WO2013059885A2 - Polypeptide constructs and uses thereof - Google Patents
Polypeptide constructs and uses thereof Download PDFInfo
- Publication number
- WO2013059885A2 WO2013059885A2 PCT/AU2012/001323 AU2012001323W WO2013059885A2 WO 2013059885 A2 WO2013059885 A2 WO 2013059885A2 AU 2012001323 W AU2012001323 W AU 2012001323W WO 2013059885 A2 WO2013059885 A2 WO 2013059885A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- antibody
- antigen
- seq
- cells
- ifn
- Prior art date
Links
- 108090000765 processed proteins & peptides Proteins 0.000 title claims abstract description 128
- 102000004196 processed proteins & peptides Human genes 0.000 title claims abstract description 95
- 229920001184 polypeptide Polymers 0.000 title claims abstract description 91
- 210000004027 cell Anatomy 0.000 claims abstract description 291
- 239000000427 antigen Substances 0.000 claims abstract description 206
- 108091007433 antigens Proteins 0.000 claims abstract description 193
- 102000036639 antigens Human genes 0.000 claims abstract description 193
- 239000003446 ligand Substances 0.000 claims abstract description 170
- 230000027455 binding Effects 0.000 claims abstract description 79
- 230000011664 signaling Effects 0.000 claims abstract description 58
- 125000003275 alpha amino acid group Chemical group 0.000 claims abstract description 34
- 238000006467 substitution reaction Methods 0.000 claims abstract description 34
- 230000014509 gene expression Effects 0.000 claims abstract description 28
- 238000012217 deletion Methods 0.000 claims abstract description 19
- 230000037430 deletion Effects 0.000 claims abstract description 19
- 206010028980 Neoplasm Diseases 0.000 claims description 154
- 201000011510 cancer Diseases 0.000 claims description 84
- 238000000034 method Methods 0.000 claims description 61
- 108090000623 proteins and genes Proteins 0.000 claims description 60
- -1 HLA- SR Proteins 0.000 claims description 55
- 238000011282 treatment Methods 0.000 claims description 53
- 102000005962 receptors Human genes 0.000 claims description 52
- 108020003175 receptors Proteins 0.000 claims description 52
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 45
- 101000777636 Homo sapiens ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Proteins 0.000 claims description 39
- 102100031585 ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Human genes 0.000 claims description 37
- 108090000978 Interleukin-4 Proteins 0.000 claims description 36
- 208000034578 Multiple myelomas Diseases 0.000 claims description 31
- 108090001005 Interleukin-6 Proteins 0.000 claims description 27
- 101000716102 Homo sapiens T-cell surface glycoprotein CD4 Proteins 0.000 claims description 22
- 102100036011 T-cell surface glycoprotein CD4 Human genes 0.000 claims description 22
- 150000001413 amino acids Chemical class 0.000 claims description 22
- 101710089372 Programmed cell death protein 1 Proteins 0.000 claims description 17
- 102100026144 Transferrin receptor protein 1 Human genes 0.000 claims description 17
- 101000934338 Homo sapiens Myeloid cell surface antigen CD33 Proteins 0.000 claims description 15
- 102100025243 Myeloid cell surface antigen CD33 Human genes 0.000 claims description 15
- 102100024217 CAMPATH-1 antigen Human genes 0.000 claims description 14
- 108010065524 CD52 Antigen Proteins 0.000 claims description 14
- 101000916644 Homo sapiens Macrophage colony-stimulating factor 1 receptor Proteins 0.000 claims description 13
- 102100028198 Macrophage colony-stimulating factor 1 receptor Human genes 0.000 claims description 13
- 239000000203 mixture Substances 0.000 claims description 13
- 101000835093 Homo sapiens Transferrin receptor protein 1 Proteins 0.000 claims description 12
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 claims description 12
- 101000946843 Homo sapiens T-cell surface glycoprotein CD8 alpha chain Proteins 0.000 claims description 10
- 102100034922 T-cell surface glycoprotein CD8 alpha chain Human genes 0.000 claims description 10
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 claims description 9
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 claims description 9
- 101000946889 Homo sapiens Monocyte differentiation antigen CD14 Proteins 0.000 claims description 9
- 102100035877 Monocyte differentiation antigen CD14 Human genes 0.000 claims description 9
- 102000017420 CD3 protein, epsilon/gamma/delta subunit Human genes 0.000 claims description 8
- 108050005493 CD3 protein, epsilon/gamma/delta subunit Proteins 0.000 claims description 8
- 108010045374 CD36 Antigens Proteins 0.000 claims description 8
- 102000006354 HLA-DR Antigens Human genes 0.000 claims description 8
- 108010058597 HLA-DR Antigens Proteins 0.000 claims description 8
- 102100032912 CD44 antigen Human genes 0.000 claims description 7
- 102100025137 Early activation antigen CD69 Human genes 0.000 claims description 7
- 101000868273 Homo sapiens CD44 antigen Proteins 0.000 claims description 7
- 101000934374 Homo sapiens Early activation antigen CD69 Proteins 0.000 claims description 7
- 101000596234 Homo sapiens T-cell surface protein tactile Proteins 0.000 claims description 7
- 102100026547 Platelet-derived growth factor receptor beta Human genes 0.000 claims description 7
- 102100035268 T-cell surface protein tactile Human genes 0.000 claims description 7
- 230000004048 modification Effects 0.000 claims description 7
- 238000012986 modification Methods 0.000 claims description 7
- 101001005269 Arabidopsis thaliana Ceramide synthase 1 LOH3 Proteins 0.000 claims description 6
- 101001005312 Arabidopsis thaliana Ceramide synthase LOH1 Proteins 0.000 claims description 6
- 102100035793 CD83 antigen Human genes 0.000 claims description 6
- 102100028801 Calsyntenin-1 Human genes 0.000 claims description 6
- 101000946856 Homo sapiens CD83 antigen Proteins 0.000 claims description 6
- 101000633784 Homo sapiens SLAM family member 7 Proteins 0.000 claims description 6
- 101000884271 Homo sapiens Signal transducer CD24 Proteins 0.000 claims description 6
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 claims description 6
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 claims description 6
- 102100038081 Signal transducer CD24 Human genes 0.000 claims description 6
- 101000668858 Spinacia oleracea 30S ribosomal protein S1, chloroplastic Proteins 0.000 claims description 6
- 101000898746 Streptomyces clavuligerus Clavaminate synthase 1 Proteins 0.000 claims description 6
- 108091008605 VEGF receptors Proteins 0.000 claims description 6
- BGFTWECWAICPDG-UHFFFAOYSA-N 2-[bis(4-chlorophenyl)methyl]-4-n-[3-[bis(4-chlorophenyl)methyl]-4-(dimethylamino)phenyl]-1-n,1-n-dimethylbenzene-1,4-diamine Chemical compound C1=C(C(C=2C=CC(Cl)=CC=2)C=2C=CC(Cl)=CC=2)C(N(C)C)=CC=C1NC(C=1)=CC=C(N(C)C)C=1C(C=1C=CC(Cl)=CC=1)C1=CC=C(Cl)C=C1 BGFTWECWAICPDG-UHFFFAOYSA-N 0.000 claims description 5
- 102100020895 Ammonium transporter Rh type A Human genes 0.000 claims description 5
- 108010008014 B-Cell Maturation Antigen Proteins 0.000 claims description 5
- 102000006942 B-Cell Maturation Antigen Human genes 0.000 claims description 5
- 102100037086 Bone marrow stromal antigen 2 Human genes 0.000 claims description 5
- 101001075525 Homo sapiens Ammonium transporter Rh type A Proteins 0.000 claims description 5
- 101000740785 Homo sapiens Bone marrow stromal antigen 2 Proteins 0.000 claims description 5
- 101000692455 Homo sapiens Platelet-derived growth factor receptor beta Proteins 0.000 claims description 5
- 108010051742 Platelet-Derived Growth Factor beta Receptor Proteins 0.000 claims description 5
- 108010014608 Proto-Oncogene Proteins c-kit Proteins 0.000 claims description 5
- 102000016971 Proto-Oncogene Proteins c-kit Human genes 0.000 claims description 5
- 108010033576 Transferrin Receptors Proteins 0.000 claims description 5
- 102000009484 Vascular Endothelial Growth Factor Receptors Human genes 0.000 claims description 5
- 102100026293 Asialoglycoprotein receptor 2 Human genes 0.000 claims description 4
- 102100028239 Basal cell adhesion molecule Human genes 0.000 claims description 4
- 102000049320 CD36 Human genes 0.000 claims description 4
- 102100027842 Fibroblast growth factor receptor 3 Human genes 0.000 claims description 4
- 101710182396 Fibroblast growth factor receptor 3 Proteins 0.000 claims description 4
- 102100030595 HLA class II histocompatibility antigen gamma chain Human genes 0.000 claims description 4
- 102100031573 Hematopoietic progenitor cell antigen CD34 Human genes 0.000 claims description 4
- 101000785948 Homo sapiens Asialoglycoprotein receptor 2 Proteins 0.000 claims description 4
- 101001082627 Homo sapiens HLA class II histocompatibility antigen gamma chain Proteins 0.000 claims description 4
- 101000777663 Homo sapiens Hematopoietic progenitor cell antigen CD34 Proteins 0.000 claims description 4
- 101000959820 Homo sapiens Interferon alpha-1/13 Proteins 0.000 claims description 4
- 101000971879 Homo sapiens Kell blood group glycoprotein Proteins 0.000 claims description 4
- 101000581981 Homo sapiens Neural cell adhesion molecule 1 Proteins 0.000 claims description 4
- 108010064593 Intercellular Adhesion Molecule-1 Proteins 0.000 claims description 4
- 102100037877 Intercellular adhesion molecule 1 Human genes 0.000 claims description 4
- 102100037792 Interleukin-6 receptor subunit alpha Human genes 0.000 claims description 4
- 102100021447 Kell blood group glycoprotein Human genes 0.000 claims description 4
- 102100035792 Kininogen-1 Human genes 0.000 claims description 4
- 108010077861 Kininogens Proteins 0.000 claims description 4
- 108010091175 Matriptase Proteins 0.000 claims description 4
- 102100027347 Neural cell adhesion molecule 1 Human genes 0.000 claims description 4
- 102100031574 Platelet glycoprotein 4 Human genes 0.000 claims description 4
- 102100037942 Suppressor of tumorigenicity 14 protein Human genes 0.000 claims description 4
- 108040006858 interleukin-6 receptor activity proteins Proteins 0.000 claims description 4
- 102100026292 Asialoglycoprotein receptor 1 Human genes 0.000 claims description 3
- 101000785944 Homo sapiens Asialoglycoprotein receptor 1 Proteins 0.000 claims description 3
- 108010041012 Integrin alpha4 Proteins 0.000 claims description 3
- 102000018967 Platelet-Derived Growth Factor beta Receptor Human genes 0.000 claims description 3
- 101000600776 Rattus norvegicus Podoplanin Proteins 0.000 claims description 3
- 239000003085 diluting agent Substances 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- 102220492437 2'-5'-oligoadenylate synthase 3_R33A_mutation Human genes 0.000 claims 4
- 102100023990 60S ribosomal protein L17 Human genes 0.000 claims 2
- 102000002698 KIR Receptors Human genes 0.000 claims 2
- 108010043610 KIR Receptors Proteins 0.000 claims 2
- 102000015736 beta 2-Microglobulin Human genes 0.000 claims 2
- 108010081355 beta 2-Microglobulin Proteins 0.000 claims 2
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 claims 2
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 claims 2
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 claims 2
- 102220467431 Protein Jade-1_R35A_mutation Human genes 0.000 claims 1
- 102000014150 Interferons Human genes 0.000 description 157
- 108010050904 Interferons Proteins 0.000 description 157
- 108020001507 fusion proteins Proteins 0.000 description 148
- 102000037865 fusion proteins Human genes 0.000 description 148
- 229940079322 interferon Drugs 0.000 description 139
- 230000000694 effects Effects 0.000 description 111
- 238000003556 assay Methods 0.000 description 99
- 230000002238 attenuated effect Effects 0.000 description 86
- 230000035772 mutation Effects 0.000 description 59
- 102000004169 proteins and genes Human genes 0.000 description 47
- 235000018102 proteins Nutrition 0.000 description 46
- 235000001014 amino acid Nutrition 0.000 description 37
- 230000008685 targeting Effects 0.000 description 36
- 102000004388 Interleukin-4 Human genes 0.000 description 34
- 229940028885 interleukin-4 Drugs 0.000 description 34
- 210000001744 T-lymphocyte Anatomy 0.000 description 32
- 230000001225 therapeutic effect Effects 0.000 description 31
- 210000004369 blood Anatomy 0.000 description 28
- 239000008280 blood Substances 0.000 description 28
- 102000004889 Interleukin-6 Human genes 0.000 description 25
- 210000002950 fibroblast Anatomy 0.000 description 25
- 229940100601 interleukin-6 Drugs 0.000 description 25
- 102100026720 Interferon beta Human genes 0.000 description 24
- 108090000467 Interferon-beta Proteins 0.000 description 24
- 125000005647 linker group Chemical group 0.000 description 24
- 102000003745 Hepatocyte Growth Factor Human genes 0.000 description 23
- 108090000100 Hepatocyte Growth Factor Proteins 0.000 description 23
- 230000001028 anti-proliverative effect Effects 0.000 description 23
- 229940024606 amino acid Drugs 0.000 description 22
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 22
- 201000006417 multiple sclerosis Diseases 0.000 description 22
- 102000003951 Erythropoietin Human genes 0.000 description 20
- 108090000394 Erythropoietin Proteins 0.000 description 20
- 230000004913 activation Effects 0.000 description 20
- 229940105423 erythropoietin Drugs 0.000 description 20
- 102000003814 Interleukin-10 Human genes 0.000 description 19
- 108090000174 Interleukin-10 Proteins 0.000 description 19
- 150000001875 compounds Chemical class 0.000 description 19
- 201000010099 disease Diseases 0.000 description 19
- OXCMYAYHXIHQOA-UHFFFAOYSA-N potassium;[2-butyl-5-chloro-3-[[4-[2-(1,2,4-triaza-3-azanidacyclopenta-1,4-dien-5-yl)phenyl]phenyl]methyl]imidazol-4-yl]methanol Chemical compound [K+].CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C2=N[N-]N=N2)C=C1 OXCMYAYHXIHQOA-UHFFFAOYSA-N 0.000 description 19
- 230000002829 reductive effect Effects 0.000 description 19
- 108090000695 Cytokines Proteins 0.000 description 18
- 238000013459 approach Methods 0.000 description 18
- 102100028226 COUP transcription factor 2 Human genes 0.000 description 17
- 108010074328 Interferon-gamma Proteins 0.000 description 17
- 229940076144 interleukin-10 Drugs 0.000 description 17
- 230000035755 proliferation Effects 0.000 description 17
- 101710188750 COUP transcription factor 2 Proteins 0.000 description 16
- 102000004127 Cytokines Human genes 0.000 description 16
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 16
- 102100040678 Programmed cell death protein 1 Human genes 0.000 description 16
- 230000004069 differentiation Effects 0.000 description 16
- 201000001441 melanoma Diseases 0.000 description 16
- 108020004414 DNA Proteins 0.000 description 15
- 102100037850 Interferon gamma Human genes 0.000 description 15
- 229940047124 interferons Drugs 0.000 description 15
- TZCPCKNHXULUIY-RGULYWFUSA-N 1,2-distearoyl-sn-glycero-3-phosphoserine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OC[C@H](N)C(O)=O)OC(=O)CCCCCCCCCCCCCCCCC TZCPCKNHXULUIY-RGULYWFUSA-N 0.000 description 14
- 201000009030 Carcinoma Diseases 0.000 description 14
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 14
- ZWZWYGMENQVNFU-UHFFFAOYSA-N Glycerophosphorylserin Natural products OC(=O)C(N)COP(O)(=O)OCC(O)CO ZWZWYGMENQVNFU-UHFFFAOYSA-N 0.000 description 14
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 14
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 14
- 230000008901 benefit Effects 0.000 description 14
- 201000000050 myeloid neoplasm Diseases 0.000 description 14
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 14
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 13
- 206010025323 Lymphomas Diseases 0.000 description 13
- 102100029981 Receptor tyrosine-protein kinase erbB-4 Human genes 0.000 description 13
- 101710100963 Receptor tyrosine-protein kinase erbB-4 Proteins 0.000 description 13
- 230000001965 increasing effect Effects 0.000 description 13
- 206010016654 Fibrosis Diseases 0.000 description 12
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 12
- 239000003814 drug Substances 0.000 description 12
- 239000012634 fragment Substances 0.000 description 12
- AQTQHPDCURKLKT-PNYVAJAMSA-N vincristine sulfate Chemical compound OS(O)(=O)=O.C([C@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C=O)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 AQTQHPDCURKLKT-PNYVAJAMSA-N 0.000 description 12
- 102000001301 EGF receptor Human genes 0.000 description 11
- 108060006698 EGF receptor Proteins 0.000 description 11
- 229960004397 cyclophosphamide Drugs 0.000 description 11
- 230000004761 fibrosis Effects 0.000 description 11
- 208000032839 leukemia Diseases 0.000 description 11
- 210000003289 regulatory T cell Anatomy 0.000 description 11
- 102220473689 Ras-related protein Rab-5A_Y89A_mutation Human genes 0.000 description 10
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 10
- 230000000840 anti-viral effect Effects 0.000 description 10
- 230000015572 biosynthetic process Effects 0.000 description 10
- 239000003795 chemical substances by application Substances 0.000 description 10
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 10
- 208000019425 cirrhosis of liver Diseases 0.000 description 10
- 230000007423 decrease Effects 0.000 description 10
- 238000002474 experimental method Methods 0.000 description 10
- 230000012010 growth Effects 0.000 description 10
- 102220535515 tRNA wybutosine-synthesizing protein 5_Y85A_mutation Human genes 0.000 description 10
- 206010006187 Breast cancer Diseases 0.000 description 9
- 208000026310 Breast neoplasm Diseases 0.000 description 9
- 239000004471 Glycine Substances 0.000 description 9
- 208000005176 Hepatitis C Diseases 0.000 description 9
- 201000009794 Idiopathic Pulmonary Fibrosis Diseases 0.000 description 9
- 102220548502 Macrophage-capping protein_Q61S_mutation Human genes 0.000 description 9
- 102220522737 Mitogen-activated protein kinase kinase kinase 6_H57Y_mutation Human genes 0.000 description 9
- 230000006907 apoptotic process Effects 0.000 description 9
- 230000004071 biological effect Effects 0.000 description 9
- 229940079593 drug Drugs 0.000 description 9
- 230000003176 fibrotic effect Effects 0.000 description 9
- 230000006870 function Effects 0.000 description 9
- 230000004927 fusion Effects 0.000 description 9
- 208000036971 interstitial lung disease 2 Diseases 0.000 description 9
- 239000000546 pharmaceutical excipient Substances 0.000 description 9
- 238000011160 research Methods 0.000 description 9
- 239000000126 substance Substances 0.000 description 9
- 210000004881 tumor cell Anatomy 0.000 description 9
- 208000003950 B-cell lymphoma Diseases 0.000 description 8
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 8
- 102220496408 Bone marrow stromal antigen 2_N65A_mutation Human genes 0.000 description 8
- 102220496875 DNA dC->dU-editing enzyme APOBEC-3C_L80A_mutation Human genes 0.000 description 8
- MWWSFMDVAYGXBV-RUELKSSGSA-N Doxorubicin hydrochloride Chemical compound Cl.O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 MWWSFMDVAYGXBV-RUELKSSGSA-N 0.000 description 8
- 102000002227 Interferon Type I Human genes 0.000 description 8
- 108010014726 Interferon Type I Proteins 0.000 description 8
- 241000699666 Mus <mouse, genus> Species 0.000 description 8
- 102220555961 Ubiquitin-like modifier-activating enzyme 6_L117A_mutation Human genes 0.000 description 8
- 241000700605 Viruses Species 0.000 description 8
- 150000001720 carbohydrates Chemical class 0.000 description 8
- 229960004630 chlorambucil Drugs 0.000 description 8
- 230000002401 inhibitory effect Effects 0.000 description 8
- 230000004044 response Effects 0.000 description 8
- 229960004641 rituximab Drugs 0.000 description 8
- 238000007920 subcutaneous administration Methods 0.000 description 8
- 231100000419 toxicity Toxicity 0.000 description 8
- 230000001988 toxicity Effects 0.000 description 8
- 208000023275 Autoimmune disease Diseases 0.000 description 7
- 108010006654 Bleomycin Proteins 0.000 description 7
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 7
- 108010035532 Collagen Proteins 0.000 description 7
- 102000008186 Collagen Human genes 0.000 description 7
- 102000003886 Glycoproteins Human genes 0.000 description 7
- 108090000288 Glycoproteins Proteins 0.000 description 7
- 108010086140 Interferon alpha-beta Receptor Proteins 0.000 description 7
- 102000007438 Interferon alpha-beta Receptor Human genes 0.000 description 7
- 102100034256 Mucin-1 Human genes 0.000 description 7
- 238000011579 SCID mouse model Methods 0.000 description 7
- 235000014633 carbohydrates Nutrition 0.000 description 7
- 229920001436 collagen Polymers 0.000 description 7
- 210000004443 dendritic cell Anatomy 0.000 description 7
- 238000011161 development Methods 0.000 description 7
- 230000018109 developmental process Effects 0.000 description 7
- 229960004679 doxorubicin Drugs 0.000 description 7
- 229940088597 hormone Drugs 0.000 description 7
- 239000005556 hormone Substances 0.000 description 7
- 238000001727 in vivo Methods 0.000 description 7
- 230000002757 inflammatory effect Effects 0.000 description 7
- 210000004072 lung Anatomy 0.000 description 7
- 210000001616 monocyte Anatomy 0.000 description 7
- 230000008506 pathogenesis Effects 0.000 description 7
- 229960004618 prednisone Drugs 0.000 description 7
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 7
- 210000002966 serum Anatomy 0.000 description 7
- 210000001519 tissue Anatomy 0.000 description 7
- 229960004528 vincristine Drugs 0.000 description 7
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 7
- 229960002110 vincristine sulfate Drugs 0.000 description 7
- 102000019034 Chemokines Human genes 0.000 description 6
- 108010012236 Chemokines Proteins 0.000 description 6
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 6
- 101000852865 Homo sapiens Interferon alpha/beta receptor 2 Proteins 0.000 description 6
- 102100036718 Interferon alpha/beta receptor 2 Human genes 0.000 description 6
- 102000013691 Interleukin-17 Human genes 0.000 description 6
- 108050003558 Interleukin-17 Proteins 0.000 description 6
- 241001529936 Murinae Species 0.000 description 6
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 6
- 102220498199 Negative regulator of P-body association_R33A_mutation Human genes 0.000 description 6
- 101710100969 Receptor tyrosine-protein kinase erbB-3 Proteins 0.000 description 6
- 102100029986 Receptor tyrosine-protein kinase erbB-3 Human genes 0.000 description 6
- 208000006265 Renal cell carcinoma Diseases 0.000 description 6
- 108700012411 TNFSF10 Proteins 0.000 description 6
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 6
- 210000000612 antigen-presenting cell Anatomy 0.000 description 6
- 210000003719 b-lymphocyte Anatomy 0.000 description 6
- 229960001561 bleomycin Drugs 0.000 description 6
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 6
- 239000000872 buffer Substances 0.000 description 6
- 230000001684 chronic effect Effects 0.000 description 6
- 229960002918 doxorubicin hydrochloride Drugs 0.000 description 6
- 210000003494 hepatocyte Anatomy 0.000 description 6
- 238000000338 in vitro Methods 0.000 description 6
- 229960000485 methotrexate Drugs 0.000 description 6
- 231100000350 mutagenesis Toxicity 0.000 description 6
- 210000000056 organ Anatomy 0.000 description 6
- 229920001223 polyethylene glycol Polymers 0.000 description 6
- 230000009467 reduction Effects 0.000 description 6
- 238000006722 reduction reaction Methods 0.000 description 6
- 230000004083 survival effect Effects 0.000 description 6
- 230000003612 virological effect Effects 0.000 description 6
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 5
- 102100021943 C-C motif chemokine 2 Human genes 0.000 description 5
- 108010022366 Carcinoembryonic Antigen Proteins 0.000 description 5
- 102100025475 Carcinoembryonic antigen-related cell adhesion molecule 5 Human genes 0.000 description 5
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 description 5
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 5
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 5
- 101000852870 Homo sapiens Interferon alpha/beta receptor 1 Proteins 0.000 description 5
- 101000874179 Homo sapiens Syndecan-1 Proteins 0.000 description 5
- 108060003951 Immunoglobulin Proteins 0.000 description 5
- 102100036714 Interferon alpha/beta receptor 1 Human genes 0.000 description 5
- 102100020990 Interferon lambda-1 Human genes 0.000 description 5
- 108010008707 Mucin-1 Proteins 0.000 description 5
- 241000699670 Mus sp. Species 0.000 description 5
- 102100035721 Syndecan-1 Human genes 0.000 description 5
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 5
- 125000003277 amino group Chemical group 0.000 description 5
- 230000010056 antibody-dependent cellular cytotoxicity Effects 0.000 description 5
- 238000006243 chemical reaction Methods 0.000 description 5
- 230000003247 decreasing effect Effects 0.000 description 5
- 210000003013 erythroid precursor cell Anatomy 0.000 description 5
- 229960005420 etoposide Drugs 0.000 description 5
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 5
- 239000012091 fetal bovine serum Substances 0.000 description 5
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 5
- 210000004024 hepatic stellate cell Anatomy 0.000 description 5
- 102000018358 immunoglobulin Human genes 0.000 description 5
- 230000001939 inductive effect Effects 0.000 description 5
- 230000003993 interaction Effects 0.000 description 5
- 229960001388 interferon-beta Drugs 0.000 description 5
- 230000003834 intracellular effect Effects 0.000 description 5
- 238000004519 manufacturing process Methods 0.000 description 5
- 239000012528 membrane Substances 0.000 description 5
- 238000013508 migration Methods 0.000 description 5
- 238000002703 mutagenesis Methods 0.000 description 5
- 239000000813 peptide hormone Substances 0.000 description 5
- 230000003389 potentiating effect Effects 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- 230000000770 proinflammatory effect Effects 0.000 description 5
- 208000005069 pulmonary fibrosis Diseases 0.000 description 5
- 230000001105 regulatory effect Effects 0.000 description 5
- 238000012216 screening Methods 0.000 description 5
- 230000019491 signal transduction Effects 0.000 description 5
- 230000007781 signaling event Effects 0.000 description 5
- 230000004936 stimulating effect Effects 0.000 description 5
- 238000002560 therapeutic procedure Methods 0.000 description 5
- 229960005267 tositumomab Drugs 0.000 description 5
- 230000004614 tumor growth Effects 0.000 description 5
- KDQAABAKXDWYSZ-PNYVAJAMSA-N vinblastine sulfate Chemical compound OS(O)(=O)=O.C([C@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 KDQAABAKXDWYSZ-PNYVAJAMSA-N 0.000 description 5
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 4
- 101150013553 CD40 gene Proteins 0.000 description 4
- 108010021064 CTLA-4 Antigen Proteins 0.000 description 4
- 108010088652 Histocompatibility Antigens Class I Proteins 0.000 description 4
- 102000008949 Histocompatibility Antigens Class I Human genes 0.000 description 4
- 208000017604 Hodgkin disease Diseases 0.000 description 4
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 4
- 241000282412 Homo Species 0.000 description 4
- 101001002470 Homo sapiens Interferon lambda-1 Proteins 0.000 description 4
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 description 4
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 description 4
- 102100026120 IgG receptor FcRn large subunit p51 Human genes 0.000 description 4
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 4
- 102100027268 Interferon-stimulated gene 20 kDa protein Human genes 0.000 description 4
- 208000007766 Kaposi sarcoma Diseases 0.000 description 4
- SNDPXSYFESPGGJ-UHFFFAOYSA-N L-norVal-OH Natural products CCCC(N)C(O)=O SNDPXSYFESPGGJ-UHFFFAOYSA-N 0.000 description 4
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 4
- 102220498197 Negative regulator of P-body association_L30A_mutation Human genes 0.000 description 4
- 102220482092 Nuclear cap-binding protein subunit 2_D114A_mutation Human genes 0.000 description 4
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 4
- 108010038512 Platelet-Derived Growth Factor Proteins 0.000 description 4
- 102000010780 Platelet-Derived Growth Factor Human genes 0.000 description 4
- 201000004681 Psoriasis Diseases 0.000 description 4
- 239000012980 RPMI-1640 medium Substances 0.000 description 4
- 102220506973 Rab11 family-interacting protein 1_R35K_mutation Human genes 0.000 description 4
- 241000700159 Rattus Species 0.000 description 4
- 102220509128 Sphingosine 1-phosphate receptor 1_R120A_mutation Human genes 0.000 description 4
- 208000005718 Stomach Neoplasms Diseases 0.000 description 4
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 4
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 4
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 4
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 230000003213 activating effect Effects 0.000 description 4
- 230000009824 affinity maturation Effects 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 230000001093 anti-cancer Effects 0.000 description 4
- 102000023732 binding proteins Human genes 0.000 description 4
- 108091008324 binding proteins Proteins 0.000 description 4
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 4
- 150000001718 carbodiimides Chemical class 0.000 description 4
- 230000011712 cell development Effects 0.000 description 4
- 230000024245 cell differentiation Effects 0.000 description 4
- 230000004540 complement-dependent cytotoxicity Effects 0.000 description 4
- 102000003675 cytokine receptors Human genes 0.000 description 4
- 108010057085 cytokine receptors Proteins 0.000 description 4
- 230000006378 damage Effects 0.000 description 4
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 4
- 238000005516 engineering process Methods 0.000 description 4
- 210000002919 epithelial cell Anatomy 0.000 description 4
- 210000002744 extracellular matrix Anatomy 0.000 description 4
- 102000006815 folate receptor Human genes 0.000 description 4
- 108020005243 folate receptor Proteins 0.000 description 4
- 206010017758 gastric cancer Diseases 0.000 description 4
- 208000005017 glioblastoma Diseases 0.000 description 4
- 239000003102 growth factor Substances 0.000 description 4
- 201000009277 hairy cell leukemia Diseases 0.000 description 4
- 230000028993 immune response Effects 0.000 description 4
- 230000006698 induction Effects 0.000 description 4
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 4
- 208000020816 lung neoplasm Diseases 0.000 description 4
- 210000004698 lymphocyte Anatomy 0.000 description 4
- 210000002540 macrophage Anatomy 0.000 description 4
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 4
- 230000001404 mediated effect Effects 0.000 description 4
- 230000005012 migration Effects 0.000 description 4
- 201000005962 mycosis fungoides Diseases 0.000 description 4
- 210000000066 myeloid cell Anatomy 0.000 description 4
- 208000008443 pancreatic carcinoma Diseases 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- FSYKKLYZXJSNPZ-UHFFFAOYSA-N sarcosine Chemical compound C[NH2+]CC([O-])=O FSYKKLYZXJSNPZ-UHFFFAOYSA-N 0.000 description 4
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 4
- 201000011549 stomach cancer Diseases 0.000 description 4
- 235000000346 sugar Nutrition 0.000 description 4
- 230000009885 systemic effect Effects 0.000 description 4
- 230000009385 viral infection Effects 0.000 description 4
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 3
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 3
- FUOOLUPWFVMBKG-UHFFFAOYSA-N 2-Aminoisobutyric acid Chemical compound CC(C)(N)C(O)=O FUOOLUPWFVMBKG-UHFFFAOYSA-N 0.000 description 3
- RTQWWZBSTRGEAV-PKHIMPSTSA-N 2-[[(2s)-2-[bis(carboxymethyl)amino]-3-[4-(methylcarbamoylamino)phenyl]propyl]-[2-[bis(carboxymethyl)amino]propyl]amino]acetic acid Chemical compound CNC(=O)NC1=CC=C(C[C@@H](CN(CC(C)N(CC(O)=O)CC(O)=O)CC(O)=O)N(CC(O)=O)CC(O)=O)C=C1 RTQWWZBSTRGEAV-PKHIMPSTSA-N 0.000 description 3
- ZHSKUOZOLHMKEA-UHFFFAOYSA-N 4-[5-[bis(2-chloroethyl)amino]-1-methylbenzimidazol-2-yl]butanoic acid;hydron;chloride Chemical compound Cl.ClCCN(CCCl)C1=CC=C2N(C)C(CCCC(O)=O)=NC2=C1 ZHSKUOZOLHMKEA-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- 208000036762 Acute promyelocytic leukaemia Diseases 0.000 description 3
- 102100023635 Alpha-fetoprotein Human genes 0.000 description 3
- 102100038080 B-cell receptor CD22 Human genes 0.000 description 3
- 102100030802 Beta-2-glycoprotein 1 Human genes 0.000 description 3
- 101710180007 Beta-2-glycoprotein 1 Proteins 0.000 description 3
- 101710155857 C-C motif chemokine 2 Proteins 0.000 description 3
- 108090000565 Capsid Proteins Proteins 0.000 description 3
- 101710197658 Capsid protein VP1 Proteins 0.000 description 3
- 102100023321 Ceruloplasmin Human genes 0.000 description 3
- 208000030939 Chronic inflammatory demyelinating polyneuropathy Diseases 0.000 description 3
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 3
- 208000011231 Crohn disease Diseases 0.000 description 3
- 239000004971 Cross linker Substances 0.000 description 3
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 3
- 201000009051 Embryonal Carcinoma Diseases 0.000 description 3
- 101710181478 Envelope glycoprotein GP350 Proteins 0.000 description 3
- 108010087819 Fc receptors Proteins 0.000 description 3
- 102000009109 Fc receptors Human genes 0.000 description 3
- 201000008808 Fibrosarcoma Diseases 0.000 description 3
- 102100041003 Glutamate carboxypeptidase 2 Human genes 0.000 description 3
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 description 3
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 3
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 3
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 3
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 3
- 101000892862 Homo sapiens Glutamate carboxypeptidase 2 Proteins 0.000 description 3
- 108090000144 Human Proteins Proteins 0.000 description 3
- 102000003839 Human Proteins Human genes 0.000 description 3
- 108091006905 Human Serum Albumin Proteins 0.000 description 3
- 102000008100 Human Serum Albumin Human genes 0.000 description 3
- 241000701044 Human gammaherpesvirus 4 Species 0.000 description 3
- 101710177940 IgG receptor FcRn large subunit p51 Proteins 0.000 description 3
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 3
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 3
- 206010061218 Inflammation Diseases 0.000 description 3
- 102100030703 Interleukin-22 Human genes 0.000 description 3
- 108010002386 Interleukin-3 Proteins 0.000 description 3
- 102000000646 Interleukin-3 Human genes 0.000 description 3
- 102000004890 Interleukin-8 Human genes 0.000 description 3
- 108090001007 Interleukin-8 Proteins 0.000 description 3
- LRQKBLKVPFOOQJ-YFKPBYRVSA-N L-norleucine Chemical compound CCCC[C@H]([NH3+])C([O-])=O LRQKBLKVPFOOQJ-YFKPBYRVSA-N 0.000 description 3
- 108090001090 Lectins Proteins 0.000 description 3
- 102000004856 Lectins Human genes 0.000 description 3
- 206010067125 Liver injury Diseases 0.000 description 3
- 241000531897 Loma Species 0.000 description 3
- 102100027754 Mast/stem cell growth factor receptor Kit Human genes 0.000 description 3
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 3
- 108010025020 Nerve Growth Factor Proteins 0.000 description 3
- 108091034117 Oligonucleotide Proteins 0.000 description 3
- 101710160107 Outer membrane protein A Proteins 0.000 description 3
- 108091008606 PDGF receptors Proteins 0.000 description 3
- 102000011653 Platelet-Derived Growth Factor Receptors Human genes 0.000 description 3
- 208000033826 Promyelocytic Acute Leukemia Diseases 0.000 description 3
- 206010039491 Sarcoma Diseases 0.000 description 3
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 3
- 108010003533 Viral Envelope Proteins Proteins 0.000 description 3
- 108010067390 Viral Proteins Proteins 0.000 description 3
- 208000036142 Viral infection Diseases 0.000 description 3
- 208000027418 Wounds and injury Diseases 0.000 description 3
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 3
- 238000009825 accumulation Methods 0.000 description 3
- 108010026331 alpha-Fetoproteins Proteins 0.000 description 3
- 230000003042 antagnostic effect Effects 0.000 description 3
- 230000003510 anti-fibrotic effect Effects 0.000 description 3
- 230000003110 anti-inflammatory effect Effects 0.000 description 3
- 230000000259 anti-tumor effect Effects 0.000 description 3
- 239000002246 antineoplastic agent Substances 0.000 description 3
- 230000033228 biological regulation Effects 0.000 description 3
- 230000008499 blood brain barrier function Effects 0.000 description 3
- 210000001218 blood-brain barrier Anatomy 0.000 description 3
- 210000001185 bone marrow Anatomy 0.000 description 3
- 229960001467 bortezomib Drugs 0.000 description 3
- 210000004556 brain Anatomy 0.000 description 3
- 210000000481 breast Anatomy 0.000 description 3
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 3
- 230000010261 cell growth Effects 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical class OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000010367 cloning Methods 0.000 description 3
- 108010017271 denileukin diftitox Proteins 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 230000003828 downregulation Effects 0.000 description 3
- 210000002889 endothelial cell Anatomy 0.000 description 3
- 208000002672 hepatitis B Diseases 0.000 description 3
- 210000005260 human cell Anatomy 0.000 description 3
- 210000004408 hybridoma Anatomy 0.000 description 3
- 229960001001 ibritumomab tiuxetan Drugs 0.000 description 3
- 210000002865 immune cell Anatomy 0.000 description 3
- 210000000987 immune system Anatomy 0.000 description 3
- 230000036039 immunity Effects 0.000 description 3
- 208000015181 infectious disease Diseases 0.000 description 3
- 208000027866 inflammatory disease Diseases 0.000 description 3
- 230000004054 inflammatory process Effects 0.000 description 3
- 230000005764 inhibitory process Effects 0.000 description 3
- 208000014674 injury Diseases 0.000 description 3
- 229940096397 interleukin-8 Drugs 0.000 description 3
- XKTZWUACRZHVAN-VADRZIEHSA-N interleukin-8 Chemical compound C([C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@@H](NC(C)=O)CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N[C@@H](CCSC)C(=O)N1[C@H](CCC1)C(=O)N1[C@H](CCC1)C(=O)N[C@@H](C)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC=1C=CC(O)=CC=1)C(=O)N[C@H](CO)C(=O)N1[C@H](CCC1)C(N)=O)C1=CC=CC=C1 XKTZWUACRZHVAN-VADRZIEHSA-N 0.000 description 3
- 239000000543 intermediate Substances 0.000 description 3
- 239000002523 lectin Substances 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 150000002632 lipids Chemical class 0.000 description 3
- 210000004185 liver Anatomy 0.000 description 3
- 208000014018 liver neoplasm Diseases 0.000 description 3
- 238000004020 luminiscence type Methods 0.000 description 3
- 201000005202 lung cancer Diseases 0.000 description 3
- 230000036210 malignancy Effects 0.000 description 3
- IXOXBSCIXZEQEQ-UHTZMRCNSA-N nelarabine Chemical compound C1=NC=2C(OC)=NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1O IXOXBSCIXZEQEQ-UHTZMRCNSA-N 0.000 description 3
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 3
- 201000002528 pancreatic cancer Diseases 0.000 description 3
- 150000003904 phospholipids Chemical class 0.000 description 3
- 230000026731 phosphorylation Effects 0.000 description 3
- 238000006366 phosphorylation reaction Methods 0.000 description 3
- YIQPUIGJQJDJOS-UHFFFAOYSA-N plerixafor Chemical compound C=1C=C(CN2CCNCCCNCCNCCC2)C=CC=1CN1CCCNCCNCCCNCC1 YIQPUIGJQJDJOS-UHFFFAOYSA-N 0.000 description 3
- 229920001282 polysaccharide Polymers 0.000 description 3
- 229920000136 polysorbate Polymers 0.000 description 3
- OGSBUKJUDHAQEA-WMCAAGNKSA-N pralatrexate Chemical compound C1=NC2=NC(N)=NC(N)=C2N=C1CC(CC#C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OGSBUKJUDHAQEA-WMCAAGNKSA-N 0.000 description 3
- 239000002243 precursor Substances 0.000 description 3
- 108020001580 protein domains Proteins 0.000 description 3
- 229930002330 retinoic acid Natural products 0.000 description 3
- OHRURASPPZQGQM-GCCNXGTGSA-N romidepsin Chemical compound O1C(=O)[C@H](C(C)C)NC(=O)C(=C/C)/NC(=O)[C@H]2CSSCC\C=C\[C@@H]1CC(=O)N[C@H](C(C)C)C(=O)N2 OHRURASPPZQGQM-GCCNXGTGSA-N 0.000 description 3
- OHRURASPPZQGQM-UHFFFAOYSA-N romidepsin Natural products O1C(=O)C(C(C)C)NC(=O)C(=CC)NC(=O)C2CSSCCC=CC1CC(=O)NC(C(C)C)C(=O)N2 OHRURASPPZQGQM-UHFFFAOYSA-N 0.000 description 3
- 108010091666 romidepsin Proteins 0.000 description 3
- 102200141681 rs6170 Human genes 0.000 description 3
- 150000003839 salts Chemical class 0.000 description 3
- 238000002741 site-directed mutagenesis Methods 0.000 description 3
- 230000000638 stimulation Effects 0.000 description 3
- 150000005846 sugar alcohols Chemical class 0.000 description 3
- 150000008163 sugars Chemical class 0.000 description 3
- 208000011580 syndromic disease Diseases 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 125000003396 thiol group Chemical group [H]S* 0.000 description 3
- 238000012546 transfer Methods 0.000 description 3
- 229960003048 vinblastine Drugs 0.000 description 3
- JXLYSJRDGCGARV-CFWMRBGOSA-N vinblastine Chemical compound C([C@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-CFWMRBGOSA-N 0.000 description 3
- 229960004982 vinblastine sulfate Drugs 0.000 description 3
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 3
- WAEXFXRVDQXREF-UHFFFAOYSA-N vorinostat Chemical compound ONC(=O)CCCCCCC(=O)NC1=CC=CC=C1 WAEXFXRVDQXREF-UHFFFAOYSA-N 0.000 description 3
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 2
- SFLSHLFXELFNJZ-QMMMGPOBSA-N (-)-norepinephrine Chemical compound NC[C@H](O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-QMMMGPOBSA-N 0.000 description 2
- AXDLCFOOGCNDST-VIFPVBQESA-N (2s)-3-(4-hydroxyphenyl)-2-(methylamino)propanoic acid Chemical compound CN[C@H](C(O)=O)CC1=CC=C(O)C=C1 AXDLCFOOGCNDST-VIFPVBQESA-N 0.000 description 2
- GAUBNQMYYJLWNF-UHFFFAOYSA-N 3-(Carboxymethylamino)propanoic acid Chemical compound OC(=O)CCNCC(O)=O GAUBNQMYYJLWNF-UHFFFAOYSA-N 0.000 description 2
- DFVFTMTWCUHJBL-UHFFFAOYSA-N 4-azaniumyl-3-hydroxy-6-methylheptanoate Chemical compound CC(C)CC(N)C(O)CC(O)=O DFVFTMTWCUHJBL-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- 102220554192 APC membrane recruitment protein 1_H34A_mutation Human genes 0.000 description 2
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 2
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 2
- 102400001318 Adrenomedullin Human genes 0.000 description 2
- 101800004616 Adrenomedullin Proteins 0.000 description 2
- 108090000672 Annexin A5 Proteins 0.000 description 2
- 102000004121 Annexin A5 Human genes 0.000 description 2
- 239000004475 Arginine Substances 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical class OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 206010003571 Astrocytoma Diseases 0.000 description 2
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 description 2
- 101000805768 Banna virus (strain Indonesia/JKT-6423/1980) mRNA (guanine-N(7))-methyltransferase Proteins 0.000 description 2
- 102100032412 Basigin Human genes 0.000 description 2
- 101000742334 Bdellovibrio phage phiMH2K Replication-associated protein VP4 Proteins 0.000 description 2
- 102100023995 Beta-nerve growth factor Human genes 0.000 description 2
- 206010005003 Bladder cancer Diseases 0.000 description 2
- 102000007350 Bone Morphogenetic Proteins Human genes 0.000 description 2
- 108010007726 Bone Morphogenetic Proteins Proteins 0.000 description 2
- 208000003174 Brain Neoplasms Diseases 0.000 description 2
- 102000004219 Brain-derived neurotrophic factor Human genes 0.000 description 2
- 108090000715 Brain-derived neurotrophic factor Proteins 0.000 description 2
- 101100314454 Caenorhabditis elegans tra-1 gene Proteins 0.000 description 2
- 102220614548 Calmodulin-3_R23A_mutation Human genes 0.000 description 2
- 101710132601 Capsid protein Proteins 0.000 description 2
- 102100032231 Caveolae-associated protein 2 Human genes 0.000 description 2
- 108050005259 Caveolae-associated protein 2 Proteins 0.000 description 2
- 102000000844 Cell Surface Receptors Human genes 0.000 description 2
- 108010001857 Cell Surface Receptors Proteins 0.000 description 2
- 206010008342 Cervix carcinoma Diseases 0.000 description 2
- 101000686790 Chaetoceros protobacilladnavirus 2 Replication-associated protein Proteins 0.000 description 2
- 101000864475 Chlamydia phage 1 Internal scaffolding protein VP3 Proteins 0.000 description 2
- 102100028757 Chondroitin sulfate proteoglycan 4 Human genes 0.000 description 2
- 206010057645 Chronic Inflammatory Demyelinating Polyradiculoneuropathy Diseases 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- 102220519361 Coatomer subunit alpha_C17S_mutation Human genes 0.000 description 2
- 206010009944 Colon cancer Diseases 0.000 description 2
- 229920000858 Cyclodextrin Polymers 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- CKLJMWTZIZZHCS-UWTATZPHSA-N D-aspartic acid Chemical compound OC(=O)[C@H](N)CC(O)=O CKLJMWTZIZZHCS-UWTATZPHSA-N 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical class OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- WHUUTDBJXJRKMK-GSVOUGTGSA-N D-glutamic acid Chemical compound OC(=O)[C@H](N)CCC(O)=O WHUUTDBJXJRKMK-GSVOUGTGSA-N 0.000 description 2
- QSJXEFYPDANLFS-UHFFFAOYSA-N Diacetyl Chemical compound CC(=O)C(C)=O QSJXEFYPDANLFS-UHFFFAOYSA-N 0.000 description 2
- 101150029707 ERBB2 gene Proteins 0.000 description 2
- 102100038132 Endogenous retrovirus group K member 6 Pro protein Human genes 0.000 description 2
- 101710091045 Envelope protein Proteins 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 102220508665 Ephrin type-A receptor 4_Q40A_mutation Human genes 0.000 description 2
- 101000803553 Eumenes pomiformis Venom peptide 3 Proteins 0.000 description 2
- 102000018233 Fibroblast Growth Factor Human genes 0.000 description 2
- 108050007372 Fibroblast Growth Factor Proteins 0.000 description 2
- 102220474004 Gamma-secretase subunit PEN-2_L26A_mutation Human genes 0.000 description 2
- 208000007465 Giant cell arteritis Diseases 0.000 description 2
- 102000034615 Glial cell line-derived neurotrophic factor Human genes 0.000 description 2
- 108091010837 Glial cell line-derived neurotrophic factor Proteins 0.000 description 2
- 102220585695 Glutamate receptor ionotropic, delta-2_D24A_mutation Human genes 0.000 description 2
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical compound O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 2
- 102000004858 Growth differentiation factor-9 Human genes 0.000 description 2
- 108090001086 Growth differentiation factor-9 Proteins 0.000 description 2
- 102100039939 Growth/differentiation factor 8 Human genes 0.000 description 2
- 102220562000 HLA class I histocompatibility antigen, A alpha chain_L15A_mutation Human genes 0.000 description 2
- 101000852023 Halorubrum pleomorphic virus 1 Envelope protein Proteins 0.000 description 2
- 101000583961 Halorubrum pleomorphic virus 1 Matrix protein Proteins 0.000 description 2
- 208000035186 Hemolytic Autoimmune Anemia Diseases 0.000 description 2
- 102100031000 Hepatoma-derived growth factor Human genes 0.000 description 2
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 2
- 101000884305 Homo sapiens B-cell receptor CD22 Proteins 0.000 description 2
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 description 2
- 101000798441 Homo sapiens Basigin Proteins 0.000 description 2
- 101000897480 Homo sapiens C-C motif chemokine 2 Proteins 0.000 description 2
- 101000916489 Homo sapiens Chondroitin sulfate proteoglycan 4 Proteins 0.000 description 2
- 101001027128 Homo sapiens Fibronectin Proteins 0.000 description 2
- 101000853000 Homo sapiens Interleukin-26 Proteins 0.000 description 2
- 101001133056 Homo sapiens Mucin-1 Proteins 0.000 description 2
- 101000623901 Homo sapiens Mucin-16 Proteins 0.000 description 2
- 101000595923 Homo sapiens Placenta growth factor Proteins 0.000 description 2
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 2
- 101000851370 Homo sapiens Tumor necrosis factor receptor superfamily member 9 Proteins 0.000 description 2
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 2
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 2
- 206010022004 Influenza like illness Diseases 0.000 description 2
- 108090001061 Insulin Proteins 0.000 description 2
- 102000004877 Insulin Human genes 0.000 description 2
- 108090000723 Insulin-Like Growth Factor I Proteins 0.000 description 2
- 102000014429 Insulin-like growth factor Human genes 0.000 description 2
- 102100040019 Interferon alpha-1/13 Human genes 0.000 description 2
- 102100040018 Interferon alpha-2 Human genes 0.000 description 2
- 108010005716 Interferon beta-1a Proteins 0.000 description 2
- 108010005714 Interferon beta-1b Proteins 0.000 description 2
- 102100026688 Interferon epsilon Human genes 0.000 description 2
- 101710147309 Interferon epsilon Proteins 0.000 description 2
- 102100022469 Interferon kappa Human genes 0.000 description 2
- 102100020989 Interferon lambda-2 Human genes 0.000 description 2
- 102100020992 Interferon lambda-3 Human genes 0.000 description 2
- 108010047761 Interferon-alpha Proteins 0.000 description 2
- 102000006992 Interferon-alpha Human genes 0.000 description 2
- 102000008070 Interferon-gamma Human genes 0.000 description 2
- 102100026018 Interleukin-1 receptor antagonist protein Human genes 0.000 description 2
- 101710144554 Interleukin-1 receptor antagonist protein Proteins 0.000 description 2
- 102100033096 Interleukin-17D Human genes 0.000 description 2
- 102000000588 Interleukin-2 Human genes 0.000 description 2
- 108010002350 Interleukin-2 Proteins 0.000 description 2
- 102100036679 Interleukin-26 Human genes 0.000 description 2
- 108010066979 Interleukin-27 Proteins 0.000 description 2
- 102100033502 Interleukin-37 Human genes 0.000 description 2
- 108010002616 Interleukin-5 Proteins 0.000 description 2
- 102000000743 Interleukin-5 Human genes 0.000 description 2
- 108010002586 Interleukin-7 Proteins 0.000 description 2
- 102000000704 Interleukin-7 Human genes 0.000 description 2
- 208000008839 Kidney Neoplasms Diseases 0.000 description 2
- SNDPXSYFESPGGJ-BYPYZUCNSA-N L-2-aminopentanoic acid Chemical compound CCC[C@H](N)C(O)=O SNDPXSYFESPGGJ-BYPYZUCNSA-N 0.000 description 2
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 2
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 2
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 2
- 239000004472 Lysine Substances 0.000 description 2
- 102000043131 MHC class II family Human genes 0.000 description 2
- 108091054438 MHC class II family Proteins 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- 102000000440 Melanoma-associated antigen Human genes 0.000 description 2
- 108050008953 Melanoma-associated antigen Proteins 0.000 description 2
- 108010052285 Membrane Proteins Proteins 0.000 description 2
- 102220608626 Methyl-CpG-binding domain protein 1_D32A_mutation Human genes 0.000 description 2
- 102220608646 Methyl-CpG-binding domain protein 1_R22K_mutation Human genes 0.000 description 2
- 101710081079 Minor spike protein H Proteins 0.000 description 2
- 102100023123 Mucin-16 Human genes 0.000 description 2
- 108010063954 Mucins Proteins 0.000 description 2
- 102000015728 Mucins Human genes 0.000 description 2
- 201000003793 Myelodysplastic syndrome Diseases 0.000 description 2
- 108010056852 Myostatin Proteins 0.000 description 2
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical class ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- 238000011789 NOD SCID mouse Methods 0.000 description 2
- 102220564245 Natural killer cells antigen CD94_S25A_mutation Human genes 0.000 description 2
- 102220498202 Negative regulator of P-body association_D35A_mutation Human genes 0.000 description 2
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 2
- 206010033128 Ovarian cancer Diseases 0.000 description 2
- 206010061535 Ovarian neoplasm Diseases 0.000 description 2
- 206010034277 Pemphigoid Diseases 0.000 description 2
- 102000015731 Peptide Hormones Human genes 0.000 description 2
- 108010038988 Peptide Hormones Proteins 0.000 description 2
- 201000007100 Pharyngitis Diseases 0.000 description 2
- 102100035194 Placenta growth factor Human genes 0.000 description 2
- 102100024616 Platelet endothelial cell adhesion molecule Human genes 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 206010036105 Polyneuropathy Diseases 0.000 description 2
- 239000004793 Polystyrene Substances 0.000 description 2
- 102220599460 Protein NPAT_F27A_mutation Human genes 0.000 description 2
- 101710188315 Protein X Proteins 0.000 description 2
- 101710118046 RNA-directed RNA polymerase Proteins 0.000 description 2
- MUPFEKGTMRGPLJ-RMMQSMQOSA-N Raffinose Natural products O(C[C@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O[C@@]2(CO)[C@H](O)[C@@H](O)[C@@H](CO)O2)O1)[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 MUPFEKGTMRGPLJ-RMMQSMQOSA-N 0.000 description 2
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 2
- 201000000582 Retinoblastoma Diseases 0.000 description 2
- 108010077895 Sarcosine Proteins 0.000 description 2
- 206010041067 Small cell lung cancer Diseases 0.000 description 2
- 208000021712 Soft tissue sarcoma Diseases 0.000 description 2
- 230000006044 T cell activation Effects 0.000 description 2
- 208000000389 T-cell leukemia Diseases 0.000 description 2
- 208000028530 T-cell lymphoblastic leukemia/lymphoma Diseases 0.000 description 2
- 206010042971 T-cell lymphoma Diseases 0.000 description 2
- MUMGGOZAMZWBJJ-DYKIIFRCSA-N Testostosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 MUMGGOZAMZWBJJ-DYKIIFRCSA-N 0.000 description 2
- 210000004241 Th2 cell Anatomy 0.000 description 2
- 102000036693 Thrombopoietin Human genes 0.000 description 2
- 108010041111 Thrombopoietin Proteins 0.000 description 2
- 102100023935 Transmembrane glycoprotein NMB Human genes 0.000 description 2
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 2
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 2
- 108060008683 Tumor Necrosis Factor Receptor Proteins 0.000 description 2
- 102100031988 Tumor necrosis factor ligand superfamily member 6 Human genes 0.000 description 2
- 102100036856 Tumor necrosis factor receptor superfamily member 9 Human genes 0.000 description 2
- MUPFEKGTMRGPLJ-UHFFFAOYSA-N UNPD196149 Natural products OC1C(O)C(CO)OC1(CO)OC1C(O)C(O)C(O)C(COC2C(C(O)C(O)C(CO)O2)O)O1 MUPFEKGTMRGPLJ-UHFFFAOYSA-N 0.000 description 2
- 108090000435 Urokinase-type plasminogen activator Proteins 0.000 description 2
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 2
- 206010046851 Uveitis Diseases 0.000 description 2
- 101710108545 Viral protein 1 Proteins 0.000 description 2
- 208000008383 Wilms tumor Diseases 0.000 description 2
- TVXBFESIOXBWNM-UHFFFAOYSA-N Xylitol Natural products OCCC(O)C(O)C(O)CCO TVXBFESIOXBWNM-UHFFFAOYSA-N 0.000 description 2
- OIPILFWXSMYKGL-UHFFFAOYSA-N acetylcholine Chemical compound CC(=O)OCC[N+](C)(C)C OIPILFWXSMYKGL-UHFFFAOYSA-N 0.000 description 2
- 229960004373 acetylcholine Drugs 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 230000010933 acylation Effects 0.000 description 2
- 238000005917 acylation reaction Methods 0.000 description 2
- 208000009956 adenocarcinoma Diseases 0.000 description 2
- UCTWMZQNUQWSLP-UHFFFAOYSA-N adrenaline Chemical compound CNCC(O)C1=CC=C(O)C(O)=C1 UCTWMZQNUQWSLP-UHFFFAOYSA-N 0.000 description 2
- ULCUCJFASIJEOE-NPECTJMMSA-N adrenomedullin Chemical compound C([C@@H](C(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)NCC(=O)N[C@@H]1C(N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)NCC(=O)N[C@H](C(=O)N[C@@H](CSSC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)[C@@H](C)O)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=CC=C1 ULCUCJFASIJEOE-NPECTJMMSA-N 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 235000004279 alanine Nutrition 0.000 description 2
- 230000029936 alkylation Effects 0.000 description 2
- 238000005804 alkylation reaction Methods 0.000 description 2
- FPIPGXGPPPQFEQ-OVSJKPMPSA-N all-trans-retinol Chemical compound OC\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-OVSJKPMPSA-N 0.000 description 2
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 2
- 230000004075 alteration Effects 0.000 description 2
- 210000002821 alveolar epithelial cell Anatomy 0.000 description 2
- 125000000539 amino acid group Chemical group 0.000 description 2
- 208000007502 anemia Diseases 0.000 description 2
- 230000033115 angiogenesis Effects 0.000 description 2
- 229940045799 anthracyclines and related substance Drugs 0.000 description 2
- 229940046836 anti-estrogen Drugs 0.000 description 2
- 230000001833 anti-estrogenic effect Effects 0.000 description 2
- 230000006023 anti-tumor response Effects 0.000 description 2
- 229940121357 antivirals Drugs 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- 206010003246 arthritis Diseases 0.000 description 2
- 208000006673 asthma Diseases 0.000 description 2
- 230000001363 autoimmune Effects 0.000 description 2
- 201000003710 autoimmune thrombocytopenic purpura Diseases 0.000 description 2
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 2
- 229960001215 bendamustine hydrochloride Drugs 0.000 description 2
- MMIMIFULGMZVPO-UHFFFAOYSA-N benzyl 3-bromo-2,6-dinitro-5-phenylmethoxybenzoate Chemical compound [O-][N+](=O)C1=C(C(=O)OCC=2C=CC=CC=2)C([N+](=O)[O-])=C(Br)C=C1OCC1=CC=CC=C1 MMIMIFULGMZVPO-UHFFFAOYSA-N 0.000 description 2
- 238000004166 bioassay Methods 0.000 description 2
- 230000008512 biological response Effects 0.000 description 2
- 229940112869 bone morphogenetic protein Drugs 0.000 description 2
- 229940077737 brain-derived neurotrophic factor Drugs 0.000 description 2
- 210000004899 c-terminal region Anatomy 0.000 description 2
- 102200002501 c.98G>A Human genes 0.000 description 2
- 230000001275 ca(2+)-mobilization Effects 0.000 description 2
- 230000021235 carbamoylation Effects 0.000 description 2
- 125000000837 carbohydrate group Chemical group 0.000 description 2
- 229960004562 carboplatin Drugs 0.000 description 2
- 150000007942 carboxylates Chemical class 0.000 description 2
- 238000000423 cell based assay Methods 0.000 description 2
- 230000030833 cell death Effects 0.000 description 2
- 238000012054 celltiter-glo Methods 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 230000005754 cellular signaling Effects 0.000 description 2
- 208000025997 central nervous system neoplasm Diseases 0.000 description 2
- 201000010881 cervical cancer Diseases 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 2
- 201000010897 colon adenocarcinoma Diseases 0.000 description 2
- 238000012875 competitive assay Methods 0.000 description 2
- 230000021615 conjugation Effects 0.000 description 2
- 230000001276 controlling effect Effects 0.000 description 2
- 239000003431 cross linking reagent Substances 0.000 description 2
- XLJMAIOERFSOGZ-UHFFFAOYSA-M cyanate Chemical compound [O-]C#N XLJMAIOERFSOGZ-UHFFFAOYSA-M 0.000 description 2
- XVOYSCVBGLVSOL-UHFFFAOYSA-N cysteic acid Chemical compound OC(=O)C(N)CS(O)(=O)=O XVOYSCVBGLVSOL-UHFFFAOYSA-N 0.000 description 2
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 2
- 235000018417 cysteine Nutrition 0.000 description 2
- 229960000684 cytarabine Drugs 0.000 description 2
- 230000016396 cytokine production Effects 0.000 description 2
- 229940127089 cytotoxic agent Drugs 0.000 description 2
- 229960003901 dacarbazine Drugs 0.000 description 2
- 230000003210 demyelinating effect Effects 0.000 description 2
- 229960002923 denileukin diftitox Drugs 0.000 description 2
- 229960003957 dexamethasone Drugs 0.000 description 2
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 2
- 229960003638 dopamine Drugs 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 229940000406 drug candidate Drugs 0.000 description 2
- 229960004137 elotuzumab Drugs 0.000 description 2
- 229940088598 enzyme Drugs 0.000 description 2
- 230000007705 epithelial mesenchymal transition Effects 0.000 description 2
- 230000010437 erythropoiesis Effects 0.000 description 2
- 239000000328 estrogen antagonist Substances 0.000 description 2
- 239000013604 expression vector Substances 0.000 description 2
- 229940126864 fibroblast growth factor Drugs 0.000 description 2
- 210000000630 fibrocyte Anatomy 0.000 description 2
- 238000000684 flow cytometry Methods 0.000 description 2
- 229960003692 gamma aminobutyric acid Drugs 0.000 description 2
- 201000006585 gastric adenocarcinoma Diseases 0.000 description 2
- 210000001035 gastrointestinal tract Anatomy 0.000 description 2
- 150000004676 glycans Chemical class 0.000 description 2
- KWIUHFFTVRNATP-UHFFFAOYSA-N glycine betaine Chemical compound C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 2
- LEQAOMBKQFMDFZ-UHFFFAOYSA-N glyoxal Chemical compound O=CC=O LEQAOMBKQFMDFZ-UHFFFAOYSA-N 0.000 description 2
- 208000035474 group of disease Diseases 0.000 description 2
- 230000002489 hematologic effect Effects 0.000 description 2
- 231100000234 hepatic damage Toxicity 0.000 description 2
- 208000006454 hepatitis Diseases 0.000 description 2
- 231100000283 hepatitis Toxicity 0.000 description 2
- 108010052188 hepatoma-derived growth factor Proteins 0.000 description 2
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 2
- 102000052645 human CD38 Human genes 0.000 description 2
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 2
- 230000001900 immune effect Effects 0.000 description 2
- 230000001976 improved effect Effects 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 229940125396 insulin Drugs 0.000 description 2
- 102000006495 integrins Human genes 0.000 description 2
- 108010044426 integrins Proteins 0.000 description 2
- 108010018844 interferon type III Proteins 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- JDNTWHVOXJZDSN-UHFFFAOYSA-N iodoacetic acid Chemical compound OC(=O)CI JDNTWHVOXJZDSN-UHFFFAOYSA-N 0.000 description 2
- 229950002884 lexatumumab Drugs 0.000 description 2
- 108010052322 limitin Proteins 0.000 description 2
- 201000007270 liver cancer Diseases 0.000 description 2
- 230000008818 liver damage Effects 0.000 description 2
- 208000019423 liver disease Diseases 0.000 description 2
- 206010025135 lupus erythematosus Diseases 0.000 description 2
- 230000003211 malignant effect Effects 0.000 description 2
- 210000004962 mammalian cell Anatomy 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 239000003550 marker Substances 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 229960004961 mechlorethamine Drugs 0.000 description 2
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical compound ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 2
- 229960001913 mecysteine Drugs 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- HEBKCHPVOIAQTA-UHFFFAOYSA-N meso ribitol Natural products OCC(O)C(O)C(O)CO HEBKCHPVOIAQTA-UHFFFAOYSA-N 0.000 description 2
- 108020004999 messenger RNA Proteins 0.000 description 2
- 150000002772 monosaccharides Chemical class 0.000 description 2
- 229940051875 mucins Drugs 0.000 description 2
- 231100000707 mutagenic chemical Toxicity 0.000 description 2
- 230000000869 mutational effect Effects 0.000 description 2
- 210000000651 myofibroblast Anatomy 0.000 description 2
- 208000004995 necrotizing enterocolitis Diseases 0.000 description 2
- 229960000801 nelarabine Drugs 0.000 description 2
- 229940053128 nerve growth factor Drugs 0.000 description 2
- 238000007857 nested PCR Methods 0.000 description 2
- 239000002858 neurotransmitter agent Substances 0.000 description 2
- 210000004967 non-hematopoietic stem cell Anatomy 0.000 description 2
- 229960002748 norepinephrine Drugs 0.000 description 2
- SFLSHLFXELFNJZ-UHFFFAOYSA-N norepinephrine Natural products NCC(O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-UHFFFAOYSA-N 0.000 description 2
- 108020004707 nucleic acids Proteins 0.000 description 2
- 102000039446 nucleic acids Human genes 0.000 description 2
- 150000007523 nucleic acids Chemical class 0.000 description 2
- 239000002773 nucleotide Substances 0.000 description 2
- 125000003729 nucleotide group Chemical group 0.000 description 2
- 201000008106 ocular cancer Diseases 0.000 description 2
- 230000009437 off-target effect Effects 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 229960000402 palivizumab Drugs 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 230000001717 pathogenic effect Effects 0.000 description 2
- 230000001575 pathological effect Effects 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 2
- 238000002823 phage display Methods 0.000 description 2
- 229940124531 pharmaceutical excipient Drugs 0.000 description 2
- OJUGVDODNPJEEC-UHFFFAOYSA-N phenylglyoxal Chemical compound O=CC(=O)C1=CC=CC=C1 OJUGVDODNPJEEC-UHFFFAOYSA-N 0.000 description 2
- 239000002953 phosphate buffered saline Substances 0.000 description 2
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 2
- 239000013612 plasmid Substances 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 229960002169 plerixafor Drugs 0.000 description 2
- 230000007824 polyneuropathy Effects 0.000 description 2
- 239000005017 polysaccharide Substances 0.000 description 2
- 229960000214 pralatrexate Drugs 0.000 description 2
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 2
- 229960001586 procarbazine hydrochloride Drugs 0.000 description 2
- 230000002062 proliferating effect Effects 0.000 description 2
- XNSAINXGIQZQOO-SRVKXCTJSA-N protirelin Chemical compound NC(=O)[C@@H]1CCCN1C(=O)[C@@H](NC(=O)[C@H]1NC(=O)CC1)CC1=CN=CN1 XNSAINXGIQZQOO-SRVKXCTJSA-N 0.000 description 2
- 230000002685 pulmonary effect Effects 0.000 description 2
- MUPFEKGTMRGPLJ-ZQSKZDJDSA-N raffinose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO[C@@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O2)O)O1 MUPFEKGTMRGPLJ-ZQSKZDJDSA-N 0.000 description 2
- 238000002708 random mutagenesis Methods 0.000 description 2
- 230000001172 regenerating effect Effects 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 229960003452 romidepsin Drugs 0.000 description 2
- 102220075008 rs139397227 Human genes 0.000 description 2
- 230000037390 scarring Effects 0.000 description 2
- 230000035945 sensitivity Effects 0.000 description 2
- 210000003491 skin Anatomy 0.000 description 2
- 208000000587 small cell lung carcinoma Diseases 0.000 description 2
- 239000012279 sodium borohydride Substances 0.000 description 2
- 229910000033 sodium borohydride Inorganic materials 0.000 description 2
- 125000006850 spacer group Chemical group 0.000 description 2
- 150000003431 steroids Chemical class 0.000 description 2
- 206010043207 temporal arteritis Diseases 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- AYEKOFBPNLCAJY-UHFFFAOYSA-O thiamine pyrophosphate Chemical compound CC1=C(CCOP(O)(=O)OP(O)(O)=O)SC=[N+]1CC1=CN=C(C)N=C1N AYEKOFBPNLCAJY-UHFFFAOYSA-O 0.000 description 2
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 2
- 206010044412 transitional cell carcinoma Diseases 0.000 description 2
- 230000014616 translation Effects 0.000 description 2
- 229960000575 trastuzumab Drugs 0.000 description 2
- 102000003298 tumor necrosis factor receptor Human genes 0.000 description 2
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 2
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 2
- 201000005112 urinary bladder cancer Diseases 0.000 description 2
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 2
- 238000002255 vaccination Methods 0.000 description 2
- 230000035899 viability Effects 0.000 description 2
- 230000029812 viral genome replication Effects 0.000 description 2
- 229960000237 vorinostat Drugs 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- 239000000811 xylitol Substances 0.000 description 2
- 235000010447 xylitol Nutrition 0.000 description 2
- HEBKCHPVOIAQTA-SCDXWVJYSA-N xylitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)CO HEBKCHPVOIAQTA-SCDXWVJYSA-N 0.000 description 2
- 229960002675 xylitol Drugs 0.000 description 2
- LUBKKVGXMXTXOZ-QGZVFWFLSA-N (+)-geodin Chemical compound COC(=O)C1=CC(=O)C=C(OC)[C@@]11C(=O)C(C(O)=C(Cl)C(C)=C2Cl)=C2O1 LUBKKVGXMXTXOZ-QGZVFWFLSA-N 0.000 description 1
- OGNSCSPNOLGXSM-UHFFFAOYSA-N (+/-)-DABA Natural products NCCC(N)C(O)=O OGNSCSPNOLGXSM-UHFFFAOYSA-N 0.000 description 1
- CWLQUGTUXBXTLF-RXMQYKEDSA-N (2r)-1-methylpyrrolidine-2-carboxylic acid Chemical compound CN1CCC[C@@H]1C(O)=O CWLQUGTUXBXTLF-RXMQYKEDSA-N 0.000 description 1
- YAXAFCHJCYILRU-RXMQYKEDSA-N (2r)-2-(methylamino)-4-methylsulfanylbutanoic acid Chemical compound CN[C@@H](C(O)=O)CCSC YAXAFCHJCYILRU-RXMQYKEDSA-N 0.000 description 1
- XLBVNMSMFQMKEY-SCSAIBSYSA-N (2r)-2-(methylamino)pentanedioic acid Chemical compound CN[C@@H](C(O)=O)CCC(O)=O XLBVNMSMFQMKEY-SCSAIBSYSA-N 0.000 description 1
- GDFAOVXKHJXLEI-GSVOUGTGSA-N (2r)-2-(methylamino)propanoic acid Chemical compound CN[C@H](C)C(O)=O GDFAOVXKHJXLEI-GSVOUGTGSA-N 0.000 description 1
- SCIFESDRCALIIM-SECBINFHSA-N (2r)-2-(methylazaniumyl)-3-phenylpropanoate Chemical compound CN[C@@H](C(O)=O)CC1=CC=CC=C1 SCIFESDRCALIIM-SECBINFHSA-N 0.000 description 1
- CYZKJBZEIFWZSR-ZCFIWIBFSA-N (2r)-3-(1h-imidazol-5-yl)-2-(methylamino)propanoic acid Chemical compound CN[C@@H](C(O)=O)CC1=CN=CN1 CYZKJBZEIFWZSR-ZCFIWIBFSA-N 0.000 description 1
- CZCIKBSVHDNIDH-LLVKDONJSA-N (2r)-3-(1h-indol-3-yl)-2-(methylamino)propanoic acid Chemical compound C1=CC=C2C(C[C@@H](NC)C(O)=O)=CNC2=C1 CZCIKBSVHDNIDH-LLVKDONJSA-N 0.000 description 1
- AKCRVYNORCOYQT-RXMQYKEDSA-N (2r)-3-methyl-2-(methylazaniumyl)butanoate Chemical compound C[NH2+][C@H](C(C)C)C([O-])=O AKCRVYNORCOYQT-RXMQYKEDSA-N 0.000 description 1
- LNSMPSPTFDIWRQ-GSVOUGTGSA-N (2r)-4-amino-2-(methylamino)-4-oxobutanoic acid Chemical compound CN[C@@H](C(O)=O)CC(N)=O LNSMPSPTFDIWRQ-GSVOUGTGSA-N 0.000 description 1
- NTWVQPHTOUKMDI-RXMQYKEDSA-N (2r)-5-(diaminomethylideneamino)-2-(methylamino)pentanoic acid Chemical compound CN[C@@H](C(O)=O)CCCNC(N)=N NTWVQPHTOUKMDI-RXMQYKEDSA-N 0.000 description 1
- KSZFSNZOGAXEGH-SCSAIBSYSA-N (2r)-5-amino-2-(methylamino)-5-oxopentanoic acid Chemical compound CN[C@@H](C(O)=O)CCC(N)=O KSZFSNZOGAXEGH-SCSAIBSYSA-N 0.000 description 1
- OZRWQPFBXDVLAH-RXMQYKEDSA-N (2r)-5-amino-2-(methylamino)pentanoic acid Chemical compound CN[C@@H](C(O)=O)CCCN OZRWQPFBXDVLAH-RXMQYKEDSA-N 0.000 description 1
- KSPIYJQBLVDRRI-NTSWFWBYSA-N (2r,3s)-3-methyl-2-(methylazaniumyl)pentanoate Chemical compound CC[C@H](C)[C@@H](NC)C(O)=O KSPIYJQBLVDRRI-NTSWFWBYSA-N 0.000 description 1
- BVAUMRCGVHUWOZ-ZETCQYMHSA-N (2s)-2-(cyclohexylazaniumyl)propanoate Chemical compound OC(=O)[C@H](C)NC1CCCCC1 BVAUMRCGVHUWOZ-ZETCQYMHSA-N 0.000 description 1
- LDUWTIUXPVCEQF-LURJTMIESA-N (2s)-2-(cyclopentylamino)propanoic acid Chemical compound OC(=O)[C@H](C)NC1CCCC1 LDUWTIUXPVCEQF-LURJTMIESA-N 0.000 description 1
- NVXKJPGRZSDYPK-JTQLQIEISA-N (2s)-2-(methylamino)-4-phenylbutanoic acid Chemical compound CN[C@H](C(O)=O)CCC1=CC=CC=C1 NVXKJPGRZSDYPK-JTQLQIEISA-N 0.000 description 1
- HOKKHZGPKSLGJE-VKHMYHEASA-N (2s)-2-(methylamino)butanedioic acid Chemical compound CN[C@H](C(O)=O)CC(O)=O HOKKHZGPKSLGJE-VKHMYHEASA-N 0.000 description 1
- FPDYKABXINADKS-LURJTMIESA-N (2s)-2-(methylazaniumyl)hexanoate Chemical compound CCCC[C@H](NC)C(O)=O FPDYKABXINADKS-LURJTMIESA-N 0.000 description 1
- HCPKYUNZBPVCHC-YFKPBYRVSA-N (2s)-2-(methylazaniumyl)pentanoate Chemical compound CCC[C@H](NC)C(O)=O HCPKYUNZBPVCHC-YFKPBYRVSA-N 0.000 description 1
- WTDHSXGBDZBWAW-QMMMGPOBSA-N (2s)-2-[cyclohexyl(methyl)azaniumyl]propanoate Chemical compound OC(=O)[C@H](C)N(C)C1CCCCC1 WTDHSXGBDZBWAW-QMMMGPOBSA-N 0.000 description 1
- IUYZJPXOXGRNNE-ZETCQYMHSA-N (2s)-2-[cyclopentyl(methyl)amino]propanoic acid Chemical compound OC(=O)[C@H](C)N(C)C1CCCC1 IUYZJPXOXGRNNE-ZETCQYMHSA-N 0.000 description 1
- NPDBDJFLKKQMCM-SCSAIBSYSA-N (2s)-2-amino-3,3-dimethylbutanoic acid Chemical compound CC(C)(C)[C@H](N)C(O)=O NPDBDJFLKKQMCM-SCSAIBSYSA-N 0.000 description 1
- KWWFNGCKGYUCLC-RXMQYKEDSA-N (2s)-3,3-dimethyl-2-(methylamino)butanoic acid Chemical compound CN[C@H](C(O)=O)C(C)(C)C KWWFNGCKGYUCLC-RXMQYKEDSA-N 0.000 description 1
- XKZCXMNMUMGDJG-AWEZNQCLSA-N (2s)-3-[(6-acetylnaphthalen-2-yl)amino]-2-aminopropanoic acid Chemical compound C1=C(NC[C@H](N)C(O)=O)C=CC2=CC(C(=O)C)=CC=C21 XKZCXMNMUMGDJG-AWEZNQCLSA-N 0.000 description 1
- LNSMPSPTFDIWRQ-VKHMYHEASA-N (2s)-4-amino-2-(methylamino)-4-oxobutanoic acid Chemical compound CN[C@H](C(O)=O)CC(N)=O LNSMPSPTFDIWRQ-VKHMYHEASA-N 0.000 description 1
- XJODGRWDFZVTKW-LURJTMIESA-N (2s)-4-methyl-2-(methylamino)pentanoic acid Chemical compound CN[C@H](C(O)=O)CC(C)C XJODGRWDFZVTKW-LURJTMIESA-N 0.000 description 1
- KSZFSNZOGAXEGH-BYPYZUCNSA-N (2s)-5-amino-2-(methylamino)-5-oxopentanoic acid Chemical compound CN[C@H](C(O)=O)CCC(N)=O KSZFSNZOGAXEGH-BYPYZUCNSA-N 0.000 description 1
- OZRWQPFBXDVLAH-YFKPBYRVSA-N (2s)-5-amino-2-(methylamino)pentanoic acid Chemical compound CN[C@H](C(O)=O)CCCN OZRWQPFBXDVLAH-YFKPBYRVSA-N 0.000 description 1
- RHMALYOXPBRJBG-WXHCCQJTSA-N (2s)-6-amino-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-6-amino-2-[[(2s)-2-[[(2s)-2-[[2-[[(2s,3r)-2-[[(2s)-2-[[2-[[2-[[(2r)-2-amino-3-phenylpropanoyl]amino]acetyl]amino]acetyl]amino]-3-phenylpropanoyl]amino]-3-hydroxybutanoyl]amino]acetyl]amino]propanoyl]amino]- Chemical compound C([C@@H](C(=O)N[C@@H]([C@H](O)C)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CCCCN)C(N)=O)NC(=O)CNC(=O)CNC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 RHMALYOXPBRJBG-WXHCCQJTSA-N 0.000 description 1
- LJRDOKAZOAKLDU-UDXJMMFXSA-N (2s,3s,4r,5r,6r)-5-amino-2-(aminomethyl)-6-[(2r,3s,4r,5s)-5-[(1r,2r,3s,5r,6s)-3,5-diamino-2-[(2s,3r,4r,5s,6r)-3-amino-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-6-hydroxycyclohexyl]oxy-4-hydroxy-2-(hydroxymethyl)oxolan-3-yl]oxyoxane-3,4-diol;sulfuric ac Chemical compound OS(O)(=O)=O.N[C@@H]1[C@@H](O)[C@H](O)[C@H](CN)O[C@@H]1O[C@H]1[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](N)C[C@@H](N)[C@@H]2O)O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)N)O[C@@H]1CO LJRDOKAZOAKLDU-UDXJMMFXSA-N 0.000 description 1
- LAQPKDLYOBZWBT-NYLDSJSYSA-N (2s,4s,5r,6r)-5-acetamido-2-{[(2s,3r,4s,5s,6r)-2-{[(2r,3r,4r,5r)-5-acetamido-1,2-dihydroxy-6-oxo-4-{[(2s,3s,4r,5s,6s)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxy}hexan-3-yl]oxy}-3,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy}-4-hydroxy-6-[(1r,2r)-1,2,3-trihydrox Chemical compound O[C@H]1[C@H](O)[C@H](O)[C@H](C)O[C@H]1O[C@H]([C@@H](NC(C)=O)C=O)[C@@H]([C@H](O)CO)O[C@H]1[C@H](O)[C@@H](O[C@]2(O[C@H]([C@H](NC(C)=O)[C@@H](O)C2)[C@H](O)[C@H](O)CO)C(O)=O)[C@@H](O)[C@@H](CO)O1 LAQPKDLYOBZWBT-NYLDSJSYSA-N 0.000 description 1
- KMOUUZVZFBCRAM-OLQVQODUSA-N (3as,7ar)-3a,4,7,7a-tetrahydro-2-benzofuran-1,3-dione Chemical compound C1C=CC[C@@H]2C(=O)OC(=O)[C@@H]21 KMOUUZVZFBCRAM-OLQVQODUSA-N 0.000 description 1
- NMWKYTGJWUAZPZ-WWHBDHEGSA-N (4S)-4-[[(4R,7S,10S,16S,19S,25S,28S,31R)-31-[[(2S)-2-[[(1R,6R,9S,12S,18S,21S,24S,27S,30S,33S,36S,39S,42R,47R,53S,56S,59S,62S,65S,68S,71S,76S,79S,85S)-47-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-amino-3-methylbutanoyl]amino]-3-methylbutanoyl]amino]-3-hydroxypropanoyl]amino]-3-(1H-imidazol-4-yl)propanoyl]amino]-3-phenylpropanoyl]amino]-4-oxobutanoyl]amino]-3-carboxypropanoyl]amino]-18-(4-aminobutyl)-27,68-bis(3-amino-3-oxopropyl)-36,71,76-tribenzyl-39-(3-carbamimidamidopropyl)-24-(2-carboxyethyl)-21,56-bis(carboxymethyl)-65,85-bis[(1R)-1-hydroxyethyl]-59-(hydroxymethyl)-62,79-bis(1H-imidazol-4-ylmethyl)-9-methyl-33-(2-methylpropyl)-8,11,17,20,23,26,29,32,35,38,41,48,54,57,60,63,66,69,72,74,77,80,83,86-tetracosaoxo-30-propan-2-yl-3,4,44,45-tetrathia-7,10,16,19,22,25,28,31,34,37,40,49,55,58,61,64,67,70,73,75,78,81,84,87-tetracosazatetracyclo[40.31.14.012,16.049,53]heptaoctacontane-6-carbonyl]amino]-3-methylbutanoyl]amino]-7-(3-carbamimidamidopropyl)-25-(hydroxymethyl)-19-[(4-hydroxyphenyl)methyl]-28-(1H-imidazol-4-ylmethyl)-10-methyl-6,9,12,15,18,21,24,27,30-nonaoxo-16-propan-2-yl-1,2-dithia-5,8,11,14,17,20,23,26,29-nonazacyclodotriacontane-4-carbonyl]amino]-5-[[(2S)-1-[[(2S)-1-[[(2S)-3-carboxy-1-[[(2S)-1-[[(2S)-1-[[(1S)-1-carboxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-1-oxopropan-2-yl]amino]-1-oxopropan-2-yl]amino]-3-(1H-imidazol-4-yl)-1-oxopropan-2-yl]amino]-5-oxopentanoic acid Chemical compound CC(C)C[C@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](Cc1c[nH]cn1)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H]1CSSC[C@H](NC(=O)[C@@H](NC(=O)[C@@H]2CSSC[C@@H]3NC(=O)[C@H](Cc4ccccc4)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](Cc4c[nH]cn4)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H]4CCCN4C(=O)[C@H](CSSC[C@H](NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](Cc4c[nH]cn4)NC(=O)[C@H](Cc4ccccc4)NC3=O)[C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](Cc3ccccc3)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N3CCC[C@H]3C(=O)N[C@@H](C)C(=O)N2)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](Cc2ccccc2)NC(=O)[C@H](Cc2c[nH]cn2)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@@H](N)C(C)C)C(C)C)[C@@H](C)O)C(C)C)C(=O)N[C@@H](Cc2c[nH]cn2)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](Cc2ccc(O)cc2)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1)C(=O)N[C@@H](C)C(O)=O NMWKYTGJWUAZPZ-WWHBDHEGSA-N 0.000 description 1
- UCTWMZQNUQWSLP-VIFPVBQESA-N (R)-adrenaline Chemical compound CNC[C@H](O)C1=CC=C(O)C(O)=C1 UCTWMZQNUQWSLP-VIFPVBQESA-N 0.000 description 1
- 229930182837 (R)-adrenaline Natural products 0.000 description 1
- OWEGMIWEEQEYGQ-UHFFFAOYSA-N 100676-05-9 Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC2C(OC(O)C(O)C2O)CO)O1 OWEGMIWEEQEYGQ-UHFFFAOYSA-N 0.000 description 1
- FPIPGXGPPPQFEQ-UHFFFAOYSA-N 13-cis retinol Natural products OCC=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-UHFFFAOYSA-N 0.000 description 1
- 101150028074 2 gene Proteins 0.000 description 1
- NHJVRSWLHSJWIN-UHFFFAOYSA-N 2,4,6-trinitrobenzenesulfonic acid Chemical compound OS(=O)(=O)C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O NHJVRSWLHSJWIN-UHFFFAOYSA-N 0.000 description 1
- 150000003923 2,5-pyrrolediones Chemical class 0.000 description 1
- WAAJQPAIOASFSC-UHFFFAOYSA-N 2-(1-hydroxyethylamino)acetic acid Chemical compound CC(O)NCC(O)=O WAAJQPAIOASFSC-UHFFFAOYSA-N 0.000 description 1
- UEQSFWNXRZJTKB-UHFFFAOYSA-N 2-(2,2-diphenylethylamino)acetic acid Chemical compound C=1C=CC=CC=1C(CNCC(=O)O)C1=CC=CC=C1 UEQSFWNXRZJTKB-UHFFFAOYSA-N 0.000 description 1
- UEJJHQNACJXSKW-UHFFFAOYSA-N 2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1C1CCC(=O)NC1=O UEJJHQNACJXSKW-UHFFFAOYSA-N 0.000 description 1
- PIINGYXNCHTJTF-UHFFFAOYSA-N 2-(2-azaniumylethylamino)acetate Chemical compound NCCNCC(O)=O PIINGYXNCHTJTF-UHFFFAOYSA-N 0.000 description 1
- XCDGCRLSSSSBIA-UHFFFAOYSA-N 2-(2-methylsulfanylethylamino)acetic acid Chemical compound CSCCNCC(O)=O XCDGCRLSSSSBIA-UHFFFAOYSA-N 0.000 description 1
- DHGYLUFLENKZHH-UHFFFAOYSA-N 2-(3-aminopropylamino)acetic acid Chemical compound NCCCNCC(O)=O DHGYLUFLENKZHH-UHFFFAOYSA-N 0.000 description 1
- OGAULEBSQQMUKP-UHFFFAOYSA-N 2-(4-aminobutylamino)acetic acid Chemical compound NCCCCNCC(O)=O OGAULEBSQQMUKP-UHFFFAOYSA-N 0.000 description 1
- KGSVNOLLROCJQM-UHFFFAOYSA-N 2-(benzylamino)acetic acid Chemical compound OC(=O)CNCC1=CC=CC=C1 KGSVNOLLROCJQM-UHFFFAOYSA-N 0.000 description 1
- KFDPCYZHENQOBV-UHFFFAOYSA-N 2-(bromomethyl)-4-nitrophenol Chemical compound OC1=CC=C([N+]([O-])=O)C=C1CBr KFDPCYZHENQOBV-UHFFFAOYSA-N 0.000 description 1
- IVCQRTJVLJXKKJ-UHFFFAOYSA-N 2-(butan-2-ylazaniumyl)acetate Chemical compound CCC(C)NCC(O)=O IVCQRTJVLJXKKJ-UHFFFAOYSA-N 0.000 description 1
- FALRKNHUBBKYCC-UHFFFAOYSA-N 2-(chloromethyl)pyridine-3-carbonitrile Chemical compound ClCC1=NC=CC=C1C#N FALRKNHUBBKYCC-UHFFFAOYSA-N 0.000 description 1
- KQLGGQARRCMYGD-UHFFFAOYSA-N 2-(cyclobutylamino)acetic acid Chemical compound OC(=O)CNC1CCC1 KQLGGQARRCMYGD-UHFFFAOYSA-N 0.000 description 1
- DICMQVOBSKLBBN-UHFFFAOYSA-N 2-(cyclodecylamino)acetic acid Chemical compound OC(=O)CNC1CCCCCCCCC1 DICMQVOBSKLBBN-UHFFFAOYSA-N 0.000 description 1
- NPLBBQAAYSJEMO-UHFFFAOYSA-N 2-(cycloheptylazaniumyl)acetate Chemical compound OC(=O)CNC1CCCCCC1 NPLBBQAAYSJEMO-UHFFFAOYSA-N 0.000 description 1
- OQMYZVWIXPPDDE-UHFFFAOYSA-N 2-(cyclohexylazaniumyl)acetate Chemical compound OC(=O)CNC1CCCCC1 OQMYZVWIXPPDDE-UHFFFAOYSA-N 0.000 description 1
- PNKNDNFLQNMQJL-UHFFFAOYSA-N 2-(cyclooctylazaniumyl)acetate Chemical compound OC(=O)CNC1CCCCCCC1 PNKNDNFLQNMQJL-UHFFFAOYSA-N 0.000 description 1
- DXQCCQKRNWMECV-UHFFFAOYSA-N 2-(cyclopropylazaniumyl)acetate Chemical compound OC(=O)CNC1CC1 DXQCCQKRNWMECV-UHFFFAOYSA-N 0.000 description 1
- PRVOMNLNSHAUEI-UHFFFAOYSA-N 2-(cycloundecylamino)acetic acid Chemical compound OC(=O)CNC1CCCCCCCCCC1 PRVOMNLNSHAUEI-UHFFFAOYSA-N 0.000 description 1
- HEPOIJKOXBKKNJ-UHFFFAOYSA-N 2-(propan-2-ylazaniumyl)acetate Chemical compound CC(C)NCC(O)=O HEPOIJKOXBKKNJ-UHFFFAOYSA-N 0.000 description 1
- AWEZYTUWDZADKR-UHFFFAOYSA-N 2-[(2-amino-2-oxoethyl)azaniumyl]acetate Chemical compound NC(=O)CNCC(O)=O AWEZYTUWDZADKR-UHFFFAOYSA-N 0.000 description 1
- MNDBDVPDSHGIHR-UHFFFAOYSA-N 2-[(3-amino-3-oxopropyl)amino]acetic acid Chemical compound NC(=O)CCNCC(O)=O MNDBDVPDSHGIHR-UHFFFAOYSA-N 0.000 description 1
- YDBPFLZECVWPSH-UHFFFAOYSA-N 2-[3-(diaminomethylideneamino)propylamino]acetic acid Chemical compound NC(=N)NCCCNCC(O)=O YDBPFLZECVWPSH-UHFFFAOYSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- WTOFYLAWDLQMBZ-UHFFFAOYSA-N 2-azaniumyl-3-thiophen-2-ylpropanoate Chemical compound OC(=O)C(N)CC1=CC=CS1 WTOFYLAWDLQMBZ-UHFFFAOYSA-N 0.000 description 1
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 description 1
- LJGHYPLBDBRCRZ-UHFFFAOYSA-N 3-(3-aminophenyl)sulfonylaniline Chemical compound NC1=CC=CC(S(=O)(=O)C=2C=C(N)C=CC=2)=C1 LJGHYPLBDBRCRZ-UHFFFAOYSA-N 0.000 description 1
- WUIABRMSWOKTOF-OYALTWQYSA-N 3-[[2-[2-[2-[[(2s,3r)-2-[[(2s,3s,4r)-4-[[(2s,3r)-2-[[6-amino-2-[(1s)-3-amino-1-[[(2s)-2,3-diamino-3-oxopropyl]amino]-3-oxopropyl]-5-methylpyrimidine-4-carbonyl]amino]-3-[(2r,3s,4s,5s,6s)-3-[(2r,3s,4s,5r,6r)-4-carbamoyloxy-3,5-dihydroxy-6-(hydroxymethyl)ox Chemical compound OS([O-])(=O)=O.N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1NC=NC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C WUIABRMSWOKTOF-OYALTWQYSA-N 0.000 description 1
- FBTSQILOGYXGMD-LURJTMIESA-N 3-nitro-L-tyrosine Chemical class OC(=O)[C@@H](N)CC1=CC=C(O)C([N+]([O-])=O)=C1 FBTSQILOGYXGMD-LURJTMIESA-N 0.000 description 1
- 101800000535 3C-like proteinase Proteins 0.000 description 1
- 101800002396 3C-like proteinase nsp5 Proteins 0.000 description 1
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 1
- AOKCDAVWJLOAHG-UHFFFAOYSA-N 4-(methylamino)butyric acid Chemical compound C[NH2+]CCCC([O-])=O AOKCDAVWJLOAHG-UHFFFAOYSA-N 0.000 description 1
- JAJQQUQHMLWDFB-UHFFFAOYSA-N 4-azaniumyl-3-hydroxy-5-phenylpentanoate Chemical compound OC(=O)CC(O)C(N)CC1=CC=CC=C1 JAJQQUQHMLWDFB-UHFFFAOYSA-N 0.000 description 1
- 102100030310 5,6-dihydroxyindole-2-carboxylic acid oxidase Human genes 0.000 description 1
- SLXKOJJOQWFEFD-UHFFFAOYSA-N 6-aminohexanoic acid Chemical compound NCCCCCC(O)=O SLXKOJJOQWFEFD-UHFFFAOYSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 102000000074 ADP-ribosyl Cyclase Human genes 0.000 description 1
- 108010080394 ADP-ribosyl Cyclase Proteins 0.000 description 1
- 102000018667 ADP-ribosyl Cyclase 1 Human genes 0.000 description 1
- 108010027122 ADP-ribosyl Cyclase 1 Proteins 0.000 description 1
- 208000030507 AIDS Diseases 0.000 description 1
- 208000002874 Acne Vulgaris Diseases 0.000 description 1
- 208000010507 Adenocarcinoma of Lung Diseases 0.000 description 1
- 208000006468 Adrenal Cortex Neoplasms Diseases 0.000 description 1
- 102100036775 Afadin Human genes 0.000 description 1
- 208000008190 Agammaglobulinemia Diseases 0.000 description 1
- 229920000936 Agarose Polymers 0.000 description 1
- 208000007848 Alcoholism Diseases 0.000 description 1
- 102100024321 Alkaline phosphatase, placental type Human genes 0.000 description 1
- 208000032671 Allergic granulomatous angiitis Diseases 0.000 description 1
- 201000004384 Alopecia Diseases 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 208000037540 Alveolar soft tissue sarcoma Diseases 0.000 description 1
- 102100022704 Amyloid-beta precursor protein Human genes 0.000 description 1
- 206010061424 Anal cancer Diseases 0.000 description 1
- 206010002383 Angina Pectoris Diseases 0.000 description 1
- 102000009840 Angiopoietins Human genes 0.000 description 1
- 108010009906 Angiopoietins Proteins 0.000 description 1
- 201000003076 Angiosarcoma Diseases 0.000 description 1
- 206010002556 Ankylosing Spondylitis Diseases 0.000 description 1
- 108010049777 Ankyrins Proteins 0.000 description 1
- 102000008102 Ankyrins Human genes 0.000 description 1
- 206010059313 Anogenital warts Diseases 0.000 description 1
- 208000003343 Antiphospholipid Syndrome Diseases 0.000 description 1
- 208000007860 Anus Neoplasms Diseases 0.000 description 1
- 208000032467 Aplastic anaemia Diseases 0.000 description 1
- 102000010565 Apoptosis Regulatory Proteins Human genes 0.000 description 1
- 108010063104 Apoptosis Regulatory Proteins Proteins 0.000 description 1
- 108091023037 Aptamer Proteins 0.000 description 1
- 108010002913 Asialoglycoproteins Proteins 0.000 description 1
- 108010011485 Aspartame Proteins 0.000 description 1
- 206010003594 Ataxia telangiectasia Diseases 0.000 description 1
- 206010003827 Autoimmune hepatitis Diseases 0.000 description 1
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 1
- 206010004146 Basal cell carcinoma Diseases 0.000 description 1
- 108010064528 Basigin Proteins 0.000 description 1
- 102000015279 Basigin Human genes 0.000 description 1
- 208000009137 Behcet syndrome Diseases 0.000 description 1
- 208000009299 Benign Mucous Membrane Pemphigoid Diseases 0.000 description 1
- 206010004272 Benign hydatidiform mole Diseases 0.000 description 1
- 206010004593 Bile duct cancer Diseases 0.000 description 1
- 208000008439 Biliary Liver Cirrhosis Diseases 0.000 description 1
- 208000033222 Biliary cirrhosis primary Diseases 0.000 description 1
- 101710189812 Bilin-binding protein Proteins 0.000 description 1
- 206010005949 Bone cancer Diseases 0.000 description 1
- 208000018084 Bone neoplasm Diseases 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 206010006143 Brain stem glioma Diseases 0.000 description 1
- 208000011691 Burkitt lymphomas Diseases 0.000 description 1
- 102100032367 C-C motif chemokine 5 Human genes 0.000 description 1
- 102100032366 C-C motif chemokine 7 Human genes 0.000 description 1
- 101710155834 C-C motif chemokine 7 Proteins 0.000 description 1
- 108090000342 C-Type Lectins Proteins 0.000 description 1
- 102000003930 C-Type Lectins Human genes 0.000 description 1
- 102100025277 C-X-C motif chemokine 13 Human genes 0.000 description 1
- 108010046080 CD27 Ligand Proteins 0.000 description 1
- 102100027207 CD27 antigen Human genes 0.000 description 1
- 102100038078 CD276 antigen Human genes 0.000 description 1
- 101710185679 CD276 antigen Proteins 0.000 description 1
- 108010063916 CD40 Antigens Proteins 0.000 description 1
- 108010029697 CD40 Ligand Proteins 0.000 description 1
- 102100032937 CD40 ligand Human genes 0.000 description 1
- 102100025221 CD70 antigen Human genes 0.000 description 1
- 210000001266 CD8-positive T-lymphocyte Anatomy 0.000 description 1
- 201000002829 CREST Syndrome Diseases 0.000 description 1
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical group C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 description 1
- 101100327819 Caenorhabditis elegans chl-1 gene Proteins 0.000 description 1
- 102100033620 Calponin-1 Human genes 0.000 description 1
- 101710197665 Capsid protein VP2 Proteins 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical compound NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 1
- 206010007279 Carcinoid tumour of the gastrointestinal tract Diseases 0.000 description 1
- 208000031229 Cardiomyopathies Diseases 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 108010023798 Charybdotoxin Proteins 0.000 description 1
- 108010055166 Chemokine CCL5 Proteins 0.000 description 1
- 108010078239 Chemokine CX3CL1 Proteins 0.000 description 1
- 102000009410 Chemokine receptor Human genes 0.000 description 1
- 108050000299 Chemokine receptor Proteins 0.000 description 1
- 208000005243 Chondrosarcoma Diseases 0.000 description 1
- 208000006332 Choriocarcinoma Diseases 0.000 description 1
- 108010062540 Chorionic Gonadotropin Proteins 0.000 description 1
- 102000011022 Chorionic Gonadotropin Human genes 0.000 description 1
- 206010008874 Chronic Fatigue Syndrome Diseases 0.000 description 1
- 208000006154 Chronic hepatitis C Diseases 0.000 description 1
- 208000006344 Churg-Strauss Syndrome Diseases 0.000 description 1
- 108020004705 Codon Proteins 0.000 description 1
- 208000015943 Coeliac disease Diseases 0.000 description 1
- 208000011038 Cold agglutinin disease Diseases 0.000 description 1
- 206010009868 Cold type haemolytic anaemia Diseases 0.000 description 1
- 206010009900 Colitis ulcerative Diseases 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- 101710184994 Complement control protein Proteins 0.000 description 1
- 102100032768 Complement receptor type 2 Human genes 0.000 description 1
- 208000000907 Condylomata Acuminata Diseases 0.000 description 1
- 206010053138 Congenital aplastic anaemia Diseases 0.000 description 1
- 206010052465 Congenital poikiloderma Diseases 0.000 description 1
- 108091035707 Consensus sequence Proteins 0.000 description 1
- 102100037364 Craniofacial development protein 1 Human genes 0.000 description 1
- 102220539676 Craniofacial development protein 1_K31A_mutation Human genes 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 208000019707 Cryoglobulinemic vasculitis Diseases 0.000 description 1
- 102000036364 Cullin Ring E3 Ligases Human genes 0.000 description 1
- BQOHYSXSASDCEA-KEOHHSTQSA-N Cyclic ADP-Ribose Chemical compound C([C@@H]1[C@H]([C@H]([C@@H](O1)N1C=2N=CN3C(C=2N=C1)=N)O)O)OP(O)(=O)OP(O)(=O)OC[C@@H]1[C@@H](O)[C@@H](O)[C@H]3O1 BQOHYSXSASDCEA-KEOHHSTQSA-N 0.000 description 1
- LVZWSLJZHVFIQJ-UHFFFAOYSA-N Cyclopropane Chemical compound C1CC1 LVZWSLJZHVFIQJ-UHFFFAOYSA-N 0.000 description 1
- 108010025905 Cystine-Knot Miniproteins Proteins 0.000 description 1
- 102100025621 Cytochrome b-245 heavy chain Human genes 0.000 description 1
- 101710205889 Cytochrome b562 Proteins 0.000 description 1
- 102100035298 Cytokine SCM-1 beta Human genes 0.000 description 1
- GUBGYTABKSRVRQ-CUHNMECISA-N D-Cellobiose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-CUHNMECISA-N 0.000 description 1
- XUJNEKJLAYXESH-UWTATZPHSA-N D-Cysteine Chemical compound SC[C@@H](N)C(O)=O XUJNEKJLAYXESH-UWTATZPHSA-N 0.000 description 1
- AGPKZVBTJJNPAG-RFZPGFLSSA-N D-Isoleucine Chemical compound CC[C@@H](C)[C@@H](N)C(O)=O AGPKZVBTJJNPAG-RFZPGFLSSA-N 0.000 description 1
- AHLPHDHHMVZTML-SCSAIBSYSA-N D-Ornithine Chemical compound NCCC[C@@H](N)C(O)=O AHLPHDHHMVZTML-SCSAIBSYSA-N 0.000 description 1
- ONIBWKKTOPOVIA-SCSAIBSYSA-N D-Proline Chemical compound OC(=O)[C@H]1CCCN1 ONIBWKKTOPOVIA-SCSAIBSYSA-N 0.000 description 1
- MTCFGRXMJLQNBG-UWTATZPHSA-N D-Serine Chemical compound OC[C@@H](N)C(O)=O MTCFGRXMJLQNBG-UWTATZPHSA-N 0.000 description 1
- 229930195711 D-Serine Natural products 0.000 description 1
- QNAYBMKLOCPYGJ-UWTATZPHSA-N D-alanine Chemical compound C[C@@H](N)C(O)=O QNAYBMKLOCPYGJ-UWTATZPHSA-N 0.000 description 1
- QNAYBMKLOCPYGJ-UHFFFAOYSA-N D-alpha-Ala Natural products CC([NH3+])C([O-])=O QNAYBMKLOCPYGJ-UHFFFAOYSA-N 0.000 description 1
- ODKSFYDXXFIFQN-SCSAIBSYSA-N D-arginine Chemical compound OC(=O)[C@H](N)CCCNC(N)=N ODKSFYDXXFIFQN-SCSAIBSYSA-N 0.000 description 1
- 229930028154 D-arginine Natural products 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Chemical class OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- 229930182847 D-glutamic acid Natural products 0.000 description 1
- ZDXPYRJPNDTMRX-GSVOUGTGSA-N D-glutamine Chemical compound OC(=O)[C@H](N)CCC(N)=O ZDXPYRJPNDTMRX-GSVOUGTGSA-N 0.000 description 1
- 229930195715 D-glutamine Natural products 0.000 description 1
- HNDVDQJCIGZPNO-RXMQYKEDSA-N D-histidine Chemical compound OC(=O)[C@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-RXMQYKEDSA-N 0.000 description 1
- 229930195721 D-histidine Natural products 0.000 description 1
- 229930182845 D-isoleucine Natural products 0.000 description 1
- ROHFNLRQFUQHCH-RXMQYKEDSA-N D-leucine Chemical compound CC(C)C[C@@H](N)C(O)=O ROHFNLRQFUQHCH-RXMQYKEDSA-N 0.000 description 1
- 229930182819 D-leucine Natural products 0.000 description 1
- KDXKERNSBIXSRK-RXMQYKEDSA-N D-lysine Chemical compound NCCCC[C@@H](N)C(O)=O KDXKERNSBIXSRK-RXMQYKEDSA-N 0.000 description 1
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 1
- QWIZNVHXZXRPDR-UHFFFAOYSA-N D-melezitose Natural products O1C(CO)C(O)C(O)C(O)C1OC1C(O)C(CO)OC1(CO)OC1OC(CO)C(O)C(O)C1O QWIZNVHXZXRPDR-UHFFFAOYSA-N 0.000 description 1
- FFEARJCKVFRZRR-SCSAIBSYSA-N D-methionine Chemical compound CSCC[C@@H](N)C(O)=O FFEARJCKVFRZRR-SCSAIBSYSA-N 0.000 description 1
- 229930182818 D-methionine Natural products 0.000 description 1
- COLNVLDHVKWLRT-MRVPVSSYSA-N D-phenylalanine Chemical compound OC(=O)[C@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-MRVPVSSYSA-N 0.000 description 1
- 229930182832 D-phenylalanine Natural products 0.000 description 1
- 229930182820 D-proline Natural products 0.000 description 1
- AYFVYJQAPQTCCC-STHAYSLISA-N D-threonine Chemical compound C[C@H](O)[C@@H](N)C(O)=O AYFVYJQAPQTCCC-STHAYSLISA-N 0.000 description 1
- 229930182822 D-threonine Natural products 0.000 description 1
- 229930182827 D-tryptophan Natural products 0.000 description 1
- QIVBCDIJIAJPQS-SECBINFHSA-N D-tryptophane Chemical compound C1=CC=C2C(C[C@@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-SECBINFHSA-N 0.000 description 1
- OUYCCCASQSFEME-MRVPVSSYSA-N D-tyrosine Chemical compound OC(=O)[C@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-MRVPVSSYSA-N 0.000 description 1
- 229930195709 D-tyrosine Natural products 0.000 description 1
- KZSNJWFQEVHDMF-SCSAIBSYSA-N D-valine Chemical compound CC(C)[C@@H](N)C(O)=O KZSNJWFQEVHDMF-SCSAIBSYSA-N 0.000 description 1
- 229930182831 D-valine Natural products 0.000 description 1
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 1
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 1
- ZBNZXTGUTAYRHI-UHFFFAOYSA-N Dasatinib Chemical compound C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1Cl ZBNZXTGUTAYRHI-UHFFFAOYSA-N 0.000 description 1
- 208000016192 Demyelinating disease Diseases 0.000 description 1
- 206010012305 Demyelination Diseases 0.000 description 1
- 241000725619 Dengue virus Species 0.000 description 1
- 201000004624 Dermatitis Diseases 0.000 description 1
- 206010012468 Dermatitis herpetiformis Diseases 0.000 description 1
- 208000008334 Dermatofibrosarcoma Diseases 0.000 description 1
- 206010057070 Dermatofibrosarcoma protuberans Diseases 0.000 description 1
- 208000008743 Desmoplastic Small Round Cell Tumor Diseases 0.000 description 1
- 206010064581 Desmoplastic small round cell tumour Diseases 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical class OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- 208000006402 Ductal Carcinoma Diseases 0.000 description 1
- 229930195710 D‐cysteine Natural products 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 206010014568 Empyema Diseases 0.000 description 1
- 208000001976 Endocrine Gland Neoplasms Diseases 0.000 description 1
- 206010014733 Endometrial cancer Diseases 0.000 description 1
- 206010014759 Endometrial neoplasm Diseases 0.000 description 1
- 241000709661 Enterovirus Species 0.000 description 1
- 241000991587 Enterovirus C Species 0.000 description 1
- 208000018428 Eosinophilic granulomatosis with polyangiitis Diseases 0.000 description 1
- 206010014967 Ependymoma Diseases 0.000 description 1
- 102400001368 Epidermal growth factor Human genes 0.000 description 1
- 101800003838 Epidermal growth factor Proteins 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- 108010066687 Epithelial Cell Adhesion Molecule Proteins 0.000 description 1
- 102000018651 Epithelial Cell Adhesion Molecule Human genes 0.000 description 1
- 102100036509 Erythropoietin receptor Human genes 0.000 description 1
- 101710102442 Erythropoietin receptor Proteins 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 1
- 208000006168 Ewing Sarcoma Diseases 0.000 description 1
- 201000001342 Fallopian tube cancer Diseases 0.000 description 1
- 208000013452 Fallopian tube neoplasm Diseases 0.000 description 1
- 201000004939 Fanconi anemia Diseases 0.000 description 1
- 108010039471 Fas Ligand Protein Proteins 0.000 description 1
- 102000008857 Ferritin Human genes 0.000 description 1
- 108050000784 Ferritin Proteins 0.000 description 1
- 238000008416 Ferritin Methods 0.000 description 1
- 101710145505 Fiber protein Proteins 0.000 description 1
- 208000001640 Fibromyalgia Diseases 0.000 description 1
- 102100037362 Fibronectin Human genes 0.000 description 1
- 102000002090 Fibronectin type III Human genes 0.000 description 1
- 229920001917 Ficoll Polymers 0.000 description 1
- 102000010451 Folate receptor alpha Human genes 0.000 description 1
- 108050001931 Folate receptor alpha Proteins 0.000 description 1
- 102000012673 Follicle Stimulating Hormone Human genes 0.000 description 1
- 108010079345 Follicle Stimulating Hormone Proteins 0.000 description 1
- 102000013818 Fractalkine Human genes 0.000 description 1
- 229930091371 Fructose Natural products 0.000 description 1
- 239000005715 Fructose Substances 0.000 description 1
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 1
- 208000022072 Gallbladder Neoplasms Diseases 0.000 description 1
- 101100226596 Gallus gallus FABP gene Proteins 0.000 description 1
- 208000005577 Gastroenteritis Diseases 0.000 description 1
- 206010017918 Gastroenteritis viral Diseases 0.000 description 1
- 201000003741 Gastrointestinal carcinoma Diseases 0.000 description 1
- 206010017993 Gastrointestinal neoplasms Diseases 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 208000032612 Glial tumor Diseases 0.000 description 1
- 206010018338 Glioma Diseases 0.000 description 1
- 206010018364 Glomerulonephritis Diseases 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 102100031132 Glucose-6-phosphate isomerase Human genes 0.000 description 1
- 108010070600 Glucose-6-phosphate isomerase Proteins 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 229930186217 Glycolipid Natural products 0.000 description 1
- 208000003807 Graves Disease Diseases 0.000 description 1
- 208000015023 Graves' disease Diseases 0.000 description 1
- 102000009465 Growth Factor Receptors Human genes 0.000 description 1
- 108010009202 Growth Factor Receptors Proteins 0.000 description 1
- 108010051696 Growth Hormone Proteins 0.000 description 1
- 208000030836 Hashimoto thyroiditis Diseases 0.000 description 1
- 208000001258 Hemangiosarcoma Diseases 0.000 description 1
- 208000002250 Hematologic Neoplasms Diseases 0.000 description 1
- 108010054147 Hemoglobins Proteins 0.000 description 1
- 102000001554 Hemoglobins Human genes 0.000 description 1
- 241000711549 Hepacivirus C Species 0.000 description 1
- 206010019663 Hepatic failure Diseases 0.000 description 1
- 206010019668 Hepatic fibrosis Diseases 0.000 description 1
- 208000033640 Hereditary breast cancer Diseases 0.000 description 1
- 101000773083 Homo sapiens 5,6-dihydroxyindole-2-carboxylic acid oxidase Proteins 0.000 description 1
- 101000823051 Homo sapiens Amyloid-beta precursor protein Proteins 0.000 description 1
- 101000858064 Homo sapiens C-X-C motif chemokine 13 Proteins 0.000 description 1
- 101000914511 Homo sapiens CD27 antigen Proteins 0.000 description 1
- 101000945318 Homo sapiens Calponin-1 Proteins 0.000 description 1
- 101000941929 Homo sapiens Complement receptor type 2 Proteins 0.000 description 1
- 101000804771 Homo sapiens Cytokine SCM-1 beta Proteins 0.000 description 1
- 101000851181 Homo sapiens Epidermal growth factor receptor Proteins 0.000 description 1
- 101000935587 Homo sapiens Flavin reductase (NADPH) Proteins 0.000 description 1
- 101001019455 Homo sapiens ICOS ligand Proteins 0.000 description 1
- 101001078158 Homo sapiens Integrin alpha-1 Proteins 0.000 description 1
- 101001078133 Homo sapiens Integrin alpha-2 Proteins 0.000 description 1
- 101000994378 Homo sapiens Integrin alpha-3 Proteins 0.000 description 1
- 101000994375 Homo sapiens Integrin alpha-4 Proteins 0.000 description 1
- 101000994369 Homo sapiens Integrin alpha-5 Proteins 0.000 description 1
- 101000994365 Homo sapiens Integrin alpha-6 Proteins 0.000 description 1
- 101001046687 Homo sapiens Integrin alpha-E Proteins 0.000 description 1
- 101001078143 Homo sapiens Integrin alpha-IIb Proteins 0.000 description 1
- 101000935043 Homo sapiens Integrin beta-1 Proteins 0.000 description 1
- 101000935040 Homo sapiens Integrin beta-2 Proteins 0.000 description 1
- 101001034828 Homo sapiens Interferon alpha-14 Proteins 0.000 description 1
- 101001034835 Homo sapiens Interferon alpha-16 Proteins 0.000 description 1
- 101001034834 Homo sapiens Interferon alpha-17 Proteins 0.000 description 1
- 101000959794 Homo sapiens Interferon alpha-2 Proteins 0.000 description 1
- 101001034833 Homo sapiens Interferon alpha-21 Proteins 0.000 description 1
- 101000959708 Homo sapiens Interferon alpha-4 Proteins 0.000 description 1
- 101000959704 Homo sapiens Interferon alpha-5 Proteins 0.000 description 1
- 101000959714 Homo sapiens Interferon alpha-6 Proteins 0.000 description 1
- 101000961126 Homo sapiens Interferon alpha-7 Proteins 0.000 description 1
- 101000999391 Homo sapiens Interferon alpha-8 Proteins 0.000 description 1
- 101001054334 Homo sapiens Interferon beta Proteins 0.000 description 1
- 101001044447 Homo sapiens Interferon kappa Proteins 0.000 description 1
- 101001002469 Homo sapiens Interferon lambda-2 Proteins 0.000 description 1
- 101001002466 Homo sapiens Interferon lambda-3 Proteins 0.000 description 1
- 101000999370 Homo sapiens Interferon omega-1 Proteins 0.000 description 1
- 101001076386 Homo sapiens Interleukin-1 family member 10 Proteins 0.000 description 1
- 101001010626 Homo sapiens Interleukin-22 Proteins 0.000 description 1
- 101000998139 Homo sapiens Interleukin-32 Proteins 0.000 description 1
- 101000998140 Homo sapiens Interleukin-36 alpha Proteins 0.000 description 1
- 101000998126 Homo sapiens Interleukin-36 beta Proteins 0.000 description 1
- 101001040964 Homo sapiens Interleukin-36 receptor antagonist protein Proteins 0.000 description 1
- 101000998122 Homo sapiens Interleukin-37 Proteins 0.000 description 1
- 101001002709 Homo sapiens Interleukin-4 Proteins 0.000 description 1
- 101001076408 Homo sapiens Interleukin-6 Proteins 0.000 description 1
- 101000777628 Homo sapiens Leukocyte antigen CD37 Proteins 0.000 description 1
- 101000804764 Homo sapiens Lymphotactin Proteins 0.000 description 1
- 101000946053 Homo sapiens Lysosomal-associated transmembrane protein 4A Proteins 0.000 description 1
- 101000798109 Homo sapiens Melanotransferrin Proteins 0.000 description 1
- 101000611936 Homo sapiens Programmed cell death protein 1 Proteins 0.000 description 1
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 1
- 101000766306 Homo sapiens Serotransferrin Proteins 0.000 description 1
- 101000837401 Homo sapiens T-cell leukemia/lymphoma protein 1A Proteins 0.000 description 1
- 101100369992 Homo sapiens TNFSF10 gene Proteins 0.000 description 1
- 101000800116 Homo sapiens Thy-1 membrane glycoprotein Proteins 0.000 description 1
- 101000652736 Homo sapiens Transgelin Proteins 0.000 description 1
- 101000610605 Homo sapiens Tumor necrosis factor receptor superfamily member 10A Proteins 0.000 description 1
- 101000795167 Homo sapiens Tumor necrosis factor receptor superfamily member 13B Proteins 0.000 description 1
- 101000679903 Homo sapiens Tumor necrosis factor receptor superfamily member 25 Proteins 0.000 description 1
- 101000611023 Homo sapiens Tumor necrosis factor receptor superfamily member 6 Proteins 0.000 description 1
- 101000823316 Homo sapiens Tyrosine-protein kinase ABL1 Proteins 0.000 description 1
- 102000002265 Human Growth Hormone Human genes 0.000 description 1
- 108010000521 Human Growth Hormone Proteins 0.000 description 1
- 239000000854 Human Growth Hormone Substances 0.000 description 1
- 241000701085 Human alphaherpesvirus 3 Species 0.000 description 1
- 241001479210 Human astrovirus Species 0.000 description 1
- 241000701024 Human betaherpesvirus 5 Species 0.000 description 1
- 101100122501 Human herpesvirus 1 (strain 17) gN gene Proteins 0.000 description 1
- 241000701806 Human papillomavirus Species 0.000 description 1
- 241000702617 Human parvovirus B19 Species 0.000 description 1
- 208000006937 Hydatidiform mole Diseases 0.000 description 1
- 208000037147 Hypercalcaemia Diseases 0.000 description 1
- 206010020983 Hypogammaglobulinaemia Diseases 0.000 description 1
- 102100034980 ICOS ligand Human genes 0.000 description 1
- 102000039996 IL-1 family Human genes 0.000 description 1
- 108091069196 IL-1 family Proteins 0.000 description 1
- 102000039989 IL-17 family Human genes 0.000 description 1
- 108091069193 IL-17 family Proteins 0.000 description 1
- 108091058536 IL1F9 Proteins 0.000 description 1
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 1
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 1
- 208000010159 IgA glomerulonephritis Diseases 0.000 description 1
- 206010021263 IgA nephropathy Diseases 0.000 description 1
- 108700005091 Immunoglobulin Genes Proteins 0.000 description 1
- 206010022078 Injection site inflammation Diseases 0.000 description 1
- 102100025323 Integrin alpha-1 Human genes 0.000 description 1
- 102100025305 Integrin alpha-2 Human genes 0.000 description 1
- 102100032819 Integrin alpha-3 Human genes 0.000 description 1
- 102100032818 Integrin alpha-4 Human genes 0.000 description 1
- 102100032817 Integrin alpha-5 Human genes 0.000 description 1
- 102100032816 Integrin alpha-6 Human genes 0.000 description 1
- 102100022341 Integrin alpha-E Human genes 0.000 description 1
- 102100025306 Integrin alpha-IIb Human genes 0.000 description 1
- 102100025304 Integrin beta-1 Human genes 0.000 description 1
- 102100025390 Integrin beta-2 Human genes 0.000 description 1
- 102000001617 Interferon Receptors Human genes 0.000 description 1
- 108010054267 Interferon Receptors Proteins 0.000 description 1
- 101710192051 Interferon alpha-1/13 Proteins 0.000 description 1
- 102100039733 Interferon alpha-14 Human genes 0.000 description 1
- 102100039728 Interferon alpha-16 Human genes 0.000 description 1
- 102100039730 Interferon alpha-17 Human genes 0.000 description 1
- 108010078049 Interferon alpha-2 Proteins 0.000 description 1
- 102100039729 Interferon alpha-21 Human genes 0.000 description 1
- 102100039949 Interferon alpha-4 Human genes 0.000 description 1
- 102100039948 Interferon alpha-5 Human genes 0.000 description 1
- 102100040007 Interferon alpha-6 Human genes 0.000 description 1
- 102100039350 Interferon alpha-7 Human genes 0.000 description 1
- 102100036532 Interferon alpha-8 Human genes 0.000 description 1
- 101710099623 Interferon lambda-1 Proteins 0.000 description 1
- 101710099622 Interferon lambda-2 Proteins 0.000 description 1
- 101710099621 Interferon lambda-3 Proteins 0.000 description 1
- 102100036479 Interferon omega-1 Human genes 0.000 description 1
- 108010079944 Interferon-alpha2b Proteins 0.000 description 1
- 102000000589 Interleukin-1 Human genes 0.000 description 1
- 108010002352 Interleukin-1 Proteins 0.000 description 1
- 102100026015 Interleukin-1 family member 10 Human genes 0.000 description 1
- 102000004551 Interleukin-10 Receptors Human genes 0.000 description 1
- 108010017550 Interleukin-10 Receptors Proteins 0.000 description 1
- 108090000177 Interleukin-11 Proteins 0.000 description 1
- 102000003815 Interleukin-11 Human genes 0.000 description 1
- 102000013462 Interleukin-12 Human genes 0.000 description 1
- 108010065805 Interleukin-12 Proteins 0.000 description 1
- 102000003816 Interleukin-13 Human genes 0.000 description 1
- 108090000176 Interleukin-13 Proteins 0.000 description 1
- 108090000172 Interleukin-15 Proteins 0.000 description 1
- 102000003812 Interleukin-15 Human genes 0.000 description 1
- 101800003050 Interleukin-16 Proteins 0.000 description 1
- 102000049772 Interleukin-16 Human genes 0.000 description 1
- 102100033101 Interleukin-17B Human genes 0.000 description 1
- 102000003810 Interleukin-18 Human genes 0.000 description 1
- 108090000171 Interleukin-18 Proteins 0.000 description 1
- 102100039879 Interleukin-19 Human genes 0.000 description 1
- 108050009288 Interleukin-19 Proteins 0.000 description 1
- 108010038453 Interleukin-2 Receptors Proteins 0.000 description 1
- 102000010789 Interleukin-2 Receptors Human genes 0.000 description 1
- 108010065637 Interleukin-23 Proteins 0.000 description 1
- 102100021596 Interleukin-31 Human genes 0.000 description 1
- 101710181613 Interleukin-31 Proteins 0.000 description 1
- 102100033501 Interleukin-32 Human genes 0.000 description 1
- 102000017761 Interleukin-33 Human genes 0.000 description 1
- 108010067003 Interleukin-33 Proteins 0.000 description 1
- 102100033474 Interleukin-36 alpha Human genes 0.000 description 1
- 102100033498 Interleukin-36 beta Human genes 0.000 description 1
- 102100033503 Interleukin-36 gamma Human genes 0.000 description 1
- 102100021150 Interleukin-36 receptor antagonist protein Human genes 0.000 description 1
- 108010002335 Interleukin-9 Proteins 0.000 description 1
- 102000000585 Interleukin-9 Human genes 0.000 description 1
- 102000015696 Interleukins Human genes 0.000 description 1
- 108010063738 Interleukins Proteins 0.000 description 1
- 206010061252 Intraocular melanoma Diseases 0.000 description 1
- 108010044023 Ki-1 Antigen Proteins 0.000 description 1
- 206010023421 Kidney fibrosis Diseases 0.000 description 1
- LKDRXBCSQODPBY-AMVSKUEXSA-N L-(-)-Sorbose Chemical compound OCC1(O)OC[C@H](O)[C@@H](O)[C@@H]1O LKDRXBCSQODPBY-AMVSKUEXSA-N 0.000 description 1
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 1
- GDFAOVXKHJXLEI-UHFFFAOYSA-N L-N-Boc-N-methylalanine Natural products CNC(C)C(O)=O GDFAOVXKHJXLEI-UHFFFAOYSA-N 0.000 description 1
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 1
- QWCKQJZIFLGMSD-VKHMYHEASA-N L-alpha-aminobutyric acid Chemical compound CC[C@H](N)C(O)=O QWCKQJZIFLGMSD-VKHMYHEASA-N 0.000 description 1
- ZGUNAGUHMKGQNY-ZETCQYMHSA-N L-alpha-phenylglycine zwitterion Chemical compound OC(=O)[C@@H](N)C1=CC=CC=C1 ZGUNAGUHMKGQNY-ZETCQYMHSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- 229930182816 L-glutamine Natural products 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- JTTHKOPSMAVJFE-VIFPVBQESA-N L-homophenylalanine Chemical compound OC(=O)[C@@H](N)CCC1=CC=CC=C1 JTTHKOPSMAVJFE-VIFPVBQESA-N 0.000 description 1
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 1
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 1
- 239000002067 L01XE06 - Dasatinib Substances 0.000 description 1
- 239000005536 L01XE08 - Nilotinib Substances 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 201000005099 Langerhans cell histiocytosis Diseases 0.000 description 1
- 206010023825 Laryngeal cancer Diseases 0.000 description 1
- 208000018142 Leiomyosarcoma Diseases 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- 102100031586 Leukocyte antigen CD37 Human genes 0.000 description 1
- 201000011062 Li-Fraumeni syndrome Diseases 0.000 description 1
- 206010061523 Lip and/or oral cavity cancer Diseases 0.000 description 1
- 206010062038 Lip neoplasm Diseases 0.000 description 1
- 239000000232 Lipid Bilayer Substances 0.000 description 1
- 102000019298 Lipocalin Human genes 0.000 description 1
- 108050006654 Lipocalin Proteins 0.000 description 1
- 208000019693 Lung disease Diseases 0.000 description 1
- 102000009151 Luteinizing Hormone Human genes 0.000 description 1
- 108010073521 Luteinizing Hormone Proteins 0.000 description 1
- 206010025282 Lymphoedema Diseases 0.000 description 1
- 102100035304 Lymphotactin Human genes 0.000 description 1
- 102100034728 Lysosomal-associated transmembrane protein 4A Human genes 0.000 description 1
- 208000001344 Macular Edema Diseases 0.000 description 1
- 206010025415 Macular oedema Diseases 0.000 description 1
- 208000004059 Male Breast Neoplasms Diseases 0.000 description 1
- PEEHTFAAVSWFBL-UHFFFAOYSA-N Maleimide Chemical compound O=C1NC(=O)C=C1 PEEHTFAAVSWFBL-UHFFFAOYSA-N 0.000 description 1
- 208000032271 Malignant tumor of penis Diseases 0.000 description 1
- 229920002774 Maltodextrin Polymers 0.000 description 1
- GUBGYTABKSRVRQ-PICCSMPSSA-N Maltose Natural products O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-PICCSMPSSA-N 0.000 description 1
- 208000025205 Mantle-Cell Lymphoma Diseases 0.000 description 1
- 208000000172 Medulloblastoma Diseases 0.000 description 1
- 102100032239 Melanotransferrin Human genes 0.000 description 1
- 102000018697 Membrane Proteins Human genes 0.000 description 1
- 208000027530 Meniere disease Diseases 0.000 description 1
- 208000002030 Merkel cell carcinoma Diseases 0.000 description 1
- 206010027406 Mesothelioma Diseases 0.000 description 1
- 108010006035 Metalloproteases Proteins 0.000 description 1
- 102000005741 Metalloproteases Human genes 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 208000003250 Mixed connective tissue disease Diseases 0.000 description 1
- 208000003445 Mouth Neoplasms Diseases 0.000 description 1
- 208000012192 Mucous membrane pemphigoid Diseases 0.000 description 1
- 108010021466 Mutant Proteins Proteins 0.000 description 1
- 102000008300 Mutant Proteins Human genes 0.000 description 1
- 208000014767 Myeloproliferative disease Diseases 0.000 description 1
- 208000009525 Myocarditis Diseases 0.000 description 1
- CZCIKBSVHDNIDH-NSHDSACASA-N N(alpha)-methyl-L-tryptophan Chemical compound C1=CC=C2C(C[C@H]([NH2+]C)C([O-])=O)=CNC2=C1 CZCIKBSVHDNIDH-NSHDSACASA-N 0.000 description 1
- WRUZLCLJULHLEY-UHFFFAOYSA-N N-(p-hydroxyphenyl)glycine Chemical compound OC(=O)CNC1=CC=C(O)C=C1 WRUZLCLJULHLEY-UHFFFAOYSA-N 0.000 description 1
- VKZGJEWGVNFKPE-UHFFFAOYSA-N N-Isobutylglycine Chemical compound CC(C)CNCC(O)=O VKZGJEWGVNFKPE-UHFFFAOYSA-N 0.000 description 1
- SCIFESDRCALIIM-UHFFFAOYSA-N N-Me-Phenylalanine Natural products CNC(C(O)=O)CC1=CC=CC=C1 SCIFESDRCALIIM-UHFFFAOYSA-N 0.000 description 1
- HOKKHZGPKSLGJE-GSVOUGTGSA-N N-Methyl-D-aspartic acid Chemical compound CN[C@@H](C(O)=O)CC(O)=O HOKKHZGPKSLGJE-GSVOUGTGSA-N 0.000 description 1
- NTWVQPHTOUKMDI-YFKPBYRVSA-N N-Methyl-arginine Chemical compound CN[C@H](C(O)=O)CCCN=C(N)N NTWVQPHTOUKMDI-YFKPBYRVSA-N 0.000 description 1
- GDFAOVXKHJXLEI-VKHMYHEASA-N N-methyl-L-alanine Chemical compound C[NH2+][C@@H](C)C([O-])=O GDFAOVXKHJXLEI-VKHMYHEASA-N 0.000 description 1
- XLBVNMSMFQMKEY-BYPYZUCNSA-N N-methyl-L-glutamic acid Chemical compound CN[C@H](C(O)=O)CCC(O)=O XLBVNMSMFQMKEY-BYPYZUCNSA-N 0.000 description 1
- YAXAFCHJCYILRU-YFKPBYRVSA-N N-methyl-L-methionine Chemical compound C[NH2+][C@H](C([O-])=O)CCSC YAXAFCHJCYILRU-YFKPBYRVSA-N 0.000 description 1
- SCIFESDRCALIIM-VIFPVBQESA-N N-methyl-L-phenylalanine Chemical compound C[NH2+][C@H](C([O-])=O)CC1=CC=CC=C1 SCIFESDRCALIIM-VIFPVBQESA-N 0.000 description 1
- AKCRVYNORCOYQT-YFKPBYRVSA-N N-methyl-L-valine Chemical compound CN[C@@H](C(C)C)C(O)=O AKCRVYNORCOYQT-YFKPBYRVSA-N 0.000 description 1
- CWLQUGTUXBXTLF-YFKPBYRVSA-N N-methylproline Chemical compound CN1CCC[C@H]1C(O)=O CWLQUGTUXBXTLF-YFKPBYRVSA-N 0.000 description 1
- BAWFJGJZGIEFAR-NNYOXOHSSA-O NAD(+) Chemical compound NC(=O)C1=CC=C[N+]([C@H]2[C@@H]([C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 BAWFJGJZGIEFAR-NNYOXOHSSA-O 0.000 description 1
- 101150054880 NASP gene Proteins 0.000 description 1
- 208000001894 Nasopharyngeal Neoplasms Diseases 0.000 description 1
- 206010061306 Nasopharyngeal cancer Diseases 0.000 description 1
- 241000687607 Natalis Species 0.000 description 1
- 206010051606 Necrotising colitis Diseases 0.000 description 1
- 101710204212 Neocarzinostatin Proteins 0.000 description 1
- 208000034176 Neoplasms, Germ Cell and Embryonal Diseases 0.000 description 1
- 102000007072 Nerve Growth Factors Human genes 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 206010029260 Neuroblastoma Diseases 0.000 description 1
- 208000009905 Neurofibromatoses Diseases 0.000 description 1
- 208000025966 Neurological disease Diseases 0.000 description 1
- 102000004108 Neurotransmitter Receptors Human genes 0.000 description 1
- 108090000590 Neurotransmitter Receptors Proteins 0.000 description 1
- 208000006964 Nevi and Melanomas Diseases 0.000 description 1
- 208000004485 Nijmegen breakage syndrome Diseases 0.000 description 1
- 241001263478 Norovirus Species 0.000 description 1
- 208000010505 Nose Neoplasms Diseases 0.000 description 1
- 108091028043 Nucleic acid sequence Proteins 0.000 description 1
- XDMCWZFLLGVIID-SXPRBRBTSA-N O-(3-O-D-galactosyl-N-acetyl-beta-D-galactosaminyl)-L-serine Chemical compound CC(=O)N[C@H]1[C@H](OC[C@H]([NH3+])C([O-])=O)O[C@H](CO)[C@H](O)[C@@H]1OC1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 XDMCWZFLLGVIID-SXPRBRBTSA-N 0.000 description 1
- 150000007930 O-acyl isoureas Chemical class 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 102000001490 Opioid Peptides Human genes 0.000 description 1
- 108010093625 Opioid Peptides Proteins 0.000 description 1
- 239000012124 Opti-MEM Substances 0.000 description 1
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 1
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 1
- 206010057444 Oropharyngeal neoplasm Diseases 0.000 description 1
- 206010068319 Oropharyngeal pain Diseases 0.000 description 1
- 102000008108 Osteoprotegerin Human genes 0.000 description 1
- 108010035042 Osteoprotegerin Proteins 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 208000002193 Pain Diseases 0.000 description 1
- 208000000821 Parathyroid Neoplasms Diseases 0.000 description 1
- 208000029082 Pelvic Inflammatory Disease Diseases 0.000 description 1
- 201000011152 Pemphigus Diseases 0.000 description 1
- 208000002471 Penile Neoplasms Diseases 0.000 description 1
- 206010034299 Penile cancer Diseases 0.000 description 1
- 208000005764 Peripheral Arterial Disease Diseases 0.000 description 1
- 208000030831 Peripheral arterial occlusive disease Diseases 0.000 description 1
- 208000031845 Pernicious anaemia Diseases 0.000 description 1
- 241000577979 Peromyscus spicilegus Species 0.000 description 1
- 208000007913 Pituitary Neoplasms Diseases 0.000 description 1
- 206010035669 Pneumonia aspiration Diseases 0.000 description 1
- RVGRUAULSDPKGF-UHFFFAOYSA-N Poloxamer Chemical compound C1CO1.CC1CO1 RVGRUAULSDPKGF-UHFFFAOYSA-N 0.000 description 1
- 102100039388 Polyamine deacetylase HDAC10 Human genes 0.000 description 1
- 101710107444 Polyamine deacetylase HDAC10 Proteins 0.000 description 1
- 206010065159 Polychondritis Diseases 0.000 description 1
- 208000007048 Polymyalgia Rheumatica Diseases 0.000 description 1
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 1
- 208000012654 Primary biliary cholangitis Diseases 0.000 description 1
- 208000037276 Primitive Peripheral Neuroectodermal Tumors Diseases 0.000 description 1
- 102100032251 Pro-thyrotropin-releasing hormone Human genes 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 108010072866 Prostate-Specific Antigen Proteins 0.000 description 1
- 102100038358 Prostate-specific antigen Human genes 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 102100035703 Prostatic acid phosphatase Human genes 0.000 description 1
- 229940079156 Proteasome inhibitor Drugs 0.000 description 1
- 208000012322 Raynaud phenomenon Diseases 0.000 description 1
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 1
- 208000033464 Reiter syndrome Diseases 0.000 description 1
- 206010038389 Renal cancer Diseases 0.000 description 1
- IWUCXVSUMQZMFG-AFCXAGJDSA-N Ribavirin Chemical compound N1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 IWUCXVSUMQZMFG-AFCXAGJDSA-N 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 208000000791 Rothmund-Thomson syndrome Diseases 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 208000004337 Salivary Gland Neoplasms Diseases 0.000 description 1
- 206010061934 Salivary gland cancer Diseases 0.000 description 1
- 206010039710 Scleroderma Diseases 0.000 description 1
- 241000239226 Scorpiones Species 0.000 description 1
- 108010071390 Serum Albumin Proteins 0.000 description 1
- 102000007562 Serum Albumin Human genes 0.000 description 1
- 208000009359 Sezary Syndrome Diseases 0.000 description 1
- 208000021388 Sezary disease Diseases 0.000 description 1
- 108010029157 Sialic Acid Binding Ig-like Lectin 2 Proteins 0.000 description 1
- 208000021386 Sjogren Syndrome Diseases 0.000 description 1
- 208000000453 Skin Neoplasms Diseases 0.000 description 1
- 206010040943 Skin Ulcer Diseases 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- 102000005157 Somatostatin Human genes 0.000 description 1
- 108010056088 Somatostatin Proteins 0.000 description 1
- 102100038803 Somatotropin Human genes 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 229930182558 Sterol Natural products 0.000 description 1
- 206010072148 Stiff-Person syndrome Diseases 0.000 description 1
- 101710106388 Structural protein VP1 Proteins 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Chemical class OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 208000031673 T-Cell Cutaneous Lymphoma Diseases 0.000 description 1
- 201000011648 T-cell childhood lymphoblastic lymphoma Diseases 0.000 description 1
- 102100028676 T-cell leukemia/lymphoma protein 1A Human genes 0.000 description 1
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 102100025237 T-cell surface antigen CD2 Human genes 0.000 description 1
- 208000020982 T-lymphoblastic lymphoma Diseases 0.000 description 1
- 108700012920 TNF Proteins 0.000 description 1
- 108700042805 TRU-015 Proteins 0.000 description 1
- 208000001106 Takayasu Arteritis Diseases 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Chemical class [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 229940123237 Taxane Drugs 0.000 description 1
- 108010017842 Telomerase Proteins 0.000 description 1
- 102100038126 Tenascin Human genes 0.000 description 1
- 108010008125 Tenascin Proteins 0.000 description 1
- 208000024313 Testicular Neoplasms Diseases 0.000 description 1
- 206010057644 Testis cancer Diseases 0.000 description 1
- 102100024554 Tetranectin Human genes 0.000 description 1
- NYTOUQBROMCLBJ-UHFFFAOYSA-N Tetranitromethane Chemical compound [O-][N+](=O)C([N+]([O-])=O)([N+]([O-])=O)[N+]([O-])=O NYTOUQBROMCLBJ-UHFFFAOYSA-N 0.000 description 1
- 102100033523 Thy-1 membrane glycoprotein Human genes 0.000 description 1
- 208000000728 Thymus Neoplasms Diseases 0.000 description 1
- 208000024770 Thyroid neoplasm Diseases 0.000 description 1
- 102000011923 Thyrotropin Human genes 0.000 description 1
- 108010061174 Thyrotropin Proteins 0.000 description 1
- 101800004623 Thyrotropin-releasing hormone Proteins 0.000 description 1
- 102000002689 Toll-like receptor Human genes 0.000 description 1
- 108020000411 Toll-like receptor Proteins 0.000 description 1
- 102400001320 Transforming growth factor alpha Human genes 0.000 description 1
- 101800004564 Transforming growth factor alpha Proteins 0.000 description 1
- 101800001690 Transmembrane protein gp41 Proteins 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 229940122618 Trypsin inhibitor Drugs 0.000 description 1
- 101710162629 Trypsin inhibitor Proteins 0.000 description 1
- 101710112927 Trypsin inhibitor 2 Proteins 0.000 description 1
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 1
- 102100040247 Tumor necrosis factor Human genes 0.000 description 1
- 108050002568 Tumor necrosis factor ligand superfamily member 6 Proteins 0.000 description 1
- 102100040113 Tumor necrosis factor receptor superfamily member 10A Human genes 0.000 description 1
- 102100029675 Tumor necrosis factor receptor superfamily member 13B Human genes 0.000 description 1
- 101710187743 Tumor necrosis factor receptor superfamily member 1A Proteins 0.000 description 1
- 102100033732 Tumor necrosis factor receptor superfamily member 1A Human genes 0.000 description 1
- 101710187830 Tumor necrosis factor receptor superfamily member 1B Proteins 0.000 description 1
- 102100033733 Tumor necrosis factor receptor superfamily member 1B Human genes 0.000 description 1
- 102100022203 Tumor necrosis factor receptor superfamily member 25 Human genes 0.000 description 1
- 102100040403 Tumor necrosis factor receptor superfamily member 6 Human genes 0.000 description 1
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 1
- 102100039094 Tyrosinase Human genes 0.000 description 1
- 108060008724 Tyrosinase Proteins 0.000 description 1
- 101150042088 UL16 gene Proteins 0.000 description 1
- 101150081727 UL32 gene Proteins 0.000 description 1
- 108090000848 Ubiquitin Proteins 0.000 description 1
- 102000044159 Ubiquitin Human genes 0.000 description 1
- 201000006704 Ulcerative Colitis Diseases 0.000 description 1
- 206010046431 Urethral cancer Diseases 0.000 description 1
- 206010046458 Urethral neoplasms Diseases 0.000 description 1
- 208000008385 Urogenital Neoplasms Diseases 0.000 description 1
- 102000003990 Urokinase-type plasminogen activator Human genes 0.000 description 1
- 102100031358 Urokinase-type plasminogen activator Human genes 0.000 description 1
- 108010061861 Uroplakins Proteins 0.000 description 1
- 102000012349 Uroplakins Human genes 0.000 description 1
- 208000002495 Uterine Neoplasms Diseases 0.000 description 1
- 201000005969 Uveal melanoma Diseases 0.000 description 1
- 108010079206 V-Set Domain-Containing T-Cell Activation Inhibitor 1 Proteins 0.000 description 1
- 102100038929 V-set domain-containing T-cell activation inhibitor 1 Human genes 0.000 description 1
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 1
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 1
- 206010047115 Vasculitis Diseases 0.000 description 1
- GXBMIBRIOWHPDT-UHFFFAOYSA-N Vasopressin Natural products N1C(=O)C(CC=2C=C(O)C=CC=2)NC(=O)C(N)CSSCC(C(=O)N2C(CCC2)C(=O)NC(CCCN=C(N)N)C(=O)NCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(CCC(N)=O)NC(=O)C1CC1=CC=CC=C1 GXBMIBRIOWHPDT-UHFFFAOYSA-N 0.000 description 1
- 108010004977 Vasopressins Proteins 0.000 description 1
- 102000002852 Vasopressins Human genes 0.000 description 1
- 208000014070 Vestibular schwannoma Diseases 0.000 description 1
- 229940122803 Vinca alkaloid Drugs 0.000 description 1
- 108010087302 Viral Structural Proteins Proteins 0.000 description 1
- 206010047642 Vitiligo Diseases 0.000 description 1
- 208000004354 Vulvar Neoplasms Diseases 0.000 description 1
- 208000033559 Waldenström macroglobulinemia Diseases 0.000 description 1
- OUUYBRCCFUEMLH-YDALLXLXSA-N [(1s)-2-[4-[bis(2-chloroethyl)amino]phenyl]-1-carboxyethyl]azanium;chloride Chemical compound Cl.OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 OUUYBRCCFUEMLH-YDALLXLXSA-N 0.000 description 1
- XJLXINKUBYWONI-DQQFMEOOSA-N [[(2r,3r,4r,5r)-5-(6-aminopurin-9-yl)-3-hydroxy-4-phosphonooxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [(2s,3r,4s,5s)-5-(3-carbamoylpyridin-1-ium-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphate Chemical compound NC(=O)C1=CC=C[N+]([C@@H]2[C@H]([C@@H](O)[C@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](OP(O)(O)=O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 XJLXINKUBYWONI-DQQFMEOOSA-N 0.000 description 1
- PWJFNRJRHXWEPT-AOOZFPJJSA-N [[(2r,3s,4r,5r)-5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [(2r,3r,4r)-2,3,4-trihydroxy-5-oxopentyl] hydrogen phosphate Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OC[C@@H](O)[C@@H](O)[C@@H](O)C=O)[C@@H](O)[C@H]1O PWJFNRJRHXWEPT-AOOZFPJJSA-N 0.000 description 1
- 206010000496 acne Diseases 0.000 description 1
- 208000004064 acoustic neuroma Diseases 0.000 description 1
- 208000017733 acquired polycythemia vera Diseases 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 230000033289 adaptive immune response Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 208000002517 adenoid cystic carcinoma Diseases 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 210000004100 adrenal gland Anatomy 0.000 description 1
- 229940009456 adriamycin Drugs 0.000 description 1
- 238000001261 affinity purification Methods 0.000 description 1
- 238000003314 affinity selection Methods 0.000 description 1
- 238000012867 alanine scanning Methods 0.000 description 1
- 206010001584 alcohol abuse Diseases 0.000 description 1
- 208000025746 alcohol use disease Diseases 0.000 description 1
- 125000003172 aldehyde group Chemical group 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 229940060516 alferon n Drugs 0.000 description 1
- 229930013930 alkaloid Natural products 0.000 description 1
- 150000003797 alkaloid derivatives Chemical class 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- 230000003281 allosteric effect Effects 0.000 description 1
- 231100000360 alopecia Toxicity 0.000 description 1
- 208000004631 alopecia areata Diseases 0.000 description 1
- DLAMVQGYEVKIRE-UHFFFAOYSA-N alpha-(methylamino)isobutyric acid Chemical compound CNC(C)(C)C(O)=O DLAMVQGYEVKIRE-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-PHYPRBDBSA-N alpha-D-galactose Chemical compound OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-PHYPRBDBSA-N 0.000 description 1
- ANBQYFIVLNNZCU-CQCLMDPOSA-N alpha-L-Fucp-(1->2)-[alpha-D-GalpNAc-(1->3)]-beta-D-Galp-(1->3)-[alpha-L-Fucp-(1->4)]-beta-D-GlcpNAc-(1->3)-beta-D-Galp Chemical compound O[C@H]1[C@H](O)[C@H](O)[C@H](C)O[C@H]1O[C@H]1[C@H](O[C@H]2[C@@H]([C@@H](O[C@@H]3[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O3)NC(C)=O)[C@@H](O)[C@@H](CO)O2)O[C@H]2[C@H]([C@H](O)[C@H](O)[C@H](C)O2)O)[C@@H](NC(C)=O)[C@H](O[C@H]2[C@H]([C@@H](CO)O[C@@H](O)[C@@H]2O)O)O[C@@H]1CO ANBQYFIVLNNZCU-CQCLMDPOSA-N 0.000 description 1
- 208000008524 alveolar soft part sarcoma Diseases 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 229940093740 amino acid and derivative Drugs 0.000 description 1
- 229940067621 aminobutyrate Drugs 0.000 description 1
- 229960002684 aminocaproic acid Drugs 0.000 description 1
- 238000012197 amplification kit Methods 0.000 description 1
- DZHSAHHDTRWUTF-SIQRNXPUSA-N amyloid-beta polypeptide 42 Chemical compound C([C@@H](C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@H](C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)NCC(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(O)=O)[C@@H](C)CC)C(C)C)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@@H](NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC(O)=O)C(C)C)C(C)C)C1=CC=CC=C1 DZHSAHHDTRWUTF-SIQRNXPUSA-N 0.000 description 1
- 230000019552 anatomical structure morphogenesis Effects 0.000 description 1
- 208000025009 anogenital human papillomavirus infection Diseases 0.000 description 1
- 201000004201 anogenital venereal wart Diseases 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 230000005809 anti-tumor immunity Effects 0.000 description 1
- 230000000890 antigenic effect Effects 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000002216 antistatic agent Substances 0.000 description 1
- 201000011165 anus cancer Diseases 0.000 description 1
- 238000003782 apoptosis assay Methods 0.000 description 1
- 125000000637 arginyl group Chemical group N[C@@H](CCCNC(N)=N)C(=O)* 0.000 description 1
- KBZOIRJILGZLEJ-LGYYRGKSSA-N argipressin Chemical compound C([C@H]1C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CSSC[C@@H](C(N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N1)=O)N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCN=C(N)N)C(=O)NCC(N)=O)C1=CC=CC=C1 KBZOIRJILGZLEJ-LGYYRGKSSA-N 0.000 description 1
- 229940014583 arranon Drugs 0.000 description 1
- 239000011668 ascorbic acid Chemical class 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- IAOZJIPTCAWIRG-QWRGUYRKSA-N aspartame Chemical compound OC(=O)C[C@H](N)C(=O)N[C@H](C(=O)OC)CC1=CC=CC=C1 IAOZJIPTCAWIRG-QWRGUYRKSA-N 0.000 description 1
- 239000000605 aspartame Substances 0.000 description 1
- 235000010357 aspartame Nutrition 0.000 description 1
- 229960003438 aspartame Drugs 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 201000009807 aspiration pneumonia Diseases 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 201000000448 autoimmune hemolytic anemia Diseases 0.000 description 1
- 208000006424 autoimmune oophoritis Diseases 0.000 description 1
- 208000036923 autoimmune primary adrenal insufficiency Diseases 0.000 description 1
- 229940003504 avonex Drugs 0.000 description 1
- 229960002170 azathioprine Drugs 0.000 description 1
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 229950007843 bavituximab Drugs 0.000 description 1
- FUKOGSUFTZDYOI-BMANNDLBSA-O beacopp protocol Chemical compound ClCCN(CCCl)P1(=O)NCCCO1.CNNCC1=CC=C(C(=O)NC(C)C)C=C1.O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1.O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1.COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3C(O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1.C([C@H](C[C@]1(C(=O)OC)C=2C(=C3C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C=O)=CC=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21.N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)C(O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1NC=NC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C FUKOGSUFTZDYOI-BMANNDLBSA-O 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 229960003270 belimumab Drugs 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- HFACYLZERDEVSX-UHFFFAOYSA-N benzidine Chemical compound C1=CC(N)=CC=C1C1=CC=C(N)C=C1 HFACYLZERDEVSX-UHFFFAOYSA-N 0.000 description 1
- CXQCLLQQYTUUKJ-ALWAHNIESA-N beta-D-GalpNAc-(1->4)-[alpha-Neup5Ac-(2->8)-alpha-Neup5Ac-(2->3)]-beta-D-Galp-(1->4)-beta-D-Glcp-(1<->1')-Cer(d18:1/18:0) Chemical compound O[C@@H]1[C@@H](O)[C@H](OC[C@H](NC(=O)CCCCCCCCCCCCCCCCC)[C@H](O)\C=C\CCCCCCCCCCCCC)O[C@H](CO)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@]2(O[C@H]([C@H](NC(C)=O)[C@@H](O)C2)[C@H](O)[C@@H](CO)O[C@]2(O[C@H]([C@H](NC(C)=O)[C@@H](O)C2)[C@H](O)[C@H](O)CO)C(O)=O)C(O)=O)[C@@H](O[C@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](CO)O1 CXQCLLQQYTUUKJ-ALWAHNIESA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QUYVBRFLSA-N beta-maltose Chemical compound OC[C@H]1O[C@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@@H]1O GUBGYTABKSRVRQ-QUYVBRFLSA-N 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 229940021459 betaseron Drugs 0.000 description 1
- 230000001588 bifunctional effect Effects 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000000035 biogenic effect Effects 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 229960000106 biosimilars Drugs 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 230000003139 buffering effect Effects 0.000 description 1
- 208000000594 bullous pemphigoid Diseases 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical class O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 102200094703 c.232G>A Human genes 0.000 description 1
- GMRQFYUYWCNGIN-NKMMMXOESA-N calcitriol Chemical compound C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@@H](CCCC(C)(C)O)C)=C\C=C1\C[C@@H](O)C[C@H](O)C1=C GMRQFYUYWCNGIN-NKMMMXOESA-N 0.000 description 1
- 229960005084 calcitriol Drugs 0.000 description 1
- 235000020964 calcitriol Nutrition 0.000 description 1
- 239000011612 calcitriol Substances 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 229940112129 campath Drugs 0.000 description 1
- 238000002619 cancer immunotherapy Methods 0.000 description 1
- 210000000234 capsid Anatomy 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical class OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 208000002458 carcinoid tumor Diseases 0.000 description 1
- 230000001756 cardiomyopathic effect Effects 0.000 description 1
- BLMPQMFVWMYDKT-NZTKNTHTSA-N carfilzomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)[C@]1(C)OC1)NC(=O)CN1CCOCC1)CC1=CC=CC=C1 BLMPQMFVWMYDKT-NZTKNTHTSA-N 0.000 description 1
- 229960002438 carfilzomib Drugs 0.000 description 1
- 108010021331 carfilzomib Proteins 0.000 description 1
- 239000005018 casein Substances 0.000 description 1
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 1
- 235000021240 caseins Nutrition 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 230000020411 cell activation Effects 0.000 description 1
- 230000021164 cell adhesion Effects 0.000 description 1
- 239000002771 cell marker Substances 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 230000012292 cell migration Effects 0.000 description 1
- 230000009087 cell motility Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 238000001516 cell proliferation assay Methods 0.000 description 1
- 239000002458 cell surface marker Substances 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 230000036755 cellular response Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 238000012412 chemical coupling Methods 0.000 description 1
- 210000000038 chest Anatomy 0.000 description 1
- 208000011654 childhood malignant neoplasm Diseases 0.000 description 1
- AWGTVRDHKJQFAX-UHFFFAOYSA-M chloro(phenyl)mercury Chemical compound Cl[Hg]C1=CC=CC=C1 AWGTVRDHKJQFAX-UHFFFAOYSA-M 0.000 description 1
- VIMWCINSBRXAQH-UHFFFAOYSA-M chloro-(2-hydroxy-5-nitrophenyl)mercury Chemical compound OC1=CC=C([N+]([O-])=O)C=C1[Hg]Cl VIMWCINSBRXAQH-UHFFFAOYSA-M 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- 230000007881 chronic fibrosis Effects 0.000 description 1
- 208000016532 chronic granulomatous disease Diseases 0.000 description 1
- 208000020403 chronic hepatitis C virus infection Diseases 0.000 description 1
- 201000005795 chronic inflammatory demyelinating polyneuritis Diseases 0.000 description 1
- 201000010002 cicatricial pemphigoid Diseases 0.000 description 1
- DERZBLKQOCDDDZ-JLHYYAGUSA-N cinnarizine Chemical compound C1CN(C(C=2C=CC=CC=2)C=2C=CC=CC=2)CCN1C\C=C\C1=CC=CC=C1 DERZBLKQOCDDDZ-JLHYYAGUSA-N 0.000 description 1
- 230000007882 cirrhosis Effects 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 230000005757 colony formation Effects 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 238000002742 combinatorial mutagenesis Methods 0.000 description 1
- 230000009137 competitive binding Effects 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 239000007859 condensation product Substances 0.000 description 1
- 230000001268 conjugating effect Effects 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 201000003278 cryoglobulinemia Diseases 0.000 description 1
- CNVQLPPZGABUCM-LIGYZCPXSA-N ctx toxin Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CC(N)=O)C(=O)N[C@H](C(=O)N[C@@H](CO)C(=O)N[C@H]1CSSC[C@H]2C(=O)N[C@H](C(N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@H]3CSSC[C@@H](C(N[C@@H](CC=4C5=CC=CC=C5NC=4)C(=O)N[C@@H](CO)C(=O)N[C@H](C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CCCNC(N)=N)NC3=O)C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)N[C@@H](CO)C(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=3NC=NC=3)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N2)C(C)C)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H]([C@@H](C)O)NC1=O)=O)CCSC)C(C)C)[C@@H](C)O)NC(=O)[C@H]1NC(=O)CC1)C1=CC=CC=C1 CNVQLPPZGABUCM-LIGYZCPXSA-N 0.000 description 1
- 238000011461 current therapy Methods 0.000 description 1
- 201000007241 cutaneous T cell lymphoma Diseases 0.000 description 1
- 208000017763 cutaneous neuroendocrine carcinoma Diseases 0.000 description 1
- 229940097362 cyclodextrins Drugs 0.000 description 1
- 230000000120 cytopathologic effect Effects 0.000 description 1
- 210000005220 cytoplasmic tail Anatomy 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 229950007409 dacetuzumab Drugs 0.000 description 1
- 229960002806 daclizumab Drugs 0.000 description 1
- 229960002448 dasatinib Drugs 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 230000005860 defense response to virus Effects 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 229940070968 depocyt Drugs 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 238000009795 derivation Methods 0.000 description 1
- 238000001212 derivatisation Methods 0.000 description 1
- 201000001981 dermatomyositis Diseases 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 229940096516 dextrates Drugs 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical group OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- FFYPMLJYZAEMQB-UHFFFAOYSA-N diethyl pyrocarbonate Chemical compound CCOC(=O)OC(=O)OCC FFYPMLJYZAEMQB-UHFFFAOYSA-N 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 150000002016 disaccharides Chemical class 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 208000037765 diseases and disorders Diseases 0.000 description 1
- 125000004119 disulfanediyl group Chemical group *SS* 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 239000003596 drug target Substances 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 150000002066 eicosanoids Chemical class 0.000 description 1
- 230000013020 embryo development Effects 0.000 description 1
- 210000003038 endothelium Anatomy 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 208000010227 enterocolitis Diseases 0.000 description 1
- 229940116977 epidermal growth factor Drugs 0.000 description 1
- 229960005139 epinephrine Drugs 0.000 description 1
- YJGVMLPVUAXIQN-UHFFFAOYSA-N epipodophyllotoxin Natural products COC1=C(OC)C(OC)=CC(C2C3=CC=4OCOC=4C=C3C(O)C3C2C(OC3)=O)=C1 YJGVMLPVUAXIQN-UHFFFAOYSA-N 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 230000009786 epithelial differentiation Effects 0.000 description 1
- 229940082789 erbitux Drugs 0.000 description 1
- 210000003743 erythrocyte Anatomy 0.000 description 1
- 201000004101 esophageal cancer Diseases 0.000 description 1
- 125000000031 ethylamino group Chemical group [H]C([H])([H])C([H])([H])N([H])[*] 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 201000008819 extrahepatic bile duct carcinoma Diseases 0.000 description 1
- 208000024519 eye neoplasm Diseases 0.000 description 1
- 230000004438 eyesight Effects 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 230000019305 fibroblast migration Effects 0.000 description 1
- 229950008085 figitumumab Drugs 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 239000004052 folic acid antagonist Substances 0.000 description 1
- 229940028334 follicle stimulating hormone Drugs 0.000 description 1
- 230000003325 follicular Effects 0.000 description 1
- 229940039573 folotyn Drugs 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 description 1
- 229930182830 galactose Natural products 0.000 description 1
- 201000010175 gallbladder cancer Diseases 0.000 description 1
- GIVLTTJNORAZON-HDBOBKCLSA-N ganglioside GM2 (18:0) Chemical compound O[C@@H]1[C@@H](O)[C@H](OC[C@H](NC(=O)CCCCCCCCCCCCCCCCC)[C@H](O)\C=C\CCCCCCCCCCCCC)O[C@H](CO)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@]2(O[C@H]([C@H](NC(C)=O)[C@@H](O)C2)[C@H](O)[C@H](O)CO)C(O)=O)[C@@H](O[C@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](CO)O1 GIVLTTJNORAZON-HDBOBKCLSA-N 0.000 description 1
- PFJKOHUKELZMLE-VEUXDRLPSA-N ganglioside GM3 Chemical compound O[C@@H]1[C@@H](O)[C@H](OC[C@@H]([C@H](O)/C=C/CCCCCCCCCCCCC)NC(=O)CCCCCCCCCCCCC\C=C/CCCCCCCC)O[C@H](CO)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@]2(O[C@H]([C@H](NC(C)=O)[C@@H](O)C2)[C@H](O)[C@H](O)CO)C(O)=O)[C@@H](O)[C@@H](CO)O1 PFJKOHUKELZMLE-VEUXDRLPSA-N 0.000 description 1
- 150000002270 gangliosides Chemical class 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 238000009650 gentamicin protection assay Methods 0.000 description 1
- 210000004602 germ cell Anatomy 0.000 description 1
- 208000003884 gestational trophoblastic disease Diseases 0.000 description 1
- 201000007116 gestational trophoblastic neoplasm Diseases 0.000 description 1
- 239000000174 gluconic acid Chemical class 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 230000013595 glycosylation Effects 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- 229940015043 glyoxal Drugs 0.000 description 1
- 239000000122 growth hormone Substances 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- ZRALSGWEFCBTJO-UHFFFAOYSA-N guanidine group Chemical group NC(=N)N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 1
- 230000003394 haemopoietic effect Effects 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 210000000777 hematopoietic system Anatomy 0.000 description 1
- 231100000753 hepatic injury Toxicity 0.000 description 1
- 208000010710 hepatitis C virus infection Diseases 0.000 description 1
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 1
- 208000025581 hereditary breast carcinoma Diseases 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 229960001340 histamine Drugs 0.000 description 1
- 125000000487 histidyl group Chemical group [H]N([H])C(C(=O)O*)C([H])([H])C1=C([H])N([H])C([H])=N1 0.000 description 1
- 201000008298 histiocytosis Diseases 0.000 description 1
- 239000000710 homodimer Substances 0.000 description 1
- 108091008039 hormone receptors Proteins 0.000 description 1
- 102000055229 human IL4 Human genes 0.000 description 1
- 229940084986 human chorionic gonadotropin Drugs 0.000 description 1
- 229940116886 human interleukin-6 Drugs 0.000 description 1
- 235000020256 human milk Nutrition 0.000 description 1
- 210000004251 human milk Anatomy 0.000 description 1
- 230000008348 humoral response Effects 0.000 description 1
- 229960000890 hydrocortisone Drugs 0.000 description 1
- 230000000148 hypercalcaemia Effects 0.000 description 1
- 208000030915 hypercalcemia disease Diseases 0.000 description 1
- 201000006866 hypopharynx cancer Diseases 0.000 description 1
- 229960000908 idarubicin Drugs 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 229960003685 imatinib mesylate Drugs 0.000 description 1
- YLMAHDNUQAMNNX-UHFFFAOYSA-N imatinib methanesulfonate Chemical compound CS(O)(=O)=O.C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 YLMAHDNUQAMNNX-UHFFFAOYSA-N 0.000 description 1
- 150000002463 imidates Chemical class 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- NBZBKCUXIYYUSX-UHFFFAOYSA-N iminodiacetic acid Chemical compound OC(=O)CNCC(O)=O NBZBKCUXIYYUSX-UHFFFAOYSA-N 0.000 description 1
- 230000036737 immune function Effects 0.000 description 1
- 230000008629 immune suppression Effects 0.000 description 1
- 230000006058 immune tolerance Effects 0.000 description 1
- 229940124622 immune-modulator drug Drugs 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 230000003308 immunostimulating effect Effects 0.000 description 1
- 230000001506 immunosuppresive effect Effects 0.000 description 1
- 238000009169 immunotherapy Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000006882 induction of apoptosis Effects 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 206010022000 influenza Diseases 0.000 description 1
- 230000015788 innate immune response Effects 0.000 description 1
- CDAISMWEOUEBRE-GPIVLXJGSA-N inositol Chemical compound O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@H](O)[C@@H]1O CDAISMWEOUEBRE-GPIVLXJGSA-N 0.000 description 1
- 229960000367 inositol Drugs 0.000 description 1
- 108010078480 insect defensin A Proteins 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 108010010648 interferon alfacon-1 Proteins 0.000 description 1
- 229960003130 interferon gamma Drugs 0.000 description 1
- 108010042414 interferon gamma-1b Proteins 0.000 description 1
- 108010080375 interferon kappa Proteins 0.000 description 1
- 108700027921 interferon tau Proteins 0.000 description 1
- 229940028894 interferon type ii Drugs 0.000 description 1
- 108090000681 interleukin 20 Proteins 0.000 description 1
- 108010074108 interleukin-21 Proteins 0.000 description 1
- 108010074109 interleukin-22 Proteins 0.000 description 1
- 102000003898 interleukin-24 Human genes 0.000 description 1
- 108090000237 interleukin-24 Proteins 0.000 description 1
- 201000002313 intestinal cancer Diseases 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 230000004068 intracellular signaling Effects 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 229940065638 intron a Drugs 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229960002358 iodine Drugs 0.000 description 1
- 229940036646 iodine-131-tositumomab Drugs 0.000 description 1
- PGLTVOMIXTUURA-UHFFFAOYSA-N iodoacetamide Chemical compound NC(=O)CI PGLTVOMIXTUURA-UHFFFAOYSA-N 0.000 description 1
- 208000002551 irritable bowel syndrome Diseases 0.000 description 1
- 208000028867 ischemia Diseases 0.000 description 1
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 1
- 229960000310 isoleucine Drugs 0.000 description 1
- 150000002540 isothiocyanates Chemical class 0.000 description 1
- 229940011083 istodax Drugs 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 208000022013 kidney Wilms tumor Diseases 0.000 description 1
- 201000010982 kidney cancer Diseases 0.000 description 1
- 238000011813 knockout mouse model Methods 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 239000000832 lactitol Substances 0.000 description 1
- 235000010448 lactitol Nutrition 0.000 description 1
- VQHSOMBJVWLPSR-JVCRWLNRSA-N lactitol Chemical compound OC[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O VQHSOMBJVWLPSR-JVCRWLNRSA-N 0.000 description 1
- 229960003451 lactitol Drugs 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 206010023841 laryngeal neoplasm Diseases 0.000 description 1
- 229960004942 lenalidomide Drugs 0.000 description 1
- GOTYRUGSSMKFNF-UHFFFAOYSA-N lenalidomide Chemical compound C1C=2C(N)=CC=CC=2C(=O)N1C1CCC(=O)NC1=O GOTYRUGSSMKFNF-UHFFFAOYSA-N 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 229940063725 leukeran Drugs 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 230000021633 leukocyte mediated immunity Effects 0.000 description 1
- 201000002364 leukopenia Diseases 0.000 description 1
- 201000011486 lichen planus Diseases 0.000 description 1
- 108020001756 ligand binding domains Proteins 0.000 description 1
- 201000002818 limb ischemia Diseases 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 201000006721 lip cancer Diseases 0.000 description 1
- 206010024627 liposarcoma Diseases 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 230000005976 liver dysfunction Effects 0.000 description 1
- 231100000835 liver failure Toxicity 0.000 description 1
- 208000007903 liver failure Diseases 0.000 description 1
- 210000005228 liver tissue Anatomy 0.000 description 1
- 210000003141 lower extremity Anatomy 0.000 description 1
- 229950004563 lucatumumab Drugs 0.000 description 1
- 201000005249 lung adenocarcinoma Diseases 0.000 description 1
- 201000005296 lung carcinoma Diseases 0.000 description 1
- 201000005243 lung squamous cell carcinoma Diseases 0.000 description 1
- 229940040129 luteinizing hormone Drugs 0.000 description 1
- 208000002502 lymphedema Diseases 0.000 description 1
- 208000019420 lymphoid neoplasm Diseases 0.000 description 1
- 108010026228 mRNA guanylyltransferase Proteins 0.000 description 1
- 201000010230 macular retinal edema Diseases 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 238000009115 maintenance therapy Methods 0.000 description 1
- 201000003175 male breast cancer Diseases 0.000 description 1
- 208000010907 male breast carcinoma Diseases 0.000 description 1
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 1
- 208000026045 malignant tumor of parathyroid gland Diseases 0.000 description 1
- 208000016847 malignant urinary system neoplasm Diseases 0.000 description 1
- 239000000845 maltitol Substances 0.000 description 1
- 235000010449 maltitol Nutrition 0.000 description 1
- VQHSOMBJVWLPSR-WUJBLJFYSA-N maltitol Chemical compound OC[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O VQHSOMBJVWLPSR-WUJBLJFYSA-N 0.000 description 1
- 229940035436 maltitol Drugs 0.000 description 1
- 229960001855 mannitol Drugs 0.000 description 1
- 229950001869 mapatumumab Drugs 0.000 description 1
- 229940087732 matulane Drugs 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- QWIZNVHXZXRPDR-WSCXOGSTSA-N melezitose Chemical compound O([C@@]1(O[C@@H]([C@H]([C@@H]1O[C@@H]1[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O1)O)O)CO)CO)[C@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O QWIZNVHXZXRPDR-WSCXOGSTSA-N 0.000 description 1
- 229960002514 melphalan hydrochloride Drugs 0.000 description 1
- 210000004779 membrane envelope Anatomy 0.000 description 1
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- 208000037819 metastatic cancer Diseases 0.000 description 1
- 208000011575 metastatic malignant neoplasm Diseases 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- CWWARWOPSKGELM-SARDKLJWSA-N methyl (2s)-2-[[(2s)-2-[[2-[[(2s)-2-[[(2s)-2-[[(2s)-5-amino-2-[[(2s)-5-amino-2-[[(2s)-1-[(2s)-6-amino-2-[[(2s)-1-[(2s)-2-amino-5-(diaminomethylideneamino)pentanoyl]pyrrolidine-2-carbonyl]amino]hexanoyl]pyrrolidine-2-carbonyl]amino]-5-oxopentanoyl]amino]-5 Chemical compound C([C@@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)OC)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CCCN=C(N)N)C1=CC=CC=C1 CWWARWOPSKGELM-SARDKLJWSA-N 0.000 description 1
- SJFKGZZCMREBQH-UHFFFAOYSA-N methyl ethanimidate Chemical compound COC(C)=N SJFKGZZCMREBQH-UHFFFAOYSA-N 0.000 description 1
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 1
- 238000012737 microarray-based gene expression Methods 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 210000002500 microbody Anatomy 0.000 description 1
- 230000002025 microglial effect Effects 0.000 description 1
- 238000010232 migration assay Methods 0.000 description 1
- 229950003734 milatuzumab Drugs 0.000 description 1
- 108010071421 milk fat globule Proteins 0.000 description 1
- 239000003226 mitogen Substances 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 238000007479 molecular analysis Methods 0.000 description 1
- 210000005087 mononuclear cell Anatomy 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 229940074923 mozobil Drugs 0.000 description 1
- 206010051747 multiple endocrine neoplasia Diseases 0.000 description 1
- 238000012243 multiplex automated genomic engineering Methods 0.000 description 1
- 231100000219 mutagenic Toxicity 0.000 description 1
- 239000003471 mutagenic agent Substances 0.000 description 1
- 230000003505 mutagenic effect Effects 0.000 description 1
- 208000029766 myalgic encephalomeyelitis/chronic fatigue syndrome Diseases 0.000 description 1
- 206010028417 myasthenia gravis Diseases 0.000 description 1
- 206010028537 myelofibrosis Diseases 0.000 description 1
- 208000025113 myeloid leukemia Diseases 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- XJODGRWDFZVTKW-ZCFIWIBFSA-N n-methylleucine Chemical compound CN[C@@H](C(O)=O)CC(C)C XJODGRWDFZVTKW-ZCFIWIBFSA-N 0.000 description 1
- 208000037830 nasal cancer Diseases 0.000 description 1
- 229960005027 natalizumab Drugs 0.000 description 1
- 210000000822 natural killer cell Anatomy 0.000 description 1
- QZGIWPZCWHMVQL-UIYAJPBUSA-N neocarzinostatin chromophore Chemical compound O1[C@H](C)[C@H](O)[C@H](O)[C@@H](NC)[C@H]1O[C@@H]1C/2=C/C#C[C@H]3O[C@@]3([C@@H]3OC(=O)OC3)C#CC\2=C[C@H]1OC(=O)C1=C(O)C=CC2=C(C)C=C(OC)C=C12 QZGIWPZCWHMVQL-UIYAJPBUSA-N 0.000 description 1
- 108010068617 neonatal Fc receptor Proteins 0.000 description 1
- 201000008026 nephroblastoma Diseases 0.000 description 1
- 208000007538 neurilemmoma Diseases 0.000 description 1
- 201000002120 neuroendocrine carcinoma Diseases 0.000 description 1
- 201000004931 neurofibromatosis Diseases 0.000 description 1
- 208000004235 neutropenia Diseases 0.000 description 1
- QOTXBMGJKFVZRD-HISDBWNOSA-O nicotinic acid-adenine dinucleotide phosphate Chemical compound [N+]1([C@@H]2O[C@@H]([C@H]([C@H]2O)O)COP(O)(=O)OP(O)(=O)OC[C@H]2O[C@H]([C@@H]([C@@H]2O)OP(O)(O)=O)N2C=3N=CN=C(C=3N=C2)N)=CC=CC(C(O)=O)=C1 QOTXBMGJKFVZRD-HISDBWNOSA-O 0.000 description 1
- 229960001346 nilotinib Drugs 0.000 description 1
- HHZIURLSWUIHRB-UHFFFAOYSA-N nilotinib Chemical compound C1=NC(C)=CN1C1=CC(NC(=O)C=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)=CC(C(F)(F)F)=C1 HHZIURLSWUIHRB-UHFFFAOYSA-N 0.000 description 1
- 238000006396 nitration reaction Methods 0.000 description 1
- 201000002674 obstructive nephropathy Diseases 0.000 description 1
- 229950005751 ocrelizumab Drugs 0.000 description 1
- XXUPLYBCNPLTIW-UHFFFAOYSA-N octadec-7-ynoic acid Chemical compound CCCCCCCCCCC#CCCCCCC(O)=O XXUPLYBCNPLTIW-UHFFFAOYSA-N 0.000 description 1
- 201000002575 ocular melanoma Diseases 0.000 description 1
- 229960002450 ofatumumab Drugs 0.000 description 1
- 229920001542 oligosaccharide Polymers 0.000 description 1
- 150000002482 oligosaccharides Chemical class 0.000 description 1
- 230000004650 oncogenic pathway Effects 0.000 description 1
- 102000027450 oncoproteins Human genes 0.000 description 1
- 108091008819 oncoproteins Proteins 0.000 description 1
- 229940100027 ontak Drugs 0.000 description 1
- 239000003399 opiate peptide Substances 0.000 description 1
- 201000005443 oral cavity cancer Diseases 0.000 description 1
- 201000005737 orchitis Diseases 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 229960003104 ornithine Drugs 0.000 description 1
- 201000006958 oropharynx cancer Diseases 0.000 description 1
- 201000008968 osteosarcoma Diseases 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 238000012261 overproduction Methods 0.000 description 1
- 229960001756 oxaliplatin Drugs 0.000 description 1
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 229940094443 oxytocics prostaglandins Drugs 0.000 description 1
- 239000003002 pH adjusting agent Substances 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 201000002530 pancreatic endocrine carcinoma Diseases 0.000 description 1
- 201000001219 parotid gland cancer Diseases 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 108010092853 peginterferon alfa-2a Proteins 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 201000001976 pemphigus vulgaris Diseases 0.000 description 1
- 229960001639 penicillamine Drugs 0.000 description 1
- 238000010647 peptide synthesis reaction Methods 0.000 description 1
- 201000006195 perinatal necrotizing enterocolitis Diseases 0.000 description 1
- KHIWWQKSHDUIBK-UHFFFAOYSA-N periodic acid Chemical compound OI(=O)(=O)=O KHIWWQKSHDUIBK-UHFFFAOYSA-N 0.000 description 1
- 210000005259 peripheral blood Anatomy 0.000 description 1
- 239000011886 peripheral blood Substances 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 201000002511 pituitary cancer Diseases 0.000 description 1
- 108010031345 placental alkaline phosphatase Proteins 0.000 description 1
- 210000004180 plasmocyte Anatomy 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 230000004983 pleiotropic effect Effects 0.000 description 1
- 208000008423 pleurisy Diseases 0.000 description 1
- 210000001778 pluripotent stem cell Anatomy 0.000 description 1
- 229960001237 podophyllotoxin Drugs 0.000 description 1
- YJGVMLPVUAXIQN-XVVDYKMHSA-N podophyllotoxin Chemical compound COC1=C(OC)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@H](O)[C@@H]3[C@@H]2C(OC3)=O)=C1 YJGVMLPVUAXIQN-XVVDYKMHSA-N 0.000 description 1
- YVCVYCSAAZQOJI-UHFFFAOYSA-N podophyllotoxin Natural products COC1=C(O)C(OC)=CC(C2C3=CC=4OCOC=4C=C3C(O)C3C2C(OC3)=O)=C1 YVCVYCSAAZQOJI-UHFFFAOYSA-N 0.000 description 1
- 229920001993 poloxamer 188 Polymers 0.000 description 1
- 229920001481 poly(stearyl methacrylate) Polymers 0.000 description 1
- 201000006292 polyarteritis nodosa Diseases 0.000 description 1
- 208000037244 polycythemia vera Diseases 0.000 description 1
- 208000005987 polymyositis Diseases 0.000 description 1
- 229940068965 polysorbates Drugs 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229960000688 pomalidomide Drugs 0.000 description 1
- UVSMNLNDYGZFPF-UHFFFAOYSA-N pomalidomide Chemical compound O=C1C=2C(N)=CC=CC=2C(=O)N1C1CCC(=O)NC1=O UVSMNLNDYGZFPF-UHFFFAOYSA-N 0.000 description 1
- 231100000683 possible toxicity Toxicity 0.000 description 1
- 230000003334 potential effect Effects 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 208000025638 primary cutaneous T-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 208000003476 primary myelofibrosis Diseases 0.000 description 1
- 230000000861 pro-apoptotic effect Effects 0.000 description 1
- 230000002206 pro-fibrotic effect Effects 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 150000003180 prostaglandins Chemical class 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 108010043671 prostatic acid phosphatase Proteins 0.000 description 1
- 239000003207 proteasome inhibitor Substances 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 238000001243 protein synthesis Methods 0.000 description 1
- 230000004850 protein–protein interaction Effects 0.000 description 1
- NGVDGCNFYWLIFO-UHFFFAOYSA-N pyridoxal 5'-phosphate Chemical compound CC1=NC=C(COP(O)(O)=O)C(C=O)=C1O NGVDGCNFYWLIFO-UHFFFAOYSA-N 0.000 description 1
- 235000007682 pyridoxal 5'-phosphate Nutrition 0.000 description 1
- 239000011589 pyridoxal 5'-phosphate Substances 0.000 description 1
- 229960001327 pyridoxal phosphate Drugs 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 102000016914 ras Proteins Human genes 0.000 description 1
- 208000002574 reactive arthritis Diseases 0.000 description 1
- 229940038850 rebif Drugs 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 238000005932 reductive alkylation reaction Methods 0.000 description 1
- 230000008439 repair process Effects 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 230000033458 reproduction Effects 0.000 description 1
- 230000008672 reprogramming Effects 0.000 description 1
- 230000004043 responsiveness Effects 0.000 description 1
- 230000000284 resting effect Effects 0.000 description 1
- 150000004492 retinoid derivatives Chemical class 0.000 description 1
- 229960003471 retinol Drugs 0.000 description 1
- 235000020944 retinol Nutrition 0.000 description 1
- 239000011607 retinol Substances 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 208000013860 rhabdoid tumor of the kidney Diseases 0.000 description 1
- 201000009410 rhabdomyosarcoma Diseases 0.000 description 1
- 201000003068 rheumatic fever Diseases 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 229960000329 ribavirin Drugs 0.000 description 1
- HZCAHMRRMINHDJ-DBRKOABJSA-N ribavirin Natural products O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1N=CN=C1 HZCAHMRRMINHDJ-DBRKOABJSA-N 0.000 description 1
- 102220237428 rs1185220863 Human genes 0.000 description 1
- 102220316641 rs146101365 Human genes 0.000 description 1
- 102220157854 rs371106595 Human genes 0.000 description 1
- 102200079760 rs3824915 Human genes 0.000 description 1
- 102200072124 rs863224863 Human genes 0.000 description 1
- 102220157938 rs886063483 Human genes 0.000 description 1
- 230000036185 rubor Effects 0.000 description 1
- 201000000306 sarcoidosis Diseases 0.000 description 1
- 229940043230 sarcosine Drugs 0.000 description 1
- 231100000241 scar Toxicity 0.000 description 1
- 206010039667 schwannoma Diseases 0.000 description 1
- CDAISMWEOUEBRE-UHFFFAOYSA-N scyllo-inosotol Natural products OC1C(O)C(O)C(O)C(O)C1O CDAISMWEOUEBRE-UHFFFAOYSA-N 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 229940076279 serotonin Drugs 0.000 description 1
- 238000001542 size-exclusion chromatography Methods 0.000 description 1
- 201000000849 skin cancer Diseases 0.000 description 1
- 201000008261 skin carcinoma Diseases 0.000 description 1
- 201000002314 small intestine cancer Diseases 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 210000001082 somatic cell Anatomy 0.000 description 1
- 230000000392 somatic effect Effects 0.000 description 1
- NHXLMOGPVYXJNR-ATOGVRKGSA-N somatostatin Chemical compound C([C@H]1C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CSSC[C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(=O)N1)[C@@H](C)O)NC(=O)CNC(=O)[C@H](C)N)C(O)=O)=O)[C@H](O)C)C1=CC=CC=C1 NHXLMOGPVYXJNR-ATOGVRKGSA-N 0.000 description 1
- 229960000553 somatostatin Drugs 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 230000009870 specific binding Effects 0.000 description 1
- 150000003408 sphingolipids Chemical class 0.000 description 1
- 206010062261 spinal cord neoplasm Diseases 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 206010041823 squamous cell carcinoma Diseases 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- INIBXSLTWQVIHS-ASACRTLUSA-O stanford v protocol Chemical compound ClCCN(C)CCCl.O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1.O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1.COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3C(O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1.C([C@H](C[C@]1(C(=O)OC)C=2C(=C3C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)=CC=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21.C([C@H](C[C@]1(C(=O)OC)C=2C(=C3C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C=O)=CC=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21.N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)C(O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1NC=NC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C INIBXSLTWQVIHS-ASACRTLUSA-O 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 238000011476 stem cell transplantation Methods 0.000 description 1
- 239000008174 sterile solution Substances 0.000 description 1
- 239000003270 steroid hormone Substances 0.000 description 1
- 150000003432 sterols Chemical class 0.000 description 1
- 235000003702 sterols Nutrition 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 229940014800 succinic anhydride Drugs 0.000 description 1
- KZNICNPSHKQLFF-UHFFFAOYSA-N succinimide Chemical class O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 1
- JJAHTWIKCUJRDK-UHFFFAOYSA-N succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate Chemical compound C1CC(CN2C(C=CC2=O)=O)CCC1C(=O)ON1C(=O)CCC1=O JJAHTWIKCUJRDK-UHFFFAOYSA-N 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 230000008093 supporting effect Effects 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 206010042863 synovial sarcoma Diseases 0.000 description 1
- 238000007910 systemic administration Methods 0.000 description 1
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 1
- 101150047061 tag-72 gene Proteins 0.000 description 1
- 239000011975 tartaric acid Chemical class 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- DKPFODGZWDEEBT-QFIAKTPHSA-N taxane Chemical class C([C@]1(C)CCC[C@@H](C)[C@H]1C1)C[C@H]2[C@H](C)CC[C@@H]1C2(C)C DKPFODGZWDEEBT-QFIAKTPHSA-N 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 229950001790 tendamistat Drugs 0.000 description 1
- 108010037401 tendamistate Proteins 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- 201000003120 testicular cancer Diseases 0.000 description 1
- 229960003604 testosterone Drugs 0.000 description 1
- 108010013645 tetranectin Proteins 0.000 description 1
- 150000004044 tetrasaccharides Chemical class 0.000 description 1
- 229960003433 thalidomide Drugs 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- 201000009377 thymus cancer Diseases 0.000 description 1
- 201000002510 thyroid cancer Diseases 0.000 description 1
- 230000009772 tissue formation Effects 0.000 description 1
- 208000037816 tissue injury Diseases 0.000 description 1
- 230000017423 tissue regeneration Effects 0.000 description 1
- 229960003989 tocilizumab Drugs 0.000 description 1
- 239000003970 toll like receptor agonist Substances 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 108700012359 toxins Proteins 0.000 description 1
- SBUXRMKDJWEXRL-ZWKOTPCHSA-N trans-body Chemical compound O=C([C@@H]1N(C2=O)[C@H](C3=C(C4=CC=CC=C4N3)C1)CC)N2C1=CC=C(F)C=C1 SBUXRMKDJWEXRL-ZWKOTPCHSA-N 0.000 description 1
- 239000012096 transfection reagent Substances 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 230000023750 transforming growth factor beta production Effects 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 108091007466 transmembrane glycoproteins Proteins 0.000 description 1
- 102000027257 transmembrane receptors Human genes 0.000 description 1
- 108091008578 transmembrane receptors Proteins 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 229940066958 treanda Drugs 0.000 description 1
- 238000011277 treatment modality Methods 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 229960001727 tretinoin Drugs 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 150000004043 trisaccharides Chemical class 0.000 description 1
- 229940073585 tromethamine hydrochloride Drugs 0.000 description 1
- 208000029387 trophoblastic neoplasm Diseases 0.000 description 1
- 239000002753 trypsin inhibitor Substances 0.000 description 1
- 125000000430 tryptophan group Chemical group [H]N([H])C(C(=O)O*)C([H])([H])C1=C([H])N([H])C2=C([H])C([H])=C([H])C([H])=C12 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 1
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 1
- 150000004917 tyrosine kinase inhibitor derivatives Chemical class 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 241001529453 unidentified herpesvirus Species 0.000 description 1
- 229940091440 uniferon Drugs 0.000 description 1
- 201000004435 urinary system cancer Diseases 0.000 description 1
- 208000019206 urinary tract infection Diseases 0.000 description 1
- 102000009816 urokinase plasminogen activator receptor activity proteins Human genes 0.000 description 1
- 108040001269 urokinase plasminogen activator receptor activity proteins Proteins 0.000 description 1
- 206010046766 uterine cancer Diseases 0.000 description 1
- 208000037965 uterine sarcoma Diseases 0.000 description 1
- 230000001982 uveitic effect Effects 0.000 description 1
- 206010046885 vaginal cancer Diseases 0.000 description 1
- 208000013139 vaginal neoplasm Diseases 0.000 description 1
- 239000004474 valine Substances 0.000 description 1
- 229960000653 valrubicin Drugs 0.000 description 1
- ZOCKGBMQLCSHFP-KQRAQHLDSA-N valrubicin Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC(OC)=C4C(=O)C=3C(O)=C21)(O)C(=O)COC(=O)CCCC)[C@H]1C[C@H](NC(=O)C(F)(F)F)[C@H](O)[C@H](C)O1 ZOCKGBMQLCSHFP-KQRAQHLDSA-N 0.000 description 1
- 229960003726 vasopressin Drugs 0.000 description 1
- 229940099039 velcade Drugs 0.000 description 1
- 229960004355 vindesine Drugs 0.000 description 1
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 1
- 201000005102 vulva cancer Diseases 0.000 description 1
- 229940051021 yellow-fever virus Drugs 0.000 description 1
- 229950009268 zinostatin Drugs 0.000 description 1
- 229940061261 zolinza Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/62—DNA sequences coding for fusion proteins
- C12N15/625—DNA sequences coding for fusion proteins containing a sequence coding for a signal sequence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/64—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent
- A61K47/642—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent the peptide or protein in the drug conjugate being a cytokine, e.g. IL2, chemokine, growth factors or interferons being the inactive part of the conjugate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/52—Cytokines; Lymphokines; Interferons
- C07K14/54—Interleukins [IL]
- C07K14/5406—IL-4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/52—Cytokines; Lymphokines; Interferons
- C07K14/54—Interleukins [IL]
- C07K14/5412—IL-6
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/52—Cytokines; Lymphokines; Interferons
- C07K14/555—Interferons [IFN]
- C07K14/56—IFN-alpha
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/52—Cytokines; Lymphokines; Interferons
- C07K14/555—Interferons [IFN]
- C07K14/565—IFN-beta
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/52—Cytokines; Lymphokines; Interferons
- C07K14/555—Interferons [IFN]
- C07K14/57—IFN-gamma
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/08—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from viruses
- C07K16/10—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from viruses from RNA viruses
- C07K16/1027—Paramyxoviridae, e.g. respiratory syncytial virus
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2887—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against CD20
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2896—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against molecules with a "CD"-designation, not provided for elsewhere
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/46—Hybrid immunoglobulins
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/33—Crossreactivity, e.g. for species or epitope, or lack of said crossreactivity
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/55—Fab or Fab'
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/565—Complementarity determining region [CDR]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/20—Fusion polypeptide containing a tag with affinity for a non-protein ligand
- C07K2319/21—Fusion polypeptide containing a tag with affinity for a non-protein ligand containing a His-tag
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/30—Non-immunoglobulin-derived peptide or protein having an immunoglobulin constant or Fc region, or a fragment thereof, attached thereto
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/33—Fusion polypeptide fusions for targeting to specific cell types, e.g. tissue specific targeting, targeting of a bacterial subspecies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/70—Fusion polypeptide containing domain for protein-protein interaction
- C07K2319/74—Fusion polypeptide containing domain for protein-protein interaction containing a fusion for binding to a cell surface receptor
Definitions
- the present invention relates to polypeptide constructs comprising mutated, attenuated polypeptide ligands attached to antibodies, wherein the antibodies direct the mutated ligands to cells that express on their surfaces the antigens to which said antibodies bind, as well as receptors for said ligands.
- the invention further relates to methods of treatment involving the use of these polypeptide constructs.
- peptide and polypeptide ligands have been described to function by interacting with a receptor on a cell surface, and thereby stimulating, inhibiting, or otherwise modulating a biological response, usually involving signal transduction pathways inside the cell that bears the said receptor.
- ligands include peptide and polypeptide hormones, cytokines, chemokines, growth factors, apoptosis- inducing factors and the like.
- Natural ligands can be either soluble or can be attached to the surface of another cell. [0004] Due to the biological activity of such ligands, some have potential use as therapeutics.
- peptide or polypeptide ligands have been approved by regulatory agencies as therapeutic products, including, for example, human growth hormone, insulin, interferon (IFN)-Cc2b, IFNcc2a, IFN , erythropoietin, G-CSF and GM-CSF. Many of these and other ligands have demonstrated potential in therapeutic applications, but have also exhibited toxicity when administered to human patients. One reason for toxicity is that most of these ligands trigger receptors on a variety of cells, including cells other than those that mediate the therapeutic effect.
- IFN interferon
- IFNcc2b when used to treat multiple myeloma its utility resides, at least in part, in its binding to type I interferon receptors on the myeloma cells, which in turn triggers reduced proliferation and hence limits disease progression.
- this IFN also binds to numerous other, normal cells within the body, triggering a variety of other cellular responses, some of which are harmful (e.g. flu-like symptoms, neutropenia, depression).
- off target activity of ligands is that many ligands are not suitable as drug candidates.
- off target activity refers to activity on the ligand's natural receptor, but on the surface of cells other than those that mediate therapeutically beneficial effects.
- interferons in particular IFNa
- IFNa interferons
- type I interferon receptors on the surface of the cancer cells which, when stimulated, initiate various signal transduction pathways leading to reduced proliferation and/or the induction of terminal differentiation or apoptosis.
- IFNa has been approved by the FDA for the treatment of several cancers including melanoma, renal cell carcinoma, B cell lymphoma, multiple myeloma, chronic myelogenous leukemia (CML) and hairy cell leukemia.
- a "direct” effect of IFNa on the tumour cells is mediated by the IFNa binding directly to the type I IFN receptor on those cells and stimulating apoptosis, terminal differentiation or reduced proliferation.
- One "indirect” effect of IFNa on non-cancer cells is to stimulate the immune system, which may produce an additional anti-cancer effect by causing the immune system to reject the tumour.
- I interferon receptor is also present on most noncancerous cells. Activation of this receptor on such cells by IFNa causes the expression of numerous pro-inflammatory cytokines and chemokines, leading to toxicity. Such toxicity prevents the dosing of IFNa to a subject at levels that exert the maximum anti-proliferative and pro-apoptotic activity on the cancer cells.
- the linkage between an antibody and IFNa may be accomplished by making a fusion protein construct.
- IDEC WOO 1/97844
- IDEC disclose a direct fusion of human IFNa to the C terminus of the heavy chain of an IgG targeting the tumour antigen CD20.
- Other groups have disclosed the use of various linkers between the C-terminus of an IgG heavy chain and the IFNa.
- US 7,456,257 discloses that the C-terminus of an antibody heavy chain constant region may be connected to IFNa via an intervening serine-glycine rich (S/G) linker of the sequence (GGGGS) n , where n may be 1, 2 or 3, and that there are no significant differences in the IFNa activity of the fusion protein construct regardless of linker length.
- S/G serine-glycine rich
- n may be 1, 2 or 3
- Blood 2010;115:2864-71) also disclose IFNcc linked to the C-terminus of the heavy chain of a cancer-targeting IgG antibody, with an intervening S/G linker, and observed that the fusion of the IgG and linker to the IFNcc reduced the activity of IFNcc on cells that did not express the corresponding antigen on the cell surface.
- the decreased IFN activity of these fusion protein constructs was modest when compared to human non-fusion protein IFNcc (free IFN a) acting on human cells, but appeared to be more significant for murine IFNcc on murine cells.
- the prior art teaches to use a potent IFN and to target this IFN to cancer cells. While this approach results in an increase in activity of the IFN against cancer cells, it does not address the issue of activity of the IFN on normal "off-target" cells.
- the human IFNcc portion of the antibody- IFN fusion protein maintained a high proportion of native IFNcc activity when exposed to human cells that do not express the corresponding antigen on their cell surfaces. This activity may lead to toxicity arising from the activation of non-cancerous, normal ("off target”) cells by the IFNcc portion of the fusion protein.
- the present inventors have found that when a peptide or polypeptide signaling ligand, having one or more mutations which substantially decrease the affinity of the ligand for its receptor, is linked to an antibody that targets the mutated ligand to target cells which display the antibody's corresponding antigen, the ligand' s activity on target antigenic) positive cells is maintained while the ligand' s activity on non-target antigen-negative cells is substantially reduced.
- the net result is a ligand signaling molecule that has a much greater potency in activation of its receptors on antigen-positive target cells compared to antigen-negative non-target cells, which provides a means for reducing toxicity arising from off-target ligand activity.
- a first aspect of the present invention provides a polypeptide
- construct comprising a peptide or polypeptide signaling ligand linked to an antibody or antigen binding portion thereof which binds to a cell surface-associated antigen, wherein the ligand comprises at least one amino acid substitution or deletion which reduces its potency on cells lacking expression of said antigen.
- the present invention provides a method of treating a tumour in a subject, comprising administering to the subject the polypeptide construct of the present invention.
- the present invention provides use of the polypeptide construct of the present invention in the treatment of cancer.
- the present invention provides a composition comprising the polypeptide construct of the present invention and a pharmaceutically acceptable carrier or diluent.
- the present invention provides method of reducing the potency of a peptide or polypeptide signaling ligand on an antigen negative cell which bears the ligand receptor whilst maintaining the potency of the ligand on an antigen positive cell which bears the ligand receptor to a greater extent when compared to the antigen negative cell, the method comprising modifying the ligand such that the ligand comprises at least one amino acid substitution or deletion which reduces its potency on the antigen negative cell and linking the modified ligand to an antibody or antigen-binding portion thereof, wherein the antibody or antigen binding portion thereof is specific for a cell surface- associated antigen on the antigen positive cell but not on the antigen negative cell.
- the present invention Unlike the linking of a non-attenuated "native" or “wild-type” human ligand to an antibody or antigen-binding portion therof, which typically results in from 1 to 15-fold higher potency of the ligand on antigen-positive compared to antigen-negative cells, the present invention demonstrates that the attachment of mutated, attenuated forms of the ligand to the same antibody is able to generate higher potency on antigen-positive cells compared to antigen negative cells.
- the signaling ligand is IFNa or IFNP and the polypeptide construct shows at least 10, at least 100, at least 1,000, at least 10,000 or at least 100,000 - fold greater selectivity towards antigen positive cells over antigen negative cells compared to free, wild-type ligand using the "off-target” assay and the "on target (ARP)” or “on target (Daudi)” assays described herein.
- the present invention also provides an antibody-attenuated ligand fusion proteins, wherein the attenuated ligand is IFNa or ⁇ and the wherein fusion protein construct, when injected into a mouse with an established human tumor, can eliminate the tumor.
- the present invention also provides an antibody-attenuated ligand fusion proteins, wherein the attenuated ligand is IFNa or ⁇ and wherein the fusion protein construct, when injected into a mouse with an established human tumor with a volume of over 500 cubic millimeters, can eliminate the tumor.
- the present invention also provides an antibody-attenuated ligand fusion proteins, wherein the attenuated ligand is IFNa or ⁇ and wherein the fusion protein construct, when injected as a single one-time treatment into a mouse with an established human tumor, can eliminate the tumor.
- cell surface-associated antigen is CD 38.
- the amino acid sequence of the signaling ligand comprising at least one amino acid substitution or deletion has greater than 90% or greater than 95%, or greater than 96%, or greater than 97%, or greater than 98% or greater than 99% sequence identity with the wild-type ligand amino acid sequence.
- the construct is a fusion protein.
- the signaling ligand is linked to the C-terminus of the heavy chain of the antibody or antigen binding portion thereof. In certain embodiments the signaling ligand is linked to the C-terminus of the light chain of the antibody or antigen binding portion thereof. In either of these embodiments, the ligand may be linked directly to the C-terminus of the heavy or light chain of the antibody or antigen binding portion thereof (ie without an intervening additional linker).
- the cell surface associated antigen is selected from class I MHC or PD-1.
- the cell surface-associated antigen is a myeloma associated antigen which is selected from the group consisting of CD38, HM1.24, CD56, CS1, CD138, CD74, IL-6R, Blys (BAFF), BCMA, HLA-SR, Kininogen, beta2
- microglobulin FGFR3, ICAM-1, matriptase, CD52, EGFR, GM2, alpha4-integrin, IFG-IR and KIR, and the ligand is an IFNa.
- the signaling ligand is selected from any one of IFNa2b, ⁇ , IL-4 or IL-6.
- the amino acid substitution or deletion may be at any one or more of amino acid positions R33, R144 or A145.
- the signaling ligand is an IFNa and the substitution is selected from the group consisting of R144A (SEQ ID NO:30), R144S (SEQ ID NO:40), R144T (SEQ ID NO:41), R144Y (SEQ ID NO:43), R144I (SEQ ID NO:35), R144L (SEQ ID NO:37), A145D (SEQ ID NO:44), A145H (SEQ ID NO:47), A145Y (SEQ ID NO:58), A145K (SEQ ID NO:49), R33A+YNS (SEQ ID NO:65), R33A (SEQ ID NO: 16) and R144A+YNS (SEQ ID NO:68).
- the antibody is selected from any one of G003, G005, G024, MOR03077, MORO3079, MORO3080, MORO3100, 38SB13, 38SB18, 38SB19, 38SB30, 38SB31, 38SB39, OKT10, X355/02, X910/12, X355/07, X913/15, R5D1, R5E8, R10A2, or an antigen binding portion thereof, or an antibody with greater than 95%, greater than 96%, greater than 97%, greater than 98% or at least 99% amino acid sequence identity with any one of R5D1, R5E8 or R10A2.
- the treatment is for a cancer in a subject selected from multiple myeloma, a leukemia or a lymphoma.
- the subject is also treated with a retinoid, such as all-trans retinoic acid.
- the tumour or cancer may be selected from multiple myeloma, non-Hodgkin's lymphoma, chronic myelogenous leukemia, chronic lymphocytic leukemia or acute myelogenous leukemia.
- the antibody may be an IgG4.
- the IgG4 comprises an S228P amino acid substitution.
- the antibody or antigen binding portion thereof may bind to a cell surface associateed antigen on virally infected cells.
- the cell surface associated antigen may be selected from a virally encoded protein, phosphatidylserine or a phosphatidylserine -binding protein.
- the construct may be used to treat Hepatitis C.
- the cell surface associated antigen is selected from CD20, CD38, CD138 or CS1.
- the tumour or cancer may be selected from multiple myeloma, melanoma, renal cell carcinoma, chronic myelogenous leukemia or hairy cell leukemia.
- the construct is G005-HC-L0-IFNa (A145D) IgG4.
- the cell surface associated antigen may be a T cell, a myeloid cell or an antigen presenting cell cell surface associated protein.
- the cell surface associated antigen may be selected from the group consisting of CD3, CD4, CD8, CD24, CD38, CD44, CD69, CD71, CD83, CD86, CD96, HLA-DR, PD-1, ICOS, CD33, CD115, CDl lc, CD14, CD52 and PD-1.
- the construct may be used to treat a disease characterized by excess inflamammation, such as an autoimmune disease.
- the signaling ligand of the polypeptide construct is an IFNP
- the at least one amino acid substitution or deletion is selected from the group consisting of R35A, R35T, E42K, M62I, G78S, A141Y, A142T, E149K, R152H.
- the IFNP may also possess a C17S or C17A substitution.
- the signaling ligand of the polypeptide construct is an IFNy.
- the cell surface associated antigen may be a tumor-associated antigen.
- the cell surface associated antigen may be selected from the group consisting of CD 14, FSP1, FAP, PDGFR alpha and PDGFR beta.
- the construct may be used to treat a disease characterized by excess fibrosis.
- the at least one amino acid substitution or deletion is selected from the group consisting of a deletion of residues A23 and D24, an S20I substitution, an A23V substitution, a D21K substitution and a D24A substitution.
- the cell surface associated antigen is selected from the group consisting of CD3, CD4, CD24, CD38, CD44, CD69, CD71, CD96, PD-1, ICOS, CD52 and PD-1.
- the cell surface associated antigen is selected from the group consisting of CD3, CD4, CD24, CD38, CD44, CD69, CD71, CD96, PD-1, ICOS, CD52 and PD-1.
- the cell surface associated antigen is selected from the group consisting of ASGR1, ASGR2, FSP1, RTI140/Ti-alpha, HTI56 and a VEGF receptor.
- the cell surface associated antigen is selected from the group consisting of CD3, CD4, CD8, CD24, CD38, CD44, CD69, CD71, CD83, CD86, CD96, HLA-DR, PD-1, ICOS, CD33, CD115, CDl lc, CD14, CD52 and PD-1.
- the cell surface associated antigen is selected from the group consisting of CD241 the product of the RCHE gene, CD117 (c-kit), CD71 (transferrin receptor), CD36 (thrombospondin receptor), CD34, CD45RO, CD45RA, CD115, CD168, CD235, CD236, CD237, CD238, CD239 and CD240.
- the signaling ligand of the polypeptide construct is an interleukin-10 and the cell surface associated antigen is selected from the group consisting of CD1 lc, CD33 or CD115, CD14, FSP1, FAP, or PDGFR (alpha or beta).
- anti-CD38 antibodies with variable regions designated X910/12, X913/15, X355/02, X355/07, R5D1, R5E8, or R10A2, with sequences set out as follows:
- Figure 1 shows a schematic of the certain embodiments of the present invention that comprise an antibody consisting of 2 heavy chains and 2 light chain, in which one or two attenuated signaling ligands is or are attached to each heavy chain or each light chain, or both.
- Figure 2 shows a schematic illustrating one possible approach for how the antibody-attenuated ligand fusion proteins of the present invention cause signaling by activating receptors on cells that display the antigen corresponding to the said antibody on their cell surfaces.
- the fusion protein activates the receptor on the same cell that the antibody is bound to, via its specific antigen.
- Figure 3 shows the amino acid sequences of the human CD38 (SEQ ID NO: 131).
- Figure 4 shows the amino acid sequences of certain exemplary signaling ligands of the present invention: (a) human IFNcc2b, ⁇ , IFN lb and IFNy; (b) IL-4 and IL-6.
- Figure 5 shows the amino acid sequences of certain antibody-attenuated ligand fusion proteins of the present invention: (a) G005-HC-L0-IFNa (A145D) IgG4; (b) nBT062-HC-L0-IFNa (A145D) IgG4; (c) G005-HC-L0-IFNP (R35A) IgG4; (d) HB95- HC-L16-IL-6 (R179E) IgGl; and (e) J110-HC-L6-IL-4 (R88Q) IgGl.
- the nomenclature for the fusion proteins is described in the examples.
- Figure 6 shows the non-antibody-antigen-targeted interferon activity of IFNcc2b, and of the antibody- IFN fusion protein constructs Rituximab-IFN 2b (Rituximab-HC-L6- IFNcc IgGl) and Palivizumab-IFNcc2b (Isotype-HC-L6-IFN IgGl) in the interferon activity assay described in the examples below as the "off-target assay.
- IFNcc equivalents refers to the molar concentration of interferon molecules, either free or attached to an antibody.
- IFN refers to free (non-fusion protein) wild-type interferon.
- Figure 7 shows the antibody-antigen-targeted interferon activity of the
- Rituximab-IFN 2b fusion protein construct (Rituximab-HC-L6-IFNa IgGl) compared with IFNcc2b in the antiproliferative assay described in the examples below as the "on target (Daudi) assay.”
- Figure 8 shows the antibody-antigen-targeted interferon activity of the
- Rituximab-IFN fusion protein construct (Rituximab-HC-L6-IFNa IgGl) compared with the non-targeted activity of Palivizumab-IFN fusion protein construct (Isotype-HC-L6- IFNa IgGl) in the "on-target (Daudi) assay" described in the examples below.
- Figure 9 shows the non-antibody-antigen-targeted interferon activity of IFNcc2b, of the antibody- IFN fusion protein constructs Rituximab-IFNcc2b (Rituximab-HC-L6-IFNa IgGl) and Palivizumab-IFNcc2b (Isotype-HC-L6-IFNcc IgGl), and of certain variants of Rituximab-IFN 2b constructs that have been mutated to reduce their interferon activity.
- the assay is described in the examples as the "off-target assay".
- Figure 10 shows the non-antibody-antigen-targeted interferon activity of the antibody- IFN fusion protein constructs Rituximab-IFNcc2b (Rituximab-HC-L6-IFNcc IgGl) and of two variants of Rituximab-IFNcc2b that were mutated to reduce interferon activity.
- the assay is described in the examples as the "off-target assay”.
- Figure 11 shows the antibody-antigen-targeted interferon activity of the antibody- IFN fusion protein construct Rituximab-IFNcc2b (Rituximab-HC-L6-IFNcc IgGl) and of variants of Rituximab-IFNa2b constructs that have been mutated to reduce their interferon activity compared to the non-targeted activity of the Palivizumab-IFNcc2b (Isotype-HC- L6-IFNCC IgGl) fusion protein constructs and compared to IFNcc2b.
- the assay is described in the examples as the "on target (Daudi) assay.”
- Figure 12 shows the antibody-antigen-targeted interferon activity of the antibody- IFN fusion protein constructs Rituximab-IFNcc2b (Rituximab-HC-L6-IFNcc IgGl) and of two variants that were mutated to reduce interferon activity.
- the assay is described in the examples as the "on target (Daudi) assay.”
- Figure 13 shows the sequences of certain novel human CD38 antibodies disclosed herein.
- Figure 14 shows the results of detection of binding of novel human anti-CD38 antibodies to a CD38 + cell line RPMI-8226 by flow cytometry.
- the x axis is the antibody concentration in micrograms/ml and the y axis represents the mean fluorescence intensity.
- Figure 15 shows the non-antibody-antigen targeted IFN activity of IFNcc2b compared with an anti-CD38-IFNcc fusion protein construct (G005-HC-L0-IFNCC IgG4), based on the anti-CD38 antibody G005.
- the assay is described in the examples as the "off- target assay.”
- Figure 16 shows the antiproliferative activity of IFNcc2b vs an anti-CD38-IFNcc fusion protein construct (G005-HC-L0-IFNcc IgG4) on the multiple myeloma cell line ARP-1 (CD38 + ).
- the assay is described in the examples as the "on target (ARP) assay.”
- Figure 17 shows the non-antibody-antigen targeted IFN activity of IFNcc2b vs various anti-CD38-IFNcc fusion protein constructs bearing point mutations in the IFN portion.
- the antibody variable regions of these fusion protein constructs were derived from antibody G005.
- the assay is described in the examples as the "off-target assay.”
- Figure 18 shows the non-antibody-antigen targeted IFN activity of IFNcc2b vs various anti-CD38-IFNcc fusion protein constructs bearing point mutations in the IFN portion.
- the antibody variable regions of these fusion proteins were derived from antibody G005. The assay is described in the examples as the "off-target assay.”
- Figure 19 shows the non-antibody-antigen targeted IFN activity of IFNcc2b vs two anti-CD38-IFNcc fusion protein constructs bearing point mutations in the IFN portion.
- the antibody variable regions of these fusion protein constructs are derived from antibody G005.
- the assay is described in the examples as the "off-target assay.”
- Figure 20 shows the antiproliferative activity of IFNcc2b vs anti-CD38-IFNcc fusion protein constructs with mutations in the IFN portion on the lymphoma cell line Daudi.
- the antibody variable regions of these fusion protein constructs are derived from antibody G005.
- the assay is described in the examples as the "on target (Daudi) assay.”
- Figure 21 shows the anti-proliferative activity of IFNcc2b and various anti-CD38- IFNa fusion protein with the A145G mutation in the IFN portion.
- Fusion protein constructs have either the 6 amino acid L6 linker or no linker (L0) and are of the IgGl or IgG4 isotype.
- the antibody variable regions of these fusion protein constructs are derived from antibody G005.
- the assay is described in the examples as the "on target (Daudi) assay”.
- Figure 22 shows the anti-proliferative activity of IFNcc2b and two anti-CD38- IFNcc fusion protein with the A145G mutation in the IFN portion.
- Both fusion protein constructs had the IFN portion linked to the C-terminus of the light chain, with either a six amino acid linker (L6) or no linker (L0).
- the antibody variable regions of these fusion protein constructs are derived from antibody G005.
- the assay is described in the examples as the "on target (Daudi) assay.”
- Figure 23 shows the antiproliferative activity on the multiple myeloma cell line ARP-1 of IFNcc2b vs anti-CD38-IFNcc fusion protein constructs with the R144A mutation in the IFN portion. The experiment compares the potency of the fusion protein constructs as a function of isotype (IgGl vs.
- the antibody variable regions of these fusion protein constructs are derived from antibody G005.
- the assay is described in the examples as the "on target (ARP) assay.”
- Figure 24 shows the antiproliferative activity on the multiple myeloma cell line ARP-1 of IFNcc2b vs anti-CD38-IFNcc fusion protein constructs with the A145G mutation in the IFN portion.
- the experiment compares the potency of the fusion protein constructs as a function of isotype (IgGl vs. IgG4) and the presence or absence of the L6 linker between the antibody heavy chain C-terminus and the N-terminus of the mutated IFN.
- the antibody variable regions of these fusion protein constructs are derived from antibody G005.
- the assay is described in the examples as the "on target (ARP) assay.”
- Figure 25 shows the non-antibody-antigen targeted IFN activity of various anti- CD38-IFNCC fusion protein constructs with different point mutations in the IFN portion.
- the antibody variable regions of these fusion protein constructs are derived from antibody G005.
- the assay is described in the examples as the "off-target assay.”
- Figure 26 shows the non-antibody-antigen targeted IFN activity of various anti- CD38-IFNCC fusion protein constructs with different point mutations in the IFN portion.
- the antibody variable regions of these fusion protein constructs are derived from antibody G005.
- the assay is described in the examples as the "off-target assay.”
- Figure 27 shows the non-antibody-antigen targeted IFN activity of various anti- CD38-IFNCC fusion protein constructs with different point mutations in the IFN portion.
- the antibody variable regions of these fusion protein constructs are derived from antibody G005.
- the assay is described in the examples as the "off-target assay.”
- Figure 28 shows the non-antibody-antigen targeted IFN activity of various anti- CD38-IFNCC fusion protein constructs with different point mutations in the IFN portion.
- the antibody variable regions of these fusion protein constructs are derived from antibody G005.
- Figure 29 shows the non-antibody-antigen targeted IFN activity of IFNcc2b vs various anti-CD38-IFNcc fusion protein constructs with different point mutations in the IFN portion.
- the antibody variable regions of these fusion protein constructs are derived from antibody G005.
- the assay is described in the examples as the "off-target assay.”
- Figure 30 shows the non-antibody-antigen targeted IFN activity of various anti- CD38-IFNCC fusion protein constructs with different point mutations in the IFN portion.
- the antibody variable regions of these fusion protein constructs are derived from antibody G005.
- the assay is described in the examples as the "off-target assay.”
- Figure 31 shows the antiproliferative activity on the multiple myeloma cell line ARP-1 of anti-CD38-IFNcc fusion protein constructs with the various mutations in the IFN portion.
- the antibody variable regions of these fusion protein constructs are derived from antibody G005.
- the assay is described in the examples as the "on target (ARP) assay.”
- Figure 32 shows the antiproliferative activity on the multiple myeloma cell line ARP-1 of IFNcc2b vs anti-CD38-IFNcc fusion protein constructs with the various mutations in the IFN portion.
- the antibody variable regions of these fusion protein constructs are derived from antibody G005.
- the assay is described in the examples as the "on target (ARP) assay.”.
- Figure 33 shows the antiproliferative activity on the multiple myeloma cell line ARP-1 of IFNcc2b vs anti-CD38-IFNcc fusion protein constructs with the R144A mutation in the IFN portion.
- the experiment compares different antibody variable regions in the context of the same mutated IFN fusion protein.
- the assay is described in the examples as the "on target (ARP) assay.”
- Figure 34 shows the antiproliferative activity on the multiple myeloma cell line ARP-1 of IFNcc2b vs anti-CD38-IFNcc fusion protein constructs with the A145D mutation in the IFN portion.
- the experiment compares different antibody variable regions in the context of the same mutated IFN fusion protein construct.
- the assay is described in the examples as the "on target (ARP) assay.”
- Figure 35 shows the non-antibody-antigen targeted IFN activity of IFNcc2b and various anti-CD38-IFNcc fusion protein constructs with the R144A mutation in the IFN portion.
- the experiment compares different antibody variable regions in the context of the same mutated IFN fusion protein construct.
- the assay is described in the examples as the "off-target assay.”
- Figure 36 shows the non-antibody-antigen targeted IFN activity of IFNcc2b and various anti-CD38-IFNcc fusion protein constructs with the A145D mutation in the IFN portion.
- the experiment compares different antibody variable regions in the context of the same mutated IFN fusion protein construct.
- the assay is described in the examples as the "off-target assay.”
- Figure 37 shows the antiproliferative activity on the multiple myeloma cell line ARP-1 of IFNcc2b vs anti-CD38-IFNcc fusion protein constructs with the A145D mutation in the IFN portion.
- the experiment compares different antibody variable regions in the context of the same mutated IFN fusion protein construct.
- the assay is described in the examples as the "on target (ARP) assay.”
- Figure 38 shows the non-antibody-antigen targeted IFN activity of IFNcc2b and various anti-CD38-IFNcc fusion protein constructs with the A145D mutation in the IFN portion.
- the experiment compares different antibody variable regions in the context of the same mutated IFN fusion protein construct.
- the assay is described in the examples as the "off-target assay.”
- Figure 39 shows the antiproliferative activity on the multiple myeloma cell line ARP-1 of two antibody- IFN fusion protein constructs with the A145D mutation in the IFN portion.
- the nBT062 antibody binds CD138 whereas the "isotype” antibody does not (it is derived from the antibody 2D 12).
- the assay is described in the examples as the "on target (ARP) assay.”
- Figure 40 shows the antiproliferative activity on the multiple myeloma cell line ARP-1 of IFNcc2b and two antibody- IFN fusion protein constructs with the A145D mutation in the IFN portion.
- the HB95 antibody binds human class I MHC (which is expressed on the ARP-1 cells) whereas the "isotype" antibody does not (it is derived from the antibody 2D12).
- Palivizumab like 2D12, does not bind to the ARP-1 cells
- (b) shows the same assay, comparing antibody-attenuated IFNa fusion protein constructs in which the antibody portion is a Fab fragment rather than a full size antibody.
- the assay is described in the examples as the "on target (ARP) assay.”
- Figure 41 shows measurements of the antiviral activity of IFNa and two antibody-IFNa fusion protein constructs with the A145D mutation in the IFN portion.
- This cytopathic effect inhibition assay utilized the cell line A549 and the EMC virus.
- the HB95 antibody binds human class I MHC (which is expressed on the A549 cells) whereas the "isotype" antibody derived from the antibody 2D 12 does not.
- Figure 42 shows the non-antibody-antigen targeted IFN activity of ⁇ , an anti- CD38-IFN fusion protein construct and an identical fusion protein construct but with the attenuating R35A mutation in the IFN portion.
- the assay is described in the examples as the "off-target assay.”
- Figure 43 shows the antiproliferative activity on the multiple myeloma cell line ARP-1 of ⁇ ⁇ , an anti-CD38-IFN fusion protein construct and an identical fusion protein construct but with the attenuating R35A mutation in the IFN portion.
- the antibody variable regions of these fusion protein constructs are derived from the antibody G005.
- the assay is described in the examples as the "on target (ARP) assay.”
- “Ifn equivalents” refers to the molar concentration of interferon molecules, either free or attached to an antibody.
- Figure 44 shows the non-antibody-antigen-targeted IL-4 activity ["off-target (HB-IL4) assay”] of IL-4 and three antibody- IL-4 fusion protein constructs: Jl 10-HC-L6- IL-4 IgGl, an anti-PDl antibody fused to wild type IL-4; Jl 10-HC-L6-IL-4 (R88Q), which is identical to the previously mentioned fusion protein construct except for the attenuating R88Q mutation in the IL-4 portion; and Isotype-HC-L6-IL-4 (R88Q), based on the 2D 12 antibody, which does not bind to any of the cells used in the assays of the present invention, and is fused to the attenuated IL-4.
- Jl 10-HC-L6- IL-4 IgGl an anti-PDl antibody fused to wild type IL-4
- Jl 10-HC-L6-IL-4 (R88Q) which is identical to the previously mentioned fusion protein construct except for the attenuating R
- IL-4 equivalents refers to the molar concentration of IL-4 molecules, either free or attached to an antibody.
- Figure 45 shows the "on target (Thl diversion) assay” comparing the activity of IL-4 and three antibody- IL-4 fusion protein constructs: Jl 10-HC-L6-IL-4 IgGl, an anti- PD1 antibody fused to wild type IL-4; J110-HC-L6-IL-4 (R88Q), which is identical to the previously mentioned fusion protein construct except for the attenuating R88Q mutation in the IL-4 portion; and Isotype-HC-L6-IL-4 (R88Q), based on the 2D 12 antibody, which does not bind to any of the cells used in the assays of the present invention, and is fused to the attenuated IL-4.
- IL-4 equivalents refers to the molar concentration of IL-4 molecules, either free or attached to an antibody.
- Figure 46 shows the "IL-6 bioassay” comparing IL-6 with various antibody-IL-6 fusion protein constructs that either do bind to the target cells (based on the HB95 antibody, which binds to class I MHC on the target cells) or do not bind the target cells (based on the isotype control antibody 2D 12), fused to either wild type IL-6 or IL-6 with the attenuating R179E mutation.
- "IL-6 equivalents” refers to the molar concentration of IL-6 molecules, either free or attached to an antibody.
- Figure 47 shows the effects of various compounds on the growth of subcutaneous H929 myeloma tumors in SCID mice. The bar labeled "treatment” shows the duration of treatment with the compounds. The "isotype” antibody was based on antibody 2D 12.
- G005 is an anti-CD38 antibody.
- Figure 48 shows the effects of various compounds on survival (Kaplan-Meier graph) of NOD-SCID mice systemically inoculated with the human myeloma cell line
- MM1S MM1S.
- treatment shows the duration of treatment with the compounds.
- G005 is an anti-CD38 antibody.
- Figure 49 shows the effects of various compounds on the growth of subcutaneous Daudi lymphoma tumors in NOD-SCID mice.
- the bar labeled “treatment” shows the duration of treatment with the compounds.
- the "isotype” antibody was based on antibody 2D 12.
- G005 is an anti-CD38 antibody.
- Figure 50 shows the effects of an anti-CD38-attenuated IFNcc fusion protein construct (G005-HC-L6-IFNCC (A145G) IgGl) and an isotype control-attenuated IFNcc fusion protein construct (Isotype-HC-L6-IFNcc (A145G) IgGl) on the growth of subcutaneous H929 myeloma tumors in SCID mice, at various doses.
- the bar labeled "treatment” shows the duration of treatment with the compounds.
- the "isotype” antibody was based on antibody 2D 12.
- Figure 51 shows the effects of anti-CD38-attenuated IFNcc fusion protein constructs vs isotype control antibody-attenuated IFNcc fusion protein constructs, on the growth of subcutaneous H929 myeloma tumors in SCID mice. IgGl is compared to IgG4 in the context of these fusion protein constructs. The bar labeled "treatment” shows the duration of treatment with the compounds. The "isotype” antibody was based on antibody 2D 12.
- Figure 52 shows the effects of an anti-CD38-attenuated IFNcc fusion protein construct (X355/02-HC-L0-IFNCC (A145D) IgG4) vs an isotype control antibody- attenuated IFNcc fusion protein constructs on the growth of subcutaneous H929 myeloma tumors in SCID mice.
- the bar labeled "treatment phase” shows the duration of treatment with the compounds.
- the "isotype” antibody was based on antibody 2D12.
- Figure 53 shows the effects of various compounds on the growth of subcutaneous H929 myeloma tumors in SCID mice.
- G005 is an anti-CD38 antibody.
- Figure 54 shows the effects of an anti-CD38-attenuated IFNcc fusion protein construct (G005-HC-L6-IFNCC (A145G) IgG4) and an isotype control-attenuated IFNcc fusion protein construct (Isotype-HC-L6-IFNcc (A145G) IgG4) on the growth of subcutaneous H929 myeloma tumors in SCID mice, with several rounds of administration each at a dose of 10 mg/kg.
- the "isotype" antibody was based on antibody 2D12.
- Figure 55 shows the effects of an anti-CD38-attenuated IFNcc fusion protein construct (G005-HC-L6-IFNCC (A145G) IgG4) on the growth of subcutaneous H929 myeloma tumors in SCID mice. Dosing (indicated by arrows) was initiated when the median tumor volume reached 730 mm .
- Figure 56 shows the inhibition of colony formation from normal human bone marrow mononuclear cells (BM MNC) by IFNa2b, an anti-CD38-attenuated IFNcc fusion protein construct (G005-HC-L0-IFNCC (A145D) IgG4) and an isotype control antibody- attenuated IFNcc fusion protein construct 2D12-HC-L0-IFNCC (A145D) IgG4.
- the antibody- attenuated IFNcc fusion protein constructs show about 10,000-fold reduced potency in this assay.
- Figure 57 shows the effects of IFNcc2b vs an antibody-attenuated IFNcc fusion protein construct (Isotype-HC-L6-IFNcc (A145G) IgGl; the isotype variable regions are based on antibody 2D 12) on cytokine production by human peripheral blood mononuclear cells (PBMCs).
- PBMCs peripheral blood mononuclear cells
- the constructs of the present invention are antibody-attenuated ligand constructs, which show an elevated antigen-specificity index with respect to activating signaling pathways due to the action of the attenuated ligand on a cell surface receptor.
- These constructs are based on the surprising discovery that, in the context of an antibody-ligand construct, the ligand portion can be mutated in such a way that the ligand activity on antigen-negative cells is dramatically attenuated, while the ligand activity on antigen- positive cells is only modestly, if at all, attenuated.
- Such constructs display one, two, three, four or five orders of magnitude greater potency on antigen-positive cells compared to antigen negative cells than does the free ligand.
- the antibody- attenuated ligand construct retains at least 1%, at least 10%, at least 20%, at least 30%, at least 40% or at least 50% of the potency on antigen-positive cells as the non- attenuated free (i.e. not attached to an antibody) ligand. In addition, in one embodiment the antibody- attenuated ligand construct retains at least 30%, at least 50%, at least 75% or at least 90% of the maximal activity of the non- attenuated free (i.e.
- maximal activity should be understood as meaning the amount of signaling activity (or downstream effect thereof) at the high, plateau portion of a dose- response curve, where further increases in the agent does not further increase the amount of response).
- Specificity is not necessarily an absolute designation but often a relative term signifying the degree of selectivity of an antibody-ligand fusion protein construct for an antigen-positive cell compared to an antigen-negative cell.
- a construct may be said to have "100-fold specificity for antigen-positive cells compared to antigen-negative cells" and this would indicate that the construct has 100-fold higher potency on cells that express the antigen compared to cells that do not.
- this degree of specificity of a construct comparing antigen-positive vs. antigen- negative cells may not be based on the absolute ratio of potency of the construct on antigen-positive vs.
- Assays for determining potency of antibody-ligand fusion constructs are exemplified in the examples and include cell based assays for proliferation, apoptosis, phosphorylation of receptors and intracellular proteins, migration, differentiation (for example, differentiation of naive CD4+ T cells into Thl, Thl7, Th2 vs. Treg cells), increases or decreases in gene expression or gene product secretion into the media, etc.
- the present invention provides a polypeptide construct comprising a peptide or polypeptide signaling ligand linked to an antibody or antigen binding portion thereof which binds to a cell surface-associated antigen wherein the ligand comprises at least one amino acid substitution or deletion which reduces its potency on cells lacking expression of said antigen.
- the present invention provides a polypeptide construct comprising IFN linked to an antibody or antigen binding portion thereof which binds to a tumour associated antigen wherein the IFN comprises at least one amino acid substitution or deletion which reduces its potency on cells lacking expression of said antigen.
- a polypeptide will be capable of exerting with high potency the IFN's anti-proliferative activity on the antigen-positive tumor cells while exerting a much lower potency on the antigen-negative, non-tumour cells within the body.
- the present invention provides a method of treating a tumour in a subject comprising administering to the subject the polypeptide construct of the present invention.
- antibody-ligand construct refers to an antibody or antigen-binding fragment thereof covalently attached to a signaling ligand that has been attenuated by one or more substitutions or deletions that reduce the ligand's potency on cells that do not express the antigen corresponding to the antibody.
- antibody broadly refers to any immunoglobulin (Ig) molecule comprised of four polypeptide chains, two heavy (H) chains and two light (L) chains, or any functional fragment, mutant, variant, or derivation thereof, which retains the essential epitope binding features of an Ig molecule.
- Ig immunoglobulin
- Such mutant, variant, or derivative antibody formats are known in the art, non-limiting embodiments of which are discussed below.
- each heavy chain is comprised of a heavy chain variable region (abbreviated herein as HCVR or VH) and a heavy chain constant region.
- the heavy chain constant region is comprised of three domains, CHI, CH2 and CH3.
- Each light chain is comprised of a light chain variable region (abbreviated herein as LCVR or VL) and a light chain constant region.
- the light chain constant region is comprised of one domain, CL.
- the VH and VL regions can be further subdivided into regions of hypervariability, termed complementarity determining regions (CDR), interspersed with regions that are more conserved, termed framework regions (FR).
- CDR complementarity determining regions
- FR framework regions
- Each VH and VL is composed of three CDRs and four FRs, arranged from amino-terminus to carboxy- terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4.
- Immunoglobulin molecules can be of any type (e.g., IgG, IgE, IgM, IgD, IgA and IgY), class (e.g., IgGl, IgG2, IgG3, IgG4, IgAl and IgA2) or subclass.
- antigen binding domain or "antigen binding portion” of an antibody, as used herein, refers to one or more fragments of an antibody or protein that retain the ability to specifically bind to an antigen (e.g., CD38). It has been shown that the antigen- binding function of an antibody can be performed by fragments of a full-length antibody. Such antibody embodiments may also be bispecific, dual specific, or multi- specific formats, specifically binding to two or more different antigens.
- binding fragments encompassed within the term "antigen-binding portion" of an antibody include (i) a Fab fragment, a monovalent fragment consisting of the VL, VH, CL and CHI domains; (ii) a F(ab')2 fragment, a bivalent fragment comprising two Fab fragments in addition to a portion of the hinge region, linked by a disulfide bridge at the hinge region; (iii) an Fd fragment consisting of the VH and CHI domains; (iv) an Fv fragment consisting of the VL and VH domains of a single arm of an antibody , (v) a domain antibody (dAb) (Ward et al.
- VL and VH are coded for by separate genes, they can be joined, using recombinant methods, by a synthetic linker that enables them to be made as a single protein chain in which the VL and VH regions pair to form monovalent molecules (known as single chain Fv (scFv); see e.g., Bird et al. 1988 Science 242 423-6; Huston et al. 1988 Proc Natl Acad Sci U S A 85 5879-83).
- scFv single chain Fv
- single chain antibodies are also intended to be encompassed within the term "antigen-binding portion" of an antibody.
- Other forms of single chain antibodies, such as diabodies, are also encompassed.
- Diabodies are bivalent, bispecific antibodies in which VH and VL domains are expressed on a single polypeptide chain, but using a linker that is too short to allow for pairing between the two domains on the same chain, thereby forcing the domains to pair with complementary domains of another chain and creating two antigen binding sites (see e.g., Holliger, P., et al, 1993, Proc. Natl. Acad. Sci. USA 90:6444-6448; Poljak, R. J., et al, 1994, Structure 2:1121-1123).
- Such antibody binding portions are known in the art (Kontermann and Dubel eds., Antibody Engineering 2001 Springer- Verlag. New York. 790 pp., ISBN 3-540- 41354-5).
- the antibody binding portion is a Fab fragment
- the antibody described herein may be may be a humanized antibody.
- the term "humanized antibody” shall be understood to refer to a protein comprising a human-like variable region, which includes CDRs from an antibody from a non-human species (e.g., mouse or rat or non-human primate) grafted onto or inserted into FRs from a human antibody (this type of antibody is also referred to a "CDR-grafted antibody”).
- Humanized antibodies also include proteins in which one or more residues of the human protein are modified by one or more amino acid substitutions and/or one or more FR residues of the human protein are replaced by corresponding non-human residues.
- Humanized antibodies may also comprise residues which are found in neither the human antibody or in the non- human antibody. Any additional regions of the protein (e.g., Fc region) are generally human. Humanization can be performed using a method known in the art, e.g.,
- humanized antibody also encompasses a super-humanized antibody, e.g., as described in US7,732,578.
- the antibody described herein may be human.
- the term "human antibody” as used herein refers to proteins having variable and, optionally, constant antibody regions found in humans, e.g. in the human germline or somatic cells or from libraries produced using such regions.
- the "human” antibodies can include amino acid residues not encoded by human sequences, e.g. mutations introduced by random or site directed mutations in vitro (in particular mutations which involve conservative substitutions or mutations in a small number of residues of the protein, e.g.
- human antibodies do not necessarily need to be generated as a result of an immune response of a human, rather, they can be generated using recombinant means (e.g., screening a phage display library) and/or by a transgenic animal (e.g., a mouse) comprising nucleic acid encoding human antibody constant and/or variable regions and/or using guided selection (e.g., as described in or US5,565,332). This term also encompasses affinity matured forms of such antibodies.
- a human protein will also be considered to include a protein comprising FRs from a human antibody or FRs comprising sequences from a consensus sequence of human FRs and in which one or more of the CDRs are random or semi-random, e.g., as described in
- the antibody portions of polypeptides of the present invention may be full length antibodies of any class, preferably IgGl, IgG2 or IgG4.
- the constant domains of such antibodies are preferably human.
- the variable regions of such antibodies may be of non- human origin or, preferably, be of human origin or be humanized.
- Antibody fragments may also be used in place of the full length antibodies.
- antibody also includes engineered antibodies. As will be appreciated there are many variations of engineered antibodies (e.g. mouse monoclonal, chimeric, humanized and human monoclonal antibodies, single chain variable antibody fragments (scFv's), minibodies, aptamers, as well as bispecific antibodies and diabodies as described above).
- engineered antibodies e.g. mouse monoclonal, chimeric, humanized and human monoclonal antibodies, single chain variable antibody fragments (scFv's), minibodies, aptamers, as well as bispecific antibodies and diabodies as described above).
- Single variable region domains are, for example, disclosed in (Ward et al., Nature 341: 544-546, 1989; Hamers-Casterman et al., Nature 363: 446-448, 1993; Davies & Riechmann, FEBS Lett. 339: 285-290, 1994).
- Minibodies are small versions of whole antibodies, which encode in a single chain the essential elements of a whole antibody.
- the minibody is comprised of the VH and VL domains of a native antibody fused to the hinge region and CH3 domain of the immunoglobulin molecule as, for example, disclosed in U.S. Patent No 5,837,821.
- the engineered antibody may comprise non- immunoglobulin derived, protein frameworks.
- non-antibody recognition protein or protein domain scaffolds that may be utilised as the antigen binding domains in the constructs of this invention.
- CTL-4 cytotoxic T lymphocyte-associated antigen 4
- human transferrin Trans-body
- a three-helix bundle from the Z-domain of Protein A (Affibody); a monomeric or trimeric human C-type lectin domain (Tetranectin); the tenth human fibronectin type III domain (AdNectin); the Kunitz- type domain of human or bovine trypsin inhibitor; insect Defensin A (IICA29), APPI (Kuntiz domains); lipocalins, FABP, Bilin-binding protein, Apoloproptein D (Anticalins); human a-crystallin or ubiquitin molecule (Affilin); trypsin inhibitor II (Microbody); a2p8 or Ankyrin repeat (repeat-motif proteins), Charybdotoxin (Scorpion toxins), Min-23, Cellulose binding
- CTL-4 cytotoxic T lymphocyte-associated antigen 4
- ligand binding proteins or protein domains that may be utilised as the ligand binding domains in this invention.
- protein domains that possess ligand binding properties include extracellular domains of receptors, PDZ modules of signalling proteins, such as Ras-binding protein AF-6, adhesion molecules, and enzymes.
- the present invention further encompasses chemical analogues of amino acids in the subject antibodies.
- chemical analogues of amino acids is useful inter alia to stabilize the molecules such as if required to be administered to a subject.
- the analogues of the amino acids contemplated herein include, but are not limited to, modifications of side chains, incorporation of unnatural amino acids and/or their derivatives during peptide, polypeptide or protein synthesis and the use of crosslinkers and other methods which impose conformational constraints on the proteinaceous molecule or their analogues.
- side chain modifications contemplated by the present invention include modifications of amino groups such as by reductive alkylation by reaction with an aldehyde followed by reduction with NaBH 4 ; amidination with methylacetimidate;
- guanidine group of arginine residues may be modified by the formation of heterocyclic condensation products with reagents such as 2,3-butanedione, phenylglyoxal and glyoxal.
- the carboxyl group may be modified by carbodiimide activation via O- acylisourea formation followed by subsequent derivatisation, for example, to a
- Sulphydryl groups may be modified by methods such as carboxymethylation with iodoacetic acid or iodoacetamide; performic acid oxidation to cysteic acid; formation of a mixed disulphides with other thiol compounds; reaction with maleimide, maleic anhydride or other substituted maleimide; formation of mercurial derivatives using 4- chloromercuribenzoate, 4-chloromercuriphenylsulphonic acid, phenylmercury chloride, 2- chloromercuri-4-nitrophenol and other mercurials; carbamoylation with cyanate at alkaline pH.
- Tryptophan residues may be modified by, for example, oxidation with N- bromosuccinimide or alkylation of the indole ring with 2-hydroxy-5-nitrobenzyl bromide or sulphenyl halides.
- Tyrosine residues on the other hand, may be altered by nitration with tetranitromethane to form a 3-nitrotyrosine derivative.
- Modification of the imidazole ring of a histidine residue may be accomplished by alkylation with iodoacetic acid derivatives or N-carbethoxylation with
- Examples of incorporating unnatural amino acids and derivatives during peptide synthesis include, but are not limited to, use of norleucine, 4-amino butyric acid, 4-amino- 3-hydroxy-5-phenylpentanoic acid, 6-aminohexanoic acid, t-butylglycine, norvaline, phenylglycine, ornithine, sarcosine, 4-amino-3-hydroxy-6-methylheptanoic acid, 2-thienyl alanine and/or D-isomers of amino acids.
- a list of unnatural amino acid, contemplated herein is shown in Table 1.
- Non-conventional Code Non-conventional Code amino acid amino acid a-aminobutyric acid Abu L-N-methylalanine Nmala a-amino-a-methylbutyrate Mgabu L-N-methylarginine Nmarg aminocyclopropane- Cpro L-N-methylasparagine Nmasn carboxylate L-N-methylaspartic acid Nmasp aminoisobutyric acid Aib L-N-methylcysteine Nmcys aminonorbornyl- Norb L-N-methylglutamine Nmgln carboxylate L-N-methylglutamic acid Nmglu cyclohexylalanine Chexa L-Nmethylhistidine Nmhis cyclopentylalanine Cpen L-N-methylisolleucine Nmile
- ADCC antibody-dependent cell-mediated cytotoxicity
- CDC complement-dependent cytotoxicity
- serum half-life biodistribution
- biodistribution binding to Fc receptors or the combination of any of these.
- This modulation can be achieved by protein-engineering, glyco-engineering or chemical methods. Depending on the therapeutic application required, it could be advantageous to either increase or decrease any of these activities.
- Mutagenesis is often performed at the DNA level, for example by error prone PCR (Thie, Voedisch et al. 2009), by gene shuffling (Kolkman and Stemmer 2001), by use of mutagenic chemicals or irradiation, by use of 'mutator' strains with error prone replication machinery (Greener 1996) or by somatic hypermutation approaches that harness natural affinity maturation machinery (Peled, Kuang et al. 2008). Mutagenesis can also be performed at the RNA level, for example by use of QP replicase (Kopsidas, Roberts et al. 2006).
- Library-based methods allowing screening for improved variant proteins can be based on various display technologies such as phage, yeast, ribosome, bacterial or mammalian cells, and are well known in the art (Benhar 2007).
- Affinity maturation can be achieved by more directed/predictive methods for example by site-directed mutagenesis or gene synthesis guided by findings from 3D protein modeling (see for example Queen, Schneider et al. 1989 or US patent 6,180,370 or US patent 5,225,539).
- Methods of increasing ADCC have been described by Ferrara, Brunker et al.
- a number of methods for modulating antibody serum half-life and biodistribution are based on modifying the interaction between antibody and the neonatal Fc receptor
- FcRn a receptor with a key role in protecting IgG from catabolism, and maintaining high serum antibody concentration.
- Dall'Acqua et al describe substitutions in the Fc region of IgGl that enhance binding affinity to FcRn, thereby increasing serum half-life
- glycoforms are known in the art and include but are not limited to those described in U.S. Pat. Nos 6,602,684; 7,326,681; 7,388,081 and WO 2008/006554.
- the polypeptide constructs or compositions of the present invention may be used to treat patients with cancer.
- Cancers contemplated herein include: a group of diseases and disorders that are characterized by uncontrolled cellular growth (e.g. formation of tumor) without any differentiation of those cells into specialized and different cells.
- Such diseases and disorders include ABL1 protooncogene, AIDS related cancers, acoustic neuroma, acute lymphocytic leukaemia, acute myeloid leukaemia, adenocystic carcinoma, adrenocortical cancer, agnogenic myeloid metaplasia, alopecia, alveolar soft-part sarcoma, anal cancer, angiosarcoma, aplastic anaemia, astrocytoma, ataxia-telangiectasia, basal cell carcinoma (skin), bladder cancer, bone cancers, bowel cancer, brain stem glioma, brain and CNS tumors, breast cancer, CNS tumors, carcinoid tumors, cervical cancer, childhood brain tumors, childhood cancer, childhood leukaemia, childhood soft tissue sarcoma, chondrosarcoma,
- choriocarcinoma chronic lymphocytic leukaemia, chronic myeloid leukaemia, colorectal cancers, cutaneous T-Cell lymphoma, dermatofibrosarcoma-protuberans, desmoplastic- small-round-cell-tumor, ductal carcinoma, endocrine cancers, endometrial cancer, ependymoma, esophageal cancer, Ewing's sarcoma, extra-hepatic bile duct cancer, eye cancer, eye: melanoma, retinoblastoma, fallopian tube cancer, fanconi anemia,
- fibrosarcoma gall bladder cancer, gastric cancer, gastrointestinal cancers, gastrointestinal- carcinoid- tumor, genitourinary cancers, germ cell tumors, gestational-trophoblastic - disease, glioma, gynaecological cancers, hematological malignancies, hairy cell leukaemia, head and neck cancer, hepatocellular cancer, hereditary breast cancer, histiocytosis, Hodgkin's disease, human papillomavirus, hydatidiform mole, hypercalcemia,
- hypopharynx cancer intraocular melanoma, islet cell cancer, Kaposi's sarcoma, kidney cancer, Langerhan's-cell-histiocytosis, laryngeal cancer, leiomyosarcoma, leukemia, Li- Fraumeni syndrome, lip cancer, liposarcoma, liver cancer, lung cancer, lymphedema, lymphoma, Hodgkin's lymphoma, non-Hodgkin's lymphoma, male breast cancer, malignant-rhabdoid-tumor-of-kidney, medulloblastoma, melanoma, merkel cell cancer, mesothelioma, metastatic cancer, mouth cancer, multiple endocrine neoplasia, mycosis fungoides, myelodysplasia syndromes, multiple myeloma, myeloproliferative disorders, nasal cancer, nasopharyngeal cancer, nephroblasto
- the tumor is selected from a group of multiple myeloma or non-hodgkin's lymphoma.
- the antibody portions of the constructs of the present invention may bind to tumour- associated antigens, i.e., cell surface antigens that are selectively expressed by cancer cells or over-expressed in cancer cells relative to most normal cells.
- tumour- associated antigens i.e., cell surface antigens that are selectively expressed by cancer cells or over-expressed in cancer cells relative to most normal cells.
- TAAs tumour- associated antigens
- Non-limiting examples of TAAs include enzyme tyrosinase; melanoma antigen GM2; alphafetoprotein (AFP); carcinoembryonic antigen (CEA); Mucin 1
- MUC1 Human epidermal growth factor receptor (Her2/Neu); T-cell
- TAAs associated with a number of different cancers are telomerase (hTERT); prostate-specific membrane antigen
- PSMA urokinase plasminogen activator and its receptor
- VEGF/VEGFR vascular endothelial growth factor and its receptor
- EMMPRIN/CD147 extracellular matrix metalloproteinase inducer
- EGFR epidermal growth factor
- PDGF/PDGFR platelet- derived growth factor and its receptor
- CD117 c-kit
- an antigen for antibody-attenuated ligand constructs for example, an antibody-attenuated interferon construct
- CD38 is a 46kDa type II transmembrane glycoprotein. It has a short N-terminal cytoplasmic tail of 20 amino acids, a single transmembrane helix and a long extracellular domain of 256 amino acids (Bergsagel, P., Blood; 85:436, 1995 and Liu, Q., Structure, 13: 1331, 2005).
- T cells CD4 and CD8 positive T cells, B cells, NK cells, monocytes, plasma cells and on a significant proportion of normal bone marrow precursor cells (Malavasi, F., Hum. Immunol. 9:9, 1984).
- lymphocytes the expression appears to be dependent on the differentiation and activation state of the cell. Resting T and B cells are negative while immature and activated lymphocytes are predominantly positive for CD38 expression (Funaro, A., J. Immunol. 145:2390, 1990). Additional studies indicate mRNA expression in non-hemopoeitic organs such as pancreas, brain, spleen and liver (Koguma, T.,
- CD38 is a multifunctional ectoenzyme that is involved in transmembrane signaling and cell adhesion. It is also known as cyclic ADP ribose hydrolase because it can transform NAD + and NADP + into cADPR, ADPR and NAADP, depending on extracellular pH. These products induce Ca 2+ -mobilization inside the cell which can lead to tyrosine phosphorylation and activation of the cell.
- CD38 is also a receptor that can interact with a ligand, CD31. Activation of receptor via CD31 leads to intracellular events including Ca 2+ mobilization, cell activation, proliferation, differentiation and migration (reviewed in Deaglio, S., Trends in Mol. Med. 14:210, 2008.)
- CD38 is expressed at high levels on multiple myeloma cells, in most cases of T- and B-lineage acute lymphoblastic leukemias, some acute myelocytic leukemias, follicular center cell lymphomas and T lymphoblastic lymphomas. (Malavasi, F., J. Clin Lab Res. 22:73, 1992). More recently, CD38 expression has become a reliable prognostic marker in B-lineage chronic lymphoblastic leukemia (B-CLL) (Ibrahim, S., Blood. 98: 181, 2001 and Durig, J., Leuk. Res. 25:927, 2002).
- B-CLL B-lineage chronic lymphoblastic leukemia
- Preferred antigens for the development of antibody-attenuated ligand fusion protein constructs which target cancer are antigens which show selective or greater expression on the cancer cells than on most other, non-transformed cells within the body.
- Non-protein examples of such antigens include, sphingolipids, ganglioside GD2 (Saleh et al, 1993, J. Immunol., 151, 3390-3398), ganglioside GD3 (Shitara et al, 1993, Cancer Immunol. Immunother. 36:373-380), ganglioside GM2 (Livingston et al, 1994, J. Clin. Oncol.
- ganglioside GM3 Hoon et al, 1993, Cancer Res. 53:5244-5250
- Lewis x Lewis x
- lewis y lewis xy carbohydrate antigens that can be displayed on proteins or glycolipids.
- protein antigens are HER-2/neu, human papillomavirus-E6 or - E7, MUC-1; KS 1/4 pan-carcinoma antigen (Perez and Walker, 1990, J. Immunol.
- ovarian carcinoma antigen CA125 (Yu et al, 1991, Cancer Res. 51(2):468-475); prostatic acid phosphate (Tailor et al, 1990, Nucl. Acids Res. 18(16):4928); prostate specific antigen (Henttu and Vihko, 1989, Biochem. Biophys. Res. Comm. 160(2):903-910; Israeli et al, 1993, Cancer Res. 53:227-230); melanoma- associated antigen p97 (Estin et al, 1989, J. Natl. Cancer Instit. 81(6):445-446); melanoma antigen gp75 (Vijayasardahl et al, 1990, J. Exp. Med.
- differentiation antigens such as human lung carcinoma antigen L6 or L20 (Hellstrom et al, 1986, Cancer Res. 46:3917-3923); antigens of fibrosarcoma; human leukemia T cell antigen-Gp37 (Bhattacharya-Chatterjee et al, 1988, J. Immunol.
- tumor-specific transplantation type of cell- surface antigen such as virally- induced tumor antigens including T-antigen, DNA tumor virus and envelope antigens of RNA tumor viruses; neoglycoproteins, breast cancer antigens such as EGFR (Epidermal growth factor receptor), polymorphic epithelial mucin (PEM) (Hilkens et al, 1992, Trends in Bio. Chem. Sci. 17:359); polymorphic epithelial mucin antigen; human milk fat globule antigen;
- TSTA tumor- specific transplantation type of cell- surface antigen
- TAG-72 Yokata et al, 1992, Cancer Res. 52:3402-3408
- CO 17-1 A CO 17-1 A
- differentiation antigens such as I(Ma) found in gastric adenocarcinomas, SSEA-1 found in myeloid cells, VEP8, VEP9, Myl, VIM-D5, Ml 8 and M39 found in breast epithelial cancers, D 156 - 22 found in colorectal cancer, TRA-1-85 (blood group H), C14 found in colonic adenocarcinoma, F3 found in lung adenocarcinoma, AH6 found in gastric cancer, Y hapten found in embryonal carcinoma cells, TL5 (blood group A), El series (blood group B) antigens found in pancreatic cancer, FC10.2 found in embryonal carcinoma cells, gastric adenocarcinoma antigen, CO-514 (blood group Le a ) found in adenocarcinoma, NS-10 found in adenocarcinomas, CO-43 (blood group Le b ), G49 found in A4
- HMW-MAA (SEQ ID NO:433), also known as melanoma chondroitin sulfate proteoglycan, is a membrane-bound protein of 2322 residues which is overexpressed on over 90% of the surgically removed benign nevi and melanoma lesions (Camploi, et. al, Crit Rev Immunol. ;24:267,2004). Accordingly it may be a potential target cell surface associated antigen.
- cancer antigens for targeting with fusion protein constructs of the present invention include (exemplary cancers are shown in parentheses): CD5 (T-cell leukemia/lymphoma), CA15-3 (carcinomas), CA19-9 (carcinomas), L6 (carcinomas), CA 242 (colorectal), placental alkaline phosphatase (carcinomas), prostatic acid phosphatase (prostate), MAGE-1 (carcinomas), MAGE-2 (carcinomas), MAGE-3 (carcinomas), MAGE -4 (carcinomas), transferrin receptor (carcinomas), p97 (melanoma), MUC1 (breast cancer), MARTI (melanoma), CD20 (non Hodgkin's lymphoma), CD52 (leukemia), CD33 (leukemia), human chorionic gonadotropin (carcinoma), CD38 (multiple myeloma), CD21 (B-cell lymphoma), CD22 (lymphoma),
- Some specific, useful antibodies include, but are not limited to, BR64 (Trail et al, 1997, Cancer Research 57: 100 105), BR96 mAb (Trail et al, 1993, Science 261:212-215), mAbs against the CD40 antigen, such as S2C6 mAb (Francisco et al., 2000, Cancer Res. 60:3225-3231) or other anti-CD40 antibodies, such as those disclosed in U.S Patent Publication Nos. 2003-0211100 and 2002-0142358; mAbs against the CD30 antigen, such as AC10 (Bowen et al., 1993, J. Immunol.
- useful antibodies can bind to a receptor or a complex of receptors expressed on a target cell.
- the receptor or receptor complex can comprise an immunoglobulin gene superfamily member, a major histocompatibility protein, a cytokine receptor, a TNF receptor superfamily member, a chemokine receptor, an integrin, a lectin, a complement control protein, a growth factor receptor, a hormone receptor or a neurotransmitter receptor.
- suitable immunoglobulin superfamily members are CD2, CD3, CD4, CD8, CD19, CD22, CD79, CD90, CD152/CTLA-4, PD-1, B7-H4, B7-H3, and ICOS.
- Non-limiting examples of suitable TNF receptor superfamily members are TACI, BCMA, CD27, CD40, CD95/Fas, CD134/0X40, CD137/4-1BB, TNFR1, TNFR2, RANK, osteoprotegerin, APO 3, Apo2/TRAIL Rl, TRAIL R2, TRAIL R3, and TRAIL R4.
- Non-limiting examples of suitable integrins are CD 11 a, CD l ib, CDl lc, CD18, CD29, CD41, CD49a, CD49b, CD49c, CD49d, CD49e, CD49f, CD103 and CD 104.
- Non-limiting examples of suitable lectins are S type, C type, and I type lectin. Examples of antibodies to CEA are shown in Table 2.
- Antibodies that bind the CD22 antigen expressed on human B cells include, for example, HD6, RFB4, UV22-2, Tol5, 4KB128 and a humanized anti-CD22 antibody (hLL2) (see, e.g., Li et al. (1989) Cell. Immunol. I l l: 85-99; Mason et al. (1987) Blood 69: 836-40; Behr et al. (1999) Clin. Cancer Res. 5: 3304s-3314s; Bonardi et al. (1993) Cancer Res. 53: 3015-3021).
- Antibodies to CD33 include, for example, HuM195 (see, e.g., Kossman et al. (1999) Clin. Cancer Res. 5: 2748-2755; US5693761) and CMA-676 (see, e.g., Sievers et al, (1999) Blood 93: 3678-3684).
- Illustrative anti-MUC-1 antibodies include, but are not limited to Mc5 (see, e.g., Peterson et al. (1997) Cancer Res. 57: 1103-1108; Ozzello et al. (1993) Breast Cancer Res. Treat. 25: 265-276), and hCTMOl (see, e.g., Van Hof et al. (1996) Cancer Res. 56: 5179- 5185).
- Illustrative anti-TAG-72 antibodies include, but are not limited to CC49 (see, e.g., Pavlinkova et al. (1999) Clin. Cancer Res. 5: 2613-2619), B72.3 (see, e.g., Divgi et al. (1994) Nucl. Med. Biol. 21: 9-15), and those disclosed in U.S. Pat. No. 5,976,531.
- Illustrative anti-HM1.24 antibodies include, but are not limited to a mouse monoclonal anti-HM1.24 and a humanized anti-HM1.24 IgGlkappa antibody (see, e.g., Ono et al. (1999) Mol. Immuno. 36: 387-395).
- the targeting moiety comprises an anti-HER2 antibody.
- the erBB 2 gene more commonly known as (Her-2/neu), is an oncogene encoding a transmembrane receptor.
- trastuzumab e.g., HERCEPTINTM; Fornier et al. (1999) Oncology (Huntingt) 13: 647-58
- TAB-250 Rosenblum et al. (1999) Clin. Cancer Res. 5: 865-874
- BACH- 250 Id.
- TAl Maier et al. (1991) Cancer Res. 51: 5361-5369
- the mAbs described in U.S. Pat. Nos. 5,772,997; 5,770,195 mAb 4D5; ATCC CRL 10463
- trastuzumab e.g., HERCEPTINTM., Fornier et al. (1999) Oncology (Huntingt) 13: 647-658
- TAB-250 Rosenblum et al.
- C6 antibodies such as C6.5, DPL5, G98A, C6MH3-B 1, B1D2, C6VLB, C6VLD, C6VLE, C6VLF, C6MH3-D7, C6MH3-D6, C6MH3-D5, C6MH3-D3, C6MH3-D2, C6MH3-D1, C6MH3-C4, C6MH3- C3, C6MH3-B9, C6MH3-B5, C6MH3-B48, C6MH3-B47, C6MH3-B46, C6MH3-B43, C6MH3-B41, C6MH3-B39, C6MH3-B34, C6MH3-B33, C6MH3-B31, C6MH3-B27, C6MH3-B25, C6MH3-B21, C6MH3-B20
- antibodies directed to various members of the epidermal growth factor receptor family are well suited for use as targeting antibodies or antigen binding portions thereof in the constructs of the present invention.
- Such antibodies include, but are not limited to anti-EGF-R antibodies as described in U.S. Pat. Nos. 5,844,093 and
- anti-EGFR family antibodies include, but are not limited to antibodies such as C6.5, C6ML3-9, C6MH3-B1, C6-B1D2, F5, HER3.A5, HER3.F4, HER3.H1, HER3.H3, HER3.E12, HER3.B12, EGFR.E12, EGFR.C10, EGFR.B11, EGFR.E8, HER4.B4, HER4.G4, HER4.F4,
- the cell surface-associated antigen may be expressed at sufficient levels on the target cell that a sufficently therapeutic amount of polypeptide construct is presented to ligand receptors on the target cell surface. Accordingly, in particular embodiments, the cell surface associated antigen is expressed at a density of greater than 12,600 copies per cell or greater than 15,000 copies per cell. Methods for determining copy number of a cell surface antigen are well known and readily available to a person of skill in the art, for example the method provided by Jilana (Am J Clin Pathol 118:560-566, 2002)
- the cell surface-associated antigen may be expressed in a configuration on the cell surface such that the polypeptide construct is abe to contact both the cell surface antigen and the ligand receptor on the target cell. Accordingly, in particular embodiments the cell surface associated antigen has an extracelluar domain having a molecular weight of less than 240kD.
- the antibody or antigen-binding portion thereof may bind to the cell surface associated antigen with sufficient affinity to facilitate ligand binding to the ligand receptor on the cell surface. Accordingly, in particular embodiments of the present invention the polypeptide constructs exhibit an antigen binding affinity, as measured by EC50, of from 50 nM, from 25 nM, from 10 nM or from 5 nM to 0.1 pM.
- the targeting antibody or antigen binding portion thereof comprises an antibody or antigen binding portion thereof that specifically or preferentially binds CD20.
- Anti-CD20 antibodies are well known to those of skill and include, but are not limited to Rituximab, Ibritumomab, and Tositumomab, AME-133v (Applied Molecular Evolution), Ocrelizumab (Roche), Ofatumumab (Genmab), TRU-015 (Trubion) and EVIMU-106 (Immunomedics).
- WO 2010/105290 discloses an antibody designated "SC104" together with a range of humanised variants which bind an antigen expressed on a range of tumour cells.
- the antibody attachment and attenuating mutation in the ligand increases the antigen- specificity index (ASI) by greater than 10-fold, preferably greater than 50-fold, preferably greater than 100-fold, preferably greater than 1000-fold, or preferably greater than 10,000 fold.
- the antigen- specificity index (ASI) is defined herein as the fold increased potency in signaling activity of the polypeptide construct of the invention relative to the free non-mutated polypeptide ligand on target antigen-positive cells multiplied by the fold decreased potency in signaling activity relative to the free non- mutated polypeptide ligand on target antigen-negative cells.
- potency in this context may be quantitatively represented by the EC50 value, which is the mathematical midpoint of a dose-response curve, in which the dose refers to the concentration of ligand or antibody-ligand construct in an assay, and response refers to the quantitative response of the cells to the signaling activity of the ligand at a particular dose.
- EC50 expressed for example in Molar units
- response refers to the quantitative response of the cells to the signaling activity of the ligand at a particular dose.
- the present inventors identified examples of two antigens where targeting attenuated IFNa to a target-expressing cell line did not exhibit an ASI that was appreciably greater than for the free, non-mutated ligand.
- the first example is demonstrated by the antigen CSPG4 (also known as HMW-MAA, high molecular weight melanoma-associated antigen).
- HMW-MAA high molecular weight melanoma-associated antigen
- the extracelluar domain of this antigen is exceptionally large (extracellular domain MW approx. 240kD -450kD depending on glycosylation). It is possible that certain antibody- IFN fusion protein constructs that bind to very large antigens may be sterically restricted from simultaneously interacting with the IFN receptors on the same cells. It is, however, possible that other antibodies that target other epitopes of this antigen may support the targeted IFN activity. Despite this possibility it is preferred that the antibody or antigen binding portion thereof of the polypeptide construct of the present invention binds an antigen wherein the extracellular domain thereof has a molecular weight of less than 240kD.
- a second example of an antibody-attenuated IFNa fusion protein construct that did not show potent activity was based on an antibody which binds to the myeloid antigen CD33.
- CD33 is expressed at a relatively low level on KG-1 cells, reported as 12,600 copies per cell (Sutherland, MAbs. 1(5): 481-490, 2009).
- Treatment of KG-1 cells with an anti-CD33 antibody-attenuated IFNa fusion protein construct failed to inhibit proliferation at all doses tested (IC50 > 76 nM).
- the relatively low copy number of this target may in some cases, depending on other factors such as epitope position, the receptor density of the IFN receptors, etc, limit the potency of the antibody-attenuated IFN fusion protein constructs. It is, however, possible that other antibodies that target other epitopes on this antigen may support the targeted IFN activity, or that other cells with low copy numbers of CD33 may nevertheless respond to such fusion protein constructs due to higher intrinsic IFN sensitivity, for example. Despite this possibility it is preferred that the antibody or antigen binding portion thereof of the polypeptide construct of the present invention binds an antigen wherein the antigen is present on the cell at a density of greater than 12,600 copies per cell, preferably greater than 15,000 copies per cell.
- Another example of an antibody-attenuated fusion protein construct in which the antibody did not provide sufficient targeting to the cancer cells was an anti-GM2 ganglioside antibody attached to an attenuated IFNa.
- the antibody was to a carbohydrate epitope and, as typical of such antibodies, had a low affinity (EC50 for binding target cells was -50 nM by flow cytometry). Therefore, preferred embodiments of the present invention show high affinity binding to their antigens, with EC50s preferably below 50 nM, more preferably below 25 nM, still more preferably below 10 nM and ideally below 5 nM.
- preferred embodiments comprise antibodies that bind to protein and peptide epitopes rather than carbohydrate epitopes.
- Multiple myeloma is of particular interest for certain embodiments of the present invention, namely fusion protein constructs comprising antibodies to multiple myeloma antigens and attenuated IFN peptides.
- Table 3 lists examples of multiple myeloma antigens and antibodies, with a reference to antibody sequences. Table 3
- Trail-R2 (DR5, Lexatumumab, ETR2-ST01, US Granted Patent # 2006, Menoret, APO-2) anti-DR5 6,872,568 Blood, 132;1356
- CD38 is of particular interest as an antibody target for fusion protein constructs of the present invention.
- Antibodies to CD38 include for example, AT13/5 (see, e.g., Ellis et al. (1995) J. Immunol. 155: 925-937), HB7, and the like.
- Table 4 discloses several known CD38 antibodies that may be used in this context: Table 4
- the term "Signaling ligand” as used herein broadly includes any ligand involved in the activation of cell signaling pathways, including any molecule capable of activating or inhibiting cell surface receptors. The term should also be understood as including reference to molecules that can pass through the lipid bilayer of the cell membrane to activate cell signaling pathways within the cell.
- polypeptide signaling ligand refers to peptide and polypeptide sequences of length 6 amino acids through 1,000 amino acids in length, which bind to particular cell surface molecules ("receptors") on certain cells and thereby transmit a signal or signals within those cells.
- Exemplary signaling ligands and polypeptide signaling ligands contemplated by the present invention include, but are not limited to cytokines, chemokines, growth factors, hormones, neurotransmitters, and apoptosis inducing factors.
- Non-limiting examples of suitable cytokines include the interleukin's IL-1, IL-2 IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-11, IL-12, IL-13, IL-14, IL-15, IL-16, IL-17, IL-18, IL-19, IL-20, IL-21, IL-22, IL-23, IL-24, IL_25, IL-26, IL-27, IL-28, IL-29, IL-30, IL-31, IL-32, IL-33, 11-35 and their subfamiles; the interferon (IFN) subfamily including Interferon type I (IFN- (IFNA1, IFNA2, IFNA4, IFNA5, IFNA6, IFNA7, IFNA8, IFNAIO, IFNA13, IFNA14, IFNA16, IFNA17, IFNA21), IFN-
- IFNWP15, IFNWP18, and IFNWP19 and IFNK Interferon type II (IFN- ⁇ ) and Interferon type III (IFN-epsilon, -kappa, -omega, -delta, -tau, and -gamma) and interferon-like molecules (limitin, IL-28A, IL-28B, and IL-29; the IL-1 family including IL-la, IL- ⁇ , the IL-1 Receptor antagonist (IL-1RA) and IL1F5, IL1F6, IL1F7, IL1F8, IL1F9 and IL1F10 and the IL-17 family including IL-17A, IL-17B, IL-17C, IL-17D, IL-17E (IL-25), and IL- 17F.
- IFNWP15, IFNWP18, and IFNWP19 and IFNK Interferon type II
- the peptide or polypeptide signaling ligand is selected from the group consisting of an IFN, IL-4 and IL-6. In an embodiment the peptide or polypeptide signaling ligand is selected from the group consisting of IFNa, IFNa2b, IFNpi, IFNpib and IFNy. Preferably the sequence of IFNa is selected from SEQ ID NOs 1 to 3, 80 to 90, 434 and 435.
- chemokines include, for example, RANTES, MCAF, MlPl-alpha, IP- 10, monocyte chemoattractant protein-1 (MCP-1 or CCL2), interleukin-8 (IL-8), CXCL13, XCL1 (lymphotactin-a), XCL2 (lymphotactin- ⁇ ) and fractalkine (CX 3 CL1).
- Non-limiting examples of growth factors include, for example, Adrenomedullin (AM), Angiopoietin (Ang), Autocrine motility factor, Bone morphogenetic proteins (BMPs), Brain-derived neurotrophic factor (BDNF), Epidermal growth factor (EGF),
- AM Adrenomedullin
- Ang Angiopoietin
- BMPs Bone morphogenetic proteins
- BDNF Brain-derived neurotrophic factor
- EGF Epidermal growth factor
- EPO Erythropoietin
- FGF Fibroblast growth factor
- G-CSF Granulocyte colony- stimulating factor
- G-CSF Granulocyte macrophage colony-stimulating factor
- GDF9 Growth differentiation factor-9
- HGF Hepatocyte growth factor
- HDGF Hepatoma-derived growth factor
- IGF Insulin-like growth factor
- GDF-8 Nerve growth factor
- NGF Nerve growth factor
- PDGF Platelet-derived growth factor
- TPO Thrombopoietin
- TPO Transforming growth factor alpha(TGF-a)
- VEGF vascular endothelial growth factor
- P1GF IL-1- Cofactor for IL-3 and IL-6
- Exemplary apoptosis inducing factors include FasL and TRAIL.
- Exemplary hormones include peptide hormones such as TRH and vasopressin, protein hormones such as insulin and growth hormone, glycoprotein hormones such as Luteinizing hormone, follicle-stimulating hormone and thyroid- stimulating hormone, Lipid and phospholipid-derived hormones such as steroid hormones e.g. testosterone and Cortisol, Sterol hormones such as calcitriol, eicosanoids such as prostaglandins.
- Non-limiting examples of suitable neurotransmitters includemonoamines and other biogenic amines: dopamine (DA), norepinephrine (noradrenaline; NE, NA), epinephrine (adrenaline), histamine, serotonin (SE, 5-HT), somatostatin, substance P, opioid peptides and acetylcholine (ACh),
- the linkage between the antibody and the ligand could be made via a simple peptide bond by creating a fusion protein between the ligand and the heavy or light chain, or both, of the antibody.
- the ligand could be attached at either the N- or C-terminus of either the heavy or the light chain of the antibody, with or without an intervening linker peptide sequence.
- the ligand is linked to the antibody or antigen binding portion thereof via a peptide bond.
- the ligand is linked to the C-terminus of the heavy chain of a human, humanized or chimeric IgGl, IgG2 or IgG4, either directly or with an intervening linker of 1 to 20 amino acids in length.
- the mutated polypeptide ligands may be attached to the antibody or antibody fragment by means of chemical conjugation, non-covalent protein-protein interactions, or by genetic fusion. Methods for conjugating the ligands described herein with antibodies may be readily accomplished by one of ordinary skill in the art. As will be readily ascertained, commonly used chemical coupling methods may be utilized to link ligands to antibodies via for example, free amino, carboxylic acid, or sulfhydryl groups. Ligands can also be linked to antibodies via Carbonyls (-CHO); these aldehyde groups can be created by oxidizing carbohydrate groups in glycoproteins.
- -CHO Carbonyls
- Some commonly used cross-linking reagents include glutaraldehyde which links protein or peptide molecules to the N-terminal or aliphatic amine groups of peptides or polypeptides, carbodiimide (EDC) which attaches proteins or peptides to the C-terminus or side chain carboxyl groups of proteins or peptides, succinimide esters (e.g. MBS, SMCC) which conjugates free amino groups and thiols from Cys residues, benzidine (BDB) which links to Tyr residues, periodate which attaches to carbohydrate groups and isothiocyanate.
- EDC carbodiimide
- BDB benzidine
- labels are attached via spacer arms of various lengths to reduce potential steric hindrance.
- a chemical linker may be used between the ligand and the antibody.
- Exemplary linker sequences will be readily ascertained by those of skill in the art, and are likely to include linkers such as C6, C7 and C12 amino modifiers and linkers comprising thiol groups.
- the antibody-ligand fusion protein constructs of the present invention have mutations or deletions in the ligand that render the ligands less active in stimulating their receptors on cells that lack cell surface expression of the antigen to which the antibody binds.
- the ligand is an interferon, examples of which are type I interferons (IFN-a (alpha), IFN- ⁇ (beta), IFN- ⁇ (kappa), IFN- ⁇ (delta), IFN- ⁇ (epsilon), IFN- ⁇ (tau), IFN- ⁇ (omega), and IFN- ⁇ (zeta, also known as limitin), type II interferons (IFN- ⁇ ) or type III interferons (IFN- ⁇ , ⁇ - ⁇ 2 and ⁇ - ⁇ 3) (Pestka, Immunological Reviews 202(l):8-32, 2004).
- type I interferons IFN-a (alpha), IFN- ⁇ (beta), IFN- ⁇ (kappa), IFN- ⁇ (delta), IFN- ⁇ (epsilon), IFN- ⁇ (tau), IFN- ⁇ (omega), and IFN- ⁇ (zeta, also known as limitin
- type II interferons IFN- ⁇
- Type I interferons all signal through the Type I interferon receptor, which is made of of IFNAR1 and IFNAR2. Signaling occurs when a type I IFN binds IFNAR1 and IFNAR2, thus bringing them together into a complex with the IFN. This initiates a cascade of intracellular events (the "signaling") which leads, among other things, to changes in the expression of numerous interferon regulated genes. Details of the intracellular signaling events triggered by activation of the type I interferon receptor is described, for example, by Platanias, (Nature Reviews 5:375-86. 2005). Type I interferons include various interferon- alphas. Known human interferon-alphas are
- IFNcclb cc2cc, 2 ⁇ , cc4b , cc5, cc6, cc7, cc8, cclO, ccl a/13, ccl4, ccl6, ccl7, ⁇ ⁇ 21, cc2c and cc4a.
- Some embodiments comprise IFNcc2b, the sequence of which, SEQ ID NO:3, is shown in Figure 4.
- IFNs have been approved in several forms for several indications, as outlined in Table 5 (which also shows lists of approved ⁇ and ys):
- NHL Leukemia, NHL, Kaposi's sarcoma
- Non-limiting examples of mutations in IFNcc2b that can be used to reduce its potency are described in Tables 6 and 7, based on the sequence of human IFNcc2b (SEQ ID NO:3):
- IFNs interferons
- YNS is sometimes used herein to represent IFNcc variants including the following mutation: H57Y, E58N and Q61S.
- the present invention also contemplates combinations of the abovementioned mutations or deletions in IFNcc.
- the invention also contemplates the combination of the constructs of the present invention with other drugs and/or in addition to other treatment regimens or modalities such as radiation therapy or surgery.
- the contracts of the present invention are used in combination with known therapeutic agents the combination may be administered either in sequence (either continuously or broken up by periods of no treatment) or concurrently or as an admixture.
- anticancer agents that may be used in this context.
- Treatment in combination is also contemplated to encompass the treatment with either the construct of the invention followed by a known treatment, or treatment with a known agent followed by treatment with the construct of the invention, for example, as maintenance therapy.
- the constructs of the present invention may be administered in combination with an alkylating agent (such as mechlorethamine, cyclophosphamide, chlorambucil, ifosfamidecysplatin, or platinum-containing alkylating-like agents such as cysplatin, carboplatin and oxaliplatin), an antimetabolite (such as a purine or pyrimidine analogue or an antifolate agent, such as azathioprine and mercaptopurine), an anthracycline (such as Daunorubicin, Doxorubicin, Epirubicin Idarubicin, Valrubicin, Mitoxantrone, or anthracycline analog), a plant alkaloid (such as a vinca alkaloid or a taxane, such as
- an alkylating agent such as mechlorethamine, cyclophosphamide, chlorambucil, ifosfamidecysp
- Vincristine, Vinblastine, Vinorelbine, Vindesine, paclitaxel or Dosetaxel a topoisomerase inhibitor (such as a type I or type II topoisomerase inhibitor), a Podophyllotoxin (such as etoposide or teniposide), or a tyrosine kinase inhibitor (such as imatinib mesylate,
- the constructs of the present invention may be administered in combination with current therapies, such as steroids such as dexamethasone, proteasome inhibitors (such as bortezomib or carfilzomib), immunomodulatory drugs (such as thalidomide, lenalidomide or pomalidomide), or induction chemotherapy followed by autologous haematopoietic stem cell transplantation, with or without other chemotherapeutic agents such as steroids, such as dexamethasone, proteasome inhibitors (such as bortezomib or carfilzomib), immunomodulatory drugs (such as thalidomide, lenalidomide or pomalidomide), or induction chemotherapy followed by autologous haematopoietic stem cell transplantation, with or without other chemotherapeutic agents such as
- the constructs of the present invention may be administered in combination with current therapeutic approaches, such as ABVD (Adriamycin (doxorubicin), bleomycin, vinblastine, and dacarbazine), or Stanford V (doxorubicin, bleomycin, vinblastine, vincristine, mechlorethamine, etoposide, prednisone), or BEACOPP (doxorubicin, bleomycin, vincristine, cyclophosphamide, procarbazine, etoposide, prednisone).
- ABVD Adriamycin (doxorubicin), bleomycin, vinblastine, and dacarbazine
- Stanford V doxorubicin, bleomycin, vinblastine, vincristine, mechlorethamine, etoposide, prednisone
- BEACOPP doxorubicin, bleomycin, vincristine, cyclophosphamide, pro
- non-Hodgkin's lymphoma or other lymphomas it is contemplated that the constructs of the present invention may be administered in combination current therapeutic approaches.
- drugs approved for non-Hodgkin lymphoma include Abitrexate (Methotrexate), Adriamycin PFS (Doxorubicin Hydrochloride), Adriamycin RDF
- Denileukin Diftitox DepoCyt (Liposomal Cytarabine), Doxorubicin Hydrochloride, DTIC-Dome (Dacarbazine), Folex (Methotrexate), Folex PFS (Methotrexate), Folotyn (Pralatrexate), Ibritumomab Tiuxetan, Istodax (Romidepsin), Leukeran (Chlorambucil), Linfolizin (Chlorambucil), Liposomal Cytarabine, Matulane (Procarbazine Hydrochloride), Methotrexate, Methotrexate LPF (Methotrexate), Mexate (Methotrexate), Mexate-AQ (Methotrexate), Mozobil (Plerixafor), Nelarabine, Neosar (Cyclophosphamide), Ontak (Denileukin Diftitox), Plerixafor, Pralatrex
- Retinoids are a family of molecules that play a major role in many biological functions including growth, vision, reproduction, epithelial cell differentiation and immune function (Meyskens, F. et al. Crit Rev Oncol Hematol 3:75, 1987, Herold, M. et al. Acta Dermatovener 74:29 1975).
- ATRA retinol all-trans retinoic acid
- the enhanced efficacy of interferon plus the induced expression of the target CD38 would indicate a combination therapy of ATRA with our anti-CD38 antibody-attenuated IFNcc in the treatment of IFN-sensitive cancers that express CD38 or may be induced by ATRA to express CD38.
- IFN-sensitive cancers that express CD38 or may be induced by ATRA to express CD38.
- Example of such cancers are multiple myeloma, non-Hodgekin's lymphoma, CML and AML.
- the mutations or deletions could also be made in the context of any of the other IFNccs or ⁇ .
- the type I IFN is an ⁇ .
- IFN- ⁇ is approved for the treatment of multiple sclerosis (MS).
- MS multiple sclerosis
- IFN- ⁇ could be attenuated by mutation or deletion and then attached to an antibody that targets cells involved in the pathogenesis of this disease.
- IFN- ⁇ is an effective drug in MS, but its use is associated with adverse events, including injection site inflammation, flu-like symptoms, leukocytopenia, liver dysfunction and depression, leading to discontinuation in a subset of patients. By directing IFN- ⁇ activity directly to pathogenic cells, these adverse events may be avoided.
- Pathogenesis of MS is thought to be initiated and progressed by a number of events, including innate activation of dendritic and microglial cells through toll-like receptors, an imbalance between pro -inflammatory and anti-inflammatory/regulatory cytokines, differentiation of CD4+ T cells into Thl and Thl7 phenotypes, activation of Thl cells by antigen presenting cells (APCs), reduction in the number of regulatory T (Treg) cells and migration of activated immune cells across the blood-brain barrier (BBB).
- events including innate activation of dendritic and microglial cells through toll-like receptors, an imbalance between pro -inflammatory and anti-inflammatory/regulatory cytokines, differentiation of CD4+ T cells into Thl and Thl7 phenotypes, activation of Thl cells by antigen presenting cells (APCs), reduction in the number of regulatory T (Treg) cells and migration of activated immune cells across the blood-brain barrier (BBB).
- APCs antigen
- the primary drivers of the clinical episodes of the disease are thought to be autoreactive, myelin- specific Thl cells (reviewed in Vogel, 2010 J Neuroimmunol 221:7; Boppana, 2011 Mt Sinai J Med 78:207; Loma, 2011 Curr Neuropharmacol 9:409).
- an attenuated version of IFN- ⁇ may be attached to an antibody targeting a cell surface marker specific for T cells, for the treatment of multiple sclerosis or other autoimmune indications where IFN- ⁇ may be effective.
- Direct effects of IFN- ⁇ on T cells include inhibition of proliferation (Rep, 1996 J Neuroimmunol 67: 111), downregulation of the co- stimulatory molecule CD40L
- an attenuated IFN- ⁇ is attached to an anti-CD3 antibody that targets all T cells, which includes CD4+, CD8+, Treg, Thl, Th2 and Thl 7 cells.
- an anti-CD3 antibody that targets all T cells, which includes CD4+, CD8+, Treg, Thl, Th2 and Thl 7 cells.
- This comprehensive approach ensures full coverage of all T cells, as all of these cell types have reported roles in MS pathogenesis and are affected by IFN- ⁇ treatment (Dhib-Jalbut, 2010 Neurology 74:S17; Prinz, 2010 Trends Mol Med 16:379; Graber, 2010 Clin Neurol Neurosurg 112:58 and Loma, 2011 Curr Neuropharmacol 9:409).
- Examples of CD3 antibodies that may be incorporated into the fusion protein constructs of the present invention are listed in Table 8.
- an attenuated IFN-P-anti-CD4 fusion protein construct presents a more restrictive approach, but would target autoreactive and regulatory T cells, including Thl and Thl7 cells and CD4 + CD25 + Treg cells.
- DCs dendritic cells
- Examples of CD4 antibodies that may be incorporated into the fusion protein constructs of the present invention are listed in Table 9.
- Markers of activated T cells including, but not limited to CD25, CD38, CD44, CD69, CD71, CD83, CD86, CD96, HLA-DR, ICOS and PD-1, also represent attractive targets for this approach, since activated T cells are thought to be the main drivers of autoreactivity resulting in demyelination in MS (Gandhi, 2010 J Neuroimmunol 221:7; Boppana, 2011 Mt Sinai J Med 78:207; Loma, 2011 Curr Neuropharmacol 9:409).
- Antibodies targeting any of these antigens could be attached to an attenuated ⁇ ⁇ .
- CD71 antibodies include BA120g (US 7736647) and various antibodies mentioned in Wang et al (Di Yi Jun Yi Da Xue Xue Bao (Academic journal of the first medical college of PLA) 22(5):409-411, 2002).
- Examples of antibodies to CD83 include 20B08, 6G05, 20D04, 11G05, 14C12, 96G08 and 95F04 (US 7,700,740).
- An example of an antibody to CD86 includes 1G10H6D10 (US 6,071,519).
- HLA-DR antibodies include HD3, HD4, HD6, HD7, HD8 and HD10 (US 7,262,278), DN1921 and DN1924 (US2005/0208048).
- PD-1 which is expressed on recently activated T cells.
- a non-antagonizing antibody could be used, such as the Jl 10 antibody discussed in further detail below.
- Examples of antibodies to ICOS include JMabs (US 6,803,039) and JMab 136 (US2011/0243929).
- an attenuated version of IFN- ⁇ can be fused to an antibody targeting cell surface markers of myeloid cells, known to contribute to MS pathogenesis by driving T cell activation and differentiation.
- the pan- myeloid markers CD33, CD115, or the dendritic cell marker CD1 lc may be targeted.
- a broad targeting approach may be preferred, for example, using antibodies against CD33 or CD115, since the exact contribution of each of the myeloid cell subsets to MS disease pathogenesis and response to IFN- ⁇ has been disputed (Prinz, 2008 Immunity 28:675; Shinohara, 2008 Immunity 29:68; Dann, 2012 Nat Neurosci 15:98).
- Antibodies to CD33 that could be used in the present invention include My9-6 (US 7,557,189), any of 14 antibodies described in US patent application US2012/0082670, or the antibody known as huM195 (US5693761).
- Antibodies to CD115 that could be used include Abl and Abl6 (US 8,206,715) or CXIIG6 (US2011/0178278).
- An example of a CD1 lc antibody that could be used according to the present invention is mab 107 (US 7,998,738, ATCC deposit number PTA-11614).
- the attenuated IFN- ⁇ could alternatively be directed to the CD 14 antigen, present primarily on macrophages. Examples of CD 14 antibodies are shown in
- targeting CD52-expressing cells would deliver IFN- ⁇ to all lymphocytes and, in addition, to monocytes and peripheral dendritic cells (Buggins, 2002 Blood 100: 1715; Ratzinger, 2003 Blood 101 : 1422), which are the key APCs responsible for proliferation and differentiation of autoreactive T cells in MS.
- This approach would direct the activity of IFN- ⁇ to the key cell types known to be directly affected by IFN- ⁇ and would facilitate its therapeutic activity in MS.
- Examples of CD52 antibodies that could be used according to the present invention include, but are not limited to DIVHv5/DIVKv2 (US 7,910,104), any of the CD52 antibodies disclosed in
- any of the above mentioned, antibody-targeted attenuated IFNP fusion protein constructs may have therapeutic activity in the context of other inflammatory and autoimmune diseases beyond multiple sclerosis, due to their common underlying immunological etiologies.
- Autoimmune diseases contemplated herein include inter alia alopecia areata, ankylosing spondylitis, antiphospholipid syndrome, autoimmune Addison's disease multiple sclerosis, autoimmune disease of the adrenal gland, autoimmune hemolytic anemia, autoimmune hepatitis, autoimmune oophoritis and orchitis, Behcet's disease, bullous pemphigoid, cardiomyopathy, celiac sprue-dermatitis, chronic fatigue syndrome (CFIDS), chronic inflammatory demyelinating, chronic inflammatory polyneuropathy, Churg-Strauss syndrome, cicatricial pemphigoid, crest syndrome, cold agglutinin disease, Crohn's disease, irritable bowel syndrome, inflammatory bowel disease, dermatitis herpetiformis, discoid lupus, essential mixed cryoglobulinemia, fibromyalgia,
- glomerulonephritis Grave's disease, Guillain-Barre, Hashimoto's thyroiditis, idiopathic pulmonary fibrosis, idiopathic thrombocytopenia purpura (ITP), IgA nephropathy, insulin dependent diabetes (Type I), lichen planus, lupus, Meniere's disease, mixed connective tissue disease, multiple sclerosis, myasthenia gravis, myocarditis, pemphigus vulgaris, pernicious anemia, polyarteritis nodosa, polychondritis, polyglancular syndromes, polymyalgia rheumatica, polymyositis and dermatomyositis, pochitis, primary
- agammaglobulinemia primary biliary cirrhosis, psoriasis, Raynaud's phenomenon, Reiter's syndrome, rheumatic fever, rheumatoid arrthritis, sarcoidosis, scleroderma, Sjogren's syndrome, stiff-man syndrome, systemic lupus erythematosus, Takayasu arteritis, temporal arteritis/giant cell arteritis, ulcerative colitis, uveitis, vasculitis and vitiligo.
- inflammatory disease conditions contemplated by the present disclosure include but are not limited to those disease and disorders which result in a response of redness, swelling, pain, and a feeling of heat in certain areas that is meant to protect tissues affected by injury or disease.
- Inflammatory diseases which can be treated using the methods of the present disclosure, include, without being limited to, acne, angina, arthritis, aspiration pneumonia, disease, empyema, gastroenteritis, inflammation, intestinal flu, NEC, necrotizing enterocolitis, pelvic inflammatory disease, pharyngitis, PID, pleurisy, raw throat, redness, rubor, sore throat, stomach flu and urinary tract infections, chronic inflammatory demyelinating polyneuropathy, chronic inflammatory demyelinating polyradiculoneuropathy, chronic inflammatory demyelinating
- **the C17S mutation was made in order to remove the unpaired cysteine in the native sequence of ⁇ ⁇
- the IFN is IFN- ⁇
- Type I IFNs can have anti-cancer activity based on a direct stimulation of the type I IFN receptor on cancer cells. This has been shown for numerous types of cancer including multiple myeloma, melanoma, B cell lymphoma, non-small cell lung cancer, renal cell carcinoma, hairy cell leukemia, chronic myelogenous leukemia, ovarian cancer, fibrosarcoma, cervical cancer, bladder cancer, astrocytoma, pancreatic cancer, etc (Borden, Cancer Research 42:4948-53, 1982; Chawla-Sarkar, Clinical Cancer Research 7: 1821-31, 2001; Morgensen, Int J.
- Type I interferons can also display anti-viral properties.
- IFNcc2b for example, has been FDA-approved for the treatment of chronic hepatitis C infections, and may have utility in treating other viral infections as well.
- Pegylated IFN-a is currently part of the standard of care regimen for hepatitis C, according to American and European guidelines, but results in side effects in over 80% of patients, often leading to discontinuation of treatment (Aman, 2012; Calvaruso, 2011).
- a type I IFN with an attenuating mutation is attached to an antibody that binds to virally infected cells.
- the antigen to be recognized by the above referenced antibody could be a viral protein that is transiently expressed on the host cell surface, or it could be an endogenous host cell-produced antigen that is exposed on the cell surface to a greater extent after viral infection than before infection.
- exemplary viral proteins that could serve as targets for the antibody portion include but are not limited to, Hepatitis C viral envelope glycoproteins, El and E2; Hepatitis B surface antigen (HBsAg); Herpes virus viral envelope
- glycoproteins (gB, gC, gE, gH, gl, gK, gL); Epstein-barr virus viral glycoprotein gp350 and viral protein BMRF-2; Human cytomegalovirus UL16; Parvovirus B19 viral capsid proteins VP 1-3; Human astro virus structural proteins, e.g. VP26, VP29 and VP32;
- Noro viruses structural protein VP1 and capsid protein VP2 Poliovirus viral capsid proteins VP0, VP1, VP2, VP3 and VP4 Rhinovirus viral capsid proteins VP1, VP2, VP3 and VP4; and dengue virus virus particle proteins capsid (C), pre-membrane/membrane (prM/M) and envelope (E).
- IFN-a activity may be targeted with an antibody that binds, directly or indirectly via an intermediate protein such as annexin V or beta2-glycoprotein 1, to phosphatidylserine (PS), a phospholipid component of the inner leaflet of cellular membranes.
- PS phosphatidylserine
- An anti-PS antibody (or alternatively a direct or indirect PS binding protein) fused with an attenuated version of IFN-a would target IFN-a activity to PS expressing virus-infected cells without displaying the systemic safety issues related to IFN-a.
- Certain tumor cells, such as lung cancer cells also express PS on their cell surfaces, so an antibody (or alternatively a direct or indirect PS binding protein) to PS, attached to an attenuated IFN, could also have use in the treatment of certain cancers.
- antibody-targeted attenuated ⁇ could also be used in much the same way as IFNcc for the targeting virally infected cells (S. V. Kotenko, G. Gallagher, V. V. Baurin et al, "IFN- ls mediate antiviral protection through a distinct class II cytokine receptor complex," Nature Immunology, vol. 4, no. 1, pp. 69-77, 2003).
- Type II IFNs namely INFy
- IFNy has anti-proliferative properties towards cancer cells (Kalvakolanu, Histol. Histopathol 15:523-37, 2000; Xu, Cancer Research 58:2832-7, 1998; Chawla-Sarkar, Apoptosis 8:237-49, 2003; Schiller, J Interferon Resarch 6:615-25, 1986).
- Sharifi has described how to make a fusion protein in which an IFNy has been fused to the C-terminus of a tumor-targeting antibody (Sharifi, Hybridoma and Hybridomics 21(6):421-32, 2002).
- IFNy By attenuating IFNy via mutation, a more cancer- selective form of IFNy may be produced.
- Two attenuating mutations in IFNy have been described by Waschutza (Eur J. Biochem. 256:303-9, 1998), namely des-(A23, D24), in which residues A23 and D24 are deleted, and des-(N25, G26), in which residues N25 and G26 are deleted.
- the des-(A23, D24) mutant has an ⁇ 18-fold reduced affinity for the IFNy receptor compared to wild type IFNy, and had a ⁇ 100-fold reduced antiviral activity compared to wild type IFNy.
- the des-(N25, G26) variant had a ⁇ 140-fold reduced affinity for the IFNy receptor compared to wild type IFNy, and had a ⁇ 10-fold reduced antiviral activity compared to wild type IFNy.
- fusion proteins comprising antibodies to tumor cell surface targets and attenuated mutants of IFNy include the following: Rituximab may be used as fusion protein with one of these attenuated IFNy using a 7 amino acid linker described by Sharifi to produce the fusion protein construct "Rituximab-HC-L7-IFNy(A[A23,D24]) IgGl," composed of SEQ ID NOS:378 (heavy chain) and 276 (light chain)).
- Such a fusion protein construct would be expected to have potent anti-proliferative activity against CD20 malignancies such as B cell lymphomas.
- CD20 malignancies such as B cell lymphomas.
- Other attenuated mutants of IFNy that may be appropriate for fusing to a cell-targeting antibody were described by Lundell (J Biol. Chem. 269(23): 16159-62, 1994), namely S20I ( ⁇ 50x reduced affinity), D21K ( ⁇ 100x reduced affinity), A23Q ( ⁇ 2,500-fold reduced binding), A23V ( ⁇ 2,000-fold reduced binding) and D24A ( ⁇ 4-fold reduced binding).
- IFNy may be used as fusions in combination with anti- CD38 antibodies, to generate the fusion protein construct "X355/02-HC-L7-IFNy(S20I) IgGl" (composed of SEQID NOS:380 (heavy chain) and 226 (light chain)) or "R10A2- HC-L7-IFNy(D21K) IgGl” (composed of SEQ ID NOS:382 (heavy chain) and 270 (light chain)).
- Targeted attenuated IFNy may also be used to treat various indications characterized by pathological fibrosis, including kidney fibrosis, liver fibrosis and idiopathic pulmonary fibrosis (IPF).
- IPF is a chronic, progressive form of lung disease, characterized by fibrosis of unknown cause, occurring primarily in older adults.
- Pulmonary fibrosis can also be induced by exposure to drugs, particles, microorganisms or irradiation. The following relates to both IPF and lung fibrosis induced by known agents and potentially for treatment of fibrosis in other types of organs, including liver and kidney.
- Fibroblasts play a key role in fibrotic diseases of the lung and their activation leads to collagen disposition, resulting in excessive scarring and destruction of the lung architecture. Yet there is little information on the origin of these pathogenic fibroblasts, though several precursor cell types have been proposed, including bone marrow progenitors, monocytes, circulating fibrocytes, and endogeneous cells, such as resident mesenchymal and epithelial cells (Stevens, 2008 Proc Am Thorac Soc 5:783; King, 2011 Lancet 378: 1949).
- CD14 + monocytes from peripheral blood are able to differentiate into fibrocytes, the precursors of fibroblasts, and this process is inhibited by interferon- ⁇ (IFN- ⁇ ).
- IFN- ⁇ interferon- ⁇
- fibroblast cell surface molecules that are enriched in fibroblasts. These include, for example, fibroblast specific protein (FSP1; Strutz, 1995 J Cell Biol 130:393), fibroblast activation protein (FAP; Park, 1999 J Biol Chem 274:36505; Acharya, 2006 Hum Pathol 37:352), and platelet derived growth factor receptors (PDGFR- ⁇ and - ⁇ ; Trojanowska, 2008 Rheumatology (Oxford) 47S5:2).
- FSP1 fibroblast specific protein
- FAP fibroblast activation protein
- PDGFR- ⁇ and - ⁇ Trojanowska, 2008 Rheumatology (Oxford) 47S5:2
- IFN- ⁇ was delivered to hepatic stellate cells, the equivalent of fibroblasts and responsible for secreting collagen in liver fibrosis, through liposomes targeting PDGFR- ⁇ , thereby enhancing the anti-fibrotic effects of IFN- ⁇ (Li, 2012 J Control Release 159:261).
- the present invention also contemplates the attenuation and antibody-based targeting of type III IFNs, including 1FNXI (IL29), 1FNX2 (IL28A), and 1FNX3 (IL28B) (S. V. Kotenko, G. Gallagher, V. V. Baurin et al., "IFN- ls mediate antiviral protection through a distinct class II cytokine receptor complex," Nature Immunology, vol. 4, no. 1, pp. 69-77, 2003., P. Sheppard, W. Kindsvogel, W. Xu, et al., "IL-28, IL-29 and their class II cytokine receptor IL-28R,” Nature Immunology, vol. 4, no.
- IFNs act through receptors composed of the IFN IR1 chain (also known as IL28R ) and the IL10R2 chain (shared with IL10, IL22, and IL26 receptor complexes [A. Lasfar, W.
- IFNiRs are expressed on most cell types and mediate similar signalling pathways as the type I IFNs.
- the antiviral activity of ⁇ IFNs has been demonstrated against several viruses including HBV and HCV (E. M. Coccia, M. Severa, E.
- Giacomini et al. "Viral infection and toll-like receptor agonists induce a differential expression of type I and ⁇ interferons in humans plasmacytoid and monocyte-derived dendritic cells," European Journal of Immunology, vol. 34, no. 3, pp. 796-805, 2004; M. D. Robek, B. S. Boyd, and F. V. Chisari, "Lambda interferon inhibits hepatitis B and C virus replication," Journal of Virology, vol. 79, no. 6, pp. 3851-3854, 2005; N. Ank, H. West, C. Bartholdy, K. Eriksson, A. R. Thomsen, and S. R.
- One aspect of the present invention is to target a mutated, attenuated for of an ⁇ towards virally infected cells, using for example the targeting antibodies describe above for the targeting of an attenuated form of IFNcc.
- Mutated, attenuated forms of an ⁇ could also be used to target cancer cells, as described in more detail for IFNcc, above.
- Non-IFN ligands are also contemplated in the present invention and may also be attenuated by mutation and then targeted to specific cell types by antibodies or fragments thereof.
- the anti-inflammatory cytokine interleukin-10 plays a central role during innate and adaptive immune responses. IL-10 forms a homodimer and binds to the IL-10 receptor complex expressed on APCs, leading to reduced expression of MHC class II and reduced production of pro-inflammatory cytokines and chemokines, thereby inhibiting T cell development and differentiation.
- IL-10 has also been implicated in inducing the proliferation of several immune cells, including B cells (Hofmann, 2012 Clin Immunol 143: 116).
- IL-10 Reduced expression of IL-10 is associated with a number of autoimmune disorders in humans and rodents, including psoriasis, inflammatory bowel disease and rheumatoid arthritis. Mice deficient in IL-10 develop chronic enterocolitis, which can be prevented by the administration of IL-10, but the clinical translation of these findings resulted in a number of failed trials in patients.
- One explanation of these failures is that the local IL-10 concentrations may be too low, even at maximum tolerable systemic administration (Herfarth, 2002 Gut 50: 146). Another explanation may be the
- Fusing attenuated IL-10 to an antibody specific for APCs would decrease systemically active biologic activity and at the same time increase the targeted local active concentrations of IL-10.
- the demonstrated pro- inflammatory effect through B cells would be decreased or eliminated.
- the production of antibody- IL 10 fusion proteins have been described previously (Schwager Arthritis Res Ther. 11(5): R142, 2009).
- IL-10 Intra-fibrotic factor-10
- fibrosis A hallmark of fibrosis is the overproduction and deposition of collagen produced by fibroblasts, resulting in scarring tissue formation.
- IL-10 directly inhibits extracellular matrix synthesis by human fibroblasts (Reitamo, 1994 J Clin Invest 94:2489) and is anti-fibrotic in a rat hepatic fibrosis model through downregulation of TGF- ⁇ (Shi, 2006 World J Gastroenterol 12:2357; Zhang, 2007 Hepatogastroenterology 54:2092).
- Clinical use of IL-10 is hampered by its short half-life and a PEGylated version has shown promising pharmacokinetic improvements and efficacy in a preclinical model of fibrosis (Mattos, 2012 J Control
- IL-10 activity through fusion with an antibody directing it to fibroblasts could result in therapeutic benefits in fibrotic diseases, including lung and liver fibrosis.
- Antibodies against fibroblast specific proteins such as fibroblast activation protein and platelet derived growth factor receptors, as described above in the description of IFN- ⁇ - targeting, could deliver attenuated IL-10 directly to fibroblasts.
- EPO erythropoietin
- EPO receptor EPO receptor
- Many studies have examined the role of EPO and EPO-R stimulation in cancer models in vitro and in vivo, and a number of studies have demonstrated a stimulatory effect on tumor growth, either directly on cancer cells, or through increased angiogenesis in the tumors (reviewed in Jelkmann, 2008 Crit Rev Oncol Hematol 67:39).
- treatment with EPO has been associated with increased tumor growth and decreased survival, leading to the recommendation and black box warning to limit and monitor the exposure of EPO in cancer patients as much as clinically feasible (Farrell, 2004 The Oncologist 9: 18;
- Erythropoiesis is a multi-step process, in which pluripotent stem cells undergo tightly controlled differentiation and proliferation steps.
- An intermediate cell type in this process is the colony-forming-unit-erythroid (CFU-E) cell, which expresses high levels of EPOR, depends on EPO for survival and appears to be the main cell type in the differentiation process with this dependency (Elliott, 2008 Exp Hematol 36: 1573).
- CFU-E colony-forming-unit-erythroid
- Targeting EPO activity to CFU-E cells using specific markers would substantially reduce the effect of EPO on cancer and other non-hematopoietic cells, while maintaining the ability to drive erythrocyte formation and increase hemoglobin levels.
- Genome-wide analysis of CFU-E cells revealed several potential candidate cellular markers, including Rh-associated glycoproteins, e.g. CD241 and members of the Rh blood group system, e.g. the product of the RCHE gene (Terszowski, 2005 Blood 105: 1937).
- CD117 c-kit
- CD71 transferrin receptor
- CD36 thrombospondin receptor
- CD117 antibodies include SR-1 (US 7,915,391) and antibodies DSM ACC 2007, 2008 and 2009 (US 5,545,533).
- EPO antigens for targeting of an attenuated EPO
- CD34 CD45RO, CD45RA, CD115, CD168, CD235, CD236, CD237, CD238, CD239 and CD240.
- Fusing EPO activity to an antibody would also greatly increase the extent of the therapeutic activity.
- the half-life of recombinant EPO is about 5 hours in humans and this would likely be increased to weeks when attenuated EPO is fused to an antibody. This approach could benefit patients treated for anemia, who are dosed typically multiple times per week, often through intravenous injections.
- the therapeutic response to EPO is primarily controlled by the length of time EPO
- TGF- ⁇ transforming growth factor ⁇
- TGF- ⁇ knockout mice die from multifocal inflammation and autoimmune disorders, suggesting an immunosuppressive effect (Shull, 1992 Nature 359:693).
- TGF- ⁇ also has been shown to induce fibrotic disease through a prominent role in extracellular matrix regulation and by promoting fibroblast migration, proliferation and activation (Rosenbloom, 2010 Ann Intern Med 152: 159; Wynn, 2011 J Exp Med
- naive T cells can be converted into Treg cells, which can suppress antigen- specific T cell expansion in vivo and prevent allergic pathogenesis in a murine asthma model (Chen, 2003 J Exp Med 198: 1875). Inflammatory responses also contribute to the transition of acute liver disease and perpetuation into chronic fibrosis and cirrhosis and TGF- ⁇ may help dampen these responses through its effect on Treg differentiation (Dooley, 2012 Cell Tissue Res 347:245). Similarly, TGF- ⁇ directed to naive T cells in inflammatory bowel disease could lead to control and suppression of inflammation (Feagins, 2010 Inflamm Bowel Dis 16: 1963).
- TGF- ⁇ specifically to CD4 + T cells may leverage the anti-inflammatory potential of TGF- ⁇ , while minimizing its pro-fibrotic properties, and could provide a novel strategy to combat autoimmune disorders.
- TGF- ⁇ could be targeted soley to activated T cells using a T cell activation marker, as described above for the discussion of ⁇ targeting.
- One attractive target along these lines could be, for example, PD-1, which is expressed on recently activated CD4 T cells.
- a non- antagonizing antibody could be used, such as the Jl 10 antibody discussed in further detail below.
- IL-4 Interleukin-4
- Th2 cells Upon activation, Th2 cells produce more IL-4, and as a result, IL-4 is considered a main driver of Th2-mediated immune responses.
- Thl/Th2 imbalance voring Thl
- autoimmune and other inflammatory diseases was first postulated in the 1980s (reviewed in Kidd, 2003 Altern Med Rev 8:223), and indeed, a role of Thl/Thl7 cells as drivers of disease in psoriasis (Ghoreschi, 2007 Clin Dermatol 25:574), certain types of inflammatory bowel disease, in particular Crohn's disease (Sanchez-Munoz, 2008 World J Gastroenterol 14:4280), or severe versus mild forms of asthma (Hansbro, 2011 Br J Pharmacol 163:81), has been documented.
- Th2 Diversion towards Th2 may provide a therapeutic benefit in certain types of diseases. Delivery of IL-4 to CD4 + T cells could accomplish this, or IL-4 activity could be targeted to macrophages to protect from immunopathology (Ghoreschi, 2007 Clin
- Attenuating mutations in IL-4 that may be exploited in the design of antibody- attenuated IL-4 fusion protein constructs of the present invention include those listed in
- Interleukin-6 may also be attenuated and targeted to specific cell types.
- a mechanism by which tumors can evade anti-tumor immunity is by recruiting Treg cells to the tumor microenvironment, resulting in tolerance at tumor sites.
- IL-6 is a cytokine involved in regulating the balance between Treg and Thl7 cells and induces the development of Thl7 cells, while it inhibits Treg differentiation (Kimura, 2010 Eur J Immunol 40: 1830).
- IL-6 by skewing the terminal differentiation of naive CD4 + T cells towards the Thl7 lineage, or reprogramming of Thl7 cells, has the potential to reverse tumor- associated immune suppression by Treg cells in the context of cancer, thereby enabling the immune system to control the tumors.
- This strategy has proven successful in a murine model of pancreatic cancer in which mice injected with tumor cells expressing IL-6 demonstrated a significant delay in tumor growth and enhanced survival, accompanied by an increase in Thl7 cells in the tumor microenvironment, compared to mice bearing tumors not expressing IL-6 (Gnerlich, 2010 J Immunol 185:4063).
- Tregs in controlling anti-tumor activity is further exemplified by a significant increase in the humoral response to peptide vaccination in glioblastoma patients after depletion of Tregs with the anti-IL-2 receptor antibody daclizumab
- Attenuated mutants of IL-6 include those listed in Table 18. Table 18
- HGF hepatocyte growth factor
- HGF has been shown to alter the fate of epithelial cells and reduce epithelial- mesenchymal transition (EMT) through its intereference with TGF- ⁇ signaling, antagonizing the process of fibroblastogenesis (Shukla, 2009 Am J Respir Cell Mol Biol 40:643).
- EMT epithelial- mesenchymal transition
- TGF- ⁇ drives conversion of HGF-producing fibroblasts into collagen-producing myofibroblasts, while HGF in turn inhibits TGF- ⁇ production by myofibroblasts (Mizuno, 2004 Am J Physiol Renal Physiol 286:F134).
- Exogeneous HGF, or mimetics activating the MET receptor act by restoring this imbalance imposed by tissue injury, and are therefore considered promising drug candidates for treating damaged tissues and fibrotic diseases (Nakamura, 2010 Proc Jpn Acad Ser B Phys Biol Sci 86:588).
- HGF vascular endocrinology 131:2540; Ishiki, 1992 Hepatology 16: 1227
- HGF subsequently demonstrated therapeutic benefits in many additional damaged organs, including pulmonary, gastrointestinal, renal and cardiovascular models of injury and fibrosis (Nakamura, 2011 J Gastroenterol Hepatol 26: 188).
- HGF In in vivo model systems of fibrosis, HGF prevents the progression of fibrotic changes and reduces collagen accumulation when administered prophylactically or therapeutically in murine lungs exposed to bleomycin (Yaekashiwa, 1997 Am J Respir Crit Care Med 156: 1937; Mizuno, 2005 FASEB J 19:580), in an obstructive nephropathy model in mice (Yang, 2003 Am J Physiol Renal Physiol 284:F349) and in liver fibrosis models in rats (Matsuda, 1997 Hepatology 26:81); HGF also prevents fibrosis in cardiomyopathic hamsters (Nakamura, 2005 Am J Physiol Heart Circ Physiol 288:H2131).
- HGF HGF-binding protein
- Liver fibrosis typically the result of chronic liver damage caused by infections or alcohol abuse, is, like fibrosis in other organs, characterized by excessive accumulation of extracellular matrix, including collagen produced by (myo) fibroblasts. Damaged hepatocytes release inflammatory cytokines and the resulting inflammatory milieu stimulates the transformation of hepatic stellate cells (HSC) into fibroblasts, producing collagen. The accumulation of extracellular matrix proteins results in scar tissue, which leads to liver cirrhosis (Bataller, 2005 J Clin Invest 115:209).
- HSC hepatic stellate cells
- HGF HGF-specific hepatocytes
- HSC HSC-specific hepatocytes
- ASGR1 a subunit of the asialoglycoprotein, used as a target for liver specific drug delivery (Stockert, 1995 Physiol Rev 75:591), or alternatively the other subunit of this receptor, ASGR2.
- FSP1 Fibroblast-specific protein
- HGF activity may be delivered to alveolar epithelial cells by attenuating it (by mutation) and attaching it to an antibody against a specific cell surface protein on these cells, such as RTI40/Tia or HTI56 (McElroy, 2004 Eur Respir J 24:664).
- Endothelial cell-specific markers including VEGF receptors (Stuttfeld, 2009 IUBMB Life 61:915) may be used for targeting blood vessels for endothelial cell layer enhancement for a number of pathologic indications, including hindlimb ischemia.
- signaling ligands are also known in the art and may, as described in the non-limiting exemplary embodiments above, be attenuated and attached to an antibody (or fragment thereof) that binds to an antigen on specific target cells, thereby allowing the ligand to generate its biological signal on those target cells to a much greater degree than it generates its signal on antigen-negative cells.
- ligands that have a direct negative effect on tumor proliferation include TNFa, TRAIL, Fas Ligand, IFNP, IFNy or IFN , which can be targeted to various tumor cell surface antigens as discussed above for INFa.
- specific mutations in various ligands are explicitly mentioned.
- mutagenesis for example, exposing the protein to UV radiation or mutagenic chemicals and selecting mutants with desired characteristics. Random mutagenesis may also be done by using doped nucleotides in oligonucleotides synthesis, or conducting a PCR reaction in conditions that enhance misincorporation of nucleotide, thereby generating mutants.
- Another technique is site- directed mutagenesis which introduces specific changes to the DNA.
- site directed mutagenesis is using mutagenic oligonucleotides in a primer extension reaction with DNA polymerase.
- This method allows for point mutation, or deletion or insertion of small stretches of DNA to be introduced at specific sites.
- the site-directed approach may be done systematically in such technique as alanine scanning mutagenesis whereby residues are systematically mutated to alanine and its effect on the peptide's activity is determined.
- Each of the amino acid residues of the peptide is analyzed in this manner to determine the important regions of the peptide.
- Another example is combinatorial mutagenesis which allows the screening of a large number of mutants for a particular characteristic.
- a few selected positions or a short stretch of DNA may be exhaustively modified to obtain a
- One approach of this technique is to excise a portion of DNA and replaced with a library of sequences containing all possible combinations at the desired mutation sites.
- the segment may be at an enzyme active site, or sequences that have structural significance or immunogenic property.
- a segment however may also be inserted randomly into the gene in order to assess the structural or functional significance of particular part of protein.
- Methods of screening mutated ligands to determine potency includes assaying for the presence of a complex between the ligand and the target.
- One form of assay involves competitive binding assays.
- the target is typically labeled.
- Free target is separated from any putative complex and the amount of free (i.e. uncomplexed) label is a measure of the binding of the agent being tested to target molecule.
- a cell free assay is a binding assay. Whilst not directly addressing function, the ability of a modulator to bind to a target molecule in a specific fashion is strong evidence of a related biological effect. For example, binding of a molecule to a target may, in and of itself, be inhibitory, due to steric, allosteric or charge-charge interactions.
- the target may be either free in solution, fixed to a support, expressed in or on the surface of a cell. Either the target or the compound may be labeled, thereby permitting determination of binding. Usually, the target will be the labeled species, decreasing the chance that the labeling will interfere with or enhance binding.
- Competitive binding formats can be performed in which one of the agents is labeled, and one may measure the amount of free label versus bound label to determine the effect on binding.
- culture may be required.
- the cell is examined using any of a number of different physiologic assays.
- molecular analysis may be performed, for example, protein expression, mRNA expression (including differential display of whole cell or polyA RNA) and others.
- Non-limiting examples of in vitro biological assays that can be used to screen protein variants are shown in the Examples below and also include apoptosis assays, migration assays, invasion assays, caspase- activation assays, cytokine production assays and the like.
- the present invention also provides compositions comprising the polypeptides of the present invention.
- compositions can further comprise at least one of any suitable auxiliary, such as, but not limited to, diluent, binder, stabiliser, buffers, salts, lipophilic solvents, preservative, adjuvant or the like.
- suitable auxiliaries are preferred.
- Non-limiting examples of, and methods of preparing such sterile solutions are well known in the art, such as, but not limited to, Gennaro, Ed., Remington's
- Pharmaceutically acceptable carriers can be routinely selected that are suitable for the mode of administration, solubility and/or stability of the antibody composition as well known in the art or as described herein.
- Pharmaceutical excipients and additives useful in the present composition include but are not limited to proteins, peptides, amino acids, lipids, and carbohydrates (e.g., sugars, including monosaccharides, di-, tri-, tetra-, and oligosaccharides; derivatised sugars such as alditols, aldonic acids, esterified sugars and the like; and polysaccharides or sugar polymers), which can be present singly or in combination, comprising alone or in combination 1-99.99% by weight or volume.
- Exemplary protein excipients include serum albumin, such as human serum albumin (HSA), recombinant human albumin (rHA), gelatin, casein, and the like.
- Representative amino acids which can also function in a buffering capacity include alanine, glycine, arginine, betaine, histidine, glutamic acid, aspartic acid, cysteine, lysine, leucine, isoleucine, valine, methionine, phenylalanine, aspartame, and the like.
- One preferred amino acid is histidine.
- a second preferred amino acid is arginine.
- Carbohydrate excipients suitable for use in the invention include, for example, monosaccharides, such as fructose, maltose, galactose, glucose, D-mannose, sorbose, and the like; disaccharides, such as lactose, sucrose, trehalose, cellobiose, and the like;
- polysaccharides such as raffinose, melezitose, maltodextrins, dextrans, starches, and the like; and alditols, such as mannitol, xylitol, maltitol, lactitol, xylitol sorbitol (glucitol), myoinositol and the like.
- alditols such as mannitol, xylitol, maltitol, lactitol, xylitol sorbitol (glucitol), myoinositol and the like.
- Preferred carbohydrate excipients for use in the present invention are mannitol, trehalose, and raffinose.
- Antibody compositions can also include a buffer or a pH adjusting agent
- the buffer is a salt prepared from an organic acid or base.
- Representative buffers include organic acid salts, such as salts of citric acid, ascorbic acid, gluconic acid, carbonic acid, tartaric acid, succinic acid, acetic acid, or phthalic acid; Tris, tromethamine hydrochloride, or phosphate buffers.
- Preferred buffers for use in the present compositions are organic acid salts, such as citrate.
- compositions of the invention can include polymeric
- excipients/additives such as polyvinylpyrrolidones, ficolls (a polymeric sugar), dextrates (e.g., cyclodextrins, such as 2-hydroxypropyl-P-cyclodextrin), polyethylene glycols, flavoring agents, antimicrobial agents, sweeteners, antioxidants, antistatic agents, surfactants (e.g., polysorbates such as "TWEEN® 20" and “TWEEN® 80”), lipids (e.g., phospholipids, fatty acids), steroids (e.g., cholesterol), and chelating agents (e.g., EDTA).
- polyvinylpyrrolidones e.g., ficolls (a polymeric sugar)
- dextrates e.g., cyclodextrins, such as 2-hydroxypropyl-P-cyclodextrin
- polyethylene glycols e.g., flavoring agents, antimicrobial agents, sweeten
- variable regions were generated from 18 (heavy chain) and 16 (light chain) DNA oligonucleotides, which were designed according to the published amino acid sequences, by PCR-based gene assembly.
- the DNA encoding the variable regions of the G005 anti-CD38 and nBT062 anti-CD 138 monoclonal antibodies were drawn from the publications by De Weers et al. (US Patent 7829673) and by Daelken et al. (WO 2009/080832), respectively, and subjected to be synthesized by Integrated DNA Technology, Inc. (Coralville, IA) after the sequence modification to eliminate rare codons and unprefered restriction sites.
- CD38 antibodies is described in the following sections.
- the DNA encoding human interferon- 2b was isolated from genomic DNA of a HEK cell line by PCR.
- the sequences of human interferon- ⁇ (IFN l, SEQ ID NO:91), human interleukin-4 (IL-4, SEQ ID NO: 119) and human interleukin-6 (IL-6, SEQ ID NO: 123) were designed from the protein sequences such as NP_002167, NP_000580 and NP_000591, respectively, and synthesized by Integrated DNA Technology, Inc. (Coralville, IA) or GenScript USA Inc. (Piscataway, NJ) using methods commonly known to those of skill in the art. Alterations of the cytokine sequences, for example the addition of linkers or point mutations, were introduced to the cytokine genes using overlap extension PCR techniques well known in the art.
- the cytokine-endoding gene fragments were then cloned into the pTT5 expression vector (Durocher, Nucleic Acids Research volume 30, number 2, pages El-9, 2002) containing either a human IgGl heavy chain complete or partial constant region (such as Swissprot accession number P01857), a human IgG4 heavy chain constant region (such as Swissprot accession number P01861 incorporating substitution S228P), human Ig kappa constant region (Swissprot accession number P01834) or human Ig lambda constant region (Swissprot accession number P0CG05) either as a naked Ig or as a cytokine gene fusion form using overlap extension PCR techniques and restriction sites according to cloning methods well known by those skilled in the art.
- Production of IgG and IgG interferon fusion protein constructs are then cloned into the pTT5 expression vector (Durocher, Nucleic Acids Research volume 30, number 2, pages El
- DNA plasmids encoding the IgGs and IgG-cytokine fusion protein constructs were prepared using Plasmid Plus Maxi kit (Qiagen, Valencia, CA) and then transfected into HEK293-6E cells (CNRC, Montreal, Canada) grown in F17 synthetic medium supplemented with 0.1% Pluronic F-68, 4 mM L-glutamine (Invitrogen, Carlsbad, CA) using a commercially available transfection reagent and OptiMEM medium (Invitrogen, Carlsbad, CA).
- the culture media was isolated and subjected to IgG affinity purification using Protein G-agarose beads (GE Healthcare, Piscataway, NJ). Purified IgG and IgG-cytokine fusion protein constructs were then concentrated and buffer-exchanged to phosphate buffered saline (PBS) pH 7.4 using Amicon Ultra centrifugal filter devices (Millipore, Billerica, MA), followed by protein concentration determination using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA).
- PBS phosphate buffered saline
- L6 SGGGGS (SEQ ID NO: 132)
- L16 SGGGGSGGGGSGGGGS (SEQ ID NO: 133)
- On target (Daudi) assay This assay was used to quantify the anti-proliferative activity of IFNs and antibody- IFN fusion protein constructs on cells that display that antigen corresponding to the antibody to which the IFN is fused, and may be used as part of the assay for calculating the antigen-sensitivity index (ASI) defined herein. Daudi cells express both CD20 and CD38 as cell surface associated antigens. The viability of cells was measured using the reagent CellTiter-Glo®, Cat #G7570, from Promega (Madison, Wisconsin). This is a luminescence-based assay that determines the viability of cells in culture based on quantitation of ATP. The signal strength is proportional to the number of viable cells in a microtiter plate well. The details of the assay are as follows:
- Daudi cells obtained from ATCC, Manassas, VA were cultured in a T75 flask (TPP, Trasadingen, Switzerland, cat# 90076) to a preferred density of between 0.5 x 10 5 and 0.8 x 10 5 viable cells/ml in RPMI 1640 (Mediatech, Inc., Manassas, VA, cat # 10-040- CV) with 10% Fetal Bovine Serum (FBS; Hyclone, Logan, UT cat# SH30070.03). Cells were harvested by centrifuging at 400g for five minutes, decanting the supernatant, and resuspending the cell pellet in RPMI 1640 + 10% FBS.
- TPP T75 flask
- FBS Fetal Bovine Serum
- ARP On target assay
- CD38 + The multiple myeloma cell line ARP-1 was a gift from Bart Barlogie MD, PhD, Director of the Myeloma Institute at the University of Arkansas Medical Center (Little Rock, AK). It is described in Hardin J. et al., (Interleukin-6 prevents dexamethasone-induced myeloma cell death. Blood; 84:3063, 1994).
- ARP-1 cells (CD38 + ) were used to test CD38 targeting antibody-IFN fusion protein constructs.
- ARP-1 was cultured to a density of 4.0 x 10 5 to 6.0 x 10 5 cells/ml. ARP-1 concentration was adjusted to 1.0 x 10 4 cells/ml prior to assay.
- Off-target assay also sometimes referred to herein as the "not-targeted” assay: The iLite assay from PBL Interferon Source (Piscataway, NJ,Cat# 51100), was performed largely as described by the manufacturer with the addition of a human IgG blocking step.
- the iLite cell line is described by the manufacturer as "a stable transfected cell line derived from a commercially available pro-monocytic human cell line characterized by the expression of MHC Class II antigens, in particular the human lymphocyte antigen (HLA- DR), on the cell surface.”
- the cell line contains a stably transfected luciferase gene, the expression of which is driven by an interferon-response element (IRE), which allows for interferon activity to be quantified based on luminescence output.
- IRE interferon-response element
- the manufacturer- supplied iLite plate (hereafter "assay plate”) and diluent were removed from the -80°C freezer and allowed to equilibrate to room temperature.
- test articles were transferred from the dilution plate to the assay plate.
- the assay plate was then incubated at 37 C with 5% C0 2 for 17 hours.
- the manufacturer-supplied assay buffer and substrate were removed from the -80 C freezer and allowed to equilibrate to room temperature for two hours.
- the manufacturer- supplied assay buffer was added to the manufacturer-supplied substrate vial and mixed well according to the manufacturer's instructions to create the "luminescence solution.” Then, 100 ⁇ of the luminescence solution was added to each well of the assay plate. The plate was shaken for 2 minutes.
- the plate was then incubated at room temperature for 5 minutes in the dark and finally read on a Victor 3V Multilabel Counter on a luminometry channel and the luminescence measured and presented as RLU.
- the data was analyzed with Graphpad Prism 5 as described for the'On-target (Daudi) assay," above.
- Figure 6 shows the interferon activity of free IFNcc2b (SEQ ID NO:3; "IFNcc” in figure) as well as IFNcc2b fused to the C-terminus of the heavy chain of two different antibodies (rituximab and palivizumab, an isotype control antibody), as acting on a the iLite cell line.
- This cell line does not display the antigen for either of these antibodies, so this assay reveals the potency of various IFNcc2b-containing proteins in the absence of antibody-antigen-based targeting.
- Isotype-HC-L6-IFNcc IgGl refers to the RSV-targeting humanized antibody Palivizumab, in which the light chain (SEQ ID NO:290) is unaltered but the IgGl class heavy chain (SEQ ID NO:291) has, attached to its C terminus, a 6 amino acid linker sequence ("L6;” SGGGGS, , SEQ ID NO: 132), followed by the sequence for IFNcc2b (SEQ ID NO:3); this heavy chain-linker- IFNcc2b sequence is shown as SEQ ID NO:294.
- free IFNcc2b showed an EC 50 for activating gene expression through an interferon response element (IRE) of 1.9 pM.
- IRE interferon response element
- the EC50 (mathematical midpoint of the dose-response curve) was used as a measure of potency in the calculations presented here. In other words, when compound A showed an EC50 that is 10-fold lower than compound B, it was said to have a 10-fold higher potency.
- IFNcc2b The modest reduction in IFNcc activity that occurred as a result of linking it to an antibody may not be sufficient to prevent the toxicity of the IFNcc component of the construct in human subjects.
- Various mutations were therefore introduced into IFNcc2b in order to reduce its activity and toxicity.
- five different mutant versions of IFNcc2b were generated and, in each case, linked to the C-terminus of the heavy chain of Rituximab via the six amino acid linker L6, which has the sequence SGGGGS (SEQ ID NO: 132).
- FIG. 9 shows the degree of reduced interferon activity for each of these Rituximab-attenuated IFNcc2b fusion protein constructs relative to free, wild type IFN 2b, on antigen-negative (i.e. CD20-negative) cells.
- Figure 10 shows the degree of reduced interferon activity for the
- the R33A version of the fusion protein construct (composed of SEQ ID NOS:436 (heavy chain) and 276 (light chain)) was attenuated to such a high degree that it showed no detectable activity in the non-targeted assay.
- the Rituximab-HC-L6- IFNcc2b (R33A+YNS) IgGl fusion protein construct exhibited a substantially greater antigen- specificity index (ASI, 1,700-fold) compared to Rituximab-HC-L6-IFNcc2b IgGl (10- to 14-fold) or free IFNcc2b (1-fold, by definition), suggesting that its off-target effects in vivo will be substantially reduced.
- CD38 constructs for expression The extracellular domains (ECD) of human and cynomolgus monkey CD38 proteins were each formatted to include a cleavable N-terminal leader sequence, an AvitagTM, a poly-histidine tag and a thrombin cleavage site to yield proteins SEQID NO: 127 and 128 respectively. These were back-translated into DNA sequences and synthesized de novo by assembly of synthetic oligonucleotides by methods known by those with skill in the art.
- Heavy and light chain variable region sequences were subcloned into variants of the vector pTT5 containing either a human IgGl heavy chain constant region (such as Swissprot accession number P01857), a human IgG4 heavy chain constant region (such as Swissprot accession number P01861 incorporating substitution S228P), human kappa constant region (Swissprot accession number P01834) or human lambda region (Swissprot accession number P0CG05) to yield full length antibody chains.
- a human IgGl heavy chain constant region such as Swissprot accession number P01857
- a human IgG4 heavy chain constant region such as Swissprot accession number P01861 incorporating substitution S228P
- human kappa constant region such as Swissprot accession number P01861 incorporating substitution S228P
- human kappa constant region such as Swissprot accession number P01834
- human lambda region Swissprot accession
- HEK293-6E cells were cultured in complete cell growth media (1 L of F17 medium (InvitrogenTM), 9 mL of Pluronic F68 (InvitrogenTM), 2mM Glutamine containing 20% (w/v) Tryptone NI (Organotechnie®) with Geneticin (50 mg/mL, InvitrogenTM) at 50 ⁇ 1/100 mL culture). The day before transfection, cells were harvested by centrifugation and resuspended in fresh media (without Geneticin). The next day DNA was mixed with a commercial transfection reagent and the DNA transfection mix added to the culture drop-wise. The culture was incubated overnight at 37°C with 5% C0 2 and 120 rpm without Geneticin.
- Transient co-expression of heavy and light chains in HEK293-6E cells generated antibodies that were subsequently purified by protein A chromatography. Briefly, supernatants derived from these transfections were adjusted to pH 7.4 before being loaded onto a HiTrap Protein A column (5 mL, GE Healthcare). The column was washed with 50 mL of lx PBS (pH 7.4). Elution was performed using 0.1 M citric acid pH 2.5. The eluted antibody was desalted using Zeba Desalting columns (Pierce) into IX PBS (pH 7.4). The antibodies were analyzed using SDS-PAGE. The concentration of the antibody was determined using the BCA assay kit (Pierce).
- Immobilized metal ion affinity chromatography was used to purify human and cynomolgous monkey CD38 extracellular domain (ED) proteins from tissue culture supernatants. Briefly, protein supernatants were diluted in binding buffer (20 mM sodium phosphate, 0.5 M NaCl, 30 mM imidazole, pH 7.4) before being loaded onto a HisTrapTM FF column (1 mL, GE Healthcare). The column was washed with 5 mL of binding buffer (pH 7.4) and elution was performed using 20 mM sodium phosphate, 0.5 M NaCl, 500 mM imidazole, pH 7.4.
- binding buffer 20 mM sodium phosphate, 0.5 M NaCl, 500 mM imidazole, pH 7.4.
- the eluted proteins were desalted and buffer exchanged using Amicon Ultra- 15 centrifugal filter unit with Ultracel-10 membrane (Millipore) into 1 X PBS (pH 7.4).
- the absorbance at 280 nm (A 2 go) of the protein was assessed using a Nanodrop spectrophotometer and readings corrected using the predicted extinction coefficients to determine protein concentrations.
- Biotinylation of antigens for phage display [0313] The AvitagTM motifs of human and cynomolgus monkey CD38 EDs were biotinylated according to manufacturer's directions (Avidity LLC, Aurora, CO). Excess unconjugated biotin was removed from the biotinylated proteins by desalting into lx PBS using a 7KD molecular weight cut off (MWCO) Zeba spin column (Thermo Scientific, Logan, UT) according to manufacturer's instructions. Successful biotinylation of CD38 ED proteins was confirmed using a combination of polyacrylamide gel electrophoresis and Western blotting.
- MWCO molecular weight cut off
- FAbs that bind to both human and cynomolgus monkey CD38 EDs were isolated from a naive phagemid library comprising approximately 2.5 x 10 11 individual human FAb fragments. Methods of generating phage antibody fragment libraries are discussed in "Phage display: A Practical Approach” (Eds. Clackson and Lowman; Oxford University Press, Oxford, UK) and “Antibody Phage Display Methods and Protocols” (Eds. O'Brien and Aitken; Humana Press Inc, NJ 07512). Briefly, antibody heavy and light chain variable regions were amplified based on RNA from donor samples.
- Antibody heavy and light chain variable regions were then inserted into phagemid vectors to generate a library of antibody fragments fused to a phage coat protein.
- the antibody library used herein was a high diversity naive phagemid library that expressed antibody fragments in the Fab format.
- Anti-CD38 FAbs were isolated from the phage display library over the course of two panning 'campaigns' (i.e. discrete phage display experiments with different reagents or panning conditions). The general protocol followed the method outlined by Marks et al. (Marks, J.D. & Bradbury, A., 2004, Methods Mol Biol, 248, 161-76).
- Each phage display campaign involved three rounds of panning. For each round, -2.5 x 10 12 phage particles were blocked by mixing 1: 1 with blocking buffer (4% skim milk in PBS, pH 7.4) and incubating for 1 hr at room temperature. The blocked phage library was then pre-depleted for any biotinylated protein tag motif binders used in panning through incubation for 45 mins with 50-200 pmols of an irrelevant antigen containing an identical biotinylated tag motif. Tag- and streptavidin-binders were captured by adding an excess (75-300 ⁇ L) of streptavidin-coated Dynabeads (Invitrogen), which were blocked as described for the library. The beads (including tag- and streptavidin-binders attached to them) were immobilized using a magnet and discarded.
- blocking buffer 4% skim milk in PBS, pH 7.4
- the blocked phage library was then pre-depleted for any biotinylated protein tag motif bind
- Library panning was conducted by mixing the blocked and pre-depleted library with 50-200 pmols of biotinylated recombinant CD38 ED in a 2 mL microcentrifuge tube and rotating for 2 hrs at room temperature. Then, 100 ⁇ L of streptavidin-coupled Dynabeads (Invitrogen, Carlsbad, CA) were added and the mixture was incubated a further 15 minutes as described previously. Non-specifically bound phage were removed using a series of washes. Each wash involved pulling the bead complexes out of the solution onto the tube wall using a magnetic rack, aspirating the supernatant and then re- suspending the beads in fresh wash buffer.
- streptavidin-coupled Dynabeads Invitrogen, Carlsbad, CA
- PBS wash buffer lx PBS with 0.5% skim milk
- PBS-T wash buffer lx PBS supplemented with 0.05% TWEEN-20 [Sigma- Aldrich, St. Louis, MO] and 0.5% skim milk.
- Phage that remained bound after the washing process were eluted from the biotinylated-CD38 ED-bead complexes by incubation with either a twenty-fold excess of non-biotinylated CD38 ED for 1 hr at room temperature or 0.5 mL of 100 mM triethylamine (TEA) (Merck
- the output phage were added to a 10 mL culture of exponentially growing TGI E. coli (2x yeast-tryptone (2YT) growth media) and allowed to infect the cells during a 30 minute incubation at 37°C without shaking, then with shaking at 250 rpm for 30 additional minutes.
- the phagemids encoding the phage display output were then rescued as phage particles following a standard protocol (Marks, J.D. & Bradbury, A., 2004, Methods Mol Biol, 248, 161-76).
- TGI cells were infected with output phage and were plated on 2YT agar (supplemented with 2% glucose and 100 ⁇ g/mL carbenicillin) at a sufficient dilution to produce discrete E. coli colonies. These colonies were used to inoculate 1 mL liquid cultures to allow expression of FAb fragments for use in screening experiments.
- FAb samples were prepared by harvesting cells by centrifugation (2,000g, 10 mins) and performing a lysozyme extraction.
- the cell pellet was resuspended in 200 of lysis buffer (160 ⁇ g/mL lysozyme, 10 ⁇ g/mL RNase A, 5 ⁇ g/mL DNase and complete protease inhibitors (Roche, Nutley, NJ)) and shaken at 400 rpm for 30 minutes at 21°C.
- lysis buffer 160 ⁇ g/mL lysozyme, 10 ⁇ g/mL RNase A, 5 ⁇ g/mL DNase and complete protease inhibitors (Roche, Nutley, NJ)
- lysis buffer 160 ⁇ g/mL lysozyme, 10 ⁇ g/mL RNase A, 5 ⁇ g/mL DNase and complete protease inhibitors (Roche, Nutley, NJ)
- lysis buffer 160 ⁇ g/mL lysozyme
- FAbs that bound to CD38 ED were detected by incubation for 30 minutes at room temperature with an anti-V5-HRP conjugated antibody (Invitrogen, Carlsbad, CA) to detect the V5 tag fused to the C-terminus of the FAb heavy chain. Plates were washed to remove unbound antibody and the assay signal developed by incubation with 50 ⁇
- RPMI-8226 obtained from the Health Protection Agency Culture Collections, Porton Down, Salisbury, SP4 0JG, UK
- flow cytometry-based assays were tested. Briefly, viable RPMI-8226 cells (2 x 10 5 , as judged by trypan blue exclusion) were incubated with each antibody or with a human IgGi isotype control antibody preparation (Sigma- Aldrich, St. Louis, MO) at various concentrations in 100 ⁇ of FACS buffer (PBS plus 1% fetal calf serum, FCS) in 96 well plates for 20 minutes on ice in the dark.
- FACS buffer PBS plus 1% fetal calf serum, FCS
- Monoclonal antibodies against human CD38 ED were generated by genetic immunization with corresponding conventional protein immunization of rats.
- the DNA sequence of human CD38 ED is provided in SEQ ID NO: 129.
- the corresponding conceptually translated protein sequence is given in SEQ ID NO: 130.
- the DNA sequence of SEQ ID NO: 129 was cloned into a plasmid for genetic
- Rats were then immunized six times with the plasmid using a Helios gene gun (Bio-Rad, Germany) according to a published procedure (Kilpatrick et ah, Hybridoma 17: 569-576, 1998).
- a Helios gene gun Bio-Rad, Germany
- Untagged human CD38 ED for this purpose was produced by removing the protein tags from SEQ ID NO: 127 by thrombin cleavage followed by purification over a size exclusion column.
- the rats were sacrificed and their lymphocytes fused with myeloma cells using polyethylene glycol (HybriMaxTM; Sigma- Aldrich, Germany), seeded at 100,000 cells per well in 96- well microtiter plates and grown in DMEM medium supplemented with 10% fetal bovine serum and HAT additive for hybridoma selection (Kilpatrick et al., 1998, supra).
- HybriMaxTM polyethylene glycol
- Viable RPMI-8226 cells (2 x 10 5 , as judged by trypan blue exclusion) were incubated with 100 ⁇ L of rat hybridoma supernatant for 20 minutes on ice in the dark. Cells were washed twice with FACS buffer (lx PBS plus 1% FCS) before incubation for 20 minutes in 100 ⁇ of FACS buffer containing anti-rat IgG-FITC conjugate (Sigma- Aldrich). After washing cells in FACS buffer, they were resuspended in FACS buffer and analysed for antibody-binding by flow cytometry on a FACS Canto (BD Biosciences, San Diego, CA) using EV, side scatter and FL-1 gating.
- FACS buffer lx PBS plus 1% FCS
- Results were expressed as mean fluorescent intensity (MFI). Of the 15 rat antibodies exhibiting positive binding to human CD38 ED by ELISA, five showed weak or negligible binding to CD38 expressed on the human myeloma cell line RPMI-8226 by FACS (Table 22).
- RNA extraction from pelleted hybridoma cells of each clone was performed using TRI reagent (Sigma- Aldrich, St. Louis, MO) according to manufacturer's directions.
- the variable regions of each antibody were amplified using Rapid Amplification of cDNA Ends (RACE) reverse transcription polymerase chain reaction (RT-PCR) methodology according to manufacturer' s directions (Clontech [Mountain View, CA] SMART RACE kit; Ambion Life Technologies [Foster City, CA] RLM-RACE kit).
- RACE Rapid Amplification of cDNA Ends
- RT-PCR reverse transcription polymerase chain reaction
- Gene-specific reverse PCR primers to amplify the rat heavy chain variable domains by 5 '-RACE were designed to anneal to the available rat heavy chain constant region sequences.
- gene specific reverse PCR primers to amplify the rat light chains were designed to anneal to the rat kappa chain constant region sequences, while further primers were designed to anneal to the rat lambda chain constant region sequences.
- 5 '-RACE PCR was performed according to manufacturer's directions (Life Technologies; Clontech) using PfuUltrall polymerase (Agilent). Following 5 '-RACE PCR, products were separated by agarose gel electrophoresis and bands of approximately the predicted size based on the location of the reverse primer in the constant region were excised from the gels. DNA was purified from agarose gel slices using a Qiaquick spin gel extraction kit (Qiagen) according to manufacturer's instructions. Insert DNA was cloned and propagated in E.coli using a StrataClone Blunt PCR Cloning Kit (Agilent, Santa Clara, CA) according to manufacturer's instructions.
- Qiaquick spin gel extraction kit Qiagen
- Vectors were constructed using the rat antibody variable region sequences grafted onto human IgGl constant sequences for the heavy chain variable region and, human kappa or lambda backbones (keeping the same light chain isotype as in the rat antibodies).
- the resulting variable region sequences of each clone are listed in Table 23.
- FCl Flow Cell 1
- FC2 or alternatively FC3 and FC4
- FCl was used as a blank throughout the experiments.
- HBS-P buffer (0.01 M HEPES pH 7.4, 0.15 M NaCl, 0.005% v/v Surfactant P20).
- flow rate 20 ⁇ /min
- 20 ⁇ of 5 ⁇ g/mL of antibody was passed over FC2.
- CD38 antigen was blocked by the addition of excess naked (e.g. without IFN or IFN variants fused to it) anti-CD38 antibody for all iLite experiments using anti-CD38-IFN fusion protein constructs; in each case, the
- concentration of blocking naked CD38 antibody used was 0.5 mg/ml. Also in each case, the same antibody clone being assayed as an IFN or IFN- variant fusion protein construct was used to block any interaction with CD38.
- Figure 15 shows the off target activity of free wild type IFNcc2b (IFNcc) (SEQ ID NO:3) vs. wild type IFNcc2b fused to the C-terminus of the CD38 antibody G005 (De Weers et al. (US Patent 7829673).
- the latter fusion protein construct (G005-HC-L0-IFNCC IgG4) was of IgG4:kappa isotype and had no intervening linker between the C-terminus of the heavy chain and the first residue of the IFNcc and is described by SEQ ID NOS: 150 (heavy chain) and 134 (light chain).
- Figure 16 shows a comparison between the same two constructs in the "on target (ARP1) assay", in which the anti-CD38 antibody was allowed to bind to CD38, which was expressed at high levels on the ARP-1 cell line.
- ASI antigen specificity index
- Table 26 characterizes the CD38 antibody G005, fused in various configurations via the C-terminus of the heavy chain to IFNa with the R144A attenuating mutation. Examples in this table are of IgGl and IgG4 isotype and either have no linker between the antibody heavy chain and the IFN, L0, or have an intervening 6 amino acid linker, L6 (composed of SEQ ID NOS: 138,140,152,146 (heavy chain) each combined with 134 (light chain)).
- the fusion protein constructs had dramatically reduced potency on antigen-negative iLite cells (a reduction of from 8,300 to 100,000 fold compared to free IFNa), but substantially maintained the potency exhibited by free IFNa on CD38 positive cells (Daudi).
- ASI Antigen Specificity Index
- Table 27 shows examples using another IFNa attenuating mutation, A145G, as a construct with the same G005 antibody in either IgGl or IgG4 isotypes, with either no linker or the L6 linker (composed of SEQ ID NOS: 142, 144, 148 (heavy chain) each combined with SEQ ID 134 (light chain)).
- the G005-HC-L6-IFNa (A145G) IgG4 construct (composed of SEQ ID NOS: 148 (heavy chain) and 134 (light chain)), for example, showed an ASI of 20,000.
- Table 28 shows examples in which the mutated IFNcc is attached to the light chain rather than the heavy chain, with either no intervening linker or the L6 linker (composed of SEQ ID NOS:210 or 208 (light chain), respectively, each combined with SEQ ID NO: 135 (heavy chain)).
- the fusion protein constructs demonstrated a high ASI of 5,900 and 7,200, respectively.
- Tables 29 and 30 demonstrate the ASI for the same fusion protein constructs but use an alternative cell line (ARP-1, a myeloma) for determining activity on CD38 + cells. Using this method, the ASIs for these fusion protein constructs ranged from 1,200-55,000. Table 29
- IFNcc any of these point mutated, attenuated versions of IFNcc could be used in the context of the present invention as antibody fusion protein constructs. Certain IFN variants may be preferred due to showing higher ASIs. Other considerations, such as expression level, immunogenicity, biophysical characteristics, etc., may also be considered in evaluating constructs for optimal utility.
- R144A, R144S, R144T, R144Y, R144I, R144L, R145G, R145D, R145H, R145Y Table 25
- R33A+YNS as illustrated in the construct comprising SEQ ID NOS:286 (heavy chain) and 276 (light chain)
- R33A as illustrated in the construct comprising SEQ ID NOS:436 (heavy chain) and 276 (light chain)
- R144A+YNS such as as illustrated in the construct comprising SEQ ID NOS:288 (heavy chain) and 276 (light chain)
- X355/02-HC-L0 IFNa (A145D) IgG4 composed of SEQ ID NOS:232 (heavy chain) and SEQ ID NO:226 (light chain);
- X355/02-HC-L0 IFNa (R144A) IgG4 composed of SEQ ID NOS:230 and SEQ ID NO:226 (light chain);
- X355/07-HC-L0 IFNa (A145D) IgG4 composed of SEQ ID NOS:240 (heavy chain) and SEQ ID NO:234 (light chain);
- X355/07-HC-L0 IFNa (R144A) IgG4 composed of SEQ ID NOS:238 and SEQ ID NO:234 (light chain);
- R5D1-HC-L0 IFNa (A145D) IgG4 composed of SEQ ID NOS:262 (heavy chain) and 258 (light chain);
- R5E8-HC-L0 IFNa (A145D) IgG4 composed of SEQ ID NOS:268 (heavy chain) and 264 (light chain); and [0354] R10A2-HC-L0 IFNa (A145D) IgG4 composed of SEQ ID NOS:274 (heavy chain) and 270 (light chain).
- These fusion protein constructs all showed high ASIs, ranging from 3,820 (X910/12-HC-L0-IFNCC (R144A) IgG4) to 166,000 (X355/02-HC-L0-IFNCC (A145D) IgG4).
- CD 138 (SEQ ID NO:432), also called Syndecan-1, is a heparin sulfate proteoglycan that is thought to function as an adhesion molecule. It is expressed on most multiple myeloma cells (Dhodapkar, Blood; 91: 2679, 1998). Fusion protein constructs consisting of mutated, attenuated IFNa and the CD138-targeting antibody nBT062 (Ikeda, Clin Can Res., 15:4028, 2009; USPTO #20090175863, composed of SEQ ID NOS:330 (heavy chain) and 326 (light chain)) were generated.
- this fusion protein construct like the anti-CD38-attenuated IFNa fusion protein construct, showed much greater anti-proliferative potency on multiple myeloma cells (ARP-1, on-target assay) than a non-targeted, isotype fusion protein (based on the antibody 2D12).
- Figure 39 shows that a 28pM concentration (4 th highest concentration tested) of nBT062-HC-L0- IFNa (A145D) shows greater anti-proliferative activity on the ARP-1 myeloma cell line than does 6 nM (highest concentration tested) of the isotype-HC-LO-IFNa (A145D) protein.
- Another antigen that has been described as a potential target for antibody therapy to treat cancer is the Class I MHC (see for example Stein, Leuk. Lymphhoma 52(2):273- 84, 2011).
- antibody W6/32 Barnstable et al. (1978), Cell 14:9-20
- ATCC HB95
- This antibody reacts with monomorphic determinants on human HLA A,B,C molecules.
- the antibody variable regions were cloned and sequenced using SMART RACE cDNA Amplification kit (Clontech, Mountain View, CA) and Mouse Ig-Primer Sets (Novagen/EMD Chemicals, San Diego, CA).
- the amino acid sequences of the heavy chain and light chain variable regions are shown as SEQ ID NOS:411 and 410, respectively.
- the chimeric version of HB95, with the murine variable regions and human IgG4 kappa constant regions, fused to IFNa with the A145D mutation (HB95-HC-L0-IFNa (A145D) IgG4, composed of SEQ ID NOS:316 (heavy chain) and 312 (light chain)) was expressed, and its activity was compared to an isotype control antibody fused in the same way to the same IFNa mutant (Isotype-HC-LO-IFNcc (A145D) IgG4, where the isotype variable regions were derived from antibody 2D 12).
- the "on-target (ARP-1)" assay was run as described above for the CD38-targeted antibodies (ARP- 1 is class I MHC-positive). The results are shown in Figure 40a.
- Figure 40b demonstrates that antibody fragments may substitute for full-length antibodies and provide similar properties, namely high ASIs.
- This figure shows the effects of various Fab-attenuated IFNa fusion protein constructs on the proliferation of ARP- 1 cells.
- Two non- ARP-1 targeted constructs "Palivizumab-HC-L6-IFNa (A145D) Fab” (composed of SEQ ID NOS:298 (heavy chain) and 290 (light chain)) and "2D12-HC-L6- IFNa (A145D) Fab” (composed of SEQ ID NOS:356 (heavy chain) and 344 (light chain)), show very low potency on this cell line (EC50's from 2,410-17,000).
- the Fab portion of the fusion protein construct does target a cell surface antigen, in this case class I MHC, as for the fusion protein construct "HB95-HC-L6-IFNa (A145D) Fab" (composed of SEQ ID NOS:320 (heavy chain) and 312 (light chain)), the potency is even higher than free, wild type IFNa.
- the antigen-targeted attenuated construct is 2,760- 19,450-fold more potent than the non-targeted attenuated constructs.
- IFNa anti-viral activity
- recombinant IFNa is an FDA- approved treatment for hepatitis C viral infections.
- IFN activity was measured using the cytopathic effect inhibition (CPE) assay as described Rubinstein (J. Virol. 37, 755-8, 1981). Briefly, 10 4 human adenocarcinoma A549 cells (ATCC, Manassas, Kansas) per well were incubated with test sample or IFN (human IFN-CC2A) overnight. Cells were then challenged with EMC virus for 48-56 hours, followed by staining with crystal violet. A visual CPE determination was performed, followed by solubilization of the crystal violet and absorbance measurement at 570 nm. Nonlinear regression analysis was performed using a 4-parameter sigmoidal fit with variable slope (GraphPad Prism).
- IFNa activity is defined as the amount of interferon required to reduce the cytopathic effect by 50%.
- the units are determined with respect to the international reference standard for human IFNcc2, provided by the National Institutes of Health (see Pestka, S. "Interferon Standards and General Abbreviations,” in Methods in Enzymology (S. Pestka, ed.), Academic Press, New York vol 119, pp. 14-23, 1986).
- the samples tested in this assay were IFNa (Intron A, inverted triangles), Anti- MHC class I targeted attenuated IFNa designated HB95-HC-L0-IFNa (R145D) IgG4 (closed squares), and istoype control (2D12)-attenuated IFNa (Isotype-HC-LO-IFNa (R145D) IgG4; triangles). Data is plotted as viability vs IFNa molar equivalents.
- Results are shown in Figure 41.
- IFNa protects A549 cells from virally induced cytopathic cell death (CPE) as expected, showing at EC50 of 0.18 pM.
- CPE virally induced cytopathic cell death
- IFN also has been shown in numerous publications (see above) to have antiproliferative activity on various types of cancer cells.
- Figure 43 shows the potency of these three proteins under conditions in which the CD38 antibody can target the ⁇ to cells is fairly similar.
- Table 34 This data is summarized in Table 34. This demonstrates that the surprising finding that attenuating mutations in an interferon that is part of an antibody- IFN fusion protein construct can disproportionally affect non-targeted vs.
- the attenuating mutation reduced the potency by only 1.4-fold under conditions when the antibody could direct the IFN to the target cells, vs. 280-fold for cells in which the fusion protein construct could not target the cell surface antigen.
- the antibody-attenuated ⁇ fusion protein construct in the present example shows an ASI of 4,630 (Table 34).
- the R147A mutation in ⁇ was also found to produce antibody- ⁇ fusion protein constructs with a significantly greater ASI than free ⁇ (data not shown). The examples below will show that this "selective attenuation" can also be observed with ligands that are structurally unrelated to IFNa and ⁇ , namely to IL-4 and IL-6.
- Table 34 The examples below will show that this "selective attenuation" can also be observed with ligands that are structurally unrelated to IFNa and ⁇ , namely to IL-4 and IL-6.
- Interleukin-4 (IL-4)
- IL-4 is a helical bundle cytokine with multiple physiological activities, including the ability to bias T helper cell development towards Th2 and away from Thl. Since Thl cells play a pathological role in certain autoimmune settings, it could be therapeutically advantageous to use IL-4 to influence T helper cell development away from Thl, i.e. to create a "Thl diversion.” To avoid side effects related to IL-4's activity on other cell types, it would be advantageous to attenuate IL-4's activity by mutating it, and then attach it to an antibody that would direct it to activated (preferentially recently activated) helper T cells.
- the antibody chosen for this purpose was Jl 10, a mouse anti-human PD-1 clone described by Iwai et.al. (Immunol Lett. 83:215-20, 2002). PD-1 (SEQ ID NO:431) is expressed on recently activated ThO cells.
- Jl 10 antibody murine variable regions and human IgGLkappa constant regions; the amino acid sequences of J 110 heavy and light chain variable regions are shown as SEQ ID NOS:409 and 408, respectively
- human IL-4 SEQ ID NO:l 19
- L6 six amino acid linker
- J110-HC-L6-IL4 IgGl protein (composed of SEQ ID NOS:304 (heavy chain) and 300 (light chain))
- Jl 10-HC- L6-IL-4 (R88Q) IgGl (composed of SEQ ID NOS:306 (heavy chain) and 300 (light chain)) was made.
- the R88Q mutation in IL-4 has been reported to reduce its potency in vitro (Kruse, EMBO Journal vol.12 no.13 pp.5121 -5129, 1993).
- HB-IL4 off-target (HB-IL4) assay
- HEK-BlueTM IL-4/IL-13 Cells are specifically designed to monitor the activation of the STAT6 pathway, which is induced by IL-4.
- the cells were generated by introducing the human STAT6 gene into HEK293 cells to obtain a fully active STAT6 signaling pathway.
- the HEK-BlueTM IL-4/IL-13 Cells stably express a reporter gene, secreted embryonic alkaline phosphatase (SEAP), under the control of the ⁇ minimal promoter fused to four STAT6 binding sites.
- SEAP embryonic alkaline phosphatase
- HEK-BlueTM IL-4/IL-13 cells induces the expression of the SEAP reporter gene. SEAP is then secreted into the media and can be quantitated using the colorimetric reagent QUANTI-BlueTM. Briefly, HEK-Blue IL4/IL13 cells (Invivogen, San Diego CA cat# hkb-stat6) were thawed and cultured in DMEM media (Mediatech, Manassas VA, cat# 10-013-CV) + 10% FBS (Hyclone, Logan UT, cat# SH30070.03) that had been heat inactivated (HI FBS).
- the "on target (Thl diversion) assay” was designed to monitor the percentage of CD4 + T cells that were of Thl phenotype, as defined by their expression of IFN- ⁇ . Thl diversion is thereby quantified by a decrease in IFN- ⁇ -positive CD4 T cells.
- the assay was performed as follows: "Loaded” Dynabeads (M450 Epoxy beads, Invitrogen Dynal, Oslo, Norway cat# 140.11) were made as described by the manufacturer with 1.0 g/10 beads anti-human CD3 epsilon antibody (R&D Systems, Minneapolis MN, cat# MABIOO), 1.0 ⁇ g/10 7 beads anti-human CD28 antibody (R&D Systems, cat# MAB342) and 3 ⁇ g/10 7 beads human IgG (R&D Systems, cat# 1-001 -A). PBMCs were obtained from the
- Naive CD4 + T cells were purified from Leukocyte Reduction System (LRS) cones using the naive CD4 + kit (Miltenyi Biotech cat# 130-094- 131) according to the manufacturer's directions. A total of 4.0 x 10 5 purified naive CD4 + T-cells were aliquoted to each well of a 24 well tissue culture plate (hereafter, the
- the experimental plate was incubated at 37°C with 5% C0 2 for a minimum of four hours. Approximately 1/3 of the volume of each well of the experimental plate was then recovered and subjected to preparation for Intra-cellular Flow Cytometry according to the instructions supplied with the abovementioned Kit and utilizing the reagents supplied with the kit.
- the cells were stained for intra-cellular interferon-gamma with an anti human interferon-gamma antibody conjugated to AF647 (eBiosciences.com, cat# 51-7319-42).
- the samples were analyzed by flow cytometry on a Becton Dickinson FACSort using Cell Quest software. Acquired samples were analyzed using FloJo Software and data were graphed using Graph Pad Prism software.
- IL4 showed an EC50 of 1.26 pM ( Figure 44; Table 35).
- the "on target (Thl diversion) assay” results are shown in Figure 45.
- Activation of the naive (ThO) CD4 cells induces PD-1 expression, so that the anti-PDl-IL-4 fusion protein constructs may target the IL-4 to them.
- free, wild type IL4 shows an EC50 of 11.4 pM.
- the anti-PDl -attenuated IL-4 fusion protein construct J110-HC-L6-IL4 (R88Q) IgGl
- HB-IL4 off-target
- the non- attenuated, PD-1 targeted fusion protein construct Jl 10-HC-L6-IL4 IgGl
- the non-targeted, attenuated IL-4 fusion protein construct (Isotype-HC-L6-IL4 R88Q) IgGl, was significantly less potent than the targeted attenuated fusion protein construct, but its potency was too low to accurately determine an EC50 in this experiment.
- Interleukin-6 (IL-6)
- IL-6 (SEQ ID NO: 123) has numerous activities on different cell types and it may be advantageous to exploit some of these activities at the expense of others. For example, by targeting newly activated CD4 + T cells (via attachment to an anti-PDl antibody, as in the example above with IL-4 targeting, for example), one may shift the T helper cell population away from the Treg pathway and in favor of the Thl7 pathway. This could be advantageous to a cancer patient.
- the "IL-6 bioassay” was performed using the HEK-BlueTM IL-6 cells (Invivogen, cat# hkb-il6), an engineered reporter cell line that monitors the activation of the JAK-
- STAT pathway by IL-6 These cells were generated by introducing the human IL-6R gene into HEK293 cells. In addition, cells were further transfected with a reporter gene expressing SEAP under the control of the ⁇ minimal promoter fused to four STAT3 binding sites. In these cells, IL-6 stimulates the activation of STAT3 and leads to the secretion of SEAP. SEAP is then monitored when using the SEAP detection medium QUANTI-BlueTM. The assay was run essentially according to the manufacturer's
- HEK-Blue IL6 cells were thawed and cultured in DMEM (Mediatech, Manassas VA, cat# 10-013-CV) + 10% FBS (Hyclone, Logan UT, cat# SH30070.03) that had been heat inactivated (HI FBS). After one passage, 200 iglm ⁇ HygroGold, (Invivogen cat# ant-hg-1) and 100 g/ml Zeocin, (Invivogen cat# ant-zn-1) was added to the culture medium. After one more passage, cells were allowed to reach 60- 80% confluence and then lifted with Cell Stripper (Mediatech, cat# 25-056-Cl).
- QUANTI-Blue Invivogen, cat# rep-qbl
- QUANTI-Blue prepared according to the manufacturer's instructions, was then aliquoted (160 ⁇ per well) into each well of a flat bottom plate (hereafter, the "assay plate”). Then, 40 ⁇ supernatant per well from the experimental plate was transferred to the wells of the assay plate. The assay plate was incubated at 37°C for 1-3 hours. Assay plate absorbance at 630nm was read on a model 1420-41 Victor 3V Multilabel Counter from Perkin-Elmer. Data was analyzed using Graph Pad Prism.
- IL-6 carrying a 16-mer linker (L16, SGGGGSGGGGSGGGGS, SEQ ID NO: 133) at the N-terminus was fused to an antibody targeting class I MHC, using the HB95 antibody (which binds to human class I MHC antigen, as described above) vs. an isotype control antibody, 2D 12 (also described above).
- ASI Antigen Specificity Index
- the non-targeting, isotype control fusion protein construct 2D12-HC-L16-IL6 IgGl (composed of SEQ ID NOS:360 (heavy chain) and 344 (light chain)), was compared to free IL-6 in the "IL-6 bioassay" described above ( Figure 46).
- SCID mice subcutaneously in immunocompromised mice was used.
- mice Eight to 12 week old CB.17 SCID mice were injected subcutaneously in the flank with 1x10 NCI-H929 tumor cells in 50% Matrigel. When average tumor size reached 120- 150 mm , mice were grouped into 5 cohorts of 10 mice each and treatment began at time zero (TO). All treatments were given by intraperitoneal injection, (i.p.) twice weekly for 5 weeks (indicated by bar under graph). All compounds were dosed at 200 ⁇ g/dose (approximately 10 mg/kg) except for Interferon-a. IFNcc2b (Intron A®, Schering Corp., Merck, Whitehouse Station, NJ) was given at 2 million units/dose. Tumor volume was measured twice weekly by caliper measurement. Endpoint was tumor volume of 2,000 mm 3 .
- the non- targeted isotype control-attenuated IFNa fusion protein construct (Isotype-HC-L6- IFNa (A145G) IgGl, composed of SEQ ID NOS:348 (heavy chain) and 344 (light chain) (open inverted triangles) did show significant activity in delaying tumor growth, presumably due to the long half-life of the antibody- IFNa fusion protein construct and resulting increased systemic exposure.
- the CD38-targeted attenuated IFNa fusion protein construct (G005-HC-L6-IFNa (A145G) IgGl, composed of SEQ ID NOS: 144 (heavy chain) and 134 (light chain)), by contrast, showed dramatic anti-tumor activity compared to the non-targeted fusion protein construct (P ⁇ 0.0001.) or the other test substances.
- the targeted anti-CD38 -attenuated IFNa fusion protein construct completely resolved tumors in all (10/10) mice to undetectable levels by day 22 with no recurrence throughout the duration of the study.
- the anti-CD38-attenuated IFNa fusion protein construct (G005-HC-L6- IFNa (A145G) IgGl) was tested in a systemic multiple myeloma model based on the cell line MM1S (Crown Bioscience Inc., Santa Clara; Greenstein, Exp Hematol.
- mice Six to 8 week old NOD-SCID mice were injected intravenously with lxlO 7 MM1S tumor cells in 0.1 ml phosphate buffered saline (PBS) 24 hours after irradiation with 200 rad ( 60 Co). Mice were grouped into 4 cohorts of 10 mice each at time zero and treatments began 7 days later. All treatments were given i.p. twice weekly for 9 weeks. All compounds were dosed at 200 ⁇ g/dose (approximately 10 mg/kg) except Interferon-a (given at 2 million units/dose). Body weights and overall health were monitored twice weekly and survival was the endpoint.
- PBS phosphate buffered saline
- Results are shown in Figure 48.
- Treatment with naked anti-CD38 antibody (G005) only slightly increased survival (MST 62 days). None of the mice in the targeted anti-CD38 -attenuated IFNa (G005-HC-L6-IFNa (A145G) IgGl) treated cohort showed signs of disease during entire study. All (10/10) mice appeared healthy at termination.
- mice Six to eight week old NOD-SCID mice were injected subcutaneously in the flank with 1x10 Daudi Burkitt's Lymphoma tumor cells in 50% Matrigel one day after irradiation with 200rad ( 60 Co). When mean tumor size reached 169 mm 3 (Day 20), mice were grouped into 5 cohorts of 10 mice each and treatment began. All treatments were given i.p. twice weekly for 4 weeks. All compounds were dosed at 200 ⁇ g/dose
- Interferon- which was given at 2 million units (MIU)/dose. Tumor volume was measured twice weekly by caliper measurement.
- Results are shown in Figure 49.
- Treatment of this Burkitt's lymphoma s.c. tumor with the naked anti-CD38 antibody (closed square) did not significantly delay tumor growth in these mice compared to vehicle (closed circles).
- the IFNa treatment did result in a significant delay in tumor growth compared to vehicle (5.5 days) however this group reached the 2,000 mm endpoint by day 40.
- the non-targeted isotype control fusion protein construct (Isotype-HC-L6-IFNcc (A145G) IgGl; open inverted triangles) showed significant activity in delaying tumor growth but this group reached the 2,000 mm endpoint on day 57.
- Anti-CD38 (G005) IgGl 2443+/- 196 81 0.575
- This xenograft experiment shows that the CD38-targeted attenuated IFNa fusion protein constructs may be effective in treating lymphomas in addition to multiple myelomas.
- mice Eight to 12 week old CB.17 SCID mice were injected subcutaneously in the flank with 1x10 NCTH929 tumor cells in 50% Matrigel. When mean tumor sizes reached 120- 150 mm , mice were grouped into 9 cohorts of 10 mice each and treatment began (time zero). All treatments were given i.p. twice weekly for 5 weeks. Two compounds, targeted anti-CD38-attenuated interferon (closed grey symbols) and non-targeted isotype control- interferon (open symbols), were compared in this study at different doses (see legend for doses). Tumor volume was measured twice weekly by caliper measurement. Endpoint was tumor volume of 2,000 mm . Results:
- Results are shown in Figure 50.
- the dose titration of anti-CD38-targeted attenuated IFNcc fusion protein construct (G005-HC-L6-IFNCC (A145G) IgGl)
- antibodies of the present invention also include those of the IgG4 isotype.
- mice Eight to 12 week old CB.17 SCID mice were injected subcutaneously in the flank with 1x10 NCTH929 tumor cells in 50% Matrigel. When average tumor size reached 120-150 mm , mice were grouped into 5 cohorts of 10 mice each and treatment began (time zero). All treatments were given i.p. twice weekly for 5 weeks. All compounds were dosed at 70 ⁇ g/dose (approximately 3.5 mg/kg). Tumor volume was measured twice weekly by caliper measurement. Endpoint was 2,000 mm . Results:
- Results are shown in Figure 51.
- This study compared the activity of the targeted vs. non-targeted fusion protein constructs in two different isotype formats; IgGl isotype (G005-HC-L6-IFNCC (A145G) IgGl (targeted, closed squares) and Isotype-HC-L6- IFNcc (A145G) IgGl (non-targeted, open squares)) and IgG4 isotype (G005-HC-L6- IFNcc (A145G) IgG4, composed of SEQ ID NOS: 148 (heavy chain) and 134 (light chain) (targeted, closed diamonds) and Isotype-HC-L6-IFNcc (A145G) IgG4, composed of SEQ ID NOS:350 (heavy chain) and 344 (light chain) (non-targeted, open diamonds)) were compared.
- mice in this study were treated at a lower dose than in previous studies where we observed 100% tumor elimination.
- the tumor volumes indicate that, surprisingly, the IgG4 format is more potent than the IgGl format in this model.
- human IgGl antibodies have greater effector function than IgG4 antibodies (Hooper, Ann Clin Lab Sci.;8:201, 1978; Ward, Ther Immunol, 2:77, 1995.)
- the IgGl format would have been at least as effective, if not more so, than the IgG4 format.
- 8/10 mice in the CD38 targeted, attenuated IFN, IgG4 treated group were tumor free whereas only 3/10 were tumor-free in the IgGl format counterpart (closed squares).
- the next example extends the observation of in vivo efficacy of an antibody- targeted IFN to a second mutated form of IFNcc in which A 145 has been mutated to aspartic acid (D).
- the experiment below utilizes a different CD38 antibody, i.e. one based on the variable regions of human antibody clone X355/02; (SEQ ID NOS:391 (VH) and 390 ( ⁇ )).
- VH variable regions of human antibody clone X355/02
- VH variable regions of human antibody clone X355/02
- VH variable regions of human antibody clone X355/02
- VH variable regions of human antibody clone X355/02
- VH variable regions of human antibody clone X355/02
- VH variable regions of human antibody clone X355/02
- ⁇ 390
- mice Eight to 12 week old CB.17 SCID mice were injected subcutaneously in the flank with 1x10 NCI-H929 tumor cells in 50% Matrigel. When average tumor size reached 120-150 mm , mice were grouped into 3 cohorts of 10 mice each and then treatment began (time zero). All treatments were given i.p. twice weekly for 5 weeks. All compounds were dosed at 60 ⁇ g/dose (approximately 3 mg/kg). Tumor volume was measured twice weekly by caliper measurement. Endpoint was tumor volume of 2,000 mm .
- Results are shown in Figure 52.
- This anti-CD38-attenuated IFNcc fusion protein construct (X355/02-HC-L0-IFNCC (A145D) IgG4, composed of SEQ ID NOS:232 (heavy chain) and 226 (light chain)) was also very effective in tumor elimination, showing that the ability of anti-CD38-attenuated IFNcc fusion protein constructs to effectively treat human myeloma in an in vivo model is not restricted to a single variable domain, IFNcc mutation or linker between the antibody and the IFN.
- the isotype control fusion protein construct showed significantly less anti-myeloma activity, consistent with the CD38-based targeting.
- the next example shows that an anti-CD38 antibody-attenuated IFNcc fusion protein construct is more effective than standard drugs used to treat multiple myeloma in the same xenograft model described above.
- mice Eight to 12 week old CB.17 SCID mice were injected subcutaneously in the flank with 1x10 NCI-H929 tumor cells in 50% Matrigel. When mean tumor sizes reached 120- 150 mm , mice were grouped into cohorts of 10 mice each and treatment began (time zero). Treatments were administered at doses and regimens described in legend. Tumor volume was measured twice weekly by caliper measurement. Endpoint was 2,000 mm . Results:
- Results are shown in Figure 53.
- the activity of the anti-CD38 targeted fusion protein construct (G005-HC-L0-IFNCC (A145D)IgG4, composed of SEQ ID NOS: 180 (heavy chain) and 134 (light chain)) was compared with standard therapies Bortezomib (Velcade), Melphalan (Alkeran), and Dexamethasone.
- the anti-CD38 targeted attenuated interferon group, 8/10 were tumor free at day 60 (closed triangles), whereas all mice in the other groups had reached endpoint by day 50.
- mice Eight to 12 week old CB.17 SCID mice were injected subcutaneously in the flank with 1x10 NCTH929 tumor cells in 50% Matrigel. When mean tumor sizes reached 120- 150 mm mice were grouped into cohorts of 10 mice each and then treatment began (TO). Treatments with the anti-CD38 antibody-attenuated Interferon fusion protein construct were administered according to following regimens: single dose on day 0 (closed triangles), two doses (on day 0 and day 3; closed squares), 4 doses (on days 0, 3, 8, and 11; closed diamonds) and 6 doses (on days 0, 3, 8, 11, 15 and 18; closed black circles).
- the vehicle treatment group is shown in grey filled circles. Tumor volume was measured twice weekly by caliper measurement. Endpoint was 2,000 mm .
- Results are shown in Figure 54.
- This study surprisingly showed that a single dose of the G005-HC-L6-IFNCC (A145G) IgG4 fusion protein construct was sufficient to eliminate established tumors in all 10/10 mice by day 15; furthermore, by day 60, no tumors had re-grown in any of the mice in this single dose group. This was true of all 4 dosing regimens with the targeted attenuated interferon. The isotype control group was only tested at the 6 dose regimen and showed considerably less activity. That a single dose of a compound can effectively cure animals of established multiple myeloma tumors is unprecedented and extremely surprising since anti-tumor therapies are typically dosed multiple times in order to observe efficacy.
- mice in this cohort showed complete tumor elimination by day 30 and no tumors had re-appeared in any of these mice by end of the study. Three of these mice had starting tumors > 1000mm . The only mouse that died from the myeloma was one which had a tumor volume of 1,800 mm at the start of treatment; it reached the 2,000 mm enpoint on the following day. This result is very surprising and no other compound has been reported to eliminate myeloma tumors of this size in any animal model.
- This effect of IFNcc on hematopoiesis can be measured ex vivo by determining the effect of IFNcc on the number of colony forming units derived from primary human bone marrow mononuclear cells.
- the IFNcc vs. the antibody- attenuated IFNcc fusion protein constructs were compared in terms of their effect on colony formation.
- Emeryville, CA Emeryville, CA
- RPMI-1640 medium 10% fetal bovine serum (FBS) (complete medium) and washed with same medium two times. After washing, cells were kept in this medium at 1.75xl0 6 cells/ml.
- Cell suspensions were diluted with MethoCult H4434 Classic medium (Stem Cell Technologies, Cat# 04434) to a final cell concentration of 0.7xl0 5 cells/ml. Cells were then mixed very well and 3 ml of this mixture was aliquoted into each tube.
- Intron A (Schering Corp. Merck, NJ) and fusion protein constructs (G005-HC-L0- IFNcc (145D) IgG4 and Isotype-HC-LO-IFNcc (145D) IgG4) were diluted in tenfold serial dilutions in complete medium and 150 ⁇ of each dilution was added to tubes containing the 3 ml of the bone marrow cells in the Methocult H4434 medium. Mixtures were plated at 1.1 ml per 35mm tissue culture dish (Stem Cell Technologies, cat#27115). Plates were then incubated in a well-humidified incubator at 37°C with 5% C0 2 for two weeks.
- Percent colony recovery for a given test substance was calculated by dividing the number of colonies per plate by the number of colonies in the plates with no added test substance. A total of three human bone marrow MNC were tested using this method.
- Both fusion protein constructs had approximately ⁇ , ⁇ fold less activity in inhibiting colony formation than wild type, free IFNa, thus confirming that the A145D mutation attenuates the IFN activity of the antibody- IFNa fusion protein constructs and suggesting that such attenuated IFN-antibody fusion protein constructs will have a superior safety profile compared to IFNa itself.
- IFNa Another activity of IFNa that can be measured ex vivo is the stimulation of cytokine and chemokine secretion.
- Normal human PBMCs were stimulated with various concentrations of IFNa vs the antibody-attenuated IFNa fusion protein construct Isotype- HC-L6-IFNa (A145G) IgGl (based on the 2D12 antibody), and measured the resulting cytokine production.
- PBMC peripheral blood mononuclear cells
- IgG- attenuated IFNa fusion protein construct Isotype-HC-L6-IFNa (A145G) IgGl; isotype antibody is 2D12
- Isotype-HC-L6-IFNa (A145G) IgGl; isotype antibody is 2D12 IgG- attenuated IFNa fusion protein construct
- Plates were then incubated overnight at 37°C in 5% C0 2 . The following day, the plates were spun down and 200 ⁇ of supernatant was collected from each well. Supernatants were kept frozen until analysis using a Luminex cytokine assay.
- Luminex Assay Using the Premix 42-plex from Millipore
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Genetics & Genomics (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Medicinal Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Immunology (AREA)
- Zoology (AREA)
- Gastroenterology & Hepatology (AREA)
- Toxicology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Virology (AREA)
- Biomedical Technology (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- General Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Wood Science & Technology (AREA)
- Pulmonology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Physics & Mathematics (AREA)
- Microbiology (AREA)
- Plant Pathology (AREA)
- Peptides Or Proteins (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Abstract
Description







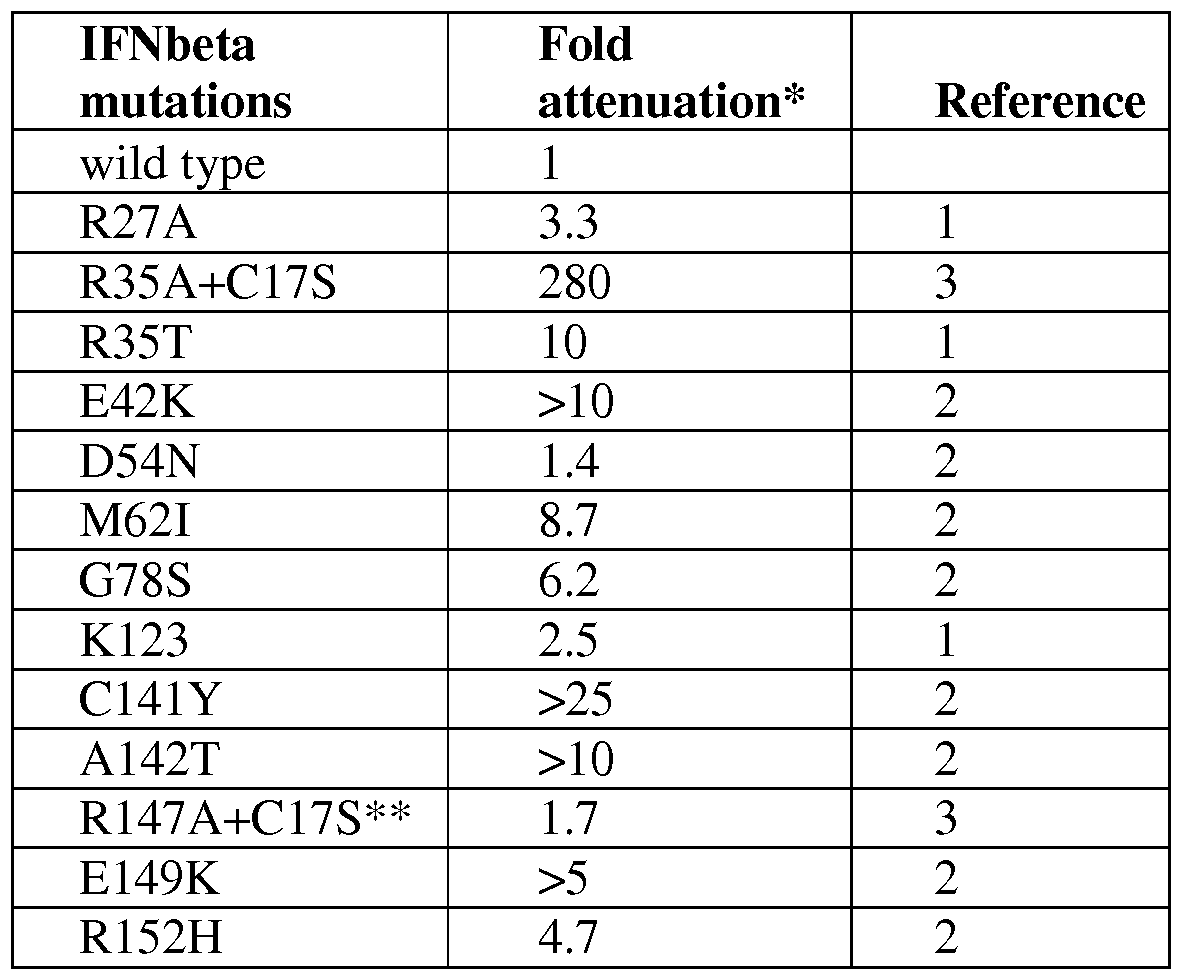






















Claims
Priority Applications (20)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP12843185.5A EP3559049A4 (en) | 2011-10-28 | 2012-10-29 | Polypeptide constructs and uses thereof |
| BR112014009925-1A BR112014009925B1 (en) | 2011-10-28 | 2012-10-29 | POLYPEPTIDE BUILDERS AND THEIR USES |
| KR1020147014249A KR102037541B1 (en) | 2011-10-28 | 2012-10-29 | Polypeptide constructs and uses thereof |
| JP2014537430A JP6184965B2 (en) | 2011-10-28 | 2012-10-29 | Polypeptide constructs and uses thereof |
| CN201280064672.8A CN104203982B (en) | 2011-10-28 | 2012-10-29 | Polypeptide construct and application thereof |
| SG11201401518TA SG11201401518TA (en) | 2011-10-28 | 2012-10-29 | Polypeptide constructs and uses thereof |
| KR1020197031072A KR102096224B1 (en) | 2011-10-28 | 2012-10-29 | Polypeptide constructs and uses thereof |
| EA201400488A EA034989B1 (en) | 2011-10-28 | 2012-10-29 | Polypeptide construct for use in treating cancer comprising attenuated alpha interferon |
| NZ623812A NZ623812B2 (en) | 2011-10-28 | 2012-10-29 | Polypeptide constructs and uses thereof |
| CN201810868213.4A CN109022465B (en) | 2011-10-28 | 2012-10-29 | Polypeptide constructs and uses thereof |
| MX2014005108A MX360741B (en) | 2011-10-28 | 2012-10-29 | Polypeptide constructs and uses thereof. |
| AU2012327877A AU2012327877B2 (en) | 2011-10-28 | 2012-10-29 | Polypeptide constructs and uses thereof |
| CA2851892A CA2851892C (en) | 2011-10-28 | 2012-10-29 | Polypeptide constructs and uses thereof |
| ZA2014/02819A ZA201402819B (en) | 2011-10-28 | 2014-04-16 | Polypeptide constructs and uses thereof |
| IL232247A IL232247B (en) | 2011-10-28 | 2014-04-24 | Polypeptide constructs and uses thereof |
| US14/262,841 US9611322B2 (en) | 2011-10-28 | 2014-04-28 | Fusions of antibodies to CD38 and attenuated interferon alpha |
| US15/194,926 US20160367695A1 (en) | 2011-10-28 | 2016-06-28 | Polypeptide constructs and uses thereof |
| US15/447,912 US10981986B2 (en) | 2011-10-28 | 2017-03-02 | Fusions of antibodies to CD38 and attenuated interferon alpha |
| AU2018200221A AU2018200221B2 (en) | 2011-10-28 | 2018-01-11 | Polypeptide constructs and uses thereof |
| IL260045A IL260045B (en) | 2011-10-28 | 2018-06-14 | Polypeptide constructs and uses thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2011904502 | 2011-10-28 | ||
| AU2011904502A AU2011904502A0 (en) | 2011-10-28 | Polypeptide constructs and uses thereof |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/262,841 Continuation US9611322B2 (en) | 2011-10-28 | 2014-04-28 | Fusions of antibodies to CD38 and attenuated interferon alpha |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2013059885A2 true WO2013059885A2 (en) | 2013-05-02 |
| WO2013059885A3 WO2013059885A3 (en) | 2014-07-17 |
Family
ID=48168677
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/AU2012/001323 WO2013059885A2 (en) | 2011-10-28 | 2012-10-29 | Polypeptide constructs and uses thereof |
Country Status (15)
| Country | Link |
|---|---|
| US (3) | US9611322B2 (en) |
| EP (1) | EP3559049A4 (en) |
| JP (3) | JP6184965B2 (en) |
| KR (2) | KR102096224B1 (en) |
| CN (2) | CN109022465B (en) |
| AU (2) | AU2012327877B2 (en) |
| BR (1) | BR112014009925B1 (en) |
| CA (1) | CA2851892C (en) |
| EA (2) | EA034989B1 (en) |
| IL (2) | IL232247B (en) |
| MX (1) | MX360741B (en) |
| NZ (1) | NZ756727A (en) |
| SG (2) | SG11201401518TA (en) |
| WO (1) | WO2013059885A2 (en) |
| ZA (1) | ZA201402819B (en) |
Cited By (82)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013107791A1 (en) | 2012-01-20 | 2013-07-25 | Vib Vzw | Targeted mutant alpha-helical bundle cytokines |
| WO2014178820A1 (en) * | 2013-04-29 | 2014-11-06 | Teva Pharmaceuticals Australia Pty Ltd. | Anti-cd38 antibodies and fusions to attenuated interferon alpha-2b |
| US8980267B2 (en) | 2012-03-03 | 2015-03-17 | Immungene Inc | Engineered antibody-interferon mutant fusion molecules |
| WO2015066450A1 (en) * | 2013-10-31 | 2015-05-07 | Sanofi | Specific anti-cd38 antibodies for treating human cancers |
| WO2015181641A3 (en) * | 2014-05-01 | 2016-01-21 | Teva Pharmaceuticals Australia Pty Ltd | Combination of lenalidomide or pomalidomide and cd38 antibody-attenuated interferon-alpha constructs, and the use thereof |
| WO2016020791A1 (en) * | 2014-08-05 | 2016-02-11 | Novartis Ag | Ckit antibody drug conjugates |
| WO2015130732A3 (en) * | 2014-02-28 | 2016-03-17 | Janssen Biotech, Inc. | Anti-cd38 antibodies for treatment of acute lymphoblastic leukemia |
| WO2016022589A3 (en) * | 2014-08-08 | 2016-04-07 | The Regents Of The University Of California | Methods for treating multiple myeloma |
| WO2016065409A1 (en) * | 2014-10-29 | 2016-05-06 | Teva Pharmaceuticals Australia Pty Ltd | INTERFERON α2B VARIANTS |
| JP2016529232A (en) * | 2013-07-19 | 2016-09-23 | ヴィブ ブイゼットダブリュー | Targeting cytokine antagonists |
| US9574012B2 (en) | 2011-07-05 | 2017-02-21 | Sanofi (China) Investment Co., Ltd. Shanghai Branch | AGR2 blocking antibody and use thereof |
| US9611322B2 (en) | 2011-10-28 | 2017-04-04 | Teva Pharmaceuticals Australia Pty Ltd | Fusions of antibodies to CD38 and attenuated interferon alpha |
| WO2017077382A1 (en) | 2015-11-06 | 2017-05-11 | Orionis Biosciences Nv | Bi-functional chimeric proteins and uses thereof |
| WO2017134305A1 (en) | 2016-02-05 | 2017-08-10 | Orionis Biosciences Nv | Bispecific signaling agents and uses thereof |
| WO2017194782A2 (en) | 2016-05-13 | 2017-11-16 | Orionis Biosciences Nv | Therapeutic targeting of non-cellular structures |
| JP2017536830A (en) * | 2014-11-26 | 2017-12-14 | ゼンコー・インコーポレイテッドXencor、 Inc. | Heterodimeric antibodies that bind to CD3 and CD38 |
| WO2018014067A1 (en) * | 2016-07-19 | 2018-01-25 | Teva Pharmaceuticals Australia Pty Ltd | Anti-cd47 combination therapy |
| WO2018014068A1 (en) * | 2016-07-19 | 2018-01-25 | Teva Pharmaceuticals Australia Pty Ltd | Attenuated type i ifn cd47 combination therapy |
| US9914759B2 (en) | 2013-07-19 | 2018-03-13 | Vib Vzw | Targeted modified TNF family members |
| US9932409B2 (en) | 2013-07-19 | 2018-04-03 | Vib Vzw | Targeted modified IL-1 family members |
| WO2018077893A1 (en) | 2016-10-24 | 2018-05-03 | Orionis Biosciences Nv | Targeted mutant interferon-gamma and uses thereof |
| WO2018144999A1 (en) | 2017-02-06 | 2018-08-09 | Orionis Biosciences, Inc. | Targeted engineered interferon and uses thereof |
| WO2018141964A1 (en) | 2017-02-06 | 2018-08-09 | Orionis Biosciences Nv | Targeted chimeric proteins and uses thereof |
| WO2018146074A1 (en) | 2017-02-07 | 2018-08-16 | Vib Vzw | Immune-cell targeted bispecific chimeric proteins and uses thereof |
| WO2018215938A1 (en) * | 2017-05-24 | 2018-11-29 | Novartis Ag | Antibody-cytokine engrafted proteins and methods of use |
| US10385135B2 (en) | 2015-11-03 | 2019-08-20 | Janssen Biotech, Inc. | Subcutaneous formulations of anti-CD38 antibodies and their uses |
| US10604580B2 (en) | 2014-09-09 | 2020-03-31 | Janssen Biotech, Inc. | Combination therapies with anti-CD38 antibodies |
| US10640542B2 (en) | 2013-07-18 | 2020-05-05 | Vib Vzw | Fusokines involving cytokines with strongly reduced receptor binding affinities |
| US10668149B2 (en) | 2015-06-22 | 2020-06-02 | Janssen Biotech, Inc. | Combination therapies for heme malignancies with anti-CD38 antibodies and survivin inhibitors |
| WO2020120730A1 (en) | 2018-12-14 | 2020-06-18 | Morphosys Ag | Antibody formulations |
| US10766965B2 (en) | 2015-05-20 | 2020-09-08 | Janssen Biotech, Inc. | Anti-CD38 antibodies for treatment of light chain amyloidosis and other CD38-positive hematological malignancies |
| US10781261B2 (en) | 2015-11-03 | 2020-09-22 | Janssen Biotech, Inc. | Subcutaneous formulations of anti-CD38 antibodies and their uses |
| US10793630B2 (en) | 2014-12-04 | 2020-10-06 | Janssen Biotech, Inc. | Anti-CD38 antibodies for treatment of acute myeloid leukemia |
| US10800851B2 (en) | 2014-02-28 | 2020-10-13 | Janssen Biotech, Inc. | Combination therapies with anti-CD38 antibodies |
| US10858451B2 (en) | 2014-03-28 | 2020-12-08 | Xencor, Inc. | Bispecific antibodies that bind to CD38 and CD3 |
| US20200399391A1 (en) * | 2019-06-10 | 2020-12-24 | Sanofi | Anti-cd38 antibodies and formulations |
| WO2021009332A1 (en) | 2019-07-18 | 2021-01-21 | Enyo Pharma | Method for decreasing adverse-effects of interferon |
| EP3618870A4 (en) * | 2017-05-01 | 2021-04-21 | The Trustees of The University of Pennsylvania | Monoclonal antibodies against alpha-synuclein fibrils |
| US11021543B2 (en) | 2015-06-24 | 2021-06-01 | Janssen Biotech, Inc. | Immune modulation and treatment of solid tumors with antibodies that specifically bind CD38 |
| US20210198375A1 (en) * | 2018-09-04 | 2021-07-01 | Nanjing Umab-Biopharma Co., Ltd. | Fusion protein and its applicaton in preparing medicine for treating tumor and/or viral infection |
| US11117975B2 (en) | 2013-04-29 | 2021-09-14 | Teva Pharmaceuticals Australia Pty Ltd | Anti-CD38 antibodies and fusions to attenuated interferon alpha-2B |
| EP3743448A4 (en) * | 2018-01-26 | 2021-11-03 | Orionis Biosciences, Inc. | Xcr1 binding agents and uses thereof |
| US11236141B2 (en) | 2016-05-13 | 2022-02-01 | Orionis Biosciences BV | Targeted mutant interferon-beta and uses thereof |
| US11248057B2 (en) | 2016-03-07 | 2022-02-15 | Vib Vzw | CD20 binding single domain antibodies |
| US11299724B2 (en) | 2016-07-14 | 2022-04-12 | Limited Liability Company Biochemical Agent | Fusion protein, polynucleotide, genetic construct, producer, preparation for regeneration of cartilage |
| US11440943B2 (en) | 2019-03-28 | 2022-09-13 | Orionis Biosciences, Inc. | Therapeutic interferon alpha 1 proteins |
| WO2022200525A1 (en) | 2021-03-26 | 2022-09-29 | Innate Pharma | Multispecific proteins comprising an nkp46-binding site, a cancer antgienge binding site fused to a cytokine for nk cell engaging |
| US11492407B2 (en) | 2016-06-14 | 2022-11-08 | Xencor, Inc. | Bispecific checkpoint inhibitor antibodies |
| US11498966B2 (en) | 2017-08-09 | 2022-11-15 | Orionis Biosciences Inc. | PD-1 and PD-L1 binding agents |
| US11518808B2 (en) | 2018-01-12 | 2022-12-06 | Amgen Inc. | Anti-PD-1 antibodies and methods of treatment |
| WO2022258691A1 (en) | 2021-06-09 | 2022-12-15 | Innate Pharma | Multispecific proteins binding to nkg2d, a cytokine receptor, a tumour antigen and cd16a |
| WO2022258662A1 (en) | 2021-06-09 | 2022-12-15 | Innate Pharma | Multispecific proteins binding to nkp46, a cytokine receptor, a tumour antigen and cd16a |
| WO2022258678A1 (en) | 2021-06-09 | 2022-12-15 | Innate Pharma | Multispecific proteins binding to nkp30, a cytokine receptor, a tumour antigen and cd16a |
| US11541103B2 (en) | 2017-08-03 | 2023-01-03 | Amgen Inc. | Interleukin-21 mutein/ anti-PD-1 antibody conjugates |
| US11566072B2 (en) | 2017-08-09 | 2023-01-31 | Orionis Biosciences, Inc. | CD8 binding agents |
| US11591401B2 (en) | 2020-08-19 | 2023-02-28 | Xencor, Inc. | Anti-CD28 compositions |
| US11618787B2 (en) | 2017-10-31 | 2023-04-04 | Janssen Biotech, Inc. | Methods of treating high risk multiple myeloma |
| US11623957B2 (en) | 2015-12-07 | 2023-04-11 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and PSMA |
| US11634506B2 (en) | 2013-01-14 | 2023-04-25 | Xencor, Inc. | Heterodimeric proteins |
| WO2023052846A3 (en) * | 2021-09-30 | 2023-05-25 | Ildong Pharmaceutical Co., Ltd. | Immunocytokine containing il-21r mutein |
| US11661455B2 (en) | 2016-02-05 | 2023-05-30 | Orionis Biosciences BV | Chimeric protein comprising an interferon alpha 2mutant and a PD-L1 binding moiety |
| US11673972B2 (en) | 2014-11-26 | 2023-06-13 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and tumor antigens |
| US11718667B2 (en) | 2013-01-14 | 2023-08-08 | Xencor, Inc. | Optimized antibody variable regions |
| US11739144B2 (en) | 2021-03-09 | 2023-08-29 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and CLDN6 |
| WO2023178253A3 (en) * | 2022-03-18 | 2023-10-19 | Igm Biosciences, Inc. | Use of a multimeric anti-pd-l1 binding molecule in combination with a monoclonal antibody therapy |
| US11814423B2 (en) | 2013-03-15 | 2023-11-14 | Xencor, Inc. | Heterodimeric proteins |
| US11859012B2 (en) | 2021-03-10 | 2024-01-02 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and GPC3 |
| US11878035B2 (en) | 2018-07-17 | 2024-01-23 | Triumvira Immunologics Usa, Inc. | T cell-antigen coupler with various construct optimizations |
| CN117430716A (en) * | 2023-10-16 | 2024-01-23 | 珠海臻谱基因科技有限公司 | Recombinant interferon drug targeting conserved HIV gp41 subunit near-membrane-end outer region and application thereof |
| US11896643B2 (en) | 2018-02-05 | 2024-02-13 | Orionis Biosciences, Inc. | Fibroblast binding agents and use thereof |
| WO2024040247A1 (en) | 2022-08-18 | 2024-02-22 | Regeneron Pharmaceuticals, Inc. | Interferon proproteins and uses thereof |
| WO2024040249A1 (en) | 2022-08-18 | 2024-02-22 | Regeneron Pharmaceuticals, Inc. | Interferon receptor agonists and uses thereof |
| US11919956B2 (en) | 2020-05-14 | 2024-03-05 | Xencor, Inc. | Heterodimeric antibodies that bind prostate specific membrane antigen (PSMA) and CD3 |
| US11945880B2 (en) | 2014-11-26 | 2024-04-02 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and tumor antigens |
| US11970545B2 (en) | 2017-10-12 | 2024-04-30 | Mcmaster University | T cell-antigen coupler with Y182T mutation and methods of uses thereof |
| US11976117B2 (en) | 2018-07-17 | 2024-05-07 | Mcmaster University | T cell-antigen coupler with various construct optimizations |
| US11981737B2 (en) | 2016-11-18 | 2024-05-14 | Hoffmann-La Roche Inc. | Anti-HLA-G antibodies and use thereof |
| US12016923B2 (en) | 2021-06-01 | 2024-06-25 | Triumvira Immunologics Usa, Inc. | Claudin 18.2 T cell-antigen couplers and uses thereof |
| US12054545B2 (en) | 2016-06-28 | 2024-08-06 | Xencor, Inc. | Heterodimeric antibodies that bind somatostatin receptor 2 |
| US12084497B2 (en) | 2018-08-08 | 2024-09-10 | Orionis Biosciences, Inc. | SIRP1α targeted chimeric proteins and uses thereof |
| US12091463B2 (en) | 2017-08-09 | 2024-09-17 | Orionis Biosciences, Inc. | Clec9A binding agents comprising a recombinant heavy-chain only antibody (VHH) |
| US12129309B2 (en) | 2022-05-04 | 2024-10-29 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and CD38 |
Families Citing this family (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1578397B1 (en) * | 2002-11-15 | 2012-12-26 | Genmab A/S | Human monoclonal antibodies against cd25 |
| EP1914242A1 (en) * | 2006-10-19 | 2008-04-23 | Sanofi-Aventis | Novel anti-CD38 antibodies for the treatment of cancer |
| BR112016018100A2 (en) * | 2014-02-07 | 2018-02-20 | Univ Mcmaster | trifunctional t-cell antigen coupler, methods and uses thereof |
| CA2988619A1 (en) * | 2015-06-12 | 2016-12-15 | Immungene, Inc. | Focused interferon immunotherapy for treatment of cancer |
| RU2611685C2 (en) * | 2015-07-20 | 2017-02-28 | Илья Владимирович Духовлинов | Humanized monoclonal antibody specific to syndecan-1 |
| CN105254717B (en) * | 2015-08-18 | 2018-08-24 | 中山大学 | The polypeptide combined with CD34 molecular specificities and its application |
| CN106065033A (en) * | 2016-03-17 | 2016-11-02 | 中国药科大学 | The preparation of a kind of cytokine fusion antibody and application thereof |
| US11499168B2 (en) * | 2016-04-25 | 2022-11-15 | Universitat Basel | Allele editing and applications thereof |
| CN106854248A (en) * | 2016-12-09 | 2017-06-16 | 中国药科大学 | A kind of preparation and its application of cell factor mutant fusion antibody |
| EA201991514A1 (en) * | 2016-12-21 | 2019-12-30 | Сефалон, Инк. | ANTIBODIES THAT SPECIALLY CONTACT THE IL-15 AND THEIR APPLICATION |
| US10767164B2 (en) | 2017-03-30 | 2020-09-08 | The Research Foundation For The State University Of New York | Microenvironments for self-assembly of islet organoids from stem cells differentiation |
| EP3648786A4 (en) * | 2017-07-03 | 2021-12-15 | Torque Therapeutics, Inc. | Fusion molecules targeting immune regulatory cells and uses thereof |
| WO2019023156A1 (en) * | 2017-07-24 | 2019-01-31 | Rutgers, The State University Of New Jersey | Phosphatidylserine targeting fusion molecules and methods for their use |
| WO2019035938A1 (en) | 2017-08-16 | 2019-02-21 | Elstar Therapeutics, Inc. | Multispecific molecules that bind to bcma and uses thereof |
| SG11202012991QA (en) * | 2018-07-10 | 2021-01-28 | Sanofi Sa | Combination therapies against cancer targeting cd38 and tgf-beta |
| US20220153801A1 (en) * | 2019-03-28 | 2022-05-19 | Orionis Biosciences, Inc. | Clec9a-based chimeric protein complexes |
| EP3958898A1 (en) * | 2019-04-23 | 2022-03-02 | Sanofi | Anti-cd38 antibodies and formulations |
| WO2021239020A1 (en) * | 2020-05-26 | 2021-12-02 | 上海邦耀生物科技有限公司 | Immunotherapy method for combining chimeric antigen receptor and type i interferon and application thereof |
| WO2022072495A2 (en) * | 2020-10-01 | 2022-04-07 | Academia Sinica | Potent neutralizing antibodies for prevention and treatment of covid-19 |
| CN115057946B (en) * | 2022-06-30 | 2023-05-16 | 四川省医学科学院·四川省人民医院 | Application of interferon in preparation of medicines for resisting broad-spectrum influenza virus and coronavirus |
| WO2024104412A1 (en) * | 2022-11-16 | 2024-05-23 | I-Mab Biopharma Co., Ltd. | Attenuated interferon proteins and fragments and multifunctional polypeptides and conjugates |
Citations (61)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1990005144A1 (en) | 1988-11-11 | 1990-05-17 | Medical Research Council | Single domain ligands, receptors comprising said ligands, methods for their production, and use of said ligands and receptors |
| US5225539A (en) | 1986-03-27 | 1993-07-06 | Medical Research Council | Recombinant altered antibodies and methods of making altered antibodies |
| EP0706799A2 (en) | 1994-09-16 | 1996-04-17 | MERCK PATENT GmbH | Immunoconjugates II |
| US5545533A (en) | 1991-05-25 | 1996-08-13 | Boehringer Mannheim Gmbh | Monoclonal antibodies against c-kit and method of detecting a malignancy using c-kit specific antibodies |
| US5558864A (en) | 1991-03-06 | 1996-09-24 | Merck Patent Gesellschaft Mit Beschrankter Haftung | Humanized and chimeric anti-epidermal growth factor receptor monoclonal antibodies |
| US5565332A (en) | 1991-09-23 | 1996-10-15 | Medical Research Council | Production of chimeric antibodies - a combinatorial approach |
| US5585089A (en) | 1988-12-28 | 1996-12-17 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| WO1997000271A1 (en) | 1995-06-14 | 1997-01-03 | The Regents Of The University Of California | Novel high affinity human antibodies to tumor antigens |
| US5677171A (en) | 1988-01-12 | 1997-10-14 | Genentech, Inc. | Monoclonal antibodies directed to the HER2 receptor |
| US5824307A (en) | 1991-12-23 | 1998-10-20 | Medimmune, Inc. | Human-murine chimeric antibodies against respiratory syncytial virus |
| US5837821A (en) | 1992-11-04 | 1998-11-17 | City Of Hope | Antibody construct |
| US5843439A (en) | 1992-11-13 | 1998-12-01 | Anderson; Darrell R. | Therapeutic application of chimeric and radiolabeled antibodies to human B lymphocyte restricted differentiation antigen for treatment of B cell lymphoma |
| US5844093A (en) | 1994-03-17 | 1998-12-01 | Merck Patent Gesellschaft Mit Beschrankter Haftung | Anti-EGFR single-chain Fvs and anti-EGFR antibodies |
| US5976531A (en) | 1990-04-19 | 1999-11-02 | The Dow Chemical Company | Composite antibodies of human subgroup IV light chain capable of binding to tag-72 |
| US6054297A (en) | 1991-06-14 | 2000-04-25 | Genentech, Inc. | Humanized antibodies and methods for making them |
| US6071519A (en) | 1995-06-07 | 2000-06-06 | Innogenetics N.V. | Immunotoxins specific for CD86 expressing cells |
| WO2000042072A2 (en) | 1999-01-15 | 2000-07-20 | Genentech, Inc. | Polypeptide variants with altered effector function |
| US6277375B1 (en) | 1997-03-03 | 2001-08-21 | Board Of Regents, The University Of Texas System | Immunoglobulin-like domains with increased half-lives |
| US6300064B1 (en) | 1995-08-18 | 2001-10-09 | Morphosys Ag | Protein/(poly)peptide libraries |
| WO2001097844A1 (en) | 2000-06-22 | 2001-12-27 | Idec Pharmaceuticals Corporation | Bispecific fusion protein and method of use for enhancing effector cell killing of target cells |
| US20020142358A1 (en) | 2000-04-28 | 2002-10-03 | Toshifumi Mikayama | Human anti-CD40 antibodies and methods of making and using same |
| US6602684B1 (en) | 1998-04-20 | 2003-08-05 | Glycart Biotechnology Ag | Glycosylation engineering of antibodies for improving antibody-dependent cellular cytotoxicity |
| US20030211100A1 (en) | 2001-11-09 | 2003-11-13 | Vahe Bedian | Antibodies to CD40 |
| US20040006215A1 (en) | 2002-01-09 | 2004-01-08 | Tibor Keler | Human monoclonal antibodies against CD30 |
| US20040071696A1 (en) | 2002-04-05 | 2004-04-15 | The Regents Of The University Of California | Bispecific single chain Fv antibody molecules and methods of use thereof |
| US6803039B2 (en) | 2000-05-18 | 2004-10-12 | Japan Tobacco Inc. | Human monoclonal antibody against a costimulatory signal transduction molecule AILIM |
| US20050208048A1 (en) | 2003-12-15 | 2005-09-22 | Dendreon Corporation | HLA-DR specific antibodies, compositions and methods |
| US20060083736A1 (en) | 2004-10-15 | 2006-04-20 | Seattle Genetics, Inc. | Anti-CD70 antibody and its use for the treatment and prevention of cancer and immune disorders |
| US20060099205A1 (en) | 2002-04-05 | 2006-05-11 | The Regents Of The University Of California | Bispecific single chain FV antibody molecules and methods of use thereof |
| US7083784B2 (en) | 2000-12-12 | 2006-08-01 | Medimmune, Inc. | Molecules with extended half-lives, compositions and uses thereof |
| US7166697B1 (en) | 1998-03-06 | 2007-01-23 | Diatech Pty. Ltd. | V-like domain binding molecules |
| WO2007029041A2 (en) | 2005-09-09 | 2007-03-15 | Imperial Innovations Limited | Interferon lambda therapy for treatment of respiratory diseases |
| US7202346B2 (en) | 2002-07-03 | 2007-04-10 | Immunogen Inc. | Antibodies to non-shed Muc1 and Muc16, and uses thereof |
| US7217797B2 (en) | 2002-10-15 | 2007-05-15 | Pdl Biopharma, Inc. | Alteration of FcRn binding affinities or serum half-lives of antibodies by mutagenesis |
| US7262278B2 (en) | 2001-10-15 | 2007-08-28 | Kirin Beer Kabushiki Kaisha | Anti-HLA-DR antibody |
| WO2008006554A2 (en) | 2006-07-11 | 2008-01-17 | Greenovation Biotech Gmbh | Glyco-engineered antibodies |
| US7326681B2 (en) | 2000-06-28 | 2008-02-05 | Glycofi, Inc. | Methods for producing modified glycoproteins |
| US7388081B2 (en) | 1998-12-09 | 2008-06-17 | Dfb Biotech, Inc. | Method for manufacturing glycoproteins having human-type glycosylation |
| US7456257B2 (en) | 2003-02-18 | 2008-11-25 | Merck Patent Gmbh | Fusion proteins of interferon alpha muteins with improved properties |
| US20090068175A1 (en) | 2002-03-01 | 2009-03-12 | Xencor, Inc. | Optimized FC Variants and Methods for Their Generation |
| US20090092599A1 (en) | 2002-09-27 | 2009-04-09 | Xencor, Inc. | Optimized Fc variants and methods for their generation |
| WO2009080832A1 (en) | 2007-12-26 | 2009-07-02 | Biotest Ag | Methods and agents for improving targeting of cd138 expressing tumor cells |
| US7557189B2 (en) | 2002-11-07 | 2009-07-07 | Immunogen Inc. | Anti-CD33 antibodies and method for treatment of acute myeloid leukemia using the same |
| US7566771B1 (en) | 1989-12-21 | 2009-07-28 | Celltech R&D Limited | Humanised antibodies |
| US7700740B2 (en) | 2001-11-21 | 2010-04-20 | Celltech R&D Ltd | Antibodies to CD83 |
| US7723485B2 (en) | 2007-05-08 | 2010-05-25 | Genentech, Inc. | Cysteine engineered anti-MUC16 antibodies and antibody drug conjugates |
| US7732578B2 (en) | 2001-07-12 | 2010-06-08 | Arrowsmith Technology Licensing Llc | Super humanized antibodies |
| US7736647B2 (en) | 2005-06-15 | 2010-06-15 | Monoclonal Antibodies Therapeutics | Anti-CD71 monoclonal antibodies and uses thereof for treating malignant tumor cells |
| WO2010105290A1 (en) | 2009-03-16 | 2010-09-23 | Cephalon Australia Pty Ltd | Humanised antibodies with anti-tumour activity |
| US7829673B2 (en) | 2005-03-23 | 2010-11-09 | Genmab A/S | Antibodies against CD38 for treatment of multiple myeloma |
| US20100297076A1 (en) | 2007-09-21 | 2010-11-25 | The Regents Of The University Of California | Targeted interferon demonstrates potent apoptotic and anti-tumor activities |
| US7910104B2 (en) | 2003-11-01 | 2011-03-22 | Merck Patent Gmbh | Modified anti-CD52 antibody |
| US7915391B2 (en) | 2006-04-24 | 2011-03-29 | Amgen Inc. | Humanized c-Kit antibody |
| US20110178278A1 (en) | 2008-03-14 | 2011-07-21 | Haegel Helene | Antibody Against the CSF-1R |
| US7998738B2 (en) | 2000-07-31 | 2011-08-16 | The General Hospital Corporation | Integrin-binding antibodies |
| US20110243929A1 (en) | 2007-05-07 | 2011-10-06 | Medlmmune, Llc | Anti-icos antibodies and their use in treatment of oncology, transplantation and autoimmune disease |
| US8039593B2 (en) | 2005-10-31 | 2011-10-18 | Duke University | Antibodies and immunotoxins that target human glycoprotein NMB |
| US8124738B2 (en) | 2005-09-26 | 2012-02-28 | Medarex, Inc. | Human monoclonal antibodies to CD70 |
| US20120082670A1 (en) | 2010-10-04 | 2012-04-05 | Boehringer Ingelheim International Gmbh | Cd33 binding agents |
| US20120100152A1 (en) | 2009-05-13 | 2012-04-26 | Genzyme Corporation | Anti-human cd52 immunoglobulins |
| US8206715B2 (en) | 2010-05-04 | 2012-06-26 | Five Prime Therapeutics, Inc. | Antibodies that bind colony stimulating factor 1 receptor (CSF1R) |
Family Cites Families (79)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB2148299B (en) | 1983-09-01 | 1988-01-06 | Hybritech Inc | Antibody compositions of therapeutic agents having an extended serum half-life |
| US4908431A (en) | 1986-01-22 | 1990-03-13 | Temple University-Of The Commonwealth System Of Higher Education | Monoclonal antibodies to human kininogen and methods of preparing same |
| US5846534A (en) | 1988-02-12 | 1998-12-08 | British Technology Group Limited | Antibodies to the antigen campath-1 |
| AU4128089A (en) | 1988-09-15 | 1990-03-22 | Rorer International (Overseas) Inc. | Monoclonal antibodies specific to human epidermal growth factor receptor and therapeutic methods employing same |
| GB9022543D0 (en) | 1990-10-17 | 1990-11-28 | Wellcome Found | Antibody production |
| US5650150A (en) * | 1990-11-09 | 1997-07-22 | Gillies; Stephen D. | Recombinant antibody cytokine fusion proteins |
| DK0628639T3 (en) | 1991-04-25 | 2000-01-24 | Chugai Pharmaceutical Co Ltd | Reconstituted human antibody to human interleukin-6 receptor |
| US6042828A (en) | 1992-09-07 | 2000-03-28 | Kyowa Hakko Kogyo Co., Ltd. | Humanized antibodies to ganglioside GM2 |
| US5441734A (en) * | 1993-02-25 | 1995-08-15 | Schering Corporation | Metal-interferon-alpha crystals |
| US5840299A (en) | 1994-01-25 | 1998-11-24 | Athena Neurosciences, Inc. | Humanized antibodies against leukocyte adhesion molecule VLA-4 |
| US20020164788A1 (en) | 1994-12-02 | 2002-11-07 | The Wellcome Foundation Limited | Humanized antibodies to CD38 |
| US5723125A (en) | 1995-12-28 | 1998-03-03 | Tanox Biosystems, Inc. | Hybrid with interferon-alpha and an immunoglobulin Fc linked through a non-immunogenic peptide |
| DE69737188T2 (en) | 1996-05-04 | 2007-10-11 | Astrazeneca Ab | MONOCLONAL ANTIBODY AGAINST CEA, CONJUGATES CONTAINING THIS ANTIBODY AND THEIR THERAPEUTIC USE IN AN ADEPT SYSTEM |
| US6417337B1 (en) | 1996-10-31 | 2002-07-09 | The Dow Chemical Company | High affinity humanized anti-CEA monoclonal antibodies |
| US6872568B1 (en) | 1997-03-17 | 2005-03-29 | Human Genome Sciences, Inc. | Death domain containing receptor 5 antibodies |
| GB9709421D0 (en) * | 1997-05-10 | 1997-07-02 | Zeneca Ltd | Chemical compounds |
| CN1269805A (en) | 1997-07-14 | 2000-10-11 | 博尔德生物技术公司 | Derivatives of growth hormone and related proteins |
| BR9915548A (en) | 1998-10-16 | 2001-08-14 | Biogen Inc | Interferon-beta fusion proteins and uses |
| US7223397B1 (en) | 1999-01-07 | 2007-05-29 | Research Development Foundation | Potentiation of anti-CD38-Immunotoxin cytotoxicity |
| US7355015B1 (en) | 1999-03-12 | 2008-04-08 | Georgetown University School Of Medicine | Matriptase, a serine protease and its applications |
| US6946129B1 (en) | 1999-06-08 | 2005-09-20 | Seattle Genetics, Inc. | Recombinant anti-CD40 antibody and uses thereof |
| WO2001058957A2 (en) | 2000-02-11 | 2001-08-16 | Lexigen Pharmaceuticals Corp. | Enhancing the circulating half-life of antibody-based fusion proteins |
| US7521047B2 (en) | 2000-05-12 | 2009-04-21 | Gpc Biotech Ag | Human polypeptides causing or leading to the killing of cells including lymphoid tumor cells |
| EP1174440A1 (en) | 2000-07-19 | 2002-01-23 | U-BISys B.V. | A selectively-expressed epitope on the human CD38 molecule detected by a phage display library-derived human scFv antibody fragment |
| BRPI0116728B1 (en) | 2001-01-05 | 2018-10-30 | Abgenix Inc | insulin-like growth factor i receptor antibodies |
| FR2823764B1 (en) | 2001-04-24 | 2003-12-12 | Genodyssee | NOVEL POLYNUCLEOTIDES AND POLYPEPTIDES OF THE IFN ALPHA-17 GENE |
| US20020193569A1 (en) | 2001-06-04 | 2002-12-19 | Idec Pharmaceuticals Corporation | Bispecific fusion protein and method of use for enhancing effector cell killing of target cells |
| EP2192130A1 (en) | 2001-07-03 | 2010-06-02 | Genentech, Inc. | Human DR4 antibodies and uses thereof |
| AR035119A1 (en) | 2001-08-16 | 2004-04-14 | Lilly Co Eli | ANTI-HTNFSF13B HUMAN ANTI-BODIES |
| DE60333732D1 (en) | 2002-03-01 | 2010-09-23 | Immunomedics Inc | INTERNALIZATION OF ANTI CD74 MONOCLONAL ANTIBODIES AND THEIR USES |
| CA2498284A1 (en) | 2002-09-09 | 2004-03-18 | Nautilus Biotech | Rational directed protein evolution using two-dimensional rational mutagenesis scanning |
| CA2498319A1 (en) * | 2002-09-09 | 2004-03-18 | Nautilus Biotech | Rational evolution of cytokines for higher stability, the cytokines and encoding nucleic acid molecules |
| US7709610B2 (en) | 2003-05-08 | 2010-05-04 | Facet Biotech Corporation | Therapeutic use of anti-CS1 antibodies |
| PT1684869E (en) | 2003-11-04 | 2011-09-16 | Novartis Vaccines & Diagnostic | Methods of therapy for b cell-related cancers |
| SI2511297T1 (en) | 2004-02-06 | 2015-07-31 | Morphosys Ag | Anti-CD38 human antibodies and uses therefor |
| ES2541489T3 (en) | 2004-02-06 | 2015-07-21 | Morphosys Ag | Human anti-CD38 antibodies and uses for them |
| US8034352B2 (en) | 2005-04-06 | 2011-10-11 | Ibc Pharmaceuticals, Inc. | Tetrameric cytokines with improved biological activity |
| WO2005086875A2 (en) | 2004-03-11 | 2005-09-22 | City Of Hope | A humanized anti-cea t84.66 antibody and uses thereof |
| US7670595B2 (en) | 2004-06-28 | 2010-03-02 | Merck Patent Gmbh | Fc-interferon-beta fusion proteins |
| EP1791868B1 (en) | 2004-07-01 | 2011-02-23 | Novo Nordisk A/S | Antibodies binding to receptors kir2dl1, -2, 3 but not kir2ds4 and their therapeutic use |
| CA2576030A1 (en) | 2004-08-09 | 2006-02-23 | Alios Biopharma Inc. | Synthetic hyperglycosylated, protease-resistant polypeptide variants, oral formulations and methods of using the same |
| DE602005026869D1 (en) * | 2004-11-12 | 2011-04-21 | Hayashibara Biochem Lab | INTERFERON PROTEIN MUTANES AND ITS APPLICATION |
| US20090123950A1 (en) | 2005-05-24 | 2009-05-14 | Morphosys Ag | Generation And Profiling Of Fully Human Hucal Gold®-Derived Therapeutic Antibodies Specific For Human CD38 |
| CN101184771A (en) | 2005-05-26 | 2008-05-21 | 先灵公司 | Interferon-igg fusion |
| EP1909822B1 (en) | 2005-06-29 | 2013-09-25 | Yeda Research And Development Co., Ltd. | Recombinant interferon alpha 2 (ifn alpha 2) mutants |
| EP2388277A3 (en) | 2005-07-18 | 2013-03-20 | Amgen, Inc | Human anti-B7RP1 neutralizing antibodies |
| US20070190068A1 (en) * | 2005-10-10 | 2007-08-16 | Richard Hart | uPAR-binding molecule-drug conjugates and uses thereof |
| BRPI0618399B1 (en) | 2005-10-12 | 2023-10-03 | Morphosys Ag | HUMAN ANTI-CD38 SPECIFIC ANTIBODY, NUCLEIC ACID COMPOSITION, EXPRESSION VECTOR, PHARMACEUTICAL COMPOSITION, USE OF THE ANTIBODY AND USE OF A PHARMACEUTICAL COMPOSITION |
| GB0525214D0 (en) | 2005-12-12 | 2006-01-18 | Bioinvent Int Ab | Biological materials and uses thereof |
| CN101045156B (en) * | 2006-03-29 | 2012-05-02 | 刘宏利 | Special target medicine and its use |
| FR2905375A1 (en) | 2006-08-29 | 2008-03-07 | Biomethodes Sa | Thermostable variants of human interferon alpha useful for preparing antiviral, antiproliferative or immunomodulatory medicaments have one or more selected amino acid substitutions |
| CA2660795C (en) | 2006-09-18 | 2014-11-18 | Xencor, Inc. | Optimized antibodies that target hm1.24 |
| EA034877B1 (en) | 2006-09-26 | 2020-04-01 | Генмаб А/С | Method of combination therapy of cd38-expressing tumors and therapeutic combination for use in said method |
| EP1914242A1 (en) | 2006-10-19 | 2008-04-23 | Sanofi-Aventis | Novel anti-CD38 antibodies for the treatment of cancer |
| DE102007001370A1 (en) | 2007-01-09 | 2008-07-10 | Curevac Gmbh | RNA-encoded antibodies |
| US11535673B2 (en) | 2007-04-05 | 2022-12-27 | President and Fellows of Harvard CoHege | Chimeric activators: quantitatively designed protein therapeutics and uses thereof |
| US20110045007A1 (en) | 2007-05-31 | 2011-02-24 | Genmab A/S | Fusion or linked proteins with extended half life |
| US8248984B2 (en) | 2007-06-20 | 2012-08-21 | I Squared Llc | System and method for interfacing devices |
| CA2695382A1 (en) * | 2007-08-01 | 2009-02-05 | The Government Of The United States Of America, As Represented By The Se Cretary, Department Of Health Of Human Services, National Institutes Of | A fold-back diabody diphtheria toxin immunotoxin and methods of use |
| WO2009017838A2 (en) * | 2007-08-01 | 2009-02-05 | Exelixis, Inc. | Combinations of jak-2 inhibitors and other agents |
| US20090076249A1 (en) | 2007-09-19 | 2009-03-19 | Michel De Weers | Antibodies against CD38 for treatment of multiple myeloma |
| WO2009073975A1 (en) | 2007-12-10 | 2009-06-18 | National Research Council Of Canada | Production of recombinant interferon proteins |
| US8092804B2 (en) | 2007-12-21 | 2012-01-10 | Medimmune Limited | Binding members for interleukin-4 receptor alpha (IL-4Rα)-173 |
| DK2238168T3 (en) | 2007-12-26 | 2014-08-25 | Biotest Ag | Means targeted to CD138 AND APPLICATIONS THEREOF |
| EP2313435A4 (en) | 2008-07-01 | 2012-08-08 | Aveo Pharmaceuticals Inc | Fibroblast growth factor receptor 3 (fgfr3) binding proteins |
| DK2580243T3 (en) | 2010-06-09 | 2020-01-13 | Genmab As | ANTIBODIES AGAINST HUMAN CD38 |
| US8909257B2 (en) | 2010-06-19 | 2014-12-09 | Qualcomm Incorporated | Positioning protocol conveyance |
| PL2621531T3 (en) | 2010-09-27 | 2017-07-31 | Morphosys Ag | Anti-cd38 antibody and lenalidomide or bortezomib for the treatment of multiple myeloma and nhl |
| CN103429261A (en) | 2010-12-22 | 2013-12-04 | 塞法隆澳大利亚股份有限公司 | Modified antibody with improved half-life |
| JOP20210044A1 (en) | 2010-12-30 | 2017-06-16 | Takeda Pharmaceuticals Co | Anti-cd38 antibodies |
| CN109022465B (en) * | 2011-10-28 | 2022-04-29 | 特瓦制药澳大利亚私人有限公司 | Polypeptide constructs and uses thereof |
| AU2013211059B8 (en) | 2012-01-20 | 2017-12-21 | Centre Hospitalier Regional Universitaire De Montpellier | Targeted mutant alpha-helical bundle cytokines |
| CA2866126A1 (en) | 2012-03-03 | 2013-09-12 | Immungene, Inc. | Engineered antibody-interferon mutant fusion molecules |
| US9783589B2 (en) | 2012-08-13 | 2017-10-10 | Immungene Inc | Engineered antibody-interferon fusion molecules for treatment of autoimmune diseases |
| CN105051620A (en) | 2013-01-25 | 2015-11-11 | 拜耳材料科技股份有限公司 | Security element having volume hologram and printed feature |
| DK3677591T5 (en) | 2013-04-29 | 2024-08-26 | Teva Pharmaceuticals Australia Pty Ltd | Anti-CD38 antibodies and fusions to attenuated interferon alpha-2b |
| US11117975B2 (en) | 2013-04-29 | 2021-09-14 | Teva Pharmaceuticals Australia Pty Ltd | Anti-CD38 antibodies and fusions to attenuated interferon alpha-2B |
| UA119352C2 (en) | 2014-05-01 | 2019-06-10 | Тева Фармасьютикалз Острейліа Пті Лтд | Combination of lenalidomide or pomalidomide and cd38 antibody-attenuated interferon-alpha constructs, and the use thereof |
| PE20170908A1 (en) | 2014-10-29 | 2017-07-12 | Teva Pharmaceuticals Australia Pty Ltd | VARIANTS OF INTERFERON a2b |
-
2012
- 2012-10-29 CN CN201810868213.4A patent/CN109022465B/en active Active
- 2012-10-29 EP EP12843185.5A patent/EP3559049A4/en active Pending
- 2012-10-29 CN CN201280064672.8A patent/CN104203982B/en active Active
- 2012-10-29 CA CA2851892A patent/CA2851892C/en active Active
- 2012-10-29 JP JP2014537430A patent/JP6184965B2/en active Active
- 2012-10-29 AU AU2012327877A patent/AU2012327877B2/en active Active
- 2012-10-29 SG SG11201401518TA patent/SG11201401518TA/en unknown
- 2012-10-29 BR BR112014009925-1A patent/BR112014009925B1/en active IP Right Grant
- 2012-10-29 SG SG10201603411WA patent/SG10201603411WA/en unknown
- 2012-10-29 WO PCT/AU2012/001323 patent/WO2013059885A2/en active Application Filing
- 2012-10-29 MX MX2014005108A patent/MX360741B/en active IP Right Grant
- 2012-10-29 NZ NZ756727A patent/NZ756727A/en unknown
- 2012-10-29 EA EA201400488A patent/EA034989B1/en not_active IP Right Cessation
- 2012-10-29 KR KR1020197031072A patent/KR102096224B1/en active IP Right Grant
- 2012-10-29 EA EA201700111A patent/EA201700111A1/en unknown
- 2012-10-29 KR KR1020147014249A patent/KR102037541B1/en active IP Right Grant
-
2014
- 2014-04-16 ZA ZA2014/02819A patent/ZA201402819B/en unknown
- 2014-04-24 IL IL232247A patent/IL232247B/en active IP Right Grant
- 2014-04-28 US US14/262,841 patent/US9611322B2/en active Active
-
2016
- 2016-06-28 US US15/194,926 patent/US20160367695A1/en not_active Abandoned
-
2017
- 2017-03-02 US US15/447,912 patent/US10981986B2/en active Active
- 2017-07-26 JP JP2017144182A patent/JP2017193580A/en active Pending
-
2018
- 2018-01-11 AU AU2018200221A patent/AU2018200221B2/en active Active
- 2018-06-14 IL IL260045A patent/IL260045B/en active IP Right Grant
-
2019
- 2019-09-02 JP JP2019159406A patent/JP7111669B2/en active Active
Patent Citations (71)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5225539A (en) | 1986-03-27 | 1993-07-06 | Medical Research Council | Recombinant altered antibodies and methods of making altered antibodies |
| US5677171A (en) | 1988-01-12 | 1997-10-14 | Genentech, Inc. | Monoclonal antibodies directed to the HER2 receptor |
| US5772997A (en) | 1988-01-12 | 1998-06-30 | Genentech, Inc. | Monoclonal antibodies directed to the HER2 receptor |
| US5770195A (en) | 1988-01-12 | 1998-06-23 | Genentech, Inc. | Monoclonal antibodies directed to the her2 receptor |
| WO1990005144A1 (en) | 1988-11-11 | 1990-05-17 | Medical Research Council | Single domain ligands, receptors comprising said ligands, methods for their production, and use of said ligands and receptors |
| US6248516B1 (en) | 1988-11-11 | 2001-06-19 | Medical Research Council | Single domain ligands, receptors comprising said ligands methods for their production, and use of said ligands and receptors |
| US5693761A (en) | 1988-12-28 | 1997-12-02 | Protein Design Labs, Inc. | Polynucleotides encoding improved humanized immunoglobulins |
| US6180370B1 (en) | 1988-12-28 | 2001-01-30 | Protein Design Labs, Inc. | Humanized immunoglobulins and methods of making the same |
| US5585089A (en) | 1988-12-28 | 1996-12-17 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| US7566771B1 (en) | 1989-12-21 | 2009-07-28 | Celltech R&D Limited | Humanised antibodies |
| US5976531A (en) | 1990-04-19 | 1999-11-02 | The Dow Chemical Company | Composite antibodies of human subgroup IV light chain capable of binding to tag-72 |
| US5558864A (en) | 1991-03-06 | 1996-09-24 | Merck Patent Gesellschaft Mit Beschrankter Haftung | Humanized and chimeric anti-epidermal growth factor receptor monoclonal antibodies |
| US5545533A (en) | 1991-05-25 | 1996-08-13 | Boehringer Mannheim Gmbh | Monoclonal antibodies against c-kit and method of detecting a malignancy using c-kit specific antibodies |
| US6054297A (en) | 1991-06-14 | 2000-04-25 | Genentech, Inc. | Humanized antibodies and methods for making them |
| US5565332A (en) | 1991-09-23 | 1996-10-15 | Medical Research Council | Production of chimeric antibodies - a combinatorial approach |
| US5824307A (en) | 1991-12-23 | 1998-10-20 | Medimmune, Inc. | Human-murine chimeric antibodies against respiratory syncytial virus |
| US5837821A (en) | 1992-11-04 | 1998-11-17 | City Of Hope | Antibody construct |
| US5843439A (en) | 1992-11-13 | 1998-12-01 | Anderson; Darrell R. | Therapeutic application of chimeric and radiolabeled antibodies to human B lymphocyte restricted differentiation antigen for treatment of B cell lymphoma |
| US5844093A (en) | 1994-03-17 | 1998-12-01 | Merck Patent Gesellschaft Mit Beschrankter Haftung | Anti-EGFR single-chain Fvs and anti-EGFR antibodies |
| EP0706799A2 (en) | 1994-09-16 | 1996-04-17 | MERCK PATENT GmbH | Immunoconjugates II |
| US6071519A (en) | 1995-06-07 | 2000-06-06 | Innogenetics N.V. | Immunotoxins specific for CD86 expressing cells |
| US5977322A (en) | 1995-06-14 | 1999-11-02 | The Regents Of The University Of California | High affinity human antibodies to tumor antigens |
| US6512097B1 (en) | 1995-06-14 | 2003-01-28 | The Regents Of The University Of California | High affinity human antibodies to tumor antigens |
| WO1997000271A1 (en) | 1995-06-14 | 1997-01-03 | The Regents Of The University Of California | Novel high affinity human antibodies to tumor antigens |
| US6300064B1 (en) | 1995-08-18 | 2001-10-09 | Morphosys Ag | Protein/(poly)peptide libraries |
| US6277375B1 (en) | 1997-03-03 | 2001-08-21 | Board Of Regents, The University Of Texas System | Immunoglobulin-like domains with increased half-lives |
| US6821505B2 (en) | 1997-03-03 | 2004-11-23 | Board Of Regents, The University Of Texas System | Immunoglobin-like domains with increased half lives |
| US7166697B1 (en) | 1998-03-06 | 2007-01-23 | Diatech Pty. Ltd. | V-like domain binding molecules |
| US6602684B1 (en) | 1998-04-20 | 2003-08-05 | Glycart Biotechnology Ag | Glycosylation engineering of antibodies for improving antibody-dependent cellular cytotoxicity |
| US7388081B2 (en) | 1998-12-09 | 2008-06-17 | Dfb Biotech, Inc. | Method for manufacturing glycoproteins having human-type glycosylation |
| WO2000042072A2 (en) | 1999-01-15 | 2000-07-20 | Genentech, Inc. | Polypeptide variants with altered effector function |
| US20020142358A1 (en) | 2000-04-28 | 2002-10-03 | Toshifumi Mikayama | Human anti-CD40 antibodies and methods of making and using same |
| US6803039B2 (en) | 2000-05-18 | 2004-10-12 | Japan Tobacco Inc. | Human monoclonal antibody against a costimulatory signal transduction molecule AILIM |
| WO2001097844A1 (en) | 2000-06-22 | 2001-12-27 | Idec Pharmaceuticals Corporation | Bispecific fusion protein and method of use for enhancing effector cell killing of target cells |
| US7326681B2 (en) | 2000-06-28 | 2008-02-05 | Glycofi, Inc. | Methods for producing modified glycoproteins |
| US7998738B2 (en) | 2000-07-31 | 2011-08-16 | The General Hospital Corporation | Integrin-binding antibodies |
| US7083784B2 (en) | 2000-12-12 | 2006-08-01 | Medimmune, Inc. | Molecules with extended half-lives, compositions and uses thereof |
| US7732578B2 (en) | 2001-07-12 | 2010-06-08 | Arrowsmith Technology Licensing Llc | Super humanized antibodies |
| US7262278B2 (en) | 2001-10-15 | 2007-08-28 | Kirin Beer Kabushiki Kaisha | Anti-HLA-DR antibody |
| US20030211100A1 (en) | 2001-11-09 | 2003-11-13 | Vahe Bedian | Antibodies to CD40 |
| US7700740B2 (en) | 2001-11-21 | 2010-04-20 | Celltech R&D Ltd | Antibodies to CD83 |
| US20040006215A1 (en) | 2002-01-09 | 2004-01-08 | Tibor Keler | Human monoclonal antibodies against CD30 |
| US20090142340A1 (en) | 2002-03-01 | 2009-06-04 | Xencor, Inc. | Optimized Fc Variants and Methods for Their Generation |
| US20090068175A1 (en) | 2002-03-01 | 2009-03-12 | Xencor, Inc. | Optimized FC Variants and Methods for Their Generation |
| US20060099205A1 (en) | 2002-04-05 | 2006-05-11 | The Regents Of The University Of California | Bispecific single chain FV antibody molecules and methods of use thereof |
| US20040071696A1 (en) | 2002-04-05 | 2004-04-15 | The Regents Of The University Of California | Bispecific single chain Fv antibody molecules and methods of use thereof |
| US7202346B2 (en) | 2002-07-03 | 2007-04-10 | Immunogen Inc. | Antibodies to non-shed Muc1 and Muc16, and uses thereof |
| US20090092599A1 (en) | 2002-09-27 | 2009-04-09 | Xencor, Inc. | Optimized Fc variants and methods for their generation |
| US7217797B2 (en) | 2002-10-15 | 2007-05-15 | Pdl Biopharma, Inc. | Alteration of FcRn binding affinities or serum half-lives of antibodies by mutagenesis |
| US7557189B2 (en) | 2002-11-07 | 2009-07-07 | Immunogen Inc. | Anti-CD33 antibodies and method for treatment of acute myeloid leukemia using the same |
| US7456257B2 (en) | 2003-02-18 | 2008-11-25 | Merck Patent Gmbh | Fusion proteins of interferon alpha muteins with improved properties |
| US7910104B2 (en) | 2003-11-01 | 2011-03-22 | Merck Patent Gmbh | Modified anti-CD52 antibody |
| US20050208048A1 (en) | 2003-12-15 | 2005-09-22 | Dendreon Corporation | HLA-DR specific antibodies, compositions and methods |
| US20060083736A1 (en) | 2004-10-15 | 2006-04-20 | Seattle Genetics, Inc. | Anti-CD70 antibody and its use for the treatment and prevention of cancer and immune disorders |
| US7829673B2 (en) | 2005-03-23 | 2010-11-09 | Genmab A/S | Antibodies against CD38 for treatment of multiple myeloma |
| US7736647B2 (en) | 2005-06-15 | 2010-06-15 | Monoclonal Antibodies Therapeutics | Anti-CD71 monoclonal antibodies and uses thereof for treating malignant tumor cells |
| WO2007029041A2 (en) | 2005-09-09 | 2007-03-15 | Imperial Innovations Limited | Interferon lambda therapy for treatment of respiratory diseases |
| US8124738B2 (en) | 2005-09-26 | 2012-02-28 | Medarex, Inc. | Human monoclonal antibodies to CD70 |
| US8039593B2 (en) | 2005-10-31 | 2011-10-18 | Duke University | Antibodies and immunotoxins that target human glycoprotein NMB |
| US7915391B2 (en) | 2006-04-24 | 2011-03-29 | Amgen Inc. | Humanized c-Kit antibody |
| WO2008006554A2 (en) | 2006-07-11 | 2008-01-17 | Greenovation Biotech Gmbh | Glyco-engineered antibodies |
| US20110243929A1 (en) | 2007-05-07 | 2011-10-06 | Medlmmune, Llc | Anti-icos antibodies and their use in treatment of oncology, transplantation and autoimmune disease |
| US7723485B2 (en) | 2007-05-08 | 2010-05-25 | Genentech, Inc. | Cysteine engineered anti-MUC16 antibodies and antibody drug conjugates |
| US20100297076A1 (en) | 2007-09-21 | 2010-11-25 | The Regents Of The University Of California | Targeted interferon demonstrates potent apoptotic and anti-tumor activities |
| US20110104112A1 (en) | 2007-09-21 | 2011-05-05 | The Regents Of The University Of California | Targeted interferons demonstrate potent apoptotic and anti-tumor activities |
| WO2009080832A1 (en) | 2007-12-26 | 2009-07-02 | Biotest Ag | Methods and agents for improving targeting of cd138 expressing tumor cells |
| US20110178278A1 (en) | 2008-03-14 | 2011-07-21 | Haegel Helene | Antibody Against the CSF-1R |
| WO2010105290A1 (en) | 2009-03-16 | 2010-09-23 | Cephalon Australia Pty Ltd | Humanised antibodies with anti-tumour activity |
| US20120100152A1 (en) | 2009-05-13 | 2012-04-26 | Genzyme Corporation | Anti-human cd52 immunoglobulins |
| US8206715B2 (en) | 2010-05-04 | 2012-06-26 | Five Prime Therapeutics, Inc. | Antibodies that bind colony stimulating factor 1 receptor (CSF1R) |
| US20120082670A1 (en) | 2010-10-04 | 2012-04-05 | Boehringer Ingelheim International Gmbh | Cd33 binding agents |
Non-Patent Citations (233)
| Title |
|---|
| "Remington's Pharmaceutical Sciences", 1990, MACK PUBLISHING CO. |
| A. LASFARW. ABUSHAHBAM. BALANK. A. COHEN-SOLAL: "Interferon lambda: a new sword in cancer immunotherapy", CLINICAL AND DEVELOPMENTAL IMMUNOLOGY, vol. 2011, 2011, pages 11 |
| AASS, N. ET AL., J. CLIN. ONCOL., vol. 23, 2005, pages 4172 |
| ABDOLLAHI, J EXP MED, 2005 |
| ACHARYA, HUM PATHOL, vol. 37, 2006, pages 352 |
| ALKAN ET AL., JOURNAL OF INTERFERON RESEARCH, vol. 4, no. 3, 1984, pages 355 - 63 |
| BARNSTABLE ET AL., CELL, vol. 14, 1978, pages 9 - 20 |
| BATALLER, J CLIN INVEST, vol. 115, 2005, pages 209 |
| BERGSAGEL, P., BLOOD, vol. 114, no. 18, 1995, pages 3864 - 71 |
| BERNHARD ET AL., SCIENCE, vol. 245, 1989, pages 301 - 304 |
| BHATTACHARYA-CHATTERJEE ET AL., J. IMMUNOL., vol. 141, 1988, pages 1398 - 1403 |
| BIRD ET AL., SCIENCE, vol. 242, 1988, pages 423 - 6 |
| BONARDI ET AL., CANCER RES., vol. 53, 1993, pages 3015 - 3021 |
| BOPPANA, MT SINAI J MED, vol. 78, 2011, pages 207 |
| BORDEN, CANCER RESEARCH, vol. 42, 1982, pages 4948 - 53 |
| BOWEN ET AL., J. IMMUNOL., vol. 151, 1993, pages 5896 - 5906 |
| BUGGINS, BLOOD, vol. 100, 2002, pages 1715 |
| BUMAL, HYBRIDOMA, vol. 7, no. 4, 1988, pages 407 - 415 |
| CAMPLOI, CRIT REV IMMUNOL., vol. 24, 2004, pages 267 |
| CANCER RES, vol. 62, 1 November 2002 (2002-11-01), pages 6132 |
| CARAGLIA, CELL DEATH AND DIFFERENTIATION, vol. 6, 1999, pages 773 - 80 |
| CHAWLA-SARKAR, APOPTOSIS, vol. 8, 2003, pages 237 - 49 |
| CHAWLA-SARKAR, CLINICAL CANCER RESEARCH, vol. 7, 2001, pages 1821 - 31 |
| CHEN, J EXP MED, vol. 198, 2003, pages 1875 |
| CHEN, J NEUROIMMUNOL, vol. 242, 2012, pages 39 |
| COL, J ET AL., CANCER RES., vol. 72, 2012, pages 1825 |
| DANN, NAT NEUROSCI, vol. 15, 2012, pages 98 |
| DAVIESRIECHMANN, FEBS LETT, vol. 339, 1994, pages 285 - 290 |
| DE ANDRADE, CURR OPIN RHEUMATOL, vol. 21, 2009, pages 649 |
| DE ANDRES, J NEUROIMMUNOL, vol. 182, 2007, pages 204 |
| DEROSE, P. ET AL., IMMUNOTHERAPY, vol. 3, 2011, pages 933 |
| DEUTER, DEV OPHTHALMOL, vol. 51, 2012, pages 90 - 7 |
| DHIB-JALBUT, NEUROLOGY, vol. 74, 2010, pages S17 |
| DHODAPKAR, BLOOD, vol. 91, 1998, pages 2679 |
| DIAZ, J AEROSOL MED PULM DRUG DELIV, vol. 25, 2012, pages 79 |
| DIVGI, NUCL. MED. BIOL., vol. 21, 1994, pages 9 - 15 |
| DOOLEY, CELL TISSUE RES, vol. 347, 2012, pages 245 |
| DURIG, J., LEUK. RES., vol. 25, 2002, pages 927 |
| DUROCHER, NUCLEIC ACIDS RESEARCH, vol. 30, no. 2, 2002, pages El-9 |
| E. L. RAMOS: "Preclinical and clinical development of pegylated interferon-lambda 1 in chronic hepatitis C", JOURNAL OF INTERFERON AND CYTOKINE RESEARCH, vol. 30, no. 8, 2010, pages 591 - 595 |
| E. M. COCCIAM. SEVERAE. GIACOMINI ET AL.: "Viral infection and toll-like receptor agonists induce a differential expression of type I and.le interferons in humans plasmacytoid and monocyte-derived dendritic cells", EUROPEAN JOURNAL OF IMMUNOLOGY, vol. 34, no. 3, 2004, pages 796 - 805, XP009076892, DOI: 10.1002/eji.200324610 |
| ELLIOTT, BIOLOGIES, vol. 6, 2012, pages 163 |
| ELLIOTT, EXP HEMATOL, vol. 36, 2008, pages 1573 |
| ELLIS ET AL., J. IMMUNOL., vol. 155, 1995, pages 925 - 937 |
| ESTIN ET AL., J. NATL. CANCER INSTIT., vol. 81, no. 6, 1989, pages 445 - 446 |
| FARRELL, THE ONCOLOGIST, vol. 9, 2004, pages 18 |
| FEAGINS, INFLAMM BOWEL DIS, vol. 16, 2010, pages 1963 |
| FEIZI, NATURE, vol. 314, 1985, pages 53 - 57 |
| FISH, DRUG DES DELIV, vol. 2, no. 3, February 1988 (1988-02-01), pages 191 - 206 |
| FOON ET AL., PROC. AM. SOC. CLIN. ONCOL., vol. 13, 1994, pages 294 |
| FORNIER ET AL., ONCOLOGY (HUNTINGT, vol. 13, 1999, pages 647 - 658 |
| FRANCISCO ET AL., CANCER RES., vol. 60, 2000, pages 3225 - 3231 |
| FRANKE ET AL., CANCER BIOTHER. RADIOPHARM., vol. 15, 2000, pages 459 - 76 |
| FRIESE, ANN NEUROL, vol. 66, 2009, pages 132 |
| FRIESE, BRAIN, vol. 128, 2005, pages 1747 |
| GANDHI, J NEUROIMMUNOL, vol. 221, 2010, pages 7 |
| GAZDAR, BLOOD, vol. 67, 1986, pages 1542 - 1549 |
| GERBER, D. ET AL., CLIN CANCER RES, vol. 17, 2011, pages 4550 |
| GHORESCHI, CLIN DERMATOL, vol. 25, 2007, pages 574 |
| GHORESCHI, NAT MED, vol. 9, 2003, pages 40 |
| GNERLICH, J IMMUNOL, vol. 185, 2010, pages 4063 |
| GRABER, CLIN NEUROL NEUROSURG, vol. 112, 2010, pages 58 |
| GREENSTEIN, EXP HEMATOL, vol. 31, no. 4, 2003, pages 271 - 82 |
| H. SCHRECKHISEK. KHUU-DUONG ET AL.: "Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes", JOURNAL OF HEPATOLOGY, vol. 44, no. 4, 2006, pages 896 - 906 |
| HALLAL-LONGO, J INTERFERON CYTOKINE RES, vol. 27, 2007, pages 865 |
| HAMERS-CASTERMAN ET AL., NATURE, vol. 363, 1993, pages 446 - 448 |
| HANSBRO, BR J PHARMACOL, vol. 163, 2011, pages 81 |
| HARDIN J. ET AL.: "Interleukin-6 prevents dexamethasone-induced myeloma cell death.", BLOOD, vol. 84, 1994, pages 3063 - 1336 |
| HE, J. ET AL., CLIN CANCER RES., vol. 15, 2009, pages 6871 |
| HELLSTROM ET AL., CANCER RES., vol. 46, 1986, pages 3917 - 3923 |
| HELLSTROM ET AL., CANCER. RES., vol. 45, 1985, pages 2210 - 2188 |
| HENTTUVIHKO, BIOCHEM. BIOPHYS. RES. COMM., vol. 160, no. 2, 1989, pages 903 - 910 |
| HERFARTH, GUT, vol. 50, 2002, pages 191 |
| HERLYN ET AL., J. CLIN. IMMUNOL., vol. 2, 1982, pages 135 |
| HEROLD, M. ET AL., ACTA DERMATOVENER, vol. 74, 1975, pages 29 |
| HILKENS ET AL., TRENDS IN BIO. CHEM. SCI., vol. 17, 1992, pages 359 |
| HOFMANN, CLIN IMMUNOL, vol. 143, 2012, pages 116 |
| HOLLIGER, P. ET AL., PROC. NATL. ACAD. SCI. USA, vol. 90, 1993, pages 6444 - 6448 |
| HOOPER, ANN CLIN LAB SCI, vol. 8, 1978, pages 201 |
| HUNIG, MED MICROBIOL IMMUNOL, vol. 199, 2010, pages 239 |
| HUSTON ET AL., PROC NATL ACAD SCI USA, vol. 85, 1988, pages 5879 - 83 |
| IBRAHIM, S., BLOOD, vol. 98, 2001, pages 181 |
| IKEDA, CLIN CAN RES, vol. 15, 2009, pages 4028 |
| IMMUNOL. LETT, vol. 83, pages 215 - 220 |
| ISHIKI, HEPATOLOGY, vol. 16, 1992, pages 1227 |
| ISRAELI ET AL., CANCER RES, vol. 53, 1993, pages 227 - 230 |
| IWAI, IMMUNOL LETT., vol. 83, 2002, pages 215 - 20 |
| JAITIN, DIEGO A,, ROISMAN, LAILA C, JAKS, EVA, GAVUTIS, MARTYNAS, PIEHLER,JACOB, VAN DER HEYDEN, JOSE, UZE, GILLES, SCHREIBER, GID: "Inquiring into the differential action of interferons (IFNs): an IFN-a2 mutant with enhanced affinity to IFNAR1 is functionally similar to IFN-β", MOL. CELL. BIOL., vol. 26, 2006, pages 1888 - 1897, XP009079017, DOI: 10.1128/MCB.26.5.1888-1897.2006 |
| JELKMANN, CRIT REV ONCOL HEMATOL, vol. 67, 2008, pages 39 |
| JILANA, AM J CLIN PATHOL, vol. 118, 2002, pages 560 - 566 |
| K. ERIKSSONA. R. THOMSENS. R. PALUDAN: "Lambda interferon (FFN-A), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo", JOURNAL OF VIROLOGY, vol. 80, no. 9, 2006, pages 4501 - 4509, XP002694469, DOI: 10.1128/JVI.80.9.4501-4509.2006 |
| KALAI M, BLOOD, vol. 89, no. 4, 15 February 1997 (1997-02-15), pages 1319 - 1333 |
| KALAI, BLOOD, vol. 89, no. 4, 1997, pages 1319 - 33 |
| KALIE, EYALJAITIN, DIEGO A.PODOPLELOVA, YULIAPIEHLER, JACOBSCHREIBER, GIDEON: "The Stability of the ternary interferon-receptor complex rather than the affinity to the individual subunits dictates differential biological activities", J. BIOL. CHEM., vol. 283, 2008, pages 32925 - 32936, XP055065215, DOI: 10.1074/jbc.M806019200 |
| KALIEEYALJAITINDIEGO A.ABRAMOVICHRENNESCHREIBERGIDEON: "An interferon a2 mutant optimized by phage display for IFNAR1 binding confers specifically enhanced antitubor activities", J. BIOL. CHEM., vol. 282, 2007, pages 11602 - 11611, XP055241192, DOI: 10.1074/jbc.M610115200 |
| KALVAKOLANU, HISTOL. HISTOPATHOL, vol. 15, 2000, pages 523 - 37 |
| KAST, R., CANCER BIOLOGY AND THERAPY, vol. 7, 2008, pages 1515 |
| KIDD, ALTERN MED REV, vol. 8, 2003, pages 223 |
| KILPATRICK ET AL., HYBRIDOMA, vol. 17, 1998, pages 569 - 576 |
| KIMURA, EUR J IMMUNOL, vol. 40, 2010, pages 1830 |
| KING, LANCET, vol. 378, 2011, pages 1949 |
| KLEIN, CANCER RES., vol. 28, 1968, pages 1300 - 1310 |
| KOCHENDERFER, BLOOD, vol. 120, 2012, pages 2417 |
| KOGUMA, T., BIOCHIM. BIOPHYS. ACTA, vol. 1223, 1994, pages 160 |
| KORPORAL, ARCH NEUROL, vol. 65, 2008, pages 1434 |
| KRUSE, EMBO JOURNAL, vol. 12, no. 13, 1993, pages 5121 - 5129 |
| KUSCHUTZ, PROC. NATL. ACAD. SCI. USA, vol. 92, 1995, pages 6552 - 6556 |
| KWIECINSKI, PLOS ONE, vol. 6, 2012, pages e24568 |
| LANDER, J MOL BIOL., vol. 299, no. 1, 26 May 2000 (2000-05-26), pages 169 - 79 |
| LAWSON, AM J RESPIR CRIT CARE MED, vol. 171, 2005, pages 899 |
| LI ET AL., CELL. IMMUNOL., vol. 111, 1989, pages 85 - 99 |
| LINDNER, J OF INTERFERON AND CYTOKINE RESEARCH, vol. 17, 1997, pages 681 - 693 |
| LIU, Q., STRUCTURE, vol. 13, 2005, pages 1331 |
| LIVINGSTON ET AL., J. CLIN. ONCOL., vol. 12, 1994, pages 1036 - 1044 |
| LOMA, CURR NEUROPHARMACOL, vol. 9, 2011, pages 409 |
| LUNDELL, J BIOL. CHEM., vol. 269, no. 23, pages 16159 - 62 |
| M. D. ROBEKB. S. BOYDF. V. CHISARI: "Lambda interferon inhibits hepatitis B and C virus replication", JOURNAL OF VIROLOGY, vol. 79, no. 6, 2005, pages 3851 - 3854, XP002637113, DOI: 10.1128/JVI.79.6.3851-3854.2005 |
| MA, WORLD J GASTROENTEROL, vol. 11, no. 10, 2005, pages 1521 - 8 |
| MADONNA, THROMB HAEMOST, vol. 107, 2012, pages 656 |
| MAIER ET AL., CANCER RES, vol. 51, 1991, pages 5361 - 5369 |
| MAIER ET AL., CANCER RES., vol. 51, no. 2, 1991, pages 5361 - 5369 |
| MALAVASI, F., HUM. IMMUNOL., vol. 9, 1984, pages 9 |
| MALAVASI, F., J. CLIN LAB RES., vol. 22, 1992, pages 73 |
| MARKS, J.D.BRADBURY, A., METHODS MOL BIOL, vol. 248, 2004, pages 161 - 76 |
| MASON ET AL., BLOOD, vol. 69, 1987, pages 836 - 40 |
| MATSUDA, HEPATOLOGY, vol. 26, 1997, pages 81 |
| MATTOS, J CONTROL RELEASE, vol. 162, 2012, pages 261 |
| MCELROY, EUR RESPIR J, vol. 24, 2004, pages 664 |
| MEHTA, MOL CANCER THER, vol. 3, no. 3, 2004, pages 345 - 52 |
| MEYSKENS, F. ET AL., CRIT REV ONCOL HEMATOL, vol. 3, 1987, pages 75 |
| MITTELMAN ET AL., J. CLIN. INVEST., vol. 86, 1990, pages 2136 - 2144 |
| MIZUNO, AM J PHYSIOL RENAL PHYSIOL, vol. 286, 2004, pages F134 |
| MIZUNO, FASEB J, vol. 19, 2005, pages 580 |
| MOL CANCER THER, vol. 9, no. 5, May 2010 (2010-05-01), pages 1274 - 1285 |
| MORABITO, F., HAEMATOLOGICA, vol. 87, 2002, pages 217 |
| MORGENSEN, INT J. CANCER, vol. 28, 1981, pages 575 - 82 |
| MOTZER, R., J. CLIN. ONCOL., vol. 18, 2000, pages 2972 |
| MURRAY, SEMIN. ONCOL., vol. 27, 2000, pages 64 - 70 |
| NAKAMURA, AM J PHYSIOL HEART CIRC PHYSIOL, vol. 288, 2005, pages H2131 |
| NAKAMURA, J GASTROENTEROL HEPATOL, vol. 26, 2011, pages 188 |
| NAKAMURA, PROC JPN ACAD SER B PHYS BIOL SCI, vol. 86, 2010, pages 588 |
| NAMADA, J CELL PHYSIOL DOI 10.1002/JCP.24143, 2012 |
| NATALI ET AL., CANCER, vol. 59, 1987, pages 55 - 63 |
| O'CONNELL, ADV THER, vol. 28, 2011, pages 986 |
| ONO ET AL., MOL. IMMUNO., vol. 36, 1999, pages 387 - 395 |
| OSTERREICHER, PROC NATL ACAD SCI USA, vol. 108, 2011, pages 308 |
| OTSUKA, BRITISH JOURNAL OF HAEMATOLOGY, vol. 103, 1998, pages 518 - 529 |
| OZZELLO ET AL., BREAST CANCER RES. TREAT., vol. 25, 1993, pages 265 - 276 |
| OZZELLO ET AL., BREAST CANCER RESEARCH AND TREATMENT, vol. 25, 1993, pages 265 - 76 |
| P. SHEPPARDW. KINDSVOGELW. XU ET AL.: "IL-28, IL-29 and their class II cytokine receptor IL-28R", NATURE IMMUNOLOGY, vol. 4, no. 1, 2003, pages 63 - 68 |
| PAN, MANJINGKALIE, EYALSCAGLIONE, BRIAN J.RAVECHE, ELIZABETH S.SCHREIBER, GIDEONLANGER, JEROME A: "Mutation of te IFNAR-1 receptor binding site of human IFN-a2 generates Tyep I IFN competitive antagonists", BIOCHEMISTRY, vol. 47, 2008, pages 12018 - 12027, XP055145910, DOI: 10.1021/bi801588g |
| PANGANIBAN, ACTA PHARMACOL SIN, vol. 32, 2011, pages 12 |
| PARK, J BIOL CHEM, vol. 274, 1999, pages 36505 |
| PAVLINKOVA ET AL., CLIN. CANCER RES., vol. 5, 1999, pages 3304s - 3314s |
| PEREZWALKER, J. IMMUNOL., vol. 142, 1990, pages 3662 - 3667 |
| PESTKA, IMMUNOLOGICAL REVIEWS, vol. 202, no. 1, 2004, pages 8 - 32 |
| PESTKA, S.: "Methods in Enzymology", vol. 119, 1986, ACADEMIC PRESS, article "Interferon Standards and General Abbreviations", pages: 14 - 23 |
| PETERSON ET AL., CANCER RES., vol. 57, 1997, pages 1103 - 1108 |
| PIEHLER, JACOBROISMAN, LAILA C.SCHREIBER, GIDEON: "New structural and functional aspects of the Type I interferon-receptor interaction revealed by comprehensive mutational analysis of the binding interface.", J. BIOL. CHEM., vol. 275, 2000, pages 40425 - 40433, XP002267030, DOI: 10.1074/jbc.M006854200 |
| PLATANIAS, NATURE REVIEWS, vol. 5, 2005, pages 375 - 86 |
| POLJAK, R. J. ET AL., STRUCTURE, vol. 2, 1994, pages 1121 - 1123 |
| PRINZ, TRENDS MOL MED, vol. 16, 2010, pages 379 |
| RAGNHAMMAR ET AL., INT. J. CANCER, vol. 53, 1993, pages 751 - 758 |
| RAN, S. ET AL., CANCER RES., vol. 62, no. 13, 2002, pages 6132 - 42 |
| RAN, S. ET AL., CLIN CANCER RES, vol. 11, 2005, pages 1551 |
| RATZINGER, BLOOD, vol. 101, 2003, pages 1422 |
| RECCHIA, F. ET AL., J. INTERFERON CYTOKINE RES., vol. 15, 1995, pages 605 |
| REIDL, L. ET AL., J IMMUNOL., vol. 14, 1991, pages 3623 |
| REITAMO, J CLIN INVEST, vol. 94, 1994, pages 2489 |
| REP, J NEUROIMMUNOL, vol. 67, 1996, pages 111 |
| ROGLIANI, THER ADV RESPIR DIS, vol. 2, 2008, pages 75 |
| ROOS, ENDOCRINOLOGY, vol. 131, 1992, pages 2540 |
| ROSENBLOOM, ANN INTERN MED, vol. 152, 2010, pages 159 |
| RUBINSTEIN, J. VIROL., vol. 37, 1981, pages 755 - 8 |
| RUNKEL ET AL., J. BIOL. CHEM., vol. 273, 1998, pages 8003 - 8 |
| RUNKEL, L.PFEFFER, L.LEWERENZ, M.MOGENSEN, K.: "Differences in Activity between a and β Type I Interferons Explored by Mutational Analysis", J. BIOL. CHEM., vol. 273, 1998, pages 8003 - 8008, XP002160242, DOI: 10.1074/jbc.273.14.8003 |
| S. V. KOTENKOG. GALLAGHERV. V. BAURIN ET AL.: "IFN-/.S mediate antiviral protection through a distinct class II cytokine receptor complex", NATURE IMMUNOLOGY, vol. 4, no. 1, 2003, pages 69 - 77 |
| SAMPSON, PLOS ONE, vol. 7, 2012, pages e31046 |
| SANCHEZ-MUNOZ, WORLD J GASTROENTEROL, vol. 14, 2008, pages 4280 |
| SARASELLA, FASEB J, vol. 22, 2008, pages 3500 |
| SCHIER ET AL., J MOL BIOL, vol. 263, 1996, pages 551 - 567 |
| SCHILLER, J INTERFERON RESARCH, vol. 6, 1986, pages 615 - 25 |
| SCHWAGER ARTHRITIS RES THER., vol. 11, no. 5, 2009, pages R142 |
| See also references of EP3559049A4 |
| SGOUROS ET AL., J. NUCL. MED., vol. 34, 1993, pages 422 - 430 |
| SHAO, J LEUKOC BIOL, vol. 83, 2008, pages 1323 |
| SHARIEF, J NEUROIMMUNOL, vol. 120, 2001, pages 199 |
| SHARIEF, J NEUROIMMUNOL, vol. 129, 2002, pages 224 |
| SHARIFI, HYBRIDOMA AND HYBRIDOMICS, vol. 21, no. 6, 2002, pages 421 - 32 |
| SHI, WORLD J GASTROENTEROL, vol. 12, 2006, pages 2357 |
| SHINKAWA T. ET AL., J BIOL CHEM, vol. 278, 2003, pages 3466 - 73 |
| SHINOHARA, IMMUNITY, vol. 28, 2008, pages 675 |
| SHITARA ET AL., CANCER IMMUNOL. IMMUNOTHER., vol. 36, 1993, pages 373 - 380 |
| SHLESINGER, VIROLOGY, vol. 125, 1983, pages 8 - 17 |
| SHUKLA, AM J RESPIR CELL MOL BIOL, vol. 40, 2009, pages 643 |
| SHULL, NATURE, vol. 359, 1992, pages 693 |
| SIEVERS ET AL., BLOOD, vol. 93, 1999, pages 3678 - 3684 |
| SLUTZKI, MICHAL, JAITIN, DIEGO A., YEHEZKEL, TUVAL BEN, SCHREIBER, GIDEON: "Variations in the unstructured C-terminal tail of interferons contribute to differential receptor binding and biological activity", J. MOL. BIOL., vol. 360, 2006, pages 1019 - 1030, XP024951243, DOI: 10.1016/j.jmb.2006.05.069 |
| SMITH, M., J. CLIN. ONCOL., vol. 10, 1992, pages 839 |
| SOARES, M. ET AL., NAT MED, vol. 14, 2008, pages 1357 |
| SOARES, M. ET AL., NAT MED., vol. 14, 2008, pages 1357 |
| STEIN, LEUK. LYMPHHOMA, vol. 52, no. 2, 2011, pages 273 - 84 |
| STEVENS, PROC AM THORAC SOC, vol. 5, 2008, pages 783 |
| STEWART, A. G.ADAIR, J. R.CATLIN, G.HYNES, C.HALL, J.DAV IES, J.DAWSON, K.PORTER, A. G.: "Chemical mutagenesis of human interferon-beta: construction, expression in E. coli, and biological activity of sodium bisulfite-induced mutations", DNA, vol. 6, 1987, pages 119 - 128 |
| STOCKERT, PHYSIOL REV, vol. 75, 1995, pages 591 |
| STRUTZ, J CELL BIOL, vol. 130, 1995, pages 393 |
| STUTTFELD, IUBMB LIFE, vol. 61, 2009, pages 915 |
| STUVE, ANN NEUROL, vol. 40, 1996, pages 853 |
| SUTHERLAND, MABS, vol. 1, no. 5, 2009, pages 481 - 490 |
| T. MARCELLOA. GRAKOUIG. BARBA-SPAETH ET AL.: "Interferons a and.le inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics", GASTROENTEROLOGY, vol. 131, no. 6, 2006, pages 1887 - 1898, XP005750989, DOI: 10.1053/j.gastro.2006.09.052 |
| TAILOR ET AL., NUCL. ACIDS RES., vol. 18, no. 16, 1990, pages 4928 |
| TELESHOVA, SCAND J IMMUNOL, vol. 51, 2000, pages 312 |
| TERSZOWSKI, BLOOD, vol. 105, 2005, pages 1937 |
| TRAIL ET AL., CANCER RESEARCH, vol. 57, 1997, pages 100 - 105 |
| TRAIL ET AL., SCIENCE, vol. 261, 1993, pages 212 - 215 |
| TROJANOWSKA, RHEUMATOLOGY (OXFORD, vol. 47S5, 2008, pages 2 |
| UHM, ANN NEUROL, vol. 46, 1999, pages 319 |
| VAN HOF ET AL., CANCER RES., vol. 56, 1996, pages 5179 - 5185 |
| VIJAYASARDAHL ET AL., J. EXP. MED., vol. 171, no. 4, 1990, pages 1375 - 1380 |
| VIROL, vol. 125, pages 8 - 17 |
| WANG YSHEN BSEBALD W., PROC. NATL. ACAD. SCI. USA, vol. 94, no. 5, 4 March 1997 (1997-03-04), pages 1657 - 62 |
| WANGDI YIJUN YIDA XUEXUE BAO ET AL., ACADEMIC JOURNAL OF THE FIRST MEDICAL COLLEGE OF PLA, vol. 22, no. 5, 2002, pages 409 - 411 |
| WARD ET AL., NATURE, vol. 341, 1989, pages 544 - 546 |
| WARD, THER IMMUNOL, vol. 2, 1995, pages 77 |
| WASCHUTZA, EUR J. BIOCHEM., vol. 256, 1998, pages 303 - 9 |
| WYNN, J EXP MED, vol. 208, 2011, pages 1339 |
| XU, CANCER RESEARCH, vol. 58, 1998, pages 2832 - 7 |
| XUAN CSTEWARD KKTIMMERMAN JMMORRISON SL: "Targeted delivery of interferon-a via fusion to anti-CD20 results in potent antitumor activity against B-cell lymphoma", BLOOD, vol. 115, 2010, pages 2864 - 71, XP002673645, DOI: 10.1182/BLOOD-2009-10-250555 |
| YAEKASHIWA, AM J RESPIR CRIT CARE MED, vol. 156, 1997, pages 1937 |
| YANG, AM J PHYSIOL RENAL PHYSIOL, vol. 284, 2003, pages F349 |
| YOKATA ET AL., CANCER RES, vol. 52, 1992, pages 3402 - 3408 |
| ZAFRANSKAYA, IMMUNOL, vol. 121, 2006, pages 29 |
| ZHANG, HEPATOGASTROENTEROLOGY, vol. 54, 2007, pages 2092 |
Cited By (166)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9574012B2 (en) | 2011-07-05 | 2017-02-21 | Sanofi (China) Investment Co., Ltd. Shanghai Branch | AGR2 blocking antibody and use thereof |
| JP2020010697A (en) * | 2011-10-28 | 2020-01-23 | テバ・ファーマシューティカルズ・オーストラリア・ピーティワイ・リミテッド | Polypeptide construct and use thereof |
| US9611322B2 (en) | 2011-10-28 | 2017-04-04 | Teva Pharmaceuticals Australia Pty Ltd | Fusions of antibodies to CD38 and attenuated interferon alpha |
| US10981986B2 (en) | 2011-10-28 | 2021-04-20 | Teva Pharmaceuticals Australia Pty Ltd | Fusions of antibodies to CD38 and attenuated interferon alpha |
| US10946070B2 (en) | 2012-01-20 | 2021-03-16 | Vib Vzw | Targeted and mutated human-interferon fusion proteins |
| US9492562B2 (en) | 2012-01-20 | 2016-11-15 | Vib Vzw | Targeted human-interferon fusion proteins |
| EP2804877B1 (en) * | 2012-01-20 | 2018-08-22 | VIB vzw | Targeted mutant alpha-helical bundle cytokines |
| US9878014B2 (en) | 2012-01-20 | 2018-01-30 | Vib Vzw | Targeted human-interferon fusion proteins |
| WO2013107791A1 (en) | 2012-01-20 | 2013-07-25 | Vib Vzw | Targeted mutant alpha-helical bundle cytokines |
| US10034919B2 (en) | 2012-01-20 | 2018-07-31 | Vib Vzw | Targeted human-interferon fusion proteins |
| US9534057B2 (en) | 2012-03-03 | 2017-01-03 | Immungene Inc | Methods of treating tumors with engineered antibody-interferon mutant fusion molecules |
| US8980267B2 (en) | 2012-03-03 | 2015-03-17 | Immungene Inc | Engineered antibody-interferon mutant fusion molecules |
| US10259854B2 (en) | 2012-03-03 | 2019-04-16 | Immungene Inc | Engineered antibody-interferon mutant fusion molecules |
| US11634506B2 (en) | 2013-01-14 | 2023-04-25 | Xencor, Inc. | Heterodimeric proteins |
| US11718667B2 (en) | 2013-01-14 | 2023-08-08 | Xencor, Inc. | Optimized antibody variable regions |
| US11814423B2 (en) | 2013-03-15 | 2023-11-14 | Xencor, Inc. | Heterodimeric proteins |
| JP2016522816A (en) * | 2013-04-29 | 2016-08-04 | テバ・ファーマシューティカルズ・オーストラリア・ピーティワイ・リミテッド | Fusion with anti-CD38 antibody and attenuated interferon alpha-2B |
| EP3677591A1 (en) * | 2013-04-29 | 2020-07-08 | Teva Pharmaceuticals Australia Pty Ltd | Anti-cd38 antibodies and fusions to attenuated interferon alpha-2b |
| US9963515B2 (en) | 2013-04-29 | 2018-05-08 | Teva Pharmaceuticals Australia Pty Ltd. | Anti-CD38 antibodies and fusions to attenuated interferon alpha-2B |
| US11117975B2 (en) | 2013-04-29 | 2021-09-14 | Teva Pharmaceuticals Australia Pty Ltd | Anti-CD38 antibodies and fusions to attenuated interferon alpha-2B |
| EA033115B1 (en) * | 2013-04-29 | 2019-08-30 | Тева Фармасьютикалз Острэйлиа Пти Лтд. | Anti-cd38 antibodies and fusion proteins with attenuated interferon alpha-2b |
| WO2014178820A1 (en) * | 2013-04-29 | 2014-11-06 | Teva Pharmaceuticals Australia Pty Ltd. | Anti-cd38 antibodies and fusions to attenuated interferon alpha-2b |
| US11358997B2 (en) | 2013-07-18 | 2022-06-14 | Vib Vzw | Fusokines involving cytokines with strongly reduced receptor binding affinities |
| US10640542B2 (en) | 2013-07-18 | 2020-05-05 | Vib Vzw | Fusokines involving cytokines with strongly reduced receptor binding affinities |
| US9932409B2 (en) | 2013-07-19 | 2018-04-03 | Vib Vzw | Targeted modified IL-1 family members |
| US10947288B2 (en) | 2013-07-19 | 2021-03-16 | Vib Vzw | Targeting of human interferon antagonists |
| US10787493B2 (en) | 2013-07-19 | 2020-09-29 | Vib Vzw | Targeted modified TNF family members |
| US9732135B2 (en) | 2013-07-19 | 2017-08-15 | Vib Vzw | Targeting of human interferon antagonists |
| US10072059B2 (en) | 2013-07-19 | 2018-09-11 | Vib Vzw | Targeting of human interferon antagonists |
| US11396532B2 (en) | 2013-07-19 | 2022-07-26 | Vib Vzw | Targeted modified TNF family members |
| JP2016529232A (en) * | 2013-07-19 | 2016-09-23 | ヴィブ ブイゼットダブリュー | Targeting cytokine antagonists |
| US9914759B2 (en) | 2013-07-19 | 2018-03-13 | Vib Vzw | Targeted modified TNF family members |
| US10407480B2 (en) | 2013-07-19 | 2019-09-10 | Vib Vzw | Targeted modified TNF family members |
| US10035835B2 (en) | 2013-07-19 | 2018-07-31 | Vib Vzw | Targeted modified TNF family members |
| WO2015066450A1 (en) * | 2013-10-31 | 2015-05-07 | Sanofi | Specific anti-cd38 antibodies for treating human cancers |
| EP3805266A1 (en) * | 2013-10-31 | 2021-04-14 | Sanofi | Specific anti-cd38 antibodies for treating human cancers |
| US12060432B2 (en) | 2014-02-28 | 2024-08-13 | Janssen Biotech, Inc. | Combination therapies with anti-CD38 antibodies |
| US11713355B2 (en) | 2014-02-28 | 2023-08-01 | Janssen Biotech, Inc. | Anti-CD38 antibodies for treatment of acute lymphoblastic leukemia |
| EA037597B1 (en) * | 2014-02-28 | 2021-04-20 | Янссен Байотек, Инк. | Anti-cd38 antibodies for treatment of acute lymphoblastic leukemia |
| US10800851B2 (en) | 2014-02-28 | 2020-10-13 | Janssen Biotech, Inc. | Combination therapies with anti-CD38 antibodies |
| WO2015130732A3 (en) * | 2014-02-28 | 2016-03-17 | Janssen Biotech, Inc. | Anti-cd38 antibodies for treatment of acute lymphoblastic leukemia |
| US10556961B2 (en) | 2014-02-28 | 2020-02-11 | Janssen Biotech, Inc. | Anti-CD38 antibodies for treatment of acute lymphoblastic leukemia |
| US9732154B2 (en) | 2014-02-28 | 2017-08-15 | Janssen Biotech, Inc. | Anti-CD38 antibodies for treatment of acute lymphoblastic leukemia |
| US11840579B2 (en) | 2014-03-28 | 2023-12-12 | Xencor, Inc. | Bispecific antibodies that bind to CD38 and CD3 |
| US10858451B2 (en) | 2014-03-28 | 2020-12-08 | Xencor, Inc. | Bispecific antibodies that bind to CD38 and CD3 |
| US10232041B2 (en) | 2014-05-01 | 2019-03-19 | Teva Pharmaceuticals Pty Ltd | Combination of lenalidomide and polypeptide construct, and uses thereof |
| JP2017515815A (en) * | 2014-05-01 | 2017-06-15 | テバ・ファーマシューティカルズ・オーストラリア・ピーティワイ・リミテッド | Combinations of lenalidomide and polypeptide constructs and uses thereof |
| WO2015181641A3 (en) * | 2014-05-01 | 2016-01-21 | Teva Pharmaceuticals Australia Pty Ltd | Combination of lenalidomide or pomalidomide and cd38 antibody-attenuated interferon-alpha constructs, and the use thereof |
| KR102412023B1 (en) | 2014-05-01 | 2022-06-21 | 테바 파마슈티컬즈 오스트레일리아 피티와이 엘티디 | Combination of lenalidomide or pomalidomide and cd38 antibody-attenuated interferon-alpha constructs, and the use thereof |
| US11253591B2 (en) | 2014-05-01 | 2022-02-22 | Teva Pharmaceuticals Australia Pty Ltd. | Combination of lenalidomide and polypeptide construct, and uses thereof |
| CN106536562B (en) * | 2014-05-01 | 2023-04-14 | 泰华制药澳大利亚公司 | Combination of lenalidomide or pomalidomide and an interferon alpha construct detoxified by an anti-CD 38 antibody, and uses thereof |
| AU2015265624B2 (en) * | 2014-05-01 | 2021-02-18 | Teva Pharmaceuticals Australia Pty Ltd | Combination of lenalidomide or pomalidomide and CD38 antibody-attenuated interferon-alpha constructs, and the use thereof |
| KR20160146770A (en) * | 2014-05-01 | 2016-12-21 | 테바 파마슈티컬즈 오스트레일리아 피티와이 엘티디 | Combination of lenalidomide or pomalidomide and cd38 antibody-attenuated interferon-alpha constructs, and the use thereof |
| EA033359B1 (en) * | 2014-05-01 | 2019-10-31 | Teva Pharmaceuticals Australia Pty Ltd | Combination of lenalidomide and polypeptide construct, and uses thereof |
| CN106536562A (en) * | 2014-05-01 | 2017-03-22 | 泰华制药澳大利亚公司 | Combination of lenalidomide or pomalidomide and CD38 antibody-attenuated interferon-alpha constructs, and the use thereof |
| US9636334B2 (en) | 2014-05-01 | 2017-05-02 | Teva Pharmaceuticals Australia Pty Ltd | Combination of lenalidomide and polypeptide construct, and uses thereof |
| EA035098B1 (en) * | 2014-07-31 | 2020-04-27 | Санофи | Method of treating relapsed and/or refractory multiple myeloma |
| US10786578B2 (en) | 2014-08-05 | 2020-09-29 | Novartis Ag | CKIT antibody drug conjugates |
| US11833215B2 (en) | 2014-08-05 | 2023-12-05 | Novartis Ag | CKIT antibody drug conjugates |
| WO2016020791A1 (en) * | 2014-08-05 | 2016-02-11 | Novartis Ag | Ckit antibody drug conjugates |
| WO2016022589A3 (en) * | 2014-08-08 | 2016-04-07 | The Regents Of The University Of California | Methods for treating multiple myeloma |
| US10604580B2 (en) | 2014-09-09 | 2020-03-31 | Janssen Biotech, Inc. | Combination therapies with anti-CD38 antibodies |
| CN107106656A (en) * | 2014-10-29 | 2017-08-29 | 梯瓦制药澳大利亚股份有限公司 | Interferon alpha 2 b variant |
| KR20170077185A (en) * | 2014-10-29 | 2017-07-05 | 테바 파마슈티컬즈 오스트레일리아 피티와이 엘티디 | INTERFERON α2b VARIANTS |
| JP2020103319A (en) * | 2014-10-29 | 2020-07-09 | テバ・ファーマシューティカルズ・オーストラリア・ピーティワイ・リミテッド | INTERFERON α 2B VARIANTS |
| US11319356B2 (en) | 2014-10-29 | 2022-05-03 | Teva Pharmaceuticals Australia Pty Ltd | Interferon alpha 2B variants |
| AU2015337858B2 (en) * | 2014-10-29 | 2020-09-24 | Teva Pharmaceuticals Australia Pty Ltd. | Interferon alpha2b variants |
| WO2016065409A1 (en) * | 2014-10-29 | 2016-05-06 | Teva Pharmaceuticals Australia Pty Ltd | INTERFERON α2B VARIANTS |
| JP2017534282A (en) * | 2014-10-29 | 2017-11-24 | テバ・ファーマシューティカルズ・オーストラリア・ピーティワイ・リミテッド | Interferon α2B variant |
| IL251822B2 (en) * | 2014-10-29 | 2023-03-01 | Teva Pharmaceuticals Australia Pty Ltd | Interferon alpha2b variants |
| EP3212216A4 (en) * | 2014-10-29 | 2018-04-18 | Teva Pharmaceuticals Australia Pty Ltd | Interferon alpha2b variants |
| US10544199B2 (en) | 2014-10-29 | 2020-01-28 | Teva Pharmaceuticals Australia Pty Ltd | Interferon alpha 2B variants |
| KR102639037B1 (en) | 2014-10-29 | 2024-02-20 | 테바 파마슈티컬즈 오스트레일리아 피티와이 엘티디 | INTERFERON α2b VARIANTS |
| EA037749B1 (en) * | 2014-10-29 | 2021-05-18 | Тева Фармасьютикалз Острэйлиа Пти Лтд | INTERFERON 2b VARIANTS |
| IL251822B (en) * | 2014-10-29 | 2022-11-01 | Teva Pharmaceuticals Australia Pty Ltd | Interferon alpha2b variants |
| JP2017536830A (en) * | 2014-11-26 | 2017-12-14 | ゼンコー・インコーポレイテッドXencor、 Inc. | Heterodimeric antibodies that bind to CD3 and CD38 |
| US11673972B2 (en) | 2014-11-26 | 2023-06-13 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and tumor antigens |
| US11859011B2 (en) | 2014-11-26 | 2024-01-02 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and tumor antigens |
| US11352442B2 (en) | 2014-11-26 | 2022-06-07 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and CD38 |
| US11945880B2 (en) | 2014-11-26 | 2024-04-02 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and tumor antigens |
| US10793630B2 (en) | 2014-12-04 | 2020-10-06 | Janssen Biotech, Inc. | Anti-CD38 antibodies for treatment of acute myeloid leukemia |
| US12091466B2 (en) | 2015-05-20 | 2024-09-17 | Janssen Biotech, Inc. | Anti-CD38 antibodies for treatment of light chain amyloidosis and other CD38-positive hematological malignancies |
| US10766965B2 (en) | 2015-05-20 | 2020-09-08 | Janssen Biotech, Inc. | Anti-CD38 antibodies for treatment of light chain amyloidosis and other CD38-positive hematological malignancies |
| US10668149B2 (en) | 2015-06-22 | 2020-06-02 | Janssen Biotech, Inc. | Combination therapies for heme malignancies with anti-CD38 antibodies and survivin inhibitors |
| US11021543B2 (en) | 2015-06-24 | 2021-06-01 | Janssen Biotech, Inc. | Immune modulation and treatment of solid tumors with antibodies that specifically bind CD38 |
| US11566079B2 (en) | 2015-11-03 | 2023-01-31 | Janssen Biotech, Inc. | Subcutaneous formulations of anti-CD38 antibodies and their uses |
| US10385135B2 (en) | 2015-11-03 | 2019-08-20 | Janssen Biotech, Inc. | Subcutaneous formulations of anti-CD38 antibodies and their uses |
| US11732051B2 (en) | 2015-11-03 | 2023-08-22 | Janssen Biotech, Inc. | Subcutaneous formulations of anti-CD38 antibodies and their uses |
| US11708419B2 (en) | 2015-11-03 | 2023-07-25 | Janssen Biotech, Inc. | Subcutaneous formulations of anti-CD38 antibodies and their uses |
| US11708420B2 (en) | 2015-11-03 | 2023-07-25 | Janssen Biotech, Inc. | Subcutaneous formulations of anti-CD38 antibodies and their uses |
| US10781261B2 (en) | 2015-11-03 | 2020-09-22 | Janssen Biotech, Inc. | Subcutaneous formulations of anti-CD38 antibodies and their uses |
| WO2017077382A1 (en) | 2015-11-06 | 2017-05-11 | Orionis Biosciences Nv | Bi-functional chimeric proteins and uses thereof |
| US11702477B2 (en) | 2015-11-06 | 2023-07-18 | Orionis Biosciences BV | Bi-functional chimeric proteins and uses thereof |
| US11623957B2 (en) | 2015-12-07 | 2023-04-11 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and PSMA |
| US10988538B2 (en) | 2016-02-05 | 2021-04-27 | Orionis Biosciences BV | Bispecific signaling agents and uses thereof |
| WO2017134305A1 (en) | 2016-02-05 | 2017-08-10 | Orionis Biosciences Nv | Bispecific signaling agents and uses thereof |
| WO2017134302A2 (en) | 2016-02-05 | 2017-08-10 | Orionis Biosciences Nv | Targeted therapeutic agents and uses thereof |
| US11661455B2 (en) | 2016-02-05 | 2023-05-30 | Orionis Biosciences BV | Chimeric protein comprising an interferon alpha 2mutant and a PD-L1 binding moiety |
| US11236166B2 (en) | 2016-02-05 | 2022-02-01 | Orionis Biosciences BV | CD8 binding agents |
| EP3998281A1 (en) | 2016-02-05 | 2022-05-18 | Orionis Biosciences BV | Cd8 binding agents |
| EP3411397A1 (en) * | 2016-02-05 | 2018-12-12 | Orionis Biosciences NV | Cd8 binding agents |
| EP3909978A1 (en) | 2016-02-05 | 2021-11-17 | Orionis Biosciences BV | Clec9a binding agents and use thereof |
| EP4421094A2 (en) | 2016-02-05 | 2024-08-28 | Orionis Biosciences BV | Targeted therapeutic agents and uses thereof |
| EP4059957A1 (en) | 2016-02-05 | 2022-09-21 | Orionis Biosciences BV | Bispecific signaling agents and uses thereof |
| US11001631B2 (en) | 2016-02-05 | 2021-05-11 | Orionis Biosciences BV | Clec9A binding agents |
| US11248057B2 (en) | 2016-03-07 | 2022-02-15 | Vib Vzw | CD20 binding single domain antibodies |
| US11753463B2 (en) | 2016-05-13 | 2023-09-12 | Orionis Biosciences BV | Therapeutic targeting of non-cellular structures |
| CN109563141A (en) * | 2016-05-13 | 2019-04-02 | 奥里尼斯生物科学公司 | To the therapeutic targeting of cellular structures |
| EP3455245A2 (en) * | 2016-05-13 | 2019-03-20 | Orionis Biosciences NV | Therapeutic targeting of non-cellular structures |
| US11236141B2 (en) | 2016-05-13 | 2022-02-01 | Orionis Biosciences BV | Targeted mutant interferon-beta and uses thereof |
| WO2017194782A2 (en) | 2016-05-13 | 2017-11-16 | Orionis Biosciences Nv | Therapeutic targeting of non-cellular structures |
| US11492407B2 (en) | 2016-06-14 | 2022-11-08 | Xencor, Inc. | Bispecific checkpoint inhibitor antibodies |
| US12054545B2 (en) | 2016-06-28 | 2024-08-06 | Xencor, Inc. | Heterodimeric antibodies that bind somatostatin receptor 2 |
| US11299724B2 (en) | 2016-07-14 | 2022-04-12 | Limited Liability Company Biochemical Agent | Fusion protein, polynucleotide, genetic construct, producer, preparation for regeneration of cartilage |
| WO2018014068A1 (en) * | 2016-07-19 | 2018-01-25 | Teva Pharmaceuticals Australia Pty Ltd | Attenuated type i ifn cd47 combination therapy |
| WO2018014067A1 (en) * | 2016-07-19 | 2018-01-25 | Teva Pharmaceuticals Australia Pty Ltd | Anti-cd47 combination therapy |
| US11618784B2 (en) | 2016-07-19 | 2023-04-04 | Teva Pharmaceuticals Australia Pty Ltd. | Anti-CD47 combination therapy |
| US11084859B2 (en) | 2016-10-24 | 2021-08-10 | Orionis Biosciences BV | Targeted mutant interferon-gamma and uses thereof |
| WO2018077893A1 (en) | 2016-10-24 | 2018-05-03 | Orionis Biosciences Nv | Targeted mutant interferon-gamma and uses thereof |
| US11981737B2 (en) | 2016-11-18 | 2024-05-14 | Hoffmann-La Roche Inc. | Anti-HLA-G antibodies and use thereof |
| US10906985B2 (en) | 2017-02-06 | 2021-02-02 | Orionis Biosciences, Inc. | Targeted engineered interferon and uses thereof |
| US11384154B2 (en) | 2017-02-06 | 2022-07-12 | Orionis Biosciences BV | Targeted chimeric proteins and uses thereof |
| WO2018141964A1 (en) | 2017-02-06 | 2018-08-09 | Orionis Biosciences Nv | Targeted chimeric proteins and uses thereof |
| EP3576765A4 (en) * | 2017-02-06 | 2020-12-02 | Orionis Biosciences, Inc. | Targeted engineered interferon and uses thereof |
| WO2018144999A1 (en) | 2017-02-06 | 2018-08-09 | Orionis Biosciences, Inc. | Targeted engineered interferon and uses thereof |
| WO2018146074A1 (en) | 2017-02-07 | 2018-08-16 | Vib Vzw | Immune-cell targeted bispecific chimeric proteins and uses thereof |
| US11246911B2 (en) | 2017-02-07 | 2022-02-15 | Vib Vzw | Immune-cell targeted bispecific chimeric proteins and uses thereof |
| EP3618870A4 (en) * | 2017-05-01 | 2021-04-21 | The Trustees of The University of Pennsylvania | Monoclonal antibodies against alpha-synuclein fibrils |
| US11220538B2 (en) | 2017-05-01 | 2022-01-11 | The Trustees Of The University Of Pennsylvania | Monoclonal antibodies against alpha-synuclein fibrils |
| US12071474B2 (en) | 2017-05-01 | 2024-08-27 | The Trustees Of The University Of Pennsylvania | Monoclonal antibodies against alpha-synuclein fibrils |
| WO2018215938A1 (en) * | 2017-05-24 | 2018-11-29 | Novartis Ag | Antibody-cytokine engrafted proteins and methods of use |
| US11541103B2 (en) | 2017-08-03 | 2023-01-03 | Amgen Inc. | Interleukin-21 mutein/ anti-PD-1 antibody conjugates |
| US12091463B2 (en) | 2017-08-09 | 2024-09-17 | Orionis Biosciences, Inc. | Clec9A binding agents comprising a recombinant heavy-chain only antibody (VHH) |
| US11498966B2 (en) | 2017-08-09 | 2022-11-15 | Orionis Biosciences Inc. | PD-1 and PD-L1 binding agents |
| US11566072B2 (en) | 2017-08-09 | 2023-01-31 | Orionis Biosciences, Inc. | CD8 binding agents |
| US11970545B2 (en) | 2017-10-12 | 2024-04-30 | Mcmaster University | T cell-antigen coupler with Y182T mutation and methods of uses thereof |
| US11618787B2 (en) | 2017-10-31 | 2023-04-04 | Janssen Biotech, Inc. | Methods of treating high risk multiple myeloma |
| US11518808B2 (en) | 2018-01-12 | 2022-12-06 | Amgen Inc. | Anti-PD-1 antibodies and methods of treatment |
| EP3743448A4 (en) * | 2018-01-26 | 2021-11-03 | Orionis Biosciences, Inc. | Xcr1 binding agents and uses thereof |
| US11896643B2 (en) | 2018-02-05 | 2024-02-13 | Orionis Biosciences, Inc. | Fibroblast binding agents and use thereof |
| US11976117B2 (en) | 2018-07-17 | 2024-05-07 | Mcmaster University | T cell-antigen coupler with various construct optimizations |
| US11878035B2 (en) | 2018-07-17 | 2024-01-23 | Triumvira Immunologics Usa, Inc. | T cell-antigen coupler with various construct optimizations |
| US12084497B2 (en) | 2018-08-08 | 2024-09-10 | Orionis Biosciences, Inc. | SIRP1α targeted chimeric proteins and uses thereof |
| US20210198375A1 (en) * | 2018-09-04 | 2021-07-01 | Nanjing Umab-Biopharma Co., Ltd. | Fusion protein and its applicaton in preparing medicine for treating tumor and/or viral infection |
| WO2020120730A1 (en) | 2018-12-14 | 2020-06-18 | Morphosys Ag | Antibody formulations |
| US11440943B2 (en) | 2019-03-28 | 2022-09-13 | Orionis Biosciences, Inc. | Therapeutic interferon alpha 1 proteins |
| US11655302B2 (en) * | 2019-06-10 | 2023-05-23 | Sanofi | Anti-CD38 antibodies and formulations |
| US20200399391A1 (en) * | 2019-06-10 | 2020-12-24 | Sanofi | Anti-cd38 antibodies and formulations |
| WO2021009332A1 (en) | 2019-07-18 | 2021-01-21 | Enyo Pharma | Method for decreasing adverse-effects of interferon |
| US11919956B2 (en) | 2020-05-14 | 2024-03-05 | Xencor, Inc. | Heterodimeric antibodies that bind prostate specific membrane antigen (PSMA) and CD3 |
| US11919958B2 (en) | 2020-08-19 | 2024-03-05 | Xencor, Inc. | Anti-CD28 compositions |
| US11591401B2 (en) | 2020-08-19 | 2023-02-28 | Xencor, Inc. | Anti-CD28 compositions |
| US11739144B2 (en) | 2021-03-09 | 2023-08-29 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and CLDN6 |
| US11859012B2 (en) | 2021-03-10 | 2024-01-02 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and GPC3 |
| WO2022200525A1 (en) | 2021-03-26 | 2022-09-29 | Innate Pharma | Multispecific proteins comprising an nkp46-binding site, a cancer antgienge binding site fused to a cytokine for nk cell engaging |
| US12016923B2 (en) | 2021-06-01 | 2024-06-25 | Triumvira Immunologics Usa, Inc. | Claudin 18.2 T cell-antigen couplers and uses thereof |
| WO2022258691A1 (en) | 2021-06-09 | 2022-12-15 | Innate Pharma | Multispecific proteins binding to nkg2d, a cytokine receptor, a tumour antigen and cd16a |
| WO2022258662A1 (en) | 2021-06-09 | 2022-12-15 | Innate Pharma | Multispecific proteins binding to nkp46, a cytokine receptor, a tumour antigen and cd16a |
| WO2022258678A1 (en) | 2021-06-09 | 2022-12-15 | Innate Pharma | Multispecific proteins binding to nkp30, a cytokine receptor, a tumour antigen and cd16a |
| WO2023052846A3 (en) * | 2021-09-30 | 2023-05-25 | Ildong Pharmaceutical Co., Ltd. | Immunocytokine containing il-21r mutein |
| WO2023178253A3 (en) * | 2022-03-18 | 2023-10-19 | Igm Biosciences, Inc. | Use of a multimeric anti-pd-l1 binding molecule in combination with a monoclonal antibody therapy |
| US12129309B2 (en) | 2022-05-04 | 2024-10-29 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and CD38 |
| WO2024040249A1 (en) | 2022-08-18 | 2024-02-22 | Regeneron Pharmaceuticals, Inc. | Interferon receptor agonists and uses thereof |
| WO2024040247A1 (en) | 2022-08-18 | 2024-02-22 | Regeneron Pharmaceuticals, Inc. | Interferon proproteins and uses thereof |
| CN117430716B (en) * | 2023-10-16 | 2024-05-31 | 珠海臻谱基因科技有限公司 | Recombinant interferon drug targeting conserved HIV gp41 subunit near-membrane-end outer region and application thereof |
| CN117430716A (en) * | 2023-10-16 | 2024-01-23 | 珠海臻谱基因科技有限公司 | Recombinant interferon drug targeting conserved HIV gp41 subunit near-membrane-end outer region and application thereof |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2018200221B2 (en) | Polypeptide constructs and uses thereof | |
| JP6929827B2 (en) | ASGPR antibody and its use | |
| CN107531784B (en) | Ultra-potent neutralization of cytokines by multispecific antibodies and uses thereof | |
| US20210244825A1 (en) | Il-21 antibodies | |
| KR20180089514A (en) | Multispecific antibody molecules with specificity for TNF-alpha, IL-17A and IL-17F | |
| NZ623812B2 (en) | Polypeptide constructs and uses thereof | |
| NZ734996B2 (en) | Polypeptide constructs and uses thereof | |
| EA040832B1 (en) | POLYPEPTIDE CONSTRUCTS AND THEIR APPLICATIONS | |
| WO2024005713A1 (en) | Bispecific antibodies for hbv surface antigen(hbsag)and cd3 and uses thereof | |
| NZ715807B2 (en) | Polypeptide constructs and uses thereof | |
| TW202421649A (en) | Fusions with cd8 antigen binding molecules for treating chronic viral infection | |
| CN117136198A (en) | Agonist anti-IL-2R antibodies and methods of use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12843185 Country of ref document: EP Kind code of ref document: A2 |
|
| ENP | Entry into the national phase |
Ref document number: 2851892 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 232247 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2014/005108 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2014537430 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2012327877 Country of ref document: AU Date of ref document: 20121029 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201400488 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: 20147014249 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012843185 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112014009925 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112014009925 Country of ref document: BR Kind code of ref document: A2 Effective date: 20140425 |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01E Ref document number: 112014009925 Country of ref document: BR Kind code of ref document: A2 |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01E Ref document number: 112014009925 Country of ref document: BR Kind code of ref document: A2 |
|
| ENPW | Started to enter national phase and was withdrawn or failed for other reasons |
Ref document number: 112014009925 Country of ref document: BR Kind code of ref document: A2 |
|
| ENP | Entry into the national phase |
Ref document number: 2012843185 Country of ref document: EP Effective date: 20140528 |
|
| ENP | Entry into the national phase |
Ref document number: 2012843185 Country of ref document: EP Effective date: 20140528 |
|
| ENP | Entry into the national phase |
Ref document number: 112014009925 Country of ref document: BR Kind code of ref document: A2 Effective date: 20140425 |