WO2023120551A1 - 自閉スペクトラム症治療のためのオピオイドの使用 - Google Patents
自閉スペクトラム症治療のためのオピオイドの使用 Download PDFInfo
- Publication number
- WO2023120551A1 WO2023120551A1 PCT/JP2022/047004 JP2022047004W WO2023120551A1 WO 2023120551 A1 WO2023120551 A1 WO 2023120551A1 JP 2022047004 W JP2022047004 W JP 2022047004W WO 2023120551 A1 WO2023120551 A1 WO 2023120551A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pharmaceutically acceptable
- acceptable salt
- buprenorphine
- pharmaceutical composition
- morphine
- Prior art date
Links
Images









Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/485—Morphinan derivatives, e.g. morphine, codeine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
Definitions
- the present invention is particularly preferred for treating and/or preventing autism spectrum disorders, fragile X syndrome, autism spectrum-like symptoms and/or disorders in social communication and/or social interactions. It relates to new pharmaceutical compositions and methods for treating spectrum disorders.
- ASD Autism Spectrum Disorder
- Behavioral therapy such as applied behavior analysis is the main treatment for ASD, but it is known that behavioral therapy requires a great deal of time and human resources, and its effectiveness decreases with age. Therefore, the number of patients who can benefit from it is very limited. Against the background described above, there is a strong demand for a therapeutic drug that is effective for the core symptoms of ASD.
- Non-Patent Document 3 a theory was proposed that ASD symptoms were caused by excessive opioid signals in the brain (Non-Patent Document 3), and then clinical trials were conducted on the therapeutic effects of Naltrexone, a ⁇ opioid receptor antagonist, on ASD. Effectiveness was confirmed for symptoms such as irritability (Non-Patent Document 4). On the other hand, analysis of ⁇ opioid receptor (MOR) knockout mice has reported that ASD-like symptoms are also induced by decreased opioid signals (Non-Patent Document 5).
- Non-Patent Document 6 There is also a report that administration of a MOR agonist increases social behavior in normal rats. As described above, the possibility that opioid signals are involved in ASD symptoms and social behavior has been suggested, but as discussed in the review paper Non-Patent Document 5, the details are unknown. It is also unclear whether it is better to lower or enhance opioid signaling in ASD treatment.
- FXS human fragile X syndrome
- Buprenorphine is a mu opioid receptor partial agonist and a kappa opioid receptor antagonist.
- formulations for transdermal administration, transmucosal administration (buccal, sublingual, suppository), and injections (intravenous, intramuscular, subcutaneous) have been approved (Non-Patent Documents 10 to 16). The contents of these documents are incorporated by reference into the present application).
- Morphine is an opioid agonist, in particular a mu opioid receptor agonist.
- formulations for oral administration formulations for transmucosal administration (suppositories), and injections (intravenous, subcutaneous, etc.) have been approved (Non-Patent Documents 17 to 22.
- Patent Document 3 describes combined use data of oxytocin and naloxone (mu opioid receptor antagonist).
- Non-Patent Document 26 describes the pharmacokinetics of sublingual administration of buprenorphine.
- Non-Patent Document 28 describes the results of evaluating the analgesic effect of buprenorphine using mice and the like.
- autism spectrum disorders fragile X syndrome, autism spectrum-like symptoms and/or disorders in social communication and/or social interactions, particularly preferably autism spectrum disorders
- New pharmaceutical compositions and methods for treatment are provided.
- the present inventors particularly preferably use the Pharmaceutical compositions and methods for treating autism spectrum disorders have been extensively investigated and suitable active ingredients have been found. Furthermore, they have found a preferred administration method, dosage, and the like for the active ingredient.
- the present invention relates to, for example, the following.
- (2A) The pharmaceutical composition according to any one of (1) and (1A) to (1C) above for treating and/or preventing core symptoms of autism spectrum disorders.
- (3) The pharmaceutical composition according to any one of (1), (2), (1A) to (1C) and (2A) above, for administration to a patient diagnosed with an autism spectrum disorder.
- buprenorphine or a pharmaceutically acceptable salt thereof and/or morphine or a pharmaceutically acceptable salt thereof is buprenorphine or a pharmaceutically acceptable salt thereof, (1) to (7), (1A)
- the pharmaceutical composition has (eg, continuously for about 30 minutes or more) buprenorphine in plasma within the range of about 4 pg/mL to about 1 ng/mL (eg, about 4 pg/mL to about 0.6 ng/mL); any of (1)-(10), (1A)-(1C), (2A), (5A)-(5C), (7A), and (10A)-(10C) above, which provides concentration Pharmaceutical composition as described.
- a) to c) below a) a pharmaceutical composition for transdermal administration containing from about 0.01 mg to about 10 mg of buprenorphine or a pharmaceutically acceptable salt thereof; b) a pharmaceutical composition for transdermal administration that releases buprenorphine or a pharmaceutically acceptable salt thereof at about 0.01 ⁇ g/h to about 10 ⁇ g/h; and c) a pharmaceutical composition for transmucosal administration containing about 0.0001 mg to 0.1 mg of buprenorphine or a pharmaceutically acceptable salt thereof; Any of the above (1) to (12), (1A) to (1C), (2A), (5A) to (5C), (7A), (10A) to (10E), and (12A ).
- (13B) a) a pharmaceutical composition for transdermal administration containing from about 0.56 mg to about 1.7 mg of buprenorphine or a pharmaceutically acceptable salt thereof; or b) a pharmaceutical composition for transdermal administration that releases buprenorphine or a pharmaceutically acceptable salt thereof at about 0.56 ⁇ g/h to about 1.7 ⁇ g/h; Any of the above (1) to (13), (1A) to (1C), (2A), (5A) to (5C), (7A), (10A) to (10E), (12A) , and (13A).
- (13C) a) a pharmaceutical composition for transdermal administration containing about 0.56 mg or about 1.7 mg of buprenorphine or a pharmaceutically acceptable salt thereof; or b) a pharmaceutical composition for transdermal administration that releases buprenorphine or a pharmaceutically acceptable salt thereof at about 0.56 ⁇ g/h or about 1.7 ⁇ g/h; Any of the above (1) to (13), (1A) to (1C), (2A), (5A) to (5C), (7A), (10A) to (10E), (12A) , (13A) and (13B).
- (16B) The above (1) to (16), (1A) to (1C), (2A), (5A) to (5C), (10A) to (10E), which are pharmaceutical compositions for transdermal administration, (12A), (13A) to (13C), and the pharmaceutical composition according to any one of (14A).
- (16C) The above (1) to (16), (1A) to (1C), (2A), (5A) to (5C), (7A), (10A) to which is a pharmaceutical composition for transmucosal administration The pharmaceutical composition according to any one of (10E), (12A), (13A)-(13C) and (14A).
- (101B) autism spectrum comprising administering buprenorphine or a derivative thereof or a pharmaceutically acceptable salt thereof and/or morphine or a derivative thereof (derivatives of morphine such as codeine) or a pharmaceutically acceptable salt thereof A method of treating and/or preventing autism spectrum disorder, fragile X syndrome, and/or autism spectrum disorder-like symptoms.
- (102) The method according to any of (101) and (101A) to (101C) above for treating and/or preventing autism spectrum disorders.
- (102A) The method according to any of (101) and (101A) to (101C) above for treating and/or preventing core symptoms of autism spectrum disorders.
- (103) The method according to any one of (101), (102), (101A) to (101C) and (102A) above, for administering to a patient diagnosed with an autism spectrum disorder.
- Buprenorphine or a pharmaceutically acceptable salt thereof and/or morphine or a pharmaceutically acceptable salt thereof is administered at a dose less than or equal to the minimum effective pain treatment dose, (101) to (105), (101A) ) to (101C), (102A), and (105A) to (105C).
- the buprenorphine or a pharmaceutically acceptable salt thereof and/or morphine or a pharmaceutically acceptable salt thereof is buprenorphine or a pharmaceutically acceptable salt thereof (101) to (107), (101A) (101C), (102A), (105A) to (105C), and (107A).
- the maximum plasma concentration of buprenorphine or a pharmaceutically acceptable salt thereof in the patient to whom it is administered is about 1 ng/mL or less (101) to (108), (101A) to (101C), ( 102A), (105A)-(105C), and (107A).
- the maximum plasma concentration of buprenorphine or a pharmaceutically acceptable salt thereof in the patient to whom it is administered is about 4 pg/mL to about 0.6 ng/mL (101) to (109), (101A) above (101C), (102A), (105A) to (105C), and (107A).
- the peak plasma concentration of buprenorphine or a pharmaceutically acceptable salt thereof in the administered patient is greater than 3.3 pg/mL and less than or equal to about 0.6 ng/mL (101) to (110) above , (101A)-(101C), (102A), (105A)-(105C), and (107A).
- the maximum plasma concentration of buprenorphine or a pharmaceutically acceptable salt thereof in the patient to whom it is administered is about 0.6 ng/mL or less (101) to (110), (101A) to (101C) above , (102A), (105A)-(105C), (107A), and (110A).
- the maximum plasma concentration of buprenorphine or a pharmaceutically acceptable salt thereof in the patient to whom it is administered is about 1 pg/mL to about 1 ng/mL (101)-(110), (101A)-( 101C), (102A), (105A)-(105C), (107A), (110A), and (110B).
- (110E) a peak plasma concentration of buprenorphine or a pharmaceutically acceptable salt thereof in the administered patient within the range of about 4 pg/mL to about 1 ng/mL (eg, about 3 pg/mL to about 0.6 ng/mL)
- the method according to any one of (101) to (110), (101A) to (101C), (102A), (105A) to (105C), (107A) and (110A) to (110D) above. .
- the maximum plasma concentration of buprenorphine or a pharmaceutically acceptable salt thereof in the patient to whom it is administered is about 0.2 ng/mL or less (101) to (110), (101A) to (101C) above , (102A), (105A)-(105C), (107A), and (110A)-(110E).
- the maximum plasma concentration of buprenorphine or a pharmaceutically acceptable salt thereof in the patient to whom it is administered is about 8 pg/mL to about 0.2 ng/mL (101) to (111), (101A) above - (101C), (102A), (105A) - (105C), (107A), and (110A) - (110E).
- the maximum plasma concentration or plasma concentration is the maximum plasma concentration or plasma concentration at steady state, (101) to (112), (101A) to (101C), (102A), ( 105A) to (105C), (107A), and (110A) to (110E).
- (113) a) to c) below: a) transdermally administering from about 0.01 mg to about 10 mg of buprenorphine or a pharmaceutically acceptable salt thereof; b) transdermal administration of buprenorphine or a pharmaceutically acceptable salt thereof at about 0.01 ⁇ g/h to about 10 ⁇ g/h, and c) administering about 0.0001 mg to 0.1 mg transmucosally of buprenorphine or a pharmaceutically acceptable salt thereof; (101) to (112), (101A) to (101C), (102A), (105A) to (105C), (107A), (110A) to (110E), and (112A), including any of ).
- (113B) a) transdermally administering about 0.56 mg to about 1.7 mg of buprenorphine or a pharmaceutically acceptable salt thereof, or b) transdermal administration of buprenorphine or a pharmaceutically acceptable salt thereof at about 0.56 ⁇ g/h to about 1.7 ⁇ g/h; Any of the above (101) to (113), (101A) to (101C), (102A), (105A) to (105C), (107A), (110A) to (110E), (112A) , and (113A).
- (113C) a) transdermally administering about 0.56 mg or about 1.7 mg of buprenorphine or a pharmaceutically acceptable salt thereof, or b) about 0.56 ⁇ g/h of buprenorphine or a pharmaceutically acceptable salt thereof or transdermal administration at about 1.7 ⁇ g/h, Any of the above (101) to (113), (101A) to (101C), (102A), (105A) to (105C), (107A), (110A) to (110E), (112A) , (113A) and (113B).
- (114) The above (101) to (113), (101A) to (101C), (102A), (105A) to (105C), wherein buprenorphine or a pharmaceutically acceptable salt thereof is administered at an administration interval of 12 hours or longer ), (107A), (110A)-(110E), (112A), and (113A)-(113C).
- (114A) The above (101) to (114), (101A) to (101C), (102A), (105A) to (105C), (107A), (110A), wherein the administration frequency is twice a day or less ) to (110E), (112A), and (113A) to (113C).
- (116A) Transdermal or transmucosal administration of buprenorphine or a pharmaceutically acceptable salt thereof, (101) to (116), (101A) to (101C), (102A), (105A) to (105C) ), (107A), (110A)-(110E), (112A), (113A)-(113C), and (114A).
- (116B) Transdermal administration of buprenorphine or a pharmaceutically acceptable salt thereof, (101) to (116), (101A) to (101C), (102A), (105A) to (105C), (107A) ), (110A)-(110E), (112A), (113A)-(113C), and (114A).
- (116C) Transmucosal administration of buprenorphine or a pharmaceutically acceptable salt thereof, (101) to (116), (101A) to (101C), (102A), (105A) to (105C), (107A) ), (110A)-(110E), (112A), (113A)-(113C), and (114A).
- Buprenorphine or a pharmaceutically acceptable salt thereof and/or morphine or a pharmaceutically acceptable salt thereof is morphine or a pharmaceutically acceptable salt thereof (101) to (107), (101A) (101C), (102A), (105A) to (105C), and (107A).
- (119A) Activates the nucleus accumbens and the medial prefrontal cortex and does not activate the dorsomedial periaqueductal gray matter, (101) to (119), (101A) to (101C), (102A), ( 105A) to (105C), (107A), (110A) to (110E), (112A), (113A) to (113C), (114A), and (116A) to (116C). .
- (201B) Buprenorphine or a derivative thereof or a pharmaceutically acceptable salt thereof and/or morphine or a derivative thereof for the treatment and/or prevention of autism spectrum disorder, fragile X syndrome, and/or autism spectrum disorder-like symptoms (as a derivative of morphine, eg codeine) or a pharmaceutically acceptable salt thereof.
- (201C) The ⁇ opioid agonist buprenorphine or a pharmaceutically acceptable salt thereof and/or morphine or a pharmaceutically acceptable salt thereof.
- (202) Buprenorphine or a pharmaceutically acceptable salt thereof and/or morphine or morphine according to any one of (201) and (201A) to (201C) above for the treatment and/or prevention of autism spectrum disorder A pharmaceutically acceptable salt.
- (202A) Buprenorphine or a pharmaceutically acceptable salt thereof and/or according to any of (201) and (201A) to (201C) above for the treatment and/or prevention of core symptoms of autism spectrum disorder Morphine or a pharmaceutically acceptable salt thereof.
- (203) Buprenorphine or a pharmaceutical formulation thereof according to any one of (201), (202), (201A) to (201C), and (202A) above for administration to a patient diagnosed with an autism spectrum disorder Acceptable salts and/or morphine or a pharmaceutically acceptable salt thereof.
- (302) Use according to any of (301) and (301A) to (301C) above for the manufacture of a pharmaceutical composition for treating and/or preventing autism spectrum disorders.
- (302A) Use according to any of (301) and (301A) to (301C) above for the manufacture of a pharmaceutical composition for the treatment and/or prevention of core symptoms of autism spectrum disorders.
- (303) Any of the above (301), (302), (301A) to (301C), and (302A) for the manufacture of a pharmaceutical composition for administration to a patient diagnosed with an autism spectrum disorder Use as described.
- (304) The above (301) to ( 303), (301A) to (301C), and (302A).
- (305B) The use according to any one of (301) to (305), (301A) to (301C), (302A) and (305A) above, wherein the pharmaceutical composition does not substantially exhibit an analgesic effect.
- (305C) The above (301 ) to (305), (301A) to (301C), (302A), (305A), and (305B).
- (306) The content of buprenorphine or a pharmaceutically acceptable salt thereof and/or morphine or a pharmaceutically acceptable salt thereof is less than or equal to the minimum effective amount for pain treatment, for the above (301)
- a pharmaceutical composition in which the content of buprenorphine or a pharmaceutically acceptable salt thereof and/or morphine or a pharmaceutically acceptable salt thereof is about 1/30 times to about 3 times the minimum effective amount for pain treatment Use according to any of (301) to (306), (301A) to (301C), (302A) and (305A) to (305C) above for the manufacture.
- (307A) For the manufacture of a pharmaceutical composition in which the content of buprenorphine or a pharmaceutically acceptable salt thereof and/or morphine or a pharmaceutically acceptable salt thereof is less than or equal to about 1/2 times the minimum effective pain treatment dose of any one of (301) to (307), (301A) to (301C), (302A) and (305A) to (305C) above.
- Buprenorphine or a pharmaceutically acceptable salt thereof and/or morphine or a pharmaceutically acceptable salt thereof is buprenorphine or a pharmaceutically acceptable salt thereof (301) to (307), (301A) - (301C), (302A), (305A) - (305C), and (307A).
- the pharmaceutical composition is a pharmaceutical composition that provides a peak plasma concentration of buprenorphine or a pharmaceutically acceptable salt thereof of about 4 pg/mL to about 0.6 ng/mL (301) to ( 309), (301A)-(301C), (302A), (305A)-(305C), and (307A).
- the pharmaceutical composition is a pharmaceutical composition that provides a maximum plasma concentration of buprenorphine or a pharmaceutically acceptable salt thereof of about 0.6 ng/mL or less (301) to (310), ( 301A) to (301C), (302A), (305A) to (305C), (307A) and (310A).
- the pharmaceutical composition comprises (eg, continuously for about 30 minutes or more) buprenorphine in the plasma within the range of about 4 pg/mL to about 1 ng/mL (eg, about 4 pg/mL to about 0.6 ng/mL); Any of the above (301) to (310), (301A) to (301C), (302A), (305A) to (305C), and (310A) to (310C), which is a pharmaceutical composition that provides concentration Use as described.
- the pharmaceutical composition provides a plasma concentration of buprenorphine within the range of about 4 pg/mL to about 1 ng/mL (eg, about 3 pg/mL to about 0.6 ng/mL); Use according to any of (301) to (310), (301A) to (301C), (302A), (305A) to (305C), (307A), and (310A) to (310D) above.
- the pharmaceutical composition is a pharmaceutical composition that provides a maximum plasma concentration of buprenorphine or a pharmaceutically acceptable salt thereof of about 0.2 ng/mL or less (301) to (310), ( 301A) to (301C), (302A), (305A) to (305C), (307A), and (310A) to (310E).
- the pharmaceutical composition is a pharmaceutical composition that provides a maximum plasma concentration of buprenorphine or a pharmaceutically acceptable salt thereof of about 8 pg/mL to about 0.2 ng/mL (301) to ( 311), (301A) to (301C), (302A), (305A) to (305C), (307A), and (310A) to (310E).
- the maximum plasma concentration or plasma concentration is the maximum plasma concentration or plasma concentration at steady state, (301) to (312), (301A) to (301C), (302A), ( 305A) to (305C), (307A), and (310A) to (310E).
- the pharmaceutical composition comprises the following a)-c): a) a pharmaceutical composition for transdermal administration containing from about 0.01 mg to about 10 mg of buprenorphine or a pharmaceutically acceptable salt thereof; b) a pharmaceutical composition for transdermal administration that releases buprenorphine or a pharmaceutically acceptable salt thereof at about 0.01 ⁇ g/h to about 10 ⁇ g/h; and c) a pharmaceutical composition for transmucosal administration containing about 0.0001 mg to 0.1 mg of buprenorphine or a pharmaceutically acceptable salt thereof; Any of the above (301) to (312), (301A) to (301C), (302A), (305A) to (305C), (307A), (310A) to (310E), and (312A ).
- the pharmaceutical composition comprises the following a)-c): a) a pharmaceutical composition for transdermal administration containing from about 0.01 mg to about 5 mg of buprenorphine or a pharmaceutically acceptable salt thereof; b) a pharmaceutical composition for transdermal administration that releases buprenorphine or a pharmaceutically acceptable salt thereof at about 0.1 ⁇ g/h to about 5 ⁇ g/h; and c) a pharmaceutical composition for transmucosal administration containing 0.001 mg to 0.075 mg of buprenorphine or a pharmaceutically acceptable salt thereof; Any of the above (301) to (313), (301A) to (301C), (302A), (305A) to (305C), (307A), (310A) to (310E), and (312A ).
- the pharmaceutical composition comprises a) a pharmaceutical composition for transdermal administration containing from about 0.56 mg to about 1.7 mg of buprenorphine or a pharmaceutically acceptable salt thereof, or b) a pharmaceutical composition for transdermal administration that releases buprenorphine or a pharmaceutically acceptable salt thereof at about 0.56 ⁇ g/h to about 1.7 ⁇ g/h; Any of the above (301) to (313), (301A) to (301C), (302A), (305A) to (305C), (307A), (310A) to (310E), (312A) , and (313A).
- the pharmaceutical composition comprises a) a pharmaceutical composition for transdermal administration containing about 0.56 mg or about 1.7 mg of buprenorphine or a pharmaceutically acceptable salt thereof, or b) a pharmaceutical composition for transdermal administration that releases buprenorphine or a pharmaceutically acceptable salt thereof at about 0.56 ⁇ g/h or about 1.7 ⁇ g/h; Any of the above (301) to (313), (301A) to (301C), (302A), (305A) to (305C), (307A), (310A) to (310E), (312A) , (313A) and (313B).
- (314) The above (301) to (313), (301A) to (301C), (302A), (305A), wherein the pharmaceutical composition is a pharmaceutical composition for administration at an administration interval of 12 hours or more. - (305C), (307A), (310A) - (310E), (312A), and (313A) - (313C). (314A) The above (301)-(314), (301A)-(301C), (302A), (305A)-, wherein the pharmaceutical composition is a pharmaceutical composition for administration twice a day or less Use according to any of (305C), (307A), (310A)-(310E), (312A), and (313A)-(313C).
- (315) The above (301)-(314), (301A)-(301C), (302A), (305A)-( 305C), (307A), (310A)-(310E), (312A), (313A)-(313C), and (314A).
- (316) The above (301) to (315), (301A) to (301C), (302A), wherein the pharmaceutical composition contains only buprenorphine or a pharmaceutically acceptable salt thereof as an active ingredient. ), (305A) to (305C), (307A), (310A) to (310E), (312A), (313A) to (313C), and (314A).
- the pharmaceutical composition is a pharmaceutical composition for transdermal administration or a pharmaceutical composition for transmucosal administration (301) to (316), (301A) to (301C), (302A), (305A) ) to (305C), (307A), (310A) to (310E), (312A), (313A) to (313C), and (314A).
- the pharmaceutical composition is a pharmaceutical composition for transdermal administration, (301) to (316), (301A) to (301C), (302A), (305A) to (305C), (307A) ), (310A) to (310E), (312A), (313A) to (313C), and (314A).
- the pharmaceutical composition is a pharmaceutical composition for transmucosal administration, (301) to (316), (301A) to (301C), (302A), (305A) to (305C), (307A) ), (310A) to (310E), (312A), (313A) to (313C), and (314A).
- Buprenorphine or a pharmaceutically acceptable salt thereof and/or morphine or a pharmaceutically acceptable salt thereof is morphine or a pharmaceutically acceptable salt thereof (301) to (307), (301A) - (301C), (302A), (305A) - (305C), and (307A).
- buprenorphine or a pharmaceutically acceptable salt thereof and/or morphine or a pharmaceutically acceptable salt thereof that includes one or more of the characteristics described in any one or more of the above items.
- compositions and methods of the present invention are useful in the treatment of various psychiatric disorders, particularly Autism Spectrum Disorders, Fragile X Syndrome, Autism Spectrum Disorder-like symptoms, and/or disorders in social communication and/or social interaction. and/or useful for prophylaxis.
- Fig. 1 shows the results of evaluating the social improvement effect of morphine using valproic acid model mice, which are ASD model mice.
- Fig. 2 shows the relationship between the plasma concentration of unchanged morphine and the improvement of social disorders and analgesic efficacy.
- Fig. 2 shows the relationship between the plasma concentration of unchanged buprenorphine, improvement of social disorder, and analgesic efficacy.
- Fig. 1 shows the results of evaluating the social improvement effect of morphine using valproic acid model mice, which are ASD model mice.
- FIG. 1 shows the results of evaluating the sustained social improvement effect of buprenorphine using valproic acid model mice.
- FMR-1 knockout mice results of evaluation of buprenorphine's social improvement effect are shown.
- valproic acid model mice the results of evaluating the effect of naloxone concomitant use on the social improvement action of buprenorphine are shown.
- the results of quantifying the activation of the nucleus accumbens (NAc) by buprenorphine using valproic acid model mice are shown.
- FIG. 2 shows the results of quantification of activation of the medial prefrontal cortex by buprenorphine using valproic acid model mice.
- Fig. 2 shows the results of quantifying the activation of dorsomedial periaqueductal gray (dmPAG) by buprenorphine using valproic acid model mice.
- dmPAG dorsomedial periaqueductal gray
- ATD autism Spectrum Disorder
- ICD International Classification of Diseases
- WHO World Health Organization
- DSM-5 American Psychiatric Association
- autism spectrum disorder includes "persistent impairment of social communication and/or social interaction” and “restricted and repetitive patterns of behavior, interests, or activities” in individuals with autism spectrum disorder.
- symptoms include, for example, persistent impairment of social communication and/or social interaction.
- autism spectrum disorder-like symptoms include those who do not meet the diagnostic criteria for autism spectrum disorder but exhibit autism spectrum disorder-like symptoms.
- autism spectrum disorder-like symptoms further include autism spectrum disorder-like symptoms associated with other diseases (eg, fragile X syndrome, Rett syndrome, tuberous sclerosis (TSC), etc.).
- “Autism spectrum disorder-like symptoms” include, for example, “impaired social communication and/or social interaction (including persistent impairment)” and “limited and repetitive patterns of behavior, interests, or activities””symptoms related to”, preferably impairments (including persistent impairments) of social communication and/or social interactions.
- prevention includes avoiding the development of symptoms. Treatment includes relief, alleviation and amelioration of symptoms.
- a "pain therapeutically effective amount” is a dose effective to treat pain.
- a "pain therapeutically effective amount” for example, formulations containing 5 mg, 10 mg, and 20 mg of buprenorphine for transdermal administration have been approved, and the release rates of buprenorphine in these formulations are 5 ⁇ g/h and 10 ⁇ g/h, respectively. , 20 ⁇ g/h (Non-Patent Documents 10, 11).
- the "effective amount for pain treatment” for example, 0.075 mg, 0.15 mg, 0.3 mg, 0.6 mg, 0.75 mg, and 0.9 mg are approved as transmucosal preparations of buprenorphine for buccal film. ing.
- 200 ⁇ g, 400 ⁇ g, 2 mg, and 8 mg have been approved as sublingual tablets (Non-Patent Documents 12-14).
- 0.2 mg and 0.4 mg have been approved as suppositories (Non-Patent Document 15), and 0.2 mg and 0.4 mg have been approved as injections of buprenorphine (Non-Patent Document 16 ).
- a minimally effective pain treatment amount includes a formulation containing about 5 mg.
- the transdermal administration formulations of buprenorphine described in Non-Patent Documents 7 and 8 are formulations that are administered once every 7 days, in one embodiment, when buprenorphine is transdermally administered, the minimum amount used for pain treatment is The dose (per day) is approximately 0.7 mg containing formulation. In one aspect, when buprenorphine is transdermally administered, the minimum effective dose for pain treatment is, for example, about 5 ⁇ g/h. Minimal doses (per day) used for pain treatment include, for example, a dose of about 0.12 mg when administered at about 5 ⁇ g/h for 24 hours.
- a minimally effective pain-treating dose when buprenorphine is administered transmucosally (eg, oromucosally), a minimally effective pain-treating dose is about 0.075 mg. In one aspect, about 0.075 mg is the daily dose. In one aspect, when morphine is administered orally, a minimally effective pain treatment amount includes about 5 mg. In one aspect, when morphine is administered transmucosally or as an injection, the minimum effective dose for pain treatment is about 10 mg.
- An "autism spectrum disorder therapeutically effective amount" is an amount effective to treat and/or prevent autism spectrum disorder, fragile X syndrome, and/or autism spectrum disorder-like symptoms. Preferably, the amount is effective for treating and/or preventing autism spectrum disorders.
- Buprenorphine is 21-cyclopropyl-7- ⁇ -[(S)-1-hydroxy-1,2,2-trimethylpropyl]-6,14-endo-ethano-6,7,8,14 tetrahydrooripavine.
- Buprenorphine has the following formula: is a compound represented by Buprenorphine or a pharmaceutically acceptable salt thereof for use in the present invention includes possible isomers of buprenorphine (e.g., keto-enol isomers, imine-enamine isomers, diastereoisomers, optical isomers, rotational isomers, racemates or mixtures thereof), prodrugs or pharmaceutically acceptable salts thereof.
- One or more hydrogen, carbon and/or other atoms of the buprenorphine of the present invention may be substituted with isotopes of hydrogen, carbon and/or other atoms, respectively.
- isotopes include hydrogen, carbon, nitrogen and oxygen as 2 H, 3 H, 11 C, 13 C, 14 C, 15 N, 18 O, 17 O respectively.
- the buprenorphine of the present invention also includes buprenorphine substituted with such isotopes.
- the isotopically substituted buprenorphine is also useful as a pharmaceutical, including all radiolabeled forms of buprenorphine.
- a "radiolabeling method" for producing the "radiolabel” is also encompassed by the present invention, and the “radiolabel” is useful as a research and/or diagnostic tool in metabolic pharmacokinetic studies, binding assays. is.
- the radiolabeled buprenorphine of the present invention can be prepared by methods well known in the art.
- a tritiated compound of buprenorphine can be prepared by introducing tritium into buprenorphine through a catalytic dehalogenation reaction with tritium. The method involves reacting a precursor in which the buprenorphine is appropriately halogenated with tritium gas in the presence or absence of a base in the presence of a suitable catalyst such as Pd/C.
- a suitable catalyst such as Pd/C.
- Other suitable methods for preparing tritiated compounds can be found in "Isotopes in the Physical and Biomedical Sciences, Vol. 1, Labeled Compounds (Part A), Chapter 6 (1987)". 14 C-labeled compounds can be prepared by using starting materials with a 14 C carbon.
- Morphine has the following formula: is a compound represented by Morphine or a pharmaceutically acceptable salt thereof for use in the present invention includes possible isomers of morphine (e.g., keto-enol isomers, imine-enamine isomers, diastereoisomers, optical isomers, rotational isomers, racemates or mixtures thereof), prodrugs or pharmaceutically acceptable salts thereof.
- morphine e.g., keto-enol isomers, imine-enamine isomers, diastereoisomers, optical isomers, rotational isomers, racemates or mixtures thereof
- prodrugs e.g., prodrugs or pharmaceutically acceptable salts thereof.
- One or more hydrogen, carbon and/or other atoms of the morphine of the present invention may be substituted with isotopes of hydrogen, carbon and/or other atoms, respectively.
- isotopes include hydrogen, carbon, nitrogen and oxygen as 2 H, 3 H, 11 C, 13 C, 14 C, 15 N, 18 O, 17 O respectively.
- the morphine of the present invention also includes morphine substituted with such isotopes.
- the isotopically substituted morphine is also useful as a pharmaceutical, including all radiolabeled forms of morphine.
- a "radiolabeling method" for producing the "radiolabel” is also encompassed by the present invention, and the “radiolabel” is useful as a research and/or diagnostic tool in metabolic pharmacokinetic studies, binding assays. is.
- the radiolabeled forms of morphine of the present invention can be prepared by methods well known in the art.
- a tritiated compound of morphine can be prepared by introducing tritium into morphine by a catalytic dehalogenation reaction with tritium.
- the method involves reacting a morphine suitably halogenated precursor with tritium gas in the presence or absence of a base in the presence of a suitable catalyst such as Pd/C.
- a suitable catalyst such as Pd/C.
- Other suitable methods for preparing tritiated compounds can be found in "Isotopes in the Physical and Biomedical Sciences, Vol. 1, Labeled Compounds (Part A), Chapter 6 (1987)".
- 14 C-labeled compounds can be prepared by using starting materials with a 14 C carbon.
- the term "pharmaceutically acceptable salt” includes basic salts such as alkali metal salts such as lithium salts, sodium salts and potassium salts; alkaline earth metal salts such as calcium salts and barium salts; salts, transition metal salts such as iron salts; magnesium salts; ammonium salts; Amine salts; aralkylamine salts such as N,N-dibenzylethylenediamine; heterocyclic aromatic amine salts such as pyridine salts, picoline salts, quinoline salts and isoquinoline salts; tetramethylammonium salts, tetraethylammonium salts, benzyltrimethylammonium salts, quaternary ammonium salts such as benzyltriethylammonium salt, benzyltributylammonium salt, methyltrioctylammonium salt and tetrabutylammonium salt; basic amino acid salts such as arg
- Buprenorphine or a pharmaceutically acceptable salt thereof and morphine or a pharmaceutically acceptable salt thereof used in the present invention may be solvates, co-crystals and/or polymorphs thereof.
- Solvates include organic solvates with any number of organic solvent molecules and hydrates with any number of water molecules.
- solvate means a solvate of the compound or a pharmaceutically acceptable salt thereof, such as monosolvate, disolvate, monohydrate, dihydrate things, etc.
- hydrate, ethanolate, methyl acetate, ethyl acetate and 2-propanol, n-propyl acetate and 2-propanol, acetonitrile, 1,2-dimethoxyethane, methylisobutyl Ketone hydrates may be mentioned, preferably hydrates, eg monohydrates, eg trihydrates.
- buprenorphine or a pharmaceutically acceptable salt thereof includes buprenorphine hydrochloride.
- morphine or a pharmaceutically acceptable salt thereof includes morphine hydrochloride (eg, morphine hydrochloride hydrate) and morphine sulfate.
- An effective amount of the compound used in the present invention is optionally mixed with various pharmaceutical additives such as excipients, binders, disintegrants, and lubricants suitable for the dosage form to prepare a pharmaceutical composition.
- various pharmaceutical additives such as excipients, binders, disintegrants, and lubricants suitable for the dosage form to prepare a pharmaceutical composition.
- the pharmaceutical composition can be made into a pharmaceutical composition for children, the elderly, critically ill patients, or surgery by appropriately changing the effective amount of the compound, dosage form and/or various pharmaceutical additives. can also
- the administration route of the pharmaceutical composition of the present invention is not particularly limited, and it can be administered either orally or parenterally.
- parenteral administration methods include transdermal, subcutaneous, intravenous, intraarterial, intramuscular, intraperitoneal, transmucosal, inhalation, nasal, ocular, ear and intravaginal administration.
- the route of administration of the pharmaceutical composition of the present invention is transdermal or transmucosal administration (e.g., transoral mucosa, sublingual, suppository, preferably transoral mucosa, sublingual), e.g., transdermal administration. .
- internal solid preparations e.g., tablets, powders, granules, capsules, pills, films, etc.
- internal liquid preparations e.g., suspensions, emulsions, elixirs, syrups, etc.
- Tablets may be sugar-coated tablets, film-coated tablets, enteric-coated tablets, sustained-release tablets, troches, sublingual tablets, buccal tablets, chewable tablets or orally disintegrating tablets, and powders and granules may be dry syrups.
- the capsules may be soft capsules, microcapsules or sustained release capsules.
- injections, drops, external preparations e.g., eye drops, nasal drops, ear drops, aerosols, inhalants, lotions, injections, coatings, mouthwashes, enemas, Any commonly used dosage form such as ointments, plasters, jellies, creams, patches, poultices, powders for external use, suppositories, etc.
- Injections may be emulsions such as O/W, W/O, O/W/O and W/O/W types.
- Pharmaceutical compositions for transdermal administration are not particularly limited. ), ointments, creams, gels, patches (eg, tapes (eg, plasters, plasters), poultices), and the like. For example, a patch.
- Embodiments of the method, agent, and pharmaceutical composition of the present invention are not particularly limited, and can be appropriately determined according to the degree of symptoms, patient's body weight, age, dosage form of the agent, and the like. Aspects of the invention are exemplified below. Embodiments of the methods, medicaments, and pharmaceutical compositions of the present invention are exemplified by the embodiments and ranges of all possible combinations of the above items and the items exemplified below. Buprenorphine or a pharmaceutically acceptable salt thereof and/or morphine or a pharmaceutically acceptable salt thereof in a dose less than the pain therapeutically effective amount (e.g., about 1/2 times or less, about 1/10 times the pain therapeutically effective amount) dose below).
- the pain therapeutically effective amount e.g., about 1/2 times or less, about 1/10 times the pain therapeutically effective amount
- Buprenorphine or a pharmaceutically acceptable salt thereof and/or morphine or a pharmaceutically acceptable salt thereof about 3 times or less (e.g., about 3 times less), about 2 times or less (e.g., about 1-fold or less, about 1/2-fold or less, about 1/3-fold or less, about 1/4-fold or less).
- Buprenorphine or a pharmaceutically acceptable salt thereof and/or morphine or a pharmaceutically acceptable salt thereof at least about 1/33 times the minimum effective pain treatment dose (e.g., a dose greater than about 1/33 times), pain It is administered at about 1/30 times or more (eg, about 1/25 times or more, about 1/20 times or more, about 1/10 times or more) of the minimally effective therapeutic dose.
- Buprenorphine or a pharmaceutically acceptable salt thereof and/or morphine or a pharmaceutically acceptable salt thereof from about 1/33 to about 3 times the pain treatment minimally effective amount (e.g., greater than about 1/33 to about 3 times) less than 1/30 times), about 1/30 to about 3 times (for example, about 1/30 to about 3 times), about 1/30 to about 2 times, about 1/30 to about 1 times, about 1 /30 times to about 1/2 times, about 1/20 times to about 3 times (for example, about 1/20 times or more and less than about 3 times), about 1/20 times to about 2 times, about 1/20 times or more about 1-fold, about 1/30-fold to about 1/2-fold, about 1/10-fold to about 3-fold (for example, about 1/10-fold to about 3-fold), about 1/10-fold to about 2-fold, It is administered at about 1/10 to about 1, about 1/10 to about 1/2 times.
- the plasma concentration (preferably the maximum plasma concentration) of buprenorphine or a pharmaceutically acceptable salt thereof is, for example, about 2 ng/mL or less, about 1.5 ng/mL or less, about 1 ng/mL or less, about 0.9 ng/mL mL or less, about 0.8 ng/mL or less, about 0.7 ng/mL or less.
- it is about 0.2 ng/mL or less, for example, about 0.18 ng/mL or less, about 0.17 ng/mL or less, about 0.15 ng/mL or less, about 0.1 ng/mL or less, about 90 pg /mL or less, about 80 pg/mL or less, about 70 pg/mL or less, about 60 pg/mL or less, about 50 pg/mL or less (the plasma concentration is expressed in buprenorphine-equivalent concentration without considering the salt portion .same as below.).
- the plasma concentration (preferably the maximum plasma concentration) of buprenorphine or a pharmaceutically acceptable salt thereof is, for example, about 0.1 pg/mL or more, about 0.5 pg/mL or more, about 1 pg/mL or more, about 2 pg/mL mL or greater, about 3 pg/mL or greater, greater than about 3 pg/mL, greater than about 3.3 pg/mL.
- the concentration is, for example, about 0.9 ng/mL or less, about 0.8 ng/mL or less, about 0.7 ng/mL or less.
- concentration is, for example, about 0.6 ng/mL or less, about 0.62 ng/mL or less, about 0.6 ng/mL or less, about 0.62 ng/mL or less, about 0.5 ng/mL or less, about 0.4 ng/mL mL or less, about 0.3 ng/mL or less.
- 0.2 ng/mL or less More preferably, for example, about 0.2 ng/mL or less, about 0.18 ng/mL or less, about 0.17 ng/mL or less, about 0.15 ng/mL or less, about 0.1 ng/mL or less, about 90 pg/mL Below, about 80 pg/mL or less, about 70 pg/mL or less, about 60 pg/mL or less, about 50 pg/mL or less, about 40 pg/mL or less.
- the plasma concentration (preferably maximum plasma concentration) of buprenorphine or a pharmaceutically acceptable salt thereof is, for example, about 1 pg/mL to about 2 ng/mL or less, about 1 pg/mL to about 1 ng/mL or less, about 1 pg/mL to about 0.6 ng/mL, about 1 pg/mL to about 0.2 ng/mL, about 1 pg/mL to about 0.1 ng/mL, from about 1 pg/mL to about 50 pg/mL, about 3 pg/mL to about 2 ng/mL or less, about 3 pg/mL to about 1 ng/mL or less, about 3 pg/mL to about 0.6 ng/mL, about 3 pg/mL to about 0.3 ng/mL, about 3 pg/mL to about 0.2 ng/mL, from about 3 pg/mL to about 0.1 ng/mL, from about 3
- an inclusive range e.g., about 3 pg/mL to about 0.6 ng/mL, greater than about 3 pg/mL to about 0.6 ng/mL less than
- the plasma concentration is the peak plasma concentration at steady state.
- the plasma concentration is the plasma concentration at steady state.
- the plasma concentration is maintained continuously for 30 minutes or more, such as 1 hour or more, such as 2 hours or more.
- the content or dosage of buprenorphine or a pharmaceutically acceptable salt thereof is, for example, about 20 mg or less, about 10 mg or less, about 7 mg or less, or about 5 mg below, about 4 mg or less, about 2.5 mg or less, about 2 mg or less, about 1 mg or less, about 0.7 mg or less, about 0.5 mg or less, about 0.4 mg or less, about 0.3 mg or less, about 0.2 mg or less; It is about 0.1 mg or less (the content or dose is expressed in terms of buprenorphine without considering the salt portion; the same shall apply hereinafter).
- the content or dosage of buprenorphine or a pharmaceutically acceptable salt thereof (e.g., the content or dosage in a pharmaceutical composition for transdermal administration) is, for example, about 0.001 mg or more, about 0.003 mg or more, about 0 0.005 mg or more, about 0.01 mg or more, about 0.03 mg or more, about 0.05 mg or more, about 0.1 mg or more, about 0.3 mg or more, about 0.5 mg or more.
- the content of buprenorphine or a pharmaceutically acceptable salt thereof in the pharmaceutical composition for transdermal administration of the present invention may be about 0.56 mg to about 1.7 mg, and may be about 0.56 mg or about 1.7 mg. Sometimes there is.
- the content or dose of buprenorphine or a pharmaceutically acceptable salt thereof is, for example, about 20 ⁇ g/h or less, about 10 ⁇ g/h or less, or about 5 ⁇ g /h or less, about 2.5 ⁇ g/h or less, about 3 ⁇ g/h or less, about 2 ⁇ g/h or less, about 1 ⁇ g/h or less, about 0.1 ⁇ g/h or less (the release rate does not consider the salt portion It is expressed in terms of buprenorphine equivalent. The same shall apply hereinafter.).
- the content or dose of buprenorphine or a pharmaceutically acceptable salt thereof is, for example, about 0.01 ⁇ g/h or more, about 0.05 ⁇ g/h Above, about 0.1 ⁇ g/h or more, about 0.5 ⁇ g/h or more, about 1 ⁇ g/h or more.
- a pharmaceutical composition for transdermal administration of buprenorphine or a pharmaceutically acceptable salt thereof of the present invention may have a release rate of about 0.56 ⁇ g/h to about 1.7 ⁇ g/h, and about 0.56 ⁇ g/h. or about 1.7 ⁇ g/h.
- the content or dosage of buprenorphine or a pharmaceutically acceptable salt thereof is, for example, about 1 mg or less, about 0.5 mg or less, about 0.2 mg Below, about 0.1 mg or less, about 0.075 mg or less, about 0.05 mg or less, about 0.01 mg or less.
- the content or dosage of buprenorphine or a pharmaceutically acceptable salt thereof is, for example, about 0.00001 mg or more, about 0.0001 mg or more, about 0 0.001 mg or more, about 0.01 mg or more.
- the content or dosage of buprenorphine or a pharmaceutically acceptable salt thereof is, for example, about 0.3 ⁇ g/kg to about 30 ⁇ g/kg (eg, greater than about 0.3 ⁇ g/kg and less than about 30 ⁇ g/kg), about 0 .5 ⁇ g/kg to about 20 ⁇ g/kg, about 1 ⁇ g/kg to about 10 ⁇ g/kg.
- a 60 kg patient for example about 18 ⁇ g to about 1.8 mg (eg, greater than about 18 ⁇ g and less than about 1.8 mg), about 30 ⁇ g to about 1.2 mg, about 60 ⁇ g to about 0.6 mg. .
- the 20 ⁇ g to about 0.8 mg when administered to a 40 kg patient, for example about 12 ⁇ g to about 1.2 mg (eg, greater than about 12 ⁇ g and less than about 1.2 mg), about 20 ⁇ g to about 0.8 mg, about 40 ⁇ g to about 0.4 mg.
- the 20 kg patient when administered to a 20 kg patient, for example about 6 ⁇ g to about 0.6 mg (eg, greater than about 6 ⁇ g and less than about 0.6 mg), about 10 ⁇ g to about 0.4 mg, about 20 ⁇ g to about 0.2 mg.
- the plasma concentration (preferably the maximum plasma concentration) of morphine or a pharmaceutically acceptable salt thereof is, for example, about 500 ng/mL or less, about 350 ng/mL or less, about 300 ng/mL or less, about 200 ng/mL or less, about 100 ng/mL or less, about 10 ng/mL or less, about 7 ng/mL or less.
- Plasma concentration (preferably maximum plasma concentration) of morphine or a pharmaceutically acceptable salt thereof is, for example, about 0.1 ng/mL or more, about 0.5 ng/mL or more, about 1 ng/mL or more, about 2 ng/mL mL or greater, about 5 ng/mL or greater, about 10 ng/mL or greater.
- the plasma concentration is the peak plasma concentration at steady state. In one embodiment, the plasma concentration is the plasma concentration at steady state. In one embodiment, the plasma concentration is maintained continuously for 30 minutes or longer (eg, 1 hour or longer, 2 hours or longer).
- the content or dosage of morphine or a pharmaceutically acceptable salt thereof is, for example, 10 mg or less, 5 mg or less, 3 mg or less, 1 mg or less, or 0.1 mg or less.
- the content or dosage of morphine or a pharmaceutically acceptable salt thereof is, for example, 0.01 mg or more, 0.05 mg or more, 0.1 mg or more, 0.5 mg or more, or 1 mg or more.
- the content or dosage of morphine or a pharmaceutically acceptable salt thereof is, for example, about 0.03 mg/kg to about 0.3 mg/kg (eg, greater than about 0.03 mg/kg and less than about 0.3 mg/kg). ), from about 0.03 mg/kg to about 0.1 mg/kg.
- about 0.03 mg/kg to about 0.1 mg/kg when administered to a 60 kg patient, for example about 1.8 mg to about 18 mg (eg, greater than about 1.8 mg and less than about 18 mg), about 1.8 mg to about 6 mg.
- about 1.2 mg to about 12 mg eg, greater than about 1.2 mg and less than about 12 mg
- about 1.2 mg to about 4 mg when administered to a 40 kg patient.
- when administered to a 20 kg patient for example about 0.6 mg to about 6 mg (eg, greater than about 0.6 mg and less than about 6 mg), about 0.6 mg to about 2 mg.
- the methods, agents, medicaments, and pharmaceutical compositions of the present invention contain a single active ingredient and may be administered alone.
- the dosing interval is about 12 hours or more (eg, about 24 hours, about 24 hours or more).
- the administration frequency is twice a day or less (eg, once a day).
- the methods, agents, medicines, pharmaceutical compositions, etc. of the present invention include guidelines for treatment and / or prevention, such as specifying the target patient or subject of the present invention as a treatment subject, dosage and administration, precautions, etc. , may include package inserts and labels that are written to give instructions to medical professionals such as doctors who are engaged in prevention or treatment, but these official documents are not limited to paper media and are provided via the Internet In addition to official documents, various other sources of information can be used to provide preventive or therapeutic guidance to physicians and the like. Therefore, it is understood that the present invention encompasses embodiments that are used based on information other than package inserts and labels. As embodiments of the methods, agents, medicaments, and pharmaceutical compositions of the present invention, the may also be used, and may be used without these.
- the pharmaceutical compositions or methods of the invention preferably have one or more of the distinguishing characteristics selected from the following.
- it is useful for treating and/or preventing autism spectrum disorders.
- it is useful for treating and/or preventing core symptoms of autism spectrum disorders.
- Buprenorphine and its pharmaceutically acceptable salts and morphine or its pharmaceutically acceptable salts can be synthesized according to known methods. (Example)
- a valproic acid model mouse (VPA model) is an ASD model mouse based on clinical evidence that administration of valproic acid during pregnancy in humans significantly increases the probability of a neonate suffering from ASD. Since ASD-like symptoms such as social disorder and cognitive dysfunction are observed, it is one of the most widely used ASD model mice.
- Previous research (Hara Y, Ago Y, Higuchi M, Hasebe S, Nakazawa T, Hashimoto H, Matsuda T, Takuma K. Oxytocin attenuates deficits in social interaction but not recognition memory in a prenatal valproic acid-induced mouse model of autism. 2017 Nov;96:130-136.
- morphine, buprenorphine, or tramadol which is an MOR agonist used as an analgesic
- tramadol which is an MOR agonist used as an analgesic
- both compounds showed a characteristic dose-dependency in which the ameliorating effect on social disorders decreased.
- Tramadol which is used as an analgesic similarly to morphine and buprenorphine and has MOR agonist activity in vivo, was not found to have an ameliorating effect on social disorders.
- morphine and buprenorphine are thought to have ameliorating effects on social disturbance in ASD and be useful as therapeutic agents for ASD.
- morphine and buprenorphine have a stronger analgesic action in the high dose range, but in terms of the social improvement action, it was surprisingly found that they tend to exhibit high efficacy, especially in the low dose range.
- buprenorphine improved social disorders at a lower dose than morphine, so it is expected as a highly safe ASD therapeutic drug with a low risk of side effects.
- Example 2 Pharmacokinetics
- ICR mice were subcutaneously administered morphine hydrochloride or buprenorphine adjusted to each dose with saline, blood was collected and the brain was removed at each time point after administration, and drug concentrations in plasma and brain were determined by LC-MS. It was measured. Doses were calculated as hydrochloride for morphine and free form for buprenorphine. Results are shown in Tables 1 and 2.
- Figures 4 and 5 show the relationship between plasma concentrations of unchanged morphine and buprenorphine, improvement of social disorders, and analgesic efficacy. 4 and 5, the left axis shows the social disorder ameliorating action measured in the same manner as in Example 1, and the right axis shows the analgesic action.
- BLQ LLOQ (lower limit of quantification: 2pg/mL) or less *: Samples were quantified after 10-fold dilution.
- Example 3 Confirmation of sustained effect
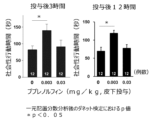
- Saline or buprenorphine was subcutaneously administered to the VPA model in the same manner as in Example 1, and the amount of social behavior was quantified for 20 minutes from 3 hours or 20 minutes from 12 hours after administration.
- the results are shown in FIG.
- the effect of 0.003 mg/kg administration of buprenorphine on improving social disorders was also observed 3 hours and 12 hours after administration.
- oxytocin which is under development as a therapeutic drug for autism spectrum disorders, has been shown to improve social disorders in a model similar to this experiment, but it has been reported that the drug effect disappears 3 hours after administration (Horm 2017 Nov;96:130-136.).
- buprenorphine has a longer lasting efficacy in improving social disorders than oxytocin, and can be a useful therapeutic drug.
- Table 2 the plasma concentration of buprenorphine at 12 hours after administration was lower than that at 3 hours after administration in each of 0.003 mg/kg and 0.03 mg/kg.
- Cmax maximum plasma concentration
- Example 4 Estimation of Preferred Buprenorphine Doses in Humans 2 and 3, buprenorphine showed a significant sociability-improving action in VPA model mice at 1 ⁇ g/kg to 10 ⁇ g/kg. From Example 2, the Cmax of buprenorphine at 0.3 ⁇ g/kg administration and 30 ⁇ g/kg administration were 26.6 pg/mL and 2360 pg/mL, respectively. Accordingly, preferred peak plasma concentrations for ASD applications in mice are within the range of about 26.6 pg/mL to about 2360 pg/mL (preferably greater than about 26.6 pg/mL and less than about 2360 pg/mL).
- the Cmax of buprenorphine at 1 ⁇ g/kg administration and 10 ⁇ g/kg administration which showed a significant social improvement effect, was 67 pg/mL (estimated) and 670 pg/mL, respectively.
- a range of intermediate concentrations includes about 67 pg/mL to 670 pg/mL.
- a latency response was observed at doses of 10 ⁇ g/kg subcutaneous administration or higher (Non-Patent Document 28), so 10 ⁇ g/kg is the minimum dose at which analgesic activity is observed.
- the Cmax at the lowest approved dose is 176 pg/mL.
- the Cmax at the lowest approved dose is 0.17 ⁇ 0.30 ng/mL.
- the Cmax at the minimum approved dose is 84 pg ⁇ 19/mL.
- a 10 ⁇ g/kg dose of buprenorphine in mice would be equivalent to a Cmax of buprenorphine in humans of approximately 84 pg/mL to 176 pg/mL.
- Preferred peak plasma concentrations for ASD applications in mice as described above are in the range of 26.6 pg/mL (at 0.3 ⁇ g/kg dose) to 2360 pg/mL (at 30 ⁇ g/kg dose) (preferably about 26 .6 pg/mL and less than about 2360 pg/mL).
- the preferred peak plasma concentration for ASD use in humans is in the range of about 3.3 pg/mL to about 620 pg/mL (preferably greater than about 3.3 pg/mL and less than about 620 pg/mL ). Furthermore, a particularly preferred peak plasma concentration in humans is estimated to be about 8.4 pg/mL to about 176 pg/mL, corresponding to the range of particularly preferred peak plasma concentrations in mice (67 pg/mL to 670 pg/mL). rice field.
- a particularly preferred dose of buprenorphine is the minimum dose approved as a pain treatment drug (for example, a transdermal dosage formulation containing 5 mg (a 7-day dosage formulation, and a daily dose of is about 0.7 mg), about 1/10 to about 1 times the dosage of transdermal administration formulations with a release rate of 5 ⁇ g/h, 75 ⁇ g transoral mucosal administration formulations, etc.).
- doses corresponding to administration of 0.3 ⁇ g/kg and administration of 30 ⁇ g/kg to mice are approximately 1/33 times and approximately 3 times the minimum doses approved as pain therapeutic drugs, respectively.
- FXS human fragile X syndrome
- ASD Non-Patent Documents 7 to 9
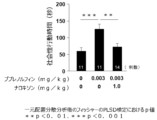
- An Fmr1 knockout mouse (Cell, 1994, 15;78(1):23-33) is a model mouse mimicking human fragile X syndrome (FXS), and is also widely used as an ASD model.
- a single dose of buprenorphine hydrochloride, a MOR partial agonist used as an analgesic, and physiological saline as a solvent was administered, and the effect on social dysfunction in Fmr1 knockout mice was examined.
- the dose of buprenorphine was calculated as the free form.
- 3 chamber test (Nature Medicine, 2017, 23; p674-677) was used.
- 3-chamber test two types of mice (Unfamiliar and Familiar) were placed in cups at the left and right ends of the three compartments, and the ratio of time spent exploring the novelty mice and the mice with low novelty was calculated. It is quantified as social function (preference for highly novel mice).
- the results are shown in FIG. Fmr1 knockout mice exhibited social impairment compared to wild-type normal B6 mice, and subcutaneous administration of buprenorphine significantly improved social impairment. This drug effect was observed at a low dose of less than 1/10 of the dose (1 mg/kg) at which the analgesic effect of buprenorphine is maximal, indicating a characteristic effect in the low dose range.
- Example 6 antagonist test
- Saline or a MOR selective antagonist, naloxone hydrochloride was subcutaneously administered as hydrochloride to the VPA model at a concentration of 1 mg/kg, and 15 minutes later, 0.003 mg/kg of buprenorphine was subcutaneously administered. Then, the amount of social behavior was quantified for 20 minutes from 1 hour after administration of buprenorphine. The results are shown in FIG. The effect of 0.003 mg/kg administration of buprenorphine on improving social impairment was antagonized by concomitant use of naloxone. From this, it was shown that buprenorphine exhibits a social disorder ameliorating effect by activating MOR.
- Example 7 Analysis of activated regions of the brain
- the number of c-Fos-positive cells in the dorsomedial periaqueductal gray (dmPAG) region was quantified as the number of activated neurons. The results are shown in Figures 9-11.
- ⁇ 1 -Opioid receptors in the dorsomedial and ventrolateral columns of the periaqueductal gray matter are critical for the enhancement of post-ictal antinociception. Synapse. 2016 Dec;70(12 ): 519-530. doi: 10.1002/syn.21926. Epub 2016 Sep 6. PMID: 27503688.) that activation of the dorsomedial periaqueductal gray matter is important for the expression of analgesia by opioids. Therefore, it is considered that a dose lower than that of buprenorphine used for analgesic purposes is preferable for improving social disorders.
- Example 8 Test to investigate the effect of low-dose buprenorphine administration on ASD symptoms The following studies are conducted to investigate the effects of ASD when low doses of buprenorphine are administered to children with autism spectrum disorders. To examine the effects of buprenorphine administration on dozens of children diagnosed with autism spectrum disorder according to DSM-5 criteria, under clinical and biological surveillance. Buprenorphine is transdermally administered to a subject using a transdermal patch at a patch dose of 0.56 mg or 1.7 mg and a release rate of about 0.56 ⁇ g/h or about 1.7 ⁇ g/h. do.
- a buprenorphine patch may be administered at 1/9 or 1/3 dose.
- Standard ASD symptom rating scales such as SRS-2 (Social Responsiveness Scale Second Edition), CARS-2 (Childhood Autism Rating Scale Second Edition), RBS-R (Repetitive Behavior Scale-Rev. ised), etc., by medication Examine the presence or absence of symptom changes.
- changes in gaze patterns in ASD patients which has been suggested as an objective symptom evaluation index, we will examine whether there are changes due to medication using a gaze measurement device.
- buprenorphine or a pharmaceutically acceptable salt thereof and/or morphine or a pharmaceutically acceptable salt thereof by any conventional route, e.g. orally, e.g., in tablet or capsule form, or parenterally, It can be administered as a pharmaceutical composition, eg in the form of an injectable solution or suspension, topically, eg in the form of a lotion, gel, ointment or cream, or in nasal or suppository form.
- a pharmaceutical composition containing the salt can be prepared in a conventional manner by mixing, granulating or coating methods.
- oral compositions can be tablets, granules, capsules containing excipients, disintegrants, binders, lubricants, etc. and active ingredients.
- injectable compositions may be in the form of solutions or suspensions, may be sterilized, and may contain preservatives, stabilizers, buffers and the like.
- compositions containing buprenorphine or a pharmaceutically acceptable salt thereof and/or morphine or a pharmaceutically acceptable salt thereof and buprenorphine or a pharmaceutically acceptable salt thereof and/or morphine or a pharmaceutically acceptable salt thereof of the present invention are useful for treating and/or preventing autism spectrum disorders, fragile X syndrome, and/or autism spectrum disorder-like symptoms. Furthermore, the pharmaceutical composition or method of the present invention has a low effective concentration and is a highly safe medicine or therapeutic method.
Landscapes
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Psychiatry (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Neurosurgery (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Emergency Medicine (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
自閉スペクトラム症、脆弱X症候群、並びに/または自閉スペクトラム症様症状を治療並びに/または予防するための、新たな医薬組成物および方法を提供する。 ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を含有する、自閉スペクトラム症、脆弱X症候群、並びに/または自閉スペクトラム症様症状の治療および/若しくは予防のための医薬組成物。
Description
本発明は、自閉スペクトラム症、脆弱X症候群、自閉スペクトラム症様症状、並びに/または社会的コミュニケーションおよび/若しくは社会的相互関係における障害を治療並びに/または予防するための、特に好ましくは自閉スペクトラム症を治療するための、新たな医薬組成物および方法に関する。
自閉スペクトラム症(ASD)は、複数の状況で社会的コミュニケーションおよび社会的相互関係における持続的障害があること、行動・興味または活動の限定された反復的な様式が2つ以上あること(情動的、反復的な身体の運動や会話、固執やこだわり、極めて限定され執着する興味、感覚刺激に対する過敏さまたは鈍感さなど)、そしてこれらの症状が発達早期から存在していることを特徴とする疾患である。世界で380万人(非特許文献1)、日本で35万人以上の患者が存在し、患者1人あたりに必要な経済的負担は年間500万円以上であるともいわれており(非特許文献2)、多くの場合、罹患期間は一生涯であることを考えるとその経済的影響は非常に大きい。しかしながら、現在のところ、ASDの中核症状に対する有効な治療薬は存在しない。ASDに対する治療としては、応用行動分析法などの行動療法が中心的であるが、行動療法には多大なる時間と人的リソースを要する上に、年齢が進むとともに効果が低下することが知られているため、その恩恵を享受できる患者は非常に限定される。以上のような背景からASDの中核症状に有効な治療薬は強く求められている。
現在ASD治療薬の標的分子として、例えばオキシトシンやバソプレシンといった社会性行動との関連が知られている分子やその受容体が注目されている。
オピオイド受容体と社会性行動については、いくつかの報告がある。1979年に脳内における過剰なオピオイドシグナルによってASD症状が引き起こされるとの説が提唱され(非特許文献3)、その後μオピオイド受容体拮抗剤であるNaltrexoneのASD治療効果について臨床試験が実施され、易刺激性などの症状については効果が確認された(非特許文献4)。その一方で、逆にオピオイドシグナルが低下することによってもASD様症状が誘発されることがμオピオイド受容体(MOR)ノックアウトマウスの解析により報告されている(非特許文献5)。また、正常ラットにおいてMORアゴニストを投与することで社会性行動が増加するとの報告も存在する(非特許文献6)。
以上のように、ASD症状や社会性行動にオピオイドシグナルが関与している可能性は示唆されているが、総説論文である非特許文献5において議論されているように、その詳細は不明であり、ASD治療においてオピオイドシグナルを低下させるのがよいのか、増強させるのがよいのかも不明である。
オピオイド受容体と社会性行動については、いくつかの報告がある。1979年に脳内における過剰なオピオイドシグナルによってASD症状が引き起こされるとの説が提唱され(非特許文献3)、その後μオピオイド受容体拮抗剤であるNaltrexoneのASD治療効果について臨床試験が実施され、易刺激性などの症状については効果が確認された(非特許文献4)。その一方で、逆にオピオイドシグナルが低下することによってもASD様症状が誘発されることがμオピオイド受容体(MOR)ノックアウトマウスの解析により報告されている(非特許文献5)。また、正常ラットにおいてMORアゴニストを投与することで社会性行動が増加するとの報告も存在する(非特許文献6)。
以上のように、ASD症状や社会性行動にオピオイドシグナルが関与している可能性は示唆されているが、総説論文である非特許文献5において議論されているように、その詳細は不明であり、ASD治療においてオピオイドシグナルを低下させるのがよいのか、増強させるのがよいのかも不明である。
X染色体の異常に起因するヒト脆弱X症候群(fragile X syndrome:FXS)の患者では、しばしばASDで見られるような社会性障害症状を呈することが知られている(非特許文献7~9)。
ブプレノルフィンは、μオピオイド受容体部分アゴニストであり、κオピオイド受容体アンタゴニストである。疼痛治療薬として、経皮投与製剤、経粘膜投与製剤(バッカル、舌下、坐剤)、注射剤(静脈内、筋肉内、皮下)の剤形が承認されている(非特許文献10~16。これらの文献の記載内容は、本出願に参照として組み込まれる)。
モルヒネは、オピオイドアゴニストであり、特にμオピオイド受容体アゴニストである。疼痛治療薬として、経口投与製剤、経粘膜投与製剤(坐剤)、注射剤(静脈内、皮下等)の剤形が承認されている(非特許文献17~22。これらの文献の記載内容は、本出願に参照として組み込まれる)。
特許文献1、2、および非特許文献23~25には、モルヒネまたはブプレノルフィンのASD治療用途について、記載も示唆もされていない。特許文献3には、オキシトシンとナロキソン(μオピオイド受容体アンタゴニスト)の併用データが記載されている。非特許文献26には、ブプレノルフィンを舌下投与したときの薬物動態が記載されている。非特許文献28には、ブプレノルフィンの鎮痛作用をマウス等を用いて評価した結果が記載されている。
モルヒネは、オピオイドアゴニストであり、特にμオピオイド受容体アゴニストである。疼痛治療薬として、経口投与製剤、経粘膜投与製剤(坐剤)、注射剤(静脈内、皮下等)の剤形が承認されている(非特許文献17~22。これらの文献の記載内容は、本出願に参照として組み込まれる)。
特許文献1、2、および非特許文献23~25には、モルヒネまたはブプレノルフィンのASD治療用途について、記載も示唆もされていない。特許文献3には、オキシトシンとナロキソン(μオピオイド受容体アンタゴニスト)の併用データが記載されている。非特許文献26には、ブプレノルフィンを舌下投与したときの薬物動態が記載されている。非特許文献28には、ブプレノルフィンの鎮痛作用をマウス等を用いて評価した結果が記載されている。
JAMA Pediatr. 2014;168(8):721-728.
Pediatrics 2014;133:e520-e529
A neurochemical theory of autism. Trends Neurosci 2: 174-177.
J Intellect Disabil Res. 2015 Apr;59(4):293-306.
Br J Pharmacol. 2018 Jul; 175(14): 2750-2769.
Vanderschuren LJ, Niesink RJ, Spruijt BM, Van Ree JM (1995b). Mu‐ and kappa‐opioid receptor‐mediated opioid effects on social play in juvenile rats. Eur J Pharmacol 276: 257-266.
Howes et al., J Psychopharmacol (2018)
RefBerry-Kravis et al., Nature Drug Dis (2018)
Castagnola et al., Fron Synaptic Neurosci (2017)
Butrans(登録商標)米国添付文書(ブプレノルフィン 5mcg/h、10mcg/h、20mcg/h経皮投与製剤)
ノルスパン(登録商標)テープ5mg、10mg、20mg 医薬品インタビューフォーム(日本)(ブプレノルフィン 5mg、10mg、20mg 経皮投与製剤)
BELBUCA(登録商標)米国添付文書(ブプレノルフィン塩酸塩 75mcg、15mcg、300mcg、450mcg、600mcg、750mcg、900mcg バッカルフィルム)
SUBTEX(登録商標)米国添付文書(ブプレノルフィン2mg、8mg 舌下錠)
Temgesic 200μg、400μg舌下錠 添付文書(ブプレノルフィン塩酸塩)
レペタン(登録商標)坐剤0.2mg、0.4mg 医薬品インタビューフォーム(日本)(ブプレノルフィン塩酸塩)
レペタン(登録商標)注0.2mg、0.4mg 医薬品インタビューフォーム(日本)(ブプレノルフィン塩酸塩)
INFUMORPH 米国添付文書(モルヒネ硫酸塩 200mg/mL、500mg/mL注射剤)
ARYMO(登録商標)ER 米国添付文書(モルヒネ硫酸塩 15mg、30mg、60mg徐放錠)
モルヒネ塩酸塩注射液10mg、50mg、200mg「タケダ」医薬品インタビューフォーム(日本)
パシーフ(登録商標)カプセル30mg、60mg、120mg 日本添付文書(モルヒネ塩酸塩水和物徐放性カプセル)
オプソ(登録商標)内服液5mg、10mg 日本添付文書(モルヒネ塩酸塩内用液剤)
アンペック(登録商標)坐剤10mg、20mg、30mg 日本添付文書(モルヒネ塩酸塩坐剤)
Social Cognitive and Affective Neuroscience, 2016, 1902-1909
Psychoneuroendocrinology (2016) 63, 43-49
Psychoneuroendocrinology (2015) 53, 10-15
AmJ Psychiatry 2016; 173:491-498; doi: 10.1176/appi.ajp.2015.15040535
Br. J. clin. Pharmac. (1982), 13, 665-673
日薬理誌 (Folia Pharmcol. Japon.) 79, 147~162 (1982)
自閉スペクトラム症、脆弱X症候群、自閉スペクトラム症様症状、並びに/または社会的コミュニケーションおよび/若しくは社会的相互関係における障害を治療並びに/または予防するための、特に好ましくは自閉症スペクトラム症を治療するための、新たな医薬組成物および方法を提供する。
本発明者らは、自閉スペクトラム症、脆弱X症候群、自閉スペクトラム症様症状、並びに/または社会的コミュニケーションおよび/若しくは社会的相互関係における障害を治療並びに/または予防するための、特に好ましくは自閉症スペクトラム症を治療するための、医薬組成物および方法を鋭意検討し、好適な有効成分を見出した。さらに、当該有効成分における好ましい投与方法、投与量等を見出した。
本発明は、例えば、以下に関する。
(1)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を含有する、自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状の治療および/または予防のための医薬組成物。
(1A)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を含有する、社会的コミュニケーションおよび/または社会的相互関係における障害を治療および/または予防するための、医薬組成物。
(1B)ブプレノルフィン若しくはその誘導体またはその製薬上許容される塩および/またはモルヒネ若しくはその誘導体(モルヒネの誘導体としては、例えばコデイン)またはその製薬上許容される塩を含有する、自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状の治療および/または予防のための医薬組成物。
(1C)μオピオイドアゴニストであるブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を含有する、自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状の治療および/または予防のための医薬組成物。
(2)自閉スペクトラム症の治療および/または予防のための、上記(1)および(1A)~(1C)のいずれかに記載の医薬組成物。
(2A)自閉スペクトラム症の中核症状の治療および/または予防のための、上記(1)および(1A)~(1C)のいずれかに記載の医薬組成物。
(3)自閉スペクトラム症と診断された患者に投与するための、上記(1)、(2)、(1A)~(1C)、および(2A)のいずれかに記載の医薬組成物。
(4)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療有効量以下の用量で投与するための、上記(1)~(3)、(1A)~(1C)、および(2A)のいずれかに記載の医薬組成物。
(5)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療有効量の約1/2倍以下の投与量で投与するための、上記(1)~(4)、(1A)~(1C)、および(2A)のいずれかに記載の医薬組成物。
(5A)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療有効量の約1/10倍以下の投与量で投与するための、上記(1)~(5)、(1A)~(1C)、および(2A)のいずれかに記載の医薬組成物。
(5B)鎮痛作用を実質的に示さない、上記(1)~(5)、(1A)~(1C)、(2A)、および(5A)のいずれかに記載の医薬組成物。
(5C)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、自閉スペクトラム症治療有効量で投与する、上記(1)~(5)、(1A)~(1C)、(2A)、(5A)、および(5B)のいずれかに記載の医薬組成物。
(6)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療最小有効量以下の投与量で投与するための上記(1)~(5)、(1A)~(1C)、(2A)、および(5A)~(5C)のいずれかに記載の医薬組成物。
(7)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療最小有効量の約1/30倍~約3倍の投与量で投与するための、上記(1)~(6)、(1A)~(1C)、(2A)、および(5A)~(5C)のいずれかに記載の医薬組成物。
(7A)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療最小有効量の約1/2倍以下の投与量で投与するための、上記(1)~(7)、(1A)~(1C)、(2A)、および(5A)~(5C)のいずれかに記載の医薬組成物。
(8)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩が、ブプレノルフィンまたはその製薬上許容される塩である、上記(1)~(7)、(1A)~(1C)、(2A)、(5A)~(5C)、および(7A)のいずれかに記載の医薬組成物。
(9)前記医薬組成物が、約1ng/mL以下のブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する、上記(1)~(8)、(1A)~(1C)、(2A)、(5A)~(5C)、および(7A)のいずれかに記載の医薬組成物。
(10)前記医薬組成物が、約4pg/mL~約0.6ng/mLのブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する、上記(1)~(9)、(1A)~(1C)、(2A)、(5A)~(5C)、および(7A)のいずれかに記載の医薬組成物。
(10A)前記医薬組成物が、3.3pg/mLより大きく、約0.6ng/mL以下のブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する、上記(1)~(10)、(1A)~(1C)、(2A)、(5A)~(5C)、および(7A)のいずれかに記載の医薬組成物。
(10B)前記医薬組成物が、約0.6ng/mL以下のブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する、上記(1)~(10)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、および(10A)のいずれかに記載の医薬組成物。
(10C)前記医薬組成物が、約1pg/mL~約1ng/mLのブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する、上記(1)~(10)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)、および(10B)のいずれかに記載の医薬組成物。
(10D)前記医薬組成物が、(例えば連続して約30分間以上)約4pg/mL~約1ng/mL(例えば約4pg/mL~約0.6ng/mL)の範囲内のブプレノルフィンの血漿中濃度を提供する、上記(1)~(10)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、および(10A)~(10C)のいずれかに記載の医薬組成物。
(10E)前記医薬組成物が、約4pg/mL~約1ng/mL(例えば約3pg/mL~約0.6ng/mL)の範囲内のブプレノルフィンの血漿中濃度を提供する、上記(1)~(10)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、および(10A)~(10D)のいずれかに記載の医薬組成物。
(11)前記医薬組成物が、約0.2ng/mL以下のブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する、上記(1)~(10)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、および(10A)~(10E)のいずれかに記載の医薬組成物。
(12)前記医薬組成物が、約8pg/mL~約0.2ng/mLのブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する、上記(1)~(11)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、および(10A)~(10E)のいずれかに記載の医薬組成物。
(12A)前記最高血漿中濃度または血漿中濃度が、定常状態における最高血漿中濃度または血漿中濃度である、上記(1)~(12)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、および(10A)~(10E)のいずれかに記載の医薬組成物。
(13)以下a)~c):
a)ブプレノルフィンまたはその製薬上許容される塩を、約0.01mg~約10mg含有する、経皮投与用医薬組成物、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.01μg/h~約10μg/hで放出する、経皮投与用医薬組成物、および、
c)ブプレノルフィンまたはその製薬上許容される塩を、約0.0001mg~0.1mg含有する、経粘膜投与用医薬組成物、
のいずれかである、上記(1)~(12)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、および(12A)のいずれかに記載の医薬組成物。
(13A)以下a)~c):
a)ブプレノルフィンまたはその製薬上許容される塩を、約0.01mg~約5mg含有する、経皮投与用医薬組成物、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.1μg/h~約5μg/hで放出する、経皮投与用医薬組成物、および、
c)ブプレノルフィンまたはその製薬上許容される塩を、0.001mg~0.075mg含有する、経粘膜投与用医薬組成物、
のいずれかである、上記(1)~(13)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、および(12A)のいずれかに記載の医薬組成物。
(13B)a)ブプレノルフィンまたはその製薬上許容される塩を、約0.56mg~約1.7mg含有する、経皮投与用医薬組成物、または、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.56μg/h~約1.7μg/hで放出する、経皮投与用医薬組成物、
のいずれかである、上記(1)~(13)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、(12A)、および(13A)のいずれかに記載の医薬組成物。
(13C)a)ブプレノルフィンまたはその製薬上許容される塩を、約0.56mgまたは約1.7mg含有する、経皮投与用医薬組成物、または、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.56μg/hまたは約1.7μg/hで放出する、経皮投与用医薬組成物、
のいずれかである、上記(1)~(13)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、(12A)、(13A)および(13B)のいずれかに記載の医薬組成物。
(14)12時間以上の投与間隔で投与するための、上記(1)~(13)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、(12A)、および(13A)~(13C)のいずれかに記載の医薬組成物。
(14A)投与回数が、1日2回以下である、上記(1)~(14)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、(12A)、および(13A)~(13C)のいずれかに記載の医薬組成物。
(15)投与後12時間以上薬効が持続する、上記(1)~(14)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、(12A)、(13A)~(13C)、および(14A)のいずれかに記載の医薬組成物。
(16)ブプレノルフィンまたはその製薬上許容される塩を、他の薬剤と併用することなく投与するための、上記(1)~(15)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、(12A)、(13A)~(13C)、および(14A)のいずれかに記載の医薬組成物。
(16A)経皮投与用医薬組成物または経粘膜投与用医薬組成物である、上記(1)~(16)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、(12A)、(13A)~(13C)、および(14A)のいずれかに記載の医薬組成物。
(16B)経皮投与用医薬組成物である、上記(1)~(16)、(1A)~(1C)、(2A)、(5A)~(5C)、(10A)~(10E)、(12A)、(13A)~(13C)、および(14A)のいずれかに記載の医薬組成物。
(16C)経粘膜投与用医薬組成物である、上記(1)~(16)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、(12A)、(13A)~(13C)、および(14A)のいずれかに記載の医薬組成物。
(17)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩が、モルヒネまたはその製薬上許容される塩である、上記(1)~(7)、(1A)~(1C)、(2A)、(5A)~(5C)、および(7A)のいずれかに記載の医薬組成物。
(18)前記医薬組成物が、約200ng/mL以下のモルヒネまたはその製薬上許容される塩の最高血漿中濃度を提供する、上記(17)記載の医薬組成物。
(19)前記医薬組成物が、約1ng/mL~約200ng/mLのモルヒネまたはその製薬上許容される塩の最高血漿中濃度を提供する、上記(17)または(18)記載の医薬組成物。
(19A)側坐核および内側前頭前皮質を活性化し、かつ、背内側水道周囲灰白質を活性化させない、上記(1)~(19)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、(12A)、(13A)~(13C)、(14A)、および(16A)~(16C)のいずれかに記載の医薬組成物。
(1A)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を含有する、社会的コミュニケーションおよび/または社会的相互関係における障害を治療および/または予防するための、医薬組成物。
(1B)ブプレノルフィン若しくはその誘導体またはその製薬上許容される塩および/またはモルヒネ若しくはその誘導体(モルヒネの誘導体としては、例えばコデイン)またはその製薬上許容される塩を含有する、自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状の治療および/または予防のための医薬組成物。
(1C)μオピオイドアゴニストであるブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を含有する、自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状の治療および/または予防のための医薬組成物。
(2)自閉スペクトラム症の治療および/または予防のための、上記(1)および(1A)~(1C)のいずれかに記載の医薬組成物。
(2A)自閉スペクトラム症の中核症状の治療および/または予防のための、上記(1)および(1A)~(1C)のいずれかに記載の医薬組成物。
(3)自閉スペクトラム症と診断された患者に投与するための、上記(1)、(2)、(1A)~(1C)、および(2A)のいずれかに記載の医薬組成物。
(4)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療有効量以下の用量で投与するための、上記(1)~(3)、(1A)~(1C)、および(2A)のいずれかに記載の医薬組成物。
(5)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療有効量の約1/2倍以下の投与量で投与するための、上記(1)~(4)、(1A)~(1C)、および(2A)のいずれかに記載の医薬組成物。
(5A)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療有効量の約1/10倍以下の投与量で投与するための、上記(1)~(5)、(1A)~(1C)、および(2A)のいずれかに記載の医薬組成物。
(5B)鎮痛作用を実質的に示さない、上記(1)~(5)、(1A)~(1C)、(2A)、および(5A)のいずれかに記載の医薬組成物。
(5C)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、自閉スペクトラム症治療有効量で投与する、上記(1)~(5)、(1A)~(1C)、(2A)、(5A)、および(5B)のいずれかに記載の医薬組成物。
(6)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療最小有効量以下の投与量で投与するための上記(1)~(5)、(1A)~(1C)、(2A)、および(5A)~(5C)のいずれかに記載の医薬組成物。
(7)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療最小有効量の約1/30倍~約3倍の投与量で投与するための、上記(1)~(6)、(1A)~(1C)、(2A)、および(5A)~(5C)のいずれかに記載の医薬組成物。
(7A)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療最小有効量の約1/2倍以下の投与量で投与するための、上記(1)~(7)、(1A)~(1C)、(2A)、および(5A)~(5C)のいずれかに記載の医薬組成物。
(8)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩が、ブプレノルフィンまたはその製薬上許容される塩である、上記(1)~(7)、(1A)~(1C)、(2A)、(5A)~(5C)、および(7A)のいずれかに記載の医薬組成物。
(9)前記医薬組成物が、約1ng/mL以下のブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する、上記(1)~(8)、(1A)~(1C)、(2A)、(5A)~(5C)、および(7A)のいずれかに記載の医薬組成物。
(10)前記医薬組成物が、約4pg/mL~約0.6ng/mLのブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する、上記(1)~(9)、(1A)~(1C)、(2A)、(5A)~(5C)、および(7A)のいずれかに記載の医薬組成物。
(10A)前記医薬組成物が、3.3pg/mLより大きく、約0.6ng/mL以下のブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する、上記(1)~(10)、(1A)~(1C)、(2A)、(5A)~(5C)、および(7A)のいずれかに記載の医薬組成物。
(10B)前記医薬組成物が、約0.6ng/mL以下のブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する、上記(1)~(10)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、および(10A)のいずれかに記載の医薬組成物。
(10C)前記医薬組成物が、約1pg/mL~約1ng/mLのブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する、上記(1)~(10)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)、および(10B)のいずれかに記載の医薬組成物。
(10D)前記医薬組成物が、(例えば連続して約30分間以上)約4pg/mL~約1ng/mL(例えば約4pg/mL~約0.6ng/mL)の範囲内のブプレノルフィンの血漿中濃度を提供する、上記(1)~(10)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、および(10A)~(10C)のいずれかに記載の医薬組成物。
(10E)前記医薬組成物が、約4pg/mL~約1ng/mL(例えば約3pg/mL~約0.6ng/mL)の範囲内のブプレノルフィンの血漿中濃度を提供する、上記(1)~(10)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、および(10A)~(10D)のいずれかに記載の医薬組成物。
(11)前記医薬組成物が、約0.2ng/mL以下のブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する、上記(1)~(10)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、および(10A)~(10E)のいずれかに記載の医薬組成物。
(12)前記医薬組成物が、約8pg/mL~約0.2ng/mLのブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する、上記(1)~(11)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、および(10A)~(10E)のいずれかに記載の医薬組成物。
(12A)前記最高血漿中濃度または血漿中濃度が、定常状態における最高血漿中濃度または血漿中濃度である、上記(1)~(12)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、および(10A)~(10E)のいずれかに記載の医薬組成物。
(13)以下a)~c):
a)ブプレノルフィンまたはその製薬上許容される塩を、約0.01mg~約10mg含有する、経皮投与用医薬組成物、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.01μg/h~約10μg/hで放出する、経皮投与用医薬組成物、および、
c)ブプレノルフィンまたはその製薬上許容される塩を、約0.0001mg~0.1mg含有する、経粘膜投与用医薬組成物、
のいずれかである、上記(1)~(12)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、および(12A)のいずれかに記載の医薬組成物。
(13A)以下a)~c):
a)ブプレノルフィンまたはその製薬上許容される塩を、約0.01mg~約5mg含有する、経皮投与用医薬組成物、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.1μg/h~約5μg/hで放出する、経皮投与用医薬組成物、および、
c)ブプレノルフィンまたはその製薬上許容される塩を、0.001mg~0.075mg含有する、経粘膜投与用医薬組成物、
のいずれかである、上記(1)~(13)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、および(12A)のいずれかに記載の医薬組成物。
(13B)a)ブプレノルフィンまたはその製薬上許容される塩を、約0.56mg~約1.7mg含有する、経皮投与用医薬組成物、または、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.56μg/h~約1.7μg/hで放出する、経皮投与用医薬組成物、
のいずれかである、上記(1)~(13)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、(12A)、および(13A)のいずれかに記載の医薬組成物。
(13C)a)ブプレノルフィンまたはその製薬上許容される塩を、約0.56mgまたは約1.7mg含有する、経皮投与用医薬組成物、または、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.56μg/hまたは約1.7μg/hで放出する、経皮投与用医薬組成物、
のいずれかである、上記(1)~(13)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、(12A)、(13A)および(13B)のいずれかに記載の医薬組成物。
(14)12時間以上の投与間隔で投与するための、上記(1)~(13)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、(12A)、および(13A)~(13C)のいずれかに記載の医薬組成物。
(14A)投与回数が、1日2回以下である、上記(1)~(14)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、(12A)、および(13A)~(13C)のいずれかに記載の医薬組成物。
(15)投与後12時間以上薬効が持続する、上記(1)~(14)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、(12A)、(13A)~(13C)、および(14A)のいずれかに記載の医薬組成物。
(16)ブプレノルフィンまたはその製薬上許容される塩を、他の薬剤と併用することなく投与するための、上記(1)~(15)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、(12A)、(13A)~(13C)、および(14A)のいずれかに記載の医薬組成物。
(16A)経皮投与用医薬組成物または経粘膜投与用医薬組成物である、上記(1)~(16)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、(12A)、(13A)~(13C)、および(14A)のいずれかに記載の医薬組成物。
(16B)経皮投与用医薬組成物である、上記(1)~(16)、(1A)~(1C)、(2A)、(5A)~(5C)、(10A)~(10E)、(12A)、(13A)~(13C)、および(14A)のいずれかに記載の医薬組成物。
(16C)経粘膜投与用医薬組成物である、上記(1)~(16)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、(12A)、(13A)~(13C)、および(14A)のいずれかに記載の医薬組成物。
(17)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩が、モルヒネまたはその製薬上許容される塩である、上記(1)~(7)、(1A)~(1C)、(2A)、(5A)~(5C)、および(7A)のいずれかに記載の医薬組成物。
(18)前記医薬組成物が、約200ng/mL以下のモルヒネまたはその製薬上許容される塩の最高血漿中濃度を提供する、上記(17)記載の医薬組成物。
(19)前記医薬組成物が、約1ng/mL~約200ng/mLのモルヒネまたはその製薬上許容される塩の最高血漿中濃度を提供する、上記(17)または(18)記載の医薬組成物。
(19A)側坐核および内側前頭前皮質を活性化し、かつ、背内側水道周囲灰白質を活性化させない、上記(1)~(19)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、(12A)、(13A)~(13C)、(14A)、および(16A)~(16C)のいずれかに記載の医薬組成物。
(101)必要とする患者にブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を投与することを含む、自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状の治療および/または予防方法。
(101A)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を投与することを含む、社会的コミュニケーションおよび/または社会的相互関係における障害を治療および/または予防する方法。
(101B)ブプレノルフィン若しくはその誘導体またはその製薬上許容される塩および/またはモルヒネ若しくはその誘導体(モルヒネの誘導体としては、例えばコデイン)またはその製薬上許容される塩を投与することを含む、自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状を治療および/または予防する方法。
(101C)μオピオイドアゴニストであるブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を投与することを含む、自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状を治療および/または予防する方法。
(102)自閉スペクトラム症の治療および/または予防のための、上記(101)および(101A)~(101C)のいずれかに記載の方法。
(102A)自閉スペクトラム症の中核症状の治療および/または予防のための、上記(101)および(101A)~(101C)のいずれかに記載の方法。
(103)自閉スペクトラム症と診断された患者に投与するための、上記(101)、(102)、(101A)~(101C)、および(102A)のいずれかに記載の方法。
(104)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療有効量以下の用量で投与することを含む、上記(101)~(103)、(101A)~(101C)、および(102A)のいずれかに記載の方法。
(105)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療有効量の約1/2倍以下の投与量で投与することを含む、上記(101)~(104)、(101A)~(101C)、および(102A)のいずれかに記載の方法。
(105A)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療有効量の約1/10倍以下の投与量で投与することを含む、上記(101)~(105)、(101A)~(101C)、および(102A)のいずれかに記載の方法。
(105B)鎮痛作用を実質的に示さない、上記(101)~(105)、(101A)~(101C)、(102A)、および(105A)のいずれかに記載の方法。
(105C)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、自閉スペクトラム症治療有効量で投与する、上記(101)~(105)、(101A)~(101C)、(102A)、(105A)、および(105B)のいずれかに記載の方法。
(106)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療最小有効量以下の投与量で投与する、上記(101)~(105)、(101A)~(101C)、(102A)、および(105A)~(105C)のいずれかに記載の方法。
(107)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療最小有効量の約1/30倍~約3倍の投与量で投与する、上記(101)~(106)、(101A)~(101C)、(102A)、および(105A)~(105C)のいずれかに記載の方法。
(107A)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療最小有効量の約1/2倍以下の投与量で投与するための、上記(101)~(107)、(101A)~(101C)、(102A)、および(105A)~(105C)のいずれかに記載の方法。
(108)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩が、ブプレノルフィンまたはその製薬上許容される塩である、上記(101)~(107)、(101A)~(101C)、(102A)、(105A)~(105C)、および(107A)のいずれかに記載の方法。
(109)投与された患者におけるブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度が、約1ng/mL以下である、上記(101)~(108)、(101A)~(101C)、(102A)、(105A)~(105C)、および(107A)のいずれかに記載の方法。
(110)投与された患者におけるブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度が、約4pg/mL~約0.6ng/mLである、上記(101)~(109)、(101A)~(101C)、(102A)、(105A)~(105C)、および(107A)のいずれかに記載の方法。
(110A)投与された患者におけるブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度が、3.3pg/mLより大きく、約0.6ng/mL以下である、上記(101)~(110)、(101A)~(101C)、(102A)、(105A)~(105C)、および(107A)のいずれかに記載の方法。
(110B)投与された患者におけるブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度が、約0.6ng/mL以下である、上記(101)~(110)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、および(110A)のいずれかに記載の方法。
(110C)投与された患者におけるブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度が、約1pg/mL~約1ng/mLである、上記(101)~(110)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)(110A)、および(110B)のいずれかに記載の方法。
(110D)投与された患者におけるブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度が、(例えば連続して約30分間以上)約4pg/mL~約1ng/mL(例えば約4pg/mL~約0.6ng/mL)の範囲内である、上記(101)~(110)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)および(110A)~(110C)のいずれかに記載の方法。
(110E)投与された患者におけるブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度が、約4pg/mL~約1ng/mL(例えば約3pg/mL~約0.6ng/mL)の範囲内である、上記(101)~(110)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)および(110A)~(110D)のいずれかに記載の方法。
(111)投与された患者におけるブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度が、約0.2ng/mL以下である、上記(101)~(110)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、および(110A)~(110E)のいずれかに記載の方法。
(112)投与された患者におけるブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度が、約8pg/mL~約0.2ng/mLである、上記(101)~(111)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、および(110A)~(110E)のいずれかに記載の方法。
(112A)前記最高血漿中濃度または血漿中濃度が、定常状態における最高血漿中濃度または血漿中濃度である、上記(101)~(112)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、および(110A)~(110E)のいずれかに記載の方法。
(113)以下a)~c):
a)ブプレノルフィンまたはその製薬上許容される塩を、約0.01mg~約10mg経皮投与する、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.01μg/h~約10μg/hで経皮投与する、および、
c)ブプレノルフィンまたはその製薬上許容される塩を、約0.0001mg~0.1mg経粘膜投与する、
のいずれかを含む、上記(101)~(112)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、および(112A)のいずれかに記載の方法。
(113A)以下a)~c):
a)ブプレノルフィンまたはその製薬上許容される塩を、約0.01mg~約5mg経皮投与する、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.1μg/h~約5μg/hで経皮投与する、および、
c)ブプレノルフィンまたはその製薬上許容される塩を、0.001mg~0.075mg経粘膜投与する、
のいずれかである、上記(101)~(113)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、および(112A)のいずれかに記載の方法。
(113B)a)ブプレノルフィンまたはその製薬上許容される塩を、約0.56mg~約1.7mg経皮投与する、または、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.56μg/h~約1.7μg/hで経皮投与する、
のいずれかである、上記(101)~(113)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、(112A)、および(113A)のいずれかに記載の方法。
(113C)a)ブプレノルフィンまたはその製薬上許容される塩を、約0.56mgまたは約1.7mg経皮投与する、または
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.56μg/hまたは約1.7μg/hで経皮投与する、
のいずれかである、上記(101)~(113)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、(112A)、(113A)および(113B)のいずれかに記載の方法。
(114)ブプレノルフィンまたはその製薬上許容される塩を12時間以上の投与間隔で投与する、上記(101)~(113)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、(112A)、および(113A)~(113C)のいずれかに記載の方法。
(114A)投与回数が、1日2回以下である、上記(101)~(114)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、(112A)、および(113A)~(113C)のいずれかに記載の方法。
(115)投与後12時間以上薬効が持続する、上記(101)~(114)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、(112A)、(113A)~(113C)、および(114A)のいずれかに記載の方法。
(116)ブプレノルフィンまたはその製薬上許容される塩を、他の薬剤と併用することなく投与する、上記(101)~(115)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、(112A)、(113A)~(113C)、および(114A)のいずれかに記載の方法。
(116A)ブプレノルフィンまたはその製薬上許容される塩を、経皮投与または経粘膜投与する、上記(101)~(116)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、(112A)、(113A)~(113C)、および(114A)のいずれかに記載の方法。
(116B)ブプレノルフィンまたはその製薬上許容される塩を、経皮投与する、上記(101)~(116)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、(112A)、(113A)~(113C)、および(114A)のいずれかに記載の方法。
(116C)ブプレノルフィンまたはその製薬上許容される塩を、経粘膜投与する、上記(101)~(116)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、(112A)、(113A)~(113C)、および(114A)のいずれかに記載の方法。
(117)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩が、モルヒネまたはその製薬上許容される塩である、上記(101)~(107)、(101A)~(101C)、(102A)、(105A)~(105C)、および(107A)のいずれかに記載の方法。
(118)モルヒネまたはその製薬上許容される塩の最高血漿中濃度が、約200ng/mL以下である、上記(117)記載の方法。
(119)モルヒネまたはその製薬上許容される塩の最高血漿中濃度が、約1ng/mL~約200ng/mLである、上記(117)または(118)記載の方法。
(119A)側坐核および内側前頭前皮質を活性化し、かつ、背内側水道周囲灰白質を活性化させない、上記(101)~(119)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、(112A)、(113A)~(113C)、(114A)、および(116A)~(116C)のいずれかに記載の方法。
(101A)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を投与することを含む、社会的コミュニケーションおよび/または社会的相互関係における障害を治療および/または予防する方法。
(101B)ブプレノルフィン若しくはその誘導体またはその製薬上許容される塩および/またはモルヒネ若しくはその誘導体(モルヒネの誘導体としては、例えばコデイン)またはその製薬上許容される塩を投与することを含む、自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状を治療および/または予防する方法。
(101C)μオピオイドアゴニストであるブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を投与することを含む、自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状を治療および/または予防する方法。
(102)自閉スペクトラム症の治療および/または予防のための、上記(101)および(101A)~(101C)のいずれかに記載の方法。
(102A)自閉スペクトラム症の中核症状の治療および/または予防のための、上記(101)および(101A)~(101C)のいずれかに記載の方法。
(103)自閉スペクトラム症と診断された患者に投与するための、上記(101)、(102)、(101A)~(101C)、および(102A)のいずれかに記載の方法。
(104)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療有効量以下の用量で投与することを含む、上記(101)~(103)、(101A)~(101C)、および(102A)のいずれかに記載の方法。
(105)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療有効量の約1/2倍以下の投与量で投与することを含む、上記(101)~(104)、(101A)~(101C)、および(102A)のいずれかに記載の方法。
(105A)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療有効量の約1/10倍以下の投与量で投与することを含む、上記(101)~(105)、(101A)~(101C)、および(102A)のいずれかに記載の方法。
(105B)鎮痛作用を実質的に示さない、上記(101)~(105)、(101A)~(101C)、(102A)、および(105A)のいずれかに記載の方法。
(105C)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、自閉スペクトラム症治療有効量で投与する、上記(101)~(105)、(101A)~(101C)、(102A)、(105A)、および(105B)のいずれかに記載の方法。
(106)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療最小有効量以下の投与量で投与する、上記(101)~(105)、(101A)~(101C)、(102A)、および(105A)~(105C)のいずれかに記載の方法。
(107)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療最小有効量の約1/30倍~約3倍の投与量で投与する、上記(101)~(106)、(101A)~(101C)、(102A)、および(105A)~(105C)のいずれかに記載の方法。
(107A)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療最小有効量の約1/2倍以下の投与量で投与するための、上記(101)~(107)、(101A)~(101C)、(102A)、および(105A)~(105C)のいずれかに記載の方法。
(108)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩が、ブプレノルフィンまたはその製薬上許容される塩である、上記(101)~(107)、(101A)~(101C)、(102A)、(105A)~(105C)、および(107A)のいずれかに記載の方法。
(109)投与された患者におけるブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度が、約1ng/mL以下である、上記(101)~(108)、(101A)~(101C)、(102A)、(105A)~(105C)、および(107A)のいずれかに記載の方法。
(110)投与された患者におけるブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度が、約4pg/mL~約0.6ng/mLである、上記(101)~(109)、(101A)~(101C)、(102A)、(105A)~(105C)、および(107A)のいずれかに記載の方法。
(110A)投与された患者におけるブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度が、3.3pg/mLより大きく、約0.6ng/mL以下である、上記(101)~(110)、(101A)~(101C)、(102A)、(105A)~(105C)、および(107A)のいずれかに記載の方法。
(110B)投与された患者におけるブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度が、約0.6ng/mL以下である、上記(101)~(110)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、および(110A)のいずれかに記載の方法。
(110C)投与された患者におけるブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度が、約1pg/mL~約1ng/mLである、上記(101)~(110)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)(110A)、および(110B)のいずれかに記載の方法。
(110D)投与された患者におけるブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度が、(例えば連続して約30分間以上)約4pg/mL~約1ng/mL(例えば約4pg/mL~約0.6ng/mL)の範囲内である、上記(101)~(110)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)および(110A)~(110C)のいずれかに記載の方法。
(110E)投与された患者におけるブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度が、約4pg/mL~約1ng/mL(例えば約3pg/mL~約0.6ng/mL)の範囲内である、上記(101)~(110)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)および(110A)~(110D)のいずれかに記載の方法。
(111)投与された患者におけるブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度が、約0.2ng/mL以下である、上記(101)~(110)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、および(110A)~(110E)のいずれかに記載の方法。
(112)投与された患者におけるブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度が、約8pg/mL~約0.2ng/mLである、上記(101)~(111)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、および(110A)~(110E)のいずれかに記載の方法。
(112A)前記最高血漿中濃度または血漿中濃度が、定常状態における最高血漿中濃度または血漿中濃度である、上記(101)~(112)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、および(110A)~(110E)のいずれかに記載の方法。
(113)以下a)~c):
a)ブプレノルフィンまたはその製薬上許容される塩を、約0.01mg~約10mg経皮投与する、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.01μg/h~約10μg/hで経皮投与する、および、
c)ブプレノルフィンまたはその製薬上許容される塩を、約0.0001mg~0.1mg経粘膜投与する、
のいずれかを含む、上記(101)~(112)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、および(112A)のいずれかに記載の方法。
(113A)以下a)~c):
a)ブプレノルフィンまたはその製薬上許容される塩を、約0.01mg~約5mg経皮投与する、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.1μg/h~約5μg/hで経皮投与する、および、
c)ブプレノルフィンまたはその製薬上許容される塩を、0.001mg~0.075mg経粘膜投与する、
のいずれかである、上記(101)~(113)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、および(112A)のいずれかに記載の方法。
(113B)a)ブプレノルフィンまたはその製薬上許容される塩を、約0.56mg~約1.7mg経皮投与する、または、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.56μg/h~約1.7μg/hで経皮投与する、
のいずれかである、上記(101)~(113)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、(112A)、および(113A)のいずれかに記載の方法。
(113C)a)ブプレノルフィンまたはその製薬上許容される塩を、約0.56mgまたは約1.7mg経皮投与する、または
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.56μg/hまたは約1.7μg/hで経皮投与する、
のいずれかである、上記(101)~(113)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、(112A)、(113A)および(113B)のいずれかに記載の方法。
(114)ブプレノルフィンまたはその製薬上許容される塩を12時間以上の投与間隔で投与する、上記(101)~(113)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、(112A)、および(113A)~(113C)のいずれかに記載の方法。
(114A)投与回数が、1日2回以下である、上記(101)~(114)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、(112A)、および(113A)~(113C)のいずれかに記載の方法。
(115)投与後12時間以上薬効が持続する、上記(101)~(114)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、(112A)、(113A)~(113C)、および(114A)のいずれかに記載の方法。
(116)ブプレノルフィンまたはその製薬上許容される塩を、他の薬剤と併用することなく投与する、上記(101)~(115)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、(112A)、(113A)~(113C)、および(114A)のいずれかに記載の方法。
(116A)ブプレノルフィンまたはその製薬上許容される塩を、経皮投与または経粘膜投与する、上記(101)~(116)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、(112A)、(113A)~(113C)、および(114A)のいずれかに記載の方法。
(116B)ブプレノルフィンまたはその製薬上許容される塩を、経皮投与する、上記(101)~(116)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、(112A)、(113A)~(113C)、および(114A)のいずれかに記載の方法。
(116C)ブプレノルフィンまたはその製薬上許容される塩を、経粘膜投与する、上記(101)~(116)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、(112A)、(113A)~(113C)、および(114A)のいずれかに記載の方法。
(117)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩が、モルヒネまたはその製薬上許容される塩である、上記(101)~(107)、(101A)~(101C)、(102A)、(105A)~(105C)、および(107A)のいずれかに記載の方法。
(118)モルヒネまたはその製薬上許容される塩の最高血漿中濃度が、約200ng/mL以下である、上記(117)記載の方法。
(119)モルヒネまたはその製薬上許容される塩の最高血漿中濃度が、約1ng/mL~約200ng/mLである、上記(117)または(118)記載の方法。
(119A)側坐核および内側前頭前皮質を活性化し、かつ、背内側水道周囲灰白質を活性化させない、上記(101)~(119)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、(112A)、(113A)~(113C)、(114A)、および(116A)~(116C)のいずれかに記載の方法。
(201)自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状の治療および/または予防に用いるための、ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩。
(201A)社会的コミュニケーションおよび/または社会的相互関係における障害の治療および/または予防に用いるための、ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩。
(201B)自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状の治療および/または予防のための、ブプレノルフィン若しくはその誘導体またはその製薬上許容される塩および/またはモルヒネ若しくはその誘導体(モルヒネの誘導体としては、例えばコデイン)またはその製薬上許容される塩。
(201C)自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状の治療および/または予防のための、μオピオイドアゴニストであるブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩。
(202)自閉スペクトラム症の治療および/または予防のための、上記(201)および(201A)~(201C)のいずれかに記載のブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩。
(202A)自閉スペクトラム症の中核症状の治療および/または予防のための、上記(201)および(201A)~(201C)のいずれかに記載のブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩。
(203)自閉スペクトラム症と診断された患者に投与するための、上記(201)、(202)、(201A)~(201C)、および(202A)のいずれかに記載のブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩。
上記(1)~(19)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、(12A)、(13A)~(13C)、(14A)、(16A)~(16C)、(101)~(119)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、(112A)、(113A)~(113C)、(114A)、および(116A)~(116C)(以下、上記項目群とする)のいずれか1項または複数に記載の特徴を1つまたは複数含む、上記(201)~(203)、(201A)~(201C)、および(202A)のいずれかに記載のブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩。
(201A)社会的コミュニケーションおよび/または社会的相互関係における障害の治療および/または予防に用いるための、ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩。
(201B)自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状の治療および/または予防のための、ブプレノルフィン若しくはその誘導体またはその製薬上許容される塩および/またはモルヒネ若しくはその誘導体(モルヒネの誘導体としては、例えばコデイン)またはその製薬上許容される塩。
(201C)自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状の治療および/または予防のための、μオピオイドアゴニストであるブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩。
(202)自閉スペクトラム症の治療および/または予防のための、上記(201)および(201A)~(201C)のいずれかに記載のブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩。
(202A)自閉スペクトラム症の中核症状の治療および/または予防のための、上記(201)および(201A)~(201C)のいずれかに記載のブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩。
(203)自閉スペクトラム症と診断された患者に投与するための、上記(201)、(202)、(201A)~(201C)、および(202A)のいずれかに記載のブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩。
上記(1)~(19)、(1A)~(1C)、(2A)、(5A)~(5C)、(7A)、(10A)~(10E)、(12A)、(13A)~(13C)、(14A)、(16A)~(16C)、(101)~(119)、(101A)~(101C)、(102A)、(105A)~(105C)、(107A)、(110A)~(110E)、(112A)、(113A)~(113C)、(114A)、および(116A)~(116C)(以下、上記項目群とする)のいずれか1項または複数に記載の特徴を1つまたは複数含む、上記(201)~(203)、(201A)~(201C)、および(202A)のいずれかに記載のブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩。
(301)自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状の治療および/または予防のための医薬組成物の製造のための、ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩の使用。
(301A)社会的コミュニケーションおよび/または社会的相互関係における障害を治療および/または予防するための医薬組成物の製造のための、ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩の使用。
(301B)自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状の治療および/または予防のための医薬組成物の製造のための、ブプレノルフィン若しくはその誘導体またはその製薬上許容される塩および/またはモルヒネ若しくはその誘導体(モルヒネの誘導体としては、例えばコデイン)またはその製薬上許容される塩の使用。
(301C)自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状の治療および/または予防のための医薬組成物の製造のための、μオピオイドアゴニストであるブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩の使用。
(302)自閉スペクトラム症の治療および/または予防のための医薬組成物の製造のための、上記(301)および(301A)~(301C)のいずれかに記載の使用。
(302A)自閉スペクトラム症の中核症状の治療および/または予防のための医薬組成物の製造のための、上記(301)および(301A)~(301C)のいずれかに記載の使用。
(303)自閉スペクトラム症と診断された患者に投与するための医薬組成物の製造のための、上記(301)、(302)、(301A)~(301C)、および(302A)のいずれかに記載の使用。
(304)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩の含有量が疼痛治療有効量以下である医薬組成物の製造のための、上記(301)~(303)、(301A)~(301C)、および(302A)のいずれかに記載の使用。
(305)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩の含有量が疼痛治療有効量の約1/2倍以下である医薬組成物の製造のための、上記(301)~(304)、(301A)~(301C)、および(302A)のいずれかに記載の使用。
(305A)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩の含有量が、疼痛治療有効量の約1/10倍以下である医薬組成物の製造のための、上記(301)~(305)、(301A)~(301C)、および(302A)のいずれかに記載の使用。
(305B)該医薬組成物が鎮痛作用を実質的に示さない、上記(301)~(305)、(301A)~(301C)、(302A)、および(305A)のいずれかに記載の使用。
(305C)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩の含有量が、自閉スペクトラム症治療有効量である医薬組成物の製造のための、上記(301)~(305)、(301A)~(301C)、(302A)、(305A)、および(305B)のいずれかに記載の使用。
(306)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩の含有量が、疼痛治療最小有効量以下である医薬組成物の製造のための、上記(301)~(305)、(301A)~(301C)、(302A)、および(305A)~(305C)のいずれかに記載の使用。
(307)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩の含有量が、疼痛治療最小有効量の約1/30倍~約3倍である医薬組成物の製造のための、上記(301)~(306)、(301A)~(301C)、(302A)、および(305A)~(305C)のいずれかに記載の使用。
(307A)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩の含有量が、疼痛治療最小有効量の約1/2倍以下である医薬組成物の製造のための、上記(301)~(307)、(301A)~(301C)、(302A)、および(305A)~(305C)のいずれかに記載の使用。
(308)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩が、ブプレノルフィンまたはその製薬上許容される塩である、上記(301)~(307)、(301A)~(301C)、(302A)、(305A)~(305C)、および(307A)のいずれかに記載の使用。
(309)前記医薬組成物が、約1ng/mL以下のブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する医薬組成物である、上記(301)~(308)、(301A)~(301C)、(302A)、(305A)~(305C)、および(307A)のいずれかに記載の使用。
(310)前記医薬組成物が、約4pg/mL~約0.6ng/mLのブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する医薬組成物である、上記(301)~(309)、(301A)~(301C)、(302A)、(305A)~(305C)、および(307A)のいずれかに記載の使用。
(310A)前記医薬組成物が、3.3pg/mLより大きく、約0.6ng/mL以下のブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する医薬組成物である、上記(301)~(310)、(301A)~(301C)、(302A)、(305A)~(305C)、および(307A)のいずれかに記載の使用。
(310B)前記医薬組成物が、約0.6ng/mL以下のブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する医薬組成物である、上記(301)~(310)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、および(310A)のいずれかに記載の使用。
(310C)前記医薬組成物が、約1pg/mL~約1ng/mLのブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する医薬組成物である、上記(301)~(310)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)、および(310B)のいずれかに記載の使用。
(310D)前記医薬組成物が、(例えば連続して約30分間以上)約4pg/mL~約1ng/mL(例えば約4pg/mL~約0.6ng/mL)の範囲内のブプレノルフィンの血漿中濃度を提供する医薬組成物である、上記(301)~(310)、(301A)~(301C)、(302A)、(305A)~(305C)、および(310A)~(310C)のいずれかに記載の使用。
(310E)前記医薬組成物が、約4pg/mL~約1ng/mL(例えば約3pg/mL~約0.6ng/mL)の範囲内のブプレノルフィンの血漿中濃度を提供する医薬組成物である、上記(301)~(310)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、および(310A)~(310D)のいずれかに記載の使用。
(311)前記医薬組成物が、約0.2ng/mL以下のブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する医薬組成物である、上記(301)~(310)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、および(310A)~(310E)のいずれかに記載の使用。
(312)前記医薬組成物が、約8pg/mL~約0.2ng/mLのブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する医薬組成物である、上記(301)~(311)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、および(310A)~(310E)のいずれかに記載の使用。
(312A)前記最高血漿中濃度または血漿中濃度が、定常状態における最高血漿中濃度または血漿中濃度である、上記(301)~(312)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、および(310A)~(310E)のいずれかに記載の使用。
(313)前記医薬組成物が、以下a)~c):
a)ブプレノルフィンまたはその製薬上許容される塩を、約0.01mg~約10mg含有する、経皮投与用医薬組成物、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.01μg/h~約10μg/hで放出する、経皮投与用医薬組成物、および、
c)ブプレノルフィンまたはその製薬上許容される塩を、約0.0001mg~0.1mg含有する、経粘膜投与用医薬組成物、
のいずれかである、上記(301)~(312)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、および(312A)のいずれかに記載の使用。
(313A)前記医薬組成物が、以下a)~c):
a)ブプレノルフィンまたはその製薬上許容される塩を、約0.01mg~約5mg含有する、経皮投与用医薬組成物、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.1μg/h~約5μg/hで放出する、経皮投与用医薬組成物、および、
c)ブプレノルフィンまたはその製薬上許容される塩を、0.001mg~0.075mg含有する、経粘膜投与用医薬組成物、
のいずれかである、上記(301)~(313)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、および(312A)のいずれかに記載の使用。
(313B)前記医薬組成物が、
a)ブプレノルフィンまたはその製薬上許容される塩を、約0.56mg~約1.7mg含有する、経皮投与用医薬組成物、または、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.56μg/h~約1.7μg/hで放出する、経皮投与用医薬組成物、
のいずれかである、上記(301)~(313)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、(312A)、および(313A)のいずれかに記載の使用。
(313C)前記医薬組成物が、
a)ブプレノルフィンまたはその製薬上許容される塩を、約0.56mgまたは約1.7mg含有する、経皮投与用医薬組成物、または、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.56μg/hまたは約1.7μg/hで放出する、経皮投与用医薬組成物、
のいずれかである、上記(301)~(313)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、(312A)、(313A)および(313B)のいずれかに記載の使用。
(314)前記医薬組成物が、12時間以上の投与間隔で投与するための医薬組成物である、上記(301)~(313)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、(312A)、および(313A)~(313C)のいずれかに記載の使用。
(314A)前記医薬組成物が、1日2回以下で投与するための医薬組成物である、上記(301)~(314)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、(312A)、および(313A)~(313C)のいずれかに記載の使用。
(315)前記医薬組成物が、投与後12時間以上薬効が持続する医薬組成物である、上記(301)~(314)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、(312A)、(313A)~(313C)、および(314A)のいずれかに記載の医薬組成物。
(316)前記医薬組成物が、有効成分としてブプレノルフィンまたはその製薬上許容される塩のみを含有する医薬組成物である、上記(301)~(315)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、(312A)、(313A)~(313C)、および(314A)のいずれかに記載の使用。
(316A)前記医薬組成物が、経皮投与用医薬組成物または経粘膜投与用医薬組成物である、上記(301)~(316)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、(312A)、(313A)~(313C)、および(314A)のいずれかに記載の使用。
(316B)前記医薬組成物が、経皮投与用医薬組成物である、上記(301)~(316)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、(312A)、(313A)~(313C)、および(314A)のいずれかに記載の使用。
(316C)前記医薬組成物が、経粘膜投与用医薬組成物である、上記(301)~(316)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、(312A)、(313A)~(313C)、および(314A)のいずれかに記載の使用。
(317)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩が、モルヒネまたはその製薬上許容される塩である、上記(301)~(307)、(301A)~(301C)、(302A)、(305A)~(305C)、および(307A)のいずれかに記載の使用。
(318)前記医薬組成物が、約200ng/mL以下のモルヒネまたはその製薬上許容される塩の最高血漿中濃度を提供する医薬組成物である、上記(317)記載の使用。
(319)前記医薬組成物が、約1ng/mL~約200ng/mLのモルヒネまたはその製薬上許容される塩の最高血漿中濃度を提供する医薬組成物である、上記(317)または(318)記載の使用。
(319A)前記医薬組成物が、側坐核および内側前頭前皮質を活性化し、かつ、背内側水道周囲灰白質を活性化させないための医薬組成物である、上記(301)~(319)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、(312A)、(313A)~(313C)、(314A)、および(316A)~(316C)のいずれかに記載の使用。
(301A)社会的コミュニケーションおよび/または社会的相互関係における障害を治療および/または予防するための医薬組成物の製造のための、ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩の使用。
(301B)自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状の治療および/または予防のための医薬組成物の製造のための、ブプレノルフィン若しくはその誘導体またはその製薬上許容される塩および/またはモルヒネ若しくはその誘導体(モルヒネの誘導体としては、例えばコデイン)またはその製薬上許容される塩の使用。
(301C)自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状の治療および/または予防のための医薬組成物の製造のための、μオピオイドアゴニストであるブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩の使用。
(302)自閉スペクトラム症の治療および/または予防のための医薬組成物の製造のための、上記(301)および(301A)~(301C)のいずれかに記載の使用。
(302A)自閉スペクトラム症の中核症状の治療および/または予防のための医薬組成物の製造のための、上記(301)および(301A)~(301C)のいずれかに記載の使用。
(303)自閉スペクトラム症と診断された患者に投与するための医薬組成物の製造のための、上記(301)、(302)、(301A)~(301C)、および(302A)のいずれかに記載の使用。
(304)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩の含有量が疼痛治療有効量以下である医薬組成物の製造のための、上記(301)~(303)、(301A)~(301C)、および(302A)のいずれかに記載の使用。
(305)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩の含有量が疼痛治療有効量の約1/2倍以下である医薬組成物の製造のための、上記(301)~(304)、(301A)~(301C)、および(302A)のいずれかに記載の使用。
(305A)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩の含有量が、疼痛治療有効量の約1/10倍以下である医薬組成物の製造のための、上記(301)~(305)、(301A)~(301C)、および(302A)のいずれかに記載の使用。
(305B)該医薬組成物が鎮痛作用を実質的に示さない、上記(301)~(305)、(301A)~(301C)、(302A)、および(305A)のいずれかに記載の使用。
(305C)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩の含有量が、自閉スペクトラム症治療有効量である医薬組成物の製造のための、上記(301)~(305)、(301A)~(301C)、(302A)、(305A)、および(305B)のいずれかに記載の使用。
(306)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩の含有量が、疼痛治療最小有効量以下である医薬組成物の製造のための、上記(301)~(305)、(301A)~(301C)、(302A)、および(305A)~(305C)のいずれかに記載の使用。
(307)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩の含有量が、疼痛治療最小有効量の約1/30倍~約3倍である医薬組成物の製造のための、上記(301)~(306)、(301A)~(301C)、(302A)、および(305A)~(305C)のいずれかに記載の使用。
(307A)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩の含有量が、疼痛治療最小有効量の約1/2倍以下である医薬組成物の製造のための、上記(301)~(307)、(301A)~(301C)、(302A)、および(305A)~(305C)のいずれかに記載の使用。
(308)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩が、ブプレノルフィンまたはその製薬上許容される塩である、上記(301)~(307)、(301A)~(301C)、(302A)、(305A)~(305C)、および(307A)のいずれかに記載の使用。
(309)前記医薬組成物が、約1ng/mL以下のブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する医薬組成物である、上記(301)~(308)、(301A)~(301C)、(302A)、(305A)~(305C)、および(307A)のいずれかに記載の使用。
(310)前記医薬組成物が、約4pg/mL~約0.6ng/mLのブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する医薬組成物である、上記(301)~(309)、(301A)~(301C)、(302A)、(305A)~(305C)、および(307A)のいずれかに記載の使用。
(310A)前記医薬組成物が、3.3pg/mLより大きく、約0.6ng/mL以下のブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する医薬組成物である、上記(301)~(310)、(301A)~(301C)、(302A)、(305A)~(305C)、および(307A)のいずれかに記載の使用。
(310B)前記医薬組成物が、約0.6ng/mL以下のブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する医薬組成物である、上記(301)~(310)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、および(310A)のいずれかに記載の使用。
(310C)前記医薬組成物が、約1pg/mL~約1ng/mLのブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する医薬組成物である、上記(301)~(310)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)、および(310B)のいずれかに記載の使用。
(310D)前記医薬組成物が、(例えば連続して約30分間以上)約4pg/mL~約1ng/mL(例えば約4pg/mL~約0.6ng/mL)の範囲内のブプレノルフィンの血漿中濃度を提供する医薬組成物である、上記(301)~(310)、(301A)~(301C)、(302A)、(305A)~(305C)、および(310A)~(310C)のいずれかに記載の使用。
(310E)前記医薬組成物が、約4pg/mL~約1ng/mL(例えば約3pg/mL~約0.6ng/mL)の範囲内のブプレノルフィンの血漿中濃度を提供する医薬組成物である、上記(301)~(310)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、および(310A)~(310D)のいずれかに記載の使用。
(311)前記医薬組成物が、約0.2ng/mL以下のブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する医薬組成物である、上記(301)~(310)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、および(310A)~(310E)のいずれかに記載の使用。
(312)前記医薬組成物が、約8pg/mL~約0.2ng/mLのブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する医薬組成物である、上記(301)~(311)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、および(310A)~(310E)のいずれかに記載の使用。
(312A)前記最高血漿中濃度または血漿中濃度が、定常状態における最高血漿中濃度または血漿中濃度である、上記(301)~(312)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、および(310A)~(310E)のいずれかに記載の使用。
(313)前記医薬組成物が、以下a)~c):
a)ブプレノルフィンまたはその製薬上許容される塩を、約0.01mg~約10mg含有する、経皮投与用医薬組成物、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.01μg/h~約10μg/hで放出する、経皮投与用医薬組成物、および、
c)ブプレノルフィンまたはその製薬上許容される塩を、約0.0001mg~0.1mg含有する、経粘膜投与用医薬組成物、
のいずれかである、上記(301)~(312)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、および(312A)のいずれかに記載の使用。
(313A)前記医薬組成物が、以下a)~c):
a)ブプレノルフィンまたはその製薬上許容される塩を、約0.01mg~約5mg含有する、経皮投与用医薬組成物、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.1μg/h~約5μg/hで放出する、経皮投与用医薬組成物、および、
c)ブプレノルフィンまたはその製薬上許容される塩を、0.001mg~0.075mg含有する、経粘膜投与用医薬組成物、
のいずれかである、上記(301)~(313)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、および(312A)のいずれかに記載の使用。
(313B)前記医薬組成物が、
a)ブプレノルフィンまたはその製薬上許容される塩を、約0.56mg~約1.7mg含有する、経皮投与用医薬組成物、または、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.56μg/h~約1.7μg/hで放出する、経皮投与用医薬組成物、
のいずれかである、上記(301)~(313)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、(312A)、および(313A)のいずれかに記載の使用。
(313C)前記医薬組成物が、
a)ブプレノルフィンまたはその製薬上許容される塩を、約0.56mgまたは約1.7mg含有する、経皮投与用医薬組成物、または、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.56μg/hまたは約1.7μg/hで放出する、経皮投与用医薬組成物、
のいずれかである、上記(301)~(313)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、(312A)、(313A)および(313B)のいずれかに記載の使用。
(314)前記医薬組成物が、12時間以上の投与間隔で投与するための医薬組成物である、上記(301)~(313)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、(312A)、および(313A)~(313C)のいずれかに記載の使用。
(314A)前記医薬組成物が、1日2回以下で投与するための医薬組成物である、上記(301)~(314)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、(312A)、および(313A)~(313C)のいずれかに記載の使用。
(315)前記医薬組成物が、投与後12時間以上薬効が持続する医薬組成物である、上記(301)~(314)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、(312A)、(313A)~(313C)、および(314A)のいずれかに記載の医薬組成物。
(316)前記医薬組成物が、有効成分としてブプレノルフィンまたはその製薬上許容される塩のみを含有する医薬組成物である、上記(301)~(315)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、(312A)、(313A)~(313C)、および(314A)のいずれかに記載の使用。
(316A)前記医薬組成物が、経皮投与用医薬組成物または経粘膜投与用医薬組成物である、上記(301)~(316)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、(312A)、(313A)~(313C)、および(314A)のいずれかに記載の使用。
(316B)前記医薬組成物が、経皮投与用医薬組成物である、上記(301)~(316)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、(312A)、(313A)~(313C)、および(314A)のいずれかに記載の使用。
(316C)前記医薬組成物が、経粘膜投与用医薬組成物である、上記(301)~(316)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、(312A)、(313A)~(313C)、および(314A)のいずれかに記載の使用。
(317)ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩が、モルヒネまたはその製薬上許容される塩である、上記(301)~(307)、(301A)~(301C)、(302A)、(305A)~(305C)、および(307A)のいずれかに記載の使用。
(318)前記医薬組成物が、約200ng/mL以下のモルヒネまたはその製薬上許容される塩の最高血漿中濃度を提供する医薬組成物である、上記(317)記載の使用。
(319)前記医薬組成物が、約1ng/mL~約200ng/mLのモルヒネまたはその製薬上許容される塩の最高血漿中濃度を提供する医薬組成物である、上記(317)または(318)記載の使用。
(319A)前記医薬組成物が、側坐核および内側前頭前皮質を活性化し、かつ、背内側水道周囲灰白質を活性化させないための医薬組成物である、上記(301)~(319)、(301A)~(301C)、(302A)、(305A)~(305C)、(307A)、(310A)~(310E)、(312A)、(313A)~(313C)、(314A)、および(316A)~(316C)のいずれかに記載の使用。
上記項目群のいずれか1項または複数に記載の特徴を1つまたは複数含む、ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩の使用。
本発明の医薬組成物および方法は、各種精神疾患、特に、自閉症スペクトラム症、脆弱X症候群、自閉スペクトラム症様症状、並びに/または社会的コミュニケーションおよび/若しくは社会的相互関係における障害の治療並びに/または予防のために有用である。
「からなる」という用語は、構成要件のみを有することを意味する。「含む」という用語は、構成要件に限定されず、記載されていない要素を排除しないことを意味する。
以下、本発明について実施形態を示しながら説明する。本明細書の全体にわたり、単数形の表現は、特に言及しない限り、その複数形の概念をも含むことが理解されるべきである。従って、単数形の冠詞(例えば、英語の場合は「a」、「an」、「the」など)は、特に言及しない限り、その複数形の概念をも含むことが理解されるべきである。
また、本明細書において使用される用語は、特に言及しない限り、当上記分野で通常用いられる意味で用いられることが理解されるべきである。したがって、他に定義されない限り、本明細書中で使用される全ての専門用語および科学技術用語は、本発明の属する分野の当業者によって一般的に理解されるのと同じ意味を有する。矛盾する場合、本明細書(定義を含めて)が優先する。
また、本明細書において使用される用語は、特に言及しない限り、当上記分野で通常用いられる意味で用いられることが理解されるべきである。したがって、他に定義されない限り、本明細書中で使用される全ての専門用語および科学技術用語は、本発明の属する分野の当業者によって一般的に理解されるのと同じ意味を有する。矛盾する場合、本明細書(定義を含めて)が優先する。
「自閉スペクトラム症(Autistic Spectrum Disorder:ASD)」とは、神経発達症群(神経発達障害群)の一種であり、自閉性障害、アスペルガー障害、特定不能の広汎性発達障害を包含する。その用語(定義)、障害の特徴、及び診断基準は、世界保健機関(WHO)の作成した「国際疾病分類(International Classification of Diseases:ICD)第11版」及び米国精神医学会による「DSM-5(登録商標)精神疾患の診断・統計マニュアル」等を参照することができる。なお、自閉スペクトラム症を的確に診断可能な基準であればよく、上記ICD-11及びDSM-5に特に限定されない。
自閉スペクトラム症の中核症状としては、自閉スペクトラム症患者における、「社会的コミュニケーションおよび/または社会的相互関係の持続的障害」および「行動、興味、または活動の限定された反復的な様式に関する症状」が挙げられ、例えば、社会的コミュニケーションおよび/または社会的相互関係の持続的障害である。
「自閉スペクトラム症様症状」としては、自閉スペクトラム症の診断基準を満たさないものの、自閉スペクトラム症様の症状を示す場合を含む。「自閉スペクトラム症様症状」としては、さらに、他の疾患(例えば脆弱X症候群、レット症候群、結節性硬化症(TSC)等)に伴う自閉スペクトラム症様の症状を含む。「自閉スペクトラム症様症状」としては、例えば、「社会的コミュニケーションおよび/または社会的相互関係の障害(持続的障害を含む)」および「行動、興味、または活動の限定された反復的な様式に関する症状」が挙げられ、好ましくは、社会的コミュニケーションおよび/または社会的相互関係の障害(持続的障害を含む)である。
自閉スペクトラム症の中核症状としては、自閉スペクトラム症患者における、「社会的コミュニケーションおよび/または社会的相互関係の持続的障害」および「行動、興味、または活動の限定された反復的な様式に関する症状」が挙げられ、例えば、社会的コミュニケーションおよび/または社会的相互関係の持続的障害である。
「自閉スペクトラム症様症状」としては、自閉スペクトラム症の診断基準を満たさないものの、自閉スペクトラム症様の症状を示す場合を含む。「自閉スペクトラム症様症状」としては、さらに、他の疾患(例えば脆弱X症候群、レット症候群、結節性硬化症(TSC)等)に伴う自閉スペクトラム症様の症状を含む。「自閉スペクトラム症様症状」としては、例えば、「社会的コミュニケーションおよび/または社会的相互関係の障害(持続的障害を含む)」および「行動、興味、または活動の限定された反復的な様式に関する症状」が挙げられ、好ましくは、社会的コミュニケーションおよび/または社会的相互関係の障害(持続的障害を含む)である。
「脆弱X症候群(Fragile X syndrome)」、および「レット症候群」の用語(定義)、障害の特徴、及び診断基準は、例えば、世界保健機関(WHO)の作成した「国際疾病分類(International Classification of Diseases:ICD)第11版」を参照することができる。
本明細書において、予防は、症状の発生の回避を含む。治療は、症状の軽減、緩和、改善を含む。
「疼痛治療有効量」とは、疼痛を治療するために有効な用量である。
「疼痛治療有効量」として、例えば、ブプレノルフィンの経皮投与製剤としては、5mg、10mg、20mg含有製剤が承認されており、これらの製剤におけるブプレノルフィンの放出速度はそれぞれ、5μg/h、10μg/h、20μg/hである(非特許文献10、11)。
「疼痛治療有効量」として、例えば、ブプレノルフィンの経粘膜投与製剤としては、例えばバッカルフィルムとして、0.075mg、0.15mg、0.3mg、0.6mg、0.75mg、0.9mgが承認されている。また、例えば舌下錠としては、200μg、400μg、2mg、8mgが承認されている(非特許文献12~14)。また、例えば坐剤としては、0.2mg、0.4mgが承認されている(非特許文献15)ブプレノルフィンの注射剤としては、0.2mg、0.4mgが承認されている(非特許文献16)。
「疼痛治療有効量」として、例えば、モルヒネの経口投与製剤としては、5mg、10mg、15mg、30mg、60mgが承認されている(非特許文献18、21)。例えば、モルヒネの注射剤としては、10mg、50mg、200mgが承認されている(非特許文献19)。例えば、モルヒネの経粘膜投与製剤(坐剤)としては、10mg、20mg、30mgが承認されている(非特許文献22)。
「疼痛治療最小有効量」とは、疼痛治療に用いられる最小用量である。例えば、疼痛治療薬として承認された最小用量である。
一つの態様として、ブプレノルフィンを経皮投与する場合、疼痛治療最小有効量としては、約5mg含有製剤が挙げられる。ここで、非特許文献7、8記載のブプレノルフィンの経皮投与製剤は7日間に1回投与する製剤であるから、一つの態様としては、ブプレノルフィンを経皮投与する場合、疼痛治療に用いられる最小用量(1日当たり)は、約0.7mg含有製剤である。
一つの態様として、ブプレノルフィンを経皮投与する場合、疼痛治療最小有効量としては、例えば約5μg/hである。疼痛治療に用いられる最小用量(1日当たり)としては、例えば、約5μg/hで24時間投与した場合の投与量、すなわち約0.12mgが挙げられる。
一つの態様として、ブプレノルフィンを経粘膜投与(例えば経口腔粘膜投与)する場合、疼痛治療最小有効量としては、約0.075mgが挙げられる。一つの態様として、約0.075mgは、1日あたりの投与量である。
一つの態様として、モルヒネを経口投与する場合、疼痛治療最小有効量としては、約5mgが挙げられる。
一つの態様として、モルヒネを経粘膜投与する場合、または注射剤の場合、疼痛治療最小有効量としては、約10mgが挙げられる。
「自閉スペクトラム症治療有効量」とは、自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状を治療および/または予防するために有効な量である。好ましくは、自閉スペクトラム症を治療および/または予防するために有効な量である。
「疼痛治療有効量」とは、疼痛を治療するために有効な用量である。
「疼痛治療有効量」として、例えば、ブプレノルフィンの経皮投与製剤としては、5mg、10mg、20mg含有製剤が承認されており、これらの製剤におけるブプレノルフィンの放出速度はそれぞれ、5μg/h、10μg/h、20μg/hである(非特許文献10、11)。
「疼痛治療有効量」として、例えば、ブプレノルフィンの経粘膜投与製剤としては、例えばバッカルフィルムとして、0.075mg、0.15mg、0.3mg、0.6mg、0.75mg、0.9mgが承認されている。また、例えば舌下錠としては、200μg、400μg、2mg、8mgが承認されている(非特許文献12~14)。また、例えば坐剤としては、0.2mg、0.4mgが承認されている(非特許文献15)ブプレノルフィンの注射剤としては、0.2mg、0.4mgが承認されている(非特許文献16)。
「疼痛治療有効量」として、例えば、モルヒネの経口投与製剤としては、5mg、10mg、15mg、30mg、60mgが承認されている(非特許文献18、21)。例えば、モルヒネの注射剤としては、10mg、50mg、200mgが承認されている(非特許文献19)。例えば、モルヒネの経粘膜投与製剤(坐剤)としては、10mg、20mg、30mgが承認されている(非特許文献22)。
「疼痛治療最小有効量」とは、疼痛治療に用いられる最小用量である。例えば、疼痛治療薬として承認された最小用量である。
一つの態様として、ブプレノルフィンを経皮投与する場合、疼痛治療最小有効量としては、約5mg含有製剤が挙げられる。ここで、非特許文献7、8記載のブプレノルフィンの経皮投与製剤は7日間に1回投与する製剤であるから、一つの態様としては、ブプレノルフィンを経皮投与する場合、疼痛治療に用いられる最小用量(1日当たり)は、約0.7mg含有製剤である。
一つの態様として、ブプレノルフィンを経皮投与する場合、疼痛治療最小有効量としては、例えば約5μg/hである。疼痛治療に用いられる最小用量(1日当たり)としては、例えば、約5μg/hで24時間投与した場合の投与量、すなわち約0.12mgが挙げられる。
一つの態様として、ブプレノルフィンを経粘膜投与(例えば経口腔粘膜投与)する場合、疼痛治療最小有効量としては、約0.075mgが挙げられる。一つの態様として、約0.075mgは、1日あたりの投与量である。
一つの態様として、モルヒネを経口投与する場合、疼痛治療最小有効量としては、約5mgが挙げられる。
一つの態様として、モルヒネを経粘膜投与する場合、または注射剤の場合、疼痛治療最小有効量としては、約10mgが挙げられる。
「自閉スペクトラム症治療有効量」とは、自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状を治療および/または予防するために有効な量である。好ましくは、自閉スペクトラム症を治療および/または予防するために有効な量である。
本明細書において、文脈が別途指図しない限り、数値Xの前に「約(about)」と記載したときは、数値Xの+/-10%の範囲を含み、かつ、数値Xの有効数字を考慮し、最も近い有効数字に四捨五入される数値を含む。
例えば、約1ng/mLは、0.9ng/mLおよび1.4ng/mLを含む。
例えば、約1ng/mLは、0.9ng/mLおよび1.4ng/mLを含む。
ブプレノルフィンは、21-cyclopropyl-7-α-[(S)-1-hydroxy-1,2,2-trimethylpropyl]-6,14-endo-ethano-6,7,8,14 tetrahydrooripavineである。
ブプレノルフィンは、以下の式:

で示される化合物である。
本発明で用いられるブプレノルフィンまたはその製薬上許容される塩は、ブプレノルフィンの可能な異性体(例えば、ケト-エノール異性体、イミン-エナミン異性体、ジアステレオ異性体、光学異性体、回転異性体、ラセミ体またはそれらの混合物等)、プロドラッグまたはその製薬上許容される塩として投与されてもよい。
ブプレノルフィンは、以下の式:
で示される化合物である。
本発明で用いられるブプレノルフィンまたはその製薬上許容される塩は、ブプレノルフィンの可能な異性体(例えば、ケト-エノール異性体、イミン-エナミン異性体、ジアステレオ異性体、光学異性体、回転異性体、ラセミ体またはそれらの混合物等)、プロドラッグまたはその製薬上許容される塩として投与されてもよい。
本発明のブプレノルフィンの一つ以上の水素、炭素および/または他の原子は、それぞれ水素、炭素および/または他の原子の同位体で置換され得る。そのような同位体の例としては、それぞれ2H、3H、11C、13C、14C、15N、18O、17Oのように、水素、炭素、窒素および酸素が包含される。本発明のブプレノルフィンは、そのような同位体で置換されたブプレノルフィンも包含する。該同位体で置換されたブプレノルフィンは、医薬品としても有用であり、ブプレノルフィンのすべての放射性標識体を包含する。また該「放射性標識体」を製造するための「放射性標識化方法」も本発明に包含され、該「放射性標識体」は、代謝薬物動態研究、結合アッセイにおける研究および/または診断のツールとして有用である。
本発明のブプレノルフィンの放射性標識体は、当該技術分野で周知の方法で調製できる。例えば、ブプレノルフィンのトリチウム標識化合物は、トリチウムを用いた触媒的脱ハロゲン化反応によって、ブプレノルフィンにトリチウムを導入することで調製できる。この方法は、適切な触媒、例えばPd/Cの存在下、塩基の存在下または非存在下で、ブプレノルフィンが適切にハロゲン置換された前駆体とトリチウムガスとを反応させることを包含する。トリチウム標識化合物を調製するための他の適切な方法は、“Isotopes in the Physical and Biomedical Sciences,Vol.1,Labeled Compounds (Part A),Chapter 6 (1987年)”を参照することができる。14C-標識化合物は、14C炭素を有する原料を用いることによって調製できる。
モルヒネは、以下の式:

で示される化合物である。
本発明で用いられるモルヒネまたはその製薬上許容される塩は、モルヒネの可能な異性体(例えば、ケト-エノール異性体、イミン-エナミン異性体、ジアステレオ異性体、光学異性体、回転異性体、ラセミ体またはそれらの混合物等)、プロドラッグまたはその製薬上許容される塩として投与されてもよい。
で示される化合物である。
本発明で用いられるモルヒネまたはその製薬上許容される塩は、モルヒネの可能な異性体(例えば、ケト-エノール異性体、イミン-エナミン異性体、ジアステレオ異性体、光学異性体、回転異性体、ラセミ体またはそれらの混合物等)、プロドラッグまたはその製薬上許容される塩として投与されてもよい。
本発明のモルヒネの一つ以上の水素、炭素および/または他の原子は、それぞれ水素、炭素および/または他の原子の同位体で置換され得る。そのような同位体の例としては、それぞれ2H、3H、11C、13C、14C、15N、18O、17Oのように、水素、炭素、窒素および酸素が包含される。本発明のモルヒネは、そのような同位体で置換されたモルヒネも包含する。該同位体で置換されたモルヒネは、医薬品としても有用であり、モルヒネのすべての放射性標識体を包含する。また該「放射性標識体」を製造するための「放射性標識化方法」も本発明に包含され、該「放射性標識体」は、代謝薬物動態研究、結合アッセイにおける研究および/または診断のツールとして有用である。
本発明のモルヒネの放射性標識体は、当該技術分野で周知の方法で調製できる。例えば、モルヒネのトリチウム標識化合物は、トリチウムを用いた触媒的脱ハロゲン化反応によって、モルヒネにトリチウムを導入することで調製できる。この方法は、適切な触媒、例えばPd/Cの存在下、塩基の存在下または非存在下で、モルヒネが適切にハロゲン置換された前駆体とトリチウムガスとを反応させることを包含する。トリチウム標識化合物を調製するための他の適切な方法は、“Isotopes in the Physical and Biomedical Sciences,Vol.1,Labeled Compounds (Part A),Chapter 6 (1987年)”を参照することができる。14C-標識化合物は、14C炭素を有する原料を用いることによって調製できる。
本明細書において「製薬上許容される塩」としては、塩基性塩として、例えば、リチウム塩、ナトリウム塩、カリウム塩等のアルカリ金属塩;カルシウム塩、バリウム塩等のアルカリ土類金属塩;亜鉛塩、鉄塩等の遷移金属塩;マグネシウム塩;アンモニウム塩;トリメチルアミン塩、トリエチルアミン塩、ジシクロヘキシルアミン塩、エタノールアミン塩、ジエタノールアミン塩、トリエタノールアミン塩、エチレンジアミン塩、メグルミン塩、プロカイン塩等の脂肪族アミン塩;N,N-ジベンジルエチレンジアミン等のアラルキルアミン塩;ピリジン塩、ピコリン塩、キノリン塩、イソキノリン塩等のヘテロ環芳香族アミン塩;テトラメチルアンモニウム塩、テトラエチルアンモニウム塩、ベンジルトリメチルアンモニウム塩、ベンジルトリエチルアンモニウム塩、ベンジルトリブチルアンモニウム塩、メチルトリオクチルアンモニウム塩、テトラブチルアンモニウム塩等の第4級アンモニウム塩;アルギニン塩、リジン塩等の塩基性アミノ酸塩等が挙げられる。酸性塩としては、例えば、塩酸塩、硫酸塩、硝酸塩、リン酸塩、炭酸塩、炭酸水素塩、臭化水素酸塩、ヨウ化水素酸塩、過塩素酸塩等の無機酸塩;ギ酸塩、酢酸塩、プロピオン酸塩、トリフルオロ酢酸塩、クエン酸塩、乳酸塩、酒石酸塩、シュウ酸塩、マレイン酸塩、フマル酸塩、コハク酸塩、マンデル酸塩、グルタル酸塩、リンゴ酸塩、安息香酸塩、フタル酸塩、アスコルビン酸塩等の有機酸塩;メタンスルホン酸塩、エタンスルホン酸塩、イセチオン酸塩、ベンゼンスルホン酸塩、p-トルエンスルホン酸塩等のスルホン酸塩;アスパラギン酸塩、グルタミン酸塩等の酸性アミノ酸塩等が挙げられる。これらの塩は、通常行われる方法によって形成させることができる。
「製薬上許容される塩」としては、酸性塩が好ましく、より好ましくは、塩酸塩または硫酸塩であり、特に好ましくは、塩酸塩である。
「製薬上許容される塩」としては、酸性塩が好ましく、より好ましくは、塩酸塩または硫酸塩であり、特に好ましくは、塩酸塩である。
本発明で用いられるブプレノルフィンまたはその製薬上許容される塩ならびにモルヒネまたはその製薬上許容される塩は、それらの溶媒和物、共結晶および/または結晶多形であってもよい。
溶媒和物は、任意の数の有機溶媒分子を配位する有機溶媒和物及び任意の数の水分子を配位する水和物を包含する。本明細書における「溶媒和物」としては、当該化合物又はその製薬上許容される塩の溶媒和物を意味し、例えば、一溶媒和物、二溶媒和物、一水和物、二水和物等が挙げられる。例えば、水和物、エタノール和物、酢酸メチル和物、酢酸エチルおよび2-プロパノール和物、酢酸n-プロピルおよび2-プロパノール和物、アセトニトリル和物、1,2-ジメトキシエタン和物、メチルイソブチルケトン和物が挙げられ、好ましくは、水和物、例えば一水和物、例えば三水和物が挙げられる。
ブプレノルフィンまたはその製薬上許容される塩の一つの態様としては、ブプレノルフィン塩酸塩が挙げられる。
モルヒネまたはその製薬上許容される塩の一つの態様としては、モルヒネ塩酸塩(例えば、モルヒネ塩酸塩水和物)およびモルヒネ硫酸塩が挙げられる。
溶媒和物は、任意の数の有機溶媒分子を配位する有機溶媒和物及び任意の数の水分子を配位する水和物を包含する。本明細書における「溶媒和物」としては、当該化合物又はその製薬上許容される塩の溶媒和物を意味し、例えば、一溶媒和物、二溶媒和物、一水和物、二水和物等が挙げられる。例えば、水和物、エタノール和物、酢酸メチル和物、酢酸エチルおよび2-プロパノール和物、酢酸n-プロピルおよび2-プロパノール和物、アセトニトリル和物、1,2-ジメトキシエタン和物、メチルイソブチルケトン和物が挙げられ、好ましくは、水和物、例えば一水和物、例えば三水和物が挙げられる。
ブプレノルフィンまたはその製薬上許容される塩の一つの態様としては、ブプレノルフィン塩酸塩が挙げられる。
モルヒネまたはその製薬上許容される塩の一つの態様としては、モルヒネ塩酸塩(例えば、モルヒネ塩酸塩水和物)およびモルヒネ硫酸塩が挙げられる。
本発明に用いられる化合物の有効量にその剤型に適した賦形剤、結合剤、崩壊剤、滑沢剤等の各種医薬用添加剤を必要に応じて混合し、医薬組成物とすることができる。さらに、該医薬組成物は、化合物の有効量、剤型及び/又は各種医薬用添加剤を適宜変更することにより、小児用、高齢者用、重症患者用又は手術用の医薬組成物とすることもできる。
本発明の医薬組成物の投与経路は、特に限定されず、経口、非経口のいずれの方法でも投与することができる。非経口投与の方法としては、経皮、皮下、静脈内、動脈内、筋肉内、腹腔内、経粘膜、吸入、経鼻、点眼、点耳、膣内投与等が挙げられる。
一つの態様として、本発明の医薬組成物の投与経路は、経皮または経粘膜投与(例えば経口腔粘膜、舌下、坐剤、好ましくは経口腔粘膜、舌下)、例えば経皮投与である。
一つの態様として、本発明の医薬組成物の投与経路は、経皮または経粘膜投与(例えば経口腔粘膜、舌下、坐剤、好ましくは経口腔粘膜、舌下)、例えば経皮投与である。
経口投与の場合は常法に従って、内用固形製剤(例えば、錠剤、散剤、顆粒剤、カプセル剤、丸剤、フィルム剤等)、内用液剤(例えば、懸濁剤、乳剤、エリキシル剤、シロップ剤、リモナーデ剤、酒精剤、芳香水剤、エキス剤、煎剤、チンキ剤等)等の通常用いられるいずれの剤型に調製して投与すればよい。錠剤は、糖衣錠、フィルムコーティング錠、腸溶性コーティング錠、徐放錠、トローチ錠、舌下錠、バッカル錠、チュアブル錠または口腔内崩壊錠であってもよく、散剤および顆粒剤はドライシロップであってもよく、カプセル剤は、ソフトカプセル剤、マイクロカプセル剤または徐放性カプセル剤であってもよい。
非経口投与の場合は、注射剤、点滴剤、外用剤(例えば、点眼剤、点鼻剤、点耳剤、エアゾール剤、吸入剤、ローション剤、注入剤、塗布剤、含嗽剤、浣腸剤、軟膏剤、硬膏剤、ゼリー剤、クリーム剤、貼付剤、パップ剤、外用散剤、坐剤等)等の通常用いられるいずれの剤型でも好適に投与することができる。注射剤は、O/W、W/O、O/W/O、W/O/W型等のエマルジョンであってもよい。
経皮投与用の医薬組成物としては、特に限定されないが、例えば、外用固形剤(例えば、外用散剤)、外用液剤(例えば、リニメント剤、ローション剤)、スプレー剤(外用エアゾール剤、ポンプスプレー剤)、軟膏剤、クリーム剤、ゲル剤、貼付剤(例えば、テープ剤(例えば、プラスター剤、硬膏剤)、パップ剤)などを含む。例えば、貼付剤である。
経皮投与用の医薬組成物としては、特に限定されないが、例えば、外用固形剤(例えば、外用散剤)、外用液剤(例えば、リニメント剤、ローション剤)、スプレー剤(外用エアゾール剤、ポンプスプレー剤)、軟膏剤、クリーム剤、ゲル剤、貼付剤(例えば、テープ剤(例えば、プラスター剤、硬膏剤)、パップ剤)などを含む。例えば、貼付剤である。
本発明の方法、剤、医薬組成物の態様としては、特に限定されず、症状の程度、患者の体重、年齢及び薬剤の投与形態等に応じて適宜決定できる。本発明の態様を以下に例示する。本発明の方法、医薬、医薬組成物の態様としては、上記の項目と、下記に例示される項目の取りうる全ての組み合わせの態様および範囲が例示される。
ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療有効量以下の用量(例えば、疼痛治療有効量の約1/2倍以下、約1/10倍以下の用量)で投与する。
ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療最小有効量の約3倍以下(例えば、約3倍未満)、約2倍以下(例えば、約1倍以下、約1/2倍以下、約1/3倍以下、約1/4倍以下)の用量で投与する。
ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療最小有効量の約1/33倍以上(例えば、約1/33倍より大きい投与量)、疼痛治療最小有効量の約1/30倍以上(例えば、約1/25倍以上、約1/20倍以上、約1/10倍以上)で投与する。
ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療最小有効量の約1/33倍~約3倍(例えば、約1/33倍より大きく約3倍未満)、約1/30倍~約3倍(例えば、約1/30倍以上約3倍未満)、約1/30倍~約2倍、約1/30倍~約1倍、約1/30倍~約1/2倍、約1/20倍~約3倍(例えば、約1/20倍以上約3倍未満)、約1/20倍~約2倍、約1/20倍~約1倍、約1/30倍~約1/2倍、約1/10倍~約3倍(例えば、約1/10倍以上約3倍未満)、約1/10倍~約2倍、約1/10倍~約1倍、約1/10倍~約1/2倍で投与する。
ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療有効量以下の用量(例えば、疼痛治療有効量の約1/2倍以下、約1/10倍以下の用量)で投与する。
ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療最小有効量の約3倍以下(例えば、約3倍未満)、約2倍以下(例えば、約1倍以下、約1/2倍以下、約1/3倍以下、約1/4倍以下)の用量で投与する。
ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療最小有効量の約1/33倍以上(例えば、約1/33倍より大きい投与量)、疼痛治療最小有効量の約1/30倍以上(例えば、約1/25倍以上、約1/20倍以上、約1/10倍以上)で投与する。
ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療最小有効量の約1/33倍~約3倍(例えば、約1/33倍より大きく約3倍未満)、約1/30倍~約3倍(例えば、約1/30倍以上約3倍未満)、約1/30倍~約2倍、約1/30倍~約1倍、約1/30倍~約1/2倍、約1/20倍~約3倍(例えば、約1/20倍以上約3倍未満)、約1/20倍~約2倍、約1/20倍~約1倍、約1/30倍~約1/2倍、約1/10倍~約3倍(例えば、約1/10倍以上約3倍未満)、約1/10倍~約2倍、約1/10倍~約1倍、約1/10倍~約1/2倍で投与する。
ブプレノルフィンまたはその製薬上許容される塩の血漿中濃度(好ましくは最高血漿中濃度)が、例えば、約2ng/mL以下、約1.5ng/mL以下、約1ng/mL以下、約0.9ng/mL以下、約0.8ng/mL以下、約0.7ng/mL以下である。好ましくは、約0.6ng/mL以下、約0.62ng/mL以下、約0.6ng/mL未満、約0.62ng/mL未満、約0.5ng/mL以下、約0.4ng/mL以下、約0.3ng/mL以下である。より好ましくは、約0.2ng/mL以下であり、例えば、約0.18ng/mL以下、約0.17ng/mL以下、約0.15ng/mL以下、約0.1ng/mL以下、約90pg/mL以下、約80pg/mL以下、約70pg/mL以下、約60pg/mL以下、約50pg/mL以下である(なお、血漿中濃度は塩部分を考慮しないブプレノルフィン換算の濃度で表記している。以下同じ。)。
ブプレノルフィンまたはその製薬上許容される塩の血漿中濃度(好ましくは最高血漿中濃度)が、例えば、約0.1pg/mL以上、約0.5pg/mL以上、約1pg/mL以上、約2pg/mL以上、約3pg/mL以上、約3pg/mLより大きい、約3.3pg/mLより大きい濃度である。好ましくは、例えば、約4pg/mL以上、約4pg/mLより大きい、約5pg/mL以上、約6pg/mL以上、約7pg/mL以上、約8pg/mL以上、約9pg/mL以上、約10pg/mL以上、約11pg/mL以上、約12pg/mL以上、約13pg/mL以上、約14pg/mL以上、約15pg/mL以上、約16pg/mL以上、約17pg/mL以上、約18pg/mL以上である。
該濃度は、例えば、約0.9ng/mL以下、約0.8ng/mL以下、約0.7ng/mL以下である。好ましくは、例えば、約0.6ng/mL以下、約0.62ng/mL以下、約0.6ng/mL未満、約0.62ng/mL未満、約0.5ng/mL以下、約0.4ng/mL以下、約0.3ng/mL以下である。より好ましくは、例えば、約0.2ng/mL以下、約0.18ng/mL以下、約0.17ng/mL以下、約0.15ng/mL以下、約0.1ng/mL以下、約90pg/mL以下、約80pg/mL以下、約70pg/mL以下、約60pg/mL以下、約50pg/mL以下、約40pg/mL以下である。
ブプレノルフィンまたはその製薬上許容される塩の血漿中濃度(好ましくは最高血漿中濃度)が、例えば、
約1pg/mL~約2ng/mL以下、約1pg/mL~約1ng/mL以下、約1pg/mL~約0.6ng/mL、約1pg/mL~約0.2ng/mL、約1pg/mL~約0.1ng/mL、約1pg/mL~約50pg/mL、
約3pg/mL~約2ng/mL以下、約3pg/mL~約1ng/mL以下、約3pg/mL~約0.6ng/mL、約3pg/mL~約0.3ng/mL、約3pg/mL~約0.2ng/mL、約3pg/mL~約0.1ng/mL、約3pg/mL~約50pg/mL、
約4pg/mL~約2ng/mL以下、約4pg/mL~約1ng/mL以下、約4pg/mL~約0.6ng/mL、約4pg/mL~約0.2ng/mL、約4pg/mL~約0.1ng/mL、約4pg/mL~約50pg/mL、
約5pg/mL~約2ng/mL以下、約5pg/mL~約1ng/mL以下、約5pg/mL~約0.6ng/mL、約5pg/mL~約0.2ng/mL、約5pg/mL~約0.1ng/mL、約5pg/mL~約50pg/mL、
約6pg/mL~約2ng/mL以下、約6pg/mL~約1ng/mL以下、約6pg/mL~約0.6ng/mL、約6pg/mL~約0.2ng/mL、約6pg/mL~約0.1ng/mL、約5pg/mL~約50pg/mL、
約8pg/mL~約2ng/mL以下、約8pg/mL~約1ng/mL以下、約8pg/mL~約0.6ng/mL、約8pg/mL~約0.2ng/mL、約8pg/mL~約0.1ng/mL、約8pg/mL~約80pg/mL、約8pg/mL~約50pg/mL、
約10pg/mL~約2ng/mL以下、約10pg/mL~約1ng/mL以下、約10pg/mL~約0.6ng/mL、約10pg/mL~約0.2ng/mL、約10pg/mL~約0.1ng/mL、約10pg/mL~約50pg/mL、
約15pg/mL~約2ng/mL以下、約15pg/mL~約1ng/mL以下、約15pg/mL~約0.6ng/mL、約15pg/mL~約0.2ng/mL、約15pg/mL~約0.1ng/mL、約15pg/mL~約50pg/mL、
約18pg/mL~約2ng/mL以下、約18pg/mL~約1ng/mL以下、約18pg/mL~約0.6ng/mL、約18pg/mL~約0.2ng/mL、約18pg/mL~約0.17ng/mL、約18pg/mL~約0.1ng/mL、約18pg/mL~約50pg/mL、
約20pg/mL~約2ng/mL以下、約20pg/mL~約1ng/mL以下、約20pg/mL~約0.6ng/mL、約20pg/mL~約0.2ng/mL、約20pg/mL~約0.1ng/mL、約20pg/mL~約50pg/mLである。
さらなる態様として、上記いずれかの血漿中濃度範囲について、両端の数値を含まない範囲(例えば、約3pg/mL~約0.6ng/mLについて、約3pg/mLより大きく、約0.6ng/mL未満)が例示される。
一つの態様として、上記血漿中濃度が、定常状態における最高血漿中濃度である。
一つの態様として、上記血漿中濃度が、定常状態における血漿中濃度である。
一つの態様として、上記血漿中濃度が、連続して30分以上、例えば1時間以上、例えば2時間以上維持される。
ブプレノルフィンまたはその製薬上許容される塩の血漿中濃度(好ましくは最高血漿中濃度)が、例えば、約0.1pg/mL以上、約0.5pg/mL以上、約1pg/mL以上、約2pg/mL以上、約3pg/mL以上、約3pg/mLより大きい、約3.3pg/mLより大きい濃度である。好ましくは、例えば、約4pg/mL以上、約4pg/mLより大きい、約5pg/mL以上、約6pg/mL以上、約7pg/mL以上、約8pg/mL以上、約9pg/mL以上、約10pg/mL以上、約11pg/mL以上、約12pg/mL以上、約13pg/mL以上、約14pg/mL以上、約15pg/mL以上、約16pg/mL以上、約17pg/mL以上、約18pg/mL以上である。
該濃度は、例えば、約0.9ng/mL以下、約0.8ng/mL以下、約0.7ng/mL以下である。好ましくは、例えば、約0.6ng/mL以下、約0.62ng/mL以下、約0.6ng/mL未満、約0.62ng/mL未満、約0.5ng/mL以下、約0.4ng/mL以下、約0.3ng/mL以下である。より好ましくは、例えば、約0.2ng/mL以下、約0.18ng/mL以下、約0.17ng/mL以下、約0.15ng/mL以下、約0.1ng/mL以下、約90pg/mL以下、約80pg/mL以下、約70pg/mL以下、約60pg/mL以下、約50pg/mL以下、約40pg/mL以下である。
ブプレノルフィンまたはその製薬上許容される塩の血漿中濃度(好ましくは最高血漿中濃度)が、例えば、
約1pg/mL~約2ng/mL以下、約1pg/mL~約1ng/mL以下、約1pg/mL~約0.6ng/mL、約1pg/mL~約0.2ng/mL、約1pg/mL~約0.1ng/mL、約1pg/mL~約50pg/mL、
約3pg/mL~約2ng/mL以下、約3pg/mL~約1ng/mL以下、約3pg/mL~約0.6ng/mL、約3pg/mL~約0.3ng/mL、約3pg/mL~約0.2ng/mL、約3pg/mL~約0.1ng/mL、約3pg/mL~約50pg/mL、
約4pg/mL~約2ng/mL以下、約4pg/mL~約1ng/mL以下、約4pg/mL~約0.6ng/mL、約4pg/mL~約0.2ng/mL、約4pg/mL~約0.1ng/mL、約4pg/mL~約50pg/mL、
約5pg/mL~約2ng/mL以下、約5pg/mL~約1ng/mL以下、約5pg/mL~約0.6ng/mL、約5pg/mL~約0.2ng/mL、約5pg/mL~約0.1ng/mL、約5pg/mL~約50pg/mL、
約6pg/mL~約2ng/mL以下、約6pg/mL~約1ng/mL以下、約6pg/mL~約0.6ng/mL、約6pg/mL~約0.2ng/mL、約6pg/mL~約0.1ng/mL、約5pg/mL~約50pg/mL、
約8pg/mL~約2ng/mL以下、約8pg/mL~約1ng/mL以下、約8pg/mL~約0.6ng/mL、約8pg/mL~約0.2ng/mL、約8pg/mL~約0.1ng/mL、約8pg/mL~約80pg/mL、約8pg/mL~約50pg/mL、
約10pg/mL~約2ng/mL以下、約10pg/mL~約1ng/mL以下、約10pg/mL~約0.6ng/mL、約10pg/mL~約0.2ng/mL、約10pg/mL~約0.1ng/mL、約10pg/mL~約50pg/mL、
約15pg/mL~約2ng/mL以下、約15pg/mL~約1ng/mL以下、約15pg/mL~約0.6ng/mL、約15pg/mL~約0.2ng/mL、約15pg/mL~約0.1ng/mL、約15pg/mL~約50pg/mL、
約18pg/mL~約2ng/mL以下、約18pg/mL~約1ng/mL以下、約18pg/mL~約0.6ng/mL、約18pg/mL~約0.2ng/mL、約18pg/mL~約0.17ng/mL、約18pg/mL~約0.1ng/mL、約18pg/mL~約50pg/mL、
約20pg/mL~約2ng/mL以下、約20pg/mL~約1ng/mL以下、約20pg/mL~約0.6ng/mL、約20pg/mL~約0.2ng/mL、約20pg/mL~約0.1ng/mL、約20pg/mL~約50pg/mLである。
さらなる態様として、上記いずれかの血漿中濃度範囲について、両端の数値を含まない範囲(例えば、約3pg/mL~約0.6ng/mLについて、約3pg/mLより大きく、約0.6ng/mL未満)が例示される。
一つの態様として、上記血漿中濃度が、定常状態における最高血漿中濃度である。
一つの態様として、上記血漿中濃度が、定常状態における血漿中濃度である。
一つの態様として、上記血漿中濃度が、連続して30分以上、例えば1時間以上、例えば2時間以上維持される。
ブプレノルフィンまたはその製薬上許容される塩の含有量または投与量(例えば、経皮投与用医薬組成物における含有量または投与量)が、例えば、約20mg以下、約10mg以下、約7mg以下、約5mg以下、約4mg以下、約2.5mg以下、約2mg以下、約1mg以下、約0.7mg以下、約0.5mg以下、約0.4mg以下、約0.3mg以下、約0.2mg以下、約0.1mg以下である(なお、含有量または投与量は塩部分を考慮しないブプレノルフィン換算量で表記している。以下同じ。)。
ブプレノルフィンまたはその製薬上許容される塩の含有量または投与量(例えば、経皮投与用医薬組成物における含有量または投与量)が、例えば、約0.001mg以上、約0.003mg以上、約0.005mg以上、約0.01mg以上、約0.03mg以上、約0.05mg以上、約0.1mg以上、約0.3mg以上、約0.5mg以上である。
本発明のブプレノルフィンまたはその製薬上許容される塩の経皮投与用医薬組成物における含有量は、約0.56mg~約1.7mgである場合もあり、約0.56mgまたは約1.7mgである場合もある。
ブプレノルフィンまたはその製薬上許容される塩の含有量または投与量(例えば、経皮投与用医薬組成物における投与量または放出速度)が、例えば、約20μg/h以下、約10μg/h以下、約5μg/h以下、約2.5μg/h以下、約3μg/h以下、約2μg/h以下、約1μg/h以下、約0.1μg/h以下である(なお、放出速度は塩部分を考慮しないブプレノルフィン換算量で表記している。以下同じ。)。
ブプレノルフィンまたはその製薬上許容される塩の含有量または投与量(例えば、経皮投与用医薬組成物における投与量または放出速度)が、例えば、約0.01μg/h以上、約0.05μg/h以上、約0.1μg/h以上、約0.5μg/h以上、約1μg/h以上である。
本発明のブプレノルフィンまたはその製薬上許容される塩の経皮投与用医薬組成物の放出速度は、約0.56μg/h~約1.7μg/hである場合もあり、約0.56μg/hまたは約1.7μg/hである場合もある。
ブプレノルフィンまたはその製薬上許容される塩の含有量または投与量(例えば、経粘膜投与用医薬組成物における含有量または投与量)が、例えば、約1mg以下、約0.5mg以下、約0.2mg以下、約0.1mg以下、約0.075mg以下、約0.05mg以下、約0.01mg以下である。
ブプレノルフィンまたはその製薬上許容される塩の含有量または投与量(例えば、経粘膜投与用医薬組成物における含有量または投与量)が、例えば、約0.00001mg以上、約0.0001mg以上、約0.001mg以上、約0.01mg以上である。
ブプレノルフィンまたはその製薬上許容される塩の含有量または投与量が、例えば約0.3μg/kg~約30μg/kg(例えば、約0.3μg/kgより大きく、約30μg/kg未満)、約0.5μg/kg~約20μg/kg、約1μg/kg~約10μg/kgである。
例えば、60kgの患者に投与する場合、例えば約18μg~約1.8mg(例えば、約18μgより大きく、約1.8mg未満)、約30μg~約1.2mg、約60μg~約0.6mgである。
例えば40kgの患者に投与する場合、例えば約12μg~約1.2mg(例えば、約12μgより大きく、約1.2mg未満)、約20μg~約0.8mg、約40μg~約0.4mgである。
例えば20kgの患者に投与する場合、例えば約6μg~約0.6mg(例えば、約6μgより大きく、約0.6mg未満)、約10μg~約0.4mg、約20μg~約0.2mgである。
ブプレノルフィンまたはその製薬上許容される塩の含有量または投与量(例えば、経皮投与用医薬組成物における含有量または投与量)が、例えば、約0.001mg以上、約0.003mg以上、約0.005mg以上、約0.01mg以上、約0.03mg以上、約0.05mg以上、約0.1mg以上、約0.3mg以上、約0.5mg以上である。
本発明のブプレノルフィンまたはその製薬上許容される塩の経皮投与用医薬組成物における含有量は、約0.56mg~約1.7mgである場合もあり、約0.56mgまたは約1.7mgである場合もある。
ブプレノルフィンまたはその製薬上許容される塩の含有量または投与量(例えば、経皮投与用医薬組成物における投与量または放出速度)が、例えば、約20μg/h以下、約10μg/h以下、約5μg/h以下、約2.5μg/h以下、約3μg/h以下、約2μg/h以下、約1μg/h以下、約0.1μg/h以下である(なお、放出速度は塩部分を考慮しないブプレノルフィン換算量で表記している。以下同じ。)。
ブプレノルフィンまたはその製薬上許容される塩の含有量または投与量(例えば、経皮投与用医薬組成物における投与量または放出速度)が、例えば、約0.01μg/h以上、約0.05μg/h以上、約0.1μg/h以上、約0.5μg/h以上、約1μg/h以上である。
本発明のブプレノルフィンまたはその製薬上許容される塩の経皮投与用医薬組成物の放出速度は、約0.56μg/h~約1.7μg/hである場合もあり、約0.56μg/hまたは約1.7μg/hである場合もある。
ブプレノルフィンまたはその製薬上許容される塩の含有量または投与量(例えば、経粘膜投与用医薬組成物における含有量または投与量)が、例えば、約1mg以下、約0.5mg以下、約0.2mg以下、約0.1mg以下、約0.075mg以下、約0.05mg以下、約0.01mg以下である。
ブプレノルフィンまたはその製薬上許容される塩の含有量または投与量(例えば、経粘膜投与用医薬組成物における含有量または投与量)が、例えば、約0.00001mg以上、約0.0001mg以上、約0.001mg以上、約0.01mg以上である。
ブプレノルフィンまたはその製薬上許容される塩の含有量または投与量が、例えば約0.3μg/kg~約30μg/kg(例えば、約0.3μg/kgより大きく、約30μg/kg未満)、約0.5μg/kg~約20μg/kg、約1μg/kg~約10μg/kgである。
例えば、60kgの患者に投与する場合、例えば約18μg~約1.8mg(例えば、約18μgより大きく、約1.8mg未満)、約30μg~約1.2mg、約60μg~約0.6mgである。
例えば40kgの患者に投与する場合、例えば約12μg~約1.2mg(例えば、約12μgより大きく、約1.2mg未満)、約20μg~約0.8mg、約40μg~約0.4mgである。
例えば20kgの患者に投与する場合、例えば約6μg~約0.6mg(例えば、約6μgより大きく、約0.6mg未満)、約10μg~約0.4mg、約20μg~約0.2mgである。
モルヒネまたはその製薬上許容される塩の血漿中濃度(好ましくは最高血漿中濃度)が、例えば、約500ng/mL以下、約350ng/mL以下、約300ng/mL以下、約200ng/mL以下、約100ng/mL以下、約10ng/mL以下、約7ng/mL以下、である。
モルヒネまたはその製薬上許容される塩の血漿中濃度(好ましくは最高血漿中濃度)が、例えば、約0.1ng/mL以上、約0.5ng/mL以上、約1ng/mL以上、約2ng/mL以上、約5ng/mL以上、約10ng/mL以上である。
一つの態様として、上記血漿中濃度が、定常状態における最高血漿中濃度である。
一つの態様として、上記血漿中濃度が、定常状態における血漿中濃度である。
一つの態様として、上記血漿中濃度が、連続して30分以上(例えば、1時間以上、2時間以上)維持される。
モルヒネまたはその製薬上許容される塩の含有量または投与量が、例えば、10mg以下、5mg以下、3mg以下、1mg以下、0.1mg以下である。
モルヒネまたはその製薬上許容される塩の含有量または投与量が、例えば、0.01mg以上、0.05mg以上、0.1mg以上、0.5mg以上、1mg以上である。
モルヒネまたはその製薬上許容される塩の含有量または投与量が、例えば約0.03mg/kg~約0.3mg/kg(例えば、約0.03mg/kgより大きく、約0.3mg/kg未満)、約0.03mg/kg~約0.1mg/kgである。
例えば、60kgの患者に投与する場合、例えば約1.8mg~約18mg(例えば、約1.8mgより大きく、約18mg未満)、約1.8mg~約6mgである。
例えば40kgの患者に投与する場合、例えば約1.2mg~約12mg(例えば、約1.2mgより大きく、約12mg未満)、約1.2mg~約4mgである。
例えば20kgの患者に投与する場合、例えば約0.6mg~約6mg(例えば、約0.6mgより大きく、約6mg未満)、約0.6mg~約2mgである。
モルヒネまたはその製薬上許容される塩の血漿中濃度(好ましくは最高血漿中濃度)が、例えば、約0.1ng/mL以上、約0.5ng/mL以上、約1ng/mL以上、約2ng/mL以上、約5ng/mL以上、約10ng/mL以上である。
一つの態様として、上記血漿中濃度が、定常状態における最高血漿中濃度である。
一つの態様として、上記血漿中濃度が、定常状態における血漿中濃度である。
一つの態様として、上記血漿中濃度が、連続して30分以上(例えば、1時間以上、2時間以上)維持される。
モルヒネまたはその製薬上許容される塩の含有量または投与量が、例えば、10mg以下、5mg以下、3mg以下、1mg以下、0.1mg以下である。
モルヒネまたはその製薬上許容される塩の含有量または投与量が、例えば、0.01mg以上、0.05mg以上、0.1mg以上、0.5mg以上、1mg以上である。
モルヒネまたはその製薬上許容される塩の含有量または投与量が、例えば約0.03mg/kg~約0.3mg/kg(例えば、約0.03mg/kgより大きく、約0.3mg/kg未満)、約0.03mg/kg~約0.1mg/kgである。
例えば、60kgの患者に投与する場合、例えば約1.8mg~約18mg(例えば、約1.8mgより大きく、約18mg未満)、約1.8mg~約6mgである。
例えば40kgの患者に投与する場合、例えば約1.2mg~約12mg(例えば、約1.2mgより大きく、約12mg未満)、約1.2mg~約4mgである。
例えば20kgの患者に投与する場合、例えば約0.6mg~約6mg(例えば、約0.6mgより大きく、約6mg未満)、約0.6mg~約2mgである。
一つの態様として、本発明の方法、剤、医薬、医薬組成物は、単一の有効成分を含有し、単独で投与されてもよい。
一つの態様として、投与間隔が、約12時間以上、(例えば、約24時間、約24時間以上)である。
一つの態様として、投与回数が、1日2回以下(例えば、1日1回)である。
一つの態様として、投与間隔が、約12時間以上、(例えば、約24時間、約24時間以上)である。
一つの態様として、投与回数が、1日2回以下(例えば、1日1回)である。
本発明の方法、剤、医薬、医薬組成物等には、本発明が標的とする患者又は被験者を治療対象として特定することや用法用量、注意事項等の治療及び/又は予防のための指針について、医師などの予防又は治療に当たる医療従事者に対して指示を与えるために記載された添付文書やラベルを含むことができるが、これらの公的書類は紙媒体に限らず、インターネットを介して提供されることができ、また、公的書類以外にも、種々の他の情報源に基づいて医師などへ予防又は治療の指針を与えることができる。したがって、本発明は、添付文書やラベル以外の情報に基づいて使用される実施形態も包含するものであることが理解される。
本発明の方法、剤、医薬、医薬組成物の態様としては、他の精神疾患薬(デジタル薬を含む)および/または他の治療法(薬を用いない各種療法を含む)と組み合わせて用いてもよく、および、これらと組み合わせずに用いてもよい。
本発明の方法、剤、医薬、医薬組成物の態様としては、他の精神疾患薬(デジタル薬を含む)および/または他の治療法(薬を用いない各種療法を含む)と組み合わせて用いてもよく、および、これらと組み合わせずに用いてもよい。
本発明の医薬組成物または方法は、好ましくは、下記から選択される1つまたは1つ以上の優れた特徴を有している。
a)自閉スペクトラム症、脆弱X症候群、自閉スペクトラム症様症状、並びに/または社会的コミュニケーションおよび/若しくは社会的相互関係における障害の治療並びに/または予防に有用である。好ましくは、自閉スペクトラム症の治療および/または予防に有用である。特に好ましくは、自閉スペクトラム症の中核症状の治療および/または予防に有用である。
b)有効薬効量および濃度が低い。よって、安全性マージンの高い医薬品となりうる。また、副作用リスクの低い医薬品となりうる。
c)疼痛治療薬として承認されている用量と、同等または好ましくはより低い用量(あるいは血漿中濃度)で薬効を示す。したがって、医薬品としての安全性が高い。また、例えば、規制薬物としての制限が少ない可能性がある。例えば、投与にあたって特別のプログラム(REMS等)を必要としない。
d)薬効の持続時間が長い。これにより、少ない投与回数での治療が可能であり、例えば、患者の服薬コンプライアンスが高いことが期待される。また、例えば患者が小児である場合、養育者の負担低減につながることが期待される。
a)自閉スペクトラム症、脆弱X症候群、自閉スペクトラム症様症状、並びに/または社会的コミュニケーションおよび/若しくは社会的相互関係における障害の治療並びに/または予防に有用である。好ましくは、自閉スペクトラム症の治療および/または予防に有用である。特に好ましくは、自閉スペクトラム症の中核症状の治療および/または予防に有用である。
b)有効薬効量および濃度が低い。よって、安全性マージンの高い医薬品となりうる。また、副作用リスクの低い医薬品となりうる。
c)疼痛治療薬として承認されている用量と、同等または好ましくはより低い用量(あるいは血漿中濃度)で薬効を示す。したがって、医薬品としての安全性が高い。また、例えば、規制薬物としての制限が少ない可能性がある。例えば、投与にあたって特別のプログラム(REMS等)を必要としない。
d)薬効の持続時間が長い。これにより、少ない投与回数での治療が可能であり、例えば、患者の服薬コンプライアンスが高いことが期待される。また、例えば患者が小児である場合、養育者の負担低減につながることが期待される。
ブプレノルフィンおよびその製薬上許容される塩ならびにモルヒネまたはその製薬上許容される塩は、公知の方法に沿って合成できる。
(実施例)
(実施例)
以下、本発明を実施例に基づいて説明するが、本発明はこれらの実施例等によりなんら限定されるものではない。
(実施例1:バルプロ酸モデルマウスによる薬効評価)
バルプロ酸モデルマウス(VPAモデル)は、ヒトにおいて妊娠中にバルプロ酸を服薬すると新生児がASDに罹患する確率が有意に上昇する臨床エビデンスに基づいたASDモデルマウスである。社会性障害や認知機能障害などのASD様症状が認められることから、最も汎用されるASDモデルマウスの1つである。
先行研究(Hara Y, Ago Y, Higuchi M, Hasebe S, Nakazawa T, Hashimoto H, Matsuda T, Takuma K. Oxytocin attenuates deficits in social interaction but not recognition memory in a prenatal valproic acid-induced mouse model of autism. Horm Behav. 2017 Nov;96:130-136. doi: 10.1016/j.yhbeh.2017.09.013. Epub 2017 Sep 28. PMID: 28942000.)に記載の通りに胎児期にバルプロ酸をICRマウスに投与し作製されたVPAモデルに対し、鎮痛薬として用いられるMORアゴニストであるモルヒネ、ブプレノルフィンもしくはトラマドールを単回投与し、VPAモデルの社会性障害に対する作用を検討した。
社会性評価としては、Social Interaction Testという方法を用いた。Social Interaction Testでは、1つのケージの中に2匹のマウスをいれ、対象マウス(Testマウス)がもう1匹のマウス(Intruderマウス)に対して、社会性行動であるsniffingを何秒行うかを定量する。Saline、モルヒネ塩酸塩、ブプレノルフィン塩酸塩もしくはトラマドール塩酸塩を皮下投与し、投与後1時間後から20分間の社会性行動量を定量化した。用量については、モルヒネおよびトラマドールについては塩酸塩として、ブプレノルフィンについてはフリー体として算出した。
結果を図1~3に示す。
モルヒネおよびブプレノルフィンについてはそれぞれの鎮痛作用が最大となる用量(8mg/kg、1mg/kg、非特許文献28)の1/10未満の低用量において有意な社会性障害改善作用が認められた。一方で、鎮痛用量に近い高用量においては両化合物とも社会性障害改善作用が低下するという特徴的な用量依存性が認められた。
また、モルヒネやブプレノルフィンと同様に鎮痛薬として用いられ生体内においてMORアゴニスト活性を有するトラマドールについては、社会性障害改善作用は認められなかった。
以上の結果より、モルヒネ及びブプレノルフィンは、ASDにおける社会性障害の改善作用を有し、ASDの治療薬として有用と考えられる。また、モルヒネ及びブプレノルフィンは、高用量域で鎮痛作用が強まるが、社会性改善作用においては、驚くべきことに、特に低用量域において高い薬効を示す傾向が見いだされた。
特にブプレノルフィンは、モルヒネと比べさらに低い用量で社会性障害を改善したことから、副作用リスクが低く、安全性の高いASD治療薬として期待される。
バルプロ酸モデルマウス(VPAモデル)は、ヒトにおいて妊娠中にバルプロ酸を服薬すると新生児がASDに罹患する確率が有意に上昇する臨床エビデンスに基づいたASDモデルマウスである。社会性障害や認知機能障害などのASD様症状が認められることから、最も汎用されるASDモデルマウスの1つである。
先行研究(Hara Y, Ago Y, Higuchi M, Hasebe S, Nakazawa T, Hashimoto H, Matsuda T, Takuma K. Oxytocin attenuates deficits in social interaction but not recognition memory in a prenatal valproic acid-induced mouse model of autism. Horm Behav. 2017 Nov;96:130-136. doi: 10.1016/j.yhbeh.2017.09.013. Epub 2017 Sep 28. PMID: 28942000.)に記載の通りに胎児期にバルプロ酸をICRマウスに投与し作製されたVPAモデルに対し、鎮痛薬として用いられるMORアゴニストであるモルヒネ、ブプレノルフィンもしくはトラマドールを単回投与し、VPAモデルの社会性障害に対する作用を検討した。
社会性評価としては、Social Interaction Testという方法を用いた。Social Interaction Testでは、1つのケージの中に2匹のマウスをいれ、対象マウス(Testマウス)がもう1匹のマウス(Intruderマウス)に対して、社会性行動であるsniffingを何秒行うかを定量する。Saline、モルヒネ塩酸塩、ブプレノルフィン塩酸塩もしくはトラマドール塩酸塩を皮下投与し、投与後1時間後から20分間の社会性行動量を定量化した。用量については、モルヒネおよびトラマドールについては塩酸塩として、ブプレノルフィンについてはフリー体として算出した。
結果を図1~3に示す。
モルヒネおよびブプレノルフィンについてはそれぞれの鎮痛作用が最大となる用量(8mg/kg、1mg/kg、非特許文献28)の1/10未満の低用量において有意な社会性障害改善作用が認められた。一方で、鎮痛用量に近い高用量においては両化合物とも社会性障害改善作用が低下するという特徴的な用量依存性が認められた。
また、モルヒネやブプレノルフィンと同様に鎮痛薬として用いられ生体内においてMORアゴニスト活性を有するトラマドールについては、社会性障害改善作用は認められなかった。
以上の結果より、モルヒネ及びブプレノルフィンは、ASDにおける社会性障害の改善作用を有し、ASDの治療薬として有用と考えられる。また、モルヒネ及びブプレノルフィンは、高用量域で鎮痛作用が強まるが、社会性改善作用においては、驚くべきことに、特に低用量域において高い薬効を示す傾向が見いだされた。
特にブプレノルフィンは、モルヒネと比べさらに低い用量で社会性障害を改善したことから、副作用リスクが低く、安全性の高いASD治療薬として期待される。
(実施例2:薬物動態)
ICRマウスにモルヒネ塩酸塩もしくはブプレノルフィンを各用量になるようにSalineで調整し皮下投与し、投与後の各時点において採血および脳の摘出を行い、血漿中および脳内の薬物濃度をLC-MSにより測定した。用量については、モルヒネについては塩酸塩として、ブプレノルフィンについてはフリー体として算出した。結果を表1および表2に示す。
図4および図5に、モルヒネ及びブプレノルフィンの未変化体の血漿中濃度と社会性障害改善と鎮痛薬効との関係を示した。
図4および5の左軸は、実施例1と同様に測定した社会性障害改善作用を示し、右軸は鎮痛作用を非特許文献28 Fig.4のデータを引用し、モルヒネ及びブプレノルフィンの未変化体の血漿中濃度と社会性障害改善と鎮痛薬効との関係をグラフ化した。横軸は、薬効評価時の投与用量から算出した血漿中未変化体濃度を示す。モルヒネ、ブプレノルフィンのいずれに関しても、社会性障害改善作用が認められる血漿中薬物濃度は、鎮痛作用が認められる濃度よりも低く、最大薬効を示すときの濃度を比較すると10倍以上乖離している。
表1に、モルヒネの未変化体およびM6G代謝物の血漿中濃度の時間推移を示す。
ICRマウスにモルヒネ塩酸塩もしくはブプレノルフィンを各用量になるようにSalineで調整し皮下投与し、投与後の各時点において採血および脳の摘出を行い、血漿中および脳内の薬物濃度をLC-MSにより測定した。用量については、モルヒネについては塩酸塩として、ブプレノルフィンについてはフリー体として算出した。結果を表1および表2に示す。
図4および図5に、モルヒネ及びブプレノルフィンの未変化体の血漿中濃度と社会性障害改善と鎮痛薬効との関係を示した。
図4および5の左軸は、実施例1と同様に測定した社会性障害改善作用を示し、右軸は鎮痛作用を非特許文献28 Fig.4のデータを引用し、モルヒネ及びブプレノルフィンの未変化体の血漿中濃度と社会性障害改善と鎮痛薬効との関係をグラフ化した。横軸は、薬効評価時の投与用量から算出した血漿中未変化体濃度を示す。モルヒネ、ブプレノルフィンのいずれに関しても、社会性障害改善作用が認められる血漿中薬物濃度は、鎮痛作用が認められる濃度よりも低く、最大薬効を示すときの濃度を比較すると10倍以上乖離している。
表1に、モルヒネの未変化体およびM6G代謝物の血漿中濃度の時間推移を示す。

*0.1mg/kg投与時の60分後 N=3のデータ(モルヒネ:23.8ng/mL、M6G:321ng/mL)は、明らかな測定エラーと考えられるため外れ値として除外し算出した。
表2に、ブプレノルフィンの未変化体の血漿中濃度の時間推移を示す。

BLQ:LLOQ (定量下限:2pg/mL)以下
* :サンプルを10倍希釈後に定量した。
(実施例3:持続作用の確認)
実施例1と同様にVPAモデルに対して、Salineもしくはブプレノルフィンを皮下投与し、投与後3時間後から20分間もしくは12時間後から20分間の社会性行動量を定量化した。結果を図6に示す。ブプレノルフィン0.003mg/kg投与による社会性障害改善効果は、投与後3時間後、12時間後においても認められた。
一方、自閉スペクトラム症の治療薬として開発中のオキシトシンは、本実験と同様のモデルで社会性障害の改善作用を示すものの、投与3時間後には薬効が消失することが報告されている(Horm Behav. 2017 Nov;96:130-136.)。
したがって、ブプレノルフィンは、オキシトシンと比べて社会性障害改善における薬効の持続性が高く、有用な治療薬となりうることが示唆された。
なお、表2に示すように、0.003mg/kg投与、0.03mg/kg投与のそれぞれにおいて、投与後12時間時点のブプレノルフィン血漿中濃度は投与後3時間時点での血漿中濃度よりも低いが、図6において、投与後3時間時点と投与後12時間時点での社会性改善作用の傾向に明確な差は認められなかった。このことは、特に最大血漿中濃度(Cmax)が社会性改善作用に影響していることを示唆している。
実施例1と同様にVPAモデルに対して、Salineもしくはブプレノルフィンを皮下投与し、投与後3時間後から20分間もしくは12時間後から20分間の社会性行動量を定量化した。結果を図6に示す。ブプレノルフィン0.003mg/kg投与による社会性障害改善効果は、投与後3時間後、12時間後においても認められた。
一方、自閉スペクトラム症の治療薬として開発中のオキシトシンは、本実験と同様のモデルで社会性障害の改善作用を示すものの、投与3時間後には薬効が消失することが報告されている(Horm Behav. 2017 Nov;96:130-136.)。
したがって、ブプレノルフィンは、オキシトシンと比べて社会性障害改善における薬効の持続性が高く、有用な治療薬となりうることが示唆された。
なお、表2に示すように、0.003mg/kg投与、0.03mg/kg投与のそれぞれにおいて、投与後12時間時点のブプレノルフィン血漿中濃度は投与後3時間時点での血漿中濃度よりも低いが、図6において、投与後3時間時点と投与後12時間時点での社会性改善作用の傾向に明確な差は認められなかった。このことは、特に最大血漿中濃度(Cmax)が社会性改善作用に影響していることを示唆している。
(実施例4:ヒトにおける好ましいブプレノルフィン投与量の推定)
図2、3より、ブプレノルフィンは、VPAモデルマウスにおいて、1μg/kg~10μg/kgで有意な社会性改善作用を示した。
実施例2より、0.3μg/kg投与時、30μ/kg投与時のブプレノルフィンのCmaxはそれぞれ、26.6pg/mL、2360pg/mLであった。したがって、マウスにおけるASD用途における好ましい最高血漿中濃度は、約26.6pg/mL~約2360pg/mLの範囲内(好ましくは、約26.6pg/mLより大きく約2360pg/mL未満)である。ここで、有意な社会性改善作用を示した1μg/kg投与時および10μg/kg投与時のブプレノルフィンのCmaxはそれぞれ、67pg/mL(推定)、670pg/mLであるから、マウスにおける特に好ましい最高血漿中濃度の範囲としては、約67pg/mL~670pg/mLを含む。
ここで、マウスにおいては、鎮痛作用評価において、10μg/kg皮下投与以上の用量で潜時反応が見られることから(非特許文献28)、10μg/kgが鎮痛作用が認められる最小用量である。
ここで、経皮投与製剤であるButrans(商標)package insertによれば、承認されている最小用量(5mg製剤、放出速度5μg/h)におけるCmaxは、176pg/mLである。また、経口腔粘膜投与製剤であるBELBUCA(登録商標)package insertによれば、承認されている最小用量(75μg)におけるCmaxは0.17±0.30ng/mLである。また、経皮投与製剤であるノルスパンテープ(登録商標)の添付文書によれば、承認されている最小用量(5mg製剤、放出速度5μg/h)におけるCmaxは、84pg±19/mLである。したがって、マウスにおけるブプレノルフィン10μg/kg投与(Cmax:670pg/mL)は、ヒトにおけるブプレノルフィンのCmaxが約84pg/mL~176pg/mL相当と考えられる。
上述のようにマウスにおけるASD用途における好ましい最高血漿中濃度は、26.6pg/mL(0.3μg/kg投与時)~2360pg/mL(30μ/kg投与時)の範囲内(好ましくは、約26.6pg/mLより大きく約2360pg/mL未満)である。比例計算することにより、ヒトにおけるASD用途における好ましい最高血漿中濃度は、約3.3pg/mL~約620pg/mLの範囲内(好ましくは、約3.3pg/mLより大きく、約620pg/mL未満)と推定された。さらに、マウスにおける特に好ましい最高血漿中濃度(67pg/mL~670pg/mL)の範囲に対応する、ヒトにおける特に好ましい最高血漿中濃度としては、約8.4pg/mL~約176pg/mLと推定された。
また、マウスにおいては、鎮痛作用が認められる最小用量である10μg/kg投与時、およびその1/10量の投与時である1μg/kg投与時において有意な社会性改善作用が認められた。したがって、別の言い方をすれば、ブプレノルフィンの特に好ましい投与量としては、疼痛治療薬として承認されている最小用量(例えば5mg含有経皮投与製剤(7日間投与製剤であり、1日あたり投与量としては約0.7mg)、放出速度5μg/hの経皮投与製剤、75μg経口腔粘膜投与製剤等)の約1/10倍~約1倍の投与量を含む。また、マウスにおける0.3μg/kg投与時、30μ/kg投与時に相当する投与量としては、それぞれ疼痛治療薬として承認されている最小用量の約1/33倍および約3倍である。
図2、3より、ブプレノルフィンは、VPAモデルマウスにおいて、1μg/kg~10μg/kgで有意な社会性改善作用を示した。
実施例2より、0.3μg/kg投与時、30μ/kg投与時のブプレノルフィンのCmaxはそれぞれ、26.6pg/mL、2360pg/mLであった。したがって、マウスにおけるASD用途における好ましい最高血漿中濃度は、約26.6pg/mL~約2360pg/mLの範囲内(好ましくは、約26.6pg/mLより大きく約2360pg/mL未満)である。ここで、有意な社会性改善作用を示した1μg/kg投与時および10μg/kg投与時のブプレノルフィンのCmaxはそれぞれ、67pg/mL(推定)、670pg/mLであるから、マウスにおける特に好ましい最高血漿中濃度の範囲としては、約67pg/mL~670pg/mLを含む。
ここで、マウスにおいては、鎮痛作用評価において、10μg/kg皮下投与以上の用量で潜時反応が見られることから(非特許文献28)、10μg/kgが鎮痛作用が認められる最小用量である。
ここで、経皮投与製剤であるButrans(商標)package insertによれば、承認されている最小用量(5mg製剤、放出速度5μg/h)におけるCmaxは、176pg/mLである。また、経口腔粘膜投与製剤であるBELBUCA(登録商標)package insertによれば、承認されている最小用量(75μg)におけるCmaxは0.17±0.30ng/mLである。また、経皮投与製剤であるノルスパンテープ(登録商標)の添付文書によれば、承認されている最小用量(5mg製剤、放出速度5μg/h)におけるCmaxは、84pg±19/mLである。したがって、マウスにおけるブプレノルフィン10μg/kg投与(Cmax:670pg/mL)は、ヒトにおけるブプレノルフィンのCmaxが約84pg/mL~176pg/mL相当と考えられる。
上述のようにマウスにおけるASD用途における好ましい最高血漿中濃度は、26.6pg/mL(0.3μg/kg投与時)~2360pg/mL(30μ/kg投与時)の範囲内(好ましくは、約26.6pg/mLより大きく約2360pg/mL未満)である。比例計算することにより、ヒトにおけるASD用途における好ましい最高血漿中濃度は、約3.3pg/mL~約620pg/mLの範囲内(好ましくは、約3.3pg/mLより大きく、約620pg/mL未満)と推定された。さらに、マウスにおける特に好ましい最高血漿中濃度(67pg/mL~670pg/mL)の範囲に対応する、ヒトにおける特に好ましい最高血漿中濃度としては、約8.4pg/mL~約176pg/mLと推定された。
また、マウスにおいては、鎮痛作用が認められる最小用量である10μg/kg投与時、およびその1/10量の投与時である1μg/kg投与時において有意な社会性改善作用が認められた。したがって、別の言い方をすれば、ブプレノルフィンの特に好ましい投与量としては、疼痛治療薬として承認されている最小用量(例えば5mg含有経皮投与製剤(7日間投与製剤であり、1日あたり投与量としては約0.7mg)、放出速度5μg/hの経皮投与製剤、75μg経口腔粘膜投与製剤等)の約1/10倍~約1倍の投与量を含む。また、マウスにおける0.3μg/kg投与時、30μ/kg投与時に相当する投与量としては、それぞれ疼痛治療薬として承認されている最小用量の約1/33倍および約3倍である。
(実施例5:FMR-1ノックアウトマウスによる薬効評価)
X染色体の異常に起因するヒト脆弱X症候群(fragile X syndrome:FXS)の患者では、しばしばASDで見られるような社会性障害症状を呈することが知られている(非特許文献7~9)。Fmr1 ノックアウトマウス(Cell, 1994, 15;78(1):23-33)は、ヒト脆弱X症候群(fragile X syndrome:FXS)を模倣したモデルマウスであり、ASDのモデルとしても汎用される。鎮痛薬として用いられるMOR部分アゴニストであるブプレノルフィン塩酸塩および溶媒の生理食塩水を単回投与し、Fmr1ノックアウトマウスの社会性機能障害に対する作用を検討した。用量については、ブプレノルフィンについてはフリー体として算出した。社会性機能評価として、3 chamber test(Nature Medicine,2017,23;p674-677)を用いた。3 chamber testは、3つのコンパートメントの左右両端に、2種類のマウス (UnfamiliarとFamiliar)をカップの中に入れて設置し、新規性の高いマウスと低いマウスを探索する時間の比を算出し、社会性機能(新規性の高いマウスへの嗜好性)として定量する。
結果を図7に示す。Fmr1 ノックアウトマウスは、野生型の正常B6マウスに比べ社会性障害を呈し、ブプレノルフィンの皮下投与によって有意な社会性障害改善作用が認められた。本薬効は、ブプレノルフィンの鎮痛作用が最大となる用量(1mg/kg)の1/10未満の低用量において認められたことから、低用量域における特徴的な効果が認められた。
X染色体の異常に起因するヒト脆弱X症候群(fragile X syndrome:FXS)の患者では、しばしばASDで見られるような社会性障害症状を呈することが知られている(非特許文献7~9)。Fmr1 ノックアウトマウス(Cell, 1994, 15;78(1):23-33)は、ヒト脆弱X症候群(fragile X syndrome:FXS)を模倣したモデルマウスであり、ASDのモデルとしても汎用される。鎮痛薬として用いられるMOR部分アゴニストであるブプレノルフィン塩酸塩および溶媒の生理食塩水を単回投与し、Fmr1ノックアウトマウスの社会性機能障害に対する作用を検討した。用量については、ブプレノルフィンについてはフリー体として算出した。社会性機能評価として、3 chamber test(Nature Medicine,2017,23;p674-677)を用いた。3 chamber testは、3つのコンパートメントの左右両端に、2種類のマウス (UnfamiliarとFamiliar)をカップの中に入れて設置し、新規性の高いマウスと低いマウスを探索する時間の比を算出し、社会性機能(新規性の高いマウスへの嗜好性)として定量する。
結果を図7に示す。Fmr1 ノックアウトマウスは、野生型の正常B6マウスに比べ社会性障害を呈し、ブプレノルフィンの皮下投与によって有意な社会性障害改善作用が認められた。本薬効は、ブプレノルフィンの鎮痛作用が最大となる用量(1mg/kg)の1/10未満の低用量において認められたことから、低用量域における特徴的な効果が認められた。
(実施例6:アンタゴニスト試験)
実施例1と同様にVPAモデルに対して、SalineもしくはMOR選択的アンタゴニスト、ナロキソン塩酸塩を塩酸塩として1mg/kgとなるように皮下投与し、その15分後にブプレノルフィン0.003mg/kgを皮下投与し、ブプレノルフィン投与後1時間後から20分間の社会性行動量を定量化した。
結果を図8に示す。ブプレノルフィン0.003mg/kg投与による社会性障害改善効果は、ナロキソン併用により拮抗された。このことから、ブプレノルフィンは、MOR活性化により社会性障害改善作用を示すことが示された。
実施例1と同様にVPAモデルに対して、SalineもしくはMOR選択的アンタゴニスト、ナロキソン塩酸塩を塩酸塩として1mg/kgとなるように皮下投与し、その15分後にブプレノルフィン0.003mg/kgを皮下投与し、ブプレノルフィン投与後1時間後から20分間の社会性行動量を定量化した。
結果を図8に示す。ブプレノルフィン0.003mg/kg投与による社会性障害改善効果は、ナロキソン併用により拮抗された。このことから、ブプレノルフィンは、MOR活性化により社会性障害改善作用を示すことが示された。
(実施例7:脳の活性化領域の解析)
Salineもしくは0.0003、0.003、0.03mg/kgのブプレノルフィンをVPAモデルに投与し、1時間後に脳を摘出し、抗c-Fos抗体により免疫染色を行い、社会性行動を促進することが知られている脳領域である側坐核(Nucleus Accumbens:NAc)、内側前頭前皮質(medial prefrontal cortex)と、社会性行動を低下させることや鎮痛作用発現に関わることが知られている脳領域である背内側水道周囲灰白質(dorsomedial periaqueductal gray:dmPAG)におけるc-Fos陽性細胞数を活性化神経細胞数として定量した。結果を図9~11に示す。
図9、10に示すように、先行研究(Coley AA, Padilla-Coreano N, Patel R, Tye KM. Valence processing in the PFC: Reconciling circuit-level and systems-level views. Int Rev Neurobiol. 2021;158:171-212. doi: 10.1016/bs.irn.2020.12.002. Epub 2021 Feb 4. PMID: 33785145.)において社会性行動の促進を司る脳部位とされる側坐核や内側前頭前皮質においては、社会性障害改善用量である0.003mg/kgにおいて活性化神経細胞数が有意に増加した。また、図10に示すように、先行研究において(Franklin TB, Silva BA, Perova Z, Marrone L, Masferrer ME, Zhan Y, Kaplan A, Greetham L, Verrechia V, Halman A, Pagella S, Vyssotski AL, Illarionova A, Grinevich V, Branco T, Gross CT. Prefrontal cortical control of a brainstem social behavior circuit. Nat Neurosci. 2017 Feb;20(2):260-270. doi:10.1038/nn.4470. Epub 2017 Jan 9. PMID:28067904; PMCID: PMC5580810.)社会性行動を抑制することが知られている脳部位である背内側水道周囲灰白質においては、0.003mg/kgでは活性化神経細胞数に変化はなかった。
一方、鎮痛作用が認められるが社会性障害改善作用が低下する0.03mg/kg投与時には、側坐核や内側前頭前皮質だけでなく、背内側水道周囲灰白質においても有意に活性化神経細胞数が増加した。
以上のことから、ブプレノルフィンを用いて社会性障害を改善させるためには、側坐核や内側前頭前皮質を活性化させるが背内側水道周囲灰白質を活性化させない用量が好ましいと考えられる。先行研究(de Freitas RL,Medeiros P,Khan AU,Coimbra NC.μ1 -Opioid receptors in the dorsomedial and ventrolateral columns of the periaqueductal grey matter are critical for the enhancement of post-ictal antinociception. Synapse. 2016 Dec;70(12):519-530. doi:10.1002/syn.21926. Epub 2016 Sep 6. PMID:27503688.)において背内側水道周囲灰白質活性化はオピオイドによる鎮痛作用発現に重要であることが知られていることから、社会性障害改善のためには、鎮痛目的で用いられるブプレノルフィンの用量より低い用量が好ましいと考えられる。
Salineもしくは0.0003、0.003、0.03mg/kgのブプレノルフィンをVPAモデルに投与し、1時間後に脳を摘出し、抗c-Fos抗体により免疫染色を行い、社会性行動を促進することが知られている脳領域である側坐核(Nucleus Accumbens:NAc)、内側前頭前皮質(medial prefrontal cortex)と、社会性行動を低下させることや鎮痛作用発現に関わることが知られている脳領域である背内側水道周囲灰白質(dorsomedial periaqueductal gray:dmPAG)におけるc-Fos陽性細胞数を活性化神経細胞数として定量した。結果を図9~11に示す。
図9、10に示すように、先行研究(Coley AA, Padilla-Coreano N, Patel R, Tye KM. Valence processing in the PFC: Reconciling circuit-level and systems-level views. Int Rev Neurobiol. 2021;158:171-212. doi: 10.1016/bs.irn.2020.12.002. Epub 2021 Feb 4. PMID: 33785145.)において社会性行動の促進を司る脳部位とされる側坐核や内側前頭前皮質においては、社会性障害改善用量である0.003mg/kgにおいて活性化神経細胞数が有意に増加した。また、図10に示すように、先行研究において(Franklin TB, Silva BA, Perova Z, Marrone L, Masferrer ME, Zhan Y, Kaplan A, Greetham L, Verrechia V, Halman A, Pagella S, Vyssotski AL, Illarionova A, Grinevich V, Branco T, Gross CT. Prefrontal cortical control of a brainstem social behavior circuit. Nat Neurosci. 2017 Feb;20(2):260-270. doi:10.1038/nn.4470. Epub 2017 Jan 9. PMID:28067904; PMCID: PMC5580810.)社会性行動を抑制することが知られている脳部位である背内側水道周囲灰白質においては、0.003mg/kgでは活性化神経細胞数に変化はなかった。
一方、鎮痛作用が認められるが社会性障害改善作用が低下する0.03mg/kg投与時には、側坐核や内側前頭前皮質だけでなく、背内側水道周囲灰白質においても有意に活性化神経細胞数が増加した。
以上のことから、ブプレノルフィンを用いて社会性障害を改善させるためには、側坐核や内側前頭前皮質を活性化させるが背内側水道周囲灰白質を活性化させない用量が好ましいと考えられる。先行研究(de Freitas RL,Medeiros P,Khan AU,Coimbra NC.μ1 -Opioid receptors in the dorsomedial and ventrolateral columns of the periaqueductal grey matter are critical for the enhancement of post-ictal antinociception. Synapse. 2016 Dec;70(12):519-530. doi:10.1002/syn.21926. Epub 2016 Sep 6. PMID:27503688.)において背内側水道周囲灰白質活性化はオピオイドによる鎮痛作用発現に重要であることが知られていることから、社会性障害改善のためには、鎮痛目的で用いられるブプレノルフィンの用量より低い用量が好ましいと考えられる。
(実施例8 低用量のブプレノルフィン投与がASDの症状に及ぼす影響を調べるための試験)
自閉スペクトラム症の小児に低用量のブプレノルフィンを投与した場合のASDの影響を調べるため、以下の試験を実施する。
DSM-5の基準により自閉スペクトラム症と診断される数十名の小児を対象とし、臨床的および生物学的監視を行いながら、ブプレノルフィン投与の効果を検証する。対象者に対して、ブプレノルフィンを経皮吸収型貼付剤により、貼付用量としては、0.56mgまたは1.7mg、放出速度としては約0.56μg/hまたは約1.7μg/hで経皮投与する。例えば、ブプレノルフィン貼付剤(貼付用量5mg、放出速度5μg/h)を1/9量または1/3量で投与すればよい。標準的なASDの症状評価スケール、例えばSRS-2(Social Responsiveness Scale Second Edition)、CARS-2(Childhood Autism Rating Scale Second Edition)、RBS-R(Repetitive Behavior Scale-Revised)等を用いて、投薬による症状変化の有無を検討する。客観的な症状評価指標としての可能性が示唆されているASD患者における視線パターン変化については、視線計測装置を用いて投薬による変化の有無を検討する。
自閉スペクトラム症の小児に低用量のブプレノルフィンを投与した場合のASDの影響を調べるため、以下の試験を実施する。
DSM-5の基準により自閉スペクトラム症と診断される数十名の小児を対象とし、臨床的および生物学的監視を行いながら、ブプレノルフィン投与の効果を検証する。対象者に対して、ブプレノルフィンを経皮吸収型貼付剤により、貼付用量としては、0.56mgまたは1.7mg、放出速度としては約0.56μg/hまたは約1.7μg/hで経皮投与する。例えば、ブプレノルフィン貼付剤(貼付用量5mg、放出速度5μg/h)を1/9量または1/3量で投与すればよい。標準的なASDの症状評価スケール、例えばSRS-2(Social Responsiveness Scale Second Edition)、CARS-2(Childhood Autism Rating Scale Second Edition)、RBS-R(Repetitive Behavior Scale-Revised)等を用いて、投薬による症状変化の有無を検討する。客観的な症状評価指標としての可能性が示唆されているASD患者における視線パターン変化については、視線計測装置を用いて投薬による変化の有無を検討する。
(製剤例)
以下に示す製剤例は例示にすぎないものであり、発明の範囲を何ら限定することを意図するものではない。
以下に示す製剤例は例示にすぎないものであり、発明の範囲を何ら限定することを意図するものではない。
ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩は、任意の従来の経路により、例えば、経口で、例えば、錠剤またはカプセル剤の形態で、または非経口で、例えば注射液剤または懸濁剤の形態で、局所で、例えば、ローション剤、ゲル剤、軟膏剤またはクリーム剤の形態で、または経鼻形態または座剤形態で医薬組成物として投与することができる。少なくとも1種の薬学的に許容される担体または希釈剤と一緒にして、遊離形態または薬学的に許容される塩の形態のブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を含む医薬組成物は、従来の方法で、混合、造粒またはコーティング法によって製造することができる。例えば、経口用組成物としては、賦形剤、崩壊剤、結合剤、滑沢剤等および有効成分等を含有する錠剤、顆粒剤、カプセル剤とすることができる。また、注射用組成物としては、溶液剤または懸濁剤とすることができ、滅菌されていてもよく、また、保存剤、安定化剤、緩衝化剤等を含有してもよい。
本発明の、ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を含有する医薬組成物、およびブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を投与する方法は、自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状の治療および/または予防に有用である。さらに、本発明の医薬組成物または方法は、有効濃度が低く、安全性の高い医薬または治療方法である。
Claims (21)
- ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を含有する、自閉スペクトラム症、脆弱X症候群、および/または自閉スペクトラム症様症状の治療および/または予防のための医薬組成物。
- 自閉スペクトラム症の治療および/または予防のための、請求項1記載の医薬組成物。
- 自閉スペクトラム症と診断された患者に投与するための、請求項1または2記載の医薬組成物。
- ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療有効量以下の用量で投与するための、請求項1~3のいずれかに記載の医薬組成物。
- ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療有効量の約1/2倍以下の投与量で投与するための、請求項1~4のいずれかに記載の医薬組成物。
- ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療最小有効量以下の投与量で投与するための、請求項1~5のいずれかに記載の医薬組成物。
- ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩を、疼痛治療最小有効量の約1/30倍~約3倍の投与量で投与するための、請求項1~6のいずれかに記載の医薬組成物。
- ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩が、ブプレノルフィンまたはその製薬上許容される塩である、請求項1~7のいずれかに記載の医薬組成物。
- 前記医薬組成物が、約1ng/mL以下のブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する、請求項8記載の医薬組成物。
- 前記医薬組成物が、約4pg/mL~約0.6ng/mLのブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する、請求項8または9記載の医薬組成物。
- 前記医薬組成物が、約0.2ng/mL以下のブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する、請求項8~10のいずれかに記載の医薬組成物。
- 前記医薬組成物が、約8pg/mL~約0.2ng/mLのブプレノルフィンまたはその製薬上許容される塩の最高血漿中濃度を提供する、請求項8~11のいずれかに記載の医薬組成物。
- 以下a)~c):
a)ブプレノルフィンまたはその製薬上許容される塩を、約0.01mg~約10mg含有する、経皮投与用医薬組成物、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.01μg/h~約10μg/hで放出する、経皮投与用医薬組成物、および、
c)ブプレノルフィンまたはその製薬上許容される塩を、約0.0001mg~0.1mg含有する、経粘膜投与用医薬組成物、
のいずれかである、請求項8~12のいずれかに記載の医薬組成物。 - a)ブプレノルフィンまたはその製薬上許容される塩を、約0.56mg~約1.7mg含有する、経皮投与用医薬組成物、または、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.56μg/h~約1.7μg/hで放出する、経皮投与用医薬組成物、
のいずれかである、請求項8~12のいずれかに記載の医薬組成物。 - a)ブプレノルフィンまたはその製薬上許容される塩を、約0.56mgまたは約1.7mg含有する、経皮投与用医薬組成物、または、
b)ブプレノルフィンまたはその製薬上許容される塩を、約0.56μg/hまたは約1.7μg/hで放出する、経皮投与用医薬組成物、
のいずれかである、請求項8~12のいずれかに記載の医薬組成物。 - 12時間以上の投与間隔で投与するための、請求項8~15のいずれかに記載の医薬組成物。
- 投与後12時間以上薬効が持続する、請求項8~16のいずれかに記載の医薬組成物。
- ブプレノルフィンまたはその製薬上許容される塩を、他の薬剤と併用することなく投与するための、請求項8~17のいずれかに記載の医薬組成物。
- ブプレノルフィン若しくはその製薬上許容される塩および/またはモルヒネ若しくはその製薬上許容される塩が、モルヒネまたはその製薬上許容される塩である、請求項1~7のいずれかに記載の医薬組成物。
- 前記医薬組成物が、約200ng/mL以下のモルヒネまたはその製薬上許容される塩の最高血漿中濃度を提供する、請求項19記載の医薬組成物。
- 前記医薬組成物が、約1ng/mL~約200ng/mLのモルヒネまたはその製薬上許容される塩の最高血漿中濃度を提供する、請求項19または20記載の医薬組成物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021-206572 | 2021-12-21 | ||
| JP2021206572 | 2021-12-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023120551A1 true WO2023120551A1 (ja) | 2023-06-29 |
Family
ID=86902678
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/047004 WO2023120551A1 (ja) | 2021-12-21 | 2022-12-21 | 自閉スペクトラム症治療のためのオピオイドの使用 |
Country Status (2)
| Country | Link |
|---|---|
| TW (1) | TW202329963A (ja) |
| WO (1) | WO2023120551A1 (ja) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013544780A (ja) * | 2010-10-07 | 2013-12-19 | カリフォルニア インスティチュート オブ テクノロジー | 自閉症の生菌療法 |
| JP2014532703A (ja) * | 2011-10-31 | 2014-12-08 | ザ・ジョンズ・ホプキンス・ユニバーシティー | 自閉症の処置のための方法および組成物 |
| JP2019089755A (ja) * | 2017-11-06 | 2019-06-13 | スタリクラ ソシエテ アノニムStalicla Sa | 自閉症を治療するための医薬組成物 |
-
2022
- 2022-12-21 WO PCT/JP2022/047004 patent/WO2023120551A1/ja unknown
- 2022-12-21 TW TW111149205A patent/TW202329963A/zh unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013544780A (ja) * | 2010-10-07 | 2013-12-19 | カリフォルニア インスティチュート オブ テクノロジー | 自閉症の生菌療法 |
| JP2014532703A (ja) * | 2011-10-31 | 2014-12-08 | ザ・ジョンズ・ホプキンス・ユニバーシティー | 自閉症の処置のための方法および組成物 |
| JP2019089755A (ja) * | 2017-11-06 | 2019-06-13 | スタリクラ ソシエテ アノニムStalicla Sa | 自閉症を治療するための医薬組成物 |
Non-Patent Citations (3)
| Title |
|---|
| SKOGLUND CHARLOTTE, LEKNES SIRI, HEILIG MARKUS: "The partial µ-opioid agonist buprenorphine in autism spectrum disorder: a case report", JOURNAL OF MEDICAL CASE REPORTS, vol. 16, no. 1, 1 December 2022 (2022-12-01), XP093074740, DOI: 10.1186/s13256-022-03384-w * |
| THORNTON, AOIFE M.; HUMPHREY, RACHEL M.; KERR, DANIEL M.; FINN, DAVID P.; ROCHE, MICHELLE: "Effects of pharmacological modulation of the endogenous opioid and cannabinoid systems on affective responding in a rat model of autism", BRAIN AND NEUROSCIENCE ADVANCES, SAGE PUBLISHING, vol. 3, 1 January 2019 (2019-01-01), pages 146, XP009547339, ISSN: 2398-2128 * |
| YAMAGUCHI, TAKU; YOSHIOKA, MITSUHIRO: "Evaluation for anxiety-related behaviors in rodents", NIHON NAIBUNPI GAKKAI ZASSHI - FOLIA ENDOCRINOLOGICA JAPONICA, NIHON NAIBUNPI GAKKAI, JP, vol. 130, no. 2, 1 January 2007 (2007-01-01), JP , pages 105 - 111, XP009547338, ISSN: 0029-0661, DOI: 10.1254/fpj.130.105 * |
Also Published As
| Publication number | Publication date |
|---|---|
| TW202329963A (zh) | 2023-08-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2021098743A (ja) | 神経疾患治療のためのデキストロメトルファンおよびキニジンを含む薬剤組成物 | |
| EP2892518B1 (en) | Compositions for treating parkinson's disease | |
| EP3209298B1 (en) | Compositions for treating insomnia | |
| EP2958573B1 (en) | Pharmaceutical formulations of nitrite and uses thereof | |
| JP6137833B2 (ja) | 脱髄性および他の神経系疾患を患っている患者における神経認知的および/または神経精神医学的障害を改善するための4−アミノピリジンの使用 | |
| US10765646B2 (en) | Methods of treating developmental encephalopathies | |
| WO2023120551A1 (ja) | 自閉スペクトラム症治療のためのオピオイドの使用 | |
| US11826321B2 (en) | Cyclobenzaprine treatment for agitation, psychosis and cognitive decline in dementia and neurodegenerative conditions | |
| WO2021161981A1 (en) | Novel medicament for treating hepatic encephalopathy | |
| TW202308653A (zh) | 以神經活性類固醇進行治療的方法 | |
| KR100740079B1 (ko) | 당뇨병성 신경 장해용 의약 조성물 | |
| WO2021047525A1 (zh) | 苯并硫代吡喃酮类化合物的盐及其制备方法和用途 | |
| Aki et al. | Toxicology in Emergency Medicine | |
| WO2023186827A1 (en) | 5-methoxy-n,n-dimethyltryptamine for the treatment of postpartum depression | |
| JP2024058212A (ja) | 自閉スペクトラム症治療のためのamg517の使用 | |
| WO2023186826A1 (en) | 5-meo-dmt for use in the treatment of postpartum depression | |
| TW202131910A (zh) | 使用mtorc1調節劑的治療方法 | |
| CN117580576A (zh) | Luvadaxistat用于治疗认知损害的用途 | |
| Kamath | Study of Anticonvulsant effect of Simvastatin in Maximal Electroshock and Pentylenetetrazole Induced Seizure Model In Albino Mice |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22911253 Country of ref document: EP Kind code of ref document: A1 |