WO2023138689A1 - 抑制LPA(Apo(a))蛋白表达的组合物和方法 - Google Patents
抑制LPA(Apo(a))蛋白表达的组合物和方法 Download PDFInfo
- Publication number
- WO2023138689A1 WO2023138689A1 PCT/CN2023/073456 CN2023073456W WO2023138689A1 WO 2023138689 A1 WO2023138689 A1 WO 2023138689A1 CN 2023073456 W CN2023073456 W CN 2023073456W WO 2023138689 A1 WO2023138689 A1 WO 2023138689A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- lpa
- dsrna
- subject
- agent
- nucleotides
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7088—Compounds having three or more nucleosides or nucleotides
- A61K31/713—Double-stranded nucleic acids or oligonucleotides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
Definitions
- Some embodiments of the present invention relate to compositions and methods useful for inhibiting the expression of LPA (Apo(a)) protein.
- Lp(a) particles are heterogeneous low-density lipoprotein particles mainly expressed in the liver (Witztum and Ginsberg, J Lipid Res. 2016 Mar;57(3):336-9). They are composed of apolipoprotein (a) (Apolipoprotein (a)) (Apo (a) or Lp (a) encoded by LPA gene) connected to LDL-like particles through ApoB polypeptide. Genetically defined high Lp(a) particle serum levels are unaffected by diet and exercise and are associated with increased risk of cardiovascular disease through an associated atherosclerotic potential (Alonso et al., Journal of the American College of Cardiology Vol. 63, No. 19, 2014).
- Lp(a) particles in patients are a highly prevalent independent genetic risk factor for coronary heart disease and aortic stenosis (Saeedi and Frohlich Clinical Diabetes and Endocrinology (2016) 2:7).
- Analysis of Lp(a) levels in multiple studies suggests that high Lp(a) levels are an independent risk factor for cardiovascular disease, stroke and other related conditions, including atherosclerotic stenosis.
- genome-wide association analysis also found LPA as a genetic risk factor for diseases such as atherosclerotic stenosis.
- Significant reductions in cardiovascular events were observed when both Lp(a) and LDL levels were lowered in hyperlipidemic patients using therapeutic lipoproteinapheresis. Accordingly, there is a need for therapeutic agents and treatments associated with these and other LPA-related diseases.
- a double-stranded ribonucleic acid (dsRNA) reagent that inhibits the expression of LPA (Apo(a)), the dsRNA reagent comprises a sense strand and an antisense strand, and the nucleotide position 2 to 18 in the antisense strand comprises an LPA RNA A region of transcript complementarity, wherein the region of complementarity comprises at least 15 contiguous nucleotides that differ from one of the antisense sequences listed in Tables 1-3 by 0, 1, 2 or 3 nucleotides, and optionally comprises a targeting ligand.
- dsRNA double-stranded ribonucleic acid
- the region complementary to the LPA RNA transcript comprises at least 15, 16, 17, 18, or 19 contiguous nucleotides that differ by no more than 3 nucleotides from one of the antisense sequences listed in Tables 1-3.
- the antisense strand of the dsRNA is at least substantially complementary to any target region of human LPA gene mRNA and is provided in one of Tables 1-3. In some embodiments, the antisense strand of the dsRNA is fully complementary to any target region of human LPA gene mRNA and is provided in one of Tables 1-3.
- the dsRNA reagent comprises any one of the sense strand sequences listed in Tables 1-3, wherein the sense strand sequence is at least substantially complementary to the antisense strand sequence in the dsRNA reagent. In certain embodiments, the dsRNA reagent comprises any one of the sense strand sequences listed in Tables 1-3, wherein the sense strand sequence is fully complementary to the antisense strand sequence in the dsRNA reagent. In some embodiments, the dsRNA agent comprises any one of the antisense strand sequences listed in Tables 1-3. In some embodiments, the dsRNA agent comprises any one of the sequences listed in Tables 1-3 as a duplex sequence.
- the dsRNA agent comprises a sense strand that differs from formula (A) by 0, 1, 2, or 3 nucleotides: 5'-Z 1 GUUAUCGAGGCACAUAZ 2 -3' formula (A), wherein Z 1 is a nucleotide sequence comprising 0-15 nucleotide motifs, and Z 2 is selected from one of A, U, C, G or is absent. In certain embodiments, Z is A.
- the Z nucleotide sequence is selected from the following motifs: A, AA, UA, GA, CA, AGA, UGA, GGA, CGA, UAGA, CAGA, AAGA, ACAGA, GACAGA, GGACAGA, UGGACAGA, AUGGACAGA, AAUGGACAGA, UAAUGGACAGA, GUAAUGGACAGA, GGUAAUGGACAGA, UGGUAAUGGACAGA, AUGG One of UAAUGGACAGA or nonexistent.
- Z is a nucleotide sequence comprising 1, 2, 3 or 4 nucleotide motifs selected from the group consisting of: A, AA, UA, GA, CA, AGA, UGA, GGA, CGA, UAGA, CAGA, AAGA, ACAGA.
- the dsRNA agent comprises an antisense strand that differs from formula (B) by 0, 1, 2, or 3 nucleotides: 5'-Z 3 UAUGUGCCUCGAUAACZ 4 -3' formula (B), wherein Z 3 is selected from one of A, U, C, G or is absent, and Z 4 is a nucleotide sequence comprising 0-15 nucleotide motifs.
- Z3 is U.
- the Z nucleotide sequence is selected from the following motifs: U, UU, UA, UC, UG, UCU, UCA, UCC, UCG, UCUC, UCUA, UCUG, UCUU, UCUGU, UCUGUC, UCUCUU, UCUCGA, UCUGUCC, UCUGUCCA, UCUGUCCAU, UCUGUCCAU, UCUGUCCAUU, UCUGUCCAUUA, UCUGUC CAUUAC, UCUGUCCAUUACC, UCUGUCCAUUACCA, UCUGUCCAUUACCAU, or not present.
- Z is a nucleotide sequence comprising 1, 2, 3 or 4 nucleotide motifs selected from the group consisting of: U, UU, UA, UC, UG, UCU, UCA, UCC, UCG, UCUC, UCUA, UCUG, UCUU.
- the dsRNA reagent pack Containing a sense strand and an antisense strand, the sense strand and the antisense strand respectively comprise a nucleotide sequence having a difference of 0, 1, 2 or 3 nucleotides from the formula (A) and formula (B) described herein, and optionally include a targeting ligand.
- the sense strand (A) and antisense strand (B) of the dsRNA agent are each no more than 35 nucleotides in length.
- the Z1 and Z4 nucleotide motifs are fully or partially complementary.
- the Z2 and Z3 nucleotide motifs are fully or partially complementary.
- the sense strand is complementary or substantially complementary to the antisense strand, and the region of complementarity is between 16 and 23 nucleotides in length. In some embodiments, the complementary region is 19-21 nucleotides in length.
- the sense strand is no more than 35 nucleotides in length, including a region complementary to the antisense strand, comprising at least 15, 16, 17, 18, or 19 nucleotides.
- the dsRNA reagent comprises a sense strand that differs from formula (C) by 0, 1, 2, or 3 nucleotides: 5'-Z 5 CCAAGCUUGGUCAUCUZ 6 -3' formula (C), wherein Z is a nucleotide sequence comprising 0-15 nucleotide motifs, and Z is selected from one of A, U, C, G or is absent.
- Z6 is A.
- the Z nucleotide sequence is selected from the following motifs: G, AG, UG, GG, CG, AUG, UUG, GUG, CUG, UUUG, CUUG, AUUG, ACUUG, AACUUG, GAACUUG, AGAACUUG, AAGAACUUG, GAAGAACUUG, GGAAGAACUUG, AGGAAGAACUUG, CAGGAAGAACUUG, ACAGGAAGAA One of CUUG, CACAGGAAGAACUUG, or nonexistent.
- Z is a nucleotide sequence comprising 1, 2, 3 or 4 nucleotide motifs selected from the group consisting of G, AG, UG, GG, CG, AUG, UUG, GUG, CUG, UUUG, CUUG, AUUG.
- the dsRNA agent comprises an antisense strand that differs from formula (D) by 0, 1, 2, or 3 nucleotides: 5'-Z 7 AGAUGACCAAGCUUGGZ 8 -3' formula (D), wherein Z 7 is selected from one of A, U, C, G or does not exist, and Z 8 is a nucleotide sequence comprising 0-15 nucleotide motifs.
- Z7 is U.
- the Z nucleotide sequence is selected from the following motifs: C, CU, CA, CC, CG, CAU, CAA, CAC, CAG, CAAC, CAAA, CAAG, CAAU, CAAGU, CAAGUU, CAACUU, CAACGA, CAAGUUC, CAAGUUCU, CAAGUUCUUC, CAAGUUCUUCC, CAAGUUCUUCCU, CAAGUUCUUCCUG, CAAGU UCUUCCUGU, CAAGUUCUUCCUGUG or does not exist.
- Z is a nucleotide sequence comprising 1, 2, 3 or 4 nucleotide motifs selected from the group consisting of: C, CU, CA, CC, CG, CAU, CAA, CAC, CAG, CAAC, CAAA, CAAG, CAAU.
- the dsRNA agent comprises a sense strand and an antisense strand comprising a nucleotide sequence represented herein that differs from formula (C) and formula (D) by 0, 1, 2, or 3 nucleotides, respectively, and optionally comprising a targeting ligand.
- the sense strand (C) and antisense strand (D) of the dsRNA agent are each no more than 35 nucleotides in length.
- the Z5 and Z8 nucleotide motifs are all or are partially complementary. In certain embodiments, the Z6 and Z7 nucleotide motifs are fully or partially complementary.
- the sense strand is complementary or substantially complementary to the antisense strand, and the region of complementarity is between 16 and 23 nucleotides in length. In some embodiments, the complementary region is 19-21 nucleotides in length. In some embodiments, wherein the sense strand is no more than 35 nucleotides in length, including a region complementary to the antisense strand, comprising at least 15, 16, 17, 18, or 19 nucleotides.
- the dsRNA reagent comprises a sense strand that differs from formula (E) by 0, 1, 2, or 3 nucleotides: 5'-Z 9 GACAGAGUUAUCGAGGZ 10 -3' formula (E), wherein Z 9 is a nucleotide sequence comprising 0-15 nucleotide motifs, and Z 10 is selected from one of A, U, C, G or is absent. In certain embodiments, Z 10 is A.
- the Z nucleotide sequence is selected from the following motifs: G, AG, UG, GG, CG, AUG, UUG, GUG, CUG, CAUG, UAUG, GAUG, AAUG, UGAUG, GUGAUG, GGUGAUG, UGGUGAUG, AUGGUGAUG, CAUGGUGAUG, CCAUGGUGAUG, ACCAUGGUGAUG, U One of ACCAUGGUGAUG, CUACCAUGGUGAUG, GCUACCAUGGUGAUG, or nonexistent.
- Z is a nucleotide sequence comprising 1, 2, 3 or 4 nucleotide motifs selected from the group consisting of G, AG, UG, GG, CG, AUG, UUG, GUG, CUG, CAUG, UAUG, GAUG, AAUG.
- the dsRNA agent comprises an antisense strand that differs from formula (F) by 0, 1, 2 or 3 nucleotides: 5'-Z 11 CCUCGAUAACUCUGUCZ 12 -3' formula (F), wherein Z 11 is selected from one of A, U, C, G or does not exist, and Z 12 is a nucleotide sequence comprising 0-15 nucleotide motifs.
- Z 11 is U.
- the Z nucleotide sequence is selected from the following motifs: C, CU, CA, CC, CG, CAU, CAA, CAC, CAG, CAUA, CAUG, CAUC, CAUU, CAUCA, CAUCAC, CAUGUU, CAUGGA, CAUCACC, CAUCACCA, CAUCACCAU, CAUCACCAUG, CAUCACCAUGG, CAUCACCAUGGU, CAUCACCAUGGU A.
- CAUCACCAUGGUAG CAUCACCAUGGUAGC or does not exist.
- Z is a nucleotide sequence comprising 1, 2, 3 or 4 nucleotide motifs selected from the group consisting of: C, CU, CA, CC, CG, CAU, CAA, CAC, CAG, CAUA, CAUG, CAUC, CAUU.
- the dsRNA agent comprises a sense strand and an antisense strand comprising a nucleotide sequence represented herein that differs from formula (E) and formula (F) by 0, 1, 2, or 3 nucleotides, respectively, and optionally comprising a targeting ligand.
- the sense strand (F) and antisense strand (F) of the dsRNA agent are each no more than 35 nucleotides in length.
- the Z9 and Z12 nucleotide motifs are fully or partially complementary.
- the Z 10 and Z 11 nucleotide motifs are fully or partially complementary.
- the sense strand is complementary or substantially complementary to the antisense strand, and the region of complementarity is between 16 and 23 nucleotides in length. in some implementations In this case, the complementary region is 19-21 nucleotides in length.
- the sense strand is no more than 35 nucleotides in length, including a region complementary to the antisense strand, comprising at least 15, 16, 17, 18, or 19 nucleotides.
- the dsRNA agent comprises at least one modified nucleotide.
- all or substantially all of the nucleotides of the antisense strand are modified nucleotides.
- at least one modified nucleotide includes: 2'-O-methyl nucleotides, 2'-fluoro nucleotides, 2'-deoxy nucleotides, 2',3'-seco nucleotide mimics, locked nucleotides, unlocked nucleic acid nucleotides (UNA), glycol nucleic acid nucleotides (GNA), 2'-F-arabino nucleotides, 2'-methoxyethyl nucleotides, abasic Nucleotides, ribitol, inverted nucleotides, inverted abasic nucleotides, inverted 2'-OMe nucleotides, inverted 2'-deoxynucleotides, 2'-amin
- the antisense strand comprises 15 or more modified nucleotides independently selected from 2'-O-methyl nucleotides and 2'-fluoro nucleotides, wherein fewer than 6 2'-fluoro nucleotides are modified nucleotides.
- the antisense strand comprises 3 or 5 2'-fluoro nucleotides, preferably, the antisense strand comprises 5 2'-fluoro nucleotides.
- the sense strand comprises 15 or more modified nucleotides independently selected from 2'-O-methyl nucleotides and 2'-fluoro nucleotides, wherein fewer than 4 2'-fluoro nucleotides are modified nucleotides. In certain embodiments, the sense strand comprises 3 2'-fluoro nucleotides.
- the antisense strand comprises 15 or more modified nucleotides independently selected from 2'-O-methyl nucleotides and 2'-fluoro nucleotides, wherein at least 16 of the modified nucleotides are 2'-O-methyl nucleotides and positions 2, 7, 12, 14 and/or 16 at the 5' end of the antisense strand are 2'-fluoro nucleotide modified nucleotides (calculated from the first paired nucleotide in the 5' of the antisense strand).
- the sense strand comprises 15 or more modified nucleotides independently selected from 2'-O-methyl nucleotides and 2'-fluoro nucleotides, wherein at least 18 of the modified nucleotides are 2'-O-methyl nucleotides and positions 9, 11 and/or 13 at the 3' end of the sense strand are 2'-fluoro nucleotide modified nucleotides (counting from the first paired nucleotide of the sense strand 3').
- the antisense strand comprises in the direction from the 5' end to the 3' end, the nucleotides at positions 2, 7, 12, 14, and 16 of the antisense strand are 2'-fluoro-modified nucleotides, counted from the first paired nucleotide at the 5' end of the antisense strand, and the nucleotides at other positions in each antisense strand are independently non-fluorine-modified nucleotides.
- the antisense strand includes positions 2, 5, 12, 14, and 18 in the direction from the 5' end to the 3' end of the antisense strand that are 2'-fluoro-modified nucleotides, counting from the first paired nucleotide 5' of the antisense strand, and each nucleotide at other positions in the antisense strand is independently a non-fluoro-modified nucleotide.
- the sense strand comprises in the direction from the 3' end to the 5' end, the nucleotides at positions 9, 11 and 13 of the sense strand are 2'-fluoro-modified nucleotides, counting from the first paired nucleotide at the 3' end of the sense strand, and each nucleotide is at its other position in the sense strand Other positions are independently non-fluorine-modified nucleotides.
- the dsRNA agent includes E-vinylphosphonate nucleotides at the 5' end of the guide strand. In certain embodiments, the dsRNA agent comprises at least one phosphorothioate internucleoside linkage.
- the sense strand comprises at least one phosphorothioate internucleoside linkage. In some embodiments, the antisense strand comprises at least one phosphorothioate internucleoside linkage. In some embodiments, the sense strand comprises 1, 2, 3, 4, 5, or 6 phosphorothioate internucleoside linkages. In some embodiments, the antisense strand comprises 1, 2, 3, 4, 5, or 6 phosphorothioate internucleoside linkages. In certain embodiments, all or substantially all nucleotides of the sense and antisense strands are modified nucleotides. In some embodiments, the modified sense strand is a modified sense strand sequence listed in Tables 2-3.
- the modified antisense strand is a modified antisense strand sequence listed in Tables 2-3.
- the sense strand is complementary or substantially complementary to the antisense strand, and the region of complementarity is between 16 and 23 nucleotides in length.
- the complementary region is 19-21 nucleotides in length.
- the complementary region is 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 or 30 nucleotides in length.
- each strand is no more than 40 nucleotides in length.
- each strand is no more than 30 nucleotides in length.
- each strand is no more than 25 nucleotides in length.
- each strand is no more than 23 nucleotides in length.
- a dsRNA agent comprises at least one modified nucleotide and further comprises one or more targeting or linking groups.
- one or more targeting groups or linking groups are conjugated to the sense strand.
- the targeting group or linking group includes N-acetyl-galactosamine (GalNAc).
- the targeting moiety in the targeting group has the following structural fragments,
- p 1 or 2.
- the targeting group has the following structure:
- the dsRNA agent comprises a targeting group conjugated to the 5'-end of the sense strand. In some embodiments, the dsRNA agent comprises a targeting group conjugated to the 3'-end of the sense strand. In some embodiments, the antisense strand comprises an inverted abasic residue at the 3'-end. In certain embodiments, the sense strand comprises one or two inverted abasic residues at the 3' and/or 5' ends. In certain embodiments, the sense strand comprises one or two isomannitol residues at the 3' and/or 5' ends. In certain embodiments, the sense strand independently comprises an isomannitol residue at each of the 3' and 5' ends.
- the sense strand independently comprises an isomannitol residue at the 3' and 5' ends, and further comprises a targeting group conjugated to the 5'-end, preferably the targeting group is the aforementioned GLS-15.
- the dsRNA reagent has two blunt ends.
- at least one strand comprises a 3' overhang that is at least 1 nucleotide long. In some embodiments, at least one strand comprises a 3' overhang that is at least 2 nucleotides long.
- a double-stranded ribonucleic acid (dsRNA) reagent that suppresses LPA (Apo(a)) expression
- the dsRNA reagent comprises a sense strand and an antisense strand, and comprises a region complementary to the LPA RNA transcript at nucleotide positions 2 to 18 in the antisense strand, the antisense strand is completely or partially complementary to the sense strand, and optionally includes a targeting ligand, wherein each strand is 14 to 30 nucleotides in length, wherein the sense strand sequence can be represented by formula (I):
- each N′ F represents a 2′-fluoro-modified nucleotide
- each N′ N1 , N ′ N2 , N′ N3 and N′ N4 independently represents a modified or unmodified nucleotide
- each N' L independently represents a modified or unmodified nucleotide but does not represent a 2'-fluoro-modified nucleotide
- n' is an integer of 0-7
- m' is an integer of 0-3.
- each N'N3 represents a 2'-fluoro-modified nucleotide
- N'N1 , N'N2 and N'N4 independently represent a modified or unmodified nucleotide but not a 2'-fluoro-modified nucleotide
- m' is 1.
- each N'N4 represents a 2'-fluoro-modified nucleotide
- N'N1 , N'N2 and N'N3 independently represent a modified or unmodified nucleotide but not a 2'-fluoro-modified nucleotide
- m' is 1.
- n' is 3 and m' is 1; or n' is 0 and m' is 0; or n' is 3 and m' is 3. In certain embodiments, there are only 3 2'-fluoro modified nucleotides in formula (I).
- the present invention relates to open-circle nucleic acid (UNA) oligomers for use in therapy.
- RNA locked nucleic acid
- UNA locked nucleic acid
- RNA is an acyclic analog of RNA in which the bond between the C2' and C3' atoms of the ribose ring has been severed.
- Incorporation of UNA has been shown to be well tolerated and in some cases even enhance the activity of siRNA gene silencing (Meghan A. et al. "Locked vs. unlocked nucleic acids (LNA vs. UNA): contrasting structures work towards common therapeutic goals". Chem. Soc. Rev., 2011, 40, 5680–568 9).
- UNA is a thermolabile modification, and replacing ribonucleotides with UNA reduces base-pairing strength and duplex stability.
- Strategically placing UNA in the seed region of the antisense strand of siRNA can reduce the off-target activity in the gene silencing mechanism mediated by microRNA (miRNA).
- miRNAs recognize target genes mainly through base pairing between the antisense seed region (2-8 from the 5' end) and target mRNA for gene suppression. Each miRNA potentially regulates a large number of genes.
- the antisense strand of siRNA loaded by the RNA-induced silencing complex (RISC) can also potentially regulate a large number of unintended genes through miRNA-mediated mechanisms.
- RISC RNA-induced silencing complex
- thermolabile nucleotides such as UNA
- UNU thermolabile nucleotides
- RNA oligonucleotides or complexes of RNA oligonucleotides contain at least one UNA nucleotide monomer in the seed region (Narendra Vaish et al. "Improved specificity of gene silencing by siRNAs containing unlocked nucleobase analog". Nucleic Acids Research, 2011, Vol. 39, No. 51823-1832).
- RNA oligonucleotides or complexes of RNA oligonucleotides according to the present technology include, but are not limited to:
- UNA is well tolerated in terms of siRNA activity. In some cases, UNA can lead to enhanced activity.
- Exemplary UNA monomers that can be used in this technical solution include, but are not limited to:
- compositions comprising any embodiment of the above-mentioned dsRNA agent aspect of the present invention.
- the composition also includes a pharmaceutically acceptable carrier.
- the composition also comprises one or more additional therapeutic agents such as HMg Co-A reductase inhibitors (statins), ezetimibe, PCSK-9 inhibitors, CTEP inhibitors, ANGPTL3-targeting therapies, AGT-targeting therapies, APOC3-targeting therapies, and niacin, or a combination of any of the foregoing.
- the compositions are packaged in kits, containers, wrappers, dispensers, pre-filled syringes, or vials.
- the composition is formulated for subcutaneous administration or is formulated for intravenous (IV) administration.
- a cell comprising any embodiment of the above-mentioned dsRNA reagent aspect of the present invention.
- the cells are mammalian cells, optionally human cells.
- a method for inhibiting the expression of LPA gene in a cell comprising: (i) preparing a cell comprising an effective amount of the above-mentioned dsRNA agent or any embodiment of the above-mentioned composition aspect.
- the method further comprises: (ii) maintaining the prepared cells for a sufficient time to obtain the degradation of the mRNA transcript of the LPA gene, thereby inhibiting the expression of the LPA gene in the cells.
- the cells are in a subject and the dsRNA agent is administered to the subject subcutaneously.
- the cells are in a subject and the dsRNA agent is administered to the subject by IV administration.
- the method further comprises assessing inhibition of the LPA gene after administration of the dsRNA agent to the subject, wherein the means for assessing include: (i) determining one or more physiological characteristics of the LPA-associated disease or disorder in the subject, and (ii) comparing the determined physiological characteristics with a baseline pre-treatment physiological characteristic of the LPA-related disease or disorder and/or a control physiological characteristic of the LPA-related disease or disorder, wherein the result of the comparison indicates the presence or absence of inhibition of LPA gene expression in the subject.
- the determined physiological characteristic is the level of Lp(a) in blood. A decrease in the level of LPA in the blood indicates a decrease in the expression of the LPA gene in the subject.
- a method of inhibiting LPA gene expression in a subject which comprises administering to the subject an effective amount of the aforementioned embodiment of the dsRNA agent or the aforementioned embodiment of the composition.
- the dsRNA agent is administered subcutaneously to the subject.
- the dsRNA agent is administered to the subject by IV administration.
- the method further comprises: assessing inhibition of the LPA gene after administration of the dsRNA agent, wherein the means for assessing comprises: (i) determining one or more physiological characteristics of an LPA-associated disease or disorder in the subject; (ii) comparing the determined physiological characteristics with a baseline pre-treatment physiological characteristic of the LPA-related disease or disorder and/or a control physiological characteristic of the LPA-related disease or disorder; wherein the result of the comparison indicates the presence or absence of inhibition of LPA gene expression in the subject.
- the determined physiological characteristic is the level of Lp(a) in blood. A decrease in the level of LPA in the blood indicates a decrease in the expression of the LPA gene in the subject.
- a method for treating a disease or condition related to LPA protein which comprises: administering to a subject an effective amount of any embodiment of the aforementioned dsRNA reagent aspect of the present invention or any embodiment of the aforementioned composition of the present invention to inhibit LPA gene expression.
- the LPA-associated disorder is cardiovascular disease, wherein said cardiovascular disease includes Berger's disease, peripheral artery disease, coronary artery disease, metabolic syndrome, acute coronary syndrome, aortic stenosis, aortic regurgitation, aortic dissection, retinal artery occlusion, cerebrovascular disease, mesenteric ischemia, superior mesenteric artery occlusion, renal artery stenosis, stable/unstable cardiac Angina, acute coronary syndrome, heterozygous or homozygous familial hypercholesterolemia, hyperapolipoprotein beta lipoproteinemia, cerebrovascular atherosclerosis, cerebrovascular disease and venous thrombosis, stroke, atherosclerosis, thrombosis, coronary heart disease or aortic stenosis and/or any other disease or pathology associated with elevated levels of Lp(a)-containing particles.
- said cardiovascular disease includes Berger's disease, peripheral artery disease, coronary artery disease, metabolic syndrome, acute coronary syndrome
- the method further comprises: administering to the subject an additional treatment regimen.
- the additional treatment regimen includes treatment of an LPA-associated disease or disorder.
- additional treatment regimens include: administering to a subject one or more LPA antisense polynucleotides of the invention; administering to a subject a non-LPA dsRNA therapeutic agent; and effecting behavioral modification in the subject.
- the non-LPA dsRNA therapeutic agent is an additional therapeutic agent such as an HMg Co-A reductase inhibitor (statin), ezetimibe, a PCSK-9 inhibitor, a CTEP inhibitor, an ANGPTL3-targeting therapy, an APOC3-targeting therapy, and niacin, or a combination of any of the foregoing.
- an HMg Co-A reductase inhibitor statin
- ezetimibe ezetimibe
- PCSK-9 inhibitor a PCSK-9 inhibitor
- CTEP inhibitor a CTEP inhibitor
- an ANGPTL3-targeting therapy an APOC3-targeting therapy
- niacin a combination of any of the foregoing.
- the dsRNA agent is administered subcutaneously to the subject. In certain embodiments, the dsRNA agent is administered to the subject by IV administration. In some embodiments, the method further comprises determining the efficacy of the administered double-stranded ribonucleic acid (dsRNA) agent in the subject.
- dsRNA double-stranded ribonucleic acid
- the means for determining the efficacy of a treatment in a subject comprises: (i) determining one or more physiological characteristics of an LPA-related disease or disorder in the subject; (ii) comparing the determined physiological characteristics with baseline pre-treatment physiological characteristics of the LPA-related disease or disorder, wherein the comparison indicates one or more of the presence, absence, and level of efficacy of administering a double-stranded ribonucleic acid (dsRNA) agent to the subject.
- the determined physiological characteristic is the level of Lp(a) in blood. A decrease in the level of LPA in the blood indicates the presence of effectiveness of administering the double-stranded ribonucleic acid (dsRNA) agent to the subject.
- a method of reducing the level of LPA protein in a subject compared to the baseline pre-treatment level of LPA protein in the subject comprising administering to the subject an effective amount of any embodiment of the aforementioned dsRNA agent aspect of the invention or any embodiment of the aforementioned composition of the invention to reduce the level of LPA gene expression.
- the dsRNA agent is administered to the subject subcutaneously or IV.
- a method of altering the physiological characteristics of an LPA-associated disease or disorder in a subject as compared to the baseline pre-treatment physiological characteristics of the LPA-associated disease or disorder in the subject comprising administering to the subject an effective amount of any embodiment of the aforementioned dsRNA agent aspect of the invention or any embodiment of the aforementioned composition of the invention to alter the physiological characteristics of the LPA-associated disease or disorder in the subject.
- the dsRNA agent is administered to the subject subcutaneously or IV.
- the physiological characteristic is Lp(a) levels in blood.
- Duplexes AV00122 to AD00484-1, AD00474-2, AV01867-AV01968 are shown in Table 1 and their sense strand sequences are shown.
- Duplexes AV00122 to AD00484-1 , AD00474-2, AV01867-AV01968 are shown in Table 1 and their antisense strand sequences are shown.
- the delivery molecules used in the in vivo studies are indicated as "GLO-0" at the 3' end of each sense strand.
- the delivery molecules used in the in vivo studies are denoted as "GLS-5" or "GLS-15” at the 5' end of each sense strand.
- mRNA sequence SEQ ID NO: 1 of human Lp(a): NM_005577.4Homo sapienslipoprotein(a)(LPA), mRNA
- Fig. 1 shows the schematic diagram of monkey serum LPA protein level
- Figure 2 shows a schematic diagram of the level of serum LPA protein in monkeys with a dose of 2 mpk of AD00480-8.
- RNAi agents capable of inhibiting LPA (Apo(a)) gene expression such as but not limited to double-stranded (ds) RNAi agents.
- Some embodiments of the invention also include compositions comprising LPA RNAi agents and methods of using the compositions.
- the LPA RNAi agents disclosed herein can be attached to delivery compounds for delivery to cells, including delivery to hepatocytes.
- a pharmaceutical composition of the invention may comprise at least one dsRNA agent and a delivery compound.
- the delivery compound is a GalNAc-containing delivery compound.
- LPA RNAi agents delivered to cells are capable of inhibiting LPA gene expression, thereby reducing the gene's LPA protein production.
- dsRNAi agents of the invention are useful in the treatment of LPA-related diseases and disorders.
- dsRNAi agents include, for example, the duplexes AV00122 to AD00484-1, AD00474-2, AV01867-AV01968 shown in Table 1.
- dsRNAi agents include duplex variants, eg, variants of duplexes AV00122 to AD00484-1, AD00474-2, AV01867-AV01968.
- reducing LPA expression in a cell or a subject is treated with Diseases or conditions associated with LPA expression.
- diseases and conditions treatable by reducing LPA expression are cardiovascular diseases, wherein said cardiovascular diseases include Berger's disease, peripheral arterial disease, coronary artery disease, metabolic syndrome, acute coronary syndrome, aortic stenosis, aortic regurgitation, aortic dissection, retinal artery occlusion, cerebrovascular disease, mesenteric ischemia, superior mesenteric artery occlusion, renal artery stenosis, stable/unstable angina, acute coronary syndrome, heterozygous or homozygous familial hypertension Cholesterolemia, hyperapolipoprotein beta lipoproteinemia, cerebrovascular atherosclerosis, cerebrovascular disease and venous thrombosis, stroke, atherosclerosis, thrombosis, coronary heart disease or aortic stenosis and/or any other disease or pathology associated
- RNAi LPA single-stranded (ssRNA) and double-stranded (dsRNA) agents to inhibit LPA gene expression, as well as compositions and methods for treating diseases and conditions caused or regulated by LPA gene expression.
- RNAi is also known in the art and may be referred to as "siRNA”.
- RNAi refers to an agent that comprises RNA and mediates targeted cleavage of RNA transcripts through the RNA-induced silencing complex (RISC) pathway.
- RISC RNA-induced silencing complex
- an RNAi target region refers to a contiguous portion of the nucleotide sequence of an RNA molecule formed during gene transcription, which includes messenger RNA (mRNA), which is a processed product of primary transcript RNA. The target portion of the sequence will be at least long enough to serve as a substrate for RNAi-directed cleavage at or near that portion.
- mRNA messenger RNA
- the target sequence can be 8-30 nucleotides long (inclusive), 10-30 nucleotides long (inclusive), 12-25 nucleotides long (inclusive), 15-23 nucleotides long (inclusive), 16-23 nucleotides long (inclusive), or 18-23 nucleotides long (inclusive), and including all shorter lengths within each stated range.
- the target sequence is 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25 or 26 nucleotides in length. In certain embodiments, the target sequence is between 9 and 26 nucleotides in length, inclusive, including all subranges and integers therebetween.
- the target sequence is 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, or 30 nucleotides in length that is completely or at least substantially complementary to at least a portion of the RNA transcript of the LPA gene.
- Some aspects of the invention include pharmaceutical compositions comprising one or more LPA dsRNA agents and a pharmaceutically acceptable carrier.
- LPA RNAi as described herein inhibits the expression of LPA protein.
- dsRNA agent refers to a composition comprising RNA or RNA-like (eg, chemically modified RNA) oligonucleotide molecules capable of degrading or inhibiting translation of a target mRNA transcript.
- RNA interference i.e., induce RNA interference by interacting with the RNA interference pathway machinery of mammalian cells (RNA-induced silencing complex or RISC)
- RISC RNA-induced silencing complex
- the dsRNA agents disclosed herein consist of a sense strand and an antisense strand, which include, but are not limited to: short interfering RNA (siRNA), RNAi agents, microRNA (miRNA), short hairpin RNA (shRNA), and Dicer substrates.
- the antisense strands of the dsRNA agents described herein are at least partially complementary to the targeted mRNA, and it is understood in the art that dsRNA duplex structures of various lengths can be used to inhibit target gene expression. For example, dsRNAs with duplex structures of 19, 20, 21, 22 and 23 base pairs are known to efficiently induce RNA interference (Elbashir et al., EMBO 2001, 20:6877-6888).
- the LPA dsRNA in certain embodiments of the invention may comprise at least one strand of at least 21 nt in length, or the duplex may have a length based on the length of one of the sequences listed in any of Tables 1-3 minus 1, 2, 3 nt, or less. A reduction of 4 nucleotides at one or both ends of the dsRNA may also be effective compared to the dsRNAs listed in Tables 1-3, respectively.
- LPA dsRNA agents may have a partial sequence of at least 15, 16, 17, 18, 19, 20 or more contiguous nucleotides from one or more of the sequences in Tables 1-3, and their ability to inhibit LPA gene expression is no more than 5%, 10%, 15%, 20%, 25% or 30% of the level of inhibition produced by a dsRNA comprising the full sequence (also referred to herein as the "parental" sequence).
- compositions and methods of the invention include single-stranded RNA in the composition and/or administer single-stranded RNA to a subject.
- the antisense strands listed in any one of Tables 1-3 can be administered as or within a composition that, when administered to a subject, reduces expression of the LPA polypeptide and/or LPA gene in the subject.
- Tables 1-3 show the antisense strand and sense strand core extension base sequences of some LPAdsRNA reagents.
- Single-stranded antisense molecules that may be included in certain compositions of the invention and/or administered in certain methods of the invention are referred to herein as “single-stranded antisense agents” or “antisense polynucleotide agents.”
- Single-stranded sense molecules that may be included in certain compositions and/or administered in certain methods of the invention are referred to herein as “single-stranded sense agents” or “sense polynucleotide agents.”
- base sequence herein refers to a polynucleotide sequence without chemical modifications or delivery compounds.
- the sense strand shown in Table 1 corresponds to the corresponding base sequence in Table 3; however, the respective chemical modification and delivery compounds are shown in the corresponding sequences in Table 3.
- Sequences disclosed herein may be assigned identifiers. For example, a single-stranded sense sequence can be identified by a "sense strand SS#"; a single-stranded antisense sequence can be identified by an "antisense strand AS#”; and a duplex comprising a sense and antisense strand can be identified by a "duplex AD#”.
- Table 1 includes the sense and antisense strands and provides the identification numbers of duplexes formed by the sense and antisense strands on the same row in Table 1.
- the antisense sequence comprises nucleobase u or nucleobase a in its first position.
- the antisense sequence comprises the nucleobase u at position 1 of the antisense sequence.
- the term "matching position" refers in a sense to the position in each strand that "pairs" with each other when the two strands act as a duplex.
- nucleobase at position 1 of the sense strand is in a "matching position" with the nucleobase at position 21 of the antisense strand.
- nucleobase position 2 of the sense strand matches position 22 of the antisense strand.
- nucleobase number 1 of the sense strand matches nucleobase number 18 of the antisense strand; and The 4th nucleobase in the sense strand matches the 15th nucleobase in the antisense strand.
- the skilled artisan will understand how to identify the position of the match between the sense and antisense strands of the duplex and paired strands.
- a column in Table 1 represents the duplex AV#, AD# of a duplex comprising sense and antisense sequences in the same table row.
- Table 1 discloses a duplex designated "Duplex AV00122" comprising the corresponding sense and antisense strand sequences.
- each row in Table 1 identifies a duplex of the invention, each containing sense and antisense sequences shown in the same row, and the assigned identifier for each duplex shown in the last column of the row.
- an RNAi agent comprising the polynucleotide sequence shown in Table 1 is administered to the subject.
- the RNAi agent administered to the subject comprises a duplex comprising at least one of the base sequences listed in Table 1 and comprising 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23 or 24 sequence modifications.
- further comprising linking the RNAi agent of the polynucleotide sequence shown in Table 1 to a delivery molecule a non-limiting example of which is a delivery compound comprising GalNAc.
- Table 1 Antisense and sense strand sequences of unmodified LPA RNAi reagents. All sequences are shown in 5' to 3' orientation. Duplex AV# or AD# is the number assigned to the duplex of both strands in the same row in the table
- Table 2 shows the antisense and sense strand sequences of certain chemically modified LPA RNAi agents of the invention.
- an RNAi agent having the polynucleotide sequence shown in Table 2 is administered to the cell and/or subject.
- an RNAi agent having the polynucleotide sequence shown in Table 2 is administered to the subject.
- the RNAi agent administered to the subject comprises a duplex noted in the first column of Table 2, And contain the sequence modifications in the sense and antisense strand sequences shown in the third column and the sixth column of the same row in Table 2, respectively.
- the sequences shown in Table 2 can be linked to (also referred to herein as "conjugated to") a compound capable of delivering the RNAi agent to cells and/or tissues of a subject.
- delivery compounds that may be used in certain embodiments of the present invention are GalNAc-containing compounds.
- the first column indicates the duplex AD# of the base sequence, corresponding to Table 1.
- the base sequence identified by the duplex AD# not only the base sequence contained in the sense and antisense strands is shown, but also the designated chemical modification shown in the same row of Table 2 is shown.
- the first row of Table 1 shows the sense and antisense base single-stranded sequences, which together constitute a duplex, identified as: duplex AV00122; and in the duplex AV00122 listed in Table 2, as a duplex, it contains the base sequences of AV00122-SS and AV00122-AS, and contains chemical modifications in the sense and antisense sequences shown in the third and sixth columns, respectively.
- "Sense Strand SS#" in column 2 of Table 2 is the assigned identifier for the sense sequence (including modifications) shown in column 3 in the same row.
- the "antisense strand AS#" in the fifth column of Table 2 is the assigned identifier for the antisense sequence (including modifications) shown in the sixth column.
- Table 3 shows the antisense and sense strand sequences of certain chemically modified LPA RNAi agents of the present invention.
- the RNAi agents shown in Table 3 are administered to cells and/or subjects.
- an RNAi agent having the polynucleotide sequence shown in Table 3 is administered to the subject.
- the RNAi agent administered to the subject comprises the duplex identified in the first column of Table 3, and comprises the sequence modification and/or delivery compound shown in the sense and antisense strand sequences, respectively, in the third and sixth columns of the same row of Table 3. This sequence was used in some of the in vivo testing studies described elsewhere herein.
- the sequence shown in Table 3 may be linked to (also referred to herein as "conjugated to") a compound for delivery, a non-limiting example of which is a GalNAc-containing compound, i.e., a delivery compound identified as "GLX-n" on the sense strand in the third column of Table 3.
- a compound for delivery a non-limiting example of which is a GalNAc-containing compound, i.e., a delivery compound identified as "GLX-n" on the sense strand in the third column of Table 3.
- GLX-n is used to indicate the attached GalNAc-containing compound, which is the compound GLS-1, GLS-2, GLS-3, GLS-4, GLS-5, GLS-6, GLS-7, GLS-8, GLS-9, GLS-10, GLS-11, GLS-12, GLS-13, GLS-14, GLS-15, GLS-16, GLO-1 , GLO-2, GLO-3, GLO-4, GLO-5, GLO-6, GLO-7, GLO-8, GLO-9, GLO-10, GLO-11, GLO-12, GLO-13, GLO-14, GLO-15, and GLO-16.
- the structure of each of these is provided elsewhere herein.
- the first column of Table 3 provides the duplex AD# of the duplex assigned to the sense and antisense sequences in that row of the table.
- duplex AD00122 is a duplex composed of sense strand AD00122-SS and antisense strand AD00122-AS.
- Each row in Table 3 provides a sense strand and an antisense strand and discloses the duplexes formed by the indicated sense and antisense strands.
- the "sense strand SS#" in the second column of Table 3 is the assigned identifier for the sense sequence (including modifications) shown in the third column of the same row.
- the "antisense strand AS#" in the fifth column of Table 3 is the assigned identifier for the antisense sequence (including modifications) shown in the sixth column.
- GLO-0 The identifier for certain linked GalNAc-containing GLO compounds is shown as GLO-0, and it is understood that another of the GLO-n or GLS-n compounds may be substituted for the compound shown as GLO-0, and the resulting compounds are also included in embodiments of the methods and/or compositions of the invention.
- Table 3 provides the antisense and sense strand sequences of the chemically modified LPA RNAi reagents used for in vivo testing. All sequences are shown 5' to 3'. These sequences were used in some of the in vivo testing studies described elsewhere herein.
- the delivery molecules used in the in vivo studies are indicated as "GLO-0" at the 3' end of each sense strand.
- the delivery molecules used in the in vivo studies are indicated as "GLS-5" or "GLS-15" at the 5' end of each sense strand.
- mismatches can be tolerated, especially if they are within the terminal regions of the dsRNA.
- Certain mismatches are better tolerated, such as those with wobble base pairs G:U and A:C (Du et el., A systematic analysis of the silencing effects of an active siRNA at all single-nucleotide mismatched target sites. Nucleic Acids Res. 2005 Mar 21; 33(5):1671-7. Doi:10.1093/nar/gki312. Nucleic Acids Res. 2005; 33(11):3698).
- the LPA dsRNA agent may contain one or more mismatches to the LPA target sequence.
- the LPA dsRNA reagents of the invention contain no mismatches.
- the LPA dsRNA reagents of the invention contain no more than 1 mismatch.
- the LPA dsRNA reagents of the invention contain no more than 2 mismatches.
- the LPA dsRNA reagents of the invention contain no more than 3 mismatches.
- the antisense strand of the LPA dsRNA agent comprises a mismatch to the LPA target sequence that is not located in the center of the region of complementarity. In some embodiments, the antisense strand of the LPA dsRNA agent comprises 1, 2, 3, 4 or more mismatches within the last 5, 4, 3, 2 or 1 nucleotides of either or both of the 5' or 3' ends of the region of complementarity.
- the methods described herein and/or methods known in the art can be used to determine whether an LPA dsRNA agent comprising a mismatch to an LPA target sequence effectively inhibits the expression of the LPA gene.
- the term "complementarity/complementarity" when used to describe the relatedness of a first nucleotide sequence (e.g., the sense strand of an LPA dsRNA agent or a target LPA mRNA) to a second nucleotide sequence (e.g., the antisense strand of an LPA dsRNA agent or a single-stranded antisense polynucleotide) means that an oligonucleotide or polynucleotide comprising the first nucleotide sequence hybridizes to an oligonucleotide or polynucleotide comprising the second nucleotide sequence [forms base-pair hydrogen bonds under mammalian physiological conditions (or similar conditions in vitro)], and forms under certain conditions Ability to double helix or double helix structure.
- Complementary sequences include Watson-Crick base pairs or non-Watson-Crick base pairs, and include natural or modified nucleotides or nucleotide mimetics, at least to the extent required for hybridization as described above. Sequence identity or complementarity is independent of modification.
- a complementary sequence within an LPA dsRNA as described herein comprises base pairing of an oligonucleotide or polynucleotide comprising a first nucleotide sequence with an oligonucleotide or polynucleotide comprising a second nucleotide sequence over the entire length of one or both nucleotide sequences.
- sequences may be referred to herein as being "fully complementary" to each other. It should be understood that in embodiments where two oligonucleotides are designed to form one or more single-stranded overhangs upon hybridization, such overhangs are not considered mismatches determined based on complementarity herein.
- an LPA dsRNA reagent comprises one oligonucleotide that is 19 nucleotides in length and another oligonucleotide that is 20 nucleotides in length, where the longer oligonucleotide contains a sequence of 19 nucleotides that is perfectly complementary to the shorter oligonucleotide, which may be referred to as "fully complementary” for the purposes described herein.
- "fully complementary” means that all (100%) of the bases in a contiguous sequence of a first polynucleotide will hybridize to the same number of bases in a contiguous sequence of a second polynucleotide.
- the contiguous sequence may comprise all or part of the first or second nucleotide sequence.
- the term “substantially complementary” means that in a hybridizing pair of nucleobase sequences, at least about 85% (but not all) of the bases in the contiguous sequence of the first polynucleotide will hybridize to the same number of bases in the contiguous sequence of the second polynucleotide.
- the term “substantially complementary” may be used to mean that the first sequence forms a duplex of up to 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 or 30 base pairs (bp) with respect to the second sequence if the two sequences, when hybridized, contain one or more mismatched base pairs, e.
- partially complementary refers to hybridizing pairs of nucleobase sequences in which at least 75% (but not all) of the bases in the contiguous sequence of the first polynucleotide hybridize to the same number of bases in the contiguous sequence of the second polynucleotide.
- "partially complementary” means that at least 76%, 77%, 78%, 79%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% of the bases in the contiguous sequence of the first polynucleotide will be compatible with the second polynucleotide. The same number of bases in the contiguous sequence hybridize.
- complementary may be used to refer to a base match between the sense and antisense strands of an LPA dsRNA agent, the antisense strand of an LPA dsRNA agent and a target LPA mRNA
- antisense strand of an LPA dsRNA agent may refer to the same sequence as an "LPA antisense polynucleotide agent".
- the term "substantially identical” or “substantial identity” when used in reference to nucleic acid sequences means that the nucleic acid sequences comprise sequences having at least about 85% or greater sequence identity, preferably at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity compared to a reference sequence.
- the percent sequence identity is determined by comparing the optimal alignment of the two sequences over the alignment window.
- Percentages are calculated by determining the number of positions at which the same nucleic acid base occurs in the two sequences to yield the number of matching positions; dividing the number of matching positions by the total number of positions in the alignment window and multiplying the result by 100 to give the percent sequence identity.
- the invention disclosed herein includes nucleotide sequences substantially identical to those disclosed herein (eg, in Tables 1-5). In some embodiments, the nucleotide sequence is identical to, or at least about 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to the sequences disclosed herein (e.g., in Tables 1-3).
- strand comprising a sequence refers to an oligonucleotide comprising a chain of nucleotides described by a sequence referred to using standard nucleotide nomenclature.
- double-stranded RNA or “dsRNA” refers to a sequence comprising a complex of RNA molecules or RNAi molecules having a hybrid double-stranded region comprising two antiparallel and substantially or fully complementary nucleic acid strands, respectively referred to as having "sense” and “antisense” orientations relative to the target LPA RNA.
- the double stranded region may be of any desired length to allow specific degradation of the target LPA RNA by the RISC pathway, but is typically 9 to 30 base pairs in length, for example 15-30 base pairs in length.
- the duplexes can be any length within this range, for example, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, or 30 base pairs, and any subranges therein, including but not limited to 15-30 base pairs, 1 5-26 bp; 15-23 bp, 15-22 bp, 15-21 bp, 15-20 bp, 15-19 bp, 15-18 bp, 15-17 bp, 18-30 bp, 18-26 bp, 18-23 bp, 18-22 bp, 18-21 bp, 1 8-20 bp, 19-30 bp, 19-26 bp, 19-23 bp, 19-22 bp, 19-21 bp, 19-20 bp, 20-30 bp
- LPA dsRNA reagents produced in cells by processing with Dicer and similar enzymes typically range in length from 19-22 base pairs.
- One strand of the double-stranded region of the LPA dsDNA agent comprises a sequence that is substantially complementary to a region of the target LPA RNA.
- the two strands forming the duplex structure can arise from a single RNA molecule with at least one self-complementary region, or can be formed from two or more separate RNA molecules.
- the molecule may have a duplex structure (referred to herein as a "hairpin loop") formed by one strand at the 3'-terminus of a single-stranded nucleotide chain and the other strand at the corresponding 5'-terminus.
- the hairpin configuration comprises at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or more unpaired nucleotides.
- the LPA dsRNA agent may comprise sense and antisense sequences with unpaired nucleotides or nucleotide analogs at one or both ends of the dsRNA agent. Ends with no unpaired nucleotides are called “blunt ends” and have no nucleotide overhangs. A dsRNA is said to be “blunt-ended” if both ends of the dsRNA reagent are blunt-ended.
- the first end of the dsRNA reagent is blunt-ended, in some embodiments, the second end of the dsRNA reagent is blunt-ended, and in some embodiments of the invention, both ends of the LPA dsRNA reagent are blunt-ended.
- the dsRNA does not have one or two blunt ends.
- a nucleotide overhang exists when the 3'-end of one strand of a dsRNA extends beyond the 5'-end of the other strand, and vice versa.
- a dsRNA may comprise an overhang of at least 1, 2, 3, 4, 5, 6 or more nucleotides.
- Nucleotide overhangs may comprise or consist of nucleotide/nucleoside analogs, including deoxynucleotides/nucleosides.
- the nucleotide overhangs are on the sense strand of the dsRNA agent, on the antisense strand of the dsRNA agent, or at both ends of the dsRNA agent, and that the nucleotides of the overhang can be present at the 5' end, the 3' end, or both ends of the antisense strand or the sense strand of the dsRNA agent.
- one or more nucleotides in the overhang are replaced with nucleoside phosphorothioate.
- the term “antisense strand” or “guide strand” refers to the strand of an LPAdsRNA agent comprising a region substantially complementary to an LPA target sequence.
- the term “sense strand” or “passenger strand” refers to the strand of the LPA dsRNA agent comprising a region substantially complementary to a region of the antisense strand of the LPA dsRNA agent.
- the RNA of the LPA RNAi agent is chemically modified for enhanced stability and/or one or more other beneficial properties.
- Nucleic acids in certain embodiments of the invention can be synthesized and/or modified by methods known in the art, see, e.g., "Current protocols in Nucleic Acid Chemistry,” Beaucage, S Let al. (Eds.), John Wiley & Sons, Inc., New York, NY, USA, which is incorporated herein by reference.
- Modifications that may be present in certain embodiments of the LPA dsRNA reagents of the invention include, for example: (a) terminal modifications, such as 5' end modifications (phosphorylation, conjugation, reverse ligation, etc.), 3' end modifications (conjugation, DNA nucleotides, reverse ligation, etc.); position or 4' position) or sugar replacement; and (d) backbone modifications, including modification or replacement of phosphodiester linkages.
- Specific examples of RNA compounds useful in certain embodiments of the LPA dsRNA agents, LPA antisense polynucleotides, and LPA sense polynucleotides of the invention include, but are not limited to, RNAs comprising modified backbones or without natural internucleoside linkages.
- RNA with backbone modifications There may be no phosphorus atom in the skeleton. RNAs that have no phosphorus atoms in their internucleoside backbone may be referred to as oligonucleotides. In certain embodiments of the invention, the modified RNA has phosphorus atoms in its internucleoside backbone.
- RNA molecule or “RNA” or “ribonucleic acid molecule” includes not only RNA molecules expressed or found in nature, but also analogs and derivatives of RNA comprising one or more ribonucleotide/ribonucleoside analogs or derivatives as described herein or known in the art.
- ribonucleoside and “ribonucleotide” are used interchangeably herein.
- RNA molecules can be modified in nucleobase structure or ribose-phosphate backbone structure (eg, as described below), and molecules comprising ribonucleoside analogs or derivatives must retain the ability to form duplexes.
- the RNA molecule may also comprise at least one modified ribonucleoside, which includes, but is not limited to, a 2'-O-methyl modified nucleoside, a nucleoside comprising a 5' phosphorothioate group, a terminal nucleoside linked to a cholesterol derivative or a dodecanoic acid didecylamide group, a locked nucleoside, an abasic nucleoside, a 2'-deoxy-2'-fluoro modified nucleoside, a 2'-amino modified nucleoside, a 2'-alkyl modified nucleoside, a morpholino nucleoside, a phosphoramidate or a nucleoside comprising a nucleoside unnatural bases, or any combination thereof.
- a 2'-O-methyl modified nucleoside a nucleoside comprising a 5' phosphorothioate group, a terminal nucleoside linked to a cholesterol derivative or a do
- the RNA molecule comprises a modified ribonucleoside in an amount of at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, or up to the full length of the ribonucleoside of the LPA dsRNA reagent molecule.
- the modification need not be the same for each of the plurality of modified ribonucleosides in such an RNA molecule.
- the dsRNA agents, LPA antisense polynucleotides and/or LPA sense polynucleotides of the invention may comprise one or more independently selected modified nucleotides and/or one or more independently selected non-phosphodiester bonds.
- independently selected is used to refer to selected elements, such as modified nucleotides, non-phosphodiester linkages, etc., and means that two or more selected elements may be identical to each other but need not be identical to each other.
- nucleotide base is a heterocyclic pyrimidine or purine compound that is a standard constituent of all nucleic acids and includes the bases that form nucleotides: adenine (a), guanine (g), cytosine (c), thymine (t), and uracil (u).
- Nucleobases can be further modified to include, but are not intended to be limited to: universal bases, hydrophobic bases, promiscuous bases, bases of enlarged size, and fluorinated bases.
- ribonucleotide or “nucleotide” may be used herein to refer to unmodified nucleotides, modified nucleotides or substituted moieties.
- guanine, cytosine, adenine and uracil can be replaced by other moieties without significantly changing the base pairing properties of oligonucleotides comprising nucleotides with such replacement moieties.
- the modified RNA contemplated for use in the methods and compositions described herein is a peptide nucleic acid (PNA) that has the ability to form a desired duplex structure and allow or mediate specific degradation of the target RNA via the RISC pathway.
- the LPA RNA interfering agent comprises a single-stranded RNA that interacts with a target LPA RNA sequence to direct cleavage of the target LPA RNA.
- Modified RNA backbones may contain, for example, phosphorothioates, chiral phosphorothioates, phosphorodithioates, phosphotriesters, aminoalkylphosphotriesters, methyl and other alkylphosphonates (including 3'-alkylenephosphonates and chiral phosphonates), phosphinates, phosphoramidates (including 3'-aminophosphoramidates and aminoalkylphosphoramidates), phosphorothioates, thioalkylphosphonates, thioalkylphosphotriesters, and boronic acid phosphates (which have normal 3'-5' linkages, and those of these 2'-5' linked analogs, and those with inverted polarity in which adjacent pairs of nucleoside units are aligned 3'-5' to 5'-3' Or 2'-5' to 5'-2' form connection).
- phosphorothioates chiral phosphorothioates, phosphorodithioates, phosphotries
- Modified RNA backbones wherein no phosphorus atoms are included have backbones formed from short chain alkyl or cycloalkyl internucleoside linkages, mixed heteroatom and alkyl or cycloalkyl internucleoside linkages, or one or more short chain heteroatom or heterocyclic internucleoside linkages.
- modified RNA backbones that do not contain phosphorus atoms are routine practice in the art, and such methods can be used to prepare certain modified LPA dsRNA agents, certain modified LPA antisense polynucleotides, and/or certain modified LPA sense polynucleotides of the invention.
- RNA mimetics are included in LPA dsRNA, LPA antisense polynucleotides and/or LPA sense polynucleotides, such as, but not limited to, replacing the sugar and internucleoside linkages (i.e., backbone) of the nucleotide units with new groups.
- the base unit is maintained to hybridize to the appropriate LPA nucleic acid target compound.
- peptide nucleic acid PNA
- PNA peptide nucleic acid
- the sugar backbone of RNA is replaced by an amide-containing backbone, especially an aminoethylglycine backbone.
- RNA mimetics are retained and bonded directly or indirectly to aza nitrogen atoms of the backbone amide moiety. Methods of making RNA mimetics are routinely practiced in the art, and such methods can be used to make certain modified LPA dsRNA agents of the invention.
- RNAs with phosphorothioate backbones and oligonucleosides with heteroatom backbones in particular -CH2 -- NH-- CH2- , --CH2 --N( CH3 )--O-- CH2 --[known as methylene (methylimino) or MMI backbone], --CH2 --O--N( CH3 )-- CH2-- , --CH2-- N ( CH3 )--N( CH3 )-- CH2- and --N( CH3 )-- CH2 ----[wherein the natural phosphodiester backbone is represented as --O--P--O-- CH2-- ].
- RNAs with phosphorothioate backbones and oligonucleotides with heteroatom backbones are routinely practiced in the art, and such methods can be used to prepare certain modified LPA dsRNA agents, certain LPA antisense polynucleotides, and/or certain LPA sense polynucleotides of the invention.
- Modified RNAs may also contain one or more substituted sugar moieties.
- the LPA dsRNA, LPA antisense polynucleotide and/or LPA sense polynucleotide of the present invention may comprise one of the following at the 2' position: OH; F; O--, S--, or N-alkyl; O--, S-- , or N-alkenyl ; O-, S-, or N -alkynyl; Exemplary suitable modifications include: O[(CH 2 ) n O] m CH 3 , O(CH 2 ) n OCH 3 , O(CH 2 ) n NH 2 , O(CH 2 ) n CH 3 , O(CH 2 ) n ONH 2 , and O(CH 2 ) n ON[(CH 2 ) n CH 3 )] 2 , where n and m range from 1 to about 10.
- the dsRNA comprises one of the following at the 2' position: C 1 to C 10 lower alkyl, substituted lower alkyl, alkaryl, aralkyl, O-alkaryl or O-aralkyl, SH, SCH 3 , OCN, Cl, Br, CN, CF 3 , OCF 3 , SOCH 3 , SO 2 CH 3 , ONO 2 , NO 2 , N 3 , NH 2 , heterocycloalkyl, heterocycloalkyl, heterocycloalkyl, Cycloalkaryl, aminoalkylamino, polyalkylamino; substituted silyl, RNA cleavage Groups, reporter groups, intercalators; groups for improving the pharmacokinetic properties of LPA dsRNA agents; or groups for improving the pharmacodynamic properties of LPA dsRNA agents, LPA antisense polynucleotides and/or LPA sense polynucleotides, and other substituents with similar properties.
- the modification includes 2'-methoxyethoxy (2'-O-- CH2CH2OCH3 , also known as 2'-O-( 2 -methoxyethyl) or 2'-MOE) (Martin et al., Helv. Chim. Acta, 1995, 78:486-504), ie, alkoxy-alkoxy.
- Another exemplary modification is 2'-dimethylaminoethoxyethoxy, an O( CH2 ) 2ON ( CH3 ) 2 group, also known as 2'-DMAOE, as described in the Examples below ; .
- Methods of making modified RNAs of those described are routinely practiced in the art, and such methods can be used to make certain modified LPA dsRNA reagents of the invention.
- modifications include 2'-methoxy (2'- OCH3 ), 2'-aminopropoxy ( 2' - OCH2CH2CH2NH2 ) and 2'-fluoro (2'-F). Similar modifications can also be made at other positions on the RNA of the LPA dsRNA reagents, LPA antisense polynucleotides, LPA sense polynucleotides and/or other positions on the LPA sense polynucleotides of the invention, particularly the 3' position of the sugar on the 3' terminal nucleotide or 2'-5' linked LPA dsRNA, LPA antisense polynucleotide or LPA sense polynucleotide, and the 5' position of the 5' terminal nucleotide.
- LPA dsRNA agents, LPA antisense polynucleotides, and/or LPA sense polynucleotides may also have sugar mimetics, eg, cyclobutyl moieties in place of pentofuranose.
- sugar mimetics eg, cyclobutyl moieties in place of pentofuranose.
- LPA dsRNA agents, LPA antisense polynucleotides, and/or LPA sense polynucleotides may include nucleobase (commonly referred to in the art simply as “bases”) modifications or substitutions.
- nucleobases commonly referred to in the art simply as “bases”
- “unmodified” or “natural” nucleobases include the purine bases adenine (A) and guanine (G), and the pyrimidine bases thymine (T), cytosine (C) and uracil (U).
- Modified nucleobases include other synthetic and natural nucleobases such as 5-methylcytosine (5-me-C), 5-hydroxymethylcytosine, xanthine, hypoxanthine, 2-aminoadenine, 6-methyl and other alkyl derivatives of adenine and guanine, 2-propyl and other alkyl derivatives of adenine and guanine, 2-thiouracil, 2-thiothymine and 2-thiocytosine, 5-halouracil and cytosine, 5- Proynyluracil and cytosine, 6-azouracil, cytosine and thymine, 5-uracil (pseudouracil), 4-thiouracil; 8-halo, 8-amino, 8-thiol, 8-thioalkyl, 8-hydroxy and other 8-substituted adenine and guanine; 5-halo, especially 5-bromo, 5-trifluoromethyl and other 5-substituted ura
- nucleobases that may be included in certain embodiments of the LPA dsRNA reagents of the invention are known in the art, see for example: Modified Nucleosides in Biochemistry, Biotechnology and Medicine, Herdewijn, P. Ed. Wiley-VCH, 2008; The Concise Encyclopedia Of Polymer Science And Engineering, pages 858-859, K roschwitz, JL, Ed. John Wiley & Sons, 1990, English et al., Angewandte Chemie, International Edition, 1991, 30, 613, Sanghvi, Y S., Chapter 15, dsRNA Research and Applications, pages 289-302, Crooke, ST and Lebleu, B., Ed., CRC Press, 1993.
- dsRNA comprising nucleobase modification and/or substitution, LPA antisense strand polynucleotide and/or LPA sense
- Methods for stranded polynucleotides are routinely practiced in the art, and such methods can be used to prepare certain modified LPA dsRNA agents, LPA sense polynucleotides and/or LPA antisense polynucleotides of the invention.
- LPA dsRNA agents, LPA antisense polynucleotides, and/or LPA sense polynucleotides of the invention include RNA modified to include one or more locked nucleic acids (LNAs).
- Locked nucleic acids are nucleotides that have a modified ribose moiety that includes an additional bridge connecting the 2' and 4' carbons. This structure effectively "locks" the ribose sugar in the 3'-endostructural conformation.
- LPA antisense polynucleotide and/or LPA sense polynucleotide can increase the stability in serum, and reduce off-target effect (Elmen, J. et al., (2005) Nucleic Acids Research 33 (1): 439-447; Mook, O R. et al., (2007) Mol Canc Ther6 (3): 833-843 ; Grunweller, A. et al., (2003) Nucleic Acids Research 31(12):3185-3193).
- dsRNA reagents comprising locked nucleic acids, LPA antisense polynucleotides and/or LPA sense polynucleotides are routinely practiced in the art, and such methods can be used to prepare certain modified LPA dsRNA reagents of the present invention.
- LPA dsRNA compounds, sense polynucleotides and/or antisense polynucleotides of the present invention comprise at least one modified nucleotide, wherein the at least one modified nucleotide comprises: 2'-O-methyl nucleotides, 2'-fluoro nucleotides, 2'-deoxy nucleotides, 2',3'-seco nucleotide mimetics, locked nucleotides, 2'-F-arabino nucleotides, 2'-methoxyethyl nucleotides, 2'-amino modified nucleotides, 2'-alkyl modified nucleotides, morpholino nucleotides and 3'-Ome nucleotides, Nucleotides comprising 5'-phosphorothioate groups, or terminal nucleotides linked to cholesterol derivatives or dodecanoic acid bis-decylamide groups, 2'-amino modified nucleo
- the LPA dsRNA compound, the 3' and 5' ends of the sense polynucleotide and/or the 3' end of the antisense polynucleotide comprise at least one modified nucleotide, wherein the at least one modified nucleotide includes: abasic nucleotide, ribitol, inverted nucleotide, inverted abasic nucleotide, inverted 2'-OMe nucleotide, inverted 2'-deoxynucleotide.
- abasic or inverted abasic nucleotides at the ends of oligonucleotides is known to those skilled in the art to enhance stability (Czauderna et al. Structural variations and stabilizing modifications of synthetic siRNAs in mammalian cells. Nucleic Acids Res. 2003; 31(11):2705-2716. /nar/gkg393).
- the LPA dsRNA comprises one or two isomannitol residues at the 3' and 5' ends of the sense strand.
- the sense strand independently comprises an isomannitol residue at the 3' and 5' ends, respectively. Inclusion of isomannitol residues has the following examples:
- isomannitol residues include, but are not limited to the following:
- isomannitol residues can also be replaced by their stereoisomers, non-limiting examples:
- the sense strand independently comprises an isomannitol residue (imann) at the 3' or and 5' end, and further comprises a targeting group conjugated at the 5'-end, for example, targeting group N-acetyl-galactosamine, preferably the above-mentioned GLS-15, and an exemplary structure is as follows:.
- the LPA dsRNA compound, antisense polynucleotide comprises at least one modified nucleotide, wherein said at least one modified nucleotide comprises ring-opening nucleic acid nucleotide (UNA) or/and diol nucleic acid nucleotide (GNA).
- UNA and GNA are thermally unstable chemical modifications, which can significantly improve the off-target spectrum of siRNA compounds (Janas, et al., Selection of GalNAc-conjugated siRNAs with limited off-target-driven rat hepatitis. Nat Commun. 2018; 9(1): 723.
- RNA of the LPA dsRNA agent, LPA antisense polynucleotide, and/or LPA sense polynucleotide of certain embodiments of the invention includes one or more ligands, moieties, or conjugates chemically linked to the RNA that enhance one or more characteristics of the LPA dsRNA agent, LPA antisense polynucleotide, and/or LPA sense polynucleotide, respectively.
- Non-limiting examples of characteristics that may be enhanced are: LPA dsRNA agent, LPA antisense polynucleotide and/or LPA sense polynucleotide activity, cellular distribution, delivery of LPA dsRNA agent, pharmacokinetic properties of LPA dsRNA agent, and cellular uptake of LPA dsRNA agent.
- the LPA dsRNA agents comprise one or more targeting groups or linking groups, which in certain embodiments of the LPA dsRNA agents of the invention are conjugated to the sense strand.
- a non-limiting example of a targeting group is a compound comprising N-acetyl-galactosamine (GalNAc).
- the LPA dsRNA reagent comprises a targeting compound conjugated to the 5'-end of the sense strand. In certain embodiments of the invention, the LPA dsRNA reagent comprises a targeting compound conjugated to the 3'-end of the sense strand. In some embodiments of the invention, the LPA dsRNA agent comprises a GalNAc-containing targeting group. In certain embodiments of the invention, the LPA dsRNA reagent does not comprise a targeting compound conjugated to either or both of the 3'-end and the 5'-end of the sense strand. In certain embodiments of the invention, the LPA dsRNA reagent does not comprise a GalNAc-containing targeting compound conjugated to either or both of the 5'-end and the 3'-end of the sense strand.
- targeting and linking agents useful in certain embodiments of the invention include, but are not limited to, lipid moieties such as cholesterol moieties (Letsinger et al., Proc. Natl. Acid. Sci. USA, 1989, 86:6553-6556), cholic acid (Manoharan et al., Biorg. Med. Chem.
- thioethers such as beryl-S-trityl mercaptan (Manoharan et al., Ann.N.Y.Acad.Sci., 1992,660:306-309; Manoharan et al., Biorg.Med.Chem.Let., 1993,3:2765-2770), thiocholesterol (Oberhauser et al., Nucl.Acids Res.,1992,20:533-538), aliphatic chains such as dodecanediol or undecyl residues (Saison-Behmoaras et al., EMBO J,1991,10:1111-1118; Kabanov et al., FEBS Lett.,1990,259:327-330; Svinarchuk et al., Biochimie, 1993,75:49-54), phospholipids such as di-hexadecyl-rac-gly
- compositions comprising LPA dsRNA agents, LPA antisense polynucleotides, and/or LPA sense polynucleotides may include ligands that alter the distribution, targeting, etc. properties of the LPA dsRNA agents.
- the ligand increases affinity for a selected target (e.g., a molecule, cell or cell type, compartment, e.g., a cell or organ compartment, tissue, organ or body region), e.g., compared to a species in which such ligand is absent.
- Ligands useful in the compositions and/or methods of the invention may be naturally occurring substances such as proteins (e.g.
- HSA human serum albumin
- LDL low density lipoprotein
- globulin Such as human serum albumin (HSA), low density lipoprotein (LDL) or globulin), carbohydrates (eg, dextran, pullulan, chitin, chitosan, inulin, cyclodextrin or hyaluronic acid) or lipids.
- Ligands may also be recombinant or synthetic molecules, such as synthetic polymers, such as synthetic polyamino acids or polyamines.
- poly amino acids examples include polystrazeminaine (PLL), polyacoline, and polyetine, polyaclamine, styrene-Malanic anhydride cluster, polymer (L-propylene-common-ethanol) co-vector, dihydrine-picotinic anhydride consecomer, N-(2-hydroxypropyl) methyl-based polymer (HMPA), polyethylene two. Alcohol (PEG), polyethylene (PVA), polyurethane, polyethylene (2-ethyl acrylics), N-isopropyl acrylic polymer, or polyphone.
- PLL polystrazeminaine
- polyacoline polyacoline
- polyetine polyaclamine
- polyaclamine polyaclamine
- styrene-Malanic anhydride cluster examples include polymer (L-propylene-common-ethanol) co-vector, dihydrine-picotinic anhydride consecomer, N-(2-hydroxypropyl) methyl-based polymer
- polyamines examples include: polyethyleneimine, polylysine (PLL), spermine, spermidine, polyamines, pseudopeptide-polyamines, peptidomimetic polyamines, dendritic polyamines, arginine, amidines, protamine, cationic lipids, cationic porphyrins, quaternary salts of polyamines, or alpha-helical peptides.
- Ligands included in the compositions and/or methods of the invention may comprise targeting groups, non-limiting examples of which are cell or tissue targeting agents, e.g., lectins, glycoproteins, lipids or proteins, e.g. antibodies that bind specific cell types such as kidney cells or hepatocytes.
- cell or tissue targeting agents e.g., lectins, glycoproteins, lipids or proteins, e.g. antibodies that bind specific cell types such as kidney cells or hepatocytes.
- Targeting groups can be thyrotropin, melanin, lectins, glycoproteins, surfactant protein A, mucin carbohydrates, polyvalent lactose, polyvalent galactose, N-acetyl-galactosamine, N-acetyl-glucosamine polyvalent mannose, polyvalent fucose, glycosylated polyamino acids, polyvalent galactose, transferrin, bisphosphonates, polyglutamate, polyaspartic acid, lipids, cholesterol, steroids, bile acids, folic acid, vitamin B12, vitamin A. Biotin or RGD peptide or RGD peptidomimetic.
- ligands include dyes, intercalators (e.g. acridine), crosslinkers (e.g. psoralen, mitomycin C), porphyrins (TPPC4, texaphyrin, Sapphyrin), polycyclic aromatic hydrocarbons (e.g. phenazine, dihydrophenazine); artificial endonucleases (e.g.
- EDTA lipophilic molecules
- lipophilic molecules such as cholesterol, cholic acid, adamantaneacetic acid, 1-pyrenebutyric acid, dihydrotestosterone, 1,3-bis-O(hexadecyl)glycerol, geranyloxyhexyl cetylglycerol, borneol, menthol, 1,3-propanediol, heptadecyl, palmitic acid, myristic acid, O3-(oleoyl)lithocholic acid, O3-(oleoyl)cholic acid, dimethoxytrityl or phenoxazine and peptide conjugates (e.g., Antenna, Tat peptide), alkylating agents, phosphates, amino groups, sulfhydryl groups, PEG (e.g., PEG-40K), MPEG, [MPEG] 2 , polyamino groups, alkyl groups, substituted alkyl
- Ligands included in the compositions and/or methods of the invention may be proteins, such as glycoproteins or peptides, such as molecules with a specific affinity for co-ligands, or antibodies, such as antibodies that bind to specific cell types such as cancer cells, endothelial cells, cardiomyocytes or bone cells.
- Ligands useful in embodiments of the compositions and/or methods of the invention may be hormones or hormone receptors.
- Ligands useful in embodiments of the compositions and/or methods of the invention may be lipids, lectins, carbohydrates, vitamins, coenzymes, polyvalent lactose, polyvalent galactose, N-acetyl-galactosamine, N-acetyl-glucosamine polyvalent mannose, or polyvalent fucose.
- Ligands useful in embodiments of the compositions and/or methods of the invention can be, for example, increasing LPA dsRNA by disrupting the cytoskeleton of the cell (e.g., by disrupting the microtubules, microfilaments and/or intermediate filaments of the cell) A substance that is taken up by a reagent into a cell.
- Non-limiting examples of such agents are: taxon, vincristine, vinblastine, cytochalasin, nocodazole, japlakinolide, latrunculin A, phalloidin, swinholide A, indanocine, and myoservin.
- ligands linked to LPA dsRNA agents of the invention are used as pharmacokinetic (PK) modulators.
- PK modulators useful in the compositions and methods of the invention include, but are not limited to: lipophilic agents, bile acids, steroids, phospholipid analogs, peptides, protein binding agents, PEGs, vitamins, cholesterol, fatty acids, cholic acids, lithocholic acids, dialkylglycerides, diacylglycerides, phospholipids, sphingolipids, naproxen, ibuprofen, vitamin E, biotin, aptamers that bind serum proteins, and the like.
- Oligonucleotides comprising many phosphorothioate linkages are also known to bind serum proteins, thus short oligonucleotides comprising multiple phosphorothioate linkages in the backbone, such as oligonucleotides of about 5 bases, 10 bases, 15 bases or 20 bases, may also be used as ligands in the compositions and/or methods of the invention.
- the LPA dsRNA agent is in the composition.
- Compositions of the invention may comprise one or more LPA dsRNA agents and optionally one or more pharmaceutically acceptable carriers, delivery agents, targeting agents, detectable labels, etc.
- Non-limiting examples of targeting agents useful according to some embodiments of the methods of the invention are agents that direct the LPA dsRNA agents of the invention to and/or enter cells to be treated. The choice of targeting agent will depend on the following factors: the nature of the LPA-associated disease or disorder, and the target cell type. In one non-limiting example, in some embodiments of the invention, it may be desirable to target and/or enter the LPA dsRNA agent to hepatocytes.
- the therapeutic agent comprises an LPA dsRNA agent with only a delivery agent, such as a delivery agent comprising N-acetylgalactosamine (GalNAc), without any additional linking elements.
- a delivery agent such as a delivery agent comprising N-acetylgalactosamine (GalNAc)
- the LPA dsRNA agent can be linked to a delivery compound comprising GalNAc, included in a composition comprising a pharmaceutically acceptable carrier, and administered to the cell or subject without any detectable label or targeting agent, etc., attached to the LPA dsRNA agent.
- LPA dsRNA reagents of the invention are administered with and/or linked to one or more delivery agents, targeting agents, labeling agents, etc.
- suitable reagents for use in the methods of the invention.
- Labeling reagents can be used in certain methods of the invention to determine the location of LPA dsRNA agents in cells and tissues, and can be used to determine the location of cells, tissues or organs of therapeutic compositions comprising LPA dsRNA agents that have been administered in the methods of the invention.
- Means for attaching and using labeling reagents such as enzymatic labels, dyes, radiolabels, etc. are well known in the art.
- the labeling reagent is linked to one or both of the sense polynucleotide and the antisense polynucleotide comprised in the LPAdsRNA reagent.
- Certain embodiments of the methods of the invention comprise delivering an LPA dsRNA agent into a cell.
- delivery means to facilitate or affect cellular uptake or absorption. Absorption or uptake of LPA dsRNA agents can occur through independent diffusion or active cellular processes, or through the use of delivery agents, targeting agents, etc. that can be associated with the LPA dsRNA agents of the invention.
- Modes of delivery suitable for use in the methods of the invention include, but are not limited to, in vivo delivery wherein the LPA dsRNA agent is injected into a tissue site or administered systemically.
- the LPA dsRNA agent is linked to a delivery agent.
- Non-limiting examples of methods that can be used to deliver LPA dsRNA agents to cells, tissues, and/or subjects include: LPA dsRNA-GalNAc conjugates, SAMiRNA technology, LNP-based delivery methods, and naked RNA delivery. These and other delivery methods have been successfully used in the art to deliver therapeutic RNAi agents for the treatment of various diseases and conditions such as, but not limited to: liver disease, acute intermittent porphyria (AIP), hemophilia, pulmonary fibrosis, and the like. Details of the various modes of delivery can be found in publications such as: Nikam, R.R. & K.R. Gore (2016) Nucleic Acid Ther, 28(4), 209-224 Aug 2018; Springer A.D. & S.F.
- LNPs lipid nanoparticles
- LNPs are commonly used to deliver LPA dsRNA agents in vivo, including therapeutic LPA dsRNA agents.
- One benefit of using LNP or other delivery agents is the increased stability of the LPA RNA agent when delivered to a subject using LNP or other delivery agents.
- LNPs comprise cationic LNPs loaded with one or more LPA RNAi molecules of the invention.
- the LNP comprising the LPA RNAi molecule is administered to the subject, the LNP and its attached LPA RNAi molecule are taken up by the cell through endocytosis, and their presence results in the release of the RNAi triggering molecule, thereby mediating the RNAi.
- a delivery agent that can be used in embodiments of the invention to deliver an LPA dsRNA agent of the invention to a cell, tissue, and/or subject is a GalNAc-containing agent that is linked to and delivers the LPA dsRNA agent of the invention to a cell, tissue, and/or subject.
- a GalNAc-containing agent that is linked to and delivers the LPA dsRNA agent of the invention to a cell, tissue, and/or subject.
- examples of certain other delivery agents comprising GalNAc that may be used in certain embodiments of the methods and compositions of the invention are disclosed in PCT application WO2020191183A1.
- a non-limiting example of a GalNAc targeting ligand that can be used in the compositions and methods of the invention to deliver an LPA dsRNA agent to a cell is a targeting ligand cluster.
- GalNAc ligands with phosphodiester linkages GLO
- GalNAc ligands with phosphorothioate linkages GLS
- the term "GLX-n" may be used herein to indicate that the attached GalNAC-containing compound is the following compound: GLS-1, GLS-2, GLS-3, GLS-4, GLS-5, GLS-6, GLS-7, GLS-8, GLS-9, GLS-10, GLS-11, GLS-12, GLS-13, GLS-14, GLS-15, GLS-16, GLO-1, GLO Any one of -2, GLO-3, GLO-4, GLO-5, GLO-6, GLO-7, GLO-8, GLO-9, GLO-10, GLO-11, GLO-12, GLO-13, GLO-14, GLO-15 and GLO-16, the structure of each is as follows, the connection position of the GalNAc targeting ligand and the RNAi agent of the present invention in the figure below is on the far right of each targeting ligand
- any of the RNAi and dsRNA molecules of the invention can be linked to GLS-1, GLS-2, GLS-3, GLS-4, GLS-5, GLS-6, GLS-7, GLS-8, GLS-9, GLS-10, GLS-11, GLS-12, GLS-13, GLS-14, GLS-15, GLS-16, GLO-1, GLO-2, GLO-3, GLO -4, GLO-5, GLO-6, GLO-7, GLO-8, GLO-9, GLO-10, GLO-11, GLO-12, GLO-13, GLO-14, GLO-15 and GLO-16, the following are the structures of GLO-1 to GLO-16 and GLS-1 to GLS-16.
- in vivo delivery may also be by beta-glucan delivery systems, such as those described in US Patent Nos. 5,032,401 and 5,607,677, and US Publication No. 2005/0281781, the entire contents of which are incorporated herein by reference.
- LPA RNAi agents can also be introduced into cells in vitro using methods known in the art such as electroporation and lipofection.
- the LPA dsRNA is delivered without a targeting agent. These RNAs can be delivered as "naked" RNA molecules.
- the LPA dsRNAs of the invention can be administered to a subject in a pharmaceutical composition comprising an RNAi agent but no targeting agent (e.g., a GalNAc targeting compound) to treat an LPA-associated disease or disorder, such as cardiovascular disease, including Berger's disease, peripheral artery disease, coronary artery disease, metabolic syndrome, acute coronary syndrome, aortic valve stenosis, aortic regurgitation, aortic dissection, retinal artery occlusion, cerebrovascular disease, mesentery in a subject Ischemia, superior mesenteric artery occlusion, renal artery stenosis, stable/unstable angina, acute coronary syndrome, heterozygous or homozygous familial hypercholesterolemia, hyperapolipoprotein beta lipoproteinemia, cerebrovascular atherosclerosis, cerebrovascular disease and venous thrombosis, stroke, atherosclerosis, thrombosis, coronary heart disease or aor
- RNAi delivery modes may be used in conjunction with embodiments of the LPA RNAi agents and methods of treatment described herein, such as, but not limited to, those described herein and those used in the art.
- the LPA dsRNA agents of the invention can be administered to a subject in an amount and in a manner effective to reduce the level of LPA polypeptide in the cell and/or the subject.
- one or more LPA dsRNA agents are administered to a cell and/or subject to treat a disease or condition associated with LPA expression.
- the methods of the invention comprise administering to a subject in need of such treatment one or more LPA dsRNA agents to alleviate a disease or condition associated with LPA expression in the subject.
- LPA dsRNA agents or LPA antisense polynucleotide agents of the invention can be administered to reduce LPA expression in one or more of cells in vitro, ex vivo, and in vivo.
- the LPA dsRNA agent or LPA antisense polynucleotide agent is delivered (eg, introduced into) a cell to reduce the level of LPA polypeptide in the cell.
- Targeting agents and methods can be used to facilitate delivery of LPA dsRNA agents or LPA antisense polynucleotide agents to specific cell types, cell subtypes, organs, spatial regions, and/or subcellular regions within cells in a subject.
- LPA dsRNA agents may be administered alone or in combination with one or more additional LPA dsRNA agents in certain methods of the invention. In some embodiments, 2, 3, 4 or more independently selected LPA dsRNA agents are administered to the subject.
- an LPA dsRNA agent is administered to a subject in combination with one or more additional therapeutic regimens for the treatment of an LPA-associated disease or disorder to treat the LPA-associated disease or disorder.
- additional treatment regimens are: administration of one or more LPA antisense polynucleotides of the invention, administration of non-LPA dsRNA therapeutics, and behavioral modification.
- the additional treatment regimen may be administered at one or more of: before, concurrently with, and after administration of the LPA dsRNA agents of the invention.
- non-LPA dsRNA therapeutics are: additional therapeutics such as HMg Co-A reductase inhibitors (statins), ezetimibe, PCSK-9 inhibitors, CTEP inhibitors, ANGPTL3-targeting therapies, APOC3-targeting therapies, and niacin, or combinations of any of the foregoing.
- Non-limiting examples of behavior modification are: dietary regimens, counseling and exercise regimens. These and other therapeutic agents and behavioral modifications are known in the art and can be used to treat an LPA disease or condition in a subject, and can also be administered to a subject in combination with one or more LPA dsRNA agents of the invention to treat an LPA disease or condition.
- LPA dsRNA agents of the invention administered to a cell or subject to treat an LPA-associated disease or disorder can act in a synergistic manner with one or more other therapeutic agents or active ingredients, thereby increasing the effectiveness of the one or more therapeutic agents or active ingredients and/or increasing the effectiveness of the LPA dsRNA agent for treating an LPA-associated disease or disorder.
- the methods of treatment of the present invention comprising administration of LPA dsRNA agents, may be used prior to the onset of an LPA-associated disease or disorder and/or when an LPA-associated disease or disorder is present, including in the early, middle, late stages of the disease or disorder, and at all times before and after any of these stages.
- the methods of the invention may also treat a subject who has previously been treated for an LPA-related disease or disorder with one or more other therapeutic agents and/or therapeutically active ingredients, wherein the one or more other therapeutic agents and/or therapeutically active ingredients were unsuccessful, minimally successful, and/or no longer successful in treating the subject's LPA-related disease or disorder.
- a vector can be used to deliver an LPA dsRNA agent into a cell.
- LPAdsRNA reagent transcription units can be contained in DNA or RNA vectors.
- the preparation and use of such transgene-encoding vectors for delivery of sequences into cells and/or subjects is well known in the art.
- Vectors that result in transient expression of LPAdsRNA e.g., at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 or more hours, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 or more weeks, may be used in the methods of the invention.
- the length of transient expression can be determined using routine methods based on factors such as, but not limited to, the particular vector construct and target cell and/or tissue selected.
- Such genetically modified can be introduced as linear constructs, circular plasmids or viral vectors, which may be integrating or non-integrating vectors.
- Transgenes can also be constructed so that they are inherited as extrachromosomal plasmids (Gassmann, et al., Proc. Natl. Acad. Sci. USA (1995) 92:1292).
- One or more single strands of the LPA dsRNA reagent can be transcribed from a promoter on the expression vector. Where two separate strands are to be expressed to produce eg dsRNA, two separate expression vectors can be co-introduced into the cell using eg transfection or infection.
- each individual strand of an LPA dsRNA agent of the invention can be transcribed by a promoter contained on the same expression vector.
- the LPA dsRNA agent is expressed as an inverted repeat polynucleotide linked by a linker polynucleotide sequence such that the LPA dsRNA agent has a stem-loop structure.
- Non-limiting examples of RNA expression vectors are DNA plasmids or viral vectors.
- Expression vectors useful in embodiments of the invention are compatible with eukaryotic cells.
- Eukaryotic expression vectors are routinely used in the art and are available from a number of commercial sources.
- Delivery of the LPA dsRNA expression vector can be systemic, such as by intravenous or intramuscular administration, by administration to target cells removed from the subject and then reintroduction of the target cells into the subject, or by any other means that permits the introduction of the desired target cells.
- Viral vector systems that may be included in embodiments of the method include, but are not limited to: (a) adenoviral vectors; (b) retroviral vectors, including but not limited to lentiviral vectors, Moloney murine leukemia virus, etc.; (c) adeno-associated viral vectors; (d) herpes simplex virus vectors; (e) SV40 vectors; Vectors or fowlpox virus vectors, such as canarypox or fowlpox virus vectors; (j) helper-dependent or gut-free adenovirus vectors.
- adenoviral vectors include, but are not limited to: (a) adenoviral vectors; (b) retroviral vectors, including but not limited to lentiviral vectors, Moloney murine leukemia virus, etc.; (c) adeno-associated viral vectors; (d) herpes simplex virus vectors; (e) SV40 vectors; Vectors or fowlp
- Constructs for recombinant expression of LPAdsRNA agents may contain regulatory elements, such as promoters, enhancers, etc., which may be selected to provide constitutive or regulated/inducible expression.
- regulatory elements such as promoters, enhancers, etc.
- Viral vector systems and the use of promoters and enhancers, etc. are routine in the art and can be used in conjunction with the methods and compositions described herein.
- Certain embodiments of the invention include the use of viral vectors to deliver LPA dsRNA agents into cells.
- a number of adenovirus-based delivery systems are routinely used in the art for delivery to, eg, the lung, liver, central nervous system, endothelial cells and muscle.
- Non-limiting examples of viral vectors that can be used in the methods of the invention are: AAV vectors, pox viruses such as vaccinia virus, modified Ankara virus (MVA), NYVAC, fowl pox such as fowl pox or canary pox virus.
- Certain embodiments of the invention include methods of delivering an LPA dsRNA agent into a cell using a carrier, and such carrier may be in a pharmaceutically acceptable carrier which may, but need not, include a sustained release matrix in which the gene delivery carrier is embedded.
- vectors for delivery of LPA dsRNA can be produced by recombinant cells, and pharmaceutical compositions of the invention can include one or more cells that produce the LPA dsRNA delivery system.
- compositions containing LPA dsRNA or ssRNA agents are provided.
- Certain embodiments of the invention include agents comprising LPA dsRNA or LPA antisense polynucleotide agents and pharmaceutically Use of a pharmaceutical composition with an acceptable carrier.
- Pharmaceutical compositions comprising LPA dsRNA agents or LPA antisense polynucleotide agents can be used in the methods of the invention to reduce LPA gene expression in cells, and can be used to treat LPA-related diseases or disorders.
- Such pharmaceutical compositions can be formulated based on the mode of delivery.
- Non-limiting examples of formulations for the mode of delivery are: compositions formulated for subcutaneous delivery, compositions formulated for systemic administration by parenteral delivery, compositions formulated for intravenous (IV) delivery, compositions formulated for intrathecal delivery, compositions formulated for direct delivery into the brain, etc.
- compositions of the invention can be administered using one or more means to deliver the LPA dsRNA agent or the LPA antisense polynucleotide agent into the cell, for example: surface (e.g., by a transdermal patch); lung, e.g., by inhalation or insufflation of a powder or aerosol, including by nebulizer; intraairway, intranasal, epidermal and transdermal, orally or parenterally.
- Parenteral administration includes intravenous, intraarterial, subcutaneous, intraperitoneal, or intramuscular injection or infusion; subcutaneous, eg, by an implanted device; or intracranial, eg, by intraparenchymal, intrathecal or intraventricular administration.
- LPA dsRNA agents or LPA antisense polynucleotide agents can also be delivered directly to target tissues, eg, directly to the liver, directly to the kidney, and the like.
- "delivering LPA dsRNA agent” or "delivering LPA antisense polynucleotide agent” into a cell respectively includes delivering LPA dsRNA agent or LPA antisense polynucleotide agent, expressing LPA dsRNA agent directly in a cell and expressing LPA dsRNA agent from an encoding vector delivered into a cell, or any suitable means that causes LPA dsRNA or LPA antisense polynucleotide agent to appear in a cell.
- the preparation and use of formulations and means for delivering inhibitory RNA are well known and routinely used in the art.
- a “pharmaceutical composition” comprises a pharmacologically effective amount of the LPA dsRNA agent or LPA antisense polynucleotide agent of the present invention and a pharmaceutically acceptable carrier.
- pharmaceutically acceptable carrier refers to a vehicle for administering a therapeutic agent.
- Such carriers include, but are not limited to, saline, buffered saline, dextrose, water, glycerol, ethanol, and combinations thereof. The term specifically excludes cell culture media.
- pharmaceutically acceptable carriers include, but are not limited to, pharmaceutically acceptable excipients such as inert diluents, disintegrants, binders, lubricants, sweeteners, flavoring agents, coloring agents and preservatives.
- suitable inert diluents include sodium and calcium carbonate, sodium and calcium phosphate and lactose, while corn starch and alginic acid are suitable disintegrants.
- Binders may include starch and gelatin, while lubricants, if present, are usually magnesium stearate, stearic acid or talc.
- Tablets may, if desired, be coated with a material such as glyceryl monostearate or glyceryl distearate to delay absorption from the gastrointestinal tract.
- a material such as glyceryl monostearate or glyceryl distearate to delay absorption from the gastrointestinal tract.
- pharmaceutically effective amount refers to the amount of the LPA dsRNA agent or LPA antisense polynucleotide agent of the invention that produces the desired pharmacological, therapeutic or prophylactic result.
- a therapeutically effective amount of a drug for treating the disease or condition is that amount required to reduce that parameter by at least 10%.
- a therapeutically effective amount of an LPA dsRNA agent or an LPA antisense polynucleotide agent can reduce LPA polypeptide levels by at least 10%.
- Pharmaceutical compositions may comprise dsRNAi agents comprising, for example, the duplexes shown in Table 1. In other embodiments, such dsRNAi agents include variants of the duplexes in Table 1.
- the methods of the invention comprise contacting a cell with an effective amount of an LPA dsRNA agent or an LPA antisense polynucleotide agent to reduce LPA gene expression in the contacted cell.
- Certain embodiments of the methods of the invention comprise administering to a subject an LPA dsRNA agent or an LPA antisense polynucleotide agent in an amount effective to reduce LPA gene expression and treat an LPA-associated disease or disorder in the subject.
- the "effective amount" used is the amount necessary or sufficient to achieve the desired biological effect.
- an effective amount of an LPA dsRNA agent or an LPA antisense polynucleotide agent to treat an LPA-associated disease or disorder can be: (i) the amount required to slow or stop the progression of the disease or disorder; (ii) reverse, reduce or eliminate one or more symptoms of the disease or disorder.
- an effective amount is that amount of an LPA dsRNA agent or LPA antisense polynucleotide agent that, when administered to a subject in need of treatment of an LPA-related disease or disorder, results in a therapeutic response for the prevention and/or treatment of the disease or disorder.
- an effective amount is an amount of an LPA dsRNA agent or LPA antisense polynucleotide agent of the invention that, when combined or co-administered with another therapeutic treatment for an LPA-associated disease or condition, results in a therapeutic response that prevents and/or treats the disease or condition.
- the biological effect of treating a subject with an LPA dsRNA agent or an LPA antisense polynucleotide agent of the invention may be an amelioration and/or complete elimination of symptoms caused by an LPA-associated disease or disorder.
- the biological effect is complete elimination of the LPA-associated disease or disorder, eg, as evidenced by a diagnostic test that indicates that the subject is free of the LPA-associated disease or disorder.
- detectable physiological symptoms include a decrease in lipid accumulation in the liver of a subject following administration of an agent of the invention.
- Other art-known means of assessing the status of an LPA-associated disease or disorder can be used to determine the effect of the agents and/or methods of the invention on an LPA-associated disease or disorder.
- an effective amount of an LPAdsRNA agent or an LPA antisense polynucleotide agent that lowers an LPA polypeptide to a level that treats an LPA-associated disease or condition is typically determined in a clinical trial that establishes an effective dose for test and control populations in blinded studies.
- an effective amount is an amount that results in a desired response, eg, an amount that reduces an LPA-associated disease or disorder in a cell, tissue, and/or in a subject suffering from the disease or disorder.
- an effective amount of an LPA dsRNA agent or LPA antisense polynucleotide agent for treating an LPA-associated disease or condition treatable by reducing the LPA polypeptide may be an amount that, when administered, reduces the amount of LPA polypeptide in a subject below the amount that would be present in the cell, tissue, and/or subject if the LPA dsRNA agent or LPA antisense polynucleotide agent were not administered.
- the level of LPA polypeptide and/or LPA gene expression present in cells, tissues and/or subjects that have not been exposed to or administered an LPA dsRNA agent or LPA antisense polynucleotide agent of the invention is referred to as a "control" amount.
- the subject's control amount is the subject's pre-treatment amount; in other words, the subject's level before administration of the LPA agent can be the subject's control level and used for comparison to its LPA polypeptide and/or LPA gene expression level after administration of the siRNA to the subject.
- the desired response may be to reduce or eliminate one or more symptoms of the disease or disorder in the cell, tissue, and/or subject. Reduction or elimination can be temporary or permanent. It will be appreciated that methods of determining LPA polypeptide, LPA gene expression, symptom assessment, clinical testing, etc. can be used to monitor the status of an LPA-associated disease or disorder. In some aspects of the invention, the desired response to treating an LPA-associated disease or disorder is to delay the onset of the disease or disorder or even prevent the onset of the disease or disorder.
- An effective amount of a compound that lowers an LPA polypeptide can also be determined by assessing the use of an LPA dsRNA assay.
- the physiological effect of the agent or the LPA antisense polynucleotide agent on the cell or subject such as the reduction of LPA-associated disease or disorder after administration.
- Assays and/or symptom monitoring in subjects can be used to determine the efficacy of the LPA dsRNA agents or LPA antisense polynucleotide agents of the invention (which can be administered in the pharmaceutical compounds of the invention), and to determine response to treatment.
- a non-limiting example is one or more serum lipid profile tests known in the art.
- one or more liver function tests known in the art can be used to determine the status of a subject's LPA-associated disease or disorder before and after treatment of the subject with an LPA dsRNA agent of the invention.
- the status of an LPA-related disease in a subject is determined using one or more tests for cholesterol accumulation in the liver known in the art.
- the disease involves cholesterol accumulation, and the test is used to determine cholesterol levels in a subject before and after treatment of the subject with an LPA dsRNA agent of the invention.
- Some embodiments of the invention include methods of determining the efficacy of a dsRNA agent or LPA antisense polynucleotide agent of the invention administered to a subject to treat an LPA-associated disease or disorder by assessing and/or monitoring one or more "physiological characteristics" of the LPA-associated disease or disorder in the subject.
- physiological characteristics of an LPA-related disease or disorder are the subject's serum LPA level, the subject's serum lipid level, the subject's low-density lipoprotein level, the subject's HDL level, the subject's LDL:HDL ratio, the subject's triglyceride level, the presence of fat in the subject's liver, physical symptoms, etc. Standard methods for determining such physiological characteristics are known in the art and include, but are not limited to, blood tests, imaging studies, physical examination, and the like.
- the amount of LPA dsRNA agent or LPA antisense polynucleotide agent administered to a subject can be modified based at least in part on such determination of a disease and/or condition state and/or physiological characteristic of the subject.
- the therapeutic amount can be varied, for example, by increasing or decreasing the amount of the LPA-dsRNA agent or LPA antisense polynucleotide agent by changing the composition in which the LPA dsRNA agent or LPA antisense polynucleotide agent is administered, by changing the route of administration, by changing the time of administration, etc.
- the effective amount of the LPA dsRNA agent or LPA antisense polynucleotide agent will vary with the particular condition being treated, the age and physical condition of the subject being treated, the severity of the condition, the duration of the treatment, the nature of co-treatments (if any), the particular route of administration, and other factors within the knowledge and expertise of a health practitioner.
- an effective amount can depend on the level of LPA polypeptide and/or the desired level of LPA gene expression effective to treat an LPA-related disease or disorder.
- a skilled artisan can empirically determine the effective amount of a particular LPA dsRNA agent or LPA antisense polynucleotide agent for use in the methods of the invention without undue experimentation.
- an effective prophylactic or therapeutic treatment regimen can be planned to effectively treat a particular subject.
- an effective amount of an LPA dsRNA agent or an LPA antisense polynucleotide agent of the invention may be an amount that produces a desired biological effect in the cell when contacted with the cell.
- LPA gene silencing can be performed constitutively or by genome engineering in any cell expressing LPA and determined by any suitable assay.
- LPA gene expression is reduced by at least 5%, 6%, 7%, 8%, 9%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, or 100%.
- LPA gene expression is reduced by 5% to 10%, 5% to 25%, 10% to 50%, 10% to 75%, 25% to 75%, 25% to 100%, or 50% to 100% by administering an LPA dsRNA agent of the invention.
- the LPA dsRNA agent and the LPA antisense polynucleotide agent are delivered in the pharmaceutical composition in an amount sufficient to inhibit expression of the LPA gene.
- the dose of LPA dsRNA agent or LPA antisense polynucleotide agent is 0.01 to 200.0 mg per kilogram of body weight of the recipient per day, typically 1 to 50 mg/kg body weight, 5 to 40 mg/kg body weight, 10 to 30 mg/kg body weight, 1 to 20 mg/kg body weight, 1 to 10 mg/kg body weight, 4 to 15 mg/kg body weight per day, inclusive.
- each single administration of LPA dsRNA agent or LPA antisense polynucleotide agent can be from about 0.01 mg/kg, 0.05 mg/kg, 0.1 mg/kg, 0.2 mg/kg, 0.3 mg/kg, 0.4 mg/kg, 0.5 mg/kg, 1 mg/kg, 1.1 mg/kg, 1.2 mg/kg, 1.3 mg/kg, 1.4 mg/kg, 1.5 mg/kg, 1.6 mg/kg , 1.7mg/kg, 1.8mg/kg, 1.9mg/kg, 2mg/kg, 2.1mg/kg, 2.2mg/kg, 2.3mg/kg, 2.4mg/kg, 2.5mg/kg, 2.6mg/kg, 2.7mg/kg, 2.8mg/kg, 2.9mg/kg, 3.0mg/kg, 3.1mg/kg, 3.2mg/kg, 3.3mg/kg, 3.4mg/kg kg, 3.5mg/kg, 3.6mg/kg,
- LPAdsRNA agent or LPA antisense polynucleotide agent delivered will depend on a variety of factors including co-treatments, number of doses and individual subject parameters including age, physical condition, size and body weight. These are factors well known to those of ordinary skill in the art and can be addressed by routine experimentation. In some embodiments, a maximum dose may be used, that is, the highest safe dose according to sound medical judgment.
- the methods of the invention may comprise administering to a subject 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 or more doses of an LPA dsRNA agent or an LPA antisense polynucleotide agent.
- the dosage of the pharmaceutical compound e.g., an LPA dsRNA-comprising agent or an LPA antisense polynucleotide-comprising agent
- compositions of the invention may be administered once daily; alternatively the LPA dsRNA agent or LPA antisense polynucleotide agent may be administered in two, three or more subdoses at appropriate intervals throughout the day, or even delivered using continuous infusion or via a controlled release formulation.
- the pharmaceutical composition of the invention is administered to the subject one or more times per day, one or more times per week, one or more times per month, or one or more times per year.
- the methods of the invention comprise administering a pharmaceutical compound alone; in combination with one or more other LPA dsRNA agents or LPA antisense polynucleotide agents; and/or in combination with other drug therapies or therapeutic activities or regimens administered to a subject suffering from an LPA-related disease or disorder.
- Pharmaceutical compounds can be administered in the form of pharmaceutical compositions.
- Pharmaceutical compositions used in the methods of the invention can be sterile and contain an amount of an LPA dsRNA agent or an LPA antisense polynucleotide agent that will reduce the level of an LPA polypeptide to a level sufficient to produce the desired response in a weight or volume unit suitable for administration to a subject.
- the dosage of a pharmaceutical composition comprising an LPA dsRNA agent or an LPA antisense polynucleotide agent administered to a subject to reduce LPA protein levels can be selected according to different parameters, in particular the mode of administration used and the state of the subject. Other factors include the length of treatment needed. If the subject does not respond adequately at the initial dose, higher doses may be employed (or effectively escalated by a different, more local route of delivery) as patient tolerance allows.
- the term "preventing” or “preventing”, when used in reference to a disease, disorder or condition thereof that would benefit from reduced expression of the LPA gene, refers to a reduction in the likelihood of a subject developing symptoms associated with such disease, disorder or condition, such as cardiovascular disease, including Berger's disease, peripheral artery disease, coronary artery disease, metabolic syndrome, acute coronary syndrome, aortic stenosis, aortic regurgitation, aortic dissection , retinal artery occlusion, cerebrovascular disease, mesenteric ischemia, superior mesenteric artery occlusion, renal artery stenosis, stable/unstable angina, acute coronary syndrome, heterozygous or homozygous familial hypercholesterolemia, hyperapolipoprotein beta lipoproteinemia, cerebrovascular atherosclerosis, cerebrovascular disease and venous thrombosis, stroke, atherosclerosis, thrombosis, coronary heart disease or aortic stenosis and
- Prophylaxis is considered effective if it reduces (eg, by at least about 10% on a scale that is clinically present with the disease or disorder), or delays the manifestation of symptoms (eg, by days, weeks, months, or years).
- LPA-associated diseases and conditions where reduction of the level of LPA polypeptide is effective in treating the disease or condition
- the methods of the invention and LPA dsRNA agents can be used for treatment to inhibit LPA expression.
- diseases and conditions that may be treated with the LPAdsRNA reagents or LPA antisense polynucleotide reagents of the invention and the methods of treatment of the invention include, but are not limited to: Berger's disease, peripheral artery disease, coronary artery disease, metabolic syndrome, acute coronary syndrome, aortic stenosis, aortic regurgitation, aortic dissection, retinal artery occlusion, cerebrovascular disease, mesenteric ischemia, superior mesenteric artery occlusion, renal artery stenosis, stable/unstable angina, acute coronary Syndrome, heterozygous or homozygous familial hypercholesterolemia, hyperapolipoprotein beta lipoproteinemia, cerebrovascular atherosclerosis, cerebrovascular
- an LPA dsRNA agent or LPA antisense polynucleotide agent of the invention can be administered to a subject at one or more times before or after diagnosis of an LPA-related disease or disorder.
- the subject is at risk of having or developing an LPA-related disease or disorder.
- a subject at risk of developing an LPA-associated disease or disorder is one who has an increased likelihood of developing an LPA-associated disease or disorder compared to a control risk of developing an LPA-associated disease or disorder.
- the level of risk is statistically significant compared to a control level of risk.
- Subjects at risk can include, for example: are or will be subjects with pre-existing diseases and/or genetic abnormalities that make the subject more susceptible to LPA-related diseases or disorders than control subjects without pre-existing diseases or genetic abnormalities; subjects with a family and/or personal history of LPA-related diseases or disorders; and subjects who have previously received treatment for LPA-related diseases or disorders.
- pre-existing diseases and/or genetic abnormalities that make a subject more susceptible to an LPA-related disease or disorder can be diseases or genetic abnormalities that, when present, have previously been determined to be associated with a higher likelihood of developing an LPA-related disease or disorder.
- LPA dsRNA agents or LPA antisense polynucleotide agents can be administered to a subject based on the medical condition of the individual subject.
- a healthcare provider to a subject can evaluate LPA levels measured in a sample obtained from the subject and determine that it is desirable to reduce the subject's LPA levels by administering an LPA dsRNA agent or an LPA antisense polynucleotide agent of the invention.
- a biological sample such as a blood or serum sample, can be obtained from a subject and the subject's LPA level determined in the sample.
- the LPA dsRNA reagent or the LPA antisense polynucleotide reagent is administered to the subject, and a blood or serum sample is obtained from the subject after administration, and the sample is used to determine LPA levels and the results compared to those determined in the subject's pre-dose (previous) sample.
- a subsequent decrease in the subject's LPA level in the sample compared to the pre-dose level indicates the efficacy of the administered LPA dsRNA agent or LPA antisense polynucleotide agent in reducing the subject's LPA level.
- Lp(a) levels in the blood can be considered a physiological characteristic of an LPA-related disorder, even if the subject has not been diagnosed with an LPA-related disorder, such as those disclosed herein.
- medical insurance The provider can monitor changes in Lp(a) levels in the subject's blood as a measure of the efficacy of the administered LPA dsRNA agent or LPA antisense polynucleotide agent of the invention.
- Certain embodiments of the methods of the invention include adjusting therapy comprising administering to a subject a dsRNA agent or an LPA antisense polynucleotide agent of the invention based at least in part on an assessment of a change in one or more physiological characteristics of an LPA-associated disease or disorder in the subject as a result of the treatment.
- the effect of a dsRNA agent or LPA antisense polynucleotide agent of the invention administered to a subject can be determined and used to help regulate the amount of a dsRNA agent or LPA antisense polynucleotide agent of the invention subsequently administered to the subject.
- a dsRNA agent or LPA antisense polynucleotide agent of the invention is administered to a subject, and the subject's blood level of Lp(a) is determined after administration; and based at least in part on the determined level, it is determined whether a higher amount of the dsRNA agent or LPA antisense polynucleotide agent is required to increase the physiological effect of the administered agent, such as reducing or further reducing the subject's blood level of Lp(a).
- a dsRNA agent or LPA antisense polynucleotide agent of the invention is administered to a subject, and the subject's blood level of Lp(a) is determined following administration, and based at least in part on the determined level, a lower amount of the dsRNA agent or LPA antisense polynucleotide agent is expected to be administered to the subject.
- some embodiments of the invention include assessing changes in one or more physiological characteristics resulting from previous treatment of a subject to adjust the amount of a dsRNA agent or LPA antisense polynucleotide agent of the invention subsequently administered to the subject.
- Some embodiments of the methods of the invention comprise 1, 2, 3, 4, 5, 6 or more determinations of a physiological characteristic of an LPA-associated disease or disorder; evaluating and/or monitoring the efficacy of administered LPA dsRNA agents or LPA antisense polynucleotide agents of the invention; and optionally using the results of the assays to adjust one or more of the dose, dosing regimen, and/or frequency of dosing in a subject treated with a dsRNA agent or LPA antisense polynucleotide agent of the invention for an LPA-associated disease or disorder.
- the desired result of administering to a subject an effective amount of a dsRNA agent or an LPA antisense polynucleotide agent of the invention is: the Lp(a) level in the blood of the subject is reduced compared to the previously determined Lp(a) level in the blood for the subject; the Lp(a) level in the blood of the subject is within the normal range.
- treating when applied to an LPA-associated disease or disorder can refer to prophylactic treatment, reducing the likelihood of a subject developing an LPA-associated disease or disorder, and can also refer to treatment to eliminate or reduce the level of an LPA-associated disease or disorder after a subject has developed an LPA-associated disease or disorder, prevent an LPA-associated disease or disorder from becoming more severe, and/or slow down an LPA-associated disease or disorder in a subject compared to a subject in the absence of therapy that reduces LPA polypeptide levels in the subject. Disease progression.
- the terms “inhibiting”, “silencing”, “reducing”, “down-regulating” and “knocking down” with respect to the expression of the LPA gene refer to altering the expression of the LPA gene, for example, by one or more of the level of RNA transcribed from the gene when a cell, cell population, tissue, organ or subject is contacted (e.g., treated) with an LPA dsRNA agent or an LPA antisense polynucleotide agent of the invention, compared to a control level of RNA transcribed from the LPA gene, a control level of LPA translated from the mRNA, respectively , the level of LPA expressed, and the LPA polypeptide translated from mRNA in a cell, cell population, tissue, organ or object, Decreased levels of protein or protein subunits.
- the control level is the level in a cell, tissue, organ, or subject
- LPA dsRNA agents or LPA antisense polynucleotide agents can be used in the methods of the invention.
- the choice of a particular mode of delivery will depend, at least in part, on the particular condition being treated and the dosage required for therapeutic efficacy.
- the methods of the invention can be practiced using any mode of administration that is medically acceptable, meaning any mode that produces effective therapeutic levels of an LPA-associated disease or disorder without causing clinically unacceptable side effects.
- LPA dsRNA agents or LPA antisense polynucleotide agents can be administered orally, enterally, mucosally, subcutaneously and/or parenterally.
- parenteral includes subcutaneous, intravenous, intrathecal, intramuscular, intraperitoneal and intrasternal injection or infusion techniques.
- Other routes include, but are not limited to, nasal (eg, via a gastric nasogastric tube), transdermal, vaginal, rectal, sublingual, and inhalation.
- the delivery routes of the present invention may include intrathecal, intraventricular or intracranial.
- the LPA dsRNA agent or the LPA antisense polynucleotide agent can be placed in a sustained release matrix and administered by placing the matrix in a subject.
- LPA dsRNA agents or LPA antisense polynucleotide agents can be delivered to cells in a subject using nanoparticles coated with delivery agents that target specific cells or organelles.
- delivery modes, methods, reagents are known in the art. Non-limiting examples of delivery methods and delivery agents are provided elsewhere herein.
- the term "delivery" in reference to an LPA dsRNA agent or LPA antisense polynucleotide agent may refer to administering one or more "naked" LPA dsRNA agent or LPA antisense polynucleotide agent sequences to a cell or subject.
- delivery refers to administering cells or objects by transfection, delivering cells comprising LPA dsRNA agents or LPA antisense polynucleotide agents to objects, delivering vectors encoding LPA dsRNA agents or LPA antisense polynucleotide agents to cells and/or objects, etc. Delivery of an LPA dsRNA agent or an LPA antisense polynucleotide agent using transfection can include administering the vector to the cell and/or subject.
- one or more LPA dsRNA agents or LPA antisense polynucleotide agents may be administered in a formulation or in a pharmaceutically acceptable solution, which typically may contain pharmaceutically acceptable concentrations of salts, buffers, preservatives, compatible carriers, adjuvants, and optionally other therapeutic ingredients.
- an LPA dsRNA agent or an LPA antisense polynucleotide agent may be formulated for simultaneous administration with another therapeutic agent.
- the LPA dsRNA agent or the LPA antisense polynucleotide agent can be administered in the form of a pharmaceutical composition.
- compositions comprise an LPA dsRNA agent or an LPA antisense polynucleotide agent and optionally a pharmaceutically acceptable carrier.
- Pharmaceutically acceptable carriers are well known to those of ordinary skill in the art.
- a pharmaceutically acceptable carrier refers to a nontoxic material that does not interfere with the effectiveness of the biological activity of the active ingredient (eg, the ability of an LPA dsRNA agent or an LPA antisense polynucleotide agent to inhibit LPA gene expression in a cell or subject).
- Various methods of administering and delivering dsRNA agents or LPA antisense polynucleotide agents for therapeutic use are known in the art and can be used in the methods of the invention.
- Pharmaceutically acceptable carriers include diluents, fillers, salts, buffers, stabilizers, solubilizers and other materials known in the art. Exemplary pharmaceutically acceptable carriers are described in US Patent No. 5,211,657, while others are known to those of skill in the art. Such formulations generally may contain salts, buffers, preservatives, compatible carriers and optionally other therapeutic agents. When used in medicine, the salt should be pharmaceutically acceptable, but non-pharmaceutically acceptable salts can be conveniently used to prepare their pharmaceutically acceptable salts, which are not excluded from the scope of the present invention.
- Such pharmacologically and pharmaceutically acceptable salts include, but are not limited to, salts prepared from the following acids: hydrochloric, hydrobromic, sulfuric, nitric, phosphoric, maleic, acetic, salicylic, citric, formic, malonic, succinic, and the like.
- pharmaceutically acceptable salts can be prepared as alkali metal or alkaline earth metal salts, such as sodium, potassium or calcium salts.
- Some embodiments of the methods of the invention comprise administering one or more LPA dsRNA agents or LPA antisense polynucleotide agents directly to the tissue.
- the tissue to which the compound is administered is a tissue in which an LPA-associated disease or disorder exists or is likely to occur, non-limiting examples of which are the liver or kidney.
- Direct tissue administration can be achieved by direct injection or other means. Many orally delivered compounds naturally enter and pass through the liver and kidneys, and some embodiments of the methods of treatment of the present invention comprise orally administering one or more LPA dsRNA agents to a subject.
- LPA dsRNA agents or LPA antisense polynucleotide agents can be administered once, or they can be administered multiple times. If administered multiple times, the LPA dsRNA agent or LPA antisense polynucleotide agent can be administered by different routes. For example, although not intended to be limiting, a first (or first few) administrations may be given subcutaneously, and one or more additional administrations may be oral and/or systemic.
- the LPA dsRNA agent or LPA antisense polynucleotide agent can be formulated for parenteral administration by injection, such as by bolus injection or continuous infusion.
- Formulations for injection may be presented in unit dosage form, eg, in ampoules or in multi-dose containers, with or without an added preservative.
- LPA dsRNA reagent preparations also referred to as pharmaceutical compositions
- Preparations for parenteral administration include sterile aqueous or non-aqueous solutions, suspensions and emulsions.
- non-aqueous solvents are propylene glycol, polyethylene glycol, vegetable oils such as olive oil, and injectable organic esters such as ethyl oleate.
- Aqueous carriers include water, alcoholic/aqueous solutions, emulsions or suspensions, including saline and buffered media.
- Parenteral vehicles include sodium chloride solution, Ringer's dextrose, dextrose and sodium chloride solution, lactated Ringer's, or fixed oils.
- Intravenous vehicles include fluid and nutrient replenishers, electrolyte replenishers (such as those based on Ringer's dextrose), and the like. Preservatives and other additives may also be present, such as antimicrobials, antioxidants, chelating agents, and inert gases, among others. Other forms of administration, such as intravenous administration, will result in lower doses. If the subject does not respond adequately at the initial dose, higher doses may be employed (or effectively escalated by a different, more local route of delivery) as tolerated by the patient. Multiple doses per day can be used as needed to achieve appropriate systemic or local levels of one or more LPA dsRNA agents or LPA antisense polynucleotide agents and to achieve appropriate reductions in LPA protein levels.
- the methods of the invention involve the use of delivery vehicles, such as biocompatible microparticles, nanoparticles Granules or implants suitable for implantation into a recipient, such as a subject.
- delivery vehicles such as biocompatible microparticles, nanoparticles Granules or implants suitable for implantation into a recipient, such as a subject.
- biodegradable implants that may be used according to this method are described in PCT Publication WO95/24929 (incorporated herein by reference), which describes biocompatible, biodegradable polymer matrices for containing biomacromolecules.
- both non-biodegradable and biodegradable polymer matrices can be used in the methods of the invention to deliver one or more LPA dsRNA agents or LPA antisense polynucleotide agents to a subject.
- the matrix can be biodegradable.
- Matrix polymers can be natural or synthetic polymers. The polymer can be selected based on the desired period of time for release, typically on the order of a few hours to a year or more. Typically, releases over a period of a few hours to a period of between three and twelve months are available.
- the polymer is optionally in the form of a hydrogel, which can absorb up to about 90% of its weight in water, and is also optionally crosslinked with multivalent ions or other polymers.
- LPA dsRNA agents or LPA antisense polynucleotide agents may in some embodiments of the invention be delivered by diffusion or by degradation of the polymer matrix using biodegradable implants.
- Exemplary synthetic polymers for this use are well known in the art.
- Biodegradable polymers and non-biodegradable polymers can be used to deliver LPA dsRNA agents or LPA antisense polynucleotide agents using methods known in the art.
- Bioadhesive polymers such as bioerodible hydrogels (H.S. Sawhney, C.P. Pathak and J.A.
- Hubell in Macromolecules, 1993, 26, 581-587) can also be used to deliver LPA dsRNA agents or LPA antisense polynucleotide agents to treat LPA-related diseases or conditions.
- Other suitable delivery systems may include timed release, delayed release or sustained release delivery systems. Such systems can avoid repeated administration of LPA dsRNA agents or LPA antisense polynucleotide agents, thereby improving convenience for subjects and healthcare professionals.
- Many types of release delivery systems are available and known to those of ordinary skill in the art. See, eg, US Patent Nos. 5,075,109, 4,452,775, 4,675,189, 5,736,152, 3,854,480, 5,133,974, and 5,407,686. Additionally, pump-based hardware delivery systems are available, some of which are also suitable for implantation.
- long-term sustained-release implants may be suitable for prophylactic treatment of subjects and subjects at risk of developing recurrent LPA-related diseases or conditions.
- long-term release refers to an implant constructed and arranged to deliver therapeutic levels of an LPA dsRNA agent or an LPA antisense polynucleotide agent for at least up to 10 days, 20 days, 30 days, 60 days, 90 days, six months, one year, or longer.
- Long term sustained release implants are well known to those of ordinary skill in the art and include some of the release systems described above.
- Therapeutic formulations of LPA dsRNA reagents or LPA antisense polynucleotide reagents can be prepared for storage by mixing the molecule or compound of desired purity with optional pharmaceutically acceptable carriers, excipients or stabilizers [Remington's Pharmaceutical Sciences 21 st edition, (2006)] either as a lyophilized formulation or as an aqueous solution.
- Acceptable carriers, excipients, or stabilizers are nontoxic to recipients at the dosages and concentrations employed, and include buffers, such as phosphates, citrates, and other organic acids; antioxidants, including ascorbic acid and methionine; preservatives, such as octadecyldimethylbenzylammonium chloride; hexamethylammonium chloride; benzalkonium chloride, benzethonium chloride; phenol, butanol, or benzyl alcohol; parabens, such as methyl or propylparaben; cyclohexanol; 3-pentanol; and m-cresol); low molecular weight (less than about 10 residues) polypeptides; proteins such as serum albumin, gelatin, or immunoglobulins; Hydrophilic polymers such as polyvinylpyrrolidone; amino acids such as glycine, glutamine, asparagine, histidine, arginine or lys
- the methods of the invention can be used in conjunction with cells, tissues, organs and/or subjects.
- the subject is a human or a vertebrate mammal including, but not limited to, dogs, cats, horses, cows, goats, mice, rats, and primates such as monkeys. Accordingly, the present invention is useful in the treatment of LPA-related diseases or conditions in both human and non-human subjects.
- the subject can be a farm animal, a zoo animal, a domesticated animal, or a non-domesticated animal, and the methods of the invention can be used in veterinary prophylactic and therapeutic regimens.
- the subject is a human and the methods of the invention are useful in human prophylactic and therapeutic regimens.
- a non-limiting example of a subject to which the present invention may be applied is a subject diagnosed, suspected of having or at risk of having a disease or condition associated with a higher than desired expression of LPA, also referred to as "elevated LPA expression level".
- diseases and disorders associated with higher than desired levels of LPA expression are described elsewhere herein.
- the methods of the invention are applicable to subjects who have been diagnosed with the disease or disorder, are associated with, or are considered to be at risk of having or developing a disease or disorder associated with higher than desired expression of LPA at the time of treatment.
- the disease or disorder associated with higher than desired LPA expression levels is an acute disease or disorder; in certain aspects of the invention, the disease or disorder associated with higher than desired LPA expression levels is a chronic disease or disorder.
- the LPA dsRNA reagents of the present invention are administered to a diagnosed cardiovascular disease, wherein the cardiovascular disease includes Berger's disease, peripheral artery disease, coronary artery disease, metabolic syndrome, acute coronary syndrome, aortic stenosis, aortic regurgitation, aortic dissection, retinal artery occlusion, cerebrovascular disease, mesenteric ischemia, superior mesenteric artery occlusion, renal artery stenosis, stable/unstable angina, acute coronary syndrome, heterozygous or pure Zygotic familial hypercholesterolemia, hyperapolipoprotein beta lipoproteinemia, cerebrovascular atherosclerosis, cerebrovascular disease and venous thrombosis, stroke, atherosclerosis, thrombosis, coronary heart disease or aortic stenosis and/or any other disease or pathology associated with elevated levels of Lp(a)-containing particles.
- the methods of the invention are applicable to subjects
- LPA-associated disease includes diseases, disorders or conditions that benefit from reduced expression of LPA. These diseases are often associated with high blood pressure.
- Non-limiting examples of LPA-related diseases include cardiovascular diseases, wherein the cardiovascular diseases include Berger's disease, peripheral artery disease, coronary artery disease, metabolic syndrome, acute coronary syndrome, aortic stenosis, aortic regurgitation, aortic dissection, retinal artery occlusion, cerebrovascular disease, mesenteric ischemia, superior mesenteric artery occlusion, renal artery stenosis, stable/unstable angina, acute coronary syndrome, heterozygous or homozygous familial hypercholesterolemia, hyperapolipoprotein beta Lipoproteinemia, cerebrovascular atherosclerosis, cerebrovascular disease and venous thrombosis, stroke, atherosclerosis, thrombosis, coronary heart disease or aortic stenosis and/or any other disease or pathology associated with elevated levels of Lp(a)-containing particles.
- cardiovascular diseases include Berger's disease, peripheral artery disease, coronary artery disease, metabolic syndrome, acute coronar
- Cells to which the methods of the present invention can be applied include in vitro, in vivo, and ex vivo cells.
- Cells may be in a subject, in culture and/or in suspension, or in any other suitable state or condition.
- the cells to which the methods of the invention may be applied may be: liver cells, hepatocytes, cardiac cells, pancreatic cells, cardiovascular cells, kidney cells or other types of vertebrate cells, including human and non-human mammalian cells.
- the cells to which the methods of the invention are applicable are healthy normal cells that are not known to be diseased cells.
- control cells are normal cells, but it is understood that cells with a disease or condition may also be used as control cells in certain circumstances, such as in the context of comparing the results of treated cells with a disease or condition to untreated cells with a disease or condition, etc.
- LPA polypeptide levels can be determined and compared to LPA polypeptide control levels.
- a control can be a predetermined value, which can take a variety of forms. It can be a single cutoff such as median or mean. It can be established based on comparing groups, for example in a group with normal levels of LPA polypeptide and a group with increased level of LPA polypeptide activity.
- Another non-limiting example of a comparison group may be a population with one or more symptoms or diagnosis of a LPA-related disease or disorder versus a population without one or more symptoms or diagnosis of a disease or disorder; a group of subjects to whom an siRNA treatment of the invention has been administered versus a group of subjects who have not been administered an siRNA treatment of the invention.
- controls can be based on apparently healthy normal individuals or apparently healthy cells in an appropriate age group.
- a control according to the invention may be a material sample tested in parallel with the experimental material. Examples include samples from control populations or control samples produced by manufacturing for testing in parallel with experimental samples.
- a control can include a cell or subject that has not been contacted or treated with an LPA dsRNA agent of the invention, in which case the control level of the LPA polypeptide can be compared to the level of the LPA polypeptide in a cell or subject contacted with an LPA dsRNA agent or an LPA antisense polynucleotide agent of the invention.
- control level may be a level of LPA polypeptide determined for a subject, wherein the level of LPA polypeptide determined for the same subject at different times is compared to the control level.
- the level of LPA is determined in a biological sample obtained from a subject who has not received LPA treatment of the present invention.
- the biological sample is a serum sample. The level of LPA polypeptide determined in the sample obtained from the subject can be used as the subject's baseline or control value.
- one or more additional serum samples can be obtained from the subject, and the level of LPA polypeptide in the subsequent one or more samples can be compared to the subject's control/baseline level. Such comparisons can be used to assess the onset, progression or regression of an LPA-related disease or disorder in a subject.
- a level of LPA polypeptide in a baseline sample obtained from a subject that is higher than the level obtained from the same subject following administration of an LPA dsRNA agent of the invention or an LPA antisense polynucleotide agent of the invention is indicative of regression of the LPA-associated disease or disorder and indicative of efficacy of the administered LPA dsRNA agent of the invention in treating the LPA-associated disease or disorder.
- one or more of the LPA polypeptide levels determined for a subject can be used as a control value and used to compare LPA polypeptide levels later in the same subject, thereby allowing assessment of changes in "baseline" LPA polypeptide levels in the subject.
- the initial level can be used as a control level for the subject, the initial LPA polypeptide level can be used to show and/or determine that the methods and compounds of the invention are capable of reducing the level of LPA polypeptide in a subject.
- LPA dsRNA agents and/or LPA antisense polynucleotide agents of the invention can be administered to a subject.
- dsRNAi agents include, for example, the duplexes shown in Table 1.
- dsRNAi agents include duplex variants, such as the duplex variants shown in Table 1.
- Efficacy of administration and treatment of the invention can be assessed by reducing the level of LPA polypeptide in a serum sample obtained from a subject by at least 0.5%, 1%, 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95% when administered and treated, compared to the pre-dose level of LPA polypeptide in a serum sample obtained from the subject at a previous time point, or compared to a non-contact control level (e.g., the level of LPA polypeptide in a control serum sample). % or more. It should be understood that the level of LPA polypeptide is related to the level of LPA gene expression.
- Certain embodiments of the methods of the invention comprise administering to a subject an LPA dsRNA and/or an LPA antisense agent of the invention in an amount effective to inhibit expression of the LPA gene, thereby reducing the level of an LPA polypeptide in the subject.
- Some embodiments of the invention include determining the presence, absence and/or amount (also referred to herein as level) of an LPA polypeptide in one or more biological samples obtained from one or more subjects.
- This assay can be used to assess the efficacy of the therapeutic methods of the invention.
- the methods and compositions of the invention can be used to determine the level of an LPA polypeptide in a biological sample obtained from a subject previously treated with administration of an LPA dsRNA agent and/or an LPA antisense agent of the invention.
- a decrease in the level of LPA polypeptide in a serum sample obtained from a subject after administration and treatment by at least 0.5%, 1%, 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95% or more is indicative of efficacy of the treatment administered to the subject when compared to the pre-administration level of the LPA polypeptide in a serum sample obtained from the subject at a previous time point, or compared to a non-contact control level (e.g., the level of LPA polypeptide in a control serum sample). level.
- the physiological characteristics of an LPA-related disease or condition determined for a subject can be used as a control result, and the determination of the physiological characteristics of the same subject at different times can be compared with the control results.
- Lp(a) levels (and/or other physiological characteristics of an LPA disease or condition) in blood are measured from subjects who have never been administered LPA treatment of the invention, which can be used as a baseline or control value for the subject. After one or more LPA dsRNA reagents are administered to the subject in the treatment method of the present invention, the Lp(a) level in the blood is measured and compared with that of the subject respectively. / baseline level for comparison.
- Such comparisons can be used to assess the onset, progression or regression of an LPA-related disease or disorder in a subject.
- a baseline LPA level obtained from a subject that is higher than the LPA level measured from the same subject after administration of an LPA dsRNA agent of the invention or an LPA antisense polynucleotide agent to the subject is indicative of regression of the LPA-associated disease or disorder and indicative of the efficacy of the administered LPA dsRNA agent of the invention in treating the LPA-associated disease or disorder.
- the values determined for a subject for one or more physiological characteristics of an LPA-associated disease or disorder may serve as control values for later comparison of the same subject's physiological characteristics, thereby allowing assessment of changes in the subject's "baseline" physiological characteristics.
- the initial physiological profile assay as a control for the subject, and show and/or determine the effectiveness of the methods and compounds of the invention for reducing the level of LPA polypeptide in an individual.
- the LPA dsRNA agents and/or LPA antisense polynucleotide agents of the invention can be administered to a subject in an amount effective to treat an LPA disease or disorder.
- Efficacy of administrations and treatments of the invention can be assessed by determining changes in one or more physiological characteristics of LPA diseases or conditions.
- the Lp(a) level in the subject's blood is reduced compared to the Lp(a) level in the blood obtained from the subject at a previous time point, or compared to the LPA level in an unexposed control, until the Lp(a) level in the subject's blood is within a normal range.
- Some embodiments of the invention include determining the presence, absence, and/or changes in physiological characteristics of an LPA-associated disease or disorder using methods such as, but not limited to: (1) measuring Lp(a) levels in the blood of a subject; (2) evaluating physiological characteristics in one or more biological samples obtained from one or more subjects; (3) or performing a physical examination of the subject. This assay can be used to assess the efficacy of the therapeutic methods of the invention.
- Kits comprising one or more LPA dsRNA reagents and/or LPA antisense polynucleotide reagents and instructions for their use in the methods of the invention are also within the scope of the invention.
- the kits of the invention may comprise one or more of LPA dsRNA reagents, LPA sense polynucleotides and LPA antisense polynucleotide reagents useful for treating LPA-related diseases or conditions.
- Kits comprising one or more LPA dsRNA agents, LPA sense polynucleotides, and LPA antisense polynucleotide agents can be prepared for use in the methods of treatment of the invention.
- kits of the invention may be packaged in aqueous media or in lyophilized form.
- a kit of the invention may comprise a carrier compartmentalized to receive hermetically therein one or more container means or series of container means (eg test tubes, vials, flasks, bottles, syringes, etc.).
- the first container device or series of container devices may contain one or more compounds, such as LPA dsRNA reagents and/or LPA sense or antisense polynucleotide reagents.
- a second container device or series of container devices may contain targeting agents, labeling agents, delivery agents, etc., which may be included as part of the LPA dsRNA agent and/or LPA antisense polynucleotide administered in embodiments of the therapeutic methods of the invention.
- kits of the invention may further comprise instructions.
- the instructions are usually in written form and will provide guidance on administering the treatment embodied by the kit and making decisions based on that treatment.
- the sense and antisense strand sequences of the siRNA are synthesized on an oligonucleotide synthesizer using well-established solid-phase synthesis methods based on phosphoramidite chemistry.
- the growth of the oligonucleotide chain is achieved by a 4-step cycle: deprotection, condensation, capping and an oxidation or sulfuration step for the addition of each nucleotide.
- the synthesis is made of controlled pore glass (CPG, ) made on a solid support.
- Monomeric phosphoramidites were purchased from commercial sources.
- Phosphoramidites with GalNAc ligand clusters (GLPA1 and GLPA2 as non-limiting examples) were synthesized according to the procedures of Examples 2-3 herein.
- Trichloroacetic acid (TCA) in 3% dichloromethane was used for deprotection of the 4,4'-dimethoxytrityl protecting group (DMT). 5-Ethylthio-1H-tetrazole was used as activator.
- I2 in THF/Py/ H2O and phenylacetyl disulfide (PADS) in pyridine/MeCN were used for the oxidation and sulfurization reactions, respectively.
- PADS phenylacetyl disulfide
- the solid support-bound oligomer was cleaved and the protecting groups removed by treatment with 1:1 volume of 40 wt% aqueous methylamine and 28% ammonium hydroxide solution.
- the crude mixture was concentrated. The remaining solid was dissolved in 1.0M NaOAc and ice-cold EtOH was added to precipitate the single-chain product as the sodium salt, which was used for annealing without further purification.
- the crude single-stranded product was further purified by ion-pair reverse-phase HPLC (IP-RP-HPLC).
- IP-RP-HPLC ion-pair reverse-phase HPLC
- the purified single-stranded oligonucleotide product from IP-RP-HPLC was converted to the sodium salt by dissolving it in 1.0 M NaOAc and precipitating by adding ice-cold EtOH. Annealing of the sense and antisense strand oligonucleotides was performed by equimolar complementation in water to form a double-stranded siRNA product, which was lyophilized to provide a fluffy white solid.
- Intermediate-A was synthesized by treating commercially available galactosamine pentaacetate with trimethylsilyl triflate (TMSOTf) in dichloromethane (DCM) as shown in Scheme 1 below. Glycosylation with Cbz-protected 2-(2-aminoethoxy)ethan-1-ol then affords compound II. The Cbz protecting group was removed by hydrogenation to provide Intermediate-A as a trifluoroacetate (TFA) salt.
- Intermediate B was synthesized based on the same protocol except using Cbz protected 2-(2-(2-aminoethoxy)ethoxy)ethan-1-ol as starting material.
- GLPA1 and GLPA2 were prepared according to Scheme 2 below. Starting from benzyl-protected propane-1,3-diamine, its alkylation with tert-butyl 2-bromoacetate affords triester compound I. Removal of the benzyl protecting group by hydrogenation affords the secondary amine compound II. Coupling of the amide with 6-hydroxyhexanoic acid affords compound III. The tert-butyl protecting group is then removed upon treatment with HCl in dioxane to yield the triacid compound IV. Amide coupling between triacid Compound IV and Intermediate-A or Intermediate-B is performed to provide Compound Va or Vb.
- the phosphoramidites GLPA1 or GLPA2 are synthesized by the phosphoritylation of compounds Va or Vb with 2-cyanoethyl N,N-diisopropylchlorophosphoramidite and a catalytic amount of 1H-tetrazole.
- the GalNAc ligand phosphoramidite compound GLPA2 was synthesized using the same procedure except using Intermediate-B.
- 1 H NMR(400MHz,CDCl 3 ):ppm ⁇ 7.94-8.18(m,1H),7.69(br s,1H),6.66-7.10(m,3H),5.35(d,J 3.5Hz,3H),5.07-5.25(m,3H),4.76-4.86(m,3H),4.01-4.31(m,10H),3.91-4.01(m,8H),3.74-3.86(m,4H),3.52-3.71(m,30H),3.42-3.50(m,6H),3.15-3.25(m,4H),2.52-2.70(m,4H),2.22-2.45(m,2H),2.15-2.22(s,9H),2.06(s,9H),1.95-2.03(m,18H),1.77(br s,2H),1.58-1.66(m,4H),1.40(m,
- GLPA15 was prepared according to Scheme 3 below.
- TBTU (327g, 1.02mol, 3.90eq.), triethylamine (212g, 2.09mol, 8.00eq.) were added to the DMF (1.0L) solution of compound 6 (100g, 261mmol.) and intermediate-A (502g, 915.mmol, 3.50eq.), and the reaction was carried out at 25°C for 1 hour.
- LCMS showed that the conversion of compound 6 was complete.
- the reaction solution was added to 4000 mL of water, and extracted with methyl tert-butyl ether (2000 mL twice) to remove impurities, and the remaining aqueous phase was extracted with dichloromethane (3000 mL twice).
- the reaction was quenched by slow addition of saturated NH4Cl (3.0 L), the layers were separated, the aqueous phase was extracted with dichloromethane (2x1000 mL) and combined with the previous organic phase. The combined organic phases were washed with a 1:1 mixture of saturated NaHCO 3 (aq) and saturated brine (3.0 L), dried over anhydrous Na 2 SO 4 , filtered and concentrated under reduced pressure.
- the crude product was dissolved in 1.5L of dichloromethane, added dropwise to methyl tert-butyl ether (7.5L), half A transparent white precipitate gradually formed during the dropwise addition.
- a targeting group comprising GalNAc also referred to herein as a GalNAc delivery compound
- a GalNAc delivery compound also referred to herein as a GalNAc delivery compound
- GLPA1 GalNAc phosphoramidite
- the method of attaching a GalNAc-containing targeting group to the 3'-end of the sense strand involved the use of a GLO-n-containing solid support (CPG).
- CPG GLO-n-containing solid support
- methods for attaching a GalNAc-containing targeting group to the 3′-end of the sense strand included: attaching the GalNAc targeting group to a CPG solid support via an ester bond, and using the resulting CPG with the attached GalNAc targeting group when synthesizing the sense strand, which resulted in the attachment of the GalNAc targeting group at the 3′-end of the sense strand.
- Other GalNAc phosphoramidite compounds (GLPAn) can also be obtained by using a reasonable corresponding intermediate, using a method similar to this article or well-known in the art, and connected to the siRNA duplex as a targeting group Suitable location.
- Embodiment 4 Synthesis of isomannitol phosphoramidite (compound 2)
- N,N-dimethylformamide (23.50kg) into a 100L glass kettle and start stirring. Control the temperature at 20-30°C, and under the protection of nitrogen, add the product of the previous step, O-benzotriazole-tetramethyluronium hexafluorophosphate (0.33kg) and N,N-diisopropylethylamine (0.13kg) into the above-mentioned 100L glass kettle through the solid addition funnel.
- the isosorbide residue (imann) can be added to the 5' end or 3' end of the oligonucleotide chain by a method well known to those skilled in the art, such as the reverse abasic (invab) process, and a targeting group can be further added.
- Huh7 cells were adjusted to a suitable density and seeded into 96-well plates. Simultaneously with inoculation, the dual fluorescent reporter vector psciCHECK2 containing the target gene was co-transfected with siRNA into Huh7 cells using Lipofectamine RNAiMax (Invitrogen-13778-150) according to the manufacturer's recommendations. Cells were transfected with test siRNA or control siRNA. siRNA was tested in triplicate at two concentrations (0.1 nM and 1.0 nM), and 48 hours after transfection, Dual- Luciferase Assay Reagent reagent detects fluorescence value.
- Lipofectamine RNAiMax Invitrogen-13778-150
- the ratio of renilla luminescence to firefly luminescence was calculated and normalized based on the ratio of control siRNA-treated samples to calculate knockdown efficiency.
- the results are shown in Table 4, and the duplex AV# used was derived from the corresponding sequence shown in Table 2.
- Table 4 provides the experimental results of in vitro studies using various LPA RNAi agents to inhibit LPA expression.
- mice infected with AAV encoding human LPA and luciferase genes were used (4 mice per group).
- AAV8 adeno-associated virus 8
- Percent knockdown was calculated by comparing the luciferase activity of pre-dose blood samples from the siRNA-treated group with those collected at the end of day 7 and normalized based on the change in luciferase activity in serum samples from the PBS-treated group. The results are shown in Table 5, and the duplex AD# used was derived from the corresponding sequence shown in Table 3.
- Table 5 provides the experimental results of the in vivo study of inhibition of LPA expression using various LPA_RNAi single 6mpk agents. Day 7, relative to remaining luciferase activity on day 0, normalized by change in PBS-treated group (mean ⁇ SD)
- mice infected with AAV encoding human LPA and luciferase genes were used (4 mice per group).
- AAV8 adeno-associated virus 8
- Luciferase activity was measured and percent knockdown was calculated by comparing the luciferase activity of pre-dose blood samples in the siRNA-treated group with those collected on days 7 and 14 and normalizing based on the change in luciferase activity in serum samples from the PBS-treated group.
- human LPA mRNA level (qPCR determination) knockdown (retention) percentage in the 14th day mouse liver of the siRNA treatment group and the PBS treatment group the results are shown in Table 6, and the duplex AD# used is derived from the sequence corresponding to that shown in Table 3.
- Table 6 Provides the experimental results of an in vivo study of inhibition of LPA expression using various LPA_RNAi single 6mpk agents. NA means not detected.
- mice infected with AAV encoding human LPA and luciferase genes were used (4 mice per group).
- AAV8 adeno-associated virus 8
- Table 7 Provides the experimental results of the in vivo study on the inhibition of LPA expression using various LPA_RNAi agents of 3mpk, 6mpk and 10mpk respectively.
- mice infected with AAV encoding human LPA and luciferase genes were used (4 mice per group).
- AAV8 adeno-associated virus 8
- Percent knockdown was calculated by comparing the luciferase activity of pre-dose blood samples in the siRNA-treated group with those collected at the end of days 7, 14, and 21 and normalizing based on the change in luciferase activity in serum samples from the PBS-treated group. The results are shown in Table 8.
- the duplex AD# used was derived from the corresponding sequence shown in Table 3.
- Table 8 Provides experimental results from an in vivo study of inhibition of LPA expression using various LPA_RNAi single 2 and 6 mpk agents, on days 7, 14 and 21, relative to luciferase activity remaining on day 0, normalized by the change in the PBS-treated group (mean ⁇ SD).
- the collected blood samples were left at room temperature for at least 30 minutes to clot, and then centrifuged at 3500 rpm for 10 minutes at 4°C.
- the collected sera (approximately 1.0 mL) were transferred into two pre-labeled polypropylene screw-cap vials (0.5 ml/vial, one for ELISA assay and the other for spare) and stored in a -80°C freezer until testing.
- Percentage LPA remaining normalized to day -14 (pre-dose), -7 (pre-dose), 1 (pre-dose) means, pre-dose of siRNA) is shown in Figure 1 .
- the collected sera (approximately 1.0 mL) were transferred into two pre-labeled polypropylene screw cap vials (0.5 ml/vial, one for ELISA assay and the other for spare) and stored in a -80°C freezer until testing.
- Percent LPA remaining (normalized to Day -14 (pre-dose), 0 (pre-dose) mean, pre-dose of siRNA) is shown in FIG. 2 .
- Huh7 cells were adjusted to a suitable density and seeded into 96-well plates. Simultaneously with inoculation, the dual fluorescent reporter vector psciCHECK2 containing the target gene was co-transfected with siRNA into Huh7 cells using Lipofectamine RNAiMax (Invitrogen-13778-150) according to the manufacturer's recommendations. Cells were transfected with test siRNA or control siRNA. siRNA was tested in triplicate at two concentrations (0.1 nM and 1.0 nM), and 48 hours after transfection, Dual- Luciferase Assay Reagent reagent detects fluorescence value.
- Lipofectamine RNAiMax Invitrogen-13778-150
- the ratio of renilla luminescence to firefly luminescence was calculated and normalized based on the ratio of control siRNA-treated samples to calculate knockdown efficiency.
- the results are shown in Table 9, and the duplex AV# used was derived from the sequence corresponding to that shown in Table 2.
- Table 9 provides the experimental results of in vitro studies using various LPA RNAi agents to inhibit LPA expression.
Abstract
本申请提供了可用于降低LPA(Apo(a))基因表达和用于治疗LPA相关疾病和病症的组合物和方法。提供了可用于降低细胞和对象中LPA表达的LPA dsRNA试剂、LPA反义多核苷酸试剂、包含LPA dsRNA试剂的组合物和包含LPA反义多核苷酸试剂的组合物。
Description
本发明的部分实施方案涉及可用于抑制LPA(Apo(a))蛋白表达的组合物和方法。
Lp(a)粒子是主要在肝中表达的异质低密度脂蛋白粒子(Witztum和Ginsberg,J Lipid Res.2016年3月;57(3):336-9)。它们由载脂蛋白(a)(Apolipoprotein(a))(Apo(a)或Lp(a)由LPA基因编码)通过ApoB多肽连接于LDL样粒子组成。遗传限定的高Lp(a)粒子血清水平不受膳食和运动的影响,并且通过相关联的动脉粥样硬化潜在性而与罹患心血管疾病的风险增加相关联(Alonso等,Journal ofthe American College of Cardiology第63卷,第19期,2014)。根据诊断学和预防医学,患者的Lp(a)粒子血清水平是冠心病和主动脉瓣狭窄的一种高度普遍的独立遗传风险因素(Saeedi和Frohlich Clinical Diabetes and Endocrinology(2016)2:7)。在多项研究中对Lp(a)水平的分析提示高Lp(a)水平是心血管疾病、中风和其他相关病症(包括动脉粥样硬化性狭窄)的独立风险因子。此外,全基因组关联分析也发现LPA作为诸如动脉粥样硬化性狭窄的疾病的遗传风险因子。当使用治疗性脂蛋白清血法降低高脂血症患者体内的Lp(a)和LDL水平两者时,观察到心血管事件显著减少。因此,需要与这些和其它LPA相关疾病相关的治疗剂和治疗。
然而,除间接标准一般性LDL降低措施之外,不存在当前被核准的特定Lp(a)粒子降低疗法。因此,当前需要用于有效治疗、预防诸如以下以及与以下相关的病症以及降低罹患诸如以下以及与以下相关的病症的风险的方法:伯格氏病(Berger’s disease)、外周动脉疾病、冠状动脉疾病、代谢综合征、急性冠脉综合征、主动脉瓣狭窄、主动脉瓣反流、主动脉夹层、视网膜动脉阻塞、脑血管疾病、肠系膜缺血、肠系膜上动脉阻塞、肾动脉狭窄、稳定型/不稳定型心绞痛、急性冠脉综合征、杂合子或纯合子家族性高胆固醇血症、高载脂蛋白β脂蛋白血症、脑血管动脉粥样硬化、脑血管疾病和静脉血栓形成、中风、动脉粥样硬化、血栓形成、冠心病或主动脉瓣狭窄和/或与含Lp(a)粒子的水平升高相关的任何其他疾病或病理以及其他尚未鉴定的相关病症、病理或综合征。本发明解决这个未满足的医学需要。
发明内容
根据本发明的一个方面,提供了一种抑制LPA(Apo(a))表达的双链核糖核酸(dsRNA)试剂,该dsRNA试剂包含正义链和反义链,在反义链中的核苷酸位置2至18处包含与LPA RNA
转录物互补的区域,其中互补区域包含与表1-3中所列出的反义序列之一相差0、1、2或3个核苷酸的至少15个连续核苷酸,并且任选地包含靶向配体。在一些实施方案中,与LPARNA转录物互补的区域包含与表1-3中所列出的反义序列之一相差不超过3个核苷酸的至少15、16、17、18或19个连续核苷酸。在某些实施方案中,dsRNA的反义链与人LPA基因mRNA的任意靶区域至少基本互补,并且在表1-3之一中提供。在一些实施方案中,dsRNA的反义链与人LPA基因mRNA的任意靶区域完全互补并且在表1-3之一中提供。在一些实施方案中,dsRNA试剂包含表1-3中所列出的任一个正义链序列,其中正义链序列与dsRNA试剂中的反义链序列至少基本上互补。在某些实施方案中,dsRNA试剂包含表1-3中列出的任一个正义链序列,其中正义链序列与dsRNA试剂中的反义链序列完全互补。在一些实施方案中,dsRNA试剂包含表1-3中列出的任一个反义链序列。在一些实施方案中,dsRNA试剂包含表1-3中作为双链体序列列出的任一个序列。在一些实施方案中,dsRNA试剂包含与式(A)相差0、1、2或3个核苷酸的正义链:5'-Z1GUUAUCGAGGCACAUAZ2-3'式(A),其中Z1为包含0-15个核苷酸基序的核苷酸序列,Z2选自A、U、C、G中的一种或者不存在。在某些实施方案中,Z2为A。在某些实施方案中,Z1核苷酸序列选自以下基序:A、AA、UA、GA、CA、AGA、UGA、GGA、CGA、UAGA、CAGA、AAGA、ACAGA、GACAGA、GGACAGA、UGGACAGA、AUGGACAGA、AAUGGACAGA、UAAUGGACAGA、GUAAUGGACAGA、GGUAAUGGACAGA、UGGUAAUGGACAGA、AUGGUAAUGGACAGA或者不存在中的一种。在某些实施方案中,Z1为包含1、2、3或4个核苷酸基序的核苷酸序列,选自以下基序:A、AA、UA、GA、CA、AGA、UGA、GGA、CGA、UAGA、CAGA、AAGA、ACAGA。在一些实施方案中,dsRNA试剂包含与式(B)相差0、1、2或3个核苷酸的反义链:5'-Z3UAUGUGCCUCGAUAACZ4-3'式(B),其中Z3选自A、U、C、G中的一种或者不存在,Z4为包含0-15个核苷酸基序的核苷酸序列。在某些实施方案中,Z3为U。在某些实施方案中,Z4核苷酸序列选自以下基序:U、UU、UA、UC、UG、UCU、UCA、UCC、UCG、UCUC、UCUA、UCUG、UCUU、UCUGU、UCUGUC、UCUCUU、UCUCGA、UCUGUCC、UCUGUCCA、UCUGUCCAU、UCUGUCCAU、UCUGUCCAUU、UCUGUCCAUUA、UCUGUCCAUUAC、UCUGUCCAUUACC、UCUGUCCAUUACCA、UCUGUCCAUUACCAU或者不存在。在某些实施方案中,Z4为包含1、2、3或4个核苷酸基序的核苷酸序列,选自以下基序:U、UU、UA、UC、UG、UCU、UCA、UCC、UCG、UCUC、UCUA、UCUG、UCUU。在一些实施方案中,dsRNA试剂包
含正义链和反义链,所述的正义链和反义链分别包括具有本文所述与式(A)和式(B)相差0、1、2或3个核苷酸表示的核苷酸序列,并且任选地包含靶向配体。在某些实施方案中,dsRNA试剂所述的正义链式(A)和反义链式(B)每条链的长度不超过35个核苷酸。在某些实施方案中,Z1和Z4核苷酸基序是完全或者部分互补的。在某些实施方案中,Z2和Z3核苷酸基序是完全或者部分互补的。在某些实施方案中,正义链与反义链互补或实质互补,并且互补区域的长度在16至23个核苷酸之间。在一些实施方案中,互补区域长度为19-21个核苷酸。在一些实施方案中,其中所述正义链的长度不超过35个核苷酸,包括与所述反义链互补的区域,包括至少15、16、17、18或19个核苷酸。在一些实施方案中,dsRNA试剂包含与式(C)相差0、1、2或3个核苷酸的正义链:5'-Z5CCAAGCUUGGUCAUCUZ6-3'式(C),其中Z5为包含0-15个核苷酸基序的核苷酸序列,Z6选自A、U、C、G中的一种或者不存在。在某些实施方案中,Z6为A。在某些实施方案中,Z5核苷酸序列选自以下基序:G、AG、UG、GG、CG、AUG、UUG、GUG、CUG、UUUG、CUUG、AUUG、ACUUG、AACUUG、GAACUUG、AGAACUUG、AAGAACUUG、GAAGAACUUG、GGAAGAACUUG、AGGAAGAACUUG、CAGGAAGAACUUG、ACAGGAAGAACUUG、CACAGGAAGAACUUG或者不存在中的一种。在某些实施方案中,Z5为包含1、2、3或4个核苷酸基序的核苷酸序列,选自以下基序:G、AG、UG、GG、CG、AUG、UUG、GUG、CUG、UUUG、CUUG、AUUG。在一些实施方案中,dsRNA试剂包含与式(D)相差0、1、2或3个核苷酸的反义链:5'-Z7AGAUGACCAAGCUUGGZ8-3'式(D),其中Z7选自A、U、C、G中的一种或者不存在,Z8为包含0-15个核苷酸基序的核苷酸序列。在某些实施方案中,Z7为U。在某些实施方案中,Z8核苷酸序列选自以下基序:C、CU、CA、CC、CG、CAU、CAA、CAC、CAG、CAAC、CAAA、CAAG、CAAU、CAAGU、CAAGUU、CAACUU、CAACGA、CAAGUUC、CAAGUUCU、CAAGUUCUU、CAAGUUCUUC、CAAGUUCUUCC、CAAGUUCUUCCU、CAAGUUCUUCCUG、CAAGUUCUUCCUGU、CAAGUUCUUCCUGUG或者不存在。在某些实施方案中,Z8为包含1、2、3或4个核苷酸基序的核苷酸序列,选自以下基序:C、CU、CA、CC、CG、CAU、CAA、CAC、CAG、CAAC、CAAA、CAAG、CAAU。在一些实施方案中,dsRNA试剂包含正义链和反义链,所述的正义链和反义链分别包括具有本文所述与式(C)和式(D)相差0、1、2或3个核苷酸表示的核苷酸序列,并且任选地包含靶向配体。在某些实施方案中,dsRNA试剂所述的正义链式(C)和反义链式(D)每条链的长度不超过35个核苷酸。在某些实施方案中,Z5和Z8核苷酸基序是完全或
者部分互补的。在某些实施方案中,Z6和Z7核苷酸基序是完全或者部分互补的。在某些实施方案中,正义链与反义链互补或实质互补,并且互补区域的长度在16至23个核苷酸之间。在一些实施方案中,互补区域长度为19-21个核苷酸。在一些实施方案中,其中所述正义链的长度不超过35个核苷酸,包括与所述反义链互补的区域,包括至少15、16、17、18或19个核苷酸。在一些实施方案中,dsRNA试剂包含与式(E)相差0、1、2或3个核苷酸的正义链:5'-Z9GACAGAGUUAUCGAGGZ10-3'式(E),其中Z9为包含0-15个核苷酸基序的核苷酸序列,Z10选自A、U、C、G中的一种或者不存在。在某些实施方案中,Z10为A。在某些实施方案中,Z9核苷酸序列选自以下基序:G、AG、UG、GG、CG、AUG、UUG、GUG、CUG、CAUG、UAUG、GAUG、AAUG、UGAUG、GUGAUG、GGUGAUG、UGGUGAUG、AUGGUGAUG、CAUGGUGAUG、CCAUGGUGAUG、ACCAUGGUGAUG、UACCAUGGUGAUG、CUACCAUGGUGAUG、GCUACCAUGGUGAUG或者不存在中的一种。在某些实施方案中,Z9为包含1、2、3或4个核苷酸基序的核苷酸序列,选自以下基序:G、AG、UG、GG、CG、AUG、UUG、GUG、CUG、CAUG、UAUG、GAUG、AAUG。在一些实施方案中,dsRNA试剂包含与式(F)相差0、1、2或3个核苷酸的反义链:5'-Z11CCUCGAUAACUCUGUCZ12-3'式(F),其中Z11选自A、U、C、G中的一种或者不存在,Z12为包含0-15个核苷酸基序的核苷酸序列。在某些实施方案中,Z11为U。在某些实施方案中,Z12核苷酸序列选自以下基序:C、CU、CA、CC、CG、CAU、CAA、CAC、CAG、CAUA、CAUG、CAUC、CAUU、CAUCA、CAUCAC、CAUGUU、CAUGGA、CAUCACC、CAUCACCA、CAUCACCAU、CAUCACCAUG、CAUCACCAUGG、CAUCACCAUGGU、CAUCACCAUGGUA、CAUCACCAUGGUAG、CAUCACCAUGGUAGC或者不存在。在某些实施方案中,Z12为包含1、2、3或4个核苷酸基序的核苷酸序列,选自以下基序:C、CU、CA、CC、CG、CAU、CAA、CAC、CAG、CAUA、CAUG、CAUC、CAUU。在一些实施方案中,dsRNA试剂包含正义链和反义链,所述的正义链和反义链分别包括具有本文所述与式(E)和式(F)相差0、1、2或3个核苷酸表示的核苷酸序列,并且任选地包含靶向配体。在某些实施方案中,dsRNA试剂所述的正义链式(F)和反义链式(F)每条链的长度不超过35个核苷酸。在某些实施方案中,Z9和Z12核苷酸基序是完全或者部分互补的。在某些实施方案中,Z10和Z11核苷酸基序是完全或者部分互补的。在某些实施方案中,正义链与反义链互补或实质互补,并且互补区域的长度在16至23个核苷酸之间。在一些实施方
案中,互补区域长度为19-21个核苷酸。在一些实施方案中,其中所述正义链的长度不超过35个核苷酸,包括与所述反义链互补的区域,包括至少15、16、17、18或19个核苷酸。
在一些实施方案中,dsRNA试剂包含至少一种修饰的核苷酸。在某些实施方案中,反义链的所有核苷酸或基本上所有核苷酸都是修饰的核苷酸。在一些实施方案中,至少一种修饰的核苷酸包括:2'-O-甲基核苷酸、2'-氟核苷酸、2'-脱氧核苷酸、2',3'-seco核苷酸模拟物、锁核苷酸、开环核酸核苷酸(unlocked nucleic acid nucleotide,UNA)、乙二醇核酸核苷酸(glycol nucleic acid nucleotide,GNA)、2'-F-阿拉伯糖核苷酸、2'-甲氧基乙基核苷酸、无碱基核苷酸、核糖醇、反向核苷酸、反向无碱基核苷酸、反向2'-OMe核苷酸、反向2'-脱氧核苷酸、2'-氨基修饰的核苷酸、2'-烷基修饰的核苷酸、吗啉代核苷酸和3'-OMe核苷酸、包括5'-硫代磷酸酯基团的核苷酸、或与胆固醇衍生物或十二烷酸双癸酰胺基团连接的末端核苷酸、2'-氨基修饰的核苷酸、氨基磷酸酯或包含核苷酸的非天然碱基。在一些实施方案中,反义链包含15个或更多个独立选自2'-O-甲基核苷酸和2'-氟核苷酸的修饰核苷酸,其中少于6个2'-氟核苷酸修饰的核苷酸。在某些实施方案中,反义链包含3个或5个2'-氟核苷酸,优选地,反义链包含5个2'-氟核苷酸。在一些实施方案中,正义链包含15个或更多个独立选自2'-O-甲基核苷酸和2'-氟核苷酸的修饰核苷酸,其中少于4个2'-氟核苷酸修饰的核苷酸。在某些实施方案中,正义链包含3个2'-氟核苷酸。在一些实施方案中,反义链包含15个或更多个独立选自2'-O-甲基核苷酸和2'-氟核苷酸的修饰核苷酸,其中至少16个修饰核苷酸是2'-O-甲基核苷酸并且位于反义链5'端的第2、7、12、14和/或16位是2'-氟核苷酸修饰的核苷酸(从反义链5'第一个配对的核苷酸开始计算)。在一些实施例中,正义链包含独立地选自2'-O-甲基核苷酸和2'-氟核苷酸的15个或更多修饰核苷酸,其中至少18个修饰核苷酸是2'-O-甲基核苷酸并且位于正义链3'端的第9、11和/或13位是2'-氟核苷酸修饰的核苷酸(从正义链3'第一个配对的核苷酸开始计算)。在一些实施例中,反义链包含在从5'末端到3'末端的方向上,反义链的第2、7、12、14和16位的核苷酸是2'-氟修饰的核苷酸,从反义链5'端第一个配对的核苷酸开始计算,并且每个反义链中其他位置的核苷酸独立地是非氟修饰的核苷酸。在一些实施方案中,反义链在从5'末端到3'末端的方向上包含位置反义链的第2、5、12、14和18个是2'-氟修饰的核苷酸,从反义链5'第一个配对的核苷酸开始计算,并且反义链中其他位置的每个核苷酸独立地是非氟修饰的核苷酸。在一些实施例中,正义链包含在从3'末端到5'末端的方向上,在正义链的位置9、11和13的核苷酸是2'-氟修饰的核苷酸,从正义链3'端第一个配对的核苷酸开始计算,并且每个核苷酸在正义链中的其
他位置独立地是非氟修饰的核苷酸。在一些实施方案中,dsRNA试剂包括在引导链的5'末端处的E-乙烯基膦酸酯核苷酸。在某些实施方案中,dsRNA试剂包含至少一个硫代磷酸酯核苷间键联。在某些实施方案中,正义链包含至少一个硫代磷酸酯核苷间键联。在一些实施方案中,反义链包含至少一个硫代磷酸酯核苷间键联。在一些实施方案中,正义链包含1、2、3、4、5或6个硫代磷酸酯核苷间键联。在一些实施方案中,反义链包含1、2、3、4、5或6个硫代磷酸酯核苷间键联。在某些实施方案中,正义链和反义链的所有或基本上所有核苷酸都是修饰的核苷酸。在一些实施方案中,修饰的正义链是表2-3中列出的修饰的正义链序列。在一些实施方案中,修饰的反义链是表2-3中列出的修饰的反义链序列。在某些实施方案中,正义链与反义链互补或基本互补,并且互补区域的长度在16至23个核苷酸之间。在一些实施方案中,互补区域的长度为19-21个核苷酸。在某些实施方案中,互补区域的长度为14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30个核苷酸。在一些实施方案中,每条链的长度不超过40个核苷酸。在一些实施方案中,每条链的长度不超过30个核苷酸。在一些实施方案中,每条链的长度不超过25个核苷酸。在一些实施方案中,每条链的长度不超过23个核苷酸。在某些实施方案中,dsRNA试剂包含至少一种修饰的核苷酸并且还包含一个或更多个靶向基团或连接基团。在一些实施方案中,一个或更多个靶向基团或连接基团与正义链缀合。在一些实施方案中,靶向基团或连接基团包括N-乙酰基-半乳糖胺(GalNAc)。在一些实施方案中,靶向基团中的靶向部分具有为以下结构片段,
p为1或者2。
在一些实施方案中,靶向基团具有以下结构:
在某些实施方案中,dsRNA试剂包含与正义链的5'-末端缀合的靶向基团。在一些实施方案中,dsRNA试剂包含与正义链的3'-末端缀合的靶向基团。在一些实施方案中,反义链在3'-末端包含一个反向无碱基残基。在某些实施方案中,正义链在3'或/和5'末端包含一个或两个反向无碱基残基。在某些实施方案中,正义链在3'或/和5'末端包含一个或两个异甘露醇残基。在某些实施方案中,正义链在3'和5'末端分别独立地包含一个异甘露醇残基。在某些实施方案中,正义链在3'和5'末端分别独立地包含一个异甘露醇残基,进一步包含5'-末端缀合的靶向基团,靶向基团优选为上述GLS-15。在一些实施方案中,dsRNA试剂具有两个平末端。在一些实施方案中,至少一条链包含至少1个核苷酸长的3'突出端。在一些实施方案中,至少一条链包含至少2个核苷酸长的3'突出端。
在某些实施方案中,一种抑制LPA(Apo(a))表达的双链核糖核酸(dsRNA)试剂,该dsRNA试剂包含正义链和反义链,在反义链中的核苷酸位置2至18处包含与LPA RNA转录物互补的区域,反义链与正义链完全或者部分互补,并且任选地包含靶向配体,其中每条链的长度为14至30个核苷酸,其中正义链序列可由式(I)表示:
5′-(N′L)n′N′LN′LN′LN′N1N′N2N′F N′LN′F N′LN′N3N′N4N′L N′LN′LN′LN′LN′L(N′L)m′-3′ (I)其中:每个N′F表示一个2'-氟修饰的核苷酸;每个N′N1,N′N2,N′N3和N′N4,独立地表示一个修饰或者未修饰的核苷酸;每个N′L独立代表修饰或未修饰的核苷酸但不代表2'-氟修饰的核苷酸,n'为0-7的整数,m'为0-3的整数。在某些实施方案中,每个N′N3代表2'-氟修饰的核苷酸,N′N1,N′N2和N′N4独立地表示修饰或未修饰的核苷酸但不代表2'-氟修饰的核苷酸,m′为1。在某些实施方案中,每个N′N4代表2'-氟修饰的核苷酸,N′N1,N′N2和N′N3独立地表示修饰或未修饰的核苷酸但不代表2'-氟修饰的核苷酸,m′为1。在某些实施方案中,n′为3,m′为1;或者n′为0,m′为0;或n′为3,m′为3。在某些实施方案中,式(I)中仅有3个2'-氟修饰的核苷酸。
在某些实施方案中,本发明涉及用于治疗的开环核酸(UNA)寡聚体。开环核酸
(unlocked nucleic acid,UNA)是RNA的无环类似物,其中核糖环的C2'和C3'原子之间的键已被切断。已经证明,掺入UNA对siRNA基因沉默活性具有良好的耐受性,在某些情况下甚至可以增强其活性(Meghan A.et al.“Locked vs.unlocked nucleic acids(LNA vs.UNA):contrasting structures work towards common therapeutic goals”.Chem.Soc.Rev.,2011,40,5680–5689)。
UNA是一种热不稳定修饰,用UNA替换核糖核苷酸会降低碱基配对强度和双链体稳定性。将UNA策略性地放置在siRNA反义链的种子区域可以降低通过microRNA(miRNA)介导的基因沉默机制中的脱靶活性。miRNA主要通过反义种子区(从5'端开始的第2-8位)与靶mRNA之间的碱基配对来识别靶基因,以进行基因抑制。每个miRNA都可能调节大量基因。RNA诱导沉默复合物(RISC)所加载的siRNA反义链也可以通过miRNA介导的机制潜在地调节大量非预期基因。因此,在siRNA的种子区域中加入热不稳定的核苷酸,如UNA,可以降低脱靶活性(Lam JK,Chow MY,Zhang Y,Leung SW.siRNA Versus miRNA as Therapeutics for Gene Silencing.Mol Ther Nucleic Acids.2015Sep15;4(9):e252.doi:10.1038/mtna.2015.23.PMID:26372022;PMCID:PMC4877448.)。具体而言,这样的RNA寡核苷酸或RNA寡核苷酸的复合物在种子区域含有至少一个UNA核苷酸单体(Narendra Vaish et al.“Improved specificity of gene silencing by siRNAs containing unlocked nucleobase analog”.Nucleic Acids Research,2011,Vol.39,No.51823–1832)。
根据本技术方案,在RNA寡核苷酸或RNA寡核苷酸的复合物中并入UNA的潜在优势包括但不限于:
1.减少脱靶活性。在siRNA种子区添加UNA会降低种子区的碱基配对强度,从而降低由micro-RNA机制引起的潜在脱靶活性。
2.UNA在siRNA活性方面具有良好的耐受性。在某些情况下,UNA可以导致活性增强。
可用于本技术方案的示例性UNA单体包括但不限于:
根据本发明的一个方面,提供了一种组合物,其包含本发明上述dsRNA试剂方面的任意实施方案。在某些实施方案中,组合物还包含药学上可接受的载体。在一些实施方案中,
组合物还包含一种或更多种另外的治疗剂,如HMg Co-A还原酶抑制剂(他汀类)、依折麦布、PCSK-9抑制剂、CTEP抑制剂、靶向ANGPTL3的疗法、靶向AGT的疗法、靶向APOC3的疗法和烟酸,或上述任何的组合。在某些实施方案中,组合物被包装在药盒、容器、包装物、分配器、预填充注射器或小瓶中。在一些实施方案中,组合物被配制用于皮下给药或被配制用于静脉内(IV)给药。
根据本发明的另一方面,提供了一种细胞,其包含本发明上述dsRNA试剂方面的任意实施方案。在一些实施方案中,细胞是哺乳动物细胞,任选地是人细胞。
根据本发明的另一方面,提供了一种抑制细胞中LPA基因表达的方法,该方法包括:(i)制备包含有效量的上述dsRNA试剂或上述组合物方面的任意实施方案的细胞。在某些实施方案中,该方法还包括:(ii)将制备的细胞维持足够的时间以获得LPA基因的mRNA转录物的降解,从而抑制细胞中LPA基因的表达。在一些实施方案中,细胞在对象体内并且dsRNA试剂经皮下施用于对象。在一些实施方案中,细胞在对象体内并且dsRNA试剂通过IV给药施用于对象。在某些实施方案中,该方法还包括在向对象施用dsRNA试剂后评估对LPA基因的抑制,其中评估的手段包括:(i)确定对象中LPA相关疾病或病症的一个或更多个生理特征,以及(ii)将所确定的生理特征与LPA相关疾病或病症的基线治疗前生理特征和/或LPA相关疾病或病症的对照生理特征进行比较,其中的比较结果指示对象中LPA基因表达的抑制存在或不存在。在一些实施方案中,所确定的生理特征是在血液中的Lp(a)水平。血液中LPA水平的降低表明对象中LPA基因表达的降低。
根据本发明的另一方面,提供了一种抑制对象中LPA基因表达的方法,其包括向对象施用有效量的前述dsRNA试剂方面的实施方案或前述组合物的实施方案。在一些实施方案中,将dsRNA试剂皮下施用于对象。在某些实施方案中,dsRNA试剂通过IV给药施用于对象。在一些实施方案中,该方法还包括:在施用dsRNA试剂后评估LPA基因的抑制,其中评估手段包括:(i)确定对象的LPA相关疾病或病症的一种或更多种生理特征;(ii)将确定的生理特征与LPA相关疾病或病症的基线治疗前生理特征和/或LPA相关疾病或病症的对照生理特征进行比较;其中比较结果指示对象中LPA基因表达的抑制存在或不存在。在一些实施方案中,所确定的生理特征是在血液中的Lp(a)水平。血液中LPA水平的降低表明对象中LPA基因表达的降低。
根据本发明的另一方面,提供了一种治疗与LPA蛋白相关之疾病或病症的方法,其包括:向对象施用有效量的本发明的前述dsRNA试剂方面的任意实施方案或本发明的前述组合物的任意实施方案,以抑制LPA基因表达。在某些实施方式中,LPA相关障碍是心血管疾病,其中所述心血管疾病包括伯格氏病(Berger’s disease)、外周动脉疾病、冠状动脉疾病、代谢综合征、急性冠脉综合征、主动脉瓣狭窄、主动脉瓣反流、主动脉夹层、视网膜动脉阻塞、脑血管疾病、肠系膜缺血、肠系膜上动脉阻塞、肾动脉狭窄、稳定型/不稳定型心
绞痛、急性冠脉综合征、杂合子或纯合子家族性高胆固醇血症、高载脂蛋白β脂蛋白血症、脑血管动脉粥样硬化、脑血管疾病和静脉血栓形成、中风、动脉粥样硬化、血栓形成、冠心病或主动脉瓣狭窄和/或与含Lp(a)粒子的水平升高相关的任何其他疾病或病理。在一些实施方案中,该方法还包括:向对象施用另外的治疗方案。在一些实施方案中,另外的治疗方案包括LPA相关疾病或病症的治疗。在某些实施方案中,另外的治疗方案包括:向对象施用一种或更多种本发明的LPA反义多核苷酸;向对象施用非LPA dsRNA治疗剂;以及在对象中进行行为改变。在一些实施方案中,非LPA dsRNA治疗剂是以下中的一种另外的治疗剂,如HMg Co-A还原酶抑制剂(他汀类)、依折麦布、PCSK-9抑制剂、CTEP抑制剂、靶向ANGPTL3的疗法、靶向APOC3的疗法和烟酸,或上述任何的组合。
在一些实施方案中,将dsRNA试剂皮下施用于对象。在某些实施方案中,dsRNA试剂通过IV给药施用于对象。在一些实施方案中,该方法还包括确定所施用的双链核糖核酸(dsRNA)试剂在对象中的功效。在一些实施方案中,确定治疗在对象中的功效的手段包括:(i)确定对象中LPA相关疾病或病症的一种或更多种生理特征;(ii)将确定的生理特征与LPA相关疾病或病症的基线治疗前生理特征进行比较,其中该比较结果指示对对象施用双链核糖核酸(dsRNA)试剂的功效存在、不存在和水平中的一种或更多种。在一些实施方案中,所确定的生理特征是在血液中的Lp(a)水平。血液中LPA水平的降低表明对对象施用双链核糖核酸(dsRNA)试剂的有效性的存在。
根据本发明的另一方面,提供了与对象中LPA蛋白的基线治疗前水平相比降低对象中LPA蛋白水平的方法,其包括向对象施用有效量的本发明的前述dsRNA试剂方面的任意实施方案或本发明的前述组合物的任意实施方案,以降低LPA基因表达的水平。在一些实施方案中,将dsRNA试剂皮下施用于对象或通过IV施用于对象。
根据本发明的另一方面,提供了与对象中LPA相关疾病或病症的基线治疗前生理特征相比改变对象中LPA相关疾病或病症的生理特征的方法,该方法包括向对象施用有效量的本发明前述dsRNA试剂方面的任意实施方案或本发明的前述组合物的任意实施方案,以改变对象中LPA相关疾病或病症的生理特征。在一些实施方案中,将dsRNA试剂皮下施用于对象或通过IV施用于对象。在某些实施方案中,生理特征是在血液中的Lp(a)水平。
序列说明
双链体AV00122至AD00484-1,AD00474-2,AV01867-AV01968显示在表1中并且显示了其正义链序列。
双链体AV00122至AD00484-1,AD00474-2,AV01867-AV01968显示在表1中并且显示了其反义链序列。
在显示于表2的序列中,化学修饰表示为:大写:2'-氟;小写:2'-OMe;硫代磷酸酯:
*。
在显示于表3的序列中,体内研究中使用的递送分子在每条正义链的3'末端表示为“GLO-0”。体内研究中使用的递送分子在每条正义链的5'末端表示为“GLS-5”或者“GLS-15”化学修饰表示为:大写:2'-氟;小写:2'-OMe;硫代磷酸酯:*;开环核酸:UNA。
以下为人Lp(a)的mRNA序列(SEQ ID NO:1):NM_005577.4Homo sapienslipoprotein(a)(LPA),mRNA
图1表示猴子血清LPA蛋白水平示意图;
图2表示2mpk剂量的AD00480-8在猴子血清LPA蛋白水平示意图。
本发明的部分实施方案包括能够抑制LPA(Apo(a))基因表达的RNAi试剂,例如但不限于双链(ds)RNAi试剂。本发明的部分实施方案还包括包含LPA RNAi试剂的组合物和使用该组合物的方法。本文公开的LPA RNAi试剂可接附于递送化合物以递送至细胞,包括递送至肝细胞。本发明的药物组合物可包含至少一种dsRNA试剂和递送化合物。在本发明的一些实施方案中,递送化合物是含GalNAc的递送化合物。递送至细胞的LPA RNAi试剂能够抑制LPA基因表达,从而降低基因的LPA蛋白产物。本发明的dsRNAi试剂可用于治疗LPA相关疾病和病症。这样的dsRNAi试剂包括例如表1中所显示的双链体AV00122至AD00484-1,AD00474-2,AV01867-AV01968。在其他一些实施方案中,这样的dsRNAi试剂包括双链体变体,例如双链体AV00122至AD00484-1,AD00474-2,AV01867-AV01968的变体。
在本发明的一些实施方案中,降低细胞或对象中LPA表达分别治疗与细胞或对象中
LPA表达相关的疾病或病症。可通过降低LPA表达治疗的疾病和病症的非限制性实例是心血管疾病,其中所述心血管疾病包括伯格氏病(Berger’s disease)、外周动脉疾病、冠状动脉疾病、代谢综合征、急性冠脉综合征、主动脉瓣狭窄、主动脉瓣反流、主动脉夹层、视网膜动脉阻塞、脑血管疾病、肠系膜缺血、肠系膜上动脉阻塞、肾动脉狭窄、稳定型/不稳定型心绞痛、急性冠脉综合征、杂合子或纯合子家族性高胆固醇血症、高载脂蛋白β脂蛋白血症、脑血管动脉粥样硬化、脑血管疾病和静脉血栓形成、中风、动脉粥样硬化、血栓形成、冠心病或主动脉瓣狭窄和/或与含Lp(a)粒子的水平升高相关的任何其他疾病或病理。
下面描述了如何制备和使用包含LPA单链(ssRNA)和双链(dsRNA)试剂的组合物来抑制LPA基因表达,以及用于治疗由LPA基因表达引起或调节的疾病和病症的组合物和方法。术语“RNAi”也是本领域已知的,并且可以被称为“siRNA”。
如本文所用,术语“RNAi”是指包含RNA并通过RNA诱导的沉默复合物(RISC)途径介导RNA转录物的靶向切割的试剂。如本领域已知的,RNAi靶区域是指在基因转录过程中形成的RNA分子的核苷酸序列的连续部分,其包括信使RNA(mRNA),它是初级转录产物RNA的加工产物。该序列的靶标部分将至少足够长以用作在该部分处或附近进行RNAi定向切割的底物。靶序列可以是8-30个核苷酸长(包括端值)、10-30个核苷酸长(包括端值)、12-25个核苷酸长(包括端值)、15-23个核苷酸长(包括端值)、16-23个核苷酸长(包括端值),或18-23个核苷酸长(包括端值),并包括每个规定范围内的所有较短长度。在本发明的一些实施方案中,靶序列为9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25或26个核苷酸长。在某些实施方案中,靶序列的长度在9到26个核苷酸之间(包括端值),包括其间的所有子范围和整数。例如,虽然不意在限制,但在本发明的某些实施方案中,靶序列为8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30个核苷酸长,该序列与LPA基因的RNA转录物的至少一部分完全或至少基本上互补。本发明的一些方面包括包含一种或更多种LPA dsRNA试剂和药学上可接受的载体的药物组合物。在本发明的某些实施方案中,如本文所述的LPA RNAi抑制LPA蛋白的表达。
如本文所用,“dsRNA试剂”是指包含RNA或RNA样(例如,化学修饰的RNA)寡核苷酸分子的组合物,其能够降解或抑制靶mRNA转录物的翻译。尽管不希望限于特定理论,但本发明的dsRNA试剂可通过RNA干扰机制起作用(即,通过与哺乳动物细胞的RNA干扰途径机制(RNA诱导的沉默复合物或RISC)相互作用来诱导产生RNA干扰),或通过任何替代机制或途径起作用。在植物、无脊椎动物和脊椎动物细胞中实现基因沉默的方法是本领域所公知的(参见例如,Sharp et al.,Genes Dev.2001,15:485;Bernstein,et al.,(2001)Nature409:363;Nykanen,et al.,(2001)Cell107:309;以及Elbashir,et al.,(2001)Genes Dev.15:188)),其各自的公开内容通过引用整体并入本文。本领域已知的基因沉默手段可与本文
提供的公开内容结合使用以实现抑制LPA的表达。
本文公开的dsRNA试剂由正义链和反义链组成,其包括但不限于:短干扰RNA(siRNA)、RNAi试剂、微RNA(miRNA)、短发夹RNA(shRNA)和切丁酶(Dicer)底物。本文描述的dsRNA试剂的反义链至少部分地与所靶向的mRNA互补,本领域能够理解,多种长度的dsRNA双链体结构可用于抑制靶基因表达。例如,已知具有19、20、21、22和23个碱基对的双链体结构的dsRNA可有效诱导RNA干扰(Elbashir et al.,EMBO 2001,20:6877-6888)。本领域还已知较短或较长的RNA双链体结构也可有效诱导RNA干扰。本发明的某些实施方案中的LPA dsRNA可以包含至少一条长度至少为21nt的链,或者双链体可以具有基于表1-3中任何列出的序列之一的长度减1、2、3nt或更短的长度。与分别在表1-3中列出的dsRNA相比,在其一端或两端减少4个核苷酸也可以是有效的。在本发明的一些实施方案中,LPA dsRNA试剂可具有来自表1-3的一个或更多个序列的至少15、16、17、18、19、20或更多个连续核苷酸的部分序列,并且它们抑制LPA基因表达的能力与由包含全序列(此处也称为“亲本”序列)的dsRNA产生的抑制水平相差不超过5%、10%、15%、20%、25%或30%。
本发明的组合物和方法的某些实施方案在组合物中包含单链RNA和/或将单链RNA施用于对象。例如,表1-3任一项中所列的反义链可以作为一种组合物或在一种组合物内,该组合物施用给对象会降低对象中LPA多肽和/或LPA基因的表达。表1-3显示了某些LPAdsRNA试剂的反义链和正义链核心延伸碱基序列。可以包含在本发明的某些组合物中和/或在本发明的某些方法中施用的单链反义分子在本文中称为“单链反义试剂”或“反义多核苷酸试剂”。可以包含在某些组合物中和/或在本发明的某些方法中施用的单链正义分子在本文中称为“单链正义试剂”或“正义多核苷酸试剂”。术语“碱基序列”在本文中是指没有化学修饰或递送化合物的多核苷酸序列。例如,表1所示的正义链对应于是表3中的相应碱基序列;但表3中的相应序列中显示了各自的化学修饰和递送化合物。在此公开的序列可以被分配标识符。例如,单链正义序列可以用“正义链SS#”来标识;单链反义序列可以用“反义链AS#”来标识;并且包含正义链和反义链的双链体可以用“双链体AD#”来标识。
表1包括正义链和反义链,并提供了由表1中同一行上的正义链和反义链形成的双链体的标识号。在本发明的某些实施方案中,反义序列在其第1位中包含核碱基u或核碱基a。在本发明的某些实施方案中,反义序列在反义序列的第1位包含核碱基u。如本文所用,术语“匹配位置”在某种意义上是指当两条链作为双链体时每条链中互相“配对”的位置。例如,在21核碱基正义链和21核碱基反义链中,正义链第1位与反义链第21位的核碱基处于“匹配位置”。在另一个非限制性实例中,对于23核碱基正义链和23核碱基反义链,正义链的第2位核碱基与反义链的22位处于匹配位置。在另一个非限制性实例中,在18核碱基正义链和18核碱基反义链中,正义链第1位核碱基与反义链第18位核碱基处于匹配位置;并且
正义链中的第4位核碱基与反义链中的第15位核碱基处于匹配位置。技术人员会理解如何识别双链和成对链的正义和反义链之间的匹配位置。
表1中的一列表示双链体的双链体AV#、AD#,该双链体在同一表格行中包含正义和反义序列。例如,表1公开了指定为“双链体AV00122”的双链体,其包含相应的正义链序列和反义链序列。因此,表1中的每一行都标识了本发明的双链体,每一个都包含显示在同一行中的正义和反义序列,每个双链体的指定标识符均显示在该行的最后一列中。
在本发明方法的一些实施方案中,向对象施用包含表1中所示多核苷酸序列的RNAi试剂。在本发明的一些实施方案中,向对象施用的RNAi试剂包括双链体,所述双链体包含表1中列出的碱基序列中的至少一个,并包含0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23或24个序列修改。在本发明方法的一些实施方案中,还包括将表1中所示多核苷酸序列的RNAi试剂连接至递送分子上,其非限制性实例是包含GalNAc的递送化合物。
表1:无修饰的LPA RNAi试剂反义链和正义链序列。所有序列均显示为5'到3'方向。双链体AV#或AD#是分配给表中同一行中两条链的双链体的编号
表2显示了本发明的某些化学修饰的LPA RNAi剂反义链和正义链序列。在本发明方法的一些实施方案中,将具有表2中所示多核苷酸序列的RNAi试剂施用于细胞和/或对象。在本发明方法的一些实施方案中,将具有表2中所示多核苷酸序列的RNAi试剂施用于对象。在本发明的一些实施方案中,向对象施用的RNAi试剂包含在表2第一列中标注的双链体,
并且分别包含显示在表2中同一行第三列和第六列的正义和反义链序列中的序列修饰。在本发明方法的一些实施方案中,表2中所示的序列可以连接到(在本文中也称为“缀合到”)能够将RNAi试剂递送至对象的细胞和/或组织的化合物上。可用于本发明的某些实施方案中的递送化合物的非限制性实例是含GalNAc的化合物。在表2中,第一列表示碱基序列的双链体AD#,与表1对应。对于双链体AD#标识的碱基序列,不仅显示正义和反义链所包含的碱基序列,而且具有表2同一行中所示的指定化学修饰。例如,表1第一行显示了正义和反义碱基单链序列,它们一起构成双链体,标识为:双链体AV00122;而表2列出的双链体AV00122中,作为双链体,其包含AV00122-SS和AV00122-AS的碱基序列,而且分别包含在第三列和第六列中显示的正义和反义序列中的化学修饰。表2第2列中的“正义链SS#”是同一行中第3列所示正义序列(包括修饰)的指定标识符。表2第五列中的“反义链AS#”是第六列中显示的反义序列(包括修饰)的指定标识符。
表2:化学修饰的LPA RNAi试剂反义链和正义链序列。所有序列都显示为5'到3'。这些序列用于本文所述的某些体外测试研究。化学修饰表示为:大写:2'-氟;小写:2'-OMe;硫代磷酸酯:*
表3显示了本发明的某些化学修饰的LPA RNAi试剂反义链和正义链序列。在本发明方法的一些实施方案中,将表3中所示的RNAi试剂施用于细胞和/或对象。在本发明方法的一些实施方案中,将具有表3中所示多核苷酸序列的RNAi试剂施用于对象。在本发明的一些实施方案中,向对象施用的RNAi试剂包含在表3的第一列中标识的双链体,并且分别包含在表3中同一行第三列和第六列的正义和反义链序列中显示的序列修饰和/或递送化合物。该序列用于本文别处描述的某些体内测试研究。在本发明方法的一些实施方案中,表3中所示的序列可以连接到(在本文中也称为“缀合到”)用于递送的化合物上,其非限制性实例是含GalNAc的化合物,即在表3中第三列的正义链上具有标识为“GLX-n”的递送化合物。如本文所用和表3所示,“GLX-n”用于表示所连接的含GalNAc的化合物,其是化合物GLS-1、GLS-2、GLS-3、GLS-4、GLS-5、GLS-6、GLS-7、GLS-8、GLS-9、GLS-10、GLS-11、GLS-12、GLS-13、GLS-14、GLS-15、GLS-16、GLO-1、GLO-2、GLO-3、GLO-4、GLO-5、GLO-6、GLO-7、GLO-8、GLO-9、GLO-10、GLO-11、GLO-12、GLO-13、GLO-14、GLO-15和GLO-16中的任一种。其中每一个的结构在本文别处提供。表3的第一列提供了分配给表中该行中的正义和反义序列的双链体的双链体AD#。例如,双链体AD00122是正义链AD00122-SS和反义链AD00122-AS构成的双链体。表3中的每一行提供了一条正义链和一条反义链,并公开了所示正义链和反义链构成的双链体。表3第二列中的“正义链SS#”是同一行第3列所示正义序列(包括修改)的指定标识符。表3第五列中的“反义链AS#”是第六列中显示的反义序列(包括修饰)的指定标识符。某些所连接的含GalNAc的GLO化合物的标识符显示为GLO-0,并且应当理解,GLO-n或GLS-n化合物中的另一种可以替代显示为GLO-0的化合物,所得化合物也包括在本发明的方法和/或组合物的实施方案中。
[根据细则91更正 17.04.2023]
表3提供了用于体内测试的化学修饰的LPA RNAi试剂反义链和正义链序列。所有序列都显示为5'到3'。这些序列用于本文别处描述的某些体内测试研究。体内研究中使用的递送分子在每条正义链的3'末端表示为“GLO-0”。体内研究中使用的递送分子在每条正义链的5'末端表示为“GLS-5”或者“GLS-15”。化学修饰表示为:大写:2'-氟;小写:2'-OMe;硫代磷酸酯:*;开环核酸:UNA;invab=反向无碱基;imann:在每条链末端时: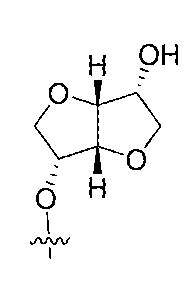 或当进一步偶联递送分子:
或当进一步偶联递送分子: 。
。
表3提供了用于体内测试的化学修饰的LPA RNAi试剂反义链和正义链序列。所有序列都显示为5'到3'。这些序列用于本文别处描述的某些体内测试研究。体内研究中使用的递送分子在每条正义链的3'末端表示为“GLO-0”。体内研究中使用的递送分子在每条正义链的5'末端表示为“GLS-5”或者“GLS-15”。化学修饰表示为:大写:2'-氟;小写:2'-OMe;硫代磷酸酯:*;开环核酸:UNA;invab=反向无碱基;imann:在每条链末端时:
错配
本领域技术人员已知,对于dsRNA的功效而言,错配是可以容忍的,尤其是错配在dsRNA的末端区域内的情况。某些错配具有更好的耐受性,例如具有摆动碱基对G:U和A:C的错配对功效的耐受性更好(Du et el.,A systematic analysis ofthe silencing effects ofan active siRNA at all single-nucleotide mismatched target sites.Nucleic Acids Res.2005 Mar 21;33(5):1671-7.Doi:10.1093/nar/gki312.Nucleic Acids Res.2005;33(11):3698)。本发明的方法和化合物的一些实施方案中,LPA dsRNA试剂可以含有一个或更多个与LPA靶序列的错配。在一些实施方案中,本发明的LPA dsRNA试剂不包含错配。在某些实施方案中,本发明的LPA dsRNA试剂包含不超过1个错配。在一些实施方案中,本发明的LPA dsRNA试剂包含不超过2个错配。在某些实施方案中,本发明的LPA dsRNA试剂包含不超过3个错配。在本发明的一些实施方案中,LPA dsRNA试剂的反义链包含与不位于互补区域中心的LPA靶序列的错配。在一些实施方案中,LPA dsRNA试剂的反义链包含1、2、3、4或更多个错配,其位于互补区域的5'或3'末端之一或两者的最末5、4、3、2或1个核苷酸内。本文所述的方法和/或本领域已知的方法可用于确定包含与LPA靶序列错配的LPA dsRNA试剂是否有效抑制LPA基因的表达。
互补性
如本文所用,除非另有说明,否则术语“互补性/互补”当用于描述第一核苷酸序列(例如,LPA dsRNA试剂正义链或靶标LPA mRNA)与第二核苷酸序列(例如,LPA dsRNA试剂反义链或单链反义多核苷酸)的相关性时,是指包含第一核苷酸序列的寡核苷酸或多核苷酸与包含第二核苷酸序列的寡核苷酸或多核苷酸杂交[在哺乳动物生理条件(或体外类似条件)下形成碱基对间氢键]、并且在某些条件下形成双螺旋或双螺旋结构的能力。其中也可以应用其他条件,例如在生物体内可能遇到的生理相关条件。技术人员将能够根据杂交核苷酸的最终应用确定最适合测试两个序列互补性的条件集。互补序列包括沃森-克里克碱基对或非沃森-克里克碱基对,并且包括天然或修饰的核苷酸或核苷酸模拟物,只要至少达到上述杂交要求的程度即可。序列同一性或互补性与修饰无关。
例如,在如本文所述的LPA dsRNA内的互补序列包含含有第一核苷酸序列的寡核苷酸或多核苷酸与包含第二核苷酸序列的寡核苷酸或多核苷酸在一个或两个核苷酸序列的全长上的碱基配对。此类序列在本文中可被称为彼此“完全互补”。应当理解,在设计两个寡核苷酸以在杂交时形成一个或更多个单链突出端的实施方案中,这种突出端在本文中不被视为基于互补性确定的错配。例如,LPA dsRNA试剂包含一个长度为19个核苷酸的寡核苷酸和另一个长度为20个核苷酸的寡核苷酸,其中较长的寡核苷酸包含与较短的寡核苷酸完全互补的19个核苷酸的序列,出于本文所述的目的,此种情况可以称为“完全互补”。因此,如本文所用,“完全互补”是指第一多核苷酸的连续序列中的所有(100%)碱基会与第二多核苷酸的连续序列中的相同数目的碱基杂交。连续序列可以包含第一或第二核苷酸序列的全部或部分。
如本文所用,术语“基本互补”是指在核碱基序列的杂交对中,第一多核苷酸的连续序列中的碱基的至少约85%(但不是全部)会与第二多核苷酸的连续序列中相同数目的碱基杂交。如果两个序列在杂交时包含一个或更多个错配碱基对,例如至少1、2、3、4或5个错配碱基对,则可以使用术语“基本上互补”来指第一序列相对于第二序列形成多达15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30个碱基对(bp)的双链体,同时保留在与其最终应用最相关的条件下杂交的能力,例如,通过RISC途径抑制LPA基因表达。术语“部分互补”在本文中可用于指核碱基序列的杂交对中,第一多核苷酸的连续序列中碱基的至少75%(但不是全部)会与第二多核苷酸的连续序列中相同数目的碱基杂交。在一些实施方案中,“部分互补”是指第一多核苷酸的连续序列中至少76%、77%、78%、79%、80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%或99%的碱基会与第二多核苷酸的连续序列中相同数量的碱基杂交。
术语“互补”、“完全互补”、“基本互补”和“部分互补”在本文中使用时可用于指LPAdsRNA试剂的正义链与反义链之间的碱基匹配、LPA dsRNA试剂的反义链与靶LPA mRNA
的序列之间的碱基匹配,或单链反义寡核苷酸与靶LPA mRNA序列之间的碱基匹配。应当理解,术语“LPA dsRNA试剂的反义链”可以指与“LPA反义多核苷酸试剂”相同的序列。
如本文所用,在提及核酸序列时使用的术语“基本相同”或“基本同一性”是指核酸序列与参考序列相比,包含具有至少约85%或更高序列同一性的序列,优选至少90%、至少91%、至少92%、至少93%、至少94%、至少95%、至少96%、至少97%、至少98%或至少99%同一性。序列同一性的百分比通过在比对窗口上比较两个序列的最佳比对来确定。百分比是通过以下方式来计算的:确定在两个序列中出现相同核酸碱基的位置数以产生匹配位置的数量;将匹配位置的数量除以比对窗口中的位置总数,然后将结果乘以100,从而得出序列同一性的百分比。本文公开的发明包括与本文公开(例如,在表1-5中)的那些基本相同的核苷酸序列。在一些实施方案中,所述核苷酸序列与本文公开(例如,在表1-3中)的序列完全相同,或具有至少约85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%或99%的同一性。
如本文所用,术语“包含序列的链”是指包含核苷酸链的寡核苷酸,所述核苷酸链由使用标准核苷酸命名法指代的序列描述。如本文所用,术语“双链RNA”或“dsRNA”指包含RNA分子或RNAi分子复合物的序列,所述分子或复合物具有包含两条反向平行且基本或完全互补的核酸链的杂交双链区,其分别被称为相对于靶LPA RNA具有“正义”和“反义”方向。双链区可以具有允许通过RISC途径特异性降解靶标LPA RNA的任何所需长度,但通常长度为9至30个碱基对,例如长度为15-30个碱基对。考虑到9到30个碱基对之间的双链体,双链体可以是此范围内的任何长度,例如,9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30个碱基对,以及其中的任何子范围,包括但不限于15-30个碱基对、15-26个碱基对;15-23碱基对、15-22碱基对、15-21碱基对、15-20碱基对、15-19碱基对、15-18碱基对、15-17碱基对、18-30个碱基对、18-26个碱基对、18-23个碱基对、18-22个碱基对、18-21个碱基对、18-20个碱基对、19-30个碱基对、19-26个碱基对、19-23个碱基对、19-22个碱基对、19-21个碱基对、19-20个碱基对、20-30个碱基对、20-26个碱基对、20-25个碱基对、20-24个碱基对、20-23个碱基对、20-22个碱基对、20-21个碱基对、21-30个碱基对、21-26个碱基对、21-25个碱基对、21-24个碱基对、21-23个碱基对或21-22个碱基对。通过用切丁酶和类似酶加工在细胞中产生的LPA dsRNA试剂的长度通常在19-22个碱基对的范围内。LPA dsDNA剂的双链区的一条链包含与靶LPARNA的区域基本互补的序列。形成双链体结构的两条链可以来自具有至少一个自身互补区的单个RNA分子,或者可以由两个或更多个单独的RNA分子形成。在双链区由单个分子形成的情况下,该分子可以具有由单链核苷酸链在3'-末端的一条链和相应的5'-末端的另一条链形成的双链体结构(本文称为“发夹环”)。在本发明的一些实施方案中,发夹构型包含至少1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20个或更多个未配对的核苷酸。当LPA dsRNA试剂的基本互补的两条链由单独的RNA分子组成
时,这些分子不需要共价连接,但也可以共价连接。当两条链通过发夹环以外的方式共价连接时,连接结构被称为“接头”。术语“siRNA”在本文中也用于指如本文所述的dsRNA试剂。
在本发明的一些实施方案中,LPA dsRNA试剂可以包含在dsRNA试剂的一个或两个末端具有未配对核苷酸或核苷酸类似物的正义和反义序列。没有未配对核苷酸的末端被称为“平末端”并且没有核苷酸突出端。如果dsRNA试剂的两端都是平末端,则dsRNA被称为“平末端的”。在本发明的一些实施方案中,dsRNA试剂的第一末端是平末端的,在一些实施方案中,dsRNA试剂的第二末端是平末端的,并且在本发明的某些实施方案中,LPA dsRNA试剂的两个末端都是平末端的。
在本发明的dsRNA试剂的一些实施方案中,dsRNA不具有一个或两个平末端。在这种情况下,在dsRNA试剂的一条链的末端有至少一个未配对的核苷酸。例如,当dsRNA一条链的3'-末端延伸超出另一条链的5'-末端时,则存在核苷酸突出端,反之亦然。dsRNA可包含至少1、2、3、4、5、6或更多个核苷酸的突出端。核苷酸突出端可包含核苷酸/核苷类似物或由其组成,包括脱氧核苷酸/核苷。应当理解,在一些实施方案中,核苷酸突出端在dsRNA试剂的正义链上、在dsRNA试剂的反义链上,或在dsRNA试剂的两端,突出端的核苷酸可存在于dsRNA的反义链或正义链的5'端、3'端或两端。在本发明的某些实施方案中,突出端中的一个或更多个核苷酸被核苷硫代磷酸酯替换。
如本文所用,术语“反义链”或“引导链”是指包含与LPA靶序列基本互补的区域的LPAdsRNA试剂的链。如本文所用,术语“正义链”或“过客链”是指包含与LPA dsRNA试剂的反义链的区域基本互补的区域的LPA dsRNA试剂的链。
修饰
在本发明的一些实施方案中,LPA RNAi剂的RNA被化学修饰以获得增强的稳定性和/或一种或更多种其他有益特性。本发明的某些实施方案中的核酸可以通过本领域公知的方法合成和/或修饰,例如,见“Current protocols in Nucleic Acid Chemistry,"Beaucage,S.L.et al.(Eds.),John Wiley&Sons,Inc.,New York,N.Y.,USA,其作为参考在此并入本文。可以存在于本发明的LPA dsRNA试剂的某些实施方案中的修饰包括例如:(a)末端修饰,例如5'端修饰(磷酸化、缀合、反向连接等)、3'端修饰(缀合、DNA核苷酸、反向连接等);(b)碱基修饰,例如用稳定碱基、去稳定碱基或与扩展的配偶体库进行碱基配对的碱基替换、缺失碱基(无碱基核苷酸)或缀合碱基;(c)糖修饰(例如,在2'位置或4'位置)或糖的替换;以及(d)骨架修饰,包括磷酸二酯键的修饰或替换。在本发明的LPA dsRNA试剂、LPA反义多核苷酸和LPA正义多核苷酸的某些实施方案中可用的RNA化合物的具体实例包括但不限于包含修饰骨架或没有天然核苷间键联的RNA。作为非限制性实例,具有骨架修饰的RNA
在骨架中可以不具有磷原子。在其核苷间骨架中没有磷原子的RNA可称为寡核苷。在本发明的某些实施方案中,修饰的RNA在其核苷间骨架中具有磷原子。
应当理解,术语“RNA分子”或“RNA”或“核糖核酸分子”不仅包括在自然界中表达或发现的RNA分子,还包括RNA的类似物和衍生物,其包含一种或更多种如本文所述或本领域已知的核糖核苷酸/核糖核苷类似物或衍生物。术语“核糖核苷”和“核糖核苷酸”在本文中可互换使用。RNA分子可以在核碱基结构或核糖-磷酸骨架结构中进行修饰(例如,如下文所述),并且包含核糖核苷类似物或衍生物的分子必须保留形成双链体的能力。作为非限制性实例,RNA分子还可包含至少一种修饰的核糖核苷,其包括但不限于2'-O-甲基修饰的核苷、包含5'硫代磷酸酯基团的核苷、与胆固醇衍生物或十二烷酸双癸酰胺基团相连的末端核苷、锁核苷、无碱基核苷、2'-脱氧-2'-氟修饰的核苷、2'-氨基修饰的核苷、2'-烷基修饰的核苷、吗啉代核苷、氨基磷酸酯或包含核苷的非天然碱基,或其任何组合。在本发明的一些实施方案中,RNA分子包含以下数量的修饰的核糖核苷:至少1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20或长达LPA dsRNA试剂分子的核糖核苷的全长。对于这种RNA分子中的多个修饰的核糖核苷中的每一个,修饰不必相同。
在一些实施方案中,本发明的dsRNA试剂、LPA反义多核苷酸和/或LPA正义多核苷酸可以包含一个或更多个独立选择的修饰核苷酸和/或一个或更多个独立选择的非磷酸二酯键。本文所用的术语“独立选择”用于指选定的要素,例如修饰的核苷酸、非磷酸二酯键等,是指两个或更多个选定的要素可以彼此相同但不必彼此相同。如本文所用,“核苷酸碱基”、“核苷酸”或“核碱基”是杂环嘧啶或嘌呤化合物,其是所有核酸的标准成分,并且包括形成核苷酸的碱基:腺嘌呤(a)、鸟嘌呤(g)、胞嘧啶(c)、胸腺嘧啶(t)和尿嘧啶(u)。核碱基可进一步修饰以包括(但不旨在限制):通用碱基、疏水性碱基、混杂碱基、尺寸扩大的碱基和氟化碱基。术语“核糖核苷酸”或“核苷酸”在本文中可用于指未修饰的核苷酸、修饰的核苷酸或替代部分。本领域技术人员将认识到,鸟嘌呤、胞嘧啶、腺嘌呤和尿嘧啶可以被其他部分替换,而不会显著改变包含带有这种替换部分的核苷酸的寡核苷酸的碱基配对特性。
在一个实施方案中,预期用于本文所述的方法和组合物中的修饰的RNA是肽核酸(PNA),其具有形成所需双链体结构并且允许或介导靶RNA经由RISC途径的特异性降解的能力。在本发明的某些实施方案中,LPA RNA干扰剂包括与靶LPA RNA序列相互作用以指导靶LPA RNA切割的单链RNA。
修饰的RNA骨架可以包含例如硫代磷酸酯、手性硫代磷酸酯、二硫代磷酸酯、磷酸三酯、氨基烷基磷酸三酯、甲基和其他烷基膦酸酯(包括3'-亚烷基膦酸酯和手性膦酸酯)、次膦酸酯、氨基磷酸酯(包括3'-氨基氨基磷酸酯和氨基烷基氨基磷酸酯)、硫代氨基磷酸酯、硫代烷基膦酸酯、硫代烷基磷酸三酯,和硼酸磷酸酯(其具有正常3'-5'连接的,以及这些的2'-5'连接类似物,以及具有倒置极性的那些,其中相邻的核苷单元对以3'-5'至5'-3'
或2'-5'至5'-2'形式连接)。还包括各种盐、混合盐和游离酸形式。制备含磷键的方法是本领域的常规实施手段,并且此类方法可用于制备本发明的某些修饰的LPA dsRNA试剂、某些修饰的LPA反义多核苷酸和/或某些修饰的LPA正义多核苷酸。
其中不包含磷原子的修饰的RNA骨架具有由短链烷基或环烷基核苷间键联、混合杂原子和烷基或环烷基核苷间键联、或一个或更多个短链杂原子或杂环核苷间键联形成的骨架。其包括具有吗啉键的那些(部分由核苷的糖部分形成);硅氧烷骨架;硫化物、亚砜和砜骨架;甲乙酰和硫甲乙酰骨架;亚甲基甲乙酰和硫甲乙酰骨架;含有烯烃的骨架;氨基磺酸盐骨架;亚甲基亚氨基和亚甲基肼基骨架;磺酸盐和磺酰胺骨架;酰胺骨架;以及其他混合有N、O、S和CH2成分的部分。制备不含磷原子的修饰的RNA骨架的方法在本领域中是常规实践,并且此类方法可用于制备本发明的某些修饰的LPA dsRNA试剂、某些修饰的LPA反义多核苷酸和/或某些修饰的LPA正义多核苷酸。
在本发明的某些实施方案中,RNA模拟物被包括在LPA dsRNA、LPA反义多核苷酸和/或LPA正义多核苷酸中,例如但不限于用新基团替换核苷酸单元的糖和核苷间键联(即骨架)。在此类实施方案中,保持碱基单位以与合适的LPA核酸靶化合物杂交。一种这样的寡聚化合物(已被证明具有优异杂交特性的RNA模拟物),被称为肽核酸(PNA)。在PNA化合物中,RNA的糖骨架被含有酰胺的骨架,特别是氨乙基甘氨酸骨架取代。核碱基被保留并直接或间接地与骨架酰胺部分的氮杂氮原子结合。制备RNA模拟物的方法是本领域常规实践的,并且此类方法可用于制备本发明的某些修饰的LPA dsRNA试剂。
本发明的一些实施方案包括具有硫代磷酸酯骨架的RNA和具有杂原子骨架的寡核苷,特别是-CH2--NH--CH2-、--CH2--N(CH3)--O--CH2--[称为亚甲基(甲基亚氨基)或MMI骨架]、--CH2--O--N(CH3)--CH2--、--CH2--N(CH3)--N(CH3)--CH2-以及--N(CH3)--CH2----[其中天然磷酸二酯骨架表示为--O--P--O--CH2--]。制备具有硫代磷酸酯骨架的RNA和具有杂原子骨架的寡核苷的方法是本领域常规实践的,并且此类方法可用于制备本发明的某些修饰的LPA dsRNA试剂、某些LPA反义多核苷酸和/或某些LPA正义多核苷酸。
修饰的RNA还可以包含一个或更多个取代的糖部分。本发明的LPA dsRNA、LPA反义多核苷酸和/或LPA正义多核苷酸可在2'位置包含以下之一:OH;F;O--、S--、或N-烷基;O--、S--或N-烯基;O-、S-或N-炔基;或O-烷基-O-烷基,其中烷基、烯基和炔基可以是取代或未取代的C1至C10烷基或C2至C10烯基和炔基。示例性的合适的修饰包括:O[(CH2)nO]mCH3、O(CH2)nOCH3、O(CH2)nNH2、O(CH2)nCH3、O(CH2)nONH2、以及O(CH2)nON[(CH2)nCH3)]2,其中n和m为1至约10。在其他实施方案中,dsRNA在2'位置包括以下之一:C1至C10低级烷基、取代的低级烷基、烷芳基、芳烷基、O-烷芳基或O-芳烷基、SH、SCH3、OCN、Cl、Br、CN、CF3、OCF3、SOCH3、SO2CH3、ONO2、NO2、N3、NH2、杂环烷基、杂环烷芳基、氨基烷基氨基、聚烷基氨基;取代的甲硅烷基、RNA裂解
基团、报告基团、嵌入剂;用于改善LPA dsRNA试剂的药代动力学特性的基团;或用于改善LPA dsRNA试剂、LPA反义多核苷酸和/或LPA正义多核苷酸的药效学特性的基团,和其他具有类似性质的取代基。在一些实施方案中,修饰包括2'-甲氧基乙氧基(2'-O--CH2CH2OCH3,也称为2'-O-(2-甲氧基乙基)或2'-MOE)(Martin et al.,Helv.Chim.Acta,1995,78:486-504),即烷氧基-烷氧基。另一种示例性修饰是2'-二甲氨基乙氧基乙氧基,即O(CH2)2ON(CH3)2基团,也称为2'-DMAOE,如下文实施例中所述;以及2'-二甲氨基乙氧基乙氧基(在本领域中也称为2'-O-二甲氨基乙氧基乙基或2'-DMAEOE),即2'-O--CH2-O--CH2--N(CH2)2。制备所描述的那些的修饰RNA的方法是本领域常规实践的,并且此类方法可用于制备本发明的某些修饰的LPA dsRNA试剂。
其他修饰包括2'-甲氧基(2'-OCH3)、2'-氨基丙氧基(2'-OCH2CH2CH2NH2)和2'-氟(2'-F)。类似的修饰也可以在本发明的LPA dsRNA试剂、LPA反义多核苷酸的RNA上的其他位置、LPA正义多核苷酸和/或LPA正义多核苷酸的其他位置,特别是3'末端核苷酸上或2'-5'连接的LPA dsRNA、LPA反义多核苷酸或LPA正义多核苷酸中的糖的3'位置、和5'末端核苷酸的5'位置进行。LPA dsRNA试剂、LPA反义多核苷酸和/或LPA正义多核苷酸也可以具有糖模拟物,例如代替呋喃戊糖的环丁基部分。制备例如所描述的那些的修饰RNA的方法是本领域常规实践的,并且此类方法可用于制备本发明的某些修饰的LPA dsRNA试剂、LPA反义多核苷酸和/或LPA正义多核苷酸。
在一些实施方案中,LPA dsRNA试剂、LPA反义多核苷酸和/或LPA正义多核苷酸可以包括核碱基(在本领域中通常简称为“碱基”)修饰或取代。如本文所用,“未修饰的”或“天然”核碱基包括嘌呤碱基腺嘌呤(A)和鸟嘌呤(G),以及嘧啶碱基胸腺嘧啶(T)、胞嘧啶(C)和尿嘧啶(U)。修饰的核碱基包括其他合成和天然核碱基,例如5-甲基胞嘧啶(5-me-C)、5-羟甲基胞嘧啶、黄嘌呤、次黄嘌呤、2-氨基腺嘌呤、6-甲基和其他腺嘌呤和鸟嘌呤的烷基衍生物、2-丙基和其他腺嘌呤和鸟嘌呤的烷基衍生物、2-硫尿嘧啶、2-硫胸腺嘧啶和2-硫胞嘧啶、5-卤尿嘧啶和胞嘧啶、5-丙炔基尿嘧啶和胞嘧啶、6-偶氮尿嘧啶、胞嘧啶和胸腺嘧啶、5-尿嘧啶(假尿嘧啶)、4-硫尿嘧啶;8-卤代、8-氨基、8-硫醇、8-硫代烷基、8-羟基以及其他8-取代的腺嘌呤和鸟嘌呤;5-卤代,特别是5-溴、5-三氟甲基和其他5-取代的尿嘧啶和胞嘧啶;7-甲基鸟嘌呤和7-甲基腺嘌呤、8-氮杂鸟嘌呤和8-氮杂腺嘌呤、7-氮杂鸟嘌呤和7-氮杂腺嘌呤以及3-氮杂鸟嘌呤和3-氮杂腺嘌呤。可以包含在本发明的LPA dsRNA试剂的某些实施方案中的另外的核碱基是本领域已知的,参见例如:Modified Nucleosides in Biochemistry,Biotechnology and Medicine,Herdewijn,P.Ed.Wiley-VCH,2008;The Concise Encyclopedia Of Polymer Science And Engineering,pages858-859,Kroschwitz,J.L,Ed.John Wiley&Sons,1990,English et al.,Angewandte Chemie,International Edition,1991,30,613,Sanghvi,Y S.,Chapter 15,dsRNA Research and Applications,pages 289-302,Crooke,S.T.and Lebleu,B.,Ed.,CRC Press,1993。制备包含核碱基修饰和/或取代的dsRNA、LPA反义链多核苷酸和/或LPA正义
链多核苷酸(例如本文所述的那些)的方法是本领域常规实践的,并且此类方法可用于制备本发明的某些修饰的LPA dsRNA试剂、LPA正义多核苷酸和/或LPA反义多核苷酸。
本发明的LPA dsRNA试剂、LPA反义多核苷酸和/或LPA正义多核苷酸的某些实施方案包括经修饰以包括一种或更多种锁核酸(LNA)的RNA。锁核酸是具有这样的修饰核糖部分的核苷酸,其包含额外的连接2'和4'碳的桥。这种结构有效地将核糖“锁定”在3'-内结构构象中。在本发明的LPA dsRNA试剂、LPA反义多核苷酸和/或LPA正义多核苷酸中添加锁核酸可以增加血清中的稳定性,并减少脱靶效应(Elmen,J.et al.,(2005)Nucleic Acids Research 33(1):439-447;Mook,O R.et al.,(2007)Mol Canc Ther6(3):833-843;Grunweller,A.et al.,(2003)Nucleic Acids Research 31(12):3185-3193)。制备包含锁核酸的dsRNA试剂、LPA反义多核苷酸和/或LPA正义多核苷酸的方法是本领域常规实施的,并且此类方法可用于制备本发明的某些修饰的LPA dsRNA试剂。本发明的LPA dsRNA化合物、正义多核苷酸和/或反义多核苷酸的某些实施方案包括至少一种修饰的核苷酸,其中所述至少一种修饰的核苷酸包含:2'-O-甲基核苷酸、2'-氟核苷酸、2'-脱氧核苷酸、2',3'-seco核苷酸模拟物、锁核苷酸、2'-F-阿拉伯糖核苷酸、2'-甲氧基乙基核苷酸、2'-氨基修饰的核苷酸、2'-烷基修饰的核苷酸、吗啉代核苷酸和3'-Ome核苷酸、包含5'-硫代磷酸酯基团的核苷酸,或与胆固醇衍生物或十二烷酸双癸酰胺基团连接的末端核苷酸、2'-氨基修饰的核苷酸、氨基磷酸酯或包含核苷酸的非天然碱基。在一些实施方案中,LPA dsRNA化合物在反义链(在本文中也称为引导链)的5’末端处包含E-乙烯基膦酸酯核苷酸。
本发明的某些实施方案中,在LPA dsRNA化合物、正义多核苷酸的3'和5'末端和/或反义多核苷酸的3'末端包含至少一种修饰的核苷酸,其中至少一种修饰的核苷酸包括:无碱基核苷酸、核糖醇、反向核苷酸、反向无碱基核苷酸、反向2'-OMe核苷酸、反向2'-脱氧核苷酸。本领域技术人员已知,在寡核苷酸末端包含无碱基或反向无碱基核苷酸可增强稳定性(Czauderna et al.Structural variations and stabilizing modifications of synthetic siRNAs in mammalian cells.Nucleic Acids Res.2003;31(11):2705-2716.doi:10.1093/nar/gkg393)。
本发明的某些实施方案中,在LPA dsRNA在正义链的3'和5'末端包含一个或两个异甘露醇残基。在某些实施方案中,正义链在3'或和5'末端分别独立地包含一个异甘露醇残基。包含异甘露醇残基具有以下示例:
其中短语“Olig”各自独立地代表多核苷酸部分。示例性异甘露醇残基(imann)包括但不限于以下:
在某些实施方案中,异甘露醇残基还可以使用其立体异构体替代,非限制性的示例性:在某些实施方案中,正义链在3'或和5'末端分别独立地包含一个异甘露醇残基(imann),进一步包含5'-末端缀合的靶向基团,例如,靶向基团N-乙酰-半乳糖胺,优选为上述GLS-15,示例性结构如下:.
其中短语“Olig”各自独立地代表多核苷酸部分。
本发明的某些实施方案中,LPA dsRNA化合物、反义多核苷酸包含至少一种修饰的核苷酸,其中所述至少一种修饰的核苷酸包含开环核酸核苷酸(UNA)或/和二醇核酸核苷酸(GNA)。本领域技术人员已知,UNA和GNA是热不稳定化学修饰,可以显著改善siRNA化合物的脱靶谱(Janas,et al.,Selection of GalNAc-conjugated siRNAs with limited off-target-driven rat hepatotoxicity.Nat Commun.2018;9(1):723.doi:10.1038/s41467-018-02989-4;Laursen et al.,Utilization of unlocked nucleic acid(UNA)to enhance siRNA performance in vitro and in vivo.Mol BioSyst.2010;6:862–70)。
可以在本发明的某些实施方案的LPA dsRNA试剂、LPA反义多核苷酸和/或LPA正义多核苷酸的RNA中包含另一种修饰,其包括分别增强LPA dsRNA试剂、LPA反义多核苷酸和/或LPA正义多核苷酸的一种或更多种特征的一种或更多种配体、部分或与RNA化学连接的缀合物。可以增强的特征的非限制性实例是:LPA dsRNA试剂、LPA反义多核苷酸和/或LPA正义多核苷酸活性、细胞分布、LPA dsRNA试剂的递送、LPA dsRNA试剂的药代动力学特性以及LPA dsRNA试剂的细胞摄取。在本发明的一些实施方案中,LPA dsRNA试剂包含一个或更多个靶向基团或连接基团,在本发明的LPA dsRNA试剂的某些实施方案中,其与正义链缀合。靶向基团的非限制性实例是包含N-乙酰基-半乳糖胺(GalNAc)的化合物。术语“靶向基团”、“靶向剂”、“连接剂”、“靶向化合物”和“靶向配体”在本文中可互换使用。在本发明的某些实施方案中,LPA dsRNA试剂包含与正义链的5'-末端缀合的靶向化合物。在本发明的某些实施方案中,LPA dsRNA试剂包含与正义链的3'-末端缀合的靶向化合物。在本发明的一些实施方案中,LPA dsRNA试剂包含含有GalNAc的靶向基团。在本发明的某些实施方案中,LPA dsRNA试剂不包含与正义链的3'-末端和5'-末端之一或两者缀合的靶向化合物。在本发明的某些实施方案中,LPA dsRNA试剂不包含与正义链的5'-末端和3'-末端之一或两者缀合的含有GalNAc的靶向化合物。
另外的靶向和连接剂是本领域众所周知的,例如,可用于本发明的某些实施方案中的靶向和连接剂包括但不限于脂质部分,例如胆固醇部分(Letsinger et al.,Proc.Natl.Acid.Sci.USA,1989,86:6553-6556)、胆酸(Manoharan et al.,Biorg.Med.Chem.Let.,1994,4:1053-1060)、硫醚,例如beryl-S-三苯甲基硫醇(Manoharan et al.,Ann.N.Y.Acad.Sci.,1992,660:306-309;Manoharan et al.,Biorg.Med.Chem.Let.,1993,3:2765-2770)、硫胆固醇(Oberhauser et al.,Nucl.Acids Res.,1992,20:533-538)、脂肪链,例如十二烷二醇或十一烷基残基(Saison-Behmoaras et al.,EMBO J,1991,10:1111-1118;Kabanov et al.,FEBS Lett.,1990,259:327-330;Svinarchuk et al.,Biochimie,1993,75:49-54)、磷脂,例如二-十六烷基-rac-甘油或三乙基-铵1,2-二-O-十六烷基-rac-甘油-3-膦酸酯(Manoharan et al.,Tetrahedron Lett.,1995,36:3651-3654;Shea et al.,Nucl.Acids Res.,1990,18:3777-3783)、聚胺或聚乙二醇链(Manoharan et al.,Nucleosides&Nucleotides,1995,14:969-973)或金刚烷乙酸(Manoharan et al.,Tetrahedron Lett.,1995,36:3651-3654)、棕榈酰部分(Mishra et al.,Biochim.Biophys.Acta,1995,1264:229-237)或十八胺或己氨基-羰氧基胆固醇部分(Crooke et al.,J.Pharmacol.Exp.Ther.,1996,277:923-937)。
包含LPA dsRNA试剂、LPA反义多核苷酸和/或LPA正义多核苷酸的组合物的某些实施方案可包含改变LPA dsRNA试剂的分布、靶向等性质的配体。在包含本发明的LPA dsRNA试剂的组合物的一些实施方案中,例如与不存在此类配体的物种相比,配体增加对选定靶标(例如分子、细胞或细胞类型、区室,例如细胞或器官区室、组织、器官或身体区域)的亲和力。在本发明的组合物和/或方法中有用的配体可以是天然存在的物质,例如蛋白质(例
如人血清白蛋白(HSA)、低密度脂蛋白(LDL)或球蛋白)、碳水化合物(例如,葡聚糖、支链淀粉、几丁质、壳聚糖、菊粉、环糊精或透明质酸)或脂质。配体也可以是重组或合成分子,例如合成聚合物,例如合成聚氨基酸或聚胺。聚氨基酸的实例是聚赖氨酸(PLL)、聚L-天冬氨酸、聚L-谷氨酸、苯乙烯-马来酸酐共聚物、聚(L-丙交酯-共-乙醇酸)共聚物、二乙烯基醚-马来酸酐共聚物、N-(2-羟丙基)甲基丙烯酰胺共聚物(HMPA)、聚乙二醇(PEG)、聚乙烯醇(PVA)、聚氨酯、聚(2-乙基丙烯酸)、N-异丙基丙烯酰胺聚合物,或聚磷嗪。多胺的示例包括:聚乙烯亚胺、聚赖氨酸(PLL)、精胺、亚精胺、多胺、假肽-多胺、拟肽多胺、树枝状多胺、精氨酸、脒、鱼精蛋白、阳离子脂质、阳离子卟啉、多胺的季盐或α螺旋肽。
本发明的组合物和/或方法中包含的配体可包含靶向基团,其非限制性实例为细胞或组织靶向剂,例如,凝集素、糖蛋白、脂质或蛋白质,例如结合特定细胞类型如肾细胞或肝细胞的抗体。靶向基团可以是促甲状腺素、促黑素、凝集素、糖蛋白、表面活性蛋白A、黏蛋白碳水化合物、多价乳糖、多价半乳糖、N-乙酰-半乳糖胺、N-乙酰-葡糖胺多价甘露糖、多价岩藻糖、糖基化聚氨基酸、多价半乳糖、转铁蛋白、双膦酸盐、聚谷氨酸盐、聚天冬氨酸、脂质、胆固醇、类固醇、胆汁酸、叶酸、维生素B12、维生素A、生物素或RGD肽或RGD肽模拟物。
配体的其他实例包括染料、嵌入剂(例如吖啶)、交联剂(例如补骨脂素、丝裂霉素C)、卟啉(TPPC4、texaphyrin、Sapphyrin)、多环芳烃(例如吩嗪、二氢吩嗪);人工核酸内切酶(例如EDTA)、亲脂性分子,例如胆固醇、胆酸、金刚烷乙酸、1-芘丁酸、二氢睾酮、1,3-双-O(十六烷基)甘油、香叶氧基己基、十六烷基甘油、冰片、薄荷醇、1,3-丙二醇、十七烷基、棕榈酸、肉豆蔻酸、O3-(油酰基)石胆酸、O3-(油酰基)胆酸、二甲氧基三苯甲基或吩噁嗪和肽缀合物(例如,触角肽、Tat肽)、烷化剂、磷酸盐、氨基、巯基、PEG(例如,PEG-40K)、MPEG、[MPEG]2、聚氨基、烷基、取代烷基、放射性标记物、酶、半抗原(例如生物素)、转运/吸收促进剂(例如阿司匹林、维生素E、叶酸)、合成核糖核酸酶(例如咪唑、双咪唑、组胺、咪唑簇、吖啶-咪唑偶联物、四氮杂大环的Eu3+复合物)、二硝基苯基、HRP或AP。
本发明的组合物和/或方法中包括的配体可以是蛋白质,例如糖蛋白或肽,例如对共配体具有特定亲和力的分子,或抗体,例如与特定细胞类型如癌细胞、内皮细胞、心肌细胞或骨细胞结合的抗体。在本发明的组合物和/或方法的实施方案中有用的配体可以是激素或激素受体。在本发明的组合物和/或方法的实施方案中有用的配体可以是脂质、凝集素、碳水化合物、维生素、辅酶、多价乳糖、多价半乳糖、N-乙酰-半乳糖胺、N-乙酰-葡糖胺多价甘露糖或多价岩藻糖。在本发明的组合物和/或方法的实施方案中有用的配体可以是例如通过破坏细胞的细胞骨架(例如,通过破坏细胞的微管、微丝和/或中间丝)而增加LPA dsRNA
试剂向细胞中的摄取的物质。此类试剂的非限制性实例是:taxon、长春新碱、长春碱、细胞松弛素、诺考达唑、japlakinolide、latrunculin A、鬼笔环肽、swinholide A、indanocine和myoservin。
在一些实施方案中,与本发明的LPA dsRNA试剂连接的配体用作药代动力学(PK)调节剂。可用于本发明的组合物和方法的PK调节剂的实例包括但不限于:亲脂剂、胆汁酸、类固醇、磷脂类似物、肽、蛋白质结合剂、PEG、维生素、胆固醇、脂肪酸、胆酸、石胆酸、二烷基甘油酯、二酰基甘油酯、磷脂、鞘脂、萘普生、布洛芬、维生素E、生物素、结合血清蛋白的适体等。还已知包含许多硫代磷酸酯键的寡核苷酸与血清蛋白结合,因此,在骨架中包含多个硫代磷酸酯键的短寡核苷酸,例如约5个碱基、10个碱基、15个碱基或20个碱基的寡核苷酸也可用作本发明的组合物和/或方法中的配体。
LPA dsRNA试剂组合物
在本发明的一些实施方案中,LPA dsRNA试剂在组合物中。本发明的组合物可包含一种或更多种LPA dsRNA试剂和任选的一种或更多种药学上可接受的载体、递送剂、靶向剂、可检测标签等,根据本发明方法的一些实施方案可用的靶向剂的非限制性实例是将本发明的LPA dsRNA试剂引导至和/或进入待治疗细胞的试剂。靶向剂的选择将取决于以下要素:LPA相关疾病或病症的性质,以及靶细胞类型。在一个非限制性实例中,在本发明的一些实施方案中,可能需要将LPA dsRNA试剂靶向至和/或进入肝细胞。应当理解,在本发明方法的一些实施方案中,治疗剂包含仅具有递送剂的LPA dsRNA试剂,例如包含N-乙酰半乳糖胺(GalNAc)的递送剂,而没有任何附加的连接元件。例如,在本发明的一些方面,LPA dsRNA试剂可以连接到包含GalNAc的递送化合物上,并且包含在含有药学上可接受载体的组合物中,并且在没有任何连接至LPA dsRNA试剂的可检测标记或靶向剂等的情况下施用至细胞或对象。
在本发明的LPA dsRNA试剂与一种或更多种递送剂、靶向剂、标记剂等一起施用和/或连接到其上的情况下,本领域技术人员能够了解并能够选择和使用适合的试剂用于本发明的方法中。在本发明的某些方法中可以使用标记试剂来确定LPA dsRNA试剂在细胞和组织中的位置,并且可用于确定已在本发明的方法中施用的包含LPA dsRNA试剂的治疗组合物的细胞、组织或器官位置。接附和使用标记试剂如酶标记、染料、放射性标记等的手段是本领域公知的。应当理解,在本发明的组合物和方法的一些实施方案中,标记试剂连接至LPAdsRNA试剂中所包含的正义多核苷酸和反义多核苷酸之一或两者。
LPA dsRNA试剂和LPA反义多核苷酸试剂的递送
本发明方法的某些实施方案包括将LPA dsRNA试剂递送到细胞中。如本文所用,术语“递送”是指促进或影响细胞摄取或吸收。LPA dsRNA试剂的吸收或摄取可通过独立的扩散或活性细胞过程来发生,或通过使用可与本发明的LPA dsRNA试剂相关的递送剂、靶向剂等来进行。适用于本发明方法的递送方式包括但不限于体内递送,其中将LPA dsRNA试剂注射到组织部位或全身给药。在本发明的一些实施方案中,LPA dsRNA试剂连接至递送剂。
可用于将LPA dsRNA试剂递送至细胞、组织和/或对象的方法的非限制性实例包括:LPA dsRNA-GalNAc缀合物、SAMiRNA技术、基于LNP的递送方法和裸RNA递送。这些和其他递送方法已在本领域成功地用于递送治疗性RNAi试剂以治疗各种疾病和病症,例如但不限于:肝病、急性间歇性卟啉症(AIP)、血友病、肺纤维化等。多种递送方式的详细信息可在出版物中找到,例如:Nikam,R.R.&K.R.Gore(2018)Nucleic Acid Ther,28(4),209-224Aug2018;Springer A.D.&S.F.Dowdy(2018)Nucleic Acid Ther.Jun1;28(3):109–118;Lee,K.et al.,(2018)Arch Pharm Res,41(9),867-874;和Nair,J.K.et al.,(2014)J.Am.Chem.Soc.136:16958-16961,其内容均以引用方式并入本文。
本发明的一些实施方案包括使用脂质纳米颗粒(LNP)将本发明的LPA dsRNA试剂递送至细胞、组织和/或对象。LNP通常用于体内递送LPA dsRNA试剂,包括治疗性LPAdsRNA试剂。使用LNP或其他递送剂的一个好处是,当使用LNP或其他递送剂递送至对象时,LPA RNA剂的稳定性增加。在本发明的一些实施方案中,LNP包含负载有一种或更多种本发明的LPA RNAi分子的阳离子LNP。将包含LPA RNAi分子的LNP施用于对象,LNP及其接附的LPA RNAi分子通过胞吞作用被细胞摄取,它们的存在导致RNAi触发分子的释放,从而介导RNAi。
[根据细则91更正 17.04.2023]
可用于本发明的实施方案以将本发明的LPA dsRNA试剂递送至细胞、组织和/或对象的递送剂的另一个非限制性实例是:包含GalNAc的试剂,其与本发明的LPA dsRNA试剂连接并将LPA dsRNA试剂递送至细胞、组织和/或对象。PCT申请WO2020191183A1中公开了可用于本发明的方法和组合物的某些实施方案中的某些包含GalNAc的其他递送剂的实例。可用于本发明的组合物和方法中以将LPA dsRNA试剂递送至细胞的GalNAc靶向配体的非限制性实例是靶向配体簇。在此提出的靶向配体簇的实例如:具有磷酸二酯键的GalNAc配体(GLO)和具有硫代磷酸酯键的GalNAc配体(GLS)。术语“GLX-n”在本文中可用于表示所连接的含GalNAC的化合物是以下化合物:GLS-1、GLS-2、GLS-3、GLS-4、GLS-5、GLS-6、GLS-7、GLS-8、GLS-9、GLS-10、GLS-11、GLS-12、GLS-13、GLS-14、GLS-15、GLS-16、GLO-1、GLO-2、GLO-3、GLO-4、GLO-5、GLO-6、GLO-7、GLO-8、GLO-9、GLO-10、GLO-11、GLO-12、GLO-13、GLO-14、GLO-15和GLO-16中的任一种,每个的结构如下所示,下图中GalNAc靶向配体与本发明的RNAi剂的连接位置在每个靶向配体的最右侧。应当理解,本发明的任何RNAi和dsRNA分子都可以连接到GLS-1、GLS-2、GLS-3、GLS-4、GLS-5、GLS-6、GLS-7、GLS-8、GLS-9、GLS-10、GLS-11、GLS-12、GLS-13、GLS-14、GLS-15、GLS-16、GLO-1、GLO-2、GLO-3、GLO-4、GLO-5、GLO-6、GLO-7、GLO-8、GLO-9、GLO-10、GLO-11、GLO-12、GLO-13、GLO-14、GLO-15和GLO-16上,以下是GLO-1到GLO-16和GLS-1到GLS-16的结构。
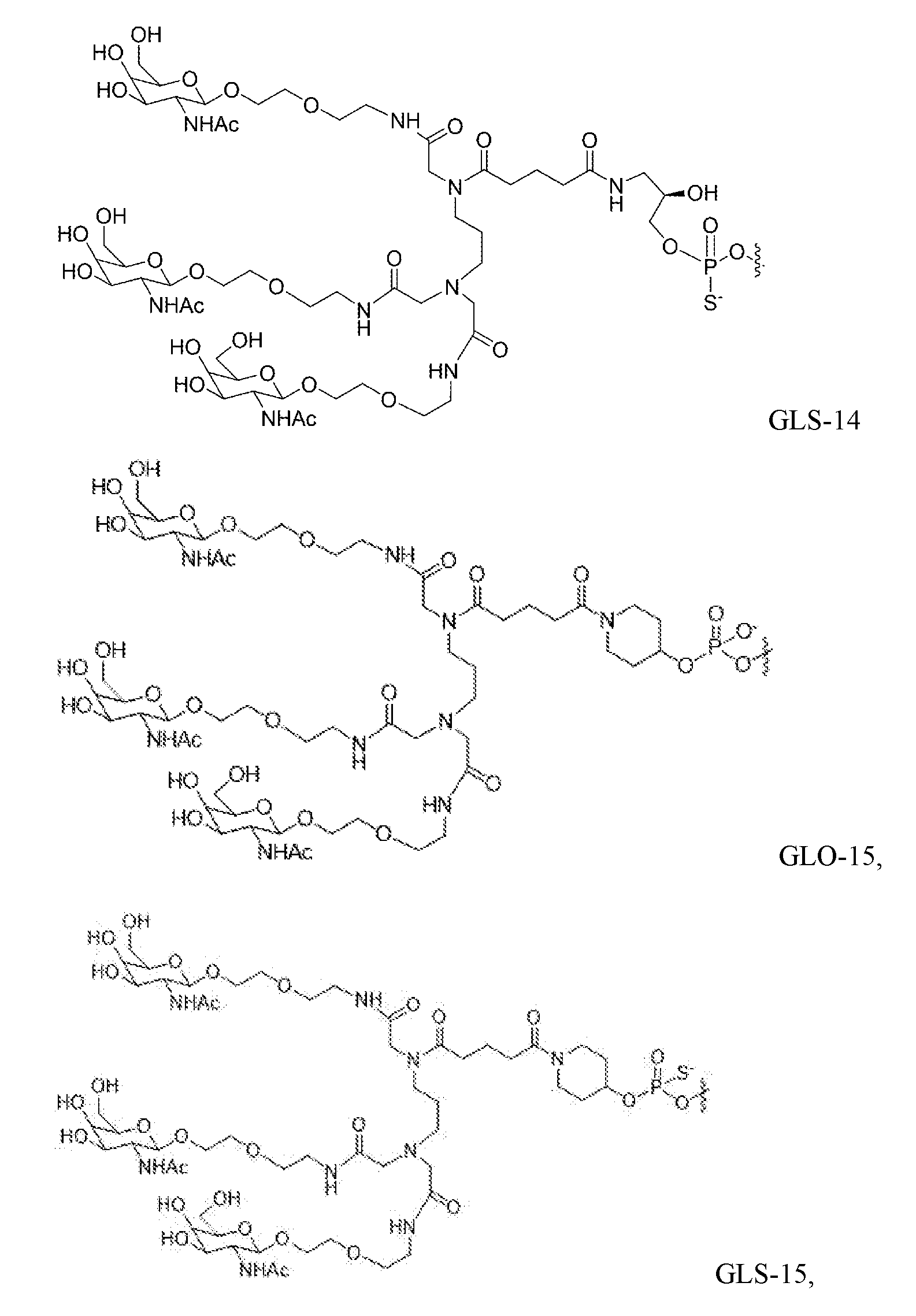
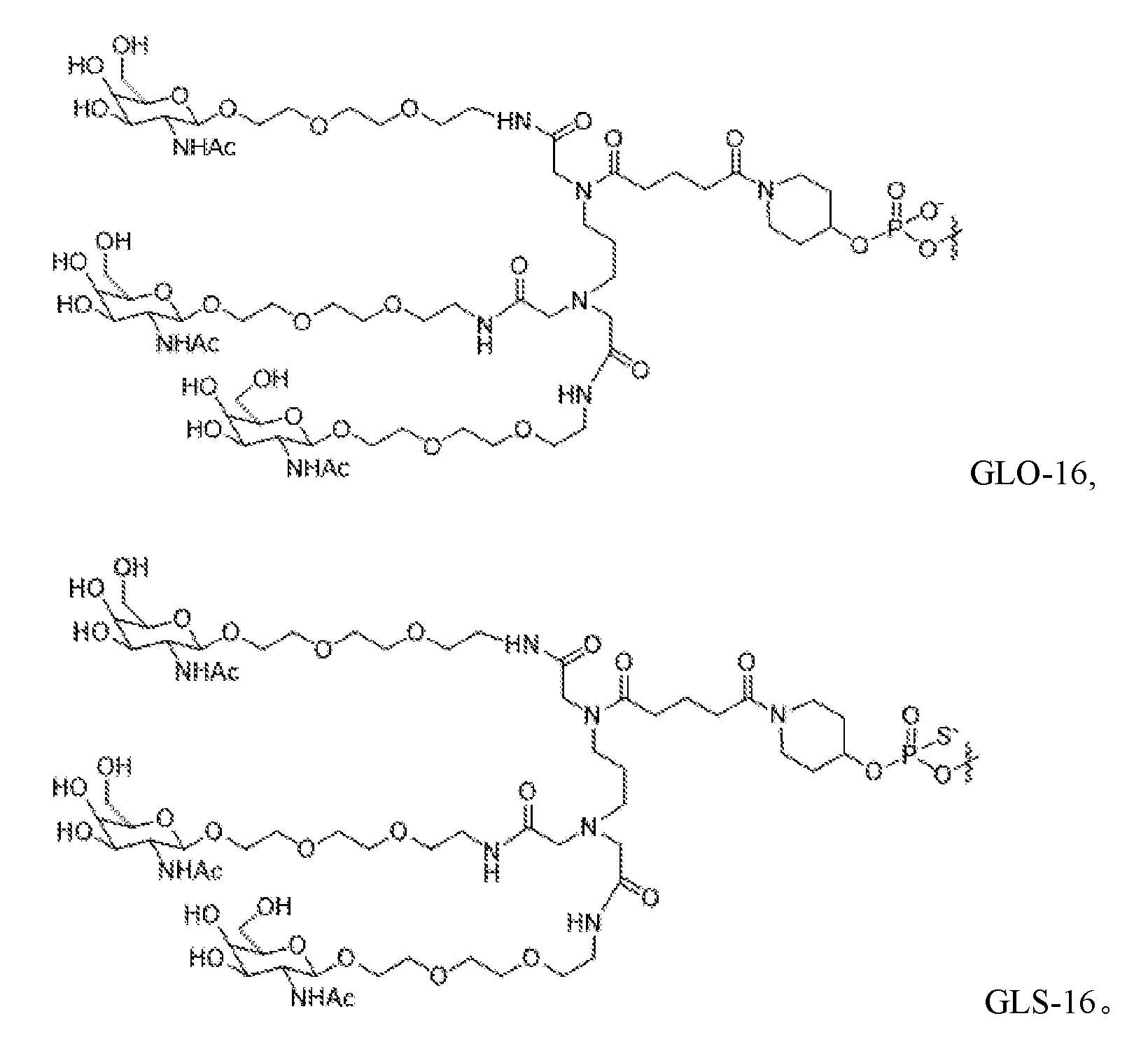
可用于本发明的实施方案以将本发明的LPA dsRNA试剂递送至细胞、组织和/或对象的递送剂的另一个非限制性实例是:包含GalNAc的试剂,其与本发明的LPA dsRNA试剂连接并将LPA dsRNA试剂递送至细胞、组织和/或对象。PCT申请WO2020191183A1中公开了可用于本发明的方法和组合物的某些实施方案中的某些包含GalNAc的其他递送剂的实例。可用于本发明的组合物和方法中以将LPA dsRNA试剂递送至细胞的GalNAc靶向配体的非限制性实例是靶向配体簇。在此提出的靶向配体簇的实例如:具有磷酸二酯键的GalNAc配体(GLO)和具有硫代磷酸酯键的GalNAc配体(GLS)。术语“GLX-n”在本文中可用于表示所连接的含GalNAC的化合物是以下化合物:GLS-1、GLS-2、GLS-3、GLS-4、GLS-5、GLS-6、GLS-7、GLS-8、GLS-9、GLS-10、GLS-11、GLS-12、GLS-13、GLS-14、GLS-15、GLS-16、GLO-1、GLO-2、GLO-3、GLO-4、GLO-5、GLO-6、GLO-7、GLO-8、GLO-9、GLO-10、GLO-11、GLO-12、GLO-13、GLO-14、GLO-15和GLO-16中的任一种,每个的结构如下所示,下图中GalNAc靶向配体与本发明的RNAi剂的连接位置在每个靶向配体的最右侧。应当理解,本发明的任何RNAi和dsRNA分子都可以连接到GLS-1、GLS-2、GLS-3、GLS-4、GLS-5、GLS-6、GLS-7、GLS-8、GLS-9、GLS-10、GLS-11、GLS-12、GLS-13、GLS-14、GLS-15、GLS-16、GLO-1、GLO-2、GLO-3、GLO-4、GLO-5、GLO-6、GLO-7、GLO-8、GLO-9、GLO-10、GLO-11、GLO-12、GLO-13、GLO-14、GLO-15和GLO-16上,以下是GLO-1到GLO-16和GLS-1到GLS-16的结构。
在本发明的一些实施方案中,体内递送也可以通过β-葡聚糖递送系统,例如美国专利No.5,032,401和5,607,677,以及美国公布No.2005/0281781中描述的那些,它们的全部内容通过引用并入本文。也可以使用本领域已知的方法例如电穿孔和脂质转染将LPA RNAi试剂体外引入细胞。在本发明方法的某些实施方案中,LPA dsRNA在没有靶向剂的情况下被递送。这些RNA可以作为“裸”RNA分子递送。作为非限制性实例,本发明的LPA dsRNA可以在包含RNAi试剂但不包含靶向剂(例如GalNAc靶向化合物)的药物组合物中施用于对象,以治疗对象的LPA相关疾病或病症,例如心血管疾病,其中所述心血管疾病包括伯格氏病(Berger’s disease)、外周动脉疾病、冠状动脉疾病、代谢综合征、急性冠脉综合征、主动脉瓣狭窄、主动脉瓣反流、主动脉夹层、视网膜动脉阻塞、脑血管疾病、肠系膜缺血、肠系膜上动脉阻塞、肾动脉狭窄、稳定型/不稳定型心绞痛、急性冠脉综合征、杂合子或纯合子家族性高胆固醇血症、高载脂蛋白β脂蛋白血症、脑血管动脉粥样硬化、脑血管疾病和静脉血栓形成、中风、动脉粥样硬化、血栓形成、冠心病或主动脉瓣狭窄和/或与含Lp(a)粒子的水平升高相关的任何其他疾病或病理。
应当理解,除了本文描述的某些递送方式之外,其他RNAi递送方式可以与本文描述的LPA RNAi试剂和治疗方法的实施方案结合使用,例如但不限于本文描述的那些和本领域中使用的那些。
本发明的LPA dsRNA试剂可以以有效降低细胞和/或对象中LPA多肽的水平的量和方式施用于对象。在本发明方法的一些实施方案中,将一种或更多种LPA dsRNA试剂施用于细胞和/或对象以治疗与LPA表达相关的疾病或病症。在一些实施方案中,本发明的方法包括向需要此类治疗的对象施用一种或更多种LPA dsRNA试剂以减轻对象中与LPA表达相关的疾病或病症。可以施用本发明的LPA dsRNA试剂或LPA反义多核苷酸试剂以降低体外、离体和体内细胞中的一种或更多种中的LPA表达。
在本发明的一些实施方案中,通过将LPA dsRNA试剂或LPA反义多核苷酸试剂递送
(例如引入)细胞中来降低细胞中LPA多肽的水平。靶向剂和方法可用于帮助将LPA dsRNA试剂或LPA反义多核苷酸剂递送至对象内的特定细胞类型、细胞亚型、器官、空间区域,和/或细胞内的亚细胞区域。LPA dsRNA试剂可以在本发明的某些方法中单独地或与一种或更多种另外的LPA dsRNA试剂组合施用。在一些实施方案中,向对象施用2、3、4或更多种独立选择的LPA dsRNA试剂。在本发明的某些实施方案中,将LPA dsRNA试剂与一种或更多种用于治疗LPA相关疾病或病症的另外的治疗方案联合施用于对象以治疗LPA相关疾病或病症。另外的治疗方案的非限制性实例是:施用一种或更多种本发明的LPA反义多核苷酸、施用非LPA dsRNA治疗剂和行为改变。可以在以下一个或更多个时间施用另外的治疗方案:在施用本发明的LPA dsRNA试剂之前、同时和之后。应当理解,本文所用的“同时”是指在零时间的5分钟内、零时间的10分钟内、零时间的30分钟内、零时间的45分钟内和零时间的60分钟内,其中“零时间”是向对象施用本发明的LPA dsRNA试剂的时间。非LPA dsRNA治疗剂的非限制性实例是:另外的治疗剂,如HMg Co-A还原酶抑制剂(他汀类)、依折麦布、PCSK-9抑制剂、CTEP抑制剂、靶向ANGPTL3的疗法、靶向APOC3的疗法和烟酸,或上述任何的组合。行为改变的非限制性实例是:饮食方案、咨询和锻炼方案。这些和其他治疗剂以及行为改变是本领域已知的,并且可用于治疗对象的LPA疾病或病症,并且还可以与一种或更多种本发明的LPA dsRNA试剂组合,向对象给药以治疗LPA疾病或病症。向细胞或对象施用以治疗LPA相关疾病或病症的本发明的LPA dsRNA试剂可以与一种或更多种其他治疗剂或活性成分以协同方式起作用,从而增加一种或更多种治疗剂或活性成分的有效性和/或增加LPA dsRNA试剂治疗LPA相关疾病或病症的有效性。
本发明的治疗方法包括施用LPA dsRNA试剂,可在LPA相关疾病或病症发作之前和/或当存在LPA相关疾病或病症时使用,包括在疾病或病症的早期、中期、晚期阶段以及任何这些阶段之前和之后的所有时间使用。本发明的方法还可以治疗先前已经接受过一种或更多种其他治疗剂和/或治疗活性成分的LPA相关疾病或病症治疗的对象,其中一种或更多种其他治疗剂和/或治疗活性成分在治疗对象的LPA相关疾病或病症方面不成功、成功性最小和/或不再成功。
载体编码的dsRNA
在本发明的某些实施方案中,可以使用载体将LPA dsRNA试剂递送到细胞中。LPAdsRNA试剂转录单位可包含在DNA或RNA载体中。用于将序列递送到细胞和/或对象中的此类编码转基因的载体的制备和使用是本领域公知的。可在本发明的方法中使用导致LPAdsRNA瞬时表达的载体,瞬时表达例如至少1、2、3、4、5、6、7、8、9、10小时或更多小时、1、2、3、4、5、6、7、8、9、10周或更多周。瞬时表达的长度可以使用基于以下要素的常规方法确定:例如但不限于所选的特定载体构建体和靶细胞和/或组织。此类转基因
可作为线性构建体、环状质粒或病毒载体引入,其可为整合或非整合载体。也可以构建转基因以使其作为染色体外质粒遗传(Gassmann,et al.,Proc.Natl.Acad.Sci.USA(1995)92:1292)。
LPA dsRNA试剂的一条或更多条单链可以从表达载体上的启动子转录。在要表达两条单独的链以产生例如dsRNA的情况下,可以使用例如转染或感染的方式将两个单独的表达载体共同引入细胞。在某些实施方案中,本发明的LPA dsRNA试剂的每条单独链都可以被包含在同一表达载体上的启动子转录。在本发明的某些实施方案中,LPA dsRNA试剂被表达为通过接头多核苷酸序列连接的反向重复多核苷酸,使得LPA dsRNA试剂具有茎环结构。
RNA表达载体的非限制性实例是DNA质粒或病毒载体。在本发明的实施方案中有用的表达载体可以与真核细胞相容。真核细胞表达载体在本领域是常规使用的,并且可从许多商业来源获得。LPA dsRNA表达载体的递送可以是全身性的,例如通过静脉内或肌肉内给药、通过给药至从对象移出的靶细胞然后将靶细胞重新引入对象,或通过允许引入所需靶细胞的任何其他方式来进行。
可包括在该方法的实施方案中的病毒载体系统包括但不限于:(a)腺病毒载体;(b)逆转录病毒载体,包括但不限于慢病毒载体、莫洛尼鼠白血病病毒等;(c)腺相关病毒载体;(d)单纯疱疹病毒载体;(e)SV40载体;(f)多瘤病毒载体;(g)乳头状瘤病毒载体;(h)小核糖核酸病毒载体;(i)痘病毒载体,例如正痘病毒载体,例如痘苗病毒载体或禽痘病毒载体,例如金丝雀痘或家禽痘病毒载体;(j)辅助依赖型或无肠腺病毒载体。用于重组表达LPAdsRNA试剂的构建体可以包含调节元件,例如启动子、增强子等,它们可以被选择以提供组成型或调节型/诱导型表达。病毒载体系统以及启动子和增强子的使用等在本领域是常规的并且可以与本文所述的方法和组合物结合使用。
本发明的某些实施方案包括使用病毒载体将LPA dsRNA试剂递送到细胞中。许多基于腺病毒的递送系统在本领域中常规用于递送至例如肺、肝、中枢神经系统、内皮细胞和肌肉。可用于本发明方法的病毒载体的非限制性实例是:AAV载体、痘病毒如痘苗病毒、改良安卡拉病毒(MVA)、NYVAC、禽痘如禽痘或金丝雀痘病毒。
本发明的某些实施方案包括使用载体将LPA dsRNA试剂递送到细胞中的方法,并且此类载体可以在药学上可接受的载体中,所述载体可以但不必包括其中嵌入基因递送载体的缓释基质。在一些实施方案中,用于递送LPA dsRNA的载体可以由重组细胞产生,并且本发明的药物组合物可以包括一种或更多种产生LPA dsRNA递送系统的细胞。
含有LPA dsRNA或ssRNA试剂的药物组合物
本发明的某些实施方案包括含有LPA dsRNA试剂或LPA反义多核苷酸试剂和药学上
可接受的载体的药物组合物的用途。包含LPA dsRNA试剂或LPA反义多核苷酸剂的药物组合物可用于本发明的方法中以降低细胞中的LPA基因表达,并可用于治疗LPA相关疾病或病症。此类药物组合物可以基于递送方式来配制。用于递送方式的制剂的非限制性实例是:配制用于皮下递送的组合物、配制用于通过肠胃外递送全身给药的组合物、配制用于静脉内(IV)递送的组合物、配制用于鞘内递送的组合物、配制用于直接递送至脑中的组合物等。可以使用一种或更多种方式施用本发明的药物组合物以将LPA dsRNA试剂或LPA反义多核苷酸试剂递送到细胞中,例如:表面(例如,通过透皮贴剂);肺部,例如通过吸入或吹入粉末或气雾剂,包括通过雾化器;气道内、鼻内、表皮和透皮、口服或肠胃外。肠胃外给药包括静脉内、动脉内、皮下、腹膜内或肌肉内注射或输注;表皮下,例如通过植入装置;或颅内,例如通过实质内;鞘内或心室内施用。LPA dsRNA试剂或LPA反义多核苷酸剂也可以直接递送至靶组织,例如直接递送至肝脏、直接递送至肾脏等。可以理解的是,“递送LPAdsRNA试剂”或“递送LPA反义多核苷酸试剂”到细胞中分别包括递送LPA dsRNA试剂或LPA反义多核苷酸剂、直接在细胞中表达LPA dsRNA试剂以及从递送到细胞中的编码载体表达LPA dsRNA试剂,或使得LPA dsRNA或LPA反义多核苷酸试剂出现在细胞中的任何合适的方式。制剂的制备和使用以及用于递送抑制性RNA的手段是本领域公知的和常规使用的。
如本文所用,“药物组合物”包含药理学有效量的本发明的LPA dsRNA试剂或LPA反义多核苷酸剂和药学上可接受的载体。术语“药学上可接受的载体”是指用于施用治疗剂的载体。此类载体包括但不限于盐水、缓冲盐水、葡萄糖、水、甘油、乙醇及其组合。该术语明确排除细胞培养基。对于口服给药的药物,药学上可接受的载体包括但不限于药学上可接受的赋形剂,例如惰性稀释剂、崩解剂、黏合剂、润滑剂、甜味剂、调味剂、着色剂和防腐剂。合适的惰性稀释剂包括碳酸钠和碳酸钙、磷酸钠和磷酸钙以及乳糖,而玉米淀粉和藻酸是合适的崩解剂。黏合剂可包括淀粉和明胶,而润滑剂(如果存在)通常是硬脂酸镁、硬脂酸或滑石粉。如果需要,片剂可以用例如单硬脂酸甘油酯或二硬脂酸甘油酯之类的材料包衣,以延迟在胃肠道中的吸收。包含在药物制剂中的试剂在下文进一步描述。如本文所用的术语,例如“药理学有效量”、“治疗有效量”和“有效量”,是指本发明的LPA dsRNA试剂或LPA反义多核苷酸试剂产生预期的药理学、治疗或预防结果的量。例如,如果与疾病或障碍相关的可测量参数至少降低10%时,则认为给定的临床治疗有效,那么用于治疗该疾病或病症的药物的治疗有效量是使该参数降低至少10%所需的量。例如,治疗有效量的LPA dsRNA试剂或LPA反义多核苷酸剂可以将LPA多肽水平降低至少10%。药物组合物可以包含这样的dsRNAi试剂,其包括例如表1中所显示的双链体。在其他一些实施方案中,这样的dsRNAi试剂包括表1中双链体的变体。
有效量
在一些方面,本发明的方法包括将细胞与有效量的LPA dsRNA试剂或LPA反义多核苷酸试剂接触以减少所接触细胞中的LPA基因表达。本发明方法的某些实施方案包括以有效降低LPA基因表达和治疗对象的LPA相关疾病或病症的量向对象施用LPA dsRNA试剂或LPA反义多核苷酸剂。就减少LPA的表达和/或用于治疗LPA相关疾病或病症而言,所使用的“有效量”是实现所需生物学效果所必需或足够的量。例如,治疗LPA相关疾病或病症的LPA dsRNA试剂或LPA反义多核苷酸试剂的有效量可以是:(i)减缓或停止疾病或病症的进展所需的量;(ii)逆转、减少或消除疾病或病症的一种或更多种症状。在本发明的一些方面,有效量是当施用于需要治疗LPA相关疾病或病症的对象时,导致疾病或病症的预防和/或治疗的治疗响应的LPA dsRNA试剂或LPA反义多核苷酸剂的量。根据本发明的一些方面,有效量是本发明的LPA dsRNA试剂或LPA反义多核苷酸试剂当与针对LPA相关疾病或病症的另一种治疗性治疗组合或共同施用时,导致预防和/或治疗该疾病或病症的治疗响应的量。在本发明的一些实施方案中,用本发明的LPA dsRNA试剂或LPA反义多核苷酸试剂治疗对象的生物学效应可以是由LPA相关疾病或病症引起的症状的改善和/或完全消除。在本发明的一些实施方案中,生物学效应是LPA相关疾病或病症的完全消除,例如通过指示对象没有LPA相关疾病或病症的诊断测试来证明。可检测的生理症状的非限制性实例包括在施用本发明的试剂后对象肝脏中脂质积累的减少。其他评估LPA相关疾病或病症状态的本领域已知方式可用于确定本发明的试剂和/或方法对LPA相关疾病或病症的影响。
通常在临床试验中确定将LPA多肽降低至治疗LPA相关疾病或病症的水平的LPAdsRNA试剂或LPA反义多核苷酸试剂的有效量,这样的临床试验在盲法研究中为测试人群与对照人群建立有效剂量。在一些实施方案中,有效量是导致所需响应的量,例如减少细胞、组织和/或患有疾病或病症的对象中的LPA相关疾病或病症的量。因此,用于治疗可通过降低LPA多肽治疗的LPA相关疾病或病症的LPA dsRNA试剂或LPA反义多核苷酸试剂的有效量可以是这样的量:当施用时,将对象中LPA多肽的量降低至低于在未施用LPA dsRNA试剂或LPA反义多核苷酸剂的情况下将存在于细胞、组织和/或对象中的量。在本发明的某些方面,存在于未接触或施用过本发明的LPA dsRNA试剂或LPA反义多核苷酸剂的细胞、组织和/或对象中的LPA多肽和/或LPA基因表达的水平被称为“对照”量。在本发明方法的一些实施方案中,对象的对照量是对象的治疗前量;换言之,对象在施用LPA试剂之前的水平可以是该对象的对照水平,并且用于与其在向对象施用siRNA后的LPA多肽和/或LPA基因表达水平相比较。在治疗LPA相关疾病或病症的情况下,期望的响应可以是减少或消除细胞、组织和/或对象中疾病或病症的一种或更多种症状。减少或消除可以是暂时的,也可以是永久性的。应当理解,可以使用确定LPA多肽、LPA基因表达、症状评估、临床测试等的方法来监测LPA相关疾病或病症的状态。在本发明的一些方面,对治疗LPA相关疾病或病症的期望响应是延迟疾病或病症的发作或甚至预防疾病或病症的发作。
降低LPA多肽的化合物的有效量也可以通过以下方式确定:评估施用LPA dsRNA试
剂或LPA反义多核苷酸试剂对细胞或对象的生理作用,例如施用后LPA相关疾病或病症的减少。对象的测定和/或症状监测可用于确定本发明的LPA dsRNA试剂或LPA反义多核苷酸剂的功效(其可以在本发明的药物化合物中给药),并确定对治疗是否有响应。一个非限制性实例是一种或更多种本领域已知的血清脂质谱测试。另一个非限制性示例是:在用本发明的LPA dsRNA试剂治疗对象之前和之后,可以使用一种或更多种本领域已知的肝功能测试来确定对象的LPA相关疾病或病症的状态。在另一个非限制性实例中,使用一种或更多种本领域已知的肝脏中胆固醇积累测试来确定对象中LPA相关疾病的状态。在该实施例中,疾病包括胆固醇积累,并且该测试用于确定在用本发明的LPA dsRNA试剂治疗对象之前和之后对象中的胆固醇水平。
本发明的一些实施方案包括确定向对象施用的本发明的dsRNA试剂或LPA反义多核苷酸试剂来治疗LPA相关疾病或病症之功效的方法,其通过评估和/或监测对象中LPA相关疾病或病症的一种或更多种“生理特征”来进行。LPA相关疾病或病症的生理特征的非限制性实例是对象的血清LPA水平、对象的血清脂质水平、对象的低密度脂蛋白水平、对象的HDL水平、对象的LDL:HDL比率、对象的甘油三酯水平、对象肝脏中存在的脂肪、身体症状等。确定这种生理特征的标准方法是本领域已知的,包括但不限于血液测试、成像研究、身体检查等。
可以理解的是,可以至少部分地基于这种对对象确定的疾病和/或病症状态和/或生理特征的测定结果来修改向对象施用的LPA dsRNA试剂或LPA反义多核苷酸剂的量。治疗量可以通过例如以下方式改变:通过改变施用LPA dsRNA试剂或LPA反义多核苷酸剂的组合物、通过改变给药途径、通过改变给药时间等,来增加或减少LPA-dsRNA试剂或LPA反义多核苷酸试剂的量。LPA dsRNA试剂或LPA反义多核苷酸剂的有效量将随着所治疗的特定病症、所治疗对象的年龄和身体状况、病情的严重程度、治疗的持续时间、共同治疗的性质(如果有的话)、具体的给药途径以及健康从业者知识和专业知识范围内的其他因素而变化。例如,有效量可取决于对治疗LPA相关疾病或病症有效的LPA多肽水平和/或LPA基因表达的所需水平。技术人员可以凭经验确定用于本发明方法的特定LPA dsRNA试剂或LPA反义多核苷酸试剂的有效量,而无需过度实验。结合本文提供的教导,通过从本发明的多种LPA dsRNA试剂或LPA反义多核苷酸试剂中进行选择,并权衡例如效力、相对生物利用度、患者体重、不良副作用的严重程度和优选的给药方式等因素,可以规划有效的预防性或治疗性治疗方案以有效治疗特定对象。如在本发明的实施方案中使用的,本发明的LPA dsRNA试剂或LPA反义多核苷酸试剂的有效量可以是当与细胞接触时在细胞中产生所需生物学效应的量。
应当认识到,LPA基因沉默可以在表达LPA的任何细胞中通过组成型或通过基因组工程进行,并通过任何合适的测定来确定。在本发明的一些实施方案中,通过施用本发明的LPA dsRNA试剂,LPA基因表达降低至少5%、6%、7%、8%、9%、10%、15%、20%、25%、
30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%或100%。在本发明的一些实施方案中,通过施用本发明的LPA dsRNA试剂,LPA基因表达减少5%至10%、5%至25%、10%至50%、10%至75%、25%至75%、25%至100%或50%至100%。
给药
LPA dsRNA试剂和LPA反义多核苷酸试剂以足以抑制LPA基因表达的剂量在药物组合物中递送。在本发明的某些实施方案中,LPA dsRNA试剂或LPA反义多核苷酸剂的剂量为每千克接受者体重每天0.01至200.0毫克,一般为每天1至50mg/kg体重、5至40mg/kg体重、10至30mg/kg体重、1至20mg/kg体重、1至10mg/kg体重、4至15mg/kg体重,包括端值。例如,LPA dsRNA试剂或LPA反义多核苷酸试剂的每单次给药可以以从约0.01mg/kg、0.05mg/kg、0.1mg/kg、0.2mg/kg、0.3mg/kg、0.4mg/kg、0.5mg/kg、1mg/kg、1.1mg/kg、1.2mg/kg、1.3mg/kg、1.4mg/kg、1.5mg/kg、1.6mg/kg、1.7mg/kg、1.8mg/kg、1.9mg/kg、2mg/kg、2.1mg/kg、2.2mg/kg、2.3mg/kg、2.4mg/kg、2.5mg/kg、2.6mg/kg、2.7mg/kg、2.8mg/kg、2.9mg/kg、3.0mg/kg、3.1mg/kg、3.2mg/kg、3.3mg/kg、3.4mg/kg、3.5mg/kg、3.6mg/kg、3.7mg/kg、3.8mg/kg、3.9mg/kg、4mg/kg、4.1mg/kg、4.2mg/kg、4.3mg/kg、4.4mg/kg、4.5mg/kg、4.6mg/kg、4.7mg/kg、4.8mg/kg、4.9mg/kg、5mg/kg、5.1mg/kg、5.2mg/kg、5.3mg/kg、5.4mg/kg、5.5mg/kg、5.6mg/kg、5.7mg/kg、5.8mg/kg、5.9mg/kg、6mg/kg、6.1mg/kg、6.2mg/kg、6.3mg/kg、6.4mg/kg、6.5mg/kg、6.6mg/kg、6.7mg/kg、6.8mg/kg、6.9mg/kg、7mg/kg、7.1mg/kg、7.2mg/kg、7.3mg/kg、7.4mg/kg、7.5mg/kg、7.6mg/kg、7.7mg/kg、7.8mg/kg、7.9mg/kg、8mg/kg、8.1mg/kg、8.2mg/kg、8.3mg/kg、8.4mg/kg、8.5mg/kg、8.6mg/kg、8.7mg/kg、8.8mg/kg、8.9mg/kg、9mg/kg、9.1mg/kg、9.2mg/kg、9.3mg/kg、9.4mg/kg、9.5mg/kg、9.6mg/kg、9.7mg/kg、9.8mg/kg、9.9mg/kg、10mg/kg、11mg/kg、12mg/kg、13mg/kg、14mg/kg、15mg/kg、16mg/kg、17mg/kg、18mg/kg、19mg/kg、20mg/kg、21mg/kg、22mg/kg、23mg/kg、24mg/kg、25mg/kg、26mg/kg、27mg/kg、28mg/kg、29mg/kg、30mg/kg、31mg/kg、32mg/kg、33mg/kg、34mg/kg、35mg/kg、36mg/kg、37mg/kg、38mg/kg、39mg/kg、40mg/kg、41mg/kg、42mg/kg、43mg/kg、44mg/kg、45mg/kg、46mg/kg、47mg/kg、48mg/kg、49mg/kg至50mg/kg体重的量来施用。
在确定本发明的LPA dsRNA试剂的递送剂量和时间时可以考虑多种因素。递送的LPAdsRNA试剂或LPA反义多核苷酸剂的绝对量将取决于多种因素,包括共同治疗、剂量数和个体对象参数,包括年龄、身体状况、体格大小和体重。这些是本领域普通技术人员众所公知的因素,并且可以通过常规实验解决。在一些实施方案中,可以使用最大剂量,即根据合理的医学判断的最高安全剂量。
在一些实施方案中,本发明的方法可包括向对象施用1、2、3、4、5、6、7、8、9、10或更多个剂量的LPA dsRNA试剂或LPA反义多核苷酸试剂。在一些情况下,可以至少每天、每隔一天、每周、每隔一周、每月等向对象施用药物化合物(例如,包含LPA dsRNA试剂或包含LPA反义多核苷酸剂)的剂量,可以每天给药一次或每天给药多于一次,例如在一个24小时周期内给药2、3、4、5或更多次。本发明的药物组合物可以每天给药一次;或者LPA dsRNA试剂或LPA反义多核苷酸试剂可以在一天中以适当的间隔以两个、三个或更多个亚剂量给药,或者甚至使用连续输注或通过控释制剂递送。在本发明方法的一些实施方案中,将本发明的药物组合物每天一次或更多次、每周一次或更多次、每月一次或更多次或每年一次或更多次施用给对象。
在某些方面,本发明的方法包括单独施用药物化合物;与一种或更多种其他LPA dsRNA试剂或LPA反义多核苷酸试剂组合;和/或与对患有LPA相关疾病或病症的对象施用的其他药物疗法或治疗活动或方案组合。药物化合物可以以药物组合物的形式给药。本发明方法中使用的药物组合物可以是无菌的,并且含有一定量的LPA dsRNA试剂或LPA反义多核苷酸试剂,其会将LPA多肽的水平降低到足以在适合施用于对象的重量或体积单位中产生所需响应的水平。可以根据不同参数选择向对象施用的包含LPA dsRNA试剂或LPA反义多核苷酸剂药物组合物的剂量以降低LPA蛋白水平,特别是根据所使用的给药方式和对象的状态来进行选择。其他因素包括所需的治疗时间。如果对象在初始剂量下的响应不足,则可以在患者耐受性允许的范围内采用更高的剂量(或通过不同的、更局部的递送途径有效地提高剂量)。
治疗
如本文所用的,术语“预防”或“进行预防”,当用于指将受益于LPA基因表达降低的疾病、病症或其病况时,是指对象发生与此类疾病、病症或病况相关的症状的可能性降低,所述与此类疾病、病症或病况相关的症状是例如心血管疾病,其中所述心血管疾病包括伯格氏病(Berger’s disease)、外周动脉疾病、冠状动脉疾病、代谢综合征、急性冠脉综合征、主动脉瓣狭窄、主动脉瓣反流、主动脉夹层、视网膜动脉阻塞、脑血管疾病、肠系膜缺血、肠系膜上动脉阻塞、肾动脉狭窄、稳定型/不稳定型心绞痛、急性冠脉综合征、杂合子或纯合子家族性高胆固醇血症、高载脂蛋白β脂蛋白血症、脑血管动脉粥样硬化、脑血管疾病和静脉血栓形成、中风、动脉粥样硬化、血栓形成、冠心病或主动脉瓣狭窄和/或与含Lp(a)粒子的水平升高相关的任何其他疾病或病理。在这样的情况下发生此类疾病的可能性被降低:例如,当个体具有一种或更多种心血管疾病风险因素,但未发生心血管疾病或仅发生严重程度较低的心血管疾病的情况下,其相对于具有相同风险因素且未接受本文所述治疗的人群而言,未能发展出相关疾病、病症或病症,或与此类疾病、病症或病症相关的症状的发展程度
降低(例如,在临床上患有该疾病或病症的量表上降低至少约10%),或延迟症状的表现(例如,延迟数天、数周、数月或数年),则被认为是有效的预防。
对于LPA相关疾病和病症,其中LPA多肽的水平的降低可有效治疗该疾病或病症,可以使用本发明的方法和LPA dsRNA试剂来治疗以抑制LPA表达。可以用本发明的LPAdsRNA试剂或LPA反义多核苷酸试剂和本发明的治疗方法治疗的疾病和病症的实例包括但不限于:伯格氏病(Berger’s disease)、外周动脉疾病、冠状动脉疾病、代谢综合征、急性冠脉综合征、主动脉瓣狭窄、主动脉瓣反流、主动脉夹层、视网膜动脉阻塞、脑血管疾病、肠系膜缺血、肠系膜上动脉阻塞、肾动脉狭窄、稳定型/不稳定型心绞痛、急性冠脉综合征、杂合子或纯合子家族性高胆固醇血症、高载脂蛋白β脂蛋白血症、脑血管动脉粥样硬化、脑血管疾病和静脉血栓形成、中风、动脉粥样硬化、血栓形成、冠心病或主动脉瓣狭窄和/或与含Lp(a)粒子的水平升高相关的任何其他疾病或病理。此类疾病和病症在本文中可称为“LPA相关疾病和病症”和“由LPA引起和/或调节的疾病和病症”。
在本发明的某些方面,可以在诊断LPA相关疾病或病症之前或之后的一个或更多个时间向对象施用本发明的LPA dsRNA试剂或LPA反义多核苷酸试剂。在本发明的一些方面,对象处于患有或发展LPA相关疾病或病症的风险中。与发展LPA相关疾病或病症的对照风险相比,有发展LPA相关疾病或病症风险的对象是发展LPA相关疾病或病症的可能性提高的对象。在本发明的一些实施方案中,与风险的对照水平相比,风险水平在统计上是显著的。有风险的对象可包括,例如:是或将是具有预先存在的疾病和/或遗传异常的对象,其使得该对象比没有预先存在的疾病或遗传异常的对照对象更易患LPA相关疾病或病症;具有LPA相关疾病或病症的家族和/或个人病史的对象;以及先前已接受LPA相关疾病或病症治疗的对象。应当理解,使对象对LPA相关疾病或病症更易感的预先存在的疾病和/或遗传异常可以是这样的疾病或遗传异常:当存在时,其先前已被确定为与发展LPA相关疾病或病症的更高可能性具有相关关系。
应当理解,可以基于个体对象的医学状况向对象施用LPA dsRNA试剂或LPA反义多核苷酸试剂。例如,为对象提供的医疗保健可以评估从对象获得的样品中测量的LPA水平,并确定通过施用本发明的LPA dsRNA试剂或LPA反义多核苷酸试剂来降低对象的LPA水平是可期望的。在一个非限制性实例中,可以从对象获得生物样品,例如血液或血清样品,并且在样品中确定对象的LPA水平。向对象施用LPA dsRNA试剂或LPA反义多核苷酸试剂,并且在给药后从对象获得血液或血清样品,并且使用该样品测定LPA水平,并将该结果与对象给药前(先前)样品中确定的结果进行比较。与给药前水平相比,随后样品中对象的LPA水平降低则表明所施用的LPA dsRNA试剂或LPA反义多核苷酸试剂在降低对象的LPA水平方面的功效。在一个非限制性实例中,血液中的Lp(a)水平可以被认为是LPA相关病症的生理特征,即使对象没有被诊断为患有LPA相关疾病,例如本文公开的疾病。医疗保健
提供者可以监测对象血液中的Lp(a)水平的变化,作为施用的本发明的LPA dsRNA试剂或LPA反义多核苷酸剂的功效的量度。
本发明方法的某些实施方案包括调整治疗,所述治疗包括至少部分地基于对对象中由治疗引起的LPA相关疾病或病症一种或更多种生理特征的变化的评估,来向对象施用本发明的dsRNA试剂或LPA反义多核苷酸试剂。例如,在本发明的一些实施方案中,可以确定对对象施用的本发明的dsRNA试剂或LPA反义多核苷酸剂的作用,并用于帮助调节随后向对象施用的本发明的dsRNA试剂或LPA反义多核苷酸剂的量。在一个非限制性实例中,对对象施用本发明的dsRNA试剂或LPA反义多核苷酸试剂,并在施用后测定对象在血液中的Lp(a)水平;并且至少部分基于所确定的水平,确定是否需要更高量的dsRNA试剂或LPA反义多核苷酸试剂以提高所施用试剂的生理作用,例如降低或进一步降低对象在血液中的Lp(a)水平。在另一个非限制性实例中,向对象施用本发明的dsRNA试剂或LPA反义多核苷酸剂,并在给药后确定对象在血液中的Lp(a)水平,并且至少部分地基于所确定的水平,预期向对象施用更低量的dsRNA试剂或LPA反义多核苷酸试剂。
因此,本发明的一些实施方案包括评估由对象先前治疗引起的一种或更多种生理特征的变化,以调整随后施用于对象的本发明的dsRNA试剂或LPA反义多核苷酸剂的量。本发明方法的一些实施方案包括对LPA相关疾病或病症的生理特征的1、2、3、4、5、6或更多次测定;评估和/或监测施用的本发明的LPA dsRNA试剂或LPA反义多核苷酸试剂的功效;并任选地使用所测定的结果来调整以下一项或更多项:本发明的dsRNA试剂或LPA反义多核苷酸试剂治疗对象中LPA相关疾病或病症的剂量、给药方案和/或给药频率。在本发明方法的一些实施方案中,向对象施用有效量的本发明的dsRNA试剂或LPA反义多核苷酸试剂的期望结果是:与为对象确定的先前在血液中的Lp(a)水平相比,对象血液中的Lp(a)水平降低;对象在血液中的Lp(a)水平处于正常范围内。
如本文所用,术语“治疗”、“治疗性”或“治疗的”当用于LPA相关疾病或病症时可指预防性治疗、降低对象发展LPA相关疾病或病症的可能性,并且也可以指在对象已经发展出LPA相关疾病或病症之后为了消除或降低LPA相关疾病或病症的水平而进行的治疗、防止LPA相关疾病或病症变得更严重,和/或与在不存在降低对象中LPA多肽水平的疗法的情况下的对象相比,减缓对象中LPA相关疾病或病症的进展。
本发明的试剂、组合物和方法的某些实施方案可用于抑制LPA基因表达。如本文所用,关于LPA基因的表达,术语“抑制”、“沉默”、“减少”、“下调”和“敲低”是指例如通过以下一种或更多种情况改变LPA基因的表达:分别与由LPA基因转录的RNA的对照水平、由mRNA翻译的LPA的对照水平相比,当细胞、细胞群、组织、器官或对象与本发明的LPA dsRNA试剂或LPA反义多核苷酸试剂接触(例如,用其处理)时,其中由基因转录的RNA的水平、表达的LPA的水平,以及由细胞、细胞群、组织、器官或对象中的mRNA翻译的LPA多肽、
蛋白质或蛋白质亚基的水平降低。在一些实施方案中,对照水平是未接触LPA dsRNA试剂或LPA反义多核苷酸试剂(例如用其处理)的细胞、组织、器官或对象中的水平。
施用方法
LPA dsRNA试剂或LPA反义多核苷酸剂的多种给药途径可用于本发明的方法。特定递送模式的选择将至少部分取决于所治疗的特定病症和治疗功效所需的剂量。一般而言,本发明的方法可以使用医学上可接受的任何给药模式来实施,这意味着产生LPA相关疾病或病症的有效治疗水平而不引起临床上不可接受的副作用的任何模式。在本发明的一些实施方案中,LPA dsRNA试剂或LPA反义多核苷酸剂可以通过口服、肠内、黏膜、皮下和/或肠胃外途径施用。术语“肠胃外”包括皮下、静脉内、鞘内、肌内、腹膜内和胸骨内注射或输注技术。其他途径包括但不限于鼻(例如,通过胃鼻管)、经皮、阴道、直肠、舌下和吸入。本发明的递送途径可包括鞘内、心室内或颅内。在本发明的一些实施方案中,LPA dsRNA试剂或LPA反义多核苷酸剂可以放置在缓释基质中并通过将基质放置在对象中来施用。在本发明的一些方面,LPA dsRNA试剂或LPA反义多核苷酸剂可以使用涂覆有靶向特定细胞或细胞器的递送剂的纳米颗粒递送至对象细胞。多种递送方式、方法、试剂是本领域已知的。递送方法和递送剂的非限制性实例在本文别处另外提供。在本发明的一些方面,关于LPA dsRNA试剂或LPA反义多核苷酸试剂的术语“递送”可以是指:向细胞或对象施用一种或更多种“裸”LPA dsRNA试剂或LPA反义多核苷酸剂序列。在本发明的某些方面,“递送”是指通过转染方式给予细胞或对象、将包含LPA dsRNA试剂或LPA反义多核苷酸试剂的细胞递送给对象、将编码LPA dsRNA试剂或LPA反义多核苷酸试剂的载体递送到细胞和/或对象等中。使用转染方式递送LPA dsRNA试剂或LPA反义多核苷酸剂可包括向细胞和/或对象施用载体。
在本发明的一些方法中,一种或更多种LPA dsRNA试剂或LPA反义多核苷酸试剂可以以制剂形式给药,也可以在药学上可接受的溶液中给药,其通常可以含有药学上可接受浓度的盐、缓冲剂、防腐剂、相容的载体、佐剂和任选的其他治疗成分。在本发明的一些实施方案中,LPA dsRNA试剂或LPA反义多核苷酸剂可以与另一种治疗剂一起配制成用于同时给药。根据本发明的方法,LPA dsRNA试剂或LPA反义多核苷酸试剂可以以药物组合物的形式给药。通常,药物组合物包含LPA dsRNA试剂或LPA反义多核苷酸剂和任选的药学上可接受的载体。药学上可接受的载体是本领域普通技术人员公知的。如本文所用,药学上可接受的载体是指不干扰活性成分生物活性(例如,LPA dsRNA试剂或LPA反义多核苷酸剂抑制细胞或对象中LPA基因表达的能力)有效性的无毒材料。施用和递送用于治疗用途的dsRNA试剂或LPA反义多核苷酸试剂的多种方法是本领域已知的并且可用于本发明的方法中。
药学上可接受的载体包括稀释剂、填充剂、盐、缓冲剂、稳定剂、增溶剂和本领域公知的其他材料。示例性的药学上可接受的载体描述于美国专利No.5,211,657中,而其他载体是本领域技术人员已知的。这种制剂通常可以含有盐、缓冲剂、防腐剂、相容的载体和任选的其他治疗剂。用于医药时,该盐应当是药学上可接受的,但非药学上可接受的盐可以方便地用于制备其药学上可接受的盐,不排除在本发明的范围之外。此类药理学和药学上可接受的盐包括但不限于由以下酸制备的盐:盐酸、氢溴酸、硫酸、硝酸、磷酸、马来酸、乙酸、水杨酸、柠檬酸、甲酸、丙二酸、琥珀酸等。此外,药学上可接受的盐可以制备为碱金属盐或碱土金属盐,例如钠盐、钾盐或钙盐。
本发明方法的一些实施方案包括将一种或更多种LPA dsRNA试剂或LPA反义多核苷酸试剂直接施用于组织。在一些实施方案中,施用化合物的组织是其中存在或可能出现LPA相关疾病或病症的组织,其非限制性实例是肝脏或肾脏。直接组织给药可以通过直接注射或其他方式实现。许多口服递送的化合物自然进入并通过肝脏和肾脏,本发明的治疗方法的一些实施方案包括向对象口服施用一种或更多种LPA dsRNA试剂。LPA dsRNA试剂或LPA反义多核苷酸剂,单独或与其他治疗剂联合,可以施用一次,或者它们可以多次施用。如果多次给药,LPA dsRNA试剂或LPA反义多核苷酸剂可以通过不同途径给药。例如,虽然不打算限制,第一次(或前几次)给药可以通过皮下方式进行,并且一次或更多次额外给药可以是口服和/或全身给药。
对于其中希望全身性施用LPA dsRNA试剂或LPA反义多核苷酸剂的本发明实施方案,可以配制LPA dsRNA试剂或LPA反义多核苷酸剂用于通过注射例如通过推注或连续输注肠胃外施用。注射制剂可以以单位剂型存在,例如安瓿或多剂量容器,其添加或不添加防腐剂。LPA dsRNA试剂制剂(也称为药物组合物)可采用油性或水性载体中的混悬液、溶液或乳液等形式,并且可含有配制剂,例如混悬剂、稳定剂和/或分散剂。
肠胃外给药的制剂包括无菌水溶液或非水溶液、混悬液和乳液。非水溶剂的例子是丙二醇、聚乙二醇、植物油如橄榄油和可注射的有机酯如油酸乙酯。水性载体包括水、酒精/水溶液、乳液或混悬液,包括盐水和缓冲介质。肠胃外载体包括氯化钠溶液、林格氏葡萄糖溶液、葡萄糖和氯化钠溶液、乳酸林格氏液或固定油。静脉内赋形剂包括流体和营养补充剂、电解质补充剂(例如基于林格氏葡萄糖溶液的那些)等。也可以存在防腐剂和其他添加剂,例如抗微生物剂、抗氧化剂、螯合剂和惰性气体等。其他形式的给药,例如静脉给药,将导致较低的剂量。如果对象在初始剂量下的反应不足,则可以在患者耐受性允许的范围内采用更高的剂量(或通过不同的、更局部的递送途径有效地提高剂量)。可以根据需要每天使用多次剂量以实现一种或更多种LPA dsRNA试剂或LPA反义多核苷酸试剂的适当全身或局部水平,并实现LPA蛋白水平适当降低。
在其他实施方案中,本发明的方法包括使用递送载体,例如生物相容性微粒、纳米颗
粒或适合植入受体例如对象的植入物。PCT公开WO95/24929(通过引用并入本文)中描述了可根据该方法使用的示例性可生物降解植入物,其描述了用于包含生物大分子的生物相容的、可生物降解的聚合物基质。
不可生物降解的和可生物降解的聚合物基质都可用于本发明的方法中,以将一种或更多种LPA dsRNA试剂或LPA反义多核苷酸试剂递送给对象。在一些实施方案中,基质可以是可生物降解的。基质聚合物可以是天然或合成聚合物。可以基于期望释放的时间段来选择聚合物,通常在几小时到一年或更长时间的数量级。通常,可以使用在几小时到三到十二个月之间的一段时间内的释放。聚合物任选地呈水凝胶形式,其可以吸收高达其重量约90%的水,并且还任选地与多价离子或其他聚合物交联。
通常,LPA dsRNA试剂或LPA反义多核苷酸试剂在本发明的一些实施方案中可以使用可生物降解的植入物通过扩散或通过聚合物基质的降解来递送。用于这种用途的示例性合成聚合物是本领域公知的。使用本领域已知的方法,可生物降解的聚合物和不可生物降解的聚合物可用于递送LPA dsRNA试剂或LPA反义多核苷酸试剂。生物黏附聚合物如可生物侵蚀的水凝胶(H.S.Sawhney,C.P.Pathak and J.A.Hubell in Macromolecules,1993,26,581-587)也可用于递送LPA dsRNA试剂或LPA反义多核苷酸试剂,以治疗LPA相关疾病或病症。其他合适的递送系统可以包括定时释放、延迟释放或持续释放递送系统。此类系统可避免重复施用LPA dsRNA试剂或LPA反义多核苷酸剂,从而提高对象和医疗保健专业人员的便利性。许多类型的释放递送系统是可用的并且是本领域普通技术人员已知的。见例如美国专利No.5,075,109、4,452,775、4,675,189、5,736,152、3,854,480、5,133,974和5,407,686。此外,可以使用基于泵的硬件输送系统,其中一些也适用于植入。
长期持续释放植入物的使用可以适用于对象的预防性治疗和具有发生复发性LPA相关疾病或病症的风险的对象。如本文所用,长期释放是指将植入物构建和布置成以至少长达10天、20天、30天、60天、90天、六个月、一年或更长时间递送治疗水平的LPA dsRNA试剂或LPA反义多核苷酸试剂。长期持续释放植入物是本领域普通技术人员众所公知的并且包括上述的一些释放系统。
LPA dsRNA试剂或LPA反义多核苷酸试剂的治疗制剂可以通过将具有所需纯度的分子或化合物与任选的药学上可接受的载体、赋形剂或稳定剂[Remington's Pharmaceutical Sciences 21st edition,(2006)]以冻干制剂或水溶液的形式混合来制备用于储存。可接受的载体、赋形剂或稳定剂在所采用的剂量和浓度下对接受者是无毒的,并且包括缓冲剂,例如磷酸盐、柠檬酸盐和其他有机酸;抗氧化剂,包括抗坏血酸和蛋氨酸;防腐剂(例如十八烷基二甲基苄基氯化铵;六甲铵氯化物;苯扎氯铵、苄索氯铵;苯酚、丁醇或苯甲醇;对羟基苯甲酸酯类,例如对羟基苯甲酸甲酯或丙酯;邻苯二酚;间苯二酚;环己醇;3-戊醇;和间甲酚);低分子量(少于约10个残基)多肽;蛋白质,例如血清白蛋白、明胶或免疫球蛋白;
亲水性聚合物,如聚乙烯吡咯烷酮;氨基酸,例如甘氨酸、谷氨酰胺、天冬酰胺、组氨酸、精氨酸或赖氨酸;单糖、二糖和其他碳水化合物,包括葡萄糖、甘露糖或糊精;螯合剂如EDTA;蔗糖、甘露糖醇、海藻糖或山梨糖醇等糖类;形成盐的反离子,如钠;金属配合物(例如,锌-蛋白质配合物);和/或非离子表面活性剂,例如或聚乙二醇(PEG)。
细胞、对象和对照
本发明的方法可以与细胞、组织、器官和/或对象结合使用。在本发明的一些方面,对象是人或脊椎动物哺乳动物,包括但不限于狗、猫、马、牛、山羊、小鼠、大鼠和灵长类动物,例如猴。因此,本发明可用于治疗人和非人对象的LPA相关疾病或病症。在本发明的一些方面,对象可以是农场动物、动物园动物、驯养动物或非驯养动物,并且本发明的方法可用于兽医预防和治疗方案。在本发明的一些实施方案中,对象是人并且本发明的方法可用于人预防和治疗方案。
可应用本发明的对象的非限制性实例是被诊断患有、怀疑患有或有风险患有与以下疾病或病症相关的疾病或病症的对象:高于期望的LPA表达,也称为“升高的LPA表达水平”。与高于期望水平的LPA表达相关的疾病和病症的非限制性实例在本文别处描述。本发明的方法可应用于在治疗时已被诊断为患有该疾病或病症的对象、与高于期望的LPA表达相关的对象,或被认为处于患有或发展与高于期望的LPA表达相关的疾病或病症的风险中的对象。在本发明的一些方面,与高于期望的LPA表达水平相关的疾病或病症是急性疾病或病症;在本发明的某些方面,与高于期望的LPA表达水平相关的疾病或病症是慢性疾病或病症。
在一个非限制性实例中,将本发明的LPA dsRNA试剂施用于被诊断心血管疾病,其中所述心血管疾病包括伯格氏病(Berger’s disease)、外周动脉疾病、冠状动脉疾病、代谢综合征、急性冠脉综合征、主动脉瓣狭窄、主动脉瓣反流、主动脉夹层、视网膜动脉阻塞、脑血管疾病、肠系膜缺血、肠系膜上动脉阻塞、肾动脉狭窄、稳定型/不稳定型心绞痛、急性冠脉综合征、杂合子或纯合子家族性高胆固醇血症、高载脂蛋白β脂蛋白血症、脑血管动脉粥样硬化、脑血管疾病和静脉血栓形成、中风、动脉粥样硬化、血栓形成、冠心病或主动脉瓣狭窄和/或与含Lp(a)粒子的水平升高相关的任何其他疾病或病理。本发明的方法可应用于在治疗时已被诊断为患有该疾病或病症的对象,或被认为有患或发展该疾病或病症的风险的对象。
在另一个非限制性实例中,将本发明的LPA dsRNA试剂施用以治疗指因肾素-血管紧张素-醛固酮系统(RAAS)激活导致或与其相关的疾病或障碍,或其症状或进展响应于RAAS
失活的疾病或障碍。术语“LPA相关疾病”包括因LPA表达降低而受益的疾病、障碍或病症。这类疾病通常与高的血压相关。LPA相关疾病的非限制实例包括心血管疾病,其中所述心血管疾病包括伯格氏病(Berger’s disease)、外周动脉疾病、冠状动脉疾病、代谢综合征、急性冠脉综合征、主动脉瓣狭窄、主动脉瓣反流、主动脉夹层、视网膜动脉阻塞、脑血管疾病、肠系膜缺血、肠系膜上动脉阻塞、肾动脉狭窄、稳定型/不稳定型心绞痛、急性冠脉综合征、杂合子或纯合子家族性高胆固醇血症、高载脂蛋白β脂蛋白血症、脑血管动脉粥样硬化、脑血管疾病和静脉血栓形成、中风、动脉粥样硬化、血栓形成、冠心病或主动脉瓣狭窄和/或与含Lp(a)粒子的水平升高相关的任何其他疾病或病理。
可应用本发明方法的细胞包括体外、体内、离体细胞。细胞可以在对象中、在培养物中和/或混悬液中,或处于任何其他合适的状态或条件中。可以应用本发明的方法的细胞可以是:肝脏细胞(liver cell)、肝细胞(hepatocyte)、心脏细胞、胰腺细胞、心血管细胞、肾细胞或其他类型的脊椎动物细胞,包括人和非人哺乳动物细胞。在本发明的某些方面,可应用本发明方法的细胞是健康的正常细胞,其未知为疾病细胞。在本发明的某些实施方案中,将本发明的方法和组合物应用于肝脏细胞、肝细胞、心脏细胞、胰腺细胞、心血管细胞和/或肾细胞的细胞。在本发明的某些方面,对照细胞是正常细胞,但应当理解,具有疾病或病症的细胞也可以在特定情况下用作对照细胞,例如在比较具有疾病或病症的经处理细胞与具有疾病或病症的未处理细胞的结果等的情况下。
根据本发明的方法,可以确定LPA多肽水平并将其与LPA多肽对照水平进行比较。对照可以是预定值,其可以采取多种形式。它可以是单个截止值,例如中位数或平均值。它可以基于比较组来建立,例如在具有正常水平的LPA多肽的组和具有增加的LPA多肽活性水平的组中。比较组的另一个非限制性实例可以是具有LPA相关疾病或病症的一种或更多种症状或诊断的群体与没有疾病或病症的一种或更多种症状或诊断的群体;已对其施用本发明的siRNA治疗的对象组与未对其施用本发明的siRNA治疗的对象组。通常,对照可以基于适当年龄组中的明显健康的正常个体或明显健康的细胞。应当理解,除了预定值之外,根据本发明的对照可以是与实验材料平行测试的材料样品。示例包括来自对照群体的样品或通过制造产生的对照样品,以用于与实验样品进行平行测试。在本发明的一些实施方案中,对照可包括未用本发明的LPA dsRNA试剂接触或处理的细胞或对象,在这种情况下,可以比较LPA多肽的对照水平以及与本发明的LPA dsRNA试剂或LPA反义多核苷酸试剂接触的细胞或对象中LPA多肽的水平。
在本发明的一些实施方案中,对照水平可以是为对象确定的LPA多肽水平,其中将在不同时间为同一对象确定的LPA多肽水平与该对照水平进行比较。在一个非限制性实例中,在从未接受过本发明的LPA治疗的对象获得的生物样品中确定LPA的水平。在一些实施方案中,生物样品是血清样品。从对象获得的样品中测定的LPA多肽水平可作为对象的基线
或对照值。在本发明的治疗方法中向对象施用一次或更多次LPA dsRNA试剂之后,可以从对象获得一个或更多个另外的血清样品,并且可以将随后的一个或更多个样品中的LPA多肽水平与对象的对照/基线水平进行比较。此类比较可用于评估对象中LPA相关疾病或病症的发作、进展或消退。例如,从对象获得的基线样品中LPA多肽的水平高于在给予对象本发明的LPA dsRNA试剂或LPA反义多核苷酸试剂后从同一对象获得的水平,则表示LPA相关疾病或病症的消退并且表示施用的本发明的LPA dsRNA试剂治疗LPA相关疾病或病症产生的功效。
在本发明的某些方面,为对象确定的LPA多肽水平中的一个或更多个值可以作为对照值,并用于稍后在同一对象中比较LPA多肽水平,从而允许评估对象中“基线”LPA多肽水平的变化。因此,将初始水平用作该对象的对照水平的情况下,初始LPA多肽水平可以用于显示和/或确定本发明的方法和化合物在对象中所能够降低对象中LPA多肽的水平。
使用本发明的方法,可以将本发明的LPA dsRNA试剂和/或LPA反义多核苷酸试剂施用于对象。这样的dsRNAi试剂包括例如表1中所显示的双链体。在其他一些实施方案中,这样的dsRNAi试剂包括双链体变体,例如表1中所显示的双链体的变体。可以如下评估本发明的施用和治疗的功效:与先前时间点从对象获得的血清样品中LPA多肽的给药前水平相比,或与非接触对照水平(例如对照血清样品中的LPA多肽水平)相比,当施用和治疗后,从对象获得的血清样品中LPA多肽的水平降低至少0.5%、1%、5%、10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或更多。应当理解,LPA多肽的水平都与LPA基因表达的水平相关。本发明方法的某些实施方案包括以有效抑制LPA基因表达的量向对象施用本发明的LPA dsRNA和/或LPA反义试剂,从而降低对象中LPA多肽的水平。
本发明的一些实施方案包括从一个或更多个对象获得的一个或更多个生物样品中确定LPA多肽的存在、不存在和/或量(本文也称为水平)。该测定可用于评估本发明的治疗方法的功效。例如,本发明的方法和组合物可用于确定生物样品中LPA多肽的水平,该生物样品获自先前用施用本发明的LPA dsRNA试剂和/或LPA反义剂治疗的对象。与先前时间点从对象获得的血清样品中LPA多肽的给药前水平相比,或与非接触对照水平(例如对照血清样品中的LPA多肽水平)相比,当施用和治疗后,从对象获得的血清样品中LPA多肽的水平降低至少0.5%、1%、5%、10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或更多,则表明给予对象的治疗的功效水平。
在本发明的一些实施方案中,针对对象确定的LPA相关疾病或病症的生理特征可以作为对照结果,并将同一对象在不同时间的生理特征的确定结果与对照结果进行比较。在一个非限制性实例中,血液中的Lp(a)水平(和/或LPA疾病或病症的其他生理特征)测量自从未给予本发明的LPA治疗的对象,其可用作对象的基线或对照值。在本发明的治疗方法中向对象施用一次或更多次LPA dsRNA试剂之后,测量血液中的Lp(a)水平并分别与对象的对照
/基线水平进行比较。此类比较可用于评估对象中LPA相关疾病或病症的发作、进展或消退。例如,从对象获得的基线LPA水平高于在对对象施用本发明的LPA dsRNA试剂或LPA反义多核苷酸试剂后从同一对象测定的LPA水平,则表示LPA相关疾病或病症的消退并且表示施用的本发明的LPA dsRNA试剂治疗LPA相关疾病或病症的功效。
在本发明的一些方面,为对象确定的LPA相关疾病或病症的一种或更多种生理特征的值可以作为对照值,用于稍后对同一对象的生理特征进行比较,从而允许评估对象“基线”生理特征的变化。因此,可以获得个体中的初始生理特征,用初始生理特征测定作为该对象的对照,并且显示和/或确定本发明的方法和化合物可用于降低个体中LPA多肽的水平的效果。使用本发明的方法,本发明的LPA dsRNA试剂和/或LPA反义多核苷酸试剂可以以治疗LPA疾病或病症的有效量施用于对象。可以通过确定LPA疾病或病症的一种或更多种生理特征的变化来评估本发明的施用和治疗的功效。在一个非限制性实例中,与先前时间点从对象获得的血液中的Lp(a)水平相比,或与非接触对照中的LPA水平进行比较,对象血液中的Lp(a)水平降低,直到对象血液中的Lp(a)水平处于正常范围内。
本发明的一些实施方案包括使用例如但不限于以下方法确定LPA相关疾病或病症的生理特征的存在、不存在和/或变化:(1)测量对象的血液中的Lp(a)水平;(2)评估从一名或更多名对象获得的一份或更多份生物样品的生理特征;(3)或对对象进行身体检查。该测定可用于评估本发明的治疗方法的功效。
药盒
包含一种或更多种LPA dsRNA试剂和/或LPA反义多核苷酸试剂及其在本发明方法中的使用说明的药盒也在本发明的范围内。本发明的药盒可包含可用于治疗LPA相关疾病或病症的LPA dsRNA试剂、LPA正义多核苷酸和LPA反义多核苷酸试剂中的一种或更多种。可以制备包含一种或更多种LPA dsRNA试剂、LPA正义多核苷酸和LPA反义多核苷酸试剂的药盒以用于本发明的治疗方法。本发明药盒的组分可以以水性介质或冻干形式包装。本发明的药盒可以包含被分隔开以在其中封闭地收纳一个或更多个容器装置或一系列容器装置(例如试管、小瓶、烧瓶、瓶子、注射器等)的载体。第一容器装置或一系列容器装置可包含一种或更多种化合物,例如LPA dsRNA试剂和/或LPA正义或反义多核苷酸试剂。第二容器装置或一系列容器装置可包含靶向剂、标记剂、递送剂等,其可作为在本发明的治疗方法的实施方案中施用的LPA dsRNA试剂和/或LPA反义多核苷酸的一部分包括在内。
本发明的药盒还可包含说明书。说明通常采用书面形式,并且将为执行由药盒体现的治疗和基于该治疗做出决定提供指导。
提供以下实施例以说明本发明实践的具体实例,其并不旨在限制本发明的范围。对本
领域普通技术人员来说明显的是,本发明可应用于多种组合物和方法。
实施例
实施例1.RNAi试剂的合成
上表2-3中所示的LPA RNAi试剂双链体是根据以下通用程序合成的:
使用基于亚磷酰胺化学的成熟固相合成方法,在寡核苷酸合成仪上合成siRNA的正义和反义链序列。寡核苷酸链的增长是通过4步循环实现的:去保护、缩合、加帽和用于添加每个核苷酸的氧化或硫化步骤。合成是在由可控多孔玻璃(CPG,)制成的固体支持物上进行的。单体亚磷酰胺购自商业来源。根据本文实施例2-3的程序合成具有GalNAc配体簇的亚磷酰胺(GLPA1和GLPA2作为非限制性实例)。对于用于体外筛选的siRNA(表2),合成是在2μmol规模下进行的;对于用于体内测试的siRNA(表3、4至5),合成规模为5μmol或更大。在GalNAc配体(作为非限制性实例的GLO-0)连接在正义链的3'-末端的情况下,使用连接有GalNAc配体的CPG固体支持物。在GalNAc配体(GLS-1或GLS-2作为非限制性实例)连接在正义链的5'-末端的情况下,将GalNAc亚磷酰胺(GLPA1或GLPA2作为非限制性实例)用于最后的偶联反应。将3%二氯甲烷中的三氯乙酸(TCA)用于4,4'-二甲氧基三苯甲基保护基(DMT)的脱保护。5-乙硫基-1H-四唑用作活化剂。THF/Py/H2O中的I2和吡啶/MeCN中的苯乙酰二硫化物(PADS)分别用于氧化和硫化反应。在最后的固相合成步骤之后,通过用1:1体积的40wt%甲胺水溶液和28%氢氧化铵溶液处理来切割固体载体结合的低聚物并去除保护基团。为了合成用于体外筛选的siRNA,将粗混合物浓缩。将剩余的固体溶解在1.0M NaOAc中,加入冰冷的EtOH以沉淀出作为钠盐的单链产物,其无需进一步纯化即可用于退火。为了合成用于体内测试的siRNA,粗单链产物通过离子对反相HPLC(IP-RP-HPLC)进一步纯化。通过将来自IP-RP-HPLC的纯化单链寡核苷酸产物溶解在1.0M NaOAc中并通过添加冰冷的EtOH进行沉淀,将其转化为钠盐。在水中通过等摩尔互补进行正义链和反义链寡核苷酸的退火,以形成双链siRNA产物,将其冻干以提供蓬松的白色固体。
实施例2.中间体-A和中间体-B的制备
如以下方案1所示,通过在二氯甲烷(DCM)中用三甲基甲硅烷基三氟甲磺酸酯(TMSOTf)处理市售的半乳糖胺五乙酸酯来合成中间体-A。然后用Cbz保护的2-(2-氨基乙氧基)乙-1-醇进行糖基化,得到化合物II。通过氢化除去Cbz保护基团以提供作为三氟乙酸盐(TFA)盐的中间体-A。除了使用Cbz保护的2-(2-(2-氨基乙氧基)乙氧基)乙-1-醇作为原料之外,中间体B基于相同的方案合成。
方案1
向化合物I(20.0g,51.4mmol)在100mL1,2-二氯乙烷(DCE)中的溶液中加入TMSOTf(17.1g,77.2mmol)。将所得反应溶液在60℃下搅拌2小时,然后在25℃下搅拌1小时;Cbz保护的2-(2-氨基乙氧基)乙-1-醇(13.5g,56.5mmol)在DCE(100mL)中用粉末分子筛(10g)干燥,在N2气氛下在0℃滴加到上述反应溶液中。在N2气氛下,将所得反应混合物在25℃下搅拌16小时。过滤反应混合物并用饱和NaHCO3(200mL)、水(200mL)和饱和盐水(200mL)洗涤。有机层经无水Na2SO4干燥,过滤并减压浓缩,得到粗产物,将其与2-甲基四氢呋喃/庚烷(5/3,v/v,1.80L)一起研磨2小时。将所得混合物过滤并干燥以得到呈白色固体状的化合物II(15.0g,产率50.3%)。
将10%Pd/C(1.50g)小心地加入到干燥和氩气吹扫的氢化瓶中,然后加入10毫升四氢呋喃(THF),然后是化合物II(15.0克,26.4毫摩尔)在THF(300毫升)和TFA(三氟乙酸,3.00克,26.4毫摩尔)中的溶液。将所得混合物脱气并用H2吹扫3次并在H2(45psi)气氛下在25℃下搅拌3小时。薄层色谱法(TLC,溶剂:DCM:MeOH=10:1)表明化合物II已完全消耗。过滤反应混合物并减压浓缩。将残余物溶解在无水DCM(500mL)中并浓缩。该过程重复3次以得到呈泡沫状白色固体的中间体-A(14.0g,产率96.5%)。1H NMR
(400MHz DMSO-d6):δppm7.90(d,J=9.29Hz,1H),7.78(br s,3H),5.23(d,J=3.26Hz,1H),4.98(dd,J=11.29,3.26Hz,1H),4.56(d,J=8.53Hz,1H),3.98-4.07(m,3H),3.79-3.93(m,2H),3.55-3.66(m,5H),2.98(br d,J=4.77Hz,2H),2.11(s,3H),2.00(s,3H),1.90(s,3H),1.76(s,3H)。
使用与合成中间体-A类似的程序合成中间体-B。1H NMR(400MHz DMSO-d6):δppm7.90(br d,J=9.03Hz,4H),5.21(d,J=3.51Hz,1H),4.97(dd,J=11.1Hz,1H),4.54(d,J=8.53Hz,1H),3.98-4.06(m,3H),3.88(dt,J=10.9Hz,1H),3.76-3.83(m,1H),3.49-3.61(m,9H),2.97(br s,2H),2.10(s,3H),1.99(s,3H),1.88(s,3H),1.78(s,3H).计算质谱C20H34N2O11:478.22;实测:479.3(M+H+)。
实施例3.GalNAc配体簇亚磷酰胺GLPA1、GLPA2和GLPA15的合成。
按照以下方案2制备GLPA1和GLPA2。从苄基保护的丙烷-1,3-二胺开始,用2-溴乙酸叔丁酯对其进行烷基化,得到三酯化合物I。通过氢化除去苄基保护基,得到仲胺化合物II。将酰胺与6-羟基己酸偶联得到化合物III。然后在用二氧六环中的HCl处理时除去叔丁基保护基以生成三酸化合物IV。进行三酸化合物IV和中间体-A或中间体-B之间的酰胺偶联以提供化合物Va或Vb。亚磷酰胺GLPA1或GLPA2是通过化合物Va或Vb与2-氰乙基N,N-二异丙基氯亚磷酰胺和催化量的1H-四唑的亚磷酸化合成的。
方案2
向N-苄基-1,3-丙二胺(5.00g,30.4mmol)在二甲基甲酰胺(DMF,100mL)中的溶液中加入2-溴乙酸叔丁酯(23.7g,121mmol);然后滴加二异丙基乙胺(DIEA,23.61g,182mmol)。将所得反应混合物在25-30℃搅拌16小时。LCMS显示N-苄基-1,3-丙二胺被完全消耗。反应混合物用H2O(500mL)稀释并用EtOAc(500mL×2)萃取。合并的有机物用饱和盐水(1L)洗涤,用无水Na2SO4干燥,过滤,减压浓缩,得到粗产物;经硅胶柱层析纯化(梯度:石油醚:乙酸乙酯20:1至5:1)。获得无色油状化合物I(12.1g,产率78.4%)。1H NMR(400MHz,CDCl3):δppm7.26-7.40(m,5H),3.79(s,2H),3.43(s,4H),3.21(s,2H),2.72(dt,J=16.9,7.34Hz,4H),1.70(quin,J=7.2Hz,2H),1.44-1.50(m,27H)。
干燥的氢化瓶用氩气吹扫3次。加入Pd/C(200mg,10%),然后加入MeOH(5mL),然后加入化合物I(1.00g,1.97mmol)在MeOH(5mL)中的溶液。反应混合物在真空下脱气并重新填充H2。这个过程重复3次。将混合物在H2(15psi)气氛下在25℃搅拌12小时。LCMS显示化合物I被完全消耗。在N2气氛下减压过滤反应混合物。减压浓缩滤液,得到黄色油状化合物II(655mg,产率79.7%),其无需进一步纯化即可用于下一步。1H NMR(400MHz,CDCl3):δppm3.44(s,4H),3.31(s,2H),2.78(t,J=7.1Hz,2H),2.68(t,J=6.9Hz,2H),1.88(br s,1H),1.69(quin,J=7.03Hz,2H),1.44-1.50(s,27H)。
化合物II(655mg,1.57mmol)、6-羟基己酸(249mg,1.89mmol)、DIEA(1.02g,7.86mmol)、1-乙基-3-(3-二甲氨基丙基)碳二亚胺盐酸盐(EDCI,904mg,4.72mmol)和1-羟基苯并三唑(HOBt,637mg,4.72mmol)的DMF(6mL)溶液的混合物脱气并用N2吹扫3次;然后在N2气氛下在25℃搅拌3小时。LCMS指示所需产物。反应混合物用H2O(10mL)稀释并用EtOAc20mL(10mL×2)萃取。合并有机物并用饱和盐水(20mL)洗涤,经无水Na2SO4干燥,过滤并浓缩以得到粗产物;将其通过硅胶柱色谱法(梯度:石油醚:乙酸乙酯从5:1至1:1)纯化,得到呈黄色油状的化合物III(650mg,产率77.8%)。1H NMR(400MHz,CDCl3):δppm3.90-3.95(s,2H),3.63(t,J=6.40Hz,2H),3.38-3.45(m,6H),2.72(t,J=6.65Hz,2H),2.40(t,J=7.28Hz,2H),1.55-1.75(m,8H),1.44(s,27H).计算质谱C27H50N2O8:530.36;实测:531.3(M+H+)。
将化合物III(5.5g,10.3mmol)在HCl/二氧六环(2M,55mL)中的混合物在25℃下搅拌3小时。LCMS显示完全消耗化合物III。过滤反应混合物,用EtOAc(50mL)洗涤,减压干燥,得到粗产物。将其溶解在CH3CN(50mL)中,真空除去挥发物。重复该过程3次以得到呈白色固体状的化合物IV(2.05g,产率54.5%)。1H NMR(400MHz,D2O):δppm4.21(s,1H),4.07(d,J=4.5Hz,4H),3.99(s,1H),3.45-3.52(m,3H),3.42(t,J=6.5Hz,1H),3.32-3.38(m,1H),3.24-3.31(m,1H),2.37(t,J=7.4Hz,1H),2.24(t,J=7.4Hz,1H),1.99(dt,J=15.5,7.53Hz,1H),1.85-1.94(m,1H),1.85-1.94(m,1H),1.39-1.56(m,4H),1.19-1.31(m,2H)。
将化合物IV(500mg,1.05mmol)、中间体-A(2.02g,3.67mmol)、DIEA(813mg,6.30mmol)、EDCI(704mg,3.67mmol)和在DMF(10毫升)中的HOBt(496mg,3.67mmol)的混合物脱气并用N2吹扫3次,然后将混合物在N2气氛下在25℃搅拌3小时。LCMS指示所需产物。通过加入H2O(10mL)淬灭反应混合物,用DCM(10mL×2)萃取。合并的有机物用10%柠檬酸(20mL)萃取。水相用饱和NaHCO3溶液中和并用DCM(10mL x2)再萃取。有机物经硫酸钠干燥,过滤并在减压下浓缩以产生呈白色固体状的化合物Va(570mg,0.281mmol,产率26.8%)。1H NMR:(400MHz,CDCl3)ppmδ7.84-8.12(m,3H),6.85-7.15(m,2H),6.66-6.81(m,1H),5.36(br d,J=2.7Hz,3H),5.11-5.27(m,3H),4.63-
4.85(m,3H),3.90-4.25(m,18H),3.37-3.75(m,28H),3.15-3.28(m,4H),2.64(br d,J=6.53Hz,2H),2.30-2.46(m,2H),2.13-2.18(m,9H),2.05(s,9H),1.94-2.03(m,18H),1.68(br s,2H),1.45(br s,2H),1.12(br t,J=7.0Hz,2H)。
向化合物Va(260mg,0.161mmol)的无水DCM(5mL)溶液中加入四唑二异丙基铵(30.3mg,0.177mmol);然后在环境温度和N2下滴加3-双(二异丙基氨基)膦酰氧基丙腈(194mg,0.645mmol)。将反应混合物在20至25℃搅拌2小时。LCMS表明化合物Va被完全消耗。冷却至-20℃后,将反应混合物在0℃加入搅拌的盐水/饱和NaHCO3(1:1,5mL)溶液。搅拌1分钟后,加入DCM(5mL)。出现分层。有机层用盐水/饱和NaHCO3水溶液(1:1,5mL)洗涤,用Na2SO4干燥,过滤并浓缩至体积约1mL。在搅拌下将残余溶液滴加到20mL甲基叔丁基醚(MTBE)中。这导致白色固体沉淀。将混合物离心,收集固体。将固体重新溶解在1mL DCM中并通过加入MTBE(20mL)沉淀。再次通过离心分离固体。将收集的固体溶解在无水CH3CN中。除去挥发物。该过程再重复两次,得到呈白色固体状的GalNAc配体亚磷酰胺化合物GLPA1(153mg,84.4μmol)。1H NMR(400MHz,CDCl3):ppmδ7.71-8.06(m,2H),6.60-7.06(m,3H),5.37(br d,J=3.0Hz,3H),5.18-5.32(m,3H),4.70-4.86(m,3H),3.92-4.25(m,18H),3.42-3.85(m,30H),3.25(m,4H),2.59-2.75(m,4H),2.27-2.44(m,2H),2.15-2.20(s,9H)2.07(s,9H),1.96-2.03(m,18H),1.65(br s,4H),1.44(br d,J=7.28Hz,2H),1.14-1.24(m,12H).31P NMR(CDCl3):ppmδ147.15.
除了使用中间体-B之外,使用相同的程序合成GalNAc配体亚磷酰胺化合物GLPA2。1H NMR(400MHz,CDCl3):ppmδ7.94-8.18(m,1H),7.69(br s,1H),6.66-7.10(m,3H),5.35(d,J=3.5Hz,3H),5.07-5.25(m,3H),4.76-4.86(m,3H),4.01-4.31(m,10H),3.91-4.01(m,8H),3.74-3.86(m,4H),3.52-3.71(m,30H),3.42-3.50(m,6H),3.15-3.25(m,4H),2.52-2.70(m,4H),2.22-2.45(m,2H),2.15-2.22(s,9H),2.06(s,9H),1.95-2.03(m,18H),1.77(br s,2H),1.58-1.66(m,4H),1.40(m,2H),1.08-1.24(m,12H).31P NMR(CDCl3):ppmδ147.12.
按照以下方案3制备GLPA15。
方案3
向中间体化合物II(275g,660mmol,1.00eq.)的二氯甲烷(2.75L)溶液中加入三乙胺(133g,1.32mol,2.00eq.),随后滴加入Cbz-Cl(169g,990mmol,1.50eq.)。反应液在25℃搅拌2小时,LCMS显示化合物II完全转化。反应液依次用NaHCO3(800mL)饱和溶液、饱和食盐水(500mL)洗涤,有机相用无水Na2SO4干燥。过滤除去干燥剂后,滤液浓缩至干。该粗品经柱层析后(SiO2,PE/EA=100/1to5/1)得到无色油状化合物5(290g,527mmol,产率75.7%)。1H NMR(400MHz in DMSO-d6):δppm7.23-7.40(m,5H),5.00-5.12(m,2H),3.86-3.95(m,2H),3.23-3.39(m,6H),2.55-2.67(m,2H),1.56-1.64(m,2H),1.31-1.46(m,27H).MS(ESI)[M+H]+m/z:551.6。
向化合物5(145g,263mmol,1.00eq)中加入HCOOH(2.9L),该溶液在60℃下搅拌12小时,LCMS显示化合物5转化完全。向反应液中加入1.5L甲苯和1.5L乙腈,减压浓缩至约500mL,随后加入甲苯/乙腈(1:1,~750mL)并浓缩至约500mL,然后加入乙腈(~1000mL)并浓缩至干,粗品用700mL乙腈在60℃粉碎2小时,过滤。收集固体,干燥得白色固体化合物6(105g,定量)。1H NMR(400MHz in DMSO-d6):δppm7.26-7.40(m,5H),5.02-5.10(m,2H),3.89-4.00(m,2H),3.36-3.45(m,4H),3.24-3.34(m,2H),2.59-2.72(m,2H),1.40(s,2H).MS(ESI)[M+H]+m/z:383.0。
向化合物6(100g,261mmol.)和中间体-A(502g,915.mmol,3.50eq.)的DMF(1.0L)溶液中加入TBTU(327g,1.02mol,3.90eq.),三乙胺(212g,2.09mol,8.00eq.),反应在25℃进行1小时,LCMS显示化合物6转化完成。将反应液加入到4000mL水中,并用甲基叔丁基醚(2000mL分两次)萃取除去杂质,剩余水相用二氯甲烷(3000mL分两次)萃取。二氯甲烷相依次用10%柠檬酸水溶液(2000mL分两次)、饱和NaHCO3(2.0L分两次),饱和盐水(2.0L)洗涤,无水Na2SO4干燥。过滤得滤液,减压浓缩得到白色固体化合物8(260g,159mmol,产率60.9%)。1H NMR(400MHz in DMSO-d6):δppm7.99-8.08(m,2H),7.93(br d,J=5.50Hz,1H),7.79-7.86(m,3H),7.26-7.39(m,5H),5.22(d,J=3.13Hz,3H),4.95-5.08(m,5H),4.54(br d,J=8.38Hz,3H),4.03(s,9H),3.81-3.93(m,5H),3.76(br d,J=4.88Hz,3H),3.44-3.62(m,10H),3.34-3.43(m,6H),3.24(br d,J=6.13Hz,7H),3.02-3.09(m,4H),2.40-2.47(m,2H),2.10(s,9H),1.99(s,9H),1.89(s,9H),1.77(s,9H),1.57-1.68(m,2H)。MS(ESI)[M+H]+m/z:816.4。
用氩气惰性化2L氢化釜并小心加入干Pd/C(9g),加入MeOH(50mL)润湿Pd/C,然后在氩气气氛下缓慢加入化合物8(90g,55.1mmol,1.00eq.)和三氟乙酸(6.29g,55.1mmol,1.00eq.)的MeOH(850mL)溶液。混合物经脱气/加H2三次置换为氢气气氛,在25℃搅拌10小时。LCMS显示化合物8转化完全,过滤除去Pd/C,滤液经减压浓缩得到化合物9(80g,产率90.2%).1H NMR(400MHz in DMSO-d6):δppm9.12(br s,2H),8.50(br t,J=5.19Hz,1H),8.10(br t,J=5.50Hz,2H),7.85-7.91(m,3H),5.22(d,J=3.25Hz,3H),4.95-5.01(m,3H),4.52-4.58(m,3H),4.03(s,9H),3.84-3.93(m,3H),3.75-3.83(m,3H),3.39-3.61(m,16H),3.23-3.32(m,6H),3.15-3.18(m,3H),2.97-3.05(m,2H),2.54-2.61(m,2H),2.10(s,9H),2.00(s,9H),1.89(s,9H),1.77-1.80(m,9H),1.70-1.76(m,2H).MS(ESI)[M+H]+m/z:749.3。
向化合物9(270g,168mmol,1.00eq.)和戊二酸酐(28.6g,252mmol,1.50eq)的二氯甲烷(2.7L)溶液中加入三乙胺(67.8g,672mmol,4.00eq),该溶液在25℃搅拌1小时,LCMS显示化合物9完全转化为化合物11。向反应液中加入4-羟基哌啶(42.4g,420mmol,2.50eq.)和TBTU(107g,335mmol,2.00eq.),并在25℃继续搅拌1小时。LCMS显示化合物11转化完全。缓慢加入饱和NH4Cl(3.0L)淬灭反应,分层,水相用二氯甲烷(2x1000mL)萃取并与先前的有机相合并。合并的有机相用饱和NaHCO3(aq)和饱和盐水的1:1混合液(3.0L)洗涤,用无水Na2SO4干燥,过滤减压浓缩。粗品溶于1.5L二氯甲烷,滴加到甲基叔丁基醚(7.5L)中,半
透明的白色沉淀在滴加过程中逐渐形成。真空过滤沉淀,收集固体真空干燥得到白色固体化合物13(207g,产率72.8%)。1H NMR(400MHz in DMSO-d6):δppm8.05(br d,J=2.00Hz,2H),7.82(br d,J=7.38Hz,3H),5.21(br s,3H),4.98(br d,J=10.26Hz,3H),4.72(br s,1H),4.54(br d,J=7.88Hz,3H),4.03(br s,9H),3.74-3.94(m,9H),3.45-3.71(m,12H),3.40(br s,6H),3.24(br s,7H),3.07(br d,J=14.13Hz,5H),2.91-3.01(m,1H),2.24-2.44(m,5H),2.20(br s,1H),2.10(s,9H),1.96-2.04(m,9H),1.89(br s,9H),1.74-1.81(m,9H),1.51-1.73(m,6H),1.07-1.36(m,3H).MS(ESI)[M+H]+m/z:848.0。
向化合物13(200g,118mmol,1.00eq.)、四唑二异丙基铵(8.08g,47.2mmol,0.40eq)的二氯甲烷(2.0L)溶液中加入3-双(二异丙基氨基)膦酰氧基丙腈,(53.3g,177mmol,1.50eq.),反应液在40℃搅拌2小时,LCMS显示化合物13转化完成。反应液用饱和NaHCO3和饱和食盐水的1:1混合液(2.0L)洗涤,经无水Na2SO4干燥,滤液浓缩后所得粗品溶于二氯甲烷(1.2L),滴加到搅拌的甲基叔丁基醚(6.0L)中,过滤悬浊液,滤饼用基叔丁基醚淋洗,收集固体进行真空干燥,将产品溶于二氯甲烷(1.0L)并浓缩至干,重复操作4次以除去残留叔丁基醚得到GLPA15(164g,产率73.3%).1H NMR(400MHz in DMSO-d6):δppm8.05(br d,J=6.50Hz,2H),7.81(br d,J=9.01Hz,3H),5.22(d,J=3.25Hz,3H),4.98(dd,J=11.26,3.25Hz,3H),4.55(brd,J=8.50Hz,3H),4.03(s,9H),3.64-3.97(m,12H),3.55-3.63(m,6H),3.50(br s,5H),3.40(br d,J=6.13Hz,6H),3.17-3.30(m,9H),3.07(br d,J=14.26Hz,4H),2.76(t,J=5.82Hz,2H),2.18-2.47(m,6H),2.10(s,9H),1.99(s,9H),1.89(s,9H),1.78(s,9H),1.52-1.74(m,6H),1.12-1.19(m,12H).31P NMR(DMSO-d6):ppmδ145.25.MS(ESI)[M+H]+m/z:1895.7。
在某些研究中,提供了用于将包含GalNAc(本文也称为GalNAc递送化合物)的靶向基团连接到正义链的5'-末端的方法,其包括在固相合成的最后一个偶联步骤中使用GalNAc亚磷酰胺(GLPA1),使用这样的合成工艺,例如在寡核苷酸链延长时使用的(即在正义链的5'末端添加核苷酸)工艺,以将其连接到正义链的5'-末端。
在一些研究中,将包含GalNAc的靶向基团连接到正义链的3'-末端的方法包括使用包含GLO-n的固体支持物(CPG)。在一些研究中,将包含GalNAc的靶向基团连接到正义链的3'-末端的方法包括:将GalNAc靶向基团通过酯键连接到CPG固体支持物上,并在合成正义链时使用带有连接的GalNAc靶向基团的所得CPG,这导致GalNAc靶向基团连接在正义链的3'-末端。其他GalNAc亚磷酰胺化合物(GLPAn)也同样可以在使用合理对应的中间体后,采用本文类似或者本领域中熟知的方法获得,并作为靶向基团连接到siRNA双链体
合适的位置。
实施例4.异甘露醇亚磷酰胺(化合物2)的合成
将在吡啶(400毫升)中的4,4'-二甲氧基三苯基氯甲烷(DMTrCl,232克,684毫摩尔,1.0当量),加入到化合物A(异甘露醇,100克,684毫摩尔,1.0当量)的吡啶(600毫升)溶液中,将混合物在25℃下搅拌16小时。LC-MS显示化合物A被完全消耗,并且检测到一个具有所需质量的主峰。将所得反应混合物用水(500毫升)稀释,用二氯甲烷(500毫升*2)萃取,合并的有机相,用盐水(500毫升)洗涤,用Na2SO4干燥并在真空中浓缩以获得残留物。残余物用柱层析(DCM/MeOH=100/1至50/1,0.1%Et3N)纯化,得到化合物B(150克,收率48.9%)的黄色固体。1H NMR:EC4783-404-P1B1_C(400MHz,DMSO-d6)δppm7.46(br d,J=7.63Hz,2H)7.28-7.37(m,6H)7.19-7.25(m,1H)6.90(br d,J=7.88Hz,4H)4.70(d,J=6.50Hz,1H)3.99-4.09(m,6H)3.88-3.96(m,2H)3.83(br dd,J=7.82,6.94Hz,1H)3.74(s,6H)3.41(br t,J=8.13Hz,1H)3.05(t,J=8.44Hz,1H)2.85(br t,J=7.50Hz,1H).
在N2大气压下,在向化合物B(80.0克,178毫摩尔,1.0当量)的二氯甲烷(5.0毫升)溶液中,在25℃下滴加2H-四氮唑(0.45M,436毫升,1.1当量),然后滴加化合物C(2-氰基乙基二异丙基氯代亚磷酰胺,80.6克,267毫摩尔,85.0毫升,1.5当量)中的二氯甲烷(200毫升)的溶液;将反应混合物在25℃下搅拌1.0小时;LC-MS显示化合物B被完全消耗,并且检测到一个具有所需质量的主峰。将所得反应混合物冷却至-20℃并倒入冰冷的坐下;NaHCO3(500毫升),用二氯甲烷(500毫升*3)提取,合并的有机层用NaHCO3/盐水=1:1(300毫升/300毫升)洗涤,用Na2SO4干燥,并在真空(35℃)中浓缩以获得残留物(100毫升)。残余物用柱层析法纯化(Al2O3,DCM/MeOH=100/1至50/1,0.1%Et3N),得到异甘露醇亚磷酰胺化合物2(77g,119mmol,收率66.5%)为白色固体。1H NMR:EC4783-423-P1B1_C(400MHz,DMSO-d6)
δppm7.22(br d,J=7.50Hz,2H)7.05-7.14(m,6H)6.96-7.02(m,1H)6.67(br dd,J=8.82,1.81Hz,4H)3.95-4.07(m,2H)3.73-3.83(m,1H)3.62-3.72(m,2H)3.48-3.53(m,6H)3.27-3.37(m,3H)3.11(s,6H)2.82(td,J=8.54,2.31Hz,1H)2.47-2.63(m,3H)2.28(br d,J=1.63Hz,3H)0.82-1.00(m,13H).
实施例5.包含异甘露醇单体的固体载体的制备
表示大孔胺甲基聚乙烯树脂载体部分
将50L玻璃釜在氮气保护下,向玻璃釜中加入二氯甲烷(19.50kg),开动搅拌。控温20~30℃,向玻璃釜中加入DMTr-imann(1.47kg),向反应釜中加入三乙胺(1.50kg)、4-二甲氨基吡啶(0.164kg),以及加入丁二酸酐(1.34kg)。体系保温20~30℃反应18h后取样,结束反应。向反应完体系中加入饱和碳酸氢钠溶液(22.50kg),搅拌10~20min后静置至分层,下层有机相转移至,上层水相用二氯甲烷萃取2次,合并有机相,采用无水硫酸钠干燥,过滤滤液,再转移旋蒸浓缩至无馏分,形成成固体灰色至类白色固体1.83kg。
在向100L玻璃釜中加入N,N-二甲基甲酰胺(23.50kg),开动搅拌。控温20~30℃,氮气保护下,通过固体加料漏斗向上述100L玻璃釜中加入上步产物、O-苯并三氮唑-四甲基脲六氟磷酸盐(0.33kg)、加入N,N-二异丙基乙胺(0.13kg),加料完毕搅拌10~30分钟后放出至50L镀锌桶待用。通过固体加料漏斗向上述100L固相合成釜中加入大孔胺甲基树脂(3.25kg)(购于天津南开和成科技有限公司,批号HA2X1209,负载量0.48mmol/g),控温20~30℃,向固相合成釜中加入N,N-二甲基甲酰胺(21.00kg+21.00kg),和上步镀锌桶待用反应液。体系于保温反应,,跟踪至固体载量≥250umol/g,载量检测方法为UV。体系用氮气压滤,滤饼用N,N-二甲基甲酰胺淋洗三次(26.00kg+26.10kg+26.00kg),滤饼留在釜中。向80L玻璃釜中加入CAP.A(4.40kg+4.42kg+4.30kg)和CAP.B(4.40kg+4.40kg+4.47kg),搅拌3~8min后待用。此操作重复进行3次进行盖帽,向固相合成釜中加入乙腈(18.00kg+18.00kg+18.00kg+17.50kg+17.50kg),氮气鼓泡10~30min后压滤。重复此操作四次,滤饼在固相合成釜中使用氮气吹扫2~4h后转移至50L压滤罐,控温15~30℃,继续干燥,将烘干后黄色至白色固体产品,重量:3.516kg。
异山梨醇残基(imann)可以通过本领域技术人员熟知方法,如反向无碱基(invab)同样的工艺方法,将其添加到寡核苷酸链的5'末端或者3'末端,并进一步添加靶向基团。
实施例6.应用Huh7细胞和双荧光报告基因载体进行LPA siRNA双链体的体外筛选
Huh7细胞调整到合适的密度,然后接种到96孔板中。根据制造商的建议,在接种的同时,使用Lipofectamine RNAiMax(Invitrogen-13778-150)将含靶基因的双荧光报告基因载体psciCHECK2与siRNA共同转染进入Huh7细胞。用测试siRNA或对照siRNA转染细胞。siRNA以两个浓度(0.1nM和1.0nM)一式三份进行测试,转染后48小时,加入Dual-Luciferase Assay Reagent试剂检测荧光值。计算海肾发光和萤火虫发光的比,并将其基于对照siRNA处理的样品的比进行归一化,以计算敲低效率。结果如表4所示,使用的双链体AV#来源于对应在表2中所示的序列。
表4提供了使用多种LPA RNAi试剂抑制LPA表达的体外研究的实验结果。
实施例7.LPA siRNA双链体的体内测试
为了评估LPA siRNA的体内活性,使用了感染了编码人LPA和萤光素酶基因的AAV的小鼠(每组4只小鼠)。在siRNA给药前14天,对雌性C57BL/6J小鼠通过静脉注射2x10^11 viral particle of编码人LPA和萤光素酶基因的腺相关病毒8(AAV8)载体的原液来进行感染。在第0天,小鼠皮下注射单剂量6mg/kg的LPA siRNA试剂或PBS。在第0天、siRNA给药前和第7天终止时收集血样。测量荧光素酶活性。通过比较siRNA处理组给药前血样的荧光素酶活性和第7天终止时收集血样的荧光素酶活性并且基于来自PBS处理组的血清样品中的荧光素酶活性变化进行归一化,以计算敲低百分比。结果如表5所示,使用的双链体AD#来源于对应在表3中所示的序列。
表5提供了使用多种LPA_RNAi单次6mpk药剂抑制LPA表达的体内研究的实验结果。第7天,相对于第0天剩余的荧光素酶活性,通过PBS处理组中的变化归一化(平均±SD)
实施例8.LPA siRNA双链体的体内测试
为了评估LPA siRNA的体内活性,使用了感染了编码人LPA和萤光素酶基因的AAV的小鼠(每组4只小鼠)。在siRNA给药前14天,对雌性C57BL/6J小鼠通过静脉注射2x10^11 viral particle of编码人LPA和萤光素酶基因的腺相关病毒8(AAV8)载体的原液来进行感染。在第0天,小鼠皮下注射单剂量6mg/kg的LPA siRNA试剂或PBS。在第0天siRNA给药前、第7天和14天终止时收集血样。测量荧光素酶活性,通过比较siRNA处理组给药前血样的荧光素酶活性与第7和14天时收集血样的荧光素酶活性并且基于来自PBS处理组的血清样品中的荧光素酶活性变化进行归一化,以计算敲低百分比。通过比较siRNA处理组和PBS处理组的第14天小鼠肝脏中人LPA mRNA水平(qPCR确定)敲低(保留)百分比,结果如表6所示,使用的双链体AD#来源于对应在表3中所示的序列。
表6.提供了使用多种LPA_RNAi单次6mpk药剂抑制LPA表达的体内研究的实验结果。
NA表示未检测。
NA表示未检测。
实施例9.LPA siRNA双链体的体内测试
为了评估LPA siRNA的体内活性,使用了感染了编码人LPA和萤光素酶基因的AAV的小鼠(每组4只小鼠)。在siRNA给药前7天,对雌性C57BL/6J小鼠通过静脉注射2x10^11
viral particle of编码人LPA和萤光素酶基因的腺相关病毒8(AAV8)载体的原液来进行感染。在第0天,小鼠皮下分别注射单剂量3mg/kg,6mg/kg和10mg/kg的LPA siRNA试剂或PBS。终止时处死小鼠。通过比较siRNA处理组和PBS处理组的第14天小鼠肝脏中人LPA mRNA水平(qPCR确定)敲低(保留)百分比,结果如表7所示,使用的双链体AD#来源于对应在表3中所示的序列。
表7.提供了使用多种LPA_RNAi分别单次3mpk、6mpk以及10mpk药剂抑制LPA表达的体内研究的实验结果。
实施例10.LPA siRNA双链体的体内测试
为了评估LPA siRNA的体内活性,使用了感染了编码人LPA和萤光素酶基因的AAV的小鼠(每组4只小鼠)。在siRNA给药前7天,对雌性C57BL/6J小鼠通过静脉注射2x10^11viral particle of编码人LPA和萤光素酶基因的腺相关病毒8(AAV8)载体的原液来进行感染。在第0天,小鼠皮下注射单剂量2mg/kg或者6mg/kg的LPA siRNA试剂或PBS。分别在第0天、siRNA给药前和第7、14和21天终止时收集血样。测量荧光素酶活性。通过比较siRNA处理组给药前血样的荧光素酶活性与第7、14和21天终止时收集血样的荧光素酶活性并且基于来自PBS处理组的血清样品中的荧光素酶活性变化进行归一化,以计算敲低百分比。结果如表8所示,使用的双链体AD#来源于对应在表3中所示的序列。
表8.提供了使用多种LPA_RNAi单次2和6mpk药剂抑制LPA表达的体内研究的实验结果,第7、14和21天,相对于第0天剩余的荧光素酶活性,通过PBS处理组中的变化归一化(平均±SD)。
实施例11.LPA siRNA双链体的体内测试
为了评估LPA siRNA的体内活性,本研究共招募雄性食蟹猴(13-22岁,体重7~9公斤),每组3只,每只动物皮下注射给予2mg/kg供试品,使用的供试品对应于表3中
所示的化合物(AD00377-1、AD00436-1、AD00480、AD00480-1、AD00480-2、AD00474-2)。禁食过夜后,在第-14天(给药前)、-7(给药前)、1(给药前)和给药后第8、15、22、29、43、50、57、64、71、78、85、92和99天进行采血。将采集的血样在室温下放置至少30分钟凝固,然后在4℃下以3500rpm的转速离心10分钟。将收集的血清(约1.0mL)转移到两个预先标记的聚丙烯螺旋盖小瓶中(0.5ml/小瓶,一个用于ELISA测定,另一个备用),储存在-80℃的冰箱中直至测试。LPA剩余百分比(标准化至第-14天(给药前)、-7(给药前)、1(给药前)均值,siRNA的预给药)如图1所示。
实施例12.LPA siRNA双链体的体内测试
为了评估LPA siRNA的体内活性,本研究共招募雄性食蟹猴(2-6岁,体重2~6公斤),每组4只,每只动物皮下注射给予生理盐水、2mg/kg的AD00480-8供试品,使用的供试品AD00480-8对应于表3中所示的化合物。禁食过夜后,在第-14天(给药前)、0(给药前)和给药后第7、14和21天进行采血。附图将采集的血样在室温下放置至少30分钟凝固,然后在4℃下以3500rpm的转速离心10分钟。将收集的血清(约1.0mL)转移到两个预先标记的聚丙烯螺旋盖小瓶中(0.5ml/小瓶,一个用于ELISA测定,另一个备用),储存在-80℃的冰箱中直至测试。LPA剩余百分比(标准化至第-14天(给药前)、0(给药前)均值,siRNA的预给药)如图2所示。
实施例13.应用Huh7细胞和双荧光报告基因载体进行LPA siRNA双链体的体外筛选
Huh7细胞调整到合适的密度,然后接种到96孔板中。根据制造商的建议,在接种的同时,使用Lipofectamine RNAiMax(Invitrogen-13778-150)将含靶基因的双荧光报告基因载体psciCHECK2与siRNA共同转染进入Huh7细胞。用测试siRNA或对照siRNA转染细胞。siRNA以两个浓度(0.1nM和1.0nM)一式三份进行测试,转染后48小时,加入Dual-Luciferase Assay Reagent试剂检测荧光值。计算海肾发光和萤火虫发光的比,并将其基于对照siRNA处理的样品的比进行归一化,以计算敲低效率。结果如表9所示,使用的双链体AV#来源于对应在表2中所示的序列。
表9提供了使用多种LPA RNAi试剂抑制LPA表达的体外研究的实验结果。
等同物
尽管本文已经描述和说明了本发明的几个实施例,但本领域普通技术人员很容易理解,用于执行此处描述的功能和/或获得结果和/或一个或更多个优点的多种其他手段和/或结构,以及这些变化和/或修改中的每一个都被认为在本发明的范围内。更一般地,本领域技术人员将容易理解,此处描述的所有参数、尺寸、材料和配置都是示例性的,并且实际参数、尺寸、材料和/或配置将取决于使用本发明教导的具体应用。本领域技术人员将认识到或能够仅使用常规实验来确定本文描述的本发明的特定实施例的许多等价物。因此,应当理解,前述实施例仅通过示例的方式呈现并且属于所附权利要求及其等效物的范围内,本发明可以以不同于具体描述和要求保护的方式实施。本发明针对在此描述的每个单独的特征、系统、物品、材料和/或方法。此外,两个或更多个此类特征、系统、物品、材料和/或方法的任何组合,如果此类特征、系统、物品、材料和/或方法相互不矛盾,则也包括在本发明的范围内。
如本文所定义和使用的所有定义应理解为对照字典定义、通过引用并入的文件中的定义和/或所定义术语的普通含义。
在说明书和权利要求中并未使用数量限定的情况,除非明确指出相反,否则应理解为“至少一个”。
说明书和权利要求书中使用的短语“和/或”应理解为表示如此结合的要素中的“一个或两个”,即这样的要素在某些情况下组合出现而在其他情况下分离出现。除了由“和/或”具体标识的要素之外,除非明确指出相反,否则可以可选地存在其他要素,无论与那些具体标识的要素相关或不相关。
本申请中引用或参考的所有参考文献、专利和专利申请和出版物均通过引用整体并入本文。
Claims (72)
- 抑制LPA(Apo(a))表达的双链核糖核酸(dsRNA)试剂,其中所述dsRNA试剂包含正义链和反义链,所述反义链中的核苷酸第2至18位包含与LPARNA转录物互补的区域,其中互补区域包含与表1-3之一中所列出的反义序列之一相差0、1、2或3个核苷酸的至少15个连续核苷酸,并且任选地包含靶向配体。
- 权利要求1所述的dsRNA试剂,其中所述与LPARNA转录物互补的区域包含至少15、16、17、18或19个连续核苷酸,其与表1-3之一中所列出的反义序列之一相差不超过3个核苷酸。
- 权利要求1或2所述的dsRNA试剂,其中所述dsRNA的反义链与人LPA基因mRNA中的任一个靶区域至少基本上互补,并且在表1-3的任一个中提供。
- 权利要求3所述的dsRNA试剂,其中所述dsRNA的反义链与人LPA基因mRNA中的任一靶区完全互补,并在表1-3的任一个中提供。
- 权利要求1所述的dsRNA试剂,其中所述dsRNA试剂包含表1-3中任一项所述的正义链序列,其中所述正义链序列与所述dsRNA试剂中的反义链序列至少基本上互补。
- 权利要求1所述的dsRNA试剂,其中所述dsRNA试剂包含表1-3中任一项所述的正义链序列,其中所述正义链序列与所述dsRNA试剂中的反义链序列完全互补。
- 权利要求1所述的dsRNA试剂,其中所述dsRNA试剂包含表1-3中任一项所列出的反义链序列。
- 权利要求1所述的dsRNA试剂,其中所述dsRNA试剂包含表1-3中任一项中作为双链体序列所列出的序列。
- 权利要求1所述的dsRNA试剂,其中所述dsRNA试剂包含至少一种修饰的核苷酸。
- 权利要求1所述的dsRNA试剂,其中所述反义链中的所有或基本上所有核苷酸是修饰的核苷酸。
- 权利要求5或6所述的dsRNA试剂,其中所述至少一种修饰的核苷酸包括:2'-O-甲基核苷酸、2'-氟核苷酸、2'-脱氧核苷酸、2'3'-seco核苷酸模拟物、锁定核苷酸、开环核酸核苷酸(UNA)、乙二醇核酸核苷酸(GNA)、2'-F-阿拉伯糖核苷酸、2'-甲氧基乙基核苷酸、无碱基核苷酸、核糖醇、反向核苷酸、反向无碱基核苷酸、反向2'-OMe核苷酸、反向2'-脱氧核苷酸、2'-氨基修饰核苷酸、2'-烷基修饰核苷酸、吗啉代核苷酸和3'-OMe核苷酸、包含5'-硫代磷酸酯基团的核苷酸,或与胆固醇衍生物或十二烷酸双癸酰胺基团连接的末端核苷酸、2'-氨基修饰的核苷酸、氨基磷酸酯,或包含核苷酸的非天然碱基。
- 权利要求9或10所述的dsRNA试剂,其中在引导链的5'末端包含E-乙烯基膦酸酯核苷酸。
- 权利要求1所述的dsRNA试剂,其中所述dsRNA试剂包含至少一个硫代磷酸酯核苷间键联。
- 权利要求1所述的dsRNA试剂,其中所述正义链包含至少一个硫代磷酸酯核苷间键联。
- 权利要求1所述的dsRNA试剂,其中所述反义链包含至少一个硫代磷酸酯核苷间键联。
- 权利要求1所述的dsRNA试剂,其中所述正义链包含1、2、3、4、5或6个硫代磷酸酯核苷间键联。
- 权利要求1所述的dsRNA试剂,其中所述反义链包含1、2、3、4、5或6个硫代磷酸酯核苷间键联。
- 权利要求1所述的dsRNA试剂,其中所述正义链和反义链的全部或基本上全部核苷酸是修饰的核苷酸。
- 权利要求1所述的dsRNA试剂,其中修饰的正义链是表2-3之一中所列出的修饰的正义链序列。
- 权利要求1所述的dsRNA试剂,其中修饰的反义链是表2-3之一中所列出的修饰的反义链序列。
- 权利要求1所述的dsRNA试剂,其中所述正义链与反义链互补或基本上互补,并且互补区域的长度为16至23个核苷酸。
- 权利要求21所述的dsRNA试剂,其中所述互补区域的长度为19至21个核苷酸。
- 权利要求1所述的dsRNA试剂,其中每条链的长度为不超过30个核苷酸。
- 权利要求1所述的dsRNA试剂,其中每条链的长度为不超过25个核苷酸。
- 权利要求1所述的dsRNA试剂,其中每条链的长度为不超过23个核苷酸。
- 权利要求1所述的dsRNA试剂,其中所述dsRNA试剂包含至少一种修饰的核苷酸,并且还包含一种或更多种靶向基团或连接基团。
- 权利要求26所述的dsRNA试剂,其中所述一种或更多种靶向基团或连接基团与所述正义链缀合。
- 权利要求26或27所述的dsRNA试剂,其中所述靶向基团或连接基团包括N-乙酰基-半乳糖胺(GalNAc)。
- 权利要求26或27所述的dsRNA试剂,其中所述靶向基团具有以下结构:
- 权利要求1所述的dsRNA试剂,其中所述dsRNA试剂包含与所述正义链的5'-末端 缀合的靶向基团。
- 权利要求1所述的dsRNA试剂,其中所述dsRNA试剂包含与所述正义链的3'-末端缀合的靶向基团。
- 权利要求1所述的dsRNA试剂,其中所述反义链在3'-末端包含一个反向无碱基残基。
- 权利要求1所述的dsRNA试剂,其中所述正义链在3'或/和5'末端包含一个或两个反向无碱基残基;或正义链在3'或/和5'末端包含一个或两个异甘露醇残基。
- 权利要求1所述的dsRNA试剂,其中所述dsRNA试剂具有两个平末端。
- 权利要求1所述的dsRNA试剂,其中至少一条链包含至少1个核苷酸的3'突出端。
- 权利要求1所述的dsRNA试剂,其中至少一条链包含至少2个核苷酸的3'突出端。
- 包含权利要求1至36中任一项所述的dsRNA试剂的组合物。
- 权利要求37所述的组合物,其还包含药学上可接受的载体。
- 权利要求38所述的组合物,还包含一种或更多种另外的治疗剂。
- 权利要求39所述的组合物,其中所述组合物被包装在药盒、容器、包装物、分配器、预填充注射器或小瓶中。
- 权利要求37所述的组合物,其中所述组合物被配制成用于皮下给药或被配制成用于静脉内(IV)给药。
- 包含权利要求1至36中任一项所述的dsRNA试剂的细胞。
- 权利要求42所述的细胞,其特征在于,所述细胞是哺乳动物细胞,任选地是人细胞。
- 抑制细胞中LPA基因表达的方法,其包括:(i)制备包含有效量的权利要求1至36中任一项所述的双链核糖核酸(dsRNA)试剂或权利要求37至41中任一项所述的组合物的细胞。
- 权利要求44所述的方法,其还包括:(ii)将权利要求44的(i)中制备的细胞维持足够的时间,以获得LPA基因的mRNA转录物的降解,从而抑制细胞中LPA基因的表达。
- 权利要求44所述的方法,其特征在于,所述细胞在对象体内并且将所述dsRNA试剂皮下施用至所述对象。
- 权利要求44所述的方法,其中所述细胞在对象体内并且通过IV施用将所述dsRNA 试剂施用至所述对象。
- 权利要求46或47所述的方法,其还包括在向所述对象施用所述dsRNA试剂之后评估对LPA基因的抑制,其中用于评估的手段包括:(i)确定对象中LPA相关疾病或病症的一种或多种生理特征,以及(ii)将所确定的生理特征与LPA相关疾病或病症的基线治疗前生理特征和/或LPA相关疾病或病症的对照生理特征进行比较,其中经过比较表明对象中LPA基因表达抑制存在或不存在中的一种或更多种。
- 权利要求48所述的方法,其中所确定的生理特征是在血液中的Lp(a)水平。
- 权利要求49所述的方法,其中所述对象在血液中的Lp(a)水平的降低表明对象中LPA基因表达的降低。
- 抑制对象中LPA基因表达的方法,所述方法包括向对象施用有效量的权利要求1至36中任一项所述的双链核糖核酸(dsRNA)试剂或权利要求37至41中任一项所述的组合物。
- 权利要求51所述的方法,其中将所述dsRNA试剂皮下施用于所述对象。
- 权利要求51所述的方法,其中所述dsRNA试剂通过IV给药施用于所述对象。
- 权利要求51至53中任一项所述的方法,其还包括在向所述对象施用所述dsRNA试剂之后评估对LPA基因的抑制,其中用于评估的手段包括:(i)确定对象中LPA相关疾病或病症的一种或多种生理特征,以及(ii)将所确定的生理特征与LPA相关疾病或病症的基线治疗前生理特征和/或LPA相关疾病或病症的对照生理特征进行比较,其中经过比较表明对象中LPA基因表达抑制存在或不存在中的一种或更多种。
- 权利要求54所述的方法,其中所确定的生理特征是在血液中的Lp(a)水平。
- 权利要求55所述的方法,其中所述对象在血液中的Lp(a)水平的降低表明对象中LPA基因表达的降低。
- 治疗及预防与LPA蛋白相关的疾病或病症的方法,所述方法包括向对象施用有效量的权利要求1至36中任一项所述的双链核糖核酸(dsRNA)试剂或权利要求37至41中任一项所述的组合物,以抑制LPA基因表达。
- 权利要求57所述的方法,其中所述疾病或病症是心血管疾病,其中所述心血管疾病包括:伯格氏病(Berger’s disease)、外周动脉疾病、冠状动脉疾病、代谢综合征、急性 冠脉综合征、主动脉瓣狭窄、主动脉瓣反流、主动脉夹层、视网膜动脉阻塞、脑血管疾病、肠系膜缺血、肠系膜上动脉阻塞、肾动脉狭窄、稳定型/不稳定型心绞痛、急性冠脉综合征、杂合子或纯合子家族性高胆固醇血症、高载脂蛋白β脂蛋白血症、脑血管动脉粥样硬化、脑血管疾病和静脉血栓形成、中风、动脉粥样硬化、血栓形成、冠心病或主动脉瓣狭窄和/或与含Lp(a)粒子的水平升高相关的任何其他疾病或病理。
- 权利要求57所述的方法,其还包括对所述对象施用另外的治疗方案。
- 权利要求59所述的方法,其中所述另外的治疗方案包括:向所述对象施用一种或更多种本发明的LPA反义多核苷酸,向所述对象施用非LPAdsRNA治疗剂,以及所述对象的行为改变。
- 权利要求60所述的方法,其中所述非LPAdsRNA治疗剂是以下中的一种或更多种另外的治疗剂,如HMg Co-A还原酶抑制剂(他汀类)、依折麦布、PCSK-9抑制剂、CTEP抑制剂、靶向ANGPTL3的疗法、靶向AGT的疗法、靶向APOC3的疗法和烟酸,或上述任何的组合。
- 权利要求57所述的方法,其中所述dsRNA试剂皮下施用于所述对象。
- 权利要求57所述的方法,其中所述dsRNA试剂通过IV给药施用于所述对象。
- 权利要求57至63中任一项所述的方法,其还包括确定所施用的双链核糖核酸(dsRNA)试剂在所述对象中的功效。
- 权利要求64所述的方法,其中确定治疗在所述对象中的功效的手段包括:(i)确定对象中LPA相关疾病或病症的一种或多种生理特征,并且(ii)将所确定的生理特征与LPA相关疾病或病症的基线治疗前生理特征进行比较,其中通过比较表明对对象施用双链核糖核酸(dsRNA)试剂的功效的存在、不存在和功效水平中的一种或更多种。
- 权利要求65所述的方法,其中所确定的生理特征是在血液中的Lp(a)水平。
- 权利要求65所述的方法,其中所述对象在血液中的Lp(a)水平的降低表示对对象施用双链核糖核酸(dsRNA)试剂存在功效。
- 与对象中LPA蛋白的基线治疗前水平相比降低对象中LPA蛋白水平的方法,所述方法包括向对象施用有效量的权利要求1至36中任一项所述的双链核糖核酸(dsRNA)试剂或权利要求37至41中任一项的组合物,以降低LPA基因表达水平。
- 权利要求68所述的方法,其中将所述dsRNA试剂皮下施用于所述对象或通过IV施用于所述对象。
- 与对象中LPA相关疾病或病症的基线治疗前生理特征相比改变对象中LPA相关疾病或病症的生理特征的方法,所述方法包括向对象施用有效量的权利要求1至36中任一项所述的双链核糖核酸(dsRNA)试剂或权利要求37至41中任一项所述的组合物,以改变对象中LPA相关疾病或病症的生理特征。
- 权利要求70所述的方法,其中将所述dsRNA试剂皮下施用于所述对象或通过IV施用于所述对象。
- 权利要求70所述的方法,其中所述生理特征是在血液中的Lp(a)水平。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2022073415 | 2022-01-24 | ||
| CNPCT/CN2022/073415 | 2022-01-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023138689A1 true WO2023138689A1 (zh) | 2023-07-27 |
Family
ID=87347910
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2023/073456 WO2023138689A1 (zh) | 2022-01-24 | 2023-01-23 | 抑制LPA(Apo(a))蛋白表达的组合物和方法 |
Country Status (2)
| Country | Link |
|---|---|
| TW (1) | TW202345865A (zh) |
| WO (1) | WO2023138689A1 (zh) |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101292034A (zh) * | 2005-09-15 | 2008-10-22 | 桑塔里斯制药公司 | 抑制apo-b100表达的rna拮抗化合物 |
| CN104302654A (zh) * | 2012-05-24 | 2015-01-21 | Isis制药公司 | 用于调节载脂蛋白(a)表达的方法和组合物 |
| CN111107853A (zh) * | 2017-09-11 | 2020-05-05 | 箭头药业股份有限公司 | 用于抑制载脂蛋白C-III (APOC3)的表达的RNAi试剂和组合物 |
| CN112423794A (zh) * | 2018-12-28 | 2021-02-26 | 苏州瑞博生物技术股份有限公司 | 一种核酸、含有该核酸的组合物与缀合物及制备方法和用途 |
| CN112921036A (zh) * | 2013-05-01 | 2021-06-08 | Ionis制药公司 | 用于调节载脂蛋白(a)表达的组合物和方法 |
-
2023
- 2023-01-18 TW TW112102207A patent/TW202345865A/zh unknown
- 2023-01-23 WO PCT/CN2023/073456 patent/WO2023138689A1/zh unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101292034A (zh) * | 2005-09-15 | 2008-10-22 | 桑塔里斯制药公司 | 抑制apo-b100表达的rna拮抗化合物 |
| CN104302654A (zh) * | 2012-05-24 | 2015-01-21 | Isis制药公司 | 用于调节载脂蛋白(a)表达的方法和组合物 |
| CN112921036A (zh) * | 2013-05-01 | 2021-06-08 | Ionis制药公司 | 用于调节载脂蛋白(a)表达的组合物和方法 |
| CN111107853A (zh) * | 2017-09-11 | 2020-05-05 | 箭头药业股份有限公司 | 用于抑制载脂蛋白C-III (APOC3)的表达的RNAi试剂和组合物 |
| CN112423794A (zh) * | 2018-12-28 | 2021-02-26 | 苏州瑞博生物技术股份有限公司 | 一种核酸、含有该核酸的组合物与缀合物及制备方法和用途 |
Non-Patent Citations (1)
| Title |
|---|
| DING YANG; WANG YAZHE; ZHOU JIANPING; GU XIAOCHEN; WANG WEI; LIU CONGYAN; BAO XIULI; WANG CHENG; LI YUANRU; ZHANG QIANG: "Direct cytosolic siRNA delivery by reconstituted high density lipoprotein for target-specific therapy of tumor angiogenesis", BIOMATERIALS, ELSEVIER, AMSTERDAM, NL, vol. 35, no. 25, 27 May 2014 (2014-05-27), AMSTERDAM, NL , pages 7214 - 7227, XP028868106, ISSN: 0142-9612, DOI: 10.1016/j.biomaterials.2014.05.009 * |
Also Published As
| Publication number | Publication date |
|---|---|
| TW202345865A (zh) | 2023-12-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20210340540A1 (en) | Chimeric double-stranded nucleic acid | |
| CN111343994B (zh) | 用于抑制血管生成素-样3 (ANGPTL3)的表达的RNAi剂和组合物以及使用方法 | |
| CN111107853A (zh) | 用于抑制载脂蛋白C-III (APOC3)的表达的RNAi试剂和组合物 | |
| JP2016185150A (ja) | Timp1およびtimp2発現の調節 | |
| CN113164509A (zh) | 用于抑制17β-HSD 13型(HSD17B13)表达的RNAi试剂、其组合物和使用方法 | |
| EP3366773A1 (en) | Nucleic acid complex | |
| US10023862B2 (en) | Organic compositions to treat beta-catenin-related diseases | |
| WO2024088190A1 (zh) | 一种抑制lpa基因表达的rna抑制剂及其应用 | |
| CN111212909A (zh) | 用于抑制去唾液酸糖蛋白受体1的表达的RNAi试剂和组合物 | |
| CN114555188A (zh) | 治疗apoc3相关疾病和病症的方法 | |
| WO2023138689A1 (zh) | 抑制LPA(Apo(a))蛋白表达的组合物和方法 | |
| WO2023088227A1 (zh) | 抑制血管紧张素原(agt)蛋白表达的组合物和方法 | |
| WO2023093896A1 (zh) | 用于抑制乙型肝炎病毒(hbv)蛋白表达的组合物和方法 | |
| WO2023202686A1 (zh) | 用于抑制黄嘌呤脱氢酶(xdh)的组合物和方法 | |
| WO2024061185A1 (zh) | 特定修饰的RNAi试剂和组合物 | |
| WO2023186056A1 (zh) | 用于抑制补体成分c3蛋白表达的组合物和方法 | |
| CN116888263A (zh) | 用于抑制血管生成素样3(angptl3)蛋白表达的组合物和方法 | |
| TW202413645A (zh) | 特定修飾的RNAi試劑和組合物 | |
| WO2023143483A1 (en) | Compositions and methods for inhibiting expression of prekallikrein (pkk) protein | |
| US20230092615A1 (en) | Compositions and methods for inhibiting expressing of methylation-controlled j-protein (mcj) | |
| TW202415389A (zh) | 用於抑制脂蛋白元C-III (APOC3)表現之RNAi試劑及組合物 | |
| WO2023245061A2 (en) | Lipid conjugates for the delivery of therapeutic agents to cns tissue | |
| CN116670278A (zh) | 用于降低z-aat蛋白水平的方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23743004 Country of ref document: EP Kind code of ref document: A1 |