WO2023085232A1 - 改良されたライブラリー調製方法 - Google Patents
改良されたライブラリー調製方法 Download PDFInfo
- Publication number
- WO2023085232A1 WO2023085232A1 PCT/JP2022/041394 JP2022041394W WO2023085232A1 WO 2023085232 A1 WO2023085232 A1 WO 2023085232A1 JP 2022041394 W JP2022041394 W JP 2022041394W WO 2023085232 A1 WO2023085232 A1 WO 2023085232A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- acid
- stranded cdna
- rna
- double
- library
- Prior art date
Links
- 238000002360 preparation method Methods 0.000 title claims abstract description 33
- 238000000034 method Methods 0.000 claims abstract description 84
- 230000002194 synthesizing effect Effects 0.000 claims abstract description 14
- 239000002299 complementary DNA Substances 0.000 claims abstract 10
- 239000002253 acid Substances 0.000 claims description 49
- 229920000642 polymer Polymers 0.000 claims description 40
- 150000003839 salts Chemical class 0.000 claims description 30
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 claims description 28
- 150000007523 nucleic acids Chemical class 0.000 claims description 27
- 102000039446 nucleic acids Human genes 0.000 claims description 19
- 108020004707 nucleic acids Proteins 0.000 claims description 19
- 238000010438 heat treatment Methods 0.000 claims description 15
- 239000011780 sodium chloride Substances 0.000 claims description 14
- 108091005804 Peptidases Proteins 0.000 claims description 13
- 150000003841 chloride salts Chemical class 0.000 claims description 12
- 102000004594 DNA Polymerase I Human genes 0.000 claims description 10
- 108010017826 DNA Polymerase I Proteins 0.000 claims description 10
- 108010067770 Endopeptidase K Proteins 0.000 claims description 9
- 239000004365 Protease Substances 0.000 claims description 9
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 claims description 8
- 235000019419 proteases Nutrition 0.000 claims description 8
- 229920001519 homopolymer Polymers 0.000 claims description 7
- 102000035195 Peptidases Human genes 0.000 claims description 5
- 108090000787 Subtilisin Proteins 0.000 claims description 4
- 108091034057 RNA (poly(A)) Proteins 0.000 claims description 3
- JSRLJPSBLDHEIO-SHYZEUOFSA-N dUMP Chemical compound O1[C@H](COP(O)(O)=O)[C@@H](O)C[C@@H]1N1C(=O)NC(=O)C=C1 JSRLJPSBLDHEIO-SHYZEUOFSA-N 0.000 claims description 3
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 88
- 239000000243 solution Substances 0.000 description 69
- 108020004414 DNA Proteins 0.000 description 42
- 235000002639 sodium chloride Nutrition 0.000 description 42
- 238000006243 chemical reaction Methods 0.000 description 40
- 239000000203 mixture Substances 0.000 description 32
- 238000003786 synthesis reaction Methods 0.000 description 30
- 238000010839 reverse transcription Methods 0.000 description 25
- 238000000746 purification Methods 0.000 description 24
- 102000004190 Enzymes Human genes 0.000 description 22
- 108090000790 Enzymes Proteins 0.000 description 22
- 229940088598 enzyme Drugs 0.000 description 22
- 102100034343 Integrase Human genes 0.000 description 21
- 230000000694 effects Effects 0.000 description 21
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 17
- 230000015572 biosynthetic process Effects 0.000 description 15
- 238000011534 incubation Methods 0.000 description 15
- 238000007481 next generation sequencing Methods 0.000 description 15
- 230000002779 inactivation Effects 0.000 description 14
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 12
- 102000016911 Deoxyribonucleases Human genes 0.000 description 11
- 108010053770 Deoxyribonucleases Proteins 0.000 description 11
- 239000002585 base Substances 0.000 description 11
- 102000053602 DNA Human genes 0.000 description 10
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 8
- GRSZFWQUAKGDAV-UHFFFAOYSA-N Inosinic acid Natural products OC1C(O)C(COP(O)(O)=O)OC1N1C(NC=NC2=O)=C2N=C1 GRSZFWQUAKGDAV-UHFFFAOYSA-N 0.000 description 7
- 238000007792 addition Methods 0.000 description 7
- 150000001413 amino acids Chemical group 0.000 description 7
- 239000000872 buffer Substances 0.000 description 7
- 239000003085 diluting agent Substances 0.000 description 7
- 235000013902 inosinic acid Nutrition 0.000 description 7
- 239000004245 inosinic acid Substances 0.000 description 7
- 229940028843 inosinic acid Drugs 0.000 description 7
- 239000000523 sample Substances 0.000 description 7
- AUHDWARTFSKSAC-HEIFUQTGSA-N (2S,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)-2-(6-oxo-1H-purin-9-yl)oxolane-2-carboxylic acid Chemical compound [C@]1([C@H](O)[C@H](O)[C@@H](CO)O1)(N1C=NC=2C(O)=NC=NC12)C(=O)O AUHDWARTFSKSAC-HEIFUQTGSA-N 0.000 description 6
- 101710203526 Integrase Proteins 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- 239000000539 dimer Substances 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- 238000003199 nucleic acid amplification method Methods 0.000 description 6
- 239000001103 potassium chloride Substances 0.000 description 6
- 235000011164 potassium chloride Nutrition 0.000 description 6
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 description 6
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 6
- 102000052510 DNA-Binding Proteins Human genes 0.000 description 5
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 5
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 5
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 5
- 230000003321 amplification Effects 0.000 description 5
- 230000000295 complement effect Effects 0.000 description 5
- SUYVUBYJARFZHO-RRKCRQDMSA-N dATP Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@H]1C[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 SUYVUBYJARFZHO-RRKCRQDMSA-N 0.000 description 5
- RGWHQCVHVJXOKC-SHYZEUOFSA-N dCTP Chemical compound O=C1N=C(N)C=CN1[C@@H]1O[C@H](CO[P@](O)(=O)O[P@](O)(=O)OP(O)(O)=O)[C@@H](O)C1 RGWHQCVHVJXOKC-SHYZEUOFSA-N 0.000 description 5
- HAAZLUGHYHWQIW-KVQBGUIXSA-N dGTP Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@H]1C[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 HAAZLUGHYHWQIW-KVQBGUIXSA-N 0.000 description 5
- 230000000593 degrading effect Effects 0.000 description 5
- 239000005547 deoxyribonucleotide Substances 0.000 description 5
- 239000012634 fragment Substances 0.000 description 5
- 230000002934 lysing effect Effects 0.000 description 5
- 229910052700 potassium Inorganic materials 0.000 description 5
- 239000011591 potassium Substances 0.000 description 5
- 239000000758 substrate Substances 0.000 description 5
- AHCYMLUZIRLXAA-SHYZEUOFSA-N Deoxyuridine 5'-triphosphate Chemical compound O1[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)C[C@@H]1N1C(=O)NC(=O)C=C1 AHCYMLUZIRLXAA-SHYZEUOFSA-N 0.000 description 4
- 102000003960 Ligases Human genes 0.000 description 4
- 108090000364 Ligases Proteins 0.000 description 4
- 230000027455 binding Effects 0.000 description 4
- 230000006037 cell lysis Effects 0.000 description 4
- 239000000356 contaminant Substances 0.000 description 4
- NHVNXKFIZYSCEB-XLPZGREQSA-N dTTP Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)C1 NHVNXKFIZYSCEB-XLPZGREQSA-N 0.000 description 4
- 125000002637 deoxyribonucleotide group Chemical group 0.000 description 4
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 4
- 229910001629 magnesium chloride Inorganic materials 0.000 description 4
- 235000011147 magnesium chloride Nutrition 0.000 description 4
- -1 polyoxyethylene lauryl ether Polymers 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 235000018102 proteins Nutrition 0.000 description 4
- 108090000623 proteins and genes Proteins 0.000 description 4
- 102000004169 proteins and genes Human genes 0.000 description 4
- 238000012163 sequencing technique Methods 0.000 description 4
- 229930183912 Cytidylic acid Natural products 0.000 description 3
- 101710116602 DNA-Binding protein G5P Proteins 0.000 description 3
- 238000011530 RNeasy Mini Kit Methods 0.000 description 3
- 101710162453 Replication factor A Proteins 0.000 description 3
- 101710176758 Replication protein A 70 kDa DNA-binding subunit Proteins 0.000 description 3
- 101710176276 SSB protein Proteins 0.000 description 3
- 238000012300 Sequence Analysis Methods 0.000 description 3
- 101710126859 Single-stranded DNA-binding protein Proteins 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 229910052925 anhydrite Inorganic materials 0.000 description 3
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 3
- 239000005018 casein Substances 0.000 description 3
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 3
- 235000021240 caseins Nutrition 0.000 description 3
- 239000013592 cell lysate Substances 0.000 description 3
- 150000001805 chlorine compounds Chemical class 0.000 description 3
- IERHLVCPSMICTF-XVFCMESISA-N cytidine 5'-monophosphate Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(O)=O)O1 IERHLVCPSMICTF-XVFCMESISA-N 0.000 description 3
- IERHLVCPSMICTF-UHFFFAOYSA-N cytidine monophosphate Natural products O=C1N=C(N)C=CN1C1C(O)C(O)C(COP(O)(O)=O)O1 IERHLVCPSMICTF-UHFFFAOYSA-N 0.000 description 3
- GYOZYWVXFNDGLU-XLPZGREQSA-N dTMP Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)C1 GYOZYWVXFNDGLU-XLPZGREQSA-N 0.000 description 3
- 239000002773 nucleotide Substances 0.000 description 3
- 125000003729 nucleotide group Chemical group 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 239000003161 ribonuclease inhibitor Substances 0.000 description 3
- GUAHPAJOXVYFON-ZETCQYMHSA-N (8S)-8-amino-7-oxononanoic acid zwitterion Chemical compound C[C@H](N)C(=O)CCCCCC(O)=O GUAHPAJOXVYFON-ZETCQYMHSA-N 0.000 description 2
- NCMVOABPESMRCP-SHYZEUOFSA-N 2'-deoxycytosine 5'-monophosphate Chemical compound O=C1N=C(N)C=CN1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)C1 NCMVOABPESMRCP-SHYZEUOFSA-N 0.000 description 2
- LTFMZDNNPPEQNG-KVQBGUIXSA-N 2'-deoxyguanosine 5'-monophosphate Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@H]1C[C@H](O)[C@@H](COP(O)(O)=O)O1 LTFMZDNNPPEQNG-KVQBGUIXSA-N 0.000 description 2
- 241000894006 Bacteria Species 0.000 description 2
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 2
- 241000131500 Chionoecetes opilio Species 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 108700020911 DNA-Binding Proteins Proteins 0.000 description 2
- 241000238557 Decapoda Species 0.000 description 2
- 241000206602 Eukaryota Species 0.000 description 2
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- 229910021380 Manganese Chloride Inorganic materials 0.000 description 2
- GLFNIEUTAYBVOC-UHFFFAOYSA-L Manganese chloride Chemical compound Cl[Mn]Cl GLFNIEUTAYBVOC-UHFFFAOYSA-L 0.000 description 2
- SEQKRHFRPICQDD-UHFFFAOYSA-N N-tris(hydroxymethyl)methylglycine Chemical compound OCC(CO)(CO)[NH2+]CC([O-])=O SEQKRHFRPICQDD-UHFFFAOYSA-N 0.000 description 2
- 101710163270 Nuclease Proteins 0.000 description 2
- 241000209094 Oryza Species 0.000 description 2
- 235000007164 Oryza sativa Nutrition 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 239000007997 Tricine buffer Substances 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 229910001514 alkali metal chloride Inorganic materials 0.000 description 2
- 229910001617 alkaline earth metal chloride Inorganic materials 0.000 description 2
- 238000000137 annealing Methods 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 239000007853 buffer solution Substances 0.000 description 2
- 238000010804 cDNA synthesis Methods 0.000 description 2
- 230000003196 chaotropic effect Effects 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- LTFMZDNNPPEQNG-UHFFFAOYSA-N deoxyguanylic acid Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1CC(O)C(COP(O)(O)=O)O1 LTFMZDNNPPEQNG-UHFFFAOYSA-N 0.000 description 2
- 238000006911 enzymatic reaction Methods 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- RQFCJASXJCIDSX-UUOKFMHZSA-N guanosine 5'-monophosphate Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H]1O RQFCJASXJCIDSX-UUOKFMHZSA-N 0.000 description 2
- 235000013928 guanylic acid Nutrition 0.000 description 2
- 239000004226 guanylic acid Substances 0.000 description 2
- 239000012139 lysis buffer Substances 0.000 description 2
- 239000011565 manganese chloride Substances 0.000 description 2
- 235000002867 manganese chloride Nutrition 0.000 description 2
- 229940099607 manganese chloride Drugs 0.000 description 2
- 238000006116 polymerization reaction Methods 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- 229940024999 proteolytic enzymes for treatment of wounds and ulcers Drugs 0.000 description 2
- 235000009566 rice Nutrition 0.000 description 2
- 108700014590 single-stranded DNA binding proteins Proteins 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 230000005758 transcription activity Effects 0.000 description 2
- 239000001226 triphosphate Substances 0.000 description 2
- 235000011178 triphosphate Nutrition 0.000 description 2
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 2
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 1
- 108020004465 16S ribosomal RNA Proteins 0.000 description 1
- 108020004463 18S ribosomal RNA Proteins 0.000 description 1
- KHWCHTKSEGGWEX-RRKCRQDMSA-N 2'-deoxyadenosine 5'-monophosphate Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@H]1C[C@H](O)[C@@H](COP(O)(O)=O)O1 KHWCHTKSEGGWEX-RRKCRQDMSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- JYCQQPHGFMYQCF-UHFFFAOYSA-N 4-tert-Octylphenol monoethoxylate Chemical compound CC(C)(C)CC(C)(C)C1=CC=C(OCCO)C=C1 JYCQQPHGFMYQCF-UHFFFAOYSA-N 0.000 description 1
- 241000713838 Avian myeloblastosis virus Species 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- LZZYPRNAOMGNLH-UHFFFAOYSA-M Cetrimonium bromide Chemical compound [Br-].CCCCCCCCCCCCCCCC[N+](C)(C)C LZZYPRNAOMGNLH-UHFFFAOYSA-M 0.000 description 1
- 241001529572 Chaceon affinis Species 0.000 description 1
- UDMBCSSLTHHNCD-UHFFFAOYSA-N Coenzym Q(11) Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(O)=O)C(O)C1O UDMBCSSLTHHNCD-UHFFFAOYSA-N 0.000 description 1
- 108700036248 MT-RNR1 Proteins 0.000 description 1
- 241001124325 Marsupenaeus japonicus Species 0.000 description 1
- 241000713869 Moloney murine leukemia virus Species 0.000 description 1
- 108091028043 Nucleic acid sequence Proteins 0.000 description 1
- 108091034117 Oligonucleotide Proteins 0.000 description 1
- 241000238070 Pandalus borealis Species 0.000 description 1
- 241000238124 Paralithodes camtschaticus Species 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical group OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 239000013614 RNA sample Substances 0.000 description 1
- 238000003559 RNA-seq method Methods 0.000 description 1
- 102000001218 Rec A Recombinases Human genes 0.000 description 1
- 108010055016 Rec A Recombinases Proteins 0.000 description 1
- 102000006382 Ribonucleases Human genes 0.000 description 1
- 108010083644 Ribonucleases Proteins 0.000 description 1
- 108020004682 Single-Stranded DNA Proteins 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 241000727771 Solenocera melantho Species 0.000 description 1
- UZMAPBJVXOGOFT-UHFFFAOYSA-N Syringetin Natural products COC1=C(O)C(OC)=CC(C2=C(C(=O)C3=C(O)C=C(O)C=C3O2)O)=C1 UZMAPBJVXOGOFT-UHFFFAOYSA-N 0.000 description 1
- 101150104425 T4 gene Proteins 0.000 description 1
- 102000008579 Transposases Human genes 0.000 description 1
- 108010020764 Transposases Proteins 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 1
- DJJCXFVJDGTHFX-UHFFFAOYSA-N Uridinemonophosphate Natural products OC1C(O)C(COP(O)(O)=O)OC1N1C(=O)NC(=O)C=C1 DJJCXFVJDGTHFX-UHFFFAOYSA-N 0.000 description 1
- FOGRQMPFHUHIGU-UHFFFAOYSA-N Uridylic acid Natural products OC1C(OP(O)(O)=O)C(CO)OC1N1C(=O)NC(=O)C=C1 FOGRQMPFHUHIGU-UHFFFAOYSA-N 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- UDMBCSSLTHHNCD-KQYNXXCUSA-N adenosine 5'-monophosphate Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H]1O UDMBCSSLTHHNCD-KQYNXXCUSA-N 0.000 description 1
- 229950006790 adenosine phosphate Drugs 0.000 description 1
- 230000006154 adenylylation Effects 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 239000003945 anionic surfactant Substances 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 230000008033 biological extinction Effects 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 239000003093 cationic surfactant Substances 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000003759 clinical diagnosis Methods 0.000 description 1
- 238000012398 clinical drug development Methods 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- KHWCHTKSEGGWEX-UHFFFAOYSA-N deoxyadenylic acid Natural products C1=NC=2C(N)=NC=NC=2N1C1CC(O)C(COP(O)(O)=O)O1 KHWCHTKSEGGWEX-UHFFFAOYSA-N 0.000 description 1
- 229960003964 deoxycholic acid Drugs 0.000 description 1
- 239000005549 deoxyribonucleoside Substances 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- KCFYHBSOLOXZIF-UHFFFAOYSA-N dihydrochrysin Natural products COC1=C(O)C(OC)=CC(C2OC3=CC(O)=CC(O)=C3C(=O)C2)=C1 KCFYHBSOLOXZIF-UHFFFAOYSA-N 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 238000012869 ethanol precipitation Methods 0.000 description 1
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 1
- 238000005194 fractionation Methods 0.000 description 1
- 238000013467 fragmentation Methods 0.000 description 1
- 238000006062 fragmentation reaction Methods 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 150000002500 ions Chemical group 0.000 description 1
- XIXADJRWDQXREU-UHFFFAOYSA-M lithium acetate Chemical compound [Li+].CC([O-])=O XIXADJRWDQXREU-UHFFFAOYSA-M 0.000 description 1
- 229940071257 lithium acetate Drugs 0.000 description 1
- MHCFAGZWMAWTNR-UHFFFAOYSA-M lithium perchlorate Chemical compound [Li+].[O-]Cl(=O)(=O)=O MHCFAGZWMAWTNR-UHFFFAOYSA-M 0.000 description 1
- 229910001486 lithium perchlorate Inorganic materials 0.000 description 1
- 229910003002 lithium salt Inorganic materials 0.000 description 1
- 159000000002 lithium salts Chemical class 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- UEGPKNKPLBYCNK-UHFFFAOYSA-L magnesium acetate Chemical compound [Mg+2].CC([O-])=O.CC([O-])=O UEGPKNKPLBYCNK-UHFFFAOYSA-L 0.000 description 1
- 239000011654 magnesium acetate Substances 0.000 description 1
- 235000011285 magnesium acetate Nutrition 0.000 description 1
- 229940069446 magnesium acetate Drugs 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 229940071125 manganese acetate Drugs 0.000 description 1
- 229940099596 manganese sulfate Drugs 0.000 description 1
- 239000011702 manganese sulphate Substances 0.000 description 1
- 235000007079 manganese sulphate Nutrition 0.000 description 1
- UOGMEBQRZBEZQT-UHFFFAOYSA-L manganese(2+);diacetate Chemical compound [Mn+2].CC([O-])=O.CC([O-])=O UOGMEBQRZBEZQT-UHFFFAOYSA-L 0.000 description 1
- SQQMAOCOWKFBNP-UHFFFAOYSA-L manganese(II) sulfate Chemical compound [Mn+2].[O-]S([O-])(=O)=O SQQMAOCOWKFBNP-UHFFFAOYSA-L 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 239000002736 nonionic surfactant Substances 0.000 description 1
- 229920002113 octoxynol Polymers 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 210000002826 placenta Anatomy 0.000 description 1
- 229940115272 polyinosinic:polycytidylic acid Drugs 0.000 description 1
- 229920000259 polyoxyethylene lauryl ether Polymers 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- 229960004109 potassium acetate Drugs 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- OTYBMLCTZGSZBG-UHFFFAOYSA-L potassium sulfate Chemical compound [K+].[K+].[O-]S([O-])(=O)=O OTYBMLCTZGSZBG-UHFFFAOYSA-L 0.000 description 1
- 229910052939 potassium sulfate Inorganic materials 0.000 description 1
- 235000011151 potassium sulphates Nutrition 0.000 description 1
- 239000013615 primer Substances 0.000 description 1
- 239000002987 primer (paints) Substances 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000002250 progressing effect Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 230000004845 protein aggregation Effects 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000011896 sensitive detection Methods 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 229960004249 sodium acetate Drugs 0.000 description 1
- NRHMKIHPTBHXPF-TUJRSCDTSA-M sodium cholate Chemical compound [Na+].C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC([O-])=O)C)[C@@]2(C)[C@@H](O)C1 NRHMKIHPTBHXPF-TUJRSCDTSA-M 0.000 description 1
- FHHPUSMSKHSNKW-SMOYURAASA-M sodium deoxycholate Chemical compound [Na+].C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC([O-])=O)C)[C@@]2(C)[C@@H](O)C1 FHHPUSMSKHSNKW-SMOYURAASA-M 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 238000010257 thawing Methods 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 150000003582 thiophosphoric acids Chemical group 0.000 description 1
- 229910021381 transition metal chloride Inorganic materials 0.000 description 1
- 125000002264 triphosphate group Chemical class [H]OP(=O)(O[H])OP(=O)(O[H])OP(=O)(O[H])O* 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- DJJCXFVJDGTHFX-XVFCMESISA-N uridine 5'-monophosphate Chemical compound O[C@@H]1[C@H](O)[C@@H](COP(O)(O)=O)O[C@H]1N1C(=O)NC(=O)C=C1 DJJCXFVJDGTHFX-XVFCMESISA-N 0.000 description 1
- 239000002888 zwitterionic surfactant Substances 0.000 description 1
Images
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6806—Preparing nucleic acids for analysis, e.g. for polymerase chain reaction [PCR] assay
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B40/00—Libraries per se, e.g. arrays, mixtures
- C40B40/04—Libraries containing only organic compounds
- C40B40/06—Libraries containing nucleotides or polynucleotides, or derivatives thereof
Definitions
- the present invention relates to a library preparation method.
- next-generation sequencing enables advanced and high-speed processing that enables simultaneous sequencing of multiple individuals, and is a powerful analysis technology that contributes to medical technology such as clinical diagnosis and drug development, and life science fields such as agricultural technology and basic research. is.
- next-generation sequencing it is usually necessary to prepare a library consisting of nucleic acid fragments with adapter sequences added to the ends that can function as binding sites for flow cells or as primer binding sites.
- RNA as a template When preparing a library using RNA as a template, a method of purifying the generated double-stranded cDNA solution and then preparing the library is carried out.
- the RT-RamDA method amplifies cDNA using RNA as a template by incubating a mixture containing template RNA, primers, DNA strand-specific RNA:DNA hybrid strandase, RNase H minus reverse transcriptase, and substrate.
- it is an amplification reverse transcription method that makes a library
- a method in which double-stranded cDNA is generated from single-stranded cDNA and the solution is purified before library preparation is started Patent Document 1, Non-Patent Document 1) Reference 1).
- the double-stranded cDNA solution contains cell-derived contaminants, enzymes, primers, dNTPs, salts, etc. used in the reverse transcription reaction, which can inhibit the reaction in the next step.
- substances in these solutions can significantly inhibit enzymatic reactions used for library preparation.
- purification work is complicated and workability is poor.
- a reduction in yield also occurs due to the presence of the purification step. For these reasons, it is desirable to simplify the purification process.
- a main object of the present invention is to provide a more efficient method for library preparation.
- the inventors of the present invention have found that even with a small amount of template RNA, a sufficient library can be obtained by directly bringing the generated double-stranded cDNA into the library preparation without purifying it. It was found that yields were obtained.
- the present invention was completed by further studies based on the findings.
- a library preparation method comprising the following steps (a), (b), and (c): (a) a step of synthesizing a single-stranded cDNA from 10 pg or more of template RNA; (b) synthesizing double-stranded cDNA from said single-stranded cDNA; and (c) preparing a library using said non-purified double-stranded cDNA.
- step (b) is performed in the presence of 12 mM or less sodium chloride.
- step (b) is performed in the presence of 12 mM or less sodium chloride.
- the nucleic acid polymer is polyinosinic acid, polycytidylic acid, polyguanylic acid, polyadenylic acid, polythymidylic acid, polyuridylic acid, polydeoxyinosinic acid, polydeoxycytidylic acid, polydeoxyguanylic acid, polydeoxyadenylic acid, polydeoxythymidylic acid, poly Item 5.
- the method according to Item 3 or 4, wherein the homopolymer is at least one selected from the group consisting of deoxyuridylic acid and salts thereof.
- nucleic acid polymer is at least one homopolymer selected from the group consisting of polyinosinic acid, polydeoxyinosinic acid, and salts thereof.
- the proteolytic enzyme comprises either proteinase K or subtilisin.
- Item 8 Item 8. The method according to any one of Items 1 to 7, wherein the template RNA is RNA extracted from 1 to 1000 cells.
- Item 9 Item 9. The method according to any one of Items 1 to 8, wherein the non-purified double-stranded cDNA in step (c) is in the form of a solution of 1 ⁇ L or less.
- Item 10 Item 10.
- step (a) is performed by the RT-RamDA method.
- step (b) is performed using Klenow fragment.
- step (c) is performed by either a transposon method or a ligation method.
- step (b) is performed in the presence of a chloride salt of 60mM or less.
- a method for efficiently preparing a library using, for example, non-purified double-stranded cDNA is provided.
- a method for preparing a library with sufficient yield without purifying double-stranded cDNA generated from a small amount of template RNA, such as about 10 pg is provided.
- the method is useful for library preparation for next generation sequencing.
- a library can be efficiently prepared even with a double-stranded cDNA solution of 1 ⁇ L or less, which is much smaller than the amount used in conventional library preparation methods. Therefore, it is possible to reduce the amount of reagents required for library preparation and to reduce costs.
- the amount of the double-stranded cDNA synthesized from the template RNA is usually more than 1 ⁇ L.
- Stranded cDNA solutions are now available for other uses and quality checks. If such other uses and quality checks can be performed, it is possible to decide whether to proceed to the next step, which is beneficial because it saves time, labor, and materials.
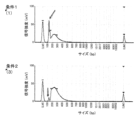
- FIG. 1 is a diagram showing the results of measuring a library with MultiNA (Shimadzu Corporation) in Example 5.
- FIG. 1 is a diagram showing the results of measuring a library with MultiNA (Shimadzu Corporation) in Example 5.
- the library preparation method preferably comprises the following steps (a), (b), and (c): (a) a step of synthesizing a single-stranded cDNA from 10 pg or more of template RNA; (b) synthesizing double-stranded cDNA from said single-stranded cDNA; and (c) preparing a library using said non-purified double-stranded cDNA.
- the template RNA may be purified RNA extracted from tissues or cells (the extracted RNA is further purified (e.g., treated by any purification means known in the art such as ethanol precipitation, column purification, etc.)). may be any RNA such as The type of cells from which template RNA is extracted is not particularly limited, and any type of cells may be used.
- the amount of template RNA is preferably 10 pg or more. Although the upper limit of the amount of template RNA is not particularly limited, it is preferably 10 ng or less, 5 ng or less, 1 ng or less, 500 pg or less, or 100 pg or less.
- a library can be efficiently prepared from the obtained double-stranded cDNA even if the amount of template RNA is small (for example, 10 pg or more and 1 ng or less).
- the amount of template RNA may be, for example, an amount contained in a small number of cells (eg, 1 to 1000, preferably 1 to 100, more preferably 1 to 10, more preferably 1). That is, RNA extracted from the above number of cells can also be used as template RNA. It is said that the standard amount of RNA contained in one cell is about 10 pg. According to the present invention, since a library can be prepared from a small amount of template RNA, it is possible to prepare a library with a sufficient yield even from one cell.
- the template RNA is preferably RNA extracted by lysing cells using a composition for cell lysis.
- the composition for cell lysis contains at least a cell lysing agent.
- Cell lysing agents include, for example, surfactants and chaotropic agents.
- Surfactants include anionic surfactants (e.g. sodium dodecyl sulfate, sodium cholate, sodium deoxycholate), cationic surfactants (e.g. cetyltrimethylammonium bromide), nonionic surfactants (e.g. , octylphenol ethoxylate, polyoxyethylene lauryl ether, polyoxyethylene stearyl ether, polyoxyethylene sorbitan monolaurate), and zwitterionic surfactants (e.g., 3-[(3-cholamidopropyl)dimethylammonio]).
- anionic surfactants e.g. sodium dodecyl sulfate, sodium cholate, sodium deoxycholate
- cationic surfactants e.g. cetyltrimethylammonium bromide
- Chaotropic agents include, for example, urea and lithium salts such as lithium perchlorate.
- Cell lysing agents are usually dissolved in water (preferably nuclease-free water) and used in the form of an aqueous solution. The cell lysing agents may be used singly or in combination of two or more.
- composition for cell lysis may contain additional components.
- Additional components include, for example, proteolytic enzymes (eg, proteinase K and subtilisin), nucleic acid polymers, RNase inhibitors, combinations of two or more thereof.
- Nucleic acid polymers include inosinic acid polymers, cytidylic acid polymers, guanylic acid polymers, adenylic acid polymers, thymidylic acid polymers, uridylic acid polymers, deoxyinosinic acid polymers, deoxycytidylic acid polymers, deoxyguanylic acid polymers, deoxyadenylic acid polymers, At least one selected from the group consisting of deoxythymidylic acid polymers and deoxyuridylic acid polymers is preferred.
- inosinic acid polymers include inosinic acid homopolymers (polyinosinic acid), inosinic acid copolymers (e.g., inosinic acid-derived structural units exceeding 50 mol%, 60 mol% or more, 70 mol% or more, 80 mol% or greater than or equal to 90 mol % but less than 100 mol %), derivatives thereof (e.g.
- the nucleic acid polymer is at least one selected from the group consisting of inosinic acid polymer, cytidylic acid polymer, guanylic acid polymer, deoxyinosinic acid polymer, deoxycytidylic acid polymer, and deoxyguanylic acid polymer. Inosinic acid polymers and/or deoxyinosinic acid polymers are more preferred.
- the nucleic acid polymer is polyinosinic acid, polycytidylic acid, polyguanylic acid, polyadenylic acid, polythymidylic acid, polyuridylic acid, polydeoxyinosinic acid, polydeoxycytidylic acid, polydeoxyguanylic acid, polydeoxyadenylic acid, polydeoxy It is preferably at least one homopolymer selected from the group consisting of thymidylic acid, polydeoxyuridylic acid, and salts thereof, and is preferably selected from the group consisting of polyinosinic acid, polydeoxyinosinic acid, and salts thereof. It is further preferred that at least one homopolymer is
- the nucleic acid polymer can be of any length, but as an example, it can be a nucleic acid polymer with a total length of 30 to 10000 bases.
- the step (a) is preferably performed in the presence of a protease and/or nucleic acid polymer.
- Library yield can be increased by synthesizing single-stranded cDNA in the presence of these components.
- One reason for this is thought to be that these components alleviate the inhibition of library preparation by substances such as cell-derived contaminants and reverse transcriptase used in the reverse transcription reaction.
- the reaction solution does not need to contain the protease and/or the nucleic acid polymer separately.
- the concentration of the protease in the synthesis reaction solution in step (a) is preferably 0.001 mg/mL or more, for example 0.001 to 0.1 mg/mL, preferably 0.001 to 0.01 mg/mL, more preferably 0.001 to 0.01 mg/mL. 0.005 mg/mL.
- the concentration of the protease can also be expressed in activity units, and is preferably 1 U/mL or more, for example 1 U/mL to 100 U/mL, preferably 1 U/mL, in the synthesis reaction solution of step (a). ⁇ 10 U/mL, more preferably 1 U/mL to 5 U/mL.
- proteolytic enzymes The activity of proteolytic enzymes is determined by definitions generally used in the art.
- the definition of activity for proteinase K is 1 unit (1 U) of the enzymatic activity that causes an increase in optical density at 275 nm equivalent to 1 microgram of tyrosine per minute under the conditions described below.
- the activity of other proteases such as subtilisin can be similarly measured.
- [reagent] (A) casein solution (substrate solution): 2.0% (20.0 g of Hammersten milk casein (Merck) is suspended and dissolved in 400 mL of 0.5% NaOH solution. Adjust the pH to 7.2 with 1 N HCl and add 500 mL of 20 mM boron containing 4 mM CaSO4 (pH 7.2).
- TCA solution TCA solution: 20% trichloroacetic acid (TCA)
- C Enzyme Diluent: Concentration 10 mM Borate Buffer in 10 mM Borate Buffer Assay Mix with 2 mM CaSO4 (pH 8.0) Casein 1.0% CaSO4 2mM [procedure] 1. Pipette 1.0 mL Substrate Solution (A) and 0.9 mL Enzyme Diluent (C) into test tubes and equilibrate at 35°C for approximately 5 minutes. 2. Add 0.1 mL of the sample to be measured and mix. 3.
- Weight activity (U/mg) (U/ml) x 1/C
- Vt total volume (4.0 mL)
- Vs sample volume (0.1 mL)
- 0.0074 extinction coefficient of tyrosine ( cm2 /microgram)
- t reaction time (10 minutes)
- df dilution factor
- C Enzyme concentration during dissolution (mg/mL)
- a 0.001 mg/mL solution is about 1 U/mL (1.2 to 1.4 U/mL) when the proteinase K of Promega is measured by the above method.
- the concentration of the nucleic acid polymer in the synthesis reaction solution in step (a) is preferably 1 ng/ ⁇ L or more, for example 1 to 10 ng/ ⁇ L, preferably 1 to 5 ng/ ⁇ L, more preferably 1 to 2 ng/ ⁇ L. be.
- the reaction for synthesizing single-stranded cDNA from template RNA is not particularly limited, and various conventionally known methods can be employed.
- step (a) is performed by reverse transcription, it typically includes a step of incubating a composition for reverse transcription (or a solution for reverse transcription) containing a protein having reverse transcription activity such as a reverse transcriptase.
- a composition for reverse transcription includes, for example, template RNA, protease and/or nucleic acid polymer, primers, deoxyribonucleotides, and reverse transcriptase.
- the composition for reverse transcription may also optionally contain other components such as a DNA polymerase.
- primers include primers specific to template RNA, oligo dT primers, random primers, and combinations of two or more of these.
- INDUSTRIAL APPLICABILITY The present invention is useful when performing reverse transcription reactions using oligo-dT primers and/or random primers, and is particularly beneficial when performing reverse transcription reactions using a combination of oligo-dT primers and random primers.
- the molar ratio of oligo dT primers to random primers is, for example, 1:5 to 1:15, preferably 1:8 to 1:12.
- Random primers include, for example, completely random primers and NSR (Not So Random) primers.
- Completely random primers are a mixture of primers with various base sequences, and each base sequence is a completely random base sequence.
- a fully random primer can contain a sequence that perfectly matches (or is perfectly complementary to) the rRNA sequence.
- completely random primers include completely random pentamers, completely random hexamers, completely random heptamers, completely random octamers, combinations thereof, and the like.
- a completely random hexamer may be a mixture of all possible base sequences (46 types) with four types of nucleotides (A, T, C, G).
- the NSR primer is a complete random primer from which the primer with a sequence completely complementary to the rRNA sequence has been removed.
- the rRNA sequences to be removed include, for example, 18S rRNA sequences, 28S rRNA sequences, 12S rRNA sequences, 16S rRNA sequences, combinations thereof.
- NSR primers include, for example, completely random hexamers excluding hexamers having a sequence completely complementary to the rRNA sequence. NSR primers can also be obtained by excluding those having a sequence completely complementary to the rRNA sequence from a set of primers such as completely random pentamers, completely random heptamers, and completely random octamers.
- the length of the primer is, for example, 5 bases or more, preferably 6 bases or more, and from the viewpoint of synthesis, it is, for example, 30 bases or less, preferably 25 bases or less, more preferably 20 bases or less.
- the concentration of the primer is not particularly limited, but is, for example, 1-10 ⁇ M, preferably 2-6 ⁇ M, more preferably 3-5 ⁇ M.
- Deoxyribonucleoside triphosphates are preferred as deoxyribonucleotides.
- Deoxyribonucleotide triphosphates include, for example, deoxycytidine triphosphate (dCTP), deoxyguanosine triphosphate (dGTP), deoxyadenosine triphosphate (dATP), deoxythymidine triphosphate (dTTP), deoxyuridine triphosphate (dUTP), these and combinations of two or more thereof.
- a mixture of dCTP, dGTP, dATP and dTTP, a mixture of dCTP, dGTP, dATP and dUTP, a mixture of dCTP, dGTP, dATP, dTTP and dUTP, and the like are preferred.
- a reverse transcriptase is any protein (enzyme) that has reverse transcription activity (RNA-dependent DNA polymerase activity), and is not particularly limited, but a polymerase that exhibits reverse transcriptase activity is preferred. Also, the reverse transcriptase preferably has low RNase H activity or no RNase H activity. Examples of reverse transcriptases include, e.g., Avian Myeloblastosis Virus reverse transcriptase (AMV-RT), Moloney Murine Leukemia Virus reverse transcriptase (MMLV-RT), human immune Virus (Human Immunovirus) reverse transcriptase (HIV-RT), EIRV-RT, RAV2-RT, C. hydrogenogormans DNA polymerase, rTth DNA polymerase, SuperScript I, SuperScript )II, variants thereof, and derivatives thereof. Of these, MMLV-RT is preferred.
- AMV-RT Avian Myeloblastosis Virus reverse transcriptase
- MMLV-RT Moloney Murine Le
- composition for reverse transcription may or may not contain the following DNA polymerases: Taq, Tbr, Tfl, Tru, Tth, Tli, Tac, Tne, Tma, Tih, Tfi, Pfu, Pwo , Kod, Bst, Sac, Sso, Poc, Pab, Mth, Pho, ES4, VENTTM, DEEPVENTTM, variants thereof.
- the composition for reverse transcription may contain an RNase inhibitor.
- RNase inhibitors are not particularly limited, and examples thereof include proteins derived from human placenta, rat lung, and pig liver.
- composition for reverse transcription may contain additives.
- Additives include, for example, buffers, salts, and combinations of two or more thereof.
- Buffers include, for example, Tris, Tricine, Bis-Tricine, Hepes, Mops, Tes, Taps, Pipes , Caps, and combinations of two or more thereof.
- the buffer is usually dissolved in water (preferably nuclease-free water) and used in the form of an aqueous solution.
- salts include chlorides (eg, lithium chloride, sodium chloride, potassium chloride, magnesium chloride, manganese chloride), acetates (eg, lithium acetate, sodium acetate, potassium acetate, magnesium acetate, manganese acetate), sulfates. (eg, potassium sulfate, magnesium sulfate, manganese sulfate), and combinations of two or more thereof.
- chlorides eg, lithium chloride, sodium chloride, potassium chloride, magnesium chloride, manganese chloride
- acetates eg, lithium acetate, sodium acetate, potassium acetate, magnesium acetate, manganese acetate
- sulfates eg, potassium sulfate, magnesium sulfate, manganese sulfate
- Conditions for incubation of the composition for reverse transcription are not particularly limited as long as single-stranded cDNA is produced from template RNA, and any conditions known in the art can be adopted.
- the incubation temperature is, for example, 30-65°C, preferably 35-60°C.
- the incubation time is, for example, 5-120 minutes, preferably 10-60 minutes.
- Amplification reverse transcription method (also known as RT-RamDA method) that amplifies cDNA using RNA as a template
- the RT-RamDA method is a nucleic acid amplification method that includes the step of incubating a mixture containing template RNA, primers, DNA strand-specific RNA:DNA hybrid strandase, RNase H minus reverse transcriptase, and a substrate.
- the complementary strand DNA (cDNA) of the template RNA is synthesized by the RNA-dependent DNA polymerase activity of RNase H-minus reverse transcriptase, and the cDNA strand among the hybrid strands of RNA and cDNA is identified as DNA strand-specific.
- Targeted RNA DNA is randomly cleaved by a hybrid chain degrading enzyme, and the cleavage site is the starting point, and the 3' cDNA strand is stripped from the RNA by the strand displacement activity of RNase H minus type reverse transcriptase, resulting in RNase H minus. A new cDNA strand is synthesized in the part stripped off by type reverse transcriptase. Details of the RT-RamDA method are described in US Patent Application Publication No. 2017/0275685, which is incorporated herein by reference in its entirety, and the like.
- step (a) When step (a) is performed by the RT-RamDA method, it includes a step of incubating the composition for reverse transcription containing the DNA strand-specific RNA:DNA hybrid chain degrading enzyme.
- the DNA strand-specific RNA:DNA hybrid strand degrading enzyme is preferably an enzyme that has the activity of cleaving the DNA strand in the hybrid strand of RNA and DNA.
- the enzyme for example, a double-strand-specific DNase and a non-specific DNase can be used. Among these, non-specific DNases are preferred in the present invention.
- double-strand-specific DNase double-strand-specific nuclease; also referred to as DSN
- enzymes derived from prokaryotes or eukaryotes can be used.
- Specific DNases or variants thereof can be used. Specific examples include the following. ⁇ Solenocera melantho DNase ⁇ Penaeus japonicus DNase ⁇ Paralithodes camtschaticus (king crab) DSN ⁇ Pandalus borealis dsDNase ⁇ Chionoecetes opilio (snow crab) DSN ⁇ Other DSN homologues
- the double-strand-specific DNase is preferably an enzyme that has DNase activity even at temperatures below 60°C.
- shrimp-derived double-strand-specific DNA degrading enzymes or modifications thereof are preferred.
- a commercially available product can be used as the double-strand-specific DNA degrading enzyme.
- Commercially available products include, for example, dsDNase (Arctic Zymes), Hl-dsDNase (Arctic Zymes), dsDNase (Thermo scientific), Shrimp Dnase, Recombinant (affymetrix), Atlantis dsDNase (Zymo Research), Thermolabile Nuclease (Roche ) and so on.
- the non-specific DNase has the activity of cleaving the DNA strand of the hybrid strand of RNA and DNA, and substantially has the activity of cleaving the RNA strand and single-stranded RNA of the hybrid strand of RNA and DNA. Enzymes that do not have the ability to cleave single-stranded DNA, but preferably have lower activity to cleave the DNA strand than the activity to cleave the hybrid strand of RNA and DNA.
- the non-specific DNase is preferably an enzyme that has DNase activity even below 60°C. Commercially available products such as DNaseI (manufactured by Thermo Fisher, DNaseI) can be used as such non-specific DNases.
- non-specific DNase enzymes derived from prokaryotes or eukaryotes can be used. Specific DNases or variants thereof can be used.
- the above variant means an enzyme obtained by altering a naturally occurring amino acid sequence. Specifically, an enzyme consisting of an amino acid sequence having a sequence identity of 80% or more (preferably 90% or more, more preferably 95% or more) with a naturally occurring amino acid sequence, and one or a number in the naturally occurring amino acid sequence An enzyme consisting of an amino acid sequence having deletions, substitutions and/or additions of amino acids (eg, 1 to 10, preferably 1 to 5, more preferably 1 to 3) amino acids.
- the composition for reverse transcription may contain a single-stranded DNA binding protein.
- Single-stranded DNA binding proteins are commonly used with DNA strand-specific RNA:DNA hybrid strandases.
- Single-stranded DNA binding proteins include, for example, T4 gene 32 protein, RecA, SSB (Single-Stranded DNA Binding Protein), and combinations of two or more thereof.
- step (a) is performed by the RT-RamDA method
- incubation of the reverse transcription composition may be performed under isothermal conditions or under thermocycling conditions.
- Isothermal conditions When incubation is performed under isothermal conditions, for example, a predetermined temperature between 25°C and less than 50°C, preferably a predetermined temperature between 30 and 45°C, more preferably between 35 and 40°C It can be carried out at a predetermined temperature, eg, 37° C. for a predetermined time (eg, 5 to 180 minutes, preferably 10 to 150 minutes). Incubation at a predetermined temperature between 25°C and 50°C may be performed in two or more steps.
- a predetermined temperature between 25°C and less than 50°C, preferably a predetermined temperature between 30 and 45°C, more preferably between 35 and 40°C
- a predetermined temperature eg, 37° C.
- a predetermined time eg, 5 to 180 minutes, preferably 10 to 150 minutes.
- Incubation at a predetermined temperature between 25°C and 50°C may be performed in two or more steps.
- a predetermined temperature between 25°C and less than 30°C for 5 to 15 minutes then at a predetermined temperature between 30°C and less than 35°C for 5 to 15 minutes, and then at a predetermined temperature between 35°C and less than 50°C.
- Incubation at temperature for a predetermined time eg, 5 to 60 minutes
- incubation at a predetermined temperature between 25°C and less than 50°C incubation at a predetermined temperature between 25°C and less than 50°C
- incubation at a predetermined temperature between, for example, 50°C and less than 100°C may be performed.
- Incubation at a predetermined temperature between 50°C and less than 100°C may be performed in two or more stages. For example, it may be incubated at a predetermined temperature between 50° C. and less than 80° C. for 5 to 15 minutes, followed by incubation at a predetermined temperature between 80 to 100° C. for 5 to 15 minutes.
- thermal cycle conditions When incubation is performed under thermal cycle conditions, for example, a predetermined temperature T1 between 20°C and less than 30°C (eg, 25°C) and a predetermined temperature T2 between 30 and 45°C (eg, 37°C ) in combination with T1 for a predetermined time (eg, 1 to 3 minutes, such as 2 minutes) and T2 for a predetermined time (eg, 1 to 3 minutes, such as 2 minutes) as one cycle, which is preferably may be repeated for 10 to 40 cycles, more preferably 15 to 35 cycles.
- a predetermined temperature T1 between 20°C and less than 30°C (eg, 25°C) and a predetermined temperature T2 between 30 and 45°C (eg, 37°C ) in combination with T1 for a predetermined time (eg, 1 to 3 minutes, such as 2 minutes) and T2 for a predetermined time (eg, 1 to 3 minutes, such as 2 minutes) as one cycle, which is preferably may be repeated for 10 to 40 cycles, more
- a predetermined temperature between 25°C and less than 30°C for a predetermined time (for example, 5 to 15 minutes), and then at a predetermined temperature between 30°C and less than 35°C. for a period of time (eg, 5-15 minutes), followed by incubation at a predetermined temperature between 35° C. and less than 50° C. for a predetermined period of time (eg, 1-5 minutes).
- a predetermined temperature between 50 ° C and less than 80 ° C for a predetermined time (eg, 5 to 15 minutes)
- a predetermined temperature between 80 to 90 ° C for a predetermined time (for example 5-15 minutes).
- Step (b) is preferably carried out in the presence of salt.
- Library yield can be increased by synthesizing double-stranded cDNA in the presence of salt.
- One reason for this is thought to be that the amount of double-stranded cDNA produced by optimizing the binding of the primer to the template was increased.
- salts include chloride salts (e.g., alkali metal chloride salts such as lithium chloride, sodium chloride, and potassium chloride; alkaline earth metal chloride salts such as magnesium chloride; transition metal chloride salts such as manganese chloride). is mentioned. Among them, alkali metal chloride salts and/or alkaline earth metal chloride salts are preferred, and at least one selected from the group consisting of magnesium chloride, potassium chloride and sodium chloride is preferred. Salts may be used singly or in combination of two or more.
- chloride salts e.g., alkali metal chloride salts such as lithium chloride, sodium chloride, and potassium chloride
- alkaline earth metal chloride salts such as magnesium chloride
- transition metal chloride salts such as manganese chloride
- salt When a salt is contained in the reaction product of step (a), salt may not be added separately to the reaction solution of step (b), or salt may be added separately to the reaction solution of step (b). good.
- the salt concentration in the synthesis reaction solution in step (b) is, for example, 60 mM or less, preferably 50 mM or less, more preferably 40 mM or less. Also, the salt concentration in the synthesis reaction solution of step (b) is, for example, 5 to 60 mM, preferably 5 to 50 mM, more preferably 5 to 40 mM. In certain embodiments, the concentration of the salt in the synthesis reaction solution in step (b) is, for example, 20 mM or less (eg, 5 to 20 mM), 15 mM or less (eg, 5 to 15 mM), or 12 mM or less (eg, 5 to 12 mM). ), etc. The concentration of the salt is preferably that of a chloride salt.
- the concentration of sodium chloride is, the better. be.
- the synthesis reaction solution in step (b) may not contain sodium chloride, and when the synthesis reaction solution in step (b) contains sodium chloride, its concentration in the synthesis reaction solution in step (b) is, for example, 5-20 mM, preferably 5-15 mM, more preferably 5-12 mM.
- the concentration of potassium chloride is, for example, 5 mM or higher (eg, 5 to 50 mM), preferably 10 mM or higher (eg, 10 to 40 mM), more preferably 15 mM or higher (eg, : 15-35 mM).
- the concentration of magnesium chloride is, for example, 20 mM or less (eg, 1 to 20 mM), preferably 15 mM or less (eg, 1 to 15 mM), more preferably 10 mM or less (eg, : 1-10 mM).
- the concentration of chloride salt is 60 mM or less (for example, 50 mM or less, preferably 40 mM or less) and the concentration of sodium chloride is 20 mM or less in the synthesis reaction solution of step (b). More preferably, the chloride salt concentration is 60 mM or less (eg, 50 mM or less, preferably 40 mM or less), the sodium chloride concentration is 20 mM or less, and the potassium chloride concentration is 5 mM or more.
- the reaction for synthesizing double-stranded cDNA from single-stranded cDNA is not particularly limited, and various conventionally known methods can be employed.
- the reaction is typically performed using a composition for synthesizing double-stranded cDNA containing an enzyme (double-stranded cDNA synthetase) that catalyzes a nucleotide polymerization reaction in the 5′ ⁇ 3′ direction in the presence of a template DNA and a primer. including the step of incubating the object.
- an enzyme double-stranded cDNA synthetase
- a composition for double-stranded cDNA synthesis includes, for example, single-stranded cDNA, salts, primers, deoxyribonucleotides, and double-stranded cDNA synthetase.
- the composition for double-strand synthesis may also optionally contain other components such as buffers.
- Double-stranded cDNA synthetase refers to an enzyme that catalyzes the polymerization reaction of nucleotides in the 5' ⁇ 3' direction, and is not particularly limited. Examples include the Klenow fragment, T4 DNA polymerase, variants thereof, and derivatives thereof. Of these, the Klenow fragment is preferred.
- primers examples include those exemplified for the reverse transcription composition.
- Conditions for incubation of the composition for double-stranded cDNA synthesis are not particularly limited as long as double-stranded cDNA is produced from single-stranded cDNA, and any conditions known in the art can be adopted. .
- it may be incubated at a predetermined temperature between 10° C. and lower than 35° C. for a predetermined time (eg, 5 to 90 minutes).
- the library preparation method preferably further includes the step of heating the double-stranded cDNA (for example, the reaction solution in step (b)). This step is preferably performed after step (b) and before the next step (c).
- the step may correspond to an inactivation treatment.
- the heating conditions are not particularly limited. minutes or longer).
- the heating temperature is preferably 80°C or higher and 95°C or lower, more preferably 80°C or higher and 90°C or lower, still more preferably 80°C or higher and 85°C or lower.
- the incubation time is preferably 5 minutes or more and 30 minutes or less, more preferably 10 minutes or more and 20 minutes or less, and still more preferably 10 minutes or more and 15 minutes or less.
- step (c) By performing such a heating step, the formation of adapter dimers can be more effectively suppressed in step (c).
- heating at such temperatures unfolds the higher-order structure of the double-stranded cDNA and improves the efficiency of enzymatic reactions (e.g., transposon reactions) in library preparation, thereby promoting the formation of adapter dimers. It is assumed that it can be suppressed more effectively.
- the library is preferably nucleic acid fragments to which sequences necessary for next-generation sequencing have been assigned. Sequences required for next-generation sequencing include flow cell binding sequences and sequences required for sequencing.
- next-generation sequencing typically refers to sequencing technology that can simultaneously perform millions to billions of massive sequencing reactions.
- next-generation sequencing include a method of parallel sequence analysis using amplification techniques such as emulsion PCR and bridge PCR, and highly sensitive detection techniques such as single-molecule observation.
- the device (sequencer) for next-generation sequencing is not particularly limited, but for example, MiSeq, HiSeq, NovaSeq (Illumina); Genetic Analyzer V2.0, Ion Proton (Thermo Fisher Scientific); MinION, PromethION (Nanopore Co.) and the like.
- Next generation sequencing is described, for example, in US Patent Application Publication 2014/178438, which is incorporated herein by reference in its entirety.
- the double-stranded cDNA used for library preparation is in the form of a solution of, for example, 5 ⁇ L or less, preferably 4 ⁇ L or less, more preferably 3 ⁇ L or less, even more preferably 2 ⁇ L or less, and particularly preferably 1 ⁇ L or less.
- the double-stranded cDNA is in the form of a solution of, for example, 0.01 ⁇ L or more, 0.05 ⁇ L or more, or 0.1 ⁇ L or more. In the present invention, a sufficient library yield can be obtained without purification even with a small amount of double-stranded cDNA.
- the library preparation method is not particularly limited.
- transposons are used to fragment and tag template DNA, and nucleic acid amplification of a library composed of tagged DNA fragments is carried out.
- two transposon terminal sequences (equivalent to adapters) and a transposase form a transposome complex, which fragments and tags template DNA in solution, which is necessary for next-generation sequencing such as PCR.
- Sequence analysis can be performed because a unique sequence is added (this is also referred to as the “transposon method”).
- Examples of commercially available kits using transposons include, but are not limited to, Illumina's Nextera XT DNA Library Preparation Kit.
- Another technique is fragmentation of template DNA (e.g., full-length DNA), end-repair of fragmented DNA, and/or adenylation of the ends of fragmented DNA or end-repaired fragmented DNA (this is referred to as "A addition ”), end-repaired and/or end-adenylated DNA fragments added with adapter sequences by ligation with T4 Ligase or the like, and library nucleic acids composed of DNA fragments to which adapters have been added Amplification has been carried out. A sequence necessary for next-generation sequencing is added to the adapter sequence, so sequence analysis can be performed (this is also referred to as a “ligation method”).
- a addition end-repaired and/or end-adenylated DNA fragments added with adapter sequences by ligation with T4 Ligase or the like, and library nucleic acids composed of DNA fragments to which adapters have been added Amplification has been carried out.
- a sequence necessary for next-generation sequencing is added to the adapter sequence, so sequence analysis
- the adapter sequence and length are not particularly limited, and may be, for example, Y-shaped adapters, stem-loop adapters, and the like.
- Examples of commercially available kits using the ligation method include, but are not limited to, GenNext (R) NGS Library Prep Kit from Toyobo Co., Ltd. and KAPA HyperPrep Kit from KAPA.
- the library preparation method is preferably the transposon method.
- purification means separation of contaminants such as the primers contained in a solution or the like from double-stranded cDNA.
- a purification method a method using magnetic beads, a method using a column, a method using phenol or phenol/chloroform, and a method using protein aggregation are known.
- the present invention is characterized by preparing a library using non-purified double-stranded cDNA that has not undergone such purification steps.
- non-purified double-stranded cDNA refers to the double-stranded cDNA synthesized in step (b) (including the double-stranded cDNA solution) itself or heated Examples include those obtained by diluting the main-stranded cDNA with a solvent such as water or a buffer solution, or the products obtained by drying and concentrating the above-mentioned double-stranded cDNA solution, but are limited to these if they have not undergone a purification process. isn't it.
- the library concentration can be increased, eg, 10 nM or higher, 15 nM or higher, 20 nM or higher, 25 nM or higher, 30 nM or higher, 35 nM or higher, or 40 nM or higher.
- the amount of adapter dimers in the library can be reduced.
- the peak intensity of the indicated region is 90% or less, 80% or less, 70% or less, 60% or less, or 50% of the maximum intensity of the region showing the library yield (region present in the range of approximately 150 bp to 5000 bp) You can:
- Example 1 Effect of Sodium Chloride Concentration on Yield
- RNA was diluted, single-stranded cDNA was synthesized by the RT-RamDA method, and double-stranded cDNA was synthesized using Klenow fragment.
- a library was prepared using the Nextera XT DNA Library Preparation Kit (Illumina) using the transposon method, and the library concentration was measured using MultiNA (Shimadzu Corporation). The procedure followed the manufacturer's instructions.
- 10 pg of RNA purified from NIH3T3 cells using RNeasy Mini Kit (Qiagen) was used as the nucleic acid fragment sample used in this example.
- 1 ng/ ⁇ L of RNA was diluted with the RNA diluent shown in Table 1 to prepare a 10 pg/ ⁇ L RNA solution.
- NSR primers (hexamers) were synthesized using Sigma's custom oligos, and were described in Ozsolak et al., Digital transcriptome profiling from attomole-level RNA samples, Genome Research, Vol. 20, 2010, pp. 519-525. They were used by mixing so as to obtain a primer composition.
- Example 2 Influence of addition amount of potassium polyinosinic acid salt and proteinase K on yield
- the following experiment was conducted to examine whether the addition amount of potassium polyinosinic acid salt and proteinase K affects the yield of the library.
- rice field RNA was diluted, single-stranded cDNA was synthesized by the RT-RamDA method, and double-stranded cDNA was synthesized using Klenow fragment.
- a library was prepared using the Nextera XT DNA Library Preparation Kit (Illumina), and the library concentration was measured using MultiNA (Shimadzu Corporation). The procedure followed the manufacturer's instructions.
- RNA purified from NIH3T3 cells using RNeasy Mini Kit As the nucleic acid fragment sample used in this example, 10 pg of RNA purified from NIH3T3 cells using RNeasy Mini Kit (Qiagen) was used. 1 ng/ ⁇ L of RNA was diluted with the RNA diluent shown in Table 6 to prepare a 10 pg/ ⁇ L RNA solution. 1 ⁇ L of this RNA solution was heat-treated at 70° C. for 1 minute and 30 seconds. Then, 2 ⁇ L of the RT-RamDA reaction solution shown in Table 7 was added, and the RT-RamDA reaction was carried out at 3 ⁇ L at 25°C for 10 minutes, 30°C for 10 minutes, 37°C for 30 minutes, 50°C for 5 minutes, and 98°C for 5 minutes.
- Example 3 Yield Improvement by Non-Purification Protocol by Addition of Potassium Polyinosinic Acid Salt
- the following experiment was carried out in order to examine whether the addition of potassium polyinosinic acid salt affects the yield of the library.
- Cells were lysed, single-stranded cDNA was synthesized by RT-RamDA method, and double-stranded cDNA was synthesized using Klenow fragment.
- a library was prepared using the Nextera XT DNA Library Preparation Kit (Illumina), and the library concentration was measured using MultiNA (Shimadzu Corporation). The procedure followed the manufacturer's instructions. Specifically, the following methods were used. Cell samples used in Examples were prepared with the following composition.
- NIH3T3 cells were reacted with a trypsin solution (Nacalai Tesque) at 37°C for 2 minutes to dissociate into single cells. Immediately after dissociation, the reaction was stopped by replacing with PBS(-). Using a FACS Melody cell sorter (BD), PI-negative cells of the dead cell fluorescent marker dye were used as a live cell fraction, and 120 cells were fractionated from this fraction into 3 ⁇ L of the Lysis Buffer shown in Table 11 below. Immediately after fractionation, centrifugation was performed and stored at -80°C.
- BD FACS Melody cell sorter
- Example 4 Evaluation of Amount of Template RNA
- RNA was diluted, single-stranded cDNA was synthesized by RT-RamDA method or conventional reverse transcription method (RT method), and double-stranded cDNA was synthesized using Klenow fragment.
- a library was prepared using the Nextera XT DNA Library Preparation Kit (Illumina), and the library concentration was measured using MultiNA (Shimadzu Corporation). The procedure followed the manufacturer's instructions.
- Mouse ES cell total RNA (Unitech Co., Ltd.) was used as the nucleic acid fragment sample used in this example.
- RNA template amount 10 pg
- 1 ⁇ L of this RNA solution was heat-treated at 70° C. for 1 minute and 30 seconds.
- Conditions 1 and 2 add 2 ⁇ L of the RT-RamDA reaction solution shown in Table 17, and condition 3 adds 2 ⁇ L of the RT reaction solution shown in Table 18, 3 ⁇ L at 25 ° C. 10 minutes, 30 ° C. 10 minutes, 37 RT-RamDA or RT reactions were performed at 30°C, 50°C for 5 minutes, and 98°C for 5 minutes.
- Example 5 Comparison of Inactivation Temperature in Synthesis of Double Strands
- RNA was diluted, single-stranded cDNA was synthesized by the RT-RamDA method, and double-stranded cDNA was synthesized using Klenow fragment. After that, they were inactivated by heating at 70°C or 80°C, then a library was prepared using the Nextera XT DNA Library Preparation Kit (Illumina), and the library concentration was measured using MultiNA (Shimadzu Corporation). The procedure followed the manufacturer's instructions. Mouse ES cell total RNA was used as the nucleic acid fragment sample used in this example.
- RNA template amount 10 pg
- RNA template amount 10 pg
- 1 ⁇ L of this RNA solution was heat-treated at 70° C. for 1 minute and 30 seconds.
- Example 6 Evaluation of Chlorides in Step (b)
- RNA was diluted, single-stranded cDNA was synthesized by the RT-RamDA method, and double-stranded cDNA was synthesized using Klenow fragment.
- a library was prepared using the Nextera XT DNA Library Preparation Kit (Illumina), and the library concentration was measured using MultiNA (Shimadzu Corporation). The procedure followed the manufacturer's instructions. Nucleic acid fragment samples used in this example were total RNA purified from human K562 cells using RNeasy mini kit (QIAGEN).
- RNA template amount 10 pg
- RNA template amount 10 pg
- 1 ⁇ L of this RNA solution was heat-treated at 70° C. for 1 minute and 30 seconds.
- 2 ⁇ L of the RT-RamDA reaction solution shown in Table 2 in Example 1 was added, and 3 ⁇ L was added at 25°C for 10 minutes, 30°C for 10 minutes, 37°C for 30 minutes, 50°C for 5 minutes, and 98°C.
- the RT-RamDA reaction was performed in minutes.
Abstract
効率的にライブラリーを調製する方法が提供される。 以下の工程(a)、(b)、及び(c)を含む、ライブラリーの調製方法が開示される: (a)10pg以上の鋳型RNAから一本鎖cDNAを合成する工程; (b)前記一本鎖cDNAから二本鎖cDNAを合成する工程;及び (c)非精製の前記二本鎖cDNAを用いてライブラリーを調製する工程。
Description
本発明は、ライブラリー調製方法に関する。
DNAやRNA等の核酸シーケンス解析の中で、短時間で膨大な塩基配列情報を得ることができる次世代シークエンシング(NGS:next-generation sequencing)技術は急速に進歩している。次世代シーケンシングは、複数個体を同時に配列決定できる高度かつ高速な処理が可能であり、臨床診断や薬剤開発等の医療技術、農業技術や基礎研究などの生命科学分野に貢献する強力な解析技術である。
次世代シークエンシングにおいては、フローセルへの結合やプライマー結合部位等として機能し得るアダプター配列を末端に付加した核酸断片から構成されるライブラリーの調製が通常必要とされている。
RNAを鋳型としてライブラリー調製を行う場合、生成された二本鎖cDNA溶液を精製した後にライブラリー調製を行う手法が実施されている。例えばRT-RamDA法は、鋳型RNA、プライマー、DNA鎖特異的RNA:DNAハイブリッド鎖分解酵素、RNase Hマイナス型逆転写酵素、及び基質を含む混合物をインキュベートする工程により、RNAを鋳型としてcDNAを増幅させる増幅逆転写法であるが、ライブラリー化する際は一本鎖cDNAから二本鎖cDNA生成し、溶液を精製した後にライブラリー調製を開始する手法が実施されている(特許文献1、非特許文献1)。
Hayashi T. et.al., Single-cell full-length total RNA sequencing uncovers dynamics of recursive splicing and enhancer RNAs, Nat.Commun., 2018
二本鎖cDNA溶液には細胞由来の夾雑物や逆転写反応に用いられる酵素、プライマー、dNTP、塩類等が含まれており、次工程の反応を阻害し得る。特にこれら溶液中の物質によりライブラリー調製に用いられる酵素反応が著しく阻害され得る。一方で、精製作業は煩雑であり、作業性が悪い。また精製工程があることにより、収量の減少が発生する。これらのことから精製工程を簡略化することが望ましい。本発明の主な目的は、より効率的にライブラリーを調製する方法を提供することにある。
本発明者等は、上記目的を達成するために鋭意検討した結果、少量の鋳型RNAであっても、生成した二本鎖cDNAを精製することなくそのままライブラリー調製に持ち込むことで十分なライブラリー収量が得られることを見出した。当該知見を基に更に検討を重ねることにより、本発明の完成に至った。
本発明は、代表的には以下の態様を包含する。
[項1]
以下の工程(a)、(b)、及び(c)を含むライブラリーの調製方法:
(a)10pg以上の鋳型RNAから一本鎖cDNAを合成する工程;
(b)前記一本鎖cDNAから二本鎖cDNAを合成する工程;及び
(c)非精製の前記二本鎖cDNAを用いてライブラリーを調製する工程。
[項2]
前記工程(b)が12mM以下の塩化ナトリウムの存在下で行われる、項1に記載の方法。
[項3]
前記工程(a)が1ng/μL以上の核酸ポリマー及び/又は1U/mL以上のタンパク質分解酵素の存在下で行われる、項1又は2に記載の方法。
[項4]
前記二本鎖cDNAを80℃以上で10分以上加熱する工程を更に含む、項1~3のいずれかに記載の方法。
[項5]
前記核酸ポリマーが、ポリイノシン酸、ポリシチジル酸、ポリグアニル酸、ポリアデニル酸、ポリチミジル酸、ポリウリジル酸、ポリデオキシイノシン酸、ポリデオキシシチジル酸、ポリデオキシグアニル酸、ポリデオキシアデニル酸、ポリデオキシチミジル酸、ポリデオキシウリジル酸、及びこれらの塩からなる群より選択される少なくとも1種のホモポリマーである、項3又は4に記載の方法。
[項6]
前記核酸ポリマーが、ポリイノシン酸、ポリデオキシイノシン酸、及びこれらの塩からなる群より選択される少なくとも1種のホモポリマーである、項3~5のいずれかに記載の方法。
[項7]
前記タンパク質分解酵素がプロテイナーゼK及びサブチリシンのいずれかを含む、項3~6のいずれかに記載の方法。
[項8]
前記鋳型RNAが1細胞~1000細胞から抽出されたRNAである、項1~7のいずれかに記載の方法。
[項9]
前記工程(c)の非精製の二本鎖cDNAが1μL以下の溶液の形態である、項1~8のいずれかに記載の方法。
[項10]
前記工程(a)がRT-RamDA法で行われる、項1~9のいずれかに記載の方法。
[項11]
前記工程(b)がクレノウフラグメントを用いて行われる、項1~10のいずれかに記載の方法。
[項12]
前記工程(c)がトランスポゾン法及びライゲーション法のいずれかで行われる、項1~11のいずれかに記載の方法。
[項13]
前記工程(b)が60mM以下の塩化物塩の存在下で行われる、請求項1~12のいずれかに記載の方法。
[項1]
以下の工程(a)、(b)、及び(c)を含むライブラリーの調製方法:
(a)10pg以上の鋳型RNAから一本鎖cDNAを合成する工程;
(b)前記一本鎖cDNAから二本鎖cDNAを合成する工程;及び
(c)非精製の前記二本鎖cDNAを用いてライブラリーを調製する工程。
[項2]
前記工程(b)が12mM以下の塩化ナトリウムの存在下で行われる、項1に記載の方法。
[項3]
前記工程(a)が1ng/μL以上の核酸ポリマー及び/又は1U/mL以上のタンパク質分解酵素の存在下で行われる、項1又は2に記載の方法。
[項4]
前記二本鎖cDNAを80℃以上で10分以上加熱する工程を更に含む、項1~3のいずれかに記載の方法。
[項5]
前記核酸ポリマーが、ポリイノシン酸、ポリシチジル酸、ポリグアニル酸、ポリアデニル酸、ポリチミジル酸、ポリウリジル酸、ポリデオキシイノシン酸、ポリデオキシシチジル酸、ポリデオキシグアニル酸、ポリデオキシアデニル酸、ポリデオキシチミジル酸、ポリデオキシウリジル酸、及びこれらの塩からなる群より選択される少なくとも1種のホモポリマーである、項3又は4に記載の方法。
[項6]
前記核酸ポリマーが、ポリイノシン酸、ポリデオキシイノシン酸、及びこれらの塩からなる群より選択される少なくとも1種のホモポリマーである、項3~5のいずれかに記載の方法。
[項7]
前記タンパク質分解酵素がプロテイナーゼK及びサブチリシンのいずれかを含む、項3~6のいずれかに記載の方法。
[項8]
前記鋳型RNAが1細胞~1000細胞から抽出されたRNAである、項1~7のいずれかに記載の方法。
[項9]
前記工程(c)の非精製の二本鎖cDNAが1μL以下の溶液の形態である、項1~8のいずれかに記載の方法。
[項10]
前記工程(a)がRT-RamDA法で行われる、項1~9のいずれかに記載の方法。
[項11]
前記工程(b)がクレノウフラグメントを用いて行われる、項1~10のいずれかに記載の方法。
[項12]
前記工程(c)がトランスポゾン法及びライゲーション法のいずれかで行われる、項1~11のいずれかに記載の方法。
[項13]
前記工程(b)が60mM以下の塩化物塩の存在下で行われる、請求項1~12のいずれかに記載の方法。
本発明によれば、例えば非精製の二本鎖cDNAを用いて効率的にライブラリーを調製する方法が提供される。特に、10pg程度という少量の鋳型RNAからでも生成した二本鎖cDNAを精製することなく十分な収量のライブラリーを調製する方法が提供される。当該方法は、次世代シークエンシングのためのライブラリー調製に有用である。当該方法では、1μL以下という従来のライブラリー調製方法で用いられていた量よりも非常に少ない量の二本鎖cDNA溶液を用いても効率よくライブラリーを調製できる。従って、ライブラリー調製に必要な試薬の量を抑えることができ、コストを抑えることも可能となる。また、鋳型RNAから合成した二本鎖cDNAの溶液の量は1μLよりも多くなる場合が通例であるが、1μL以下の二本鎖cDNA溶液をライブラリー調製に用いることにより、余剰量の二本鎖cDNA溶液を他の用途やクオリティチェックに使用可能となる。このような他の用途やクオリティチェックが行えると次の工程に進むか判断することができ、時間や労力、資材などの節約にもなるので有益である。
以下、本発明の実施形態を示しつつ、本発明について更に詳説する。
<ライブラリー調製方法>
一実施形態において、ライブラリーの調製方法は、以下の工程(a)、(b)、及び(c)を含むことが好ましい:
(a)10pg以上の鋳型RNAから一本鎖cDNAを合成する工程;
(b)前記一本鎖cDNAから二本鎖cDNAを合成する工程;及び
(c)非精製の前記二本鎖cDNAを用いてライブラリーを調製する工程。
一実施形態において、ライブラリーの調製方法は、以下の工程(a)、(b)、及び(c)を含むことが好ましい:
(a)10pg以上の鋳型RNAから一本鎖cDNAを合成する工程;
(b)前記一本鎖cDNAから二本鎖cDNAを合成する工程;及び
(c)非精製の前記二本鎖cDNAを用いてライブラリーを調製する工程。
1.工程(a)
鋳型RNAとしては、組織や細胞から抽出されたRNA(抽出されたRNAは、更に精製(例えば、エタノール沈殿、カラム精製等の当該分野で公知の任意の精製手段により処理)された精製RNAであってもよい)等の任意のRNAであり得る。鋳型RNAを抽出する細胞の種類は特に制限されず、いかなる種類の細胞であってもよい。鋳型RNA量は10pg以上であることが好ましい。鋳型RNA量の上限は特に制限されないが、10ng以下、5ng以下、1ng以下、500pg以下、又は100pg以下であることが好ましい。本発明では鋳型RNA量が少量(例えば10pg以上1ng以下)であっても得られた二本鎖cDNAから効率的にライブラリーを調製することができる。鋳型RNA量は、例えば、少数(例えば1~1000個、好ましくは1~100個、より好ましくは1~10個、より好ましくは1個)の細胞に含まれる量であってもよい。すなわち、上記の数の細胞から抽出されたRNAを鋳型RNAとして使用することもできる。なお、1細胞に含まれるRNA量は標準的には約10pg程度とも言われている。本発明によれば、少量の鋳型RNAからライブラリー調製できるので、1細胞からでも十分な収量のライブラリーを調製することが可能である。
鋳型RNAとしては、組織や細胞から抽出されたRNA(抽出されたRNAは、更に精製(例えば、エタノール沈殿、カラム精製等の当該分野で公知の任意の精製手段により処理)された精製RNAであってもよい)等の任意のRNAであり得る。鋳型RNAを抽出する細胞の種類は特に制限されず、いかなる種類の細胞であってもよい。鋳型RNA量は10pg以上であることが好ましい。鋳型RNA量の上限は特に制限されないが、10ng以下、5ng以下、1ng以下、500pg以下、又は100pg以下であることが好ましい。本発明では鋳型RNA量が少量(例えば10pg以上1ng以下)であっても得られた二本鎖cDNAから効率的にライブラリーを調製することができる。鋳型RNA量は、例えば、少数(例えば1~1000個、好ましくは1~100個、より好ましくは1~10個、より好ましくは1個)の細胞に含まれる量であってもよい。すなわち、上記の数の細胞から抽出されたRNAを鋳型RNAとして使用することもできる。なお、1細胞に含まれるRNA量は標準的には約10pg程度とも言われている。本発明によれば、少量の鋳型RNAからライブラリー調製できるので、1細胞からでも十分な収量のライブラリーを調製することが可能である。
一実施形態において、鋳型RNAは、細胞溶解用組成物を用いて細胞を溶解して抽出したRNAであることが好ましい。
細胞溶解用組成物は、少なくとも細胞溶解剤を含む。細胞溶解剤としては、例えば、界面活性剤、カオトロピック剤などが挙げられる。界面活性剤には、アニオン性界面活性剤(例えば、ドデシル硫酸ナトリウム、コール酸ナトリウム、デオキシコール酸ナトリウム)、カチオン性界面活性剤(例えば、臭化セチルトリメチルアンモニウム)、ノニオン性界面活性剤(例えば、オクチルフェノールエトキシレート、ポリオキシエチレンラウリルエーテル、ポリオキシエチレンステアリルエーテル、ポリオキシエチレンソルビタンモノラウレート)、及び両イオン性界面活性剤(例えば、3-[(3-コールアミドプロピル)ジメチルアンモニオ]-1-プロパンスルホン酸)が含まれる。カオトロピック剤としては、例えば、尿素、過塩素酸リチウム塩などのリチウム塩が挙げられる。細胞溶解剤は、通常、水(好ましくはヌクレアーゼフリー水)に溶解され、水溶液の形態で使用される。細胞溶解剤は1種を単独で又は2種以上を組み合わせて使用してもよい。
細胞溶解用組成物は、追加の成分を含んでいてもよい。追加の成分としては、例えば、タンパク質分解酵素(例えば、プロテイナーゼKやサブチリシン)、核酸ポリマー、RNase阻害剤、これら2種以上の組合せが挙げられる。
核酸ポリマーとしては、イノシン酸ポリマー、シチジル酸ポリマー、グアニル酸ポリマー、アデニル酸ポリマー、チミジル酸ポリマー、ウリジル酸ポリマー、デオキシイノシン酸ポリマー、デオキシシチジル酸ポリマー、デオキシグアニル酸ポリマー、デオキシアデニル酸ポリマー、デオキシチミジル酸ポリマー、及びデオキシウリジル酸ポリマーからなる群より選択される少なくとも一種が好ましい。ここで、例えば、イノシン酸ポリマーは、イノシン酸ホモポリマー(ポリイノシン酸)、イノシン酸コポリマー(例えば、イノシン酸由来の構成単位が50モル%超、60モル%以上、70モル%以上、80モル%以上、又は90モル%以上であり、100モル%未満であるコポリマー)、これらの誘導体(例えば、少なくとも一部の塩基部分に、例えば、フッ素原子、臭素原子、ヨウ素原子、アルキル基、アミノ基、メルカプト基などの官能基が導入されたもの、及び/又は、少なくとも一部のリン酸部分がチオリン酸部分に置き換わったもの)、及びこれらの塩(例えば、ナトリウム塩、カリウム塩などのアルカリ金属塩)を包含する意味で用い、ポリイノシン酸とポリシチジル酸とがアニールしたポリ(I:C)等と呼ばれる二本鎖のポリマーも包含する意味で用いる。他のシチジル酸ポリマー等についても同様の意味で用いる。
一実施形態において、核酸ポリマーは、イノシン酸ポリマー、シチジル酸ポリマー、グアニル酸ポリマー、デオキシイノシン酸ポリマー、デオキシシチジル酸ポリマー、及びデオキシグアニル酸ポリマーからなる群より選択される少なくとも一種であることが好ましく、イノシン酸ポリマー及び/又はデオキシイノシン酸ポリマーであることが更に好ましい。
一実施形態において、核酸ポリマーは、ポリイノシン酸、ポリシチジル酸、ポリグアニル酸、ポリアデニル酸、ポリチミジル酸、ポリウリジル酸、ポリデオキシイノシン酸、ポリデオキシシチジル酸、ポリデオキシグアニル酸、ポリデオキシアデニル酸、ポリデオキシチミジル酸、ポリデオキシウリジル酸、及びこれらの塩からなる群より選択される少なくとも1種のホモポリマーであることが好ましく、ポリイノシン酸、ポリデオキシイノシン酸、及びこれらの塩からなる群より選択される少なくとも1種のホモポリマーであることが更に好ましい。
核酸ポリマーは任意の長さであり得るが、一例として全長が30~10000塩基長である核酸ポリマーであり得る。
工程(a)は、タンパク質分解酵素及び/又は核酸ポリマーの存在下で行うことが好ましい。これらの成分の存在下で一本鎖cDNAを合成することによりライブラリー収量を増加させることができる。これは、細胞由来の夾雑物や逆転写反応に使用される逆転写酵素などの物質によるライブラリー調製への阻害をこれらの成分が緩和することが一因と考えられる。
タンパク質分解酵素及び/又は核酸ポリマーが鋳型RNAの調製に用いられ得る細胞溶解用組成物に含まれる場合、反応溶液には、別途タンパク質分解酵素及び/又は核酸ポリマーを添加しなくてもよい。
タンパク質分解酵素の濃度は、工程(a)の合成反応液中において0.001mg/mL以上であることが好ましく、例えば0.001~0.1mg/mL、好ましくは0.001~0.01mg/mL、より好ましくは0.001~0.005mg/mLである。タンパク質分解酵素の濃度は、活性単位でも表わすことができ、工程(a)の合成反応液中において、1U/mL以上であることが好ましく、例えば1U/mL~100U/mL、好ましくは1U/mL~10U/mL、より好ましくは1U/mL~5U/mLである。
タンパク質分解酵素の活性は、当該分野で一般的に用いられている定義により決定されるものである。例えば、プロテイナーゼKの活性定義は以下に記載する条件下において275nmでの光学密度が1分あたり1マイクログラムのチロシンに相当する増加を引き起こす酵素活性を1ユニット(1U)とする。なお、サブチリシン等の他のタンパク質分解酵素の活性も同様にして測定することができる。
[試薬]
(A)カゼイン溶液(基質溶液):
2.0%(20.0gのハマーステンミルクカゼイン(Merck)を400mLの0.5%NaOH溶液に懸濁して溶解する。1N HClでpHを7.2に調整し、4mM CaSO4(pH 7.2)を含む500mLの20mMホウ酸緩衝液を加えて水にて1000mLにする)
(B)TCAソリューション(TCA溶液):20%トリクロロ酢酸(TCA)
(C)酵素希釈液:2mM CaSO4(pH 8.0)を含む10mMホウ酸緩衝液
アッセイ混合物中の濃度
ホウ酸緩衝液 10 mM
カゼイン 1.0%
CaSO4 2mM
[手順]
1. 1.0mLの基質溶液(A)と0.9mLの酵素希釈液(C)を試験管にピペットで入れ、35℃で約5分間平衡化する。
2. 測定対象サンプル 0.1mLを加えて混合する。
3. 35℃で10分後、2.0mLのTCA溶液(B)を加えて反応を停止する。
4. 35℃でさらに30分間インキュベートする。
5. 14000rpmにて遠心し、上清を275nmの吸光度を測定する(OD test)。
同時に、35℃で10分間インキュベートした後、最初に基質溶液を2.0mLのTCA溶液(B)と混合し、続いて測定対象サンプルを添加してブランクを調製し、同じ手順4-5をする (OD blank)。
活性は、次の式を使用して計算する。
Volume activity(U/mL)={OD(OD test-OD blank)×Vt×df}/{0.0074×1.0×t×Vs}=OD×541×df
Weight activity(U / mg)=(U / ml)×1 / C
Vt:総量(4.0mL)
Vs:サンプル量(0.1mL)
0.0074:チロシンの吸光係数(cm2/マイクログラム)
t:反応時間(10分)
df:希釈係数
C:溶解中の酵素濃度(mg/mL)
上記方法にてプロメガ社のプロテイナーゼKを測定すると0.001mg/mL溶液は約1U/mL(1.2~1.4U/mL)となる。
[試薬]
(A)カゼイン溶液(基質溶液):
2.0%(20.0gのハマーステンミルクカゼイン(Merck)を400mLの0.5%NaOH溶液に懸濁して溶解する。1N HClでpHを7.2に調整し、4mM CaSO4(pH 7.2)を含む500mLの20mMホウ酸緩衝液を加えて水にて1000mLにする)
(B)TCAソリューション(TCA溶液):20%トリクロロ酢酸(TCA)
(C)酵素希釈液:2mM CaSO4(pH 8.0)を含む10mMホウ酸緩衝液
アッセイ混合物中の濃度
ホウ酸緩衝液 10 mM
カゼイン 1.0%
CaSO4 2mM
[手順]
1. 1.0mLの基質溶液(A)と0.9mLの酵素希釈液(C)を試験管にピペットで入れ、35℃で約5分間平衡化する。
2. 測定対象サンプル 0.1mLを加えて混合する。
3. 35℃で10分後、2.0mLのTCA溶液(B)を加えて反応を停止する。
4. 35℃でさらに30分間インキュベートする。
5. 14000rpmにて遠心し、上清を275nmの吸光度を測定する(OD test)。
同時に、35℃で10分間インキュベートした後、最初に基質溶液を2.0mLのTCA溶液(B)と混合し、続いて測定対象サンプルを添加してブランクを調製し、同じ手順4-5をする (OD blank)。
活性は、次の式を使用して計算する。
Volume activity(U/mL)={OD(OD test-OD blank)×Vt×df}/{0.0074×1.0×t×Vs}=OD×541×df
Weight activity(U / mg)=(U / ml)×1 / C
Vt:総量(4.0mL)
Vs:サンプル量(0.1mL)
0.0074:チロシンの吸光係数(cm2/マイクログラム)
t:反応時間(10分)
df:希釈係数
C:溶解中の酵素濃度(mg/mL)
上記方法にてプロメガ社のプロテイナーゼKを測定すると0.001mg/mL溶液は約1U/mL(1.2~1.4U/mL)となる。
核酸ポリマーの濃度は、工程(a)の合成反応液中において1ng/μL以上であることが好ましく、例えば1~10ng/μL、好ましくは1~5ng/μL、より好ましくは1~2ng/μLである。
鋳型RNAから一本鎖cDNAを合成する反応は、特に制限されず、従来より公知の各種の方法を採用することができる。
1-1.逆転写反応
鋳型RNAから一本鎖cDNAを合成する反応の一例としては、逆転写反応が挙げられる。工程(a)を逆転写反応で行う場合、典型的には、逆転写酵素等の逆転写活性を有するタンパク質を含む逆転写用組成物(又は逆転写反応用溶液)をインキュベーションする工程を含む。
鋳型RNAから一本鎖cDNAを合成する反応の一例としては、逆転写反応が挙げられる。工程(a)を逆転写反応で行う場合、典型的には、逆転写酵素等の逆転写活性を有するタンパク質を含む逆転写用組成物(又は逆転写反応用溶液)をインキュベーションする工程を含む。
逆転写用組成物は、例えば、鋳型RNA、タンパク質分解酵素及び/又は核酸ポリマー、プライマー、デオキシリボヌクレオチド、及び逆転写酵素を含む。また、逆転写用組成物は、DNAポリメラーゼ等の他の成分等も任意に含み得る。
プライマーとしては、例えば、鋳型RNAに対する特異的なプライマー、オリゴdTプライマー、ランダムプライマー、これら2種以上の組合せが挙げられる。本発明は、オリゴdTプライマー及び/又はランダムプライマーを用いる逆転写反応を行う場合に有益であり、オリゴdTプライマー及びランダムプライマーの組合せを用いて逆転写反応を行う場合に特に有益である。オリゴdTプライマーとランダムプライマーのモル比は、例えば1:5~1:15、好ましくは1:8~1:12である。
ランダムプライマーとしては、例えば、完全ランダムプライマー、NSR(Not So Random)プライマーが挙げられる。
完全ランダムプライマーとは、種々の塩基配列を有するプライマーの混合物であり、各塩基配列は完全にランダムな塩基配列である。完全ランダムプライマーは、rRNA配列と完全に一致する(又は完全に相補的な)配列を含み得る。完全ランダムプライマーとしては、完全ランダムペンタマー、完全ランダムヘキサマー、完全ランダムヘプタマー、完全ランダムオクタマー、これらの組合せなどが例示できる。例えば、完全ランダムヘキサマーは、4種類のヌクレオチド(A、T、C、G)で可能な全ての塩基配列(46種類)の混合物であってもよい。
NSRプライマーとは、完全ランダムプライマーから、rRNA配列と完全に相補的な配列を有するプライマーを除去したものである。除去するrRNA配列としては、例えば、18S rRNA配列、28S rRNA配列、12S rRNA配列、16S rRNA配列、これらの組合せが挙げられる。
NSRプライマーとしては、例えば、完全ランダムヘキサマーからrRNA配列と完全に相補的な配列を有するヘキサマーを除外したものが挙げられる。また、完全ランダムペンタマー、完全ランダムヘプタマー、完全ランダムオクタマーなどのプライマーセットからrRNA配列と完全に相補的な配列を有するものを除外したものをNSRプライマーとすることもできる。
プライマーの長さは、アニーリングの観点から、例えば5塩基以上、好ましくは6塩基以上であり、合成の観点から、例えば30塩基以下、好ましくは25塩基以下、より好ましくは20塩基以下である。
逆転写用組成物において、プライマーの濃度は、特に制限されないが、例えば1~10μM、好ましくは2~6μM、より好ましくは3~5μMである。
デオキシリボヌクレオチドとしては、デオキシリボヌクレオシドトリホスフェートが好ましい。デオキシリボヌクレオチドトリホスフェートとしては、例えば、デオキシシチジントリホスフェート(dCTP)、デオキシグアノシントリホスフェート(dGTP)、デオキシアデノシントリホスフェート(dATP)、デオキシチミジントリホスフェート(dTTP)、デオキシウリジントリホスフェート(dUTP)、これらの誘導体、これら2種以上の組合せが挙げられる。これらのうち、dCTP、dGTP、dATP、及びdTTPの混合物、dCTP、dGTP、dATP、及びdUTPの混合物、dCTP、dGTP、dATP、dTTP、及びdUTPの混合物等が好ましい。
逆転写酵素は、逆転写活性(RNA依存性DNAポリメラーゼ活性)を有する任意のタンパク質(酵素)をいい、特に制限されないが、逆転写酵素活性を示すポリメラーゼが好ましい。また、逆転写酵素は、RNase H活性が低いか、又はRNase H活性がないものが好ましい。逆転写酵素の例としては、例えば、トリ骨髄芽球ウイルス(Avian Myeloblastosis Virus)逆転写酵素(AMV-RT)、モロニーネズミ白血病ウイルス(Moloney Murine Leukemia Virus)逆転写酵素(MMLV-RT)、ヒト免疫ウイルス(Human Immunovirus)逆転写酵素(HIV-RT)、EIRV-RT、RAV2-RT、C.ヒドロゲノホルマンス(C.hydrogenogormans)DNAポリメラーゼ、rTthDNAポリメラーゼ、スーパースクリプト(SuperScript)I、スーパースクリプト(SuperScript)II、これらの変異体、及びこれらの誘導体が挙げられる。これらのうち、MMLV-RTが好ましい。
逆転写用組成物は、下記のDNAポリメラーゼを含んでいてもよいし、含まなくてもよい:Taq、Tbr、Tfl、Tru、Tth、Tli、Tac、Tne、Tma、Tih、Tfi、Pfu、Pwo、Kod、Bst、Sac、Sso、Poc、Pab、Mth、Pho、ES4、VENT(商標)、DEEPVENT(商標)、これらの変異体。
逆転写用組成物は、RNase阻害剤を含んでいてもよい。RNase阻害剤は、特に制限されず、例えば、ヒト胎盤由来、ラット肺由来、又はブタ肝臓由来のタンパク質が挙げられる。
逆転写用組成物は、添加剤を含んでいてもよい。添加剤としては、例えば、緩衝剤、塩、これら2種以上の組合せが挙げられる。
緩衝剤としては、例えば、トリス(Tris)、トリシン(Tricine)、ビス-トリシン(Bis-Tricine)、ヘペス(Hepes)、モプス(Mops)、テス(Tes)、タプス(Taps)、ピペス(Pipes)、キャプス(Caps)、これら2種以上の組合せが挙げられる。緩衝剤は、通常、水(好ましくはヌクレアーゼフリー水)に溶解され、水溶液の形態で使用される。
塩としては、例えば、塩化物(例えば、塩化リチウム、塩化ナトリウム、塩化カリウム、塩化マグネシウム、塩化マンガン)、酢酸塩(例えば、酢酸リチウム、酢酸ナトリウム、酢酸カリウム、酢酸マグネシウム、酢酸マンガン)、硫酸塩(例えば、硫酸カリウム、硫酸マグネシウム、硫酸マンガン)、これら2種以上の組合せが挙げられる。
逆転写用組成物のインキュベーションの条件は、鋳型RNAから一本鎖cDNAが生成される限り特に制限されず、当該分野において知られている、いずれの条件も採用することができる。インキュベーションの温度は、例えば30~65℃、好ましくは35~60℃である。インキュベーションの時間は、例えば5~120分、好ましくは10~60分である。
1-2.RNAを鋳型としてcDNAを増幅させる増幅逆転写法(RT-RamDA法とも言う)
RT-RamDA法とは、鋳型RNA、プライマー、DNA鎖特異的RNA:DNAハイブリッド鎖分解酵素、RNase Hマイナス型逆転写酵素、及び基質を含む混合物をインキュベートする工程を含む、核酸の増幅方法である。RT-RamDA法では、RNase Hマイナス型逆転写酵素のRNA依存性DNAポリメラーゼ活性により鋳型RNAの相補鎖DNA(cDNA)を合成し、RNAとcDNAとのハイブリッド鎖のうちのcDNA鎖をDNA鎖特異的RNA:DNAハイブリッド鎖分解酵素により無作為に切断し、前記の切断部位が起点となり、RNase Hマイナス型逆転写酵素の鎖置換活性により3’側のcDNA鎖がRNAから剥がされ、RNase Hマイナス型逆転写酵素により剥がされた部分に新たなcDNA鎖が合成される。RT-RamDA法の詳細については、米国特許出願公開公報2017/0275685(参照することによりその全体が本明細書に組み込まれる)等に記載されている。
RT-RamDA法とは、鋳型RNA、プライマー、DNA鎖特異的RNA:DNAハイブリッド鎖分解酵素、RNase Hマイナス型逆転写酵素、及び基質を含む混合物をインキュベートする工程を含む、核酸の増幅方法である。RT-RamDA法では、RNase Hマイナス型逆転写酵素のRNA依存性DNAポリメラーゼ活性により鋳型RNAの相補鎖DNA(cDNA)を合成し、RNAとcDNAとのハイブリッド鎖のうちのcDNA鎖をDNA鎖特異的RNA:DNAハイブリッド鎖分解酵素により無作為に切断し、前記の切断部位が起点となり、RNase Hマイナス型逆転写酵素の鎖置換活性により3’側のcDNA鎖がRNAから剥がされ、RNase Hマイナス型逆転写酵素により剥がされた部分に新たなcDNA鎖が合成される。RT-RamDA法の詳細については、米国特許出願公開公報2017/0275685(参照することによりその全体が本明細書に組み込まれる)等に記載されている。
工程(a)をRT-RamDA法で行う場合、DNA鎖特異的RNA:DNAハイブリッド鎖分解酵素を含む前記逆転写用組成物をインキュベーションする工程を含む。DNA鎖特異的RNA:DNAハイブリッド鎖分解酵素は、RNAとDNAとのハイブリッド鎖中のDNA鎖を切断する活性を有する酵素であることが好ましい。当該酵素としては、例えば、二本鎖特異的DNA分解酵素、非特異的DNA分解酵素を使用することができる。本発明ではこれらのうち、非特異的DNA分解酵素が好ましい。
二本鎖特異的DNA分解酵素(二本鎖特異的ヌクレアーゼ;DSNとも言う)は、原核生物又は真核生物に由来する酵素を使用することができるが、好ましくは、甲殻類由来の二本鎖特異的DNA分解酵素又はその改変体を使用することができる。具体的な例としては、以下のものが挙げられる。
・ Solenocera melantho(ナミクダヒゲエビ)DNase
・ Penaeus japonicus(クルマエビ)DNase
・ Paralithodes camtschaticus(タラバガニ)DSN
・ Pandalus borealis(ホッコクアカエビ)dsDNase
・ Chionoecetes opilio(ズワイガニ)DSN
・ その他のDSNホモログ
・ Solenocera melantho(ナミクダヒゲエビ)DNase
・ Penaeus japonicus(クルマエビ)DNase
・ Paralithodes camtschaticus(タラバガニ)DSN
・ Pandalus borealis(ホッコクアカエビ)dsDNase
・ Chionoecetes opilio(ズワイガニ)DSN
・ その他のDSNホモログ
二本鎖特異的DNA分解酵素は、好ましくは、60℃未満でもDNA分解活性を有する酵素である。上記の中では、エビ由来の二本鎖特異的DNA分解酵素又はその改変体が好ましい。
二本鎖特異的DNA分解酵素としては、市販品を使用することができる。市販品としては、例えば、dsDNase(ArcticZymes社)、Hl-dsDNase(ArcticZymes社)、dsDNase(Thermo scientific社)、Shrimp Dnase、Recombinant(affymetrix社)、Atlantis dsDNase(Zymo Research社)、Thermolabile Nuclease(Roche社)などを挙げることができる。
非特異的DNA分解酵素としては、RNAとDNAとのハイブリッド鎖のDNA鎖を切断する活性を有し、RNAとDNAとのハイブリッド鎖のRNA鎖、一本鎖RNAを切断する活性を実質的に有さず、好ましくは、一本鎖DNAを切断する活性がRNAとDNAとのハイブリッド鎖のDNA鎖を切断する活性に比較して低くなる酵素を挙げることができる。非特異的DNA分解酵素は、好ましくは、60℃未満でもDNA分解活性を有する酵素である。このような非特異的DNA分解酵素としては、市販品を使用することもでき、例えば、DNaseI(Thermo Fisher社製、DNaseI)等を用いることが可能である。
非特異的DNA分解酵素は、原核生物又は真核生物に由来する酵素を使用することができるが、好ましくは、ほ乳類由来の非特異的DNA分解酵素又はその改変体、より好ましくはウシ由来の非特異的DNA分解酵素又はその改変体を使用することができる。
上記改変体とは、天然由来のアミノ酸配列を改変することによって得られる酵素を意味する。具体的には、天然由来のアミノ酸配列と80%以上(好ましくは90%以上、より好ましくは95%以上)の配列同一性を有するアミノ酸配列からなる酵素、並びに天然由来のアミノ酸配列において1又は数個(例えば、1~10個、好ましくは1~5個、より好ましくは1~3個)のアミノ酸の欠失、置換、及び/又は付加を有するアミノ酸配列からなる酵素である。
工程(a)をRT-RamDA法で行う場合、前記逆転写用組成物は、一本鎖DNA結合タンパク質を含んでいてもよい。一本鎖DNA結合タンパク質は、通常、DNA鎖特異的RNA:DNAハイブリッド鎖分解酵素とともに使用される。一本鎖DNA結合タンパク質としては、例えば、T4ジーン32プロテイン、RecA、SSB(Single-Stranded DNA Binding Protein)、これら2種以上の組合せが挙げられる。
工程(a)をRT-RamDA法で行う場合、前記逆転写用組成物のインキュベーションは、等温条件で行ってもよいし、熱サイクル条件で行ってもよい。
(1)等温条件
インキュベーションを等温条件で行う場合、例えば25℃以上50℃未満の間の所定の温度、好ましくは30~45℃の間の所定の温度、より好ましくは35~40℃の間の所定の温度、例えば37℃で所定の時間(例えば5~180分、好ましくは10~150分)行うことができる。
25℃以上50℃未満の間の所定の温度でのインキュベーションは、2以上の段階に分けて行ってもよい。例えば25℃以上30℃未満の間の所定の温度で5~15分、次いで30℃以上35℃未満の間の所定の温度で5~15分、次いで35℃以上50℃未満の間の所定の温度で所定の時間(例えば5~60分)インキュベーションしてもよい。
25℃以上50℃未満の間の所定の温度でのインキュベーションの後、例えば50℃以上100℃未満の間の所定の温度でインキュベーションしてもよい。50℃以上100℃未満の間の所定の温度でのインキュベーションは2以上の段階に分けて行ってもよい。例えば50℃以上80℃未満の間の所定の温度で5~15分、次いで80~100℃の間の所定の温度で5~15分インキュベーションしてもよい。
インキュベーションを等温条件で行う場合、例えば25℃以上50℃未満の間の所定の温度、好ましくは30~45℃の間の所定の温度、より好ましくは35~40℃の間の所定の温度、例えば37℃で所定の時間(例えば5~180分、好ましくは10~150分)行うことができる。
25℃以上50℃未満の間の所定の温度でのインキュベーションは、2以上の段階に分けて行ってもよい。例えば25℃以上30℃未満の間の所定の温度で5~15分、次いで30℃以上35℃未満の間の所定の温度で5~15分、次いで35℃以上50℃未満の間の所定の温度で所定の時間(例えば5~60分)インキュベーションしてもよい。
25℃以上50℃未満の間の所定の温度でのインキュベーションの後、例えば50℃以上100℃未満の間の所定の温度でインキュベーションしてもよい。50℃以上100℃未満の間の所定の温度でのインキュベーションは2以上の段階に分けて行ってもよい。例えば50℃以上80℃未満の間の所定の温度で5~15分、次いで80~100℃の間の所定の温度で5~15分インキュベーションしてもよい。
(2)熱サイクル条件
インキュベーションを熱サイクル条件で行う場合、例えば20℃以上30℃未満の間の所定の温度T1(例えば25℃)と30~45℃の間の所定の温度T2(例えば37℃)とを組み合わせて、T1で所定の時間(例えば1~3分、一例として2分)とT2で所定の時間(例えば1~3分、一例として2分)とを1サイクルとして、これを好ましくは10~40サイクル、より好ましくは15~35サイクル繰り返して行ってもよい。なお、上記の熱サイクルに先立って、例えば25℃以上30℃未満の間の所定の温度で所定の時間(例えば5~15分)、次いで30℃以上35℃未満の間の所定の温度で所定の時間(例えば5~15分)、次いで35℃以上50℃未満の間の所定の温度で所定の時間(例えば1~5分)インキュベーションしてもよい。また、上記の熱サイクルの後、例えば50℃以上80℃未満の間の所定の温度で所定の時間(例えば5~15分)、次いで80~90℃の間の所定の温度で所定の時間(例えば5~15分)インキュベーションしてもよい。
インキュベーションを熱サイクル条件で行う場合、例えば20℃以上30℃未満の間の所定の温度T1(例えば25℃)と30~45℃の間の所定の温度T2(例えば37℃)とを組み合わせて、T1で所定の時間(例えば1~3分、一例として2分)とT2で所定の時間(例えば1~3分、一例として2分)とを1サイクルとして、これを好ましくは10~40サイクル、より好ましくは15~35サイクル繰り返して行ってもよい。なお、上記の熱サイクルに先立って、例えば25℃以上30℃未満の間の所定の温度で所定の時間(例えば5~15分)、次いで30℃以上35℃未満の間の所定の温度で所定の時間(例えば5~15分)、次いで35℃以上50℃未満の間の所定の温度で所定の時間(例えば1~5分)インキュベーションしてもよい。また、上記の熱サイクルの後、例えば50℃以上80℃未満の間の所定の温度で所定の時間(例えば5~15分)、次いで80~90℃の間の所定の温度で所定の時間(例えば5~15分)インキュベーションしてもよい。
2.工程(b)
工程(b)は、塩の存在下で行うことが好ましい。塩の存在下で二本鎖cDNAを合成することによりライブラリー収量を増加させることができる。これは、プライマーの鋳型への結合が最適化されたことで生成される二本鎖cDNAが増加したことが一因と考えられる。
工程(b)は、塩の存在下で行うことが好ましい。塩の存在下で二本鎖cDNAを合成することによりライブラリー収量を増加させることができる。これは、プライマーの鋳型への結合が最適化されたことで生成される二本鎖cDNAが増加したことが一因と考えられる。
塩としては、例えば、塩化物塩(例えば、塩化リチウム、塩化ナトリウム、塩化カリウム等のアルカリ金属塩化物塩;塩化マグネシウム等のアルカリ土類金属塩化物塩;塩化マンガン等の遷移金属塩化物塩)が挙げられる。中でも、アルカリ金属塩化物塩及び/又はアルカリ土類金属塩化物塩が好ましく、塩化マグネシウム、塩化カリウム、及び塩化ナトリウムからなる群より選択される少なくとも一種が好ましい。塩は1種を単独で又は2種以上を組み合わせて用いてもよい。
塩が工程(a)の反応産物に含まれる場合、工程(b)の反応溶液に別途塩を添加しなくてもよいし、又は更に工程(b)の反応溶液に別途塩を添加してもよい。
塩の濃度は、工程(b)の合成反応液中において、例えば60mM以下、好ましくは50mM以下、より好ましくは40mM以下である。また、塩の濃度は、工程(b)の合成反応液中において、例えば5~60mM、好ましくは5~50mM、より好ましくは5~40mMである。特定の実施形態では、工程(b)の合成反応液中における塩の濃度は、例えば20mM以下(例:5~20mM)、15mM以下(例:5~15mM、又は12mM以下(例:5~12mM)などであってもよい。当該塩の濃度は、塩化物塩の濃度であることが好ましい。
工程(b)の合成反応液中の塩のうち、塩化ナトリウムの濃度は少ない方がよく、工程(b)の合成反応液中において、例えば20mM以下、好ましくは15mM以下、より好ましくは12mM以下である。工程(b)の合成反応液は塩化ナトリウムを含まなくてもよいし、工程(b)の合成反応液が塩化ナトリウムを含む場合、その濃度は、工程(b)の合成反応液中において、例えば5~20mM、好ましくは5~15mM、より好ましくは5~12mMである。
工程(b)の合成反応液中の塩のうち、塩化カリウムの濃度は、例えば5mM以上(例:5~50mM)、好ましくは10mM以上(例:10~40mM)、より好ましくは15mM以上(例:15~35mM)である。
工程(b)の合成反応液中の塩のうち、塩化マグネシウムの濃度は、例えば20mM以下(例:1~20mM)、好ましくは15mM以下(例:1~15mM)、より好ましくは10mM以下(例:1~10mM)である。
特定の実施形態では、工程(b)の合成反応液中において、塩化物塩の濃度が60mM以下(例えば50mM以下、好ましくは40mM以下)であり、且つ、塩化ナトリウムの濃度が20mM以下であることが好ましく、塩化物塩の濃度が60mM以下(例えば50mM以下、好ましくは40mM以下)であり、塩化ナトリウムの濃度が20mM以下であり、且つ、塩化カリウムの濃度が5mM以上であることがより好ましい。
工程(b)の合成反応液中の塩のうち、塩化ナトリウムの濃度は少ない方がよく、工程(b)の合成反応液中において、例えば20mM以下、好ましくは15mM以下、より好ましくは12mM以下である。工程(b)の合成反応液は塩化ナトリウムを含まなくてもよいし、工程(b)の合成反応液が塩化ナトリウムを含む場合、その濃度は、工程(b)の合成反応液中において、例えば5~20mM、好ましくは5~15mM、より好ましくは5~12mMである。
工程(b)の合成反応液中の塩のうち、塩化カリウムの濃度は、例えば5mM以上(例:5~50mM)、好ましくは10mM以上(例:10~40mM)、より好ましくは15mM以上(例:15~35mM)である。
工程(b)の合成反応液中の塩のうち、塩化マグネシウムの濃度は、例えば20mM以下(例:1~20mM)、好ましくは15mM以下(例:1~15mM)、より好ましくは10mM以下(例:1~10mM)である。
特定の実施形態では、工程(b)の合成反応液中において、塩化物塩の濃度が60mM以下(例えば50mM以下、好ましくは40mM以下)であり、且つ、塩化ナトリウムの濃度が20mM以下であることが好ましく、塩化物塩の濃度が60mM以下(例えば50mM以下、好ましくは40mM以下)であり、塩化ナトリウムの濃度が20mM以下であり、且つ、塩化カリウムの濃度が5mM以上であることがより好ましい。
一本鎖cDNAから二本鎖cDNAを合成する反応は、特に制限されず、従来より公知の各種の方法を採用することができる。当該反応は、典型的には、鋳型DNA及びプライマーの存在下で5´→3´方向へのヌクレオチドの重合反応を触媒する酵素(二本鎖cDNA合成酵素)を含む二本鎖cDNA合成用組成物をインキュベーションする工程を含む。
二本鎖cDNA合成用組成物は、例えば、一本鎖cDNA、塩、プライマー、デオキシリボヌクレオチド、及び二本鎖cDNA合成酵素を含む。また、二本鎖合成用組成物は、緩衝剤等の他の成分等も任意に含み得る。
二本鎖cDNA合成酵素は、5´→3´方向へのヌクレオチドの重合反応を触媒する酵素のことをいい、特に制限されない。例えば、クレノウフラグメント、T4 DNAポリメラーゼ、これらの変異体、及びこれらの誘導体が挙げられる。これらのうち、クレノウフラグメントが好ましい。
プライマー、デオキシリボヌクレオチド、及び緩衝剤としては、それぞれ、前記逆転写用組成物で例示したものが挙げられる。
二本鎖cDNA合成用組成物のインキュベーションの条件は、一本鎖cDNAから二本鎖cDNAが生成される限り特に制限されず、当該分野において知られている、いずれの条件も採用することができる。例えば10℃以上35℃未満の間の所定の温度で所定の時間(例えば5~90分)インキュベーションしてもよい。
一実施形態において、ライブラリーの調製方法は、前記二本鎖cDNA(例えば工程(b)の反応溶液)を加熱する工程を更に含むことが好ましい。当該工程は、工程(b)の後、次の工程(c)の前に実施することが好ましい。当該工程は、不活化処理に相当し得る。加熱条件(不活化の条件)は特に限定されず、例えば、70℃以上(好ましくは75℃以上、更に好ましくは80℃以上)の所定の温度で所定の時間(例えば5分以上、好ましくは10分以上)インキュベーションしてもよい。加熱温度(不活化のための温度)は好ましくは80℃以上95℃以下、より好ましくは80℃以上90℃以下、更に好ましくは80℃以上85℃以下である。またインキュベーション時間は、好ましくは5分以上30分以下、より好ましくは10分以上20分以下、更に好ましくは10分以上15分以下である。このような加熱工程を行うことで、工程(c)において、アダプターダイマーの形成をより効果的に抑制できる。理論に束縛されないが、このような温度で加熱することで二本鎖cDNAの高次構造が解かれ、ライブラリー調製における酵素反応効率(例えばトランスポゾン反応)が向上することで、アダプターダイマーの形成をより効果的に抑制できると推測される。
3.工程(c)
ライブラリーは、次世代シーケンシングに必要な配列が付与されている核酸断片であることが好ましい。次世代シーケンシングに必要な配列としてはフローセル結合配列やシーケンシングに必要な配列を含む。
ライブラリーは、次世代シーケンシングに必要な配列が付与されている核酸断片であることが好ましい。次世代シーケンシングに必要な配列としてはフローセル結合配列やシーケンシングに必要な配列を含む。
ここで、次世代シーケンシング(NGS解析、次世代シーケンサー解析等ともいう)とは、典型的には、数百万から数十億もの膨大なシーケンシング反応を同時に実施できる配列決定技術をいう。次世代シーケンシングとしては、例えば、エマルジョンPCRやブリッジPCR等の増幅技術や1分子観察等の高感度検出技術を用いて並列的に配列解析する方法が挙げられる。次世代シーケンシングを行う装置(シークエンサー)としては、特に限定されないが、例えば、MiSeq、HiSeq、NovaSeq(イルミナ社);Genetic Analyzer V2.0、Ion Proton(サーモフィッシャーサイエンティフィック社);MinION、PromethION(ナノポア社)等が挙げられる。次世代シーケンシングについては、例えば米国特許出願公開公報2014/178438(参照することによりその全体が明細書に組み込まれる)等に記載されている。
ライブラリー調製に用いられる二本鎖cDNAは、例えば5μL以下、好ましくは4μL以下、より好ましくは3μL以下、さらに好ましくは2μL以下、特に好ましくは1μL以下の溶液の形態である。当該二本鎖cDNAは、例えば0.01μL以上、0.05μL以上、又は0.1μL以上の溶液の形態である。本発明では、少量の二本鎖cDNAであっても精製することなく十分なライブラリー収量を得ることができる。
ライブラリーの調製方法は、特に制限されない。例えばトランスポゾンを用いて鋳型DNAを断片化及びタグ化し、タグ化されたDNA断片から構成されるライブラリーの核酸増幅を行うことが行われている。例えば2つのトランスポゾン末端配列(アダプターに相当)及びトランスポザーゼがトランスポソーム複合体を形成し、トランスポソーム複合体は、溶液中の鋳型DNAをフラグメント化及びタグ化して、PCR等で次世代シーケンシングに必要な配列が付加されるため配列解析を行うことが可能である(これを、「トランスポゾン法」ともいう)。トランスポゾンを使用した市販のキットとしては、例えば、illumina社のNextera XT DNA Library Preparation Kitが挙げられるが、これに限定されない。
また別の手法としては鋳型DNA(例えば、全長DNA)の断片化、断片化したDNAの末端修復及び/又は断片化DNA若しくは末端修復した断片化DNAの末端のアデニル化(これを、「A付加」ともいう)、末端修復及び/又は末端アデニル化を行ったDNA断片へのアダプター配列をT4 Ligase等にてライゲーションすることで付加し、並びにアダプターを付加したDNA断片から構成されるライブラリーの核酸増幅を行うことが行われている。アダプター配列には次世代シーケンシングに必要な配列が付加されるため配列解析を行うことが可能である(これを、「ライゲーション法」ともいう)。アダプターの配列及び長さは、特に限定されず、例えば、Y字アダプター、ステムループアダプターなどでもよい。ライゲーション法を使用した市販のキットとしては、例えば、東洋紡株式会社のGenNext(R) NGS Library Prep KitやKAPA社のKAPA HyperPrep Kitが挙げられるが、これらに限定されない。
一実施形態において、ライブラリー調製方法は、トランスポゾン法であることが好ましい。
従来、ライブラリー調製を行う前に二本鎖cDNA溶液の精製を行い溶液中のプライマー、アダプター、dNTP、塩類を除去することが一般的である。ここで、「精製」とは、溶液等に含まれる前記のプライマーのような夾雑物質と、二本鎖cDNAとを分離することをいう。精製方法としては磁性ビーズを用いた方法、カラムを用いた方法、フェノール又はフェノール・クロロホルムを用いた方法、タンパク質凝集作用を用いた方法が知られている。本発明は、このような精製工程を経ていない非精製の二本鎖cDNAを用いてライブラリーを調製することを特徴としている。なお、前記工程(b)により合成された二本鎖cDNAの溶液において加熱等の方法により夾雑物質を分解等する行為は、本発明でいう精製には該当しない。本発明において、「非精製の二本鎖cDNA」とは、前記工程(b)により合成された二本鎖cDNA(二本鎖cDNA溶液の状態の場合を含む)そのもの又は加熱したもの、前記二本鎖cDNAを水、緩衝液等の溶媒で希釈したもの、或いは前記二本鎖cDNA溶液を乾燥させて濃縮したもの等が挙げられるが、精製工程を経ていなければ、これらに限定されるものではない。本発明では、二本鎖cDNA溶液の精製を省略しても、十分なライブラリー収量を得ることができる。本発明では、ライブラリー濃度を増大することができ、例えば10nM以上、15nM以上、20nM以上、25nM以上、30nM以上、35nM以上、又は40nM以上にすることができる。また、本発明では、ライブラリー中のアダプターダイマーの量を低減することができ、例えば、MultiNA(島津製作所)にてライブラリーを測定したとき、100bp以上150bp未満の領域に存在するアダプターダイマー発生を示す領域のピークの強度を、ライブラリー収量を示す領域(概ね150bp以上5000bp以下の範囲に存在する領域)の最大強度の90%以下、80%以下、70%以下、60%以下、又は50%以下にすることができる。
以下、実施例に基づいて本発明をより詳細に説明するが、本発明はこれらの実施例に限定されるものではない。
実施例1.塩化ナトリウムの濃度による収量への影響
本実施例では、塩化ナトリウムの濃度によって、ライブラリー収量への影響があるか検討するため、以下の実験を行った。
RNAを希釈し、RT-RamDA法により一本鎖cDNAの合成を行い、クレノウフラグメントを用いて二本鎖cDNAを合成した。その後トランスポゾン法を使用しているNextera XT DNA Library Preparation Kit(illumina社)を用いてライブラリーを調製しMultiNA(島津製作所)にてライブラリー濃度を測定した。手順は取扱い説明書に従った。
本実施例で用いる核酸断片サンプルは、NIH3T3細胞をRNeasy Mini Kit(Qiagen社)を用いて精製したRNA 10pgを用いた。1ng/μLのRNAを表1に示したRNA希釈液にて希釈し10pg/μLのRNA溶液を調製した。
本実施例では、塩化ナトリウムの濃度によって、ライブラリー収量への影響があるか検討するため、以下の実験を行った。
RNAを希釈し、RT-RamDA法により一本鎖cDNAの合成を行い、クレノウフラグメントを用いて二本鎖cDNAを合成した。その後トランスポゾン法を使用しているNextera XT DNA Library Preparation Kit(illumina社)を用いてライブラリーを調製しMultiNA(島津製作所)にてライブラリー濃度を測定した。手順は取扱い説明書に従った。
本実施例で用いる核酸断片サンプルは、NIH3T3細胞をRNeasy Mini Kit(Qiagen社)を用いて精製したRNA 10pgを用いた。1ng/μLのRNAを表1に示したRNA希釈液にて希釈し10pg/μLのRNA溶液を調製した。
このRNA溶液1μLを70℃1分30秒で熱処理を行った。その後、表2に示したRT-RamDA反応液2μLを添加し、3μLにて25℃10分、30℃10分、37℃30分、50℃5分、及び98℃5分でRT-RamDA反応を行った。RT-RamDA反応後、表3に示した二本鎖合成反応液2μLを添加し、5μLにて16℃60分で二本鎖合成し、80℃15分で加熱(不活化)を実施した。その後、表4に示した非精製プロトコルにてライブラリー調製を実施し、ライブラリー濃度をMultiNAにて測定した結果を表5に示す。



なお、NSRプライマー(ヘキサマー)は、Sigma社カスタムオリゴにて合成し、Ozsolak et al., Digital transcriptome profiling from attomole-levelRNA samples, Genome Research, Vol. 20, 2010, pp. 519-525に記載されたプライマー組成となるように混合して用いた。


塩化物塩の濃度が50mM以下(特に40mM以下)であって、塩化ナトリウム濃度が特に12mM以下である場合に収量が増加し、十分なライブラリー収量が得られた。
実施例2.ポリイノシン酸カリウム塩とプロテイナーゼKの添加量による収量への影響
本実施例では、ポリイノシン酸カリウム塩とプロテイナーゼKの添加量によって、ライブラリー収量への影響があるか検討するため、以下の実験を行った。
RNAを希釈し、RT-RamDA法により一本鎖cDNAの合成を行い、クレノウフラグメントを用いて二本鎖cDNAを合成した。その後Nextera XT DNA Library Preparation Kit(illumina社)を用いてライブラリーを調製しMultiNA(島津製作所)にてライブラリー濃度を測定した。手順は取扱い説明書に従った。
本実施例で用いる核酸断片サンプルは、NIH3T3細胞をRNeasy Mini Kit(Qiagen社)を用いて精製したRNA10pgを用いた。1ng/μLのRNAを表6に示したRNA希釈液にて希釈し10pg/μLのRNA溶液を調製した。
このRNA溶液1μLを70℃1分30秒で熱処理を行った。その後、表7に示したRT-RamDA反応液2μLを添加し、3μLにて25℃10分、30℃10分、37℃30分、50℃5分、及び98℃5分でRT-RamDA反応を行った。RT-RamDA反応後、表8に示した2本鎖合成反応液2μLを添加し、5μLにて16℃60分で二本鎖合成し、80℃15分での加熱(不活化)を実施した。その後、表9に示した非精製プロトコルにてライブラリー調製を実施し、ライブラリー濃度をMultiNAにて測定した結果を表10に示す。
本実施例では、ポリイノシン酸カリウム塩とプロテイナーゼKの添加量によって、ライブラリー収量への影響があるか検討するため、以下の実験を行った。
RNAを希釈し、RT-RamDA法により一本鎖cDNAの合成を行い、クレノウフラグメントを用いて二本鎖cDNAを合成した。その後Nextera XT DNA Library Preparation Kit(illumina社)を用いてライブラリーを調製しMultiNA(島津製作所)にてライブラリー濃度を測定した。手順は取扱い説明書に従った。
本実施例で用いる核酸断片サンプルは、NIH3T3細胞をRNeasy Mini Kit(Qiagen社)を用いて精製したRNA10pgを用いた。1ng/μLのRNAを表6に示したRNA希釈液にて希釈し10pg/μLのRNA溶液を調製した。
このRNA溶液1μLを70℃1分30秒で熱処理を行った。その後、表7に示したRT-RamDA反応液2μLを添加し、3μLにて25℃10分、30℃10分、37℃30分、50℃5分、及び98℃5分でRT-RamDA反応を行った。RT-RamDA反応後、表8に示した2本鎖合成反応液2μLを添加し、5μLにて16℃60分で二本鎖合成し、80℃15分での加熱(不活化)を実施した。その後、表9に示した非精製プロトコルにてライブラリー調製を実施し、ライブラリー濃度をMultiNAにて測定した結果を表10に示す。





プロテイナーゼK濃度が約1U(1.2~1.4U)/mL(=0.001mg/mL)以上で収量が増加、またポリイノシン酸カリウム塩濃度が1ng/μL以上で収量が増加し十分なライブラリー収量が得られた。
実施例3.ポリイノシン酸カリウム塩添加による非精製プロトコルによる収量改善
本実施例では、ポリイノシン酸カリウム塩の添加によって、ライブラリー収量への影響があるか検討するため、以下の実験を行った。
細胞を溶解し、RT-RamDA法により一本鎖cDNAの合成を行い、クレノウフラグメントを用いて二本鎖cDNAを合成した。その後Nextera XT DNA Library Preparation Kit(illumina社)を用いてライブラリーを調製しMultiNA(島津製作所)にてライブラリー濃度を測定した。手順は取扱い説明書に従った。
具体的には、以下の手法により行った。
実施例で用いる細胞サンプルは、以下の組成にて調製を行った。NIH3T3細胞をトリプシン溶液(ナカライテスク社)にて37℃2分反応させ、1細胞へ乖離した。解離後は直ちにPBS(-)へ置換し反応を止めた。FACS Melodyセルソーター(BD社)を用いて死細胞蛍光マーカー色素のPI陰性細胞を生細胞分画とし、この分画から120細胞を3μLの以下に示す表11に示したLysis Bufferに分取した。分取後は直ちに遠心を行い、-80℃にて保存した。RT-RamDA反応に使用する際は、融解後に117μLのLysis Bufferを加え、細胞溶解液1μLを1細胞溶解液として用いた。
この1細胞溶解液1μLを70℃1分30秒で熱処理を行った。その後、表12に示したRT-RamDA反応液2μLを添加し、3μLにて25℃10分、30℃10分、37℃30分、50℃5分、及び98℃5分でRT-RamDA反応を行った。RT-RamDA反応後、表13に示した2本鎖合成反応液2μLを添加し、5μLにて16℃60分で二本鎖合成し、80℃15分で加熱(不活化)を実施した。その後、表14に示した精製及び非精製プロトコルにてライブラリー調製を実施し、ライブラリー濃度をMultiNAにて測定した結果を表15に示す。
本実施例では、ポリイノシン酸カリウム塩の添加によって、ライブラリー収量への影響があるか検討するため、以下の実験を行った。
細胞を溶解し、RT-RamDA法により一本鎖cDNAの合成を行い、クレノウフラグメントを用いて二本鎖cDNAを合成した。その後Nextera XT DNA Library Preparation Kit(illumina社)を用いてライブラリーを調製しMultiNA(島津製作所)にてライブラリー濃度を測定した。手順は取扱い説明書に従った。
具体的には、以下の手法により行った。
実施例で用いる細胞サンプルは、以下の組成にて調製を行った。NIH3T3細胞をトリプシン溶液(ナカライテスク社)にて37℃2分反応させ、1細胞へ乖離した。解離後は直ちにPBS(-)へ置換し反応を止めた。FACS Melodyセルソーター(BD社)を用いて死細胞蛍光マーカー色素のPI陰性細胞を生細胞分画とし、この分画から120細胞を3μLの以下に示す表11に示したLysis Bufferに分取した。分取後は直ちに遠心を行い、-80℃にて保存した。RT-RamDA反応に使用する際は、融解後に117μLのLysis Bufferを加え、細胞溶解液1μLを1細胞溶解液として用いた。
この1細胞溶解液1μLを70℃1分30秒で熱処理を行った。その後、表12に示したRT-RamDA反応液2μLを添加し、3μLにて25℃10分、30℃10分、37℃30分、50℃5分、及び98℃5分でRT-RamDA反応を行った。RT-RamDA反応後、表13に示した2本鎖合成反応液2μLを添加し、5μLにて16℃60分で二本鎖合成し、80℃15分で加熱(不活化)を実施した。その後、表14に示した精製及び非精製プロトコルにてライブラリー調製を実施し、ライブラリー濃度をMultiNAにて測定した結果を表15に示す。





非精製プロトコルにおいてポリイノシン酸を添加することで収量が増加し、十分なライブラリー収量が得られた。なお、条件1の精製プロトコルでは5μLの二本鎖cDNAを使用しているのに対し、条件2及び3の非精製プロトコルでは、その1/5量の1μLの二本鎖cDNAを使用してライブラリー調製を行っている。しかしながら、特に条件3の場合において、条件1の場合と比較して1/2程度の量までライブラリー収量が高められていた。従って、本発明により効率的なライブラリー調製が可能となることがわかった。
実施例4.鋳型RNA量の評価
本実施例では、本発明によりライブラリー調製を行う際に必要となる鋳型RNA量について検討するため、以下の実験を行った。
RNAを希釈し、RT-RamDA法又は通常の逆転写方法(RT法)により一本鎖cDNAの合成を行い、クレノウフラグメントを用いて二本鎖cDNAを合成した。その後Nextera XT DNA Library Preparation Kit(illumina社)を用いてライブラリーを調製しMultiNA(島津製作所)にてライブラリー濃度を測定した。手順は取扱い説明書に従った。
本実施例で用いる核酸断片サンプルは、マウスES細胞total RNA(ユニーテック株式会社)を用いた。1ng/μLの前記RNAを表16に示したRNA希釈液にて希釈し10pg/μLのRNA溶液を調製した(RNA鋳型量10pg)。
このRNA溶液1μLを70℃1分30秒で熱処理を行った。条件1、2は表17に示したRT-RamDA反応液2μLを添加し、条件3は表18に示したRT反応液2μLを添加し、3μLにて25℃10分、30℃10分、37℃30分、50℃5分、及び98℃5分でRT-RamDA反応又はRT反応を行った。RT-RamDA反応またはRT反応後、表19に示した2本鎖合成反応液2μLを添加し、5μLにて16℃60分で二本鎖合成し、80℃15分で不活化を実施した。その後、表20に示した非精製プロトコルにてライブラリー調製を実施し、ライブラリー濃度をMultiNAにて測定した結果を表21に示す。
本実施例では、本発明によりライブラリー調製を行う際に必要となる鋳型RNA量について検討するため、以下の実験を行った。
RNAを希釈し、RT-RamDA法又は通常の逆転写方法(RT法)により一本鎖cDNAの合成を行い、クレノウフラグメントを用いて二本鎖cDNAを合成した。その後Nextera XT DNA Library Preparation Kit(illumina社)を用いてライブラリーを調製しMultiNA(島津製作所)にてライブラリー濃度を測定した。手順は取扱い説明書に従った。
本実施例で用いる核酸断片サンプルは、マウスES細胞total RNA(ユニーテック株式会社)を用いた。1ng/μLの前記RNAを表16に示したRNA希釈液にて希釈し10pg/μLのRNA溶液を調製した(RNA鋳型量10pg)。
このRNA溶液1μLを70℃1分30秒で熱処理を行った。条件1、2は表17に示したRT-RamDA反応液2μLを添加し、条件3は表18に示したRT反応液2μLを添加し、3μLにて25℃10分、30℃10分、37℃30分、50℃5分、及び98℃5分でRT-RamDA反応又はRT反応を行った。RT-RamDA反応またはRT反応後、表19に示した2本鎖合成反応液2μLを添加し、5μLにて16℃60分で二本鎖合成し、80℃15分で不活化を実施した。その後、表20に示した非精製プロトコルにてライブラリー調製を実施し、ライブラリー濃度をMultiNAにて測定した結果を表21に示す。






条件1及び3においてtotal RNA10pgから十分な収量が得られている。また条件1よりも条件2は収量が多くプロテイナーゼKとポリイノシン酸カリウム塩により収量が増加したと考えられる。本実施例の結果から、鋳型RNA量が少なくとも10pg以上あれば、本発明により、精製工程を経ずに十分な量のライブラリー調製が可能であることが分かった。
実施例5.二本鎖合成における不活化温度の比較
本実施例では、二本鎖合成後の不活化温度が収量に与える影響を確認するため、以下の実験を行った。
RNAを希釈し、RT-RamDA法により一本鎖cDNAの合成を行い、クレノウフラグメントを用いて二本鎖cDNAを合成した。その後70℃又は80℃での加熱による不活化を行い、続いてNextera XT DNA Library Preparation Kit(illumina社)を用いてライブラリーを調製しMultiNA(島津製作所)にてライブラリー濃度を測定した。手順は取扱い説明書に従った。
本実施例で用いる核酸断片サンプルは、マウスES細胞total RNAを用いた。1ng/μLの前記RNAを表22に示したRNA希釈液にて希釈し10pg/μLのRNA溶液を調製した(RNA鋳型量10pg)。
このRNA溶液1μLを70℃1分30秒で熱処理を行った。表23に示したRT-RamDA反応液2μLを添加し、3μLにて25℃10分、30℃10分、37℃30分、50℃5分、及び98℃5分でRT-RamDA反応を行った。RT-RamDA反応後、表24に示した2本鎖合成反応液2μLを添加し、5μLにて16℃60分で二本鎖合成し、その後70℃10分(条件1)又は80℃15分(条件2)で加熱して不活化を実施した(n=2)。その後、表25に示した非精製プロトコルにてライブラリー調製を実施し、ライブラリー濃度をMultiNAにて測定した結果を表26に示す。また代表的な測定結果を図1に示す(条件1は(1)の結果、条件2は(3)の結果)。
本実施例では、二本鎖合成後の不活化温度が収量に与える影響を確認するため、以下の実験を行った。
RNAを希釈し、RT-RamDA法により一本鎖cDNAの合成を行い、クレノウフラグメントを用いて二本鎖cDNAを合成した。その後70℃又は80℃での加熱による不活化を行い、続いてNextera XT DNA Library Preparation Kit(illumina社)を用いてライブラリーを調製しMultiNA(島津製作所)にてライブラリー濃度を測定した。手順は取扱い説明書に従った。
本実施例で用いる核酸断片サンプルは、マウスES細胞total RNAを用いた。1ng/μLの前記RNAを表22に示したRNA希釈液にて希釈し10pg/μLのRNA溶液を調製した(RNA鋳型量10pg)。
このRNA溶液1μLを70℃1分30秒で熱処理を行った。表23に示したRT-RamDA反応液2μLを添加し、3μLにて25℃10分、30℃10分、37℃30分、50℃5分、及び98℃5分でRT-RamDA反応を行った。RT-RamDA反応後、表24に示した2本鎖合成反応液2μLを添加し、5μLにて16℃60分で二本鎖合成し、その後70℃10分(条件1)又は80℃15分(条件2)で加熱して不活化を実施した(n=2)。その後、表25に示した非精製プロトコルにてライブラリー調製を実施し、ライブラリー濃度をMultiNAにて測定した結果を表26に示す。また代表的な測定結果を図1に示す(条件1は(1)の結果、条件2は(3)の結果)。





表26及び図1の結果に示されるように、70℃で不活化を行った条件1よりも、80℃で不活化を行った条件2の収量が多い。また、図1に示されるように、80℃で不活化した条件2ではアダプターダイマーの形成を示すピーク(矢印)が大幅に小さくなり、アダプターダイマーの形成が著しく抑えられた。本実施例の結果から、80℃以上の加熱処理で二本鎖合成後の不活化を行うことでダイマー発生がより効果的に抑制され、ライブラリーの収量が向上することが明らかとなった。
実施例6.工程(b)における塩化物の評価
本実施例では、工程(b)における塩化物の種類等について検討するため、以下の実験を行った。
RNAを希釈し、RT-RamDA法により一本鎖cDNAの合成を行い、クレノウフラグメントを用いて二本鎖cDNAを合成した。その後Nextera XT DNA Library Preparation Kit(illumina社)を用いてライブラリーを調製しMultiNA(島津製作所)にてライブラリー濃度を測定した。手順は取扱い説明書に従った。
本実施例で用いる核酸断片サンプルは、ヒトK562細胞からRNeasy mini kit(QIAGEN社)を用いて精製したtotal RNAを用いた。1ng/μLの前記RNAを実施例1中の表1に示したRNA希釈液にて希釈し10pg/μLのRNA溶液を調製した(RNA鋳型量10pg)。
このRNA溶液1μLを70℃1分30秒で熱処理を行った。熱処理後、実施例1中の表2に示したRT-RamDA反応液2μLを添加し、3μLにて25℃10分、30℃10分、37℃30分、50℃5分、及び98℃5分でRT-RamDA反応を行った。RT-RamDA反応後、表27に示した2本鎖合成反応液2μLを添加し、5μLにて16℃60分で二本鎖合成し、80℃15分で不活化を実施した。その後、実施例1中の表4に示した非精製プロトコルにてライブラリー調製を実施し、ライブラリー濃度をMultiNAにて測定した結果を表28に示す。
本実施例では、工程(b)における塩化物の種類等について検討するため、以下の実験を行った。
RNAを希釈し、RT-RamDA法により一本鎖cDNAの合成を行い、クレノウフラグメントを用いて二本鎖cDNAを合成した。その後Nextera XT DNA Library Preparation Kit(illumina社)を用いてライブラリーを調製しMultiNA(島津製作所)にてライブラリー濃度を測定した。手順は取扱い説明書に従った。
本実施例で用いる核酸断片サンプルは、ヒトK562細胞からRNeasy mini kit(QIAGEN社)を用いて精製したtotal RNAを用いた。1ng/μLの前記RNAを実施例1中の表1に示したRNA希釈液にて希釈し10pg/μLのRNA溶液を調製した(RNA鋳型量10pg)。
このRNA溶液1μLを70℃1分30秒で熱処理を行った。熱処理後、実施例1中の表2に示したRT-RamDA反応液2μLを添加し、3μLにて25℃10分、30℃10分、37℃30分、50℃5分、及び98℃5分でRT-RamDA反応を行った。RT-RamDA反応後、表27に示した2本鎖合成反応液2μLを添加し、5μLにて16℃60分で二本鎖合成し、80℃15分で不活化を実施した。その後、実施例1中の表4に示した非精製プロトコルにてライブラリー調製を実施し、ライブラリー濃度をMultiNAにて測定した結果を表28に示す。


条件1および2において、total RNA10pgから十分な収量が得られている。また条件1と条件2では収量にほとんど差はなく、工程(b)において、塩化ナトリウムに替えて塩化カリウムを添加する場合であっても、本発明により十分な量のライブラリー調製が可能であることが分かった。
Claims (13)
- 以下の工程(a)、(b)、及び(c)を含むライブラリーの調製方法:
(a)10pg以上の鋳型RNAから一本鎖cDNAを合成する工程;
(b)前記一本鎖cDNAから二本鎖cDNAを合成する工程;及び
(c)非精製の前記二本鎖cDNAを用いてライブラリーを調製する工程。 - 前記工程(b)が12mM以下の塩化ナトリウムの存在下で行われる、請求項1に記載の方法。
- 前記工程(a)が1ng/μL以上の核酸ポリマー及び/又は1U/mL以上のタンパク質分解酵素の存在下で行われる、請求項1に記載の方法。
- 前記二本鎖cDNAを80℃以上で10分以上加熱する工程を更に含む、請求項1に記載の方法。
- 前記核酸ポリマーが、ポリイノシン酸、ポリシチジル酸、ポリグアニル酸、ポリアデニル酸、ポリチミジル酸、ポリウリジル酸、ポリデオキシイノシン酸、ポリデオキシシチジル酸、ポリデオキシグアニル酸、ポリデオキシアデニル酸、ポリデオキシチミジル酸、ポリデオキシウリジル酸、及びこれらの塩からなる群より選択される少なくとも1種のホモポリマーである、請求項3に記載の方法。
- 前記核酸ポリマーが、ポリイノシン酸、ポリデオキシイノシン酸、及びこれらの塩からなる群より選択される少なくとも1種のホモポリマーである、請求項3に記載の方法。
- 前記タンパク質分解酵素がプロテイナーゼK及びサブチリシンのいずれかを含む、請求項3に記載の方法。
- 前記鋳型RNAが1細胞~1000細胞から抽出されたRNAである、請求項1~7のいずれかに記載の方法。
- 前記工程(c)の非精製の二本鎖cDNAが1μL以下の溶液の形態である、請求項1~7のいずれかに記載の方法。
- 前記工程(a)がRT-RamDA法で行われる、請求項1~7のいずれかに記載の方法。
- 前記工程(b)がクレノウフラグメントを用いて行われる、請求項1~7のいずれかに記載の方法。
- 前記工程(c)がトランスポゾン法及びライゲーション法のいずれかで行われる、請求項1~7のいずれかに記載の方法。
- 前記工程(b)が60mM以下の塩化物塩の存在下で行われる、請求項1~7のいずれかに記載の方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021-183612 | 2021-11-10 | ||
| JP2021183612 | 2021-11-10 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023085232A1 true WO2023085232A1 (ja) | 2023-05-19 |
Family
ID=86336073
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/041394 WO2023085232A1 (ja) | 2021-11-10 | 2022-11-07 | 改良されたライブラリー調製方法 |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2023085232A1 (ja) |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020184551A1 (ja) * | 2019-03-13 | 2020-09-17 | 東洋紡株式会社 | 核酸の生成および増幅 |
-
2022
- 2022-11-07 WO PCT/JP2022/041394 patent/WO2023085232A1/ja active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020184551A1 (ja) * | 2019-03-13 | 2020-09-17 | 東洋紡株式会社 | 核酸の生成および増幅 |
Non-Patent Citations (1)
| Title |
|---|
| HAYASHI TETSUTARO, OZAKI HARUKA, SASAGAWA YOHEI, UMEDA MANA, DANNO HIROKI, NIKAIDO ITOSHI: "Single-cell full-length total RNA sequencing uncovers dynamics of recursive splicing and enhancer RNAs", NATURE COMMUNICATIONS, vol. 9, no. 1, 12 February 2018 (2018-02-12), XP093016358, DOI: 10.1038/s41467-018-02866-0 * |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20210253625A1 (en) | Methods of library construction for polynucleotide sequencing | |
| US9255291B2 (en) | Oligonucleotide ligation methods for improving data quality and throughput using massively parallel sequencing | |
| JP2021509587A (ja) | シトシン修飾の、亜硫酸水素塩非含有、塩基分解能特定 | |
| EP3837379B1 (en) | Method of nucleic acid enrichment using site-specific nucleases followed by capture | |
| US20160257985A1 (en) | Degradable adaptors for background reduction | |
| JPH05505111A (ja) | Rnaレプリカーゼのdna依存性rnaポリメラーゼ活性を用いる核酸増幅 | |
| US20200048693A1 (en) | Method for nucleic acid amplification | |
| JP2013516192A (ja) | 等温核酸増幅のための材料および方法 | |
| WO2016135300A1 (en) | Efficiency improving methods for gene library generation | |
| KR20220024778A (ko) | 차세대 시퀀싱 라이브러리를 제조하기 위한 핵산 표지화에 유용한 올리고뉴클레오티드-테더된 트리포스페이트 뉴클레오티드 | |
| EP1854882A1 (en) | Method for obtaining subtraction polynucleotide | |
| US20220154180A1 (en) | Production and amplification of nucleic acids | |
| WO2023085232A1 (ja) | 改良されたライブラリー調製方法 | |
| JPWO2002034907A1 (ja) | 1本鎖核酸の合成方法 | |
| CN112534062A (zh) | 可切割合作引物和使用所述可切割合作引物扩增核酸序列的方法 | |
| WO2019023243A1 (en) | METHODS AND COMPOSITIONS FOR SELECTING AND AMPLIFYING DNA TARGETS IN A SINGLE REACTION MIXTURE | |
| KR20230124636A (ko) | 멀티플렉스 반응에서 표적 서열의 고 감응성 검출을위한 조성물 및 방법 | |
| US11725305B2 (en) | Rapid library construction for high throughput sequencing | |
| JP2023038069A (ja) | 長時間の核酸増幅反応の改善 | |
| JP2022183762A (ja) | 核内ノンコーディングrnaの抽出方法 | |
| JP2024026695A (ja) | 非特異的な核酸増幅を抑制する方法 | |
| JP7016511B2 (ja) | 核酸合成法 | |
| WO2023107899A2 (en) | A method of capturing crispr endonuclease cleavage products | |
| KR20230080464A (ko) | 전사된 핵산을 생성하는 방법 및 수단 | |
| WO2024006552A1 (en) | Ambient temperature nucleic acid amplification and detection |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22892731 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023559618 Country of ref document: JP |