WO2015109272A1 - Material and method of manufacture of electrodes and porous filters formed of ice-templated graphene-oxide and carbon nanotube composite, and applications thereof - Google Patents
Material and method of manufacture of electrodes and porous filters formed of ice-templated graphene-oxide and carbon nanotube composite, and applications thereof Download PDFInfo
- Publication number
- WO2015109272A1 WO2015109272A1 PCT/US2015/011882 US2015011882W WO2015109272A1 WO 2015109272 A1 WO2015109272 A1 WO 2015109272A1 US 2015011882 W US2015011882 W US 2015011882W WO 2015109272 A1 WO2015109272 A1 WO 2015109272A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- carbon
- graphene oxide
- composition
- graphene
- freeze
- Prior art date
Links
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 title claims abstract description 238
- 239000002041 carbon nanotube Substances 0.000 title claims abstract description 76
- 229910021393 carbon nanotube Inorganic materials 0.000 title claims abstract description 71
- 238000000034 method Methods 0.000 title claims abstract description 69
- 239000002131 composite material Substances 0.000 title claims abstract description 22
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 17
- 239000000463 material Substances 0.000 title abstract description 66
- 229910021389 graphene Inorganic materials 0.000 claims abstract description 96
- 239000011148 porous material Substances 0.000 claims abstract description 71
- 239000000203 mixture Substances 0.000 claims abstract description 66
- 239000002134 carbon nanofiber Substances 0.000 claims abstract description 58
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical class C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 claims abstract description 43
- 230000002829 reductive effect Effects 0.000 claims abstract description 38
- 238000010438 heat treatment Methods 0.000 claims abstract description 23
- 230000001590 oxidative effect Effects 0.000 claims abstract description 10
- 239000012298 atmosphere Substances 0.000 claims abstract description 9
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 114
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 69
- 238000005266 casting Methods 0.000 claims description 41
- 239000005720 sucrose Substances 0.000 claims description 40
- 239000002002 slurry Substances 0.000 claims description 39
- 229930006000 Sucrose Natural products 0.000 claims description 38
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 claims description 36
- 239000002245 particle Substances 0.000 claims description 16
- 239000000126 substance Substances 0.000 claims description 14
- 239000002121 nanofiber Substances 0.000 claims description 11
- 229920002678 cellulose Polymers 0.000 claims description 8
- 239000001913 cellulose Substances 0.000 claims description 8
- 238000006243 chemical reaction Methods 0.000 claims description 7
- 238000000746 purification Methods 0.000 claims description 4
- 230000009467 reduction Effects 0.000 abstract description 15
- 238000011065 in-situ storage Methods 0.000 abstract description 2
- 229920002749 Bacterial cellulose Polymers 0.000 description 65
- 239000005016 bacterial cellulose Substances 0.000 description 65
- 239000004964 aerogel Substances 0.000 description 53
- 239000000523 sample Substances 0.000 description 44
- 239000011230 binding agent Substances 0.000 description 41
- 230000008014 freezing Effects 0.000 description 38
- 238000007710 freezing Methods 0.000 description 38
- 235000019441 ethanol Nutrition 0.000 description 35
- 238000003763 carbonization Methods 0.000 description 26
- 229920001046 Nanocellulose Polymers 0.000 description 23
- 238000000137 annealing Methods 0.000 description 19
- 239000000017 hydrogel Substances 0.000 description 18
- 239000000243 solution Substances 0.000 description 18
- 239000002904 solvent Substances 0.000 description 18
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 17
- 229920001661 Chitosan Polymers 0.000 description 17
- 238000002474 experimental method Methods 0.000 description 17
- 239000000725 suspension Substances 0.000 description 17
- 238000006722 reduction reaction Methods 0.000 description 15
- 238000012360 testing method Methods 0.000 description 15
- 241000208140 Acer Species 0.000 description 12
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 12
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 12
- 229910052799 carbon Inorganic materials 0.000 description 12
- 238000012512 characterization method Methods 0.000 description 11
- 238000001816 cooling Methods 0.000 description 11
- 238000004108 freeze drying Methods 0.000 description 11
- 238000002360 preparation method Methods 0.000 description 11
- 239000002011 CNT10 Substances 0.000 description 10
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 10
- 239000010949 copper Substances 0.000 description 10
- 229910052802 copper Inorganic materials 0.000 description 10
- 239000002048 multi walled nanotube Substances 0.000 description 10
- 230000008569 process Effects 0.000 description 10
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 9
- 239000000499 gel Substances 0.000 description 9
- 239000002071 nanotube Substances 0.000 description 9
- 239000006188 syrup Substances 0.000 description 9
- 235000020357 syrup Nutrition 0.000 description 9
- 239000013078 crystal Substances 0.000 description 8
- 238000010612 desalination reaction Methods 0.000 description 8
- 238000005259 measurement Methods 0.000 description 8
- 238000002156 mixing Methods 0.000 description 8
- 239000000661 sodium alginate Substances 0.000 description 8
- 229940005550 sodium alginate Drugs 0.000 description 8
- 239000003795 chemical substances by application Substances 0.000 description 7
- 229910003460 diamond Inorganic materials 0.000 description 7
- 239000010432 diamond Substances 0.000 description 7
- 239000002270 dispersing agent Substances 0.000 description 7
- 238000007669 thermal treatment Methods 0.000 description 7
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 6
- 229910052786 argon Inorganic materials 0.000 description 6
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 6
- 239000005539 carbonized material Substances 0.000 description 6
- 210000004027 cell Anatomy 0.000 description 6
- 229910002804 graphite Inorganic materials 0.000 description 6
- 239000010439 graphite Substances 0.000 description 6
- 235000010355 mannitol Nutrition 0.000 description 6
- 229910052757 nitrogen Inorganic materials 0.000 description 6
- 239000001301 oxygen Substances 0.000 description 6
- 229910052760 oxygen Inorganic materials 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- 238000011084 recovery Methods 0.000 description 6
- 150000003839 salts Chemical class 0.000 description 6
- 238000004626 scanning electron microscopy Methods 0.000 description 6
- 239000007787 solid Substances 0.000 description 6
- 235000000346 sugar Nutrition 0.000 description 6
- 229920001817 Agar Polymers 0.000 description 5
- 239000008272 agar Substances 0.000 description 5
- 239000003575 carbonaceous material Substances 0.000 description 5
- 238000005336 cracking Methods 0.000 description 5
- 239000006185 dispersion Substances 0.000 description 5
- 239000000835 fiber Substances 0.000 description 5
- 239000011268 mixed slurry Substances 0.000 description 5
- 239000003607 modifier Substances 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 238000012545 processing Methods 0.000 description 5
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- 229930195725 Mannitol Natural products 0.000 description 4
- 229910000831 Steel Inorganic materials 0.000 description 4
- 239000000654 additive Substances 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 238000012669 compression test Methods 0.000 description 4
- 238000010586 diagram Methods 0.000 description 4
- 239000007789 gas Substances 0.000 description 4
- 238000001879 gelation Methods 0.000 description 4
- 239000011521 glass Substances 0.000 description 4
- 150000004676 glycans Chemical class 0.000 description 4
- 239000001963 growth medium Substances 0.000 description 4
- 150000002500 ions Chemical class 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 239000000594 mannitol Substances 0.000 description 4
- 230000003287 optical effect Effects 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 229920001282 polysaccharide Polymers 0.000 description 4
- 239000005017 polysaccharide Substances 0.000 description 4
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 4
- 239000004810 polytetrafluoroethylene Substances 0.000 description 4
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 4
- 235000010413 sodium alginate Nutrition 0.000 description 4
- 239000010959 steel Substances 0.000 description 4
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 3
- 238000001069 Raman spectroscopy Methods 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- 229940041514 candida albicans extract Drugs 0.000 description 3
- 210000002421 cell wall Anatomy 0.000 description 3
- 239000003086 colorant Substances 0.000 description 3
- 230000006835 compression Effects 0.000 description 3
- 238000007906 compression Methods 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- 239000008367 deionised water Substances 0.000 description 3
- 229910021641 deionized water Inorganic materials 0.000 description 3
- 150000002016 disaccharides Chemical class 0.000 description 3
- 239000002149 hierarchical pore Substances 0.000 description 3
- 239000001257 hydrogen Substances 0.000 description 3
- 229910052739 hydrogen Inorganic materials 0.000 description 3
- 238000011835 investigation Methods 0.000 description 3
- 230000003647 oxidation Effects 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- 229920000642 polymer Polymers 0.000 description 3
- 230000001105 regulatory effect Effects 0.000 description 3
- 238000001223 reverse osmosis Methods 0.000 description 3
- 238000001878 scanning electron micrograph Methods 0.000 description 3
- 238000011218 seed culture Methods 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 239000012137 tryptone Substances 0.000 description 3
- 230000000007 visual effect Effects 0.000 description 3
- 239000012138 yeast extract Substances 0.000 description 3
- 101100327837 Arabidopsis thaliana CHLH gene Proteins 0.000 description 2
- 229920000049 Carbon (fiber) Polymers 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 241000589216 Komagataeibacter hansenii Species 0.000 description 2
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- XOJVVFBFDXDTEG-UHFFFAOYSA-N Norphytane Natural products CC(C)CCCC(C)CCCC(C)CCCC(C)C XOJVVFBFDXDTEG-UHFFFAOYSA-N 0.000 description 2
- CVRALZAYCYJELZ-UHFFFAOYSA-N O-(4-bromo-2,5-dichlorophenyl) O-methyl phenylphosphonothioate Chemical compound C=1C=CC=CC=1P(=S)(OC)OC1=CC(Cl)=C(Br)C=C1Cl CVRALZAYCYJELZ-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 230000003466 anti-cipated effect Effects 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 239000007900 aqueous suspension Substances 0.000 description 2
- 229960005070 ascorbic acid Drugs 0.000 description 2
- 239000003990 capacitor Substances 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 239000004917 carbon fiber Substances 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 239000003638 chemical reducing agent Substances 0.000 description 2
- 238000009646 cryomilling Methods 0.000 description 2
- 238000005520 cutting process Methods 0.000 description 2
- 230000007547 defect Effects 0.000 description 2
- 238000002242 deionisation method Methods 0.000 description 2
- 238000011038 discontinuous diafiltration by volume reduction Methods 0.000 description 2
- 230000001747 exhibiting effect Effects 0.000 description 2
- 239000013505 freshwater Substances 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 238000003384 imaging method Methods 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- CSJDCSCTVDEHRN-UHFFFAOYSA-N methane;molecular oxygen Chemical compound C.O=O CSJDCSCTVDEHRN-UHFFFAOYSA-N 0.000 description 2
- 150000002772 monosaccharides Chemical class 0.000 description 2
- 239000004033 plastic Substances 0.000 description 2
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 2
- 238000010926 purge Methods 0.000 description 2
- 239000013535 sea water Substances 0.000 description 2
- 239000002109 single walled nanotube Substances 0.000 description 2
- 238000005245 sintering Methods 0.000 description 2
- 238000007711 solidification Methods 0.000 description 2
- 230000008023 solidification Effects 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 238000009827 uniform distribution Methods 0.000 description 2
- DUFCMRCMPHIFTR-UHFFFAOYSA-N 5-(dimethylsulfamoyl)-2-methylfuran-3-carboxylic acid Chemical compound CN(C)S(=O)(=O)C1=CC(C(O)=O)=C(C)O1 DUFCMRCMPHIFTR-UHFFFAOYSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- 229920000936 Agarose Polymers 0.000 description 1
- 238000012935 Averaging Methods 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- XMWRBQBLMFGWIX-UHFFFAOYSA-N C60 fullerene Chemical class C12=C3C(C4=C56)=C7C8=C5C5=C9C%10=C6C6=C4C1=C1C4=C6C6=C%10C%10=C9C9=C%11C5=C8C5=C8C7=C3C3=C7C2=C1C1=C2C4=C6C4=C%10C6=C9C9=C%11C5=C5C8=C3C3=C7C1=C1C2=C4C6=C2C9=C5C3=C12 XMWRBQBLMFGWIX-UHFFFAOYSA-N 0.000 description 1
- 239000004966 Carbon aerogel Substances 0.000 description 1
- 229920002101 Chitin Polymers 0.000 description 1
- 241000581364 Clinitrachus argentatus Species 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- 239000002211 L-ascorbic acid Substances 0.000 description 1
- 235000000069 L-ascorbic acid Nutrition 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 241000199919 Phaeophyceae Species 0.000 description 1
- 238000001237 Raman spectrum Methods 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 229910008760 WITec Inorganic materials 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 238000005452 bending Methods 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 239000007833 carbon precursor Substances 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 239000000919 ceramic Substances 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 230000001010 compromised effect Effects 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 239000000356 contaminant Substances 0.000 description 1
- OPQARKPSCNTWTJ-UHFFFAOYSA-L copper(ii) acetate Chemical compound [Cu+2].CC([O-])=O.CC([O-])=O OPQARKPSCNTWTJ-UHFFFAOYSA-L 0.000 description 1
- 238000007872 degassing Methods 0.000 description 1
- 238000001739 density measurement Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000007599 discharging Methods 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 238000002389 environmental scanning electron microscopy Methods 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000010419 fine particle Substances 0.000 description 1
- 238000007667 floating Methods 0.000 description 1
- 239000012520 frozen sample Substances 0.000 description 1
- 229910003472 fullerene Inorganic materials 0.000 description 1
- 238000007306 functionalization reaction Methods 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 239000011881 graphite nanoparticle Substances 0.000 description 1
- 230000035876 healing Effects 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 239000001307 helium Substances 0.000 description 1
- 229910052734 helium Inorganic materials 0.000 description 1
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 1
- 230000000887 hydrating effect Effects 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 238000005470 impregnation Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 229960004903 invert sugar Drugs 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- WJZHMLNIAZSFDO-UHFFFAOYSA-N manganese zinc Chemical compound [Mn].[Zn] WJZHMLNIAZSFDO-UHFFFAOYSA-N 0.000 description 1
- 238000011326 mechanical measurement Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 239000013586 microbial product Substances 0.000 description 1
- 238000001000 micrograph Methods 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 229910052754 neon Inorganic materials 0.000 description 1
- GKAOGPIIYCISHV-UHFFFAOYSA-N neon atom Chemical compound [Ne] GKAOGPIIYCISHV-UHFFFAOYSA-N 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 238000000879 optical micrograph Methods 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 238000005191 phase separation Methods 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 239000012286 potassium permanganate Substances 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 238000009877 rendering Methods 0.000 description 1
- 230000000284 resting effect Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- -1 sorbitol Chemical class 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 238000004544 sputter deposition Methods 0.000 description 1
- 230000003068 static effect Effects 0.000 description 1
- 238000000859 sublimation Methods 0.000 description 1
- 230000008022 sublimation Effects 0.000 description 1
- 150000005846 sugar alcohols Chemical class 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 238000009864 tensile test Methods 0.000 description 1
- 238000009997 thermal pre-treatment Methods 0.000 description 1
- 229940099259 vaseline Drugs 0.000 description 1
- 239000011800 void material Substances 0.000 description 1
- 229920003169 water-soluble polymer Polymers 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 239000002023 wood Substances 0.000 description 1
- 230000037303 wrinkles Effects 0.000 description 1
- 229910052724 xenon Inorganic materials 0.000 description 1
- FHNFHKCVQCLJFQ-UHFFFAOYSA-N xenon atom Chemical compound [Xe] FHNFHKCVQCLJFQ-UHFFFAOYSA-N 0.000 description 1
- 238000004383 yellowing Methods 0.000 description 1
- SZKTYYIADWRVSA-UHFFFAOYSA-N zinc manganese(2+) oxygen(2-) Chemical compound [O--].[O--].[Mn++].[Zn++] SZKTYYIADWRVSA-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B35/00—Shaped ceramic products characterised by their composition; Ceramics compositions; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products
- C04B35/515—Shaped ceramic products characterised by their composition; Ceramics compositions; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products based on non-oxide ceramics
- C04B35/52—Shaped ceramic products characterised by their composition; Ceramics compositions; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products based on non-oxide ceramics based on carbon, e.g. graphite
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/05—Preparation or purification of carbon not covered by groups C01B32/15, C01B32/20, C01B32/25, C01B32/30
-
- C—CHEMISTRY; METALLURGY
- C02—TREATMENT OF WATER, WASTE WATER, SEWAGE, OR SLUDGE
- C02F—TREATMENT OF WATER, WASTE WATER, SEWAGE, OR SLUDGE
- C02F1/00—Treatment of water, waste water, or sewage
- C02F1/46—Treatment of water, waste water, or sewage by electrochemical methods
- C02F1/469—Treatment of water, waste water, or sewage by electrochemical methods by electrochemical separation, e.g. by electro-osmosis, electrodialysis, electrophoresis
- C02F1/4691—Capacitive deionisation
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B35/00—Shaped ceramic products characterised by their composition; Ceramics compositions; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products
- C04B35/515—Shaped ceramic products characterised by their composition; Ceramics compositions; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products based on non-oxide ceramics
- C04B35/52—Shaped ceramic products characterised by their composition; Ceramics compositions; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products based on non-oxide ceramics based on carbon, e.g. graphite
- C04B35/528—Shaped ceramic products characterised by their composition; Ceramics compositions; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products based on non-oxide ceramics based on carbon, e.g. graphite obtained from carbonaceous particles with or without other non-organic components
- C04B35/532—Shaped ceramic products characterised by their composition; Ceramics compositions; Processing powders of inorganic compounds preparatory to the manufacturing of ceramic products based on non-oxide ceramics based on carbon, e.g. graphite obtained from carbonaceous particles with or without other non-organic components containing a carbonisable binder
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B38/00—Porous mortars, concrete, artificial stone or ceramic ware; Preparation thereof
- C04B38/06—Porous mortars, concrete, artificial stone or ceramic ware; Preparation thereof by burning-out added substances by burning natural expanding materials or by sublimating or melting out added substances
- C04B38/061—Porous mortars, concrete, artificial stone or ceramic ware; Preparation thereof by burning-out added substances by burning natural expanding materials or by sublimating or melting out added substances by melting out
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES, LIGHT-SENSITIVE OR TEMPERATURE-SENSITIVE DEVICES OF THE ELECTROLYTIC TYPE
- H01G11/00—Hybrid capacitors, i.e. capacitors having different positive and negative electrodes; Electric double-layer [EDL] capacitors; Processes for the manufacture thereof or of parts thereof
- H01G11/22—Electrodes
- H01G11/30—Electrodes characterised by their material
- H01G11/32—Carbon-based
- H01G11/36—Nanostructures, e.g. nanofibres, nanotubes or fullerenes
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES, LIGHT-SENSITIVE OR TEMPERATURE-SENSITIVE DEVICES OF THE ELECTROLYTIC TYPE
- H01G11/00—Hybrid capacitors, i.e. capacitors having different positive and negative electrodes; Electric double-layer [EDL] capacitors; Processes for the manufacture thereof or of parts thereof
- H01G11/22—Electrodes
- H01G11/30—Electrodes characterised by their material
- H01G11/32—Carbon-based
- H01G11/38—Carbon pastes or blends; Binders or additives therein
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/13—Electrodes for accumulators with non-aqueous electrolyte, e.g. for lithium-accumulators; Processes of manufacture thereof
- H01M4/133—Electrodes based on carbonaceous material, e.g. graphite-intercalation compounds or CFx
-
- C—CHEMISTRY; METALLURGY
- C02—TREATMENT OF WATER, WASTE WATER, SEWAGE, OR SLUDGE
- C02F—TREATMENT OF WATER, WASTE WATER, SEWAGE, OR SLUDGE
- C02F1/00—Treatment of water, waste water, or sewage
- C02F1/46—Treatment of water, waste water, or sewage by electrochemical methods
- C02F1/461—Treatment of water, waste water, or sewage by electrochemical methods by electrolysis
- C02F1/46104—Devices therefor; Their operating or servicing
- C02F1/46109—Electrodes
- C02F2001/46133—Electrodes characterised by the material
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B2111/00—Mortars, concrete or artificial stone or mixtures to prepare them, characterised by specific function, property or use
- C04B2111/00474—Uses not provided for elsewhere in C04B2111/00
- C04B2111/00793—Uses not provided for elsewhere in C04B2111/00 as filters or diaphragms
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B2111/00—Mortars, concrete or artificial stone or mixtures to prepare them, characterised by specific function, property or use
- C04B2111/00474—Uses not provided for elsewhere in C04B2111/00
- C04B2111/00844—Uses not provided for elsewhere in C04B2111/00 for electronic applications
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B2111/00—Mortars, concrete or artificial stone or mixtures to prepare them, characterised by specific function, property or use
- C04B2111/90—Electrical properties
- C04B2111/94—Electrically conducting materials
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B2235/00—Aspects relating to ceramic starting mixtures or sintered ceramic products
- C04B2235/02—Composition of constituents of the starting material or of secondary phases of the final product
- C04B2235/50—Constituents or additives of the starting mixture chosen for their shape or used because of their shape or their physical appearance
- C04B2235/52—Constituents or additives characterised by their shapes
- C04B2235/5208—Fibers
- C04B2235/5216—Inorganic
- C04B2235/524—Non-oxidic, e.g. borides, carbides, silicides or nitrides
- C04B2235/5248—Carbon, e.g. graphite
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B2235/00—Aspects relating to ceramic starting mixtures or sintered ceramic products
- C04B2235/02—Composition of constituents of the starting material or of secondary phases of the final product
- C04B2235/50—Constituents or additives of the starting mixture chosen for their shape or used because of their shape or their physical appearance
- C04B2235/52—Constituents or additives characterised by their shapes
- C04B2235/5208—Fibers
- C04B2235/526—Fibers characterised by the length of the fibers
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B2235/00—Aspects relating to ceramic starting mixtures or sintered ceramic products
- C04B2235/02—Composition of constituents of the starting material or of secondary phases of the final product
- C04B2235/50—Constituents or additives of the starting mixture chosen for their shape or used because of their shape or their physical appearance
- C04B2235/52—Constituents or additives characterised by their shapes
- C04B2235/5208—Fibers
- C04B2235/5264—Fibers characterised by the diameter of the fibers
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B2235/00—Aspects relating to ceramic starting mixtures or sintered ceramic products
- C04B2235/02—Composition of constituents of the starting material or of secondary phases of the final product
- C04B2235/50—Constituents or additives of the starting mixture chosen for their shape or used because of their shape or their physical appearance
- C04B2235/52—Constituents or additives characterised by their shapes
- C04B2235/5284—Hollow fibers, e.g. nanotubes
- C04B2235/5288—Carbon nanotubes
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B2235/00—Aspects relating to ceramic starting mixtures or sintered ceramic products
- C04B2235/60—Aspects relating to the preparation, properties or mechanical treatment of green bodies or pre-forms
- C04B2235/606—Drying
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B2235/00—Aspects relating to ceramic starting mixtures or sintered ceramic products
- C04B2235/65—Aspects relating to heat treatments of ceramic bodies such as green ceramics or pre-sintered ceramics, e.g. burning, sintering or melting processes
- C04B2235/652—Reduction treatment
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B2235/00—Aspects relating to ceramic starting mixtures or sintered ceramic products
- C04B2235/70—Aspects relating to sintered or melt-casted ceramic products
- C04B2235/74—Physical characteristics
- C04B2235/77—Density
-
- C—CHEMISTRY; METALLURGY
- C04—CEMENTS; CONCRETE; ARTIFICIAL STONE; CERAMICS; REFRACTORIES
- C04B—LIME, MAGNESIA; SLAG; CEMENTS; COMPOSITIONS THEREOF, e.g. MORTARS, CONCRETE OR LIKE BUILDING MATERIALS; ARTIFICIAL STONE; CERAMICS; REFRACTORIES; TREATMENT OF NATURAL STONE
- C04B2235/00—Aspects relating to ceramic starting mixtures or sintered ceramic products
- C04B2235/70—Aspects relating to sintered or melt-casted ceramic products
- C04B2235/96—Properties of ceramic products, e.g. mechanical properties such as strength, toughness, wear resistance
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
Definitions
- the present document relates to the field of ice-templated carbon-based materials.
- Graphene a material comprised of monatomic sheets of two dimensional, sp2-hybridized carbon atoms arranged in a hexagonal pattern, has been investigated intensely since its discovery in 2004. It is hypothesized that graphene, and carbon nanotubes formed of graphene sheets folded into cylinders, may have properties allowing them to form useful materials. Carbon nanofibers are also expected to have interesting properties. Among other properties of graphene, nanotubes, and nanofibers are a degree of electrical conductivity. Carbon nanotubes and nanofibers also have good tensile strength within each fiber or particle; for these nanotubes and fibers to be useful in large objects, however, they must be linked into a composite structure.
- a slurry - a suspension of fine particles in a solvent carrier such as water - may be freeze-east.
- a phase separation occurs during solidification; ice crystals grow by solidifying the water carrier of the slurry, while concentrating between them solutes and solid particulates of the aqueous slurry.
- the solutes and solid particles containing a desired material or material precursor may include a binder.
- the ice crystals template a three-dimensional (3D) micro structure that consists of the solutes and particles that coalesce, producing a material with an ordered, self-assembled, hierarchical pore structure.
- the crystalized solvent carrier is then removed - with water as solvent-carrier this is typically done by sublimation such as during freeze- drying.
- Freeze-east objects may be subjected to further processing, such as a thermal treatment of the material that frequently results in a desirable property profile with favorable structural, mechanical, electrical, thermal, optical or other properties.
- Further processing may include annealing, sintering and carbonization, where castings are heated to a temperature that is high enough to fuse or bond particles together, and in some systems to burn out or to transform or to reduce the binder phase used during casting, yet low enough to avoid the complete melting of the particles. Annealing has been done with freeze-east objects formed with metal powders.
- Capacitive deionization is one method of purifying brackish water into a less-salty water that does not require high pressures and the corresponding energy intensity of other desalination methods such as reverse osmosis (RO) or the high heat of distillation.
- RO reverse osmosis
- CDI operates by applying a voltage across a brackish water feed to draw ions out of solution and into the carbon electrodes.
- CDI does not rely on forcing water through a membrane, so at low salt content levels (less than 6000 mg/L) even the basic method is three times more efficient than RO.
- the electrode surfaces act as electrochemical capacitors, so CDI can be conceptualized as a process of charging and discharging these capacitors. The energy lost during discharge can be re-captured, increasing system efficiency by 80-90% in some instances. Such efficiencies would allow CDI to compete with RO, perhaps even at the salt concentration level of seawater.
- Electrodes Once the electrodes become charged with separated ions, voltage on the electrodes may be reversed and the channels briefly backflushed to remove ions from the system.
- Carbon-based electrodes are regularly used in CDI due to their high electrical conductivity, low-cost, and impressive specific surface areas. Materials with high specific surface have many more locations to store ions, and thus larger desalination capacities in each cycle.
- Carbon-based electrodes are used in a number of other applications, including lithium batteries, zinc-manganese (Le-Clanche and Alkaline) batteries,
- Materials with controllable porosity are also of use in filtration, both of water and other substances, and may potentially be doped with catalysts for use in other applications.
- a composition of matter, and method of manufacture, of a material having a porous mass including a bound and reduced composite of grapheme oxide that in situ has either fully or partially been reduced to graphene, and either carbon nanotubes or carbon nanofibers, or both.
- the mass is directionally porous, with pores having an average length over 10,000 microns and an average cross sectional area less than 2500 square microns and more than 25 square microns.
- a heat treatment at between 400 and 1500°C in non-oxidizing atmosphere is used to reduce graphene oxide to graphene while binding the graphene to the carbon nanotubes or nanofibers, in an alternative embodiment graphene oxide is reduced chemically.
- Graphene is present at a ratio between 2: 1 and 1:4 in proportion to carbon nanotubes or nanofibers.
- the material can be made with porosity with a typical range of about 75-99% percent.
- a composition of matter includes a porous mass, the porous mass includes a bound and reduced composite of graphene fully or partially reduced from graphene oxide, and a carbon structure selected from the group consisting of carbon nanotubes and carbon nanofibers.
- a method of manufacture of a porous mass includes: preparing a slurry comprising water, graphene oxide, and a carbon structure selected from the group consisting of carbon nanotubes, carbon nanofibers, and activated carbon; freeze-casting the slurry, and sublimating the water, to form a green casting; and reducing graphene oxide of the green casting, thereby binding particles of the graphene oxide and carbon structure to form the porous mass.
- a water purification apparatus includes a first and a second electrode, at least one electrode including a porous mass comprising a bound and reduced composite comprising graphene fully or partially reduced from graphene oxide, and a carbon structure selected from the group consisting of carbon nanotubes and carbon nanofibers, the first and second electrodes adjacent to a channel; a power supply coupled to apply a voltage difference between the first and second electrode; apparatus configured to supply salty water to the channel; and apparatus configured to receive water from the channel.
- An electrochemical apparatus including a first and a second electrode, at least one electrode including a composition of matter comprising a porous mass comprising a bound and reduced composite comprising graphene fully or partially reduced from graphene oxide, and a carbon structure selected from the group consisting of carbon nanotubes and carbon nanofibers; the porous mass of the composition of matter impregnated with a first chemical composition; the apparatus configured to permit the first chemical composition to enter into chemical reactions that provide an electric current between the first and second electrodes.
- Fig. 1 is a flowchart of a process for making an embodiment of a directionally-porous, annealed, graphene-oxide / carbon nanotube composite material.
- FIG. 2 is a diagram illustrating an apparatus adapted to directionally freeze- casting an aqueous material, as is used for forming the directionally-porous, annealed, graphene-oxide / carbon nanotube composite material.
- FIG. 3 is a flowchart of material preparation with Graphene Oxide
- Fig. 4 is a flowchart of material preparation with Graphene Oxide and Carbon Nanotubes or Carbon Nanofibers with and without cellulose nanofibers.
- Fig. 4A is an SEM micrograph of a section taken perpendicular to the freezing direction, showing pore structure.
- Fig. 4B is an SEM micrograph of a section taken parallel to the freezing direction, showing pore structure with pores extending entirely across the micrograph from top left to lower right.
- Fig. 5 is a flowchart of material preparation with graphene oxide and carbon nanofibers, with and without ethanol and copper acetate additives.
- Fig. 6 is a diagram of a capacitive desalination device where carbon electrodes prepared according to the described method may prove useful.
- Fig. 7 is a diagram indicating density versus strength for one embodiment of the annealed or annealed, freeze-east, carbon materials as made by the method herein.
- Fig. 8 is a diagram indicating stress versus strain for an embodiment of the sintered or annealed, freeze-east, carbon materials as made by the method herein.
- Fig. 9 is an illustration from Ramen spectroscopy of graphene oxide reduced to graphene at different temperatures, indicating a potential optimum between 900 and 1100 Celsius.
- Fig. 10 is a plot of Young's modulus on the Y axis, with density in mg/cm 3 on the X axis, for some of the sample scaffolds made.
- the materials have an overall porosity and pore structure that can be carefully controlled.
- the pore structure can be highly aligned with a honeycomb-like structure, isotropic, or a combination of the two.
- the porosity can be homogeneous and uniform or graded.
- the hierarchical 3D architecture of the material and its properties can be tailored to exhibit significant stiffness, strength, toughness, and resilience.
- a directionally-porous, sintered / annealed, graphene-oxide / carbon nanotube composite material is formed by a method 100 (Fig. 1) that begins with preparing 102 a slurry of graphene oxide (GO), carbon nanotubes (CNT), carbon nanofibers (CNFs), or activated carbon, freeze modifiers and binders that may include one or more of a water soluble polymer such as chitosan, alginate, agar, agarose, gelatin, starch, nanocellulose fibers (NF), Ethyl Alcohol (Ethanol), ascorbic acid, or sugar (sucrose), and water.
- a water soluble polymer such as chitosan, alginate, agar, agarose, gelatin, starch, nanocellulose fibers (NF), Ethyl Alcohol (Ethanol), ascorbic acid, or sugar (sucrose), and water.
- Graphene Oxide Dispersion in Water (Graphene Laboratories Inc., Calverton, NY, USA, also known as Graphene Supermarket) at a concentration of 5g/L, and a carbon-oxygen ratio of 4: 1 was used.
- GO is an oxygenated form of graphene, featuring carboxyl, hydroxyl, and carbonyl groups.
- GO is synthesized by oxidizing graphite, using nitric acid or a combination of potassium permanganate and sulfuric acid. GO can be readily reduced to graphene at large quantities using thermal and chemical methods.
- the GO used here was an aqueous dispersion of 0.5- 5 micron diameter flakes, with a thickness of 1 atomic layer in at least 60% of flakes.
- a reduced form of this material has displayed a Brunauer-Emmett- Teller (BET) surface area as high as 833 m /g.
- BET Brunauer-Emmett- Teller
- graphene oxide is used to refer to samples prepared with GO, even those that have been treated thermally and may be fully or partially reduced to graphene (G).
- G graphene
- RGO reduced GO
- Thermal reduction of GO occurs when it is heated to between 600- 1200C in an inert or non-oxidizing atmosphere such as argon, removing resident water molecules and oxygen containing groups from the GO sheets and varies with temperature.
- aromatic carbon sources ethylene or radical carbon
- straight carbon nanotubes - JC121 (JEIO Tech, South Korea) were added to increase surface area, electrical conductivity, and improve mechanical properties.
- Carbon nanotubes are tubular structures made of sp2 bonded carbon atoms. Carbon nanotubes are hydrophobic and exhibit ⁇ - ⁇ interaction, often leading to clumping and poor dispersion when added to water. Carbon nanotubes have diameters on the order of tens of nanometers, and can have lengths up to one micrometer.
- the carbon nanotubes used here are all of the MWCNT variety, with a pristine surface area of 300-310 m7e. 'Curly' Carbon Nanotubes (Curly CNT)
- Carbon Nanofibers (CNFs)
- the carbon nanofibers used in the reported study belong, like multi- walled carbon nanotubes, to the structural family of fullerenes.
- PR-24-XT-PS carbon nanofibers were purchased from Pyrograf ® Products (Inc.) and are pyrolitically stripped carbon nanofibers.
- MWCNTs and CNFs The difference between both, MWCNTs and CNFs, is their structures.
- the CNFs used have an average diameter of 100 nm and an average length of 70 ⁇ .
- Cellulose nano-fibrils also referred to as nanocellulose
- Cellulose nano-fibrils are a wood-based material.
- Nanocellulose is used as a binder.
- Chitosan solution 2.4% (w/v) was prepared by dissolving low molecular weight chitosan powder (448869, Sigma Aldrich, St. Louis, MO, USA) into a mixture of acetic acid and water. The resulting solution was then vigorously shaken, and placed on a bench top bottle roller for at least 24 hours to ensure complete mixing. Chitosan acts simultaneously a binder and dispersant when freeze casting MWCNTs.
- Ethanol EMD Millipore EX0280— 3 Denatured Ethyl Alcohol 95% was used as a dispersing agent, particularly in the CNT hybrid preparations.
- L-Ascobic Acid (LAA) in some embodiments is used both as a binder and as a chemical reducing agent capable of reducing some of the GO to G. and encouraging formation of covalent chemical bonds between G and CNT or CNF in the cast scaffolds
- sucrose was used as a binder.
- maple sap and maple syrup was used as a binder.
- Maple syrup was certified organic, grade- A amber, maple syrup from Mt. Cube Farm, Orford, NH were used. Maple sap was also obtained from Mt. Cube Farm.
- Dispersants and freeze modifiers such as ethanol and chitosan
- some additional solid carbon material were chosen, and other binders and dispersants were added for some trials.
- the carbonaceous slurries are prepared as 10 mL samples in 50 mL plastic cups. First, dry powder carbon nanotubes or nanofibers were weighed with a precision balance to within 1% accuracy. Samples with standard carbon nanotubes were prepared with 100 mg - 400 mg in the lOmL sample. Curly carbon nano tubes were prepared with 50-150 mg of solids in the 10 mL of liquid. Any additional binders, such as cellulose nanofibers, in the recipe tested were added to the slurry. lOmL of water or 10 mL of GO- water suspension was added.
- ethanol 1-2 mL was added as dispersant in many instances in order to aid dispersion of CNT which tend to remain aggregated or entangled and poorly dispersed in solution. We believe that the ethanol is fully sublimated during freeze- drying, and thus does not remain in the final scaffold. In experiments with other carbon sources, ethanol was used in some experiments as a freeze- modifier to increase final pore sizes.
- the cup is sealed, and the sealed cup is then shear mixed for two minutes at 3000 rpm.
- the mixed slurry is then placed 104 immediately in the freeze-casting mold 202 (Fig. 2).
- LAA with a mass ratio of 3.33: 1 LAA to GO was added to a 30mL vial.
- concentration ratios ranging from 0.5: 1 to 100: 1 were used to check differences between gels and resultant scaffolds, and corroborate that 3.33: 1 provided the best gelation.
- 5-25 mL GO aqueous solution was added to the 30mL vial, and the solution was mixed vigorously with a magnetic stir bar for several minutes until the solution appeared of uniform composition with no LAA remaining unabsorbed, at which point the stir bar was removed.
- the solution was then left undisturbed for between 1 to 48 h at temperatures between 25-80°C, during which time gelation occurred, forming an RGO hydrogel.
- the RGO hydrogels were subjected to solvent exchange, in order to chemically remove LAA from the hydrogel.
- the hydrogel was left in the 30 mL vial and ethanol was added until the vial was full. In the first few of these ethanol baths, a slight yellowing of the solution surrounding the gel indicated that solvent exchange was indeed eliminating LAA from the hydrogel.
- the ethanol was decanted and replaced with fresh ethanol every few hours or as necessary until visual mixing of water and ethanol was no longer evident in the vial and bubbles stopped emerging from the surface of the hydrogel. At this point, the vial was rinsed several times with deionized (DI) water and then filled with DI water.
- DI deionized
- ethanol infused hydrogels floated in DI water and the same procedure was used with water as with ethanol, decanting and replacing the water every few hours until the hydrogel sank, stopped bubbling, there was no more visual mixing of ethanol and water, and the smell of ethanol was undetectable when the container was opened after an hour or more of resting.
- gelation was attempted in the PTFE molds used for freezing. In this instance, a layer of parafilm was laid over a copper base plate and the base plate then taped on securely to the bottom of the mold, ensuring that water could not enter or exit and that the bottom would not fall off. A magnetic stirrer was placed in the mold and then removed after mixing.
- a copper base plate and parafilm layer were added to the top of mold in the same manner used with the bottom, and the entire contraption was heated according to the same outlines pertaining to gels in vials. Solvent exchange was conducted by removing the top base plate and placing the whole contraption in an ethanol and then water bath until the reactions came to an end. A small strip of tape was placed over the open end of the mold to ensure that the sample did not come out while floating in water.
- RGO hydrogels were prepared in standard, 10 mL mixing cups.
- gel preparation was identical to production in a vial, except that solvent exchange was conducted in a large beaker, since almost no solvent could fit in the mixing jar.
- the mold as illustrated in Fig. 2, is a Teflon tube 202 placed on a cooled plate 203 that closes the tube and is attached to a copper cold finger 204 with a top end adjacent to the bottom end of the tube.
- the cold finger 204 top end is temperature regulated by using feedback from a thermocouple 206 attached to the cooled plate applied to an electric heater 208 attached to the finger.
- the cold finger is cooled by immersing its lower end into liquid nitrogen 210.
- a layer of Vaseline is applied around the edge of the copper base plate to form a seal between the edge of the base plate and the bottom of the mold.
- the copper base plate is then placed on top of a copper rod, or "cold finger," which stretches down into a liquid nitrogen bath.
- the temperature at the top of the copper rod is monitored by a thermocouple and temperature clines can be regulated via a PID controller and a powerful heating element near the top of the cold finger.
- a thermocouple Prior to freezing, the top of the rod was maintained at a constant 5°C, and samples placed on top were precooled to this temperature before being subjected to a determined cooling rate of either 1 or 10 0 °C min - " 1 to a minimum temperature of -150°C.
- the ice phase (not shown) nucleates at the cold plate, and grows along the thermal gradient and upwards through the mold, concentrating solute and particles between the crystals thereby ice-templating it, thus forming a material with a hierarchical architecture whose pores are filled with ice.
- samples are fully frozen, they are removed from the cold finger, the samples are punched out of the mold using an Arbor press. The green casting is then freeze- dried to remove the ice phase.
- each mold has a tubular shape with an inner diameter of 18.8 mm (3/4 inch) and a height of 40 mm.
- each sample is dehydrated for 48-72 h in a FreeZone 4.5 Liter Benchtop Freeze Dry System (Labconco, Kansas City, MO).
- Samples containing sucrose, maple sap, or maple syrup are placed in a glass desiccation chamber with silica gel desiccant (-3+8mesh granules, Alfa Aesar, Ward Hill, MA) with relative humidity 0.0 in order to keep atmospheric moisture from hydrating the sugar until such a time as they could be subjected to heat treatment or acclimated for mechanical testing.
- silica gel desiccant -3+8mesh granules, Alfa Aesar, Ward Hill, MA
- the graphene oxide By heating the graphene oxide to a temperature in the range of about 490°C to 1000 °C, and in particular embodiments to 498 °C and 1000 °C, many of the carboxyl and hydroxyl groups forming the outer hydrophilic portion of the graphene oxide flakes are removed, which results in reduced graphene.
- the samples are thermally reduced in an inert atmosphere, such as argon, by placing them in graphite boats and inserting them in a tube furnace which is heated at 5 °C per minute from room temperature to 1000 °C, before holding them at this temperature for two hours.
- the temperature of the furnace is increased at a heating rate of 5°C per minute to 498°C in argon gas, and maintained there for two hours. After two hours the furnace was shut off, and allowed to cool to room temperature unassisted (at an approximate rate of 5 °C).
- the GO thermal reduction and annealing may be done by heating the graphene oxide in various alternative non- oxidizing or reducing atmospheres, for example Argon, Hydrogen, Nitrogen, Xenon, Helium, Neon, blends of the aforementioned non-oxidizing gasses such as Forming gas (a mixture of hydrogen and nitrogen), or ultra-high vacuum should suffice to prevent oxidation of the material.
- non-oxidizing gasses such as Forming gas (a mixture of hydrogen and nitrogen), or ultra-high vacuum should suffice to prevent oxidation of the material.
- several heating sources capable of reaching 1000°C are known and usable including microwave, flash light, laser, plasma, electric current, field assisted, or other kinds of furnaces.
- other temperatures are used in the 400 to 2000 °C range.
- the wire is a 220 ⁇ diameter diamond encrusted steel wire, and samples are cut using a wire speed of 0.7 m s-1. Each sample is mounted on a ceramic plate using CrystalbondTM with the unidirectional pores perpendicular to the face of the plate, then cut to provide repeatable data points from several heights along the sample. 5mm cubes are cut at standard heights above the bottom of samples before heat treatment, with the cube centered at 10, 20, and 30 mm from the bottom of the sample. For each height, four cubes are prepared and at least three subjected to mechanical and structural
- SEM Scanning electron microscopy
- a Leica Optical microscope was used for preliminary visual investigation of the samples, such as pore alignment and orientation. Representative optical micrographs were taken of each sample after each cut at several magnifications, providing a basic understanding of the sample and its hierarchical pore structure.
- At least some electrical conductivity is required for use of the freeze-east and annealed material produced by the method described above as an electrode.
- the four point probe method (SIGNATONE, Gilroy, CA) was used to perform conductivity measurements. Measurements were taken on at least 5 different points on samples and averaged.
- a disadvantage of this method applied to porous materials such as those of this study is that the probes affect the micro structure of the samples. However, because the probe diameter is about one magnitude larger than the typical pore diameters of the carbon aerogel materials under investigation, reproducible results were obtained.
- a first series of experimental samples using Graphene Oxide and a selected binder in a ratio of 0.24: 1 to graphene oxide mass ratio of graphene solid was performed using maple syrup, maple sap, sucrose, and chitosan as binders.
- the graphene was the 5 g per liter suspension in water previously described, which also provided the liquid phase (water) for the slurry.
- sucrose resulted in particularly promising, stable green bodies for heat treatment followed by mechanical testing.
- Stable freeze cast graphene and GO structures depended on the freezing rate. Faster cooling rates of 10°C/min led to significantly more stable samples than l°C/min, the 10°C/min rate was used for all samples subjected to further characterization. A cooling rate to 10°C/min led to successfully lyophilized samples in every case, even though those frozen from GO alone and GO + LAA, GO+sap, GO+syrup, and GO+chitosan were all too fragile to further analyze. Alone among these, GO+sucrose samples were relatively easy to handle. GO+activated carbon also gave scaffolds, although these were not analyzed.
- Un-annealed GO+sucrose samples had striking coloration of various shades between gold and translucent white, and after annealing were a slightly reflective gray.
- Shrinkage due to reduction of GO was observed by both the hydrogel samples during chemical reduction (GO-LAA) and the GO-sucrose samples during thermal reduction or annealing.
- Hydrogels exhibit significant shrinkage over time, of about 30-40% volume.
- GO-LAA Gels that have expelled water and are thus smaller also have a lower porosity and less water contained in the gel when freezing. Additionally, gels shrank so that they were smaller than the PTFE molds, meaning they were not in direct contact with the sides of the insulating molds during freezing. Cracking and issues solvent exchange arose in many samples and were addressed.
- Aerogels with ratios of 10: 1 LAA:GO were brittle compared to those frozen with the ratio of 3.33: 1, while gels with a ratio of 1: 1 fell apart in the lyophilizer. Greater shrinkage exhibited by samples with ratios of 10: 1 and 100: 1 combined with brittleness resulting from residual LAA made samples too dense and small for desired characteristics. 3.33: 1 was chosen as the best mass ratio of LAA to GO.
- Shrinkage exhibited in hydrogels meant that they were no longer flush with PTFE molds during freezing, the gap was filled with water.
- sucrose added as a binder that through thermal treatment can be converted to carbon performed better.
- sucrose far outperformed any of the other alternatives, producing more robust scaffolds with distinct anisotropic features and a well- aligned pore structure.
- GO + sucrose aerogels fabricated and tested according to the flowchart of Fig. 3 displayed large discrete streaks of differing golden, translucent colors; these colors are caused by light reflecting on the GO sheets of different orientations as templated by the ice crystals and illustrate that the pores are continuous along the entire length of the sample with the excellent pore alignment. The pores parallel the direction of ice crystal growth, the uniform colors indicate well aligned GO sheets.
- sucrose samples created with 0.05g per 10 mL were structurally sound and exhibited good micro structure. Lower amounts of sucrose are preferable because of a resulting lower density and because thermally converted sucrose has lower electrical properties than GO or graphene.
- the green body GO + sucrose samples resulted in low- density samples (25.0 mg/cm ) with very high overall porosity (98.9%), good pore alignment, and an elastic modulus between 144-318 kPa. As expected, green body samples did not exhibit the same elastic recovery as the annealed material..
- Samples of GO+sucrose were initially subjected to thermal annealing at 200 C for 2 hours in an Ar atmosphere. This temperature was chosen because it is just above the caramelization temperature of sucrose and because 200°C is a typical thermal treatment temperature for GO and RGO structures for partial reduction. However, exposure to this tempereature may have melted the sucrose. The samples had lost all structure when removed from the furnace.
- Annealing temperatures of 600-800C were chosen for the next set of experiment. These higher target temperatures meant that the furnace would only be in the sucrose melting regime for a short period of time.
- the cell wall surfaces have a wrinkled appearance, that likely is due to the thermal reduction of the GO material. These wrinkles, which may belie defects, might reduce electron mobility and thus electrical conductivity, though it has been suggested that thermally reduced GO can be 'healed' to an extent with the proper carbon source.
- Table 2 Material properties of GO + sucrose before and after annealing.
- Composition 1 (Green Body): Slurries were prepared using sucrose (EMD Chemicals, Gibbstown, NJ) as binding agent. Graphene Oxide solution (5g/L aqueous dispersion, flake size 0.5-5 ⁇ , 4: 1 C:0 ratio, Graphene Supermarket, Calverton, NY) was used for the carbonaceous component. 10 mL of Graphene Oxide solution was added to 50 mg sucrose. The solution was mixed on a high shear SpeedMixer (DAC 150 FVZ-K, FlackTek, Landrum, SC) at a speed of 3000 RPM for 30s for mixing and degassing. Samples were frozen on the freeze cast setup depicted in Fig.
- sucrose EMD Chemicals, Gibbstown, NJ
- Graphene Oxide solution 5g/L aqueous dispersion, flake size 0.5-5 ⁇ , 4: 1 C:0 ratio, Graphene Supermarket, Calverton, NY
- Composition 2 (Annealed): Samples from Composition 1 were subjected to a temperature of 800°C for 2 hrs in an argon atmosphere heated at a rate of 4°C/min.
- Samples were placed in a graphite crucible to prevent oxidation during heat treatment.
- Samples were prepared in 5 mm cubes with a diamond wire saw (WELL Diamond Wire Saws, Inc., Norcross, GA). It is expected that a range from 600-2000C would give similar results. All gases preventing oxidation during thermal treatment would be expected to be appropriate, and a temperature ramp rate from 0.1-100 times the rate used would be expected to work, the ramps could include holding times at lower temperatures for thermal pre- treatment of the samples.
- Table 3 GO- sucrose pore structure before and after annealing.
- At least 3 samples per layer of at least 3 corks per freezing run were tested parallel and perpendicular to the Freezing Direction (FD), in the case of the electrical measurements both with the probe parallel to the long pore axis and with the probe perpendicular to the long pore axis gave a 4 Point Probe measurement averaging 11.3 Siemens.
- FD Freezing Direction
- GO-sucrose and GO-LAA produced 3D RGO aerogels exhibiting high porosities (98.6-99.42%), low densities (12.1-31.1 g/cm ), uniform aligned hierarchical pore structure, and anisotropic mechanical properties.
- sucrose as a binder and carbon precursor in freeze casting applications is introduced herewith.
- GO+sucrose samples showed great promise for thermal reduction.
- the limited elastic recovery and resilience seen in the first generations of these samples may be improved upon and better mechanically strong and resilient scaffolds will result.
- GO+CNT+ nanocellulose were fabricated from 10 ml of the GO suspension with 2% wt. % nanocellulose and either 1% carbon nano fibers or 1, 2, or 4 wt. % CNTs, with or without 1 or 2 wt. % chitosan or 2 % ethanol, according to the flowchart of Fig. 4.
- the GO-CNT material was sectioned perpendicular to the axis of freezing, and then imaged with a scanning electron microscope to visualize its pore structure, as shown in Fig. 4A. Pores averaged 917 square microns in cross section. Pores are enlarged along the axis of freezing with most pores having lengths over 10,000 microns, and many pores extend through the entire 35mm of a freeze-east sample, as illustrated in Fig. 4B. Other
- pore sizes ranging from 1494 to 2022 square microns, with pore aspect ratios ranging from 1.4 to 4.5 and porosity of 97 to 98.5 percent. It is expected that pore sizes may be adjusted by altering the rate of freezing during the freeze-casting step of the process, as well as the Ethanol content of the slurry, pore size gradients or variations along the length of the sample may further be controlled and adjusted by, for example, altering material composition, applied cooling rate or applying thermal property variations along the freeze-casting mold during the freezing process.
- conductivity of the GO-CNF materials ranged from 7 to 15.6 Siemens/meter.
- Slurry Preparation - Carbonaceous slurries are prepared by mixing the components according to a specific composition into a 50-mL SpeedMixer cup. After all of the components are transferred into the cup, the cup containing the slurry is shear-mixed with a SpeedMixer at approximately 3,000 rpm for 2 minutes. The following compositions are attempted.
- the freezing process starts at 2°C and a cooling rate of 10°C is applied until a temperature of -150°C is achieved. The entire freezing process takes approximately 40 minutes.
- the frozen samples are freeze dried in the FreeZone 4.5 Liter Freeze Dry System (Labcono, Kansas City, MO, USA) running at less than 0.01 mBar for approximately 72 hours to sublime the ice.
- Freeze dried samples are annealed, or thermally reduced, in a tube furnace (Thermo Scientific Linberg/Blue MTM 1,500°C General-Purpose Tube Furnace) at 1,000°C under constant Argon purge at 10 psi gauge.
- a sample is placed in a graphite sample holder, which is then being placed inside the tube furnace.
- the tube furnace is first evacuated using a vacuum pump to approximately -70 psi gauge.
- Argon gas is filled into the tube at 10 psi gauge.
- the evacuation and purging process is repeated 3 times to minimize residual oxygen inside the chamber.
- the temperature is raised at 5°C/min to 1,000°C held at 1,000°C for 2 hours, finally, the furnace cooled unassisted to room temperature at a rate of approximately 5°C/min.
- sucrose - a common disaccharide - was demonstrated as functioning as a binder, other sugars (including monosaccharides and disaccharides) and sugar alcohols (including sorbitol, or mannitol) will suffice.
- sugar alcohols including sorbitol, or mannitol
- ethanol was demonstrated as a freeze modifier in our experiments, other common low-molecular- weight alcohols such as methanol, propanol, isopropanol, or butanol will also modify freeze casting, and that methanol and propanol in particular will serve to increase pore sizes.
- nanocellulose fibers as a binder
- other polysaccharides such as chitin or starch will also function as binders.
- fibrous polypeptides such as gelatin, to also serve as a binder.
- scaffolds may be built with slurry of graphene oxide and both straight and curly nanotubes, or with slurry of graphene oxide and both nanotubes and nanofibers. Further, it is anticipated that the slurry may or may not contain a binder, and where a binder is used the binder may include Certain specific combinations of features that are anticipated include the following:
- a composition of matter designated A includes a porous mass, the porous mass includes a bound and reduced composite of graphene fully or partially reduced from graphene oxide, and a carbon structure selected from carbon nanotubes and carbon nanofibers.
- composition of matter designated AA including the composition designated A wherein the mass is directionally porous.
- a composition of matter designated AB including the composition designated A or AA wherein the porous mass has been formed from graphene oxide reduced by heat treatment in non-oxidizing atmosphere, where graphene and unreduced graphene oxide is present at ratios of between 1: 1 and 1:4 in proportion by weight to the carbon structure.
- a composition of matter designated AC including the composition designated A, AA, or AB wherein at least a a plurality of pores of the porous mass extend in an axis at least 10,000 microns, and have an average cross sectional area measured perpendicular to the axis is between 25 and 2500 square microns.
- a composition of matter designated AD including the composition designated A, AA, AB, or AC wherein the average cross sectional area of the pores is between 80 and 1000 square microns.
- a composition of matter designated AE including the composition designated A, AA, AB, AC, or AD the porous mass having porosity between 75 and 99 percent.
- a composition of matter designated AF including the composition designated A, AA, AB, AC, AD, or AE wherein the porous mass has been formed from graphene oxide reduced by heat treatment in non-oxidizing atmosphere, wherein graphene and unreduced graphene oxide is present at ratios of between 1:2 and 1:4 in proportion by weight to the carbon structure.
- a composition of matter designated AG including the composition designated A, AA, AB, AC, AD, AE, or AF having an electrical conductivity of at least 7 siemens/meter.
- a composition of matter designated AH including the composition designated AG having an electrical conductivity between 7 and 50 siemens/meter.
- a composition of matter designated AI including the composition designated A, AA, AB, AC, AD, AE, AF, AG, or AH wherein the carbon structure comprises carbon nanotubes.
- a composition of matter designated AJ including the composition designated A, AA, AB, AC, AD, AE, AF, AG, or AH wherein the carbon structure comprises carbon nanofibers.
- a composition of matter designated AK including the composition designated AI or AJ, further comprising a second carbon structure selected from graphite nanoparticles and activated carbon nanoparticles.
- a method of manufacture designated B of a porous mass including:
- preparing a slurry comprising water, graphene oxide, and a carbon structure selected from carbon nanotubes, carbon nanofibers, and activated carbon; freeze-casting the slurry, and sublimating the water, to form a green casting; and reducing graphene oxide of the green casting, thereby binding particles of the graphene oxide and carbon structure to form the porous mass.
- a method designated BA including the method designated B wherein reducing graphene oxide of the green casting is performed by heating the green casting to a temperature between 400 and 1500 degrees Celsius.
- a method designated BB including the method designated BA or B wherein the graphene oxide includes primarily 0.5- 5 micron diameter flakes, with a thickness of 1 atomic layer in at least 60% of flakes.
- a method designated BC including the method designated B, BA or BB wherein the slurry further includes ethanol.
- a method designated BD including the method designated B, BA or BB wherein the carbon structure includes straight carbon nanotubes.
- a method designated BE including the method designated B, BA or BB wherein the carbon structure includes curly carbon nanotubes.
- a method designated BF including the method designated B, BA or BB wherein the carbon structure comprises carbon nanofibers.
- a method designated BGA including the method designated BG wherein the polysaccharide includes cellulose nanofibers.
- a method designated BGB including the method designated BG wherein the polysaccharide includes cellulose nanofibers.
- a method designated BH including the method designated B, BA, BB,
- the slurry further includes a sugar.
- a method designated BHA including the method designated BH wherein the sugar includes sucrose.
- a method designated BI including the method designated B, BA, BB, BC,
- slurry further includes starch
- a water purification apparatus designated C including a first and a second electrode, at least one electrode including a composition of matter according to those designated A, AA, AB, AC, AD, AE, AF, AG, AH, AI or AJ, the first and second electrodes adjacent to a channel; a power supply coupled to apply a voltage difference between the first and second electrode; apparatus configured to supply salty water to the channel; and apparatus configured to receive water from the channel.
- An electrochemical apparatus designated D including a first and a second electrode, at least one electrode including a composition of matter according to those designated A, AA, AB, AC, AD, AE, AF, AG, AH, AI or AJ; the porous mass of the composition of matter impregnated with a first chemical composition; the apparatus configured to permit the first chemical composition to enter into chemical reactions that provide an electric current between the first and second electrodes.
- D-Mannitol (white to off-white powder), tryptone (product of New Zealand), yeast extract (molecular genetics powder) and agar (product of Morocco) were obtained from Fisher Scientific (Waltham, MA).
- Sodium alginate (from brown algae) was purchased from Sigma- Aldrich (St. Louis, MO). All chemicals were directly used without any purification.
- Gluconacetobacter hansenii (ATCC® 23769TM) was obtained from the American Type Culture Collection (ATCC, Manassas, VA) for bacterial cellulose culture.
- Gluconacetobacter hansenii ATCC ® 23769TM
- ATCC ® 23769TM was used as the model strain and maintained on agar plates containing 25 g/L D-mannitol, 5 g/L yeast extract and 5 g/L tryptone and 20 g/L agar.
- the mannitol culture medium used for BC production consisted of 25 g/L D- mannitol, 5 g/L yeast extract and 5 g/L tryptone.
- the strain from the agar plate was inoculated into a conical flask containing mannitol culture medium as the seed culture.
- the initial pH value of the medium was adjusted to 5.0 and was not regulated during the culture.
- the seed culture was incubated at 30°C and 130 rpm on a rotary shaker for 2 days, and 6 mL of this seed culture was inoculated into a 100-mL culture medium in 600-mL conical flask for production of BC.
- the cultivation was carried out at initial pH 5.0 and 30°C in a static incubator for 10 days.
- the BC pellicles produced on the surface of mannitol culture medium were harvested and washed successively with water and 1% (w/v), aqueous NaOH at 90°C for 15 min, and then washed with deionized water to remove all microbial product contaminants and obtain purified pellicles.
- the BC pellicles were initially broken down into particles by cryomill (Retsch, Model 2013, Germany) via pre-cooling step at 5 Hz for 20 min and cryomilling step at 30Hz for 30 min. The whole procedure was carried out at liquid nitrogen temperature (- 96°C). The obtained BC particles could maintain BC's unique network structure with nanometer- scale pores.
- the BC particles was then mixed with a binder, sodium alginate solution (4.8 wt%) by a shear mixer (SpeedMixerTM DAC 150 FVZ-K, FlackTek Inc, Landrum, SC) at 3000 rpm for 2 min, to form homogenous BC-sodium alginate mixed slurries (5.0 wt BC and 0.9 wt sodium alginate in the slurries) prior to freeze casting.
- a shear mixer SpeedMixerTM DAC 150 FVZ-K, FlackTek Inc, Landrum, SC
- the BC-sodium alginate mixed slurries were freezed by a custom-made freeze-caster at a speed of 10°C/min from 2°C to -150°C. After the BC-sodium alginate mixed slurries were completely frozen, the frozen materials were then lyophilized by a FreeZone 4.5 L lyophilizer (Labconco, Kansas City, MS) for 3 days to obtain the freeze-east BC aerogels.
- the freeze-casting process can generate micrometer- scale porous structure for the aerogels while the BC pellicles maintain their nanometer- scale porous structure.
- the freeze-east BC aerogels with unique structure include two-lengths- scale porosity with high specific surface area.
- the freeze-east BC aerogels were placed in cylindrical graphite boats for carbonization in a high vacuum furnace (ABAR 90, ABAR Corporation).
- the carbonization process is performed in 3 steps: (1) heat from room temperature (RT) to carbonization temperatures at 1000°C, 1100°C and 1200°C respectively at a heating rate 10°C/min; (2) carbonize at 1000°C, 1100°C and 1200°C respectively for lhr; (3) cool down to room temperature at a cooling rate 10°C/min.
- the cylindrical frozen BC-sodium alginate mixed slurries after freeze casting were cut initially by a saw and then smoothed by a permanent steel knife (Delaware Diamond Knives, Inc. Wilmington, DE) at a -10°C cold room.
- the purpose for the step was to form uniform and smooth cross sections both perpendicular and parallel to the direction of freeze casting for the freeze-east BC aerogels.
- the cut samples were then placed for 3 days in the FreeZone 4.5 L lyophilizer to freeze-dry the freeze-east BC aerogels.
- the freeze-dried BC aerogels with smooth cross sections and carbonized aerogels were observed using scanning electron microscopy (SEM, FEI XL-30 FEG ESEM, Hillsboro, OR), and the accelerating voltages were set between 2 to 5 KV.
- SEM scanning electron microscopy
- the mean value of the pore area from the cross sections (cut perpendicular to the freeze direction) in the freeze-east BC aerogels and carbonized freeze-east aerogels were measured on SEM images using ImageJ (ImageJ, U.S. National Institutes of Health, Bethesda, MD).
- freeze-east BC aerogels and carbonized freeze-east BC aerogels were cut into cubes with 5x5x5 mm3 dimension from topic, middle and bottom layers of the scaffolds by a Well 4240 saw (WELL Diamond Wire Saws, Inc., Norcross, GA) for specific surface area characterization.
- the diameter of diamond- decorated steel wire for the saw was 220 ⁇ , and the wire speed was 0.6 m/s. All the cubes were first degassed at 100°C and specific area values for them were measured through nitrogen adsorption by Brenauer- Emmett-Teller (BET) system.
- the electrical conductivity ( ⁇ ) measurement for all samples from top, middle and bottom layers of carbonized freeze-east BC aerogel scaffolds was conducted using a standard Four- Point probe test.
- a four point probe (Signatone, Model S-301-4, Gilroy, CA) used is a simple apparatus for measuring the resistivity (the inverse of electrical conductivity) of samples by passing a current through two outer probes and measuring the voltage through the inner two probes.
- the Four-Point probe tests are ideal for measurement of resistivity of sheet and bulk.
- a corrected formula was used to calculate the electrical conductivity of carbonized freeze-east BC aerogels.
- the Raman spectra for the carbonized freeze-east BC aerogel samples processed at different carbonization temperatures were obtained using Confocal Raman Microscope CRM 200 (WITec GmbH, Ulm, Germany) with a 514.4 nm laser source.
- the range of wavenumber for the spectra was chosen from 800 to 2000cm "1 .
- Freeze-east BC aerogels and carbonized freeze-east BC aerogels were cut into cubes with 5x5x5 mm dimension from topic, middle and bottom layers of the scaffolds by the Well saw for mechanical properties characterization.
- the diamond- decorated steel wire diameter for the saw was 220 ⁇ , and the wire speed was 0.6 m/s.
- the compression tests were then conducted on an Instron 5948 (Instron, Norwood, MA) with a 5 N load cell at ambient conditions.
- the cross-head speed for all the measurements was 0.05 mm/s (strain rate 0.01/s).
- Two test directions were used for the mechanical test, including the test parallel to the freezing direction and the test perpendicular to the freezing direction.
- BC is usually grown in the form of pellicles of several tens of millimeter in diameter and few millimeter in thickness.
- the pellicles consist of a network of BC- nano fibers, which typically are less than 100 nm in diameter and have pore diameters that range from several tens to several hundred nanometers.
- the BC pellicles were cryomilled into micrometer- scale BC flakes with edge lengths in the range of 20-100 ⁇ and a well- maintained nanometer- scale pore structure. The cryomilling may be omitted in some embodiments.
- the mean density of freeze-east BC aerogels before carbonization was about 0.055 g/cm. 3
- the diameter and height of carbonized freeze-east BC aerogel were about 50% of their original values for freeze-east BC aerogel due to the significant dimensional shrinkage during carbonization. However, because during carbonization elements and side- groups other than carbon are mostly removed the mean density of the freeze-east BC aerogels after carbonization was 0.052 g/cm ⁇ very similar to its density before carbonization.
- the freeze-east aerogels exhibit a hierarchical architecture with a honeycomb-like, uniform distribution of micro meter- scale pores that have mean long and short pore axes in the range of 15-40 ⁇ and 8-20 ⁇ , respectively, perpendicular to the freezing direction and run through the entire sample parallel to the freezing direction.
- the nanoporosity in the cell wall material ranges from several tens to several hundreds of nanometers in diameter.
- the freeze-east aerogels retained the hierarchical architecture from before carbonication.
- the aerogels had a honeycomb-like, uniform distribution of micro meter- scale pores that had mean long and short pore axes in the range of 8-20 ⁇ and 4-10 ⁇ , respectively, perpendicular to the freezing direction and run through the entire sample parallel to the freezing direction, which is about half of the value before carbonization.
- the nanoporosity size in the cell wall material was, due to the material shrinkage, difficult to measure after carbonization.
- the electrical conductivity of carbonized freeze-east BC aerogel processed at 1200°C can be up to 1.68+0.04 S/cm at a density of 0.055 g/cm 3 , which was significantly higher than the carbonized freeze-east BC aerogels processed at 1000°C (0.70+0.02 S/cm) and 1100°C (0.71+0.02 S/cm) whose density was in the range of 0.044-0.046 g/cm 3 .
- Carbonized freeze-east BC aerogels processed above 1200°C turned into are structure with poor integrity and lower electrical conductivity.
- the Young's modulus of the not yet carbonized material ranged from 476 to 646 kPa, the yield strength ranged from 27.3 to 37.1 kPa, and the toughness from 24.2 to 42.2 kJ/m for densities ranging from 0.0520 to 0.0570 g/cm , respectively.
- the Young's modulus of the not yet carbonized material ranged from 196 to 268 kPa, the yield strength from 12.3 to 15 kPa, and the toughness from 18.6 to 25.2 kJ/m 3 for densities ranging from 0.052 to 0.057 respectively.
- the mechanical anisotropy which is the ratio of the property values measured parallel to freezing direction divided by the values determined perpendicular to the perpendicular to the freezing direction ranged from 2.43 to 2.41 for the not yet carbonized material.
- the Young's modulus of the carbonized material ranged from 373 to 633 kPa, the yield strength from 33.9 to 37.7 kPa, and the toughness from 19.8 to 26.0 kJ/m 3 for densities ranging from 0.049 to 0.052
- the Young's modulus of the carbonized material ranged from 332 to 507 kPa, the yield strength from 20.1 to 36.5 kPa, and the toughness from 12.3 to 21.5 kJ/m 3 for densities ranging from 0.049 to 0.052 respectively.
- the mechanical anisotropy which is the ratio of the property values measured parallel to freezing direction divided by the values determined perpendicular to the perpendicular to the freezing direction ranged from 1.12 to 1.25 for the carbonized material.
- the materials can be suitable for many applications, like bone tissue engineering, drug delivery, biocatalysis reactions.
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Ceramic Engineering (AREA)
- Materials Engineering (AREA)
- Power Engineering (AREA)
- Structural Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Microelectronics & Electronic Packaging (AREA)
- General Chemical & Material Sciences (AREA)
- Electrochemistry (AREA)
- Manufacturing & Machinery (AREA)
- Life Sciences & Earth Sciences (AREA)
- Molecular Biology (AREA)
- Water Supply & Treatment (AREA)
- Environmental & Geological Engineering (AREA)
- Hydrology & Water Resources (AREA)
- Inorganic Chemistry (AREA)
- Crystallography & Structural Chemistry (AREA)
- Nanotechnology (AREA)
- Health & Medical Sciences (AREA)
- Analytical Chemistry (AREA)
- Carbon And Carbon Compounds (AREA)
Abstract
A composition of matter, and method of manufacture, of a material having a porous mass including a bound and reduced composite of graphene formed by in-situ reduction of graphene oxide, and either carbon nanotubes or carbon nanofibers. In particular embodiments, the mass is directionally porous, with pores having average length over 10,000 microns and average cross sectional area less than 2500 square microns and more than 25 square microns. In embodiments, heat treatment at between 400 and 2000 degrees Celsius in non-oxidizing atmosphere is used to reduce graphene oxide to graphene while binding the graphene to the carbon nanotubes or nanofibers, in an alternative embodiment graphene oxide is reduced chemically. Graphene and/or graphene oxide is present at a ratio between 2:1 and 1:4 in proportion to carbon nanotubes or nanofibers. The material can be made with porosity 75 to 99 percent.
Description
MATERIAL AND METHOD OF MANUFACTURE OF ELECTRODES AND POROUS FILTERS FORMED OF ICE-TEMPLATED GRAPHENE-OXIDE AND CARBON NANOTUBE COMPOSITE, AND APPLICATIONS THEREOF
RELATED APPLICATIONS
[0001] The present application claims priority to United States Provisional Patent Application 61/928,920 filed 17 January 2014, and to United States Provisional Patent Application 62/087,565, filed 4 December 2014. The entire contents of the above-referenced parent applications are incorporated herein by reference.
GOVERNMENT INTEREST
[0002] The invention was made with government support under grant 1200408 awarded by the Civil, Mechanical and Manufacturing Innovation (CMMI) Division of the National Science Foundation and grant number DE-AC07-05ID14517 (NEUP10-848) awarded by the Department of Energy. The government has certain rights in the invention.
FIELD
[0003] The present document relates to the field of ice-templated carbon-based materials.
BACKGROUND
[0004] Graphene, a material comprised of monatomic sheets of two dimensional, sp2-hybridized carbon atoms arranged in a hexagonal pattern, has been investigated intensely since its discovery in 2004. It is hypothesized that graphene, and carbon nanotubes formed of graphene sheets folded into cylinders, may have properties allowing them to form useful materials. Carbon nanofibers are also expected to have interesting properties. Among other properties of graphene, nanotubes, and nanofibers are a degree of electrical conductivity. Carbon nanotubes and nanofibers also have good tensile strength within each fiber or particle; for these nanotubes and fibers to be useful in large objects, however, they must be linked into a composite structure.
[0005] A slurry - a suspension of fine particles in a solvent carrier such as water - may be freeze-east. In freeze casting, a phase separation occurs during solidification; ice crystals grow by solidifying the water carrier of the slurry, while concentrating between them solutes and solid particulates of the aqueous slurry. The solutes and solid particles containing a desired material or material precursor may include a binder. The ice crystals template a three-dimensional (3D) micro structure that consists of the solutes and particles that coalesce,
producing a material with an ordered, self-assembled, hierarchical pore structure. The crystalized solvent carrier is then removed - with water as solvent-carrier this is typically done by sublimation such as during freeze- drying.
[0006] Freeze-east objects may be subjected to further processing, such as a thermal treatment of the material that frequently results in a desirable property profile with favorable structural, mechanical, electrical, thermal, optical or other properties. Further processing may include annealing, sintering and carbonization, where castings are heated to a temperature that is high enough to fuse or bond particles together, and in some systems to burn out or to transform or to reduce the binder phase used during casting, yet low enough to avoid the complete melting of the particles. Annealing has been done with freeze-east objects formed with metal powders.
[0007] Most water on earth, including brackish water or seawater, has more salt than is desirable for use in agriculture or for human consumption; fresh water of low salt content is often a scarce resource. Desalination or deionization is occasionally used to remove salts from salty or brackish water to provide less-salty water for agriculture and for human consumption. Current methods of desalination require significant energy, restricting use of desalination to areas where salt water and energy are readily available and cheap, and fresh water is particularly scarce and expensive; reductions in energy cost for desalination could permit more widespread use of desalination.
[0008] Capacitive deionization (CDI) is one method of purifying brackish water into a less-salty water that does not require high pressures and the corresponding energy intensity of other desalination methods such as reverse osmosis (RO) or the high heat of distillation.
[0009] CDI operates by applying a voltage across a brackish water feed to draw ions out of solution and into the carbon electrodes. CDI does not rely on forcing water through a membrane, so at low salt content levels (less than 6000 mg/L) even the basic method is three times more efficient than RO. In addition, the electrode surfaces act as electrochemical capacitors, so CDI can be conceptualized as a process of charging and discharging these capacitors. The energy lost during discharge can be re-captured, increasing system efficiency by 80-90% in some instances. Such efficiencies would allow CDI to compete with RO, perhaps even at the salt concentration level of seawater.
[0010] Once the electrodes become charged with separated ions, voltage on the electrodes may be reversed and the channels briefly backflushed to remove ions from the system.
[0011] Carbon-based electrodes are regularly used in CDI due to their high electrical conductivity, low-cost, and impressive specific surface areas. Materials with high specific surface have many more locations to store ions, and thus larger desalination capacities in each cycle.
[0012] Carbon-based electrodes are used in a number of other applications, including lithium batteries, zinc-manganese (Le-Clanche and Alkaline) batteries,
supercapacitors, and others, these often require controllable porosity and density, as well as good conductivity.
[0013] Materials with controllable porosity are also of use in filtration, both of water and other substances, and may potentially be doped with catalysts for use in other applications.
SUMMARY
[0014] A composition of matter, and method of manufacture, of a material having a porous mass including a bound and reduced composite of grapheme oxide that in situ has either fully or partially been reduced to graphene, and either carbon nanotubes or carbon nanofibers, or both. In particular embodiments, the mass is directionally porous, with pores having an average length over 10,000 microns and an average cross sectional area less than 2500 square microns and more than 25 square microns. In embodiments, a heat treatment at between 400 and 1500°C in non-oxidizing atmosphere is used to reduce graphene oxide to graphene while binding the graphene to the carbon nanotubes or nanofibers, in an alternative embodiment graphene oxide is reduced chemically. Graphene is present at a ratio between 2: 1 and 1:4 in proportion to carbon nanotubes or nanofibers. The material can be made with porosity with a typical range of about 75-99% percent.
[0015] A composition of matter includes a porous mass, the porous mass includes a bound and reduced composite of graphene fully or partially reduced from graphene oxide, and a carbon structure selected from the group consisting of carbon nanotubes and carbon nanofibers.
[0016] A method of manufacture of a porous mass includes: preparing a slurry comprising water, graphene oxide, and a carbon structure selected from the group consisting of carbon nanotubes, carbon nanofibers, and activated carbon; freeze-casting the slurry, and sublimating the water, to form a green casting; and reducing graphene oxide of the green casting, thereby binding particles of the graphene oxide and carbon structure to form the porous mass.
[0017] A water purification apparatus includes a first and a second electrode, at least one electrode including a porous mass comprising a bound and reduced composite comprising graphene fully or partially reduced from graphene oxide, and a carbon structure selected from the group consisting of carbon nanotubes and carbon nanofibers, the first and second electrodes adjacent to a channel; a power supply coupled to apply a voltage difference between the first and second electrode; apparatus configured to supply salty water to the channel; and apparatus configured to receive water from the channel.
[0018] An electrochemical apparatus including a first and a second electrode, at least one electrode including a composition of matter comprising a porous mass comprising a bound and reduced composite comprising graphene fully or partially reduced from graphene oxide, and a carbon structure selected from the group consisting of carbon nanotubes and carbon nanofibers; the porous mass of the composition of matter impregnated with a first chemical composition; the apparatus configured to permit the first chemical composition to enter into chemical reactions that provide an electric current between the first and second electrodes.
BRIEF DESCRIPTION OF THE DRAWINGS
[0019] Fig. 1 is a flowchart of a process for making an embodiment of a directionally-porous, annealed, graphene-oxide / carbon nanotube composite material.
[0020] Fig. 2 is a diagram illustrating an apparatus adapted to directionally freeze- casting an aqueous material, as is used for forming the directionally-porous, annealed, graphene-oxide / carbon nanotube composite material.
[0021] Fig. 3 is a flowchart of material preparation with Graphene Oxide and
Sucrose binder.
[0022] Fig. 4 is a flowchart of material preparation with Graphene Oxide and Carbon Nanotubes or Carbon Nanofibers with and without cellulose nanofibers.
[0023] Fig. 4A is an SEM micrograph of a section taken perpendicular to the freezing direction, showing pore structure.
[0024] Fig. 4B is an SEM micrograph of a section taken parallel to the freezing direction, showing pore structure with pores extending entirely across the micrograph from top left to lower right.
[0025] Fig. 5 is a flowchart of material preparation with graphene oxide and carbon nanofibers, with and without ethanol and copper acetate additives.
[0026] Fig. 6 is a diagram of a capacitive desalination device where carbon electrodes prepared according to the described method may prove useful.
[0027] Fig. 7 is a diagram indicating density versus strength for one embodiment of the annealed or annealed, freeze-east, carbon materials as made by the method herein.
[0028] Fig. 8 is a diagram indicating stress versus strain for an embodiment of the sintered or annealed, freeze-east, carbon materials as made by the method herein.
[0029] Fig. 9 is an illustration from Ramen spectroscopy of graphene oxide reduced to graphene at different temperatures, indicating a potential optimum between 900 and 1100 Celsius.
[0030] Fig. 10 is a plot of Young's modulus on the Y axis, with density in mg/cm3 on the X axis, for some of the sample scaffolds made.
DETAILED DESCRIPTION OF THE EMBODIMENTS
[0031] Several series of experiments were run using different combinations and a range of concentrations of carbon fibers or carbon nanotubes and various binders, additives as freeze modifiers, freeze casting conditions and heat treatment conditions. The experiments reported here resulted in materials of desirable structure, porosity, pore morphology, density, mechanical properties including resilience, and electrical conductivity that we expect to be useful in a variety of applications. All were made in a similar manner, beginning by first preparing and then freeze-casting a slurry to form a green body. Many of our green bodies were then heat-treated to carbonize some constituent materials, and to improve bonding between particles, for convenience herein we refer to the heat treatment as annealing although terms like carbonization and sintering are also used in the art to describe similar heat treatment. We produced low density aerogels from carbonaceous materials such as carbon nanofibers, carbon nanotubes, graphene oxide and reduced graphene oxide. The materials have an overall porosity and pore structure that can be carefully controlled. The pore structure can be highly aligned with a honeycomb-like structure, isotropic, or a combination of the two. The porosity can be homogeneous and uniform or graded. The hierarchical 3D architecture of the material and its properties can be tailored to exhibit significant stiffness, strength, toughness, and resilience.
Slurry Compositions
[0032] A directionally-porous, sintered / annealed, graphene-oxide / carbon nanotube composite material is formed by a method 100 (Fig. 1) that begins with preparing 102 a slurry of graphene oxide (GO), carbon nanotubes (CNT), carbon nanofibers (CNFs), or activated carbon, freeze modifiers and binders that may include one or more of a water
soluble polymer such as chitosan, alginate, agar, agarose, gelatin, starch, nanocellulose fibers (NF), Ethyl Alcohol (Ethanol), ascorbic acid, or sugar (sucrose), and water.
Graphene Oxide (GO)
[0033] Highly Concentrated Graphene Oxide Dispersion in Water (Graphene Laboratories Inc., Calverton, NY, USA, also known as Graphene Supermarket) at a concentration of 5g/L, and a carbon-oxygen ratio of 4: 1 was used. GO is an oxygenated form of graphene, featuring carboxyl, hydroxyl, and carbonyl groups. GO is synthesized by oxidizing graphite, using nitric acid or a combination of potassium permanganate and sulfuric acid. GO can be readily reduced to graphene at large quantities using thermal and chemical methods. The GO used here was an aqueous dispersion of 0.5- 5 micron diameter flakes, with a thickness of 1 atomic layer in at least 60% of flakes. A reduced form of this material has displayed a Brunauer-Emmett- Teller (BET) surface area as high as 833 m /g.
[0034] Throughout this document the term graphene oxide (GO) is used to refer to samples prepared with GO, even those that have been treated thermally and may be fully or partially reduced to graphene (G). We will not assume that the GO has been fully reduced to G, although it is known that reduced GO (RGO) is often very similar in structure and composition to G. Thermal reduction of GO occurs when it is heated to between 600- 1200C in an inert or non-oxidizing atmosphere such as argon, removing resident water molecules and oxygen containing groups from the GO sheets and varies with temperature. Recent findings suggest that aromatic carbon sources (ethylene or radical carbon), can also donate carbons to defects in RGO, improving electrical conductivity by "healing" the RGO. It is also possible to only partially thermally reduce GO.
Carbon Additives
'Straight' Carbon Nanotubes (Straight CNT)
[0035] In some embodiments, straight carbon nanotubes - JC121 (JEIO Tech, South Korea) were added to increase surface area, electrical conductivity, and improve mechanical properties. Carbon nanotubes are tubular structures made of sp2 bonded carbon atoms. Carbon nanotubes are hydrophobic and exhibit π-π interaction, often leading to clumping and poor dispersion when added to water. Carbon nanotubes have diameters on the order of tens of nanometers, and can have lengths up to one micrometer. Though single- walled carbon nanotubes exist (SWCNT), in many instances it is simpler and more cost- effective to use multi- walled carbon nanotubes (MWCNT). The carbon nanotubes used here are all of the MWCNT variety, with a pristine surface area of 300-310 m7e.
'Curly' Carbon Nanotubes (Curly CNT)
[0036] A second variety of CNTs, JC142 (JEIO tech, South Korea) was explored. These nanotubes are curly, which increases the surface area over the standard straight CNTs described above. These 'curly' MWCNTs were used in the production of many of the scaffolds for this study. The JC142 nanotubes have a bulk density of 0.01 g/mL and a pristine surface area of 500-510 m7e.
Carbon Nanofibers (CNFs)
[0037] The carbon nanofibers used in the reported study belong, like multi- walled carbon nanotubes, to the structural family of fullerenes. PR-24-XT-PS carbon nanofibers were purchased from Pyrograf ® Products (Inc.) and are pyrolitically stripped carbon nanofibers. The difference between both, MWCNTs and CNFs, is their structures. The CNFs used have an average diameter of 100 nm and an average length of 70 μιη.
Activated Carbon Powder
[0038] Activated carbon powder, DARCO(R) KB-G, obtained from Sigma Aldritch.
Binders
[0039] Many, but not all, of our experiments have used a binder for green body stability and strength during freeze-casting, freeze- drying, and handling prior to annealing.
Cellulose Nano-Fibrils (Nanocellulose)
[0040] Cellulose nano-fibrils (also referred to as nanocellulose) were purchased from the University of Maine Process Development Center (Orono, ME, USA), and are 3.1% solids by volume, grade CNF. Cellulose nano-fibrils are a wood-based material.
Nanocellulose is used as a binder.
Chitosan
[0041] Chitosan solution 2.4% (w/v) was prepared by dissolving low molecular weight chitosan powder (448869, Sigma Aldrich, St. Louis, MO, USA) into a mixture of acetic acid and water. The resulting solution was then vigorously shaken, and placed on a bench top bottle roller for at least 24 hours to ensure complete mixing. Chitosan acts simultaneously a binder and dispersant when freeze casting MWCNTs. We tried this "all-in- one" agent in our study of CNTs, using the ratio of chitosan: MWCNT resulting from preparing Chitosan solutions (1 wt.%) by dissolving chitosan flakes (0.5 g) in 50 mL of an aqueous solution of acetic acid (0.05 M, pH 5.5) and then dispersing MWCNTs (30 mg) in
1.5 mL of the 1 wt % chitosan solution (1 wt.%), sonicated and stirred. The MWCNT contents at the stock water suspension was 2 wt. %.
Ethyl Alcohol
[0042] Ethanol (EMD Millipore EX0280— 3 Denatured Ethyl Alcohol 95% was used as a dispersing agent, particularly in the CNT hybrid preparations.
L- Ascorbic Acid
[0043] L-Ascobic Acid (LAA) in some embodiments is used both as a binder and as a chemical reducing agent capable of reducing some of the GO to G. and encouraging formation of covalent chemical bonds between G and CNT or CNF in the cast scaffolds
Maple Syrup, Maple Sap, or Sucrose
[0044] In some experiments, sucrose was used as a binder. In other experiments maple sap and maple syrup was used as a binder. Maple syrup was certified organic, grade- A amber, maple syrup from Mt. Cube Farm, Orford, NH were used. Maple sap was also obtained from Mt. Cube Farm.
Slurry
[0045] A variety of recipes were used to produce the samples studied from the above-listed ingredients. After a combination of materials was selected, forming a 'recipe,' the preparation methods included:
1. Selecting one of each (base solvent, carbon fiber or nano tube, binder (if any)).
a. A base solvent selected from the GO suspension, Deionized water (DW), and mixtures of the two was selected and placed in a plastic cup. b. Selected carbon was added.
c. Chitosan and/or nanocellulose fibers were added, if required by the specific recipe,
2. Dispersants and freeze modifiers (such as ethanol and chitosan) and some additional solid carbon material were chosen, and other binders and dispersants were added for some trials.
Slurry Preparation
For embodiments not using LAA
[0046] The carbonaceous slurries are prepared as 10 mL samples in 50 mL plastic cups. First, dry powder carbon nanotubes or nanofibers were weighed with a precision balance to within 1% accuracy. Samples with standard carbon nanotubes were prepared with
100 mg - 400 mg in the lOmL sample. Curly carbon nano tubes were prepared with 50-150 mg of solids in the 10 mL of liquid. Any additional binders, such as cellulose nanofibers, in the recipe tested were added to the slurry. lOmL of water or 10 mL of GO- water suspension was added. 1-2 mL of ethanol was added as dispersant in many instances in order to aid dispersion of CNT which tend to remain aggregated or entangled and poorly dispersed in solution. We believe that the ethanol is fully sublimated during freeze- drying, and thus does not remain in the final scaffold. In experiments with other carbon sources, ethanol was used in some experiments as a freeze- modifier to increase final pore sizes.
[0047] The cup is sealed, and the sealed cup is then shear mixed for two minutes at 3000 rpm. The mixed slurry is then placed 104 immediately in the freeze-casting mold 202 (Fig. 2).
Embodiments containing LAA
[0048] First, LAA with a mass ratio of 3.33: 1 LAA to GO was added to a 30mL vial. Several other concentration ratios ranging from 0.5: 1 to 100: 1 were used to check differences between gels and resultant scaffolds, and corroborate that 3.33: 1 provided the best gelation. After adding the corresponding mass of LAA according to that ratio, 5-25 mL GO aqueous solution was added to the 30mL vial, and the solution was mixed vigorously with a magnetic stir bar for several minutes until the solution appeared of uniform composition with no LAA remaining unabsorbed, at which point the stir bar was removed. The solution was then left undisturbed for between 1 to 48 h at temperatures between 25-80°C, during which time gelation occurred, forming an RGO hydrogel.
[0049] After gelation and reduction, the RGO hydrogels were subjected to solvent exchange, in order to chemically remove LAA from the hydrogel. The hydrogel was left in the 30 mL vial and ethanol was added until the vial was full. In the first few of these ethanol baths, a slight yellowing of the solution surrounding the gel indicated that solvent exchange was indeed eliminating LAA from the hydrogel. The ethanol was decanted and replaced with fresh ethanol every few hours or as necessary until visual mixing of water and ethanol was no longer evident in the vial and bubbles stopped emerging from the surface of the hydrogel. At this point, the vial was rinsed several times with deionized (DI) water and then filled with DI water. The ethanol infused hydrogels floated in DI water, and the same procedure was used with water as with ethanol, decanting and replacing the water every few hours until the hydrogel sank, stopped bubbling, there was no more visual mixing of ethanol and water, and the smell of ethanol was undetectable when the container was opened after an hour or more of resting.
[0050] Alternatively, gelation was attempted in the PTFE molds used for freezing. In this instance, a layer of parafilm was laid over a copper base plate and the base plate then taped on securely to the bottom of the mold, ensuring that water could not enter or exit and that the bottom would not fall off. A magnetic stirrer was placed in the mold and then removed after mixing. A copper base plate and parafilm layer were added to the top of mold in the same manner used with the bottom, and the entire contraption was heated according to the same outlines pertaining to gels in vials. Solvent exchange was conducted by removing the top base plate and placing the whole contraption in an ethanol and then water bath until the reactions came to an end. A small strip of tape was placed over the open end of the mold to ensure that the sample did not come out while floating in water.
[0051] Additionally, RGO hydrogels were prepared in standard, 10 mL mixing cups. In this case, gel preparation was identical to production in a vial, except that solvent exchange was conducted in a large beaker, since almost no solvent could fit in the mixing jar.
Freeze casting
[0052] The mold, as illustrated in Fig. 2, is a Teflon tube 202 placed on a cooled plate 203 that closes the tube and is attached to a copper cold finger 204 with a top end adjacent to the bottom end of the tube. The cold finger 204 top end is temperature regulated by using feedback from a thermocouple 206 attached to the cooled plate applied to an electric heater 208 attached to the finger. The cold finger is cooled by immersing its lower end into liquid nitrogen 210. A layer of Vaseline is applied around the edge of the copper base plate to form a seal between the edge of the base plate and the bottom of the mold. The copper base plate is then placed on top of a copper rod, or "cold finger," which stretches down into a liquid nitrogen bath. The temperature at the top of the copper rod is monitored by a thermocouple and temperature clines can be regulated via a PID controller and a powerful heating element near the top of the cold finger. Prior to freezing, the top of the rod was maintained at a constant 5°C, and samples placed on top were precooled to this temperature before being subjected to a determined cooling rate of either 1 or 10 0 °C min -"1 to a minimum temperature of -150°C. As freezing progresses 106 of slurry 212 in the freeze-casting mold 202, the ice phase (not shown) nucleates at the cold plate, and grows along the thermal gradient and upwards through the mold, concentrating solute and particles between the crystals thereby ice-templating it, thus forming a material with a hierarchical architecture whose pores are filled with ice.
[0053] When samples are fully frozen, they are removed from the cold finger, the samples are punched out of the mold using an Arbor press. The green casting is then freeze- dried to remove the ice phase.
[0054] The samples are freeze cast with a cooling rate rate of either 1° C/ min or 10° C/ min applied to the copper plates that seals the mold bottom. In a particular embodiment, each mold has a tubular shape with an inner diameter of 18.8 mm (3/4 inch) and a height of 40 mm.
Freeze-Drying or Lyophylization
[0055] Once fully frozen, each sample is dehydrated for 48-72 h in a FreeZone 4.5 Liter Benchtop Freeze Dry System (Labconco, Kansas City, MO).
Storage before Annealing
[0056] Samples containing sucrose, maple sap, or maple syrup are placed in a glass desiccation chamber with silica gel desiccant (-3+8mesh granules, Alfa Aesar, Ward Hill, MA) with relative humidity 0.0 in order to keep atmospheric moisture from hydrating the sugar until such a time as they could be subjected to heat treatment or acclimated for mechanical testing.
GO Reduction And Annealing
[0057] By heating the graphene oxide to a temperature in the range of about 490°C to 1000 °C, and in particular embodiments to 498 °C and 1000 °C, many of the carboxyl and hydroxyl groups forming the outer hydrophilic portion of the graphene oxide flakes are removed, which results in reduced graphene. The samples are thermally reduced in an inert atmosphere, such as argon, by placing them in graphite boats and inserting them in a tube furnace which is heated at 5 °C per minute from room temperature to 1000 °C, before holding them at this temperature for two hours. In another particular embodiment, the temperature of the furnace is increased at a heating rate of 5°C per minute to 498°C in argon gas, and maintained there for two hours. After two hours the furnace was shut off, and allowed to cool to room temperature unassisted (at an approximate rate of 5 °C).
[0058] It is expected that a full reduction of graphene oxide to graphene during heating would result in a mass loss of graphene oxide-graphene particles of up to 35% because of the release of bound oxygen from graphene oxide as carbon dioxide and water vapor. Release of the bound oxygen also permits bonding of forming graphene structures to nearby carbon structures such as nanotubes and nanofibers.
[0059] It is expected that in alternative embodiments the GO thermal reduction and annealing may be done by heating the graphene oxide in various alternative non- oxidizing or reducing atmospheres, for example Argon, Hydrogen, Nitrogen, Xenon, Helium, Neon, blends of the aforementioned non-oxidizing gasses such as Forming gas (a mixture of hydrogen and nitrogen), or ultra-high vacuum should suffice to prevent oxidation of the material. Moreover, several heating sources capable of reaching 1000°C are known and usable including microwave, flash light, laser, plasma, electric current, field assisted, or other kinds of furnaces. Further, in alternative embodiments other temperatures are used in the 400 to 2000 °C range.
Analysis techniques
[0060] After the samples are removed from the lyophilizer and subjected to furnace treatment if called for, the samples are cut using a diamond wire saw (WELL
Diamond Wire Saws, Inc., Norcross, GA). The wire is a 220 μιη diameter diamond encrusted steel wire, and samples are cut using a wire speed of 0.7 m s-1. Each sample is mounted on a ceramic plate using Crystalbond™ with the unidirectional pores perpendicular to the face of the plate, then cut to provide repeatable data points from several heights along the sample. 5mm cubes are cut at standard heights above the bottom of samples before heat treatment, with the cube centered at 10, 20, and 30 mm from the bottom of the sample. For each height, four cubes are prepared and at least three subjected to mechanical and structural
investigation. In the case of annealed sucrose-bound samples, shrinkage was observed as expected before and after thermal treatment. Because of initial size restrictions of the graphite boat, the entire green body could not be annealed but had to be cut prior to thermal treatment. In order to ensure a fair comparisons, the a volume that corresponds to a green body volume of -10 mm x 10 mm x 7 to 32mm of sample height) was heat treated and then appropriately cut to match its green-body counterpart height, which correspond identically to the 5 mm cubes cut at 10, 20, and 30 mm from the green body.
Density
[0061] To obtain a density measurement, the samples are weighed on a high precision balance (+0.01 mg; XP105 Delta Range, Mettler Toledo Inc., Columbus, OH), volume is measured in an optical microscope or with a caliper (+0.01 mm), if appropriate, and density is calculated from these.
[0062] Scanning electron microscopy (SEM) was conducted with a Zeiss Supra 50VP (Carl Zeiss NTS LLC, Peabody, MA, USA), at accelerating voltages between 5 keV
and 15 keV. For these carbonaceous scaffolds, sputter coating was optional for SEM imaging, as the samples were already conductive.
[0063] A Leica Optical microscope was used for preliminary visual investigation of the samples, such as pore alignment and orientation. Representative optical micrographs were taken of each sample after each cut at several magnifications, providing a basic understanding of the sample and its hierarchical pore structure.
Mechanical Characterization
[0064] Compression tests were conducted on an Instron 5948 (Instron, Norwood, MA) with a 5 N load cell and a cross-head speed of 0.05 mm s-1, corresponding to a strain rate of 1% s-1. Young's modulus and yield strength were determined from the stress-strain curves obtained. The cubes were tested parallel to the freezing direction, thus parallel to the long pore axis. At least three cubes from each layer of three sample were tested.
Electrical Characterization
[0065] At least some electrical conductivity is required for use of the freeze-east and annealed material produced by the method described above as an electrode. The four point probe method (SIGNATONE, Gilroy, CA) was used to perform conductivity measurements. Measurements were taken on at least 5 different points on samples and averaged.
[0066] A disadvantage of this method applied to porous materials such as those of this study is that the probes affect the micro structure of the samples. However, because the probe diameter is about one magnitude larger than the typical pore diameters of the carbon aerogel materials under investigation, reproducible results were obtained.
Experimental Recipes Used
GO - binder, GO - LAA
[0067] A first series of experimental samples using Graphene Oxide and a selected binder in a ratio of 0.24: 1 to graphene oxide mass ratio of graphene solid was performed using maple syrup, maple sap, sucrose, and chitosan as binders. The graphene was the 5 g per liter suspension in water previously described, which also provided the liquid phase (water) for the slurry. Of the binders used in this first series of experiments, sucrose resulted in particularly promising, stable green bodies for heat treatment followed by mechanical testing.
GO-CNT, GO-CNT-nanocellulose
[0068] A second series of experiments showed that Graphene- Oxide (GO)- Carbon Nanotube (CNT) blends with and without nanocellulose as a binder also resulted in green bodies that were sufficiently stable for freeze- drying, heat treatment, sample preparation, structural characterization and mechanical testing. In this series, Chitosan proved less successful as a dispersant and binder. Some particular recipes experimented with include, with the last column indicating whether samples survived lyophylization:
50, 100 mg JC 142 CNT 10 mL GO susp. 0.2 mL chit some
50, 100 mg JC 142 CNT 10 mL GO susp. 0.1 mL chit - some
50, 100 mg JC 142 CNT 10 mL GO susp. 0.2 mL eth yes
150, 200 mg JC 142 CNT 10 mL GO susp. 0.2 mL eth yes
150, 200 mg JC 121 CNT 10 mL GO susp. 0.2 mL eth yes
100, 200 mg JC 121 CNT lO mL DI 0.1 mL chit - none
100, 200 mg JC 121 CNT lO mL DI 0.2 mL chit none
200, 400, 600, 800 mg AC lO mL DI 0.2 mL nanocell. none
100 mg JC 142 CNT 10 mL GO susp. 0.2 mL nanocell. yes
Best results with Carbon Nanotubes were obtained with the following recipes:
50 & 100 mg JC 142 CNT 10 mL GO suspension, 0.2 mL ethanol; and
100 mg JC 142 CNT 10 mL GO suspension, 0.2 mL nanocellulose, and 0.2 mL ethanol.
Recipes also producing strong materials of good conductivity with CNF were:
10 mL GO suspension, 1% wt % CNF, 0.2 mL nanocellulose; and
10 mL GO suspension, 1% wt % CNF, 0.2 mL ethanol.
GO-CNF, GO-CNF-Ethanol
[0069] A third series of experiments performed with Graphene- Oxide - Carbon Nanofiber blends with and without ethanol, also sufficed to form lyophilized and freeze-east material with sufficient mechanical stability to undergo post-lyophilization cutting, annealing, testing and characterization.
Results
[0070] Certain recipes, GO+ sucrose, as well as GO+CNT and GO+CNT+ nanocellulose, along with GO-NF, and GO-NF-Ethanol produced lyophilized and freeze-east material having sufficient mechanical stability to undergo post-lyophilization cutting, testing and characterization. Samples from these recipes were also reduced and annealed (or carbonized) using the method described above.
GO-Binder (including Sucrose)
[0071] Stable freeze cast graphene and GO structures depended on the freezing rate. Faster cooling rates of 10°C/min led to significantly more stable samples than l°C/min, the 10°C/min rate was used for all samples subjected to further characterization. A cooling rate to 10°C/min led to successfully lyophilized samples in every case, even though those frozen from GO alone and GO + LAA, GO+sap, GO+syrup, and GO+chitosan were all too fragile to further analyze. Alone among these, GO+sucrose samples were relatively easy to handle. GO+activated carbon also gave scaffolds, although these were not analyzed.
[0072] Un-annealed GO+sucrose samples had striking coloration of various shades between gold and translucent white, and after annealing were a slightly reflective gray. Shrinkage due to reduction of GO was observed by both the hydrogel samples during chemical reduction (GO-LAA) and the GO-sucrose samples during thermal reduction or annealing. The loss of oxygen-containing groups from GO during reduction or corresponds to about a 30% loss of volume, which was observed in both types of samples after reduction
[0073] Hydrogels exhibit significant shrinkage over time, of about 30-40% volume. GO-LAA Gels that have expelled water and are thus smaller also have a lower porosity and less water contained in the gel when freezing. Additionally, gels shrank so that they were smaller than the PTFE molds, meaning they were not in direct contact with the sides of the insulating molds during freezing. Cracking and issues solvent exchange arose in many samples and were addressed.
[0074] GO-LAA Gels that had shrunk significantly proved more suitable for freeze casting than those that had not, likely because better bonding between particles.
Despite slightly higher densities and lower porosities, these samples exhibited aligned porous structure and resulted in more stable and resilient aerogels. Gels created in polymer containers adhered to the polymer walls during hydrogel formation, even to the point that gels would tear apart during shrinkage, sticking to the polymer walls in large pieces rather than separating easily from the walls, as in glass vials. Because of this and the fact that hydrogels having lost -30% of their volume were easy to remove from glass vials, glass vials were used and hydrogels were allowed to complete their volume reduction before any further experimentation.
[0075] Aerogels with ratios of 10: 1 LAA:GO were brittle compared to those frozen with the ratio of 3.33: 1, while gels with a ratio of 1: 1 fell apart in the lyophilizer. Greater shrinkage exhibited by samples with ratios of 10: 1 and 100: 1 combined with
brittleness resulting from residual LAA made samples too dense and small for desired characteristics. 3.33: 1 was chosen as the best mass ratio of LAA to GO.
[0076] Attempts to freeze structures prior to LAA removal by solvent exchange resulted in poor structural integrity, and attempts at solvent exchange after freezing and lyophilization were not successful. Completion of solvent exchange proved to be very important to the resultant aerogel, as large cracks, pervasive smaller cracks, and poor structural stability compromised samples that did not finish solvent exchange. Even in some of the best samples analyzed, we suspect that solvent exchange was not 100% effective.
[0077] Varying degrees of cracking were observed. Even after discovering that both too much LAA and incomplete solvent exchange led to large cracks compromising structural integrity, samples frozen with proper solvent exchange still exhibited cracks, although they were smaller and did not compromise the integrity of the samples. Even so, these cracks weaken mechanical properties, introduce irregularities that can lower electrical conductivity, skew porosity measurements by adding unwanted macroporosity, and were generally not desirable and still needed to be removed from the samples.
[0078] Shrinkage exhibited in hydrogels meant that they were no longer flush with PTFE molds during freezing, the gap was filled with water.
[0079] Forces generated by the formation and presence of the ice phase within the scaffold as well as others generated and imposed during processing by the materials surrounding the sample are likely causes for the crack formation.
[0080] The solution to this came about unintentionally. The bottoms of the hydrogels were slightly concave, which meant that the thermal contact with the copper base plate was not uniform across the bottom of the sample. This could have heat flow
implications and cause fanning, cracking, or generally jeopardize uniformity and stability of the samples and their porous structures. A solution for these issues was the addition of a small amount (~5 drops) of DI water was pipette onto the base plate before placing the sample into the mold. This was to ensure that the hydrogel, which was inserted into the mold hydrated, would remain fully hydrated, and that ice crystals could grow uninterrupted along the thermal gradient from the mold bottom, at which solidification had started. The solution to the cracking problem was discovered when, for one sample, water was added to approximately half the sample height. When this sample was removed from the mold, the water was frozen to about half the height of the sample and found to protect it from cracking, while above this height, the sample was found, after lyophilization, to have cracked. The bottom half of that sample further exhibited excellent elastic properties, resilience and shape
recovery upon deformation and was the best sample prepared up to that point. After this discovery, the mold was filled with water to the full height of the sample and resultant aerogels were void of cracks, exhibiting a high electrical conductivity (160 S/cm), a Young's modulus of 65 kPa), 100% recovery after 10 cycles of compression to 80% strain at low density (31.1 mg/cm ), high porosity (98.6% overall), and a hierarchical 3D structure with regular lamellar spacing, see table 1.

Table 1: Experimental results compared to literature values.
Binder Results
Green-Body Results
[0081] Samples frozen with sap, syrup, chitosan, and no additional binder created successful 3D structures that maintained form upon lyophilization. However, these samples were fragile. The syrup samples did not lyophilize well, likely because of their high sugar content. Of the, samples manufactured those made from grapheme + maple sap frozen with a 10°C/min cooling rate provided the best structures. None of these binders were found to be ideally suited for this application with graphene, GO, and freeze casting.
[0082] Sucrose, added as a binder that through thermal treatment can be converted to carbon performed better. As a binder, sucrose far outperformed any of the other alternatives, producing more robust scaffolds with distinct anisotropic features and a well- aligned pore structure. GO + sucrose aerogels fabricated and tested according to the flowchart of Fig. 3 displayed large discrete streaks of differing golden, translucent colors; these colors are caused by light reflecting on the GO sheets of different orientations as
templated by the ice crystals and illustrate that the pores are continuous along the entire length of the sample with the excellent pore alignment. The pores parallel the direction of ice crystal growth, the uniform colors indicate well aligned GO sheets. These findings were further supported by optical microscope and SEM imaging.
[0083] Sucrose samples created with 0.05g per 10 mL were structurally sound and exhibited good micro structure. Lower amounts of sucrose are preferable because of a resulting lower density and because thermally converted sucrose has lower electrical properties than GO or graphene. The green body GO + sucrose samples resulted in low- density samples (25.0 mg/cm ) with very high overall porosity (98.9%), good pore alignment, and an elastic modulus between 144-318 kPa. As expected, green body samples did not exhibit the same elastic recovery as the annealed material..
[0084] Samples of GO+sucrose were taken to the Advanced Photon Source at Argonne National Laboratory for tomographic characterization. Pore alignment was confirmed, and 3D reconstructions of the samples provide excellent proof of a highly regular, hierarchical porous architecture.
Annealed results
[0085] Samples of GO+sucrose were initially subjected to thermal annealing at 200 C for 2 hours in an Ar atmosphere. This temperature was chosen because it is just above the caramelization temperature of sucrose and because 200°C is a typical thermal treatment temperature for GO and RGO structures for partial reduction. However, exposure to this tempereature may have melted the sucrose. The samples had lost all structure when removed from the furnace.
[0086] Annealing temperatures of 600-800C were chosen for the next set of experiment. These higher target temperatures meant that the furnace would only be in the sucrose melting regime for a short period of time.
[0087] Expected shrinkage occurred (-40%) due to volume loss from both sucrose and GO during conversion. 69% mass reduction also occurred, indicating good thermal conversion of sucrose . The resulting scaffold had an anisotropic, honeycomb-like pore structure, a density of 12.9 mg/cm and 99.4% porosity, and good mechanical properties 12.1 mg/cm . Electrical conductivity was not measured. When compressed to 80% on the Instron, annealed GO+sucrose samples did not exhibit the same elastic behavior and elastic recovery that the chemically reduced aerogels had, but some. Pore morphology remained similar, while pore size was reduced in keeping with the volume reduction. The cell wall
surfaces have a wrinkled appearance, that likely is due to the thermal reduction of the GO material. These wrinkles, which may belie defects, might reduce electron mobility and thus electrical conductivity, though it has been suggested that thermally reduced GO can be 'healed' to an extent with the proper carbon source.

Table 2: Material properties of GO + sucrose before and after annealing.
[0088] In another experiment, Composition 1 (Green Body): Slurries were prepared using sucrose (EMD Chemicals, Gibbstown, NJ) as binding agent. Graphene Oxide solution (5g/L aqueous dispersion, flake size 0.5-5μιη, 4: 1 C:0 ratio, Graphene Supermarket, Calverton, NY) was used for the carbonaceous component. 10 mL of Graphene Oxide solution was added to 50 mg sucrose. The solution was mixed on a high shear SpeedMixer (DAC 150 FVZ-K, FlackTek, Landrum, SC) at a speed of 3000 RPM for 30s for mixing and degassing. Samples were frozen on the freeze cast setup depicted in Fig. 2 at a rate of - 10°C/min. Samples were lyophilized for 48+ hours and then stored in a container with relative humidity 0.0 until testing. It is expected that a range of 0.1-100 times the amount of binder used, as well as other monosaccharides, disaccharides, invert sugar, or combinations of these would produce the same results.
[0089] Composition 2 (Annealed): Samples from Composition 1 were subjected to a temperature of 800°C for 2 hrs in an argon atmosphere heated at a rate of 4°C/min.
Samples were placed in a graphite crucible to prevent oxidation during heat treatment.
Samples were prepared in 5 mm cubes with a diamond wire saw (WELL Diamond Wire Saws, Inc., Norcross, GA). It is expected that a range from 600-2000C would give similar results. All gases preventing oxidation during thermal treatment would be expected to be appropriate, and a temperature ramp rate from 0.1-100 times the rate used would be expected to work, the ramps could include holding times at lower temperatures for thermal pre- treatment of the samples.
Material Structure

Table 3 GO- sucrose pore structure before and after annealing.
Mechanical Properties
[0090] Compression tests were conducted on an Instron 5948 (Instron, Norwood, MA) with a 5 N load cell and a cross-head speed of 0.05 mm s-1, corresponding to a strain rate of 1% s-1. Young's modulus and yield strength were determined from stress-strain curves obtained. The cubes were tested applying the force parallel to the direction of the long axis of the honeycomb-like pores. At least three cubes from each of the layers of each sample were tested. Cubes containing sucrose were dehydrated via storage in a container with relative humidity 0.0 prior to testing.
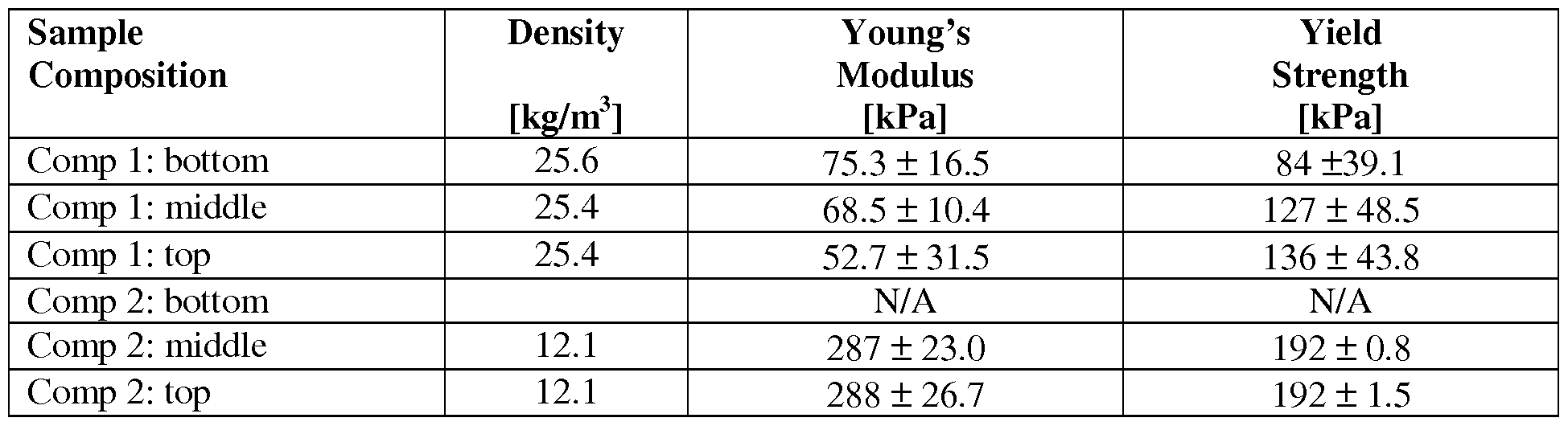
Table 4 - GO- sucrose mechanica properties
Electrical and Thermal Properties
[0091] At least 3 samples per layer of at least 3 corks per freezing run were tested parallel and perpendicular to the Freezing Direction (FD), in the case of the electrical measurements both with the probe parallel to the long pore axis and with the probe perpendicular to the long pore axis gave a 4 Point Probe measurement averaging 11.3 Siemens.
GO - binder, GO - LAA Conclusions
[0092] GO-sucrose and GO-LAA produced 3D RGO aerogels exhibiting high porosities (98.6-99.42%), low densities (12.1-31.1 g/cm ), uniform aligned hierarchical pore structure, and anisotropic mechanical properties.
[0093] High resilience (>80% elastic recovery after compression to 80% strain) exhibited by GO+LAA combined with high density and extreme electrical conductivity of 160 S/cm set these samples apart. Different concentrations of GO solution would help achieve a better adjustable density. While a successful process for the freeze casting of GO+LAA hydrogels has been developed, there is no doubt that the process can be further improved.
[0094] The use of sucrose as a binder and carbon precursor in freeze casting applications is introduced herewith. In addition, GO+sucrose samples showed great promise for thermal reduction. The limited elastic recovery and resilience seen in the first generations of these samples may be improved upon and better mechanically strong and resilient scaffolds will result.
[0095] The use of a carbon source to heal thermally reduced GO matrices formed through freeze casting and the use of Zirconium Acetate, which changes the preferential crystal growth orientation of the ice crystals to favor growth in a hexagonal pore instead of lamellar pores are proposed.
GO+CNT and GO+CNT+ nanocellulose
[0096] Particular experiments at making these materials GO+CNT and
GO+CNT+ nanocellulose, were fabricated from 10 ml of the GO suspension with 2% wt. % nanocellulose and either 1% carbon nano fibers or 1, 2, or 4 wt. % CNTs, with or without 1 or 2 wt. % chitosan or 2 % ethanol, according to the flowchart of Fig. 4.
[0097] See above for a list of recipes tested.
Best results with Carbon Nanotubes were encountered with the following recipes:
50 mg JC 142 CNT 10 mL GO suspension, 0.2 mL ethanol; and
100 mg JC 142 CNT 10 mL GO suspension, 0.2 mL nanocellulose, and 0.2 mL ethanol. Recipes also producing strong materials of good conductivity with CNF were:
10 mL GO suspension, 1% wt % CNF, 0.2 mL nanocellulose; and
10 mL GO suspension, 1% wt % CNF, 0.2 mL ethanol.
[0098] In Table 5, COMP 3 = GO + lOOmg. CNT (no binder), and COMP 4 = GO + lOOmg. CNT + nanocellulose binder:

Table 5, Density of annealed GO-CNT and Go-CNT-nanocellulose lOOmg CNT material. Porosity
[0099] The GO-CNT material was sectioned perpendicular to the axis of freezing, and then imaged with a scanning electron microscope to visualize its pore structure, as shown in Fig. 4A. Pores averaged 917 square microns in cross section. Pores are enlarged along the axis of freezing with most pores having lengths over 10,000 microns, and many pores extend through the entire 35mm of a freeze-east sample, as illustrated in Fig. 4B. Other
embodiments gave average pore sizes ranging from 1494 to 2022 square microns, with pore aspect ratios ranging from 1.4 to 4.5 and porosity of 97 to 98.5 percent. It is expected that pore sizes may be adjusted by altering the rate of freezing during the freeze-casting step of the process, as well as the Ethanol content of the slurry, pore size gradients or variations along the length of the sample may further be controlled and adjusted by, for example, altering material composition, applied cooling rate or applying thermal property variations along the freeze-casting mold during the freezing process.
[0100] For annealed material, conductivity of the GO-CNF materials ranged from 7 to 15.6 Siemens/meter.
Mechanical measurements
[0101] Density of material fabricated from 10 ml of the GO suspension with 1% carbon nanofibers with either 2% ethanol or 2% chitosan was 15-25 mg/cc. Some other mechanical properties of the GO-CNF material produced is illustrated in the plot of Fig. 7 and curve of Fig. 8.
Mechanical Properties

Table 6, mechanical properties of GO-CNT, GO-CNT-nanocellulose material with and without annealing (reduced lines are annealed material).
GO-CNT and GO-CNT-Ethanol with 50mg CNT Results
[0102] Slurry Preparation - Carbonaceous slurries are prepared by mixing the components according to a specific composition into a 50-mL SpeedMixer cup. After all of the components are transferred into the cup, the cup containing the slurry is shear-mixed with a SpeedMixer at approximately 3,000 rpm for 2 minutes. The following compositions are attempted.
Comp 5. lOmL GO suspension + 50 mg JC121 CNT + 0.2 niL 95% EtOH
Comp. 6. lOmL GO suspension + 50 mg JC142 CNT + 0.2 niL 95% EtOH
Freeze-casting
[0103] Slurries are transferred into a PTFE mold that is sealed with a copper bottom plate. The inner diameter of the mold is 18.5 mm (3/4 inch). Then, the mold
(containing the slurries) is placed on the cold finger of the freeze caster. The freezing process starts at 2°C and a cooling rate of 10°C is applied until a temperature of -150°C is achieved. The entire freezing process takes approximately 40 minutes.
Freeze-drying
[0104] The frozen samples are freeze dried in the FreeZone 4.5 Liter Freeze Dry System (Labcono, Kansas City, MO, USA) running at less than 0.01 mBar for approximately 72 hours to sublime the ice.
Annealing
[0105] Freeze dried samples are annealed, or thermally reduced, in a tube furnace (Thermo Scientific Linberg/Blue M™ 1,500°C General-Purpose Tube Furnace) at 1,000°C
under constant Argon purge at 10 psi gauge. First, a sample is placed in a graphite sample holder, which is then being placed inside the tube furnace. The tube furnace is first evacuated using a vacuum pump to approximately -70 psi gauge. Then, Argon gas is filled into the tube at 10 psi gauge. The evacuation and purging process is repeated 3 times to minimize residual oxygen inside the chamber. For thermal treatment, the temperature is raised at 5°C/min to 1,000°C held at 1,000°C for 2 hours, finally, the furnace cooled unassisted to room temperature at a rate of approximately 5°C/min.
Material Structure
Table 7, Pore structure of 50mg GO-CNT, GO-CNT-Ethanol samples

Mechanical Properties

Table 8, mechanical properties of 50mg CNT: GO-CNT, GO-CNT-Ethanol samples
Electrical Properties

Table 9, electrical properties of GO-CNT, GO-CNT-Ethanol with 50mg CNT
GO-CNF-nanocellulose and GO-CNF-Ethanol Embodiments
[0106] Embodiments having carbon nanofibers instead of carbon nanotubes were also tested. Recipes also producing strong materials of good conductivity with CNF and according to the flowchart of Fig. 5 were:
[0107] 10 mL GO suspension, 1% wt % CNF, 0.2 mL nanocellulose; and
[0108] 10 mL GO suspension, 1% wt % CNF, 0.2 mL ethanol.

Table 10, results obtained with GO-CNF, annealed at lOOOC and unannealed, with either nanocellulose or ethanol.
GO-CNF
Young's Yield Toughness Electrical
comositions
Composition Modulus Strength Conductivity
[kPa] [kPa] [S/m]
GO-CNF-nc 36.95 + 3.11 + 5.31 + 0.0011 +
12.56 0.34 1.11 0.0002
GO-CNF-
41.42 + 3.42 + 6.44 + 7.0405 +
nc- 10.23 0.41 0.66 0.3859
carbonized
GO-CNF- 136.56 + 11.93 + 10.43 + 0.0031 +
eth 23,26 0.95 0.75 0.0007
GO-CNF-
148.07 + 7.92 + 8.14 + 15.6413 +
eth- 26.63 0.42 0.27 0.4253
carbonized
Table 11, more results obtained with GO-CNF, annealed at lOOOC and unannealed, with either nanocellulose or ethanol.
Alternative Embodiments of Graphene-Nanotube/Nanofiber Composites
[0109] While in our experiments, a majority of samples with aligned pores had average cross sectional pore area of between 80 and 1000 square microns, some of our experiments demonstrated cross sectional pore area of up to 2500 square microns. We expect to be able to produce product with selected average cross sectional pore areas between 25 and 2500 square microns. While it is difficult to cut samples along the axis of freezing precisely enough to show a total length of pores along that axis with the SEM, it was apparent that a majority of pores in our samples were at least 10,000 microns long in that axis, and in fact most pores extended through the entire 35 millimeters of freeze-east samples. It is this length and directionality of pores that permit easy impregnation of pores with chemical pastes, such as manganese dioxide paste for use as a cathode of a zinc-manganese dioxide dry cell, including alkaline cells; it is expected that the electrically conductive composites herein described are useful in a variety of electrochemical cells including some forms of lithium batteries. These scaffolds may also be of use in storing hydrogen.
[0110] It is expected that a similar composite formed with nonaligned pores having size between 25 and 2500 square microns in cross section can be formed by freeze- casting similar slurry by cooling without directional cooling.
[0111] While we demonstrated conductivity of our composites in the range from 7 to just over 30 siemens/meter with material having porosity in the 97-98.5% range, we expect that by slightly decreasing porosity we can produce material with conductivity as high as 50 siemens/meter; a range of 7 to 50 seems possible.
[0112] While we demonstrated successful scaffolds fabricated with mass ratios of 1: 1 to 2: 1 of graphene oxide to carbon structures such as carbon nanotubes and carbon nanofibers, we note that loss of oxygen during annealing from 80-20 carbon-oxygen graphene oxide gives a weight loss of up to 35 percent, thus producing scaffolds having mass ratios of reduced graphene oxide to carbon structures in our various experiments in the range of 0.7: 1 to nearly 2: 1. We expect to be able to produce material having mass ratios of reduced graphene oxide to carbon structures of nanotube and nanofibers in the range of 1:2 to 4: 1. With sufficient ethanol solvent, or another dispersant, or particle functionalization, we expect to be able to make a slurry of high enough concentration to produce material with porosity as low as 90%, and thus expect to be able to manufacture material with porosity between 90% and 99%.
[0113] We have made some scaffolds with added activated carbon, and some with fine graphite powder, in addition to graphene oxide. With such an additive, we expect to be able to make material with porosity as low as 75%, rendering our method adaptable to the porosity range of 75 to 99 percent.
[0114] It is expected that the It is also expected that, where sucrose - a common disaccharide - was demonstrated as functioning as a binder, other sugars (including monosaccharides and disaccharides) and sugar alcohols (including sorbitol, or mannitol) will suffice. Further, it is expected that, while ethanol was demonstrated as a freeze modifier in our experiments, other common low-molecular- weight alcohols such as methanol, propanol, isopropanol, or butanol will also modify freeze casting, and that methanol and propanol in particular will serve to increase pore sizes.
[0115] While we demonstrated nanocellulose fibers as a binder, we expect that other polysaccharides such as chitin or starch will also function as binders. Similarly, we expect fibrous polypeptides such as gelatin, to also serve as a binder.
[0116] While we demonstrated carbonization and annealing at temperatures between 495 and 1000 degrees Celsius, we expect that similar processing at other temperatures in the range from 400 to 1500 degrees Celsius will also give product; at lower temperatures in this range it may be necessary to extend annealing beyond 2 hours, and higher temperatures in this range may produce better electrical conductivity. We also expect that some particular mixtures may survive processing and/or benefit from as much as 2000 degrees Celsius.
[0117] We also note that an optimum temperature for certain graphene-oxide- based materials, according to Fig. 9, may lie between 900 and 1100 Celsius.
COMBINATIONS
[0118] The features of the present scaffolds may be present in many combinations in product. For example, scaffolds may be built with slurry of graphene oxide and both straight and curly nanotubes, or with slurry of graphene oxide and both nanotubes and nanofibers. Further, it is anticipated that the slurry may or may not contain a binder, and where a binder is used the binder may include Certain specific combinations of features that are anticipated include the following:
[0119] A composition of matter designated A includes a porous mass, the porous mass includes a bound and reduced composite of graphene fully or partially reduced from graphene oxide, and a carbon structure selected from carbon nanotubes and carbon nanofibers.
[0120] A composition of matter designated AA including the composition designated A wherein the mass is directionally porous.
[0121] A composition of matter designated AB including the composition designated A or AA wherein the porous mass has been formed from graphene oxide reduced by heat treatment in non-oxidizing atmosphere, where graphene and unreduced graphene oxide is present at ratios of between 1: 1 and 1:4 in proportion by weight to the carbon structure.
[0122] A composition of matter designated AC including the composition designated A, AA, or AB wherein at least a a plurality of pores of the porous mass extend in an axis at least 10,000 microns, and have an average cross sectional area measured perpendicular to the axis is between 25 and 2500 square microns.
[0123] A composition of matter designated AD including the composition designated A, AA, AB, or AC wherein the average cross sectional area of the pores is between 80 and 1000 square microns.
[0124] A composition of matter designated AE including the composition designated A, AA, AB, AC, or AD the porous mass having porosity between 75 and 99 percent.
[0125] A composition of matter designated AF including the composition designated A, AA, AB, AC, AD, or AE wherein the porous mass has been formed from graphene oxide reduced by heat treatment in non-oxidizing atmosphere, wherein graphene and unreduced graphene oxide is present at ratios of between 1:2 and 1:4 in proportion by weight to the carbon structure.
[0126] A composition of matter designated AG including the composition designated A, AA, AB, AC, AD, AE, or AF having an electrical conductivity of at least 7 siemens/meter.
[0127] A composition of matter designated AH including the composition designated AG having an electrical conductivity between 7 and 50 siemens/meter.
[0128] A composition of matter designated AI including the composition designated A, AA, AB, AC, AD, AE, AF, AG, or AH wherein the carbon structure comprises carbon nanotubes.
[0129] A composition of matter designated AJ including the composition designated A, AA, AB, AC, AD, AE, AF, AG, or AH wherein the carbon structure comprises carbon nanofibers.
[0130] A composition of matter designated AK including the composition designated AI or AJ, further comprising a second carbon structure selected from graphite nanoparticles and activated carbon nanoparticles.
[0131] A method of manufacture designated B of a porous mass including:
preparing a slurry comprising water, graphene oxide, and a carbon structure selected from carbon nanotubes, carbon nanofibers, and activated carbon; freeze-casting the slurry, and sublimating the water, to form a green casting; and reducing graphene oxide of the green casting, thereby binding particles of the graphene oxide and carbon structure to form the porous mass.
[0132] A method designated BA including the method designated B wherein reducing graphene oxide of the green casting is performed by heating the green casting to a temperature between 400 and 1500 degrees Celsius.
[0133] A method designated BB including the method designated BA or B wherein the graphene oxide includes primarily 0.5- 5 micron diameter flakes, with a thickness of 1 atomic layer in at least 60% of flakes.
[0134] A method designated BC including the method designated B, BA or BB wherein the slurry further includes ethanol.
[0135] A method designated BD including the method designated B, BA or BB wherein the carbon structure includes straight carbon nanotubes.
[0136] A method designated BE including the method designated B, BA or BB wherein the carbon structure includes curly carbon nanotubes.
[0137] A method designated BF including the method designated B, BA or BB wherein the carbon structure comprises carbon nanofibers.
[0138] A method designated BG including the method designated B, BA, BB, BC, BD, BE, or BF wherein the slurry further includes a polysaccharide.
[0139] A method designated BGA including the method designated BG wherein the polysaccharide includes cellulose nanofibers.
[0140] A method designated BGB including the method designated BG wherein the polysaccharide includes cellulose nanofibers.
[0141] A method designated BH including the method designated B, BA, BB,
BC, BD, BE, or BF wherein the slurry further includes a sugar.
[0142] A method designated BHA including the method designated BH wherein the sugar includes sucrose.
[0143] A method designated BI including the method designated B, BA, BB, BC,
BD, BE, or BF wherein the slurry further includes starch.
[0144] A water purification apparatus designated C including a first and a second electrode, at least one electrode including a composition of matter according to those designated A, AA, AB, AC, AD, AE, AF, AG, AH, AI or AJ, the first and second electrodes adjacent to a channel; a power supply coupled to apply a voltage difference between the first and second electrode; apparatus configured to supply salty water to the channel; and apparatus configured to receive water from the channel.
[0145] An electrochemical apparatus designated D including a first and a second electrode, at least one electrode including a composition of matter according to those designated A, AA, AB, AC, AD, AE, AF, AG, AH, AI or AJ; the porous mass of the composition of matter impregnated with a first chemical composition; the apparatus configured to permit the first chemical composition to enter into chemical reactions that provide an electric current between the first and second electrodes.
[0146] The apparatus designated D wherein the first chemical composition is impregnated into pores of the porous mass, the pores extending in an axis at least 10,000 microns, and having an average cross sectional area measured perpendicular to the axis between 25 and 2500 square microns.
Bacterial Cellulose (BC)
Bacterial Cellulose Production
[0147] D-Mannitol (white to off-white powder), tryptone (product of New Zealand), yeast extract (molecular genetics powder) and agar (product of Morocco) were obtained from Fisher Scientific (Waltham, MA). Sodium alginate (from brown algae) was
purchased from Sigma- Aldrich (St. Louis, MO). All chemicals were directly used without any purification. Gluconacetobacter hansenii (ATCC® 23769TM) was obtained from the American Type Culture Collection (ATCC, Manassas, VA) for bacterial cellulose culture. Gluconacetobacter hansenii, ATCC® 23769™, was used as the model strain and maintained on agar plates containing 25 g/L D-mannitol, 5 g/L yeast extract and 5 g/L tryptone and 20 g/L agar. The mannitol culture medium used for BC production consisted of 25 g/L D- mannitol, 5 g/L yeast extract and 5 g/L tryptone.
Bacterial Cellulose Pellicle Production
[0148] For the bacterial cellulose pellicle production, the strain from the agar plate was inoculated into a conical flask containing mannitol culture medium as the seed culture. The initial pH value of the medium was adjusted to 5.0 and was not regulated during the culture. The seed culture was incubated at 30°C and 130 rpm on a rotary shaker for 2 days, and 6 mL of this seed culture was inoculated into a 100-mL culture medium in 600-mL conical flask for production of BC. The cultivation was carried out at initial pH 5.0 and 30°C in a static incubator for 10 days. After incubation, the BC pellicles produced on the surface of mannitol culture medium were harvested and washed successively with water and 1% (w/v), aqueous NaOH at 90°C for 15 min, and then washed with deionized water to remove all microbial product contaminants and obtain purified pellicles.
Preparation of BC Flakes and BC- Sodium Alginate Composite Slurries
[0149] The BC pellicles were initially broken down into particles by cryomill (Retsch, Model 2013, Germany) via pre-cooling step at 5 Hz for 20 min and cryomilling step at 30Hz for 30 min. The whole procedure was carried out at liquid nitrogen temperature (- 96°C). The obtained BC particles could maintain BC's unique network structure with nanometer- scale pores. The BC particles was then mixed with a binder, sodium alginate solution (4.8 wt%) by a shear mixer (SpeedMixer™ DAC 150 FVZ-K, FlackTek Inc, Landrum, SC) at 3000 rpm for 2 min, to form homogenous BC-sodium alginate mixed slurries (5.0 wt BC and 0.9 wt sodium alginate in the slurries) prior to freeze casting.
Preparation of Freeze-east BC Aerogels by Directional Freeze Casting
[0150] The BC-sodium alginate mixed slurries were freezed by a custom-made freeze-caster at a speed of 10°C/min from 2°C to -150°C. After the BC-sodium alginate mixed slurries were completely frozen, the frozen materials were then lyophilized by a FreeZone 4.5 L lyophilizer (Labconco, Kansas City, MS) for 3 days to obtain the freeze-east
BC aerogels. The freeze-casting process can generate micrometer- scale porous structure for the aerogels while the BC pellicles maintain their nanometer- scale porous structure. Thus, the freeze-east BC aerogels with unique structure include two-lengths- scale porosity with high specific surface area.
Carbonization of Freeze-east BC Aerogels
[0151] The freeze-east BC aerogels were placed in cylindrical graphite boats for carbonization in a high vacuum furnace (ABAR 90, ABAR Corporation). The carbonization process is performed in 3 steps: (1) heat from room temperature (RT) to carbonization temperatures at 1000°C, 1100°C and 1200°C respectively at a heating rate 10°C/min; (2) carbonize at 1000°C, 1100°C and 1200°C respectively for lhr; (3) cool down to room temperature at a cooling rate 10°C/min.
Structural Characterization
[0152] The cylindrical frozen BC-sodium alginate mixed slurries after freeze casting were cut initially by a saw and then smoothed by a permanent steel knife (Delaware Diamond Knives, Inc. Wilmington, DE) at a -10°C cold room. The purpose for the step was to form uniform and smooth cross sections both perpendicular and parallel to the direction of freeze casting for the freeze-east BC aerogels. The cut samples were then placed for 3 days in the FreeZone 4.5 L lyophilizer to freeze-dry the freeze-east BC aerogels. The freeze-dried BC aerogels with smooth cross sections and carbonized aerogels were observed using scanning electron microscopy (SEM, FEI XL-30 FEG ESEM, Hillsboro, OR), and the accelerating voltages were set between 2 to 5 KV. The mean value of the pore area from the cross sections (cut perpendicular to the freeze direction) in the freeze-east BC aerogels and carbonized freeze-east aerogels were measured on SEM images using ImageJ (ImageJ, U.S. National Institutes of Health, Bethesda, MD).
Specific Surface Area Characterization
[0153] The freeze-east BC aerogels and carbonized freeze-east BC aerogels were cut into cubes with 5x5x5 mm3 dimension from topic, middle and bottom layers of the scaffolds by a Well 4240 saw (WELL Diamond Wire Saws, Inc., Norcross, GA) for specific surface area characterization. The diameter of diamond- decorated steel wire for the saw was 220 μιη, and the wire speed was 0.6 m/s. All the cubes were first degassed at 100°C and specific area values for them were measured through nitrogen adsorption by Brenauer- Emmett-Teller (BET) system.
Electrical Conductivity
[0154] The electrical conductivity (σ) measurement for all samples from top, middle and bottom layers of carbonized freeze-east BC aerogel scaffolds was conducted using a standard Four- Point probe test. A four point probe (Signatone, Model S-301-4, Gilroy, CA) used is a simple apparatus for measuring the resistivity (the inverse of electrical conductivity) of samples by passing a current through two outer probes and measuring the voltage through the inner two probes. Usually, the Four-Point probe tests are ideal for measurement of resistivity of sheet and bulk. For the samples thickness equal or higher than half the probe spacing, a corrected formula was used to calculate the electrical conductivity of carbonized freeze-east BC aerogels.
Raman Spectroscopy
[0155] The Raman spectra for the carbonized freeze-east BC aerogel samples processed at different carbonization temperatures (1000, 1100, and 1200°C) were obtained using Confocal Raman Microscope CRM 200 (WITec GmbH, Ulm, Germany) with a 514.4 nm laser source. The range of wavenumber for the spectra was chosen from 800 to 2000cm"1.
Mechanical Properties
[0156] Freeze-east BC aerogels and carbonized freeze-east BC aerogels were cut into cubes with 5x5x5 mm dimension from topic, middle and bottom layers of the scaffolds by the Well saw for mechanical properties characterization. The diamond- decorated steel wire diameter for the saw was 220 μιη, and the wire speed was 0.6 m/s. The compression tests were then conducted on an Instron 5948 (Instron, Norwood, MA) with a 5 N load cell at ambient conditions. The cross-head speed for all the measurements was 0.05 mm/s (strain rate 0.01/s). Two test directions were used for the mechanical test, including the test parallel to the freezing direction and the test perpendicular to the freezing direction. The reason for choosing compression test instead of tensile test is that cellular materials tend to be used for more applications when they were loaded in compression and bending rather than in tension. Young's modulus, yield strength and toughness were obtained from compressive stress-strain curves from the tests. The toughness values were evaluated by the area up to 60% strain under compressive stress-strain curves.
Results
BC Pellicle Structure
[0157] BC is usually grown in the form of pellicles of several tens of millimeter in diameter and few millimeter in thickness. The pellicles consist of a network of BC- nano fibers, which typically are less than 100 nm in diameter and have pore diameters that range from several tens to several hundred nanometers. For freeze casting, the BC pellicles were cryomilled into micrometer- scale BC flakes with edge lengths in the range of 20-100 μιη and a well- maintained nanometer- scale pore structure. The cryomilling may be omitted in some embodiments.
Density of Freeze-east BC Aerogels before Carbonization
[0158] The mean density of freeze-east BC aerogels before carbonization was about 0.055 g/cm.3
Density of Freeze-east BC Aerogels before Carbonization
[0159] The diameter and height of carbonized freeze-east BC aerogel were about 50% of their original values for freeze-east BC aerogel due to the significant dimensional shrinkage during carbonization. However, because during carbonization elements and side- groups other than carbon are mostly removed the mean density of the freeze-east BC aerogels after carbonization was 0.052 g/cm\ very similar to its density before carbonization.
Pore size and morphology of Freeze-east BC Aerogels before Carbonization
[0160] Before carbonization, the freeze-east aerogels exhibit a hierarchical architecture with a honeycomb-like, uniform distribution of micro meter- scale pores that have mean long and short pore axes in the range of 15-40 μιη and 8-20 μιη, respectively, perpendicular to the freezing direction and run through the entire sample parallel to the freezing direction. The nanoporosity in the cell wall material ranges from several tens to several hundreds of nanometers in diameter.
Pore size and morphology of Freeze-east BC Aerogels after Carbonization
[0161] After carbonization, the freeze-east aerogels retained the hierarchical architecture from before carbonication. The aerogels had a honeycomb-like, uniform distribution of micro meter- scale pores that had mean long and short pore axes in the range of 8-20 μιη and 4-10 μιη, respectively, perpendicular to the freezing direction and run through
the entire sample parallel to the freezing direction, which is about half of the value before carbonization. The nanoporosity size in the cell wall material was, due to the material shrinkage, difficult to measure after carbonization.
Electrical Conductivity for Carbonized Freeze- cast BC Aerogels
[0162] The electrical conductivity of carbonized freeze-east BC aerogel processed at 1200°C can be up to 1.68+0.04 S/cm at a density of 0.055 g/cm3, which was significantly higher than the carbonized freeze-east BC aerogels processed at 1000°C (0.70+0.02 S/cm) and 1100°C (0.71+0.02 S/cm) whose density was in the range of 0.044-0.046 g/cm3.
Carbonized freeze-east BC aerogels processed above 1200°C turned into are structure with poor integrity and lower electrical conductivity.
[0163] Raman spectroscopy performed on carbonized freeze-east BC aereogels processed at different temperatures in the range of 1000-1200°C additionally indicated, through the shift of the intensities of the D peak at 1330 cm"1 and G peak at 1590 cm"1 that higher temperature carbonization resulted in a higher R value, which represents the intensity ratio of D peak and G peak (R=ID/IG) and is frequently used as an indicator of the degree of carbonization. This higher degree of carbonization, will likely have contributed to the higher electrical conductivity in addition to the higher density of the material carbonized at 1200°C.
[0164] These results illustrate that several mechanisms exist by which the electrical conductivity can be tailored for a given application.
[0165] The electrical conductivity values for the carbonized freeze-east BC aerogel in the study was considerably higher than those previously reports for carbonized BC aerogel materials, which had a typical range of 0.20-0.41 S/cm. However, density values were not reported for these literature values, which makes a fair comparison difficult.
Mechanical Properties of Freeze-east BC Aerogels before Carbonization
[0166] The mechanical properties of the not yet carbonized freeze-east BC aerogels were tested in two directions: parallel and perpendicular to the freezing direction.
[0167] Parallel to the freezing direction, the Young's modulus of the not yet carbonized material ranged from 476 to 646 kPa, the yield strength ranged from 27.3 to 37.1 kPa, and the toughness from 24.2 to 42.2 kJ/m for densities ranging from 0.0520 to 0.0570 g/cm , respectively.
[0168] Perpendicular to the freezing direction, the Young's modulus of the not yet carbonized material ranged from 196 to 268 kPa, the yield strength from 12.3 to 15 kPa, and
the toughness from 18.6 to 25.2 kJ/m 3 for densities ranging from 0.052 to 0.057
 respectively.
respectively.
[0169] The mechanical anisotropy, which is the ratio of the property values measured parallel to freezing direction divided by the values determined perpendicular to the perpendicular to the freezing direction ranged from 2.43 to 2.41 for the not yet carbonized material.
Mechanical Properties of Freeze-east BC Aerogels after Carbonization
[0170] The mechanical properties of carbonized freeze-east BC aerogels were tested in two directions: parallel and perpendicular to the freezing direction.
[0171] Parallel to the freezing direction, the Young's modulus of the carbonized material ranged from 373 to 633 kPa, the yield strength from 33.9 to 37.7 kPa, and the toughness from 19.8 to 26.0 kJ/m 3 for densities ranging from 0.049 to 0.052

respectively.
[0172] Perpendicular to the freezing direction, the Young's modulus of the carbonized material ranged from 332 to 507 kPa, the yield strength from 20.1 to 36.5 kPa, and the toughness from 12.3 to 21.5 kJ/m 3 for densities ranging from 0.049 to 0.052
 respectively.
respectively.
[0173] The mechanical anisotropy, which is the ratio of the property values measured parallel to freezing direction divided by the values determined perpendicular to the perpendicular to the freezing direction ranged from 1.12 to 1.25 for the carbonized material.
[0174] Young's modulus and yield strength tested parallel to the freezing direction for the carbonized freeze-east BC aerogels didn't change significantly, but the toughness values decreased significantly. However, Young's modulus and yield strength from the test direction perpendicular to the freezing direction for the carbonized freeze-east BC aerogels increased significantly, and the toughness decreased.
Mechanical properties of freeze-east BC aerogels

Mechanical properties of carbonized BC freeze-east BC aerogels

Design Requirements and suitable applications:
Freeze cast BC aerogel
[0175] A variety of material applications, especially in biomedical and catalysis areas, require the fabrication of highly porous scaffolds with superior properties in high surface area, low density, decent mechanical properties, and excellent biodegradability and biocompatibility. The materials can be suitable for many applications, like bone tissue engineering, drug delivery, biocatalysis reactions. In addition, due to its unique porous
structures, it can be good candidates as matrixes for making biorobots (microscale pore can fit bacteria while nanoscale pore to fit virus etc.)
Conclusion
[0176] While the invention has been particularly shown and described with reference to particular embodiments thereof, it will be understood by those skilled in the art that various other changes in the form and details may be made without departing from the spirit and scope of the invention. It is to be understood that various changes may be made in adapting the invention to different embodiments without departing from the broader inventive concepts disclosed herein and comprehended by the claims that follow.
Claims
1. A composition of matter comprising a porous mass comprising a bound and reduced composite comprising graphene fully or partially reduced from graphene oxide, and a carbon structure selected from the group consisting of carbon nanotubes and carbon nano fibers.
2. The composition of matter of claim 1 wherein the mass is directionally porous.
3. The composition of matter of claim 2 wherein the porous mass has been formed from graphene oxide reduced by heat treatment in non-oxidizing atmosphere, where reduced graphene oxide is present at ratios of between 1: 1 and 1:4 in proportion by weight to the carbon structure.
4. The composition of matter of claim 3 wherein at least a plurality of pores of the porous mass extend in an axis at least 10,000 microns, and have an average cross sectional area measured perpendicular to the axis is between 25 and 2500 square microns.
5. The composition of matter of claim 4 wherein the average cross sectional area of the pores is between 80 and 1000 square microns.
6. The compositon of matter of claim 1, 2, 4, or 5, the porous mass having porosity between 75 and 99 percent.
7. The composition of matter of claim 6 wherein the porous mass has been formed from graphene oxide reduced by heat treatment in non-oxidizing atmosphere, wherein graphene and unreduced graphene oxide is present at ratios of between 1:2 and 1:4 in proportion by weight to the carbon structure.
8. The composition of matter of claim 7 having an electrical conductivity of at least 7 siemens/meter.
9. The composition of matter of claim 7 having an electrical conductivity between 7 and 50 siemens/meter.
10. The composition of matter of claim 9 wherein the carbon structure comprises carbon nanotubes.
11. The composition of matter of claim 9 wherein the carbon structure comprises carbon nanofibers.
12. A method of manufacture of a porous mass comprising:
Preparing a slurry comprising water, graphene oxide, and a carbon structure selected from the group consisting of carbon nanotubes, carbon nanofibers, and activated carbon;
Freeze-casting the slurry, and sublimating the water, to form a green casting; and Reducing graphene oxide of the green casting, thereby binding particles of the
graphene oxide and carbon structure to form the porous mass.
13. The method of claim 12 wherein reducing graphene oxide of the green casting is performed by heating the green casting to a temperature between 400 and 1500 degrees Celsius.
14. The method of claim 13 wherein the graphene oxide comprises 0.5- 5 micron diameter flakes, with a thickness of 1 atomic layer in at least 60% of flakes.
15. The method of claim 12, 13, or 14 wherein the slurry further comprises ethanol.
16. The method of claim 15 wherein the carbon structure comprises straight carbon nanotubes.
16. The method of claim 15 wherein the carbon structure comprises curly carbon nanotubes.
17. The method of claim 15 wherein the carbon structure comprises carbon nanofibers.
18. The method of claim 12, 13, or 14 wherein the slurry further comprises cellulose nanofibers.
19. The method of claim 18 wherein the carbon structure comprises straight carbon nanotubes.
20. The method of claim 18 wherein the carbon structure comprises curly carbon nanotubes.
21. The method of claim 18 wherein the carbon structure comprises carbon nano fibers.
22. The method of claim 14 or 15 wherein the slurry further comprises sucrose.
23. A water purification apparatus comprising
a first and a second electrode, at least one electrode comprising a porous mass
comprising an annealed graphene oxide composite, the composite further comprising a carbon structure selected from the group consisting of carbon nanotubes and carbon nanofibers, the first and second electrodes adjacent to a channel;
a power supply coupled to apply a voltage difference between the first and second electrode;
apparatus configured to supply salty water to the channel; and
apparatus configured to receive water from the channel.
24. An electrochemical apparatus comprising
a first and a second electrode, at least the first electrode comprising a porous mass comprising an annealed graphene oxide composite, the composite further comprising a carbon structure selected from the group consisting of carbon nanotubes and carbon nanofibers;
the porous mass impregnated with a first chemical composition;
the apparatus configured to permit the first chemical composition to enter into
chemical reactions that provide an electric current between the first and second electrodes.
25. The apparatus of claim 24 wherein the first chemical composition is impregnated into pores of the porous mass, the pores extending in an axis at least 10,000 microns, and having an average cross sectional area measured perpendicular to the axis between 25 and 2500 square microns.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201461928920P | 2014-01-17 | 2014-01-17 | |
| US61/928,920 | 2014-01-17 | ||
| US201462087565P | 2014-12-04 | 2014-12-04 | |
| US62/087,565 | 2014-12-04 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015109272A1 true WO2015109272A1 (en) | 2015-07-23 |
Family
ID=53543526
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2015/011882 WO2015109272A1 (en) | 2014-01-17 | 2015-01-17 | Material and method of manufacture of electrodes and porous filters formed of ice-templated graphene-oxide and carbon nanotube composite, and applications thereof |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2015109272A1 (en) |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017031403A1 (en) * | 2015-08-20 | 2017-02-23 | University Of Virginia Patent Foundation | Method of forming graphene/metal-oxide hybrid reinforced composites and product thereof |
| WO2017217832A1 (en) * | 2016-06-17 | 2017-12-21 | 한국기계연구원 | Method for producing carbon aerogel precursor, and carbon aerogel precursor and carbon aerogel produced thereby |
| CN107500292A (en) * | 2017-09-25 | 2017-12-22 | 江苏苏利精细化工股份有限公司 | A kind of method that graphene oxide is modified cocoanut active charcoal |
| KR101812946B1 (en) | 2016-03-22 | 2017-12-28 | 재단법인대구경북과학기술원 | Graphene-Reduced Graphene Oxide Composite and Method for Preparing the Same |
| CN109020547A (en) * | 2018-09-03 | 2018-12-18 | 大同宇林德石墨设备股份有限公司 | A kind of preparation method of the graphite electrode nipple containing graphene |
| WO2019010565A1 (en) | 2017-07-10 | 2019-01-17 | Noel John Alexander | Phase-change material and method for producing same |
| CN109397474A (en) * | 2018-10-12 | 2019-03-01 | 中国科学院金属研究所 | The construction method and refrigerating plant of construction material with microcosmic orienting stephanoporate structure |
| CN109417209A (en) * | 2016-07-01 | 2019-03-01 | 日本电信电话株式会社 | Battery and the method for manufacturing its anode |
| WO2019071033A1 (en) * | 2017-10-05 | 2019-04-11 | The Board Of Trustees Of The Leland Stanford Junior University | Conductive graphene/carbon nanofiber composite scaffold, its use for neural tissue engineering and a method of preparation thereof |
| EP3529207A4 (en) * | 2016-10-18 | 2020-04-29 | SABIC Global Technologies B.V. | Methods for producing carbon material-graphene composite films |
| CN111129449A (en) * | 2019-12-02 | 2020-05-08 | 深圳石墨烯创新中心有限公司 | Graphene/carbon/ferroferric oxide nanocomposite and preparation method and application thereof |
| CN111252754A (en) * | 2020-03-12 | 2020-06-09 | 浙江大学 | A kind of graphene aerogel pore regulation method and a kind of graphene gradient aerogel |
| EP3663259A4 (en) * | 2017-08-04 | 2021-05-05 | Nippon Telegraph and Telephone Corporation | Cellulose nanofiber carbon and method of producing the same |
| EP3782956A4 (en) * | 2018-04-16 | 2022-01-12 | Nippon Telegraph And Telephone Corporation | PROCESS FOR THE PRODUCTION OF BACTERIA-PRODUCED CELLULOSIC CARBON |
| CN114597360A (en) * | 2022-03-02 | 2022-06-07 | 江西省纳米技术研究院 | Composite positive electrode material with array orientation hole structure, preparation method and application thereof |
| CN114590794A (en) * | 2022-03-09 | 2022-06-07 | 中国科学技术大学 | Compressible carbon nanofiber aerogel, and preparation method and application thereof |
| CN114890815A (en) * | 2022-05-07 | 2022-08-12 | 常州富烯科技股份有限公司 | Method for preparing carbon fiber reinforced graphene foam block and related product |
| CN115818628A (en) * | 2022-09-30 | 2023-03-21 | 中国人民解放军陆军装甲兵学院 | Three-dimensional graphene foam, preparation method and application thereof, and wave-absorbing material |
| WO2023097008A1 (en) * | 2021-11-23 | 2023-06-01 | The Trustees Of Princeton University | Method, system, and devices for water, organics, and/or mineral recovery |
| CN117558897A (en) * | 2023-12-01 | 2024-02-13 | 浙江大学 | Preparation method of silicon/carbon thick electrode and its application in lithium-ion battery |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100317790A1 (en) * | 2009-03-03 | 2010-12-16 | Sung-Yeon Jang | Graphene composite nanofiber and preparation method thereof |
| US20120031852A1 (en) * | 2009-04-06 | 2012-02-09 | Sa Envitech S.R.L. | Graphene based electrodes for electrochemical reactions, and electrooxidation process for the removal of contaminants from liquids using said electrodes |
| US20120034385A1 (en) * | 2010-08-06 | 2012-02-09 | Delta Electronics, Inc. | Manufacturing process for porous material |
| WO2012109665A1 (en) * | 2011-02-13 | 2012-08-16 | Indiana University Research And Technology Corporation | High surface area nano-structured graphene composites and capac!tive devices incorporating the same |
| CN102674315A (en) * | 2012-04-25 | 2012-09-19 | 浙江大学 | Graphene-carbon nano tube composite all-carbon ultra-light elastic aerogel and preparation method thereof |
| CN102941042A (en) * | 2012-10-25 | 2013-02-27 | 北京理工大学 | Graphene/metal oxide hybrid aerogel, preparation method and applications thereof |
| WO2013153255A1 (en) * | 2012-04-11 | 2013-10-17 | Kangas Veijo | Carbon nanotube - polysaccharide composite |
| WO2013180661A1 (en) * | 2012-06-01 | 2013-12-05 | National University Of Singapore | Synthesis of three-dimensional graphene foam: use as supercapacitors |
-
2015
- 2015-01-17 WO PCT/US2015/011882 patent/WO2015109272A1/en active Application Filing
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100317790A1 (en) * | 2009-03-03 | 2010-12-16 | Sung-Yeon Jang | Graphene composite nanofiber and preparation method thereof |
| US20120031852A1 (en) * | 2009-04-06 | 2012-02-09 | Sa Envitech S.R.L. | Graphene based electrodes for electrochemical reactions, and electrooxidation process for the removal of contaminants from liquids using said electrodes |
| US20120034385A1 (en) * | 2010-08-06 | 2012-02-09 | Delta Electronics, Inc. | Manufacturing process for porous material |
| WO2012109665A1 (en) * | 2011-02-13 | 2012-08-16 | Indiana University Research And Technology Corporation | High surface area nano-structured graphene composites and capac!tive devices incorporating the same |
| WO2013153255A1 (en) * | 2012-04-11 | 2013-10-17 | Kangas Veijo | Carbon nanotube - polysaccharide composite |
| CN102674315A (en) * | 2012-04-25 | 2012-09-19 | 浙江大学 | Graphene-carbon nano tube composite all-carbon ultra-light elastic aerogel and preparation method thereof |
| WO2013180661A1 (en) * | 2012-06-01 | 2013-12-05 | National University Of Singapore | Synthesis of three-dimensional graphene foam: use as supercapacitors |
| CN102941042A (en) * | 2012-10-25 | 2013-02-27 | 北京理工大学 | Graphene/metal oxide hybrid aerogel, preparation method and applications thereof |
Cited By (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10781142B2 (en) | 2015-08-20 | 2020-09-22 | University Of Virginia Patent Foundation | Method of forming graphene/metal-oxide hybrid reinforced composites and product thereof |
| WO2017031403A1 (en) * | 2015-08-20 | 2017-02-23 | University Of Virginia Patent Foundation | Method of forming graphene/metal-oxide hybrid reinforced composites and product thereof |
| KR101812946B1 (en) | 2016-03-22 | 2017-12-28 | 재단법인대구경북과학기술원 | Graphene-Reduced Graphene Oxide Composite and Method for Preparing the Same |
| KR20170142935A (en) * | 2016-06-17 | 2017-12-28 | 한국기계연구원 | Producing method of carbon aerogel and carbon aerogel made by the same |
| KR20170142937A (en) * | 2016-06-17 | 2017-12-28 | 한국기계연구원 | Nano fiber of carbon aerogel and graphene composite and manufacturing method of the same |
| KR20170142936A (en) * | 2016-06-17 | 2017-12-28 | 한국기계연구원 | A carbon airgel containing carbon particles on its surface and producing method of the same |
| US11203003B2 (en) | 2016-06-17 | 2021-12-21 | Korea Institute Of Materials Science | Method of preparing carbon aerogel precursor, carbon aerogel precursor prepared thereby, and carbon aerogel |
| WO2017217832A1 (en) * | 2016-06-17 | 2017-12-21 | 한국기계연구원 | Method for producing carbon aerogel precursor, and carbon aerogel precursor and carbon aerogel produced thereby |
| KR101963139B1 (en) * | 2016-06-17 | 2019-03-28 | 한국기계연구원 | Producing method of carbon aerogel and carbon aerogel made by the same |
| KR101963128B1 (en) * | 2016-06-17 | 2019-04-01 | 한국기계연구원 | Nano fiber of carbon aerogel and graphene composite and manufacturing method of the same |
| KR101963133B1 (en) * | 2016-06-17 | 2019-07-31 | 한국기계연구원 | A carbon airgel containing carbon particles on its surface and producing method of the same |
| US11876207B2 (en) | 2016-07-01 | 2024-01-16 | Nippon Telegraph And Telephone Corporation | Battery and method of manufacturing cathode of the same |
| CN109417209A (en) * | 2016-07-01 | 2019-03-01 | 日本电信电话株式会社 | Battery and the method for manufacturing its anode |
| EP3518338A4 (en) * | 2016-07-01 | 2020-06-24 | Nippon Telegraph And Telephone Corporation | BATTERY AND METHOD FOR PRODUCING POSITIVE ELECTRODE FOR BATTERY |
| EP3529207A4 (en) * | 2016-10-18 | 2020-04-29 | SABIC Global Technologies B.V. | Methods for producing carbon material-graphene composite films |
| JP2020527178A (en) * | 2017-07-10 | 2020-09-03 | アレクサンダー ノエル,ジョン | Phase change material and method for manufacturing it |
| WO2019010565A1 (en) | 2017-07-10 | 2019-01-17 | Noel John Alexander | Phase-change material and method for producing same |
| US11795360B2 (en) | 2017-07-10 | 2023-10-24 | Mary Anne White | Phase-change material and method for producing same |
| US11396619B2 (en) | 2017-07-10 | 2022-07-26 | Mary Anne White | Phase-change material and method for producing same |
| EP4299548A3 (en) * | 2017-07-10 | 2024-04-03 | Noël, John Alexander | Phase-change material and method for producing same |
| EP3652266A4 (en) * | 2017-07-10 | 2021-03-31 | Noël, John Alexander | PHASE CHANGE MATERIAL AND METHOD OF ITS MANUFACTURING |
| US11319209B2 (en) | 2017-08-04 | 2022-05-03 | Nippon Telegraph And Telephone Corporation | Method of producing cellulose-nanofiber carbon |
| EP3663259A4 (en) * | 2017-08-04 | 2021-05-05 | Nippon Telegraph and Telephone Corporation | Cellulose nanofiber carbon and method of producing the same |
| CN107500292A (en) * | 2017-09-25 | 2017-12-22 | 江苏苏利精细化工股份有限公司 | A kind of method that graphene oxide is modified cocoanut active charcoal |
| US20200222591A1 (en) * | 2017-10-05 | 2020-07-16 | The Board Of Trustees Of The Leland Stanford Junior University | Conductive Graphene/Carbon Nanofiber Composite Scaffold, its use for neural tissue engineering and a method of preparation thereof |
| WO2019071033A1 (en) * | 2017-10-05 | 2019-04-11 | The Board Of Trustees Of The Leland Stanford Junior University | Conductive graphene/carbon nanofiber composite scaffold, its use for neural tissue engineering and a method of preparation thereof |
| US12285544B2 (en) | 2017-10-05 | 2025-04-29 | The Board Of Trustees Of The Leland Stanford Junior University | Conductive graphene/carbon nanofiber composite scaffold, its use for neural tissue engineering and a method of preparation thereof |
| EP3782956A4 (en) * | 2018-04-16 | 2022-01-12 | Nippon Telegraph And Telephone Corporation | PROCESS FOR THE PRODUCTION OF BACTERIA-PRODUCED CELLULOSIC CARBON |
| US11472707B2 (en) | 2018-04-16 | 2022-10-18 | Nippon Telegraph And Telephone Corporation | Method for manufacturing bacterium-produced cellulose carbon |
| CN109020547A (en) * | 2018-09-03 | 2018-12-18 | 大同宇林德石墨设备股份有限公司 | A kind of preparation method of the graphite electrode nipple containing graphene |
| CN109397474B (en) * | 2018-10-12 | 2021-01-08 | 中国科学院金属研究所 | Construction method of building material with microcosmic directional porous structure and refrigerating device |
| CN109397474A (en) * | 2018-10-12 | 2019-03-01 | 中国科学院金属研究所 | The construction method and refrigerating plant of construction material with microcosmic orienting stephanoporate structure |
| CN111129449A (en) * | 2019-12-02 | 2020-05-08 | 深圳石墨烯创新中心有限公司 | Graphene/carbon/ferroferric oxide nanocomposite and preparation method and application thereof |
| CN111252754B (en) * | 2020-03-12 | 2020-12-29 | 浙江大学 | A kind of graphene aerogel pore regulation method and a kind of graphene gradient aerogel |
| CN111252754A (en) * | 2020-03-12 | 2020-06-09 | 浙江大学 | A kind of graphene aerogel pore regulation method and a kind of graphene gradient aerogel |
| WO2023097008A1 (en) * | 2021-11-23 | 2023-06-01 | The Trustees Of Princeton University | Method, system, and devices for water, organics, and/or mineral recovery |
| CN114597360B (en) * | 2022-03-02 | 2023-12-08 | 江西省纳米技术研究院 | Composite positive electrode material with array orientation hole structure, preparation method and application thereof |
| CN114597360A (en) * | 2022-03-02 | 2022-06-07 | 江西省纳米技术研究院 | Composite positive electrode material with array orientation hole structure, preparation method and application thereof |
| CN114590794A (en) * | 2022-03-09 | 2022-06-07 | 中国科学技术大学 | Compressible carbon nanofiber aerogel, and preparation method and application thereof |
| CN114890815A (en) * | 2022-05-07 | 2022-08-12 | 常州富烯科技股份有限公司 | Method for preparing carbon fiber reinforced graphene foam block and related product |
| CN115818628A (en) * | 2022-09-30 | 2023-03-21 | 中国人民解放军陆军装甲兵学院 | Three-dimensional graphene foam, preparation method and application thereof, and wave-absorbing material |
| CN115818628B (en) * | 2022-09-30 | 2024-01-30 | 中国人民解放军陆军装甲兵学院 | Three-dimensional graphene foam, preparation method and application thereof, and wave-absorbing material |
| CN117558897A (en) * | 2023-12-01 | 2024-02-13 | 浙江大学 | Preparation method of silicon/carbon thick electrode and its application in lithium-ion battery |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2015109272A1 (en) | Material and method of manufacture of electrodes and porous filters formed of ice-templated graphene-oxide and carbon nanotube composite, and applications thereof | |
| KR101963139B1 (en) | Producing method of carbon aerogel and carbon aerogel made by the same | |
| Lu et al. | Chemical synthesis of carbon materials with intriguing nanostructure and morphology | |
| CN104661959B (en) | graphene-based material | |
| Whitby et al. | Geometric control and tuneable pore size distribution of buckypaper and buckydiscs | |
| Shehzad et al. | Three-dimensional macro-structures of two-dimensional nanomaterials | |
| CA2962468C (en) | Porous carbon films | |
| Jiang et al. | Enhanced room temperature hydrogen storage capacity of hollow nitrogen-containing carbon spheres | |
| Sudeep et al. | Covalently interconnected three-dimensional graphene oxide solids | |
| Wang et al. | Self-assembly of graphene into three-dimensional structures promoted by natural phenolic acids | |
| US20180251377A1 (en) | Aerogels | |
| Zhao et al. | Easy synthesis of ordered meso/macroporous carbon monolith for use as electrode in electrochemical capacitors | |
| Chen et al. | Aligned macroporous TiO2/chitosan/reduced graphene oxide (rGO) composites for photocatalytic applications | |
| Yu et al. | Porous tubular carbon nanorods with excellent electrochemical properties | |
| KR20140087022A (en) | Method for preparing a silicon/carbon composite material, resulting material, and electrode, in particular negative electrode, including said material | |
| KR20140143756A (en) | Aerogel based on doped graphene | |
| Qiu et al. | Excellent Specific Mechanical and Electrical Properties of Anisotropic Freeze‐Cast Native and Carbonized Bacterial Cellulose‐Alginate Foams | |
| CN113527753B (en) | Bio-based foam material prepared at normal pressure and preparation method and application thereof | |
| RU2577273C1 (en) | Method of producing of aerogels based on multilayer carbon nanotubes | |
| Lee et al. | Fabrication of Li2TiO3 pebbles by a freeze drying process | |
| CN111389358A (en) | Preparation method of modified nitrogen-doped carbon aerogel | |
| CN111164716A (en) | Graphene-containing electrode, method for producing the same, and power storage device using the same | |
| EP2842910B1 (en) | Method of preparing graphene paper | |
| JP2016191014A (en) | Functional porous material containing carbon nanotubes | |
| JP5388051B2 (en) | Mesoporous carbon (MC-MCM-48) and method for producing the same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15737075 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 15737075 Country of ref document: EP Kind code of ref document: A1 |