WO2014053208A1 - 7-azaindol-2,7-naphthyridin-derivat zur behandlung von tumoren - Google Patents
7-azaindol-2,7-naphthyridin-derivat zur behandlung von tumoren Download PDFInfo
- Publication number
- WO2014053208A1 WO2014053208A1 PCT/EP2013/002696 EP2013002696W WO2014053208A1 WO 2014053208 A1 WO2014053208 A1 WO 2014053208A1 EP 2013002696 W EP2013002696 W EP 2013002696W WO 2014053208 A1 WO2014053208 A1 WO 2014053208A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- methyl
- pyrrolo
- naphthyridin
- treatment
- Prior art date
Links
- GJTKPIYJTYMWMY-UHFFFAOYSA-N Cc([nH]c1c2cccn1)c2-c(c1c2cncc1)cnc2N Chemical compound Cc([nH]c1c2cccn1)c2-c(c1c2cncc1)cnc2N GJTKPIYJTYMWMY-UHFFFAOYSA-N 0.000 description 1
- QUDRYDPGDJQBAY-UHFFFAOYSA-N Cc([n](c1ncccc11)S(c2ccccc2)(=O)=O)c1-c(c1c2cncc1)cnc2N Chemical compound Cc([n](c1ncccc11)S(c2ccccc2)(=O)=O)c1-c(c1c2cncc1)cnc2N QUDRYDPGDJQBAY-UHFFFAOYSA-N 0.000 description 1
- 0 Cc([n]1*)c(*)c2c1nccc2 Chemical compound Cc([n]1*)c(*)c2c1nccc2 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4375—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a six-membered ring having nitrogen as a ring heteroatom, e.g. quinolizines, naphthyridines, berberine, vincamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Definitions
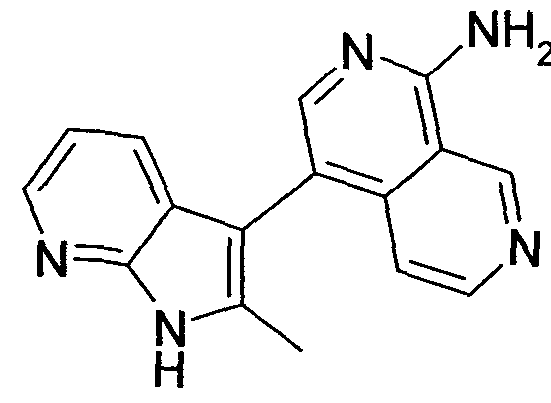
- the invention relates to the compound 4- (2-methyl-1H-pyrrolo [2,3-b] pyridin-3-yl) - [2,7] naphthyridin-1-ylamine
- the invention had the object of finding new compounds with valuable properties, in particular those that can be used for the production of medicaments.
- the compound according to the invention can therefore be used for the control and / or treatment of tumors, tumor growth and / or tumor metastases.
- the antiproliferative effect can be tested in a proliferation assay / vitality assay.
- carcinomas such as carcinomas (eg, the lungs, pancreas, thyroid, urinary bladder, or bladder) Colon), myeloid diseases (eg myeloid leukemia) or
- Adenomas eg villous colon adenoma
- the tumors also include monocytic leukemia, brain, urogenital, lymphatic, gastric, laryngeal and lung carcinomas, including lung adenocarcinoma and small cell lung carcinoma, pancreatic and / or breast carcinoma.
- the compound is also useful in the treatment of immunodeficiency induced by HIV-1 (Human Immunodeficiency Virus type 1).
- Cancerous hyperproliferative disorders include brain, lung, squamous, bladder, stomach, pancreatic, liver, kidney, colorectal, breast, head, neck, esophageal, gynecological, thyroid, lymphoma, chronic leukemia and acute leukemia.
- cancerous cell growth is a disease that is an object of the present invention.
- the present invention is therefore the compound of the invention as a drug and / or drug in the treatment and / or prophylaxis of the diseases mentioned and the use of the compound of the invention for the preparation of a pharmaceutical for the treatment and / or prophylaxis of the diseases mentioned as well as a method for Treatment of said
- the compound of the invention is administered to a patient with a hyperproliferative disorder, e.g. To inhibit tumor growth, to reduce inflammation associated with lymphoproliferative disease, to inhibit graft rejection or neurological damage due to
- the present compound is useful for Tissue repair, etc.
- the present compound is useful for Tissue repair, etc.
- Prevention of proliferation / vitality is achieved by administration of the compound of the invention prior to the development of the evident disease, e.g. To prevent tumor growth.
- the compound is used to treat persistent diseases by stabilizing or ameliorating the clinical symptoms of the patient.
- the host or patient may be of any mammalian species, e.g. A primate species, especially humans; Rodents, including mice, rats and hamsters; Rabbits; Horses, cattle, dogs, cats, etc. Animal models are of interest for experimental studies, providing a model for the treatment of human disease.
- the susceptibility of a particular cell to treatment with the compound of the invention can be determined by testing in vitro.
- Compound is incubated at various concentrations for a time sufficient to allow the active agents to induce cell death or to inhibit cell proliferation, cell vitality or migration, usually between about one hour and one week.
- cultured cells from a biopsy sample can be used. The amount of cells remaining after treatment are then determined.
- the dose will vary depending on the specific compound used, the specific disease, the patient status, etc. Typically, a therapeutic dose will be sufficient to substantially reduce the undesirable cell population in the target tissue while increasing the viability of the patient
- Treatment is generally continued until there is a significant reduction, e.g. B. at least about 50% reduction in cell load and can be continued until essentially no more unwanted cells are detected in the body.
- a significant reduction e.g. B. at least about 50% reduction in cell load and can be continued until essentially no more unwanted cells are detected in the body.
- diseases associated with deregulation of cell proliferation and cell death apoptosis.
- the ailments of interest include, but are not limited to, the following conditions.
- the compound of the invention is useful in the treatment of a series
- Occlusive transplant vascular diseases of interest include atherosclerosis, coronary vascular disease after transplantation, vein graft stenosis, perinastomotic prosthetic restenosis, restenosis after angioplasty or stent placement, and the like.
- the compound according to the invention also acts as a regulator, modulator or inhibitor of protein kinases, in particular of the serine / threonine kinase type, which include, among others, the phosphoinositide-dependent kinase 1 (PDK 1).
- PDK1 phosphoinositide-dependent kinase 1
- PDK1 phosphorylates and activates a subset of the AGC protein kinase family, including PKB, SGK, S6K and PKC isoforms. These kinases are involved in the PI3K signaling pathway and control basic cellular functions such as survival, growth, and differentiation. PDK1 is thus an important regulator of diverse metabolic, proliferative and life-sustaining effects.
- the compound of the invention also exhibits TGF ⁇ receptor I kinase inhibiting properties.
- TGF-ß1 Inhibitors of the intracellular TGF- ⁇ signaling pathway are suitable treatments for fibroproliferative disorders.
- Fibroproliferative disorders specifically include renal disorders associated with unregulated TGF- ⁇ activity, and severe fibrosis, including glomerulonephritis (GN), such as mesangial proliferative GN, immune GN, and crescent GN.
- Other renal conditions include diabetic nephropathy, renal interstitial fibrosis, renal fibrosis in transplant patients receiving cyclosporin, and nephropathy associated with HIV.
- Collagen vascular disorders include progressive systemic sclerosis, polymyositis, scleroderma, dermatomyositis, eosinophilic fascitis, morphea, or those associated with the incidence of Raynaud's syndrome.
- Pulmonary fibrosis caused by excessive TGF- ⁇ activity includes adult respiratory distress syndrome, idiopathic pulmonary fibrosis, and interstitial pulmonary fibrosis, often with
- autoimmune disorders such as systemic lupus erythematosus and scleroderma, chemical contact or allergies.
- Another autoimmune disorder associated with fibroproliferative properties is rheumatoid arthritis.
- Ocular disorders associated with a fibroproliferative condition include proliferative vitreoretinopathy associated with a
- Retinal repair surgery cataract extraction with intraocular lens implantation, and post-glaucoma drainage surgery, and is associated with TGF- ⁇ 1 overproduction.
- TGF- ⁇ 1 is a ligand of the TGF- ⁇ receptor family consisting of heterodimeric cell membrane-containing proteins having an extracellular receptor portion and an intracellular kinase domain.
- Members are Type I and Type II Rezptoren; see also Hinck FEBS Lett. 2012 http://dx.doi.Org/10.1016/j.febslet.2012.05.028
- Signal transduction of the ligands TGF- ⁇ 1, - ⁇ 2 and - ⁇ 3 via their respective receptors is known to be involved in cell cycle arrest in epithelial and hematopoietic cells, control of mesenchymal cell proliferation and differentiation, in wound healing, production
- TGF-B1 When TGF-B1 binds to a type II receptor, the corresponding type I receptor associates and becomes phosphorylated. This complex phosphorylates a receptor-regulated 4s Smad protein (R-Smad), which subsequently associates with Smad4, migrates to the nucleus and, by activating transcription, leads to a change in cell behavior.
- R-Smad receptor-regulated 4s Smad protein
- TGF- ⁇ type I receptor also called ALK5 (activin receptor-like kinase 5) or T ⁇ R-1
- ALK5 activin receptor-like kinase 5
- T ⁇ R-1 TGF- ⁇ type I receptor
- the compound according to the invention represents a selection from WO
- the compound according to the invention has a markedly higher activity than the structurally closest compounds from WO 2012/04007.
- WO 2005/095400 A1 describes other azaindole derivatives as protein kinase inhibitors.
- Pyridinonyl derivatives are known as PDK1 inhibitors for combating cancer from WO 2008/005457.
- heterocyclic compounds described as PDK1 inhibitors for the fight against cancer are described as PDK1 inhibitors for the fight against cancer.
- A1 pyrrolopyridine derivatives are described as PDK1 inhibitors for combating cancer.
- ⁇ and TBK1 are serine / threonine kinases that have high homologies with each other and with other IkB kinases. Both kinases play an integral role in the innate immune system.
- Double-stranded RNA viruses are recognized by the Toll-like receptors 3 and 4, as well as the RNA helicases RIG-I and MDA-5, and lead to activation of the TR1F-TBK1 / IKKE-IRF3 signaling cascade, resulting in a type I interferon Answer leads.
- Protein kinase-mediated diseases are characterized by abnormal activity or hyperactivity of such protein kinases.
- Abnormal activity involves either: (1) expression in cells that usually do not express these protein kinases; (2) increased kinase expression leading to unwanted cell proliferation such as cancer; (3) increased kinase activity resulting in undesired cell proliferation, such as cancer, and / or hyperactivity of the corresponding protein kinases.
- Hyperactivity refers to either amplification of the gene encoding a particular protein kinase or the generation of an activity level that can be correlated with a cell proliferative disorder (ie, as the kinase level increases, the severity of one or more symptoms of the cell proliferative disorder increases). The bioavailability of a protein kinase may also be due to the
- the major cancers that can be treated using the compound of the invention include colorectal cancer,
- small cell lung cancer non-small cell lung cancer, multiple myeloma as well as renal cell carcinoma and endometrial carcinoma, especially cancers in which PTEN is mutated, and the like.
- a. Breast cancer, prostate cancer and glioblastoma.
- the compound of the present invention can be used to achieve additive or synergistic effects in certain existing cancer chemotherapies and radiation and / or to restore the efficacy of certain existing cancer chemotherapies and radiation.
- the compound according to the invention is also understood as meaning the hydrates and solvates of this compound, furthermore pharmaceutically usable derivatives.
- the invention also relates to the salts, and the hydrates and solvates of this compound.
- Solvates of the compound are understood to mean additions of inert solvent molecules to the compounds which form due to their mutual attraction. Solvates are e.g. Mono or dihydrate or alcoholates.
- the invention also encompasses the solvates of the salts of the compound according to the invention.
- compositions are understood, for example, as the salts of the compound of the invention as well as so-called prodrug compounds.
- Prodrug derivative is understood to mean the z. B. alkyl or acyl groups, sugars or oligopeptides modified inventive compound which is rapidly cleaved in the organism to the active compound of the invention.
- the term "effective amount” means the amount of a drug or pharmaceutical agent that elicits a biological or medical response in a tissue, system, animal, or human, such as is sought or sought by a researcher or physician.
- terapéuticaally effective amount means an amount that, as compared to a corresponding subject who has not received that amount, results in:
- terapéuticaally effective amount also includes the amounts effective to increase normal physiological function.
- the invention provides the compound 4- (2-methyl-1H-pyrrolo [2,3-b] pyridin-3-yl) - [2,7] naphthyridin-1-ylamine and its salts and a process for the preparation of this compound and their pharmaceutically acceptable salts and tautomers, characterized in that
- R is Br or I and R 2 is an aza-indole protecting group, reacted with 4,4,5,5-tetramethyl-1,3,2-dioxaborolane, and the intermediately formed boronic pinacol ester in a Suzuki reaction with a compound of formula III
- R 2 is an aza-indole protecting group, and then the protecting group R 2 is split off from the compound of formula IV, and / or 4- (2-methyl-1H-pyrrolo [2,3-b] pyridine-3 -yl) - [2,7] naphthyridin-1-ylamine converts to one of its salts.
- radicals R 1 and R 2 have the meanings given for the formulas II, II and IV, unless expressly stated otherwise.
- R 1 is Br or I, preferably I.
- R 2 represents an azaindole protective group, preferably tert-butyloxycarbonyl or benzenesulphonyl, more preferably benzenesulphonyl.
- the benzenesulfonyl protecting group may also be replaced by other sulfonyl or oxycarbonyl protecting groups known to those skilled in the art.
- the cleavage of alkyl or Arylsulfonyl phenomenon carried out with alkali metal hydroxide and primary alcohols under standard conditions.
- the compound 4- (2-methyl-1H-pyrrolo [2,3-b] pyridin-3-yl) - [2,7] naphthyridin-1-ylamine can preferably be obtained by dissolving in a
- X is preferably Cl, Br or I.
- the reaction time is between a few minutes and 14 days depending on the conditions used, the reaction temperature between about -30 ° and 140 °, normally between 0 ° and 110 °, in particular between about 70 ° and about 100 °.
- Suitable inert solvents are e.g. Hydrocarbons, such as hexane, petroleum ether, benzene, toluene or xylene; chlorinated hydrocarbons such as trichlorethylene, 1,2-dichloroethane, carbon tetrachloride, chloroform or dichloromethane; Alcohols such as methanol, ethanol, isopropanol, n-propanol, n-butanol or tert-butanol; Ethers, such as diethyl ether, diisopropyl ether,
- Hydrocarbons such as hexane, petroleum ether, benzene, toluene or xylene
- chlorinated hydrocarbons such as trichlorethylene, 1,2-dichloroethane, carbon tetrachloride, chloroform or dichloromethane
- Alcohols such as methanol, ethanol, isopropanol, n-propan
- Tetrahydrofuran (THF) or dioxane Tetrahydrofuran (THF) or dioxane; Glycol ethers, such as ethylene glycol monomethyl or monoethyl ether (methyl glycol or ethyl glycol), ethylene glycol dimethyl ether (diglyme); Ketones such as acetone or butanone; Amides such as acetamide, dimethylacetamide or dimethylformamide (DMF); Nitrites such as acetonitrile; Sulfoxides such as dimethylsulfoxide (DMSO); Carbon disulphide; Carboxylic acids such as formic acid or acetic acid; Nitro compounds such as nitromethane or nitrobenzene; Esters such as ethyl acetate or mixtures of said solvents.
- Glycol ethers such as ethylene glycol monomethyl or monoethyl ether (methyl glycol or ethyl glycol), ethylene glycol dimethyl ether (diglyme); Ke
- dimethoxyethane diglyme, methanol and / or dioxane.
- Pharmaceutical salts and other forms are particularly preferred.
- the said compound of the invention can be used in its final non-salt form.
- the present invention also encompasses the use of this compound in the form of its pharmaceutically acceptable salts derived from various organic and
- inorganic acids and bases can be derived by art-known procedures.
- Pharmaceutically acceptable salt forms of the compound according to the invention are mostly prepared conventionally.
- the acid addition salts can be formed by adding the
- Ethansulfonat, toluenesulfonate and benzenesulfonate, and other organic acids and their corresponding salts such as acetate, trifluoroacetate, tartrate, maleate, succinate, citrate, benzoate, salicylate, ascorbate and the like.
- pharmaceutically acceptable acid addition salts of the compound of the invention include the following: acetate, adipate, alginate, arginate, aspartate, benzoate, benzenesulfonate (besylate), bisulfate, bisulfite, bromide, butyrate, camphorate, camphorsulfonate, caprylate,
- the compound of the present invention containing basic nitrogen-containing groups can be reacted with agents such as (C C) alkyl halides, e.g.
- aryl- (C 1 -C 4 ) alkyl halides eg benzyl chloride and phenethyl bromide, quaternize.
- Preferred pharmaceutical salts include acetate, trifluoroacetate, besylate, citrate, fumarate, gluconate, hemisuccinate, hippurate, hydrochloride, hydrobromide, isethionate, mandelate, meglumine, nitrate, oleate, phosphonate, pivalate, sodium phosphate, stearate, Sulfate, sulfosalicylate, tartrate, thiomalate, tosylate and tromethamine, but no
- the acid addition salts of the compound of the invention are prepared by contacting the free base form with a sufficient amount of the desired acid to form the salt in a conventional manner.
- the free base can be regenerated by contacting the salt form with a base and isolating the free base in a conventional manner.
- the free base forms differ, in a sense, from their corresponding salt forms with respect to certain physical ones Properties such as solubility in polar solvents; however, in the context of the invention, the salts otherwise correspond to their respective free base forms.
- a compound according to the invention contains more than one group which can form such pharmaceutically acceptable salts, the invention also encompasses multiple salts.
- Typical multiple salt forms include, for example, bitartrate, diacetate, difumarate, dimeglumine, diphosphate, disodium and trihydrochloride, but this is not intended to be limiting.
- pharmaceutically acceptable salt as used herein means an active ingredient containing the compound of the invention in the form of one of its salts, particularly when that salt form is the active ingredient compared to the free form of the active ingredient or any other salt form of the active ingredient previously used, imparts improved pharmacokinetic properties.
- acceptable salt form of the active ingredient may provide this drug with a desired pharmacokinetic property that it has not previously possessed, and may even positively affect the pharmacodynamics of that drug in terms of its therapeutic efficacy in the body.
- the invention furthermore relates to medicaments containing 4- (2-methyl-1H-pyrrolo [2,3-b] pyridin-3-yl) - [2,7] naphthyridin-1-ylamine and / or its
- compositions may be presented in the form of dosage units containing a predetermined amount of active ingredient per unit dose.
- a moiety may contain, for example, 0.5 mg to 1 g, preferably 1 mg to 700 mg, more preferably 5 mg to 100 mg of the compound of the invention, depending on the condition of the disease being treated, the route of administration and the age, weight and condition of the patient, or pharmaceutical formulations may be presented in the form of dosage units containing a predetermined amount of active ingredient per unit dose.
- Preferred dosage unit formulations are those containing a daily or partial dose as indicated above or a corresponding fraction thereof of an active ingredient. Furthermore, such pharmaceutical formulations with one of the im
- compositions may be administered by any suitable route, for example, oral (including buccal or sublingual), rectal, nasal, topical (including buccal, sublingual or transdermal), vaginal or parenteral (including subcutaneous, intramuscular, intravenous or intradermal). Ways, adapt.
- Such formulations can be prepared by any method known in the pharmaceutical art, for example, by bringing the active ingredient together with the carrier (s) or excipient (s).
- compositions adapted for oral administration may be administered as separate units, e.g. Capsules or tablets; Powder or granules; Solutions or suspensions in aqueous or non-aqueous liquids; edible foams or foam foods; or oil-in-water liquid emulsions or water-in-oil liquid emulsions.
- the active ingredient component in the case of oral administration in the form of a tablet or capsule, can be mixed with an oral, non-toxic and pharmaceutically acceptable inert carrier such as ethanol, glycerine, water and the like. combine. Powders are prepared by comminuting the compound to a suitable fine size and mixing it with a similarly comminuted pharmaceutical carrier such as an edible carbohydrate such as starch or mannitol. A flavor, preservative, dispersant and dye may also be present.
- an oral, non-toxic and pharmaceutically acceptable inert carrier such as ethanol, glycerine, water and the like.
- Powders are prepared by comminuting the compound to a suitable fine size and mixing it with a similarly comminuted pharmaceutical carrier such as an edible carbohydrate such as starch or mannitol.
- a flavor, preservative, dispersant and dye may also be present.
- Capsules are made by preparing a powder mix as described above and filling shaped gelatin casings therewith. Sliding and
- Lubricants such as e.g. fumed silica, talc, magnesium stearate, calcium stearate or polyethylene glycol in solid form can be added to the powder mixture before the filling process.
- a disintegrants or solubilizers e.g. Agar-agar, calcium carbonate or sodium carbonate may also be added to improve the availability of the drug after ingestion of the capsule.
- Suitable binding, lubricating and disintegrants as well as dyes can also be incorporated into the mixture.
- suitable binders include starch,
- Gelatin natural sugars, e.g. Glucose or beta-lactose, corn sweeteners, natural and synthetic gums, e.g. Acacia, tragacanth or sodium alginate, carboxymethylcellulose, polyethylene glycol, waxes, etc.
- the lubricants used in these dosage forms include
- the disintegrating agents include, but are not limited to, starch, methyl cellulose, agar, bentonite, xanthan gum and the like.
- the tablets are formulated by, for example, preparing a powder mixture, granulating or dry pressing, adding a lubricant and a disintegrating agent and pressing the whole into tablets.
- a powder mixture is prepared by dissolving the appropriately comminuted compound with a diluent or a base as described above, and optionally with a binder, e.g. Carboxymethylcellulose, an alginate, gelatin or polyvinylpyrrolidone, a dissolution reducer, e.g. Paraffin, a resorption accelerator, such as a quaternary salt and / or an absorbent, e.g.
- a binder e.g. Carboxymethylcellulose, an alginate, gelatin or polyvinylpyrrolidone
- a dissolution reducer e.g. Paraffin
- a resorption accelerator such as a quaternary salt and / or an absorbent, e.g.
- Bentonite, kaolin or dicalcium phosphate is mixed.
- the powder mixture can be granulated by wetting it with a binder such as syrup, starch paste, Acadia slime, or solutions of cellulosic or polymeric materials and pressing it through a sieve.
- a binder such as syrup, starch paste, Acadia slime, or solutions of cellulosic or polymeric materials and pressing it through a sieve.
- the powder mixture can be run through a tabletting machine to produce non-uniformly shaped lumps which are in granules
- the granules may be greased by the addition of stearic acid, a stearate salt, talc or mineral oil to prevent sticking to the tablet molds. The greased mixture is then compressed into tablets.
- the compound of the invention may also be combined with a free-flowing inert carrier and then compressed directly into tablets without carrying out the granulation or dry-pressing steps.
- Protective layer consisting of a shellac sealant, a layer of sugar or polymeric material and a glossy layer of wax may be present. Dyes can be added to these coatings in order to differentiate between different dosage units.
- Oral fluids e.g. Solution, syrups and elixirs may be prepared in unit dosage form such that a given quantity contains a predetermined amount of the compound.
- Syrups can be prepared by dissolving the compound in an appropriate taste aqueous solution while preparing elixirs using a non-toxic alcoholic vehicle.
- Suspensions can be formulated by dispersing the compound in a non-toxic vehicle.
- Solubilizers and emulsifiers e.g. ethoxylated isostearyl alcohols and polyoxyethylene sorbitol ethers, preservatives, flavoring additives such as e.g. Peppermint oil or natural sweeteners or saccharin or other artificial sweeteners, etc. can also be added.
- the unit dosage formulations for oral administration may optionally be encapsulated in microcapsules.
- the formulation can also be prepared so that the release is prolonged or retarded, such as by coating or embedding particulate material in polymers, wax, etc.
- the compound of the invention and salts and tautomers thereof can also be in the form of Liposomenzu brieflysystemen, such as small
- Liposomes can be different
- Phospholipids e.g. Cholesterol, stearylamine or phosphatidylcholines.
- the compound of the invention as well as the salts and tautomers thereof can also be delivered using monoclonal antibodies as individual carriers to which the compound molecules are coupled.
- the compounds can also be coupled with soluble polymers as targeted drug carriers.
- Such polymers may include polyvinylpyrrolidone, pyran copolymer, polyhydroxypropylmethacrylamidephenol, polyhydroxyethylaspartamidephenol or polyethyleneoxidepolylysine substituted with palmitoyl radicals.
- the compounds can be attached to a class of biodegradable polymers suitable for the controlled release of a drug, e.g.
- Polylactic acid Polyepsilon-caprolactone, polyhydroxybutyric acid,
- Polyorthoesters polyacetals, polydihydroxypyrans, polycyanoacrylates and cross-linked or amphipathic block copolymers of hydrogels, be coupled.
- Formulations may be presented as discrete plasters for prolonged, intimate contact with the epidermis of the recipient.
- the drug may be delivered from the patch by iontophoresis as generally described in Pharmaceutical Research, 3 (6), 318 (1986).
- Pharmaceutical compounds adapted for topical administration may be formulated as ointments, creams, suspensions, lotions, powders, solutions, pastes, gels, sprays, aerosols or oils.
- the formulations are preferably applied as a topical ointment or cream.
- the active ingredient may be either paraffinic or water-miscible
- Cream base can be used.
- the active ingredient can be formulated into a cream with an oil-in-water cream base or a water-in-oil base.
- the pharmaceutical formulations adapted for topical application to the eye include eye drops wherein the active ingredient is dissolved or suspended in a suitable carrier, especially an aqueous solvent.
- Formulations include lozenges, lozenges and mouthwashes.
- compositions adapted for rectal administration may be presented in the form of suppositories or enemas.
- compositions adapted for nasal administration in which the vehicle is a solid contain a coarse powder having a particle size, for example, in the range of 20-500 microns, which is administered in the manner in which snuff is received, i. by Schneilinhalation via the nasal passages from a container held close to the nose with the powder.
- Administration as a nasal spray or nasal drops with a liquid as a carrier substance comprise solutions of active substance in water or oil.
- a liquid as a carrier substance comprise solutions of active substance in water or oil.
- Formulations include fine particulate dusts or nebulas containing various types of pressurized dosing dispenser
- Aerosols, nebulizers or insufflators can be generated.
- compositions adapted for vaginal administration may be used as pessaries, tampons, creams, gels, pastes, foams or
- compositions adapted for parenteral administration include aqueous and nonaqueous sterile injection solutions containing antioxidants, buffers, bacteriostats and solutes which render the formulation isotonic with the blood of the recipient to be treated; and aqueous and non-aqueous sterile suspensions which may contain suspending agents and thickeners.
- the formulations may be administered in single or multiple dose containers, e.g. sealed
- Suspensions can be made from sterile powders, granules and tablets.
- formulations may include other means conventional in the art with respect to the particular type of formulation; for example, formulations suitable for oral administration may contain flavorings.
- a therapeutically effective amount of the compound of the invention will depend on a number of factors including, for example, age and age Weight of the animal, the exact disease state that requires treatment, and its severity, the nature of the formulation and the route of administration, and is ultimately determined by the doctor or veterinarian.
- an effective amount of a compound of the invention for the treatment of neoplastic growth, eg, colon or breast carcinoma is generally in the range of 0.1 to 100 mg / kg body weight of the recipient (mammal) per day, and more typically in the range of 1 to 10 mg / kg body weight per day.
- the actual amount per day would usually be between 70 and 700 mg, this amount as a single dose per day or more commonly in a number of divided doses (such as two, three, four, five or six) per Day can be given so that the total daily dose is the same.
- An effective amount of a salt or tautomer thereof may be determined as a proportion of the effective amount of the compound of the invention per se. It can be assumed that similar dosages are suitable for the treatment of the other disease states mentioned above.
- the invention furthermore relates to medicaments comprising 4- (2-methyl-1H-pyrrolo [2,3-b] pyridin-3-yl) - [2,7] naphthyridin-1-ylamine and / or its
- the invention is also a set (kit), consisting of separate packages of
- the kit contains suitable containers, such as boxes or boxes, individual bottles, bags or ampoules.
- suitable containers such as boxes or boxes, individual bottles, bags or ampoules.
- the set can eg separate ampoules in which in each case an effective amount of 4- (2-methylm H-pyrrolo [2,3-b] pyridin-3-yl) - [2,7] naphthyridin-1-ylamine and / or their
- the present compound is useful as a pharmaceutical agent for mammals, especially for humans, in the treatment and
- the invention further provides 4- (2-methyl-1H-pyrrolo [2,3-b] pyridin-3-yl) - [2,7] naphthyridin-1-ylamine and its pharmaceutically acceptable salts and tautomers for use in treatment tumors, tumor growth, tumor metastases and / or AIDS.
- the invention further provides 4- (2-methyl-1H-pyrrolo [2,3-b] pyridin-3-yl) - [2,7] naphthyridin-1-ylamine and its pharmaceutically acceptable salts and tautomers, for use with Treatment of fibrosis, restenosis, HIV infection, Alzheimer's disease, atherosclerosis and / or to promote wound healing.
- the present invention encompasses the use of 4- (2-methyl-1H-pyrrolo [2,3-b] pyridin-3-yl) - [2,7] naphthyridin-1-ylamine and / or its
- carcinomas for the treatment are from the brain carcinoma, genitourinary tract carcinoma, carcinoma of the lymphatic system, gastric carcinoma, laryngeal carcinoma and lung carcinoma colorectal cancer.
- Another group of preferred forms of cancer are monocytic leukemia, lung adenocarcinoma, small cell lung carcinoma, pancreatic cancer, glioblastoma and breast carcinoma.
- a pharmaceutical composition for the treatment and / or control of a tumorigenic disease in a mammal which process comprises administering to a diseased mammal in need of such treatment a therapeutically effective amount of a compound of the invention.
- the therapeutic amount depends on the particular disease and can be determined by the skilled person without great effort.
- a disease wherein the disease is a solid tumor.
- the solid tumor is preferably selected from the group of squamous cell tumors, bladder, stomach, kidney, head and neck, esophagus, cervix, thyroid, intestine, liver, brain, prostate, Urogenital tract, lymphatic system, stomach, larynx and / or lungs.
- the solid tumor is furthermore preferably selected from the group of lung adenocarcinoma, small cell lung carcinoma, pancreatic cancer, glioblastoma, colon carcinoma and breast carcinoma.
- a tumor of the blood and immune system preferably for the treatment of a tumor selected from the group of acute myelotic leukemia, the
- the invention furthermore relates to the use of the compound according to the invention for the treatment of bone pathologies, the bone pathology originating from the group osteosarcoma, osteoarthritis and rickets.
- the present compound is also suitable for combination with known anticancer agents.
- known anticancer agents include the following: estrogen receptor modulators, androgen receptor modulators, retinoid receptor modulators, cytotoxic agents, antiproliferative agents, prenyl protein transferase inhibitors, HMG-CoA reductase inhibitors, HIV protease inhibitors, reverse transcriptase inhibitors and other angiogenesis inhibitors.
- the present compounds are particularly useful for co-administration with radiotherapy.
- Estrogen Receptor Modulators refers to compounds that inhibit the binding of estrogen to the
- estrogen receptor modulators include
- “Androgen receptor modulators” refers to compounds that interfere with or inhibit the binding of androgens to the receptor, regardless of how this is done. “Androgen receptor modulators include, for example, finasteride and other 5a-reductase inhibitors, nilutamide, flutamide, bicalutamide , Liarozole and abiraterone acetate.
- Retinoid receptor modulators refers to compounds that interfere with or inhibit the binding of retinoids to the receptor. depending on how this happens.
- retinoid receptor modulators include, for example, bexarotene, tretinoin, 13-cis retinoic acid, 9-cis retinoic acid, ⁇ -difluoromethylornithine, ILX23-7553, trans-N- (4'-hydroxyphenyl) retinamide and N-4-carboxyphenylretinamide.
- Cytotoxic agents refers to compounds that cause cell death or interfere with cell myosis, primarily through direct action on cell function, including alkylating agents, tumor necrosis factors, intercalators, microtubulin inhibitors, and topoisomerase inhibitors.
- the cytotoxic agents include, for example, tirapazimine, Sertenef, cachectin, ifosfamide, tasonermine, lonidamine, carboplatin, altretamine, prednimustine, dibromodulcite, ranimustine, fotemustine, nedaplatin, oxaliplatin, temozolomide, heptaplatin, estramustine, improvisulfan-tosylate, trofosfamide, nimustine, dibrosylamine.
- MEN 10755 and 4-desmethoxy-3-desamino-3-aziridinyl-4-methylsulfonyl-daunorubicin see WO 00/50032, but this is not intended to be limiting.
- microtubulin inhibitors include, for example, paclitaxel, vindesine sulfate, 3 ⁇ 4'-didehydro-4'-deoxy-8'-norvincaleukoblastin, docetaxol, rhizoxin, dolastatin, mivobulinisethionate, auristatin, cemadotin, RPR109881,
- BMS184476 vinflunine, cryptophycin, 2,3,4,5,6-pentafluoro-N- (3-fluoro-4-methoxyphenyl) benzenesulfonamide, anhydrovinblastine, N, N-dimethyl-L-valyl-L-valyl-N-methyl -L-valyl-L-prolyl-L-proline t-butylamide, TDX258 and BMS188797.
- Topoisomerase inhibitors are for example topotecan, hycaptamine,
- Antiproliferative agents include antisense RNA and DNA oligonucleotides such as G3139, ODN698, RVASKRAS, GEM231, and INX3001, as well as antimetabolites such as enocitabine, carmofur, tegafur, pentostatin,
- Doxifluridine trimetrexate, fludarabine, capecitabine, galocitabine, cytarabine ocfosfate, fosteabin sodium hydrate, raltitrexed, paltitrexide, emitefur, tiazofurin, decitabine, nolatrexed, pemetrexed, nelzarabine, 2'-deoxy-2'-methylidene cytidine, 2'-fluoromethylene -2'-deoxycytidine, N- [5- (2,3-dihydrobenzofuryl) -sulfonyl-N-diS-dichlorophenyl urea, N6- [4-deoxy-4- [N 2 - [2 (E), 4 (E. ) -tetradecadienoyl] glycylamino] -L-glycero-BL-mannoheptopyranosyl] adenine,
- antiproliferative agents other monoclonal antibodies against growth factors than those already mentioned under the “angiogenesis inhibitors”, such as Trastuzu-mab, as well as tumor suppressor genes, such as p53, which can be delivered via recombinant virus-mediated gene transfer (see, eg, US Pat. No. 6,069,134).
- Rhizoxin (Fujisawa) D 24851 (ASTA Medica)
- Epothilone B Novartis
- ZD 6126 AstraZeneca
- Auristatin PE (Teikoku NeuroPharma)
- Bs BMS 247550 (BMS) BNP-7787 (BioNumerik) BMS 184476 (BMS) CA-4 prodrug (OXiGENE) BMS 188797 (BMS) Dolastatin-10 (NrH)
- Taxoprexin (Protarga) CA-4 (OXiGENE)
- Thymidylate pemetrexed (Eli Lilly) Nolatrexed (Eximias) synthase ZD-9331 (BTG) CoFactor TM (BioKeys)
- Histone acetyl trans-Tacedinalin Pfizer pivaloyloxymethyl butyrate ferase inhibitors SAHA (Aton Pharma) (titanium)
- TNF-alpha-virulizine (Lorus Therapeutic? Revimid (Celgene) Agonists / anti-CDC-394 (Celgene)
- CTL MGV Synchrovax Vaccines
- CapCell TM CYP450- (Reducing Agent, Zambon) Stimulant, Bavarian Nordic)
- R-Flurbiprofen NF-kappaB-GCS-IOO (gal3 antagonist, inhibitor, Encore)
- PCK-3145 apoptosis-bortezomib (proteasome promoter, Procyon)
- PT-100 growth factor (differentiator, NIH)
- MX6 apoptosis promoter, midostaurin (PKC inhibitor, MAXIA)
- Bryostatin-1 (PKC promoter, ILEX Oncology) stimulant, GPC Biotech) Urocidin (apoptosis-CDA-II (apoptosis promoter, promoter, Bioniche)
- Particularly preferred is 4- (2-methyl-1H-pyrrolo [2,3-b] pyridin-3-yl) - [2,7] naphthyridin-1-ylamine and its pharmaceutically acceptable salts and / or tautomers with immunomodulators, preferably combined with anti-PDL-1 or IL-12.
- the invention further provides 4- (2-methyl-1H-pyrrolo [2,3-b] pyridin-3-yl) - [2,7] naphthyridin-1-ylamine and / or its physiologically acceptable salts and tautomers Use for the treatment of tumors, wherein a therapeutically effective amount of a compound of formula I is administered in combination with a compound of the group of immunomodulators.
- the invention further provides 4- (2-methyl-1H-pyrrolo [2,3-b] pyridin-3-yl) - [2,7] naphthyridin-1-ylamine and / or its physiologically acceptable salts and tautomers for use for the treatment of tumors, wherein a therapeutically effective amount of a compound of the formula I in combination with radiotherapy and a compound from the group of
- Immunomodulators is administered.
- the cells are seeded in suitable cell density in microtiter plates (96-well format) and the test substances are added in the form of a concentration series. After four more days of culture in serum-containing medium, tumor cell proliferation / tumor cell vitality can be determined by means of an Alamarblue test system.
- colon carcinoma cell lines For example, commercially available colon carcinoma cell lines, cell lines of the ovary, cell lines of the prostate, or breast cell lines.
- the cells are cultured in medium. At intervals of several days, the cells are detached from the culture dishes with the aid of trypsin solution and seeded in fresh medium at a suitable dilution. The cells are cultured at 37 ° C and 10% C0 2 . 2.2. Sowing the cells
- a defined number of cells are incubated per culture / well in a volume of 180 ⁇ culture medium in microtiter plates (96 well).
- test substances are dissolved, for example, in DMSO and then used in the cell culture medium in appropriate concentration (if appropriate a dilution series). The dilution levels may vary depending on
- test substances are in corresponding
- Test substances to the cells can be made on the same day as the Aussat of the cells. For this purpose, from the predilution plate each 20 ⁇
- the cells are cultured for a further 4 days at 37 ° C and 10% CO 2 .
- microtiter plates are incubated for a further seven hours in a CO2 incubator (at 37 ° C. and 10% CO 2).
- the plates are measured on a reader with a fluorescence filter at a wavelength of 540 nm.
- the plates can be easily shaken just before the measurement.
- the absorbance value of the medium control (no use of cells and test substances) is subtracted from all other extinction values.
- the Controls (cells without test substance) are set equal to 100 percent and all other absorbance values related thereto (expressed as% of control, for example):
- IC 50 values 50% inhibition
- RS1 statistical programs
- the test substances are in corresponding
- the radioactivity (decays per minute) of the blank (no use of test substance in the presence of staurosporine) is different from all others
- the controls (kinase activity without test substance) are set equal to 100 percent and all others Radioactivity values (after deduction of the blank) are expressed in relation to them (expressed as% of control, for example).
- IC 5 o values (50% inhibition) is carried out with the help of statistical programs such as RS1.
- IC 50 data according to the invention is carried out with the help of statistical programs such as RS1.
- the kinase assay is performed as a 384-well flashplate assay.
- 3 3 p_ATP / well are added in a total volume of 50 ⁇ M (10 mM MOPS, 10 mM magnesium acetate, 0.1 mM EGTA, 1 mM dithiothreitol, 0.02% Brij35, 0.1% BSA, 0.1% BioStab, pH 7.5) without or with test substance for 20 min at 30 ° C incubated.
- the reaction is stopped with 25 .mu.l 200 mM EDTA solution, filtered off with suction after 30 min at room temperature and the wells washed 3 times with 100 .mu.l 0.9% NaCl solution.
- the kinase assay is performed as a 384-well flashplate assay.
- 0.6 nM TANK binding kinase (TBK1), 800 nM biotinylated MELK-derived peptide (biotin-Ah-Ah-AKPKGNKDYHLQTCCGSLAYRRR) and 10 ⁇ ATP (with 0.25 ⁇ 33 P-ATP / well) are in a total volume of 50 ⁇ (10 mM MOPS, 10 mM magnesium acetate, 0.1 mM EGTA, 1 mM DTT, 0.02% Brij35, 0.1% BSA, pH 7.5) with or without test substance incubated for 120 min at 30 ° C. The reaction becomes 25 ⁇ ! Stopped 200 mM EDTA solution, filtered off with suction at room temperature after 30 min and the wells washed 3 times with 100 .mu. ⁇ 0.9% NaCl solution.
- the ability of the inhibitors to abrogate TGF-beta mediated growth inhibition is tested.
- Cells of the lung epithelial cell line MvILu are seeded in defined cell density in a 96-well microtiter plate and cultured overnight under standard conditions. On the following day, the medium with medium containing 0.5% FCS and 1 ng / ml TGF-beta, replaced and the test substances in defined
- Concentrations usually in the form of serial dilutions with 5-fold steps added.
- the concentration of the solvent DMSO is constant at 0.5%.
- crystal violet staining of the cells occurs.
- absorbance at 550 nm is measured spectrophotometrically. It can be used as a quantitative measure of the existing adherent cells and thus of cell proliferation during culture.
- the wells are washed 3 times with 100 ⁇ l of 0.9% aqueous NaCl solution and the remaining radioactivity in a TopCount solution.
- Device Perkin-Elmer
- the IC50 values are calculated using RS1 software.
- the radioactivity (decays per minute) of the blank (no use of test substance in the presence of 100 nM staurosporine) is subtracted from all other radioactivity values.
- the controls (kinase activity without test substance) are set equal to 100 percent and all other radioactivity values (after deduction of the blank value) are related thereto (expressed as% of control, for example).
- IC 50 values concentration of the test substance with 50% inhibition
- RS1 statistical programs
- IC 5 o data of compounds according to the invention are given in Table 2.
- “usual workup” means adding water if necessary, adjusting to pH values between 2 and 10, if necessary, depending on the constitution of the final product, extracting with ethyl acetate or dichloromethane, separating, drying organic phase over sodium sulfate, evaporated and purified by chromatography on silica gel and / or by crystallization.
- the preparation of the compound according to the invention is carried out by Pd-catalyzed cross-coupling of starting material 1 (4-bromo [2,7] naphthyridin-1-ylamine) with starting material 2 (1-benzenesulfonyl) -methyl-S ⁇ SS-tetramethylKI.S ⁇ ldioxa- borolan-2-yl) -1H-pyrrolo [2,3-b] pyridine) and subsequent cleavage of the benzenesulfonyl group with alcohols under basic conditions.
- the compound is prepared from 2H- [2,7] naphthyridin-1-one CAS 67988-50-5, the hydrobromide CAS 950746-19-7 or the hydrochloride CAS 369648-60- 2.
- 4-methyl-nicotinonitrile CAS 5444-01-9 is reacted with DMF-acetal (eg CAS 4637-24-5 [dimethyl]) to give 4 - ((E) -2-dimethylamino-vinyl) nicotinonitrile CAS 36106 -34-0 which is cyclized to [2,7] naphthyridin-1-ylamine.
- the suspension obtained is dissolved in 500 ml of water and the pH is adjusted to pH 7-8 with 0 500 ml of 25% aqueous ammonia solution.
- THF / trifluoroethanol (1: 1 vol) is heated at reflux for 20 h.
- the mixture is cooled, the solvent is removed and purified via flash chromatography over 220 g of silica with a methanol gradient in ethyl acetate at 150 ml / min with UV detection at 254 nm.
- Example A Injection glasses
- Disodium hydrogen phosphate is adjusted to pH 6.5 in 2 l of bidistilled water with 2N hydrochloric acid, filtered sterile, filled into injection jars, lyophilized under sterile conditions and sealed in a sterile manner. Each injection jar contains 5 mg of active ingredient.
- a mixture of 20 g of the compound according to the invention is melted with 100 g of soya lecithin and 1400 g of cocoa butter, poured into molds and allowed to cool.
- Each suppository contains 20 mg of active ingredient.
- a solution of 1 g of the compound according to the invention, 9.38 g of NaH 2 PO 4 .2H 2 O, 28.48 g of Na 2 HPO 4 .12H 2 O and 0.1 g of benzalkonium chloride in 940 ml of bidistilled water is prepared. Adjust to pH 6.8, make up to 1 liter and sterilize by irradiation. This solution can be used in the form of eye drops.
- 500 mg of the compound according to the invention are mixed with 99.5 g of Vaseline under aseptic conditions.
- a mixture of 1 kg of the compound of the invention, 4 kg lactose, 1, 2 kg of potato starch, 0.2 kg of talc and 0.1 kg of magnesium stearate is compressed in the usual way to tablets, such that each tablet 10 mg
- Tablets are pressed analogously to Example E, which are then coated in the usual way with a coating of sucrose, potato starch, talc, tragacanth and dye.
- Example H Ampoules
- a solution of 1 kg of the compound according to the invention in 60 l of bidistilled water is sterile filtered, filled into ampoules, lyophilized under sterile conditions and sealed sterile. Each vial contains 10 mg of active ingredient.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oncology (AREA)
- Virology (AREA)
- Communicable Diseases (AREA)
- Molecular Biology (AREA)
- Tropical Medicine & Parasitology (AREA)
- AIDS & HIV (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
Description







Claims

Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP13759449.5A EP2903992A1 (de) | 2012-10-02 | 2013-09-09 | 7-azaindol-2,7-naphthyridin-derivat zur behandlung von tumoren |
| CN201380051232.3A CN104736543A (zh) | 2012-10-02 | 2013-09-09 | 用于治疗肿瘤的7-氮杂吲哚-2,7-二氮杂萘衍生物 |
| JP2015534920A JP2015531381A (ja) | 2012-10-02 | 2013-09-09 | 腫瘍の処置のための7−アザインドール−2,7−ナフチリジン誘導体 |
| US14/432,767 US9382244B2 (en) | 2012-10-02 | 2013-09-09 | 7-azaindole-2,7-naphthyridine derivative for the treatment of tumours |
| AU2013327282A AU2013327282A1 (en) | 2012-10-02 | 2013-09-09 | 7-azaindol-2,7-naphthyridine derivative for the treatment of tumors |
| CA 2886886 CA2886886A1 (en) | 2012-10-02 | 2013-09-09 | 7-azaindole-2,7-naphthyridine derivative for the treatment of tumours |
| IL237976A IL237976A0 (en) | 2012-10-02 | 2015-03-26 | A derivative of 7-azaindole-2,7-naphthyridine for the treatment of tumors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102012019369.6 | 2012-10-02 | ||
| DE102012019369.6A DE102012019369A1 (de) | 2012-10-02 | 2012-10-02 | 7-Azaindolderivat |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014053208A1 true WO2014053208A1 (de) | 2014-04-10 |
Family
ID=49123818
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2013/002696 WO2014053208A1 (de) | 2012-10-02 | 2013-09-09 | 7-azaindol-2,7-naphthyridin-derivat zur behandlung von tumoren |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US9382244B2 (de) |
| EP (1) | EP2903992A1 (de) |
| JP (1) | JP2015531381A (de) |
| CN (1) | CN104736543A (de) |
| AR (1) | AR092772A1 (de) |
| AU (1) | AU2013327282A1 (de) |
| CA (1) | CA2886886A1 (de) |
| DE (1) | DE102012019369A1 (de) |
| IL (1) | IL237976A0 (de) |
| WO (1) | WO2014053208A1 (de) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020083662A1 (en) | 2018-10-23 | 2020-04-30 | Basf Se | Tricyclic pesticidal compounds |
| WO2021209265A1 (en) | 2020-04-14 | 2021-10-21 | Basf Se | Tricyclic pesticidal compounds |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SG11202002886PA (en) * | 2017-09-28 | 2020-04-29 | Cstone Pharmaceuticals Suzhou Co Ltd | Fused ring derivative as a2a receptor inhibitor |
| CA3102645A1 (en) * | 2018-06-07 | 2019-12-12 | Disarm Therapeutics, Inc. | Inhibitors of sarm1 |
| CN115944739B (zh) * | 2022-12-30 | 2023-12-19 | 深圳开悦生命科技有限公司 | Rna解旋酶dhx33抑制剂在制备用于治疗黑色素瘤的药物中的应用 |
Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6069134A (en) | 1991-03-06 | 2000-05-30 | Board Of Regents, The University Of Texas System | Methods and compositions comprising DNA damaging agents and p53 |
| WO2000050032A1 (en) | 1999-02-25 | 2000-08-31 | Pharmacia & Upjohn S.P.A. | Antitumour synergistic composition |
| WO2005095400A1 (en) | 2004-03-30 | 2005-10-13 | Vertex Pharmaceuticals Incorporated | Azaindoles useful as inhibitors of jak and other protein kinases |
| WO2006106326A1 (en) | 2005-04-06 | 2006-10-12 | Astrazeneca Ab | Substituted heterocycles and their use as chk1, pdk1 and pak inhibitors |
| WO2007076423A2 (en) | 2005-12-22 | 2007-07-05 | Smithkline Beecham Corporation | INHIBITORS OF Akt ACTIVITY |
| WO2008005457A2 (en) | 2006-06-30 | 2008-01-10 | Sunesis Pharmaceuticals | Pyridinonyl pdk1 inhibitors |
| WO2008079988A2 (en) | 2006-12-22 | 2008-07-03 | Novartis Ag | Quinazolines for pdk1 inhibition |
| WO2008112217A1 (en) | 2007-03-13 | 2008-09-18 | Merck & Co., Inc. | Inhibitors of janus kinases and/or 3-phosphoinositide-dependent protein kinase-1 |
| WO2008124849A2 (en) | 2007-04-10 | 2008-10-16 | Sgx Pharmaceuticals, Inc. | Pyrrolo-pyridine kinase modulators |
| WO2008156726A1 (en) | 2007-06-20 | 2008-12-24 | Merck & Co., Inc. | Inhibitors of janus kinases |
| WO2009054941A1 (en) | 2007-10-25 | 2009-04-30 | Merck & Co., Inc. | Therapeutic compounds |
| WO2012104007A2 (de) | 2011-02-01 | 2012-08-09 | Merck Patent Gmbh | 7-azaindolderivate |
-
2012
- 2012-10-02 DE DE102012019369.6A patent/DE102012019369A1/de not_active Withdrawn
-
2013
- 2013-09-09 US US14/432,767 patent/US9382244B2/en not_active Expired - Fee Related
- 2013-09-09 CN CN201380051232.3A patent/CN104736543A/zh active Pending
- 2013-09-09 AU AU2013327282A patent/AU2013327282A1/en not_active Abandoned
- 2013-09-09 JP JP2015534920A patent/JP2015531381A/ja active Pending
- 2013-09-09 EP EP13759449.5A patent/EP2903992A1/de not_active Withdrawn
- 2013-09-09 CA CA 2886886 patent/CA2886886A1/en active Pending
- 2013-09-09 WO PCT/EP2013/002696 patent/WO2014053208A1/de active Application Filing
- 2013-10-02 AR ARP130103564A patent/AR092772A1/es unknown
-
2015
- 2015-03-26 IL IL237976A patent/IL237976A0/en unknown
Patent Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6069134A (en) | 1991-03-06 | 2000-05-30 | Board Of Regents, The University Of Texas System | Methods and compositions comprising DNA damaging agents and p53 |
| WO2000050032A1 (en) | 1999-02-25 | 2000-08-31 | Pharmacia & Upjohn S.P.A. | Antitumour synergistic composition |
| WO2005095400A1 (en) | 2004-03-30 | 2005-10-13 | Vertex Pharmaceuticals Incorporated | Azaindoles useful as inhibitors of jak and other protein kinases |
| WO2006106326A1 (en) | 2005-04-06 | 2006-10-12 | Astrazeneca Ab | Substituted heterocycles and their use as chk1, pdk1 and pak inhibitors |
| WO2007076423A2 (en) | 2005-12-22 | 2007-07-05 | Smithkline Beecham Corporation | INHIBITORS OF Akt ACTIVITY |
| WO2008005457A2 (en) | 2006-06-30 | 2008-01-10 | Sunesis Pharmaceuticals | Pyridinonyl pdk1 inhibitors |
| WO2008079988A2 (en) | 2006-12-22 | 2008-07-03 | Novartis Ag | Quinazolines for pdk1 inhibition |
| WO2008112217A1 (en) | 2007-03-13 | 2008-09-18 | Merck & Co., Inc. | Inhibitors of janus kinases and/or 3-phosphoinositide-dependent protein kinase-1 |
| WO2008124849A2 (en) | 2007-04-10 | 2008-10-16 | Sgx Pharmaceuticals, Inc. | Pyrrolo-pyridine kinase modulators |
| WO2008156726A1 (en) | 2007-06-20 | 2008-12-24 | Merck & Co., Inc. | Inhibitors of janus kinases |
| WO2009054941A1 (en) | 2007-10-25 | 2009-04-30 | Merck & Co., Inc. | Therapeutic compounds |
| WO2012104007A2 (de) | 2011-02-01 | 2012-08-09 | Merck Patent Gmbh | 7-azaindolderivate |
Non-Patent Citations (11)
| Title |
|---|
| C.KORHERR ET AL., PNAS, vol. 103, 2006, pages 4240 - 4245 |
| CUNHA, BLOOD, vol. 117, no. 26, 30 June 2011 (2011-06-30), pages 6999 |
| D.A.BARBIE ET AL., NATURE LETTERS, 2009, pages 1 - 5 |
| HINCK, FEBS LETT., 2012, Retrieved from the Internet <URL:http://dx.doi.org/10.1016/j.febslet.2012.05.028> |
| INT. J. PHARM., vol. 115, 1995, pages 61 - 67 |
| J.S. BOEHM ET AL., CELL, vol. 129, 2007, pages 1065 - 1079 |
| PHARMACEUTICAL RESEARCH, vol. 3, no. 6, 1986, pages 318 |
| REVIEW VON MASSAGUE ANNU. REV. BIOCHEM., vol. 67, 1998, pages 753 - 91 |
| S.F.EDDY ET AL., CANCER RES., vol. 65, no. 24, 2005, pages 11375 - 11383 |
| S.I. CUNHA; K. PIETRAS, BLOOD, vol. 117, no. 26, 2011, pages 6999 - 7006 |
| Y.CHIEN ET AL., CELL, vol. 127, 2006, pages 157 - 170 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020083662A1 (en) | 2018-10-23 | 2020-04-30 | Basf Se | Tricyclic pesticidal compounds |
| WO2021209265A1 (en) | 2020-04-14 | 2021-10-21 | Basf Se | Tricyclic pesticidal compounds |
Also Published As
| Publication number | Publication date |
|---|---|
| CN104736543A (zh) | 2015-06-24 |
| AR092772A1 (es) | 2015-04-29 |
| US20150252041A1 (en) | 2015-09-10 |
| AU2013327282A1 (en) | 2015-05-14 |
| EP2903992A1 (de) | 2015-08-12 |
| IL237976A0 (en) | 2015-05-31 |
| JP2015531381A (ja) | 2015-11-02 |
| DE102012019369A1 (de) | 2014-04-03 |
| US9382244B2 (en) | 2016-07-05 |
| CA2886886A1 (en) | 2014-04-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2753615B1 (de) | Benzonitrilderivate als kinasehemmer | |
| EP2193118B1 (de) | Piperidin- und piperazinderivate zur behandlung von tumoren | |
| EP2414327B1 (de) | Heterocyclische verbindungen als autotaxin-inhibitoren | |
| EP2193122B1 (de) | Imidazolderivate | |
| EP2209777B1 (de) | Thiazolderivate zur behandlung von krebs | |
| WO2010060532A1 (de) | Benzo-naphtyridin verbindungen als inhibitoren von autotaxin | |
| DE102006060598A1 (de) | Tetrahydrobenzoisoxazole | |
| EP2033959B1 (de) | Tetrahydropyranochinolinderivate | |
| DE102011111400A1 (de) | Bicyclische heteroaromatische Verbindungen | |
| WO2006125555A2 (de) | Chinazolinone | |
| AU2018317789B2 (en) | Stable lyophilisates comprising 5,10-methylene-(6R)-tetrahydrofolic acid and a dicarboxylic acid | |
| EP1891076B1 (de) | Substituierte tetrahydrochinoline | |
| WO2014053208A1 (de) | 7-azaindol-2,7-naphthyridin-derivat zur behandlung von tumoren | |
| EP2121700B1 (de) | Substituierte tetrahydrochinoline | |
| EP1891013B1 (de) | Tetrahydrochinolinderivate | |
| DE102007013854A1 (de) | Tetrahydrochinoline | |
| WO2008113452A1 (de) | Substituierte tetrahydropyrrolochinoline | |
| EP1747214A2 (de) | Dihydrobenzothiophene |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13759449 Country of ref document: EP Kind code of ref document: A1 |
|
| REEP | Request for entry into the european phase |
Ref document number: 2013759449 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013759449 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 237976 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: 2886886 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2015534920 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14432767 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2013327282 Country of ref document: AU Date of ref document: 20130909 Kind code of ref document: A |