EP2464658B1 - Detoxified escherichia coli immunogens - Google Patents
Detoxified escherichia coli immunogens Download PDFInfo
- Publication number
- EP2464658B1 EP2464658B1 EP10752175.9A EP10752175A EP2464658B1 EP 2464658 B1 EP2464658 B1 EP 2464658B1 EP 10752175 A EP10752175 A EP 10752175A EP 2464658 B1 EP2464658 B1 EP 2464658B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- orf3526
- protein
- amino acids
- antigen
- seq
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 241000588724 Escherichia coli Species 0.000 title claims description 153
- 108090000623 proteins and genes Proteins 0.000 claims description 178
- 102000004169 proteins and genes Human genes 0.000 claims description 165
- 150000001413 amino acids Chemical class 0.000 claims description 144
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 137
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 135
- 229920001184 polypeptide Polymers 0.000 claims description 132
- 239000000203 mixture Substances 0.000 claims description 104
- 239000002671 adjuvant Substances 0.000 claims description 101
- 230000002163 immunogen Effects 0.000 claims description 90
- 239000000427 antigen Substances 0.000 claims description 67
- 102000036639 antigens Human genes 0.000 claims description 67
- 108091007433 antigens Proteins 0.000 claims description 67
- 229960005486 vaccine Drugs 0.000 claims description 57
- 238000012217 deletion Methods 0.000 claims description 56
- 230000037430 deletion Effects 0.000 claims description 56
- 125000001429 N-terminal alpha-amino-acid group Chemical group 0.000 claims description 53
- 230000035772 mutation Effects 0.000 claims description 40
- 210000004027 cell Anatomy 0.000 claims description 37
- 238000000034 method Methods 0.000 claims description 37
- 230000028993 immune response Effects 0.000 claims description 36
- PRAKJMSDJKAYCZ-UHFFFAOYSA-N dodecahydrosqualene Natural products CC(C)CCCC(C)CCCC(C)CCCCC(C)CCCC(C)CCCC(C)C PRAKJMSDJKAYCZ-UHFFFAOYSA-N 0.000 claims description 34
- 125000001433 C-terminal amino-acid group Chemical group 0.000 claims description 30
- 230000004572 zinc-binding Effects 0.000 claims description 25
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 claims description 24
- 229920000053 polysorbate 80 Polymers 0.000 claims description 24
- YYGNTYWPHWGJRM-UHFFFAOYSA-N (6E,10E,14E,18E)-2,6,10,15,19,23-hexamethyltetracosa-2,6,10,14,18,22-hexaene Chemical compound CC(C)=CCCC(C)=CCCC(C)=CCCC=C(C)CCC=C(C)CCC=C(C)C YYGNTYWPHWGJRM-UHFFFAOYSA-N 0.000 claims description 23
- TUHBEKDERLKLEC-UHFFFAOYSA-N squalene Natural products CC(=CCCC(=CCCC(=CCCC=C(/C)CCC=C(/C)CC=C(C)C)C)C)C TUHBEKDERLKLEC-UHFFFAOYSA-N 0.000 claims description 23
- BHEOSNUKNHRBNM-UHFFFAOYSA-N Tetramethylsqualene Natural products CC(=C)C(C)CCC(=C)C(C)CCC(C)=CCCC=C(C)CCC(C)C(=C)CCC(C)C(C)=C BHEOSNUKNHRBNM-UHFFFAOYSA-N 0.000 claims description 22
- 229940031439 squalene Drugs 0.000 claims description 22
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 claims description 12
- 231100000419 toxicity Toxicity 0.000 claims description 11
- 230000001988 toxicity Effects 0.000 claims description 11
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 claims description 10
- PRXRUNOAOLTIEF-ADSICKODSA-N Sorbitan trioleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCC\C=C/CCCCCCCC)[C@H]1OC[C@H](O)[C@H]1OC(=O)CCCCCCC\C=C/CCCCCCCC PRXRUNOAOLTIEF-ADSICKODSA-N 0.000 claims description 10
- 102000036435 Zincins Human genes 0.000 claims description 9
- 108091007139 Zincins Proteins 0.000 claims description 9
- 230000000694 effects Effects 0.000 claims description 9
- 239000013612 plasmid Substances 0.000 claims description 9
- 239000003995 emulsifying agent Substances 0.000 claims description 8
- 239000007764 o/w emulsion Substances 0.000 claims description 8
- BCCRXDTUTZHDEU-VKHMYHEASA-N Gly-Ser Chemical group NCC(=O)N[C@@H](CO)C(O)=O BCCRXDTUTZHDEU-VKHMYHEASA-N 0.000 claims description 7
- 230000007423 decrease Effects 0.000 claims description 6
- 229940124856 vaccine component Drugs 0.000 claims description 6
- 230000004075 alteration Effects 0.000 claims description 4
- 102000040430 polynucleotide Human genes 0.000 claims description 4
- 108091033319 polynucleotide Proteins 0.000 claims description 4
- 239000002157 polynucleotide Substances 0.000 claims description 4
- 108091005804 Peptidases Proteins 0.000 claims description 3
- 239000004365 Protease Substances 0.000 claims description 3
- 239000004147 Sorbitan trioleate Substances 0.000 claims description 3
- 230000003197 catalytic effect Effects 0.000 claims description 3
- 230000001939 inductive effect Effects 0.000 claims description 3
- 235000019337 sorbitan trioleate Nutrition 0.000 claims description 3
- 229960000391 sorbitan trioleate Drugs 0.000 claims description 3
- 241000606768 Haemophilus influenzae Species 0.000 claims description 2
- 241000293871 Salmonella enterica subsp. enterica serovar Typhi Species 0.000 claims description 2
- 241000607626 Vibrio cholerae Species 0.000 claims description 2
- 125000001500 prolyl group Chemical group [H]N1C([H])(C(=O)[*])C([H])([H])C([H])([H])C1([H])[H] 0.000 claims description 2
- 229940118696 vibrio cholerae Drugs 0.000 claims description 2
- 125000003275 alpha amino acid group Chemical group 0.000 claims 4
- 241000193738 Bacillus anthracis Species 0.000 claims 1
- 241000588832 Bordetella pertussis Species 0.000 claims 1
- 241000589969 Borreliella burgdorferi Species 0.000 claims 1
- 241001647372 Chlamydia pneumoniae Species 0.000 claims 1
- 241000606153 Chlamydia trachomatis Species 0.000 claims 1
- 241000193449 Clostridium tetani Species 0.000 claims 1
- 241000194032 Enterococcus faecalis Species 0.000 claims 1
- 241000194031 Enterococcus faecium Species 0.000 claims 1
- 241000590002 Helicobacter pylori Species 0.000 claims 1
- 241000588748 Klebsiella Species 0.000 claims 1
- 241000589242 Legionella pneumophila Species 0.000 claims 1
- 241000186779 Listeria monocytogenes Species 0.000 claims 1
- 241000588655 Moraxella catarrhalis Species 0.000 claims 1
- 241000187479 Mycobacterium tuberculosis Species 0.000 claims 1
- 241000588650 Neisseria meningitidis Species 0.000 claims 1
- 241000605862 Porphyromonas gingivalis Species 0.000 claims 1
- 241000589517 Pseudomonas aeruginosa Species 0.000 claims 1
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 claims 1
- 241000606701 Rickettsia Species 0.000 claims 1
- 241000191940 Staphylococcus Species 0.000 claims 1
- 241000191967 Staphylococcus aureus Species 0.000 claims 1
- 241001147691 Staphylococcus saprophyticus Species 0.000 claims 1
- 241000193985 Streptococcus agalactiae Species 0.000 claims 1
- 241000193998 Streptococcus pneumoniae Species 0.000 claims 1
- 241000193996 Streptococcus pyogenes Species 0.000 claims 1
- 241000589884 Treponema pallidum Species 0.000 claims 1
- 241000607447 Yersinia enterocolitica Species 0.000 claims 1
- 241000607479 Yersinia pestis Species 0.000 claims 1
- 241000606834 [Haemophilus] ducreyi Species 0.000 claims 1
- 229940065181 bacillus anthracis Drugs 0.000 claims 1
- 229940038705 chlamydia trachomatis Drugs 0.000 claims 1
- 229940032049 enterococcus faecalis Drugs 0.000 claims 1
- 210000002615 epidermis Anatomy 0.000 claims 1
- 229940045808 haemophilus influenzae type b Drugs 0.000 claims 1
- 229940037467 helicobacter pylori Drugs 0.000 claims 1
- 229940115932 legionella pneumophila Drugs 0.000 claims 1
- 229940031000 streptococcus pneumoniae Drugs 0.000 claims 1
- 229940098232 yersinia enterocolitica Drugs 0.000 claims 1
- 235000018102 proteins Nutrition 0.000 description 128
- 108020004707 nucleic acids Proteins 0.000 description 60
- 102000039446 nucleic acids Human genes 0.000 description 60
- 150000007523 nucleic acids Chemical class 0.000 description 60
- 239000012634 fragment Substances 0.000 description 59
- 239000000839 emulsion Substances 0.000 description 44
- 210000004899 c-terminal region Anatomy 0.000 description 39
- 239000013598 vector Substances 0.000 description 39
- -1 aromatic amino acids Chemical class 0.000 description 37
- 235000001014 amino acid Nutrition 0.000 description 32
- 229940024606 amino acid Drugs 0.000 description 30
- 230000000688 enterotoxigenic effect Effects 0.000 description 30
- 230000000369 enteropathogenic effect Effects 0.000 description 27
- 238000002649 immunization Methods 0.000 description 27
- 238000004519 manufacturing process Methods 0.000 description 23
- 239000003921 oil Substances 0.000 description 23
- 235000019198 oils Nutrition 0.000 description 23
- 230000004083 survival effect Effects 0.000 description 20
- 230000001580 bacterial effect Effects 0.000 description 19
- 230000001717 pathogenic effect Effects 0.000 description 19
- 229930182490 saponin Natural products 0.000 description 19
- 235000017709 saponins Nutrition 0.000 description 19
- 150000007949 saponins Chemical class 0.000 description 19
- 108020004414 DNA Proteins 0.000 description 18
- 239000002773 nucleotide Substances 0.000 description 18
- 125000003729 nucleotide group Chemical group 0.000 description 18
- 150000003839 salts Chemical class 0.000 description 18
- 108091034117 Oligonucleotide Proteins 0.000 description 17
- 239000001397 quillaja saponaria molina bark Substances 0.000 description 17
- 241000699670 Mus sp. Species 0.000 description 16
- 239000000872 buffer Substances 0.000 description 16
- GVJHHUAWPYXKBD-UHFFFAOYSA-N d-alpha-tocopherol Natural products OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 16
- 206010012735 Diarrhoea Diseases 0.000 description 15
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 15
- 230000003053 immunization Effects 0.000 description 15
- 239000013615 primer Substances 0.000 description 15
- 239000004094 surface-active agent Substances 0.000 description 15
- 241000282414 Homo sapiens Species 0.000 description 14
- 150000001875 compounds Chemical class 0.000 description 14
- 208000015181 infectious disease Diseases 0.000 description 14
- 239000002245 particle Substances 0.000 description 14
- 230000001965 increasing effect Effects 0.000 description 13
- 208000019206 urinary tract infection Diseases 0.000 description 13
- GVJHHUAWPYXKBD-IEOSBIPESA-N α-tocopherol Chemical compound OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 description 13
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 12
- ILRRQNADMUWWFW-UHFFFAOYSA-K aluminium phosphate Chemical compound O1[Al]2OP1(=O)O2 ILRRQNADMUWWFW-UHFFFAOYSA-K 0.000 description 12
- 238000009472 formulation Methods 0.000 description 12
- 239000006166 lysate Substances 0.000 description 12
- 238000006467 substitution reaction Methods 0.000 description 12
- 241000894006 Bacteria Species 0.000 description 11
- 229940037003 alum Drugs 0.000 description 11
- 239000003814 drug Substances 0.000 description 11
- 239000011732 tocopherol Substances 0.000 description 11
- 229930003799 tocopherol Natural products 0.000 description 11
- 229910019142 PO4 Inorganic materials 0.000 description 10
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 10
- 230000029662 T-helper 1 type immune response Effects 0.000 description 10
- 210000001744 T-lymphocyte Anatomy 0.000 description 10
- 229940001007 aluminium phosphate Drugs 0.000 description 10
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 10
- 230000000295 complement effect Effects 0.000 description 10
- 239000002158 endotoxin Substances 0.000 description 10
- 230000005847 immunogenicity Effects 0.000 description 10
- 230000003308 immunostimulating effect Effects 0.000 description 10
- 238000002255 vaccination Methods 0.000 description 10
- 206010040047 Sepsis Diseases 0.000 description 9
- 230000001154 acute effect Effects 0.000 description 9
- 238000009396 hybridization Methods 0.000 description 9
- 229920006008 lipopolysaccharide Polymers 0.000 description 9
- 239000003550 marker Substances 0.000 description 9
- 239000000463 material Substances 0.000 description 9
- 238000000746 purification Methods 0.000 description 9
- 102000004127 Cytokines Human genes 0.000 description 8
- 108090000695 Cytokines Proteins 0.000 description 8
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 8
- 241000124008 Mammalia Species 0.000 description 8
- 102100040247 Tumor necrosis factor Human genes 0.000 description 8
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 8
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 8
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical class NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 8
- 229940035032 monophosphoryl lipid a Drugs 0.000 description 8
- 231100000252 nontoxic Toxicity 0.000 description 8
- 230000003000 nontoxic effect Effects 0.000 description 8
- 235000021317 phosphate Nutrition 0.000 description 8
- 229920000136 polysorbate Polymers 0.000 description 8
- 229940068968 polysorbate 80 Drugs 0.000 description 8
- 239000002243 precursor Substances 0.000 description 8
- 230000004044 response Effects 0.000 description 8
- 239000000523 sample Substances 0.000 description 8
- 235000010384 tocopherol Nutrition 0.000 description 8
- 229960001295 tocopherol Drugs 0.000 description 8
- 230000029069 type 2 immune response Effects 0.000 description 8
- 239000003981 vehicle Substances 0.000 description 8
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 7
- 108091028043 Nucleic acid sequence Proteins 0.000 description 7
- 206010034133 Pathogen resistance Diseases 0.000 description 7
- 108010076504 Protein Sorting Signals Proteins 0.000 description 7
- 229920004890 Triton X-100 Polymers 0.000 description 7
- 239000013504 Triton X-100 Substances 0.000 description 7
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 7
- 238000006243 chemical reaction Methods 0.000 description 7
- 239000013604 expression vector Substances 0.000 description 7
- 239000000499 gel Substances 0.000 description 7
- 229910052500 inorganic mineral Inorganic materials 0.000 description 7
- 235000010755 mineral Nutrition 0.000 description 7
- 239000011707 mineral Substances 0.000 description 7
- 239000010452 phosphate Substances 0.000 description 7
- 239000003053 toxin Substances 0.000 description 7
- 231100000765 toxin Toxicity 0.000 description 7
- 108700012359 toxins Proteins 0.000 description 7
- 230000003612 virological effect Effects 0.000 description 7
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 6
- 108020004705 Codon Proteins 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- NYHBQMYGNKIUIF-UUOKFMHZSA-N Guanosine Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O NYHBQMYGNKIUIF-UUOKFMHZSA-N 0.000 description 6
- 208000032759 Hemolytic-Uremic Syndrome Diseases 0.000 description 6
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 6
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 6
- 229910021502 aluminium hydroxide Inorganic materials 0.000 description 6
- 230000003321 amplification Effects 0.000 description 6
- 238000010171 animal model Methods 0.000 description 6
- 239000013592 cell lysate Substances 0.000 description 6
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 6
- 239000003599 detergent Substances 0.000 description 6
- JXTPJDDICSTXJX-UHFFFAOYSA-N n-Triacontane Natural products CCCCCCCCCCCCCCCCCCCCCCCCCCCCCC JXTPJDDICSTXJX-UHFFFAOYSA-N 0.000 description 6
- 238000003199 nucleic acid amplification method Methods 0.000 description 6
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 6
- 238000012552 review Methods 0.000 description 6
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 6
- 239000000243 solution Substances 0.000 description 6
- 229940032094 squalane Drugs 0.000 description 6
- 230000001225 therapeutic effect Effects 0.000 description 6
- 208000004429 Bacillary Dysentery Diseases 0.000 description 5
- 201000004538 Bacteriuria Diseases 0.000 description 5
- 108091026890 Coding region Proteins 0.000 description 5
- 206010017915 Gastroenteritis shigella Diseases 0.000 description 5
- 239000007993 MOPS buffer Substances 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- RVGRUAULSDPKGF-UHFFFAOYSA-N Poloxamer Chemical compound C1CO1.CC1CO1 RVGRUAULSDPKGF-UHFFFAOYSA-N 0.000 description 5
- 206010037596 Pyelonephritis Diseases 0.000 description 5
- 229930182558 Sterol Natural products 0.000 description 5
- 241000700605 Viruses Species 0.000 description 5
- 238000013459 approach Methods 0.000 description 5
- 210000003719 b-lymphocyte Anatomy 0.000 description 5
- 235000012000 cholesterol Nutrition 0.000 description 5
- 230000001684 chronic effect Effects 0.000 description 5
- 238000010367 cloning Methods 0.000 description 5
- 201000003146 cystitis Diseases 0.000 description 5
- 230000034994 death Effects 0.000 description 5
- 201000010099 disease Diseases 0.000 description 5
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 5
- 238000002474 experimental method Methods 0.000 description 5
- 238000001415 gene therapy Methods 0.000 description 5
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 5
- 230000000968 intestinal effect Effects 0.000 description 5
- BPHPUYQFMNQIOC-NXRLNHOXSA-N isopropyl beta-D-thiogalactopyranoside Chemical compound CC(C)S[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O BPHPUYQFMNQIOC-NXRLNHOXSA-N 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- 239000011159 matrix material Substances 0.000 description 5
- 239000011859 microparticle Substances 0.000 description 5
- 239000002736 nonionic surfactant Substances 0.000 description 5
- 229920002113 octoxynol Polymers 0.000 description 5
- 206010034674 peritonitis Diseases 0.000 description 5
- 150000003904 phospholipids Chemical class 0.000 description 5
- 229920000642 polymer Polymers 0.000 description 5
- 230000010076 replication Effects 0.000 description 5
- 201000005113 shigellosis Diseases 0.000 description 5
- 239000011780 sodium chloride Substances 0.000 description 5
- 235000003702 sterols Nutrition 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- 230000009885 systemic effect Effects 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- 229960000984 tocofersolan Drugs 0.000 description 5
- 235000015112 vegetable and seed oil Nutrition 0.000 description 5
- 239000000277 virosome Substances 0.000 description 5
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 4
- 206010056396 Asymptomatic bacteriuria Diseases 0.000 description 4
- 241000701022 Cytomegalovirus Species 0.000 description 4
- 108010065805 Interleukin-12 Proteins 0.000 description 4
- 108700020354 N-acetylmuramyl-threonyl-isoglutamine Proteins 0.000 description 4
- 101500027983 Rattus norvegicus Octadecaneuropeptide Proteins 0.000 description 4
- 229910052782 aluminium Inorganic materials 0.000 description 4
- 230000000890 antigenic effect Effects 0.000 description 4
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 208000001848 dysentery Diseases 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- 150000002191 fatty alcohols Chemical class 0.000 description 4
- 238000001476 gene delivery Methods 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 235000010445 lecithin Nutrition 0.000 description 4
- 239000000787 lecithin Substances 0.000 description 4
- 229940067606 lecithin Drugs 0.000 description 4
- 231100000636 lethal dose Toxicity 0.000 description 4
- GZQKNULLWNGMCW-PWQABINMSA-N lipid A (E. coli) Chemical class O1[C@H](CO)[C@@H](OP(O)(O)=O)[C@H](OC(=O)C[C@@H](CCCCCCCCCCC)OC(=O)CCCCCCCCCCCCC)[C@@H](NC(=O)C[C@@H](CCCCCCCCCCC)OC(=O)CCCCCCCCCCC)[C@@H]1OC[C@@H]1[C@@H](O)[C@H](OC(=O)C[C@H](O)CCCCCCCCCCC)[C@@H](NC(=O)C[C@H](O)CCCCCCCCCCC)[C@@H](OP(O)(O)=O)O1 GZQKNULLWNGMCW-PWQABINMSA-N 0.000 description 4
- 230000001404 mediated effect Effects 0.000 description 4
- 238000002156 mixing Methods 0.000 description 4
- 230000004048 modification Effects 0.000 description 4
- 238000012986 modification Methods 0.000 description 4
- 238000012544 monitoring process Methods 0.000 description 4
- 230000016379 mucosal immune response Effects 0.000 description 4
- 229940066429 octoxynol Drugs 0.000 description 4
- 229940046166 oligodeoxynucleotide Drugs 0.000 description 4
- 229920001983 poloxamer Polymers 0.000 description 4
- 229920000056 polyoxyethylene ether Polymers 0.000 description 4
- 239000002987 primer (paints) Substances 0.000 description 4
- 230000000306 recurrent effect Effects 0.000 description 4
- 238000011160 research Methods 0.000 description 4
- 238000001262 western blot Methods 0.000 description 4
- 208000031729 Bacteremia Diseases 0.000 description 3
- 108010039939 Cell Wall Skeleton Proteins 0.000 description 3
- MIKUYHXYGGJMLM-GIMIYPNGSA-N Crotonoside Natural products C1=NC2=C(N)NC(=O)N=C2N1[C@H]1O[C@@H](CO)[C@H](O)[C@@H]1O MIKUYHXYGGJMLM-GIMIYPNGSA-N 0.000 description 3
- NYHBQMYGNKIUIF-UHFFFAOYSA-N D-guanosine Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(CO)C(O)C1O NYHBQMYGNKIUIF-UHFFFAOYSA-N 0.000 description 3
- 235000001815 DL-alpha-tocopherol Nutrition 0.000 description 3
- 239000011627 DL-alpha-tocopherol Substances 0.000 description 3
- 241000121399 Escherichia coli IHE3034 Species 0.000 description 3
- 101000880367 Escherichia coli O157:H7 Metalloprotease StcE Proteins 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 108010074328 Interferon-gamma Proteins 0.000 description 3
- 108010002350 Interleukin-2 Proteins 0.000 description 3
- 102000000588 Interleukin-2 Human genes 0.000 description 3
- 108090000978 Interleukin-4 Proteins 0.000 description 3
- 108010002616 Interleukin-5 Proteins 0.000 description 3
- 108090001005 Interleukin-6 Proteins 0.000 description 3
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 3
- 229920002884 Laureth 4 Polymers 0.000 description 3
- 206010058780 Meningitis neonatal Diseases 0.000 description 3
- 241001644525 Nastus productus Species 0.000 description 3
- 230000024932 T cell mediated immunity Effects 0.000 description 3
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical class O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 3
- 101710204001 Zinc metalloprotease Proteins 0.000 description 3
- 239000004411 aluminium Substances 0.000 description 3
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 3
- 159000000013 aluminium salts Chemical class 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 239000000227 bioadhesive Substances 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 230000030833 cell death Effects 0.000 description 3
- 210000004520 cell wall skeleton Anatomy 0.000 description 3
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 3
- 235000018417 cysteine Nutrition 0.000 description 3
- 229940104302 cytosine Drugs 0.000 description 3
- 238000010828 elution Methods 0.000 description 3
- 229940029575 guanosine Drugs 0.000 description 3
- 230000036039 immunity Effects 0.000 description 3
- 239000002955 immunomodulating agent Substances 0.000 description 3
- 229940121354 immunomodulator Drugs 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 238000003780 insertion Methods 0.000 description 3
- 230000037431 insertion Effects 0.000 description 3
- 229940062711 laureth-9 Drugs 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- 239000002502 liposome Substances 0.000 description 3
- 210000002540 macrophage Anatomy 0.000 description 3
- 238000002493 microarray Methods 0.000 description 3
- 230000000813 microbial effect Effects 0.000 description 3
- 230000003232 mucoadhesive effect Effects 0.000 description 3
- 229910052759 nickel Inorganic materials 0.000 description 3
- 229920002114 octoxynol-9 Polymers 0.000 description 3
- 239000008363 phosphate buffer Substances 0.000 description 3
- 239000002953 phosphate buffered saline Substances 0.000 description 3
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 3
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 3
- ONJQDTZCDSESIW-UHFFFAOYSA-N polidocanol Chemical compound CCCCCCCCCCCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO ONJQDTZCDSESIW-UHFFFAOYSA-N 0.000 description 3
- 229960000502 poloxamer Drugs 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 239000013641 positive control Substances 0.000 description 3
- 238000001556 precipitation Methods 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 230000000069 prophylactic effect Effects 0.000 description 3
- 230000002685 pulmonary effect Effects 0.000 description 3
- BXNMTOQRYBFHNZ-UHFFFAOYSA-N resiquimod Chemical compound C1=CC=CC2=C(N(C(COCC)=N3)CC(C)(C)O)C3=C(N)N=C21 BXNMTOQRYBFHNZ-UHFFFAOYSA-N 0.000 description 3
- 238000012216 screening Methods 0.000 description 3
- 238000010845 search algorithm Methods 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 238000000527 sonication Methods 0.000 description 3
- 238000001179 sorption measurement Methods 0.000 description 3
- 239000007921 spray Substances 0.000 description 3
- 150000003432 sterols Chemical class 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 239000006228 supernatant Substances 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- 125000002640 tocopherol group Chemical class 0.000 description 3
- 235000019149 tocopherols Nutrition 0.000 description 3
- 238000013518 transcription Methods 0.000 description 3
- 230000035897 transcription Effects 0.000 description 3
- 230000009466 transformation Effects 0.000 description 3
- 238000011282 treatment Methods 0.000 description 3
- GPRLSGONYQIRFK-MNYXATJNSA-N triton Chemical compound [3H+] GPRLSGONYQIRFK-MNYXATJNSA-N 0.000 description 3
- 241000701161 unidentified adenovirus Species 0.000 description 3
- 241001430294 unidentified retrovirus Species 0.000 description 3
- 239000012646 vaccine adjuvant Substances 0.000 description 3
- 229940124931 vaccine adjuvant Drugs 0.000 description 3
- 235000004835 α-tocopherol Nutrition 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- AXTGOJVKRHFYBT-XAZAIFFQSA-N (1r,2r,3r,6s,7s,8r)-3-(hydroxymethyl)-2,3,5,6,7,8-hexahydro-1h-pyrrolizine-1,2,6,7-tetrol Chemical compound O[C@@H]1[C@@H](O)CN2[C@H](CO)[C@@H](O)[C@H](O)[C@H]21 AXTGOJVKRHFYBT-XAZAIFFQSA-N 0.000 description 2
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical class OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 2
- VQFKFAKEUMHBLV-BYSUZVQFSA-N 1-O-(alpha-D-galactosyl)-N-hexacosanoylphytosphingosine Chemical compound CCCCCCCCCCCCCCCCCCCCCCCCCC(=O)N[C@H]([C@H](O)[C@H](O)CCCCCCCCCCCCCC)CO[C@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O VQFKFAKEUMHBLV-BYSUZVQFSA-N 0.000 description 2
- HNLXNOZHXNSSPN-UHFFFAOYSA-N 2-[2-[2-[2-[2-[2-[2-[4-(2,4,4-trimethylpentan-2-yl)phenoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethanol Chemical compound CC(C)(C)CC(C)(C)C1=CC=C(OCCOCCOCCOCCOCCOCCOCCO)C=C1 HNLXNOZHXNSSPN-UHFFFAOYSA-N 0.000 description 2
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 2
- TZYVRXZQAWPIAB-FCLHUMLKSA-N 5-amino-3-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-4h-[1,3]thiazolo[4,5-d]pyrimidine-2,7-dione Chemical compound O=C1SC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O TZYVRXZQAWPIAB-FCLHUMLKSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- 241000251468 Actinopterygii Species 0.000 description 2
- 101710132601 Capsid protein Proteins 0.000 description 2
- 102000053642 Catalytic RNA Human genes 0.000 description 2
- 108090000994 Catalytic RNA Proteins 0.000 description 2
- LZZYPRNAOMGNLH-UHFFFAOYSA-M Cetrimonium bromide Chemical compound [Br-].CCCCCCCCCCCCCCCC[N+](C)(C)C LZZYPRNAOMGNLH-UHFFFAOYSA-M 0.000 description 2
- IELOKBJPULMYRW-NJQVLOCASA-N D-alpha-Tocopheryl Acid Succinate Chemical compound OC(=O)CCC(=O)OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C IELOKBJPULMYRW-NJQVLOCASA-N 0.000 description 2
- 241000305071 Enterobacterales Species 0.000 description 2
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 2
- 238000000729 Fisher's exact test Methods 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- 108010070675 Glutathione transferase Proteins 0.000 description 2
- 102000005720 Glutathione transferase Human genes 0.000 description 2
- JZNWSCPGTDBMEW-UHFFFAOYSA-N Glycerophosphorylethanolamin Natural products NCCOP(O)(=O)OCC(O)CO JZNWSCPGTDBMEW-UHFFFAOYSA-N 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 208000019637 Infantile Diarrhea Diseases 0.000 description 2
- 229930010555 Inosine Natural products 0.000 description 2
- UGQMRVRMYYASKQ-KQYNXXCUSA-N Inosine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(O)=C2N=C1 UGQMRVRMYYASKQ-KQYNXXCUSA-N 0.000 description 2
- 102100037850 Interferon gamma Human genes 0.000 description 2
- 108090000174 Interleukin-10 Proteins 0.000 description 2
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 2
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 2
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 2
- 102000004083 Lymphotoxin-alpha Human genes 0.000 description 2
- 108090000542 Lymphotoxin-alpha Proteins 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- 102000005741 Metalloproteases Human genes 0.000 description 2
- 108010006035 Metalloproteases Proteins 0.000 description 2
- 108091061960 Naked DNA Proteins 0.000 description 2
- GOWLTLODGKPXMN-MEKRSRHXSA-N OM-174 Chemical compound O1[C@H](OP(O)(O)=O)[C@H](NC(=O)C[C@H](O)CCCCCCCCCCC)[C@@H](O)[C@H](O)[C@H]1CO[C@H]1[C@H](NC(=O)C[C@H](CCCCCCCCCCC)OC(=O)CCCCCCCCCCC)[C@@H](O)[C@H](OP(O)(O)=O)[C@@H](CO)O1 GOWLTLODGKPXMN-MEKRSRHXSA-N 0.000 description 2
- 102000035195 Peptidases Human genes 0.000 description 2
- 108091093037 Peptide nucleic acid Proteins 0.000 description 2
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 2
- GOOHAUXETOMSMM-UHFFFAOYSA-N Propylene oxide Chemical compound CC1CO1 GOOHAUXETOMSMM-UHFFFAOYSA-N 0.000 description 2
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 2
- 241000219287 Saponaria Species 0.000 description 2
- 241000710960 Sindbis virus Species 0.000 description 2
- NWGKJDSIEKMTRX-AAZCQSIUSA-N Sorbitan monooleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O NWGKJDSIEKMTRX-AAZCQSIUSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- 238000003917 TEM image Methods 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- 239000007983 Tris buffer Substances 0.000 description 2
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 2
- 206010046865 Vaccinia virus infection Diseases 0.000 description 2
- 108010067390 Viral Proteins Proteins 0.000 description 2
- 229930003427 Vitamin E Natural products 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- 230000000274 adsorptive effect Effects 0.000 description 2
- 238000012382 advanced drug delivery Methods 0.000 description 2
- 238000001042 affinity chromatography Methods 0.000 description 2
- 150000005215 alkyl ethers Chemical class 0.000 description 2
- NWMHDZMRVUOQGL-CZEIJOLGSA-N almurtide Chemical compound OC(=O)CC[C@H](C(N)=O)NC(=O)[C@H](C)NC(=O)CO[C@@H]([C@H](O)[C@H](O)CO)[C@@H](NC(C)=O)C=O NWMHDZMRVUOQGL-CZEIJOLGSA-N 0.000 description 2
- 230000000692 anti-sense effect Effects 0.000 description 2
- 230000005875 antibody response Effects 0.000 description 2
- 230000027455 binding Effects 0.000 description 2
- 229920001400 block copolymer Polymers 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 229910000389 calcium phosphate Inorganic materials 0.000 description 2
- 235000011010 calcium phosphates Nutrition 0.000 description 2
- 159000000007 calcium salts Chemical class 0.000 description 2
- 235000013877 carbamide Nutrition 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 125000002091 cationic group Chemical group 0.000 description 2
- AXTGOJVKRHFYBT-UHFFFAOYSA-N causarine Natural products OC1C(O)CN2C(CO)C(O)C(O)C21 AXTGOJVKRHFYBT-UHFFFAOYSA-N 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 235000013339 cereals Nutrition 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 239000007979 citrate buffer Substances 0.000 description 2
- 239000002299 complementary DNA Substances 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 239000012228 culture supernatant Substances 0.000 description 2
- 229940099418 d- alpha-tocopherol succinate Drugs 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 2
- 229940042396 direct acting antivirals thiosemicarbazones Drugs 0.000 description 2
- 239000003623 enhancer Substances 0.000 description 2
- 229940013317 fish oils Drugs 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 230000004927 fusion Effects 0.000 description 2
- 108020001507 fusion proteins Proteins 0.000 description 2
- 102000037865 fusion proteins Human genes 0.000 description 2
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 2
- 238000010353 genetic engineering Methods 0.000 description 2
- 229930182470 glycoside Natural products 0.000 description 2
- 239000011544 gradient gel Substances 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 210000002443 helper t lymphocyte Anatomy 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- HOPZBJPSUKPLDT-UHFFFAOYSA-N imidazo[4,5-h]quinolin-2-one Chemical class C1=CN=C2C3=NC(=O)N=C3C=CC2=C1 HOPZBJPSUKPLDT-UHFFFAOYSA-N 0.000 description 2
- DOUYETYNHWVLEO-UHFFFAOYSA-N imiquimod Chemical compound C1=CC=CC2=C3N(CC(C)C)C=NC3=C(N)N=C21 DOUYETYNHWVLEO-UHFFFAOYSA-N 0.000 description 2
- 230000036737 immune function Effects 0.000 description 2
- 210000000987 immune system Anatomy 0.000 description 2
- 230000004957 immunoregulator effect Effects 0.000 description 2
- 230000001976 improved effect Effects 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 229960003786 inosine Drugs 0.000 description 2
- JYJIGFIDKWBXDU-MNNPPOADSA-N inulin Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)OC[C@]1(OC[C@]2(OC[C@]3(OC[C@]4(OC[C@]5(OC[C@]6(OC[C@]7(OC[C@]8(OC[C@]9(OC[C@]%10(OC[C@]%11(OC[C@]%12(OC[C@]%13(OC[C@]%14(OC[C@]%15(OC[C@]%16(OC[C@]%17(OC[C@]%18(OC[C@]%19(OC[C@]%20(OC[C@]%21(OC[C@]%22(OC[C@]%23(OC[C@]%24(OC[C@]%25(OC[C@]%26(OC[C@]%27(OC[C@]%28(OC[C@]%29(OC[C@]%30(OC[C@]%31(OC[C@]%32(OC[C@]%33(OC[C@]%34(OC[C@]%35(OC[C@]%36(O[C@@H]%37[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O%37)O)[C@H]([C@H](O)[C@@H](CO)O%36)O)[C@H]([C@H](O)[C@@H](CO)O%35)O)[C@H]([C@H](O)[C@@H](CO)O%34)O)[C@H]([C@H](O)[C@@H](CO)O%33)O)[C@H]([C@H](O)[C@@H](CO)O%32)O)[C@H]([C@H](O)[C@@H](CO)O%31)O)[C@H]([C@H](O)[C@@H](CO)O%30)O)[C@H]([C@H](O)[C@@H](CO)O%29)O)[C@H]([C@H](O)[C@@H](CO)O%28)O)[C@H]([C@H](O)[C@@H](CO)O%27)O)[C@H]([C@H](O)[C@@H](CO)O%26)O)[C@H]([C@H](O)[C@@H](CO)O%25)O)[C@H]([C@H](O)[C@@H](CO)O%24)O)[C@H]([C@H](O)[C@@H](CO)O%23)O)[C@H]([C@H](O)[C@@H](CO)O%22)O)[C@H]([C@H](O)[C@@H](CO)O%21)O)[C@H]([C@H](O)[C@@H](CO)O%20)O)[C@H]([C@H](O)[C@@H](CO)O%19)O)[C@H]([C@H](O)[C@@H](CO)O%18)O)[C@H]([C@H](O)[C@@H](CO)O%17)O)[C@H]([C@H](O)[C@@H](CO)O%16)O)[C@H]([C@H](O)[C@@H](CO)O%15)O)[C@H]([C@H](O)[C@@H](CO)O%14)O)[C@H]([C@H](O)[C@@H](CO)O%13)O)[C@H]([C@H](O)[C@@H](CO)O%12)O)[C@H]([C@H](O)[C@@H](CO)O%11)O)[C@H]([C@H](O)[C@@H](CO)O%10)O)[C@H]([C@H](O)[C@@H](CO)O9)O)[C@H]([C@H](O)[C@@H](CO)O8)O)[C@H]([C@H](O)[C@@H](CO)O7)O)[C@H]([C@H](O)[C@@H](CO)O6)O)[C@H]([C@H](O)[C@@H](CO)O5)O)[C@H]([C@H](O)[C@@H](CO)O4)O)[C@H]([C@H](O)[C@@H](CO)O3)O)[C@H]([C@H](O)[C@@H](CO)O2)O)[C@@H](O)[C@H](O)[C@@H](CO)O1 JYJIGFIDKWBXDU-MNNPPOADSA-N 0.000 description 2
- 230000005865 ionizing radiation Effects 0.000 description 2
- 231100000518 lethal Toxicity 0.000 description 2
- 230000001665 lethal effect Effects 0.000 description 2
- 230000029226 lipidation Effects 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 210000001806 memory b lymphocyte Anatomy 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 125000001446 muramyl group Chemical group N[C@@H](C=O)[C@@H](O[C@@H](C(=O)*)C)[C@H](O)[C@H](O)CO 0.000 description 2
- GKTNLYAAZKKMTQ-UHFFFAOYSA-N n-[bis(dimethylamino)phosphinimyl]-n-methylmethanamine Chemical compound CN(C)P(=N)(N(C)C)N(C)C GKTNLYAAZKKMTQ-UHFFFAOYSA-N 0.000 description 2
- 210000004897 n-terminal region Anatomy 0.000 description 2
- 239000013642 negative control Substances 0.000 description 2
- 235000019488 nut oil Nutrition 0.000 description 2
- 229960005030 other vaccine in atc Drugs 0.000 description 2
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 2
- 150000008104 phosphatidylethanolamines Chemical class 0.000 description 2
- 229920001606 poly(lactic acid-co-glycolic acid) Polymers 0.000 description 2
- 230000008488 polyadenylation Effects 0.000 description 2
- 229920002851 polycationic polymer Polymers 0.000 description 2
- 229920002503 polyoxyethylene-polyoxypropylene Polymers 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 230000002335 preservative effect Effects 0.000 description 2
- 150000003230 pyrimidines Chemical class 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 238000010188 recombinant method Methods 0.000 description 2
- 230000003362 replicative effect Effects 0.000 description 2
- 229950010550 resiquimod Drugs 0.000 description 2
- 108091092562 ribozyme Proteins 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 239000010686 shark liver oil Substances 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 238000007619 statistical method Methods 0.000 description 2
- 230000000638 stimulation Effects 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 2
- 239000013589 supplement Substances 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- 150000003505 terpenes Chemical class 0.000 description 2
- 150000003584 thiosemicarbazones Chemical class 0.000 description 2
- 230000002103 transcriptional effect Effects 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- 238000012250 transgenic expression Methods 0.000 description 2
- XETCRXVKJHBPMK-MJSODCSWSA-N trehalose 6,6'-dimycolate Chemical compound C([C@@H]1[C@H]([C@H](O)[C@@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](COC(=O)C(CCCCCCCCCCC3C(C3)CCCCCCCCCCCCCCCCCC)C(O)CCCCCCCCCCCCCCCCCCCCCCCCC)O2)O)O1)O)OC(=O)C(C(O)CCCCCCCCCCCCCCCCCCCCCCCCC)CCCCCCCCCCC1CC1CCCCCCCCCCCCCCCCCC XETCRXVKJHBPMK-MJSODCSWSA-N 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 2
- 150000003672 ureas Chemical class 0.000 description 2
- 208000007089 vaccinia Diseases 0.000 description 2
- 229940046009 vitamin E Drugs 0.000 description 2
- 235000019165 vitamin E Nutrition 0.000 description 2
- 239000011709 vitamin E Substances 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- 239000002076 α-tocopherol Substances 0.000 description 2
- AXTGOJVKRHFYBT-PPOMMRCSSA-N (1r,2r,3r,6s,7r,8r)-3-(hydroxymethyl)-2,3,5,6,7,8-hexahydro-1h-pyrrolizine-1,2,6,7-tetrol Chemical compound O[C@H]1[C@@H](O)CN2[C@H](CO)[C@@H](O)[C@H](O)[C@H]21 AXTGOJVKRHFYBT-PPOMMRCSSA-N 0.000 description 1
- AXTGOJVKRHFYBT-JKVYXWIHSA-N (1r,2r,3s,6s,7s,8r)-3-(hydroxymethyl)-2,3,5,6,7,8-hexahydro-1h-pyrrolizine-1,2,6,7-tetrol Chemical compound O[C@@H]1[C@@H](O)CN2[C@@H](CO)[C@@H](O)[C@H](O)[C@H]21 AXTGOJVKRHFYBT-JKVYXWIHSA-N 0.000 description 1
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 1
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 description 1
- YHQZWWDVLJPRIF-JLHRHDQISA-N (4R)-4-[[(2S,3R)-2-[acetyl-[(3R,4R,5S,6R)-3-amino-4-[(1R)-1-carboxyethoxy]-5-hydroxy-6-(hydroxymethyl)oxan-2-yl]amino]-3-hydroxybutanoyl]amino]-5-amino-5-oxopentanoic acid Chemical compound C(C)(=O)N([C@@H]([C@H](O)C)C(=O)N[C@H](CCC(=O)O)C(N)=O)C1[C@H](N)[C@@H](O[C@@H](C(=O)O)C)[C@H](O)[C@H](O1)CO YHQZWWDVLJPRIF-JLHRHDQISA-N 0.000 description 1
- UGXDVELKRYZPDM-XLXQKPBQSA-N (4r)-4-[[(2s,3r)-2-[[(2r)-2-[(2r,3r,4r,5r)-2-acetamido-4,5,6-trihydroxy-1-oxohexan-3-yl]oxypropanoyl]amino]-3-hydroxybutanoyl]amino]-5-amino-5-oxopentanoic acid Chemical compound OC(=O)CC[C@H](C(N)=O)NC(=O)[C@H]([C@H](O)C)NC(=O)[C@@H](C)O[C@@H]([C@H](O)[C@H](O)CO)[C@@H](NC(C)=O)C=O UGXDVELKRYZPDM-XLXQKPBQSA-N 0.000 description 1
- DNIAPMSPPWPWGF-GSVOUGTGSA-N (R)-(-)-Propylene glycol Chemical compound C[C@@H](O)CO DNIAPMSPPWPWGF-GSVOUGTGSA-N 0.000 description 1
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 1
- PORPENFLTBBHSG-MGBGTMOVSA-N 1,2-dihexadecanoyl-sn-glycerol-3-phosphate Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(O)=O)OC(=O)CCCCCCCCCCCCCCC PORPENFLTBBHSG-MGBGTMOVSA-N 0.000 description 1
- TZCPCKNHXULUIY-RGULYWFUSA-N 1,2-distearoyl-sn-glycero-3-phosphoserine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OC[C@H](N)C(O)=O)OC(=O)CCCCCCCCCCCCCCCCC TZCPCKNHXULUIY-RGULYWFUSA-N 0.000 description 1
- PXFBZOLANLWPMH-UHFFFAOYSA-N 16-Epiaffinine Natural products C1C(C2=CC=CC=C2N2)=C2C(=O)CC2C(=CC)CN(C)C1C2CO PXFBZOLANLWPMH-UHFFFAOYSA-N 0.000 description 1
- HCQFIHRPZXGXCK-UHFFFAOYSA-N 1h-benzimidazol-2-amine;1h-quinolin-2-one Chemical compound C1=CC=C2NC(N)=NC2=C1.C1=CC=C2NC(=O)C=CC2=C1 HCQFIHRPZXGXCK-UHFFFAOYSA-N 0.000 description 1
- FKMHSNTVILORFA-UHFFFAOYSA-N 2-[2-(2-dodecoxyethoxy)ethoxy]ethanol Chemical compound CCCCCCCCCCCCOCCOCCOCCO FKMHSNTVILORFA-UHFFFAOYSA-N 0.000 description 1
- GOJUJUVQIVIZAV-UHFFFAOYSA-N 2-amino-4,6-dichloropyrimidine-5-carbaldehyde Chemical group NC1=NC(Cl)=C(C=O)C(Cl)=N1 GOJUJUVQIVIZAV-UHFFFAOYSA-N 0.000 description 1
- PFCLMNDDPTZJHQ-XLPZGREQSA-N 2-amino-7-[(2r,4s,5r)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-1h-pyrrolo[2,3-d]pyrimidin-4-one Chemical compound C1=CC=2C(=O)NC(N)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PFCLMNDDPTZJHQ-XLPZGREQSA-N 0.000 description 1
- VDCRFBBZFHHYGT-IOSLPCCCSA-N 2-amino-9-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-enyl-3h-purine-6,8-dione Chemical compound O=C1N(CC=C)C=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O VDCRFBBZFHHYGT-IOSLPCCCSA-N 0.000 description 1
- QCDWFXQBSFUVSP-UHFFFAOYSA-N 2-phenoxyethanol Chemical compound OCCOC1=CC=CC=C1 QCDWFXQBSFUVSP-UHFFFAOYSA-N 0.000 description 1
- LYFYWXLKKQIOKO-UHFFFAOYSA-N 3,3-diaminopentan-1-ol Chemical compound CCC(N)(N)CCO LYFYWXLKKQIOKO-UHFFFAOYSA-N 0.000 description 1
- RHKWIGHJGOEUSM-UHFFFAOYSA-N 3h-imidazo[4,5-h]quinoline Chemical class C1=CN=C2C(N=CN3)=C3C=CC2=C1 RHKWIGHJGOEUSM-UHFFFAOYSA-N 0.000 description 1
- XZIIFPSPUDAGJM-UHFFFAOYSA-N 6-chloro-2-n,2-n-diethylpyrimidine-2,4-diamine Chemical compound CCN(CC)C1=NC(N)=CC(Cl)=N1 XZIIFPSPUDAGJM-UHFFFAOYSA-N 0.000 description 1
- PXBWLHQLSCMJEM-IOSLPCCCSA-N 9-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)-2-methyloxolan-2-yl]-3h-purin-6-one Chemical compound C1=NC2=C(O)N=CN=C2N1[C@]1(C)O[C@H](CO)[C@@H](O)[C@H]1O PXBWLHQLSCMJEM-IOSLPCCCSA-N 0.000 description 1
- 208000030507 AIDS Diseases 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 108010042708 Acetylmuramyl-Alanyl-Isoglutamine Proteins 0.000 description 1
- 241000710929 Alphavirus Species 0.000 description 1
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 1
- 235000007319 Avena orientalis Nutrition 0.000 description 1
- 244000075850 Avena orientalis Species 0.000 description 1
- 241000271566 Aves Species 0.000 description 1
- 244000063299 Bacillus subtilis Species 0.000 description 1
- 235000014469 Bacillus subtilis Nutrition 0.000 description 1
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 238000009010 Bradford assay Methods 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 102100021935 C-C motif chemokine 26 Human genes 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 108090000565 Capsid Proteins Proteins 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 102100023321 Ceruloplasmin Human genes 0.000 description 1
- 229920001661 Chitosan Polymers 0.000 description 1
- 206010008631 Cholera Diseases 0.000 description 1
- 208000017667 Chronic Disease Diseases 0.000 description 1
- 101710094648 Coat protein Proteins 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- 108010060123 Conjugate Vaccines Proteins 0.000 description 1
- AERBNCYCJBRYDG-UHFFFAOYSA-N D-ribo-phytosphingosine Natural products CCCCCCCCCCCCCCC(O)C(O)C(N)CO AERBNCYCJBRYDG-UHFFFAOYSA-N 0.000 description 1
- 102000053602 DNA Human genes 0.000 description 1
- 238000000018 DNA microarray Methods 0.000 description 1
- 239000003155 DNA primer Substances 0.000 description 1
- 238000011238 DNA vaccination Methods 0.000 description 1
- 229940032024 DPT vaccine Drugs 0.000 description 1
- 208000005156 Dehydration Diseases 0.000 description 1
- 241000702421 Dependoparvovirus Species 0.000 description 1
- 101100041687 Drosophila melanogaster san gene Proteins 0.000 description 1
- 229920005682 EO-PO block copolymer Polymers 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 101710146739 Enterotoxin Proteins 0.000 description 1
- 206010066919 Epidemic polyarthritis Diseases 0.000 description 1
- 244000140063 Eragrostis abyssinica Species 0.000 description 1
- 235000014966 Eragrostis abyssinica Nutrition 0.000 description 1
- 241001337814 Erysiphe glycines Species 0.000 description 1
- 241000206602 Eukaryota Species 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 241000710198 Foot-and-mouth disease virus Species 0.000 description 1
- 208000000666 Fowlpox Diseases 0.000 description 1
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 1
- 241000287828 Gallus gallus Species 0.000 description 1
- 108700007698 Genetic Terminator Regions Proteins 0.000 description 1
- 108010068370 Glutens Proteins 0.000 description 1
- ZWZWYGMENQVNFU-UHFFFAOYSA-N Glycerophosphorylserin Natural products OC(=O)C(N)COP(O)(=O)OCC(O)CO ZWZWYGMENQVNFU-UHFFFAOYSA-N 0.000 description 1
- 102100021181 Golgi phosphoprotein 3 Human genes 0.000 description 1
- 241000700721 Hepatitis B virus Species 0.000 description 1
- 229940124872 Hepatitis B virus vaccine Drugs 0.000 description 1
- 241000724675 Hepatitis E virus Species 0.000 description 1
- 108700005492 His(29)- cholera holotoxin Proteins 0.000 description 1
- 108091006054 His-tagged proteins Proteins 0.000 description 1
- 102000018713 Histocompatibility Antigens Class II Human genes 0.000 description 1
- 108010027412 Histocompatibility Antigens Class II Proteins 0.000 description 1
- 101000897493 Homo sapiens C-C motif chemokine 26 Proteins 0.000 description 1
- 101000957351 Homo sapiens Myc-associated zinc finger protein Proteins 0.000 description 1
- 101000669447 Homo sapiens Toll-like receptor 4 Proteins 0.000 description 1
- 102000008100 Human Serum Albumin Human genes 0.000 description 1
- 108091006905 Human Serum Albumin Proteins 0.000 description 1
- 241000701806 Human papillomavirus Species 0.000 description 1
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical class C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 102000008070 Interferon-gamma Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010002352 Interleukin-1 Proteins 0.000 description 1
- 108010002586 Interleukin-7 Proteins 0.000 description 1
- 108010063738 Interleukins Proteins 0.000 description 1
- 102000015696 Interleukins Human genes 0.000 description 1
- 229920001202 Inulin Polymers 0.000 description 1
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 1
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 1
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- 102000003960 Ligases Human genes 0.000 description 1
- 108090000364 Ligases Proteins 0.000 description 1
- 102000004895 Lipoproteins Human genes 0.000 description 1
- 108090001030 Lipoproteins Proteins 0.000 description 1
- SJNZALDHDUYDBU-IHRRRGAJSA-N Lys-Arg-Lys Chemical compound NCCCC[C@H](N)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CCCCN)C(O)=O SJNZALDHDUYDBU-IHRRRGAJSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 102000043129 MHC class I family Human genes 0.000 description 1
- 108091054437 MHC class I family Proteins 0.000 description 1
- 102000043131 MHC class II family Human genes 0.000 description 1
- 108091054438 MHC class II family Proteins 0.000 description 1
- 102000007651 Macrophage Colony-Stimulating Factor Human genes 0.000 description 1
- 108010046938 Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 101710125418 Major capsid protein Proteins 0.000 description 1
- 101710175625 Maltose/maltodextrin-binding periplasmic protein Proteins 0.000 description 1
- 241000712079 Measles morbillivirus Species 0.000 description 1
- 201000009906 Meningitis Diseases 0.000 description 1
- 241001092142 Molina Species 0.000 description 1
- 208000019209 Monosomy 22 Diseases 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 102100038750 Myc-associated zinc finger protein Human genes 0.000 description 1
- GUVMFDICMFQHSZ-UHFFFAOYSA-N N-(1-aminoethenyl)-1-[4-[[5-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-3-[[5-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-3-[[5-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-3-[[5-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-3-[[5-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[hydroxy-[[3-[hydroxy-[[3-hydroxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy]phosphinothioyl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy]phosphinothioyl]oxyoxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxyoxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxyoxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxyoxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxyoxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxyoxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(2-amino-6-oxo-1H-purin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(2-amino-6-oxo-1H-purin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxyoxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxyoxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-[[[2-[[[2-[[[5-(2-amino-6-oxo-1H-purin-9-yl)-2-[[[5-(4-amino-2-oxopyrimidin-1-yl)-2-[[hydroxy-[2-(hydroxymethyl)-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-3-yl]oxyphosphinothioyl]oxymethyl]oxolan-3-yl]oxy-hydroxyphosphinothioyl]oxymethyl]oxolan-3-yl]oxy-hydroxyphosphinothioyl]oxymethyl]-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-3-yl]oxy-hydroxyphosphinothioyl]oxymethyl]-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-3-yl]oxy-hydroxyphosphinothioyl]oxymethyl]oxolan-2-yl]-5-methylimidazole-4-carboxamide Chemical group CC1=C(C(=O)NC(N)=C)N=CN1C1OC(COP(O)(=S)OC2C(OC(C2)N2C(N=C(N)C=C2)=O)COP(O)(=S)OC2C(OC(C2)N2C(NC(=O)C(C)=C2)=O)COP(O)(=S)OC2C(OC(C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP(O)(=S)OC2C(OC(C2)N2C(N=C(N)C=C2)=O)COP(O)(=S)OC2C(OC(C2)N2C(NC(=O)C(C)=C2)=O)CO)C(OP(O)(=S)OCC2C(CC(O2)N2C(N=C(N)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(N=C(N)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(NC(=O)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(NC(=O)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP(O)(=S)OCC2C(CC(O2)N2C(NC(=O)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(N=C(N)C=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP(O)(=S)OCC2C(CC(O2)N2C(N=C(N)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(N=C(N)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(N=C(N)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(N=C(N)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP(O)(=S)OCC2C(CC(O2)N2C(NC(=O)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(N=C(N)C=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP(O)(=S)OCC2C(CC(O2)N2C(NC(=O)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(NC(=O)C(C)=C2)=O)O)C1 GUVMFDICMFQHSZ-UHFFFAOYSA-N 0.000 description 1
- 108700015872 N-acetyl-nor-muramyl-L-alanyl-D-isoglutamine Proteins 0.000 description 1
- 241000588654 Neisseria cinerea Species 0.000 description 1
- 241000588649 Neisseria lactamica Species 0.000 description 1
- 208000006816 Neonatal Sepsis Diseases 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 102000048850 Neoplasm Genes Human genes 0.000 description 1
- 108700019961 Neoplasm Genes Proteins 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- 238000000636 Northern blotting Methods 0.000 description 1
- 241000714209 Norwalk virus Species 0.000 description 1
- 101710163270 Nuclease Proteins 0.000 description 1
- 101710141454 Nucleoprotein Proteins 0.000 description 1
- 108010038807 Oligopeptides Proteins 0.000 description 1
- 102000015636 Oligopeptides Human genes 0.000 description 1
- 240000007594 Oryza sativa Species 0.000 description 1
- 235000007164 Oryza sativa Nutrition 0.000 description 1
- 206010031252 Osteomyelitis Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 201000005702 Pertussis Diseases 0.000 description 1
- 244000046052 Phaseolus vulgaris Species 0.000 description 1
- 235000010627 Phaseolus vulgaris Nutrition 0.000 description 1
- 206010035664 Pneumonia Diseases 0.000 description 1
- 241000276498 Pollachius virens Species 0.000 description 1
- 229920002732 Polyanhydride Polymers 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 101710083689 Probable capsid protein Proteins 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 241000725643 Respiratory syncytial virus Species 0.000 description 1
- 108091028664 Ribonucleotide Proteins 0.000 description 1
- 241000710942 Ross River virus Species 0.000 description 1
- 241000702670 Rotavirus Species 0.000 description 1
- 235000019485 Safflower oil Nutrition 0.000 description 1
- 241000293869 Salmonella enterica subsp. enterica serovar Typhimurium Species 0.000 description 1
- 241000209056 Secale Species 0.000 description 1
- 235000007238 Secale cereale Nutrition 0.000 description 1
- 241000710961 Semliki Forest virus Species 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 244000000231 Sesamum indicum Species 0.000 description 1
- 235000003434 Sesamum indicum Nutrition 0.000 description 1
- 241000144290 Sigmodon hispidus Species 0.000 description 1
- 241000580858 Simian-Human immunodeficiency virus Species 0.000 description 1
- 244000044822 Simmondsia californica Species 0.000 description 1
- 235000004433 Simmondsia californica Nutrition 0.000 description 1
- 108020004682 Single-Stranded DNA Proteins 0.000 description 1
- 240000002493 Smilax officinalis Species 0.000 description 1
- 235000008981 Smilax officinalis Nutrition 0.000 description 1
- 229920002125 Sokalan® Polymers 0.000 description 1
- 244000061456 Solanum tuberosum Species 0.000 description 1
- 235000002595 Solanum tuberosum Nutrition 0.000 description 1
- 238000002105 Southern blotting Methods 0.000 description 1
- 101001060868 Strawberry mild yellow edge-associated virus Helicase Proteins 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 206010051379 Systemic Inflammatory Response Syndrome Diseases 0.000 description 1
- 108091008874 T cell receptors Proteins 0.000 description 1
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 1
- WPMWEFXCIYCJSA-UHFFFAOYSA-N Tetraethylene glycol monododecyl ether Chemical compound CCCCCCCCCCCCOCCOCCOCCOCCO WPMWEFXCIYCJSA-UHFFFAOYSA-N 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-N Thiophosphoric acid Chemical class OP(O)(S)=O RYYWUUFWQRZTIU-UHFFFAOYSA-N 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- 208000034841 Thrombotic Microangiopathies Diseases 0.000 description 1
- 102000008235 Toll-Like Receptor 9 Human genes 0.000 description 1
- 108010060818 Toll-Like Receptor 9 Proteins 0.000 description 1
- 102100039360 Toll-like receptor 4 Human genes 0.000 description 1
- 235000019714 Triticale Nutrition 0.000 description 1
- 235000021307 Triticum Nutrition 0.000 description 1
- 244000098338 Triticum aestivum Species 0.000 description 1
- VQQVWGVXDIPORV-UHFFFAOYSA-N Tryptanthrine Natural products C1=CC=C2C(=O)N3C4=CC=CC=C4C(=O)C3=NC2=C1 VQQVWGVXDIPORV-UHFFFAOYSA-N 0.000 description 1
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 1
- 241000710959 Venezuelan equine encephalitis virus Species 0.000 description 1
- ZBNRGEMZNWHCGA-PDKVEDEMSA-N [(2r)-2-[(2r,3r,4s)-3,4-bis[[(z)-octadec-9-enoyl]oxy]oxolan-2-yl]-2-hydroxyethyl] (z)-octadec-9-enoate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](O)[C@H]1OC[C@H](OC(=O)CCCCCCC\C=C/CCCCCCCC)[C@H]1OC(=O)CCCCCCC\C=C/CCCCCCCC ZBNRGEMZNWHCGA-PDKVEDEMSA-N 0.000 description 1
- ZWELIJXAKMASLK-UGKPPGOTSA-N [(2r,3r,4r,5r)-4-acetyloxy-5-(5-amino-2-oxo-[1,3]thiazolo[4,5-d]pyrimidin-3-yl)-2-(hydroxymethyl)oxolan-3-yl] acetate Chemical compound CC(=O)O[C@@H]1[C@H](OC(=O)C)[C@@H](CO)O[C@H]1N1C(=O)SC2=CN=C(N)N=C21 ZWELIJXAKMASLK-UGKPPGOTSA-N 0.000 description 1
- NKVLDFAVEWLOCX-GUSKIFEASA-N [(2s,3r,4s,5r,6r)-3-[(2s,3r,4s,5r,6s)-5-[(2s,3r,4s,5r)-4-[(2s,3r,4r)-3,4-dihydroxy-4-(hydroxymethyl)oxolan-2-yl]oxy-3,5-dihydroxyoxan-2-yl]oxy-3,4-dihydroxy-6-methyloxan-2-yl]oxy-4,5-dihydroxy-6-methyloxan-2-yl] (4ar,5r,6as,6br,9s,10s,12ar)-10-[(2r,3r,4s, Chemical compound O([C@H]1[C@H](O)CO[C@H]([C@@H]1O)O[C@H]1[C@H](C)O[C@H]([C@@H]([C@@H]1O)O)O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](C)O[C@H]1OC(=O)[C@]12CCC(C)(C)CC1C1=CCC3[C@@]([C@@]1(C[C@H]2O)C)(C)CCC1[C@]3(C)CC[C@@H]([C@@]1(C)C=O)O[C@@H]1O[C@@H]([C@H]([C@H](O[C@H]2[C@@H]([C@@H](O)[C@H](O)CO2)O)[C@H]1O[C@H]1[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O1)O)O)C(=O)NCCCCCCCCCCCC)[C@@H]1OC[C@](O)(CO)[C@H]1O NKVLDFAVEWLOCX-GUSKIFEASA-N 0.000 description 1
- ATBOMIWRCZXYSZ-XZBBILGWSA-N [1-[2,3-dihydroxypropoxy(hydroxy)phosphoryl]oxy-3-hexadecanoyloxypropan-2-yl] (9e,12e)-octadeca-9,12-dienoate Chemical compound CCCCCCCCCCCCCCCC(=O)OCC(COP(O)(=O)OCC(O)CO)OC(=O)CCCCCCC\C=C\C\C=C\CCCCC ATBOMIWRCZXYSZ-XZBBILGWSA-N 0.000 description 1
- 230000021736 acetylation Effects 0.000 description 1
- 238000006640 acetylation reaction Methods 0.000 description 1
- 101150067771 acfD gene Proteins 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000001464 adherent effect Effects 0.000 description 1
- 230000000240 adjuvant effect Effects 0.000 description 1
- 238000001261 affinity purification Methods 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- IAJILQKETJEXLJ-QTBDOELSSA-N aldehydo-D-glucuronic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-QTBDOELSSA-N 0.000 description 1
- 125000003342 alkenyl group Chemical group 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- 229940087168 alpha tocopherol Drugs 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- PYKYMHQGRFAEBM-UHFFFAOYSA-N anthraquinone Natural products CCC(=O)c1c(O)c2C(=O)C3C(C=CC=C3O)C(=O)c2cc1CC(=O)OC PYKYMHQGRFAEBM-UHFFFAOYSA-N 0.000 description 1
- 230000000840 anti-viral effect Effects 0.000 description 1
- 238000010913 antigen-directed enzyme pro-drug therapy Methods 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 239000000074 antisense oligonucleotide Substances 0.000 description 1
- 238000012230 antisense oligonucleotides Methods 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 238000013528 artificial neural network Methods 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 229960001230 asparagine Drugs 0.000 description 1
- 235000009582 asparagine Nutrition 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 150000008366 benzophenones Chemical class 0.000 description 1
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 239000012620 biological material Substances 0.000 description 1
- 239000012472 biological sample Substances 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- OWMVSZAMULFTJU-UHFFFAOYSA-N bis-tris Chemical compound OCCN(CCO)C(CO)(CO)CO OWMVSZAMULFTJU-UHFFFAOYSA-N 0.000 description 1
- 238000006664 bond formation reaction Methods 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 108010006025 bovine growth hormone Proteins 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 210000004970 cd4 cell Anatomy 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 210000002421 cell wall Anatomy 0.000 description 1
- 230000007969 cellular immunity Effects 0.000 description 1
- 230000036755 cellular response Effects 0.000 description 1
- 210000003850 cellular structure Anatomy 0.000 description 1
- 239000004464 cereal grain Substances 0.000 description 1
- 230000005591 charge neutralization Effects 0.000 description 1
- 150000001841 cholesterols Chemical class 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 235000012716 cod liver oil Nutrition 0.000 description 1
- 239000003026 cod liver oil Substances 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 229940031670 conjugate vaccine Drugs 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 238000012937 correction Methods 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 239000002577 cryoprotective agent Substances 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 1
- 210000004292 cytoskeleton Anatomy 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 239000005547 deoxyribonucleotide Substances 0.000 description 1
- 125000002637 deoxyribonucleotide group Chemical group 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 229940008099 dimethicone Drugs 0.000 description 1
- 239000004205 dimethyl polysiloxane Substances 0.000 description 1
- 235000013870 dimethyl polysiloxane Nutrition 0.000 description 1
- 229960005097 diphtheria vaccines Drugs 0.000 description 1
- ZGSPNIOCEDOHGS-UHFFFAOYSA-L disodium [3-[2,3-di(octadeca-9,12-dienoyloxy)propoxy-oxidophosphoryl]oxy-2-hydroxypropyl] 2,3-di(octadeca-9,12-dienoyloxy)propyl phosphate Chemical compound [Na+].[Na+].CCCCCC=CCC=CCCCCCCCC(=O)OCC(OC(=O)CCCCCCCC=CCC=CCCCCC)COP([O-])(=O)OCC(O)COP([O-])(=O)OCC(OC(=O)CCCCCCCC=CCC=CCCCCC)COC(=O)CCCCCCCC=CCC=CCCCCC ZGSPNIOCEDOHGS-UHFFFAOYSA-L 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 229910000397 disodium phosphate Inorganic materials 0.000 description 1
- 235000019800 disodium phosphate Nutrition 0.000 description 1
- NLEBIOOXCVAHBD-QKMCSOCLSA-N dodecyl beta-D-maltoside Chemical compound O[C@@H]1[C@@H](O)[C@H](OCCCCCCCCCCCC)O[C@H](CO)[C@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 NLEBIOOXCVAHBD-QKMCSOCLSA-N 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 239000012636 effector Substances 0.000 description 1
- 206010014665 endocarditis Diseases 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 239000000147 enterotoxin Substances 0.000 description 1
- 231100000655 enterotoxin Toxicity 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 150000002195 fatty ethers Chemical class 0.000 description 1
- 239000013020 final formulation Substances 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 244000078673 foodborn pathogen Species 0.000 description 1
- 231100000221 frame shift mutation induction Toxicity 0.000 description 1
- 230000037433 frameshift Effects 0.000 description 1
- 238000002523 gelfiltration Methods 0.000 description 1
- 238000011239 genetic vaccination Methods 0.000 description 1
- 229940097043 glucuronic acid Drugs 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 235000021312 gluten Nutrition 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 230000013595 glycosylation Effects 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 108010037896 heparin-binding hemagglutinin Proteins 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 125000000487 histidyl group Chemical group [H]N([H])C(C(=O)O*)C([H])([H])C1=C([H])N([H])C([H])=N1 0.000 description 1
- 229930186900 holotoxin Natural products 0.000 description 1
- 230000028996 humoral immune response Effects 0.000 description 1
- 230000004727 humoral immunity Effects 0.000 description 1
- 229920002674 hyaluronan Polymers 0.000 description 1
- 229960003160 hyaluronic acid Drugs 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- 229960002751 imiquimod Drugs 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 238000003119 immunoblot Methods 0.000 description 1
- 230000002998 immunogenetic effect Effects 0.000 description 1
- 230000016784 immunoglobulin production Effects 0.000 description 1
- 230000002584 immunomodulator Effects 0.000 description 1
- 239000003022 immunostimulating agent Substances 0.000 description 1
- 229940029583 inactivated polio vaccine Drugs 0.000 description 1
- 230000000415 inactivating effect Effects 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 238000011081 inoculation Methods 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 229960003130 interferon gamma Drugs 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 229940047122 interleukins Drugs 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 229940065638 intron a Drugs 0.000 description 1
- 229940029339 inulin Drugs 0.000 description 1
- JXDYKVIHCLTXOP-UHFFFAOYSA-N isatin Chemical class C1=CC=C2C(=O)C(=O)NC2=C1 JXDYKVIHCLTXOP-UHFFFAOYSA-N 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 229960000310 isoleucine Drugs 0.000 description 1
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 1
- 150000002545 isoxazoles Chemical class 0.000 description 1
- 238000005304 joining Methods 0.000 description 1
- 229940119170 jojoba wax Drugs 0.000 description 1
- KQYACACELNVFOY-UHFFFAOYSA-N kazuarin-6-O-alpha-D-glucoside Natural products C1N2C(CO)C(O)C(O)C2C(O)C1OC1OC(CO)C(O)C(O)C1O KQYACACELNVFOY-UHFFFAOYSA-N 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 230000021633 leukocyte mediated immunity Effects 0.000 description 1
- 229940059904 light mineral oil Drugs 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 230000005923 long-lasting effect Effects 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 229940041323 measles vaccine Drugs 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- 239000000693 micelle Substances 0.000 description 1
- 239000004005 microsphere Substances 0.000 description 1
- JMUHBNWAORSSBD-WKYWBUFDSA-N mifamurtide Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCCCCCCCCCC)COP(O)(=O)OCCNC(=O)[C@H](C)NC(=O)CC[C@H](C(N)=O)NC(=O)[C@H](C)NC(=O)[C@@H](C)O[C@H]1[C@H](O)[C@@H](CO)OC(O)[C@@H]1NC(C)=O JMUHBNWAORSSBD-WKYWBUFDSA-N 0.000 description 1
- 229960005225 mifamurtide Drugs 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 238000001823 molecular biology technique Methods 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 1
- 235000019796 monopotassium phosphate Nutrition 0.000 description 1
- DNIAPMSPPWPWGF-UHFFFAOYSA-N monopropylene glycol Natural products CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 210000003097 mucus Anatomy 0.000 description 1
- 229940095293 mumps vaccine Drugs 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 229920000847 nonoxynol Polymers 0.000 description 1
- 230000009871 nonspecific binding Effects 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- 239000010466 nut oil Substances 0.000 description 1
- 235000016709 nutrition Nutrition 0.000 description 1
- 230000035764 nutrition Effects 0.000 description 1
- 235000014571 nuts Nutrition 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-N octanoic acid Chemical compound CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 1
- 229940098514 octoxynol-9 Drugs 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 239000007935 oral tablet Substances 0.000 description 1
- 229940096978 oral tablet Drugs 0.000 description 1
- 125000000913 palmityl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- UNEIHNMKASENIG-UHFFFAOYSA-N para-chlorophenylpiperazine Chemical compound C1=CC(Cl)=CC=C1N1CCNCC1 UNEIHNMKASENIG-UHFFFAOYSA-N 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 230000007918 pathogenicity Effects 0.000 description 1
- 239000013610 patient sample Substances 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 208000008494 pericarditis Diseases 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 229940066827 pertussis vaccine Drugs 0.000 description 1
- 229960005323 phenoxyethanol Drugs 0.000 description 1
- 150000003905 phosphatidylinositols Chemical class 0.000 description 1
- 150000008300 phosphoramidites Chemical class 0.000 description 1
- PJNZPQUBCPKICU-UHFFFAOYSA-N phosphoric acid;potassium Chemical compound [K].OP(O)(O)=O PJNZPQUBCPKICU-UHFFFAOYSA-N 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- AERBNCYCJBRYDG-KSZLIROESA-N phytosphingosine Chemical compound CCCCCCCCCCCCCC[C@@H](O)[C@@H](O)[C@@H](N)CO AERBNCYCJBRYDG-KSZLIROESA-N 0.000 description 1
- 229940033329 phytosphingosine Drugs 0.000 description 1
- 229920000435 poly(dimethylsiloxane) Polymers 0.000 description 1
- 239000002745 poly(ortho ester) Substances 0.000 description 1
- 229920001610 polycaprolactone Polymers 0.000 description 1
- 239000004632 polycaprolactone Substances 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229920005617 polyoxidonium Polymers 0.000 description 1
- 229940051841 polyoxyethylene ether Drugs 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 229950008882 polysorbate Drugs 0.000 description 1
- 229940068977 polysorbate 20 Drugs 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 230000008092 positive effect Effects 0.000 description 1
- 239000001103 potassium chloride Substances 0.000 description 1
- 235000011164 potassium chloride Nutrition 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 244000144977 poultry Species 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- 201000007094 prostatitis Diseases 0.000 description 1
- 239000003223 protective agent Substances 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 230000001698 pyrogenic effect Effects 0.000 description 1
- 150000003233 pyrroles Chemical class 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- 150000003252 quinoxalines Chemical class 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 238000011552 rat model Methods 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 238000003259 recombinant expression Methods 0.000 description 1
- 230000002829 reductive effect Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 108091008146 restriction endonucleases Proteins 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- 239000002336 ribonucleotide Substances 0.000 description 1
- 125000002652 ribonucleotide group Chemical group 0.000 description 1
- 235000009566 rice Nutrition 0.000 description 1
- 229960003131 rubella vaccine Drugs 0.000 description 1
- 239000003813 safflower oil Substances 0.000 description 1
- 235000005713 safflower oil Nutrition 0.000 description 1
- 238000007127 saponification reaction Methods 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 208000013223 septicemia Diseases 0.000 description 1
- 229940069764 shark liver oil Drugs 0.000 description 1
- 239000013605 shuttle vector Substances 0.000 description 1
- 238000002741 site-directed mutagenesis Methods 0.000 description 1
- 239000000344 soap Substances 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 210000004872 soft tissue Anatomy 0.000 description 1
- 229940035044 sorbitan monolaurate Drugs 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 229940084106 spermaceti Drugs 0.000 description 1
- 239000012177 spermaceti Substances 0.000 description 1
- 125000004079 stearyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 239000008362 succinate buffer Substances 0.000 description 1
- 150000003890 succinate salts Chemical class 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 150000005846 sugar alcohols Chemical class 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 235000020238 sunflower seed Nutrition 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 229960002766 tetanus vaccines Drugs 0.000 description 1
- RTKIYNMVFMVABJ-UHFFFAOYSA-L thimerosal Chemical compound [Na+].CC[Hg]SC1=CC=CC=C1C([O-])=O RTKIYNMVFMVABJ-UHFFFAOYSA-L 0.000 description 1
- 229960004906 thiomersal Drugs 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 description 1
- 229940104230 thymidine Drugs 0.000 description 1
- 150000003611 tocopherol derivatives Chemical class 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 238000010361 transduction Methods 0.000 description 1
- 230000026683 transduction Effects 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 150000003918 triazines Chemical class 0.000 description 1
- LADGBHLMCUINGV-UHFFFAOYSA-N tricaprin Chemical compound CCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCC)COC(=O)CCCCCCCCC LADGBHLMCUINGV-UHFFFAOYSA-N 0.000 description 1
- 239000013638 trimer Substances 0.000 description 1
- 201000008827 tuberculosis Diseases 0.000 description 1
- 241000712461 unidentified influenza virus Species 0.000 description 1
- 238000011144 upstream manufacturing Methods 0.000 description 1
- 239000004474 valine Substances 0.000 description 1
- 229940021648 varicella vaccine Drugs 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000013603 viral vector Substances 0.000 description 1
- 239000000304 virulence factor Substances 0.000 description 1
- 230000007923 virulence factor Effects 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000010698 whale oil Substances 0.000 description 1
- 241000228158 x Triticosecale Species 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 150000003772 α-tocopherols Chemical class 0.000 description 1
Images
















Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/02—Bacterial antigens
- A61K39/025—Enterobacteriales, e.g. Enterobacter
- A61K39/0258—Escherichia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/195—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from bacteria
- C07K14/24—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from bacteria from Enterobacteriaceae (F), e.g. Citrobacter, Serratia, Proteus, Providencia, Morganella, Yersinia
- C07K14/245—Escherichia (G)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/48—Hydrolases (3) acting on peptide bonds (3.4)
- C12N9/50—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25)
- C12N9/52—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from bacteria or Archaea
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y304/00—Hydrolases acting on peptide bonds, i.e. peptidases (3.4)
- C12Y304/24—Metalloendopeptidases (3.4.24)
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- This invention relates to immunisation against pathogenic Escherichia coli strains.
- E. coli strains have traditionally been classified as either commensal or pathogenic, and pathogenic strains are then sub-classified as intestinal or extraintestinal strains.
- Pathogenic E . coli are discussed in more detail in reference 1, and fall into a number of different pathotypes i.e. a group of E. coli strains that cause a common disease using a common set of virulence factors.
- Pathotyping of strains is a routine technique that can be performed genotypically or phenotypically.
- One recent genotype-based pathotyping method [2] uses a DNA microarray.
- enteropathogenic EEC
- EHEC enterohaemorrhagic
- AEC enteroaggregative
- EIEC enteroinvasive
- ETEC enterotoxigenic
- DAEC diffusely adherent
- the extraintestinal pathogenic strains (or 'ExPEC' strains [3,4]) of E. coli include uropathogenic (UPEC) strains, neonatal meningitis (NMEC) strains, and septicemia-associated strains (SEPEC).
- UPEC uropathogenic
- NMEC neonatal meningitis
- SEPEC septicemia-associated strains
- ExPEC is the most common cause of urinary tract infections and one of the leading causes of neonatal meningitis and neonatal sepsis in humans, which can lead to serious complications and death.
- Other types of extraintestinal infections include osteomyelitis, pulmonary, intra-abdominal, soft tissue, and intravascular device-associated infections.
- Another ExPEC pathotype outside humans is avian pathogenic (APEC), causing extraintestinal infections in poultry.
- APEC avian pathogenic
- ExPEC vaccines have been based on cell lysates or on cellular structures.
- SOLCOUROVACTM includes ten different heat-killed bacteria including six ExPEC strains.
- URO-VAXOMTM is an oral tablet vaccine containing lyophilised bacterial lysates of 18 selected E. coli strains.
- Baxter Vaccines developed a UTI vaccine based on pili from 6 to 10 different strains.
- Medlmmune developed a product called MEDI 516 based on the FimH adhesin complex.
- references 5 and 6 disclose specific immunogens from ExPEC strains that can be used as the basis of defined vaccines against both NMEC and UPEC strains.
- intestinal pathotypes e.g. EAEC, EIEC, EPEC and ETEC strains
- AcfD accessory colonization factor D
- Reference 5 discloses the sequence from NMEC strain IHE3034, and the present invention is based on variants of the ExPEC 'AcfD precursor' (orf3526) that have been identified in further pathotypes, including APEC, UPEC, EAEC, EIEC, EPEC and ETEC strains. Unlike the disclosure of reference 5, these variants can be particularly useful for treating intestinal pathotypes. Thus the invention provides such variants, together with their use in immunising patients against E. coli infections.
- this disclosure includes fragments and mutants of the AcfD (orf3526) protein of all E. coli pathotypes where the fragment has increased solubility as compared to the full length while raising a substantially similar immune response in a subject as that raised by the full length protein. Further, this disclosure includes fragments and mutants of the AcfD (orf3526) protein of all E. coli pathotypes where the fragment has decreased toxicity as compared to the full length protein while raising a substantially similar immune response in a subject as that raised by the full length protein. In addition, this disclosure includes fragments and mutants of the AcfD (orf3526) protein of all E. coli pathotypes where the fragment raises a substantially similar immune response in a subject as that raised by the full length protein while having improved characteristics with regard to purification when expressed in E. coli.
- the invention provides an immunogenic polypeptide comprising an E. coli AcfD (orf3526) polypeptide comprising a mutation relative to the E. coli AcfD (orf3526) protein which decreases the toxicity of the immunogenic polypeptide as compared to the E. coli AcfD (orf3526) protein wherein the mutation is selected from a deletion of all or a portion of the zincin metalloprotease domain and a point mutation in zincin metalloprotease domain which reduces the protease activity and wherein the immunogenic polypeptide raises a substantially similar immune response in a subject as the E. coli AcfD (orf3526) protein.
- the E. coli AcfD (orf3526) protein may have an amino acid sequence selected from the group consisting of SEQ ID NOs:I-19.
- Exemplary mutations that decrease the toxicity include a deletion of all or a portion of the zincin metalloprotease domain and a point mutation in zincin metalloprotease domain which reduces the protease activity.
- the point mutation is a mutation of a zinc binding residue or a mutation of a catalytic residue.
- a preferred point mutation is substitution of amino acid number 1305 based upon alignment with SEQ ID NO: 1.
- Exemplary deletions include removal of at least the last 100 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 200 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 300 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 400 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 500 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 600 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 700 C-terminal amino acids of the E.
- coli AcfD (orf3526) protein at least the last 750 C-terminal amino acids of the E. coli AcfD (orf3526) protein, or at least the last 758 C-terminal amino acids of the E. coli AcfD (orf3526) protein or does not comprise at least the first 100 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 200 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 300 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 400 N-terminal amino acids of the E.
- coli AcfD (orf3526) protein at least the first 500 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 600 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 700 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 750 N-terminal amino acids of the E. coli AcfD (orf3526) protein, or at least the first 760 N-terminal amino acids of the E. coli AcfD (orf3526) protein.
- the invention provides an immunogenic polypeptide comprising an E. coli AcfD (orf3526) polypeptide wherein the immunogenic polypeptide comprises an amino acid sequence that comprises:
- polypeptides include other detoxified variants of SEQ ID NOs 20 to 38 and 58 to 76, including allelic variants, polymorphic forms, homologs, orthologs, paralogs, mutants, etc.
- the value of a may be selected from 85%, 87.5%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, 99.5% or more.
- the foregoing immunogenic polypeptides may further contain a deletion relative to the E. coli AcfD (orf3526) protein which increases solubility of the immunogenic polypeptide as compared to the E . coli AcfD (orf3526) protein while the immunogenic polypeptide still raises a substantially similar immune response in a subject as the E. coli AcfD (orf3526) protein.
- Exemplary deletions that increase the solubility include removal of substantially all of the N-terminal amino acids up to the gly-ser region, removal of all or a part of the N-terminal proline-rich repeat, or both.
- coli AcfD (orf3526) protein at least the first 40 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 50 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 60 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 70 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 80 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 90 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, or at least the first 94 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein.
- the invention also provides an immunogenic polypeptide fragment of an E. coli AcfD (orf3526) polypeptide wherein the immunogenic polypeptide fragment comprises an amino acid sequence that comprises:
- polypeptides include other detoxified variants of SEQ ID NOs 77 to 95, including allelic variants, polymorphic forms, homologs, orthologs, paralogs, mutants, etc.
- the value of a may be selected from 85%, 87.5%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, 99.5% or more.
- the immunogenic polypeptide fragment in certain embodiments will not comprise at least the first 10 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 20 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 25 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 30 N-terminal amino acids of the E. coli AcfD (orf3526) protein, or at least the first 33 N-terminal amino acids of the E. coli AcfD (orf3526) protein.
- the immunogenic polypeptide fragment in certain embodiments will not comprise at least the last 125 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 150 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 175 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 200 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 210 C-terminal amino acids of the E. coli AcfD (orf3526) protein, or at least the last 217 C-terminal amino acids of the E. coli AcfD (orf3526) protein.
- the invention additionally provides an immunogenic composition that comprises an immunogenic polypeptide fragment of an E. coli AcfD (orf3526) polypeptide wherein the immunogenic polypeptide fragment comprises an amino acid sequence that comprises at least a% sequence identity to any one of SEQ ID NOs: 77 to 95, wherein the immunogenic polypeptide fragment raises a substantially similar immune response in a subject as the E. coli AcfD (orf3526) protein and wherein the immunogenic polypeptide fragment does not comprise at least the last 125 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 150 C-terminal amino acids of the E.
- E. coli AcfD (orf3526) protein at least the last 175 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 200 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 210 C-terminal amino acids of the E. coli AcfD (orf3526) protein, or at least the last 217 C-terminal amino acids of the E. coli AcfD (orf3526) protein.
- polypeptides include other variants of SEQ ID NOs 77 to 95, including allelic variants, polymorphic forms, homologs, orthologs, paralogs, mutants, etc.
- the value of a may be selected from 50%, 60%, 65%, 70%, 75%, 80%, 85%, 87.5%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, 99.5% or more.
- the immunogenic polypeptide fragment of the immunogenic composition in certain embodiments will not comprise at least the first 10 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 20 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 25 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 30 N-terminal amino acids of the E. coli AcfD (orf3526) protein, or at least the first 33 N-terminal amino acids of the E. coli AcfD (orf3526) protein.
- the immunogenic composition may further comprise an adjuvant which may in certain embodiments comprise 1-12% by volume of a metabolizable oil, and 0.2% to 2.5% by weight of an emulsifying agent, wherein the metabolizable oil and the emulsifying agent are present in the form of an oil-in-water emulsion having oil droplets substantially all of which are less than 1 micron in diameter; 4-5% by volume of squalene, and (b) about 1% of an emulsifying agent comprising polyoxyethylenesorbitan monooleate and sorbitan trioleate, wherein the squalene and the emulsifying agent are present in the form of an oil-in-water emulsion having oil droplets substantially all of which are less than 1 micron in diameter; or MF59 (TM).
- an adjuvant may in certain embodiments comprise 1-12% by volume of a metabolizable oil, and 0.2% to 2.5% by weight of an emulsifying agent, wherein the metabolizable oil
- the foregoing detoxified immunogenic polypeptides and immunogenic polypeptide fragments preferably retain at least one epitope or immunogenic fragment of SEQ ID NOs 1 to 19.
- An epitope within a fragment may be a B-cell epitope and/or a T-cell epitope.
- Such epitopes can be identified empirically ( e . g . using PEPSCAN [9,10] or similar methods), or they can be predicted ( e . g .
- Epitopes are the parts of an antigen that are recognised by and bind to the antigen binding sites of antibodies or T-cell receptors, and they may also be referred to as "antigenic determinants”.
- the foregoing detoxified immunogenic polypeptides and immunogenic polypeptide fragments include, without limitation, immunogenic polypeptides that, when administered to a subject in a suitable composition which can include an adjuvant (including without limitation any of the adjuvants listed or discussed in the section "Immunogenic compositions and medicaments" below), or a suitable carrier coupled to the polypeptide, induces an antibody or T-cell mediated immune response that recognizes the isolated full length polypeptide SEQ ID NOs 1 to 19, respectively, from which the immunogenic polypeptide is derived.
- an adjuvant including without limitation any of the adjuvants listed or discussed in the section "Immunogenic compositions and medicaments" below
- the foregoing detoxified immunogenic polypeptides and immunogenic polypeptide fragments include, without limitation, immunogenic polypeptides that, when administered to a subject in a suitable composition which can include an adjuvant (including without limitation any of the adjuvants listed or discussed in the section "Immunogenic compositions and medicaments" below), or a suitable carrier coupled to the polypeptide, induces an antibody or T-cell mediated immune response that recognizes the isolated full length polypeptide SEQ ID NOs 1 to 19, respectively, from which the immunogenic fragment is derived.
- an adjuvant including without limitation any of the adjuvants listed or discussed in the section "Immunogenic compositions and medicaments" below
- a suitable carrier coupled to the polypeptide induces an antibody or T-cell mediated immune response that recognizes the isolated full length polypeptide SEQ ID NOs 1 to 19, respectively, from which the immunogenic fragment is derived.
- the foregoing detoxified immunogenic polypeptides and immunogenic polypeptide fragments include, without limitation, immunogenic polypeptides that, when administered to a subject in a suitable composition which can include an adjuvant (including without limitation any of the adjuvants listed or discussed in the section "Immunogenic compositions and medicaments" below), or a suitable carrier coupled to the polypeptide, induces an antibody or T-cell mediated immune response that recognizes the isolated full length polypeptide SEQ ID NOs 1 to 19, respectively, from which the immunogenic fragment is derived.
- an adjuvant including without limitation any of the adjuvants listed or discussed in the section "Immunogenic compositions and medicaments" below
- a suitable carrier coupled to the polypeptide induces an antibody or T-cell mediated immune response that recognizes the isolated full length polypeptide SEQ ID NOs 1 to 19, respectively, from which the immunogenic fragment is derived.
- a detoxified polypeptide of the invention may, compared to any one of SEQ ID NOs 1 to 19, include one or more (e.g. 1, 2, 3, 4, 5, 6, 7, 8, 9, etc.) amino acid substitutions, such as conservative substitutions (i.e. substitutions of one amino acid with another which has a related side chain).
- Genetically-encoded amino acids are generally divided into four families: (1) acidic i.e. aspartate, glutamate; (2) basic i.e. lysine, arginine, histidine; (3) non-polar i.e . alanine, valine, leucine, isoleucine, proline, phenylalanine, methionine, tryptophan; and (4) uncharged polar i.e.
- glycine asparagine, glutamine, cysteine, serine, threonine, tyrosine. Phenylalanine, tryptophan, and tyrosine are sometimes classified jointly as aromatic amino acids. In general, substitution of single amino acids within these families does not have a major effect on the biological activity.
- a detoxified polypeptide may include one or more (e.g. 1, 2, 3, 4, 5, 6, 7, 8, 9, etc.) single amino acid deletions relative to any one of SEQ ID NOs 1 to 19.
- a polypeptides may include one or more (e.g. 1, 2, 3, 4, 5, 6, 7, 8, 9, etc.) insertions ( e.g . each of 1, 2, 3, 4 or 5 amino acids) relative to any one of SEQ ID NOs 1 to 19.
- a detoxified polypeptide or immunogenic polypeptide fragment of the invention comprises a sequence that is not identical to a complete one of SEQ ID NOs 1 to 19 (e.g. when it comprises a sequence listing with ⁇ 100% sequence identity thereto, or when it comprises a fragment thereof) it is preferred that the polypeptide can elicit an antibody that recognises a polypeptide consisting of the complete SEQ ID sequence i.e. the antibody binds to one or more of said SEQ ID NOs 1 to 19.
- Such antibody may bind specifically to SEQ ID NOs 1 to 19, respectively while not binding to non-AcfD (orf3526) proteins with affinity significantly higher than the antibody's non-specific affinity to human serum albumin as a non-specific binding reference standard.
- Polypeptides used with the invention can take various forms (e.g . native, fusions, glycosylated, non-glycosylated, lipidated, non-lipidated, phosphorylated, non-phosphorylated, myristoylated, non-myristoylated, monomeric, multimeric, particulate, denatured, etc.).
- a polypeptide of the invention may have a lipidated N-terminal cysteine (e.g . Cys-24 of SEQ ID NOs: 1 to 19).
- Polypeptides used with the invention can be prepared by various means ( e . g . recombinant expression, purification from cell culture, chemical synthesis, etc.). Recombinantly-expressed proteins are preferred.
- Polypeptides used with the invention are preferably provided in purified or substantially purified form i.e. substantially free from other polypeptides ( e.g. free from naturally-occurring polypeptides), particularly from other E. coli or host cell polypeptides, and are generally at least about 50% pure (by weight), and usually at least about 90% pure i.e. less than about 50%, and more preferably less than about 10% ( e.g. 5%) of a composition is made up of other expressed polypeptides.
- the antigens in the compositions are separated from the whole organism with which the molecule is expressed.
- Polypeptides used with the invention are preferably E. coli polypeptides. Such polypeptides may be further selected from NMEC, APEC, UPEC, EAEC, ETEC, EPEC and ETEC E. coli polypeptides.
- polypeptide refers to amino acid polymers of any length.
- the polymer may be linear or branched, it may comprise modified amino acids, and it may be interrupted by non-amino acids.
- the terms also encompass an amino acid polymer that has been modified naturally or by intervention; for example, disulfide bond formation, glycosylation, lipidation, acetylation, phosphorylation, or any other manipulation or modification, such as conjugation with a labeling component.
- polypeptides containing one or more analogs of an amino acid including, for example, unnatural amino acids, etc.
- Polypeptides can occur as single chains or associated chains.
- polypeptides comprising a sequence -P-Q- or -Q-P-, wherein: -P- is an amino acid sequence as defined above and -Q- is not a sequence as defined above i.e. the invention provides fusion proteins.
- the N-terminus codon of -P- is not ATG, but this codon is not present at the N-terminus of a polypeptide, it will be translated as the standard amino acid for that codon rather than as a Met. Where this codon is at the N-terminus of a polypeptide, however, it will be translated as Met.
- an oligomeric protein comprising a polypeptide of the invention.
- the oligomer may be a dimer, a trimer, a tetramer, etc.
- the oligomer may be a homo-oligomer or a hetero-oligomer.
- Polypeptides in the oligomer may be covalently or non-covalently associated.
- Comparison of the immune response raised in a subject by the polypeptide with the immune response raised by the full length protein may be carried out use by any means available to one of skill in the art.
- One simple method as used in the examples below involves immunization of a model subject such as mouse and then challenge with a lethal dose of E. coli.
- a lethal dose of E. coli For proper comparison, one of skill in the art would naturally select the same adjuvant such as Freund's complete adjuvant.
- the immunogenic polypeptide fragments of the present invention will raise a substantially similar immune response in a subject (i.e., will provide substantially the same protection against the lethal challenge) if, for example, the polypeptide provides at least 70% of the protection provided by the full length protein, at least 80% of the protection provided by the full length protein, at least 85% of the protection provided by the full length protein, at least 90% of the protection provided by the full length protein, at least 95% of the protection provided by the full length protein, at least 97% of the protection provided by the full length protein, at least 98% of the protection provided by the full length protein, or at least 99% of the protection provided by the full length protein.
- the full length AcfD (orf3526) protein against which the immunogenic polypeptide fragment would be compared may be any representative E. coli AcfD (orf3526) protein including without limitation SEQ ID NOs 1-19.
- the AcfD (orf3526) protein will be the corresponding full length protein from which the immunogenic polypeptide fragment is obtained.
- the invention also provides a process for producing a polypeptide of the invention, comprising the step of culturing a host cell transformed with nucleic acid of the invention under conditions which induce polypeptide expression.
- the polypeptide may then be purified e . g . from culture supernatants.
- the invention provides an E. coli cell, containing a plasmid that encodes a polypeptide of the invention.
- the chromosome of the E. coli cell may include a homolog of AcfD (orf3526), or such a homolog may be absent, but in both cases the polypeptide of the invention can be expressed from the plasmid.
- the plasmid may include a gene encoding a marker, etc.
- heterologous host may be prokaryotic (e . g . a bacterium) or eukaryotic. Suitable hosts include, but are not limited to, Bacillus subtilis, Vibrio cholerae, Salmonella typhi, Salmonella typhimurium, Neisseria lactamica, Neisseria cinerea, Mycobacteria (e.g. M.tuberculosis), yeasts, etc.
- the invention provides a process for producing a polypeptide of the invention, comprising the step of synthesising at least part of the polypeptide by chemical means.
- any and all of the foregoing proteins, polypeptides, hybrid polypeptides, epitopes and immunogenic fragments may be in any one of a number of forms including, without limitation, recombinant, isolated or substantially purified (from materials co-existing with such proteins, polypeptides, hybrid polypeptides, epitopes and immunogenic fragments in their natural state).
- the invention also provides nucleic acid encoding polypeptides and hybrid polypeptides of the invention. It also provides nucleic acid comprising a nucleotide sequence that encodes one or more polypeptides or hybrid polypeptides of the invention.
- the invention also provides nucleic acid comprising nucleotide sequences having sequence identity to such nucleotide sequences. Identity between sequences is preferably determined by the Smith-Waterman homology search algorithm as described above. Such nucleic acids include those using alternative codons to encode the same amino acid.
- the invention also provides nucleic acid which can hybridize to these nucleic acids.
- Hybridization reactions can be performed under conditions of different "stringency”. Conditions that increase stringency of a hybridization reaction of widely known and published in the art (e.g. page 7.52 of reference 211).
- Examples of relevant conditions include (in order of increasing stringency): incubation temperatures of 25°C, 37°C, 50°C, 55°C and 68°C; buffer concentrations of 10 x SSC, 6 x SSC, 1 x SSC, 0.1 x SSC (where SSC is 0.15 M NaCl and 15 mM citrate buffer) and their equivalents using other buffer systems; formamide concentrations of 0%, 25%, 50%, and 75%; incubation times from 5 minutes to 24 hours; 1, 2, or more washing steps; wash incubation times of 1, 2, or 15 minutes; and wash solutions of 6 x SSC, 1 x SSC, 0.1 x SSC, or de-ionized water.
- Hybridization techniques and their optimization are well known in the art (e.g. see refs 28, 29, 211, 213, etc.].
- nucleic acid of the invention hybridizes to a target under low stringency conditions; in other embodiments it hybridizes under intermediate stringency conditions; in preferred embodiments, it hybridizes under high stringency conditions.
- An exemplary set of low stringency hybridization conditions is 50°C and 10 x SSC.
- An exemplary set of intermediate stringency hybridization conditions is 55°C and 1 x SSC.
- An exemplary set of high stringency hybridization conditions is 68°C and 0.1 x SSC.
- the invention includes nucleic acid comprising sequences complementary to these sequences (e.g. for antisense or probing, or for use as primers).
- Nucleic acids of the invention can be used in hybridisation reactions (e . g . Northern or Southern blots, or in nucleic acid microarrays or 'gene chips') and amplification reactions (e . g . PCR, SDA, SSSR, LCR, TMA, NASBA, etc.) and other nucleic acid techniques.
- hybridisation reactions e . g . Northern or Southern blots, or in nucleic acid microarrays or 'gene chips'
- amplification reactions e . g PCR, SDA, SSSR, LCR, TMA, NASBA, etc.
- Nucleic acid according to the invention can take various forms (e . g . single-stranded, double-stranded, vectors, primers, probes, labelled etc.). Nucleic acids of the invention may be circular or branched, but will generally be linear. Unless otherwise specified or required, any embodiment of the invention that utilizes a nucleic acid may utilize both the double-stranded form and each of two complementary single-stranded forms which make up the double-stranded form. Primers and probes are generally single-stranded, as are antisense nucleic acids.
- Nucleic acids of the invention are preferably provided in purified or substantially purified form i.e. substantially free from other nucleic acids (e.g. free from naturally-occurring nucleic acids), particularly from other E. coli or host cell nucleic acids, generally being at least about 50% pure (by weight), and usually at least about 90% pure. Nucleic acids of the invention are preferably E . coli nucleic acids.
- Nucleic acids of the invention may be prepared in many ways e.g. by chemical synthesis (e.g. phosphoramidite synthesis of DNA) in whole or in part, by digesting longer nucleic acids using nucleases (e.g. restriction enzymes), by joining shorter nucleic acids or nucleotides (e.g. using ligases or polymerases), from genomic or cDNA libraries, etc.
- nucleases e.g. restriction enzymes
- ligases or polymerases e.g. using ligases or polymerases
- Nucleic acid of the invention may be attached to a solid support (e . g . a bead, plate, filter, film, slide, microarray support, resin, etc.). Nucleic acid of the invention may be labelled e.g. with a radioactive or fluorescent label, or a biotin label. This is particularly useful where the nucleic acid is to be used in detection techniques e . g . where the nucleic acid is a primer or as a probe.
- a solid support e . g . a bead, plate, filter, film, slide, microarray support, resin, etc.
- Nucleic acid of the invention may be labelled e.g. with a radioactive or fluorescent label, or a biotin label. This is particularly useful where the nucleic acid is to be used in detection techniques e . g . where the nucleic acid is a primer or as a probe.
- nucleic acid includes in general means a polymeric form of nucleotides of any length, which contain deoxyribonucleotides, ribonucleotides, and/or their analogs. It includes DNA, RNA, DNA/RNA hybrids. It also includes DNA or RNA analogs, such as those containing modified backbones (e . g . peptide nucleic acids (PNAs) or phosphorothioates) or modified bases.
- PNAs modified backbones
- the invention includes mRNA, tRNA, rRNA, ribozymes, DNA, cDNA, recombinant nucleic acids, branched nucleic acids, plasmids, vectors, probes, primers, etc. Where nucleic acid of the invention takes the form of RNA, it may or may not have a 5' cap.
- Nucleic acids of the invention may be part of a vector i.e. part of a nucleic acid construct designed for transduction/transfection of one or more cell types.
- Vectors may be, for example, "cloning vectors” which are designed for isolation, propagation and replication of inserted nucleotides, "expression vectors” which are designed for expression of a nucleotide sequence in a host cell, "viral vectors” which is designed to result in the production of a recombinant virus or virus-like particle, or “shuttle vectors", which comprise the attributes of more than one type of vector.
- Preferred vectors are plasmids, as mentioned above.
- a "host cell” includes an individual cell or cell culture which can be or has been a recipient of exogenous nucleic acid.
- Host cells include progeny of a single host cell, and the progeny may not necessarily be completely identical (in morphology or in total DNA complement) to the original parent cell due to natural, accidental, or deliberate mutation and/or change.
- Host cells include cells transfected or infected in vivo or in vitro with nucleic acid of the invention.
- nucleic acid is DNA
- U in a RNA sequence
- T in the DNA
- RNA RNA
- T in a DNA sequence
- complement or “complementary” when used in relation to nucleic acids refers to Watson-Crick base pairing.
- the complement of C is G
- the complement of G is C
- the complement of A is T (or U)
- the complement of T is A.
- bases such as I (the purine inosine) e . g . to complement pyrimidines (C or T).
- Nucleic acids of the invention can be used, for example: to produce polypeptides; as hybridization probes for the detection of nucleic acid in biological samples; to generate additional copies of the nucleic acids; to generate ribozymes or antisense oligonucleotides; as single-stranded DNA primers or probes; or as triple-strand forming oligonucleotides.
- the invention provides a process for producing nucleic acid of the invention, wherein the nucleic acid is synthesised in part or in whole using chemical means.
- the invention provides vectors comprising nucleotide sequences of the invention (e . g . cloning or expression vectors) and host cells transformed with such vectors.
- Nucleic acid amplification according to the invention may be quantitative and/or real-time.
- nucleic acids are preferably at least 7 nucleotides in length (e.g. 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 45, 50, 55, 60, 65, 70, 75, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 225, 250,275, 300 nucleotides or longer).
- nucleic acids are preferably at most 500 nucleotides in length (e.g. 450, 400, 350, 300, 250, 200, 150, 140, 130, 120, 110, 100, 90, 80, 75, 70, 65, 60, 55, 50, 45, 40, 39, 38, 37, 36, 35, 34, 33, 32, 31, 30, 29, 28, 27, 26, 25, 24, 23, 22, 21, 20, 19, 18, 17, 16, 15 nucleotides or shorter).
- Primers and probes of the invention, and other nucleic acids used for hybridization are preferably between 10 and 30 nucleotides in length ( e . g . 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, or 30 nucleotides).
- Polypeptides of the invention are useful as active ingredients (immunogens) in immunogenic compositions, and such compositions may be useful as vaccines.
- Vaccines according to the invention may either be prophylactic (i . e . to prevent infection) or therapeutic (i.e. to treat infection), but will typically be prophylactic.
- Immunogenic compositions will be pharmaceutically acceptable. They will usually include components in addition to the antigens e.g. they typically include one or more pharmaceutical carrier(s), excipient(s) and/or adjuvant(s). A thorough discussion of carriers and excipients is available in ref.208. Thorough discussions of vaccine adjuvants are available in refs. 30 and 31.
- compositions will generally be administered to a mammal in aqueous form. Prior to administration, however, the composition may have been in a non-aqueous form. For instance, although some vaccines are manufactured in aqueous form, then filled and distributed and administered also in aqueous form, other vaccines are lyophilised during manufacture and are reconstituted into an aqueous form at the time of use. Thus a composition of the invention may be dried, such as a lyophilised formulation.
- the composition may include preservatives such as thiomersal or 2-phenoxyethanol. It is preferred, however, that the vaccine should be substantially free from (i.e. less than 5 ⁇ g/ml) mercurial material e . g . thiomersal-free. Vaccines containing no mercury are more preferred. Preservative-free vaccines are particularly preferred.
- a composition may include a temperature protective agent.
- a physiological salt such as a sodium salt.
- Sodium chloride (NaCl) is preferred, which may be present at between 1 and 20 mg/ml e . g . about 10 ⁇ 2mg/ml NaCl.
- Other salts that may be present include potassium chloride, potassium dihydrogen phosphate, disodium phosphate dehydrate, magnesium chloride, calcium chloride, etc.
- Compositions will generally have an osmolality of between 200 mOsm/kg and 400 mOsm/kg, preferably between 240-360 mOsm/kg, and will more preferably fall within the range of 290-310 mOsm/kg.
- Compositions may include one or more buffers.
- Typical buffers include: a phosphate buffer; a Tris buffer; a borate buffer; a succinate buffer; a histidine buffer (particularly with an aluminum hydroxide adjuvant); or a citrate buffer. Buffers will typically be included in the 5-20mM range.
- the pH of a composition will generally be between 5.0 and 8.1, and more typically between 6.0 and 8.0 e.g. 6.5 and 7.5, or between 7.0 and 7.8.
- the composition is preferably sterile.
- the composition is preferably non-pyrogenic e . g . containing ⁇ 1 EU (endotoxin unit, a standard measure) per dose, and preferably ⁇ 0.1 EU per dose.
- the composition is preferably gluten free.
- the composition may include material for a single immunisation, or may include material for multiple immunisations (i.e. a 'multidose' kit).
- a preservative is preferred in multidose arrangements.
- the compositions may be contained in a container having an aseptic adaptor for removal of material.
- Human vaccines are typically administered in a dosage volume of about 0.5ml, although a half dose (i . e . about 0.25ml) may be administered to children.
- Immunogenic compositions of the invention may also comprise one or more immunoregulatory agents.
- one or more of the immunoregulatory agents include one or more adjuvants.
- the adjuvants may include a TH1 adjuvant and/or a TH2 adjuvant, further discussed below.
- Adjuvants which may be used in compositions of the invention include, but are not limited to:
- Mineral containing compositions suitable for use as adjuvants in the invention include mineral salts, such as aluminium salts and calcium salts (or mixtures thereof).
- Calcium salts include calcium phosphate ( e . g . the "CAP" particles disclosed in ref. 32).
- Aluminum salts include hydroxides, phosphates, sulfates, etc., with the salts taking any suitable form (e.g. gel, crystalline, amorphous, etc.). Adsorption to these salts is preferred.
- the mineral containing compositions may also be formulated as a particle of metal salt [33].
- the adjuvants known as aluminum hydroxide and aluminum phosphate may be used. These names are conventional, but are used for convenience only, as neither is a precise description of the actual chemical compound which is present ( e . g . see chapter 9 of reference 30).
- the invention can use any of the "hydroxide” or "phosphate” adjuvants that are in general use as adjuvants.
- the adjuvants known as "aluminium hydroxide” are typically aluminium oxyhydroxide salts, which are usually at least partially crystalline.
- the adjuvants known as "aluminium phosphate” are typically aluminium hydroxyphosphates, often also containing a small amount of sulfate (i.e. aluminium hydroxyphosphate sulfate). They may be obtained by precipitation, and the reaction conditions and concentrations during precipitation influence the degree of substitution of phosphate for hydroxyl in the salt.
- a fibrous morphology (e . g . as seen in transmission electron micrographs) is typical for aluminium hydroxide adjuvants.
- the pI of aluminium hydroxide adjuvants is typically about 11 i.e. the adjuvant itself has a positive surface charge at physiological pH.
- Adsorptive capacities of between 1.8-2.6 mg protein per mg Al +++ at pH 7.4 have been reported for aluminium hydroxide adjuvants.
- Aluminium phosphate adjuvants generally have a PO 4 /Al molar ratio between 0.3 and 1.2, preferably between 0.8 and 1.2, and more preferably 0.95 ⁇ 0.1.
- the aluminium phosphate will generally be amorphous, particularly for hydroxyphosphate salts.
- a typical adjuvant is amorphous aluminium hydroxyphosphate with PO 4 /Al molar ratio between 0.84 and 0.92, included at 0.6mg Al 3+ /ml.
- the aluminium phosphate will generally be particulate (e.g. plate-like morphology as seen in transmission electron micrographs). Typical diameters of the particles are in the range 0.5-20 ⁇ m ( e . g . about 5-10 ⁇ m) after any antigen adsorption.
- Adsorptive capacities of between 0.7-1.5 mg protein per mg Al +++ at pH 7.4 have been reported for aluminium phosphate adjuvants.
- Suspensions of aluminium salts used to prepare compositions of the invention may contain a buffer (e . g . a phosphate or a histidine or a Tris buffer), but this is not always necessary.
- the suspensions are preferably sterile and pyrogen-free.
- a suspension may include free aqueous phosphate ions e . g . present at a concentration between 1.0 and 20 mM, preferably between 5 and 15 mM, and more preferably about 10 mM.
- the suspensions may also comprise sodium chloride.
- the invention can use a mixture of both an aluminium hydroxide and an aluminium phosphate.
- the concentration of Al +++ in a composition for administration to a patient is preferably less than 10mg/ml e.g. ⁇ 5 mg/ml, ⁇ 4 mg/ml, ⁇ 3 mg/ml, ⁇ 2 mg/ml, ⁇ 1 mg/ml, etc.
- a preferred range is between 0.3 and 1mg/ml.
- a maximum of 0.85mg/dose is preferred.
- Oil emulsion compositions suitable for use as adjuvants in the invention include squalene-water emulsions, such as MF59 [Chapter 10 of ref. 30; see also ref. 34] (5% Squalene, 0.5% Tween 80, and 0.5% Span 85, formulated into submicron particles using a microfluidizer). Complete Freund's adjuvant (CFA) and incomplete Freund's adjuvant (IFA) may also be used.
- CFA Complete Freund's adjuvant
- IFA incomplete Freund's adjuvant
- oil-in-water emulsion adjuvants typically include at least one oil and at least one surfactant, with the oil(s) and surfactant(s) being biodegradable (metabolisable) and biocompatible.
- the oil droplets in the emulsion are generally less than 5 ⁇ m in diameter, and ideally have a sub-micron diameter, with these small sizes being achieved with a microfluidiser to provide stable emulsions. Droplets with a size less than 220nm are preferred as they can be subjected to filter sterilization.
- the emulsion can comprise oils such as those from an animal (such as fish) or vegetable source.
- Sources for vegetable oils include nuts, seeds and grains. Peanut oil, soybean oil, coconut oil, and olive oil, the most commonly available, exemplify the nut oils.
- Jojoba oil can be used e . g . obtained from the jojoba bean.
- Seed oils include safflower oil, cottonseed oil, sunflower seed oil, sesame seed oil and the like. In the grain group, corn oil is the most readily available, but the oil of other cereal grains such as wheat, oats, rye, rice, teff, triticale and the like may also be used.
- 6-10 carbon fatty acid esters of glycerol and 1,2-propanediol may be prepared by hydrolysis, separation and esterification of the appropriate materials starting from the nut and seed oils.
- Fats and oils from mammalian milk are metabolizable and may therefore be used in the practice of this invention.
- the procedures for separation, purification, saponification and other means necessary for obtaining pure oils from animal sources are well known in the art.
- Most fish contain metabolizable oils which may be readily recovered. For example, cod liver oil, shark liver oils, and whale oil such as spermaceti exemplify several of the fish oils which may be used herein.
- a number of branched chain oils are synthesized biochemically in 5-carbon isoprene units and are generally referred to as terpenoids.
- Shark liver oil contains a branched, unsaturated terpenoids known as squalene, 2,6,10,15,19,23-hexamethyl-2,6,10,14,18,22-tetracosahexaene, which is particularly preferred herein.
- Squalane the saturated analog to squalene
- Fish oils, including squalene and squalane are readily available from commercial sources or may be obtained by methods known in the art. Other preferred oils are the tocopherols (see below). Mixtures of oils can be used.
- Surfactants can be classified by their 'HLB' (hydrophile/lipophile balance). Preferred surfactants of the invention have a HLB of at least 10, preferably at least 15, and more preferably at least 16.
- the invention can be used with surfactants including, but not limited to: the polyoxyethylene sorbitan esters surfactants (commonly referred to as the Tweens), especially polysorbate 20 and polysorbate 80; copolymers of ethylene oxide (EO), propylene oxide (PO), and/or butylene oxide (BO), sold under the DOWFAXTM tradename, such as linear EO/PO block copolymers; octoxynols, which can vary in the number of repeating ethoxy (oxy-1,2-ethanediyl) groups, with octoxynol-9 (Triton X-100, or t-octylphenoxypolyethoxyethanol) being of particular interest; (octylphenoxy)polyethoxy
- Non-ionic surfactants are preferred.
- Preferred surfactants for including in the emulsion are Tween 80 (polyoxyethylene sorbitan monooleate), Span 85 (sorbitan trioleate), lecithin and Triton X-100.
- surfactants can be used e.g. Tween 80/Span 85 mixtures.
- a combination of a polyoxyethylene sorbitan ester such as polyoxyethylene sorbitan monooleate (Tween 80) and an octoxynol such as t-octylphenoxypolyethoxyethanol (Triton X-100) is also suitable.
- Another useful combination comprises laureth 9 plus a polyoxyethylene sorbitan ester and/or an octoxynol.
- Preferred amounts of surfactants are: polyoxyethylene sorbitan esters (such as Tween 80) 0.01 to 1%, in particular about 0.1 %; octyl- or nonylphenoxy polyoxyethanols (such as Triton X-100, or other detergents in the Triton series) 0.001 to 0.1 %, in particular 0.005 to 0.02%; polyoxyethylene ethers (such as laureth 9) 0.1 to 20 %, preferably 0.1 to 10 % and in particular 0.1 to 1 % or about 0.5%.
- polyoxyethylene sorbitan esters such as Tween 80
- octyl- or nonylphenoxy polyoxyethanols such as Triton X-100, or other detergents in the Triton series
- polyoxyethylene ethers such as laureth 9
- Preferred emulsion adjuvants have an average droplets size of ⁇ 1 ⁇ m e . g . ⁇ 750nm, ⁇ 500nm, ⁇ 400nm, ⁇ 300nm, ⁇ 250nm, ⁇ 220nm, ⁇ 200nm, or smaller. These droplet sizes can conveniently be achieved by techniques such as microfluidisation.
- oil-in-water emulsion adjuvants useful with the invention include, but are not limited to:
- an emulsion may be mixed with antigen extemporaneously, at the time of delivery, and thus the adjuvant and antigen may be kept separately in a packaged or distributed vaccine, ready for final formulation at the time of use.
- an emulsion is mixed with antigen during manufacture, and thus the composition is packaged in a liquid adjuvanted form.
- the antigen will generally be in an aqueous form, such that the vaccine is finally prepared by mixing two liquids.
- the volume ratio of the two liquids for mixing can vary ( e . g . between 5:1 and 1:5) but is generally about 1:1. Where concentrations of components are given in the above descriptions of specific emulsions, these concentrations are typically for an undiluted composition, and the concentration after mixing with an antigen solution will thus decrease.
- composition includes a tocopherol
- any of the ⁇ , ⁇ , ⁇ , ⁇ , ⁇ or ⁇ tocopherols can be used, but ⁇ -tocopherols are preferred.
- the tocopherol can take several forms e . g . different salts and/or isomers. Salts include organic salts, such as succinate, acetate, nicotinate, etc. D- ⁇ -tocopherol and DL- ⁇ -tocopherol can both be used.
- Tocopherols are advantageously included in vaccines for use in elderly patients ( e . g . aged 60 years or older) because vitamin E has been reported to have a positive effect on the immune response in this patient group [48].
- a preferred ⁇ -tocopherol is DL- ⁇ -tocopherol, and the preferred salt of this tocopherol is the succinate.
- the succinate salt has been found to cooperate with TNF-related ligands in vivo.
- Saponin formulations may also be used as adjuvants in the invention.
- Saponins are a heterogeneous group of sterol glycosides and triterpenoid glycosides that are found in the bark, leaves, stems, roots and even flowers of a wide range of plant species. Saponin from the bark of the Quillaia saponaria Molina tree have been widely studied as adjuvants. Saponin can also be commercially obtained from Smilax ornata (sarsaprilla), Gypsophilla paniculata (brides veil), and Saponaria officianalis (soap root).
- Saponin adjuvant formulations include purified formulations, such as QS21, as well as lipid formulations, such as ISCOMs. QS21 is marketed as StimulonTM .
- Saponin compositions have been purified using HPLC and RP-HPLC. Specific purified fractions using these techniques have been identified, including QS7, QS17, QS18, QS21, QH-A, QH-B and QH-C.
- the saponin is QS21.
- a method of production of QS21 is disclosed in ref. 50.
- Saponin formulations may also comprise a sterol, such as cholesterol [51].
- ISCOMs immunostimulating complexs
- phospholipid such as phosphatidylethanolamine or phosphatidylcholine.
- Any known saponin can be used in ISCOMs.
- the ISCOM includes one or more of QuilA, QHA & QHC. ISCOMs are further described in refs. 51-53.
- the ISCOMS may be devoid of additional detergent [54].
- Virosomes and virus-like particles can also be used as adjuvants in the invention.
- These structures generally contain one or more proteins from a virus optionally combined or formulated with a phospholipid. They are generally non-pathogenic, non-replicating and generally do not contain any of the native viral genome.
- the viral proteins may be recombinantly produced or isolated from whole viruses.
- viral proteins suitable for use in virosomes or VLPs include proteins derived from influenza virus (such as HA or NA), Hepatitis B virus (such as core or capsid proteins), Hepatitis E virus, measles virus, Sindbis virus, Rotavirus, Foot-and-Mouth Disease virus, Retrovirus, Norwalk virus, human Papilloma virus, HIV, RNA-phages, QB-phage (such as coat proteins), GA-phage, fr-phage, AP205 phage, and Ty (such as retrotransposon Ty protein p1).
- influenza virus such as HA or NA
- Hepatitis B virus such as core or capsid proteins
- Hepatitis E virus measles virus
- Sindbis virus Rotavirus
- Foot-and-Mouth Disease virus Retrovirus
- Norwalk virus Norwalk virus
- human Papilloma virus HIV
- RNA-phages such as coat proteins
- GA-phage f-phage
- fr-phage AP
- Adjuvants suitable for use in the invention include bacterial or microbial derivatives such as non-toxic derivatives of enterobacterial lipopolysaccharide (LPS), Lipid A derivatives, immunostimulatory oligonucleotides and ADP-ribosylating toxins and detoxified derivatives thereof.
- LPS enterobacterial lipopolysaccharide
- Lipid A derivatives Lipid A derivatives
- immunostimulatory oligonucleotides and ADP-ribosylating toxins and detoxified derivatives thereof.
- Non-toxic derivatives of LPS include monophosphoryl lipid A (MPL) and 3-O-deacylated MPL (3dMPL).
- 3dMPL is a mixture of 3 de-O-acylated monophosphoryl lipid A with 4, 5 or 6 acylated chains.
- a preferred "small particle" form of 3 De-O-acylated monophosphoryl lipid A is disclosed in ref. 64. Such "small particles" of 3dMPL are small enough to be sterile filtered through a 0.22 ⁇ m membrane [64].
- Other non-toxic LPS derivatives include monophosphoryl lipid A mimics, such as aminoalkyl glucosaminide phosphate derivatives e . g . RC-529 [65,66].
- Lipid A derivatives include derivatives of lipid A from Escherichia coli such as OM-174.
- OM-174 is described for example in refs. 67 & 68.
- Immunostimulatory oligonucleotides suitable for use as adjuvants in the invention include nucleotide sequences containing a CpG motif (a dinucleotide sequence containing an unmethylated cytosine linked by a phosphate bond to a guanosine). Double-stranded RNAs and oligonucleotides containing palindromic or poly(dG) sequences have also been shown to be immunostimulatory.
- the CpG's can include nucleotide modifications/analogs such as phosphorothioate modifications and can be double-stranded or single-stranded.
- References 69, 70 and 71 disclose possible analog substitutions e . g . replacement of guanosine with 2'-deoxy-7-deazaguanosine.
- the adjuvant effect of CpG oligonucleotides is further discussed in refs. 72-77.
- the CpG sequence may be directed to TLR9, such as the motif GTCGTT or TTCGTT [78].
- the CpG sequence may be specific for inducing a Th1 immune response, such as a CpG-A ODN, or it may be more specific for inducing a B cell response, such a CpG-B ODN.
- CpG-A and CpG-B ODNs are discussed in refs. 79-81.
- the CpG is a CpG-A ODN.
- the CpG oligonucleotide is constructed so that the 5' end is accessible for receptor recognition.
- two CpG oligonucleotide sequences may be attached at their 3' ends to form "immunomers". See, for example, refs. 78 & 82-84.
- a useful CpG adjuvant is CpG7909, also known as ProMuneTM (Coley Pharmaceutical Group, Inc.). Another is CpG1826.
- TpG sequences can be used [85], and these oligonucleotides may be free from unmethylated CpG motifs.
- the immunostimulatory oligonucleotide may be pyrimidine-rich. For example, it may comprise more than one consecutive thymidine nucleotide ( e . g . TTTT, as disclosed in ref. 85), and/or it may have a nucleotide composition with >25% thymidine ( e . g .
- oligonucleotides may be free from unmethylated CpG motifs.
- Immunostimulatory oligonucleotides will typically comprise at least 20 nucleotides. They may comprise fewer than 100 nucleotides.
- an adjuvant used with the invention may comprise a mixture of (i) an oligonucleotide ( e . g . between 15-40 nucleotides) including at least one (and preferably multiple) CpI motifs ( i . e . a cytosine linked to an inosine to form a dinucleotide), and (ii) a polycationic polymer, such as an oligopeptide ( e . g . between 5-20 amino acids) including at least one (and preferably multiple) Lys-Arg-Lys tripeptide sequence(s).
- an oligonucleotide e . g . between 15-40 nucleotides
- CpI motifs i . e . a cytosine linked to an inosine to form a dinucleotide
- a polycationic polymer such as an oligopeptide ( e . g . between 5-20 amino acids) including at least one
- the oligonucleotide may be a deoxynucleotide comprising 26-mer sequence 5'-(IC) 13 -3' (SEQ ID NO: 110).
- the polycationic polymer may be a peptide comprising 11-mer amino acid sequence KLKLLLLLKLK (SEQ ID NO:111).
- Bacterial ADP-ribosylating toxins and detoxified derivatives thereof may be used as adjuvants in the invention.
- the protein is derived from E. coli (E. coli heat labile enterotoxin "LT"), cholera ("CT"), or pertussis ("PT").
- LT E. coli heat labile enterotoxin
- CT cholera
- PT pertussis
- the use of detoxified ADP-ribosylating toxins as mucosal adjuvants is described in ref. 87 and as parenteral adjuvants in ref. 88.
- the toxin or toxoid is preferably in the form of a holotoxin, comprising both A and B subunits.
- the A subunit contains a detoxifying mutation; preferably the B subunit is not mutated.
- the adjuvant is a detoxified LT mutant such as LT-K63, LT-R72, and LT-G192.
- LT-K63 LT-K63
- LT-R72 LT-G192.
- a useful CT mutant is or CT-E29H [97].
- Numerical reference for amino acid substitutions is preferably based on the alignments of the A and B subunits of ADP-ribosylating toxins set forth in ref. 98.
- Human immunomodulators suitable for use as adjuvants in the invention include cytokines, such as interleukins (e.g. IL-1, IL-2, IL-4, IL-5, IL-6, IL-7, IL-12 [99], etc.) [100], interferons (e.g. interferon- ⁇ ), macrophage colony stimulating factor, and tumor necrosis factor.
- cytokines such as interleukins (e.g. IL-1, IL-2, IL-4, IL-5, IL-6, IL-7, IL-12 [99], etc.) [100], interferons (e.g. interferon- ⁇ ), macrophage colony stimulating factor, and tumor necrosis factor.
- interferons e.g. interferon- ⁇
- macrophage colony stimulating factor e.g. interferon- ⁇
- tumor necrosis factor e.g. tumor necrosis factor.
- a preferred immunomodulator is IL-12.
- Bioadhesives and mucoadhesives may also be used as adjuvants in the invention.
- Suitable bioadhesives include esterified hyaluronic acid microspheres [101] or mucoadhesives such as cross-linked derivatives of poly(acrylic acid), polyvinyl alcohol, polyvinyl pyrollidone, polysaccharides and carboxymethylcellulose. Chitosan and derivatives thereof may also be used as adjuvants in the invention [102].
- Microparticles may also be used as adjuvants in the invention.
- Microparticles i.e. a particle of ⁇ 100nm to ⁇ 150 ⁇ m in diameter, more preferably ⁇ 200nm to ⁇ 30 ⁇ m in diameter, and most preferably ⁇ 500nm to ⁇ 10 ⁇ m in diameter
- materials that are biodegradable and non-toxic e . g . a poly( ⁇ -hydroxy acid), a polyhydroxybutyric acid, a polyorthoester, a polyanhydride, a polycaprolactone, etc.
- a negatively-charged surface e . g . with SDS
- a positively-charged surface e . g . with a cationic detergent, such as CTAB
- liposome formulations suitable for use as adjuvants are described in refs. 103-105.
- Adjuvants suitable for use in the invention include polyoxyethylene ethers and polyoxyethylene esters [106]. Such formulations further include polyoxyethylene sorbitan ester surfactants in combination with an octoxynol [107] as well as polyoxyethylene alkyl ethers or ester surfactants in combination with at least one additional non-ionic surfactant such as an octoxynol [108].
- Preferred polyoxyethylene ethers are selected from the following group: polyoxyethylene-9-lauryl ether (laureth 9), polyoxyethylene-9-steoryl ether, polyoxytheylene-8-steoryl ether, polyoxyethylene-4-lauryl ether, polyoxyethylene-35-lauryl ether, and polyoxyethylene-23-lauryl ether.
- a phosphazene such as poly[di(carboxylatophenoxy)phosphazene] ("PCPP") as described, for example, in references 109 and 110, may be used.
- PCPP poly[di(carboxylatophenoxy)phosphazene]
- muramyl peptides suitable for use as adjuvants in the invention include N-acetyl-muramyl-L-threonyl-D-isoglutamine (thr-MDP), N-acetyl-normuramyl-L-alanyl-D-isoglutamine (nor-MDP), and N-acetylmuramyl-L-alanyl-D-isoglutaminyl-L-alanine-2-(1'-2'-dipalmitoyl- sn- glycero-3-hydroxyphosphoryioxy)-ethylamine MTP-PE).
- thr-MDP N-acetyl-muramyl-L-threonyl-D-isoglutamine
- nor-MDP N-acetyl-normuramyl-L-alanyl-D-isoglutamine
- imidazoquinolone compounds suitable for use adjuvants in the invention include Imiquimod ("R-837”) [111,112], Resiquimod ("R-848”) [113], and their analogs; and salts thereof ( e . g . the hydrochloride salts). Further details about immunostimulatory imidazoquinolines can be found in references 114 to 118.
- Substituted ureas useful as adjuvants include compounds of formula I, II or III, or salts thereof: as defined in reference 119, such as 'ER 803058', 'ER 803732', 'ER 804053', ER 804058', 'ER 804059', 'ER 804442', 'ER 804680', 'ER 804764', ER 803022 or 'ER 804057' e . g .:
- the invention may also comprise combinations of aspects of one or more of the adjuvants identified above.
- the following adjuvant compositions may be used in the invention: (1) a saponin and an oil-in-water emulsion [148]; (2) a saponin (e . g . QS21) + a non-toxic LPS derivative (e.g. 3dMPL) [149]; (3) a saponin (e.g. QS21) + a non-toxic LPS derivative (e.g. 3dMPL) + a cholesterol; (4) a saponin (e.g.
- RibiTM adjuvant system (RAS), (Ribi Immunochem) containing 2% squalene, 0.2% Tween 80, and one or more bacterial cell wall components from the group consisting of monophosphorylipid A (MPL), trehalose dimycolate (TDM), and cell wall skeleton (CWS), preferably MPL + CWS (DetoxTM); and (8) one or more mineral salts (such as an aluminum salt) + a non-toxic derivative of LPS (such as 3dMPL).
- MPL monophosphorylipid A
- TDM trehalose dimycolate
- CWS cell wall skeleton
- LPS such as 3dMPL
- aluminium hydroxide and/or aluminium phosphate adjuvant are particularly preferred, and antigens are generally adsorbed to these salts.
- Calcium phosphate is another preferred adjuvant.
- Other preferred adjuvant combinations include combinations of Th1 and Th2 adjuvants such as CpG & alum or resiquimod & alum.
- a combination of aluminium phosphate and 3dMPL may be used.
- compositions of the invention may elicit both a cell mediated immune response as well as a humoral immune response.
- This immune response will preferably induce long lasting (e . g . neutralising) antibodies and a cell mediated immunity that can quickly respond upon exposure to pnuemococcus.
- CD8 T cells Two types of T cells, CD4 and CD8 cells, are generally thought necessary to initiate and/or enhance cell mediated immunity and humoral immunity.
- CD8 T cells can express a CD8 co-receptor and are commonly referred to as Cytotoxic T lymphocytes (CTLs).
- CTLs Cytotoxic T lymphocytes
- CD8 T cells are able to recognized or interact with antigens displayed on MHC Class I molecules.
- CD4 T cells can express a CD4 co-receptor and are commonly referred to as T helper cells.
- CD4 T cells are able to recognize antigenic peptides bound to MHC class II molecules.
- the CD4 cells Upon interaction with a MHC class II molecule, the CD4 cells can secrete factors such as cytokines. These secreted cytokines can activate B cells, cytotoxic T cells, macrophages, and other cells that participate in an immune response.
- Helper T cells or CD4+ cells can be further divided into two functionally distinct subsets: TH1 phenotype and TH2 phenotypes which differ in their cytokine and effector function.
- Activated TH1 cells enhance cellular immunity (including an increase in antigen-specific CTL production) and are therefore of particular value in responding to intracellular infections.
- Activated TH1 cells may secrete one or more of IL-2, IFN- ⁇ , and TNF- ⁇ .
- a TH1 immune response may result in local inflammatory reactions by activating macrophages, NK (natural killer) cells, and CD8 cytotoxic T cells (CTLs).
- a TH1 immune response may also act to expand the immune response by stimulating growth of B and T cells with IL-12.
- TH1 stimulated B cells may secrete IgG2a.
- Activated TH2 cells enhance antibody production and are therefore of value in responding to extracellular infections.
- Activated TH2 cells may secrete one or more of IL-4, IL-5, IL-6, and IL-10.
- a TH2 immune response may result in the production of IgG1, IgE, IgA and memory B cells for future protection.
- An enhanced immune response may include one or more of an enhanced TH 1 immune response and a TH2 immune response.
- a TH 1 immune response may include one or more of an increase in CTLs, an increase in one or more of the cytokines associated with a TH1 immune response (such as IL-2, IFN- ⁇ , and TNF- ⁇ ), an increase in activated macrophages, an increase in NK activity, or an increase in the production of IgG2a.
- the enhanced TH1 immune response will include an increase in IgG2a production.
- a TH1 immune response may be elicited using a TH1 adjuvant.
- a TH1 adjuvant will generally elicit increased levels of IgG2a production relative to immunization of the antigen without adjuvant.
- TH1 adjuvants suitable for use in the invention may include for example saponin formulations, virosomes and virus like particles, non-toxic derivatives of enterobacterial lipopolysaccharide (LPS), immunostimulatory oligonucleotides.
- LPS enterobacterial lipopolysaccharide
- Immunostimulatory oligonucleotides such as oligonucleotides containing a CpG motif, are preferred TH1 adjuvants for use in the invention.
- a TH2 immune response may include one or more of an increase in one or more of the cytokines associated with a TH2 immune response (such as IL-4, IL-5, IL-6 and IL-10), or an increase in the production of IgG1, IgE, IgA and memory B cells.
- the enhanced TH2 immune resonse will include an increase in IgG1 production.
- a TH2 immune response may be elicited using a TH2 adjuvant.
- a TH2 adjuvant will generally elicit increased levels of IgG1 production relative to immunization of the antigen without adjuvant.
- TH2 adjuvants suitable for use in the invention include, for example, mineral containing compositions, oil-emulsions, and ADP-ribosylating toxins and detoxified derivatives thereof. Mineral containing compositions, such as aluminium salts are preferred TH2 adjuvants for use in the invention.
- the invention includes a composition comprising a combination of a TH1 adjuvant and a TH2 adjuvant.
- a composition elicits an enhanced TH1 and an enhanced TH2 response, i.e., an increase in the production of both IgG1 and IgG2a production relative to immunization without an adjuvant.
- the composition comprising a combination of a TH1 and a TH2 adjuvant elicits an increased TH1 and/or an increased TH2 immune response relative to immunization with a single adjuvant (i.e., relative to immunization with a TH1 adjuvant alone or immunization with a TH2 adjuvant alone).
- the immune response may be one or both of a TH1 immune response and a TH2 response.
- immune response provides for one or both of an enhanced TH1 response and an enhanced TH2 response.
- the enhanced immune response may be one or both of a systemic and a mucosal immune response.
- the immune response provides for one or both of an enhanced systemic and an enhanced mucosal immune response.
- the mucosal immune response is a TH2 immune response.
- the mucosal immune response includes an increase in the production of IgA.
- the compositions of the invention may be prepared in various forms.
- the compositions may be prepared as injectables, either as liquid solutions or suspensions.
- Solid forms suitable for solution in, or suspension in, liquid vehicles prior to injection can also be prepared ( e . g . a lyophilised composition or a spray-freeze dried composition).
- the composition may be prepared for topical administration e . g . as an ointment, cream or powder.
- the composition may be prepared for oral administration e . g . as a tablet or capsule, as a spray, or as a syrup (optionally flavoured).
- the composition may be prepared for pulmonary administration e . g .
- kits may comprise one or more antigens in liquid form and one or more lyophilised antigens.
- kits may comprise two vials, or it may comprise one ready-filled syringe and one vial, with the contents of the syringe being used to reactivate the contents of the vial prior to injection.
- Immunogenic compositions used as vaccines comprise an immunologically effective amount of antigen(s), as well as any other components, as needed.
- 'immunologically effective amount' it is meant that the administration of that amount to an individual, either in a single dose or as part of a series, is effective for treatment or prevention. This amount varies depending upon the health and physical condition of the individual to be treated, age, the taxonomic group of individual to be treated (e.g. non-human primate, primate, etc.), the capacity of the individual's immune system to synthesise antibodies, the degree of protection desired, the formulation of the vaccine, the treating doctor's assessment of the medical situation, and other relevant factors. It is expected that the amount will fall in a relatively broad range that can be determined through routine trials.
- the invention also provides a method for raising an immune response in a mammal comprising the step of administering an effective amount of a composition of the invention.
- the immune response is preferably protective and preferably involves antibodies and/or cell-mediated immunity.
- the method may raise a booster response.
- the invention also provides a polypeptide of the invention for use as a medicament e . g . for use in raising an immune response in a mammal.
- the invention also provides the use of a polypeptide of the invention in the manufacture of a medicament for raising an immune response in a mammal.
- the invention also provides a delivery device pre-filled with an immunogenic composition of the invention.
- the mammal By raising an immune response in the mammal by these uses and methods, the mammal can be protected against E. coli infection, including ExPEC and non-ExPEC strains.
- the invention is particularly useful for providing broad protection against pathogenic E. coli, including intestinal pathotypes such as EPEC, EAEC, EIEC, ETEC and DAEC pathotypes.
- the mammal may be protected against diseases including, but not limited to peritonitis, pyelonephritis, cystitis, endocarditis, prostatitis, urinary tract infections (UTIs), meningitis (particularly neonatal meningitis), sepsis (or SIRS), dehydration, pneumonia, diarrhea (infantile, travellers', acute, persistent, etc.), bacillary dysentery, hemolytic uremic syndrome (HUS), pericarditis, bacteriuria, etc.
- diseases including, but not limited to peritonitis, pyelonephritis, cystitis, endocarditis, prostatitis, urinary tract infections (UTIs), meningitis (particularly neonatal meningitis), sepsis (or SIRS), dehydration, pneumonia, diarrhea (infantile, travellers', acute, persistent, etc.), bacillary dysentery, hemolytic uremic syndrome (HUS), pericarditis, bacter
- SEQ ID NO: 21, 30, 35, 40, 49, 54, 59, 68, and 73 and their other detoxified variants are particularly useful for immunising against the EAEC pathotype, and thus for preventing diarrhea (both acute and chronic).
- SEQ ID NO: 22, 28, 36, 41, 47, 55, 56, 60, 66, 74, and 75 and their other detoxified variants are particularly useful for immunising against the UPEC pathotype, and thus for preventing UTIs including, but not limited to, pyelonephritis, cystitis (both acute and recurrent), peritonitis, catheter-associated UTIs, prostatisis, and bacteriuria (including asymptomatic bacteriuria).
- SEQ ID NO: 23, 42, and 61 and their other detoxified variants are particularly useful for immunising against the EIEC pathotype, and thus for preventing dysentery (in particular bacillary dysentery) and HUS (e.g. in children).
- SEQ ID NO: 24, 27, 29, 43, 46, 48, 62, 65, and 67 and their other detoxified variants are particularly useful for immunising against the ETEC pathotype, and thus for preventing diarrhea (including travellers' and infant diarrhea).
- SEQ ID NO: 25, 26, 33, 34, 45, 53, 63, 64, and 72 and their other detoxified variants are particularly useful for immunising against the EAEC pathotype, and thus for preventing diarrhea (both acute and chronic).
- SEQ ID NO: 79, 85, 93, and 94 and their other detoxified variants are particularly useful for immunising against the UPEC pathotype, and thus for preventing UTIs including, but not limited to, pyelonephritis, cystitis (both acute and recurrent), peritonitis, catheter-associated UTIs, prostatisis, and bacteriuria (including asymptomatic bacteriuria).
- SEQ ID NO: 80 and their other detoxified variants are particularly useful for immunising against the EIEC pathotype, and thus for preventing dysentery (in particular bacillary dysentery) and HUS (e.g. in children).
- SEQ ID NO: 81, 84, and 86 and their other detoxified variants are particularly useful for immunising against the ETEC pathotype, and thus for preventing diarrhea (including travellers' and infant diarrhea).
- SEQ ID NOs: 82, 83, and 91 and their other detoxified variants are particularly useful for immunising against the EPEC pathotype, and thus for preventing diarrhea (including infantile diarrhea).
- SEQ ID NO: 21, 30, 35, 40, 49, 54, 59, 68, and 73 and their other variants are particularly useful for immunising against the EAEC pathotype, and thus for preventing diarrhea (both acute and chronic).
- SEQ ID NO: 22, 28, 36, 41, 47, 55, 56, 60, 66, 74, and 75 and their other variants are particularly useful for immunising against the UPEC pathotype, and thus for preventing UTIs including, but not limited to, pyelonephritis, cystitis (both acute and recurrent), peritonitis, catheter-associated UTIs, prostatisis, and bacteriuria (including asymptomatic bacteriuria).
- SEQ ID NO: 23, 42, and 61 and their other variants are particularly useful for immunising against the EIEC pathotype, and thus for preventing dysentery (in particular bacillary dysentery) and HUS ( e . g . in children).
- SEQ ID NO: 24, 27, 29, 43, 46, 48, 62, 65, and 67 and their other variants are particularly useful for immunising against the ETEC pathotype, and thus for preventing diarrhea (including travellers' and infant diarrhea).
- SEQ ID NO: 25, 26, 33, 34, 45, 53, 63, 64, and 72 and their other variants are particularly useful for immunising against the EAEC pathotype, and thus for preventing diarrhea (both acute and chronic).
- SEQ ID NO: 79, 85, 93, and 94 and their other are particularly useful for immunising against the UPEC pathotype, and thus for preventing UTIs including, but not limited to, pyelonephritis, cystitis (both acute and recurrent), peritonitis, catheter-associated UTIs, prostatisis, and bacteriuria (including asymptomatic bacteriuria).
- UTIs including, but not limited to, pyelonephritis, cystitis (both acute and recurrent), peritonitis, catheter-associated UTIs, prostatisis, and bacteriuria (including asymptomatic bacteriuria).
- SEQ ID NO: 80 and their other variants are particularly useful for immunising against the EIEC pathotype, and thus for preventing dysentery (in particular bacillary dysentery) and HUS ( e.g. in children).
- SEQ ID NO: 81, 84, and 86 and their other variants are particularly useful for immunising against the ETEC pathotype, and thus for preventing diarrhea (including travellers' and infant diarrhea).
- SEQ ID NOs: 82, 83, and 91 and their other variants are particularly useful for immunising against the EPEC pathotype, and thus for preventing diarrhea (including infantile diarrhea).
- the mammal is preferably a human, but may be e.g. a cow, a pig, a chicken, a cat or a dog, as E. coli disease is also problematic in these species [4].
- the human is preferably a child ( e.g. a toddler or infant) or a teenager; where the vaccine is for therapeutic use, the human is preferably a teenager or an adult.
- a vaccine intended for children may also be administered to adults e.g. to assess safety, dosage, immunogenicity, etc.
- One way of checking efficacy of therapeutic treatment involves monitoring E. coli infection after administration of the compositions of the invention.
- One way of checking efficacy of prophylactic treatment involves monitoring immune responses, systemically (such as monitoring the level of IgG1 and IgG2a production) and/or mucosally (such as monitoring the level of IgA production), against the antigens in the compositions of the invention after administration of the composition.
- antigen-specific serum antibody responses are determined post-immunisation but pre-challenge whereas antigen-specific mucosal antibody responses are determined post-immunisation and post-challenge.
- Another way of assessing the immunogenicity of the compositions of the present invention is to express the proteins recombinantly for screening patient sera or mucosal secretions by immunoblot and/or microarrays. A positive reaction between the protein and the patient sample indicates that the patient has mounted an immune response to the protein in question. This method may also be used to identify immunodominant antigens and/or epitopes within antigens.
- the efficacy of vaccine compositions can also be determined in vivo by challenging animal models of E. coli infection, e.g., guinea pigs or mice, with the vaccine compositions.
- E. coli infection e.g., guinea pigs or mice
- a murine model of ExPEC and lethal sepsis is described in reference 152.
- a cotton rat model is disclosed in ref. 153
- compositions of the invention will generally be administered directly to a patient.
- Direct delivery may be accomplished by parenteral injection (e.g. subcutaneously, intraperitoneally, intravenously, intramuscularly, or to the interstitial space of a tissue), or mucosally, such as by rectal, oral ( e.g. tablet, spray), vaginal, topical, transdermal or transcutaneous, intranasal, ocular, aural, pulmonary or other mucosal administration.
- Novel direct delivery forms can also include transgenic expression of the polypeptides disclosed herein in foods, e.g., transgenic expression in a potato.
- the invention may be used to elicit systemic and/or mucosal immunity, preferably to elicit an enhanced systemic and/or mucosal immunity.
- the enhanced systemic and/or mucosal immunity is reflected in an enhanced TH1 and/or TH2 immune response.
- the enhanced immune response includes an increase in the production of IgG1 and/or IgG2a and/or IgA.
- Dosage can be by a single dose schedule or a multiple dose schedule. Multiple doses may be used in a primary immunisation schedule and/or in a booster immunisation schedule. In a multiple dose schedule the various doses may be given by the same or different routes e.g. a parenteral prime and mucosal boost, a mucosal prime and parenteral boost, etc. Multiple doses will typically be administered at least 1 week apart ( e.g. about 2 weeks, about 3 weeks, about 4 weeks, about 6 weeks, about 8 weeks, about 10 weeks, about 12 weeks, about 16 weeks, etc. ).
- Vaccines of the invention may be used to treat both children and adults.
- a human patient may be less than 1 year old, 1-5 years old, 5-15 years old, 15-55 years old, or at least 55 years old.
- Preferred patients for receiving the vaccines are the elderly (e.g. ⁇ 50 years old, ⁇ 60 years old, and preferably ⁇ 65 years), the young ( e.g. ⁇ 5 years old), hospitalised patients, healthcare workers, armed service and military personnel, pregnant women, the chronically ill, or immunodeficient patients.
- the vaccines are not suitable solely for these groups, however, and may be used more generally in a population.
- Vaccines of the invention are particularly useful for patients who are expecting a surgical operation, or other hospital in-patients. They are also useful in patients who will be catheterized. They are also useful in adolescent females ( e.g. aged 11-18) and in patients with chronic urinary tract infections.
- Vaccines of the invention may be administered to patients at substantially the same time as ( e.g. during the same medical consultation or visit to a healthcare professional or vaccination centre) other vaccines e.g. at substantially the same time as a measles vaccine, a mumps vaccine, a rubella vaccine, a MMR vaccine, a varicella vaccine, a MMRV vaccine, a diphtheria vaccine, a tetanus vaccine, a pertussis vaccine, a DTP vaccine, a conjugated H.
- other vaccines e.g. at substantially the same time as a measles vaccine, a mumps vaccine, a rubella vaccine, a MMR vaccine, a varicella vaccine, a MMRV vaccine, a diphtheria vaccine, a tetanus vaccine, a pertussis vaccine, a DTP vaccine, a conjugated H.
- influenzae type b vaccine an inactivated poliovirus vaccine, a hepatitis B virus vaccine, a meningococcal conjugate vaccine (such as a tetravalent A-C-W135-Y vaccine), a respiratory syncytial virus vaccine, etc.
- the immunogenic compositions described above include polypeptide antigens.
- the polypeptide antigens can be replaced by nucleic acids (typically DNA) encoding those polypeptides, to give compositions, methods and uses based on nucleic acid immunisation.
- Nucleic acid immunisation is now a developed field ( e.g. see references 154 to 161 etc. ).
- the nucleic acid encoding the immunogen is expressed in vivo after delivery to a patient and the expressed immunogen then stimulates the immune system.
- the active ingredient will typically take the form of a nucleic acid vector comprising: (i) a promoter; (ii) a sequence encoding the immunogen, operably linked to the promoter; and optionally (iii) a selectable marker.
- Preferred vectors may further comprise (iv) an origin of replication; and (v) a transcription terminator downstream of and operably linked to (ii).
- (i) & (v) will be eukaryotic and (iii) & (iv) will be prokaryotic.
- Preferred promoters are viral promoters e.g. from cytomegalovirus (CMV).
- the vector may also include transcriptional regulatory sequences (e.g. enhancers) in addition to the promoter and which interact functionally with the promoter.
- Preferred vectors include the immediate-early CMV enhancer/promoter, and more preferred vectors also include CMV intron A.
- the promoter is operably linked to a downstream sequence encoding an immunogen, such that expression of the immunogen-encoding sequence is under the promoter's control.
- a marker preferably functions in a microbial host (e.g. in a prokaryote, in a bacteria, in a yeast).
- the marker is preferably a prokaryotic selectable marker (e.g. transcribed under the control of a prokaryotic promoter).
- prokaryotic selectable marker e.g. transcribed under the control of a prokaryotic promoter.
- typical markers are antibiotic resistance genes.
- the vector of the disclosure is preferably an autonomously replicating episomal or extrachromosomal vector, such as a plasmid.
- the vector of the disclosure preferably comprises an origin of replication. It is preferred that the origin of replication is active in prokaryotes but not in eukaryotes.
- Preferred vectors thus include a prokaryotic marker for selection of the vector, a prokaryotic origin of replication, but a eukaryotic promoter for driving transcription of the immunogen-encoding sequence.
- the vectors will therefore (a) be amplified and selected in prokaryotic hosts without polypeptide expression, but (b) be expressed in eukaryotic hosts without being amplified. This arrangement is ideal for nucleic acid immunization vectors.
- the vector of the disclosure may comprise a eukaryotic transcriptional terminator sequence downstream of the coding sequence. This can enhance transcription levels.
- the vector of the invention preferably comprises a polyadenylation sequence.
- a preferred polyadenylation sequence is from bovine growth hormone.
- the vector of the disclosure may comprise a multiple cloning site
- the vector may comprise a second eukaryotic coding sequence.
- the vector may also comprise an IRES upstream of said second sequence in order to permit translation of a second eukaryotic polypeptide from the same transcript as the immunogen.
- the immunogen-coding sequence may be downstream of an IRES.
- the vector of the disclosure may comprise unmethylated CpG motifs e.g. unmethylated DNA sequences which have in common a cytosine preceding a guanosine, flanked by two 5' purines and two 3' pyrimidines. In their unmethylated form these DNA motifs have been demonstrated to be potent stimulators of several types of immune cell.
- unmethylated CpG motifs e.g. unmethylated DNA sequences which have in common a cytosine preceding a guanosine, flanked by two 5' purines and two 3' pyrimidines. In their unmethylated form these DNA motifs have been demonstrated to be potent stimulators of several types of immune cell.
- Vectors may be delivered in a targeted way.
- Receptor-mediated DNA delivery techniques are described in, for example, references 162 to 167.
- Therapeutic compositions containing a nucleic acid are administered in a range of about 100ng to about 200mg of DNA for local administration in a gene therapy protocol. Concentration ranges of about 500 ng to about 50 mg, about 1 ⁇ g to about 2 mg, about 5 ⁇ g to about 500 ⁇ g, and about 20 ⁇ g to about 100 ⁇ g of DNA can also be used during a gene therapy protocol.
- Factors such as method of action (e.g. for enhancing or inhibiting levels of the encoded gene product) and efficacy of transformation and expression are considerations which will affect the dosage required for ultimate efficacy.
- Vectors can be delivered using gene delivery vehicles.
- the gene delivery vehicle can be of viral or non-viral origin (see generally references 168 to 171).
- Viral-based vectors for delivery of a desired nucleic acid and expression in a desired cell are well known in the art.
- Exemplary viral-based vehicles include, but are not limited to, recombinant retroviruses (e.g. references 172 to 182), alphavirus-based vectors (e.g. Sindbis virus vectors, Semliki forest virus (ATCC VR-67; ATCC VR-1247), Ross River virus (ATCC VR-373; ATCC VR-1246) and Venezuelan equine encephalitis virus (ATCC VR-923; ATCC VR-1250; ATCC VR 1249; ATCC VR-532); hybrids or chimeras of these viruses may also be used), poxvirus vectors (e.g.
- vaccinia fowlpox, canarypox, modified vaccinia Ankara, etc.
- adenovirus vectors e.g. see refs. 183 to 188.
- AAV adeno-associated virus
- Non-viral delivery vehicles and methods can also be employed, including, but not limited to, polycationic condensed DNA linked or unlinked to killed adenovirus alone [ e.g. 189], ligand-linked DNA [190], eukaryotic cell delivery vehicles cells [ e.g. refs. 191 to 195] and nucleic charge neutralization or fusion with cell membranes. Naked DNA can also be employed. Exemplary naked DNA introduction methods are described in refs. 196 and 197. Liposomes (e.g. immunoliposomes) that can act as gene delivery vehicles are described in refs. 198 to 202. Additional approaches are described in references 203 & 204.
- non-viral delivery suitable for use includes mechanical delivery systems such as the approach described in ref. 204.
- the coding sequence and the product of expression of such can be delivered through deposition of photopolymerized hydrogel materials or use of ionizing radiation [ e.g. refs. 205 & 206].
- Other conventional methods for gene delivery that can be used for delivery of the coding sequence include, for example, use of hand-held gene transfer particle gun [207] or use of ionizing radiation for activating transferred genes [205 & 206].
- Delivery DNA using PLG ⁇ poly(lactide-co-glycolide) ⁇ microparticles is a particularly preferred method e.g. by adsorption to the microparticles, which are optionally treated to have a negatively-charged surface (e.g. treated with SDS) or a positively-charged surface (e.g. treated with a cationic detergent, such as CTAB).
- a negatively-charged surface e.g. treated with SDS
- a positively-charged surface e.g. treated with a cationic detergent, such as CTAB
- composition comprising X may consist exclusively of X or may include something additional e.g. X + Y.
- GI numbering is used herein.
- a GI number, or "GenInfo Identifier” is a series of digits assigned consecutively to each sequence record processed by NCBI when sequences are added to its databases. The GI number bears no resemblance to the accession number of the sequence record.
- a sequence is updated (e.g. for correction, or to add more annotation or information) then it receives a new GI number. Thus the sequence associated with a given GI number is never changed.
- references to a percentage sequence identity between two amino acid sequences means that, when aligned, that percentage of amino acids are the same in comparing the two sequences.
- This alignment and the percent homology or sequence identity can be determined using software programs known in the art, for example those described in section 7.7.18 of ref. 216.
- a preferred alignment is determined by the Smith-Waterman homology search algorithm using an affine gap search with a gap open penalty of 12 and a gap extension penalty of 2, BLOSUM matrix of 62.
- the Smith-Waterman homology search algorithm is disclosed in ref. 217.
- isolated means altered “by the hand of man” from its natural state, i.e., if it occurs in nature, it has been changed or removed from its original environment, or both.
- a polynucleotide or a polypeptide naturally present in a living organism is not “isolated” when in such living organism, but the same polynucleotide or polypeptide separated from the coexisting materials of its natural state is "isolated,” as the term is used in this disclosure.
- a polynucleotide or polypeptide that is introduced into an organism by transformation, genetic manipulation or by any other recombinant method would be understood to be “isolated” even if it is still present in said organism, which organism may be living or non-living, except where such transformation, genetic manipulation or other recombinant method produces an organism that is otherwise indistinguishable from the naturally occurring organism.
- SEQ ID NOs: 1-19 full length amino acid sequences 1 Amino acid sequence from NMEC strain IHE3034 2 Amino acid sequence from EAEC strain 101-1 (GI: 83587587) 3 Amino acid sequence from UPEC strain 536 (GI: 110643204) 4 Amino acid sequence from EIEC strain 53638 (GI: 75515237) 5 Amino acid sequence from ETEC strain B7A (GI: 75227618) 6 Amino acid sequence from EPEC strain E110019 (GI: 75239450) 7 Amino acid sequence from EPEC strain E22 (GI: 75259912) 8 Amino acid sequence from ETEC strain E24377A (GI: 157156747) 9 Amino acid sequence from UPEC strain F11 (GI: 75241179) 10 Amino acid sequence from ETEC strain H10407 11 Amino acid sequence from EAEC strain 042 12 Amino acid sequence from commensal strain HS (GI: 157162442) 13 Amino
- AcfD accessory colonization factor D
- Orf3526 amino acid SEQ ID NO: 2 herein
- N-terminal amino acids are 100% conserved, and these include the signal peptide (aa 1-23) and the N-terminus cysteine of the native lipoprotein.
- strains had a frameshift mutation in the AcfD (orf3526) gene, resulting in no expression of the polypeptide.
- the acfD gene was totally absent from strains CFT073, EDL933, Sakai and B171.
- Figure 2 shows the % identity between the amino acid sequences.
- the labels are SEQ ID NOs, except for MG1655, RS218, DH10B, APECO1 and UTI89 where the strain name is used.
- the lowest level of identity (boxed in Figure 2 ) was 85.9%, between SEQ ID NOs: 2 and 4 (both ExPEC strains).
- the AcfD (orf3526) sequence from strain IHE3034 was cloned and expressed from a plasmid as a recombinant His-tagged protein without a leader peptide, in an E. coli host. Protein was purified and analysed. Gel filtration showed a much higher molecular weight than predicted based solely on the amino acid sequence. Gel analysis in the absence of DTT, but not in its presence, shows higher molecular weight forms of the protein ( Figure 3 ). Thus the protein is likely to form oligomers.
- Sera raised against AcfD were used in western blots against total cell lysates ( Figure 4(A) ) or culture supernatants precipitated with 60% TCA ( Figure 4(B) ).
- the sera recognised a ⁇ 150kDa protein in lysates from both pathogenic and commensal strains. They did not react with this band in lysates from CFT073 or from an AcfD (orf3526) knockout mutant of IHE3034. Reactivity with proteins in the supernatants indicates that the protein may be secreted.
- CD1 mice (5 weeks old) were immunized sub-cutaneously using 20 ⁇ g of the antigen plus Freund's adjuvant (or other adjuvant as indicated below). The mice were inoculated at 0, 21, and 35 days. Fourteen days after the third inoculation, the mice were challenged with a lethal dose (LD80) of a pathogenic strain of E. coli. Blood was collected from the tail 24 hours after challenge to evaluate bacteremia. The mortality was monitored for four days post-challenge. The protection rate may be calculated as (%dead in control group (no immunization) - %dead in experimental group (immunized))/ %dead in control group x 100. TABLE 1 Candidates Sepsis animal model Survival with vaccination (%) Survival without vaccination (%) P value 20 ⁇ g 3526/alum 64/78 (82) 9/80 (11) 0.0001
- the AcfD precursor (orf3526) is an effective candidate for use as a vaccine or vaccine component.
- the AcfD precursor (orf3526) was tested for its ability to provide cross protection against other pathogenic strains of E. coli using the animal sepsis model as indicated above. The results of these tests are indicated in Table 2 below (with the results from Table 1 included for comparison purposes).
- the AcfD precursor (orf3526) is an effective candidate for use as a vaccine or vaccine component not just against the strain from which the protein was derived, but also against related strains, thus increasing the utility.
- the response to the AcfD precursor (orf3526) and the response to the three immunogenic fragments tested below (3526A, 3526B, and 3526C), one would expect that these three fragments would provide similar cross protection as well.
- AcfD (orf3526) protein displayed low solubility even though the protein is a secreted protein.
- removal of the N-terminus of AcfD (orf3526) through the gly-ser linker or gly-ser region significantly increased solubility when expressed at 25° C (See pK1- ⁇ G3526 Figure 5(B) ).
- removal of the N-terminus of AcfD (orf3526) through the proline rich region significantly increased solubility when expressed at 25° C (See pK1-AP3526 Figure 5(B) ).
- the fragments were purified.
- the purified fragments were used in three immunization experiments in mice, adjuvanted with Freund's complete adjuvant. Immunized mice were then challenged with a sublethal dose of E. coli. Immunization with AcfD (orf3526) with the N-terminus through the gly-ser linker or gly-ser region removed protected 100% of the mice from death, whereas death occurred in 90% of the animals in the non-immunized control group. Immunization with AcfD (orf3526) with the N-terminus through the proline rich region removed protected 90% of the mice from death, whereas death occurred in 90% of the animals in the non-immunized control group.
- Bacteria with one of the three constructs expressing his-tagged variants of AcfD (orf3526) were cultured in 30 ml of medium and induced to express the AcfD (orf3526) variant at 25° C (AcfD (orf3526) without the leader peptide (3526), AcfD (orf3526) with the N-terminus removed through the gly-ser linker or gly-ser region ( ⁇ G3526), and AcfD (orf3526) with the N-terminus removed through the proline rich region ( ⁇ P3526).
- the bacteria were harvested and lysed by sonication.
- the soluble fractions were isolated and loaded on an IMAC column.
- a zinc binding motif was identified in the AcfD (orf3526) protein (See Figure 8 ).
- the zincin zinc metalloprotease motif is also found in StcE ( S ecreted pro t ease of C 1 esterase inhibitor from E HEC) which is a protein secreted by EHEC that is involved in the pathogenesis of such E. coli.
- StcE HE VG H N YGLG H SEQ ID NO: 96
- AcfD (orf3526) HE VG H N AAETP (SEQ ID NO: 97)
- AcfD (orf3526) similarly is a zinc metalloprotease involved in the pathogenicity of the E. coli pathotypes in which it is expressed and secreted. Therefore, inactivation of the toxicity of AcfD (orf3526) was sought.
- One of skill in the art would have no difficulty in modifying AcfD (orf3526) to inactivate the metalloprotease.
- the Glu residue of the motif is known to be most important to catalytic activity while the two His residues coordinate the Zinc
- the Glu was mutated to Ala as an exemplary mutation inactivating the metalloprotease activity - SEQ ID NOs: 58-76 (See, e.g., Microbiology and Molecular Biology Reviews, September 1998, p. 597-635, Vol. 62, No. 3 ).
- two additional detoxified variants were designed: one with the C-terminus deleted - SEQ ID NOs: 20-38, and one with the N-terminus deleted - SEQ ID NOs: 39-57.
- HBMEC human bone marrow endothelial cells
- CHO cells Chinese hamster ovary cells
- the TNF- ⁇ treated cells showed an average of ⁇ 22.5% cell death (average of three experiments) while the highest dose of AcfD (orf3526) protein showed only an average of ⁇ 5% cell death (average of three experiments) as compared to the negative control which showed only an average of ⁇ 2.5% cell death (average of three experiments).
- the effect of increasing doses of AcfD (orf3526) protein (1 ⁇ g/ml, 10 ⁇ g/ml, and 100 ⁇ g/ml) was compared to TNF- ⁇ (as a positive control) by measuring the change in potential difference over time due to the alteration of the cytoskeleton of the CHO cells.
- TNF- ⁇ as a positive control
- Mutants were obtained using GeneTailor site-directed mutagenesis system (Invitrogen). Genes were cloned in pET-21b vectors (Novagen) and transformed in DH5 ⁇ -T1 chemically competent cells for propagation (Invitrogen). BL21 (DE3) chemically competent cells were used for expression. All candidates were cloned and expressed without the signal sequence and as his-tag fusion proteins, being purified by affinity chromatography.
- a construct was designed that combined the ⁇ G3526 N-terminal deletion that improved solubility with deletion of the C-terminus through the zinc motif ( ⁇ G3526C).
- Exemplary constructs are provided as SEQ ID NOs: 77 to 95.
- a non-his-tagged expression vector for the ⁇ G3526C ( E. coli strain IHE3034 - SEQ ID NO:77) construct was obtained by PCR using the primers indicated below ( ⁇ G3526A/C_For and ⁇ G3526C_NatRev).
- the amplified PCR fragment was digested with NdeI and XhoI and ligated into a pET-24b(+) vector (Novagen) and transformed in DH5 ⁇ -T1 chemically competent cells for propagation (Invitrogen).
- BL21 (DE3) chemically competent cells were used for expression.
- a his-tagged expression vector for the ⁇ G3526C ( E. coli strain IHE3034 - SEQ ID NO:77) construct was obtained using Polymerase Incomplete Primer Extension (PIPE) using the primers indicated below ( ⁇ G3526A/C_For and ⁇ G3526C_Nat for I-PCR and p-pet1 and pet3 for V-PCR of the pET-21b vector (Novagen)).
- the expression vector was transformed in DH5 ⁇ -T1 chemically competent cells for propagation (Invitrogen).
- BL21 (DE3) chemically competent cells were used for expression.
- the his-tagged version of ⁇ G3526C was expressed (See, e.g. , Figure 10 ) without the signal sequence and purified by affinity chromatography based upon the his-tag.
- the fragment was purified.
- the purified fragment was then tested as follows. Two groups of 8 CD1 mice (4 weeks old) were immunized s.c. with three doses of antigen ⁇ G3526C (either 2 ⁇ g or 20 ⁇ g), formulated in Alum at days 0, 31 and 35. 14 days after last immunization (11 weeks old mice) mice were infected i.p. with pathogenic E. coli strain IHE3034. Blood was collected from the tail after 24 hours to evaluate bacteremia. Mortality was monitored for 4 days after the infection. Protection was calculated as % survival at day 4 and statistical analysis was carried out by Fisher's exact test.
- the purified fragment was further tested as follows.
- CD1 mice (4 weeks old) were immunized s.c. with three doses of antigen ⁇ G3526C (either 10 ⁇ g or 20 ⁇ g), formulated in Alum at days 0, 31 and 35. 14 days after last immunization (11 weeks old mice) mice were infected i.p. with the pathogenic E. coli strain as indicated on Table 5. Blood was collected from the tail after 24 hours to evaluate bacteremia. Mortality was monitored for 4 days after the infection. Protection was calculated as % survival at day 4 and statistical analysis was carried out by Fisher's exact test. Results of the challenge are shown in Table 5 below.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Microbiology (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Gastroenterology & Hepatology (AREA)
- Biophysics (AREA)
- Mycology (AREA)
- Biomedical Technology (AREA)
- Biotechnology (AREA)
- Epidemiology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- General Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Peptides Or Proteins (AREA)
- Enzymes And Modification Thereof (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Description
- This invention relates to immunisation against pathogenic Escherichia coli strains.
- E. coli strains have traditionally been classified as either commensal or pathogenic, and pathogenic strains are then sub-classified as intestinal or extraintestinal strains. Pathogenic E. coli are discussed in more detail in
reference 1, and fall into a number of different pathotypes i.e. a group of E. coli strains that cause a common disease using a common set of virulence factors. Pathotyping of strains is a routine technique that can be performed genotypically or phenotypically. One recent genotype-based pathotyping method [2] uses a DNA microarray. - Among intestinal strains at least six well-described pathotypes are known: enteropathogenic (EPEC), enterohaemorrhagic (EHEC), enteroaggregative (EAEC), enteroinvasive (EIEC), enterotoxigenic (ETEC) and diffusely adherent (DAEC).
- The extraintestinal pathogenic strains (or 'ExPEC' strains [3,4]) of E. coli include uropathogenic (UPEC) strains, neonatal meningitis (NMEC) strains, and septicemia-associated strains (SEPEC). ExPEC is the most common cause of urinary tract infections and one of the leading causes of neonatal meningitis and neonatal sepsis in humans, which can lead to serious complications and death. Other types of extraintestinal infections include osteomyelitis, pulmonary, intra-abdominal, soft tissue, and intravascular device-associated infections. Another ExPEC pathotype outside humans is avian pathogenic (APEC), causing extraintestinal infections in poultry.
- Most previous ExPEC vaccines have been based on cell lysates or on cellular structures. SOLCOUROVAC™ includes ten different heat-killed bacteria including six ExPEC strains. URO-VAXOM™ is an oral tablet vaccine containing lyophilised bacterial lysates of 18 selected E. coli strains. Baxter Vaccines developed a UTI vaccine based on pili from 6 to 10 different strains. Medlmmune developed a product called MEDI 516 based on the FimH adhesin complex. In contrast,
references - It is an object of the invention to provide further and better antigens for use in immunisation against pathogenic E. coli strains, and more particularly against intestinal pathotypes (e.g. EAEC, EIEC, EPEC and ETEC strains) as well as ExPEC pathotypes and in particular antigen that have been detoxified so as to enable their use as components for immunisation or that have been shortened without reducing the immune response raised to as to improve the expression and purification.
- One of the many antigens disclosed in
reference 5 is annotated as the accessory colonization factor D ("AcfD") precursor (orf3526) (SEQ ID NOs: 7051 & 7052 therein; SEQ ID NO: 1 herein).Reference 5 discloses the sequence from NMEC strain IHE3034, and the present invention is based on variants of the ExPEC 'AcfD precursor' (orf3526) that have been identified in further pathotypes, including APEC, UPEC, EAEC, EIEC, EPEC and ETEC strains. Unlike the disclosure ofreference 5, these variants can be particularly useful for treating intestinal pathotypes. Thus the invention provides such variants, together with their use in immunising patients against E. coli infections. In addition, this disclosure includes fragments and mutants of the AcfD (orf3526) protein of all E. coli pathotypes where the fragment has increased solubility as compared to the full length while raising a substantially similar immune response in a subject as that raised by the full length protein. Further, this disclosure includes fragments and mutants of the AcfD (orf3526) protein of all E. coli pathotypes where the fragment has decreased toxicity as compared to the full length protein while raising a substantially similar immune response in a subject as that raised by the full length protein. In addition, this disclosure includes fragments and mutants of the AcfD (orf3526) protein of all E. coli pathotypes where the fragment raises a substantially similar immune response in a subject as that raised by the full length protein while having improved characteristics with regard to purification when expressed in E. coli. - The invention provides an immunogenic polypeptide comprising an E. coli AcfD (orf3526) polypeptide comprising a mutation relative to the E. coli AcfD (orf3526) protein which decreases the toxicity of the immunogenic polypeptide as compared to the E. coli AcfD (orf3526) protein wherein the mutation is selected from a deletion of all or a portion of the zincin metalloprotease domain and a point mutation in zincin metalloprotease domain which reduces the protease activity and wherein the immunogenic polypeptide raises a substantially similar immune response in a subject as the E. coli AcfD (orf3526) protein.
- The E. coli AcfD (orf3526) protein may have an amino acid sequence selected from the group consisting of SEQ ID NOs:I-19.
- Exemplary mutations that decrease the toxicity include a deletion of all or a portion of the zincin metalloprotease domain and a point mutation in zincin metalloprotease domain which reduces the protease activity. In certain cases, the point mutation is a mutation of a zinc binding residue or a mutation of a catalytic residue. A preferred point mutation is substitution of amino acid number 1305 based upon alignment with SEQ ID NO: 1.
- Exemplary deletions include removal of at least the last 100 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 200 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 300 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 400 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 500 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 600 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 700 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 750 C-terminal amino acids of the E. coli AcfD (orf3526) protein, or at least the last 758 C-terminal amino acids of the E. coli AcfD (orf3526) protein or does not comprise at least the first 100 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 200 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 300 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 400 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 500 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 600 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 700 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 750 N-terminal amino acids of the E. coli AcfD (orf3526) protein, or at least the first 760 N-terminal amino acids of the E. coli AcfD (orf3526) protein.
- The invention provides an immunogenic polypeptide comprising an E. coli AcfD (orf3526) polypeptide wherein the immunogenic polypeptide comprises an amino acid sequence that comprises:
- (a) the amino acid sequence selected from the group consisting of
SEQ ID NOs 20 to 38, and 58 to 76; - (b) at least a% sequence identity to any one of SEQ ID NOs: 20 to 38 and 58 to 76; or
- (c) has 1, 2, 3, 4, 5, 6, 7, 8, 9 or 10 single amino acid alterations (deletions, insertions, substitutions), which may be at separate locations or may be contiguous, as compared to
SEQ ID NOs 20 to 38 and 58 to 76; - (d) when aligned with any of SEQ ID NOs: 20 to 38, and 58 to 76 using a pairwise alignment algorithm, each moving window of x amino acids from N-terminus to C-terminus (such that for an alignment that extends to p amino acids, where p>x, there are p-x+1 such windows) has at least x·y identical aligned amino acids, where: x is 30; y is 0.75; and if x·y is not an integer then it is rounded up to the nearest integer,
- The preferred pairwise alignment algorithm is the Needleman-Wunsch global alignment algorithm [7], using default parameters (e.g. with Gap opening penalty = 10.0, and with Gap extension penalty = 0.5, using the EBLOSUM62 scoring matrix). This algorithm is conveniently implemented in the needle tool in the EMBOSS package [8].
- These polypeptides include other detoxified variants of
SEQ ID NOs 20 to 38 and 58 to 76, including allelic variants, polymorphic forms, homologs, orthologs, paralogs, mutants, etc. - The value of a may be selected from 85%, 87.5%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, 99.5% or more.
- The foregoing immunogenic polypeptides may further contain a deletion relative to the E. coli AcfD (orf3526) protein which increases solubility of the immunogenic polypeptide as compared to the E. coli AcfD (orf3526) protein while the immunogenic polypeptide still raises a substantially similar immune response in a subject as the E. coli AcfD (orf3526) protein.
- Exemplary deletions that increase the solubility include removal of substantially all of the N-terminal amino acids up to the gly-ser region, removal of all or a part of the N-terminal proline-rich repeat, or both. The immunogenic polypeptide of
claim 7, wherein the deletion is removal of at least the first 20 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 20 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 30 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 38 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 40 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 50 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 60 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 70 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 80 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 90 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, or at least the first 94 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein. - The invention also provides an immunogenic polypeptide fragment of an E. coli AcfD (orf3526) polypeptide wherein the immunogenic polypeptide fragment comprises an amino acid sequence that comprises:
- (a) the amino acid sequence selected from the group consisting of SEQ ID NOs 77 to 95;
- (b) at least a% sequence identity to any one of SEQ ID NOs: 77 to 95; or
- (c) has 1, 2, 3, 4, 5, 6, 7, 8, 9 or 10 single amino acid alterations (deletions, insertions, substitutions), which may be at separate locations or may be contiguous, as compared to SEQ ID NOs: 77 to 95;
- (d) when aligned with any of SEQ ID NOs: 77 to 95 using a pairwise alignment algorithm, each moving window of x amino acids from N-terminus to C-terminus (such that for an alignment that extends to p amino acids, where p>x, there are p-x+1 such windows) has at least xy identical aligned amino acids, where: x is selected from; y is selected from 0.75; and if x·y is not an integer then it is rounded up to the nearest integer,
- The preferred pairwise alignment algorithm is the Needleman-Wunsch global alignment algorithm [7], using default parameters (e.g. with Gap opening penalty = 10.0, and with Gap extension penalty = 0.5, using the EBLOSUM62 scoring matrix). This algorithm is conveniently implemented in the needle tool in the EMBOSS package [8].
- These polypeptides include other detoxified variants of SEQ ID NOs 77 to 95, including allelic variants, polymorphic forms, homologs, orthologs, paralogs, mutants, etc.
- The value of a may be selected from 85%, 87.5%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, 99.5% or more.
- The immunogenic polypeptide fragment in certain embodiments will not comprise at least the first 10 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 20 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 25 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 30 N-terminal amino acids of the E. coli AcfD (orf3526) protein, or at least the first 33 N-terminal amino acids of the E. coli AcfD (orf3526) protein.
- The immunogenic polypeptide fragment in certain embodiments (which may be combined with the preceding embodiments) will not comprise at least the last 125 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 150 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 175 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 200 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 210 C-terminal amino acids of the E. coli AcfD (orf3526) protein, or at least the last 217 C-terminal amino acids of the E. coli AcfD (orf3526) protein.
- The invention additionally provides an immunogenic composition that comprises an immunogenic polypeptide fragment of an E. coli AcfD (orf3526) polypeptide wherein the immunogenic polypeptide fragment comprises an amino acid sequence that comprises at least a% sequence identity to any one of SEQ ID NOs: 77 to 95, wherein the immunogenic polypeptide fragment raises a substantially similar immune response in a subject as the E. coli AcfD (orf3526) protein and wherein the immunogenic polypeptide fragment does not comprise at least the last 125 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 150 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 175 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 200 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 210 C-terminal amino acids of the E. coli AcfD (orf3526) protein, or at least the last 217 C-terminal amino acids of the E. coli AcfD (orf3526) protein.
- The preferred pairwise alignment algorithm is the Needleman-Wunsch global alignment algorithm [7], using default parameters (e.g. with Gap opening penalty = 10.0, and with Gap extension penalty = 0.5, using the EBLOSUM62 scoring matrix). This algorithm is conveniently implemented in the needle tool in the EMBOSS package [8].
- These polypeptides include other variants of SEQ ID NOs 77 to 95, including allelic variants, polymorphic forms, homologs, orthologs, paralogs, mutants, etc.
- The value of a may be selected from 50%, 60%, 65%, 70%, 75%, 80%, 85%, 87.5%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, 99.5% or more.
- The immunogenic polypeptide fragment of the immunogenic composition in certain embodiments will not comprise at least the first 10 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 20 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 25 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 30 N-terminal amino acids of the E. coli AcfD (orf3526) protein, or at least the first 33 N-terminal amino acids of the E. coli AcfD (orf3526) protein.
- The immunogenic composition may further comprise an adjuvant which may in certain embodiments comprise 1-12% by volume of a metabolizable oil, and 0.2% to 2.5% by weight of an emulsifying agent, wherein the metabolizable oil and the emulsifying agent are present in the form of an oil-in-water emulsion having oil droplets substantially all of which are less than 1 micron in diameter; 4-5% by volume of squalene, and (b) about 1% of an emulsifying agent comprising polyoxyethylenesorbitan monooleate and sorbitan trioleate, wherein the squalene and the emulsifying agent are present in the form of an oil-in-water emulsion having oil droplets substantially all of which are less than 1 micron in diameter; or MF59 (TM).
- The foregoing detoxified immunogenic polypeptides and immunogenic polypeptide fragments preferably retain at least one epitope or immunogenic fragment of
SEQ ID NOs 1 to 19. An epitope within a fragment may be a B-cell epitope and/or a T-cell epitope. Such epitopes can be identified empirically (e.g. using PEPSCAN [9,10] or similar methods), or they can be predicted (e.g. using the Jameson-Wolf antigenic index [11], matrix-based approaches [12], MAPITOPE [13], TEPITOPE [14,15], neural networks [16], OptiMer & EpiMer [17, 18], ADEPT [19], Tsites [20], hydrophilicity [21], antigenic index [22] or the methods disclosed in references 23-27, etc.). Epitopes are the parts of an antigen that are recognised by and bind to the antigen binding sites of antibodies or T-cell receptors, and they may also be referred to as "antigenic determinants". - The foregoing detoxified immunogenic polypeptides and immunogenic polypeptide fragments include, without limitation, immunogenic polypeptides that, when administered to a subject in a suitable composition which can include an adjuvant (including without limitation any of the adjuvants listed or discussed in the section "Immunogenic compositions and medicaments" below), or a suitable carrier coupled to the polypeptide, induces an antibody or T-cell mediated immune response that recognizes the isolated full length polypeptide
SEQ ID NOs 1 to 19, respectively, from which the immunogenic polypeptide is derived. - The foregoing detoxified immunogenic polypeptides and immunogenic polypeptide fragments include, without limitation, immunogenic polypeptides that, when administered to a subject in a suitable composition which can include an adjuvant (including without limitation any of the adjuvants listed or discussed in the section "Immunogenic compositions and medicaments" below), or a suitable carrier coupled to the polypeptide, induces an antibody or T-cell mediated immune response that recognizes the isolated full length polypeptide
SEQ ID NOs 1 to 19, respectively, from which the immunogenic fragment is derived. - The foregoing detoxified immunogenic polypeptides and immunogenic polypeptide fragments include, without limitation, immunogenic polypeptides that, when administered to a subject in a suitable composition which can include an adjuvant (including without limitation any of the adjuvants listed or discussed in the section "Immunogenic compositions and medicaments" below), or a suitable carrier coupled to the polypeptide, induces an antibody or T-cell mediated immune response that recognizes the isolated full length polypeptide
SEQ ID NOs 1 to 19, respectively, from which the immunogenic fragment is derived. - A detoxified polypeptide of the invention may, compared to any one of
SEQ ID NOs 1 to 19, include one or more (e.g. 1, 2, 3, 4, 5, 6, 7, 8, 9, etc.) amino acid substitutions, such as conservative substitutions (i.e. substitutions of one amino acid with another which has a related side chain). Genetically-encoded amino acids are generally divided into four families: (1) acidic i.e. aspartate, glutamate; (2) basic i.e. lysine, arginine, histidine; (3) non-polar i.e. alanine, valine, leucine, isoleucine, proline, phenylalanine, methionine, tryptophan; and (4) uncharged polar i.e. glycine, asparagine, glutamine, cysteine, serine, threonine, tyrosine. Phenylalanine, tryptophan, and tyrosine are sometimes classified jointly as aromatic amino acids. In general, substitution of single amino acids within these families does not have a major effect on the biological activity. - A detoxified polypeptide may include one or more (e.g. 1, 2, 3, 4, 5, 6, 7, 8, 9, etc.) single amino acid deletions relative to any one of
SEQ ID NOs 1 to 19. Similarly, a polypeptides may include one or more (e.g. 1, 2, 3, 4, 5, 6, 7, 8, 9, etc.) insertions (e.g. each of 1, 2, 3, 4 or 5 amino acids) relative to any one ofSEQ ID NOs 1 to 19. - In general, when a detoxified polypeptide or immunogenic polypeptide fragment of the invention comprises a sequence that is not identical to a complete one of
SEQ ID NOs 1 to 19 (e.g. when it comprises a sequence listing with <100% sequence identity thereto, or when it comprises a fragment thereof) it is preferred that the polypeptide can elicit an antibody that recognises a polypeptide consisting of the complete SEQ ID sequence i.e. the antibody binds to one or more of saidSEQ ID NOs 1 to 19. Such antibody may bind specifically toSEQ ID NOs 1 to 19, respectively while not binding to non-AcfD (orf3526) proteins with affinity significantly higher than the antibody's non-specific affinity to human serum albumin as a non-specific binding reference standard. - Polypeptides used with the invention can take various forms (e.g. native, fusions, glycosylated, non-glycosylated, lipidated, non-lipidated, phosphorylated, non-phosphorylated, myristoylated, non-myristoylated, monomeric, multimeric, particulate, denatured, etc.). For instance, a polypeptide of the invention may have a lipidated N-terminal cysteine (e.g. Cys-24 of SEQ ID NOs: 1 to 19).
- Polypeptides used with the invention can be prepared by various means (e.g. recombinant expression, purification from cell culture, chemical synthesis, etc.). Recombinantly-expressed proteins are preferred.
- Polypeptides used with the invention are preferably provided in purified or substantially purified form i.e. substantially free from other polypeptides (e.g. free from naturally-occurring polypeptides), particularly from other E. coli or host cell polypeptides, and are generally at least about 50% pure (by weight), and usually at least about 90% pure i.e. less than about 50%, and more preferably less than about 10% (e.g. 5%) of a composition is made up of other expressed polypeptides. Thus the antigens in the compositions are separated from the whole organism with which the molecule is expressed.
- Polypeptides used with the invention are preferably E. coli polypeptides. Such polypeptides may be further selected from NMEC, APEC, UPEC, EAEC, ETEC, EPEC and ETEC E. coli polypeptides.
- The term "polypeptide" refers to amino acid polymers of any length. The polymer may be linear or branched, it may comprise modified amino acids, and it may be interrupted by non-amino acids. The terms also encompass an amino acid polymer that has been modified naturally or by intervention; for example, disulfide bond formation, glycosylation, lipidation, acetylation, phosphorylation, or any other manipulation or modification, such as conjugation with a labeling component. Also included are, for example, polypeptides containing one or more analogs of an amino acid (including, for example, unnatural amino acids, etc.), as well as other modifications known in the art. Polypeptides can occur as single chains or associated chains.
- Herein disclosed is polypeptides comprising a sequence -P-Q- or -Q-P-, wherein: -P- is an amino acid sequence as defined above and -Q- is not a sequence as defined above i.e. the invention provides fusion proteins. Where the N-terminus codon of -P- is not ATG, but this codon is not present at the N-terminus of a polypeptide, it will be translated as the standard amino acid for that codon rather than as a Met. Where this codon is at the N-terminus of a polypeptide, however, it will be translated as Met. Examples of -Q- moieties include, but are not limited to, histidine tags (i.e. His n where n = 3, 4, 5, 6, 7, 8, 9, 10 or more), a maltose-binding protein, or glutathione-S-transferase (GST).
- Further disclosed is an oligomeric protein comprising a polypeptide of the invention. The oligomer may be a dimer, a trimer, a tetramer, etc. The oligomer may be a homo-oligomer or a hetero-oligomer. Polypeptides in the oligomer may be covalently or non-covalently associated.
- Comparison of the immune response raised in a subject by the polypeptide with the immune response raised by the full length protein may be carried out use by any means available to one of skill in the art. One simple method as used in the examples below involves immunization of a model subject such as mouse and then challenge with a lethal dose of E. coli. For proper comparison, one of skill in the art would naturally select the same adjuvant such as Freund's complete adjuvant. In such a test the immunogenic polypeptide fragments of the present invention will raise a substantially similar immune response in a subject (i.e., will provide substantially the same protection against the lethal challenge) if, for example, the polypeptide provides at least 70% of the protection provided by the full length protein, at least 80% of the protection provided by the full length protein, at least 85% of the protection provided by the full length protein, at least 90% of the protection provided by the full length protein, at least 95% of the protection provided by the full length protein, at least 97% of the protection provided by the full length protein, at least 98% of the protection provided by the full length protein, or at least 99% of the protection provided by the full length protein.
- The full length AcfD (orf3526) protein against which the immunogenic polypeptide fragment would be compared (for solubility, toxicity and immune response raised) may be any representative E. coli AcfD (orf3526) protein including without limitation SEQ ID NOs 1-19. The AcfD (orf3526) protein will be the corresponding full length protein from which the immunogenic polypeptide fragment is obtained.
- The invention also provides a process for producing a polypeptide of the invention, comprising the step of culturing a host cell transformed with nucleic acid of the invention under conditions which induce polypeptide expression. The polypeptide may then be purified e.g. from culture supernatants.
- The invention provides an E. coli cell, containing a plasmid that encodes a polypeptide of the invention. The chromosome of the E. coli cell may include a homolog of AcfD (orf3526), or such a homolog may be absent, but in both cases the polypeptide of the invention can be expressed from the plasmid. The plasmid may include a gene encoding a marker, etc. These and other details of suitable plasmids are given below.
- Although expression of the polypeptides of the invention may take place in an E. coli strain, the invention will usually use a heterologous host for expression. The heterologous host may be prokaryotic (e.g. a bacterium) or eukaryotic. Suitable hosts include, but are not limited to, Bacillus subtilis, Vibrio cholerae, Salmonella typhi, Salmonella typhimurium, Neisseria lactamica, Neisseria cinerea, Mycobacteria (e.g. M.tuberculosis), yeasts, etc.
- The invention provides a process for producing a polypeptide of the invention, comprising the step of synthesising at least part of the polypeptide by chemical means.
- Any and all of the foregoing proteins, polypeptides, hybrid polypeptides, epitopes and immunogenic fragments may be in any one of a number of forms including, without limitation, recombinant, isolated or substantially purified (from materials co-existing with such proteins, polypeptides, hybrid polypeptides, epitopes and immunogenic fragments in their natural state).
- The invention also provides nucleic acid encoding polypeptides and hybrid polypeptides of the invention. It also provides nucleic acid comprising a nucleotide sequence that encodes one or more polypeptides or hybrid polypeptides of the invention.
- The invention also provides nucleic acid comprising nucleotide sequences having sequence identity to such nucleotide sequences. Identity between sequences is preferably determined by the Smith-Waterman homology search algorithm as described above. Such nucleic acids include those using alternative codons to encode the same amino acid.
- The invention also provides nucleic acid which can hybridize to these nucleic acids. Hybridization reactions can be performed under conditions of different "stringency". Conditions that increase stringency of a hybridization reaction of widely known and published in the art (e.g. page 7.52 of reference 211). Examples of relevant conditions include (in order of increasing stringency): incubation temperatures of 25°C, 37°C, 50°C, 55°C and 68°C; buffer concentrations of 10 x SSC, 6 x SSC, 1 x SSC, 0.1 x SSC (where SSC is 0.15 M NaCl and 15 mM citrate buffer) and their equivalents using other buffer systems; formamide concentrations of 0%, 25%, 50%, and 75%; incubation times from 5 minutes to 24 hours; 1, 2, or more washing steps; wash incubation times of 1, 2, or 15 minutes; and wash solutions of 6 x SSC, 1 x SSC, 0.1 x SSC, or de-ionized water. Hybridization techniques and their optimization are well known in the art (e.g. see
refs - In some embodiments, nucleic acid of the invention hybridizes to a target under low stringency conditions; in other embodiments it hybridizes under intermediate stringency conditions; in preferred embodiments, it hybridizes under high stringency conditions. An exemplary set of low stringency hybridization conditions is 50°C and 10 x SSC. An exemplary set of intermediate stringency hybridization conditions is 55°C and 1 x SSC. An exemplary set of high stringency hybridization conditions is 68°C and 0.1 x SSC.
- The invention includes nucleic acid comprising sequences complementary to these sequences (e.g. for antisense or probing, or for use as primers).
- Nucleic acids of the invention can be used in hybridisation reactions (e.g. Northern or Southern blots, or in nucleic acid microarrays or 'gene chips') and amplification reactions (e.g. PCR, SDA, SSSR, LCR, TMA, NASBA, etc.) and other nucleic acid techniques.
- Nucleic acid according to the invention can take various forms (e.g. single-stranded, double-stranded, vectors, primers, probes, labelled etc.). Nucleic acids of the invention may be circular or branched, but will generally be linear. Unless otherwise specified or required, any embodiment of the invention that utilizes a nucleic acid may utilize both the double-stranded form and each of two complementary single-stranded forms which make up the double-stranded form. Primers and probes are generally single-stranded, as are antisense nucleic acids.
- Nucleic acids of the invention are preferably provided in purified or substantially purified form i.e. substantially free from other nucleic acids (e.g. free from naturally-occurring nucleic acids), particularly from other E. coli or host cell nucleic acids, generally being at least about 50% pure (by weight), and usually at least about 90% pure. Nucleic acids of the invention are preferably E. coli nucleic acids.
- Nucleic acids of the invention may be prepared in many ways e.g. by chemical synthesis (e.g. phosphoramidite synthesis of DNA) in whole or in part, by digesting longer nucleic acids using nucleases (e.g. restriction enzymes), by joining shorter nucleic acids or nucleotides (e.g. using ligases or polymerases), from genomic or cDNA libraries, etc.
- Nucleic acid of the invention may be attached to a solid support (e.g. a bead, plate, filter, film, slide, microarray support, resin, etc.). Nucleic acid of the invention may be labelled e.g. with a radioactive or fluorescent label, or a biotin label. This is particularly useful where the nucleic acid is to be used in detection techniques e.g. where the nucleic acid is a primer or as a probe.
- The term "nucleic acid" includes in general means a polymeric form of nucleotides of any length, which contain deoxyribonucleotides, ribonucleotides, and/or their analogs. It includes DNA, RNA, DNA/RNA hybrids. It also includes DNA or RNA analogs, such as those containing modified backbones (e.g. peptide nucleic acids (PNAs) or phosphorothioates) or modified bases. Thus the invention includes mRNA, tRNA, rRNA, ribozymes, DNA, cDNA, recombinant nucleic acids, branched nucleic acids, plasmids, vectors, probes, primers, etc. Where nucleic acid of the invention takes the form of RNA, it may or may not have a 5' cap.
- Nucleic acids of the invention may be part of a vector i.e. part of a nucleic acid construct designed for transduction/transfection of one or more cell types. Vectors may be, for example, "cloning vectors" which are designed for isolation, propagation and replication of inserted nucleotides, "expression vectors" which are designed for expression of a nucleotide sequence in a host cell, "viral vectors" which is designed to result in the production of a recombinant virus or virus-like particle, or "shuttle vectors", which comprise the attributes of more than one type of vector. Preferred vectors are plasmids, as mentioned above. A "host cell" includes an individual cell or cell culture which can be or has been a recipient of exogenous nucleic acid. Host cells include progeny of a single host cell, and the progeny may not necessarily be completely identical (in morphology or in total DNA complement) to the original parent cell due to natural, accidental, or deliberate mutation and/or change. Host cells include cells transfected or infected in vivo or in vitro with nucleic acid of the invention.
- Where a nucleic acid is DNA, it will be appreciated that "U" in a RNA sequence will be replaced by "T" in the DNA. Similarly, where a nucleic acid is RNA, it will be appreciated that "T" in a DNA sequence will be replaced by "U" in the RNA.
- The term "complement" or "complementary" when used in relation to nucleic acids refers to Watson-Crick base pairing. Thus the complement of C is G, the complement of G is C, the complement of A is T (or U), and the complement of T (or U) is A. It is also possible to use bases such as I (the purine inosine) e.g. to complement pyrimidines (C or T).
- Nucleic acids of the invention can be used, for example: to produce polypeptides; as hybridization probes for the detection of nucleic acid in biological samples; to generate additional copies of the nucleic acids; to generate ribozymes or antisense oligonucleotides; as single-stranded DNA primers or probes; or as triple-strand forming oligonucleotides.
- The invention provides a process for producing nucleic acid of the invention, wherein the nucleic acid is synthesised in part or in whole using chemical means.
- The invention provides vectors comprising nucleotide sequences of the invention (e.g. cloning or expression vectors) and host cells transformed with such vectors.
- Nucleic acid amplification according to the invention may be quantitative and/or real-time.
- For certain embodiments of the invention, nucleic acids are preferably at least 7 nucleotides in length (e.g. 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 45, 50, 55, 60, 65, 70, 75, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 225, 250,275, 300 nucleotides or longer).
- For certain embodiments of the invention, nucleic acids are preferably at most 500 nucleotides in length (e.g. 450, 400, 350, 300, 250, 200, 150, 140, 130, 120, 110, 100, 90, 80, 75, 70, 65, 60, 55, 50, 45, 40, 39, 38, 37, 36, 35, 34, 33, 32, 31, 30, 29, 28, 27, 26, 25, 24, 23, 22, 21, 20, 19, 18, 17, 16, 15 nucleotides or shorter).
- Primers and probes of the invention, and other nucleic acids used for hybridization, are preferably between 10 and 30 nucleotides in length (e.g. 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, or 30 nucleotides).
- Polypeptides of the invention are useful as active ingredients (immunogens) in immunogenic compositions, and such compositions may be useful as vaccines. Vaccines according to the invention may either be prophylactic (i.e. to prevent infection) or therapeutic (i.e. to treat infection), but will typically be prophylactic.
- Immunogenic compositions will be pharmaceutically acceptable. They will usually include components in addition to the antigens e.g. they typically include one or more pharmaceutical carrier(s), excipient(s) and/or adjuvant(s). A thorough discussion of carriers and excipients is available in ref.208. Thorough discussions of vaccine adjuvants are available in refs. 30 and 31.
- Compositions will generally be administered to a mammal in aqueous form. Prior to administration, however, the composition may have been in a non-aqueous form. For instance, although some vaccines are manufactured in aqueous form, then filled and distributed and administered also in aqueous form, other vaccines are lyophilised during manufacture and are reconstituted into an aqueous form at the time of use. Thus a composition of the invention may be dried, such as a lyophilised formulation.
- The composition may include preservatives such as thiomersal or 2-phenoxyethanol. It is preferred, however, that the vaccine should be substantially free from (i.e. less than 5µg/ml) mercurial material e.g. thiomersal-free. Vaccines containing no mercury are more preferred. Preservative-free vaccines are particularly preferred.
- To improve thermal stability, a composition may include a temperature protective agent.
- To control tonicity, it is preferred to include a physiological salt, such as a sodium salt. Sodium chloride (NaCl) is preferred, which may be present at between 1 and 20 mg/ml e.g. about 10±2mg/ml NaCl. Other salts that may be present include potassium chloride, potassium dihydrogen phosphate, disodium phosphate dehydrate, magnesium chloride, calcium chloride, etc.
- Compositions will generally have an osmolality of between 200 mOsm/kg and 400 mOsm/kg, preferably between 240-360 mOsm/kg, and will more preferably fall within the range of 290-310 mOsm/kg.
- Compositions may include one or more buffers. Typical buffers include: a phosphate buffer; a Tris buffer; a borate buffer; a succinate buffer; a histidine buffer (particularly with an aluminum hydroxide adjuvant); or a citrate buffer. Buffers will typically be included in the 5-20mM range.
- The pH of a composition will generally be between 5.0 and 8.1, and more typically between 6.0 and 8.0 e.g. 6.5 and 7.5, or between 7.0 and 7.8.
- The composition is preferably sterile. The composition is preferably non-pyrogenic e.g. containing <1 EU (endotoxin unit, a standard measure) per dose, and preferably <0.1 EU per dose. The composition is preferably gluten free.
- The composition may include material for a single immunisation, or may include material for multiple immunisations (i.e. a 'multidose' kit). The inclusion of a preservative is preferred in multidose arrangements. As an alternative (or in addition) to including a preservative in multidose compositions, the compositions may be contained in a container having an aseptic adaptor for removal of material.
- Human vaccines are typically administered in a dosage volume of about 0.5ml, although a half dose (i.e. about 0.25ml) may be administered to children.
- Immunogenic compositions of the invention may also comprise one or more immunoregulatory agents. Preferably, one or more of the immunoregulatory agents include one or more adjuvants. The adjuvants may include a TH1 adjuvant and/or a TH2 adjuvant, further discussed below.
- Adjuvants which may be used in compositions of the invention include, but are not limited to:
- Mineral containing compositions suitable for use as adjuvants in the invention include mineral salts, such as aluminium salts and calcium salts (or mixtures thereof). Calcium salts include calcium phosphate (e.g. the "CAP" particles disclosed in ref. 32). Aluminum salts include hydroxides, phosphates, sulfates, etc., with the salts taking any suitable form (e.g. gel, crystalline, amorphous, etc.). Adsorption to these salts is preferred. The mineral containing compositions may also be formulated as a particle of metal salt [33].
- The adjuvants known as aluminum hydroxide and aluminum phosphate may be used. These names are conventional, but are used for convenience only, as neither is a precise description of the actual chemical compound which is present (e.g. see
chapter 9 of reference 30). The invention can use any of the "hydroxide" or "phosphate" adjuvants that are in general use as adjuvants. The adjuvants known as "aluminium hydroxide" are typically aluminium oxyhydroxide salts, which are usually at least partially crystalline. The adjuvants known as "aluminium phosphate" are typically aluminium hydroxyphosphates, often also containing a small amount of sulfate (i.e. aluminium hydroxyphosphate sulfate). They may be obtained by precipitation, and the reaction conditions and concentrations during precipitation influence the degree of substitution of phosphate for hydroxyl in the salt. - A fibrous morphology (e.g. as seen in transmission electron micrographs) is typical for aluminium hydroxide adjuvants. The pI of aluminium hydroxide adjuvants is typically about 11 i.e. the adjuvant itself has a positive surface charge at physiological pH. Adsorptive capacities of between 1.8-2.6 mg protein per mg Al+++ at pH 7.4 have been reported for aluminium hydroxide adjuvants.
- Aluminium phosphate adjuvants generally have a PO4/Al molar ratio between 0.3 and 1.2, preferably between 0.8 and 1.2, and more preferably 0.95±0.1. The aluminium phosphate will generally be amorphous, particularly for hydroxyphosphate salts. A typical adjuvant is amorphous aluminium hydroxyphosphate with PO4/Al molar ratio between 0.84 and 0.92, included at 0.6mg Al3+/ml. The aluminium phosphate will generally be particulate (e.g. plate-like morphology as seen in transmission electron micrographs). Typical diameters of the particles are in the range 0.5-20µm (e.g. about 5-10µm) after any antigen adsorption. Adsorptive capacities of between 0.7-1.5 mg protein per mg Al+++ at pH 7.4 have been reported for aluminium phosphate adjuvants.
- The point of zero charge (PZC) of aluminium phosphate is inversely related to the degree of substitution of phosphate for hydroxyl, and this degree of substitution can vary depending on reaction conditions and concentration of reactants used for preparing the salt by precipitation. PZC is also altered by changing the concentration of free phosphate ions in solution (more phosphate = more acidic PZC) or by adding a buffer such as a histidine buffer (makes PZC more basic). Aluminium phosphates used according to the invention will generally have a PZC of between 4.0 and 7.0, more preferably between 5.0 and 6.5 e.g. about 5.7.
- Suspensions of aluminium salts used to prepare compositions of the invention may contain a buffer (e.g. a phosphate or a histidine or a Tris buffer), but this is not always necessary. The suspensions are preferably sterile and pyrogen-free. A suspension may include free aqueous phosphate ions e.g. present at a concentration between 1.0 and 20 mM, preferably between 5 and 15 mM, and more preferably about 10 mM. The suspensions may also comprise sodium chloride.
- The invention can use a mixture of both an aluminium hydroxide and an aluminium phosphate. In this case there may be more aluminium phosphate than hydroxide e.g. a weight ratio of at least 2:1 e.g. ≥5:1, ≥6:1, ≥7:1, ≥8:1, ≥9:1, etc.
- The concentration of Al+++ in a composition for administration to a patient is preferably less than 10mg/ml e.g. ≤5 mg/ml, ≤4 mg/ml, ≤3 mg/ml, ≤2 mg/ml, ≤1 mg/ml, etc. A preferred range is between 0.3 and 1mg/ml. A maximum of 0.85mg/dose is preferred.
- Oil emulsion compositions suitable for use as adjuvants in the invention include squalene-water emulsions, such as MF59 [
Chapter 10 of ref. 30; see also ref. 34] (5% Squalene, 0.5% Tween 80, and 0.5% Span 85, formulated into submicron particles using a microfluidizer). Complete Freund's adjuvant (CFA) and incomplete Freund's adjuvant (IFA) may also be used. - Various oil-in-water emulsion adjuvants are known, and they typically include at least one oil and at least one surfactant, with the oil(s) and surfactant(s) being biodegradable (metabolisable) and biocompatible. The oil droplets in the emulsion are generally less than 5µm in diameter, and ideally have a sub-micron diameter, with these small sizes being achieved with a microfluidiser to provide stable emulsions. Droplets with a size less than 220nm are preferred as they can be subjected to filter sterilization.
- The emulsion can comprise oils such as those from an animal (such as fish) or vegetable source. Sources for vegetable oils include nuts, seeds and grains. Peanut oil, soybean oil, coconut oil, and olive oil, the most commonly available, exemplify the nut oils. Jojoba oil can be used e.g. obtained from the jojoba bean. Seed oils include safflower oil, cottonseed oil, sunflower seed oil, sesame seed oil and the like. In the grain group, corn oil is the most readily available, but the oil of other cereal grains such as wheat, oats, rye, rice, teff, triticale and the like may also be used. 6-10 carbon fatty acid esters of glycerol and 1,2-propanediol, while not occurring naturally in seed oils, may be prepared by hydrolysis, separation and esterification of the appropriate materials starting from the nut and seed oils. Fats and oils from mammalian milk are metabolizable and may therefore be used in the practice of this invention. The procedures for separation, purification, saponification and other means necessary for obtaining pure oils from animal sources are well known in the art. Most fish contain metabolizable oils which may be readily recovered. For example, cod liver oil, shark liver oils, and whale oil such as spermaceti exemplify several of the fish oils which may be used herein. A number of branched chain oils are synthesized biochemically in 5-carbon isoprene units and are generally referred to as terpenoids. Shark liver oil contains a branched, unsaturated terpenoids known as squalene, 2,6,10,15,19,23-hexamethyl-2,6,10,14,18,22-tetracosahexaene, which is particularly preferred herein. Squalane, the saturated analog to squalene, is also a preferred oil. Fish oils, including squalene and squalane, are readily available from commercial sources or may be obtained by methods known in the art. Other preferred oils are the tocopherols (see below). Mixtures of oils can be used.
- Surfactants can be classified by their 'HLB' (hydrophile/lipophile balance). Preferred surfactants of the invention have a HLB of at least 10, preferably at least 15, and more preferably at least 16. The invention can be used with surfactants including, but not limited to: the polyoxyethylene sorbitan esters surfactants (commonly referred to as the Tweens), especially
polysorbate 20 andpolysorbate 80; copolymers of ethylene oxide (EO), propylene oxide (PO), and/or butylene oxide (BO), sold under the DOWFAX™ tradename, such as linear EO/PO block copolymers; octoxynols, which can vary in the number of repeating ethoxy (oxy-1,2-ethanediyl) groups, with octoxynol-9 (Triton X-100, or t-octylphenoxypolyethoxyethanol) being of particular interest; (octylphenoxy)polyethoxyethanol (IGEPAL CA-630/NP-40); phospholipids such as phosphatidylcholine (lecithin); nonylphenol ethoxylates, such as the Tergitol™ NP series; polyoxyethylene fatty ethers derived from lauryl, cetyl, stearyl and oleyl alcohols (known as Brij surfactants), such as triethyleneglycol monolauryl ether (Brij 30); and sorbitan esters (commonly known as the SPANs), such as sorbitan trioleate (Span 85) and sorbitan monolaurate. Non-ionic surfactants are preferred. Preferred surfactants for including in the emulsion are Tween 80 (polyoxyethylene sorbitan monooleate), Span 85 (sorbitan trioleate), lecithin and Triton X-100. - Mixtures of surfactants can be used e.g.
Tween 80/Span 85 mixtures. A combination of a polyoxyethylene sorbitan ester such as polyoxyethylene sorbitan monooleate (Tween 80) and an octoxynol such as t-octylphenoxypolyethoxyethanol (Triton X-100) is also suitable. Another useful combination compriseslaureth 9 plus a polyoxyethylene sorbitan ester and/or an octoxynol. - Preferred amounts of surfactants (% by weight) are: polyoxyethylene sorbitan esters (such as Tween 80) 0.01 to 1%, in particular about 0.1 %; octyl- or nonylphenoxy polyoxyethanols (such as Triton X-100, or other detergents in the Triton series) 0.001 to 0.1 %, in particular 0.005 to 0.02%; polyoxyethylene ethers (such as laureth 9) 0.1 to 20 %, preferably 0.1 to 10 % and in particular 0.1 to 1 % or about 0.5%.
- Preferred emulsion adjuvants have an average droplets size of <1µm e.g. ≤750nm, ≤500nm, ≤400nm, ≤300nm, ≤250nm, ≤220nm, ≤200nm, or smaller. These droplet sizes can conveniently be achieved by techniques such as microfluidisation.
- Specific oil-in-water emulsion adjuvants useful with the invention include, but are not limited to:
- A submicron emulsion of squalene,
Tween 80, and Span 85. The composition of the emulsion by volume can be about 5% squalene, about 0.5% polysorbate 80 and about 0.5% Span 85. In weight terms, these ratios become 4.3% squalene, 0.5% polysorbate 80 and 0.48% Span 85. This adjuvant is known as 'MF59' [35-37], as described in more detail inChapter 10 of ref. 38 andchapter 12 of ref. 39. The MF59 emulsion advantageously includes citrate ions e.g. 10mM sodium citrate buffer. - An emulsion of squalene, a tocopherol, and
Tween 80. The emulsion may include phosphate buffered saline. It may also include Span 85 (e.g. at 1 %) and/or lecithin. These emulsions may have from 2 to 10% squalene, from 2 to 10% tocopherol and from 0.3 to 3% Tween 80, and the weight ratio of squalene:tocopherol is preferably≤1 as this provides a more stable emulsion. Squalene andTween 80 may be present volume ratio of about 5:2. One such emulsion can be made by dissolvingTween 80 in PBS to give a 2% solution, then mixing 90ml of this solution with a mixture of (5g of DL-α-tocopherol and 5ml squalene), then microfluidising the mixture. The resulting emulsion may have submicron oil droplets e.g. with an average diameter of between 100 and 250nm, preferably about 180nm. - An emulsion of squalene, a tocopherol, and a Triton detergent (e.g. Triton X-100). The emulsion may also include a 3d-MPL (see below). The emulsion may contain a phosphate buffer.
- An emulsion comprising a polysorbate (e.g. polysorbate 80), a Triton detergent (e.g. Triton X-100) and a tocopherol (e.g. an α-tocopherol succinate). The emulsion may include these three components at a mass ratio of about 75:11:10 (e.g. 750µg/
ml polysorbate 80, 110µg/ml Triton X-100 and 100µg/ml α-tocopherol succinate), and these concentrations should include any contribution of these components from antigens. The emulsion may also include squalene. The emulsion may also include a 3d-MPL (see below). The aqueous phase may contain a phosphate buffer. - An emulsion of squalane,
polysorbate 80 and poloxamer 401 ("Pluronic™ L121"). The emulsion can be formulated in phosphate buffered saline, pH 7.4. This emulsion is a useful delivery vehicle for muramyl dipeptides, and has been used with threonyl-MDP in the "SAF-1" adjuvant [40] (0.05-1% Thr-MDP, 5% squalane, 2.5% Pluronic L121 and 0.2% polysorbate 80). It can also be used without the Thr-MDP, as in the "AF" adjuvant [41] (5% squalane, 1.25% Pluronic L121 and 0.2% polysorbate 80). Microfluidisation is preferred. - An emulsion comprising squalene, an aqueous solvent, a polyoxyethylene alkyl ether hydrophilic nonionic surfactant (e.g. polyoxyethylene (12) cetostearyl ether) and a hydrophobic nonionic surfactant (e.g. a sorbitan ester or mannide ester, such as sorbitan monoleate or 'Span 80'). The emulsion is preferably thermoreversible and/or has at least 90% of the oil droplets (by volume) with a size less than 200 nm [42]. The emulsion may also include one or more of: alditol; a cryoprotective agent (e.g. a sugar, such as dodecylmaltoside and/or sucrose); and/or an alkylpolyglycoside. Such emulsions may be lyophilized.
- An emulsion of squalene, poloxamer 105 and Abil-Care [43]. The final concentration (weight) of these components in adjuvanted vaccines are 5% squalene, 4% poloxamer 105 (pluronic polyol) and 2% Abil-Care 85 (Bis-PEG/PPG-16/16 PEG/PPG-16/16 dimethicone; caprylic/capric triglyceride).
- An emulsion having from 0.5-50% of an oil, 0.1-10% of a phospholipid, and 0.05-5% of a non-ionic surfactant. As described in reference 44, preferred phospholipid components are phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine, phosphatidylinositol, phosphatidylglycerol, phosphatidic acid, sphingomyelin and cardiolipin. Submicron droplet sizes are advantageous.
- A submicron oil-in-water emulsion of a non-metabolisable oil (such as light mineral oil) and at least one surfactant (such as lecithin,
Tween 80 or Span 80). Additives may be included, such as QuilA saponin, cholesterol, a saponin-lipophile conjugate (such as GPI-0100, described in reference 45, produced by addition of aliphatic amine to desacylsaponin via the carboxyl group of glucuronic acid), dimethyidioctadecylammonium bromide and/or N,N-dioctadecyl-N,N-bis (2-hydroxyethyl)propanediamine. - An emulsion in which a saponin (e.g. QuilA or QS21) and a sterol (e.g. a cholesterol) are associated as helical micelles [46].
- An emulsion comprising a mineral oil, a non-ionic lipophilic ethoxylated fatty alcohol, and a non-ionic hydrophilic surfactant (e.g. an ethoxylated fatty alcohol and/or polyoxyethylene-polyoxypropylene block copolymer) [47].
- An emulsion comprising a mineral oil, a non-ionic hydrophilic ethoxylated fatty alcohol, and a non-ionic lipophilic surfactant (e.g. an ethoxylated fatty alcohol and/or polyoxyethylene-polyoxypropylene block copolymer) [47].
- In some embodiments an emulsion may be mixed with antigen extemporaneously, at the time of delivery, and thus the adjuvant and antigen may be kept separately in a packaged or distributed vaccine, ready for final formulation at the time of use. In other embodiments an emulsion is mixed with antigen during manufacture, and thus the composition is packaged in a liquid adjuvanted form. The antigen will generally be in an aqueous form, such that the vaccine is finally prepared by mixing two liquids. The volume ratio of the two liquids for mixing can vary (e.g. between 5:1 and 1:5) but is generally about 1:1. Where concentrations of components are given in the above descriptions of specific emulsions, these concentrations are typically for an undiluted composition, and the concentration after mixing with an antigen solution will thus decrease.
- Where a composition includes a tocopherol, any of the α, β, γ, δ, ε or ξ tocopherols can be used, but α-tocopherols are preferred. The tocopherol can take several forms e.g. different salts and/or isomers. Salts include organic salts, such as succinate, acetate, nicotinate, etc. D-α-tocopherol and DL-α-tocopherol can both be used. Tocopherols are advantageously included in vaccines for use in elderly patients (e.g. aged 60 years or older) because vitamin E has been reported to have a positive effect on the immune response in this patient group [48]. They also have antioxidant properties that may help to stabilize the emulsions [49]. A preferred α-tocopherol is DL-α-tocopherol, and the preferred salt of this tocopherol is the succinate. The succinate salt has been found to cooperate with TNF-related ligands in vivo.
- Saponin formulations may also be used as adjuvants in the invention. Saponins are a heterogeneous group of sterol glycosides and triterpenoid glycosides that are found in the bark, leaves, stems, roots and even flowers of a wide range of plant species. Saponin from the bark of the Quillaia saponaria Molina tree have been widely studied as adjuvants. Saponin can also be commercially obtained from Smilax ornata (sarsaprilla), Gypsophilla paniculata (brides veil), and Saponaria officianalis (soap root). Saponin adjuvant formulations include purified formulations, such as QS21, as well as lipid formulations, such as ISCOMs. QS21 is marketed as Stimulon™.
- Saponin compositions have been purified using HPLC and RP-HPLC. Specific purified fractions using these techniques have been identified, including QS7, QS17, QS18, QS21, QH-A, QH-B and QH-C. Preferably, the saponin is QS21. A method of production of QS21 is disclosed in ref. 50. Saponin formulations may also comprise a sterol, such as cholesterol [51].
- Combinations of saponins and cholesterols can be used to form unique particles called immunostimulating complexs (ISCOMs) [
chapter 23 of ref. 30]. ISCOMs typically also include a phospholipid such as phosphatidylethanolamine or phosphatidylcholine. Any known saponin can be used in ISCOMs. Preferably, the ISCOM includes one or more of QuilA, QHA & QHC. ISCOMs are further described in refs. 51-53. Optionally, the ISCOMS may be devoid of additional detergent [54]. - A review of the development of saponin based adjuvants can be found in refs. 55 & 56.
- Virosomes and virus-like particles (VLPs) can also be used as adjuvants in the invention. These structures generally contain one or more proteins from a virus optionally combined or formulated with a phospholipid. They are generally non-pathogenic, non-replicating and generally do not contain any of the native viral genome. The viral proteins may be recombinantly produced or isolated from whole viruses. These viral proteins suitable for use in virosomes or VLPs include proteins derived from influenza virus (such as HA or NA), Hepatitis B virus (such as core or capsid proteins), Hepatitis E virus, measles virus, Sindbis virus, Rotavirus, Foot-and-Mouth Disease virus, Retrovirus, Norwalk virus, human Papilloma virus, HIV, RNA-phages, QB-phage (such as coat proteins), GA-phage, fr-phage, AP205 phage, and Ty (such as retrotransposon Ty protein p1). VLPs are discussed further in refs. 57-62. Virosomes are discussed further in, for example, ref. 63
- Adjuvants suitable for use in the invention include bacterial or microbial derivatives such as non-toxic derivatives of enterobacterial lipopolysaccharide (LPS), Lipid A derivatives, immunostimulatory oligonucleotides and ADP-ribosylating toxins and detoxified derivatives thereof.
- Non-toxic derivatives of LPS include monophosphoryl lipid A (MPL) and 3-O-deacylated MPL (3dMPL). 3dMPL is a mixture of 3 de-O-acylated monophosphoryl lipid A with 4, 5 or 6 acylated chains. A preferred "small particle" form of 3 De-O-acylated monophosphoryl lipid A is disclosed in ref. 64. Such "small particles" of 3dMPL are small enough to be sterile filtered through a 0.22µm membrane [64]. Other non-toxic LPS derivatives include monophosphoryl lipid A mimics, such as aminoalkyl glucosaminide phosphate derivatives e.g. RC-529 [65,66].
- Lipid A derivatives include derivatives of lipid A from Escherichia coli such as OM-174. OM-174 is described for example in refs. 67 & 68.
- Immunostimulatory oligonucleotides suitable for use as adjuvants in the invention include nucleotide sequences containing a CpG motif (a dinucleotide sequence containing an unmethylated cytosine linked by a phosphate bond to a guanosine). Double-stranded RNAs and oligonucleotides containing palindromic or poly(dG) sequences have also been shown to be immunostimulatory.
- The CpG's can include nucleotide modifications/analogs such as phosphorothioate modifications and can be double-stranded or single-stranded.
References 69, 70 and 71 disclose possible analog substitutions e.g. replacement of guanosine with 2'-deoxy-7-deazaguanosine. The adjuvant effect of CpG oligonucleotides is further discussed in refs. 72-77. - The CpG sequence may be directed to TLR9, such as the motif GTCGTT or TTCGTT [78]. The CpG sequence may be specific for inducing a Th1 immune response, such as a CpG-A ODN, or it may be more specific for inducing a B cell response, such a CpG-B ODN. CpG-A and CpG-B ODNs are discussed in refs. 79-81. Preferably, the CpG is a CpG-A ODN.
- Preferably, the CpG oligonucleotide is constructed so that the 5' end is accessible for receptor recognition. Optionally, two CpG oligonucleotide sequences may be attached at their 3' ends to form "immunomers". See, for example, refs. 78 & 82-84.
- A useful CpG adjuvant is CpG7909, also known as ProMune™ (Coley Pharmaceutical Group, Inc.). Another is CpG1826. As an alternative, or in addition, to using CpG sequences, TpG sequences can be used [85], and these oligonucleotides may be free from unmethylated CpG motifs. The immunostimulatory oligonucleotide may be pyrimidine-rich. For example, it may comprise more than one consecutive thymidine nucleotide (e.g. TTTT, as disclosed in ref. 85), and/or it may have a nucleotide composition with >25% thymidine (e.g. >35%, >40%, >50%, >60%, >80%, etc.). For example, it may comprise more than one consecutive cytosine nucleotide (e.g. CCCC, as disclosed in ref. 85), and/or it may have a nucleotide composition with >25% cytosine (e.g. >35%, >40%, >50%, >60%, >80%, etc.). These oligonucleotides may be free from unmethylated CpG motifs. Immunostimulatory oligonucleotides will typically comprise at least 20 nucleotides. They may comprise fewer than 100 nucleotides.
- A particularly useful adjuvant based around immunostimulatory oligonucleotides is known as IC-31™ [86]. Thus an adjuvant used with the invention may comprise a mixture of (i) an oligonucleotide (e.g. between 15-40 nucleotides) including at least one (and preferably multiple) CpI motifs (i.e. a cytosine linked to an inosine to form a dinucleotide), and (ii) a polycationic polymer, such as an oligopeptide (e.g. between 5-20 amino acids) including at least one (and preferably multiple) Lys-Arg-Lys tripeptide sequence(s). The oligonucleotide may be a deoxynucleotide comprising 26-mer sequence 5'-(IC)13-3' (SEQ ID NO: 110). The polycationic polymer may be a peptide comprising 11-mer amino acid sequence KLKLLLLLKLK (SEQ ID NO:111).
- Bacterial ADP-ribosylating toxins and detoxified derivatives thereof may be used as adjuvants in the invention. Preferably, the protein is derived from E. coli (E. coli heat labile enterotoxin "LT"), cholera ("CT"), or pertussis ("PT"). The use of detoxified ADP-ribosylating toxins as mucosal adjuvants is described in ref. 87 and as parenteral adjuvants in ref. 88. The toxin or toxoid is preferably in the form of a holotoxin, comprising both A and B subunits. Preferably, the A subunit contains a detoxifying mutation; preferably the B subunit is not mutated. Preferably, the adjuvant is a detoxified LT mutant such as LT-K63, LT-R72, and LT-G192. The use of ADP-ribosylating toxins and detoxified derivatives thereof, particularly LT-K63 and LT-R72, as adjuvants can be found in refs. 89-96. A useful CT mutant is or CT-E29H [97]. Numerical reference for amino acid substitutions is preferably based on the alignments of the A and B subunits of ADP-ribosylating toxins set forth in ref. 98.
- Human immunomodulators suitable for use as adjuvants in the invention include cytokines, such as interleukins (e.g. IL-1, IL-2, IL-4, IL-5, IL-6, IL-7, IL-12 [99], etc.) [100], interferons (e.g. interferon-γ), macrophage colony stimulating factor, and tumor necrosis factor. A preferred immunomodulator is IL-12.
- Bioadhesives and mucoadhesives may also be used as adjuvants in the invention. Suitable bioadhesives include esterified hyaluronic acid microspheres [101] or mucoadhesives such as cross-linked derivatives of poly(acrylic acid), polyvinyl alcohol, polyvinyl pyrollidone, polysaccharides and carboxymethylcellulose. Chitosan and derivatives thereof may also be used as adjuvants in the invention [102].
- Microparticles may also be used as adjuvants in the invention. Microparticles (i.e. a particle of ~100nm to ~150µm in diameter, more preferably ~200nm to ~30µm in diameter, and most preferably ~500nm to ~10µm in diameter) formed from materials that are biodegradable and non-toxic (e.g. a poly(α-hydroxy acid), a polyhydroxybutyric acid, a polyorthoester, a polyanhydride, a polycaprolactone, etc.), with poly(lactide-co-glycolide) are preferred, optionally treated to have a negatively-charged surface (e.g. with SDS) or a positively-charged surface (e.g. with a cationic detergent, such as CTAB).
- Examples of liposome formulations suitable for use as adjuvants are described in refs. 103-105.
- Adjuvants suitable for use in the invention include polyoxyethylene ethers and polyoxyethylene esters [106]. Such formulations further include polyoxyethylene sorbitan ester surfactants in combination with an octoxynol [107] as well as polyoxyethylene alkyl ethers or ester surfactants in combination with at least one additional non-ionic surfactant such as an octoxynol [108]. Preferred polyoxyethylene ethers are selected from the following group: polyoxyethylene-9-lauryl ether (laureth 9), polyoxyethylene-9-steoryl ether, polyoxytheylene-8-steoryl ether, polyoxyethylene-4-lauryl ether, polyoxyethylene-35-lauryl ether, and polyoxyethylene-23-lauryl ether.
- A phosphazene, such as poly[di(carboxylatophenoxy)phosphazene] ("PCPP") as described, for example, in
references 109 and 110, may be used. - Examples of muramyl peptides suitable for use as adjuvants in the invention include N-acetyl-muramyl-L-threonyl-D-isoglutamine (thr-MDP), N-acetyl-normuramyl-L-alanyl-D-isoglutamine (nor-MDP), and N-acetylmuramyl-L-alanyl-D-isoglutaminyl-L-alanine-2-(1'-2'-dipalmitoyl-sn-glycero-3-hydroxyphosphoryioxy)-ethylamine MTP-PE).
- Examples of imidazoquinolone compounds suitable for use adjuvants in the invention include Imiquimod ("R-837") [111,112], Resiquimod ("R-848") [113], and their analogs; and salts thereof (e.g. the hydrochloride salts). Further details about immunostimulatory imidazoquinolines can be found in references 114 to 118.
- Substituted ureas useful as adjuvants include compounds of formula I, II or III, or salts thereof:



- Further adjuvants that may be used with the invention include:
- An aminoalkyl glucosaminide phosphate derivative, such as RC-529 [120,121].
- A thiosemicarbazone compound, such as those disclosed in reference 122. Methods of formulating, manufacturing, and screening for active compounds are also described in reference 122. The thiosemicarbazones are particularly effective in the stimulation of human peripheral blood mononuclear cells for the production of cytokines, such as TNF-α.
- A tryptanthrin compound, such as those disclosed in reference 123. Methods of formulating, manufacturing, and screening for active compounds are also described in reference 123. The thiosemicarbazones are particularly effective in the stimulation of human peripheral blood mononuclear cells for the production of cytokines, such as TNF-α.
- A nucleoside analog, such as: (a) Isatorabine (ANA-245; 7-thia-8-oxoguanosine):

- Compounds disclosed in reference 128, including: Acylpiperazine compounds, Indoledione compounds, Tetrahydraisoquinoline (THIQ) compounds, Benzocyclodione compounds, Aminoazavinyl compounds, Aminobenzimidazole quinolinone (ABIQ) compounds [129,130], Hydrapthalamide compounds, Benzophenone compounds, Isoxazole compounds, Sterol compounds, Quinazilinone compounds, Pyrrole compounds [131], Anthraquinone compounds, Quinoxaline compounds, Triazine compounds, Pyrazalopyrimidine compounds, and Benzazole compounds [132].
- Compounds containing lipids linked to a phosphate-containing acyclic backbone, such as the TLR4 antagonist E5564 [133,134]:
- A polyoxidonium polymer [135,136] or other N-oxidized polyethylene-piperazine derivative.
- Methyl inosine 5'-monophosphate ("MIMP") [137].
- A polyhydroxlated pyrrolizidine compound [138], such as one having formula:

- A CD1d ligand, such as an α-glycosylceramide [139-146] (e.g. α-galactosylceramide), phytosphingosine-containing α-glycosylceramides, OCH, KRN7000 [(2S,3S,4R)-1-O-(α-D-galactopyranosyl)-2-(N-hexacosanoylamino)-1,3,4-octadecanetriol], CRONY-101, 3"-O-sulfo-galactosylceramide, etc.
- A gamma inulin [147] or derivative thereof, such as algammulin.

- The invention may also comprise combinations of aspects of one or more of the adjuvants identified above. For example, the following adjuvant compositions may be used in the invention: (1) a saponin and an oil-in-water emulsion [148]; (2) a saponin (e.g. QS21) + a non-toxic LPS derivative (e.g. 3dMPL) [149]; (3) a saponin (e.g. QS21) + a non-toxic LPS derivative (e.g. 3dMPL) + a cholesterol; (4) a saponin (e.g. QS21) + 3dMPL + IL-12 (optionally + a sterol) [150]; (5) combinations of 3dMPL with, for example, QS21 and/or oil-in-water emulsions [151]; (6) SAF, containing 10% squalane, 0.4
% Tween 80™, 5% pluronic-block polymer L121, and thr-MDP, either microfluidized into a submicron emulsion or vortexed to generate a larger particle size emulsion. (7) Ribi™ adjuvant system (RAS), (Ribi Immunochem) containing 2% squalene, 0.2% Tween 80, and one or more bacterial cell wall components from the group consisting of monophosphorylipid A (MPL), trehalose dimycolate (TDM), and cell wall skeleton (CWS), preferably MPL + CWS (Detox™); and (8) one or more mineral salts (such as an aluminum salt) + a non-toxic derivative of LPS (such as 3dMPL). - Other substances that act as immunostimulating agents are disclosed in
chapter 7 of ref. 30. - The use of an aluminium hydroxide and/or aluminium phosphate adjuvant is particularly preferred, and antigens are generally adsorbed to these salts. Calcium phosphate is another preferred adjuvant. Other preferred adjuvant combinations include combinations of Th1 and Th2 adjuvants such as CpG & alum or resiquimod & alum. A combination of aluminium phosphate and 3dMPL may be used.
- The compositions of the invention may elicit both a cell mediated immune response as well as a humoral immune response. This immune response will preferably induce long lasting (e.g. neutralising) antibodies and a cell mediated immunity that can quickly respond upon exposure to pnuemococcus.
- Two types of T cells, CD4 and CD8 cells, are generally thought necessary to initiate and/or enhance cell mediated immunity and humoral immunity. CD8 T cells can express a CD8 co-receptor and are commonly referred to as Cytotoxic T lymphocytes (CTLs). CD8 T cells are able to recognized or interact with antigens displayed on MHC Class I molecules.
- CD4 T cells can express a CD4 co-receptor and are commonly referred to as T helper cells. CD4 T cells are able to recognize antigenic peptides bound to MHC class II molecules. Upon interaction with a MHC class II molecule, the CD4 cells can secrete factors such as cytokines. These secreted cytokines can activate B cells, cytotoxic T cells, macrophages, and other cells that participate in an immune response. Helper T cells or CD4+ cells can be further divided into two functionally distinct subsets: TH1 phenotype and TH2 phenotypes which differ in their cytokine and effector function.
- Activated TH1 cells enhance cellular immunity (including an increase in antigen-specific CTL production) and are therefore of particular value in responding to intracellular infections. Activated TH1 cells may secrete one or more of IL-2, IFN-γ, and TNF-β. A TH1 immune response may result in local inflammatory reactions by activating macrophages, NK (natural killer) cells, and CD8 cytotoxic T cells (CTLs). A TH1 immune response may also act to expand the immune response by stimulating growth of B and T cells with IL-12. TH1 stimulated B cells may secrete IgG2a.
- Activated TH2 cells enhance antibody production and are therefore of value in responding to extracellular infections. Activated TH2 cells may secrete one or more of IL-4, IL-5, IL-6, and IL-10. A TH2 immune response may result in the production of IgG1, IgE, IgA and memory B cells for future protection.
- An enhanced immune response may include one or more of an
enhanced TH 1 immune response and a TH2 immune response. - A
TH 1 immune response may include one or more of an increase in CTLs, an increase in one or more of the cytokines associated with a TH1 immune response (such as IL-2, IFN-γ, and TNF-β), an increase in activated macrophages, an increase in NK activity, or an increase in the production of IgG2a. Preferably, the enhanced TH1 immune response will include an increase in IgG2a production. - A TH1 immune response may be elicited using a TH1 adjuvant. A TH1 adjuvant will generally elicit increased levels of IgG2a production relative to immunization of the antigen without adjuvant. TH1 adjuvants suitable for use in the invention may include for example saponin formulations, virosomes and virus like particles, non-toxic derivatives of enterobacterial lipopolysaccharide (LPS), immunostimulatory oligonucleotides. Immunostimulatory oligonucleotides, such as oligonucleotides containing a CpG motif, are preferred TH1 adjuvants for use in the invention.
- A TH2 immune response may include one or more of an increase in one or more of the cytokines associated with a TH2 immune response (such as IL-4, IL-5, IL-6 and IL-10), or an increase in the production of IgG1, IgE, IgA and memory B cells. Preferably, the enhanced TH2 immune resonse will include an increase in IgG1 production.
- A TH2 immune response may be elicited using a TH2 adjuvant. A TH2 adjuvant will generally elicit increased levels of IgG1 production relative to immunization of the antigen without adjuvant. TH2 adjuvants suitable for use in the invention include, for example, mineral containing compositions, oil-emulsions, and ADP-ribosylating toxins and detoxified derivatives thereof. Mineral containing compositions, such as aluminium salts are preferred TH2 adjuvants for use in the invention.
- Preferably, the invention includes a composition comprising a combination of a TH1 adjuvant and a TH2 adjuvant. Preferably, such a composition elicits an enhanced TH1 and an enhanced TH2 response, i.e., an increase in the production of both IgG1 and IgG2a production relative to immunization without an adjuvant. Still more preferably, the composition comprising a combination of a TH1 and a TH2 adjuvant elicits an increased TH1 and/or an increased TH2 immune response relative to immunization with a single adjuvant (i.e., relative to immunization with a TH1 adjuvant alone or immunization with a TH2 adjuvant alone).
- The immune response may be one or both of a TH1 immune response and a TH2 response. Preferably, immune response provides for one or both of an enhanced TH1 response and an enhanced TH2 response.
- The enhanced immune response may be one or both of a systemic and a mucosal immune response. Preferably, the immune response provides for one or both of an enhanced systemic and an enhanced mucosal immune response. Preferably the mucosal immune response is a TH2 immune response. Preferably, the mucosal immune response includes an increase in the production of IgA.
- E. coli can cause disease at a number of anatomical locations [4] and so the compositions of the invention may be prepared in various forms. For example, the compositions may be prepared as injectables, either as liquid solutions or suspensions. Solid forms suitable for solution in, or suspension in, liquid vehicles prior to injection can also be prepared (e.g. a lyophilised composition or a spray-freeze dried composition). The composition may be prepared for topical administration e.g. as an ointment, cream or powder. The composition may be prepared for oral administration e.g. as a tablet or capsule, as a spray, or as a syrup (optionally flavoured). The composition may be prepared for pulmonary administration e.g. as an inhaler, using a fine powder or a spray. The composition may be prepared as a suppository or pessary. The composition may be prepared for nasal, aural or ocular administration e.g. as drops. The composition may be in kit form, designed such that a combined composition is reconstituted just prior to administration to a patient. Such kits may comprise one or more antigens in liquid form and one or more lyophilised antigens.
- Where a composition is to be prepared extemporaneously prior to use (e.g. where a component is presented in lyophilised form) and is presented as a kit, the kit may comprise two vials, or it may comprise one ready-filled syringe and one vial, with the contents of the syringe being used to reactivate the contents of the vial prior to injection.
- Immunogenic compositions used as vaccines comprise an immunologically effective amount of antigen(s), as well as any other components, as needed. By 'immunologically effective amount', it is meant that the administration of that amount to an individual, either in a single dose or as part of a series, is effective for treatment or prevention. This amount varies depending upon the health and physical condition of the individual to be treated, age, the taxonomic group of individual to be treated (e.g. non-human primate, primate, etc.), the capacity of the individual's immune system to synthesise antibodies, the degree of protection desired, the formulation of the vaccine, the treating doctor's assessment of the medical situation, and other relevant factors. It is expected that the amount will fall in a relatively broad range that can be determined through routine trials.
- The invention also provides a method for raising an immune response in a mammal comprising the step of administering an effective amount of a composition of the invention. The immune response is preferably protective and preferably involves antibodies and/or cell-mediated immunity. The method may raise a booster response.
- The invention also provides a polypeptide of the invention for use as a medicament e.g. for use in raising an immune response in a mammal.
- The invention also provides the use of a polypeptide of the invention in the manufacture of a medicament for raising an immune response in a mammal.
- The invention also provides a delivery device pre-filled with an immunogenic composition of the invention.
- By raising an immune response in the mammal by these uses and methods, the mammal can be protected against E. coli infection, including ExPEC and non-ExPEC strains. The invention is particularly useful for providing broad protection against pathogenic E. coli, including intestinal pathotypes such as EPEC, EAEC, EIEC, ETEC and DAEC pathotypes. Thus the mammal may be protected against diseases including, but not limited to peritonitis, pyelonephritis, cystitis, endocarditis, prostatitis, urinary tract infections (UTIs), meningitis (particularly neonatal meningitis), sepsis (or SIRS), dehydration, pneumonia, diarrhea (infantile, travellers', acute, persistent, etc.), bacillary dysentery, hemolytic uremic syndrome (HUS), pericarditis, bacteriuria, etc.
- SEQ ID NO: 21, 30, 35, 40, 49, 54, 59, 68, and 73 and their other detoxified variants are particularly useful for immunising against the EAEC pathotype, and thus for preventing diarrhea (both acute and chronic).
- SEQ ID NO: 22, 28, 36, 41, 47, 55, 56, 60, 66, 74, and 75 and their other detoxified variants are particularly useful for immunising against the UPEC pathotype, and thus for preventing UTIs including, but not limited to, pyelonephritis, cystitis (both acute and recurrent), peritonitis, catheter-associated UTIs, prostatisis, and bacteriuria (including asymptomatic bacteriuria).
- SEQ ID NO: 23, 42, and 61 and their other detoxified variants are particularly useful for immunising against the EIEC pathotype, and thus for preventing dysentery (in particular bacillary dysentery) and HUS (e.g. in children).
- (SEQ ID NO: 24, 27, 29, 43, 46, 48, 62, 65, and 67 and their other detoxified variants are particularly useful for immunising against the ETEC pathotype, and thus for preventing diarrhea (including travellers' and infant diarrhea).
- SEQ ID NO: 25, 26, 33, 34, 45, 53, 63, 64, and 72 and their other detoxified variants are particularly useful for immunising against the EAEC pathotype, and thus for preventing diarrhea (both acute and chronic).
- SEQ ID NO: 79, 85, 93, and 94 and their other detoxified variants are particularly useful for immunising against the UPEC pathotype, and thus for preventing UTIs including, but not limited to, pyelonephritis, cystitis (both acute and recurrent), peritonitis, catheter-associated UTIs, prostatisis, and bacteriuria (including asymptomatic bacteriuria).
- SEQ ID NO: 80 and their other detoxified variants are particularly useful for immunising against the EIEC pathotype, and thus for preventing dysentery (in particular bacillary dysentery) and HUS (e.g. in children).
- SEQ ID NO: 81, 84, and 86 and their other detoxified variants are particularly useful for immunising against the ETEC pathotype, and thus for preventing diarrhea (including travellers' and infant diarrhea).
- SEQ ID NOs: 82, 83, and 91 and their other detoxified variants are particularly useful for immunising against the EPEC pathotype, and thus for preventing diarrhea (including infantile diarrhea).
- SEQ ID NO: 21, 30, 35, 40, 49, 54, 59, 68, and 73 and their other variants are particularly useful for immunising against the EAEC pathotype, and thus for preventing diarrhea (both acute and chronic).
- SEQ ID NO: 22, 28, 36, 41, 47, 55, 56, 60, 66, 74, and 75 and their other variants are particularly useful for immunising against the UPEC pathotype, and thus for preventing UTIs including, but not limited to, pyelonephritis, cystitis (both acute and recurrent), peritonitis, catheter-associated UTIs, prostatisis, and bacteriuria (including asymptomatic bacteriuria).
- SEQ ID NO: 23, 42, and 61 and their other variants are particularly useful for immunising against the EIEC pathotype, and thus for preventing dysentery (in particular bacillary dysentery) and HUS (e.g. in children).
- SEQ ID NO: 24, 27, 29, 43, 46, 48, 62, 65, and 67 and their other variants are particularly useful for immunising against the ETEC pathotype, and thus for preventing diarrhea (including travellers' and infant diarrhea).
- SEQ ID NO: 25, 26, 33, 34, 45, 53, 63, 64, and 72 and their other variants are particularly useful for immunising against the EAEC pathotype, and thus for preventing diarrhea (both acute and chronic).
- SEQ ID NO: 79, 85, 93, and 94 and their other are particularly useful for immunising against the UPEC pathotype, and thus for preventing UTIs including, but not limited to, pyelonephritis, cystitis (both acute and recurrent), peritonitis, catheter-associated UTIs, prostatisis, and bacteriuria (including asymptomatic bacteriuria).
- SEQ ID NO: 80 and their other variants are particularly useful for immunising against the EIEC pathotype, and thus for preventing dysentery (in particular bacillary dysentery) and HUS (e.g. in children).
- SEQ ID NO: 81, 84, and 86 and their other variants are particularly useful for immunising against the ETEC pathotype, and thus for preventing diarrhea (including travellers' and infant diarrhea).
- SEQ ID NOs: 82, 83, and 91 and their other variants are particularly useful for immunising against the EPEC pathotype, and thus for preventing diarrhea (including infantile diarrhea).
- The mammal is preferably a human, but may be e.g. a cow, a pig, a chicken, a cat or a dog, as E. coli disease is also problematic in these species [4]. Where the vaccine is for prophylactic use, the human is preferably a child (e.g. a toddler or infant) or a teenager; where the vaccine is for therapeutic use, the human is preferably a teenager or an adult. A vaccine intended for children may also be administered to adults e.g. to assess safety, dosage, immunogenicity, etc.
- One way of checking efficacy of therapeutic treatment involves monitoring E. coli infection after administration of the compositions of the invention. One way of checking efficacy of prophylactic treatment involves monitoring immune responses, systemically (such as monitoring the level of IgG1 and IgG2a production) and/or mucosally (such as monitoring the level of IgA production), against the antigens in the compositions of the invention after administration of the composition. Typically, antigen-specific serum antibody responses are determined post-immunisation but pre-challenge whereas antigen-specific mucosal antibody responses are determined post-immunisation and post-challenge.
- Another way of assessing the immunogenicity of the compositions of the present invention is to express the proteins recombinantly for screening patient sera or mucosal secretions by immunoblot and/or microarrays. A positive reaction between the protein and the patient sample indicates that the patient has mounted an immune response to the protein in question. This method may also be used to identify immunodominant antigens and/or epitopes within antigens.
- The efficacy of vaccine compositions can also be determined in vivo by challenging animal models of E. coli infection, e.g., guinea pigs or mice, with the vaccine compositions. A murine model of ExPEC and lethal sepsis is described in reference 152. A cotton rat model is disclosed in ref. 153
- Compositions of the invention will generally be administered directly to a patient. Direct delivery may be accomplished by parenteral injection (e.g. subcutaneously, intraperitoneally, intravenously, intramuscularly, or to the interstitial space of a tissue), or mucosally, such as by rectal, oral (e.g. tablet, spray), vaginal, topical, transdermal or transcutaneous, intranasal, ocular, aural, pulmonary or other mucosal administration. Novel direct delivery forms can also include transgenic expression of the polypeptides disclosed herein in foods, e.g., transgenic expression in a potato.
- The invention may be used to elicit systemic and/or mucosal immunity, preferably to elicit an enhanced systemic and/or mucosal immunity.
- Preferably the enhanced systemic and/or mucosal immunity is reflected in an enhanced TH1 and/or TH2 immune response. Preferably, the enhanced immune response includes an increase in the production of IgG1 and/or IgG2a and/or IgA.
- Dosage can be by a single dose schedule or a multiple dose schedule. Multiple doses may be used in a primary immunisation schedule and/or in a booster immunisation schedule. In a multiple dose schedule the various doses may be given by the same or different routes e.g. a parenteral prime and mucosal boost, a mucosal prime and parenteral boost, etc. Multiple doses will typically be administered at least 1 week apart (e.g. about 2 weeks, about 3 weeks, about 4 weeks, about 6 weeks, about 8 weeks, about 10 weeks, about 12 weeks, about 16 weeks, etc.).
- Vaccines of the invention may be used to treat both children and adults. Thus a human patient may be less than 1 year old, 1-5 years old, 5-15 years old, 15-55 years old, or at least 55 years old. Preferred patients for receiving the vaccines are the elderly (e.g. ≥50 years old, ≥60 years old, and preferably ≥65 years), the young (e.g. ≤5 years old), hospitalised patients, healthcare workers, armed service and military personnel, pregnant women, the chronically ill, or immunodeficient patients. The vaccines are not suitable solely for these groups, however, and may be used more generally in a population.
- Vaccines of the invention are particularly useful for patients who are expecting a surgical operation, or other hospital in-patients. They are also useful in patients who will be catheterized. They are also useful in adolescent females (e.g. aged 11-18) and in patients with chronic urinary tract infections.
- Vaccines of the invention may be administered to patients at substantially the same time as (e.g. during the same medical consultation or visit to a healthcare professional or vaccination centre) other vaccines e.g. at substantially the same time as a measles vaccine, a mumps vaccine, a rubella vaccine, a MMR vaccine, a varicella vaccine, a MMRV vaccine, a diphtheria vaccine, a tetanus vaccine, a pertussis vaccine, a DTP vaccine, a conjugated H. influenzae type b vaccine, an inactivated poliovirus vaccine, a hepatitis B virus vaccine, a meningococcal conjugate vaccine (such as a tetravalent A-C-W135-Y vaccine), a respiratory syncytial virus vaccine, etc.
- The immunogenic compositions described above include polypeptide antigens. In all cases, however, the polypeptide antigens can be replaced by nucleic acids (typically DNA) encoding those polypeptides, to give compositions, methods and uses based on nucleic acid immunisation. Nucleic acid immunisation is now a developed field (e.g. see references 154 to 161 etc.).
- The nucleic acid encoding the immunogen is expressed in vivo after delivery to a patient and the expressed immunogen then stimulates the immune system. The active ingredient will typically take the form of a nucleic acid vector comprising: (i) a promoter; (ii) a sequence encoding the immunogen, operably linked to the promoter; and optionally (iii) a selectable marker. Preferred vectors may further comprise (iv) an origin of replication; and (v) a transcription terminator downstream of and operably linked to (ii). In general, (i) & (v) will be eukaryotic and (iii) & (iv) will be prokaryotic.
- Preferred promoters are viral promoters e.g. from cytomegalovirus (CMV). The vector may also include transcriptional regulatory sequences (e.g. enhancers) in addition to the promoter and which interact functionally with the promoter. Preferred vectors include the immediate-early CMV enhancer/promoter, and more preferred vectors also include CMV intron A. The promoter is operably linked to a downstream sequence encoding an immunogen, such that expression of the immunogen-encoding sequence is under the promoter's control.
- Where a marker is used, it preferably functions in a microbial host (e.g. in a prokaryote, in a bacteria, in a yeast). The marker is preferably a prokaryotic selectable marker (e.g. transcribed under the control of a prokaryotic promoter). For convenience, typical markers are antibiotic resistance genes.
- The vector of the disclosure is preferably an autonomously replicating episomal or extrachromosomal vector, such as a plasmid.
- The vector of the disclosure preferably comprises an origin of replication. It is preferred that the origin of replication is active in prokaryotes but not in eukaryotes.
- Preferred vectors thus include a prokaryotic marker for selection of the vector, a prokaryotic origin of replication, but a eukaryotic promoter for driving transcription of the immunogen-encoding sequence. The vectors will therefore (a) be amplified and selected in prokaryotic hosts without polypeptide expression, but (b) be expressed in eukaryotic hosts without being amplified. This arrangement is ideal for nucleic acid immunization vectors.
- The vector of the disclosure may comprise a eukaryotic transcriptional terminator sequence downstream of the coding sequence. This can enhance transcription levels. Where the coding sequence does not have its own, the vector of the invention preferably comprises a polyadenylation sequence. A preferred polyadenylation sequence is from bovine growth hormone.
- The vector of the disclosure may comprise a multiple cloning site
- In addition to sequences encoding the immunogen and a marker, the vector may comprise a second eukaryotic coding sequence. The vector may also comprise an IRES upstream of said second sequence in order to permit translation of a second eukaryotic polypeptide from the same transcript as the immunogen. Alternatively, the immunogen-coding sequence may be downstream of an IRES.
- The vector of the disclosure may comprise unmethylated CpG motifs e.g. unmethylated DNA sequences which have in common a cytosine preceding a guanosine, flanked by two 5' purines and two 3' pyrimidines. In their unmethylated form these DNA motifs have been demonstrated to be potent stimulators of several types of immune cell.
- Vectors may be delivered in a targeted way. Receptor-mediated DNA delivery techniques are described in, for example, references 162 to 167. Therapeutic compositions containing a nucleic acid are administered in a range of about 100ng to about 200mg of DNA for local administration in a gene therapy protocol. Concentration ranges of about 500 ng to about 50 mg, about 1µg to about 2 mg, about 5µg to about 500µg, and about 20µg to about 100µg of DNA can also be used during a gene therapy protocol. Factors such as method of action (e.g. for enhancing or inhibiting levels of the encoded gene product) and efficacy of transformation and expression are considerations which will affect the dosage required for ultimate efficacy. Where greater expression is desired over a larger area of tissue, larger amounts of vector or the same amounts re-administered in a successive protocol of administrations, or several administrations to different adjacent or close tissue portions may be required to effect a positive therapeutic outcome. In all cases, routine experimentation in clinical trials will determine specific ranges for optimal therapeutic effect.
- Vectors can be delivered using gene delivery vehicles. The gene delivery vehicle can be of viral or non-viral origin (see generally references 168 to 171).
- Viral-based vectors for delivery of a desired nucleic acid and expression in a desired cell are well known in the art. Exemplary viral-based vehicles include, but are not limited to, recombinant retroviruses (e.g. references 172 to 182), alphavirus-based vectors (e.g. Sindbis virus vectors, Semliki forest virus (ATCC VR-67; ATCC VR-1247), Ross River virus (ATCC VR-373; ATCC VR-1246) and Venezuelan equine encephalitis virus (ATCC VR-923; ATCC VR-1250; ATCC VR 1249; ATCC VR-532); hybrids or chimeras of these viruses may also be used), poxvirus vectors (e.g. vaccinia, fowlpox, canarypox, modified vaccinia Ankara, etc.), adenovirus vectors, and adeno-associated virus (AAV) vectors (e.g. see refs. 183 to 188). Administration of DNA linked to killed adenovirus [189] can also be employed.
- Non-viral delivery vehicles and methods can also be employed, including, but not limited to, polycationic condensed DNA linked or unlinked to killed adenovirus alone [e.g. 189], ligand-linked DNA [190], eukaryotic cell delivery vehicles cells [e.g. refs. 191 to 195] and nucleic charge neutralization or fusion with cell membranes. Naked DNA can also be employed. Exemplary naked DNA introduction methods are described in refs. 196 and 197. Liposomes (e.g. immunoliposomes) that can act as gene delivery vehicles are described in refs. 198 to 202. Additional approaches are described in references 203 & 204.
- Further non-viral delivery suitable for use includes mechanical delivery systems such as the approach described in ref. 204. Moreover, the coding sequence and the product of expression of such can be delivered through deposition of photopolymerized hydrogel materials or use of ionizing radiation [e.g. refs. 205 & 206]. Other conventional methods for gene delivery that can be used for delivery of the coding sequence include, for example, use of hand-held gene transfer particle gun [207] or use of ionizing radiation for activating transferred genes [205 & 206].
- Delivery DNA using PLG {poly(lactide-co-glycolide)} microparticles is a particularly preferred method e.g. by adsorption to the microparticles, which are optionally treated to have a negatively-charged surface (e.g. treated with SDS) or a positively-charged surface (e.g. treated with a cationic detergent, such as CTAB).
- The practice of the present invention will employ, unless otherwise indicated, conventional methods of chemistry, biochemistry, molecular biology, immunology and pharmacology, within the skill of the art. Such techniques are explained fully in the literature. See, e.g., references 208-215, etc.
- The term "comprising" encompasses "including" as well as "consisting" e.g. a composition "comprising" X may consist exclusively of X or may include something additional e.g. X + Y.
- The term "about" in relation to a numerical value x means, for example, x±10%.
- "GI" numbering is used herein. A GI number, or "GenInfo Identifier", is a series of digits assigned consecutively to each sequence record processed by NCBI when sequences are added to its databases. The GI number bears no resemblance to the accession number of the sequence record. When a sequence is updated (e.g. for correction, or to add more annotation or information) then it receives a new GI number. Thus the sequence associated with a given GI number is never changed.
- References to a percentage sequence identity between two amino acid sequences means that, when aligned, that percentage of amino acids are the same in comparing the two sequences. This alignment and the percent homology or sequence identity can be determined using software programs known in the art, for example those described in section 7.7.18 of ref. 216. A preferred alignment is determined by the Smith-Waterman homology search algorithm using an affine gap search with a gap open penalty of 12 and a gap extension penalty of 2, BLOSUM matrix of 62. The Smith-Waterman homology search algorithm is disclosed in ref. 217.
- One of skill in the art would understand that "isolated" means altered "by the hand of man" from its natural state, i.e., if it occurs in nature, it has been changed or removed from its original environment, or both. For example, a polynucleotide or a polypeptide naturally present in a living organism is not "isolated" when in such living organism, but the same polynucleotide or polypeptide separated from the coexisting materials of its natural state is "isolated," as the term is used in this disclosure. Further, a polynucleotide or polypeptide that is introduced into an organism by transformation, genetic manipulation or by any other recombinant method would be understood to be "isolated" even if it is still present in said organism, which organism may be living or non-living, except where such transformation, genetic manipulation or other recombinant method produces an organism that is otherwise indistinguishable from the naturally occurring organism.
-
-
Figure 1 shows a CLUSTALW alignment of SEQ ID NOs: 2 to 16. The N-terminal regions that may be removed to increase solubility while maintaining substantially the same immunogenicity are shown at the bottom of the alignment. The N-terminal region up to the gly-ser linker or gly-ser region is denoted with "G" and the proline-rich region is denoted with "P."Figure 2 shows the amino acid identity between pairs of sequences. -
Figure 3 shows gel analysis of purified protein, with high MW bands visible in the absence of DTT. -
Figure 4 shows the Western Blot of pathogenic and non pathogenic E. coli strains using an anti-AcfD (orf3526) serum. Panel (A) is a Western Blot of the total cell lysate. Panel (B) is a Western Blot of the supernatant from the culture. The lanes in each of panel (A) and (B) from left to right are as follows: Lane M - marker proteins with the molecular weight in kDa of each marker protein shown along the left side of panel (A); 1 - IHE3034; 2 - CFT073; 3 - 536; 4-BL21; 5 - MG 1655; 6 - W3110; 7 - NISSLE1917; 8 - IHE3034ΔAcfD. As observed from the analysis, pathogenic strains (IHE3034,lane 1; 536, lane 3) express and secrete AcfD (orf3526) while non-pathogenic strains (MG1655,lane 5; W3110,lane 6; Nissle 1917, lane 7) express the protein but its secretion is defective. Strains CFT073 (lane 2) and IHE3034ΔAcfD (lane 8) are used as negative control, since they don't harbor the AcfD (orf3526) gene. BL21 strain (lane 4) is a lab strain used as positive control, since it expresses and secretes AcfD (orf3526). -
Figure 5 shows a comparison of solubility of the AcfD (orf3526) protein and various fragments of the protein. Panel (A) is an SDS-PAGE gradient gel (4-12% MOPS buffer) of samples at 37° C comparing the pellet (left lane for each protein or fragment) to the supernatant (right lane for each protein or fragment. The lanes from left to right are: Molecular weight markers (191 kDa, 97 kDa and 64 kDa bands are labelled), control bacteria transformed with the pET expression vector with no insert, bacterial expression of 3526 (his tag , leader peptide removed), bacterial expression of L3526 (his tag, full length), bacterial expression of L3526-2stop (full length with a stop codon before His tag), bacterial expression of ΔG3526 (his tag, removal of the N-terminus of AcfD (orf3526) to the flexible glycine-serine linker), and bacterial expression of ΔP3526 (his tag + removal of the N-terminus of AcfD (orf3526) through the proline rich region). Panel (B) is an SDS-PAGE gradient gel (4-12% MOPS buffer) of samples at 25° C following the same order for the lanes as panel (A). -
Figure 6 shows a comparison of expression and purification of the AcfD (orf3526) protein and various fragments of the protein. Panel (A) is an SDS-PAGE gel (12% MOPS buffer) of samples at from bacteria expressing 3526 (his tag, leader peptide removed), cultured at 25° C comparing fractions from various stages of the purification. The lanes from left to right are: M: Molecular weight markers (191 kDa, 97 kDa and 64 kDa bands are labelled), TOT: total bacterial lysate, INS: insoluble fraction of bacterial lysate, SM: soluble fraction of bacterial lysate, FT: flow through from Nickel column; E1, E2, and E3 three elutions with 500mM imidazole buffer. Panel (B) is an SDS-PAGE gel (12% MOPS buffer) of samples at from bacteria expressing ΔG3526 (his tag , removal of the N-terminus of AcfD (orf3526) to the flexible glycine-serine linker) cultured at 25° C comparing fractions from various stages of the purification. The lanes from left to right are: M: Molecular weight markers (191 kDa, 97 kDa and 64 kDa bands are labelled), TOT: total bacterial lysate, INS: insoluble fraction of bacterial lysate, SM: soluble fraction of bacterial lysate, FT: flow through from Nickel column; E1, E2, and E3 three elutions with 500mM imidazole buffer. Panel (C) is an SDS-PAGE gel (12% MOPS buffer) of samples from bacteria expressing ΔP3526 (his tag, removal of the N-terminus of AcfD (orf3526) through the proline rich region), cultured at 25° C comparing fractions from various stages of the purification. The lanes from left to right are: M: Molecular weight markers (191 kDa, 97 kDa and 64 kDa bands are labelled), TOT: total bacterial lysate, INS: insoluble fraction of bacterial lysate, SM: soluble fraction of bacterial lysate, FT: flow through from Nickel column; E1, E2, and E3 three elutions with 500mM imidazole buffer. -
Figure 7 shows the amino acid identity between additional pairs of sequences. Sixteen Enterohemorrhagic E. coli (EHEC) were not found to have AcfD (orf3526) gene (not shown). The sequences (where represented) from left to right or top to bottom are as follows: 10 non-pathogenic or commensal strains (1: a commensal E. coli strain, 2: DH10B strain, 3: MG1655 strain, 4: W3110 strain (SEQ ID NO:14); 5: HS strain (SEQ ID NO:13); 9: another commensal E. coli strain; and 10: yet another commensal E. coli strain); three NMEC strains (1: NMEC strain RS218; 2: NMEC strain IHE3034 (SEQ ID NO:2); and 3: NMEC strain S88 (SEQ ID NO:141)); one APEC strain (1: APEC O1 strain); six UPEC strains (2: UPEC strain 536 (SEQ ID NO:4); 3: UTI89; 4: UPEC strain F11 (SEQ ID NO:10); 5: UPEC strain IAI39 (SEQ ID NO:133); and 6: UPEC strain UMN026 (SEQ ID NO:137)); three EAEC strains (1: EAEC strain 101-1 (SEQ ID NO:3); 2: EAEC strain 042 (SEQ ID NO:12; and 3: EAEC strain 55989 (SEQ ID NO:129)); one EIEC strain (1: EIEC strain 53638 (SEQ ID NO:5)); four EPEC strains (2: EPEC strain E22 (SEQ ID NO: 8)); 3: EPEC strain E2348/69 (SEQ ID NO:16); and 4: EPEC strain E110019 (SEQ ID NO:7)); three ETEC strains (1: ETEC strain B7A (SEQ ID NO:6); 2: ETEC strain E24377A (SEQ ID NO:9); and 3: ETEC strain H10407 (SEQ ID NO:11)); and one antibiotic resistant strain (1: antibiotic-resistant strain SECEC (SEQ ID NO:15)). -
Figure 8 shows the sequence motifs identified in AcfD (orf3526) including from left to right: a lipidation motif; a proline rich region, a WxxxE motif, a zinc binding motif, and an RGD domain. -
Figure 9 shows the family relationships of the various zinc metalloproteases. The zincins are highlighted with a grey box as the zincin motif is the motif found in AcfD (orf3526) (Seefigure 8 ). -
Figure 10 shows an SDS-PAGE (4-12% gradient, Bis-Tris) of orf3526C expression. The lanes are labelled across the top as follows: (1) Total cell lysate - no IPTG, 6 hours at 25°C; (2) Total cell lysate - 1 mM IPTG, 3 hours at 25°C; (3) Total cell lysate - 1 mM IPTG, 6 hours at 25°C; (M) Markers (sizes are indicated in kDa along the left side of the gel); (4) soluble fraction after sonication - 1 mM IPTG, 3 hours at 25°C; and (5) soluble fraction after sonication - 1 mM IPTG, 6 hours at 25°C. -
SEQ ID Description SEQ ID NOs: 1-19 = full length amino acid sequences 1 Amino acid sequence from NMEC strain IHE3034 2 Amino acid sequence from EAEC strain 101-1 (GI: 83587587) 3 Amino acid sequence from UPEC strain 536 (GI: 110643204) 4 Amino acid sequence from EIEC strain 53638 (GI: 75515237) 5 Amino acid sequence from ETEC strain B7A (GI: 75227618) 6 Amino acid sequence from EPEC strain E110019 (GI: 75239450) 7 Amino acid sequence from EPEC strain E22 (GI: 75259912) 8 Amino acid sequence from ETEC strain E24377A (GI: 157156747) 9 Amino acid sequence from UPEC strain F11 (GI: 75241179) 10 Amino acid sequence from ETEC strain H10407 11 Amino acid sequence from EAEC strain 042 12 Amino acid sequence from commensal strain HS (GI: 157162442) 13 Amino acid sequence from commensal strain W3110 (GI: 89109748) 14 Amino acid sequence from antibiotic-resistant strain SECEC 15 Amino acid sequence from EPEC strain E2348/69 16 Amino acid sequence from EAEC strain 55989 17 Amino acid sequence from UPEC strain IAI39 18 Amino acid sequence from UPEC strain UMN026 19 Amino acid sequence from NMEC strain S88 SEQ ID NOs: 20-38 = C-terminal deletion amino acid sequences 20 Amino acid sequence from NMEC strain IHE3034 with C-terminal deletion 21 Amino acid sequence from EAEC strain 101-1 (GI: 83587587) with C-terminal deletion 22 Amino acid sequence from UPEC strain 536 (GI: 110643204) with C-terminal deletion 23 Amino acid sequence from EIEC strain 53638 (GI: 75515237) with C-terminal deletion 24 Amino acid sequence from ETEC strain B7A (GI: 75227618) with C-terminal deletion 25 Amino acid sequence from EPEC strain E110019 (GI: 75239450) with C-terminal deletion 26 Amino acid sequence from EPEC strain E22 (GI: 75259912) with C-terminal deletion 27 Amino acid sequence from ETEC strain E24377A (GI: 157156747) with C-terminal deletion 28 Amino acid sequence from UPEC strain F11 (GI: 75241179) with C-terminal deletion 29 Amino acid sequence from ETEC strain H10407 with C-terminal deletion 30 Amino acid sequence from EAEC strain O42 with C-terminal deletion 31 Amino acid sequence from commensal strain HS (GI: 157162442) with C-terminal deletion 32 Amino acid sequence from commensal strain W3110 (GI: 89109748) with C-terminal deletion 33 Amino acid sequence from antibiotic-resistant strain SECEC with C-terminal deletion 34 Amino acid sequence from EPEC strain E2348/69 with C-terminal deletion 35 Amino acid sequence from EAEC strain 55989 with C-terminal deletion 36 Amino acid sequence from UPEC strain IAI39 with C-terminal deletion 37 Amino acid sequence from UPEC strain UMN026 with C-terminal deletion 38 Amino acid sequence from NMEC strain S88 with C-terminal deletion SEQ ID NOs: 39-57 = N-terminal deletion amino acid sequences 39 Amino acid sequence from NMEC strain IHE3034 with N-terminal deletion 40 Amino acid sequence from EAEC strain 101-1 (GI: 83587587) with N-terminal deletion 41 Amino acid sequence from UPEC strain 536 (GI: 110643204) with N-terminal deletion 42 Amino acid sequence from EIEC strain 53638 (GI: 75515237) with N-terminal deletion 43 Amino acid sequence from ETEC strain B7A (GI: 75227618) with N-terminal deletion 44 Amino acid sequence from EPEC strain E110019 (GI: 75239450) with N-terminal deletion 45 Amino acid sequence from EPEC strain E22 (GI: 75259912) with N-terminal deletion 46 Amino acid sequence from ETEC strain E24377A (GI: 157156747) with N-terminal deletion 47 Amino acid sequence from UPEC strain F11 (GI: 75241179) with N-terminal deletion 48 Amino acid sequence from ETEC strain H10407 with N-terminal deletion 49 Amino acid sequence from EAEC strain O42 with N-terminal deletion 50 Amino acid sequence from commensal strain HS (GI: 157162442) with N-terminal deletion 51 Amino acid sequence from commensal strain W3110 (GI: 89109748) with N-terminal deletion 52 Amino acid sequence from antibiotic-resistant strain SECEC with N-terminal deletion 53 Amino acid sequence from EPEC strain E2348/69 with N-terminal deletion 54 Amino acid sequence from EAEC strain 55989 with N-terminal deletion 55 Amino acid sequence from UPEC strain IAI39 with N-terminal deletion 56 Amino acid sequence from UPEC strain UMN026 with N-terminal deletion 57 Amino acid sequence from NMEC strain S88 with N-terminal deletion SEQ ID NOs: 58-76 = E1305A point mutation amino acid sequences 58 Amino acid sequence from NMEC strain IHE3034 with an E1305A point mutation 59 Amino acid sequence from EAEC strain 101-1 (GI: 83587587) with an E1305A point mutation 60 Amino acid sequence from UPEC strain 536 (GI: 110643204) with an E1305A point mutation 61 Amino acid sequence from EIEC strain 53638 (GI: 75515237) with an E1305A point mutation 62 Amino acid sequence from ETEC strain B7A (GI: 75227618) with an E1305A point mutation 63 Amino acid sequence from EPEC strain E110019 (GI: 75239450) with an E1305A point mutation 64 Amino acid sequence from EPEC strain E22 (GI: 75259912) with an E1305A point mutation 65 Amino acid sequence from ETEC strain E24377A (GI: 157156747) with an E1305A point mutation 66 Amino acid sequence from UPEC strain F11 (GI: 75241179) with an E1305A point mutation 67 Amino acid sequence from ETEC strain H10407 with an E1305A point mutation 68 Amino acid sequence from EAEC strain O42 with an E1305A point mutation 69 Amino acid sequence from commensal strain HS (GI: 157162442) with an E1305A point mutation 70 Amino acid sequence from commensal strain W3110 (GI: 89109748) with an E1305A point mutation 71 Amino acid sequence from antibiotic-resistant strain SECEC with an E1305A point mutation 72 Amino acid sequence from EPEC strain E2348/69 with an E1305A point mutation 73 Amino acid sequence from EAEC strain 55989 with an E1305A point mutation 74 Amino acid sequence from UPEC strain IAI39 with an E1305A point mutation 75 Amino acid sequence from UPEC strain UMN026 with an E1305A point mutation 76 Amino acid sequence from NMEC strain S88 with an E1305A point mutation SEQ ID NOs: 77-95 = orf3526C fragment amino acid sequences 77 Amino acid sequence from NMEC strain IHE3034 with the N-terminal ΔG region and the C-terminal portion including the zinc-binding motif deleted 78 Amino acid sequence from EAEC strain 101-1 (GI: 83587587) with the N-terminal ΔG region and the C-terminal portion including the zinc-binding motif deleted 79 Amino acid sequence from UPEC strain 536 (GI: 110643204) with the N-terminal ΔG region and the C-terminal portion including the zinc-binding motif deleted 80 Amino acid sequence from EIEC strain 53638 (GI: 75515237) with the N-terminal ΔG region and the C-terminal portion including the zinc-binding motif deleted 81 Amino acid sequence from ETEC strain B7A (GI: 75227618) with the N-terminal ΔG region and the C-terminal portion including the zinc-binding motif deleted 82 Amino acid sequence from EPEC strain E110019 (GI: 75239450 with the N-terminal ΔG region and the C-terminal portion including the zinc-binding motif deleted 83 Amino acid sequence from EPEC strain E22 (GI: 75259912) with the N-terminal ΔG region and the C-terminal portion including the zinc-binding motif deleted 84 Amino acid sequence from ETEC strain E24377A (GI: 157156747) with the N-terminal ΔG region and the C-terminal portion including the zinc-binding motif deleted 85 Amino acid sequence from UPEC strain F11 (GI: 75241179) with the N-terminal ΔG region and the C-terminal portion including the zinc-binding motif deleted 86 Amino acid sequence from ETEC strain H10407 with the N-terminal ΔG region and the C-terminal portion including the zinc-binding motif deleted 87 Amino acid sequence from EAEC strain O42 with the N-terminal ΔG region and the C-terminal portion including the zinc-binding motif deleted 88 Amino acid sequence from commensal strain HS (GI: 157162442) with the N-terminal ΔG region and the C-terminal portion including the zinc-binding motif deleted 89 Amino acid sequence from commensal strain W3110 (GI: 89109748) with the N-terminal ΔG region and the C-terminal portion including the zinc-binding motif deleted 90 Amino acid sequence from antibiotic-resistant strain SECEC with the N-terminal ΔG region and the C-terminal portion including the zinc-binding motif deleted 91 Amino acid sequence from EPEC strain E2348/69 with the N-terminal ΔG region and the C-terminal portion including the zinc-binding motif deleted 92 Amino acid sequence from EAEC strain 55989 with the N-terminal ΔG region and the C-terminal portion including the zinc-binding motif deleted 93 Amino acid sequence from UPEC strain IAI39 with the N-terminal ΔG region and the C-terminal portion including the zinc-binding motif deleted 94 Amino acid sequence from UPEC strain UMN026 with the N-terminal ΔG region and the C-terminal portion including the zinc-binding motif deleted 95 Amino acid sequence from NMEC strain S88 with the N-terminal ΔG region and the C-terminal portion including the zinc-binding motif deleted 96/97 Zinc binding motifs 98-109 Primers 110-111 IC31 sequences - One of the antigens disclosed in
reference 5 is annotated as accessory colonization factor D (AcfD) precursor (orf3526) (amino acid SEQ ID NO: 2 herein) from NMEC strain IHE3034. This protein has been expressed and purified, and it confers protection against ExPEC strains in a sepsis animal model. - Sequences were obtained for the orthologs in various other E. coli strains. The amino acid sequence seen in IHE3034 was also seen in strains APECO1 and UTI89, but 14 extra sequences were found (SEQ ID NOs: 3 to 16).
Figure 1 shows an alignment of SEQ ID NOs: 2 to 16. The - 30 N-terminal amino acids are 100% conserved, and these include the signal peptide (aa 1-23) and the N-terminus cysteine of the native lipoprotein.
- Some strains had a frameshift mutation in the AcfD (orf3526) gene, resulting in no expression of the polypeptide. The acfD gene was totally absent from strains CFT073, EDL933, Sakai and B171.
-
Figure 2 shows the % identity between the amino acid sequences. The labels are SEQ ID NOs, except for MG1655, RS218, DH10B, APECO1 and UTI89 where the strain name is used. The lowest level of identity (boxed inFigure 2 ) was 85.9%, between SEQ ID NOs: 2 and 4 (both ExPEC strains). - The AcfD (orf3526) sequence from strain IHE3034 was cloned and expressed from a plasmid as a recombinant His-tagged protein without a leader peptide, in an E. coli host. Protein was purified and analysed. Gel filtration showed a much higher molecular weight than predicted based solely on the amino acid sequence. Gel analysis in the absence of DTT, but not in its presence, shows higher molecular weight forms of the protein (
Figure 3 ). Thus the protein is likely to form oligomers. - Sera raised against AcfD (orf3526) were used in western blots against total cell lysates (
Figure 4(A) ) or culture supernatants precipitated with 60% TCA (Figure 4(B) ). The sera recognised a ∼150kDa protein in lysates from both pathogenic and commensal strains. They did not react with this band in lysates from CFT073 or from an AcfD (orf3526) knockout mutant of IHE3034. Reactivity with proteins in the supernatants indicates that the protein may be secreted. - CD1 mice (5 weeks old) were immunized sub-cutaneously using 20 µg of the antigen plus Freund's adjuvant (or other adjuvant as indicated below). The mice were inoculated at 0, 21, and 35 days. Fourteen days after the third inoculation, the mice were challenged with a lethal dose (LD80) of a pathogenic strain of E. coli. Blood was collected from the
tail 24 hours after challenge to evaluate bacteremia. The mortality was monitored for four days post-challenge. The protection rate may be calculated as (%dead in control group (no immunization) - %dead in experimental group (immunized))/ %dead in control group x 100.TABLE 1 Candidates Sepsis animal model Survival with vaccination (%) Survival without vaccination (%) P value 20µg 3526/ alum 64/78 (82) 9/80 (11) 0.0001 - Thus, the AcfD precursor (orf3526) is an effective candidate for use as a vaccine or vaccine component.
- To further demonstrate the utility of the AcfD precursor (orf3526) as a candidate for a vaccine or a vaccine component, the AcfD precursor (orf3526) was tested for its ability to provide cross protection against other pathogenic strains of E. coli using the animal sepsis model as indicated above. The results of these tests are indicated in Table 2 below (with the results from Table 1 included for comparison purposes).
TABLE 2 20µg 3526/alum Challenge Strain (percent identity to IHE3034) Sepsis animal model Survival with vaccination (%) Survival without vaccination (%) P value IHE3034 (100) 64/78 (82) 9/80 (11) 0.0001 9855/93 (87) 9/16 (56) 4/15 (26) 0.14 BK658 (98) 4/8 (50) 1/8 (12.5) 0.28 B616 (100C) 6/8 (78) 0/8 (0) 0.007 536 (86) 6/7 (85) 3/8 (37.5) 0.11 - Thus, the AcfD precursor (orf3526) is an effective candidate for use as a vaccine or vaccine component not just against the strain from which the protein was derived, but also against related strains, thus increasing the utility. Based upon the similarity between the response to the AcfD precursor (orf3526) and the response to the three immunogenic fragments tested below (3526A, 3526B, and 3526C), one would expect that these three fragments would provide similar cross protection as well.
- Unexpectedly, AcfD (orf3526) protein displayed low solubility even though the protein is a secreted protein. As shown in
Figure 5(B) , removal of the N-terminus of AcfD (orf3526) through the gly-ser linker or gly-ser region significantly increased solubility when expressed at 25° C (See pK1-ΔG3526Figure 5(B) ). Similarly removal of the N-terminus of AcfD (orf3526) through the proline rich region significantly increased solubility when expressed at 25° C (See pK1-AP3526Figure 5(B) ). - To confirm that both fragments had substantially the same immunogenicity as the full length AcfD (orf3526), the fragments were purified. The purified fragments were used in three immunization experiments in mice, adjuvanted with Freund's complete adjuvant. Immunized mice were then challenged with a sublethal dose of E. coli. Immunization with AcfD (orf3526) with the N-terminus through the gly-ser linker or gly-ser region removed protected 100% of the mice from death, whereas death occurred in 90% of the animals in the non-immunized control group. Immunization with AcfD (orf3526) with the N-terminus through the proline rich region removed protected 90% of the mice from death, whereas death occurred in 90% of the animals in the non-immunized control group.
- Bacteria with one of the three constructs expressing his-tagged variants of AcfD (orf3526) were cultured in 30 ml of medium and induced to express the AcfD (orf3526) variant at 25° C (AcfD (orf3526) without the leader peptide (3526), AcfD (orf3526) with the N-terminus removed through the gly-ser linker or gly-ser region (ΔG3526), and AcfD (orf3526) with the N-terminus removed through the proline rich region (ΔP3526). The bacteria were harvested and lysed by sonication. The soluble fractions were isolated and loaded on an IMAC column. The column was washed three times with 20mM imidazole buffer. The AcfD (orf3526) variants were then eluted with three washes of 500 mM imidazole buffer. As shown in
Figure 6 , removal of the N-terminus of AcfD (orf3526) through the gly-ser linker or gly-ser region significantly increased solubility and yield of purified protein. The yield obtained was estimated by Bradford assay to be as follows: 0.18 mg of 3526 and 2.34 mg ΔG3526. - A zinc binding motif was identified in the AcfD (orf3526) protein (See
Figure 8 ). The zincin zinc metalloprotease motif is also found in StcE (Secreted protease of C1 esterase inhibitor from EHEC) which is a protein secreted by EHEC that is involved in the pathogenesis of such E. coli.StcE HEVGHNYGLGH (SEQ ID NO: 96) AcfD (orf3526) HEVGHNAAETP (SEQ ID NO: 97) - Given that this motif is conserved in all variants of AcfD (orf3526) and EHEC was the only pathotype in which AcfD (orf3526) was not identified, it was surmised that AcfD (orf3526) similarly is a zinc metalloprotease involved in the pathogenicity of the E. coli pathotypes in which it is expressed and secreted. Therefore, inactivation of the toxicity of AcfD (orf3526) was sought. One of skill in the art would have no difficulty in modifying AcfD (orf3526) to inactivate the metalloprotease. As the Glu residue of the motif is known to be most important to catalytic activity while the two His residues coordinate the Zinc, the Glu was mutated to Ala as an exemplary mutation inactivating the metalloprotease activity - SEQ ID NOs: 58-76 (See, e.g., Microbiology and Molecular Biology Reviews, September 1998, p. 597-635, Vol. 62, No. 3). In addition, two additional detoxified variants were designed: one with the C-terminus deleted - SEQ ID NOs: 20-38, and one with the N-terminus deleted - SEQ ID NOs: 39-57.
- Two different cell lines were tested for their ability to assay toxicity of the AcfD (orf3526) protein: human bone marrow endothelial cells (HBMEC) and Chinese hamster ovary cells (CHO cells). For HBMEC, the effect of increasing doses of AcfD (orf3526) protein (0.1 µg/ml, 1 µg/ml, and 10 µg/ml) was compared to TNF-α (as a positive control). The TNF-α treated cells showed an average of ∼22.5% cell death (average of three experiments) while the highest dose of AcfD (orf3526) protein showed only an average of ∼5% cell death (average of three experiments) as compared to the negative control which showed only an average of ∼2.5% cell death (average of three experiments). For CHO cells, the effect of increasing doses of AcfD (orf3526) protein (1 µg/ml, 10 µg/ml, and 100 µg/ml) was compared to TNF-α (as a positive control) by measuring the change in potential difference over time due to the alteration of the cytoskeleton of the CHO cells. At the highest level of AcfD (orf3526) protein test, a significant decrease was detected after 46 hours, but the decrease in response to TNF-α was notably greater.
- Thus, while a preferred test for toxicity with a strong signal has not been identified, the fragments identified below still have significant utility. As discussed above, these three fragment should provide a similar degree of cross protection as the full length version tested above. Furthermore, commensal E. coli is a preferred strain for expression of AcfD (orf3526) protein. However, as indicated
Figure 4 , commensal E coli express a version of AcfD (orf3526) protein, but do not secrete the protein. Thus, expression of a fragment of an AcfD (orf3526) protein provides the advantage of allowing separation of the fragment from the endogenous AcfD (orf3526) protein based upon the size difference without needing an affinity purification tag. - Mutants were obtained using GeneTailor site-directed mutagenesis system (Invitrogen). Genes were cloned in pET-21b vectors (Novagen) and transformed in DH5α-T1 chemically competent cells for propagation (Invitrogen). BL21 (DE3) chemically competent cells were used for expression. All candidates were cloned and expressed without the signal sequence and as his-tag fusion proteins, being purified by affinity chromatography.
-
3526A 3526F GGAATTCCATATGTGTGATGGTGGTGGTTCA NdeI 98 3526AR CCGCTCGAGGTTGTTCACCACCGATTT XhoI 99 3526B 3526BF GGAATTCCATATGGATCCGCAAGGGTATCC NdeI 100 3526R CCGCTCGAGCTCGGCAGACATCTTATG XhoI 101 -
Sample Oligo name Nucleotide sequence (5' -3') 3526E1305A 3526GTF1 ACGACTGGCTGATTTGGCACGCAGTCGGTCATA 102 3526GTR1 GTGCCAAATCAGCCAGTCGTTCAGCGGCGT 103 - To confirm that both fragments and the point mutation had substantially the same immunogenicity as the full length AcfD (orf3526), the fragments were purified. The purified fragments were used in three immunization experiments in mice, adjuvanted with Freund's complete adjuvant. Immunized mice were then challenged with a lethal dose of E. coli. Results of the challenge are shown in Table 3 below.
TABLE 3 Candidates Sepsis animal model Survival with vaccination (%) Survival without vaccination (%) P value 3526A (AcfD - C-term deletion) 7/8 (87.5) 1/8 (12.5) 0.01 3526B (AcfD - N-term deletion) 6/8 (75) 1/8 (12.5) 0.04 - As an additional confirmation, a construct was designed that combined the ΔG3526 N-terminal deletion that improved solubility with deletion of the C-terminus through the zinc motif (ΔG3526C). Exemplary constructs are provided as SEQ ID NOs: 77 to 95.
- A non-his-tagged expression vector for the ΔG3526C (E. coli strain IHE3034 - SEQ ID NO:77) construct was obtained by PCR using the primers indicated below (ΔG3526A/C_For and ΔG3526C_NatRev). The amplified PCR fragment was digested with NdeI and XhoI and ligated into a pET-24b(+) vector (Novagen) and transformed in DH5α-T1 chemically competent cells for propagation (Invitrogen). BL21 (DE3) chemically competent cells were used for expression.
- A his-tagged expression vector for the ΔG3526C (E. coli strain IHE3034 - SEQ ID NO:77) construct was obtained using Polymerase Incomplete Primer Extension (PIPE) using the primers indicated below (ΔG3526A/C_For and ΔG3526C_Nat for I-PCR and p-pet1 and pet3 for V-PCR of the pET-21b vector (Novagen)). The expression vector was transformed in DH5α-T1 chemically competent cells for propagation (Invitrogen). BL21 (DE3) chemically competent cells were used for expression.
- The his-tagged version of ΔG3526C was expressed (See, e.g.,
Figure 10 ) without the signal sequence and purified by affinity chromatography based upon the his-tag. -
ΔG3526C ΔG3526A/C_For (NdeI) gaaggagatatacatatgGATACGCCGTCTGTAGATTCTGG ΔG3526C_NatRev (XhoI) gtggtggtgctcgag TTACCAAATCAGCCAGTCGTTCAGC -
ΔG3526C ΔG3526A/C_For (NdeI) gaaggagatatacatatgGATACGCCGTCTGTAGATTCTGG ΔG3526C_Rev (XhoI) gtggtggtgctcgagCCAAATCAGCCAGTCGTTCAGC -
pET-21b p-pet1 catatgatatctccttcttaaagTTAAACAAAATTATTTCTAG p-pet3 CTCGAGCACCACCACCAC - To confirm that the additional fragment had substantially the same immunogenicity as the full length AcfD (orf3526), the fragment was purified. The purified fragment was then tested as follows. Two groups of 8 CD1 mice (4 weeks old) were immunized s.c. with three doses of antigen ΔG3526C (either 2 µg or 20 µg), formulated in Alum at
days day 4 and statistical analysis was carried out by Fisher's exact test. Results of the challenge are shown in Table 4 below.TABLE 4 Candidate: Sepsis animal model ΔG3526C Survival with vaccination (%) Survival without vaccination (%) P value 2 µg/ alum 13/16 (81) 0/7 (0) 0.0005 20 µg/alum 15/16 (93) 0/7 (0) 0.0001 - To further confirm the utility of the additional fragment in providing protection against multiple strains, the purified fragment was further tested as follows. CD1 mice (4 weeks old) were immunized s.c. with three doses of antigen ΔG3526C (either 10 µg or 20 µg), formulated in Alum at
days day 4 and statistical analysis was carried out by Fisher's exact test. Results of the challenge are shown in Table 5 below.TABLE 5 Challenge Strain Candidate ΔG3526C with Alum (infectious dose) 20 µg/ alum 10 µg/alum IHE3034 Survival: 15/16 (93%) Survival: 6/8 (75%) (1 x 107 CFU) PE: 93% PE: 75% 536 Survival: 5/8 (62.5%) Survival: 6/8 (75%) (1 x 107 CFU i.v.) PE: 50% PE: 66% 9855/93 Survival: 23/47 (49%) (1 x 107 CFU) PE: 39% B616 Survival: 6/8 (75%) Survival: 22/24 (91%) (2.5 x 107 CFU) PE: 66% PE: 87% UR41/S Survival: 4/8 (50%) (2.5 x 107 CFU) PE: 50% UR40/R Survival: 5/8 (62.5%) % (5 x 106 CFU) PE: 62.5% IN22/R Survival: 7/8 (87.5%) (1 x 107 CFU) PE: 80% -
- [1] Kaper et al. (2004) Nat Rev Microbiol. 2(2):123-40.
- [2] Anjum et al. (2007) Appl Environ Microbiol 73 :5692-7.
- [3] Russo & Johnson (2000) J Infect Dis 181:1753-1754.
- [4] Smith et al. (2007) Foodborne Pathogens And Disease 4:134-63.
- [5]
WO2006/089264 . - [6]
WO2006/091517 . - [7] Needleman & Wunsch (1970) J. Mol. Biol. 48, 443-453.
- [8] Rice et al. (2000) Trends Genet 16:276-277.
- [9] Geysen et al. (1984) PNAS USA 81:3998-4002.
- [10] Carter (1994) Methods Mol Biol 36:207-23.
- [11] Jameson, BA et al. 1988, CABIOS 4(1):181-186.
- [12] Raddrizzani & Hammer (2000) Brief Bioinform 1(2):179-89.
- [13] Bublil et al. (2007) Proteins 68(1):294-304.
- [14] De Lalla et al. (1999) J. Immunol. 163:1725-29.
- [15] Kwok et al. (2001) Trends Immunol 22:583-88.
- [16] Brusic et al. (1998) Bioinformatics 14(2):121-30
- [17] Meister et al. (1995) Vaccine 13(6):581-91.
- [18] Roberts et al. (1996) AIDS Res Hum Retroviruses 12(7):593-610.
- [19] Maksyutov & Zagrebelnaya (1993) Comput Appl Biosci 9(3):291-7.
- [20] Feller & de la Cruz (1991) Nature 349(6311):720-1.
- [2]] Hopp (1993) Peptide Research 6:183-190.
- [22] Welling et al. (1985) FEBS Lett. 188:215-218.
- [23] Davenport et al. (1995) Immunogenetics 42:392-297.
- [24] Tsurui & Takahashi (2007) J Pharmacol Sci. 105(4):299-316.
- [25] Tong et al. (2007) Brief Bioinform. 8(2):96-108.
- [26] Schirle et al. (2001) J Immunol Methods. 257(1-2):1-16.
- [27] Chen et al. (2007) Amino Acids 33(3):423-8.
- [28]
US patent 5,707,829 - [29] Current Protocols in Molecular Biology (F.M. Ausubel et al. eds., 1987) .
- [30] Vaccine Design: The Subunit and Adjuvant Approach (eds. Powell & Newman) Plenum Press 1995 (ISBN 0-306-44867-X).
- [31] Vaccine Adjuvants: Preparation Methods and Research Protocols (Volume 42 of Methods in Molecular Medicine series). ISBN: 1-59259-083-7. Ed. O'Hagan.
- [32]
US patent 6355271 . - [33]
WO00/23105 - [34]
WO90/14837 - [35]
WO90/14837 - [36] Podda & Del Giudice (2003) Expert Rev Vaccines 2:197-203.
- [37] Podda (2001) Vaccine 19: 2673-2680.
- [38] Vaccine Design: The Subunit and Adjuvant Approach (eds. Powell & Newman) Plenum Press 1995 (ISBN 0-306-44867-X).
- [39] Vaccine Adjuvants: Preparation Methods and Research Protocols (Volume 42 of Methods in Molecular Medicine series). ISBN: 1-59259-083-7. Ed. O'Hagan.
- [40] Allison & Byars (1992) Res Immunol 143:519-25.
- [41] Hariharan et al. (1995) Cancer Res 55:3486-9.
- [42]
US-2007/014805 . - [43] Suli et al. (2004) Vaccine 22(25-26):3464-9.
- [44]
WO95/11700 - [45]
US patent 6,080,725 . - [46]
WO2005/097181 . - [47]
WO2006/113373 . - [48] Han et al. (2005) Impact of Vitamin E on Immune Function and Infectious Diseases in the Aged at Nutrition, Immune functions and Health EuroConference, Paris, 9-10 June 2005.
- [49]
US- 6630161 . - [50]
US 5,057,540 . - [51]
WO96/33739 - [52]
EP-A-0109942 . - [53]
WO96/11711 - [54]
WO00/07621 - [55] Barr et al. (1998) Advanced Drug Delivery Reviews 32:247-271.
- [56] Sjolanderet et al. (1998) Advanced Drug Delivery Reviews 32:321-338.
- [57] Niikura et al. (2002) Virology 293:273-280.
- [58] Lenz et al. (2001) J Immunol 166:5346-5355.
- [59] Pinto et al. (2003) J Infect Dis 188:327-338.
- [60] Gerber et al. (2001) J Virol 75:4752-4760.
- [61]
WO03/024480 - [62]
WO03/024481 - [63] Gluck et al. (2002) Vaccine 20:B10-B16.
- [64]
EP-A-0689454 . - [65] Johnson et al. (1999) Bioorg Med Chem Lett 9:2273-2278.
- [66] Evans et al. (2003) Expert Rev Vaccines 2:219-229.
- [67] Meraldi et al. (2003) Vaccine 21:2485-2491.
- [68] Pajak et al. (2003) Vaccine 21:836-842.
- [69] Kandimalla et al. (2003) Nucleic Acids Research 31:2393-2400.
- [70]
WO02/26757 - [71]
WO99/62923 - [72] Krieg (2003) Nature Medicine 9:831-835.
- [73] McCluskie et al. (2002) FEMS Immunology and Medical Microbiology 32:179-185.
- [74]
WO98/40100 - [75]
US 6,207,646 . - [76]
US 6,239,116 . - [77]
US 6,429,199 . - [78] Kandimalla et al. (2003) Biochemical Society Transactions 31 (part 3):654-658.
- [79] Blackwell et al. (2003) J Immunol 170:4061-4068.
- [80] Krieg (2002) Trends Immunol 23:64-65.
- [81]
WO01/95935 - [82] Kandimalla et al. (2003) BBRC 306:948-953.
- [83] Bhagat et al. (2003) BBRC 300:853-861.
- [84]
WO03/035836 - [85]
WO01/22972 - [86] Schellack et al. (2006) Vaccine 24:5461-72.
- [87]
WO95/17211 - [88]
WO98/42375 - [89] Beignon et al. (2002) Infect Immun 70:3012-3019.
- [90] Pizza et al. (2001) Vaccine 19:2534-2541.
- [91] Pizza et al. (2000) Int J Med Microbiol 290:455-461.
- [92] Scharton-Kersten et al. (2000) Infect Immun 68:5306-5313.
- [93] Ryan et al. (1999) Infect Immun 67:6270-6280.
- [94] Partidos et al. (1999) Immunol Lett 67:209-216.
- [95] Peppoloni et al. (2003) Expert Rev Vaccines 2:285-293.
- [96] Pine et al. (2002) J Control Release 85:263-270.
- [97] Tebbey et al. (2000) Vaccine 18:2723-34.
- [98] Domenighini et al. (1995) Mol Microbiol 15:1165-1167.
- [99]
WO99/40936 - [100]
WO99/44636 - [101] Singh et al] (2001) J Cont Release 70:267-276.
- [102]
WO99/27960 - [103]
US 6,090,406 . - [104]
US 5,916,588 . - [105]
EP-A-0626169 . - [106]
WO99/52549 - [107]
WO01/21207 - [108]
WO01/21152 - [109] Andrianov et al. (1998) Biomaterials 19:109-115.
- [110] Payne et al. (1998) Adv Drug Delivery Review 31:185-196.
- [111]
US 4,680,338 . - [112]
US 4,988,815 . - [113]
WO92/15582 - [114] Stanley (2002) Clin Exp Dermatol 27:571-577.
- [115] Wu et al. (2004) Antiviral Res. 64(2):79-83.
- [116] Vasilakos et al. (2000) Cell Immunol. 204(1):64-74.
- [117]
US patents 4689338 ,4929624 ,5238944 ,5266575 ,5268376 ,5346905 ,5352784 ,5389640 ,5395937 ,5482936 ,5494916 ,5525612 ,6083505 ,6440992 ,6627640 ,6656938 ,6660735 ,6660747 ,6664260 ,6664264 ,6664265 ,6667312 ,6670372 ,6677347 ,6677348 ,6677349 ,6683088 ,6703402 ,6743920 ,6800624 ,6809203 ,6888000 and6924293 . - [118] Jones (2003) Curr Opin Investig Drugs 4:214-218.
- [119]
WO03/011223 - [120] Johnson et al. (1999) Bioorg Med Chem Lett 9:2273-2278.
- [121] Evans et al. (2003) Expert Rev Vaccines 2:219-229.
- [122]
WO2004/060308 . - [123]
WO2004/064759 . - [124]
US 6,924,271 . - [125]
US2005/0070556 . - [126]
US 5,658,731 . - [127]
US patent 5,011,828 . - [128]
WO2004/87153 - [129]
US 6,605,617 . - [130]
WO02/18383 - [131]
WO2004/018455 . - [132]
WO03/082272 - [133] Wong et al. (2003) J Clin Pharmacol 43(7):735-42.
- [134]
US2005/0215517 . - [135] Dyakonova et al. (2004) Int Immunopharmacol 4(13):1615-23.
- [136]
FR-2859633 - [137] Signorelli & Hadden (2003) Int Immunopharmacol 3(8):1177-86.
- [138]
WO2004/064715 . - [139] De Libero et al, Nature Reviews Immunology, 2005, 5: 485-496
- [140]
US patent 5,936,076 . - [141] Oki et al, J. Clin. Investig., 113: 1631-1640
- [142]
US2005/0192248 - [143] Yang et al, Angew. Chem. Int. Ed., 2004, 43: 3818-3822
- [144]
WO2005/102049 - [145] Goff et al, J. Am. Chem., Soc., 2004, 126: 13602-13603
- [146]
WO03/105769 - [147] Cooper (1995) Pharm Biotechnol 6:559-80.
- [148]
WO99/11241 - [149]
WO94/00153 - [150]
WO98/57659 - [151] European patent applications
0835318 ,0735898 and0761231 . - [152] Durant et al. (2007) Infect Immun 75:1916-25.
- [153]
WO02/081653 - [154] Donnelly et al. (1997) Annu Rev Immunol 15:617-648.
- [155] Strugnell et al. (1997) Immunol Cell Biol 75(4):364-369.
- [156] Cui (2005) Adv Genet 54:257-89.
- [157] Robinson & Torres (1997) Seminars in Immunol 9:271-283.
- [158] Brunham et al. (2000) J Infect Dis 181 Suppl 3:S538-43.
- [159] Svanholm et al. (2000) Scand J Immunol 51(4):345-53.
- [160] DNA Vaccination - Genetic Vaccination (1998) eds. Koprowski et al. (ISBN 3540633928).
- [161] Gene Vaccination : Theory and Practice (1998) ed. Raz (ISBN 3540644288).
- [162] Findeis et al., Trends Biotechnol. (1993) 11:202
- [163] Chiou et al. (1994) Gene Therapeutics: Methods And Applications Of Direct Gene Transfer. ed. Wolff
- [164] Wu et al., J. Biol. Chem. (1988) 263:621
- [165] Wu et al., J. Biol. Chem. (1994) 269:542
- [166] Zenke et al., Proc. Natl. Acad. Sci. (USA) (1990) 87:3655
- [167] Wu et al., J. Biol. Chem. (1991) 266:338
- [168] Jolly, Cancer Gene Therapy (1994) 1:51
- [169] Kimura, Human Gene Therapy (1994) 5:845
- [170] Connelly, Human Gene Therapy (1995) 1:185
- [171] Kaplitt, Nature Genetics (1994) 6:148
- [172]
WO 90/07936 - [173]
WO 94/03622 - [174]
WO 93/25698 - [175]
WO 93/25234 - [176]
US patent 5,219,740 . - [177]
WO 93/11230 - [178]
WO 93/10218 - [179]
US patent 4,777,127 . - [180]
GB Patent No. 2,200,651 - [181]
EP-A-0345242 . - [182]
WO 91/02805 - [183]
WO 94/12649 - [184]
WO 93/03769 - [185]
WO 93/19191 - [186]
WO 94/28938 - [187]
WO 95/11984 - [188]
WO 95/00655 - [189] Curiel, Hum. Gene Ther. (1992) 3:147
- [190] Wu, J. Biol. Chem. (1989) 264:16985
- [191]
US patent 5,814,482 . - [192]
WO 95/07994 - [193]
WO 96/17072 - [194]
WO 95/30763 - [195]
WO 97/42338 - [196]
WO 90/11092 - [197]
US patent 5,580,859 - [198]
US patent 5,422,120 - [199]
WO 95/13796 - [200]
WO 94/23697 - [201]
WO 91/14445 - [202]
EP-0524968 . - [203] Philip, Mol. Cell Biol. (1994) 14:2411
- [204] Woffendin, Proc. Natl. Acad. Sci. (1994) 91:11581
- [205]
US patent 5,206,152 . - [206]
WO 92/11033 - [207]
US patent 5,149,655 . - [208] Gennaro (2000) Remington: The Science and Practice of Pharmacy. 20th edition, ISBN: 0683306472.
- [209] Methods In Enzymology (S. Colowick and N. Kaplan, eds., Academic Press, Inc.)
- [210] Handbook of Experimental Immunology, Vols. I-IV (D.M. Weir and C.C. Blackwell, eds, 1986, Blackwell Scientific Publications)
- [211] Sambrook et al. (2001) Molecular Cloning: A Laboratory Manual, 3rd edition (Cold Spring Harbor Laboratory Press).
- [212] Handbook of Surface and Colloidal Chemistry (Birdi, K.S. ed., CRC Press, 1997)
- [213] Ausubel et al. (eds) (2002) Short protocols in molecular biology, 5th edition (Current Protocols).
- [214] Molecular Biology Techniques: An Intensive Laboratory Course, (Ream et al., eds., 1998, Academic Press)
- [215] PCR (Introduction to Biotechniques Series), 2nd ed. (Newton & Graham eds., 1997, Springer Verlag)
- [216] Current Protocols in Molecular Biology (F.M. Ausubel et al., eds., 1987)
- [217] Smith & Waterman (1981) Adv. Appl. Math. 2: 482-489.
-
-
SEQ ID NO: 1

-
SEQ ID NO: 2

-
SEQ ID NO: 3


-
SEQ ID NO: 4

-
SEQ ID NO: 5

-
SEQ ID NO: 6


-
SEQ ID NO: 7

-
SEQ ID NO: 8

-
SEQ ID NO: 9


-
SEQ ID NO: 10

-
SEQ ID NO: 11

-
SEQ ID NO: 12


-
SEQ ID NO: 13

-
SEQ ID NO: 14

-
SEQ ID NO: 15

-
SEQ ID NO: 16

-
SEQ ID NO: 17


-
SEQ ID NO: 18

-
SEQ ID NO: 19

-
SEQ ID NO: 20

-
SEQ ID NO: 21


-
SEQ ID NO: 22

-
SEQ ID NO: 23

-
SEQ ID NO: 24

-
SEQ ID NO: 25

-
SEQ ID NO: 26


-
SEQ ID NO: 27

-
SEQ ID NO: 28

-
SEQ ID NO: 29

-
SEQ ID NO: 30

-
SEQ ID NO: 31

-
SEQ ID NO: 32

-
SEQ ID NO: 33

-
SEQ ID NO: 34

-
SEQ ID NO: 35

-
SEQ ID NO: 36

-
SEQ ID NO: 37


-
SEQ ID NO: 38

-
SEQ ID NO: 39

-
SEQ ID NO: 40

-
SEQ ID NO: 41

-
SEQ ID NO: 42


-
SEQ ID NO: 43

-
SEQ ID NO: 44

-
SEQ ID NO: 45

-
SEQ ID NO: 46

-
SEQ ID NO: 47

-
SEQ ID NO: 48

-
SEQ ID NO: 49

-
SEQ ID NO: 50

-
SEQ ID NO: 51

-
SEQ ID NO: 52

-
SEQ ID NO: 53


-
SEQ ID NO: 54

-
SEQ ID NO: 55

-
SEQ ID NO: 56

-
SEQ ID NO: 57

-
SEQ ID NO: 58


-
SEQ ID NO: 59

-
SEQ ID NO: 60

-
SEQ ID NO: 61


-
SEQ ID NO: 62

-
SEQ ID NO: 63

-
SEQ ID NO: 64

-
SEQ ID NO: 65

-
SEQ ID NO: 66


-
SEQ ID NO: 67

-
SEQ ID NO: 68

-
SEQ ID NO: 69


-
SEQ ID NO: 70
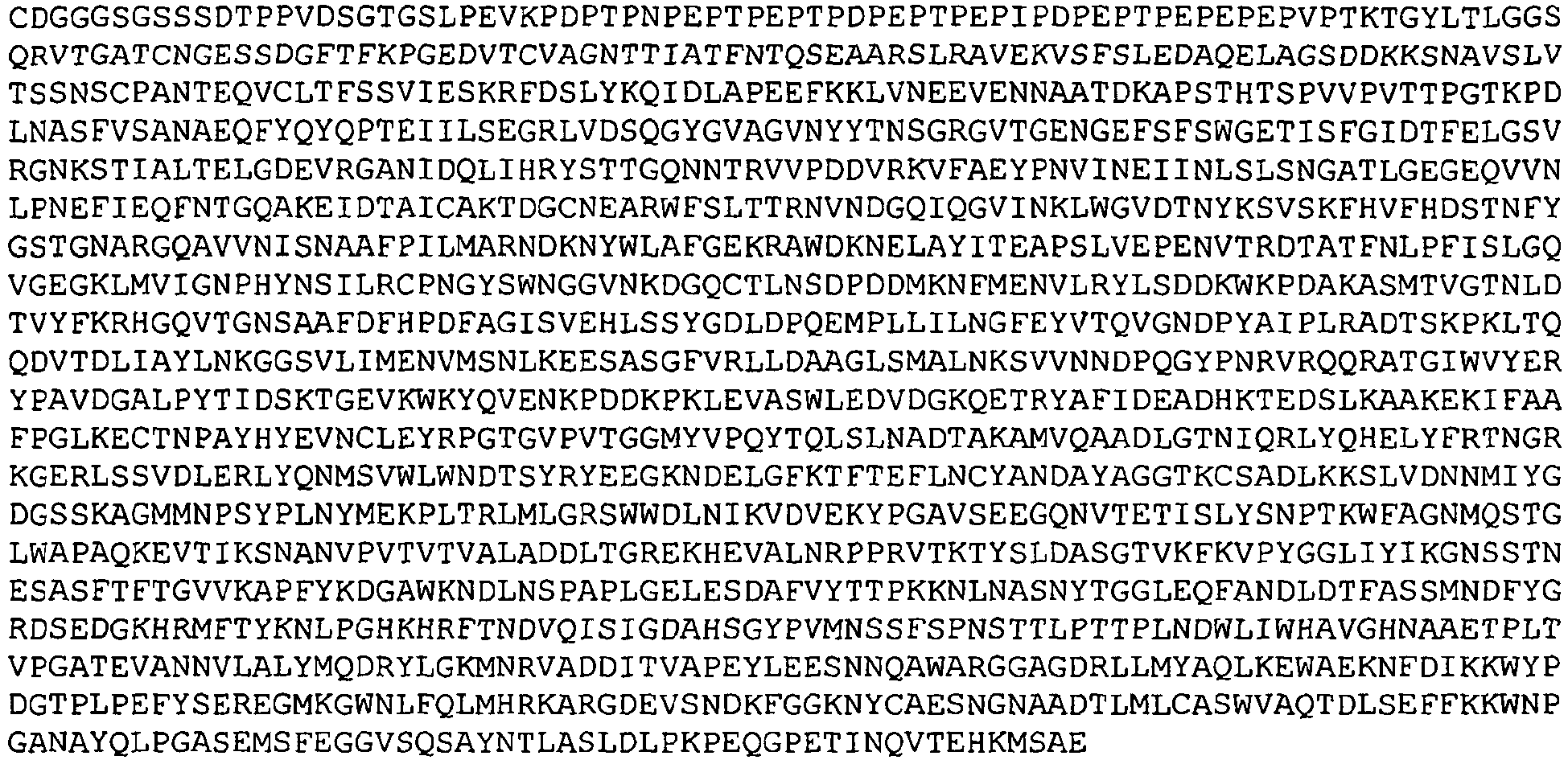
-
SEQ ID NO: 71

-
SEQ ID NO: 72


-
SEQ ID NO: 73

-
SEQ ID NO: 74

-
SEQ ID NO: 75


-
SEQ ID NO: 76

-
SEQ ID NO: 77

-
SEQ ID NO: 78


-
SEQ ID NO: 79

-
SEQ ID NO: 80

-
SEQ ID NO: 81


- SEQ ID NO: 82
-
SEQ ID NO: 83

-
SEQ ID NO: 84
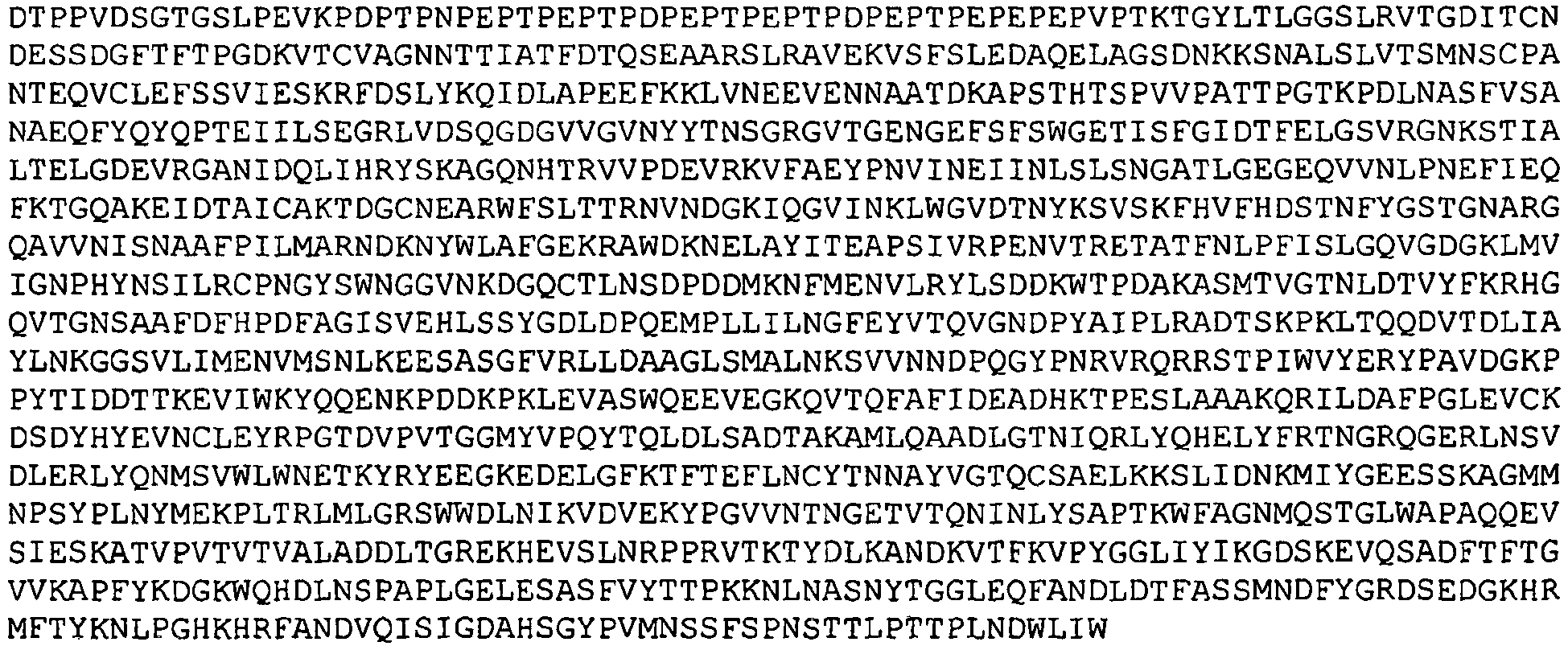
-
SEQ ID NO: 85


-
SEQ ID NO: 86

-
SEQ ID NO: 87

-
SEQ ID NO: 88


-
SEQ ID NO: 89

- SEQ ID NO: 90
-
SEQ ID NO: 91


-
SEQ ID NO: 92
-
SEQ ID NO: 93

-
SEQ ID NO: 94

-
SEQ ID NO: 95


- SEQ ID NO: 96
HEVGHNYGLGH - SEQ ID NO: 97
HEVGHNAAETP - SEQ ID NO: 98
GGAATTCCATATGTGTGATGGTGGTGGTTCA - SEQ ID NO: 99
CCGCTCGAGGTTGTTCACCACCGATTT - SEQ ID NO: 100
GGAATTCCATATGGATCCGCAAGGGTATCC - SEQ ID NO: 101
CCGCTCGAGCTCGGCAGACATCTTATG - SEQ ID NO: 102
ACGACTGGCTGATTTGGCACGCAGTCGGTCATA - SEQ ID NO: 103
GTGCCAAATCAGCCAGTCGTTCAGCGGCGT - SEQ ID NO: 104
GAAGGAGATATACATATGGATACGCCGTCTGTAGATTCTGG - SEQ ID NO: 105
GTGGTGGTGCTCGAGTTACCAAATCAGCCAGTCGTTCAGC - SEQ ID NO: 106
GAAGGAGATATACATATGGATACGCCGTCTGTAGATTCTGG - SEQ ID NO: 107
GTGGTGGTGCTCGAGCCAAATCAGCCAGTCGTTCAGC - SEQ ID NO: 108
CATATGATATCTCCTTCTTAAAGTTAAACAAAATTATTTCTAG - SEQ ID NO: 109
CTCGAGCACCACCACCAC - SEQ ID NO: 110
ICICICICICICICICICICICICIC - SEQ ID NO: 111
KLKLLLLLKLK
Claims (21)
- An immunogenic polypeptide comprising an E. coli AcfD (orf3526) polypeptide comprising a mutation relative to the E. coli AcfD (orf3526) protein which decreases the toxicity of the immunogenic polypeptide as compared to the E. coli AcfD (orf3526) protein wherein the mutation is selected from a deletion of all or a portion of the zincin metalloprotease domain and a point mutation in zincin metalloprotease domain which reduces the protease activity and wherein the immunogenic polypeptide raises a substantially similar immune response in a subject as the E. coli AcfD (orf3526) protein.
- The immunogenic polypeptide of claim 1, wherein the E. coli AcfD (orf3526) protein has an amino acid sequence selected from the group consisting of SEQ ID NOs:1-19.
- The immunogenic polypeptide of any of claims 1-2, wherein the point mutation is a mutation of a zinc binding residue or a mutation of a catalytic residue.
- The immunogenic polypeptide of any of claims 1-3, wherein the zinc binding residue is amino acid number 1305 based upon alignment with SEQ ID NO: 1.
- The immunogenic polypeptide of any of claims 1-4, wherein the immunogenic polypeptide does not comprise at least the last 100 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 200 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 300 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 400 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 500 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 600 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 700 C-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the last 750 C-terminal amino acids of the E. coli AcfD (orf3526) protein, or at least the last 758 C-terminal amino acids of the E. coli AcfD (orf3526) protein or does not comprise at least the first 100 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 200 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 300 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 400 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 500 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 600 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 700 N-terminal amino acids of the E. coli AcfD (orf3526) protein, at least the first 750 N-terminal amino acids of the E. coli AcfD (orf3526) protein, or at least the first 760 N-terminal amino acids of the E. coli AcfD (orf3526) protein.
- The immunogenic polypeptide of any of claims 1-4 wherein the immunogenic polypeptide comprises an amino acid sequence that comprises:(a) the amino acid sequence selected from the group consisting of SEQ ID NOs 20 to 38, 58 to 76 and 77 to 95;(b) from 1 to 10 single amino acid alterations compared to SEQ ID NOs: 20 to 38, 58 to 76 and 77 to 95;(c) at least 85% sequence identity to any one of SEQ ID NOs: 20 to 38, 58 to 76 and 77 to 95; or(d) when aligned with any of SEQ ID NOs: 20 to 38, 58 to 76 and 77 to 95 using a pairwise alignment algorithm, each moving window of x amino acids from N terminus to C terminus has at least x·y identical aligned amino acids, where x is 30 and y is 0.75,
wherein the immunogenic polypeptide raises a substantially similar immune response in a subject as the E. coli AcfD (orf3526) protein. - The immunogenic polypeptide any of claims 1-6, wherein the immunogenic polypeptide further contains a deletion relative to the E. coli AcfD (orf3526) protein which increases solubility of the immunogenic polypeptide as compared to the E. coli AcfD (orf3526) protein wherein the deletion is removal of substantially all of the N-terminal amino acids up to the gly-ser region, removal of all or a part of the N-terminal proline-rich repeat, or both.
- The immunogenic polypeptide of claim 7, wherein the deletion is removal of at least the first 20 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 20 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 30 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 38 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 40 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 50 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 60 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 70 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 80 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, at least the first 90 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein, or at least the first 94 N-terminal amino acids as compared to the E. coli AcfD (orf3526) protein.
- The immunogenic polypeptide of any of claims 1-8 wherein the immunogenic polypeptide is isolated, purified, or recombinant.
- The immunogenic polypeptide of any of claims 1-9 further comprising an adjuvant.
- A polynucleotide encoding the immunogenic polypeptide of any of claims 1-7.
- An E. coli cell, containing a plasmid that encodes the immunogenic polypeptide of any of claims 1-7.
- A vaccine component comprising the immunogenic polypeptide of claim 9.
- A vaccine comprising the vaccine component of claim 13.
- The vaccine of claim 14 further comprising an adjuvant.
- The vaccine of claim 14 or claim 15 further comprising an additional vaccine component selected from: a Neisseria meningitidis antigen, a Streptococcus pneumoniae antigen, a Streptococcus pyogenes antigen, a Moraxella catarrhalis antigen, a Bordetella pertussis antigen, a Staphylococcus aureus antigen, a Staphylococcus epidermis antigen, a Clostridium tetani antigen, a Cornynebacterium diphtheriae antigen, a Haemophilus influenzae type B (Hib) antigen, a Pseudomonas aeruginosa antigen, a Legionella pneumophila antigen, a Streptococcus agalactiae antigen, a Neiserria gonorrhoeae antigen, a Chlamydia trachomatis antigen, a Treponema pallidum antigen, a Haemophilus ducreyi antigen, a Enterococcus faecalis antigen, a Enterococcus faecium antigen, a Helicobacter pylori antigen, a Staphylococcus saprophyticus antigen, a Yersinia enterocolitica antigen, an additional E. coli antigen, a Bacillus anthracis antigen, a Yersinia pestis antigen, a Mycobacterium tuberculosis antigen, a Rickettsia antigen, a Listeria monocytogenes antigen, a Chlamydia pneumoniae antigen, a Vibrio cholerae antigen, a Salmonella typhi antigen, a Borrelia burgdorferi antigen, a Porphyromonas gingivalis antigen, and a Klebsiella antigen.
- An immunogenic composition comprising an immunogenic polypeptide of any of claims 1 to 9.
- The immunogenic composition of claim 17 further comprising an adjuvant.
- The immunogenic composition of claim 18 wherein the adjuvant comprises (a) 1-12% by volume of a metabolizable oil, and (b) 0.2% to 2.5% by weight of an emulsifying agent, wherein the metabolizable oil and the emulsifying agent are present in the form of an oil-in-water emulsion having oil droplets substantially all of which are less than 1 micron in diameter.
- The immunogenic composition of claim 18 wherein adjuvant comprises (a) 4-5% by volume of squalene, and (b) about 1% of an emulsifying agent comprising polyoxyethylenesorbitan monooleate and sorbitan trioleate, wherein the squalene and the emulsifying agent are present in the form of an oil-in-water emulsion having oil droplets substantially all of which are less than 1 micron in diameter.
- A vaccine in accordance with any one of claims 14-16, or an immunogenic composition in accordance with any one of claims17-20 for use in a method of inducing an immune response against E. coli, wherein the method comprises administering said vaccine or immunogenic composition to a subject.
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| SI201030830T SI2464658T1 (en) | 2009-07-16 | 2010-07-16 | Detoxified escherichia coli immunogens |
| EP14187138.4A EP2837386B1 (en) | 2009-07-16 | 2010-07-16 | Detoxified Escherichia coli immunogens |
| PL10752175T PL2464658T3 (en) | 2009-07-16 | 2010-07-16 | Detoxified escherichia coli immunogens |
| CY20141101080T CY1115849T1 (en) | 2009-07-16 | 2014-12-23 | ESCHERICHIA COLI DETOXINATED IMMUNOGENES |
| HRP20141270AT HRP20141270T1 (en) | 2009-07-16 | 2014-12-30 | Detoxified escherichia coli immunogens |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US22621909P | 2009-07-16 | 2009-07-16 | |
| US29065409P | 2009-12-29 | 2009-12-29 | |
| PCT/IB2010/002043 WO2011007257A1 (en) | 2009-07-16 | 2010-07-16 | Detoxified escherichia coli immunogens |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP14187138.4A Division EP2837386B1 (en) | 2009-07-16 | 2010-07-16 | Detoxified Escherichia coli immunogens |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP2464658A1 EP2464658A1 (en) | 2012-06-20 |
| EP2464658B1 true EP2464658B1 (en) | 2014-10-01 |
Family
ID=43126814
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP14187138.4A Active EP2837386B1 (en) | 2009-07-16 | 2010-07-16 | Detoxified Escherichia coli immunogens |
| EP10752175.9A Active EP2464658B1 (en) | 2009-07-16 | 2010-07-16 | Detoxified escherichia coli immunogens |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP14187138.4A Active EP2837386B1 (en) | 2009-07-16 | 2010-07-16 | Detoxified Escherichia coli immunogens |
Country Status (17)
| Country | Link |
|---|---|
| US (2) | US8871214B2 (en) |
| EP (2) | EP2837386B1 (en) |
| JP (1) | JP2012532626A (en) |
| CN (1) | CN102770443A (en) |
| AU (1) | AU2010272243A1 (en) |
| CA (1) | CA2768343A1 (en) |
| DK (1) | DK2464658T3 (en) |
| ES (2) | ES2670799T3 (en) |
| HR (1) | HRP20141270T1 (en) |
| IL (1) | IL217539A0 (en) |
| MX (1) | MX2012000734A (en) |
| PL (1) | PL2464658T3 (en) |
| PT (1) | PT2464658E (en) |
| SG (1) | SG178035A1 (en) |
| SI (1) | SI2464658T1 (en) |
| WO (1) | WO2011007257A1 (en) |
| ZA (1) | ZA201200833B (en) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9511130B2 (en) | 2011-09-14 | 2016-12-06 | Glaxosmithkline Biologicals Sa | Escherichia coli vaccine combination |
| WO2014102694A1 (en) * | 2012-12-24 | 2014-07-03 | Novartis Ag | Novel mucinase for use in therapy or prophylaxis |
| EP3153519A1 (en) * | 2015-10-06 | 2017-04-12 | Syddansk Universitet | Glycosylated yghj polypeptides from enterotoxigenic escherichia coli (etec) |
| KR20190139209A (en) * | 2017-02-13 | 2019-12-17 | 알렉산드레 에듀아르도 노윌 | Immunogenic Compositions for Modulating the Immune System and Methods of Treating Bacterial Infections in Individuals |
| US11260119B2 (en) | 2018-08-24 | 2022-03-01 | Pfizer Inc. | Escherichia coli compositions and methods thereof |
| WO2024083912A1 (en) | 2022-10-19 | 2024-04-25 | Glyprovac Aps | Glycosylated yghj polypeptides from uropathogenic e. coli |
Family Cites Families (130)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SE8205892D0 (en) | 1982-10-18 | 1982-10-18 | Bror Morein | IMMUNOGENT MEMBRANE PROTEIN COMPLEX, SET FOR PREPARATION AND USE THEREOF |
| IL73534A (en) | 1983-11-18 | 1990-12-23 | Riker Laboratories Inc | 1h-imidazo(4,5-c)quinoline-4-amines,their preparation and pharmaceutical compositions containing certain such compounds |
| US6090406A (en) | 1984-04-12 | 2000-07-18 | The Liposome Company, Inc. | Potentiation of immune responses with liposomal adjuvants |
| US5916588A (en) | 1984-04-12 | 1999-06-29 | The Liposome Company, Inc. | Peptide-containing liposomes, immunogenic liposomes and methods of preparation and use |
| US4777127A (en) | 1985-09-30 | 1988-10-11 | Labsystems Oy | Human retrovirus-related products and methods of diagnosing and treating conditions associated with said retrovirus |
| US4680338A (en) | 1985-10-17 | 1987-07-14 | Immunomedics, Inc. | Bifunctional linker |
| US5011828A (en) | 1985-11-15 | 1991-04-30 | Michael Goodman | Immunostimulating guanine derivatives, compositions and methods |
| GB8702816D0 (en) | 1987-02-07 | 1987-03-11 | Al Sumidaie A M K | Obtaining retrovirus-containing fraction |
| US5219740A (en) | 1987-02-13 | 1993-06-15 | Fred Hutchinson Cancer Research Center | Retroviral gene transfer into diploid fibroblasts for gene therapy |
| US5057540A (en) | 1987-05-29 | 1991-10-15 | Cambridge Biotech Corporation | Saponin adjuvant |
| US5206152A (en) | 1988-04-08 | 1993-04-27 | Arch Development Corporation | Cloning and expression of early growth regulatory protein genes |
| US5422120A (en) | 1988-05-30 | 1995-06-06 | Depotech Corporation | Heterovesicular liposomes |
| AP129A (en) | 1988-06-03 | 1991-04-17 | Smithkline Biologicals S A | Expression of retrovirus gag protein eukaryotic cells |
| AU627226B2 (en) | 1988-08-25 | 1992-08-20 | Liposome Company, Inc., The | Influenza vaccine and novel adjuvants |
| US5238944A (en) | 1988-12-15 | 1993-08-24 | Riker Laboratories, Inc. | Topical formulations and transdermal delivery systems containing 1-isobutyl-1H-imidazo[4,5-c]quinolin-4-amine |
| WO1990007936A1 (en) | 1989-01-23 | 1990-07-26 | Chiron Corporation | Recombinant therapies for infection and hyperproliferative disorders |
| US5703055A (en) | 1989-03-21 | 1997-12-30 | Wisconsin Alumni Research Foundation | Generation of antibodies through lipid mediated DNA delivery |
| EP0465529B1 (en) | 1989-03-21 | 1998-04-29 | Vical, Inc. | Expression of exogenous polynucleotide sequences in a vertebrate |
| US4929624A (en) | 1989-03-23 | 1990-05-29 | Minnesota Mining And Manufacturing Company | Olefinic 1H-imidazo(4,5-c)quinolin-4-amines |
| JPH0832638B2 (en) | 1989-05-25 | 1996-03-29 | カイロン コーポレイション | Adjuvant formulation comprising submicron oil droplet emulsion |
| EP0487587A1 (en) | 1989-08-18 | 1992-06-03 | Chiron Corporation | Recombinant retroviruses delivering vector constructs to target cells |
| US5585362A (en) | 1989-08-22 | 1996-12-17 | The Regents Of The University Of Michigan | Adenovirus vectors for gene therapy |
| US4988815A (en) | 1989-10-26 | 1991-01-29 | Riker Laboratories, Inc. | 3-Amino or 3-nitro quinoline compounds which are intermediates in preparing 1H-imidazo[4,5-c]quinolines |
| NZ237464A (en) | 1990-03-21 | 1995-02-24 | Depotech Corp | Liposomes with at least two separate chambers encapsulating two separate biologically active substances |
| US5658731A (en) | 1990-04-09 | 1997-08-19 | Europaisches Laboratorium Fur Molekularbiologie | 2'-O-alkylnucleotides as well as polymers which contain such nucleotides |
| US5149655A (en) | 1990-06-21 | 1992-09-22 | Agracetus, Inc. | Apparatus for genetic transformation |
| CA2098849C (en) | 1990-12-20 | 2007-07-10 | Ralph R. Weichselbaum | Control of gene expression by ionizing radiation |
| US5389640A (en) | 1991-03-01 | 1995-02-14 | Minnesota Mining And Manufacturing Company | 1-substituted, 2-substituted 1H-imidazo[4,5-c]quinolin-4-amines |
| DK0582581T3 (en) | 1991-03-01 | 1999-11-08 | Minnesota Mining & Mfg | 1,2-substituted 1H-imidazo [4,5-c] quinoline-4-amines |
| AU663725B2 (en) | 1991-08-20 | 1995-10-19 | United States Of America, Represented By The Secretary, Department Of Health And Human Services, The | Adenovirus mediated transfer of genes to the gastrointestinal tract |
| US5936076A (en) | 1991-08-29 | 1999-08-10 | Kirin Beer Kabushiki Kaisha | αgalactosylceramide derivatives |
| US5268376A (en) | 1991-09-04 | 1993-12-07 | Minnesota Mining And Manufacturing Company | 1-substituted 1H-imidazo[4,5-c]quinolin-4-amines |
| US5266575A (en) | 1991-11-06 | 1993-11-30 | Minnesota Mining And Manufacturing Company | 2-ethyl 1H-imidazo[4,5-ciquinolin-4-amines |
| WO1993010218A1 (en) | 1991-11-14 | 1993-05-27 | The United States Government As Represented By The Secretary Of The Department Of Health And Human Services | Vectors including foreign genes and negative selective markers |
| GB9125623D0 (en) | 1991-12-02 | 1992-01-29 | Dynal As | Cell modification |
| FR2688514A1 (en) | 1992-03-16 | 1993-09-17 | Centre Nat Rech Scient | Defective recombinant adenoviruses expressing cytokines and antitumour drugs containing them |
| IL105325A (en) | 1992-04-16 | 1996-11-14 | Minnesota Mining & Mfg | Immunogen/vaccine adjuvant composition |
| JPH07507689A (en) | 1992-06-08 | 1995-08-31 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | Specific tissue targeting methods and compositions |
| CA2137361A1 (en) | 1992-06-10 | 1993-12-23 | W. French Anderson | Vector particles resistant to inactivation by human serum |
| PL170980B1 (en) | 1992-06-25 | 1997-02-28 | Smithkline Beecham Biolog | Vaccine |
| GB2269175A (en) | 1992-07-31 | 1994-02-02 | Imperial College | Retroviral vectors |
| EP0911413A3 (en) | 1992-12-03 | 2000-11-15 | Genzyme Corporation | Minimal adenovirus-based gene therapy vector |
| US5395937A (en) | 1993-01-29 | 1995-03-07 | Minnesota Mining And Manufacturing Company | Process for preparing quinoline amines |
| ES2162139T5 (en) | 1993-03-23 | 2008-05-16 | Smithkline Beecham Biologicals S.A. | VACCINE COMPOSITIONS CONTAINING MONOFOSFORIL-LIPIDO TO 3-O-DISABLED. |
| CA2161225C (en) | 1993-04-22 | 2003-07-01 | Sinil Kim | Cyclodextrin liposomes encapsulating pharmacologic compounds and methods for their use |
| WO1995000655A1 (en) | 1993-06-24 | 1995-01-05 | Mc Master University | Adenovirus vectors for gene therapy |
| JPH09500128A (en) | 1993-07-15 | 1997-01-07 | ミネソタ マイニング アンド マニュファクチャリング カンパニー | Imidazo [4,5-c] pyridin-4-amine |
| US5352784A (en) | 1993-07-15 | 1994-10-04 | Minnesota Mining And Manufacturing Company | Fused cycloalkylimidazopyridines |
| US6015686A (en) | 1993-09-15 | 2000-01-18 | Chiron Viagene, Inc. | Eukaryotic layered vector initiation systems |
| EP0711829A3 (en) | 1993-09-15 | 1997-07-09 | Viagene Inc | Recombinant alphavirus vectors |
| WO1995011984A2 (en) | 1993-10-25 | 1995-05-04 | Canji, Inc. | Recombinant adenoviral vector and methods of use |
| AU5543294A (en) | 1993-10-29 | 1995-05-22 | Pharmos Corp. | Submicron emulsions as vaccine adjuvants |
| KR100241300B1 (en) | 1993-11-16 | 2000-03-02 | Sheldon A. Schaffer | Multivescular liposomes with controlled release of encapsulated biologically active substances |
| GB9326174D0 (en) | 1993-12-22 | 1994-02-23 | Biocine Sclavo | Mucosal adjuvant |
| GB9326253D0 (en) | 1993-12-23 | 1994-02-23 | Smithkline Beecham Biolog | Vaccines |
| ATE381624T1 (en) | 1994-05-09 | 2008-01-15 | Oxford Biomedica Ltd | RETROVIRAL VECTORS WITH REDUCED RECOMBINATION RATE |
| US6429199B1 (en) | 1994-07-15 | 2002-08-06 | University Of Iowa Research Foundation | Immunostimulatory nucleic acid molecules for activating dendritic cells |
| US6207646B1 (en) | 1994-07-15 | 2001-03-27 | University Of Iowa Research Foundation | Immunostimulatory nucleic acid molecules |
| US6239116B1 (en) | 1994-07-15 | 2001-05-29 | University Of Iowa Research Foundation | Immunostimulatory nucleic acid molecules |
| AUPM873294A0 (en) | 1994-10-12 | 1994-11-03 | Csl Limited | Saponin preparations and use thereof in iscoms |
| AU4594996A (en) | 1994-11-30 | 1996-06-19 | Chiron Viagene, Inc. | Recombinant alphavirus vectors |
| US5482936A (en) | 1995-01-12 | 1996-01-09 | Minnesota Mining And Manufacturing Company | Imidazo[4,5-C]quinoline amines |
| UA56132C2 (en) | 1995-04-25 | 2003-05-15 | Смітклайн Бічем Байолоджікалс С.А. | Vaccine composition (variants), method for stabilizing qs21 providing resistance against hydrolysis (variants), method for manufacturing vaccine |
| GB9513261D0 (en) | 1995-06-29 | 1995-09-06 | Smithkline Beecham Biolog | Vaccines |
| US5707829A (en) | 1995-08-11 | 1998-01-13 | Genetics Institute, Inc. | DNA sequences and secreted proteins encoded thereby |
| DE69739286D1 (en) | 1996-05-06 | 2009-04-16 | Oxford Biomedica Ltd | RECOMBINATION-INCOMPATIBLE RETROVIRAL VECTORS |
| EP0942964A2 (en) | 1996-11-22 | 1999-09-22 | Human Genome Sciences, Inc. | Thermostable polymerases having altered fidelity |
| DK1005368T3 (en) | 1997-03-10 | 2010-01-04 | Ottawa Hospital Res Inst | Use of nucleic acids containing non-methylated CpG dinucleotide in combination with alum as adjuvants |
| US6818222B1 (en) | 1997-03-21 | 2004-11-16 | Chiron Corporation | Detoxified mutants of bacterial ADP-ribosylating toxins as parenteral adjuvants |
| US6080725A (en) | 1997-05-20 | 2000-06-27 | Galenica Pharmaceuticals, Inc. | Immunostimulating and vaccine compositions employing saponin analog adjuvants and uses thereof |
| GB9712347D0 (en) | 1997-06-14 | 1997-08-13 | Smithkline Beecham Biolog | Vaccine |
| ES2298316T3 (en) | 1997-09-05 | 2008-05-16 | Glaxosmithkline Biologicals S.A. | WATER OIL EMULSIONS CONTAINING SAPONINS. |
| GB9725084D0 (en) | 1997-11-28 | 1998-01-28 | Medeva Europ Ltd | Vaccine compositions |
| AU759391B2 (en) | 1998-02-12 | 2003-04-10 | Wyeth Holdings Corporation | Pneumococcal and meningococcal vaccines formulated with interleukin-12 |
| US6303114B1 (en) | 1998-03-05 | 2001-10-16 | The Medical College Of Ohio | IL-12 enhancement of immune responses to T-independent antigens |
| WO1999052549A1 (en) | 1998-04-09 | 1999-10-21 | Smithkline Beecham Biologicals S.A. | Adjuvant compositions |
| CN1306438A (en) | 1998-05-07 | 2001-08-01 | 科里克萨有限公司 | Adjuvant compsn. and methods for its use |
| US6562798B1 (en) | 1998-06-05 | 2003-05-13 | Dynavax Technologies Corp. | Immunostimulatory oligonucleotides with modified bases and methods of use thereof |
| US6110929A (en) | 1998-07-28 | 2000-08-29 | 3M Innovative Properties Company | Oxazolo, thiazolo and selenazolo [4,5-c]-quinolin-4-amines and analogs thereof |
| GB9817052D0 (en) | 1998-08-05 | 1998-09-30 | Smithkline Beecham Biolog | Vaccine |
| EP1126876B1 (en) | 1998-10-16 | 2007-03-21 | GlaxoSmithKline Biologicals S.A. | Adjuvant systems and vaccines |
| US20030130212A1 (en) | 1999-01-14 | 2003-07-10 | Rossignol Daniel P. | Administration of an anti-endotoxin drug by intravenous infusion |
| US6551600B2 (en) | 1999-02-01 | 2003-04-22 | Eisai Co., Ltd. | Immunological adjuvant compounds compositions and methods of use thereof |
| DK1150918T3 (en) | 1999-02-03 | 2004-12-20 | Biosante Pharmaceuticals Inc | Process for the preparation of therapeutic calcium phosphate particles |
| US6331539B1 (en) | 1999-06-10 | 2001-12-18 | 3M Innovative Properties Company | Sulfonamide and sulfamide substituted imidazoquinolines |
| EP1221971A2 (en) | 1999-09-24 | 2002-07-17 | SmithKline Beecham Biologics SA | Use of the combination of polyoxyethylene sorbitan ester and octoxynol as adjuvant and its use in vaccines |
| CZ20021045A3 (en) | 1999-09-24 | 2002-08-14 | Smithkline Beecham Biologicals S. A. | Auxiliary preparation |
| AP1775A (en) | 1999-09-25 | 2007-08-28 | Univ Iowa Res Found | Immunostimulatory nucleic acids. |
| US20010044416A1 (en) | 2000-01-20 | 2001-11-22 | Mccluskie Michael J. | Immunostimulatory nucleic acids for inducing a Th2 immune response |
| WO2001066572A2 (en) | 2000-03-10 | 2001-09-13 | Institut National De La Sante Et De La Recherche Medicale (I.N.S.E.R.M.) | Polynucleotides isolated from e. coli of nature b2/d+ a-, and uses thereof |
| DE60140456D1 (en) | 2000-09-01 | 2009-12-24 | Novartis Vaccines & Diagnostic | AZA HETEROCYCLIC DERIVATIVES AND ITS THERAPEUTIC USE |
| EA006711B1 (en) | 2000-09-11 | 2006-02-24 | Чирон Корпорейшн | Quinolinone derivatives as tyrosine kinase inhibitors |
| EP1322656B1 (en) | 2000-09-26 | 2008-01-16 | Idera Pharmaceuticals, Inc. | Modulation of immunostimulatory activity of immunostimulatory oligonucleotide analogs by positional chemical changes |
| US6677347B2 (en) | 2000-12-08 | 2004-01-13 | 3M Innovative Properties Company | Sulfonamido ether substituted imidazoquinolines |
| US6660747B2 (en) | 2000-12-08 | 2003-12-09 | 3M Innovative Properties Company | Amido ether substituted imidazoquinolines |
| US6664260B2 (en) | 2000-12-08 | 2003-12-16 | 3M Innovative Properties Company | Heterocyclic ether substituted imidazoquinolines |
| US6660735B2 (en) | 2000-12-08 | 2003-12-09 | 3M Innovative Properties Company | Urea substituted imidazoquinoline ethers |
| US6664264B2 (en) | 2000-12-08 | 2003-12-16 | 3M Innovative Properties Company | Thioether substituted imidazoquinolines |
| UA74852C2 (en) | 2000-12-08 | 2006-02-15 | 3M Innovative Properties Co | Urea-substituted imidazoquinoline ethers |
| US6664265B2 (en) | 2000-12-08 | 2003-12-16 | 3M Innovative Properties Company | Amido ether substituted imidazoquinolines |
| US6667312B2 (en) | 2000-12-08 | 2003-12-23 | 3M Innovative Properties Company | Thioether substituted imidazoquinolines |
| US6677348B2 (en) | 2000-12-08 | 2004-01-13 | 3M Innovative Properties Company | Aryl ether substituted imidazoquinolines |
| WO2002077183A2 (en) | 2001-03-21 | 2002-10-03 | Elitra Pharmaceuticals, Inc. | Identification of essential genes in microorganisms |
| WO2002081653A2 (en) | 2001-04-03 | 2002-10-17 | Biosynexus Incorporated | An animal model for enteric pathogens |
| EP1450856B1 (en) | 2001-09-14 | 2009-11-11 | Cytos Biotechnology AG | Packaging of immunostimulatory cpg into virus-like particles: method of preparation and use |
| AU2002347404A1 (en) | 2001-09-14 | 2003-04-01 | Cytos Biotechnology Ag | In vivo activation of antigen presenting cells for enhancement of immune responses induced by virus like particles |
| WO2003035836A2 (en) | 2001-10-24 | 2003-05-01 | Hybridon Inc. | Modulation of immunostimulatory properties of oligonucleotide-based compounds by optimal presentation of 5' ends |
| EA008380B1 (en) | 2001-11-27 | 2007-04-27 | Анадис Фармасьютикалз, Инк. | 3-β-D-RIBOFURANOSYLTHIAZOLO[4,5-d] PYRIMIDINE NUCLEOSIDES AND USES THEREOF |
| US7321033B2 (en) | 2001-11-27 | 2008-01-22 | Anadys Pharmaceuticals, Inc. | 3-B-D-ribofuranosylthiazolo [4,5-d] pyrimidine nucleosides and uses thereof |
| US6677349B1 (en) | 2001-12-21 | 2004-01-13 | 3M Innovative Properties Company | Sulfonamide and sulfamide substituted imidazoquinolines |
| US20030165870A1 (en) | 2002-03-01 | 2003-09-04 | Blattner Frederick R. | Novel sequences of E. coli CFT073 |
| EP1342784A1 (en) | 2002-03-06 | 2003-09-10 | Mutabilis S.A. | ExPEC-specific proteins, genes encoding them and uses thereof |
| CA2480638C (en) | 2002-03-29 | 2013-02-12 | Chiron Corporation | Substituted benzazoles and use thereof as raf kinase inhibitors |
| US6743920B2 (en) | 2002-05-29 | 2004-06-01 | 3M Innovative Properties Company | Process for imidazo[4,5-c]pyridin-4-amines |
| AU2003251518B2 (en) | 2002-06-13 | 2009-07-02 | New York University | Synthetic C-glycolipid and its use for treating cancer infectious diseases and autoimmune diseases |
| FR2842210B1 (en) | 2002-07-09 | 2012-12-14 | Mutabilis | PATHOGENICITY DETERMINANTS FOR USE AS TARGETS FOR PREPARING AND CONTROLLING BACTERIAL INFECTIONS AND / OR SYSTEMIC DISSEMINATION |
| AU2003268184A1 (en) | 2002-08-23 | 2004-03-11 | Chiron Corporation | Pyrrole based inhibitors of glycogen synthase kinase 3 |
| EP1587473A4 (en) | 2002-12-27 | 2008-08-13 | Novartis Vaccines & Diagnostic | Thiosemicarbazones as anti-virals and immunopotentiators |
| ES2391770T3 (en) | 2003-01-21 | 2012-11-29 | Novartis Vaccines And Diagnostics, Inc. | Use of triptantrin compounds for immune potentiation |
| GB0301554D0 (en) | 2003-01-23 | 2003-02-26 | Molecularnature Ltd | Immunostimulatory compositions |
| US7893096B2 (en) | 2003-03-28 | 2011-02-22 | Novartis Vaccines And Diagnostics, Inc. | Use of small molecule compounds for immunopotentiation |
| RU2236257C1 (en) | 2003-09-15 | 2004-09-20 | Косяков Константин Сергеевич | Synthetic immunogen for therapy and prophylaxis of addiction with narcotic and psychoactive substances |
| US7771726B2 (en) | 2003-10-08 | 2010-08-10 | New York University | Use of synthetic glycolipids as universal adjuvants for vaccines against cancer and infectious diseases |
| EP1732384A4 (en) | 2004-03-31 | 2008-04-23 | Univ New York State Res Found | Novel synthetic c-glycolipids, their synthesis and use to treat infections, cancer and autoimmune diseases |
| NZ550152A (en) | 2004-04-05 | 2009-04-30 | Pfizer Prod Inc | Microfluidized oil-in-water emulsions and vaccine compositions |
| NZ560929A (en) | 2005-02-18 | 2009-12-24 | Novartis Vaccines & Diagnostic | Immunogens from uropathogenic escherichia coli |
| HUE027400T2 (en) | 2005-02-18 | 2016-10-28 | Glaxosmithkline Biologicals Sa | Proteins and nucleic acids from meningitis/sepsis-associated escherichia coli |
| US7691368B2 (en) | 2005-04-15 | 2010-04-06 | Merial Limited | Vaccine formulations |
| US8703095B2 (en) | 2005-07-07 | 2014-04-22 | Sanofi Pasteur S.A. | Immuno-adjuvant emulsion |
| ITMI20081249A1 (en) * | 2008-07-09 | 2010-01-09 | Novartis Vaccines & Diagnostic | ESCHERICHIA COLI IMMUNOGENES WITH IMPROVED SOLUBILITY. |
-
2010
- 2010-07-16 CN CN2010800406180A patent/CN102770443A/en active Pending
- 2010-07-16 MX MX2012000734A patent/MX2012000734A/en not_active Application Discontinuation
- 2010-07-16 EP EP14187138.4A patent/EP2837386B1/en active Active
- 2010-07-16 JP JP2012520121A patent/JP2012532626A/en not_active Withdrawn
- 2010-07-16 SG SG2012002994A patent/SG178035A1/en unknown
- 2010-07-16 WO PCT/IB2010/002043 patent/WO2011007257A1/en active Application Filing
- 2010-07-16 PL PL10752175T patent/PL2464658T3/en unknown
- 2010-07-16 ES ES14187138T patent/ES2670799T3/en active Active
- 2010-07-16 CA CA2768343A patent/CA2768343A1/en not_active Abandoned
- 2010-07-16 AU AU2010272243A patent/AU2010272243A1/en not_active Abandoned
- 2010-07-16 SI SI201030830T patent/SI2464658T1/en unknown
- 2010-07-16 EP EP10752175.9A patent/EP2464658B1/en active Active
- 2010-07-16 PT PT107521759T patent/PT2464658E/en unknown
- 2010-07-16 DK DK10752175.9T patent/DK2464658T3/en active
- 2010-07-16 ES ES10752175.9T patent/ES2526996T3/en active Active
- 2010-07-16 US US13/384,224 patent/US8871214B2/en active Active
-
2012
- 2012-01-15 IL IL217539A patent/IL217539A0/en unknown
- 2012-02-02 ZA ZA2012/00833A patent/ZA201200833B/en unknown
-
2014
- 2014-10-27 US US14/525,033 patent/US10058600B2/en active Active
- 2014-12-30 HR HRP20141270AT patent/HRP20141270T1/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| ZA201200833B (en) | 2014-08-27 |
| HRP20141270T1 (en) | 2015-03-13 |
| SG178035A1 (en) | 2012-03-29 |
| IL217539A0 (en) | 2012-02-29 |
| PL2464658T3 (en) | 2015-03-31 |
| ES2526996T3 (en) | 2015-01-19 |
| CA2768343A1 (en) | 2011-01-20 |
| US8871214B2 (en) | 2014-10-28 |
| MX2012000734A (en) | 2012-01-27 |
| EP2837386B1 (en) | 2018-03-07 |
| US20120207777A1 (en) | 2012-08-16 |
| DK2464658T3 (en) | 2014-12-15 |
| CN102770443A (en) | 2012-11-07 |
| EP2464658A1 (en) | 2012-06-20 |
| US20150110830A1 (en) | 2015-04-23 |
| US10058600B2 (en) | 2018-08-28 |
| ES2670799T3 (en) | 2018-06-01 |
| WO2011007257A1 (en) | 2011-01-20 |
| PT2464658E (en) | 2015-01-14 |
| EP2837386A1 (en) | 2015-02-18 |
| AU2010272243A1 (en) | 2012-03-08 |
| SI2464658T1 (en) | 2015-02-27 |
| JP2012532626A (en) | 2012-12-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9101560B2 (en) | Escherichia coli immunogens with improved solubility | |
| EP2451833B1 (en) | Conserved escherichia coli immunogens | |
| US20110300171A1 (en) | Factor h binding protein immunogens | |
| US10058600B2 (en) | Detoxified Escherichia coli immunogens | |
| US20200360504A1 (en) | Pseudomonas antigens and antigen combinations | |
| AU2013203188A1 (en) | Detoxified Escherichia coli immunogens | |
| AU2013203108A1 (en) | Escherichia coli immunogens with improved solubility |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| 17P | Request for examination filed |
Effective date: 20120130 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AL AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO SE SI SK SM TR |
|
| DAX | Request for extension of the european patent (deleted) | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: DE Ref document number: 1172040 Country of ref document: HK |
|
| 17Q | First examination report despatched |
Effective date: 20130429 |
|
| GRAP | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOSNIGR1 |
|
| INTG | Intention to grant announced |
Effective date: 20140414 |
|
| GRAS | Grant fee paid |
Free format text: ORIGINAL CODE: EPIDOSNIGR3 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AL AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO SE SI SK SM TR |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: REF Ref document number: 689513 Country of ref document: AT Kind code of ref document: T Effective date: 20141015 Ref country code: CH Ref legal event code: EP |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R096 Ref document number: 602010019260 Country of ref document: DE Effective date: 20141113 |
|
| REG | Reference to a national code |
Ref country code: DK Ref legal event code: T3 Effective date: 20141211 Ref country code: CH Ref legal event code: NV Representative=s name: E. BLUM AND CO. AG PATENT- UND MARKENANWAELTE , CH |
|
| REG | Reference to a national code |
Ref country code: RO Ref legal event code: EPE |
|
| REG | Reference to a national code |
Ref country code: HR Ref legal event code: TUEP Ref document number: P20141270 Country of ref document: HR |
|
| REG | Reference to a national code |
Ref country code: SE Ref legal event code: TRGR |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: T3 Ref country code: PT Ref legal event code: SC4A Free format text: AVAILABILITY OF NATIONAL TRANSLATION Effective date: 20141216 |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FG2A Ref document number: 2526996 Country of ref document: ES Kind code of ref document: T3 Effective date: 20150119 |
|
| REG | Reference to a national code |
Ref country code: EE Ref legal event code: FG4A Ref document number: E010043 Country of ref document: EE Effective date: 20141222 |
|
| REG | Reference to a national code |
Ref country code: NO Ref legal event code: T2 Effective date: 20141001 |
|
| REG | Reference to a national code |
Ref country code: GR Ref legal event code: EP Ref document number: 20140402614 Country of ref document: GR Effective date: 20150128 |
|
| REG | Reference to a national code |
Ref country code: HR Ref legal event code: T1PR Ref document number: P20141270 Country of ref document: HR |
|
| REG | Reference to a national code |
Ref country code: LT Ref legal event code: MG4D |
|
| REG | Reference to a national code |
Ref country code: PL Ref legal event code: T3 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IS Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20150201 Ref country code: LT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20141001 |
|
| REG | Reference to a national code |
Ref country code: SK Ref legal event code: T3 Ref document number: E 17976 Country of ref document: SK |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R097 Ref document number: 602010019260 Country of ref document: DE |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed |
Effective date: 20150702 |
|
| REG | Reference to a national code |
Ref country code: HU Ref legal event code: AG4A Ref document number: E024097 Country of ref document: HU |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: MC Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20141001 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: PLFP Year of fee payment: 7 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: MT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20141001 |
|
| REG | Reference to a national code |
Ref country code: NO Ref legal event code: CHAD Owner name: GLAXOSMITHKLINE BIOLOGICALS SA, BE |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SM Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20141001 |
|
| REG | Reference to a national code |
Ref country code: EE Ref legal event code: HC1A Ref document number: E010043 Country of ref document: EE Ref country code: CH Ref legal event code: NV Representative=s name: ISLER AND PEDRAZZINI AG, CH |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: PLFP Year of fee payment: 8 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R082 Ref document number: 602010019260 Country of ref document: DE Representative=s name: MUELLER-BORE & PARTNER PATENTANWAELTE PARTG MB, DE Ref country code: DE Ref legal event code: R081 Ref document number: 602010019260 Country of ref document: DE Owner name: GLAXOSMITHKLINE BIOLOGICALS S.A., BE Free format text: FORMER OWNER: NOVARTIS AG, 4056 BASEL, CH Ref country code: DE Ref legal event code: R082 Ref document number: 602010019260 Country of ref document: DE Representative=s name: HOFFMANN - EITLE PATENT- UND RECHTSANWAELTE PA, DE |
|
| REG | Reference to a national code |
Ref country code: LU Ref legal event code: PD Owner name: GLAXOSMITHKLINE BIOLOGICALS SA; BE Free format text: FORMER OWNER: NOVARTIS AG Effective date: 20170508 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R082 Ref document number: 602010019260 Country of ref document: DE Representative=s name: HOFFMANN - EITLE PATENT- UND RECHTSANWAELTE PA, DE |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R082 Ref document number: 602010019260 Country of ref document: DE Representative=s name: HOFFMANN - EITLE PATENT- UND RECHTSANWAELTE PA, DE |
|
| REG | Reference to a national code |
Ref country code: NO Ref legal event code: CREP Representative=s name: BRYN AARFLOT AS, POSTBOKS 449 SENTRUM, 0104 OSLO |
|
| REG | Reference to a national code |
Ref country code: SK Ref legal event code: PC4A Ref document number: E 17976 Country of ref document: SK Owner name: GLAXOSMITHKLINE BIOLOGICALS S.A., RIXENSART, BE Free format text: FORMER OWNER: NOVARTIS AG, BASEL, CH Effective date: 20170524 |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: 732E Free format text: REGISTERED BETWEEN 20170921 AND 20170927 |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: PC2A Owner name: GLAXOSMITHKLINE BIOLOGICALS S.A. Effective date: 20171124 Ref country code: BE Ref legal event code: FP Effective date: 20141223 Ref country code: BE Ref legal event code: PD Owner name: GLAXOSMITHKLINE BIOLOGICALS SA; BE Free format text: DETAILS ASSIGNMENT: CHANGE OF OWNER(S), AFFECTATION / CESSION; FORMER OWNER NAME: NOVARTIS AG Effective date: 20170801 |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: PC Ref document number: 689513 Country of ref document: AT Kind code of ref document: T Owner name: GLAXOSMITHKLINE BIOLOGICALS S.A., BE Effective date: 20180305 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: PLFP Year of fee payment: 9 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: MK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20141001 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: AL Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20141001 |
|
| REG | Reference to a national code |
Ref country code: EE Ref legal event code: GB1A Ref document number: E010043 Country of ref document: EE |
|
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: WD Ref document number: 1172040 Country of ref document: HK |
|
| REG | Reference to a national code |
Ref country code: SI Ref legal event code: SP73 Owner name: GLAXOSMITHKLINE BIOLOGICALS SA; BE Effective date: 20190121 |
|
| REG | Reference to a national code |
Ref country code: HR Ref legal event code: ODRP Ref document number: P20141270 Country of ref document: HR Payment date: 20190711 Year of fee payment: 10 |
|
| REG | Reference to a national code |
Ref country code: HR Ref legal event code: ODRP Ref document number: P20141270 Country of ref document: HR Payment date: 20200713 Year of fee payment: 11 |
|
| REG | Reference to a national code |
Ref country code: HR Ref legal event code: ODRP Ref document number: P20141270 Country of ref document: HR Payment date: 20210715 Year of fee payment: 12 |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: PD Owner name: GLAXOSMITHKLINE BIOLOGICALS S.A.; BE Free format text: DETAILS ASSIGNMENT: CHANGE OF OWNER(S), ASSIGNMENT; FORMER OWNER NAME: NOVARTIS AG Effective date: 20211222 |
|
| REG | Reference to a national code |
Ref country code: HU Ref legal event code: GB9C Owner name: GLAXOSMITHKLINE BIOLOGICALS S.A., BE Free format text: FORMER OWNER(S): NOVARTIS AG, CH Ref country code: HU Ref legal event code: FH1C Free format text: FORMER REPRESENTATIVE(S): LENGYEL ZSOLT, DANUBIA SZABADALMI ES JOGI IRODA KFT., HU Representative=s name: DR. KOCSOMBA NELLI UEGYVEDI IRODA, HU Ref country code: CH Ref legal event code: PK Free format text: BERICHTIGUNGEN |
|
| REG | Reference to a national code |
Ref country code: HR Ref legal event code: ODRP Ref document number: P20141270 Country of ref document: HR Payment date: 20220713 Year of fee payment: 13 |
|
| REG | Reference to a national code |
Ref country code: HU Ref legal event code: HC9C Owner name: GLAXOSMITHKLINE BIOLOGICALS S.A., BE Free format text: FORMER OWNER(S): NOVARTIS AG, CH |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: RO Payment date: 20230627 Year of fee payment: 14 Ref country code: PT Payment date: 20230621 Year of fee payment: 14 Ref country code: NO Payment date: 20230622 Year of fee payment: 14 Ref country code: NL Payment date: 20230622 Year of fee payment: 14 Ref country code: IT Payment date: 20230620 Year of fee payment: 14 Ref country code: IE Payment date: 20230622 Year of fee payment: 14 Ref country code: FR Payment date: 20230621 Year of fee payment: 14 Ref country code: EE Payment date: 20230620 Year of fee payment: 14 Ref country code: DK Payment date: 20230622 Year of fee payment: 14 Ref country code: CZ Payment date: 20230623 Year of fee payment: 14 |
|
| REG | Reference to a national code |
Ref country code: HR Ref legal event code: ODRP Ref document number: P20141270 Country of ref document: HR Payment date: 20230718 Year of fee payment: 14 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: TR Payment date: 20230622 Year of fee payment: 14 Ref country code: SK Payment date: 20230623 Year of fee payment: 14 Ref country code: SE Payment date: 20230622 Year of fee payment: 14 Ref country code: PL Payment date: 20230622 Year of fee payment: 14 Ref country code: LV Payment date: 20230620 Year of fee payment: 14 Ref country code: LU Payment date: 20230620 Year of fee payment: 14 Ref country code: GR Payment date: 20230622 Year of fee payment: 14 Ref country code: FI Payment date: 20230622 Year of fee payment: 14 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: BE Payment date: 20230622 Year of fee payment: 14 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: GB Payment date: 20230620 Year of fee payment: 14 Ref country code: ES Payment date: 20230801 Year of fee payment: 14 Ref country code: CY Payment date: 20230621 Year of fee payment: 14 Ref country code: CH Payment date: 20230801 Year of fee payment: 14 Ref country code: BG Payment date: 20230703 Year of fee payment: 14 Ref country code: AT Payment date: 20230622 Year of fee payment: 14 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: SI Payment date: 20230705 Year of fee payment: 14 Ref country code: HU Payment date: 20230630 Year of fee payment: 14 Ref country code: HR Payment date: 20230718 Year of fee payment: 14 Ref country code: DE Payment date: 20230620 Year of fee payment: 14 |