立平均值的统计学显著性设定为p<0.05。2个样本平均值的比较使用学生t检验确定。3个或更多个平均值的比较使用单因素或双因素方差分析和Bonferroni事后检验。所有方差的度量以平均值标准误的形式呈现。使用线性回归比较体外和体内数据的相关性,以确定是否前者在本研究所用的Hu-PBL-NSG小鼠模型中对人异体移植排斥具有预测价值。例如,体外异常增殖的百分率增加可对用于体内阶段的PBMC匹配对进行回归(输出为移植后4周报告蛋白荧光的水平)。此处成功的结果是高R2值和负斜率系数,表明eHLA-G表达导致移植物植入增加。这样的解释取决于选择用于体外和体内研究的克隆中eHLA-G表达和报告蛋白阳性之间的高相关。
实施例9 eHLA-G稳定转染进入完全分化的成纤维细胞的免疫抑制和免疫原性测定
通过核转染将eHLA-G(EF-1α)-GFP转基因和对照构建体转染进入人新生儿真皮成纤维细胞(一种已经完全分化的细胞类型)。人新生儿真皮成纤维细胞(HFD)购自ATCC并在含10%FBS和1%PS(Invitrogen)的Iscove改良的Dulbecco培养基(IMDM)(ATCC)中培养。当细胞达到80%汇集时,通过与0.25%胰蛋白酶-EDTA(Invitrogen)在37℃孵育3分钟收获细胞。将细胞计数,将0.5×106个细胞离心并重悬于人真皮成纤维细胞核转染溶液(产品目录号No.VPD-1001,Lonza,Walkersville,MD)中。将辅助质粒和转座子质粒加入所述细胞悬液中,并设定程序U-020进行核转染,参照生产商的操作手册(Lonza)。然后将细胞铺板于6孔平板中并在加湿的37℃/5%CO2中孵育24h。24h后,使用1μg/ml嘌呤霉素(Sigma)选择稳定转染的细胞7天。将稳定GFP阳性的细胞在500ng/ml嘌呤霉素中维持培养,并通过流式细胞仪检测HLA-G和GFP表达。将稳定转染子保持在培养物中用于下述的外周血单核细胞(PBMC)增殖测定和NK-92细胞毒性测定。
PBMC增殖测定对用eHLA-G(EF-1α)-GFP转基因(“HFD-m1-GFP”细胞)或单独的GFP对照构建体(“HFD-G0-GFP”细胞)稳定转染的人真皮成纤维细胞测定其抑制PBMC增殖的能力。使用丝裂霉素C(10μg/ml 2.5h)使HFD-G0-GFP和-mG1-GFP细胞失活,在96孔板中以3.0×103/孔接种,并使其粘附24h。存在6μg/ml PHA条件下将1×105PBMC一式三份加入相应的HFD细胞中。单独的HFD-G0-GFP和–mG1-GFP细胞用于MTT-OD校正。(“MTT”是浅黄色底物试剂,可被活细胞裂解产生深蓝色formazan产物。该过程需要活性线粒体,并且即使是刚死亡的细胞也不能够裂解显著量的MTT。因此,MTT可提供可被用于增殖或细胞毒性测定的比色测定。不含PHA的PBMC和含6μg/ml PHA的PBMC作为对照。共培养物孵育3天并使用MTT试剂评价PBMC增殖,使用下述公式:%PBMC增殖=[(HFD/PBMC/PHA的OD570–HFD的OD570)/(PBMC/PHA的OD570)]×10。如图22所示,HFD-mG1-GFP克隆“mG1-R1”抑制PBMC增殖超过对照和其他克隆,表明外源HLA-G表达可为分化的细胞例如成纤维细胞提供免疫抑制和/或减低的免疫原性。
NK-92细胞毒性测定基本上参照上文所述进行测定。简言之,2.5×103个靶细胞(即HFD-m1-GFP细胞或者HFD-G0-GFP细胞)与NK-92细胞以3:1、10:1、30:1的E:T比例在CTL介质中共孵育7小时。K562-WT细胞作为NK-92细胞毒性的阳性对照。使用CytoTox96细胞毒性测定试剂盒测定细胞毒性。特异性裂解的百分数如下确定:%特异性裂解=[(实验中LDH释放-效应子自发释放-靶自发释放)/(靶最大释放-靶自发释放)]×100。如下表4中所示,HFD-mG1-GFP克隆“mG1-R1”和“mG1-#1”抑制NK-92细胞毒性超过对照和其他克隆,表明外源HLA-G表达可为分化的细胞例如成纤维细胞提供免疫抑制和/或减低的免疫原性。
表4:NK-92对HFD-G0和HFD-G1靶细胞的细胞毒性







Claims (57)
1.一种遗传修饰的哺乳动物细胞,其与不含所述遗传修饰的哺乳动物细胞相比具有减低的免疫原性和/或改进的免疫抑制,其中:
(i)所述遗传修饰的哺乳动物细胞含有外源核酸,所述外源核酸包括:(a)编码由SEQID NO:2的氨基酸序列构成的HLA-G蛋白的核酸序列;和(b)由SEQ ID NO:3的序列构成的3’非翻译区(UTR);
(ii)编码的HLA-G蛋白由所述遗传修饰的哺乳动物细胞表达至少7周;和
(iii)所述遗传修饰的哺乳动物细胞不是胚胎干细胞或多能干细胞。
2.如权利要求1所述的遗传修饰的哺乳动物细胞,其中所述HLA-G蛋白表达至少20周。
3.如权利要求2所述的遗传修饰的哺乳动物细胞,其中所述HLA-G蛋白表达至少50周。
4.如权利要求1所述的遗传修饰的哺乳动物细胞,还包含延伸因子-1α(EF-1α)启动子,其中编码HLA-G蛋白的核酸序列可操作地连接到EF-1α启动子。
5.如权利要求4所述的遗传修饰的哺乳动物细胞,其中所述EF-1α启动子包含SEQ IDNO:6序列。
6.如权利要求1所述的遗传修饰的哺乳动物细胞,其中表达的HLA-G存在于所述遗传修饰的哺乳动物细胞的细胞表面上。
7.如权利要求1所述的遗传修饰的哺乳动物细胞,其中所述遗传修饰的哺乳动物细胞是人类细胞。
8.如权利要求1所述的遗传修饰的哺乳动物细胞,其中所述遗传修饰的哺乳动物细胞是表皮祖细胞、胰腺祖细胞、造血干细胞、角质形成细胞、成纤维细胞、肝细胞、间质干细胞、心肌细胞、神经干细胞、神经元或星形胶质细胞。
9.如权利要求1所述的遗传修饰的哺乳动物细胞,其中所述遗传修饰的哺乳动物细胞是非胚胎干细胞。
10.如权利要求1所述的遗传修饰的哺乳动物细胞,其中所述遗传修饰的哺乳动物细胞是祖细胞。
11.如权利要求1所述的遗传修饰的哺乳动物细胞,其中所述遗传修饰的哺乳动物细胞是由非胚胎干细胞或祖细胞体外分化产生的细胞。
12.如权利要求1所述的遗传修饰的哺乳动物细胞,其中所述遗传修饰的哺乳动物细胞是完全分化的细胞。
13.如权利要求1所述的遗传修饰的哺乳动物细胞,其中所述遗传修饰的哺乳动物细胞是人间质干细胞或人胚胎表皮祖细胞。
14.如权利要求1所述的遗传修饰的哺乳动物细胞,其中所述遗传修饰的哺乳动物细胞是人真皮成纤维细胞。
15.如权利要求1所述的遗传修饰的哺乳动物细胞,其中与不含所述遗传修饰的哺乳动物细胞相比,所述遗传修饰的哺乳动物细胞的减低的免疫原性和/或改进的免疫抑制是通过下述任意方法确定的:(1)与不含所述遗传修饰的哺乳动物细胞相比,所述遗传修饰的哺乳动物细胞的NK-92细胞毒性减低,(2)与不含所述遗传修饰的哺乳动物细胞相比,所述遗传修饰的哺乳动物细胞的体外外周血单核细胞增殖减低,和(3)在人源化NSG小鼠中,与不含所述遗传修饰的哺乳动物细胞相比,由所述遗传修饰的哺乳动物细胞引起的肿瘤形成的大小和重量增加。
16.一种人工组织,其含有权利要求1所述的遗传修饰的哺乳动物细胞。
17.一种皮肤移植、修复或再生组合物,其含有权利要求9-14任一项所述的遗传修饰的哺乳动物细胞。
18.如权利要求1所述的遗传修饰的哺乳动物细胞,其中所述外源核酸是表达载体。
19.如权利要求18所述的遗传修饰的哺乳动物细胞,其中所述哺乳动物表达载体是转座子载体。
20.如权利要求18所述的遗传修饰的哺乳动物细胞,其中所述载体还含有编码报告蛋白的核酸序列。
21.一种分离的核酸,其包括:(i)编码由SEQ ID NO:2构成的氨基酸序列的第一核酸序列;和(ii)由SEQ ID NO:3构成的第二核酸序列。
22.一种哺乳动物表达载体,其包含权利要求21所述的分离的核酸和可操作地连接到所述第一核酸序列的启动子,其中所述启动子在干细胞或者由干细胞分化产生的细胞中不是沉默的。
23.如权利要求22所述的哺乳动物表达载体,其中所述启动子包含中国仓鼠EF-1α启动子的核酸序列。
24.如权利要求22所述的哺乳动物表达载体,还包含编码报告蛋白的第三核酸序列。
25.如权利要求22所述的哺乳动物表达载体,其中所述哺乳动物表达载体是转座子载体。
26.一种遗传修饰的哺乳动物细胞,其包含权利要求23的哺乳动物表达载体,其中所述遗传修饰的哺乳动物细胞是非胚胎干细胞或多能干细胞。
27.含权利要求1所述的遗传修饰的哺乳动物细胞的群的细胞或组织组合物在制备用于治疗需要细胞或组织修复的受试者的药物中的用途,其中与所述遗传修饰的哺乳动物细胞的群相比,所述受试者具有至少1种不匹配的经典I类HLA或II类HLA分子,其中将所述遗传修饰的哺乳动物细胞注射、植入或移植至受试者,并且其中所述遗传修饰的哺乳动物细胞的群与不含所述遗传修饰的相同类型细胞相比显示出减低的免疫原性和/或改进的免疫抑制。
28.如权利要求27所述的遗传修饰的哺乳动物细胞的用途,其中所述至少1种不匹配的经典I类HLA或II类HLA分子选自HLA-A、HLA-B、HLA-C、HLA-DP、HLA-DQ和HLA-DR。
29.如权利要求27所述的遗传修饰的哺乳动物细胞的用途,其中所述受试者与所述遗传修饰的哺乳动物细胞的HLA-A、HLA-B、HLA-C、HLA-DP、HLA-DQ和HLA-DR的不匹配。
30.如权利要求27所述的遗传修饰的哺乳动物细胞的用途,其中所述减低的免疫原性和/或改进的免疫抑制通过NK-92细胞毒性测定、人源化NSG肿瘤生长测定和/或外周血单核细胞增殖测定来确定。
31.如权利要求27所述的遗传修饰的哺乳动物细胞的用途,其中所述遗传修饰的哺乳动物细胞的群包含人真皮成纤维细胞群。
32.如权利要求27所述的遗传修饰的哺乳动物细胞的用途,其中所述遗传修饰的哺乳动物细胞的群包含人表皮祖细胞群。
33.如权利要求27所述的遗传修饰的哺乳动物细胞的用途,其中所述遗传修饰的哺乳动物细胞的群包含人胚胎干细胞群。
34.如权利要求27所述的遗传修饰的哺乳动物细胞的用途,其中所述遗传修饰的哺乳动物细胞的群包含人间质干细胞群。
35.如权利要求27所述的遗传修饰的哺乳动物细胞的用途,其中所述遗传修饰的哺乳动物细胞的群包含体外分化的遗传修饰的人胚胎干细胞。
36.如权利要求27所述的遗传修饰的哺乳动物细胞的用途,其中所述遗传修饰的哺乳动物细胞的群不被受试者的免疫系统排斥至少2、4、6、8、10、12、14、16、18、20、24、36、48或52周。
37.一种生产遗传修饰的哺乳动物细胞的方法,所述遗传修饰的哺乳动物细胞与不含所述遗传修饰的哺乳动物细胞相比具有减低的免疫原性和/或改进的免疫抑制,所述方法包括:
用外源核酸遗传修饰哺乳动物细胞,所述外源核酸包括:
a.编码由SEQ ID NO:2的氨基酸序列构成的HLA-G蛋白的核酸序列;和
b.由SEQ ID NO:3的核苷酸序列构成的3’非翻译区(UTR);
其中,所述遗传修饰的哺乳细胞的减低的免疫原性和/或改进的免疫抑制通过以下任一方法测定:
i.与不含所述遗传修饰的哺乳动物细胞相比,所述遗传修饰的哺乳动物细胞的自然杀伤细胞NK-92细胞毒性减低,
ii.与不含所述遗传修饰的哺乳动物细胞相比,所述遗传修饰的哺乳动物细胞的体外外周血单核细胞增殖减少,或
iii.在人源化NOD scidγNSG小鼠中,与不含所述遗传修饰的哺乳动物细胞相比,所述遗传修饰的哺乳细胞的肿瘤形成的大小和重量增加。
38.权利要求37的方法,其中所述遗传修饰的哺乳动物细胞表达HLA-G蛋白至少7周。
39.权利要求37的方法,其中所述遗传修饰的哺乳动物细胞表达HLA-G蛋白至少20周。
40.权利要求37的方法,其中所述遗传修饰的哺乳动物细胞表达HLA-G蛋白至少50周。
41.权利要求37的方法,其中所述遗传修饰的哺乳动物细胞在所述遗传修饰的哺乳动物细胞的细胞表面上表达HLA-G蛋白。
42.权利要求37的方法,其中所述遗传修饰的哺乳动物细胞是人细胞。
43.权利要求37的方法,其中所述遗传修饰的哺乳动物细胞选自表皮祖细胞、胰腺祖细胞、造血干细胞、角质形成细胞、成纤维细胞、肝细胞、间质干细胞、心肌细胞、神经干细胞、神经元或星形胶质细胞。
44.如权利要求37所述的方法,其中所述遗传修饰的哺乳动物细胞是干细胞。
45.如权利要求37所述的方法,其中所述遗传修饰的哺乳动物细胞是祖细胞。
46.如权利要求37所述的方法,其中所述遗传修饰的哺乳动物细胞是由干细胞或祖细胞体外分化产生的细胞。
47.如权利要求37所述的方法,其中所述遗传修饰的哺乳动物细胞是完全分化的细胞。
48.权利要求37的方法,其中所述遗传修饰的哺乳动物细胞选自人胚胎干细胞、人间质干细胞、人胚胎表皮祖细胞和人真皮成纤维细胞。
49.权利要求37的方法,其中所述外源核酸序列还包含延伸因子-1α(EF-1α)启动子,其中编码HLA-G蛋白的核酸序列与EF-1α启动子可操作地连接。
50.权利要求49的方法,其中EF-1α启动子包含SEQ ID NO:6的序列。
51.如权利要求37所述的方法,其中所述外源核酸是表达载体。
52.权利要求51的方法,其中所述表达载体是转座子载体。
53.权利要求51的方法,其中所述表达载体还包含编码报告蛋白的核酸序列。
54.一种生产人造组织、皮肤移植物、皮肤修复物或皮肤再生组合物的方法,所述组合物包含通过权利要求37的方法生产的遗传修饰的哺乳动物细胞。
55.权利要求37的方法,其中用外源核酸遗传修饰哺乳动物细胞包括用外源核酸瞬时转染哺乳动物细胞。
56.权利要求37的方法,其中用外源核酸遗传修饰哺乳动物细胞包括用外源核酸稳定转染哺乳动物细胞。
57.权利要求37的方法,其中使用抗生素抗性或流式细胞术来选择遗传修饰的哺乳动物细胞。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202110063720.2A CN113151179A (zh) | 2012-07-31 | 2013-07-30 | Hla g修饰的细胞及方法 |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261677739P | 2012-07-31 | 2012-07-31 | |
| US61/677,739 | 2012-07-31 | ||
| PCT/US2013/052767 WO2014022423A2 (en) | 2012-07-31 | 2013-07-30 | Hla g-modified cells and methods |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202110063720.2A Division CN113151179A (zh) | 2012-07-31 | 2013-07-30 | Hla g修饰的细胞及方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN105612176A CN105612176A (zh) | 2016-05-25 |
| CN105612176B true CN105612176B (zh) | 2021-01-19 |
Family
ID=50028654
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201380051169.3A Active CN105612176B (zh) | 2012-07-31 | 2013-07-30 | Hla g修饰的细胞及方法 |
| CN202110063720.2A Pending CN113151179A (zh) | 2012-07-31 | 2013-07-30 | Hla g修饰的细胞及方法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202110063720.2A Pending CN113151179A (zh) | 2012-07-31 | 2013-07-30 | Hla g修饰的细胞及方法 |
Country Status (10)
| Country | Link |
|---|---|
| US (4) | US9714280B2 (zh) |
| EP (4) | EP3805261A1 (zh) |
| JP (4) | JP6297559B2 (zh) |
| KR (4) | KR101923632B1 (zh) |
| CN (2) | CN105612176B (zh) |
| AU (2) | AU2013296564C1 (zh) |
| CA (1) | CA2882028C (zh) |
| ES (2) | ES2837487T3 (zh) |
| SG (1) | SG11201500799YA (zh) |
| WO (1) | WO2014022423A2 (zh) |
Families Citing this family (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140134195A1 (en) | 2011-04-20 | 2014-05-15 | University Of Washington Through Its Center For Commercialization | Beta-2 microglobulin-deficient cells |
| KR101923632B1 (ko) * | 2012-07-31 | 2018-12-03 | 이스케이프 테라퓨틱스, 인코퍼레이티드 | Hla g-변형된 세포 및 방법 |
| US10927384B2 (en) | 2014-04-09 | 2021-02-23 | Dna Twopointo Inc. | DNA vectors, transposons and transposases for eukaryotic genome modification |
| EP3670660A1 (en) | 2014-04-09 | 2020-06-24 | Dna Twopointo Inc. | Enhanced nucleic acid constructs for eukaryotic gene expression |
| WO2016086132A1 (en) * | 2014-11-26 | 2016-06-02 | Accelerated Biosciences Corp. | Induced hepatocytes and uses thereof |
| US9888673B2 (en) | 2014-12-10 | 2018-02-13 | Regents Of The University Of Minnesota | Genetically modified cells, tissues, and organs for treating disease |
| CA3001312A1 (en) | 2015-10-08 | 2017-04-13 | Dna2.0, Inc. | Dna vectors, transposons and transposases for eukaryotic genome modification |
| KR20190017985A (ko) * | 2016-06-14 | 2019-02-20 | 리전츠 오브 더 유니버스티 오브 미네소타 | 질환을 치료하기 위한 유전적으로 변형된 세포, 조직, 및 장기 |
| WO2018022683A1 (en) | 2016-07-26 | 2018-02-01 | The University Of North Carolina At Chapel Hill | Vector-mediated immune tolerance in the eye |
| CN110268050A (zh) * | 2017-01-10 | 2019-09-20 | 综合医院公司 | 经修饰的t细胞和它们的使用方法 |
| CN110869494A (zh) * | 2017-06-12 | 2020-03-06 | 西奈卫生系统公司 | 无需全身性免疫遏制的同种异体移植物耐受 |
| CN111886027A (zh) * | 2017-12-28 | 2020-11-03 | 磨石肿瘤生物技术公司 | 靶向共同抗原的抗原结合蛋白 |
| US10883084B2 (en) | 2018-10-05 | 2021-01-05 | Xenotherapeutics, Inc. | Personalized cells, tissues, and organs for transplantation from a humanized, bespoke, designated-pathogen free, (non-human) donor and methods and products relating to same |
| WO2020120666A1 (en) | 2018-12-14 | 2020-06-18 | Promethera Biosciences S.A./N.V. | Liver progenitor cells expressing hla-g, and method for obtaining these cells compositions comprising said cells and their use |
| JP7313898B2 (ja) | 2019-05-13 | 2023-07-25 | キヤノン株式会社 | ポリゴンミラー、画像形成装置、光偏光器および光走査装置 |
| WO2021162731A1 (en) * | 2020-02-10 | 2021-08-19 | Rubius Therapeutics, Inc. | Engineered erythroid cells including hla-g polypeptides and methods of use thereof |
| WO2021241658A1 (ja) * | 2020-05-26 | 2021-12-02 | 株式会社ヘリオス | 低免疫原性細胞 |
| WO2022046675A1 (en) * | 2020-08-23 | 2022-03-03 | Applied Stemcell, Inc. | Hla-f-modified cells and methods |
| US11459372B2 (en) | 2020-11-30 | 2022-10-04 | Crispr Therapeutics Ag | Gene-edited natural killer cells |
| CN113234664A (zh) * | 2021-05-11 | 2021-08-10 | 澳门大学 | 一种胰腺祖细胞的制备方法及其应用 |
| AU2022371442A1 (en) * | 2021-10-21 | 2024-04-18 | Vertex Pharmaceuticals Incorporated | Hypoimmune cells |
Family Cites Families (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0432216A1 (en) | 1988-09-01 | 1991-06-19 | Whitehead Institute For Biomedical Research | Recombinant retroviruses with amphotropic and ecotropic host ranges |
| US5464764A (en) | 1989-08-22 | 1995-11-07 | University Of Utah Research Foundation | Positive-negative selection methods and vectors |
| US5534423A (en) | 1993-10-08 | 1996-07-09 | Regents Of The University Of Michigan | Methods of increasing rates of infection by directing motion of vectors |
| US6686198B1 (en) | 1993-10-14 | 2004-02-03 | President And Fellows Of Harvard College | Method of inducing and maintaining neuronal cells |
| US6013517A (en) | 1994-05-09 | 2000-01-11 | Chiron Corporation | Crossless retroviral vectors |
| US5650135A (en) | 1994-07-01 | 1997-07-22 | The Board Of Trustees Of The Leland Stanford Junior University | Non-invasive localization of a light-emitting conjugate in a mammal |
| US5843780A (en) | 1995-01-20 | 1998-12-01 | Wisconsin Alumni Research Foundation | Primate embryonic stem cells |
| US5741657A (en) | 1995-03-20 | 1998-04-21 | The Regents Of The University Of California | Fluorogenic substrates for β-lactamase and methods of use |
| US5744320A (en) | 1995-06-07 | 1998-04-28 | Promega Corporation | Quenching reagents and assays for enzyme-mediated luminescence |
| US6982431B2 (en) | 1998-08-31 | 2006-01-03 | Molecular Devices Corporation | Sample analysis systems |
| US6031094A (en) | 1998-07-23 | 2000-02-29 | The Regents Of The University Of California | Beta-lactam substrates and uses thereof |
| US7005252B1 (en) | 2000-03-09 | 2006-02-28 | Wisconsin Alumni Research Foundation | Serum free cultivation of primate embryonic stem cells |
| IL158514A0 (en) | 2001-04-23 | 2004-05-12 | Amaxa Gmbh | Buffer solution for electroporation and a method comprising the use of the same |
| CA2487058C (en) | 2002-05-17 | 2015-04-14 | Mount Sinai School Of Medicine Of New York University | Mesoderm and definitive endoderm cell populations |
| WO2004072232A2 (en) | 2003-01-31 | 2004-08-26 | Promega Corporation | Covalent tethering of functional groups to proteins |
| US7960166B2 (en) | 2003-05-21 | 2011-06-14 | The General Hospital Corporation | Microfabricated compositions and processes for engineering tissues containing multiple cell types |
| ES2354160T3 (es) * | 2003-10-21 | 2011-03-10 | Merck Serono Sa | Secuencia de adn mínima que actúa como un aislante de cromatina, y su uso en la expresión de proteínas. |
| US8673634B2 (en) | 2003-11-13 | 2014-03-18 | Massachusetts Eye & Ear Infirmary | Method for the treatment of hearing loss |
| CA2581422C (en) | 2004-11-08 | 2012-05-22 | Chromagenics B.V. | Selection of host cells expressing protein at high levels |
| US20070036773A1 (en) * | 2005-08-09 | 2007-02-15 | City Of Hope | Generation and application of universal T cells for B-ALL |
| CN101330935B (zh) * | 2005-10-21 | 2013-11-13 | 细胞研究私人有限公司 | 自脐带羊膜分离和培养干/祖细胞及其分化的细胞的应用 |
| US8278104B2 (en) | 2005-12-13 | 2012-10-02 | Kyoto University | Induced pluripotent stem cells produced with Oct3/4, Klf4 and Sox2 |
| EP4223769A3 (en) | 2005-12-13 | 2023-11-01 | Kyoto University | Nuclear reprogramming factor |
| WO2007087539A2 (en) * | 2006-01-24 | 2007-08-02 | The University Of Chicago | Snp binding site for micrornas in hla-g |
| WO2007091078A2 (en) * | 2006-02-10 | 2007-08-16 | Axordia Limited | Polypeptide conjugate comprising hla-g and uses thereof, stem cells transfected with hla-g |
| EP2087107A2 (en) | 2006-10-30 | 2009-08-12 | Promega Corporation | Mutant hydrolase proteins with enhanced kinetics and functional expression |
| US8647871B2 (en) * | 2007-03-30 | 2014-02-11 | Escape Therapeutics, Inc. | Endogenous expression of HLA-G and/or HLA-E by mesenchymal cells |
| US8592211B2 (en) | 2009-03-20 | 2013-11-26 | The Rockefeller University | Enhanced PiggyBac transposon and methods for transposon mutagenesis |
| US8048675B1 (en) | 2010-05-12 | 2011-11-01 | Ipierian, Inc. | Integration-free human induced pluripotent stem cells from blood |
| KR101923632B1 (ko) | 2012-07-31 | 2018-12-03 | 이스케이프 테라퓨틱스, 인코퍼레이티드 | Hla g-변형된 세포 및 방법 |
-
2013
- 2013-07-30 KR KR1020157005165A patent/KR101923632B1/ko active IP Right Grant
- 2013-07-30 ES ES18211100T patent/ES2837487T3/es active Active
- 2013-07-30 EP EP20196775.9A patent/EP3805261A1/en not_active Withdrawn
- 2013-07-30 AU AU2013296564A patent/AU2013296564C1/en active Active
- 2013-07-30 ES ES13825511T patent/ES2716577T3/es active Active
- 2013-07-30 CN CN201380051169.3A patent/CN105612176B/zh active Active
- 2013-07-30 KR KR1020197036122A patent/KR102184947B1/ko active IP Right Grant
- 2013-07-30 KR KR1020207033804A patent/KR102366081B1/ko active IP Right Grant
- 2013-07-30 KR KR1020187034048A patent/KR102055438B1/ko active IP Right Grant
- 2013-07-30 EP EP18211100.5A patent/EP3483178B1/en active Active
- 2013-07-30 CN CN202110063720.2A patent/CN113151179A/zh active Pending
- 2013-07-30 CA CA2882028A patent/CA2882028C/en active Active
- 2013-07-30 JP JP2015525519A patent/JP6297559B2/ja active Active
- 2013-07-30 SG SG11201500799YA patent/SG11201500799YA/en unknown
- 2013-07-30 EP EP13825511.2A patent/EP2880054B1/en active Active
- 2013-07-30 EP EP21209423.9A patent/EP4015530A1/en active Pending
- 2013-07-30 WO PCT/US2013/052767 patent/WO2014022423A2/en active Application Filing
-
2015
- 2015-01-28 US US14/608,004 patent/US9714280B2/en active Active
-
2017
- 2017-06-16 US US15/625,964 patent/US10502738B2/en active Active
-
2018
- 2018-02-21 JP JP2018028816A patent/JP6502544B2/ja active Active
- 2018-08-23 AU AU2018220087A patent/AU2018220087B2/en active Active
-
2019
- 2019-03-20 JP JP2019052699A patent/JP7294838B2/ja active Active
- 2019-10-22 US US16/659,918 patent/US20200041512A1/en active Pending
-
2020
- 2020-03-27 US US16/832,321 patent/US20200309776A1/en active Pending
-
2021
- 2021-04-14 JP JP2021068195A patent/JP7379410B2/ja active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7379410B2 (ja) | Hla g改変された細胞および方法 | |
| US20240091274A1 (en) | TRANSPLANTED CELL PROTECTION VIA Fc SEQUESTRATION | |
| WO2021146222A1 (en) | Modification of blood type antigens | |
| CN113316637A (zh) | 依靠人工反式激活物的选择 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| TA01 | Transfer of patent application right |
Effective date of registration: 20191224 Address after: Delaware, USA Applicant after: AgX treatment Co., Ltd Address before: California, USA Applicant before: ESCAPE THERAPEUTICS INC |
|
| TA01 | Transfer of patent application right | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |