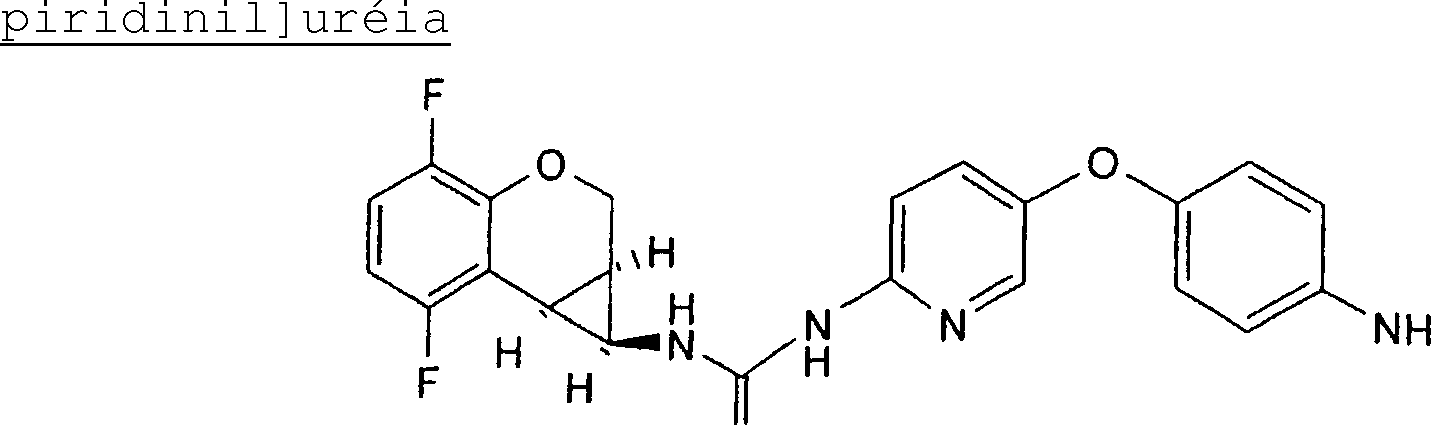
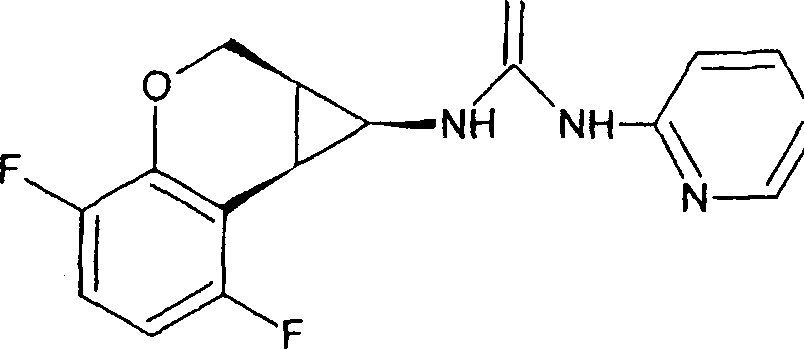
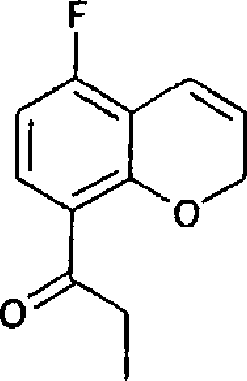
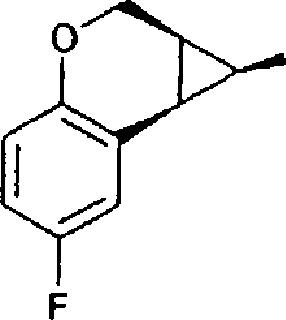
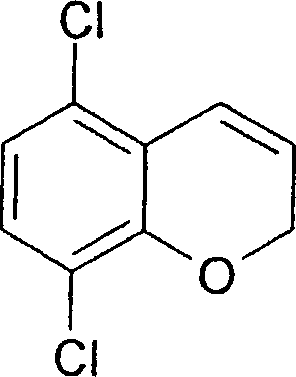
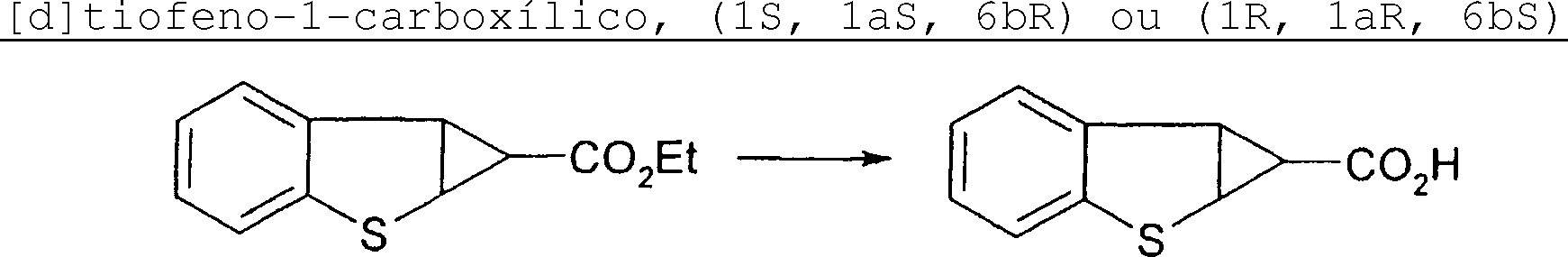(54) Título: COMPOSTO N-[(lS,lAR,7BR)-4,7-DIFLUORO-l,lA,2,7B-TETRAIDROCICLOPROPA[C] CROMEN-l-IL]-N-[5-(4-(SULFONAMIDO)FENOXI)-2-PIRIDINIL]UREIA, COMPOSIÇÃO FARMACÊUTICA E USO DO DITO COMPOSTO PARA PROFILAXIA OU TRATAMENTO DE INFECÇÕES POR HIV-1 (51) Int.CI.: C07D 213/75; C07D 405/12; C07D 409/12; A61K 31/425; A61K 31/426; A61K 31/44; A61P 31/18 (30) Prioridade Unionista: 08/01/2004 SE 0400021-2, 09/03/2004 SE 0400585.6 (73) Titular(es): MEDIVIR AB (72) Inventor(es): CHRISTIAN SUND; NATHALIE ROUE; STEFAN LINDSTRÕM; DMITRY ANTONOV; CHRISTER SAHLBERG; KATARINA JANSSON (85) Data do Início da Fase Nacional: 03/07/2006
COMPOSTO Ν-[(IS,laR,7bR)-4,7-DIFLUORO-l, la, 2, 7bTETRAIDROCICLOPROPA[c]CROMEN-l-IL]-Ν'-[5-(4-(SULFONAMIDO)FENÓXI) -2-PIRIDINIL]UREIA, COMPOSIÇÃO FARMACÊUTICA E USO DO DITO COMPOSTO PARA PROFILAXIA OU TRATAMENTO DE INFECÇÕES POR HIV-1
CAMPO TÉCNICO
A invenção relaciona-se a inibidores da transcriptase reversa não nucleotídicos (NNRTIs) ativos contra HIV-1 e tendo uma resistência e perfil farmacocinético aumentado. A invenção ainda relaciona-se a novos intermediários na síntese de tais compostos e o uso de compostos em composições e métodos antivirais.
ANTECEDENTE DA INVENÇÃO
Nosso pedido PCT W002/070516 & W003/020705 previamente depositado reivindica novos NNRTIs de fórmula I
onde ;
Ri é O, S;
R2 é um heterociclo contendo nitrogênio opcionalmente substituído, tal como piridil;
R3 é H, alquilCi-C3;
R4-R7 são independentemente selecionados de H, alquilCi-C6, alquenilC2-C6, alquinilC2-C6, haloalquilCi-C6, de 23/07/2018, pág. 10/150 alcanoilCi-C6, haloalcanoilCi-C6, alcóxiCi-C6, haloalcóxiCiC6, alcóxiCi-C6alquilCi-C6, haloalquilóxiCi-C6alquilCi-C6, hidroxialquilCi-C6, aminoalquilCi-C6, carboxialquilCi-C6, cianoalquilCi-C6, amino, carbóxi, carbamoil, ciano, halo, hidróxi, ceto e afins;
X é - (CH2) n-D-(CH2)m- ou X é -(CRaRb)cD é -NR8-, -O-, -S-, -S(=O)- ou —S(=O)2—
Rs é H, alquilCi-C3
Ra e Rb são independentemente H, alquilCi-C3, OH ou Ra e Rb juntos são =0 nem são independentemente 0 ou 1;
C é 1, 2 ou 3 e sais farmaceuticamente aceitáveis e prófármacos.
Exemplo 20 de W002/070516 revela o composto
Que é exposta para ter um EDso de 7 nM contra o HIV selvagem (HIViub) .
Nosso co-pendente, mas como os dados prioritários não publicados do pedido PCT W004/021969 revelam compostos geralmente de fórmula I acima, mas em que R2 é pirid-2-il substituído na posição 5 com um grupo de fórmula -(CHRn)PE-(CHRii) q-Rio onde E é -CH2-, -CHOH-, -C=O-, -NRg-, -O-, -S-,
-S(=O)2-;
de 23/07/2018, pág. 11/150
P e q são independentemente 0, 1 ou 2, onde p+q <
2;
Rio é um anel monocíclico que é opcionalmente morfoiinometi1substituído com halo, ciano, morfolinoceto-; e
Rn é independentemente H, alquilCi-C3, alquilCi-C3 substituído por halo ou hidróxi.
Embora os NNRTIs uréia e tiouréia revelados nos documentos acima são sensivelmente ativos contra transcriptase reversa, especialmente a do HIV-1, a natureza do vírus HIV com sua ausência extrema de fidelidade replicativa e conseqüente tendência ao desenvolvimento rápido de resistência instiga uma demanda para agentes antiretrovirais adicionais com aumentado desempenho antiviral contra a problemática de mutantes de escape de drogas, notavelmente nas posições RT 100, 103 e/ou 181.
BREVE DESCRIÇÃO DA INVENÇÃO ou
Em acordo com um primeiro aspecto da invenção são providos compostos de fórmula Z:
onde ;
A é CH ou N;
Ri é um substituinte para um átomo de carbono no anel contendo A selecionado de -S(=O)pRa, onde Ra é alquil-Ci-C4, -ORx, -NRxRx, -NHNRxRx, de 23/07/2018, pág. 12/150
NHNHC(=O)ORx, -NRxOH;
-C(=O)=Rb, onde Rb é alquil-Ci-C4, ORx, -NRxRx, -NRxNRxRx,
-NH-alquil-Ci-Cs-C(=O)ORx;
-NRxRc, onde Rc é H, alquilC1-C4, -NRxRx; -C(=O)Rd, -CN,
S(=O)pRx onde Rd é alquilC1-C4, -ORx, -NRxRx alquil-C1-C3-O-alquil-C1-C3-C (=O) ORx; alquil-C1-C3-COORx;
alquil-C1-C3-OH ou éteres ou ésteres de alquilC1-C4 dos mesmos;
-(O-alquil-C1-C3)q-O-Rx;
Um anel aromático de 5 ou 6 membros tendo 1-3 heteroátomos;
p e q são independentemente selecionados de 1 ou
2;
Rx é independentemente selecionado de H, alquilC1C4, ou acetil; ou um par de Rx pode junto com o átomo N adjacente formar um anel pirrolidina, piperidina, piperazina ou morfolina;
R2 é um substituinte para um átomo de carbono no anel contendo A e é H, halo, ciano, alquilC1-C4, haloalquilC1-C4;
L é -O-, -S(=O)r- ou -CH2-, onde r é 0, 1 ou 2;
R3 é H, alquilC1-C3;
R4-R7 são independentemente selecionados de H, alquilC1-C6, alquenilC2-Ce, alquinilC2-C6, haloalquilC1-Ce, de 23/07/2018, pág. 13/150 alcanoilCi-Ce, haloalcanoilCi-Ce, alcóxiCi-Ce, haloalcóxiCiCe, alquilóxiCi-CealquilCi-Ce, haloalquilóxiCi-CealquilCi-Ce, hidróxialquilCi-Ce, aminoalquilCi-C6, carboxialquilCi-C6, cianoalquilCi-C6, amino, carbóxi, carbamoil, ciano, halo, hidróxi, ceto;
X é -(CR8R8')n-D-(CR8R8')m-;
T é O ou S;
D é uma ligação, -NR9-, -O-, -S-, -S(=O)- ou S(=O)2-;
n e m são independentemente 0, i ou 2, desde que eles não sejam ambos 0 quando D é uma ligação;
R8 e R8' são independentemente H, alquilCi-C3, haloalquilCi-C3, hidróxi, ou R8 e R8^ juntos com seu átomo de C adjacente é -C(=O)R9 é independentemente H, alquilCi-C3; e sais farmaceuticamente aceitáveis e pró-fármacos dos mesmos, com a condição que Ri como -C(=O)Rb não é morfolinoceto-.
O valor correntemente preferido para T é O, que é um derivado de uréia, embora T como S (isto é um derivado de tiouréia) é também altamente potente.
O valor correntemente preferido para R3 é H.
Preferivelmente R4 é hidrogênio, halo, haloalquilCi-C3, ou hidróxi, especialmente flúor.
Preferivelmente R5 é halo, alquil Ci-3 carbonil, haloalquilCi-C3, alquilóxiCi-3 ou H, especialmente flúor e mais preferivelmente H.
de 23/07/2018, pág. 14/150
Preferivelmente 4a é hidrogênio, halo, haloalquilCi-C3, alquilóxiC1-C3, alquil C1-3 carbonil, ciano ou etinil, especialmente metóxi ou flúor e mais preferivelmente H.
Preferivelmente R7 é hidrogênio, haloalquilC1-C3, halo, alquilóxiC1-3, ou alquil C1-3 carbonil, mais preferivelmente flúor.
Preferivelmente R5 e R6 são H e R4 e R7 são halo, mais preferivelmente ambos são flúor. Configurações alternativas preferidas incluem aquelas em que R5 e R6 são H, R4 é flúor e R7 é acetil ou ciano.
Um valor conveniente para pelo menos um de R4-R7 é haloalquilC1-C3, tal como -CF2H, -CFH2, -CH2CF3 ou -CF2F3, e especialmente -CF3.
Grupos -S(=O)pRa protegidos para R1 incluem aqueles em que p é 2 ou especialmente 1, e em que Ra é alquil, tal como ciclopropil, metilciclopropil, e mais preferivelmente metil. Grupos preferidos, por conseguinte incluem metilsulfonil ou metilsulfinil.
Grupos -S(=O)pNRxRx protegidos adicionais incluem aqueles em que Rx são H ou Me ou em que um é H e o outro Me, ciclopropil ou metilciclopropil, mais preferivelmente NH2. Grupos preferidos, por conseguinte incluem sulfonamida.
Grupos -C=(O)-Rb protegidos para R1 incluem aqueles onde Rb é NRxRx ou NHRxRx, especialmente Nmetilcarboxamida, hidrazinocarbonil e -C(=O)NHNHC(=O)Me. Grupos -C(=O)-Rb preferidos adicionais incluem -C-(=O)NRx'N-morfolino, -C(=O)NRx'-N-piperidino, -C(=O)NRx'-Nde 23/07/2018, pág. 15/150 pirrolidino, -C(=O)NRx'-N-piperazino, onde Rx é metil, acetil ou preferivelmente H.
Grupos -NRxRc protegidos para Ri incluem aqueles em que Rx é H ou Me e aqueles em que Rc é -C(=O)Rd, onde Rd é alquil e S(=O)pRx, especialmente ciclopropilamida e acetamida.
pirimidinil, isoxazolil,
Grupos -alquilCi-C3-COORx protegidos para Ri incluem carboxietil e ésteres alquilCi-C2 dos mesmos.
Grupos -alquil-C1-C3-ORx protegidos para R1 incluem hidroxietil e éteres e ésteres alquilCi-C2 dos mesmos.
Grupos -(O-alquilCi-C3)-O-Rx protegidos para Ri incluem espécies contendo etóxi especialmente 2(metoxietóxi)etóxi.
Exemplos de anéis heteroatômicos para Ri incluem furil, tienil, piranil, pirrolil, pirrolinil, pirrolidinil, pirazolil, pirazolinil, pirazolidinil, imidazolil, imidazolinil, imidazolidinil, piridil, piridazinil, oxazolil, isoxazolidinil, tiazolil, pirazinil, oxazolidinil, tiazolidinil, isotiazolil, isotiazolidinil, especialmente anéis de 5 membros tais como tiazolil, tiadiazolil, pirazolil, diazolil e mais preferivelmente triazolil.
O valor correntemente preferido para L é -O-.
Os compostos de fórmula Z podem ser administrados como uma mistura racêmica, mas preferivelmente a fração ciclopropil intermedeia a função (tio)uréia, X e o anel fenil (denotado T abaixo) é pelo menos 75% tal como cerca de 90% enantiomericamente puro com respeito à conformação:
de 23/07/2018, pág. 16/150
Isômeros óticos preferidos dos compostos de fórmula I mostram um valor de rotação ótica negativo. Tais isômeros, por exemplo quando X é -O-CH2-, tendem a eluir menos rapidamente de um cromatograma quiral, por exemplo AGP
150x10 mm quiral, 5 pm; Crom Tech LTD Colomn, taxa de vôo 4 mL/min, fase móvel vol 89% 10 mM HOAc/NH4OAc em acetonitrila. Nas bases das análises preliminares de cristalografia de raio-X uma configuração absoluta presentemente favorecida parece ser:
O valor correntemente preferido para D é -O-.
Valores convenientemente para nem incluem 1:0 e 1:1. Valores preferidos de n:m incluem 0:2 e especialmente 0:1,
|
que é um derivado cromano. Convenientemente cada Rs e Rs |
é |
|
H. Alternativamente, |
no caso onde n é |
0 e m é 1, R |
s é |
|
vantajosamente H e Rs' |
é OH. |
|
|
|
Compostos |
particularmente |
preferidos |
têm |
|
estereoquimica correspondente a (IS, laR, |
7bR)-l, la, 2, |
7b- |
|
tetraidrociclopropa[c] |
cromen-l-il. Em |
consideração |
a |
Petição 870180063303, de 23/07/2018, pág. 17/150 claridade é notada que a estrutura

A expressão alquilCi-Cn onde n é 3 ou 4 ou alquil reduzido inclui tais grupos como metil, etil, n-propil, isopropil, ciclopropil, n-butil, s-butil, t-butil, ciclopropil, metilciclopropil e afins. O termo halo referese a cloro, bromo, flúor e iodo, especialmente flúor. Alcóxi Ci-Cn refere-se a grupos tais como metóxi, etóxi, propóxi, cicloporpóxi, t-butóxi e afins. Alquenil C2-Cn refere-se a grupos tais como vinil, l-propen-2-il, l-buten-4-il, 1penten-5-il, 1-buten-l-il e afins. Alquiltio Ci-Cn inclui metiltio, etiltio, t-butiltio e afins. Alcanoilóxi Ci-Cn inclui acetóxi, propionóxi, fornilóxi, butirilóxi e afins. Alquenóxi C2-Cn inclui etenilóxi, propenilóxi, isobutoxieteenil e afins. HaloalquilCi-Cn (incluindo substituintes complexos contendo esta fração tal como haloalquilóxiCi-Cn) inclui alquis como definidos aqui substituídos 1 a 3 vezes por um halogênio incluindo trifluormetil, 2-dicloroetil, 3,3-difluorpropil e afins. O termo amina inclui grupos tais como NH2, NHMe, (NMe)2 que podem ser opcionalmente substituídos com halogênio, de 23/07/2018, pág. 18/150 acilóxiCi-C7, alquilCi-Ce, alcóxiCi-Ce, nitro, carbóxi, carbamoil, carbamoilóxi, ciano, metilsulfonilamino e afins.
Carbóxi, carboximetil e carbamoil incluem o correspondente alquilC1-Ce farmaceuticamente aceitável e ésteres aril.
Pró-fármacos dos compostos de fórmula I são aqueles compostos que seguindo a administração ao paciente liberam um composto de fórmula I in vivo. Pró-fármacos típicos são éteres farmaceuticamente aceitáveis e especialmente ésteres (incluindo ésteres de fosfato) quando qualquer R4-R7 ou R1 ou R2 representam uma função hidróxi, amidas farmaceuticamente aceitáveis ou carbamatos quando qualquer dos substituintes R1 ou R2 ou R4-R7 representam uma função amina ou ésteres farmaceuticamente aceitáveis quando o substituinte R1, R2 ou R4-R7 representam uma função carbóxi. Ésteres farmaceuticamente aceitáveis incluem ésteres alquil, incluindo acetil, etanoil, butiril, tbutiril, e pivaloil, ésteres fostato e ésteres sulfônicos (isto é aqueles derivados de RSO2OH, onde R é alquil reduzido ou aril). Ésteres farmaceuticamente aceitáveis incluem éteres alquil reduzido e éteres revelados em WO00/47561, especialmente metoxiaminoacil e atoxiaminoacil.
Os compostos de fórmula Z podem formar sais que formam uma aspecto adicional da invenção. Sais farmaceuticamente aceitáveis apropriados dos compostos de fórmula I incluem sais de ácidos orgânicos, especialmente ácidos carboxílicos, incluindo mas não limitados a acetato, trifluoracetato, lactato, gluconato, citrato, tartarato, maleato, malato, pantotenato, isotionato, adipato, alginato, de 23/07/2018, pág. 19/150 aspartato, benzoato, butirato, digluconato, ciclopentanato, glucoeptanato, glicerofosfato, oxalato, heptanoato, hexanoato, fumarato, nicotinato, palmoato, pectinato, 3fenilpropionato, picrato, pivalato, proprionato, tartrato, lactobionato, pivolato, canforato, undecanoato e succinato, ácidos sulfônicos orgânicos tais como metanosulfonato, etanosulfonato, 2-hidroxietano sulfonato, canforsulfonato,
2-naftalenosulfonato, benzenosulfonato, pclorobenzenosulfonato e p-toluenosulfonato; e ácidos inorgânicos tais como hidrocloreto, hidrobrometo, hidroiodeto, sulfato, bisulfato, hemisulfato, tiocianato, persulfato, ácidos fosfórico e sulfônico.
Grupo protetor hidróxi como aqui utilizado referese a um substituinte que protege grupos hidroxil contra reações indesejáveis durante os procedimentos sintéticos tais como aqueles grupos de O-proteção demonstrados em Greene, “Protective Groups in Organic Synthesis, (John Wiley & Sons, Nova Iorque (1981)) . Grupos hidróxi protetores compreendem metil éteres substituídos, por exemplo, metoximetil, benziloximetil, 2-metoxietoximetil, 2(trimetilsilil)etoximetil, t-butil e outros éteres alquil reduzidos, tais como isopropil, etil e especialmente metil, benzil e trifenilmetil; éteres tetraidropiranil; éteres etil substituídos, por exemplo, 2,2,2-tricloetil; silil éteres, por exemplo, trimetilsilil, t-butildimetilsilil e tbutildifenilsilil; e ésteres preparados pela reação do grupo hidroxil com um ácido carboxílico, por exemplo, acetato, propionato, benzoato a afins.
de 23/07/2018, pág. 20/150
Similarmente, grupo N-protetor como aqui utilizado refere-se àqueles grupos N-protetores convencionais revelados em Greene, “Protective Groups in Organic Synthesis, John Wiley & Sons Nova Iorque 1981.
A invenção ademais provê composições farmacêuticas compreendendo os compostos da invenção e veículos ou diluentes farmaceuticamente aceitáveis dos mesmos. Aspectos adicionais da invenção provêem métodos de inibição de HIV compreendendo administração de um composto de fórmula Z a um paciente afligido com HIV-1 ou exposto ao mesmo. O HIV-1 pode compreender um mutante de escape de droga, tal como uma cepa de HIV compreendendo as mutações nas mutações 100, 103 e/ou 181, especialmente os mutantes K103N e/ou L100I.
A invenção também se estende ao uso de compostos de fórmula Z na terapia, tal como na preparação de um medicamento para o tratamento de infecções por HIV.
Nas condições de tratamento causado por HIV, os compostos de fórmula Z são preferivelmente administrados em uma quantidade para alcançar um nível plasmático de cerca de 100 a 5000 nM, tal como 300 a 2000 nM. Este corresponde a uma taxa de dosagem, dependendo da biodisponibilidade da formulação, na ordem de 0,01 a 10 mg/kg/dia, preferivelmente 0,1 a 2 mg/kg/dia. Uma taxa de dosagem típica para um adulto normal será de cerca de 0,05 a 5 g por dia, preferivelmente 0,1 a 2 g tal como 500-750 mg, em uma a quatro unidades de dose por dia. Como com todos os farmacêuticos, taxas de dosagens variarão com o tamanho e condição metabólica do paciente, bem como com a severidade da infecção e pode ser de 23/07/2018, pág. 21/150 necessário o ajuste de medicações concomitantes.
Mantendo a prática usual com inibidores de HIV é vantajoso co-administrar de um a três antivirais para proporcionar respostas sinergísticas e assegurar modelos de resistência complementares. Como antivirais adicionais podem incluiu-se AZT, ddl, ddC, D4T, 3TC, DAPD, alovudina, abacavir, adefovir, adefovir dipivoxil, bis-POC-PMPA, GW420 867X, foscamet, hidroxiuréia, Hoechst-Bayer HBY 097, efavirenz, trovirdina, MIV-150, capravirine, nevirapina, delaviridina, tipranavir, emtricitabina, PFA, H2G (omaciclovir), MIV-606 (estearato de valomaciclovir), TMC126, TMC-125, TMC-120, efavirenz, DMP-450, lovirida, ritonavir, (incluindo caletra), lopinavir, saquinavir, lasinavir, indinavir, amprenavir, fosfato de amprenavir, nelfinavir e afins, tipicamente em taxas molares refletindo suas respectivas atividades e biodisponibilidades. Geralmente tal taxa será na ordem de 25:1 a 1:25, relativa ao composto de fórmula I, mas pode ser menor, por exemplo, no caso dos antagonistas do citocromo p450 tal como ritonavir.
Compostos da invenção são tipicamente preparados como segue:
Esquema 1 de 23/07/2018, pág. 22/150
NHn
COOH
Re (a) DPPA, Et3N, tolueno; (b) 2-aminopiridina substituída. (c) HC1 aquoso, dioxano; (d) 2-piridil isotiocianato substituído.
Compostos de fórmula geral (I), em que T é 0 (uréia) ou S (tiouréia), Re é a fração (substituída) oxifenil ou oxipiridil, ou o análogo tio, sulfino, sulfona ou metileno de tais éteres e R3 é H, são preparados por ácido métodos mostrados no Esquema ciclopropanocarboxilico 1-Esguema-l é convertido em acil azida e aquecida a 120°C para induzir rearranjo Curtius e prover o isocianato 2-Esguema-l. A uréia 3-Esguema-l é obtida pelo acoplamento do isocianato com a relevante 210 aminopiridina substituída. Hidrólise do isocianato como no passo (c) que resulta na ciclopropilamina 4-Esquema-l, seguida pela reação com um 2-piridil isotiocianato provê a tiouréia 5-Esguema-l. O isotiocianato pode ser preparado opcionalmente do anel 2-aminopiridina substituído por 15 métodos conhecidos, tal como tratamento com tiofosgeno ou tiocarbonildiimidazol.
Variantes R3 de fórmula I são preparados correspondentemente utilizando a aminopiridina substituída
Petição 870180063303, de 23/07/2018, pág. 23/150 por amina apropriada isto é, 2-(N-metilamino)piridina para R3 como metil. Muitas 2-aminopiridinas são comercialmente disponíveis e outras são descritas na literatura ou prontamente derivadas das mesmas, por exemplo, aquelas mostradas no Esquema 2. Compostos T=S podem alternativamente ser preparados do isotiocianato correspondente ao 2-Esquema 2A ou da amina 3, 3a-Esquema 2 e amina-R2 em conjunção com um RC(=S)R' ambos descritos em WO 9303022.
Esquema 2A
(a) base, SMF, calor; (b) redução; (c) oxidação; (d) base, catalisador
Cu, calor
A preparação de 5-0- ou 5-S-substituída-2aminopiridinas adequadas são delineadas no Esquema 2A. 1Petição 870180063303, de 23/07/2018, pág. 24/150
Esquema-2A com substituintes R1 e R2 apropriados, ou precursores (synthons) desses substituintes, é reagido no passo (a) com 5-bromo-2-nitropiridina e uma base, tais como NAH ou Cs2CO3, para proporcionar a substituição do brometo e dar o composto nitro no 2-Esquema-2A. O grupo nitro é então reduzido à amina no passo (b), tipicamente por hidrogenação em pressão atmosférica em presença de catalisadores tais como Pd ou níquel Raney. Transformação de precursores para os substituintes R1 , R2 desejados podem ser feitos no composto nitro do 2-Esquema-2A antes do passo de redução (b) . No caso do sulfanil no 2-Esquema-2A, agentes oxidantes diferentes, por exemplo o peróxido de hidrogênio, convertem o grupo sulfeto a S=(O)r no passo (c), seguido por redução do grupo nitro para dar 5-Esquema-2A. Os compostos tio do 3A-Esquema-2A podem também ser preparados diretamente como no passo (d) por acoplamento de 2- amino-5-bromopiridina com o tiol 1a-Esquema-2A em presença de catalisadores cobre, por exemplo por aquecimento a 150°C com Cu ou Cul em DMF com um base tal como K2CO3.

de 23/07/2018, pág. 25/150
Esquema 2B
(a) NH3, EtOH, calor; (b) Et3SiH, TFA, H2SO4
A preparação das 5-metil-2-aminopiridinas substituídas são delineadas no Esquema 2B. A metanona do 1Esquema-2B com os substituintes Ri e R2 apropriados, ou precursores (synthons) desses substituintes, é reagida no passo (a) com amônia para propiciar a substituição do cloreto e dar o composto amino do 2-Esquema-2B. 0 grupo ceto é então reduzido a CH2 no passo (b) para dar o 3-Esquema-2B.
Esquema 3
Petição 870180063303, de 23/07/2018, pág. 26/150
(a) diazoacetato de etila, catalisador, CH2CI2; (b) cromatografia e então refluxo com LiOH, H2O, MeOH; (c) refluxo com LiOH, H2O, MeOH e então cromatografia; (d) ta, NaOH, H2O, MeOH e então refluxo com LiOH,
H20, MeOH.
Compostos de fórmula gerial (I), em que T é 0 (uréia) ou S (tiouréia), R1' e R2' são R1 e R2, protegidos como necessário com grupos protetores de amino hidroxil, carbóxi convencionais, ou synthons convencionais para R2/R2, R3 é Η, X é -D-CH2, e em que a fração ciclopropil tem a configuração relativa
X
AT
H são preparados por métodos mostrados no Esquema 3. Ciclopropanação de dupla ligação em cromeno l-Esquema-3 com diazoacetato de etila é catalizado por sais de cobre ou ródio (II) tais como Cul, (CuOTf) 2-benzeno, e Rh2(Oac)4 em solventes tais como diclorometano, 1,2-dicloroetano ou clorofórmio. A reação provê uma mistura diastereomérica dos ésteres de etila do ácido ciclopropanocarboxílico no 2Esquema-3, com toda a configuração cis relativa, e seu isômero 3-Esquema-3. Separação por cromatografia em coluna
Petição 870180063303, de 23/07/2018, pág. 27/150 dos diastereoisômeros cis e trans pode ser consumada neste estágio, seguido por hidrólise do 2-Esquema-3 isolado, tal como por refluxo em LiOH metanólico aquoso, para produzir uma mistura racêmica de todo o ácido ciclopropanocarboxílico cis 4-Esquema-3, como descrito no passo (b). Alternativamente, a mistura diastereômica de ésteres de etila pode ser sujeita a hidrólise, e separação conduzida em mistura de ácidos ciclopropanocarboxílicos para prover o isolamento de todo o isômero cis, como no passo (c). Passo (d) envolve o isolamento do éster de etila cis 2-Esquema-3 que pode também ser feito por hidrólise seletiva do 3Esquema-3 trans em temperaturas menores, tal como tratamento com NaOH metanólico aquoso em temperatura ambiente. O éster de etila cis isolado pode então ser hidrolizado na maneira usual para o ácido ciclopropanocarboxílico 4-Esquema-3. O ácido ciclopropanocarboxílico é sujeito a métodos delineados no Esquema 1 para obter a uréia ou tiouréia 5-Esquema-3. Os cromenos 1-Esquema-3 são preparados por métodos mostrados nos Esquemas 4, 5 e 6.
Embora este esquema 3 tem sido ilustrado com uma variante D=O será aparente que manipulações correspondentes serão disponíveis para as variantes D=S, S=O; S(=O)2 e D=NR8. Quando R8 é H, o nitrogênio é tipicamente protegido com um grupo protetor amino secundário convencional, tal como aqueles descritos em Greene & Wuts Protective Groups in Organic Synthesis 2a ed. Wiley NI (1991) .
Esquema 4 de 23/07/2018, pág. 28/150
(a) 3-bromopropina, K2CO3, acetona; (b) Ν,Ν-dietilanilina ou PEG-200,
2252C
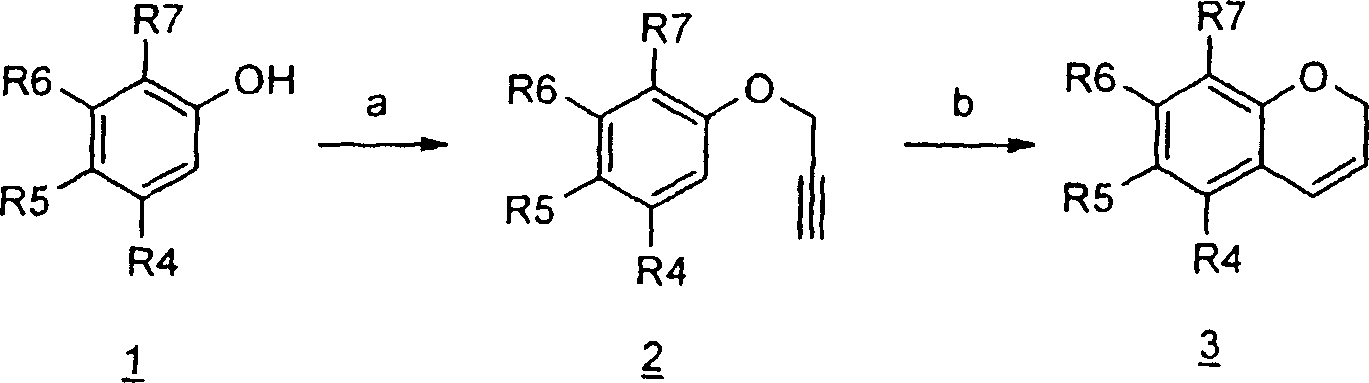
Esquema 4 descreve a preparação de cromenos, incluindo muitos dos fenóis di-substituídos disponíveis comercialmente, tais como aqueles em que o modelo de substituição no anel benzeno é como se segue: R4 e R7 são halo; R4 e R6 são halo, R5 e R7 são halo, R4 é halo e R7 é alquil C1-3 carbonil; e R4 é hidróxi enquanto R5 é alquil C1-3 carbonil. Reação do fenol di-substituído l-Esquema-4 disponível com 3-bromopropina em presença de uma base, tal como K2CO3 em acetona ou NaH em DMF, resultam em substituição nucleofílica do haleto para prover o éter 2Esquema-4. O fechamento do anel pode ser consumado por aquecimento em N, N-dimetilanilina ou polietileno glicol para produzir o cromeno 3-Esquema-4.
Esquema 5
1a
NaBH4, EtOH; (b) ácido p-toluenossulfônico, tolueno, refluxo;
Esquema 5 descreve a preparação de cromenos, utilizados como material de partida no Esquema 3, a partir das cromanonas apropriadamente substituídas, que são
Petição 870180063303, de 23/07/2018, pág. 29/150 facilmente acessados a partir de cromanonas comercialmente disponíveis, por exemplo, aqueles em que uma das posições em
R4 a R7 é substituído com halo ou alcóxiCi-3. Conversão do grupo carbonil em 4-cromanona la-Esquema-5 e o álcool correspondente por um agente redutor adequado tal como boroidreto de sódio em etanol provê 2-Esguema-5. O refluxo de álcool com pequenas quantidades de ácido, tal como p-TsOH em tolueno, causa desidratação de 2-Esguema-5 para a cromeno l-Esquema-3 desejado. Manipulações correspondentes serão disponíveis para outros variantes D. Por exemplo o 2H-1benzotiopirano correspondente é facilmente preparado a partir de tiocroman-4-onas (substituída) disponíveis pela reação com um resultante tal como um hidreto metálico, por exemplo hidreto de alumínio lítio em um solvente orgânico tal como éter, seguido por desidratação tal como refluxo com um ácido, por exemplo, sulfato do ácido de potássio ou afins.
Esquema 6
Petição 870180063303, de 23/07/2018, pág. 30/150 (c) C12P [Pcy3] 2Ru=CHPh, CH2C12 (d) Ph3P+CH=CH2Br-, DBU
Cromenos, para uso como material de partida no Esquema 3, são preparados a partir de o-hidroxibenzaldeídos como demonstrado por métodos delineados no Esquema 6. Reação de l-Esquema-6 com brometo de alquila em presença de um base, tal como K2CO3 em acetona, resulta em substituição nucleofílica do haleto para prover o éter 2-Esguema-6. A reação de wittig transforma o grupo aldeído em oleofina e provê 3-Esquema-6. 0 par de dupla ligações terminais podem passar por metátese intramolecularmente por tratamento com um catalisador tal como o catalisador complexo rutênio de Grubb no passo (c) para produzir o cromeno. Alternativamente l-Esquema-6 pode ser ciclisado diretamente como mostrado no passo (d) na legenda abaixo.
Esquema 7
(a) Pd(0), DPPP, Et3N, (CH3)3SiC=CH; (b) Pd(0), butil vinil éter, DMF;
(c) Pd(0), Zn(CN)2, DMF; (d) NaOH, H2O, MeOH
Petição 870180063303, de 23/07/2018, pág. 31/150
Pd (Ο) catalisou o acoplamento de triflato 1Esquema-7 levando a trifluormetanosulfonilóxi e substituição do grupo a introdução de outros substituintes em Rõ . Então, o Esquema 7 provê a preparação da síntese de intermediários para uso no esquema 3 para dar a uréia ou tiouréia 5-Esquema-3 em que R6 é ciano, etinil, ou alquil C1-3 carbonil.
Esquema 8
(a) BuLi/ZnCl2, THF; Pd(OAc)2, BrCH=CHCOOEt; DIBAL (b) TsNHN=CHCOCl; PhNMe2, Net3, CH2C12
Petição 870180063303, de 23/07/2018, pág. 32/150 (c) Rh2(5-R-MEPY)4, abs diclorometano sem gás (d) 30% HBr, AcOH (e) NaOH, H2O (f) NaOH, CO2, I-PrI/DMSO (g) IPrOH, HCl, DEAD, PPha, THF (h) NaOH, MeOH:H2O (i) 1.BBra, CH2Cl2 2. CH3CN 3.NaOH, água (j) 1. BuLi/ZnCL2, THF; Pd(OAc) 2. cpd 9-Esquema-8 3. reagente de Jones (ácido crômico, ácido sulfúrico em acetona)
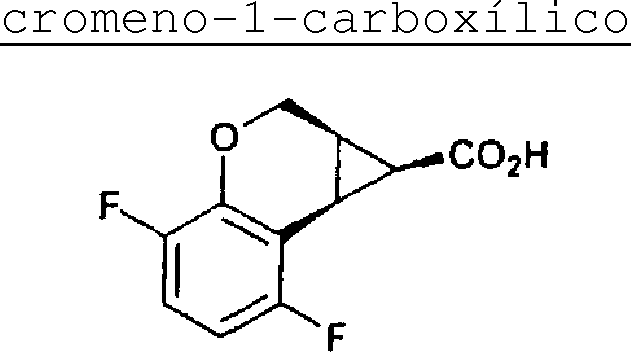
Rotas convenientes para compostos em que X é -CH2O- são descritos no Esquema 8, onde Ra e Rb são substituintes R4-R7 opcionais, que são adequadamente protegidos com grupos de proteção convencionais como necessário e Rc é um éster alquil reduzido. Fenol 1-Esquema8 substituído opcionalmente que é hidróxi-protegido com um grupo protegido tal como metil, MOM e afins é reagido com uma base tal como BuLi ou afins em um solvente tal como THF ou afins e transformados para sal de zinco por adição de cloreto de zinco ou afins. Um catalisador tal como Pd(OAc)2 ou afins é adicionado com um acrilato adicionado tal como alquil-cis-3-haloacrilato reduzido, por exemplo,
BrCH=CHCOOEt ou afins. A mistura reacional é arrefecida e um agente redutor tal como DIBAL ou afins é adicionado em porções e extinta para produzir 2-Esquema-8. Uma hidrazona tal como a p-toluenosulfonilidrazona do cloreto de ácido glioxílico ou afins e uma base tal como N,N-dimetilanilina ou afins é adicionada em um solvente tal como CH2Cl2 ou afins seguida pela adição de outra base tal como Et3N ou de 23/07/2018, pág. 33/150 afins para produzir 3-Esquema-8. O produto de reação é dissolvido em um solvente tal como diclorometano ou afins que é preferivelmente sem gás. Um catalisador de Doyle quiral tal como Rh2(5-R-MEPy)4 (US 5175311, disponível Aldrich ou Johnson Matthey), ou afins é adicionado para produzir 4-Esquema-8 em um alto excesso enantiomérico tal como maior que 80, preferivelmente maior que 90% ee. Preferivelmente, este composto é primeiro reagido com BBr3 em diclorometano seguido pela adição de acetonitrila à mistura reacional e finalmente sodiohidróxido é adicionado para dar 6-Esquema-8. Alternativamente, este produto (4Esquema-8) é um anel aberto com um eletrófilo preferivelmente HBr ou afins sob em conjunto com um ácido tal como AcOH ou afins. Sob condições ácidas um fechamento de anel espontâneo acontece, para formar cromenona 5Esquema-8. Quando sujeita a condições básicas tal como NaOH ou afins, a cromenona se rearranja para formar o ácido cromenciclopropilcarboxílico 6-Esquema-8. Alternativamente, 4-Esquema-8, por exemplo quando o grupo protetor fenólico é MOM, pode ser sujeito a condições básicas tais como NaOH, dióxido de carbono e um haleto alquil reduzido tal como iPrl em um solvente tal como DMSO para abrir a lactona e produzir o éster alquil 7-Esquema-8. A substituição do grupo protetor hidróxi e fechamento do anel com a fração hidroximetil livre ocorrem em condições acídicas tal como iPrOH/HCl ou afins seguidos por DEAD; PPH3 em um solvente orgânico tal como THF ou afins. Alternativamente, em um método convergente, composto 1-Esquema-8 é reagido com BuLi e transformado em um de 23/07/2018, pág. 34/150 sal de zinco. Esse sal reagido com o ciclopropiliodeto, 9Esguema-8 em uma reação catalisada por paládio para dar após a reação o composto reagente de Jones 4-Esquema-8. Este ácido carboxílico é sucessivamente convertido em isocianato como demonstrado no Esquema 1 e subseqüentemente em heteroariluréia ou heteroariltiouréia de Fórmula Z.
Variantes R3 de fórmula Z são preparados correspondentemente utilizando piridoxi ou feniloxipiridina (substituída) substituída por amina isto é, derivados 5substituído-2-(N-metilamino)piridina para R3 como metil.
Compostos em que é um alquileno opcionalmente substituído são convenientemente preparados pelo esquema 9:
Esquema 9
1b ~ 2 (a) NaBH4, EtOH; (b) ácido p-toluenosulfônico, tolueno, refluxo;
Esquema 9 descreve a preparação de tetralinas, indanos e homólogos, utilizados como material de partida nos esquemas acima a partir de tetralonas monosubstituídas conhecidas etc, em que posições R4 a R7 substituídas, por exemplo com halo ou alcóxiCi-3. Conversão do grupo carbonil em 1-tetralona lb-Esquema-9 no álcool correspondente por um agente redutor adequado tal boroidreto de sódio em etanol provê 2-Esquema-9. O álcool em refluxo com pequenas quantidades de ácido, tal como p-TsOH em tolueno, causa de 23/07/2018, pág. 35/150 desidratação de 2-Esquema-9 para a tetralina 1-Esquema-9 desejada. Reações correspondentes são aplicadas para n=1 ou .
Enquanto é possível para o agente ativo ser administrado sozinho, é preferível apresentá-lo como parte de uma formulação farmacêutica. Tal formulação compreenderá o agente ativo definido acima com um ou mais veículos aceitáveis ou excipientes e opcionalmente outros ingredientes terapêuticos. O(s) veículo(s) deve(m) ser aceitável(s) no senso de ser(em) compatível(is) com outros ingredientes da formulação e não deletério(s) para o recipiente.
As formulações incluem àquelas adequadas para administração retal, nasal tópica (incluindo bucal e sublingual), mas preferivelmente a formulação é uma formulação administrada oralmente. As formulações podem convenientemente estar presentes na forma de dosagem única, por exemplo, comprimidos e cápsulas de liberação prolongada, e podem ser preparadas por qualquer método bem conhecido pelos versados na técnica de farmácia.
Tais métodos incluem o passo de levar a associação do agente ativo acima definido com o veículo. Em geral, as formulações são preparadas uniformemente e intimamente levando a associação do agente ativo com veículos líquidos ou finamente veículos sólidos finamente divididos ou ambos, e então se necessário modelagem do produto. A invenção estende-se a métodos para preparar uma composição farmacêutica compreendendo um composto de fórmula Z ou seu de 23/07/2018, pág. 36/150 sal farmaceuticamente aceitável em conjunção ou associação com um veículo ou carreador farmaceuticamente aceitável. Se a fabricação de formulações farmacêuticas envolve uma íntima mistura de excipientes farmacêuticos e o ingrediente ativo na forma de sal, então é sempre preferido o uso de excipientes que são não-básicos naturalmente, isto é, ou acídicos ou neutros. Formulações para administração oral na presente invenção podem ser apresentada como unidades discretas tais como cápsulas, selos ou comprimidos contendo uma quantidade pré-determinada do ingrediente ativo; como um pó ou grânulos; como uma solução ou um suspensão do agente ativo em um líquido aquoso ou um líquido não aquoso; ou uma emulsão líquida óleo em água ou uma emulsão líquida em água e como um bolo etc.
Sem levar em consideração composições para administração oral (por exemplo comprimidos e cápsulas), o termo veículo adequado inclui veículos tal como excipientes comuns por exemplo, agentes ligantes, por exemplo xarope, acácia, gelatina, sorbitol, tragacanto, polivinilpirrolidona (Povidona), metilcelulose, etilcelulose, carboximetilcelulose de sódio, hidroxipropilmetilcelulose, sucrose e amido; enchimentos e carreadores, por exemplo amido de milho, gelatina, lactose, sucrose, celulose microcristalina, caolin, manitol, fosfato dicálcio, cloreto de cálcio e ácido algínico; e lubrificantes tais como estearato de magnésio, estearato de sódio e outros estearatos metálicos, ácido esteárico, estearato de glicerol, silicone fluido, ceras talco, óleos e sílica de 23/07/2018, pág. 37/150 coloidal. Agentes flavorizantes tais como hortelã-pimenta, óleo de gaultéria, sabor de cereja ou afins podem também ser utilizados. Pode ser desejado adicionar um agente colorante para fazer uma forma de dosagem facilmente identificável. Comprimidos podem, também, serem revestidos por métodos bem conhecidos na técnica. Um comprimido pode ser feito por compressão ou moldagem, opcionalmente com um ou mais ingredientes acessórios. Comprimidos compressos podem ser preparados por compressão em uma máquina adequada do agente ativo em uma forma de escoamento livre tal como um pó ou grânulos, opcionalmente misturados com um ligante, lubrificante, diluente inerte, conservante, agente dispersante ou ativo de superfície. Comprimidos moldados podem ser feitos por moldagem em uma máquina adequada do composto em pó umidificado com um diluente líquido inerte. Os comprimidos podem ser opcionalmente revestidos ou marcados e podem ser formulados ou para prover liberação baixa ou controlada do agente ativo.
Outras formulações adequadas para administração oral incluem pastilhas compreendendo o agente ativo em uma base com sabor, usualmente sucrose e acácia ou tragacanto; pastilhas compreendendo o agente ativo em uma base inerte tal como gelatina e glicerina, ou sucrose e acácia; e antisépticos bucais compreendendo o agente ativo em um veículo líquido adequado.
DESCRIÇÃO DETALHADA
Vários aspectos da invenção serão agora ilustrados por meio de exemplo somente com referência aos exemplos de 23/07/2018, pág. 38/150 seguintes não limitantes.
Exemplo 1
N-[(IS,laR,7bR)-4,7-difluor-l,la,2,7b-tetraidrociclopropa[c] cromen-l-il]-N-[5-(4-(sulfonamido)fenóxi)-2-piridinil]uréia
5-(4-(N-t-butilsulfonamido)fenóxi)-2nitropiridina
Para uma solução de 4-hidróxi-(N-tbutil )benzenosulfonamida (3,01g, 13,2 mmol) em DMF (48 mL) , carbonato de césio (5,67 g, 17,4 mmol) foi adicionado, seguido pela adição de 5-bromo-2-nitro piridina (2,36g, 11,6 mmol) e a mistura foi agitada a 50°C por 12 horas. A suspensão foi filtrada e o solvente evaporado e então o resíduo extraído entre NaHCCb aquoso saturado e cloreto de metileno. A fase orgânica foi seca sobre sulfato de sódio e evaporada. A mistura resultante foi purificada por cromatografía em coluna de sílica gel (0 - tè - 1% gradiente de EtOH / cloreto de metileno) para dar 3,47 g de material do qual 70% foi o composto titulo (LC-MS, API-ES+:352,4; Cale. 351,38) e cerca de 30% foi 2-(4-(N-tbutilsulfonamido)fenóxi)-5-bromopiridina (LC-MS, APIES+:386,3; Cale. 385,24) como um produto secundário.
1H-RMN (CDCls) : 8,37 (d, 1H) , 8,31 (d, 1H) , 7,98 (d,2H),
7,54 (dd, 1H), 7,20 (d,2H), 4,51 (s 1H), 1,28 (s, 9H).
b)_5 - ( 4 - (N-t-butil sulf onamido) fenóxi) - 2piridinamina de 23/07/2018, pág. 39/150
A mistura do produto obtida no Exemplo la (3,47 g) foi dissolvida em etanol (70 mL) e acetato de etila (18 mL). Então 10% de paládio em carvão (680 mg) foi adicionado e a suspensão negra foi hidrogenada com agitação sobre pressão de hidrogênio normal por 1½ hora. O catalisador foi filtrado e o filtrado foi evaporado. O resíduo resultante foi purificado por cromatografia em coluna de sílica gel (0-10% gradiente de EtOH / cloreto de metileno) para dar 2,42 g do composto titulo (57% produzidos nos dois passos). (LC-MS,
API-ES+:322,0 ; Cale. 320,41).
|
!H-RMN (de |
-DMSO) |
> : 7,77 (d, |
1H) , |
7,73 (d, |
2H), 7,20 |
(dd, 1H), |
|
7,39 (s, |
1H) , |
7,25 (dd, |
1H) , |
6,99 (d, |
1H) , 6,50 |
(d, 1H), |
|
5,97 (Br, |
s 1H) |
, 1,06 (s, |
9H) . |
|
|
|
|
|
c) |
N- [ (IS,laR,7bR) |
-4,7-difluor-l,la,2, |
7b-tetra- |
idrociclopropa[c]cromen-l-il]-N-[5-(4-(N-t-butilsulfonami do)fenóxi)-2-piridinil]uréia
O
Ácido (lS,laR,7bS)-4,7-difluor-l,la,2,7b, tetraidrociclopropa[c]cromeno-l-carboxílico, preparado como mostrado em W002/705163 (68 mg, 0,301 mmol), 5-(4-(N-tbutilsulfonamido)fenóxi)-2-piridinamina (109 mg, 0.0.341 mmol) e trietilamino (47 mL, 0,341 mmol) foram misturados de 23/07/2018, pág. 40/150
|
juntos em |
tolueno |
seco |
(2 mL) |
e |
atmosfera de |
argônio foi |
|
introduzida |
. Então |
DPPA ( |
7 4 mL, |
0, |
341 mmo1) foi |
adicionado e |
|
a solução |
reacional foi |
agitada |
a 110°C por |
3 horas. A |
mistura reacional foi trabalhada por extrações entre cloreto de metileno e 5% de ácido cítrico seguido por NaHCCb aquoso saturado. Cromatografia em coluna de sílica gel (1-2% gradiente de EtOH/cloreto de metileno) deu 143 mg de material que foi adicionalmente purificado por cromatografia CCD preparativa (10% MeOH/CHCÍ3) para finalmente dar 100 mg do produto puro como pós branco (81% produção).
(LC-MS, API-ES+:545, 0 ; Cale. 544, 48).
| 4H-RMN |
(CDCls) |
: 9,29 (Br s, 1H), 7,85 (d, 2H) |
, 7,64 |
(d, |
1H) , |
|
7,62 ( |
s, 1H), |
7,29 (dd, 1H), 6,96 (d, 2H), 6 |
,79 (d |
tr, |
1H) , |
|
6,70 ( |
d, 1H), |
6,59 (d tr, 1H), 4,52 (s, 1H) , |
4,47 ( |
dd, |
1H) , |
|
4,33 i |
(dd, 1H) |
, 3,79 (q, 1H), 2,62 (tr, 1H) , |
1,98 |
(m, |
1H) , |
|
1,26 ( |
s, 9H). |
|
|
|
|
|
|
d) |
N-[(IS,laR,7bR)-4,7-difluor-l, |
la,2,7t |
)-tetrai- |
drociclopropa[c]cromen-l-il]-N-[5-(4-(sulfonamido)fenóxi)-2
N-[(IS,laR,7bR)-4,7-difluor-l,la,2,7b-tetraidro ciclopropa[c]cromen-l-il]-N'-[5-(4-(sulfonamido)fenóxi)- 2 piridinil]uréia seco (36 mg, 0,066mmol) foi dissolvido em 1% de solução ácido/acetonitrola (5,8mL) e a solução reacional foi agitada por 30 minutos em temperatura ambiente. A reação de 23/07/2018, pág. 41/150 foi extinta com uma quantidade pequena de piridina e a acetonitrila foi removida por evaporação. O resíduo foi trabalhado por extrações entre cloreto de metileno e NaHCO3 aquoso saturado. A fase orqânica foi seca através de sulfato de sódio e evaporada. A cromatoqrafia em coluna de sílica qel (1-4% qradiente EtOH/cloreto de metileno) deu 26 mq de produto puro como um pó branco (71% produção)
| 1H-RMN (de-DMSO |
): 9,41 (s |
, 1H) |
, 8,06 (Br, s |
1H) , |
7, |
77 |
(d, |
|
2H) , |
7,73 (d, |
1H) , 7,52 |
(dd, |
1H) , 7,32 (d, |
1H) , |
7, |
29 |
(s, |
|
1H) , |
7,05 (d, |
2H), 6,79 |
(d tr, |
, 1H), 7,02 (d |
tr, |
1H) |
r |
4,32 |
|
(dd, |
1H), 4,28 |
(dd, 1H) , |
3,51 |
(q, 1H), 2,47 |
(tr, |
1H) |
r |
2,00 |
(m, 1H) .
Exemplo 2
N-[(IS,laR,7bR)-4,7-difluor-l,la,2,7b-tetraidrociclopropa[c] cromen-l-il]-N - [ 5 - ( 4-(N-metilcarboxiamido)fenóxi)-2piridinil]uréia
5-(4-N-metilcarboxiamido)fenóxi)-2a
Tert-butóxido de potássio (191 mq, 1,70 mmol) foi adicionado a uma solução de 4-hidróxi-N-metilbenzamida (257 mq, 1,70 mmol) em DMF (2,5 mL) e a mistura foi aqitada por 1 hora em temperatura ambiente. Então a mistura foi aquecida a 65°C e 5-bromo-2-nitro piridina (305mq, 1,50 mmol) foi adicionada e a mistura foi aqitada a 65°C por 12 horas. Então o solvente foi evaporado e o resíduo extraído entre de 23/07/2018, pág. 42/150 água e cloreto de metileno. A fase orgânica foi seca sobre sulfato de sódio e evaporada. A mistura resultante foi purificada por cromatografia em coluna de sílica gel (011/2% de gradiente de EtOH/cloreto de metileno) para dar 358 mg de material do qual cerca de 60% foi o composto título (LC-MS, API-ES+:307,8; Cale. 307,15) como um produto secundário.
1H-RMN (de-DMSO) : 8,46 (Br q , 1H) , 8,45 (d, 1H) , 8,34 (d,
1H) , 7,93 (d, 2H), 7,71 (dd, 1H) , 7,28 (d, 2H) , 2,78, 2,77 (2 x s, 3H).
b)_5 - ( 4 - (N-metilearboxiamido) fenóxi) - 2piridinamina
H2N
A mistura do produto obtida no passo a) (358 mg) foi dissolvida em etanol (10 mL) . Então 10% de paládio em carvão (110 mg) foi adicionada e a suspensão negra foi hidrogenada com agitação sob pressão de hidrogênio normal por 11/2 hora. O catalisador foi filtrado e o filtrado foi evaporado. O resíduo resultante foi purificado por cromatografia em coluna de sílica gel (2-6% gradiente EtOH/cloreto de metileno) para dar 118 mg do composto título (32% produção em dois passos). (LC-MS, API-ES+:244,4; Cale.
243,27) 1H-RMN (CDCls) : 7,93 (d, 1H) , 7,71 (d, 2H) , 7,21 (dd, 1H) ,
7,25 (dd, 1H), 6,94 (d, 2H), 6,55 (d, 1H), 6,01 (br, s 1H),
4,41 (br s, 2H), 3,01 (2 x s, 3H) .
de 23/07/2018, pág. 43/150
c)_Ν-[(IS,laR,7bR)-4,7-difluor-l,la,2,7b-tetraidrociclopropa[c]cromen-l-il]-N - [ 5-(4-(N-metilcarboxiamido) fenóxi)-2-piridinil]uréia
O
O composto titulo foi sintetizado analogamente ao Exemplo 1 5-(4-(N-metilcarboxiamido)fenóxi)-2-piridinamina (37mg, 0,15 mmol). Cromatografia em coluna de sílica gel (02% gradiente de EtOH/cloreto de metileno) deu 41 mg de produto puro como pó branco (65% produção) . (LC-MS, APIES+:467,1; Cale. 466,45) 1H-RMN (CDCls) : 9,33 (br s, 1H) , 7,99 (s, 1H), 7,75 (d, 2H),
7,61 (d, 1H) , 7,28 (dd, 1H), 6,93 (d, 2H), 6,78 (d tr, 1H),
6,72 (d, 1H) , 6,57 (d tr, 1H) , 6,07 (br q, 1H) , 4,45 (dd,
1H) , 4,33 (dd, 1H) , 3,78 (q, 1H) , 3,03 (d, 3H) , 3,66 (tr,
2H), 2,61 (tr, lh), 2,01-1,95 (m, 1H).
Exemplo 3
N-[(IS,laR,7bR)-4,7-difluor-l,la,2,7b-tetraidrociclopropa[c] cromen-l-il]-N -[5-(4-(N-metilsulfonamido)fenóxi)-2piridinil]uréia
a)_5 - ( 4 - (N-met il sul fonamido) fenóxi) - 2nitropiridina
O2N
O u
^S' w
O de 23/07/2018, pág. 44/150
O composto titulo foi sintetizado analogamente ao Exemplo la a partir de N-(4-hidroxifenil)metanosulfonamida (150 mg, 0,802 mmol). Cromatografia em coluna de sílica gel (0-9,75% gradiente de EtOH/cloreto de metileno) deu 63 mg do material do qual >90% foi o composto titulo (LC-MS, APIES+:308,0; Cale. 307,25). (2-(4-(N-metilsulfonamido)fenóxi)5-bromopiridina (LC-MS, API-ES+:307, 8, 308,8; Cale. 307, 15) foi formado como um produto secundário).
1H-RMN (de-DMSO) : 8,52 (d, 1H) , 8,36 (d, 1H) , 7,85 (d, 2H) ,
2,83 (dd, 1H) , 7,48 (q, 1H) , 7,40 (d, 2H) , 2,43, 4, 42 (2 x s, 3H).
b) 5-(4-(N-metilsulfonamido)fenóxi)-2-piridinamina
O composto titulo foi sintetizado analogamente ao Exemplo 2b) a partir de 5-(4-(N-metilsulfonamido)fenóxi)-2nitropiridina (63 mg, 0,204 mmol). Filtração e evaporação deram 73 mg de produto bruto. (LC-MS, API-ES+:280,0; Cale.
279,34) 1H-RMN (de-DMSO): 7,78 (d, 1H) , 7,70 (d, 2H) , 7,25 (dd, 1H) ,
7,02 (d, 2H), 6,50 (dd, 1H), 5,97 (s 1H), 2,37, 2,36 (2 x s,
3H) .
c)_N-[(IS, laR, 7bR)-4,7-difluor-l,la,2,7b-tetraidrociclopropa[c]cromen-l-il]-N-[5- (4-(N-metilsulfonamido) fenóxi)-2-piridinil]uréia de 23/07/2018, pág. 45/150
Ο
Ο composto titulo foi sintetizado analogamente ao Exemplo lc) a partir de 5-(4-(N-metilsulfonamido)fenóxi)-2piridinamina (76 mg, 0,204 mmol). Cromatografia em coluna de sílica gel (1-21/2% gradiente de EtOH/cloreto de metileno) deu frações puras contendo 36 mg do produto puro como pó branco (40% produção). (LC-MS, API-ES+:503,0 ; Cale. 502,32)
| 1H-RMN (CDCI3) : |
9,47 (br |
s, 1H), 8,68 (s, |
1H) , |
7,85 (s, |
1H) , |
|
7,57 (d, 1H), |
7,30-7,27 |
(m, 3H), 6,83-6, |
77 (m, |
2H), 6, |
56 (d |
|
tr, 1H), 4,45 |
(dd, 1H) , |
4,32 (dd, 1H) , |
3,81 |
(g, 1H), |
3,02 |
|
(s, 3H), 2,60 |
(tr, 1H), |
1,99-1,93 (m, 1H) |
|
|
|
Exemplo 4
N-[(IS,laR,7bR)-4,7-difluor-l,la,2,7b-tetraidrociclopropa[c] cromen-l-il]-N-[5-(4-aminofenóxi)-2-piridinil]uréia
a)_5 - ( 4 - (N-t-butoxicarbonilamino) fenóxi) - 2nitropiridina
O composto titulo foi sintetizado analogamente ao Exemplo la) a partir de 4-(N—t-butóxi-carbonilamino)fenol (581 mg, 2,78 mmol) . Cromatografia em coluna de sílica gel (0-2% gradiente de EtOH/cloreto de metileno) deu 704 mg do material do gual cerca de 50% foi o composto titulo. (LC-MS, API-ES+:332,0; Cale. 331,25). A outra metade consistiu de de 23/07/2018, pág. 46/150 (2-(4-(N-t-butοχicarbonilamino)fenóxi)-5-bromopiridina (LCMS, API-ES+:364,9, 366,0; Cale. 363,15), que foi formado como um produto secundário.
1H-RMN (de-DMSO) : 9,47 (br s, 1H) , 8,35 (d, 1H) , 8,29 (d,
1H) , 7,55 (d, 2H) , 7,51 (dd, 1H) , 7,15 (d, 2H) , 1,47 (s,
9H) .
b)_5 - ( 4 - (N-t-butoxic ar boni lamino) fenóxi) - 2piridinamina
O composto titulo foi sintetizado analoqamente ao Exemplo 2b) a partir da mistura obtida do passo a), contendo (5-(4-(N-t-butoxicarbonilamino)fenóxi-2-nitropiridina (total 704 mq). Após a reação, o resíduo resultante após filtração e evaporação foi purificado por cromatoqrafia em coluna de sílica qel (2-10% qradiente de EtOH/cloreto de metileno) para dar 418 mq do composto titulo (57% produção em dois passos).
(LC-MS, API-ES+: 302 , 0 ; Cale. 301,35).
1H-RMN (CDCls) : 7,69 (d, 1H) , 7,32 (d, 2H) , 7,31 (d, 1H) ,
6,90 (d, 2H) , 6,68 (d, 1H) , 6,47 (br, s 1H) , 4,98 (br s,
2H), 1,51 (s, 9H).
c)_N-[(IS, laR, 7bR)-4,7-difluor-l,la,2,7b-tetraidrociclopropa[c]cromen-l-il]-N-[5-(4-(N-t-butoxicarbonil amino)fenóxi)-2-piridinil]uréia de 23/07/2018, pág. 47/150
H
Ο composto titulo foi sintetizado analogamente ao Exemplo lc) a partir de 5-(4-(N-t-butoxicarbonilamino) fenóxi)-2-piridinamina (418 mg, 1,39 mmol). Cromatografia em coluna de sílica gel (1-4% gradiente EtOH/cloreto de
|
5 |
metileno) deu |
479 |
mg |
do produto como um |
pó brando |
(74% |
|
|
produção) . |
|
|
|
|
|
|
|
(LC-MS, API-ES+ |
:525, |
i; |
Cale. 524,30). |
|
|
|
|
1H-RMN (CDCI3) : |
9,32 |
(br s, 1H) , 7,34 (d, |
2H), 7,20 |
(dd, |
|
|
1H) , 6,88 (d, |
2H) , |
6, |
79 (d tr, 1H), 6,58 |
(d tr, 1H) , |
6, 45 |
|
10 |
(s, 1H), 4,41 |
(dd, |
1H) , 4,34 (dd, 1H) , 3, |
75 (q, 1H) , |
2,59 |
|
|
(tr, 1H), 1,98- |
1,93 |
(m |
, 1H), 1,52 (s, 9H). |
|
|
|
|
d) |
|
|
N-[(IS,laR,7bR)-4,7-difluor-l, la, |
2, 7b- |
tetraidrociclopropa[c]cromen-l-il]-N-[5-(4-aminofenóxi)-2 -
N-[(IS,laR,7bR)-4,7-diflúor-1,la,2,7b-tetraidro ciclopropa[c]cromen-l-il]-N'-[5-(4-(N-t-butoxicarbonilamino) fenóxi)-2-piridinil]uréia seca (242 mg, 0,46 mmol) foi dissolvida em cloreto de metileno (2 mL) e então 1M HC1 / AcOH (4,6 mL) foi adicionado e a solução reacional foi
Petição 870180063303, de 23/07/2018, pág. 48/150 agitada por 60 minutos em temperatura ambiente. As substâncias voláteis foram removidas por evaporação. O resíduo foi trabalhado por extrações entre cloreto de metileno e NaHCCb aquoso saturado. A fase orgânica foi seca através de sulfato de sódio e evaporada. Cromatografia em coluna de sílica gel (1-3% gradiente EtOH/cloreto de metileno) deu 139 mg de produto puro como pó branco (71% produção).
| 1H-RMN |
(CDC13) |
: 9,33 (br |
s, 1H), 7,44 (d, |
1H) , |
7,24 |
(s, |
1H) |
|
7,17 ( |
dd, |
1H) |
, 6,79 (d, |
2H), 6,77 (d tr, |
1H) , |
6,68 |
(d, |
2H) |
|
6,60-6 |
,54 |
(m, |
2H), 4,40 |
(dd, 1H) , 4,35 |
(dd, |
1H) , |
3, 73 |
(q |
|
1H) , 3 |
,61 |
(br |
s, 2H), 2, |
57 (tr, 1H) , 1,98- |
-1,92 |
(m, |
1H) . |
|
Exemplo 5
N-[(IS,laR,7bR)-4,7-difluor-l,la,2,7b-tetraidrociclopropa[c] cromen-l-il]-Lh-[5-(4-(metilsulfon)fenóxi)-2-piridinil]uréia
a) 5-(4-(metilsulfon)fenóxi)-2-nitropiridina
O composto titulo foi sintetizado analogamente ao Exemplo la) a partir de 4-hidroxifenil metil silfona (288 mg, 1,67 mmol). Cromatografia em coluna de sílica gel (0-2% gradiente EtOH/cloreto metileno) deu 300 mg de material com mais que 90% do composto titulo (LC-MS, API-ES+:353, 0 (m + AcO_) ; Cale. 294,29). Uma pequena porcentagem de contaminante 2-(4-(metilsulfon)fenóxi)-5-bromopiridina (LCMS, API-ES+:327, 9, 330,0; Cale. 328,19) estava presente.
1H-RMN (CDCls) : 8,41 (d, 1H) , 8,33 (d, 1H) , 8,04 (d, 2H) , de 23/07/2018, pág. 49/150
7,58 (dd, 1H), 7,27 (d, 2H), 3,10 (s, 3H).
b) 5-(4-(metilsulfon)fenóxi)-2-piridinamina
H2N
O
N
O
O composto título foi sintetizado analogamente ao Exemplo 2b) a partir da mistura obtida no passo a) , contendo (5-(4-(metilsulfon)fenóxi)-2-nitropiridina (300 mg). Este material foi dissolvido com aquecimento em uma mistura de acetato de etila (10 mL) , isopropanol (3 mL) e metanol (3 mL). Após a reação, que foi continuada em temperatura ambiente, o resíduo resultante após filtração e evaporação foi purificado por cromatografia em coluna de sílica gel (24% gradiente EtOH/cloreto de metileno) para dar uma fração pura contendo 160 mg do composto título (LC-MS, APIES+:265,0; Cale. 264,31).
c)_N-[(IS, laR,7bR)-4,7-difluor-l,la,2,7b-tetraidrociclopropa[c]cromen-l-il]-EH-[5-(4-(metilsulfon)fenóxi)-2
-piridinil]uréia
O composto título foi sintetizado analogamente ao Exemplo lc) a partir de 5-(4-(metilsulfon)fenóxi)-2piridinamina (31 mg, 0,118 mmol). Cromatografia em coluna de sílica gel (1-3% gradiente EtOH/cloreto de metileno) seguido por CCD preparativa (10% MeOH/CHC13) deu 10,7 mg de produto de 23/07/2018, pág. 50/150 puro como pó branco (19% produção).
(LC-MS, API-ES+:488,0; Cale. 487,48).
| 1H-RMN |
(CDC13 |
): 9,41 (br |
s, 1H), 8,99 (s, |
1H) |
, 7,91 (d, |
2H) , |
|
7,68 ( |
:d, 1H) |
, 7,31 (dd, |
1H) , 7,04 (d, |
2H) , |
6,87 (d, |
1H) , |
|
5 6,80 ( |
d tr, |
1H), 6,58 ( |
d tr 1H), 4,48 |
(dd, |
1H), 4,32 |
(dd, |
|
1H) , 3 |
,82 (q, |
1H), 2,62 |
(tr, 1H), 2,01-1, |
95 ( |
m, 1H). |
|
Exemplo 6
N-[(IS,laR,7bR)-4,7-diflúor-l,la,2,7b-tetraidrociclopropa[c] cromen-l-il]-LR-[5-(4-(2-hidroxietil)fenóxi)-2-piridinil] uréia
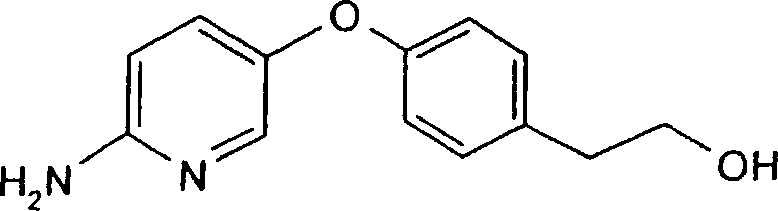
a) 5-(4-(2-hidroxietil)fenóxi)-2-nitropiridina
O título composto foi sintetizado analogamente ao Exemplo 2a a partir de 2-(4-hidroxifenil)etilálcool (2354 mg, 1,70 mmol). Cromatografia em coluna de sílica gel (0-2% gradiente EtOH/cloreto de metileno) deu 237 mg de material com mais que 80% do composto título (LC-MS, API-ES+:319,0 (m+AcO_) ; Cale. 260,25). Cerca de 10-15% de contaminante 2(4-(2-hidroxietil)fenóxi)-5-bromopiridina (LC-MS, API-ES+:
294,0, 295,25; Cale. 295,25) estava presente.
0 b) 5-(4-(2-hidroxietil)fenóxi)-2-piridinamina
O composto título foi sintetizado analogamente ao Exemplo 2b a partir da mistura obtida a partir do passo a),
Petição 870180063303, de 23/07/2018, pág. 51/150 contendo (5-(4-(2-hidroxietil)fenóxi)-2-nitropiridina (197 mg) . O resíduo resultante após filtração e evaporação foi purificado por cromatografia em coluna de sílica gel (2-10% gradiente EtOH/cloreto de metileno) para dar uma fração pura contendo 65 mg do composto titulo (LC-MS, API-ES+:231,1;
Cale. 230,27) .
c)_N-[(IS,laR,7bR)-4,7-diflúor-l,la,2,7 b-tetraidrociclopropa [c]cromen-l-il]-LR-[5-(4-(2-hidroxietil)fenóxi)
-2-piridinil]uréia
O composto titulo foi sintetizado analogamente ao Exemplo lc) a partir 5-(4-(2-hidroxietil)fenóxi)-2piridinamina (62 mg, 0,282 mmol). Cromatografia em coluna de sílica gel (0-4% gradiente EtOH/cloreto de metileno) deu frações, a partir das quais 8 mg de produto puro como pó branco foi obtido, e adicionalmente algumas frações misturadas.
(LC-MS, API-ES+:454,2; Cale. 453,45).
1H-RMN (CDCls) : 9,38 (br s, 1H), 8,28 (s, 1H), 7,54 (d, 1H) ,
|
7, |
24 |
(dd, |
1H) , |
7,20 |
(d, |
2H) , |
6,88 ( |
Χ, 2H) , 6,77 (d |
tr, |
1H) , |
|
6, |
71 |
(d, |
1H) , |
6,56 ( |
d tr, 1H) |
, 4,43 |
(dd, 1H), 4,34 |
(dd, |
1H) , |
|
3, |
87 |
(t, |
2H) , |
3, 77 |
(q, |
1H) , |
2,86 |
(t, 2H), 2,59 ( |
:tr, |
1H) , |
|
1, |
98- |
1,93 |
(m, |
lH)m 1 |
,51 |
(br, |
1H) . |
|
|
|
Exemplo 7
N-[(IS,laR,7bR)-4,7-diflúor-l,la,2,7b-tetraidrociclopropa[c] de 23/07/2018, pág. 52/150 cromen-l-il]-N^-(5-(4-(2-(2-metoxietóxi)etóxi)fenóxi)-2piridinil]uréia
Composto titulo foi sintetizado analogamente ao
Exemplo 2a) a partir de 4-(2-(2-metóxi-etóxi)etóxi)fenol (300 mg, 1,42 mmol) . Cromatografia em coluna de sílica gel (0-1/2% gradiente EtOH/cloreto de metileno) deu 173 mg de material com mais que 70% do composto titulo (LC-MS, API10 ES+:335,1; Cale. 334,33). Cerca de 20-30% de contaminante 2(4-(2-(2-metoxietóxi)etóxi)fenóxi)-5-bromopiridina (LC-MS,
O composto titulo foi sintetizado analogamente ao
Exemplo 2b) a partir da mistura obtida do Exemplo 22, contendo 5-(4-(2-(2-metoxietóxi)etóxi)fenóxi)-2nitropiridina (173 mg). O resíduo resultante após filtração e evaporação foi purificado por cromatografia em coluna de sílica gel (0-6% gradiente de EtOH/cloreto de metileno) para dar uma fração pura contendo 92 mg (60% produção) do composto titulo (LC-MS, API-ES+:305,1; Cale. 304,35).
Petição 870180063303, de 23/07/2018, pág. 53/150
| 1H-RMN (CDCI3) |
: 7,85 (d, |
1H) , 7,16 (dd, |
1H) , |
6,90-6,85 |
(m- |
|
4H) , 6,68 (d, |
1H) , 6,50 |
(d, 1H) , 4,40 |
(br, |
2H) , 4,11 |
(t, |
|
2H), 3,85 (t, |
2H), 3,72 ( |
t, 2H), 3,58 (t, |
2H) , |
3,39 (s, |
3H) . |
c)_N-[(IS, laR, 7bR)-4,7-diflúor-l,la,2,7b-tetraidrociclopropa[c]cromen-l-il]-N-[5-(4-(2-(2-metoxietóxi) etóxi)fenóxi)-2-piridinil]uréia
O composto titulo foi sintetizado analogamente ao Exemplo lc) a partir de 5-(4-(2-(2-metoxietóxi)etóxi) fenóxi)-2-piridinamina (46 mg, 0,15 mmol). Cromatografía em coluna de sílica gel (0-11/2% gradiente de EtOH/cloreto de metileno) deu frações, das quais 14 mg do produto puro como pó branco foi obtido, e adicionalmente algumas frações misturadas (~40 mg) (LC-MS, API-ES+:528,1; Cale. 527,53).
|
!H |
-RMN |
( |
CDCls) : |
9,35 (br s, |
1H), 7,82 |
(s, |
1H) , |
7, 48 |
(d, |
1H) , |
|
7, |
18 < |
:d, |
1H) , |
6,89 (m, 4H) |
, 6,77 (d |
tr, |
1H) , |
6,63 |
(d, |
1H) , |
|
6, |
57 ( |
d |
tr 1H) |
, 4,42 (dd, |
1H), 4,35 |
(dd, |
1H) , |
4, 14 |
(t, |
2H) , |
|
3, |
87 ( |
t, |
1H) , |
3,75 (q, 1H) , |
3,74 (t, |
2H) , |
3,59 |
(t, |
2H) , |
3,40 |
(s, 3H), 2,58 (tr, 1H), 1, 98-1, 92 (m, 1H) .
Exemplo 8
5-((6 - [ ({ [(IS,laR,7bR)-4,7-diflúor-l,la,2,7b,tetraidrociclopropa[c]cromen-l-il]amino}carbonil)amino]-3-piridinil}óxi)N-metil-2-piridinocarboxamida
a)_N-metil-5- [ ( 6-nitro-3-piridinil) óxi] -2piridinacarboxamida de 23/07/2018, pág. 54/150
Ácido 5- [ (6-nitro-3-piridinil)óxi]-2piridinacarboxílico (26 mg. 1 mmol) foi deixado em refluxo em cloreto de tionil (10 mL) por uma noite. O excesso de cloreto de tionil foi evaporado e o cloreto ácido bruto foi extinto com metil amina aquosa para dar N-metil-5-[(6-nitro3-piridinil)óxi]-2-piridinacarboxamida (190 mg, 70%).
1H-RMN (CDCls) : 8,4 (d. 1H) , 8,32 (d, 1H) , 8,31 (d, 1H) ,
7,42 (br s, 1H), 7,55 (m, 2H).
b) 5-({6-[({[(IS,laR,7bR)-4,7-diflúor-1,la,2,7b, tetraidrociclopropa[c]cromen-l-il]amino}carbonil)amino]- 3 piridinil}óxi)-N-metil-2-piridinocarboxamida
N-metil-5-[(6-nitro-3-piridinil)óxi]-2- piridina carboxamida (190 mg, 0,7 mmol) foi dissolvida em Metanol (20 mL) . A mistura foi hidrogenada utilizando Ra/Ni sob atmosfera de hidrogênio. Quando o material de partida foi consumado segundo a CCD (éter), a mistura foi filtrada através de celite e concentrada sob pressão reduzida.
Ao produto bruto foi adicionado ácido
Petição 870180063303, de 23/07/2018, pág. 55/150 (IS,laR,7bS)-4,7-diflúor-1,la,2,7b, tetraidrociclopropa [c] cromeno-l-carboxílico (0,170 mg, 0,76 mmol) e a mistura foi co-evaporada com Tolueno (10 mL) para a metade do volume.
Difenilfosforil azida (179 pL, 0,76 mmol), e trietil amina (106 pL, 0,76 mmol) foi adicionada. A mistura foi então deixada em refluxo por 4 h sob atmosfera de argônio. O solvente foi então removido sob pressão reduzida e o produto bruto foi dissolvido em acetato de etila e lavado com porções pequenas de ácido hidroclórico aquoso (0,0lM), hidrogênio carbonato de sódio saturado e água. Purificação por cromatografia em flash (1% metanol em éter) deu o composto desejado (158 mg, 48%).
1H-RMN (CDCls) : 9,27 (br s, 1H), 8,25 (d, 1H), 8,17 (d, 1H) ,
8,16-8,10 (br s, 1H), 7, 88-7, 82 (m, 1H), 7,67 (d, 1H) , 7,31 (dd, 1H) , 7,26 (dd, 1H) , 6,83-6, 75 (m, 2H) , 6,61-6,55 (m,
1H) , 4,48 (dd, 1H) , 4,32 (dd, 1H) , 3,81 (q, 1H) , 3,02 (D,
3H), 2,62 (t, 1H), 2,02-1,94 (m, 1H).
Exemplo 9
4-((6 - [ ( { [ (IS,laR,7bR)-4,7-diflúor-l,la,2,7b,tetraidrociclopropa [ c ] cromen-l-il]amino}carbonil)amino]-3-piridinil}óxi) benzamida
a) 4 - [ (6-nitro-3-piridinil)óxi]benzamida <x^^CONH2
4-hidroxibenzamida (150 mg, 1,1 mmol) e carbonato de césio (394 mg, 1,21 mmol) foram dissolvidos em dimetilformamida (7 mL) . 5-bromo-2-nitropiridina (244 mg, de 23/07/2018, pág. 56/150
1,21 mmol) foi então adicionada. A mistura foi deixada a 50 graus até o material de partida ser consumado segundo a CCD (1% metanol em éter). Purificação por cromatografia em flash produziu 4-[(6-nitro-3-piridinil)óxi]benzamida (110 mg,
38%) .
1H-RMN (CDCls) : 8,37 (d, 1H) , 8,29 (d, 1H) , 7,94 (m, 2H) ,
7,51 (dd, 1H), 7,18 (m, 2H).
b) 4-((6- [ ( { [(IS, laR, 7bR)-4,7-diflúor-1,la,2,7b, tetraidrociclopropa[c]cromen-l-il]amino}carbonil)amino]- 3 piridinil}óxi)benzamida
O
Este composto foi preparado essencialmente pelo mesmo procedimento como descrito para o Exemplo 8, começando com 4-[(6-nitro-3-piridinil)óxi]benzamida (100 mg, 0,38 mmol) e ácido (IS,laR,7bS)-4,7-diflúor-1,la,2,7b, tetraidrociclopropa[c]cromeno-l-carboxílico (65 mg, 0,29 mmol) para dar 20 mg do composto titulo puro (12%) .
| 1H-RMN (CDCI3 + MeOD) |
: 7, 82 |
(m, 2H) |
, 7,63 (d, 1H), |
7,30 |
(dd, |
|
1H) , 6,96 (d, 2H) , |
6,90-6, |
76 (m, |
2H) , 6,62-5,59 |
(m, |
1H) , |
|
4,45 (dd, 1H), 4,35 |
(dd, 1H) |
, 2,6 ( |
X, 1H), 2,0-1,92 |
(m, |
1H) . |
Exemplo 10
5-((6-[({[(IS,laR,7bR)-4,7-diflúor-l,la,2,7b,tetraidrociclopropa [c]cromen-l-il]amino}carbonil)amino]-3-piridinil}óxi)de 23/07/2018, pág. 57/150
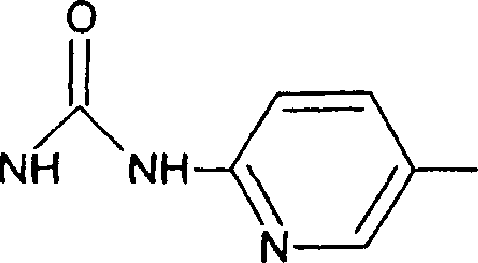
2-piridinacarboxamida
a)_5- [ ( 6-nitro-3-piridinil) óxi] -2piridinacarboxamida
o2n fY ^n<^conh2
Ácido 5- [ (6-nitro-3-piridinil)óxi]-2piridinacarboxílico (lOOmg, lmmol) foi deixado em refluxo em cloreto de tionil (5 mL) por uma noite. O excesso de cloreto de tionil foi evaporado e o cloreto ácido bruto foi extinto com metil amina aquosa para dar 5-[(6-nitro-3piridinil)óxi]-2-piridinacarboxamida (60 mf, 60%).
1H-RMN (DMSO): 8,60 (d, 1H) , 8,57 (d, 1H) , 8,13 (s, 1H) ,
8,11 (br s, 1H), 7,67 (br s, 1H).
b) 5-((6- [ ({ [(IS,laR,7bR)-4,7-diflúor-1,la,2,7b, tetraidrociclopropa[c]cromen-l-il]amino}carbonil)amino]- 3 piridiniljóxi)-2-piridinacarboxamida
Este composto foi preparado essencialmente pelo mesmo procedimento como descrito para o Exemplo 8, começando com 5-[(6-nitro-3-piridinil)óxi]-2-piridinacarboxamida (60 mg, 0,38 mmol) e ácido (IS, laR, 7bS)-4,7-diflúor-1,la,2,7btetraidrociclopropa[c]cromeno-l-carboxílico (65 mg, 0,29
Petição 870180063303, de 23/07/2018, pág. 58/150
|
mmol |
) para dar |
0 composto titulo |
puro (18 |
mg) |
(18%) |
1 . |
|
| 1H-RMN (CDCI3 + |
MeOD): 9,35 (br |
s, |
1H) , |
8,28 |
(d, |
1H) , |
8,17 |
|
(d, |
1H) , 7,75 1 |
(br s, 1H), 7,69 |
(d, |
1H) , |
7,33 |
(dd, |
1H) , |
7,27 |
|
(dd, |
1H), 6,96 |
(d, 1H), 6,8 (m, |
1H) |
, 6,58 |
(m, |
1H) , |
4, 48 |
(dd, |
|
1H) , |
4,32 (dd, |
1H), 3,80 (q, 1H), |
2,62 |
(t, |
1H) , |
2,02- |
-1,96 |
|
(m, |
1H) . |
|
|
|
|
|
|
|
O CH,
N O CH. H 3
Exemplo 11
N-[(IS,laS,7bS)-4,7-diflúor-l,la,2,7b,tetraidrociclopropa[c] cromen-l-il]-N-{5-[4-(hidrazinocarbonil)fenóxi]-2piridinil}uréia
a)_tert-butil_2 - [ 4 - (benzilóxi) benzoil ] hidrazinocarboxilato
BzO
Uma mistura de ácido 4-benziloxibenzóico (0,780 g, 3,42 mmol), tert-butil carbazato (0,443 g, 3,35 mmol)m Et3N (0,5 mL) , hidrocloreto de (1-(3-dimetilaminopropil)-3etilcarbodiimida (1,05 g, 5,47 mmol) e hidrato de 1hidroxibenzotriazol (0,778 g, 5,76 mmol) em N,Ndimetilformamida (27 mL) foi aditado em temperatura ambiente por 2 dias. A reação foi concentrada e diluída em diclorometano. A fase orgânica foi lavada duas vezes com água, seca com MgSCh e concentrada. O resíduo foi purificado por cromatografia em coluna (sílica gel, 5% MeOH em CH2CI2) e carboxilato de tert-butil 2-[4-(benzilóxi)benzoil] hidrazina (0,998 g, produção: 85%) foi identificada por de 23/07/2018, pág. 59/150 espectroscopia de RMN.
1H-RMN (CDCls) : 8,21 (s, 1H) , 7,76 (d, 2H) , 7,37 (m, 5H) ,
6,95 (d, 2H), 6,76 (s, 1H), 5,08 (s, 2H), 4,48 (s, 9H).
b)_tert-but il_2 - [ 4 - (hidroxibenzoil) hidrazinocarboxilato
Uma solução de tert-butil 2—[4— (benzilóxi)benzoil]hidrazinocarboxilato (975 mg, 2,85 mmol) em presença de quantidade catalítica de Pd-C 10% em etanol (40 mL) é hidrogenado por 3 horas. Após filtração em celite, o resíduo é purificado em cromatografia em coluna (sílica gel, 10% MeOH em CH2CI2) e tert-butil 2—[4 — (hidroxibenzoil)hidrazinocarboxilato (0,688 g, produção: 96%) foi identificado por espectroscopia de RMN.
!H-RMN (CD3OD) : 7,73 (d, 2H) , 6,82 (d, 2H) , 4,84 (s, 2H) ,
1,48 (s, 9H).
c)_tert-butil_2-{4-[ (6-nitro-3-piridinil)óxi] benzoil}hidrazinocarboxilato
A uma mistura de tert-butil 2—[4— (hidroxibenzoil)hidrazinocarboxilato (0,688 g, 2,73 mmol) e 5-bromo-2-nitropiridina (0,554 mg, 2,73 mmol) e carbonato de de 23/07/2018, pág. 60/150 césio (1,33 g, 4,08 mmol) em N,N-dimetilformamida (7 mL) foi agitada por uma noite a 80°C. A solução foi concentrada e o resíduo foi tomado com diclorometano e água. A fase orgânica foi seca em MgSCg e concentrada. O produto bruto foi purificado por cromatografia em coluna (sílica gel, 5% de MeOH em CH2CI2) , para dar 736 mg de mistura de nitropiridina e bromopiridina.
d)_tert-but il_2-{4-[ ( 6-amino-3-piridinil) óxi ] benzoil}hidrazinocarboxilato
A mistura de nitropiridina e bromopiridina (0,700 g) em presença de quantidade catalítica de Pd-C 10% em etanol (20 mL) e EtOAc (20 mL) foi hidrogenada por 1 hora. Após filtração em celite, o resíduo foi purificado em cromatografia em coluna (sílica gel, 5% MeOH em CH2CI2) e tert-butil 2 —{4 —[(6-nitro-3-piridinil)óxi]benzoil} hidrazinocarboxilato (0,326 g, produção: 35%) foi identificado por espectroscopia de RMN.
1H-RMN (CD3OD) : 7,73 (d, 2H) , 6,64 (d, 1H) , 7,17 (dd, 1H) ,
6,86 (dd, 2H), 6,55 (d, 1H), 4,74 (s, 4H), 1,39 (s, 9H).
e) tert butil 2-[ 4-({6-[({ [ (IS,laS,7bS)-4,7diflúor-1,la,2,7b,tetraidrociclopropa[c]cromen-l-il]amino} 3-piridinil}óxi)benzoil]hidrazinocarboxilato de 23/07/2018, pág. 61/150
Uma mistura de ácido quiral (155 mg, 0,687 mmol), tert-butii 2-{4-[(6-amino-3-piridinil)óxi]benzoil} hidrazinocarboxilato (267 mg, 0,776 mmol), difenilfosforil azida (0,162 mL, 0, 756 mmol) em tolueno (10 mL) foi deixado 5 em refluxo por 4 horas. A solução foi reduzida e o resíduo foi diluído em diclorometano e lavado uma vez com HC1 (0,00lN) e solução salina. A fase orgânica foi seca com MgSCh e evaporada. O resíduo foi purificado em cromatografia em coluna (sílica gel, 5% MeOH em CH2CI2) para dar o composto titulo (0,227 g, produção: 52%).
1H-RMN (CDCls) : 9,36 (s, 1H) , 9,05 (s, 1H) , 8,99 (s, 1H) ,
7,80 (d, 2H), 7,62 (d, 1H), 7,28 (s, 1H), 7,25 (d, 1H), 6,89 (d, 1H), 6,86 (d, 2H), 6,78 (m, 1H), 6,55 (m, 1H), 4,43 (dd,
1H) , 4,30 (dd, 1H) , 3,76 (m, 1H) , 2,59 (m, 1H) , 1,95 (m,
1H) , 1, 46 (s, 1H) .
f)_N-[(IS, laS, 7bS)-4,7-diflúor-l,la,2,7b,tetraidrociclopropa[c]cromen-l-il]-th-{5-[4 - (hidrazinocarbonil) fenóxi]-2-piridinil}uréia
Petição 870180063303, de 23/07/2018, pág. 62/150
O tert butil 2-[4-({6-[({[(IS,laS,7bS)-4,7diflúor-1,la,2,7b,tetraidrociclopropa[c]cromen-l-il]amino} 3- piridinil}óxi)benzoil]hidrazinocarboxilato (49 mg, 0,089 mmol) em uma mistura de diclorometano (0,5mL) e trifluoracético (0,5 mL) foi agitada em temperatura ambiente por 30 minutos. A reação foi concentrada e purificada em cromatografia em coluna (sílica gel, 2% MeOH em CH2C12) para dar o composto N-[(IS,laS,7bS)-4,7-diflúor-l,la, 2, 7b, tetraidrociclopropa [c]cromen-l-il]-N'-{5-[4 - (hidrazinocarbonil) fenóxi]-2-piridinil}uréia (17,6 mg, produção: 42%).
íH-RMN (CD3OD) : 7,80 (d, 2H) , 7,62 (d, 1H) , 7,39 (dd, 1H) ,
6,97 (d, 3H), 6,83 (m, 1H), 6,62 (m, 1H), 4,41 (m, 1H) , 4,29 (dd, 1H), 3,61 (m, 1H), 2,59 (t, 1H), 2,02 (m, 1H).
Exemplo 12
4- ((6-[({ [ (IS,laS,7bS)-4,7-diflúor-l,la,2,7b,tetraidrociclopropa [c]cromen-l-il]amino}carbonil)amino]-3-piridinil}óxi)N-ciclopropilbenzamida
a) 4-(benzilóxi)-N-ciclopropilbenzamida
4-(benzilóxi)-N-ciclopropilbenzamida (0,774 g,
Petição 870180063303, de 23/07/2018, pág. 63/150
83%) foi sintetizada analogamente ao Exemplo 11a a partir do ácido 4-benziloxibenzóico (0,759g).
1H-RMN (CDCls) : 7,70 (d, 2H) , 7,38 (m, 5H) , 6,97 (d, 2H) ,
6,17 (s, 1H) , 5,10 (s, 2H) , 2,88 (m, 1H), 0,85 (m, 2H), 0,6 (m, 2H) .
b) N-ciclopropil-4-hidroxibenzamida
N-ciclopropil-4-hidroxibenzamida (0,332 g, 68%) foi sintetizado analogamente ao Exemplo llb a partir de 4-(benzilóxi)-N-ciclopropilbenzamida (0,774 g).
íH-RMN (CD3OD) : 8,26 (s, 1H) , 7,67 (d, 2H) , 6,80 (d, 2H) ,
4,88 (s, 1H), 2,79 (m, 1H), 0,75 (m, 2H), 0,60 (m, 2H).
c)_N-ciclopropil-4-[(6-nitro-3-piridinil)óxi] benzamida
A mistura nitropiridina e bromopiridina foi sintetizada analogamente ao Exemplo 11c a partir de Nciclopropil-4-hidroxibenzamida (0,330 g).
íH-RMN (CD3OD) : 8,33 (d, 1H) , 8,32 (d, 1H) , 7,92 (d, 2H) ,
7,66 (dd, 1H) , 7,24 (d, 2H) , 2,85 (m, 1H) , 0,81 (m, 2H) ,
0,64 (m, 2H).
d)_4-[(6-amino-3-piridinil)óxi]-N-ciclopropilbenzamida de 23/07/2018, pág. 64/150
4-[(6-amino-3-piridinil)óxi]-N-ciclopropilbenz
amida (0,128 g, 25%) foi sintetizada analogamente ao Exemplo lld a partir da mistura de nitropiridina e bromopiridina.
| 1H-RMN (CDCI3) |
: 7,92 ( |
:s, ih), 7,68 |
(d, |
2H) , 7,20 |
(d, 1H), |
|
6,92 (d, 2H), |
6,54 (d, |
1H), 6,12 (s, |
1H) |
, 4,41 (s, |
2H), 2,89 |
|
(m, 1H), 0,81 |
(m, 2H), |
0,64 (m, 2H). |
|
|
|
|
e) |
4-({6-[ |
({[(IS,laS,7bS) |
'-4, |
7-diflúor-l |
,la,2,7b, |
tetraidrociclopropa[c]cromen-l-il]amino}carbonil)amino]- 3 -
4-({6-[ ({ [(IS,laS,7bS)-4,7-diflúor - 1,1a,2,7b, tetraidrociclopropa[c]cromen-l-il]amino}carbonil)amino]-3 piridinil}óxi)-N-ciclopropilbenzamida (0,090 g, 38%) foi sintetizada analogamente ao Exemplo lie a partir de 4—[(6— amino-3-piridinil)óxi]-N-ciclopropilbenzamida (0,128) .
1H-RMN (CDCls) : 9,36 (s, 1H) , 8,56 (s, 1H) , 7,73 (d, 2H) ,
7,62 (d, 1H) , 7,27 (dd, 1H) , 6,91 (m, 3H) , 6,78 (m, 1H) ,
6,57 (m, 1H) , 6,23 (s, 1H) , 4,45 (dd, 1H) , 4,33 (dd, 1H) ,
3,72 (m, 1H), 2,90 (m, 1H), 2,60 (t, 1H), 1,97 (m, 1H), 0,87 (m, 2H), 0,63 (m, 2H).
Exemplo 13 de 23/07/2018, pág. 65/150
N-[4-({6-[({[(lS,laS,7bS)-4,7-diflúor-l,la,2,7b, tetraidrociclopropa [c]cromen-l-il]amino}carbonil)amino]-3-piridinil} óxi)fenil]acetamida
a) N-{4-[(6-nitro-3-piridinil)óxi]fenil}acetamida
0 composto titulo foi sintetizado analogamente ao
Exemplo 11c a partir de N-(4-hidroxifenil)acetamida. iH-RMN (DMSO-de): 10,05 (s, 1H) , 8,36 (d, 1H), 8,29 (d, 1H),
7,67 (d, 2H), 7,54 (dd, lh), 7,18 (d, 2H), 2,03 (s, 3H).
b) N-{4-[(6-amino-3-piridinil)óxi]fenil}acetamida
O composto titulo foi sintetizado analogamente ao
Exemplo lld a partir de N-{4-[(6-nitro-3-piridinil)óxi] fenil}acetamida.
1H-RMN (CDCls) : 7,87 (d. 1H) , 7,41 (d, 2H) , 7,29 (s, 1H) ,
7,17 (dd, 1H) , 6,89 (d, 2H) , 6,51 (d, 1H) , 4,47 (s, 2H) ,
2,14 (s, 3H).
c) N-[4-({6- [ ( { [(IS, laS, 7bS)-4,7-diflúor-l,la,2,
7b,tetraidrociclopropa[c]cromen-l-il]amino}carbonil)amino]-3 -piridinil}óxi)fenil]acetamida
Petição 870180063303, de 23/07/2018, pág. 66/150
O composto título foi sintetizado analogamente ao Exemplo lie a partir de N-{4-[(6-amino-3-piridinil) óxi]fenil}acetamida.
| 1H-RMN |
(CDC13) |
: 9,39 |
(s, |
1H), 8,82 |
(s, |
1H) , |
7,54 |
(m, |
3H) |
|
7, 47 ( |
:d, 2H), |
7,20 |
(dd, |
1H), 6,88 |
(d, |
2H) , |
6, 79 |
(m, |
2H) |
|
6,55 ( |
m, 1H), |
4, 42 |
(dd, |
1H), 4,32 |
(dd, |
1H) , |
3, 76 |
(m, |
1H) |
2,57 (t, 1H), 2,04 (m, 1H).
Exemplo 14
N-[(IS,laR,7bR)-4,7-diflúor-l,la,2,7b,tetraidrociclopropa[c] cromen-l-il]-N-{5-[4-(1H-1,2,4-triazol-l-il)fenóxi]piridin2-il}uréia
a)_2-nitro-5-[4-(1H-1,2,4-triazol-l-il)fenóxi] piridina
Carbonato de césio (1,3 g, 4,03 mmol) foi misturado com 3 mL de dimetilformamida seca, 4-(lH-l,2,4triazol-l-il)fenol (0,5 g, 3,1 mmol) e 5-bromo-2nitropiridina (0,63g, 3,1 mmol) e a mistura reacional foi aquecida a 70°C em um frasco fechado. A mistura reacional foi então misturada com 40 mL de água e extraída em cloreto de metileno (3x20 mL) . Extrato orgânico foi lavado com água e solução salina, seco sobre sulfato de magnésio, e concentrado por evaporação rotatória. O sólido marromesverdeado resultante foi lavado cuidadosamente com cloreto de 23/07/2018, pág. 67/150 de metileno para dar 280 mg do composto desejado (32% produção).
1H-RMN (DMSO-d6): 9,3 (s, 1H), 8,47 (d, 1H), 8,35 (d, 1H),
8,24 (s, 1H), 7,98 (d, 2H), 7,71 (dd, 1H), 7,45 (d, 2H).
b) _5-[4-(1H-1,2,4-triazol-1-il)fenóxi]piridin-2amina
2-Nitro-5-[4-(1H-1,2,4 - triazol - 1 - il) fenóxi] piridina (100 mg, 0,35 mmol) foi misturado com 15-20 mL de etanol e borbulhado com argônio. Cerca de 20 mg de Pd/C foi adicionado à mistura reacional e gás hidrogênio foi aplicado em pressão normal e temperatura ambiente por 3-12 h. A reação foi monitorada por CCD. Após a reação estar completa, a mistura reacional foi borbulhada com argônio, filtrada através de Celite e a solução obtida foi concentrada por evaporação rotatória para dar 42 mg da aminopiridina desejada após purificação por cromatografia em coluna de sílica (EtOAc/EtOH 100:1). Produção 47%.
1H-RMN (CDCl3): 8,4 (s, 1H), 8,01 (s, 1H), 7,85 (d, ~1H),
7,50 (d, 2H), 7,35 (s, 1H), 6,96 (d, 2H), 6,48 (d, 1H), 4,55 (br s, 2H).
c) N-[(1S,1aR,7bR)-4,7-diflúor-1,1a,2, 7b,tetraidrociclopropa[c]cromen-1-il]-N-{5-[4-(1H-1,2, 4-triazol-1 il)fenóxi]piridin-2-il}uréia de 23/07/2018, pág. 68/150
Ácido (IS, laR, 7bS)-4,7-diflúor-l,la,2,7b, tetraidrociclopropa[c]cromeno-l-carboxílico (33 mg, 0,15 mmol, -95% ee) foi misturada com tolueno (1,5 mL) , trietilamina (l,leq), 5-(3-fluorfenil)-2-aminopiridina (1,1 eq) , DPPA (l,leq) e borbulhado com argônio por cerca de 5 minutos. A mistura reacional foi então aquecida em agitação a 110°C por 3 h em um frasco fechado. A mistura reacional foi concentrada por evaporação rotatória e purificada por cromatografía em coluna de sílica (30 g de YMC sílica, etilacetato/hexano 1:1). O produto desejado foi obtido como um pó bege-branco (40 mg, produção 57,5%) .
1H-RMN (CDCls): 9,42 (br s, 1H) , 9,35 (br s, 1H) , 8,52 (s,
1H) , 8,10 (s, 1H) , 7,65 (m, 3H) , 7,30 (dd, 1H) , 7,03 (d,
2H), 6,87 (d, 1H), 6,80 (m, 1H), 6,65 (d tr, 1H), 4,45 (dd,
1H) , 4,33 (dd, 1H) , 3,80 (q, 1H) , 2,60 (br tr, 1H) , 1,942,00 (m, 1H).
Braços Esquerdos Adicionais
Os seguintes braços esquerdos são acoplados a qualquer dos braços direito analogamente aos Exemplos 1 a .
Exemplo 15 de 23/07/2018, pág. 69/150
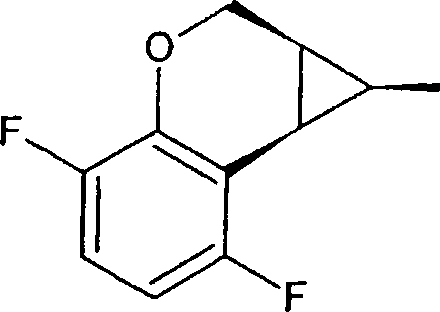
a) éster etil do acido (±)cis-1,la,2,7b-Tetraidrociclopropa[c]cromeno-l-carboxílico
Para uma mistura de 2H-cromeno (4,89 g, 37 mmol) e (CuOTf)2-benzeno (186 mg, 0,37 mmol) em 1,2-dicloroetano (80 mL) a 20°C, foi adicionado gota a gota (3 h) uma solução de diazoacetato de etila (8,44 g, 74 mmol) em 1,2-dicloroetano (20 mL) . Após 15 minutos a 20°C, a mistura reacional foi lavada com H2O (100 mL). A fase H2O foi lavada cm CH2C12 (50 mL) e o solvente das fases orgânicas combinadas foram removidas sob pressão reduzida. O produto bruto foi cromatografado em coluna (sílica gel, 20^50% EtOAc em hexano) , para dar 1,96 g (24%) de éster etil do acido ±cis1,la,2,7b-tetraidro-ciclopropa[c]cromeno-l-carboxílico e 3,87 g (48%) de éster etil do acido ±trans-l,la,2,7bTetraidro-ciclopropa[c]cromeno-l-carboxílico como um subproduto.
| 4H· |
-RMN |
(CDCls) : |
7,26 |
(d, |
1H) , |
7,10 |
(dd, |
1H) , |
6,90 |
(dd, |
1H) |
|
6, |
78 ( |
d, 1H), |
4, 49 |
(dd, |
1H) , |
4,20 |
(dd, |
1H) , |
3,97 |
(q, |
2H) |
|
2, |
44 ( |
:dd, 1H), |
2, 14 |
(dd, |
1H) |
, 2,07-1,95 |
(m, |
1H) , |
1,02 |
(t |
3H) .
b) ácido (±)cis-l,la,2, 7b-Tetraidro-ciclopropa [ c] cromeno-l-carboxílico de 23/07/2018, pág. 70/150
Uma mistura de éster etil ácido (±)cis-1,la,2,7bTetraidro-ciclopropa[c]cromeno-l-carboxílico (1,96 g, 9,0 mmol), LiOH (539 mg, 22,5 mmol), H2O (10 mL) e MeOH (20 mL) foi aquecida em refluxo por 2 h. A mistura reacional foi concentrada para cerca de 10 mL, HC1 4N foi adicionado gota a gota dando um precipitado branco. A mistura reacional foi extraída com CH2CI2 (3x15 mL) a o solvente das fases orgânicas combinados foram removidos sob pressão reduzida. O produto bruto foi cristalizado a partir de EtOAc/hexano, para dar 435 mg (25%) de ácido (±)cis-1,la,2,7b-tetraidro-
|
ciclopropa |
[c]cromeno-l-carboxílico |
como |
um sólido |
branco. |
| 1H-RMN (CDCI3) : |
9,80 (br s, 1H) , |
7,22 |
(d, 1H), |
7, 10 < |
:dd, |
|
1H), 6,89 |
(dd, |
1H) , 6,77 (d, 1H), |
4, 45 |
(dd, 1H) , |
4,22 ( |
:dd, |
|
1H), 4,45 |
(dd, |
1H) , 2,14-1,98 (m, |
2H) . |
|
|
|
Exemplo 16
a) éster etil do acido (±)-cis-1,la,3,7b-tetraidro -2-oxa-ciclopropa[a]naftaleno-l-carboxílico
Ester etil do acido (±)-cis-1,la,3,7b-Tetraidro-2oxa-ciclopropa[a]naftaleno-l-carboxílico foi sintetizado analogamente ao Exemplo 15a a partir de lH-isocromeno (3,57 de 23/07/2018, pág. 71/150 g, 27 mmol), para dar 910 mg (15%) de éster etil do acido (±)cis-1,la,3,7b-tetraidro-2-oxa-ciclopropa[a]naftaleno- 1 carboxílico 1H-RMN (CDCls) : 7,34 (d, 1H) , 7,25 (dd, lh) , 7,18 (dd, 1H) ,
7,03 (d, 1H) , 4,81 (d, 1H) , 4,51 (d, 1H) , 4,28 (dd, 1H) ,
3,95 (q, 2H), 2,43 (dd, 1H), 2,05 (dd, 1H), 1,04 (t, 3H).
b) ácido_(±)cis-1,la,3,7b-Tetraidro-2-oxaciclopropa[a]naftaleno-1-carboxílico
ácido (±)cis-1,la,3,7b-Tetraidro-2-oxa-ciclopropa [a]naftaleno-l-carboxílico foi sintetizado analogamente ao Exemplo 15b a partir de éster etil do acido (±)-cis-1,la, 3,7b-tetraidro-2-oxa-ciclopropa[a]naftaleno- 1 - carboxílico (436 mg, 2 mmol), para dar 86 mg (22%) de ácido (±)cis-1,la, 3,7b-Tetraidro-2-oxa-ciclopropa[a]naftaleno- 1 - carboxílico como um sólido branco. O produto bruto foi cromatografado em coluna (sílica gel, 1^5% MeOH em CH2CI2) .
1H-RMN (CDCls) : 8,50 (br s, 1H) , 7,39 (d, 1H) , 7,30 (dd,
1H) , 7,21 (dd, 1H) , 7,07 (d, 1H) , 4,87 (d, 1H) , 4,57 (d,
1H), 4,38 (dd, 1H), 2,59 (dd, 1H), 2,15 (dd, 1H).
O produto do passo b.
Exemplo 17 (±)-cis-l-(5-Ciano-piridin-2-il)-3-(7-hidróxi-6-propionil1,la,2,7b-tetraidro-ciclopropa[c]cromen-l-il)uréia.
a) 1-(2-Hidróxi-4-prop-2-inilóxi-fenil)-propan-1ona de 23/07/2018, pág. 72/150
Ο, /==3
ΗΟ \\ //
Uma mistura de 2'-4'-diidroxi-propiofenona (24,9 g, 0,15 mol), 3-bromo-propina (24,2 g, 0,20 mol) e K2CO3 (20,7 g, 0,15 mol) em acetona (500 mL) foi deixada em refluxo por 12 h. A mistura reacional foi deixada para chegar na temperatura ambiente sob pressão reduzida. O produto bruto foi purificado por cromatografia em coluna (sílica gel, 0->2% MeOH em H2O), para dar 26,2 g (85%) de 1(2-hidróxi-4-prop-2-inilóxi-fenil)-propen-l-ona.
íH-RMN (CDCls) : 12,80 (s, 1H) , 7,69 (d, 1H) , 6,52 (m, 2H) , 4,72 (d, 2H), 2,96 (q, 2H), 2,56 (t, 1H), 1,23 (t, 3H).
3b) 1-(5-Hidróxi-2H-cromen-6-il)-propan-lona.
Uma mistura de
1-(2-hidróxi-4-prop-2-inilóxifenil)-propan-l-ona (19,8 g, 97 mmol) e N,N-dietilanilina (100 mL) foram aquecidas em refluxo por 3 h. A mistura reacional foi concentrada sob pressão reduzida. O produto bruto foi purificado por cromatografia em coluna (sílica gel, 5Pl0% EtOAc em Hexano) e depois disso recristalizado a partir de EtOAc/Hexano, para dar 8,91 g (45%) de l-(5hidróxi-2H-cromen-6-il)propan-l-ona.
íH-RMN (CDCls) : 13,00 (s, 1H) , 7,49 (d, 1H) , 6,75 (dt, 1H) , de 23/07/2018, pág. 73/150
|
6,27 |
(d, 1H), |
5,67 |
(dt, |
1H) , |
4,86 (dd, 2H) , 2,90 (q, 2H) , |
|
1,19 |
(t, 3H).
3c) |
éster |
etil |
do |
ácido 7-Hidróxi-6-propionil- |
1,la,2,7b-tetraidro-ciclopropa[c]cromeno-l-carboxilico
A uma mistura de 1-(5-hidróxi-2H-cromen-6-il)propan-l-ona (511 mg, 2,5 mmol) e (Rh(II)Ac2)2 (11 mg, 0,025 mmol) em 1,2-dicloroetano (8 mL) a 20°C, foi adicionado gota a gota (3h) uma solução de diazoacetato de etila (571 mg, 5 mmol) em 1,2-dicloroetano (2 mL). Após 15 minutos a 20°C, a mistura reacional foi lavada com H2O (10 mL). A fase H2O foi lavada com CH2CI2 (10 mL) e o solvente das fases combinadas orgânicas foram removidos sob pressão reduzida. O produto bruto foi purificado por cromatografia em coluna (sílica gel, 1^5% MeOH em CH2CI2) , para dar 300 mg (41%) de éster etil do ácido 7-hidróxi-6-propionil-l,la,2,7b, tetraidro-
|
ciclopropa[c]cromeno-l-carboxilico |
(uma |
mistura de |
isômeros |
|
cis e tran 33/6 4) . |
|
|
|
| 1H-RMN (CDCls) : 13,13-13,07 (m, |
1H) , |
7,57-7,49 ( |
>, 1H), |
|
6,41-6,38 (m, 1H), 4,65-3,92 (m, |
4H) , |
3,01-1,95 |
(m, 5H), |
|
1,29-1,08 (m, 6H). |
|
|
|
|
3d) ácido (±)-cis-7-Hidróxi-6- |
-propionil-1, |
la,2,7b- |
tetraidro-ciclopropa[c]cromeno-l-carboxilico de 23/07/2018, pág. 74/150
Ácido (±)-cis-7-Hidróxi-6-propionil-l,la,2,7btetraidro-ciclopropa[c]cromeno-l-carboxílico foi sintetizado analogamente ao Exemplo 16b a partir de éster etil do ácido 7-Hidróxi-6-propionil-l,la,2,7b- tetraidro - ciclopropa [c] cromeno-l-carboxílico (299 mg, 1,03 mmol, uma mistura de isômeros cis e trans 33/64), para dar 39,3 mg (15%) de ácido (±)-cis-7-hidróxi-6-propionil-l,la,2,7b-tetraidro-ciclopropa [c]cromeno-l-carboxílico como um sólido branco e ácido (±)trans - 7-hidróxi-6-propionil-l,la,2,7b-tetraidro-ciclopropa [c]cromeno-l-carboxílico como um subproduto. O produto bruto foi purificado por cromatografia em coluna (sílica gel,
1^5% MeOH em CH2C12) .
1H-RMN (DMSO-de): 7,67 (d, 1H) , 6,35 (d, 1H), 4,57 (dd, 1H),
4,36 (dd, 1H) , 2,98 (q, 2H) , 2,55-2, 46 (m, 1H) , 2,18-2,00 (m, 2H), 1,10 (t, 3H).
Exemplo 18
a) 1-(2-Hidróxi-4-prop-2-inilóxi-fenil)-etanona
HO
1-(2-Hidróxi-4-prop-2-inilóxi-fenil)-etanona foi sintetizada analogamente ao Exemplo 17a a partir de 1-(2,4diidróxi-f enil)-etanona (20 g, 131 mmol), para dar 22 g (88%) de 1-(2-hidróxi-4-prop-2-inilóxi-fenil)-etanona.
de 23/07/2018, pág. 75/150 1H-RMN (CDCls) : 12,70 (s, 1H) , 7,66 (d, 1H) , 6,52 (m, 2H) ,
4,72 (d, 2H), 2,58-2,55 (m, 4H).
b) 1-(5-hidróxi-2H-cromen-6-il)-etanona
1-(5-Hidróxi-2H-cromen-6-il)-etanona foi sintetizado analogamente ao Exemplo 46b a partir de l-(2hidróxi-4-prop-2-inilóxi-fenil)-etanona (17 g, 89 mmol), para dar 6,0 g (35%) de 1-(5-hidróxi-2H-cromen-6-il)etanona.
!H-RMN (CDCls) : 12,92 (s, 1H) , 7,51 (d, 1H), 6,79 (dt, 1H) ,
6,32 (d, 1H), 5,71 (dt, 1H), 4,89 (dd, 2H), 2,55 (s, 3H).
c) éster etil do ácido 6-Acetil-7-Hidróxi1,la,2,7b-tetraidro-ciclopropa[c]cromeno-l-carboxílico
Éster etil do ácido 6-Acetil-7-hidróxi-l,la,2,7btetraidro-ciclopropa[c]cromeno-l-carboxílico (uma mistura de isômeros cis e trans 40/60) foi sintetizada analogamente ao Exemplo 17c a partir de 1-(5-hidróxi-2H-cromen-6-il)etanona.
!H-RMN (CDCls) : 13,05-12, 97 (m, 1H) , 7,54-7, 47 (m, 1H) ,
6,43-6,33 (m-lH)m 4,63-3,94 (m, 4H) , 3,02-1, 96 (m, 6H) ,
1,31-1,08 (m, 3H).
de 23/07/2018, pág. 76/150
d) ácido 6-Acetil-7-hidróxi-l,la,2,7b-tetraidrociclopropa[c]cromeno-l-carboxílico
Acido 6-Acetil-7-hidróxi-l,la,2,7b-tetraidrociclopropa[c]cromeno-l-carboxílico foi sintetizado analogamente ao Exemplo 15b a partir de éster etil do ácido 6-acetil-7-hidróxi-l,la,2,7b-tetraidro-ciclopropa[c]cromenol-carboxí lico (2 g, 8,1 mmol, uma mistura de isômeros cis e trans 40/60), para dar 300 mg (17%) de ácido 6-acetil-7hidróxi-1,la,2,7b-tetraidro - ciclopropa [c] cromeno - 1 carboxílico (uma mistura de isômeros cis e trans 40/60) . O produto bruto foi purificado por cromatografia em coluna (sílica gel, 1^5% MeOH em CH2CI2) .
1H-RMN (CDCls) : 7,55-7, 45 (m, 1H) , 6,45-6,30 (m, 1H) , 4,654,00 (m, 2H), 3,05-1,95 (m, 6H).
Exemplo 19
5a) 1-(4-Flúor-2-prop-2-inilóxi-fenil)propan-l-ona
A uma mistura de NaH (95%, 278 mg, 11 mmol) em DMF (20 mL) a 0 °C, foi adicionado 1-(4-flúor-2-hidróxifenil)propan-l-ona (1, 68 g, 10 mmol) em DMF (5 mL). Após 15 minutos a 0°C, foi adicionado à mistura reacional 3-bromode 23/07/2018, pág. 77/150 propina (3,02 mg, 20 mmol). Após lh a 0°C, permitiu-se gue a mistura reacional atingisse a temperatura ambiente. A mistura reacional foi extraída com H2O (100 mL). A fase H2O foi lavada com Et2Ü (3x100 mL) e o solvente das fases orgânicas combinados removidas sob pressão reduzida. O produto bruto foi purificado por cromatografia em coluna (sílica gel, CH2CI2), para dar 1,40 g (68%) de 1-(4-Flúor-2prop-2-inilóxi-fenil)propan-l-ona.
1H-RMN (CDCls) : 7,64 (dd, 1H) , 6,69 (dd, 1H) , 6,60 (ddd,
1H), 4,68 (d, 2H), 2,85 (g, 2H), 2,58 (t, 1H), 1,03 (t, 3H).
b) 1-(5-Flúor-2H-cromen-8-il)propan-l-ona
1-(5-Flúor-2H-cromen-8-il)propan-l-ona foi sintetizado analogamente ao Exemplo 17b a partir de l-(4flúor-2-prop-2-inilóxi-fenil)-propan-l-oma (1,34 g, 6,5 mmol), para dar 619 mg (46%) de 1-(5-flúor-2H-cromen-8il)propan-l-ona.
!H-RMN (CDCls) : 7,60 (dd, 1H), 6,67-6,58 (m, 2H), 5,86 (dt,
1H), 4,76 (dd, 2H), 2,93 (g, 2H), 1,23 (t, 3H).
c) éster etil do acido (±)-cis-7-Flúor-4-propionil —1,la,2,7b-tetraidro-ciclopropa[c]cromeno-l-carboxílico de 23/07/2018, pág. 78/150
Ester etil do acido (±)-cis-7-Flúor-4-propionil1,la,2,7b-tetraidro-ciclopropa[c]cromeno-l-carboxílico foi sintetizado de acordo com o método 17c) a partir de 1(5flúor-2H-cromen-8-il)-propan-l-ona (619 mg, 3 mmol), para dar 142 mg (16%) de éster etil do acido (±)-cis-7-flúor-4propionil-1,la,2,7b-tetraidro-ciclopropa [c] cromeno - 1 carboxílico e éster etil do acido (±)-trans-7-flúor-4propionil-1,la,2,7b-tetraidro-ciclopropa [c] cromeno - 1 carboxílico como um subproduto.
1H-RMN (CDCls) : 7,59 (dd, 1H) , 6,65 (m, 1H) , 4,50-4, 46 (m,
2H) , 3,95 (q, 2H) , 2,89 (q, 2H) , 2,57 (dd, 1H) , 2,20 (dd,
1H), 1,13-1,03 (m, 1H), 1,12-1,01 (m, 6H).
d) acido (±)-cis-7-Flúor-4-propionil-l,la,2,7btetraidro-ciclopropa[c]cromeno-l-carboxílico
Ácido (±)-cis-7-Flúor-4-propionil-l,la, 2, 7btetraidro-ciclopropa[c]cromeno-l-carboxílico foi sintetizado analogamente ao Exemplo 15b a partir de éster etil do acido (±)-cis-7-flúor-4-propionil-l,la,2,7b-tetraidro - ciclopropa [c]cromeno-l-carboxílico (140,3 mg, 0,48 mmol), para dar 83 mg (65%) de ácido (±)-cis-7-flúor-4-propionil-l,la,2,7btetraidro-ciclopropa[c]cromeno-l-carboxílico como um sólido de 23/07/2018, pág. 79/150 branco. O produto bruto foi purificado por cromatografía em coluna (sílica gel, 1^5% MeOH em CH2CÍ2) .
1H-RMN (DMSO-d6): 12,15 (br s, 1H), 7,46 (dd, 1H), 6,78 (dd,
1H), 4,57 (dd, 1H), 4,43 (dd, 1H), 2,93-2,80 (m- 2H)m 2,55 (dd, 1H), 2,24 (dd, 1H), 2,20-2,10 (m, 1H), 1,02 (t- 3H).
Exemplo 20
a) 6-Flúor-2-hidróxi-3-metóxi-benzaldeído
1M de tricloreto de boro em diclorometano (25 mL; 25mmol) foi adicionado a uma solução de 6-flúor-2-hidróxi-3dimetóxi-benzaldeído [Cantrell, Amanda S.; Engelhardt, Per; Hoegberg, Marita; Jaskunas, S. Richard; Johansson, Nils Gunnar; e colaboradores; J. Méd. Chem.; 39; 21. 1996; 42614274] (4,26 g; 23 mmol) em diclorometano (30 mL) mantendo a reação à temperatura de -70°C. A mistura reacional foi agitada em temperatura ambiente por uma noite e hidrolizada com água. A fase orgânica foi separada, lavada com água e evaporada a vácuo. O resíduo foi cromatografado (sílica gel, EA:Hex, 5:1) para dar 3,72 g (94%) de 6-flúor-2-hidróxi-3metóxi-benzaldeído como cristais amarelos.
1H-RMN (CDCl3): 11,61 (s, 1H), 10,23 (s, 1H), 7,02 (dd, 1H),
6,55 (app. T, 1H), 3,87 (s, 3H).
b) 5-Flúor-8-metóxi-2H-cromeno.
6-Flúor-2-hidróxi-3-metóxi-benzaldeído(3,32 g, 19 mmol) foi dissolvido em acetonitrila (20 mL) e DBU (2,97 mL, mmol) foi adicionado seguido por brometo de viniltrifenilfosfina (7,2 g, 19 mmol). A mistura reacional foi aquecida sob refluxo por 48h., diluída e extraída com éter (3x50 mL) . A fase orgânica foi lavada com água, 10% de de 23/07/2018, pág. 80/150 hidróxido de sódio, água e solução salina e evaporada a vácuo. O resíduo foi submetido à cromatografia em coluna (sílica gel), EA:Hex 1:20) produzindo 1,2 g de 5-Flúor-8metóxi-2H-cromeno (34%).
1H-RMN (CDCls): 6,65 (m, 2H), 6,54 (t, 1H), 5,83 (dt, 1H),
4,88 (dd, 2H), 3,83 (s, 3H).
c) éster etil do ácido (±)-cis-7-Flúor-4-metóxi1,1a,2,7b-tetraidro-ciclopropa[c]cromeno-1-carboxílico
O composto título foi sintetizado analogamente ao exemplo 17c a partir de 5-flúor-8-metóxi-2H-cromeno.
1H-RMN (CDCls): 6,7-6,5 (m, 2H), 4,48 (m, 2H), 3,99 (m, 2H),
3,80 (s, 3H), 2,57 (app. T, 1H), 2,20 (app.t, 1H), 2,05 (m,
1H), 1,08 (t, 3H).
d) _ácido_(±) -cis-7-flúor-4-metóxi-1,1a, 2, 7btetraidro-ciclopropa[c]cromeno-1-carboxílico
O composto título foi sintetizado analogamente ao exemplo 15b a partir do éster etil do ácido (±)-cis- 7 flúor-4-metóxi-1,1a,2,7b-tetraidro-ciclopropa[c]cromeno- 1 carboxílico.
1H-RMN (CDCl3): 6,7-6,5 (m, 2H), 4,48 (m, 2H), 3,80 (s, 3H), 2,61 (aap. T, 1H), 2,17 (aap. T, 1H), 2,06 (m, 1H).
e) (±)-cis-1-(5-Ciano-piridin-2-il)-3-(7-flúor-4metóxi-1,1a,2,7b-tetraidro-ciclopropa[c]cromen-1-il)-uréia.
O composto título foi sintetizado analogamente ao Exemplo 15c a partir do ácido (±)-cis-7-flúor-4-metóxi1,1a,2,7b-tetraidro-ciclopropa[c]cromeno-1-carboxílico (62 mg, 0,17 mmol). Produção 38 mg (40%).
1H-RMN (CDCl3): 10,06 (br. s, 1H), 9,40 (br. d, 1H), 8,11 de 23/07/2018, pág. 81/150
|
(d, |
1H), |
7,70 (dd, |
1H), |
6,91 |
(d, 1H), 6,68 |
(m, |
2H), 4,48 |
|
(dd, |
1H) , |
4,28 (dd, |
1H), |
3,90 |
-3,72 (m, 4H), |
2,64 |
(app. T, |
|
1H), 1,96 |
(m, 1H). |
|
|
|
|
|
Exemplo 21
a) 1-Cloro-4-flúor-2-prop-2-inilóxi-benzeno
O composto título foi sintetizado analogamente ao exemplo 15a) a partir do 2-cloro-5-fluorfenol (2,5 g). Produção 2,8 g (90%).
1H-RMN (CDCl3): 7,32 (dd, 1H), 6,85 (dd, 1h), 6,68 (m, 1H),
4,77 (d, 2H), 2,58 (t, 1H).
b) 5-Flúor-8-cloro-2H-cromeno
O composto título foi sintetizado analogamente ao Exemplo 15b) a partir do 1-cloro-4-flúor-2-prop-2-inilóxi benzeno (2,8 g). Produção 0,97 g (35%).
| 1H-RMN (CDCl3) |
: 7,09 |
(dd, |
1H) |
, 6,63 |
(dt, 1H), 6,56 |
(t, 1H), |
|
5,84 (dt, 1H) |
, 4,95 |
(dd, |
2H). |
|
|
|
|
c) |
éster |
etil |
do |
ácido |
(±)-cis-7-Flúor- |
4-cloro- |
1,1a,2,7b-tetraidro-ciclopropa[c]cromeno-1-carboxílico
O composto título foi sintetizado analogamente ao
Exemplo 15c) a partir do 5-flúor-8-cloro-2H-cromeno.
| 1H-RMN (CDCl3) |
: 7,14 (dd, |
1H), 6,60 (t, |
1H), 4,51 |
(m, 2H), |
|
4,04 (m, 2H), |
2,60 (app. |
T, 1H), 2,23 (t, |
1H), 2,09 |
(m, 1H), |
|
1,08 (t, 3H). |
|
|
|
|
|
d) |
ácido |
(±)-cis-7-Flúor |
-4-cloro-1, |
1a,2,7b- |
tetraidro-ciclopropa[c]cromeno-1-carboxílico
O composto título foi sintetizado analogamente ao
Exemplo 15d) a partir do éster etil do ácido (±)-cis -7-flúor-4-cloro-1,1a,2,7b-tetraidro-ciclopropa[c]cromeno-1de 23/07/2018, pág. 82/150 carboxílico (850 mg) . Produção 43 mg (96%) .
1H-RMN (CDCls): 8,86 (br. s, 1H), 7,13 (dd, 1H), 6,59 (t,
1H), 4,50 (m, 2H), 2,63 (t, 1H), 2,23-2,05 (m, 2H).
Exemplo 22
a) _4-formil-3-hidróxi-fenil éster do ácido triflúor-metanossulfônico
Uma solução de triflico anidro (1,77 mL, 10,5 mmol) em diclorometano (10 mL) foi adicionada a uma mistura de 2,4-ddidroxibenzaldeído (1,38 g, 10 mmol) e piridina (0,85 mL, 10,5 mmol) em diclorometano (30 mL) a -70°C. O banho de gelo seco foi removido e a mistura reacional foi agitada por 2 h em temperatura ambiente. A mistura reacional foi diluída com diclorometano, lavada com água, solução salina e evaporada a vácuo. O produto bruto foi purificado por cromatografia em coluna (sílica gel, EA:Hex, 1:6) para dar 1,55 g de 4- formil-3-hidróxi-fenil éster do ácido triflúor-metanossulfônico (57%).
1H-RMN (CDCls): 11,28 (s, 1H), 9,93 (s, 1H), 7,67 (d, 1H),
6,95 (m, 2H).
b) _3-alilóxi-4-formil-fenil éster do ácido triflúor-metanossulfônico
Carbonato de potássio (1,6 g, 11,5 mmol) e brometo de alil (1 mL, 11,5 mmol) foram adicionados a uma solução de
4- formil-3-hidróxi-fenil éster do ácido triflúormetanossulfônico (1,55 g, 5,7 mmol) em acetona (50 mL). A mistura reacional foi agitada a 55°C por 2h, filtrada e evaporada a vácuo. O resíduo foi cromatografado (sílica gel, EA:Hex, 1:20) para dar 1,3 g (73%) de 3-alilóxi-4de 23/07/2018, pág. 83/150 formil-fenil éster do ácido triflúor-metanossulfônico.
1H-RMN (CDCls): 10,47 (s, 1H), 7,93 (d, 1H), 6,95 (d, 1H),
6,90 (s, 1H), 6,05 (m, 1H), 5,47 (d, 1H), 5,40 (d, 1H), 4,69 (d, 2H).
c)_3-alilóxi-4-vinil-fenil éster do ácido triflúor-metanossulfônico
Brometo de metilfenilfosfônio (1,95 g, 5,45 mmol) foi adicionado a uma suspensão de hidreto de sódio (60% em óleo) (0,25 g, 6,3 mmol) em THF (35 mL) a 0°C e esta foi agitada por 30 minutos em temperatura ambiente. À solução acima, foi adicionada uma solução de 3-alilóxi-4-formilfenil éster do ácido triflúor-metanossulfônico (1,3 g, 4,2 mmol) em THF (15 mL), e a mistura reacional foi agitada por 2 h em temperatura ambiente. A mistura reacional foi diluída com hexano e extraída com água. Fase orgânica foi lavada com solução salina e evaporada. Cromatografia em coluna de sílica gel (EA:Hex, 1:20) proporcionou o 3-alilóxi-4-vinilfenil éster do ácido triflúor-metanossulfônico (0,68 g,
53%) .
| 1H-RMN (CDCl3) |
: 7,51 (d, 1H), |
7,02 (dd, 1H), 6,85 |
(dd, |
1H) |
|
6,77 (d, 1H), |
6,05 (m, 1H), |
5,76 (dd, 1H), 5,43 |
(m, |
1H) |
|
5,32 (m, 2H), |
4,58 (dt, 2H). |
|
|
|
|
d) |
2H-cromen-7-il |
éster do ácido |
triflúor |
metanossulfônico
A uma solução de 3-alilóxi-4-vinil-fenil éster do ácido triflúor-metanossulfônico (0,68 g, 2,2 mmol) em diclorometano (5 mL) foi adicionado o catalisador Ru (catalisador de Grubb) (36 mg, 2 mol%), e a mistura de 23/07/2018, pág. 84/150 reacional foi agitada por 2 h em temperatura ambiente. Após este período, a reação foi completada (GC) e a mistura reacional foi utilizada no próximo passo sem qualquer aperfeiçoamento. Amostra analítica foi obtida após remoção do solvente por cromatografia em coluna de sílica gel (EA:Hex, 1:20) .
1H-RMN (CDCls): 6,97 (d, 1H), 6,76 (dd, 1H), 6,68 (d, 1h),
6,39 (dt, 1H), 5,81 (dt, 1H), 4,98 (dd, 2H).
e)_éster_etil_do_ácido_(±) -cis-5trifluormetanosulfonilóxi-1,1a,2,7b-tetraidro-ciclopropa [c] cromeno-1-carboxílico
Rh(OAc)2 (19 mg, 2 mol%) foi adicionado à solução acima (10d) e a solução de EDA (0,44 mL. 4,4 mmol) em 1 mL de diclorometano foi adicionada com uma bomba de seringa por 5 h em temperatura ambiente. Quando a reação foi completada (GC) o diclorometano foi evaporado, o resíduo foi dissolvido em acetato de etila e lavado com solução de cloreto de amônio saturada e solução salina. A fase orgânica foi evaporada e a mistura bruta de isômeros cis- e trans(1:1.3) foi separada por cromatografia em coluna (sílica gel, EA:Hex, 1:6) para dar o,4 g (50%) de éster etil do ácido (±)-cis-5-trifluormetanosulfonilóxi-1,1a,2,7btetraidro-ciclopropa[c]cromeno-1-carboxílico.
| 1H-RMN (CDCl3): 7,29 (d, 1H), 6,82 |
(dd, |
1H), |
6,73 |
(d, |
1H), |
|
4,51 (dd, 1H), 4,29 (dd, 1H), 3,98 |
(m, |
2H), |
2,45 |
(t, |
1H), |
|
2,19 (t, 1H), 2,05 (m, 1H), 1,03 (t, |
3H). |
|
|
|
|
f) éster etil do ácido (±)-cis-5-Ciano-1,1a,2,7btetraidro-ciclopropa[c]cromeno-1-carboxílico de 23/07/2018, pág. 85/150
Éster etil do ácido (±)-cis-5trifluormetanosulfonilóxi-1,1a,2,7b-tetraidro-ciclopropa [c] cromeno-1-carboxílico (154 mg, 0,42 mmol), Pd(OAc)2 (9 mg, 10 mol%) e PPh3 (44 mg, 40 mol%) foram misturados em DMF (4 mL) e um fluxo de nitrogênio gentilmente passado através da mistura reacional por 10 minutos. Zn(CN)2 (74 mg, 0,63 mmol) foi adicionado, o frasco foi fechado e a mistura reacional foi agitada a 120 °C por uma noite. A mistura reacional foi diluída com acetato de etila e extraída com cloreto de amônio saturado. A fase orgânica foi evaporada e o resíduo cromatografado (sílica gel, EA:Hex 1:5) para dar 53 mg (52%) de éster etil do ácido (±)-cis-5-Ciano-1,1a,2,7b-tetraidrociclopropa[c]cromeno-1-carboxílico.
| 1H-RMN (CDCl3) |
: 7,33 |
(d, 1H), 7,19 |
(dd, 1H), 7,05 (d, |
1H), |
|
4,50 (dd, 1H) |
, 4,25 |
(dd, 1H), 3,99 |
(q, 2H), 2,46 (t, |
1H), |
|
2,25 (t, 1h), |
2,11 (m |
, 1H), 1,06 (t, |
3H). |
|
|
g) |
ácido |
(±)-cis-5-Ciano-1,1a,2,7b-tetraidro- |
ciclopropa[c]cromeno-1-carboxílico
Éster etil do ácido (±)-cis-5-Ciano-1,1a,2,7btetraidro-ciclopropa[c]cromeno-1-carboxílico (53 mg, 0,22 mmol) e NaOH (35 mg, 0,88 mmol) foram dissolvidos em uma mistura de metanol e água (1:1) (5 mL) . A mistura reacional foi agitada a 60°C por 30 minutos. Metanol foi evaporado a vácuo e 20 mL de água foi adicionada. A solução resultante foi extraída com éter. A fase aquosa concentrada, acidificada com 1M de HCl para o pH~2 e extraída com éter. A fase orgânica foi lavada com solução salina e evaporada para dar 42 mg (90%) de ácido (±)-cis-5-Ciano-1,1a,2,7bde 23/07/2018, pág. 86/150 tetraidro-ciclopropa[c]cromeno-1-carboxílico.
1H-RMN (CDCls): 7,33 (d, 1H), 7,19 (dd, 1H), 7,06 (d, 1H),
4,51 (dd, 1H), 4,31 (dd, 1H), 2,53 (app. T, 1H), 2,27 (app.
t., 1H), 2,16 (m, 1H).
Exemplo 23
a) éster etil do ácido (±)-cis-5-trimetilsilaniletinil-1,1a,2,7b-tetraidro-ciclopropa[c] cromeno - 1carboxílico
Éster etil do ácido (±)-cis-5-trifluormetanosulfonilóxi-1,1a,2,7b-tetraidro-ciclopropa[c]cromeno - 1 carboxílico (152 mg, 0,41 mmol), DPPP (38 mg, 20 mol%), Pd(dba)2 (24 mg, 10 mol%), CuI (3 mg, 4 mol%) foram misturados em 3 mL de trietilamína e um fluxo de nitrogênio foi gentilmente passado através da mistura por 10 minutos. Trimetilsilil-acetileno (0,088 mL, 0,62 mmol) foi adicionado, o frasco foi fechado e a mistura reacional foi agitada a 120°C por uma noite. A mistura reacional foi diluída com acetato de etila, lavada com água, solução salina e evaporada. O resíduo foi purificado por cromatografia em coluna de sílica gel (EA:Hex, 1:15) para dar 0,1 g (77%) de éster etil do ácido (±)-cis-5trimetilsilaniletinil-1,1a,2,7b - tetraidro - ciclopropa [c] cromeno-1-carboxílico.
| 1H-RMN (CDCl3): 7,15 (d, 1H), 7,01 |
(dd, |
1H), |
6,88 |
(d, |
1H), |
|
4,47 (dd, 1H), 4,16 (dd, 1H), 3,96 |
(q, |
2H), |
2,38 |
(t, |
1H), |
|
2,13 (t, 1H), 2,01 (m, 1H), 1,04 (t, |
3H), |
0,22 |
(s, |
9H). |
|
b)_ácido_(±)-cis-5-Etinil-1,1a,2,7b-tetraidrociclopropa[c]cromeno-1-carboxílico de 23/07/2018, pág. 87/150
Éster etil do ácido (±)-cis-5-trimetilsilaniletinil-1,1a,2,7b-tetraidro-ciclopropa[c] cromeno - 1carboxílico (0,1g, 0,32 mmol) e hidróxido de sódio (0,076 g, 1,9 mmol) foram dissolvidos em mistura de metanol:água (1:1) (5 mL) . A mistura reacional foi aquecida a 60°C por 5 h, então ela foi acidificada com 1M de HCl para pH~2 e extraída com éter. A fase orgânica foi lavada com solução salina e evaporada para dar 66 mg (97%) de ácido (±)-cis-5-etinil1,1a,2,7b-tetraidro-ciclopropa[c]cromeno-1-carboxílico.
| 1H-RMN (CDCl3): 7,17 (d, |
1H), |
7,03 |
(dd, 1H), |
6,91 (d,1H), |
|
4,45 (dd, 1H), 4,23 (dd, |
1H), |
3,02 |
(s, 1H), |
2,46 (t, 1H), |
|
2,13 (t, 1H), 2,07 (m, 1H) |
. |
|
|
|
Exemplo 24 (±)-cis-1-(5-Acetil-1,1a,2,7b-tetraidro-ciclopropa[c]cromen1-il)-3-(5-ciano-piridin-2-il)-uréia
a) éster etil do ácido (±)-cis-1-(5-Acetil-1,1a,2, 7b-tetraidro-ciclopropa[c]cromeno-1-carboxílico
Éster etil do ácido (±)-cis-5-trifluormetnosulfonilóxi-1,1a,2,7b-tetraidro-ciclopropa[c]cromeno - 1 carboxílico (117 mg, 0,32 mmol), DPPP (7,3 mg, 50 mol%), Pd(OAc)2 (2 mg, 25 mol%) e trietilamina (0,09 mL, 0,64 mmol) foram misturados em DMF (3 mL) e um fluxo de nitrogênio foi gentilmente passado através da mistura reacional por 10 minutos. Butil vinil éter (0,21 mL, 1,6 mmol) foi adicionado, o frasco foi fechado e a mistura reacional foi agitada a 100°C por 2h. 5% de HCl (5 mL) foi adicionado e a mistura reacional foi agitada em temperatura ambiente por 30 minutos. A mistura resultante foi extraída com acetato de de 23/07/2018, pág. 88/150 etila.
A fase orgânica foi lavada com cloreto de amônio saturado e evaporado. O resíduo foi purificado por cromatografia em coluna de sílica gel (EA:Hex, 1:5) para dar 76 mg (91%) de éster etil do ácido (±)-cis-5-acetil1,1a,2,7b-tetraidro-ciclopropa[c]cromeno-1-carboxílico.
1H-RMN (CDCls): 7,52 (dd, 1H), 7,37 (d, 1H), 7,34 (d, 1H), 4,52 (d, 1H), 4,26 (dd, 1H), 2,55 (s, 3H), 2,53 (t, 1H), 2,25 (t, 1H), 2,13 (m, 1H).
Exemplo 25
Ácido (±)-cis-5-Metóxi-1,1a,2,7b-tetraidro-ciclopropa[c] cromeno -1-carboxílico
O composto título foi sintetizado analogamente ao exemplo 22 a partir de 2-hidróxi-4-metoxibenzaldeído.
Exemplo 26
a) N-.Acetil-1,2-diidroquinolina.
Quinolina (19,37 g, 150 mmol) foi dissolvida em dietiléter anidro (500 mL) e arrefecido a 0°C sob atmosfera inerte. DIBAL, 1,5 M em tolueno (100 mL, 150 mmol) foi adicionado gota a gota por 2 horas e a mistura reacional foi agitada a 0°C por 30 minutos. Anidrido acético (500 mL) foi adicionado gota a gota por 30 minutos e a mistura reacional foi agitada a 0°C para 30 minutos. H2O foi adicionada cautelosamente. A mistura reacional foi extraída com dietil éter e concentrada para dar N-acetil-1,2-diidroquinolina (11,5 g, 44%).
b) éster etil do ácido (±)-cis-(N-acetil-1,1a,2,7b -tetraidro-ciclopropa[c]quinolina)-1-carboxílico de 23/07/2018, pág. 89/150
Éster etil do ácido (±)-cis-(N-acetil-1,la,2,7btetraidro-ciclopropa[c]quinolina)-1-carboxílico foi preparado de acordo com o procedimento descrito no exemplo 15a a partir de N-acetil-1,2-diidroquinolina (10 g, 58 mmol) . O produto foi purificado por cromatografia em coluna de sílica (EtOAc/hexano 5%^50%) para dar éster etil do ácido (±)-cis-(N-acetil-1,la,2,7b-tetraidro-ciclopropa[c] quinolina)-1-carboxílico (2,0 g, 13 %).
c) ácido (±)-cis-(N-acetil-1,la,2,7b-tetraidrociclopropa[c]quinolina)-1-carboxílico
Ácido (±)-cis-(N-acetil-1,la,2,7b-tetraidrociclopropa[c]quinolina)-1-carboxílico (425 mg, 24%) foi preparado de acordo com o procedimento descrito no exemplo 15b, a partir de éster etil do ácido (±)-cis-(N-acetil1,la,2,7b-tetraidro-ciclopropa[c]quinolina)- 1 - carboxílico (2,0 mg, 7,7 mmo 1) .
Exemplo 27
a) 2,4-Diflúor-2-propiniloxibenzeno.
O 2,5-difluorfenol comercialmente disponível (20 g, 0,15 mol), K2CO3 (53 g, 0,38 mol) e 3-bromopropina comercialmente disponível (45 g, 0,38 mol) foram dissolvidos em acetona (300 mL), deixado em refluxo por uma noite, arrefecido e filtrado. O solvente foi removido e o produto bruto, dissolvido em éter e lavado com água e solução de 23/07/2018, pág. 90/150 salina. A fase orgânica foi evaporada e o produto bruto foi re-dissolvido em uma pequena quantidade de éter e filtrado através de uma coluna de AI2O3 básico. Evaporação e secagem deram 20 g (80%) de 2,4-diflúor-2-prop-inilóxi-benzeno.
b) 5,8-Diflúor-2H-cromeno.
2,4-diflúor-2-propiniloxibenzeno (20 g, 0,12 mol) foi dissolvido em Ν,Ν-dietil anilina (100 mL) e aquecido sob atmosfera de argônio a 225 °C com um banho de óleo por 6-8 h. Éter (150 mL) foi adicionado e a anilina foi removida por extração utilizando 2M de HCl(aq). Purificação por cromatografia (sílica gel, n-hexano) deu 5,8-diflúor-2Hcromeno 5,8 g (29%) .
c) éster etil do ácido (±)-cis-4,7-Diflúor1,la,2,7b-tetraidro-ciclopropa[c]cromeno-l-carboxílico
CO2Et
5, 8-Diflúor-2H-cromeno (5 g, 0,03 mol), (Rh(II)Ac2)2 (0,39 g, 0, 00089 mol) foi dissolvida em 1,2dicloroetano (60 mL) ou clorofórmio livre de etanol.
Diazoacetato de etila (9,4 mL, 0089 mL) no mesmo solvente foi adicionado gota a gota por um período de aproximadamente h sob atmosfera de N2. O solvente foi então removido a vácuo e a mistura foi tomada em acetato de etila, lavado com de 23/07/2018, pág. 91/150
NaHCCb (aq), água e solução salina e o solvente removido. O produto (33% cis, 66% trans) foi purificado por cromatografia (0^10% acetato de etila em n-hexano) para dar 2,2 g do composto titulo (30%) .
d)_ácido_cis-4, 7-Diflúor-l, la, 2, 7b-tetraidrociclopropa[c]cromeno-l-carboxílico
co2h
Ester etil do ácido cis-4,7-Diflúor-1,la,2,7btetraidro-ciclopropa[c]cromeno-l-carboxílico (2g, 0,008 mol) foi aquecido em 1M LiOH em metanol-água (25%) a 80° por 2 h. O volume foi reduzido à metade e acidifiçado. Extração com éter seguido por cromatografia (sílica gel, éter) deram o composto titulo puro (35%).
Exemplo 28
Intermediários adicionais
a) 6-Fluorcroman-4-ol φτ0Η
F
6-Fluorcroman-4-ona (10 g, 61 mmol) foi dissolvida em etanol (100 mL), NaBH4 (excesso) foi adicionado e arrefecido em banho de gelo. A mistura foi então deixada em temperatura ambiente por 2h, seguido por refluxo por 4 h. Purificação por cromatografia (sílica gel, éter-hexano, 1:5) deu 8 g (80%) de 6-flúor-croman-4-ol puro.
de 23/07/2018, pág. 92/150
b) 6-Flúor-2H-cromeno
F
6-Fluorcroman-4-ol (8 g, 48 mmol) e ácido tolueno4-silfônico (lg) foram dissolvidos em tolueno e deixados em refluxo por uma noite, com subseqüente remoção de água. A mistura foi então arrefecida e lavada com NaHCCç (aq) e purificada por cromatografia (sílica gel, n-hexano) para dar 4,2 g (52%) de 6-flúor-2H-cromeno puro.
c) éster etil do ácido (±)-cis-6-Flúor-l,la,2,7btetraidro-ciclopropa[c]cromeno-l-carboxílico
CO?Et
Este composto foi preparado analogamente ao éster etil do ácido (±)-cis-4,7-Diflúor-1,la,2,7b-tetraidrociclopropa[c]cromeno-l-carboxílico mas utilizando 6-flúor2H-cromeno para dar 1,9 (29%) do composto titulo.
d)_ácido_cis-6-Flúor-l, la, 2, 7b-tetraidro15 ciclopropa[c]cromeno-l-carboxílico
co2h
Petição 870180063303, de 23/07/2018, pág. 93/150
Este composto foi preparado analogamente ao ácido cis-4,7-diflúor-l,la,2,7b-tetraidro-ciclopropa[c]cromeno-1 carboxílico mas utilizando éster etil do ácido cis-6-flúor1,la,2,7b-tetraidro-ciclopropa[c]cromeno-1-carboxílico (1,9 5 g, 8 mmol) para dar 350 mg (21%) de ácido cis-6-flúor1,la,2,7b-tetraidro-ciclopropa[c]cromeno-1-carboxílico puro.
e) 1-Bromo-4-flúor-2-prop-2-inilóxi-benzeno
Este composto foi preparado analogamente ao 2,4diflúor-2-porp-inilóxi-benzeno mas utilizando o 2-bromo-510 fluorfenol (15 g, 78 mmol) para dar o -bromo-4-flúor-2-prop2-inilóxi-benzeno 15,6 g (87%).
f) 2-Bromo-4-flúor-1-prop-2-inilóxi-benzeno
BrEste composto foi preparado analogamente ao 2,4diflúor-2-prop-inilóxi-benzeno mas utilizando o 2-bromo-415 flúor-fenol (15 g, 78 mmol) para dar o 2-bromo-4-f lúor-1prop-2-inilóxi-benzeno 15 g (84%).
g) 1,3-diflúor-5-prop-2-inilóxi-benzeno
Petição 870180063303, de 23/07/2018, pág. 94/150
ΛΓ
Este composto foi preparado analogamente ao 2,4diflúor-2-propiniloxibenzeno mas utilizando o 3,5-diflúorfenol (14 g, 107 mmol) para dar o 1,3-diflúor-5-prop-2-
|
inilóxi- |
-benzeno 12 g (67%) . |
|
5 |
h) 8-Bromo-6-flúor-2H-cromeno |
|
diflúor- |
O
F
Este composto fio preparado analogamente ao 5,8-
-2H-cromeno mas utilizando (15 g, 65 mmol) de 2- |
|
bromo-4- |
-flúor-l-prop-2-iniloxibenzeneto dando o composto |
|
título |
(7 g, 46%) . |
|
10 |
i) 8-Bromo-5-flúor-2H-cromeno |
|
diflúor- |
Este composto fio preparado analogamente ao 5,8-
-2H-cromeno mas utilizando (15 g, 65 mmol) de 1- |
|
bromo-4- |
-flúor-2-prop-2-iniloxibenzeo dando o composto título |
|
(3,7 g, |
25%) . |
|
15 |
j) 5,7-Diflúor-2H-cromeno |
Petição 870180063303, de 23/07/2018, pág. 95/150
Este composto fio preparado analogamente ao 5,8diflúor-2H-cromeno mas utilizando (18 g, 107 mmol) de 1,3diflúor-5-prop-2-iniloxibenzeno e PEG-200 como solvente dando o composto titulo (4 g, 23%).
k) éster etil do ácido(±)-cis-4-Bromo-6-flúor1,la,2,7b-tetraidro-ciclopropa[c]cromeno-l-carboxílico
CO2Et
Este composto foi preparado analogamente ao éster etil do ácido (±)-cis-4,7-diflúor-l,la,2,7b-tetraidrociclopropa[c]cromeno-l-carboxílico mas utilizando 5 g (22 mmol) de 8-bromo 6-flúor-2H-cromeno para dar 1,9 g (30%) de éster etil do ácido cis-6-flúor-1,la,2,7b-tetraidrociclopropa[c]cromeno-l-carboxílico.
1) éster etil do ácido±-cis-4-Bromo-7-flúor1,la,2,7b-tetraidro-ciclopropa[c]cromeno-l-carboxílico
Este composto foi preparado analogamente ao éster etil do ácido ±-cis-4,7-diflúor-l,la,2,7b-tetraidrociclopropa[c]cromeno-l-carboxílico mas utilizando 3,5 g (15,3 mmol) de 8-bromo-5-f lúor-2H-cromeno para dar 1,6 g
Petição 870180063303, de 23/07/2018, pág. 96/150 (33%) de éster etil do ácido ±-cis-4-Bromo-7-flúor1,la,2,7b-tetraidro-ciclopropa[c]cromeno-l-carboxilico.
m) éster etil do ácido ±-cis-5,7-diflúor1,la,2,7b-tetraidro-ciclopropa[c]cromeno-l-carboxilico
CO2Et
Este composto foi preparado analogamente ao éster etil do ácido ±-cis-4,7-diflúor-1,la,2,7b-tetraidrociclopropa[c]cromeno-l-carboxilico mas utilizando 2 g (12 mmol) de 5,7-diflúor-2H-cromeno para dar 0,9 g (29%) de éster etil do ácido ±-cis-5,7-diflúor-1,la,2,7b-tetraidrociclopropa[c]cromeno-l-carboxilico.
Exemplo 29
a) Resolução do ácido cis-7-flúor-4-cloro-l,la,2,
7b-tetraidro-ciclopropa[c]cromeno-l-carboxilico racêmico.
Foi dissolvido 0,32 g (1,32 mmol) do ácido cis -7-flúor-4-cloro-l,la,2,7b-tetraidro-ciclopropa[c]cromeno-lcarboxilico racêmico em acetonitrila quente (50 mL) e (IR, 2R)-2-benziloxiciclopentilamina (0,25 g, 1,32 mmol) foi adicionada. A solução resultante foi deixada para cristalização. Após poucas horas o liquido mãe foi decantado e cristais foram lavados com acetonitrila. A segunda cristalização da acetonitrila deu 92 mg do sal diastereomérico puro. O sal foi tratado com 1M HCl e a mistura resultante foi extraída com acetato de etila. A fase orgânica foi lavada com água, solução salina e evaporada para dar 0,05 g de ácido cis-7-flúor-4-cloro-l,la,2,7bde 23/07/2018, pág. 97/150 tetraidro-ciclopropa[c]cromeno-l-carboxílico enantiomérico. Exemplo 30 ±-cis-N-(5-ciano-2-piridinil)-N -(4,7-dicloro-l,la,2,7btetraidrociclopropa[c]cromen-l-il)uréia
a) 1,4-dicloro-2-(2-propinilóxi)benzeno
Cl
Cl
2,5-Diclorofenol (8 g, 49 mmol) foi misturado com carbonato de potássio (13, 6, 98 mmol) e 80 % de solução de brometo de propargil em tolueno (11 mL, 98 mmol) em acetona (100 mL) e agitada por uma noite em temperatura ambiente. O precipitado foi removido por filtração e lavado com acetona. A solução de acetona obtida foi concentrada por evaporação rotatória e mantida a vácuo por 5 h. O produto foi obtido como óleo amarelo com produção guantitativa. Foi utilizado transformações sem purificação adicional.
b) 5,8-dicloro-2H-cromeno
Foi retirado o gás do 1,4-dicloro-2-(2propinilóxi)benzeno e este foi aguecido em agitação com argônio por 4 h a 224°C. A mistura reacional foi então destilada no aparato Kugelrohr (150-175°C/4.lxlCh2 mbar)
Petição 870180063303, de 23/07/2018, pág. 98/150 para dar 3,58 g do produto desejado como um sólido branco. Produção de 36% a partir de diclorofenol.
b) ±-cis-etil-4,7-dicloro-l,la,2,7b-tetraidrociclopropa[c]cromeno-l-carboxílato
5,8-dicloro-2H-cromeno (3,15 g, 16 mmol), (Rh(II)Ac2)2 (30 mg, 0,1 mol%) foi dissolvido em cloreto de metileno seco sem gás (3 mL). Diazoacetato de etila (3 mL, 2 eq) no mesmo solvente foi adicionado por uma seringa na taxa de fluxo de 0,4 mL/h por um período de aproximadamente 5 h sob atmosfera N2. A mistura reacional foi então lavada com
NH4CI (aq), água e solução salina e o solvente removido. O produto (45% cis, 55% trans) foi purificado por cromatografia em sílica (200 g, acetato de etila/n-hexano 1:15) para dar 0,9 g do produto cis puro (racemato). Produção 20%. M+=287.
| 1H-RMN |
(CDCls) : 7, 15 |
(d, 1H |
, J=8,5Hz), |
6,91 |
(d, |
1H, |
|
J=8,8Hz) |
, 4,59 (dd, 1H, Ji=12, |
02, 02 = 7,03), |
4, 48 |
(dd, |
1H, |
|
Ji=12,02 |
, J2=4,10), |
4,07-3,94 |
(m, 3H) , |
2,62 |
(t, |
1H, |
|
J=8,8Hz) |
, 2,27 (t, 1H, |
J=8,36Hz |
), 2,20-2,12 ( |
m, 1H), |
1,1 |
(t, |
|
3H) . |
|
|
|
|
|
|
|
|
d) ácido |
±-cis-4, 7- |
-dicloro-1,la, |
2,7b-tetraidro- |
ciclopropa[c]cromeno-l-carboxílico de 23/07/2018, pág. 99/150
±-cis-Etil-4,7-dicloro - 1,1a,2,7b - tetraidro ciclopropa[c]cromeno-l-carboxílato foi misturado com metanol (3 mL) e solução aquosa de NaOH (1,5 eq., 3 mL) e aquecido em agitação por 1,5 h a 60°C. A extração da mistura reacional básica em hexano mostrou que não há nenhum material de partida presente. A mistura reacional foi acidificada com excesso de 3M de solução de HCl (pH=l) . O precipitado formado foi coletado por sucção e lavado com água. O sólido branco obtido foi seco a vácuo (produção
80%) .
Exemplo 59A
a) 5-cloro-2-fluorfenol
F /k /OH
Cl
5-Cloro-2-fluoranilina (10 g, 68 mmol) foi dissolvida em 6M de ácido sulfúrico e arrefecido em banho de gelo/salina para -5°C. A solução de NaNCh (5,2 g, 76 mmol) em quantidade mínima de água foi adicionada gota a gota e a suspensão agitada em temperatura não maior que -2°C. Após a adição, a solução amarela formada foi deixada em agitação por 30 minutos adicionais em arrefecimento. CuSCh foi dissolvido em água (80 mL) e misturado com ácido sufúrico de 23/07/2018, pág. 100/150 (32 mL) . O sal diazônio foi adicionado gota a gota ao sulfato de cobre pré-aquecido (160°C) e o produto foi removido do frasco de reação por destilação a vapor. A reação levou cerca de 2 h para ser completada. A solução água/phnol foi extraída em éter, lavada com solução salina e seca em Na2SC>4. A concentração deu 4g de fenol bruto (40%) .
b) 4-cloro-l-flúor-2-(2-propinilóxi)benzeno
Cl
4-cloro-l-flúor-2-(2-propinilóxi)benzeno foi sintetizado analogamente ao Exemplo 33a a partir de 4-cloro1-fluorfenol (4 g, 27 mmol) para dar 4,6 g do produto (purificado por cromatografia em coluna de sílica, acetato de etila/n-hexano 1:15) como óleo amarelo. Produção 90%.
c) 5-cloro-8-flúor-2H-cromeno
Cl
F
5-cloro-8-flúor-2H-cromeno foi sintetizado analogamente ao Exemplo 33b) a partir de 4-cloro-l-flúor-2(2-propinil [ oxi) benzeno (4,6 g, 25 mmol) para dar 1 g do produto (purificado por cromatografia em coluna de alumina, acetato de etila/n-hexano 1:15) como óleo incolor. Produção
22% .
d) etil ±-cis-7-cloro-4-flúor-1,la,2,7b-tetraidrode 23/07/2018, pág. 101/150 ciclopropa[c]cromeno-l-carboxílato
Etil ±-cis-7-cloro-4-flúor-1,la,2,7b-tetraidrociclopropa[c]cromeno-l-carboxílato foi sintetizado analogamente ao Exemplo 33c a partir do 5-cloro-8-flúor-2Hcromeno (1 g, 5,4 mmol) para dar 360 mg do produto ±-cis (purificado por cromatografia em coluna de sílica, acetato de etila/n-hexano 1:20) como um sólido branco. Produção 25%.
e)_ácido_±-cis-7-cloro-4-flúor-1, la, 2, 7btetraidro-ciclopropa[c]cromeno-l-carboxílico
Ácido ±-cis-7-cloro-4-flúor-1,la,2,7b-tetraidrociclopropa[c]cromeno-l-carboxílico foi sintetizado analogamente ao Exemplo 33d a partir de etil ±-cis-7-cloro4-flúor-1,la,2,7b-tetraidro - ciclopropa [c] cromeno - 1 carboxílato (360 mg, 1,3 mmol) para dar 259 mg de ácido ±cis (80%) .
Exemplo 31
N-[(1S, laR, 7bR) ou (IR, laS, 7bS)-1,la,2,7b, tetraidrocloropropa[c]-[1]benzotiopiran-l-il]-Eb-(5-ciano-2piridinil)uréia
a) 3,4-diidro-2H-l-benzotiopiran-4-ol de 23/07/2018, pág. 102/150
Uma solução de t iocroman-4-ona (9g) em éter (27 mL) foi adicionada vagarosamente a uma mistura de hidreto de alumínio lítio (0,53 g) em éter (54 mL) . Após o final da adição, a mistura foi deixada em refluxo por 2 horas. A mistura reacional foi arrefecida e gelo foi adicionado, seguido por água e por uma solução de 20% de H2SO4. A fase aquosa foi lavada duas vezes com éter. A fase etérea foi lavada duas vezes com NaOH 2N, e uma vez com água, seco com MgSO4 e evaporado. O óleo claro (8,9 g) foi cristalizado após poucas horas. Rdt=97%.
b) 2H-l-benzotiopurano e 4H-l-benzotiopirano
4-Tiocromanol (8,9g) e sulfato de potássio ácido (0,89g) foram colocados em um frasco e evacuado para 1 mm. O frasco foi colocado em um banho quente a 90°C até o álcool liquefeito. O agitador magnético foi iniciado e o banho vagarosamente chegou a 120°C. A desidratação foi rápida e a mistura do produto e água destilada foi coletada em um recipiente arrefecido com gelo. O produto foi tomado em éter e seco. O produto bruto (7g, Rdt=88%) não foi purificado. A RMN mostrou a presença de 10% de 4H-l-benzotiopirano.
c) Etil éster do ácido 1,la,2,7b-tetraidroPetição 870180063303, de 23/07/2018, pág. 103/150
Diazoacetato de etila foi adicionado vagarosamente a 500 mg de tiocromeno a 140°C. A reação foi seguida por cromatografia gasosa e parada quando todo o material de partida foi consumido (cerca de 7 horas) . O resíduo foi purificado por cromatografia flash (5% éter em hexano). O isômero cis (46,5 mg, Rdt=6%) foi indentifiçado por espectroscopia de RMN.
d)_ácido_1, la, 2, 7b-tetraidro-ciclopropa [c] [ 1 ] benzotiopiran-l-carboxílico, (IS, laR, 7bR) ou (IR, laS,
7bS)
Uma mistura do isômero cis (46,5 mg), LiOH (4ea., 19 mg) em 5 mL de metanol /25% H2O foi deixada em refluxo por 1 hora. Após evaporação do solvente a vácuo, o resíduo foi dissolvido em água e lavado com éter. A fase aquosa foi acidificada com HCI concentrado, e extraída duas vezes com diclorometano. Após secagem, a fase orgânica foi evaporada e deu o ácido desejado (30 mg). Rdt=73%.
Exemplo 32
Ácido (IS, laR, 7bS)-4,7-diflúor-l,la,2,7b-tetraidrociclopropa[c]cromeno-l-carboxílico de 23/07/2018, pág. 104/150
a) (2Z)-3-(3,6-diflúor-2-metoxifenil)-2-propen-l-
Solução de BuLi (2,5 m) em hexano (9,6 mL; 0,024 mol) foi adicionada a uma solução em agitação de 2,55 dif luoranisol (2,88 g, 0,02 mol) em THF seco (30 mL) a 70167C, seguido após 2 h por solução de cloreto de zinco (3,6 g; 0, 026 mol) em THF seco (50 mL) . Permitiu-se que a temperatura reação alcançasse a temperatura ambiente e então a agitação foi mantida em temperatura ambiente por 30 minutos. Pd(OAc)2 (8 mg; 0,2 mol%) foi adicionado, seguido por cis-3-bromoacrilato de etila (3,58 g; 0,02 mol) . A mistura reacional foi colocada em um banho de óleo préaquecido e aquecida em refluxo por lh. A mistura reacional resultante foi resfriada para -78°C e 60 mL (0,06 mol) de
DIBAL (1 M de solução em hexanos) foram adicionadas gota a gota. A agitação foi continuada a -78°C por 2 h e 1 h em temperatura ambiente. A reação foi extinta com água e todos os sólidos foram dissolvidos por adição de HCI. A fase orgânica foi diluída com éter, separada, lavada com 5N de
HCI, solução salina e evaporada a vácuo. 0 resíduo foi destilado em Kugelrohr (l,5xl0_2 mbar, 150 C) para dar 3,7 g (92%) de ( 2Z)-3-(3,6-diflúor-2-metoxifenil)-2-propen-l-ol bruto, que contém ~6% de outros regioisômeros. 0 produto
Petição 870180063303, de 23/07/2018, pág. 105/150 bruto foi utilizado no próximo passo sem purificação adicional.
1H-RMN (CDCls): 7,00 (m, 1H) ; 6,77 (m, 1H) , 6,31 (app. D, 1H) , 6,12 (app. Dt, 1H) ; 4,08 (br. t, 2H) , 3,89 (d, 3H) ;
1, 80 (br, t, 1H) .
b)_diazoacetato_de_(2Z)-3-(3,6-diflúor-2metoxifenil)prop-2-enil
A p-toluenosulfonilhidrazona do cloreto do ácido glioxílico (5,16 g; 0,02 mol) foi adicionado a uma solução de (2Z)-3-(3,6-diflúor-2-metoxifenil)-2-propen-l-ol (3,6 g;
0,018 mol) em CH2CI2 seco (50 mL) a -5°C, e N,Ndimetilanilina (2,5 mL; 0,02 mol) foi adicionada vagarosamente. Após agitação por 30 minutos a -5°C, Et3N (12 mL; 0,09 mol) foi adicionado vagarosamente. A mistura resultante foi agitada por 15 minutos a -5°C e então por 30 minutos em temperatura ambiente, depois do gue água (~50 mL) foi adicionada. A fase orgânica foi separada, lavada com água, solução salina e concentrada a vácuo. Cromatografia de flash (sílica, EA:Hex; 1:15) deu 3,86 g (80%) de produto como um sólido amarelo.
1H-RMN (CDCls): 7,00 (m, 1H) ; 6,76 (m, 1H) ; 6,41 (app. D, J=12,2 Hz; 1H) ; 6,00 (app. Dt, J=12,2; 6,10 Hz; 1H) ; 4,71 (br. S; 1H); 4,67 (dt, 2H); 3,89 (d, 3H).
de 23/07/2018, pág. 106/150
c) (IS, 5R, 6S)-6-(3,6-diflúor-2-metoxifenil)-3oxabiciclo[3.1.0]hexan-2-ona.
Rh2(5-R-MEPY)4^ abs. sem gás diclorometano
Diazoacetato de ( 2Z)-3-(3,6-diflúor-2-metoxifenil) prop-2-enil (3,45 g, 0,013 mol) foi dissolvido em 100 mL de diclorometano sem gás seco e adicionado gota a gota à solução de catalisador Doyle quiral (Aldrich, também disponível de Johnsson Matthey, 10 mg, 0,1 mol%) em 50 mL de diclorometano com atmosfera de argônio em temperatura ambiente por um período de ~6h. A cor azul inicial torna-se verde oliva no final da adição. A mistura reacional foi concentrada a vácuo e o produto bruto foi purificado por cromatografía flash (sílica. EA;Hex, 1:5^1:1) para dar 2,72 g (88%) de (IS, 5R, 6S)-6-(3,6-diflúor-2-metoxifenil)-3oxabiciclo[3.1.0]hexan-2-ona como um sólido incolor. A pureza enantiomérica pode ser verificada neste estágio utilizando uma coluna Chiracel OD, 10% de IPA em hexano -94% ee.
1H-RMN (CDCls): 7,00 (m, 1H) ; 6,72 (m, 1H) ; 4,33 (dd, 1H) ;
4,10 (d, 1H); 4,02 (d, 3H); 2,66 (m, 2H); 2,37 (t, 1H).
d)_(IS, laR, 7bS)-1-(bromometil)-4,7-diflúorla,7b-diidrociclopropa[c]cromeno-2(1H)-ona.
de 23/07/2018, pág. 107/150
% HBr/AcOH
Br (IS, 5R, 6S)-6-(3,6-diflúor-2-metoxifenil)-3oxabiciclo[3.1.0]hexan-2-ona (130 mg, 0,55 mmol) foi misturada com 1,2 mL de HBr/AcOH 30% (6 mmol) e aquecida em um frasco fechado em agitação por cerca de 4 h a 90°C. A mistura reacional foi então arrefecida, misturada com água e extraída em dietil éter (3x20 mL). O extraído éter foi lavado com solução de bicarbonato de sódio saturada e solução salina. Seco sobre sulfato de magnésio. Dando a concentração de 160 mg do material sólido branco. Produção
98%.
4H-RMN (CDCls): 7,08 (m, 1H) ; 6,88 (m, 1H) ; 3,44 (dd, 1H) ;
3,06 (t, 1H); 2,96 (dd, 1H); 2,64 (dd, 1H); 2,46 (m, 1H).
e) ácido (IS, laR, 7bS)-4,7-diflúor-l,la, 2,7btetraidrociclopropa[c]cromeno-l-carboxílico.
NaOH
H2O
O
VOH
Mw=226 (IS, laR, 7bS)-1-(bromometil)-4,7-diflúor-la,7bdiidrociclopropa[c]cromeno-2(1H)-ona (360 mg, ln2 mmol) foi misturado com a solução de NaOH (0,1 g, 2,5 mmol) em 5 mL de
Petição 870180063303, de 23/07/2018, pág. 108/150
100 água e aquecida em agitação por 1 h a 90°C. Após completar a reação, a mistura foi arrefecida e extraída em dietil éter (2x20 mL) . A fase aquosa foi acidificada com HC1 concentrado. O precipitado formado foi coletado por filtração para dar 180 mg do produto puro. O liquido mãe foi extraído em éter e lavado com solução salina, seca sobre sulfato de magnésio. A concentração deu 70 mg adicionais do produto (contendo mais de 15% de impurezas) . Produção total de cerca de 92%.
1H-RMN (CDCls): 6,86 (m, 1H) , 6,54 (m, lh) ; 4,48 (m, 2H) ;
2,62 (t, 1H); 2,20 (t, 1H); 2,11 (m, 1H).
Exemplo 33
a) ácido cis etil éter la, 6b-diidro-lHbenzo[b]ciclopropa[d]tiofeno-l-carboxílico, (IS, laS, 6bR)
CO2Et é adicionado vagarosamente a reação foi verificada por após 7 horas. O resíduo é flash (5% éter em hexano). O
Diazoacetato de etila g de tiofeno a 140°C. A cromatografia gasosa e parada purificado por cromatografia de isômero cis (917 mg, Rdt = 6%) foi identificado por espectroscopia de RMN.
Referência: Badger G. M. e colaboradores, J. Chem. Soc.,
1958, 1179-1184.
Badger G. M. e colaboradores, J. Che, . Soc., 1958, 477725 4779.
Petição 870180063303, de 23/07/2018, pág. 109/150
101
b) ácido cis la, 6b-diidro-lH-benzo[b]ciclopropa
Uma mistura do isômero cis (443 mg), LiOH (193 mg) em 15 mL de metanol / 25% H2O é deixada em refluxo por 1 hora. Após evaporação do solvente a vácuo, o resíduo é dissolvido em água e lavado com éter. A fase aquosa é acidificada com HC1 concentrado, e extraído duas vezes com diclorometano. Após secagem, a fase orgânica é evaporada e dada o ácido desejado (313,6 mg). Rdt=81%.
Exemplo 34 (IS, 5R, 6S)-6-(3,6-diflúor-2-metoxifenil)-2-metóxi-3oxabiciclo[3.1.0]hexano
a) Iodo-3- oxabiciclo[3.1.0]hexan-2-ona
O
O composto titulo é sintetizado na estereoquímica 15 representada como descrito em Doyle J Amer Chem Soc 117 (21)
5763-5775 (1993) .
b) Iodo-2-metóxi-oxabiciclo[3.1.0]hexano
Petição 870180063303, de 23/07/2018, pág. 110/150
102
I
Ο
DIBAL
HO
MeOH, H+
MeO
O composto titulo é sintetizado na estereoquímica representada como descrito em Martin e colaboradores Tett
Lett 39 1521-1524 (1998) .
c) (IS, 5R, 6S)-6-(3,6-diflúor-2-metoxifenil)-2metóxi-3-oxabiciclo[3.1.0]hexano
MeO
2,4-difluoranisol (90 mg, 0,62 mmol) foi dissolvido em THF anidro, sem gás, (7 mL) e arrefecido a 78°C sobre N2. nBuLi, 2,5 M em hexano, (0,30 mL, 0,77 mmol) foi adicionado e a mistura reacional foi agitada a -78°C por 2 horas. ZnCÍ2 (150 mg, 1,1 mmol), como uma solução em THF anidro (7 mL), foi adicionado e a mistura reacional foi deixada para aquecer até a temperatura ambiente por 2 horas. Iodo-2-metóxi-3-oxabiciclohexano (150 mg, 0,63 mmol), Pd(OAc)2 (1,5 mg, 6,2 μηοοί), e Tris(2,4-di-tertbutilfenil) fosfito ligante (40 mg, 62 μηοοί) foram misturados em THF anidro (7 mL) e adicionado à mistura reacional. A mistura reacional foi aquecida em refluxo por 3 dias e extinta com H2O. Éter dietil foi adicionado e as camadas foram separadas, a camada orgânica foi lavada com H2O e NaCl aquoso saturado, seca sobre MgSCh, filtrada e concentrada de 23/07/2018, pág. 111/150
103 para dar o composto título, de outra maneira denotado 2,4di-flúor-5-(ciclopropilacetal)anisol. Cromatografia em coluna de sílica gel (EtOAc/Hexano 1:3) deu (4) 50 mg, 31%. 1H-RMN (CDCls) δ (ppm): 6,88-6,94 (m, 1H, ArH), 6,68-6,73 (m, 1H, ArH), 4,82 (s, 1H, CHOCH3) , 3,97-3,98 (m, 1H, CHOCH)
3,94 (s, 3H, OCH3), 3,79-3,81 (m, 1H, CHOCH) 3,30 (s, 3H,
OCH3) , 2,13-2,19 (m, 2H, 2x CH-ciclopropil), 1,89 (tr,
J=8lHz, 1H, CH ciclopropil).
Exemplo 35 ácido cis-4,7-Diflúor-l,la,2,7b-tetraidro-ciclopropa[c]
Solução de BBr3 1M em CH2CI2 (5,8 mL; 5,8 mmol 2,1 eq) foi adicionada a lactona de partida, (IS, 5R, 6S)-6(3,6-diflúor-2-metoxifenil)-3-oxabiciclo[3.1.0]hexan-2 - ona do exemplo 42c) (0,66 g; 2,75 mmol) a 0°C. A mistura reacional foi agitada a 0°C por lh. Acetonitrila (5,8 mL) foi adicionada e a agitação foi continuada por 3 h a 0°C. A mistura reacional foi extinta por adição de água e a fase orgânica foi separada. A fase aquosa foi extraída com CH2CI2 e as fases orgânicas combinadas foram evaporadas. NaOH (0,33 g; 8,25 mmol; 3 eq) em água (~5 mL) foi adicionado ao resíduo resultante e agitado a 80°C por 45 minutos. A mistura reacional foi extraída com éter para remover nenhuma das impurezas acídicas. O éter residual na fase aquosa foi evaporado a vácuo e HCI concentrado foi adicionado para o pH de 23/07/2018, pág. 112/150
104 de ~3. Após ~1h o sólido foi filtrado, produzindo 0,497 g (80%) do ácido final bruto como sólido acastanhado. O ácido bruto foi dissolvido em 6 mL de EWH/H2O (40/60 v/v) e tratado com carbono ativado. A solução quente foi filtrada e deixada para cristalização. Produção 0,4 g (64%).
1H-RMN (CDCl3): 10,32 (br s, ~1H), 7,68 (d, 2H), 7,37 (s,
|
1H), |
7,32 |
(d, |
2H), 6,96 |
(s, 1H), 6,87 |
(m, 1H), |
6,62 (dt, |
|
1H), |
4,44 |
(dd, |
1H), 4,33 |
(dd, 1H), 3,53 |
(m, 1H), |
2,56 (m, |
|
~1H), |
1,96 |
(m, |
1H). LC-MS |
:M+ 434. |
|
|
|
|
|
|
|
Exemplo 36 |
|
|
|
|
|
a) |
éster |
etil do |
ácido |
1,1a,66a- |
tetraidrociclopropa[a]indeno-1-carboxílico.
Indeno é diluído em 100 mL de dicloroetano. Cerca de 10 mg de CuI e cerca de 10 mg de Od(OAc)2 é adicionado. Da mistura resultante, 25 mL é gota a gota adicionada a 25 mL de etildiazoacetato e é deixada em refluxo por 30 minutos. A solução é filtrada através de Al2O3 que é eluído com um gradiente de EtOAc/hexano. O eluato é evaporado vigorosamente a 100°C, 2mmHg para produzir o composto título (36 g).
b) 1,1a,66a-tetraidrociclopropa[a]indeno-1-amino.
O produto do passo a) é posto em ebulição com cerca de 50g de NaOH em 200 mL 10:1MeOH:H2O por 2 horas. A mistura é diluída com água, lavada com dicloroetano, evaporada com HOAc, extraída com dicloroetano, lavada com água, seca com sulfato, filtrada e evaporada para produzir 25 g de ácido, 95% puro. DPPA 275,2 δ=1,128 10 mL, 46.5 mmol TEA 7,1 mL 1,1 ee e 7,3 g de ácido (massa 174,12, 0,9 ee) é de 23/07/2018, pág. 113/150
105 misturado em 200 mL de tolueno e deixado em refluxo por cerca de 2 horas. O produto é evaporado e dissolvido em dioxano 200 mL, 25 mL de HCl (aq) e 25 mL de água são adicionados e a mistura agitada por 60 minutos em temperatura ambiente. A solução é particionada com ácido/base em água/diclorometano. A fase orgânica é seca, filtrada e evaporada. O produto é cromatografado através de uma coluna de sílica 60 para produzir 660 mg de cis amina 85% pura, PM 145,11.
Exemplo 37 ±cis-l-(5-Ciano-piridin-2-il)-3-(1,1a,6,6a-tetraidrociclopropa[a]indeno-l-il)-uréia.
a) éster etil do ácido ±cis-l,la,6,6a-tetraidrociclopropa[a]indeno-1-carboxílico.
Para uma mistura de indeno (11,6 g, 100 mmol) e Cu2Br2 (0,10 g, 0,35 mmol) em 1,2-dicloroetano (200 mL) a 80°C, foi adicionado gota a gota (3h) uma solução de diazoacetato de etila (17,1 g, 150 mmol) em 1,2-dicloroetano (35 mL) . Após 15 minutos a 80°C, a mistura reacional foi lavada com H2O (200 mL). A fase aquosa foi lavada com CH2CI2 (50 mL) e o solvente das fases orgânicas combinadas foi removido sob pressão reduzida. O produto bruto foi cromatografado em coluna (sílica gel, 5^10% EtOAc em Hexano), para dar 3,63 g (18%) de éster etil do ácido ±cis1,la,6,6a-tetraidro-ciclopropa[a]indeno-l-carboxílico e 6,68 de 23/07/2018, pág. 114/150
106 g (33%) de éster etil do ácido ±trans-l,la,6,6a-tetraidrociclopropa[a]indeno-l-carboxílico como um subproduto.
1H-RMN (CDCls): 7,30-7, 05 (m, 4H) , 3,81 (q, 2H) , 3,36 (d,
1H) , 3,18 (dd, 1H) , 2,92 (m, 1H) , 2,24 (m, 1H) , 1,99 (dd,
1H), 0,92 (t, 3H) .
b) ácido ±cis-l,la,6,6a-Tetraidro-ciclopropa[a] indeno-l-carboxílico
Crí
Ácido ±cis-l,la,6,6a-Tetraidro-ciclopropa[a]indeno -1-carboxílico foi sintetizado a partir do éster etil do ácido ±cis-l,la,6,6a-tetraidro-ciclopropa[a]indeno-lcarboxílico (3,53 g, 15,5 mmol), LiOH (539 mg, 22,5 mmol), H2O (10 mL) e MOH (20 mL) que foram aquecidos em refluxo por 2h, concentrados e acidificados para precipitar 1,62 g (62%) do ácido ±cis-l,la,6,6a-Tetraidro-ciclopropa[a]indeno-1-
|
carboxílico |
como um |
sólido branco. 0 |
produto |
nao |
foi |
|
cristalizado. |
|
|
|
|
|
|
| 1H-RMN (CDCI3 |
): 10,95 |
(br s, 1H) , 7,35-7, |
02 |
(m, |
4H) , |
3,29 |
|
(d, 1), 3,14 |
(dd, 1H), |
2,96 (m, 1H) , 2,27 ι |
(m, |
1H) , |
1,91 |
(dd, |
1H) .
A mistura reacional foi concentrada sob pressão reduzida, benzeno (20 mL) foi adicionado e a mistura reacional foi lavada com HCl IN (30 mL) , H2O (30 mL) e solução salina (30 mL) . O solvente das fases orgânicas foi removido sob pressão reduzida. O produto bruto foi cromatograf ado em coluna (sílica gel, 4^5% MeOH em CH2CI2) , de 23/07/2018, pág. 115/150
107 para dar 25 mg (5%) de ±cis-l-(5-Ciano-piridin-2-il)-3(1, la, 6,6a-tetraidro-ciclopropa[a]indeno-l-il)-uréia.
1H-RMN (DMSO-de): 9,58 (s, 1H), 8,18 (d, 1H), 7,96 (dd, 1H) ,
7, 40-7,25 (m, 3H) , 7,17-7,05 (m, 3H) , 3,27-3,13 (m, 2H) ,
2,80-2,73 (m, 2H), 2,05 (dd, 1H).
Exemplo 38 ±cis-l-(5-Ciano-piridin-2-il)-3-(la,2,3,7b-tetraidrociclopropa [a]naftalen-l-il)-uréia. a) éster etil do ácido la,2,3,7b-tetraidro-lHciclopropa[a]naftaleno-1-carbóxilico
Éster etil do ácido la,2,3,7b-tetraidro-lHciclopropa[a]naftaleno-l-carboxílico foi sintetizado analogamente ao Exemplo 37 a partir de 1,2-diidronaftaleno (3,91 g, 30 mmol), para dar 688 mg (11%) de éster etil do ácido la,2,3,7b-tetraidro-lH-ciclopropa[a]naftaleno-lcarboxílico (uma mistura de isômeros cis e trans 56/39) .
1H-RMN (CDCls) : 7,35-6,95 (m, 4H) , 4,30-3,85 (m, 2H) , 2,901,00 (m, 10H).
b)_ácido_la, 2,3, 7b-Tetraidro-lH-ciclopropa [a]
OH
Acido la,2,3,7b-Tetraidro-lH-ciclopropa[a] naftaleno-l-carboxílico foi sintetizado analogamente ap de 23/07/2018, pág. 116/150
108
Exemplo 37b a partir de éster etil do ácido la,2,3,7btetraidro-lH-ciclopropa[a]naftaleno-l-carboxílico (688 mg,
3,18 mmol, uma mistura de isômeros cis e trans 56/39), para dar 540 mg (90%) do ácido la,2,3,7b-tetraidro-lHciclopropa[a]naftaleno-l-carboxílico (uma mistura de
|
isômeros |
cis e trans |
56/39) . 0 produto nao |
foi |
cristalizado. |
| 1H-RMN ( |
CDCls) : 11,36 |
(br s, 1H) , 7,30-6, |
95 1 |
>, 4H), 2,80- |
|
1,65 (m, |
7H) . |
|
|
|
|
|
|
Exemplo 68 |
|
|
|
|
a) Éster |
etil ácido la, |
2,3, |
4,8b-Hexaidro- |
benzo[a]ciclopropa[c]ciclohepteno-1-carbóxilico.
Éster etil ácido la,2,3,4,8b-Hexaidrobenzo[a]ciclopropa[c]ciclohepteno-l-carboxílico foi sintetizado analogamente ao Exemplo 37a a partir de 6,7diidro-5H-benzocicloeptano (4,40g, 30,5 mmol), para dar 3,43 g (49%) de éster etil ácido la,2,3,4,8b-Hexaidrobenzo[a]ciclopropa[c]ciclohepteno-l-carboxílico (uma mistura de isômeros cis e trans 1/10) .
1H-RMN (CDCls) : 7, 40-6, 90 (m, 4H) , 4,30-4, 00 (m, 2H), 3,300,50 (m, 12H).
b) ácido la,2,3,4,8b-Hexaidro-benzo[a]ciclopropa [ c]ciclohepteno-l-carboxílico.
de 23/07/2018, pág. 117/150
109
Ácido la, 2,3,4,8b-Hexaidro-benzo[a]ciclopropa[c] ciclohepteno-l-carboxílico foi sintetizado analogamente ao Exemplo 37 a partir do éster etil ácido la,2,3,4,8bHexaidro-benzo[a]ciclopropa[c]ciclohepteno - 1 - carboxílico (3,43 g, 14,9 mmol, uma mistura de isômeros cis e trans 1/10), para dar 2,81 g (93%) do ácido la,2,3,4,8b-Hexaidrobenzo[a]ciclopropa[c]ciclohepteno-l-carboxílico (uma mistura de isômeros cis e trans 1/10) . O produto não foi cristalizado.
íH-RMN (CDCls) : 10,76 (br s, 1H) , 7, 40-7, 00 (m, 4H) , 3,300,50 (m, 9H).
Exemplo 40
a) 6-metóxi-l,2,3,4-tetraidronaftalen-l-ol
6-Metoxitetralona (10 g, 0,057 mol) foi misturada com 150 mL de etanol seco e boroidreto de sódio (1,2 eq) foi adicionado por porções à mistura agitada. A mistura reacional foi deixada em agitação em temperatura ambiente por 15 h. A mistura reacional foi então concentrada por evaporação rotatória, misturada com 100 mL de água e aquecida por lh a 45°C. A mistura resultante foi extraída em éter dietil (3x80 mL) . O combinado orgânico extraído foi de 23/07/2018, pág. 118/150
110 seco sobre Na2SO4 e concentrado por evaporação rotatória para dar 10,39 g de óleo amarelo no próximo passo sem purificação adicional.
b) 7-metóxi-l,2-diidronaftaleno
6-metóxi-l,2,3,4-tetraidronaftalen-l-ol bruto (10,3 g, 0,058 mmol) foi dissolvido em 100 mL de tolueno e aquecido em um banho de óleo (115 °C) . Ácido ptolilsulfônico (20 mg) foi adicionado à mistura reacional e
|
esta foi |
mantida em |
refluxo por cerca |
de lh. |
a reação |
|
monitorada |
por GC. A |
mistura reacional foi |
então |
arrefecida |
|
e lavada |
com solução |
de NaHCCb saturada, |
água |
e solução |
|
salina e |
a camada |
orgânica foi seca |
sobre |
NaSO4. A |
|
concentração dada foi |
8,87 g de óleo marrom claro |
. Produção |
|
96%. |
|
|
|
|
c) Etil-5-metóxi-la,2,3,7b,tetraidro-lH-ciclopropa [a]naftaleno-1-carboxilato
7-Metóxi-l,2-diidronaftaleno (8,8g, 0,055 mol) foi misturado com 10 mL de cloreto de metileno absoluto sem gás e 20 mg de acetato de ródio (apr. 0,1 mol%) . A mistura foi borbulhada com nitrogênio e diazoacetato de etila (2 eq, 50% de solução em cloreto de metileno absoluto sem gás) foi adicionado vagarosamente através de seringa (taca de fluxo de 23/07/2018, pág. 119/150
111 de cerca de 1 mL/hora) para a solução agitada em temperatura ambiente. A evolução do gás começou na adição. A reação foi monitorada por GC. Quantidade adicional de catalisador foi adicionada durante a reação (cerca de 20 mg) . Taxa-GC de isômeros cis/trans foi 21:48.
Após a reação estar completa, de acordo com os dados GC, a mistura reacional foi lavada com solução de NH4C1 saturada e solução salina. A solução de cloreto de metileno foi seca sobre Na2So4. A concentração dada foi de 13g de produto bruto como óleo amarelo. Purificado por cromatografia em coluna de silica (200g, acetato de etila/hexano 1:20). Somente o isômero trans foi obtido na forma pura. A forma cis requerida não pode ser purificada pela técnica utilizada. Frações que foram mais enriquecidas com o produto requerido foram combinadas (200 mg, proporção cis/trans 70:30 de acordo com GC) e utilizadas para transformações adicionais.
d)_ácido_5-metóxi-la, 2,3, 7b,tetraidro-lHciclopropa[a]naftaleno-1-carboxílico
Etil-5-metóxi-la,2,3,7b,tetraidro-lH-ciclopropa[a] naftaleno-l-carboxilato (0,2 g, 0,8 mmol) foi dissolvido em 2 mL de metanol e a solução de hidróxido de sódio (0,2 g, 50 mmol) em 2 mL de água foi adicionada à mistura reacional e agitada em temperatura ambiente por uma noite. A extração da de 23/07/2018, pág. 120/150
112 mistura reacional básica em hexano mostrou que não há nenhum material de partida presente. A mistura reacional foi acidificada com excesso de solução de HC1 3M (pH=l), e extraído em acetato de etila (3x15 mL) . Os extratos combinados foram lavados com água e solução salina, secos sobre Na2SO4 e concentrados por evaporação rotatória para dar 0,15 g de mistura de ácidos cis/trans como sólido branco.
Exemplo 41
7-metóxi-3,4-diidro-l(2H)-naftalenol foi sintetizado analogamente ao Exemplo 69a a partir de 7metóxi-1,2,3,4-tetraidro-l-naftalenona (5g, 28 mmol), para dar cerca de 5 g do produto bruto (produção quantitativa), que foi utilizado no próximo passo sem purificação adicional.
b) 6-metóxi-l,2-diidronaftaleno .0
6-Metóxi-l,2-diidronaftaleno foi sintetizado analogamente ao Exemplo 40b a partir do 7-metóxi-l,2,3,4tetraidro-l-naf talenol para dar 4,4 g do produto como um óleo marrom amarelado (96% produção a partir de 7-metóxi1,2,3,4-tetraidro-l-naftalenona).
c) Etil 6-metóxi-la,2,3,7b-tetraidro-lH-ciclopropa de 23/07/2018, pág. 121/150
113 [a]naftaleno-l-carboxilato
COOEt
Etil 6-metóxi-la,2,3,7b-tetraidro-lH-ciclopropa[a] naftaleno-l-carboxilato foi sintetizado analogamente ao Exemplo 38 a partir de 6-metóxi-l,2-diidronaftaleno (4,4 g, 0,28 mmol) em uma taca de adição de 0,7 mL/h para dar 9,68g do produto bruto como um óleo laranja-marrom. Purificado por cromatografia em coluna de sílica gel (200 g, acetato de etila/ hexano 1:10). Três frações foram coletadas: fração enriquecida com isômero cis (75% por GC) - 0,16 g, fração misturada - 1,76 g, e fração contendo isômero trans puro lg. Produção total 45%.
d)_Ácido_6-metóxi-la, 2,3, 7b-tetraidro-lHciclopropa[a]naftaleno-1-carboxílico
COOH
Ácido 6-metóxi-la,2,3,7b-tetraidro-lH-ciclopropa [a]naftaleno-l-carboxilico foi sintetizado analogamente ao Exemplo 69b) a partir do etil 6-metóxi-la,2,3,7b-tetraidrolH-ciclopropa[a]naftaleno-l-carboxilato (0,16 g, 0,65 mmol) para dar 0,1 g do produto como cristais brancos. Produção
71% .
Exemplo 42
a) 7,8-diidro-2-naftalenol de 23/07/2018, pág. 122/150
114
7-MetóxiOl,2-diidronaftaleno (6,4 g, 40 mmol) foi dissolvido em DMF absoluto e borbulhado com argônio, etiltiolato de sódio (2,5 eq) foi adicionado e a mistura reacional foi aquecida em agitação a 160°C por cerca de 4h. A reação foi monitora por GC. A mistura reacional foi diluída com água, acidificada com HCl 3M e extraída em etilacetato. Extrato orgânico foi lavado com água e solução salina, seca sobre Na2SC>4 e concentrada por evaporação rotatória. Purificação por cromatografia em coluna de sílica (200 g, acetato de etila/hexano) deu 5,36 g do fenol desejado. Produção 92%.
b)_trif luormetanosulf onato de_7, 8-diidro-2naftalenil
O
7,8-Diidro-2-naftalenol (5,3 g, 37 mmol) foi misturado com trietilamina (6,2 mL, 44 mmol) em cloreto de metileno absoluto e arrefecido sob nitrogênio no banho de gelo/solução salina. Anidreto tríflico (7,4 mL, 44 mmol) foi adicionado à solução em agitação através de seringa durante 10 min. Permitiu-se que a temperatura aumentasse vagarosamente até a temperatura ambiente. A mistura reacional foi então lavada com água e solução salina e seca de 23/07/2018, pág. 123/150
115 sobre Na2SC>4. 0 produto bruto foi purificado por cromatografia em coluna de sílica. Obteve-se 9 g de liquido marrom. Produção 88%.
c) Etil 5-{ [(trifluormetil)sufonil]óxi}-la,2,3, 7btetraidro-lH-ciclopropa[a]naftaleno-l-carboxilato
O
O
Etil 5-{ [ (trifluormetil)sufonil]óxi}-la,2,3,7btetraidro-lH-ciclopropa[a]naftaleno-l-carboxilato foi sintetizado analogamente ao exemplo 40 a partir de trifluormetanosulfonato de 7,8-diidro-2-naftalenil (9g, 32 mmol) em uma taxa de adição de 1 mL/h para dar 13 g de produto bruto como um óleo laranja-marrom. Purificado por cromatografia em coluna de sílica (200 g, acetato de etila / hexano 1:15) . Fração enriquecida com isômero cis (80% por GC) - 0,64g foi coletada e utilizada para transformações adicionais.
d) Etil 5-ciano-la,2,3,7b-tetraidro-lH-ciclopropa [a]naftaleno-l-carboxilato
Etil 5-{ [ (trifluormetil)sufonil]óxi}-la,2,3, 7btetraidro-lH-ciclopropa[a]naftaleno-l-carboxilato (0,2g, 0,5
Petição 870180063303, de 23/07/2018, pág. 124/150
116 mmol) foi misturado com Zn (CN)2 (0,82 mmol) e Pd(Ph3P)4 (56 mg, 10 mol%) em DMF (4 mL), borbulhao com argônio por 5 minutos e aquecido em agitação em um frasco fechado por 14 h a 100°C. A reação foi monitorada por GC. A mistura reacional foi concentrada por evaporação rotatória, misturada com
NH4CI saturado e extraído em acetato de etila (3x15 mL) . Extrato orgânico foi lavado com água e solução salina, seco sobre Na2SC>4. Concentração deu 0,12g de produto como um óleo (produção 90%).
d) ácido 5-ciano-la,2,3,7b-tetraidro-lH-ciclopropa [a]naftaleno-l-carboxílico
Ácido 5-ciano-la,2,3,7b-tetraidro-lH-ciclopropa[a] naftaleno-l-carboxílico foi sintetizado analogamente ao Exemplo 69d a partir de etil 5-ciano-la,2,3,7b-tetraidro-lH15 ciclopropa[a]naftaleno-l-carboxilato (0,12 g, 0,5 mmol) para
|
dar 0,lg do produto |
como |
cristais brancos. Produção |
94% . |
| 1H-RMN (DMSO-de) : 9, |
70 ( |
br s, 1H) , |
8,32 |
(br s, |
1H) , 8,0. |
|
(dd, 1H), 7,46-7,63 |
(m, |
4H), 7,32 i |
(br s, |
1H), 3,18 |
-3,10 (m |
|
2H) , 2, 76-2,65 (m, |
1H) , |
2,62-2,51 |
(m, |
1H) , 2,34 |
(t, 1H) |
2,01-1,80 (br m, 2H), 1,78-1,69 (br m, 1H).
Exemplo 42A
a)_Etil_5- [ (trimet ilsilil) etinil]-la,2,3, 7 btetraidro-lH-ciclopropa[a]naftaleno-l-carboxilato
Petição 870180063303, de 23/07/2018, pág. 125/150
117
Etil 5-{ [ (trifluormetil)sulfonil]óxi}-la,2,3, 7 btetraidro-lH-ciclopropa[a]naftaleno-l-carboxilato (0,2 g, 0,5 mmol) foi misturado com trimetilsililacetielno (0,2 mL,
1,37 mmol), DPP (35 mg, 10 mol%), Pd(dba)2 (30 mg, 10 mol%) 5 e Cul (3 mg) em Et3N (2,5 mL) , borbulhado com argônio por 5 minutos e aquecido em agitação em um frasco fechado por 14 h a 95°C. A reação foi monitorada por GC. A mistura reacional foi concentrada por evaporação rotatória, misturada com
NH4CI saturado e extraída em acetato de etila (3x15 mL) . 10 Extrato orgânico foi lavado com água e solução salina, seco sob Na2SC>4. A concentração dada foi 0,15 g de produto como um óleo (produção 87%).
b) ácido_5-etinil-la, 2,3, 7b-tetraidro-lHciclopropa[a]naftaleno-1-carbóxilico
Etil 5-[(trimetilsilil)etinil]-la,2,3,7btetraidro-lH-ciclopropa[a]naftaleno-l-carboxilato (0,2 g, 0,64 mmol) foi adicionado em 4 mL de metanol e a solução de hidróxido de sódio (0,05 g, lm2 mmol) em 2 mL de água foi adicionada à mistura reacional e agitada em aquecimento a
Petição 870180063303, de 23/07/2018, pág. 126/150
118
65°C por 6 horas. A extração da mistura reacional básica em hexano mostrou que nenhum material de partida estava presente. A mistura reacional foi acidificada com excesso de solução de HCI 3M (pH=l), e extraída em acetato de etila (3x15 mL) . Os extratos combinados foram lavados com água e solução salina, secos sobre Na2SO4, e concentrado por evaporação rotatória para dar 0,12 g da mistura dos ácidos cis/trans (85:15) como sólido branco. Produção 88%.
Exemplo 43
a)_5, 8-diflúor-4-metil-3,4-diidro-l (2H) naftalenona
1,4-Difluorbenzeno (22 mL, 210 mmol) foi misturada com γ-valerolactona (4 mL, 42 mmol) e AICI3 (28 g, 210 mmol) foi adicionado por porções à mistura reacional agitada. A mistura reacional foi então deixada em refluxo e agitação por 16 h (banho de óleo 110°C) . A mistura reacional foi arrefecida (banho de gelo/solução salina) e gelo/HCl concentrado foi adicionado e agitado até uma mistura homogênea ser obtida. A mistura reacional foi então extraída em cloreto de metileno, lavada com água (4xl0mL) e solução de bicarbonato de sódio (3x100 mL) . O extrato orgânico foi seco sobre Na2SO4. A concentração por evaporação rotatória deu 6,7 g de produto como um pó amarelo. Produção 81%.
Petição 870180063303, de 23/07/2018, pág. 127/150
119
b)
5,8-diflúor-4-metil-1,2,3,4-tetraidro-lnaftalenol
F OH
5,8-diflúor-4-metil-1,2,3,4-tetraidro-l-naftalenol foi sintetizado analogamente ao Exemplo 69a a partir de 5,85 diflúor-4-metil-3,4-diidro-l(2H)-naftalenona para dar 1,8 g de produto bruto, que foi utilizado no próximo passo sem purificação adicional.
c) 5,8-diflúor-l-metil-1,2-diidronaftaleno
5,8-diflúor-l-metil-1,2-diidronaftaleno foi sintetizado analogamente ao Exemplo 40b a partir de 5,8diflúor-4-metil-1,2,3,4-tetraidro-l-naftalenol (1,8 g, 9,1 mmol) para dar 1,5 g do produto como um óleo marrom amarelo (90% produção a partir de 5,8-diflúor-4-metil-3,4-diidro1(2H)-naftalenona).
d) Etil 4,7-diflúor-3-metil-la,2,3,7b-tetraidrolH-ciclopropa[a]naftaleno-1-carboxilato.
Petição 870180063303, de 23/07/2018, pág. 128/150
120
COOEt
Etil 4,7-diflúor-3-metil-la,2,3,7b-tetraidro-lHciclopropa[a]naftaleno-l-carboxilato foi sintetizado analogamente ao Exemplo 40c a partir de 5,8-diflúor-l-metil1,2-diidronaftaleno (3,5 g, 19 mmol) em uma taxa de adição de 0,5 mL/h para dar o produto como óleo amarelo-marrom. Purificado por cromatografia em coluna de sílica (220 g, acetato de etila/hexano 1:15) para dar 5,2 g da mistura dos ésteres diastereoméricos junto com dímeros de EDA como óleo incolor (proporção GC: anti- 45%; 40% /trans:cis/, sin- 11%; 2,3%/ trans:cis).
e) ácido +/-anti-cis-4,7-diflúor-3-metil-la,2,3, 7b-tetraidro-lH-ciclopropa[a]naftaleno-1-carbóxilico.
Etil 4,7-diflúor-la,2,3,7b-tetraidro-lH-ciclopropa [a]naftaleno-l-carboxilato (5,25 g, 20 mmol, -50:50 mistura de isômeros cis e trans) foi dissolvido em 2,5 mL de metanol e a solução de hidróxido de sódio (0,4 g, 10 mmol) em 2,5 mL de água foi adicionado à mistura reacional e agitada em temperatura ambiente por uma noite. A mistura reacional foi extraída em hexano (3x30 mL) . Os extratos combinados foram de 23/07/2018, pág. 129/150
121 lavados com água e solução salina, secos sobre Na2SC>4 e concentrados por evaporação rotatória para dar 1,12 g de cis ésteres como óleo incolor (mistura de ésteres de etil e metil - 94% de acordo com GC). A mistura obtida foi dissolvida em 1,5 mL de metanol e a solução de hidróxido de sódio (0,2 g, 5 mmol) em 1,5 mL de água foi adicionado à mistura reacional e agitada a 95°C por 40 minutos. A mistura reacional foi acidificada com excesso de solução de HCl 3M (pH=l), e extraída em acetato de etila (3x15 mL). Os extratos combinados foram lavados com água e solução salina, secos sobre Na2SO4 e concentrados por evaporação rotatória para dar 0,93 g anti-ácido +/-cis como cristais ligeiramente laranjas.
Exemplo 44
a) 4,7-diflúor-3-metil-l-indanona
4,7-diflúor-3-metil-l-indanona foi sintetizada analogamente ao Exemplo 43a a partir de butirolactona (4 mL, 52 mmol) para dar 7,19 g de pó amarelo (85:15 mistura da correspondente indanona e tertralona de acordo com GC). O produto foi purificado por cromatografia em coluna de sílica (200 g, acetato de etila / hexano) para dar 3,7 g (produção 40%) do produto puro junto com fração misturada e fração contendo tetralona pura.
b) 4,7-diflúor-3-metil-l-indanol de 23/07/2018, pág. 130/150
122
4,7-diflúor-3-metil-l-indanol foi sintetizado analogamente ao Exemplo 40 a partir da 4,7-diflúor-3-metil1-indanona (3,7 g, 20 mmol), para dar cerca de 3,75 g de produto bruto (produção quantitativa) , que foi utilizada no próximo passo sem purificação adicional.
c) 4,7-Diflúor-l-metil-lH-indeno
4,7-Diflúor-l-metil-lH-indeno foi sintetizado analogamente ao Exemplo 37 a partir de 4,7-diflúor-3-metill-indanol (3,75 g, 9,1 mmol) para dar 2,36 g do produto como um líquido bege (produção 70%).
d)_Etil_2,5-dif lúor-6-metil-1, la, 6,6atetraidrociclopropa[a]indeno-l-carboxilato
Etil 2,5-diflúor-6-metil-1,la,6,6atetraidrociclopropa[a]indeno-l-carboxilato foi sintetizado 15 analogamente ao Exemplo 40c a partir de 4,7-diflúor-l-metilPetição 870180063303, de 23/07/2018, pág. 131/150
123 lH-indeno (1,32 g, 7,9 mmol) em uma taxa de adição 0,4 mL/h para dar o produto bruto como óleo amarelo-marrom. Purificado por cromatografia em coluna de sílica (100 g, acetato de etila/hexano 1:15) para dar 0,61 g da mistura de ésteres diastereoméricos cis- e trans-ésteres como óleo incolor (proporção cis/trans: 84:16 de acordo com RMN) . Produção 30%.
e) ácido +/-anti-cis-2,5-diflúor-6-metil-1,la,6,6a
-tetraidrociclopropa[a]indeno-l-carboxílico
ácido + /-anti-cis-2,5-diflúor-6-metil-1, la, 6,6atetraidrociclopropa[a]indeno-l-carboxílico foi sintetizado analogamente ao acima a partir de etil 2,5-diflúor-6-metil1,la,6,6a-tetraidrociclopropa[a]indeno-1-carbóxilato (0,61 g, 2,4 mmol) passo a passo, primeiro com hidrólise 20 mol. % de NaOH e então com o excesso de NaOH em aquecimento para dar 380 mg do produto como cristais brancos. Produção 70% (apr.quantitativa se calculado para isômero cis de partida).
Exemplo 45
a) 5,8-diflúor-3,4-diidro-l(2H)-naftalenona
Petição 870180063303, de 23/07/2018, pág. 132/150
124
5,8-Diflúor-3,4-diidro-l(2H)-naftalenona foi sintetizada junto com 4,7-diflúor-3-metil-l-indanona de acordo como procedimento descrito no Exemplo 44a. Separado por cromatografia em coluna de sílica, 0,77 g do produto puro foi obtido. Produção 8%.
b) 5,8-diflúor-l,2,3,4-tetraidro-l-naftalenol
5, 8-diflúor-l,2,3,4-tetraidro-l-naftalenol foi sintetizado analogamente ao Exemplo 40a a partir da 5,8diflúor-3,4-diidro-l(2H)-naftalenona (0,77 g, 4,2 mmol), para dar o produto bruto (produção quantitativa), que foi utilizado no próximo passo sem purificação adicional.
c) 5,8-diflúor-l,2-diidronaftaleno
5,8-diflúor-l,2-diidronaftaleno foi sintetizado analogamente ao Exemplo 40b a partir de 5,8-diflúor-l,2,3,415 tetraidro-l-naftalenol para dar 0,67 g do produto bruto como
Petição 870180063303, de 23/07/2018, pág. 133/150
125 um líquido acastanhado (produção de 90% a partir de 5,8diflúor-3,4-diidro-l(2H)-naftalenona).
Quantidade adicional de produto foi também obtida a partir da mistura de 5,8-diflúor-3,4-diidro-l(2H)naftalenona e 4,7-diflúor-3-metil-l-indanona por redução seguida de desidratação. A mistura do indeno e naftaleno correspondente é fácil de separar por cromatografia em coluna de sílice (acetato de etila/hexano 1:20).
d)_Etil_4, 7-diflúor-la,2,3, 7b,tetraidro-lH, ciclopropa[a]naftaleno-l-carboxilato
Etil 4,7-diflúor-la,2,3,7b,tetraidro-lH, ciclopropa[a]naftaleno-l-carboxilato foi sintetizado analogamente ao Exemplo 40c a partir do 5,8-diflúor-1,2diidronaftaleno (0,7 g, 4,2 mmol) em uma taxa de adição de 0,4 mL/h para dar o produto bruto como um óleo amarelomarrom. Purificado por cromatografia em coluna de sílica (100 g, acetato de etila/hexano 1:15) para dar 0,45 g da mistura de cis- e trans-ésteres como um óleo incolor (proporção cis/trans: 33:67 de acordo com GC) . ácido 4,7diflúor-la,2,3,7b-tetraidro-lH-ciclopropa[a]naftaleno-lcarboxílico
e)_ácido_4, 7-diflúor-la, 2,3, 7b-tetraidro-lHciclopropa[a]naftaleno-1-carboxílico de 23/07/2018, pág. 134/150
126
Acido 4,7-diflúor-la,2,3,7b-tetraidro-lHciclopropa[a]naftaleno-l-carboxílico foi sintetizado analogamente ao Exemplo 43e a partir de etil 4,7-diflúorla,2,3,7b,tetraidro-lH, ciclopropa[a]naftaleno-l-carboxilato (0,45 g, 1,8 mmol) por um primeiro passo de hidrólise com excesso de NaOH em temperatura ambiente e então com excesso de NaOH em aquecimento (60°C, 1,5 h) para dar 80 mg do produto como cristais brancos (proporção cis~trans 78:22 de acordo com HPLC).
Exemplo 46
a) 6-Bromoindeno
Este composto foi preparado analogamente aos Exemplos 40a e 40b a partir de 5-bromo-l-indanona (4,0 g, 18,8 mmol) para dar 2,4 g (65%) de 6-bromoindeno.
b)_(±) -cis-Etil_4-bromo-l, la, 6,6atetraidrociclopropa[a]indeno-l-carboxilato
Petição 870180063303, de 23/07/2018, pág. 135/150
127
Este composto foi preparado analogamente ap Exemplo 50c a partir de 6-bromoindeno (1,95 g, 10 mmol). Purificação em sílica gel começando com hexanos seguido por hecanos com 2% de dietil éter e finalmente hexanos com 5% de dietil éter proporcionando 670 mg (24%) de cis-éter.
c)_ácido_(±) -cis-4-bromo-l, la, 6,6atetraidrociclopropa[a]indeno-l-carboxílico
Este ácido foi sintetizado analogamente ao Exemplo 40d começando com 330 mg (1,77 mmol) do composto a partir do Exemplo 75b para dar 232 mg (79%) de ácido (±)-cis-4-bromo1,la,6,6a-tetraidrociclopropa[a]indeno-l-carboxílico.
Exemplo 47
a)_(±) -cis-Etil_4-ciano-l, la, 6,6atetraidrociclopropa[a]indeno-l-carboxilato
Este composto foi preparado analogamente ao Exemplo 42d a partir de (±)-cis-etil 4-bromo-l,la, 6,6atetraidrociclopropa[a]indeno-l-carboxilato (200 mg, 0,7 mmol) para dar, após purificação em dílica gel utilizando hexanos com 10% de acetato de etila como o eluente, 73 mg (46%) de (±)-cis-etil 4-ciano-l,la,6,6a-tetraidrociclopropa [a]indeno-l-carboxilato.
b)_ácido_(±) -cis-4-ciano-l, la, 6,6ade 23/07/2018, pág. 136/150
128 tetraidrociclopropa[a]indeno-l-carboxílico
Este ácido foi sintetizado analogamente ao Exemplo 40d começando com 73 mg (0,32 mmol) do composto do Exemplo 47a para dar 59 mg (95%) de ácido (±)-cis-4-ciano-l,la,6,6atetraidrociclopropa[a]indeno-l-carboxílico
Exemplo 48
a) 4,7-Diflúor-l-indanona
Ácido 2,5-difluorcinâmico (5,0 mg, 27,2 mmol) foi dissolvido em 25 mL de etanol e uma quantidade catalítica de
Pd 10% em carbono foi adicionada. A mistura reacional foi hidrogenada em pressão normal por um período de 3 horas. Filtração através de celite e evaporação do solvente proporcionou o ácido 3-(2,5-difluorfenil)-propiônico bruto.
Este ácido foi dissolvido em 75 mL de tolueno e 5 mL de cloreto de tionil foi adicionado. A mistura reacional foi aquecida a +110°C por um período de 2 horas. Evaporação do solvente proporcionou o cloreto de 3-(2,5-difluorfenil)propionil bruto, que foi dissolvido em 25 mL de dissulfito de carbono e adicionado gota a gota a uma suspensão de 4 g de cloreto de alumínio em 100 mL de dissulfito de carbono. A mistura reacional foi deixada em refluxo por 2 horas e deu de 23/07/2018, pág. 137/150
129 após o trabalho e recristalização a partir de etanol 975 mg (22%) de 4,7-diflúor-l-indanona.
b) 4,7-Difluorindeno
composto foi preparado analogamente aos Exemplos 40a e 40b a partir de 4, 7-dif lúor-l-indanona (975 mg, 5,8 mmol) para dar 475 mg (54%) de 4,7-difluorindeno.
c)_(±) -cis-Etil_2,5-diflúor-l,la,6,6atetraidrociclopropa[a]indeno-l-carboxilato
Este composto foi preparado analogamente ao 10 Exemplo 40c a partir de 4, 7-dif luorindeno (475 mg, 3,13 mmol). Purificação em sílica gel partindo de hexanos seguido por hexanos com 2% de dietil éter e finalmente hexanos com 5% de dietil éter proporcionou 205 mg de cis-éster contaminado com 22% de trans-éster.
d)_ácido_(±) -cis-2,5-diflúor-l, la, 6,6atetraidrociclopropa[a]indeno-l-carboxílico
Petição 870180063303, de 23/07/2018, pág. 138/150
130
Este ácido foi sintetizado analogamente ao Exemplo 40d começando com 205 mg de cis-éster a partir do Exemplo 77c para dar 120 mg de ácido (±)-cis-2,5-diflúor-1,la,6,6atetraidrociclopropa[a]indeno-l-carboxilico contendo uma fração menor do ácido trans correspondente.
Exemplo 49
4-[ [6-[ [[[(IS, laS, 7bS)-4,7-diflúor-1,la,2,7b-tetraidrociclopropa[c][1]benzopiran-l-il]amino]carbonil]amino]-3piridinil]-N-(4-morfolinil)-benzamida
a) N-(4-morfolinil)-4-(fenilmetóxi)-benzamida
Uma mistura de ácido 4-benziloxibenzóico (0,5 g, 2,19 mmol), 4-aminomorfolina (0,2 mL, 2,13 mmol), Et3N (0,316 mL) , hidrocloreto de 1-(3-dimetilaminopropil)-3etilcarbodiimida (0,671 g, 3,5 mmol) e hidrato de 115 hidroxibenzotriazol (0,5 g, 3,7 mmol) em N,Ndimetilformamida (17 mL) foi agitado em temperatura ambiente por 2 dias. A reação foi concentrada e diluída em diclorometano. A fase orgânica foi lavada duas vezes com água, seca com MgSCg e concentrada. O resíduo foi purificado em cromatografia em coluna (sílica gel, 5% MeOH em CH2CI2) e
Petição 870180063303, de 23/07/2018, pág. 139/150
131
Ν-(4-morfolinil)-4-(fenilmetóxi)-benzamida (0,615 g, produção 90%) foi identificada por espectroscopia de RMN. 1H-RMN (CD3OD) : 7,99 (s, 1H) , 7,78 (d, J=8,6 Hz, 2H) , 7,45 (m, 2H), 7,39 (m, 2H), 7,33 (m, 1H), 7,07 (d, J=8,6 Hz, 2H),
5,16 (s, 2H), 3,82 (m, 4H), 2,91 (m, 4H).
lb) 4-hidróxi-N-(4-morfolinil)-benzamida
4-hidróxi-N-(4-morfolinil)-benzamida (0,288 g, 66%) foi sintetizado analogamente ao Exemplo 11b a partir de N-(4-morfolinil)-4-(fenilmetóxi)-benzamida (0,615g).
!H-RMN (CD3OD) : 7,67 (d, J=8,6Hz, 2H) , 6,81 (d, J= 8,6Hz,
2H), 3,80 (m, 4H), 2,8 (m, 4H).
lc) N-(4-morfolinil)-4-[(6-nitro-3-piridinil)óxi]benzamida
A mistura nitropiridina e bromopiridina (0,328 g) 15 foi sintetizada analogamente ao Exemplo 11c a partir de 4hidróxi-N-(4-morfolinil)-benzamida (0,288g).
ld) 4-[(6-amino-3-piridinil)óxi]-N-(4-morfolinil)benzamida
Petição 870180063303, de 23/07/2018, pág. 140/150
132
4-[(6-amino-3-piridinil)óxi] - N - (4-morfolinil)benzamida (0,234 g, 57%) foi sintetizado analogamente ao Exemplo lld a partir da mistura de nitropiridina e bromopiridina (0,328g).
íH-RMN (CD3OD) : 7,77 (d, J=8,2Hz, 2H) , 7,73 (d, J=2,73Hz,
1H) , 7,28 (m, 1H), 6,95 (d, J=8,2Hz, 2H), 6,65 (d, J=8,6Hz,
1H) , 3,80 (m, 4H), 2,89 (m, 4H) .
le) 4-[ [6-[ [ [ [ (IS, laS, 7bS)-4,7-diflúor-l,la,2,7b
-tetraidrociclopropa[c] [1] benzopiran-l-il]amino]carboníl] amino]-3-piridinil]-N-(4-morfolinil)-benzamida
4-[[6-[ [ [ [ (IS, laS, 7bS)-4, 7-diflúor-l,la,2,7btetraidrociclopropa[c][1]benzopiran-1 - il] amino] carboníl] amino]-3-piridinil]-N-(4-morfolinil)-benzamida (0.015 g, 21%) foi sintetizada analogamente ao Exemplo lie a partir de
4-[ (6-amino-3-piridinil)óxi]-N- (4 - morfolinil) - benzamida (0,041 g).
| 1H-RMN (CD3OD): |
7,82 |
(d, |
J=8, |
6 Hz, |
2H) , |
7,63 |
(d, J=2,73 Hz, |
|
1H) , 7,40 (m, |
1H) , |
6,98 |
(d, |
J=8,6 |
Hz, |
3H) , |
6,84 (m, 1H) , |
|
6,63 (m, 1H) , |
4, 42 |
(d, |
J=ll |
,3 Hz, |
1H) |
, 4,29 |
(dd, J=ll,7, |
Petição 870180063303, de 23/07/2018, pág. 141/150
133
2,73 Hz, 1H), 3,80 (m, 4H) , 3,62 (t, J=7,2 Hz, 1H), 2,91 (m,
4H), 2,6 (t, J=8,4 Hz, 1H), 2,03 (m, 1H).
(LC-MS, API-ES+: 538,2; Cale 537,5)
Exemplo 50 l-(4,7-Diflúor-l,la,2,7b-tetraidro-ciclopropa[c]cromen-lil ) -3- [ 5- ( 4-metanosulfinil-fenóxi)-piridin-2-il]-uréia
a) 4-metanosulfinil-fenol;
H2WO4 (0,029 g, 0,114 mmol) foi agitado em H2O (10 mL) . NaOH 50% (0, 040 mL) foi primeiro adicionado (pH>12) e então AcOH (0,040 mL) para alcançar pH 5. 4-metanosulfinilfenol (4g, 0,029 mol) foi adicionado e a mistura reacional foi aquecida a 65°C. H2O2 30% em H2O (3 mL) foi adicionado em porções por 10 minutos. A mistura reacional foi deixada em agitação em temperatura ambiente por 1 h. NaHSCb 50% foi 15 adicionado para extinguir a reação. Cloreto de metileno foi adicionado e o composto foi lavado com solução salina e purificado por cromatografia (0^10% EtOH em cloreto de metileno) para dar 1,9 g de 4-metanosulfinil-fenol (1) (42%) e 1,5 g de 4-metanosulfonil-fenol (2) (30%) .
0 b) 5-(4-Metanosulfinil-fenóxi)-2-nitro-piridina:
Petição 870180063303, de 23/07/2018, pág. 142/150
134
A uma solução de 4-metanosulfinil-fenol (1,52 g, 9,7 mmol) em DMF (30 mL) carbonato de césio (4,2 g, 12,9 mmol) foi adicionado, seguido por adição de 50bromo-2nitropiridina (1,75 g, 8,6 mmol) e a mistura foi agitada a
50°C por uma noite. A suspensão foi filtrada e evaporada + co-evaporada com tolueno. O composto foi purificado por cromatografia (0—>10% EtOH em cloreto de metileno) para dar 1,5 g (56%) de 5-(4-Metanosulfinil-fenóxi)-2-nitro-piridina.
c) 5-(4-Metanosulfinil-fenóxi)-piridin-2-ilamina:
5-(4-Metanosulfinil-fenóxi)-2-nitro-piridina (1,27 g, 4,56 mmol) foi dissolvida em EtOH (30 mL) e EtOAc (8 mL). Pd/C (10%) (400 mg) foi adicionado e o grupo nitro foi reduzido à amina por hidrogenação em pressão atmosférica por 3 horas. O catalisador foi retirado por filtrado e o filtrado foi evaporado para dar 0.6 g de 5-(4Metanosulfinil-fenóxi)-piridin-2-ilamina.
d)_1-(4, 7-Diflúor-l,la,2,7b-tetraidro-ciclopropa [c]cromen-l-il)-3-[5-(4-metanosulfinil-fenóxi)-piridin-2-il]
-uréia
Petição 870180063303, de 23/07/2018, pág. 143/150
135
- (4-Metanosulfinil-fenóxi) - piridin-2-ilamina (0,049 g, 0,197 mmol) foi dissolvido em tolueno (2 mL). O ácido (IS, laR, 7bS)-4,7-diflúor-1,la,2,7btetraidrociclopropa[c]cromeno-l-carboxílico, preparado como mostrado em W002/705163 (0,041 g, 0,0179 mmol), DPPA (0,04 mL, 0,189 mmol), e TEA (0,025 mL, 0,180 mmol) foram adicionados. A mistura reacional foi aguecida a 110°C e foi deixada em agitação na mesma temperatura por uma noite. A mistura reacional foi trabalhada por extrações entre cloreto de metileno e 5% de ácido cítrico seguido por NaHCCç aguoso saturado. Cromatografia em coluna de sílica gel (5% de MeOH em clorofórmio) deu 25 mg (30%) dei-(4,7-Diflúor-1,la,2,7btetraidro-ciclopropa[c]cromen-l-il)-3-[5-(4-metanosulfinil fenóxi)-piridin-2-il]-uréia.
1H-RMN (CDCls) : 9,30 (br s, 1H) , 7,65 (m, 2H), 7,30 (m, 2H),
7,05 (m, 2H) , 6, 80-6, 70 (m, 2H) , 6,60 (d tr, 1H) , 4,47 (dd,
1H) , 4,32 (dd, lh) , 3,80 (g, 1H) , 2,75 (s, 3H) , 2,60 (tr,
1H) , 1, 99 (m, 1H) .
Resultados Biológicos
Guia extensivo em ensaios de testes de compostos no nivel enzimático e em cultura celular, incluindo o isolamento e/ou seleção de cepas HIV mutantes e RT mutante são encontrados em DAIDS Virology Manual for HIV Laboratories cedido pela Divisão de AIDS, NIAIS EUA, 1997. Estudos de resistência, incluindo racional para vários tipos de 23/07/2018, pág. 144/150
136 de mutantes de escape de drogas são descritos no HIV Resistance Collaborative Group Data Analysis Plan for Resistance Studies, revisado em 31 de Agosto de 1999.
Compostos da invenção são testados para atividade HIV, por exemplo, utilizando determinações múltiplas com XTT em células MT-4 (Weislow e colaboradores, J Nat Câncer Inst 1989, vol 81 n° 8, 577 et seq), preferivelmente incluindo determinações na presença de 40-50% do soro humano para indicar a contribuição da proteína ligante. Em resumo, o ensaio XTT usa linhagem de célula T humana, células MT4 crescendo em meio RPMI 1640 suplementado com 10% de soro fetal bovino (ou 40-50% de soro humano apropriado), penicilina e estreptomicina, plaqueadas em microplacas de 96 poços (2x104 células/poço) infectadas com 10-20 TCID50 por poço de HIViiib (selvagem) ou vírus mutante, tal como aqueles tendo mutações em RT IIe 100, Cys 181 ou Asn 103. Compostos testados serialmente diluídos são adicionados aos respectivos pólos e a cultura incubada a 37°C em uma atmosfera enriquecida com CO2 e a viabilidade das células é determinada no dia cinco ou seis com corante vital XTT.
Resultados são tipicamente apresentados como ED50 μΜ.
Compostos são preferivelmente potentes contra o vírus selvagem e vírus HIV mutante, especialmente vírus compreendendo mutações de escape de droga. Mutações de escape de droga são aquelas que aparecem em pacientes devido à pressão seletiva de um aniviral anterior e que confere resistência aumentada a este antiviral. O Data Analysis Plan citado acima resume mutantes de escape de droga relevantes de 23/07/2018, pág. 145/150
137 para cada uma das classes de antiviral correntemente no mercado. Clones de escape de droga são facilmente isolados de pacientes HIV que não possuem êxito em uma terapia particular. Alternativamente a preparação das mutações RT, em um antecedente genético conhecido é mostrado em WO97/27319, WO99/61658 e WO00/73511 que, também, mostra o uso de tais mutantes no perfil de sensibilidade.
K103 N é um mutante de escape de droga particularmente relevante no contexto da terapia NNRTI e compostos da invenção preferivelmente têm um baixo ED50 contra este mutante e ainda mais preferivelmente o duplo mutante L100I, K103 especialmente em ensaios mimetizando a presença do soro humano.
Ensaios de transcriptase reversa convenientes usam transcriptase reversa como chave nas mutações de escape de droga preparadas amplamente como descrito em Unge e colaboradores Eur. J. Biochem, 269, 1670-1677 (2002). In.
Por exemplo, um mutante K103N é preparado utilizando esta metodologia e os iniciadores
CATCCCGCAGGGTTAAAAAAGAACAAATCAGTAACAGTACTGGATG
CATCCAGTACTGTTACTGATTTGTICTTTTTTACCCTGCGGGATG
O mutante L100I/K103N pe preparado por mutação de L 100 na enzima k103N:
CCACATCCCGCAGGGATTAAAAAGAACAAATCAGTAAC
GTTACTGATTTGTTCTTTTTAATCCCTGCGGATGTGG
Mutações são feitas no clone HIVRT DH 10 cDNA clonado no vetor de expressão pET11d. As mutações são geradas por amplificação do DNA mutado com a ajuda da enzima de 23/07/2018, pág. 146/150
138
Pfu. Clonagem foi então feita em células E. coli TOP10 e expressão de enzima mutada foi feita em células E. coli BL21(DE3) após indução com IPTG.
O ensaio de transcriptase reversa de HIV-1 utilizou um SPA (Ensaio de proximidade de cintilação), sistema contando com fluomicrosferas revestidas com receptor molecular de estreptavidina (Flashplates, PerkinElmer Life Science) que é capaz de ligar a ligantes rediomarcados na solução de reação. Neste ensaio, o iniciador biotinilado (5'-GTC ATA GCT GTT TCC TG-3') é pré-anelado com um padrão de DNA heterólogo (sintetizado por GENSE) dando uma seqüência de 5'-CG UCU GGC AUU GCG AGC GGA UAA CAA UUU CAC
ACA GGA AAC AGC UAU GAC-3' em um ambiente livre de RNAase.
Transcriptase reversa de HIV-1 (tal como L100I+K103N) catalizada pela atividade DNA dependente de RNA foi medida em presença de 50 mM de Tris-HCl pH=8.0, 80mM de KCl, 10 mM de Mg2Cl2, 10 mM de Ditiotreitol, 5 mg/mL de BSA e 0,05% Nonidet P40 onde a incorporação de dGTP marcado com trítio (Amersham, 35Ci/mmol) e 11gM de dNTP (dATP, dCTP e dTTP) foi monitorada em temperatura ambiente. A concentração de dGTP foi utilizada em um valor de Km de 0,25 gM, 10 nM de padrão de RNA foi utilizado e RT mutante (tal como L100I+K103N) foi utilizado a 180 ng/mL em 100 μl de volume de reação para 120 minutos de reação.
Compostos da invenção foram testados para atividade HIV contra a problemática mutante L100I, K103N em ensaio in vitro como delineado abaixo. Para referência, o mais próximo composto anterior na técnica, cis-1-(4,7de 23/07/2018, pág. 147/150
139 diflúor-1,1a,2,7b-tetraidro-ciclopropa[c]cromen-1-il0-3-(5fenóxi-piridin-2-il)-uréia, exemplo 20 da WO02/070516 como descrito acima, foi testado no mesmo sistema.
|
Exemplo |
Substituinte para fenil (ou piridil) |
IC50 nM |
|
1 |
Sulfonamido |
29 |
|
2 |
N-metilcarboxiamido |
26 |
|
3 |
N-metilsulfonamido |
60 |
|
4 |
Amino |
70 |
|
5 |
Metilsulfonil |
40 |
|
8 |
N-metilcarboxamida (em piridil) |
70 |
|
9 |
Amida |
28 |
|
10 |
Carboxamida |
80 |
|
11 |
Hidrazinocarbonil |
10 |
|
12 |
Ciclopropilamida |
27 |
|
13 |
Acetamida |
40 |
|
14 |
Triazolil |
60 |
|
Técnica |
nil |
600 |
|
anterior |
|
|
É então aparente que a adição do substituinte no braço direito, de acordo com a invenção dramaticamente aumenta a atividade contra a problemática do duplo mutante de escape L100I & K103N.
Petição 870180063303, de 23/07/2018, pág. 148/150