WO2016128335A1 - Novel 2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid derivatives for the treatment and prophylaxis of hepatitis b virus infection - Google Patents
Novel 2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid derivatives for the treatment and prophylaxis of hepatitis b virus infection Download PDFInfo
- Publication number
- WO2016128335A1 WO2016128335A1 PCT/EP2016/052585 EP2016052585W WO2016128335A1 WO 2016128335 A1 WO2016128335 A1 WO 2016128335A1 EP 2016052585 W EP2016052585 W EP 2016052585W WO 2016128335 A1 WO2016128335 A1 WO 2016128335A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methoxy
- oxo
- thienyl
- dihydrobenzo
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC=C(C(C=C(CC1=C)[C@@]2C(*)=C(*)C(C)=C(C)C2C(*)C1[Al])=O)C(O)=O Chemical compound CC=C(C(C=C(CC1=C)[C@@]2C(*)=C(*)C(C)=C(C)C2C(*)C1[Al])=O)C(O)=O 0.000 description 8
- WVDDGKGOMKODPV-UHFFFAOYSA-N OCc1ccccc1 Chemical compound OCc1ccccc1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
Definitions
- Novel 2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid derivatives for the treatment and prophylaxis of hepatitis B virus infection
- the present invention relates to organic compounds useful for therapy and/or prophylaxis in a mammal, and in particular to HBsAg (HBV Surface antigen) inhibitors and HBV DNA production inhibitors useful for treating HBV infection.
- HBsAg HBV Surface antigen
- the present invention relates to novel 2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid derivatives having pharmaceutical activity, their manufacture, pharmaceutical compositions containing them and their potential use as medicaments.
- the present invention relates to compounds of formula I
- R 1 to R 5 and Ar are as described below, or to pharmaceutically acceptable salts, or to enantiomers thereof.
- the hepatitis B virus is an enveloped, partially double- stranded DNA virus.
- the compact 3.2 kb HBV genome consists of four overlapping open reading frames (ORF), which encode for the core, polymerase (Pol), envelope and X-proteins.
- ORF open reading frames
- the Pol ORF is the longest and the envelope ORF is located within it, while the X and core ORFs overlap with the Pol ORF.
- the lifecycle of HBV has two main events: 1) generation of closed circular DNA (cccDNA) from relaxed circular (RC DNA), and 2) reverse transcription of pregenomic RNA (pgRNA) to produce RC DNA. Prior to the infection of host cells, the HBV genome exists within the virion as RC DNA.
- HBV virions are able to gain entry into host cells by no n- specifically binding to the negatively charged proteoglycans present on the surface of human hepatocytes (Schulze, A., P. Gripon & S. Urban. Hepatology, 46, (2007), 1759-68) and via the specific binding of HBV surface antigens (HBsAg) to the hepatocyte sodium- taurocholate cotransporting polypeptide (NTCP) receptor (Yan, H. et al. / Virol, 87, (2013), 7977-91).
- HBV surface antigens HBV surface antigens
- NTCP hepatocyte sodium- taurocholate cotransporting polypeptide
- ImpP/Impa nuclear transport receptors Inside the nucleus, host DNA repair enzymes convert the RC DNA into cccDNA. cccDNA acts as the template for all viral mRNAs and as such, is responsible for HBV persistence in infected individuals.
- the transcripts produced from cccDNA are grouped into two categories; Pregenomic RNA (pgRNA) and subgenomic RNA. Subgenomic transcripts encode for the three envelopes (L, M and S) and X proteins, and pgRNA encodes for Pre-Core, Core, and Pol proteins (Quasdorff, M. & U. Protzer. / Viral Hepat, 17, (2010), 527- 36).
- HBV viral replication and antigens production leads to the inhibition of HBV viral replication and antigens production (Mao, R. et al. PLoS Pathog, 9, (2013), el003494; Mao, R. et al. / Virol, 85, (2011), 1048-57).
- IFN-a was shown to inhibit HBV replication and viral HBsAg production by decreasing the transcription of pgRNA and subgenomic RNA from the HBV covalently closed circular DNA (cccDNA) minichromosome.
- HBV viral mRNAs are capped and polyadenylated, and then exported to the cytoplasm for translation.
- the assembly of new virons is initiated and nascent pgRNA is packaged with viral Pol so that reverse transcription of pgRNA, via a single stranded DNA intermediate, into RC DNA can commence.
- the mature nucleocapsids containing RC DNA are enveloped with cellular lipids and viral L, M, and S proteins and then the infectious HBV particles are then released by budding at the intracellular membrane (Locarnini, S. Semin Liver Dis, (2005), 25 Suppl 1, 9-19).
- non-infectious particles are also produced that greatly outnumber the infectious virions.
- These empty, enveloped particles (L, M and S) are referred to as subviral particles.
- subviral particles share the same envelope proteins and as infectious particles, it has been surmised that they act as decoys to the host immune system and have been used for HBV vaccines.
- the S, M, and L envelope proteins are expressed from a single ORF that contains three different start codons. All three proteins share a 226aa sequence, the S-domain, at their C- termini. M and L have additional pre-S domains, Pre-S2 and Pre-S2 and Pre-Sl, respectively. However, it is the S-domain that has the HBsAg epitope (Lambert, C. & R. Prange. Virol J, (2007), 4, 45).
- HBV Hepatitis B virus
- the secretion of antiviral cytokines in response to HBV infection by the hepatocytes and/or the intra-hepatic immune cells plays a central role in the viral clearance of infected liver.
- chronically infected patients only display a weak immune response due to various escape strategies adopted by the virus to counteract the host cell recognition systems and the subsequent antiviral responses.
- HBV empty subviral particles SVPs, HBsAg
- CHB chronically infected patients
- HBsAg has been reported to suppress the function of immune cells such as monocytes, dendritic cells (DCs) and natural killer (NK) cells by direct interaction (Op den Brouw et al. Immunology, (2009b), 126, 280-9; Woltman et al. PLoS One, (2011), 6, el5324; Shi et al. / Viral Hepat.
- HBsAg quantification is a significant biomarker for prognosis and treatment response in chronic hepatitis B.
- Current therapy such as Nucleos(t)ide analogues are molecules that inhibit HBV DNA synthesis but are not directed at reducing HBsAg level.
- Nucleos(t)ide analogs even with prolonged therapy, have demonstrated rates of HBsAg clearance comparable to those observed naturally (between - ⁇ %- 2%) (Janssen et al. Lancet, (2005), 365, 123-9; Marcellin et al. N. Engl. J.
- the present invention relates to compounds of formula I
- R J and R" are independently selected from Ci_ 6 alkyl, haloCi_ 6 alkyl, hydrogen, halogen, cyano, amino, Ci_ 6 alkylamino, diCi_ 6 alkylamino, pyrrolidinyl and OR 6 ;
- R is hydrogen or Ci_ 6 alkyl
- R 6 is hydrogen; Ci_ 6 alkyl; haloCi_ 6 alkyl; C3_ 7 cycloalkylCi- 6 alkyl; phenylCi- 6 alkyl;
- heterocycloalkylCi- 6 alkyl wherein heterocycloalkyl is N-containing monocyclic heterocycloalkyl
- Ar is phenyl; phenyl substituted by one, two or three groups independently selected from Ci_ 6alkyl, hydroxy, halogen and thienyl; thienyl substituted by one, two or three groups independently selected from hydroxy, halogen and phenylC i_6 alkoxy; benzothiophenyl; benzothiophenyl substituted by one, two or three groups independently selected from Ci_ 6 alkyl, hydroxy, halogen and phenylC 1- 6 alkoxy; pyridinyl; pyridinyl substituted by one, two or three groups
- Ci_ 6 alkyl alone or in combination signifies a saturated, linear- or branched chain alkyl group containing 1 to 6, particularly 1 to 4 carbon atoms, for example methyl, ethyl, propyl, isopropyl, 1-butyl, 2-butyl, ie/t-butyl and the like.
- Particular "Ci_ 6 alkyl” groups are methyl, ethyl, isopropyl and ie/t-butyl.
- C3_ 7 cycloalkyl refers to a saturated carbon ring containing from 3 to 7 carbon atoms, particularly from 3 to 6 carbon atoms, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and the like.
- Particular "C 3 _ 7 cycloalkyl” groups are cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
- Ci_ 6 alkoxy alone or in combination signifies a group Ci_ 6 alkyl-0-, wherein the "C h alky! is as defined above; examples for Ci- 6 alkoxy are methoxy, ethoxy, propoxy, iso- propoxy, w-butoxy, zsobutoxy, 2-butoxy, ie/t-butoxy and the like. Particular "Ci_ 6 alkoxy” groups are methoxy and ethoxy and more particularly methoxy.
- halogen means fluorine, chlorine, bromine or iodine.
- haloCi- 6 alkyl denotes a Ci_ 6 alkyl group wherein at least one of the hydrogen atoms of the C 1-6 alkyl group has been replaced by same or different halogen atoms, particularly fluoro atoms.
- haloCi- 6 alkyl include monofluoro-, difluoro- or trifluoro-methyl, - ethyl or -propyl, for example 3,3,3-trifluoropropyl, 3,3- difluoropropyl, 2-fluoroethyl, 2,2- difluoroethyl, 2,2,2-trifluoroethyl, fluoromethyl, difluoromethyl or trifluoromethyl.
- Particular "halo C 1 - 6 alkyl” group is difluoromethyl or trifluoromethyl.
- monocyclic heterocycloalkyl is a monovalent saturated or partly unsaturated monocyclic ring system of 4 to 7 ring atoms, comprising 1, 2, or 3 ring heteroatoms selected from N, O and S, the remaining ring atoms being carbon. Examples for monocyclic
- heterocycloalkyl are aziridinyl, oxiranyl, azetidinyl, oxetanyl, pyrrolidinyl, 2-oxo-pyrrolidinyl, tetrahydrofuranyl, tetrahydro-thienyl, pyrazolidinyl, imidazolidinyl, oxazolidinyl, isoxazolidinyl, thiazolidinyl, piperidinyl, tetrahydropyranyl, tetrahydrothiopyranyl, piperazinyl, morpholinyl, thiomorpholinyl, l,l-dioxo-thiomorpholin-4-yl, azepanyl, diazepanyl, homopiperazinyl, or oxazepanyl.
- Particular "monocyclic heterocycloalkyl" groups are piperidinyl, morpholinyl, 2- oxo-
- N-containing monocyclic heterocycloalkyl is a "monocyclic heterocycloalkyl" as defined above wherein at least one of the heteroatoms is N.
- Examples for 'W-containing monocyclic heterocycloalkyl” are aziridinyl, azetidinyl, pyrrolidinyl, 2-oxo-pyrrolidinyl, pyrazolidinyl, imidazolidinyl, oxazolidinyl, isoxazolidinyl, thiazolidinyl, piperidinyl, piperazinyl, morpholinyl, thio morpholinyl, l,l-dioxo-thiomorpholin-4-yl, azepanyl, diazepanyl,
- homopiperazinyl or oxazepanyl.
- Particular 'W-containing monocyclic heterocycloalkyl” groups are piperidinyl, morpholinyl, 2-oxo-pyrrolidinyl and pyrrolidinyl.
- amino denotes a group of the formula -NR'R" wherein R' and R" are independently hydrogen, Ci_ 6 alkyl, Ci_ 6 alkoxy, C 3 _ 7 cycloalkyl, heteroC 3 _ 7 cycloalkyl, aryl or heteroaryl.
- R' and R" together with the nitrogen to which they are attached, can form a heteroC 3 _ 7 cycloalkyl.
- carbonyl alone or in combination refers to the group -C(O)-.
- cyano alone or in combination refers to the group -CN.
- Ci_ 6 alkylsulfanyl denotes the group -S-R', wherein R' is a Ci_ 6 alkyl group as defined above.
- Ci_ 6 alkylsulfonyl denotes a group -S0 2 -R', wherein R' is a Ci_ 6 alkyl group as defined above.
- Examples of Ci_ 6 alkylsulfonyl include methylsulfonyl and ethylsulfonyl.
- enantiomer denotes two stereoisomers of a compound which are non- superimpo sable mirror images of one another.
- diastereomer denotes a stereoisomer with two or more centers of chirality and whose molecules are not mirror images of one another. Diastereomers have different physical properties, e.g. melting points, boiling points, spectral properties, and reactivities.
- the compounds according to the present invention may exist in the form of their
- pharmaceutically acceptable salts refers to conventional acid-addition salts or base-addition salts that retain the biological effectiveness and properties of the compounds of formula I and are formed from suitable non-toxic organic or inorganic acids or organic or inorganic bases.
- Acid-addition salts include for example those derived from inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, sulfamic acid, phosphoric acid and nitric acid, and those derived from organic acids such as /7-toluenesulfonic acid, salicylic acid, methane sulfonic acid, oxalic acid, succinic acid, citric acid, malic acid, lactic acid, fumaric acid, and the like.
- Base-addition salts include those derived from ammonium, potassium, sodium and, quaternary ammonium hydroxides, such as for example, tetramethyl ammonium hydroxide.
- the chemical modification of a pharmaceutical compound into a salt is a technique well known to pharmaceutical chemists in order to obtain improved physical and chemical stability, hygroscopicity, flowability and solubility of compounds. It is for example described in Bastin R.J., et al., Organic Process Research & Development 2000, 4, 427-435; or in Ansel, H., et al., In: Pharmaceutical Dosage Forms and Drug Delivery Systems, 6th ed. (1995), pp. 196 and 1456-1457. Particular are the sodium salts of the compounds of formula I.
- Racemates can be separated according to known methods into the enantiomers.
- diastereomeric salts which can be separated by crystallization are formed from the racemic mixtures by reaction with an optically active acid such as e.g. D- or L-tartaric acid, mandelic acid, malic acid, lactic acid or camphor sulfonic acid.
- the present invention provides i) novel compounds having the general formula I:
- R 1 , R 2 , R 3 and R 4 are independently selected from C 1-6 alkyl, haloC 1-6 alkyl, hydrogen, halogen, cyano, amino, Ci_ 6 alkylamino, diCi_ 6 alkylamino, pyrrolidinyl and OR 6 ;
- R 5 is hydrogen or C 1-6 alkyl
- R 6 is hydrogen; C 1-6 alkyl; haloC 1-6 alkyl; C 3- 7cycloalkylC 1-6 alkyl; phenylC 1-6 alkyl;
- Ar is phenyl; phenyl substituted by one, two or three groups independently selected from Ci_ 6alkyl, hydroxy, halogen and thienyl; thienyl substituted by one, two or three groups independently selected from hydroxy, halogen and phenylCi- 6 alkoxy; benzothiophenyl; benzothiophenyl substituted by one, two or three groups independently selected from Ci_ 6 alkyl, hydroxy, halogen and phenylC 1 - 6 alkoxy; pyridinyl; pyridinyl substituted by one, two or three groups
- R 1 is hydrogen
- R is hydrogen, halogen or Ci- 6 alkoxy
- R 3 is Ci- 6 alkyl, pyrrolidinyl or OR 6 ;
- R 4 is C 1 - 6 alkyl or hydrogen
- R 5 is hydrogen
- R 6 is Ci- 6 alkyl, haloCi- 6 alkyl, C3_ 7 cycloalkylCi_ 6 alkyl, phenylCi- 6 alkyl, hydroxyCi- 6 alkyl, Ci- 6 alkoxyCi- 6 alkyl, carboxyCi_ 6 alkyl, Ci_ 6 alkylsulfanylCi_ 6 alkyl, Ci_ 6 alkylsulfonylCi_ 6alkyl, cyanoCi_ 6 alkyl, aminoCi- 6 alkyl, Ci_ 6 alkylcarbonylaminoCi_ 6 alkyl, Ci_
- Ar is phenyl; phenyl substituted by Ci_ 6 alkyl, Ci- 6 alkoxy, hydroxy, halogen or phenylCi- 6alkoxy; thienyl; thienyl substituted by Ci_ 6 alkyl; or benzothiophenyl;
- Another embodiment of present invention is (iii) a compound of formula I as defined above, or pharmaceutically acceptable salts, or enantiomers thereof, wherein R 1 is hydrogen, R 4 is hydrogen, R 5 is hydrogen, and all remaining substituents have the significances given herein before.
- Another embodiment of present invention is (iv) a compound of formula I as defined above, or pharmaceutically acceptable salts, or enantiomers thereof, wherein R is Ci_ 6 alkoxy, and all remaining substituents have the significances given herein before.
- a further embodiment of present invention is (v) a compound of formula I as defined above, or pharmaceutically acceptable salts, or enantiomers thereof, wherein R is methoxy, and all remaining substituents have the significances given herein before.
- Another embodiment of present invention is (vi) a compound of formula I as defined above, or pharmaceutically acceptable salts, or enantiomers thereof, wherein R 3 is pyrrolidinyl or OR 6 , wherein R 6 is Ci_ 6 alkyl, haloCi_ 6 alkyl, C3_ 7 cycloalkylCi_ 6 alkyl, hydro xyCi_ 6 alky 1, Ci_ 6 alkoxyCi_ 6 alkyl, carboxyCi_ 6 alkyl, Ci_ 6 alkylsulfanylCi_ 6 alkyl, cyanoCi_ 6 alkyl, aminoCi_ 6 alkyl, Ci_ 6 alkylcarbonylaminoCi- 6 alkyl, Ci_ 6 alkylsulf
- FIG. 1 Further embodiment of present invention is (vii) a compound of formula I as defined above, or pharmaceutically acceptable salts, or enantiomers thereof, wherein R 3 is OR 6 , wherein R 6 is methyl, isobutyl, trifluoroethyl, cyclopropylmethyl, cyanopropyl, hydroxypropyl, hydroxyhexyl, hydroxydimethylpropyl, methoxypropyl, carboxypropyl, methylsulfanylpropyl, aminohexyl, methylcarbonylaminohexyl, methylsulfonylaminohexyl or methoxycarbonylaminohexyl, and all remaining substituents have the significances given herein before.
- Another embodiment of present invention is (viii) a compound of formula I as defined above, or pharmaceutically acceptable salts, or enantiomers thereof, wherein Ar is phenyl, phenyl substituted by Ci_ 6 alkyl or halogen, thienyl or thienyl substituted by Ci_ 6 alkyl, and all remaining substituents have the significances given herein before.
- Further embodiment of present invention is (ix) a compound of formula I as defined above, or pharmaceutically acceptable salts, or enantiomers thereof, wherein Ar is phenyl; phenyl substituted by methyl, fluoro or chloro; thienyl or thienyl substituted by methyl, and all remaining substituents have the significances given herein before.
- Another embodiment of present invention is (x) a compound of formula I, wherein
- R 1 is hydrogen
- R is Ci_ 6 alkoxy
- R 3 is pyrrolidinyl or OR 6 ;
- R 4 is hydrogen
- R 5 is hydrogen
- R 6 is C h alky!, haloCi_ 6 alkyl, C3_ 7 cycloalkylCi- 6 alkyl, hydroxyCi_ 6 alkyl, Ci_ 6 alkoxyCi- 6 alkyl or Ci- 6 alkylsulfanylCi_ 6 alkyl;
- Ar is phenyl substituted by C 1-6 alkyl, or thienyl
- a further embodiment of present invention is (x) a compound of formula I, wherein R 1 is hydrogen;
- R is methoxy
- R 3 is pyrrolidinyl or OR 6 ;
- R 4 is hydrogen
- R 5 is hydrogen
- R 6 is isobutyl, trifluoroethyl, cyclopropylmethyl, hydroxydimethylpropyl, methoxypropyl or methylsulfanylpropyl;
- Ar is phenyl substituted by methyl or thienyl
- the compounds of the present invention can be prepared by any conventional means. Suitable processes for synthesizing these compounds as well as their starting materials are provided in the schemes below and in the examples. All substituents, in particular, R 1 to R 5 and Ar are as defined above unless otherwise indicated. Furthermore, and unless explicitly otherwise stated, all reactions, reaction conditions, abbreviations and symbols have the meanings well known to a person of ordinary skill in organic chemistry.
- XIV FT is C ⁇ alkyl
- the compound of formula I can be prepared according to Scheme 1. Coupling reaction of bromo-benzene II with ketone III affords Compound IV.
- the reaction can be carried out in the presence of a Pd catalyst such as Pd 2 (dba)3 , Pd(PPh 3 ) 4 or PdCl 2 (PPh 3 ) 2 , a ligand such as Xantphos, and a suitable base such as i-BuONa, Na 2 C0 3 or Cs 2 C0 3 , in a suitable solvent such as THF, toluene or 1,4-dioxane at a temperature between room temperature and 130 °C.
- a Pd catalyst such as Pd 2 (dba)3 , Pd(PPh 3 ) 4 or PdCl 2 (PPh 3 ) 2
- a ligand such as Xantphos
- a suitable base such as i-BuONa, Na 2 C0 3 or Cs 2 C0 3
- Compound XI is treated with POCI 3 in a suitable solvent such as acetonitrile or DCM at a temperature between room temperature and 100 °C to give Compound XII.
- Compound XII reacts with Ci_ 6 alkyl 2-(ethoxymethylene)-3-oxo-butanoate in a solvent such as ethanol to give Compound XIII.
- Compound XIV is obtained by dehydrogenation of compound XIII by using p- chloranil.
- Hydrolyzation of Compound XIV with a base such as lithium hydroxide or sodium hydroxide in a suitable solvent such as THF/H 2 0, EtOH/H 2 0 or MeOH/H 2 0 affords the compound of formula I.
- Q 1 is halogen, -0-S(0) 2 CH 3 or -0-S(0) 2 -(4-CH 3 -Ph).
- Ft 7 is C ⁇ alkyl
- the compound of formula 1-1 can be prepared according to Scheme 2. Debenzylation of Compound XIV-1 by hydrogenation is carried out in the presence of Pd/C in a solvent such as ethanol, THF, methanol to afford Compound XIV-2. Then Compound XIV-2 reacts with halides, mesylates or tosylates in the presence of a base such as K 2 C0 3 in a solvent such as acetone or DMF to give XIV-3. Hydrolyzation of Compound XIV with a base such as lithium hydroxide or sodium hydroxide in a suitable solvent such as THF/H 2 0, EtOH/H 2 0 or MeOH/H 2 0 affords the compound of formula 1-1.
- This invention also relates to a process for the preparation of a compound of formula I comprising
- step (a) and step (b) a base such as lithium hydroxide or sodium hydroxide can be for example used.
- a compound of formula I when manufactured according to the above process is also an object of the invention.
- the invention also relates to a compound of formula I for use as therapeutically active substance.
- compositions or medicaments containing the compounds of the invention and a therapeutically inert carrier, diluent or excipient, as well as methods of using the compounds of the invention to prepare such compositions and medicaments.
- compounds of formula I may be formulated by mixing at ambient temperature at the appropriate pH, and at the desired degree of purity, with physiologically acceptable carriers, i.e., carriers that are non-toxic to recipients at the dosages and concentrations employed into a galenical administration form.
- the pH of the formulation depends mainly on the particular use and the concentration of compound, but preferably ranges anywhere from about 3 to about 8.
- a compound of formula I is formulated in an acetate buffer, at pH 5.
- the compounds of formula I are sterile.
- the compound may be stored, for example, as a solid or amorphous composition, as a lyophilized formulation or as an aqueous solution.
- compositions are formulated, dosed, and administered in a fashion consistent with good medical practice.
- Factors for consideration in this context include the particular disorder being treated, the particular mammal being treated, the clinical condition of the individual patient, the cause of the disorder, the site of delivery of the agent, the method of administration, the scheduling of administration, and other factors known to medical practitioners.
- the "effective amount" of the compound to be administered will be governed by such considerations, and is the minimum amount necessary to inhibit HBsAg. For example, such amount may be below the amount that is toxic to normal cells, or the mammal as a whole.
- the pharmaceutically effective amount of the compound of the invention administered parenterally per dose will be in the range of about 0.01 to 100 mg/kg, alternatively about 0.01 to 100 mg/kg of patient body weight per day, with the typical initial range of compound used being 0.3 to 15 mg/kg/day.
- oral unit dosage forms such as tablets and capsules, preferably contain from about 0.1 to about 1000 mg of the compound of the invention.
- the compounds of the invention may be administered by any suitable means, including oral, topical (including buccal and sublingual), rectal, vaginal, transdermal, parenteral, subcutaneous, intraperitoneal, intrapulmonary, intradermal, intrathecal and epidural and intranasal, and, if desired for local treatment, intralesional administration.
- Parenteral infusions include intramuscular, intravenous, intraarterial, intraperitoneal, or subcutaneous administration.
- the compounds of the present invention may be administered in any convenient administrative form, e.g., tablets, powders, capsules, solutions, dispersions, suspensions, syrups, sprays, suppositories, gels, emulsions, patches, etc.
- Such compositions may contain components conventional in pharmaceutical preparations, e.g., diluents, carriers, pH modifiers, sweeteners, bulking agents, and further active agents.
- a typical formulation is prepared by mixing a compound of the present invention and a carrier or excipient. Suitable carriers and excipients are well known to those skilled in the art and are described in detail in, e.g., Ansel, Howard C, et al., Ansel's Pharmaceutical Dosage Forms and Drug Delivery Systems. Philadelphia: Lippincott, Williams & Wilkins, 2004;
- the formulations may also include one or more buffers, stabilizing agents, surfactants, wetting agents, lubricating agents, emulsifiers, suspending agents, preservatives, antioxidants, opaquing agents, glidants, processing aids, colorants, sweeteners, perfuming agents, flavoring agents, diluents and other known additives to provide an elegant presentation of the drug (i.e., a compound of the present invention or pharmaceutical composition thereof) or aid in the manufacturing of the pharmaceutical product (i.e., medicament).
- buffers stabilizing agents, surfactants, wetting agents, lubricating agents, emulsifiers, suspending agents, preservatives, antioxidants, opaquing agents, glidants, processing aids, colorants, sweeteners, perfuming agents, flavoring agents, diluents and other known additives to provide an elegant presentation of the drug (i.e., a compound of the present invention or pharmaceutical composition thereof) or aid in the manufacturing
- An example of a suitable oral dosage form is a tablet containing about 0.1 to 1000 mg of the compound of the invention compounded with about 0 to 2000 mg anhydrous lactose, about 0 to 2000 mg sodium croscarmellose, about 0 to 2000 mg polyvinylpyrrolidone (PVP) K30, and about 0 to 2000 mg magnesium stearate.
- the powdered ingredients are first mixed together and then mixed with a solution of the PVP.
- the resulting composition can be dried, granulated, mixed with the magnesium stearate and compressed to tablet form using conventional equipment.
- An example of an aerosol formulation can be prepared by dissolving the compound, for example 0.1 to 1000 mg, of the invention in a suitable buffer solution, e.g. a phosphate buffer, adding a tonicifier, e.g. a salt such sodium chloride, if desired.
- the solution may be filtered, e.g., using a 0.2 micron filter, to remove impurities and contaminants.
- An embodiment therefore, includes a pharmaceutical composition comprising a compound of Formula I, or a stereoisomer or pharmaceutically acceptable salt thereof.
- a pharmaceutical composition comprising a compound of Formula I, or a stereoisomer or pharmaceutically acceptable salt thereof, together with a pharmaceutically acceptable carrier or excipient.
- the following example A and B illustrate typical compositions of the present invention, but serve merely as representative thereof.
- a compound of formula I can be used in a manner known per se as the active ingredient for the production of tablets of the following composition:
- a compound of formula I can be used in a manner known per se as the active ingredient for the production of capsules of the following composition:
- the compounds of the invention can inhibit HBsAg production or secretion and inhibit HBV gene expression. Accordingly, the compounds of the invention are useful for the treatment or prophylaxis of HBV infection.
- the invention relates to the use of a compound of formula I for the inhibition of HBsAg production or secretion.
- the invention relates to the use of a compound of formula I for the inhibition of HBV DNA production.
- the invention relates to the use of a compound of formula I for the inhibition of HBV gene expression.
- the invention relates to the use of a compound of formula I for the treatment or prophylaxis of HBV infection.
- the invention relates in particular to the use of a compound of formula I for the
- Another embodiment includes a method for the treatment or prophylaxis of HBV infection, which method comprises administering an effective amount of a compound of Formula I, a stereoisomer, tautomer, prodrug, conjugates or pharmaceutically acceptable salt thereof.
- the compounds of the invention can be combined with other anti HBV agents such as interferon alpha- 2b, interferon alpha-2a, and interferon alphacon-1 (pegylated and unpegylated), ribavirin, lamivudine (3TC), entecavir, tenofovir, telbivudine (LdT), adefovir, or other emerging anti HBV agents such as HBV RNA replication inhibitor, HBsAg secretion inhibitors, HBV capsid inhibitors, antisense oligomer, siRNA, HBV therapeutic vaccine, HBV prophylactic vaccine, HBV antibody therapy (monoclonal or polyclonal) and TLR 2, 3, 7, 8 and 9 agonists for the treatment or prophylaxis of HBV.
- anti HBV agents such as interferon alpha- 2b, interferon alpha-2a, and interferon alphacon-1 (pegylated and unpegylated), ribavirin,
- DIPEA N,N-diisopropylethylamine
- HCMV human cytomegalovirus
- HIV human immunodeficiency
- HSV herpes simplex virus
- HPV human papillomavirus
- Pd(PPh 3 ) 4 tetrakis(triphenylphosphine)palladium
- Pd(dppf)Cl 2 [l,l'-bis(diphenylphosphino)ferrocene]dichloropalladium(II) PE or Pet: petroleum ether
- Acidic condition A: 0.1% formic acid in H 2 O; B: 0.1% formic acid in acetonitrile;
- Mass spectra generally only ions which indicate the parent mass are reported, and unless otherwise stated the mass ion quoted is the positive mass ion (M+H) + .
- the microwave assisted reactions were carried out in a Biotage Initiator Sixty or CEM Discover.
- Step 2 Preparation of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-phenyl- ethanone
- Step 3 Preparation of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-phenyl- ethanamine
- Step 4 Preparation of N-[2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-phenyl- ethyl] formamide
- Step 6 Preparation of ethyl 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-phenyl- l,6,7,llb-tetrahydrobenzo[a] uinolizine-3-carboxylate
- Step 7 Preparation of ethyl 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-phenyl-6,7- dihydrobenzo[a]quinolizin -3-carboxylate
- Step 8 Preparation of 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-phenyl-6,7- dihydrobenzo[a] quinolizine-3-carbox lic acid
- Step 2 Preparation of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(3- thienyl)ethanamine
- Step 3 Preparation of N-[2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(3- thienyl)ethyl] formamide
- Step 5 Preparation of ethyl 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-(3-thienyl)- l,6,7,llb-tetrahydrobenzo a]quinolizine-3-carboxylate
- Step 6 Preparation of ethyl 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-(3-thienyl)-
- Step 7 Preparation of 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-(3-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carbox lic acid
- Step 2 Preparation of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(2- thienyl)ethanamine
- Step 3 Preparation of N-[2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(2- thienyl)ethyl] formamide
- Step 5 Preparation of ethyl 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-(2-thienyl)- l,6,7,llb-tetrahydrobenzo inolizine-3-carboxylate
- Step 6 Preparation of ethyl 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-(2-thienyl)- 6,7-dihydrobenzo[a]quinolizin -3-carboxylate
- Step 7 Preparation of 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine- -carboxylic acid
- Step 1 Preparation of l-(4-benzyloxyphenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]ethanone
- Step 2 Preparation of l-(4-benzyloxyphenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]ethanamine
- Step 3 Preparation of N-[l-(4-benzyloxyphenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phen l]ethyl]formamide
- Step 5 Preparation of ethyl 6-(4-benzyloxyphenyl)-10-methoxy-9-(3- methoxypropoxy)-2-oxo-l 6,7,llb-tetrahydrobenzo[a]quinolizine-3-carboxylate
- Step 6 Preparation of ethyl 6-(4-benzyloxyphenyl)-10-methoxy-9-(3- methoxypropoxy)-2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylate
- Step 7 Preparation of ethyl 6-(4-hydroxyphenyl)-10-methoxy-9-(3-methoxypropoxy)-
- Step 8 Preparation of 6-(4-hydroxyphenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo-
- Step 1 Preparation of ethyl 10-methoxy-6-(4-methoxyphenyl)-9-(3-methoxypropoxy)- 2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylate
- Step 2 Preparation of 10-methoxy-6-(4-methoxyphenyl)-9-(3-methoxypropoxy)-2- oxo-6,7-dihydrobenzo[a]quinolizine-3-carbox lic acid
- Example 7 10-methoxy-9-(3-methoxypropoxy)-6-(4-methyl-2-thienyl)-2-oxo-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid
- Step 1 Preparation of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(4-methyl-2- thienyl)ethanone
- Step 2 Preparation of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(4-methyl-2- thienyl)ethanamine
- Step 3 Preparation of N-[2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(4-methyl-2- thienyl)ethyl] formamide
- Step 4 Preparation of 7-methoxy-6-(3-methoxypropoxy)-3-(4-methyl-2-thienyl)-3,4- dihydroisoquinoline
- Step 5 Preparation of ethyl 10-methoxy-9-(3-methoxypropoxy)-6-(4-methyl-2- thienyl)-2-oxo-l,6,7,llb-tetrahydrobenzo[a]quinolizine-3-carboxylate
- Step 6 Preparation of ethyl 10-methoxy-9-(3-methoxypropoxy)-6-(4-methyl-2- thienyl)-2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylate
- Step 7 Preparation of 10-methoxy-9-(3-methoxypropoxy)-6-(4-methyl-2-thienyl)-2- oxo-6,7-dihydrobenzo[a] quinolizine-3-carboxylic acid
- Example 8 6-(3-chlorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2- dihydrobenzo[a] quinolizine-3-carbox lic acid
- Step 1 Preparation of l-(3-chlorophenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]ethanone
- Step 2 Preparation of l-(3-chlorophenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]ethanamine
- Step 3 Preparation of N-[l-(3-chlorophenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]ethyl]formamide
- Step 5 Preparation of ethyl 6-(3-chlorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2- oxo-l,6,7,llb-tetrahydrobenzo a]quinolizine-3-carboxylate
- Step 6 Preparation of ethyl 6-(3-chlorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2- oxo-6,7-dihydrobenzo[a]quinolizine-3-carbox late
- Step 7 Preparation of 6-(3-chlorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo- 6,7-dihydrobenzo[a] quinolizine-3-carboxylic acid
- Step 1 Preparation of l-(4-fluorophenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]ethanone
- Step 2 Preparation of l-(4-fluorophenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]ethanamine
- Step 3 Preparation of N-[l-(4-fiuorophenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]eth l]formamide
- Step 4 Preparation of 3-(4-fiuorophenyl)-7-methoxy-6-(3-methoxypropoxy)-3,4- dihydroisoquinoline
- Step 5 Preparation of ethyl 6-(4-fiuorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2- oxo-l,6,7,llb-tetrahydrobenzo[a]quinolizine-3-carboxylate
- Step 6 Preparation of ethyl 6-(4-fluorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2- oxo-6,7-dihydrobenzo[a] quinolizine-3-carboxylate
- Step 7 Preparation of 6-(4-fluorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo-
- the filter cake was purified by prep-HPLC to give 6-(4-fluorophenyl)-10-methoxy-9-(3- methoxypropoxy)-2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid (22 mg) as a white solid.
- Step 1 Preparation of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(o- tolyl)ethanone
- Step 3 Preparation of N-[2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(o- tol l)ethy 1] formamide
- Step 5 Preparation of ethyl 10-methoxy-9-(3-methoxypropoxy)-6-(o-tolyl)-2-oxo- l,6,7,llb-tetrahydrobenzo[a]quinolizine-3-carboxylate
- Step 6 Preparation of ethyl 10-methoxy-9-(3-methoxypropoxy)-6-(o-tolyl)-2-oxo-6,7- dihydrobenzo[a]quinolizine-3-carboxylate
- Step 7 Preparation of 10-methoxy-9-(3-methoxypropoxy)-6-(o-tolyl)-2-oxo-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
- Step 2 Preparation of 2-(3-methoxy-2-methyl-phenyl)-l-(2-thienyl)ethanone
- l-(2-thienyl)ethanone 8.97 g, 71.1 mmol
- i-BuONa 7.89 g, 82.1 mmol
- Pd 2 (dba) 3 (1 g, 1.09 mmol
- Xantphos 633 mg, 1.09 mmol
- Step 4 Preparation of N-[2-(3-methoxy-2-methyl-phenyl)-l-(2- thienyl)ethyl] formamide
- Step 6 Preparation of ethyl 9-methoxy-8-methyl-2-oxo-6-(2-thienyl)-l,6,7,llb- tetrahydrobenzo[a]quinolizine-3-carbox late
- Step 7 Preparation of ethyl 9-methoxy-8-methyl-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate
- Step 8 Preparation of 9-methoxy-8-methyl-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
- Step 6 Preparation of ethyl 9-benzyloxy-10-methoxy-2-oxo-6-(2-thienyl)-l,6,7,llb- tetrahydrobenzo[a]quinolizine-3-carboxylate
- Step 7 Preparation of ethyl 9-benzyloxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate
- Step 8 Preparation of 9-benzyloxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid
- Step 1 Preparation of ethyl 9-hydroxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate
- Step 2 Preparation of ethyl 9,10-dimethoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate
- Step 3 Preparation of 9,10-dimethoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
- Step 1 Preparation of ethyl 9-isobutoxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate
- Step 2 Preparation of 9-isobutoxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid
- Step 1 Preparation of ethyl 9-(cyclopropylmethoxy)-10-methoxy-2-oxo-6-(2-thienyl)- 6,7-dihydrobenzo[a]quinolizine-3-carboxylate
- Step 2 Preparation of 9-(cyclopropylmethoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
- Step 1 Preparation of l-(3-bromopropyl)pyrazole
- Step 2 Preparation of ethyl 10-methoxy-2-oxo-9-(3-pyrazol-l-ylpropoxy)-6-(2- thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate
- Step 3 Preparation of 10-methoxy-2-oxo-9-(3-pyrazol-l-ylpropoxy)-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid
- Step 1 Preparation of ethyl 9-(3-hydroxypropoxy)-10-methoxy-2-oxo-6-(2-thienyl)- 6,7-dihydrobenzo[a]quinolizine-3-carboxylate
- Step 2 Preparation of 9-(3-hydroxypropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
- Step 1 Preparation of ethyl 9-(3-bromopropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylate
- Step 2 Preparation of ethyl 10-methoxy-2-oxo-9-[3-(l-piperidyl)propoxy]-6-(2- thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate
- Step 3 Preparation of 10-methoxy-2-oxo-9-[3-(l-piperidyl)propoxy]-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid formate
- Step 1 Preparation of ethyl 9-(3-cyanopropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate
- Step 2 Preparation of 9-(3-cyanopropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
- Step 1 Preparation of ethyl 9-(2-bromoethoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carbox late
- Step 2 Preparation of 10-methoxy-2-oxo-9-[2-(2-oxopyrrolidin-l-yl)ethoxy]-6-(2- thienyl)-6,7-dihydrobenzo[a] quinolizine-3-carboxylic acid
- Step 1 Preparation of ethyl 9-[6-(tert-butoxycarbonylamino)hexoxy]-10-methoxy-2-
- Step 1 Preparation of ethyl 9-(6-aminohexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylate hydrochloride
- Step 2 Preparation of 9-(6-aminohexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizin -3-carboxylic acid formate
- Step 1 Preparation of ethyl 9-(6-acetamidohexoxy)-10-methoxy-2-oxo-6-(2-thienyl)- 6,7-dihydrobenzo[a]quinolizine-3-carbox late
- Step 2 Preparation of 9-(6-acetamidohexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizin -3-carboxylic acid
- Step 1 Preparation of ethyl 9-[6-(methanesulfonamido)hexoxy]-10-methoxy-2-
- Step 2 Preparation of 9-[6-(methanesulfonamido)hexoxy]-10-methoxy-2-oxo-6-(2- thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid
- Step 1 Preparation of ethyl 9-(6-hydroxyhexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate
- Step 2 Preparation of 9-(6-hydroxyhexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
- Step 1 Preparation of ethyl 9-(3-hydroxy-2,2-dimethyl-propoxy)-10-methoxy-2-oxo-6- (2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate
- Step 2 Preparation of 9-(3-hydroxy-2,2-dimethyl-propoxy)-10-methoxy-2-oxo-6-(2- thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid
- Step 1 Preparation of ethyl 10-methoxy-2-oxo-6-(2-thienyl)-9-(2,2,2-trifluoroethoxy)- 6,7-dihydrobenzo[a]quinolizine-3-carboxylate
- Step 2 Preparation of 10-methoxy-2-oxo-6-(2-thienyl)-9-(2,2,2-trifluoroethoxy)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
- Example 28 10-methoxy-9-(3-morpholinopropoxy)-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylate
- Step 1 Preparation of ethyl 10-methoxy-9-(3-morpholinopropoxy)-2- thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate
- Step 2 Preparation of 10-methoxy-9-(3-morpholinopropoxy)-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate
- Step 1 Preparation of l-(3-brom ropyl)-l,2,4-triazole
- Step 2 Preparation of ethyl 10-methoxy-2-oxo-6-(2-thienyl)-9-[3-(l,2,4-triazol-l- yl)propoxy]-6,7-dihydrobenzo[a]quinolizine-3-carboxylate
- Step 3 Preparation of 10-methoxy-2-oxo-6-(2-thienyl)-9-[3-(l,2,4-triazol-l- yl)propoxy]-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid
- Step 1 Preparation of ethyl 9-(4-ethoxy-4-oxo-butoxy)-10-methoxy-2-oxo-6-(2- thienyl)-6,7-dihydrobenzo[a] quinolizine-3-carboxylate
- Step 2 Preparation of 9-(3-carboxypropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid
- Step 2 Preparation of ethyl 10-methoxy-9-(3-methylsulfanylpropoxy)-2-oxo-6-(2- thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate
- Step 3 Preparation of 10-methoxy-9-(3-methylsulfanylpropoxy)-2-oxo-6-(2-thienyl)- 6,7-dihydrobenzo[a] quinolizine-3-carboxylic acid
- Step 2 Preparation of ethyl 10-methoxy-9-(3-methylsulfonylpropoxy)-2-oxo-6-(2- thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate
- Step 3 Preparation of 10-methoxy-9-(3-methylsulfonylpropoxy)-2-oxo-6-(2-thienyl)- 6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid
- Step 1 Preparation of ethyl 10-methoxy-2-oxo-6-(2-thienyl)-9-
- Step 2 Preparation of ethyl 10-methoxy-9-methyl-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylate
- Step 3 Preparation of 10-methoxy-9-methyl-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid
- Step 1 Preparation of ethyl 9-ethyl-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate
- Step 2 Preparation of 9-ethyl-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
- Step 1 Preparation of ethyl 10-methoxy-2-oxo-9-propyl-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate
- Step 2 Preparation of 10-methoxy-2-oxo-9-propyl-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
- Step 1 Preparation of ethyl 10-methoxy-2-oxo-9-pyrrolidin-l-yl-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate
- Step 2 Preparation of 10-methoxy-2-oxo-9-pyrrolidin-l-yl-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
- Step 2 Preparation of l-(benzothiophen-2-yl)-2-[4-chloro-3-(3- methoxypropoxy)phenyl]ethanone
- Step 3 Preparation of l-(benzothiophen-2-yl)-2-[4-chloro-3-(3- methoxypropoxy)phenyl]ethanamine
- Step 4 Preparation of N-[l-(benzothiophen-2-yl)-2-[4-chloro-3-(3- methoxypropoxy)phenyl]ethyl]formamide o
- Step 6 Preparation of ethyl 6-(benzothiophen-2-yl)-10-chloro-9-(3-methoxypropoxy)-
- Step 7 Preparation of ethyl 6-(benzothiophen-2-yl)-10-chloro-9-(3-methoxypropoxy)- 2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carbox late
- Step 8 Preparation of 6-(benzothiophen-2-yl)-10-chloro-9-(3-methoxypropoxy)-2- oxo-6,7-dihydrobenzo[a] quinolizine-3-carbox lic acid
- HepG2.2.15 cells (Acs et al. Proc Natl Acad Sci U S A, 84, (1987), 4641-4), a
- constitutively HBV-expressing cell line were cultured in DMEM+Glutamax-I medium
- HepG2.2.15 cells were seeded in duplicate into white, 96-well plates at 1.5 x 10 4 cells/well.
- the cells were treated with a three-fold serial dilution series of the compounds in DMSO.
- the final DMSO concentration in all wells was 1% and DMSO was used as no drug control.
- the HBsAg chemiluminescence immunoassay (CLIA) kit (Autobio Diagnostics Co., Zhengzhou, China, Catalog number: CL0310-2) was used to measure the levels of secreted HBV antigens semi-quantitatively.
- CLIA HBsAg chemiluminescence immunoassay
- 50 ⁇ ⁇ of the supernatant was transferred to the CLIA assay plate and 50 ⁇ ⁇ of enzyme conjugate reagent was added into each well. The plates were sealed and gently agitated for 1 hour at room temperature.
- the HBsAg chemiluminescence immunoassay (CLIA) kit Autobio Diagnostics Co., Zhengzhou, China, Catalog number: CL0310-2
- Luminance was measured using a luminometer (Mithras LB 940 Multimode Microplate Reader) after 10 minutes incubation. Dose- response curves were generated and the IC 50 value was extrapolated by using the E-WorkBook Suite (ID Business Solutions Ltd., Guildford, UK). The IC 50 was defined as the compound concentration (or conditioned media log dilution) at which HBsAg secretion was reduced by 50% compared to the no drug control.
- the compounds of the present invention were tested for their activity to inhibit HBsAg as described herein.
- the Examples were tested in the above assay and found to have ICso below 25.0 ⁇ .
- Particular compounds of formula I were found to have ICso below 0.100 ⁇ . More Particular compounds of formula I were found to have ICso below 0.010 ⁇ .
- Results of HBsAg assay are given in Table 1.
- the assay employs real-time qPCR (TaqMan) to directly measure extracellular HBV DNA copy number.
- HepG2.2.15 cells were plated in 96- well micro titer plates. Only the interior wells were utilized to reduce "edge effects" observed during cell culture, the exterior wells were filled with complete medium to help minimize sample evaporation. On the following day, the
- HepG2.2.15 cells were washed and the medium was replaced with complete medium containing various concentrations of a test compound in triplicate. 3TC was used as the positive control, while media alone was added to cells as a negative control (virus control, VC). Three days later, the culture medium was replaced with fresh medium containing the appropriately diluted drug. Six days following the initial administration of the test compound, the cell culture supernatant was collected, treated with pronase and then used in a real-time qPCR/TaqMan assay to determine HBV DNA copy numbers. Antiviral activity was calculated from the reduction in HBV DNA levels (IC 50 ).
- the compounds of the present invention were tested for their activity to anti HBV DNA production as described herein.
- the Examples were tested in the above assay and found to have ICso below 25.0 ⁇ .
- Particular compounds of formula I were found to have ICso below 0.10 ⁇ .
- Results of HBV DNA assay are given in Table 2.
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Virology (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Molecular Biology (AREA)
- Gastroenterology & Hepatology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
The invention provides novel compounds having the general formula (I), wherein R1, R2, R3, R4, R5 and Ar are as described herein, compositions including compounds and methods of using the compounds.
Description
Novel 2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid derivatives for the treatment and prophylaxis of hepatitis B virus infection
The present invention relates to organic compounds useful for therapy and/or prophylaxis in a mammal, and in particular to HBsAg (HBV Surface antigen) inhibitors and HBV DNA production inhibitors useful for treating HBV infection. FIELD OF THE INVENTION
The present invention relates to novel 2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid derivatives having pharmaceutical activity, their manufacture, pharmaceutical compositions containing them and their potential use as medicaments.
The present invention relates to compounds of formula I
o o

wherein R1 to R5 and Ar are as described below, or to pharmaceutically acceptable salts, or to enantiomers thereof.
The hepatitis B virus (HBV) is an enveloped, partially double- stranded DNA virus. The compact 3.2 kb HBV genome consists of four overlapping open reading frames (ORF), which encode for the core, polymerase (Pol), envelope and X-proteins. The Pol ORF is the longest and the envelope ORF is located within it, while the X and core ORFs overlap with the Pol ORF. The lifecycle of HBV has two main events: 1) generation of closed circular DNA (cccDNA) from relaxed circular (RC DNA), and 2) reverse transcription of pregenomic RNA (pgRNA) to produce RC DNA. Prior to the infection of host cells, the HBV genome exists within the virion as RC DNA. It has been determined that HBV virions are able to gain entry into host cells by no n- specifically binding to the negatively charged proteoglycans present on the surface of human hepatocytes (Schulze, A., P. Gripon & S. Urban. Hepatology, 46, (2007), 1759-68) and via the specific binding of HBV surface antigens (HBsAg) to the hepatocyte sodium-
taurocholate cotransporting polypeptide (NTCP) receptor (Yan, H. et al. / Virol, 87, (2013), 7977-91). Once the virion has entered the cell, the viral cores and the encapsidated RC DNA are transported by host factors, via a nuclear localization signal, into the nucleus through the
ImpP/Impa nuclear transport receptors. Inside the nucleus, host DNA repair enzymes convert the RC DNA into cccDNA. cccDNA acts as the template for all viral mRNAs and as such, is responsible for HBV persistence in infected individuals. The transcripts produced from cccDNA are grouped into two categories; Pregenomic RNA (pgRNA) and subgenomic RNA. Subgenomic transcripts encode for the three envelopes (L, M and S) and X proteins, and pgRNA encodes for Pre-Core, Core, and Pol proteins (Quasdorff, M. & U. Protzer. / Viral Hepat, 17, (2010), 527- 36). Inhibition of HBV gene expression or HBV RNA synthesis leads to the inhibition of HBV viral replication and antigens production (Mao, R. et al. PLoS Pathog, 9, (2013), el003494; Mao, R. et al. / Virol, 85, (2011), 1048-57). For instance, IFN-a was shown to inhibit HBV replication and viral HBsAg production by decreasing the transcription of pgRNA and subgenomic RNA from the HBV covalently closed circular DNA (cccDNA) minichromosome. (Belloni, L. et al. J Clin Invest, 122, (2012), 529-37; Mao, R. et al. / Virol, 85, (2011), 1048-57). All HBV viral mRNAs are capped and polyadenylated, and then exported to the cytoplasm for translation. In the cytoplasm, the assembly of new virons is initiated and nascent pgRNA is packaged with viral Pol so that reverse transcription of pgRNA, via a single stranded DNA intermediate, into RC DNA can commence. The mature nucleocapsids containing RC DNA are enveloped with cellular lipids and viral L, M, and S proteins and then the infectious HBV particles are then released by budding at the intracellular membrane (Locarnini, S. Semin Liver Dis, (2005), 25 Suppl 1, 9-19). Interestingly, non-infectious particles are also produced that greatly outnumber the infectious virions. These empty, enveloped particles (L, M and S) are referred to as subviral particles.
Importantly, since subviral particles share the same envelope proteins and as infectious particles, it has been surmised that they act as decoys to the host immune system and have been used for HBV vaccines. The S, M, and L envelope proteins are expressed from a single ORF that contains three different start codons. All three proteins share a 226aa sequence, the S-domain, at their C- termini. M and L have additional pre-S domains, Pre-S2 and Pre-S2 and Pre-Sl, respectively. However, it is the S-domain that has the HBsAg epitope (Lambert, C. & R. Prange. Virol J, (2007), 4, 45).
The control of viral infection needs a tight surveillance of the host innate immune system which could respond within minutes to hours after infection to impact on the initial growth of the virus and limit the development of a chronic and persistent infection. Despite the available
current treatments based on IFN and nucleos(t)ide analogues, the Hepatitis B virus (HBV) infection remains a major health problem worldwide which concerns an estimated 350 million chronic carriers who have a higher risk of liver cirrhosis and hepatocellular carcinoma.
The secretion of antiviral cytokines in response to HBV infection by the hepatocytes and/or the intra-hepatic immune cells plays a central role in the viral clearance of infected liver. However, chronically infected patients only display a weak immune response due to various escape strategies adopted by the virus to counteract the host cell recognition systems and the subsequent antiviral responses.
Many observations showed that several HBV viral proteins could counteract the initial host cellular response by interfering with the viral recognition signaling system and subsequently the interferon (IFN) antiviral activity. Among these, the excessive secretion of HBV empty subviral particles (SVPs, HBsAg) may participate to the maintenance of the immunological tolerant state observed in chronically infected patients (CHB). The persistent exposure to HBsAg and other viral antigens can lead to HBV- specific T-cell deletion or to progressive functional impairment (Kondo et al. Journal of Immunology (1993), 150, 4659-4671; Kondo et al. Journal of Medical Virology (2004), 74, 425-433; Fisicaro et al. Gastroenterology, (2010), 138, 682-93;). Moreover HBsAg has been reported to suppress the function of immune cells such as monocytes, dendritic cells (DCs) and natural killer (NK) cells by direct interaction (Op den Brouw et al. Immunology, (2009b), 126, 280-9; Woltman et al. PLoS One, (2011), 6, el5324; Shi et al. / Viral Hepat.
(2012), 19, e26-33; Kondo et al. ISRN Gasteroenterology, (2013), Article ID 935295).
HBsAg quantification is a significant biomarker for prognosis and treatment response in chronic hepatitis B. However the achievement of HBsAg loss and seroconversion is rarely observed in chronically infected patients but remains the ultimate goal of therapy. Current therapy such as Nucleos(t)ide analogues are molecules that inhibit HBV DNA synthesis but are not directed at reducing HBsAg level. Nucleos(t)ide analogs, even with prolonged therapy, have demonstrated rates of HBsAg clearance comparable to those observed naturally (between - \%- 2%) (Janssen et al. Lancet, (2005), 365, 123-9; Marcellin et al. N. Engl. J. Med., (2004), 351, 1206-17; Buster et al. Hepatology, (2007), 46, 388-94). Therefore, targeting HBsAg together with HBV DNA levels in CHB patients may significantly improve CHB patient immune reactivation and remission (Wieland, S. F. & F. V. Chisari. / Virol, (2005), 79, 9369-80; Kumar et al. / Virol, (2011), 85, 987-95; Woltman et al. PLoS One, (2011), 6, el5324; Op den Brouw et al. Immunology, (2009b), 126, 280-9).
SUMMARY OF THE INVENTION
The present invention relates to compounds of formula I

wherein
2 3 4
RJ and R" are independently selected from Ci_6alkyl, haloCi_6alkyl, hydrogen, halogen, cyano, amino, Ci_6alkylamino, diCi_6alkylamino, pyrrolidinyl and OR6;
R is hydrogen or Ci_6alkyl;
R6 is hydrogen; Ci_6alkyl; haloCi_6alkyl; C3_7cycloalkylCi-6alkyl; phenylCi-6alkyl;
hydroxyCi_6alkyl; Ci-6alkoxyCi_6alkyl; carboxyCi-6alkyl; Ci_6alkylsulfanylCi_6 alkyl; Ci_ 6alkylsulfonylCi_6alkyl; cyanoCi_6alkyl; aminoCi_6alkyl; Ci_6alkylaminoCi_6 alkyl; diCi_ 6 alkylaminoC i _6 alkyl; C i _6 alkylcarbonylaminoC i _6 alkyl; C i _6 alkylsulfonylaminoC i_6 alkyl; Ci-6alkoxycarbonylaminoCi-6alkyl; pyrazolylCi_6alkyl; triazolylCi_6 alkyl or
heterocycloalkylCi-6alkyl, wherein heterocycloalkyl is N-containing monocyclic heterocycloalkyl;
Ar is phenyl; phenyl substituted by one, two or three groups independently selected from Ci_ 6alkyl,
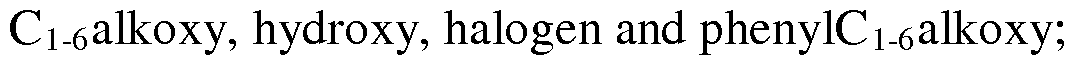 hydroxy, halogen and thienyl; thienyl substituted by one, two or three groups independently selected from
hydroxy, halogen and thienyl; thienyl substituted by one, two or three groups independently selected from
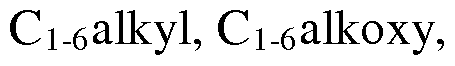 hydroxy, halogen and phenylC i_6 alkoxy; benzothiophenyl; benzothiophenyl substituted by one, two or three groups independently selected from Ci_6alkyl,
hydroxy, halogen and phenylC i_6 alkoxy; benzothiophenyl; benzothiophenyl substituted by one, two or three groups independently selected from Ci_6alkyl,
 hydroxy, halogen and phenylC 1-6 alkoxy; pyridinyl; pyridinyl substituted by one, two or three groups
hydroxy, halogen and phenylC 1-6 alkoxy; pyridinyl; pyridinyl substituted by one, two or three groups
independently selected from Ci-6 alkyl, Ci-6alkoxy, hydroxy, halogen and phenylCi- 6alkoxy; pyrimidinyl; pyrimidinyl substituted by one, two or three groups independently selected from Ci_6alkyl, Ci-6alkoxy, hydroxy, halogen and phenylCi-6 alkoxy; pyrrolyl; pyrrolyl substituted by one, two or three groups independently selected from Ci_6alkyl, Ci_ 6alkoxy, hydroxy, halogen and phenylCi-6 alkoxy; pyrazolyl; pyrazolyl substituted by one, two or three groups independently selected from Ci-6 alkyl, Ci-6alkoxy, hydroxy, halogen and phenylC 1-6 alkoxy; thiazolyl; or thiazolyl substituted by one, two or three groups
independently selected from C1-6 alkyl, Ci-6alkoxy, hydroxy, halogen and phenylCi- 6alkoxy;
or pharmaceutically acceptable salts, or enantiomers thereof.
DETAILED DESCRIPTION OF THE INVENTION DEFINITIONS
As used herein, the term "Ci_6alkyl" alone or in combination signifies a saturated, linear- or branched chain alkyl group containing 1 to 6, particularly 1 to 4 carbon atoms, for example methyl, ethyl, propyl, isopropyl, 1-butyl, 2-butyl, ie/t-butyl and the like. Particular "Ci_6alkyl" groups are methyl, ethyl, isopropyl and ie/t-butyl.
The term "C3_7cycloalkyl", alone or in combination, refers to a saturated carbon ring containing from 3 to 7 carbon atoms, particularly from 3 to 6 carbon atoms, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and the like. Particular "C3_ 7cycloalkyl" groups are cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
The term "Ci_6alkoxy" alone or in combination signifies a group Ci_6alkyl-0-, wherein the "Chalky!" is as defined above; examples for Ci-6alkoxy are methoxy, ethoxy, propoxy, iso- propoxy, w-butoxy, zsobutoxy, 2-butoxy, ie/t-butoxy and the like. Particular "Ci_6alkoxy" groups are methoxy and ethoxy and more particularly methoxy.
The term "halogen" means fluorine, chlorine, bromine or iodine.
The term "haloCi-6 alkyl" denotes a Ci_6alkyl group wherein at least one of the hydrogen atoms of the C1-6 alkyl group has been replaced by same or different halogen atoms, particularly fluoro atoms. Examples of "haloCi-6 alkyl" include monofluoro-, difluoro- or trifluoro-methyl, - ethyl or -propyl, for example 3,3,3-trifluoropropyl, 3,3- difluoropropyl, 2-fluoroethyl, 2,2- difluoroethyl, 2,2,2-trifluoroethyl, fluoromethyl, difluoromethyl or trifluoromethyl. Particular "halo C 1-6 alkyl" group is difluoromethyl or trifluoromethyl.
The term "monocyclic heterocycloalkyl" is a monovalent saturated or partly unsaturated monocyclic ring system of 4 to 7 ring atoms, comprising 1, 2, or 3 ring heteroatoms selected from N, O and S, the remaining ring atoms being carbon. Examples for monocyclic
heterocycloalkyl are aziridinyl, oxiranyl, azetidinyl, oxetanyl, pyrrolidinyl, 2-oxo-pyrrolidinyl, tetrahydrofuranyl, tetrahydro-thienyl, pyrazolidinyl, imidazolidinyl, oxazolidinyl, isoxazolidinyl, thiazolidinyl, piperidinyl, tetrahydropyranyl, tetrahydrothiopyranyl, piperazinyl, morpholinyl,
thiomorpholinyl, l,l-dioxo-thiomorpholin-4-yl, azepanyl, diazepanyl, homopiperazinyl, or oxazepanyl. Particular "monocyclic heterocycloalkyl" groups are piperidinyl, morpholinyl, 2- oxo-pyrrolidinyl, and pyrrolidinyl.
The term "N-containing monocyclic heterocycloalkyl" is a "monocyclic heterocycloalkyl" as defined above wherein at least one of the heteroatoms is N. Examples for 'W-containing monocyclic heterocycloalkyl" are aziridinyl, azetidinyl, pyrrolidinyl, 2-oxo-pyrrolidinyl, pyrazolidinyl, imidazolidinyl, oxazolidinyl, isoxazolidinyl, thiazolidinyl, piperidinyl, piperazinyl, morpholinyl, thio morpholinyl, l,l-dioxo-thiomorpholin-4-yl, azepanyl, diazepanyl,
homopiperazinyl, or oxazepanyl. Particular 'W-containing monocyclic heterocycloalkyl" groups are piperidinyl, morpholinyl, 2-oxo-pyrrolidinyl and pyrrolidinyl.
The term "amino" denotes a group of the formula -NR'R" wherein R' and R" are independently hydrogen, Ci_6alkyl, Ci_6alkoxy, C3_7cycloalkyl, heteroC3_7cycloalkyl, aryl or heteroaryl. Alternatively, R' and R", together with the nitrogen to which they are attached, can form a heteroC3_7cycloalkyl.
The term "carbonyl" alone or in combination refers to the group -C(O)-.
The term "cyano" alone or in combination refers to the group -CN.
The term "Ci_6alkylsulfanyl" denotes the group -S-R', wherein R' is a Ci_6alkyl group as defined above.
The term "Ci_6alkylsulfonyl" denotes a group -S02-R', wherein R' is a Ci_6alkyl group as defined above. Examples of Ci_6alkylsulfonyl include methylsulfonyl and ethylsulfonyl.
The term "enantiomer" denotes two stereoisomers of a compound which are non- superimpo sable mirror images of one another.
The term "diastereomer" denotes a stereoisomer with two or more centers of chirality and whose molecules are not mirror images of one another. Diastereomers have different physical properties, e.g. melting points, boiling points, spectral properties, and reactivities.
The compounds according to the present invention may exist in the form of their
pharmaceutically acceptable salts. The term "pharmaceutically acceptable salt" refers to conventional acid-addition salts or base-addition salts that retain the biological effectiveness and properties of the compounds of formula I and are formed from suitable non-toxic organic or inorganic acids or organic or inorganic bases. Acid-addition salts include for example those derived from inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, sulfamic acid, phosphoric acid and nitric acid, and those derived from organic acids such as /7-toluenesulfonic acid, salicylic acid, methane sulfonic acid, oxalic acid, succinic acid,
citric acid, malic acid, lactic acid, fumaric acid, and the like. Base-addition salts include those derived from ammonium, potassium, sodium and, quaternary ammonium hydroxides, such as for example, tetramethyl ammonium hydroxide. The chemical modification of a pharmaceutical compound into a salt is a technique well known to pharmaceutical chemists in order to obtain improved physical and chemical stability, hygroscopicity, flowability and solubility of compounds. It is for example described in Bastin R.J., et al., Organic Process Research & Development 2000, 4, 427-435; or in Ansel, H., et al., In: Pharmaceutical Dosage Forms and Drug Delivery Systems, 6th ed. (1995), pp. 196 and 1456-1457. Particular are the sodium salts of the compounds of formula I.
Compounds of the general formula I which contain one or several chiral centers can either be present as racemates, diastereomeric mixtures, or optically active single isomers. The racemates can be separated according to known methods into the enantiomers. Particularly, diastereomeric salts which can be separated by crystallization are formed from the racemic mixtures by reaction with an optically active acid such as e.g. D- or L-tartaric acid, mandelic acid, malic acid, lactic acid or camphor sulfonic acid.
INHIBITORS OF HBsAg
The present invention provides i) novel compounds having the general formula I:

R1, R2, R3 and R4 are independently selected from C1-6alkyl, haloC1-6alkyl, hydrogen, halogen, cyano, amino, Ci_6alkylamino, diCi_6alkylamino, pyrrolidinyl and OR6;
R5 is hydrogen or C1-6alkyl;
R6 is hydrogen; C1-6alkyl; haloC1-6alkyl; C3-7cycloalkylC1-6alkyl; phenylC1-6alkyl;
hydroxyC1-6alkyl; C1-6alkoxyC1-6alkyl; carboxyC1-6alkyl; C1-6alkylsulfanylC1-6alkyl; Ci_
6alkylsulfonylC1-6alkyl; cyanoC1-6alkyl; aminoC1-6alkyl; C1-6alkylaminoC1-6alkyl; diCi_ 6 alkylaminoC i -6 alkyl; C i -6 alkylcarbonylaminoC i -6 alkyl; C i -6 alkylsulfonylaminoC 1-6 alkyl; C1-6alkoxycarbonylaminoC1-6alkyl; pyrazolylC1-6alkyl; triazolylCi-6 alkyl or
heterocycloalkylCi-6alkyl, wherein heterocycloalkyl is N-containing monocyclic heterocycloalkyl;
Ar is phenyl; phenyl substituted by one, two or three groups independently selected from Ci_ 6alkyl,
 hydroxy, halogen and thienyl; thienyl substituted by one, two or three groups independently selected from
hydroxy, halogen and thienyl; thienyl substituted by one, two or three groups independently selected from
 hydroxy, halogen and phenylCi-6 alkoxy; benzothiophenyl; benzothiophenyl substituted by one, two or three groups independently selected from Ci_6alkyl,
hydroxy, halogen and phenylCi-6 alkoxy; benzothiophenyl; benzothiophenyl substituted by one, two or three groups independently selected from Ci_6alkyl,
 hydroxy, halogen and phenylC 1-6 alkoxy; pyridinyl; pyridinyl substituted by one, two or three groups
hydroxy, halogen and phenylC 1-6 alkoxy; pyridinyl; pyridinyl substituted by one, two or three groups
independently selected from Chalky!, Ci-6alkoxy, hydroxy, halogen and phenylCi- 6alkoxy; pyrimidinyl; pyrimidinyl substituted by one, two or three groups independently selected from Chalky!, Ci-6alkoxy, hydroxy, halogen and phenylCi-6 alkoxy; pyrrolyl; pyrrolyl substituted by one, two or three groups independently selected from Chalky!, Ci_ 6alkoxy, hydroxy, halogen and phenylCi-6 alkoxy; pyrazolyl; pyrazolyl substituted by one, two or three groups independently selected from Chalky!, Ci-6alkoxy, hydroxy, halogen and phenylCi-6 alkoxy; thiazolyl; or thiazolyl substituted by one, two or three groups independently selected from Chalky!, Ci-6alkoxy, hydroxy, halogen and phenylCi- 6 alkoxy;
or pharmaceutically acceptable salts, or enantiomers thereof. Further embodiment of present invention is (ii) a compound of formula I, wherein
R1 is hydrogen;
R is hydrogen, halogen or Ci-6alkoxy;
R3 is Ci-6alkyl, pyrrolidinyl or OR6;
R4 is C 1-6 alkyl or hydrogen;
R5 is hydrogen;
R6 is Ci-6alkyl, haloCi-6 alkyl, C3_7cycloalkylCi_6alkyl, phenylCi-6 alkyl, hydroxyCi-6 alkyl, Ci-6alkoxyCi-6alkyl, carboxyCi_6 alkyl, Ci_6alkylsulfanylCi_6 alkyl, Ci_6alkylsulfonylCi_ 6alkyl, cyanoCi_6 alkyl, aminoCi-6 alkyl, Ci_6alkylcarbonylaminoCi_6alkyl, Ci_
6 alkylsulfonylaminoC i _6 alkyl, C i _6 alkoxycarbonylaminoC i _6 alkyl, pyrazolylC i _6 alkyl, triazolylC 1-6 alkyl, piperidylCi-6 alkyl, morpholinylCi-6 alkyl or 2-oxopyrrolidinylCi_6 alkyl;
Ar is phenyl; phenyl substituted by Ci_6alkyl, Ci-6alkoxy, hydroxy, halogen or phenylCi- 6alkoxy; thienyl; thienyl substituted by Ci_6 alkyl; or benzothiophenyl;
or pharmaceutically acceptable salts, or enantiomers thereof.
Another embodiment of present invention is (iii) a compound of formula I as defined above, or pharmaceutically acceptable salts, or enantiomers thereof, wherein R1 is hydrogen, R4 is hydrogen, R5 is hydrogen, and all remaining substituents have the significances given herein before.
Another embodiment of present invention is (iv) a compound of formula I as defined above, or pharmaceutically acceptable salts, or enantiomers thereof, wherein R is Ci_6alkoxy, and all remaining substituents have the significances given herein before.
A further embodiment of present invention is (v) a compound of formula I as defined above, or pharmaceutically acceptable salts, or enantiomers thereof, wherein R is methoxy, and all remaining substituents have the significances given herein before. Another embodiment of present invention is (vi) a compound of formula I as defined above, or pharmaceutically acceptable salts, or enantiomers thereof, wherein R3 is pyrrolidinyl or OR6, wherein R6 is Ci_6alkyl, haloCi_6alkyl, C3_7cycloalkylCi_6alkyl, hydro xyCi_6 alky 1, Ci_6alkoxyCi_ 6alkyl, carboxyCi_6alkyl, Ci_6alkylsulfanylCi_6alkyl, cyanoCi_6alkyl, aminoCi_6alkyl, Ci_ 6alkylcarbonylaminoCi-6alkyl, Ci_6alkylsulfonylaminoCi_6alkyl or Ci_6alkoxycarbonylaminoCi_ 6alkyl, and all remaining substituents have the significances given herein before.
Further embodiment of present invention is (vii) a compound of formula I as defined above, or pharmaceutically acceptable salts, or enantiomers thereof, wherein R3 is OR6, wherein R6 is methyl, isobutyl, trifluoroethyl, cyclopropylmethyl, cyanopropyl, hydroxypropyl, hydroxyhexyl, hydroxydimethylpropyl, methoxypropyl, carboxypropyl, methylsulfanylpropyl, aminohexyl, methylcarbonylaminohexyl, methylsulfonylaminohexyl or methoxycarbonylaminohexyl, and all remaining substituents have the significances given herein before.
Another embodiment of present invention is (viii) a compound of formula I as defined above, or pharmaceutically acceptable salts, or enantiomers thereof, wherein Ar is phenyl, phenyl substituted by Ci_6alkyl or halogen, thienyl or thienyl substituted by Ci_6alkyl, and all remaining substituents have the significances given herein before.
Further embodiment of present invention is (ix) a compound of formula I as defined above, or pharmaceutically acceptable salts, or enantiomers thereof, wherein Ar is phenyl; phenyl substituted by methyl, fluoro or chloro; thienyl or thienyl substituted by methyl, and all remaining substituents have the significances given herein before.
Another embodiment of present invention is (x) a compound of formula I, wherein
R1 is hydrogen;
R is Ci_6alkoxy;
R3 is pyrrolidinyl or OR6;
R4 is hydrogen;
R5 is hydrogen;
R6 is Chalky!, haloCi_6alkyl, C3_7cycloalkylCi-6alkyl, hydroxyCi_6alkyl, Ci_6alkoxyCi-6alkyl or Ci-6alkylsulfanylCi_6alkyl;
Ar is phenyl substituted by C1-6alkyl, or thienyl;
or pharmaceutically acceptable salts, or enantiomers thereof.
A further embodiment of present invention is (x) a compound of formula I, wherein R1 is hydrogen;
R is methoxy;
R3 is pyrrolidinyl or OR6;
R4 is hydrogen;
R5 is hydrogen;
R6 is isobutyl, trifluoroethyl, cyclopropylmethyl, hydroxydimethylpropyl, methoxypropyl or methylsulfanylpropyl;
Ar is phenyl substituted by methyl or thienyl;
or pharmaceutically acceptable salts, or enantiomers thereof.
Particular compounds of formula I according to the invention are the following:
10-Methoxy-9-(3-methoxypropoxy)-2-oxo-6-phenyl-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid;
10-Methoxy-9-(3-methoxypropoxy)-2-oxo-6-(3-thienyl)-6,7-dihydrobenzo[a]quinolizine- 3-carboxylic acid;
10-Methoxy-9-(3-methoxypropoxy)-2-oxo-6-(2-thienyl)-6,7-di ydrobenzo[a]quinolizine- 3-carboxylic acid;
6-(4-Hydroxyphenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
10-Methoxy-6-(4-methoxyphenyl)-9-(3-methoxypropoxy)-2-oxo-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
6-(4-Benzyloxyphenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
10-Methoxy-9-(3-methoxypropoxy)-6-(4-methyl-2-thienyl)-2-oxo-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid;
6-(3-Chlorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
6-(4-Fluorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
10-Methoxy-9-(3-methoxypropoxy)-6-(o-tolyl)-2-oxo-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid;
9-Methoxy-8-methyl-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid;
9-Benzyloxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid;
9,10-Dimethoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid; 9-Isobutoxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid;
9- (Cyclopropylmethoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid;
10- Methoxy-2-oxo-9-(3-pyrazol-l-ylpropoxy)-6-(2-thienyl)-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
9-(3-Hydroxypropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine- 3-carboxylic acid;
10-Methoxy-2-oxo-9-[3-(l-piperidyl)propoxy]-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid formate;
9-(3-Cyanopropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid;
10-Methoxy-2-oxo-9-[2-(2-oxopyrrolidin-l-yl)ethoxy]-6-(2-thienyl)-6,7- di ydrobenzo [a] quino lizine- 3 -carboxylic acid ;
9-[6-(ieri-Butoxycarbonylamino)hexoxy]-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
9-(6-Arrdnohexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid formate;
9-(6-Acetamidohexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine- 3-carboxylic acid;
9-[6-(Methanesulfonamido)hexoxy]-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid;
9-(6-Hydroxyhexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid;
9- (3-Hydroxy-2,2-dimethyl-propoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
10-Methoxy-2-oxo-6-(2-thienyl)-9-(2,2,2-trifluoroethoxy)-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
10- Methoxy-9-(3-morpholinopropoxy)-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate;
10-Methoxy-2-oxo-6-(2-thienyl)-9-[3-(l,2,4-triazol-l-yl)propoxy]-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid;
9- (3-Carboxypropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine- 3-carboxylic acid;
10- Methoxy-9-(3-methylsulfanylpropoxy)-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
10-Methoxy-9-(3-methylsulfonylpropoxy)-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
10-Methoxy-9-methyl-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid;
9- Ethyl-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid;
10- Methoxy-2-oxo-9-propyl-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid;
10-Methoxy-2-oxo-9-pyrrolidin-l-yl-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid;
6-(Benzothiophen-2-yl)-10-chloro-9-(3-methoxypropoxy)-2-oxo-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
or pharmaceutically acceptable salts, or enantiomers thereof.
More particularly, the invention relates to the following compounds of formula I:
10-Methoxy-9-(3-methoxypropoxy)-2-oxo-6-(3-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid;
10-Methoxy-9-(3-methoxypropoxy)-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine- 3-carboxylic acid;
10-Methoxy-9-(3-methoxypropoxy)-6-(o-tolyl)-2-oxo-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid;
9-Isobutoxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid;
9-(Cyclopropylmethoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
9- (3-Hydroxy-2,2-dimethyl-propoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
10- Methoxy-2-oxo-6-(2-thienyl)-9-(2,2,2-trifluoroethoxy)-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
10-Methoxy-9-(3-methylsulfanylpropoxy)-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
10-Methoxy-2-oxo-9-pyrrolidin-l-yl-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid;
or pharmaceutically acceptable salts, or enantiomers thereof.
SYNTHESIS
The compounds of the present invention can be prepared by any conventional means. Suitable processes for synthesizing these compounds as well as their starting materials are provided in the schemes below and in the examples. All substituents, in particular, R1 to R5 and Ar are as defined above unless otherwise indicated. Furthermore, and unless explicitly otherwise stated, all reactions, reaction conditions, abbreviations and symbols have the meanings well known to a person of ordinary skill in organic chemistry.
General synthetic route for Compounds I (Scheme 1)
Scheme 1

XIV FT is C^alkyl
The compound of formula I can be prepared according to Scheme 1. Coupling reaction of bromo-benzene II with ketone III affords Compound IV. The reaction can be carried out in the presence of a Pd catalyst such as Pd2(dba)3 , Pd(PPh3)4 or PdCl2(PPh3)2, a ligand such as Xantphos, and a suitable base such as i-BuONa, Na2C03 or Cs2C03, in a suitable solvent such as THF, toluene or 1,4-dioxane at a temperature between room temperature and 130 °C.
Reductive amination of Compound IV affords Compound V. Compound V is heated with ethyl
formate or formic acid in a solvent such as ethanol or dioxane to afford Compound XI.
Compound XI is treated with POCI3 in a suitable solvent such as acetonitrile or DCM at a temperature between room temperature and 100 °C to give Compound XII. Compound XII reacts with Ci_6alkyl 2-(ethoxymethylene)-3-oxo-butanoate in a solvent such as ethanol to give Compound XIII. Compound XIV is obtained by dehydrogenation of compound XIII by using p- chloranil. Hydrolyzation of Compound XIV with a base such as lithium hydroxide or sodium hydroxide in a suitable solvent such as THF/H20, EtOH/H20 or MeOH/H20 affords the compound of formula I. General synthetic route for Compounds 1-1 (Scheme 2)
Scheme 2

XIV-2
XIV-1
R6Q1 , base, solvent
_R

1-1 XIV-3
Q1 is halogen, -0-S(0)2CH3 or -0-S(0)2-(4-CH3-Ph).
Ft7 is C^alkyl
The compound of formula 1-1 can be prepared according to Scheme 2. Debenzylation of Compound XIV-1 by hydrogenation is carried out in the presence of Pd/C in a solvent such as ethanol, THF, methanol to afford Compound XIV-2. Then Compound XIV-2 reacts with halides, mesylates or tosylates in the presence of a base such as K2C03 in a solvent such as acetone or DMF to give XIV-3. Hydrolyzation of Compound XIV with a base such as lithium hydroxide or sodium hydroxide in a suitable solvent such as THF/H20, EtOH/H20 or MeOH/H20 affords the compound of formula 1-1.
This invention also relates to a process for the preparation of a compound of formula I comprising
(a) hydrolysis of a compound of formula (A)

wherein R 1 to R 7 and Ar are defined above unless otherwise indicated.
In step (a) and step (b) a base such as lithium hydroxide or sodium hydroxide can be for example used.
A compound of formula I when manufactured according to the above process is also an object of the invention.
PHARMACEUTICAL COMPOSITIONS AND ADMINISTRATION
The invention also relates to a compound of formula I for use as therapeutically active substance.
Another embodiment provides pharmaceutical compositions or medicaments containing the compounds of the invention and a therapeutically inert carrier, diluent or excipient, as well as methods of using the compounds of the invention to prepare such compositions and medicaments. In one example, compounds of formula I may be formulated by mixing at ambient temperature at the appropriate pH, and at the desired degree of purity, with physiologically acceptable carriers, i.e., carriers that are non-toxic to recipients at the dosages and concentrations employed into a
galenical administration form. The pH of the formulation depends mainly on the particular use and the concentration of compound, but preferably ranges anywhere from about 3 to about 8. In one example, a compound of formula I is formulated in an acetate buffer, at pH 5. In another embodiment, the compounds of formula I are sterile. The compound may be stored, for example, as a solid or amorphous composition, as a lyophilized formulation or as an aqueous solution.
Compositions are formulated, dosed, and administered in a fashion consistent with good medical practice. Factors for consideration in this context include the particular disorder being treated, the particular mammal being treated, the clinical condition of the individual patient, the cause of the disorder, the site of delivery of the agent, the method of administration, the scheduling of administration, and other factors known to medical practitioners. The "effective amount" of the compound to be administered will be governed by such considerations, and is the minimum amount necessary to inhibit HBsAg. For example, such amount may be below the amount that is toxic to normal cells, or the mammal as a whole.
In one example, the pharmaceutically effective amount of the compound of the invention administered parenterally per dose will be in the range of about 0.01 to 100 mg/kg, alternatively about 0.01 to 100 mg/kg of patient body weight per day, with the typical initial range of compound used being 0.3 to 15 mg/kg/day. In another embodiment, oral unit dosage forms, such as tablets and capsules, preferably contain from about 0.1 to about 1000 mg of the compound of the invention.
The compounds of the invention may be administered by any suitable means, including oral, topical (including buccal and sublingual), rectal, vaginal, transdermal, parenteral, subcutaneous, intraperitoneal, intrapulmonary, intradermal, intrathecal and epidural and intranasal, and, if desired for local treatment, intralesional administration. Parenteral infusions include intramuscular, intravenous, intraarterial, intraperitoneal, or subcutaneous administration.
The compounds of the present invention may be administered in any convenient administrative form, e.g., tablets, powders, capsules, solutions, dispersions, suspensions, syrups, sprays, suppositories, gels, emulsions, patches, etc. Such compositions may contain components conventional in pharmaceutical preparations, e.g., diluents, carriers, pH modifiers, sweeteners, bulking agents, and further active agents.
A typical formulation is prepared by mixing a compound of the present invention and a carrier or excipient. Suitable carriers and excipients are well known to those skilled in the art and are described in detail in, e.g., Ansel, Howard C, et al., Ansel's Pharmaceutical Dosage Forms and Drug Delivery Systems. Philadelphia: Lippincott, Williams & Wilkins, 2004;
Gennaro, Alfonso R., et al. Remington: The Science and Practice of Pharmacy. Philadelphia: Lippincott, Williams & Wilkins, 2000; and Rowe, Raymond C. Handbook of Pharmaceutical Excipients. Chicago, Pharmaceutical Press, 2005. The formulations may also include one or more buffers, stabilizing agents, surfactants, wetting agents, lubricating agents, emulsifiers, suspending agents, preservatives, antioxidants, opaquing agents, glidants, processing aids, colorants, sweeteners, perfuming agents, flavoring agents, diluents and other known additives to provide an elegant presentation of the drug (i.e., a compound of the present invention or pharmaceutical composition thereof) or aid in the manufacturing of the pharmaceutical product (i.e., medicament).
An example of a suitable oral dosage form is a tablet containing about 0.1 to 1000 mg of the compound of the invention compounded with about 0 to 2000 mg anhydrous lactose, about 0 to 2000 mg sodium croscarmellose, about 0 to 2000 mg polyvinylpyrrolidone (PVP) K30, and about 0 to 2000 mg magnesium stearate. The powdered ingredients are first mixed together and then mixed with a solution of the PVP. The resulting composition can be dried, granulated, mixed with the magnesium stearate and compressed to tablet form using conventional equipment. An example of an aerosol formulation can be prepared by dissolving the compound, for example 0.1 to 1000 mg, of the invention in a suitable buffer solution, e.g. a phosphate buffer, adding a tonicifier, e.g. a salt such sodium chloride, if desired. The solution may be filtered, e.g., using a 0.2 micron filter, to remove impurities and contaminants.
An embodiment, therefore, includes a pharmaceutical composition comprising a compound of Formula I, or a stereoisomer or pharmaceutically acceptable salt thereof. In a further embodiment includes a pharmaceutical composition comprising a compound of Formula I, or a stereoisomer or pharmaceutically acceptable salt thereof, together with a pharmaceutically acceptable carrier or excipient.
The following example A and B illustrate typical compositions of the present invention, but serve merely as representative thereof.
Example A
A compound of formula I can be used in a manner known per se as the active ingredient for the production of tablets of the following composition:
Per tablet
Active ingredient 200 mg
Microcrystalline cellulose 155 mg
Corn starch 25 mg
Talc 25 mg
Hydro xypropylmethylcellulose 20 mg
425 mg Example B
A compound of formula I can be used in a manner known per se as the active ingredient for the production of capsules of the following composition:
Per capsule
Active ingredient 100.0 mg
Corn starch 20.0 mg
Lactose 95.0 mg
Talc 4.5 mg
Magnesium stearate 0.5 mg
220.0 mg
INDICATIONS AND METHODS OF TREATMENT
The compounds of the invention can inhibit HBsAg production or secretion and inhibit HBV gene expression. Accordingly, the compounds of the invention are useful for the treatment or prophylaxis of HBV infection. The invention relates to the use of a compound of formula I for the inhibition of HBsAg production or secretion.
The invention relates to the use of a compound of formula I for the inhibition of HBV DNA production.
The invention relates to the use of a compound of formula I for the inhibition of HBV gene expression. The invention relates to the use of a compound of formula I for the treatment or prophylaxis of HBV infection.
The use of a compound of formula I for the preparation of medicaments useful in the treatment or prophylaxis diseases that are related to HBV infection is an object of the invention.
The invention relates in particular to the use of a compound of formula I for the
preparation of a medicament for the treatment or prophylaxis of HBV infection.
Another embodiment includes a method for the treatment or prophylaxis of HBV infection, which method comprises administering an effective amount of a compound of Formula I, a stereoisomer, tautomer, prodrug, conjugates or pharmaceutically acceptable salt thereof.
COMBINATION THERAPY
The compounds of the invention can be combined with other anti HBV agents such as interferon alpha- 2b, interferon alpha-2a, and interferon alphacon-1 (pegylated and unpegylated), ribavirin, lamivudine (3TC), entecavir, tenofovir, telbivudine (LdT), adefovir, or other emerging anti HBV agents such as HBV RNA replication inhibitor, HBsAg secretion inhibitors, HBV capsid inhibitors, antisense oligomer, siRNA, HBV therapeutic vaccine, HBV prophylactic vaccine, HBV antibody therapy (monoclonal or polyclonal) and TLR 2, 3, 7, 8 and 9 agonists for the treatment or prophylaxis of HBV.
EXAMPLES
The invention will be more fully understood by reference to the following examples. They should not, however, be construed as limiting the scope of the invention.
Abbreviations used herein are as follows:
microliter
μιη: micrometer
μΜ: micromoles per liter
Ar: argon
DIPEA: N,N-diisopropylethylamine
DME: 1 ,2-dimethoxyethane
DMF: dimethylformamide
OMSO-d6: deuterated dimethylsulfoxide
EtOAc: ethyl acetate
h or hr: hour
hrs: hours
IC50: the half maximal inhibitory concentration
HCMV: human cytomegalovirus
HIV: human immunodeficiency
HSV: herpes simplex virus
HPV: human papillomavirus
HPLC: high performance liquid chromatography
LC/MS: Liquid chromatography/mass spectrometry
m-CPBA: m-chloroperoxybenzoic acid
MeOH: methanol
METHANOL-d4 perdeuteromethanol
M: molarity
mg: milligram
MHz: megahertz
min: minute
mL: milliliter
mM: millimoles per liter
mm: millimeter
mmol: millimole
MS (ESI): mass spectroscopy (electron spray ionization)
NMR: nuclear magnetic resonance
N2: nitrogen
rt: room temperature
Pd/C: palladium on activated carbon
Pd(PPh3)4: tetrakis(triphenylphosphine)palladium
Pd(dppf)Cl2: [l,l'-bis(diphenylphosphino)ferrocene]dichloropalladium(II)
PE or Pet: petroleum ether
prep-HPLC: preparative high performance liquid chromatography
TFA: trifluoro acetic acid
TLC: thin layer chromatography
δ: chemical shift
Xantphos: 9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene
Pd2(dba)3 : tris(dibenzylideneacetone)dipalladium(0)
i-BuONa: sodium i-butoxide GENERAL EXPERIMENTAL CONDITIONS
Intermediates and final compounds were purified by flash chromatography using one of the following instruments: i) Biotage SPl system and the Quad 12/25 Cartridge module, ii) ISCO combi-flash chromatography instrument. Silica gel Brand and pore size: i) KP-SIL 60 A, particle size: 40-60 μ m; ii) CAS registry NO: Silica Gel: 63231-67-4, particle size: 47-60 micron silica gel: iii) ZCX from Qingdao Haiyang Chemical Co.. Ltd. pore: 200-300 or 300-400.
Intermediates and final compounds were purified by preparative 11 PLC on reversed phase column using X Bridge™ Perp Ci8 ( 5 μιη. OBD™ 30 x 100 mm) column or SunFire™ Perp Ci8 (5 μιη, OBD™ 30 x 100 mm) column.
LC/MS spectra were obtained using an Acquity Ultra Performance LC - 3100 Mass Detector or Acquity Ultra Performance LC - SQ Detector. Standard LC/MS conditions were as follows (running time 3 minutes):
Acidic condition: A: 0.1% formic acid in H2O; B: 0.1% formic acid in acetonitrile;
Basic condition: A: 0.05% ΝΗ3·Η2Ο in H2O; B: acetonitrile;
Neutral condition: A: I LO: B: acetonitrile.
Mass spectra (MS): generally only ions which indicate the parent mass are reported, and unless otherwise stated the mass ion quoted is the positive mass ion (M+H)+.
The microwave assisted reactions were carried out in a Biotage Initiator Sixty or CEM Discover.
NMR Spectra were obtained using Bruker Avance 400MHz.
All reactions involving air-sensitive reagents were performed under an argon atmosphere.
Reagents were used as received from commercial suppliers without further purification unless otherwise noted.
PREPARATIVE EXAMPLES
Example 1: 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-phenyl-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
o o

Step 1: Preparation of 4-bromo-l-methoxy-2-(3-methoxypropoxy)benzene

To a solution of 5-bromo-2-methoxy-phenol (10.0 g, 49.5 mmol) in MeCN (100 mL) was added CS2CO3 (48.1 g, 148 mmol) and l-bromo-3-methoxy-propane (11.3 g, 74 mmol). The mixture was refluxed for 16 hrs, and then filtered. The filtrate was concentrated under reduced pressure, and the residue was purified by trituration with PE to give 4-bromo-l-methoxy-2-(3- methoxypropoxy)benzene (10.5 g) as a white solid.
Step 2: Preparation of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-phenyl- ethanone

A mixture of 4-bromo-l-methoxy-2-(3-methoxypropoxy)benzene (10.0 g, 36.4 mmol), 1- phenylethanone (5.7 g, 47 mmol), i-BuONa (5.24 g, 55 mmol), Xantphos (840 mg, 1.5 mmol) and Pd2(dba)3 (660 mg, 0.73 mmol) in THF (100 mL) was heated with stirring at 80 °C for 5 hrs under nitrogen atmosphere. The mixture was cooled to rt and filtered. The filtrate was
concentrated under reduced pressure. The residue was purified by the flash column
chromatography to afford 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-phenyl-ethanone (9.5 g) as a yellow solid.
Step 3: Preparation of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-phenyl- ethanamine

To a solution of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]- l-phenyl-ethanone (9.5 g, 30.2 mmol) in MeOH (100 mL) was added NH4OAc (17.2 g, 223 mmol) at 18 °C. After being stirred for 1 hr, the resulting mixture was cooled to 0 °C. Then to the cooled mixture was added NaB¾CN (4 g, 63.6 mmol) at 0 °C. The resulting mixture was then heated with stirring at 50 °C for 12 hrs. The resulting reaction mixture was concentrated under reduced pressure. The residue was diluted with H20 (200 mL) and extracted with DCM (300 mL). The organic layer was washed with brine (200 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure to afford 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]- l-phenyl-ethanamine (13.4 g) as a yellow oil, which was used directly in the next step without further purification.
Step 4: Preparation of N-[2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-phenyl- ethyl] formamide

To a solution of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]- l-phenyl-ethanamine (13.0 g, 41.3 mmol) in dioxane (150 mL) was added formic acid (13.3 g, 289 mmol). The mixture was heated with stirring at 120 °C for 12 hrs. After being cooled to rt, the resulting mixture was concentrated under reduced pressure. The residue was diluted with EtOAc (300 mL). The organic mixture was washed with H20 (200 mL) and brine (200 mL, 2 times), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to give N-[2-[4-methoxy-3-(3-methoxypropoxy)phenyl]- l-phenyl- ethyl] formamide (2.2 g, yield: 16%) as a yellow solid.
Step 5: Preparation of 7-methoxy-6-(3-methoxypropoxy)-3-phenyl-3,4- dihydroisoquinoline

To a solution of N-[2-[4-methoxy-3-(3-methoxypropoxy)phenyi]- l-phenyl-
ethyl]formamide (2.0 g, 5.8 mmol) in DCM (20 mL) was added P0C13 (2.29 g, 14.9 mmol). After being heated with stirring at 40 °C for 12 hrs, the resulting mixture was basified by ammonia water and extracted with DCM (200 mL). The organic layer was washed with brine, dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to give 7-methoxy-6-(3-methoxypropoxy)-3-phenyl-3,4- dihydroisoquinoline (500 mg) as a green solid.
Step 6: Preparation of ethyl 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-phenyl- l,6,7,llb-tetrahydrobenzo[a] uinolizine-3-carboxylate

A solution of 7-methoxy-6-(3-methoxypropoxy)-3-phenyl-3,4-dihydroisoquinoline (500 mg, 1.54 mmol) and ethyl 2-(ethoxymethylene)-3-oxo-butanoate (688 mg, 3.69 mmol) in EtOH (5 mL) was heated with stirring at 100 °C for 48 hrs. After being cooled to rt, the resulting mixture was concentrated under reduced pressure to give crude ethyl 10-methoxy-9-(3- methoxypropoxy)-2-oxo-6-phenyl- 1,6,7, llb-tetrahydrobenzo[a]quinolizine-3-carboxylate, which was used directly in the next step without further purification.
Step 7: Preparation of ethyl 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-phenyl-6,7- dihydrobenzo[a]quinolizin -3-carboxylate

To a solution of ethyl 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-phenyl-l,6,7,llb- tetrahydrobenzo[a]quinolizine-3-carboxylate (916 mg, 1.97 mmol) in DME (10 mL) was added /7-chloranil (486 mg, 1.97 mmol). The resulting mixture was heated with stirring at 70 °C for 3 hrs under nitrogen atmosphere. Then the mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was dissolved in DCM. The resulting solution was washed with NaHC03 aqueous solution and brine, dried over anhydrous Na2S04 and concentrated under reduced pressure to afford crude ethyl 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-phenyl-6,7-
dihydrobenzo[a]quinolizine-3-carboxylate (1.2 g) as a yellow solid, which was used directly in the next step without further purification.
Step 8: Preparation of 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-phenyl-6,7- dihydrobenzo[a] quinolizine-3-carbox lic acid

To a solution of ethyl 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-phenyl-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (1.02 g, 2.2 mmol) in MeOH (10 mL) was added 2 M NaOH aqueous solution (2.2 mL). The mixture was stirred at 18 °C for 16 hrs, and then concentrated under reduced pressure. The residue was acidified with 2 M hydrochloric acid to pH=3. Then the resulting aqueous mixture was extracted with DCM. The organic layer was dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by prep-HPLC to afford 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-phenyl-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid (5 mg) as a pale yellow solid. 1H NMR (400 MHz, DMSO-<ft5): δ 1.87 - 1.98 (m, 2 H) 3.21 (s, 3 H) 3.42 (t, 3 H) 3.66 (dd, 1 H) 3.85 (s, 3 H) 4.00 (t, 2 H) 6.08 (br. s., 1 H) 6.93 (s, 1 H) 6.98 (d, 2 H) 7.19 - 7.33 (m, 3 H) 7.51 (s, 1 H) 7.58 (s, 1 H) 8.82 (s, 1 H). MS obsd. (ESI+) [(M+H)+]: 435.
Example 2: 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-(3-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carbox lic acid

Step 1: Preparation of 2-[4-methoxy-3-(3-methoxypropoxy)phi
thienyl)ethanone

A mixture of 4-bromo- l-methoxy-2-(3-methoxypropoxy)benzene (16.5 g, 60 mmol), l-(3- thienyl)ethanone (9.84 g, 78 mmol), i-BuONa (8.64 g, 90 mmol), Xantphos (1.39 g, 2.4 mmol) and Pd2(dba)3 (1.1 g, 1.2 mmol) in THF (200 mL) was heated at 100 °C for 1 hr. After being cooled to rt, the mixture was filtered. The filtrate was concentrated under reduced pressure and the residue was purified by the flash column chromatography to afford 2-[4-methoxy-3-(3- methoxypropoxy)phenyl]- l-(3-thienyl)ethanone (6.8 g) as a brown oil.
Step 2: Preparation of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(3- thienyl)ethanamine

To a solution of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]- l-(3-thienyl)ethanone (6.8 g, 21.2 mmol) in MeOH (80 mL) was added NH4OAc (16.4 g, 212 mmol) at rt. After being stirred for 1 hr, the resulting mixture was cooled to 0 °C. Then to the cooled mixture was added
NaBH3CN (2.67 g, 42.5 mmol) at 0 °C. Then the mixture was heated and stirred at 50 °C for 12 hrs. The mixture was concentrated under reduced pressure. The residue was diluted with H20 (200 mL). The aqueous mixture was extracted with DCM (300 mL). The organic layer was washed with brine (200 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure to afford crude 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]- l-(3-thienyl)ethanamine (6.82 g) as a yellow oil, which was used directly in the next step without further purification.
Step 3: Preparation of N-[2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(3- thienyl)ethyl] formamide

To a solution of crude 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]- l-(3- thienyl)ethanamine (6.82 g, 21.2 mmol) in dioxane (80 mL) was added formic acid (9.77 g, 212 mmol). Then the mixture was heated at 120 °C for 12 hrs. After being cooled to rt, the mixture
was concentrated under reduced pressure. The residue was diluted with EtOAc (300 mL). The organic mixture was washed with H20 (200 mL) and brine (200 mL, 2 times), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to give N-[2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(3- thienyl) ethyl] formamide (5.0 g) as a yellow solid.
Step 4: Preparation of 7-methoxy-6-(3-methoxypropoxy)-3-(3-thienyl)-3,4- dihydroisoquinoline

To a solution of N-[2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(3- thienyl)ethyl] formamide (5.0 g, 14.3 mmol) in DCM (50 mL) was added POCl3 (4.51 g, 29.3 mmol). The mixture was heated at 40 °C for 12 hrs and then basified with ammonia water. The resulting mixture was extracted with DCM (200 mL). The organic layer was dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to give 7-methoxy-6-(3-methoxypropoxy)-3-(3-thienyl)-3,4- dihydroisoquinoline (3.0 g) as a yellow solid.
Step 5: Preparation of ethyl 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-(3-thienyl)- l,6,7,llb-tetrahydrobenzo a]quinolizine-3-carboxylate

A mixture of 7-methoxy-6-(3-methoxypropoxy)-3-(3-thienyl)-3,4-dihydroisoquinoline (610 mg, 1.84 mmol) and ethyl 2-(ethoxymethylene)-3-oxo-butanoate (1.03 g, 5.52 mmol) in EtOH (10 mL) was heated at 100 °C for 48 hrs. After being cooled to rt, the mixture was concentrated under reduced pressure to give crude ethyl 10-methoxy-9-(3-methoxypropoxy)-2- oxo-6-(3-thienyl)-l,6,7,llb-tetrahydrobenzo[a]quinolizine-3-carboxylate, which was used directly in the next step without further purification.
Step 6: Preparation of ethyl 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-(3-thienyl)-
6,7-dihydrobenzo[a]quinolizine-3-carboxylate

To a solution of ethyl 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-(3-thienyl)-l,6,7,llb- tetrahydrobenzo[a]quinolizine-3-carboxylate (800 mg, 1.70 mmol) in DME (10 mL) was added /7-chloranil (375 mg, 1.53 mmol). The mixture was heated at 70 °C for 3 hrs under nitrogen atmosphere. Then the mixture was filtered. The filter cake was dried under reduced pressure to afford ethyl 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-(3-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (250 mg) as a yellow solid.
Step 7: Preparation of 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-(3-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carbox lic acid

To a solution of ethyl 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-(3-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (250 mg, 532 μιηοΐ) in EtOH (3 mL) was added 2 M NaOH aqueous solution (0.8 mL). The mixture was stirred at rt for 0.5 hr and then concentrated under reduced pressure to remove most of EtOH. The residue was acidified by 1 M hydrochloric acid and then filtered. The filter cake was purified by column chromatography to give 10- methoxy-9-(3-methoxypropoxy)-2-oxo-6-(3-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid (110 mg) as a pale yellow solid. 1H NMR (400 MHz, DMSO-J6): δ 1.97 (q, 2 H) 3.26 (s, 3 H) 3.36 - 3.45 (m, 1 H) 3.49 (t, 2 H) 3.59 (dd, 1 H) 3.87 (s, 3 H) 4.10 (t, 2 H) 5.99 (br. s., 1 H) 6.83 (d, 1 H) 6.99 (s, 1 H) 7.11 (br. s., 1 H) 7.26 - 7.55 (m, 3 H) 8.74 (br. s., 1 H). MS obsd. (ESI+) [(M+H)+]: 442.
Example 3: 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid

Step 1: Preparation of 2-[4-methoxy-3-(3-methoxypropoxy)phi
thienyl)ethanone
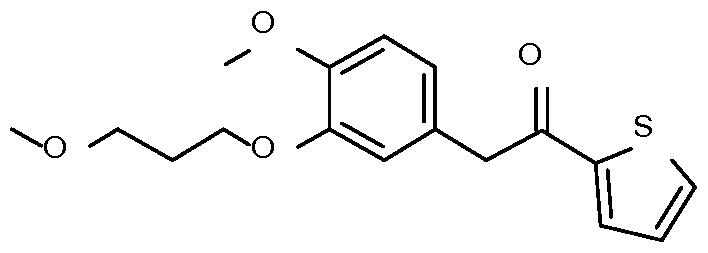
A mixture of 4-bromo-l-methoxy-2-(3-methoxypropoxy)benzene (5.0 g, 18.2 mmol), l-(2- thienyl)ethanone (3.43 g, 27.2 mmol), Xantphos (440 mg), Pd2(dba)3 (340 mg) and i-BuONa (2.61 g, 27.2 mmol) in THF (50 mL) was heated at 70 °C for 5 hrs. After being cooled to rt, the mixture was filtered. The filtrate was concentrated under reduced pressure and the residue was purified by column chromatography to give 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(2- thienyl)ethanone (2.7 g) as a yellow solid.
Step 2: Preparation of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(2- thienyl)ethanamine

To a solution of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(2-thienyl)ethanone (2.7 g,
8.43 mmol) in MeOH (40 mL) was added NH4OAc (6.50 g, 84.3 mmol). After the mixture was stirred for 20 min, to the resulting mixture was added NaBH3CN (1.06 g, 16.8 mmol). The resulting mixture was heated and stirred at 40 °C for 24 hrs and then concentrated under reduced pressure. The residue was diluted with DCM. The organic mixture was washed with water and brine, dried over anhydrous Na2S04 and concentrated under reduced pressure to give crude 2- [4- methoxy-3-(3-methoxypropoxy)phenyl]-l-(2-thienyl)ethanamine (2.2 g) as a yellow oil, which was used directly in the next step without further purification.
Step 3: Preparation of N-[2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(2- thienyl)ethyl] formamide
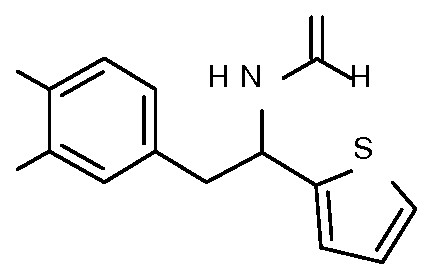
To a solution of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(2-thienyl)ethanamine (crude 2.2 g) in dioxane (30 mL) was added formic acid (1.54 g, 33.6 mmol). The mixture was refluxed for 24 hrs. After being cooled to rt, the mixture was concentrated under reduced pressure. The residue was diluted with DCM. The organic mixture was washed with water and brine, dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to give N-[2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l- (2-thienyl)ethyl]formamide (800 mg) as a gray solid.
Step 4: Preparation of 7-methoxy-6-(3-methoxypropoxy)-3-(2-thienyl)-3,4- dihydroisoquinoline

To a solution of N-[2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(2- thienyl)ethyl]formamide (750 mg, 2.15 mmol) in DCM (10 mL) was added POCl3 (493 mg, 3.22 mmol). The mixture was heated and stirred at 40 °C for 2 hrs, and then poured into ammonia water. The resulting mixture was extracted with DCM. The organic layer was washed with brine, dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to give 7-methoxy-6-(3-methoxypropoxy)-3-(2-thienyl)-3,4- dihydroisoquinoline (400 mg) as a gray solid.
Step 5: Preparation of ethyl 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-(2-thienyl)- l,6,7,llb-tetrahydrobenzo inolizine-3-carboxylate

A mixture of 7-methoxy-6-(3-methoxypropoxy)-3-(2-thienyl)-3,4-dihydroisoquinoline (50 mg, 0.15 mmol) and ethyl 2-(ethoxymethylene)-3-oxo-butanoate (84 mg, 0.45 mmol) in EtOH (1
mL) was refluxed for 24 hrs. After being cooled to rt, the mixture was concentrated under reduced pressure to give crude ethyl 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-(2-thienyl)- l,6,7,llb-tetrahydrobenzo[a]quinolizine-3-carboxylate (70 mg), which was used directly in the next step without further purification.
Step 6: Preparation of ethyl 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-(2-thienyl)- 6,7-dihydrobenzo[a]quinolizin -3-carboxylate

To a solution of ethyl 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-(2-thienyl)-l,6,7,llb- tetrahydrobenzo[a]quinolizine-3-carboxylate (crude 70 mg, 0.15 mmol) in DME (1 mL) was added /?-chloranil (37 mg, 0.15 mmol). The resulting mixture was refluxed for 3 hrs, and then filtered. The filtrate was concentrated under reduced pressure. The residue was diluted with DCM. The organic mixture was washed with NaHC03 aqueous solution and brine, dried over anhydrous Na2S04 and concentrated under reduced pressure to give crude ethyl 10-methoxy-9- (3-methoxypropoxy)-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate (60 mg), which was used directly in the next step without further purification.
Step 7: Preparation of 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine- -carboxylic acid

To a mixture of ethyl 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (crude 60 mg, 0.15 mmol) in MeOH (1 mL) was added 2 M NaOH aqueous solution (0.15 mL, 0.3 mmol). After being stirred at rt for 16 hrs, the resulting mixture was concentrated under reduced pressure to remove most of MeOH. The residue was acidified with 2 M hydrochloric acid to pH=3, and then extracted with DCM. The organic layer was dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by prep-HPLC to give 10-methoxy-9-(3-methoxypropoxy)-2-oxo-6-(2-
thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid (2.4 mg) as a gray solid. 1H NMR (400 MHz, DMSO-<¾5): δ: 8.99 (s, 1H), 7.50-7.53 (m, 2H), 7.33-7.38 (m, 1H), 7.08 (s, 1H), 7.03 (s, 1H), 6.86-7.01 (m, 1H), 6.35 (s, 1H), 4.08 (t, 2H), 3.87 (s, 3H), 3.66-3.70 (m, 1H), 3.46 (t, 2H), 3.43-3.45 (m, 2H), 3.24 (s, 3H), 1.94-2.01 (m, 2H). MS obsd. (ESI+) [(M+H)+]: 442.
Example 4: 6-(4-hydroxyphenyl)-10-methoxy-9-(3-methoxypropoxy)-2- dihydrobenzo[a] quinolizine-3-carbox lic acid

Step 1: Preparation of l-(4-benzyloxyphenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]ethanone

To a mixture of 4-bromo-l-methoxy-2-(3-methoxypropoxy)benzene (15 g, 54.5 mmol) in THF (150 mL) was added l-(4-benzyloxyphenyl)ethanone (16 g, 70.9 mmol), i-BuONa (7.86 g, 81.8 mmol), Pd2(dba)3 (1 g, 1.09 mmol) and Xantphos (1.26 g, 2.18 mmol). The mixture was heated at 50 °C for 4 hrs. After being cooled to rt, the mixture was concentrated under reduced pressure, and the residue was dissolved in DCM. The organic solution was washed with H20 and brine, dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to give l-(4-benzyloxyphenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]ethanone (17 g) as a yellow solid.
Step 2: Preparation of l-(4-benzyloxyphenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]ethanamine

To a solution of l-(4-benzyloxyphenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]ethanone (17 g, 40.4 mmol) in MeOH (170 mL) was added NH4OAc (31.2 g, 404 mmol) at 70 °C. After being heated for 1 hr, the resulting mixture was cooled to 0 °C. Then to the resulting mixture was added NaBH3CN (5.08 g, 80.9 mmol) at 0 °C. The resulting mixture was heated at 80 °C for 12 hrs and then concentrated under reduced pressure to remove most of MeOH. The residue was diluted with H20 (100 mL) and extracted with DCM (500 mL). The organic layer was washed with brine (100 mL), dried over anhydrous Na2S04 and
concentrated under reduced pressure to afford crude l-(4-benzyloxyphenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]ethanamine (24 g) as a yellow oil, which was used directly in the next step without further purification.
Step 3: Preparation of N-[l-(4-benzyloxyphenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phen l]ethyl]formamide

To a mixture of l-(4-benzyloxyphenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]ethanamine (24.0 g, 56.9 mmol) in dioxane (250 mL) was added formic acid (13.1 g, 285 mmol). After being heated at 120 °C for 12 hrs, the mixture was concentrated under reduced pressure to remove most of dioxane. The residue was partitioned between H20 (200 mL) and DCM (600 mL). The organic layer was separated, washed with brine (200 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to give N-[l-(4-benzyloxyphenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl] ethyl] formamide (13.0 g) as a light yellow solid.
Step 4: Preparation of 3-(4-benzyloxyphenyl)-7-methoxy-6-(3-methoxypropoxy)-3,4- dihydroisoquinoline

To a mixture of N-[l-(4-benzyloxyphenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl] ethyl] formamide (11.0 g, 24.5 mmol) in dry DCM (120 mL) was added POCI3 (5.75 g, 37.5 mmol) drop-wise at 0 °C. Then the mixture was refluxed for 12 hrs. After being cooled to rt, the mixture was poured into a solution of ammonia water (30 mL) in H20 (20 mL). Then the mixture was extracted with DCM (300 mL). The organic layer was washed with brine (150 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to give 3-(4-benzyloxyphenyl)-7-methoxy-6-(3- methoxypropoxy)-3,4-dihydroisoquinoline (8.4 g) as a yellow solid.
Step 5: Preparation of ethyl 6-(4-benzyloxyphenyl)-10-methoxy-9-(3- methoxypropoxy)-2-oxo-l 6,7,llb-tetrahydrobenzo[a]quinolizine-3-carboxylate

A solution of 3-(4-benzyloxyphenyl)-7-methoxy-6-(3-methoxypropoxy)-3,4- dihydroisoquinoline (4.0 g, 9.27 mmol) and ethyl 2-(ethoxymethylene)-3-oxo-butanoate (5.18 g, 27.8 mmol) in EtOH (50 mL) was heated in a 100 °C oil bath for 12 hrs. After being cooled to rt, the mixture was concentrated under reduced pressure to afford crude ethyl 6- (4- benzyloxyphenyl)- 10-methoxy-9-(3-methoxypropoxy)-2-oxo- 1,6,7, 1 lb- tetrahydrobenzo[a]quinolizine-3-carboxylate, which was used directly in the next step without further purification.
Step 6: Preparation of ethyl 6-(4-benzyloxyphenyl)-10-methoxy-9-(3- methoxypropoxy)-2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylate
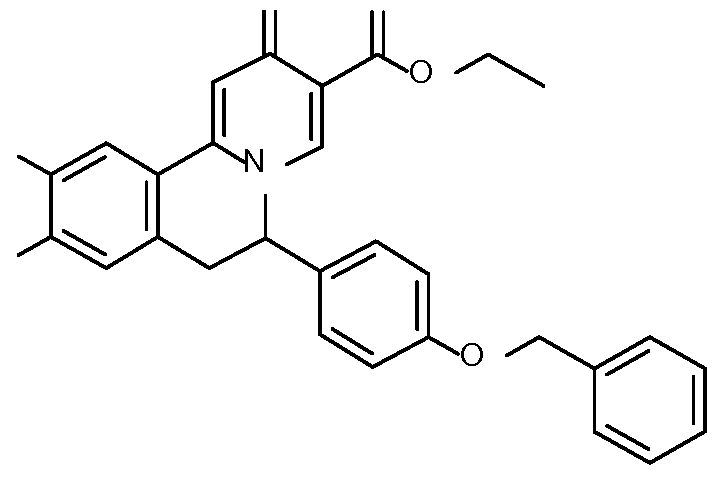
A mixture of ethyl 6-(4-benzyloxyphenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo- l,6,7,llb-tetrahydrobenzo[a]quinolizine-3-carboxylate (5.3 g, 9.27 mmol) and /?-chloranil (1.6 g, 6.49 mmol) in DME (60 mL) was heated at 80 °C for 3 hrs. After being cooled to rt, the mixture was partitioned between H20 (150 mL) and DCM (300 mL). The organic layer was separated, washed with brine (150 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to afford ethyl 6-(4- benzyloxyphenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo-6,7-dihydrobenzo[a]quinolizine-3- carboxylate (4 g) as a black solid.
Step 7: Preparation of ethyl 6-(4-hydroxyphenyl)-10-methoxy-9-(3-methoxypropoxy)-
2-oxo-6,7-dihydrobenzo[a] uinolizine-3-carboxylate

To a solution of ethyl 6-(4-benzyloxyphenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo- 6,7-dihydrobenzo[a]quinolizine-3-carboxylate (700 mg, 1.23 mmol) in EtOH (10 mL) was added Pd/C (50 mg). Then the mixture was heated to 30 °C and stirred at 30 °C under hydrogen atmosphere (30 psi) for 12 hrs. After being cooled to rt, the mixture was concentrated under reduced pressure to give crude ethyl 6-(4-hydroxyphenyl)-10-methoxy-9-(3-methoxypropoxy)-2- oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylate (650 mg) as a black solid, which was used directly in the next step without purification.
Step 8: Preparation of 6-(4-hydroxyphenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo-
6,7-dihydrobenzo[a] quinolizine-3-carboxylic acid

To a solution of ethyl 6-(4-hydroxyphenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (200 mg, 417 μιηοΐ) in MeOH (5 mL) was added 2 M NaOH aqueous solution (0.625 mL, 1.25 mmol). After being stirred at 25 °C for 12 hrs, the resulting mixture was concentrated. The residue was diluted with H20 (20 mL), acidified with 1 M hydrochloric acid to pH=3, and extracted with DCM (100 mL). The organic layer was washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by prep-HPLC to give 6-(4-hydroxyphenyl)-10-methoxy-9-(3- methoxypropoxy)-2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid (8.2 mg) as a white solid. 1H NMR (400 MHz, METHANOL- d4): δ: 1.94 - 2.10 (m, 2 H), 3.07 - 3.30 (m, 3 H), 3.33 - 3.37 (m, 1 H), 3.42 - 3.53 (m, 1 H), 3.55 (t, 2 H), 3.88 (br. s., 3 H), 4.08 (br. s., 2 H), 5.51 (br. s., 1 H), 6.64 (br. s., 2 H), 6.74 - 7.15 (m, 4 H), 7.32 (br. s., 1 H), 8.24 (br. s., 1 H). MS obsd. (ESI+) [(M+H)+]: 452.
Example 5: 10-methoxy-6-(4-methoxyphenyl)-9-(3-methoxypropoxy)-2- dihydrobenzo[a] quinolizine-3-carboxylic acid

Step 1: Preparation of ethyl 10-methoxy-6-(4-methoxyphenyl)-9-(3-methoxypropoxy)- 2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylate
o o

Step 2: Preparation of 10-methoxy-6-(4-methoxyphenyl)-9-(3-methoxypropoxy)-2- oxo-6,7-dihydrobenzo[a]quinolizine-3-carbox lic acid

To a solution of ethyl 10-methoxy-6-(4-methoxyphenyl)-9-(3-methoxypropoxy)-2-oxo-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (350 mg, 790 μιηοΐ) in MeOH (5 mL) was added 2 M NaOH aqueous solution (1.19 mL, 2.37 mmol). After being stirred at 25 °C for 12 hrs, the resulting reaction mixture was diluted with H20 (20 mL), acidified with 1 M hydrochloric acid to pH=3 and extracted with DCM (100 mL). The organic layer was washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by prep-HPLC to afford 10-methoxy-6-(4-methoxyphenyl)-9-(3-methoxypropoxy)-2- oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid (27 mg) as a yellow solid. 1H NMR (400 MHz, CDC13): δ: 2.12 (q, 2 H), 3.28 (dd, 1 H), 3.34 (s, 3 H), 3.55 (t, 3 H), 3.60 (d, 1 H), 3.77 (s, 3 H) 3.94 (s, 3 H), 4.13 (t, 2 H), 5.39 (s, 1 H), 6.69 (s, 1 H), 6.79 - 6.86 (m, 2 H), 6.88 - 6.95 (m, 2 H), 7.15 (s, 1 H) ,7.20 (s, 1 H), 8.45 (s, 1 H). MS obsd. (ESI+) [(M+H)+]: 466.
Example 6: 6-(4-benzyloxyphenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid

To a solution of ethyl 6-(4-benzyloxyphenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo- 6,7-dihydrobenzo[a]quinolizine-3-carboxylate (200 mg, 351 μηιοΐ, from step 6 of Example 4) in MeOH (5 mL) was added 2 M NaOH aqueous solution (0.526 mL, 1.05 mmol). After being stirred at 25 °C for 12 hrs, the resulting mixture was diluted with H20 (20 mL), acidified with 1 M hydrochloric acid to pH=3 and extracted with DCM (100 mL). The organic layer was washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by prep-HPLC to afford 6-(4-benzyloxyphenyl)-10-methoxy-9-(3- methoxypropoxy)-2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid (31 mg) as a white solid. . 1H NMR (400 MHz, CDC13): δ: 2.12 (q, 2 H), 3.28 (dd, 1 H), 3.34 (s, 3 H), 3.55 (t, 3 H), 3.93 (s, 3 H), 4.13 (t, 2 H), 5.02 (s, 2 H), 5.40 (br. s., 1 H), 6.69 (s, 1 H), 6.82 - 6.96 (m, 4 H), 7.17 (d, 2 H), 7.31 - 7.45 (m, 5 H), 8.46 (s, 1 H). MS obsd. (ESI+) [(M+H)+]: 542.
Example 7: 10-methoxy-9-(3-methoxypropoxy)-6-(4-methyl-2-thienyl)-2-oxo-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid
o o

Step 1: Preparation of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(4-methyl-2- thienyl)ethanone
O
 A mixture of 4-bromo- l-methoxy-2-(3-methoxypropoxy)benzene (5.0 g, 18.2 mmol), l-(4- methyl-2-thienyl)ethanone (3.82 g, 27.2 mmol), i-BuONa (2.80 g, 29.1 mmol), Pd2(dba)3 (350 mg) and Xantphos (440 mg) in THF (60 mL) was heated at 80 °C for 3 hrs. After being cooled to rt, the mixture was partitioned between DCM (200 mL) and H20 (50 mL). The organic layer was separated, washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to give 2-[4-methoxy-3- (3-methoxypropoxy)phenyl]- l-(4-methyl-2-thienyl)ethanone (3.7 g) as a yellow solid.
A mixture of 4-bromo- l-methoxy-2-(3-methoxypropoxy)benzene (5.0 g, 18.2 mmol), l-(4- methyl-2-thienyl)ethanone (3.82 g, 27.2 mmol), i-BuONa (2.80 g, 29.1 mmol), Pd2(dba)3 (350 mg) and Xantphos (440 mg) in THF (60 mL) was heated at 80 °C for 3 hrs. After being cooled to rt, the mixture was partitioned between DCM (200 mL) and H20 (50 mL). The organic layer was separated, washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to give 2-[4-methoxy-3- (3-methoxypropoxy)phenyl]- l-(4-methyl-2-thienyl)ethanone (3.7 g) as a yellow solid.
Step 2: Preparation of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(4-methyl-2- thienyl)ethanamine

To a mixture of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]- l-(4-methyl-2- thienyl)ethanone (3.7 g, 11.1 mmol) in MeOH (50 mL) was added NH4OAc (8.53 g, 111 mmol). The mixture was heated at 40 °C for 1 hr. Then to the resulting mixture was added NaBH3CN (1.39 g, 22.2 mmol). The resulting mixture was heated at 70 °C for 48 hrs, and then concentrated under reduced pressure. The residue was diluted with DCM (200 mL). The organic mixture was separated, washed with water (100 mL) and brine (100 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to give crude 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]- l-(4-methyl-2-thienyl)ethanamine (2.5 g) as a yellow oil.
Step 3: Preparation of N-[2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(4-methyl-2- thienyl)ethyl] formamide

To a mixture of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]- l-(4-methyl-2- thienyl)ethanamine (2.5 g, 7.46 mmol) in dioxane (30 mL) was added formic acid (1.72 g, 37.3 mmol). The mixture was heated at reflux for 24 hrs. After being cooled to rt, the mixture was concentrated under reduced pressure and the residue was diluted with DCM (100 mL). Then the
organic mixture was separated, washed with water (50 mL) and brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to give N-[2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(4-methyl-2- thienyl) ethyl] formamide (1.3 g) as a yellow oil.
Step 4: Preparation of 7-methoxy-6-(3-methoxypropoxy)-3-(4-methyl-2-thienyl)-3,4- dihydroisoquinoline

To a mixture of N-[2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(4-methyl-2- thienyl)ethyl] formamide (700 mg, 1.93 mmol) in DCM (10 mL) was added POCl3 (442 mg, 2.89 mmol). The mixture was heated under reflux for 2 hrs. After being cooled to rt, the mixture was partitioned between ammonia water (20 mL) and DCM (100 mL). The organic layer was separated, washed with water (30 mL) and brine (30 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to give 7-methoxy-6-(3-methoxypropoxy)-3-(4-methyl-2-thienyl)-3,4-dihydroisoquinoline (390 mg) as a white solid.
Step 5: Preparation of ethyl 10-methoxy-9-(3-methoxypropoxy)-6-(4-methyl-2- thienyl)-2-oxo-l,6,7,llb-tetrahydrobenzo[a]quinolizine-3-carboxylate

A mixture of 7-methoxy-6-(3-methoxypropoxy)-3-(4-methyl-2-thienyl)-3,4- dihydroisoquinoline (390 mg, 1.13 mmol) and ethyl 2-(ethoxymethylene)-3-oxo-butanoate (630 mg, 3.39 mmol) in EtOH (7 mL) was heated under reflux for 24 hrs. After being cooled to rt, the mixture was concentrated under reduced pressure to give crude ethyl 10-methoxy-9-(3- methoxypropoxy)-6-(4-methyl-2-thienyl)-2-oxo- 1,6,7, llb-tetrahydrobenzo[a]quinolizine-3- carboxylate (1.0 g), which was used directly in the next step without further purification.
Step 6: Preparation of ethyl 10-methoxy-9-(3-methoxypropoxy)-6-(4-methyl-2-
thienyl)-2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylate

A mixture of ethyl 10-methoxy-9-(3-methoxypropoxy)-6-(4-methyl-2-thienyl)-2-oxo- l,6,7,llb-tetrahydrobenzo[a]quinolizine-3-carboxylate (crude 1 g) and /?-chloranil (277 mg, 1.13 mmol) in DME (10 mL) was heated under reflux for 3 hrs. Then the mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was diluted with DCM. Then the organic mixture was washed with NaHC03 aqueous solution and brine, dried over anhydrous Na2S04 and concentrated under reduced pressure to give crude ethyl 10-methoxy-9-(3- methoxypropoxy)-6-(4-methyl-2-thienyl)-2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylate (1 g), which was used directly in the next step without further purification.
Step 7: Preparation of 10-methoxy-9-(3-methoxypropoxy)-6-(4-methyl-2-thienyl)-2- oxo-6,7-dihydrobenzo[a] quinolizine-3-carboxylic acid
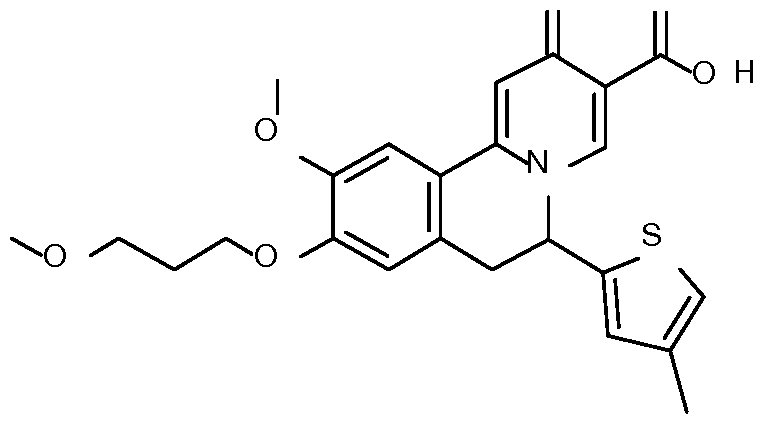
To a mixture of crude ethyl 10-methoxy-9-(3-methoxypropoxy)-6-(4-methyl-2-thienyl)-2- oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylate (1 g) in MeOH (10 mL) was added 2 M
NaOH aqueous solution (1.1 mL). The mixture was stirred at 15 °C for 16 hrs. The mixture was concentrated under reduced pressure. The residue was acidified with 1 M hydrochloric acid to pH=3. The resulting aqueous mixture was extracted with DCM. The organic layer was dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was treated with MeOH (5 mL), and then filtered. The filter cake was purified by prep-HPLC to give 10- methoxy-9-(3-methoxypropoxy)-6-(4-methyl-2-thienyl)-2-oxo-6,7-dihydrobenzo[a]quinolizine- 3-carboxylic acid (64 mg) as a white solid. 1H NMR (400 MHz, DMSO-J6): δ 8.93 (s, 1H), 7.50-7.53 (m, 2H), 7.06 (s, 1H), 6.91 (s, 1H), 6.82 (s, 1H), 6.26 (s, 1H), 4.07 (t, / = 6.4 Hz, 2H),
3.87 (s, 3H), 3.66-3.70 (m, 1H), 3.46 (t, / = 6.4 Hz, 2H), 3.30-3.31 (m, 1H), 3.24 (s, 3H), 2.07 (s, 3H), 1.94-2.01 (m, 2H). MS obsd. (ESI+) [(M+H)+]: 456.
Example 8: 6-(3-chlorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2- dihydrobenzo[a] quinolizine-3-carbox lic acid

Step 1: Preparation of l-(3-chlorophenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]ethanone

A mixture of 4-bromo-l-methoxy-2-(3-methoxypropoxy)benzene (10.0 g, 36.3 mmol), 1- (3-chlorophenyl)ethanone (7.3 g, 47.2 mmol), i-BuONa (5.24 g, 54.5 mmol), Pd2(dba)3 (1.33 g, 1.45 mmol) and Xantphos (841 mg, 1.45 mmol) in THF (100 mL) was heated at 60 °C for 12 hrs under nitrogen atmosphere. After being cooled to rt, the reaction mixture was concentrated under reduced pressure. The residue was diluted with DCM (600 mL). The organic mixture was washed with water and brine, dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to give l-(3-chlorophenyl)-2-[4- methoxy-3-(3-methoxypropoxy)phenyl]ethanone (9.0 g) as a light yellow oil.
Step 2: Preparation of l-(3-chlorophenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]ethanamine

To a mixture of l-(3-chlorophenyl)-2-[4-methoxy-3-(3-methoxypropoxy)phenyl]ethanone (9.0 g, 25.8 mmol) in MeOH (100 mL) was added NH4OAc (13.9 g, 181 mmol) at 24 °C. After the mixture was stirred for 1 hr, to the mixture was added NaBH3CN (14.2 g, 226 mmol) at 0 °C .
The resulting mixture was heated at 70 °C for 12 hrs, then cooled to rt, and concentrated under reduced pressure to remove most of MeOH. The residue was partitioned between H20 (100 mL) and DCM (500 mL). The organic layer was washed with brine (100 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure to afford crude l-(3-chlorophenyl)-2-[4- methoxy-3-(3-methoxypropoxy)phenyl]ethanamine (9.0 g) as a yellow oil, which was used directly in the next step without further purification.
Step 3: Preparation of N-[l-(3-chlorophenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]ethyl]formamide

To a mixture of l-(3-chlorophenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]ethanamine (9.0 g, 33.4 mmol) in dioxane (120 mL) was added formic acid (5.92 g, 129 mmol) at 24 °C. The mixture was refluxed for 12 hrs, then cooled to rt, and concentrated under reduced pressure to remove most of dioxane. The residue was partitioned between H20 (100 mL) and DCM (500 mL). The organic layer was washed with brine (100 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to afford N-[l-(3-chlorophenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl] ethyl] formamide (2.0 g) as a light yellow solid.
Step 4: Preparation of 3-(3-chlorophenyl)-7-methoxy-6-(3-methoxypropoxy)-3,4- dihydroisoquinoline

To a mixture of N-[l-(3-chlorophenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl] ethyl] formamide (1.0 g, 2.65 mmol) in DCM (10 mL) was added POCI3 (608 mg, 3.97 mmol). The mixture was heated under reflux for 2 hrs. After being cooled to rt, the mixture was partitioned between ammonia water (20 mL) and DCM (100 mL). The organic layer was separated, washed with water (30 mL) and brine (30 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to give 3-(3-chlorophenyl)-7-methoxy-6-(3-methoxypropoxy)-3,4-
dihydroisoquinoline (600 mg) as a brown oil.
Step 5: Preparation of ethyl 6-(3-chlorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2- oxo-l,6,7,llb-tetrahydrobenzo a]quinolizine-3-carboxylate

A mixture of 3-(3-chlorophenyl)-7-methoxy-6-(3-methoxypropoxy)-3,4- dihydroisoquinoline (600 mg, 1.67 mmol) and ethyl 2-(ethoxymethylene)-3-oxo-butanoate (930 mg, 5.00 mmol) in EtOH (10 mL) was heated under reflux for 48 hrs. After being cooled to rt, the mixture was concentrated under reduced pressure to give crude ethyl 6-(3-chlorophenyl)-10- methoxy-9-(3-methoxypropoxy)-2-oxo- 1,6,7, 1 lb-tetrahydrobenzo[a]quinolizine-3-carboxylate (1.3 g), which was used directly in the next step without further purification.
Step 6: Preparation of ethyl 6-(3-chlorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2- oxo-6,7-dihydrobenzo[a]quinolizine-3-carbox late
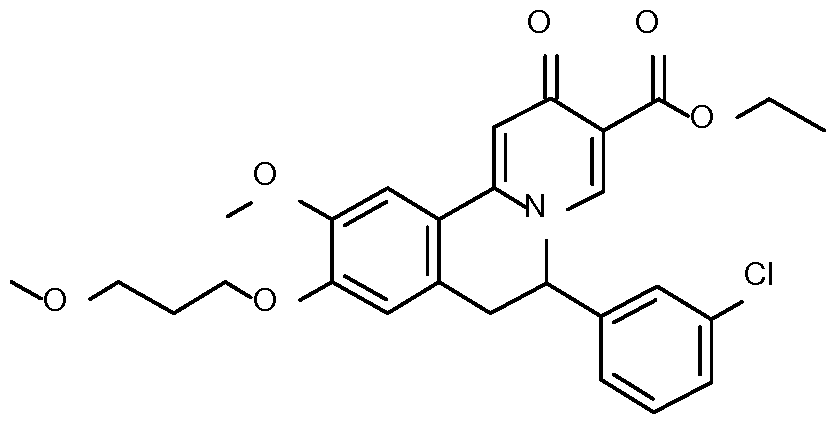
A mixture of crude ethyl 6-(3-chlorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo- l,6,7,llb-tetrahydrobenzo[a]quinolizine-3-carboxylate (1.3 g) and /?-chloranil (410 mg, 1.67 mmol) in DME (15 mL) was heated under reflux for 3 hrs. Then the mixture was filtered. The filtrate was concentrated under reduced pressure and the residue was diluted with DCM. Then the organic mixture was washed with NaHC03 aqueous solution and brine, dried over anhydrous Na2S04 and concentrated under reduced pressure to give crude ethyl 6-(3-chlorophenyl)-10- methoxy-9-(3-methoxypropoxy)-2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylate (1.4 g), which was used directly in the next step without further purification.
Step 7: Preparation of 6-(3-chlorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo- 6,7-dihydrobenzo[a] quinolizine-3-carboxylic acid

To a mixture of ethyl 6-(3-chlorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (crude 700 mg) in MeOH (7 mL) was added 2 M NaOH aqueous solution (0.8 mL). The mixture was stirred at 15 °C for 16 hrs, and then concentrated under reduced pressure. The residue was acidified with 1 M hydrochloric acid to pH=3. The resulting aqueous mixture was extracted with DCM. The organic layer was dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was treated with MeOH (5 mL) and then filtered. The filter cake was recrystallized with DMSO/MeCN to give 6- (3-chlorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid (36 mg) as a white solid. 1H NMR (400 MHz, DMSO-J6): δ 8.85 (s, 1H), 7.58 (s, 1H), 7.52 (s, 1H), 7.23-7.35 (m, 2H), 7.18 (s, 1H), 6.94 (s, 1H), 6.80 (d, 1H), 6.08 (d, 1H), 4.02 (t, 2H), 3.85 (s, 3H), 3.66-3.70 (m, 1H), 3.42-3.50 (m, 3H), 3.21 (s, 3H), 1.94-2.01 (m, 2H). MS obsd. (ESI+) [(M+H)+]: 470. Example 9: 6-(4-fluorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo-6,7- dihydrobenzo[a] quinolizine-3-carbox lic acid

Step 1: Preparation of l-(4-fluorophenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]ethanone

A mixture of 4-bromo-l-methoxy-2-(3-methoxypropoxy)benzene (8.0 g, 29.1 mmol), l-(4-
fluorophenyl)ethanone (6.02 g, 43.6 mmol), i-BuONa (4.47 g, 46.5 mmol), Pd2(dba)3 (550 mg), Xantphos (700 mg) in THF (100 mL) was heated at 80 °C for 3 hrs. After being cooled to rt, the mixture was concentrated under reduced pressure. The residue was partitioned between DCM (200 mL) and H20 (50 mL). The organic layer was washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to give l-(4-fluorophenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]ethanone (8.9 g) as a yellow solid.
Step 2: Preparation of l-(4-fluorophenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]ethanamine

To a mixture of l-(4-fluorophenyl)-2-[4-methoxy-3-(3-methoxypropoxy)phenyl]ethanone (8.9 g, 26.8 mmol) in MeOH (100 mL) was added NH4OAc (20.6 g, 268 mmol). The mixture was stirred at 30 °C for 1 hr. Then to the mixture was added NaBH3CN (3.38 g, 53.6 mmol). The resulting mixture was heated at 40 °C for 16 hrs, and then concentrated under reduced pressure. The residue was diluted with DCM (200 mL). The organic mixture was washed with water (100 mL), brine (100 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to give l-(4-fluorophenyl)-2-[4-methoxy-3- (3-methoxypropoxy)phenyl]ethanamine (6.0 g) as a yellow oil.
Step 3: Preparation of N-[l-(4-fiuorophenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]eth l]formamide

To a mixture of l-(4-fluorophenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl]ethanamine (6.0 g, 18.0 mmol) in dioxane (80 mL) was added formic acid (4.14 g, 90.0 mmol). The mixture was heated under reflux for 48 hrs. After being cooled to rt, the mixture was concentrated under reduced pressure. The residue was diluted with DCM (100 mL), and the organic mixture was washed with water (50 mL) and brine (50 mL), dried
over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to give N-[l-(4-fluorophenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl] ethyl] formamide (2.6 g) as a white solid.
Step 4: Preparation of 3-(4-fiuorophenyl)-7-methoxy-6-(3-methoxypropoxy)-3,4- dihydroisoquinoline

To a mixture of N-[l-(4-fluorophenyl)-2-[4-methoxy-3-(3- methoxypropoxy)phenyl] ethyl] formamide (1.0 g, 2.77 mmol) in DCM (10 mL) was added POCI3 (636 mg, 4.16 mmol). The mixture was heated under reflux for 2 hrs. After being cooled to rt, the mixture was partitioned between ammonia water (20 mL) and DCM (100 mL). The organic layer was washed with water (30 mL) and brine (30 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to give 3-(4-fluorophenyl)-7-methoxy-6-(3-methoxypropoxy)-3,4-dihydroisoquinoline (650 mg) as a yellow oil.
Step 5: Preparation of ethyl 6-(4-fiuorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2- oxo-l,6,7,llb-tetrahydrobenzo[a]quinolizine-3-carboxylate

A mixture of 3-(4-fluorophenyl)-7-methoxy-6-(3-methoxypropoxy)-3,4- dihydroisoquinoline (650 mg, 1.89 mmol) and ethyl 2-(ethoxymethylene)-3-oxo-butanoate (1.06 g, 5.69 mmol) in EtOH (10 mL) was heated under reflux for 24 hrs. After being cooled to rt, the mixture was concentrated under reduced pressure to give crude ethyl 6-(4-fluorophenyl)-10- methoxy-9-(3-methoxypropoxy)-2-oxo- 1,6,7, 1 lb-tetrahydrobenzo[a]quinolizine-3-carboxylate (1.2 g), which was used directly in the next step without further purification.
Step 6: Preparation of ethyl 6-(4-fluorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2- oxo-6,7-dihydrobenzo[a] quinolizine-3-carboxylate

A mixture of ethyl 6-(4-fluorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo- l,6,7,llb-tetrahydrobenzo[a]quinolizine-3-carboxylate (crude 1.2 g) and /?-chloranil (410 mg, 1.67 mmol) in DME (15 mL) was heated under reflux for 3 hrs. Then the mixture was filtered, and the filtrate was concentrated under reduced pressure. The residue was diluted with DCM. The organic mixture was washed with NaHC03 aqueous solution and brine, dried over anhydrous Na2S04 and concentrated under reduced pressure to give crude ethyl 6-(4- fluorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo-6,7-dihydrobenzo[a]quinolizine-3- carboxylate (1.4 g), which was used directly in the next step without further purification.
Step 7: Preparation of 6-(4-fluorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo-
6,7-dihydrobenzo[a] quinolizine-3-carboxylic acid

To a mixture of ethyl 6-(4-fluorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (crude 700 mg) in MeOH (7 mL) was added 2 M NaOH aqueous solution (0.8 mL). The mixture was stirred at 15 °C for 16 hrs, and then concentrated under reduced pressure. The residue was acidified with 1 M hydrochloric acid to pH=3, and extracted with DCM. The organic layer was dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was treated with MeOH (5 mL), and then filtered. The filter cake was purified by prep-HPLC to give 6-(4-fluorophenyl)-10-methoxy-9-(3- methoxypropoxy)-2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid (22 mg) as a white solid. 1H NMR (400 MHz, DMSO-<¾5): δ: 8.82 (s, IH), 7.50-7.53 (m, 2H), 7.06-7.23 (m, 4H), 6.94 (s, IH), 6.08 (s, IH), 4.02 (t, 2H), 3.86 (s, 3H), 3.66-3.70 (m, IH), 3.42-3.50 (m, 4H), 3.22 (s, 3H), 1.94-2.01 (m, 2H). MS obsd. (ESI+) [(M+H)+]: 454.
Example 10: 10-methoxy-9-(3-methoxypropoxy)-6-(o-tolyl)-2- dihydrobenzo[a] quinolizine-3-carboxylic acid
o o

Step 1: Preparation of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(o- tolyl)ethanone

A mixture of 4-bromo-l-methoxy-2-(3-methoxypropoxy)benzene (41.0 g, 155 mmol), 1- (o-tolyl)ethanone (17.5 g, 202 mmol ), i-BuONa (27.0 g, 280 mmol ), Pd2(dba)3 (11.4 g, 12.5 mmol ) and Xantphos (3.61 g, 6.23 mmol ) in THF (500 mL) was heated at 50 °C for 12 hrs. After being cooled to rt, the mixture was concentrated under reduced pressure. The residue was purified by column chromatography to give 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(o- tolyl)ethanone (38.0 g) as a light yellow oil.
Step 2: Preparation of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(o- tolyl)ethanamine

To a solution of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(o-tolyl)ethanone (10.5 g, 32 mmol) in MeOH (120 mL) was added NH4OAc (17.25 g, 224 mmol) at 24 °C. After the mixture was stirred for 1 hr, to the mixture was added NaBH3CN (2.61 g, 2.61 mmol) at 0 °C. The resulting mixture was heated under reflux for 12 hrs, then cooled to rt and concentrated under reduced pressure. The residue was partitioned between H20 (100 mL) and DCM (500 mL). The organic layer was washed with brine (100 mL), dried over anhydrous Na2S04 and
concentrated under reduced pressure to afford 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(o-
tolyl)ethanamine (11.0 g, crude) as yellow oil, which was used directly in next step without further purification.
Step 3: Preparation of N-[2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(o- tol l)ethy 1] formamide

To a solution of 2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(c>-tolyl)ethanamine (11.0 g, 33.4 mmol) in dioxane (120 mL) was added formic acid (7.68 g, 147 mmol). The mixture was heated under reflux for 12 hrs. After being cooled to rt, the mixture was concentrated under reduced pressure. The residue was partitioned between H20 (100 mL) and DCM (500 mL). The organic layer was separated, washed with brine (100 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to afford N-[2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(o-tolyl)ethyl]formamide (5.0 g) as a light yellow solid.
Step 4: Preparation of 7-methoxy-6-(3-methoxypropoxy)-3-(o-tolyl)-3,4- dihydroisoquinoline

To a stirred solution of N-[2-[4-methoxy-3-(3-methoxypropoxy)phenyl]-l-(o- tolyl)ethyl] formamide (2.0 g, 5.6 mmol) in DCM (40 mL) was added POCl3 (1.7 g, 11.1 mmol). The reaction mixture was stirred at 30 °C for 8 hrs, and then poured into a stirred mixture of DCM and ammonia water. The organic layer was separated, washed with brine, dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by the flash column chromatography to afford 7-methoxy-6-(3-methoxypropoxy)-3-(o-tolyl)-3,4- dihydroisoquinoline (1.12 g) as a yellow oil.
Step 5: Preparation of ethyl 10-methoxy-9-(3-methoxypropoxy)-6-(o-tolyl)-2-oxo- l,6,7,llb-tetrahydrobenzo[a]quinolizine-3-carboxylate
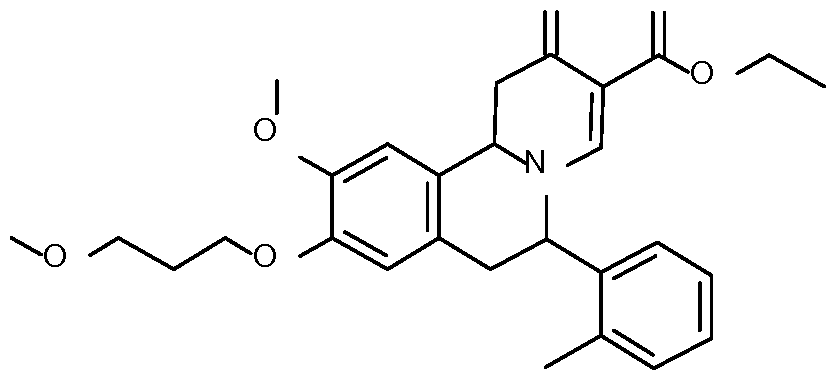
A mixture of 7-methoxy-6-(3-methoxypropoxy)-3-(o-tolyl)-3,4-dihydroisoquinoline (500 mg, 1.47 mmol) and ethyl 2-(ethoxymethylene)-3-oxo-butanoate (82 mg, 4.42 mmol) in EtOH (10 mL) was refluxed for 72 hrs. After being cooled to rt, the mixture was concentrated under reduced pressure to give crude ethyl 10-methoxy-9-(3-methoxypropoxy)-6-(o-tolyl)-2-oxo- l,6,7,llb-tetrahydrobenzo[a]quinolizine-3-carboxylate (750 mg), which was used directly in the next step without further purification.
Step 6: Preparation of ethyl 10-methoxy-9-(3-methoxypropoxy)-6-(o-tolyl)-2-oxo-6,7- dihydrobenzo[a]quinolizine-3-carboxylate
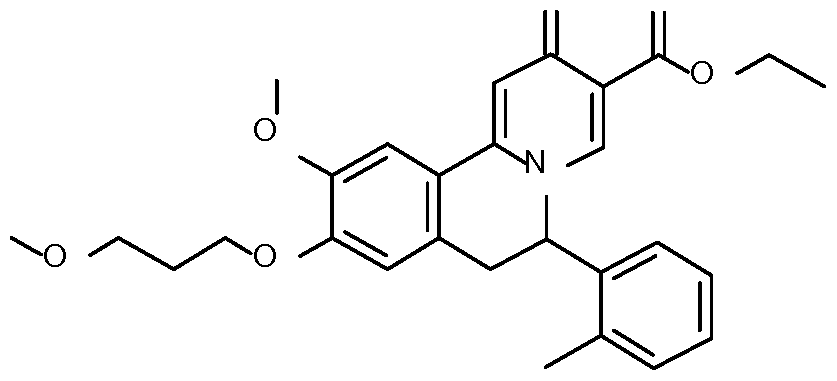
A mixture of ethyl 10-methoxy-9-(3-methoxypropoxy)-6-(o-tolyl)-2-oxo-l,6,7,llb- tetrahydrobenzo[a]quinolizine-3-carboxylate (750 mg, 1.56 mmol) and /?-chloranil (385 mg, 1.356 mmol) in DME (10 mL) was heated at 80 °C for 3 hrs. Then the reaction mixture was filtered. The filtrate was concentrated under reduced pressure to give crude ethyl 10-methoxy-9- (3-methoxypropoxy)-6-(o-tolyl)-2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylate (703 mg), which was used directly in the next step without further purification.
Step 7: Preparation of 10-methoxy-9-(3-methoxypropoxy)-6-(o-tolyl)-2-oxo-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
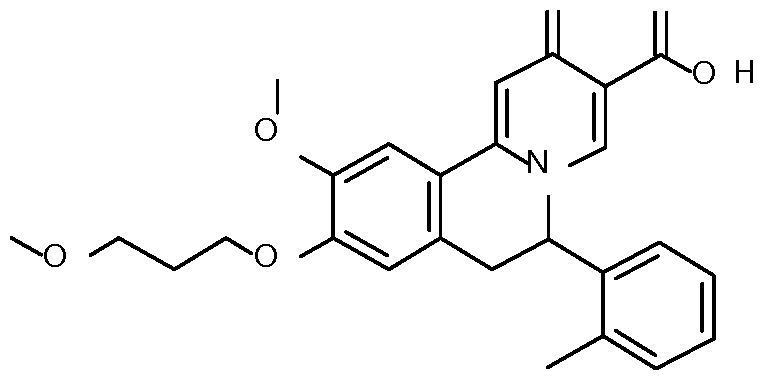
A mixture of crude ethyl 10-methoxy-9-(3-methoxypropoxy)-6-(o-tolyl)-2-oxo-6,7-
dihydrobenzo[a]quinolizine-3-carboxylate (703 mg, 1.47 mmol) and NaOH (235 mg) in a mixture solvent of H20 (3 mL) and MeOH (10 mL) was heated at 30 °C for 16 hrs. The mixture was concentrated under reduced pressure. The residue was diluted with H20 and acidified with 1 M hydrochloric acid to pH=3-4. The resulting mixture was filtered. The filter cake was recrystallized with MeOH, and then purified by prep-HPLC to afford 10-methoxy-9-(3- methoxypropoxy)-6-(o-tolyl)-2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid (51 mg) as a white solid. 1H NMR (400 MHz, MeOD): δ 1.95 - 2.07 (m, 2 H), 2.53 (s, 3 H), 3.29 (s, 3 H), 3.35 (d, 1 H) ,3.53 (td, 2 H), 3.62 (dd, 1 H), 3.95 (s, 3 H), 4.02 - 4.11 (m, 2 H), 4.61 (s, 1 H), 6.06 (br. s., 1 H), 6.56 (d, 1 H), 6.82 (s, 1 H), 7.00 (t, 1 H), 7.18 (t, 1 H), 7.24 - 7.31 (m, 1 H), 7.42 (s, 1 H), 7.51 (s, 1 H), 8.54 (s, 1 H). MS obsd. (ESI+) [(M+H)+]: 450.
Example 11: 9-methoxy-8-methyl-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine- 3-carboxylic acid

Step 1: Preparation of 4-bromo-l-methoxy-2-(3-methoxypropoxy)benzene
To a solution of 3-bromo-2-methyl-phenol (11.4 g, 60.95 mmol) in DMF (150 mL) was added K2C03 (25.3 g, 183 mmol) and iodomethane (26.0 g, 183 mmol). The mixture was stirred at 25 °C for 12 hrs, and then concentrated under reduced pressure. The residue was diluted with EtOAc (200 mL). The organic mixture was washed with water and brine, dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to give 4-bromo-l-methoxy-2-(3-methoxypropoxy)benzene (11.0 g) as a colorless oil.
Step 2: Preparation of 2-(3-methoxy-2-methyl-phenyl)-l-(2-thienyl)ethanone
 To a solution of 4-bromo-l-methoxy-2-(3-methoxypropoxy)benzene (11.0 g, 54.7 mmol) in THF (120 mL) was added l-(2-thienyl)ethanone (8.97 g, 71.1 mmol), i-BuONa (7.89 g, 82.1 mmol), Pd2(dba)3 (1 g, 1.09 mmol) and Xantphos (633 mg, 1.09 mmol ) under nitrogen atmosphere. The mixture was heated at 50 °C for 4 hrs, and then concentrated under reduced pressure. The residue was diluted with DCM (600 mL). The resulting organic mixture was washed with water and brine, dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to afford 2-(3-methoxy-2-methyl- phenyl)-l-(2-thienyl)ethanone (13.0 g) as a brown oil.
To a solution of 4-bromo-l-methoxy-2-(3-methoxypropoxy)benzene (11.0 g, 54.7 mmol) in THF (120 mL) was added l-(2-thienyl)ethanone (8.97 g, 71.1 mmol), i-BuONa (7.89 g, 82.1 mmol), Pd2(dba)3 (1 g, 1.09 mmol) and Xantphos (633 mg, 1.09 mmol ) under nitrogen atmosphere. The mixture was heated at 50 °C for 4 hrs, and then concentrated under reduced pressure. The residue was diluted with DCM (600 mL). The resulting organic mixture was washed with water and brine, dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to afford 2-(3-methoxy-2-methyl- phenyl)-l-(2-thienyl)ethanone (13.0 g) as a brown oil.
Step 3: Preparation of 2-(3-methoxy-2-methyl-phenyl)-l-(2-thienyl)ethanamine

To a solution of 2-(3-methoxy-2-methyl-phenyl)-l-(2-thienyl)ethanone (13.0 g, 52.8 mmol) in MeOH (130 mL) was added NH4OAc (40.7 g, 528 mmol) at 70 °C. The mixture was heated at 70 °C for 1 hr, and then cooled to 0 °C. To this mixture was added NaBH3CN (6.63 g, 105 mmol) at 0 °C. Then the resulting mixture was heated at 80 °C for 12 hrs. After being cooled to rt, the mixture was concentrated under reduced pressure to remove most of MeOH. The residue was partitioned between H20 (100 mL) and DCM (500 mL). The organic layer was separated, washed with brine (100 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to afford 2-(3-methoxy-2-methyl- phenyl)-l-(2-thienyl)ethanamine (6.4 g, crude) as a yellow solid.
Step 4: Preparation of N-[2-(3-methoxy-2-methyl-phenyl)-l-(2- thienyl)ethyl] formamide

To a solution of 2-(3-methoxy-2-methyl-phenyl)-l-(2-thienyl)ethanamine (5.4 g, 21.8 mmol) in dioxane (60 mL) was added formic acid (10.05 g, 218 mmol). The mixture was heated at 120 °C for 12 hrs. After being cooled to rt, the mixture was partitioned between H20 (200 mL) and DCM (600 mL). The organic layer was washed with brine (200 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to afford N-[2-(3-methoxy-2-methyl-phenyl)-l-(2-thienyl)ethyl]formamide (3.4
g) as a brown oil.
Step 5: Preparation of 6-methoxy-5-methyl-3-(2-thienyl)-3,4-dihydroisoquinoline

To a mixture of N-[2-(3-methoxy-2-methyl-phenyl)-l-(2-thienyl)ethyl]formamide (2.9 g, 10.53 mmol) in dry DCM (40 mL) was added POCl3 (5.41 g, 35.3 mmol) drop-wise at 0 °C. Then the mixture was heated under reflux for 12 hrs. After being cooled to rt, the mixture was poured into a solution of ammonia water (30 mL) in H20 (100 mL). Then the mixture was extracted with DCM (200 mL). The organic layer was washed with brine (100 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure to afford crude 6-methoxy-5- methyl-3-(2-thienyl)-3,4-dihydroisoquinoline (4.0 g) as a yellow oil, which was used directly in the next step without further purification.
Step 6: Preparation of ethyl 9-methoxy-8-methyl-2-oxo-6-(2-thienyl)-l,6,7,llb- tetrahydrobenzo[a]quinolizine-3-carbox late

A solution of 6-methoxy-5-methyl-3-(2-thienyl)-3,4-dihydroisoquinoline (2.0 g, 7.77 mmol) and ethyl 2-(ethoxymethylene)-3-oxo-butanoate (4.34 g, 23.3 mmol) in EtOH (30 mL) was heated at 100 °C for 48 hrs. After being cooled to rt, the mixture was concentrated under reduced pressure to give crude ethyl 9-methoxy-8-methyl-2-oxo-6-(2-thienyl)- 1,6,7, 11b- tetrahydrobenzo[a]quinolizine-3-carboxylate, which was used directly in the next step without further purification.
Step 7: Preparation of ethyl 9-methoxy-8-methyl-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate

Step 8: Preparation of 9-methoxy-8-methyl-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
o o

To a solution of ethyl 9-methoxy-8-methyl-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (200 mg, 506 μιηοΐ) in MeOH (2 mL) was added 2 M NaOH aqueous solution (0.76 mL, 1.5 mmol). The mixture was stirred at 25 °C for 12 hrs, then diluted with H20 (20 mL) and acidified with 1 M hydrochloric acid to pH=3. The mixture was extracted with DCM (100 mL). The organic layer was washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by prep- HPLC to afford 9-methoxy-8-methyl-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid (12 mg) as a yellow solid. 1H NMR (400 MHz, CDC13): δ: 2.22 (s, 3 H), 3.37 - 3.69 (m, 2 H), 3.93 (s, 3 H), 5.73 (br. s., 1 H), 6.81 (d, 1 H), 6.85 - 6.91 (m, 1 H), 6.95 (d, 1 H), 7.12 (s, 1 H), 7.18 (d, 1 H), 7.66 (d, 1 H), 8.59 (s, 1 H). MS obsd. (ESI+) [(M+H)+]: 368.
Example 12: 9-benzyloxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
o o

Step 1: Preparation of 2-benzyloxy-4-bromo-l-methoxy-benzene

A mixture of 5-bromo-2-methoxy-phenol (100.0 g, 0.49 mol), K2C03 (102.0 g, 0.74 mol) and bromomethylbenzene (101.0 g, 0.59 mol) in acetone (1.5 L) was heated under reflux for 16 hrs. After being cooled to rt, the mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was purified by trituration (PE, 300 mL) to give 2-benzyloxy-4- bromo-l-methoxy-benzene (140.0 g) as a white solid.
Step 2: Preparation of 2-(3-benzyloxy-4-methoxy-phenyl)-l-(2-thienyl)ethanone
A mixture of 2-benzylo xy-4-bro mo- 1-methoxy- benzene (50.0 g, 0.17 mol), l-(2- thienyl)ethanone (32.3 g, 0.26 mol), i-BuONa (29.5 g, 0.31 mol), Xantphos (4.9 g, 8.53 mmol) and Pd2(dba)3 (3.9 g, 4.26 mmol) in THF (800 ml) was heated at 60 °C for 5 hrs under nitrogen atmosphere. After being cooled to rt, the mixture was filtered. The filtrate was concentrated under reduced pressure. The residue was purified by the flash column chromatography to afford 2-(3-benzyloxy-4-methoxy-phenyl)-l-(2-thienyl)ethanone (31.0 g) as a light yellow solid.
Step 3: Preparation of 2-(3-benzyloxy-4-methoxy-phenyl)-l-(2-thienyl)ethanamine

A mixture of 2-(3-benzyloxy-4-methoxy-phenyl)-l-(2-thienyl)ethanone (31.0 g, 0.092 mol) and NH4OAC (49.4 g, 0.64 mol) in MeOH (500 ml) was refluxed for 6 hrs. The resulting mixture was cooled to rt, and then to the mixture was added NaBH3CN (8.6 g, 0.14 mol) portion wise. The resulting mixture was refluxed for 16 hrs. After being cooled to rt, the resulting reaction mixture was concentrated under reduced pressure, and the residue was partitioned between EtOAc and H20. The organic layer was washed with brine, dried over anhydrous Na2S04 and concentrated under reduced pressure to give 2-(3-benzyloxy-4-methoxy-phenyl)-l-(2- thienyl)ethanamine (38.0 g), which was used directly in the next step without further purification.
Step 4: Preparation of N-[2-(3-benzyloxy-4-methoxy-phenyl)-l-(2- thienyl)ethyl] formamide

A mixture of 2-(3-benzyloxy-4-methoxy-phenyl)- l-(2-thienyl)ethanamine (85.0 g, 0.25 mol) and formic acid (57.6 g, 1.25 mol) in 1,4-dioxane (850 ml) was refluxed for 48 hrs. The resulting mixture was cooled to rt and concentrated under reduced pressure. The residue was partitioned between EtOAc and saturated NaHC03 aqueous solution. The organic layer was separated, washed with brine, dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to afford N-[2-(3-benzyloxy-4- methoxy-phenyl)- l-(2-thienyl)ethyl]formamide (41.5 g) as a yellow oil.
Step 5: Preparation of 6-benzyloxy-7-methoxy-3-(2-thienyl)-3,4-dihydroisoquinoline
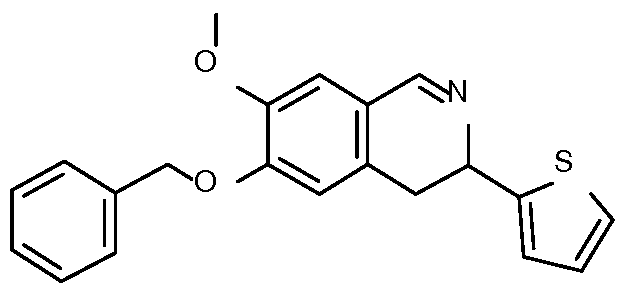
To a stirred solution of N-[2-(3-benzyloxy-4-methoxy-phenyl)- l-(2- thienyl)ethyl] formamide (35.0 g, 0.095 mol) in DCM (400 ml) was added POCl3 (31.59 g, 0.21 mol) at rt. The mixture was heated at 50 °C for 1 hr. After being cooled to rt, the mixture was poured into a stirred mixture of ammonia water and DCM. The organic phase was separated, washed with brine, dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by the flash column chromatography to afford 6-benzyloxy-7-methoxy-3- (2-thienyl)-3,4-dihydroisoquinoline (25.3 g) as a light yellow solid.
Step 6: Preparation of ethyl 9-benzyloxy-10-methoxy-2-oxo-6-(2-thienyl)-l,6,7,llb- tetrahydrobenzo[a]quinolizine-3-carboxylate

A mixture of 6-benzyloxy-7-methoxy-3-(2-thienyl)-3,4-dihydroisoquinoline (17.2 g, 0.049
mol) and ethyl 2-(ethoxymethylene)-3-oxo-butanoate (27.5 g, 0.15 mol) in EtOH (200 ml) was heated under reflux for 72 hrs. After being cooled to rt, the mixture was concentrated under reduced pressure to give crude ethyl 9-benzyloxy- 10-methoxy-2-oxo-6-(2-thienyl)-l,6,7,llb- tetrahydrobenzo[a]quinolizine-3-carboxylate (24.0 g), which was used directly in the next step without further purification.
Step 7: Preparation of ethyl 9-benzyloxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate

A mixture of ethyl 9-benzyloxy-10-methoxy-2-oxo-6-(2-thienyl)-l,6,7,llb- tetrahydrobenzo[a]quinolizine-3-carboxylate (14.6 g, 0.029 mol) and /?-chloranil (7.3 g, 0.029 mol) in DME (150 ml) was heated at 80 °C for 3 hrs. The mixture was cooled to rt and filtered. The filter cake was dried to afford ethyl 9-benzyloxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (7.0 g, yield: 48%) as a light yellow solid.
Step 8: Preparation of 9-benzyloxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid

A mixture of ethyl 9-benzyloxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (100 mg, 0.21 mmol) and NaOH (16 mg) in a mixture solvent of H20 (0.2 mL) and THF (5 mL) was stirred at room temperature for 16 hrs. The mixture was concentrated under reduced pressure. The residue was diluted with H20 and acidified with 1 M hydrochloric acid to pH=2-3. Then the mixture was filtered. The filter cake was dried under reduced pressure to afford 9-benzyloxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid (54 mg) as a yellow solid. 1H NMR (400 MHz, DMSO-J6) δ: δ 3.30 - 3.39 (m, 1 H), 3.73 (dd, 1 H), 3.88 (s, 3 H), 5.16 (s, 2 H), 6.35 (d, 1 H), 6.87 - 6.94 (m, 1 H), 7.02 (d, 1 H), 7.20 (s, 1 H), 7.32 - 7.43 (m, 4 H), 7.44 - 7.50 (m, 2 H), 7.54
(d, 2 H), 8.99 (s, 1 H). MS obsd. (ESI+) [(M+H)+]: 460.
Example 13: 9,10-dimethoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid

Step 1: Preparation of ethyl 9-hydroxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate

A solution of ethyl 9-benzyloxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (5.0 g, 0.01 mol, from step 7 of Example 12) and thioanisole (13.66 g, 0.11 mmol) in TFA (50 mL) was stirred at room temperature for 16 hrs. The resulting mixture was partitioned between DCM and H20. The organic layer was separated, washed with saturated NaHC03 aqueous solution and brine, dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was recrystallized from DCM/hexane to afford ethyl 9-hydroxy- 10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate (3.50 g) as a yellow solid.
Step 2: Preparation of ethyl 9,10-dimethoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate

A mixture of ethyl 9-hydroxy- 10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (210 mg, 0.53 mmol), K2C03 (132 mg, 0.95 mmol)
and iodomethane (113 mg, 0.79 mmol) in acetone (5 mL) was heated at 50 °C for 16 hrs. The mixture was concentrated under reduced pressure and the residue was partitioned between EtOAc and H20. The organic layer was separated, dried over anhydrous Na2S04 and
concentrated under reduced pressure. The residue was recrystallized from a mixture solvent (PE: EA=1: 1) to afford ethyl 9,10-dimethoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylate (200 mg) as a light yellow solid.
Step 3: Preparation of 9,10-dimethoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid

A mixture of ethyl 9, 10-dimethoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylate (100 mg, 0.24 mmol) and NaOH (20 mg) in MeOH (2 mL) and H20 (0.2 mL) was stirred at room temperature for 16 hrs. The resulting mixture was concentrated under reduced pressure. The residue was diluted with H20 and acidified by 1 M hydrochloric acid to pH=2-3. The mixture was filtered. The filter cake was dried under reduced pressure to afford 9,10- dimethoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid (46 mg) as a white solid. 1H NMR (400 MHz, DMSO-J6) δ: 3.37 - 3.39 (m, 1 H), 3.73 (dd, 1 H), 3.85 (d, 6 H), 6.35 (d, 1 H), 6.90 (t, 1 H), 6.99 - 7.12 (m, 2 H), 7.36 (d, 1 H), 7.51 (d, 2 H), 8.98 (s, 1 H). MS obsd. (ESI+) [(M+H)+]: 384. Example 14: 9-isobutoxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
o o

Step 1: Preparation of ethyl 9-isobutoxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate

A mixture of ethyl 9-hydroxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (150 mg, 0.38 mmol), l-bromo-2-methyl-propane (78 mg, 0.57 mmol) and K2C03 (78 mg, 0.57 mmol) in DMF (5 ml) was heated at 50 °C for 16 hrs. The mixture was partitioned between H20 and DCM. The organic layer was separated, washed with brine, dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was recrystallized from PE/EtOAc to afford ethyl 9-isobutoxy-10-methoxy-2-oxo-6-(2-thienyl)- 6,7-dihydrobenzo[a]quinolizine-3-carboxylate (110 mg) as a light yellow solid.
Step 2: Preparation of 9-isobutoxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid

A mixture of ethyl 9-isobutoxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (80 mg, 0.18 mmol) and NaOH (14 mg) in MeOH (2 mL) and H20 (0.2 mL) was stirred at room temperature for 16 hrs. The mixture was
concentrated under reduced pressure. The residue was diluted with H20 and acidified with 1 M hydrochloric acid to pH=2-3. The resulting mixture was filtered. The filter cake was dried under reduced pressure to afford 9-isobutoxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid (38 mg) as a white solid. 1H NMR (400 MHz, DMSO-J6) δ: 0.98 (d, 6 H), 1.98 - 2.12 (m, 1 H), 3.37 (br. s., 1 H), 3.70 (dd, 1 H), 3.80 (d, 2 H), 3.88 (s, 3 H), 6.27 - 6.39 (m, 1 H), 6.88 - 6.93 (m, 1 H), 7.02 (d, 1 H), 7.07 (s, 1 H), 7.36 (d, 1 H), 7.50 (d, 2 H), 8.98 (s, 1 H). MS obsd. (ESI+) [(M+H)+]: 426.
Example 15: 9-(cyclopropylmethoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid

Step 1: Preparation of ethyl 9-(cyclopropylmethoxy)-10-methoxy-2-oxo-6-(2-thienyl)- 6,7-dihydrobenzo[a]quinolizine-3-carboxylate

A mixture of ethyl 9-hydroxy- 10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (150 mg, 0.38 mmol), bromomethylcyclopropane (76 mg, 0.57 mmol) and K2CO3 (78 mg, 0.57 mmol) in DMF (5 ml) was stirred at room temperature for 16 hrs. The resulting mixture was partitioned between H20 and DCM. The organic layer was washed with brine, dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was recrystallized from PE/EA to afford ethyl 9-(cyclopropylmethoxy)-10-methoxy-2- oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate (140 mg) as a light yellow solid.
Step 2: Preparation of 9-(cyclopropylmethoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid

A mixture of ethyl 9-(cyclopropylmethoxy)- 10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (70 mg, 0.16 mmol) and NaOH (12 mg) in MeOH (2 mL) and H20 (0.1 mL) was stirred at room temperature for 16 hrs. The resulting mixture was concentrated under reduced pressure. The residue was diluted with H20 and acidified with 1 M hydrochloric acid to pH=2-3. The mixture was filtered and the filter cake was dried under reduced pressure to afford 9-(cyclopropylmethoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid. 1H NMR (400 MHz, DMSO-J6) δ: 0.26 - 0.39 (m,
2 H), 0.53 - 0.64 (m, 2 H), 1.17 - 1.33 (m, 1 H), 3.31 (s, 1 H), 3.70 (dd, 1 H), 3.81 - 3.95 (m, 5 H), 6.34 (d, 1 H), 6.90 (dd, 1 H), 6.98 - 7.07 (m, 2 H), 7.35 (dd, 1 H), 7.51 (d, 2 H), 8.98 (s, 1 H). MS obsd. (ESI+) [(M+H)+] : 424.
Example 16: 10-methoxy-2-oxo-9-(3-pyrazol-l-ylpropoxy)-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid

Step 1: Preparation of l-(3-bromopropyl)pyrazole
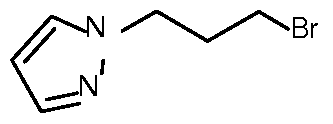
To a solution of lH-pyrazole (1 g, 14.7 mmol) in acetone (20 mL) was added K2CO3 (4.06 g, 29.4 mmol) and 1,3-dibromopropane (14.8 g, 73.4 mmol). The mixture was heated at 30 °C for 12 hrs. The mixture was concentrated under reduced pressure and the residue was partitioned between H20 (150 mL) and EtOAc (200 mL). The organic layer was separated, washed with brine (150 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to afford compound l-(3-bromopropyl)pyrazole (500 mg) as a yellow oil.
Step 2: Preparation of ethyl 10-methoxy-2-oxo-9-(3-pyrazol-l-ylpropoxy)-6-(2- thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate

A solution of compound ethyl 9-hydroxy- 10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (150 mg, 377 μιηοΐ) in DMF (3 mL) was added K2CO3 (104 mg, 754 μιηοΐ) and l-(3-bromopropyl)pyrazole (85.6 mg, 453 μιηοΐ). The mixture was heated at 30 °C for 12 hrs and then partitioned between H20 (50 mL) and EtOAc (100 mL). The organic layer was separated, washed with brine (50 mL), dried over anhydrous Na2S04 and
concentrated under reduced pressure. The residue was purified by column chromatography to afford crude ethyl 10-methoxy-2-oxo-9-(3-pyrazol-l-ylpropoxy)-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (100 mg) as a yellow oil.
Step 3: Preparation of 10-methoxy-2-oxo-9-(3-pyrazol-l-ylpropoxy)-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid
o o

To a solution of ethyl 10-methoxy-2-oxo-9-(3-pyrazol-l-ylpropoxy)-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (100 mg, 198 μιηοΐ) in MeOH (2 mL) was added 2 M NaOH aqueous solution (0.197 mL, 395 μιηοΐ). The mixture was stirred at 30 °C for 12 hrs, and then concentrated under reduced pressure. The residue was diluted with H20 (20 mL) and acidified with 1 M hydrochloric acid to pH=3. Then the resulting mixture was extracted with DCM (100 mL). The organic layer was washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by prep-HPLC to afford 10-methoxy-2-oxo-9-(3-pyrazol-l-ylpropoxy)-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid (13.8 mg) as a white solid. 1H NMR (400 MHz, DMSO-J6) δ: 2.25 (t, 2 H), 3.23 - 3.28 (m, 1 H), 3.70 (dd, 1 H), 3.90 (s, 3 H), 3.94 - 4.07 (m, 2 H), 4.27 (t, 2 H), 6.23 (t, 1 H), 6.33 (d, 1 H), 6.90 (dd, 1 H), 7.02 (s, 2 H), 7.36 (d, 1 H), 7.44 (d, 1 H), 7.52 (d, 2 H), 7.72 (d, 1 H), 8.98 (s, 1 H). MS obsd. (ESI+) [(M+H)+]: 478.
Example 17: 9-(3-hydroxypropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
o o

Step 1: Preparation of ethyl 9-(3-hydroxypropoxy)-10-methoxy-2-oxo-6-(2-thienyl)- 6,7-dihydrobenzo[a]quinolizine-3-carboxylate

To a solution of compound ethyl 9-hydroxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (150 mg, 377 μιηοΐ) in DMF (3 mL) was added K2CO3 (78 mg, 566 μιηοΐ) and 3-bromopropan-l-ol (79 mg, 566 μιηοΐ). The mixture was heated at 30 °C for 12 hrs, and then partitioned between H20 (50 mL) and EtOAc (100 mL). The organic layer was separated, washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure to afford crude ethyl 9-(3-hydroxypropoxy)-10-methoxy-2- oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate (200 mg) as brown oil, which was used directly in the next step without further purification.
Step 2: Preparation of 9-(3-hydroxypropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid

To a solution of ethyl 9-(3-hydroxypropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (200 mg, 439 μιηοΐ) in MeOH (2 mL) was added 2 M NaOH aqueous solution (0.439 mL, 878 μιηοΐ). The mixture was stirred at 30 °C for 12 hrs, and then concentrated under reduced pressure. The residue was diluted with H20 (20 mL) and acidified with 1 M hydrochloric acid to pH=3. Then the resulting mixture was extracted with DCM (100 mL). The organic layer was washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by prep-HPLC to afford 9-(3-hydroxypropoxy)- 10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine- 3-carboxylic acid (7.9 mg) as a yellow solid. 1H NMR (400 MHz, DMSO-J6) δ: 1.89 (t, 2 H), 3.33 (br. s., 1 H), 3.55 (t, 2 H), 3.71 (d, 1 H), 3.87 (s, 3 H), 4.09 (t, 2 H), 4.60 (br. s., 1 H), 6.33 (br. s., 1 H), 6.82 - 6.94 (m, 1 H), 6.96 - 7.13 (m, 2 H), 7.35 (d, 1 H), 7.50 (br. s., 2 H), 8.97 (br. s., 1 H). MS obsd. (ESI+) [(M+H)+]: 428.
Example 18: 10-methoxy-2-oxo-9-[3-(l-piperidyl)propoxy]-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid formate
o o

Step 1: Preparation of ethyl 9-(3-bromopropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylate

A mixture of ethyl 9-hydroxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (400 mg, 1.0 mmol), 1,3-dibromopropane (244 mg, 1.21 mmol) and K2CO3 (209 mg, 1.51 mmol) in DMF (7 mL) was stirred at room temperature for 16 hrs. The mixture was partitioned between H20 and DCM. The organic layer was separated, washed with brine, dried over anhydrous Na2S04 and concentrated under reduced pressure to give crude ethyl 9-(3-bromopropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (500 mg), which was used directly in the next step without further purification.
Step 2: Preparation of ethyl 10-methoxy-2-oxo-9-[3-(l-piperidyl)propoxy]-6-(2- thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate

A mixture of ethyl 9-(3-bromopropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (200 mg, 0.39 mmol), piperidine (164 mg, 1.93 mmo and K2CO3 (267 mg, 1.93 mmol) in DMF (5 mL) was stirred at room temperature for 16 hrs. The mixture was partitioned between H20 and DCM. The organic layer was separated, washed
with brine, dried over anhydrous Na2S04 and concentrated under reduced pressure to give crude ethyl 10-methoxy-2-oxo-9-[3-(l-piperidyl)propoxy]-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (140 mg), which was used directly in the next step without further purification.
Step 3: Preparation of 10-methoxy-2-oxo-9-[3-(l-piperidyl)propoxy]-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid formate

A mixture of ethyl 10-methoxy-2-oxo-9-[3-(l-piperidyl)propoxy]-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (140 mg, 0.27 mmol) and NaOH (21 mg) in MeOH (3 mL) and H20 (0.3 mL) was stirred at room temperature for 16 hrs. The mixture was
concentrated under reduced pressure. The residue was diluted with H20 and acidified with 1 M hydrochloric acid to pH=2-3. The resulting mixture was filtered. The filter cake was dried and purified by prep-HPLC (formic acid as additive) to afford 10-methoxy-2-oxo-9-[3-(l- piperidyl)propoxy]-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid formate (23 mg) as a white solid. 1H NMR (400 MHz, DMSO-J6) δ: 1.39 (d, 2 H), 1.46 - 1.57 (m, 4 H), 1.91 (t, 2 H), 2.36 - 2.48 (m, 6 H), 3.34 (s., 1 H), 3.72 (dd, 1 H), 3.88 (s, 3 H), 4.07 (t, 2 H), 6.34 (d, 1 H), 6.85 - 6.95 (m, 1 H), 7.00 - 7.11 (m, 2 H), 7.36 (d, 1 H), 7.45 - 7.56 (m, 2 H), 8.21 (s, 1 H), 8.98 (s, 1 H). MS obsd. (ESI+) [(M+H)+]: 495. Example 19: 9-(3-cyanopropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid

Step 1: Preparation of ethyl 9-(3-cyanopropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate

To a solution of compound ethyl 9-hydroxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (150 mg, 377 μιηοΐ) in DMF (3 mL) was added K2CO3 (78 mg, 566 μιηοΐ) and 4-bromobutanenitrile (84 mg, 566 μιηοΐ). Then the mixture was stirred at 30 °C for 12 hrs. The mixture partitioned between H20 (50 mL) and EtOAc (100 mL). The organic layer was separated, washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure to afford crude ethyl 9-(3-cyanopropoxy)-10-methoxy-2- oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate (190 mg) as brown oil, which was used directly in the next step without further purification.
Step 2: Preparation of 9-(3-cyanopropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid

A solution of ethyl 9-(3-cyanopropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (190 mg, 409 μιηοΐ) in THF (2 mL) was added 2 M LiOH aqueous solution (0.409 mL, 818 μιηοΐ). The mixture was stirred at 30 °C for 12 hrs, and then concentrated under reduced pressure. The residue was diluted with H20 (20 mL) and acidified with 1 M hydrochloric acid to pH=3. Then the resulting mixture was extracted with DCM (100 mL). The organic layer was washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by prep-HPLC to afford 9-(3-cyanopropoxy)- 10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid (32.5 mg) as a white solid. 1H NMR (400 MHz, DMSO-J6) δ: 1.97 - 2.15 (m, 2 H), 2.65 (t, 2 H), 3.42 - 3.49 (m, 1 H), 3.72 (dd, 1 H), 3.89 (s, 3 H), 4.10 (t, 2 H), 6.35 (br. s., 1 H), 6.91 (dd, 1 H), 7.03 (br. s., 1 H), 7.11 (s, 1 H), 7.30 - 7.41 (m, 1 H), 7.53 (d, 2 H), 8.99 (s, 1 H). MS obsd. (ESI+) [(M+H)+]: 437.
Example 20: 10-methoxy-2-oxo-9-[2-(2-oxopyrrolidin-l-yl)ethoxy]-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
o o

Step 1: Preparation of ethyl 9-(2-bromoethoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carbox late

A mixture of ethyl 9-hydroxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (100 mg, 0.25 mmol), 1,2-dibromoethane (57 mg, 0.30 mmol) and K2CO3 (53 mg, 0.38 mmol) in DMF (3 ml) was stirred at room temperature for 16 hrs. The mixture was partitioned between H20 and DCM. The organic layer was separated, washed with brine, dried over anhydrous Na2S04 and concentrated under reduced pressure to give crude ethyl 9-(2-bromoethoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (130 mg), which was used directly in the next step without further purification.
Step 2: Preparation of 10-methoxy-2-oxo-9-[2-(2-oxopyrrolidin-l-yl)ethoxy]-6-(2- thienyl)-6,7-dihydrobenzo[a] quinolizine-3-carboxylic acid

To a cooled and stirred solution of pyrrolidin-2-one (290 mg, 3.41 mmol) in DMF (2 mL) was added NaH (50 mg, 1.25 mmol) at 0 °C. After the addition, the resulting mixture was warmed up to room temperature and stirred for 10 min. Then to the mixture was added ethyl 9- (2-bromoethoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate
(130 mg, 0.25 mmol) and the resulting mixture was stirred at rt for 16 hrs. The reaction was quenched by addition of saturated NH4C1 aqueous solution, and then the mixture was extracted with EtOAc. The organic layer was washed with brine, dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by prep-HPLC to afford 10- methoxy-2-oxo-9-[2-(2-oxopyrrolidin-l-yl)ethoxy]-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid (7.0 mg) as a white solid. 1H NMR (400 MHz, DMSO-J6) δ: 1.91 (quin, 2 H), 2.15 - 2.26 (m, 2 H), 3.31 - 3.32 (m, 1 H), 3.48 (t, 2 H), 3.56 (t, 2 H), 3.71 (dd, 1 H), 3.88 (s, 3 H), 4.13 (t, 2 H), 6.34 (d, 1 H), 6.90 (dd, 1 H), 7.01 (d, 1 H), 7.10 (s, 1 H), 7.36 (d, 1 H), 7.52 (d, 2 H), 8.92 - 9.03 (m, 1 H). MS obsd. (ESI+) [(M+H)+]: 481.
Example 21: 9-[6-(tert-butoxycarbonylamino)hexoxy]-10-methoxy-2-oxo-6-(2- thienyl)-6,7-dihydrobenzo[a] quinolizine-3-carboxylic acid

Step 1: Preparation of ethyl 9-[6-(tert-butoxycarbonylamino)hexoxy]-10-methoxy-2-
-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate

To a solution of ethyl 9-hydroxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (120 mg, 0.3 mmol) in DMF (3 mL) was added K2CO3 (83 mg, 0.6 mmol) and ie/t-butyl (6-bromohexyl)carbamate (101 mg, 0.36 mmol). The mixture was stirred at 30 °C for 12 hrs, and then partitioned between H20 (50 mL) and EtOAc (100 mL). The organic layer was separated, washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure to afford crude ethyl 9-[6-(ieri- butoxycarbonylamino)hexoxy]-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (140 mg) as a yellow oil, which was used directly in the next step without further purification.
Step 2: Preparation of 9-[6-(tert-butoxycarbonylamino)hexoxy]-10-methoxy-2-
(2-thienyl)-6,7-dihydrobenzo[a] quinolizine-3-carboxylic acid

To a solution of crude ethyl 9-[6-(ieri-butoxycarbonylamino)hexoxy]-10-methoxy-2-oxo- 6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate (70 mg, 0.117 mmol) in MeOH (1 mL) was added 2 M NaOH aqueous solution (0.117 mL, 0.234 mmol). The mixture was stirred at 30 °C for 2 hrs, and then diluted with H20 (20 mL). The resulting mixture was acidified with 1 M hydrochloric acid to pH=3, and extracted with DCM (100 mL). The organic layer was washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by prep-HPLC to afford 9-[6-(ieri- butoxycarbonylamino)hexoxy]-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid (18.1 mg) as white solid. 1H NMR (400 MHz, DMSO-J6) δ: 1.36 (s, 15 H) 1.71 (d, 2 H) 2.81 - 2.98 (m, 2 H) 3.11 - 3.28 (m, 1 H) 3.70 (d, 1 H) 3.87 (s, 3 H) 4.01 (t, 2 H) 6.33 (br. s., 1 H) 6.77 (br. s., 1 H) 6.84 - 6.95 (m, 1 H) 6.96 - 7.12 (m, 2 H) 7.35 (d, 1 H) 7.50 (br. s., 2 H) 8.96 (br. s., 1 H). MS obsd. (ESI+) [(M+H)+]: 569.
Example 22: 9-(6-aminohexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizin -3-carboxylic acid formate

Step 1: Preparation of ethyl 9-(6-aminohexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylate hydrochloride
o o

Step 2: Preparation of 9-(6-aminohexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizin -3-carboxylic acid formate

To a solution of ethyl 9-(6-aminohexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate hydrochloride (60 mg, 0.112 mmol) in MeOH (1 mL) was added 2 M NaOH (0.112 mL, 0.224 mmol). The mixture was stirred at 30 °C for 2 hrs, and then concentrated under reduced pressure. The residue was diluted with H20 (20 mL) and acidified with 1 M hydrochloric acid to pH=3. Then the mixture was extracted with DCM (60 mL). The organic layer was washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by prep-HPLC (formic acid as additive) to afford compound 9-(6-aminohexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid formate (12 mg) as a yellow solid. 1H NMR (400 MHz, DMSO-J6) δ: 1.38 (br. s., 4 H), 1.48 - 1.63 (m, 2 H), 1.64 - 1.82 (m, 2 H), 2.74 (t, 2 H), 3.31 (br. s., 1 H), 3.72 (d, 1 H), 3.87 (s, 3 H), 4.01 (t, 2 H), 6.33 (br. s., 1 H), 6.90 (dd, 1 H), 6.95 - 7.11 (m, 2 H), 7.35 (d, 1 H), 7.45 - 7.52 (m, 1 H), 8.40 (br. s., 1 H), 8.95 (br. s., 1 H). MS obsd. (ESI+) [(M+H)+]: 469.
Example 23: 9-(6-acetamidohexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid
o o

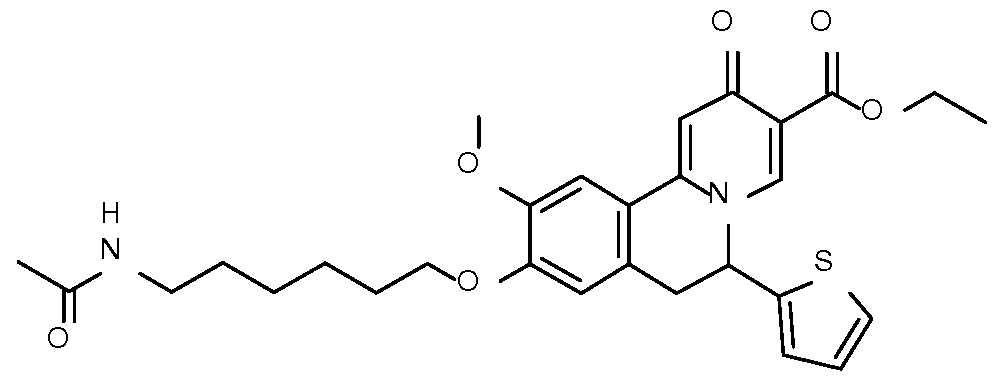
To a solution of ethyl 9-(6-aminohexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate hydrochloride (80 mg, 0.15 mmol) in DCM (3 mL) was added DIPEA (97 mg, 0.75 mmol) and acetyl chloride (95 mg, 0.75 mmol). The mixture was stirred at 30 °C for 12 hrs, and then diluted with H20 (20 mL). The resulting mixture was extracted with DCM (60 mL). The organic layer was washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure to give crude ethyl 9-(6- acetamidohexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylate (100 mg) as a yellow oil, which was used directly in the next step without further purification.
Step 2: Preparation of 9-(6-acetamidohexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizin -3-carboxylic acid

To a solution of ethyl 9-(6-acetamidohexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (100 mg, 0.185 mmol) in MeOH (2 mL) was added 2 M NaOH aqueous solution (0.186 mL, 0.371 mmol). The mixture was stirred at 30 °C for 2 hrs, and then concentrated under reduced pressure. The residue was diluted with H20 (20 mL) and acidified with 1 M hydrochloric acid to pH=3. Then the mixture was extracted with DCM (60 mL). The organic layer was washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by prep-HPLC to afford 9-(6- acetamidohexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid (17.5 mg) as a white solid. 1H NMR (400 MHz, DMSO-J6) δ: 1.25 - 1.48 (m, 6 H), 1.66 - 1.76 (m, 2 H), 1.77 (s, 3 H), 3.01 (q, 2 H), 3.39 - 3.47 (m, 1 H), 3.71 (dd, 1 H), 3.87 (s,
3 H), 4.02 (t, 2 H), 6.34 (d, 1 H), 6.90 (dd, 1 H), 7.00 - 7.11 (m, 2 H), 7.36 (ddl H), 7.51 (d, 2 H), 7.78 (br. s., 1 H), 8.98 (s, 1 H). MS obsd. (ESI+) [(M+H)+]: 511.
Example 24: 9-[6-(methanesulfonamido)hexoxy]-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid

Step 1: Preparation of ethyl 9-[6-(methanesulfonamido)hexoxy]-10-methoxy-2-
(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate
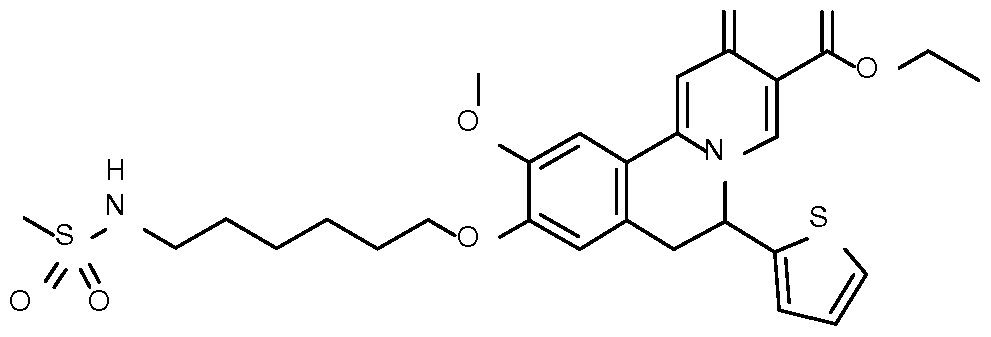
To a solution of ethyl 9-(6-aminohexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate hydrochloride (100 mg, 0.187 mmol) in DCM (3 mL) was added DIPEA (121 mg, 0.937 mmol) and methane sulfonyl chloride (107 mg, 937 μιηοΐ). The mixture was stirred at 30 °C for 12 hrs, and then concentrated under reduced pressure. The residue was partitioned between H20 (20 mL) and DCM (60 mL). The organic layer was separated, washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure to afford crude ethyl 9-[6-(methanesulfonamido)hexoxy]-10-methoxy-2-oxo-6- (2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate (100 mg) as a yellow oil, which was used directly in the next step without further purification.
Step 2: Preparation of 9-[6-(methanesulfonamido)hexoxy]-10-methoxy-2-oxo-6-(2- thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid

To a solution of ethyl 9-[6-(methanesulfonamido)hexoxy]-10-methoxy-2-oxo-6-(2-
thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate (100 mg, 0.174 mmol) in MeOH (2 mL) was added 2 M NaOH (0.174 mL, 0.348 mmol). The mixture was stirred at 30 °C for 2 hrs, then diluted with H20 (20 mL) and acidified with 1 M hydrochloric acid to pH=3. Then the resulting mixture was extracted with DCM (60 mL). The organic layer was washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by prep-HPLC to afford 9-[6-(methanesulfonamido)hexoxy]-10-methoxy-2-oxo-6-(2- thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid (53.3 mg) as a yellow solid. 1H NMR (400 MHz, DMSO-<¾5) δ: 1.32 - 1.53 (m, 6 H), 1.66 - 1.81 (m, 2 H), 2.87 (s, 3 H), 2.92 (q, 2 H),3.40 - 3.48 (m, 1 H), 3.71 (dd, 1 H), 3.87 (s, 3 H), 4.02 (t, 2 H), 6.34 (d, 1 H), 6.85 - 6.96 (m, 2 H), 6.98 - 7.11 (m, 2 H), 7.36 (dd, 1 H), 7.50 (d, 2 H) ,8.98 (s, 1 H). MS obsd. (ESI+) [(M+H)+]: 547.
Example 25: 9-(6-hydroxyhexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid

Step 1: Preparation of ethyl 9-(6-hydroxyhexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate

To a solution of compound ethyl 9-hydroxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (150 mg, 0.377 mmol) in DMF (3 mL) was added K2C03 (104 mg, 754 μπιοΐ) and 6-bromohexan-l-ol (102 mg, 0.566 mmol). The mixture was stirred at 30 °C for 12 hrs, and then partitioned between H20 (50 mL) and EtOAc (100 mL). The organic layer was separated, washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure to give ethyl 9-(6-hydroxyhexoxy)-10-methoxy-2-oxo-6-(2- thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate (90 mg), which was used directly in the
next step without further purification.
Step 2: Preparation of 9-(6-hydroxyhexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
o o

To a solution of ethyl 9-(6-hydroxyhexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (90 mg, 0.181 mmol) in MeOH (2 mL) was added 2 M NaOH aqueous solution (0.181 mL, 0.362 mmol). The mixture was stirred at 30 °C for 12 hrs, then diluted with H20 (20 mL) and acidified with 1 M hydrochloric acid to pH=3. The resulting mixture was extracted with DCM (100 mL). The organic layer was washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by prep-HPLC to afford 9-(6-hydroxyhexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid (48 mg) as white solid. 1H NMR (400 MHz, DMSO-<¾5) δ: 1.15 - 1.55 (m, 6 H), 1.73 (br. s., 2 H), 3.12 - 3.30 (m, 1 H), 3.73 (br. s., 1 H), 3.86 (br. s., 3 H), 4.01 (br. s., 2 H), 4.39 (br. s., 2 H), 6.33 (br. s., 1 H), 6.90 (d, 1 H), 7.04 (d, 2 H), 7.35 (br. s., 1 H), 7.50 (br. s., 2 H), 8.97 (br. s., 1 H). MS obsd. (ESI+) [(M+H)+]: 470.
Example 26: 9-(3-hydroxy-2,2-dimethyl-propoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
o o

Step 1: Preparation of ethyl 9-(3-hydroxy-2,2-dimethyl-propoxy)-10-methoxy-2-oxo-6- (2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate
o o

Step 2: Preparation of 9-(3-hydroxy-2,2-dimethyl-propoxy)-10-methoxy-2-oxo-6-(2- thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid

A mixture of ethyl 9-(3-hydroxy-2,2-dimethyl-propoxy)-10-methoxy-2-oxo-6-(2-thienyl)- 6,7-dihydrobenzo[a]quinolizine-3-carboxylate (130 mg, 0.27 mmol) and NaOH (22 mg) in H20 (0.3 mL) and MeOH (2 mL) was stirred at room temperature for 16 hrs. The mixture was concentrated under reduced pressure. The residue was diluted with H20 and acidified with 1 M hydrochloric acid to pH=2-3. The resulting mixture was filtered. The filter cake was purified by prep-HPLC to afford 9-(3-hydroxy-2,2-dimethyl-propoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid (52 mg) as a yellow solid. 1H NMR (400 MHz, DMSO-J6) δ: 0.93 (s, 6 H), 3.29 (br. s., 2 H), 3.32 - 3.33 (m, 1 H), 3.66 - 3.74 (m, 1 H), 3.74 - 3.79 (m, 2 H), 3.88 (s, 3 H), 6.34 (d, 1 H), 6.90 (dd, 1 H), 7.01 (d, 1 H), 7.06 (s, 1 H), 7.32 - 7.38 (m, 1 H), 7.50 (d, 2 H), 8.97 (s, 1 H). MS obsd. (ESI+) [(M+H)+]: 456.
Example 27: 10-methoxy-2-oxo-6-(2-thienyl)-9-(2,2,2-trifluoroethoxy)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid

Step 1: Preparation of ethyl 10-methoxy-2-oxo-6-(2-thienyl)-9-(2,2,2-trifluoroethoxy)-
6,7-dihydrobenzo[a]quinolizine-3-carboxylate

A mixture of ethyl 9-hydroxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (150 mg, 0.38 mmol), l,l,l-trifluoro-2-iodo-ethane (119 mg, 0.57 mmol) and K2C03 (78 mg, 0.57 mmol) in DMF (5 ml) was heated at 70 °C for 6 hrs. The mixture was partitioned between H20 and DCM. The organic layer was separated, washed with brine, dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was recrystallized from PE/EA to afford ethyl 10-methoxy-2-oxo-6-(2-thienyl)-9-(2,2,2- trifluoroethoxy)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate (100 mg) as a white solid.
Step 2: Preparation of 10-methoxy-2-oxo-6-(2-thienyl)-9-(2,2,2-trifluoroethoxy)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid

A mixture of ethyl 10-methoxy-2-oxo-6-(2-thienyl)-9-(2,2,2-trifluoroethoxy)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (70 mg, 0.15 mmol) and NaOH (12 mg) in H20 (0.1 mL) and THF (2 mL) was stirred at room temperature for 16 hrs. The mixture was concentrated under reduced pressure. The residue was diluted with H20 and acidified with 1 M hydrochloric acid to pH=2-3. The resulting mixture was filtered. The filter cake was purified by prep-HPLC to afford 10-methoxy-2-oxo-6-(2-thienyl)-9-(2,2,2-trifluoroethoxy)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid (25 mg) as a white solid. 1H NMR (400 MHz, DMSO-J6) δ: 3.30 (s, 1 H), 3.75 (dd, 1 H), 3.90 (s, 3 H), 4.82 (q, 2 H) 6.37 (d, 1 H), 6.90 (dd, 1 H), 7.02 (d, 1 H), 7.21 (s, 1 H), 7.32 - 7.42 (m, 1 H), 7.60 (d, 2 H), 9.02 (s, 1 H). MS obsd. (ESI+) [(M+H)+]: 452.
Example 28: 10-methoxy-9-(3-morpholinopropoxy)-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylate

Step 1: Preparation of ethyl 10-methoxy-9-(3-morpholinopropoxy)-2- thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate

A mixture of ethyl 9-(3-bromopropoxy)- 10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (130 mg, 0.25 mmol, step 1 of Example 18), morpholine (109 mg, 1.25 mmol) and K2CO3 (173 mg, 1.25 mmol) in DMF (3 ml) was stirred at room temperature for 16 hrs. The mixture was partitioned between H20 and DCM. The organic layer was washed with brine, dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by flash column chromatography to give ethyl 10-methoxy-9- (3-morpholinopropoxy)-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate (100 mg) as a yellow oil.
Step 2: Preparation of 10-methoxy-9-(3-morpholinopropoxy)-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate

A mixture of ethyl 10-methoxy-9-(3-morpholinopropoxy)-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (100 mg, 0.27 mmol) and NaOH (15 mg) in MeOH (2 mL) and H20 (0.2 mL) was stirred at room temperature for 16 hrs. The mixture was
concentrated under reduced pressure. The residue was diluted with H20 and acidified with 1 M hydrochloric acid to pH=2-3. The resulting mixture was filtered. The filter cake was purified by prep-HPLC to afford 10-methoxy-9-(3-morpholinopropoxy)-2-oxo-6-(2-thienyl)-6,7-
dihydrobenzo[a]quinolizine-3-carboxylate (7 mg) as a white solid. 1H NMR (400 MHz, DMSO- d6) δ: 1.90 (quin, 2 H), 2.35 (m, 4 H), 2.41 (t, 2 H), 3.33 (m, 1 H), 3.56 (t, 4 H), 3.70 (dd, 1 H), 3.87 (s, 3 H), 4.07 (t, 2 H), 6.32 (m, 1 H), 6.91 (dd, 1 H), 6.97 - 7.13 (m, 2 H), 7.29 - 7.40 (m, 1 H), 7.45 - 7.54 (m, 1 H), 8.33 (s, 1 H), 8.94 (s, 1 H). MS obsd. (ESI+) [(M+H)+]: 497.
Example 29: 10-methoxy-2-oxo-6-(2-thienyl)-9-[3-(l,2,4-triazol-l-yl)propoxy]-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid

Step 1: Preparation of l-(3-brom ropyl)-l,2,4-triazole

To a solution of lH-l,2,4-triazole (1 g, 14.5 mmol) in DMF (10 mL) was added NaH (596 mg, 14.9 mmol) and the mixture was stirred for 15 min. Then to the mixture was added 1,3- dibromopropane (14.6 g, 72.4 mmol). The resulting mixture was stirred at 30 °C for 12 hrs, and then partitioned between H20 (150 mL) and EtOAc (200 mL). The organic layer was separated, washed with brine (150 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to afford crude l-(3- bromopropyl)-l,2,4-triazole (300 mg) as a yellow oil.
Step 2: Preparation of ethyl 10-methoxy-2-oxo-6-(2-thienyl)-9-[3-(l,2,4-triazol-l- yl)propoxy]-6,7-dihydrobenzo[a]quinolizine-3-carboxylate

To a solution of ethyl 9-hydroxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (150 mg, 0.377 mmol) in DMF (3 mL) was added K2C03 (104 mg, 754 μιηοΐ) and l-(3-bromopropyl)-l,2,4-triazole (107 mg, 0.566 mmol). The mixture was stirred at 30 °C for 12 hrs, and then partitioned between H20 (50 mL) and EtOAc
(100 mL). The organic layer was washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure to afford crude ethyl 10-methoxy-2-oxo-6-(2-thienyl)-9-[3- (l,2,4-triazol-l-yl)propoxy]-6,7-dihydrobenzo[a]quinolizine-3-carboxylate (200 mg) as a brown oil, which was used directly in the next step without further purification.
Step 3: Preparation of 10-methoxy-2-oxo-6-(2-thienyl)-9-[3-(l,2,4-triazol-l- yl)propoxy]-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid

To a solution of ethyl 10-methoxy-2-oxo-6-(2-thienyl)-9-[3-(l,2,4-triazol-l-yl)propoxy]- 6,7-dihydrobenzo[a]quinolizine-3-carboxylate (200 mg, 0.395 mmol) in MeOH (2 mL) was added 2 M NaOH aqueous solution (0.395 mL, 0.790 mmol). The mixture was stirred at 30 °C for 12 hrs, and then concentrated under reduced pressure. The residue was diluted with H20 (20 mL) and acidified with 1 M hydrochloric acid to pH=3. The resulting mixture was extracted with DCM (100 mL). The organic layer was washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by prep-HPLC to afford 10-methoxy-2-oxo-6-(2-thienyl)-9-[3-(l,2,4-triazol-l-yl)propoxy]-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid (61 mg) as a white solid. 1H NMR (400 MHz, DMSO-J6) δ: 2.28 (t, 2 H), 3.23 - 3.31 (m, 1 H), 3.71 (dd, 1 H), 3.90 (s, 3 H), 4.05 (d, 2 H), 4.35 (t, 2 H), 6.34 (d, 1 H), 6.91 (dd, 1 H), 6.99 - 7.08 (m, 2 H), 7.36 (dd, 1 H), 7.53 (d, 2 H), 7.98 (s, 1 H), 8.53 (s, 1 H), 8.99 (s, 1 H). MS obsd. (ESI+) [(M+H)+]: 479.
Example 30: 9-(3-carboxypropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid

Step 1: Preparation of ethyl 9-(4-ethoxy-4-oxo-butoxy)-10-methoxy-2-oxo-6-(2- thienyl)-6,7-dihydrobenzo[a] quinolizine-3-carboxylate

A mixture of ethyl 9-hydroxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (100 mg, 0.25 mmol), ethyl 4-bromobutanoate (74 mg, 0.38 mmol) and K2CO3 (52 mg, 0.38 mmol) in DMF (3 mL) was stirred at room temperature for 16 hrs. The mixture was partitioned between H20 and DCM. The organic layer was separated, washed with brine, dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was recrystallized from PE/EA to afford ethyl 9-(4-ethoxy-4-oxo-butoxy)-10-methoxy-2- oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate (60 mg) as a white solid.
Step 2: Preparation of 9-(3-carboxypropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid

A mixture of ethyl 9-(4-ethoxy-4-oxo-butoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (60 mg, 0.12 mmol) and NaOH (20 mg) in MeOH (2 mL) and H20 (0.1 mL) was stirred at room temperature for 16 hrs. The mixture was
concentrated under reduced pressure. The residue was diluted with H20 and acidified with 1 M hydrochloric acid to pH=2-3. The resulting mixture was filtered, and the filter cake was dried under reduced pressure to afford 9-(3-carboxypropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid (26 mg) as a white solid. ΧΗ NMR (400 MHz, DMSO-J6) δ: 1.96 (t, 2 H), 2.39 (t, 2 H), 3.36 (m, 1 H), 3.71 (d, 1 H), 3.88 (s, 3 H), 4.05 (br. s., 2 H), 6.35 (br. s., 1 H), 6.90 (br. s., 1 H), 7.05 (d, 2 H), 7.35 (d, 1 H), 7.51 (d, 2 H), 8.98 (s, 1 H), 12.17 (br. s., 1 H). MS obsd. (ESI+) [(M+H)+]: 456.
Example 31: 10-methoxy-9-(3-methylsulfanylpropoxy)-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
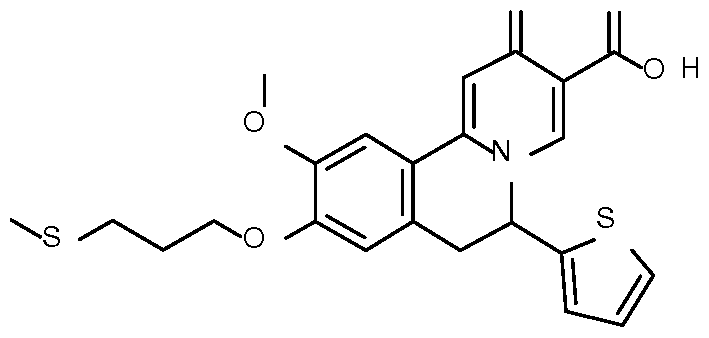
Step 1: Preparation of 3-methylsulfanylpropyl methanesulfonate

To a solution of 3-methylsulfanylpropan-l-ol (500 mg, 4.71 mmol) in DCM (5 mL) was added Et3N (2.38 g, 23.5 mmol) and methanesulfonyl chloride (2.86 g, 25.0 mmol). The mixture was stirred at 30 °C for 12 hrs, and then partitioned between H20 (20 mL) and DCM (60 mL). The organic layer was separated, washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure to afford crude 3-methylsulfanylpropyl methanesulfonate (600 mg) as a yellow oil, which was used directly in the next step without further purification.
Step 2: Preparation of ethyl 10-methoxy-9-(3-methylsulfanylpropoxy)-2-oxo-6-(2- thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate

To a solution of ethyl 9-hydroxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (100 mg, 0.251 mmol) in DMF (3 mL) was added K2C03 (69.6 mg, 0.503 mmol) and 3-methylsulfanylpropyl methanesulfonate (69.6 mg, 0.377 mmol). Then the mixture was stirred at 30 °C for 12 hrs, and then partitioned between H20 (50 mL) and EtOAc (100 mL). The organic layer was separated, washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure to afford crude ethyl 10- methoxy-9-(3-methylsulfanylpropoxy)-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylate (120 mg) as a yellow oil, which was used directly in the next step without further purification.
Step 3: Preparation of 10-methoxy-9-(3-methylsulfanylpropoxy)-2-oxo-6-(2-thienyl)- 6,7-dihydrobenzo[a] quinolizine-3-carboxylic acid

To a solution of ethyl 10-methoxy-9-(3-methylsulfanylpropoxy)-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (120 mg, 0.247 mmol) in MeOH (1 mL) was added 2 M NaOH aqueous solution (0.247 mL, 0.494 mmol). The mixture was stirred at 30 °C for 2 hrs, and then concentrated under reduced pressure. The residue was diluted with H20 (20 mL) and acidified with 1 M hydrochloric acid to pH=3. Then the resulting mixture was extracted with DCM (60 mL). The organic layer was washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by prep-HPLC to afford 10- methoxy-9-(3-methylsulfanylpropoxy)-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid (14.9 mg) as a white solid. 1H NMR (400 MHz, DMSO-J6) δ: 1.94 - 2.03 (m, 2 H), 2.04 - 2.10 (m, 3 H), 2.61 (t, 2 H), 3.41 - 3.52 (m, 1 H), 3.72 (dd, 1 H), 3.88 (s, 3 H), 4.11 (t, 2 H), 6.34 (d, 1 H), 6.90 (dd, 1 H), 6.99 - 7.15 (m, 2 H), 7.36 (dd, 1 H), 7.51 (d, 2 H), 8.98 (s, 1 H). MS obsd. (ESI+) [(M+H)+]: 458. Example 32: 10-methoxy-9-(3-methylsulfonylpropoxy)-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carbox lic acid
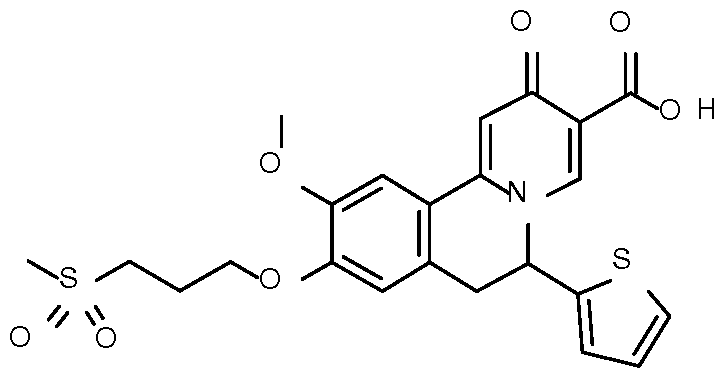
Step 1: Preparation of 3-methylsulfonylpropyl methanesulfonate
o o
To a solution of 3-methylsulfonylpropan-l-ol (200 mg, 1.45 mmol) in DCM (5 mL) was added Et3N (732 mg, 23.5 mmol) and methanesulfonyl chloride (2.84 g, 24.9 mmol). The mixture was stirred at 30 °C for 12 hrs, and then partitioned between H20 (20 mL) and DCM (60 mL). The organic layer was separated, washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure to afford crude 3-methylsulfonylpropyl
methane sulfonate (300 mg) as a yellow oil, which was used directly in the next step without further purification.
Step 2: Preparation of ethyl 10-methoxy-9-(3-methylsulfonylpropoxy)-2-oxo-6-(2- thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate

To a solution of ethyl 9-hydroxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (100 mg, 0.251 mmol) in DMF (3 mL) was added K2CO3 (69.6 mg, 0.503 mmol) and 3-methylsulfonylpropyl methane sulfonate (81.6 mg, 0.377 mmol). The mixture was stirred at 30 °C for 12 hrs, and then partitioned between H20 (50 mL) and EtOAc (100 mL). The organic layer was separated, washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure to afford crude ethyl 10-methoxy-9- (3-methylsulfonylpropoxy)-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate (90 mg) as a yellow oil, which was used directly in the next step without further purification.
Step 3: Preparation of 10-methoxy-9-(3-methylsulfonylpropoxy)-2-oxo-6-(2-thienyl)- 6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid

To a solution of ethyl 10-methoxy-9-(3-methylsulfonylpropoxy)-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (90 mg, 0.174 mmol) in MeOH (1 mL) was added 2 M NaOH aqueous solution (0.174 mL, 0.348 mmol). The mixture was stirred at 30 °C for 2 hrs, and then concentrated under reduced pressure. The residue was diluted with H20 (20 mL) and acidified with 1 M hydrochloric acid to pH=3. The resulting mixture was extracted with DCM (60 mL). The organic layer was washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by prep-HPLC to afford 10- methoxy-9-(3-methylsulfonylpropoxy)-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid (14.4 mg) as a yellow solid. 1H NMR (400 MHz, DMSO-J6) δ: 2.12 - 2.25 (m, 2 H) 3.03 (s, 3 H), 3.23 - 3.30 (m, 2 H), 3.34 (br. s., 1 H), 3.72 (dd, 1 H), 3.89 (s, 3 H), 4.16 (t, 2 H),
6.35 (br. s., 1 H), 6.86 - 6.94 (m, 1 H), 6.97 - 7.13 (m, 2 H), 7.35 (d, 1 H), 7.52 (d, 2 H), 8.98 (s, 1 H). MS obsd. (ESI+) [(M+H)+]: 490.
Example 33: 10-methoxy-9-methyl-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid

Step 1: Preparation of ethyl 10-methoxy-2-oxo-6-(2-thienyl)-9-
(trifluoromethylsulfonyloxy)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate
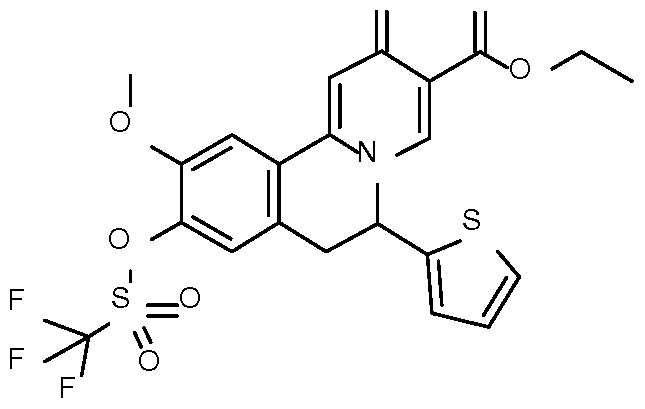
Step 2: Preparation of ethyl 10-methoxy-9-methyl-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylate
o o

Step 3: Preparation of 10-methoxy-9-methyl-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid

A mixture of ethyl 10-methoxy-9-methyl-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (310 mg, 0.78 mmol) and NaOH (63 mg) in MeOH (3 mL) and H20 (0.8 mL) was stirred at room temperature for 16 hrs. The mixture was
concentrated under reduced pressure. The residue was diluted with H20 and acidified with 1 M hydrochloric acid to pH=2-3. The resulting mixture was filtered. The filter cake was purified by prep-HPLC to afford 10-methoxy-9-methyl-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine- 3-carboxylic acid (26 mg) as a light yellow solid. 1H NMR (400 MHz, DMSO-J6) δ: 2.20 (s, 3 H), 3.31 (m, 1 H), 3.68-3.71 (m, 1 H), 3.91 (s, 3 H), 6.37 (br. s., 1 H), 6.90 (dd, 1 H), 7.00 (br. s., 1 H), 7.24 (s, 1 H), 7.36 (d, 1 H), 7.50 (s, 1 H) 7.62 (s, 1 H,) 9.02 (s, 1 H). MS obsd. (ESI+)
[(M+H)+]: 368.
Example 34: 9-ethyl-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine- 3-carboxylic acid
o o
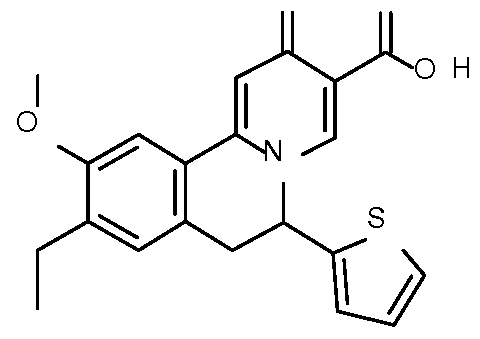
Step 1: Preparation of ethyl 9-ethyl-10-methoxy-2-oxo-6-(2-thienyl)-6,7-
dihydrobenzo[a]quinolizine-3-carboxylate

A mixture of ethyl 10-methoxy-2-oxo-6-(2-thienyl)-9-(trifluoromethylsulfonyloxy)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (200 mg, 0.38 mmol, from step 1 of Example 33), ethylboronic acid (42 mg, 0.57 mmol), K2C03 (157 mg, 1.13 mmol) and Pd(dppf)Ci2-CH2Cl2 (31 mg, 0.04 mmol) in THF/H20 (4 mL/1 mL) was heated at 60 °C for 12 hrs under nitrogen atmosphere. The mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography to afford crude ethyl 9-ethyl-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (140 mg) as a yellow oil, which was used directly in the next step without further purification.
Step 2: Preparation of 9-ethyl-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
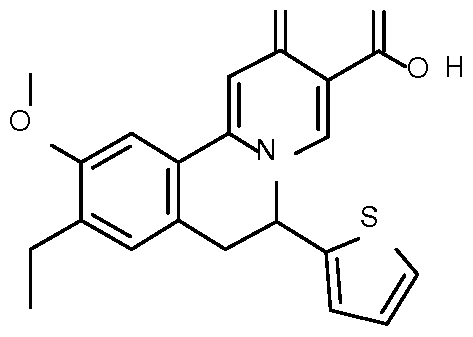
A mixture of ethyl 9-ethyl-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (140 mg, 0.34 mmol) and NaOH (27 mg) in H20 (0.3 mL) and MeOH (2 mL) was stirred at room temperature for 16 hrs. The mixture was
concentrated under reduced pressure. The residue was diluted with H20 and acidified with 1 M hydrochloric acid to pH=2-3. The resulting mixture was filtered. The filter cake was purified by prep-HPLC to afford 9-ethyl-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid (21 mg) as a light yellow solid. 1H NMR (400 MHz, DMSO-J6) δ: 1.15 (t, 3 H), 2.55 - 2.71 (m, 2 H), 3.37 (m, 1 H), 3.68-3.73 (m, 1 H),3.92 (s, 3 H), 6.37 (br. s., 1 H), 6.91 (dd, 1 H), 7.01 (br. s., 1 H), 7.24 (s, 1 H), 7.36 (d, 1 H,) 7.51 (s, 1 H), 7.62 (s, 1 H), 9.02 (s, 1 H). MS obsd. (ESI+) [(M+H)+]: 382. Example 35: 10-methoxy-2-oxo-9-propyl-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
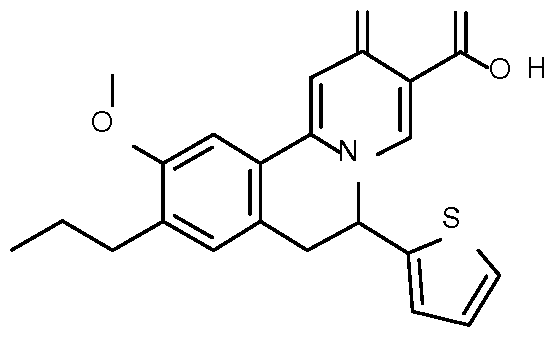
Step 1: Preparation of ethyl 10-methoxy-2-oxo-9-propyl-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate

A mixture of ethyl 10-methoxy-2-oxo-6-(2-thienyl)-9-(trifluoromethylsulfonyloxy)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (200 mg, 0.38 mmol), propylboronic acid (50 mg, 0.57 mmol), K2C03 (157 mg, 1.13 mmol) and Pd(dppf)Ci2-CH2Cl2 (31 mg, 0.04 mmol) in THF/H20 (4 mL/1 mL) was heated at 70 °C for 12 hrs under nitrogen atmosphere. The mixture was concentrated under reduced pressure. The residue was purified by flash column
chromatography to afford crude ethyl 10-methoxy-2-oxo-9-propyl-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (100 mg) as a brown solid, which was used directly in the next step without further purification.
Step 2: Preparation of 10-methoxy-2-oxo-9-propyl-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid

A mixture of ethyl 10-methoxy-2-oxo-9-propyl-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (100 mg, 0.24 mmol) and NaOH (19 mg) in H20 (0.2 mL) and MeOH (2 mL) was stirred at room temperature for 16 hrs. The mixture was
concentrated under reduced pressure. The residue was diluted with H20 and acidified with 1 M hydrochloric acid to pH=2-3. The resulting mixture was filtered. The filter cake was purified by prep-HPLC to afford 10-methoxy-2-oxo-9-propyl-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine- 3-carboxylic acid (15 mg) as a white solid. 1H NMR (400 MHz, DMSO-<¾5) δ: 0.89 (t, 3 H), 1.56
(sxt, 2 H), 2.53 - 2.62 (m, 2 H), 3.31 - 3.33 (m, 1 H), 3.65 - 3.74 (m, 1 H), 3.90 (s, 3 H), 6.36 (br. s., 1 H), 6.90 (dd, H), 7.02 (br. s., 1 H), 7.21 (s, 1 H), 7.35 (d, 1 H), 7.50 (s, 1 H), 7.61 (s, 1 H), 9.02 (s, 1 H). MS obsd. (ESI+) [(M+H)+]: 396. Example 36: 10-methoxy-2-oxo-9-pyrrolidin-l-yl-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid

Step 1: Preparation of ethyl 10-methoxy-2-oxo-9-pyrrolidin-l-yl-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate

To a solution of ethyl 10-methoxy-2-oxo-6-(2-thienyl)-9-(trifluoromethylsulfonyloxy)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate (100 mg, 0.189 mmol) in NMP (20 mL) was added pyrrolidine (67 mg, 0.944 mmol). The reaction vessel was sealed and heated in microwave reactor at 150 °C for 1 hr. Then the resulting mixture was purified by column chromatography to give crude product (3 g) as yellow oil, which was partitioned between H20 (50 mL) and EtOAc (100 mL). The organic layer was separated, washed with brine (50 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure to afford ethyl 10-methoxy-2-oxo-9- pyrrolidin-l-yl-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylate (60 mg) as a brown oil, which was used directly in the next step without further purification.
Step 2: Preparation of 10-methoxy-2-oxo-9-pyrrolidin-l-yl-6-(2-thienyl)-6,7- dihydrobenzo[a] quinolizine-3-carboxylic acid
o o

concentrated under reduced pressure. The residue was purified by prep-HPLC to afford 10- methoxy-2-oxo-9-pyrrolidin-l-yl-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid (13.8 mg) as a yellow solid. 1H NMR (400 MHz, DMSO-<¾5) δ: 1.78 - 1.92 (m, 4 H), 3.24 (br. s., 1 H) 3.44 (d, 4 H), 3.63 (dd, 1 H), 3.84 (s, 3 H), 6.29 (d, 1 H), 6.54 (s, 1 H), 6.91 (dd, 1 H), 7.02 (d, 1 H), 7.26 - 7.42 (m, 3 H), 8.90 (s, 1 H). MS obsd. (ESI+) [(M+H)+]: 423.
Example 37: 6-(benzothiophen-2-yl)-10-chloro-9-(3-methoxypropoxy)-2- dihydrobenzo[a] quinolizine- -carboxylic acid

Step 1: Preparation of 4-bromo-l-chloro-2-(3-methoxypropoxy)benzene

A mixture of 5-bromo-2-chloro-phenol (22.0 g, 106 mmol), l-bromo-3-methoxy-propane (19.5 g, 127 mmol) and K2C03 (30 g, 212 mmol) in DMF (50 mL) was heated at 50 °C for 3 hrs. Then the mixture was partitioned between ethyl acetate and water. The organic layer was separated, dried over anhydrous Na2S04 and concentrated under reduced pressure to give crude 4-bromo-l-chloro-2-(3-methoxypropoxy)benzene (30.0 g), which was used directly in the next step without further purification.
Step 2: Preparation of l-(benzothiophen-2-yl)-2-[4-chloro-3-(3- methoxypropoxy)phenyl]ethanone

To a mixture of crude 4-bromo-l-chloro-2-(3-methoxypropoxy)benzene (1.11 g, 4 mmol), tris(dibenzylideneacetone)dipalladium(0) (37 mg, 0.04 mmol), Xantphos (46 mg, 0.08 mmol) and i-BuONa (768 mg, 8 mmol) in THF (10 mL) was added l-(benzothiophen-2-yl)ethanone (1.41 g, 8 mmol). The resulting mixture was heated at 60 °C for 30 minutes under microwave. After being cooled to rt, the mixture was partitioned between water and EtOAc. The separated aqueous layer was extracted with EtOAc for three times. The combined organic layers were washed with brine, dried over anhydrous Na2S04, and concentrated under reduced pressure. The residue was purified by column chromatography to afford l-(benzothiophen-2-yl)-2-[4-chloro-3- (3-methoxypropoxy)phenyl]ethanone (1.41 g) as a brown oil.
Step 3: Preparation of l-(benzothiophen-2-yl)-2-[4-chloro-3-(3- methoxypropoxy)phenyl]ethanamine

To a mixture of l-(benzothiophen-2-yl)-2-[4-chloro-3-(3- methoxypropoxy)phenyl]ethanone (770 mg, 2 mmol) and ammonium acetate (2.31 g, 30 mmol) in methanol (10 mL) and dichloroethane (5 mL) was added NaB¾CN (252 mg, 4 mmol). The resulting mixture was heated at 65 °C for 16 hrs. After being cooled to rt, the reaction mixture was basified by 2 M NaOH aqueous solution to pH=12, and then diluted with water. The resulting mixture was extracted with DCM for three times. The combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated under reduced pressure to give crude l-(benzothiophen-2-yl)-2-[4-chloro-3-(3-methoxypropoxy)phenyl]ethanamine (739 mg) as a brown oil, which was used directly in the next step without further purification.
Step 4: Preparation of N-[l-(benzothiophen-2-yl)-2-[4-chloro-3-(3- methoxypropoxy)phenyl]ethyl]formamide
o

A mixture of crude l-(benzothiophen-2-yl)-2-[4-chloro-3-(3- methoxypropoxy)phenyl]ethanamine (739 mg, 2 mmol) and formic acid (0.1 mL) in ethyl formate (10 mL) was heated at 90 °C for 2 hrs. After being cooled to rt, the mixture was concentrated under reduced pressure to give N-[l-(benzothiophen-2-yl)-2-[4-chloro-3-(3- methoxypropoxy)phenyl] ethyl] formamide (756 mg) as a brown oil, which was used directly in the next step without further purification.
Step 5: Preparation of 3-(benzothiophen-2-yl)-7-chloro-6-(3-methoxypropoxy)-3,4- dihydroisoquinoline
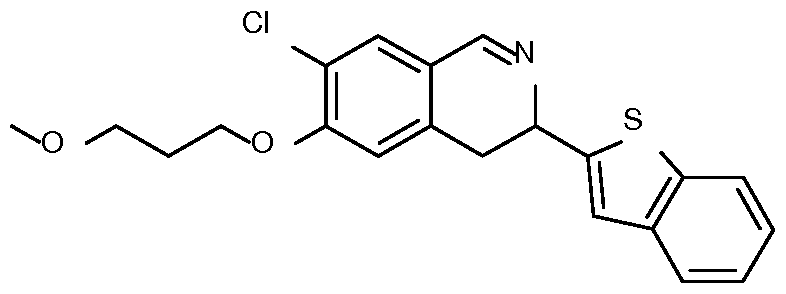
To a solution of N-[l-(benzothiophen-2-yl)-2-[4-chloro-3-(3- methoxypropoxy)phenyl] ethyl] formamide (756 mg, 1.9 mmol) in CH3CN (10 mL) was added POCI3 (291 mg, 1.9 mmol). The mixture was heated at 80 °C for 2 hrs. After being cooled to rt, the mixture was concentrated under reduced pressure. The residue was dissolved in CH3CN (10 mL) and then basified by ammonia water to pH=10 at 0 °C. The resulting mixture was extracted with DCM. The organic layer was washed with brine, dried over anhydrous Na2S04 and concentrated under reduced pressure to give crude 3-(benzothiophen-2-yl)-7-chloro-6-(3- methoxypropoxy)-3,4-dihydroisoquinoline (727 mg) as a brown oil, which was used directly in the next step without further purification.
Step 6: Preparation of ethyl 6-(benzothiophen-2-yl)-10-chloro-9-(3-methoxypropoxy)-
2-oxo-l,6,7,llb-tetrahydrobenzo[a]quinolizine-3-carboxylate
o o

Step 7: Preparation of ethyl 6-(benzothiophen-2-yl)-10-chloro-9-(3-methoxypropoxy)- 2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carbox late

A mixture of crude ethyl 6-(benzothiophen-2-yl)-10-chloro-9-(3-methoxypropoxy)-2-oxo-
1,6,7, l lb-tetrahydrobenzo[a]quinolizine-3-carboxylate (1.70 g, 1.9 mmol) and /?-chloranil (467 mg, 1.9 mmol) in DME (10 mL) was heated at 70 °C for 3 hrs. After being cooled to rt, the mixture was partitioned between DCM and water. The organic layer was washed with saturated NaHC03 aqueous solution (50 mL) 5 times and brine, dried over anhydrous Na2S04, and concentrated under reduced pressure to give crude ethyl 6-(benzothiophen-2-yl)-10-chloro-9-(3- methoxypropoxy)-2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylate (2.01 g) as a brown oil, which was used directly in next step without further purification.
Step 8: Preparation of 6-(benzothiophen-2-yl)-10-chloro-9-(3-methoxypropoxy)-2- oxo-6,7-dihydrobenzo[a] quinolizine-3-carbox lic acid
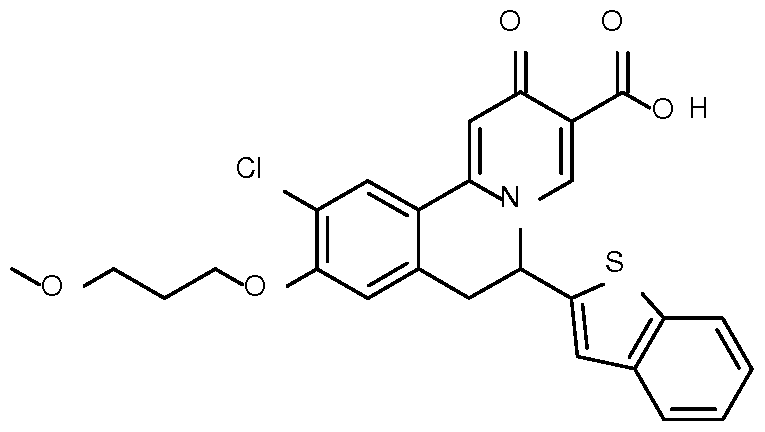
To a mixture of crude ethyl 6-(benzothiophen-2-yl)-10-chloro-9-(3-methoxypropoxy)-2- oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylate (2.01 g, 1.9 mmol) in methanol (16 mL) and H20 (4 mL) was added LiOH H20 (958 mg, 22.8 mmol). The resulting mixture was stirred at rt overnight, and then acidified by 1 M hydrochloric acid to pH=2-3. The mixture was extracted
with DCM for three times. The combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was purified by column chromatography to give crude product as a brown solid, which was recrystallized from diethyl ether and EtOH to afford 6-(benzothiophen-2-yl)-10-chloro-9-(3-methoxypropoxy)-2- oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid (80 mg) as a light-brown solid. 1HNMR (400MHz, OMSO-d6) δ: 8.95 (s, 1H), 8.14 (s, 1H), 7.81 - 7.76 (m, 1H), 7.75 - 7.69 (m, 1H), 7.37 (s, 1H), 7.34 - 7.28 (m, 2H), 7.24 (s, 1H), 6.40 (s, 1H), 4.23 - 4.09 (m, 2H), 3.85 - 3.75 (m, 1H), 3.60 - 3.50 (m, 1H), 3.49 - 3.41 (m, 2H), 3.20 (m, 3H), 2.00 - 1.88 (m, 2H). MS obsd. (ESI+) [(M+H)+]: 496.
BIOLOGICAL EXAMPLES
Example 38 materials and methods
HBV cell line
HepG2.2.15 cells (Acs et al. Proc Natl Acad Sci U S A, 84, (1987), 4641-4), a
constitutively HBV-expressing cell line were cultured in DMEM+Glutamax-I medium
(Invitrogen, Carlsbad, CA, USA), supplemented with 10% fetal bovine serum (Invitrogen) and G418 (Invitrogen) at a final concentration of 200 mg/L and maintained in 5% C02 at 37°C.
HBsAg Assay
HepG2.2.15 cells were seeded in duplicate into white, 96-well plates at 1.5 x 104 cells/well.
The cells were treated with a three-fold serial dilution series of the compounds in DMSO. The final DMSO concentration in all wells was 1% and DMSO was used as no drug control.
The HBsAg chemiluminescence immunoassay (CLIA) kit (Autobio Diagnostics Co., Zhengzhou, China, Catalog number: CL0310-2) was used to measure the levels of secreted HBV antigens semi-quantitatively. For the detection 50 μΕΛνεΙΙ culture supernatant was used and HBsAg was quantified using HBsAg chemiluminescence immunoassay (CLIA) kit (Autobio Diagnostics Co., Zhengzhou, China, Catalog number: CL0310-2), 50 μΐ^ of the supernatant was transferred to the CLIA assay plate and 50 μΐ^ of enzyme conjugate reagent was added into each well. The plates were sealed and gently agitated for 1 hour at room temperature. The
supernatant-enzyme-mixture was discarded and wells were washed 6 times with 300 μΐ^ of PBS. The residual liquid was removed by plating the CLIA plate right side down on absorbent tissue paper. 25 μΐ^ of substrates A and B were added to each well. Luminance was measured using a
luminometer (Mithras LB 940 Multimode Microplate Reader) after 10 minutes incubation. Dose- response curves were generated and the IC50 value was extrapolated by using the E-WorkBook Suite (ID Business Solutions Ltd., Guildford, UK). The IC50 was defined as the compound concentration (or conditioned media log dilution) at which HBsAg secretion was reduced by 50% compared to the no drug control.
The compounds of the present invention were tested for their activity to inhibit HBsAg as described herein. The Examples were tested in the above assay and found to have ICso below 25.0 μΜ. Particular compounds of formula I were found to have ICso below 0.100 μΜ. More Particular compounds of formula I were found to have ICso below 0.010 μΜ. Results of HBsAg assay are given in Table 1.
Table 1 : Activity data of particular compounds

HBV DNA assay
The assay employs real-time qPCR (TaqMan) to directly measure extracellular HBV DNA copy number. HepG2.2.15 cells were plated in 96- well micro titer plates. Only the interior wells were utilized to reduce "edge effects" observed during cell culture, the exterior wells were filled with complete medium to help minimize sample evaporation. On the following day, the
HepG2.2.15 cells were washed and the medium was replaced with complete medium containing
various concentrations of a test compound in triplicate. 3TC was used as the positive control, while media alone was added to cells as a negative control (virus control, VC). Three days later, the culture medium was replaced with fresh medium containing the appropriately diluted drug. Six days following the initial administration of the test compound, the cell culture supernatant was collected, treated with pronase and then used in a real-time qPCR/TaqMan assay to determine HBV DNA copy numbers. Antiviral activity was calculated from the reduction in HBV DNA levels (IC50).
The compounds of the present invention were tested for their activity to anti HBV DNA production as described herein. The Examples were tested in the above assay and found to have ICso below 25.0 μΜ. Particular compounds of formula I were found to have ICso below 0.10 μΜ. Results of HBV DNA assay are given in Table 2.
Table 2: Anti HBV DNA production activity in HepG2.2.15 cells
Example No. IC50 (μΜ)
7 <0.032
8 <0.032
15 <0.032
Claims
1. A compound of formula I

R1, R2, R3 and R4 are independently selected from Ci_6alkyl, haloCi_6alkyl, hydrogen, halogen, cyano, amino, Ci_6alkylamino, diCi_6alkylamino, pyrrolidinyl and OR6;
R5 is hydrogen or Ci_6alkyl;
R6 is hydrogen; Ci-6alkyl; haloCi_6alkyl; C3_7cycloalkylCi-6alkyl; phenylCi-6alkyl;
hydroxyCi_6alkyl; Ci-6alkoxyCi_6alkyl; carboxyCi_6alkyl; Ci_6alkylsulfanylCi-6alkyl; Ci_ 6alkylsulfonylCi_6alkyl; cyanoCi_6alkyl; aminoCi_6alkyl; Ci_6alkylaminoCi-6alkyl; diCi_ 6 alkylaminoC i _6 alkyl; C i _6 alkylcarbonylaminoC i _6 alkyl; C i _6 alkylsulfonylaminoC i_6 alkyl; Ci_6alkoxycarbonylaminoCi-6alkyl; pyrazolylCi_6alkyl; triazolylCi_6 alkyl or
heterocycloalkylCi-6alkyl, wherein heterocycloalkyl is N-containing monocyclic heterocycloalkyl;
Ar is phenyl; phenyl substituted by one, two or three groups independently selected from Ci_ 6alkyl,
 hydroxy, halogen and thienyl; thienyl substituted by one, two or three groups independently selected from
hydroxy, halogen and thienyl; thienyl substituted by one, two or three groups independently selected from
 hydroxy, halogen and phenylC i_6 alkoxy; benzothiophenyl; benzothiophenyl substituted by one, two or three groups independently selected from Ci_6alkyl,
hydroxy, halogen and phenylC i_6 alkoxy; benzothiophenyl; benzothiophenyl substituted by one, two or three groups independently selected from Ci_6alkyl,
 hydroxy, halogen and phenylC 1-6 alkoxy; pyridinyl; pyridinyl substituted by one, two or three groups
hydroxy, halogen and phenylC 1-6 alkoxy; pyridinyl; pyridinyl substituted by one, two or three groups
independently selected from Ci-6 alkyl, Ci-6alkoxy, hydroxy, halogen and phenylCi- 6alkoxy; pyrimidinyl; pyrimidinyl substituted by one, two or three groups independently selected from Ci_6alkyl, Ci-6alkoxy, hydroxy, halogen and phenylCi-6 alkoxy; pyrrolyl; pyrrolyl substituted by one, two or three groups independently selected from Ci_6alkyl, Ci_ 6alkoxy, hydroxy, halogen and phenylCi-6 alkoxy; pyrazolyl; pyrazolyl substituted by one, two or three groups independently selected from Ci-6 alkyl, Ci-6alkoxy, hydroxy, halogen and phenylC 1-6 alkoxy; thiazolyl; or thiazolyl substituted by one, two or three groups
independently selected from C1-6alkyl,
 hydroxy, halogen and phenylCi- 6alkoxy;
hydroxy, halogen and phenylCi- 6alkoxy;
or pharmaceutically acceptable salts, or enantiomers thereof.
2. A compound according to claim 1, wherein
R1 is hydrogen;
R is hydrogen, halogen or Ci-6alkoxy;
R3 is Chalky!, pyrrolidinyl or OR6;
R4 is C 1-6 alkyl or hydrogen;
R5 is hydrogen;
R6 is Ci_6alkyl, haloCi_6 alkyl, C3_7cycloalkylCi-6alkyl, phenylCi_6 alkyl, hydroxyCi_6 alkyl, Ci-6alkoxyCi_6alkyl, carboxyCi_6 alkyl, Ci_6alkylsulfanylCi_6 alkyl, Ci_6alkylsulfonylCi_ 6alkyl, cyanoCi_6 alkyl, aminoCi-6 alkyl, Ci_6alkylcarbonylaminoCi_6alkyl, Ci_
6 alkylsulfonylaminoC i -6 alkyl, C i -6 alkoxycarbonylaminoC i -6 alkyl, pyrazolylC i -6 alkyl, triazolylC 1-6 alkyl, piperidylCi-6 alkyl, morpholinylCi-6 alkyl or 2-oxopyrrolidinylCi_6 alkyl;
Ar is phenyl; phenyl substituted by Ci_6alkyl, Ci-6alkoxy, hydroxy, halogen or phenylCi- 6alkoxy; thienyl; thienyl substituted by C1-6 alkyl; or benzothiophenyl;
or pharmaceutically acceptable salts, or enantiomers thereof.
3. A compound according to claim 1 or 2, or pharmaceutically acceptable salts, or
enantiomers thereof, wherein R1 is hydrogen, R4 is hydrogen, R5 is hydrogen.
4. A compound according to any one of claims 1 to 3, or pharmaceutically acceptable salts, or enantiomers thereof, wherein R is Ci_6alkoxy.
5. A compound according to any one of claims 1 to 4, or pharmaceutically acceptable salts, or enantiomers thereof, wherein R is methoxy.
6. A compound according to any one of claims 1 to 5, or pharmaceutically acceptable salts, or enantiomers thereof, wherein R3 is pyrrolidinyl or OR6, wherein R6 is Ci_6alkyl, haloCi_6 alkyl, C3-7cycloalkylCi_6alkyl, hydro xyCi_6 alkyl, Ci_6alkoxyCi_6alkyl,
 Ci_
Ci_
6alkylsulfanylCi_6alkyl, cyanoCi_6 alkyl, aminoCi-6 alkyl, Ci-6alkylcarbonylaminoCi_6alkyl, Ci_ 6alkylsulfonylaminoCi_6alkyl or Ci_6alkoxycarbonylaminoCi_6alkyl.
7. A compound according to any one of claims 1 to 6, or pharmaceutically acceptable salts, or enantiomers thereof, wherein R3 is OR6, wherein R6 is methyl, isobutyl, trifluoroethyl, cyclopropylmethyl, cyanopropyl, hydroxypropyl, hydroxyhexyl, hydroxydimethylpropyl, methoxypropyl, carboxypropyl, methylsulfanylpropyl, aminohexyl, methylcarbonylaminohexyl, methylsulfonylaminohexyl or methoxycarbonylaminohexyl.
8. A compound according to any one of claims 1 to 7, or pharmaceutically acceptable salts, or enantiomers thereof, wherein Ar is phenyl, phenyl substituted by Ci_6alkyl or halogen, thienyl or thienyl substituted by Ci_6alkyl.
9. A compound according to any one of claims 1 to 8, or pharmaceutically acceptable salts, or enantiomers thereof, wherein Ar is phenyl; phenyl substituted by methyl, fluoro or chloro;
thienyl or thienyl substituted by methyl.
10. A compound according to claim 1 or 2, wherein
R1 is hydrogen;
R2 is Ci-6alkoxy;
R3 is pyrrolidinyl or OR6;
R4 is hydrogen;
R5 is hydrogen;
R6 is Ci_6alkyl, haloCi_6alkyl, C3_7cycloalkylCi_6alkyl, hydroxyCi_6alkyl, Ci_6alkoxyCi_6alkyl or Ci-6alkylsulfanylCi-6alkyl;
Ar is phenyl substituted by Ci_6alkyl, or thienyl;
or ph armaceutically acceptable salts, or enantiomers thereof.
11. A compound according to claim 1, 2 or 10, wherein
R1 is hydrogen;
R2 is methoxy;
R3 is pyrrolidinyl or OR6;
R4 is hydrogen;
R5 is hydrogen;
R6 is isobutyl, trifluoroethyl, cyclopropylmethyl, hydroxydimethylpropyl, methoxypropyl or methylsulfanylpropyl;
Ar is phenyl substituted by methyl or thienyl;
or pharmaceutically acceptable salts, or enantiomers thereof.
12. A compound according to claim 1, selected from
10-Methoxy-9-(3-methoxypropoxy)-2-oxo-6-phenyl-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid;
10-Methoxy-9-(3-methoxypropoxy)-2-oxo-6-(3-thienyl)-6,7-dihydrobenzo[a]quinolizine- 3-carboxylic acid;
10-Methoxy-9-(3-methoxypropoxy)-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine- 3-carboxylic acid;
6-(4-Hydroxyphenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo-6,7- dihydrobenzo [a] quino lizine- 3-carboxylic acid ;
10-Methoxy-6-(4-methoxyphenyl)-9-(3-methoxypropoxy)-2-oxo-6,7- dihydrobenzo [a] quino lizine- 3-carboxylic acid ;
6-(4-Benzyloxyphenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo-6,7- dihydrobenzo [a] quino lizine- 3-carboxylic acid ;
10-Methoxy-9-(3-methoxypropoxy)-6-(4-methyl-2-thienyl)-2-oxo-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid;
6-(3-Chlorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo-6,7- dihydrobenzo [a] quino lizine- 3-carboxylic acid ;
6-(4-Fluorophenyl)-10-methoxy-9-(3-methoxypropoxy)-2-oxo-6,7- dihydrobenzo [a] quino lizine- 3-carboxylic acid ;
10-Methoxy-9-(3-methoxypropoxy)-6-(o-tolyl)-2-oxo-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid;
9-Methoxy-8-methyl-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid;
9-Benzyloxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid;
9,10-Dimethoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid; 9-Isobutoxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid;
9- (Cyclopropylmethoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- di ydrobenzo [a] quino lizine- 3 -carboxylic acid ;
10- Methoxy-2-oxo-9-(3-pyrazol-l-ylpropoxy)-6-(2-thienyl)-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
9-(3-Hydroxypropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine- 3-carboxylic acid;
10-Methoxy-2-oxo-9-[3-(l-piperidyl)propoxy]-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid formate;
9- (3-Cyanopropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid;
10- Methoxy-2-oxo-9-[2-(2-oxopyrrolidin-l-yl)ethoxy]-6-(2-thienyl)-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
9-[6-(ieri-Butoxycarbonylamino)hexoxy]-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
9-(6-Aminohexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid formate;
9-(6-Acetamidohexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine- 3-carboxylic acid;
9-[6-(Methanesulfonamido)hexoxy]-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid;
9-(6-Hydroxyhexoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid;
9- (3-Hydroxy-2,2-dimethyl-propoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
10-Methoxy-2-oxo-6-(2-thienyl)-9-(2,2,2-trifluoroethoxy)-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
10- Methoxy-9-(3-morpholinopropoxy)-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo[a]quinolizine-3-carboxylate;
10-Methoxy-2-oxo-6-(2-thienyl)-9-[3-(l,2,4-triazol-l-yl)propoxy]-6,7- dihydrobenzo[a]quinolizine-3-carboxylic acid;
9-(3-Carboxypropoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine- 3-carboxylic acid;
10-Methoxy-9-(3-methylsulfanylpropoxy)-2-oxo-6-(2-thienyl)-6,7- di ydrobenzo [a] quino lizine- 3 -carboxylic acid ;
10-Methoxy-9-(3-methylsulfonylpropoxy)-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
10-Methoxy-9-methyl-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid;
9- Ethyl-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid;
10- Methoxy-2-oxo-9-propyl-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid;
10-Methoxy-2-oxo-9-pyrrolidin-l-yl-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid;
6-(Benzothiophen-2-yl)-10-chloro-9-(3-methoxypropoxy)-2-oxo-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
or pharmaceutically acceptable salts, or enantiomers thereof.
13. A compound according to claim 1, selected from
10-Methoxy-9-(3-methoxypropoxy)-2-oxo-6-(3-thienyl)-6,7-dihydrobenzo[a]quinolizine- 3-carboxylic acid;
10-Methoxy-9-(3-methoxypropoxy)-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine- 3-carboxylic acid;
10-Methoxy-9-(3-methoxypropoxy)-6-(o-tolyl)-2-oxo-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid;
9-Isobutoxy-10-methoxy-2-oxo-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid;
9-(Cyclopropylmethoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
9- (3-Hydroxy-2,2-dimethyl-propoxy)-10-methoxy-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
10-Methoxy-2-oxo-6-(2-thienyl)-9-(2,2,2-trifluoroethoxy)-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
10- Methoxy-9-(3-methylsulfanylpropoxy)-2-oxo-6-(2-thienyl)-6,7- dihydrobenzo [a] quino lizine- 3 -carboxylic acid ;
10-Methoxy-2-oxo-9-pyrrolidin-l-yl-6-(2-thienyl)-6,7-dihydrobenzo[a]quinolizine-3- carboxylic acid;
or pharmaceutically acceptable salts, or enantiomers thereof.
14. A process for the preparation of a compound according to any one of claims 1 to 13 comprising
(a) hydrolysis of a compound of formula (A)

(b) hydrolysis of a compound of formula (B)
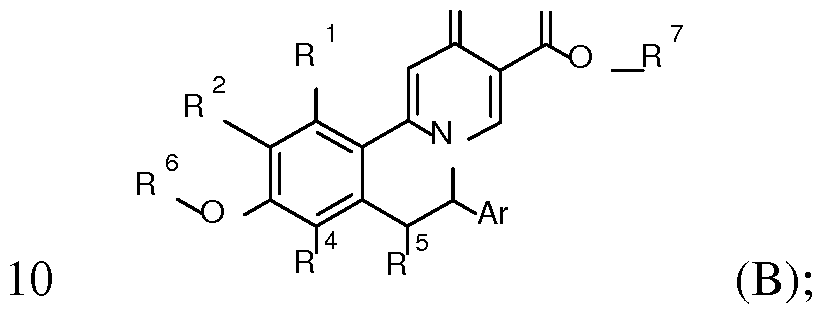
wherein R 1 to R 7 and Ar are defined as in any one of claims 1 to 11.
15. A compound according to any one of claims 1 to 13 for use as therapeutically active substance.
16. A pharmaceutical composition comprising a compound in accordance with any one of claims 1 to 13 and a therapeutically inert carrier.
17. The use of a compound according to any one of claims 1 to 13 for the treatment or prophylaxis of HBV infection.
18. The use of a compound according to any one of claims 1 to 13 for the preparation of a medicament for the treatment or prophylaxis of HBV infection.
19. The use of a compound according to any one of claims 1 to 13 for the inhibition of HBsAg production or secretion, or for the the inhibition of HBsAg production or secretion.
20. A compound according to any one of claims 1 to 13 for the treatment or prophylaxis of HBV infection.
21. A compound according to any one of claims 1 to 13, when manufactured according to a process of claim 14.
22. A method for the treatment or prophylaxis of HBV infection, which method comprises administering an effective amount of a compound as defined in any one of claims 1 to 13.
23. The invention as hereinbefore described.
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201680008772.7A CN107207505B (en) | 2015-02-11 | 2016-02-08 | Treat and prevent hepatitis b virus infected 2- oxo -6,7- dihydrobenzo [a] quinolizine -3- formic acid derivates |
| HK18103815.7A HK1244281B (en) | 2015-02-11 | 2016-02-08 | 2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid derivatives for the treatment and prophylaxis of hepatitis b virus infection |
| EP16703311.7A EP3256471B1 (en) | 2015-02-11 | 2016-02-08 | Novel 2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid derivatives for the treatment and prophylaxis of hepatitis b virus infection |
| JP2017542045A JP6435054B2 (en) | 2015-02-11 | 2016-02-08 | Novel 2-oxo-6,7-dihydrobenzo [a] quinolidine-3-carboxylic acid derivatives for the treatment and prevention of hepatitis B virus infection |
| US15/674,810 US10093671B2 (en) | 2015-02-11 | 2017-08-11 | 2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid derivatives for the treatment and prophylaxis of hepatitis B virus infection |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CNPCT/CN2015/072741 | 2015-02-11 | ||
| CN2015072741 | 2015-02-11 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/674,810 Continuation US10093671B2 (en) | 2015-02-11 | 2017-08-11 | 2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid derivatives for the treatment and prophylaxis of hepatitis B virus infection |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016128335A1 true WO2016128335A1 (en) | 2016-08-18 |
Family
ID=55310820
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2016/052585 Ceased WO2016128335A1 (en) | 2015-02-11 | 2016-02-08 | Novel 2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid derivatives for the treatment and prophylaxis of hepatitis b virus infection |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US10093671B2 (en) |
| EP (1) | EP3256471B1 (en) |
| JP (1) | JP6435054B2 (en) |
| CN (1) | CN107207505B (en) |
| HK (1) | HK1244281B (en) |
| WO (1) | WO2016128335A1 (en) |
Cited By (123)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017140821A1 (en) | 2016-02-19 | 2017-08-24 | Novartis Ag | Tetracyclic pyridone compounds as antivirals |
| US20170342068A1 (en) | 2016-05-27 | 2017-11-30 | Gilead Sciences, Inc. | Compounds for the treatment of hepatitis b virus infection |
| US9845322B2 (en) | 2015-05-04 | 2017-12-19 | Hoffmann-La Roche Inc. | Tetrahydropyridopyrimidines and tetrahydropyridopyridines for the treatment and prophylaxis of hepatitis B virus infection |
| WO2018045144A1 (en) | 2016-09-02 | 2018-03-08 | Gilead Sciences, Inc. | Toll like receptor modulator compounds |
| WO2018045150A1 (en) | 2016-09-02 | 2018-03-08 | Gilead Sciences, Inc. | 4,6-diamino-pyrido[3,2-d]pyrimidine derivaties as toll like receptor modulators |
| WO2018073753A1 (en) * | 2016-10-18 | 2018-04-26 | Novartis Ag | Fused tetracyclic pyridone compounds as antivirals |
| WO2018083136A1 (en) | 2016-11-03 | 2018-05-11 | F. Hoffmann-La Roche Ag | Novel tetrahydroisoquinolines and terahydronaphthyridines for the treatment and prophylaxis of hepatitis b virus infection |
| WO2018083081A1 (en) | 2016-11-03 | 2018-05-11 | F. Hoffmann-La Roche Ag | Novel tetrahydropyridopyrimidines for the treatment and prophylaxis of hepatitis b virus infection |
| WO2018083106A1 (en) | 2016-11-03 | 2018-05-11 | F. Hoffmann-La Roche Ag | Novel tetrahydropyridopyrimidines for the treatment and prophylaxis of hepatitis b virus infection |
| WO2018085619A1 (en) | 2016-11-07 | 2018-05-11 | Arbutus Biopharma, Inc. | Substituted pyridinone-containing tricyclic compounds, and methods using same |
| WO2018087345A1 (en) | 2016-11-14 | 2018-05-17 | F. Hoffmann-La Roche Ag | COMBINATION THERAPY OF AN HBsAg INHIBITOR, A NUCLEOS(T)IDE ANALOGUE AND AN INTERFERON |
| WO2018144390A1 (en) | 2017-01-31 | 2018-08-09 | Gilead Sciences, Inc. | Crystalline forms of tenofovir alafenamide |
| WO2018154466A1 (en) * | 2017-02-21 | 2018-08-30 | Glaxosmithkline Intellectual Property Development Limited | Dihydroquinolizinones as antivirals |
| WO2018161960A1 (en) | 2017-03-09 | 2018-09-13 | 福建广生堂药业股份有限公司 | Hepatitis b virus surface antigen inhibitor |
| CN108530449A (en) * | 2017-05-22 | 2018-09-14 | 河南春风医药科技有限公司 | For treating or preventing hepatitis b virus infected compound and the preparation method and application thereof |
| WO2018172852A1 (en) | 2017-03-21 | 2018-09-27 | Arbutus Biopharma Corporation | Substituted dihydroindene-4-carboxamides and analogs thereof, and methods using same |
| WO2018195321A1 (en) | 2017-04-20 | 2018-10-25 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2018219356A1 (en) * | 2017-06-01 | 2018-12-06 | Sunshine Lake Pharma Co., Ltd. | Fused tricyclic compounds and uses thereof in medicine |
| CN108976223A (en) * | 2017-06-01 | 2018-12-11 | 广东东阳光药业有限公司 | Fused tricyclic class compound and its application in drug |
| US10150740B2 (en) | 2015-07-28 | 2018-12-11 | Hoffmann-La Roche Inc. | 6,7-dihydropyrido[2,1-A]phthalazin-2-ones for the treatment and prophylaxis of hepatitis B virus infection |
| US10189846B2 (en) | 2016-06-10 | 2019-01-29 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| US10239872B2 (en) | 2016-07-29 | 2019-03-26 | Newave Pharmaceutical Inc. | Therapeutic agents for the treatment of HBV infection |
| US10301312B2 (en) | 2017-04-27 | 2019-05-28 | Novartis Ag | Fused indazole pyridone compounds as antivirals |
| WO2019110352A1 (en) * | 2017-12-04 | 2019-06-13 | Galapagos Nv | 2-oxo-5h-chromeno[4,3-b]pyridines for use in the treatment of hepatitis b |
| WO2019123285A1 (en) * | 2017-12-20 | 2019-06-27 | Novartis Ag | Fused tricyclic pyrazolo-dihydropyrazinyl-pyridone compounds as antivirals |
| US10336751B2 (en) | 2015-07-21 | 2019-07-02 | Hoffmann-La Roche Inc. | Tricyclic 4-pyridone-3-carboxylic acid derivatives for the treatment and prophylaxis of hepatitis B virus infection |
| EP3492467A4 (en) * | 2016-07-29 | 2019-07-24 | Ginkgo Pharma Co., Ltd. | ISOQUINOLINONE COMPOUND AND USE THEREOF IN THE PREPARATION OF AN ANTIVIRAL DRUG |
| WO2019160882A1 (en) | 2018-02-13 | 2019-08-22 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2019165374A1 (en) | 2018-02-26 | 2019-08-29 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds as hbv replication inhibitors |
| US10428070B2 (en) | 2017-12-06 | 2019-10-01 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| WO2019193543A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotides |
| WO2019193542A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotides |
| WO2019193533A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'2'-cyclic dinucleotides |
| WO2019195181A1 (en) | 2018-04-05 | 2019-10-10 | Gilead Sciences, Inc. | Antibodies and fragments thereof that bind hepatitis b virus protein x |
| US10442804B2 (en) | 2017-02-02 | 2019-10-15 | Gilead Sciences, Inc. | Compounds for the treatment of hepatitis B virus infection |
| WO2019200247A1 (en) | 2018-04-12 | 2019-10-17 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the hepatitis b virus genome |
| WO2019204609A1 (en) | 2018-04-19 | 2019-10-24 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2019211799A1 (en) | 2018-05-03 | 2019-11-07 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide |
| CN110475775A (en) * | 2017-03-31 | 2019-11-19 | 富士胶片株式会社 | 4- pyridinone compounds or its salt, pharmaceutical composition and agent comprising 4- pyridinone compounds |
| WO2020014643A1 (en) | 2018-07-13 | 2020-01-16 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2020023710A1 (en) | 2018-07-27 | 2020-01-30 | Arbutus Biopharma Corporation | Substituted tetrahydrocyclopenta[c]pyrroles, substituted dihydropyrrolizines, analogues thereof, and methods using same |
| WO2020028097A1 (en) | 2018-08-01 | 2020-02-06 | Gilead Sciences, Inc. | Solid forms of (r)-11-(methoxymethyl)-12-(3-methoxypropoxy)-3,3-dimethyl-8-0x0-2,3,8,13b-tetrahydro-1h-pyrido[2,1-a]pyrrolo[1,2-c] phthalazine-7-c arboxylic acid |
| WO2020061435A1 (en) | 2018-09-21 | 2020-03-26 | Enanta Pharmaceuticals, Inc. | Functionalized heterocycles as antiviral agents |
| CN110950860A (en) * | 2018-09-26 | 2020-04-03 | 广东东阳光药业有限公司 | Condensed tricyclic compounds and their application in medicine |
| WO2020086556A1 (en) | 2018-10-24 | 2020-04-30 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2020092528A1 (en) | 2018-10-31 | 2020-05-07 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds having hpk1 inhibitory activity |
| WO2020092621A1 (en) | 2018-10-31 | 2020-05-07 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds as hpk1 inhibitors |
| US10662416B2 (en) | 2016-10-14 | 2020-05-26 | Precision Biosciences, Inc. | Engineered meganucleases specific for recognition sequences in the hepatitis B virus genome |
| WO2020123674A1 (en) | 2018-12-12 | 2020-06-18 | Arbutus Biopharma Corporation | Substituted arylmethylureas and heteroarylmethylureas, analogues thereof, and methods using same |
| US10702528B2 (en) | 2015-07-13 | 2020-07-07 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| US10723733B2 (en) | 2017-12-06 | 2020-07-28 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| US10729688B2 (en) | 2018-03-29 | 2020-08-04 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| WO2020178770A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotides and prodrugs thereof |
| WO2020178768A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide as sting modulator |
| WO2020178769A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotides and prodrugs thereof |
| WO2020214652A1 (en) | 2019-04-17 | 2020-10-22 | Gilead Sciences, Inc. | Solid forms of a toll-like receptor modulator |
| WO2020214663A1 (en) | 2019-04-17 | 2020-10-22 | Gilead Sciences, Inc. | Solid forms of a toll-like receptor modulator |
| WO2020214716A1 (en) | 2019-04-17 | 2020-10-22 | Gilead Sciences, Inc. | 2-imino-5-oxo-imidazolidine inhibitors of hiv protease |
| WO2020221074A1 (en) | 2019-04-30 | 2020-11-05 | 南京海璞医药科技有限公司 | Compound containing fused ring, use thereof and pharmaceutical composition containing same |
| WO2020237025A1 (en) | 2019-05-23 | 2020-11-26 | Gilead Sciences, Inc. | Substituted exo-methylene-oxindoles which are hpk1/map4k1 inhibitors |
| WO2020263830A1 (en) | 2019-06-25 | 2020-12-30 | Gilead Sciences, Inc. | Flt3l-fc fusion proteins and methods of use |
| WO2021011891A1 (en) | 2019-07-18 | 2021-01-21 | Gilead Sciences, Inc. | Long-acting formulations of tenofovir alafenamide |
| WO2021034804A1 (en) | 2019-08-19 | 2021-02-25 | Gilead Sciences, Inc. | Pharmaceutical formulations of tenofovir alafenamide |
| US10934306B2 (en) | 2016-03-07 | 2021-03-02 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| US10966999B2 (en) | 2017-12-20 | 2021-04-06 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3′3′ cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| WO2021067181A1 (en) | 2019-09-30 | 2021-04-08 | Gilead Sciences, Inc. | Hbv vaccines and methods treating hbv |
| US11007208B2 (en) | 2015-09-16 | 2021-05-18 | Gilead Sciences, Inc. | Methods for treating arenaviridae and coronaviridae virus infections |
| WO2021113765A1 (en) | 2019-12-06 | 2021-06-10 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the hepatitis b virus genome |
| WO2021130638A1 (en) | 2019-12-24 | 2021-07-01 | Carna Biosciences, Inc. | Diacylglycerol kinase modulating compounds |
| US11058678B2 (en) | 2018-01-22 | 2021-07-13 | Enanta Pharmaceuticals, Inc. | Substituted heterocycles as antiviral agents |
| WO2021154687A1 (en) | 2020-01-27 | 2021-08-05 | Gilead Sciences, Inc. | Methods for treating sars cov-2 infections |
| WO2021188959A1 (en) | 2020-03-20 | 2021-09-23 | Gilead Sciences, Inc. | Prodrugs of 4'-c-substituted-2-halo-2'-deoxyadenosine nucleosides and methods of making and using the same |
| WO2021207049A1 (en) | 2020-04-06 | 2021-10-14 | Gilead Sciences, Inc. | Inhalation formulations of 1'-cyano substituted carbanucleoside analogs |
| WO2021228222A1 (en) | 2020-05-15 | 2021-11-18 | 福建广生堂药业股份有限公司 | Combination for treating hepatitis b |
| US11198693B2 (en) | 2018-11-21 | 2021-12-14 | Enanta Pharmaceuticals, Inc. | Functionalized heterocycles as antiviral agents |
| US11203610B2 (en) | 2017-12-20 | 2021-12-21 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2′3′ cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| WO2021262826A2 (en) | 2020-06-24 | 2021-12-30 | Gilead Sciences, Inc. | 1'-cyano nucleoside analogs and uses thereof |
| US11236111B2 (en) | 2019-06-03 | 2022-02-01 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| US11236108B2 (en) | 2019-09-17 | 2022-02-01 | Enanta Pharmaceuticals, Inc. | Functionalized heterocycles as antiviral agents |
| WO2022031894A1 (en) | 2020-08-07 | 2022-02-10 | Gilead Sciences, Inc. | Prodrugs of phosphonamide nucleotide analogues and their pharmaceutical use |
| US11260070B2 (en) | 2017-03-14 | 2022-03-01 | Gilead Sciences, Inc. | Methods of treating feline coronavirus infections |
| WO2022046631A1 (en) | 2020-08-24 | 2022-03-03 | Gilead Sciences, Inc. | Phospholipid compounds and uses thereof |
| WO2022047065A2 (en) | 2020-08-27 | 2022-03-03 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US11266681B2 (en) | 2017-07-11 | 2022-03-08 | Gilead Sciences, Inc. | Compositions comprising an RNA polymerase inhibitor and cyclodextrin for treating viral infections |
| US11266666B2 (en) | 2014-10-29 | 2022-03-08 | Gilead Sciences, Inc. | Methods for treating Filoviridae virus infections |
| WO2022081973A1 (en) | 2020-10-16 | 2022-04-21 | Gilead Sciences, Inc. | Phospholipid compounds and uses thereof |
| WO2022087149A2 (en) | 2020-10-22 | 2022-04-28 | Gilead Sciences, Inc. | Interleukin-2-fc fusion proteins and methods of use |
| US11472808B2 (en) | 2019-06-04 | 2022-10-18 | Enanta Pharmaceuticals, Inc. | Substituted pyrrolo[1,2-c]pyrimidines as hepatitis B antiviral agents |
| US11491169B2 (en) | 2020-05-29 | 2022-11-08 | Gilead Sciences, Inc. | Remdesivir treatment methods |
| US11492353B2 (en) | 2010-07-22 | 2022-11-08 | Gilead Sciences, Inc. | Methods and compounds for treating Paramyxoviridae virus infections |
| WO2022241134A1 (en) | 2021-05-13 | 2022-11-17 | Gilead Sciences, Inc. | COMBINATION OF A TLR8 MODULATING COMPOUND AND ANTI-HBV siRNA THERAPEUTICS |
| WO2022251318A1 (en) | 2021-05-26 | 2022-12-01 | Gilead Sciences, Inc. | Phospholipid formulations of 1'-cyano substituted carba-nucleoside analogs |
| WO2022271659A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271684A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271650A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271677A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2023023527A1 (en) | 2021-08-18 | 2023-02-23 | Gilead Sciences, Inc. | Phospholipid compounds and methods of making and using the same |
| US11597742B2 (en) | 2017-05-01 | 2023-03-07 | Gilead Sciences, Inc. | Crystalline forms of (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f] [1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy) (phenoxy) phosphoryl)amino)propanoate |
| US11596611B2 (en) | 2017-08-28 | 2023-03-07 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| US11613553B2 (en) | 2020-03-12 | 2023-03-28 | Gilead Sciences, Inc. | Methods of preparing 1′-cyano nucleosides |
| US11738019B2 (en) | 2019-07-11 | 2023-08-29 | Enanta Pharmaceuticals, Inc. | Substituted heterocycles as antiviral agents |
| WO2023167944A1 (en) | 2022-03-02 | 2023-09-07 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| WO2023168254A1 (en) | 2022-03-03 | 2023-09-07 | Gilead Sciences, Inc. | Antiviral compounds and methods of making and using the same |
| WO2023167938A1 (en) | 2022-03-02 | 2023-09-07 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| WO2023168194A1 (en) | 2022-03-03 | 2023-09-07 | Gilead Sciences, Inc. | Antiviral compounds and methods of making and using the same |
| US11760755B2 (en) | 2019-06-04 | 2023-09-19 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| US11802125B2 (en) | 2020-03-16 | 2023-10-31 | Enanta Pharmaceuticals, Inc. | Functionalized heterocyclic compounds as antiviral agents |
| WO2023239665A1 (en) | 2022-06-06 | 2023-12-14 | Gilead Sciences, Inc. | Methods for treatment of viral infections including sars-cov-2 |
| CN117279909A (en) * | 2021-04-04 | 2023-12-22 | 巴鲁克斯布伦博格研究所 | Novel liver-selective polyadenylation polymerase inhibitors and methods of using them |
| WO2024006461A1 (en) | 2022-06-30 | 2024-01-04 | Gilead Sciences, Inc. | Solid forms of a nucleoside analogue and uses thereof |
| WO2024006376A1 (en) | 2022-06-29 | 2024-01-04 | Gilead Sciences, Inc. | Solid forms of a nucleoside analogue and uses thereof |
| RU2823673C1 (en) * | 2019-09-19 | 2024-07-29 | Фуцзянь Акейлинк Биотекнолоджи Ко., Лтд. | Method of producing crystalline form of hepatitis b virus surface antigen inhibitor |
| WO2024173458A1 (en) | 2023-02-16 | 2024-08-22 | Gilead Sciences, Inc. | Phospholipid compounds and methods of making and using the same |
| US12171755B2 (en) | 2017-10-25 | 2024-12-24 | Children's Medical Center Corporation | PAPD5 inhibitors and methods of use thereof |
| WO2025049493A1 (en) | 2023-08-31 | 2025-03-06 | Gilead Sciences, Inc. | Antiviral compounds and methods of making and using the same |
| WO2025049699A1 (en) | 2023-08-31 | 2025-03-06 | Gilead Sciences, Inc. | Antiviral compounds and methods of making and using the same |
| WO2025054278A1 (en) | 2023-09-06 | 2025-03-13 | Gilead Sciences, Inc. | Solid forms of a nucleoside analogue and uses thereof |
| US12357577B1 (en) | 2024-02-02 | 2025-07-15 | Gilead Sciences, Inc. | Pharmaceutical formulations and uses thereof |
| WO2025240246A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with ribavirin |
| WO2025240243A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with bulevirtide and an inhibitory nucleic acid targeting hepatitis b virus |
| WO2025240244A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies comprising bulevirtide and lonafarnib for use in the treatment of hepatitis d virus infection |
| WO2025240242A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with ribavirin |
| US12486234B2 (en) | 2019-04-24 | 2025-12-02 | Children's Medical Center Corporation | PAPD5 inhibitors and methods of use thereof |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MA45496A (en) * | 2016-06-17 | 2019-04-24 | Hoffmann La Roche | NUCLEIC ACID MOLECULES FOR PADD5 OR PAD7 MRNA REDUCTION FOR TREATMENT OF HEPATITIS B INFECTION |
| WO2019069293A1 (en) * | 2017-10-05 | 2019-04-11 | Glaxosmithkline Intellectual Property Development Limited | Chemical compounds |
| CN118048356A (en) | 2017-10-16 | 2024-05-17 | 豪夫迈·罗氏有限公司 | Nucleic acid molecules that reduce PAPD5 and PAPD7 mRNA for treating hepatitis B infection |
| EP3694856B1 (en) * | 2017-11-22 | 2023-10-25 | Sunshine Lake Pharma Co., Ltd. | Fused tricyclic compounds and uses thereof in medicine |
| PE20210346A1 (en) | 2018-07-03 | 2021-02-25 | Hoffmann La Roche | OLIGONUCLEOTIDES TO MODULATE THE EXPRESSION OF TAU |
| CN110862390B (en) | 2018-08-28 | 2023-05-09 | 广东东阳光药业有限公司 | Fused tricyclic compounds and their application in medicine |
| KR20210069680A (en) | 2018-09-30 | 2021-06-11 | 선샤인 레이크 파르마 컴퍼니 리미티드 | Fused tetracyclic compounds and their use in drugs |
| KR102623169B1 (en) * | 2019-09-19 | 2024-01-11 | 푸지엔 에이키링크 바이오테크놀로지 컴퍼니 리미티드 | Crystalline form of hepatitis B surface antigen inhibitor and uses thereof |
| CN111943946B (en) * | 2020-08-19 | 2022-07-01 | 中国医学科学院医药生物技术研究所 | Dihydroquinazinone carboxylic acid compound containing nitrogen heterocyclic segment and application thereof |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103450184A (en) * | 2012-05-29 | 2013-12-18 | 上海壹志医药科技有限公司 | Salt of scoulerine derivatives |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS60197684A (en) * | 1984-03-21 | 1985-10-07 | Dainippon Pharmaceut Co Ltd | Benzo(a)quinolizine derivative and its salt |
| WO2015113990A1 (en) * | 2014-01-30 | 2015-08-06 | F. Hoffmann-La Roche Ag | Novel dihydroquinolizinones for the treatment and prophylaxis of hepatitis b virus infection |
| CA2948080A1 (en) * | 2014-05-13 | 2015-11-19 | F. Hoffmann-La Roche Ag | Novel dihydroquinolizinones for the treatment and prophylaxis of hepatitis b virus infection |
| US9637485B2 (en) * | 2014-11-03 | 2017-05-02 | Hoffmann-La Roche Inc. | 6,7-dihydrobenzo[a]quinolizin-2-one derivatives for the treatment and prophylaxis of hepatitis B virus infection |
-
2016
- 2016-02-08 HK HK18103815.7A patent/HK1244281B/en unknown
- 2016-02-08 CN CN201680008772.7A patent/CN107207505B/en active Active
- 2016-02-08 EP EP16703311.7A patent/EP3256471B1/en active Active
- 2016-02-08 WO PCT/EP2016/052585 patent/WO2016128335A1/en not_active Ceased
- 2016-02-08 JP JP2017542045A patent/JP6435054B2/en active Active
-
2017
- 2017-08-11 US US15/674,810 patent/US10093671B2/en active Active
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103450184A (en) * | 2012-05-29 | 2013-12-18 | 上海壹志医药科技有限公司 | Salt of scoulerine derivatives |
Non-Patent Citations (1)
| Title |
|---|
| Y-.R. WU: "Two New Quaternary Alkaloids and Anti-Hepatitis B Virus Active Constituents from Corydalis saxicola", PLANTA MEDICA, 2007, pages 787 - 791, XP055160353, Retrieved from the Internet <URL:https://www.thieme-connect.de/products/ejournals/pdf/10.1055/s-2007-981549.pdf> [retrieved on 20150107], DOI: 10.1055/s-2007-98154 * |
Cited By (201)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11492353B2 (en) | 2010-07-22 | 2022-11-08 | Gilead Sciences, Inc. | Methods and compounds for treating Paramyxoviridae virus infections |
| US11266666B2 (en) | 2014-10-29 | 2022-03-08 | Gilead Sciences, Inc. | Methods for treating Filoviridae virus infections |
| US11344565B2 (en) | 2014-10-29 | 2022-05-31 | Gilead Sciences, Inc. | Methods for the preparation of ribosides |
| US9845322B2 (en) | 2015-05-04 | 2017-12-19 | Hoffmann-La Roche Inc. | Tetrahydropyridopyrimidines and tetrahydropyridopyridines for the treatment and prophylaxis of hepatitis B virus infection |
| US10702528B2 (en) | 2015-07-13 | 2020-07-07 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| US10336751B2 (en) | 2015-07-21 | 2019-07-02 | Hoffmann-La Roche Inc. | Tricyclic 4-pyridone-3-carboxylic acid derivatives for the treatment and prophylaxis of hepatitis B virus infection |
| US10150740B2 (en) | 2015-07-28 | 2018-12-11 | Hoffmann-La Roche Inc. | 6,7-dihydropyrido[2,1-A]phthalazin-2-ones for the treatment and prophylaxis of hepatitis B virus infection |
| US11382926B2 (en) | 2015-09-16 | 2022-07-12 | Gilead Sciences, Inc. | Methods for treating Arenaviridae and Coronaviridae virus infections |
| US11007208B2 (en) | 2015-09-16 | 2021-05-18 | Gilead Sciences, Inc. | Methods for treating arenaviridae and coronaviridae virus infections |
| US9845325B2 (en) | 2016-02-19 | 2017-12-19 | Novartis Ag | Tetracyclic pyridone compounds as antivirals |
| US10093673B2 (en) | 2016-02-19 | 2018-10-09 | Novartis Ag | Tetracyclic pyridone compounds as antivirals |
| WO2017140821A1 (en) | 2016-02-19 | 2017-08-24 | Novartis Ag | Tetracyclic pyridone compounds as antivirals |
| US10934306B2 (en) | 2016-03-07 | 2021-03-02 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| US12054493B2 (en) | 2016-03-07 | 2024-08-06 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| US20170342068A1 (en) | 2016-05-27 | 2017-11-30 | Gilead Sciences, Inc. | Compounds for the treatment of hepatitis b virus infection |
| US10189846B2 (en) | 2016-06-10 | 2019-01-29 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| US10640511B2 (en) | 2016-06-10 | 2020-05-05 | Enant Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| JP6991213B2 (en) | 2016-07-29 | 2022-01-14 | グアンジョウ・ルペン・ファーマシューティカル・カンパニー・リミテッド | New therapeutic agents for the treatment of HBV infection |
| EP3492467A4 (en) * | 2016-07-29 | 2019-07-24 | Ginkgo Pharma Co., Ltd. | ISOQUINOLINONE COMPOUND AND USE THEREOF IN THE PREPARATION OF AN ANTIVIRAL DRUG |
| JP2019527731A (en) * | 2016-07-29 | 2019-10-03 | ニュウェーブ ファーマシューティカル インコーポレーテッド | Novel therapeutics for the treatment of HBV infection |
| US10501456B2 (en) | 2016-07-29 | 2019-12-10 | Newave Pharmaceutical Inc. | Therapeutic agents for the treatment of HBV infection |
| US10239872B2 (en) | 2016-07-29 | 2019-03-26 | Newave Pharmaceutical Inc. | Therapeutic agents for the treatment of HBV infection |
| WO2018045144A1 (en) | 2016-09-02 | 2018-03-08 | Gilead Sciences, Inc. | Toll like receptor modulator compounds |
| WO2018045150A1 (en) | 2016-09-02 | 2018-03-08 | Gilead Sciences, Inc. | 4,6-diamino-pyrido[3,2-d]pyrimidine derivaties as toll like receptor modulators |
| US11274285B2 (en) | 2016-10-14 | 2022-03-15 | Precision Biosciences, Inc. | Engineered meganucleases specific for recognition sequences in the Hepatitis B virus genome |
| US10662416B2 (en) | 2016-10-14 | 2020-05-26 | Precision Biosciences, Inc. | Engineered meganucleases specific for recognition sequences in the hepatitis B virus genome |
| WO2018073753A1 (en) * | 2016-10-18 | 2018-04-26 | Novartis Ag | Fused tetracyclic pyridone compounds as antivirals |
| WO2018083136A1 (en) | 2016-11-03 | 2018-05-11 | F. Hoffmann-La Roche Ag | Novel tetrahydroisoquinolines and terahydronaphthyridines for the treatment and prophylaxis of hepatitis b virus infection |
| WO2018083081A1 (en) | 2016-11-03 | 2018-05-11 | F. Hoffmann-La Roche Ag | Novel tetrahydropyridopyrimidines for the treatment and prophylaxis of hepatitis b virus infection |
| WO2018083106A1 (en) | 2016-11-03 | 2018-05-11 | F. Hoffmann-La Roche Ag | Novel tetrahydropyridopyrimidines for the treatment and prophylaxis of hepatitis b virus infection |
| US11124495B2 (en) | 2016-11-03 | 2021-09-21 | Hoffmann-La Roche, Inc. | Tetrahydroisoquinolines and terahydronaphthyridines for the treatment of hepatitis B virus infection |
| WO2018085619A1 (en) | 2016-11-07 | 2018-05-11 | Arbutus Biopharma, Inc. | Substituted pyridinone-containing tricyclic compounds, and methods using same |
| WO2018087345A1 (en) | 2016-11-14 | 2018-05-17 | F. Hoffmann-La Roche Ag | COMBINATION THERAPY OF AN HBsAg INHIBITOR, A NUCLEOS(T)IDE ANALOGUE AND AN INTERFERON |
| EP4424374A2 (en) | 2017-01-31 | 2024-09-04 | Gilead Sciences, Inc. | Crystalline forms of tenofovir alafenamide |
| WO2018144390A1 (en) | 2017-01-31 | 2018-08-09 | Gilead Sciences, Inc. | Crystalline forms of tenofovir alafenamide |
| US10442804B2 (en) | 2017-02-02 | 2019-10-15 | Gilead Sciences, Inc. | Compounds for the treatment of hepatitis B virus infection |
| WO2018154466A1 (en) * | 2017-02-21 | 2018-08-30 | Glaxosmithkline Intellectual Property Development Limited | Dihydroquinolizinones as antivirals |
| WO2018161960A1 (en) | 2017-03-09 | 2018-09-13 | 福建广生堂药业股份有限公司 | Hepatitis b virus surface antigen inhibitor |
| US11008331B2 (en) | 2017-03-09 | 2021-05-18 | Fujian Cosunter Pharmaceutical Co., Ltd. | Hepatitis B virus surface antigen inhibitor |
| AU2018232071B2 (en) * | 2017-03-09 | 2021-07-08 | Fujian Akeylink Biotechnology Co., Ltd. | Hepatitis B virus surface antigen inhibitor |
| EA038122B1 (en) * | 2017-03-09 | 2021-07-09 | Фуцзянь Косантер Фармасьютикал Ко., Лтд. | Hepatitis b virus surface antigen inhibitor |
| KR20190117799A (en) * | 2017-03-09 | 2019-10-16 | 푸젠 코선터 파마슈티컬 컴퍼니 리미티드 | Surface Antigen Inhibitors of Hepatitis B Virus |
| JP2020509075A (en) * | 2017-03-09 | 2020-03-26 | フーチェン・コサンター・ファーマスーティカル・カンパニー・リミテッドFujian Cosunter Pharmaceutical Co., Ltd. | Hepatitis B virus surface antigen inhibitor |
| CN109071564A (en) * | 2017-03-09 | 2018-12-21 | 福建广生堂药业股份有限公司 | HBsAg Inhibitors |
| CN109071564B (en) * | 2017-03-09 | 2019-08-27 | 福建广生堂药业股份有限公司 | HBV surface antigen inhibitor |
| EA038122B9 (en) * | 2017-03-09 | 2021-09-14 | Фуцзянь Косантер Фармасьютикал Ко., Лтд. | Hepatitis b virus surface antigen inhibitor |
| KR102087397B1 (en) | 2017-03-09 | 2020-03-11 | 푸젠 코선터 파마슈티컬 컴퍼니 리미티드 | Surface Antigen Inhibitors of Hepatitis B Virus |
| US11260070B2 (en) | 2017-03-14 | 2022-03-01 | Gilead Sciences, Inc. | Methods of treating feline coronavirus infections |
| WO2018172852A1 (en) | 2017-03-21 | 2018-09-27 | Arbutus Biopharma Corporation | Substituted dihydroindene-4-carboxamides and analogs thereof, and methods using same |
| CN110475775A (en) * | 2017-03-31 | 2019-11-19 | 富士胶片株式会社 | 4- pyridinone compounds or its salt, pharmaceutical composition and agent comprising 4- pyridinone compounds |
| EP3604301A4 (en) * | 2017-03-31 | 2020-02-05 | FUJIFILM Corporation | 4-PYRIDONE COMPOUND OR SALT THEREOF, PHARMACEUTICAL COMPOSITION AND FORMULATION COMPRISING SAME |
| US11161830B2 (en) | 2017-03-31 | 2021-11-02 | Fujifilm Corporation | 4-pyridone compound or salt thereof, and pharmaceutical composition and formulation including same |
| JPWO2018181883A1 (en) * | 2017-03-31 | 2020-01-16 | 富士フイルム株式会社 | 4-pyridone compound or salt thereof, pharmaceutical composition and agent containing the same |
| CN110475775B (en) * | 2017-03-31 | 2023-05-09 | 富士胶片株式会社 | 4-pyridone compound or salt thereof, pharmaceutical composition and agent comprising 4-pyridone compound |
| EP4026835A2 (en) | 2017-04-20 | 2022-07-13 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2018195321A1 (en) | 2017-04-20 | 2018-10-25 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| US10301312B2 (en) | 2017-04-27 | 2019-05-28 | Novartis Ag | Fused indazole pyridone compounds as antivirals |
| US10975078B2 (en) | 2017-04-27 | 2021-04-13 | Novartis Ag | Fused indazole pyridone compounds as antivirals |
| US11597742B2 (en) | 2017-05-01 | 2023-03-07 | Gilead Sciences, Inc. | Crystalline forms of (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f] [1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy) (phenoxy) phosphoryl)amino)propanoate |
| US12030906B2 (en) | 2017-05-01 | 2024-07-09 | Gilead Sciences, Inc. | Crystalline forms of (s)-2-ethylbutyl 2-(((s)-(((2r,3s,4r,5r)-5-(4-aminopyrrolo[2,1-f] [1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy) (phenoxy) phosphoryl)amino)propanoate |
| CN108530449A (en) * | 2017-05-22 | 2018-09-14 | 河南春风医药科技有限公司 | For treating or preventing hepatitis b virus infected compound and the preparation method and application thereof |
| CN108976223B (en) * | 2017-06-01 | 2020-08-07 | 广东东阳光药业有限公司 | Condensed tricyclic compounds and their application in medicine |
| CN110066278B (en) * | 2017-06-01 | 2021-06-08 | 广东东阳光药业有限公司 | Fused tricyclic compound and application thereof in medicines |
| CN110066278A (en) * | 2017-06-01 | 2019-07-30 | 广东东阳光药业有限公司 | Fused tricyclic class compound and its application in drug |
| WO2018219356A1 (en) * | 2017-06-01 | 2018-12-06 | Sunshine Lake Pharma Co., Ltd. | Fused tricyclic compounds and uses thereof in medicine |
| CN108976223A (en) * | 2017-06-01 | 2018-12-11 | 广东东阳光药业有限公司 | Fused tricyclic class compound and its application in drug |
| US10966970B2 (en) | 2017-06-01 | 2021-04-06 | Sunshine Lake Pharma Co., Ltd. | Fused tricyclic compounds and uses thereof in medicine |
| US11975017B2 (en) | 2017-07-11 | 2024-05-07 | Gilead Sciences, Inc. | Compositions comprising an RNA polymerase inhibitor and cyclodextrin for treating viral infections |
| US11266681B2 (en) | 2017-07-11 | 2022-03-08 | Gilead Sciences, Inc. | Compositions comprising an RNA polymerase inhibitor and cyclodextrin for treating viral infections |
| US12011425B2 (en) | 2017-08-28 | 2024-06-18 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| US11596611B2 (en) | 2017-08-28 | 2023-03-07 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| US12496282B2 (en) | 2017-08-28 | 2025-12-16 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| US12171755B2 (en) | 2017-10-25 | 2024-12-24 | Children's Medical Center Corporation | PAPD5 inhibitors and methods of use thereof |
| CN111448197A (en) * | 2017-12-04 | 2020-07-24 | 加拉帕戈斯股份有限公司 | 2-oxo-5H-benzopyrano[4,3-b]pyridine for the treatment of hepatitis B |
| WO2019110352A1 (en) * | 2017-12-04 | 2019-06-13 | Galapagos Nv | 2-oxo-5h-chromeno[4,3-b]pyridines for use in the treatment of hepatitis b |
| US10723733B2 (en) | 2017-12-06 | 2020-07-28 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| US10428070B2 (en) | 2017-12-06 | 2019-10-01 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| JP2021507906A (en) * | 2017-12-20 | 2021-02-25 | ノバルティス アーゲー | Fusion tricyclic pyrazolo-dihydropyrazinyl-pyridone compound as an antiviral agent |
| WO2019123285A1 (en) * | 2017-12-20 | 2019-06-27 | Novartis Ag | Fused tricyclic pyrazolo-dihydropyrazinyl-pyridone compounds as antivirals |
| US11203610B2 (en) | 2017-12-20 | 2021-12-21 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2′3′ cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| US10966999B2 (en) | 2017-12-20 | 2021-04-06 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3′3′ cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| CN111433210A (en) * | 2017-12-20 | 2020-07-17 | 诺华股份有限公司 | Fused tricyclic pyrazolo-dihydropyrazinyl-pyridone compounds as antiviral agents |
| US11234977B2 (en) | 2017-12-20 | 2022-02-01 | Novartis Ag | Fused tricyclic pyrazolo-dihydropyrazinyl-pyridone compounds as antivirals |
| US11058678B2 (en) | 2018-01-22 | 2021-07-13 | Enanta Pharmaceuticals, Inc. | Substituted heterocycles as antiviral agents |
| WO2019160882A1 (en) | 2018-02-13 | 2019-08-22 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| EP4227302A1 (en) | 2018-02-13 | 2023-08-16 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2019165374A1 (en) | 2018-02-26 | 2019-08-29 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds as hbv replication inhibitors |
| US10729688B2 (en) | 2018-03-29 | 2020-08-04 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| WO2019195181A1 (en) | 2018-04-05 | 2019-10-10 | Gilead Sciences, Inc. | Antibodies and fragments thereof that bind hepatitis b virus protein x |
| US11149052B2 (en) | 2018-04-06 | 2021-10-19 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2′3′-cyclic dinucleotides |
| US11292812B2 (en) | 2018-04-06 | 2022-04-05 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3′3′-cyclic dinucleotides |
| WO2019193542A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotides |
| WO2019193533A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'2'-cyclic dinucleotides |
| WO2019193543A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotides |
| WO2019200247A1 (en) | 2018-04-12 | 2019-10-17 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the hepatitis b virus genome |
| US11788077B2 (en) | 2018-04-12 | 2023-10-17 | Precision Biosciences, Inc. | Polynucleotides encoding optimized engineered meganucleases having specificity for a recognition sequence in the Hepatitis B virus genome |
| US11142750B2 (en) | 2018-04-12 | 2021-10-12 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the Hepatitis B virus genome |
| EP4600247A2 (en) | 2018-04-19 | 2025-08-13 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2019204609A1 (en) | 2018-04-19 | 2019-10-24 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2019211799A1 (en) | 2018-05-03 | 2019-11-07 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide |
| WO2020014643A1 (en) | 2018-07-13 | 2020-01-16 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| EP4234030A2 (en) | 2018-07-13 | 2023-08-30 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2020023710A1 (en) | 2018-07-27 | 2020-01-30 | Arbutus Biopharma Corporation | Substituted tetrahydrocyclopenta[c]pyrroles, substituted dihydropyrrolizines, analogues thereof, and methods using same |
| WO2020028097A1 (en) | 2018-08-01 | 2020-02-06 | Gilead Sciences, Inc. | Solid forms of (r)-11-(methoxymethyl)-12-(3-methoxypropoxy)-3,3-dimethyl-8-0x0-2,3,8,13b-tetrahydro-1h-pyrido[2,1-a]pyrrolo[1,2-c] phthalazine-7-c arboxylic acid |
| WO2020061435A1 (en) | 2018-09-21 | 2020-03-26 | Enanta Pharmaceuticals, Inc. | Functionalized heterocycles as antiviral agents |
| US10865211B2 (en) | 2018-09-21 | 2020-12-15 | Enanta Pharmaceuticals, Inc. | Functionalized heterocycles as antiviral agents |
| US11377450B2 (en) | 2018-09-21 | 2022-07-05 | Enanta Pharmaceuticals, Inc. | Functionalized heterocycles as antiviral agents |
| CN110950860A (en) * | 2018-09-26 | 2020-04-03 | 广东东阳光药业有限公司 | Condensed tricyclic compounds and their application in medicine |
| CN110950860B (en) * | 2018-09-26 | 2023-03-31 | 广东东阳光药业有限公司 | Fused tricyclic compound and application thereof in medicines |
| WO2020086556A1 (en) | 2018-10-24 | 2020-04-30 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2020092621A1 (en) | 2018-10-31 | 2020-05-07 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds as hpk1 inhibitors |
| EP4371987A1 (en) | 2018-10-31 | 2024-05-22 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds as hpk1 inhibitors |
| WO2020092528A1 (en) | 2018-10-31 | 2020-05-07 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds having hpk1 inhibitory activity |
| US11891393B2 (en) | 2018-11-21 | 2024-02-06 | Enanta Pharmaceuticals, Inc. | Functionalized heterocycles as antiviral agents |
| US11198693B2 (en) | 2018-11-21 | 2021-12-14 | Enanta Pharmaceuticals, Inc. | Functionalized heterocycles as antiviral agents |
| US12264159B2 (en) | 2018-11-21 | 2025-04-01 | Enanta Pharmaceuticals, Inc. | Functionalized heterocycles as antiviral agents |
| WO2020123674A1 (en) | 2018-12-12 | 2020-06-18 | Arbutus Biopharma Corporation | Substituted arylmethylureas and heteroarylmethylureas, analogues thereof, and methods using same |
| US12318403B2 (en) | 2019-03-07 | 2025-06-03 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2′3′-cyclic dinucleotides and prodrugs thereof |
| WO2020178768A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide as sting modulator |
| WO2020178769A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotides and prodrugs thereof |
| US11766447B2 (en) | 2019-03-07 | 2023-09-26 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3′3′-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide as sting modulator |
| WO2020178770A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotides and prodrugs thereof |
| EP4458416A2 (en) | 2019-04-17 | 2024-11-06 | Gilead Sciences, Inc. | Solid forms of a toll-like receptor modulator |
| WO2020214716A1 (en) | 2019-04-17 | 2020-10-22 | Gilead Sciences, Inc. | 2-imino-5-oxo-imidazolidine inhibitors of hiv protease |
| WO2020214663A1 (en) | 2019-04-17 | 2020-10-22 | Gilead Sciences, Inc. | Solid forms of a toll-like receptor modulator |
| WO2020214652A1 (en) | 2019-04-17 | 2020-10-22 | Gilead Sciences, Inc. | Solid forms of a toll-like receptor modulator |
| US12486234B2 (en) | 2019-04-24 | 2025-12-02 | Children's Medical Center Corporation | PAPD5 inhibitors and methods of use thereof |
| WO2020221074A1 (en) | 2019-04-30 | 2020-11-05 | 南京海璞医药科技有限公司 | Compound containing fused ring, use thereof and pharmaceutical composition containing same |
| US11661420B2 (en) | 2019-04-30 | 2023-05-30 | Nanjing Hepo Pharmaceutical Co., Ltd | Compound containing fused ring, use thereof and pharmaceutical composition containing same |
| WO2020237025A1 (en) | 2019-05-23 | 2020-11-26 | Gilead Sciences, Inc. | Substituted exo-methylene-oxindoles which are hpk1/map4k1 inhibitors |
| US11236111B2 (en) | 2019-06-03 | 2022-02-01 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| US11760755B2 (en) | 2019-06-04 | 2023-09-19 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| US11472808B2 (en) | 2019-06-04 | 2022-10-18 | Enanta Pharmaceuticals, Inc. | Substituted pyrrolo[1,2-c]pyrimidines as hepatitis B antiviral agents |
| WO2020263830A1 (en) | 2019-06-25 | 2020-12-30 | Gilead Sciences, Inc. | Flt3l-fc fusion proteins and methods of use |
| US11738019B2 (en) | 2019-07-11 | 2023-08-29 | Enanta Pharmaceuticals, Inc. | Substituted heterocycles as antiviral agents |
| WO2021011891A1 (en) | 2019-07-18 | 2021-01-21 | Gilead Sciences, Inc. | Long-acting formulations of tenofovir alafenamide |
| WO2021034804A1 (en) | 2019-08-19 | 2021-02-25 | Gilead Sciences, Inc. | Pharmaceutical formulations of tenofovir alafenamide |
| US11236108B2 (en) | 2019-09-17 | 2022-02-01 | Enanta Pharmaceuticals, Inc. | Functionalized heterocycles as antiviral agents |
| RU2823673C1 (en) * | 2019-09-19 | 2024-07-29 | Фуцзянь Акейлинк Биотекнолоджи Ко., Лтд. | Method of producing crystalline form of hepatitis b virus surface antigen inhibitor |
| WO2021067181A1 (en) | 2019-09-30 | 2021-04-08 | Gilead Sciences, Inc. | Hbv vaccines and methods treating hbv |
| EP4458975A2 (en) | 2019-09-30 | 2024-11-06 | Gilead Sciences, Inc. | Hbv vaccines and methods treating hbv |
| WO2021113765A1 (en) | 2019-12-06 | 2021-06-10 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the hepatitis b virus genome |
| EP4567109A2 (en) | 2019-12-06 | 2025-06-11 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the hepatitis b virus genome |
| US12410418B2 (en) | 2019-12-06 | 2025-09-09 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the Hepatitis B virus genome |
| EP4445902A2 (en) | 2019-12-24 | 2024-10-16 | Carna Biosciences, Inc. | Diacylglycerol kinase modulating compounds |
| WO2021130638A1 (en) | 2019-12-24 | 2021-07-01 | Carna Biosciences, Inc. | Diacylglycerol kinase modulating compounds |
| US11660307B2 (en) | 2020-01-27 | 2023-05-30 | Gilead Sciences, Inc. | Methods for treating SARS CoV-2 infections |
| WO2021154687A1 (en) | 2020-01-27 | 2021-08-05 | Gilead Sciences, Inc. | Methods for treating sars cov-2 infections |
| US12012431B2 (en) | 2020-03-12 | 2024-06-18 | Gilead Sciences, Inc. | Methods of preparing 1′-cyano nucleosides |
| US11613553B2 (en) | 2020-03-12 | 2023-03-28 | Gilead Sciences, Inc. | Methods of preparing 1′-cyano nucleosides |
| US11802125B2 (en) | 2020-03-16 | 2023-10-31 | Enanta Pharmaceuticals, Inc. | Functionalized heterocyclic compounds as antiviral agents |
| WO2021188959A1 (en) | 2020-03-20 | 2021-09-23 | Gilead Sciences, Inc. | Prodrugs of 4'-c-substituted-2-halo-2'-deoxyadenosine nucleosides and methods of making and using the same |
| US11701372B2 (en) | 2020-04-06 | 2023-07-18 | Gilead Sciences, Inc. | Inhalation formulations of 1'-cyano substituted carba-nucleoside analogs |
| WO2021207049A1 (en) | 2020-04-06 | 2021-10-14 | Gilead Sciences, Inc. | Inhalation formulations of 1'-cyano substituted carbanucleoside analogs |
| WO2021228222A1 (en) | 2020-05-15 | 2021-11-18 | 福建广生堂药业股份有限公司 | Combination for treating hepatitis b |
| US11491169B2 (en) | 2020-05-29 | 2022-11-08 | Gilead Sciences, Inc. | Remdesivir treatment methods |
| US11903953B2 (en) | 2020-05-29 | 2024-02-20 | Gilead Sciences, Inc. | Remdesivir treatment methods |
| US11975012B2 (en) | 2020-05-29 | 2024-05-07 | Gilead Sciences, Inc. | Remdesivir treatment methods |
| US12404289B2 (en) | 2020-06-24 | 2025-09-02 | Gilead Sciences, Inc. | 1′-cyano nucleoside analogs and uses thereof |
| WO2021262826A2 (en) | 2020-06-24 | 2021-12-30 | Gilead Sciences, Inc. | 1'-cyano nucleoside analogs and uses thereof |
| US11939347B2 (en) | 2020-06-24 | 2024-03-26 | Gilead Sciences, Inc. | 1′-cyano nucleoside analogs and uses thereof |
| WO2022031894A1 (en) | 2020-08-07 | 2022-02-10 | Gilead Sciences, Inc. | Prodrugs of phosphonamide nucleotide analogues and their pharmaceutical use |
| EP4537828A2 (en) | 2020-08-24 | 2025-04-16 | Gilead Sciences, Inc. | Phospholipid compounds and uses thereof |
| WO2022046631A1 (en) | 2020-08-24 | 2022-03-03 | Gilead Sciences, Inc. | Phospholipid compounds and uses thereof |
| US12297226B2 (en) | 2020-08-27 | 2025-05-13 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US11926645B2 (en) | 2020-08-27 | 2024-03-12 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| WO2022047065A2 (en) | 2020-08-27 | 2022-03-03 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| EP4424372A2 (en) | 2020-08-27 | 2024-09-04 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US11814406B2 (en) | 2020-08-27 | 2023-11-14 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| WO2022081973A1 (en) | 2020-10-16 | 2022-04-21 | Gilead Sciences, Inc. | Phospholipid compounds and uses thereof |
| WO2022087149A2 (en) | 2020-10-22 | 2022-04-28 | Gilead Sciences, Inc. | Interleukin-2-fc fusion proteins and methods of use |
| EP4320117A4 (en) * | 2021-04-04 | 2025-02-26 | Baruch S. Blumberg Institute | Novel hepatoselective polyadenylating polymerases inhibitors and their method of use |
| CN117279909A (en) * | 2021-04-04 | 2023-12-22 | 巴鲁克斯布伦博格研究所 | Novel liver-selective polyadenylation polymerase inhibitors and methods of using them |
| WO2022241134A1 (en) | 2021-05-13 | 2022-11-17 | Gilead Sciences, Inc. | COMBINATION OF A TLR8 MODULATING COMPOUND AND ANTI-HBV siRNA THERAPEUTICS |
| WO2022251318A1 (en) | 2021-05-26 | 2022-12-01 | Gilead Sciences, Inc. | Phospholipid formulations of 1'-cyano substituted carba-nucleoside analogs |
| WO2022271677A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271659A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271684A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271650A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2023023527A1 (en) | 2021-08-18 | 2023-02-23 | Gilead Sciences, Inc. | Phospholipid compounds and methods of making and using the same |
| US11780844B2 (en) | 2022-03-02 | 2023-10-10 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| WO2023167938A1 (en) | 2022-03-02 | 2023-09-07 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US12448383B2 (en) | 2022-03-02 | 2025-10-21 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| WO2023167944A1 (en) | 2022-03-02 | 2023-09-07 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US12180217B2 (en) | 2022-03-02 | 2024-12-31 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US11851438B2 (en) | 2022-03-02 | 2023-12-26 | Gilead Sciences, Inc. | 1′-cyano nucleoside analogs and methods for treatment of viral infections |
| US11845755B2 (en) | 2022-03-02 | 2023-12-19 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| WO2023168254A1 (en) | 2022-03-03 | 2023-09-07 | Gilead Sciences, Inc. | Antiviral compounds and methods of making and using the same |
| WO2023168194A1 (en) | 2022-03-03 | 2023-09-07 | Gilead Sciences, Inc. | Antiviral compounds and methods of making and using the same |
| WO2023239665A1 (en) | 2022-06-06 | 2023-12-14 | Gilead Sciences, Inc. | Methods for treatment of viral infections including sars-cov-2 |
| WO2024006376A1 (en) | 2022-06-29 | 2024-01-04 | Gilead Sciences, Inc. | Solid forms of a nucleoside analogue and uses thereof |
| WO2024006461A1 (en) | 2022-06-30 | 2024-01-04 | Gilead Sciences, Inc. | Solid forms of a nucleoside analogue and uses thereof |
| WO2024173458A1 (en) | 2023-02-16 | 2024-08-22 | Gilead Sciences, Inc. | Phospholipid compounds and methods of making and using the same |
| WO2025049699A1 (en) | 2023-08-31 | 2025-03-06 | Gilead Sciences, Inc. | Antiviral compounds and methods of making and using the same |
| WO2025049493A1 (en) | 2023-08-31 | 2025-03-06 | Gilead Sciences, Inc. | Antiviral compounds and methods of making and using the same |
| WO2025054278A1 (en) | 2023-09-06 | 2025-03-13 | Gilead Sciences, Inc. | Solid forms of a nucleoside analogue and uses thereof |
| US12357577B1 (en) | 2024-02-02 | 2025-07-15 | Gilead Sciences, Inc. | Pharmaceutical formulations and uses thereof |
| WO2025240246A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with ribavirin |
| WO2025240243A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with bulevirtide and an inhibitory nucleic acid targeting hepatitis b virus |
| WO2025240244A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies comprising bulevirtide and lonafarnib for use in the treatment of hepatitis d virus infection |
| WO2025240242A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with ribavirin |
Also Published As
| Publication number | Publication date |
|---|---|
| US20170342069A1 (en) | 2017-11-30 |
| HK1244281B (en) | 2020-02-07 |
| EP3256471B1 (en) | 2018-12-12 |
| CN107207505A (en) | 2017-09-26 |
| CN107207505B (en) | 2018-12-14 |
| JP6435054B2 (en) | 2018-12-05 |
| US10093671B2 (en) | 2018-10-09 |
| EP3256471A1 (en) | 2017-12-20 |
| JP2018505198A (en) | 2018-02-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3256471B1 (en) | Novel 2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid derivatives for the treatment and prophylaxis of hepatitis b virus infection | |
| EP3325477B1 (en) | Novel tricyclic 4-pyridone-3-carboxylic acid derivatives for the treatment and prophylaxis of hepatitis b virus infection | |
| EP3143020B1 (en) | Dihydroquinolizinones for the treatment and prophylaxis of hepatitis b virus infection | |
| US10150740B2 (en) | 6,7-dihydropyrido[2,1-A]phthalazin-2-ones for the treatment and prophylaxis of hepatitis B virus infection | |
| US10053461B2 (en) | Tetracyclic 4-oxo-pyridine-3-carboxylic acid derivatives for the treatment and prophylaxis of hepatitis B virus infection | |
| JP6420491B2 (en) | Novel 6,7-dihydrobenzo [a] quinolizin-2-one derivatives for the treatment or prevention of hepatitis B virus infection | |
| HK1244281A1 (en) | 2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid derivatives for the treatment and prophylaxis of hepatitis b virus infection | |
| CA2931329A1 (en) | Novel dihydroquinolizinones for the treatment and prophylaxis of hepatitis b virus infection | |
| KR20160097371A (en) | Novel dihydroquinolizinones for the treatment and prophylaxis of hepatitis b virus infection | |
| HK1251221B (en) | Tricyclic 4-pyridone-3-carboxylic acid derivatives for the treatment and prophylaxis of hepatitis b virus infection |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16703311 Country of ref document: EP Kind code of ref document: A1 |
|
| REEP | Request for entry into the european phase |
Ref document number: 2016703311 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2017542045 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |