WO2013047509A1 - 新規ホスファチジルイノシトール3キナーゼ阻害剤及び医薬組成物 - Google Patents
新規ホスファチジルイノシトール3キナーゼ阻害剤及び医薬組成物 Download PDFInfo
- Publication number
- WO2013047509A1 WO2013047509A1 PCT/JP2012/074542 JP2012074542W WO2013047509A1 WO 2013047509 A1 WO2013047509 A1 WO 2013047509A1 JP 2012074542 W JP2012074542 W JP 2012074542W WO 2013047509 A1 WO2013047509 A1 WO 2013047509A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- pi3k
- equation
- formula
- cancer
- Prior art date
Links
- 0 CCC(C(N)=Cl)NC([C@](*)NC(C[C@](C=CCCS)[U]C(C(*C)*C(C)C)=[U])=Cl)=N Chemical compound CCC(C(N)=Cl)NC([C@](*)NC(C[C@](C=CCCS)[U]C(C(*C)*C(C)C)=[U])=Cl)=N 0.000 description 4
- ZGXGRQXAWONMPL-WGQQNVHESA-N CC(C)C[C@H](C(NCC(O[C@@H](CC(N[C@@H]1C(C)C)=O)/C=C/CCSSCC[C@H]2NC1=O)=O)=O)NC2=O Chemical compound CC(C)C[C@H](C(NCC(O[C@@H](CC(N[C@@H]1C(C)C)=O)/C=C/CCSSCC[C@H]2NC1=O)=O)=O)NC2=O ZGXGRQXAWONMPL-WGQQNVHESA-N 0.000 description 1
Images























Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/12—Cyclic peptides with only normal peptide bonds in the ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/06—Tripeptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/10—Tetrapeptides
- C07K5/1002—Tetrapeptides with the first amino acid being neutral
- C07K5/1005—Tetrapeptides with the first amino acid being neutral and aliphatic
- C07K5/101—Tetrapeptides with the first amino acid being neutral and aliphatic the side chain containing 2 to 4 carbon atoms, e.g. Val, Ile, Leu
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/12—Cyclic peptides with only normal peptide bonds in the ring
- C07K5/123—Tripeptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Definitions
- the present invention relates to a novel phosphatidylinositol 3-kinase inhibitor and a pharmaceutical composition containing the same.
- Phosphatidylinositol 3-kinase (hereinafter referred to as “PI3K”) phosphorylates the 3-position phosphate group of the inositol ring of the phospholipid phosphatidylinositol 4,5-biphosphate (PIP2) present on the cell membrane.
- PIP3 phosphatidylinositol 3,4,5-triphosphate
- Non-patent Document 2 Cantley, L. C. Science 296, 1655-1657, doi: 10.1126 / science. 296.5573.1655 (2002)).
- PI3K is known to be associated with cancer.
- PI3K forms a heterodimer composed of a catalytic subunit and a regulatory subunit.
- the PIK3CA gene encodes p110 ⁇ , a catalytic subunit of class IA PI3K.
- high-frequency gene amplification and gain-of-function point mutations have been reported in various cancer types such as breast cancer and colorectal cancer (Non-Patent Document 3: Samuels et al. Science 304). , 554 (2004); Non-Patent Document 4: Ikenoue et al. Cancer Res 65, 4562 (2005); Non-Patent Document 5: Kang et al., Proc. Natl. Acad. Sci. USA 102, 802 (2005)) .
- PTEN Phosphatase and tensin homologue deleted on chromosome 10
- PIP3 Phosphatase and tensin homologue deleted on chromosome 10
- PIP3 Phosphatase and tensin homologue deleted on chromosome 10
- Non-patent Document 8 Han et al. Cancer Res 60, 3147 (2000). These mutations are thought to result in abnormal constitutive activation of the PI3K / AKT pathway and transmission of cancer cell survival signals.
- Non-Patent Document 9 KongK & Yamori, Current Medicinal Chemistry 16, 2839-2854 (2009 )).
- Histone acetylation is one of the important mechanisms of epigenetic regulation (Non-Patent Document 10: Carew et al., Giles, Cancer Let 269, 7-17 (2008)). It has been found that inhibition of acetylase (hereinafter referred to as “HDAC”) causes a change in gene expression, which is accompanied by cell differentiation and apoptosis. For this reason, HDAC inhibitors are attracting attention as new cancer molecule targeted drugs.
- HDAC acetylase
- Depsipeptide compounds are generic terms for peptides in which one or more amide bonds (—CONHR—) are substituted with ester bonds (—COOR).
- FK228 FR901228, also called romidepsin
- FR901228 is a compound isolated as a fermentation product from Chromobacterium violaceum
- Non-patent Document 12 Furumai et al. Cancer Res 62, 4916 (2002).
- FK228 is in clinical trials as an anti-cancer agent, and together with HDAC inhibitor suberoylanilide hydroxamic acid (SAHA), FK228 has been approved by the US FDA as a therapeutic agent for cutaneous T-cell lymphoma. is recieving.
- SAHA HDAC inhibitor suberoylanilide hydroxamic acid
- depsipeptide refers to FK228 in a narrow sense, but is used as a term having the above-mentioned meaning in this specification.
- Non-patent Document 13 Wozniak et al. Haematologica 95, 613 (2010 )
- HDAC inhibitors per se affect the PI3K / AKT pathway
- Non-Patent Document 14 Hanker et al., J Molecular Signaling 4, 5 (2009)
- Non-Patent Document 15 Graham et al. al. Clinical Cancer Res 12, 223 (2006)).
- TSA trichostatin A
- the present invention aims to provide a novel PI3K inhibitor. Furthermore, the development of drugs that inhibit both PI3K and HDAC is considered to make a great contribution to overcoming refractory cancer. Therefore, an object of the present invention is to provide a substance having both a PI3K inhibitory action and an HDAC inhibitory action. Finally, an object of the present invention is to provide a novel anticancer pharmaceutical composition containing these substances, particularly an anticancer pharmaceutical composition that is effective even for intractable cancer.
- Non-patent Document 18 Tugendreich et al., Genome Research) 11, 1899 (2001);
- Non-patent document 19 Rodriguez-Escudero et al. Biochemical Journal 390, 613 (2005);
- Non-patent document 20 Cid et al.
- Non-Patent Document 19 Non-Patent Document 21: The 15th Annual Meeting of the Japanese Molecular Target Society of Japan, Abstract, May 31, 2011, Title No. P10-5 “Search for new PI3K inhibitors using budding yeast as a screening tool” ) was used to screen a compound library.
- depsipeptide compounds that are HDAC inhibitors, that is, FK228 and its analogs were selected as candidate substances.
- the inventors of the present invention evaluated PI3K inhibitory activity in vitro and confirmed that these compounds have direct inhibitory activity of PI3K at a concentration of ⁇ M order. Further, in Western blot analysis using cultured human cells, these compounds were found to suppress phosphorylated AKT and downstream molecules of the AKT pathway. Furthermore, from the results of the MTT assay, it was found that these compounds exert a strong cell killing effect on cells resistant to HDAC inhibitors in a specific concentration range. Then, it was confirmed that the cell death was apoptosis, and that the apoptosis was induced by the HDAC / PI3K double inhibitory activity of the depsipeptide compounds, thereby completing the present invention.
- a phosphatidylinositol 3 kinase (PI3K) inhibitor comprising a depsipeptide compound represented by the following formula 1 or a physiologically acceptable salt thereof:
- Formula 1 (In the formula, A represents —CONH— or —CH (OH) —, and R 1 , R 2 and R 3 are the same or different and represent a hydrogen atom, a lower alkyl group, a lower alkylidene group, substituted or unsubstituted.
- a pharmaceutical composition for treating refractory cancer comprising the PI3K inhibitor according to any one of [1] to [3] as an active ingredient; [5] The pharmaceutical composition for treating refractory cancer according to [4], wherein the PI3K inhibitor is administered at a dose of about 1 mg / day to 10,000 mg / day per kg of body weight.
- a novel PI3K inhibitor is provided.
- This novel PI3K inhibitor is a compound having a completely different structure from conventionally known PI3K inhibitors.
- the PI3K inhibitor of the present invention also has an HDAC inhibitory activity and can also act as an HDAC inhibitor.
- the PI3K inhibitor can also be used against cancer cells resistant to an HDAC inhibitor. It exerts cell-killing activity by inducing apoptosis. Therefore, the PI3K inhibitor of the present invention can provide a pharmaceutical composition that is effective against cancer cells that are resistant to conventional anticancer agents including HDAC inhibitors.
- the PI3K inhibitor of the present invention has been found to have the possibility of inhibiting other kinases in addition to HDAC inhibitory activity, and is presumed to be advantageous in controlling cancer cells. It may be a multi-target drug.
- the PI3K inhibitor of the present invention is used as a molecular target drug that acts specifically on cancer, it is considered to have relatively little effect on cells and tissues other than cancer cells.
- FK228 is relatively light even in cases where no side effects are caused even by long-term administration or in the case of any side effects, and can be suppressed by using in combination with a drug that suppresses this.
- the FK228 analog is considered to have lower side effects than FK228 as will be described later, and therefore the pharmaceutical composition of the present invention may cause serious side effects even when used at a high dose to the living body. Is low.
- FIG. 1 is a diagram showing a synthetic route of a compound of the present invention.
- Boc tert-butoxycarbonyl
- TBS tert-butyldimethylsilyl
- Tr trityl
- PMB p-methoxybenzyl
- FIG. 2A is a diagram showing the measurement of PI3K inhibitory activity of FK228 and its analogs.
- FIG. 2B shows the PI3K inhibition curve of LY294002 measured in the same manner.
- the vertical axis shows the inhibition rate for PI3K activity.
- FIG. 2C shows a PI3K inhibition curve of FK228 measured in the same manner.
- the vertical axis shows the inhibition rate for PI3K activity.
- FIG. 2D shows the PI3K inhibition curve of FK-A5 measured in the same manner.
- FIG. 3 is a diagram showing evaluation of suppression of AKT pathway by Western blotting. Panel A represents changes in phosphorylated AKT over time of compound treatment.
- the figure shows the results of detecting phosphorylated (p-) AKT and AKT by Western blotting after 10 ⁇ M LY294002, FK228 or FK-A5 was allowed to act on PC3 cells for 5 minutes, 30 minutes or 180 minutes, respectively.
- Panel B represents inhibition of the AKT pathway due to changes in FK228 and FK-A5 concentrations.
- PC3 cells were treated with FK228 and FK-A5 at the concentrations ( ⁇ M) shown in the figure for 180 minutes, and using the collected samples, phosphorylated AKT (Ser-473, Thr-308) and AKT, phosphorylated GSK- 3 ⁇ (Ser-9), phosphorylated mTOR (Ser-2448), phosphorylated p70S6K (Thr-389), phosphorylated 4E-BP1 (Thr-37 / 46), phosphorylated MEK1 / 2 (Ser-217 / 221) 2 shows the results of Western blot analysis in which the expression level of phosphorylated ERK1 / 2 (Thr-202 / Tyr-204) was detected.
- FIG. 4A is a diagram showing the results of confirming the expression of HDAC2 and HDAC1 in HCT116 cells, RKO cells, and CO115 cells by Western blotting.
- FIG. 4B is a diagram showing the evaluation of the cytocidal effect of FK228 and FK-A5 in HCT116 cells.
- LY294002 (50 ⁇ M), SAHA (2.5 ⁇ M), combined use of LY294002 (50 ⁇ M) and SAHA (2.5 ⁇ M) at the same concentration (SAHA + LY), and FK228, FK-A5, FK228 or FK-A5 at the concentrations shown in the figure.
- SAHA + LY SAHA + LY
- FK228, FK-A5, FK228 or FK-A5 at the concentrations shown in the figure
- FIG. 4C is a diagram showing the evaluation of the cytocidal effect of FK228 and FK-A5 in RKO cells, measured in the same manner as in FIG. 4B.
- FIG. 4D is a diagram showing the evaluation of the cell killing effect of FK228 and FK-A5 in CO115 cells, measured in the same manner as in FIG. 4A.
- FIG. 5A is a diagram showing the results of FACS analysis of HCT116 cells cultured for 24 hours in the presence of FK228 or FK228 and LY294002 (50 ⁇ M) at the concentrations shown in the figure.
- FIG. 5B is a diagram showing the results of analysis performed in the same manner as FIG. 5A using FK-A5 instead of FK228.
- FIG. 5C is a diagram showing the results of analyzing the results of FIG. 5B for FK-A5 and calculating the percentages of the G2 / M, S, G1 / G0, and subG1 fractions.
- FIG. 5B is a diagram showing the results of analysis performed in the same manner as FIG. 5A using FK-A5 instead of FK228.
- FIG. 5C is a diagram showing the results of analyzing the results of FIG. 5B for FK-A5 and calculating the percentages of the G2 / M, S, G1 / G0, and subG1 fractions.
- FIG. 5D shows the expression of PARP, cleared PARP, phosphorylated AKT (S473), AKT, acetylated histone H3, and acetylated histone H4 when HCT116 cells were treated with SAHA and FK-228 at the concentrations shown in the figure. It is a figure showing the result of having performed Western blot analysis.
- FIG. 6 is a diagram showing evaluation of suppression of the AKT pathway by Western blotting performed in the same manner as the experiment shown in FIG. 3 panel B. From the left, it shows suppression of the AKT pathway due to changes in the concentrations of FK-A11, FK-A5, and FK228.
- FIG. 7A is a diagram showing the evaluation of the cytocidal effect of FK-A11 in HCT116 cells.
- LY294002 (“LY”; 50 ⁇ M), SAHA (2.5 ⁇ M), LY294002 (50 ⁇ M) and SAHA (2.5 ⁇ M) at the same concentration (SAHA + LY), and FK-A11 at the concentration shown in the figure.
- FIG. 7B is a diagram showing the evaluation of the cell killing effect of FK-A11 in RKO cells, measured in the same manner as in FIG. 7A.
- FIG. 7C is a diagram showing the evaluation of the cell killing effect of FK-A11 in CO115 cells, measured in the same manner as in FIG. 7A.
- FIG. 7D is a diagram showing an evaluation of the cell killing effect of FK-A11 in normal cells KMST6 cells (non-tumor, fibroblasts), measured in the same manner as in FIG. 7A.
- FIG. 7B is a diagram showing the evaluation of the cell killing effect of FK-A11 in RKO cells, measured in the same manner as in FIG. 7A.
- FIG. 7C is a diagram showing the evaluation of the cell killing effect of FK-A11 in CO115 cells, measured in the same manner as in FIG. 7A.
- FIG. 7D is a diagram showing an evaluation of the cell killing effect of FK-A11 in normal cells KMST6 cells (non
- FIG. 8A is a view showing a comparison of cell killing effects of FK228, FK-5, and FK-A11 in HCT116 cells, measured in the same manner as FIG. 7A. Bars with horizontal lines indicate controls (DMSO, SAHA, LY, or SAHA + LY), bars with diagonal lines indicate FK228, black bars indicate FK-5, and white bars indicate FK-A11.
- FIG. 8B is a diagram showing an evaluation of the cell killing effect of FK228, FK-5, and FK-A11 in RKO cells, measured in the same manner as in FIG. 7A. Bars with horizontal lines indicate controls (DMSO, SAHA, LY, or SAHA + LY), bars with diagonal lines indicate FK228, black bars indicate FK-5, and white bars indicate FK-A11.
- FIG. 8C is a diagram showing the evaluation of the cell killing effect of FK228, FK-5, and FK-A11 in CO115 cells, measured in the same manner as in FIG. 7A. Bars with horizontal lines indicate controls (DMSO, SAHA, LY, or SAHA + LY), bars with diagonal lines indicate FK228, black bars indicate FK-5, and white bars indicate FK-A11.
- the PI3K inhibitor of the present invention is characterized by comprising a depsipeptide compound represented by the following formula 1 or a physiologically acceptable salt thereof.
- A represents —CONH— or —CH (OH) —
- R 1 , R 2 and R 3 are the same or different and represent a hydrogen atom, a lower alkyl group, a lower alkylidene group, substituted or unsubstituted.
- R 1 is a hydrogen atom, a lower alkyl group, a lower alkylidene group or a substituted or unsubstituted aralkyl group
- R 2 is a lower alkyl group
- R 3 is a hydrogen atom or a lower alkyl group.
- the “lower alkyl group” includes methyl, ethyl, n-propyl, i-propyl, cyclopropyl, n-butyl, s-butyl, t-butyl, cyclobutyl, n-pentyl, i-pentyl, 2 And straight-chain or branched alkyl groups having 1 to 6 carbon atoms such as -methylbutyl, cyclopentyl, n-hexyl and cyclohexyl.
- Examples of the “lower alkylidene group” include linear or branched alkylidene groups having 1 to 6 carbon atoms such as methylene, ethylidene, propylidene, cyclopropylidene, butylidene, pentylidene and hexylidene.
- Examples of the “aryl group” include phenyl, naphthyl, pyridinyl, furanyl and the like, and examples of the substituent include a hydroxyl group, a hydroxyl group protected with a protecting group, an amino group, and an amino group protected with a protecting group.
- aralkyl group examples include benzyl, 1-phenylethyl, naphthylmethyl, pyridinylmethyl and the like, and the substituents include a hydroxyl group, a hydroxyl group protected with a protecting group, an amino group, and an amino group protected with a protecting group. Is mentioned.
- Particularly preferable compounds include the following compounds.
- R 3 is a hydrogen atom and / or R 1 is a substituted or unsubstituted aralkyl group.
- physiologically acceptable salts include inorganic salts (sodium salt, potassium salt, lithium salt; calcium salt, magnesium salt; aluminum salt, iron salt). , Zinc salts, copper salts, nickel salts, cobalt salts; ammonium salts, etc.), and various organic salts, hydrohalide salts, inorganic acid salts, organic acid salts, amino acid salts, and the like.
- one or more of these depsipeptide compounds or salts can be appropriately selected and used.
- FK228 may be obtained by isolating and purifying a natural product produced by a microorganism, and may be produced by semi-synthesis or total synthesis by a conventionally known method.
- a conventionally known method for the synthesis of the compound of the present invention, for example, Narita et al. Chemistry-A European Journal 15, 11174-11186 (2009) (Non-patent Document 22); Takizawa et al. Chemical Communication, 1677-1679 (2008) (Non-patent Document 23); Takizawa et al. Et Heterocycles 76, 275-290 (2008) (Non-Patent Document 24).
- the cancers to be treated by the pharmaceutical composition of the present invention are various cancers that are known to be effective for FK228, and are particularly refractory cancers. Specifically, skin cancer, mesothelioma, lung cancer, stomach cancer, liver cancer, colon cancer, breast cancer, esophageal cancer, pancreatic cancer, uterine cancer (cervical cancer, intimal cancer), ovary Cancer such as cancer, skin cancer, urological cancer, head and neck cancer, cancer of unknown primary origin, hematopoietic tumor (leukemia, lymphoma), bone and soft tissue sarcoma, etc. Low one is mentioned.
- Non-patent Document 3 the effectiveness of this drug is expected.
- the pharmaceutical composition of the present invention can be appropriately produced by a method known in the art using the PI3K inhibitor of the present invention and various additives known in the pharmaceutical industry.
- the administration route of the pharmaceutical composition of the present invention can be selected from known routes such as oral, nasal, sublingual, instillation, transdermal, injection, enteral, intrarectal and the like. Preferably, it is oral or injection.
- the pharmaceutical composition of the present invention can be used for oral preparations (tablets, granules, capsules, powders, etc.), injections, suppositories, patches, drops, gargles, eye drops, lozenges, etc. Can be used in Methods for producing such various preparations are well known to those skilled in the art.
- various excipients, disintegrants, lubricants, binders, surfactants, fluidity promoters, colorants, fragrances and the like used in the pharmaceutical industry can be used as additives. As appropriate, it can be used in the production of the pharmaceutical composition of the present invention.
- the pharmaceutical composition of the present invention can contain other known pharmaceutically active ingredients.
- an antiemetic, a digestive agent, other anticancer agents, etc. are mentioned.
- composition of the present invention can be combined with a known drug delivery system.
- examples thereof include liposomes, inorganic nanoparticles, inorganic-organic hybrid nanoparticles, and implants.
- the dosage and dosage form of the pharmaceutical composition of the present invention can be individually determined according to the cancer to be treated, the method of administration, the age, weight, disease state, etc. of the patient to be administered.
- the active ingredient is about 1 mg / day to 10,000 mg / day, more preferably about 10 mg / day to about 5,000 mg / day, more preferably about 20 mg / day to about 5,000 mg / kg of active ingredient.
- Daily, most preferably about 50 mg / day to about 5,000 mg / day divided into once a day or 2-3 times a day for administration to a patient in need of treatment with the pharmaceutical composition of the invention can do.
- the screening system used in the present invention is based on the cell proliferation disorder caused by expressing human p110 ⁇ in budding yeast. Basically, the method of Rodriguez-Escudero et al. 19: Biochemical Journal 390, 613 (2005)). However, in this screening, seven ABC transporter genes and two transcription factor genes involved in activation of ABC transporter were knocked out instead of wild-type strain YPH499, which is commonly used as a budding yeast.
- the drug-sensitive strain AD1-9 (MAT alpha, yor1, snq2, pdr5, pdr10, pdr11, ycf1, pdr3, pdr15, pdr1, his1, ura3) was used (Non-patent Document 25: Rogers et al. J. of Mol. Microbiol. Biotechnol. 3, 207-214 (2001)).
- the AD1-9 strain is available from Nader Nourizad (Stanford Genome Technology Center Stanford University, CA, USA).
- Non-patent Document 26 Alani et al., Genetics 116, 541 (1987)
- the LEU2 gene disruption AD1-9 strain was prepared and used.
- the transformed strain AD1-9 was cultured at 30 ° C. in the minimal complete medium SC-UL without uracil and leucine or SGal-UL in which 2% glucose was replaced with 2% galactose.
- AD1-9 used for screening it was observed at 5 ⁇ M. That is, it was confirmed that AD1-9 used for screening was about 10 times more sensitive to LY294002 than YPH499.
- the above system was used to screen a library of compounds provided by the Ministry of Education, Culture, Sports, Science and Technology's Cancer Specific Area Research / Integrated Cancer Chemotherapy Information Support Team. Each compound was added to SGal-UL liquid medium at a concentration of 0.5 ⁇ M and 5 ⁇ M, and the AD1-9 transformant was shake-cultured for 24 hours, and A 600 was measured. DMSO and LY294002 were subjected to the same screening as controls. It performs three independent repeated experiments, the average value of the A 600, and enumerated in order of decreasing its value, the compound becomes higher were selected as compounds to restore a cell proliferation disorder by pi 10a.
- Table 2 Structural formula of FK228 and its analogs and 50% inhibitory concentration (IC 50 ) against HDAC1 and HDAC6
- the ratio of IC 50 for HDAC1 and HDAC6 is believed to be associated with risk of developing an effect on the heart. Any analog is expected to have such a low risk compared to FK228.
- step (A) an aldehyde derivative having a side chain R 1 was reacted with ethyl acetate to obtain an adduct.
- step (B) protection of the hydroxyl group of the adduct, deprotection of the amino group and transesterification were sequentially performed to give an amino ester derivative.
- step (C) the amino ester derivative and a carboxylic acid derivative prepared from cysteine were condensed to give peptide derivative (I).
- step (D) a carboxylic acid derivative known in the literature and an amino acid ester derivative having a side chain R 2 were condensed to obtain a peptide derivative (II).
- step (E) the two peptide derivatives (I) and (II) obtained above were condensed to obtain a tripeptide derivative.
- step (F) intramolecular lactonization, disulfide bond formation, and hydroxyl group deprotection were sequentially performed to synthesize depsipeptide compounds.
- step (G) a tripeptide derivative having side chains R 1 and R 3 that can be prepared using a conventional route was condensed with a carboxylic acid derivative (II) to give a tetrapeptide derivative.
- step (H) intramolecular lactonization, disulfide bond formation, and hydroxyl group deprotection were sequentially performed to complete the synthesis of depsipeptide compounds.
- Each compound was mixed with 21 nM PI3K, 1000 nM phosphatidylinositol, 50 ⁇ M ATP, 5 mM MgCl 2 in assay buffer (20 mM HEPES, 2 mM DTT, 25 ⁇ M sodium cholate, 75 mM NaCl, 20 ⁇ M cantharidin) at room temperature. After 5 hours of reaction, a mobility shift assay was performed. The test was repeated twice. The inhibition rate was calculated as follows: As a control, the kinase reaction was evaluated by the product / substrate + product value calculated by MSA (mobility shift assay), and this was defined as 0% inhibition rate. The value of product / substrate + product when a drug was added thereto was calculated in the same manner, and the inhibition rate was calculated from the difference from the control value.
- FIG. 2A The result is shown in FIG. 2A. All compounds showed direct inhibitory activity against PI3K. Among them, SP-5, FK-A1, FK-A2, FK-A3, FK-A5 and FK-A6 showed a higher inhibition rate than FK228, and the strongest inhibitory activity was observed in FK- In A5, the PI3K activity inhibition rate was 66.8%.
- FK228 and FK-A5 inhibited PI3K activity in a concentration-dependent manner.
- the IC 50 of LY294002, FK228, and FK-A5 were 0.7 ⁇ M, 57.1 ⁇ M, and 26.2 ⁇ M, respectively. Inhibitory activity was observed in the range of 5 to 500 ⁇ M for FK228 and 1 to 300 ⁇ M for FK-A5. It was also found that FK-A5 inhibits PI3K more strongly than FK228. In addition, in the inhibition curve of PI3K of FK228, the inhibition rate at 10 ⁇ M was 6.5% (FIG. 2C).
- FK-A5 and FK-A6 had the strongest PI3K inhibitory activity among FK228 and its analogs (FIG. 2B).
- FK-A5 and FK-A6 are stereoisomers and have a characteristic structure with a benzyl (phenylmethyl) group at the 7-position (Table 2). This feature may contribute to the higher inhibitory activity of FK-A5 and FK-A6 than FK228.
- the docking simulation uses the trade name “eHiTS” (Simulated Biomolecular Systems) as simulation software, and the crystal structure data of a complex of PI3K (p110 ⁇ H1047R / p85 ⁇ ) -wortmannin as a three-dimensional structure of PI3K as a template (Protein Data Bank Identification Code: 3HHM).
- eHiTS Simulated Biomolecular Systems
- 3HHM Protein Data Bank Identification Code
- PC3 cells were obtained from the Medical Cell Resource Center, Institute of Aging Medicine, Tohoku University. The cells were cultured at 37 ° C. in the presence of 5% CO 2 using RPMI 1640 medium containing 10% of inactivated fetal bovine serum.
- Western blotting was performed as follows. After collecting various cells treated with each compound, Lysis buffer: 500 mM Tris-HCl pH 7.5, 100 mM NaCl, 2 mM EDTA, 1 mM sodium orthovanadate, 1% NP-40, It was dissolved in 1% protease inhibitor cocktail (Protease Inhibitor cocktail, manufactured by SIGMA-ALDRICH), and the supernatant was collected after centrifugation. The sample was electrophoresed on a 12.5% polyacrylamide gel, and PVDF membrane (Immobilon-FL , Millipore).
- Lysis buffer 500 mM Tris-HCl pH 7.5, 100 mM NaCl, 2 mM EDTA, 1 mM sodium orthovanadate, 1% NP-40, It was dissolved in 1% protease inhibitor cocktail (Protease Inhibitor cocktail, manufactured by SIGMA-ALDRICH), and the supernatant was collected after centrifugation. The sample was electrophoresed
- Anti-AKT antibody (“total AKT” or “AKT”), anti-phosphorylated AKT (Ser-473) antibody (“p-AKT (S473)”), anti-phosphorylated AKT (Thr-308) antibody (“p-AKT”) (T308) "), anti-phosphorylated GSK-3 ⁇ (Ser-9) antibody (" p-GSK-3 ⁇ "), anti-phosphorylated mTOR (Ser-2448) antibody (" p-mTOR "), anti-phosphorylated p70S6K (Thr-389) antibody (“p-p70S6K”), anti-phosphorylated 4E-BP1 (Thr-37 / 46) antibody (“p-4EBP1”), anti-phosphorylated MEK1 / 2 (Ser-217 / 221) antibody (“P-MEK1 / 2"), anti-phosphorylated ERK1 / 2 (Thr-202 / Tyr-204) antibody (“p-ERK1 / 2”) (all polyclonal antibodies, Cell Signal ing Technology), anti
- Both FK228 and FK-A5 are phosphorylated AKT (Ser-473, Thr-308) in a concentration-dependent manner, and phosphorylated GSK-3 ⁇ (Ser-9), phosphorylated mTOR (Ser-308) downstream of the signal transduction pathway. 2448), the expression levels of phosphorylated p70S6K (Thr-389) and phosphorylated 4E-BP1 (Thr-37 / 46) were suppressed. In addition, phosphorylated MEK1 / 2 in the RAS-MAP pathway was not suppressed, but phosphorylated ERK1 / 2 was slightly suppressed.
- FK228 and FK-A5 suppress the AKT pathway through inhibition of PI3K.
- these compounds since it inhibits phosphorylated ERK1 / 2, these compounds may have an inhibitory activity against other kinases besides PI3K, or may inhibit ERK1 / 2 through the action of other proteins.
- HCT116 colon cancer
- CO115 RKO
- ATCC American Type Culture Collection
- CO115 was obtained from John M. Mariadason (Ludwig Institute for Cancer Research, Melbourne, Australia).
- the cells were cultured at 37 ° C. in the presence of 5% CO 2 using RPMI 1640 medium containing 10% of inactivated fetal bovine serum.
- HCT116, RKO, and CO115 are all microsatellite instability (MSI) positive cells, and HCT116 is sensitive to HDAC inhibitors, while RKO and CO115 are resistant to HDAC inhibitors.
- RKO and CO115 the HDAC2 protein was deleted as a result of the frameshift mutation in the A 9 (9 repeats of adenine) sequence in exon 1 of both HDAC2 alleles.
- Reported to be resistant to HDAC inhibitors TSA and SAHA due to increased apoptotic protease-activating factor 1 (APAF1) expression level, resulting in abnormal regulation of apoptosis (Non-patent document 29: Ropero et al. Nature 200, 6; Non-patent document 30: Hanigan et al. Gastroenterology 135, 1654-1664. E1652 (2008)). Therefore, first, the expression level of HDAC in these cells was confirmed by Western blot.
- APAF1 apoptotic protease-activating factor 1
- HDAC2 Human Cell Signaling Technology
- HDAC1 anti-HDAC1 polyclonal antibody
- FIG. 4A The result is shown in FIG. 4A. Unlike conventional reports, weak expression of HDAC2 was also observed in RKO and CO115. Regarding RKO, it has been reported that there are variations in the expression of HDAC2 (Non-patent Document 31: Ree et al., Nature et al. Genetics 40, 812-813 (2008)). Although HDAC1 was expressed in all cells, the expression level in CO115 was slightly low.
- LY294002 50 ⁇ M
- SAHA purchased from Cayman Chemical Company; 2.5 ⁇ M
- LY294002 50 ⁇ M
- Medium containing + SAHA 2.5 ⁇ M
- FK228 or FK-A5 5 nM, 50 nM, 500 nM, 5 ⁇ M or 50 ⁇ M, respectively
- FK228 or FK-A5 5 nM, 50 nM or 500 nM, respectively
- LY294002 50 ⁇ M
- Cell Counting Kit-8 Cell Counting Kit-8
- WST-8 water-soluble tetrazolium salt WST-8 as a coloring reagent
- WST-8 generates formazan by intracellular dehydrogenase.
- the amount of formazan dye and the number of living cells are in a proportional relationship, and the absorbance of formazan at 450 nm was measured with a microplate reader (trade name “SpectraMax M2e”, MolecularMdevices) to count the number of living cells.
- FACS analysis fluorescence-activated cell sorting analysis
- the cells were collected, fixed with ethanol, stained with propidium iodide / PBS solution, and analyzed using a trade name “Cytomics FC500 Flow Cytometry System” (Beckman Coulter). The cell cycle fraction was calculated using the trade name “Multicycle software” (Phenomix Flow Systems).
- Non-patent Document 32 Xu et al. Oncogene 26, 5541-5552 (2007).
- Non-patent Document 33 Richon et al. Proc. Natl. Acad. Sci. 97, 10014 ( 2000);
- Non-Patent Document 34 Nakajima et al. Exp. Cell Res. 241, 126-133 (1998);
- Non-Patent Document 35 Kumagai et al. Int. J. of Cancer 121, 656-665 (2007)) .
- LY294002 caused G0 / G1 phase arrest and apoptosis induction was shown to be weak, but LY294002 also induced G0 / G1 phase arrest for HCT116 cells in this experiment (FIG. 5A-C).
- SAHA + LY294002 the combined use of SAHA + LY294002 and the combined use of low concentrations of FK228 and FK-A5 + LY294002
- the subG1 fraction was markedly increased to 20-40%, and strong apoptosis was induced (FIGS. 5A to C).
- HCT116 cells were treated with predetermined concentrations of SAHA and FK-228 for 24 hours, and analyzed for expression of PARP, cleared PARP, phosphorylated AKT (S473), AKT, acetylated histone H3, and acetylated histone H4.
- FIG. 5D Induction of apoptosis by FK228 was supported by an increase in Cleared PARP in HCT116 cells. Similar to the results of FCAS analysis, Cleaved PARP increased most at high concentrations of FK228 (5 ⁇ M), indicating strong apoptosis induction. At the same time, it was also found that a high concentration of FK228 (5 ⁇ M) suppressed phosphorylated AKT and increased the expression of acetylated histones.
- FK-A11 was shown to suppress AKT phosphorylation at a lower concentration than FK228 and FK-A5.
- FK-A11 10 nM, 50 nM, 500 nM, 5 ⁇ M. Or used at 50 ⁇ M.
- FIGS. 8A-C A comparison of the results of FK228, FK-A5, and FK-A11 is shown in FIGS. 8A-C. From these results, it became clear that FK-A11 having a very strong inhibitory activity against p110 ⁇ exhibits a stronger cell growth inhibitory effect than any of FK228 and FK-A5 against any cancer cell line. .
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Gastroenterology & Hepatology (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Peptides Or Proteins (AREA)
Abstract
Description
また、PI3Kとは逆反応を触媒する脱リン酸化酵素であるPTEN(Phosphatase and tensin homologue deleted on chromosome 10)は、シグナルメッセンジャーであるPIP3を減少させることによって細胞の増殖を抑制する(非特許文献6:Maehama et al., J. Biol. Chem. 273, 13375-13378 (1998))。PTEN遺伝子については、子宮内膜がんや悪性黒色腫など多くのがん種で欠失や点突然変異が見られ(非特許文献7:Salmena et al., Cell 133, 403-414 (2008))、その点突然変異のほとんどで脱リン酸化酵素活性が低下していることが報告されている(非特許文献8:Han et al. Cancer Res 60, 3147 (2000))。
これらの変異の結果、PI3K/AKT経路の異常な恒常的活性化が生じ、がん細胞の生存シグナルが伝達されると考えられている。
なお、用語「デプシペプチド」は、狭義ではFK228を指すが、本明細書においては上記の意義を有する用語として使用する。
HDAC阻害剤がそれ自体でPI3K/AKT経路に影響するか否かに関しては議論がある(非特許文献14:Hanker et al., J Molecular Signaling 4, 5 (2009);非特許文献15:Graham et al. Clinical Cancer Res 12, 223 (2006))。古典的なHDAC阻害剤であるトリコスタチンA(trichostatin A;TSA)は、プロテインホスファターゼ1(protein phosphatase 1)を介してリン酸化AKTを抑えることが報告されている(非特許文献16:Chen et al., J Biol Chem 280, 38879 (2005))。
〔1〕 以下の式1で示されるデプシペプチド類化合物又は生理学的に許容可能なその塩からなるホスファチジルイノシトール3キナーゼ(PI3K)阻害剤

(式中、Aは、-CONH-又は-CH(OH)-を表し、R1、R2及びR3は、同一又は異なって、水素原子、低級アルキル基、低級アルキリデン基、置換もしくは無置換のアリール基又は置換もしくは無置換のアラルキル基を表す。);
〔2〕 式1において、R3が水素原子である、前記〔1〕記載のPI3K阻害剤;
〔3〕 式1において、R1が置換もしくは無置換のアラルキル基である、前記〔1〕又は〔2〕記載のPI3K阻害剤;
〔4〕 前記〔1〕~〔3〕のいずれか1項記載のPI3K阻害剤を有効成分として含有することを特徴とする、難治性がんの治療用医薬組成物;
〔5〕 前記PI3K阻害剤が体重1kgあたり約1mg/日~10,000mg/日の投与用量で投与されることを特徴とする、前記〔4〕記載の難治性がんの治療用医薬組成物;
〔6〕 前記PI3K阻害剤が、以下の式2~20のいずれかで示されるデプシペプチド類化合物又は生理学的に許容可能なその塩である、前記〔4〕又は〔5〕記載の難治性がんの治療用医薬組成物




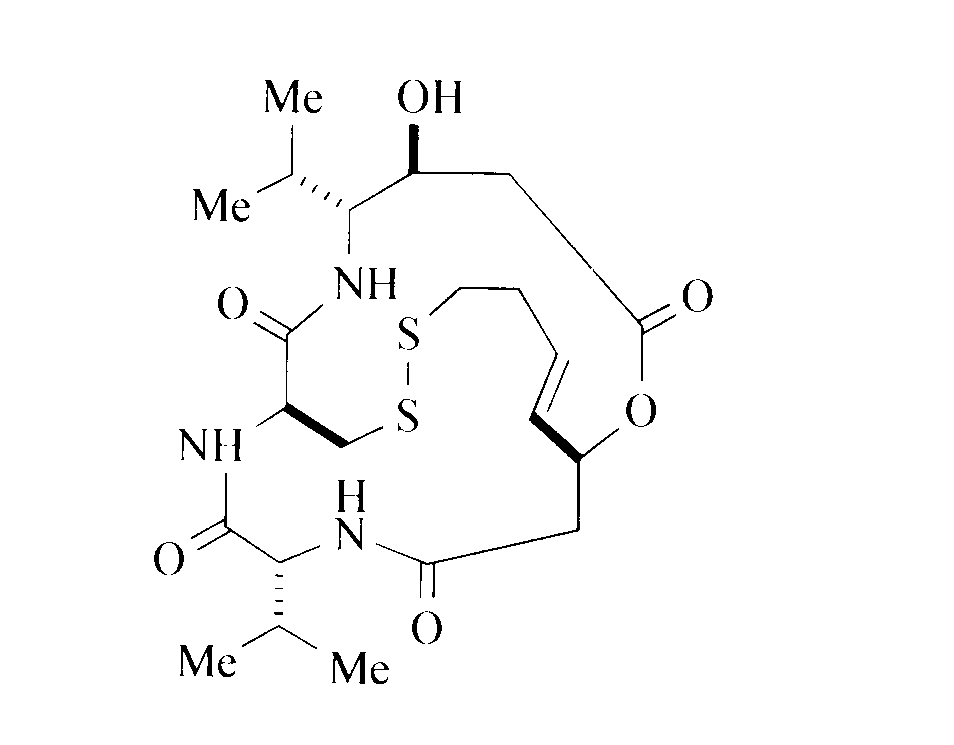














〔7〕 以下の式1で示されるデプシペプチド類化合物又は生理学的に許容可能なその塩

(式中、Aは、-CONH-又は-CH(OH)-を表し、R1、R2及びR3は、同一又は異なって、水素原子、低級アルキル基、低級アルキリデン基、置換もしくは無置換のアリール基又は置換もしくは無置換のアラルキル基を表す;但し、A、R1、R2及びR3の組合せが、A=-CONH-かつR1=エチリデン基かつR2=R3=イソプロピル基、A=-CH(OH)- かつR1=イソプロピル基かつR2=メチル基かつR3=水素原子、A=-CH(OH)- かつR1=sec-ブチル基かつR2=メチル基かつR3=水素原子、A=-CH(OH)- かつR1=R2=イソプロピル基かつR3=水素原子、A=-CONH- かつR1=エチリデン基かつR2=イソプロピル基かつR3=水素原子、A=-CONH- かつR1=R3=水素原子かつR2=イソプロピル基、A=-CONH- かつR1=メチル基かつR2=イソプロピル基かつR3=水素原子、及び、A=-CONH- かつR1=ベンジル基かつR2=イソプロピル基かつR3=水素原子、のいずれかである化合物を除く);
が提供される。
本発明のPI3K阻害剤は、HDAC阻害活性のほか、他のキナーゼをも阻害する可能性を有していることが見出され、がん細胞を制御する上で有利に働くことが推測される多分子標的の薬剤である可能性がある。
また、本発明のPI3K阻害剤は、がん特異的に作用する分子標的薬として使用されるものであるから、がん細胞以外の細胞や組織に与える影響は比較的少ないと考えられ、実際、FK228については、長期投与によっても何ら副作用を生じない例や、何らかの副作用を生じた場合であっても比較的軽く、これを抑える薬剤との併用によって抑えることが可能であることがわかっている。FK228の類縁体については、後述するようにFK228よりも副作用が低いと考えられ、したがって、本発明の医薬組成物は、生体に対して高用量で使用しても重篤な副作用が起こる可能性が低い。




















R3が水素原子のもの、及び/又はR1が置換もしくは無置換のアラルキル基のものがさらに好ましい。
一般的には、有効成分として体重1kgあたり約1mg/日~10,000mg/日、より好ましくは約10mg/日~約5,000mg/日、さらに好ましくは約20mg/日~約5,000mg/日、最も好ましくは約50mg/日~約5,000mg/日を1日1回あるいは1日2~3回に分割して、本発明の医薬組成物による治療が必要とされている患者に投与することができる。
本発明において用いたスクリーニング系は、出芽酵母にヒトのp110αを発現させることにより生じる細胞増殖障害に基づいており、基本的に、Rodriguez-Escuderoらの方法(非特許文献19:Biochemical Journal 390, 613 (2005))に記載された方法にしたがってスクリーニングを行った。ただし、このスクリーニングにおいては、出芽酵母として、一般的に使用されている野生型株YPH499の代わりに、7つのABCトランスポーター遺伝子及びABCトランスポーターの活性化に関与する2つの転写因子の遺伝子をノックアウトした薬剤感受性株AD1-9(MAT alpha, yor1, snq2, pdr5, pdr10, pdr11, ycf1, pdr3, pdr15, pdr1, his1, ura3)を用いた(非特許文献25:Rogers et al. J. of Mol. Microbiol. Biotechnol. 3, 207-214 (2001))。AD1-9株は、Nader Nourizad(Stanford Genome Technology Center Stanford University, CA, USA)より入手可能である。



その後の実験に使用したFK228及びFK228類縁体は、全て、本発明者の東北薬科大学医薬合成教室の加藤正教授のグループによって公知の技術にしたがって合成されたものをDMSOに溶解して用いた。
各化合物の合成経路及び詳しい合成方法は、図1及びNarita et al. Chemistry-A European Journal 15, 11174-11186 (2009)(非特許文献22);Takizawa et al. Chemical Communication, 1677-1679 (2008) (非特許文献23); Takizawa et al. Heterocycles 76, 275-290 (2008) (非特許文献24)に記載のとおりである。
段階(D)において、文献既知のカルボン酸誘導体と側鎖R2を有するアミノ酸エステル誘導体を縮合させて、ペプチド誘導体(II)を得た。
段階(E)において、上記で得られた2種類のペプチド誘導体(I)及び(II)を縮合させて、トリペプチド誘導体を得た。段階(F)において、分子内ラクトン化、ジスルフィド結合形成、水酸基の脱保護を順次行い、デプシペプチド類化合物の合成を行った。
一方、段階(G)において、従来の経路を用いて調製可能な側鎖R1及びR3を有するトリペプチド誘導体とカルボン酸誘導体(II)を縮合させて、テトラペプチド誘導体を生じさせた。段階(H)において、分子内ラクトン化、ジスルフィド結合形成、水酸基の脱保護を順次行い、デプシペプチド類化合物の合成を完了した。
FK228、SP-1、SP-2、SP-3、FK-A1、FK-A2、FK-A3、FK-A4、FK-A5、FK-A6及びSP-5の計11種類の化合物について、20μMの濃度におけるPI3K阻害活性の評価を行った。
阻害率は、以下のようにして計算した:
コントロールとして、キナーゼ反応をMSA(モビリティ シフト アッセイ)で算出された産生物/基質+産生物の値で評価し、それを0%阻害率とした。そこに薬剤が添加された場合の産生物/基質+産生物の値を同様に算出し、コントロール値との差から阻害率を算出した。
なお、FK228のPI3Kの阻害曲線において10μMでの阻害率は6.5%であった(図2C)。
従来のPI3K阻害剤は、すべてp110触媒サブユニットのATP結合部位に結合するとされている(非特許文献27:Walker et al. Molecular Cell 6, 909-919 (2000))。FK228も同じようにATP結合部位に結合するかを検証した。
このことは、上記3.に記載した実験において、異なるATP濃度条件下(50μM及び500μM)でFK228のPI3K阻害活性を測定することにより確認された。結果を図2Eに示す。FK228のPI3K阻害活性は、高濃度のATP存在下で低減した。標準的ATP濃度(50μM)及び高ATP濃度(500μM)でのIC50値は、それぞれ57.2μM及び125.0μMであった。これらの結果は、FK228がATP競合的なPI3K阻害剤であること、すなわちATP結合部位に結合することを示唆する。
PTENが欠失しており、PI3Kの下流のAKTが恒常的に活性化しているPC3(前立腺がん)細胞を用いて(非特許文献28:Grunwald et al. Cancer Res 62, 6141 (2002))、FK228及びFK-A5によるAKTのリン酸化の抑制を評価した。
二次抗体としては、商品名「Alexa Fluor680IgG」(Invitrogen)を用いた。商品名「Odyssey Infrared Imaging system」(LI-COR)を用いて目的とするタンパク質の発現を検出した。
LY294002と同様に、FK228、FK-A5ともに5分から180分という短時間で、全AKTの発現レベルは変化させずに、リン酸化AKT(Ser-473、Thr-308)の発現レベルを減少させた。
FK228は、HDAC1に対して1.6~3.6nMのIC50をもつ強力なHDAC阻害剤であり(非特許文献12:Furumai et al. Cancer Res 62, 4916 (2002);非特許文献22:Narita et al. Chemistry-A European Journal 15, 11174-11186 (2009))、多くのヒトがん細胞株に対してnMの範囲で50%成長阻害を発揮することが示されている。ここまでの実験結果より、FK228及びFK-A5がPI3K阻害活性を示すのはμMの範囲であるため、HDAC阻害剤に抵抗性の細胞を用いてFK228及びFK-A5の殺細胞効果を検討した。
HDAC阻害剤に感受性であるHCT116細胞では、SAHA、低濃度(5nM~500nM程度のレベル)のFK228、FK-A5により約50%(43~70%)の細胞死を生じた。これに対し、RKO、CO115細胞においては、HDAC2の弱発現が認められたものの、SAHA、50nM程度までのFK228及びFK-A5に対して抵抗性(6~15%の細胞死)であった。LY294002に対してはいずれの細胞も感受性(50~60%の細胞死)であったが、SAHAとLY294002との併用に対しては、HCT116、RKOでは殺細胞効果の増強(相加効果)が認められた一方、CO115の場合は増強が認められなかった。
PI3K阻害活性が発揮される高濃度(μMレベル)のFK228及びFK-A5は、いずれの細胞においても極めて強く細胞数を減少させた。
FK228及びFK-A5の投与による細胞死の特徴を解析するため、まず、細胞周期解析としてFACS解析(fluorescence-activated cell sorting analysis)を行った。
HCT116細胞を6穴プレートに2×105個ずつ播き、24時間の前培養後、各薬剤存在下でさらに24時間培養し、SAHA、LY294002、FK228、FK-A5を上記6.に記載した実験と同条件で作用させた。細胞を回収し、エタノールで固定後、ヨウ化プロピジウム/PBS溶液で染色し、商品名「Cytomics FC500 Flow Cytometry System」(Beckman Coulter)を用いて解析した。細胞周期分画は商品名「Multicycle software」(Phenomix Flow Systems)を用いて算出した。
上記2.と同様にして、さらにデプシペプチド類化合物FK-A7~A13及びFK-A17を合成し、特性を調べた。表3に、それらの化合物の構造式、及び上記3.と同様にして測定したPI3K(p110α/p85α)に対する50%阻害濃度(IC50)を、比較のためFK228、SAHA、LY294002とともに示す。表3中、FK228、SAHAのHDAC1に対する50%阻害濃度(IC50)は、文部科学省がん特定領域研究・統合がん化学療法基盤情報支援班により評価された。



上記5.と同様にして、FK-A11によるAKTのリン酸化の抑制を評価し、FK-A5、FK228と比較した。ただし、作用時間は180分とし、一次抗体としては、抗リン酸化AKT(Ser-473)抗体(「p-AKT(S473)」)、抗リン酸化AKT(Thr-308)抗体(「p-AKT(T308)」)、抗AKT抗体(「AKT」)、抗β-アクチンモノクローナル抗体(「B-actin」)を使用した。
上記6.と同様にして、MTTアッセイによりFK-A11の殺細胞効果を検討した。ただし、ヒト培養細胞としては、大腸がん(HCT116、CO115、RKO)の細胞株、及び正常細胞として非腫瘍性線維芽細胞株KMST6を使用し、FK-A11は、5nM、50nM、500nM、5μM又は50μMで使用した。
HCT116、RKO細胞では、低濃度(5nM~500nM程度のレベル)のFK-A11により顕著な細胞死を生じた。これに対し、HDAC阻害剤に対して抵抗性であるCO115細胞においては、50nM程度までのFK-A11に対して抵抗性であったが、PI3K阻害活性が発揮されると推測される500nMからは強い細胞死を生じた。一方、その濃度においては、非がん細胞であるKMST6は顕著な細胞死を生じなかった。
これらの結果から、p110αに対する阻害活性が非常に強いFK-A11は、いずれのがん細胞株に対しても、FK228、FK-A5よりも強力な細胞増殖抑制効果を示すことが明らかになった。
Claims (7)
- 以下の式1で示されるデプシペプチド類化合物又は生理学的に許容可能なその塩からなるホスファチジルイノシトール3キナーゼ(PI3K)阻害剤。

(式中、Aは、-CONH-又は-CH(OH)-を表し、R1、R2及びR3は、同一又は異なって、水素原子、低級アルキル基、低級アルキリデン基、置換もしくは無置換のアリール基又は置換もしくは無置換のアラルキル基を表す。) - 式1において、R3が水素原子である、請求項1記載のPI3K阻害剤。
- 式1において、R1が置換もしくは無置換のアラルキル基である、請求項1又は2記載のPI3K阻害剤。
- 請求項1~3のいずれか1項記載のPI3K阻害剤を有効成分として含有することを特徴とする、難治性がんの治療用医薬組成物。
- 前記PI3K阻害剤が体重1kgあたり約1mg/日~10,000mg/日の投与用量で投与されることを特徴とする、請求項4記載の難治性がんの治療用医薬組成物。
- 前記PI3K阻害剤が、以下の式2~20のいずれかで示されるデプシペプチド類化合物又は生理学的に許容可能なその塩である、請求項4又は5記載の難治性がんの治療用医薬組成物。



















- 以下の式1で示されるデプシペプチド類化合物又は生理学的に許容可能なその塩。

(式中、Aは、-CONH-又は-CH(OH)-を表し、R1、R2及びR3は、同一又は異なって、水素原子、低級アルキル基、低級アルキリデン基、置換もしくは無置換のアリール基又は置換もしくは無置換のアラルキル基を表す;但し、A、R1、R2及びR3の組合せが、A=-CONH-かつR1=エチリデン基かつR2=R3=イソプロピル基、A=-CH(OH)- かつR1=イソプロピル基かつR2=メチル基かつR3=水素原子、A=-CH(OH)- かつR1=sec-ブチル基かつR2=メチル基かつR3=水素原子、A=-CH(OH)- かつR1=R2=イソプロピル基かつR3=水素原子、A=-CONH- かつR1=エチリデン基かつR2=イソプロピル基かつR3=水素原子、A=-CONH- かつR1=R3=水素原子かつR2=イソプロピル基、A=-CONH- かつR1=メチル基かつR2=イソプロピル基かつR3=水素原子、及び、A=-CONH- かつR1=ベンジル基かつR2=イソプロピル基かつR3=水素原子、のいずれかである化合物を除く)
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP12835791.0A EP2762148B1 (en) | 2011-09-30 | 2012-09-25 | Depsipeptide as phosphatidylinositol-3-kinase inhibitor and use thereof for treating cancer |
| JP2013536294A JP6214397B2 (ja) | 2011-09-30 | 2012-09-25 | 新規ホスファチジルイノシトール3キナーゼ阻害剤及び医薬組成物 |
| US14/348,799 US9963482B2 (en) | 2011-09-30 | 2012-09-25 | Phosphatidylinositol-3-kinase inhibitor and pharmaceutical composition |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011-217378 | 2011-09-30 | ||
| JP2011217378 | 2011-09-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013047509A1 true WO2013047509A1 (ja) | 2013-04-04 |
Family
ID=47995541
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/074542 WO2013047509A1 (ja) | 2011-09-30 | 2012-09-25 | 新規ホスファチジルイノシトール3キナーゼ阻害剤及び医薬組成物 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US9963482B2 (ja) |
| EP (1) | EP2762148B1 (ja) |
| JP (1) | JP6214397B2 (ja) |
| WO (1) | WO2013047509A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014115859A3 (ja) * | 2013-01-25 | 2014-10-16 | オンコリスバイオファーマ株式会社 | 分子標的併用腫瘍治療・予防薬 |
| WO2017122822A1 (ja) * | 2016-01-13 | 2017-07-20 | 国立大学法人東北大学 | デプシペプチド類化合物の製造中間体及びその製造方法 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113861267B (zh) * | 2021-10-25 | 2023-06-27 | 深圳湾实验室坪山生物医药研发转化中心 | 一种缩酯环肽类化合物lzg-pku-h及其合成方法和应用 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0285296A (ja) | 1988-07-26 | 1990-03-26 | Fujisawa Pharmaceut Co Ltd | Fr901228物質およびその製造法 |
| JPH0479892A (ja) | 1990-07-24 | 1992-03-13 | Fujisawa Pharmaceut Co Ltd | Fr901228物質の製造方法 |
| JP2003516418A (ja) * | 1999-12-08 | 2003-05-13 | エクサイト セラピーズ, インコーポレイテッド | 免疫抑制剤として使用するためのデプシペプチドおよびその同族体 |
| JP2008542347A (ja) | 2005-06-02 | 2008-11-27 | ユニバーシティ、オブ、サウサンプトン | Hdac阻害剤としてのfk228誘導体 |
| JP2009519224A (ja) | 2005-11-18 | 2009-05-14 | グロスター ファーマシューティカルズ, インコーポレイテッド | Hdacインヒビターfk228の代謝産物誘導体 |
| JP2010510300A (ja) * | 2006-11-22 | 2010-04-02 | カルス セラピューティクス リミテッド | デプシペプチドおよびその治療的使用 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0285296U (ja) | 1988-12-14 | 1990-07-04 | ||
| WO2009141657A1 (en) | 2008-05-22 | 2009-11-26 | Karus Therapeutics Limited | Depsipeptides and their therapeutic use |
| GB0905970D0 (en) | 2009-04-06 | 2009-05-20 | Karus Therapeutics Ltd | Depsipeptides and their therapeutic use |
-
2012
- 2012-09-25 WO PCT/JP2012/074542 patent/WO2013047509A1/ja active Application Filing
- 2012-09-25 EP EP12835791.0A patent/EP2762148B1/en active Active
- 2012-09-25 US US14/348,799 patent/US9963482B2/en active Active
- 2012-09-25 JP JP2013536294A patent/JP6214397B2/ja active Active
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0285296A (ja) | 1988-07-26 | 1990-03-26 | Fujisawa Pharmaceut Co Ltd | Fr901228物質およびその製造法 |
| JPH0479892A (ja) | 1990-07-24 | 1992-03-13 | Fujisawa Pharmaceut Co Ltd | Fr901228物質の製造方法 |
| JP2003516418A (ja) * | 1999-12-08 | 2003-05-13 | エクサイト セラピーズ, インコーポレイテッド | 免疫抑制剤として使用するためのデプシペプチドおよびその同族体 |
| JP2008542347A (ja) | 2005-06-02 | 2008-11-27 | ユニバーシティ、オブ、サウサンプトン | Hdac阻害剤としてのfk228誘導体 |
| JP2009519224A (ja) | 2005-11-18 | 2009-05-14 | グロスター ファーマシューティカルズ, インコーポレイテッド | Hdacインヒビターfk228の代謝産物誘導体 |
| JP2010510300A (ja) * | 2006-11-22 | 2010-04-02 | カルス セラピューティクス リミテッド | デプシペプチドおよびその治療的使用 |
Non-Patent Citations (73)
| Title |
|---|
| "Search for novel PI3K inhibitor by using budding yeast as screening tool", SERIES NO. P10-5, ABSTRACT OF THE 15TH MEETING OF THE JAPANESE ASSOCIATION FOR MOLECULAR TARGET THERAPY OF CANCER, 31 May 2011 (2011-05-31) |
| "Search for novel PI3K inhibitor by using budding yeast as screening tool", SERIES NO. PI 0-5, ABSTRACT OF THE 15TH MEETING OF THE JAPANESE ASSOCIATION FOR MOLECULAR TARGET THERAPY OF CANCER, 31 May 2011 (2011-05-31) |
| ALANI ET AL., GENETICS, vol. 116, 1987, pages 541 |
| ALANI, E.; CAO, L.; KLECKNER, N., GENETICS, vol. 116, 1987, pages 541 |
| CANTLEY, L. C., SCIENCE, vol. 296, 2002, pages 1655 - 1657 |
| CAREW ET AL., GILES, CANCER LET, vol. 269, 2008, pages 7 - 17 |
| CAREW, J. S.; GILES, F. J.; NAWROCKI, S. T., CANCER LETTERS, vol. 269, 2008, pages 7 - 17 |
| CHEN ET AL., J BIOL CHEM, vol. 280, 2005, pages 38879 |
| CHEN, C. S.; WENG, S. C.; TSENG, P. H.; LIN, H. P., JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 280, 2005, pages 38879 |
| CID ET AL., ONCOGENE, vol. 27, 2008, pages 5431 - 5442 |
| CID, V. ET AL., ONCOGENE, vol. 27, 2008, pages 5431 - 5442 |
| FRUMAN ET AL., ANNUAL REV BIOCHEM, vol. 67, 1998, pages 481 - 507 |
| FRUMAN, D. A.; MEYERS, R. E.; CANTLEY, L. C., ANNUAL REVIEW OF BIOCHEMISTRY, vol. 67, 1998, pages 481 - 507 |
| FURUMAI ET AL., CANCER RES, vol. 62, 2002, pages 4916 |
| FURUMAI, R. ET AL., CANCER RESEARCH, vol. 62, 2002, pages 4916 |
| GRAHAM ET AL., CLINICAL CANCER RES, vol. 12, 2006, pages 223 |
| GRAHAM, C. ET AL., CLINICAL CANCER RESEARCH, vol. 12, 2006, pages 223 |
| GRUNWALD ET AL., CANCER RES, vol. 62, 2002, pages 6141 |
| GRUNWALD, V. ET AL., CANCER RESEARCH, vol. 62, 2002, pages 6141 |
| HAN ET AL., CANCER RES, vol. 60, 2000, pages 3147 |
| HAN, S. Y. ET AL., CANCER RESEARCH, vol. 60, 2000, pages 3147 |
| HANIGAN ET AL., GASTROENTEROLOGY, vol. 135, 2008, pages 1654 - 1664 |
| HANIGAN, C. L. ET AL., GASTROENTEROLOGY, vol. 135, 2008, pages 1654 - 1664 |
| HANKER ET AL., J MOLECULAR SIGNALING, vol. 4, 2009, pages 5 |
| HANKER, A. B.; HEALY, K. D.; NICHOLS, J.; DER, C. J., JOURNAL OF MOLECULAR SIGNALING, vol. 4, 2009, pages 5 |
| HIROKAWA Y. ET AL.: "Signal therapy of NF1- deficient tumor xenograft in mice by the anti- PAK1 drug FK228.", CANCER BIOLOGY & THERAPY, vol. 4, no. 4, 2005, pages 379 - 381, XP055149017 * |
| HIROSHI MARUTA ET AL.: "Kazokusei Shuyo to Genome Shinkei Sen'ishusho (NF) no Signal Ryoho", GENOME MEDICINE, vol. 4, no. 2, 2004, pages 185 - 192, XP008173986 * |
| IKENOUE ET AL., CANCER RES, vol. 65, 2005, pages 4562 |
| IKENOUE, T. ET AL., CANCER RESEARCH, vol. 65, 2005, pages 4562 |
| KANG ET AL., PROC. NATL. ACAD. SCI. USA, vol. 102, 2005, pages 802 |
| KANG, S.; BADER, A. G.; VOGT, P. K., PROC. NATL. ACAD. SCI. USA, vol. 102, 2005, pages 802 |
| KODANI ET AL., ONCOLOGY REPORTS, vol. 13, 2005, pages 477 - 483 |
| KODANI, M. ET AL., ONCOLOGY REPORTS, vol. 13, 2005, pages 477 - 483 |
| KONG, D.; YAMORI, T., CURRENT MEDICINAL CHEMISTRY, vol. 16, 2009, pages 2839 - 2854 |
| KONG; YAMORI, CURRENT MEDICINAL CHEMISTRY, vol. 16, 2009, pages 2839 - 2854 |
| KUMAGAI ET AL., INT. J. OF CANCER, vol. 121, 2007, pages 656 - 665 |
| KUMAGAI, T. ET AL., INTERNATIONAL JOURNAL OF CANCER, vol. 121, 2007, pages 656 - 665 |
| MAEHAMA ET AL., J. BIOL. CHEM., vol. 273, 1998, pages 13375 - 13378 |
| MAEHAMA, T.; DIXON, J. E., THE JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 273, 1998, pages 13375 - 13378 |
| MASAHIRO KOTANI ET AL.: "Haisengan Baiyo Saibokabu ni Okeru FK228(FR901228) no Akt Yokusei Sayo to amrubicin tono Sojo Koka", JAPANESE JOURNAL OF LUNG CANCER, vol. 43, no. 5, 2003, pages 581, XP055158644 * |
| NAKAJIMA ET AL., EXP. CELL RES., vol. 241, 1998, pages 126 - 133 |
| NAKAJIMA, H.; KIM, Y. B.; TERANO, H.; YOSHIDA, M.; HORINOUCHI, S., EXPERIMENTAL CELL RESEARCH, vol. 241, 1998, pages 126 - 133 |
| NARITA ET AL., CHEMISTRY-A EUROPEAN JOURNAL, vol. 15, 2009, pages 11174 - 11186 |
| NARITA, K. ET AL., CHEMISTRY-A EUROPEAN JOURNAL, vol. 15, 2009, pages 11174 - 11186 |
| REE ET AL., NATURE GENETICS, vol. 40, 2008, pages 812 - 813 |
| REE, A. H.; FOLKVORD, S.; FLATMARK, K., NATURE GENETICS, vol. 40, 2008, pages 812 - 813 |
| RICHON ET AL., PROC. NATL. ACAD. SCI., vol. 97, 2000, pages 10014 |
| RICHON, V. M.; SANDHOFF, T. W.; RIFKIND, R. A.; MARKS, P. A., PROC. NATL. ACAD. SCI., vol. 97, 2000, pages 10014 |
| RODRIGUEZ-ESCUDERO ET AL., BIOCHEMICAL JOURNAL, vol. 390, 2005, pages 613 |
| RODRIGUEZ-ESCUDERO, BIOCHEMICAL JOURNAL, vol. 390, 2005, pages 613 |
| RODRIGUEZ-ESCUDERO, I. ET AL., BIOCHEMICAL JOURNAL, vol. 390, 2005, pages 613 |
| ROGERS ET AL., J. OF MOL. MICROBIOL. BIOTECHNOL., 2001, pages 207 - 214 |
| ROGERS, B. ET AL., JOURNAL OF MOLECULAR MICROBIOLOGY AND BIOTECHNOLOGY, vol. 3, 2001, pages 207 - 214 |
| ROPERO ET AL., NATURE, vol. 200, pages 6 |
| ROPERO, S. ET AL., NATURE, vol. 200, pages 6 |
| SALMENA ET AL., CELL, vol. 133, 2008, pages 403 - 414 |
| SALMENA, L.; CARRACEDO, A.; PANDOLFI, P. P., CELL, vol. 133, 2008, pages 403 - 414 |
| SAMUELS ET AL., SCIENCE, vol. 304, 2004, pages 554 |
| SAMUELS, Y. ET AL., SCIENCE, vol. 304, 2004, pages 554 |
| See also references of EP2762148A4 |
| TAKIZAWA ET AL., CHEMICAL COMMUNICATION, 2008, pages 1677 - 1679 |
| TAKIZAWA ET AL., HETEROCYCLES, vol. 76, 2008, pages 275 - 290 |
| TUGENDREICH ET AL., GENOME RESEARCH, vol. 11, 2001, pages 1899 |
| TUGENDREICH, S. ET AL., GENOME RESEARCH, vol. 11, 2001, pages 1899 |
| UEDA ET AL., J. ANTIBIOTICS, vol. 47, 1994, pages 301 |
| UEDA, H. ET AL., THE JOURNAL OF ANTIBIOTICS, vol. 47, 1994, pages 301 |
| WALKER ET AL., MOLECULAR CELL, vol. 6, 2000, pages 909 - 919 |
| WALKER, E. H. ET AL., MOLECULAR CELL, vol. 6, 2000, pages 909 - 919 |
| WANG C. ET AL.: "Thailandepsins: bacterial products with potent histone deacetylase inhibitory activities and broad-spectrum antiproliferative activities", JOURNAL OF NATURAL PRODUCTS, vol. 74, no. 10, 27 July 2011 (2011-07-27), pages 2031 - 2038, XP055149021, Retrieved from the Internet <URL:http://pubs.acs.org/doi/abs/10.1021/np200324x> * |
| WOZNIAK ET AL., HAEMATOLOGICA, vol. 95, 2010, pages 613 |
| WOZNIAK, M. B. ET AL., HAEMATOLOGICA, vol. 95, 2010, pages 613 |
| XU ET AL., ONCOGENE, vol. 26, 2007, pages 5541 - 5552 |
| XU, W.; PARMIGIANI, R.; MARKS, P., ONCOGENE, vol. 26, 2007, pages 5541 - 5552 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014115859A3 (ja) * | 2013-01-25 | 2014-10-16 | オンコリスバイオファーマ株式会社 | 分子標的併用腫瘍治療・予防薬 |
| WO2017122822A1 (ja) * | 2016-01-13 | 2017-07-20 | 国立大学法人東北大学 | デプシペプチド類化合物の製造中間体及びその製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2762148A1 (en) | 2014-08-06 |
| EP2762148B1 (en) | 2017-12-27 |
| US20140235821A1 (en) | 2014-08-21 |
| EP2762148A4 (en) | 2015-10-28 |
| US9963482B2 (en) | 2018-05-08 |
| JP6214397B2 (ja) | 2017-10-18 |
| JPWO2013047509A1 (ja) | 2015-03-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11723947B2 (en) | Anti-senescence compounds and uses thereof | |
| Lee et al. | Inhibition of cell growth and induction of apoptosis by Antrodia camphorata in HER-2/neu-overexpressing breast cancer cells through the induction of ROS, depletion of HER-2/neu, and disruption of the PI3K/Akt signaling pathway | |
| US20100092479A1 (en) | Compositions and methods for treatment of viral diseases | |
| TW200303920A (en) | Method for identification of tumor targeting enzymes | |
| CN103108651A (zh) | Cyp3a药物代谢的抑制 | |
| TWI697329B (zh) | 血液癌症治療用之醫藥及其用途 | |
| US11260062B2 (en) | Pharmaceutical composition for treatment of lung cancer comprising glucocorticoid-based compound | |
| JP6214397B2 (ja) | 新規ホスファチジルイノシトール3キナーゼ阻害剤及び医薬組成物 | |
| Lee et al. | A novel pyrazolo [3, 4-d] pyrimidine induces heme oxygenase-1 and exerts anti-inflammatory and neuroprotective effects | |
| Li et al. | Bioactive peptides sensitize cells to anticancer effects of oxaliplatin in human colorectal cancer xenografts in nude mice | |
| Mun et al. | Synthesis and antitumor activity of (−)-bassianolide in MDA-MB 231 breast cancer cells through cell cycle arrest | |
| She et al. | Discovery of novel organoarsenicals as robust thioredoxin reductase inhibitors for oxidative stress mediated cancer therapy | |
| CN109862903A (zh) | 治疗性sall4肽 | |
| WO2021178663A1 (en) | Compositions and methods for treatment of platinum-based chemotherapeutic resistant tumors | |
| CN109718374B (zh) | Irf3抑制剂在制备yap过度激活的癌症的治疗或预防药物中的用途 | |
| KR101921676B1 (ko) | Tesk1 억제제를 포함하는 항암제 내성 억제용 조성물 및 tesk1 억제제의 스크리닝 방법 | |
| US20230270815A1 (en) | Apoptosis inducing peptide (SSTP1) | |
| US11091517B2 (en) | Peptide derivative and pharmaceutical composition containing same | |
| CN111139299B (zh) | Josd2蛋白在制备治疗恶性肿瘤药物中的应用 | |
| Isgor et al. | IN VITRO AND IN SILICO CYTOTOXICITY EVALUATION OF SOME ISATIN MANNICH BASES ON HUMAN MELANOMA CELLS | |
| KR102152977B1 (ko) | 암 예방 또는 치료용 펩타이드 및 이를 포함하는 약학적 조성물 | |
| US20220220153A1 (en) | B-catenin/b-cell lymphoma 9 protein-protein interaction inhibiting peptidomimetics | |
| Xue et al. | Lycorine (Lycoris radiata)-A unique natural medicine on breast cancer | |
| JP2009001494A (ja) | 非典型的なプロリンリッチ配列結合阻害剤 | |
| Meerod et al. | Cytotoxic stress caused by azalamellarin D (AzaD) interferes with cellular protein translation by targeting the nutrient-sensing kinase mTOR |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12835791 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2013536294 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14348799 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012835791 Country of ref document: EP |