JP6297559B2 - Hlag改変された細胞および方法 - Google Patents
Hlag改変された細胞および方法 Download PDFInfo
- Publication number
- JP6297559B2 JP6297559B2 JP2015525519A JP2015525519A JP6297559B2 JP 6297559 B2 JP6297559 B2 JP 6297559B2 JP 2015525519 A JP2015525519 A JP 2015525519A JP 2015525519 A JP2015525519 A JP 2015525519A JP 6297559 B2 JP6297559 B2 JP 6297559B2
- Authority
- JP
- Japan
- Prior art keywords
- cells
- hla
- cell
- genetically modified
- nucleic acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 102100028967 HLA class I histocompatibility antigen, alpha chain G Human genes 0.000 title claims description 178
- 238000000034 method Methods 0.000 title description 42
- 101100395318 Homo sapiens HLA-G gene Proteins 0.000 title 1
- 210000004027 cell Anatomy 0.000 claims description 354
- 108010024164 HLA-G Antigens Proteins 0.000 claims description 177
- 150000007523 nucleic acids Chemical group 0.000 claims description 75
- 210000004962 mammalian cell Anatomy 0.000 claims description 71
- 210000000130 stem cell Anatomy 0.000 claims description 62
- 230000002829 reductive effect Effects 0.000 claims description 47
- 108091028043 Nucleic acid sequence Proteins 0.000 claims description 43
- 108090000623 proteins and genes Proteins 0.000 claims description 40
- 102000010292 Peptide Elongation Factor 1 Human genes 0.000 claims description 39
- 108010077524 Peptide Elongation Factor 1 Proteins 0.000 claims description 39
- 239000013604 expression vector Substances 0.000 claims description 38
- 230000005847 immunogenicity Effects 0.000 claims description 35
- 108020004707 nucleic acids Proteins 0.000 claims description 32
- 102000039446 nucleic acids Human genes 0.000 claims description 32
- 230000001506 immunosuppresive effect Effects 0.000 claims description 31
- 102000004169 proteins and genes Human genes 0.000 claims description 30
- 210000001519 tissue Anatomy 0.000 claims description 29
- 239000013598 vector Substances 0.000 claims description 29
- 206010062016 Immunosuppression Diseases 0.000 claims description 25
- 230000004069 differentiation Effects 0.000 claims description 25
- 241000699670 Mus sp. Species 0.000 claims description 24
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 claims description 24
- 239000000203 mixture Substances 0.000 claims description 23
- 108020005345 3' Untranslated Regions Proteins 0.000 claims description 21
- 125000003275 alpha amino acid group Chemical group 0.000 claims description 19
- 210000001671 embryonic stem cell Anatomy 0.000 claims description 19
- 231100000135 cytotoxicity Toxicity 0.000 claims description 16
- 230000003013 cytotoxicity Effects 0.000 claims description 16
- 210000002950 fibroblast Anatomy 0.000 claims description 16
- 230000001965 increasing effect Effects 0.000 claims description 16
- 108091036066 Three prime untranslated region Proteins 0.000 claims description 13
- 238000012239 gene modification Methods 0.000 claims description 13
- 238000000338 in vitro Methods 0.000 claims description 13
- 230000005017 genetic modification Effects 0.000 claims description 12
- 235000013617 genetically modified food Nutrition 0.000 claims description 12
- 210000003958 hematopoietic stem cell Anatomy 0.000 claims description 11
- 210000003491 skin Anatomy 0.000 claims description 11
- 231100000263 cytotoxicity test Toxicity 0.000 claims description 10
- 238000002784 cytotoxicity assay Methods 0.000 claims description 9
- 241000699802 Cricetulus griseus Species 0.000 claims description 8
- 210000002510 keratinocyte Anatomy 0.000 claims description 8
- 210000001178 neural stem cell Anatomy 0.000 claims description 8
- 210000002569 neuron Anatomy 0.000 claims description 8
- 125000003729 nucleotide group Chemical group 0.000 claims description 8
- 210000002901 mesenchymal stem cell Anatomy 0.000 claims description 7
- 239000002773 nucleotide Substances 0.000 claims description 7
- 102100028972 HLA class I histocompatibility antigen, A alpha chain Human genes 0.000 claims description 6
- 108010075704 HLA-A Antigens Proteins 0.000 claims description 6
- 108010010378 HLA-DP Antigens Proteins 0.000 claims description 6
- 102000015789 HLA-DP Antigens Human genes 0.000 claims description 6
- 238000003556 assay Methods 0.000 claims description 6
- 238000001516 cell proliferation assay Methods 0.000 claims description 6
- 102100028976 HLA class I histocompatibility antigen, B alpha chain Human genes 0.000 claims description 5
- 102100028971 HLA class I histocompatibility antigen, C alpha chain Human genes 0.000 claims description 5
- 108010058607 HLA-B Antigens Proteins 0.000 claims description 5
- 108010052199 HLA-C Antigens Proteins 0.000 claims description 5
- 108010062347 HLA-DQ Antigens Proteins 0.000 claims description 5
- 108010058597 HLA-DR Antigens Proteins 0.000 claims description 5
- 102000006354 HLA-DR Antigens Human genes 0.000 claims description 5
- 108091023045 Untranslated Region Proteins 0.000 claims description 5
- 210000001130 astrocyte Anatomy 0.000 claims description 5
- 230000002500 effect on skin Effects 0.000 claims description 5
- 210000000987 immune system Anatomy 0.000 claims description 5
- 210000004413 cardiac myocyte Anatomy 0.000 claims description 4
- 210000003494 hepatocyte Anatomy 0.000 claims description 4
- 230000009467 reduction Effects 0.000 claims description 4
- 230000008439 repair process Effects 0.000 claims description 4
- 210000005260 human cell Anatomy 0.000 claims description 3
- 230000004614 tumor growth Effects 0.000 claims description 3
- 230000004663 cell proliferation Effects 0.000 claims description 2
- 238000011069 regeneration method Methods 0.000 claims description 2
- 230000005740 tumor formation Effects 0.000 claims description 2
- 230000008629 immune suppression Effects 0.000 claims 1
- 230000008929 regeneration Effects 0.000 claims 1
- 230000014509 gene expression Effects 0.000 description 106
- 239000005090 green fluorescent protein Substances 0.000 description 60
- 108700019146 Transgenes Proteins 0.000 description 59
- 230000035772 mutation Effects 0.000 description 35
- 108010043121 Green Fluorescent Proteins Proteins 0.000 description 28
- 102000004144 Green Fluorescent Proteins Human genes 0.000 description 28
- 206010028980 Neoplasm Diseases 0.000 description 21
- 230000000694 effects Effects 0.000 description 19
- 210000004271 bone marrow stromal cell Anatomy 0.000 description 16
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 15
- 102100040445 Keratin, type I cytoskeletal 14 Human genes 0.000 description 14
- 239000002609 medium Substances 0.000 description 14
- 238000011282 treatment Methods 0.000 description 14
- 101710183391 Keratin, type I cytoskeletal 14 Proteins 0.000 description 13
- 238000000684 flow cytometry Methods 0.000 description 13
- 239000003550 marker Substances 0.000 description 13
- 210000001778 pluripotent stem cell Anatomy 0.000 description 13
- 210000001626 skin fibroblast Anatomy 0.000 description 13
- 238000002054 transplantation Methods 0.000 description 13
- 230000027455 binding Effects 0.000 description 12
- 201000010099 disease Diseases 0.000 description 12
- 239000013612 plasmid Substances 0.000 description 11
- 108090000765 processed proteins & peptides Proteins 0.000 description 11
- 241000700605 Viruses Species 0.000 description 10
- 208000027418 Wounds and injury Diseases 0.000 description 10
- 238000002474 experimental method Methods 0.000 description 10
- 210000004263 induced pluripotent stem cell Anatomy 0.000 description 10
- 230000002269 spontaneous effect Effects 0.000 description 10
- 230000017423 tissue regeneration Effects 0.000 description 10
- 230000003612 virological effect Effects 0.000 description 10
- 230000000735 allogeneic effect Effects 0.000 description 9
- 150000001413 amino acids Chemical class 0.000 description 9
- 230000006378 damage Effects 0.000 description 9
- 108010048367 enhanced green fluorescent protein Proteins 0.000 description 9
- 238000001727 in vivo Methods 0.000 description 9
- 229920001184 polypeptide Polymers 0.000 description 9
- 102000004196 processed proteins & peptides Human genes 0.000 description 9
- 230000009261 transgenic effect Effects 0.000 description 9
- 101000935043 Homo sapiens Integrin beta-1 Proteins 0.000 description 8
- 102100025304 Integrin beta-1 Human genes 0.000 description 8
- 108700011259 MicroRNAs Proteins 0.000 description 8
- 102000011755 Phosphoglycerate Kinase Human genes 0.000 description 8
- 101001099217 Thermotoga maritima (strain ATCC 43589 / DSM 3109 / JCM 10099 / NBRC 100826 / MSB8) Triosephosphate isomerase Proteins 0.000 description 8
- 230000030279 gene silencing Effects 0.000 description 8
- 239000001963 growth medium Substances 0.000 description 8
- 208000014674 injury Diseases 0.000 description 8
- 239000002679 microRNA Substances 0.000 description 8
- 230000004048 modification Effects 0.000 description 8
- 238000012986 modification Methods 0.000 description 8
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 description 8
- 238000002560 therapeutic procedure Methods 0.000 description 8
- 238000001890 transfection Methods 0.000 description 8
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 7
- 108700028369 Alleles Proteins 0.000 description 7
- 241001465754 Metazoa Species 0.000 description 7
- 102000008579 Transposases Human genes 0.000 description 7
- 108010020764 Transposases Proteins 0.000 description 7
- 239000000427 antigen Substances 0.000 description 7
- 108091007433 antigens Proteins 0.000 description 7
- 102000036639 antigens Human genes 0.000 description 7
- 230000008859 change Effects 0.000 description 7
- 230000009089 cytolysis Effects 0.000 description 7
- 230000037430 deletion Effects 0.000 description 7
- 238000012217 deletion Methods 0.000 description 7
- 238000009826 distribution Methods 0.000 description 7
- 239000012636 effector Substances 0.000 description 7
- 238000010166 immunofluorescence Methods 0.000 description 7
- 238000003780 insertion Methods 0.000 description 7
- 230000037431 insertion Effects 0.000 description 7
- 230000014759 maintenance of location Effects 0.000 description 7
- 210000002237 B-cell of pancreatic islet Anatomy 0.000 description 6
- 101000937544 Homo sapiens Beta-2-microglobulin Proteins 0.000 description 6
- 108060001084 Luciferase Proteins 0.000 description 6
- 239000005089 Luciferase Substances 0.000 description 6
- 102100027881 Tumor protein 63 Human genes 0.000 description 6
- 238000013459 approach Methods 0.000 description 6
- 238000001473 dynamic force microscopy Methods 0.000 description 6
- 210000002242 embryoid body Anatomy 0.000 description 6
- 102000047279 human B2M Human genes 0.000 description 6
- 238000009396 hybridization Methods 0.000 description 6
- 239000012212 insulator Substances 0.000 description 6
- 230000002147 killing effect Effects 0.000 description 6
- 108020004999 messenger RNA Proteins 0.000 description 6
- 210000000822 natural killer cell Anatomy 0.000 description 6
- 230000035755 proliferation Effects 0.000 description 6
- 241000894007 species Species 0.000 description 6
- 239000000758 substrate Substances 0.000 description 6
- 101150024418 HLA-G gene Proteins 0.000 description 5
- 241000699666 Mus <mouse, genus> Species 0.000 description 5
- 208000018737 Parkinson disease Diseases 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 210000004369 blood Anatomy 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- 201000011510 cancer Diseases 0.000 description 5
- 238000002659 cell therapy Methods 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- 206010012601 diabetes mellitus Diseases 0.000 description 5
- 238000011156 evaluation Methods 0.000 description 5
- 238000011577 humanized mouse model Methods 0.000 description 5
- 230000002163 immunogen Effects 0.000 description 5
- 230000037361 pathway Effects 0.000 description 5
- 238000004064 recycling Methods 0.000 description 5
- 210000002966 serum Anatomy 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- 241001430294 unidentified retrovirus Species 0.000 description 5
- 208000024827 Alzheimer disease Diseases 0.000 description 4
- 241000282693 Cercopithecidae Species 0.000 description 4
- 101710087047 Cytoskeleton-associated protein 4 Proteins 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 4
- 108010010803 Gelatin Proteins 0.000 description 4
- WZUVPPKBWHMQCE-UHFFFAOYSA-N Haematoxylin Chemical compound C12=CC(O)=C(O)C=C2CC2(O)C1C1=CC=C(O)C(O)=C1OC2 WZUVPPKBWHMQCE-UHFFFAOYSA-N 0.000 description 4
- 101000994365 Homo sapiens Integrin alpha-6 Proteins 0.000 description 4
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 4
- 102100032816 Integrin alpha-6 Human genes 0.000 description 4
- 208000012902 Nervous system disease Diseases 0.000 description 4
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 4
- 101710140697 Tumor protein 63 Proteins 0.000 description 4
- 108010007093 dispase Proteins 0.000 description 4
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 4
- 239000008273 gelatin Substances 0.000 description 4
- 229920000159 gelatin Polymers 0.000 description 4
- 235000019322 gelatine Nutrition 0.000 description 4
- 235000011852 gelatine desserts Nutrition 0.000 description 4
- 230000006058 immune tolerance Effects 0.000 description 4
- 230000006872 improvement Effects 0.000 description 4
- 208000015181 infectious disease Diseases 0.000 description 4
- CDAISMWEOUEBRE-UHFFFAOYSA-N inositol Chemical compound OC1C(O)C(O)C(O)C(O)C1O CDAISMWEOUEBRE-UHFFFAOYSA-N 0.000 description 4
- 230000000670 limiting effect Effects 0.000 description 4
- 230000001404 mediated effect Effects 0.000 description 4
- 201000006417 multiple sclerosis Diseases 0.000 description 4
- 210000000107 myocyte Anatomy 0.000 description 4
- 229950010131 puromycin Drugs 0.000 description 4
- 238000003757 reverse transcription PCR Methods 0.000 description 4
- 230000036560 skin regeneration Effects 0.000 description 4
- 208000020431 spinal cord injury Diseases 0.000 description 4
- 238000010186 staining Methods 0.000 description 4
- 230000002459 sustained effect Effects 0.000 description 4
- 230000008685 targeting Effects 0.000 description 4
- FWBHETKCLVMNFS-UHFFFAOYSA-N 4',6-Diamino-2-phenylindol Chemical compound C1=CC(C(=N)N)=CC=C1C1=CC2=CC=C(C(N)=N)C=C2N1 FWBHETKCLVMNFS-UHFFFAOYSA-N 0.000 description 3
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 3
- 241000713800 Feline immunodeficiency virus Species 0.000 description 3
- 102100024785 Fibroblast growth factor 2 Human genes 0.000 description 3
- 108090000379 Fibroblast growth factor 2 Proteins 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- 208000009329 Graft vs Host Disease Diseases 0.000 description 3
- 102100028970 HLA class I histocompatibility antigen, alpha chain E Human genes 0.000 description 3
- 101000986085 Homo sapiens HLA class I histocompatibility antigen, alpha chain E Proteins 0.000 description 3
- 108020004684 Internal Ribosome Entry Sites Proteins 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- FDJKUWYYUZCUJX-AJKRCSPLSA-N N-glycoloyl-beta-neuraminic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@@H]1O[C@](O)(C(O)=O)C[C@H](O)[C@H]1NC(=O)CO FDJKUWYYUZCUJX-AJKRCSPLSA-N 0.000 description 3
- FDJKUWYYUZCUJX-UHFFFAOYSA-N N-glycolyl-beta-neuraminic acid Natural products OCC(O)C(O)C1OC(O)(C(O)=O)CC(O)C1NC(=O)CO FDJKUWYYUZCUJX-UHFFFAOYSA-N 0.000 description 3
- 241000700159 Rattus Species 0.000 description 3
- 241000713311 Simian immunodeficiency virus Species 0.000 description 3
- 206010043276 Teratoma Diseases 0.000 description 3
- 238000011316 allogeneic transplantation Methods 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 230000001472 cytotoxic effect Effects 0.000 description 3
- 208000035475 disorder Diseases 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- 238000001476 gene delivery Methods 0.000 description 3
- 208000024908 graft versus host disease Diseases 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 230000010354 integration Effects 0.000 description 3
- 208000019423 liver disease Diseases 0.000 description 3
- 230000004770 neurodegeneration Effects 0.000 description 3
- 208000015122 neurodegenerative disease Diseases 0.000 description 3
- 210000004248 oligodendroglia Anatomy 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 230000004224 protection Effects 0.000 description 3
- 238000011084 recovery Methods 0.000 description 3
- 239000000523 sample Substances 0.000 description 3
- 239000012679 serum free medium Substances 0.000 description 3
- 239000000243 solution Substances 0.000 description 3
- 238000003153 stable transfection Methods 0.000 description 3
- 238000007920 subcutaneous administration Methods 0.000 description 3
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 3
- HJCMDXDYPOUFDY-WHFBIAKZSA-N Ala-Gln Chemical compound C[C@H](N)C(=O)N[C@H](C(O)=O)CCC(N)=O HJCMDXDYPOUFDY-WHFBIAKZSA-N 0.000 description 2
- 208000023275 Autoimmune disease Diseases 0.000 description 2
- 102100024505 Bone morphogenetic protein 4 Human genes 0.000 description 2
- 241000283707 Capra Species 0.000 description 2
- 208000005623 Carcinogenesis Diseases 0.000 description 2
- 208000024172 Cardiovascular disease Diseases 0.000 description 2
- 108091026890 Coding region Proteins 0.000 description 2
- 108010035532 Collagen Proteins 0.000 description 2
- 102000008186 Collagen Human genes 0.000 description 2
- 108091035707 Consensus sequence Proteins 0.000 description 2
- 206010011878 Deafness Diseases 0.000 description 2
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 2
- 206010015548 Euthanasia Diseases 0.000 description 2
- 108090000331 Firefly luciferases Proteins 0.000 description 2
- 241000710198 Foot-and-mouth disease virus Species 0.000 description 2
- 101001035782 Gallus gallus Hemoglobin subunit beta Proteins 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 101000762379 Homo sapiens Bone morphogenetic protein 4 Proteins 0.000 description 2
- 241000713772 Human immunodeficiency virus 1 Species 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 108010025815 Kanamycin Kinase Proteins 0.000 description 2
- 241000713666 Lentivirus Species 0.000 description 2
- 206010024604 Lipoatrophy Diseases 0.000 description 2
- 241000714177 Murine leukemia virus Species 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- 208000025966 Neurological disease Diseases 0.000 description 2
- 108700026244 Open Reading Frames Proteins 0.000 description 2
- YHIPILPTUVMWQT-UHFFFAOYSA-N Oplophorus luciferin Chemical compound C1=CC(O)=CC=C1CC(C(N1C=C(N2)C=3C=CC(O)=CC=3)=O)=NC1=C2CC1=CC=CC=C1 YHIPILPTUVMWQT-UHFFFAOYSA-N 0.000 description 2
- 208000001132 Osteoporosis Diseases 0.000 description 2
- 108010052090 Renilla Luciferases Proteins 0.000 description 2
- 208000028990 Skin injury Diseases 0.000 description 2
- 208000006011 Stroke Diseases 0.000 description 2
- 241000282887 Suidae Species 0.000 description 2
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 2
- 208000025865 Ulcer Diseases 0.000 description 2
- 206010052428 Wound Diseases 0.000 description 2
- 230000001154 acute effect Effects 0.000 description 2
- 210000000577 adipose tissue Anatomy 0.000 description 2
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 2
- 125000000539 amino acid group Chemical group 0.000 description 2
- 206010003246 arthritis Diseases 0.000 description 2
- 208000027115 auditory system disease Diseases 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 210000000481 breast Anatomy 0.000 description 2
- 230000036952 cancer formation Effects 0.000 description 2
- 231100000504 carcinogenesis Toxicity 0.000 description 2
- 230000000747 cardiac effect Effects 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 206010008118 cerebral infarction Diseases 0.000 description 2
- 208000026106 cerebrovascular disease Diseases 0.000 description 2
- 238000010367 cloning Methods 0.000 description 2
- 229920001436 collagen Polymers 0.000 description 2
- 239000003636 conditioned culture medium Substances 0.000 description 2
- 231100000433 cytotoxic Toxicity 0.000 description 2
- 231100000895 deafness Toxicity 0.000 description 2
- 230000007547 defect Effects 0.000 description 2
- 210000002986 dental sac Anatomy 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 238000010586 diagram Methods 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 238000010494 dissociation reaction Methods 0.000 description 2
- 230000005593 dissociations Effects 0.000 description 2
- 210000005064 dopaminergic neuron Anatomy 0.000 description 2
- 210000002472 endoplasmic reticulum Anatomy 0.000 description 2
- YQGOJNYOYNNSMM-UHFFFAOYSA-N eosin Chemical compound [Na+].OC(=O)C1=CC=CC=C1C1=C2C=C(Br)C(=O)C(Br)=C2OC2=C(Br)C(O)=C(Br)C=C21 YQGOJNYOYNNSMM-UHFFFAOYSA-N 0.000 description 2
- 239000003797 essential amino acid Substances 0.000 description 2
- 235000020776 essential amino acid Nutrition 0.000 description 2
- 208000030533 eye disease Diseases 0.000 description 2
- 238000002073 fluorescence micrograph Methods 0.000 description 2
- 102000034287 fluorescent proteins Human genes 0.000 description 2
- 108091006047 fluorescent proteins Proteins 0.000 description 2
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 2
- 210000000442 hair follicle cell Anatomy 0.000 description 2
- 230000035876 healing Effects 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 208000016354 hearing loss disease Diseases 0.000 description 2
- 208000019622 heart disease Diseases 0.000 description 2
- 201000005787 hematologic cancer Diseases 0.000 description 2
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 2
- 230000002440 hepatic effect Effects 0.000 description 2
- 206010021093 hypospadias Diseases 0.000 description 2
- 238000003384 imaging method Methods 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 102000006639 indoleamine 2,3-dioxygenase Human genes 0.000 description 2
- 108020004201 indoleamine 2,3-dioxygenase Proteins 0.000 description 2
- CDAISMWEOUEBRE-GPIVLXJGSA-N inositol Chemical compound O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@H](O)[C@@H]1O CDAISMWEOUEBRE-GPIVLXJGSA-N 0.000 description 2
- 210000004153 islets of langerhan Anatomy 0.000 description 2
- 210000003292 kidney cell Anatomy 0.000 description 2
- 208000017169 kidney disease Diseases 0.000 description 2
- 210000000265 leukocyte Anatomy 0.000 description 2
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 2
- 230000007774 longterm Effects 0.000 description 2
- 208000002780 macular degeneration Diseases 0.000 description 2
- 108010082117 matrigel Proteins 0.000 description 2
- 230000001394 metastastic effect Effects 0.000 description 2
- 206010061289 metastatic neoplasm Diseases 0.000 description 2
- 238000001000 micrograph Methods 0.000 description 2
- 238000000386 microscopy Methods 0.000 description 2
- 230000000869 mutational effect Effects 0.000 description 2
- 210000000963 osteoblast Anatomy 0.000 description 2
- 208000028169 periodontal disease Diseases 0.000 description 2
- 230000002085 persistent effect Effects 0.000 description 2
- 239000008194 pharmaceutical composition Substances 0.000 description 2
- 238000002135 phase contrast microscopy Methods 0.000 description 2
- 239000013600 plasmid vector Substances 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 230000002062 proliferating effect Effects 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 210000000844 retinal pigment epithelial cell Anatomy 0.000 description 2
- 229930002330 retinoic acid Natural products 0.000 description 2
- 230000001177 retroviral effect Effects 0.000 description 2
- 239000002356 single layer Substances 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 210000003014 totipotent stem cell Anatomy 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 238000010361 transduction Methods 0.000 description 2
- 230000026683 transduction Effects 0.000 description 2
- 230000005945 translocation Effects 0.000 description 2
- 231100000397 ulcer Toxicity 0.000 description 2
- 238000011144 upstream manufacturing Methods 0.000 description 2
- 230000029663 wound healing Effects 0.000 description 2
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- KSXTUUUQYQYKCR-LQDDAWAPSA-M 2,3-bis[[(z)-octadec-9-enoyl]oxy]propyl-trimethylazanium;chloride Chemical compound [Cl-].CCCCCCCC\C=C/CCCCCCCC(=O)OCC(C[N+](C)(C)C)OC(=O)CCCCCCC\C=C/CCCCCCCC KSXTUUUQYQYKCR-LQDDAWAPSA-M 0.000 description 1
- RNIPJYFZGXJSDD-UHFFFAOYSA-N 2,4,5-triphenyl-1h-imidazole Chemical compound C1=CC=CC=C1C1=NC(C=2C=CC=CC=2)=C(C=2C=CC=CC=2)N1 RNIPJYFZGXJSDD-UHFFFAOYSA-N 0.000 description 1
- 108020003589 5' Untranslated Regions Proteins 0.000 description 1
- 102000007469 Actins Human genes 0.000 description 1
- 108010085238 Actins Proteins 0.000 description 1
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 1
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 1
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 1
- 241000059559 Agriotes sordidus Species 0.000 description 1
- 102100027211 Albumin Human genes 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 241000714230 Avian leukemia virus Species 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- 101710164563 Beta-catenin-like protein 1 Proteins 0.000 description 1
- 102100026189 Beta-galactosidase Human genes 0.000 description 1
- 108010045123 Blasticidin-S deaminase Proteins 0.000 description 1
- 208000019838 Blood disease Diseases 0.000 description 1
- 241000714266 Bovine leukemia virus Species 0.000 description 1
- 102000004657 Calcium-Calmodulin-Dependent Protein Kinase Type 2 Human genes 0.000 description 1
- 108010003721 Calcium-Calmodulin-Dependent Protein Kinase Type 2 Proteins 0.000 description 1
- 241000282994 Cervidae Species 0.000 description 1
- 235000004391 Chenopodium capitatum Nutrition 0.000 description 1
- 244000038022 Chenopodium capitatum Species 0.000 description 1
- 108010077544 Chromatin Proteins 0.000 description 1
- 108020004705 Codon Proteins 0.000 description 1
- 208000014526 Conduction disease Diseases 0.000 description 1
- 208000032170 Congenital Abnormalities Diseases 0.000 description 1
- IGXWBGJHJZYPQS-SSDOTTSWSA-N D-Luciferin Chemical compound OC(=O)[C@H]1CSC(C=2SC3=CC=C(O)C=C3N=2)=N1 IGXWBGJHJZYPQS-SSDOTTSWSA-N 0.000 description 1
- 238000000116 DAPI staining Methods 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- 102000012410 DNA Ligases Human genes 0.000 description 1
- 108010061982 DNA Ligases Proteins 0.000 description 1
- 241000702421 Dependoparvovirus Species 0.000 description 1
- 108090000204 Dipeptidase 1 Proteins 0.000 description 1
- 108010044266 Dopamine Plasma Membrane Transport Proteins Proteins 0.000 description 1
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 206010016654 Fibrosis Diseases 0.000 description 1
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 1
- 241000713813 Gibbon ape leukemia virus Species 0.000 description 1
- 102000053171 Glial Fibrillary Acidic Human genes 0.000 description 1
- 101710193519 Glial fibrillary acidic protein Proteins 0.000 description 1
- 206010019663 Hepatic failure Diseases 0.000 description 1
- SQUHHTBVTRBESD-UHFFFAOYSA-N Hexa-Ac-myo-Inositol Natural products CC(=O)OC1C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C1OC(C)=O SQUHHTBVTRBESD-UHFFFAOYSA-N 0.000 description 1
- 208000017604 Hodgkin disease Diseases 0.000 description 1
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 1
- 101000756632 Homo sapiens Actin, cytoplasmic 1 Proteins 0.000 description 1
- 101000868279 Homo sapiens Leukocyte surface antigen CD47 Proteins 0.000 description 1
- 101001111338 Homo sapiens Neurofilament heavy polypeptide Proteins 0.000 description 1
- 101001098352 Homo sapiens OX-2 membrane glycoprotein Proteins 0.000 description 1
- 241000725303 Human immunodeficiency virus Species 0.000 description 1
- 102000004157 Hydrolases Human genes 0.000 description 1
- 108090000604 Hydrolases Proteins 0.000 description 1
- 206010061598 Immunodeficiency Diseases 0.000 description 1
- 208000029462 Immunodeficiency disease Diseases 0.000 description 1
- 102100034343 Integrase Human genes 0.000 description 1
- 108010002350 Interleukin-2 Proteins 0.000 description 1
- 206010048858 Ischaemic cardiomyopathy Diseases 0.000 description 1
- 108010066321 Keratin-14 Proteins 0.000 description 1
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 1
- HXEACLLIILLPRG-YFKPBYRVSA-N L-pipecolic acid Chemical compound [O-]C(=O)[C@@H]1CCCC[NH2+]1 HXEACLLIILLPRG-YFKPBYRVSA-N 0.000 description 1
- 102100023981 Lamina-associated polypeptide 2, isoform alpha Human genes 0.000 description 1
- 108091026898 Leader sequence (mRNA) Proteins 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- 101710097668 Leucine aminopeptidase 2 Proteins 0.000 description 1
- 102100032913 Leukocyte surface antigen CD47 Human genes 0.000 description 1
- 101500022510 Lithobates catesbeianus GnRH-associated peptide 2 Proteins 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 241000713862 Moloney murine sarcoma virus Species 0.000 description 1
- 208000034578 Multiple myelomas Diseases 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- OVBPIULPVIDEAO-UHFFFAOYSA-N N-Pteroyl-L-glutaminsaeure Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-UHFFFAOYSA-N 0.000 description 1
- 229930193140 Neomycin Natural products 0.000 description 1
- 241000772415 Neovison vison Species 0.000 description 1
- 102100024007 Neurofilament heavy polypeptide Human genes 0.000 description 1
- 102100038553 Neurogenin-3 Human genes 0.000 description 1
- 101710096141 Neurogenin-3 Proteins 0.000 description 1
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 1
- 102100037589 OX-2 membrane glycoprotein Human genes 0.000 description 1
- 238000012408 PCR amplification Methods 0.000 description 1
- 241000282577 Pan troglodytes Species 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- 102000012288 Phosphopyruvate Hydratase Human genes 0.000 description 1
- 108010022181 Phosphopyruvate Hydratase Proteins 0.000 description 1
- 206010035226 Plasma cell myeloma Diseases 0.000 description 1
- 102100037935 Polyubiquitin-C Human genes 0.000 description 1
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 1
- 108010076504 Protein Sorting Signals Proteins 0.000 description 1
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 1
- 101100247004 Rattus norvegicus Qsox1 gene Proteins 0.000 description 1
- 241000712909 Reticuloendotheliosis virus Species 0.000 description 1
- 241000714474 Rous sarcoma virus Species 0.000 description 1
- 101150086694 SLC22A3 gene Proteins 0.000 description 1
- 238000012300 Sequence Analysis Methods 0.000 description 1
- 102100033928 Sodium-dependent dopamine transporter Human genes 0.000 description 1
- 208000032383 Soft tissue cancer Diseases 0.000 description 1
- 238000000692 Student's t-test Methods 0.000 description 1
- 102000001435 Synapsin Human genes 0.000 description 1
- 108050009621 Synapsin Proteins 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- 108010056354 Ubiquitin C Proteins 0.000 description 1
- 108010051583 Ventricular Myosins Proteins 0.000 description 1
- 108700005077 Viral Genes Proteins 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 230000006978 adaptation Effects 0.000 description 1
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 1
- 238000003277 amino acid sequence analysis Methods 0.000 description 1
- AVKUERGKIZMTKX-NJBDSQKTSA-N ampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 AVKUERGKIZMTKX-NJBDSQKTSA-N 0.000 description 1
- 229960000723 ampicillin Drugs 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 238000003491 array Methods 0.000 description 1
- 102000015736 beta 2-Microglobulin Human genes 0.000 description 1
- 108010081355 beta 2-Microglobulin Proteins 0.000 description 1
- 108010005774 beta-Galactosidase Proteins 0.000 description 1
- SQVRNKJHWKZAKO-UHFFFAOYSA-N beta-N-Acetyl-D-neuraminic acid Natural products CC(=O)NC1C(O)CC(O)(C(O)=O)OC1C(O)C(O)CO SQVRNKJHWKZAKO-UHFFFAOYSA-N 0.000 description 1
- 102000006635 beta-lactamase Human genes 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000008499 blood brain barrier function Effects 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- 210000001218 blood-brain barrier Anatomy 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 238000003570 cell viability assay Methods 0.000 description 1
- 208000015114 central nervous system disease Diseases 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 210000003483 chromatin Anatomy 0.000 description 1
- 230000007882 cirrhosis Effects 0.000 description 1
- 208000019425 cirrhosis of liver Diseases 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000013377 clone selection method Methods 0.000 description 1
- 238000003501 co-culture Methods 0.000 description 1
- 238000007398 colorimetric assay Methods 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 238000003271 compound fluorescence assay Methods 0.000 description 1
- 230000001010 compromised effect Effects 0.000 description 1
- 230000001143 conditioned effect Effects 0.000 description 1
- 238000012937 correction Methods 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 238000005202 decontamination Methods 0.000 description 1
- 230000003588 decontaminative effect Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 210000003027 ear inner Anatomy 0.000 description 1
- 210000003981 ectoderm Anatomy 0.000 description 1
- 210000002308 embryonic cell Anatomy 0.000 description 1
- 210000001900 endoderm Anatomy 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000007159 enucleation Effects 0.000 description 1
- ZMMJGEGLRURXTF-UHFFFAOYSA-N ethidium bromide Chemical compound [Br-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CC)=C1C1=CC=CC=C1 ZMMJGEGLRURXTF-UHFFFAOYSA-N 0.000 description 1
- 229960005542 ethidium bromide Drugs 0.000 description 1
- 230000017188 evasion or tolerance of host immune response Effects 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 238000005562 fading Methods 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 1
- 238000000799 fluorescence microscopy Methods 0.000 description 1
- 238000012632 fluorescent imaging Methods 0.000 description 1
- 229960000304 folic acid Drugs 0.000 description 1
- 235000019152 folic acid Nutrition 0.000 description 1
- 239000011724 folic acid Substances 0.000 description 1
- 229960003692 gamma aminobutyric acid Drugs 0.000 description 1
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 1
- 238000001502 gel electrophoresis Methods 0.000 description 1
- 238000012226 gene silencing method Methods 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 210000005046 glial fibrillary acidic protein Anatomy 0.000 description 1
- 208000014951 hematologic disease Diseases 0.000 description 1
- 208000018706 hematopoietic system disease Diseases 0.000 description 1
- 210000003897 hepatic stem cell Anatomy 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- 231100000283 hepatitis Toxicity 0.000 description 1
- 230000006801 homologous recombination Effects 0.000 description 1
- 238000002744 homologous recombination Methods 0.000 description 1
- 210000005104 human peripheral blood lymphocyte Anatomy 0.000 description 1
- 108010002685 hygromycin-B kinase Proteins 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 230000037451 immune surveillance Effects 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 230000007813 immunodeficiency Effects 0.000 description 1
- 238000010820 immunofluorescence microscopy Methods 0.000 description 1
- 238000003364 immunohistochemistry Methods 0.000 description 1
- 230000002480 immunoprotective effect Effects 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 229940125721 immunosuppressive agent Drugs 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000011503 in vivo imaging Methods 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 229960000367 inositol Drugs 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- HXEACLLIILLPRG-RXMQYKEDSA-N l-pipecolic acid Natural products OC(=O)[C@H]1CCCCN1 HXEACLLIILLPRG-RXMQYKEDSA-N 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 208000007903 liver failure Diseases 0.000 description 1
- 231100000835 liver failure Toxicity 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 238000003670 luciferase enzyme activity assay Methods 0.000 description 1
- 238000004020 luminiscence type Methods 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 210000005075 mammary gland Anatomy 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 210000003716 mesoderm Anatomy 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 210000003470 mitochondria Anatomy 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 210000002161 motor neuron Anatomy 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 210000002894 multi-fate stem cell Anatomy 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- VMGAPWLDMVPYIA-HIDZBRGKSA-N n'-amino-n-iminomethanimidamide Chemical compound N\N=C\N=N VMGAPWLDMVPYIA-HIDZBRGKSA-N 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 229960004927 neomycin Drugs 0.000 description 1
- 210000001577 neostriatum Anatomy 0.000 description 1
- 210000004498 neuroglial cell Anatomy 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 230000009871 nonspecific binding Effects 0.000 description 1
- 210000000535 oligodendrocyte precursor cell Anatomy 0.000 description 1
- 238000001543 one-way ANOVA Methods 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- 230000008488 polyadenylation Effects 0.000 description 1
- 108091033319 polynucleotide Chemical group 0.000 description 1
- 102000040430 polynucleotide Human genes 0.000 description 1
- 239000002157 polynucleotide Chemical group 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 238000010149 post-hoc-test Methods 0.000 description 1
- 230000001323 posttranslational effect Effects 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 101710082686 probable leucine aminopeptidase 2 Proteins 0.000 description 1
- 210000004129 prosencephalon Anatomy 0.000 description 1
- 230000005892 protein maturation Effects 0.000 description 1
- 230000020978 protein processing Effects 0.000 description 1
- 108010045647 puromycin N-acetyltransferase Proteins 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 108010054624 red fluorescent protein Proteins 0.000 description 1
- 230000001172 regenerating effect Effects 0.000 description 1
- 230000001373 regressive effect Effects 0.000 description 1
- BOLDJAUMGUJJKM-LSDHHAIUSA-N renifolin D Natural products CC(=C)[C@@H]1Cc2c(O)c(O)ccc2[C@H]1CC(=O)c3ccc(O)cc3O BOLDJAUMGUJJKM-LSDHHAIUSA-N 0.000 description 1
- 230000001718 repressive effect Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 210000003583 retinal pigment epithelium Anatomy 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 239000011435 rock Substances 0.000 description 1
- 102200007391 rs794726878 Human genes 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 231100000735 select agent Toxicity 0.000 description 1
- SQVRNKJHWKZAKO-OQPLDHBCSA-N sialic acid Chemical compound CC(=O)N[C@@H]1[C@@H](O)C[C@@](O)(C(O)=O)OC1[C@H](O)[C@H](O)CO SQVRNKJHWKZAKO-OQPLDHBCSA-N 0.000 description 1
- 230000009870 specific binding Effects 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 210000000278 spinal cord Anatomy 0.000 description 1
- 230000010473 stable expression Effects 0.000 description 1
- 238000009168 stem cell therapy Methods 0.000 description 1
- 238000009580 stem-cell therapy Methods 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 210000000115 thoracic cavity Anatomy 0.000 description 1
- 230000036962 time dependent Effects 0.000 description 1
- 238000003151 transfection method Methods 0.000 description 1
- 239000012096 transfection reagent Substances 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 238000007492 two-way ANOVA Methods 0.000 description 1
- 238000005199 ultracentrifugation Methods 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 238000011179 visual inspection Methods 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
- 150000003952 β-lactams Chemical class 0.000 description 1
Images





















Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/569—Immunoassay; Biospecific binding assay; Materials therefor for microorganisms, e.g. protozoa, bacteria, viruses
- G01N33/56966—Animal cells
- G01N33/56977—HLA or MHC typing
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K67/00—Rearing or breeding animals, not otherwise provided for; New or modified breeds of animals
- A01K67/027—New or modified breeds of vertebrates
- A01K67/0271—Chimeric vertebrates, e.g. comprising exogenous cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/28—Bone marrow; Haematopoietic stem cells; Mesenchymal stem cells of any origin, e.g. adipose-derived stem cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/33—Fibroblasts
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/37—Digestive system
- A61K35/407—Liver; Hepatocytes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/44—Vessels; Vascular smooth muscle cells; Endothelial cells; Endothelial progenitor cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/70503—Immunoglobulin superfamily
- C07K14/70539—MHC-molecules, e.g. HLA-molecules
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0603—Embryonic cells ; Embryoid bodies
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/566—Immunoassay; Biospecific binding assay; Materials therefor using specific carrier or receptor proteins as ligand binding reagents where possible specific carrier or receptor proteins are classified with their target compounds
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
- G01N33/6893—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids related to diseases not provided for elsewhere
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2207/00—Modified animals
- A01K2207/12—Animals modified by administration of exogenous cells
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2227/00—Animals characterised by species
- A01K2227/10—Mammal
- A01K2227/105—Murine
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2267/00—Animals characterised by purpose
- A01K2267/02—Animal zootechnically ameliorated
- A01K2267/025—Animal producing cells or organs for transplantation
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2510/00—Genetically modified cells
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/435—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans
- G01N2333/705—Assays involving receptors, cell surface antigens or cell surface determinants
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/435—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans
- G01N2333/705—Assays involving receptors, cell surface antigens or cell surface determinants
- G01N2333/71—Assays involving receptors, cell surface antigens or cell surface determinants for growth factors; for growth regulators
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2800/00—Detection or diagnosis of diseases
- G01N2800/24—Immunology or allergic disorders
- G01N2800/245—Transplantation related diseases, e.g. graft versus host disease
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Immunology (AREA)
- Biomedical Technology (AREA)
- Cell Biology (AREA)
- Zoology (AREA)
- General Health & Medical Sciences (AREA)
- Biotechnology (AREA)
- Medicinal Chemistry (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Developmental Biology & Embryology (AREA)
- Genetics & Genomics (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Virology (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Epidemiology (AREA)
- Hematology (AREA)
- Wood Science & Technology (AREA)
- Microbiology (AREA)
- Urology & Nephrology (AREA)
- Environmental Sciences (AREA)
- General Engineering & Computer Science (AREA)
- Biophysics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Physics & Mathematics (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Food Science & Technology (AREA)
- Analytical Chemistry (AREA)
- General Physics & Mathematics (AREA)
- Pathology (AREA)
- Biodiversity & Conservation Biology (AREA)
Description
A.外因性HLA−Gを発現する遺伝子改変された哺乳動物細胞
本明細書において説明されるように、外因性HLA−Gを発現する広範な哺乳動物細胞型(HLA−G改変された細胞)が生成され得る。そのような細胞型は、分化全能性細胞、胚性幹細胞(例えば、ヒト胚性幹細胞)、誘導多能性幹細胞(例えば、ヒト誘導多能性幹細胞)、多分化能幹細胞、表皮前駆細胞、間葉系幹細胞、膵臓β細胞前駆細胞、膵臓β細胞、心臓前駆細胞、心筋細胞、肝前駆細胞、肝細胞、筋細胞前駆細胞、筋細胞、腎細胞、骨芽細胞、造血前駆細胞、歯小嚢細胞、毛包細胞、網膜色素上皮細胞、神経幹細胞、ニューロン、星状膠細胞、乏突起膠細胞、内耳細胞、および線維芽細胞(ヒト皮膚線維芽細胞(HFD)を含む)を含むが、これらに限定されない。いくつかの実施形態において、HLA−G改変された細胞は、免疫系細胞型を有する細胞ではない。そのような哺乳動物細胞は、例えばヒト、マウス、ラット、サル、またはブタを含むいくつかの種の1つから得ることができる。本質的に、任意の細胞型が、本明細書に記載のコンストラクトでトランスフェクトされ、次いでHLA−G発現について、およびそのような発現が低減された免疫原性および/または改善された免疫抑制をどのように改変された細胞に付与し得るかについて試験されてもよい。
本明細書に記載のようなHLA改変された哺乳動物細胞を生成するために使用される単離された核酸(例えば、哺乳動物プラスミド発現ベクター)は、野生型HLA−G導入遺伝子により駆動される細胞表面発現と比べて増加した細胞表面発現および/またはHLA−Gの分泌を駆動する、強化されたHLA−G「eHLA−G」導入遺伝子を含有する。そのような導入遺伝子は、典型的には、少なくとも3つの別個の成分、すなわちプロモーターおよび5’非翻訳領域(5’UTR)配列、コード配列、ならびに3’非翻訳領域(3’UTR)配列を含む。
MVVMAPRTLFLLLSGALTLTETWAGSHSMRYFSAAVSRPGRGEPRFIAMGYVDDTQFVRFDSDSACPRMEPRAPWVEQEGPEYWEEETRNTKAHAQTDRMNLQTLRGYYNQSEASSHTLQWMIGCDLGSDGRLLRGYEQYAYDGKDYLALNEDLRSWTAADTAAQISKRKCEAANVAEQRRAYLEGTCVEWLHRYLENGKEMLQRADPPKTHVTHHPVFDYEATLRCWALGFYPAEIILTWQRDGEDQTQDVELVETRPAGDGTFQKWAAVVVPSGEEQRYTCHVQHEGLPEPLMLRWKQSSLPTIPIMGIVAGLVVLAAVVTGAAVAAVLWRKKSSD
MVVMAPRTLFLLLSGALTLTETWAGSHSMRYFSAAVSRPGRGEPRFIAMGYVDDTQFVRFDSDSACPRMEPRAPWVEQEGPEYWEEETRNTKAHAQTDRMNLQTLRGYYNQSEASSHTLQWMIGCDLGSDGRLLRGYEQYAYDGKDYLALNEDLRSWTAADTAAQISKRKCEAANVAEQRRAYLEGTCVEWLHRYLENGKEMLQRADPPKTHVTHHPVFDYEATLRCWALGFYPAEIILTWQRDGEDQTQDVELVETRPAGDGTFQKWAAVVVPSGEEQRYTCHVQHEGLPEPLMLRWKQSSLPTIPIMGIVAGLVVLAAVVTGAAVAAVLWRAASSD
TGTGAAACAGCTGCCCTGTGTGGGACTGAGTGGCAAGTCCCTTTGTGACTTCAAGAACCCTGACTTCTCTTTGTGCAGAGACCAGCCCAACCCTGTGCCCACCATGACCCTCTTCCTCATGCTGAACTGCATTCCTTCCCCAATCACCTTTCCTGTTCCAGAAAAGGGGCTGGGATGTCTCCGTCTCTGTCTCAAATTTGTGGTCCACTGAGCTATAACTTACTTCTGTATTAAAATTAGAATCTGAGTG
配列番号:4 HLA 3’UTRにおける14−bp挿入配列
ATTTGTTCATGCCT
配列番号:5(+14 BP HLA−G 3’UTR変異体)
TGTGAAACAGCTGCCCTGTGTGGGACTGAGTGGCAAGatttgttcatgcctTCCCTTTGTGACTTCAAGAACCCTGACTTCTCTTTGTGCAGAGACCAGCCCAACCCTGTGCCCACCATGACCCTCTTCCTCATGCTGAACTGCATTCCTTCCCCAATCACCTTTCCTGTTCCAGAAAAGGGGCTGGGATGTCTCCGTCTCTGTCTCAAATTTGTGGTCCACTGAGCTATAACTTACTTCTGTATTAAAATTAGAATCTGAGTG
本明細書において、1種以上のHLA−G改変された哺乳動物細胞型を含む、またはそれを使用して生成された薬学的組成物、局所用組成物、細胞移植片、および人工組織が包含される。本明細書において示されるように、eHLA−G改変されたhESCは、hEEPへの誘導分化にもかかわらず、安定で持続的なHLA−G発現を示した。さらに、安定で持続的なHLA−G発現は、低減された免疫原性および/または改善された免疫抑制を有する遺伝子改変された細胞を提供した。さらに、本明細書において、完全に分化した細胞であるeHLA−G改変されたヒト皮膚線維芽細胞もまた、低減された免疫原性および/または改善された免疫抑制を提供する安定で持続的なHLA−G発現を有することが示される。したがって、本明細書に記載のeHLA−Gコンストラクトは、遺伝子改変された多能性もしくは多分化能細胞の誘導分化によるか、または完全に分化した細胞の遺伝子改変によるかに関わらず、任意の型の普遍的なドナー細胞を生成するために使用され得る。
細胞治療処置
本明細書に記載の様式で外因性HLA−Gを安定に発現するように改変された細胞は、低減された免疫原性および/または増加した免疫抑制を有するため、これらの形質により、改質された細胞は、普遍的または改善されたドナー細胞または組織として機能することができる。これは、細胞に提供される、HLA−Gが媒介した免疫原性の低減および/または免疫抑制の改善が、多くの損傷、疾患、または障害において、ドナー細胞と受容者との間の古典的ヒト白血球抗原(HLA)クラスIおよびクラスII分子の種類を一致させる必要性を低減または排除し得るためである。
ヒト軟組織癌アレイを、まず、おそらくは免疫監視機構の回避により、転移状態への癌の進行と共に増加する発現を示す候補免疫寛容遺伝子を求めてスクリーニングした。データは、正常な転移と、以前に免疫寛容に関与したいくつかの遺伝子の発現レベルとの間の強い正の相関を明らかにした。MSCが免疫寛容および同種移植片受容を誘導し得るかどうかを評価するために、MSCをRT−PCRによりクロススクリーニングし、これらの候補遺伝子およびいくつかの抗原HLAの発現レベルを検査した。表1は、継代1のMSCが、CD200、CD47、およびインドールアミン2,3−ジオキシゲナーゼ(IDO)の他に、HLAクラスIa、HLA−GおよびIIマーカーを発現したことを示している。MSCの集団は、攻撃的転移能を有する癌株であるJEG−3に見られるものよりも少ないものの、中程度のレベルでHLA−Gを発現することが見出された。

別段の指定がない限り、全ての組織培養試薬は、Life Technologiesからのものであった。ESC成長培地は、20%ノックアウト血清代替物、0.1mM MEM非必須アミノ酸、1mM GlutaMax、0.1mM β−メルカプトエタノール(Sigma)を添加したDMEM/F12(1:1)を含有する。有糸分裂不活性化されたマウス胚性線維芽細胞(MEF)(CF−1、ATCC)を、5×104細胞/cm2の密度で播種し、18〜24時間インキュベートすることにより、ESC成長培地を調整した。調整後、4ng/ml bFGFを添加し、完全調整培地を滅菌濾過した。hESCを、5〜6日毎に(1:3または1:4分割で)、Matrigel被覆プレート上で、細胞コロニーを除去するために1mg/mlディスパーゼを使用して継代培養した。K14+/p63+hEEPを、Metallo et al(上記参照)の方法に従って生成した。簡単に述べると、hESCを6ウェルプレートで4日間培養し、次いで、1μMのオールトランス型レチノイン酸(Sigma)および25ng/mlのBMP4を含有する無条件のhESC成長培地を含む、2ml/ウェルの分化培地で処理した。7日間の毎日の培地交換後、細胞をディスパーゼで処理し、遠心分離し、ケラチノサイト合成無血清培地(DSFM)中に再懸濁させ、ゼラチン被覆プレート上に1:3の分割比で播種した。DSFMは、3〜4週間、一日おきに交換する。次いで、トリプシン処理により細胞を継代培養し、遠心分離し、洗浄し、cm2当たり10,000細胞で、DSFM中のゼラチン被覆組織培養プレート上に播種した。ケラチノサイト合成無血清培地中で14日後、表皮シート構造の形成を特徴とするような表皮分化の初期の兆候が、顕微鏡法により観察された。4週間の培養後、表皮シート内の細胞は、立方体形態を有する典型的な表皮分化表現型を示した。
1)HLA−GのER回収モチーフの突然変異(K334A/K335A)、および2)HLA−Gの3’UTR microRNA結合部位の突然変異の複数の改変を組み合わせることにより、新規HLA−Gコンストラクトを設計した。ウイルス遺伝子送達系は、依然として深刻な規制の課題であるため、最近H1 hESCにおいて90%のトランスフェクション効率を達成することが示された(Lacoste et al(2009),Cell Stem Cell,5:332−342.)トランスポゾンベースの非ウイルスアプローチである、PiggyBacシステムを使用した。このシステムは、トランスポゾンを含有するドナープラスミド(図1A)およびトランスポザーセを発現するヘルパープラスミド(図2)を必要とする。ヘルパープラスミドを生成するために、ePiggyBacコドンヒト化トランスポザーセcDNAをカスタム合成し(GeneArt)、次いでPGKプロモーターの下流側およびSV40ポリアデニル化シグナル配列(pA)の上流側をpBluescript(Stratagene)でクローニングした。eHLA−G発現カセットのために、M−U3/Rプロモーター、MSCVプロモーター、およびチャイニーズハムスターEF1α(CHEF−1α)プロモーターを含む複数のプロモーターを比較したが、その配列を配列番号:6として以下に示す。
(配列番号:6)−CHEF−1αプロモーターの一実施形態
GGATGGCGGGGCTGACGTCGGGAGGTGGCCTCCACGGGAAGGGACACCCGGATCTCGACACAGCCTTGGCAGTGGAGTCAGGAAGGGTAGGACAGATTCTGGACGCCCTCTTGGCCAGTCCTCACCGCCCCACCCCCGATGGAGCCGAGAGTAATTCATACAAAAGGAGGGATCGCCTTCGCCCCTGGGAATCCCAGGGACCGTCGCTAAATTCTGGCCGGCCTCCCAGCCCGGAACCGCTGTGCCCGCCCAGCGCGGCGGGAGGAGCCTGCGCCTAGGGCGGATCGCGGGTCGGCGGGAGAGCACAAGCCCACAGTCCCCGGCGGTGGGGGAGGGGCGCGCTGAGCGGGGGCCCGGGAGCCAGCGCGGGGCAAACTGGGAAAGTGGTGTCGTGTGCTGGCTCCGCCCTCTTCCCGAGGGTGGGGGAGAACGGTATAAAAGTGCGGTAGTCGCGTTGGACGTTCTTTTTCGCAACGGGTTTGCCGTCAGAACGCAGGTGAGTGGCGGGTGTGGCCTCCGCGGGCCCGGGCTCCCTCCTTTGAGCGGGGTCGGACCGCCGTGCGGGTGTCGTCGGCCGGGCTTCTCTGCGAGCGTTCCCGCCCTGGATGGCGGGCTGTGCGGGAGGGCGAGGGGGGGAGGCCTGGCGGCGGCCCCGGAGCCTCGCCTCGTGTCGGGCGTGAGGCCTAGCGTGGCTTCCGCCCCGCCGCGTGCCACCGCGGCCGCGCTTTGCTGTCTGCCCGGCTGCCCTCGATTGCCTGCCCGCGGCCCGGGCCAACAAAGGGAGGGCGTGGAGCTGGCTGGTAGGGAGCCCCGTAGTCCGCATGTCGGGCAGGGAGAGCGGCAGCAGTCGGGGGGGGGACCGGGCCCGCCCGTCCCGCAGCACATGTCCGACGCCGCCTGGACGGGTAGCGGCCTGTGTCCTGATAAGGCGGCCGGGCGGTGGGTTTTAGATGCCGGGTTCAGGTGGCCCCGGGTCCCGGCCCGGTCTGGCCAGTACCCCGTAGTGGCTTAGCTCCGAGGAGGGCGAGCCCGCCCGCCCGGCACCAGTTGCGTGCGCGGAAAGATGGCCGCTCCCGGGCCCTGTAGCAAGGAGCTCAAAATGGAGGACGCGGCAGCCCGGCGGAGCGGGGCGGGTGAGTCACCCACACAAAGGAAGAGGGCCTTGCCCCTCGCCGGCCGCTGCTTCCTGTGACCCCGTGGTGTACCGGCCGCACTTCAGTCACCCCGGGCGCTCTTTCGGAGCACCGCTGGCCTCCGCTGGGGGAGGGGATCTGTCTAATGGCGTTGGAGTTTGCTCACATTTGGTGGGTGGAGACTGTAGCCAGGCCAGCCTGGCCATGGAAGTAATTCTTGGAATTTGCCCATTTTGAGTTTGGAGCGAAGCTGATTGACAAAGCTGCTTAGCCGTTCAAAGGTATTCTTCGAACTTTTTTTTTAAGGTGTTGTGAAAACCACCG
(配列番号:7)HSF要素の一実施形態
GAGCTCACGGGGACAGCCCCCCCCCAAAGCCCCCAGGGATGTAATTACGTCCCTCCCCCGCTAGGGGGCAGCAGCGAGCCGCCCGGGGCTCCGCTCCGGTCCGGCGCTCCCCCCGCATCCCCGAGCCGGCAGCGTGCGGGGACAGCCCGGGCACGGGGAAGGTGGCACGGGATCGCTTTCCTCTGAACGCTTCTCGCTGCTCTTTGAGCCTGCAGACACCTGGGGGATACGGGGAAAAAGCTT
トランスフェクトされた細胞を、CMおよびY−27632中、6cm皿当たり2×103細胞で24時間播種し、次いでCMのみに変更することにより、eHLA−Gトランスフェクション効率を決定した。次いで、培地を7日間毎日交換し、実施例5で説明されるように、コロニーを生細胞染色および免疫蛍光顕微鏡法により評価した。各クローンに対して、3つの高倍率視野を計数し、レポータータンパク質+/eHLA−G+hESCのパーセンテージを計算した。この実験の結果を、図3〜4に示す。Lacoste et al(上記参照)に記載のように、プラスミドレスキュー戦略を使用してeHLA−G挿入部位を決定した。簡単に説明すると、ゲノムDNAを、トランスジェニックhESCクローンから単離し、BamHI/BglII/NotIで消化し、T4 DNAリガーゼで、16℃で一晩、低濃度で自己連結させ、100%イソプロパノールで沈殿させ、70%エタノールで洗浄してから、DH10B大腸菌中で転換し、アンピシリン上で選択した。SplinkTA PCRを使用して、eHLA−Gコピー数を決定した。核型安定性を評価するために、標準的なGバンド法を20継代毎に行った。eHLA−Gおよびレポータータンパク質遺伝子サイレンシングを、フローサイトメトリーを使用して10継代毎に評価した。解離および分析の前に、hESCを10μM Y−27632で1時間処理した。次いで、細胞を、0.25%トリプシン−EDTAで、37℃で5分間解離させ、CMおよびY−27632中で洗浄し、0.1%BSAおよび0.5mM EDTAを含有する氷冷PBS中に再懸濁させ、次いでBD FACS Canto IIフローサイトメーター(Becton Dickinson)を使用して分析した。図8および9に示されるように、継代16において、本質的にEGFPまたはeHLA−G発現のサイレンシングは観察されなかった。以下の免疫細胞化学的検出により、20継代毎に多能性を評価した:1)多能性マーカーOct3/4、SSEA−4、Sox2およびNanog(図5および6)、2)レポータータンパク質+/eHLA−G+胚様体形成(図7)、ならびに3)分化したトランスジェニックhESCの内胚葉マーカーGata 6、中胚葉マーカー筋アクチン、および外胚葉マーカーニューロフィラメント重鎖(データは示さず)(全てLeica CTR6500蛍光顕微鏡を使用)。特に指定がない限り、すべての抗体は、Abcamからのものであった。
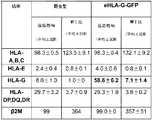
生細胞染色を使用して、トランスジェニックウイルス野生型hESCおよびhEEPにおける細胞表面HLAクラスIa、HLA−E、eHLA−G、およびHLAクラスII発現を検出した。簡単に述べると、細胞を回収して冷PBS中で洗浄し、10%ヤギ血清および3%BSAを含有するPBS中で、4℃で60分間、対応する1度のmAbで染色し、洗浄し、1%パラホルムアルデヒドで10分間固定し、続いて4℃で30分間、FITCと複合体化したヤギ抗マウスIgGで染色した。対照アリコートをアイソタイプ適合IgGで染色し、標的細胞への非特異的結合を評価した。特異的結合のみを達成するための最適条件を決定するために、各抗体(HLA−Gの場合MEMG/9、HLA−Eの場合MEM−E/08、HLAクラスIaの場合Bu8、およびHLAクラスIIの場合HKB1(Abbiotec、San Diego、CA))を、まずいくつかの希釈で試験した。染色後、細胞をスライドガラス上に塗抹し、空気乾燥させ、次いでDAPI(Vector Laboratories)を含有する抗退色媒体を載せた。Leica CTR6500蛍光顕微鏡下で速やかにスライドを観察した。図5および10に示されるように、HLA−A、B、C;HLA−E、HLA−DP、DQ、DR、およびβ−ミクログロブリンの発現は、野生型およびeHLA−G改変されたヒトES細胞において同様であり、一方、約7倍高いレベルのHLA−G発現が、eHLA−G改変されたヒトES細胞において観察された。
eHLA−Gを発現するhESCを培養し、実施例2に記載のようにhEEPに分化させた。NK−92細胞(CRL−2407、American Type Culture Collection、Manassas、VA)を、12.5%FBS、12.5%ウマ血清、0.2mMイノシトール、0.1mM β−メルカプトエタノール、0.02mM葉酸および100IU/ml組み換えIL−2(Sigma)を添加した最小必須培地アルファ培地(α−MEM、Invitrogen)中で、37℃で、5%CO2加湿インキュベータ内で培養した。
最近、ヒト末梢血リンパ球(Hu−PBL−NSG)を有するが、野生型免疫不全NSGマウスではないヒト化マウスモデルが、移植後1〜2週間以内にミスマッチヒト膵島を拒絶することが示された(King et al(2008),Clin Immunol,126:303−314)。移植片対宿主病(GVHD)は4〜5週で開始するが、安楽死の基準が満たされるまで移植片の生存を監視する。より低レベルのGVHDのみが存在する場合、我々は観察枠を拡張することができる。NSGマウス(6週齢の雌)を、Jackson Laboratoryから購入し、Institutional Animal Care and Use CommitteeのガイドラインおよびGuide for the Care and Use of Laboratory Animals(Institute of Laboratory Animal Resources、National Research Council、National Academy of Sciences)における推奨に従って取り扱う。機能性ヒト化NSGマウスを、約20×106ヒトPBMCのNSGマウスへの静脈内注射により生成した(Pearson et al(2008),Curr Protoc Immunol,Ch.15:Unit15.21、およびKing et al(上記参照)に従う)。約4週目で、EDTA被覆毛細管(Drummond Scientific)およびEDTA処理された1.5ml管(Eppendorf)を使用して、麻酔されたマウスの後眼窩静脈叢から血液を採取することにより、生着を検証した。次いで、ヒトCD45陽性のために、King et al(上記参照)に従い、FACS分析により細胞を処理した。4週目に血液中0.1%に達するヒトCD45+細胞のレベルは、生着の成功とみなされ、同種移植拒絶試験を可能とする。
細胞が非ヒト化NSGマウスに移植されることを除いて実施例8で説明されたのと同様のプロトコルを使用して、hEEP腫瘍形成に対するeHLA−G効果の影響を、トランスジェニックG+および非トランスジェニックG−−hEEPを注射することにより評価する。9ヶ月または安楽死基準の適合のいずれか早い方までの、月単位での生きた動物の縦方向の蛍光画像化および体重評価を使用して、全部で14匹のマウスを監視する。次いで、マウスを致死させ、注射された胸部乳腺を、4%中性緩衝ホルムアルデヒド中に回収する。固定組織を70%エタノールに移し、パラフィン中に包埋する。切片をヘマトキシリンおよびエオシンで染色し、蛍光レポータータンパク質顕微鏡観察用に処理する。組織切片は、レポータータンパク質陰性であることが判明した場合は、注射部位における非PBMCの存在を確認するために役立つ、ヒトβ2ミクログロブリンおよびCD45に特異的なmAbを用いて染色される。
ヌクレオフェクションを使用して、eHLA−G(EF−1α)−GFP導入遺伝子および対照コンストラクトを、ヒト新生児皮膚線維芽細胞(すでに完全に分化している細胞型)にトランスフェクトした。ヒト新生児皮膚線維芽細胞(HFD)は、ATCCから購入し、10%FBSおよび1%PS(Invitrogen)を添加したイスコフ改変ダルベッコ培地(IMDM)(ATCC)中で培養した。細胞が80%のコンフルエンスに達した時に、0.25%トリプシン−EDTA(Invitrogen)で、37℃で3分間インキュベートすることにより細胞を回収した。細胞を計数し、0.5×106を遠心分離して、ヒト皮膚線維芽細胞ヌクレオフェクション溶液(カタログ番号VPD−1001、Lonza、Walkersville、MD)中に再懸濁させた。ヘルパーおよびトランスポゾンプラスミドを細胞懸濁液に添加し、製造者のプロトコル(Lonza)に従い、プログラム設定U−020でヌクレオフェクションを行った。次いで、細胞を6ウェルプレート内に播種し、加湿37℃/5%CO2中で24時間インキュベートした。24時間後、安定にトランスフェクトされた細胞を、1μg/mlピューロマイシン(Sigma)で7日間選択した。安定なGFP陽性細胞を、500ng/mlピューロマイシン中に維持し、HLA−GおよびGFP発現をフローサイトメトリーで検出した。安定なトランスフェクタントを、後述のような末梢血単核細胞(PBMC)増殖アッセイおよびNK−92細胞毒性アッセイにおける使用のために、培養物中に維持した。
Claims (34)
- 遺伝子改変を有さない哺乳動物細胞と比較して、低減された免疫原性および/または改善された免疫抑制を有する、前記遺伝子改変された哺乳動物細胞であって、
(i)前記遺伝子改変された哺乳動物細胞は、(a)配列番号:2のアミノ酸配列を含むHLA−Gタンパク質をコードする核酸配列と、(b)配列番号:3のヌクレオチド配列を含む3’非翻訳領域(UTR)とを含む、外因性核酸を含み、
(ii)コードされたHLA−Gタンパク質は、少なくとも7週間、前記遺伝子改変された哺乳動物細胞により発現される、遺伝子改変された哺乳動物細胞。 - 前記HLA−Gタンパク質は、少なくとも20週間発現される、請求項1の遺伝子改変された哺乳動物細胞。
- 前記HLA−Gタンパク質は、少なくとも50週間発現される、請求項2に記載の遺伝子改変された哺乳動物細胞。
- 延長因子−1アルファ(EF−1α)プロモーターをさらに含み、前記HLA−Gタンパク質をコードする前記核酸配列は、作用可能に前記EF−1αプロモーターに結合する、請求項1に記載の遺伝子改変された哺乳動物細胞。
- 前記EF−1αプロモーターは、配列番号:6の配列を含む、請求項4に記載の遺伝子改変された哺乳動物細胞。
- 前記発現されたHLA−Gが、前記遺伝子改変された哺乳動物細胞の細胞表面上に存在する、請求項1に記載の遺伝子改変された細胞。
- 前記改変された哺乳動物細胞は、ヒト細胞である、請求項1に記載の遺伝子改変された哺乳動物細胞。
- 前記遺伝子改変された細胞は、幹細胞、前駆細胞、前記幹細胞もしくは前記前駆細胞のin vitro分化により得られる細胞、完全に分化した細胞、表皮前駆細胞、膵臓前駆細胞、造血幹細胞、ケラチノサイト、線維芽細胞、肝細胞、間葉系幹細胞、心筋細胞、神経幹細胞、ニューロン、または星状膠細胞である、請求項1に記載の遺伝子改変された哺乳動物細胞。
- 前記遺伝子改変された細胞は、ヒト胚性幹細胞、ヒト間葉系幹細胞、またはヒト胚性表皮前駆細胞である、請求項1に記載の遺伝子改変された哺乳動物細胞。
- 前記遺伝子改変された細胞は、ヒト皮膚線維芽細胞である、請求項1に記載の遺伝子改変された哺乳動物細胞。
- 前記遺伝子改変を有さない前記哺乳動物細胞と比較して、前記遺伝子改変された細胞の前記低減された免疫原性および/または改善された免疫抑制が、(1)前記遺伝子改変を有さない前記哺乳動物細胞と比較した、前記遺伝子改変された細胞のNK−92細胞毒性の低減、(2)前記遺伝子改変を有さない前記哺乳動物細胞と比較した、前記遺伝子改変された細胞のin vitro末梢血単核細胞増殖の低減、ならびに(3)ヒト化NSGマウスにおける、前記遺伝子改変を有さない前記哺乳動物細胞と比較した、前記遺伝子改変された細胞による腫瘍形成のサイズおよび重量の増加により決定される、請求項1に記載の遺伝子改変された細胞。
- 請求項1に記載の遺伝子改変された哺乳動物細胞を含む人工組織。
- 請求項9または請求項10に記載の遺伝子改変された哺乳動物細胞を含む皮膚移植、修復、または再生組成物。
- 前記外因性核酸は、発現ベクターである、請求項1に記載の遺伝子改変された哺乳動物細胞。
- 前記哺乳動物発現ベクターは、トランスポゾンベクターである、請求項14に記載の遺伝子改変された哺乳動物細胞。
- 前記ベクターは、レポータータンパク質をコードする核酸配列をさらに含む、請求項14に記載の遺伝子改変された哺乳動物細胞。
- 単離された核酸であって、
(i)配列番号:2のアミノ酸配列をコードする第1の核酸配列と、
(ii)配列番号:3のヌクレオチド配列を含む3’非翻訳領域を含み、前記第1の核酸配列に作用可能に結合した第2の核酸配列とを含む、単離された核酸。 - 前記第2の核酸配列は、配列番号:4を含まない、請求項17に記載の単離された核酸。
- 請求項17に記載の単離された核酸および前記第1の核酸配列に作用可能に結合したプロモーターを含む哺乳動物発現ベクターであって、前記プロモーターは、幹細胞内、または前記幹細胞の分化により生成された細胞内でサイレンシングされていない、哺乳動物発現ベクター。
- 前記プロモーターは、チャイニーズハムスターEF−1αプロモーターの核酸配列を含む、請求項19に記載の哺乳動物発現ベクター。
- レポータータンパク質をコードする第3の核酸配列をさらに含む、請求項19に記載の哺乳動物発現ベクター。
- 前記哺乳動物発現ベクターは、トランスポゾンベクターである、請求項19に記載の哺乳動物発現ベクター。
- (a)チャイニーズハムスターEF−1αプロモーター、(b)前記プロモーターに作用可能に結合し、配列番号:2のアミノ酸配列を含むヒトHLA−Gをコードする核酸配列、および(c)配列番号:3を含む3’UTR配列を含む、哺乳動物発現ベクター。
- 請求項23に記載の哺乳動物発現ベクターを含む、遺伝子改変された哺乳動物細胞。
- 必要とする対象に投与される、請求項1に記載の遺伝子改変された細胞の集団を含む細胞または組織組成物であって、前記対象に注入され、埋め込まれ、または移植され、前記対象は、前記遺伝子改変された細胞の集団と比較して、少なくとも1つのミスマッチ古典的HLAクラスIまたはHLAクラスII分子を有し、前記遺伝子改変された細胞の集団は、前記遺伝子改変を有さない同じ型の細胞と比較して、低減された免疫原性および/または改善された免疫抑制を示す、組成物。
- 前記少なくとも1つのミスマッチ古典的HLAクラスIまたはHLAクラスII分子は、HLA−A、HLA−B、HLA−C、HLA−DP、HLA−DQ、およびHLA−DRからなる群から選択される、請求項25に記載の組成物。
- 前記対象は、HLA−A、HLA−B、HLA−C、HLA−DP、HLA−DQ、およびHLA−DRに関して、前記遺伝子改変された細胞とのマッチを有さない、請求項25に記載の組成物。
- 前記低減された免疫原性および/または改善された免疫抑制は、NK−92細胞毒性アッセイ、ヒト化NSG腫瘍成長アッセイ、および/または末梢血単核細胞増殖アッセイにより決定される、請求項25に記載の組成物。
- 前記遺伝子改変された細胞の集団は、ヒト皮膚線維芽細胞の集団を含む、請求項25に記載の組成物。
- 前記遺伝子改変された細胞の集団は、ヒト表皮前駆細胞の集団を含む、請求項25に記載の組成物。
- 前記遺伝子改変された細胞の集団は、ヒト胚性幹細胞の集団を含む、請求項25に記載の組成物。
- 前記遺伝子改変された細胞の集団は、ヒト間葉系幹細胞の集団を含む、請求項25に記載の組成物。
- 前記遺伝子改変された細胞の集団は、in vitroで分化した遺伝子改変されたヒト胚性幹細胞を含む、請求項25に記載の組成物。
- 前記遺伝子改変された細胞の集団は、少なくとも2、4、6、8、10、12、14、16、18、20、24、36、48、または52週間、前記対象の免疫系により拒絶されない、請求項25に記載の組成物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261677739P | 2012-07-31 | 2012-07-31 | |
| US61/677,739 | 2012-07-31 | ||
| PCT/US2013/052767 WO2014022423A2 (en) | 2012-07-31 | 2013-07-30 | Hla g-modified cells and methods |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2018028816A Division JP6502544B2 (ja) | 2012-07-31 | 2018-02-21 | Hla g改変された細胞および方法 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2015529457A JP2015529457A (ja) | 2015-10-08 |
| JP2015529457A5 JP2015529457A5 (ja) | 2016-09-15 |
| JP6297559B2 true JP6297559B2 (ja) | 2018-03-20 |
Family
ID=50028654
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015525519A Active JP6297559B2 (ja) | 2012-07-31 | 2013-07-30 | Hlag改変された細胞および方法 |
| JP2018028816A Active JP6502544B2 (ja) | 2012-07-31 | 2018-02-21 | Hla g改変された細胞および方法 |
| JP2019052699A Active JP7294838B2 (ja) | 2012-07-31 | 2019-03-20 | Hla g改変された細胞および方法 |
| JP2021068195A Active JP7379410B2 (ja) | 2012-07-31 | 2021-04-14 | Hla g改変された細胞および方法 |
Family Applications After (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2018028816A Active JP6502544B2 (ja) | 2012-07-31 | 2018-02-21 | Hla g改変された細胞および方法 |
| JP2019052699A Active JP7294838B2 (ja) | 2012-07-31 | 2019-03-20 | Hla g改変された細胞および方法 |
| JP2021068195A Active JP7379410B2 (ja) | 2012-07-31 | 2021-04-14 | Hla g改変された細胞および方法 |
Country Status (10)
| Country | Link |
|---|---|
| US (4) | US9714280B2 (ja) |
| EP (4) | EP3483178B1 (ja) |
| JP (4) | JP6297559B2 (ja) |
| KR (4) | KR101923632B1 (ja) |
| CN (2) | CN113151179A (ja) |
| AU (2) | AU2013296564C1 (ja) |
| CA (1) | CA2882028C (ja) |
| ES (2) | ES2716577T3 (ja) |
| SG (1) | SG11201500799YA (ja) |
| WO (1) | WO2014022423A2 (ja) |
Families Citing this family (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140134195A1 (en) | 2011-04-20 | 2014-05-15 | University Of Washington Through Its Center For Commercialization | Beta-2 microglobulin-deficient cells |
| WO2014022423A2 (en) * | 2012-07-31 | 2014-02-06 | Hantash Basil M | Hla g-modified cells and methods |
| US10233454B2 (en) | 2014-04-09 | 2019-03-19 | Dna2.0, Inc. | DNA vectors, transposons and transposases for eukaryotic genome modification |
| EP3129487B1 (en) * | 2014-04-09 | 2020-10-07 | Dna Twopointo Inc. | Enhanced nucleic acid constructs for eukaryotic gene expression |
| JP6836990B2 (ja) * | 2014-11-26 | 2021-03-03 | アクセラレイテッド・バイオサイエンシズ・コーポレーション | 誘導肝細胞およびそれらの使用 |
| EP3229586A4 (en) | 2014-12-10 | 2018-10-24 | Regents of the University of Minnesota | Genetically modified cells, tissues, and organs for treating disease |
| HUE056009T2 (hu) | 2015-10-08 | 2022-01-28 | Dna Twopointo Inc | DNS vektorok, transzpozonok és transzpozázok eukarióta genom módosítására |
| EP3468356A4 (en) * | 2016-06-14 | 2020-02-26 | Regents Of The University Of Minnesota | GENETICALLY MODIFIED CELLS, TISSUES AND ORGANS FOR THE TREATMENT OF A DISEASE |
| JP2019521688A (ja) | 2016-07-26 | 2019-08-08 | ザ ユニバーシティ オブ ノース カロライナ アット チャペル ヒル | 眼におけるベクター介在の免疫寛容 |
| CA3048910A1 (en) * | 2017-01-10 | 2018-07-19 | The General Hospital Corporation | Modified t cells and methods of their use |
| EP3638258A4 (en) * | 2017-06-12 | 2021-09-08 | Sinai Health System | ALLOGRAFT TOLERANCE WITHOUT REQUIRING SYSTEMIC IMMUNE SUPPRESSION |
| CA3086923A1 (en) * | 2017-12-28 | 2019-07-04 | Gritstone Oncology, Inc. | Antigen-binding proteins targeting shared antigens |
| US10883084B2 (en) | 2018-10-05 | 2021-01-05 | Xenotherapeutics, Inc. | Personalized cells, tissues, and organs for transplantation from a humanized, bespoke, designated-pathogen free, (non-human) donor and methods and products relating to same |
| TW202035682A (zh) | 2018-12-14 | 2020-10-01 | 比利時商普羅米修亞生物科技股份有限公司 | 表現hla-g之肝先驅細胞及取得包含該等細胞之此等細胞組成物之方法與其用途 |
| SG11202106169SA (en) * | 2018-12-14 | 2021-07-29 | Promethera Therapeutics Sa | Cell composition comprising liver progenitor cells expressing hla-e |
| JP7313898B2 (ja) | 2019-05-13 | 2023-07-25 | キヤノン株式会社 | ポリゴンミラー、画像形成装置、光偏光器および光走査装置 |
| US20210246426A1 (en) * | 2020-02-10 | 2021-08-12 | Rubius Therapeutics, Inc. | Engineered Erythroid Cells Including HLA-G Polypeptides and Methods of Use Thereof |
| CA3185067A1 (en) * | 2020-05-26 | 2021-12-02 | Kouichi Tamura | Hypoimmunogenic cells |
| CN114746152A (zh) * | 2020-08-23 | 2022-07-12 | 应用干细胞有限公司 | Hla-f修饰的细胞和方法 |
| EP4251741A1 (en) | 2020-11-30 | 2023-10-04 | CRISPR Therapeutics AG | Gene-edited natural killer cells |
| CN113234664B (zh) * | 2021-05-11 | 2024-05-10 | 澳门大学 | 一种胰腺祖细胞的制备方法及其应用 |
| CA3234231A1 (en) * | 2021-10-21 | 2023-04-27 | Vertex Pharmaceuticals Incorporated | Hypoimmune cells |
Family Cites Families (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA1339354C (en) | 1988-09-01 | 1997-08-26 | The Whitehead Institute For Biomedical Research | Recombinant retroviruses with amphotropic and ecotropic host ranges |
| US5464764A (en) | 1989-08-22 | 1995-11-07 | University Of Utah Research Foundation | Positive-negative selection methods and vectors |
| US5534423A (en) | 1993-10-08 | 1996-07-09 | Regents Of The University Of Michigan | Methods of increasing rates of infection by directing motion of vectors |
| US6686198B1 (en) | 1993-10-14 | 2004-02-03 | President And Fellows Of Harvard College | Method of inducing and maintaining neuronal cells |
| US6013517A (en) | 1994-05-09 | 2000-01-11 | Chiron Corporation | Crossless retroviral vectors |
| US5650135A (en) | 1994-07-01 | 1997-07-22 | The Board Of Trustees Of The Leland Stanford Junior University | Non-invasive localization of a light-emitting conjugate in a mammal |
| US5843780A (en) | 1995-01-20 | 1998-12-01 | Wisconsin Alumni Research Foundation | Primate embryonic stem cells |
| US5741657A (en) | 1995-03-20 | 1998-04-21 | The Regents Of The University Of California | Fluorogenic substrates for β-lactamase and methods of use |
| US5744320A (en) | 1995-06-07 | 1998-04-28 | Promega Corporation | Quenching reagents and assays for enzyme-mediated luminescence |
| US6982431B2 (en) | 1998-08-31 | 2006-01-03 | Molecular Devices Corporation | Sample analysis systems |
| US6031094A (en) | 1998-07-23 | 2000-02-29 | The Regents Of The University Of California | Beta-lactam substrates and uses thereof |
| US7005252B1 (en) | 2000-03-09 | 2006-02-28 | Wisconsin Alumni Research Foundation | Serum free cultivation of primate embryonic stem cells |
| CN1302115C (zh) | 2001-04-23 | 2007-02-28 | 埃麦克萨有限公司 | 电穿孔用缓冲溶液及上述溶液的使用方法 |
| AU2003304106C1 (en) | 2002-05-17 | 2010-10-28 | Mount Sinai School Of Medicine Of New York University | Mesoderm and definitive endoderm cell populations |
| WO2004072232A2 (en) | 2003-01-31 | 2004-08-26 | Promega Corporation | Covalent tethering of functional groups to proteins |
| US7960166B2 (en) | 2003-05-21 | 2011-06-14 | The General Hospital Corporation | Microfabricated compositions and processes for engineering tissues containing multiple cell types |
| ES2354160T3 (es) * | 2003-10-21 | 2011-03-10 | Merck Serono Sa | Secuencia de adn mínima que actúa como un aislante de cromatina, y su uso en la expresión de proteínas. |
| US8673634B2 (en) | 2003-11-13 | 2014-03-18 | Massachusetts Eye & Ear Infirmary | Method for the treatment of hearing loss |
| JP5291341B2 (ja) * | 2004-11-08 | 2013-09-18 | クロマジェニックス ベー ヴェー | タンパク質を高レベルで発現する宿主細胞の選定 |
| US20070036773A1 (en) * | 2005-08-09 | 2007-02-15 | City Of Hope | Generation and application of universal T cells for B-ALL |
| CN103555655B (zh) * | 2005-10-21 | 2018-02-27 | 细胞研究私人有限公司 | 自脐带羊膜分离和培养干/祖细胞及其分化的细胞的应用 |
| US8048999B2 (en) | 2005-12-13 | 2011-11-01 | Kyoto University | Nuclear reprogramming factor |
| US8278104B2 (en) | 2005-12-13 | 2012-10-02 | Kyoto University | Induced pluripotent stem cells produced with Oct3/4, Klf4 and Sox2 |
| US20090156532A1 (en) * | 2006-01-24 | 2009-06-18 | The University Of Chicago | SNP BINDING SITE FOR microRNAs IN HLA-G |
| WO2007091078A2 (en) * | 2006-02-10 | 2007-08-16 | Axordia Limited | Polypeptide conjugate comprising hla-g and uses thereof, stem cells transfected with hla-g |
| WO2008054821A2 (en) | 2006-10-30 | 2008-05-08 | Promega Corporation | Mutant hydrolase proteins with enhanced kinetics and functional expression |
| WO2008121894A2 (en) * | 2007-03-30 | 2008-10-09 | Escape Therapeutics, Inc. | Endogenous expression of hla-g and/or hla-e by mesenchymal cells |
| EP2362912A1 (en) * | 2008-11-14 | 2011-09-07 | Life Technologies Corporation | Compositions and methods for engineering cells |
| US8592211B2 (en) | 2009-03-20 | 2013-11-26 | The Rockefeller University | Enhanced PiggyBac transposon and methods for transposon mutagenesis |
| US8048675B1 (en) | 2010-05-12 | 2011-11-01 | Ipierian, Inc. | Integration-free human induced pluripotent stem cells from blood |
| WO2014022423A2 (en) | 2012-07-31 | 2014-02-06 | Hantash Basil M | Hla g-modified cells and methods |
| CN103333861A (zh) * | 2013-06-03 | 2013-10-02 | 浙江省台州医院 | 特异性表达hla-g1抗原的细胞株k562 |
| CN111808819A (zh) * | 2020-05-26 | 2020-10-23 | 台州恩泽医疗中心(集团) | 表达hla-g3异构体标准蛋白的细胞株及其应用 |
-
2013
- 2013-07-30 WO PCT/US2013/052767 patent/WO2014022423A2/en active Application Filing
- 2013-07-30 ES ES13825511T patent/ES2716577T3/es active Active
- 2013-07-30 KR KR1020157005165A patent/KR101923632B1/ko active IP Right Grant
- 2013-07-30 ES ES18211100T patent/ES2837487T3/es active Active
- 2013-07-30 EP EP18211100.5A patent/EP3483178B1/en active Active
- 2013-07-30 CN CN202110063720.2A patent/CN113151179A/zh active Pending
- 2013-07-30 AU AU2013296564A patent/AU2013296564C1/en active Active
- 2013-07-30 KR KR1020197036122A patent/KR102184947B1/ko active IP Right Grant
- 2013-07-30 JP JP2015525519A patent/JP6297559B2/ja active Active
- 2013-07-30 CA CA2882028A patent/CA2882028C/en active Active
- 2013-07-30 EP EP21209423.9A patent/EP4015530A1/en active Pending
- 2013-07-30 EP EP13825511.2A patent/EP2880054B1/en active Active
- 2013-07-30 SG SG11201500799YA patent/SG11201500799YA/en unknown
- 2013-07-30 EP EP20196775.9A patent/EP3805261A1/en not_active Withdrawn
- 2013-07-30 KR KR1020207033804A patent/KR102366081B1/ko active IP Right Grant
- 2013-07-30 CN CN201380051169.3A patent/CN105612176B/zh active Active
- 2013-07-30 KR KR1020187034048A patent/KR102055438B1/ko active IP Right Grant
-
2015
- 2015-01-28 US US14/608,004 patent/US9714280B2/en active Active
-
2017
- 2017-06-16 US US15/625,964 patent/US10502738B2/en active Active
-
2018
- 2018-02-21 JP JP2018028816A patent/JP6502544B2/ja active Active
- 2018-08-23 AU AU2018220087A patent/AU2018220087B2/en active Active
-
2019
- 2019-03-20 JP JP2019052699A patent/JP7294838B2/ja active Active
- 2019-10-22 US US16/659,918 patent/US11977073B2/en active Active
-
2020
- 2020-03-27 US US16/832,321 patent/US20200309776A1/en active Pending
-
2021
- 2021-04-14 JP JP2021068195A patent/JP7379410B2/ja active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7379410B2 (ja) | Hla g改変された細胞および方法 | |
| US20230272429A1 (en) | Modification of blood type antigens | |
| JP2020532291A (ja) | 二段階相同組換え修復によるスカーレスゲノム編集 | |
| EP4090349A2 (en) | Transplanted cell protection via inhibition of polymorphonuclear cells |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20160728 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20160728 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20170613 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20170831 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20170905 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20180123 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20180221 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6297559 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313113 |
|
| R371 | Transfer withdrawn |
Free format text: JAPANESE INTERMEDIATE CODE: R371 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313113 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |