WO2023090411A1 - 心疾患または骨格筋の疾患の治療用医薬 - Google Patents
心疾患または骨格筋の疾患の治療用医薬 Download PDFInfo
- Publication number
- WO2023090411A1 WO2023090411A1 PCT/JP2022/042796 JP2022042796W WO2023090411A1 WO 2023090411 A1 WO2023090411 A1 WO 2023090411A1 JP 2022042796 W JP2022042796 W JP 2022042796W WO 2023090411 A1 WO2023090411 A1 WO 2023090411A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pharmaceutical composition
- skeletal muscle
- disease
- composition according
- formula
- Prior art date
Links
Images







Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4184—1,3-Diazoles condensed with carbocyclic rings, e.g. benzimidazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/06—Antiarrhythmics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
Definitions
- the present invention relates to new pharmaceutical compositions for treating and/or preventing at least one of heart disease, heart disease complications, skeletal muscle diseases and skeletal muscle pathologies.
- Triglyceride deposit cardiomyovasculopathy (hereinafter also referred to as TGCV) is a new disease unit that was first reported in 2008. It is an intractable disease that causes severe heart failure and arrhythmia as a result of its accumulation (Non-Patent Document 1). Deficiency of ATGL (Adipose triglyceride lipase), which is an essential enzyme for intracellular neutral lipolysis, is known to be the cause of TGCV. Long-chain fatty acids, which normally serve as an energy source in the heart, cannot be metabolized intracellularly by TGCV, and accumulate as TG in cardiovascular vessels, resulting in the heart falling into a so-called obesity state (Non-Patent Document 2).
- ATGL Adipose triglyceride lipase
- BMI body mass index
- ACC is an enzyme that carboxylates acetyl-CoA to malonyl-CoA and is involved in fatty acid metabolism.
- ACC has two isoforms, acetyl-CoA carboxylase 1 (hereinafter referred to as ACC1) and acetyl-CoA carboxylase 2 (hereinafter referred to as ACC2).
- ACC2 is mainly expressed in heart and skeletal muscle, and malonyl-CoA produced by ACC2 inhibits fatty acid oxidation by inhibiting carnitine palmitoyltransferase I (CPT-I).
- ACC2-deficient mice decreased amounts of malonyl-CoA in the heart and skeletal muscle cause continuous fatty acid oxidation, resulting in weight loss regardless of increased food intake. Furthermore, it has also been reported that ACC2-deficient mice acquire resistance to diabetes and obesity induced by administration of a high-fat/high-carbohydrate diet. These findings suggest that ACC2 is involved in diseases such as diabetes and obesity, and that inhibitors thereof will serve as antidiabetic and antiobesity drugs. On the other hand, since ACC1-deficient mice are lethal in the fetal stage, a selective inhibitor that inhibits ACC2 without inhibiting ACC1 is desired (Non-Patent Document 3).
- Patent Documents 2 and 3 describe methods of treating nonalcoholic fatty liver disease using thienopyrimidine derivatives that have inhibitory activity against both ACC1 and ACC2.
- the compound shown below is known as firsocostat (a dual inhibitor of ACC1/2) and is being developed for indications such as non-alcoholic steatohepatitis.
- firsocostat is designed to be selectively taken up by the liver via a transporter.
- topline results from the 48-week Phase II ATLAS trial were announced on December 16, 2019.
- a Phase II ATLAS trial is investigating cilofexor (a selective non-steroidal FXR agonist) 30 mg, firsocostat 20 mg, and selonsertib (an ASK1 inhibitor) 18 in patients with advanced fibrosis (F3-F4) due to NASH
- cilofexor a selective non-steroidal FXR agonist
- firsocostat 20 mg a selective non-steroidal FXR agonist
- selonsertib an ASK1 inhibitor
- Elevated plasma triglyceride concentrations have also been reported in NAFLD patients receiving MK-4074, another dual inhibitor of ACC1/2. Elevated plasma triglyceride concentrations are known to increase the incidence of cardiovascular events, which are reported to be the most common cause of death in patients with NASH. NAFLD is an independent risk factor for cardiovascular disease, and progression to NASH has been reported to increase the risk of cardiovascular disease. The ATLAS trial reported increased plasma triglyceride concentrations in NASH patients who received a combination of firsocostat and cilofexor.
- Patent Document 4 describes dual inhibitors of ACC1/2.
- the compound shown below known as PF-05175157 (a dual inhibitor of ACC1/2), reduced platelet concentrations upon repeated administration to healthy subjects. It has been reported that this is due to decreased platelet production due to inhibition of fatty acid synthesis mediated by ACC1 inhibition in the bone marrow.
- Patent Documents 5 and 6 describe benzimidazole derivatives that specifically inhibit ACC2. Furthermore, Patent Document 7 describes that benzimidazole derivatives are useful as therapeutic agents for NASH. Further, Patent Documents 8 to 20 and Non-Patent Documents 4 to 6 describe compounds similar to benzimidazole derivatives contained in the pharmaceutical composition of the present invention as ACC inhibitors.
- Patent Documents 21 to 26 describe that an ACC inhibitor is useful as a therapeutic agent for sarcopenia, but do not describe the benzimidazole derivative contained in the pharmaceutical composition of the present invention.
- Non-Patent Document 6 describes that a specific ACC2 inhibitor reduces skeletal muscle intracellular lipid levels and improves insulin resistance and hyperglycemia. Moreover, the benzimidazole derivative contained in the pharmaceutical composition of the present invention is not described.
- An object of the present invention is to provide an anti-cardiac disease, heart disease complication, skeletal muscle disease, and skeletal muscle pathology that have excellent selective ACC2 inhibitory action and are not accompanied by side effects such as increased plasma triglycerides or decreased platelet concentration.
- An object is to provide a pharmaceutical composition for treating and/or preventing at least one. More preferably, the objective is to provide a pharmaceutical composition for the treatment and/or prevention of triglyceride-accumulating cardiomyoangiopathy or skeletal muscle diseases or conditions caused by triglyceride accumulation in skeletal muscle cells.
- the present inventors found that among the compounds having ACC2 selective inhibitory activity described in Patent Documents 5 and 6, specific compounds (high selectivity of ACC2 and , a compound with good metabolic stability) is effective for the treatment and/or prevention of at least one of heart disease, heart disease complication, skeletal muscle disease and skeletal muscle condition, and plasma triglyceride elevation or platelet concentration The inventors have found that there is no side effect such as reduction, and have completed the present invention.
- the ACC2 selective inhibitor contained in the pharmaceutical composition of the present invention can avoid side effects due to ACC1 inhibition and inhibit systemic ACC2.
- the ACC2 selective inhibitor contained in the pharmaceutical composition of the present invention causes thrombocytopenia, unlike an ACC1/2 dual inhibitor and a liver-selective ACC1/2 dual inhibitor such as firsocostat. It does not increase the plasma triglyceride concentration, and can exhibit a metabolic improvement effect based on systemic ACC2 inhibition.
- the present invention relates to the following.
- R 1 is a haloalkyl or non-aromatic carbocyclic group, R2 is a hydrogen atom or halogen, R3 is halogen;
- Ring A has the formula: is a group represented by -L 1 - is -O-(CH 2 )-, -(CH 2 ) 2 -, -(CH 2 )-(CF 2 )- or -(CF 2 )-(CH 2 )- (where left is attached to ring A, and the right bond is the formula: It binds to the group represented by ) and R4 is alkyl or haloalkyl; R5 is alkylcarbonyl or carbamoyl.
- Ring A is of the formula: The pharmaceutical composition according to any one of the above items [1] to [3], which is a group represented by [5] The pharmaceutical composition according to any one of the above items [1] to [4], wherein -L 1 - is -O-(CH 2 )- or -(CH 2 ) 2 -. [6] The pharmaceutical composition according to any one of the above items [1] to [5], wherein R 4 is alkyl. [7] The pharmaceutical composition according to any one of the above items [1] to [6], wherein R5 is methylcarbonyl or carbamoyl.
- a compound of formula (I) or a pharmaceutically acceptable salt thereof is represented by the following formula:
- [10] Caused by the accumulation of neutral fat in at least one of cardiomyocytes, vascular smooth muscle cells, endothelial cells, skeletal muscle cells, polymorphonuclear leukocytes, renal tubular epithelial cells, and pancreatic islet cells
- the disease is at least one disease selected from the group consisting of heart failure, angina pectoris, myocardial infarction, cardiomyopathy, arrhythmia, coronary artery disease, skeletal muscle myopathy, chronic kidney disease and impaired glucose tolerance.
- a pharmaceutical composition as described in [12] The pharmaceutical composition according to any one of the above items [1] to [11], wherein the administration of the pharmaceutical composition is not accompanied by the side effect of increasing plasma triglycerides.
- R 1 is a haloalkyl or non-aromatic carbocyclic group, R2 is a hydrogen atom or halogen, R3 is halogen;
- Ring A has the formula: is a group represented by -L 1 - is -O-(CH 2 )-, -(CH 2 ) 2 -, -(CH 2 )-(CF 2 )- or -(CF 2 )-(CH 2 )- (where left is attached to ring A, and the right bond is the formula: It binds to the group represented by ) and R4 is alkyl or haloalkyl; R5 is alkylcarbonyl or carbamoyl.
- Ring A is of the formula: The pharmaceutical composition according to any one of the above items [14] to [16], which is a group represented by [18] The pharmaceutical composition according to any one of the above items [14] to [17], wherein -L 1 - is -O-(CH 2 )- or -(CH 2 ) 2 -.
- a compound of formula (I) or a pharmaceutically acceptable salt thereof is represented by the following formula: The pharmaceutical composition according to the above item [14], which is a compound selected from the group consisting of or a pharmaceutically acceptable salt thereof. [22] The pharmaceutical composition according to any one of the above items [14] to [21], wherein the skeletal muscle disease or condition is caused by the accumulation of neutral fat in skeletal muscle cells (e.g., myosteatosis). .
- the pharmaceutical composition of the present invention containing a compound of formula (I) or a pharmaceutically acceptable salt thereof is used to treat at least one of heart disease, heart disease complication, skeletal muscle disease and skeletal muscle pathology, especially It is effective for the treatment and/or prevention of triglyceride accumulation myocardiovasculopathy or skeletal muscle diseases or conditions caused by accumulation of triglycerides in skeletal muscle cells. In addition, it has high safety without side effects such as increased plasma triglycerides or decreased platelet concentration.
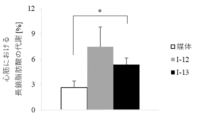
- FIG. 1 shows long-chain fatty acid metabolism in the myocardium when compound I-12 and compound I-13 were administered to Atgl knockout mice.
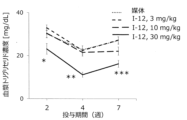
- FIG. 2 shows plasma triglyceride concentrations in mice fed an ultra-high fat, choline-deficient, methionine-reduced diet following administration of compound I-12.
- FIG. 3 shows plasma triglyceride concentrations in mice fed an ultra-high-fat, choline-deficient, methionine-reduced diet following administration of compound I-13.
- FIG. 4 shows the intracellular lipid levels in skeletal muscle when compound I-13 was administered to SD rats.
- FIG. 5 shows intracellular lipid levels in skeletal muscle when compound I-12 was administered to leptin receptor-deficient mice.
- FIG. 1 shows long-chain fatty acid metabolism in the myocardium when compound I-12 and compound I-13 were administered to Atgl knockout mice.
- FIG. 2 shows plasma triglyceride concentrations in mice fed an ultra-high fat, choline-de
- FIG. 6 shows the intracellular lipid levels in skeletal muscle when compound I-12 was administered to diabetic model rats.
- FIG. 7 shows intracellular lipid levels in skeletal muscle when compound I-14 was administered to high-fat diet-fed mice.
- FIG. 8 shows skeletal muscle intracellular lipid levels and skeletal muscle extracellular lipid levels when wild-type mice and ACC2 knockout mice were loaded with a high-fat diet.
- FIG. 9 shows locomotor activity in Atgl knockout mice treated with compound I-13.
- FIG. 10 shows locomotor activity in Atgl knockout mice and Atgl and ACC2 double knockout mice.
- Halogen includes a fluorine atom, a chlorine atom, a bromine atom, and an iodine atom. Fluorine and chlorine atoms are particularly preferred.
- Alkyl includes a linear or branched hydrocarbon group having 1 to 15 carbon atoms, preferably 1 to 10 carbon atoms, more preferably 1 to 6 carbon atoms, still more preferably 1 to 4 carbon atoms. do. For example, methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, n-pentyl, isopentyl, neopentyl, n-hexyl, isohexyl, n-heptyl, isoheptyl, n-octyl , isooctyl, n-nonyl, n-decyl and the like.
- alkyl examples include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl and n-pentyl. More preferred embodiments include methyl, ethyl, n-propyl, isopropyl and tert-butyl. A particularly preferred embodiment is methyl.
- Haloalkyl means a group in which one or more arbitrary hydrogen atoms of the above “alkyl” are substituted with the above "halogen".
- Alkylcarbonyl means a group in which the above “alkyl” is bonded to a carbonyl group. Examples include methylcarbonyl, ethylcarbonyl, propylcarbonyl, isopropylcarbonyl, tert-butylcarbonyl, isobutylcarbonyl, sec-butylcarbonyl, pentylcarbonyl, isopentylcarbonyl, hexylcarbonyl and the like. Preferred embodiments of "alkylcarbonyl” include methylcarbonyl, ethylcarbonyl, n-propylcarbonyl and the like. A more preferred embodiment is methylcarbonyl.
- aromatic carbocyclic group means a monocyclic or bicyclic or more cyclic aromatic hydrocarbon group. Examples include phenyl, naphthyl, anthryl, phenanthryl and the like. A preferred embodiment of the "aromatic carbocyclic group” is phenyl.
- Aromatic carbocyclic ring means a ring derived from the above “aromatic carbocyclic group”.
- Non-aromatic carbocyclic group means a monocyclic or bicyclic or more ring saturated cyclic hydrocarbon group or cyclic non-aromatic unsaturated hydrocarbon group.
- the "non-aromatic carbocyclic group” having two or more rings includes a monocyclic or non-aromatic carbocyclic group having two or more rings condensed with the above “aromatic carbocyclic group”.
- the “non-aromatic carbocyclic group” also includes a group that forms a bridge or a spiro ring as shown below.
- the monocyclic non-aromatic carbocyclic group preferably has 3 to 16 carbon atoms, more preferably 3 to 12 carbon atoms, still more preferably 3 to 6 carbon atoms, and particularly preferably 3 to 4 carbon atoms. be.
- Examples include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl, cyclodecyl, cyclopropenyl, cyclobutenyl, cyclopentenyl, cyclohexenyl, cycloheptenyl, cyclohexadienyl and the like.
- the bicyclic or more non-aromatic carbocyclic group preferably has 8 to 20 carbon atoms, more preferably 8 to 16 carbon atoms.
- Examples include indanyl, indenyl, acenaphthyl, tetrahydronaphthyl, fluorenyl and the like.
- Non-aromatic carbocyclic ring means a ring derived from the above “non-aromatic carbocyclic group”.
- “Aromatic heterocyclic group” means a monocyclic or bicyclic or more aromatic cyclic group having one or more heteroatoms which are the same or different and are arbitrarily selected from O, S and N in the ring. do.
- An aromatic heterocyclic group with two or more rings includes a monocyclic or an aromatic heterocyclic group with two or more rings condensed with the ring in the above "aromatic carbocyclic group", and the bond is You may have it in any ring.
- the monocyclic aromatic heterocyclic group is preferably 5- to 8-membered, more preferably 5- or 6-membered.
- Five-membered aromatic heterocyclic groups include, for example, pyrrolyl, imidazolyl, pyrazolyl, triazolyl, tetrazolyl, furyl, thienyl, isoxazolyl, oxazolyl, oxadiazolyl, isothiazolyl, thiazolyl, thiadiazolyl and the like.
- 6-membered aromatic heterocyclic groups include pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl and the like.
- the bicyclic aromatic heterocyclic group is preferably 8- to 10-membered, more preferably 9- or 10-membered.
- indolyl isoindolyl, indazolyl, indolizinyl, quinolinyl, isoquinolinyl, cinnolinyl, phthalazinyl, quinazolinyl, naphthyridinyl, quinoxalinyl, purinyl, pteridinyl, benzimidazolyl, benzisoxazolyl, benzoxazolyl, benzoxadiazolyl, benzisothiazolyl.
- Ryl benzothiazolyl, benzothiadiazolyl, benzofuryl, isobenzofuryl, benzothienyl, benzotriazolyl, imidazopyridyl, triazolopyridyl, imidazothiazolyl, pyrazinopyridazinyl, oxazolopyridyl, thiazolopyridyl, etc. is mentioned.
- the aromatic heterocyclic group having 3 or more rings is preferably 13- to 15-membered. Examples include carbazolyl, acridinyl, xanthenyl, phenothiazinyl, phenoxathiinyl, phenoxazinyl, dibenzofuryl and the like.
- Aromatic heterocyclic ring means a ring derived from the above “aromatic heterocyclic group”.
- Non-aromatic heterocyclic group means a monocyclic or bicyclic or more non-aromatic cyclic group having one or more heteroatoms in the ring that are the same or different and arbitrarily selected from O, S and N.
- a bicyclic or more non-aromatic heterocyclic group is a monocyclic or bicyclic or more non-aromatic heterocyclic group, the above "aromatic carbocyclic group", “non-aromatic carbocyclic group”, and / Or a ring in which each ring in the "aromatic heterocyclic group” is condensed, and a ring in the above "aromatic heterocyclic group” is condensed to a monocyclic or bicyclic or more non-aromatic carbocyclic group and the bond may be in any ring.
- non-aromatic heterocyclic group also includes a group that forms a bridge or a spiro ring as shown below.
- the monocyclic non-aromatic heterocyclic group is preferably 3- to 8-membered, more preferably 5- or 6-membered.
- Three-membered non-aromatic heterocyclic groups include, for example, thiiranyl, oxiranyl, aziridinyl.
- Examples of 4-membered non-aromatic heterocyclic groups include oxetanyl and azetidinyl.
- Examples of 5-membered non-aromatic heterocyclic groups include oxathiolanyl, thiazolidinyl, pyrrolidinyl, pyrrolinyl, imidazolidinyl, imidazolinyl, pyrazolidinyl, pyrazolinyl, tetrahydrofuryl, dihydrothiazolyl, tetrahydroisothiazolyl, dioxolanyl, dioxolyl, thiolanyl, and the like. mentioned.
- 6-membered non-aromatic heterocyclic groups include, for example, dioxanyl, thianyl, piperidyl, piperazinyl, morpholinyl, morpholino, thiomorpholinyl, thiomorpholino, dihydropyridyl, tetrahydropyridyl, tetrahydropyranyl, dihydrooxazinyl, tetrahydropyridazinyl hexahydropyrimidinyl, dioxazinyl, thiinyl, thiazinyl and the like.
- Seven-membered non-aromatic heterocyclic groups include, for example, hexahydroazepinyl, tetrahydrodiazepinyl, oxepanyl.
- the non-aromatic heterocyclic group having two or more rings is preferably 8- to 20-membered, more preferably 8- to 10-membered. Examples include indolinyl, isoindolinyl, chromanyl, isochromanyl and the like.
- Non-aromatic heterocyclic ring means a ring derived from the above “non-aromatic heterocyclic group”.
- Disease means a disease/disorder/syndrome/symptom in various stages from presymptomatic to progressive (including symptoms and signs of disease, including prodromal symptoms and other signs of disease).
- Phathology means a state of low performance compared to the individual's potential and goals.
- Increasing skeletal muscle function means improving an individual's skeletal muscle function.
- a pharmaceutical composition that increases skeletal muscle function also includes prophylactic uses (applications) of the pharmaceutical composition.
- “Promoting skeletal muscle recovery” means improving skeletal muscle function in individuals with skeletal muscle diseases or conditions.
- the compound of the present invention has a fatty acid metabolism action in the myocardium, it is particularly limited if it is a disease in which triglycerides are accumulated in the tissue or cells of the myocardium or blood vessels of the heart or a disease caused by triglyceride accumulation.
- a disease in which triglycerides are accumulated in the tissue or cells of the myocardium or blood vessels of the heart or a disease caused by triglyceride accumulation.
- selected from the group consisting of diseases in which blood vessels are constricted or occluded due to accumulation of triglycerides in blood vessels, diseases caused by said constriction or obstruction, and diseases caused by triglyceride accumulation in myocardium is preferred.
- diseases exhibiting at least one finding selected from the group consisting of myocardial hypertrophy, coronary artery diffuse stenosis, and coronary artery afferent stenosis are preferred.
- "heart disease and heart disease complications" of the present invention include, for example, at least one disease selected from the group consisting of heart failure, angina pectoris, myocardial infarction, cardiomyopathy, arrhythmia and coronary artery disease.
- the compounds of the present invention contain neutral fat in at least one of cardiomyocytes, vascular smooth muscle cells, endothelial cells, skeletal muscle cells, polymorphonuclear leukocytes, renal tubular epithelial cells, and pancreatic islet cells. It is also effective for a disease caused by accumulation, ie, "triglyceridal cardiomyovascular disease (TGCV)".
- TGCV triglyceridal cardiomyovascular disease
- "Neutral fat accumulation myocardiovasculopathy” includes, for example, at least one selected from the group consisting of heart failure, angina pectoris, myocardial infarction, cardiomyopathy, arrhythmia, coronary artery disease, skeletal muscle myopathy, chronic kidney disease and impaired glucose tolerance.
- TGCV triglyceridal cardiomyovascular disease
- Atherosclerosis caused by the accumulation of neutral fat in blood vessels.
- it has a therapeutic and preventive effect in cases in which improvement of symptoms is not observed with cholesterol-lowering drugs.
- compositions of the present invention comprise compounds for treating and/or preventing diseases or conditions of skeletal muscle.
- Said compound or a pharmaceutically acceptable salt thereof is represented by Formula (I).
- the "skeletal muscle disease or condition” is caused by accumulation of triglycerides in skeletal muscle cells.
- the "skeletal muscle disease or condition” is myosteatosis. Myosteatosis refers to changes in muscle quality due to the accumulation of triglycerides in skeletal muscle cells.
- the "skeletal muscle disease or condition” may relate to liver disease. Non-limiting examples of such liver disease include advanced or late stage liver disease.
- the "skeletal muscle disease or condition” is skeletal muscle myopathy. Skeletal muscle myopathy is characterized by progressive degeneration of muscle tissue due to the presence of abnormal or insufficient structural support proteins. In some embodiments, skeletal muscle myopathy may be associated with muscle fiber breakdown and/or loss of muscle strength.
- the skeletal muscle myopathy can be a hereditary myopathy or an acquired myopathy.
- the skeletal muscle myopathy is congenital myopathy, mitochondrial myopathy, metabolic myopathy, muscular dystrophy, toxic myopathy, endocrine myopathy, infectious myopathy, critical illness myopathy, myopathy caused by electrolyte imbalance, and It can be any combination thereof.
- the "skeletal muscle disease or condition" is sarcopenia. Sarcopenia is a type of muscle atrophy that involves a decrease in muscle fiber size and number. In some embodiments, sarcopenia can be associated with loss of one or more of muscle strength, muscle mass, and muscle function.
- the pathology and/or symptoms of a "skeletal muscle disease or condition" are attributable to ACC2 activity.
- ACC2 activity is associated with triglyceride accumulation in skeletal muscle cells.
- ACC2 activity contributes to a condition selected from the group consisting of myosteatosis, skeletal muscle myopathy, sarcopenia, and any combination thereof.
- ACC2 activity contributes to a condition selected from the group consisting of diabetes, obesity, and cirrhosis.
- ACC2 activity simultaneously contributes to a combination of two or more conditions selected from the group consisting of diabetes, obesity, cirrhosis, myosteatosis, skeletal muscle myopathy, and sarcopenia.
- the "skeletal muscle disease or condition" is associated with a condition selected from the group consisting of diabetes, obesity, and cirrhosis.
- myosteatosis is concurrently associated with diabetes, obesity, or cirrhosis.
- skeletal muscle myopathy is concurrently associated with diabetes, obesity, or cirrhosis.
- sarcopenia is concurrently associated with diabetes, obesity, or cirrhosis.
- the "skeletal muscle disease or condition" is associated with liver cirrhosis.
- myosteatosis is associated with cirrhosis.
- the skeletal muscle myopathy is associated with liver cirrhosis.
- sarcopenia is associated with cirrhosis.
- the pharmaceutical compositions of the invention maintain or increase skeletal muscle function in a subject. In certain embodiments, the pharmaceutical compositions of the invention promote skeletal muscle recovery in a subject with a skeletal muscle disease or condition. In certain embodiments, "skeletal muscle function" and “skeletal muscle recovery” can be determined using manual muscle testing. This determination involves measuring muscle performance against resistance and rating that performance on a 0-5 scale.
- muscle performance is measured if it corresponds to the following conditions: (1) the muscle can only be moved slightly, such as cramping, and the muscle cannot move through its full range of motion; (2) able to move the entire range of motion of the muscle under zero gravity, (3) able to move the entire range of motion of the muscle under normal gravity, and (4) able to move the entire range of motion of the muscle under the presence of some resistance.
- muscles that can be measured include shoulder abductors, elbow flexors, elbow extensors, wrist extensors, finger flexors, intrinsic hand muscles, hip flexors, knee extensors, dorsi flexors, big toe extensors muscles, and plantar flexors.
- "skeletal muscle function” and “skeletal muscle recovery” comprise handgrip test, chairstand test, walking speed test, short physical performance battery (SPPB), and time up and go test (TUG).
- skeletal muscle function and skeletal muscle recovery can be determined using standard tests, including
- skeletal muscle function and skeletal muscle recovery can be determined using imaging studies including dual-energy X-ray absorptiometry (DEXA or DXA) and bioelectrical impedance analysis (BIA) techniques.
- skeletal muscle function and recovery is determined using tests including blood tests, electromyography (EMG), magnetic resonance imaging (MRI), genetic testing, and muscle biopsy. can do.
- EMG electromyography
- MRI magnetic resonance imaging
- genetic testing genetic testing
- muscle biopsy can do.
- "skeletal muscle function" and "skeletal muscle recovery” can be determined using frailty index.
- frailty index For example, the liver frailty index (LFI) can be used.
- the LFI includes three performance-based tests (grip strength, chairstand, and balance).
- grip strength is measured in kilograms using a handheld dynamometer on the subject's dominant hand.
- An average of three trials is calculated for analysis.
- the Timed Chair Stand measures the number of seconds it takes for a subject to complete standing up from a chair five times with their arms crossed over their chest.
- the balance test measures the number of seconds a subject can maintain balance in three positions (legs side-to-side, semi-tandem, and tandem) for up to 10 seconds each. Any one or more of three factors can be used in the evaluation.
- R 1 is a haloalkyl or non-aromatic carbocyclic group.
- R 1 is preferably a non-aromatic carbocyclic group.
- R 1 is more preferably a monocyclic non-aromatic carbocyclic group, particularly preferably cyclopropyl or cyclobutyl.
- Another preferred embodiment of R 1 is preferably 2,2-difluoroethyl.
- R2 is a hydrogen atom or halogen.
- R 2 is preferably a hydrogen atom or a fluorine atom.
- R2 is more preferably a hydrogen atom.
- R3 is halogen.
- R 3 is preferably a fluorine atom or a chlorine atom.
- R 3 is more preferably a fluorine atom.
- R4 is alkyl or haloalkyl.
- R4 is preferably alkyl.
- R4 is more preferably methyl.
- Another preferred embodiment of R 4 is preferably monofluoromethyl.
- R5 is alkylcarbonyl or carbamoyl.
- R5 is preferably methylcarbonyl or carbamoyl.
- Ring A has the formula: is a group represented by Ring A preferably has the formula: is a group represented by Ring A is more preferably of the formula: is a group represented by Ring A is more preferably of the formula: is a group represented by Ring A is more preferably of the formula: is a group represented by
- -L 1 - is -O-(CH 2 )-, -(CH 2 ) 2 -, -(CH 2 )-(CF 2 )- or -(CF 2 )-(CH 2 )- (wherein The left bond is attached to ring A and the right bond is of the formula: It binds to the group represented by ).
- -L 1 - is preferably -O-(CH 2 )- or -(CH 2 ) 2 -.
- the pharmaceutical composition for treating and/or preventing at least one of heart disease, heart disease complication, skeletal muscle disease and skeletal muscle pathology of the present invention comprises formula (I) as an active ingredient: (In the formula, each symbol has the same meaning as above) It is characterized by being a pharmaceutical composition containing a compound represented by or a pharmaceutically acceptable salt thereof.
- the compound represented by formula (I) can be synthesized according to known methods, for example, the methods described in WO2015/056782 and WO2016/159082.
- the compounds of formula (I) are not limited to any particular isomer, but include all possible isomers (e.g. keto-enol isomers, imine-enamine isomers, diastereoisomers, optical isomers , rotamers, etc.), racemates or mixtures thereof.
- One or more hydrogen, carbon and/or other atoms of the compounds of formula (I) may be substituted with isotopes of hydrogen, carbon and/or other atoms, respectively.
- isotopes include 2 H, 3 H, 11 C, 13 C, 14 C, 15 N, 18 O, 17 O , 31 P, 32 P, 35 S, 18 F, 123 I and Hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur, fluorine, iodine and chlorine are included, as is 36 Cl.
- the compounds of formula (I) also include such isotopically substituted compounds.
- the isotopically substituted compounds are also useful as pharmaceuticals and include all radiolabeled compounds of formula (I).
- a "radiolabeling method" for producing the "radiolabel” is also encompassed by the present invention, and the “radiolabel” is useful as a research and/or diagnostic tool in metabolic pharmacokinetic studies, binding assays. is.
- Radiolabeled compounds of formula (I) can be prepared by methods well known in the art.
- a tritium-labeled compound of formula (I) can be prepared by introducing tritium into a specific compound of formula (I) through a catalytic dehalogenation reaction using tritium.
- This method comprises reacting a suitably halogenated precursor of a compound of formula (I) with tritium gas in the presence or absence of a base, in the presence of a suitable catalyst such as Pd/C.
- a suitable catalyst such as Pd/C.
- 14 C-labeled compounds can be prepared by using starting materials with a 14 C carbon.
- Pharmaceutically acceptable salts of the compound represented by formula (I) include, for example, the compound represented by formula (I) and an alkali metal (e.g., lithium, sodium, potassium, etc.), alkaline earth metal (e.g., calcium, barium, etc.), magnesium, transition metals (e.g., zinc, iron, etc.), ammonia, organic bases (e.g., trimethylamine, triethylamine, dicyclohexylamine, ethanolamine, diethanolamine, triethanolamine, meglumine, ethylenediamine, pyridine, picoline, quinoline, etc.) and salts with amino acids, or inorganic acids (e.g., hydrochloric acid, sulfuric acid, nitric acid, carbonic acid, hydrobromic acid, phosphoric acid, hydroiodic acid, etc.), and organic acids (e.g., formic acid, acetic acid, propionic acid) , trifluoroacetic acid, citric acid, lactic acid, tarta
- the compound represented by formula (I) or a pharmaceutically acceptable salt thereof contained in the pharmaceutical composition of the present invention forms solvates (e.g., hydrates, etc.), co-crystals and/or crystal polymorphs. and the invention also includes various such solvates, co-crystals and polymorphs.
- a "solvate” may be coordinated with any number of solvent molecules (eg, water molecules, etc.) to a compound of formula (I).
- solvent molecules eg, water molecules, etc.
- the compound represented by formula (I) or a pharmaceutically acceptable salt thereof When the compound represented by formula (I) or a pharmaceutically acceptable salt thereof is left in the air, it may absorb water, attach adsorbed water, or form a hydrate. Also, the compound of formula (I) or a pharmaceutically acceptable salt thereof may be recrystallized to form polymorphs.
- “Co-crystal” means that a compound or salt of formula (I) and a counter molecule are present in the same crystal lattice, and may
- a compound represented by Formula (I) or a pharmaceutically acceptable salt thereof contained in the pharmaceutical composition of the present invention may form a prodrug, and the present invention includes various such prodrugs. It also includes pharmaceutical compositions.
- Prodrugs are derivatives of the compounds of the invention having groups which are chemically or metabolically degradable, and which, upon solvolysis or under physiological conditions, become pharmaceutically active compounds of the invention in vivo.
- a prodrug is a compound that undergoes enzymatic oxidation, reduction, hydrolysis, or the like under physiological conditions in vivo and is converted into a compound represented by formula (I), or a compound that is hydrolyzed by gastric acid or the like to form formula (I). It includes compounds that are converted to the indicated compounds, and the like. Methods for selecting and preparing suitable prodrug derivatives are described, for example, in "Design of Prodrugs, Elsevier, Amsterdam, 1985". A prodrug may itself have activity.
- the compound of the present invention Since the compound of the present invention has an excellent selective ACC2 inhibitory action, it is used as a therapeutic and/or preventive agent for heart disease, heart disease complications, skeletal muscle disease and skeletal muscle pathology, triglyceride accumulation myocardial angiopathy or It is also useful as a therapeutic and/or preventive agent for skeletal muscle diseases or conditions caused by accumulation of neutral fat in skeletal muscle cells. Furthermore, the compound of the present invention is useful as a medicine, and preferably has one or more of the following excellent characteristics. a) It has a weak inhibitory effect on CYP enzymes (eg, CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4, etc.).
- CYP enzymes eg, CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4, etc.
- b) shows good pharmacokinetics such as high bioavailability and moderate clearance; c) high metabolic stability; d) Does not exhibit irreversible inhibitory action on CYP enzymes (eg, CYP3A4) within the concentration range of the measurement conditions described herein. e) not mutagenic; f) low cardiovascular risk; g) exhibit high solubility; h) without side effects of elevated plasma triglycerides; i) insulin resistance is improved; j) without the side effect of lowering platelet concentration.
- CYP enzymes eg, CYP3A4
- the pharmaceutical composition of the present invention can be administered orally or parenterally.
- parenteral administration methods include transdermal, subcutaneous, intravenous, intraarterial, intramuscular, intraperitoneal, transmucosal, inhalation, nasal, ocular, ear and intravaginal administration.
- internal solid preparations e.g., tablets, powders, granules, capsules, pills, films, etc.
- internal liquid preparations e.g., suspensions, emulsions, elixirs, syrups, etc.
- Tablets may be sugar-coated tablets, film-coated tablets, enteric-coated tablets, sustained-release tablets, troches, sublingual tablets, buccal tablets, chewable tablets or orally disintegrating tablets, and powders and granules may be dry syrups.
- the capsules may be soft capsules, microcapsules or sustained release capsules.
- injections In the case of parenteral administration, injections, drops, external preparations (e.g., eye drops, nasal drops, ear drops, aerosols, inhalants, lotions, injections, coatings, gargles, enemas, Any commonly used dosage form such as ointments, plasters, jellies, creams, patches, poultices, powders for external use, suppositories, etc.) can be suitably administered. Injections may be emulsions such as O/W, W/O, O/W/O and W/O/W types.
- a pharmaceutical composition can be prepared by mixing an effective amount of the compound of the present invention with various pharmaceutical additives such as excipients, binders, disintegrants, and lubricants suitable for the dosage form, if necessary. Furthermore, by appropriately changing the effective amount, dosage form and/or various pharmaceutical additives of the compound of the present invention, the pharmaceutical composition can be used as a pharmaceutical composition for children, the elderly, critically ill patients, or for surgery. You can also For example, a pediatric pharmaceutical composition can be used for newborns (less than 4 weeks after birth), infants (4 weeks after birth to less than 1 year old), infants (1 to 7 years old), children (7 to 15 years old), or Patients between the ages of 15 and 18 may be administered. For example, geriatric pharmaceutical compositions may be administered to patients 65 years of age or older.
- the dosage of the pharmaceutical composition of the present invention is preferably set in consideration of the patient's age, body weight, type and degree of disease, administration route, etc., but when administered orally, it is usually 0.05 to 100 mg / kg/day, preferably within the range of 0.1 to 10 mg/kg/day. In the case of parenteral administration, it is generally 0.005 to 10 mg/kg/day, preferably 0.01 to 1 mg/kg/day, although it varies greatly depending on the route of administration. It may be administered once to several times a day.
- Formula (I) contained in the pharmaceutical composition of the present invention (wherein each symbol has the same meaning as above) or a pharmaceutically acceptable salt thereof enhances or enhances the therapeutic and/or preventive action of the compound for heart disease or complications of heart disease.
- a pharmaceutically acceptable salt thereof enhances or enhances the therapeutic and/or preventive action of the compound for heart disease or complications of heart disease.
- diuretics, ACE inhibitors, angiotensin II receptor blockers, angiotensin receptor/neprilysin inhibitors, aldosterone antagonists, beta-blockers, SGLT2 inhibitors and vasodilators for the purpose of reducing the dose of the compound can be used
- at least one compound or a pharmaceutically acceptable salt thereof selected from the group consisting of furosemide, enalapril, candesartan cilexetil, sacubitril/valsartan, spironolactone, carvedilol, dapagliflozin and nitroglycerin (herein
- Formula (I) contained in the pharmaceutical composition of the present invention (wherein each symbol has the same meaning as above) or a pharmaceutically acceptable salt thereof is used for the treatment of skeletal muscle diseases or conditions (e.g., liver cirrhosis associated with sarcopenia) and/or Alternatively, it can be used in combination with testosterone replacement therapy and/or amino acid supplementation (for example, branched-chain amino acid supplementation), myostatin-neutralizing antibody, etc., for the purpose of enhancing preventive action or reducing the dosage of the compound.
- Myostatin-neutralizing antibodies include domaglozumab, landoglozumab, stamulumab, and the like.
- the compound of the present invention may be used in combination with at least one compound selected from the group consisting of testosterone, branched-chain amino acids, and myostatin-neutralizing antibodies, or a pharmaceutically acceptable salt thereof (hereinafter referred to as concomitant drug 2).
- concomitant drug 2 a pharmaceutically acceptable salt thereof
- the timing of administering the compound of the present invention and the concomitant drug 2 are not limited, and they may be administered to the subject at the same time, consecutively, or at different times.
- the compound of the present invention and the concomitant drug 2 may be administered as two or more formulations containing each active ingredient, or may be administered as a single formulation containing those active ingredients.
- the compound of the present invention can be used in combination with at least one therapy (hereinafter referred to as combination therapy 1) selected from the group consisting of testosterone replacement therapy and amino acid supplementation (e.g., branched-chain amino acid supplementation).
- combination therapy 1 selected from the group consisting of testosterone replacement therapy and amino acid supplementation (e.g., branched-chain amino acid supplementation).
- the timing of administration of the compound of the present invention and the timing of implementation of combination therapy 1 are not limited.
- the compound of the present invention may be administered to an administration subject simultaneously with or continuously with combination therapy 1, or may be administered with a time lag from combination therapy 1.
- the dose of concomitant drug 1 or concomitant drug 2 can be appropriately selected based on the clinically used dose.
- the compounding ratio of the compound of the present invention and the concomitant drug 1 or concomitant drug 2 can be appropriately selected depending on the subject of administration, administration route, target disease, symptom, combination, and the like. For example, when the subject of administration is a human, 0.01 to 100 parts by weight of the concomitant drug may be used per 1 part by weight of the compound of the present invention.
- Preparation Example 1 Preparation of Recombinant Human ACC2 A cDNA encoding human ACC2 protein (27 amino acid residues to 2458 amino acid residues from the N-terminus) was cloned from a human kidney cDNA library (Clontech), and His- After introducing the tag sequence, it was inserted into pFastBac1 (Invitrogen). Following the protocol of the Bac-to-Bac baculovirus expression system (Invitrogen), a recombinant baculovirus was prepared and then infected with Sf-9 cells to express the human ACC2 protein. The recovered cells were disrupted, filtered, and subjected to Ni affinity chromatography and anion exchange chromatography. Fractions containing human ACC2 protein were collected to obtain recombinant human ACC2.
- Preparation Example 2 Preparation of Recombinant Human ACC1 A cDNA encoding the human ACC1 protein (1 amino acid residue to 2346 amino acid residues from the N-terminus) was cloned from a human liver cDNA library (BioChain), and a myc tag was added to the 3′ end. and His-tag sequences were introduced, and then inserted into pIEXBAC3 (Novagen). According to the protocol of FlashBACGOLD (Oxford Expression Technologies), recombinant baculovirus was prepared and then infected with Sf-9 cells to express human ACC1 protein. The recovered cells were disrupted, filtered, and subjected to Ni affinity chromatography and anion exchange chromatography. Fractions containing human ACC1 protein were collected to obtain recombinant human ACC1.
- Test Example 1 Measurement of human ACC1 and ACC2 inhibitory activity Preincubation was performed for 1 hour in potassium citrate, 4 mM reduced glutathione, 1.5 mg/ml bovine serum albumin). Then, 5 ⁇ L of the pre-incubated enzyme solution and substrate solution (50 mM HEPES-KOH (pH 7.4), 1 mM ATP, 0.2 ⁇ L of each compound solution of the present invention (DMSO) were dispensed into a 384-well microplate. 8 mM acetyl-CoA, 25-50 mM potassium hydrogencarbonate) (5 ⁇ L) was added, and after centrifugation and shaking, the mixture was incubated in a humidified box at room temperature for 1-3 hours.
- Test Example 1 The results of Test Example 1 are shown below.
- Test Example 2 Metabolic Stability Test Commercially available pooled human liver microsomes and the compound of the present invention are allowed to react for a certain period of time, the residual rate is calculated by comparing the reacted sample and the unreacted sample, and the degree of metabolism of the compound of the present invention in the liver is evaluated. bottom.
- the compound of the present invention in the centrifugal supernatant was quantified by LC/MS/MS or solid phase extraction (SPE)/MS, and the amount of the compound remaining after the reaction was calculated assuming that the amount of the compound at the time of 0 minute reaction was 100%. Calculated.
- Test Example 2 The results of Test Example 2 are shown below.
- Preparation Example 3 Preparation of Atgl Knockout Mouse Atgl (adipose triglyceride lipase) knockout mouse is known as a nonclinical model of TGCV. Atgl knockout mice lacking the Atgl gene (KO mice) were produced by known methods.
- Test Example 3 Effect on Long Chain Fatty Acid Metabolic Ability in Atgl Knockout Mouse Myocardium 0.5% HPMC of the compound of the present invention was administered to male Atgl knockout mice aged 6 to 7 weeks at a dose of 30 or 45 mg/kg/10 mL. It was suspended in an aqueous solution and orally administered on the day before and on the day of measuring long-chain fatty acid metabolic capacity. On the day of measurement of long-chain fatty acid metabolic capacity, iodine-123- ⁇ methyl iodophenyl-pen-tadecanoic acid (BMIPP) was administered at 14.6 MBq/mouse through the tail vein.
- BMIPP iodine-123- ⁇ methyl iodophenyl-pen-tadecanoic acid
- mice were anesthetized and subjected to imaging by single-photon emission computed tomography (SPECT)/CT, and the radioactivity of the administered BMIPP in the myocardium was measured.
- Metabolism of long-chain fatty acids in myocardium was calculated as washout rate by the following formula. [(BMIPP uptake (1 h) - remaining BMIPP (4 h)) / BMIPP uptake (1 h)] (result)
- compound I-13 45 mg/kg/10 mL showed a significant improvement effect on the ability to metabolize long-chain fatty acids (p ⁇ 0.05, Welch's t test).
- the results of Test Example 3 are shown in FIG.
- Test Example 4 Evaluation of Cardiac Lipid Level in Atgl Knockout Mice
- Atgl knockout mice were administered a vehicle or the compounds of the present invention, and then their hearts were harvested.
- the collected heart was homogenized, tissue lipids were extracted using the Folch method described in Folch et al. Hitech) was used to calculate the amounts of triglycerides (TG) in the heart and free fatty acids (NEFA) in the heart per tissue, and the effects of the compounds of the present invention on the amount of lipids in the heart muscle were evaluated.
- Autocera (registered trademark) TG or Autocera (registered trademark) NEFA (Sekisui Medical) was used for each reagent.
- Test Example 5 Evaluation of Cardiac Function in Atgl Knockout Mice
- Ejection fraction Left Ventricular Ejection Fraction: LVEF
- Vevo770 Imaging System
- Test Example 6 Evaluation of Survival Period in Atgl Knockout Mice
- the vehicle or the compound of the present invention was repeatedly administered to Atgl knockout mice, and then the effect on survival period was evaluated. .
- Test Example 7 Effect on platelet count 0.5% of the compound of the present invention was administered to 6-week-old male Crl:CD (SD) rats under non-fasting conditions at a dose of 50-600 mg/kg/day. It was suspended in a methylcellulose solution and orally administered repeatedly for 4 days. Five days after the start of administration, blood was collected from the abdominal vena cava under anesthesia, and plasma was collected. Platelet counts in plasma were assessed by an automated hemocytometer.
- Test Example 8 Evaluation of plasma triglyceride concentration in mice fed an ultra-high-fat choline-deficient methionine-reduced diet 6-week-old male C57BL/6JJcl mice were fed an ultra-high-fat choline-deficient methionine-reduced diet (60 kcal% fat content, lard, choline Deficiency, methionine reduction (0.1%)), and at the same time, the compound of the present invention was suspended in a vehicle (0.5% methylcellulose aqueous solution) so that the dosage was 3 to 45 mg/kg/10 mL, and oral repeated administration ( bid) was performed for 8 weeks. Blood was collected from the tail vein during the repeated administration period, and biochemical parameters in plasma were measured.
- Test Example 9 CYP inhibition test Using commercially available pooled human liver microsomes, O-deethylation of 7-ethoxyresorufin (CYP1A2), tolbutamide and Using methyl-hydroxylation (CYP2C9), 4'-hydroxylation of mephenytoin (CYP2C19), O-demethylation of dextromethorphan (CYP2D6), and hydroxylation of terfenadine (CYP3A4) as indicators, the amount of each metabolite produced was The degree of inhibition by the compounds of the present invention was evaluated.
- reaction conditions are as follows: substrate, 0.5 ⁇ mol/L ethoxyresorufin (CYP1A2), 100 ⁇ mol/L tolbutamide (CYP2C9), 50 ⁇ mol/L S-mephenytoin (CYP2C19), 5 ⁇ mol/L dextromethorphan (CYP2D6), 1 ⁇ mol/L terfenadine (CYP3A4); reaction time, 15 minutes; reaction temperature, 37°C; enzyme, pooled human liver microsomes 0.2 mg protein/mL; concentration of the compound of the present invention, 1, 5, 10, 20 ⁇ mol/L (4 points) .
- resorufin CYP1A2 metabolite
- CYP1A2 metabolite resorufin in the centrifugation supernatant was quantified using a fluorescence multi-label counter or LC/MS/MS, tolbutamide hydroxide (CYP2C9 metabolite), mephenytoin 4'-hydroxylation. body (CYP2C19 metabolite), dextrorphan (CYP2D6 metabolite), terfenadine alcohol (CYP3A4 metabolite) were quantified by LC/MS/MS.
- IC50 was calculated by inverse estimation.
- CYP3A4 Fluorescence MBI Test is a test to investigate enhancement of CYP3A4 inhibition of the compounds of the present invention by metabolic reaction.
- 7-benzyloxytrifluoromethylcoumarin (7-BFC) is debenzylated by the CYP3A4 enzyme (E. coli expressed enzyme) to yield the fluorescent metabolite 7-hydroxytrifluoromethylcoumarin (7-HFC).
- CYP3A4 inhibition was evaluated using the 7-HFC production reaction as an index.
- reaction conditions were as follows: substrate, 5.6 ⁇ mol/L 7-BFC; pre-reaction time, 0 or 30 minutes; reaction time, 15 minutes; reaction temperature, 25°C (room temperature); 62.5 pmol / mL during pre-reaction, 6.25 pmol / mL during reaction (10-fold dilution); concentration of the compound of the present invention, 0.625, 1.25, 2.5, 5, 10, 20 ⁇ mol / L (6 points ).
- K-Pi buffer pH 7.4
- the control (100%) was obtained by adding only DMSO, which is the solvent in which the compound of the present invention was dissolved, to the reaction system, and the residual activity (%) was calculated when each concentration of the compound of the present invention was added.
- DMSO the solvent in which the compound of the present invention was dissolved
- the residual activity (%) was calculated when each concentration of the compound of the present invention was added.
- Test Example 11 CYP3A4 (MDZ) MBI Test Regarding the CYP3A4 inhibition of the compounds of the present invention, this is a test to evaluate the mechanism based inhibition (MBI) ability from the enhancement of the inhibitory action resulting from the metabolic reaction of the compounds of the present invention. CYP3A4 inhibition was evaluated using pooled human liver microsomes as an index of 1-hydroxylation of midazolam (MDZ).
- Reaction conditions were as follows: substrate, 10 ⁇ mol/L MDZ; pre-reaction time, 0 or 30 minutes; substrate metabolism reaction time, 2 minutes; reaction temperature, 37°C; 0.05 mg/mL at the time of reaction (at 10-fold dilution); concentrations at the time of pre-reaction of the compound of the present invention, 1, 5, 10, and 20 ⁇ mol/L (4 points).
- a control (100%) was obtained by adding only DMSO, which is a solvent in which the compound was dissolved instead of the compound of the present invention, to the reaction solution, and the residual activity (%) was calculated when the compound of the present invention was added at each concentration, IC was calculated by inverse estimation with a logistic model using concentration and inhibition rate.
- the IC with 0 min of preincubation/IC with 30 min of preincubation was taken as the Shifted IC value, and if the Shifted IC was 1.5 or more, it was taken as Positive, and if the Shifted IC was 1.0 or less, it was taken as Negative.
- Administration method Oral administration was forcibly administered into the stomach using an oral probe. Intravenous administration was administered through the tail vein using a syringe with an injection needle. (6) Evaluation item: Blood was collected over time, and the concentration of the compound of the present invention in plasma was measured using LC/MS/MS. (7) Statistical analysis: Concerning the transition of the concentration of the compound of the present invention in plasma, the area under the plasma concentration-time curve (AUC) was calculated by the moment analysis method. The bioavailability (BA) of the compounds of the invention was calculated. Note that the dilution concentration and dilution solvent were changed as necessary.
- Test Example 14 Fluctuation Ames Test The mutagenicity of the compound of the present invention is evaluated. 20 ⁇ L of cryopreserved Salmonella typhimurium (Salmonella typhimurium TA98 strain, TA100 strain) was inoculated into 10 mL liquid nutrient medium (2.5% Oxoid nutrient broth No. 2) and cultured at 37° C. for 10 hours before shaking. For the TA98 strain, 7.70 to 8.00 mL of the bacterial solution was centrifuged (2000 ⁇ g, 10 minutes) to remove the culture medium.
- Salmonella typhimurium Salmonella typhimurium TA98 strain, TA100 strain
- Micro F buffer K 2 HPO 4 : 3.5 g/L, KH 2 PO 4 : 1 g/L, (NH 4 ) 2 SO 4 : 1 g/L, Tricitrate Sodium dihydrate: 0.25 g/L, MgSO 4 7H 2 0: 0.1 g/L), and 120 mL of Exposure medium (biotin: 8 ⁇ g/mL, histidine: 0.2 ⁇ g/mL, Glucose: MicroF buffer containing 8 mg/mL).
- Exposure medium biotin: 8 ⁇ g/mL, histidine: 0.2 ⁇ g/mL
- Glucose MicroF buffer containing 8 mg/mL.
- 3.10 to 3.42 mL of bacterial solution was added to 120 to 130 mL of Exposure medium to prepare a test bacterial solution.
- DMSO solution of the compound of the present invention (several dilutions from the highest dose of 50 mg/mL to 2- to 3-fold common ratio), DMSO as a negative control, and 50 ⁇ g/mL of 4- Nitroquinoline-1-oxide in DMSO, 0.25 ⁇ g/mL 2-(2-furyl)-3-(5-nitro-2-furyl)acrylamide in DMSO for strain TA100, TA98 in metabolic activation conditions 12 ⁇ L of 40 ⁇ g/mL 2-aminoanthracene DMSO solution for the strain, 12 ⁇ L of 20 ⁇ g/mL 2-aminoanthracene DMSO solution for the TA100 strain, and 588 ⁇ L of the test bacterial solution (under metabolic activation conditions, 498 ⁇ L of the test bacterial solution and S9 90 ⁇ L of the mixture) was mixed and cultured with shaking at 37° C.
- Test Example 15 hERG test CHO cells expressing human ether-a-go-go related gene (hERG) channels were used for the purpose of evaluating the risk of electrocardiographic QT interval prolongation of the compounds of the present invention.
- analysis software (QPatch Assay software; Sophion Bioscience A/S) was used to measure the absolute value of the maximum tail current based on the current value at the retained membrane potential. Furthermore, the maximum tail current after application of the compound of the present invention relative to the maximum tail current after application of the vehicle was calculated as the inhibition rate, and the effect of the compound of the present invention on I Kr was evaluated. Note that the dilution concentration and dilution solvent were changed as necessary.
- Test Example 16 Ames test The mutagenicity of the compounds of the present invention was evaluated by the Ames test using Salmonella typhimurium strains TA98, TA100, TA1535, TA1537 and Escherichia coli WP2uvrA strains as test strains. To 0.1 mL of DMSO solution of the compound of the present invention, 0.5 mL of S9mix under metabolic activation conditions, and 0.5 mL of phosphate buffer and 0.1 mL of test bacterial solution under non-metabolic activation conditions were mixed, and histidine and biotin were mixed. , or overlaid on minimal glucose agar plates with 2 mL of tryptophan-containing soft overlay agar.
- negative controls DMSO
- positive controls (2-(2-furyl)-3-(5-nitro-2-furyl)acrylamide, sodium azide, 9-aminoacridine, or 2-aminoanthracene) was similarly performed.
- DMSO negative controls
- positive controls (2-(2-furyl)-3-(5-nitro-2-furyl)acrylamide, sodium azide, 9-aminoacridine, or 2-aminoanthracene
- a positive (+) was determined when the number of revertant colonies increased in a concentration-dependent manner and was at least twice the number of colonies in the negative control group. Note that the dilution concentration and dilution solvent were changed as necessary.
- Test Example 17 Photohemolytic test The compound of the present invention is dissolved at the desired concentration, and placed on a microplate to prepare a 0.1 to 0.0008% concentration erythrocyte suspension (2.5 v/v%) prepared from defibrinated sheep blood. ) and photoirradiation in the UVA and UVB range (10 J/cm 2 , 290-400 nm) with UV fluorescent lamps (GL20SE lamp, Sankyo Denki and FL20S-BLB lamp, Panasonic). After the end of light irradiation, the mixed solution is sampled and centrifuged.
- UVA and UVB range 10 J/cm 2 , 290-400 nm
- UV fluorescent lamps GL20SE lamp, Sankyo Denki and FL20S-BLB lamp, Panasonic
- the absorbance (540 or 630 nm) of the supernatant is measured, and determination is made based on the absorbance.
- Absorbance at 540 and 630 nm serve as indicators of biomembrane damage (% photohemolysis rate) and lipid membrane peroxidation (methemoglobin production), respectively.
- Test Example 18 P-gp Substrate Test
- the compound of the present invention was added to one side of Transwell (registered trademark, CORNING) in which human MDR1-expressing cells or parental cells were monolayer cultured, and allowed to react for a certain period of time.
- Transwell registered trademark, CORNING
- Efflux ratios (ER values) of MDR1-expressing cells and parental cells were compared to determine whether the compounds of the present invention are P-gp substrates.
- SPE solid phase extraction
- the composition of the JP-1 liquid is as follows. Water was added to 2.0 g of sodium chloride and 7.0 mL of hydrochloric acid to make 1000 mL.
- the composition of the JP-2 liquid is as follows. 1 volume of water was added to 1 volume of 3.40 g of potassium dihydrogen phosphate and 3.55 g of anhydrous disodium hydrogen phosphate dissolved in water to make up to 1000 mL.
- Test Example 20 Powder solubility test An appropriate amount of the compound of the present invention is placed in a suitable container, JP-1 solution (sodium chloride 2.0 g, hydrochloric acid 7.0 mL is added with water to make 1000 mL), JP-2 solution (1 volume of water was added to 1 volume of 3.40 g of potassium dihydrogen phosphate and 3.55 g of anhydrous disodium hydrogen phosphate dissolved in water to make 1000 mL), 20 mmol / L sodium taurocholate (TCA) / JP- Liquid 2 (1.08 g of TCA was added with liquid JP-2 to make 100 mL) was added in 200 ⁇ L portions.
- JP-1 solution sodium chloride 2.0 g, hydrochloric acid 7.0 mL is added with water to make 1000 mL
- JP-2 solution (1 volume of water was added to 1 volume of 3.40 g of potassium dihydrogen phosphate and 3.55 g of anhydrous disodium hydrogen phosphate dissolved in water to make 1000
- the compound of the present invention was added as appropriate. After sealing and shaking at 37° C. for 1 hour, filtration was performed, and 100 ⁇ L of methanol was added to 100 ⁇ L of each filtrate to perform two-fold dilution. Dilution ratios were changed as needed. Check for air bubbles and deposits, seal and shake. The compounds of the present invention were quantified using HPLC with an absolute calibration curve method. Note that the dilution concentration and dilution solvent were changed as necessary.
- Test Example 21 Visual Solubility Test About 5 mg of the compound is weighed into 3 micro test tubes, and each medium (water for injection, normal saline, 0.5% glucose solution) is added so that the compound concentration is 20%. After stirring with a vortex, the presence or absence of dissolution is visually confirmed. If dissolved, the solubility in the medium is >20%. Each medium (water for injection, saline solution, glucose solution) is further added to the test solution to prepare a test solution with a compound concentration of 10%. If dissolved, the solubility in that medium is 20% to 10%. Similarly, test up to 5% concentration, 2.5% concentration, and 1% concentration, and if it does not dissolve at 1% concentration, the solubility in that medium shall be ⁇ 1%. Measure and record the pH of the 1% test solution. Note that the dilution concentration and the dilution solvent may be changed as necessary.
- Test Example 22 pKa measurement (capillary electrophoresis method, CE method) It is a separation method that uses capillary zone electrophoresis technology and free migration of each sample component in a buffer solution containing electrolytes. After injecting a compound solution into a fused silica capillary filled with a buffer solution adjusted to pH 2.5 to 11.5, when a high voltage (Inlet side +, Outlet side -) is applied to the capillary, the compound It migrates at a rate that reflects its ionization state at pH (faster for +charged compounds, slower for -charged compounds). The difference between the migration time of this compound and that of a neutral molecule (DMSO) was plotted against pH and fitted to calculate the pKa.
- DMSO neutral molecule
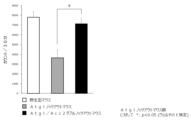
- Test Example 23 Evaluation of skeletal muscle intracellular lipids (IMCL) in SD rats
- vehicle Vehicle
- compound I-13 MR spectroscopy
- tibialis anterior muscle were measured using veterinary MRI (Varian MRI System 7T/210, Agilent Technologies) under isoflurane anesthesia 24 hours after a single oral dose of 20 mg/kg).
- the measurement data was analyzed using MRS data quantitative analysis software (LCModel), and the amount of IMCL was calculated as the creatine ratio.
- LCModel MRS data quantitative analysis software
- Compound I-13 (20 mg/kg) showed a significant IMCL reduction effect in SD rats.
- the results of Test Example 23 are shown in FIG.
- Test Example 24 Evaluation of skeletal muscle intracellular lipids (IMCL) in diabetic model mice
- leptin receptor-deficient mice Leprdb/ Leprdb, db/db; db/db mice
- animal MRI Varian MRI System 7T/ 210, Agilent Technologies
- MRS MR spectroscopy
- Test Example 25 Evaluation of skeletal muscle intracellular lipids (IMCL) in diabetic model rats
- IMCL skeletal muscle intracellular lipids
- ZDF rats spontaneous diabetes model
- Test Example 26 Evaluation of skeletal muscle intracellular lipid (IMCL) and skeletal muscle extracellular lipid (EMCL) in high-fat diet-loaded mice
- IMCL skeletal muscle intracellular lipid
- EMCL skeletal muscle extracellular lipid
- Test Example 27 Evaluation of skeletal muscle intracellular lipid (IMCL) and skeletal muscle extracellular lipid (EMCL) in ACC2 knockout mice ACC2 deficiency affects skeletal muscle intracellular lipid (IMCL) and skeletal muscle extracellular lipid (EMCL)
- animal MRI Varian MRI System 7T/210, Agilent Technologies
- MRS MR spectroscopy
- the measurement data was analyzed using MRS data quantitative analysis software (LCModel), and the IMCL amount and EMCL amount were calculated as the creatine ratio.
- LCModel MRS data quantitative analysis software
- ACC2 knockout mice showed a significant decrease in IMCL compared to wild-type mice before and after high-fat diet loading. On the other hand, there was no significant difference in EMCL between wild-type and ACC2 knockout mice.
- the results of Test Example 27 are shown in FIG.
- Test Example 28 Evaluation of spontaneous locomotor capacity in Atgl knockout mice
- Atgl knockout mice were repeatedly administered a vehicle or the compound of the present invention at 12 weeks of age.
- Locomotor activity was measured using SCANET MV-40 (MELQUEST). Mice were placed in cages placed in the device, and motor activity was measured for 30 minutes.
- Compound I-13 (45 mg/kg/10 mL) showed a significant ameliorating effect on the decreased locomotor activity observed in Atgl knockout mice (p ⁇ 0.05, Welch's t test).
- the results of Test Example 28 are shown in FIG.
- Test Example 29 Evaluation of spontaneous locomotor ability in Atgl knockout mice and double knockout mice of Atgl and ACC2
- 12-week-old wild-type mice, Atgl knockout mice, and Atgl were tested.
- ACC2 double knockout mice locomotor activity was measured using SCANET MV-40 (MELQUEST). Mice were placed in cages placed in the device, and motor activity was measured for 30 minutes. (result) Deficiency of ACC showed a significant ameliorating effect on the decreased locomotor activity observed in Atgl knockout mice (p ⁇ 0.05, Welch's t test). The results of Test Example 29 are shown in FIG.
- compositions of the present invention can be administered by any conventional route, in particular enterally, e.g. orally, e.g. in the form of tablets or capsules, or parenterally, e.g. in the form of injection solutions or suspensions. , topically, for example, in the form of lotions, gels, ointments or creams, or in nasal or suppository form.
- Pharmaceutical compositions of the present invention comprising a compound in free form or in pharmaceutically acceptable salt form together with at least one pharmaceutically acceptable carrier or diluent can be mixed, It can be manufactured by a granulation or coating method.
- oral compositions can be tablets, granules, capsules containing excipients, disintegrants, binders, lubricants, etc. and active ingredients.
- injectable compositions may be in the form of solutions or suspensions, may be sterilized, and may contain preservatives, stabilizers, buffers and the like.
- the method of treating and/or preventing at least one of heart disease, heart disease complication, skeletal muscle disease and skeletal muscle pathology, and the therapeutic pharmaceutical composition used therefor of the present invention are represented by the active ingredient formula (I): It is believed that administration of a predetermined amount of the compound or a pharmaceutically acceptable salt thereof to a patient will exhibit excellent therapeutic effects. In addition, administration of the compound represented by formula (I) or a pharmaceutically acceptable salt thereof does not cause side effects such as elevation of plasma triglycerides or reduction of platelet concentration, and can be applied very safely, and can be administered for a long period of time. Therefore, the therapeutic and/or preventive method and therapeutic pharmaceutical composition of the present invention are excellent therapeutic and/or preventive methods and therapeutic pharmaceutical compositions.
Abstract
優れたACC2選択的阻害作用を有し、血漿トリグリセリド上昇または血小板濃度の低下などの副作用を伴わない、心疾患、心疾患合併症、骨格筋の疾患および骨格筋の病態の少なくとも1つを治療および/または予防するための医薬組成物を提供する。 式(I): 化1 (式中、R1はハロアルキルまたは非芳香族炭素環式基であり、 R2は水素原子またはハロゲンであり、 R3はハロゲンであり、 環Aは、式: 化2 で示される基であり、 -L1-は-O-(CH2)-、-(CH2)2-等であり、 R4はアルキルまたはハロアルキルであり、 R5はアルキルカルボニルまたはカルバモイルである。)で示される化合物またはその製薬上許容される塩を含有する、心疾患、心疾患合併症、骨格筋の疾患および骨格筋の病態の少なくとも1つを治療および/または予防するための、医薬組成物。
Description
本発明は、心疾患、心疾患合併症、骨格筋の疾患および骨格筋の病態の少なくとも1つを治療および/または予防するための、新たな医薬組成物に関する。
中性脂肪蓄積心筋血管症(Triglyceride deposit cardiomyovasculopathy、以下TGCVともいう。)は、2008年に初めて報告された新規疾患単位であり、心筋細胞や冠状動脈硬化巣に中性脂肪(Triglyceride、TG)が蓄積する結果、重症心不全、不整脈を来す難病である(非特許文献1)。TGCVの原因として、これまでのところ、細胞内中性脂肪分解の必須酵素であるATGL(Adipose triglyceride lipase)の欠損が知られている。正常では心臓においてエネルギー源となる長鎖脂肪酸を、TGCVでは細胞内で代謝できず、TGとして心血管に蓄積してしまう結果、心臓がいわば肥満状態に陥る(非特許文献2)。心筋細胞や、血管平滑筋細胞に「異所性に」TGが蓄積することを特徴とする病態であり、生理的貯蔵組織における中性脂肪蓄積の多寡を反映すると考えられるBMI(body mass index)、体重、肥満等とは必ずしも相関を認めず、また細胞内代謝異常に基づくため、血清TG値とも直接的な関連はない。
TGCVの治療法は研究段階にあるが、最近、中鎖脂肪酸であるカプリン酸が、強力な細胞内TG含量低下活性を持ち、心筋又は血管の組織又は細胞において中性脂肪が蓄積している糖尿病性心血管合併症の予防又は治療に有用であることが報告されている(特許文献1)。
しかし、これまでにTGCV治療薬は上市されておらず、本疾患の治療方法の確立は重要な臨床課題である。
しかし、これまでにTGCV治療薬は上市されておらず、本疾患の治療方法の確立は重要な臨床課題である。
ACCは、アセチル-CoAをカルボキシル化してマロニル-CoAに変換する酵素であり、脂肪酸の代謝に関与する。ACCには、アセチル-CoAカルボキシラーゼ1(以下、ACC1という)及びアセチル-CoAカルボキシラーゼ2(以下、ACC2という)の2つのアイソフォームが存在する。
ACC2は、おもに心臓や骨格筋で発現しており、ACC2によって産生されるマロニル-CoAはカルニチンパルミトイルトランスフェラーゼI(CPT-I)を阻害することにより脂肪酸の酸化を阻害する。
ACC2欠損マウスにおいて、心臓や骨格筋におけるマロニル-CoA量の低下により、継続的な脂肪酸の酸化が起こっており、食餌量の増加にかかわらず、体重の減少が見られる。さらに、ACC2欠損マウスは高脂肪/高炭水化物の餌の投与によって誘発される糖尿病や肥満に対して耐性を獲得していることも報告されている。
以上の知見から、ACC2は糖尿病や肥満症などの疾患に関与しており、その阻害剤は抗糖尿病薬や抗肥満薬となることが示唆される。
一方、ACC1欠損マウスは胎児期において致死的であることから、ACC1を阻害することなくACC2を阻害する選択的な阻害剤が望まれている(非特許文献3)。
ACC2は、おもに心臓や骨格筋で発現しており、ACC2によって産生されるマロニル-CoAはカルニチンパルミトイルトランスフェラーゼI(CPT-I)を阻害することにより脂肪酸の酸化を阻害する。
ACC2欠損マウスにおいて、心臓や骨格筋におけるマロニル-CoA量の低下により、継続的な脂肪酸の酸化が起こっており、食餌量の増加にかかわらず、体重の減少が見られる。さらに、ACC2欠損マウスは高脂肪/高炭水化物の餌の投与によって誘発される糖尿病や肥満に対して耐性を獲得していることも報告されている。
以上の知見から、ACC2は糖尿病や肥満症などの疾患に関与しており、その阻害剤は抗糖尿病薬や抗肥満薬となることが示唆される。
一方、ACC1欠損マウスは胎児期において致死的であることから、ACC1を阻害することなくACC2を阻害する選択的な阻害剤が望まれている(非特許文献3)。
特許文献2および3には、ACC1及びACC2の両方に対し阻害活性を有するチエノピリミジン誘導体を用いて非アルコール性脂肪肝疾患を処置する方法が記載されている。例えば、以下に示される化合物は、firsocostat(ACC1/2のデュアル阻害剤)として知られており、非アルコール性脂肪性肝炎などを適応として開発されている。firsocostatはトランスポーターを介して肝臓に選択的に取り込まれるようにデザインされている。

firsocostatについては、2019年12月16日に48週間にわたる第II相ATLAS試験のトップラインの成績が発表されている。第II相ATLAS試験は、NASHによる線維化が進行(F3-F4)した患者を対象として、cilofexor(選択的非ステロイド系FXR作動薬) 30 mg、firsocostat 20 mg、およびselonsertib(ASK1阻害薬) 18 mgによる単独療法と2剤併用療法の安全性および有効性を評価する、無作為化、二重盲検、プラセボ対照試験である。その結果、いずれのレジメンでも、有効性の主要評価項目であるNASHが悪化することなく、1 ステージ以上の線維化の改善を達成した患者の割合に統計学的に有意な増加は認められなかったと報告されている。先に実施された12週間にわたる第II相試験では、firsocostatを投与されたNAFLD患者で血漿トリグリセリド濃度が上昇したと報告されている。血漿トリグリセリド濃度の上昇は別のACC1/2のデュアル阻害剤であるMK-4074を投与したNAFLD患者でも報告されている。血漿トリグリセリド濃度の上昇は心血管イベントの発生を増加させることが知られており、NASH患者の死亡原因として最も多いのは心血管イベントであると報告されている。NAFLDは心血管病の独立したリスク因子であり、NASHに病態が進行することで心血管病のリスクが増大することが報告されている。ATLAS試験ではfirsocostatとcilofexorを併用したNASH患者で血漿トリグリセリド濃度が上昇することが報告されている。
firsocostatについては、2019年12月16日に48週間にわたる第II相ATLAS試験のトップラインの成績が発表されている。第II相ATLAS試験は、NASHによる線維化が進行(F3-F4)した患者を対象として、cilofexor(選択的非ステロイド系FXR作動薬) 30 mg、firsocostat 20 mg、およびselonsertib(ASK1阻害薬) 18 mgによる単独療法と2剤併用療法の安全性および有効性を評価する、無作為化、二重盲検、プラセボ対照試験である。その結果、いずれのレジメンでも、有効性の主要評価項目であるNASHが悪化することなく、1 ステージ以上の線維化の改善を達成した患者の割合に統計学的に有意な増加は認められなかったと報告されている。先に実施された12週間にわたる第II相試験では、firsocostatを投与されたNAFLD患者で血漿トリグリセリド濃度が上昇したと報告されている。血漿トリグリセリド濃度の上昇は別のACC1/2のデュアル阻害剤であるMK-4074を投与したNAFLD患者でも報告されている。血漿トリグリセリド濃度の上昇は心血管イベントの発生を増加させることが知られており、NASH患者の死亡原因として最も多いのは心血管イベントであると報告されている。NAFLDは心血管病の独立したリスク因子であり、NASHに病態が進行することで心血管病のリスクが増大することが報告されている。ATLAS試験ではfirsocostatとcilofexorを併用したNASH患者で血漿トリグリセリド濃度が上昇することが報告されている。
特許文献4には、ACC1/2のデュアル阻害剤が記載されている。例えば、以下に示される化合物は、PF-05175157(ACC1/2のデュアル阻害剤)として知られており、健常者への反復投与によって血小板濃度を低下させた。

これは骨髄のACC1阻害を介する脂肪酸合成の抑制によって血小板の生成が低下したためであると報告されている。
これは骨髄のACC1阻害を介する脂肪酸合成の抑制によって血小板の生成が低下したためであると報告されている。
特許文献5および6には、ACC2を特異的に阻害するベンズイミダゾール誘導体が記載されている。更にベンズイミダゾール誘導体がNASHの治療剤として有用なことが特許文献7に記載されている。
また特許文献8~20および非特許文献4~6には本発明の医薬組成物に含有されるベンズイミダゾール誘導体に類似した化合物が、ACC阻害剤として記載されている。
また特許文献8~20および非特許文献4~6には本発明の医薬組成物に含有されるベンズイミダゾール誘導体に類似した化合物が、ACC阻害剤として記載されている。
特許文献21~26には、ACC阻害剤が、サルコペニアの治療剤として有用なことが記載されているが、本発明の医薬組成物に含有されるベンズイミダゾール誘導体については記載されていない。
非特許文献6には特定のACC2阻害剤が、骨格筋細胞内脂質量を減少させること、インスリン抵抗性および高血糖を改善することが記載されている。また、本発明の医薬組成物に含有されるベンズイミダゾール誘導体については記載されていない。
非特許文献6には特定のACC2阻害剤が、骨格筋細胞内脂質量を減少させること、インスリン抵抗性および高血糖を改善することが記載されている。また、本発明の医薬組成物に含有されるベンズイミダゾール誘導体については記載されていない。
N. Engl. J. Med., 359(22), 2396-2398, 2008
J. Atheroscler. Thromb., 16(5), 702-705, 2009
PNAS August 23, 2005 102 (34) 12011-12016
Molecular Diversity (2013), 17(1), 139-149
Journal of Medicinal Chemistry (2010), 53(24), 8679-8687
J Pharmacol Exp Ther 372: 256-263, 2020
本発明の課題は、優れたACC2選択的阻害作用を有し、血漿トリグリセリド上昇または血小板濃度の低下などの副作用を伴わない、心疾患、心疾患合併症、骨格筋の疾患および骨格筋の病態の少なくとも1つを治療および/または予防するための医薬組成物を提供することにある。より好ましくは中性脂肪蓄積心筋血管症または骨格筋細胞における中性脂肪の蓄積によって引き起こされる骨格筋の疾患または病態の治療および/または予防のための医薬組成物を提供することにある。
本発明者らは、前記課題を解決するために検討を重ねた結果、特許文献5および6に記載のACC2選択的阻害作用を有する化合物のうち、特定の化合物(ACC2の選択性が高く、かつ、代謝安定性が優れた化合物)が心疾患、心疾患合併症、骨格筋の疾患および骨格筋の病態の少なくとも1つの治療および/または予防に有効であり、かつ、血漿トリグリセリド上昇または血小板濃度の低下などの副作用を伴わないことを見出し、本発明を完成するに至った。本発明の医薬組成物に含有されるACC2選択的阻害剤は、ACC1阻害による副作用を回避し、全身のACC2を阻害することができる。その結果、本発明の医薬組成物に含有されるACC2選択的阻害剤はACC1/2のデュアル阻害剤及び、firsocostatのような肝臓選択的ACC1/2のデュアル阻害剤とは異なり、血小板低下を引き起こさず、血漿トリグリセリド濃度を上昇させず、全身のACC2阻害に基づく代謝改善作用を発揮できる。本発明は、以下に関する。
[1]
式(I):

(式中、R1はハロアルキルまたは非芳香族炭素環式基であり、
R2は水素原子またはハロゲンであり、
R3はハロゲンであり、
環Aは、式:

で示される基であり、
-L1-は-O-(CH2)-、-(CH2)2-、-(CH2)-(CF2)-または-(CF2)-(CH2)-(ここで、左の結合手は環Aに結合し、右の結合手は式:

で示される基に結合する。)であり、
R4はアルキルまたはハロアルキルであり、
R5はアルキルカルボニルまたはカルバモイルである。)で示される化合物(本明細書において、「本発明化合物」と称することもある)またはその製薬上許容される塩を含有する、心疾患および心疾患合併症の少なくとも1つを治療および/または予防するための、医薬組成物。
[2]
R1が非芳香族炭素環式基である、上記項目[1]記載の医薬組成物。
[3]
R2が水素原子である、上記項目[1]または[2]記載の医薬組成物。
[4]
環Aが、式:

で示される基である、上記項目[1]~[3]のいずれかに記載の医薬組成物。
[5]
-L1-が-O-(CH2)-または-(CH2)2-である、上記項目[1]~[4]のいずれかに記載の医薬組成物。
[6]
R4がアルキルである、上記項目[1]~[5]のいずれかに記載の医薬組成物。
[7]
R5がメチルカルボニルまたはカルバモイルである、上記項目[1]~[6]のいずれかに記載の医薬組成物。
[8]
式(I)で示される化合物またはその製薬上許容される塩が、下式:

からなる群から選択される化合物またはその製薬上許容される塩である、上記項目[1]記載の医薬組成物。
[9]
前記心疾患および心疾患合併症が、心不全、狭心症、心筋梗塞、心筋症、不整脈、冠動脈疾患および骨格筋ミオパチーからなる群より選択される少なくとも1つの疾患である上記項目[1]~[8]のいずれかに記載の記載の医薬組成物。
[10]
心筋細胞内、血管平滑筋細胞内、内皮細胞内、骨格筋細胞内、多型核白血球、腎尿細管上皮細胞内および膵島細胞内の少なくとも1つに中性脂肪が蓄積されることにより引き起こされる疾患を治療および/または予防するための、上記項目[1]~[8]のいずれかに記載の化合物を含有する医薬組成物。
[11]
前記疾患が、心不全、狭心症、心筋梗塞、心筋症、不整脈、冠動脈疾患、骨格筋ミオパチー、慢性腎臓病および耐糖能障害からなる群より選択される少なくとも1つの疾患である、[10]記載の記載の医薬組成物。
[12]
前記医薬組成物の投与によって、血漿トリグリセリド上昇の副作用を伴わない、上記項目[1]~[11]のいずれかに記載の医薬組成物。
[13]
前記医薬組成物の投与によって、血小板濃度の低下の副作用を伴わない、上記項目[1]~[11]のいずれかに記載の医薬組成物。
[14]
式(I):

(式中、R1はハロアルキルまたは非芳香族炭素環式基であり、
R2は水素原子またはハロゲンであり、
R3はハロゲンであり、
環Aは、式:

で示される基であり、
-L1-は-O-(CH2)-、-(CH2)2-、-(CH2)-(CF2)-または-(CF2)-(CH2)-(ここで、左の結合手は環Aに結合し、右の結合手は式:

で示される基に結合する。)であり、
R4はアルキルまたはハロアルキルであり、
R5はアルキルカルボニルまたはカルバモイルである。)で示される化合物またはその製薬上許容される塩を含有する、骨格筋の疾患または病態を治療および/または予防するための、医薬組成物。
[15]
R1が非芳香族炭素環式基である、上記項目[14]記載の医薬組成物。
[16]
R2が水素原子である、上記項目[14]または[15]記載の医薬組成物。
[17]
環Aが、式:

で示される基である、上記項目[14]~[16]のいずれかに記載の医薬組成物。
[18]
-L1-が-O-(CH2)-または-(CH2)2-である、上記項目[14]~[17]のいずれかに記載の医薬組成物。
[19]
R4がアルキルである、上記項目[14]~[18]のいずれかに記載の医薬組成物。
[20]
R5がメチルカルボニルまたはカルバモイルである、上記項目[14]~[19]のいずれかに記載の医薬組成物。
[21]
式(I)で示される化合物またはその製薬上許容される塩が、下式:

からなる群から選択される化合物またはその製薬上許容される塩である、上記項目[14]記載の医薬組成物。
[22]
前記骨格筋の疾患または病態が、骨格筋細胞における中性脂肪の蓄積によって引き起こされるもの(例えば、ミオステアトーシス)である、上記項目[14]~[21]のいずれかに記載の医薬組成物。
[23]
前記骨格筋の疾患または病態が、骨格筋ミオパチーまたはサルコペニアである、上記項目[14]~[22]のいずれかに記載の医薬組成物。
[24]
前記骨格筋の疾患または病態の病理および/または症候がACC2活性の寄与によるものである、上記項目[14]~[23]のいずれかに記載の医薬組成物。
[25]
前記ACC2活性が糖尿病、肥満および肝硬変からなる群から選択される状態に寄与する、上記項目[24]記載の医薬組成物。
[26]
前記骨格筋の疾患または病態が、糖尿病、肥満および肝硬変からなる群から選択される状態と関連している、上記項目[14]~[25]のいずれかに記載の医薬組成物。
[27]
前記骨格筋の疾患または病態が、肝硬変と関連している、上記項目[26]記載の医薬組成物。
[28]
前記医薬組成物が、前記骨格筋の疾患または病態を有する対象の骨格筋機能を維持または増加させる、上記項目[14]~[27]のいずれかに記載の医薬組成物。
[29]
前記医薬組成物が、前記骨格筋の疾患または病態を有する対象の骨格筋の回復を促進する、上記項目[14]~[27]のいずれかに記載の医薬組成物。
[30]
前記医薬組成物が、対象の骨格筋機能を維持または増加させる、上記項目[14]~[27]のいずれかに記載の医薬組成物。
式(I):
(式中、R1はハロアルキルまたは非芳香族炭素環式基であり、
R2は水素原子またはハロゲンであり、
R3はハロゲンであり、
環Aは、式:
で示される基であり、
-L1-は-O-(CH2)-、-(CH2)2-、-(CH2)-(CF2)-または-(CF2)-(CH2)-(ここで、左の結合手は環Aに結合し、右の結合手は式:
で示される基に結合する。)であり、
R4はアルキルまたはハロアルキルであり、
R5はアルキルカルボニルまたはカルバモイルである。)で示される化合物(本明細書において、「本発明化合物」と称することもある)またはその製薬上許容される塩を含有する、心疾患および心疾患合併症の少なくとも1つを治療および/または予防するための、医薬組成物。
[2]
R1が非芳香族炭素環式基である、上記項目[1]記載の医薬組成物。
[3]
R2が水素原子である、上記項目[1]または[2]記載の医薬組成物。
[4]
環Aが、式:
で示される基である、上記項目[1]~[3]のいずれかに記載の医薬組成物。
[5]
-L1-が-O-(CH2)-または-(CH2)2-である、上記項目[1]~[4]のいずれかに記載の医薬組成物。
[6]
R4がアルキルである、上記項目[1]~[5]のいずれかに記載の医薬組成物。
[7]
R5がメチルカルボニルまたはカルバモイルである、上記項目[1]~[6]のいずれかに記載の医薬組成物。
[8]
式(I)で示される化合物またはその製薬上許容される塩が、下式:
からなる群から選択される化合物またはその製薬上許容される塩である、上記項目[1]記載の医薬組成物。
[9]
前記心疾患および心疾患合併症が、心不全、狭心症、心筋梗塞、心筋症、不整脈、冠動脈疾患および骨格筋ミオパチーからなる群より選択される少なくとも1つの疾患である上記項目[1]~[8]のいずれかに記載の記載の医薬組成物。
[10]
心筋細胞内、血管平滑筋細胞内、内皮細胞内、骨格筋細胞内、多型核白血球、腎尿細管上皮細胞内および膵島細胞内の少なくとも1つに中性脂肪が蓄積されることにより引き起こされる疾患を治療および/または予防するための、上記項目[1]~[8]のいずれかに記載の化合物を含有する医薬組成物。
[11]
前記疾患が、心不全、狭心症、心筋梗塞、心筋症、不整脈、冠動脈疾患、骨格筋ミオパチー、慢性腎臓病および耐糖能障害からなる群より選択される少なくとも1つの疾患である、[10]記載の記載の医薬組成物。
[12]
前記医薬組成物の投与によって、血漿トリグリセリド上昇の副作用を伴わない、上記項目[1]~[11]のいずれかに記載の医薬組成物。
[13]
前記医薬組成物の投与によって、血小板濃度の低下の副作用を伴わない、上記項目[1]~[11]のいずれかに記載の医薬組成物。
[14]
式(I):
(式中、R1はハロアルキルまたは非芳香族炭素環式基であり、
R2は水素原子またはハロゲンであり、
R3はハロゲンであり、
環Aは、式:
で示される基であり、
-L1-は-O-(CH2)-、-(CH2)2-、-(CH2)-(CF2)-または-(CF2)-(CH2)-(ここで、左の結合手は環Aに結合し、右の結合手は式:
で示される基に結合する。)であり、
R4はアルキルまたはハロアルキルであり、
R5はアルキルカルボニルまたはカルバモイルである。)で示される化合物またはその製薬上許容される塩を含有する、骨格筋の疾患または病態を治療および/または予防するための、医薬組成物。
[15]
R1が非芳香族炭素環式基である、上記項目[14]記載の医薬組成物。
[16]
R2が水素原子である、上記項目[14]または[15]記載の医薬組成物。
[17]
環Aが、式:
で示される基である、上記項目[14]~[16]のいずれかに記載の医薬組成物。
[18]
-L1-が-O-(CH2)-または-(CH2)2-である、上記項目[14]~[17]のいずれかに記載の医薬組成物。
[19]
R4がアルキルである、上記項目[14]~[18]のいずれかに記載の医薬組成物。
[20]
R5がメチルカルボニルまたはカルバモイルである、上記項目[14]~[19]のいずれかに記載の医薬組成物。
[21]
式(I)で示される化合物またはその製薬上許容される塩が、下式:
からなる群から選択される化合物またはその製薬上許容される塩である、上記項目[14]記載の医薬組成物。
[22]
前記骨格筋の疾患または病態が、骨格筋細胞における中性脂肪の蓄積によって引き起こされるもの(例えば、ミオステアトーシス)である、上記項目[14]~[21]のいずれかに記載の医薬組成物。
[23]
前記骨格筋の疾患または病態が、骨格筋ミオパチーまたはサルコペニアである、上記項目[14]~[22]のいずれかに記載の医薬組成物。
[24]
前記骨格筋の疾患または病態の病理および/または症候がACC2活性の寄与によるものである、上記項目[14]~[23]のいずれかに記載の医薬組成物。
[25]
前記ACC2活性が糖尿病、肥満および肝硬変からなる群から選択される状態に寄与する、上記項目[24]記載の医薬組成物。
[26]
前記骨格筋の疾患または病態が、糖尿病、肥満および肝硬変からなる群から選択される状態と関連している、上記項目[14]~[25]のいずれかに記載の医薬組成物。
[27]
前記骨格筋の疾患または病態が、肝硬変と関連している、上記項目[26]記載の医薬組成物。
[28]
前記医薬組成物が、前記骨格筋の疾患または病態を有する対象の骨格筋機能を維持または増加させる、上記項目[14]~[27]のいずれかに記載の医薬組成物。
[29]
前記医薬組成物が、前記骨格筋の疾患または病態を有する対象の骨格筋の回復を促進する、上記項目[14]~[27]のいずれかに記載の医薬組成物。
[30]
前記医薬組成物が、対象の骨格筋機能を維持または増加させる、上記項目[14]~[27]のいずれかに記載の医薬組成物。
式(I)で示される化合物またはその製薬上許容される塩を含有する本発明の医薬組成物は、心疾患、心疾患合併症、骨格筋の疾患および骨格筋の病態の少なくとも1つ、特に中性脂肪蓄積心筋血管症または骨格筋細胞における中性脂肪の蓄積によって引き起こされる骨格筋の疾患もしくは病態の治療および/または予防に有効であるという優れた効果を奏するものである。また、血漿トリグリセリド上昇または血小板濃度の低下などの副作用を伴わない、高い安全性を有する。
以下に本明細書において用いられる各用語の意味を説明する。各用語は特に断りのない限り、単独で用いられる場合も、または他の用語と組み合わせて用いられる場合も、同一の意味で用いられる。
「からなる」という用語は、構成要件のみを有することを意味する。
「含む」という用語は、構成要件に限定されず、記載されていない要素を排除しないことを意味する。
以下、本発明について実施形態を示しながら説明する。本明細書の全体にわたり、単数形の表現は、特に言及しない限り、その複数形の概念をも含むことが理解されるべきである。従って、単数形の冠詞(例えば、英語の場合は「a」、「an」、「the」など)は、特に言及しない限り、その複数形の概念をも含むことが理解されるべきである。
また、本明細書において使用される用語は、特に言及しない限り、当上記分野で通常用いられる意味で用いられることが理解されるべきである。したがって、他に定義されない限り、本明細書中で使用される全ての専門用語および科学技術用語は、本発明の属する分野の当業者によって一般的に理解されるのと同じ意味を有する。矛盾する場合、本明細書(定義を含めて)が優先する。
「からなる」という用語は、構成要件のみを有することを意味する。
「含む」という用語は、構成要件に限定されず、記載されていない要素を排除しないことを意味する。
以下、本発明について実施形態を示しながら説明する。本明細書の全体にわたり、単数形の表現は、特に言及しない限り、その複数形の概念をも含むことが理解されるべきである。従って、単数形の冠詞(例えば、英語の場合は「a」、「an」、「the」など)は、特に言及しない限り、その複数形の概念をも含むことが理解されるべきである。
また、本明細書において使用される用語は、特に言及しない限り、当上記分野で通常用いられる意味で用いられることが理解されるべきである。したがって、他に定義されない限り、本明細書中で使用される全ての専門用語および科学技術用語は、本発明の属する分野の当業者によって一般的に理解されるのと同じ意味を有する。矛盾する場合、本明細書(定義を含めて)が優先する。
「ハロゲン」とは、フッ素原子、塩素原子、臭素原子、およびヨウ素原子を包含する。特にフッ素原子および塩素原子が好ましい。
「アルキル」とは、炭素数1~15、好ましくは炭素数1~10、より好ましくは炭素数1~6、さらに好ましくは炭素数1~4の直鎖又は分枝状の炭化水素基を包含する。例えば、メチル、エチル、n-プロピル、イソプロピル、n-ブチル、イソブチル、sec-ブチル、tert-ブチル、n-ペンチル、イソペンチル、ネオペンチル、n-ヘキシル、イソヘキシル、n-へプチル、イソヘプチル、n-オクチル、イソオクチル、n-ノニル、n-デシル等が挙げられる。
「アルキル」の好ましい態様として、メチル、エチル、n-プロピル、イソプロピル、n-ブチル、イソブチル、sec-ブチル、tert-ブチル、n-ペンチルが挙げられる。さらに好ましい態様として、メチル、エチル、n-プロピル、イソプロピル、tert-ブチルが挙げられる。特に好ましい態様として、メチルが挙げられる。
「アルキル」の好ましい態様として、メチル、エチル、n-プロピル、イソプロピル、n-ブチル、イソブチル、sec-ブチル、tert-ブチル、n-ペンチルが挙げられる。さらに好ましい態様として、メチル、エチル、n-プロピル、イソプロピル、tert-ブチルが挙げられる。特に好ましい態様として、メチルが挙げられる。
「ハロアルキル」とは、上記「アルキル」の1以上の任意の水素原子が上記「ハロゲン」で置換された基を意味する。例えば、モノフルオロメチル、モノフルオロエチル、モノフルオロプロピル、2,2-ジフルオロエチル、2,2,3,3,3-ペンタフルオロプロピル、モノクロロメチル、トリフルオロメチル、トリクロロメチル、2,2,2-トリフルオロエチル、2,2,2-トリクロロエチル、1,2-ジブロモエチル、1,1,1-トリフルオロプロパン-2-イル等が挙げられる。
「アルキルカルボニル」とは、上記「アルキル」がカルボニル基に結合した基を意味する。例えば、メチルカルボニル、エチルカルボニル、プロピルカルボニル、イソプロピルカルボニル、tert-ブチルカルボニル、イソブチルカルボニル、sec-ブチルカルボニル、ペンチルカルボニル、イソペンチルカルボニル、へキシルカルボニル等が挙げられる。「アルキルカルボニル」の好ましい態様として、メチルカルボニル、エチルカルボニル、n-プロピルカルボニル等が挙げられる。さらに好ましい態様として、メチルカルボニルが挙げられる。
「芳香族炭素環式基」とは、単環または2環以上の、環状芳香族炭化水素基を意味する。例えば、フェニル、ナフチル、アントリル、フェナントリル等が挙げられる。
「芳香族炭素環式基」の好ましい態様として、フェニルが挙げられる。
「芳香族炭素環式基」の好ましい態様として、フェニルが挙げられる。
「芳香族炭素環」とは、上記「芳香族炭素環式基」から導かれる環を意味する。
「非芳香族炭素環式基」とは、単環または2環以上の、環状飽和炭化水素基または環状非芳香族不飽和炭化水素基を意味する。2環以上の「非芳香族炭素環式基」は、単環または2環以上の非芳香族炭素環式基に、上記「芳香族炭素環式基」における環が縮合したものも包含する。
さらに、「非芳香族炭素環式基」は、以下のように架橋している基、またはスピロ環を形成する基も包含する。

単環の非芳香族炭素環式基としては、炭素数3~16が好ましく、より好ましくは炭素数3~12、さらに好ましくは炭素数3~6であり、特に好ましくは炭素数3~4である。例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、シクロノニル、シクロデシル、シクロプロペニル、シクロブテニル、シクロペンテニル、シクロヘキセニル、シクロヘプテニル、シクロヘキサジエニル等が挙げられる。
2環以上の非芳香族炭素環式基としては、炭素数8~20が好ましく、より好ましくは炭素数8~16である。例えば、インダニル、インデニル、アセナフチル、テトラヒドロナフチル、フルオレニル等が挙げられる。
さらに、「非芳香族炭素環式基」は、以下のように架橋している基、またはスピロ環を形成する基も包含する。
単環の非芳香族炭素環式基としては、炭素数3~16が好ましく、より好ましくは炭素数3~12、さらに好ましくは炭素数3~6であり、特に好ましくは炭素数3~4である。例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、シクロノニル、シクロデシル、シクロプロペニル、シクロブテニル、シクロペンテニル、シクロヘキセニル、シクロヘプテニル、シクロヘキサジエニル等が挙げられる。
2環以上の非芳香族炭素環式基としては、炭素数8~20が好ましく、より好ましくは炭素数8~16である。例えば、インダニル、インデニル、アセナフチル、テトラヒドロナフチル、フルオレニル等が挙げられる。
「非芳香族炭素環」とは、上記「非芳香族炭素環式基」から導かれる環を意味する。
「芳香族複素環式基」とは、O、SおよびNから任意に選択される同一または異なるヘテロ原子を環内に1以上有する、単環または2環以上の、芳香族環式基を意味する。
2環以上の芳香族複素環式基は、単環または2環以上の芳香族複素環式基に、上記「芳香族炭素環式基」における環が縮合したものも包含し、該結合手はいずれの環に有していても良い。
単環の芳香族複素環式基としては、5~8員が好ましく、より好ましくは5員または6員である。5員芳香族複素環式基としては、例えば、ピロリル、イミダゾリル、ピラゾリル、トリアゾリル、テトラゾリル、フリル、チエニル、イソオキサゾリル、オキサゾリル、オキサジアゾリル、イソチアゾリル、チアゾリル、チアジアゾリル等が挙げられる。6員芳香族複素環式基としては、例えば、ピリジル、ピリダジニル、ピリミジニル、ピラジニル、トリアジニル等が挙げられる。
2環の芳香族複素環式基としては、8~10員が好ましく、より好ましくは9員または10員である。例えば、インドリル、イソインドリル、インダゾリル、インドリジニル、キノリニル、イソキノリニル、シンノリニル、フタラジニル、キナゾリニル、ナフチリジニル、キノキサリニル、プリニル、プテリジニル、ベンズイミダゾリル、ベンズイソオキサゾリル、ベンズオキサゾリル、ベンズオキサジアゾリル、ベンズイソチアゾリル、ベンゾチアゾリル、ベンゾチアジアゾリル、ベンゾフリル、イソベンゾフリル、ベンゾチエニル、ベンゾトリアゾリル、イミダゾピリジル、トリアゾロピリジル、イミダゾチアゾリル、ピラジノピリダジニル、オキサゾロピリジル、チアゾロピリジル等が挙げられる。
3環以上の芳香族複素環式基としては、13~15員が好ましい。例えば、カルバゾリル、アクリジニル、キサンテニル、フェノチアジニル、フェノキサチイニル、フェノキサジニル、ジベンゾフリル等が挙げられる。
2環以上の芳香族複素環式基は、単環または2環以上の芳香族複素環式基に、上記「芳香族炭素環式基」における環が縮合したものも包含し、該結合手はいずれの環に有していても良い。
単環の芳香族複素環式基としては、5~8員が好ましく、より好ましくは5員または6員である。5員芳香族複素環式基としては、例えば、ピロリル、イミダゾリル、ピラゾリル、トリアゾリル、テトラゾリル、フリル、チエニル、イソオキサゾリル、オキサゾリル、オキサジアゾリル、イソチアゾリル、チアゾリル、チアジアゾリル等が挙げられる。6員芳香族複素環式基としては、例えば、ピリジル、ピリダジニル、ピリミジニル、ピラジニル、トリアジニル等が挙げられる。
2環の芳香族複素環式基としては、8~10員が好ましく、より好ましくは9員または10員である。例えば、インドリル、イソインドリル、インダゾリル、インドリジニル、キノリニル、イソキノリニル、シンノリニル、フタラジニル、キナゾリニル、ナフチリジニル、キノキサリニル、プリニル、プテリジニル、ベンズイミダゾリル、ベンズイソオキサゾリル、ベンズオキサゾリル、ベンズオキサジアゾリル、ベンズイソチアゾリル、ベンゾチアゾリル、ベンゾチアジアゾリル、ベンゾフリル、イソベンゾフリル、ベンゾチエニル、ベンゾトリアゾリル、イミダゾピリジル、トリアゾロピリジル、イミダゾチアゾリル、ピラジノピリダジニル、オキサゾロピリジル、チアゾロピリジル等が挙げられる。
3環以上の芳香族複素環式基としては、13~15員が好ましい。例えば、カルバゾリル、アクリジニル、キサンテニル、フェノチアジニル、フェノキサチイニル、フェノキサジニル、ジベンゾフリル等が挙げられる。
「芳香族複素環」とは、上記「芳香族複素環式基」から導かれる環を意味する。
「非芳香族複素環式基」とは、O、SおよびNから任意に選択される同一または異なるヘテロ原子を環内に1以上有する、単環または2環以上の、非芳香族環式基を意味する。2環以上の非芳香族複素環式基は、単環または2環以上の非芳香族複素環式基に、上記「芳香族炭素環式基」、「非芳香族炭素環式基」、および/または「芳香族複素環式基」におけるそれぞれの環が縮合したもの、さらに、単環または2環以上の非芳香族炭素環式基に、上記「芳香族複素環式基」における環が縮合したものも包含し、該結合手はいずれの環に有していても良い。
さらに、「非芳香族複素環式基」は、以下のように架橋している基、またはスピロ環を形成する基も包含する。

単環の非芳香族複素環式基としては、3~8員が好ましく、より好ましくは5員または6員である。
3員非芳香族複素環式基としては、例えば、チイラニル、オキシラニル、アジリジニルが挙げられる。4員非芳香族複素環式基としては、例えば、オキセタニル、アゼチジニルが挙げられる。5員非芳香族複素環式基としては、例えば、オキサチオラニル、チアゾリジニル、ピロリジニル、ピロリニル、イミダゾリジニル、イミダゾリニル、ピラゾリジニル、ピラゾリニル、テトラヒドロフリル、ジヒドロチアゾリル、テトラヒドロイソチアゾリル、ジオキソラニル、ジオキソリル、チオラニル等が挙げられる。6員非芳香族複素環式基としては、例えば、ジオキサニル、チアニル、ピペリジル、ピペラジニル、モルホリニル、モルホリノ、チオモルホリニル、チオモルホリノ、ジヒドロピリジル、テトラヒドロピリジル、テトラヒドロピラニル、ジヒドロオキサジニル、テトラヒドロピリダジニル、ヘキサヒドロピリミジニル、ジオキサジニル、チイニル、チアジニル等が挙げられる。7員非芳香族複素環式基としては、例えば、ヘキサヒドロアゼピニル、テトラヒドロジアゼピニル、オキセパニルが挙げられる。
2環以上の非芳香族複素環式基としては、8~20員が好ましく、より好ましくは8~10員である。例えば、インドリニル、イソインドリニル、クロマニル、イソクロマニル等が挙げられる。
さらに、「非芳香族複素環式基」は、以下のように架橋している基、またはスピロ環を形成する基も包含する。
単環の非芳香族複素環式基としては、3~8員が好ましく、より好ましくは5員または6員である。
3員非芳香族複素環式基としては、例えば、チイラニル、オキシラニル、アジリジニルが挙げられる。4員非芳香族複素環式基としては、例えば、オキセタニル、アゼチジニルが挙げられる。5員非芳香族複素環式基としては、例えば、オキサチオラニル、チアゾリジニル、ピロリジニル、ピロリニル、イミダゾリジニル、イミダゾリニル、ピラゾリジニル、ピラゾリニル、テトラヒドロフリル、ジヒドロチアゾリル、テトラヒドロイソチアゾリル、ジオキソラニル、ジオキソリル、チオラニル等が挙げられる。6員非芳香族複素環式基としては、例えば、ジオキサニル、チアニル、ピペリジル、ピペラジニル、モルホリニル、モルホリノ、チオモルホリニル、チオモルホリノ、ジヒドロピリジル、テトラヒドロピリジル、テトラヒドロピラニル、ジヒドロオキサジニル、テトラヒドロピリダジニル、ヘキサヒドロピリミジニル、ジオキサジニル、チイニル、チアジニル等が挙げられる。7員非芳香族複素環式基としては、例えば、ヘキサヒドロアゼピニル、テトラヒドロジアゼピニル、オキセパニルが挙げられる。
2環以上の非芳香族複素環式基としては、8~20員が好ましく、より好ましくは8~10員である。例えば、インドリニル、イソインドリニル、クロマニル、イソクロマニル等が挙げられる。
「非芳香族複素環」とは、上記「非芳香族複素環式基」から導かれる環を意味する。
「疾患」とは、(疾患の前駆症状及び他の徴候を含めて、疾患の症状及びサインを含む)発症前段階から進行段階までさまざまな段階における疾患/障害/症候群/症状を意味する。
「病態」とは、個体の潜在能力及び目標に比べて低いパフォーマンスである状態を意味する。
「骨格筋機能を増加させる」とは、個体の骨格筋機能を改善することを意味する。また、「骨格筋機能を増加させる」医薬組成物という文言には、該医薬組成物の予防的な使用(用途)も含まれる。
「骨格筋の回復を促進する」とは、骨格筋の疾患または病態を有する個体の骨格筋機能を改善することを意味する。
(本発明の医薬組成物の心疾患および心疾患合併症に対する用途)
本発明化合物は、心筋の脂肪酸代謝作用を有することから、心筋、心臓の血管の組織内又は細胞内において中性脂肪が蓄積している疾患や中性脂肪蓄積により引き起こされる疾患であれば特に限定されないが、血管における中性脂肪の蓄積に起因して血管が狭窄又は閉塞している疾患、該狭窄又は閉塞に起因する疾患および心筋における中性脂肪の蓄積に起因する疾患からなる群より選択される少なくとも1つの疾患が好ましい。例えば臨床的には、心筋の肥大、冠動脈のびまん性狭窄および求心性狭窄からなる群より選択される少なくとも1つの所見がみられる疾患が好ましい。
従って、本願発明の「心疾患および心疾患合併症」としては、例えば、心不全、狭心症、心筋梗塞、心筋症、不整脈および冠動脈疾患からなる群より選択される少なくとも1つの疾患が挙げられる。
本発明化合物は、心筋の脂肪酸代謝作用を有することから、心筋、心臓の血管の組織内又は細胞内において中性脂肪が蓄積している疾患や中性脂肪蓄積により引き起こされる疾患であれば特に限定されないが、血管における中性脂肪の蓄積に起因して血管が狭窄又は閉塞している疾患、該狭窄又は閉塞に起因する疾患および心筋における中性脂肪の蓄積に起因する疾患からなる群より選択される少なくとも1つの疾患が好ましい。例えば臨床的には、心筋の肥大、冠動脈のびまん性狭窄および求心性狭窄からなる群より選択される少なくとも1つの所見がみられる疾患が好ましい。
従って、本願発明の「心疾患および心疾患合併症」としては、例えば、心不全、狭心症、心筋梗塞、心筋症、不整脈および冠動脈疾患からなる群より選択される少なくとも1つの疾患が挙げられる。
更に本発明化合物は、心筋細胞内、血管平滑筋細胞内、内皮細胞内、骨格筋細胞内、多型核白血球、腎尿細管上皮細胞内および膵島細胞内等の少なくとも1つに中性脂肪が蓄積されることにより引き起こされる疾患、すなわち「中性脂肪蓄積心筋血管症(TGCV)」にも有効である。「中性脂肪蓄積心筋血管症」としては、例えば、心不全、狭心症、心筋梗塞、心筋症、不整脈、冠動脈疾患、骨格筋ミオパチー、慢性腎臓病および耐糖能障害からなる群より選択される少なくとも1つの疾患が挙げられる。
また、血管に中性脂肪が蓄積することで引き起こされる、粥状(アテローム性)動脈硬化にも有効である。特にコレステロール低下剤で症状の改善がみられない症例にも治療・予防効果がある。
(本発明の医薬組成物の骨格筋の疾患または病態に対する用途)
本発明の医薬組成物は、骨格筋の疾患または病態を治療および/または予防するための化合物を含む。該化合物またはその薬学的に許容される塩は、式(I)によって表される。
本発明の医薬組成物は、骨格筋の疾患または病態を治療および/または予防するための化合物を含む。該化合物またはその薬学的に許容される塩は、式(I)によって表される。
特定の実施形態では、「骨格筋の疾患または病態」は、骨格筋細胞における中性脂肪の蓄積によって引き起こされる。いくつかの実施形態では、「骨格筋の疾患または病態」は、ミオステアトーシスである。ミオステアトーシスは、骨格筋細胞における中性脂肪の蓄積による筋肉の質の変化を意味する。いくつかの実施形態では、「骨格筋の疾患または病態」は、肝疾患に関連し得る。当該肝疾患の非限定的な例としては、進行性または後期の肝疾患が挙げられる。
特定の実施形態では、「骨格筋疾患または病態」は骨格筋ミオパチーである。骨格筋ミオパチーは、存在する異常または不十分な構造支持タンパク質による筋肉組織の進行性変性を特徴としている。いくつかの実施形態では、骨格筋ミオパチーは、筋線維の崩壊および/または筋力の喪失に関連し得る。いくつかの実施形態では、骨格筋ミオパチーは、遺伝性ミオパチーまたは後天性ミオパチーであり得る。いくつかの実施形態では、骨格筋ミオパチーは、先天性ミオパチー、ミトコンドリアミオパチー、代謝性ミオパチー、筋ジストロフィー、中毒性ミオパチー、内分泌ミオパチー、感染性ミオパチー、重症疾患ミオパチー、電解質の不均衡によって引き起こされるミオパチー、およびそれらの任意の組み合わせであり得る。
特定の実施形態では、「骨格筋の疾患または病態」はサルコペニアである。サルコペニアは、筋線維の大きさと数の減少を含む一種の筋萎縮症である。いくつかの実施形態では、サルコペニアは、筋力、筋量、および筋機能のうちの1つまたは複数の喪失に関連し得る。
特定の実施形態では、「骨格筋疾患または病態」の病理および/または症候はACC2活性の寄与するものである。いくつかの実施形態では、ACC2活性は、骨格筋細胞における中性脂肪の蓄積と関連している。特定の実施形態では、ACC2活性は、ミオステアトーシス、骨格筋ミオパチー、サルコペニア、およびそれらの任意の組み合わせからなる群から選択される病態に寄与する。
特定の実施形態では、ACC2活性は、糖尿病、肥満、および肝硬変からなる群から選択される病態に寄与する。特定の実施形態では、ACC2活性は、糖尿病、肥満、肝硬変、ミオステアトーシス、骨格筋ミオパチー、およびサルコペニアからなる群から選択される2つ以上の病態の組み合わせに同時に寄与する。
特定の実施形態では、「骨格筋の疾患または病態」は、糖尿病、肥満、および肝硬変からなる群から選択される病態に関連する。特定の実施形態では、ミオステアトーシスは、糖尿病、肥満、または肝硬変と同時に関連する。特定の実施形態では、骨格筋ミオパチーは、糖尿病、肥満、または肝硬変と同時に関連する。特定の実施形態では、サルコペニアは、糖尿病、肥満、または肝硬変と同時に関連する。
特定の実施形態では、「骨格筋の疾患または病態」は肝硬変に関連する。特定の実施形態では、ミオステアトーシスは肝硬変に関連する。特定の実施形態では、骨格筋ミオパチーは肝硬変に関連する。特定の実施形態では、サルコペニアは肝硬変に関連する。
特定の実施形態では、「骨格筋疾患または病態」は骨格筋ミオパチーである。骨格筋ミオパチーは、存在する異常または不十分な構造支持タンパク質による筋肉組織の進行性変性を特徴としている。いくつかの実施形態では、骨格筋ミオパチーは、筋線維の崩壊および/または筋力の喪失に関連し得る。いくつかの実施形態では、骨格筋ミオパチーは、遺伝性ミオパチーまたは後天性ミオパチーであり得る。いくつかの実施形態では、骨格筋ミオパチーは、先天性ミオパチー、ミトコンドリアミオパチー、代謝性ミオパチー、筋ジストロフィー、中毒性ミオパチー、内分泌ミオパチー、感染性ミオパチー、重症疾患ミオパチー、電解質の不均衡によって引き起こされるミオパチー、およびそれらの任意の組み合わせであり得る。
特定の実施形態では、「骨格筋の疾患または病態」はサルコペニアである。サルコペニアは、筋線維の大きさと数の減少を含む一種の筋萎縮症である。いくつかの実施形態では、サルコペニアは、筋力、筋量、および筋機能のうちの1つまたは複数の喪失に関連し得る。
特定の実施形態では、「骨格筋疾患または病態」の病理および/または症候はACC2活性の寄与するものである。いくつかの実施形態では、ACC2活性は、骨格筋細胞における中性脂肪の蓄積と関連している。特定の実施形態では、ACC2活性は、ミオステアトーシス、骨格筋ミオパチー、サルコペニア、およびそれらの任意の組み合わせからなる群から選択される病態に寄与する。
特定の実施形態では、ACC2活性は、糖尿病、肥満、および肝硬変からなる群から選択される病態に寄与する。特定の実施形態では、ACC2活性は、糖尿病、肥満、肝硬変、ミオステアトーシス、骨格筋ミオパチー、およびサルコペニアからなる群から選択される2つ以上の病態の組み合わせに同時に寄与する。
特定の実施形態では、「骨格筋の疾患または病態」は、糖尿病、肥満、および肝硬変からなる群から選択される病態に関連する。特定の実施形態では、ミオステアトーシスは、糖尿病、肥満、または肝硬変と同時に関連する。特定の実施形態では、骨格筋ミオパチーは、糖尿病、肥満、または肝硬変と同時に関連する。特定の実施形態では、サルコペニアは、糖尿病、肥満、または肝硬変と同時に関連する。
特定の実施形態では、「骨格筋の疾患または病態」は肝硬変に関連する。特定の実施形態では、ミオステアトーシスは肝硬変に関連する。特定の実施形態では、骨格筋ミオパチーは肝硬変に関連する。特定の実施形態では、サルコペニアは肝硬変に関連する。
特定の実施形態では、本発明の医薬組成物は、対象の骨格筋機能を維持または増加させる。特定の実施形態では、本発明の医薬組成物は、骨格筋の疾患または病態を有する対象の骨格筋の回復を促進する。
特定の実施形態では、「骨格筋機能」および「骨格筋の回復」は、徒手筋力テストを使用して決定することができる。この決定には、抵抗に対する筋肉のパフォーマンスの測定と、そのパフォーマンスを0~5のスケールで評価することが含まれる。特定の実施形態では、筋肉のパフォーマンスは以下の状態に該当するか否かを測定される;(1)痙攣のように筋肉をわずかにしか動かせず、筋肉の可動域全体を動かすことができない、(2)無重力下において筋肉の可動域全体を動かすことができる、(3)通常の重力下において筋肉の可動域全体を動かすことができる、(4)ある程度の抵抗存在下で筋肉の可動域全体を動かすことができる、(5)強い抵抗存在下で筋肉の可動域全体を動かすことができる。特定の実施形態では、測定可能な筋肉には、肩関節外転筋、肘屈筋、肘伸筋、手首伸筋、指屈筋、手内在筋、股関節屈筋、膝伸筋、背屈筋、足親指伸筋、および足底屈筋が含まれる。
特定の実施形態では、「骨格筋機能」および「骨格筋の回復」は、ハンドグリップ試験、チェアスタンド試験、ウォーキングスピード試験、ショート フィジカル パフォーマンス バッテリー(SPPB)、およびタイムアップ アンド ゴー試験(TUG)を含む標準的な試験を使用して決定することができる。特定の実施形態では、骨格筋機能および骨格筋の回復は、二重エネルギーX線吸収測定法(DEXAまたはDXA)および生体電気インピーダンス分析(BIA)法を含む画像検査を使用して決定することができる。特定の実施形態では、骨格筋機能および骨格筋の回復は、血液検査、筋電図検査(EMG)、磁気共鳴画像法(MRI)、遺伝子検査、および筋生検を含む検査を使用して決定することができる。
特定の実施形態では、「骨格筋機能」および「骨格筋の回復」は、虚弱指数を用いて決定することができる。例えば、肝虚弱指数(LFI)を使用することができる。LFIには、パフォーマンスに基づく3つのテスト (握力、チェアスタンド、バランス) が含まれる。たとえば、握力は、被験者の利き手にハンドヘルド動力計を使用してキログラム単位で測定される。分析のために3回の試行の平均を計算する。たとえば、タイムドチェアスタンドでは、被験者が腕を胸の上で組んだ状態で椅子から5回立ち上がるのを完了するのにかかる秒数が測定される。たとえば、バランステストでは、被験者が3つの姿勢 (足を左右、セミタンデム、および縦(タンデム)に並べる) で、それぞれ最大10秒間として、バランスを保つことができる秒数が測定される。3つの要因のいずれか1つまたは複数を評価に使用することができる。
特定の実施形態では、「骨格筋機能」および「骨格筋の回復」は、徒手筋力テストを使用して決定することができる。この決定には、抵抗に対する筋肉のパフォーマンスの測定と、そのパフォーマンスを0~5のスケールで評価することが含まれる。特定の実施形態では、筋肉のパフォーマンスは以下の状態に該当するか否かを測定される;(1)痙攣のように筋肉をわずかにしか動かせず、筋肉の可動域全体を動かすことができない、(2)無重力下において筋肉の可動域全体を動かすことができる、(3)通常の重力下において筋肉の可動域全体を動かすことができる、(4)ある程度の抵抗存在下で筋肉の可動域全体を動かすことができる、(5)強い抵抗存在下で筋肉の可動域全体を動かすことができる。特定の実施形態では、測定可能な筋肉には、肩関節外転筋、肘屈筋、肘伸筋、手首伸筋、指屈筋、手内在筋、股関節屈筋、膝伸筋、背屈筋、足親指伸筋、および足底屈筋が含まれる。
特定の実施形態では、「骨格筋機能」および「骨格筋の回復」は、ハンドグリップ試験、チェアスタンド試験、ウォーキングスピード試験、ショート フィジカル パフォーマンス バッテリー(SPPB)、およびタイムアップ アンド ゴー試験(TUG)を含む標準的な試験を使用して決定することができる。特定の実施形態では、骨格筋機能および骨格筋の回復は、二重エネルギーX線吸収測定法(DEXAまたはDXA)および生体電気インピーダンス分析(BIA)法を含む画像検査を使用して決定することができる。特定の実施形態では、骨格筋機能および骨格筋の回復は、血液検査、筋電図検査(EMG)、磁気共鳴画像法(MRI)、遺伝子検査、および筋生検を含む検査を使用して決定することができる。
特定の実施形態では、「骨格筋機能」および「骨格筋の回復」は、虚弱指数を用いて決定することができる。例えば、肝虚弱指数(LFI)を使用することができる。LFIには、パフォーマンスに基づく3つのテスト (握力、チェアスタンド、バランス) が含まれる。たとえば、握力は、被験者の利き手にハンドヘルド動力計を使用してキログラム単位で測定される。分析のために3回の試行の平均を計算する。たとえば、タイムドチェアスタンドでは、被験者が腕を胸の上で組んだ状態で椅子から5回立ち上がるのを完了するのにかかる秒数が測定される。たとえば、バランステストでは、被験者が3つの姿勢 (足を左右、セミタンデム、および縦(タンデム)に並べる) で、それぞれ最大10秒間として、バランスを保つことができる秒数が測定される。3つの要因のいずれか1つまたは複数を評価に使用することができる。
(本発明の医薬組成物に含有される式(I)で示される化合物)
式(I)で示される化合物における、R1、R2、R3、R4、R5、L1および環Aの好ましい態様を以下に示す。式(I)で示される化合物としては、以下に示される具体例のすべての組み合わせの態様が例示される。
式(I)で示される化合物における、R1、R2、R3、R4、R5、L1および環Aの好ましい態様を以下に示す。式(I)で示される化合物としては、以下に示される具体例のすべての組み合わせの態様が例示される。
R1は、ハロアルキルまたは非芳香族炭素環式基である。
R1は、好ましくは、非芳香族炭素環式基である。
R1は、より好ましくは、単環の非芳香族炭素環式基であり、特に好ましくは、シクロプロピルまたはシクロブチルである。
R1の別の好ましい態様として、好ましくは、2,2-ジフルオロエチルが挙げられる。
R1は、好ましくは、非芳香族炭素環式基である。
R1は、より好ましくは、単環の非芳香族炭素環式基であり、特に好ましくは、シクロプロピルまたはシクロブチルである。
R1の別の好ましい態様として、好ましくは、2,2-ジフルオロエチルが挙げられる。
R2は、水素原子またはハロゲンである。
R2は、好ましくは、水素原子またはフッ素原子である。
R2は、より好ましくは、水素原子である。
R2は、好ましくは、水素原子またはフッ素原子である。
R2は、より好ましくは、水素原子である。
R3は、ハロゲンである。
R3は、好ましくは、フッ素原子または塩素原子である。
R3は、より好ましくは、フッ素原子である。
R3は、好ましくは、フッ素原子または塩素原子である。
R3は、より好ましくは、フッ素原子である。
R4は、アルキルまたはハロアルキルである。
R4は、好ましくは、アルキルである。
R4は、より好ましくは、メチルである。
R4の別の好ましい態様として、好ましくは、モノフルオロメチルが挙げられる。
R4は、好ましくは、アルキルである。
R4は、より好ましくは、メチルである。
R4の別の好ましい態様として、好ましくは、モノフルオロメチルが挙げられる。
R5は、アルキルカルボニルまたはカルバモイルである。
R5は、好ましくは、メチルカルボニルまたはカルバモイルである。
R5は、好ましくは、メチルカルボニルまたはカルバモイルである。
環Aは、式:

で示される基である。
環Aは、好ましくは、式:

で示される基である。
環Aは、より好ましくは、式:

で示される基である。
環Aは、さらに好ましくは、式:

で示される基である。
で示される基である。
環Aは、好ましくは、式:
で示される基である。
環Aは、より好ましくは、式:
で示される基である。
環Aは、さらに好ましくは、式:
で示される基である。
-L1-は、-O-(CH2)-、-(CH2)2-、-(CH2)-(CF2)-または-(CF2)-(CH2)-(ここで、左の結合手は環Aに結合し、右の結合手は式:

で示される基に結合する。)である。
-L1-は、好ましくは、-O-(CH2)-または-(CH2)2-である。
で示される基に結合する。)である。
-L1-は、好ましくは、-O-(CH2)-または-(CH2)2-である。
式(I)で示される化合物としては、特に以下に示される化合物、またはその製薬上許容される塩が好ましい。

本発明の心疾患、心疾患合併症、骨格筋の疾患および骨格筋の病態の少なくとも1つを治療および/または予防するための医薬組成物は、有効成分として式(I):

(式中、各記号は前記と同意義)
で示される化合物またはその製薬上許容される塩を含有する医薬組成物であることを特徴とする。
(式中、各記号は前記と同意義)
で示される化合物またはその製薬上許容される塩を含有する医薬組成物であることを特徴とする。
式(I)で示される化合物は、公知の方法、例えば、国際公開第2015/056782号および国際公開第2016/159082号に記載の方法に従って合成することができる。
式(I)で示される化合物は、特定の異性体に限定するものではなく、全ての可能な異性体(例えば、ケト-エノール異性体、イミン-エナミン異性体、ジアステレオ異性体、光学異性体、回転異性体等)、ラセミ体またはそれらの混合物を含む。
式(I)で示される化合物の一つ以上の水素、炭素および/または他の原子は、それぞれ水素、炭素および/または他の原子の同位体で置換され得る。そのような同位体の例としては、それぞれ2H、3H、11C、13C、14C、15N、18O、17O、31P、32P、35S、18F、123Iおよび36Clのように、水素、炭素、窒素、酸素、リン、硫黄、フッ素、ヨウ素および塩素が包含される。式(I)で示される化合物は、そのような同位体で置換された化合物も包含する。該同位体で置換された化合物は、医薬品としても有用であり、式(I)で示される化合物のすべての放射性標識体を包含する。また該「放射性標識体」を製造するための「放射性標識化方法」も本発明に包含され、該「放射性標識体」は、代謝薬物動態研究、結合アッセイにおける研究および/または診断のツールとして有用である。
式(I)で示される化合物の放射性標識体は、当該技術分野で周知の方法で調製できる。例えば、式(I)で示されるトリチウム標識化合物は、トリチウムを用いた触媒的脱ハロゲン化反応によって、式(I)で示される特定の化合物にトリチウムを導入することで調製できる。この方法は、適切な触媒、例えばPd/Cの存在下、塩基の存在下または非存在下で、式(I)で示される化合物が適切にハロゲン置換された前駆体とトリチウムガスとを反応させることを包含する。トリチウム標識化合物を調製するための他の適切な方法は、“Isotopes in the Physical and Biomedical Sciences,Vol.1,Labeled Compounds (Part A),Chapter 6 (1987年)”を参照することができる。14C-標識化合物は、14C炭素を有する原料を用いることによって調製できる。
式(I)で示される化合物の製薬上許容される塩としては、例えば、式(I)で示される化合物と、アルカリ金属(例えば、リチウム、ナトリウム、カリウム等)、アルカリ土類金属(例えば、カルシウム、バリウム等)、マグネシウム、遷移金属(例えば、亜鉛、鉄等)、アンモニア、有機塩基(例えば、トリメチルアミン、トリエチルアミン、ジシクロヘキシルアミン、エタノールアミン、ジエタノールアミン、トリエタノールアミン、メグルミン、エチレンジアミン、ピリジン、ピコリン、キノリン等)およびアミノ酸との塩、または無機酸(例えば、塩酸、硫酸、硝酸、炭酸、臭化水素酸、リン酸、ヨウ化水素酸等)、および有機酸(例えば、ギ酸、酢酸、プロピオン酸、トリフルオロ酢酸、クエン酸、乳酸、酒石酸、シュウ酸、マレイン酸、フマル酸、コハク酸、マンデル酸、グルタル酸、リンゴ酸、安息香酸、フタル酸、アスコルビン酸、ベンゼンスルホン酸、p-トルエンスルホン酸、メタンスルホン酸、エタンスルホン酸、トリフルオロ酢酸等)との塩が挙げられる。これらの塩は、通常行われる方法によって形成させることができる。
本発明の医薬組成物に含有される式(I)で示される化合物またはその製薬上許容される塩は、溶媒和物(例えば、水和物等)、共結晶および/または結晶多形を形成する場合があり、本発明はそのような各種の溶媒和物、共結晶および結晶多形も包含する。「溶媒和物」は、式(I)で示される化合物に対し、任意の数の溶媒分子(例えば、水分子等)と配位していてもよい。式(I)で示される化合物またはその製薬上許容される塩を、大気中に放置することにより、水分を吸収し、吸着水が付着する場合や、水和物を形成する場合がある。また、式(I)で示される化合物またはその製薬上許容される塩を、再結晶することで結晶多形を形成する場合がある。「共結晶」は、式(I)で示される化合物または塩とカウンター分子が同一結晶格子内に存在することを意味し、任意の数のカウンター分子を含んでいても良い。
本発明の医薬組成物に含有される式(I)で示される化合物またはその製薬上許容される塩は、プロドラッグを形成する場合があり、本発明はそのような各種のプロドラッグを含有する医薬組成物も包含する。プロドラッグは、化学的又は代謝的に分解できる基を有する本発明化合物の誘導体であり、加溶媒分解により又は生理学的条件下でインビボにおいて薬学的に活性な本発明化合物となる化合物である。プロドラッグは、生体内における生理条件下で酵素的に酸化、還元、加水分解等を受けて式(I)で示される化合物に変換される化合物、胃酸等により加水分解されて式(I)で示される化合物に変換される化合物等を包含する。適当なプロドラッグ誘導体を選択する方法および製造する方法は、例えば “Design of Prodrugs, Elsevier, Amsterdam, 1985”に記載されている。プロドラッグは、それ自身が活性を有する場合がある。
本発明化合物は、優れたACC2選択的阻害作用を有するため、心疾患、心疾患合併症、骨格筋の疾患および骨格筋の病態の治療剤及び/又は予防剤や中性脂肪蓄積心筋血管症または骨格筋細胞における中性脂肪の蓄積によって引き起こされる骨格筋の疾患または病態の治療剤及び/又は予防剤としても有用である。
さらに本発明化合物は、医薬としての有用性を備えており、好ましくは、下記のいずれか、または複数の優れた特徴を有している。
a)CYP酵素(例えば、CYP1A2、CYP2C9、CYP2C19、CYP2D6、CYP3A4等)に対する阻害作用が弱い。
b)高いバイオアベイラビリティー、適度なクリアランス等良好な薬物動態を示す。
c)代謝安定性が高い。
d)CYP酵素(例えば、CYP3A4)に対し、本明細書に記載する測定条件の濃度範囲内で不可逆的阻害作用を示さない。
e)変異原性を有さない。
f)心血管系のリスクが低い。
g)高い溶解性を示す。
h)血漿トリグリセリド上昇の副作用を伴わない。
i)インスリン抵抗性が改善される。
j)血小板濃度の低下の副作用を伴わない。
さらに本発明化合物は、医薬としての有用性を備えており、好ましくは、下記のいずれか、または複数の優れた特徴を有している。
a)CYP酵素(例えば、CYP1A2、CYP2C9、CYP2C19、CYP2D6、CYP3A4等)に対する阻害作用が弱い。
b)高いバイオアベイラビリティー、適度なクリアランス等良好な薬物動態を示す。
c)代謝安定性が高い。
d)CYP酵素(例えば、CYP3A4)に対し、本明細書に記載する測定条件の濃度範囲内で不可逆的阻害作用を示さない。
e)変異原性を有さない。
f)心血管系のリスクが低い。
g)高い溶解性を示す。
h)血漿トリグリセリド上昇の副作用を伴わない。
i)インスリン抵抗性が改善される。
j)血小板濃度の低下の副作用を伴わない。
本発明の医薬組成物は、経口的、非経口的のいずれの方法でも投与することができる。非経口投与の方法としては、経皮、皮下、静脈内、動脈内、筋肉内、腹腔内、経粘膜、吸入、経鼻、点眼、点耳、膣内投与等が挙げられる。
経口投与の場合は常法に従って、内用固形製剤(例えば、錠剤、散剤、顆粒剤、カプセル剤、丸剤、フィルム剤等)、内用液剤(例えば、懸濁剤、乳剤、エリキシル剤、シロップ剤、リモナーデ剤、酒精剤、芳香水剤、エキス剤、煎剤、チンキ剤等)等の通常用いられるいずれの剤型に調製して投与すればよい。錠剤は、糖衣錠、フィルムコーティング錠、腸溶性コーティング錠、徐放錠、トローチ錠、舌下錠、バッカル錠、チュアブル錠または口腔内崩壊錠であってもよく、散剤および顆粒剤はドライシロップであってもよく、カプセル剤は、ソフトカプセル剤、マイクロカプセル剤または徐放性カプセル剤であってもよい。
非経口投与の場合は、注射剤、点滴剤、外用剤(例えば、点眼剤、点鼻剤、点耳剤、エアゾール剤、吸入剤、ローション剤、注入剤、塗布剤、含嗽剤、浣腸剤、軟膏剤、硬膏剤、ゼリー剤、クリーム剤、貼付剤、パップ剤、外用散剤、坐剤等)等の通常用いられるいずれの剤型でも好適に投与することができる。注射剤は、O/W、W/O、O/W/O、W/O/W型等のエマルジョンであってもよい。
本発明化合物の有効量にその剤型に適した賦形剤、結合剤、崩壊剤、滑沢剤等の各種医薬用添加剤を必要に応じて混合し、医薬組成物とすることができる。さらに、該医薬組成物は、本発明化合物の有効量、剤型および/または各種医薬用添加剤を適宜変更することにより、小児用、高齢者用、重症患者用または手術用の医薬組成物とすることもできる。例えば、小児用医薬組成物は、新生児(出生後4週未満)、乳児(出生後4週~1歳未満)、幼児(1歳以上7歳未満)、小児(7歳以上15歳未満)若しくは15歳~18歳の患者に投与されうる。例えば、高齢者用医薬組成物は、65歳以上の患者に投与されうる。
本発明の医薬組成物の投与量は、患者の年齢、体重、疾病の種類や程度、投与経路等を考慮した上で設定することが望ましいが、経口投与する場合、通常0.05~100mg/kg/日であり、好ましくは0.1~10mg/kg/日の範囲内である。非経口投与の場合には投与経路により大きく異なるが、通常0.005~10mg/kg/日であり、好ましくは0.01~1mg/kg/日の範囲内である。これを1日1回~数回に分けて投与すれば良い。
本発明の医薬組成物に含有される式(I):

(式中、各記号は前記と同意義である。)で示される化合物またはその製薬上許容される塩は、該化合物の心疾患もしくは心疾患合併症の治療および/または予防作用の増強または該化合物の投与量の低減等を目的として、利尿薬、ACE阻害薬、アンジオテンシンII受容体拮抗薬、アンジオテンシン受容体/ネプリライシン阻害薬、アルドステロン拮抗薬、β遮断薬、SGLT2阻害剤および血管拡張薬と組み合わせて用いることができる。例えば、フロセミド、エナラプリル、カンデサルタンシレキセチル、サクビトリル/バルサルタン、スピロノラクトン、カルベジロール、ダパグリフロジンおよびニトログリセリンからなる群より選択される少なくとも一つの化合物またはその製薬上許容される塩(以下、併用薬剤1と称する)と組み合わせて用いることができる。この際、本発明化合物と併用薬剤1の投与時期は限定されず、これらを投与対象に対し、同時にまたは連続的に投与してもよいし、時間差をおいて投与してもよい。さらに、本発明化合物と併用薬剤1とは、それぞれの活性成分を含む2種類以上の製剤として投与されてもよいし、それらの活性成分を含む単一の製剤として投与されてもよい。
(式中、各記号は前記と同意義である。)で示される化合物またはその製薬上許容される塩は、該化合物の心疾患もしくは心疾患合併症の治療および/または予防作用の増強または該化合物の投与量の低減等を目的として、利尿薬、ACE阻害薬、アンジオテンシンII受容体拮抗薬、アンジオテンシン受容体/ネプリライシン阻害薬、アルドステロン拮抗薬、β遮断薬、SGLT2阻害剤および血管拡張薬と組み合わせて用いることができる。例えば、フロセミド、エナラプリル、カンデサルタンシレキセチル、サクビトリル/バルサルタン、スピロノラクトン、カルベジロール、ダパグリフロジンおよびニトログリセリンからなる群より選択される少なくとも一つの化合物またはその製薬上許容される塩(以下、併用薬剤1と称する)と組み合わせて用いることができる。この際、本発明化合物と併用薬剤1の投与時期は限定されず、これらを投与対象に対し、同時にまたは連続的に投与してもよいし、時間差をおいて投与してもよい。さらに、本発明化合物と併用薬剤1とは、それぞれの活性成分を含む2種類以上の製剤として投与されてもよいし、それらの活性成分を含む単一の製剤として投与されてもよい。
本発明の医薬組成物に含有される式(I):

(式中、各記号は前記と同意義である。)で示される化合物またはその製薬上許容される塩は、該化合物の骨格筋の疾患または病態(例えば、サルコペニアを伴う肝硬変)の治療および/または予防作用の増強または該化合物の投与量の低減等を目的として、テストステロン補充療法および/若しくはアミノ酸補給(例えば、分岐鎖アミノ酸補給)、ミオスタチン中和抗体等と組み合わせて用いることができる。ミオスタチン中和抗体としては、ドマグロズマブ、ランドグロズマブ、スタムルマブ等が挙げられる。
例えば、本発明化合物は、テストステロン、分岐鎖アミノ酸およびミオスタチン中和抗体からなる群より選択される少なくとも一つの化合物もしくはその製薬上許容される塩(以下、併用薬剤2と称する)と組み合わせて用いることができる。この際、本発明化合物と併用薬剤2の投与時期は限定されず、これらを投与対象に対し、同時にまたは連続的に投与してもよいし、時間差をおいて投与してもよい。さらに、本発明化合物と併用薬剤2とは、それぞれの活性成分を含む2種類以上の製剤として投与されてもよいし、それらの活性成分を含む単一の製剤として投与されてもよい。
また、本発明化合物は、テストステロン補充療法およびアミノ酸補給(例えば、分岐鎖アミノ酸補給)からなる群より選択される少なくとも一つの療法(以下、併用療法1と称する)と組み合わせて用いることができる。この際、本発明化合物の投与時期と併用療法1の実施時期は限定されない。本発明化合物を投与対象に対し、併用療法1と同時にまたは連続的に投与してもよいし、併用療法1と時間差をおいて投与してもよい。
(式中、各記号は前記と同意義である。)で示される化合物またはその製薬上許容される塩は、該化合物の骨格筋の疾患または病態(例えば、サルコペニアを伴う肝硬変)の治療および/または予防作用の増強または該化合物の投与量の低減等を目的として、テストステロン補充療法および/若しくはアミノ酸補給(例えば、分岐鎖アミノ酸補給)、ミオスタチン中和抗体等と組み合わせて用いることができる。ミオスタチン中和抗体としては、ドマグロズマブ、ランドグロズマブ、スタムルマブ等が挙げられる。
例えば、本発明化合物は、テストステロン、分岐鎖アミノ酸およびミオスタチン中和抗体からなる群より選択される少なくとも一つの化合物もしくはその製薬上許容される塩(以下、併用薬剤2と称する)と組み合わせて用いることができる。この際、本発明化合物と併用薬剤2の投与時期は限定されず、これらを投与対象に対し、同時にまたは連続的に投与してもよいし、時間差をおいて投与してもよい。さらに、本発明化合物と併用薬剤2とは、それぞれの活性成分を含む2種類以上の製剤として投与されてもよいし、それらの活性成分を含む単一の製剤として投与されてもよい。
また、本発明化合物は、テストステロン補充療法およびアミノ酸補給(例えば、分岐鎖アミノ酸補給)からなる群より選択される少なくとも一つの療法(以下、併用療法1と称する)と組み合わせて用いることができる。この際、本発明化合物の投与時期と併用療法1の実施時期は限定されない。本発明化合物を投与対象に対し、併用療法1と同時にまたは連続的に投与してもよいし、併用療法1と時間差をおいて投与してもよい。
併用薬剤1または併用薬剤2の投与量は、臨床上用いられている用量を基準として適宜選択することができる。また、本発明化合物と併用薬剤1または併用薬剤2の配合比は、投与対象、投与ルート、対象疾患、症状、組み合わせ等により適宜選択することができる。例えば、投与対象がヒトである場合、本発明化合物1重量部に対し、併用薬剤を0.01~100重量部用いればよい。
以下に実施例および試験例を挙げて本発明をさらに詳しく説明するが、本発明はこれらにより限定されるものではない。
調製例1:リコンビナントヒトACC2の調製
ヒトACC2蛋白質(N末より27アミノ酸残基~2458アミノ酸残基)をコードするcDNAをヒト腎臓cDNAライブラリー(クロンテック社)よりクローニングし、5’末端にHis-tag配列を導入後、pFastBac1(インビトロジェン社)に挿入した。Bac-to-Bacバキュロウイルス発現システム(Invitrogen社)のプロトコールに従い、組換えバキュロウィルスを作製後、Sf-9細胞に感染させ、ヒトACC2蛋白質を発現させた。回収した細胞を破砕し、フィルターろ過後、Niアフィニティクロマトグラフィー及び陰イオン交換クロマトグラフィーに供した。ヒトACC2蛋白質が含まれている画分を回収し、リコンビナントヒトACC2を得た。
ヒトACC2蛋白質(N末より27アミノ酸残基~2458アミノ酸残基)をコードするcDNAをヒト腎臓cDNAライブラリー(クロンテック社)よりクローニングし、5’末端にHis-tag配列を導入後、pFastBac1(インビトロジェン社)に挿入した。Bac-to-Bacバキュロウイルス発現システム(Invitrogen社)のプロトコールに従い、組換えバキュロウィルスを作製後、Sf-9細胞に感染させ、ヒトACC2蛋白質を発現させた。回収した細胞を破砕し、フィルターろ過後、Niアフィニティクロマトグラフィー及び陰イオン交換クロマトグラフィーに供した。ヒトACC2蛋白質が含まれている画分を回収し、リコンビナントヒトACC2を得た。
調製例2:リコンビナントヒトACC1の調製
ヒトACC1蛋白質(N末より1アミノ酸残基~2346アミノ酸残基)をコードするcDNAをヒト肝臓cDNAライブラリー(BioChain社)よりクローニングし、3’末端にmycタグ及びHis-tag配列を導入後、pIEXBAC3(ノバジェン社)に挿入した。FlashBACGOLD(オックスフォードエクスプレッションテクノロジーズ社)のプロトコールに従い、組換えバキュロウィルスを作製後、Sf-9細胞に感染させ、ヒトACC1蛋白質を発現させた。回収した細胞を破砕し、フィルターろ過後、Niアフィニティクロマトグラフィー及び陰イオン交換クロマトグラフィーに供した。ヒトACC1蛋白質が含まれている画分を回収し、リコンビナントヒトACC1を得た。
ヒトACC1蛋白質(N末より1アミノ酸残基~2346アミノ酸残基)をコードするcDNAをヒト肝臓cDNAライブラリー(BioChain社)よりクローニングし、3’末端にmycタグ及びHis-tag配列を導入後、pIEXBAC3(ノバジェン社)に挿入した。FlashBACGOLD(オックスフォードエクスプレッションテクノロジーズ社)のプロトコールに従い、組換えバキュロウィルスを作製後、Sf-9細胞に感染させ、ヒトACC1蛋白質を発現させた。回収した細胞を破砕し、フィルターろ過後、Niアフィニティクロマトグラフィー及び陰イオン交換クロマトグラフィーに供した。ヒトACC1蛋白質が含まれている画分を回収し、リコンビナントヒトACC1を得た。
試験例1:ヒトACC1及びACC2阻害活性の測定
上記の調製例により得たリコンビナントヒトACC1及びリコンビナントヒトACC2を、アッセイ緩衝液(50mM HEPES-KOH(pH 7.4)、10mM 塩化マグネシウム、6~10mM クエン酸カリウム、4mM 還元型グルタチオン、1.5mg/ml 牛血清アルブミン)中で1時間プレインキュベーションを行った。ついで、0.2μLの各々の本発明化合物溶液(DMSO)を分注した384穴マイクロプレートに、プレインキュベーションした酵素溶液5μLと基質溶液(50mM HEPES-KOH(pH 7.4)、1mM ATP、0.8mM アセチルCoA、25~50mM 炭酸水素カリウム)5μLを添加し、遠心、振とう後、湿潤箱中で室温、1~3時間インキュベーションした。インキュベーション後にEDTAの添加により酵素反応を停止し、その後、MALDIターゲットプレート上でCHCA(α-cyano-4-hydroxy cinnamic acid)マトリックスと共結晶させ、マトリックス支援レーザー脱離イオン化-飛行時間型質量分析計(MALDI-TOF MS)を用いて、リフレクターネガティブモードで測定を行った。基質のアセチルCoA(AcCoA)と反応産物であるマロニルCoA(MalCoA)の脱プロトン化イオンを検出し、それぞれのシグナル強度を用いてマロニルCoA又はスクシニルCoAへの変換率Intensity of [MalCoA-H]―/(Intensity of [MalCoA-H]―+Intensity of [AcCoA-H]―)を算出した。各化合物濃度における酵素反応の阻害率から50%阻害濃度(IC50値)を算出した。なお、アッセイ緩衝液中のクエン酸カリウム濃度、基質溶液中の炭酸水素カリウム濃度及びインキュベーションの時間は、使用する酵素のロット毎に上記の濃度又は反応時間内で調整した。
上記の調製例により得たリコンビナントヒトACC1及びリコンビナントヒトACC2を、アッセイ緩衝液(50mM HEPES-KOH(pH 7.4)、10mM 塩化マグネシウム、6~10mM クエン酸カリウム、4mM 還元型グルタチオン、1.5mg/ml 牛血清アルブミン)中で1時間プレインキュベーションを行った。ついで、0.2μLの各々の本発明化合物溶液(DMSO)を分注した384穴マイクロプレートに、プレインキュベーションした酵素溶液5μLと基質溶液(50mM HEPES-KOH(pH 7.4)、1mM ATP、0.8mM アセチルCoA、25~50mM 炭酸水素カリウム)5μLを添加し、遠心、振とう後、湿潤箱中で室温、1~3時間インキュベーションした。インキュベーション後にEDTAの添加により酵素反応を停止し、その後、MALDIターゲットプレート上でCHCA(α-cyano-4-hydroxy cinnamic acid)マトリックスと共結晶させ、マトリックス支援レーザー脱離イオン化-飛行時間型質量分析計(MALDI-TOF MS)を用いて、リフレクターネガティブモードで測定を行った。基質のアセチルCoA(AcCoA)と反応産物であるマロニルCoA(MalCoA)の脱プロトン化イオンを検出し、それぞれのシグナル強度を用いてマロニルCoA又はスクシニルCoAへの変換率Intensity of [MalCoA-H]―/(Intensity of [MalCoA-H]―+Intensity of [AcCoA-H]―)を算出した。各化合物濃度における酵素反応の阻害率から50%阻害濃度(IC50値)を算出した。なお、アッセイ緩衝液中のクエン酸カリウム濃度、基質溶液中の炭酸水素カリウム濃度及びインキュベーションの時間は、使用する酵素のロット毎に上記の濃度又は反応時間内で調整した。
試験例1の結果を以下に示す。

試験例2:代謝安定性試験
市販のプールドヒト肝ミクロソームと本発明化合物を一定時間反応させ、反応サンプルと未反応サンプルの比較により残存率を算出し、本発明化合物が肝で代謝される程度を評価した。
市販のプールドヒト肝ミクロソームと本発明化合物を一定時間反応させ、反応サンプルと未反応サンプルの比較により残存率を算出し、本発明化合物が肝で代謝される程度を評価した。
ヒト肝ミクロソーム0.5mgタンパク質/mLを含む0.2mLの緩衝液(50mmol/L Tris-HCl pH7.4、150mmol/L 塩化カリウム、10mmol/L 塩化マグネシウム)中で、1mmol/L NADPH存在下で37℃、0分あるいは30分間反応させた(酸化的反応)。反応後、メタノール/アセトニトリル=1/1(v/v)溶液の100μLに反応液50μLを添加、混合し、3000rpmで15分間遠心した。その遠心上清中の本発明化合物をLC/MS/MSまたは固相抽出(SPE)/MSにて定量し、0分反応時の化合物量を100%として反応後の本発明化合物の残存量を計算した。
試験例2の結果を以下に示す。

調整例3:Atglノックアウトマウスの作製
Atgl(adipose triglyceride lipase)ノックアウトマウスはTGCVの非臨床モデルとして知られている。Atgl遺伝子を欠損したAtglノックアウトマウス(KOマウス)をそれぞれ公知の方法により作製した。
Atgl(adipose triglyceride lipase)ノックアウトマウスはTGCVの非臨床モデルとして知られている。Atgl遺伝子を欠損したAtglノックアウトマウス(KOマウス)をそれぞれ公知の方法により作製した。
試験例3:Atglノックアウトマウス心筋における長鎖脂肪酸代謝能への効果
6-7週齢、雄性のAtglノックアウトマウスに本発明化合物を30又は45mg/kg/10mLの投与量となるように0.5% HPMC水溶液に懸濁し、長鎖脂肪酸代謝能を測定する前日および当日に経口投与を行った。長鎖脂肪酸代謝能の測定当日、iodine-123-β methyl iodophenyl-pen-tadecanoic acid(BMIPP)を14.6 MBq/マウスで尾静脈より投与を行った。投与1時間後と4時間後に、マウスを麻酔下で、single-photon emission computed tomography(SPECT)/CT による撮像を行い、投与されたBMIPPの心筋における放射活性を測定した。心筋における長鎖脂肪酸の代謝は以下の式によりwash out rateとして計算した。
[(BMIPP uptake(1 h) - remaining BMIPP(4 h))/ BMIPP uptake(1 h)]
(結果)
本評価系において、化合物I-12(30mg/kg/10mL)は長鎖脂肪酸代謝能の改善傾向を示した(p=0.096、ウェルチの t 検定)。また、化合物I-13(45mg/kg/10mL)は、長鎖脂肪酸代謝能の有意な改善作用を示した(p<0.05、ウェルチの t 検定)。試験例3の結果を図1に示す。
6-7週齢、雄性のAtglノックアウトマウスに本発明化合物を30又は45mg/kg/10mLの投与量となるように0.5% HPMC水溶液に懸濁し、長鎖脂肪酸代謝能を測定する前日および当日に経口投与を行った。長鎖脂肪酸代謝能の測定当日、iodine-123-β methyl iodophenyl-pen-tadecanoic acid(BMIPP)を14.6 MBq/マウスで尾静脈より投与を行った。投与1時間後と4時間後に、マウスを麻酔下で、single-photon emission computed tomography(SPECT)/CT による撮像を行い、投与されたBMIPPの心筋における放射活性を測定した。心筋における長鎖脂肪酸の代謝は以下の式によりwash out rateとして計算した。
[(BMIPP uptake(1 h) - remaining BMIPP(4 h))/ BMIPP uptake(1 h)]
(結果)
本評価系において、化合物I-12(30mg/kg/10mL)は長鎖脂肪酸代謝能の改善傾向を示した(p=0.096、ウェルチの t 検定)。また、化合物I-13(45mg/kg/10mL)は、長鎖脂肪酸代謝能の有意な改善作用を示した(p<0.05、ウェルチの t 検定)。試験例3の結果を図1に示す。
試験例4:Atglノックアウトマウスにおける心臓中脂質量評価
本発明化合物が心臓中脂質に与える影響を調べるため、Atglノックアウトマウスに媒体(Vehicle)又は本発明化合物を投与した後に心臓を採取した。採取した心臓をホモジナイズし、Folchら(J Biol Chem. 1957 May;226(1):497-509.)等に記載のFolch法を用いて組織中脂質を抽出し、日立自動分析装置7180(日立ハイテク)を用いて組織当たりの心臓中中性脂肪(TG)および心臓中遊離脂肪酸(NEFA)量を算出し、本発明化合物による心筋中脂質量への影響を評価した。試薬はそれぞれについて、オートセラ(登録商標) TG又はオートセラ(登録商標)NEFA(積水メディカル)を使用した。
本発明化合物が心臓中脂質に与える影響を調べるため、Atglノックアウトマウスに媒体(Vehicle)又は本発明化合物を投与した後に心臓を採取した。採取した心臓をホモジナイズし、Folchら(J Biol Chem. 1957 May;226(1):497-509.)等に記載のFolch法を用いて組織中脂質を抽出し、日立自動分析装置7180(日立ハイテク)を用いて組織当たりの心臓中中性脂肪(TG)および心臓中遊離脂肪酸(NEFA)量を算出し、本発明化合物による心筋中脂質量への影響を評価した。試薬はそれぞれについて、オートセラ(登録商標) TG又はオートセラ(登録商標)NEFA(積水メディカル)を使用した。
試験例5:Atglノックアウトマウスにおける心機能評価
本発明化合物が心機能に与える影響を調べるため、Atglノックアウトマウスに媒体(Vehicle)又は本発明化合物を反復投与した後に、心臓の収縮力を示す左室駆出率(Left Ventricular Ejection Fraction:LVEF)を超音波高解像度イメージングシステム Vevo770(Primetech)を用いて評価した。
本発明化合物が心機能に与える影響を調べるため、Atglノックアウトマウスに媒体(Vehicle)又は本発明化合物を反復投与した後に、心臓の収縮力を示す左室駆出率(Left Ventricular Ejection Fraction:LVEF)を超音波高解像度イメージングシステム Vevo770(Primetech)を用いて評価した。
試験例6:Atglノックアウトマウスにおける生存期間評価
本発明化合物が生存期間に与える影響を調べるため、Atglノックアウトマウスに媒体(Vehicle)又は本発明化合物を反復投与した後に、生存期間に与える影響を評価した。
本発明化合物が生存期間に与える影響を調べるため、Atglノックアウトマウスに媒体(Vehicle)又は本発明化合物を反復投与した後に、生存期間に与える影響を評価した。
試験例7:血小板数への影響
6週齢、雄性のCrl:CD(SD)ラットに非絶食下で本発明化合物を50~600 mg/kg/dayの投与量となるように0.5%メチルセルロース溶液に懸濁し、4日間反復経口投与を行った。投与開始5日目に麻酔下で腹部大静脈より採血を行い、血漿を採取した。血漿中の血小板数を自動血球計数装置により評価した。
6週齢、雄性のCrl:CD(SD)ラットに非絶食下で本発明化合物を50~600 mg/kg/dayの投与量となるように0.5%メチルセルロース溶液に懸濁し、4日間反復経口投与を行った。投与開始5日目に麻酔下で腹部大静脈より採血を行い、血漿を採取した。血漿中の血小板数を自動血球計数装置により評価した。
化合物I-12、化合物I-13はともに評価に用いた投与量域内でSDラットの血小板数に影響を与えなかった。試験例7の結果を以下に示す。

データ: 平均値±標準偏差

データ: 平均値±標準偏差
データ: 平均値±標準偏差
データ: 平均値±標準偏差
試験例8:超高脂肪コリン欠乏メチオニン減量飼料給餌マウスにおける血漿中トリグリセリド濃度評価
6週齢、雄性のC57BL/6JJclマウスに超高脂肪コリン欠乏メチオニン減量飼料(60kcal% 脂肪含有量、ラード使用、コリン欠乏、メチオニン減量(0.1%))を給餌すると同時に本発明化合物を3~45mg/kg/10mLの投与量となるように媒体(0.5% メチルセルロース水溶液)に懸濁し、経口反復投与(b.i.d.)を8週間行った。反復投与期間中に尾静脈より採血を行い、血漿中の生化学パラメーターを測定した。
(結果)
化合物I-12、化合物I-13投与群はともに媒体投与群と比較して、血漿中トリグリセリド濃度が低値であった。図2、3および表5、6に結果を示す。

データ: 平均値±標準誤差
媒体投与群に対して *: p<0.05, **: p<0.01, ***:p<0.001(ダネット検定)

データ: 平均値±標準誤差
媒体投与群に対して *: p<0.05, **: p<0.01, ***:p<0.001(ダネット検定)
6週齢、雄性のC57BL/6JJclマウスに超高脂肪コリン欠乏メチオニン減量飼料(60kcal% 脂肪含有量、ラード使用、コリン欠乏、メチオニン減量(0.1%))を給餌すると同時に本発明化合物を3~45mg/kg/10mLの投与量となるように媒体(0.5% メチルセルロース水溶液)に懸濁し、経口反復投与(b.i.d.)を8週間行った。反復投与期間中に尾静脈より採血を行い、血漿中の生化学パラメーターを測定した。
(結果)
化合物I-12、化合物I-13投与群はともに媒体投与群と比較して、血漿中トリグリセリド濃度が低値であった。図2、3および表5、6に結果を示す。
データ: 平均値±標準誤差
媒体投与群に対して *: p<0.05, **: p<0.01, ***:p<0.001(ダネット検定)
データ: 平均値±標準誤差
媒体投与群に対して *: p<0.05, **: p<0.01, ***:p<0.001(ダネット検定)
試験例9:CYP阻害試験
市販のプールドヒト肝ミクロソームを用いて、ヒト主要CYP5分子種(CYP1A2、2C9、2C19、2D6、3A4)の典型的基質代謝反応である7-エトキシレゾルフィンのO-脱エチル化(CYP1A2)、トルブタミドのメチル-水酸化(CYP2C9)、メフェニトインの4’-水酸化(CYP2C19)、デキストロメトルファンのO脱メチル化(CYP2D6)、テルフェナジンの水酸化(CYP3A4)を指標とし、それぞれの代謝物生成量が本発明化合物によって阻害される程度を評価した。
市販のプールドヒト肝ミクロソームを用いて、ヒト主要CYP5分子種(CYP1A2、2C9、2C19、2D6、3A4)の典型的基質代謝反応である7-エトキシレゾルフィンのO-脱エチル化(CYP1A2)、トルブタミドのメチル-水酸化(CYP2C9)、メフェニトインの4’-水酸化(CYP2C19)、デキストロメトルファンのO脱メチル化(CYP2D6)、テルフェナジンの水酸化(CYP3A4)を指標とし、それぞれの代謝物生成量が本発明化合物によって阻害される程度を評価した。
反応条件は以下のとおり:基質、0.5μmol/L エトキシレゾルフィン(CYP1A2)、100μmol/L トルブタミド(CYP2C9)、50μmol/L S-メフェニトイン(CYP2C19)、5μmol/L デキストロメトルファン(CYP2D6)、1μmol/L テルフェナジン(CYP3A4);反応時間、15分;反応温度、37℃;酵素、プールドヒト肝ミクロソーム0.2mg タンパク質/mL;本発明化合物濃度、1、5、10、20μmol/L(4点)。
96穴プレートに50mmol/L Hepes緩衝液中に各5種の基質、ヒト肝ミクロソーム、本発明化合物を上記組成で加え、補酵素であるNADPHを添加して、指標とする代謝反応を開始した。37℃、15分間反応した後、メタノール/アセトニトリル=1/1(V/V)溶液を添加することで反応を停止した。3000rpm、15分間の遠心後、遠心上清中のレゾルフィン(CYP1A2代謝物)を蛍光マルチラベルカウンタまたはLC/MS/MSで定量し、トルブタミド水酸化体(CYP2C9代謝物)、メフェニトイン4’水酸化体(CYP2C19代謝物)、デキストロルファン(CYP2D6代謝物)、テルフェナジンアルコール体(CYP3A4代謝物)をLC/MS/MSで定量した。
本発明化合物の代わりに化合物を溶解した溶媒であるDMSOのみを反応溶液に添加したものをコントロール(100%)とし、残存活性(%)を算出し、濃度と抑制率を用いて、ロジスティックモデルによる逆推定によりIC50を算出した。
試験例10:CYP3A4蛍光MBI試験
CYP3A4蛍光MBI試験は、代謝反応による本発明化合物のCYP3A4阻害の増強を調べる試験である。CYP3A4酵素(大腸菌発現酵素)により7-ベンジルオキシトリフルオロメチルクマリン(7-BFC)が脱ベンジル化されて、蛍光を発する代謝物7-ハイドロキシトリフルオロメチルクマリン(7-HFC)が生じる。7-HFC生成反応を指標としてCYP3A4阻害を評価した。
CYP3A4蛍光MBI試験は、代謝反応による本発明化合物のCYP3A4阻害の増強を調べる試験である。CYP3A4酵素(大腸菌発現酵素)により7-ベンジルオキシトリフルオロメチルクマリン(7-BFC)が脱ベンジル化されて、蛍光を発する代謝物7-ハイドロキシトリフルオロメチルクマリン(7-HFC)が生じる。7-HFC生成反応を指標としてCYP3A4阻害を評価した。
反応条件は以下のとおり:基質、5.6μmol/L 7-BFC;プレ反応時間、0または30分;反応時間、15分;反応温度、25℃(室温);CYP3A4含量(大腸菌発現酵素)、プレ反応時62.5pmol/mL、反応時6.25pmol/mL(10倍希釈時);本発明化合物濃度、0.625、1.25、2.5、5、10、20μmol/L(6点)。
96穴プレートにプレ反応液としてK-Pi緩衝液(pH7.4)中に酵素、本発明化合物溶液を上記のプレ反応の組成で加え、別の96穴プレートに基質とK-Pi緩衝液で1/10希釈されるようにその一部を移行し、補酵素であるNADPHを添加して指標とする反応を開始し(プレ反応無)、所定の時間反応後、アセトニトリル/0.5mol/L Tris(トリスヒドロキシアミノメタン)=4/1(V/V)を加えることによって反応を停止した。また残りのプレ反応液にもNADPHを添加しプレ反応を開始し(プレ反応有)、所定時間プレ反応後、別のプレートに基質とK-Pi緩衝液で1/10希釈されるように一部を移行し指標とする反応を開始した。所定の時間反応後、アセトニトリル/0.5mol/L Tris(トリスヒドロキシアミノメタン)=4/1(V/V)を加えることによって反応を停止した。それぞれの指標反応を行ったプレートを蛍光プレートリーダーで代謝物である7-HFCの蛍光値を測定した。(Ex=420nm、Em=535nm)。なお,希釈濃度や希釈溶媒は、必要に応じて変更した。
本発明化合物を溶解した溶媒であるDMSOのみを反応系に添加したものをコントロール(100%)とし、本発明化合物をそれぞれの濃度添加したときの残存活性(%)を算出し、濃度と抑制率を用いて、ロジスティックモデルによる逆推定によりIC50を算出した。IC50値の差が5μmol/L以上の場合を(+)とし、3μmol/L以下の場合を(-)とした。
試験例11:CYP3A4(MDZ)MBI試験
本発明化合物のCYP3A4阻害に関して、本発明化合物の代謝反応に起因した阻害作用の増強からMechanism based inhibition(MBI)能を評価する試験である。プールドヒト肝ミクロソームを用いてミダゾラム(MDZ)の1-水酸化反応を指標としてCYP3A4阻害を評価した。
本発明化合物のCYP3A4阻害に関して、本発明化合物の代謝反応に起因した阻害作用の増強からMechanism based inhibition(MBI)能を評価する試験である。プールドヒト肝ミクロソームを用いてミダゾラム(MDZ)の1-水酸化反応を指標としてCYP3A4阻害を評価した。
反応条件は以下のとおり:基質、10μmol/L MDZ;プレ反応時間、0または30分;基質代謝反応時間、2分;反応温度、37℃;プールドヒト肝ミクロソーム、プレ反応時0.5mg/mL、反応時0.05mg/mL(10倍希釈時);本発明化合物プレ反応時の濃度、1、5、10、20μmol/L(4点)。
96穴プレートにプレ反応液としてK-Pi緩衝液(pH7.4)中にプールドヒト肝ミクロソーム、本発明化合物溶液を上記のプレ反応の組成で加え、別の96穴プレートに基質を含むK-Pi緩衝液で1/10希釈されるようにその一部を移行し、補酵素であるNADPHを添加して指標とする反応を開始し(Preincubataion 0min)、所定の時間反応後、メタノール/アセトニトリル=1/1(V/V)溶液を加えることによって反応を停止した。また残りのプレ反応液にもNADPHを添加しプレ反応を開始し(Preincubataion 30min)、所定時間プレ反応後、別のプレートに基質を含むK-Pi緩衝液で1/10希釈されるように一部を移行し指標とする反応を開始した。所定の時間反応後、メタノール/アセトニトリル=1/1(V/V)溶液を加えることによって反応を停止した。それぞれの指標反応を行ったプレートを3000rpm、15分間の遠心後、遠心上清中の1-水酸化ミダゾラム をLC/MS/MSで定量した。なお、希釈濃度や希釈溶媒は、必要に応じて変更した。
本発明化合物の代わりに化合物を溶解した溶媒であるDMSOのみを反応液に添加したものをコントロール(100%)とし、本発明化合物をそれぞれの濃度添加したときの残存活性(%)を算出し、濃度と阻害率を用いて、ロジスティックモデルによる逆推定によりICを算出した。Preincubataion 0minのIC/Preincubataion 30minのICをShifted IC値とし,Shifted ICが1.5以上であればPositiveとし、Shifted ICが1.0以下であればNegativeとした。
試験例12:BA試験
経口吸収性の検討実験材料と方法
(1)使用動物:マウスあるいはラットを使用した。
(2)飼育条件:マウスあるいはラットは、固形飼料および滅菌水道水を自由摂取させた。
(3)投与量、群分けの設定:所定の投与量で経口投与および静脈内投与した。以下のように群を設定した。(化合物ごとで投与量は変更有)
経口投与 2~60μmol/kgあるいは1~30mg/kg(n=2~3)
静脈内投与 1~30μmol/kgあるいは0.5~10mg/kg(n=2~3)
(4)投与液の調製:経口投与は溶液または懸濁液として投与した。静脈内投与は可溶化して投与した。
(5)投与方法:経口投与は、経口ゾンデにより強制的に胃内に投与した。静脈内投与は、注射針を付けたシリンジにより尾静脈から投与した。
(6)評価項目:経時的に採血し、血漿中本発明化合物濃度をLC/MS/MSを用いて測定した。
(7)統計解析:血漿中本発明化合物濃度推移について、モーメント解析法により血漿中濃度‐時間曲線下面積(AUC)を算出し、経口投与群と静脈内投与群の投与量比およびAUC比から本発明化合物のバイオアベイラビリティ(BA)を算出した。
なお、希釈濃度や希釈溶媒は、必要に応じて変更した。
経口吸収性の検討実験材料と方法
(1)使用動物:マウスあるいはラットを使用した。
(2)飼育条件:マウスあるいはラットは、固形飼料および滅菌水道水を自由摂取させた。
(3)投与量、群分けの設定:所定の投与量で経口投与および静脈内投与した。以下のように群を設定した。(化合物ごとで投与量は変更有)
経口投与 2~60μmol/kgあるいは1~30mg/kg(n=2~3)
静脈内投与 1~30μmol/kgあるいは0.5~10mg/kg(n=2~3)
(4)投与液の調製:経口投与は溶液または懸濁液として投与した。静脈内投与は可溶化して投与した。
(5)投与方法:経口投与は、経口ゾンデにより強制的に胃内に投与した。静脈内投与は、注射針を付けたシリンジにより尾静脈から投与した。
(6)評価項目:経時的に採血し、血漿中本発明化合物濃度をLC/MS/MSを用いて測定した。
(7)統計解析:血漿中本発明化合物濃度推移について、モーメント解析法により血漿中濃度‐時間曲線下面積(AUC)を算出し、経口投与群と静脈内投与群の投与量比およびAUC比から本発明化合物のバイオアベイラビリティ(BA)を算出した。
なお、希釈濃度や希釈溶媒は、必要に応じて変更した。
試験例13:クリアランス評価試験
実験材料と方法
(1)使用動物:SDラットを使用した。
(2)飼育条件:SDラットは、固形飼料および滅菌水道水を自由摂取させた。
(3)投与量、群分けの設定:静脈内投与を所定の投与量により投与した。以下のように群を設定した。
静脈内投与 1μmol/kg(n=2)
(4)投与液の調製:ジメチルスルホキシド/プロピレングリコール=1/1溶媒を用いて可溶化して投与した。
(5)投与方法:注射針を付けたシリンジにより尾静脈から投与した。
(6)評価項目:経時的に採血し、血漿中本発明化合物濃度をLC/MS/MSを用いて測定した。
(7)統計解析:血漿中本発明化合物濃度推移について、モーメント解析法により全身クリアランス(CLtot)を算出した。なお、希釈濃度や希釈溶媒は、必要に応じて変更した。
実験材料と方法
(1)使用動物:SDラットを使用した。
(2)飼育条件:SDラットは、固形飼料および滅菌水道水を自由摂取させた。
(3)投与量、群分けの設定:静脈内投与を所定の投与量により投与した。以下のように群を設定した。
静脈内投与 1μmol/kg(n=2)
(4)投与液の調製:ジメチルスルホキシド/プロピレングリコール=1/1溶媒を用いて可溶化して投与した。
(5)投与方法:注射針を付けたシリンジにより尾静脈から投与した。
(6)評価項目:経時的に採血し、血漿中本発明化合物濃度をLC/MS/MSを用いて測定した。
(7)統計解析:血漿中本発明化合物濃度推移について、モーメント解析法により全身クリアランス(CLtot)を算出した。なお、希釈濃度や希釈溶媒は、必要に応じて変更した。
試験例14:Fluctuation Ames Test
本発明化合物の変異原性を評価する。
凍結保存しているネズミチフス菌(Salmonella typhimurium TA98株、TA100株)20μLを10mL液体栄養培地(2.5% Oxoid nutrient broth No.2)に接種し37℃にて10時間、振盪前培養した。TA98株は7.70~8.00mLの菌液を遠心(2000×g、10分間)して培養液を除去した。遠心に用いた菌液と同容量のMicro F緩衝液(K2HPO4:3.5g/L、KH2PO4:1g/L、(NH4)2SO4:1g/L、クエン酸三ナトリウム二水和物:0.25g/L、MgSO4・7H20:0.1g/L)に菌を懸濁し、120mLのExposure培地(ビオチン:8μg/mL、ヒスチジン:0.2μg/mL、グルコース:8mg/mLを含むMicroF緩衝液)に添加した。TA100株は3.10~3.42mLの菌液をExposure培地120~130mLに添加し試験菌液を調製した。本発明化合物DMSO溶液(最高用量50mg/mLから2~3倍公比で数段階希釈)、陰性対照としてDMSO、陽性対照として非代謝活性化条件ではTA98株に対しては50μg/mLの4-ニトロキノリン-1-オキシドDMSO溶液、TA100株に対しては0.25μg/mLの2-(2-フリル)-3-(5-ニトロ-2-フリル)アクリルアミドDMSO溶液、代謝活性化条件ではTA98株に対して40μg/mLの2-アミノアントラセンDMSO溶液、TA100株に対しては20μg/mLの2-アミノアントラセンDMSO溶液それぞれ12μLと試験菌液588μL(代謝活性化条件では試験菌液498μLとS9 mix 90μLの混合液)を混和し、37℃にて90分間、振盪培養した。本発明化合物を曝露した菌液460μLを、Indicator培地(ビオチン:8μg/mL、ヒスチジン:0.2μg/mL、グルコース:8mg/mL、ブロモクレゾールパープル:37.5μg/mLを含むMicroF緩衝液)2300μLに混和し、50μLずつマイクロプレート48ウェル/用量に分注し、37℃にて3日間、静置培養した。アミノ酸(ヒスチジン)合成酵素遺伝子の突然変異によって増殖能を獲得した菌を含むウェルは、pH変化により紫色から黄色に変色するため、1用量あたり48ウェル中の黄色に変色した菌増殖ウェルを計数し、陰性対照群と比較して評価した。変異原性が陰性のものを(-)、陽性のものを(+)として示した。なお、希釈濃度や希釈溶媒は、必要に応じて変更した。
本発明化合物の変異原性を評価する。
凍結保存しているネズミチフス菌(Salmonella typhimurium TA98株、TA100株)20μLを10mL液体栄養培地(2.5% Oxoid nutrient broth No.2)に接種し37℃にて10時間、振盪前培養した。TA98株は7.70~8.00mLの菌液を遠心(2000×g、10分間)して培養液を除去した。遠心に用いた菌液と同容量のMicro F緩衝液(K2HPO4:3.5g/L、KH2PO4:1g/L、(NH4)2SO4:1g/L、クエン酸三ナトリウム二水和物:0.25g/L、MgSO4・7H20:0.1g/L)に菌を懸濁し、120mLのExposure培地(ビオチン:8μg/mL、ヒスチジン:0.2μg/mL、グルコース:8mg/mLを含むMicroF緩衝液)に添加した。TA100株は3.10~3.42mLの菌液をExposure培地120~130mLに添加し試験菌液を調製した。本発明化合物DMSO溶液(最高用量50mg/mLから2~3倍公比で数段階希釈)、陰性対照としてDMSO、陽性対照として非代謝活性化条件ではTA98株に対しては50μg/mLの4-ニトロキノリン-1-オキシドDMSO溶液、TA100株に対しては0.25μg/mLの2-(2-フリル)-3-(5-ニトロ-2-フリル)アクリルアミドDMSO溶液、代謝活性化条件ではTA98株に対して40μg/mLの2-アミノアントラセンDMSO溶液、TA100株に対しては20μg/mLの2-アミノアントラセンDMSO溶液それぞれ12μLと試験菌液588μL(代謝活性化条件では試験菌液498μLとS9 mix 90μLの混合液)を混和し、37℃にて90分間、振盪培養した。本発明化合物を曝露した菌液460μLを、Indicator培地(ビオチン:8μg/mL、ヒスチジン:0.2μg/mL、グルコース:8mg/mL、ブロモクレゾールパープル:37.5μg/mLを含むMicroF緩衝液)2300μLに混和し、50μLずつマイクロプレート48ウェル/用量に分注し、37℃にて3日間、静置培養した。アミノ酸(ヒスチジン)合成酵素遺伝子の突然変異によって増殖能を獲得した菌を含むウェルは、pH変化により紫色から黄色に変色するため、1用量あたり48ウェル中の黄色に変色した菌増殖ウェルを計数し、陰性対照群と比較して評価した。変異原性が陰性のものを(-)、陽性のものを(+)として示した。なお、希釈濃度や希釈溶媒は、必要に応じて変更した。
試験例15:hERG試験
本発明化合物の心電図QT間隔延長リスク評価を目的として、human ether-a-go-go related gene (hERG)チャネルを発現させたCHO細胞を用いて、心室再分極過程に重要な役割を果たす遅延整流K+電流(IKr)への本発明化合物の作用を検討した。
全自動パッチクランプシステム(QPatch;Sophion Bioscience A/S)を用い、ホールセルパッチクランプ法により、細胞を-80mVの膜電位に保持し、-50mVのリーク電位を与えた後、+20mVの脱分極刺激を2秒間、さらに-50mVの再分極刺激を2秒間与えた際に誘発されるIKrを記録した。ジメチルスルホキシドを0.1%に調整した細胞外液(NaCl:145 mmol/L、KCl:4 mmol/L、CaCl2:2 mmol/L、MgCl2:1 mmol/L、グルコース:10 mmol/L、HEPES(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid、4-(2-ヒドロキシエチル)-1-ピペラジンエタンスルホン酸):10mmol/L、pH=7.4)を媒体とし、媒体及び本発明化合物を目的の濃度で溶解させた細胞外液をそれぞれ室温条件下で、7分以上細胞に適用させた。得られたIKrから、解析ソフト(QPatch Assay software;Sophion Bioscience A/S)を使用して、保持膜電位における電流値を基準に最大テール電流の絶対値を計測した。さらに、媒体適用後の最大テール電流に対する本発明化合物適用後の最大テール電流を阻害率として算出し、本発明化合物のIKrへの影響を評価した。なお、希釈濃度や希釈溶媒は、必要に応じて変更した。
本発明化合物の心電図QT間隔延長リスク評価を目的として、human ether-a-go-go related gene (hERG)チャネルを発現させたCHO細胞を用いて、心室再分極過程に重要な役割を果たす遅延整流K+電流(IKr)への本発明化合物の作用を検討した。
全自動パッチクランプシステム(QPatch;Sophion Bioscience A/S)を用い、ホールセルパッチクランプ法により、細胞を-80mVの膜電位に保持し、-50mVのリーク電位を与えた後、+20mVの脱分極刺激を2秒間、さらに-50mVの再分極刺激を2秒間与えた際に誘発されるIKrを記録した。ジメチルスルホキシドを0.1%に調整した細胞外液(NaCl:145 mmol/L、KCl:4 mmol/L、CaCl2:2 mmol/L、MgCl2:1 mmol/L、グルコース:10 mmol/L、HEPES(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid、4-(2-ヒドロキシエチル)-1-ピペラジンエタンスルホン酸):10mmol/L、pH=7.4)を媒体とし、媒体及び本発明化合物を目的の濃度で溶解させた細胞外液をそれぞれ室温条件下で、7分以上細胞に適用させた。得られたIKrから、解析ソフト(QPatch Assay software;Sophion Bioscience A/S)を使用して、保持膜電位における電流値を基準に最大テール電流の絶対値を計測した。さらに、媒体適用後の最大テール電流に対する本発明化合物適用後の最大テール電流を阻害率として算出し、本発明化合物のIKrへの影響を評価した。なお、希釈濃度や希釈溶媒は、必要に応じて変更した。
試験例16:Ames試験
サルモネラ菌(Salmonella typhimurium)TA98株、TA100株、TA1535株、TA1537株および大腸菌(Escherichia coli)WP2uvrA株を試験菌株としたAmes試験により、本発明化合物の変異原性を評価した。本発明化合物のDMSO溶液0.1mLに、代謝活性化条件ではS9mixを0.5mL、非代謝活性化条件ではリン酸緩衝液を0.5mLと試験菌液0.1mLを混和し、ヒスチジン及びビオチン、またはトリプトファン含有の重層用軟寒天2mLと共に最少グルコース寒天平板に重層した。同時に、陰性対照物質(DMSO)および陽性対照物質(2-(2-フリル)-3-(5-ニトロ-2-フリル)アクリルアミド、アジ化ナトリウム、9-アミノアクリジン、または2-アミノアントラセン)についても同様に実施した。37℃で48時間培養した後、出現した復帰変異コロニーを計数し、陰性対照群と比較して評価した。復帰変異コロニー数が濃度依存的に増加し、かつ陰性対照群のコロニー数の2倍以上となる場合を陽性(+)と判断した。なお、希釈濃度や希釈溶媒は、必要に応じて変更した。
サルモネラ菌(Salmonella typhimurium)TA98株、TA100株、TA1535株、TA1537株および大腸菌(Escherichia coli)WP2uvrA株を試験菌株としたAmes試験により、本発明化合物の変異原性を評価した。本発明化合物のDMSO溶液0.1mLに、代謝活性化条件ではS9mixを0.5mL、非代謝活性化条件ではリン酸緩衝液を0.5mLと試験菌液0.1mLを混和し、ヒスチジン及びビオチン、またはトリプトファン含有の重層用軟寒天2mLと共に最少グルコース寒天平板に重層した。同時に、陰性対照物質(DMSO)および陽性対照物質(2-(2-フリル)-3-(5-ニトロ-2-フリル)アクリルアミド、アジ化ナトリウム、9-アミノアクリジン、または2-アミノアントラセン)についても同様に実施した。37℃で48時間培養した後、出現した復帰変異コロニーを計数し、陰性対照群と比較して評価した。復帰変異コロニー数が濃度依存的に増加し、かつ陰性対照群のコロニー数の2倍以上となる場合を陽性(+)と判断した。なお、希釈濃度や希釈溶媒は、必要に応じて変更した。
試験例17:光溶血試験
本発明化合物を目的の濃度で溶解させ、マイクロプレート上において、ヒツジ脱繊維血から調製した0.1~0.0008%濃度の赤血球浮遊液(2.5v/v%)と混合し、紫外線蛍光ランプ(GL20SEランプ、三共電気およびFL20S―BLBランプ、パナソニック)を用いてUVAおよびUVB領域での光照射(10 J/cm2、290~400nm)を行う。光照射終了後の混合液を採取し、遠心を行う。遠心後の上清を採取しマイクロプレートに移した後、上清の吸光度(540または630nm)を測定、吸光度を基にした判定を行う。540および630nmでの吸光度は、それぞれ生体膜損傷(光溶血率%)および脂質膜過酸化(メトヘモグロビン産生)の指標とする。光溶血率が10%未満であり、かつ、630nmでの吸光度の変化量が0.05未満の場合を(-)とし、光溶血率が10%以上または630nmでの吸光度の変化量が0.05以上の場合を(+)とする。
本発明化合物を目的の濃度で溶解させ、マイクロプレート上において、ヒツジ脱繊維血から調製した0.1~0.0008%濃度の赤血球浮遊液(2.5v/v%)と混合し、紫外線蛍光ランプ(GL20SEランプ、三共電気およびFL20S―BLBランプ、パナソニック)を用いてUVAおよびUVB領域での光照射(10 J/cm2、290~400nm)を行う。光照射終了後の混合液を採取し、遠心を行う。遠心後の上清を採取しマイクロプレートに移した後、上清の吸光度(540または630nm)を測定、吸光度を基にした判定を行う。540および630nmでの吸光度は、それぞれ生体膜損傷(光溶血率%)および脂質膜過酸化(メトヘモグロビン産生)の指標とする。光溶血率が10%未満であり、かつ、630nmでの吸光度の変化量が0.05未満の場合を(-)とし、光溶血率が10%以上または630nmでの吸光度の変化量が0.05以上の場合を(+)とする。
試験例18:P-gp基質試験
ヒトMDR1発現細胞または親細胞を単層培養したトランスウェル(登録商標、CORNING社)の片側に本発明化合物を添加し、一定時間反応させた。MDR1発現細胞と親細胞についてApical側からBasolateral側方向(A→B)とBasolateral側からApical側方向(B→A)の膜透過係数を算出し,MDR1発現細胞と親細胞のEfflux Ratio(ER;B→AとA→Bの膜透過係数の比)値を算出した。MDR1発現細胞と親細胞のEfflux Ratio(ER値)を比較し、本発明化合物がP-gp基質であるか否かを判断した。
ヒトMDR1発現細胞または親細胞を単層培養したトランスウェル(登録商標、CORNING社)の片側に本発明化合物を添加し、一定時間反応させた。MDR1発現細胞と親細胞についてApical側からBasolateral側方向(A→B)とBasolateral側からApical側方向(B→A)の膜透過係数を算出し,MDR1発現細胞と親細胞のEfflux Ratio(ER;B→AとA→Bの膜透過係数の比)値を算出した。MDR1発現細胞と親細胞のEfflux Ratio(ER値)を比較し、本発明化合物がP-gp基質であるか否かを判断した。
試験例19:溶解性試験
本発明化合物の溶解度は、1%DMSO添加条件下で決定した。DMSOにて10mmol/L化合物溶液を調製した。本発明化合物溶液2μLをそれぞれJP-1液、JP-2液198μLに添加した。室温で1時間振盪させた後、混液を吸引濾過した。濾液をメタノール/水=1/1(V/V)またはアセトニトリル/メタノール/水=1/1/2(V/V/V)にて10または100倍希釈し、絶対検量線法によりLC/MSまたは固相抽出(SPE)/MSを用いて濾液中濃度を測定した。なお、希釈濃度や希釈溶媒は、必要に応じて変更した。
本発明化合物の溶解度は、1%DMSO添加条件下で決定した。DMSOにて10mmol/L化合物溶液を調製した。本発明化合物溶液2μLをそれぞれJP-1液、JP-2液198μLに添加した。室温で1時間振盪させた後、混液を吸引濾過した。濾液をメタノール/水=1/1(V/V)またはアセトニトリル/メタノール/水=1/1/2(V/V/V)にて10または100倍希釈し、絶対検量線法によりLC/MSまたは固相抽出(SPE)/MSを用いて濾液中濃度を測定した。なお、希釈濃度や希釈溶媒は、必要に応じて変更した。
JP-1液の組成は、以下の通りである。
塩化ナトリウム2.0g、塩酸7.0mLに水を加えて1000mLとした。
JP-2液の組成は、以下の通りである。
リン酸二水素カリウム3.40gおよび無水リン酸水素二ナトリウム3.55gを水に溶かし1000mLとしたもの1容量に水1容量を加えた。
塩化ナトリウム2.0g、塩酸7.0mLに水を加えて1000mLとした。
JP-2液の組成は、以下の通りである。
リン酸二水素カリウム3.40gおよび無水リン酸水素二ナトリウム3.55gを水に溶かし1000mLとしたもの1容量に水1容量を加えた。
試験例20:粉末溶解度試験
適当な容器に本発明化合物を適量入れ、各容器にJP-1液(塩化ナトリウム2.0g、塩酸7.0mLに水を加えて1000mLとする)、JP-2液(リン酸二水素カリウム3.40gおよび無水リン酸水素二ナトリウム3.55gを水に溶かし1000mLとしたもの1容量に水1容量を加えた)、20mmol/L タウロコール酸ナトリウム(TCA)/JP-2液(TCA1.08gにJP-2液を加え100mLとした)を200μLずつ添加した。試験液添加後に全量溶解した場合には、適宜、本発明化合物を追加した。密閉して37℃で1時間振とう後に濾過し、各濾液100μLにメタノール100μLを添加して2倍希釈を行った。希釈倍率は、必要に応じて変更した。気泡および析出物がないかを確認し、密閉して振とうした。絶対検量線法によりHPLCを用いて本発明化合物を定量した。なお、希釈濃度や希釈溶媒は、必要に応じて変更した。
適当な容器に本発明化合物を適量入れ、各容器にJP-1液(塩化ナトリウム2.0g、塩酸7.0mLに水を加えて1000mLとする)、JP-2液(リン酸二水素カリウム3.40gおよび無水リン酸水素二ナトリウム3.55gを水に溶かし1000mLとしたもの1容量に水1容量を加えた)、20mmol/L タウロコール酸ナトリウム(TCA)/JP-2液(TCA1.08gにJP-2液を加え100mLとした)を200μLずつ添加した。試験液添加後に全量溶解した場合には、適宜、本発明化合物を追加した。密閉して37℃で1時間振とう後に濾過し、各濾液100μLにメタノール100μLを添加して2倍希釈を行った。希釈倍率は、必要に応じて変更した。気泡および析出物がないかを確認し、密閉して振とうした。絶対検量線法によりHPLCを用いて本発明化合物を定量した。なお、希釈濃度や希釈溶媒は、必要に応じて変更した。
試験例21:目視溶解性試験
化合物約5mgを微量試験管3本に秤量し、各媒体(注射用水、生食注、0.5%ブドウ糖液)を化合物濃度20%になるように添加する。ボルテックスにて撹拌後、目視にて溶解の有無を確認する。溶解していればその媒体での溶解度を>20%とする。それら試験液に各媒体(注射用水、生食注、ブドウ糖液)を更に加えて化合物濃度10%の試験液を調製し、ボルテックスにて撹拌後、目視にて溶解の有無を確認する。溶解していればその媒体での溶解度を20%~10%とする。同様に5%濃度、2.5%濃度、1%濃度まで試験をし、1%濃度で溶解しない場合はその媒体での溶解度を<1%とする。1%濃度の試験液でのpHを測定し、記録する。なお、希釈濃度や希釈溶媒は、必要に応じて変更してもよい。
化合物約5mgを微量試験管3本に秤量し、各媒体(注射用水、生食注、0.5%ブドウ糖液)を化合物濃度20%になるように添加する。ボルテックスにて撹拌後、目視にて溶解の有無を確認する。溶解していればその媒体での溶解度を>20%とする。それら試験液に各媒体(注射用水、生食注、ブドウ糖液)を更に加えて化合物濃度10%の試験液を調製し、ボルテックスにて撹拌後、目視にて溶解の有無を確認する。溶解していればその媒体での溶解度を20%~10%とする。同様に5%濃度、2.5%濃度、1%濃度まで試験をし、1%濃度で溶解しない場合はその媒体での溶解度を<1%とする。1%濃度の試験液でのpHを測定し、記録する。なお、希釈濃度や希釈溶媒は、必要に応じて変更してもよい。
試験例22:pKa測定(キャピラリー電気泳動法 (capillary electrophoresis法,CE法)の測定方法)
キャピラリーゾーン電気泳動技術を用いた手法で,電解質を含む緩衝液中での各試料成分の自由泳動を利用した分離方法である。
pH2.5~11.5に調製した緩衝液が充填されたフューズドシリカキャピラリーに、化合物溶液を注入した後、キャピラリーに高電圧 (Inlet側+,Outlet側-) をかけると、化合物は緩衝液pHにおけるイオン化状態を反映した速度 (+チャージした化合物は速く、-チャージした化合物は遅く) で移動する。この化合物の移動時間と中性分子 (DMSO) の移動時間との差をpHに対してプロットし、フィッティングをかけてpKaを算出した。測定条件を以下に示す。
使用装置:Beckman P/ACEシステムMDQ PDA
泳動液:pH2.5~11.5 Buffer (10vol% MeOH含有)
サンプル溶液:Blank DMSO 10μL+注用水90μL混合
Sample 10mM DMSO stock solution 4μL + DMSO 6μL + 注用水 90μL
(メソッド)
キャピラリー :Fused silica capillary (BECKMAN COULTER,内径50 μm,全長30.2 cm,有効長20.0 cm)
印加電圧 :10kV (331 V/cm)
印加空気圧 :0.7 psi
キャピラリー温度 :25℃
電気浸透流マーカー :DMSO
検出 :紫外部多波長吸光検出 (測定波長;215 nm,238 nm)
試料注入 :加圧法 (0.5 psi,5 sec)
キャピラリーゾーン電気泳動技術を用いた手法で,電解質を含む緩衝液中での各試料成分の自由泳動を利用した分離方法である。
pH2.5~11.5に調製した緩衝液が充填されたフューズドシリカキャピラリーに、化合物溶液を注入した後、キャピラリーに高電圧 (Inlet側+,Outlet側-) をかけると、化合物は緩衝液pHにおけるイオン化状態を反映した速度 (+チャージした化合物は速く、-チャージした化合物は遅く) で移動する。この化合物の移動時間と中性分子 (DMSO) の移動時間との差をpHに対してプロットし、フィッティングをかけてpKaを算出した。測定条件を以下に示す。
使用装置:Beckman P/ACEシステムMDQ PDA
泳動液:pH2.5~11.5 Buffer (10vol% MeOH含有)
サンプル溶液:Blank DMSO 10μL+注用水90μL混合
Sample 10mM DMSO stock solution 4μL + DMSO 6μL + 注用水 90μL
(メソッド)
キャピラリー :Fused silica capillary (BECKMAN COULTER,内径50 μm,全長30.2 cm,有効長20.0 cm)
印加電圧 :10kV (331 V/cm)
印加空気圧 :0.7 psi
キャピラリー温度 :25℃
電気浸透流マーカー :DMSO
検出 :紫外部多波長吸光検出 (測定波長;215 nm,238 nm)
試料注入 :加圧法 (0.5 psi,5 sec)
試験例23:SDラットにおける骨格筋細胞内脂質(IMCL)の評価
化合物I-13が骨格筋細胞内脂質(IMCL)に与える影響を調べるため、SDラットに媒体(Vehicle)又は化合物I-13(20 mg/kg)を単回経口投与した24時間後に、イソフルラン麻酔下で動物用MRI(Varian MRI System 7T/210, Agilent Technologies)を用いて前脛骨筋のMRスペクトロスコピー(MRS)データを測定した。測定データはMRSデータ定量解析ソフト(LCModel)を用いて解析し、クレアチン比としてIMCL量を算出した。
(結果)
化合物I-13(20 mg/kg)は、SDラットにおいて有意なIMCLの減少作用を示した。試験例23の結果を図4に示す。
化合物I-13が骨格筋細胞内脂質(IMCL)に与える影響を調べるため、SDラットに媒体(Vehicle)又は化合物I-13(20 mg/kg)を単回経口投与した24時間後に、イソフルラン麻酔下で動物用MRI(Varian MRI System 7T/210, Agilent Technologies)を用いて前脛骨筋のMRスペクトロスコピー(MRS)データを測定した。測定データはMRSデータ定量解析ソフト(LCModel)を用いて解析し、クレアチン比としてIMCL量を算出した。
(結果)
化合物I-13(20 mg/kg)は、SDラットにおいて有意なIMCLの減少作用を示した。試験例23の結果を図4に示す。
試験例24:糖尿病モデルマウスにおける骨格筋細胞内脂質(IMCL)の評価
化合物I-12が糖尿病モデルマウスの骨格筋細胞内脂質(IMCL)に与える影響を調べるため、レプチン受容体欠損マウス(Leprdb/Leprdb, db/db;db/dbマウス)に媒体(Vehicle)又は化合物I-12(15 mg/kg)を単回経口投与した6時間後に、イソフルラン麻酔下で動物用MRI(Varian MRI System 7T/210, Agilent Technologies)を用いて前脛骨筋のMRスペクトロスコピー(MRS)データを測定した。測定データはMRSデータ定量解析ソフト(LCModel)を用いて解析し、クレアチン比としてIMCL量を算出した。
(結果)
化合物I-12(15 mg/kg)は、db/dbマウスにおいて有意なIMCLの減少作用を示した。試験例24の結果を図5に示す。
化合物I-12が糖尿病モデルマウスの骨格筋細胞内脂質(IMCL)に与える影響を調べるため、レプチン受容体欠損マウス(Leprdb/Leprdb, db/db;db/dbマウス)に媒体(Vehicle)又は化合物I-12(15 mg/kg)を単回経口投与した6時間後に、イソフルラン麻酔下で動物用MRI(Varian MRI System 7T/210, Agilent Technologies)を用いて前脛骨筋のMRスペクトロスコピー(MRS)データを測定した。測定データはMRSデータ定量解析ソフト(LCModel)を用いて解析し、クレアチン比としてIMCL量を算出した。
(結果)
化合物I-12(15 mg/kg)は、db/dbマウスにおいて有意なIMCLの減少作用を示した。試験例24の結果を図5に示す。
試験例25:糖尿病モデルラットにおける骨格筋細胞内脂質(IMCL)の評価
化合物I-12が糖尿病モデルラットの骨格筋細胞内脂質(IMCL)に与える影響を調べるため、自然発症糖尿病モデル(ZDFラット)に媒体(Vehicle)又は化合物I-12を1日2回経口投与し(1回目午前中投与:7 mg/kg;2回目午後投与:10 or 23 mg/kg)、初回投与から24時間後に、イソフルラン麻酔下で動物用MRI(Varian MRI System 7T/210, Agilent Technologies)を用いて前脛骨筋のMRスペクトロスコピー(MRS)データを測定した。測定データはMRSデータ定量解析ソフト(LCModel)を用いて解析し、クレアチン比としてIMCL量を算出した。
(結果)
化合物I-12は、ZDFラットにおいて有意なIMCLの減少作用を示した。試験例25の結果を図6に示す。
化合物I-12が糖尿病モデルラットの骨格筋細胞内脂質(IMCL)に与える影響を調べるため、自然発症糖尿病モデル(ZDFラット)に媒体(Vehicle)又は化合物I-12を1日2回経口投与し(1回目午前中投与:7 mg/kg;2回目午後投与:10 or 23 mg/kg)、初回投与から24時間後に、イソフルラン麻酔下で動物用MRI(Varian MRI System 7T/210, Agilent Technologies)を用いて前脛骨筋のMRスペクトロスコピー(MRS)データを測定した。測定データはMRSデータ定量解析ソフト(LCModel)を用いて解析し、クレアチン比としてIMCL量を算出した。
(結果)
化合物I-12は、ZDFラットにおいて有意なIMCLの減少作用を示した。試験例25の結果を図6に示す。
試験例26:高脂肪食負荷マウスにおける骨格筋細胞内脂質(IMCL)及び骨格筋細胞外脂質(EMCL)の評価
化合物I-14が高脂肪食負荷マウスの骨格筋細胞内脂質(IMCL)および骨格筋細胞外脂質(EMCL)に与える影響を調べるため、高脂肪食を27週間負荷したC57BL/6マウス(DIOマウス)に媒体(Vehicle)又は化合物I-14(5 mg/kg)を1日1回、5週間反復経口投与した後、イソフルラン麻酔下で動物用MRI(Varian MRI System 7T/210, Agilent Technologies)を用いて前脛骨筋のMRスペクトロスコピー(MRS)データを測定した。測定データはMRSデータ定量解析ソフト(LCModel)を用いて解析し、クレアチン比としてIMCL量およびEMCL量を算出した。
(結果)
化合物I-14は、DIOマウスにおいて有意なIMCLの減少作用を示した。一方でEMCLには有意な影響は認められなかった。試験例26の結果を図7に示す。
化合物I-14が高脂肪食負荷マウスの骨格筋細胞内脂質(IMCL)および骨格筋細胞外脂質(EMCL)に与える影響を調べるため、高脂肪食を27週間負荷したC57BL/6マウス(DIOマウス)に媒体(Vehicle)又は化合物I-14(5 mg/kg)を1日1回、5週間反復経口投与した後、イソフルラン麻酔下で動物用MRI(Varian MRI System 7T/210, Agilent Technologies)を用いて前脛骨筋のMRスペクトロスコピー(MRS)データを測定した。測定データはMRSデータ定量解析ソフト(LCModel)を用いて解析し、クレアチン比としてIMCL量およびEMCL量を算出した。
(結果)
化合物I-14は、DIOマウスにおいて有意なIMCLの減少作用を示した。一方でEMCLには有意な影響は認められなかった。試験例26の結果を図7に示す。
試験例27:ACC2ノックアウトマウスの骨格筋細胞内脂質(IMCL)及び骨格筋細胞外脂質(EMCL)の評価
ACC2の欠損が骨格筋細胞内脂質(IMCL)および骨格筋細胞外脂質(EMCL)に与える影響を調べるため、野生型マウス及びACC2ノックアウトマウスにおいて、高脂肪食負荷前、及び高脂肪食を3日間、12週間負荷した後、イソフルラン麻酔下で動物用MRI(Varian MRI System 7T/210, Agilent Technologies)を用いて前脛骨筋のMRスペクトロスコピー(MRS)データを測定した。測定データはMRSデータ定量解析ソフト(LCModel)を用いて解析し、クレアチン比としてIMCL量およびEMCL量を算出した。
(結果)
ACC2ノックアウトマウスでは、高脂肪食負荷前及び高脂肪食負荷後において、野生型マウスと比べ有意なIMCLの減少を示した。一方でEMCLは野生型マウスとACC2ノックアウトマウスで有意な差は認められなかった。試験例27の結果を図8に示す。
ACC2の欠損が骨格筋細胞内脂質(IMCL)および骨格筋細胞外脂質(EMCL)に与える影響を調べるため、野生型マウス及びACC2ノックアウトマウスにおいて、高脂肪食負荷前、及び高脂肪食を3日間、12週間負荷した後、イソフルラン麻酔下で動物用MRI(Varian MRI System 7T/210, Agilent Technologies)を用いて前脛骨筋のMRスペクトロスコピー(MRS)データを測定した。測定データはMRSデータ定量解析ソフト(LCModel)を用いて解析し、クレアチン比としてIMCL量およびEMCL量を算出した。
(結果)
ACC2ノックアウトマウスでは、高脂肪食負荷前及び高脂肪食負荷後において、野生型マウスと比べ有意なIMCLの減少を示した。一方でEMCLは野生型マウスとACC2ノックアウトマウスで有意な差は認められなかった。試験例27の結果を図8に示す。
試験例28:Atglノックアウトマウスにおける自発性運動能評価
本発明化合物が自発性運動能に与える影響を調べるため、Atglノックアウトマウスに媒体(Vehicle)又は本発明化合物を反復投与した後に12週齢において、SCANET MV-40(MELQUEST)を用いて自発性運動能を測定した。装置内に置かれたケージ内にマウスを静置し、30分間の運動能を測定した。
(結果)
化合物I-13(45mg/kg/10mL)は、Atglノックアウトマウスにおいて認められた自発性運動能の低下に対し有意な改善作用を示した(p<0.05、ウェルチの t 検定)。試験例28の結果を図9に示す。
本発明化合物が自発性運動能に与える影響を調べるため、Atglノックアウトマウスに媒体(Vehicle)又は本発明化合物を反復投与した後に12週齢において、SCANET MV-40(MELQUEST)を用いて自発性運動能を測定した。装置内に置かれたケージ内にマウスを静置し、30分間の運動能を測定した。
(結果)
化合物I-13(45mg/kg/10mL)は、Atglノックアウトマウスにおいて認められた自発性運動能の低下に対し有意な改善作用を示した(p<0.05、ウェルチの t 検定)。試験例28の結果を図9に示す。
試験例29:Atglノックアウトマウス、およびAtglとACC2のダブルノックアウトマウスにおける自発性運動能評価
ACC2の欠損が自発性運動能に与える影響を調べるため、12週齢の野生型マウス、Atglノックアウトマウス、AtglとACC2のダブルノックアウトマウスにおいて、SCANET MV-40(MELQUEST)を用いて自発性運動能を測定した。装置内に置かれたケージ内にマウスを静置し、30分間の運動能を測定した。
(結果)
ACCの欠損は、Atglノックアウトマウスにおいて認められた自発性運動能の低下に対し有意な改善作用を示した(p<0.05、ウェルチの t 検定)。試験例29の結果を図10に示す。
ACC2の欠損が自発性運動能に与える影響を調べるため、12週齢の野生型マウス、Atglノックアウトマウス、AtglとACC2のダブルノックアウトマウスにおいて、SCANET MV-40(MELQUEST)を用いて自発性運動能を測定した。装置内に置かれたケージ内にマウスを静置し、30分間の運動能を測定した。
(結果)
ACCの欠損は、Atglノックアウトマウスにおいて認められた自発性運動能の低下に対し有意な改善作用を示した(p<0.05、ウェルチの t 検定)。試験例29の結果を図10に示す。
以下に示す製剤例は例示にすぎないものであり、発明の範囲を何ら限定することを意図するものではない。
本発明の医薬組成物は、任意の従来の経路により、特に、経腸、例えば、経口で、例えば、錠剤またはカプセル剤の形態で、または非経口で、例えば注射液剤または懸濁剤の形態で、局所で、例えば、ローション剤、ゲル剤、軟膏剤またはクリーム剤の形態で、または経鼻形態または座剤形態で投与することができる。少なくとも1種の薬学的に許容される担体または希釈剤と一緒にして、遊離形態または薬学的に許容される塩の形態の化合物を含む本発明の医薬組成物は、従来の方法で、混合、造粒またはコーティング法によって製造することができる。例えば、経口用組成物としては、賦形剤、崩壊剤、結合剤、滑沢剤等および有効成分等を含有する錠剤、顆粒剤、カプセル剤とすることができる。また、注射用組成物としては、溶液剤または懸濁剤とすることができ、滅菌されていてもよく、また、保存剤、安定化剤、緩衝化剤等を含有してもよい。
本発明の医薬組成物は、任意の従来の経路により、特に、経腸、例えば、経口で、例えば、錠剤またはカプセル剤の形態で、または非経口で、例えば注射液剤または懸濁剤の形態で、局所で、例えば、ローション剤、ゲル剤、軟膏剤またはクリーム剤の形態で、または経鼻形態または座剤形態で投与することができる。少なくとも1種の薬学的に許容される担体または希釈剤と一緒にして、遊離形態または薬学的に許容される塩の形態の化合物を含む本発明の医薬組成物は、従来の方法で、混合、造粒またはコーティング法によって製造することができる。例えば、経口用組成物としては、賦形剤、崩壊剤、結合剤、滑沢剤等および有効成分等を含有する錠剤、顆粒剤、カプセル剤とすることができる。また、注射用組成物としては、溶液剤または懸濁剤とすることができ、滅菌されていてもよく、また、保存剤、安定化剤、緩衝化剤等を含有してもよい。
本発明の心疾患、心疾患合併症、骨格筋の疾患および骨格筋の病態の少なくとも1つの治療および/または予防方法、およびそれに用いる治療用医薬組成物は、活性成分の式(I)で示される化合物またはその製薬上許容される塩の所定量を、患者に対して投与することにより、優れた治療効果を示すと考えられる。また、式(I)で示される化合物またはその製薬上許容される塩の投与による血漿トリグリセリド上昇または血小板濃度の低下などの副作用は認められず、極めて安全に適用することができ、長期間の投与にも適していることから、本発明の治療および/または予防方法および治療用医薬組成物は、きわめて優れた治療および/または予防方法および治療用医薬組成物である。
Claims (29)
- 式(I):

(式中、R1はハロアルキルまたは非芳香族炭素環式基であり、
R2は水素原子またはハロゲンであり、
R3はハロゲンであり、
環Aは、式:

で示される基であり、
-L1-は-O-(CH2)-、-(CH2)2-、-(CH2)-(CF2)-または-(CF2)-(CH2)-(ここで、左の結合手は環Aに結合し、右の結合手は式:

で示される基に結合する。)であり、
R4はアルキルまたはハロアルキルであり、
R5はアルキルカルボニルまたはカルバモイルである。)で示される化合物またはその製薬上許容される塩を含有する、心疾患および心疾患合併症の少なくとも1つを治療および/または予防するための、医薬組成物。 - R1が非芳香族炭素環式基である、請求項1記載の医薬組成物。
- R2が水素原子である、請求項1または2記載の医薬組成物。
- 環Aが、式:

で示される基である、請求項1~3のいずれかに記載の医薬組成物。 - -L1-が-O-(CH2)-または-(CH2)2-である、請求項1~4のいずれかに記載の医薬組成物。
- R4がアルキルである、請求項1~5のいずれかに記載の医薬組成物。
- R5がメチルカルボニルまたはカルバモイルである、請求項1~6のいずれかに記載の医薬組成物。
- 式(I)で示される化合物またはその製薬上許容される塩が、下式:

からなる群から選択される化合物またはその製薬上許容される塩である、請求項1記載の医薬組成物。 - 前記心疾患および心疾患合併症が、心不全、狭心症、心筋梗塞、心筋症、不整脈、冠動脈疾患および骨格筋ミオパチーからなる群より選択される少なくとも1つの疾患である、請求項1~8のいずれかに記載の記載の医薬組成物。
- 心筋細胞内、血管平滑筋細胞内、内皮細胞内、骨格筋細胞内、多型核白血球、腎尿細管上皮細胞内および膵島細胞内の少なくとも1つに中性脂肪が蓄積されることにより引き起こされる疾患を治療および/または予防するための、請求項1~8のいずれかに記載の化合物を含有する医薬組成物。
- 前記疾患が、心不全、狭心症、心筋梗塞、心筋症、不整脈、冠動脈疾患、骨格筋ミオパチー、慢性腎臓病および耐糖能障害からなる群より選択される少なくとも1つの疾患である、請求項10記載の記載の医薬組成物。
- 前記医薬組成物の投与によって、血漿トリグリセリド上昇の副作用を伴わない、請求項1~11のいずれかに記載の医薬組成物。
- 前記医薬組成物の投与によって、血小板濃度の低下の副作用を伴わない、請求項1~11のいずれかに記載の医薬組成物。
- 式(I):

(式中、R1はハロアルキルまたは非芳香族炭素環式基であり、
R2は水素原子またはハロゲンであり、
R3はハロゲンであり、
環Aは、式:

で示される基であり、
-L1-は-O-(CH2)-、-(CH2)2-、-(CH2)-(CF2)-または-(CF2)-(CH2)-(ここで、左の結合手は環Aに結合し、右の結合手は式:

で示される基に結合する。)であり、
R4はアルキルまたはハロアルキルであり、
R5はアルキルカルボニルまたはカルバモイルである。)で示される化合物またはその製薬上許容される塩を含有する、骨格筋の疾患または病態を治療および/または予防するための、医薬組成物。 - R1が非芳香族炭素環式基である、請求項14記載の医薬組成物。
- R2が水素原子である、請求項14または15記載の医薬組成物。
- 環Aが、式:

で示される基である、請求項14~16のいずれかに記載の医薬組成物。 - -L1-が-O-(CH2)-または-(CH2)2-である、請求項14~17のいずれかに記載の医薬組成物。
- R4がアルキルである、請求項14~18のいずれかに記載の医薬組成物。
- R5がメチルカルボニルまたはカルバモイルである、請求項14~19のいずれかに記載の医薬組成物。
- 式(I)で示される化合物またはその製薬上許容される塩が、下式:

からなる群から選択される化合物またはその製薬上許容される塩である、請求項14記載の医薬組成物。 - 前記骨格筋の疾患または病態が、骨格筋細胞における中性脂肪の蓄積によって引き起こされるものである、請求項14~21のいずれかに記載の医薬組成物。
- 前記骨格筋の疾患または病態が、骨格筋ミオパチーまたはサルコペニアである、請求項14~22のいずれかに記載の医薬組成物。
- 前記骨格筋の疾患または病態の病理および/または症候がACC2活性の寄与によるものである、請求項14~23のいずれかに記載の医薬組成物。
- 前記ACC2活性が糖尿病、肥満および肝硬変からなる群から選択される状態に寄与する、請求項24記載の医薬組成物。
- 前記骨格筋の疾患または病態が、糖尿病、肥満および肝硬変からなる群から選択される状態と関連している、請求項14~25のいずれかに記載の医薬組成物。
- 前記骨格筋の疾患または病態が、肝硬変と関連している、請求項26記載の医薬組成物。
- 前記医薬組成物が、前記骨格筋の疾患または病態を有する対象の骨格筋機能を維持または増加させる、請求項14~27のいずれかに記載の医薬組成物。
。 - 前記医薬組成物が、前記骨格筋の疾患または病態を有する対象の骨格筋の回復を促進する、請求項14~27のいずれかに記載の医薬組成物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021188247 | 2021-11-19 | ||
| JP2021-188247 | 2021-11-19 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023090411A1 true WO2023090411A1 (ja) | 2023-05-25 |
Family
ID=86397177
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/042796 WO2023090411A1 (ja) | 2021-11-19 | 2022-11-18 | 心疾患または骨格筋の疾患の治療用医薬 |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2023090411A1 (ja) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015056782A1 (ja) * | 2013-10-17 | 2015-04-23 | 塩野義製薬株式会社 | 新規アルキレン誘導体 |
| WO2016159082A1 (ja) * | 2015-03-30 | 2016-10-06 | 塩野義製薬株式会社 | 9員縮合環誘導体 |
| WO2021235508A1 (ja) * | 2020-05-21 | 2021-11-25 | 塩野義製薬株式会社 | 脂肪性肝疾患の治療用医薬 |
-
2022
- 2022-11-18 WO PCT/JP2022/042796 patent/WO2023090411A1/ja unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015056782A1 (ja) * | 2013-10-17 | 2015-04-23 | 塩野義製薬株式会社 | 新規アルキレン誘導体 |
| WO2016159082A1 (ja) * | 2015-03-30 | 2016-10-06 | 塩野義製薬株式会社 | 9員縮合環誘導体 |
| WO2021235508A1 (ja) * | 2020-05-21 | 2021-11-25 | 塩野義製薬株式会社 | 脂肪性肝疾患の治療用医薬 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI501949B (zh) | 硫乙酸酯/鹽組合物及其使用方法 | |
| CN104470923B (zh) | 选择性PI3K δ抑制剂 | |
| CN107921035B (zh) | 高苯丙氨酸血症及其治疗 | |
| EP3653625B1 (en) | Fused ring derivative having mgat-2 inhibitory activity | |
| JP2017526726A (ja) | ヘテロ環化合物およびその用途 | |
| UA120353C2 (uk) | Модулятори натрієвого каналу для лікування болю і діабету | |
| KR20170095302A (ko) | 선택적 s1p1 수용체 아고니스트를 포함하는 약학 조합물 | |
| US20070185195A1 (en) | Medicinal compositions containing 6-hydroxybenzbromarone or salts thereof | |
| WO2014160430A1 (en) | Small molecule lrrk2 and erk5 inhibitors | |
| JP2020111571A (ja) | Mgat2阻害活性を有する縮合環誘導体を含有する医薬組成物 | |
| WO2023090411A1 (ja) | 心疾患または骨格筋の疾患の治療用医薬 | |
| WO2021235508A1 (ja) | 脂肪性肝疾患の治療用医薬 | |
| JPWO2017110841A1 (ja) | Mgat2阻害活性を有する非芳香族複素環誘導体 | |
| RU2560175C2 (ru) | Антикоагулянтные соединения и их применение | |
| US10266490B2 (en) | Radioprotector compounds | |
| BR112020010120A2 (pt) | compostos derivados de pirimidina e sua composição farmacêutica | |
| US11974988B2 (en) | Neuroprotective compositions and methods of using the same | |
| CN117321050A (zh) | 修饰的膜铁转运蛋白抑制剂 | |
| TW201818964A (zh) | 使用色胺酸羥化酶抑制劑之方法 | |
| BR112021012428A2 (pt) | Derivado de dihidropirazolopirazinona tendo atividade inibitória de mgat2 | |
| JP6999331B2 (ja) | 新規糖尿病治療剤 | |
| JP2022500445A (ja) | Gabaa受容体リガンド | |
| US20230134958A1 (en) | New medical treatment | |
| US10689394B2 (en) | Thieno[2,3-b]pyridine derivative, quinoline derivative, and use thereof | |
| CA3137019A1 (en) | New medical use |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22895692 Country of ref document: EP Kind code of ref document: A1 |