WO2013075785A1 - 3-cyanaryl-1h-pyrrolo[2,3-b]pyridin-derivate - Google Patents
3-cyanaryl-1h-pyrrolo[2,3-b]pyridin-derivate Download PDFInfo
- Publication number
- WO2013075785A1 WO2013075785A1 PCT/EP2012/004543 EP2012004543W WO2013075785A1 WO 2013075785 A1 WO2013075785 A1 WO 2013075785A1 EP 2012004543 W EP2012004543 W EP 2012004543W WO 2013075785 A1 WO2013075785 A1 WO 2013075785A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pyrrolo
- pyridin
- het
- tetrahydropyran
- pyrazol
- Prior art date
Links
- WDGWPLVSZGIYDX-UHFFFAOYSA-N COc(cc1)nc2c1c(-c(cc1C#N)ccc1OC1CCOCC1)c[nH]2 Chemical compound COc(cc1)nc2c1c(-c(cc1C#N)ccc1OC1CCOCC1)c[nH]2 WDGWPLVSZGIYDX-UHFFFAOYSA-N 0.000 description 1
- XFBRAQSVXDSRGM-UHFFFAOYSA-N N#Cc1cc(-c(c2cc(Br)cnc22)c[n]2S(c2ccccc2)(=O)=O)ccc1OC1CCOCC1 Chemical compound N#Cc1cc(-c(c2cc(Br)cnc22)c[n]2S(c2ccccc2)(=O)=O)ccc1OC1CCOCC1 XFBRAQSVXDSRGM-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
Definitions
- the present invention relates to pyridine compounds capable of inhibiting one or more kinases.
- the compounds find use in the treatment of a variety of disorders including cancer, septic shock, primary open angle glaucoma (POAG), hyperplasia, rheumatoid arthritis, psoriasis, atherosclerosis, retinopathy, osteoarthritis, endometriosis, chronic inflammation and / or neurodegenerative Diseases such as Alzheimer's disease.
- the present invention relates to compounds and the use of compounds in which the inhibition, regulation and / or modulation of the signal transduction of receptor kinases plays a role, further pharmaceutical
- compositions containing these compounds as well as the use of the compounds for the treatment of kinase-related diseases.
- protein kinases regulate almost every cell process, including metabolism, cell proliferation, cell differentiation and cell survival, they are attractive targets for therapeutic intervention in various disease states.
- cell cycle control and angiogenesis in which protein kinases play a key role, are cell events associated with numerous disease states, such as cancer , Inflammatory diseases, abnormal angiogenesis and related diseases, atherosclerosis, macular degeneration, diabetes, obesity, and pain are, but are not limited to.
- the present invention relates to compounds and to the use of compounds in which the inhibition, regulation and / or modulation of signal transduction of TBK1 and ⁇ plays a role.
- One of the major mechanisms by which cell regulation is effected is through the transduction of extracellular signals across the membrane, which in turn modulate biochemical pathways in the cell.
- Protein phosphorylation is a process by which intracellular signals are propagated from molecule to molecule, ultimately resulting in a cellular response.
- These signal transduction cascades are highly regulated and often overlap, as evidenced by the presence of many protein kinases as well as phosphatases.
- Phosphorylation of proteins occurs predominantly with serine, threonine or tyrosine residues, and protein kinases were therefore classified according to their specificity of the phosphorylation site, ie the serine-AThreonin kinases and tyrosine kinases. Since phosphorylation is such a widespread process in cells, and as cell phenotypes are largely influenced by the activity of these pathways, it is presently believed that a number of disease states and / or diseases may be due to either aberrant activation or functional mutations in the molecular components of Kinase cascades are attributed. Consequently, the characterization of these proteins and
- ⁇ and TBK1 are serine / threonine kinases that have high homologies with each other and with other IkB kinases. Both kinases play an integral role in the innate immune system. Double-stranded RNA viruses are recognized by the Toll-like receptors 3 and 4, as well as the RNA helicases RiG-1 and MDA-5, and lead to activation of the TRIF-TBK1 / IKKs-IRF3 signaling cascade, resulting in a Type I interferon Answer leads.
- Protein kinase-mediated diseases are characterized by an abnormal activity or hyperactivity of such protein kinases.
- Abnormal activity involves either: (1) expression in cells, usually these
- Hyperactivity refers to either amplification of the gene encoding a particular protein kinase, or the
- an activity level associated with a cell proliferation disease ie, as the kinase level increases, the severity of one or more symptoms of cell proliferation disease increases.
- the bioavailability of a protein kinase may also be affected by the presence or absence of a set of binding proteins of that kinase.
- ⁇ and TBK1 are highly homologous Ser / Thr kinases that play a crucial role in the innate immune response through the induction of type 1 interferons and other cytokines. These kinases are stimulated in response to viral / bacterial infection.
- the immune response to viral and bacterial infections involves the binding of antigens such as bacterial lipopolysaccharide (LPS), viral double-stranded RNA (dsRNA) to Toll-like receptors, and subsequent activation of the TBK1 pathway.
- LPS bacterial lipopolysaccharide
- dsRNA viral double-stranded RNA
- Toll-like receptors Toll-like receptors
- ⁇ and TBK1 have also been linked to cancer. It has been shown that ⁇ cooperates with activated MEK to transform human cells. In addition, ⁇ is often amplified / overexpressed in breast cancer cell lines and patient-derived tumors. TBK1 is induced under hypoxic conditions and expressed in significant levels in many solid tumors. Furthermore, TBKI is required to support oncogenic Ras transformation, and TBK1 kinase activity is increased in transformed cells and required for their survival in culture. It has also been found that TBK1 and NF-kB signaling are essential in KRAS mutant tumors. TBK1 has been identified as a synthetic lethal partner of oncogenic KRAS.
- WO 2011/046970 A1 describes the use of TBK1 and / or ⁇ inhibitors for the treatment of various diseases, such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), Sjörgrens syndrome, Aicardi-Goutieres syndrome Lupus chilblain, retinal vasculopathy and cerebral leukodystrophy (RVCL), systemic sclerosis, myositis, psoriasis, chronic obstructive pulmonary disease (CPD), inflammatory bowel disease (IBD), obesity, insulin resistance, type 2 diabetes (NIDDM), metabolic syndrome, cancers,
- RA rheumatoid arthritis
- SLE systemic lupus erythematosus
- Sjörgrens syndrome Aicardi-Goutieres syndrome Lupus chilblain
- retinal vasculopathy and cerebral leukodystrophy RVCL
- systemic sclerosis myositis, p
- the compounds of the invention or a pharmaceutically acceptable salt thereof are administered for the treatment of cancer including solid carcinomas such as carcinomas (eg, the lungs, pancreas, thyroid, bladder, or colon), myeloid diseases (eg myeloid leukemia) or adenomas (eg villous colonic adenoma).
- solid carcinomas such as carcinomas (eg, the lungs, pancreas, thyroid, bladder, or colon)
- myeloid diseases eg myeloid leukemia
- adenomas eg villous colonic adenoma
- the tumors further include monocytic leukemia, brain, urogenital, lymphatic, gastric, laryngeal and lung carcinomas, including lung adenocarcinoma and small cell lung carcinoma, pancreatic and / or breast carcinoma.
- the compounds are also useful in the treatment of immunodeficiency induced by HIV (Human Immunodeficiency Virus Type 1).
- Cancerous hyperproliferative disorders include brain, lung, squamous, bladder, stomach, pancreatic, liver, kidney, colorectal, breast, head, neck, esophageal, gynecological, thyroid, lymphoma
- cancerous cell growth is a disease that is an object of the present invention.
- the present invention therefore relates to compounds according to the invention as medicaments and / or active pharmaceutical ingredients in the treatment and / or prophylaxis of said diseases and the use of compounds according to the invention for the preparation of a
- the compounds according to the invention have an antiproliferative action.
- the compounds of the invention are administered to a patient with a hyperproliferative disorder, e.g. To inhibit tumor growth, to reduce inflammation associated with a lymphoproliferative disorder, to inhibit graft rejection or neurological damage due to tissue repair, etc.
- the present compounds are useful for prophylactic or therapeutic purposes.
- the term "treating" is used to refer to both the prevention of disease and the treatment of pre-existing conditions
- Prevention of proliferation / vitality is achieved by administration of the compounds of the invention prior to the development of the apparent disease, e.g. Prevention of Tumor Growth
- the compounds are used to treat persistent diseases by stabilizing or ameliorating the clinical symptoms of the patient.
- the host or patient may be of any mammalian species, e.g. A primate species, especially humans; Rodents, including mice, rats and
- Animal models are of interest for experimental studies, providing a model for the treatment of human disease.
- the susceptibility of a particular cell to treatment with the compounds of the invention can be determined by testing in vitro.
- a culture of the cell is incubated with a compound of the invention at various concentrations for a period of time sufficient to allow the active agents to induce cell death or to inhibit cell proliferation, cell vitality or migration, usually between about one hour and one week.
- cultured cells from a biopsy sample can be used. be. The amount of cells remaining after treatment are then determined.
- the dose will vary depending on the specific compound used, the specific disease, the patient status, etc. Typically, a therapeutic dose will be sufficient to substantially reduce the unwanted cell population in the target tissue while maintaining patient viability. Treatment is generally continued until there is a significant reduction, e.g. B. at least about 50% reduction in cell load and can be continued until essentially no more unwanted cells are detected in the body.
- the compounds of the present invention are useful in the treatment of a variety of conditions in which proliferation and / or migration of smooth muscle cells and / or inflammatory cells into the intimal layer of a vessel results in limited blood flow to that vessel, e.g. In neointimal occlusive lesions. Too occlusive
- Transplant vascular diseases of interest include atherosclerosis, coronary vascular disease after transplantation, vein graft stenosis, pert-anastomotic prosthetic restenosis, restenosis after angioplasty or stent placement, and the like.
- the compounds of the invention may be used to achieve additive or synergistic effects in certain existing cancer chemotherapies and radiation and / or to restore the efficacy of certain existing cancer chemotherapies and radiation.
- method refers to modes of operation, means, techniques, and procedures to accomplish a given task, including those modes of operation, means, techniques, and procedures that are either known to those skilled in the chemical, pharmacological, biological, biochemical, and medical arts are or from him easily known practices, means, techniques and
- Procedures may be developed, but are not limited thereto.
- the term "administration" refers to a method for bringing together a compound of the present invention and a target kinase such that the compound directs the enzyme activity of the kinase either directly, i. by interaction with the kinase itself, or indirectly, i. by interaction with another molecule on which the catalytic activity of the kinase depends can influence.
- administration may be either in vitro, i. in the test tube, or in vivo, i. in cells or tissues of a living organism.
- treating herein includes overriding, largely inhibiting, slowing or reversing the progression of a disease or disorder, substantially ameliorating the clinical symptoms of a disease or disorder, or largely preventing the occurrence of the clinical symptoms of a disease or disorder.
- prevent means a method to block an organism from acquiring a disorder or disease at all.
- a therapeutically effective amount may be first calculated from cell culture assays.
- a dose can be formulated to achieve a circulatory concentration range comprising the IC50 or IC 100 as determined in cell cultures. This information can be used to more accurately determine useful doses for humans.
- Initial dosages can also be calculated from in vivo data. Based on these initial guidelines, one of ordinary skill in the art could determine an effective dosage for humans.
- the toxicity and therapeutic efficacy of the compounds described herein can be determined according to standard pharmaceutical procedures on cell cultures or experimental animals by, for example, LD50 and ED50 certainly.
- the dose ratio between toxic and therapeutic effects is the therapeutic index, which can be expressed as the ratio between LD50 and ED50.
- Compounds having a high therapeutic index are preferred.
- the data obtained from these cell culture assays and animal studies can be used to formulate a dosage range that is non-toxic to human use.
- the dosage of such compounds is preferably in bloodstream concentration ranges that include the ED50 with little or no toxicity. Within this range, the dosage may vary depending on the dosage form used and the route of administration used.
- the exact formulation, route of administration, and dosage may be chosen by the individual physician, taking into account the condition of the patient (see, eg, Fingl et al., 1975, The Pharmacological Basis of Therapeutics, Chapter 1, page 1).
- Dosage amount and interval can be adjusted individually to provide plasma levels of active compound sufficient to produce a therapeutic
- Typical patient doses for oral administration are in the range of about 50-2000 mg / kg / day, generally about 100-000 mg / kg / day, preferably about 150-700 mg / kg / day, and most preferably about 250-400 mg / kg / day. 500 mg / kg / day.
- therapeutically effective serum levels are achieved by administering multiple doses per day.
- the effective local concentration of the drug may not be related to the plasma concentration. The skilled artisan will be able to optimize therapeutically effective local dosages without undue experimentation.
- Preferred diseases or disorders for the prevention, treatment and / or study of which the compounds described herein are useful are cell proliferative disorders, particularly cancer such as papilloma, blastoglioma, Kaposi's sarcoma, melanoma, lung cancer, ovarian cancer, prostate cancer, squamous cell carcinoma, astrocytoma, Head cancer, cervical, skin, liver, bladder, breast, lung, uterine, prostate but not limited to, rectal cancer, thyroid cancer, pancreatic cancer, gastric cancer, hepatocellular carcinoma, leukemia, lymphoma, Hodgkin's disease, and Burkitt's disease.
- cancer such as papilloma, blastoglioma, Kaposi's sarcoma, melanoma, lung cancer, ovarian cancer, prostate cancer, squamous cell carcinoma, astrocytoma, Head cancer, cervical, skin, liver, bladder, breast, lung, uterine, prostate but not limited to, rectal cancer
- benzonitrile derivatives are described in WO 2011/046970 A1 as TBK1 and / or ⁇ inhibitors.
- Pyrrolopyrimidines have been described as ⁇ and TBK1 inhibitors in WO 2010/100431.
- the invention relates to compounds of the formula I.
- R 1 is H, A or Cyc
- R 3 is H, Hal, A, OR 6 , N (R 6 ) 2> 0 [C (R 6 ) 2 ] m N (R 6 ) 2 , 0 [C (R 6 ) 2 ] n Het 2 ,
- Ar is unsubstituted or mono-, di- or trisubstituted by Hal, A, [C (R 6 ) 2 ] p OR 6 ,
- A is unbranched or branched alkyl having 1-0 C atoms, wherein one or two non-adjacent CH and / or CH 2 groups may be replaced by N, O and / or S atoms and / or also 1 -7 H atoms can be replaced by F and / or Cl,
- n 0, 1 or 2
- p 0, 1, 2, 3 or 4
- the invention also relates to the optically active forms (stereoisomers), salts, the enantiomers, the racemates, the diastereomers and the hydrates and solvates of these compounds.
- Solvates of the compounds are understood to mean additions of inert solvent molecules to the compounds that form themselves due to their mutual attraction. Solvates are, for example, mono- or dihydrate or alcoholates.
- the invention of course also the solvates of the salts.
- compositions are understood, for example, as the salts of the compounds according to the invention as well as so-called prodrug compounds. Under prodrug derivatives is understood with z.
- sugars or oligopeptides modified compounds of formula I which are rapidly cleaved in the organism to the active compounds of the invention.
- biodegradable polymer derivatives of the compounds of the invention as z. In Int. J. Pharm. 115, 61-67 (1995).
- the term "effective amount” means the amount of a drug or pharmaceutical agent which elicits a biological or medical response in a tissue, system, animal or human, e.g. sought or desired by a researcher or physician.
- terapéuticaally effective amount means an amount that, as compared to a corresponding subject who has not received that amount, results in:
- terapéuticaally effective amount also includes the amounts effective to increase normal physiological function.
- the invention also provides the use of mixtures of the compounds of formula I, e.g. Mixtures of two diastereomers, e.g. in the ratio 1: 1, 1: 2, 1: 3, 1: 4, 1: 5, 1: 10, 1: 100 or 1: 1000.
- the invention relates to the compounds of the formula I and their salts and to a process for the preparation of compounds of the formula I and their pharmaceutically usable salts, tautomers and stereoisomers, characterized in that a) a compound of the formula II
- L denotes a boronic acid radical or a boronic acid ester group, reacts, and then splits off Q, or b) releasing them from one of their functional derivatives by treatment with a solvolyzing or hydrogenolysing agent, and / or converting a base or acid of formula I into one of its salts.
- A is alkyl, is unbranched (linear) or branched and has 1, 2, 3, 4, 5, 6, 7, 8, 9 or 10 C atoms.
- A is preferably methyl, furthermore ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl or tert-butyl, furthermore also pentyl, 1-, 2- or 3-methylbutyl, 1, 1, 1, 2 - or 2,2-dimethylpropyl, 1-ethylpropyl, hexyl, 1-, 2-, 3- or 4-methylpentyl, 1, 1-, 1, 2-, 1, 3-, 2,2-, 2,3 or 3,3-dimethylbutyl, 1- or 2-ethylbutyl, 1-ethyl-1-methylpropyl, 1-ethyl-2-methylpropyl, 1, 1, 2 or 1, 2,2-trimethyl-propyl, more preferably, for example Trifluoromethyl.
- one or two CH and / or CH 2 groups can also be replaced by N, O or S
- Atoms be replaced.
- A means e.g. also 2-methoxyethyl.
- a particularly preferably A is unbranched or branched alkyl having 1-8 C
- Atoms in which one or two non-adjacent CH and / or CH 2 groups may be replaced by N and / or O atoms and / or 1-7 H atoms may be replaced by F.
- Cycloalkyl (cyclic alkyl) means cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or cycloheptyl.
- R is preferably H or A.
- R 2 is preferably 0 [C (R 6 ) 2 ] n Het or O [C (R 6 ) 2 ] n Cyc.
- R 3 is preferably H, Hal, O [C (R 6 ) 2 ] n Het 2 , Ar or Het 2 .
- R 4 particularly preferably denotes H.
- R 5 particularly preferably denotes H.
- R 6 is preferably H or methyl.
- Ar is, for example, phenyl, o-, m- or p-tolyl, o-, m- or p-ethylphenyl, o-, m- or p-propylphenyl, o-, m- or p-isopropylphenyl, o-, m- or p-tert-butylphenyl, o-, m- or p-trifluoromethylphenyl, o-, m- or p-fluorophenyl, o-, m- or p-bromophenyl, o-, m- or p-chlorophenyl, o-, m- or p-hydroxyphenyl, o-, m- or p-methoxyphenyl, o-, m- or p-methylsulfonylphenyl, o-, m- or p-nitrophenyl, o-,
- Het 1 is preferably unsubstituted or substituted by S (0) n A, S (0) n Ar or A dihydropyrrolyl, pyrrolidinyl, azetidinyl, oxetanyl, tetrahydroimidazolyl, dihydropyrazolyl, tetrahydropyrazolyi, tetrahydrofuranyl, dihydropyridyl, tetrahydropyridyl, piperidinyl, morpholinyl , Hexahydropyridazinyl, hexahydropyrimidinyl, [1,3] dioxolanyl, tetrahydropyranyl or piperazinyl.
- Het 1 particularly preferably denotes unsubstituted or pyrrolidinyl, oxetanyl, piperidinyl, morpholinyl which is unsubstituted or monosubstituted by S (O) n A, S (0) n Ar or A,
- Het 2 is preferably unsubstituted or monosubstituted by A, [C (R 6 ) 2 ] p OR 6 or [C (R 6 ) 2 ] P Het 1 dihydropyrrolyl, pyrrolidinyl, azetidinyl, tetrahydroimidazolyl, tetrahydrofuranyl, dihydropyrazolyl, tetrahydro ' dropyrazolyl, dihydropyridyl, tetrahydropyridyl, dihydropyranyl, tetrahydropyranyl, piperidinyl, morpholinyl, hexa-hydropyridazinyl, hexahydropyrimidinyl, [1, 3] dioxolanyl, piperazinyl, furyl, thienyl, pyrrolyl, imidazolyl, pyrazolyl, oxazolyl, isoxazo
- Het 2 more preferably means unsubstituted or simply through A
- Hal preferably denotes F, Cl or Br, but also I, particularly preferably F or Cl.
- the compounds of the formula I can possess one or more chiral centers and therefore occur in different stereoisomeric forms.
- Formula I encompasses all these forms.
- dihydropyrrolyl substituted dihydropyrrolyl, pyrrolidinyl, azetidinyl, oxetanyl, tetrahydroimidazolyl, dihydropyrazolyl, tetrahydropyrazolyl, tetrahydrofuranyl, dihydropyridyl, tetrahydropyridyl, piperidinyl, morpholinyl, hexahydropyridazinyl, hexahydropyrimidinyl,
- A is unbranched or branched alkyl having 1-8 C atoms, wherein one or two non-adjacent CH and / or CH 2 groups may be replaced by N and / or O atoms and / or 1-7 H atoms may be replaced by F;
- S (O) n A, S (O) n Ar or A dihydropyrrolyl, pyrrolidinyl, azetidinyl, oxetanyl, tetrahydroimidazolyl, dihydropyrazolyl, tetrahydropyrazolyl, tetrahydrofuranyl, dihydropyridyl, tetrahydropyridyl, piperidinyl, morpholinyl, hexahydropyridazinyl, hexahydropyrimidinyl,
- branched or branched alkyl having 1-8 C atoms, wherein one or two non-adjacent CH and / or CH 2 groups may be replaced by N and / or O atoms and / or 1-7 H atoms may be replaced by F,
- n 0, 1 or 2
- the compounds of the formula II and of the formula III are generally known. If they are new, they can be produced by methods known per se.
- Y preferably denotes I.
- Q preferably denotes BOC or phenylsulfonyl.
- L is preferably
- the reaction takes place under conditions known to those skilled in the art as Suzuki conditions or Suzuki reaction.
- the reaction time is between a few minutes and 14 days depending on the conditions used, the reaction temperature between about -10 ° and 160 °, usually between 20 ° and 150 °, more preferably between 40 ° and 100 ° C.
- Suitable inert solvents are e.g. Hydrocarbons such as hexane, petroleum ether, benzene, toluene or xylene; chlorinated hydrocarbons such as trichlorethylene, 1, 2-dichloroethane, carbon tetrachloride, chloroform or dichloromethane; Alcohols such as methanol, ethanol, isopropanol, n-propanol, n-butanol or tert-butanol; Ethers such as diethyl ether, diisopropyl ether, tetrahydrofuran (THF) or dioxane; Glycol ethers such as ethylene glycol monomethyl or monoethyl ether, ethylene glycol dimethyl ether (diglyme); Ketones such as acetone or butanone; Amides, such as acetamide, dimethylacetamide or dimethylformamide (DMF); Nitrites such as acetonitrile
- DMF and water are particularly preferred.
- a standard method for ether cleavage e.g. of a methyl ether, is the use of boron tribromide.
- Hydrogenolytically removable groups such as the cleavage of a benzyl ether, z.
- a catalyst e.g., a noble metal catalyst such as palladium, conveniently on a support such as carbon.
- Suitable solvents are those given above, in particular z.
- alcohols such as methanol or ethanol or amides such as DMF.
- the hydrogenolysis is usually carried out at temperatures between about 0 and 100 ° and pressures between about 1 and 200 bar, preferably at 20-30 ° and 1-10 bar.
- esters can be saponified with acetic acid or with NaOH or KOH in water, water-THF or water-dioxane at temperatures between 0 and 100 °. Alkylations on the nitrogen are carried out under standard conditions, as known to those skilled in the art.
- the compounds of formula I can be further obtained by liberating them from their functional derivatives by solvolysis, in particular hydrolysis, or by hydrogenolysis.
- Preferred starting materials for the solvolysis or hydrogenolysis are those which contain, instead of one or more free amino and / or hydroxyl groups corresponding protected amino and / or hydroxy groups, preferably those which instead of an H atom, which is connected to an N atom is to carry an amino protecting group, for.
- those corresponding to formula I but containing an NHR 'group (where R' represents an amino-protecting group, e.g., BOC or CBZ) instead of an NH 2 group.
- amino protecting group is well known and refers to groups which are capable of protecting (blocking) an amino group from chemical reactions, but which are readily removable after the desired chemical reaction has been carried out elsewhere in the molecule. Typical of such groups are in particular unsubstituted or substituted acyl, aryl, aralkoxymethyl or aralkyl groups. Moreover, because the amino protecting groups are removed after the desired reaction (or reaction sequence), their type and size is not critical; however, preference is given to those having 1-20, in particular 1-8 C atoms.
- acyl group is to be understood in the broadest sense in the context of the present process.
- acyl groups derived from aliphatic, araliphatic, aromatic or heterocyclic carboxylic acids or sulfonic acids, and in particular alkoxycarbonyl, aryloxycarbonyl and especially aralkoxycarbonyl groups.
- acyl groups are alkanoyl such as acetyl, propionyl, butyryl; Aralkanoyl, such as phenylacetyl; Aroyl such as benzoyl or toluyl; Aryloxyalkanoyl such as POA; Alkoxycarbonyl such as methoxycarbonyl, ethoxycarbonyl, 2,2,2-trichloroethoxycarbonyl, BOC, 2-iodoethoxycarbonyl; Aralkyloxycarbonyl such as CBZ ("carbobenzoxy"), 4-methoxybenzyloxycarbonyl, F OC; Arylsulfonyl such as phenylsulfonyl, Mtr, Pbf or Pmc.
- Preferred amino protecting groups are BOC and Mtr, furthermore CBZ, Fmoc, benzyl and acetyl.
- Reactions preferably protected by an indole protecting group Preferred are BOC or phenylsulfonyl. This protective group will be at the end of
- the phenylsulfonyl group is preferably cleaved off with trifluoroethanol or methanol in THF with the addition of an organic or inorganic base.
- Suitable bases are organic bases, such as DIPEA, triethylamine, dimethylamine, pyridine or quinoline. Preference is also the addition of an alkali or
- hydroxy protecting group is also well known and refers to groups which are suitable for protecting a hydroxy group from chemical reactions, but which are readily removable after the desired chemical reaction has been carried out at other sites on the molecule. Typical of such groups are the abovementioned unsubstituted or substituted aryl, aralkyl or acyl groups, and also alkyl groups.
- the nature and size of the hydroxy protecting groups is not critical because they are removed after the desired chemical reaction or reaction sequence; preferred are groups having 1-20, in particular 1-10 C-atoms. examples for
- Hydroxy protecting groups are i.a. Tetrahydropyranyl, tert-butoxycarbonyl, benzyl, p-nitrobenzoyl, p-toluenesulfonyl, tert-butyl and acetyl, with benzyl and tert-butyl being particularly preferred.
- Suitable inert solvents are preferably organic, for example carboxylic acids such as acetic acid, ethers such as tetrahydrofuran or dioxane, amides such as DMF, halogenated hydrocarbons such as dichloromethane, and also alcohols such as methanol, ethanol or isopropanol, and water.
- carboxylic acids such as acetic acid
- ethers such as tetrahydrofuran or dioxane
- amides such as DMF
- halogenated hydrocarbons such as dichloromethane
- alcohols such as methanol, ethanol or isopropanol, and water.
- reaction temperatures for the cleavage are suitably between about 0 and about 50 °, preferably between 15 and 30 ° (room temperature).
- the groups BOC, OBut, Pbf, Pmc and Mtr can, for. B. preferably cleaved with TFA in dichloromethane or with about 3 to 5 N HCl in dioxane at 15-30 °, the FMOC group with an about 5- to 50% solution of dimethylamine, diethylamine or piperidine in DMF at 15- 30 °.
- Hydrogenolytically removable protecting groups e.g CBZ or benzyl
- a catalyst e.g., a noble metal catalyst such as palladium, conveniently on a support such as carbon.
- Suitable solvents are those given above, in particular z.
- alcohols such as methanol or ethanol or amides such as DMF.
- the hydrogenolysis is usually carried out at temperatures between about 0 and 100 ° and pressures between about 1 and 200 bar, preferably at 20-30 ° and 1-10 bar.
- Hydrogenolysis of the CBZ group succeeds z.
- the invention furthermore relates to the compounds of the formula II
- Q is tert-butoxycarbonyl or phenylsulfonyl
- R is H, A or Cyc
- Ar is unsubstituted or substituted by [C (R 6 ) 2 ] p Het 1 or CN
- A is unbranched or branched alkyl having 1-8 C atoms, in which one or two nonadjacent CH and / or CH 2 groups may be replaced by N and / or O atoms and / or also 1-7 H Atoms can be replaced by F,
- n 0, 1 or 2
- p 0, 1, 2, 3 or 4
- the abovementioned compounds according to the invention can be used in their final non-salt form.
- the present invention also encompasses the use of these compounds in the form of their pharmaceutically acceptable salts, which can be derived from various organic and inorganic acids and bases according to procedures known in the art.
- Pharmaceutically acceptable salt forms of the compounds of formula I are for the most part prepared conventionally. If the compound of the formula I contains a carboxylic acid group, one of its suitable salts can be formed by reacting the compound with a suitable base to give the corresponding base addition salt.
- Such bases include, for example, alkali metal hydroxides, including potassium hydroxide, sodium hydroxide and lithium hydroxide; Alkaline earth metal hydroxides such as barium hydroxide and calcium hydroxide; Alkali metal alcoholates, e.g. Potassium ethanolate and sodium propanolate; and various organic bases such as piperidine, diethanolamine and N-methylglutamine.
- alkali metal hydroxides including potassium hydroxide, sodium hydroxide and lithium hydroxide
- Alkaline earth metal hydroxides such as barium hydroxide and calcium hydroxide
- Alkali metal alcoholates e.g. Potassium ethanolate and sodium propanolate
- various organic bases such as piperidine, diethanolamine and N-methylglutamine.
- acid addition salts can be formed by reacting these compounds with pharmaceutically acceptable organic and inorganic acids, e.g.
- Hydrogen halides such as hydrogen chloride, hydrogen bromide or hydrogen iodide, other mineral acids and their corresponding salts such as sulfate, nitrate or phosphate and the like, and alkyl and monoarylsulfonates such as ethanesulfonate, toluenesulfonate and benzenesulfonate, and other organic acids and their corresponding salts such as acetate, trifluoroacetate, tartrate, maleate , Succinate, citrate, benzoate, salicylate, ascorbate and the like.
- pharmaceutically acceptable acid addition salts of the compounds of formula I include the following: acetate, adipate, alginate, arginate, aspartate, benzoate,
- Benzenesulfonate (besylate), bisulfate, bisulfite, bromide, butyrate, camphorate, camphorsulfonate, caprylate, chloride, chlorobenzoate, citrate, cyclopentaneproprionate, digluconate, dihydrogenphosphate, dinitrobenzoate, dodecylsulphate, ethanesulphonate, fumarate, galacterate (from mucic acid), galacturonate, glucoheptanoate , Gluconate, glutamate, glycerophosphate, hemisuccinate, hemisulfate, heptanoate, hexanoate, hippurate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxyethanesulfonate, iodide, isethionate, Isobutyrate, lactate, lactobionate, malate, maleate, malonate, mandelate, meta
- the base salts of the compounds according to the invention include aluminum, ammonium, calcium, copper, iron (III), iron (II), lithium, magnesium, manganese (III), manganese (II), potassium , Sodium and zinc salts, but this should not be limiting.
- Preferred among the above salts are ammonium; the alkali metal salts sodium and potassium, and the alkaline earth metal salts calcium and magnesium.
- Salts of compounds of formula I derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary and tertiary amines, substituted amines, including naturally occurring substituted amines, cyclic amines and basic ion exchange resins, e.g.
- Arginine betaine, caffeine, chloroprocaine, choline, ⁇ , ⁇ '-dibenzylethylenediamine (benzathine), dicyclohexylamine, diethanolamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, Histidine, hydrabamine, iso-propylamine, lidocaine, lysine, meglumine, N-methyl-D-glucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethanolamine, triethylamine, trimethylamine, tripropylamine and tris- (hydroxymethyl) - methylamine (tromethamine), which is not intended to be limiting.
- Compounds of the present invention containing basic nitrogen-containing groups can be reacted with agents such as (CC 4 ) alkyl halides, eg, methyl, ethyl, isopropyl, and tert-butyl chloride, bromide, and iodide; Di (C 1 -C 4 ) alkyl sulfates, eg, dimethyl diethyl and diamyl sulfate; (C 10 -C 8 ) alkyl halides, eg decyl, dodecyl, lauryl, myristyl and stearyl chloride, bromide and iodide; and aryl (CC 4 ) alkyl halides, eg, benzyl chloride and phenethyl bromide, quaternize. With Such salts can be prepared both water-soluble and oil-soluble compounds of the invention.
- agents such as (CC 4 ) alkyl halides, e
- Preferred pharmaceutical salts include acetate, trifluoroacetate, besylate, citrate, fumarate, maleate, gluconate, hemisuccinate, hippurate, hydrochloride, hydrobromide, isethionate, mandelate, meglumine, nitrate, oleate, phosphonate, pivalate, sodium phosphate, Stearate, sulfate, sulfosalicylate, tartrate, thiomalate, tosylate and tromethamine, but this is not intended to be limiting.
- the acid addition salts of basic compounds of formula I are prepared by contacting the free base form with a sufficient amount of the desired acid to form the salt in a conventional manner.
- the free base can be regenerated by contacting the salt form with a base and isolating the free base in a conventional manner.
- the free base forms in some sense differ from their corresponding salt forms in terms of certain physical properties such as solubility in polar solvents; however, in the context of the invention, the salts otherwise correspond to their respective free base forms.
- the pharmaceutically acceptable base addition salts of the compounds of formula I are formed with metals or amines such as alkali metals and alkaline earth metals or organic amines.
- metals are sodium, potassium, magnesium and calcium.
- Preferred organic amines are ⁇ , ⁇ '-dibenzylethylenediamine, chloroprocaine, choline, diethanolamine, ethylenediamine, N-methyl-D-glucamine and procaine.
- the base addition salts of acidic compounds of the invention are prepared by contacting the free acid form with a sufficient amount of the desired base to form the salt in a conventional manner.
- the free acid can be regenerated by contacting the salt form with an acid and isolating the free acid in a conventional manner.
- the free Acid forms in some sense differ from their corresponding salt forms in terms of certain physical properties such as solubility in polar solvents; However, in the context of the invention, the salts otherwise correspond to their respective free acid forms.
- a compound according to the invention contains more than one group which can form such pharmaceutically acceptable salts, the invention also encompasses multiple salts.
- Typical multiple salt forms include, for example, bitartrate, diacetate, difumarate, dimeglumine, diphosphate, disodium and trihydrochloride, but this is not intended to be limiting.
- the term "pharmaceutically acceptable salt” in the present context means an active ingredient which contains a compound of the formula I in the form of one of its salts, especially if this salt form is the active ingredient in the Imparts improved pharmacokinetic properties to the free form of the active ingredient or any other salt form of the active ingredient which has previously been used.
- the pharmaceutically acceptable salt form of the active substance may also first impart a desired pharmacokinetic property to this active ingredient which it has not previously possessed, and may even positively influence the pharmacodynamics of this active ingredient in terms of its therapeutic efficacy in the body.
- the invention furthermore relates to medicaments comprising at least one compound of the formula I and / or pharmaceutically usable salts, tautomers and stereoisomers thereof, including mixtures thereof in all ratios, and optionally excipients and / or adjuvants.
- compositions may be presented in the form of dosage units containing a predetermined amount of active ingredient per unit dose.
- a unit may, for example, 0.5 mg to 1 g, preferably 1 mg to 700 mg, particularly preferably 5 mg to 100 mg of a compound according to the invention depending on the disease condition being treated, the route of administration and the age, weight and condition of the patient, or pharmaceutical formulations may be presented in the form of dosage units containing a predetermined amount of active ingredient per unit dose.
- Preferred dosage unit formulations are those containing a daily or partial dose as indicated above or a corresponding fraction of an active ingredient.
- such pharmaceutical formulations can be prepared by any of the methods well known in the pharmaceutical art.
- compositions may be administered by any suitable route, for example, oral (including buccal or sublingual), rectal, nasal, topical (including buccal, sublingual or transdermal), vaginal or parenteral (including subcutaneous, intramuscular, intravenous or intradermal). Ways, adapt.
- Such formulations may be prepared by any method known in the pharmaceutical art, for example, by bringing the active ingredient together with the carrier (s) or excipients.
- compositions adapted for oral administration may be administered as separate units, e.g. Capsules or tablets; Powder or granules; Solutions or suspensions in aqueous or non-aqueous liquids;
- the active ingredient component in the case of oral administration in the form of a tablet or capsule, can be mixed with an oral, non-toxic and pharmaceutically acceptable inert carrier such as ethanol, glycerine, water and the like. combine. Powders are prepared by comminuting the compound to a suitable fine size and mixing it with a similarly comminuted pharmaceutical excipient, such as an edible carbohydrate, such as wise starch or mannitol is mixed. A flavor, preservative, dispersant and dye may also be present.
- an oral, non-toxic and pharmaceutically acceptable inert carrier such as ethanol, glycerine, water and the like.
- Powders are prepared by comminuting the compound to a suitable fine size and mixing it with a similarly comminuted pharmaceutical excipient, such as an edible carbohydrate, such as wise starch or mannitol is mixed.
- a flavor, preservative, dispersant and dye may also be present.
- Capsules are made by preparing a powder mix as described above and filling shaped gelatin casings therewith.
- Lubricants such as e.g. highly disperse silica, talc, magnesium stearate, calcium stearate or polyethylene glycol in solid form can be added to the powder mixture before the filling process.
- a disintegrants or solubilizers e.g. Agar-agar, calcium carbonate or sodium carbonate may also be added to improve the availability of the drug after ingestion of the capsule.
- suitable bonding, lubricating and disintegrants as well as dyes can also be incorporated into the mixture.
- Suitable binders include starch, gelatin, natural sugars such as glucose or beta-lactose, corn sweeteners, natural and synthetic gums such as acacia, tragacanth or sodium alginate, carboxymethyl cellulose, polyethylene glycol, waxes, and the like.
- the lubricants used in these dosage forms include sodium oleate, sodium stearate, magnesium stearate, sodium benzoate, sodium acetate, sodium chloride, and the like.
- the disintegrating agents include, but are not limited to, starch, methyl cellulose, agar, bentonite, xanthan gum and the like.
- the tablets are formulated by, for example, preparing a powder mixture, granulating or dry pressing, adding a lubricant and a disintegrating agent and pressing the whole into tablets.
- a powder mixture is prepared by dissolving the appropriately comminuted compound with a diluent or base as described above and optionally with a binder such as carboxymethyl cellulose, an alginate, gelatin or polyvinylpyrrolidone, a dissolution reducer such as paraffin, a resorption accelerator, such as a quaternary salt and / or an absorbent, such as bentonite, kaolin or dicalcium phosphate.
- the powder mixture can be granulated by adding it with a binder, such as syrup, starch paste, Acadia slime or solutions of cellulose or Wets polymer material and pressed through a sieve.
- the powder mixture can be run through a tabletting machine to produce non-uniformly shaped lumps which are broken up into granules.
- the granules may be greased by the addition of stearic acid, a stearate salt, talc or mineral oil to adhere to the
- the compounds of the invention can also be used with a
- a transparent or opaque protective layer consisting of a shellac sealant, a layer of sugar or polymeric material, and a glossy layer of wax may be present. Dyes can be added to these coatings in order to differentiate between different dosage units.
- Oral fluids e.g. Solution, syrups and elixirs may be prepared in unit dosage form such that a given quantity contains a predetermined amount of the compound.
- Syrups can be prepared by dissolving the compound in an appropriate taste aqueous solution while preparing elixirs using a non-toxic alcoholic vehicle.
- Suspensions can be formulated by dispersing the compound in a non-toxic vehicle.
- Solubilizers and emulsifiers e.g. ethoxylated isostearyl alcohols and polyoxyethylene sorbitol ethers, preservatives, flavoring additives such as e.g. Peppermint oil or natural sweeteners or saccharin or other artificial sweeteners, etc. can also be added.
- the unit dosage formulations for oral administration may optionally be encapsulated in microcapsules.
- the formulation may also be prepared to prolong or retard the release, such as by coating or embedding particulate material in polymers, wax, and the like.
- the compounds of formula I as well as their pharmaceutically acceptable salts, tautomers and stereoisomers can also be administered in the form of liposome delivery systems, such as small unilamellar vesicles, large unilamellar vesicles and multilamellar vesicles.
- Liposomes can be formed from various phospholipids such as cholesterol, stearylamine or phosphatidylcholines.
- the compounds of formula I as well as their pharmaceutically acceptable salts, tautomers and stereoisomers can also be delivered using monoclonal antibodies as individual carriers to which the compound molecules are coupled.
- the compounds can also be coupled with soluble polymers as targeted drug carriers.
- Such polymers may include polyvinylpyrrolidone, pyran copolymer, polyhydroxypropylmethacrylamidephenol, polyhydroxyethylaspartamidephenol or polyethyleneoxidepolylysine substituted with palmitoyl radicals.
- the compounds can be attached to a class of biodegradable polymers suitable for the controlled release of a drug, e.g.
- Polylactic acid Polyepsilon caprolactone, polyhydroxybutyric acid, polyorthoesters, polyacetals, polydihydroxypyrans, polycyanoacrylates and cross-linked or amphipathic block copolymers of hydrogels.
- compositions adapted for transdermal administration may be presented as discrete patches for prolonged, intimate contact with the epidermis of the recipient.
- the drug may be delivered from the patch by iontophoresis as generally described in Pharmaceutical Research, 3 (6), 318 (1986).
- compositions adapted for topical administration may be formulated as ointments, creams, suspensions, lotions, powders, solutions, pastes, gels, sprays, aerosols or oils.
- the formulations are preferably applied as a topical ointment or cream.
- the active ingredient can be used with either a paraffinic or water miscible cream base.
- the active ingredient can be formulated into a cream with an oil-in-water cream base or a water-in-oil base.
- Formulations include eye drops wherein the active ingredient is dissolved or suspended in a suitable carrier, especially an aqueous solvent.
- compositions adapted for topical application in the mouth include lozenges, troches and mouthwashes.
- compositions adapted for rectal administration may be presented in the form of suppositories or enemas.
- compositions adapted for nasal administration in which the vehicle is a solid contain a coarse powder having a particle size, for example, in the range of 20-500 microns, which is administered in the manner in which snuff is received, i. by rapid inhalation via the nasal passages from a container held close to the nose with the powder.
- compositions adapted for administration by inhalation include fine particulate dusts or mists produced by various types of pressurized dosing dispensers with aerosols, nebulizers or nebulizers
- Insufflators can be generated.
- Pharmaceutical formulations adapted for vaginal administration may be presented as pessaries, tampons, creams, gels, pastes, foams or spray formulations.
- compositions adapted for parenteral administration include aqueous and nonaqueous sterile injection solutions containing antioxidants, buffers, bacteriostats and solutes which render the formulation isotonic with the blood of the recipient to be treated; and aqueous and non-aqueous sterile suspensions which may contain suspending agents and thickeners.
- the formulations may be administered in single or multiple dose containers, e.g. sealed vials and vials, and stored in the freeze-dried (lyophilized) state so that only the addition of the sterile carrier liquid, e.g. Water for injections, needed immediately before use.
- Injection solutions and suspensions prepared by formulation may consist of sterile powders, granules and
- formulations may include other means conventional in the art with respect to the particular type of formulation; so for example for the oral
- a therapeutically effective amount of a compound of formula I depends on a number of factors, including e.g. the age and weight of the animal, the exact condition of the disease requiring treatment, as well as its severity, the nature of the formulation and the route of administration, and will ultimately be determined by the doctor or veterinarian.
- an effective amount of a compound of the invention is useful for the treatment of neoplastic growth, e.g. Colon or breast carcinoma, generally in the range of 0.1 to 100 mg / kg body weight of the recipient
- (Mammal) per day and more typically in the range of 1 to 10 mg / kg of body weight per day.
- the actual amount per day is usually between 70 and 700 mg, which may be given as a single dose per day or more commonly in a number of divided doses (such as two, three, four, five or six) per day, so that
- Total daily dose is the same.
- An effective amount of a salt or solvate or a physiologically functional derivative thereof can be determined as a proportion of the effective amount of the compound of the invention per se. It can be assumed that similar dosages are suitable for the treatment of the other disease states mentioned above.
- the invention furthermore relates to medicaments comprising at least one compound of the formula I and / or pharmaceutically acceptable salts, tautomers and stereoisomers thereof, including mixtures thereof in all ratios, and at least one further active pharmaceutical ingredient.
- the invention is also a set (kit), consisting of separate
- the kit contains suitable containers, such as boxes or boxes, individual bottles, bags or ampoules.
- the set may e.g. containing separate ampoules each containing an effective amount of a compound of formula I and / or its pharmaceutically acceptable salts, tautomers and stereoisomers, including mixtures thereof in all proportions,
- the invention relates to the compounds of formula I for use in the treatment of cancer, septic shock, primary open angle glaucoma (POAG), hyperplasia, rheumatoid arthritis, psoriasis, atherosclerosis, retinopathy, osteoarthritis, endometriosis, chronic inflammation and / or neurodegenerative diseases such as Alzheimer's disease ,
- the invention relates to the use of compounds of formula I for the manufacture of a medicament for the treatment of cancer, septic shock, primary open angle glaucoma (POAG), hyperplasia, rheumatoid arthritis, psoriasis, atherosclerosis, retinopathy, osteoarthritis, endometriosis, chronic inflammation and / or neurodegenerative Diseases such as Alzheimer's disease.
- POAG primary open angle glaucoma
- hyperplasia rheumatoid arthritis
- psoriasis psoriasis
- atherosclerosis retinopathy
- osteoarthritis retinopathy
- endometriosis chronic inflammation
- neurodegenerative Diseases such as Alzheimer's disease.
- the invention relates to a method of treating a mammal suffering from a disease arising from cancer, septic shock, primary open angle glaucoma (POAG), hyperplasia, rheumatoid arthritis, psoriasis, atherosclerosis, retinopathy, osteoarthritis, endometriosis, chronic inflammation and / or neurodegenerative diseases such as Alzheimer's disease, the method comprising administering to a mammal a therapeutically effective amount of a compound of formula I.
- POAG primary open angle glaucoma
- the invention further relates to the compounds of formula I for use in the treatment of cancer, septic shock, primary open angle glaucoma (POAG), hyperplasia, atherosclerosis, retinopathy, osteoarthritis, endometriosis, chronic inflammation, neurodegenerative diseases, rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), Sjörgrens Syndrome, Aicardi-Goutieres Syndrome Lupus Chilblain, Retinal Vasculopathy, Cerebral Leukodystrophy (RVCL), Systemic Sclerosis, Myositis, Psoriasis, Chronic Obstructive Pulmonary Disease (CPD), Inflammatory Bowel Disease (IBD), Obesity, Insulin Resistance, Type 2 diabetes (NIDDM) and / or metabolic syndrome
- POAG primary open angle glaucoma
- POAG primary open angle glaucoma
- hyperplasia atherosclerosis
- retinopathy retinopathy
- the host or patient may be of any mammalian species, e.g. A primate species, especially humans; Rodents, including mice, rats and hamsters; Rabbits; Horses, cattle, dogs, cats, etc. Animal models are of interest for experimental studies, providing a model for the treatment of human disease.
- the susceptibility of a particular cell to treatment with the compounds of the invention can be determined by testing in vitro.
- a culture of the cell is combined with a compound of the invention at various concentrations for a period of time sufficient to allow the active agents, such as anti-IgM, to induce a cellular response, such as expression of a surface marker, usually between about one hour and one week.
- a cellular response such as expression of a surface marker
- cultured cells from blood or a biopsy sample can be used. The amount of surface marker expressed is assessed by flow cytometry, with specific
- Antibodies are used that recognize the marker.
- the dose will vary depending on the specific compound used, the specific disease, the patient status, etc. Typically, one therapeutic dose will be sufficient to treat the unwanted cell population in the human body
- the treatment is generally continued until there is a significant reduction, e.g. At least about 50% reduction in cell load and can be continued until essentially no more unwanted cells are detected in the body.
- Suitable models or model systems have been developed by various scientists, eg Cell culture models (eg Khwaja et al., EMBO, 1997, 16, 2783-93) and models of transgenic animals (eg White et al., Oncogene, 2001, 20, 7064-7072).
- interacting compounds can be used to modulate the signal (eg, Stephens et al., Biochemical J., 2000, 351, 95-105).
- the compounds according to the invention can also be used as reagents for testing kinase-dependent signal transduction pathways in animals and / or cell culture models or in the clinical diseases mentioned in this application.
- kinase activity is a technique well known to those skilled in the art.
- Generic Assay Systems for Determining Kinase Activity with Substrates e.g. Histone (eg Alessi et al., FEBS Lett. 1996, 399, 3, pages 333-338) or the myelin basic protein are described in the literature (eg Campos-Gonzalez, R. and Glenney, Jr., JR 1992, J. Bio / Chem. 267, page 14535).
- Non-radioactive ELISA assay use specific phospho-antibodies (Phospho-AK).
- Phospho-AK binds only the phosphorylated substrate. This binding is detectable by chemiluminescence with a second peroxidase-conjugated anti-sheep antibody (Ross et al., 2002, Biochem. J.).
- the present invention encompasses the use of the compounds of the formula I and / or their physiologically acceptable salts, tautomers and solvates for the manufacture of a medicament for the treatment or prevention of cancer.
- Preferred carcinomas for the treatment are from the brain carcinoma, genitourinary tract carcinoma, carcinoma of the lymphatic system, gastric carcinoma, laryngeal carcinoma and lung carcinoma colorectal cancer.
- Another group of preferred forms of cancer are monocytic leukemia, lung adenocarcinoma, small cell lung carcinoma, pancreatic cancer, glioblastoma and breast carcinoma.
- a therapeutically effective amount of a compound of the invention is administered.
- the therapeutic amount depends on the particular disease and can be determined by the skilled person without great effort.
- a disease wherein the cancerous disease is a solid tumor.
- the solid tumor is preferably selected from the group of squamous cell tumors, bladder, stomach, kidney, head and neck,
- the solid tumor is furthermore preferably selected from the group lung adenocarcinoma, small cell lung carcinoma, pancreatic cancer,
- Glioblastomas colon carcinoma and breast carcinoma.
- a tumor of the blood and immune system preferably for the treatment of a tumor selected from the group of acute myelotic leukemia, the chronic myeloid Leukemia, acute lymphocytic leukemia and / or chronic lymphocytic leukemia.
- the invention furthermore relates to the use of the compounds according to the invention for the treatment of bone pathologies, the bone pathology originating from the group osteosarcoma, osteoarthritis and rickets.
- the compounds of formula I may also be coadministered with other well-known therapeutics selected for their particular suitability for the condition being treated.
- the present compounds are also useful for combination with known anticancer agents.
- known anticancer agents include the following:
- the present compounds are particularly suitable for co-administration with radiotherapy.
- Estrogen receptor modulators refers to compounds that interfere with or inhibit the binding of estrogen to the receptor, regardless of how it is done. "The estrogen receptor modulators include, for example, tamoxifen, raloxifene, idoxifen, LY353381, LY1 17081, toremifene,
- Fulvestrant 4- [7- (2,2-dimethyl-1-oxopropoxy-4-methyl-2- [4- [2- (1-piperidinyl) ethoxy] phenyl] -2H-1-benzopyran-3-yl ] phenyl-2,2-dimethylpropanoate, 4,4'-dihydroxybenzophenone-2,4-dinitrophenylhydrazone and SH646, but this is not intended to be limiting.
- Androgen receptor modulators refers to compounds that interfere with or inhibit the binding of androgens to the receptor, regardless of how this occurs, and the androgen receptor modulators include, for example, finasteride and other 5a-reductase inhibitors, nilutamide, flutamide, bicalutamide , Liarozole and abiraterone acetate.
- Retinoid receptor modulators refers to compounds that interfere with or inhibit the binding of retinoids to the receptor, independently 'How this happens.
- Such retinoid receptor modulators include, for example, bexarotene, tretinoin, 13-cis-retinoic acid, 9-cis-retinoic acid, a-difluoromethyl-ornithine, ILX23-7553, trans-N- (4'-hydroxyphenyl) -retinamide and N-4-carboxyphenyl- retinamide.
- Cytotoxic agents refers to compounds that cause cell death primarily through direct action on cell function, or that inhibit or interfere with cell myosis, including alkylating agents, tumor necrosis factors, intercalators, microtubulin inhibitors, and topoisomerase inhibitors.
- the cytotoxic agents include, for example, tirapazimine, Sertenef, cachectin, ifosfamide, tasonermine, lonidamine, carboplatin, altretamine, prednimustine, dibromodulcite, ranimustine, fotemustine, nedaplatin, oxaliplatin, temozolomide, heptaplatin, estramustine, improvisulfan-tosylate, trofosfamide, nimustine, dibrospidium chloride, Pumitepa, Lobaplatin, Satraplatin, Profiromycin, Cisplatin, Irofulvene, Dexifosfamide, cis-Amine dichloro (2-methylpyridine) platinum, Benzylguanine, Glufosfamide, GPX100, (trans, trans, trans) -bis-mu (hexane-1, 6 -diamine) -mu- [di
- microtubulin inhibitors include, for example, paclitaxel, vindesine sulfate, 3 ', 4-dideshydro-4'-deoxy-8'-norvincaleukoblastin, docetaxol, rhizoxin, dolastatin, mivobulinisethionate, auristatin, cemadotin, RPR109881, BMS184476, vinflunine , Cryptophycin, 2,3,4,5,6-pentafluoro-N- (3-fluoro-4-methoxyphenyl) benzenesulfonamide, anhydrovinblastine, N, N-dimethyl-L-valyl-L-valyl-N-methyl-L- valyl-L-prolyl-L-proline t-butylamide, TDX258 and BMS 88797.
- paclitaxel vindesine sulfate
- 3 ' 4-dideshydro-4'-deoxy-8'-
- Topoisomerase inhibitors are, for example, topotecan, hycaptamine, irinotecan, rubitecane, 6-ethoxypropionyl-3 ', 4'-O-exo-benzylidene-chartreusine, 9-methoxy-N, N-dimethyl-5-nitropyrazolo [3,4; 5-kl] acridine-2- (6H) propanamine, 1-amino-9-ethyl-5-fluoro-2,3-dihydro-9-hydroxy-4-methyl-1H, 12H-benzo [de] pyrano [ 3 ', 4': b, 7] indolizino [1, 2b] - quinoline-10, 13 (9H, 15H) -dione, lurtotecan, 7- [2- (N-isopropylamino) ethyl] - (20S) -camptothecin, BNP1350, BNPI1 100, BN80915,
- Antiproliferative agents include antisense RNA and DNA oligonucleotides such as G3139, ODN698, RVASKRAS, GEM231 and ⁇ 300, and antimetabolites such as enocitabine, carmofur, tegafur, pentostatin, doxifluridine, trimetrexate, fludarabine, capecitabine, galocitabine, Cytarabine ocfosfate, Fosteabin Sodium Hydrate, Raltitrexed, Paltitrexide, Emitefur, Tiazofurin, Decitabine, Nolatrexed, Pemetrexed, Nelzarabine, 2'-Deoxy-2'-met yiidencytidine, 2'-Fluoromethylene-2'-deoxycytidine, N- [5- (2,3-Dihydrobenzofuryl) sulfonyl] -N '- (3,4-dichlorophenyl) urea,
- antiproliferative agents also include other monoclonal antibodies against growth factors than those already mentioned under the “angiogenesis inhibitors”, such as trastuzumab, as well as tumor-suppressing genes, such as p53, which can be delivered via recombinant virus-mediated gene transfer (see eg US Pat 6,069, 134). Test for the inhibition of ⁇
- the kinase assay is performed as a 384-well flashplate assay (e.g., for topcount measurement).
- the kinase assay is performed as a 384-well flashplate assay (e.g., for topcount measurement).
- Tinned binding kinase (TBK1), 800 nM biotinylated MELK-derived peptide (biotin-Ah-Ah-AKPKGNKDYHLQTCCGSLAYRRR) and 10 ⁇ M ATP (spiked with 0.25 pCi 33 P-ATP / well) are added in a total volume of 50 pl (10mM MOPS, 10mM Mg-acetate, 0.1mM EGTA, 1mM DTT, 0.02% Brij35, 0.1% BSA, pH 7.5) or without test compound incubated at 30 ° C for 120 min. The reaction is stopped with 25 ⁇ M 200 mM EDTA.
- TBK1 and ⁇ are mainly known as key substances in the innate immune response
- recent findings indicate a role for TBK1 and ⁇ in Ras-induced oncogenic transformation.
- TBK1 was identified as a RalB effector in the pathway of the Ras-like (Ral) guanine nucleotide exchange factor (GEF) required for Ras-induced transformation.
- GEF Ras-like guanine nucleotide exchange factor
- TB 1 directly activates IRF3, which homodimerizes upon phosphorylation and shifts to the nucleus, where it activates processes related to inflammation, immune regulation, cell survival, and proliferation.
- This assay was developed to evaluate the potency / potency of ⁇ 1 / ⁇ inhibitor compounds based on immunocytochemical detection of nuclear localized phospho-IRF3, a target immediately after TBK1.
- dsRNA double-stranded RNA
- TLR3 toll-like receptor 3
- MDA-MB-468 cells are detached with HyQ-Tase, counted and placed on a 384 well TC-bottomed tablet with a density of 10,000 cells per well in a total volume of 35 ⁇ l complete medium seeded. Alternatively, the cells are seeded directly from frozen glass vials.
- 3rd day The primary antibody is washed away, an AlexaFluor488 conjugated secondary added, the cells are contrast stained with propidium iodide, followed by image acquisition on an IMX Ultra High Content Reader.
- Plating medium culture medium:
- Plates 384-well bottom cell culture plates with black / transparent
- Place plate on ice Permeabilize cells Rapidly add 100 ⁇ -20 ° C cold eOH
- Block unspecific binding Add 30 ⁇ L of 0% goat serum in PBS / 2% BSA
- Image capture on IMX Ultra (Metaexpress 3.1 scan settings TBK_10x_pin8) Image Analysis (Metaexpress 3.1. ⁇ Cell scoring>, TBK1 Cell Scoring) Data analysis and reporting with assay explorer
- reaction mixture is heated to 80 ° C and stirred at this temperature for 18 hours.
- the reaction mixture is cooled to room temperature and treated with water.
- the resulting precipitate is filtered off with suction, washed with water and dried under reduced pressure: 5- [1- (benzenesulfonyl) -5-bromopyrrolo [2,3-b] pyridin-3-yl] -2-tetrahydropyran-4-yloxy benzonitrile as light gray crystals; HPLC / MS (A): 3.32 min, [M + H] 538/540;
- the reaction mixture is heated to 80 ° C and stirred at this temperature for 4 days.
- the reaction mixture is cooled to room temperature and partitioned between water and dichloromethane.
- the organic phase is dried over sodium sulfate and evaporated.
- the residue is chromatographed on a silica gel column with cyclohexane / ethyl acetate: 5- [1- (benzenesulfonyl) -5- (4-cyanophenyl) pyrrolo [2,3-b] pyridin-3-yl] -2-tetrahydropyran-4 -yloxybenzonitrile as colorless crystals; HPLC / S (A): 3.55 min, [+ H] 561.
- reaction mixture 18 Sodium bicarbonate and 1 ml of water, the reaction mixture 18
- reaction mixture is stirred for 2 hours at room temperature, evaporated and then chromatographed on a silica gel column with ethyl acetate / methanol as eluent: 5- [1- (benzenesulfonyl) -5- (2-morpholinoethoxy) pyrrolo [2,3-b] pyridine-3 -yl] -2-tetrahydropyran-4-yloxybenzonitrile as a white foam; HPLC / MS (A): 2.13 min, [M + H] 589.
- reaction mixture is evaporated and the residue on a silica gel column with dichloromethane / methanol as the eluent chromatographed: 5- [5- (2-morpholinoethoxy) -1H-pyrrolo [2,3-b] pyridin-3-yl] -2-tetrahydropyran-4-yloxybenzonitrile as a light yellow powder; HPLC / MS (A): 1.75 min, [M + H] 449;
- Example A Injection jars
- a solution of 100 g of an active ingredient according to the invention and 5 g of disodium hydrogen phosphate is adjusted to pH 6.5 in 2 l of bidistilled water with 2N hydrochloric acid, filtered sterile, filled into injection jars, lyophilized under sterile conditions and closed under sterile conditions. Each injection jar contains 5 mg of active ingredient.
- Example C A mixture of 20 g of an active ingredient according to the invention is melted with 100 g of soya lecithin and 1400 g of cocoa butter, poured into molds and allowed to cool. Each suppository contains 20 mg of active ingredient.
- Example C Solution
- a solution of 1 g of an active ingredient according to the invention, 9.38 g of NaH2P04 ⁇ 2 H2O, 28.48 g Na2HP04 x 12 H2O and 0.1 g of benzalkonium chloride in 940 ml of double-distilled water is prepared. Adjust to pH 6.8, make up to 1 liter and sterilize by irradiation. This solution can be used in the form of eye drops.
- 500 mg of an active ingredient according to the invention are mixed with 99.5 g of Vaseline under aseptic conditions.
- a mixture of 1 kg of active ingredient, 4 kg of lactose, 1, 2 kg of potato starch, 0.2 kg of talc and 0.1 kg of magnesium stearate is compressed in the usual way into tablets, such that each tablet contains 10 mg of active ingredient.
- Tablets are pressed analogously to Example E, which are then coated in the usual way with a coating of sucrose, potato starch, talc, tragacanth and dye.
- a solution of 1 kg of an active ingredient according to the invention in 60 l of bidistilled water is sterile filtered, filled into ampoules, lyophilized under sterile conditions and sealed sterile. Each vial contains 10 mg of active ingredient.
Abstract
Description














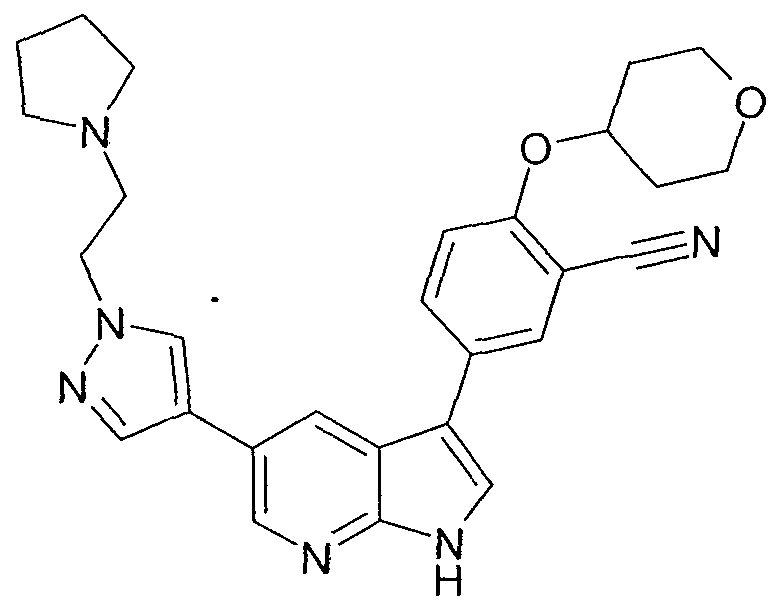

















Claims




Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2012342891A AU2012342891B2 (en) | 2011-11-22 | 2012-10-30 | 3-Cyanoaryl-1H-pyrrolo[2.3-b]pyridine derivatives |
| US14/359,705 US9102675B2 (en) | 2011-11-22 | 2012-10-30 | 3-Cyanoaryl-1H-pyrrolo[2,3-B] pyridine derivatives |
| JP2014542726A JP6096797B2 (ja) | 2011-11-22 | 2012-10-30 | 3−シアノアリール−1H−ピラゾロ[2.3−b]ピリジン誘導体 |
| CA2856357A CA2856357C (en) | 2011-11-22 | 2012-10-30 | 3-cyanoaryl-1h-pyrrolo[2,3-b]pyridine derivatives |
| EP12780423.5A EP2794602B1 (de) | 2011-11-22 | 2012-10-30 | 3-cyanaryl-1h-pyrazolo-(2,3-b-)pyridinderivate |
| CN201280056772.6A CN103946223B (zh) | 2011-11-22 | 2012-10-30 | 3‑氰基芳基‑1H‑吡咯并[2,3‑b]吡啶衍生物 |
| ES12780423.5T ES2661256T3 (es) | 2011-11-22 | 2012-10-30 | Derivados de 3-cianaril-1H-pirrolo[2,3-b]piridina |
| IL232717A IL232717B (en) | 2011-11-22 | 2014-05-20 | 3-cyanoaryl-h1-pyrrolo[b-3,2]pyridine derivatives |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102011119127A DE102011119127A1 (de) | 2011-11-22 | 2011-11-22 | 3-Cyanaryl-1H-pyrrolo[2.3-b]pyridin-Derivate |
| DE102011119127.9 | 2011-11-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013075785A1 true WO2013075785A1 (de) | 2013-05-30 |
Family
ID=47115757
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2012/004543 WO2013075785A1 (de) | 2011-11-22 | 2012-10-30 | 3-cyanaryl-1h-pyrrolo[2,3-b]pyridin-derivate |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US9102675B2 (de) |
| EP (1) | EP2794602B1 (de) |
| JP (1) | JP6096797B2 (de) |
| CN (1) | CN103946223B (de) |
| AR (1) | AR088925A1 (de) |
| AU (1) | AU2012342891B2 (de) |
| CA (1) | CA2856357C (de) |
| DE (1) | DE102011119127A1 (de) |
| ES (1) | ES2661256T3 (de) |
| IL (1) | IL232717B (de) |
| WO (1) | WO2013075785A1 (de) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015187684A1 (en) * | 2014-06-03 | 2015-12-10 | Gilead Sciences, Inc. | Tank-binding kinase inhibitor compounds |
| US9433622B2 (en) | 2013-02-21 | 2016-09-06 | Case Western Reserve University | Pyrimidine compounds useful in the treatment of diseases mediated by IKKE and/or TBK1 mechanisms |
| WO2017102091A1 (en) | 2015-12-18 | 2017-06-22 | Bayer Pharma Aktiengesellschaft | Heteroarylbenzimidazole compounds |
| WO2017207534A1 (en) | 2016-06-03 | 2017-12-07 | Bayer Pharma Aktiengesellschaft | Substituted heteroarylbenzimidazole compounds |
| DE102016113714A1 (de) | 2016-07-26 | 2018-02-01 | Rosa Karl | Transfektionsverfahren mit nicht-viralen Genliefersystemen |
| US10040781B2 (en) | 2014-09-26 | 2018-08-07 | Gilead Sciences, Inc. | Tank-binding kinase inhibitor compounds |
| US10316049B2 (en) | 2015-12-17 | 2019-06-11 | Gilead Sciences, Inc. | Tank-binding kinase inhibitor compounds |
| WO2020210366A1 (en) * | 2019-04-09 | 2020-10-15 | Plexxikon Inc. | Condensed azines for ep300 or cbp modulation and indications therefor |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20180021702A (ko) * | 2015-06-29 | 2018-03-05 | 메르크 파텐트 게엠베하 | TBK/IKKε 억제제 화합물 및 이의 용도 |
| GB201702947D0 (en) | 2017-02-23 | 2017-04-12 | Domainex Ltd | Novel compounds |
Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6069134A (en) | 1991-03-06 | 2000-05-30 | Board Of Regents, The University Of Texas System | Methods and compositions comprising DNA damaging agents and p53 |
| WO2000050032A1 (en) | 1999-02-25 | 2000-08-31 | Pharmacia & Upjohn S.P.A. | Antitumour synergistic composition |
| WO2004055005A1 (en) | 2002-12-17 | 2004-07-01 | Astrazeneca Ab | Novel compounds having selective inhibiting effect at gsk3 |
| WO2005095400A1 (en) | 2004-03-30 | 2005-10-13 | Vertex Pharmaceuticals Incorporated | Azaindoles useful as inhibitors of jak and other protein kinases |
| WO2006015123A1 (en) | 2004-07-27 | 2006-02-09 | Sgx Pharmaceuticals, Inc. | Pyrrolo-pyridine kinase modulators |
| WO2007129044A1 (en) | 2006-05-03 | 2007-11-15 | Astrazeneca Ab | Thiazole derivatives and their use as anti-tumour agents |
| WO2008124848A1 (en) * | 2007-04-10 | 2008-10-16 | Sgx Pharmaceuticals, Inc. | Fused ring heterocycle kinase modulators |
| WO2009030890A1 (en) | 2007-09-03 | 2009-03-12 | University Court Of The University Of Dundee | Pyrimidine compounds for the treatment of cancer, septic shock and/or primary open angle glaucoma |
| WO2009054941A1 (en) | 2007-10-25 | 2009-04-30 | Merck & Co., Inc. | Therapeutic compounds |
| WO2009053737A2 (en) | 2007-10-25 | 2009-04-30 | Astrazeneca Ab | Pyridine and pyrazine derivatives useful in the treatment of cell proliferative disorders |
| WO2009122180A1 (en) | 2008-04-02 | 2009-10-08 | Medical Research Council | Pyrimidine derivatives capable of inhibiting one or more kinases |
| WO2010100431A1 (en) | 2009-03-04 | 2010-09-10 | Medical Research Council Technology | Pyrrolopyrimidines used as kinase inhibitors |
| WO2011008915A1 (en) | 2009-07-15 | 2011-01-20 | Abbott Laboratories | Pyrrolopyridine inhibitors of kinases |
| WO2011046970A1 (en) | 2009-10-12 | 2011-04-21 | Myrexis, Inc. | Amino - pyrimidine compounds as inhibitors of tbkl and/or ikk epsilon |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9718913D0 (en) * | 1997-09-05 | 1997-11-12 | Glaxo Group Ltd | Substituted oxindole derivatives |
| MY145634A (en) * | 2003-12-29 | 2012-03-15 | Bristol Myers Squibb Co | Pyrrolotriazine compounds as kinase inhibitors |
| CA2628455A1 (en) * | 2005-11-10 | 2007-05-24 | Schering Corporation | Imidazopyrazines as protein kinase inhibitors |
-
2011
- 2011-11-22 DE DE102011119127A patent/DE102011119127A1/de not_active Withdrawn
-
2012
- 2012-10-30 CN CN201280056772.6A patent/CN103946223B/zh not_active Expired - Fee Related
- 2012-10-30 AU AU2012342891A patent/AU2012342891B2/en not_active Ceased
- 2012-10-30 JP JP2014542726A patent/JP6096797B2/ja not_active Expired - Fee Related
- 2012-10-30 EP EP12780423.5A patent/EP2794602B1/de not_active Not-in-force
- 2012-10-30 WO PCT/EP2012/004543 patent/WO2013075785A1/de active Application Filing
- 2012-10-30 US US14/359,705 patent/US9102675B2/en not_active Expired - Fee Related
- 2012-10-30 ES ES12780423.5T patent/ES2661256T3/es active Active
- 2012-10-30 CA CA2856357A patent/CA2856357C/en active Active
- 2012-11-21 AR ARP120104361A patent/AR088925A1/es not_active Application Discontinuation
-
2014
- 2014-05-20 IL IL232717A patent/IL232717B/en active IP Right Grant
Patent Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6069134A (en) | 1991-03-06 | 2000-05-30 | Board Of Regents, The University Of Texas System | Methods and compositions comprising DNA damaging agents and p53 |
| WO2000050032A1 (en) | 1999-02-25 | 2000-08-31 | Pharmacia & Upjohn S.P.A. | Antitumour synergistic composition |
| WO2004055005A1 (en) | 2002-12-17 | 2004-07-01 | Astrazeneca Ab | Novel compounds having selective inhibiting effect at gsk3 |
| WO2005095400A1 (en) | 2004-03-30 | 2005-10-13 | Vertex Pharmaceuticals Incorporated | Azaindoles useful as inhibitors of jak and other protein kinases |
| WO2006015123A1 (en) | 2004-07-27 | 2006-02-09 | Sgx Pharmaceuticals, Inc. | Pyrrolo-pyridine kinase modulators |
| WO2007129044A1 (en) | 2006-05-03 | 2007-11-15 | Astrazeneca Ab | Thiazole derivatives and their use as anti-tumour agents |
| WO2008124848A1 (en) * | 2007-04-10 | 2008-10-16 | Sgx Pharmaceuticals, Inc. | Fused ring heterocycle kinase modulators |
| WO2009030890A1 (en) | 2007-09-03 | 2009-03-12 | University Court Of The University Of Dundee | Pyrimidine compounds for the treatment of cancer, septic shock and/or primary open angle glaucoma |
| WO2009054941A1 (en) | 2007-10-25 | 2009-04-30 | Merck & Co., Inc. | Therapeutic compounds |
| WO2009053737A2 (en) | 2007-10-25 | 2009-04-30 | Astrazeneca Ab | Pyridine and pyrazine derivatives useful in the treatment of cell proliferative disorders |
| WO2009122180A1 (en) | 2008-04-02 | 2009-10-08 | Medical Research Council | Pyrimidine derivatives capable of inhibiting one or more kinases |
| WO2010100431A1 (en) | 2009-03-04 | 2010-09-10 | Medical Research Council Technology | Pyrrolopyrimidines used as kinase inhibitors |
| WO2011008915A1 (en) | 2009-07-15 | 2011-01-20 | Abbott Laboratories | Pyrrolopyridine inhibitors of kinases |
| WO2011046970A1 (en) | 2009-10-12 | 2011-04-21 | Myrexis, Inc. | Amino - pyrimidine compounds as inhibitors of tbkl and/or ikk epsilon |
Non-Patent Citations (18)
| Title |
|---|
| ALESSI ET AL., FEBS LETT., vol. 399, no. 3, 1996, pages 333 - 338 |
| C.KORHERR ET AL., PNAS, vol. 103, 2006, pages 4240 - 4245 |
| CAMPOS-GONZÄLEZ, R.; GLENNEY, JR., J.R., J. BIOL. CHEM., vol. 267, 1992, pages 14535 |
| D.A. BARBIE ET AL., NATURE, 2009, pages 1 - 5 |
| D.A.BARBIE ET AL., NATURE LETTERS, 2009, pages 1 - 5 |
| INT. J. PHARM., vol. 115, 1995, pages 61 - 67 |
| J.S. BOEHM ET AL., CELL, vol. 129, 2007, pages 1065 - 1079 |
| KHWAJA ET AL., EMBO, vol. 16, 1997, pages 2783 - 93 |
| PHARMACEUTICAL RESEARCH, vol. 3, no. 6, 1986, pages 318 |
| ROSS ET AL., BIOCHEM. J., 2002 |
| S.F.EDDY ET AL., CANCER RES., vol. 65, no. 24, 2005, pages 11375 - 11383 |
| SILLS ET AL., J. OF BIOMOLECULAR SCREENING, 2002, pages 191 - 214 |
| SORG ET AL., J. OF. BIOMOLECULAR SCREENING, vol. 7, 2002, pages 11 - 19 |
| STEPHENS ET AL., BIOCHEMICAL J., vol. 351, 2000, pages 95 - 105 |
| WEINSTEIN-OPPENHEIMER ET AL., PHARMA. &. THERAP., vol. 88, 2000, pages 229 - 279 |
| WHITE ET AL., ONCOGENE, vol. 20, 2001, pages 7064 - 7072 |
| Y.CHIEN ET AL., CELL, vol. 127, 2006, pages 157 - 170 |
| Y.-H.OU ET AL., MOLECULAR CELL, vol. 41, 2011, pages 458 - 470 |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9433622B2 (en) | 2013-02-21 | 2016-09-06 | Case Western Reserve University | Pyrimidine compounds useful in the treatment of diseases mediated by IKKE and/or TBK1 mechanisms |
| WO2015187684A1 (en) * | 2014-06-03 | 2015-12-10 | Gilead Sciences, Inc. | Tank-binding kinase inhibitor compounds |
| CN106459044A (zh) * | 2014-06-03 | 2017-02-22 | 吉利德科学公司 | Tank‑结合激酶抑制剂化合物 |
| US10259811B2 (en) | 2014-06-03 | 2019-04-16 | Gilead Sciences, Inc. | Tank-binding kinase inhibitor compounds |
| JP2017516814A (ja) * | 2014-06-03 | 2017-06-22 | ギリアード サイエンシーズ, インコーポレイテッド | Tank結合キナーゼインヒビター化合物 |
| EA031757B1 (ru) * | 2014-06-03 | 2019-02-28 | Джилид Сайэнс, Инк. | Соединения, ингибирующие tank-связывающую киназу |
| KR101926245B1 (ko) | 2014-06-03 | 2018-12-06 | 길리애드 사이언시즈, 인코포레이티드 | Tank-결합 키나제 억제제 화합물 |
| US10072001B2 (en) | 2014-06-03 | 2018-09-11 | Gilead Sciences, Inc. | Tank-binding kinase inhibitor compounds |
| US10040781B2 (en) | 2014-09-26 | 2018-08-07 | Gilead Sciences, Inc. | Tank-binding kinase inhibitor compounds |
| US10253019B2 (en) | 2014-09-26 | 2019-04-09 | Gilead Sciences, Inc. | Tank-binding kinase inhibitor compounds |
| US10316049B2 (en) | 2015-12-17 | 2019-06-11 | Gilead Sciences, Inc. | Tank-binding kinase inhibitor compounds |
| WO2017102091A1 (en) | 2015-12-18 | 2017-06-22 | Bayer Pharma Aktiengesellschaft | Heteroarylbenzimidazole compounds |
| US10894784B2 (en) | 2015-12-18 | 2021-01-19 | Bayer Pharma Aktiengesellschaft | Heteroarylbenzimidazole compounds |
| WO2017207534A1 (en) | 2016-06-03 | 2017-12-07 | Bayer Pharma Aktiengesellschaft | Substituted heteroarylbenzimidazole compounds |
| WO2018019341A1 (de) | 2016-07-26 | 2018-02-01 | Karl Rosa | Transfektionsverfahren mit nicht-viralen genliefersystemen |
| DE102016113714A1 (de) | 2016-07-26 | 2018-02-01 | Rosa Karl | Transfektionsverfahren mit nicht-viralen Genliefersystemen |
| WO2020210366A1 (en) * | 2019-04-09 | 2020-10-15 | Plexxikon Inc. | Condensed azines for ep300 or cbp modulation and indications therefor |
| US11446287B2 (en) | 2019-04-09 | 2022-09-20 | Opna Immuno-Oncology Sa | Compounds and methods for EP300 or CBP modulation and indications therefor |
Also Published As
| Publication number | Publication date |
|---|---|
| DE102011119127A1 (de) | 2013-05-23 |
| AR088925A1 (es) | 2014-07-16 |
| ES2661256T3 (es) | 2018-03-28 |
| AU2012342891A8 (en) | 2014-07-31 |
| JP2014534246A (ja) | 2014-12-18 |
| JP6096797B2 (ja) | 2017-03-15 |
| EP2794602A1 (de) | 2014-10-29 |
| CN103946223A (zh) | 2014-07-23 |
| IL232717B (en) | 2018-08-30 |
| AU2012342891A1 (en) | 2014-07-03 |
| IL232717A0 (en) | 2014-08-03 |
| AU2012342891B2 (en) | 2016-10-13 |
| US20140323481A1 (en) | 2014-10-30 |
| CA2856357A1 (en) | 2013-05-30 |
| US9102675B2 (en) | 2015-08-11 |
| EP2794602B1 (de) | 2017-11-29 |
| CA2856357C (en) | 2019-10-01 |
| CN103946223B (zh) | 2017-01-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2794602B1 (de) | 3-cyanaryl-1h-pyrazolo-(2,3-b-)pyridinderivate | |
| EP2291375B1 (de) | Pyrrolopyridinyl-pyrimidin-2-yl-amin-derivate | |
| EP2155745B1 (de) | 6-(Pyrrolopyridinyl)- Pyrimidin-2-yl-Amin-Derivate und ihre Verwendung zur Behandlung von Krebs und Aids | |
| DE102011112978A1 (de) | Benzonitrilderivate | |
| EP2670748B1 (de) | 7-azaindolderivate | |
| EP2454253B1 (de) | Aminopyridinderivate zur behandlung von tumoren und entzündungskrankheiten | |
| EP2635581B1 (de) | 7-([1,2,3]triazol-4-yl)-pyrrolo[2,3-b]pyrazinderivate | |
| EP2427461A1 (de) | 3-(ý1,2,3¨triazol-4-yl)-pyrroloý2,3-b¨pyridinderivate | |
| EP2231664A1 (de) | 4-(pyrroloý2,3-c¨pyridine-3-yl)-pyrimidin-2-amin-derivative | |
| EP2516442B1 (de) | Pyrrolo[2,3-d]pyrazin-7-yl-Pyrimidin-Verbindungen | |
| EP2663566A1 (de) | 5-([1,2,3]triazol-4-yl)-7h-pyrrolo[2,3-d]pyrimidinderivate | |
| EP2516431B1 (de) | Pyrrolopyridinyl-pyrimidin-2-yl-amin-derivate | |
| DE102012019369A1 (de) | 7-Azaindolderivat | |
| WO2012072200A1 (de) | 3-hetaryl-substituierte pyrrolo[2,3-b]pyridin-derivative als pdk1 - inhibitoren | |
| DE102008025751A1 (de) | 4-(1H-Pyrrolo[2,3-b]pyridin-3-yl)-pyridin-2-ylamin-derivate | |
| EP2121682A1 (de) | 4-(pyrrolopyridinyl)-pyrimidin-2-yl-amin-derivate | |
| EP2723736B1 (de) | Zur Behandlung von Krebserkrankungen geeignete 7-Azaindolderivate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12780423 Country of ref document: EP Kind code of ref document: A1 |
|
| REEP | Request for entry into the european phase |
Ref document number: 2012780423 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012780423 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2856357 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 232717 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: 2014542726 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14359705 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2012342891 Country of ref document: AU Date of ref document: 20121030 Kind code of ref document: A |