WO2024257756A1 - 活性炭 - Google Patents
活性炭 Download PDFInfo
- Publication number
- WO2024257756A1 WO2024257756A1 PCT/JP2024/021177 JP2024021177W WO2024257756A1 WO 2024257756 A1 WO2024257756 A1 WO 2024257756A1 JP 2024021177 W JP2024021177 W JP 2024021177W WO 2024257756 A1 WO2024257756 A1 WO 2024257756A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- activated carbon
- reaction gas
- amount
- vitamin
- surface area
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images



Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/62—Selection of inactive substances as ingredients for active masses, e.g. binders, fillers
- H01M4/624—Electric conductive fillers
- H01M4/625—Carbon or graphite
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/30—Active carbon
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/30—Active carbon
- C01B32/312—Preparation
- C01B32/318—Preparation characterised by the starting materials
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/30—Active carbon
- C01B32/312—Preparation
- C01B32/336—Preparation characterised by gaseous activating agents
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/30—Active carbon
- C01B32/312—Preparation
- C01B32/342—Preparation characterised by non-gaseous activating agents
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/30—Active carbon
- C01B32/354—After-treatment
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES, LIGHT-SENSITIVE OR TEMPERATURE-SENSITIVE DEVICES OF THE ELECTROLYTIC TYPE
- H01G11/00—Hybrid capacitors, i.e. capacitors having different positive and negative electrodes; Electric double-layer [EDL] capacitors; Processes for the manufacture thereof or of parts thereof
- H01G11/04—Hybrid capacitors
- H01G11/06—Hybrid capacitors with one of the electrodes allowing ions to be reversibly doped thereinto, e.g. lithium ion capacitors [LIC]
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES, LIGHT-SENSITIVE OR TEMPERATURE-SENSITIVE DEVICES OF THE ELECTROLYTIC TYPE
- H01G11/00—Hybrid capacitors, i.e. capacitors having different positive and negative electrodes; Electric double-layer [EDL] capacitors; Processes for the manufacture thereof or of parts thereof
- H01G11/22—Electrodes
- H01G11/24—Electrodes characterised by structural features of the materials making up or comprised in the electrodes, e.g. form, surface area or porosity; characterised by the structural features of powders or particles used therefor
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES, LIGHT-SENSITIVE OR TEMPERATURE-SENSITIVE DEVICES OF THE ELECTROLYTIC TYPE
- H01G11/00—Hybrid capacitors, i.e. capacitors having different positive and negative electrodes; Electric double-layer [EDL] capacitors; Processes for the manufacture thereof or of parts thereof
- H01G11/22—Electrodes
- H01G11/30—Electrodes characterised by their material
- H01G11/32—Carbon-based
- H01G11/34—Carbon-based characterised by carbonisation or activation of carbon
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES, LIGHT-SENSITIVE OR TEMPERATURE-SENSITIVE DEVICES OF THE ELECTROLYTIC TYPE
- H01G11/00—Hybrid capacitors, i.e. capacitors having different positive and negative electrodes; Electric double-layer [EDL] capacitors; Processes for the manufacture thereof or of parts thereof
- H01G11/22—Electrodes
- H01G11/30—Electrodes characterised by their material
- H01G11/32—Carbon-based
- H01G11/42—Powders or particles, e.g. composition thereof
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES, LIGHT-SENSITIVE OR TEMPERATURE-SENSITIVE DEVICES OF THE ELECTROLYTIC TYPE
- H01G11/00—Hybrid capacitors, i.e. capacitors having different positive and negative electrodes; Electric double-layer [EDL] capacitors; Processes for the manufacture thereof or of parts thereof
- H01G11/22—Electrodes
- H01G11/30—Electrodes characterised by their material
- H01G11/50—Electrodes characterised by their material specially adapted for lithium-ion capacitors, e.g. for lithium-doping or for intercalation
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/13—Electrodes for accumulators with non-aqueous electrolyte, e.g. for lithium-accumulators; Processes of manufacture thereof
- H01M4/133—Electrodes based on carbonaceous material, e.g. graphite-intercalation compounds or CFx
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/62—Selection of inactive substances as ingredients for active masses, e.g. binders, fillers
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/86—Inert electrodes with catalytic activity, e.g. for fuel cells
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/12—Surface area
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
Definitions
- the present invention relates to activated carbon for use in electricity storage devices.
- aqueous electrolyte batteries such as lead-carbon batteries
- non-aqueous electrolyte batteries such as lithium-ion batteries, lithium-sulfur batteries, and sodium-ion batteries
- all-solid-state batteries as well as electric double-layer capacitors and fuel cells
- Patent Document 1 describes porous carbon whose pores are filled with sulfur.
- electric double layer capacitors which are one type of energy storage device, have superior output and life characteristics compared to batteries because they use a capacity (electric double layer capacity) that is obtained only from the physical adsorption and desorption of ions without involving a chemical reaction.
- a capacity electric double layer capacity
- Patent Document 2 discloses that when activated carbon derived from soft carbon with a specific range of specific surface area, particle size, and amount of surface functional groups is used in an electric double layer capacitor, it provides output and durability while maintaining the capacitance of the electric double layer capacitor.
- Patent Document 3 also discloses that the use of activated carbon with a specific range for the ratio of the D peak height to the G peak height in the Raman spectrum and the specific surface area in an electric double layer capacitor can increase the electrical capacitance per electrode.
- Patent Document 1 the pores of the porous carbon are filled with sulfur to facilitate the movement of electrons, thereby increasing the capacity.
- the sulfur filling rate is increased to increase the capacity, the pores are blocked, making it difficult for the electrolyte to diffuse, which may increase the resistance of the electricity storage device.
- Patent Documents 2 and 3 the capacitance of the electricity storage device is improved by adjusting parameters such as the specific surface area.
- Patent Documents 2 and 3 do not disclose anything about reducing the resistance of the electricity storage device by adjusting the parameters of the activated carbon.
- the present invention aims to provide activated carbon for electricity storage devices that is useful for improving the performance of electricity storage devices.
- the present invention includes the following preferred embodiments.
- the specific surface area calculated from the nitrogen adsorption/desorption isotherm by the BET method is 1150 m 2 /g to 2200 m 2 /g;
- the amount of vitamin B12 adsorbed when contacted with a 300 ppm aqueous vitamin B12 solution for 24 hours is 10.2 mg/mL to 50 mg/mL.
- the present invention provides activated carbon for electricity storage devices that is useful for improving the performance of electricity storage devices.
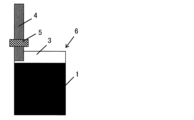
- FIG. 2 shows a sheet-like electrode composition. This is a diagram showing a current collector (etched aluminum foil) coated with a conductive adhesive.
- FIG. 1 shows a polarizable electrode in which a sheet-shaped electrode composition is bonded to a current collector and an aluminum tab is ultrasonically welded to the electrode.
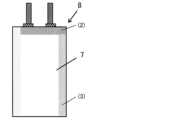
- FIG. 2 is a diagram showing a bag-shaped exterior sheet.
- FIG. 1 is a diagram showing an electric double layer capacitor.
- the activated carbon of the present invention has a specific surface area of 1150 m 2 /g to 2200 m 2 /g calculated by the BET method from the nitrogen adsorption/desorption isotherm, an adsorption amount of vitamin B12 of 10.2 mg/mL to 50 mg/mL when contacted with a 300 ppm aqueous vitamin B12 solution for 24 hours, and a chlorine content of 2500 ppm or less.
- the present inventors have studied a method for improving the performance of electricity storage devices such as non-aqueous electrolyte batteries and electric double layer capacitors, and have found that in the prior art, attention has been paid only to the micropores through which the adsorbate such as an electrolyte is finally adsorbed, and the mesopores corresponding to the entrances when the adsorbate is adsorbed in the activated carbon, and the characteristics of the electricity storage device have been improved by appropriately adjusting the specific surface area, mesopore and macropore volumes, etc. of a carbonaceous material such as activated carbon, but the pores through which the adsorbate moves from the mesopores to the micropores have not been taken into consideration.
- the present inventors have found that by appropriately adjusting the volume of the pores corresponding to the size of the path through which the adsorbate diffuses in the activated carbon before the adsorbate that has entered from the mesopores is adsorbed by the micropores, the adsorbate can move smoothly, thereby realizing low resistance.
- the present inventors have also found that the volume of the pores corresponding to the above-mentioned path of the adsorbate is expressed by the adsorption amount of vitamin B12. Therefore, according to the present invention, it is possible to provide an activated carbon that is particularly useful for reducing the resistance of an electricity storage device.
- the activated carbon of the present invention when used in an electric double layer capacitor, the electrolyte is adsorbed into the micropores without clogging the path during diffusion, and as a result, the capacitance can be improved. Therefore, in this specification, when the power storage device is an electric double layer capacitor, its performance includes, for example, resistance and capacitance, and the performance of the electric double layer capacitor is improved or increased, for example, meaning that the resistance is reduced and the capacitance is increased. In this specification, when the power storage device is a non-aqueous electrolyte battery, its performance can include, for example, resistance and discharge capacity.
- electrolytes that can be used in electricity storage devices include, for example, in the case of lithium-sulfur batteries, LiPF 6 , LiBF 4 , LiTFSI (lithium bis(trifluoromethanesulfonyl)imide), LiFSI (lithium bis(fluorosulfonyl)imide), and the like.
- tetraethylammonium salts such as TEABF 4 (tetraethylammonium tetrafluoroborate) and TEAPF 6 (tetraethylammonium hexafluorophosphate); triethylmethylammonium salts such as TEMABF 4 (triethylmethylammonium tetrafluoroborate); tetraethylphosphonium salts such as TEPBF 4 (tetraethylphosphonium tetrafluoroborate); and ionic liquids such as EMIBF 4 .
- TEABF 4 tetraethylammonium tetrafluoroborate
- TEAPF 6 tetraethylammonium hexafluorophosphate
- triethylmethylammonium salts such as TEMABF 4 (triethylmethylammonium tetrafluoroborate)
- the activated carbon of the present invention has a specific surface area calculated by the BET method from the nitrogen adsorption/desorption isotherm (hereinafter also referred to as "BET specific surface area") of 1150 m 2 /g to 2200 m 2 /g.
- BET specific surface area calculated by the BET method from the nitrogen adsorption/desorption isotherm (hereinafter also referred to as "BET specific surface area") of 1150 m 2 /g to 2200 m 2 /g.
- the BET specific surface area is 1150 m 2 /g to 2200 m 2 /g
- the capacitance or discharge capacity of the electricity storage device containing the activated carbon of the present invention tends to be high and the resistance tends to be low.
- the BET specific surface area When the BET specific surface area is less than 1150 m 2 /g, the average pore size becomes relatively small, and the electrolyte is less likely to diffuse in the pores during charging and discharging, so that the resistance tends to increase. In addition, the durability of the electricity storage device containing the activated carbon of the present invention tends to decrease with the increase in resistance. On the other hand, when the BET specific surface area exceeds 2200 m 2 /g, the bulk density of the activated carbon decreases, and the capacitance or discharge capacity per unit volume tends to decrease.
- the BET specific surface area is preferably from 1200 m 2 /g to 2180 m 2 /g, from 1250 m 2 /g to 2160 m 2 /g, from 1300 m 2 /g to 2140 m 2 /g, from 1350 m 2 /g to 2120 m 2 /g or from 1400 m 2 /g to 2100 m 2 /g.
- the BET specific surface area is also preferably 1400 m 2 /g to 2200 m 2 /g, more preferably 1500 m 2 /g to 2100 m 2 /g, even more preferably 1550 m 2 /g to 2000 m 2 /g, still more preferably 1600 m 2 /g to 1950 m 2 /g, particularly preferably 1650 m 2 /g to 1900 m 2 /g, and particularly preferably 1750 m 2 /g to 1900 m 2 /g.
- the capacitance or discharge capacity of the electricity storage device containing the activated carbon of the present invention is more likely to be increased and the resistance is more likely to be lower.
- the BET specific surface area can be adjusted to within the above range by appropriately adjusting, for example, the type of raw material of the activated carbon, the temperature and/or atmosphere and/or time in the activation step during the production of the activated carbon, the number of activation steps, the temperature and/or atmosphere and/or time in the heat treatment step, etc.
- the BET specific surface area can be determined, for example, by the method described in the Examples below.
- the amount of vitamin B12 adsorbed by the activated carbon of the present invention when it is contacted with a 300 ppm aqueous solution of vitamin B12 for 24 hours (hereinafter, sometimes simply referred to as the "adsorbed amount of vitamin B12") is 10.2 mg/mL to 50 mg/mL.
- the amount of vitamin B12 adsorbed is 10.2 mg/mL or more, the capacitance or discharge capacity of the electricity storage device containing the activated carbon of the present invention is likely to be high and the resistance is likely to be low.
- the amount of vitamin B12 adsorbed represents the volume of the pores corresponding to the size of the path through which the electrolyte diffuses within the activated carbon.
- the amount of vitamin B12 adsorbed is less than 10.2 mg/mL, the volume of the pores that can serve as the diffusion path of the electrolyte becomes small, so that the electrolyte is less likely to diffuse within the pores during charging and discharging, and the resistance tends to increase.
- the durability of the electricity storage device containing the activated carbon of the present invention tends to decrease with the increase in resistance.
- the amount of vitamin B12 adsorbed exceeds 50 mg/mL, the volume of the pores becomes large, so that the capacitance or discharge capacity per unit volume tends to decrease.
- the adsorption amount of vitamin B12 is preferably 10.5 mg/mL to 50 mg/mL, 10.8 mg/mL to 50 mg/mL, 11.0 mg/mL to 50 mg/mL, 11.5 mg/mL to 50 mg/mL, or 12.0 mg/mL to 50 mg/mL, more preferably 13 mg/mL to 50 mg/mL, even more preferably 15 mg/mL to 40 mg/mL, even more preferably 21 mg/mL to 30 mg/mL, and particularly preferably 24 mg/mL to 28 mg/mL.
- the capacitance or discharge capacity of the electricity storage device containing the activated carbon of the present invention tends to be higher and the resistance tends to be lower.
- the adsorption amount of vitamin B12 can be adjusted within the above range by appropriately adjusting, for example, the temperature and/or atmosphere and/or time of the activation step during the production of the activated carbon; the number of activation steps; the temperature and/or atmosphere and/or time of the heat treatment step, etc.
- the amount of vitamin B12 adsorbed can be determined, for example, by the method described in the Examples below.
- the ratio of the amount of adsorption when in contact with a 300 ppm vitamin B12 aqueous solution for 2 hours to the amount of adsorption when in contact with the aqueous solution for 24 hours is preferably 58% or more, more preferably 60% or more, even more preferably 62% or more, even more preferably 65% or more, particularly preferably 70% or more, and particularly preferably 76% or more.
- the ratio of the amount of adsorption when in contact with vitamin B12 for 2 hours to the amount of adsorption when in contact with vitamin B12 for 24 hours is equal to or greater than the lower limit, the adsorption and desorption of the electrolyte in the pores during charging and discharging is fast, and the electrolyte is more easily diffused, resulting in excellent rate characteristics and lower resistance.
- the ratio is preferably 90% or less, more preferably 85% or less, and even more preferably 82% or less. In addition, 58% to 90% is preferable, 60 to 85% is more preferable, and 65 to 82%, 70 to 82%, and 76 to 82% are also suitable embodiments.
- the ratio can be adjusted to within the above range by appropriately adjusting, for example, the temperature and/or atmosphere and/or time of the activation step during the production of activated carbon; the number of activation steps; the temperature and/or atmosphere and/or time of the heat treatment step, etc.
- the activated carbon of the present invention has a ratio of the adsorption amount of vitamin B12 when contacted with a 300 ppm aqueous solution of vitamin B12 for 24 hours to the BET specific surface area [(adsorption amount of vitamin B12 when contacted with a 300 ppm aqueous solution of vitamin B12 for 24 hours)/(BET specific surface area)] of preferably 0.00810 (mg/mL)/(m 2 /g) to 0.02470 (mg/mL)/(m 2 /g), more preferably 0.00820 (mg/mL)/(m 2 /g) to 0.02440 (mg/mL)/(m 2 /g), more preferably 0.00830 (mg/mL)/(m 2 /g) to 0.02410 (mg/mL)/(m 2 /g), and even more preferably 0.00840 (mg/mL)/(m 2 /g).
- the capacitance or discharge capacity of the electricity storage device containing the activated carbon of the present invention is more likely to be increased and the resistance is more likely to be reduced.
- the activated carbon of the present invention has a chlorine content of 2500 ppm or less.
- the chlorine content is 2500 ppm or less, the capacitance or discharge capacity of the electricity storage device containing the activated carbon of the present invention is likely to be high and the resistance is likely to be low.
- the chlorine content exceeds 2500 ppm, an SEI film is formed by the generation of chlorides, and the pores are blocked, so that the capacitance or discharge capacity is reduced, the resistance is increased, and the durability is reduced.
- the chlorine content is preferably 1000 ppm or less, more preferably 400 ppm or less, even more preferably 100 ppm or less, even more preferably 50 ppm or less, particularly preferably 35 ppm or less, and extremely preferably 30 ppm or less.
- the chlorine content is equal to or less than the upper limit, the resistance is likely to be lower.
- the lower the content of chlorine, which is an impurity, the more preferable it is, and the lower limit of the chlorine content may be 0 ppm, and is preferably 1 ppm or more.
- the chlorine content can be adjusted within the above range by appropriately adjusting, for example, the type of raw material of the activated carbon; the type and/or concentration and/or time of the acid in the washing step during the production of the activated carbon; the temperature and/or atmosphere and/or time of the heat treatment step, etc.
- the chlorine content can be determined by fluorescent X-ray analysis, for example, by the method described in the Examples below.
- the activated carbon has a specific surface area of 1150 m 2 /g to 2200 m 2 /g (preferably 1400 m 2 /g to 2200 m 2 /g) calculated by the BET method from the nitrogen adsorption/desorption isotherm, an adsorption amount of vitamin B12 of 13 mg/mL to 50 mg/mL when contacted with a 300 ppm aqueous vitamin B12 solution for 24 hours, and a chlorine content of 2500 ppm or less.
- the activated carbon has a specific surface area of 1150 m 2 /g to 2200 m 2 /g (preferably 1400 m 2 /g to 2200 m 2 /g) calculated by the BET method from the nitrogen adsorption/desorption isotherm, an adsorption amount of vitamin B12 when contacted with a 300 ppm aqueous solution of vitamin B12 for 24 hours is 10.2 mg/mL to 50 mg/mL, a ratio of the adsorption amount of vitamin B12 when contacted with a 300 ppm aqueous solution of vitamin B12 for 24 hours to the specific surface area calculated by the BET method from the nitrogen adsorption/desorption isotherm is 0.00810 [(mg/mL)/(m 2 /g)] to 0.02470 [(mg/mL)/(m 2 /g)], and a chlorine content of 2500 ppm or less.
- an electrical storage device contains the activated carbon of one of these
- the intensity ratio D/G of the D band peak near 1360 cm -1 to the G band peak near 1580 cm -1 is preferably 1.15 to 1.40, more preferably 1.19 to 1.39, and even more preferably 1.21 to 1.35 or 1.21 to 1.32.
- the D band peak is a peak caused by the disorder and defects of the graphite structure
- the G band peak is a peak derived from the graphite structure.
- the D band peak near 1360 cm -1 is usually observed in the range of 1300 cm -1 to 1375 cm -1 , preferably 1330 cm -1 to 1370 cm -1 .
- the G band peak near 1580 cm -1 is usually observed in the range of 1560 cm -1 to 1615 cm -1 , preferably 1565 cm -1 to 1610 cm -1 .
- the value of D/G is related to the crystallinity of the activated carbon, and when the value of D/G is equal to or greater than the lower limit, the crystallinity is not too low, so that the amount of amorphous material is not too large, and the resistance of an electric device containing the activated carbon of the present invention is likely to be low.
- the value of D/G is equal to or less than the upper limit, the crystallinity of the activated carbon is not too high, so that the carbon edge is not likely to decrease due to the development of the graphite structure, and the coordination sites of the electrolyte are not likely to decrease. Therefore, the resistance of an electric device containing the activated carbon of the present invention, particularly at low temperatures, is likely to be low.
- the value of D/G can be adjusted within the above range, for example, by appropriately adjusting the temperature and/or atmosphere and/or time of the activation step during the production of the activated carbon; the number of activation steps; the temperature and/or time and/or atmosphere of the heat treatment step, etc.
- the value of D/G can be determined, for example, by the method described in the Examples below.
- the activated carbon of the present invention when the specific surface area is 1150 m 2 /g to 2200 m 2 /g, it is considered that the micropores into which the electrolyte is adsorbed are relatively developed, and the electrostatic capacity or discharge capacity is likely to be higher when used in an electricity storage device. In addition to the above, when the micropores are developed, the adsorption rate is faster, so that the activated carbon tends to have excellent adsorption performance. This means that the activated carbon of a preferred embodiment of the present invention is not only suitable for electricity storage devices, but can also be useful as an adsorbent.
- the adsorption amount of chloroform is preferably 0.35 mg/g or more, more preferably 0.40 mg/g or more, more preferably 0.45 mg/g or more, and even more preferably 0.48 mg/g or more.
- the amount of chloroform adsorption can be adjusted to within the above range by, for example, appropriately adjusting the temperature and/or atmosphere and/or time of the activation step in the production of activated carbon, the number of activation steps, the temperature and/or time and/or atmosphere of the heat treatment step, etc.
- the amount of chloroform adsorption can be determined, for example, by the method described in the Examples below.
- the activated carbon of the present invention can be produced, for example, by a process comprising carbonizing a plant-derived carbon precursor, activating, washing, and heat treating the precursor to obtain activated carbon.
- a plant-derived carbon precursor is first carbonized.
- the plant that is the raw material for the plant-derived carbon precursor is not particularly limited, but examples include coconut shells, coffee beans, tea leaves, sugar cane, fruits (e.g., mandarin oranges or bananas), straw, broadleaf trees, coniferous trees, bamboo, and rice husks. These plants can be used alone or in combination of two or more types, but coconut shells are preferred because they are available in large quantities and are commercially advantageous.
- the coconut that is the raw material for the coconut shells is not particularly limited, but examples include palm (oil palm), coconut palm, salak, and omiya palm.
- these plants can be calcined to obtain them in the form of char (e.g., coconut shell char).
- Char generally refers to a powdered solid rich in carbon that is produced when coal is heated without melting or softening, but here it also refers to a powdered solid rich in carbon that is produced when organic matter is heated without melting or softening.
- char can be produced by burning (carbonizing) the plant raw material at a temperature of 300°C or higher, for example, about 400 to 800°C, in an atmosphere of an inert gas such as nitrogen, carbon dioxide, helium, argon, carbon monoxide, or fuel exhaust gas, a mixture of these inert gases, or a mixture of these inert gases with other gases that mainly contain these inert gases.
- an inert gas such as nitrogen, carbon dioxide, helium, argon, carbon monoxide, or fuel exhaust gas, a mixture of these inert gases, or a mixture of these inert gases with other gases that mainly contain these inert gases.
- the carbon precursor has a BET specific surface area of preferably 100 to 800 m 2 /g, more preferably 200 to 700 m 2 /g, and even more preferably 300 to 600 m 2 /g.
- BET specific surface area of the carbon precursor is within the above range, it is easy to adjust the pore structure to the desired one.
- the activation method is not particularly limited and can be selected according to the desired contact ratio between the carbon precursor and the reaction gas described below.
- the contact ratio between the carbon precursor and the reaction gas is to be high (e.g., about 70 to 100%), for example, a batch-type vertical fluidized activation furnace may be used.
- a continuous horizontal rotary kiln may be used.
- the partial pressure of the water vapor is usually 10 to 60%, preferably 20 to 55%, and more preferably 30 to 50%.
- the partial pressure of the water vapor is within the above range, activation is likely to proceed sufficiently and a rapid activation reaction is likely to be suppressed.
- the mixed gas is preferably supplied so that the amount of catalytic reaction gas per carbon precursor, calculated by the following formula, is 0.1 to 10.0 mL/min/kg.
- the activation after the secondary activation described below it may be preferable to supply the catalytic reaction gas per carbon precursor at 3.0 to 10.0 mL/min/kg.
- the contact ratio between the carbon precursor and the reaction gas is 15% or more.
- the contact ratio between the carbon precursor and the reaction gas is 100% because the reaction gas flows uniformly from the hearth to the top, and in the case of a horizontal furnace, it can be calculated by the ratio of the volume of the carbon precursor to the volume of the furnace.
- the activation temperature in the primary activation is usually 700 to 1100° C., preferably 800 to 1000° C.
- the activation time and the temperature rise rate are not particularly limited, and can be appropriately adjusted depending on the type, shape, size, etc. of the plant-derived carbon precursor selected. It is preferable to carry out the primary activation until the BET specific surface area of the primary activated carbon obtained after the primary activation reaches about 1000 to 2000 m 2 /g.
- the washing can be performed by immersing the primary activated carbon in a washing solution containing an acid.
- the washing solution include mineral acids and organic acids.
- mineral acids include hydrochloric acid and sulfuric acid.
- organic acids include formic acid, acetic acid, propionic acid, oxalic acid, tartaric acid, citric acid, and other saturated carboxylic acids; and benzoic acid and terephthalic acid, and other aromatic carboxylic acids.
- the acid used in the washing solution is preferably a mineral acid, and more preferably hydrochloric acid. After washing with the acid, it is preferable to further wash with water or the like to remove excess acid, and this operation can reduce the load on the activation equipment after the secondary activation.
- the cleaning solution can usually be prepared by mixing an acid with an aqueous solution.
- the aqueous solution include water and mixtures of water and water-soluble organic solvents.
- the water-soluble organic solvent include alcohols such as methanol, ethanol, propylene glycol, and ethylene glycol.
- the concentration of the acid in the cleaning solution is not particularly limited, and may be adjusted appropriately depending on the type of acid used.
- the acid concentration of the cleaning solution is preferably 0.1 to 2.0 normal, more preferably 0.2 to 1.5 normal, and even more preferably 0.3 to 1.0 normal. It is preferable that the acid concentration in the cleaning solution is within the above range, since it is possible to efficiently remove impurities contained in the primary activated carbon.
- the pH of the cleaning solution is not particularly limited and may be adjusted as appropriate depending on the type of acid used and the object to be removed.
- the temperature of the cleaning solution when soaking the primary activated carbon is not particularly limited, but is preferably 0 to 98°C, more preferably 10 to 95°C, and even more preferably 15 to 90°C. If the temperature of the cleaning solution when soaking the primary activated carbon is within the above range, it is preferable because cleaning can be performed in a practical time with reduced strain on the equipment.
- the method for washing the primary activated carbon is not particularly limited as long as it is possible to immerse the primary activated carbon in the washing solution, and may be a method in which washing solution is continuously added, retained for a specified time, and immersed while removing the solution, or a method in which the primary activated carbon is immersed in the washing solution, retained for a specified time, drained, and then new washing solution is added and the immersion-draining process is repeated.
- the washing solution may be entirely renewed, or may be partially renewed.
- the time for which the primary activated carbon is immersed in the washing solution may be appropriately adjusted depending on the acid used, the acid concentration, the treatment temperature, etc.
- the washing time is not particularly limited, but from the viewpoints of the economic efficiency of the reaction equipment and the structural retention of the activated carbon, it is preferably 0.05 to 4 hours, and more preferably 0.1 to 3 hours.
- the mass ratio between the cleaning solution and the primary activated carbon may be adjusted as appropriate depending on the type, concentration, and temperature of the cleaning solution used.
- the mass of the primary activated carbon to be immersed relative to the mass of the cleaning solution is usually 0.1 to 50 mass%, preferably 1 to 20 mass%, and more preferably 1.5 to 10 mass%. Within the above range, impurities dissolved in the cleaning solution are less likely to precipitate from the cleaning solution, and redeposition to the primary activated carbon is easily suppressed.
- the atmosphere in which cleaning is performed is not particularly limited and may be selected appropriately depending on the method used for cleaning, but is usually performed in the air.
- Cleaning may be performed once or multiple times using one type of cleaning solution, or multiple times using a combination of two or more types of cleaning solutions.
- Washing can remove impurities that may be present in the activated carbon. These impurities are brought about by the plant-derived carbon precursors, such as alkali metals, such as lithium, sodium, and potassium; alkaline earth metals, such as beryllium, magnesium, and calcium; and transition metals, such as iron, copper, and nickel.
- alkali metals such as lithium, sodium, and potassium
- alkaline earth metals such as beryllium, magnesium, and calcium
- transition metals such as iron, copper, and nickel.
- the primary activated carbon may be dried. Drying is an operation for removing moisture and other substances adsorbed to the primary activated carbon.
- the primary activated carbon may be heated to remove moisture and other substances adsorbed to the primary activated carbon.
- drying may be performed by means of, for example, reduced pressure, reduced pressure heating, freezing, or the like to remove moisture and other substances adsorbed to the primary activated carbon.
- the drying temperature is preferably 100 to 330°C, more preferably 110 to 300°C, and even more preferably 120 to 250°C.
- the drying time depends on the drying temperature used, but from the viewpoint of removing the moisture adsorbed on the primary activated carbon, it is preferably 0.1 hours or more, more preferably 0.5 hours or more, and even more preferably 1 hour or more. From the viewpoint of economy, it is preferably 24 hours or less, more preferably 12 hours or less, and even more preferably 6 hours or less.
- Drying can be performed under normal pressure or reduced pressure. When drying is performed under normal pressure, it is preferable to perform it under an inert gas atmosphere such as nitrogen gas or argon gas, or under an air atmosphere with a dew point of -20°C or lower.
- an inert gas atmosphere such as nitrogen gas or argon gas
- an air atmosphere with a dew point of -20°C or lower or under an air atmosphere with a dew point of -20°C or lower.
- Secondary activation may be performed after the above-mentioned washing and drying.
- the amount of contact reaction gas per primary activated carbon is preferably supplied so as to be 5 to 30 mL/min/kg, from the viewpoint of easily increasing the capacitance or discharge capacity of the electricity storage device containing the activated carbon of the present invention and easily decreasing the resistance.
- the activation temperature, contact ratio, water vapor partial pressure, and other conditions may be within the same range as those of the above-mentioned primary activation.
- the BET specific surface area of the secondary activated carbon obtained after the secondary activation reaches about 1150 to 2200 m 2 /g (preferably 1400 to 2200 m 2 /g, more preferably 1600 to 2200 m 2 /g).
- a tertiary activation may be performed, or even a higher activation may be performed.
- washing and drying may be performed between each activation after the secondary activation. From an economical standpoint, it is preferable to perform up to the secondary activation.
- the conditions for the tertiary activation and the higher activation may be appropriately adjusted depending on the desired properties of the activated carbon.
- Heat treatment increases crystallinity, which tends to lower resistance and also makes it easier to reduce the amount of impurities in the activated carbon.
- the heat treatment method is not particularly limited, but can be carried out in the same manner as the primary activation described above, and can be selected according to the desired contact ratio between the activated carbon and the reaction gas after activation, which will be described later.
- the partial pressure of the water vapor is 15% or less. If the partial pressure of the water vapor is within the above range, the crystallinity is high and therefore the resistance is likely to be low.
- the mixed gas is preferably supplied so that the amount of contact reaction gas per activated carbon after activation, calculated by the following formula, is 0.1 to 5 mL/min/kg.
- the amount of contact reaction gas per activated carbon after activation is within the above range, pores of a size expressed by the adsorption amount of vitamin B12 are more likely to develop, and the capacitance or discharge capacity of an electricity storage device containing the activated carbon of the present invention is more likely to be increased and the resistance is more likely to be reduced.
- the contact ratio between the activated carbon and the reaction gas after activation is 20% or more.
- the contact ratio between the activated carbon and the reaction gas after activation is 100% because the reaction gas flows uniformly from the hearth to the top, and in the case of a horizontal furnace, it can be calculated by the ratio of the volume of the activated carbon after activation to the volume of the furnace.
- the heat treatment temperature is usually 600 to 1000°C, preferably 700 to 900°C.
- the heat treatment time and temperature rise rate are not particularly limited, but from the viewpoint of reducing the load on the reaction equipment and maintaining the structure of the activated carbon, the heat treatment time is preferably 0.5 to 24 hours, more preferably 1 to 12 hours, and the temperature rise rate is preferably 10 to 60°C/min, more preferably 20 to 50°C/min.
- the activated carbon after heat treatment may be pulverized.
- the pulverization method there are no particular limitations on the pulverization method, but known pulverization methods such as a bead mill, ball mill, hammer mill, roll mill, rod mill, or jet mill, or a combination of these, can be used.
- the average particle size of the pulverized activated carbon it is preferably 100 ⁇ m or less, more preferably 90 ⁇ m or less, and preferably 1 ⁇ m or more, more preferably 2 ⁇ m or more.
- the average particle size can be measured, for example, using a particle size/particle size distribution measuring device.
- the activated carbon obtained by pulverization may be classified. For example, by removing particles with a particle size of 1 ⁇ m or less, activated carbon particles with a narrow particle size distribution can be obtained.
- the classification method is not particularly limited, but examples include classification using a sieve, wet classification, and dry classification.
- wet classifiers include classifiers that utilize the principles of gravity classification, inertia classification, hydraulic classification, centrifugal classification, etc.
- dry classifiers include classifiers that utilize the principles of sedimentation classification, mechanical classification, centrifugal classification, etc. From the viewpoint of economy, it is preferable to use a dry classification device.
- the activated carbon of the present invention can be suitably used as an electrode material for various electricity storage devices such as electric double layer capacitors, lithium ion batteries, and lithium sulfur batteries.
- it is suitable as an electrode material for electric double layer capacitors, and by using the activated carbon of the present invention, an electric double layer capacitor that can achieve both high capacitance and low resistance can be obtained.
- an electricity storage device with excellent performance at low temperatures can be obtained.
- an electrode material containing the activated carbon of the present invention can be provided.
- an electrode containing the electrode material, and an electricity storage device containing the electrode can be provided.
- the electrode of the present invention is characterized by using the activated carbon of the present invention.
- the electrode of the present invention can be manufactured, for example, by using an electrode material obtained by kneading the activated carbon of the present invention as a raw material with components such as a conductive agent (also called a conductive assistant), a binder, and a solvent, coating and drying the kneaded product, adding a solvent to the electrode material to prepare a paste, applying the paste to a current collector such as aluminum foil, drying and removing the solvent, and placing the paste in a mold and press-molding it.
- a conductive agent also called a conductive assistant
- a binder also called a conductive assistant
- Examples of the conductive agent that can be used include carbon black, acetylene black, and Ketjen black.
- the binder that can be used include fluorine-based polymer compounds such as polytetrafluoroethylene and polyvinylidene fluoride, carboxymethyl cellulose, styrene-butadiene rubber, petroleum pitch, and phenolic resin.
- solvent examples include water; alcohols such as methanol and ethanol; saturated hydrocarbons such as hexane and heptane; aromatic hydrocarbons such as toluene, xylene, and mesitylene; ketones such as acetone and ethyl methyl ketone; esters such as methyl acetate and ethyl acetate; amides such as N,N-dimethylformamide and N,N-diethylformamide; and cyclic amides such as N-methylpyrrolidone and N-ethylpyrrolidone.
- alcohols such as methanol and ethanol
- saturated hydrocarbons such as hexane and heptane
- aromatic hydrocarbons such as toluene, xylene, and mesitylene
- ketones such as acetone and ethyl methyl ketone
- esters such as methyl acetate and ethyl acetate
- amides such as N
- the electric storage device of the present invention is characterized by using the above-mentioned electrodes.
- the electric double layer capacitor generally has a structure in which the main components are electrodes, an electrolyte, and a separator, and the separator is disposed between a pair of electrodes.
- Examples of the electrolyte include an electrolyte in which an amidine salt is dissolved in an organic solvent such as propylene carbonate, ethylene carbonate, or methyl ethyl carbonate, an electrolyte in which a quaternary ammonium salt of perchloric acid is dissolved, an electrolyte in which a tetrafluoroborate or a hexafluorophosphate of an alkali metal such as quaternary ammonium or lithium is dissolved, and an electrolyte in which a quaternary phosphonium salt is dissolved.
- an organic solvent such as propylene carbonate, ethylene carbonate, or methyl ethyl carbonate
- an electrolyte in which a quaternary ammonium salt of perchloric acid is dissolved
- an electrolyte in which a tetrafluoroborate or a hexafluorophosphate of an alkali metal such as quaternary ammonium or lithium
- the separator examples include a nonwoven fabric, cloth, or microporous film mainly composed of cellulose, glass fiber, or a polyolefin such as polyethylene or polypropylene.
- the electric double layer capacitor can be manufactured, for example, by disposing these main components by a method conventionally used in the field.
- the absorbance at 550 nm of the 300 ppm aqueous solution of vitamin B12 before adsorption and the vitamin B12 filtrate after adsorption treatment used in the adsorption test were measured, and the concentrations of the aqueous solution of vitamin B12 before adsorption and the vitamin B12 filtrate after adsorption treatment were calculated based on a calibration curve prepared in advance.
- the activated carbon obtained in the examples and comparative examples was pulverized, vacuum dried at 120°C for 3 hours or more, and then cooled to room temperature in a desiccator.
- the obtained activated carbon was placed in a polyethylene container of 15 cm3 or more, the back side was pressed with a plankton net, the measurement surface was covered with a polypropylene film, and the container was placed in a sample holder for the top irradiation method.
- the measurement was performed using "Primus II" manufactured by Rigaku Corporation, under a helium gas atmosphere, with an output of 3 kW, a scan speed of 8 deg/min, and an X-ray source of 30 kV and 100 mA.
- the chlorine content was calculated from the detected elements and their X-ray intensities by SQX (Scan Quant X) analysis: Standardless Fundamental Parameter Analysis by Rigaku Corporation.
- ⁇ Ratio of D-band peak to G-band peak in Raman spectroscopy> The activated carbon obtained in the examples and comparative examples was pulverized, vacuum dried at 120°C for 3 hours or more, and then cooled to room temperature in a desiccator.
- Raman spectra were measured using a HORIBA Ltd. "XploRA PLUS" with a laser wavelength of 532 nm as a light source. The test was performed on randomly sampled particles in each sample. The measurement conditions were a wavelength range of 500 to 2000 cm -1 , an exposure time of 10 seconds, and 50 cumulative times, and the average value of a total of five points was calculated as the measured value.
- the D/G value was the intensity ratio ID/IG (D band peak intensity/G band peak intensity) of the peaks of the D band near 1360 cm -1 and the G band near 1580 cm -1 .
- ⁇ Chloroform adsorption amount> An arbitrary amount of pulverized activated carbon was added to 100 mL of chloroform solution adjusted to an initial concentration of about 0.100 mg/L, and then the solution was shaken at 160 rpm for 2 hours at about 25° C. Then, the solution was left to stand in a thermostatic chamber at 25° C. for 1 hour, and the equilibrium adsorption amount of chloroform was calculated.
- the adsorption amount A (mg/g) was calculated from the chloroform concentration Co (mg/L) of the blank without activated carbon, the chloroform concentration C (mg/L) of the test water with activated carbon, and the mass W (mg) of the activated carbon added, using the following formula:
- the chloroform concentration was calculated by the headspace method using ECD gas chromatography.
- A (Co-C) ⁇ 1000 ⁇ 0.1/W
- a power approximation formula was calculated from the adsorption amounts A obtained at three different test water concentrations, and the adsorption amount at a test water concentration of 0.01 mg/L was calculated as the equilibrium adsorption amount.
- Example 1 1 kg of char (specific surface area: about 400 m2 /g) made from coconut shells from the Philippines was placed in a fluidized bed activation furnace heated to 900°C, and propane combustion gas + steam (steam partial pressure: 35%) was introduced at a rate of 0.75 mL/min (temperature-corrected reaction gas amount: 3.22 mL/min, contact ratio between char and reaction gas: 100%, contact reaction gas amount per char: 3.22 mL/min/kg), and primary activation was performed until the specific surface area reached 1550 m2 /g.
- the char was then pickled with hydrochloric acid (concentration: 0.5 N, diluent: ion-exchanged water) at a temperature of 70°C for 30 minutes, thoroughly washed with ion-exchanged water, and dried.
- hydrochloric acid concentration: 0.5 N, diluent: ion-exchanged water
- This primary activated and washed carbon was placed in a heat treatment furnace, and propane combustion gas + water vapor (water vapor partial pressure: 10%) was introduced at a rate of 0.70 mL/min (temperature-corrected reaction gas amount: 2.62 mL/min, contact ratio between primary activated carbon and reaction gas: 100%, contact reaction gas amount per primary activated carbon: 2.62 mL/min/kg), and heat treatment was performed at 750°C to obtain a specific surface area of 1641 m2 /g, thereby obtaining activated carbon.
- the temperature-corrected reaction gas amount, the catalytic reaction gas amount per char, and the catalytic reaction gas amount per activated carbon were calculated according to the following formulas.
- Example 2 Activated carbon was obtained in the same manner as in Example 1, except that the amount of propane combustion gas + steam introduced was 1.05 mL/min (temperature-corrected reaction gas amount was 4.51 mL/min, contact ratio between char and reaction gas was 100%, and contact reaction gas amount per char was 4.51 mL/min/kg) and primary activation was performed until the specific surface area reached 1648 m2 /g, and that the amount of propane combustion gas + steam introduced was 0.55 mL/min (temperature-corrected reaction gas amount was 2.06 mL/min, contact ratio between activated carbon and reaction gas was 100%, and contact reaction gas amount per activated carbon was 2.06 mL/min/kg) and heat treatment was performed at 750°C until the specific surface area reached 1737 m2 /g.
- Example 3 Activated carbon was obtained in the same manner as in Example 1, except that the amount of propane combustion gas + steam was introduced at 1.00 mL/min (temperature-corrected reaction gas amount: 4.30 mL/min, contact ratio between char and reaction gas: 100%, contact reaction gas amount per char: 4.30 mL/min/kg) and primary activation was performed until the specific surface area reached 1788 m2 /g, and the amount of propane combustion gas + steam was introduced at 0.60 mL/min (temperature-corrected reaction gas amount: 2.25 mL/min, contact ratio between activated carbon and reaction gas: 100%, contact reaction gas amount per activated carbon: 2.25 mL/min/kg) and heat treatment was performed at 750°C until the specific surface area reached 1858 m2 /g.
- propane combustion gas + steam was introduced at 1.00 mL/min (temperature-corrected reaction gas amount: 4.30 mL/min, contact ratio between char and reaction gas: 100%, contact reaction gas amount per char:
- Example 4 Activated carbon was obtained in the same manner as in Example 1, except that the amount of propane combustion gas + steam was introduced at 0.95 mL/min (temperature-corrected reaction gas amount: 4.08 mL/min, contact ratio between char and reaction gas: 100%, contact reaction gas amount per char: 4.08 mL/min/kg) and primary activation was performed until the specific surface area reached 1,862 m 2 /g, and that the amount of propane combustion gas + steam was introduced at 0.55 mL/min (temperature-corrected reaction gas amount: 2.06 mL/min, contact ratio between activated carbon and reaction gas: 100%, contact reaction gas amount per activated carbon: 2.06 mL/min/kg) and heat treatment was performed at 750°C until the specific surface area reached 1,940 m 2 /g.
- Example 5 1 kg of char (specific surface area: about 400 m 2 /g) made from coconut shells from the Philippines was put into a rotary kiln heated to 900°C, and propane combustion gas + steam (steam partial pressure: 35%) was introduced at a rate of 0.50 mL/min (temperature-corrected reaction gas amount: 2.15 mL/min, contact ratio between char and reaction gas: 15%, contact reaction gas amount per char: 0.32 mL/min/kg), and primary activation was performed until the specific surface area reached 1676 m 2 /g. The contact ratio with the reaction gas was calculated from the ratio of the char volume to the volume of the furnace.
- the char was pickled with hydrochloric acid (concentration: 0.5 N, diluent: ion-exchanged water) at a temperature of 70°C for 30 minutes, and then thoroughly washed with ion-exchanged water and dried.
- This primary activated and washed coal was placed in a fluidized bed activation furnace heated to 950°C, and propane combustion gas + steam (steam partial pressure: 30%) was introduced at a rate of 2.50 mL/min (temperature-corrected reaction gas amount: 11.20 mL/min, contact ratio between char and reaction gas: 100%, contact reaction gas amount per char: 11.20 mL/min/kg), and secondary activation was performed until the specific surface area reached 2079 m2 /g.
- coal was pickled at a temperature of 70°C for 30 minutes using hydrochloric acid (concentration: 0.5 N, diluent: ion-exchanged water), and then thoroughly washed with ion-exchanged water and dried.
- hydrochloric acid concentration: 0.5 N
- diluent ion-exchanged water
- This secondary activated and washed carbon was placed in a heat treatment furnace, and propane combustion gas + water vapor (water vapor partial pressure: 15%) was introduced at a rate of 0.75 mL/min (temperature-corrected reaction gas amount: 2.81 mL/min, contact ratio between activated carbon and reaction gas: 100%, contact reaction gas amount per activated carbon: 2.81 mL/min/kg), and heat treatment was carried out at 750°C until the specific surface area reached 2108 m2 /g, thereby obtaining activated carbon.
- Example 6 Activated carbon was obtained in the same manner as in Example 1, except that the amount of propane combustion gas + steam was introduced at 0.95 mL/min (temperature-corrected reaction gas amount: 4.08 mL/min, contact ratio between char and reaction gas: 100%, contact reaction gas amount per char: 4.08 mL/min/kg) and primary activation was performed until the specific surface area reached 1407 m2 /g, and that the amount of propane combustion gas + steam was introduced at 0.55 mL/min (temperature-corrected reaction gas amount: 2.06 mL/min, contact ratio between activated carbon and reaction gas: 100%, contact reaction gas amount per activated carbon: 2.06 mL/min/kg) and heat treatment was performed at 750°C until the specific surface area reached 1482 m2 /g.
- Example 7 Activated carbon was obtained in the same manner as in Example 1, except that the amount of propane combustion gas + steam was introduced at 1.00 mL/min (temperature-corrected reaction gas amount: 4.30 mL/min, contact ratio between char and reaction gas: 100%, contact reaction gas amount per char: 4.30 mL/min/kg) and primary activation was performed until the specific surface area reached 1250 m2 /g, and the amount of propane combustion gas + steam was introduced at 0.60 mL/min (temperature-corrected reaction gas amount: 2.25 mL/min, contact ratio between activated carbon and reaction gas: 100%, contact reaction gas amount per activated carbon: 2.25 mL/min/kg) and heat treatment was performed at 750°C until the specific surface area reached 1320 m2 /g.
- Example 8 Activated carbon was obtained in the same manner as in Example 1, except that the amount of propane combustion gas + steam was introduced at 0.95 mL/min (temperature-corrected reaction gas amount: 4.08 mL/min, contact ratio between char and reaction gas: 100%, contact reaction gas amount per char: 4.08 mL/min/kg) and primary activation was performed until the specific surface area reached 1208 m2 /g, and the amount of propane combustion gas + steam was introduced at 0.55 mL/min (temperature-corrected reaction gas amount: 2.06 mL/min, contact ratio between activated carbon and reaction gas: 100%, contact reaction gas amount per activated carbon: 2.06 mL/min/kg) and heat treatment was performed at 750°C until the specific surface area reached 1283 m2 /g.
- ⁇ Comparative Example 1> 100 g of pine sawdust (moisture content 48% by mass) was mixed with 97.8 g of 85% by mass phosphoric acid aqueous solution (dry solids mass of phosphoric acid/dry solids mass of sawdust 1.6). This mixture was heated while stirring in a circulation dryer heated to a temperature of 175°C. The resulting mixture was placed in a rotary kiln heated to 300°C and oxidized by holding it under air for 3 hours. This oxidized product was subjected to primary activation at 500°C under nitrogen until the specific surface area reached 1097 m2 /g. It was then thoroughly washed with ion-exchanged water and dried.
- This primary activated and washed carbon was placed in a heat treatment furnace, and propane combustion gas + water vapor (water vapor partial pressure: 10%) was introduced at a rate of 0.35 mL/min (temperature-corrected reaction gas amount: 1.31 mL/min, contact ratio between activated carbon and reaction gas: 100%, contact reaction gas amount per activated carbon: 1.31 mL/min/kg), and heat treatment was performed at 750°C to obtain a specific surface area of 1,146 m2 /g, thereby obtaining activated carbon.
- ⁇ Comparative Example 2 1 kg of char (specific surface area: about 400 m2 /g) made from coconut shells from the Philippines was placed in a fluidized bed activation furnace heated to 900°C, and propane combustion gas + steam (steam partial pressure: 35%) was introduced at a rate of 0.65 mL/min (temperature-corrected reaction gas amount: 2.79 mL/min, contact ratio between char and reaction gas: 100%, contact reaction gas amount per char: 2.79 mL/min/kg), and primary activation was performed until the specific surface area reached 1621 m2 /g.
- the char was then pickled with hydrochloric acid (concentration: 0.5 N, diluent: ion-exchanged water) at a temperature of 70°C for 30 minutes, thoroughly washed with ion-exchanged water and dried to obtain activated carbon.
- hydrochloric acid concentration: 0.5 N, diluent: ion-exchanged water
- the char was then pickled with hydrochloric acid (concentration: 0.5 N, diluent: ion-exchanged water) at a temperature of 70°C for 30 minutes, thoroughly washed with ion-exchanged water and dried to obtain activated carbon.
- hydrochloric acid concentration: 0.5 N, diluent: ion-exchanged water
- the contact ratio with the reaction gas was calculated from the ratio of the char volume to the volume of the furnace.
- the char was pickled with hydrochloric acid (concentration: 0.5 N, diluent: ion-exchanged water) at a temperature of 70°C for 30 minutes, and then thoroughly washed with ion-exchanged water and dried.
- This primary activated and washed coal was placed in a fluidized bed activation furnace heated to 950°C, and propane combustion gas + steam (steam partial pressure: 15%) was introduced at a rate of 1.85 mL/min (temperature-corrected reaction gas amount: 8.29 mL/min, contact ratio between char and reaction gas: 100%, contact reaction gas amount per char: 8.29 mL/min/kg), and secondary activation was performed until the specific surface area reached 2195 m2 /g.
- coal was pickled with hydrochloric acid (concentration: 0.5 N, diluent: ion-exchanged water) at a temperature of 70°C for 30 minutes, and then thoroughly washed with ion-exchanged water and dried.
- hydrochloric acid concentration: 0.5 N, diluent: ion-exchanged water
- This secondary activated and washed carbon was placed in a heat treatment furnace, and propane combustion gas + water vapor (water vapor partial pressure: 10%) was introduced at a rate of 0.45 mL/min (temperature-corrected reaction gas amount: 1.69 mL/min, contact ratio between activated carbon and reaction gas: 100%, contact reaction gas amount per activated carbon: 1.69 mL/min/kg), and heat treatment was carried out at 750°C until the specific surface area reached 2221 m2 /g, thereby obtaining activated carbon.
- Activated carbon was obtained in the same manner as in Comparative Example 2, except that the oxidation treatment product was subjected to primary activation under nitrogen at 500°C until the specific surface area reached 1548 m2 /g, and propane combustion gas + water vapor (water vapor partial pressure: 10%) was introduced at a rate of 0.5 mL/min (temperature-corrected reaction gas amount: 1.87 mL/min, contact ratio between activated carbon and reaction gas: 100%, contact reaction gas amount per activated carbon: 1.87 mL/min/kg), and heat treatment was performed at 750°C until the specific surface area reached 1601 m2 /g.
- the char was then pickled with hydrochloric acid (concentration: 0.5 N, diluent: ion-exchanged water) at a temperature of 70°C for 30 minutes, thoroughly washed with ion-exchanged water, and dried.
- hydrochloric acid concentration: 0.5 N, diluent: ion-exchanged water
- This primary activated and washed carbon was placed in a heat treatment furnace, and propane combustion gas + water vapor (water vapor partial pressure: 10%) was introduced at a rate of 0.30 mL/min (temperature-corrected reaction gas amount: 1.12 mL/min, contact ratio between activated carbon and reaction gas: 100%, contact reaction gas amount per activated carbon: 1.12 mL/min/kg), and heat treatment was performed at 750°C to obtain a specific surface area of 1,353 m2 /g, thereby obtaining activated carbon.
- the activated carbon obtained in the examples and comparative examples was pulverized to an average particle size of 6 ⁇ m.
- the pulverized activated carbon, conductive assistant, and binder were dried in advance at 120° C. under reduced pressure (0.1 KPa or less) for 16 hours or more before use.
- the activated carbon, conductive assistant, and binder were weighed and kneaded so that the ratio of (mass of activated carbon): (mass of conductive assistant): (mass of binder) was 81:9:10.
- As the conductive assistant conductive carbon black "Denka Black Granular" manufactured by Denka Co., Ltd.
- a conductive adhesive 2 "HITASOL GA-715" manufactured by Hitachi Chemical Co., Ltd. was applied to an etched aluminum foil 3 manufactured by Hosen Co., Ltd. to a coating thickness of 100 ⁇ m. Then, as shown in Fig. 3, the etched aluminum foil 3 to which the conductive adhesive 2 was applied was adhered to a sheet-like electrode composition 1 that had been cut in advance. Then, a tab 4 with an aluminum sealant 5 manufactured by Hosen Co., Ltd. was welded to the etched aluminum foil 3 using an ultrasonic welding machine. After welding, the product was vacuum dried at 120°C to obtain a polarizable electrode 6 equipped with an aluminum current collector.
- an aluminum laminated resin sheet manufactured by Hosen Co., Ltd. was cut into a rectangle (200 mm long x 60 mm wide), folded in half, and one side ((1) in Figure 4) was heat-pressed while the remaining two sides were left open to prepare a bag-shaped exterior sheet 7.
- a laminate was made by stacking two of the polarizable electrodes 6 described above via a cellulose separator "TF-40" (not shown) manufactured by Nippon Kodoshi Co., Ltd. This laminate was inserted into the exterior sheet 7, and the polarizable electrode 6 was fixed by heat-pressing one side ((2) in Figure 5) where the tab 4 contacts.
- the obtained electric double layer capacitor 8 was charged at a constant current of 50 mA per electrode surface area at ⁇ 30° C. using a “CAPACITOR TESTER PFX2411” manufactured by Kikusui Electronics Co., Ltd., to a final voltage of 3.0 V, and then supplementary charged at a constant voltage of 3.0 V for 30 minutes, and discharged at 25 mA after the supplementary charging was completed.
- the obtained discharge curve data was calculated by an energy conversion method to obtain the capacitance (F). Specifically, after charging, the capacitor was discharged until the voltage became zero, and the capacitance (F) was calculated from the discharge energy discharged at this time. Then, the capacitance was divided by the electrode volume to obtain the capacitance (F/cc).
- Resistance measurements were performed at -30°C using an electrochemical measurement device (VMP3 manufactured by BioLogic), with a constant voltage AC impedance measurement method, with an amplitude of 20 mV centered around 0 V, and at frequencies from 4 mHz to 1 MHz.
- VMP3 electrochemical measurement device
- a Cole-Cole plot was obtained showing the relationship between complex impedance, with the real part on the horizontal axis and the imaginary part on the vertical axis. From this plot, the resistance of the cable, etc.
- the electrode interface resistance (the point of contact with the real axis of the semicircle, the diameter of the arc) and the electrode diffusion resistance (the resistance value obtained by excluding the interface resistance from the point of contact between the tangent of the low frequency side curve and the real axis) were obtained.
- the electric double layer capacitors equipped with electrodes containing activated carbon obtained in Examples 1 to 8 had low resistance and high capacitance.
- the electric double layer capacitors equipped with electrodes containing activated carbon obtained in Comparative Examples 1 to 6 had insufficient resistance or capacitance. Therefore, it was found that the activated carbon of the present invention is useful for improving the performance of electricity storage devices.
- Electrode composition 2 Conductive adhesive 3 Etched aluminum foil 4 Tab 5 Sealant 6 Polarizable electrode 7 Bag-shaped exterior sheet 8 Electric double layer capacitor (1) One side that is thermocompression bonded (2) One side that is in contact with the tab (3) Remaining side of the bag-shaped exterior sheet
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Power Engineering (AREA)
- Materials Engineering (AREA)
- Inorganic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Electrochemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Microelectronics & Electronic Packaging (AREA)
- Carbon And Carbon Compounds (AREA)
- Electric Double-Layer Capacitors Or The Like (AREA)
- Solid-Sorbent Or Filter-Aiding Compositions (AREA)
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2025527935A JPWO2024257756A1 (https=) | 2023-06-15 | 2024-06-11 | |
| CN202480038886.0A CN121358689A (zh) | 2023-06-15 | 2024-06-11 | 活性炭 |
| KR1020257033895A KR20250168308A (ko) | 2023-06-15 | 2024-06-11 | 활성탄 |
| EP24823372.8A EP4729479A1 (en) | 2023-06-15 | 2024-06-11 | Activated carbon |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2023098631 | 2023-06-15 | ||
| JP2023-098631 | 2023-06-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024257756A1 true WO2024257756A1 (ja) | 2024-12-19 |
Family
ID=93852015
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2024/021177 Ceased WO2024257756A1 (ja) | 2023-06-15 | 2024-06-11 | 活性炭 |
Country Status (6)
| Country | Link |
|---|---|
| EP (1) | EP4729479A1 (https=) |
| JP (1) | JPWO2024257756A1 (https=) |
| KR (1) | KR20250168308A (https=) |
| CN (1) | CN121358689A (https=) |
| TW (1) | TW202508964A (https=) |
| WO (1) | WO2024257756A1 (https=) |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003290654A (ja) * | 2002-04-01 | 2003-10-14 | Kuraray Co Ltd | 活性炭素繊維及びその製造方法、並びに浄水器用カートリッジ及び浄水器 |
| JP2010168238A (ja) * | 2009-01-21 | 2010-08-05 | Kansai Coke & Chem Co Ltd | 高純度化活性炭の製造方法、及び該製造方法によって得られた電気二重層キャパシタ |
| JP2012092013A (ja) | 2002-04-11 | 2012-05-17 | Showa Denko Kk | 活性炭、分極性電極及び電気二重層キャパシタ |
| JP2013118191A (ja) | 2008-03-12 | 2013-06-13 | Toyota Motor Engineering & Manufacturing North America Inc | 硫黄−炭素材料 |
| WO2015146459A1 (ja) | 2014-03-27 | 2015-10-01 | Jx日鉱日石エネルギー株式会社 | 活性炭、活性炭の製造方法および活性炭の処理方法 |
| JP2017009501A (ja) * | 2015-06-24 | 2017-01-12 | 株式会社キャタラー | 炭素系材料の塩素含有量の分析方法及び塩素を除去した炭素系材料の製造方法 |
| JP6829796B1 (ja) * | 2019-04-26 | 2021-02-10 | 株式会社クラレ | 炭素質材料及びその製造方法、並びに浄水用フィルター及び浄水器 |
| JP7060772B1 (ja) * | 2020-10-23 | 2022-04-26 | 株式会社クラレ | 炭素質材料及びその製造方法、並びに、含フッ素有機化合物除去材、浄水用フィルター及び浄水器 |
-
2024
- 2024-06-11 CN CN202480038886.0A patent/CN121358689A/zh active Pending
- 2024-06-11 WO PCT/JP2024/021177 patent/WO2024257756A1/ja not_active Ceased
- 2024-06-11 EP EP24823372.8A patent/EP4729479A1/en active Pending
- 2024-06-11 KR KR1020257033895A patent/KR20250168308A/ko active Pending
- 2024-06-11 JP JP2025527935A patent/JPWO2024257756A1/ja active Pending
- 2024-06-13 TW TW113121811A patent/TW202508964A/zh unknown
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003290654A (ja) * | 2002-04-01 | 2003-10-14 | Kuraray Co Ltd | 活性炭素繊維及びその製造方法、並びに浄水器用カートリッジ及び浄水器 |
| JP2012092013A (ja) | 2002-04-11 | 2012-05-17 | Showa Denko Kk | 活性炭、分極性電極及び電気二重層キャパシタ |
| JP2013118191A (ja) | 2008-03-12 | 2013-06-13 | Toyota Motor Engineering & Manufacturing North America Inc | 硫黄−炭素材料 |
| JP2010168238A (ja) * | 2009-01-21 | 2010-08-05 | Kansai Coke & Chem Co Ltd | 高純度化活性炭の製造方法、及び該製造方法によって得られた電気二重層キャパシタ |
| WO2015146459A1 (ja) | 2014-03-27 | 2015-10-01 | Jx日鉱日石エネルギー株式会社 | 活性炭、活性炭の製造方法および活性炭の処理方法 |
| JP2017009501A (ja) * | 2015-06-24 | 2017-01-12 | 株式会社キャタラー | 炭素系材料の塩素含有量の分析方法及び塩素を除去した炭素系材料の製造方法 |
| JP6829796B1 (ja) * | 2019-04-26 | 2021-02-10 | 株式会社クラレ | 炭素質材料及びその製造方法、並びに浄水用フィルター及び浄水器 |
| JP7060772B1 (ja) * | 2020-10-23 | 2022-04-26 | 株式会社クラレ | 炭素質材料及びその製造方法、並びに、含フッ素有機化合物除去材、浄水用フィルター及び浄水器 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN121358689A (zh) | 2026-01-16 |
| KR20250168308A (ko) | 2025-12-02 |
| EP4729479A1 (en) | 2026-04-22 |
| TW202508964A (zh) | 2025-03-01 |
| JPWO2024257756A1 (https=) | 2024-12-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2214236B1 (en) | Nonaqueous lithium-type storage element | |
| US11823837B2 (en) | Carbonaceous material for electric double layer capacitors and method for producing same | |
| CN108604504B (zh) | 非水系锂型蓄电元件 | |
| JP5931326B2 (ja) | 電気二重層キャパシタ用活性炭 | |
| KR101958645B1 (ko) | 수증기 활성화를 이용한 부분 결정성 다공성 활성탄의 제조방법 및 상기 부분 결정성 다공성 활성탄을 이용한 슈퍼커패시터의 제조방법 | |
| US12002948B2 (en) | Immobilized selenium in a porous carbon with the presence of oxygen, a method of making, and uses of immobilized selenium in a rechargeable battery | |
| Sangprasert et al. | Making use of the inherent nitrogen content of spent coffee grounds to create nanostructured activated carbon for supercapacitor and lithium-ion battery applications | |
| CN111415821A (zh) | 非水系锂蓄电元件的制造方法 | |
| US11870059B2 (en) | Immobilized selenium in a porous carbon with the presence of oxygen, a method of making, and uses of immobilized selenium in a rechargeable battery | |
| JP7545418B2 (ja) | 炭素質材料およびその製造方法、電気二重層キャパシタ用電極材料 | |
| EP3025360A1 (en) | Electrodes of electrochemical double layer capacitor containing co2 activated coconut char | |
| JP2025100629A (ja) | 炭素質材料、その製造方法、電気二重層キャパシタ用電極活物質、電気二重層キャパシタ用電極および電気二重層キャパシタ | |
| US20250125356A1 (en) | Carbonaceous material, method for producing same, electrode active material for electrochemical device, electrode for electrochemical device, and electrochemical device | |
| JP2013080780A (ja) | 非水系リチウム型蓄電素子用負極材料、及びそれを用いた非水系リチウム型蓄電素子 | |
| Thumkaew et al. | Sugarcane waste-derived activated carbon for lithium-sulfur batteries with enhanced performance by thiourea doping | |
| WO2022168847A1 (ja) | 非水電解質二次電池用正極添加剤、それを含む非水電解質二次電池用正極活物質組成物、非水電解質二次電池用正極およびこれを備える非水電解質二次電池 | |
| WO2024257756A1 (ja) | 活性炭 | |
| WO2021154332A1 (en) | High-performance supercapacitors from biomass-derived carbon | |
| WO2025178126A1 (ja) | 炭素質材料、電極材料、電極、蓄電デバイス、および炭素質材料の製造方法 | |
| JP5367974B2 (ja) | 電気二重層キャパシタ用電極材料、および該電極材料用添加材 | |
| US12215030B2 (en) | Carbonaceous material, method for producing same, electrode active material for electrochemical device, electrode for electrochemical device, and electrochemical device | |
| JP2009158532A (ja) | 炭素質電極材およびこれを用いた蓄電装置 | |
| JP2006100725A (ja) | 電気二重層キャパシタ用有機電解液及び電気二重層キャパシタ | |
| JP2024011759A (ja) | 非水系リチウム蓄電素子 | |
| JP2016056041A (ja) | 窒素含有炭素材料の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24823372 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2025527935 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2025527935 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 1020257033895 Country of ref document: KR Free format text: ST27 STATUS EVENT CODE: A-0-1-A10-A15-NAP-PA0105 (AS PROVIDED BY THE NATIONAL OFFICE) |
|
| WWE | Wipo information: entry into national phase |
Ref document number: KR1020257033895 Country of ref document: KR Ref document number: 1020257033895 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020257033895 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202517128294 Country of ref document: IN |
|
| WWP | Wipo information: published in national office |
Ref document number: 202517128294 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2024823372 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2024823372 Country of ref document: EP Effective date: 20260115 |
|
| ENP | Entry into the national phase |
Ref document number: 2024823372 Country of ref document: EP Effective date: 20260115 |
|
| ENP | Entry into the national phase |
Ref document number: 2024823372 Country of ref document: EP Effective date: 20260115 |
|
| ENP | Entry into the national phase |
Ref document number: 2024823372 Country of ref document: EP Effective date: 20260115 |
|
| WWP | Wipo information: published in national office |
Ref document number: 2024823372 Country of ref document: EP |