WO2014178309A1 - ジメチルスルホキシドの精製法 - Google Patents
ジメチルスルホキシドの精製法 Download PDFInfo
- Publication number
- WO2014178309A1 WO2014178309A1 PCT/JP2014/061306 JP2014061306W WO2014178309A1 WO 2014178309 A1 WO2014178309 A1 WO 2014178309A1 JP 2014061306 W JP2014061306 W JP 2014061306W WO 2014178309 A1 WO2014178309 A1 WO 2014178309A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- dimethyl sulfoxide
- water
- distillation
- distilled
- purifying
- Prior art date
Links
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 title claims abstract description 269
- 238000000034 method Methods 0.000 title claims abstract description 49
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims abstract description 70
- 239000002994 raw material Substances 0.000 claims abstract description 22
- 238000004821 distillation Methods 0.000 claims description 55
- 238000002835 absorbance Methods 0.000 claims description 23
- 238000000746 purification Methods 0.000 claims description 14
- 239000011259 mixed solution Substances 0.000 claims description 10
- 238000002156 mixing Methods 0.000 claims description 8
- 239000012153 distilled water Substances 0.000 claims description 5
- 239000008399 tap water Substances 0.000 claims description 5
- 235000020679 tap water Nutrition 0.000 claims description 5
- 229910021642 ultra pure water Inorganic materials 0.000 claims description 5
- 239000012498 ultrapure water Substances 0.000 claims description 5
- 238000004458 analytical method Methods 0.000 claims description 4
- 239000000243 solution Substances 0.000 claims description 4
- 238000004817 gas chromatography Methods 0.000 description 10
- 239000000126 substance Substances 0.000 description 8
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 238000001577 simple distillation Methods 0.000 description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 230000000052 comparative effect Effects 0.000 description 4
- 239000012535 impurity Substances 0.000 description 4
- 238000011403 purification operation Methods 0.000 description 4
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 238000000691 measurement method Methods 0.000 description 3
- 239000003960 organic solvent Substances 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 101000671819 Homo sapiens Ubiquitin carboxyl-terminal hydrolase 36 Proteins 0.000 description 2
- 102100040109 Ubiquitin carboxyl-terminal hydrolase 36 Human genes 0.000 description 2
- 239000003905 agrochemical Substances 0.000 description 2
- 238000000998 batch distillation Methods 0.000 description 2
- 230000005587 bubbling Effects 0.000 description 2
- 239000012159 carrier gas Substances 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 239000007810 chemical reaction solvent Substances 0.000 description 2
- 238000001944 continuous distillation Methods 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 239000001307 helium Substances 0.000 description 2
- 229910052734 helium Inorganic materials 0.000 description 2
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 238000000199 molecular distillation Methods 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 230000009965 odorless effect Effects 0.000 description 2
- 239000010409 thin film Substances 0.000 description 2
- 238000005292 vacuum distillation Methods 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 1
- 239000012459 cleaning agent Substances 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 239000012776 electronic material Substances 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000010408 film Substances 0.000 description 1
- 239000008235 industrial water Substances 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 239000010453 quartz Substances 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N silicon dioxide Inorganic materials O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C315/00—Preparation of sulfones; Preparation of sulfoxides
- C07C315/06—Separation; Purification; Stabilisation; Use of additives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C317/00—Sulfones; Sulfoxides
- C07C317/02—Sulfones; Sulfoxides having sulfone or sulfoxide groups bound to acyclic carbon atoms
- C07C317/04—Sulfones; Sulfoxides having sulfone or sulfoxide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
Definitions
- Example 1 In a 200 mL round bottom flask, 50 g of general industrial quality dimethyl sulfoxide having a purity of 99.9% by weight (absorbance at 275 nm is 0.22, odor intensity is 3) and 50 g of ion-exchanged water equivalent to A4 of JIS-K0557 (100 parts by weight / 100 parts by weight of dimethyl sulfoxide) was added and mixed. A rectifying tube having a diameter of 3 cm and a height of 15 cm filled with 10 cm of Raschig rings (length 5 mm, outer diameter 5 mm, inner diameter 3 mm) was placed in the mouth of the round bottom flask, and distilled under reduced pressure at 6.7 kPa.
- Example 18 In Example 13, the mixing amount of water was changed as described in Table 4, and the internal temperature of the round bottom flask was changed from 80 ° C. to 110 ° C. without using a rectification tube in the second distillation operation. The same purification operation as in Example 13 was performed except that the film was distilled by heating. The quality of the purified dimethyl sulfoxide obtained is listed in Table 4.
Abstract
Description
(1)原料ジメチルスルホキシド100重量部に対し、水35重量部以上を混合した溶液を蒸留することを特徴とするジメチルスルホキシドの精製法。
(2)精製されたジメチルスルホキシドの275nmにおける吸光度を、0.01~0.20にすることを特徴とする(1)に記載のジメチルスルホキシドの精製法。
(3)前記水がイオン交換水、蒸留水または超純水であることを特徴とする(1)または(2)に記載のジメチルスルホキシドの精製法。
(4)前記水の25℃の電気伝導率が50mS/m以下であることを特徴とする(1)~(3)のいずれかに記載のジメチルスルホキシドの精製法。
(5)前記混合した溶液の蒸留を1段で行い、水を留去してジメチルスルホキシドを得ることを特徴とする(1)~(4)のいずれかに記載のジメチルスルホキシドの精製法。
(6)前記混合した溶液に1段目の蒸留を行い、水を留去させ、粗精製したジメチルスルホキシドを得、さらに粗精製したジメチルスルホキシドに2段目の蒸留を行い、精製したジメチルスルホキシドを得ることを特徴とする(1)~(4)のいずれかに記載のジメチルスルホキシドの精製法。
(7)上記(1)~(6)のいずれかに記載の精製法により得られたジメチルスルホキシドであって、ガスクロマトグラフ(GC)分析での純度が99.996%以上であることを特徴とする高純度ジメチルスルホキシド。
臭気強度0: 無臭
臭気強度1: やっと感知できるにおい
臭気強度2: 何のにおいか判る弱いにおい
臭気強度3: 楽に感知できるにおい
臭気強度4: 強いにおい
臭気強度5: 強烈なにおい
以下の条件のガスクロマトグラフ(島津社製GC-2010)により、原料ジメチルスルホキシドの化学純度を測定した。
分析装置 :GC(島津社製GC-2010)
キャリアガス :ヘリウム (カラム流量1.7mL/分)
分析カラム :RESTEK社製Rxt-1 (15m×0.32mm×3μm)
カラム温度 :100℃(15分)→(10℃/分)→170℃(20分)
注入口温度 :210℃
注入量 :1μL
検出器 :FID
検出器温度 :220℃
以下の条件のガスクロマトグラフ(Agilent社製7890A)により質量分析を行い、保持時間4.3分に検出した不明不純物を測定した。
分析装置 :GC(Agilent社製7890A)
キャリアガス :ヘリウム (カラム流量2.0mL/分)
分析カラム :RESTEK社製Stabilwax (30m×0.32mm×0.5μm)
カラム温度 :35℃(3分)→(7℃/分)→130℃(10分)→(7℃/分)→160℃→(15℃/分)→250℃(8分)
注入口温度 :200℃
注入量 :1μL
検出器 :FID
検出器温度 :270℃
ジメチルスルホキシドを事前に窒素バブリングを30分間実施し、その後10分以内に、紫外吸光光度計(島津社製UV-1800)を使用し、以下の条件で275nmの吸光度を測定した。
分析装置 :紫外吸光光度計
セル :石英、幅1cm
ブランク :水
ジメチルスルホキシドの臭気を、下記に示す6段階の臭気強度表示法に基づき3人のパネラーが官能評価した。30mLサンプル瓶に、ジメチルスルホキシドを15mL入れサンプルとした。パネラー3人が同じサンプルの臭気を、室温下でサンプル瓶に鼻を近づけて嗅ぎ、下記に示す0~5の6段階の基準で判定し、3人の判定値のうち多数の値をサンプルの臭気強度とした。
臭気強度0: 無臭
臭気強度1: やっと感知できるにおい
臭気強度2: 何のにおいか判る弱いにおい
臭気強度3: 楽に感知できるにおい
臭気強度4: 強いにおい
臭気強度5: 強烈なにおい
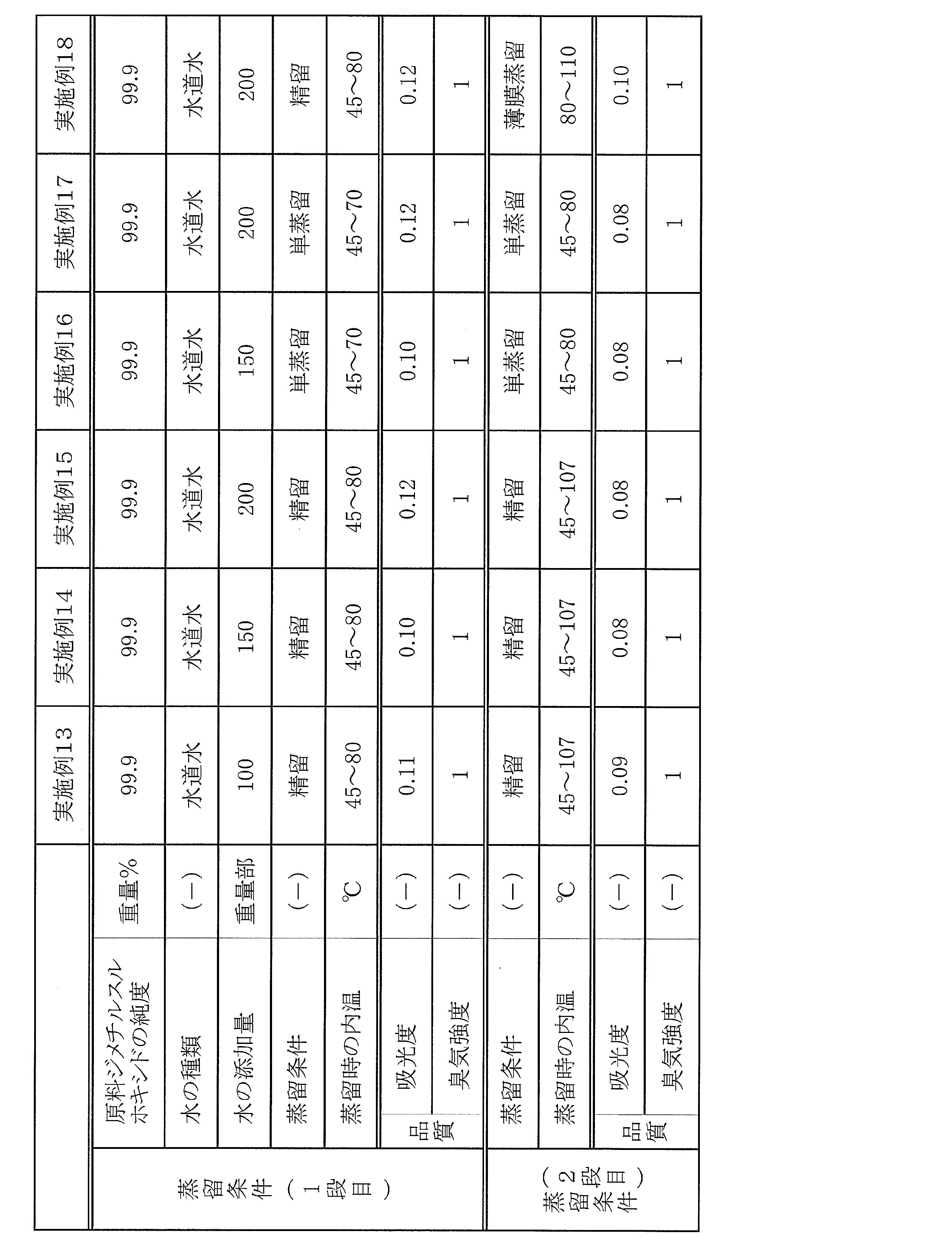
200mL丸底フラスコに純度99.9重量%の一般工業品質のジメチルスルホキシド(275nmにおける吸光度が0.22、臭気強度が3)50gとJIS-K0557のA4相当のイオン交換水50g(100重量部/ジメチルスルホキシド100重量部)を仕込み、混合した。この丸底フラスコの口にラシヒリング(長さ5mm、外径5mm、内径3mm)を10cm充填した直径3cm、高さ15cmの精留管を設置し、6.7kPaで減圧蒸留した。丸底フラスコの内温45℃から加熱を開始し、温度が107℃に到達した時点で受器を交換し、40gの精製ジメチルスルホキシドを得た。この時の蒸留収率は80%であった。得られた精製ジメチルスルホキシドの275nmにおける吸光度は0.12であり、臭気強度は1であった。これらの結果を表1にまとめて示す。なお、表中の水の添加量の単位は、ジメチルスルホキシドを100重量部にしたときの重量部である。
水の混合量を、表1に記載するように変更したことを除き、実施例1と同様の精製操作を行った。実施例2,3,4および5の蒸留収率は、81%,80%,70%および79%であった。得られた精製ジメチルスルホキシドの品質を、表1に記載する。
実施例1において、添加する水の種類を、JIS-K0557のA4相当の蒸留水または超純水に変更したことを除き、実施例1と同様の精製操作を行った。実施例6および7の蒸留収率は、79%および81%であった。得られた精製ジメチルスルホキシドの品質を、表1に記載する。

比較例1,2および4は、表2に記載するように水の添加量を変更したことを除き、実施例1と同様の操作を行った。また比較例3は、水の代わりにメタノール(ナカライテスク社製特級)を添加したことを除き、実施例1と同様の操作を行った。比較例1,2,3および4の蒸留収率は、85%,95%,95%および79%であった。得られたジメチルスルホキシドの品質は表2の通りである。

200mL丸底フラスコに純度99.9重量%の一般工業品質のジメチルスルホキシド(275nmにおける吸光度が0.22、臭気強度が3)50gと水道水50g(100重量部/ジメチルスルホキシド100重量部)を仕込み、混合した。使用した水道水の電気伝導率は13mS/m(25℃)であった。この丸底フラスコの口にラシヒリング(長さ5mm、外径5mm、内径3mm)を10cm充填した直径3cm、高さ15cmの精留管を設置し、6.7kPaで減圧蒸留した。丸底フラスコの内温45℃から80℃へ加熱し、留出液の含水率が0.1%以下に達した時点で蒸留を停止した。水を除去して残ったジメチルスルホキシドの275nmにおける吸光度は0.11であり、臭気強度は1であった。これらの結果を表3にまとめて示す。なお、表中の水の添加量の単位は、ジメチルスルホキシドを100重量部にしたときの重量部である。
水の混合量を、表3に記載するように変更したことを除き、実施例8と同様の蒸留操作を行った。得られたジメチルスルホキシドの品質を、表3に記載する。
実施例8において、精留管を使用せず、丸底フラスコの内温を45℃から70℃へ加熱して単蒸留したことおよび水の混合量を表3に記載するように変更したことを除き、実施例8と同様の蒸留操作を行った。得られたジメチルスルホキシドの品質を、表3に記載する。

200mL丸底フラスコに純度99.9重量%の一般工業品質のジメチルスルホキシド(275nmにおける吸光度が0.22、臭気強度が3)50gと水道水50g(100重量部/ジメチルスルホキシド100重量部)を仕込んで混合溶液にした。この混合溶液を1段目の蒸留操作として、上記の丸底フラスコの口にラシヒリング(長さ5mm、外径5mm、内径3mm)を10cm充填した直径3cm、高さ15cmの精留管を設置し、6.7kPaで減圧蒸留した。丸底フラスコの内温45℃から80℃へ加熱し、留出液の含水率が0.1%以下に達した時点で蒸留を停止した。水を除去して得られた粗精製しジメチルスルホキシドの275nmにおける吸光度は0.11であり、臭気強度は1であった。
実施例13において、水の混合量を、表4に記載するように変更したことを除き、実施例13と同様の1段目および2段目の蒸留操作を行った。1段目の蒸留により得られた粗精製しジメチルスルホキシドの275nmにおける吸光度は0.10、臭気強度は1であり、GC分析による化学純度は、99.9957%であった。また、2段目の蒸留により得られた精製ジメチルスルホキシドの275nmにおける吸光度は0.08、臭気強度は1であり、GC分析による化学純度は、99.9989%であった。これらの結果を表4に記載する。
実施例13において、水の混合量を、表4に記載するように変更したことを除き、実施例13と同様の1段目および2段目の蒸留操作を行った。1段目の蒸留により得られた粗精製しジメチルスルホキシドの275nmにおける吸光度は0.12、臭気強度は1であり、GC分析による化学純度は、99.9957%であった。また、2段目の蒸留により得られた精製ジメチルスルホキシドの275nmにおける吸光度は0.08、臭気強度は1であり、GC分析による化学純度は、99.9980%であった。これらの結果を表4に記載する。
実施例13において、水の混合量を表4に記載するように変更したこと、1段目の蒸留操作において精留管を使用せず、丸底フラスコの内温を45℃から70℃へ加熱して単蒸留したこと、および2段目の蒸留操作において精留管を使用せず、丸底フラスコの内温を45℃から80℃へ加熱して単蒸留したことを除き、実施例13と同様の精製操作を行った。得られた精製ジメチルスルホキシドの品質を、表4に記載する。
実施例16において、水の混合量を、表4に記載するように変更したことを除き、実施例16と同様の1段目および2段目の蒸留操作を行った。得られた精製ジメチルスルホキシドの品質を、表4に記載する。
実施例13において、水の混合量を表4に記載するように変更したこと、および2段目の蒸留操作において精留管を使用せず、丸底フラスコの内温を80℃から110℃へ加熱して薄膜蒸留したことを除き、実施例13と同様の精製操作を行った。得られた精製ジメチルスルホキシドの品質を、表4に記載する。
Claims (7)
- 原料ジメチルスルホキシド100重量部に対し、水35重量部以上を混合した溶液を蒸留することを特徴とするジメチルスルホキシドの精製法。
- 精製されたジメチルスルホキシドの275nmにおける吸光度を、0.01~0.20にすることを特徴とする請求項1に記載のジメチルスルホキシドの精製法。
- 前記水がイオン交換水、蒸留水、超純水または水道水であることを特徴とする請求項1または2に記載のジメチルスルホキシドの精製法。
- 前記水の25℃の電気伝導率が50mS/m以下であることを特徴とする請求項1~3のいずれかに記載のジメチルスルホキシドの精製法。
- 前記混合した溶液の蒸留を1段で行い、水を留去してジメチルスルホキシドを得ることを特徴とする請求項1~4のいずれかに記載のジメチルスルホキシドの精製法。
- 前記混合した溶液に1段目の蒸留を行い、水を留去させ、粗精製したジメチルスルホキシドを得、さらに粗精製したジメチルスルホキシドに2段目の蒸留を行い、精製したジメチルスルホキシドを得ることを特徴とする請求項1~4のいずれかに記載のジメチルスルホキシドの精製法。
- 請求項1~6のいずれかに記載の精製法により得られたジメチルスルホキシドであって、ガスクロマトグラフ(GC)分析での純度が99.996%以上であることを特徴とする高純度ジメチルスルホキシド。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/786,235 US20160083340A1 (en) | 2013-04-30 | 2014-04-22 | Method of purifying dimethyl sulfoxide |
| EP14791804.9A EP2993166A4 (en) | 2013-04-30 | 2014-04-22 | PROCESS FOR PURIFYING DIMETHYL SULFOXIDE |
| JP2014530040A JPWO2014178309A1 (ja) | 2013-04-30 | 2014-04-22 | ジメチルスルホキシドの精製法 |
| CA2908035A CA2908035A1 (en) | 2013-04-30 | 2014-04-22 | Method for purifying dimethyl sulfoxide |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013095375 | 2013-04-30 | ||
| JP2013-095375 | 2013-04-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014178309A1 true WO2014178309A1 (ja) | 2014-11-06 |
Family
ID=51843442
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/061306 WO2014178309A1 (ja) | 2013-04-30 | 2014-04-22 | ジメチルスルホキシドの精製法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20160083340A1 (ja) |
| EP (1) | EP2993166A4 (ja) |
| JP (1) | JPWO2014178309A1 (ja) |
| CA (1) | CA2908035A1 (ja) |
| WO (1) | WO2014178309A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015145359A (ja) * | 2014-01-06 | 2015-08-13 | 東レ・ファインケミカル株式会社 | ジメチルスルホキシドの精製方法 |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109406700A (zh) * | 2018-08-30 | 2019-03-01 | 国网吉林省电力有限公司电力科学研究院 | 作业场所空气中二甲基亚砜浓度的检测方法 |
| CN112321468A (zh) * | 2020-10-16 | 2021-02-05 | 浙江巨化技术中心有限公司 | 一种从醚唑类药物合成过程中的废水中分离二甲基亚砜的方法 |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3358036A (en) | 1964-03-12 | 1967-12-12 | Merck & Co Inc | Purification of dimethyl sulfoxide |
| GB1222453A (en) * | 1968-06-28 | 1971-02-17 | Glanzstoff Ag | Process for obtaining pure dimethyl sulphoxide |
| JPS63270659A (ja) * | 1987-04-30 | 1988-11-08 | Daikin Ind Ltd | ジメチルスルホキシドとヨウ素とを分離する方法 |
| JPH0312128B2 (ja) | 1984-02-27 | 1991-02-19 | Chugai Ro Kogyo Kk | |
| JPH0329737B2 (ja) * | 1981-03-20 | 1991-04-25 | ||
| JPH0912534A (ja) * | 1995-06-23 | 1997-01-14 | Toray Fine Chem Co Ltd | Dmsoの回収方法 |
| JP2006096763A (ja) * | 2005-11-18 | 2006-04-13 | Toray Fine Chemicals Co Ltd | Dmsoの回収方法 |
| JP2006151821A (ja) * | 2004-11-25 | 2006-06-15 | Toray Ind Inc | 溶媒の回収方法 |
| JP2009096792A (ja) * | 2007-09-28 | 2009-05-07 | Toray Ind Inc | ジメチルスルホキシドの精製方法 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6042640A (en) * | 1998-06-29 | 2000-03-28 | Xerox Corporation | Deodorization of sulfur-containing solvents by oxidation |
| US20090005601A1 (en) * | 2007-06-29 | 2009-01-01 | Gaylord Chemical Company Llc | Process for preparing low malodor dimethyl sulfoxide |
-
2014
- 2014-04-22 CA CA2908035A patent/CA2908035A1/en not_active Abandoned
- 2014-04-22 US US14/786,235 patent/US20160083340A1/en not_active Abandoned
- 2014-04-22 EP EP14791804.9A patent/EP2993166A4/en not_active Withdrawn
- 2014-04-22 JP JP2014530040A patent/JPWO2014178309A1/ja active Pending
- 2014-04-22 WO PCT/JP2014/061306 patent/WO2014178309A1/ja active Application Filing
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3358036A (en) | 1964-03-12 | 1967-12-12 | Merck & Co Inc | Purification of dimethyl sulfoxide |
| GB1222453A (en) * | 1968-06-28 | 1971-02-17 | Glanzstoff Ag | Process for obtaining pure dimethyl sulphoxide |
| JPH0329737B2 (ja) * | 1981-03-20 | 1991-04-25 | ||
| JPH0312128B2 (ja) | 1984-02-27 | 1991-02-19 | Chugai Ro Kogyo Kk | |
| JPS63270659A (ja) * | 1987-04-30 | 1988-11-08 | Daikin Ind Ltd | ジメチルスルホキシドとヨウ素とを分離する方法 |
| JPH0912534A (ja) * | 1995-06-23 | 1997-01-14 | Toray Fine Chem Co Ltd | Dmsoの回収方法 |
| JP2006151821A (ja) * | 2004-11-25 | 2006-06-15 | Toray Ind Inc | 溶媒の回収方法 |
| JP2006096763A (ja) * | 2005-11-18 | 2006-04-13 | Toray Fine Chemicals Co Ltd | Dmsoの回収方法 |
| JP2009096792A (ja) * | 2007-09-28 | 2009-05-07 | Toray Ind Inc | ジメチルスルホキシドの精製方法 |
Non-Patent Citations (2)
| Title |
|---|
| "ALDRICH Chemistry", 2009, JAPAN, pages 1090 - 1092, XP008181439, Retrieved from the Internet <URL:sigma-aldrich.com> * |
| See also references of EP2993166A4 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015145359A (ja) * | 2014-01-06 | 2015-08-13 | 東レ・ファインケミカル株式会社 | ジメチルスルホキシドの精製方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2993166A1 (en) | 2016-03-09 |
| EP2993166A4 (en) | 2016-12-07 |
| US20160083340A1 (en) | 2016-03-24 |
| CA2908035A1 (en) | 2014-11-06 |
| JPWO2014178309A1 (ja) | 2017-02-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2662966T3 (es) | Procedimiento para procesar soluciones de cloruro de magnesio | |
| NZ587962A (en) | Purification of acetic acid from wood acetylation process | |
| MX2009005012A (es) | Una forma pura de rapamicina y procedimiento para la recuperacion y purificacion de la misma. | |
| WO2014178309A1 (ja) | ジメチルスルホキシドの精製法 | |
| CN103467475B (zh) | 1,8-桉叶素的纯化方法 | |
| MX2012012912A (es) | Procedimiento para recuperar etanol con descargaslaterales para regular las concentracioes de alcoholes de c3+. | |
| JP6266453B2 (ja) | ジメチルスルホキシドの精製方法 | |
| MX2007010403A (es) | Proceso de purificacion. | |
| EP2370388B1 (en) | Method for purifying acetone | |
| WO2018003974A1 (ja) | 補酵素q10の製造方法 | |
| JP2008308500A (ja) | 高純度酢酸ブチルの製造方法 | |
| JP6266420B2 (ja) | ジメチルスルホキシドの精製方法 | |
| PH12019501803A1 (en) | Acetic acid production method | |
| RU2010134125A (ru) | Способ получения фуллерена c60 | |
| Landis | The use of mixed-mode ion-exchange solid phase extraction to characterize pharmaceutical drug degradation | |
| CA2957812C (en) | Process for the recovery of carboxylic acid and wood treatment process | |
| JP2006514087A (ja) | 蒸留による1,3−プロパンジオールの精製 | |
| Polevaya et al. | Optimization of synthesis conditions of 2, 3-dimethylbutadiene | |
| JP6562379B2 (ja) | 脂肪酸クロライドの製造方法 | |
| RU2621054C2 (ru) | Способ разделения монохлоруксусной кислоты и дихлоруксусной кислоты экстракционной дистилляцией с использованием органического растворителя | |
| CN103360219A (zh) | 一种高纯度丙泊酚的合成方法 | |
| CN110922308B (zh) | 一种含有亚甲氧桥基的液晶分子的工业化制备色谱分离纯化方法 | |
| WO2014175187A1 (ja) | ジメチルスルホキシドの精製方法 | |
| JP6118589B2 (ja) | 3−メチル−1,3−ブタンジオールの製造方法 | |
| EP2948422A1 (en) | Process for the production of 2-alkyl-3-butyn-2-ols |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2014530040 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14791804 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2908035 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014791804 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14786235 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |