WO2024024938A1 - デラマニド含有固体分散体 - Google Patents
デラマニド含有固体分散体 Download PDFInfo
- Publication number
- WO2024024938A1 WO2024024938A1 PCT/JP2023/027723 JP2023027723W WO2024024938A1 WO 2024024938 A1 WO2024024938 A1 WO 2024024938A1 JP 2023027723 W JP2023027723 W JP 2023027723W WO 2024024938 A1 WO2024024938 A1 WO 2024024938A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- component
- solid dispersion
- dispersion according
- range
- mass ratio
- Prior art date
Links
- 239000007962 solid dispersion Substances 0.000 title claims abstract description 97
- XDAOLTSRNUSPPH-XMMPIXPASA-N delamanid Chemical compound C([C@]1(C)OC2=NC(=CN2C1)[N+]([O-])=O)OC(C=C1)=CC=C1N(CC1)CCC1OC1=CC=C(OC(F)(F)F)C=C1 XDAOLTSRNUSPPH-XMMPIXPASA-N 0.000 title abstract description 21
- 229960003496 delamanid Drugs 0.000 title abstract description 20
- 229920003132 hydroxypropyl methylcellulose phthalate Polymers 0.000 claims abstract description 23
- 229940031704 hydroxypropyl methylcellulose phthalate Drugs 0.000 claims abstract description 21
- 239000002202 Polyethylene glycol Substances 0.000 claims abstract description 17
- 229920001223 polyethylene glycol Polymers 0.000 claims abstract description 17
- 229920000578 graft copolymer Polymers 0.000 claims abstract description 13
- 229920001577 copolymer Polymers 0.000 claims abstract description 12
- 229920002554 vinyl polymer Polymers 0.000 claims abstract description 7
- GVJHHUAWPYXKBD-UHFFFAOYSA-N (±)-α-Tocopherol Chemical compound OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 claims description 46
- -1 acetate-polyethylene Chemical group 0.000 claims description 21
- 239000000203 mixture Substances 0.000 claims description 17
- 239000008194 pharmaceutical composition Substances 0.000 claims description 16
- 229930003427 Vitamin E Natural products 0.000 claims description 15
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 claims description 15
- 229960000984 tocofersolan Drugs 0.000 claims description 15
- 229940046009 vitamin E Drugs 0.000 claims description 15
- 235000019165 vitamin E Nutrition 0.000 claims description 15
- 239000011709 vitamin E Substances 0.000 claims description 15
- 239000011627 DL-alpha-tocopherol Substances 0.000 claims description 14
- 235000001815 DL-alpha-tocopherol Nutrition 0.000 claims description 14
- 238000002360 preparation method Methods 0.000 claims description 14
- 238000004519 manufacturing process Methods 0.000 claims description 13
- 238000010438 heat treatment Methods 0.000 claims description 10
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 claims description 10
- 239000007787 solid Substances 0.000 claims description 9
- 239000002775 capsule Substances 0.000 claims description 8
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 claims description 8
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 claims description 8
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 claims description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 claims description 2
- 150000003900 succinic acid esters Chemical class 0.000 claims 1
- AOBORMOPSGHCAX-DGHZZKTQSA-N tocofersolan Chemical group OCCOC(=O)CCC(=O)OC1=C(C)C(C)=C2O[C@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C AOBORMOPSGHCAX-DGHZZKTQSA-N 0.000 claims 1
- 150000001875 compounds Chemical class 0.000 abstract description 11
- 229920000639 hydroxypropylmethylcellulose acetate succinate Polymers 0.000 abstract description 7
- 229920002689 polyvinyl acetate Polymers 0.000 abstract description 3
- 239000011118 polyvinyl acetate Substances 0.000 abstract description 3
- JBKVHLHDHHXQEQ-UHFFFAOYSA-N epsilon-caprolactam Chemical compound O=C1CCCCCN1 JBKVHLHDHHXQEQ-UHFFFAOYSA-N 0.000 abstract 2
- 239000002245 particle Substances 0.000 description 24
- GVJHHUAWPYXKBD-IEOSBIPESA-N α-tocopherol Chemical group OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 description 19
- 238000000034 method Methods 0.000 description 17
- 239000003826 tablet Substances 0.000 description 17
- 239000000047 product Substances 0.000 description 14
- 239000000654 additive Substances 0.000 description 12
- 238000009472 formulation Methods 0.000 description 10
- 239000003963 antioxidant agent Substances 0.000 description 7
- 235000006708 antioxidants Nutrition 0.000 description 7
- 230000000052 comparative effect Effects 0.000 description 7
- 238000009474 hot melt extrusion Methods 0.000 description 7
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 6
- TVXBFESIOXBWNM-UHFFFAOYSA-N Xylitol Natural products OCCC(O)C(O)C(O)CCO TVXBFESIOXBWNM-UHFFFAOYSA-N 0.000 description 6
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 6
- 238000000354 decomposition reaction Methods 0.000 description 6
- 239000008187 granular material Substances 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- HEBKCHPVOIAQTA-UHFFFAOYSA-N meso ribitol Natural products OCC(O)C(O)C(O)CO HEBKCHPVOIAQTA-UHFFFAOYSA-N 0.000 description 6
- 239000002994 raw material Substances 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 239000000811 xylitol Substances 0.000 description 6
- 235000010447 xylitol Nutrition 0.000 description 6
- 229960002675 xylitol Drugs 0.000 description 6
- HEBKCHPVOIAQTA-SCDXWVJYSA-N xylitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)CO HEBKCHPVOIAQTA-SCDXWVJYSA-N 0.000 description 6
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 5
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 5
- 239000004480 active ingredient Substances 0.000 description 5
- 230000003078 antioxidant effect Effects 0.000 description 5
- 239000007894 caplet Substances 0.000 description 5
- 238000004898 kneading Methods 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- SPSPIUSUWPLVKD-UHFFFAOYSA-N 2,3-dibutyl-6-methylphenol Chemical compound CCCCC1=CC=C(C)C(O)=C1CCCC SPSPIUSUWPLVKD-UHFFFAOYSA-N 0.000 description 4
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 4
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 4
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 4
- 239000004698 Polyethylene Substances 0.000 description 4
- 229930006000 Sucrose Natural products 0.000 description 4
- 210000004369 blood Anatomy 0.000 description 4
- 239000008280 blood Substances 0.000 description 4
- 238000001125 extrusion Methods 0.000 description 4
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 4
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 4
- 235000010355 mannitol Nutrition 0.000 description 4
- 238000002844 melting Methods 0.000 description 4
- 230000008018 melting Effects 0.000 description 4
- 239000011812 mixed powder Substances 0.000 description 4
- 238000000465 moulding Methods 0.000 description 4
- 229920000573 polyethylene Polymers 0.000 description 4
- 229920000642 polymer Polymers 0.000 description 4
- 229950008882 polysorbate Drugs 0.000 description 4
- 229920000136 polysorbate Polymers 0.000 description 4
- 239000012488 sample solution Substances 0.000 description 4
- 210000002966 serum Anatomy 0.000 description 4
- 229960002920 sorbitol Drugs 0.000 description 4
- 239000000454 talc Substances 0.000 description 4
- 229910052623 talc Inorganic materials 0.000 description 4
- 229940033134 talc Drugs 0.000 description 4
- 235000012222 talc Nutrition 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 4
- IUVCFHHAEHNCFT-INIZCTEOSA-N 2-[(1s)-1-[4-amino-3-(3-fluoro-4-propan-2-yloxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]ethyl]-6-fluoro-3-(3-fluorophenyl)chromen-4-one Chemical compound C1=C(F)C(OC(C)C)=CC=C1C(C1=C(N)N=CN=C11)=NN1[C@@H](C)C1=C(C=2C=C(F)C=CC=2)C(=O)C2=CC(F)=CC=C2O1 IUVCFHHAEHNCFT-INIZCTEOSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 3
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 3
- 241000282472 Canis lupus familiaris Species 0.000 description 3
- ZAKOWWREFLAJOT-CEFNRUSXSA-N D-alpha-tocopherylacetate Chemical compound CC(=O)OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C ZAKOWWREFLAJOT-CEFNRUSXSA-N 0.000 description 3
- 229930195725 Mannitol Natural products 0.000 description 3
- RVGRUAULSDPKGF-UHFFFAOYSA-N Poloxamer Chemical compound C1CO1.CC1CO1 RVGRUAULSDPKGF-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- XTXRWKRVRITETP-UHFFFAOYSA-N Vinyl acetate Chemical compound CC(=O)OC=C XTXRWKRVRITETP-UHFFFAOYSA-N 0.000 description 3
- 235000010724 Wisteria floribunda Nutrition 0.000 description 3
- 230000000996 additive effect Effects 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 235000014113 dietary fatty acids Nutrition 0.000 description 3
- 239000006185 dispersion Substances 0.000 description 3
- 239000000194 fatty acid Substances 0.000 description 3
- 229930195729 fatty acid Natural products 0.000 description 3
- 235000011187 glycerol Nutrition 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- JEIPFZHSYJVQDO-UHFFFAOYSA-N iron(III) oxide Inorganic materials O=[Fe]O[Fe]=O JEIPFZHSYJVQDO-UHFFFAOYSA-N 0.000 description 3
- YOBAEOGBNPPUQV-UHFFFAOYSA-N iron;trihydrate Chemical compound O.O.O.[Fe].[Fe] YOBAEOGBNPPUQV-UHFFFAOYSA-N 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 239000000594 mannitol Substances 0.000 description 3
- 229960001855 mannitol Drugs 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 239000004014 plasticizer Substances 0.000 description 3
- 229960000502 poloxamer Drugs 0.000 description 3
- 229920001983 poloxamer Polymers 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 238000000634 powder X-ray diffraction Methods 0.000 description 3
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 235000010356 sorbitol Nutrition 0.000 description 3
- 229940032147 starch Drugs 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 238000003860 storage Methods 0.000 description 3
- 239000004094 surface-active agent Substances 0.000 description 3
- 239000006188 syrup Substances 0.000 description 3
- 235000020357 syrup Nutrition 0.000 description 3
- 229940042585 tocopherol acetate Drugs 0.000 description 3
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 3
- 229920003169 water-soluble polymer Polymers 0.000 description 3
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 2
- 235000001809 DL-alpha-tocopherylacetate Nutrition 0.000 description 2
- 239000011626 DL-alpha-tocopherylacetate Substances 0.000 description 2
- 229920000881 Modified starch Polymers 0.000 description 2
- 241000187479 Mycobacterium tuberculosis Species 0.000 description 2
- 239000008118 PEG 6000 Substances 0.000 description 2
- 229920002584 Polyethylene Glycol 6000 Polymers 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- MSCCTZZBYHQMQJ-AZAGJHQNSA-N Tocopheryl nicotinate Chemical compound C([C@@](OC1=C(C)C=2C)(C)CCC[C@H](C)CCC[C@H](C)CCCC(C)C)CC1=C(C)C=2OC(=O)C1=CC=CN=C1 MSCCTZZBYHQMQJ-AZAGJHQNSA-N 0.000 description 2
- 238000002835 absorbance Methods 0.000 description 2
- VJHCJDRQFCCTHL-UHFFFAOYSA-N acetic acid 2,3,4,5,6-pentahydroxyhexanal Chemical compound CC(O)=O.OCC(O)C(O)C(O)C(O)C=O VJHCJDRQFCCTHL-UHFFFAOYSA-N 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 229920001400 block copolymer Polymers 0.000 description 2
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 2
- 239000008116 calcium stearate Substances 0.000 description 2
- 235000013539 calcium stearate Nutrition 0.000 description 2
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 2
- 229950008138 carmellose Drugs 0.000 description 2
- 239000004359 castor oil Substances 0.000 description 2
- 235000019438 castor oil Nutrition 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 238000007334 copolymerization reaction Methods 0.000 description 2
- 239000007884 disintegrant Substances 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 239000003814 drug Substances 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 239000007941 film coated tablet Substances 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 2
- 229960003943 hypromellose Drugs 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 239000004570 mortar (masonry) Substances 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 239000008188 pellet Substances 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 2
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 238000010298 pulverizing process Methods 0.000 description 2
- 229920005604 random copolymer Polymers 0.000 description 2
- 235000012239 silicon dioxide Nutrition 0.000 description 2
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- 238000005063 solubilization Methods 0.000 description 2
- 230000007928 solubilization Effects 0.000 description 2
- 239000000243 solution Substances 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 150000005846 sugar alcohols Chemical class 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 2
- URAYPUMNDPQOKB-UHFFFAOYSA-N triacetin Chemical compound CC(=O)OCC(OC(C)=O)COC(C)=O URAYPUMNDPQOKB-UHFFFAOYSA-N 0.000 description 2
- 239000002076 α-tocopherol Substances 0.000 description 2
- 235000004835 α-tocopherol Nutrition 0.000 description 2
- QIJRTFXNRTXDIP-UHFFFAOYSA-N (1-carboxy-2-sulfanylethyl)azanium;chloride;hydrate Chemical compound O.Cl.SCC(N)C(O)=O QIJRTFXNRTXDIP-UHFFFAOYSA-N 0.000 description 1
- JLPULHDHAOZNQI-ZTIMHPMXSA-N 1-hexadecanoyl-2-(9Z,12Z-octadecadienoyl)-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCC\C=C/C\C=C/CCCCC JLPULHDHAOZNQI-ZTIMHPMXSA-N 0.000 description 1
- OALHHIHQOFIMEF-UHFFFAOYSA-N 3',6'-dihydroxy-2',4',5',7'-tetraiodo-3h-spiro[2-benzofuran-1,9'-xanthene]-3-one Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC(I)=C(O)C(I)=C1OC1=C(I)C(O)=C(I)C=C21 OALHHIHQOFIMEF-UHFFFAOYSA-N 0.000 description 1
- IELOKBJPULMYRW-IKTKBOKFSA-N 4-oxo-4-[[(2S)-2,5,7,8-tetramethyl-2-[(4S,8S)-4,8,12-trimethyltridecyl]-3,4-dihydrochromen-6-yl]oxy]butanoic acid Chemical group CC(C)CCC[C@H](C)CCC[C@H](C)CCC[C@@](C)(CC1)Oc(c(C)c2C)c1c(C)c2OC(CCC(O)=O)=O IELOKBJPULMYRW-IKTKBOKFSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 108010011485 Aspartame Proteins 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- GUBGYTABKSRVRQ-DCSYEGIMSA-N Beta-Lactose Chemical compound OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-DCSYEGIMSA-N 0.000 description 1
- SGHZXLIDFTYFHQ-UHFFFAOYSA-L Brilliant Blue Chemical compound [Na+].[Na+].C=1C=C(C(=C2C=CC(C=C2)=[N+](CC)CC=2C=C(C=CC=2)S([O-])(=O)=O)C=2C(=CC=CC=2)S([O-])(=O)=O)C=CC=1N(CC)CC1=CC=CC(S([O-])(=O)=O)=C1 SGHZXLIDFTYFHQ-UHFFFAOYSA-L 0.000 description 1
- 239000004255 Butylated hydroxyanisole Substances 0.000 description 1
- MCRNHLQVPJEMSQ-UHFFFAOYSA-N C(C=CC(=O)O)(=O)O.C(CCCCCCCCCCCCCCCCC)[Na] Chemical compound C(C=CC(=O)O)(=O)O.C(CCCCCCCCCCCCCCCCC)[Na] MCRNHLQVPJEMSQ-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- PTHCMJGKKRQCBF-UHFFFAOYSA-N Cellulose, microcrystalline Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC)C(CO)O1 PTHCMJGKKRQCBF-UHFFFAOYSA-N 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 229920002785 Croscarmellose sodium Polymers 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- IELOKBJPULMYRW-NJQVLOCASA-N D-alpha-Tocopheryl Acid Succinate Chemical compound OC(=O)CCC(=O)OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C IELOKBJPULMYRW-NJQVLOCASA-N 0.000 description 1
- CIWBSHSKHKDKBQ-DUZGATOHSA-N D-araboascorbic acid Natural products OC[C@@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-DUZGATOHSA-N 0.000 description 1
- GZIFEOYASATJEH-UHFFFAOYSA-N D-delta tocopherol Natural products OC1=CC(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1 GZIFEOYASATJEH-UHFFFAOYSA-N 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 239000004386 Erythritol Substances 0.000 description 1
- UNXHWFMMPAWVPI-UHFFFAOYSA-N Erythritol Natural products OCC(O)C(O)CO UNXHWFMMPAWVPI-UHFFFAOYSA-N 0.000 description 1
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 1
- 229930091371 Fructose Natural products 0.000 description 1
- 239000005715 Fructose Substances 0.000 description 1
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- QAQJMLQRFWZOBN-LAUBAEHRSA-N L-ascorbyl-6-palmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](O)[C@H]1OC(=O)C(O)=C1O QAQJMLQRFWZOBN-LAUBAEHRSA-N 0.000 description 1
- 239000011786 L-ascorbyl-6-palmitate Substances 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- WINXNKPZLFISPD-UHFFFAOYSA-M Saccharin sodium Chemical compound [Na+].C1=CC=C2C(=O)[N-]S(=O)(=O)C2=C1 WINXNKPZLFISPD-UHFFFAOYSA-M 0.000 description 1
- 102100025490 Slit homolog 1 protein Human genes 0.000 description 1
- 101710123186 Slit homolog 1 protein Proteins 0.000 description 1
- 102100027340 Slit homolog 2 protein Human genes 0.000 description 1
- 101710133576 Slit homolog 2 protein Proteins 0.000 description 1
- 239000004376 Sucralose Substances 0.000 description 1
- QEKBRBCVWVLFHH-QAKUKHITSA-L Tocopherol calcium succinate Chemical compound [Ca+2].[O-]C(=O)CCC(=O)OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C.[O-]C(=O)CCC(=O)OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C QEKBRBCVWVLFHH-QAKUKHITSA-L 0.000 description 1
- DOOTYTYQINUNNV-UHFFFAOYSA-N Triethyl citrate Chemical compound CCOC(=O)CC(O)(C(=O)OCC)CC(=O)OCC DOOTYTYQINUNNV-UHFFFAOYSA-N 0.000 description 1
- YKTSYUJCYHOUJP-UHFFFAOYSA-N [O--].[Al+3].[Al+3].[O-][Si]([O-])([O-])[O-] Chemical compound [O--].[Al+3].[Al+3].[O-][Si]([O-])([O-])[O-] YKTSYUJCYHOUJP-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- ZUAAPNNKRHMPKG-UHFFFAOYSA-N acetic acid;butanedioic acid;methanol;propane-1,2-diol Chemical compound OC.CC(O)=O.CC(O)CO.OC(=O)CCC(O)=O ZUAAPNNKRHMPKG-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 229940087168 alpha tocopherol Drugs 0.000 description 1
- 229920005603 alternating copolymer Polymers 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- WMGSQTMJHBYJMQ-UHFFFAOYSA-N aluminum;magnesium;silicate Chemical compound [Mg+2].[Al+3].[O-][Si]([O-])([O-])[O-] WMGSQTMJHBYJMQ-UHFFFAOYSA-N 0.000 description 1
- WLDHEUZGFKACJH-UHFFFAOYSA-K amaranth Chemical compound [Na+].[Na+].[Na+].C12=CC=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(O)=C1N=NC1=CC=C(S([O-])(=O)=O)C2=CC=CC=C12 WLDHEUZGFKACJH-UHFFFAOYSA-K 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 229960004977 anhydrous lactose Drugs 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 235000010385 ascorbyl palmitate Nutrition 0.000 description 1
- 239000000605 aspartame Substances 0.000 description 1
- 235000010357 aspartame Nutrition 0.000 description 1
- IAOZJIPTCAWIRG-QWRGUYRKSA-N aspartame Chemical compound OC(=O)C[C@H](N)C(=O)N[C@H](C(=O)OC)CC1=CC=CC=C1 IAOZJIPTCAWIRG-QWRGUYRKSA-N 0.000 description 1
- 229960003438 aspartame Drugs 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 229940067573 brown iron oxide Drugs 0.000 description 1
- 235000019282 butylated hydroxyanisole Nutrition 0.000 description 1
- 229940043253 butylated hydroxyanisole Drugs 0.000 description 1
- CZBZUDVBLSSABA-UHFFFAOYSA-N butylated hydroxyanisole Chemical compound COC1=CC=C(O)C(C(C)(C)C)=C1.COC1=CC=C(O)C=C1C(C)(C)C CZBZUDVBLSSABA-UHFFFAOYSA-N 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- FUFJGUQYACFECW-UHFFFAOYSA-L calcium hydrogenphosphate Chemical compound [Ca+2].OP([O-])([O-])=O FUFJGUQYACFECW-UHFFFAOYSA-L 0.000 description 1
- 239000000378 calcium silicate Substances 0.000 description 1
- 229910052918 calcium silicate Inorganic materials 0.000 description 1
- OYACROKNLOSFPA-UHFFFAOYSA-N calcium;dioxido(oxo)silane Chemical compound [Ca+2].[O-][Si]([O-])=O OYACROKNLOSFPA-UHFFFAOYSA-N 0.000 description 1
- 229960001777 castor oil Drugs 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 239000007910 chewable tablet Substances 0.000 description 1
- 229960004106 citric acid Drugs 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 238000000975 co-precipitation Methods 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 238000010924 continuous production Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 229960001681 croscarmellose sodium Drugs 0.000 description 1
- 229960000913 crospovidone Drugs 0.000 description 1
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 1
- 229960001305 cysteine hydrochloride Drugs 0.000 description 1
- 229940099418 d- alpha-tocopherol succinate Drugs 0.000 description 1
- 238000004925 denaturation Methods 0.000 description 1
- 230000036425 denaturation Effects 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 235000019700 dicalcium phosphate Nutrition 0.000 description 1
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 1
- VZFDRQUWHOVFCA-UHFFFAOYSA-L disodium;2-sulfanylbutanedioate Chemical compound [Na+].[Na+].[O-]C(=O)CC(S)C([O-])=O VZFDRQUWHOVFCA-UHFFFAOYSA-L 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 229940088679 drug related substance Drugs 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 235000010350 erythorbic acid Nutrition 0.000 description 1
- 239000004318 erythorbic acid Substances 0.000 description 1
- UNXHWFMMPAWVPI-ZXZARUISSA-N erythritol Chemical compound OC[C@H](O)[C@H](O)CO UNXHWFMMPAWVPI-ZXZARUISSA-N 0.000 description 1
- 235000019414 erythritol Nutrition 0.000 description 1
- 229940009714 erythritol Drugs 0.000 description 1
- 239000007888 film coating Substances 0.000 description 1
- 238000009501 film coating Methods 0.000 description 1
- 239000003205 fragrance Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 238000012812 general test Methods 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000001087 glyceryl triacetate Substances 0.000 description 1
- 235000013773 glyceryl triacetate Nutrition 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 229940026239 isoascorbic acid Drugs 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 229940031703 low substituted hydroxypropyl cellulose Drugs 0.000 description 1
- 229960003511 macrogol Drugs 0.000 description 1
- HCWCAKKEBCNQJP-UHFFFAOYSA-N magnesium orthosilicate Chemical compound [Mg+2].[Mg+2].[O-][Si]([O-])([O-])[O-] HCWCAKKEBCNQJP-UHFFFAOYSA-N 0.000 description 1
- 239000000395 magnesium oxide Substances 0.000 description 1
- CPLXHLVBOLITMK-UHFFFAOYSA-N magnesium oxide Inorganic materials [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 1
- 235000012245 magnesium oxide Nutrition 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 235000019792 magnesium silicate Nutrition 0.000 description 1
- 229910052919 magnesium silicate Inorganic materials 0.000 description 1
- AXZKOIWUVFPNLO-UHFFFAOYSA-N magnesium;oxygen(2-) Chemical compound [O-2].[Mg+2] AXZKOIWUVFPNLO-UHFFFAOYSA-N 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 238000000691 measurement method Methods 0.000 description 1
- 150000004667 medium chain fatty acids Chemical class 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 229920000191 poly(N-vinyl pyrrolidone) Polymers 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 229940068984 polyvinyl alcohol Drugs 0.000 description 1
- 235000013809 polyvinylpolypyrrolidone Nutrition 0.000 description 1
- 229920000523 polyvinylpolypyrrolidone Polymers 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 229960004063 propylene glycol Drugs 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- 239000008213 purified water Substances 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 1
- 238000010008 shearing Methods 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000010288 sodium nitrite Nutrition 0.000 description 1
- 229960000819 sodium nitrite Drugs 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229940001482 sodium sulfite Drugs 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- GNBVPFITFYNRCN-UHFFFAOYSA-M sodium thioglycolate Chemical compound [Na+].[O-]C(=O)CS GNBVPFITFYNRCN-UHFFFAOYSA-M 0.000 description 1
- 229940046307 sodium thioglycolate Drugs 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 229940083466 soybean lecithin Drugs 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000001694 spray drying Methods 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 235000019408 sucralose Nutrition 0.000 description 1
- BAQAVOSOZGMPRM-QBMZZYIRSA-N sucralose Chemical compound O[C@@H]1[C@@H](O)[C@@H](Cl)[C@@H](CO)O[C@@H]1O[C@@]1(CCl)[C@@H](O)[C@H](O)[C@@H](CCl)O1 BAQAVOSOZGMPRM-QBMZZYIRSA-N 0.000 description 1
- 239000007940 sugar coated tablet Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000007939 sustained release tablet Substances 0.000 description 1
- 239000012085 test solution Substances 0.000 description 1
- 229960002622 triacetin Drugs 0.000 description 1
- 239000001069 triethyl citrate Substances 0.000 description 1
- VMYFZRTXGLUXMZ-UHFFFAOYSA-N triethyl citrate Natural products CCOC(=O)C(O)(C(=O)OCC)C(=O)OCC VMYFZRTXGLUXMZ-UHFFFAOYSA-N 0.000 description 1
- 235000013769 triethyl citrate Nutrition 0.000 description 1
- UFTFJSFQGQCHQW-UHFFFAOYSA-N triformin Chemical compound O=COCC(OC=O)COC=O UFTFJSFQGQCHQW-UHFFFAOYSA-N 0.000 description 1
- UJMBCXLDXJUMFB-UHFFFAOYSA-K trisodium;5-oxo-1-(4-sulfonatophenyl)-4-[(4-sulfonatophenyl)diazenyl]-4h-pyrazole-3-carboxylate Chemical compound [Na+].[Na+].[Na+].[O-]C(=O)C1=NN(C=2C=CC(=CC=2)S([O-])(=O)=O)C(=O)C1N=NC1=CC=C(S([O-])(=O)=O)C=C1 UJMBCXLDXJUMFB-UHFFFAOYSA-K 0.000 description 1
- 238000001195 ultra high performance liquid chromatography Methods 0.000 description 1
- GZIFEOYASATJEH-VHFRWLAGSA-N δ-tocopherol Chemical compound OC1=CC(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1 GZIFEOYASATJEH-VHFRWLAGSA-N 0.000 description 1
Images
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/22—Heterocyclic compounds, e.g. ascorbic acid, tocopherol or pyrrolidones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/32—Macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. carbomers, poly(meth)acrylates, or polyvinyl pyrrolidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
- A61K47/38—Cellulose; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
- A61P31/06—Antibacterial agents for tuberculosis
Definitions
- the present invention relates to a solid dispersion containing delamanid, a method for producing the solid dispersion, and the like.
- all documents described in this specification are incorporated herein by reference.
- Delamanid has the following formula: (2R)-2-methyl-6-nitro-2-[(4- ⁇ 4-[4-(trifluoromethoxy)phenoxy]piperidin-1-yl ⁇ phenoxy)methyl]-2, 3-dihydroimidazo[2,1-b]oxazole. Delamanid has excellent bactericidal activity against Mycobacterium tuberculosis, multidrug-resistant Mycobacterium tuberculosis, and atypical acid-fast bacteria (Patent Document 1). Moreover, delamanid is known as a poorly water-soluble compound with low solubility in water, and studies have been made to improve solubility and oral absorption (Patent Documents 2 and 3).
- Solubilization methods to improve the solubility of poorly water-soluble compounds include microparticulation (nanoparticles), metastable forms (crystalline polymorphs), salts, cocrystals, amorphous forms, surfactants, oils, etc.
- a variety of techniques have been used, including solubilization.
- the amorphous form is a highly practical formulation technology, and one approach to improving the instability of the amorphous form is solid dispersion technology in which it is complexed with a carrier such as a polymer.
- a solid dispersion is a "dispersion of one or more active ingredients in a solid, inert carrier or matrix thereof.”
- Methods for producing solid dispersions include coprecipitation, spray drying, heated melt extrusion, and other methods.
- Hot melt extrusion is a continuous process (easily scaled up), a solvent-free method (useful from an environmental and cost perspective as no solvent treatment is required and no solvent residue), and different drug delivery systems. (including granules, pellets, sustained release tablets, suppositories, stents, ophthalmic inserts, and transdermal and transmucosal delivery systems).
- process parameters in HME depend on thermodynamic and rheological properties, high processing temperatures are generally required, and there is a risk of thermal deterioration of the drug. There are issues such as the concern that crystalline drugs may remain if the concentration is lowered.
- Delamanide is a compound with poor thermal stability (melting point: approximately 195°C (decomposition)), and as its melting point and decomposition point are close to each other, decomposition products are generated in HME, which requires high-temperature treatment, and its production is considered difficult. .
- One object of the present invention is to provide a solid dispersion containing delamanid, a method for producing the solid dispersion, a pharmaceutical composition containing the solid dispersion, and an oral solid preparation.
- the present invention includes, for example, the subject matter described in the following sections.
- Item 1. A solid dispersion containing the following components (a), (b), and (c).
- (a) Delamanide (b) At least one member selected from the group consisting of polyvinylcaprolactam-polyvinylacetate-polyethylene glycol graft copolymer and vinylpyrrolidone-vinyl acetate copolymer (c) Hydroxypropylmethylcellulose phthalate and hydroxypropylmethylcellulose acetate At least one species selected from the group consisting of succinate2.
- Item 2. The solid dispersion according to Item 1, wherein the mass ratio of component (a) to component (b) is within the range of 1:1 to 1:20.
- Item 3. Item 3.
- component (b) comprises a vinylpyrrolidone-vinyl acetate copolymer.
- Item 11 The solid dispersion according to any one of Items 1 to 10, wherein component (c) comprises hydroxypropyl methylcellulose phthalate.
- Item 12. Item 12. The solid dispersion according to any one of Items 1 to 11, wherein component (b) comprises a polyvinylcaprolactam-polyvinylacetate-polyethylene glycol graft copolymer and component (c) comprises hydroxypropyl methylcellulose phthalate.
- Item 13 13.
- An oral solid preparation comprising the solid dispersion according to any one of Items 1 to 16.
- Item 19. The oral solid preparation according to item 18, which is a tablet or capsule.
- Section 20. Item 17.
- Section 21. Item 17.
- Section 22. Item 17.
- Section 24. 13 The solid dispersion according to item 9, 11, or 12, wherein the mass ratio of component (a), component (b), and component (c) is about 1:4:3.
- Section 25. 14 The solid dispersion according to item 10, 11, or 13, wherein the mass ratio of component (a), component (b), and component (c) is about 1:2:2.
- the solid dispersion containing delamanide included in the present invention has excellent solubility and the like. Further, according to the method for producing a delamanide solid dispersion included in the present invention, it is possible to produce the solid dispersion with less burden on the environment and at low cost.
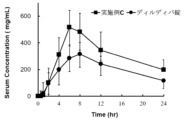
- FIG. 1 is a graph showing the change in serum concentration over time after administering a capsule containing the solid dispersion of Example C to a dog.
- the solid dispersion included in the present invention includes (a) delamanid, (b) polyvinylcaprolactam-polyvinyl acetate-polyethylene glycol graft copolymer and vinylpyrrolidone. - at least one selected from the group consisting of vinyl acetate copolymers, and (c) at least one selected from the group consisting of hydroxypropyl methyl cellulose phthalate and hydroxypropyl methyl cellulose acetate succinate.
- delamanid is preferably in microcrystalline or amorphous form. Specifically, it is preferable that no crystalline peak exists in the powder X-ray diffraction pattern. Powder X-ray diffraction can be measured according to "General Test Methods 2.58 Powder X-ray Diffraction Measurement Method" of the Japanese Pharmacopoeia (18th revision), and may be measured, for example, under the following conditions.
- Measuring device X'Pert PRO MPD (Spectris Co., Ltd.)
- Optical system Concentrated optical system (transmission method) Goniometer radius: 240 mm Tube voltage, tube current: 45 kV, 40 mA Incident side slit: solar slit, solar 0.04 rad Divergence slit 1/2 deg
- Light receiving side slit Solar slit large solar 0.04 rad Anti-scatter slit 5.5 mm Measurement range: 2 ⁇ 3 ⁇ 40 deg Operation speed: 1.11 deg/sec.
- Sampling interval 0.02 deg/step Wobbled scan: Step count 5, step size 0.02 deg
- Delamanid can be produced by a conventional method, for example, the method described in JP-A No. 2004-149527, US Patent Application Publication No. 2006/094767, and the like.
- Component (b) The present inventors have discovered that by combining component (a) and component (b), the two components become compatible, making heating melt-kneading possible, ensuring fluidity in the melt-kneading machine, and solid dispersion. It has been found that a solid dispersion can be obtained which can realize good manufacturability and has a solubility higher than that of delamanide drug substance (jet mill pulverized product).
- the polyvinylcaprolactam-polyvinyl acetate-polyethylene glycol graft copolymer contains at least one polyvinylcaprolactam block and at least one polyvinyl acetate block. , at least one polyethylene glycol block.
- the polyethylene glycol can be, for example, PEG6000.
- the ratio of polyvinylcaprolactam blocks/polyvinyl acetate blocks/polyethylene glycol blocks may be, for example, about 57/30/13.
- component (b1) has the following formula: (In the formula, l, m, and n are each independently an integer of 1 or more.) It may also be a copolymer represented by
- the average molecular weight of component (b1) may be, for example, 10,000 g/mol or more, 50,000 g/mol or more, or 90,000 g/mol or more.
- the average molecular weight of component (b1) can be, for example, 1,000,000 g/mol or less, 500,000 g/mol or less, 200,000 g/mol or less, or 140,000 g/mol or less.
- the average molecular weight of component (b1) may be within the range of any combination of the lower limit and the upper limit, for example within the range of 10,000 to 1,000,000 g/mol, preferably within the range of 90,000 to 140,000 g/mol.
- component (b1) preferably comprises SoluplusTM from BASF.
- the vinylpyrrolidone-vinyl acetate copolymer (hereinafter sometimes referred to as component (b2)) is a polymer of at least vinylpyrrolidone (usually 1-vinyl-2-pyrrolidone) and vinyl acetate.
- the copolymerization ratio (mass ratio) of vinyl pyrrolidone and vinyl acetate is, for example, within the range of 1:9 to 9:1, within the range of 2:8 to 8:2, or within the range of 3:7 to 7:3. It can be within.
- the copolymerization ratio (mass ratio) of vinyl pyrrolidone and vinyl acetate may be, for example, within the range of 4:6 to 8:2, or within the range of 5:5 to 7:3.
- Component (b2) is not particularly limited in the arrangement of monomers, and may be any of random copolymers, alternating copolymers, block copolymers, and graft copolymers. In one embodiment, component (b2) is preferably a random copolymer.
- the average molecular weight of component (b2) may be, for example, 10000 g/mol or more, 20000 g/mol or more, 30000 g/mol or more, 40000 g/mol or more, or 45000 g/mol or more.
- the average molecular weight of component (b2) can be, for example, 1000000 g/mol or less, 500000 g/mol or less, 100000 g/mol or less, or 70000 g/mol or less.
- the average molecular weight of component (b2) may be within the range of any combination of the lower limit and the upper limit, for example within the range of 10,000 to 1,000,000 g/mol, preferably within the range of 45,000 to 70,000 g/mol.
- component (b2) preferably comprises a copolypidone, for example KollidonTM VA64 from BASF.
- component (b) preferably includes component (b1). In another embodiment, component (b) preferably comprises component (b2). In yet another embodiment, component (b) is preferably a combination of components (b1) and (b2). In this combination, the mass ratio of component (b1) to component (b2) is not particularly limited, but is, for example, in the range of 0.5:1 to 10:1, preferably in the range of 1:1 to 6:1. It is within.
- Component (c) The present inventors have discovered that by further combining components (a) and (b) with component (c), a solid dispersion with even better solubility can be obtained.
- hydroxypropyl methyl cellulose phthalate (hereinafter sometimes referred to as component (c1)) is a polymer compound in which phthalic acid is half-ester bonded to hydroxypropyl cellulose.
- Hydroxypropyl methylcellulose phthalate is preferably "hypromellose phthalate" described in the monograph (chemicals, etc.) of the Japanese Pharmacopoeia (18th revision).
- Hydroxypropyl methylcellulose phthalate is commercially available, for example, from Shin-Etsu Chemical Co., Ltd. under the trade names HPMCP (trademark) HP-55, HP-55S, and HP-50. In the present invention, any of these commercially available products can be used.
- the hydroxypropyl methylcellulose phthalate is preferably substituted with at least 21 to 35% by weight (eg, 21 to 27% by weight, or 27 to 35% by weight) of carboxybenzoyl groups at its hydroxyl groups.
- the hydroxyl groups of the hydroxypropyl methylcellulose phthalate are preferably substituted with 18 to 24% by weight of methoxy groups, 5 to 10% by weight of hydroxypropoxy groups, and 21 to 35% by weight of carboxybenzoyl groups.
- hydroxypropyl methyl cellulose acetate succinate (hereinafter sometimes referred to as component (c2)) is a polymer compound in which acetic acid and succinic acid are ester-bonded to hydroxypropyl cellulose.
- Hydroxypropyl methylcellulose acetate succinate is preferably "hypromellose acetate succinate" described in the monograph (chemicals, etc.) of the Japanese Pharmacopoeia (18th revision). Hydroxypropyl methylcellulose acetate succinate is commercially available, for example, from Shin-Etsu Chemical Co., Ltd. under the trade names Shin-Etsu AQOAT (trademark) AS-LF, AS-MF, and AS-HF.
- Hydroxypropyl methylcellulose acetate succinate has its hydroxyl groups substituted with 20 to 26% by mass of methoxy groups, 5 to 10% by mass of hydroxypropoxy groups, 5 to 14% by mass of acetyl groups, and 4 to 18% by mass of succinoyl groups. is preferred.
- component (c) preferably includes component (c1) (for example, component (c1) alone or a combination of component (c1) and component (c2)).
- the content of component (a) may be, for example, 1% by mass or more, 5% by mass or more, or 10% by mass or more.
- the content of component (a) may be, for example, 30% by weight or less, 25% by weight or less, or 20% by weight or less.
- the content of component (a) may be, for example, within the range of any combination of the lower limit and the upper limit, for example, within the range of 1 to 30% by mass.
- the mass ratio of component (a) and component (b) is not particularly limited.
- the mass ratio of component (a) and component (b) may be less than 1:1 (for example, within the range of 1:0.1 to 1:0.9), but is usually 1:1 or more (
- the ratio of component (b)/component (a) is 1/1 or more), preferably 1:1.5 or more, more preferably 1:2 or more.
- the mass ratio of component (a) and component (b) is, for example, 1:20 or less (meaning that component (b)/component (a) is 20/1 or less), 1:15 or less, 1 :10 or less, 1:8 or less, or 1:5 or less.
- the mass ratio of component (a) and component (b) is within the range of any combination of the above lower limit and the above upper limit, for example, within the range of 1:1 to 1:20, preferably 1:1 to 1: 8, more preferably within the range of 1:1 to 1:5, still more preferably within the range of 1:2 to 1:5, even more preferably within the range of 1:2 to 1:4, Or about 1:2 or about 1:4.
- component (b) contains component (b1)
- the mass ratio of component (a) to component (b) is preferably about 1:4
- component (b) contains component (b2) Preferably, the mass ratio of component (a) to component (b) is about 1:2.
- the mass ratio of component (a) to component (c) may be less than 1:1 (for example, within the range of 1:0.1 to 1:0.9), but is usually 1:1 or more (
- the ratio of component (c)/component (a) is 1/1 or more), preferably 1:1.5 or more, more preferably 1:2 or more.
- the mass ratio of component (a) and component (c) is, for example, 1:10 or less (meaning that component (c)/component (a) is 10/1 or less), 1:8 or less, or
- the ratio may be 1:5 or less.
- the mass ratio of component (a) to component (c) is within the range of any combination of the above lower limit and the above upper limit, for example, within the range of 1:1 to 1:10, preferably 1:1 to 1: 8, more preferably within the range of 1:1 to 1:5, still more preferably within the range of 1:1 to 1:4, even more preferably within the range of 1:2 to 1:4.
- the ratio is particularly preferably about 1:2 or about 1:3.
- the mass ratio of component (a) to component (c) is preferably about 1:3, and when component (b) contains component (b2), Preferably, the mass ratio of component (a) to component (c) is about 1:2.
- the mass ratio of component (a) to component (b) to component (c) is preferably in the range of 1:1 to 8:1 to 8, more preferably 1:1 to 5. : within the range of 1 to 5, more preferably within the range of 1:1.5 to 5:1.5 to 5, even more preferably within the range of 1:2 to 4:2 to 4, or The ratio is about 1:2:2 or about 1:4:3.
- component (b) contains component (b1)
- the mass ratio of component (a), component (b), and component (c) is preferably about 1:4:3
- component (b) contains component (b1).
- the mass ratio of component (a), component (b), and component (c) is preferably about 1:2:2.
- the solid dispersion of the present invention may further contain other components.
- Other components include, for example, antioxidants, pH-independent water-soluble polymer compounds other than component (b), plasticizers, and the like.
- Other components can be used alone or in combination of two or more.
- antioxidants include vitamin E, dibutylated hydroxytoluene, butylated hydroxyanisole, soybean lecithin, ascorbyl palmitate, cysteine hydrochloride, ascorbic acid, citric acid, erythorbic acid, sodium nitrite, sodium sulfite, sodium thioglycolate, Examples include sodium thiomalate.
- the antioxidant preferably comprises vitamin E, and comprises vitamin E and dibutylhydroxytoluene (e.g., the weight ratio of vitamin E to dibutylhydroxytoluene is from 15:1 to 1:1). is also preferable. According to this embodiment, it is possible to suppress the generation of decomposition products during storage (particularly during storage at a high temperature such as about 60° C.).
- vitamin E examples include d- ⁇ -tocopherol, d- ⁇ -tocopherol, d- ⁇ -tocopherol acetate, d- ⁇ -tocopherol succinate, dl- ⁇ -tocopherol, dl- ⁇ -tocopherol acetate, and dl succinate.
- - contains natural and synthetic vitamin E such as ⁇ -tocopherol, dl- ⁇ -tocopherol calcium succinate, and dl- ⁇ -tocopherol nicotinate.
- Preferred vitamin E is dl- ⁇ -tocopherol, dl- ⁇ -tocopherol acetate, dl- ⁇ -tocopherol succinate and dl- ⁇ -tocopherol nicotinate, and a more preferred vitamin E is dl- ⁇ -tocopherol.
- the amount of the antioxidant (for example, vitamin E) added to the solid dispersion of the present invention is usually 0.001 part by mass or more, preferably 0.01 part by mass or more, more preferably 0.01 part by mass or more, per 1 part by mass of delamanide. is 0.03 parts by mass or more.
- the amount of the antioxidant (for example, vitamin E) is usually 1 part by mass or less, preferably 0.8 part by mass or less, more preferably 0.5 part by mass or less, per 1 part by mass of delamanide.
- the amount of the antioxidant is within the range of any combination of the lower limit and the upper limit, for example, usually within the range of 0.001 to 1 part by mass per 1 part by mass of delamanide, It is preferably within the range of 0.01 to 0.8 parts by weight, more preferably within the range of 0.03 to 0.5 parts by weight.
- pH-independent water-soluble polymer compounds other than component (b) include hydroxypropylcellulose, hydroxypropylmethylcellulose, polyvinylpyrrolidone, polyethylene glycol, and cyclodextrin.
- plasticizer examples include triethyl citrate, glycerin, glycerin fatty acid ester, medium chain fatty acid triglyceride, triacetin, castor oil, propylene glycol, polysorbate, and the like.
- component (b) can also be blended into the solid dispersion in the same amount as the antioxidant.
- component (b) such as a pH-independent water-soluble polymer compound and a plasticizer, can also be blended into the solid dispersion in the same amount as the antioxidant.
- the solid dispersion of the present invention preferably does not contain xylitol, more preferably does not contain xylitol, sorbitol, and mannitol, and even more preferably does not contain sugar alcohols.
- the solid dispersion of the present invention preferably does not contain a poloxamer (a block copolymer consisting of a polyoxypropylene chain and two polyoxyethylene chains sandwiching it).
- a poloxamer a block copolymer consisting of a polyoxypropylene chain and two polyoxyethylene chains sandwiching it.
- the solid dispersion of the present invention preferably does not contain xylitol, sorbitol, mannitol, and poloxamer.
- the solid dispersion of the present invention is preferably a heated melt extrudate or a pulverized product thereof, and is preferably produced by the production method described below.
- the pulverized material is usually a particle, and is sometimes called a solid dispersion particle, a molten granule, or the like.
- the size of the pulverized product is not particularly limited, but is, for example, a size that passes through a sieve with an opening of 500 ⁇ m (30 mesh) or 425 ⁇ m (36 mesh), preferably a size of 355 ⁇ m (42 mesh).
- the particle size may be adjusted using any two types of sieves among the sieves described above, and the size of the pulverized product is, for example, 25 to 500 ⁇ m, preferably 53 to 150 ⁇ m, and more preferably 75 to 106 ⁇ m.
- the method for producing a solid dispersion included in the present invention comprises a composition containing components (a), (b), and (c). It is preferable to include a step of hot melt extrusion (HME).
- the composition may contain components other than components (a), (b), and (c), and may also contain "other components” described in the "solid dispersion” section above.
- HME is typically a method characterized by uniformly mixing raw materials, heating and melting, extruding, and then cooling, using commonly used methods and equipment (e.g., a stirrer with a heat source, a kneader, etc.).
- an extruder having a screw in a barrel (cylinder) for example, a single-screw extruder, a twin-screw extruder, a twin-screw extruder, etc.
- a twin-screw extruder is preferred.
- An extruder consists of a hopper (the input structure), a motor (which controls the rotation of the screw), a screw (the primary power source for shearing and moving the material), a barrel (which houses the screw and provides temperature control), and a die. (exit) (controls the shape and size of the extrudate).
- Raw materials are introduced from the hopper of the extruder into the device maintained at an appropriate heating and melting temperature, and by rotating the screw, the solid materials of the raw materials are melted and uniformly kneaded.
- the raw materials can be preliminarily mixed before being introduced into the apparatus, if necessary.
- Conditions such as pressure, temperature, powder supply rate, die diameter, screw shape, screw rotation speed, etc. in the manufacturing method of the present invention vary depending on the extruder model, etc., but they must be adjusted so that the temperature is below the decomposition temperature of the raw material. It is preferable to combine them.
- the heating temperature needs to be set appropriately in view of stability issues such as decomposition and denaturation of raw materials.
- the heating temperature is preferably 190°C or lower, more preferably 185°C or lower, even more preferably 180°C or lower.
- the heating temperature is usually 150°C or higher, preferably 155°C or higher, and more preferably 160°C or higher.
- the heating temperature may be within the range of any combination of the lower limit and the upper limit, for example, within the range of 150 to 190°C, preferably within the range of 160 to 190°C.
- the screw rotation speed is preferably 10 rpm or more, more preferably 15 rpm or more, and still more preferably 20 rpm or more.
- the screw rotation speed is preferably 250 rpm or less, more preferably 200 rpm or less, still more preferably 150 rpm or less, even more preferably 100 rpm or less.
- the screw rotation speed may be within the range of any combination of the lower limit and the upper limit, for example, 20 to 250 rpm.
- a preferred method for producing the solid dispersion of the present invention includes a step of preparing a composition (mixture) by mixing component (a), component (b), component (c), and optionally an additive; It includes a step of heating and melt extruding.
- the composition is preferably supplied to a twin-screw extruder and processed at a screw rotation speed of 20 to 250 rpm and a processing temperature of 160 to 190°C.
- the method for producing a solid dispersion of the present invention further includes a step of pulverizing the heated melt extrudate.
- Solid dispersion particles having an arbitrary particle size can be easily obtained by pulverization using a suitable pulverizer, and can be used as they are as powders, fine granules, granules, etc.
- Pharmaceutical compositions or oral solid preparations, etc. containing the solid dispersion of the present invention are prepared by adding appropriate pharmaceutical formulation components (additives) to the solid dispersion (or solid dispersion particles) of the present invention and forming the formulation. It can also be made into an oral formulation.
- the pharmaceutical composition included in the present invention contains the above solid dispersion.
- the amount of the solid dispersion described above in the pharmaceutical composition of the present invention is not particularly limited, but may be, for example, within the range of 50 to 90% by weight.
- the pharmaceutical composition of the present invention may further contain other active ingredients and/or additives in addition to the solid dispersion described above.
- the additives are no particular limitations on the additives as long as they are pharmaceutically acceptable, such as excipients, disintegrants, binders, flow agents, lubricants, preservatives, stabilizers, and isotonic agents.
- additives can be used alone or in combination of two or more.
- the amount of other active ingredients and/or additives in the pharmaceutical composition of the invention is not particularly limited, but may be, for example, within the range of 10 to 50% by weight.
- excipients include lactose, anhydrous lactose, refined white sugar, white sugar, D-mannitol, D-sorbitol, xylitol, erythritol, dextrin, crystalline cellulose, microcrystalline cellulose, corn starch, potato starch, anhydrous calcium hydrogen phosphate. etc.
- disintegrant examples include sodium carboxymethyl starch, carmellose, carmellose calcium, carmellose sodium, croscarmellose sodium, sodium starch glycolate, crospovidone, low-substituted hydroxypropyl cellulose, partially pregelatinized starch, and the like. It will be done.
- binder examples include hydroxypropylcellulose, hydroxypropylmethylcellulose, polyvinylpyrrolidone, pregelatinized starch, syrup, starch syrup, and the like.
- fluidizing agent examples include light anhydrous silicic acid, calcium silicate, synthetic aluminum silicate, hydrated silicon dioxide, calcium stearate, magnesium aluminate metasilicate, and talc.
- lubricant examples include magnesium stearate, calcium stearate, magnesium silicate, magnesium oxide, talc, hydrogenated oil, sucrose fatty acid ester, stearyl sodium fumarate, and the like.
- coating agent examples include hydroxypropyl methylcellulose, polyvinyl alcohol, polysorbate, macrogol, and talc.
- coloring agent examples include yellow iron sesquioxide, brown iron oxide, iron sesquioxide, titanium oxide, food blue No. 1, food red No. 2, food red No. 3, food yellow No. 4, and the like.
- suspending agent examples include polysorbate, polyethylene glycol, gum arabic, glycerin, sucrose fatty acid ester, gelatin, and the like.
- sweeteners examples include aspartame, sucralose, saccharin, sodium saccharin, starch syrup, and fructose.
- surfactant examples include sodium lauryl sulfate, polysorbate, polyoxyethylene hydrogenated castor oil, and the like.
- the form of the pharmaceutical composition of the present invention is not particularly limited.
- it can be preferably used in the form of solid (granules, tablets, capsules, etc.), pellets, or liquid (suspension, etc.).
- the above-mentioned solid dispersion and, if necessary, additives can be filled into capsules and used as capsules.
- the solid dispersion described above and, if necessary, additives may be mixed and compressed to form a tablet.
- the solid dispersion described above can also be used as a suspension by dispersing it in water.
- the oral preparation included in the present invention contains the above solid dispersion.
- the amount of the above-mentioned solid dispersion in the oral preparation of the present invention is not particularly limited, but may be, for example, within the range of 50 to 90% by weight.
- the oral preparation of the present invention may further contain other active ingredients and/or additives.
- the additive those exemplified as the "additive” described in the "Pharmaceutical composition” section above can be used.
- the amount of other active ingredients and/or additives in the oral preparation of the present invention is not particularly limited, but may be, for example, within the range of 10 to 50% by weight.
- oral preparations of the present invention include oral solid preparations such as tablets, capsules, powders, and granules.
- the oral formulation of the invention is preferably a tablet or capsule. Tablets include, for example, plain tablets, film-coated tablets, sugar-coated tablets, caplet tablets, and chewable tablets.
- Examples A1 to A11 Delamanid (hammer milled product, Otsuka Pharmaceutical Co., Ltd.), polyvinylcaprolactam-polyvinylacetate-polyethylene glycol graft copolymer (Soluplus (trademark), BASF), and hypromellose phthalate (HPMCP (trademark) HP -50, Shin-Etsu Chemical Co., Ltd.) and, in some cases, dl- ⁇ -tocopherol (a type of vitamin E, so hereinafter referred to as VE) (Fuji Film Wako Pure Chemical Industries, Ltd.), in some cases.
- VE dl- ⁇ -tocopherol
- BHT dibutylhydroxytoluene
- This molded body was cooled to room temperature, pulverized using a pulverizer (tablet pulverizer HTF-35, Daido Kako Co., Ltd.), and passed through a 53 ⁇ m sieve to obtain particles (solid dispersion particles).
- a pulverizer tablet pulverizer HTF-35, Daido Kako Co., Ltd.
- Example A12 Particles were prepared in the same manner as in Example A11, except that hydroxypropyl methylcellulose phthalate was changed to hydroxypropyl methylcellulose acetate succinate (Shin-Etsu AQOATTM AS-LF, Shin-Etsu Chemical Co., Ltd.). Created.
- Example A4 When the same procedure as in Example A1 was performed except that the polyvinylcaprolactam-polyvinyl acetate-polyethylene glycol graft copolymer and dl- ⁇ -tocopherol were not added, delamanide was decomposed during the molding process and a molded article was not produced. could not.
- Examples B1 to B5 Delamanid (hammer milled product, Otsuka Pharmaceutical Co., Ltd.), vinylpyrrolidone-vinyl acetate copolymer (Kollidon (trademark) VA64, BASF), hydroxypropyl methylcellulose phthalate (HPMCP (trademark) HP-50, Shin-Etsu Chemical) Kogyo Co., Ltd.) and dl- ⁇ -tocopherol (VE) (Fuji Film Wako Pure Chemical Industries, Ltd.) in the amounts shown in Table 5 were placed in a polyethylene bag and mixed for several minutes.
- Kollidon trademark
- HPMCP hydroxypropyl methylcellulose phthalate
- VE dl- ⁇ -tocopherol
- This mixed powder was mixed using a twin-screw extruder (Pharma 11 twin-screw extruder, Thermo Fisher Scientific Co., Ltd.) equipped with a die with a diameter of 1 mm ⁇ , and the barrel temperature of the kneading section was set at 160 to 170°C. Then, molding was performed at an extrusion speed of 40 rpm to obtain a rod-shaped molded product.
- This molded body was cooled to room temperature, pulverized using a pulverizer (tablet pulverizer HTF-35, Daido Kako Co., Ltd.), and passed through a 53 ⁇ m sieve to obtain particles (solid dispersion particles).
- Example C 40 g of delamanid (hammer mill product, Otsuka Pharmaceutical Co., Ltd.), 160 g of polyvinylcaprolactam-polyvinylacetate-polyethylene glycol graft copolymer (Soluplus (trademark), BASF), and hydroxypropyl methylcellulose phthalate (HPMCP (trademark) HP -50 (Shin-Etsu Chemical Co., Ltd.) and 9.6 g of dl- ⁇ -tocopherol (VE) (Fuji Film Wako Pure Chemical Industries, Ltd.) were placed in a polyethylene bag and mixed for several minutes.
- delamanid hammer mill product, Otsuka Pharmaceutical Co., Ltd.
- 160 g polyvinylcaprolactam-polyvinylacetate-polyethylene glycol graft copolymer
- HP -50 Shin-Etsu Chemical Co., Ltd.
- VE dl- ⁇ -tocopherol
- This mixed powder was mixed using a twin-screw extruder (Pharma 11 twin-screw extruder, Thermo Fisher Scientific Co., Ltd.) equipped with a die with a diameter of 1 mm ⁇ , and the barrel temperature of the kneading section was set at 180°C. Molding treatment was performed at an extrusion speed of 40 rpm to obtain a rod-shaped molded body. This molded body was cooled to room temperature, pulverized using a pulverizer (tablet pulverizer HTF-35, Daido Kako Co., Ltd.), and passed through a 53 ⁇ m sieve to obtain particles (solid dispersion particles).
- a twin-screw extruder Pulharma 11 twin-screw extruder, Thermo Fisher Scientific Co., Ltd.
- Molding treatment was performed at an extrusion speed of 40 rpm to obtain a rod-shaped molded body.
- This molded body was cooled to room temperature, pulverized
- Test Example 1 (Solubility)
- Method 1 or 2 The solubility of the particles of Examples A1 to A12 and Comparative Examples A1 to A3 was investigated using Method 1 or 2 below.
- Method 1 100 mL of a 0.3% by mass aqueous sodium lauryl sulfate solution was placed in a 100 mL beaker and stirred with a magnetic stirrer (rotation speed: 500 rpm).
- a magnetic stirrer rotating speed: 500 rpm
- particles of Examples and Comparative Examples were weighed out in an agate mortar so that the amount of delamanid was 5 mg, and several mL of the test solution in the beaker was added. After dispersing for about 1 minute, the dispersion was poured into a beaker.
- Method 2 A sample solution was prepared in the same manner as Method 1, except that a 0.1% by mass sodium lauryl sulfate aqueous solution was used as the test liquid, and the absorbance at a wavelength of 335 nm was measured.
- Table 5 shows the solubility results of the particles of Examples B1 to B5.
- Example 2 Purity of delamanid
- the particles obtained in Examples A8 to A10 were placed in a glass container, the outer layer was sealed with an aluminum bag, and the particles were stored for two weeks in an environment of 60° C.
- the purity of delamanid was examined using the particles as samples.
- the purity of delamanide was determined by connecting a column (ACQUITY UPLC (trademark) BEH C18, Nippon Waters Co., Ltd.) to a UPLC (ultra high performance liquid chromatography, ACQUITY UPLC/TUV system, Nippon Waters Co., Ltd.), using a sample cooler 15.
- the column oven was set at 40°C and the detector was set at a measurement wavelength of 254 nm.
- the obtained blood was centrifuged at 3000 rpm for 10 minutes to obtain serum.
- the concentration of delamanide in this serum was measured by LC-MS. The results are shown in Figure 1.
- each component other than magnesium stearate was mixed in a polyethylene bag, and then magnesium stearate was added and further mixed.
- the mixed powder was compressed into tablets using a rotary tablet press (Clean Press, Kikusui Seisakusho Co., Ltd.) equipped with a caplet (18.0 x 10.8 mm) mortar at a compression pressure of 20 kN, and each tablet contained 100 mg of delamanide.
- a caplet containing the following was prepared.
- caplet tablets of Formulation Examples 5, 7, and 16 were coated with a film coating solution in which hydroxypropyl methylcellulose, PEG 6000, titanium oxide, talc, and yellow iron sesquioxide of the formulation shown in Table 9 were dissolved and suspended in purified water. Caplets were sprayed in a machine (Powrec Coater PRC-GTX mini, Powrec Co., Ltd.) to prepare film-coated tablets.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Oncology (AREA)
- Organic Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Communicable Diseases (AREA)
- Engineering & Computer Science (AREA)
- Pulmonology (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
下記の成分(a)、(b)、及び(c)を含む固体分散体が開示される。 (a)デラマニド (b)ポリビニルカプロラクタム-ポリビニルアセテート-ポリエチレングリコールグラフト共重合体及びビニルピロリドン-ビニルアセテート共重合体からなる群より選ばれた少なくとも1種 (c)ヒドロキシプロピルメチルセルロースフタレート及びヒドロキシプロピルメチルセルロースアセテートサクシネートからなる群より選ばれた少なくとも1種
Description
本発明は、デラマニドを含有する固体分散体、及び当該固体分散体の製造方法等に関する。なお、本明細書に記載される全ての文献は参照により本明細書に組み込まれる。
デラマニドは、下記式:

で示される、(2R)-2-メチル-6-ニトロ-2-[(4-{4-[4-(トリフルオロメトキシ)フェノキシ]ピぺリジン-1-イル}フェノキシ)メチル]-2,3-ジヒドロイミダゾ[2,1-b]オキサゾールである。デラマニドは、結核菌、多剤耐性結核菌、及び非定型抗酸菌に対して優れた殺菌作用を有している(特許文献1)。また、デラマニドは、水に対する溶解性が低い難水溶性化合物として知られており、溶解性を改善し、経口吸収性を改善する検討がなされている(特許文献2及び3)。
難水溶性化合物の溶解性を改善するための可溶化手法として、微粒子化(ナノ粒子化)、準安定形(結晶多形)、塩、共結晶、非晶質、界面活性剤や油等を用いた可溶化等の様々な手法が利用されている。中でも、非晶質形態は実用性の高い製剤技術であり、非晶質形態の不安定性を改善するアプローチとして、ポリマーなどの担体と複合化させる固体分散体技術がある。固体分散体は、「固体状態で不活性な担体またはそのマトリックス中に、1種又はそれ以上の活性成分が分散したもの」である。固体分散体の製法としては、共沈法、噴霧乾燥法、加熱溶融押出法などの方法がある。加熱溶融押出法(Hot Melt Extrusion、HME)は、連続プロセス(スケールアップが容易)、無溶媒法(溶媒の処理が不要、溶媒の残留が無いなど環境・コスト面で有用)、異なる薬物送達システム(顆粒、ペレット、徐放性錠剤、座薬、ステント、眼科用インサート、並びに経皮及び経粘膜送達システムを含む)に適用可能などの多くの利点を有している。しかし、HMEにおけるプロセスパラメーターは、熱力学的及びレオロジー特性に依存していることから一般的に高い処理温度が必要となり、薬物の熱劣化が生じるリスクがあること、そのリスクを抑えるために処理温度を下げると結晶性薬物の残留が懸念されることなどの課題がある。デラマニドは熱安定性の悪い(融点:約195℃(分解))化合物であり、融点と分解点が近いため、高温処理が必要なHMEでは分解物が生成し、製造は困難であると考えられる。
本発明は、デラマニドを含む固体分散体、当該固体分散体を製造する方法、当該固体分散体を含む医薬組成物及び経口固形製剤を提供することが1つの課題である。
本発明者らは、上記課題を解決すべく鋭意研究を重ねた結果、デラマニドと特定のビニル系ポリマーと特定のセルロース化合物とを組み合わせることにより、溶解性等に優れた固体分散体が得られることを見出した。本発明者らは、当該知見に基づいて更に検討を重ねて、本発明を完成させた。
本発明は例えば以下の項に記載の主題を包含する。
項1.
下記の成分(a)、(b)、及び(c)を含む固体分散体。
(a)デラマニド
(b)ポリビニルカプロラクタム-ポリビニルアセテート-ポリエチレングリコールグラフト共重合体及びビニルピロリドン-ビニルアセテート共重合体からなる群より選ばれた少なくとも1種
(c)ヒドロキシプロピルメチルセルロースフタレート及びヒドロキシプロピルメチルセルロースアセテートサクシネートからなる群より選ばれた少なくとも1種
項2.
成分(a)と成分(b)との質量比が1:1~1:20の範囲内である、項1に記載の固体分散体。
項3.
成分(a)と成分(b)との質量比が1:1~1:8の範囲内である、項1又は2に記載の固体分散体。
項4.
成分(a)と成分(b)との質量比が1:1~1:5の範囲内である、項1~3のいずれかに記載の固体分散体。
項5.
成分(a)と成分(c)との質量比が1:1~1:10の範囲内である、項1~4のいずれかに記載の固体分散体。
項6.
成分(a)と成分(c)との質量比が1:1~1:8の範囲内である、項1~5のいずれかに記載の固体分散体。
項7.
成分(a)と成分(c)との質量比が1:1~1:5の範囲内である、項1~6のいずれかに記載の固体分散体。
項8.
成分(a)と成分(b)との質量比が1:1~1:5の範囲内であり、成分(a)と成分(c)との質量比が1:1~1:5の範囲内である、項1~7のいずれかに記載の固体分散体。
項9.
成分(b)がポリビニルカプロラクタム-ポリビニルアセテート-ポリエチレングリコールグラフト共重合体を含む、項1~8のいずれかに記載の固体分散体。
項10.
成分(b)がビニルピロリドン-ビニルアセテート共重合体を含む、項1~9のいずれかに記載の固体分散体。
項11.
成分(c)がヒドロキシプロピルメチルセルロースフタレートを含む、項1~10のいずれかに記載の固体分散体。
項12.
成分(b)がポリビニルカプロラクタム-ポリビニルアセテート-ポリエチレングリコールグラフト共重合体を含み、成分(c)がヒドロキシプロピルメチルセルロースフタレートを含む、項1~11のいずれかに記載の固体分散体。
項13.
成分(b)がビニルピロリドン-ビニルアセテート共重合体を含み、成分(c)がヒドロキシプロピルメチルセルロースフタレートを含む、項1~12のいずれかに記載の固体分散体。
項14.
ビタミンEを更に含む、項1~13のいずれかに記載の固体分散体。
項15.
ビタミンEがdl-α-トコフェロールである、項14に記載の固体分散体。
項16.
加熱溶融押出物又はその粉砕物である、項1~15のいずれかに記載の固体分散体。
項17.
項1~16のいずれかに記載の固体分散体を含む医薬組成物。
項18.
項1~16のいずれかに記載の固体分散体を含む経口固形製剤。
項19.
錠剤又はカプセル剤である、項18に記載の経口固形製剤。
項20.
成分(a)、(b)、及び(c)を含む組成物を加熱溶融押出する工程を含む、項1~16のいずれかに記載の固体分散体を製造する方法。
項21.
キシリトールを含まない、項1~16のいずれかに記載の固体分散体。
項22.
糖アルコールを含まない、項1~16のいずれかに記載の固体分散体。
項23.
キシリトール、ソルビトール、マンニトール、及びポロキサマーを含まない、項1~16のいずれかに記載の固体分散体。
項24.
成分(a)と成分(b)と成分(c)との質量比が、約1:4:3である、項9、11、又は12に記載の固体分散体。
項25.
成分(a)と成分(b)と成分(c)との質量比が、約1:2:2である、項10、11、又は13に記載の固体分散体。
項1.
下記の成分(a)、(b)、及び(c)を含む固体分散体。
(a)デラマニド
(b)ポリビニルカプロラクタム-ポリビニルアセテート-ポリエチレングリコールグラフト共重合体及びビニルピロリドン-ビニルアセテート共重合体からなる群より選ばれた少なくとも1種
(c)ヒドロキシプロピルメチルセルロースフタレート及びヒドロキシプロピルメチルセルロースアセテートサクシネートからなる群より選ばれた少なくとも1種
項2.
成分(a)と成分(b)との質量比が1:1~1:20の範囲内である、項1に記載の固体分散体。
項3.
成分(a)と成分(b)との質量比が1:1~1:8の範囲内である、項1又は2に記載の固体分散体。
項4.
成分(a)と成分(b)との質量比が1:1~1:5の範囲内である、項1~3のいずれかに記載の固体分散体。
項5.
成分(a)と成分(c)との質量比が1:1~1:10の範囲内である、項1~4のいずれかに記載の固体分散体。
項6.
成分(a)と成分(c)との質量比が1:1~1:8の範囲内である、項1~5のいずれかに記載の固体分散体。
項7.
成分(a)と成分(c)との質量比が1:1~1:5の範囲内である、項1~6のいずれかに記載の固体分散体。
項8.
成分(a)と成分(b)との質量比が1:1~1:5の範囲内であり、成分(a)と成分(c)との質量比が1:1~1:5の範囲内である、項1~7のいずれかに記載の固体分散体。
項9.
成分(b)がポリビニルカプロラクタム-ポリビニルアセテート-ポリエチレングリコールグラフト共重合体を含む、項1~8のいずれかに記載の固体分散体。
項10.
成分(b)がビニルピロリドン-ビニルアセテート共重合体を含む、項1~9のいずれかに記載の固体分散体。
項11.
成分(c)がヒドロキシプロピルメチルセルロースフタレートを含む、項1~10のいずれかに記載の固体分散体。
項12.
成分(b)がポリビニルカプロラクタム-ポリビニルアセテート-ポリエチレングリコールグラフト共重合体を含み、成分(c)がヒドロキシプロピルメチルセルロースフタレートを含む、項1~11のいずれかに記載の固体分散体。
項13.
成分(b)がビニルピロリドン-ビニルアセテート共重合体を含み、成分(c)がヒドロキシプロピルメチルセルロースフタレートを含む、項1~12のいずれかに記載の固体分散体。
項14.
ビタミンEを更に含む、項1~13のいずれかに記載の固体分散体。
項15.
ビタミンEがdl-α-トコフェロールである、項14に記載の固体分散体。
項16.
加熱溶融押出物又はその粉砕物である、項1~15のいずれかに記載の固体分散体。
項17.
項1~16のいずれかに記載の固体分散体を含む医薬組成物。
項18.
項1~16のいずれかに記載の固体分散体を含む経口固形製剤。
項19.
錠剤又はカプセル剤である、項18に記載の経口固形製剤。
項20.
成分(a)、(b)、及び(c)を含む組成物を加熱溶融押出する工程を含む、項1~16のいずれかに記載の固体分散体を製造する方法。
項21.
キシリトールを含まない、項1~16のいずれかに記載の固体分散体。
項22.
糖アルコールを含まない、項1~16のいずれかに記載の固体分散体。
項23.
キシリトール、ソルビトール、マンニトール、及びポロキサマーを含まない、項1~16のいずれかに記載の固体分散体。
項24.
成分(a)と成分(b)と成分(c)との質量比が、約1:4:3である、項9、11、又は12に記載の固体分散体。
項25.
成分(a)と成分(b)と成分(c)との質量比が、約1:2:2である、項10、11、又は13に記載の固体分散体。
本発明に包含されるデラマニドを含む固体分散体は、溶解性等に優れている。また、本発明に包含されるデラマニド固体分散体の製造方法によれば、環境への負荷が少なく安価な方法での製造が可能である。
本明細書において、「約」とは、誤差、例えば、後に続く数値の±10%以内、±5%以内、又は±1%以内の変動を包含することを意味する。
本明細書において、「含む」とは、「実質的にからなる」及び「からなる」という概念を包含する。
以下、本発明の各実施形態について、さらに詳細に説明する。
[固体分散体]
本発明に包含される固体分散体(以下、本発明の固体分散体と表記することがある)は、(a)デラマニド、(b)ポリビニルカプロラクタム-ポリビニルアセテート-ポリエチレングリコールグラフト共重合体及びビニルピロリドン-ビニルアセテート共重合体からなる群より選ばれた少なくとも1種、並びに(c)ヒドロキシプロピルメチルセルロースフタレート及びヒドロキシプロピルメチルセルロースアセテートサクシネートからなる群より選ばれた少なくとも1種を含む。
[固体分散体]
本発明に包含される固体分散体(以下、本発明の固体分散体と表記することがある)は、(a)デラマニド、(b)ポリビニルカプロラクタム-ポリビニルアセテート-ポリエチレングリコールグラフト共重合体及びビニルピロリドン-ビニルアセテート共重合体からなる群より選ばれた少なくとも1種、並びに(c)ヒドロキシプロピルメチルセルロースフタレート及びヒドロキシプロピルメチルセルロースアセテートサクシネートからなる群より選ばれた少なくとも1種を含む。
(成分(a))
本発明の固体分散体において、デラマニドは、微結晶又は非晶質の形態であることが好ましい。具体的には、粉末X線回折パターンにおいて結晶性ピークが存在しないことが好ましい。粉末X線回折は、日本薬局方(第十八改正)の「一般試験法 2.58 粉末X線回折測定法」に従って測定することができ、例えば、下記の条件で測定してもよい。
測定装置:X’Pert PRO MPD(スペクトリス(株))
光学系:集中光学系(透過法)
ゴニオメータ半径:240 mm
管電圧、管電流:45 kV、40 mA
入射側スリット:ソーラースリット、ソーラー0.04 rad
ダイバージェンススリット 1/2 deg
受光側スリット:ソーラースリット ラージソーラー 0.04 rad
アンチスキャッタースリット 5.5 mm
測定範囲:2θ 3~40 deg
操作速度:1.11 deg/sec.
サンプリング間隔:0.02 deg/step
Wobbled scan:ステップ回数5、ステップサイズ0.02 deg
本発明の固体分散体において、デラマニドは、微結晶又は非晶質の形態であることが好ましい。具体的には、粉末X線回折パターンにおいて結晶性ピークが存在しないことが好ましい。粉末X線回折は、日本薬局方(第十八改正)の「一般試験法 2.58 粉末X線回折測定法」に従って測定することができ、例えば、下記の条件で測定してもよい。
測定装置:X’Pert PRO MPD(スペクトリス(株))
光学系:集中光学系(透過法)
ゴニオメータ半径:240 mm
管電圧、管電流:45 kV、40 mA
入射側スリット:ソーラースリット、ソーラー0.04 rad
ダイバージェンススリット 1/2 deg
受光側スリット:ソーラースリット ラージソーラー 0.04 rad
アンチスキャッタースリット 5.5 mm
測定範囲:2θ 3~40 deg
操作速度:1.11 deg/sec.
サンプリング間隔:0.02 deg/step
Wobbled scan:ステップ回数5、ステップサイズ0.02 deg
デラマニドは、慣用の方法、例えば、特開2004-149527号公報、米国特許出願公開2006/094767号公報等に記載の方法により製造することができる。
(成分(b))
本発明者らは、成分(a)と成分(b)とを組み合わせることにより、両成分の相溶が進み、加熱溶融混練が可能になり、溶融混練機内での流動性が確保され、固体分散体の良好な製造性を実現でき、デラマニド原薬(ジェットミル粉砕品)以上の溶解性を示す固体分散体が得られることを見出した。
本発明者らは、成分(a)と成分(b)とを組み合わせることにより、両成分の相溶が進み、加熱溶融混練が可能になり、溶融混練機内での流動性が確保され、固体分散体の良好な製造性を実現でき、デラマニド原薬(ジェットミル粉砕品)以上の溶解性を示す固体分散体が得られることを見出した。
成分(b)のうち、ポリビニルカプロラクタム-ポリビニルアセテート-ポリエチレングリコールグラフト共重合体(以下、成分(b1)と表記することがある)は、少なくとも1つのポリビニルカプロラクタムブロックと、少なくとも1つのポリビニルアセテートブロックと、少なくとも1つのポリエチレングリコールブロックとを含む。前記ポリエチレングリコールは、例えば、PEG6000であり得る。ポリビニルカプロラクタムブロック/ポリビニルアセテートブロック/ポリエチレングリコールブロックの比率は、例えば、約57/30/13であってもよい。
一実施形態において、成分(b1)は、下記式:

(式中、l、m、及びnは、それぞれ独立して、1以上の整数である。)
で表される共重合体であってもよい。
で表される共重合体であってもよい。
成分(b1)の平均分子量は、例えば、10000g/mol以上、50000g/mol以上、又は90000g/mol以上であり得る。成分(b1)の平均分子量は、例えば、1000000g/mol以下、500000g/mol以下、200000g/mol以下、又は140000g/mol以下であり得る。成分(b1)の平均分子量は、前記下限と前記上限とを任意に組み合わせた範囲内、例えば、10000~1000000g/molの範囲内、好ましくは90000~140000g/molの範囲内であり得る。
一実施形態において、成分(b1)は、BASF社のSoluplus(商標)を含むことが好ましい。
成分(b)のうち、ビニルピロリドン-ビニルアセテート共重合体(以下、成分(b2)と表記することがある)は、少なくともビニルピロリドン(通常、1-ビニル-2-ピロリドン)及びビニルアセテートを重合成分とする共重合体である限り、特に限定されない。ビニルピロリドンとビニルアセテートとの共重合比(質量比)は、例えば、1:9~9:1の範囲内、2:8~8:2の範囲内、又は3:7~7:3の範囲内であり得る。ビニルピロリドンとビニルアセテートとの共重合比(質量比)は、例えば、4:6~8:2の範囲内、又は5:5~7:3の範囲内であり得る。
成分(b2)は、単量体の配列に特に制限はなく、ランダム共重合体、交互共重合体、ブロック共重合体、及びグラフト共重合体のいずれであってもよい。一実施形態において、成分(b2)は、ランダム共重合体であることが好ましい。
成分(b2)の平均分子量は、例えば、10000g/mol以上、20000g/mol以上、30000g/mol以上、40000g/mol以上、又は45000g/mol以上であり得る。成分(b2)の平均分子量は、例えば、1000000g/mol以下、500000g/mol以下、100000g/mol以下、又は70000g/mol以下であり得る。成分(b2)の平均分子量は、前記下限と前記上限とを任意に組み合わせた範囲内、例えば、10000~1000000g/molの範囲内、好ましくは45000~70000g/molの範囲内であり得る。
一実施形態において、成分(b2)は、コポリピドン、例えば、BASF社のKollidon(商標)VA64を含むことが好ましい。
一実施形態において、成分(b)は、成分(b1)を含むことが好ましい。別の実施形態において、成分(b)は、成分(b2)を含むことが好ましい。更に別の実施形態において、成分(b)は、成分(b1)と成分(b2)との組合せが好ましい。当該組合せにおいて、成分(b1)と成分(b2)との質量比は、特に制限されないが、例えば、0.5:1~10:1の範囲内、好ましくは1:1~6:1の範囲内である。
(成分(c))
本発明者らは、成分(a)及び(b)に更に成分(c)を組み合わせることにより、より一層溶解性に優れた固体分散体が得られることを見出した。
本発明者らは、成分(a)及び(b)に更に成分(c)を組み合わせることにより、より一層溶解性に優れた固体分散体が得られることを見出した。
成分(c)のうち、ヒドロキシプロピルメチルセルロースフタレート(以下、成分(c1)と表記することがある)は、ヒドロキシプロピルセルロースにフタル酸がハーフエステル結合した高分子化合物である。ヒドロキシプロピルメチルセルロースフタレートは、日本薬局方(第十八改正)の医薬品各条(化学薬品等)に記載の「ヒプロメロースフタル酸エステル」であることが好ましい。ヒドロキシプロピルメチルセルロースフタレートとしては、例えば、信越化学工業(株)から商品名HPMCP(商標)HP-55、HP-55S、及びHP-50として市販されている。本発明では、これらの市販品をいずれも使用することができる。一実施形態において、ヒドロキシプロピルメチルセルロースフタレートは、その水酸基が少なくともカルボキシベンゾイル基21~35質量%(例えば、21~27質量%、又は27~35質量%)で置換されていることが好ましい。当該実施形態において、ヒドロキシプロピルメチルセルロースフタレートは、その水酸基がメトキシ基18~24質量%、ヒドロキシプロポキシ基5~10質量%、及びカルボキシベンゾイル基21~35質量%で置換されていることが好ましい。
成分(c)のうち、ヒドロキシプロピルメチルセルロースアセテートサクシネート(以下、成分(c2)と表記することがある)は、ヒドロキシプロピルセルロースに酢酸及びコハク酸がエステル結合した高分子化合物である。ヒドロキシプロピルメチルセルロースアセテートサクシネートは、日本薬局方(第十八改正)の医薬品各条(化学薬品等)に記載の「ヒプロメロース酢酸エステルコハク酸エステル」であることが好ましい。ヒドロキシプロピルメチルセルロースアセテートサクシネートとしては、例えば、信越化学工業(株)から商品名Shin-Etsu AQOAT(商標)AS-LF、AS-MF、及びAS-HFとして市販されている。本発明では、これらの市販品をいずれも使用することができる。ヒドロキシプロピルメチルセルロースアセテートサクシネートは、その水酸基がメトキシ基20~26質量%、ヒドロキシプロポキシ基5~10質量%、アセチル基5~14質量%、及びサクシノイル基4~18質量%で置換されていることが好ましい。
一実施形態において、成分(c)は、成分(c1)を含むこと(例えば、成分(c1)単独、又は、成分(c1)と成分(c2)との組合せ)が好ましい。
本発明の固体分散体において、成分(a)の含有率は、例えば、1質量%以上、5質量%以上、又は10質量%以上であり得る。成分(a)の含有率は、例えば、30質量%以下、25質量%以下、又は20質量%以下であり得る。成分(a)の含有率は、例えば、前記下限と前記上限とを任意に組み合わせた範囲内、例えば、1~30質量%の範囲内であり得る。
成分(a)と成分(b)との質量比は、特に制限されない。成分(a)と成分(b)との質量比は、1:1未満(例えば、1:0.1~1:0.9の範囲内)であってもよいが、通常1:1以上(成分(b)/成分(a)が1/1以上であることを意味する)であり、好ましくは1:1.5以上であり、より好ましくは1:2以上である。成分(a)と成分(b)との質量比は、例えば、1:20以下(成分(b)/成分(a)が20/1以下であることを意味する)、1:15以下、1:10以下、1:8以下、又は1:5以下であり得る。成分(a)と成分(b)との質量比は、前記下限と前記上限とを任意に組み合わせた範囲内、例えば、1:1~1:20の範囲内、好ましくは1:1~1:8の範囲内、より好ましくは1:1~1:5の範囲内、更に好ましくは1:2~1:5の範囲内であり、更により好ましくは1:2~1:4の範囲内、或いは約1:2又は約1:4である。成分(b)が成分(b1)を含む場合、成分(a)と成分(b)との質量比は約1:4であることが好ましく、成分(b)が成分(b2)を含む場合、成分(a)と成分(b)との質量比は約1:2であることが好ましい。
成分(a)と成分(c)との質量比は、1:1未満(例えば、1:0.1~1:0.9の範囲内)であってもよいが、通常1:1以上(成分(c)/成分(a)が1/1以上であることを意味する)であり、好ましくは1:1.5以上、より好ましくは1:2以上である。成分(a)と成分(c)との質量比は、例えば、1:10以下(成分(c)/成分(a)が10/1以下であることを意味する)、1:8以下、又は1:5以下であり得る。成分(a)と成分(c)との質量比は、前記下限と前記上限とを任意に組み合わせた範囲内、例えば、1:1~1:10の範囲内、好ましくは1:1~1:8の範囲内、より好ましくは1:1~1:5の範囲内であり、更に好ましくは1:1~1:4の範囲内であり、更により好ましくは1:2~1:4の範囲内、特に好ましくは約1:2又は約1:3である。成分(b)が成分(b1)を含む場合、成分(a)と成分(c)との質量比は約1:3であることが好ましく、成分(b)が成分(b2)を含む場合、成分(a)と成分(c)との質量比は約1:2であることが好ましい。
一実施形態において、成分(a)と成分(b)と成分(c)との質量比は、好ましくは1:1~8:1~8の範囲内であり、より好ましくは1:1~5:1~5の範囲内であり、更に好ましくは1:1.5~5:1.5~5の範囲内であり、更により好ましくは1:2~4:2~4の範囲内、或いは約1:2:2又は約1:4:3である。成分(b)が成分(b1)を含む場合、成分(a)と成分(b)と成分(c)との質量比は約1:4:3であることが好ましく、成分(b)が成分(b2)を含む場合、成分(a)と成分(b)と成分(c)との質量比は約1:2:2であることが好ましい。
(その他の成分)
本発明の固体分散体は、その他の成分を更に配合することができる。その他の成分としては、例えば、抗酸化剤、成分(b)以外のpH非依存型水溶性高分子化合物、可塑剤等が挙げられる。その他の成分は1種単独で又は2種以上を組み合わせて使用することができる。
本発明の固体分散体は、その他の成分を更に配合することができる。その他の成分としては、例えば、抗酸化剤、成分(b)以外のpH非依存型水溶性高分子化合物、可塑剤等が挙げられる。その他の成分は1種単独で又は2種以上を組み合わせて使用することができる。
抗酸化剤としては、例えば、ビタミンE、ジブチルヒドロキシトルエン、ブチルヒドロキシアニソール、大豆レシチン、パルミチン酸アスコルビル、塩酸システイン、アスコルビン酸、クエン酸、エリソルビン酸、亜硝酸ナトリウム、亜硫酸ナトリウム、チオグリコール酸ナトリウム、チオリンゴ酸ナトリウム等が挙げられる。一実施形態において、抗酸化剤は、ビタミンEを含むことが好ましく、ビタミンE及びジブチルヒドロキシトルエン(例えば、ビタミンEとジブチルヒドロキシトルエンの質量比は15:1~1:1である)を含むことも好ましい。当該実施形態によれば、保存時(特に、約60℃のような高温での保存時)の分解物の生成を抑制することができる。
ビタミンEには、例えば、d-α-トコフェロール、d-δ-トコフェロール、酢酸d-α-トコフェロール、コハク酸d-α-トコフェロール、dl-α-トコフェロール、酢酸dl-α-トコフェロール、コハク酸dl-α-トコフェロール、コハク酸dl-α-トコフェロールカルシウム、ニコチン酸dl-α-トコフェロール等の天然及び合成ビタミンE等が含まれる。好ましいビタミンEは、dl-α-トコフェロール、酢酸dl-α-トコフェロール、コハク酸dl-α-トコフェロール及びニコチン酸dl-α-トコフェロールであり、より好ましいビタミンEは、dl-α-トコフェロールである。
本発明の固体分散体に配合される抗酸化剤(例えば、ビタミンE)の量は、デラマニド1質量部に対して、通常0.001質量部以上、好ましくは0.01質量部以上、より好ましくは0.03質量部以上である。前記抗酸化剤(例えば、ビタミンE)の量は、デラマニド1質量部に対して、通常1質量部以下、好ましくは0.8質量部以下、より好ましくは0.5質量部以下である。前記抗酸化剤(例えば、ビタミンE)の量は、前記下限と前記上限とを任意に組み合わせた範囲内、例えば、デラマニド1質量部に対して、通常0.001~1質量部の範囲内、好ましくは0.01~0.8重量部の範囲内、より好ましくは0.03~0.5質量部の範囲内である。
成分(b)以外のpH非依存型水溶性高分子化合物としては、例えば、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ポリビニルピロリドン、ポリエチレングリコール、シクロデキストリン等が挙げられる。
可塑剤としては、例えば、クエン酸トリエチル、グリセリン、グリセリン脂肪酸エステル、中鎖脂肪酸トリグリセライド、トリアセチン、ヒマシ油、プロピレングリコール、ポリソルベート等が挙げられる。
成分(b)以外のpH非依存型水溶性高分子化合物、可塑剤等のその他の成分も、抗酸化剤と同様の量を固体分散体に配合することができる。
一実施形態において、本発明の固体分散体は、キシリトールを含まないことが好ましく、キシリトール、ソルビトール、及びマンニトールを含まないことがより好ましく、糖アルコールを含まないことが更に好ましい。
一実施形態において、本発明の固体分散体は、ポロキサマー(ポリオキシプロピレン鎖と、それを挟む2個のポリオキシエチレン鎖からなるブロック共重合体)を含まないことが好ましい。
一実施形態において、本発明の固体分散体は、キシリトール、ソルビトール、マンニトール、及びポロキサマーを含まないことが好ましい。
本発明の固体分散体は、加熱溶融押出物又はその粉砕物であることが好ましく、後述の製造方法により製造されるものであることが好ましい。前記粉砕物は、通常粒子であり、固体分散体粒子、溶融顆粒等と呼ぶこともある。前記粉砕物のサイズは、特に制限されないが、例えば、500μmの目開き(30メッシュ)又は425μmの目開き(36メッシュ)の篩を通過するサイズであり、好ましくは355μmの目開き(42メッシュ)、300μmの目開き(50メッシュ)、250μmの目開き(60メッシュ)、212μmの目開き(70メッシュ)、180μmの目開き(83メッシュ)、160μmの目開き(93メッシュ)、150μmの目開き(100メッシュ)、125μmの目開き(119メッシュ)、106μmの目開き(140メッシュ)、100μmの目開き(149メッシュ)、90μmの目開き(166メッシュ)、75μmの目開き(200メッシュ)、63μmの目開き(235メッシュ)、53μmの目開き(280メッシュ)、45μmの目開き(330メッシュ)、38μmの目開き(390メッシュ)、32μmの目開き(440メッシュ)、又は25μmの目開き(500メッシュ)の篩を通過するサイズである。前記の篩のうち、任意の2種類の篩により粒度を調整してもよく、前記粉砕物のサイズは、例えば、25~500μm、好ましくは53~150μm、より好ましくは75~106μmである。
[固体分散体の製造方法]
本発明に包含される固体分散体の製造方法(以下、本発明の固体分散体の製造方法と表記することがある)は、成分(a)、(b)、及び(c)を含む組成物を加熱溶融押出(Hot Melt Extrusion、HME)する工程を含むことが好ましい。前記組成物は、成分(a)、(b)、及び(c)以外の成分を含んでいてもよく、上記「固体分散体」の項目に記載の「その他の成分」を含んでいてもよい。HMEは、典型的には、原料を均一に混合して加熱融解し、押出し、その後冷却することを特徴とする方法であり、慣用されている方法及び装置(例えば、熱源を有する攪拌機、混練機等)を用いて行うことができる。
本発明に包含される固体分散体の製造方法(以下、本発明の固体分散体の製造方法と表記することがある)は、成分(a)、(b)、及び(c)を含む組成物を加熱溶融押出(Hot Melt Extrusion、HME)する工程を含むことが好ましい。前記組成物は、成分(a)、(b)、及び(c)以外の成分を含んでいてもよく、上記「固体分散体」の項目に記載の「その他の成分」を含んでいてもよい。HMEは、典型的には、原料を均一に混合して加熱融解し、押出し、その後冷却することを特徴とする方法であり、慣用されている方法及び装置(例えば、熱源を有する攪拌機、混練機等)を用いて行うことができる。
HMEは、例えば、バレル(シリンダ)内にスクリューを有する押出機(例えば、単軸押出機、2軸押出機、2軸型エクストルーダー等)を使用することができる。中でも好ましくは2軸型エクストルーダーである。
押出機は、ホッパー(投入口構造)、モーター(スクリューの回転を制御する)、スクリュー(材料のせん断及び移動の一次電源装置)、バレル(スクリューを収納して温度制御を提供する)、及びダイ(出口)(押出成形品の形及びサイズを制御する)の5つの主要な部分を含む。
押出機のホッパーから、適切な加熱融解温度に維持した装置内部に原料を投入し、スクリューを回転させることにより、原料の固形物質が融解し、均一に混練される。あるいは必要に応じて装置内部に投入する前に、原料を予備的に混合することもできる。
本発明の製造方法における圧力、温度、粉体供給速度、ダイ口径、スクリューの形状、スクリュー回転数などの条件は、押出機の機種等によっても異なるが、原料の分解温度以下になるようにそれぞれを組み合わせることが好ましい。特に、原料の分解や変性等の安定性の問題から加熱温度は適切に設定される必要がある。加熱温度は、好ましくは190℃以下、より好ましくは185℃以下、更に好ましくは180℃以下である。加熱温度は、通常150℃以上、好ましくは155℃以上、より好ましくは160℃以上である。加熱温度は、前記下限と前記上限とを任意に組み合わせた範囲内、例えば、150~190℃の範囲内、好ましくは160~190℃の範囲内であり得る。スクリュー回転数は、好ましくは10rpm以上、より好ましくは15rpm以上、更に好ましくは20rpm以上である。スクリュー回転数は、好ましくは250rpm以下であり、より好ましくは200rpm以下、更に好ましくは150rpm以下、更により好ましくは100rpm以下である。スクリュー回転数は、前記下限と前記上限とを任意に組み合わせた範囲内、例えば、20~250rpmの範囲内であり得る。
本発明の固体分散体の好ましい製造方法は、成分(a)と成分(b)と成分(c)と必要により添加剤とを混合して組成物(混合物)を調製する工程、及び前記組成物を加熱溶融押出する工程を含む。前記加熱溶融押出工程では、組成物を2軸型エクストルーダーに供給し、スクリュー回転数20~250rpm、及び処理温度160~190℃で処理することが好ましい。
本発明の固体分散体の製造方法は、加熱溶融押出物を粉砕する工程を更に含むことが好ましい。適当な粉砕機を用いて粉砕すれば任意の粒子径を持った固体分散体粒子を簡単に得ることができ、そのまま散剤、細粒剤、顆粒剤等として使用することができる。本発明の固体分散体(又は固体分散体粒子)に、適宜医薬製剤化成分(添加剤)を加え、製剤化する工程を経て、本発明の固体分散体を含む医薬組成物又は経口固形製剤等の経口製剤とすることもできる。
[医薬組成物]
本発明に包含される医薬組成物(以下、本発明の医薬組成物と表記することがある)は、上記の固体分散体を含む。本発明の医薬組成物中の上記の固体分散体の量は、特に制限されないが、例えば、50~90質量%の範囲内であり得る。本発明の医薬組成物は、上記の固体分散体に加えて、他の活性成分及び/又は添加剤を更に含んでいてもよい。添加剤としては、薬学的に許容し得るものであれば特に制限はなく、例えば、賦形剤、崩壊剤、結合剤、流動化剤、滑沢剤、保存剤、安定化剤、等張化剤、溶解補助剤、コーティング剤、着色剤、懸濁化剤、界面活性剤、甘味料、香料、防腐剤、分散剤、pH調整剤等が挙げられる。添加剤は1種単独で又は2種以上を組み合わせて使用することができる。本発明の医薬組成物中の他の活性成分及び/又は添加剤の量は、特に制限されないが、例えば、10~50質量%の範囲内であり得る。
本発明に包含される医薬組成物(以下、本発明の医薬組成物と表記することがある)は、上記の固体分散体を含む。本発明の医薬組成物中の上記の固体分散体の量は、特に制限されないが、例えば、50~90質量%の範囲内であり得る。本発明の医薬組成物は、上記の固体分散体に加えて、他の活性成分及び/又は添加剤を更に含んでいてもよい。添加剤としては、薬学的に許容し得るものであれば特に制限はなく、例えば、賦形剤、崩壊剤、結合剤、流動化剤、滑沢剤、保存剤、安定化剤、等張化剤、溶解補助剤、コーティング剤、着色剤、懸濁化剤、界面活性剤、甘味料、香料、防腐剤、分散剤、pH調整剤等が挙げられる。添加剤は1種単独で又は2種以上を組み合わせて使用することができる。本発明の医薬組成物中の他の活性成分及び/又は添加剤の量は、特に制限されないが、例えば、10~50質量%の範囲内であり得る。
賦形剤としては、例えば、乳糖、無水乳糖、精製白糖、白糖、D-マンニトール、D-ソルビトール、キシリトール、エリスリトール、デキストリン、結晶セルロース、微結晶セルロース、トウモロコシデンプン、バレイショデンプン、無水リン酸水素カルシウム等が挙げられる。
崩壊剤としては、例えば、カルボキシメチルスターチナトリウム、カルメロース、カルメロースカルシウム、カルメロースナトリウム、クロスカルメロースナトリウム、デンプングリコール酸ナトリウム、クロスポビドン、低置換度ヒドロキシプロピルセルロース、部分アルファ-化デンプン等が挙げられる。
結合剤としては、例えば、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ポリビニルピロリドン、アルファー化デンプン、シロップ、水あめ等が挙げられる。
流動化剤としては、例えば、軽質無水ケイ酸、ケイ酸カルシウム、合成ケイ酸アルミニウム、含水二酸化ケイ素、ステアリン酸カルシウム、メタケイ酸アルミン酸マグネシウム、タルク等が挙げられる。
滑沢剤としては、例えば、ステアリン酸マグネシウム、ステアリン酸カルシウム、ケイ酸マグネシウム、酸化マグネシウム、タルク、硬化油、ショ糖脂肪酸エステル、フマル酸ステアリルナトリウム等が挙げられる。
コーティング剤としては、例えば、ヒドロキシプロピルメチルセルロース、ポリビニルアルコール、ポリソリベート、マクロゴール、タルク等が挙げられる。
着色剤としては、例えば、黄色三二酸化鉄、褐色酸化鉄、三二酸化鉄、酸化チタン、食用青色1号、食用赤色2号、食用赤色3号、食用黄色4号等が挙げられる。
懸濁化剤としては、例えば、ポリソリベート、ポリエチレングリコール、アラビアゴム、グリセリン、ショ糖脂肪酸エステル、ゼラチン等が挙げられる。
甘味剤としては、例えば、アスパルテーム、スクラロース、サッカリン、サッカリンナトリウム、水アメ、果糖等が挙げられる。
界面活性剤としては、例えば、ラウリル硫酸ナトリウム、ポリソリベート、ポリオキシエチレン硬化ヒマシ油等が挙げられる。
本発明の医薬組成物の形態は、特に制限されない。例えば、固形状(粒状、錠剤、カプセル剤等)、ペレット状、又は液状(懸濁液等)の形態として好ましく用いることができる。本発明の医薬組成物の一例として、上記の固体分散体と必要により添加剤とをカプセルに充填してカプセル剤として用いることができる。本発明の医薬組成物の別の例として、上記の固体分散体と必要により添加剤とを混合して打錠し、錠剤として用いることもできる。本発明の医薬組成物の更に別の例として、上記の固体分散体を水に分散させることで懸濁液として用いることもできる。
[経口製剤]
本発明に包含される経口製剤(以下、本発明の経口製剤と表記することがある)は、上記の固体分散体を含む。本発明の経口製剤中の上記の固体分散体の量は、特に制限されないが、例えば、50~90質量%の範囲内であり得る。本発明の経口製剤は、上記の固体分散体に加えて、他の活性成分及び/又は添加剤を更に含んでいてもよい。添加剤としては、上記「医薬組成物」の項目に記載の「添加剤」として例示したものを使用することができる。本発明の経口製剤中の他の活性成分及び/又は添加剤の量は、特に制限されないが、例えば、10~50質量%の範囲内であり得る。本発明の経口製剤としては、例えば、錠剤、カプセル剤、散剤、顆粒剤等の経口固形製剤が挙げられる。一実施形態において、本発明の経口製剤は、錠剤又はカプセル剤であることが好ましい。錠剤には、例えば、素錠、フィルムコーティング錠、糖衣錠、カプレット錠、及びチュアブル錠等が含まれる。
本発明に包含される経口製剤(以下、本発明の経口製剤と表記することがある)は、上記の固体分散体を含む。本発明の経口製剤中の上記の固体分散体の量は、特に制限されないが、例えば、50~90質量%の範囲内であり得る。本発明の経口製剤は、上記の固体分散体に加えて、他の活性成分及び/又は添加剤を更に含んでいてもよい。添加剤としては、上記「医薬組成物」の項目に記載の「添加剤」として例示したものを使用することができる。本発明の経口製剤中の他の活性成分及び/又は添加剤の量は、特に制限されないが、例えば、10~50質量%の範囲内であり得る。本発明の経口製剤としては、例えば、錠剤、カプセル剤、散剤、顆粒剤等の経口固形製剤が挙げられる。一実施形態において、本発明の経口製剤は、錠剤又はカプセル剤であることが好ましい。錠剤には、例えば、素錠、フィルムコーティング錠、糖衣錠、カプレット錠、及びチュアブル錠等が含まれる。
上述した本発明の各実施形態について説明した各構成(性質、構造、機能等)は、本発明に包含される主題を特定するにあたり、どのように組み合わせられてもよい。すなわち、本発明には、本明細書に記載される組み合わせ可能な各構成のあらゆる組み合わせからなる主題が全て包含される。
以下、本発明をより具体的に説明するが、本発明は下記の例に限定されるものではない。
[実施例A1~A11]
デラマニド(ハンマーミル粉砕品、大塚製薬(株))と、ポリビニルカプロラクタム-ポリビニルアセテート-ポリエチレングリコールグラフト共重合体(Soluplus(商標)、BASF社)と、ヒプロメロースフタル酸エステル(HPMCP(商標)HP-50、信越化学工業(株))と、場合によりdl-α-トコフェロール(ビタミンEの一種であるため、以下VEと表記することがある)(富士フィルム和光純薬(株))と、場合によりジブチルヒドロキシトルエン(BHT)とを表1~4に示す量でポリエチレン袋に入れ、数分間混合した。
この混合粉体を、口径1mmφのダイを装着した二軸エクストルーダー(Pharma 11 二軸スクリューエクストルーダー、サーモフィッシャーサイエンティフィック(株))を用いて混練部のバレル温度を160~180℃に設定し、40~80rpmの押出速度で成形処理を行い、棒状の成形体を得た。この成形体を室温で冷まし、粉砕機(錠剤粉砕機HTF-35、大同化工(株))を用いて微粉砕し、53μmの篩にかけて粒子(固体分散体粒子)を得た。
デラマニド(ハンマーミル粉砕品、大塚製薬(株))と、ポリビニルカプロラクタム-ポリビニルアセテート-ポリエチレングリコールグラフト共重合体(Soluplus(商標)、BASF社)と、ヒプロメロースフタル酸エステル(HPMCP(商標)HP-50、信越化学工業(株))と、場合によりdl-α-トコフェロール(ビタミンEの一種であるため、以下VEと表記することがある)(富士フィルム和光純薬(株))と、場合によりジブチルヒドロキシトルエン(BHT)とを表1~4に示す量でポリエチレン袋に入れ、数分間混合した。
この混合粉体を、口径1mmφのダイを装着した二軸エクストルーダー(Pharma 11 二軸スクリューエクストルーダー、サーモフィッシャーサイエンティフィック(株))を用いて混練部のバレル温度を160~180℃に設定し、40~80rpmの押出速度で成形処理を行い、棒状の成形体を得た。この成形体を室温で冷まし、粉砕機(錠剤粉砕機HTF-35、大同化工(株))を用いて微粉砕し、53μmの篩にかけて粒子(固体分散体粒子)を得た。
[実施例A12]
ヒドロキシプロピルメチルセルロースフタレートをヒドロキシプロピルメチルセルロースアセテートサクシネート(Shin-Etsu AQOAT(商標)AS-LF、信越化学工業(株))に変更した点を除き、実施例A11と同様に操作することにより、粒子を作製した。
ヒドロキシプロピルメチルセルロースフタレートをヒドロキシプロピルメチルセルロースアセテートサクシネート(Shin-Etsu AQOAT(商標)AS-LF、信越化学工業(株))に変更した点を除き、実施例A11と同様に操作することにより、粒子を作製した。
[比較例A1]
ヒドロキシプロピルメチルセルロースフタレート及びdl-α-トコフェロール(VE)を添加しない点を除き、実施例A1と同様に操作することにより、粒子を作製した。
ヒドロキシプロピルメチルセルロースフタレート及びdl-α-トコフェロール(VE)を添加しない点を除き、実施例A1と同様に操作することにより、粒子を作製した。
[比較例A2]
ヒドロキシプロピルメチルセルロースフタレート及びdl-α-トコフェロール(VE)を添加しない点を除き、実施例A3と同様に操作することにより、粒子を作製した。
ヒドロキシプロピルメチルセルロースフタレート及びdl-α-トコフェロール(VE)を添加しない点を除き、実施例A3と同様に操作することにより、粒子を作製した。
[比較例A3]
ヒドロキシプロピルメチルセルロースフタレート、dl-α-トコフェロール(VE)、及びジブチルヒドロキシトルエン(BHT)を添加しない点を除き、実施例A11と同様に操作することにより、粒子を作製した。
ヒドロキシプロピルメチルセルロースフタレート、dl-α-トコフェロール(VE)、及びジブチルヒドロキシトルエン(BHT)を添加しない点を除き、実施例A11と同様に操作することにより、粒子を作製した。
[比較例A4]
ポリビニルカプロラクタム-ポリビニルアセテート-ポリエチレングリコールグラフト共重合体及びdl-α-トコフェロールを添加しない点を除き、実施例A1と同様に操作したところ、成形処理においてデラマニドが分解し、成形体を作製することができなかった。
ポリビニルカプロラクタム-ポリビニルアセテート-ポリエチレングリコールグラフト共重合体及びdl-α-トコフェロールを添加しない点を除き、実施例A1と同様に操作したところ、成形処理においてデラマニドが分解し、成形体を作製することができなかった。
[実施例B1~B5]
デラマニド(ハンマーミル粉砕品、大塚製薬(株))と、ビニルピロリドン-ビニルアセテート共重合体(Kollidon(商標)VA64、BASF社)と、ヒドロキシプロピルメチルセルロースフタレート(HPMCP(商標)HP-50、信越化学工業(株))と、dl-α-トコフェロール(VE)(富士フィルム和光純薬(株))とを表5に示す量でポリエチレン袋に入れ、数分間混合した。
この混合粉体を、口径1mmφのダイを装着した二軸エクストルーダー(Pharma 11 二軸スクリューエクストルーダー、サーモフィッシャーサイエンティフィック(株))を用いて混練部のバレル温度を160~170℃に設定し、40rpmの押出速度で成形処理を行い、棒状の成形体を得た。この成形体を室温で冷まし、粉砕機(錠剤粉砕機HTF-35、大同化工(株))を用いて微粉砕し、53μmの篩にかけて粒子(固体分散体粒子)を得た。
デラマニド(ハンマーミル粉砕品、大塚製薬(株))と、ビニルピロリドン-ビニルアセテート共重合体(Kollidon(商標)VA64、BASF社)と、ヒドロキシプロピルメチルセルロースフタレート(HPMCP(商標)HP-50、信越化学工業(株))と、dl-α-トコフェロール(VE)(富士フィルム和光純薬(株))とを表5に示す量でポリエチレン袋に入れ、数分間混合した。
この混合粉体を、口径1mmφのダイを装着した二軸エクストルーダー(Pharma 11 二軸スクリューエクストルーダー、サーモフィッシャーサイエンティフィック(株))を用いて混練部のバレル温度を160~170℃に設定し、40rpmの押出速度で成形処理を行い、棒状の成形体を得た。この成形体を室温で冷まし、粉砕機(錠剤粉砕機HTF-35、大同化工(株))を用いて微粉砕し、53μmの篩にかけて粒子(固体分散体粒子)を得た。
[実施例C]
デラマニド(ハンマーミル粉砕品、大塚製薬(株))40gと、ポリビニルカプロラクタム-ポリビニルアセテート-ポリエチレングリコールグラフト共重合体(Soluplus(商標)、BASF社)160gと、ヒドロキシプロピルメチルセルロースフタレート(HPMCP(商標)HP-50、信越化学工業(株))120gと、dl-α-トコフェロール(VE)(富士フィルム和光純薬(株))9.6gとをポリエチレン袋に入れ、数分間混合した。
この混合粉体を、口径1mmφのダイを装着した二軸エクストルーダー(Pharma 11 二軸スクリューエクストルーダー、サーモフィッシャーサイエンティフィック(株))を用いて混練部のバレル温度を180℃に設定し、40rpmの押出速度で成形処理を行い、棒状の成形体を得た。この成形体を室温で冷まし、粉砕機(錠剤粉砕機HTF-35、大同化工(株))を用いて微粉砕し、53μmの篩にかけて粒子(固体分散体粒子)を得た。
デラマニド(ハンマーミル粉砕品、大塚製薬(株))40gと、ポリビニルカプロラクタム-ポリビニルアセテート-ポリエチレングリコールグラフト共重合体(Soluplus(商標)、BASF社)160gと、ヒドロキシプロピルメチルセルロースフタレート(HPMCP(商標)HP-50、信越化学工業(株))120gと、dl-α-トコフェロール(VE)(富士フィルム和光純薬(株))9.6gとをポリエチレン袋に入れ、数分間混合した。
この混合粉体を、口径1mmφのダイを装着した二軸エクストルーダー(Pharma 11 二軸スクリューエクストルーダー、サーモフィッシャーサイエンティフィック(株))を用いて混練部のバレル温度を180℃に設定し、40rpmの押出速度で成形処理を行い、棒状の成形体を得た。この成形体を室温で冷まし、粉砕機(錠剤粉砕機HTF-35、大同化工(株))を用いて微粉砕し、53μmの篩にかけて粒子(固体分散体粒子)を得た。
[試験例1(溶解性)]
下記の方法1又は2により、実施例A1~A12並びに比較例A1~A3の粒子の溶解性を調べた。
(方法1)
100mLのビーカーに0.3質量%ラウリル硫酸ナトリウム水溶液100mLを入れ、マグネットスターラーで撹拌した(回転数:500rpm)。別途、メノウ乳鉢に、実施例及び比較例の粒子をデラマニドの量が5mgになるように量り取り、ビーカー中の試験液を数mL加えた。約1分間分散させた後、分散液をビーカー中に投入した。分散液投入から5分後、10分後、20分後、及び30分後に、ビーカー中の液6mLをサンプリングし、この液を孔径0.5μm以下のメンブランフィルターで濾過した。初めの濾液2mLを除き、後の濾液4mLを試料溶液とした。試料溶液の波長335nmでの吸光度を測定した。
(方法2)
試験液として0.1質量%ラウリル硫酸ナトリウム水溶液を用いる点を除き、方法1と同様に試料溶液を作製し、波長335nmでの吸光度を測定した。
下記の方法1又は2により、実施例A1~A12並びに比較例A1~A3の粒子の溶解性を調べた。
(方法1)
100mLのビーカーに0.3質量%ラウリル硫酸ナトリウム水溶液100mLを入れ、マグネットスターラーで撹拌した(回転数:500rpm)。別途、メノウ乳鉢に、実施例及び比較例の粒子をデラマニドの量が5mgになるように量り取り、ビーカー中の試験液を数mL加えた。約1分間分散させた後、分散液をビーカー中に投入した。分散液投入から5分後、10分後、20分後、及び30分後に、ビーカー中の液6mLをサンプリングし、この液を孔径0.5μm以下のメンブランフィルターで濾過した。初めの濾液2mLを除き、後の濾液4mLを試料溶液とした。試料溶液の波長335nmでの吸光度を測定した。
(方法2)
試験液として0.1質量%ラウリル硫酸ナトリウム水溶液を用いる点を除き、方法1と同様に試料溶液を作製し、波長335nmでの吸光度を測定した。
実施例A1~A12並びに比較例A1~A3の粒子の溶解性の結果を表1~4に示す。




実施例B1~B5の粒子の溶解性の結果を表5に示す。

[試験例2(デラマニドの純度)]
実施例A8~A10で得られた粒子を、ガラス容器に入れ、外層をアルミ袋で密閉後、60℃環境下で2週間保存した粒子をサンプルとして、デラマニドの純度を調べた。デラマニドの純度は、UPLC(超高速液体クロマトグラフィー、ACQUITY UPLC/TUVシステム、日本ウォーターズ(株))にカラム(ACQUITY UPLC(商標) BEH C18、日本ウォーターズ(株))を接続して、サンプルクーラー15℃、カラムオーブン40℃及び検出器を測定波長254nmに設定した。1.0mg/mLになるように調製した試料溶液を2.0μL注入し,isocratic modeで分析を行った。測定後のチャートを解析し、デラマニドの純度を、面積百分率で示した。結果を表6に示す。
実施例A8~A10で得られた粒子を、ガラス容器に入れ、外層をアルミ袋で密閉後、60℃環境下で2週間保存した粒子をサンプルとして、デラマニドの純度を調べた。デラマニドの純度は、UPLC(超高速液体クロマトグラフィー、ACQUITY UPLC/TUVシステム、日本ウォーターズ(株))にカラム(ACQUITY UPLC(商標) BEH C18、日本ウォーターズ(株))を接続して、サンプルクーラー15℃、カラムオーブン40℃及び検出器を測定波長254nmに設定した。1.0mg/mLになるように調製した試料溶液を2.0μL注入し,isocratic modeで分析を行った。測定後のチャートを解析し、デラマニドの純度を、面積百分率で示した。結果を表6に示す。

表6からdl-α-トコフェロールを配合することにより、デラマニドの高温での保存安定性が向上することが確認された。
[試験例3(イヌの血清中濃度)]
雄性ビーグル犬(約30月齢、体重8~12kg、ノーサンビーグル、(株)ナルク)に、実施例Cの粒子をデラマニドとして50mgとなるように充填したゼラチンカプセルを強制経口投与した直後に、水40mLを強制投与した。投与30分前に飼料CD-5(日本クレア(株))を約50g給餌し、最後の採血まで絶食とした。また、デラマニドを50mg含む市販錠剤(デルディバ(登録商標)錠50mg)をゼラチンカプセルに入れたものを用いて、同様の検討を行った。
採血時点は、投与前、投与後1、2、4、6、8、12及び24時間とし、採血量は約1mLとした(n=6)。得られた血液を10分間3000rpmで遠心分離し、血清を得た。この血清中のデラマニド濃度をLC-MSにより測定した。結果を図1に示す。
雄性ビーグル犬(約30月齢、体重8~12kg、ノーサンビーグル、(株)ナルク)に、実施例Cの粒子をデラマニドとして50mgとなるように充填したゼラチンカプセルを強制経口投与した直後に、水40mLを強制投与した。投与30分前に飼料CD-5(日本クレア(株))を約50g給餌し、最後の採血まで絶食とした。また、デラマニドを50mg含む市販錠剤(デルディバ(登録商標)錠50mg)をゼラチンカプセルに入れたものを用いて、同様の検討を行った。
採血時点は、投与前、投与後1、2、4、6、8、12及び24時間とし、採血量は約1mLとした(n=6)。得られた血液を10分間3000rpmで遠心分離し、血清を得た。この血清中のデラマニド濃度をLC-MSにより測定した。結果を図1に示す。
[製剤例1~16]
製剤例1~16の処方を表7及び8に示す。
製剤例1~16の処方を表7及び8に示す。


表7及び8の処方に従って、ステアリン酸マグネシウム以外の各成分をポリエチレン袋で混合後、ステアリン酸マグネシウムを添加して更に混合した。混合粉末をカプレット(18.0×10.8mm)の臼杵を付けたロータリー打錠機(クリーンプレス、(株)菊水製作所)を用い、打錠圧20kNで打錠し、1錠あたり100mgのデラマニドを含有するカプレット錠を調製した。
製剤例5、7及び16のカプレット錠に対して、表9の処方のヒドロキシプロピルメチルセルロース、PEG6000、酸化チタン、タルク、黄色三二酸化鉄を精製水に溶解及び懸濁させたフィルムコーティング液をフィルムコーティング機(パウレックコーターPRC-GTXmini、(株)パウレック)内でカプレット錠に対して噴霧し、フィルムコーティング錠を調製した。

製剤例5、7及び16のカプレット錠に対して、表9の処方のヒドロキシプロピルメチルセルロース、PEG6000、酸化チタン、タルク、黄色三二酸化鉄を精製水に溶解及び懸濁させたフィルムコーティング液をフィルムコーティング機(パウレックコーターPRC-GTXmini、(株)パウレック)内でカプレット錠に対して噴霧し、フィルムコーティング錠を調製した。
Claims (20)
- 下記の成分(a)、(b)、及び(c)を含む固体分散体。
(a)デラマニド
(b)ポリビニルカプロラクタム-ポリビニルアセテート-ポリエチレングリコールグラフト共重合体及びビニルピロリドン-ビニルアセテート共重合体からなる群より選ばれた少なくとも1種
(c)ヒドロキシプロピルメチルセルロースフタレート及びヒドロキシプロピルメチルセルロースアセテートサクシネートからなる群より選ばれた少なくとも1種 - 成分(a)と成分(b)との質量比が1:1~1:20の範囲内である、請求項1に記載の固体分散体。
- 成分(a)と成分(b)との質量比が1:1~1:8の範囲内である、請求項1に記載の固体分散体。
- 成分(a)と成分(b)との質量比が1:1~1:5の範囲内である、請求項1に記載の固体分散体。
- 成分(a)と成分(c)との質量比が1:1~1:10の範囲内である、請求項1に記載の固体分散体。
- 成分(a)と成分(c)との質量比が1:1~1:8の範囲内である、請求項1に記載の固体分散体。
- 成分(a)と成分(c)との質量比が1:1~1:5の範囲内である、請求項1に記載の固体分散体。
- 成分(a)と成分(b)との質量比が1:1~1:5の範囲内であり、成分(a)と成分(c)との質量比が1:1~1:5の範囲内である、請求項1に記載の固体分散体。
- 成分(b)がポリビニルカプロラクタム-ポリビニルアセテート-ポリエチレングリコールグラフト共重合体を含む、請求項1に記載の固体分散体。
- 成分(b)がビニルピロリドン-ビニルアセテート共重合体を含む、請求項1に記載の固体分散体。
- 成分(c)がヒドロキシプロピルメチルセルロースフタレートを含む、請求項1に記載の固体分散体。
- 成分(b)がポリビニルカプロラクタム-ポリビニルアセテート-ポリエチレングリコールグラフト共重合体を含み、成分(c)がヒドロキシプロピルメチルセルロースフタレートを含む、請求項1に記載の固体分散体。
- 成分(b)がビニルピロリドン-ビニルアセテート共重合体を含み、成分(c)がヒドロキシプロピルメチルセルロースフタレートを含む、請求項1に記載の固体分散体。
- ビタミンEを更に含む、請求項1に記載の固体分散体。
- ビタミンEがdl-α-トコフェロールである、請求項14に記載の固体分散体。
- 加熱溶融押出物又はその粉砕物である、請求項1~15のいずれかに記載の固体分散体。
- 請求項1~16のいずれかに記載の固体分散体を含む医薬組成物。
- 請求項1~16のいずれかに記載の固体分散体を含む経口固形製剤。
- 錠剤又はカプセル剤である、請求項18に記載の経口固形製剤。
- 成分(a)、(b)、及び(c)を含む組成物を加熱溶融押出する工程を含む、請求項1~16のいずれかに記載の固体分散体を製造する方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2022-122027 | 2022-07-29 | ||
| JP2022122027 | 2022-07-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024024938A1 true WO2024024938A1 (ja) | 2024-02-01 |
Family
ID=89706652
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2023/027723 WO2024024938A1 (ja) | 2022-07-29 | 2023-07-28 | デラマニド含有固体分散体 |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2024024938A1 (ja) |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019240104A1 (ja) * | 2018-06-11 | 2019-12-19 | 大塚製薬株式会社 | デラマニド含有組成物 |
| WO2020122243A1 (ja) * | 2018-12-14 | 2020-06-18 | 富士フイルム株式会社 | 医薬組成物及びその製造方法 |
-
2023
- 2023-07-28 WO PCT/JP2023/027723 patent/WO2024024938A1/ja unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019240104A1 (ja) * | 2018-06-11 | 2019-12-19 | 大塚製薬株式会社 | デラマニド含有組成物 |
| WO2020122243A1 (ja) * | 2018-12-14 | 2020-06-18 | 富士フイルム株式会社 | 医薬組成物及びその製造方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3914252B1 (en) | Pharmaceutical compositions of nilotinib | |
| AU2021277731B2 (en) | Improved formulations of deferasirox and methods of making the same | |
| EP3130354B1 (en) | Posaconazole pharmaceutical composition and preparation method, application and pharmaceutical preparation thereof | |
| JP5794650B2 (ja) | 難溶性薬物の溶解性改善製剤 | |
| EP1849830B1 (en) | Finely divided composition containing poorly water soluble substance | |
| US20100143459A1 (en) | Pharmaceutical dosage form for oral administration of tyrosine kinase inhibitor | |
| JP6666490B2 (ja) | Cgrp活性化合物の錠剤製剤 | |
| US12036315B2 (en) | Pharmaceutical composition for oral administration comprising enzalutamide | |
| WO2015124995A1 (en) | Solid dosage forms of rivaroxaban | |
| WO2014125352A1 (en) | Pharmaceutical compositions comprising tadalafil | |
| CA3008634A1 (en) | Pharmaceutical compositions comprising phenylaminopyrimidine derivative | |
| JP4926319B2 (ja) | 固体医薬剤形のための粉末形態の可溶化賦形剤 | |
| WO2024024938A1 (ja) | デラマニド含有固体分散体 | |
| AU2014215920B2 (en) | Solid dispersion comprising amorphous cilostazol | |
| JP2020090470A (ja) | アジルサルタン及びアムロジピンを含有する医薬組成物及びその製造方法 | |
| US9775832B2 (en) | Pharmaceutical composition for oral administration | |
| AU2018419112A1 (en) | Instant release pharmaceutical preparation of anticoagulant and preparation method therefor | |
| RU2792098C1 (ru) | Способ получения фармацевтической композиции с такролимусом (варианты) и фармацевтическая композиция, полученная указанными способами | |
| IE981008A1 (en) | Microparticles Containing Water Insoluble Active Agents | |
| WO2024033703A1 (en) | Amorphous solid dispersions comprising naporafenib | |
| WO2012097867A1 (en) | Cladribine particles and pharmaceutical compositions comprising them |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23846668 Country of ref document: EP Kind code of ref document: A1 |