WO2012102405A1 - 縮環化合物 - Google Patents
縮環化合物 Download PDFInfo
- Publication number
- WO2012102405A1 WO2012102405A1 PCT/JP2012/052009 JP2012052009W WO2012102405A1 WO 2012102405 A1 WO2012102405 A1 WO 2012102405A1 JP 2012052009 W JP2012052009 W JP 2012052009W WO 2012102405 A1 WO2012102405 A1 WO 2012102405A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- methyl
- pharmaceutically acceptable
- lower alkyl
- Prior art date
Links
- 0 CC1=C(C)*(*)*2*1C*2 Chemical compound CC1=C(C)*(*)*2*1C*2 0.000 description 10
- VLVGSTUHSCEXSF-UHFFFAOYSA-N CC(C)N(c(ccc(C(O)=O)c1)c1N1Cc2c(C)cccc2C)C1=O Chemical compound CC(C)N(c(ccc(C(O)=O)c1)c1N1Cc2c(C)cccc2C)C1=O VLVGSTUHSCEXSF-UHFFFAOYSA-N 0.000 description 1
- ZAGLHAIDTVHBTF-UHFFFAOYSA-N CC1(C)c(ccc(CC(O)=O)c2)c2N(Cc2c(C)cccc2C)C1 Chemical compound CC1(C)c(ccc(CC(O)=O)c2)c2N(Cc2c(C)cccc2C)C1 ZAGLHAIDTVHBTF-UHFFFAOYSA-N 0.000 description 1
- BOQICOKUEJLKMY-UHFFFAOYSA-N CCC(C(O)=O)c1ccc(c(C)n[n]2Cc(c(Cl)ccc3)c3Cl)c2c1 Chemical compound CCC(C(O)=O)c1ccc(c(C)n[n]2Cc(c(Cl)ccc3)c3Cl)c2c1 BOQICOKUEJLKMY-UHFFFAOYSA-N 0.000 description 1
- UCAJMKKVUWHJLK-UHFFFAOYSA-N CCc1c[n](Cc2c(C)cccc2C)c2c1ccc(C(O)=O)c2 Chemical compound CCc1c[n](Cc2c(C)cccc2C)c2c1ccc(C(O)=O)c2 UCAJMKKVUWHJLK-UHFFFAOYSA-N 0.000 description 1
- MCCFANCRJLAPLA-UHFFFAOYSA-N CCc1n[n](Cc(c(Cl)ccc2)c2Cl)c2cc(Br)cnc12 Chemical compound CCc1n[n](Cc(c(Cl)ccc2)c2Cl)c2cc(Br)cnc12 MCCFANCRJLAPLA-UHFFFAOYSA-N 0.000 description 1
- XPWVNZPABFXTKE-UHFFFAOYSA-N CCc1n[n](Cc(cccc2Cl)c2Cl)c2cc(C(O)=O)cnc12 Chemical compound CCc1n[n](Cc(cccc2Cl)c2Cl)c2cc(C(O)=O)cnc12 XPWVNZPABFXTKE-UHFFFAOYSA-N 0.000 description 1
- MMQFTZZVWPKEGP-UHFFFAOYSA-N CCc1n[n](Cc2c(C)cccc2C)c2c1ccc(CC(O)=O)c2 Chemical compound CCc1n[n](Cc2c(C)cccc2C)c2c1ccc(CC(O)=O)c2 MMQFTZZVWPKEGP-UHFFFAOYSA-N 0.000 description 1
- LYUUAYGWFYONQK-HWKANZROSA-N COC(/C=C/c1cc([nH]cc2)c2cc1)=O Chemical compound COC(/C=C/c1cc([nH]cc2)c2cc1)=O LYUUAYGWFYONQK-HWKANZROSA-N 0.000 description 1
- RCXKFLGEPNCNOO-UHFFFAOYSA-N Cc1c(C[n](cc2)c3c2ccc(CC(O)=O)c3)c(C)ccc1 Chemical compound Cc1c(C[n](cc2)c3c2ccc(CC(O)=O)c3)c(C)ccc1 RCXKFLGEPNCNOO-UHFFFAOYSA-N 0.000 description 1
- KUESGFCXMXSZHG-UHFFFAOYSA-N Cc1c(C[n]2c(cc(cc3)C(OCc4c(C)cccc4C)=O)c3nc2)c(C)ccc1 Chemical compound Cc1c(C[n]2c(cc(cc3)C(OCc4c(C)cccc4C)=O)c3nc2)c(C)ccc1 KUESGFCXMXSZHG-UHFFFAOYSA-N 0.000 description 1
- SWMXDKYPDBYEAM-UHFFFAOYSA-N Cc1c(C[n]2ncc3c2cc(CC#N)cc3)c(C)ccc1 Chemical compound Cc1c(C[n]2ncc3c2cc(CC#N)cc3)c(C)ccc1 SWMXDKYPDBYEAM-UHFFFAOYSA-N 0.000 description 1
- GKHAUIOQLHPUMA-UHFFFAOYSA-N Cc1c(C[n]2ncc3c2cc(CC(O)=O)cc3)c(C)ccc1 Chemical compound Cc1c(C[n]2ncc3c2cc(CC(O)=O)cc3)c(C)ccc1 GKHAUIOQLHPUMA-UHFFFAOYSA-N 0.000 description 1
- RNKRTUZHQIFNHZ-UHFFFAOYSA-N Cc1c[n](Cc(c(Cl)ccc2)c2Cl)c2c1ccc(CC(O)=O)c2 Chemical compound Cc1c[n](Cc(c(Cl)ccc2)c2Cl)c2c1ccc(CC(O)=O)c2 RNKRTUZHQIFNHZ-UHFFFAOYSA-N 0.000 description 1
- BGEBSRRTTLVBFL-UHFFFAOYSA-N Cc1cc(ccc(C(OC)=O)c2)c2[nH]1 Chemical compound Cc1cc(ccc(C(OC)=O)c2)c2[nH]1 BGEBSRRTTLVBFL-UHFFFAOYSA-N 0.000 description 1
- GEOWXZIVYNJEQH-UHFFFAOYSA-N Cc1ccc(C[n](cc2)c3c2ccc(CC(O)=O)c3)cc1 Chemical compound Cc1ccc(C[n](cc2)c3c2ccc(CC(O)=O)c3)cc1 GEOWXZIVYNJEQH-UHFFFAOYSA-N 0.000 description 1
- SZQMNXYFSFKUNX-UHFFFAOYSA-N Cc1cccc(C)c1CN(c(cc(cc1)C(OC)=O)c1N1)C1=O Chemical compound Cc1cccc(C)c1CN(c(cc(cc1)C(OC)=O)c1N1)C1=O SZQMNXYFSFKUNX-UHFFFAOYSA-N 0.000 description 1
- UWKOVYZHOSMSSW-UHFFFAOYSA-N Cc1n[n](Cc(c(C)ccc2)c2Cl)c2c1ccc(CC(O)=O)c2 Chemical compound Cc1n[n](Cc(c(C)ccc2)c2Cl)c2c1ccc(CC(O)=O)c2 UWKOVYZHOSMSSW-UHFFFAOYSA-N 0.000 description 1
- KKEHRCXNWIVIHK-UHFFFAOYSA-N Cc1n[n](Cc(c(C2CC2)ccc2)c2Cl)c2cc(-c3nnn[nH]3)cnc12 Chemical compound Cc1n[n](Cc(c(C2CC2)ccc2)c2Cl)c2cc(-c3nnn[nH]3)cnc12 KKEHRCXNWIVIHK-UHFFFAOYSA-N 0.000 description 1
- LIBJAOLOVPPUES-UHFFFAOYSA-N Cc1n[n](Cc(c(Cl)ccc2)c2Cl)c2c1ccc(C=C)c2 Chemical compound Cc1n[n](Cc(c(Cl)ccc2)c2Cl)c2c1ccc(C=C)c2 LIBJAOLOVPPUES-UHFFFAOYSA-N 0.000 description 1
- WFWSAVGCERGIJZ-UHFFFAOYSA-N Cc1n[n](Cc(c(Cl)ccc2)c2Cl)c2c1ccc(CC#N)c2 Chemical compound Cc1n[n](Cc(c(Cl)ccc2)c2Cl)c2c1ccc(CC#N)c2 WFWSAVGCERGIJZ-UHFFFAOYSA-N 0.000 description 1
- YDGPTJQLYSGPNO-UHFFFAOYSA-N Cc1n[n](Cc(c(Cl)ccc2)c2Cl)c2c1ccc(CCC(N)=O)c2 Chemical compound Cc1n[n](Cc(c(Cl)ccc2)c2Cl)c2c1ccc(CCC(N)=O)c2 YDGPTJQLYSGPNO-UHFFFAOYSA-N 0.000 description 1
- NHBXXRDRLRAQLQ-UHFFFAOYSA-N Cc1n[n](Cc(c(Cl)ccc2)c2Cl)c2c1ccc(Cl)n2 Chemical compound Cc1n[n](Cc(c(Cl)ccc2)c2Cl)c2c1ccc(Cl)n2 NHBXXRDRLRAQLQ-UHFFFAOYSA-N 0.000 description 1
- UQMSUPQIXMHKGT-UHFFFAOYSA-N Cc1n[n](Cc(c(Cl)ccc2)c2Cl)c2c1ccc(O)c2 Chemical compound Cc1n[n](Cc(c(Cl)ccc2)c2Cl)c2c1ccc(O)c2 UQMSUPQIXMHKGT-UHFFFAOYSA-N 0.000 description 1
- IZKAMFDIMWTAFJ-UHFFFAOYSA-N Cc1n[n](Cc(c(Cl)ccc2)c2Cl)c2c1ncc(C(O)=O)c2 Chemical compound Cc1n[n](Cc(c(Cl)ccc2)c2Cl)c2c1ncc(C(O)=O)c2 IZKAMFDIMWTAFJ-UHFFFAOYSA-N 0.000 description 1
- TUQCQIZHYQBYPT-UHFFFAOYSA-N Cc1n[n](Cc(c(Cl)ccc2)c2Cl)c2cc(CC(C(O)=O)O)ccc12 Chemical compound Cc1n[n](Cc(c(Cl)ccc2)c2Cl)c2cc(CC(C(O)=O)O)ccc12 TUQCQIZHYQBYPT-UHFFFAOYSA-N 0.000 description 1
- PFQADMQBKHLZBV-UHFFFAOYSA-N Cc1n[n](Cc2c(C)cccc2C)c2c1ccc(C(O)=O)c2 Chemical compound Cc1n[n](Cc2c(C)cccc2C)c2c1ccc(C(O)=O)c2 PFQADMQBKHLZBV-UHFFFAOYSA-N 0.000 description 1
- LUJBEFQVVXDCPL-UHFFFAOYSA-N O=Cc(c(F)c1)ncc1Br Chemical compound O=Cc(c(F)c1)ncc1Br LUJBEFQVVXDCPL-UHFFFAOYSA-N 0.000 description 1
- ZWSOQYHSRVCXNO-UHFFFAOYSA-N OC(Cc1cc([n](Cc(cc(cc2)Cl)c2Cl)cc2)c2cc1)=O Chemical compound OC(Cc1cc([n](Cc(cc(cc2)Cl)c2Cl)cc2)c2cc1)=O ZWSOQYHSRVCXNO-UHFFFAOYSA-N 0.000 description 1
- JUQQTUGGDSEYTP-UHFFFAOYSA-N OC(Cc1cc([n](Cc(cc2OCOc2c2)c2Cl)cc2)c2cc1)=O Chemical compound OC(Cc1cc([n](Cc(cc2OCOc2c2)c2Cl)cc2)c2cc1)=O JUQQTUGGDSEYTP-UHFFFAOYSA-N 0.000 description 1
- GLJJVTHQMSVDGC-UHFFFAOYSA-N OC(Cc1cc([n](Cc(cccc2)c2F)cc2)c2cc1)=O Chemical compound OC(Cc1cc([n](Cc(cccc2)c2F)cc2)c2cc1)=O GLJJVTHQMSVDGC-UHFFFAOYSA-N 0.000 description 1
- HMPMZDNCLNHGTG-UHFFFAOYSA-N OC(Cc1cc([n](Cc2cc(Cl)ccc2)cc2)c2cc1)=O Chemical compound OC(Cc1cc([n](Cc2cc(Cl)ccc2)cc2)c2cc1)=O HMPMZDNCLNHGTG-UHFFFAOYSA-N 0.000 description 1
- AXHXYIWJWSDZJJ-UHFFFAOYSA-N OC(Cc1cc([n](Cc2ccncc2)cc2)c2cc1)=O Chemical compound OC(Cc1cc([n](Cc2ccncc2)cc2)c2cc1)=O AXHXYIWJWSDZJJ-UHFFFAOYSA-N 0.000 description 1
- MVXVYAKCVDQRLW-UHFFFAOYSA-O c1c[nH]c2c1ccc[nH+]2 Chemical compound c1c[nH]c2c1ccc[nH+]2 MVXVYAKCVDQRLW-UHFFFAOYSA-O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/04—Drugs for disorders of the urinary system for urolithiasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/06—Antigout agents, e.g. antihyperuricemic or uricosuric agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/08—Indoles; Hydrogenated indoles with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/12—Radicals substituted by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/30—Indoles; Hydrogenated indoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/54—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings condensed with carbocyclic rings or ring systems
- C07D231/56—Benzopyrazoles; Hydrogenated benzopyrazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/06—Benzimidazoles; Hydrogenated benzimidazoles with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/06—Benzimidazoles; Hydrogenated benzimidazoles with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 2
- C07D235/08—Radicals containing only hydrogen and carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/06—Benzimidazoles; Hydrogenated benzimidazoles with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 2
- C07D235/10—Radicals substituted by halogen atoms or nitro radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/06—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
Definitions
- the present invention relates to a condensed ring compound useful in the field of medicine. More specifically, the present invention relates to a fused ring compound having URAT1 inhibitory activity and useful in the field of treatment of diseases involving blood uric acid, and a URAT1 inhibitor, a blood uric acid level-lowering agent and a pharmaceutical comprising the compound. Relates to the composition.
- Uric acid is the final metabolite of purines in humans.
- Purine bodies are produced by degradation of intracellular nucleic acids, ATP, which is a bioenergy source, and absorption by meals.
- Purine is metabolized to uric acid via hypoxanthine and xanthine. Since primates including humans are deficient in uric acid oxidase (uricase), uric acid is the final metabolite of purines. In many other mammals, uric acid is oxidized by uricase and metabolized to allantoin.
- uric acid is oxidized by uricase and metabolized to allantoin.
- Non-patent Document 1 About 98% of uric acid is present in the body fluid in the form of sodium urate (Non-patent Document 1). Since the solubility of sodium urate under physiological pH conditions is 6.4 mg / dL (Non-patent Document 1), a blood uric acid level exceeding the solubility in body fluids, 7 mg / dL or more is defined as hyperuricemia (Non-Patent Document 2). When hyperuricemia persists, urate crystals are precipitated and deposited in body fluids, causing ventilatory arthritis, gouty kidneys, gouty nodules, urinary calculi, and renal dysfunction (Non-patent Document 3).
- Non-Patent Documents 4, 5, 6, 7 Is known to increase the incidence of cardiovascular and cerebrovascular disorders.
- Non-patent Document 8 It has been reported that hyperuricemia is present in over 20% of adult males in Japan and is still increasing due to westernization of lifestyle habits.
- Non-patent Document 9 As the type classification of hyperuricemia, it has been reported that uric acid production excess type is 12%, uric acid excretion reduced type is 60%, and mixed type is 25% (Non-Patent Document 9), and uric acid excretion reduced type is 60%.
- 85% which is a combination of 25% and 25%, shows a decrease in uric acid excretion, suggesting the importance of reduced uric acid excretion to the pathogenesis of hyperuricemia.
- Uric acid is mainly excreted from the kidney. In humans, about 70% is excreted from the kidney, and 30% is excreted by extrarenal treatment such as in bile, saliva, and sweat. After 100% filtration of uric acid in the glomeruli of the kidney, most of the uric acid is reabsorbed in the proximal tubule and about 10% is excreted in the terminal urine (Non-patent Documents 3 and 10). ), Uric acid excretion is suggested to be strongly regulated by reabsorption.
- URAT1 Since uric acid exists as an organic acid under physiological pH conditions, transporters that reabsorb uric acid were expected to be highly similar to the organic anion transporter family.
- URAT1 was identified as a transporter that is present in tubules and reabsorbs uric acid (Non-patent Document 11).
- URAT1 is a 12-transmembrane transporter belonging to the SLC family. According to Northern blot analysis, URAT1 gene expression is restricted to the kidney in adults and fetuses. By observation of immunohistochemically stained images using anti-human URAT1 antibody, URAT1 protein is present on the proximal tubular lumen side.
- Non-patent Document 11 uric acid uptake is observed, and it has been confirmed that URAT1 is a transporter that transports uric acid. Furthermore, it has been clarified that a decrease in function due to mutation of the URAT1 gene causes renal hypouricemia, and the importance of URAT1 for uric acid excretion has been clarified (Non-Patent Documents 11 and 12).
- Non-patent Document 13 Benzbromarone and probenecid, which are currently used uric acid excretion promoters, have been shown to inhibit the uric acid transport activity of URAT1, and the importance of URAT1 in uric acid excretion has also been revealed pharmacologically.
- a drug that inhibits URAT1 can reduce blood uric acid level by suppressing reabsorption of uric acid in the proximal tubule and enhancing uric acid excretion. It is useful as a therapeutic or prophylactic agent for the pathological condition, ie, hyperuricemia, gout nodule, gout arthritis, gout kidney, urinary calculus, and renal dysfunction. It is also useful as a therapeutic or prophylactic agent for hypertension, hyperlipidemia, impaired glucose tolerance, obesity, coronary artery disease, and cerebrovascular disorder associated with hyperuricemia.
- Patent Document 1 discloses a compound having the following general formula.
- Patent Document 2 discloses a compound having the following general formula.
- Patent Document 3 discloses a compound having the following general formula.
- Patent Document 4 discloses a compound of the following general formula as a PDE5 (phosphodiesterase 5) inhibitor.
- Patent Document 5 discloses a compound having the following general formula as a PDE5 inhibitor.
- Patent Document 6 discloses a compound of the following general formula as a 17 ⁇ HSD (17 ⁇ -hydroxysteroid dehydrogenase) type 5 inhibitor.
- R 1 is a lower alkyl group which may be substituted with a cycloalkyl group, a cycloalkyl group, a halo lower alkyl group, a hydroxy lower alkyl group, a lower alkoxy lower alkyl group, a lower alkoxycarbonyl group, a lower alkylsulfonyl group or a general formula:
- Q 1 represents a single bond or a lower alkylene group (wherein one or more methylene groups constituting the lower alkylene group are each independently a carbonyl group, a sulfinyl group or a sulfonyl group, and the entire methylene group is May be substituted and / or the hydrogen constituting the methylene group may be substituted with a lower alkyl group);
- a 1 represents an aryl group or a heteroaryl group which may be substituted with 1 to 3 substituents selected from
- W 1 represents a group represented by the general formula: —C (R aa ) (R ab ) — or a group represented by the general formula: — (C ⁇ O) —
- W 2 represents a general formula: A group represented by —C (R ba ) (R bb ) —, a group represented by the general formula: — (C ⁇ O) — or a group represented by the general formula: —N (R bc ) —
- R a and R b each independently represent a hydrogen atom, a substituent selected from the ⁇ Substituent group M> described later, or a group represented by the general formula: -Q 2 -A 2
- R aa and R ab each independently represent a hydrogen atom, a substituent selected from the following ⁇ Substituent group N>, or a group represented by the general formula: -Q 2 -A 2 , or R a
- R ba and R bb are each independently a hydrogen atom, a halogen atom, an amino group, a lower alkylamino group, a di-lower alkylamino group, a hydroxy-lower alkylamino group, a lower alkylsulfonylamino group, a lower alkoxycarbonylamino group, A substituent selected from ⁇ Substituent group N> or a group represented by the general formula: -Q 2 -A 2 , or R ba and R bb together form a lower alkylene group
- one or more methylene groups constituting the lower alkylene group are each independently represented by an oxygen atom, a carbonyl group, a vinylene group or a general formula: —N (R c ) —.
- the entire methylene group may be replaced with a group, and / or hydrogen constituting the methylene group may be replaced with a hydroxyl group, a lower alkyl group or a halogen atom.
- R bc is selected from the group consisting of a hydrogen atom, a lower alkyl group, a cycloalkyl group, a halo lower alkyl group, a lower alkoxycarbonyl group, a carbamoyl group, a mono-lower alkylcarbamoyl group, a di-lower alkylcarbamoyl group, and a lower alkanoyl group.
- Q 2 represents a single bond, a lower alkylene group or a lower alkenylene group (wherein one or more methylene groups constituting the lower alkylene group are each independently an oxygen atom, a nitrogen atom or a carbonyl group).
- the entire methylene group may be replaced and / or the hydrogen constituting the methylene group may be replaced by a halogen atom, a cyano group, a hydroxyl group or a lower alkyl group);
- a 2 represents a cycloalkyl group, an aliphatic heterocyclic group, an aryl group or a heteroaryl group, which may be substituted with 1 to 3 substituents selected from the ⁇ Substituent group L> (wherein Any two substituents adjacent to each other on the aryl group or heteroaryl group may be combined to form a lower alkylenedioxy group);
- W 3 , W 4 and W 5 are each independently a nitrogen atom or a halogen atom, a hydroxyl group, a cyano group, a lower alkyl group, a cycloalkyl group, a halo lower alkyl group, a lower alkoxy group and a halo lower alkoxy group.
- X represents a single bond, oxygen atom, sulfur atom, sulfinyl group, sulfonyl group, carbonyl group, lower alkenylene group, lower alkynylene group or a group represented by the general formula: —N (R X ) — (where, R X is a hydrogen atom or a lower alkyl group.);
- R c represents a hydrogen atom, a lower alkyl group, a halo lower alkyl group or a lower alkanoyl group
- Z is a hydroxyl group, COOR 2 , CONR 3 R 4 , SO 3 R 2 , SO 3 NR 3 R 4 , 5-tetrazolyl group, 5-oxo-1,2,4-oxadiazolyl group, 2-oxo-1,3 , 4-oxadiazolyl group, 5-imino-4,5-dihydro-1,3,4-oxadiazolyl group, 2-thioxo-1,3,4-oxadiazolyl group or 5-oxo-1,2,4-thiadiazolyl group Represents;
- R 2 , R 3 and R 4 each independently represent a hydrogen atom or a lower alkyl group; ⁇ Substituent Group L>, ⁇ Substituent Group M> and ⁇ Substituent Group N> Is defined as ⁇ Substituent group L
- the compound represented by the above formula (I) includes all enantiomers and diastereomers that can exist as well as the racemic form of the compound.
- the present invention is also selected from the group consisting of hyperuricemia in mammals (especially humans), gouty nodules, acute draft arthritis, chronic draft arthritis, gout kidney, urolithiasis, renal dysfunction, coronary artery disease and ischemic heart disease
- the present invention relates to a method for treating or preventing a pathological condition involving blood uric acid, which comprises administering to the mammal a therapeutically effective amount of a compound of formula (I).
- the present invention is selected from the group consisting of hyperuricemia in mammals (particularly humans), gout nodules, acute ventilatory arthritis, chronic ventilatory arthritis, gout kidney, urolithiasis, renal dysfunction, coronary artery disease and ischemic heart disease
- a method for treating or preventing a disease state involving blood uric acid, wherein the URAT1 inhibitor, a blood uric acid level-lowering agent or a pharmaceutical composition comprising the compound of formula (I) is therapeutically effective in the mammal. It relates to a method characterized by administering an amount.
- the present invention relates to a URAT1 inhibitor comprising a compound of formula (I) as an active ingredient.
- the present invention further relates to a blood uric acid level-lowering agent containing a compound of formula (I) as an active ingredient.
- the present invention provides a blood uric acid selected from the group consisting of hyperuricemia, gouty nodule, acute ventilatory arthritis, chronic ventilatory arthritis, gout kidney, urolithiasis, renal dysfunction, coronary artery disease and ischemic heart disease.
- the present invention relates to a pharmaceutical composition for treating or preventing a disease state involved, which comprises the compound (I) as an active ingredient.
- the compound represented by the formula (I) of the present invention and pharmaceutically acceptable salts and esters of the compound have an excellent URAT1 inhibitory action as shown in the examples described later, and therefore promote uric acid excretion. Therefore, the compound represented by the formula (I) of the present invention and pharmaceutically acceptable salts and esters of the compound can reduce blood uric acid level, for example, hyperuricemia, gouty nodule, acute ventilatory arthritis It is useful as a therapeutic or prophylactic agent for pathological conditions involving blood uric acid such as chronic gout arthritis, gout kidney, urinary calculus, renal dysfunction, coronary artery disease or ischemic heart disease.
- halogen atom in the above formula (I) include a fluorine atom, a chlorine atom, a bromine atom and an iodine atom.
- the “lower alkyl group” in the above formula (I) means a linear or branched alkyl group having 1 to 6 carbon atoms, such as a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, Isobutyl group, sec-butyl group, tert-butyl group, pentyl group, isopentyl group, isoamyl group, neopentyl group, 1,1-dimethylpropyl group, 1-methylbutyl group, 2-methylbutyl group, 1,2-dimethylpropyl group Hexyl group, isohexyl group, 1-methylpentyl group, 2-methylpentyl group, 3-methylpentyl group, 1,1-dimethylbutyl group, 1,2-dimethylbutyl group, 2,2-dimethylbutyl group, 1 , 3-dimethylbutyl group, 2,3-dimethylbutyl group,
- cycloalkyl group in the above formula (I) means a 3- to 8-membered aliphatic cyclic group, for example, a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexyl group, a cycloheptyl group, and a cyclooctyl group.
- halo lower alkyl group in the above formula (I) is the above “lower alkyl group” substituted at one or more, preferably 1 to 3, the same or different halogen atoms at any substitutable position.
- the “lower alkoxy group” in the above formula (I) means a group in which a hydrogen atom of a hydroxyl group is substituted with the “lower alkyl group”.
- halo-lower alkoxy group in the above formula (I) is the above-mentioned “lower alkoxy group” substituted at one or more, preferably 1 to 3, the same or different halogen atoms at any substitutable position.
- Examples thereof include an ethoxy group, 1,2-dichloroethoxy group, bromomethoxy group and iodomethoxy group.
- hydroxy lower alkyl group in the above formula (I) means the “lower alkyl group” substituted at one or more, preferably 1 or 2, hydroxyl groups at any substitutable position. Hydroxymethyl group, 1-hydroxyethyl group, 1-hydroxypropyl group, 2-hydroxyethyl group, 2-hydroxypropyl group, 2-hydroxy-1-methylethyl group, 1-hydroxy-1-methylethyl group, 1, Examples include 2-dihydroxyethyl group and 3-hydroxypropyl group.
- lower alkoxy lower alkyl group in the above formula (I) is the above-mentioned “lower alkoxy group” which is substituted with 1 or 2 or more, preferably 1 or 2 identical or different “lower alkoxy groups” at any substitutable position.
- Lower alkyl group for example, methoxymethyl group, ethoxymethyl group, 2-methoxyethyl group, 2-ethoxyethyl group, 1-methoxy-1-methylethyl group, 1,2-dimethoxyethyl group and 3-methoxy A propyl group etc. are mentioned.
- the “lower alkoxycarbonyl group” in the above formula (I) means a group in which the “lower alkoxy group” and a carbonyl group are bonded, that is, an alkoxycarbonyl group having 2 to 7 carbon atoms, such as a methoxycarbonyl group.
- the “lower alkanoyl group” in the above formula (I) means a group in which the lower alkyl group and a carbonyl group are bonded, that is, an alkanoyl group having 2 to 7 carbon atoms, such as an acetyl group, a propionyl group, a butyryl group, Examples include isobutyryl group, valeryl group, isovaleryl group, and pivaloyl group.
- the “lower alkylthio group” in the above formula (I) means a group in which the “lower alkyl group” and a sulfur atom are bonded, that is, an alkylthio group having 1 to 6 carbon atoms, such as a methylthio group or an ethylthio group.
- an alkylthio group having 1 to 6 carbon atoms such as a methylthio group or an ethylthio group.
- the “lower alkylsulfonyl group” in the above formula (I) means a group in which the “lower alkyl group” and a sulfonyl group are bonded.
- the “lower alkylamino group” in the above formula (I) means an amino group N-monosubstituted by the above “lower alkyl group”, for example, an N-methylamino group, an N-ethylamino group, an N— Examples thereof include a propylamino group, an N-isopropylamino group, an N-butylamino group, an N-sec-butylamino group, and an N-tert-butylamino group.
- the “di-lower alkylamino group” in the above formula (I) means an amino group N, N-disubstituted by the same or different “lower alkyl group”, for example, N, N-dimethylamino group N, N-diethylamino group, N, N-dipropylamino group, N, N-diisopropylamino group, N-methyl-N-ethylamino group, N-methyl-N-propylamino group and N-methyl-N -An isopropylamino group etc. are mentioned.
- hydroxy lower alkylamino group in the above formula (I) means an amino group N-monosubstituted or N, N-disubstituted, preferably N-monosubstituted by the “hydroxy lower alkyl group”. Examples thereof include a hydroxymethylamino group, a 2-hydroxyethylamino group, a 1-hydroxy-1-methylethylamino group, a 1,2-dihydroxyethylamino group, and a 3-hydroxypropylamino group.
- the “mono-lower alkylcarbamoyl group” in the above formula (I) means a group in which the above-mentioned “lower alkyl group” is N-monosubstituted on the nitrogen atom of the carbamoyl group, and N-methylcarbamoyl group, N-ethylcarbamoyl group Group, N-propylcarbamoyl group, N-isopropylcarbamoyl group, N-butylcarbamoyl group, N-sec-butylcarbamoyl group, N-tert-butylcarbamoyl group and the like.
- di-lower alkylcarbamoyl group in the above formula (I) means a group in which the same or different “lower alkyl group” as described above is N, N-disubstituted on the nitrogen atom of the carbamoyl group.
- the “di-lower alkylcarbamoyl group” is a 5- to 8-membered member formed by combining the nitrogen atom constituting the carbamoyl group with the same or different “lower alkyl group” bonded to the nitrogen atom.
- the “lower alkanoylamino group” in the above formula (I) means a group in which the “lower alkanoyl group” and an amino group or the “lower alkylamino group” are bonded, for example, an N-acetylamino group, N -Propanoylamino group, N-butanoylamino group, N-pentanoylamino group, N-pivaloyl group, N-methyl-N-acetylamino group, N-methyl-N-propanoylamino group, N-methyl- N-butanoylamino group, N-methyl-N-pentanoylamino group, N-ethyl-N-acetylamino group, N-ethyl-N-propanoylamino group, N-ethyl-N-butanoylamino group and And N-ethyl-N-pentanoylamino group.
- the “lower alkoxycarbonylamino group” in the above formula (I) means a group in which the “lower alkoxycarbonyl group” is bonded to an amino group or the “lower alkylamino group”, such as a methoxycarbonylamino group, Ethoxycarbonylamino group, propoxycarbonylamino group, isopropoxycarbonylamino group, butoxycarbonylamino group, isobutoxycarbonylamino group, sec-butoxycarbonylamino group, tert-butoxycarbonylamino group, pentyloxycarbonylamino group, neopentyloxy Examples thereof include a carbonylamino group, a hexyloxycarbonylamino group, an isohexyloxycarbonylamino group, an N-methyl-methoxycarbonylamino group, and an N-methyl-ethoxycarbonylamino group.
- the “lower alkylsulfonylamino group” in the above formula (I) means a group in which the “lower alkylsulfonyl group” and the amino group or the “lower alkylamino group” are bonded, for example, a methylsulfonylamino group, Ethylsulfonylamino group, propylsulfonylamino group, isopropylsulfonylamino group, butylsulfonylamino group, sec-butylsulfonylamino group, tert-butylsulfonylamino group, N-methyl-methylsulfonylamino group, N-methyl-ethylsulfonylamino group Group, N-methyl-propylsulfonylamino group, N-methyl-isopropylsulfonylamino group, N-methyl-butylsulfonylamino
- aryl group examples include a phenyl group, a naphthyl group, a biphenyl group, and an anthryl group.
- heteroaryl group in the above formula (I) contains 1 or 2 or more, preferably 1 to 4 heteroatoms, which are the same or different from the group consisting of oxygen atom, nitrogen atom and sulfur atom.
- the “aliphatic heterocyclic group” in the above formula (I) is a 5-membered or 6-membered member containing 1 or 2 or more heteroatoms selected from the group consisting of an oxygen atom, a nitrogen atom and a sulfur atom.
- a saturated or unsaturated aliphatic heterocyclic group which is a condensed ring composed of 2 to 3 rings containing the heteroatom, such as azetidyl group, pyrrolidinyl group, piperidinyl group, pyrazinyl Group, morpholino group, tetrahydrofuranyl group, imidazolidinyl group, thiomorpholino group, tetrahydroquinolyl group, tetrahydroisoquinolyl group and the like.
- the “aryloxy group” in the above formula (I) means a group in which an oxygen atom is bonded to the “aryl group”, and examples thereof include a phenoxy group, a naphthalen-1-yloxy group, and a naphthalen-2-yloxy group. Can be mentioned.
- heteroaryloxy group in the above formula (I) means a group in which an oxygen atom is bonded to the “heteroaryl group”, and examples thereof include a furan-2-yloxy group, a furan-3-yloxy group, and thiophene.
- the “lower alkylene group” in the above formula (I) means a linear or branched alkylene group having 1 to 6 carbon atoms, such as a methylene group, an ethylene group, a trimethylene group, a tetramethylene group, or a pentamethylene group. And a hexamethylene group.
- the “lower alkenylene group” in the above formula (I) means a divalent group formed by removing one hydrogen atom from each chain end of the “lower alkenyl group”, for example, a vinylene group and A propenylene group etc. are mentioned.
- the “lower alkynylene group” in the above formula (I) means a divalent group formed by removing one hydrogen atom from each chain end of the “lower alkynyl group”. For example, an ethynylene group and And propynylene group.
- the “lower alkylenedioxy group” in the above formula (I) means a group formed by bonding both ends of the “lower alkylene group” to an oxygen atom, for example, methylenedioxy group, ethylenedioxy group. Group and propylenedioxy group.
- the “lower alkenyl group” in the above formula (I) means a linear or branched alkenyl group having 2 to 6 carbon atoms, such as a vinyl group, 1-propenyl group, allyl group, isopropenyl group, 3 -Butenyl group, 2-butenyl group, 1-butenyl group, 1-methyl-2-propenyl group, 1-methyl-1-propenyl group, 1-ethyl-1-ethenyl group, 2-methyl-2-propenyl group, Examples include a 2-methyl-1-propenyl group, a 3-methyl-2-butenyl group, and a 4-pentenyl group.
- the “lower alkynyl group” in the above formula (I) means a straight-chain or branched alkynyl group having 2 to 6 carbon atoms, such as ethynyl group, 1-propynyl group, 2-propynyl group, 3-butynyl. Group, 2-butynyl group, 1-butynyl group, 1-methyl-2-propynyl group, 1-ethyl-2-propynyl group, 1-methyl-2-butynyl group and 4-pentynyl group.
- the “aralkyl group” in the above formula (I) means the “lower alkyl group” which is substituted with one or two, preferably 1 or 2, of the “aryl group” at any substitutable position. Examples include benzyl group, 1-phenylethyl group, 2-phenylethyl group, 1-naphthylmethyl group, 2-naphthylmethyl group and the like.
- any substitutable position means a substitutable hydrogen atom on a carbon atom, nitrogen atom, oxygen atom and / or sulfur atom, and the substitution of the hydrogen atom is chemically permissible. Means the site of what results in a stable compound.
- R 1 is a lower alkyl group, cycloalkyl group, halo lower alkyl group, hydroxy lower alkyl group, lower alkoxy lower alkyl group, lower alkoxycarbonyl group, lower alkoxy group which may be substituted with a cycloalkyl group, lower An alkylsulfonyl group or a group represented by the general formula: -Q 1 -A 1 .
- R 1 is preferably, for example, a lower alkyl group that may be substituted with a cycloalkyl group, a cycloalkyl group, a lower alkylsulfonyl group, or a group represented by the general formula: -Q 1 -A 1 , and the like. Is a group represented by the general formula: -Q 1 -A 1 .
- the “lower alkyl group which may be substituted with a cycloalkyl group” for R 1 is an unsubstituted lower alkyl group or one or two or more, preferably 1 or 2 of the above-described “ Means a lower alkyl group substituted by ⁇ cycloalkyl group '', for example, methyl group, ethyl group, propyl group, isopropyl group, butyl group, isobutyl group, sec-butyl group, tert-butyl group, pentyl group, isopentyl group, Isoamyl group, neopentyl group, 1,1-dimethylpropyl group, 1-methylbutyl group, 2-methylbutyl group, 1,2-dimethylpropyl group, hexyl group, isohexyl group, 1-methylpentyl group, 2-methylpentyl group, 3-methylpentyl group, 1,1-dimethylbutyl group, 1,
- isopropyl group isobutyl group, cyclopropylmethyl group A cyclobutylmethyl group, a cyclopentylmethyl group, a cyclohexylmethyl group, and the like.
- the cycloalkyl group represented by R 1 is preferably, for example, a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, or a cyclohexyl group.
- the halo-lower alkyl group for R 1 is preferably, for example, a fluoromethyl group, a difluoromethyl group, a trifluoromethyl group, a 2,2,2-trifluoroethyl group, or the like.
- the hydroxy lower alkyl group for R 1 is preferably, for example, a hydroxymethyl group or a 2-hydroxyethyl group.
- the lower alkoxy lower alkyl group for R 1 is preferably, for example, a methoxymethyl group or an ethoxymethyl group.
- the lower alkoxycarbonyl group for R 1 is preferably, for example, a methoxycarbonyl group, an ethoxycarbonyl group, a propoxycarbonyl group, an isopropoxycarbonyl group, a butoxycarbonyl group, an isobutoxycarbonyl group, a tert-butoxycarbonyl group, or a pentyloxycarbonyl group. is there.
- the lower alkylsulfonyl group for R 1 is, for example, preferably a methanesulfonyl group or an ethanesulfonyl group.
- Q 1 represents a single bond or a lower alkylene group (wherein one or more methylene groups constituting the lower alkylene group are each independently a carbonyl group, a sulfinyl group or a sulfonyl group, and the entire methylene group is And / or hydrogen constituting the methylene group may be substituted with a lower alkyl group).
- the lower alkylene group for Q 1 is preferably, for example, a methylene group, an ethylene group or a trimethylene group.
- One or two or more methylene groups constituting the lower alkylene group of Q 1 may each independently be replaced by the entire methylene group with a carbonyl group, a sulfinyl group or a sulfonyl group, and / or
- the constituent hydrogen may be substituted with a lower alkyl group, and as such a substituted or substituted group, for example, a group selected from the following formulas is preferable.
- a 1 represents an aryl group or a heteroaryl group which may be substituted with 1 to 3 substituents selected from the ⁇ Substituent group L> (here, on the aryl group or heteroaryl group) Any two substituents adjacent to each other may be taken together to form a lower alkylenedioxy group.)
- ⁇ Substituent group L> is a halogen atom, hydroxyl group, nitro group, cyano group, formyl group, amino group, carboxyl group, lower alkyl group, halo lower alkyl group, cycloalkyl group, lower alkoxy group, halo lower group.
- Alkoxy group hydroxy lower alkyl group, lower alkoxy lower alkyl group, lower alkoxycarbonyl group, lower alkanoyl group, lower alkylthio group, lower alkylsulfonyl group, lower alkylamino group, di-lower alkylamino group, carbamoyl group, mono-lower alkylcarbamoyl A group consisting of a group, a di-lower alkylcarbamoyl group, a lower alkanoylamino group, a lower alkylsulfonylamino group, a lower alkoxycarbonylamino group, an aralkyl group, an aryloxy group, a heteroaryloxy group, and a lower alkenyl group.
- the aryl group for A 1 is preferably, for example, a phenyl group, a naphthyl group, or a biphenyl group.
- the heteroaryl group of A 1 is, for example, imidazolyl group, furyl group, thienyl group, pyrazolyl group, thiazolyl group, isothiazolyl group, oxazolyl group, isoxazolyl group, 1,2,4-oxadiazolyl group, 1,3,4-oxadiazolyl group , Pyridyl group, pyrazinyl group, pyrimidinyl group, benzofuranyl group, quinolyl group, benzothienyl group, etc., more preferably pyridyl group, quinolyl group, thienyl group, pyrazolyl group, thiazolyl group, isoxazolyl group, benzothienyl group, etc., more preferably Pyridyl group, isoxazoly
- a 1 "any two substituents adjacent to each other on the aryl group or heteroaryl group may be combined to form a lower alkylenedioxy group” means that the aryl group or heteroaryl group A group in which any two substituents adjacent to each other above form a lower alkylenedioxy group, for example, a benzo [1,3] dioxolyl group and a 2,3-dihydro-benzo [1,4 A dioxynyl group and the like are preferable.
- a 1 is, for example, a phenyl group, 2-fluorophenyl group, 3-fluorophenyl group, 4-fluorophenyl group, 2,3-difluorophenyl group, 2,4-difluorophenyl group, 2,5-difluorophenyl.
- W 1 represents a nitrogen atom or a group represented by the general formula: ⁇ C (R a ) —
- W 2 represents a nitrogen atom or a group represented by the general formula: ⁇ C (R b ) —.
- W 1 represents a group represented by the general formula: —C (R aa ) (R ab ) — or a group represented by the general formula: — (C ⁇ O) —
- R a and R b each independently represent a hydrogen atom, a substituent selected from ⁇ Substituent group M>, or a group represented by the general formula: -Q 2 -A 2 .
- ⁇ Substituent group M> is a halogen atom, a hydroxyl group, a nitro group, a cyano group, a formyl group, an amino group, a carboxyl group, a lower alkyl group, a halo lower alkyl group, a cycloalkyl group, a lower alkoxy group, a halo lower group.
- Alkoxy group hydroxy lower alkyl group, lower alkoxy lower alkyl group, lower alkoxycarbonyl group, lower alkanoyl group, lower alkylthio group, lower alkylsulfonyl group, lower alkylamino group, di-lower alkylamino group, carbamoyl group, mono-lower alkylcarbamoyl A group consisting of a group, a di-lower alkylcarbamoyl group, a lower alkanoylamino group, a lower alkylsulfonylamino group and a lower alkoxycarbonylamino group.
- R a and R b include, for example, a hydrogen atom, a halogen atom, a hydroxyl group, a cyano group, a formyl group, a carboxyl group, a lower alkyl group, a halo lower alkyl group, a cycloalkyl group, a hydroxy lower alkyl group, a lower alkoxy lower alkyl group, Preferred are a lower alkoxycarbonyl group, a lower alkanoyl group and a group represented by the general formula: -Q 2 -A 2 , more preferably a hydrogen atom, a halogen atom, a cyano group, a lower alkyl group, a halo-lower alkyl group.
- R aa and R ab are each independently a hydrogen atom, a substituent selected from ⁇ Substituent group N>, or a group represented by the general formula: -Q 2 -A 2 .
- ⁇ Substituent group N> is a hydroxyl group, a cyano group, a formyl group, a carboxyl group, a lower alkyl group, a halo lower alkyl group, a cycloalkyl group, a lower alkoxy group, a halo lower alkoxy group, a hydroxy lower alkyl group, a lower group.
- It is a group consisting of an alkoxy lower alkyl group, a lower alkoxycarbonyl group, a lower alkanoyl group, a carbamoyl group, a mono-lower alkylcarbamoyl group and a di-lower alkylcarbamoyl group.
- R aa and R ab may be combined to form a lower alkylene group (wherein one or more methylene groups constituting the lower alkylene group are each independently an oxygen atom, carbonyl
- the entire methylene group may be replaced by a group, vinylene group or a group represented by the general formula: —N (R c ) — and / or hydrogen constituting the methylene group is a hydroxyl group, a lower alkyl group or a halogen atom. It may be substituted with an atom).
- one or more methylene groups constituting the lower alkylene group are each independently an oxygen atom, a carbonyl group, a vinylene group or a group represented by the general formula: —N (R c ) —.
- the entire group may be replaced and / or the hydrogen constituting the methylene group may be replaced with a hydroxyl group, a lower alkyl group or a halogen atom.
- a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, and a cyclohexyl group are preferable.
- R aa and R ab for example, a hydrogen atom, a lower alkyl group, a halo lower alkyl group, a cycloalkyl group, a lower alkoxy group, and a group represented by the general formula: -Q 2 -A 2 are preferable.
- R ba and R bb are each independently a hydrogen atom, a halogen atom, an amino group, a lower alkylamino group, a di-lower alkylamino group, a hydroxy-lower alkylamino group, a lower alkylsulfonylamino group, a lower alkoxycarbonylamino group, A substituent selected from ⁇ Substituent group N> or a group represented by the general formula: -Q 2 -A 2 is represented.
- R ba and R bb may be combined to form a lower alkylene group (wherein one or more methylene groups constituting the lower alkylene group are each independently an oxygen atom, carbonyl
- the entire methylene group may be replaced with a group, vinylene group or a group represented by the general formula: —N (R c ) — and / or may be substituted with a hydroxyl group, a lower alkyl group or a halogen atom.
- R ba and R bb may be combined to form a lower alkylene group (wherein one or more methylene groups constituting the lower alkylene group are each independently an oxygen atom, carbonyl
- the entire methylene group may be replaced with a group, vinylene group or a group represented by the general formula: —N (R c ) — and / or may be substituted with a hydroxyl group, a lower alkyl group or a halogen atom.
- R c —N (R
- one or more methylene groups constituting the lower alkylene group are each independently an oxygen atom, a carbonyl group, a vinylene group or a group represented by the general formula: —N (R c ) —.
- the entire group may be replaced and / or the hydrogen constituting the methylene group may be replaced with a hydroxyl group, a lower alkyl group or a halogen atom.
- a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexyl group and the like are preferable.
- R ba and R bb include a hydrogen atom, a halogen atom, a lower alkylamino group, a di-lower alkylamino group, a lower alkyl group, a halo-lower alkyl group, a cycloalkyl group, a lower alkoxy group, a lower alkoxycarbonyl group, and a lower alkanoyl.
- a group, a carbamoyl group, a mono-lower alkylcarbamoyl group, a di-lower alkylcarbamoyl group, a group represented by the general formula: -Q 2 -A 2 , and the like are preferable.
- R bc is selected from the group consisting of a hydrogen atom, a lower alkyl group, a cycloalkyl group, a halo lower alkyl group, a lower alkoxycarbonyl group, a carbamoyl group, a mono-lower alkylcarbamoyl group, a di-lower alkylcarbamoyl group, and a lower alkanoyl group.
- a substituent or a group represented by the general formula: -Q 2 -A 2 is represented.
- R bc is preferably, for example, a hydrogen atom, a lower alkyl group, a halo lower alkyl group, a lower alkanoyl group, or a group represented by the general formula: -Q 2 -A 2 .
- Q 2 represents a single bond, a lower alkylene group or a lower alkenylene group, wherein one or two or more methylene groups constituting the lower alkylene group are each independently an oxygen atom, a nitrogen atom or a carbonyl group.
- the entire methylene group may be replaced and / or the hydrogen constituting the methylene group may be replaced with a halogen atom, a cyano group, a hydroxyl group or a lower alkyl group.
- the lower alkylene group for Q 2 for example, a methylene group, an ethylene group, a trimethylene group and the like are preferable.
- One or two or more methylene groups constituting the lower alkylene group of Q 2 may each independently replace the entire methylene group with an oxygen atom, a nitrogen atom or a carbonyl group, and / or
- the constituent hydrogen may be substituted with a halogen atom, a cyano group, a hydroxyl group or a lower alkyl group.
- a substituted or substituted group for example, a group selected from the following formulas is preferable.
- Q 2 is more preferably a single bond, a methylene group, or a group selected from the following.
- a 2 represents a cycloalkyl group, an aliphatic heterocyclic group, an aryl group, or a heteroaryl group, which may be substituted with 1 to 3 substituents selected from the ⁇ Substituent group L> (wherein Any two substituents adjacent to each other on the aryl group or heteroaryl group may be combined to form a lower alkylenedioxy group.
- aryl group for A 2 for example, a phenyl group, a naphthyl group, a biphenyl group and the like are preferable.
- heteroaryl group for A 2 include imidazolyl, furyl, thienyl, pyrazolyl, thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, 1,2,4-oxadiazolyl, 1,3,4-oxadiazolyl Group, pyridyl group, pyrazinyl group, pyrimidinyl group, benzofuranyl group, quinolyl group and the like are preferable, pyridyl group and quinolyl group are more preferable, and pyridyl group is more preferable.
- a 2 "any two substituents adjacent to each other on the aryl group or heteroaryl group may be combined to form a lower alkylenedioxy group” means that the aryl group or heteroaryl group It means that any two substituents adjacent to each other above together form a lower alkylenedioxy group.
- a benzo [1,3] dioxolyl group and a 2,3-dihydro-benzo [1,4] dioxinyl group are preferable.
- a 2 represents, for example, a phenyl group, 2-fluorophenyl group, 3-fluorophenyl group, 4-fluorophenyl group, 2,3-difluorophenyl group, 2,4-difluorophenyl group, 2,5-difluorophenyl.
- W 3 , W 4 and W 5 are each independently a nitrogen atom or a halogen atom, a hydroxyl group, a cyano group, a lower alkyl group, a cycloalkyl group, a halo lower alkyl group, a lower alkoxy group and a halo lower alkoxy group.
- a methine group optionally having a substituent selected from the group consisting of a halogen atom, a hydroxyl group, a cyano group, a lower alkyl group, a cycloalkyl group, a halo lower alkyl group, a lower alkoxy group, and a halo lower alkoxy group” An unsubstituted methine group or a methine group having a substituent.
- the substituent can be selected from the group consisting of a halogen atom, a hydroxyl group, a cyano group, a lower alkyl group, a cycloalkyl group, a halo lower alkyl group, a lower alkoxy group, and a halo lower alkoxy group.
- the halogen atom for the substituent is, for example, preferably a fluorine atom or a chlorine atom.
- the lower alkyl group for the substituent is, for example, preferably a methyl group or an ethyl group.
- the cycloalkyl group for the substituent is, for example, preferably a cyclopropyl group.
- the halo-lower alkyl group for the substituent is, for example, preferably a fluoromethyl group, a difluoromethyl group, a trifluoromethyl group.
- the lower alkoxy group for the substituent is, for example, preferably a methoxy group and an ethoxy group.
- the halo-lower alkoxy group for the substituent is, for example, preferably a difluoromethoxy group and a trifluoromethoxy group.
- W 1 , W 2 , W 3 , W 4 and W 5 are 0 to 4, preferably 0 to 3, particularly preferably 0 to W 1 , W 2 , W 3 , W 4 and W 5.
- Two are nitrogen atoms.
- X is a single bond, oxygen atom, sulfur atom, sulfinyl group, sulfonyl group, carbonyl group, lower alkenylene group, lower alkynylene group or a group represented by the general formula: —N (R X ) — (where, R X is a hydrogen atom or a lower alkyl group.)
- the lower alkenylene group for X for example, a vinylene group is preferred.
- the lower alkynylene group for X is, for example, preferably an ethynylene group.
- X is preferably, for example, a single bond, an oxygen atom, a carbonyl group, a vinylene group, or a group represented by the general formula: —N (R X ) —.
- the lower alkyl group for R X is, for example, preferably a methyl group, an ethyl group, a propyl group, and more preferably a methyl group.
- (CR Yi R Yi ′ ) n are preferable (where n is an integer of 1 to 3, and i is an integer of 1 to n).
- n 3
- (CR Y1 R Y1 ′ ) ⁇ (CR Y2 R Y2 ′ ) — (CR Y3 R Y3 ′ ) is represented).
- R Y1 , R Y1 ′ , R Y2 , R Y2 ′ , R Y3 , R Y3 ′ , R Y4 , R Y4 ′ , R Y5 , R Y5 ′ , R Y6, and R Y6 ′ are each independently a hydrogen atom , A halogen atom or a substituent selected from ⁇ Substituent group N>.
- R Y1 , R Y1 ′ , R Y2 , R Y2 ′ , R Y3 , R Y3 ′ , R Y4 , R Y4 ′ , R Y5 , R Y5 ′ , R Y6, and R Y6 ′ are each independently, for example, A hydrogen atom, a halogen atom, a hydroxyl group, a cyano group, a lower alkyl group, a lower alkoxy group and the like are preferable.
- the lower alkyl group for the substituent is, for example, preferably a methyl group or an ethyl group.
- the lower alkoxy group for the substituent is, for example, preferably a methoxy group and an ethoxy group.
- R X R Y1 , R Y1 ′ , R Y2 , R Y2 ′ , R Y3 , R Y3 ′ , R Y4 , R Y4 ′ , R Y5 , R Y5 ′ , R Y6, and R Y6 ′ , the following two (I) R Y1 and R Y1 ′ , (ii) R Y2 and R Y2 ′ , (iii) R Y3 and R Y3 ′ , (iv) R Y4 and R Y4 ′ , (v) R Y5 And R Y5 ′ , (vi) R Y6 and R Y6 ′ , (vii) R X and R Y1 , (viii) R X and R Y2 , (ix) R X and R Y3 , (x) R Y1 and R Y2 , (Xi) R Y1
- R Y1 and R Y1 ′ together form a lower alkylene group means that three-membered by bonding the carbon atom substituted by R Y1 and R Y1 ′ via the lower alkylene group. Forms a 7-membered cyclosaturated carbocyclic ring.
- a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexyl group, etc. are mentioned, A cyclopropyl group etc. are suitable.
- R Y2 and R Y2 ′ together form a lower alkylene group means that a three-membered group is formed by bonding a carbon atom substituted by R Y2 and R Y2 ′ via a lower alkylene group. Forms a 7-membered cyclosaturated carbocyclic ring.
- a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexyl group, etc. are mentioned, A cyclopropyl group etc. are suitable.
- R Y3 and R Y3 ′ together form a lower alkylene group means that a three-membered group is formed by bonding a carbon atom substituted by R Y3 and R Y3 ′ via a lower alkylene group. Forms a 7-membered cyclosaturated carbocyclic ring.
- a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexyl group, etc. are mentioned, A cyclopropyl group etc. are suitable.
- R Y4 and R Y4 ′ together form a lower alkylene group means that a three-membered group is formed by bonding a carbon atom substituted by R Y4 and R Y4 ′ via a lower alkylene group. Forms a 7-membered cyclosaturated carbocyclic ring.
- a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexyl group, etc. are mentioned, A cyclopropyl group etc. are suitable.
- R Y5 and R Y5 ′ together form a lower alkylene group means that a three-membered structure is formed by bonding a carbon atom substituted by R Y5 and R Y5 ′ via a lower alkylene group. Forms a 7-membered cyclosaturated carbocyclic ring.
- a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexyl group, etc. are mentioned, A cyclopropyl group etc. are suitable.
- R Y6 and R Y6 ′ together form a lower alkylene group means that a three-membered group is formed by bonding a carbon atom substituted by R Y6 and R Y6 ′ via a lower alkylene group. Forms a 7-membered cyclosaturated carbocyclic ring.
- a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexyl group, etc. are mentioned, A cyclopropyl group etc. are suitable.
- R X and R Y1 together form a lower alkylene group means that R X and R Y1 are bonded via a lower alkylene group to form a 3- to 7-membered cyclosaturated carbon. To form a ring. For example, it is represented by the following formula.
- R X and R Y2 together form a lower alkylene group means that R X and R Y2 are bonded via a lower alkylene group to form a 3- to 7-membered cyclosaturated carbon. To form a ring. For example, it is represented by the following formula.
- R X and R Y3 together form a lower alkylene group means that R X and R Y3 are bonded via a lower alkylene group to form a 3- to 7-membered cyclosaturated carbon. To form a ring. For example, it is represented by the following formula.
- R Y1 and R Y2 together form a lower alkylene group means that R Y1 and R Y2 are bonded via a lower alkylene group to form a 3- to 7-membered cyclosaturated carbon. To form a ring. For example, it is represented by the following formula.
- R Y1 and R Y3 together form a lower alkylene group means that R Y1 and R Y3 are bonded via a lower alkylene group to form a 3- to 7-membered cyclosaturated carbon. To form a ring. For example, it is represented by the following formula.
- R Y1 and R Y4 together form a lower alkylene group means that R Y1 and R Y4 are bonded via a lower alkylene group to form a 3- to 7-membered cyclosaturated carbon. To form a ring. For example, it is represented by the following formula.
- R Y2 and R Y3 together form a lower alkylene group means that R Y2 and R Y3 are bonded via a lower alkylene group to form a 3- to 7-membered cyclosaturated carbon. To form a ring. For example, it is represented by the following formula.
- R Y2 and R Y4 together form a lower alkylene group means that R Y2 and R Y4 are bonded via a lower alkylene group to form a 3- to 7-membered cyclosaturated carbon. To form a ring. For example, it is represented by the following formula.
- R Y2 and R Y5 together form a lower alkylene group means that R Y2 and R Y5 are bonded via a lower alkylene group to form a 3- to 7-membered cyclosaturated carbon. To form a ring. For example, it is represented by the following formula.
- One or more methylene groups constituting the lower alkylene group forming the above (i) to (xv) are each independently an oxygen atom, a carbonyl group, a vinylene group or a general formula: —N (R c ) —
- the whole methylene group may be replaced with hydrogen and / or the hydrogen constituting the methylene group may be replaced with a hydroxyl group, a lower alkyl group or a halogen atom.
- R c is a hydrogen atom, a lower alkyl group, a halo lower alkyl group or a lower alkanoyl group.
- Z is a hydroxyl group, COOR 2 , CONR 3 R 4 , SO 3 R 2 , SO 3 NR 3 R 4 , 5-tetrazolyl group, 5-oxo-1,2,4-oxadiazolyl group, 2-oxo-1,3 , 4-oxadiazolyl group, 5-imino-4,5-dihydro-1,3,4-oxadiazolyl group, 2-thioxo-1,3,4-oxadiazolyl group or 5-oxo-1,2,4-thiadiazolyl group It is.
- R 2 , R 3 and R 4 are each independently a hydrogen atom or a lower alkyl group.
- Preferred embodiments of the present invention can also be expressed as the following (1) to (9).
- a compound of the above formula (I) or a pharmaceutically acceptable salt or ester of the compound is a compound of the above formula (I) or a pharmaceutically acceptable salt or ester of the compound.
- W 3 , W 4 and W 5 are each independently a methine group which may have a nitrogen atom or a substituent selected from the group consisting of a halogen atom and a lower alkyl group.
- R 1 is a group represented by the general formula: -Q 1 -A 1 , or a pharmaceutically acceptable salt or ester of the compound.
- the aryl group or heteroaryl group of A 1 is a phenyl group, a naphthyl group, a pyridyl group, a pyridanidyl group, a pyrazinyl group, a pyrimidinyl group, a quinolyl group, an isoquinolyl group, or a benzothienyl group according to (7) above A compound or a pharmaceutically acceptable salt or ester of the compound.
- (9) ⁇ Substituent group L> is a group consisting of a hydroxyl group, a halogen atom, a cyano group, a methyl group, an ethyl group, a cyclopropyl group, a trifluoromethyl group, a hydroxymethyl group, a methoxy group, and a trifluoromethoxy group.
- the compound according to (8) above, or a pharmaceutically acceptable salt or ester of the compound is a group consisting of a hydroxyl group, a halogen atom, a cyano group, a methyl group, an ethyl group, a cyclopropyl group, a trifluoromethyl group, a hydroxymethyl group, a methoxy group, and a trifluoromethoxy group.
- the compounds of the present invention may have asymmetric centers, chiral axes, and chiral planes.
- the compounds of the invention can occur as racemates, racemic mixtures and as individual diastereomers.
- all possible isomers, including optical isomers, and mixtures thereof are all included in the present invention.
- the compounds disclosed herein may exist as tautomers, and both tautomeric forms are within the scope of the invention, even if only one tautomeric structure is depicted. It is intended to be conjugated by.
- N-oxides of the compounds represented by the above formula (I) can be formed on any available nitrogen atom.
- N-oxides can be formed by conventional means, for example by reacting a compound of formula (I) with oxone in the presence of wet alumina.
- the “salt” of the compound of the present invention means a conventional pharmaceutically acceptable one.
- salts of acid addition salts in the group or basic heteroaryl group for example, a base addition salt in the carboxyl group, hydroxyl group or acidic heteroaryl group in the case of having an acidic heteroaryl group such as a carboxyl group, hydroxyl group or tetrazolyl group, or the amino in the case of having an amino group or a basic heteroaryl group.
- Examples of the base addition salt include alkali metal salts such as sodium salts and potassium salts; alkaline earth metal salts such as calcium salts and magnesium salts; ammonium salts; and trimethylamine salts, triethylamine salts, dicyclohexylamine salts, ethanol, and the like.
- Examples include amine salts, diethanolamine salts, triethanolamine salts, procaine salts, and organic amine salts such as N, N′-dibenzylethylenediamine salt.
- the acid addition salt examples include inorganic acid salts such as hydrochloride, sulfate, nitrate, phosphate and perchlorate; for example, maleate, fumarate, tartrate, citrate, ascorbate, Examples thereof include organic acid salts such as trifluoroacetate salts; and sulfonic acid salts such as methanesulfonate, isethionate, benzenesulfonate, and p-toluenesulfonate.
- inorganic acid salts such as hydrochloride, sulfate, nitrate, phosphate and perchlorate
- organic acid salts such as trifluoroacetate salts
- sulfonic acid salts such as methanesulfonate, isethionate, benzenesulfonate, and p-toluenesulfonate.
- esters when having a carboxyl group, it means a conventional pharmaceutically acceptable one in the carboxyl group.
- an ester with a lower alkyl group such as methyl group, ethyl group, propyl group, isopropyl group, butyl group, sec-butyl group, tert-butyl group, pentyl group, isopentyl group, neopentyl group, cyclopropyl group, cyclobutyl group, Esters with cycloalkyl groups such as cyclopentyl groups, esters with aralkyl groups such as benzyl groups and phenethyl groups, esters with lower alkenyl groups such as allyl groups and 2-butenyl groups, methoxymethyl groups, 2-methoxyethyl groups, Esters with lower alkoxy lower alkyl groups such as 2-ethoxyethyl group, esters with lower alkano
- the method for producing a pharmaceutically acceptable salt of the compound according to the present invention can be carried out by appropriately combining methods usually used in the field of synthetic organic chemistry. Specific examples include neutralization titration of a free solution of the compound according to the present invention with an alkaline solution or an acidic solution.
- the method for producing the ester of the compound according to the present invention can be carried out by appropriately combining methods usually used in the field of synthetic organic chemistry. Specifically, it can be produced by esterifying a free carboxy group according to a conventional method.
- the “pharmaceutically acceptable salt” of the present invention includes a solvate with a pharmaceutically acceptable solvent such as water or ethanol.
- the production method of the compound of the present invention will be specifically described.
- the present invention is not limited to these production methods.
- the order of the reaction can be appropriately changed.
- the reaction may be performed from a process or a site that seems reasonable.
- a substituent conversion (substituent conversion or further modification) step may be appropriately inserted between the steps.
- protection and deprotection may be appropriately performed.
- reagents other than the illustrated reagent can be used suitably.
- microwave irradiation may be used as necessary.
- raw material compounds whose production methods are not described are commercially available, or compounds that can be easily prepared by combining known synthetic reactions.
- the compound obtained in each step can be isolated and purified by a conventional method such as crystallization, recrystallization, column chromatography, preparative HPLC, etc. The process can be proceeded to.
- room temperature means 1 to 40 ° C.
- Schemes 1-8 below are general synthetic methods for compounds of formula (I).
- Scheme 1 Method for producing compound of formula (I) from compound of formula (II)
- the protecting group PG 2 of the above formula (II) is not particularly limited as long as it has the function, but for example, a lower alkyl group such as methyl group, ethyl group, propyl group, isopropyl group, tert-butyl group, etc.
- a halo lower alkyl group such as 2,2,2-trichloroethyl group; a lower alkenyl group such as an allyl group; a benzyl group, a p-methoxybenzyl group, a p-nitrobenzyl group, a benzhydryl group, a trityl group, etc.
- a methyl group, an ethyl group, a tert-butyl group, an allyl group, a benzyl group, a p-methoxybenzyl group, a benzhydryl group, and the like are preferable.
- the method for removing the protecting group varies depending on the kind of the protecting group and the stability of the target compound (I). For example, the method described in the literature [Protective Groups in Organic Synthesis], 3rd edition, T.W. W.
- Formula (II) [wherein R 1 , W 1 , W 2 , W 3 , W 4 , W 5 , X and Y are as defined above, and PG 2 is a protecting group.
- the compound of the above formula (III) [wherein W 1 , W 2 , W 3 , W 4 , W 5 , X and Y are as defined above, and PG 2 is a protecting group].
- a compound represented by the above formula (IV), wherein R 1 has the same meaning as described above, and L represents a leaving group. ] Can be obtained by an alkylation reaction with the compound.
- the leaving group L of the above formula (IV) is not particularly limited as long as it can be eliminated by the reaction with the compound (III) to produce the compound (II).
- the amount of compound (IV) to be used is generally 1 to 10 mol, preferably 1 to 3 mol, relative to 1 mol of compound (III).
- Examples of the base used include sodium carbonate, potassium carbonate, sodium hydrogen carbonate, sodium hydride, potassium hydroxide and the like, and potassium carbonate, sodium hydride, potassium hydroxide and the like are preferable.
- the amount of the base is usually 1 to 10 mol, preferably 1 to 5 mol, per 1 mol of compound (III).
- the reaction temperature is usually 0 ° C. to 160 ° C., preferably 25 ° C. to 100 ° C.
- the reaction time is usually 1 hour to 24 hours, preferably 1 hour to 12 hours.
- the reaction solvent is not particularly limited as long as it does not hinder the reaction, but solvents such as dimethylformamide, N-methyl-2-pyrrolidone, tetrahydrofuran, 1,4-dioxane, acetone, methyl ethyl ketone, and acetonitrile are preferable.
- Formula (III) [wherein W 1 , W 2 , W 3 , W 4 , W 5 , X and Y are as defined above, and PG 2 represents a protecting group.
- the compound of the above formula (V) [wherein W 1 , W 2 , W 3 , W 4 , W 5 , X and Y are as defined above, and PG 1 and PG 2 represent a protecting group]. It can be obtained by removing the protecting group PG 1 of the compound represented by the formula:
- the protecting group PG 1 of the above formula (V) is not particularly limited as long as it has the function.
- Aralkyl groups such as a group, p-nitrobenzyl group, benzhydryl group, trityl group; lower alkanoyl groups such as formyl group, acetyl group, propionyl group, butyryl group, pivaloyl group; such as benzoyl group; such as phenylacetyl group, phenoxyacetyl Arylalkanoyl groups such as a group; lower alkoxycarbonyl groups such as methoxycarbonyl group, ethoxycarbonyl group, propyloxycarbonyl group, tert-butoxycarbonyl group; for example benzyloxycarbonyl group, p-nitrobenzyloxycarbonyl group, phenethyloxyca Aralkyloxycarbonyl groups such as bonyl groups; lower alkylsilyl groups such as trimethylsilyl groups and tert-butyldimethylsilyl groups; tetrahydropyranyl groups; such as
- Examples thereof include arylsulfonyl groups such as benzenesulfonyl group and p-toluenesulfonyl group, and tert-butoxycarbonyl group, methylsulfonyl group and p-toluenesulfonyl group are particularly preferable.
- the method for removing the protecting group varies depending on the kind of the protecting group and the stability of the target compound (III). For example, the method described in the literature [Protective Groups in Organic Synthesis], 3rd edition, T.W. W.
- a lower alkyl group such as a methyl group, an ethyl group, a propyl group, an isopropyl group, or a tert-butyl group.
- a halo lower alkyl group such as 2,2,2-trichloroethyl group; a lower alkenyl group such as an allyl group; a benzyl group, a p-methoxybenzyl group, a p-nitrobenzyl group, a benzhydryl group, a trityl group, etc.
- a methyl group, an ethyl group, a tert-butyl group, an allyl group, a benzyl group, a p-methoxybenzyl group, a benzhydryl group, and the like are preferable.
- the method for introducing a protecting group varies depending on the type of the protecting group and the stability of the compound. For example, the method described in the literature [Protective Groups in Organic Synthesis, 3rd edition, T.A. W. According to Green (TW Greene, John Wiley & Sons (1999)) or a similar method.
- 2-methyl-2-butene is usually 1 to 20 mol, preferably 1 to 10 mol
- sodium dihydrogen phosphate is usually 1 to 5 mol, preferably 1 mol, per 1 mol of compound (VII). 1 to 3 mol
- sodium chlorite is usually used in an amount of 1 to 10 mol, preferably 1 to 5 mol.
- the reaction temperature is usually 0 ° C. to 100 ° C., preferably 0 ° C. to 40 ° C.
- the reaction time is usually 1 hour to 24 hours, preferably 1 hour to 6 hours.
- the reaction solvent is not particularly limited as long as it does not hinder the reaction, but water or a mixed solvent of water and a water-soluble solvent such as tert-butanol or acetonitrile is preferable.
- Formula (VII) [wherein W 1 , W 2 , W 3 , W 4 , W 5 , X and Y are as defined above, and PG 1 represents a protecting group.
- the compound of the above formula (VIII) [wherein W 1 , W 2 , W 3 , W 4 , W 5 , X and Y are as defined above, and PG 1 represents a protecting group]. It can obtain by the oxidation reaction of the compound represented. For example, it can be synthesized by allowing osmium tetroxide and sodium periodate to act in a tert-butanol-water mixed solvent.
- osmium tetroxide is usually 0.0001 to 1 mol, preferably 0.01 to 1 mol, and sodium periodate is usually 1 to 10 mol, preferably 1 to 1 mol. Use 5 moles.
- the reaction temperature is usually 0 ° C. to 100 ° C., preferably 0 ° C. to 40 ° C.
- the reaction time is usually 1 hour to 24 hours, preferably 1 hour to 12 hours.
- the reaction solvent is not particularly limited as long as it does not hinder the reaction, but a mixed solvent of water and a water-soluble solvent such as tert-butanol, dioxane or acetone is preferable.
- Formula (VIII) [wherein W 1 , W 2 , W 3 , W 4 , W 5 , X and Y are as defined above, and PG 1 represents a protecting group.
- Compounds of] the above formula (IX) [wherein, W 1, W 2, W 3, W 4 and W 5 are as defined above, PG 1 represents a protecting group, the X L is halogen atom .
- the above formula (X) wherein X and Y are as defined above, and M is boron, tin or the like. ] Can be obtained by a coupling reaction.
- compound (IX) having a halogen atom and (HO) 2 B—X—Y—CH ⁇ CH 2 or ( n-Bu) 3 Sn—X—Y—CH ⁇ CH 2 can be reacted to give compound (VIII).
- compound (X) is generally used in 1 to 10 mol, preferably 1 to 3 mol, per 1 mol of compound (IX).
- Examples of the compound (X) include tributyl vinyl tin and tributyl allyl tin.
- Examples of the base used include sodium carbonate, potassium carbonate, cesium carbonate, cesium fluoride, potassium fluoride, sodium fluoride and lithium chloride.
- the amount of the base to be used is generally 1 to 10 mol, preferably 1 to 3 mol, per 1 mol of compound (IX).
- Examples of the palladium catalyst used include Pd (PPh 3 ) 4 , Pd (OAc) 2 , Pd 2 (dba) 3, and PdCl 2 (PPh 3 ) 2 .
- the amount of the palladium catalyst to be used is usually 0.01 to 0.5 mol, preferably 0.05 to 0.2 mol, per 1 mol of compound (IX).
- Examples of the phosphine ligand used include PPh 3 , P (o-tol) 3 , P (tert-Bu) 3 , 2- [di (tert-butyl) phosphino] -1,1′-biphenyl, 2- [ Di (tert-butyl) phosphino] -2'-dimethylamino-1,1'-biphenyl, 2- [dicyclohexylphosphino] -1,1'-biphenyl, 2- [dicyclohexylphosphino] -2'-dimethyl Amino-1,1′-biphenyl and the like can be mentioned.
- the reaction temperature is usually 0 ° C.
- reaction solvent is not particularly limited as long as it does not hinder the reaction, but solvents such as dimethylformamide, tetrahydrofuran, 1,4-dioxane, acetonitrile, toluene and the like are preferable.
- Scheme 8 Method for producing compound of formula (IX) from compound of formula (XI) Formula (IX) [wherein W 1 , W 2 , W 3 , W 4 and W 5 are as defined above, PG 1 represents a protecting group, and X L represents a halogen atom. ]
- PG 1 represents a protecting group
- X L represents a halogen atom.
- Compound (XI) is a commercially available compound that can be purchased or a compound that is known in the literature.
- the protecting group PG 1 of the above formula (IX) is not particularly limited as long as it has the function, but for example, benzyl group, p-methoxybenzyl group, 3,4-dimethoxybenzyl group, o-nitrobenzyl group.
- Aralkyl groups such as a group, p-nitrobenzyl group, benzhydryl group, trityl group; lower alkanoyl groups such as formyl group, acetyl group, propionyl group, butyryl group, pivaloyl group; such as benzoyl group; Arylalkanoyl groups such as alkenyl groups; lower alkoxycarbonyl groups such as methoxycarbonyl group, ethoxycarbonyl group, propyloxycarbonyl group, tert-butoxycarbonyl group; Aralkyloxycarbonyl groups such as sulfonyl groups; lower alkylsilyl groups such as trimethylsilyl groups and tert-butyldimethylsilyl groups; tetrahydropyranyl groups; trimethylsilylethoxymethyl groups; lower alkyl groups such as methylsulfonyl groups and ethylsulfonyl groups.
- Examples thereof include arylsulfonyl groups such as benzenesulfonyl group and p-toluenesulfonyl group, with tert-butoxycarbonyl group, methylsulfonyl group and p-toluenesulfonyl group being preferred.
- the method for introducing a protecting group varies depending on the type of the protecting group and the stability of the compound. For example, the method described in the literature [Protective Groups in Organic Synthesis, 3rd edition, T.A. W. According to Green (TW Greene, John Wiley & Sons (1999)) or a similar method.
- the halogenating agent is generally used in an amount of 1 to 10 mol, preferably 1 to 3 mol, per 1 mol of compound (XII).
- the halogenating agent include N-chlorosuccinamide and N-bromosuccinamide.
- the reaction temperature is usually 0 ° C. to 100 ° C., preferably 0 ° C. to 25 ° C.
- the reaction solvent is not particularly limited as long as it does not hinder the reaction, but solvents such as tetrahydrofuran, 1,4-dioxane, chloroform and acetonitrile are preferable.
- the compound (XIII) having a halogen atom is reacted with R b —B (OH) 2 or the like in the presence of a base and a palladium catalyst (and further, if necessary, a phosphine ligand).
- (XV) can be obtained.
- R b —B (OH) 2 is usually used in an amount of 1 to 10 mol, preferably 1 to 3 mol, per 1 mol of compound (XIII).
- R b —B (OH) 2 examples include commercially available arylboron derivatives such as phenylboric acid, phenylboric acid ester, dialkylphenylborane, heteroarylboron derivatives, vinylboron derivatives, allylboron derivatives, lower alkylboron derivatives and cyclolower Alkyl boron derivatives can be used. Moreover, you may manufacture the target boron derivative by the well-known method, the method according to this, or the method which combined these and the conventional method. Examples of the base used include sodium carbonate, potassium carbonate, cesium carbonate, cesium fluoride, potassium fluoride and sodium fluoride.
- the amount of the base to be used is generally 1 to 10 mol, preferably 1 to 3 mol, per 1 mol of compound (XIII).
- Examples of the palladium catalyst used include Pd (PPh 3 ) 4 , Pd (OAc) 2 , Pd 2 (dba) 3, and PdCl 2 (PPh 3 ) 2 .
- the amount of the palladium catalyst to be used is generally 0.01 to 0.5 mol, preferably 0.05 to 0.2 mol, per 1 mol of compound (XIII).
- Examples of the phosphine ligand used include PPh 3 , P (o-tol) 3 , P (tert-Bu) 3 , 2- [di (tert-butyl) phosphino] -1,1′-biphenyl, 2- [ Di (tert-butyl) phosphino] -2'-dimethylamino-1,1'-biphenyl, 2- [dicyclohexylphosphino] -1,1'-biphenyl, 2- [dicyclohexylphosphino] -2'-dimethyl Amino-1,1′-biphenyl and the like can be mentioned.
- the reaction temperature is usually 0 ° C.
- reaction solvent is not particularly limited as long as it does not hinder the reaction, but solvents such as dimethylformamide, tetrahydrofuran, 1,4-dioxane, acetonitrile, toluene and the like are preferable.
- the protecting group PG 2 of the above formula (III-1) is not particularly limited as long as it has the function, but for example, a lower group such as a methyl group, an ethyl group, a propyl group, an isopropyl group, a tert-butyl group, etc.
- an aralkyl group such as a group, and a methyl group, an ethyl group, a tert-butyl group, an allyl group, a benzyl group, a p-methoxybenzyl group, a benzhydryl group, and the like are particularly preferable.
- the method for introducing a protecting group varies depending on the type of the protecting group and the stability of the compound. For example, the method described in the literature [Protective Groups in Organic Synthesis, 3rd edition, T.A. W. According to Green (TW Greene, John Wiley & Sons (1999)) or a similar method.
- Formula (XVI) [wherein W 1 , W 2 , W 3 , W 4 and W 5 are as defined above. ] Is a compound of the above formula (XVII) wherein W 1 , W 2 , W 3 , W 4 and W 5 are as defined above, and PG 1 is a protecting group. It can be obtained by hydrolysis of the compound represented by More specifically, it can be obtained by hydrolyzing the cyano group of the compound of the above formula (XVII) and deprotecting the protecting group PG 1 .
- the protecting group PG 1 of the compound represented by the formula (XVII) is not particularly limited as long as it has the function, but for example, a formyl group, an acetyl group, a propionyl group, a butyryl group, a pivaloyl group, etc.
- Lower alkanoyl group for example, benzoyl group;
- arylalkanoyl group such as phenylacetyl group, phenoxyacetyl group;
- lower alkoxycarbonyl group such as methoxycarbonyl group, ethoxycarbonyl group, propyloxycarbonyl group, tert-butoxycarbonyl group;
- Aralkyloxycarbonyl groups such as oxycarbonyl group, p-nitrobenzyloxycarbonyl group and phenethyloxycarbonyl group;
- lower alkylsilyl groups such as trimethylsilyl group and tert-butyldimethylsilyl group;
- Lower alkylsulfonyl groups such as sulfonyl group, ethylsulfonyl group and the like; and arylsulfonyl groups such as benzenesulfonyl group and p-toluenesulfonyl
- the hydrolysis reaction is performed by hydrolysis using an acid or a base, that is, for example, 0.01 mol to a large excess of acid, preferably acetic acid, formic acid, sulfuric acid, phthalic acid, hydrochloric acid, or the like, or 0.01 mol to a large excess. It is carried out by a method in which a base, preferably sodium hydroxide or potassium hydroxide is allowed to act.
- the reaction temperature is usually 0 ° C. to 200 ° C., preferably 0 ° C. to 160 ° C.
- the reaction solvent is not particularly limited as long as it does not hinder the reaction, but methanol, ethanol, 1-propanol, 2-propanol, 1-butanol, 2-butanol, tert-butanol, tetrahydrofuran, 1,4-dioxane, Solvents such as cyclohexane, 1,3-dimethylbenzene and toluene are preferred.
- the formula (XVII) [wherein, W 1, W 2, W 3, W 4 and W 5 are as defined above, PG 1 is a protecting group. ] Is a compound of the above formula (XVIII) wherein W 1 , W 2 , W 3 , W 4 and W 5 are as defined above, PG 1 is a protecting group, and L is a leaving group. . ] It can obtain by cyanation of the compound represented.
- the leaving group L of the above formula (XVIII) is not particularly limited as long as it has the function.
- a halogen atom chlorine atom, bromine atom, etc.
- p-toluenesulfonyloxy group examples include a methanesulfonyloxy group and ethanesulfonyloxy group, and a bromine atom, a chlorine atom, a p-toluenesulfonyloxy group, a methanesulfonyloxy group, and the like are preferable.
- the cyanide is generally used in an amount of 1 mol to 10 mol, preferably 1 mol to 3 mol, per 1 mol of compound (XVIII).
- Examples of the cyanide used include lithium cyanide, sodium cyanide, potassium cyanide, tetraethyl cyanide, trimethylsilyl cyanide and tetrabutyl cyanide.
- the reaction temperature is usually 0 ° C. to 200 ° C., preferably 0 ° C. to 160 ° C.
- the reaction solvent is not particularly limited as long as it does not hinder the reaction, but N, N-dimethylformamide, N, N-dimethylacetamide, dimethyl sulfoxide, methanol, ethanol, 1-propanol, 2-propanol, 1-butanol Solvents such as 2-butanol, tert-butanol, tetrahydrofuran, dimethoxyethane, 1,4-dioxane, cyclohexane, 1,3-dimethylbenzene and toluene are preferred.
- Formula (XVIII) [wherein W 1 , W 2 , W 3 , W 4 and W 5 are as defined above, PG 1 is a protecting group, and L is a leaving group. ]
- the leaving group L of the above formula (XVIII) is not particularly limited as long as it has the function.
- a halogen atom chlorine atom, bromine atom, etc.
- p-toluenesulfonyloxy group p-toluenesulfonyloxy group
- benzenesulfonyloxy group examples include a methanesulfonyloxy group and ethanesulfonyloxy group, and a bromine atom, a chlorine atom, a p-toluenesulfonyloxy group, a methanesulfonyloxy group, and the like are preferable.
- sulfonic acid chloride for example, 1 mol to 10 mol, preferably 1 mol to 3 mol of sulfonic acid chloride is used as a compound that provides a leaving group with respect to 1 mol of compound (XIX).
- sulfonic acid chloride used include methanesulfonyl chloride, ethanesulfonyl chloride, p-toluenesulfonyl chloride, phenylsulfonyl chloride and the like.
- base used include triethylamine and diisopropylethylamine.
- the reaction temperature is usually 0 ° C. to 200 ° C., preferably 0 ° C. to 25 ° C.
- the reaction solvent is not particularly limited as long as it does not hinder the reaction, but a solvent such as chloroform, dichloromethane, tetrahydrofuran, dimethoxyethane, 1,4-dioxane, cyclohexane, 1,3-dimethylbenzene and toluene is preferable.
- the compound of the above formula (XVIII) can also be obtained by an Appel reaction in which a halogenating agent such as carbon tetrabromide or carbon tetrachloride and triphenylphosphine are allowed to act on the compound represented by the above formula (XIX).
- the halogenating agent is generally used in an amount of 1 mol to 10 mol, preferably 1 mol to 3 mol, per 1 mol of compound (XIX).
- the halogenating agent used include carbon tetrabromide, carbon tetrachloride, hexachloroacetone, hexabromoacetone, triphosgene, lithium bromide, methane iodide, bromine and iodine.
- triphenylphosphine is generally used in an amount of 1 mol to 10 mol, preferably 1 mol to 3 mol, per 1 mol of compound (XIX).
- the reaction temperature is usually 0 ° C.
- the reaction solvent is not particularly limited as long as it does not hinder the reaction, but a solvent such as chloroform, dichloromethane, tetrahydrofuran, dimethoxyethane, 1,4-dioxane, cyclohexane, 1,3-dimethylbenzene, acetonitrile and toluene is preferable. .
- Formula (XIX) [wherein W 1 , W 2 , W 3 , W 4 and W 5 are as defined above, and PG 1 is a protecting group. ] Is a compound of the above formula (XX) wherein W 1 , W 2 , W 3 , W 4 and W 5 are as defined above, and PG 1 and PG 2 are protecting groups. It can obtain by reducing the compound represented by this. More specifically, a compound of formula (XIX) can be obtained by reacting a compound of formula (XX) having an ester with a reducing agent such as lithium aluminum hydride.
- the protecting group PG 2 of the above formula (XX) is not particularly limited as long as it has the function.
- a halo lower alkyl group such as 2,2,2-trichloroethyl group; a lower alkenyl group such as an allyl group; a benzyl group, a p-methoxybenzyl group, a p-nitrobenzyl group, a benzhydryl group, a trityl group, etc. And particularly preferred are methyl, ethyl, tert-butyl, allyl, benzyl, p-methoxybenzyl and benzhydryl groups.
- the reducing agent is generally used in an amount of 1 mol to 10 mol, preferably 1 mol to 3 mol, per 1 mol of compound (XX).
- Examples of the reducing agent used include lithium aluminum hydride, diisobutylaluminum hydride, lithium triethylborohydride, sodium bis (2-methoxyethoxy) aluminum hydride and borohydride.
- the reaction temperature is usually ⁇ 100 ° C. to 200 ° C., preferably 0 ° C. to 25 ° C.
- the reaction solvent is not particularly limited as long as it does not hinder the reaction, but diethyl ether, tetrahydrofuran, 1,4-dioxane, dimethoxyethane, cyclohexane, 1,3-dimethylbenzene, toluene, methanol, ethanol, 1-propanol A solvent such as 2-propanol is preferred.
- Formula (XX) [wherein W 1 , W 2 , W 3 , W 4 and W 5 are the same as defined above, and PG 1 and PG 2 are protecting groups. ] Is a compound of the above formula (XXI) wherein W 1 , W 2 , W 3 , W 4 and W 5 are as defined above, and PG 2 is a protecting group. ] Can be obtained by protecting with a protecting group PG 1 .
- the protecting group PG 1 of the above formula (XX) is not particularly limited as long as it has the function, but for example, benzyl group, p-methoxybenzyl group, 3,4-dimethoxybenzyl group, o-nitrobenzyl group.
- Aralkyl groups such as a group, p-nitrobenzyl group, benzhydryl group, trityl group; lower alkanoyl groups such as formyl group, acetyl group, propionyl group, butyryl group, pivaloyl group; such as benzoyl group; Arylalkanoyl groups such as alkenyl groups; lower alkoxycarbonyl groups such as methoxycarbonyl group, ethoxycarbonyl group, propyloxycarbonyl group and tert-butoxycarbonyl group; for example benzyloxycarbonyl group, p-nitrobenzyloxycarbonyl group, phenethyloxy Aralkyloxycarbonyl groups such as sulfonyl groups; lower alkylsilyl groups such as trimethylsilyl groups and tert-butyldimethylsilyl groups; tetrahydropyranyl groups; trimethylsilylethoxymethyl groups; lower alkyl groups
- Examples thereof include arylsulfonyl groups such as benzenesulfonyl group and p-toluenesulfonyl group, with tert-butoxycarbonyl group, methylsulfonyl group and p-toluenesulfonyl group being preferred.
- the method for introducing a protecting group varies depending on the type of the protecting group and the stability of the compound. For example, the method described in the literature [Protective Groups in Organic Synthesis, 3rd edition, T.A. W. According to Green (TW Greene, John Wiley & Sons (1999)) or a similar method.
- Examples of the compound of the formula (XXI) include methyl indole-6-carboxylate, ethyl indole-6-carboxylate, benzyl indole-6-carboxylate, tert-butyl indole-6-carboxylate, 3-methyl- Methyl 1H-indole-6-carboxylate, ethyl 3-methyl-1H-indole-6-carboxylate, methyl 3-ethyl-1H-indole-6-carboxylate, 3-isopropyl-1H-indole-6-carboxylic acid Methyl, 1H-pyrrolo [3,2-b] pyridine-6-carboxylate, methyl indazole-6-carboxylate, ethyl indazole-6-carboxylate, methyl 3-methylindazole-6-carboxylate, etc.
- a commercially available product is used, or a known method or
- the compound represented by the formula (II) is a compound in which the group represented by X is a single bond and the group represented by Y is a group represented by CR Y1 R Y1 ′ This is another synthesis method of the compound of the formula (II-1).
- compound (II-1) is reacted with the compound (XXIII) having a halogen atom in the presence of a base and a palladium catalyst (and optionally a phosphine ligand), to thereby react the compound (II-1). ) Can be obtained.
- compound (XXIII) is generally used in 1 to 10 mol, preferably 1 to 3 mol, per 1 mol of compound (XXII).
- Examples of compound (XXIII) include methyl acetate, ethyl acetate, tert-butyl acetate, methyl propionate, ethyl propionate, tert-butyl propionate and methyl isobutyrate.
- Examples of the base used include lithium dicyclohexylamide, sodium dicyclohexylamide, lithium hexamethyldisilazide, sodium hexamethyldisilazide, potassium hexamethyldisilazide, and lithium diisopropylamide.
- the amount of the base used is generally 1 to 10 mol, preferably 1 to 3 mol, per 1 mol of compound (XXII).
- the palladium catalyst used include Pd (PPh 3 ) 4 , Pd (OAc) 2 , Pd (dba) 2 , Pd 2 (dba) 3 , and PdCl 2 (PPh 3 ) 2 .
- the amount of the palladium catalyst to be used is generally 0.01 to 0.5 mol, preferably 0.05 to 0.2 mol, per 1 mol of compound (XXII).
- the phosphine ligand used include PPh 3 , P (o-tol) 3 , P (tert-Bu) 3 , 2- [di (tert-butyl) phosphino] -1,1′-biphenyl, 2- [ Di (tert-butyl) phosphino] -2'-dimethylamino-1,1'-biphenyl, 1,2,3,4,5-pentaphenyl-1 '-[di (tert-butyl) phosphino] ferrocene, Examples include-[dicyclohexylphosphino] -1,1'-biphenyl, 2- [dicyclohexylphosphino] -2'-dimethylamino-1,1'-biphenyl, and the like
- the reaction temperature is usually 0 ° C. to 80 ° C., preferably 0 ° C. to 25 ° C.
- the reaction solvent is not particularly limited as long as it does not hinder the reaction, but a solvent such as N, N-dimethylformamide, tetrahydrofuran, 1,4-dioxane, cyclohexane, 1,3-dimethylbenzene, acetonitrile or toluene is preferable. .
- Formula (XXII) [wherein R 1 , W 1 , W 2 , W 3 , W 4 and W 5 are as defined above, and X L is a halogen atom.
- Compounds of] the above formula (XXIV) [in the formula, W 1, W 2, W 3, W 4 and W 5 have the same meanings as those defined above, X L is a halogen atom.
- the leaving group L of the above formula (IV) is not particularly limited as long as it can be eliminated by the reaction with the compound (IV) to produce the compound (XXII).
- the amount of compound (IV) to be used is generally 1 to 10 mol, preferably 1 to 3 mol, per 1 mol of compound (XXIV).
- the base used include sodium carbonate, potassium carbonate, cesium carbonate, sodium hydrogen carbonate, cesium fluoride, sodium hydride and potassium hydroxide, and potassium carbonate, sodium hydride, potassium hydroxide and the like are preferable.
- the amount of the base is generally 1 to 10 mol, preferably 1 to 5 mol, per 1 mol of compound (XXIV).
- the reaction temperature is usually 0 ° C. to 160 ° C., preferably 25 ° C. to 100 ° C.
- the reaction time is usually 1 hour to 24 hours, preferably 1 hour to 12 hours.
- the reaction solvent is not particularly limited as long as it does not hinder the reaction, but N, N-dimethylformamide, N, N-dimethylacetamide, N-methyl-2-pyrrolidone, chloroform, dichloromethane, tetrahydrofuran, 1,4- Solvents such as dioxane, acetone, methyl ethyl ketone and acetonitrile are preferred.
- Examples of the compound of the formula (XXIV) include 6-bromoindole, 6-bromo-3-methylindole, 6-bromo-3-ethylindole, 6-bromoindazole, 6-bromo-2,3-dihydro- 1H-indole, 6-bromo-2,3-dihydro-3-methyl-1H-indole-2-one, 6-bromo-2,3-dihydro-3-methyl-1H-indole, 6-bromo-2, 3-dihydro-3,3-dimethyl-1H-indole-2-one, 6-bromo-2,3-dihydro-3,3-dimethyl-1H-indole, 6-bromo-3-methyl-1H-indazole, 6-bromo-3-methyl-1H-indazole, 6-bromo-3-methyl-1H-indazole, 6-bromo-3-methyl-1H-indazole, 6-bromo-3-ethyl-1H-indazo
- Examples of the catalyst used include 5% palladium carbon, 10% palladium carbon, 20% palladium hydroxide, Raney nickel, platinum and platinum oxide.
- the amount of the catalyst used is generally 0.01 to 1 mol, preferably 0.05 to 0.2 mol, per 1 mol of compound (III-3).
- the reaction temperature is usually 0 ° C. to 200 ° C., preferably 25 ° C. to 80 ° C.
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction, but methanol, ethanol, 1-propanol, 2-propanol, ethyl acetate, dimethylformamide, dimethylacetamide, tetrahydrofuran, 1,4-dioxane, toluene and the like. A solvent is preferred.
- compound (III-3) is reacted with the compound (XXIV) having a halogen atom in the presence of a base and a palladium catalyst (and optionally a phosphine ligand), to thereby react the compound (III-3). ) Can be obtained.
- compound (XXV) is generally used in 1 to 10 mol, preferably 1 to 3 mol, per 1 mol of compound (XXIV).
- Examples of the compound (XXV) include methyl acrylate, ethyl acrylate, propyl acrylate, tert-butyl acrylate, methyl 2-butenoate, methyl 3-butenoate, acrylic acid, 2-butenoic acid and 3-butenoic acid.
- Examples of the base used include triethylamine, sodium acetate, potassium acetate, sodium hydrogen carbonate, sodium carbonate, cesium fluoride, potassium fluoride and potassium carbonate.
- the amount of the base to be used is generally 1 to 10 mol, preferably 1 to 3 mol, per 1 mol of compound (XXIV).
- Examples of the palladium catalyst used include Pd (PPh 3 ) 4 , Pd (OAc) 2 , Pd 2 (dba) 3 , and PdCl 2 (PPh 3 ) 2 .
- the amount of the palladium catalyst to be used is generally 0.01 to 0.5 mol, preferably 0.05 to 0.2 mol, per 1 mol of compound (XXIV).
- phosphine ligand As the phosphine ligand used, PPh 3 , P (o-tol) 3 , P (2-furyl) 3 , P (tert-Bu) 3 , 2- [di (tert-butyl) phosphino] -1, 1'-biphenyl, 2- [di (tert-butyl) phosphino] -2'-dimethylamino-1,1'-biphenyl, 2- [dicyclohexylphosphino] -1,1'-biphenyl, 2- [dicyclohexyl, Phosphino] -2'-dimethylamino-1,1'-biphenyl and the like.
- the reaction temperature is usually 0 ° C. to 200 ° C., preferably 25 ° C. to 160 ° C.
- the reaction solvent is not particularly limited as long as it does not hinder the reaction, but a solvent such as N, N-dimethylformamide, N, N-dimethylacetamide, chloroform, dichloromethane, tetrahydrofuran, 1,4-dioxane, acetonitrile, toluene, etc. Is preferred.
- Examples of the compound of the formula (XXIV) include 6-bromoindole, 6-bromo-3-methylindole, 6-bromo-3-ethylindole, 6-bromoindazole, 6-bromo-2,3-dihydro- 1H-indole, 6-bromo-2,3-dihydro-3-methyl-1H-indole-2-one, 6-bromo-2,3-dihydro-3-methyl-1H-indole, 6-bromo-2, 3-dihydro-3,3-dimethyl-1H-indole-2-one, 6-bromo-2,3-dihydro-3,3-dimethyl-1H-indole, 6-bromo-3-methyl-1H-indazole, 6-bromo-3-methyl-1H-indazole, 6-bromo-3-methyl-1H-indazole, 6-bromo-3-methyl-1H-indazole, 6-bromo-3-ethyl-1H-indazo
- Schemes 21 to 23 are methods for synthesizing a compound of the formula (I) in which Z is a 5-tetrazolyl group (formula (I-1)).
- 1 to 10 mol, preferably 1 to 3 mol of azide is generally used with respect to 1 mol of compound (XXVI).
- the azide include alkali metal azides such as lithium azide, sodium azide, and potassium azide, trialkyltin azides such as trioctyltin azide, and hydrazoic acid.
- the salt used include ammonium chloride, zinc chloride, zinc bromide and aluminum chloride.
- the amount of the salt to be used is generally 1 to 10 mol, preferably 1 to 3 mol, per 1 mol of compound (XXVI).
- the reaction temperature is usually 0 ° C. to 200 ° C., preferably 100 ° C. to 170 ° C.
- the reaction solvent is not particularly limited as long as it does not hinder the reaction, but solvents such as dimethylformamide, water, dimethylacetamide, N-methyl-2-pyrrolidone, tetrahydrofuran, 1,4-dioxane and toluene are preferable.
- the dehydrating agent is generally used in an amount of 1 to 10 mol, preferably 1 to 3 mol, per 1 mol of compound (XXVII).
- the dehydrating agent include thionyl chloride, oxalyl chloride, cyanuric chloride, phosphorus pentoxide, phosphorus pentachloride, acetic anhydride and phosphorus oxychloride.
- the reaction temperature is usually 0 ° C. to 200 ° C., preferably 0 ° C. to 25 ° C.
- the reaction solvent is not particularly limited as long as it does not hinder the reaction, but solvents such as dichloromethane, chloroform, tetrahydrofuran, 1,4-dioxane, acetonitrile, toluene and the like are preferable.
- the halogenating agent is generally used in an amount of 1 to 10 mol, preferably 1 to 3 mol, per 1 mol of compound (I).
- the halogenating agent include thionyl chloride, oxalyl chloride, phosphorus trichloride, phosphorus pentachloride and sulfuryl chloride.
- the reaction temperature is usually 0 ° C. to 200 ° C., preferably 0 ° C. to 25 ° C.
- the reaction solvent is not particularly limited as long as it does not hinder the reaction, but solvents such as dichloromethane, chloroform, tetrahydrofuran, 1,3-dimethylbenzene, 1,4-dioxane, toluene and the like are preferable.
- Scheme 24 below is a method for synthesizing a compound represented by formula (XXVI), wherein X is a single bond and Y is a single bond (formula (XXVI-1)).
- Formula (XXVI-1) [wherein R 1 is as defined above, and W 1 , W 2 , W 3 , W 4 and W 5 are as defined above.
- Compounds of] the above formula (XXII) [In the formula, R 1 are as defined above, W 1, W 2, W 3, W 4 and W 5 have the same meanings as those defined above, X L is a halogen atom is there. And a cyanide such as zinc cyanide. More specifically, compound (XXVI-1) is obtained by reacting halogenated compound (XXII) with cyanide in the presence of a palladium catalyst (and optionally a phosphine ligand). Can do.
- 1 to 10 mol, preferably 1 to 3 mol of cyanide is generally used with respect to 1 mol of compound (XXII).
- the cyanide include zinc cyanide, sodium cyanide and potassium cyanide.
- the palladium catalyst used include Pd (PPh 3 ) 4 , Pd (OAc) 2 , Pd (dba) 2, Pd 2 (dba) 3 , and PdCl 2 (PPh 3 ) 2 .
- the amount of the palladium catalyst to be used is generally 0.01 to 0.5 mol, preferably 0.05 to 0.2 mol, per 1 mol of compound (XXII).
- Examples of the phosphine ligand used include PPh 3 , P (o-tol) 3 , P (tert-Bu) 3 , 2- [di (tert-butyl) phosphino] -1,1′-biphenyl, 2- [ Di (tert-butyl) phosphino] -2'-dimethylamino-1,1'-biphenyl, 2- [dicyclohexylphosphino] -1,1'-biphenyl, 2- [dicyclohexylphosphino] -2'-dimethyl Amino-1,1′-biphenyl and the like can be mentioned. If necessary, a reducing agent such as zinc can be added to this reaction.
- the reaction temperature is usually 0 ° C. to 200 ° C., preferably 25 ° C. to 130 ° C.
- the reaction solvent is not particularly limited as long as it does not hinder the reaction, but solvents such as dimethylformamide, tetrahydrofuran, dimethoxyethane, 1,4-dioxane, acetonitrile, and toluene are preferable.
- Scheme 25 is another method for synthesizing a compound of formula (I).
- Compound (I) can be obtained by a method in which 0.01 mol to a large excess of base, preferably potassium hydroxide, sodium hydroxide, calcium hydroxide or the like is allowed to act.
- the reaction temperature is usually 0 ° C. to 200 ° C., preferably 0 ° C. to 160 ° C.
- the reaction solvent is not particularly limited as long as it does not hinder the reaction, but methanol, ethanol, 1-propanol, 2-propanol, 1-butanol, 2-butanol, tert-butanol, tetrahydrofuran, 1,4-dioxane, Solvents such as cyclohexane, 1,3-dimethylbenzene and toluene are preferred.
- the alcohol solvent used for the solvolysis using a base is not particularly limited as long as it does not hinder the reaction, but methanol, ethanol, 1-propanol, 2-propanol, 1-butanol, 2-butanol and tert- Butanol and the like are preferable.
- Scheme 26 is a general method for producing a compound of formula (II) wherein X is a carbonyl group (formula (II-2)).
- Formula (II-2) [wherein R 1 , W 1 , W 2 , W 3 , W 4 , W 5 and Y are as defined above, and PG 2 represents a protecting group.
- the compound of the above formula (XXVIII) [wherein R 1 , W 1 , W 2 , W 3 , W 4 and W 5 are as defined above, and M is boron, tin or the like. It can obtain by the coupling reaction of the compound and acid chloride which are represented. More specifically, compound (II-2) can be obtained by reacting compound (XXVIII) with acid chloride in the presence of a palladium catalyst (and optionally a phosphine ligand and a base). .
- acid chloride is generally used in 1 to 10 mol, preferably 1 to 3 mol, per 1 mol of compound (XXVIII).
- the acid chloride include succinic acid monomethyl chloride, succinic acid monoethyl chloride, and ethyl chloroglyoxylate.
- PG 2 in the formula (II-2) is a group derived from acid chloride, and examples thereof include a methyl group and an ethyl group.
- the base used include triethylamine and diisopropylethylamine.
- the amount of the base to be used is generally 1 to 10 mol, preferably 1 to 3 mol, per 1 mol of compound (XXVIII).
- Examples of the palladium catalyst used include Pd (PPh 3 ) 4 , Pd (OAc) 2 , Pd (dba) 2, Pd 2 (dba) 3 , and PdCl 2 (PPh 3 ) 2 .
- the amount of the palladium catalyst to be used is generally 0.01 to 0.5 mol, preferably 0.05 to 0.2 mol, per 1 mol of compound (XXVIII).
- Examples of the phosphine ligand used include PPh 3 , P (o-tol) 3 , P (tert-Bu) 3 , 2- [di (tert-butyl) phosphino] -1,1′-biphenyl, 2- [ Di (tert-butyl) phosphino] -2'-dimethylamino-1,1'-biphenyl, 2- [dicyclohexylphosphino] -1,1'-biphenyl, 2- [dicyclohexylphosphino] -2'-dimethyl Amino-1,1′-biphenyl and the like can be mentioned.
- the reaction temperature is usually 0 ° C.
- reaction solvent is not particularly limited as long as it does not hinder the reaction, but solvents such as dimethylformamide, tetrahydrofuran, 1,4-dioxane, acetonitrile, toluene and the like are preferable.
- compound (XXVIII) is obtained by reacting compound (XXII) having a halogen atom with compound (XXIX) in the presence of a palladium catalyst (further necessary, a phosphine ligand and a base). Obtainable.
- compound (XXIX) is generally used in 1 to 10 mol, preferably 1 to 3 mol, per 1 mol of compound (XXII).
- Examples of compound (XXIX) include bis (trimethyltin), bis (triethyltin), bis (tributyltin), and bispinacolatodiboron.
- the base used include potassium acetate and trimethylamine.
- the amount of the base used is generally 1 to 10 mol, preferably 1 to 3 mol, per 1 mol of compound (XXII).
- Examples of the palladium catalyst used include Pd (PPh 3 ) 4 , Pd (OAc) 2 , Pd 2 (dba) 3 , and PdCl 2 (PPh 3 ) 2 .
- the amount of the palladium catalyst to be used is generally 0.01 to 0.5 mol, preferably 0.05 to 0.2 mol, per 1 mol of compound (XXII).
- Examples of the phosphine ligand used include PPh 3 , P (o-tol) 3 , P (tert-Bu) 3 , 2- [di (tert-butyl) phosphino] -1,1′-biphenyl, 2- [ Di (tert-butyl) phosphino] -2'-dimethylamino-1,1'-biphenyl, 2- [dicyclohexylphosphino] -1,1'-biphenyl, 2- [dicyclohexylphosphino] -2'-dimethyl Amino-1,1′-biphenyl and the like can be mentioned.
- the reaction temperature is usually 0 ° C.
- reaction solvent is not particularly limited as long as it does not hinder the reaction, but solvents such as dimethylformamide, tetrahydrofuran, dimethoxyethane, 1,4-dioxane, acetonitrile, and toluene are preferable.
- the following scheme 28 is represented by the formula (II) where X is a single bond and Y is —CF 2 — (CH 2 ) p — (wherein p is an integer of 0 to 5).
- the fluorinating agent is generally used in an amount of 1 to 10 mol, preferably 1 to 3 mol, per 1 mol of compound (II-2).
- Fluorinating agents include diethylaminosulfur trifluoride (DAST), bis (2-methoxyethyl) aminosulfur trifluoride, 1,1,2,2-tetrafluoroethyl-N, N-dimethylamine, diethylaminodifluorosulfinium Examples thereof include tetrafluoroborate, morpholinodifluorosulfinium tetrafluoroborate, 4-tert-butyl-2,6-dimethylphenylsulfur trifluoride and the like.
- the reaction temperature is usually 0 ° C. to 200 ° C., preferably 25 ° C. to 130 ° C.
- the reaction solvent is not particularly limited as long as it does not hinder the reaction, but solvents such as dichloromethane, tetrahydrofuran, 1,4-dioxane, 1,2-dimethoxyethane, acetonitrile, toluene and the like are preferable.
- the URAT1 inhibitor of the present invention the blood uric acid level-lowering agent, and the pharmaceutical composition for treating or preventing a pathological condition involving blood uric acid are described.
- URAT1 represents uric acid transporter 1 (Uric acid transporter 1).
- inhibiting URAT1 means to inhibit the function of URAT1 as a uric acid transporter to eliminate or attenuate its activity. For example, based on the conditions of Example 187 described below, It means specifically inhibiting the function of URAT1.
- a “URAT1 inhibitor” includes a compound of the formula (I) (including a pharmaceutically acceptable salt or ester form of the compound), and functions as a uric acid transporter of URAT1. It means a drug that inhibits the activity and loses or attenuates its activity.
- a “blood uric acid level-lowering agent” includes a compound of the formula (I) (including a pharmaceutically acceptable salt or ester form of the compound) and inhibits URAT1 Means a drug that lowers the blood uric acid level.
- reducing blood uric acid levels refers to uric acid (including urate) in blood (including serum or plasma) by inhibiting the function of URAT1 as a uric acid transporter.
- high blood uric acid level means that the serum uric acid level is 6 mg / dL or higher, preferably 7 mg / dL or higher, more preferably 8 mg / dL or higher.
- pharmaceutical composition for treating or preventing a pathological condition involving blood uric acid refers to a compound of formula (I) (in the form of a pharmaceutically acceptable salt or ester of the compound) And a therapeutic composition for treating or preventing a pathological condition involving blood uric acid by inhibiting URAT1.
- ⁇ pathological condition involving blood uric acid '' refers to a pathological condition involving the above-mentioned ⁇ high blood uric acid level '', such as hyperuricemia, gouty nodule, acute draft arthritis, chronic draft arthritis, Examples include gout kidney, urinary calculus, renal dysfunction, coronary artery disease, and ischemic heart disease.
- a pharmaceutical composition for treating or preventing a URAT1 inhibitor, a blood uric acid level-lowering agent, and a disease state involving blood uric acid can be provided as a preparation.
- “Formulation” includes oral and parenteral preparations. Examples of oral preparations include tablets, capsules, powders and granules, while parenteral preparations include sterilized liquid preparations such as solutions or suspensions, specifically injections. Intravenous injections and intravenous infusions are preferable.
- the “formulation” according to the present invention may usually contain a therapeutically effective amount of the compound according to the present invention together with a pharmaceutically acceptable carrier or diluent.
- This formulation technique is considered common knowledge and well known to those skilled in the art.
- it can be formulated for oral formulation, intravenous infusion or injection in a number of ways well known to those skilled in the art, together with pharmaceutically acceptable carriers or diluents.
- “Pharmaceutically acceptable carrier or diluent” includes excipients (eg, fats, beeswax, semi-solid and liquid polyols, natural or hardened oils, etc.); water (eg, distilled water, especially injectable distillation) Water, etc.), physiological saline, alcohol (eg, ethanol), glycerol, polyol, glucose aqueous solution, mannitol, vegetable oil, etc .; additives (eg, bulking agents, disintegrants, binders, lubricants, wetting agents, stable Agents, emulsifiers, dispersants, preservatives, sweeteners, colorants, seasonings or fragrances, thickeners, diluents, buffer substances, solvents or solubilizers, drugs to achieve storage effects, osmotic pressure Salt for changing, coating agent, or antioxidant).
- excipients eg, fats, beeswax, semi-solid and liquid polyols, natural
- the preparation according to the present invention can be selected from various forms.
- oral preparations such as tablets, capsules, powders, granules or liquids
- sterilized liquid parenteral preparations such as solutions or suspensions, suppositories, ointments and the like can be mentioned.
- the preparation according to the present invention may be a solid preparation or a liquid preparation.
- the solid preparation can be produced as it is in the form of a tablet, capsule, granule or powder, but can also be produced using an appropriate carrier (additive).
- carriers include sugars such as lactose or glucose; starches such as corn, wheat or rice; fatty acids such as stearic acid; inorganic salts such as magnesium aluminate metasilicate or anhydrous calcium phosphate
- a synthetic polymer such as polyvinyl pyrrolidone or polyalkylene glycol; a fatty acid salt such as calcium stearate or magnesium stearate; an alcohol such as stearyl alcohol or benzyl alcohol; a methyl cellulose, carboxymethyl cellulose, ethyl cellulose or hydroxypropyl methyl cellulose; Synthetic cellulose derivatives; and other commonly used additives such as gelatin, talc, vegetable oil, gum arabic and the like.
- solid preparations such as tablets, capsules, granules, and powders are generally based on the weight of the whole preparation, for example, 0.1 to 100% by weight of the compound represented by the above formula (I), preferably May contain 5 to 98% by mass as an active ingredient.
- the liquid preparation uses appropriate additives usually used in liquid preparations such as water, alcohols, and plant-derived oils such as soybean oil, peanut oil, sesame oil, and the like, suspensions, syrups, injections, It is manufactured in the form of an infusion (intravenous infusion).
- appropriate additives usually used in liquid preparations such as water, alcohols, and plant-derived oils such as soybean oil, peanut oil, sesame oil, and the like, suspensions, syrups, injections, It is manufactured in the form of an infusion (intravenous infusion).
- suitable solvents or diluents for parenteral administration in the form of intramuscular injection, intravenous injection or subcutaneous injection include, for example, distilled water for injection, aqueous lidocaine hydrochloride (for intramuscular injection), and physiological saline.
- aqueous lidocaine hydrochloride for intramuscular injection
- physiological saline for example, physiological saline.
- injections may take a form in which the active ingredient is dissolved in advance, or the active ingredient in powder form or with an appropriate carrier (additive) added.
- These injection solutions can contain, for example, 0.1 to 10% by mass of an active ingredient based on the mass of the entire preparation.
- suspensions for oral administration and liquids such as syrups can contain, for example, 0.1 to 10% by mass of an active ingredient based on the mass of the whole preparation.
- the present compound, the URAT1 inhibitor of the present invention, the blood uric acid level-lowering agent and the pharmaceutical composition for treating or preventing the pathological condition involving uric acid are other pharmaceutical compositions or drugs (hereinafter also referred to as concomitant drugs). Can be used together.
- Combination means that a plurality of medicines are used in combination as active ingredients.
- use as a combination agent use as a kit, use in combination characterized by being separately administered by the same or different administration routes, and the like.
- the administration time of the compound of the present invention, the URAT1 inhibitor of the present invention, the blood uric acid level-lowering agent and the pharmaceutical composition for treating or preventing the pathological condition involving blood uric acid and the concomitant drug is not limited. It may be administered to the administration subject at the same time or may be administered with a time difference.
- the dose of the concomitant drug may be in accordance with the clinically used dose, and may be appropriately selected depending on the administration subject, age and weight of the administration subject, symptoms, administration time, dosage form, administration method, combination, etc. it can.
- the administration form of the concomitant drug is not particularly limited, and the pharmaceutical composition for treating a pathological condition involving the URAT1 inhibitor, blood uric acid level reducing agent or blood uric acid of the present invention is combined with the concomitant drug at the time of administration. It only has to be.
- concomitant drug examples include “therapeutic and / or prophylactic agent for hyperuricemia”, “therapeutic and / or prophylactic agent for gout arthritis”, “therapeutic and / or prophylactic agent for gout kidney”, “urine” Remedy and / or preventive for calculus ",” therapeutic and / or preventive for hypertension or hypertensive complications ",” therapeutic and / or preventive for hyperlipidemia or hyperlipidemic complications ", “Therapeutic and / or preventive agent for diabetes or diabetic complications”, “Therapeutic and / or preventive agent for obesity or obesity complications”, “Therapeutic agent for primary disease causing secondary hyperuricemia and And / or a prophylactic agent ”,“ a therapeutic and / or prophylactic agent for renal failure, cardiovascular disorder, and cerebrovascular disorder caused by hyperuricemia ”, and“ nucleic acid metabolism antagonist ”.
- These 1 to 3 combination drugs can be used in combination with the URAT1 inhibitor of the present invention, a blood uric acid level-lowering agent,
- Examples of the “remedy and / or preventive for hyperuricemia” include uric acid production inhibitors such as xanthine oxidase inhibitors, uric acid excretion promoters and the like. Specifically, allopurinol, probenecid, bucolome, febuxostat, FYX-051 (4- (5-pyridin-4-yl-1H- [1,2,4] triazol-3-yl) pyridine-2-carbonitrile ), Benzbromarone, oxypurinol and the like.
- Examples of the “therapeutic and / or prophylactic agent for gout arthritis” include non-steroidal anti-inflammatory drugs such as indomethacin, naproxen, fenbufen, pranoprofen, oxaprozin, colchicine, and corticosteroids.
- non-steroidal anti-inflammatory drugs such as indomethacin, naproxen, fenbufen, pranoprofen, oxaprozin, colchicine, and corticosteroids.
- uric acid production inhibitors such as xanthine oxidase inhibitors, uric acid excretion promoters, citric acid preparations, urine alkalizing agents such as sodium bicarbonate, and the like.
- uric acid production inhibitors such as xanthine oxidase inhibitors, uric acid excretion promoters, citric acid preparations, urine alkalizing agents such as sodium bicarbonate, and the like.
- FYX-051 (4- (5-pyridin-4-yl-1H- [1,2,4] triazol-3-yl) pyridine-2-carbonitrile )
- Benzbromarone oxypurinol and the like.
- Examples of the “therapeutic agent and / or preventive agent for urinary calculus” include citrate preparations and urine alkalizing agents such as sodium bicarbonate.
- therapeutic and / or preventive agent for hypertension or hypertension complications include, for example, loop diuretics, angiotensin converting enzyme inhibitors, angiotensin II receptor antagonists, Ca antagonists, ⁇ blockers, ⁇ , ⁇ blockers And alpha blockers.
- furosemide sustained release agent captopril, captopril sustained release agent, enalapril maleate, alacepril, delapril hydrochloride, cilazapril, lisinopril, benazepril hydrochloride, imidapril hydrochloride, temocapril hydrochloride, quinapril hydrochloride, trandolapril, perindopril Erbumin, losartan potassium, candesartan cilexetil, nicardipine hydrochloride, nicardipine hydrochloride sustained release agent, nilvadipine, nifedipine, nifedipine hydrochloride sustained release agent, benidipine hydrochloride, diltiazem hydrochloride, diltiazem hydrochloride sustained release agent, nisoldipine, nitrendipine, manidipine hydrochlor
- Examples of “therapeutic and / or prophylactic agents for hyperlipidemia or hyperlipidemia complications” include, for example, HMG-CoA reductase inhibitors, anion exchange resins, probucol, nicotinic acid preparations, fibrates, Examples include eicosapentaenoic acid preparations. More specifically, for example, lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, cerivastatin, colestimide, cholestyramine, niceritrol, nicomol, fenofibrate, bezafibrate, clinofibrate, clofibrate, ethyl icosapentate, etc. .
- Examples of “therapeutic and / or preventive agent for diabetes or diabetic complications” include, for example, insulin preparations, sulfonylurea drugs, insulin secretagogues, sulfonamide drugs, biguanide drugs, ⁇ -glucosidase inhibitors, insulin resistance improving drugs, Examples include dipeptidyl peptidase IV inhibitors, angiotensin converting enzyme inhibitors, aldose reductase inhibitors, and antiarrhythmic agents.
- insulin More specifically, for example, insulin, chlorpropamide, glibenclamide, glipizide, tolbutamide, glyclopyramide, acetohexamide, glimepiride, tolazamide, gliclazide, nateglinide, glybsol, metformin hydrochloride, buformin hydrochloride, voglibose, acarbose, pioglitazone hydrochloride , Sitagliptin phosphate, vildagliptin, alogliptin benzoate mexiletine, epalrestat and the like.
- Examples of the “therapeutic and / or preventive agent for obesity or obesity complications” include, for example, mazindol, acarbose, voglibose and epalestat.
- therapeutic and / or prophylactic agent for primary disease causing hypouria excretion type secondary hyperuricemia include, for example, chronic kidney disease, polycystic kidney disease, pregnancy toxemia, lead nephropathy, hyperlactic acid blood Symptom, Down's syndrome, sarcoidosis, type I glycogenosis (via hyperlactic acidemia), and therapeutic or preventive agents such as dehydration.
- Examples of “therapeutic and / or preventive agent for renal failure, cardiovascular disorder, and cerebrovascular disorder caused by hyperuricemia” include, for example, loop diuretic (eg, furosemide), citric acid preparation, sodium bicarbonate, cation Exchange resin, aluminum hydroxide, alphacalcidol, ⁇ -blocker (eg, propranolol hydrochloride), angiotensin converting enzyme inhibitor (eg, captopril), cardiotonic agent (eg, digoxin), angina pectoris (eg, isosorbide nitrate) ), Ca antagonist (eg, diltiazem hydrochloride), uric acid production inhibitor (eg, allopurinol), amino acid preparation, hyperammonemia ameliorating agent, antiarrhythmic agent (eg, mexiletine), anemia agent (eg, mepithiostan, Erythropoietin), and other "therapeutic agents for hypertension or hypertensive complications and / or "Pre
- nucleic acid metabolism antagonist examples include azathioprine, mizoribine, mycophenolic acid and the like.
- the compound of the present invention, the URAT1 inhibitor of the present invention, the blood uric acid level-lowering agent, and the pharmaceutical composition for treating or preventing a pathological condition involving uric acid are all drugs that increase the blood uric acid level. It can also be used in combination to lower the blood uric acid level.
- Examples of “drugs that increase blood uric acid levels” include nucleic acid metabolism antagonists, antihypertensive diuretics (eg furosemide, thiazide diuretics), antituberculosis drugs (eg pyrazinamide, ethambutol), anti-inflammatory analgesics (eg salicylic acid), high Examples include lipemia drugs (for example, nicotinic acid), asthma drugs (for example, theophylline), immunosuppressants (for example, cyclosporine), hepatitis C drugs (for example, ribavirin), ethanol, and the like.
- nucleic acid metabolism antagonists include nucleic acid metabolism antagonists, antihypertensive diuretics (eg furosemide, thiazide diuretics), antituberculosis drugs (eg pyrazinamide, ethambutol), anti-inflammatory analgesics (eg salicylic acid), high Examples include lipemia drugs (for example,
- CombiPrep Pro C18 manufactured by YMC was used for the column, and 0.1% trifluoroacetic acid aqueous solution and 0.1% trifluoroacetic acid acetonitrile solution were used for the mobile phase.
- step (4) The compound [4-4] (2.4 g) obtained in step (4) was dissolved in ethanol (12 mL), 5N-aqueous sodium hydroxide solution (12 mL) was added at room temperature, and the mixture was heated to reflux for 6 hours. The reaction mixture was returned to room temperature, ethanol was concentrated under reduced pressure, and the obtained aqueous phase was washed with diethyl ether. Thereafter, concentrated hydrochloric acid (5 mL) was added dropwise at 0 ° C. to make it acidic, and the resulting precipitate was collected by filtration.
- step (2) To a solution of compound [52-2] (110 mg) obtained in step (2) in ethanol (3 mL) was added 3N aqueous sodium hydroxide solution (3 mL) at room temperature, and the mixture was heated to reflux for 15 hours. The reaction mixture was acidified with 1N hydrochloric acid, and the precipitated solid was collected by filtration to give the titled compound (81 mg) as a white solid.
- the obtained residue was dissolved in N, N-dimethylformamide (10 mL), potassium carbonate (421 mg) and methyl iodide (0.5 mL) were added at room temperature, and the mixture was stirred at room temperature for 10 min. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was dried over anhydrous sodium sulfate and filtered, and the filtrate was concentrated under reduced pressure. To a solution of the obtained residue in methanol (3 mL) was added 5% palladium on carbon (184 mg), and the mixture was stirred at room temperature for 1 hour in a hydrogen atmosphere.
- Lithium aluminum hydride (126 mg) was added at 0 ° C. to a tetrahydrofuran (10 mL) solution of the compound [61-1] (265 mg) obtained in the step (1) of Example 61, followed by stirring at room temperature for 10 minutes. Water was added to the reaction mixture, 1N-hydrochloric acid was added, and the mixture was extracted with chloroform. The obtained organic layer was dried over anhydrous sodium sulfate and filtered, and the filtrate was concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography to give the titled compound (179 mg) as a white solid.
- the obtained residue was dissolved in tetrahydrofuran (20 mL), a solution of lithium aluminum hydride (518 mg) in tetrahydrofuran (20 mL) was added at 0 ° C., and the mixture was stirred for 10 min. Water was added to the reaction mixture, 1N-hydrochloric acid was added, and the mixture was extracted with chloroform. The obtained organic layer was dried over anhydrous sodium sulfate and filtered, and the filtrate was concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography to give the titled compound (1.6 g) as a white solid.
- the obtained residue was dissolved in dimethyl sulfoxide (20 mL), sodium cyanide (516 mg) was added at room temperature, and the mixture was stirred at room temperature for 20 hr. Water was added to the reaction mixture, and the mixture was extracted with chloroform. The obtained organic layer was dried over anhydrous sodium sulfate and filtered, and the filtrate was concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography to give the titled compound (1.3 g) as a white solid.
- 1,4-Dioxane (3 mL) and water (1.5 mL) were added to the resulting residue, and the mixture was stirred at 100 ° C. for 18 hours.
- the reaction mixture was concentrated under reduced pressure, water was added, and the mixture was extracted with ethyl acetate.
- the obtained organic layer was dried over anhydrous sodium sulfate and filtered, and the filtrate was concentrated under reduced pressure.
- the obtained residue was purified by silica gel column chromatography to give the titled compound (589 mg) as a brown oil.
- step (2) The compound [69-2] (589 mg) obtained in step (2) was dissolved in acetic acid (5 mL), iron powder (691 mg) was added, and the mixture was stirred at 100 ° C. for 8 hr. The reaction mixture was returned to room temperature, insolubles were filtered, and the filtrate was concentrated under reduced pressure. Water was added to the residue and extracted with ethyl acetate. The obtained organic layer was dried over anhydrous sodium sulfate and filtered, and the filtrate was concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography to give the titled compound (303 mg) as a brown solid.
- benzimidazole compound (105 mg) was dissolved in N-methyl-2-pyrrolidone (2.3 mL), potassium carbonate (227 mg) and 2,6-dimethylbenzyl chloride (139 mg) were added, and then microwave reaction was performed. It was made to react at 160 degreeC for 30 minutes with an apparatus. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography to give 118 mg (79%) of a mixture of the title compounds [73-2A] and [73-2B] as a yellow solid. ESI-MSfound: 363 [M + H] +
- step (2) A mixture (118 mg) of the compounds [73-2A] and [73-2B] obtained in step (2) was dissolved in tetrahydrofuran (3.3 mL), 1N-sodium hydroxide (1.7 mL) was added, and The mixture was stirred at 120 ° C. for 20 minutes using a microwave reactor. 1N-hydrochloric acid was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The obtained residue was purified by COOH column chromatography to give the titled compound (13 mg) as a pale yellow solid.
- the obtained crude carboxylic acid compound (3.8 g) was dissolved in N, N-dimethylformamide (70 mL), sodium hydrogen carbonate (2.3 g) and methyl iodide (4.2 mL) were added, and then 60 ° C. For 2 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain a crude ester as a light brown oil.
- the obtained crude ester was dissolved in chloroform (80 mL), trifluoroacetic acid (80 mL) was added at room temperature, and the mixture was stirred at room temperature for 2 hours.
- the solvent was concentrated under reduced pressure, an aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate.
- the obtained organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure.
- the obtained residue was purified by silica gel column chromatography to give the titled compound [74-3A] (0.8 g) as a brown solid.
- the title compound [74-3B] (0.8 g) was obtained as a brown solid.
- step (3) The compound [74-3A] (30 mg) obtained in step (3) was dissolved in N, N-dimethylformamide (1.6 mL), cesium carbonate (163 mg) and benzyl bromide (37 mL) were added at room temperature, The mixture was stirred at 90 ° C. for 30 minutes in a microwave reactor. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography to give the titled compound (30 mg) as a white solid.
- step (4) The compound [74-4] (30 mg) obtained in step (4) was dissolved in tetrahydrofuran (1 mL), 1N-sodium hydroxide (0.5 mL) was added, and the mixture was stirred at room temperature for 24 hours. 1N-hydrochloric acid was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The obtained residue was dissolved in methanol, 1M potassium hydroxide-methanol solution (92.4 ⁇ L) was added, and the solvent was concentrated under reduced pressure.
- the obtained organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure.
- the residue was dissolved in ethanol (75 mL) and tin (11.5 g) was added at room temperature. Concentrated hydrochloric acid (45 mL) was added at 0 ° C., and the mixture was heated to reflux for 2 hours. The reaction mixture was cooled to room temperature, water was added, and the mixture was extracted with ethyl acetate.
- the obtained organic layer was washed with saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure.
- step (1) The compound [78-1] (4.9 g) obtained in step (1) was suspended in toluene (22 mL), borane / dimethyl sulfide complex (4.4 mL) was added at 0 ° C., and the mixture was heated to reflux for 2 hours. .
- the reaction mixture was ice-cooled, 5N-aqueous sodium hydroxide solution (8 mL), 8N-aqueous sodium hydroxide solution (8 mL) and ethyl acetate (8 mL) were sequentially added, and the mixture was stirred at room temperature for 2.5 hr. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate.
- Example 78 Compound [78] (26 mg) obtained in Example 78 was added to an optically active column (Daicel Chemical Industries, Ltd. Chiralcel (registered trademark) OD (CHRALCEL OD), 2 cm ⁇ 25 cm); 0.1% trifluoroacetic acid, hexane / isopropyl alcohol.
- an optically active column (Daicel Chemical Industries, Ltd. Chiralcel (registered trademark) OD (CHRALCEL OD), 2 cm ⁇ 25 cm); 0.1% trifluoroacetic acid, hexane / isopropyl alcohol.
- the obtained oil was dissolved in tetrahydrofuran (2 mL) and methanol (2 mL), 3N-sodium hydroxide (1 mL) was added at room temperature, and the mixture was reacted at 160 ° C. for 10 minutes using a microwave reactor. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was dried over anhydrous sodium sulfate and filtered, and the filtrate was concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography to give the titled compound (194 mg) as a white solid.
- Example 82 the title compound (38 mg) was obtained as a white solid from the compound [64-3] (50 mg) obtained in Step (3) of Example 64 and 2-chloro-6-fluorobenzyl chloride (72 ⁇ L).
- 1 H-NMR 400 MHz, CD 3 OD
- step (2) The compound [89-2] (99 mg) obtained in step (2) was suspended in methanol (2 mL) and ethyl acetate (2 mL), 5% palladium carbon (38 mg) was added at room temperature, For 3 hours. After palladium carbon was filtered off, the filtrate was concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography to give the titled compound (101 mg) as a colorless oil.
- step (3) The compound [89-3] (94.5 mg) obtained in step (3) is dissolved in methanol (2 mL), 1N-aqueous sodium hydroxide solution (1 mL) is added, and then 160 ° C. using a microwave reactor. For 10 minutes. 2N-hydrochloric acid was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was dried over anhydrous sodium sulfate and filtered, and the filtrate was concentrated under reduced pressure. The obtained residue was dissolved in N, N-dimethylformamide (1 mL), potassium carbonate (48 mg) and methyl iodide (18 ⁇ L) were added at room temperature, and the mixture was stirred at room temperature for 2 days.
- Methyl indole-6-carboxylate (497 mg) was dissolved in dichloromethane (15 mL), aluminum chloride (831 mg) and 2-bromopropane (0.27 mL) were added at room temperature, and the mixture was stirred at room temperature for 1 hr. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was dried over anhydrous sodium sulfate and filtered, and the filtrate was concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography to give the titled compound (65 mg) as a white solid.
- N-chlorosuccinimide (1.2 g) was added to a solution of 6-indolecarbonitrile (1.0 g) in methanol (10 mL) at room temperature, and the mixture was stirred at room temperature for 16 hours. Saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was dried over anhydrous sodium sulfate and filtered, and the filtrate was concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography to give the titled compound (1.3 g) as a white solid.
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Urology & Nephrology (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pain & Pain Management (AREA)
- Vascular Medicine (AREA)
- Immunology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Indole Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description
尿酸ナトリウムの生理的pH条件での溶解度は6.4mg/dLであるため(非特許文献1)、体液中で溶解度を超える血中尿酸値である、7mg/dL以上が高尿酸血症と定義される(非特許文献2)。
高尿酸血症が持続すると、体液中に尿酸塩結晶が析出、沈着し、通風関節炎、痛風腎、痛風結節、尿路結石、腎機能障害などを引き起こす(非特許文献3)。
また近年、高尿酸血症は高血圧、高脂血症、耐糖能異常、肥満などの生活習慣病と高率に合併することが知られ(非特許文献4、5、6、7)、こうした合併症が、心血管障害、脳血管障害の発症率を高くすることが知られている。
さらに、URAT1遺伝子の変異による機能低下が、腎性低尿酸血症をおこすことが明らかとなり、URAT1の尿酸排泄に対する重要性が明らかにされている(非特許文献11、12)。
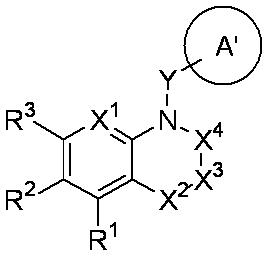




したがって、優れたURAT1阻害作用を有する新規化合物や、血中尿酸が関与する疾患の治療薬や予防薬の提供が課題となっていた。
即ち、本発明は、
式(I):

R1は、シクロアルキル基で置換されてもよい低級アルキル基、シクロアルキル基、ハロ低級アルキル基、ヒドロキシ低級アルキル基、低級アルコキシ低級アルキル基、低級アルコキシカルボニル基、低級アルキルスルホニル基又は一般式:-Q1-A1で表される基を表し;
Q1は、単結合又は低級アルキレン基を表し(ここで、該低級アルキレン基を構成する1若しくは2以上のメチレン基は、それぞれ独立して、カルボニル基、スルフィニル基若しくはスルホニル基でメチレン基全体が置き換えられていてもよく、及び/又は、メチレン基を構成する水素が低級アルキル基で置換されていてもよい。);
A1は、後記の<置換基群L>から選択される1乃至3個の置換基で置換されていてもよい、アリール基又はヘテロアリール基を表し(ここで、当該アリール基又はヘテロアリール基上のお互いに隣接する任意の二つの置換基が一緒になって低級アルキレンジオキシ基を形成してもよい。);
- - - - は、二重結合又は単結合を表し;
- - - - が二重結合である場合、
W1は、窒素原子又は一般式:=C(Ra)-で表される基を表し、かつ
W2は、窒素原子又は一般式:=C(Rb)-で表される基を表し;
- - - - が単結合である場合、
W1は、一般式:-C(Raa)(Rab)-で表される基又は一般式:-(C=O)-で表される基を表し、かつ
W2は、一般式:-C(Rba)(Rbb)-で表される基、一般式:-(C=O)-で表される基又は一般式:-N(Rbc)-で表される基を表し;
Ra及びRbは、それぞれ独立して、水素原子、後記の<置換基群M>から選択される置換基又は一般式:-Q2-A2で表される基を表し;
Raa及びRabは、それぞれ独立して、水素原子、後記の<置換基群N>から選択される置換基又は一般式:-Q2-A2で表される基を表すか、或いは、Raa及びRabが一緒になって低級アルキレン基を形成してもよく(ここで、該低級アルキレン基を構成する1乃至2以上のメチレン基は、それぞれ独立して、酸素原子、カルボニル基、ビニレン基若しくは一般式:-N(Rc)-で表される基でメチレン基全体が置き換えられていてもよく、及び/又は、メチレン基を構成する水素が水酸基、低級アルキル基若しくはハロゲン原子で置換されていてもよい。);
Rba及びRbbは、それぞれ独立して、水素原子、ハロゲン原子、アミノ基、低級アルキルアミノ基、ジ低級アルキルアミノ基、ヒドロキシ低級アルキルアミノ基、低級アルキルスルホニルアミノ基、低級アルコキシカルボニルアミノ基、<置換基群N>から選択される置換基又は一般式:-Q2-A2で表される基を表すか、或いは、Rba及びRbbが一緒になって低級アルキレン基を形成してもよく(ここで、該低級アルキレン基を構成する1乃至2以上のメチレン基は、それぞれ独立して、酸素原子、カルボニル基、ビニレン基若しくは一般式:-N(Rc)-で表される基でメチレン基全体が置き換えられていてもよく、及び/又は、メチレン基を構成する水素が水酸基、低級アルキル基若しくはハロゲン原子で置換されていてもよい。);
Rbcは、水素原子、低級アルキル基、シクロアルキル基、ハロ低級アルキル基、低級アルコキシカルボニル基、カルバモイル基、モノ低級アルキルカルバモイル基、ジ低級アルキルカルバモイル基及び低級アルカノイル基からなる群から選択される基又は一般式:-Q2-A2で表される基を表し;
Q2は、単結合、低級アルキレン基又は低級アルケニレン基を表し(ここで、該低級アルキレン基を構成する1若しくは2以上のメチレン基は、それぞれ独立して、酸素原子、窒素原子若しくはカルボニル基でメチレン基全体が置き換えられていてもよく、及び/又は、メチレン基を構成する水素がハロゲン原子、シアノ基、水酸基若しくは低級アルキル基で置換されていてもよい。);
A2は、<置換基群L>から選択される1乃至3個の置換基で置換されていてもよい、シクロアルキル基、脂肪族複素環基、アリール基又はヘテロアリール基を表し(ここで、当該アリール基又はヘテロアリール基上のお互いに隣接する任意の二つの置換基が一緒になって低級アルキレンジオキシ基を形成してもよい。);
W3、W4及びW5は、それぞれ独立して、窒素原子、又は、ハロゲン原子、水酸基、シアノ基、低級アルキル基、シクロアルキル基、ハロ低級アルキル基、低級アルコキシ基及びハロ低級アルコキシ基からなる群から選択される置換基を有してもよいメチン基を表し;但し、W1、W2、W3、W4及びW5のうち0個乃至4個が窒素原子であり;
Xは、単結合、酸素原子、硫黄原子、スルフィニル基、スルホニル基、カルボニル基、低級アルケニレン基、低級アルキニレン基又は一般式:-N(RX)-で表される基を表し(ここで、RXは、水素原子又は低級アルキル基である。);
Yは、単結合又は(CRYiRYi')nであり(ここで、nは、1乃至6のいずれかの整数であり、iは、1乃至nのいずれかの整数であり、(CRYiRYi')nは、n=1のとき(CRY1RY1')を表し、n=2のとき(CRY1RY1')-(CRY2RY2')を表し、n=3のとき(CRY1RY1')-(CRY2RY2')-(CRY3RY3')を表し、n=4のとき(CRY1RY1')-(CRY2RY2')-(CRY3RY3')-(CRY4RY4')を表し、n=5のとき(CRY1RY1')-(CRY2RY2')-(CRY3RY3')-(CRY4RY4')-(CRY5RY5')を表し、n=6のとき(CRY1RY1')-(CRY2RY2')-(CRY3RY3')-(CRY4RY4')-(CRY5RY5')-(CRY6RY6')を表し(ここで、RY1、RY1'、RY2、RY2'、RY3、RY3'、RY4、RY4'、RY5、RY5'、RY6及びRY6'は、それぞれ独立して、水素原子、ハロゲン原子又は<置換基群N>から選択される置換基であるか、
或いは、RX、RY1、RY1'、RY2、RY2'、RY3、RY3'、RY4、RY4'、RY5、RY5'、RY6及びRY6'に関して、以下の2個の基の組み合わせ:「RY1とRY1'」、「RY2とRY2'」、「RY3とRY3'」、「RY4とRY4'」、「RY5とRY5'」、「RY6とRY6'」、「RXとRY1」、「RXとRY2」、「RXとRY3」、「RY1とRY2」、「RY1とRY3」、「RY1とRY4」、「RY2とRY3」、「RY2とRY4」又は「RY2とRY5」は、当該組み合わせを構成する2個の基が一緒になって、低級アルキレン基を形成してもよく、ここで、該低級アルキレン基を構成する1若しくは2以上のメチレン基は、それぞれ独立して、酸素原子、カルボニル基、ビニレン基若しくは一般式:-N(Rc)-でメチレン基全体が置き換えられていてもよく、及び/又は、メチレン基を構成する水素が水酸基、低級アルキル基若しくはハロゲン原子で置換されていてもよい。);
Rcは、水素原子、低級アルキル基、ハロ低級アルキル基又は低級アルカノイル基を表し;
Zは、水酸基、COOR2、CONR3R4、SO3R2、SO3NR3R4、5-テトラゾリル基、5-オキソ-1,2,4-オキサジアゾリル基、2-オキソ-1,3,4-オキサジアゾリル基、5-イミノ-4,5-ジヒドロ-1,3,4-オキサジアゾリル基、2-チオキソ-1,3,4-オキサジアゾリル基又は5-オキソ-1,2,4-チアジアゾリル基を表し;
ここでR2、R3及びR4は、それぞれ独立して、水素原子又は低級アルキル基を表し; <置換基群L>、<置換基群M>及び<置換基群N>は、下記のように定義される。
<置換基群L>:
ハロゲン原子、水酸基、ニトロ基、シアノ基、ホルミル基、アミノ基、カルボキシル基、低級アルキル基、ハロ低級アルキル基、シクロアルキル基、低級アルコキシ基、ハロ低級アルコキシ基、ヒドロキシ低級アルキル基、低級アルコキシ低級アルキル基、低級アルコキシカルボニル基、低級アルカノイル基、低級アルキルチオ基、低級アルキルスルホニル基、低級アルキルアミノ基、ジ低級アルキルアミノ基、カルバモイル基、モノ低級アルキルカルバモイル基、ジ低級アルキルカルバモイル基、低級アルカノイルアミノ基、低級アルキルスルホニルアミノ基、低級アルコキシカルボニルアミノ基、アラルキル基、アリールオキシ基、ヘテロアリールオキシ基及び低級アルケニル基
<置換基群M>:
ハロゲン原子、水酸基、ニトロ基、シアノ基、ホルミル基、アミノ基、カルボキシル基、低級アルキル基、ハロ低級アルキル基、シクロアルキル基、低級アルコキシ基、ハロ低級アルコキシ基、ヒドロキシ低級アルキル基、低級アルコキシ低級アルキル基、低級アルコキシカルボニル基、低級アルカノイル基、低級アルキルチオ基、低級アルキルスルホニル基、低級アルキルアミノ基、ジ低級アルキルアミノ基、カルバモイル基、モノ低級アルキルカルバモイル基、ジ低級アルキルカルバモイル基、低級アルカノイルアミノ基、低級アルキルスルホニルアミノ基及び低級アルコキシカルボニルアミノ基
<置換基群N>:
水酸基、シアノ基、ホルミル基、カルボキシル基、低級アルキル基、ハロ低級アルキル基、シクロアルキル基、低級アルコキシ基、ハロ低級アルコキシ基、ヒドロキシ低級アルキル基、低級アルコキシ低級アルキル基、低級アルコキシカルボニル基、低級アルカノイル基、カルバモイル基、モノ低級アルキルカルバモイル基及びジ低級アルキルカルバモイル基]
で示される化合物又は該化合物の薬学的に許容できる塩若しくはエステルである。
更に本発明は、哺乳類(特にヒト)における高尿酸血症、痛風結節、急性通風関節炎、慢性通風関節炎、痛風腎、尿路結石、腎機能障害、冠動脈疾患及び虚血性心疾患からなる群より選択される血中尿酸が関与する病態を治療または予防する方法であって、当該哺乳類に、式(I)の化合物を含むURAT1阻害剤、血中尿酸値低下剤又は医薬組成物の治療上の有効量を投与することを特徴とする方法、に関する。
更に本発明は、式(I)の化合物を有効成分として含有する血中尿酸値低下剤に関する。
更に本発明は、高尿酸血症、痛風結節、急性通風関節炎、慢性通風関節炎、痛風腎、尿路結石、腎機能障害、冠動脈疾患及び虚血性心疾患からなる群より選択される血中尿酸が関与する病態の治療薬または予防するための医薬組成物であって、(I)の化合物を有効成分として含むことを特徴とする医薬組成物に関する。
上記式(I)中の「ハロゲン原子」としては、フッ素原子、塩素原子、臭素原子及びヨウ素原子等が挙げられる。
また、「ジ低級アルキルカルバモイル基」には、カルバモイル基を構成する窒素原子と、該窒素原子に結合した同一の、又は異なる前記「低級アルキル基」とが一緒になって形成する5乃至8員の単環、又は該単環とベンゼン環若しくはピリジン環とが縮合して形成される双環も含まれ、例えば下記式で表される基が挙げられる。

R1のハロ低級アルキル基は、例えばフルオロメチル基、ジフルオロメチル基、トリフルオロメチル基及び2,2,2-トリフルオロエチル基等が好適である。
R1のヒドロキシ低級アルキル基は、例えばヒドロキシメチル基及び2-ヒドロキシエチル基等が好適である。
R1の低級アルコキシカルボニル基は、例えばメトキシカルボニル基、エトキシカルボニル基、プロポキシカルボニル基、イソプロポキシカルボニル基、ブトキシカルボニル基、イソブトキシカルボニル基、tert-ブトキシカルボニル基及びペンチルオキシカルボニル基等が好適である。
R1の低級アルキルスルホニル基は、例えばメタンスルホニル基及びエタンスルホニル基等が好適である。
Q1の低級アルキレン基を構成する1若しくは2以上のメチレン基は、それぞれ独立して、カルボニル基、スルフィニル基若しくはスルホニル基でメチレン基全体が置き換えられていてもよく、及び/又は、メチレン基を構成する水素が低級アルキル基で置換されていてもよく、そのような置き換え又は置換された基としては、例えば下記式から選択される基が好適である。

A1のヘテロアリール基は、例えばイミダゾリル基、フリル基、チエニル基、ピラゾリル基、チアゾリル基、イソチアゾリル基、オキサゾリル基、イソキサゾリル基、1,2,4-オキサジアゾリル基、1,3,4-オキサジアゾリル基、ピリジル基、ピラジニル基、ピリミジニル基、ベンゾフラニル基、キノリル基、ベンゾチエニル基等、より好ましくはピリジル基、キノリル基、チエニル基、ピラゾリル基、チアゾリル基、イソキサゾリル基、ベンゾチエニル基等、更に好ましくはピリジル基、イソキサゾリル基、キノリル基、ベンゾチエニル基等が好適である。
- - - - が二重結合である場合、
W1は、窒素原子又は一般式:=C(Ra)-で表される基を表し、かつ
W2は、窒素原子又は一般式:=C(Rb)-で表される基を表し、
- - - - が単結合である場合、
W1は、一般式:-C(Raa)(Rab)-で表される基、又は一般式:-(C=O)-で表される基を表し、かつ
W2は、一般式:-C(Rba)(Rbb)-で表される基、一般式:-(C=O)-で表される基又は一般式:-N(Rbc)-で表される基を表す。


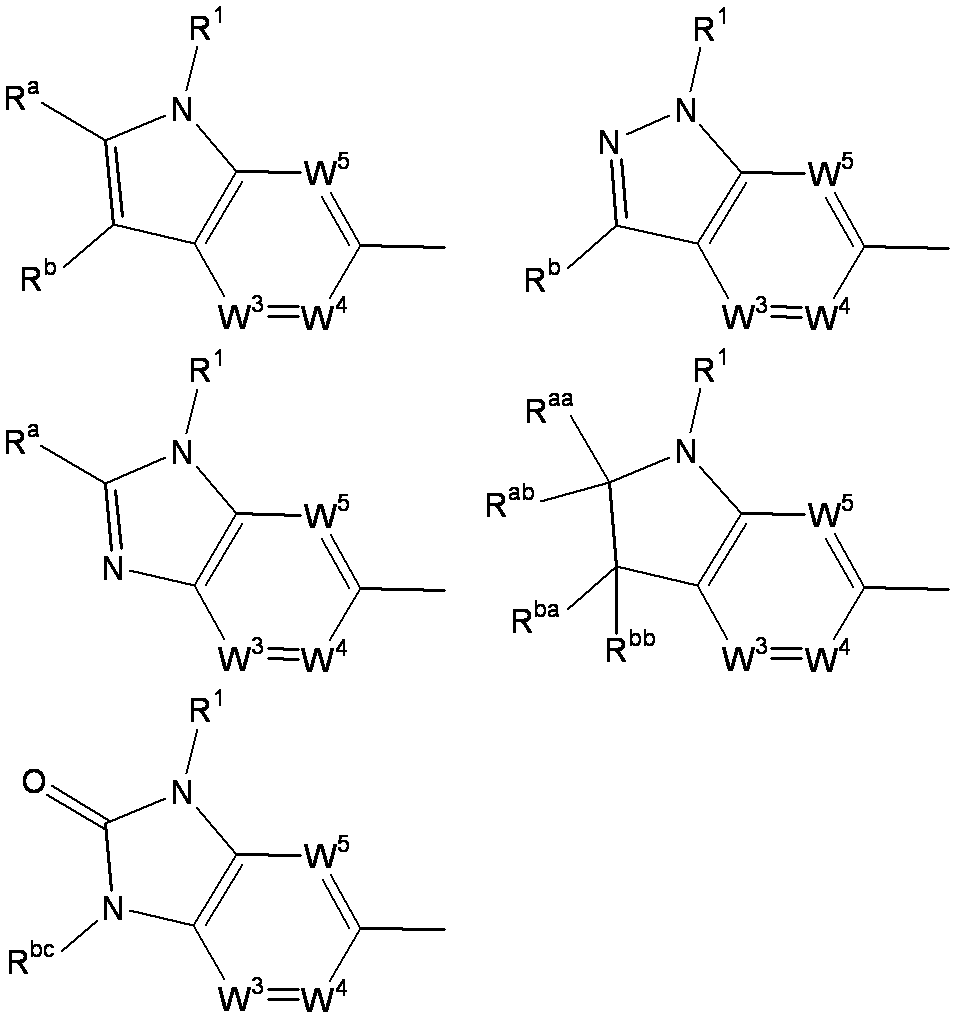
ここで、<置換基群M>は、ハロゲン原子、水酸基、ニトロ基、シアノ基、ホルミル基、アミノ基、カルボキシル基、低級アルキル基、ハロ低級アルキル基、シクロアルキル基、低級アルコキシ基、ハロ低級アルコキシ基、ヒドロキシ低級アルキル基、低級アルコキシ低級アルキル基、低級アルコキシカルボニル基、低級アルカノイル基、低級アルキルチオ基、低級アルキルスルホニル基、低級アルキルアミノ基、ジ低級アルキルアミノ基、カルバモイル基、モノ低級アルキルカルバモイル基、ジ低級アルキルカルバモイル基、低級アルカノイルアミノ基、低級アルキルスルホニルアミノ基及び低級アルコキシカルボニルアミノ基からなる群である。
ここで、<置換基群N>は、水酸基、シアノ基、ホルミル基、カルボキシル基、低級アルキル基、ハロ低級アルキル基、シクロアルキル基、低級アルコキシ基、ハロ低級アルコキシ基、ヒドロキシ低級アルキル基、低級アルコキシ低級アルキル基、低級アルコキシカルボニル基、低級アルカノイル基、カルバモイル基、モノ低級アルキルカルバモイル基及びジ低級アルキルカルバモイル基からなる群である。
Q2の低級アルキレン基を構成する1若しくは2以上のメチレン基は、それぞれ独立して、酸素原子、窒素原子若しくはカルボニル基でメチレン基全体が置き換えられていてもよく、及び/又は、メチレン基を構成する水素がハロゲン原子、シアノ基、水酸基若しくは低級アルキル基で置換されていてもよい。そのような置き換え又は置換された基としては、例えば下記式から選択される基が好適である。
A2のヘテロアリール基としては、例えばイミダゾリル基、フリル基、チエニル基、ピラゾリル基、チアゾリル基、イソチアゾリル基、オキサゾリル基、イソオキサゾリル基、1,2,4-オキサジアゾリル基、1,3,4-オキサジアゾリル基、ピリジル基、ピラジニル基、ピリミジニル基、ベンゾフラニル基及びキノリル基等が好適であり、より好ましくはピリジル基及びキノリル基等、更に好ましくはピリジル基等である。
該置換基の低級アルキル基としては、例えばメチル基及びエチル基等が好適である。
該置換基のシクロアルキル基としては、例えばシクロプロピル基等が好適である。
該置換基のハロ低級アルキル基としては、例えばフルオロメチル基、ジフルオロメチル基及びトリフルオロメチル基等が好適である。
該置換基の低級アルコキシ基はとして、例えばメトキシ基及びエトキシ基等が好適である。
該置換基のハロ低級アルコキシ基としては、例えばジフルオロメトキシ基及びトリフルオロメトキシ基等が好適である。
Xの低級アルキニレン基としては、例えばエチニレン基等が好適である。
該置換基の低級アルキル基としては、例えばメチル基及びエチル基等が好適である。
該置換基の低級アルコキシ基としては、例えばメトキシ基及びエトキシ基等が好適である。


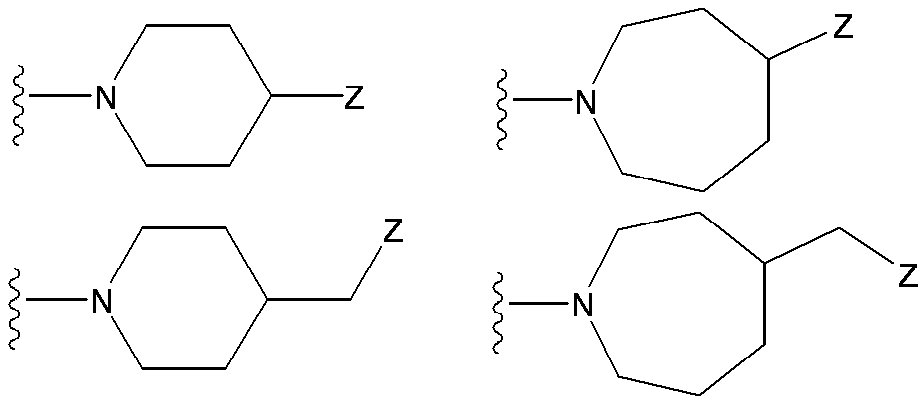

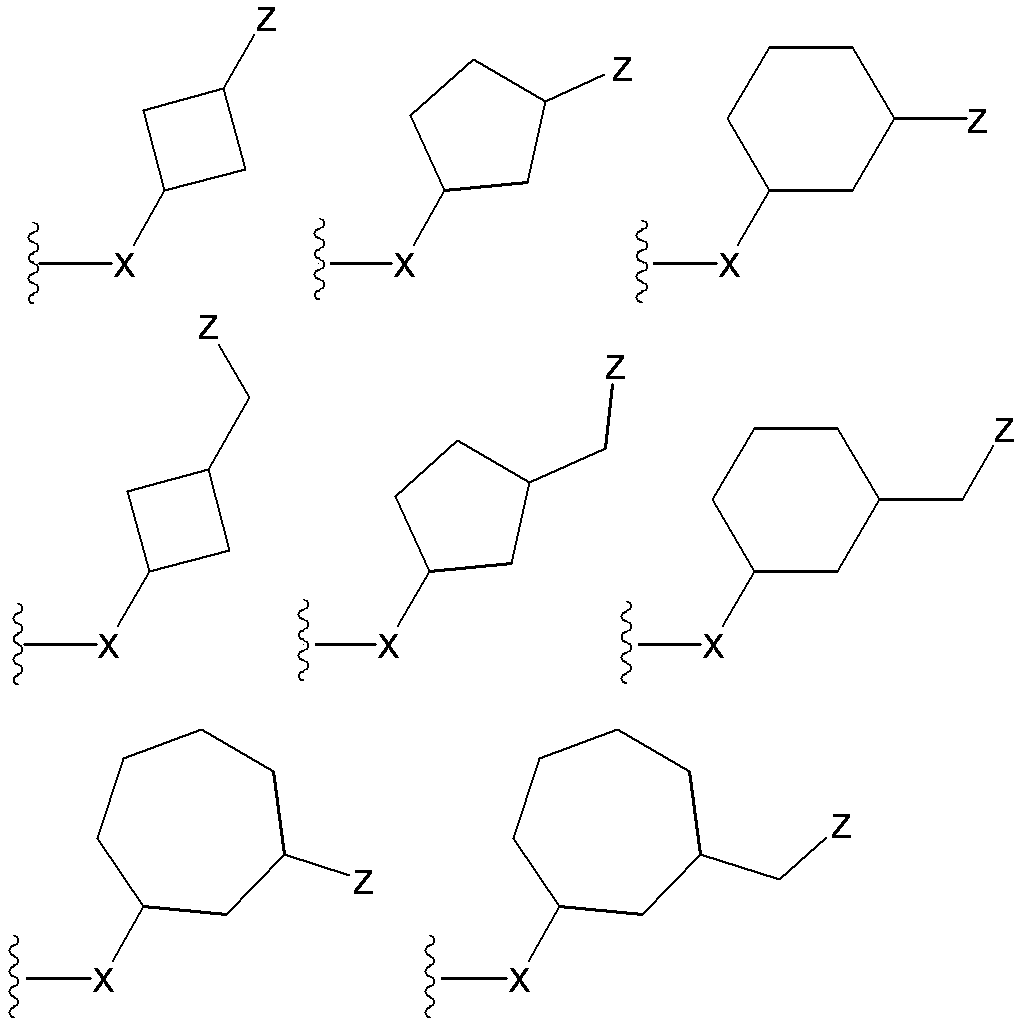


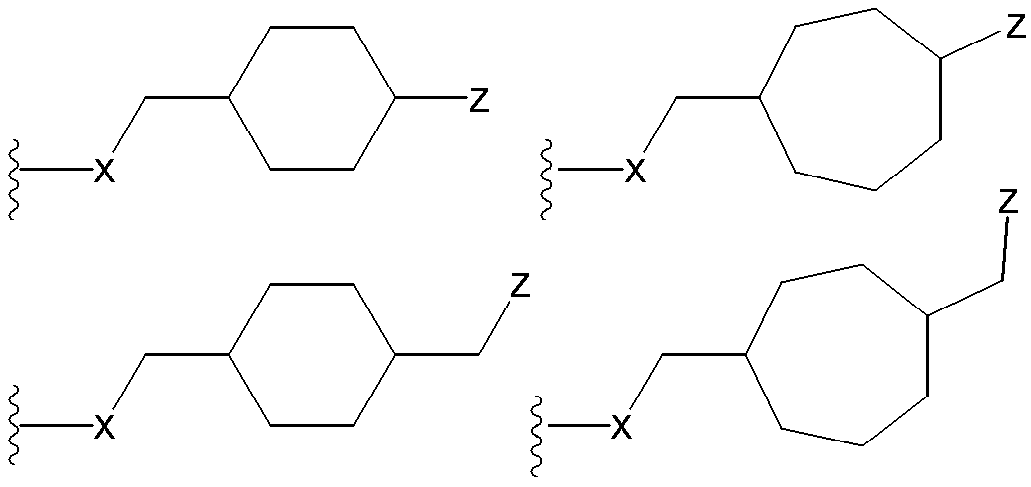
(1)Xが単結合、Yが単結合のとき、一般式(I)は、下記式のように表される。
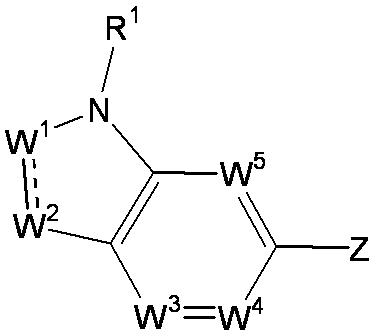
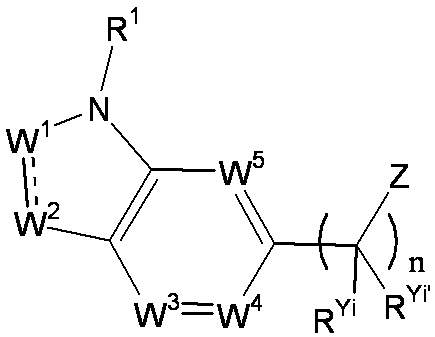
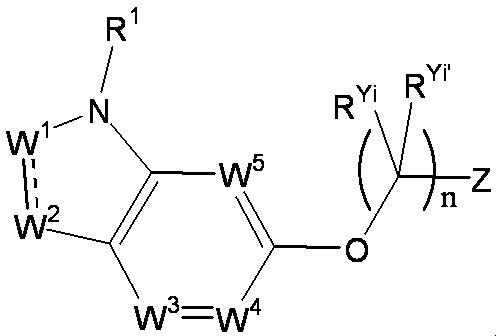
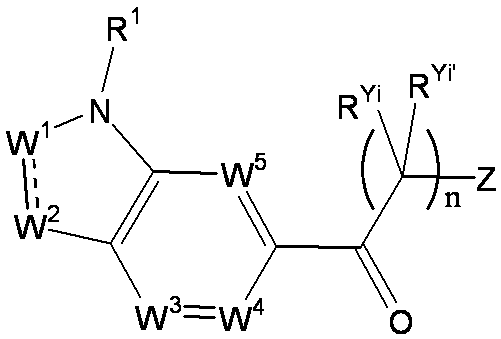

(1)下記部分構造:

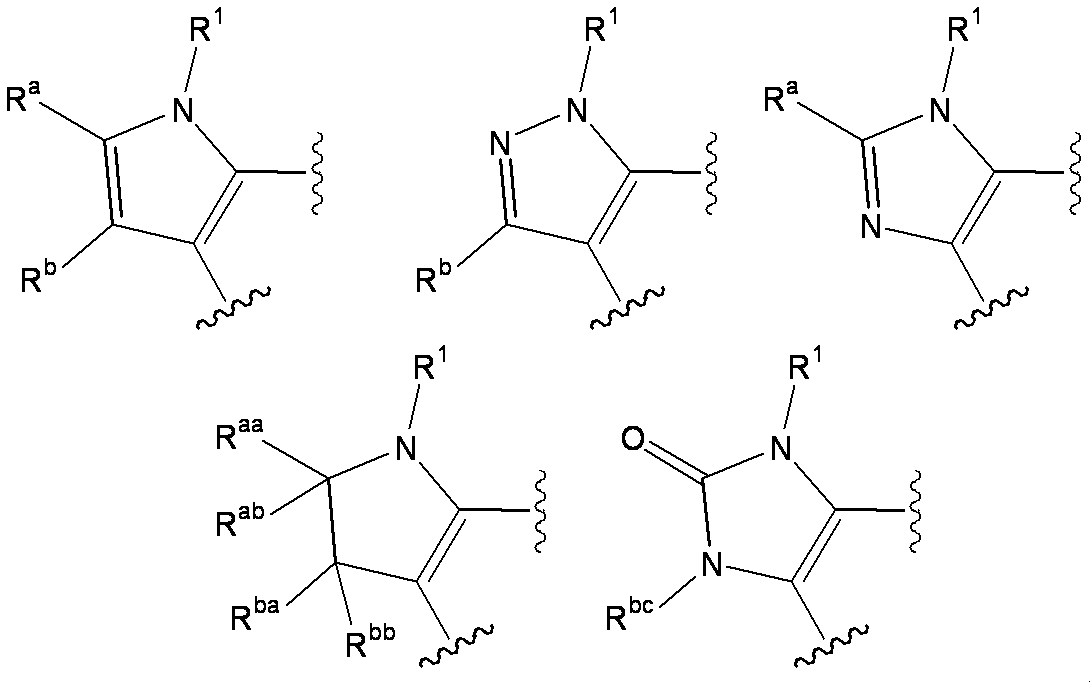
(a)1-(2,6-ジメチルベンジル)-3-メチル-1H-インドール-6-カルボン酸(実施例61)
(b)2-[1-(2,6-ジメチルベンジル)-3-メチル-1H-インドール-6-イル]酢酸(実施例64)
(c)2-[1-(2,6-ジメチルベンジル)-3-メチル-1H-インダゾール-6-イル]酢酸(実施例66)
(d)2-[3-クロロ-1-(2,6-ジメチルベンジル)-1H-インドール-6-イル]酢酸(実施例68)
(e)2-[1-(2,6-ジメチルベンジル)-2,3-ジヒドロ-1H-インドール-6-イル]酢酸(実施例75)
(f)(3-RS)-2-[1-(2,6-ジメチルベンジル)-2,3-ジヒドロ-3-メチル-1H-インドール-6-イル]酢酸(実施例78)
(g)2-[1-(2,6-ジメチルベンジル)-2,3-ジヒドロ-3,3-ジメチル-1H-インドール-6-イル]酢酸(実施例80)
(h)2-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インドール-6-イル]酢酸(実施例82)
(i)2-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]酢酸(実施例97)
(j)2-[1-(2-クロロ-6-メチルベンジル)-3-メチル-1H-インダゾール-6-イル]酢酸(実施例102)
(k)3-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]プロピオン酸(実施例122)
(l)1-(2,6-ジクロロベンジル)-3-メチル-6-(1H-テトラゾール-5-イルメチル)-1H-インダゾール(実施例134)
(m)2-[1-(2,6-ジクロロベンジル)-3-エチル-1H-インダゾール-6-イル]酢酸(実施例140)
(n)1-(2-クロロ-6-メチルベンジル)-3-メチル-6-(1H-テトラゾール-5-イル)-1H-ピラゾロ[4,3-b]ピリジン(実施例160)
(o)1-(2-クロロ-6-シクロプロピルベンジル)-3-メチル-6-(1H-テトラゾール-5-イル)-1H-ピラゾロ[4,3-b]ピリジン(実施例164)
(p)3-クロロ-1-(2,6-ジクロロベンジル)-1H-ピラゾロ[4,3-b]ピリジン-6-カルボン酸(実施例172)
(q)3-クロロ-1-(2-クロロ-6-メチルベンジル)-1H-ピラゾロ[4,3-b]ピリジン-6-カルボン酸(実施例174)
(r)3-クロロ-1-(2-クロロ-6-メチルベンジル)-6-(1H-テトラゾール-5-イル)-1H-ピラゾロ[4,3-b]ピリジン(実施例177)
(s)[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]ジフルオロ酢酸(実施例183)
並びに、上記化合物の薬学的に許容できる塩及びエステル等がより好適である。
本発明の化合物は、ラセミ体として、ラセミ混合物として、及び個々のジアステレオマーとして生じ得る。
また、光学異性体を含む、全ての可能な異性体、及びそれらの混合物は全て、本発明に含まれる。
さらに、本明細書において開示された化合物は、互変異性体として存在してよく、たとえ一方の互変異性体構造のみが描かれている場合でも、双方の互変異性体型が本発明の範囲によって抱合されることが意図されている。
また、各工程間に適宜置換基変換(置換基の変換又は更なる修飾)工程が挿入されていてもよい。反応性官能基がある場合は、適宜保護、脱保護を行えばよい。また、反応の進行を促進するために、例示した試薬以外の試薬を適宜用いることができる。各反応の加熱には、必要に応じて、マイクロウェーブの照射を利用してもよい。また、製造方法未記載の原料化合物は市販されているか、又は既知の合成反応を組み合わせて容易に調製可能な化合物である。
各工程で得られる化合物は、結晶化、再結晶化、カラムクロマトグラフィー、分取HPLC等の慣用される常法で単離及び精製することができるが、場合によっては、単離精製せず次の工程に進むことができる。
以下の製造方法において、「室温」とは1乃至40℃を意味する。
スキーム1:式(II)の化合物から式(I)の化合物の製造法

ここで、上記式(II)の保護基PG2は、その機能を有するものであれば特に限定されないが、例えばメチル基、エチル基、プロピル基、イソプロピル基、tert-ブチル基等の低級アルキル基;例えば2,2,2-トリクロロエチル基等のハロ低級アルキル基や;例えばアリル基等の低級アルケニル基;例えばベンジル基、p-メトキシベンジル基、p-ニトロベンジル基、ベンズヒドリル基、トリチル基等のアラルキル基等が挙げられ、特にメチル基、エチル基、tert-ブチル基、アリル基、ベンジル基、p-メトキシベンジル基、ベンズヒドリル基等が好ましい。
保護基の除去法は、当該保護基の種類及び目的化合物(I)の安定性等により異なるが、例えば文献記載の方法[プロテクティブ・グループス・イン・オーガニック・シンセシス(Protective Groups in Organic Synthesis)、第3版、T.W.グリーン(T.W.Greene)著、John Wiley & Sons社(1999年)参照]又はそれに準じる方法に従って、例えば酸又は塩基を用いる加溶媒分解、即ち、例えば0.01モル乃至大過剰の酸、好ましくはトリフルオロ酢酸、ギ酸、塩酸等、又は等モル乃至大過剰の塩基、好ましくは水酸化カリウム、水酸化カルシウム等を作用させる方法、水素化金属錯体等を用いる化学的還元又はパラジウム-炭素触媒、ラネーニッケル触媒等を用いる接触還元等により行われる。

上記式(IV)の脱離基Lは、上記化合物(III)との反応により脱離して化合物(II)を生成するものであれば特に限定されないが、脱離基としては、ハロゲン原子(塩素原子、臭素原子等)、p-トルエンスルホニル基やメタンスルホニル基等が挙げられ、臭素原子、塩素原子やp-トルエンスルホニル基等が好ましい。
用いられる化合物(IV)の量は、化合物(III)1モルに対して、通常1乃至10モル、好ましくは1乃至3モルである。
用いられる塩基としては、炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム、水素化ナトリウムや水酸化カリウム等が挙げられ、炭酸カリウム、水素化ナトリウムや水酸化カリウム等が好ましい。
塩基の量は、化合物(III)1モルに対して、通常1乃至10モル、好ましくは1乃至5モルである。
反応温度は、通常0℃乃至160℃であり、好ましくは25℃乃至100℃である。
反応時間は、通常1時間乃至24時間であり、好ましくは1時間乃至12時間である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、ジメチルホルムアミド、N-メチル-2-ピロリドン、テトラヒドロフラン、1,4-ジオキサン、アセトン、メチルエチルケトンやアセトニトリル等の溶媒が好ましい。
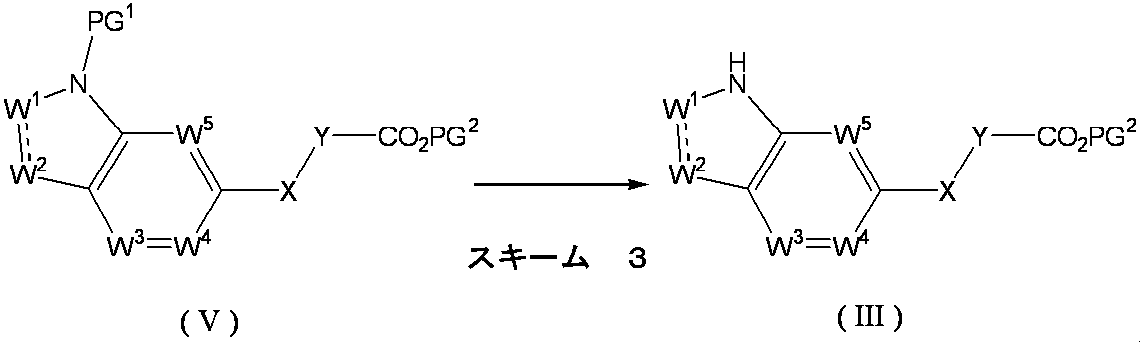
ここで、上記式(V)の保護基PG1は、その機能を有するものであれば特に限定されないが、例えばベンジル基、p-メトキシベンジル基、3,4-ジメトキシベンジル基、o-ニトロベンジル基、p-ニトロベンジル基、ベンズヒドリル基、トリチル基等のアラルキル基;例えばホルミル基、アセチル基、プロピオニル基、ブチリル基、ピバロイル基等の低級アルカノイル基;例えばベンゾイル基;例えばフェニルアセチル基、フェノキシアセチル基等のアリールアルカノイル基;例えばメトキシカルボニル基、エトキシカルボニル基、プロピルオキシカルボニル基、tert-ブトキシカルボニル基等の低級アルコキシカルボニル基;例えばベンジルオキシカルボニル基、p-ニトロベンジルオキシカルボニル基、フェネチルオキシカルボニル基等のアラルキルオキシカルボニル基;例えばトリメチルシリル基、tert-ブチルジメチルシリル基等の低級アルキルシリル基;例えばテトラヒドロピラニル基;例えばトリメチルシリルエトキシメチル基;例えばメチルスルホニル基、エチルスルホニル基等の低級アルキルスルホニル基等や;例えばベンゼンスルホニル基、p-トルエンスルホニル基等のアリールスルホニル基等が挙げられ、特にtert-ブトキシカルボニル基、メチルスルホニル基やp-トルエンスルホニル基等が好ましい。
保護基の除去法は、当該保護基の種類及び目的化合物(III)の安定性等により異なるが、例えば文献記載の方法[プロテクティブ・グループス・イン・オーガニック・シンセシス(Protective Groups in Organic Synthesis)、第3版、T.W.グリーン(T.W.Greene)著、John Wiley & Sons社(1999年)参照]又はそれに準じる方法に従って、例えば酸又は塩基を用いる加溶媒分解、即ち、例えば0.01モル乃至大過剰の酸、好ましくはトリフルオロ酢酸、ギ酸、塩酸等;等モル乃至大過剰の塩基、好ましくは水酸化カリウム、水酸化カルシウム等を作用させる方法や;水素化金属錯体等を用いる化学的還元又はパラジウム-炭素触媒、ラネーニッケル触媒等を用いる接触還元等により行われる。

ここで、上記式(V)の保護基PG2は、その機能を有するものであれば特に限定されないが、例えばメチル基、エチル基、プロピル基、イソプロピル基、tert-ブチル基等の低級アルキル基;例えば2,2,2-トリクロロエチル基等のハロ低級アルキル基;例えばアリル基等の低級アルケニル基や;例えばベンジル基、p-メトキシベンジル基、p-ニトロベンジル基、ベンズヒドリル基、トリチル基等のアラルキル基等が挙げられ、特にメチル基、エチル基、tert-ブチル基、アリル基、ベンジル基、p-メトキシベンジル基やベンズヒドリル基等が好ましい。
保護基の導入法は、当該保護基の種類及び化合物の安定性等により異なるが、例えば文献記載の方法[プロテクティブ・グループス・イン・オーガニック・シンセシス(Protective Groups in Organic Synthesis)、第3版、T.W.グリーン(T.W.Greene)著、John Wiley & Sons社(1999年)参照]又はそれに準じる方法に従って合成できる。

当該反応では、化合物(VII)1モルに対して、2-メチル-2-ブテンを通常1乃至20モル、好ましくは1乃至10モル、リン酸二水素ナトリウムを通常1乃至5モル、好ましくは1乃至3モル、亜塩素酸ナトリウムを通常1乃至10モル、好ましくは1乃至5モル用いる。
反応温度は、通常0℃乃至100℃であり、好ましくは0℃乃至40℃である。
反応時間は、通常1時間乃至24時間であり、好ましくは1時間乃至6時間である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、水または水とtert-ブタノール若しくはアセトニトリル等の水溶性溶媒との混合溶媒が好ましい。
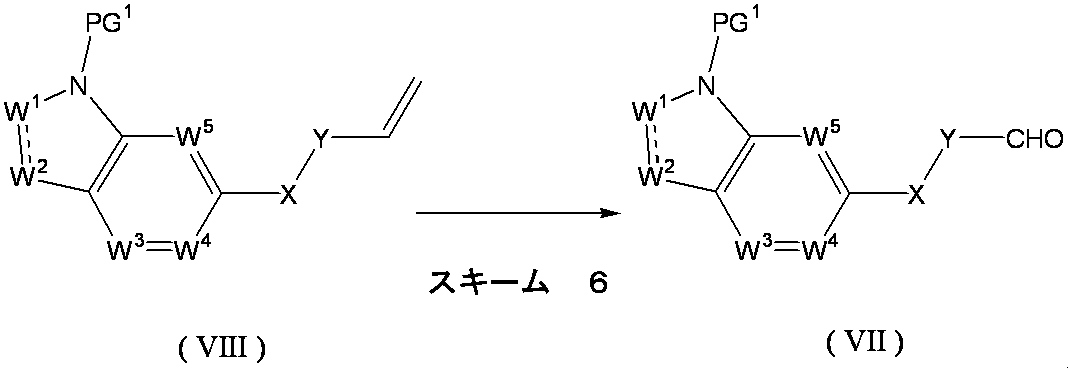
当該反応では、化合物(VIII)1モルに対して、四酸化オスミウムを通常0.0001乃至1モル、好ましくは0.01乃至1モル、過ヨウ素酸ナトリウムを通常1乃至10モル、好ましくは1乃至5モル用いる。
反応温度は、通常0℃乃至100℃であり、好ましくは0℃乃至40℃である。
反応時間は、通常1時間乃至24時間であり、好ましくは1時間乃至12時間である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、水とtert-ブタノール、ジオキサン又はアセトンなどの水溶性溶媒との混合溶媒が好ましい。

当該反応では、化合物(IX)1モルに対して、化合物(X)を通常1乃至10モル、好ましくは1乃至3モル用いる。
化合物(X)としては、例えばトリブチルビニルスズや、トリブチルアリルスズ等が挙げられる。
用いられる塩基としては、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、フッ化セシウム、フッ化カリウム、フッ化ナトリウムや塩化リチウム等が挙げられる。
用いられる塩基の量は、化合物(IX)1モルに対して、通常1乃至10モル、好ましくは1乃至3モルである。
用いられるパラジウム触媒としては、例えば、Pd(PPh3)4、Pd(OAc)2、Pd2(dba)3やPdCl2(PPh3)2等が挙げられる。
用いられるパラジウム触媒の量は、化合物(IX)1モルに対して、通常0.01乃至0.5モル、好ましくは0.05乃至0.2モルである。
用いられるホスフィン配位子としては、PPh3、P(o-tol)3、P(tert-Bu)3、2-[ジ(tert-ブチル)ホスフィノ]-1,1’-ビフェニル、2-[ジ(tert-ブチル)ホスフィノ]-2’-ジメチルアミノ-1,1’-ビフェニル、2-[ジシクロヘキシルホスフィノ]-1,1’-ビフェニルや、2-[ジシクロヘキシルホスフィノ]-2’-ジメチルアミノ-1,1’-ビフェニル等が挙げられる。
反応温度は、通常0℃乃至200℃であり、好ましくは25℃乃至130℃である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、ジメチルホルムアミド、テトラヒドロフラン、1,4-ジオキサン、アセトニトリルやトルエン等の溶媒が好ましい。

化合物(XI)は、購入可能な市販化合物又は文献等で公知の化合物である。
ここで、上記式(IX)の保護基PG1は、その機能を有するものであれば特に限定されないが、例えばベンジル基、p-メトキシベンジル基、3,4-ジメトキシベンジル基、o-ニトロベンジル基、p-ニトロベンジル基、ベンズヒドリル基、トリチル基等のアラルキル基;例えばホルミル基、アセチル基、プロピオニル基、ブチリル基、ピバロイル基等の低級アルカノイル基;例えばベンゾイル基;例えばフェニルアセチル基、フェノキシアセチル基等のアリールアルカノイル基;例えばメトキシカルボニル基、エトキシカルボニル基、プロピルオキシカルボニル基、tert-ブトキシカルボニル基等の低級アルコキシカルボニル基;例えばベンジルオキシカルボニル基、p-ニトロベンジルオキシカルボニル基、フェネチルオキシカルボニル基等のアラルキルオキシカルボニル基;例えばトリメチルシリル基、tert-ブチルジメチルシリル基等の低級アルキルシリル基;例えばテトラヒドロピラニル基;例えばトリメチルシリルエトキシメチル基;例えばメチルスルホニル基、エチルスルホニル基等の低級アルキルスルホニル基等や;例えばベンゼンスルホニル基、p-トルエンスルホニル基等のアリールスルホニル基等が挙げられ、特にtert-ブトキシカルボニル基、メチルスルホニル基やp-トルエンスルホニル基等が好ましい。
保護基の導入法は、当該保護基の種類及び化合物の安定性等により異なるが、例えば文献記載の方法[プロテクティブ・グループス・イン・オーガニック・シンセシス(Protective Groups in Organic Synthesis)、第3版、T.W.グリーン(T.W.Greene)著、John Wiley & Sons社(1999年)参照]又はそれに準じる方法に従って合成できる。

当該反応では、化合物(XII)1モルに対して、ハロゲン化剤を通常1乃至10モル、好ましくは1乃至3モル用いる。
ハロゲン化剤としては、例えばN-クロロスクシンアミドやN-ブロモスクシンアミド等が挙げられる。
反応温度は、通常0℃乃至100℃であり、好ましくは0℃乃至25℃である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、テトラヒドロフラン、1,4-ジオキサン、クロロホルムやアセトニトリル等の溶媒が好ましい。

当該反応では、化合物(XIII)1モルに対して、Rb-B(OH)2を通常1乃至10モル、好ましくは1乃至3モル用いる。
Rb-B(OH)2としては、例えばフェニルホウ酸、フェニルホウ酸エステル、ジアルキルフェニルボラン等の市販のアリールホウ素誘導体、ヘテロアリールホウ素誘導体、ビニルホウ素誘導体、アリルホウ素誘導体、低級アルキルホウ素誘導体やシクロ低級アルキルホウ素誘導体を用いることができる。また、公知の方法、これに準じた方法、又はこれらと常法とを組み合わせた方法により目的とするホウ素誘導体を製造してもよい。
用いられる塩基としては、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、フッ化セシウム、フッ化カリウムやフッ化ナトリウム等が挙げられる。
用いられる塩基の量は、化合物(XIII)1モルに対して、通常1乃至10モル、好ましくは1乃至3モルである。
用いられるパラジウム触媒としては、例えば、Pd(PPh3)4、Pd(OAc)2、Pd2(dba)3やPdCl2(PPh3)2等が挙げられる。
用いられるパラジウム触媒の量は、化合物(XIII)1モルに対して、通常0.01乃至0.5モル、好ましくは0.05乃至0.2モルである。
用いられるホスフィン配位子としては、PPh3、P(o-tol)3、P(tert-Bu)3、2-[ジ(tert-ブチル)ホスフィノ]-1,1’-ビフェニル、2-[ジ(tert-ブチル)ホスフィノ]-2’-ジメチルアミノ-1,1’-ビフェニル、2-[ジシクロヘキシルホスフィノ]-1,1’-ビフェニルや、2-[ジシクロヘキシルホスフィノ]-2’-ジメチルアミノ-1,1’-ビフェニル等が挙げられる。
反応温度は、通常0℃乃至200℃であり、好ましくは25℃乃至130℃である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、ジメチルホルムアミド、テトラヒドロフラン、1,4-ジオキサン、アセトニトリルやトルエン等の溶媒が好ましい。

ここで、上記式(III-1)の保護基PG2は、その機能を有するものであれば特に限定されないが、例えばメチル基、エチル基、プロピル基、イソプロピル基、tert-ブチル基等の低級アルキル基;例えば2,2,2-トリクロロエチル基等のハロ低級アルキル基;例えばアリル基等の低級アルケニル基や;例えばベンジル基、p-メトキシベンジル基、p-ニトロベンジル基、ベンズヒドリル基、トリチル基等のアラルキル基等が挙げられ、特にメチル基、エチル基、tert-ブチル基、アリル基、ベンジル基、p-メトキシベンジル基やベンズヒドリル基等が好ましい。
保護基の導入法は、当該保護基の種類及び化合物の安定性等により異なるが、例えば文献記載の方法[プロテクティブ・グループス・イン・オーガニック・シンセシス(Protective Groups in Organic Synthesis)、第3版、T.W.グリーン(T.W.Greene)著、John Wiley & Sons社(1999年)参照]又はそれに準じる方法に従って合成できる。

ここで、上記式(XVII)で表される化合物の保護基PG1は、その機能を有するものであれば特に限定されないが、例えばホルミル基、アセチル基、プロピオニル基、ブチリル基、ピバロイル基等の低級アルカノイル基;例えばベンゾイル基;例えばフェニルアセチル基、フェノキシアセチル基等のアリールアルカノイル基;例えばメトキシカルボニル基、エトキシカルボニル基、プロピルオキシカルボニル基、tert-ブトキシカルボニル基等の低級アルコキシカルボニル基;例えばベンジルオキシカルボニル基、p-ニトロベンジルオキシカルボニル基、フェネチルオキシカルボニル基等のアラルキルオキシカルボニル基;例えばトリメチルシリル基、tert-ブチルジメチルシリル基等の低級アルキルシリル基;例えばメチルスルホニル基、エチルスルホニル基等の低級アルキルスルホニル基等や;例えばベンゼンスルホニル基、p-トルエンスルホニル基等のアリールスルホニル基等が挙げられ、特にtert-ブトキシカルボニル基、メチルスルホニル基やp-トルエンスルホニル基等が好ましい。
本加水分解反応は、酸又は塩基を用いる加水分解、即ち、例えば0.01モル乃至大過剰の酸、好ましくは酢酸、ギ酸、硫酸、フタル酸や塩酸等、又は0.01モル乃至大過剰の塩基、好ましくは水酸化ナトリウムや水酸化カリウム等を作用させる方法により行われる。
反応温度は、通常0℃乃至200℃であり、好ましくは0℃乃至160℃である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、メタノール、エタノール、1-プロパノール、2-プロパノール、1-ブタノール、2-ブタノール、tert-ブタノール、テトラヒドロフラン、1,4-ジオキサン、シクロヘキサン、1,3-ジメチルベンゼンやトルエン等の溶媒が好ましい。
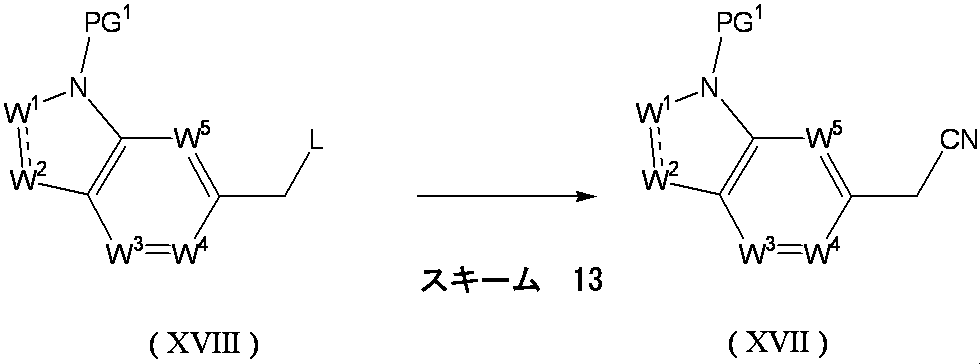
上記式(XVIII)の脱離基Lとしては、その機能を有するものであれば特に限定されないが、例えばハロゲン原子(塩素原子、臭素原子等)、p-トルエンスルホニルオキシ基、ベンゼンスルホニルオキシ基、メタンスルホニルオキシ基やエタンスルホニルオキシ等が挙げられ、臭素原子、塩素原子、p-トルエンスルホニルオキシ基やメタンスルホニルオキシ基等が好ましい。
当該反応では、化合物(XVIII)1モルに対して、シアン化物を通常1モル乃至10モル、好ましくは1モル乃至3モル用いる。
用いられるシアン化物としては、シアン化リチウム、シアン化ナトリウム、シアン化カリウム、テトラエチルシアニド、トリメチルシリルシアニドやテトラブチルシアニド等が挙げられる。
反応温度は、通常0℃乃至200℃であり、好ましくは0℃乃至160℃である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、ジメチルスルホキシド、メタノール、エタノール、1-プロパノール、2-プロパノール、1-ブタノール、2-ブタノール、tert-ブタノール、テトラヒドロフラン、ジメトキシエタン、1,4-ジオキサン、シクロヘキサン、1,3-ジメチルベンゼンやトルエン等の溶媒が好ましい。
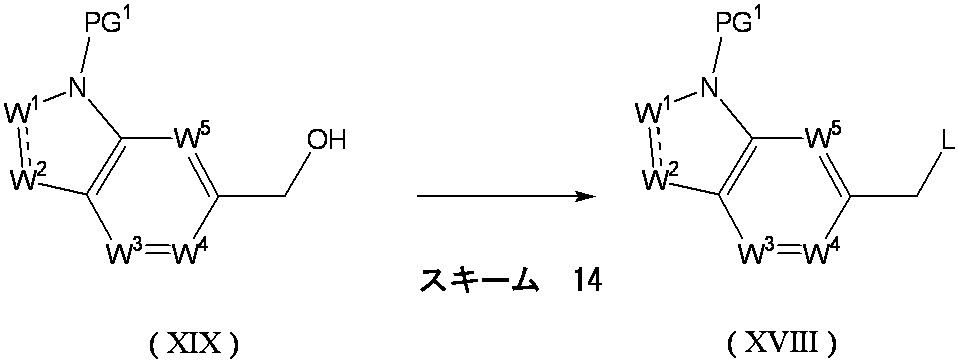
上記式(XVIII)の脱離基Lとしては、その機能を有するものであれば特に限定されないが、例えばハロゲン原子(塩素原子、臭素原子等)、p-トルエンスルホニルオキシ基、ベンゼンスルホニルオキシ基、メタンスルホニルオキシ基やエタンスルホニルオキシ等が挙げられ、臭素原子、塩素原子、p-トルエンスルホニルオキシ基やメタンスルホニルオキシ基等が好ましい。
当該反応では、化合物(XIX)1モルに対して、脱離基をもたらす化合物として、例えばスルホン酸クロリドを通常1モル乃至10モル、好ましくは1モル乃至3モル用いる。
用いられるスルホン酸クロリドとしては、メタンスルホニルクロリド、エタンスルホニルクロリド、p-トルエンスルホニルクロリドやフェニルスルホニルクロリド等が挙げられる。
用いられる塩基としては、トリエチルアミンやジイソプロピルエチルアミン等が挙げられる。
反応温度は、通常0℃乃至200℃であり、好ましくは0℃乃至25℃である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、クロロホルム、ジクロロメタン、テトラヒドロフラン、ジメトキエタン、1,4-ジオキサン、シクロヘキサン、1,3-ジメチルベンゼンやトルエン等の溶媒が好ましい。
また上記式(XVIII)の化合物は、上記式(XIX)で表される化合物に四臭化炭素、四塩化炭素等のハロゲン化剤及びトリフェニルホスフィンを作用させるアッペル反応により得ることもできる。
当該反応では、化合物(XIX)1モルに対して、ハロゲン化剤を通常1モル乃至10モル、好ましくは1モル乃至3モル用いる。
用いられるハロゲン化剤としては、四臭化炭素、四塩化炭素、ヘキサクロロアセトン、ヘキサブロモアセトン、トリホスゲン、臭化リチウム、ヨウ化メタン、臭素やヨウ素等が挙げられる。
当該反応では、化合物(XIX)1モルに対して、トリフェニルホスフィンを通常1モル乃至10モル、好ましくは1モル乃至3モル用いる。
反応温度は、通常0℃乃至200℃であり、好ましくは0℃乃至25℃である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、クロロホルム、ジクロロメタン、テトラヒドロフラン、ジメトキシエタン、1,4-ジオキサン、シクロヘキサン、1,3-ジメチルベンゼン、アセトニトリルやトルエン等の溶媒が好ましい。
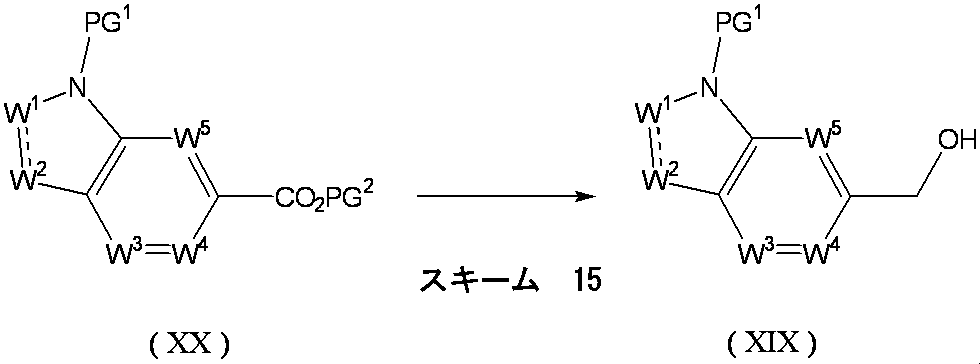
ここで、上記式(XX)の保護基PG2は、その機能を有するものであれば特に限定されないが、例えばメチル基、エチル基、プロピル基、イソプロピル基、tert-ブチル基等の低級アルキル基;例えば2,2,2-トリクロロエチル基等のハロ低級アルキル基;例えばアリル基等の低級アルケニル基や;例えばベンジル基、p-メトキシベンジル基、p-ニトロベンジル基、ベンズヒドリル基、トリチル基等のアラルキル基等が挙げられ、特にメチル基、エチル基、tert-ブチル基、アリル基、ベンジル基、p-メトキシベンジル基やベンズヒドリル基等が好ましい。
当該反応では、化合物(XX)1モルに対して、還元剤を通常1モル乃至10モル、好ましくは1モル乃至3モル用いる。
用いられる還元剤としては、水素化リチウムアルミニウム、水素化ジイソブチルアルミニウム、水素化トリエチルホウ素リチウム、水素化ビス(2-メトキシエトキシ)アルミニウムナトリウムや水素化ホウ素等が挙げられる。
反応温度は、通常-100℃乃至200℃であり、好ましくは0℃乃至25℃である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、ジエチルエーテル、テトラヒドロフラン、1,4-ジオキサン、ジメトキシエタン、シクロヘキサン、1,3-ジメチルベンゼン、トルエン、メタノール、エタノール、1-プロパノールや2-プロパノール等の溶媒が好ましい。

ここで、上記式(XX)の保護基PG1は、その機能を有するものであれば特に限定されないが、例えばベンジル基、p-メトキシベンジル基、3,4-ジメトキシベンジル基、o-ニトロベンジル基、p-ニトロベンジル基、ベンズヒドリル基、トリチル基等のアラルキル基;例えばホルミル基、アセチル基、プロピオニル基、ブチリル基、ピバロイル基等の低級アルカノイル基;例えばベンゾイル基;例えばフェニルアセチル基、フェノキシアセチル基等のアリールアルカノイル基;例えばメトキシカルボニル基、エトキシカルボニル基、プロピルオキシカルボニル基、tert-ブトキシカルボニル基等の低級アルコキシカルボニル基;例えばベンジルオキシカルボニル基、p-ニトロベンジルオキシカルボニル基、フェネチルオキシカルボニル基等のアラルキルオキシカルボニル基;例えばトリメチルシリル基、tert-ブチルジメチルシリル基等の低級アルキルシリル基;例えばテトラヒドロピラニル基;例えばトリメチルシリルエトキシメチル基;例えばメチルスルホニル基、エチルスルホニル基等の低級アルキルスルホニル基等や;例えばベンゼンスルホニル基、p-トルエンスルホニル基等のアリールスルホニル基等が挙げられ、特にtert-ブトキシカルボニル基、メチルスルホニル基やp-トルエンスルホニル基等が好ましい。
保護基の導入法は、当該保護基の種類及び化合物の安定性等により異なるが、例えば文献記載の方法[プロテクティブ・グループス・イン・オーガニック・シンセシス(Protective Groups in Organic Synthesis)、第3版、T.W.グリーン(T.W.Greene)著、John Wiley & Sons社(1999年)参照]又はそれに準じる方法に従って合成できる。
なお、式(XXI)の化合物としては、例えばインドール-6-カルボン酸メチル、インドール-6-カルボン酸エチル、インドール-6-カルボン酸ベンジル、インドール-6-カルボン酸tert-ブチル、3-メチル-1H-インドール-6-カルボン酸メチル、3-メチル-1H-インドール-6-カルボン酸エチル、3-エチル-1H-インドール-6-カルボン酸メチル、3-イソプロピル-1H-インドール-6-カルボン酸メチル、1H-ピロロ[3,2-b]ピリジン-6-カルボン酸メチル、インダゾール-6-カルボン酸メチル、インダゾール-6-カルボン酸エチル、3-メチルインダゾール-6-カルボン酸メチル等が挙げられ、式(XXI)の化合物は市販品を用いるか、公知の方法若しくは実施例記載の方法又はそれに準じる方法を必要に応じ適宣組み合わせることにより製造することができる。

当該反応では、化合物(XXII)1モルに対して、化合物(XXIII)を通常1乃至10モル、好ましくは1乃至3モル用いる。
化合物(XXIII)としては、例えば酢酸メチル、酢酸エチル、酢酸tert-ブチル、プロピオン酸メチル、プロピオン酸エチル、プロピオン酸tert-ブチルやイソ酪酸メチル等が挙げられる。
用いられる塩基としては、リチウムジシクロヘキシルアミド、ナトリウムジシクロヘキシルアミド、リチウムヘキサメチルジシラジド、ナトリウムヘキサメチルジシラジド、カリウムヘキサメチルジシラジドやリチウムジイソプロピルアミド等が挙げられる。
用いられる塩基の量は、化合物(XXII)1モルに対して、通常1乃至10モル、好ましくは1乃至3モルである。
用いられるパラジウム触媒としては、例えば、Pd(PPh3)4、Pd(OAc)2、Pd(dba)2、Pd2(dba)3や、PdCl2(PPh3)2等が挙げられる。
用いられるパラジウム触媒の量は、化合物(XXII)1モルに対して、通常0.01乃至0.5モル、好ましくは0.05乃至0.2モルである。
用いられるホスフィン配位子としては、PPh3、P(o-tol)3、P(tert-Bu)3、2-[ジ(tert-ブチル)ホスフィノ]-1,1’-ビフェニル、2-[ジ(tert-ブチル)ホスフィノ]-2’-ジメチルアミノ-1,1’-ビフェニル、1,2,3,4,5-ペンタフェニル-1’-[ジ(tert-ブチル)ホスフィノ]フェロセン、2-[ジシクロヘキシルホスフィノ]-1,1’-ビフェニルや、2-[ジシクロヘキシルホスフィノ]-2’-ジメチルアミノ-1,1’-ビフェニル等が挙げられる。
反応温度は、通常0℃乃至80℃であり、好ましくは0℃乃至25℃である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、N,N-ジメチルホルムアミド、テトラヒドロフラン、1,4-ジオキサン、シクロヘキサン、1,3-ジメチルベンゼン、アセトニトリルやトルエン等の溶媒が好ましい。

上記式(IV)の脱離基Lは、上記化合物(IV)との反応により脱離して化合物(XXII)を生成するものであれば特に限定されないが、脱離基としては、ハロゲン原子(塩素原子、臭素原子等)、p-トルエンスルホニルオキシ基、ベンゼンスルホニルオキシ基、エタンスルホニルオキシ基やメタンスルホニルオキシ基等が挙げられ、臭素原子、塩素原子やp-トルエンスルホニルオキシ基等が好ましい。
用いられる化合物(IV)の量は、化合物(XXIV)1モルに対して、通常1乃至10モル、好ましくは1乃至3モルである。
用いられる塩基としては、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、炭酸水素ナトリウム、フッ化セシウム、水素化ナトリウムや水酸化カリウム等が挙げられ、炭酸カリウム、水素化ナトリウムや水酸化カリウム等が好ましい。
塩基の量は、化合物(XXIV)1モルに対して、通常1乃至10モル、好ましくは1乃至5モルである。
反応温度は、通常0℃乃至160℃であり、好ましくは25℃乃至100℃である。
反応時間は、通常1時間乃至24時間であり、好ましくは1時間乃至12時間である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、N-メチル-2-ピロリドン、クロロホルム、ジクロロメタン、テトラヒドロフラン、1,4-ジオキサン、アセトン、メチルエチルケトンやアセトニトリル等の溶媒が好ましい。
なお、式(XXIV)の化合物としては、例えば6-ブロモインドール、6-ブロモ-3-メチルインドール、6-ブロモ-3-エチルインドール、6-ブロモインダゾール、6-ブロモ-2,3-ジヒドロ-1H-インドール、6-ブロモ-2,3-ジヒドロ-3-メチル-1H-インドール-2-オン、6-ブロモ-2,3-ジヒドロ-3-メチル-1H-インドール、6-ブロモ-2,3-ジヒドロ-3,3-ジメチル-1H-インドール-2-オン、6-ブロモ-2,3-ジヒドロ-3,3-ジメチル-1H-インドール、6-ブロモ-3-メチル-1H-インダゾール、6-ブロモ-3-エチル-1H-インダゾール、6-ブロモ-3-プロピル-1H-インダゾール、6-ブロモ-3-イソプロピル-1H-インダゾール、6-ブロモ-3-シクロプロピル-1H-インダゾール、6-ブロモ-1H-インダゾール-3-カルボニトリル、6-クロロ-3-メチル-1H-ピラゾロ[3,4-b]ピリジン、6-ブロモ-3-メチル-1H-ピラゾロ[4,3-b]ピリジン、6-ブロモ-3-エチル-1H-ピラゾロ[4,3-b]ピリジン、6-ブロモ-3-プロピル-1H-ピラゾロ[4,3-b]ピリジン、6-ブロモ-3-シクロプロピル-1H-ピラゾロ[4,3-b]ピリジン、6-ブロモ-3-クロロ-1H-ピラゾロ[4,3-b]ピリジンや、6-クロロ-5-メトキシ-3-メチル-1H-インダゾール等が挙げられる。式(XXIV)の化合物は市販品を用いるか、公知の方法若しくは実施例記載の方法又はそれに準じる方法を必要に応じ適宣組み合わせることにより製造することができる。
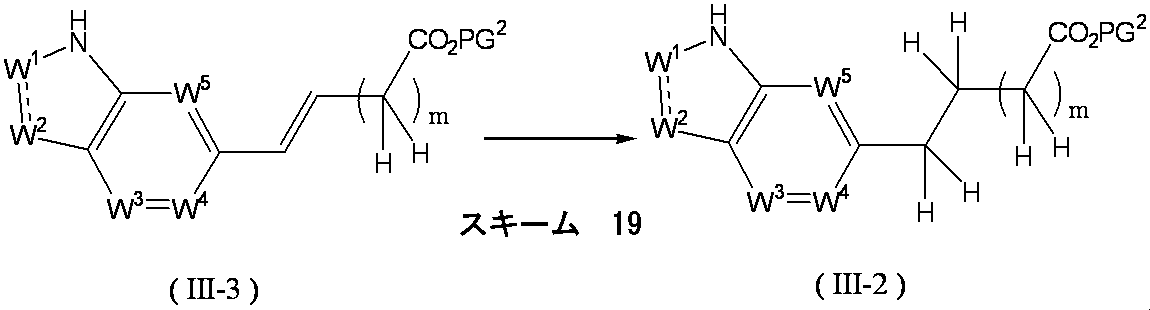
用いられる触媒としては、例えば、5%パラジウム炭素、10%パラジウム炭素、20%水酸化パラジウム、ラニーニッケル、白金や酸化白金等が挙げられる。
用いられる触媒の量は、化合物(III-3)1モルに対して、通常0.01乃至1モル、好ましくは0.05乃至0.2モルである。
反応温度は、通常0℃乃至200℃であり、好ましくは25℃乃至80℃である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、メタノール、エタノール、1-プロパノール、2-プロパノール、酢酸エチル、ジメチルホルムアミド、ジメチルアセトアミド、テトラヒドロフラン、1,4-ジオキサンやトルエン等の溶媒が好ましい。
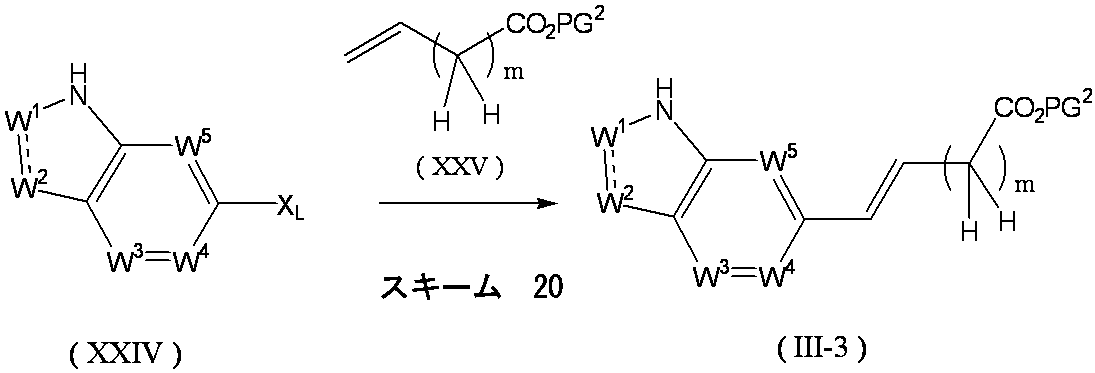
当該反応では、化合物(XXIV)1モルに対して、化合物(XXV)を通常1乃至10モル、好ましくは1乃至3モル用いる。
化合物(XXV)としては、例えばアクリル酸メチル、アクリル酸エチル、アクリル酸プロピル、アクリル酸tert-ブチル、2-ブテン酸メチル、3-ブテン酸メチル、アクリル酸、2-ブテン酸や3-ブテン酸等が挙げられる。
用いられる塩基としては、トリエチルアミン、酢酸ナトリウム、酢酸カリウム、炭酸水素ナトリウム、炭酸ナトリウム、フッ化セシウム、フッ化カリウムや炭酸カリウム等が挙げられる。
用いられる塩基の量は、化合物(XXIV)1モルに対して、通常1乃至10モル、好ましくは1乃至3モルである。
用いられるパラジウム触媒としては、例えば、Pd(PPh3)4、Pd(OAc)2、Pd2(dba)3や、PdCl2(PPh3)2等が挙げられる。
用いられるパラジウム触媒の量は、化合物(XXIV)1モルに対して、通常0.01乃至0.5モル、好ましくは0.05乃至0.2モルである。
用いられるホスフィン配位子としては、PPh3、P(o-tol)3、P(2-furyl)3、P(tert-Bu)3、2-[ジ(tert-ブチル)ホスフィノ]-1,1’-ビフェニル、2-[ジ(tert-ブチル)ホスフィノ]-2’-ジメチルアミノ-1,1’-ビフェニル、2-[ジシクロヘキシルホスフィノ]-1,1’-ビフェニルや、2-[ジシクロヘキシルホスフィノ]-2’-ジメチルアミノ-1,1’-ビフェニル等が挙げられる。
反応温度は、通常0℃乃至200℃であり、好ましくは25℃乃至160℃である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、クロロホルム、ジクロロメタン、テトラヒドロフラン、1,4-ジオキサン、アセトニトリルやトルエン等の溶媒が好ましい。
なお、式(XXIV)の化合物としては、例えば6-ブロモインドール、6-ブロモ-3-メチルインドール、6-ブロモ-3-エチルインドール、6-ブロモインダゾール、6-ブロモ-2,3-ジヒドロ-1H-インドール、6-ブロモ-2,3-ジヒドロ-3-メチル-1H-インドール-2-オン、6-ブロモ-2,3-ジヒドロ-3-メチル-1H-インドール、6-ブロモ-2,3-ジヒドロ-3,3-ジメチル-1H-インドール-2-オン、6-ブロモ-2,3-ジヒドロ-3,3-ジメチル-1H-インドール、6-ブロモ-3-メチル-1H-インダゾール、6-ブロモ-3-エチル-1H-インダゾール、6-ブロモ-3-プロピル-1H-インダゾール、6-ブロモ-3-イソプロピル-1H-インダゾール、6-ブロモ-3-シクロプロピル-1H-インダゾール、6-ブロモ-1H-インダゾール-3-カルボニトリル、6-クロロ-3-メチル-1H-ピラゾロ[3,4-b]ピリジン、6-ブロモ-3-メチル-1H-ピラゾロ[4,3-b]ピリジン、6-ブロモ-3-エチル-1H-ピラゾロ[4,3-b]ピリジン、6-ブロモ-3-プロピル-1H-ピラゾロ[4,3-b]ピリジン、6-ブロモ-3-シクロプロピル-1H-ピラゾロ[4,3-b]ピリジン、6-ブロモ-3-クロロ-1H-ピラゾロ[4,3-b]ピリジンや、6-クロロ-5-メトキシ-3-メチル-1H-インダゾール等が挙げられる。式(XXIV)の化合物は市販品を用いるか、公知の方法若しくは実施例記載の方法又はそれに準じる方法を必要に応じ適宣組み合わせることにより製造することができる。

当該反応では、化合物(XXVI)1モルに対して、アジ化物を通常1乃至10モル、好ましくは1乃至3モル用いる。
アジ化物としては、例えばアジ化リチウム、アジ化ナトリウム、アジ化カリウム等のアルカリ金属アジド、トリオクチルアジ化スズ等のトリアルキルスズアジド又はアジ化水素酸等が挙げられる。
用いられる塩としては、塩化アンモニウム、塩化亜鉛、臭化亜鉛や塩化アルミニウム等が挙げられる。
用いられる塩の量は、化合物(XXVI)1モルに対して、通常1乃至10モル、好ましくは1乃至3モルである。
反応温度は、通常0℃乃至200℃であり、好ましくは100℃乃至170℃である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、ジメチルホルムアミド、水、ジメチルアセトアミド、N-メチル-2-ピロリドン、テトラヒドロフラン、1,4-ジオキサンやトルエン等の溶媒が好ましい。

当該反応では、化合物(XXVII)1モルに対して、脱水剤を通常1乃至10モル、好ましくは1乃至3モル用いる。
脱水剤としては、例えば塩化チオニル、塩化オキザリル、シアヌル酸クロリド、五酸化リン、五塩化リン、無水酢酸やオキシ塩化リン等が挙げられる。
反応温度は、通常0℃乃至200℃であり、好ましくは0℃乃至25℃である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、ジクロロメタン、クロロホルム、テトラヒドロフラン、1,4-ジオキサン、アセトニトリルやトルエン等の溶媒が好ましい。

当該反応では、化合物(I)1モルに対して、ハロゲン化剤を通常1乃至10モル、好ましくは1乃至3モル用いる。
ハロゲン化剤としては、例えば塩化チオニル、オキザリルクロリド、三塩化リン、五塩化リンや塩化スルフリル等が挙げられる。
反応温度は、通常0℃乃至200℃であり、好ましくは0℃乃至25℃である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、ジクロロメタン、クロロホルム、テトラヒドロフラン、1,3-ジメチルベンゼン、1,4-ジオキサンやトルエン等の溶媒が好ましい。

当該反応では、化合物(XXII)1モルに対して、シアン化物を通常1乃至10モル、好ましくは1乃至3モル用いる。
シアン化物としては、例えばシアン化亜鉛、シアン化ナトリウムやシアン化カリウム等が挙げられる。
用いられるパラジウム触媒としては、例えば、Pd(PPh3)4、Pd(OAc)2、Pd(dba)2、Pd2(dba)3や、PdCl2(PPh3)2等が挙げられる。
用いられるパラジウム触媒の量は、化合物(XXII)1モルに対して、通常0.01乃至0.5モル、好ましくは0.05乃至0.2モルである。
用いられるホスフィン配位子としては、PPh3、P(o-tol)3、P(tert-Bu)3、2-[ジ(tert-ブチル)ホスフィノ]-1,1’-ビフェニル、2-[ジ(tert-ブチル)ホスフィノ]-2’-ジメチルアミノ-1,1’-ビフェニル、2-[ジシクロヘキシルホスフィノ]-1,1’-ビフェニルや、2-[ジシクロヘキシルホスフィノ]-2’-ジメチルアミノ-1,1’-ビフェニル等が挙げられる。
本反応には、必要に応じて亜鉛等の還元剤を添加することができる。
反応温度は、通常0℃乃至200℃であり、好ましくは25℃乃至130℃である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、ジメチルホルムアミド、テトラヒドロフラン、ジメトキシエタン、1,4-ジオキサン、アセトニトリルやトルエン等の溶媒が好ましい。
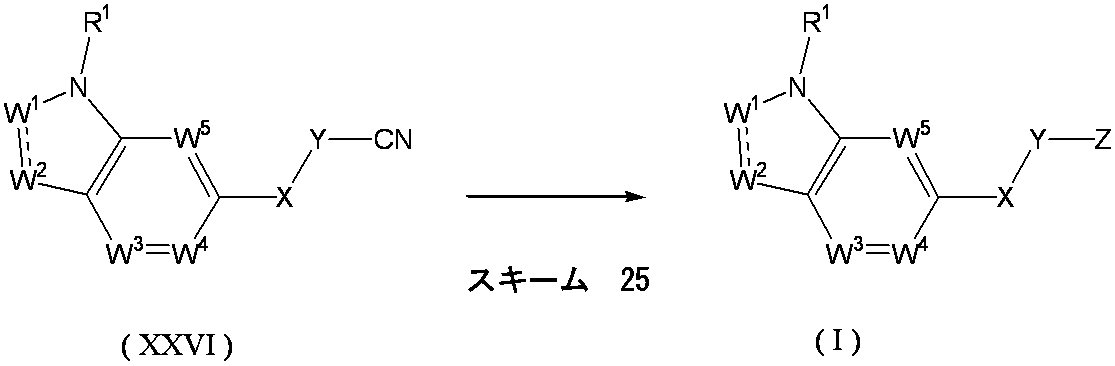
反応温度は、通常0℃乃至200℃であり、好ましくは0℃乃至160℃である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、メタノール、エタノール、1-プロパノール、2-プロパノール、1-ブタノール、2-ブタノール、tert-ブタノール、テトラヒドロフラン、1,4-ジオキサン、シクロヘキサン、1,3-ジメチルベンゼンやトルエン等の溶媒が好ましい。
塩基を用いた加溶媒分解に用いるアルコール溶媒としては、反応に支障が無いものであれば特に限定されないが、メタノール、エタノール、1-プロパノール、2-プロパノール、1-ブタノール、2-ブタノールやtert-ブタノール等が好ましい。
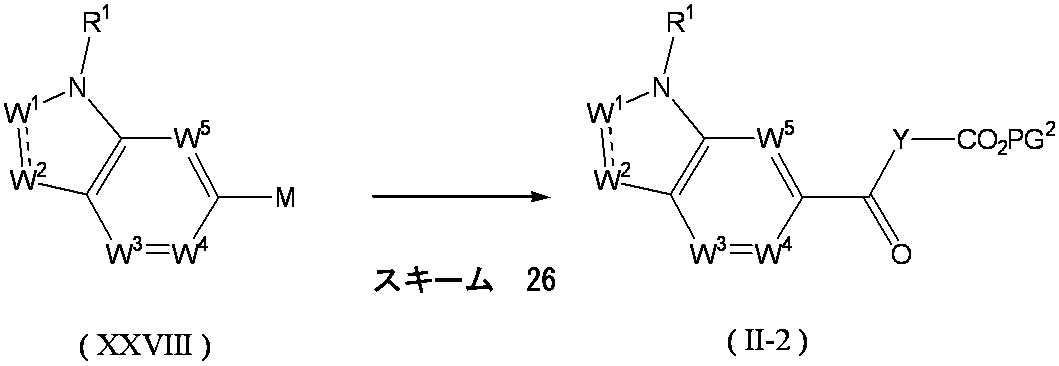
当該反応では、化合物(XXVIII)1モルに対して、酸クロリドを通常1乃至10モル、好ましくは1乃至3モル用いる。
酸クロリドとしては、例えばコハク酸モノメチルクロリド、コハク酸モノエチルクロリドやクロログリオキシル酸エチル等が挙げられる。
式(II-2)中のPG2は、酸クロリドに由来する基であり、例えばメチル基やエチル基等が挙げられる。
用いられる塩基としては、トリエチルアミンやジイソプロピルエチルアミン等が挙げられる。
用いられる塩基の量は、化合物(XXVIII)1モルに対して、通常1乃至10モル、好ましくは1乃至3モルである。
用いられるパラジウム触媒としては、例えば、Pd(PPh3)4、Pd(OAc)2、Pd(dba)2、Pd2(dba)3や、PdCl2(PPh3)2等が挙げられる。
用いられるパラジウム触媒の量は、化合物(XXVIII)1モルに対して、通常0.01乃至0.5モル、好ましくは0.05乃至0.2モルである。
用いられるホスフィン配位子としては、PPh3、P(o-tol)3、P(tert-Bu)3、2-[ジ(tert-ブチル)ホスフィノ]-1,1’-ビフェニル、2-[ジ(tert-ブチル)ホスフィノ]-2’-ジメチルアミノ-1,1’-ビフェニル、2-[ジシクロヘキシルホスフィノ]-1,1’-ビフェニルや、2-[ジシクロヘキシルホスフィノ]-2’-ジメチルアミノ-1,1’-ビフェニル等が挙げられる。
反応温度は、通常0℃乃至200℃であり、好ましくは25℃乃至130℃である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、ジメチルホルムアミド、テトラヒドロフラン、1,4-ジオキサン、アセトニトリルやトルエン等の溶媒が好ましい。
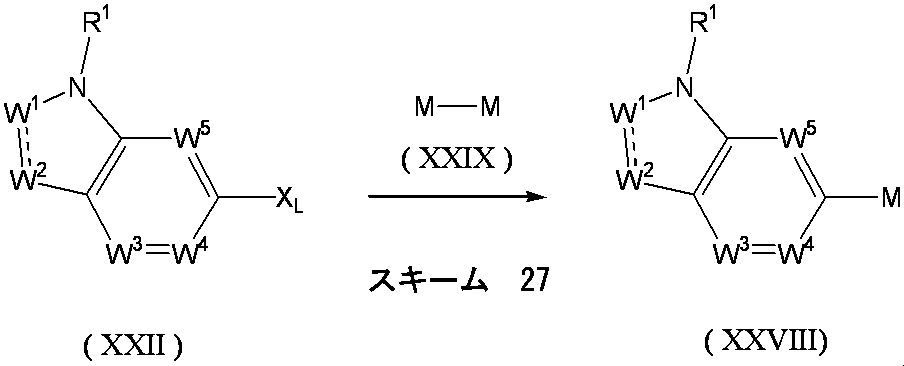
当該反応では、化合物(XXII)1モルに対して、化合物(XXIX)を通常1乃至10モル、好ましくは1乃至3モル用いる。
化合物(XXIX)としては、例えばビス(トリメチルスズ)、ビス(トリエチルスズ)、ビス(トリブチルスズ)やビスピナコラトジボロン等が挙げられる。
用いられる塩基としては、酢酸カリウムやトリメチルアミン等が挙げられる。
用いられる塩基の量は、化合物(XXII)1モルに対して、通常1乃至10モル、好ましくは1乃至3モルである。
用いられるパラジウム触媒としては、例えば、Pd(PPh3)4、Pd(OAc)2、Pd2(dba)3や、PdCl2(PPh3)2等が挙げられる。
用いられるパラジウム触媒の量は、化合物(XXII)1モルに対して、通常0.01乃至0.5モル、好ましくは0.05乃至0.2モルである。
用いられるホスフィン配位子としては、PPh3、P(o-tol)3、P(tert-Bu)3、2-[ジ(tert-ブチル)ホスフィノ]-1,1’-ビフェニル、2-[ジ(tert-ブチル)ホスフィノ]-2’-ジメチルアミノ-1,1’-ビフェニル、2-[ジシクロヘキシルホスフィノ]-1,1’-ビフェニルや、2-[ジシクロヘキシルホスフィノ]-2’-ジメチルアミノ-1,1’-ビフェニル等が挙げられる。
反応温度は、通常0℃乃至200℃であり、好ましくは25℃乃至130℃である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、ジメチルホルムアミド、テトラヒドロフラン、ジメトキシエタン、1,4-ジオキサン、アセトニトリルやトルエン等の溶媒が好ましい。

当該反応では、化合物(II-2)1モルに対して、フッ素化剤を通常1乃至10モル、好ましくは1乃至3モル用いる。
フッ素化剤としては、ジエチルアミノ硫黄トリフルオリド(DAST)、ビス(2-メトキシエチル)アミノ硫黄トリフルオリド、1,1,2,2-テトラフルオロエチル-N,N-ジメチルアミン、ジエチルアミノジフルオロスルフィニウムテトラフルオロボレート、モルホリノジフルオロスルフィニウムテトラフルオロボレートや、4-tert-ブチル-2,6-ジメチルフェニル硫黄トリフルオリド等が挙げられる。
反応温度は、通常0℃乃至200℃であり、好ましくは25℃乃至130℃である。
反応溶媒は、反応に支障が無いものであれば特に限定されないが、ジクロロメタン、テトラヒドロフラン、1,4-ジオキサン、1,2-ジメトキシエタン、アセトニトリルやトルエン等の溶媒が好ましい。
「製剤」には、経口製剤及び非経口製剤が含まれる。経口製剤としては、例えば、錠剤、カプセル剤、散剤や顆粒剤などであり、一方、非経口製剤としては、例えば、溶液若しくは懸濁液等の殺菌した液状の製剤、具体的には、注射剤や点滴剤などであり、好ましくは、静脈内注射剤や静脈内点滴剤である。
固体の製剤は、そのまま錠剤、カプセル剤、顆粒剤又は粉末の形態として製造することもできるが、適当な担体(添加物)を使用して製造することもできる。そのような担体(添加物)としては、例えば乳糖若しくはブドウ糖等の糖類;例えばトウモロコシ、小麦若しくは米等の澱粉類;例えばステアリン酸等の脂肪酸;例えばメタケイ酸アルミン酸マグネシウム若しくは無水リン酸カルシウム等の無機塩;例えばポリビニルピロリドン若しくはポリアルキレングリコール等の合成高分子;例えばステアリン酸カルシウム若しくはステアリン酸マグネシウム等の脂肪酸塩;例えばステアリルアルコール若しくはベンジルアルコール等のアルコール類;例えばメチルセルロース、カルボキシメチルセルロース、エチルセルロース若しくはヒドロキシプロピルメチルセルロース等の合成セルロース誘導体や;その他、ゼラチン、タルク、植物油、アラビアゴム等通常用いられる添加物等が挙げられる。
シリカゲルカラムクロマトグラフィーには、バイオタージ(Biotage)社製Biotage(登録商標)SNAP Cartridge KP-Silシリカゲルプレパックドカラム又は富士シリシア化学社製Chromatorex(登録商標)Q-PACK COOHシリカゲルプレパックドカラムを用いた。逆相分取液体クロマトグラフィーは、YMC社製CombiPrep Pro C18をカラムに用い、0.1%トリフルオロ酢酸水溶液及び0.1%トリフルオロ酢酸アセトニトリル溶液を移動相に用いた。
s:シングレット
d:ダブレット
t:トリプレット
q:カルテット
dd:ダブル ダブレット
dt:ダブル トリプレット
td:トリプル ダブレット
tt:トリプル トリプレット
ddd:ダブル ダブル ダブレット
ddt:ダブル ダブル トリプレット
dtd:ダブル トリプル ダブレット
tdd:トリプル ダブル ダブレット
tq:トリプル カルテット
m:マルチプレット
br:ブロード
DMSO-d6:重ジメチルスルホキシド
CDCl3:重クロロホルム
CD3OD:重メタノール
tBu:tert-ブチル基
1-ベンジル-1H-インドール-6-カルボン酸[1](以下、化合物[1]という)の合成

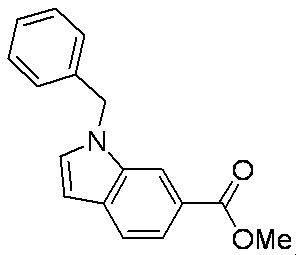
得られた残渣をシリカゲルカラムクロマトグラフィーにて精製することにより表題化合物(681mg)を無色油状物質として得た。
1H-NMR(400MHz,CDCl3)δ:8.09(1H,s),7.81(1H,dd,J=8.3,1.5Hz),7.66(1H,d,J=8.3Hz),7.33-7.25(4H,m),7.12-7.09(2H,m),6.59(1H,dd,J=3.2,0.7Hz),5.38(2H,s),3.91(3H,s).
工程(1)で得た化合物[1-1](681mg)のエタノール(30mL)溶液に1N-水酸化ナトリウム水溶液(10mL)を加えた後、50℃で18時間攪拌した。反応混合物を室温に戻し、1N-塩酸を加えて酸性にし、クロロホルムで抽出した。得られた有機層を無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィーにて精製することにより表題化合物(453mg)を白色固体として得た。
1H-NMR(400MHz,CDCl3)δ:8.17(1H,s),7.87(1H,dd,J=8.3,1.5Hz),7.69(1H,d,J=8.5Hz),7.35-7.24(3H,m),7.26(1H,d,J=0.5Hz),7.13(2H,d,J=7.1Hz),6.61(1H,d,J=3.2Hz),5.41(2H,s).
1-(2,6-ジメチルベンジル)-1H-インドール-6-カルボン酸[2](以下、化合物[2]という)の合成
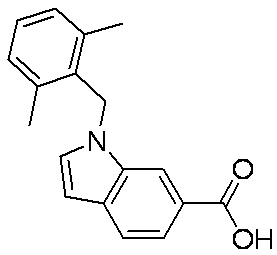

1H-NMR(400MHz,CDCl3)δ:8.30(1H,s),7.84(1H,dd,J=8.3,1.5Hz),7.65(1H,d,J=8.3Hz),7.23(1H,dd,J=8.4,6.8Hz),7.14(2H,d,J=7.6Hz),6.74(1H,d,J=3.2Hz),6.44(1H,dd,J=3.2,1.0Hz),5.32(2H,s),3.98(3H,s),2.26(6H,s).
1H-NMR(400MHz,CDCl3)δ:8.37(1H,s),7.91(1H,dd,J=8.4,1.3Hz),7.68(1H,d,J=8.3Hz),7.22(1H,dd,J=8.4,7.2Hz),7.14(2H,d,J=7.6Hz),6.78(1H,d,J=3.2Hz),6.47(1H,dd,J=3.0,0.9Hz),5.34(2H,s),2.27(6H,s).
1-(2,4,6-トリメチルベンジル)-1H-インドール-6-カルボン酸[3](以下、化合物[3]という)の合成

1H-NMR(400MHz,CDCl3)δ:8.38(1H,s),7.92(1H,dd,J=8.4,1.3Hz),7.68(1H,d,J=8.3Hz),6.96(2H,s),6.80(1H,d,J=3.2Hz),6.46(1H,dd,J=3.0,0.6Hz),5.30(2H,s),2.33(3H,s),2.23(6H,s).
2-(1-ベンジル-1H-インドール-6-イル)酢酸[4](以下、化合物[4]という)の合成
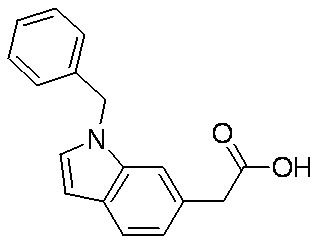

1H-NMR(400MHz,CDCl3)δ:8.69(1H,s),7.93(1H,dd,J=8.2,1.3Hz),7.80(2H,d,J=8.3Hz),7.71(1H,d,J=3.7Hz),7.56(1H,d,J=8.3Hz),7.24(2H,d,J=8.3Hz),6.69(1H,d,J=3.4Hz),3.97(3H,s),2.35(3H,s).

1H-NMR(400MHz,CDCl3)δ:7.99(1H,s),7.77(2H,d,J=10.0Hz),7.57(1H,d,J=2.9Hz),7.51(1H,d,J=8.1Hz),7.25-7.20(3H,m),6.64(1H,d,J=3.4Hz),4.79(2H,s),2.34(3H,s),1.80-1.70(1H,brs).

1H-NMR(400MHz,CDCl3)δ:8.03(1H,s),7.77(2H,d,J=8.5Hz),7.58(1H,d,J=3.7Hz),7.51(1H,d,J=8.1Hz),7.26(1H,dd,J=8.1,1.5Hz),7.23(2H,d,J=8.8Hz),6.64(1H,d,J=3.7Hz),4.73(2H,s),2.35(3H,s).

1H-NMR(400MHz,CDCl3)δ:7.95(1H,s),7.77(2H,d,J=8.3Hz),7.59(1H,d,J=3.7Hz),7.53(1H,d,J=8.1Hz),7.25(2H,d,J=7.8Hz),7.20(1H,dd,J=7.9,1.8Hz),6.65(1H,d,J=3.7Hz),3.87(2H,s),2.35(3H,s).
得られた固体をクロロホルム-メタノール混合溶媒に溶解し、無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮することにより表題化合物(980mg)を無色油状物質として得た。
1H-NMR(400MHz,CD3OD)δ:10.40(1H,s),7.47(1H,d,J=7.3Hz),7.30(1H,s),7.20-7.15(1H,m),6.93(1H,dd,J=8.2,2.1Hz),6.39(1H,s),3.65(2H,s).

1H-NMR(400MHz,CD3OD)δ:8.25-8.00(1H,br),7.59(1H,d,J=8.1Hz),7.33(1H,d,J=0.7Hz),7.19(1H,t,J=2.8Hz),7.04(1H,dd,J=8.1,1.2Hz),6.53(1H,s),3.73(2H,s),3.69(3H,s).
工程(6)で得た化合物[4-6](49mg)をアセトニトリル(2mL)に溶解し、粉末状の水酸化カリウム(19mg)及びベンジルクロリド(0.033mL)を室温で加えた後、マイクロウェーブ反応装置を用いて140℃で20分反応させた。この反応混合物にメタノール(1mL)及び1N-水酸化ナトリウム水溶液(1mL)を加え、マイクロウェーブ反応装置を用いて140℃で10分反応させた。反応混合物に4N-塩酸を加えて酸性にした後、酢酸エチルで抽出した。得られた有機層を無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィーにて精製することにより表題化合物(42mg)を白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:7.44(1H,d,J=8.3Hz),7.31(1H,s),7.28-7.10(6H,m),7.03(1H,dd,J=8.3,1.5Hz),6.41(1H,dd,J=3.2,1.0Hz),5.36(2H,s),3.53(2H,s).
ESI-MSfound:266[M+H]+
2-[1-(2,6-ジメチルベンジル)-1H-インドール-6-イル]酢酸[5](以下、化合物[5]という)の合成
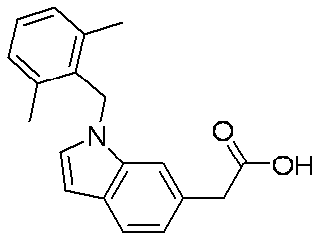
1H-NMR(400MHz,CD3OD)δ:7.52-7.44(2H,m),7.21-7.14(1H,m),7.10(2H,d,J=7.6Hz),7.00(1H,dd,J=8.2,1.1Hz),6.56(1H,d,J=3.2Hz),6.32(1H,d,J=3.2Hz),5.29(2H,s),3.72(2H,s),2.24(6H,s).
ESI-MSfound:294[M+H]+
2-[1-(2-メチルベンジル)-1H-インドール-6-イル]酢酸[6](以下、化合物[6]という)の合成

1H-NMR(400MHz,CD3OD)δ:7.51(1H,d,J=8.1Hz),7.23(1H,s),7.21-7.12(2H,m),7.08-7.02(2H,m),6.98(1H,dd,J=8.3,1.7Hz),6.68(1H,d,J=7.8Hz),6.45(1H,dd,J=3.2,0.7Hz),5.34(2H,s),3.64(2H,s),2.29(3H,s).
ESI-MSfound:280[M+H]+
2-[1-(2-クロロベンジル)-1H-インドール-6-イル]酢酸[7](以下、化合物[7]という)の合成

1H-NMR(400MHz,CD3OD)δ:7.53(1H,d,J=8.1Hz),7.43(1H,dd,J=8.1,1.2Hz),7.26-7.20(3H,m),7.12(1H,td,J=7.5,1.1Hz),7.00(1H,dd,J=8.2,1.3Hz),6.62(1H,dd,J=7.7,1.1Hz),6.50(1H,dd,J=3.2,0.7Hz),5.47(2H,s),3.63(2H,s).
ESI-MSfound:300[M+H]+
2-{1-[2-(トリフルオロメチル)ベンジル]-1H-インドール-6-イル}酢酸[8](以下、化合物[8]という)の合成
1H-NMR(400MHz,CD3OD)δ:7.73(1H,d,J=6.1Hz),7.50(1H,d,J=7.8Hz),7.42-7.30(2H,m),7.20-7.10(2H,m),7.08(1H,d,J=7.6Hz),6.60-6.48(2H,m),5.59(2H,s),3.50(2H,s).
ESI-MSfound:334[M+H]+
2-[1-(2-フルオロベンジル)-1H-インドール-6-イル]酢酸[9](以下、化合物[9]という)の合成

ESI-MSfound:284[M+H]+
2-[1-(3-フルオロベンジル)-1H-インドール-6-イル]酢酸[10](以下、化合物[10]という)の合成

ESI-MSfound:284[M+H]+
2-[1-(4-フルオロベンジル)-1H-インドール-6-イル]酢酸[11](以下、化合物[11]という)の合成

ESI-MSfound:284[M+H]+
2-[1-(2-クロロ-6-フルオロベンジル)-1H-インドール-6-イル]酢酸[12](以下、化合物[12]という)の合成

ESI-MSfound:318[M+H]+
2-[1-(2-クロロ-4-フルオロベンジル)-1H-インドール-6-イル]酢酸[13](以下、化合物[13]という)の合成
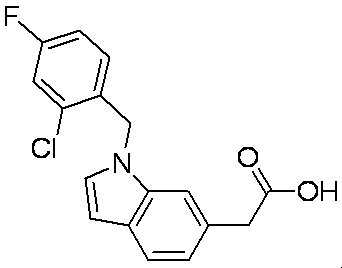
ESI-MSfound:318[M+H]+
2-[1-(2-クロロ-5-フルオロベンジル)-1H-インドール-6-イル]酢酸[14](以下、化合物[14]という)の合成
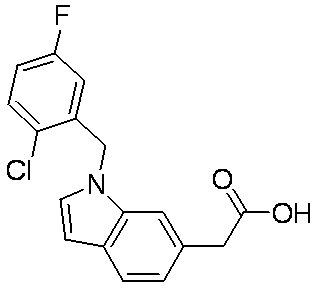
ESI-MSfound:318[M+H]+
2-[1-(3-クロロベンジル)-1H-インドール-6-イル]酢酸[15](以下、化合物[15]という)の合成
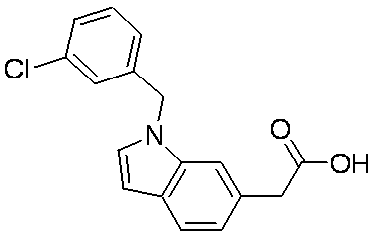
ESI-MSfound:300[M+H]+
2-[1-(4-クロロベンジル)-1H-インドール-6-イル]酢酸[16](以下、化合物[16]という)の合成

ESI-MSfound:300[M+H]+
2-[1-(2,6-ジクロロベンジル)-1H-インドール-6-イル]酢酸[17](以下、化合物[17]という)の合成

ESI-MSfound:334[M+H]+
2-[1-(2,3-ジクロロベンジル)-1H-インドール-6-イル]酢酸[18](以下、化合物[18]という)の合成
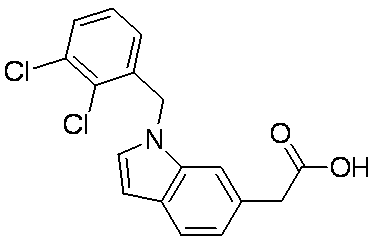
ESI-MSfound:334[M+H]+
2-{1-[(6-クロロベンゾ[d][1,3]ジオキソル-5-イル)メチル]-1H-インドール-6-イル}酢酸[19](以下、化合物[19]という)の合成

ESI-MSfound:344[M+H]+
2-[1-(2,4-ジクロロベンジル)-1H-インドール-6-イル]酢酸[20](以下、化合物[20]という)の合成

ESI-MSfound:334[M+H]+
2-[1-(2,5-ジクロロベンジル)-1H-インドール-6-イル]酢酸[21](以下、化合物[21]という)の合成

ESI-MSfound:334[M+H]+
2-{1-[2-フルオロ-6-(トリフルオロメチル)ベンジル]-1H-インドール-6-イル}酢酸[22](以下、化合物[22]という)の合成

ESI-MSfound:352[M+H]+
2-{1-[4-フルオロ-2-(トリフルオロメチル)ベンジル]-1H-インドール-6-イル}酢酸[23](以下、化合物[23]という)の合成

ESI-MSfound:352[M+H]+
2-[1-(4-エチルベンジル)-1H-インドール-6-イル]酢酸[24](以下、化合物[24]という)の合成

ESI-MSfound:294[M+H]+
2-[1-(2,4-ジフルオロベンジル)-1H-インドール-6-イル]酢酸[25](以下、化合物[25]という)の合成

ESI-MSfound:302[M+H]+
2-[1-(2,6-ジフルオロベンジル)-1H-インドール-6-イル]酢酸[26](以下、化合物[26]という)の合成

ESI-MSfound:302[M+H]+
2-[1-(2,5-ジフルオロベンジル)-1H-インドール-6-イル]酢酸[27](以下、化合物[27]という)の合成
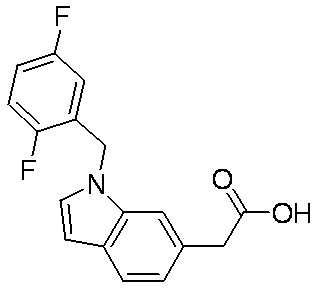
ESI-MSfound:302[M+H]+
2-[1-(2,3-ジフルオロベンジル)-1H-インドール-6-イル]酢酸[28](以下、化合物[28]という)の合成

ESI-MSfound:302[M+H]+
2-{1-[3-(トリフルオロメチル)ベンジル]-1H-インドール-6-イル}酢酸[29](以下、化合物[29]という)の合成

ESI-MSfound:334[M+H]+
2-{1-[4-(トリフルオロメチル)ベンジル]-1H-インドール-6-イル}酢酸[30](以下、化合物[30]という)の合成
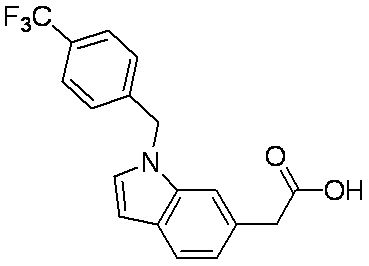
ESI-MSfound:334[M+H]+
2-[1-(2,4-ジメチルベンジル)-1H-インドール-6-イル]酢酸[31](以下、化合物[31]という)の合成

ESI-MSfound:294[M+H]+
2-[1-(2,5-ジメチルベンジル)-1H-インドール-6-イル]酢酸[32](以下、化合物[32]という)の合成

ESI-MSfound:294[M+H]+
2-[1-(3-メチルベンジル)-1H-インドール-6-イル]酢酸[33](以下、化合物[33]という)の合成
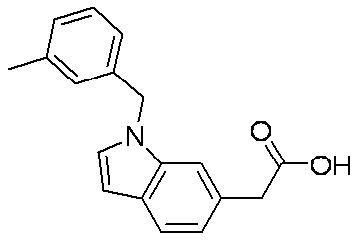
ESI-MSfound:280[M+H]+
2-[1-(4-メチルベンジル)-1H-インドール-6-イル]酢酸[34](以下、化合物[34]という)の合成

ESI-MSfound:280[M+H]+
2-[1-(ピリジン-4-イルメチル)-1H-インドール-6-イル]酢酸[35](以下、化合物[35]という)の合成

ESI-MSfound:267[M+H]+
2-[1-(2-メトキシベンジル)-1H-インドール-6-イル]酢酸[36](以下、化合物[36]という)の合成

ESI-MSfound:296[M+H]+
2-[1-(3-メトキシベンジル)-1H-インドール-6-イル]酢酸[37](以下、化合物[37]という)の合成

ESI-MSfound:296[M+H]+
2-[1-(4-メトキシベンジル)-1H-インドール-6-イル]酢酸[38](以下、化合物[38]という)の合成
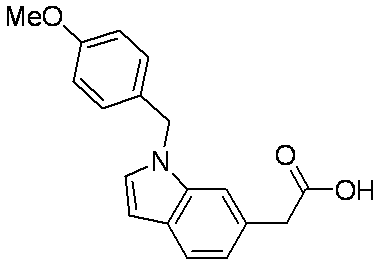
ESI-MSfound:296[M+H]+
2-[1-(ナフタレン-1-イルメチル)-1H-インドール-6-イル]酢酸[39](以下、化合物[39]という)の合成

ESI-MSfound:316[M+H]+
2-[1-(ナフタレン-2-イルメチル)-1H-インドール-6-イル]酢酸[40]以下、化合物[40]という)の合成

ESI-MSfound:316[M+H]+
2-{1-[(6-クロロピリジン-3-イル)メチル]-1H-インドール-6-イル}酢酸[41](以下、化合物[41]という)の合成

ESI-MSfound:301[M+H]+
2-{1-[(6-メチルピリジン-2-イル)メチル]-1H-インドール-6-イル}酢酸[42](以下、化合物[42]という)の合成
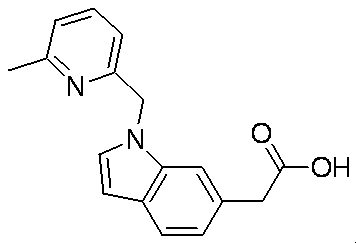
ESI-MSfound:281[M+H]+
2-[1-(ビフェニル-2-イルメチル)-1H-インドール-6-イル]酢酸[43](以下、化合物[43]という)の合成
ESI-MSfound:342[M+H]+
2-[1-(ビフェニル-3-イルメチル)-1H-インドール-6-イル]酢酸[44](以下、化合物[44]という)の合成

ESI-MSfound:342[M+H]+
2-[1-(ビフェニル-4-イルメチル)-1H-インドール-6-イル]酢酸[45](以下、化合物[45]という)の合成

ESI-MSfound:342[M+H]+
2-[1-(3-フェノキシベンジル)-1H-インドール-6-イル]酢酸[46](以下、化合物[46]という)の合成
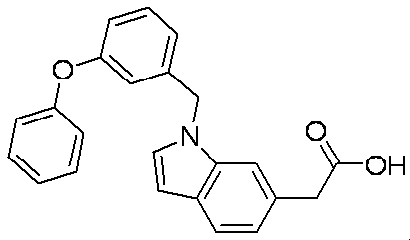
ESI-MSfound:358[M+H]+
2-[1-(3-フェニルプロピル)-1H-インドール-6-イル]酢酸[47](以下、化合物[47]という)の合成

ESI-MSfound:294[M+H]+
2-(1-イソプロピル-1H-インドール-6-イル)酢酸[48](以下、化合物[48]という)の合成

ESI-MSfound:218[M+H]+
2-(1-イソブチル-1H-インドール-6-イル)酢酸[49](以下、化合物[49]という)の合成

ESI-MSfound:232[M+H]+
2-[1-(シクロヘキシルメチル)-1H-インドール-6-イル]酢酸[50](以下、化合物[50]という)の合成

ESI-MSfound:272[M+H]+
1-(2,6-ジメチルベンジル)-1H-ベンズイミダゾール-6-カルボン酸[51](以下、化合物[51]という)の合成


1H-NMR(400MHz,CDCl3)δ:8.19(1H,d,J=2.2Hz),8.01(1H,dd,J=8.5,1.7Hz),7.80(1H,d,J=8.5Hz),7.52(1H,s),7.24-7.22(2H,m),7.13-7.11(4H,m),5.49(2H,s),5.31(2H,s),2.48(6H,s),2.26(6H,s).
1H-NMR(400MHz,DMSO-d6)δ:8.16(1H,d,J=1.2Hz),7.92(1H,s),7.83(1H,dd,J=8.5,1.5Hz),7.71(1H,d,J=8.5Hz),7.22(1H,dd,J=8.3,6.8Hz),7.14(2H,d,J=7.6Hz),5.52(2H,s),2.26(6H,s).
2-[1-(2,6-ジメチルベンジル)-1H-ベンズイミダゾール-6-イル]酢酸[52](以下、化合物[52]という)の合成


1H-NMR(400MHz,CDCl3)δ:7.78(1H,d,J=8.3Hz),7.55(1H,s),7.38(1H,s),7.29-7.26(2H,m),7.15(2H,d,J=7.3Hz),5.28(2H,s),4.88(2H,d,J=4.9Hz),2.29(6H,s),1.95(1H,s).

1H-NMR(400MHz,CDCl3)δ:7.79(1H,d,J=7.6Hz),7.49(1H,s),7.43(1H,s),7.27(1H,t,J=7.1Hz),7.21-7.15(3H,m),5.29(2H,s),3.93(2H,s),2.29(6H,s).
反応混合物に1N-塩酸を加えて酸性にし、析出した固体を濾取することにより表題化合物(81mg)を白色固体として得た。
1H-NMR(400MHz,DMSO-d6)δ:7.63(1H,s),7.57(1H,d,J=8.3Hz),7.43(1H,s),7.21(1H,dd,J=8.2,6.7Hz),7.13-7.09(3H,m),5.38(2H,s),3.64(2H,s),2.24(6H,s).
1-(2,6-ジメチルベンジル)-1H-インダゾール-6-カルボン酸[53](以下、化合物[53]という)の合成
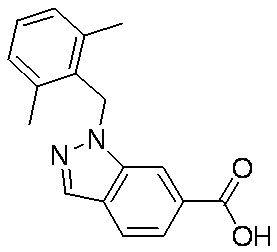

1H-NMR(400MHz,CDCl3)δ:8.02(1H,d,J=1.0Hz),7.87-7.86(1H,m),7.77(1H,dd,J=8.4,1.3Hz),7.71(1H,dd,J=8.5,0.7Hz),7.24(1H,dd,J=8.2,6.7Hz),7.14(2H,d,J=7.6Hz),7.08(1H,dd,J=8.1,7.1Hz),6.96(2H,d,J=7.6Hz),5.59(2H,s),5.42(2H,s),2.44(6H,s),2.25(6H,s).
工程(1)で得られた化合物[53-1](110mg)より実施例51の工程(2)の方法に準じて表題化合物(51mg)を白色固体として得た。
1H-NMR(400MHz,DMSO-d6)δ:8.33(1H,d,J=1.2Hz),8.06(1H,d,J=1.0Hz),7.77(1H,dd,J=8.4,0.6Hz),7.70(1H,dd,J=8.3,1.2Hz),7.14-7.12(1H,m),7.04(2H,d,J=7.6Hz),5.62(2H,s),2.28(6H,s).
2-[1-(2,6-ジメチルベンジル)-1H-インダゾール-6-イル]酢酸[54](以下、化合物[54]という)の合成

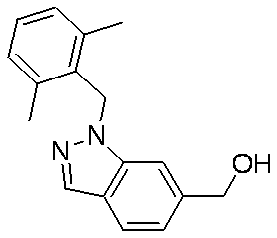
1H-NMR(400MHz,CDCl3)δ:7.96(1H,s),7.69(1H,d,J=8.1Hz),7.27-7.26(1H,m),7.19-7.17(1H,m),7.11-7.09(3H,m),5.54(2H,s),4.79(2H,d,J=5.9Hz),2.32(6H,s),1.82(1H,t,J=5.6Hz).
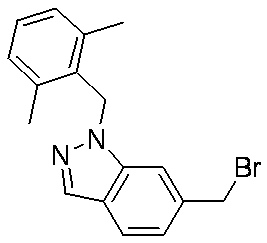
1H-NMR(400MHz,CDCl3)δ:7.97(1H,d,J=1.0Hz),7.69(1H,d,J=8.3Hz),7.26-7.25(1H,m),7.22-7.16(2H,m),7.11(2H,d,J=7.3Hz),5.54(2H,s),4.60(2H,s),2.33(6H,s).
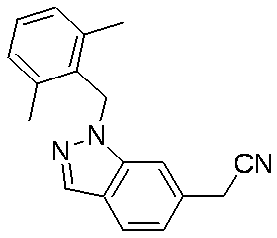
1H-NMR(400MHz,CDCl3)δ:7.98(1H,d,J=1.0Hz),7.71(1H,dd,J=8.3,0.7Hz),7.22-7.18(2H,m),7.11(2H,d,J=7.6Hz),7.02(1H,dd,J=8.5,1.7Hz),5.57(2H,s),3.84(2H,s),2.32(6H,s).
1H-NMR(400MHz,DMSO-d6)δ:7.94(1H,s),7.65(1H,d,J=8.3Hz),7.60(1H,s),7.11(1H,dd,J=8.3,6.6Hz),7.04-7.03(3H,m),5.49(2H,s),3.65(2H,s),2.27(6H,s).
(E)-3-[1-(2,6-ジメチルベンジル)-1H-インドール-6-イル]アクリル酸[55](以下、化合物[55]という)の合成

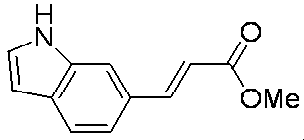
1H-NMR(400MHz,CDCl3)δ:8.33(1H,s),7.83(1H,d,J=15.6Hz),7.64(1H,d,J=6.3Hz),7.55(1H,s),7.36(1H,d,J=8.3Hz),7.31-7.29(1H,m),6.58(1H,t,J=3.9Hz),6.46(1H,dd,J=15.9,1.5Hz),3.82(3H,s).
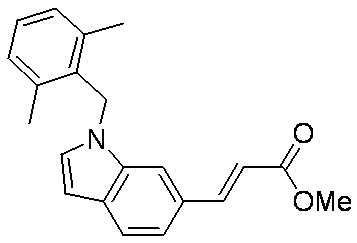
1H-NMR(400MHz,CDCl3)δ:7.88(1H,d,J=15.9Hz),7.61-7.59(2H,m),7.36(1H,dd,J=8.7,1.3Hz),7.21(1H,dd,J=8.7,6.2Hz),7.12-7.09(2H,m),6.68(1H,d,J=3.4Hz),6.49(1H,d,J=15.9Hz),6.40(1H,d,J=3.2Hz),5.23(2H,s),3.81(3H,s),2.24(6H,s).
工程(2)で得られた化合物[55-2](96mg)のメタノール(3mL)溶液に1N-水酸化ナトリウム水溶液(3mL)を室温で加えた後、60℃で18時間攪拌した。反応混合物を室温に戻し、1N-塩酸を加えて酸性にし、析出した固体を濾取することにより表題化合物(81mg)を黄色固体として得た。
1H-NMR(400MHz,CDCl3)δ:7.99(1H,d,J=15.4Hz),7.65-7.64(2H,m),7.41(1H,dd,J=8.3,1.7Hz),7.26-7.22(1H,m),7.14(2H,d,J=7.8Hz),6.71(1H,d,J=3.2Hz),6.52(1H,d,J=15.6Hz),6.43(1H,d,J=3.2Hz),5.28(2H,s),2.27(6H,s).
3-[1-(2,6-ジメチルベンジル)-1H-インドール-6-イル]プロピオン酸[56](以下、化合物[56]という)の合成
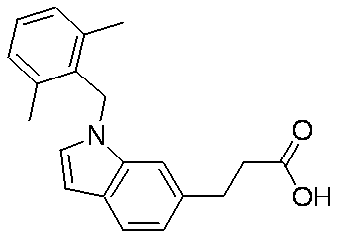

1H-NMR(400MHz,CDCl3)δ:8.14(1H,s),7.57(1H,d,J=8.3Hz),7.23(1H,s),7.17-7.16(1H,m),6.98(1H,dd,J=8.1,1.5Hz),6.53-6.51(1H,m),3.68(3H,s),3.07(2H,t,J=7.8Hz),2.70(2H,t,J=7.8Hz).

1H-NMR(400MHz,CDCl3)δ:7.57(1H,d,J=8.1Hz),7.34(1H,s),7.23(1H,dd,J=8.2,6.7Hz),7.14(2H,d,J=7.6Hz),7.02(1H,dd,J=8.1,1.5Hz),6.57(1H,d,J=3.2Hz),6.38(1H,dd,J=3.2,0.7Hz),5.23(2H,s),3.72(3H,s),3.15(2H,t,J=7.9Hz),2.76(2H,t,J=7.9Hz),2.28(6H,s).
工程(2)で得られた化合物[56-2](75mg)のメタノール(3mL)溶液に1N-水酸化ナトリウム水溶液(3mL)を室温で加えた後、60℃で18時間攪拌した。反応混合物を室温に戻し、1N-塩酸を加えて酸性にし、析出した固体を濾取することにより表題化合物(61mg)を赤色固体として得た。
1H-NMR(400MHz,CDCl3)δ:7.56(1H,d,J=8.3Hz),7.33(1H,s),7.22(1H,dd,J=8.1,6.8Hz),7.12(2H,d,J=7.6Hz),7.02(1H,dd,J=8.1,1.5Hz),6.57(1H,d,J=3.2Hz),6.37(1H,dd,J=3.2,0.7Hz),5.22(2H,s),3.15(2H,t,J=7.9Hz),2.80(2H,t,J=7.8Hz),2.26(6H,s).
3-[1-(2,6-ジメチルベンジル)-1H-インドール-6-イル]-3-ヒドロキシプロピオン酸[57](以下、化合物[57]という)の合成

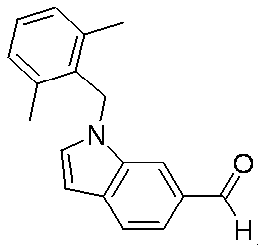
1H-NMR(400MHz,CDCl3)δ:10.11(1H,s),8.08(1H,s),7.73(1H,d,J=8.1Hz),7.68(1H,dd,J=8.2,1.3Hz),7.24(1H,dd,J=7.4,6.5Hz),7.14(2H,d,J=7.6Hz),6.83(1H,d,J=3.2Hz),6.48(1H,d,J=2.9Hz),5.34(2H,s),2.27(6H,s).

1H-NMR(400MHz,CDCl3)δ:7.62-7.59(2H,m),7.22(1H,dd,J=8.1,6.8Hz),7.13-7.11(3H,m),6.59(1H,d,J=3.2Hz),6.38(1H,dd,J=3.2,0.7Hz),5.35-5.31(1H,m),5.24(2H,s),4.22(2H,q,J=7.1Hz),3.30(1H,s),2.92-2.80(2H,m),2.26(6H,s),1.29(3H,t,J=7.2Hz).
工程(2)で得られた化合物[57-2](84mg)のメタノール(1mL)溶液に1N-水酸化ナトリウム水溶液(1mL)を室温で加えた後、60℃で18時間攪拌した。反応混合物を室温に戻し、1N-塩酸を加えて酸性にし、析出した固体を濾取することにより表題化合物(70mg)を白色固体として得た。
1H-NMR(400MHz,CDCl3)δ:7.62(1H,d,J=8.1Hz),7.57(1H,s),7.22(1H,dd,J=8.1,7.1Hz),7.15-7.11(3H,m),6.61(1H,d,J=3.2Hz),6.39(1H,d,J=3.4Hz),5.36(1H,dd,J=8.7,3.5Hz),5.25(2H,s),2.98(1H,dd,J=16.3,9.3Hz),2.89(1H,dd,J=16.5,3.3Hz),2.26(6H,s).
6-[1-(2,6-ジメチルベンジル)-1H-インドール-6-イル]ヘキサン酸[58](以下、化合物[58]という)の合成
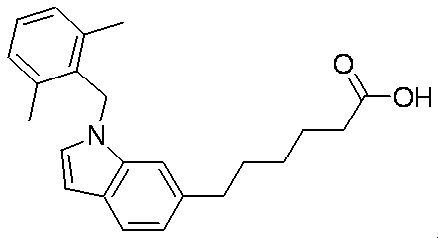
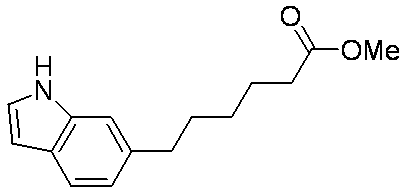
得られた有機層を無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮した。得られた残渣のメタノール(3mL)溶液に5%パラジウム炭素(184mg)を加え、水素雰囲気下、室温で1時間攪拌した。パラジウム炭素を濾別した後、濾液を減圧濃縮することにより表題化合物(182mg)を無色液体として得た。
1H-NMR(400MHz,CDCl3)δ:8.07(1H,s),7.55(1H,d,J=8.1Hz),7.19(1H,s),7.16-7.15(1H,m),6.96(1H,d,J=8.1Hz),6.51(1H,s),3.66(3H,s),2.72(2H,t,J=7.6Hz),2.31(2H,t,J=7.6Hz),1.72-1.63(4H,m),1.42-1.35(2H,m).

1H-NMR(400MHz,CDCl3)δ:7.53(1H,d,J=8.1Hz),7.27(1H,s),7.22-7.20(1H,m),7.11(2H,d,J=7.3Hz),6.98(1H,d,J=8.8Hz),6.53(1H,d,J=3.7Hz),6.35(1H,d,J=3.4Hz),5.21(2H,s),3.67(3H,s),2.78(2H,t,J=7.7Hz),2.33(2H,t,J=7.7Hz),2.26(6H,s),1.77-1.66(4H,m),1.46-1.39(2H,m).
工程(2)で得られた化合物[58-2](169mg)のメタノール(3mL)溶液に1N-水酸化ナトリウム水溶液(3mL)を室温で加えた後、60℃で2時間攪拌した。反応混合物を室温に戻し、1N-塩酸を加えて酸性にし、析出した固体を濾取することにより表題化合物(120mg)を赤色固体として得た。
1H-NMR(400MHz,CDCl3)δ:7.55(1H,d,J=8.1Hz),7.28(1H,s),7.24-7.20(1H,m),7.12(2H,d,J=7.6Hz),6.99(1H,dd,J=8.2,1.3Hz),6.55(1H,d,J=3.2Hz),6.36(1H,d,J=3.2Hz),5.22(2H,s),2.80(2H,t,J=7.7Hz),2.38(2H,t,J=7.4Hz),2.26(6H,s),1.79-1.68(4H,m),1.50-1.42(2H,m).
4-[1-(2,6-ジメチルベンジル)-1H-インドール-6-イル]-4-オキソブタン酸[59](以下、化合物[59]という)の合成

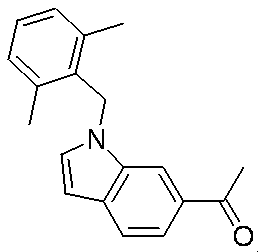
1H-NMR(400MHz,CDCl3)δ:8.20(1H,d,J=3.7Hz),7.76(1H,dd,J=8.3,1.5Hz),7.66(1H,dd,J=8.3,0.7Hz),7.25-7.21(1H,m),7.13(2H,d,J=7.6Hz),6.78(1H,d,J=2.9Hz),6.44(1H,dd,J=3.2,0.7Hz),5.32(2H,s),2.70(3H,s),2.26(6H,s).
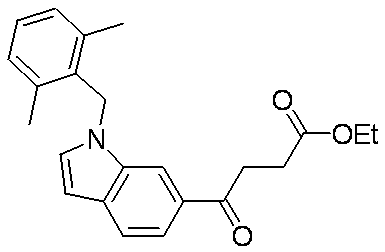
1H-NMR(400MHz,CDCl3)δ:8.23(1H,s),7.80(1H,dd,J=8.4,1.6Hz),7.66(1H,dd,J=8.3,0.7Hz),7.26-7.21(1H,m),7.13(2H,d,J=7.3Hz),6.77(1H,d,J=3.4Hz),6.44(1H,dd,J=3.2,0.7Hz),5.32(2H,s),4.19(2H,q,J=7.1Hz),3.45(2H,t,J=6.8Hz),2.81(2H,t,J=6.8Hz),2.26(6H,s),1.29(3H,t,J=7.2Hz).
工程(2)で得られた化合物[59-2](496mg)のメタノール(10mL)溶液に1N-水酸化ナトリウム水溶液(10mL)を加えた後、60℃で2時間攪拌した。
反応混合物を室温に戻し、1N-塩酸を加えて酸性にし、析出した固体を濾取することにより表題化合物(425mg)を白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:8.25(1H,s),7.76(1H,dd,J=8.3,1.5Hz),7.63(1H,d,J=8.3Hz),7.23-7.19(1H,m),7.13(2H,d,J=7.6Hz),6.87(1H,d,J=3.2Hz),6.46(1H,d,J=3.2Hz),5.44(2H,d,J=2.0Hz),3.41(2H,t,J=6.3Hz),2.73(2H,t,J=6.5Hz),2.26(6H,s).
4-[1-(2,6-ジメチルベンジル)-1H-インドール-6-イル]ブタン酸[60](以下、化合物[60]という)の合成

反応液を室温に戻した後、反応混合物に水を加え、クロロホルムで抽出した。得られた有機層を無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィーにて精製することにより表題化合物(35mg)を黄色固体として得た。
1H-NMR(400MHz,CDCl3)δ:7.55(1H,d,J=8.8Hz),7.28(1H,s),7.23-7.19(1H,m),7.11(2H,d,J=7.8Hz),6.99(1H,d,J=7.8Hz),6.54(1H,d,J=3.2Hz),6.35(1H,d,J=3.2Hz),5.21(2H,s),2.85(2H,t,J=7.3Hz),2.44(2H,t,J=7.3Hz),2.26(6H,s),2.11-2.03(2H,m).
1-(2,6-ジメチルベンジル)-3-メチル-1H-インドール-6-カルボン酸[61](以下、化合物[61]という)の合成

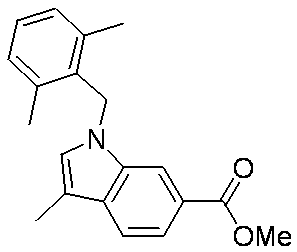
1H-NMR(400MHz,CDCl3)δ:8.24(1H,dd,J=1.3,0.6Hz),7.83(1H,dd,J=8.3,1.5Hz),7.58(1H,dd,J=8.4,0.6Hz),7.23(1H,dd,J=8.3,6.8Hz),7.13(2H,d,J=7.6Hz),6.51(1H,s),5.26(2H,s),3.97(3H,s),2.26(6H,s),2.23(3H,s).
工程(1)で得られた化合物[61-1](77mg)のメタノール(1mL)溶液に1N-水酸化ナトリウム水溶液(1mL)を室温で加えた後、60℃で10時間攪拌した。反応混合物を室温に戻し、1N-塩酸を加えて酸性にし、析出した固体を濾取することにより表題化合物(73mg)を黄色固体として得た。
1H-NMR(400MHz,CDCl3)δ:8.32(1H,s),7.91(1H,dd,J=8.3,1.5Hz),7.62(1H,d,J=8.5Hz),7.24-7.22(1H,m),7.14(2H,d,J=7.6Hz),6.55(1H,d,J=1.0Hz),5.29(2H,s),5.28(6H,s),2.24(3H,s).
ESI-MSfound:294[M+H]+
1-(2,6-ジメチルベンジル)-3-メチル-1H-インドール-6-カルボン酸カリウム[62](以下、化合物[62]という)の合成

1H-NMR(400MHz,CD3OD)δ:8.19(1H,s),7.75(1H,d,J=8.3,1.5Hz),7.45(1H,dd,J=8.3,0.7Hz),7.19-7.17(1H,m),7.10(2H,d,J=7.6Hz),6.40(1H,s),5.30(2H,s),2.24(6H,s),2.19(3H,s).
ESI-MSfound:294[M-K+2H]+
[1-(2,6-ジメチルベンジル)-3-メチル-1H-インドール-6-イル]メタノール[63](以下、化合物[63]という)の合成
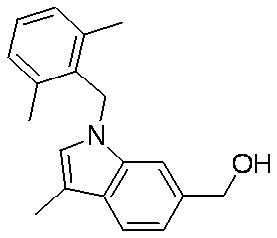
1H-NMR(400MHz,CDCl3)δ:7.56(1H,d,J=7.8Hz),7.48(1H,s),7.24-7.20(1H,m),7.15-7.11(3H,m),6.37(1H,s),5.19(2H,s),4.86(2H,d,J=5.6Hz),2.27(6H,s),2.22(3H,s),1.66(1H,t,J=6.0Hz).
2-[1-(2,6-ジメチルベンジル)-3-メチル-1H-インドール-6-イル]酢酸[64](以下、化合物[64]という)の合成


1H-NMR(400MHz,CDCl3)δ:7.98(1H,s),7.75(2H,d,J=8.3Hz),7.44(1H,d,J=8.1Hz),7.31-7.26(2H,m),7.21(2H,d,J=8.1Hz),4.81(2H,d,J=5.1Hz),2.34(3H,s),2.24(3H,s),1.77(1H,t,J=5.4Hz).
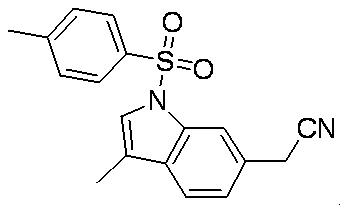
1H-NMR(400MHz,CDCl3)δ:7.93(1H,s),7.74(2H,d,J=8.5Hz),7.45(1H,d,J=8.1Hz),7.32(1H,s),7.23-7.21(3H,m),3.86(2H,s),2.34(3H,s),2.24(3H,s).

1H-NMR(400MHz,CDCl3)δ:7.89(1H,s),7.53(1H,d,J=7.8Hz),7.28(1H,s),7.05(1H,d,J=8.1Hz),6.96(1H,s),3.74(2H,s),3.69(3H,s),2.33(3H,s).

1H-NMR(400MHz,CDCl3)δ:7.53(1H,d,J=7.6Hz),7.38(1H,s),7.24-7.21(1H,m),7.13(2H,d,J=7.3Hz),7.07(1H,d,J=7.6Hz),6.34(1H,s),5.18(2H,s),3.81(2H,s),3.72(3H,s),2.27(6H,s),2.21(3H,s).
工程(4)で得られた化合物[64-4](268mg)のメタノール(5mL)溶液に1N-水酸化ナトリウム水溶液(5mL)を室温で加えた後、60℃で2時間攪拌した。反応混合物を室温に戻し、1N-塩酸を加えて酸性にし、析出した固体を濾取することにより表題化合物(226mg)を黄色固体として得た。
1H-NMR(400MHz,CDCl3)δ:7.53(1H,d,J=8.1Hz),7.37(1H,s),7.24-7.20(1H,m),7.12(2H,d,J=8.1Hz),7.07(1H,d,J=8.1Hz),6.34(1H,s),5.17(2H,s),3.82(2H,s),2.26(6H,s),2.20(3H,s).ESI-MSfound:308[M+H]+
2-[1-(2,6-ジメチルベンジル)-3-メチル-1H-インドール-6-イル]酢酸カリウム[65](以下、化合物[65]という)の合成
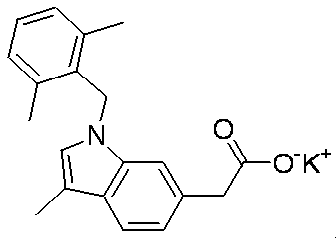
1H-NMR(400MHz,CD3OD)δ:7.46(1H,s),7.37(1H,d,J=8.1Hz),7.19-7.15(1H,m),7.09(2H,d,J=7.6Hz),7.06(1H,dd,J=8.1,1.5Hz),6.23-6.22(1H,m),5.22(2H,s),3.61(2H,s),2.23(6H,s),2.15(3H,s).
ESI-MSfound:308[M-K+2H]+
2-[1-(2,6-ジメチルベンジル)-3-メチル-1H-インダゾール-6-イル]酢酸[66](以下、化合物[66]という)の合成


1H-NMR(400MHz,CDCl3)δ:7.47(1H,d,J=8.8Hz),7.22-7.14(2H,m),7.11-7.09(3H,m),5.45(2H,s),2.51(3H,s),2.33(6H,s).

1H-NMR(400MHz,CDCl3)δ:7.52(1H,d,J=7.8Hz),7.18-7.15(1H,m),7.07(2H,d,J=7.3Hz),6.91(1H,dd,J=8.3,1.2Hz),6.75(1H,s),5.96-5.85(1H,m),5.47(2H,s),5.07-5.02(2H,m),3.40(2H,d,J=6.6Hz),2.52(3H,s),2.33(6H,s).
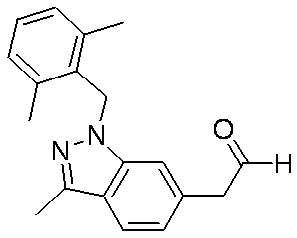
1H-NMR(400MHz,CDCl3)δ:9.68(1H,t,J=2.4Hz),7.60(1H,d,J=7.8Hz),7.20-7.16(1H,m),7.08(2H,d,J=7.8Hz),6.90(1H,d,J=8.3Hz),6.74(1H,s),5.49(2H,s),3.68(2H,d,J=2.0Hz),2.53(3H,s),2.32(6H,s).
工程(3)で得られた化合物[66-3](149mg)のtert-ブタノール(10mL)と水(5mL)溶液に2-メチル-2-ブテン(0.25mL)、リン酸二水素ナトリウム2水和物(86mg)及び亜塩素酸ナトリウム(175mg)を室温で加えて、室温で1時間攪拌した。反応混合物に塩酸を加え、クロロホルムで抽出し、得られた有機層を無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィーにて精製することにより表題化合物(126mg)を白色固体として得た。
1H-NMR(400MHz,CDCl3)δ:7.58(1H,d,J=8.3Hz),7.18-7.15(1H,m),7.07(2H,d,J=7.3Hz),7.00(1H,dd,J=8.3,1.5Hz),6.88(1H,s),5.49(2H,s),3.68(2H,s),2.53(3H,s),2.33(6H,s).
ESI-MSfound:309[M+H]+
2-(1-ベンジル-3-クロロ-1H-インドール-6-イル)酢酸[67](以下、化合物[67]という)の合成

反応混合物に4N-塩酸を加えて酸性にした後、酢酸エチルで抽出した。得られた有機層を無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィーにて精製することにより表題化合物(19mg)を赤色固体として得た。
1H-NMR(400MHz,CD3OD)δ:7.47(1H,d,J=8.3Hz),7.31-7.22(5H,m),7.18-7.12(2H,m),7.07(1H,dd,J=8.2,1.1Hz),5.33(2H,s),3.66(2H,s).
ESI-MSfound:300[M+H]+
2-[3-クロロ-1-(2,6-ジメチルベンジル)-1H-インドール-6-イル]酢酸[68](以下、化合物[68]という)の合成

1H-NMR(400MHz,CD3OD)δ:7.51(1H,s),7.46(1H,d,J=8.1Hz),7.21(1H,t,J=7.7Hz),7.17-7.08(3H,m),6.51(1H,s),5.30(2H,s),3.74(2H,s),2.25(6H,s).
ESI-MSfound:328[M+H]+
1-(2,6-ジメチルベンジル)-2-メチル-1H-インドール-6-カルボン酸[69](以下、化合物[69]という)の合成


1H-NMR(400MHz,CDCl3)δ:8.62(1H,s),8.15(1H,dd,J=7.8,1.2Hz),7.45(1H,d,J=7.8Hz),3.96(3H,s),2.67(3H,s).
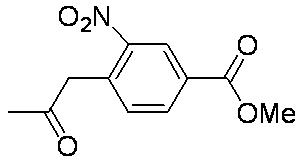
反応混合物を減圧濃縮することにより赤褐色固体を得た。得られた固体をクロロホルム(6mL)に溶解し、ピリジン(0.46mL)及び塩化アセチル(0.36mL)を室温で加えた後、室温で3日間攪拌した。反応混合物に水を加え、減圧濃縮した。得られた残渣に1,4-ジオキサン(3mL)及び水(1.5mL)を加えた後、100℃で18時間攪拌した。反応混合物を減圧濃縮後、水を加え、酢酸エチルで抽出した。得られた有機層を無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィーにて精製することにより表題化合物(589mg)を褐色油状物質として得た。
1H-NMR(400MHz,CDCl3)δ:8.75(1H,d,J=1.2Hz),8.24(1H,dd,J=7.8,2.0Hz),7.38(1H,d,J=7.8Hz),4.20(2H,s),3.97(3H,s),2.35(3H,s).

1H-NMR(400MHz,CDCl3)δ:8.21-8.20(1H,br),8.04(1H,s),7.77(1H,dd,J=8.5,1.5Hz),7.51(1H,d,J=8.3Hz),6.28-6.27(1H,m),3.92(3H,s),2.49(3H,s).
1H-NMR(400MHz,DMSO-d6)δ:7.74(1H,s),7.54(1H,d,J=8.3Hz),7.46(1H,d,J=8.3Hz),7.15-7.10(1H,m),7.04(2H,d,J=7.8Hz),6.33(1H,s),5.45(2H,s),2.28(3H,s),2.08(6H,s).
ESI-MSfound:294[M+H]+
1-ベンジル-2-メチル-1H-インドール-6-カルボン酸[70](以下、化合物[70]という)の合成

1H-NMR(400MHz,CD3OD)δ:7.98(1H,s),7.70(1H,dd,J=8.3,1.2Hz),7.51(1H,d,J=8.3Hz),7.29-7.19(3H,m),6.96(2H,d,J=7.3Hz),6.38(1H,s),5.47(2H,s),2.38(3H,s).
ESI-MSfound:266[M+H]+
2-メチル-1-(2-メチルベンジル)-1H-インドール-6-カルボン酸[71](以下、化合物[71]という)の合成

1H-NMR(400MHz,CD3OD)δ:7.86(1H,s),7.71(1H,dd,J=8.5,1.2Hz),7.53(1H,d,J=7.6Hz),7.22(1H,d,J=7.3Hz),7.11(1H,t,J=7.6Hz),6.93(1H,t,J=7.6Hz),6.42(1H,s),6.05(1H,d,J=7.3Hz),5.39(2H,s),2.45(3H,s),2.35(3H,s).
ESI-MSfound:280[M+H]+
1-(2-クロロベンジル)-2-メチル-1H-インドール-6-カルボン酸[72](以下、化合物[72]という)の合成
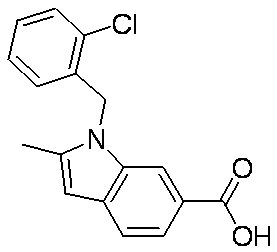
1H-NMR(400MHz,CD3OD)δ:7.87(1H,s),7.72(1H,d,J=7.8Hz),7.54(1H,d,J=7.8Hz),7.47(1H,d,J=7.1Hz),7.24(1H,t,J=7.6Hz),7.08(1H,t,J=6.8Hz),6.44(1H,s),6.18(1H,d,J=6.8Hz),5.51(2H,s),2.38(3H,s).
ESI-MSfound:300[M+H]+
1-(2,6-ジメチルベンジル)-2-(トリフルオロメチル)-1H-ベンゾイミダゾール-6-カルボン酸[73](以下、化合物[73]という)の合成
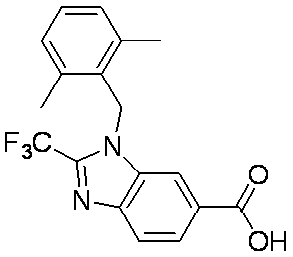
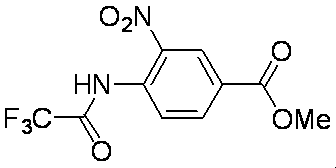
1H-NMR(400MHz,CDCl3)δ:11.58(1H,s),8.99(1H,d,J=2.0Hz),8.87(1H,d,J=8.8Hz),8.40(1H,dd,J=8.8,2.0Hz),4.00(3H,s).
ESI-MSfound:291[M-H]-

ESI-MSfound:363[M+H]+
1H-NMR(400MHz,CDCl3)δ:7.97(1H,dd,J=8.5,1.5Hz),7.85(1H,d,J=8.5Hz),7.29-7.23(2H,m),7.15(2H,d,J=7.6Hz),5.64(2H,s),2.25(6H,s).
ESI-MSfound:349[M+H]+
2-(1-ベンジル-2,3-ジヒドロ-1H-インドール-6-イル)酢酸カリウム[74](以下、化合物[74]という)の合成

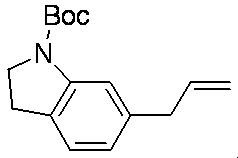
1H-NMR(400MHz,CDCl3)δ:7.72(1H,brs),7.05(1H,d,J=7.4Hz),6.76(1H,d,J=7.4Hz),6.02-5.89(1H,m),5.16-4.98(2H,m),4.03-3.91(2H,m),3.36(2H,d,J=6.6Hz),3.04(2H,t,J=8.5Hz),1.56(9H,brs).
ESI-MSfound:204[M-tBu+H]+

1H-NMR(400MHz,CDCl3)δ:9.73(1H,t,J=2.0Hz),7.78(1H,brs),7.13(1H,d,J=7.4Hz),6.76(1H,d,J=7.4Hz),3.99(2H,t,J=8.7Hz),3.64(2H,d,J=2.0Hz),3.08(2H,t,J=8.7Hz),1.56(9H,brs).
ESI-MSfound:206[M-tBu+H]+
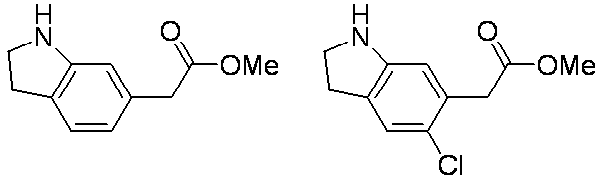
得られた粗カルボン酸体(3.8g)をN,N-ジメチルホルムアミド(70mL)に溶解し、炭酸水素ナトリウム(2.3g)及びヨウ化メチル(4.2mL)を加えた後、60℃で2時間攪拌した。反応混合物に水を加え、酢酸エチルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮することにより粗エステル体を淡褐色油状物として得た。
得られた粗エステル体をクロロホルム(80mL)に溶解し、トリフルオロ酢酸(80mL)を室温で加えた後、室温で2時間攪拌した。溶媒を減圧濃縮した後、反応混合物に炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィーにて精製することにより表題化合物[74-3A](0.8g)を褐色固体として得た。また表題化合物[74-3B](0.8g)を褐色固体として得た。
[74-3A]
1H-NMR(400MHz,CDCl3)δ:7.05(1H,d,J=7.4Hz),6.59(1H,d,J=7.4Hz),6.57(1H,s),3.68(3H,s),3.56(2H,t,J=8.4Hz),3.52(2H,s),3.00(2H,t,J=8.4Hz).
ESI-MSfound:192[M-Boc+H]+
[74-3B]
1H-NMR(400MHz,CDCl3)δ:7.09(1H,s),6.54(1H,s),3.71(3H,s),3.67(2H,s),3.57(2H,t,J=8.4Hz),3.00(2H,t,J=8.4Hz).
ESI-MSfound:226[M-Boc+H]+
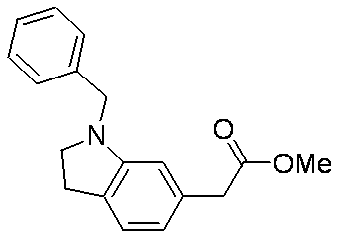
1H-NMR(400MHz,CDCl3)δ:7.37-7.27(5H,m),7.03(1H,d,J=7.3Hz),6.57(1H,dd,J=7.3,1.0Hz),6.44(1H,d,J=1.0Hz),4.26(2H,s),3.67(3H,s),3.53(2H,s),3.31(2H,t,J=8.4Hz),2.94(2H,t,J=8.4Hz).
ESI-MSfound:282[M+H]+
反応混合物に1N-塩酸を加え、酢酸エチルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮した。得られた残渣をメタノールに溶解し1M水酸化カリウム-メタノール溶液(92.4μL)を加え、溶媒を減圧濃縮した。得られた残渣を酢酸エチルに懸濁させ、固体を濾取することにより表題化合物(23mg)を白色固体として得た。
1H-NMR(400MHz,DMSO-d6)δ:7.37-7.22(5H,m),6.85(1H,d,J=7.3Hz),6.47(1H,d,J=1.2Hz),6.42(1H,dd,J=7.3,1.2Hz),4.19(2H,s),3.15(2H,t,J=8.3Hz),3.00(2H,s),2.79(2H,t,J=8.3Hz).
ESI-MSfound:268[M-K+2H]+
2-[1-(2,6-ジメチルベンジル)-2,3-ジヒドロ-1H-インドール-6-イル]酢酸[75](以下、化合物[75]という)の合成
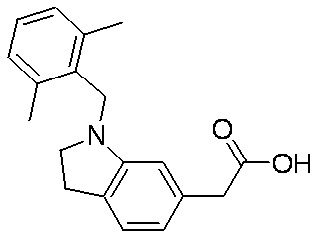

1H-NMR(400MHz,CDCl3)δ:7.12(1H,dd,J=8.4,6.5Hz),7.06-7.00(3H,m),6.57(1H,dd,J=7.4,1.1Hz),6.53(1H,d,J=1.1Hz),4.21(2H,s),3.71(3H,s),3.59(2H,s),3.11(2H,t,J=8.3Hz),2.82(2H,t,J=8.3Hz),2.38(6H,s).
ESI-MSfound:310[M+H]+
1H-NMR(400MHz,CDCl3)δ:7.11(1H,dd,J=8.5,6.7Hz),7.07-7.01(3H,m),6.58(1H,dd,J=6.7,1.2Hz),6.52(1H,d,J=1.2Hz),4.21(2H,s),3.62(2H,s),3.11(2H,t,J=8.2Hz),2.82(2H,t,J=8.2Hz),2.38(6H,s).
ESI-MSfound:296[M+H]+
2-[1-(2,6-ジメチルベンジル)-2,3-ジヒドロ-1H-インドール-6-イル]酢酸カリウム[76](以下、化合物[76]という)の合成
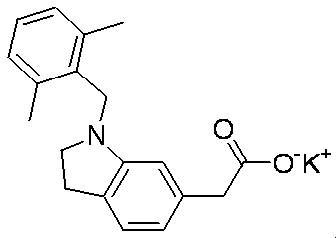
1H-NMR(400MHz,DMSO-d6)δ:7.12-7.01(3H,m),6.89(1H,d,J=7.2Hz),6.60(1H,s),6.46(1H,d,J=7.2Hz),4.14(2H,s),3.25(2H,s),2.98(2H,t,J=8.2Hz),2.71(2H,t,J=8.2Hz),2.34(6H,s).
ESI-MSfound:296[M-K+2H]+
2-[5-クロロ-1-(2,6-ジメチルベンジル)-2,3-ジヒドロ-1H-インドール-6-イル]酢酸[77](以下、化合物[77]という)の合成


1H-NMR(400MHz,CDCl3)δ:7.12(1H,dd,J=8.3,6.6Hz),7.05-7.04(3H,m),6.46(1H,s),4.18(2H,s),3.73(3H,s),3.72(2H,s),3.11(2H,t,J=8.1Hz),2.82(2H,t,J=8.1Hz),2.37(6H,s).
ESI-MSfound:344[M+H]+
1H-NMR(400MHz,DMSO-d6)δ:7.12(1H,dd,J=8.3,6.6Hz),7.07-7.02(3H,m),6.45(1H,s),4.18(2H,s),3.77(2H,s),3.12(2H,t,J=8.3Hz),2.82(2H,t,J=8.3Hz),2.37(6H,s).
ESI-MSfound:330[M+H]+
(3-RS)-2-[1-(2,6-ジメチルベンジル)-2,3-ジヒドロ-3-メチル-1H-インドール-6-イル]酢酸[78](以下、化合物[78]という)の合成


1H-NMR(400MHz,CDCl3)δ:8.48(1H,brs),7.17(1H,dd,J=7.9,1.3Hz),7.08(1H,d,J=7.9Hz),7.07(1H,s),3.41(1H,q,J=7.8Hz),1.48(3H,d,J=7.8Hz).
ESI-MSfound:226[M+H]+
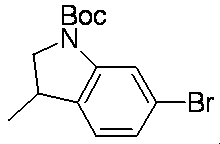
1H-NMR(400MHz,CDCl3)δ:8.03(1H,s),7.07(1H,dd,J=7.9,1.6Hz),6.96(1H,d,J=8.3Hz),4.19-4.08(1H,m),3.57-3.43(1H,m),3.39-3.27(1H,m),1.53(9H,brs),1.30(3H,d,J=6.8Hz).
ESI-MSfound:256[M-tBu+H]+

1H-NMR(400MHz,CDCl3)δ:7.01(1H,d,J=7.3Hz),6.62(1H,d,J=7.3Hz),6.57(1H,s),3.73-3.64(2H,m),3.68(3H,s),3.53(2H,s),3.39-3.27(1H,m),3.11(1H,t,J=8.7Hz),1.30(3H,d,J=6.8Hz).
ESI-MSfound:206[M+H]+

1H-NMR(400MHz,CDCl3)δ:7.12(1H,dd,J=8.3,6.6Hz),7.07-7.02(2H,m),6.99(1H,d,J=7.3Hz),6.60(1H,d,J=7.3Hz),6.52(1H,s),4.30(1H,d,J=12.9Hz),4.10(1H,d,J=12.9Hz),3.71(3H,s),3.59(2H,s),3.24(1H,t,J=8.4Hz),3.13(1H,tq,J=8.3,6.6Hz),2.67(1H,t,J=8.2Hz),2.38(6H,s),1.21(3H,d,J=6.6Hz).
ESI-MSfound:324[M+H]+
工程(4)で得た化合物[78-4](46mg)から実施例77の工程(2)の方法に準じて表題化合物(26mg)を白色固体として得た。
1H-NMR(400MHz,CDCl3)δ:7.12(1H,dd,J=8.2,6.5Hz),7.07-7.02(2H,m),7.00(1H,d,J=7.3Hz),6.61(1H,d,J=7.3Hz),6.51(1H,s),4.30(1H,d,J=12.9Hz),4.10(1H,d,J=12.9Hz),3.62(2H,s),3.25(1H,t,J=8.4Hz),3.14(1H,td,J=14.8,7.3Hz),2.67(1H,t,J=8.3Hz),2.37(6H,s),1.21(3H,d,J=6.6Hz).
ESI-MSfound:310[M+H]+
(3R*)-2-[1-(2,6-ジメチルベンジル)-2,3-ジヒドロ-3-メチル-1H-インドール-6-イル]酢酸[79A](以下、化合物[79A]という)及び(3S*)-2-[1-(2,6-ジメチルベンジル)-2,3-ジヒドロ-3-メチル-1H-インドール-6-イル]酢酸[79B](以下、化合物[79B]という)の合成
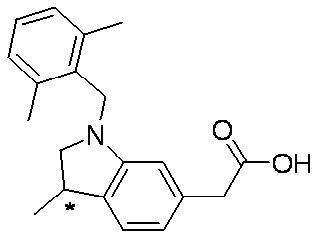
化合物[79A]
1HNMR、ESI-MSは化合物[78]と同じ。
化合物[79B]
1HNMR、ESI-MSは化合物[78]と同じ。
2-[1-(2,6-ジメチルベンジル)-2,3-ジヒドロ-3,3-ジメチル-1H-インドール-6-イル]酢酸[80](以下、化合物[80]という)の合成

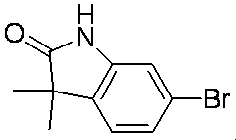
1H-NMR(400MHz,CDCl3)δ:7.65(1H,brs),7.18(1H,dd,J=7.8,2.0Hz),7.06(1H,d,J=2.0Hz),7.05(1H,d,J=7.8Hz),1.38(6H,s).
ESI-MSfound:240[M+H]+

1H-NMR(400MHz,CDCl3)δ:8.05(1H,s),7.08(1H,dd,J=1.7,7.8Hz),6.94(1H,d,J=7.8Hz),3.70(2H,s),1.57(9H,s),1.30(6H,s).
ESI-MSfound:270[M-tBu+H]+

1H-NMR(400MHz,CDCl3)δ:6.97(1H,d,J=7.3Hz),6.63(1H,d,J=7.6Hz),6.57(1H,s),3.68(3H,s),3.53(2H,s),3.31(2H,s),1.29(6H,s).
ESI-MSfound:220[M+H]+
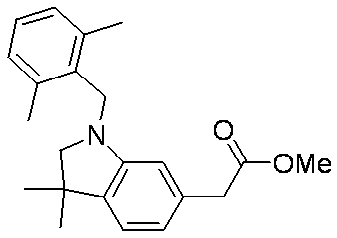
1H-NMR(400MHz,CDCl3)δ:7.12(1H,dd,J=8.2,6.6Hz),7.08-7.02(2H,m),6.95(1H,d,J=7.6Hz),6.60(1H,d,J=7.3Hz),6.52(1H,s),4.22(2H,s),3.71(3H,s),3.59(2H,s),2.84(2H,s),2.39(6H,s),1.19(6H,s).
ESI-MSfound:338[M+H]+
1H-NMR(400MHz,CDCl3)δ:7.12(1H,dd,J=8.2,6.7Hz),7.07-7.02(2H,m),6.96(1H,d,J=7.6Hz),6.62(1H,d,J=7.3Hz),6.51(1H,s),4.22(2H,s),3.62(2H,s),2.84(2H,s),2.37(6H,s),1.19(6H,s).
ESI-MSfound:324[M+H]+
2,3-ジヒドロ-1-(2,6-ジメチルベンジル)-1H-インドール-6-カルボン酸[81](以下、化合物[81]という)の合成


1H-NMR(400MHz,CDCl3)δ:7.96(1H,brs),7.09(1H,d,J=7.6Hz),7.01-6.92(1H,brm),6.69(1H,dd,J=17.6,10.8Hz),5.80-5.66(1H,brm),5.19(1H,d,J=10.8Hz),4.06-3.91(2H,brm),3.07(2H,t,J=8.7Hz),1.57(9H,s).
ESI-MSfound:190[M-tBu+H]+

1H-NMR(400MHz,CDCl3)δ:7.41(1H,dd,J=7.6,1.5Hz),7.26(1H,d,J=1.5Hz),7.14(1H,d,J=7.6Hz),3.87(3H,s),3.61(2H,t,J=8.5Hz),3.07(2H,t,J=8.5Hz).
ESI-MSfound:178[M+H]+

1H-NMR(400MHz,CDCl3)δ:7.40(1H,dd,J=7.6,1.5Hz),7.23(1H,d,J=1.5Hz),7.15-7.03(4H,m),4.27(2H,s),3.91(3H,s),3.15(2H,t,J=8.3Hz),2.89(2H,t,J=8.3Hz),2.39(6H,s).
ESI-MSfound:296[M+H]+
工程(3)で得た化合物[81-3](55mg)をテトラヒドロフラン(1.8mL)に溶解し、1N-水酸化ナトリウム(1.0mL)を加えた後、マイクロウェーブ反応装置を用いて120℃で20分間攪拌した後、反応混合物に1N-塩酸を加え、酢酸エチルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィーにて精製することにより表題化合物(42mg)を白色固体として得た。
1H-NMR(400MHz,CDCl3)δ:7.49(1H,d,J=7.6Hz),7.28(1H,s),7.17-7.10(2H,m),7.08-7.03(2H,m),4.28(2H,s),3.17(2H,t,J=8.2Hz),2.91(2H,t,J=8.2Hz),2.40(6H,s).
ESI-MSfound:282[M+H]+
2-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インドール-6-イル]酢酸[82](以下、化合物[82]という)の合成

1H-NMR(400MHz,CD3OD)δ:7.48-7.32(5H,m),6.99(1H,dd,J=8.1,1.5Hz),6.61(1H,d,J=1.0Hz),5.50(2H,s),3.70(2H,s),2.20(3H、s).
ESI-MSfound:348[M+H]+
2-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インドール-6-イル]酢酸カリウム[83](以下、化合物[83]という)の合成
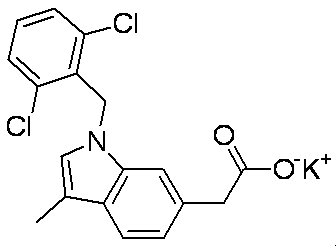
1H-NMR(400MHz,CD3OD)δ:7.52-7.30(5H,m),7.05(1H,d,J=8.1Hz),6.51(1H,s),5.48(2H,s),3.61(2H,s),2.18(3H、s).
ESI-MSfound:348[M-K+2H]+
2-[1-(2,3-ジクロロベンジル)-3-メチル-1H-インドール-6-イル]酢酸[84](以下、化合物[84]という)の合成

1H-NMR(400MHz,CD3OD)δ:7.44(1H,d,J=8.1Hz),7.40(1H,d,J=7.8Hz),7.17(1H,s),7.13-7.03(2H,m),6.93(1H,s),6.39(1H,d,J=7.6Hz),5.42(2H,s),3.52(2H,s),2.30(3H、s).
ESI-MSfound:348[M+H]+
2-[1-(2,5-ジメチルベンジル)-3-メチル-1H-インドール-6-イル]酢酸[85](以下、化合物[85]という)の合成

1H-NMR(400MHz,CD3OD)δ:7.41(1H,d,J=8.1Hz),7.24(1H,s),7.07-7.02(2H,m),6.95(1H,d,J=7.1Hz),6.73(1H,d,J=1.0Hz),6.54(1H,s),5.21(2H,s),3.54(2H,s),2.27(3H、s),2.23(3H,s),2.14(3H,s).
ESI-MSfound:308[M+H]+
2-[1-(2-クロロ-6-フルオロベンジル)-3-メチル-1H-インドール-6-イル]酢酸[86](以下、化合物[86]という)の合成
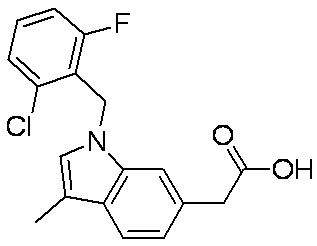
1H-NMR(400MHz,CD3OD)δ:7.46-7.26(4H,m),7.15(1H,t,J=8.8Hz),6.97(1H,d,J=8.1Hz),6.85(1H,s),5.41(2H,s),3.68(2H,s),2.22(3H,s).
ESI-MSfound:332[M+H]+
2-[1-(2-クロロベンジル)-3-メチル-1H-インドール-6-イル]酢酸[87](以下、化合物[87]という)の合成
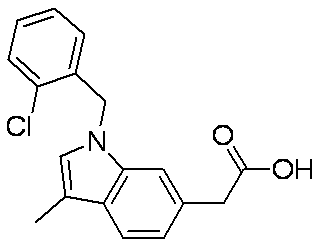
1H-NMR(400MHz,CD3OD)δ:7.48(1H,d,J=8.1Hz),7.42(1H,d,J=8.1Hz),7.22(1H,t,J=7.2Hz),7.14(1H,s),7.10(1H,t,J=7.3Hz),7.02-6.95(2H,m),6.60(1H,d,J=7.6Hz),5.39(2H,s),3.63(2H,s),2.30(3H,s).
ESI-MSfound:314[M+H]+
2-{1-[(6-クロロベンゾ[d][1,3]ジオキソル-5-イル)メチル]-3-メチル-1H-インドール-6-イル}酢酸[88](以下、化合物[88]という)の合成
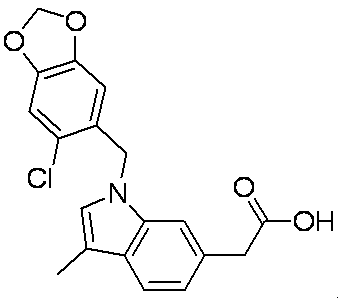
1H-NMR(400MHz,CD3OD)δ:7.47(1H,d,J=8.1Hz),7.17(1H,s),7.00(1H,d,J=8.1Hz),6.96(1H,s),6.92(1H,s),6.05(1H,s),5.89(2H,s),5.28(2H,s),3.65(2H,s),2.30(3H,s).
ESI-MSfound:358[M+H]+
3-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インドール-6-イル]プロピオン酸[89](以下、化合物[89]という)の合成


1H-NMR(400MHz,CDCl3)δ:10.09(1H,s),8.47(1H,s),7.81-7.76(3H,m),7.57(1H,d,J=8.1Hz),7.52(1H,s),7.26-7.23(2H,m),2.35(3H,s),2.28(3H,s).

1H-NMR(400MHz,CDCl3)δ:8.12(1H,s),7.81(1H,d,J=16.1Hz),7.74(2H,d,J=8.3Hz),7.44(2H,s),7.37(1H,s),7.22(2H,d,J=8.3Hz),6.50(1H,d,J=16.1Hz),3.83(3H,s),2.34(3H,s),2.24(3H,s).
(3)3-(3-メチル-1-トシル-1H-インドール-6-イル)プロピオン酸メチル[89-3](以下、化合物[89-3]という)の合成

1H-NMR(400MHz,CDCl3)δ:7.82(1H,s),7.73(2H,d,J=8.3Hz),7.35(1H,d,J=8.1Hz),7.26-7.18(3H,m),7.08(1H,d,J=7.8Hz),3.68(3H,s),3.07(2H,t,J=7.7Hz),2.67(2H,t,J=7.8Hz),2.33(3H,s),2.21(3H,s).
1H-NMR(400MHz,CDCl3)δ:7.88-7.73(1H,br),7.49(1H,d,J=8.1Hz),7.17(1H,s),6.97(1H,d,J=7.3Hz),6.92(1H,s),3.67(3H,s),3.06(2H,t,J=7.8Hz),2.68(2H,t,J=7.8Hz),2.31(3H,s).
工程(4)で得た化合物[89-4](41mg)及び2,6-ジクロロベンジルクロリド(60mg)から実施例82の方法に準じて表題化合物(8.9mg)を淡褐色固体として得た。
1H-NMR(400MHz,CD3OD)δ:7.47(2H,d,J=8.1Hz),7.41-7.33(3H,m),6.93(1H,d,J=8.1Hz),6.59(1H,s),5.47(2H,s),3.03(2H,t,J=7.7Hz),2.65(2H,t,J=7.7Hz),2.19(3H,s).
ESI-MSfound:362[M+H]+
3-アセチル-1-(2,6-ジメチルベンジル)-1H-インドール-6-カルボン酸[90](以下、化合物[90]という)の合成


1H-NMR(400MHz,CD3OD)δ:8.34(1H,s),8.29(1H,dd,J=8.5,0.7Hz),8.15(1H,dd,J=1.5,0.7Hz),7.87(1H,dd,J=8.5,1.5Hz),3.92(3H,s),2.54(3H,s).

1H-NMR(400MHz,CDCl3)δ:8.41(1H,d,J=8.1Hz),8.32(1H,s),8.03(1H,dd,J=8.4,1.3Hz),7.31-7.29(1H,m),7.26(1H,s),7.19(2H,d,J=7.6Hz),5.34(2H,s),3.99(3H,s),2.39(3H,s),2.28(6H,s).
工程(2)で得た化合物[90-2](132mg)をメタノール(3mL)に溶解し、1N-水酸化ナトリウム水溶液(3mL)を室温で加えた後、50℃で20時間攪拌した。反応混合物に1N-塩酸を加えて酸性とし、析出した固体を濾取することにより表題化合物(160mg)を白色固体として得た。
1H-NMR(400MHz,CDCl3)δ:8.46(1H,d,J=8.3Hz),8.39(1H,s),8.10(1H,dd,J=8.5,1.2Hz),7.32-7.30(2H,m),7.20(2H,d,J=7.3Hz),5.37(2H,s),2.40(3H,s),2.29(6H,s).
1-(2,6-ジメチルベンジル)-3-エチル-1H-インドール-6-カルボン酸[91](以下、化合物[91]という)の合成
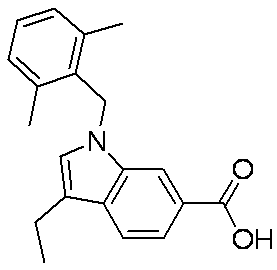

1H-NMR(400MHz,CDCl3)δ:8.12(1H,d,J=0.7Hz),7.81(1H,dd,J=8.3,1.5Hz),7.63(1H,d,J=8.3Hz),7.15(1H,t,J=1.2Hz),3.94(3H,s),2.80(2H,q,J=7.5Hz),1.34(3H,t,J=7.6Hz).

1H-NMR(400MHz,CDCl3)δ:8.24(1H,s),7.82(1H,dd,J=8.4,1.3Hz),7.62(1H,d,J=8.3Hz),7.24-7.22(1H,m),7.14(2H,d,J=7.6Hz),6.52(1H,s),5.26(2H,s),3.97(3H,s),2.68(2H,q,J=7.6Hz),2.26(6H,s),1.21(3H,t,J=7.6Hz).
工程(2)で得た化合物[91-2](184mg)から、実施例90の工程(3)の方法に準じて表題化合物(160mg)を白色固体として得た。
1H-NMR(400MHz,CDCl3)δ:8.32(1H,s),7.90(1H,dd,J=8.4,1.3Hz),7.65(1H,d,J=8.3Hz),7.28-7.22(1H,m),7.14(2H,d,J=7.6Hz),6.56(1H,s),5.29(2H,s),2.70(2H,q,J=7.6Hz),2.28(6H,s),1.22(3H,t,J=7.6Hz).
ESI-MSfound:308[M+H]+
1-(2,6-ジメチルベンジル)-3-イソプロピル-1H-インドール-6-カルボン酸[92](以下、化合物[92]という)の合成


1H-NMR(400MHz,CDCl3)δ:8.34(1H,s),8.13(1H,d,J=0.7Hz),7.82-7.80(1H,m),7.68(1H,d,J=8.3Hz),7.13(1H,dd,J=2.4,0.7Hz),3.94(3H,s),3.28-3.18(1H,m),1.36(6H,d,J=8.1Hz).

1H-NMR(400MHz,CDCl3)δ:8.24(1H,s),7.82(1H,d,J=8.3Hz),7.67(1H,d,J=8.3Hz),7.25-7.23(1H,m),7.14(2H,d,J=7.6Hz),6.50(1H,s),5.26(2H,s),3.97(3H,s),3.16-3.09(1H,m),2.27(6H,s),1.25(6H,d,J=6.8Hz).
工程(2)で得た化合物[92-2](68mg)から実施例90の工程(3)の方法に準じて表題化合物(30mg)を白色固体として得た。
1H-NMR(400MHz,CDCl3)δ:8.34(1H,s),7.92-7.90(1H,m),7.71(1H,d,J=8.3Hz),7.27-7.23(1H,m),7.15(2H,d,J=7.6Hz),6.56(1H,s),5.29(2H,d,J=3.4Hz),3.19-3.12(1H,m),2.29(6H,s),1.27(6H,d,J=7.1Hz).
ESI-MSfound:322[M+H]+
2-{1-[(5-クロロチオフェン-2-イル)メチル]-1H-インドール-6-イル}酢酸[93](以下、化合物[93]という)の合成

ESI-MSfound:306[M+H]+
3-クロロ-1-(2,6-ジメチルベンジル)-6-(1H-テトラゾール-5-イル)-1H-インドール[94](以下、化合物[94]という)の合成


1H-NMR(400MHz,CDCl3)δ:8.55-8.45(1H,br),7.75-7.70(2H,m),7.44(1H,dd,J=8.3,1.2Hz),7.40(1H,d,J=2.7Hz).

1H-NMR(400MHz,CDCl3)δ:7.82(1H,d,J=0.5Hz),7.69(1H,d,J=8.3Hz),7.44(1H,dd,J=8.3,1.2Hz),7.30-7.15(1H,m),7.15(2H,d,J=7.6Hz),6.75(1H,s),5.25(2H,s),2.26(6H,s).
工程(2)で得られた化合物[94-2](164mg)のN,N-ジメチルホルムアミド(2mL)溶液に塩化アンモニウム(47mg)及びアジ化ナトリウム(45mg)を室温で加えた後、100℃で17時間攪拌した。更に塩化アンモニウム(55mg)及びアジ化ナトリウム(45mg)を加え、100℃で24時間攪拌した。反応混合物に水を加え、酢酸エチルで抽出した。得られた有機層を無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィーにて精製することにより表題化合物(10mg)を褐色固体として得た。
1H-NMR(400MHz,CD3OD)δ:8.35(1H,s),7.82(1H,dd,J=8.3,1.5Hz),7.71(1H,d,J=8.3Hz),7.24(1H,dd,J=8.4,6.7Hz),7.16(2H,d,J=7.8Hz),6.71(1H,s),5.43(2H,s),2.28(6H,s).
ESI-MSfound:338[M+H]+
1-(2,6-ジメチルベンジル)-1H-ピロロ[3,2-b]ピリジン-6-カルボン酸[95](以下、化合物[95]という)の合成
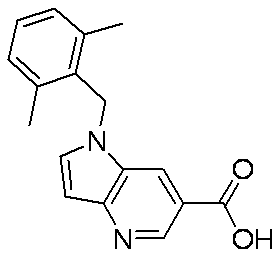
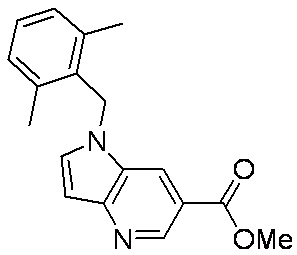
1H-NMR(400MHz,CDCl3)δ:9.14(1H,s),8.45(1H,s),7.25(1H,t,J=7.3Hz),7.15(2H,d,J=7.3Hz),7.02(1H,d,J=3.0Hz),6.67(1H,d,J=3.0Hz),5.33(2H,s),4.00(3H,s),2.26(6H,s).
ESI-MSfound:295[M+H]+
工程(1)で得た化合物[95-1](21mg)のテトラヒドロフラン(1mL)及びメタノール(1mL)溶液に1N-水酸化ナトリウム水溶液(0.5mL)を加えた後、マイクロウェーブ反応装置を用いて140℃で30分間反応させた。反応混合物に1N-塩酸を加え、酢酸エチルで抽出した。得られた有機層を無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮することにより、表題化合物(13mg)を淡褐色固体として得た。
1H-NMR(400MHz,DMSO-d6)δ:13.01(1H,brs),8.91(1H,d,J=1.7Hz),8.53-8.51(1H,m),7.28(1H,d,J=3.4Hz),7.23-7.17(1H,m),7.14-7.10(2H,m),6.62(1H,dd,J=3.4,1.7Hz),5.48(2H,s),2.20(6H,s).
ESI-MSfound:281[M+H]+
1-(2,6-ジメチルベンジル)-1H-ピロロ[2,3-b]ピリジン-6-カルボン酸[96](以下、化合物[96]という)の合成
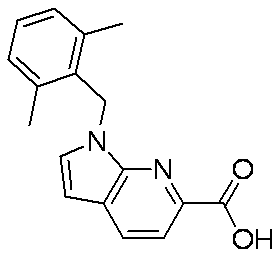

1H-NMR(400MHz,CDCl3)δ:13.1(1H,s),8.23(1H,d,J=6.1Hz),7.70(1H,d,J=7.8Hz),7.42(1H,d,J=3.2Hz),7.06(1H,dd,J=7.8,6.3Hz),6.56(1H,d,J=3.2Hz).

1H-NMR(400MHz,CDCl3)δ:8.08(1H,d,J=8.1Hz),7.71(1H,dd,J=3.5,2.6Hz),7.52(1H,d,J=8.1Hz),6.65(1H,dd,J=3.7,2.0Hz).
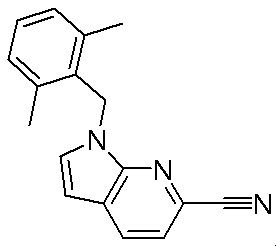
1H-NMR(400MHz,CDCl3)δ:7.98(1H,d,J=7.8Hz),7.49(1H,d,J=8.1Hz),7.22(1H,dd,J=8.2,7.0Hz),7.12(2H,d,J=7.6Hz),6.96(1H,d,J=3.4Hz),6.46(1H,d,J=3.7Hz),5.49(2H,s),2.27(6H,s).
工程(3)で得た化合物[96-3](66mg)のエタノール(2mL)溶液に3N-水酸化ナトリウム水溶液(2mL)を室温で加えた後、1時間加熱還流した。反応混合物に1N-塩酸を加え、析出した固体を濾取することにより表題化合物(15mg)を白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:8.08(1H,d,J=8.1Hz),7.96(1H,d,J=8.1Hz),7.20-7.18(1H,m),7.11(2H,d,J=7.6Hz),6.97(1H,d,J=3.7Hz),6.51(1H,d,J=3.7Hz),5.62(2H,s),2.25(6H,s).
2-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]酢酸[97](以下、化合物[97]という)の合成


1H-NMR(400MHz,CDCl3)δ:7.55(1H,s),7.47(1H,d,J=8.5Hz),7.38(2H,d,J=8.1Hz),7.25(1H,d,J=5.9Hz),7.22-7.20(1H,m),5.66(2H,s),2.50(3H,s).

1H-NMR(400MHz,CDCl3)δ:7.53-7.52(1H,m),7.37-7.36(2H,m),7.25-7.18(2H,m),6.95(1H,d,J=8.1Hz),6.03-5.94(1H,m),5.69(2H,s),5.12-5.10(2H,m),3.51-3.49(2H,m),2.51(3H,s).

1H-NMR(400MHz,CDCl3)δ:9.77(1H,t,J=2.3Hz),7.62(1H,d,J=8.3Hz),7.37(2H,d,J=8.1Hz),7.25-7.23(2H,m),6.95(1H,d,J=8.3Hz),5.70(2H,s),3.80(2H,d,J=2.2Hz),2.52(3H,s).
工程(3)で得た化合物[97-3](1.03g)から実施例66の工程(4)の方法に準じて表題化合物(1.06g)を白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:7.62(1H,d,J=8.3Hz),7.46-7.44(3H,m),7.35-7.33(1H,m),7.09(1H,d,J=8.3Hz),5.72(2H,s),3.73(2H,s),2.46(3H,s).
ESI-MSfound:349[M+H]+
2-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]酢酸カリウム[98](以下、化合物[98]という)の合成

1H-NMR(400MHz,CD3OD)δ:7.56(1H,d,J=8.3Hz),7.47-7.43(3H,m),7.35-7.31(1H,m),7.15(1H,d,J=8.1Hz),5.70(2H,s),3.61(2H,s),2.44(3H,s).
ESI-MSfound:349[M-K+2H]+
2-[1-(2,3-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]酢酸[99](以下、化合物[99]という)の合成

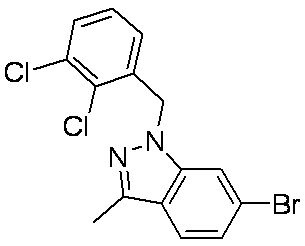
1H-NMR(400MHz,CDCl3)δ:7.84-7.82(2H,m),7.70(1H,d,J=8.4Hz),7.40(1H,d,J=8.1Hz),7.08-7.04(1H,m),6.57-6.54(1H,m),5.73(2H,s),2.65(3H,s).
工程(1)で得た化合物[99-1]から実施例66の工程(2)~(4)の方法に準じて表題化合物(50mg)を白色固体として得た。
1H-NMR(400MHz,CDCl3)δ:7.65(1H,d,J=8.3Hz),7.34(1H,d,J=7.3Hz),7.17(1H,s),7.09(1H,d,J=8.3Hz),7.02-7.00(1H,m),6.50(1H,d,J=7.8Hz),5.62(2H,s),3.74(2H,s),2.59(3H,s).
ESI-MSfound:349[M+H]+
2-[1-(2,3-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]酢酸カリウム[100](以下、化合物[100]という)の合成

1H-NMR(400MHz,CD3OD)δ:7.63(1H,d,J=8.3Hz),7.43(1H,d,J=7.6Hz),7.34(1H,s),7.18(1H,d,J=8.3Hz),7.11-7.10(1H,m),6.45(1H,d,J=7.8Hz),5.66(2H,s),3.57(2H,s),2.54(3H,s).
ESI-MSfound:349[M-K+2H]+
2-[1-(2,5-ジメチルベンジル)-3-メチル-1H-インダゾール-6-イル]酢酸[101](以下、化合物[101]という)の合成

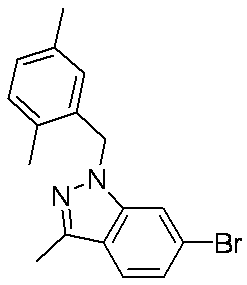
1H-NMR(400MHz,CDCl3)δ:7.59(1H,d,J=4.1Hz),7.06-7.04(2H,m),6.98-6.96(2H,m),6.61(1H,s),5.44(2H,s),2.58(3H,s),2.31(3H,s),2.19(3H,s).
工程(1)で得た化合物[101-1]から実施例66の工程(2)~(4)の方法に準じて表題化合物(59mg)を白色固体として得た。
1H-NMR(400MHz,CDCl3)δ:7.64(1H,d,J=8.3Hz),7.13(1H,s),7.06-7.04(2H,m),6.98-6.96(1H,m),6.60(1H,s),5.46(2H,s),3.72(2H,s),2.59(3H,s),2.29(3H,s),2.17(3H,s).
ESI-MSfound:309[M+H]+
2-[1-(2-クロロ-6-メチルベンジル)-3-メチル-1H-インダゾール-6-イル]酢酸[102](以下、化合物[102]という)の合成


1H-NMR(400MHz,CDCl3)δ:7.69(1H,s),7.51(1H,d,J=8.5Hz),7.27(1H,d,J=6.8Hz),5.61(2H,s),3.54(2H,t,J=8.2Hz),2.55(3H,s),0.89(2H,t,J=8.2Hz),0.00(9H,s).

1H-NMR(400MHz,CDCl3)δ:7.59(1H,d,J=8.1Hz),7.40(1H,s),7.10(1H,d,J=8.1Hz),5.64(2H,s),3.66(2H,s),3.55(2H,t,J=8.2Hz),2.55(3H,s),1.44(9H,s),0.89(2H,t,J=8.3Hz),0.00(9H,s).

1H-NMR(400MHz,CDCl3)δ:7.76(1H,s),7.68(1H,d,J=8.5Hz),7.18(1H,d,J=8.3Hz),3.82(2H,s),2.66(3H,s).

1H-NMR(400MHz,CDCl3)δ:7.63(1H,d,J=8.3Hz),7.34(1H,s),7.07(1H,d,J=8.3Hz),3.76(2H,s),3.70(3H,s),2.57(3H,s).
工程(4)で得た化合物[102-4](19.4mg)のN,N-ジメチルホルムアミド(0.5mL)溶液に炭酸カリウム(20.7mg)及び2-クロロ-6-メチルベンジルクロリド(25.0mg)を室温で加えた後、室温で20時間攪拌した。反応混合物に水を加え、酢酸エチルで抽出した。得られた有機層を無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィーにて精製し無色油状物質として得た。得られた無色油状物質にメタノール(0.2mL)及びテトラヒドロフラン(0.2mL)を加え、1N-水酸化ナトリウム水溶液(0.2mL)を室温で加えた後、室温で40分間攪拌した。反応混合物に1N-塩酸を加えて酸性にし、クロロホルムで抽出した。得られた有機層を無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮した。得られた残渣を逆相分取液体クロマトグラフィーにて精製することにより表題化合物(6.0mg)を白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:7.60(1H,d,J=8.3Hz),7.35(1H,s),7.30(1H,d,J=7.6Hz),7.21(1H,t,J=7.7Hz),7.15(1H,d,J=7.3Hz),7.05(1H,d,J=8.3Hz),5.61(2H,s),3.70(2H,s),2.46(3H,s),2.29(3H,s).
ESI-MSfound:329[M+H]+
2-[1-(2-クロロ-6-メチルベンジル)-3-メチル-1H-インダゾール-6-イル]酢酸カリウム[103](以下、化合物[103]という)の合成

1H-NMR(400MHz,CD3OD)δ:7.56(1H,d,J=8.3Hz),7.39(1H,s),7.29(1H,d,J=7.8Hz),7.20(1H,t,J=7.8Hz),7.15-7.13(2H,m),5.60(2H,s),3.60(2H,s),2.45(3H,s),2.28(3H,s).
ESI-MSfound:329[M-K+2H]+
2-[1-(2-クロロ-6-フルオロベンジル)-3-メチル-1H-インダゾール-6-イル]酢酸[104](以下、化合物[104]という)の合成

1H-NMR(400MHz,CDCl3)δ:7.59(1H,d,J=8.3Hz),7.39(1H,s),7.23-7.19(2H,m),7.07-6.98(2H,m),5.64(2H,s),3.79(2H,s),2.53(3H,s).
ESI-MSfound:333[M+H]+
2-[1-(3、5-ジメチルイソオキサゾール-4-イルメチル)-3-メチル-1H-インダゾール-6-イル]酢酸[105](以下、化合物[105]という)の合成
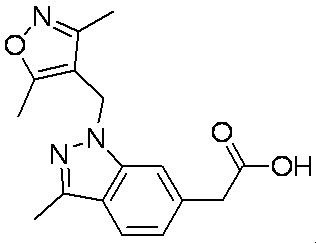
1H-NMR(400MHz,CDCl3)δ:7.59(1H,d,J=8.3Hz),7.19(1H,s),7.05(1H,d,J=8.1Hz),5.21(2H,s),3.75(2H,s),2.52(3H,s),2.34(3H,s),2.12(3H,s).
ESI-MSfound:300[M+H]+
2-{1-[(5-クロロベンゾ[b]チオフェン-3-イル)メチル]-3-メチル-1H-インダゾール-6-イル}酢酸[106](以下、化合物[106]という)の合成

1H-NMR(400MHz,CDCl3)δ:7.84(1H,s),7.72(1H,d,J=8.5Hz),7.63(1H,d,J=8.3Hz),7.31-7.23(2H,m),7.13(1H,s),7.06(1H,d,J=8.3Hz),5.68(2H,s),3.74(2H,s),2.59(3H,s).
ESI-MSfound:371[M+H]+
2-[3-メチル-1-(ナフタレン-1-イルメチル)-1H-インダゾール-6-イル]酢酸[107](以下、化合物[107]という)の合成

1H-NMR(400MHz,CDCl3)δ:8.13(1H,d,J=8.1Hz),7.88(1H,d,J=7.3Hz),7.78(1H,d,J=8.1Hz),7.66(1H,d,J=8.3Hz),7.55(1H,t,J=8.4Hz),7.51(1H,t,J=8.4Hz),7.33(1H,t,J=7.7Hz),7.16(1H,s),7.07(1H,d,J=7.8Hz),6.92(1H,d,J=6.8Hz),6.02(2H,s),3.70(2H,s),2.62(3H,s).
ESI-MSfound:331[M+H]+
2-[3-メチル-1-(2,4,6-トリメチルベンジル)-1H-インダゾール-6-イル]酢酸[108](以下、化合物「108」という)の合成

1H-NMR(400MHz,CDCl3)δ:7.56(1H,d,J=8.3Hz),6.99(1H,d,J=8.3Hz),6.91(1H,s),6.88(2H,s),5.43(2H,s),3.67(2H,s),2.51(3H,s),2.27(9H,s).
ESI-MSfound:323[M+H]+
2-[1-(2-クロロ-6-シアノベンジル)-3-メチル-1H-インダゾール-6-イル]酢酸[109](以下、化合物[109]という)の合成

1H-NMR(400MHz,CD3OD)δ:7.78(1H,d,J=7.6Hz),7.74(1H,d,J=8.1Hz),7.63(1H,d,J=8.1Hz),7.54-7.52(2H,m),7.11(1H,d,J=8.3Hz),5.73(2H,s),3.77(2H,s),2.46(3H,s).
ESI-MSfound:340[M+H]+
2-[1-(2-クロロ-6-シアノベンジル)-3-メチル-1H-インダゾール-6-イル]酢酸カリウム[110](以下、化合物[110]という)の合成

1H-NMR(400MHz,CD3OD)δ:7.78(1H,d,J=7.8Hz),7.74(1H,d,J=8.1Hz),7.57(1H,d,J=8.3Hz),7.54-7.50(2H,m),7.17(1H,d,J=8.3Hz),5.72(2H,s),3.63(2H,s),2.45(3H,s).
ESI-MSfound:340[M-K+2H]+
2-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]プロピオン酸[111](以下、化合物[111]という)の合成


1H-NMR(400MHz,CDCl3)δ:7.55(1H,d,J=8.3Hz),7.36(2H,d,J=8.1Hz),7.29(1H,s),7.22(1H,t,J=8.1Hz),7.03(1H,d,J=8.3Hz),5.69(2H,s),3.63(2H,s),2.50(3H,s),1.43(9H,s).
工程(1)で得た化合物[111-1](45.3mg)及びヨウ化メチル(9.1μL)をテトラヒドロフラン(0.6mL)に溶解し、カリウムビス(トリメチルシリル)アミド0.5Mトルエン溶液(0.3mL)を0℃で加えた後、室温で9時間攪拌した。反応混合物に飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。得られた有機層を無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィーにて精製した。精製した化合物に水(50μL)及びトリフルオロ酢酸(500μL)を室温で加えた後、室温で1.5時間攪拌した。反応混合物に水を加え、酢酸エチルで抽出した。得られた有機層を無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮した。得られた残渣を逆相分取液体クロマトグラフィーにて精製することにより表題化合物(13.7mg)を白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:7.61(1H,d,J=8.3Hz),7.44(2H,d,J=4.9Hz),7.42(1H,s),7.33(1H,t,J=8.1Hz),7.11(1H,d,J=8.3Hz),5.72(2H,s),3.84(1H,q,J=7.1Hz),2.46(3H,s),1.50(3H,d,J=7.1Hz).
ESI-MSfound:364[M+H]+
2-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]-2-メチルプロピオン酸[112](以下、化合物[112]という)の合成
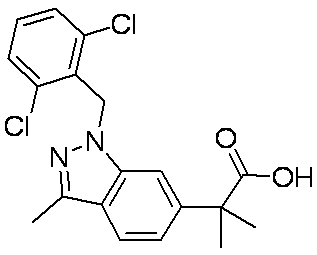
1H-NMR(400MHz,CDCl3)δ:7.58(1H,d,J=8.5Hz),7.34(2H,d,J=8.1Hz),7.31(1H,s),7.20(1H,t,J=8.0Hz),7.15(1H,d,J=8.4Hz),5.77(2H,s),2.53(3H,s),1.61(6H,s).
ESI-MSfound:378[M+H]+
2-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]酪酸[113](以下、化合物[113]という)の合成

1H-NMR(400MHz,CDCl3)δ:7.62(1H,d,J=8.3Hz),7.32(2H,d,J=8.1Hz),7.26(1H,s),7.18(1H,t,J=8.1Hz),7.12(1H,d,J=8.3Hz),5.80(2H,s),3.56(1H,t,J=7.6Hz),2.56(3H,s),2.16-2.05(1H,m),1.87-1.76(1H,m),0.89(3H,t,J=7.3Hz).
ESI-MSfound:378[M+H]+
2-(3-メチル-1-フェネチル-1H-インダゾール-6-イル)酢酸[114](以下、化合物[114]という)の合成

1H-NMR(400MHz,CDCl3)δ:7.59(1H,d,J=8.3Hz),7.26-7.18(3H,m),7.12(2H,d,J=6.8Hz),7.03-7.00(2H,m),4.51(2H,t,J=7.4Hz),3.72(2H,s),3.15(2H,t,J=7.4Hz),2.58(3H,s).
ESI-MSfound:295[M+H]+
2-[3-メチル-1-(キノリン-8-イルメチル)-1H-インダゾール-6-イル]酢酸[115](以下、化合物[115]という)の合成

1H-NMR(400MHz,CD3OD)δ:8.98(1H,d,J=2.7Hz),8.32(1H,d,J=6.8Hz),7.82(1H,d,J=8.1Hz),7.68(1H,d,J=8.3Hz),7.56(1H,dd,J=8.3Hz,4.1Hz),7.45(1H,s),7.40(1H,t,J=7.7Hz),7.10(1H,t,J=8.3Hz),6.98(1H,d,J=6.8Hz),6.23(2H,s),3.67(2H,s),2.57(3H,s).
ESI-MSfound:332[M+H]+
2-[3-メチル-1-(キノリン-8-イルメチル)-1H-インダゾール-6-イル]酢酸カリウム[116](以下、化合物[116]という)の合成
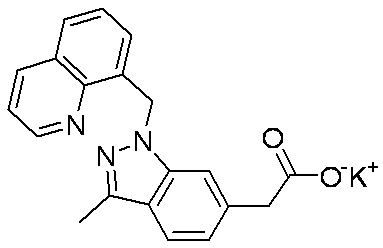
1H-NMR(400MHz,CD3OD)δ:8.99(1H,d,J=2.8),8.32(1H,d,J=7.2Hz),7.81(1H,d,J=8.3Hz),7.64(1H,d,J=8.3Hz),7.56(1H,dd,J=8.3Hz,4.4Hz),7.41(1H,s),7.37(1H,t,J=7.7Hz),7.17(1H,d,J=8.3Hz),6.85(1H,d,J=7.1Hz),6.23(2H,s),3.54(2H,s),2.57(3H,s).
ESI-MSfound:332[M-K+2H]+
1-(2,6-ジメチルベンジル)-3-メチル-1H-インダゾール-6-カルボン酸[117](以下、化合物[117]という)の合成
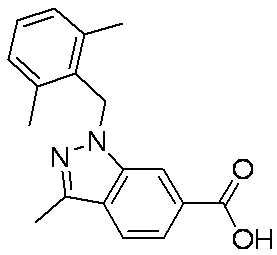

1H-NMR(400MHz,CDCl3)δ:7.77(1H,s),7.73(1H,d,J=8.3Hz),7.63(1H,d,J=8.3Hz),7.20-7.18(1H,m),7.10(2H,d,J=7.6Hz),5.55(2H,s),4.36(2H,q,J=7.1Hz),2.55(3H,s),2.35(6H,s),1.40(3H,t,J=7.2Hz).
工程(1)で得た化合物[117-1](636mg)のエタノール(10mL)溶液に1N-水酸化ナトリウム水溶液(10mL)を室温で加えた後、70℃で2時間攪拌した。反応混合物に1N塩酸を加え、析出した固体を濾取することにより表題化合物(568mg)を白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:8.04(1H,s),7.73(2H,s),7.15-7.13(1H,m),7.07(2H,d,J=7.6Hz),5.59(2H,s),2.52(3H,s),2.29(6H,s).
ESI-MSfound:295[M+H]+
1-(2,6-ジメチルベンジル)-3-メチル-1H-インダゾール-6-カルボン酸カリウム[118](以下、化合物[118]という)の合成
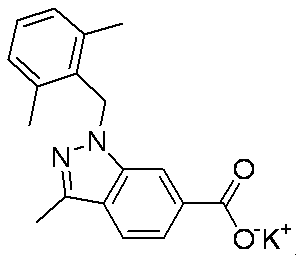
1H-NMR(400MHz,CD3OD)δ:8.06(1H,s),7.73(1H,d,J=8.3Hz),7.62(1H,d,J=8.5Hz),7.12-7.11(1H,m),7.05(2H,d,J=7.3Hz),5.53(2H,s),2.48(3H,s),2.29(6H,s).
ESI-MSfound:295[M+K-2H]+
1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-カルボン酸[119](以下、化合物[119]という)の合成

1H-NMR(400MHz,CD3OD)δ:8.28(1H,s),7.78-7.75(2H,m),7.45(2H,d,J=7.8Hz),7.36-7.34(1H,m),5.81(2H,s),2.50(3H,s).
ESI-MSfound:335[M+H]+
1-(2,3-ジクロロベンジル)-3-メチル-1H-インダゾール-6-カルボン酸[120](以下、化合物[120]という)の合成

1H-NMR(400MHz,CD3OD)δ:8.17(1H,s),7.82(2H,s),7.47(1H,d,J=7.1Hz),7.18-7.16(1H,m),6.66(1H,d,J=7.6Hz),5.75(2H,s),2.59(3H,s).
3-[1-(2,6-ジメチルベンジル)-3-メチル-1H-インダゾール-6-イル]プロピオン酸[121](以下、化合物[121]という)の合成


1H-NMR(400MHz,CDCl3)δ:7.64(1H,d,J=15.9Hz),7.60(1H,d,J=8.3Hz),7.27-7.26(1H,m),7.21(1H,t,J=7.3Hz),7.10(2H,d,J=7.6Hz),6.96(1H,s),6.37(1H,d,J=15.9Hz),5.52(2H,s),3.81(3H,s),2.54(3H,s),2.33(6H,s).
ESI-MSfound:335[M+H]+

1H-NMR(400MHz,CDCl3)δ:7.52(1H,d,J=8.3Hz),7.19-7.15(1H,m),7.08(2H,d,J=7.6Hz),6.91(1H,d,J=8.1Hz),6.75(1H,s),5.47(2H,s),3.65(3H,s),2.96(2H,t,J=7.8Hz),2.58(2H,t,J=7.8Hz),2.51(3H,s)2.33(6H,s).
ESI-MSfound:337[M+H]+
工程(2)で得られた化合物[121-2](13mg)から実施例117の工程(2)の方法に準じて表題化合物(13mg)を白色固体として得た。
1H-NMR(400MHz,CDCl3)δ:7.52(1H,d,J=8.3Hz),7.19-7.15(1H,m),7.07(2H,d,J=7.6Hz),6.91(1H,d,J=8.1Hz),6.72(1H,s),5.48(2H,s),2.97(2H,t,J=7.7Hz),2.62(2H,t,J=7.7Hz),2.51(3H,s),2.31(6H,s).
ESI-MSfound:323[M+H]+
3-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]プロピオン酸[122](以下、化合物[122]という)の合成

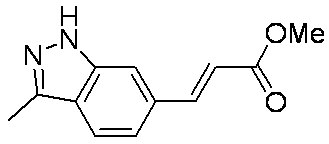
1H-NMR(400MHz,CDCl3)δ:9.91(1H,br),7.81(1H,d,J=15.9Hz),7.67(1H,d,J=8.3Hz),7.54(1H,s),7.36(1H,d,J=8.3Hz),6.52(1H,d,J=15.9Hz),3.83(3H,s),2.57(3H,s).
ESI-MSfound:217[M+H]+
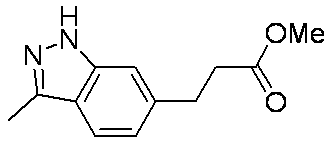
1H-NMR(400MHz,CDCl3)δ:9.72(1H,br),7.59(1H,d,J=8.3Hz),7.24(1H,s),7.00(1H,d,J=8.1Hz),3.67(3H,s),3.08(2H,t,J=7.7Hz),2.69(2H,t,J=7.8Hz),2.56(3H,s).
ESI-MSfound:219[M+H]+
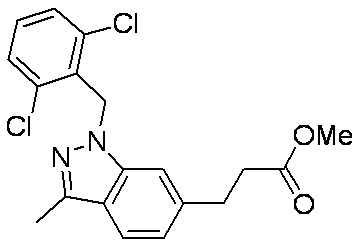
1H-NMR(400MHz,CDCl3)δ:7.53(1H,d,J=8.3Hz),7.37(2H,d,J=8.1Hz),7.24-7.20(1H,m),7.18(1H,s),6.95(1H,d,J=8.3Hz),5.69(2H,s),3.67(3H,s),3.07(2H,t,J=7.7Hz),2.68(2H,t,J=7.8Hz),2.50(3H,s).
ESI-MSfound:337[M+H]+
工程(3)で得られた化合物[122-3](871mg)から実施例117の工程(2)の方法に準じて表題化合物(680mg)を黄白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:7.57(1H,d,J=8.1Hz),7.44(2H,d,J=8.1Hz),7.34(1H,s),7.33(1H,t,J=7.2Hz),7.02(1H,d,J=8.3Hz),5.70(2H,s),3.04(2H,t,J=7.7Hz),2.65(2H,t,J=7.7Hz),2.44(3H,s).
ESI-MSfound:363[M+H]+
3-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]プロピオン酸カリウム[123](以下、化合物[123]という)の合成
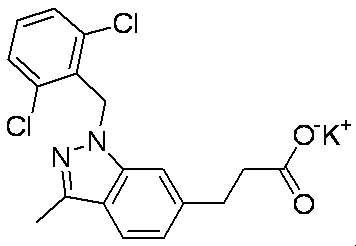
1H-NMR(400MHz,CD3OD)δ:7.55(1H,d,J=8.3Hz),7.44(2H,d,J=8.1Hz),7.36(1H,s),7.36-7.32(1H,m),7.05(1H,d,J=8.3Hz),5.71(2H,s),3.04(2H,t,J=8.1Hz),2.50(2H,t,J=8.2Hz),2.44(3H,s).
ESI-MSfound:363[M-K+2H]+
3-{1-[(5-クロロベンゾ[b]チオフェン-3-イル)メチル]-3-メチル-1H-インダゾール-6-イル}プロピオン酸[124](以下、化合物[124]という)の合成
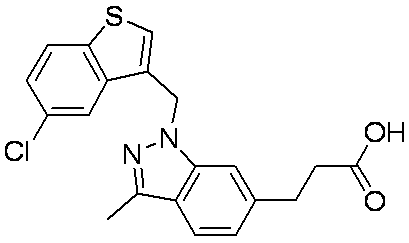
1H-NMR(400MHz,CDCl3)δ:7.85(1H,s),7.71(1H,d,J=8.5Hz),7.58(1H,d,J=7.8Hz),7.29(1H,dd,J=8.7,1.6Hz),7.14(1H,s),7.13(1H,s),6.99(1H,d,J=8.3Hz),5.66(2H,s),3.05(2H,t,J=7.7Hz),2.70(2H,t,J=7.7Hz),2.58(3H,s).
ESI-MSfound:385[M+H]+
3-[1-(2-クロロ-6-シアノベンジル)-3-メチル-1H-インダゾール-6-イル]プロピオン酸[125](以下、化合物[125]という)の合成
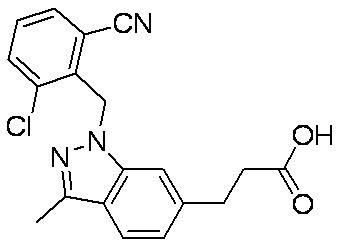
1H-NMR(400MHz,CD3OD)δ:7.78(1H,d,J=7.6Hz),7.74(1H,d,J=8.1Hz),7.59(1H,d,J=8.3Hz),7.54-7.50(1H,m),7.45(1H,s),7.06(1H,d,J=8.3Hz),5.72(2H,s),3.08(2H,t,J=7.7Hz),2.68(2H,t,J=7.6Hz),2.44(3H,s).
ESI-MSfound:354[M+H]+
3-[1-(2-クロロ-6-シアノベンジル)-3-メチル-1H-インダゾール-6-イル]プロピオン酸カリウム[126](以下、化合物[126]という)の合成

1H-NMR(400MHz,CD3OD)δ:7.78(1H,d,J=7.8Hz),7.74(1H,d,J=8.3Hz),7.57(1H,d,J=8.3Hz),7.54-7.50(1H,m),7.45(1H,s),7.08(1H,d,J=8.3Hz),5.72(2H,s),3.06(2H,t,J=8.1Hz),2.52(2H,t,J=8.1Hz),2.44(3H,s).
ESI-MSfound:354[M-K+2H]+
3-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]-3-ヒドロキシプロピオン酸[127](以下、化合物[127]という)の合成

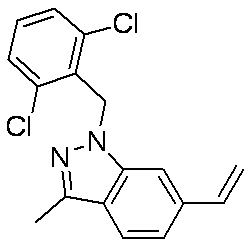
1H-NMR(400MHz,CDCl3)δ:7.55(1H,d,J=8.3Hz),7.46-7.21(5H,m),6.81(1H,dd,J=17.4,10.9Hz),5.79(1H,d,J=17.6Hz),5.71(2H,s),5.29(1H,d,J=11.0Hz),2.51(3H,s).

1H-NMR(400MHz,CDCl3)δ:10.1(1H,s),7.92(1H,s),7.75(1H,d,J=8.3Hz),7.64(1H,d,J=8.3Hz),7.39(2H,d,J=8.1Hz),7.29-7.25(1H,m),5.81(2H,s),2.56(3H,s).
工程(2)で得た化合物[127-2](105mg)のベンゼン(5mL)溶液に亜鉛粉末(40mg)、及びブロモ酢酸エチル(0.1mL)を室温で加えた後、2時間加熱還流した。反応混合物に水を加え、酢酸エチルで抽出した。得られた有機層を無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィーにて精製することにより黄色固体を得た。得られた黄色固体をエタノール(3mL)に溶解し、1N-水酸化ナトリウム水溶液(3mL)を室温で加えた後、60℃で2時間攪拌した。反応混合物に1N塩酸を加え、析出した固体を濾取することにより表題化合物(108mg)を白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:7.65(1H,dd,J=8.3Hz),7.57(1H,s),7.44(2H,d,J=8.1Hz),7.35-7.33(1H,m),7.18(1H,d,J=8.3Hz),5.73(2H,s),5.24-5.22(1H,m),2.74-2.73(2H,m),2.46(3H,s).
[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]ヒドロキシ酢酸[128](以下、化合物[128]という)の合成
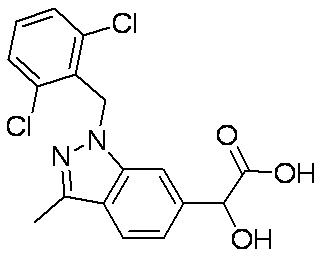
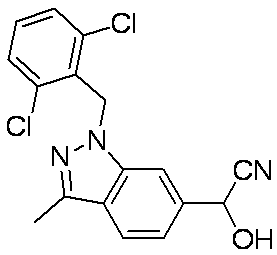
1H-NMR(400MHz,CDCl3)δ:7.70(1H,d,J=8.3Hz),7.56(1H,s),7.38(2H,d,J=8.1Hz),7.30-7.20(2H,m),5.75(2H,s),5.66(1H,d,J=6.6Hz),2.54(3H,s),1.60-1.50(1H,br).
工程(1)で得た化合物[128-1](24mg)を酢酸(1mL)に溶解し、濃塩酸(1mL)を室温で加え、60℃で5時間攪拌した。反応混合物に水を加え、酢酸エチルで抽出した。得られた有機層を無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮した。得られた残渣を逆相分取液体クロマトグラフィーにて精製することにより表題化合物(19mg)を白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:7.65(2H,d,J=9.8Hz),7.42(2H,d,J=8.1Hz),7.34-7.26(2H,m),5.73(2H,s),5.29(1H,s),2.46(3H,s).
ESI-MSfound:365[M+H]+
[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]ヒドロキシ酢酸カリウム[129](以下、化合物[129]という)の合成

1H-NMR(400MHz,CD3OD)δ:7.67(1H,s),7.59(1H,d,J=8.3Hz),7.43(2H,d,J=7.8Hz),7.36-7.28(2H,m),5.71(2H,s),4.99(1H,s),2.44(3H,s).
ESI-MSfound:365[M-K+2H]+
4-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]-4-オキソ酪酸[130](以下、化合物[130]という)の合成


1H-NMR(400MHz,CDCl3)δ:7.59(1H,d,J=7.8Hz),7.37-7.36(3H,m),7.23-7.21(1H,m),7.16(1H,d,J=7.8Hz),5.77(2H,s),2.53(3H,s),1.54-1.48(6H,m),1.39-1.28(12H,m),0.95-0.87(9H,m).

1H-NMR(400MHz,CDCl3)δ:8.06(1H,s),7.73-7.68(2H,m),7.39(2H,d,J=8.1Hz),7.28-7.25(1H,m),5.79(2H,s),4.19(2H,q,J=7.2Hz),3.37(2H,t,J=6.6Hz),2.79(2H,t,J=6.7Hz),2.55(3H,s),1.29(3H,t,J=7.2Hz).
工程(2)で得た化合物[130-2](136mg)のメタノール(3mL)溶液に1N-水酸化ナトリウム水溶液(3mL)を室温で加えた後、60℃で2時間攪拌した。反応混合物に1N-塩酸を加え、析出した固体を濾取することにより表題化合物(119mg)を白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:8.25(1H,s),7.76(2H,s),7.46(2H,d,J=7.8Hz),7.38-7.34(1H,m),5.85(2H,s),3.39(2H,t,J=6.5Hz),2.72(2H,t,J=6.3Hz),2.50(3H,s).
ESI-MSfound:391[M+H]+
2-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]アセトアミド[131](以下、化合物[131]という)の合成
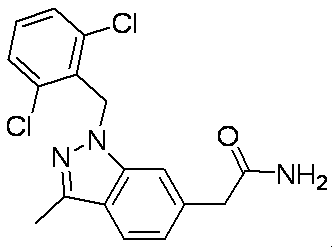
1H-NMR(400MHz,CD3OD)δ:7.61(1H,d,J=8.1Hz),7.47(1H,s),7.43(2H,d,J=7.8Hz),7.33(1H,dd,J=8.8,7.3Hz),7.09(1H,d,J=8.3Hz),5.72(2H,s),4.90-4.80(2H,brs),3.65(2H,s),2.45(3H,s).
ESI-MSfound:348[M+H]+
3-[1-(2、6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]プロピオンアミド[132](以下、化合物[132]という)の合成

1H-NMR(400MHz,CDCl3)δ:7.54(1H,d,J=8.3Hz),7.36(2H,d,J=8.1Hz),7.26-7.20(2H,m),6.97(1H,d,J=8.3Hz),5.69(2H,s),5.26(2H,s),3.09(2H,t,J=7.6Hz),2.57(2H,t,J=7.6Hz),2.50(3H,s).
ESI-MSfound:362[M+H]+
4-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]酪酸[133](以下、化合物[133]という)の合成


1H-NMR(400MHz,CDCl3)δ:9.90-9.80(1H,brs),7.58(1H,d,J=8.3Hz),7.33(1H,s),7.23(1H,d,J=8.5Hz),6.58(1H,d,J=15.9Hz),6.41-6.33(1H,m),3.73(3H,s),3.28(2H,d,J=6.6Hz),2.57(3H,s).
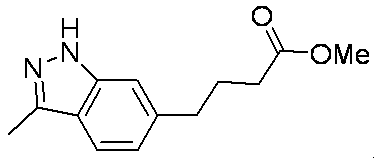
1H-NMR(400MHz,CDCl3)δ:9.78(1H,brs),7.58(1H,d,J=8.3Hz),7.21(1H,s),6.99(1H,d,J=8.1Hz),3.66(3H,s),2.78(2H,t,J=7.6Hz),2.57(3H,s),2.35(2H,t,J=7.4Hz),2.05-1.97(2H,m).
工程(2)で得た化合物[133-2](76mg)及び2,6-ジクロロベンジルクロリド(98mg)から実施例102の工程(5)の方法に準じて表題化合物(61mg)を白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:7.58(1H,d,J=8.3Hz),7.44(2H,d,J=8.1Hz),7.34-7.32(1H,m),7.28(1H,s),7.00(1H,d,J=8.3Hz),5.71(2H,s),2.78(2H,t,J=7.6Hz),2.45(3H,s),2.29(2H,t,J=7.3Hz),1.99-1.92(2H,m).
1-(2,6-ジクロロベンジル)-3-メチル-6-(1H-テトラゾール-5-イルメチル)-1H-インダゾール[134](以下、化合物[134]という)の合成

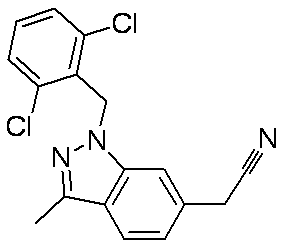
1H-NMR(400MHz,CDCl3)δ:7.61(1H,d,J=8.3Hz),7.38(2H,d,J=8.1Hz),7.34(1H,s),7.26-7.23(1H,m),7.00(1H,d,J=8.1Hz),5.73(2H,s),3.87(2H,s),2.52(3H,s).
ESI-MSfound:330[M+H]+
工程(1)で得られた化合物[134-1](112mg)をN,N-ジメチルホルムアミド(1.7mL)に溶解し、塩化アンモニウム(73mg)及びアジ化ナトリウム(66mg)を室温で加えた後、マイクロウェーブ反応装置を用いて160℃で2時間攪拌した。放冷後、反応混合物に6N-塩酸を加え、酢酸エチルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィーにて精製することにより表題化合物(77mg)を白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:7.64(1H,d,J=8.3Hz),7.41(2H,d,J=7.8Hz),7.33(1H,t,J=8.1Hz),7.28(1H,s),7.05(1H,d,J=8.3Hz),5.70(2H,s),4.44(2H,s),2.46(3H,s).
ESI-MSfound:373[M+H]+
1-(2,6-ジクロロベンジル)-3-メチル-6-[2-(1H-テトラゾール-5-イル)エチル]-1H-インダゾール[135](以下、化合物[135]という)の合成

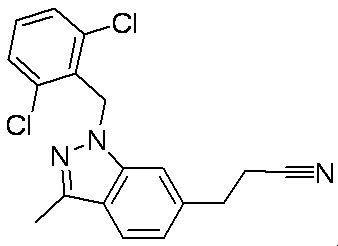
1H-NMR(400MHz,CDCl3)δ:7.58(1H,d,J=8.1Hz),7.37(2H,d,J=8.1Hz),7.26-7.23(2H,m),6.96(1H,d,J=8.1Hz),5.71(2H,s),3.07(2H,t,J=7.3Hz),2.66(2H,t,J=7.3Hz),2.51(3H,s).
工程(1)で得た化合物[135-1](40.7mg)から実施例134の工程(2)の方法に準じて表題化合物(9.2mg)を白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:7.60(1H,d,J=8.3Hz),7.43(2H,d,J=7.8Hz),7.33-7.25(2H,m),7.00(1H,d,J=8.1Hz),5.66(2H,s),3.31-3.22(4H,m),2.44(3H,s).
ESI-MSfound:387[M+H]+
5-{[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]メチル}-1,3,4-オキサジアゾール-2(3H)-オン[136](以下、化合物[136]という)の合成
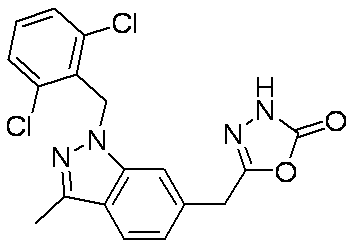

1H-NMR(400MHz,CDCl3)δ:7.61(1H,d,J=8.1Hz),7.37(2H,d,J=8.0Hz),7.33(1H,s),7.23-7.08(2H,m),7.04(1H,d,J=8.1Hz),6,39(1H,s),5.69(2H,s),3.76(2H,s),2.50(3H,s),1.45(9H,s).
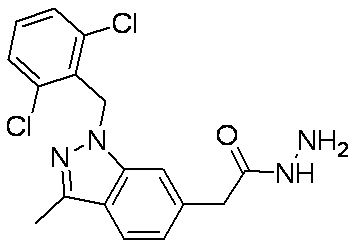
1H-NMR(400MHz,CDCl3)δ:7.60(1H,d,J=8.1Hz),7.37(2H,d,J=8.1Hz),7.24-7.20(2H,m),6.98(1H,d,J=8.1Hz),6.59(1H,s),5.70(2H,s),3.84(2H,s),3.69(2H,s),2.51(3H,s).
工程(2)で得た[136-2](84.0mg)のテトラヒドロフラン(1.7mL)溶液にN,N-ジイソプロピルエチルアミン(0.15mL)及び1,1’-カルボニルジイミダゾール(84.2mg)を室温で加えた後、2.5時間攪拌した。反応混合物を減圧濃縮した後に、得られた残渣を逆相分取液体クロマトグラフィーにて精製することにより表題化合物(9.2mg)を白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:7.65(1H,d,J=8.3Hz),7.44(2H,d,J=7.8Hz),7.36(1H,s),7.35-7.32(1H,m),7.07(1H,d,J=8.3Hz),5.74(2H,s),4.03(2H,s),2.47(3H,s).
ESI-MSfound:389[M+H]+
5-{[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]メチル}-1,3,4-オキサジアゾール-2(3H)-チオン[137](以下、化合物[137]という)の合成

1H-NMR(400MHz,CD3OD)δ:7.56(1H,d,J=8.0Hz),7.35(2H,d,J=7.6Hz),7.27(1H,s),7.26-7.23(1H,m),6.98(1H,d,J=8.4Hz),5.64(2H,s),4.08(2H,s),2.37(3H,s).
ESI-MSfound:405[M+H]+
2-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イルオキシ]酢酸[138](以下、化合物[138]という)の合成


1H-NMR(400MHz,CDCl3)δ:7.52(1H,d,J=9.3Hz),6.80-6.78(2H,m),3.86(3H,s),2.54(3H,s).
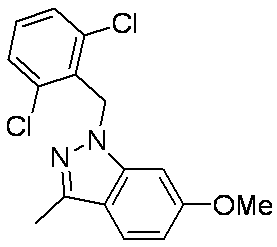
1H-NMR(400MHz,CDCl3)δ:7.46(1H,d,J=8.8Hz),7.37(2H,d,J=8.1Hz),,7.25-7.21(1H,m),6.74(1H,dd,J=8.7,1.8Hz),6.69(1H,s),5.67(2H,s),3.81(3H,s),2.49(3H,s).
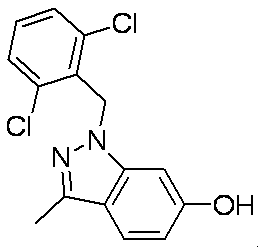
1H-NMR(400MHz,CD3OD)δ:7.46-7.44(3H,m),7.35-7.31(1H,m),6.70(1H,s),6.68-6.65(1H,m),5.60(2H,s),2.40(3H,s).
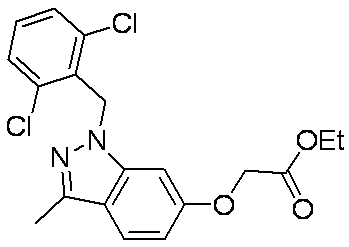
1H-NMR(400MHz,CDCl3)δ:7.50(1H,d,J=8.8Hz),7.37(2H,d,J=8.1Hz),7.24-7.22(1H,m),6.81(1H,dd,J=8.8,2.0Hz),6.67(1H,d,J=1.7Hz),5.65(2H,s),4.63(2H,s),4.28(2H,q,J=7.2Hz),2.48(3H,s),1.30(3H,t,J=7.2Hz).
工程(4)で得た化合物[138-4](89mg)から実施例117の工程(2)の方法に準じて表題化合物(72mg)を白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:7.55(1H,d,J=9.3Hz),7.44(2H,d,J=8.1Hz),7.36-7.32(1H,m),6.83(2H,d,J=7.1Hz),5.68(2H,s),4.68(2H,s),2.43(3H,s).
[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]メタンスルホン酸[139](以下、化合物[139]という)の合成

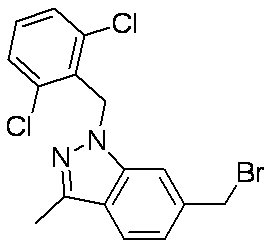
1H-NMR(400MHz,CDCl3)δ:7.59(1H,d,J=8.3Hz),7.40-7.36(3H,m),7.23(1H,d,J=8.1Hz),7.14(1H,d,J=8.3Hz),5.70(2H,s),4.63(2H,s),2.50(3H,s).
工程(1)で得た化合物[139-1](119mg)のジメチスルホキシド(2mL)及び水(5mL)溶液に亜硫酸ナトリウム(49mg)を加えて、130℃で1時間攪拌した。反応混合物に水を加え、クロロホルムで抽出した。得られた有機層を無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィーにて精製することにより表題化合物(6mg)を白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:7.65-7.63(2H,brm),7.45(2H,d,J=7.8Hz),7.37-7.33(1H,m),7.29(1H,s),5.76(2H,s),4.22(2H,s),2.49(3H,s).
ESI-MSfound:385[M+H]+
2-[1-(2,6-ジクロロベンジル)-3-エチル-1H-インダゾール-6-イル]酢酸[140](以下、化合物[140]という)の合成


1H-NMR(400MHz,CDCl3)δ:7.39-7.16(3H,m),4.96-4.85(1H,m),1.88(1H,d,J=2.9Hz),1.84-1.70(2H,m),0.94(3H,t,J=7.4Hz).

1H-NMR(400MHz,CDCl3)δ:7.77(1H,t,J=8.2Hz),7.40-7.30(2H、m),3.02-2.94(2H,m),1.20(3H,t,J=7.2Hz).

1H-NMR(400MHz,CDCl3)δ:10.50-9.50(1H,br),7.61(1H,s),7.57(1H,d,J=8.5Hz),7.28-7.21(1H,m),3.00(2H,q,J=7.6Hz),1.41(3H,t,J=7.6Hz).

1H-NMR(400MHz,CDCl3)δ:7.51(2H,d,J=8.3Hz),7.37(2H,d,J=8.1Hz),7.23(1H,d,J=8.3Hz),7.18(1H,d,J=9.8Hz),5.68(2H,s),2.92(2H,q,J=7.6Hz),1.32(3H,t,J=7.6Hz).

1H-NMR(400MHz,CDCl3)δ:7.57(1H,d,J=8.3Hz),7.35(2H,d,J=8.1Hz),7.26-7.18(1H,m),7.12(1H,s),6.93(1H,d,J=8.1Hz),6.04-5.90(1H,m),5.70(2H,s),5.12-5.05(2H,m),3.48(2H,d,J=6.6Hz),2.93(2H,q,J=7.6Hz),1.33(3H,t,J=7.6Hz).

工程(6)で得られた化合物[140-6](71mg)から実施例66の工程(4)の方法に準じて表題化合物(56mg)を褐色固体として得た。
1H-NMR(400MHz,CD3OD)δ:7.65(1H,d,J=8.3Hz),7.48-7.40(3H,m),7.34(1H,dd,J=8.8,7.3Hz),7.07(1H,d,J=7.6Hz),5.73(2H,s),3.72(2H,s),2.90(2H,q,J=7.6Hz),1.30(3H,t,J=7.7Hz).
ESI-MSfound:363[M+H]+
2-[1-(2,6-ジクロロベンジル)-3-エチル-1H-インダゾール-6-イル]酢酸カリウム[141](以下、化合物[141]という)の合成
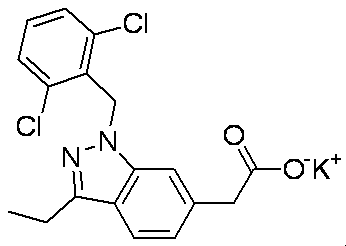
1H-NMR(400MHz,CD3OD)δ:7.60(1H,d,J=8.1Hz),7.46-7.38(3H,m),7.34-7.30(1H,m),7.13(1H,d,J=8.1Hz),5.71(2H,s),3.61(2H,s),2.88(2H,q,J=7.5Hz),1.28(3H,t,J=7.6Hz).
ESI-MSfound:363[M-K+2H]+
2-[1-(2,6-ジメチルベンジル)-3-エチル-1H-インダゾール-6-イル]酢酸[142](以下、化合物[142]という)の合成


1H-NMR(400MHz,CDCl3)δ:7.46(1H,d,J=8.5Hz),7.21-7.08(5H,m),5.45(2H,s),2.93(2H,q,J=7.6Hz),2.32(6H,s),1.35(3H,t,J=7.6Hz).
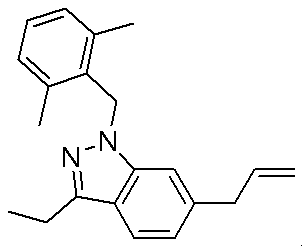
1H-NMR(400MHz,CDCl3)δ:7.55(1H,d,J=8.3Hz),7.16(1H,t,J=7.6Hz),7.06(2H,d,J=7.6Hz),6.89(1H,d,J=8.3Hz),6.71(1H,s),5.95-5.82(1H,m),5.49(2H,s),5.10-5.00(2H,m),3.37(2H,d,J=6.8Hz),2.93(2H,q,J=7.6Hz),2.33(6H,s),1.35(3H,t,J=7.6Hz).
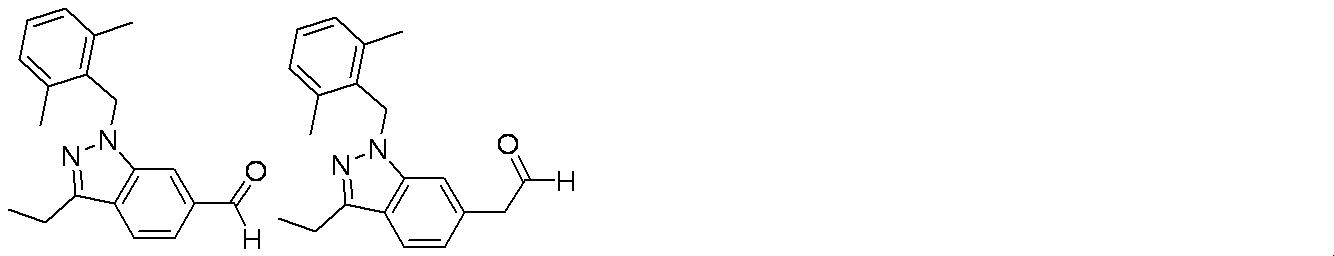
[142-3A]
1H-NMR(400MHz,CDCl3)δ:9.91(1H,s),7.76(1H,d,J=8.3Hz),7.58(1H,d,J=8.3Hz),7.38(1H,s),7.23-7.19(1H,m),7.11(2H,d,J=7.6Hz),5.61(2H,s),2.99(2H,q,J=7.6Hz),2.34(6H,s),1.38(3H,t,J=7.6Hz).
[142-3B]
1H-NMR(400MHz,CDCl3)δ:9.67(1H,s),7.64(1H,d,J=8.3Hz),7.20-7.16(1H,m),7.08(2H,d,J=7.6Hz),6.88(1H,d,J=7.8Hz),6.70(1H,s),5.51(2H,s),3.66(2H,d,J=2.0Hz),2.96(2H,q,J=7.6Hz),2.32(6H,s),1.36(3H,t,J=7.6Hz).
工程(3)で得た化合物[142-3B](51mg)から、実施例66の工程(4)の方法に準じて表題化合物(35mg)を白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:7.64(1H,d,J=8.3Hz),7.17-7.00(5H,m),5.50(2H,s),3.64(2H,s),2.92(2H,q,J=7.6Hz),2.27(6H,s),1.31(3H,t,J=7.6Hz).
ESI-MSfound:323[M+H]+
1-(2,6-ジメチルベンジル)-3-エチル-1H-インダゾール-6-カルボン酸[143](以下、化合物[143]という)の合成

1H-NMR(400MHz,DMSO-d6)δ:8.21(1H,s),7.80(1H,d,J=8.3Hz),7.63(1H,d,J=8.5Hz),7.14-7.03(3H,m),5.56(2H,s),2.87(2H,q,J=7.5Hz),2.29(6H,s),1.24(3H,t,J=7.6Hz).
ESI-MSfound:309[M+H]+
1-(2,6-ジクロロベンジル)-3-エチル-1H-インダゾール-6-カルボン酸[144](以下、化合物[144]という)の合成
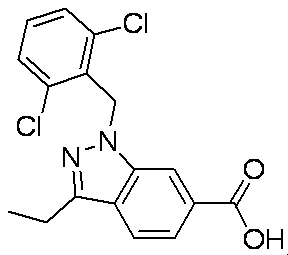
1H-NMR(400MHz,CD3OD)δ:8.25(1H,s),7.79-7.75(2H,m),7.45(2H,d,J=8.1Hz),7.35(1H,t,J=8.1Hz),5.82(2H,s),2.94(2H,q,J=7.6Hz),1.31(3H,t,J=7.6Hz).
ESI-MSfound:349[M+H]+
3-[1-(2,6-ジクロロベンジル)-3-エチル-1H-インダゾール-6-イル]プロピオン酸[145](以下、化合物[145]という)の合成
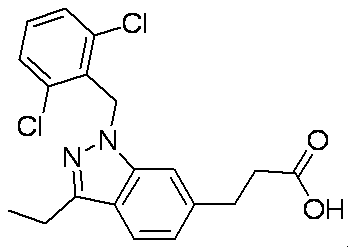

1H-NMR(400MHz,CDCl3)δ:7.77(1H,d,J=16.1Hz),7.65(1H,d,J=8.5Hz),7.44-7.34(3H,m),7.33-7.20(2H,m),6.47(1H,d,J=16.1Hz),5.76(2H,s),3.83(3H,s),2.95(2H,q,J=7.6Hz),1.34(3H,t,J=7.6Hz).

1H-NMR(400MHz,CDCl3)δ:7.57(1H,d,J=8.3Hz),7.36(2H,d,J=7.8Hz),7.30-7.18(1H,m),7.14(1H,s),6.94(1H,d,J=8.3Hz),5.70(2H,s),3.67(3H,s),3.05(2H,t,J=7.8Hz),2.92(2H,q,J=7.6Hz),2.66(2H,t,J=7.7Hz),1.31(3H,t,J=7.6Hz).
工程(2)で得た化合物[145-2](40mg)から、実施例117の工程(2)記載の方法に準じて表題化合物(39mg)を白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:7.62(1H,d,J=8.1Hz),7.44(2H,d,J=8.1Hz),7.35(1H,d,J=7.6Hz),7.31(1H,s),7.02(1H,d,J=8.1Hz),5.72(2H,s),3.04(2H,t,J=7.6Hz),2.89(2H,q,J=7.6Hz),2.65(2H,t,J=7,7Hz),1.29(3H,t,J=7.6Hz).
ESI-MSfound:377[M+H]+
(E)-3-[1-(2,6-ジクロロベンジル)-3-エチル-1H-インダゾール-6-イル]アクリル酸[146](以下、化合物[146]という)の合成

1H-NMR(400MHz,CD3OD)δ:7.85-7.65(3H,m),7.50-7.30(4H,m),6.55(1H,d,J=16.1Hz),5.80(2H,s),2.92(2H,q,J=7.6Hz),1.31(3H,t,J=7.7Hz).
ESI-MSfound:375[M+H]+
2-[1-(2,6-ジメチルベンジル)-3-イソプロピル-1H-インダゾール-6-イル]酢酸[147](以下、化合物[147]という)の合成


1H-NMR(400MHz,CDCl3)δ:7.64-7.61(2H,m),7.24(1H,dd,J=8.5,1.7Hz),3.45-3.35(1H,m),1.45(6H,d,J=6.8Hz).
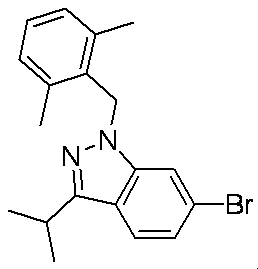
1H-NMR(400MHz,CDCl3)δ:7.56(1H,dd,J=8.5,0.5Hz),7.21-7.17(1H,m),7.12-7.02(4H,m),5.48(2H,s),3.39-3.28(1H,m),2.33(6H,s),1.41(6H,d,J=7.1Hz).
工程(2)で得た化合物[147-2](50mg)から実施例66の工程(2)~(4)の方法に準じての表題化合物(41mg)を白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:7.69(1H,dd,J=8.4,0.6Hz),7.14-7.11(1H,m),7.06-7.04(3H,m),7.00(1H,dd,J=8.3,1.5Hz),5.50(2H,s),3.61(2H,s),3.38-3.29(1H,m),2.28(6H,s),1.39(6H,d,J=6.8Hz).
ESI-MSfound:337[M+H]+
2-[3-シクロプロピル-1-(2,6-ジクロロベンジル)-1H-インダゾール-6-イル]酢酸[148](以下、化合物[148]という)の合成


1H-NMR(400MHz,CDCl3)δ:7.70-7.35(1H,m),7.40-7.20(2H,m),2.60-2.50(1H,m),1.43-1.25(2H,m),1.11-1.05(2H,m).

1H-NMR(400MHz,CDCl3)δ:10.0-9.50(1H,br),7.65-7.55(2H,m),7.23(1H,d,J=8.5,1.2Hz),2.24-2.16(1H,m),1.10-1.00(4H,m).

1H-NMR(400MHz,CDCl3)δ:7.51-7.49(2H,m),7.40-7.30(2H,m),7.27-7.10(2H,m),5.63(2H,s),2.16-2.08(1H,m),1.10-0.99(4H,m).
工程(3)で得られた化合物[148-3](286mg)から実施例66の工程(2)~(4)の方法に準じて表題化合物(28mg)を褐色固体として得た。
1H-NMR(400MHz,CD3OD)δ:7.58(1H,d,J=7.3Hz),7.45-7.36(3H,m),7.31(1H,dd,J=8.8,7.6Hz),7.10(1H,d,J=7.3Hz),5.67(2H,s),3.59(2H,s),2.17-2.11(1H,m),0.94(4H,d,J=5.4Hz).
ESI-MSfound:375[M+H]+
3-クロロ-1-(2,6-ジメチルベンジル)-1H-インダゾール-6-カルボン酸[149](以下、化合物[149]という)の合成


1H-NMR(400MHz,CDCl3)δ:7.79(1H,d,J=8.8Hz),7.65(2H,d,J=8.1Hz),7.26-7.23(1H,m),7.14(2H,d,J=7.6Hz),7.06-7.02(1H,m),6.92(2H,d,J=7.6Hz),5.54(2H,s),5.40(2H,s),2.42(6H,s),2.25(6H,s).
工程(1)で得た化合物[149-1](24mg)から実施例117の工程(2)の方法に準じて表題化合物(15mg)を白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:8.19(1H,s),7.84(1H,d,J=7.8Hz),7.71(1H,d,J=8.5Hz),7.18-7.14(1H,m),7.08(2H,d,J=7.3Hz),5.64(2H,s),2.31(6H,s).
ESI-MSfound:315[M+H]+
3-シアノ-1-(2,6-ジクロロベンジル)-1H-インダゾール-6-カルボン酸[150](以下、化合物[150]という)の合成

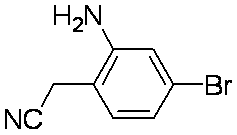
1H-NMR(400MHz,CDCl3)δ:7.06(1H,d,J=8.1Hz),6.94-6.91(2H,m),3.75(2H,brs),3.52(2H,s).

1H-NMR(400MHz,CD3OD)δ:7.91(1H,s),7.76(1H,d,J=8.8Hz),7.49(1H,d,J=8.8Hz).
工程(2)で得た化合物[150-2](570mg)及び2、6-ジクロロベンジルクロリド(747mg)から実施例66の方法に準じて表題化合物(118mg)を白色固体として得た。
1H-NMR(400MHz,DMSO-d6)δ:13.31(1H,brs),8.67(1H,s),7.97(1H,d,J=8.5Hz),7.92(1H,d,J=8.5Hz),7.55(2H,d,J=7.8Hz),7.48-7.44(1H,m),6.04(2H,s).
1-(2,6-ジクロロベンジル)-3-メチル-1H-ピラゾロ[3,4-b]ピリジン-6-カルボン酸[151](以下、化合物[151]という)の合成


1H-NMR(400MHz,CDCl3)δ:7.65(1H,d,J=8.0Hz),7.34(1H,d,J=8.0Hz),3.51(3H,s),3.40(3H,s).
ESI-MSfound:235[M+H]+

1H-NMR(400MHz,CDCl3)δ:7.93(1H,d,J=8.1Hz),7.37(1H,d,J=8.1Hz),2.71(3H,s).
ESI-MSfound:190[M+H]+
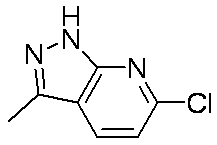
1H-NMR(400MHz,CD3OD)TM:10.15(1H,brs),7.97(1H,d,J=8.3Hz),7.15(1H,d,J=8.3Hz),2.58(3H,s).
ESI-MSfound:168[M+H]+

1H-NMR(400MHz,CDCl3)δ:7.89(1H,d,J=8.3Hz),7.39-7.31(2H,m),7.26-7.18(1H,m),7.09(1H,d,J=8.3Hz),5.83(2H,s),2.47(3H,s).
ESI-MSfound:326[M+H]+

1H-NMR(400MHz,CDCl3)δ:8.09(1H,d,J=8.1Hz),7.46(1H,d,J=8.1Hz),7.37(2H,d,J=8.1Hz),7.25(1H,d,J=8.3Hz),5.90(2H,s),2.53(3H,s).
ESI-MSfound:317[M+H]+
工程(5)で得た化合物[151-5](38mg)のテトラヒドロフラン(0.5mL)及びメタノール(0.5mL)溶液に3N-水酸化ナトリウム溶液(0.6mL)を加えた後、マイクロウェーブ反応装置を用いて150℃で5分間反応させた。反応混合物に1M塩酸(0.46mL)を加え、溶媒を減圧濃縮した。得られた残渣を逆相分取液体クロマトグラフィーにて精製することにより、表題化合物(34mg)を白色固体として得た。
1H-NMR(400MHz,DMSO-d6)δ:13.42(1H,s),8.37(1H,d,J=8.3Hz),7.86(1H,d,J=8.3Hz),7.52(1H,d,J=8.7Hz),7.52(1H,d,J=7.4Hz),7.42(1H,dd,J=8.7,7.4Hz),5.82(2H,s),2.44(3H,s).
ESI-MSfound:336[M+H]+
1-(2,6-ジクロロベンジル)-3-メチル-6-(1H-テトラゾール-5-イルメチル)-1H-ピラゾロ[3,4-b]ピリジン[152](以下、化合物[152]という)の合成


1H-NMR(400MHz,CDCl3)δ:7.98(1H,d,J=8.1Hz),7.35(2H,d,J=7.8Hz),7.24-7.20(1H,m),7.17(1H,d,J=8.1Hz),5.86(2H,s),4.04(2H,s),2.50(3H,s).
ESI-MSfound:331[M+H]+
工程(1)で得た化合物[152-1](28mg)から実施例134の工程(2)の方法に準じて表題化合物(4.4mg)を黄白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:8.11(1H,d,J=8.0Hz),7.37(2H,d,J=7.6Hz),7.29(1H,dd,J=8.8,7.1Hz),7.21(1H,d,J=8.3Hz),5.79(2H,s),4.64(2H,s),2.45(3H,s).
ESI-MSfound:374[M+H]+
1-(2,6-ジクロロベンジル)-3-メチル-1H-ピラゾロ[4,3-b]ピリジン-6-カルボン酸[153](以下、化合物[153]という)の合成
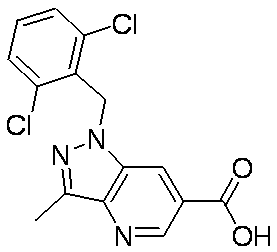

1H-NMR(400MHz,CDCl3)δ:8.56(1H,s),7.74(1H,d,J=9.6Hz),2.68(3H,s).

1H-NMR(400MHz,CD3OD)δ:8.51(1H,d,J=1.7Hz),8.16(1H,d,J=2.0Hz),2.59(3H,s).
ESI-MSfound:212[M+H]+

1H-NMR(400MHz,CDCl3)δ:8.51(1H,d,J=1.7Hz),7.80(1H,d,J=1.7Hz),7.39(2H,d,J=7.8Hz),7.29-7.26(1H,m),5.69(2H,s),2.60(3H,s).
ESI-MSfound:370[M+H]+

1H-NMR(400MHz,CDCl3)δ:8.69(1H,d,J=1.5Hz),7.93(1H,d,J=1.5Hz),7.41(2H,d,J=8.1Hz),7.33-7.29(1H,m),5.80(2H,s),2.65(3H,s).
ESI-MSfound:317[M+H]+
工程(4)で得られた化合物[153-4](44mg)のエタノール(3.5mL)溶液に3N-水酸化ナトリウム水溶液(3.5mL)を室温で加えた後、110℃で15分間攪拌した。放冷後、3N-塩酸を加え、クロロホルムで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮した。得られた残渣を逆相分取液体クロマトグラフィーにて精製することにより表題化合物(28mg)を白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:9.05(1H,s),8.64(1H,s),7.46(2H,d,J=7.8Hz),7.38-7.34(1H,m),5.87(2H,s),2.56(3H,s).
ESI-MSfound:336[M+H]+
1-(2,6-ジクロロベンジル)-3-メチル-1H-ピラゾロ[4,3-b]ピリジン-6-カルボン酸カリウム[154](以下、化合物[154]という)の合成
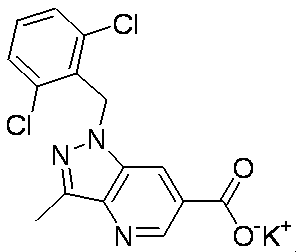
1H-NMR(400MHz,CD3OD)δ:9.06(1H,s),8.55(1H,s),7.44(2H,d,J=7.8Hz),7.36-7.32(1H,m),5.83(2H,s),2.54(3H,s).
ESI-MSfound:336[M+H]+
(E)-3-[1-(2,6-ジクロロベンジル)-3-メチル-1H-ピラゾロ[4,3-b]ピリジン-6-イル]アクリル酸[155](以下、化合物[155]という)の合成
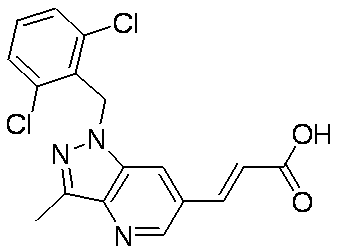

1H-NMR(400MHz,CDCl3)δ:8.68(1H,s),7.79(1H,d,J=16.1Hz),7.70(1H,s),7.41(2H,d,J=8.1Hz),7.31-7.26(1H,m),6.55(1H,d,J=16.1Hz),5.77(2H,s),3.84(3H,s),2.63(3H,s).
ESI-MSfound:376[M+H]+
工程(1)で得た化合物[155-1](25mg)から実施例117の工程(2)の方法に準じて表題化合物(9.0mg)を白色固体として得た。
1H-NMR(400MHz,CDCl3)δ:8.65(1H,s),7.76(1H,d,J=16.3Hz),7.71(1H,s),7.39(2H,d,J=8.1Hz),7.29-7.26(1H,m),6.52(1H,d,J=15.9Hz),5.76(2H,s),2.61(3H,s).
ESI-MSfound:362[M+H]+
1-(2,6-ジクロロベンジル)-3-メチル-6-(1H-テトラゾール-5-イル)-1H-ピラゾロ[4,3-b]ピリジン[156](以下、化合物[156]という)の合成

1H-NMR(400MHz,CD3OD)δ:9.14(1H,d,J=1.5Hz),8.74(1H,d,J=1.5Hz),7.47(2H,d,J=8.1Hz),7.37(1H,dd,J=8.8,7.3Hz),5.89(2H,s),2.58(3H,s).
ESI-MSfound:360[M+H]+
5-[1-(2,6-ジクロロベンジル)-3-メチル-1H-ピラゾロ[4,3-b]ピリジン-6-イル]-1H-テトラゾール-1-イド=カリウム[157](以下、化合物[157]という)の合成
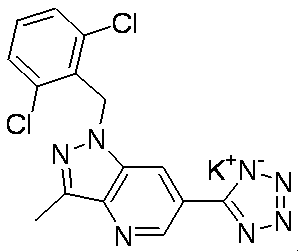
1H-NMR(400MHz,CD3OD)δ:9.17(1H,s),8.69(1H,s),7.46(2H,d,J=7.8Hz),7.38-7.34(1H,m),5.88(2H,s),2.57(3H,s).
ESI-MSfound:360[M-K+2H]+
1-(2,3-ジクロロベンジル)-3-メチル-6-(1H-テトラゾール-5-イル)-1H-ピラゾロ[4,3-b]ピリジン[158](以下、化合物[158]という)の合成
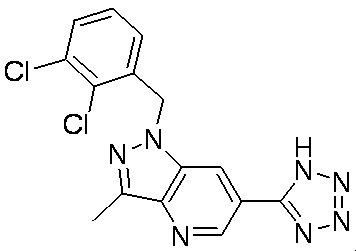
1H-NMR(400MHz,CD3OD)δ:9.19(1H,d,J=1.5Hz),8.64(1H,d,J=1.5Hz),7.50(1H,d,J=8.3Hz),7.25-7.21(1H,m),6.88(1H,d,J=7.6Hz),6.82(2H,s)2.66(3H,s).
ESI-MSfound:360[M+H]+
5-[1-(2,3-ジクロロベンジル)-3-メチル-1H-ピラゾロ[4,3-b]ピリジン-6-イル]-1H-テトラゾール-1-イド=カリウム[159](以下、化合物[159]という)の合成

1H-NMR(400MHz,CD3OD)δ:9.25(1H,d,J=1.5Hz),8.55(1H,d,J=1.5Hz),7.48(1H,d,J=8.1Hz),7.23-7.19(1H,m),6.81(1H,d,J=7.6Hz),5.80(2H,s)2.65(3H,s).
ESI-MSfound:360[M-K+2H]+
1-(2-クロロ-6-メチルベンジル)-3-メチル-6-(1H-テトラゾール-5-イル)-1H-ピラゾロ[4,3-b]ピリジン[160](以下、化合物[160]という)の合成

1H-NMR(400MHz,CD3OD)δ:9.12(1H,d,J=1.5Hz),8.69(1H,d,J=1.5Hz),7.31(1H,d,J=7.2Hz),7.28-7.25(2H,m),5.79(2H,s),2.59(3H,s),2.49(3H,s).
ESI-MSfound:340[M+H]+
5-[1-(2-クロロ-6-メチルベンジル)-3-メチル-1H-ピラゾロ[4,3-b]ピリジン-6-イル]-1H-テトラゾール-1-イド=カリウム[161](以下、化合物[161]という)の合成
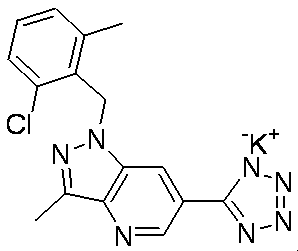
1H-NMR(400MHz,CDCl3)δ:9.15(1H,d,J=1.2Hz),8.64(1H,s),7.30(1H,d,J=7.1Hz),7.27-7.22(2H,m),5.76(2H,s),2.58(3H,s),2.48(3H,s).
ESI-MSfound:340[M+K-2H]+
3-メチル-1-(ナフタレン-1-イル)メチル-6-(1H-テトラゾール-5-イル)-1H-ピラゾロ[4,3-b]ピリジン[162](以下、化合物[162]という)の合成

1H-NMR(400MHz,CD3OD)δ:9.20(1H,d,J=1.2Hz),8.52(1H,d,J=1.2Hz),8.25(1H,d,J=8.3Hz),7.90(1H,d,J=6.8Hz),7.84(1H,d,J=8.3Hz),7.56-7.51(2H,m),7.43-7.39(1H,m),7.16(1H,d,J=6.8Hz),6.16(2H,s),2.67(3H,s).
1-(2,5-ジメチルベンジル)-3-メチル-6-(1H-テトラゾール-5-イル)-1H-ピラゾロ[4,3-b]ピリジン[163](以下、化合物[163]という)の合成

1H-NMR(400MHz,CD3OD)δ:9.17(1H,d,J=1.7Hz),8.48(1H,d,J=1.5Hz),7.07(1H,d,J=7.8Hz),7.00(1H,d,J=7.3Hz),6.72(1H,s),5.62(2H,s),2.65(3H,s),2.31(3H,s),2.19(3H,s).
1-(2-クロロ-6-シクロプロピルベンジル)-3-メチル-6-(1H-テトラゾール-5-イル)-1H-ピラゾロ[4,3-b]ピリジン[164](以下、化合物[164]という)の合成

1H-NMR(400MHz,CD3OD)δ:9.12(1H,d,J=1.5Hz),8.68(1H,d,J=1.5Hz),7.27-7.26(2H,m),7.13(1H,d,J=6.8Hz),6.00(2H,s),2.59(3H,s),2.24-2.17(1H,m),0.92-0.89(2H,m),0.70-0.68(2H,m).
ESI-MSfound:366[M+H]+
5-[1-(2-クロロ-6-シクロプロピルベンジル)-3-メチル-1H-ピラゾロ[4,3-b]ピリジン-6-イル]-1H-テトラゾール-1-イド=カリウム[165](以下、化合物[165]という)の合成

1H-NMR(400MHz,CD3OD)δ:9.19(1H,d,J=1.5Hz),8.60(1H,d,J=1.5Hz),7.32-7.25(2H,m),7.12(1H,d,J=7.3Hz),5.96(2H,s),2.57(3H,s),2.18-2.16(1H,m),0.90-0.86(2H,m),0.68-0.66(2H,m).
ESI-MSfound:366[M-K+2H]+
1-(2,6-ジシクロプロピルベンジル)-3-メチル-6-(1H-テトラゾール-5-イル)-1H-ピラゾロ[4,3-b]ピリジン[166](以下、化合物[166]という)の合成

1H-NMR(400MHz,CDCl3)δ:9.55(1H,s),8.48(1H,s),7.05(2H,d,J=7.3Hz),6.18(2H,s),2.78(3H,s),1.91-1.87(2H,m),0.90-0.86(4H,m),0.69-0.66(4H,m).
ESI-MSfound:372[M+H]+
1-(2,6-ジクロロベンジル)-3-エチル-1H-ピラゾロ[4,3-b]ピリジン-6-カルボン酸[167](以下、化合物[167]という)の合成

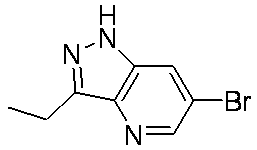
1H-NMR(400MHz,CDCl3)δ:9.87(1H,s),8.58(1H,d,J=1.7Hz),7.95(1H,d,J=1.7Hz),3.12(2H,q,J=7.6Hz),1.45(3H,t,J=7.6Hz).

1H-NMR(400MHz,CDCl3)δ:8.51(1H,d,J=1.7Hz),7.77(1H,d,J=1.7Hz),7.40(2H,d,J=8.1Hz),7.29-7.27(1H,m),5.71(2H,s),3.04(2H,q,J=7.6Hz),1.39(3H,t,J=7.6Hz).
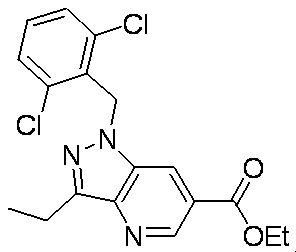
1H-NMR(400MHz,CDCl3)δ:9.11(1H,s),8.32(1H,s),7.40(2H,d,J=8.1Hz),7.30-7.26(1H,m),5.81(2H,s),4.44(2H,q,J=7.2Hz),3.10(2H,q,J=7.6Hz),1.44-1.40(6H,m).
1H-NMR(400MHz,CD3OD)δ:9.04(1H,d,J=1.5Hz),8.64(1H,d,J=1.5Hz),7.47(2H,d,J=7.8Hz),7.38-7.36(1H,m),5.89(2H,s),3.03(2H,q,J=7.5Hz),1.34(3H,t,J=7.6Hz).
1-(2,6-ジメチルベンジル)-3-エチル-1H-ピラゾロ[4,3-b]ピリジン-6-カルボン酸[168](以下、化合物[168]という)の合成

1H-NMR(400MHz,CD3OD)δ:8.99(1H,d,J=1.5Hz),8.20(1H,d,J=1.5Hz),7.20-7.16(1H,m),7.09(2H,d,J=7.3Hz),5.67(2H,s),3.05(2H,q,J=7.6Hz),2.31(6H,s),1.37(3H,t,J=7.6Hz).
ESI-MSfound:310[M+H]+
1-(2,3-ジクロロベンジル)-3-エチル-1H-ピラゾロ[4,3-b]ピリジン-6-カルボン酸[169](以下、化合物[169]という)の合成

1H-NMR(400MHz,CD3OD)δ:9.06(1H,d,J=1.5Hz),8.58(1H,d,J=1.5Hz),7.48(1H,d,J=7.8Hz),7.22-7.18(1H,m),6.84(1H,d,J=7.3Hz),5.79(2H,s),3.09(2H,q,J=7.6Hz),1.40(3H,t,J=7.6Hz).
ESI-MSfound:350[M+H]+
1-(2-クロロ-6-メチルベンジル)-3-エチル-1H-ピラゾロ[4,3-b]ピリジン-6-カルボン酸[170](以下、化合物[170]という)の合成

1H-NMR(400MHz,CDCl3)δ:9.02(1H,d,J=1.2Hz),8.54(1H,d,J=1.5Hz),7.32(1H,d,J=7.3Hz),7.28-7.24(1H,m),7.22(1H,d,J=6.8Hz),5.79(2H,s),3.04(2H,q,J=7.6Hz),2.45(3H,s),1.36(3H,t,J=7.6Hz).
ESI-MSfound:330[M+H]+
1-(2,6-ジクロロベンジル)-3-エチル-6-(1H-テトラゾール-5-イル)-1H-ピラゾロ[4,3-b]ピリジン[171](以下、化合物[171]という)の合成
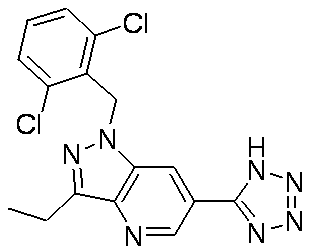
1H-NMR(400MHz,CD3OD)δ:9.12(1H,s),8.72(1H,d,J=1.5Hz),7.47(2H,d,J=7.8Hz),7.39-7.35(1H,m),5.90(2H,s),3.04(2H,q,J=7.6Hz),1.36(3H,t,J=7.6Hz).
ESI-MSfound:374[M+H]+
3-クロロ-1-(2,6-ジクロロベンジル)-1H-ピラゾロ[4,3-b]ピリジン-6-カルボン酸[172](以下、化合物[172]という)の合成

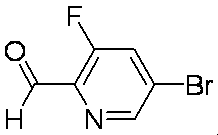
1H-NMR(400MHz,CDCl3)δ:10.17(1H,s),8.69(1H,s),7.80(1H,dd,J=9.1,1.3Hz).

1H-NMR(400MHz,CDCl3)δ:10.17(1H,br),8.65(1H,d,J=1.7Hz),8.31(1H,s),8.04(1H,s).
ESI-MSfound:198[M+H]+

1H-NMR(400MHz,CDCl3)δ:9.99(1H,br),8.69(1H,d,J=2.0Hz),8.02(1H,d,J=2.0Hz).
ESI-MSfound:232[M+H]+

1H-NMR(400MHz,CDCl3)δ:8.62(1H,d,J=1.5Hz),7.93(1H,d,J=1.5Hz),7.41(2H,d,J=8.1Hz),7.32-7.28(1H,m),5.72(2H,s).
ESI-MSfound:390[M+H]+

1H-NMR(400MHz,CDCl3)δ:8.79(1H,d,J=1.5Hz),8.06(1H,d,J=1.5Hz),7.43(2H,d,J=7.8Hz),7.36-7.32(1H,m),5.84(2H,s).
ESI-MSfound:337[M+H]+
1H-NMR(400MHz,DMSO-d6)δ:9.08(1H,s),8.94(1H,s),7.57(2H,d,J=8.1Hz),7.49-7.45(1H,m),5.95(2H,s).
ESI-MSfound:356[M+H]+
3-クロロ-1-(2,6-ジクロロベンジル)-1H-ピラゾロ[4,3-b]ピリジン-6-カルボン酸カリウム[173](以下、化合物[173]という)の合成

1H-NMR(400MHz,DMSO-d6)δ:9.06(1H,s),8.56(1H,s),7.56(2H,d,J=8.1Hz),7.47-7.43(1H,m),5.82(2H,s).
ESI-MSfound:356[M-K+2H]+
3-クロロ-1-(2-クロロ-6-メチルベンジル)-1H-ピラゾロ[4,3-b]ピリジン-6-カルボン酸[174](以下、化合物[174]という)の合成


1H-NMR(400MHz,CDCl3)δ:8.59(1H,d,J=1.5Hz),7.82(1H,d,J=1.5Hz),7.32(1H,d,J=8.1Hz),7.26-7.24(1H,m),7.18(1H,d,J=7.3Hz),5.64(2H,s),2.45(3H,s).
ESI-MSfound:370[M+H]+
1H-NMR(400MHz,CDCl3)δ:8.76(1H,s),7.95(1H,s),7.35-7.19(3H,m),5.74(2H,s),2.47(3H,s).
ESI-MSfound:317[M+H]+
工程(2)で得られた化合物[174-2](38mg)から実施例153の工程(5)の方法に準じて表題化合物(35mg)を白色固体として得た。
1H-NMR(400MHz,DMSO-d6)δ:9.07(1H,s),8.93(1H,s),7.37-7.26(3H,m),5.84(2H,s),2.39(3H,s).
ESI-MSfound:336[M+H]+
3-クロロ-1-(2,6-ジクロロベンジル)-6-(1H-テトラゾール-5-イル)-1H-ピラゾロ[4,3-b]ピリジン[175](以下、化合物[175]という)の合成

1H-NMR(400MHz,DMSO-d6)δ:9.24(1H,s),9.05(1H,s),7.58(2H,d,J=8.1Hz),7.50-7.46(1H,m),5.94(2H,s).
ESI-MSfound:380[M+H]+
5-[3-クロロ-1-(2,6-ジクロロベンジル)-1H-ピラゾロ[4,3-b]ピリジン-6-イル]-1H-テトラゾール-1-イド=カリウム[176](以下、化合物[176]という)の合成

1H-NMR(400MHz,DMSO-d6)δ:9.27(1H,s),8.73(1H,s),7.57(2H,d,J=8.1Hz),7.48-7.44(1H,m),5.88(2H,s).
ESI-MSfound:380[M+H]+
3-クロロ-1-(2-クロロ-6-メチルベンジル)-6-(1H-テトラゾール-5-イル)-1H-ピラゾロ[4,3-b]ピリジン[177](以下、化合物[177]という)の合成

1H-NMR(400MHz,DMSO-d6)δ:9.22(1H,s),9.06(1H,s),7.38-7.28(3H,m),5.84(2H,s),2.44(3H,s).
ESI-MSfound:360[M+H]+
5-[3-クロロ-1-(2-クロロ-6-メチルベンジル)-1H-ピラゾロ[4,3-b]ピリジン-6-イル]-1H-テトラゾール-1-イド=カリウム[178](以下、化合物[178]という)の合成

1H-NMR(400MHz,DMSO-d6)δ:9.26(1H,s),8.73(1H,s),7.38-7.26(3H,m),5.78(2H,s),2.41(3H,s).
ESI-MSfound:360[M-K+2H]+
1-(2,6-ジクロロベンジル)-5-メトキシ-3-メチル-1H-インダゾール-6-カルボン酸[179](以下、化合物[179]という)の合成

1H-NMR(400MHz,CDCl3)δ:10.31(1H,s),7.46(1H,d,J=9.0Hz),7.31(1H,d,J=5.9Hz),3.93(3H,s).

1H-NMR(400MHz,CDCl3)δ:7.40(1H,d,J=9.8Hz),7.38(1H,d,J=6.1Hz),3.92(3H,s),2.64(3H,d,J=5.4Hz).

1H-NMR(400MHz,CDCl3)δ:7.61(1H,s),7.37(2H,d,J=8.1Hz),7.26-7.22(1H,m),6.97(1H,s),5.64(2H,s),3.93(3H,s),2.49(3H,s).
ESI-MSfound:399[M+H]+

1H-NMR(400MHz,CDCl3)δ:7.60(1H,s),7.39(2H,d,J=8.1Hz),7.29-7.25(1H,m),7.01(1H,s),5.71(2H,s),3.96(3H,s),2.51(3H,s).
ESI-MSfound:346[M+H]+
工程(4)で得た化合物[179-4](23mg)のテトラヒドロフラン(0.5mL)及びメタノール(0.5mL)溶液に3N-水酸化ナトリウム水溶液(0.6mL)を加えた後、マイクロウェーブ反応装置を用いて150℃で30分間反応させた。反応混合物に5%硫酸水素カリウム水溶液を加え、酢酸エチルで抽出した。得られた有機層を無水硫酸ナトリウムで乾燥後、濾過し、濾液を減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィーにて精製することにより、表題化合物(13mg)を白色固体として得た。
1H-NMR(400MHz,CDCL3)δ:11.25(1H,brs),8.41(1H,s),7.38(2H,d,J=7.8Hz),7.27-7.23(1H,m),7.14(1H,s),5.74(2H,s),4.13(3H,s),2.52(3H,s).
ESI-MSfound:365[M+H]+
1-(2,6-ジクロロベンジル)-5-ヒドロキシ-3-メチル-1H-インダゾール-6-カルボン酸[180](以下、化合物[180]という)の合成
1H-NMR(400MHz,CD3OD)δ:8.17(1H,s),7.45(1H,d,J=7.7Hz),7.45(1H,d,J=8.4Hz),7.34(1H,dd,J=8.4,7.7Hz),7.07(1H,s),5.73(2H,s),2.43(3H,s).
ESI-MSfound:351[M+H]+
3-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]-2-ヒドロキシプロピオン酸[181](以下、化合物[181]という)の合成

1H-NMR(400MHz,CD3OD)δ:7.52(1H,d,J=7.6Hz),7.41-7.39(3H,m),7.30(1H,d,J=7.6Hz),7.09(1H,d,J=8.1Hz),5.62(2H,s),4.32(1H,s),3.34-3.30(1H,m),3.01-2.98(1H,m),2.40(3H,s).
1-(2,6-ジクロロベンジル)-3-エチル-6-(1H-テトラゾール-5-イル)-1H-インダゾール[182](以下、化合物[182]という)の合成

1H-NMR(400MHz,DMSO-d6)δ:8.44(1H,s),7.98(1H,d,J=8.3Hz),7.76(1H,d,J=8.3Hz),7.55(2H,d,J=7.8Hz),7.46-7.42(1H,m),5.79(2H,s)2.88(2H,q,J=7.6Hz),1.25(3H,t,J=7.6Hz).
ESI-MSfound:373[M+H]+
[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]ジフルオロ酢酸[183](以下、化合物[183]という)の合成


1H-NMR(400MHz,CDCl3)δ:8.10(1H,s),7.72(2H,s),7.38(2H,d,J=8.1Hz),7.27-7.25(1H,m),5.81(2H,s),4.48(2H,q,J=7.2Hz),2.56(3H,s),1.45(3H,t,J=7.1Hz).

1H-NMR(400MHz,CDCl3)δ:7.70-7.68(2H,m),7.38(2H,d,J=8.1Hz),7.32(1H,d,J=9.0Hz),7.25-7.23(1H,m),5.75(2H,s),4.29(2H,q,J=7.2Hz),2.53(3H,s),1.29(3H,t,J=7.1Hz).
工程(2)で得た化合物[183-2](96mg)のエタノール(2mL)溶液に1N-水酸化ナトリウム水溶液(2mL)を室温で加えた後、60℃で1時間攪拌した。反応混合物に1N-塩酸を加え、析出した固体を濾取することにより表題化合物(89mg)を白色固体として得た。
1H-NMR(400MHz,CD3OD)δ:7.81-7.79(2H,m),7.45(2H,d,J=8.1Hz),7.36-7.34(2H,m),5.81(2H,s),2.50(3H,s).
ESI-MSfound:385[M+H]+
[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]ジフルオロ酢酸カリウム[184](以下、化合物[184]という)の合成

1H-NMR(400MHz,CD3OD)δ:7.88(1H,s),7.71(1H,d,J=8.5Hz),7.44-7.42(3H,m),7.35-7.33(1H,m),5.77(2H,s),2.48(3H,s).
ESI-MSfound:385[M-K+2H]+
3-[1-(2-クロロ-6-シクロプロピルベンジル)-3-メチル-1H-インダゾール-6-イル]プロピオン酸[185](以下、化合物[185]という)の合成

1H-NMR(400MHz,CDCl3)δ:7.53(1H,d,J=8.3Hz),7.29(1H,d,J=8.1Hz),7.21-7.17(1H,m),7.07(1H,s),6.97(1H,d,J=7.6Hz),6.94(1H,d,J=8.4Hz),5.83(2H,s),3.03(2H,t,J=7.7Hz),2.68(2H,t,J=7.6Hz),2.51(3H,s),2.10-2.03(1H,m),0.87-0.82(2H,m),0.61-0.58(2H,m).
ESI-MSfound:369[M+H]+
3-(2,6-ジメチルベンジル)-1-イソプロピル-2-オキソ-2,3-ジヒドロ-1H-ベンゾイミダゾール-5-カルボン酸[186](以下、化合物[186]という)の合成


1H-NMR(400MHz,CDCl3)δ:11.58(1H,s),8.99(1H,d,J=2.0Hz),8.87(1H,d,J=8.8Hz),8.40(1H,dd,J=8.8,2.0Hz),4.00(3H,s).
ESI-MSfound:291[M-H]-

1H-NMR(400MHz,CDCl3)δ:8.71(1H,d,J=2.0Hz),8.29(1H,dd,J=8.1,2.0Hz),7.42(1H,d,J=8.1Hz),3.99(3H,s),1.39(18H,s).
ESI-MSfound:396[M-H]-

1H-NMR(400MHz,CDCl3)δ:10.28(1H,brs),7.87(1H,dd,J=8.8,2.0Hz),7.84(1H,d,J=2.0Hz),7.79(1H,d,J=8.8Hz),3.93(3H,s),1.72(9H,s).
ESI-MSfound:291[M-H]-

1H-NMR(400MHz,DMSO-d6)δ:11.40(1H,brs),7.60(1H,d,J=8.5Hz),7.13(1H,dd,J=8.1,6.8Hz),7.10(1H,s),7.08-7.03(3H,m),5.05(2H,s),3.74(3H,s),2.29(6H,s).
ESI-MSfound:309[M-H]-

1H-NMR(400MHz,CDCl3)δ:7.73(1H,dd,J=8.3,1.5Hz),7.18-7.04(6H,m),5.15(2H,s),4.77(1H,septet,J=7.1Hz),3.82(3H,s),2.36(6H,s),1.56(6H,d,J=7.1Hz).
ESI-MSfound:353[M+H]+
工程(5)で得た化合物[186-5](10mg)のテトラヒドロフラン(2mL)溶液に1N-水酸化ナトリウム水溶液(0.5mL)を加えた後、マイクロウェーブ反応装置を用いて130℃で45分間反応させた。反応混合物に1M-塩酸を加え、酢酸エチルで抽出した。得られた有機層を減圧濃縮した。得られた残渣を逆相分取液体クロマトグラフィーにて精製することにより表題化合物(3mg)を白色固体として得た。
ESI-MSfound:339[M+H]+
本実施例では、実施例化合物がURAT1阻害活性を有することを評価した。
ヒトURAT1完全長cDNAを発現ベクターpcDNA5/FRT/V5-His TOPO(登録商標)(インビトロジェン社)に導入した。得られた発現プラスミドを、Lipofectamine LTX(インビトロジェン社)を用いたリポソーム法により、チャイニーズハムスター卵巣細胞(以下、CHO細胞という)に導入し、ハイグロマイシンを含む選択培地で培養することにより、ヒトURAT1安定発現細胞株を作製した。






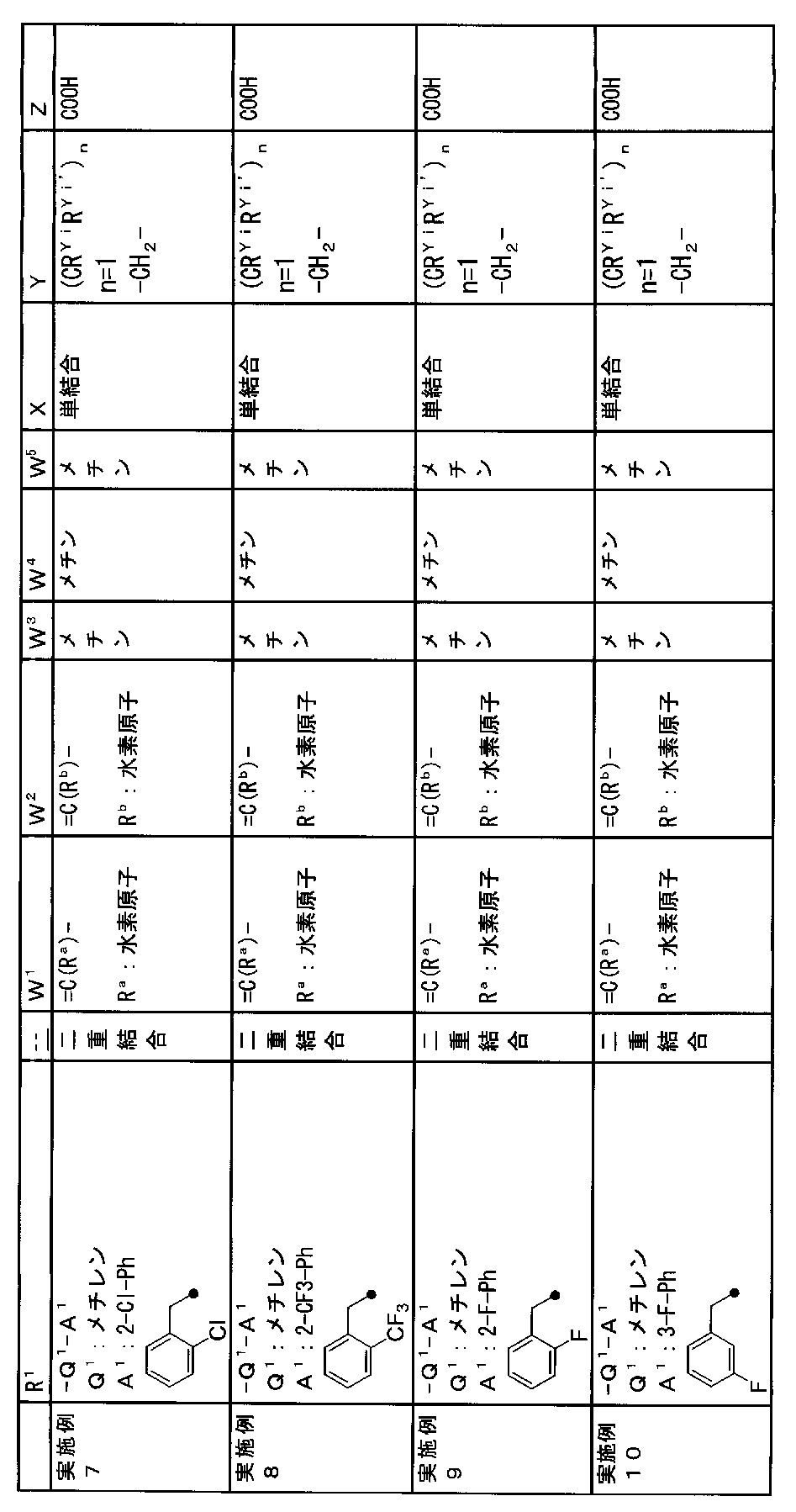




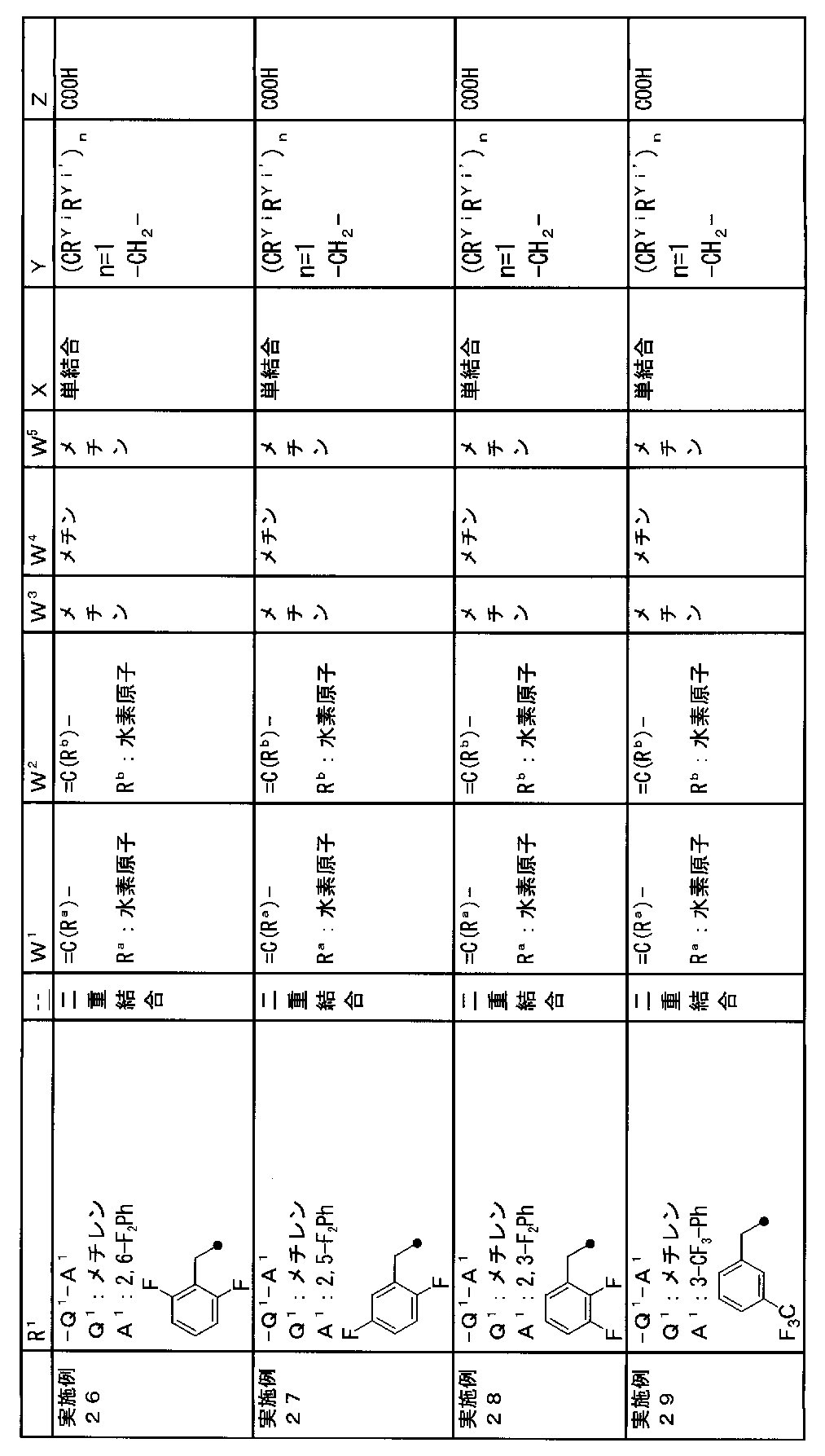


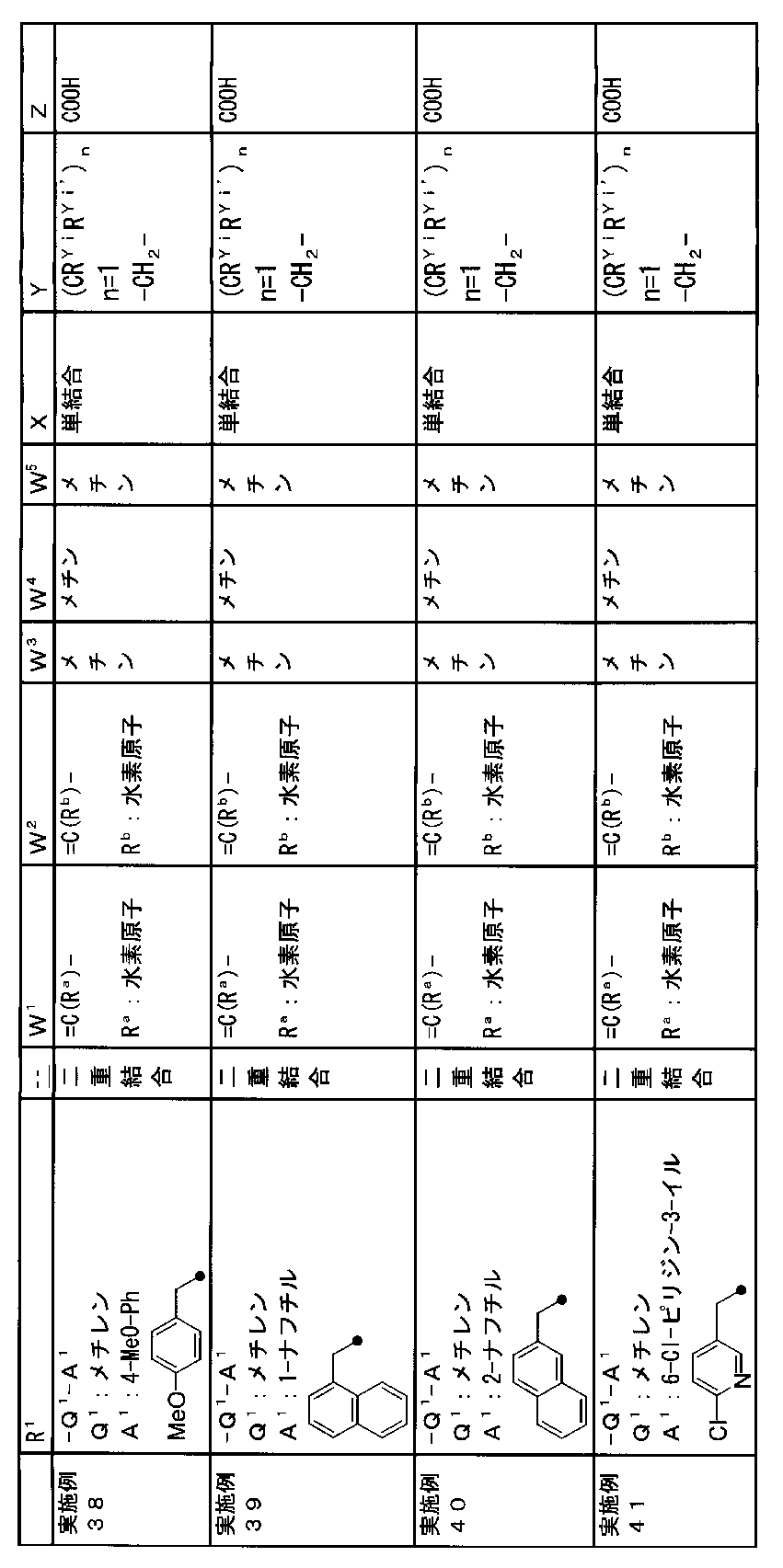
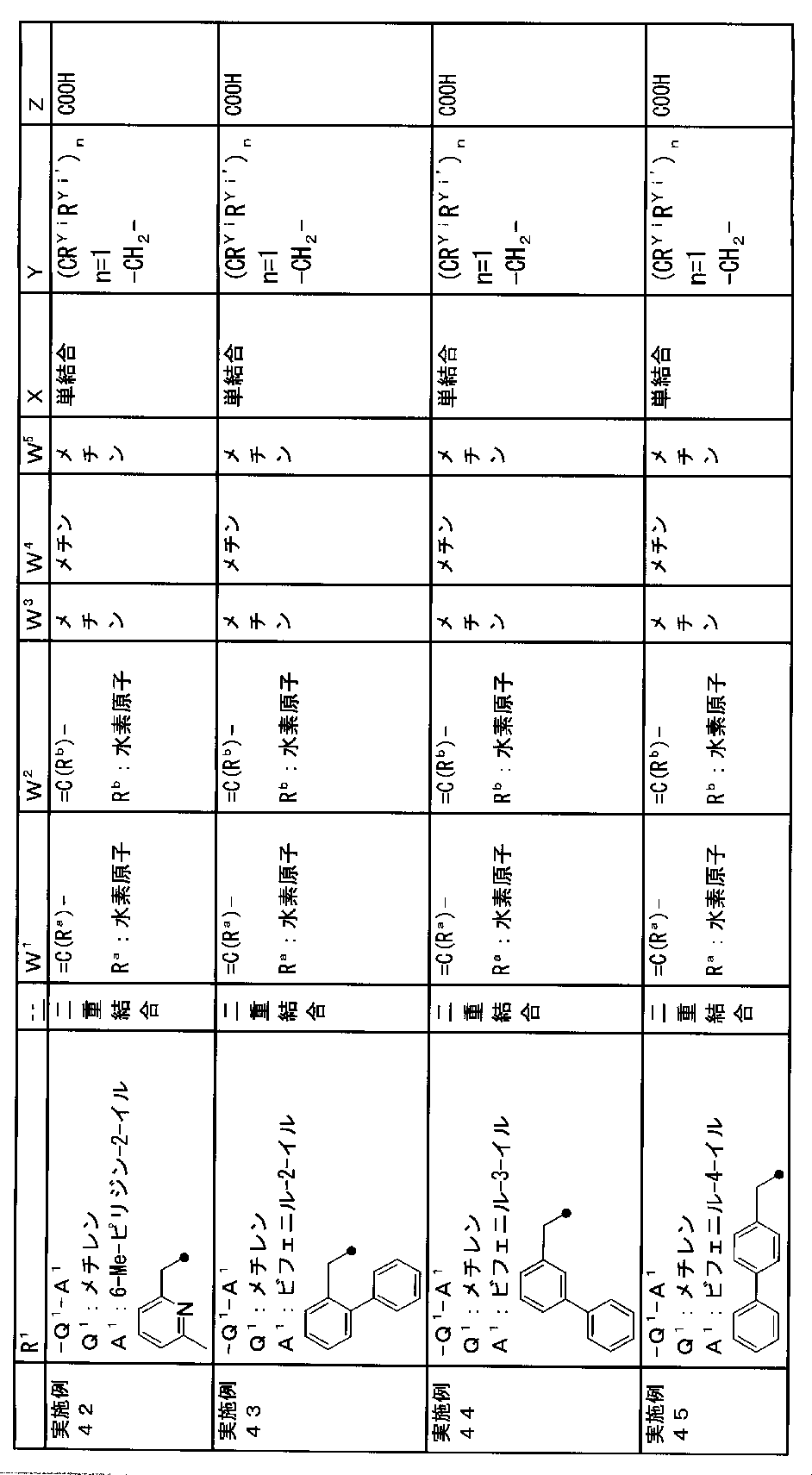
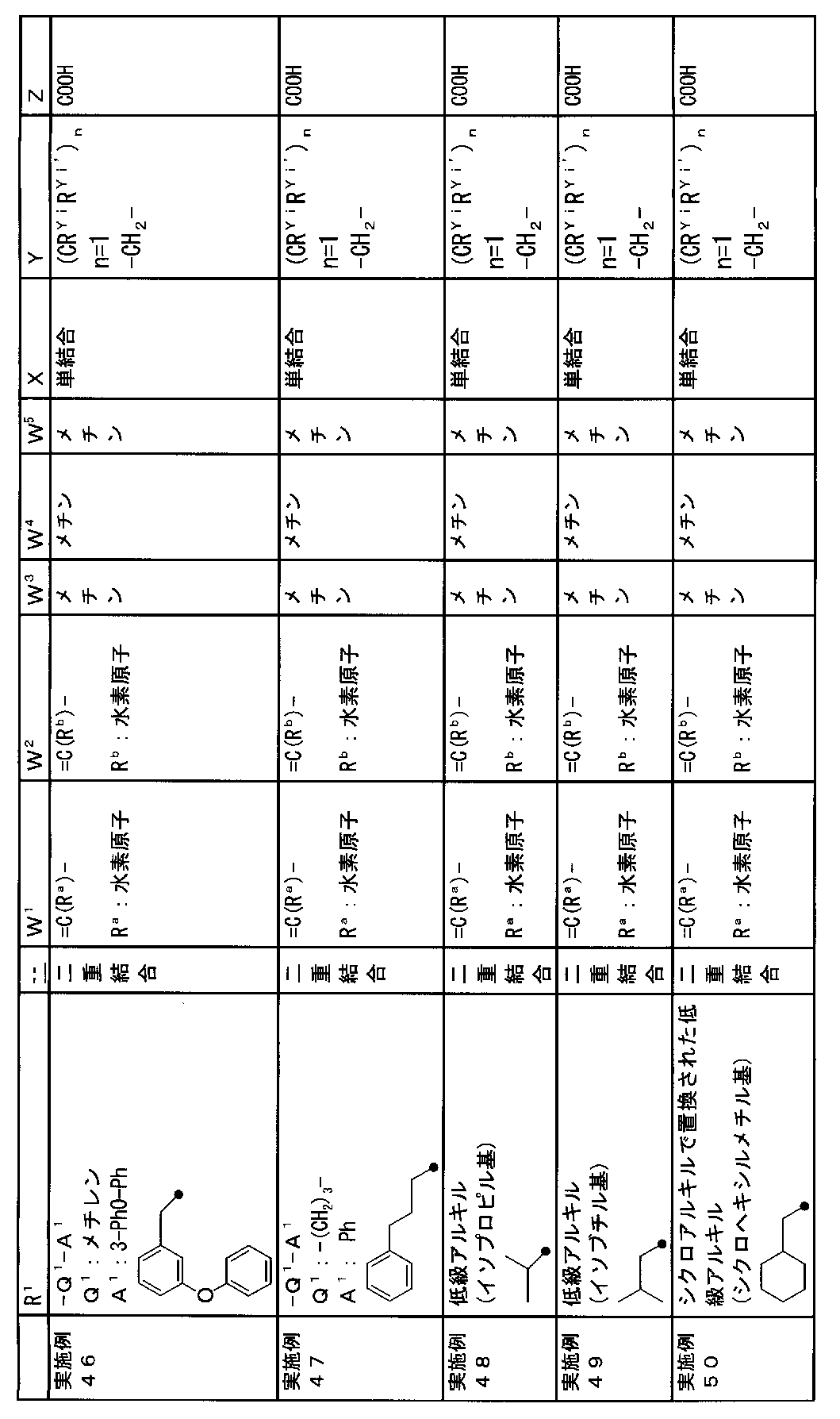


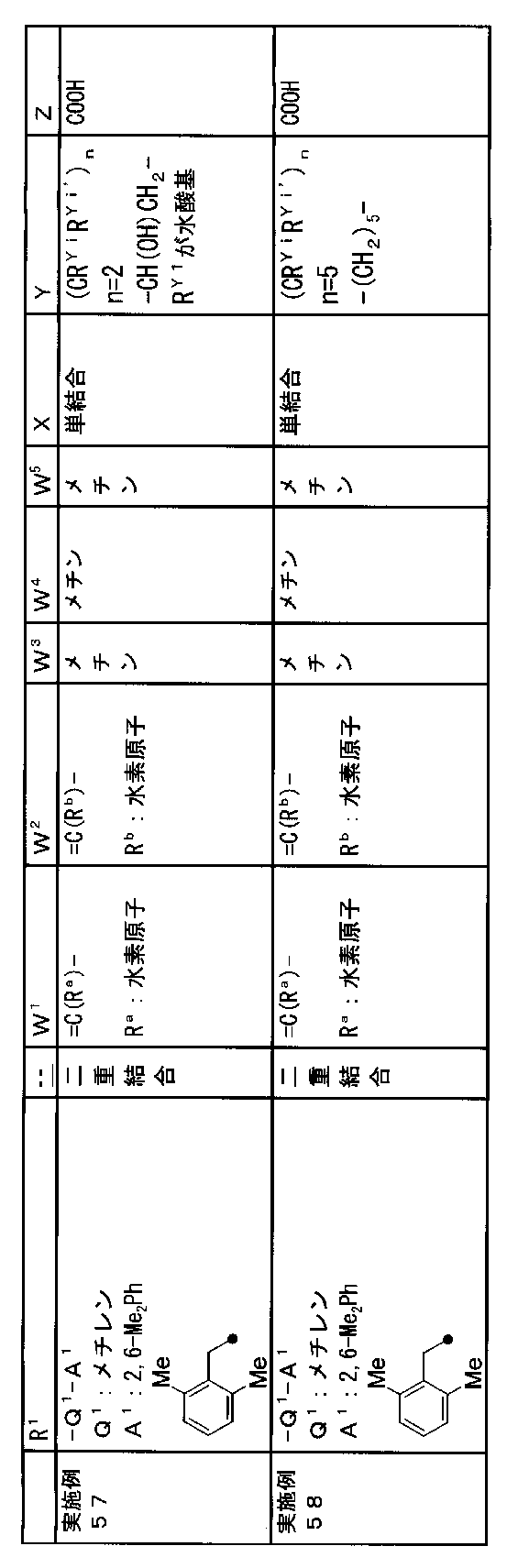
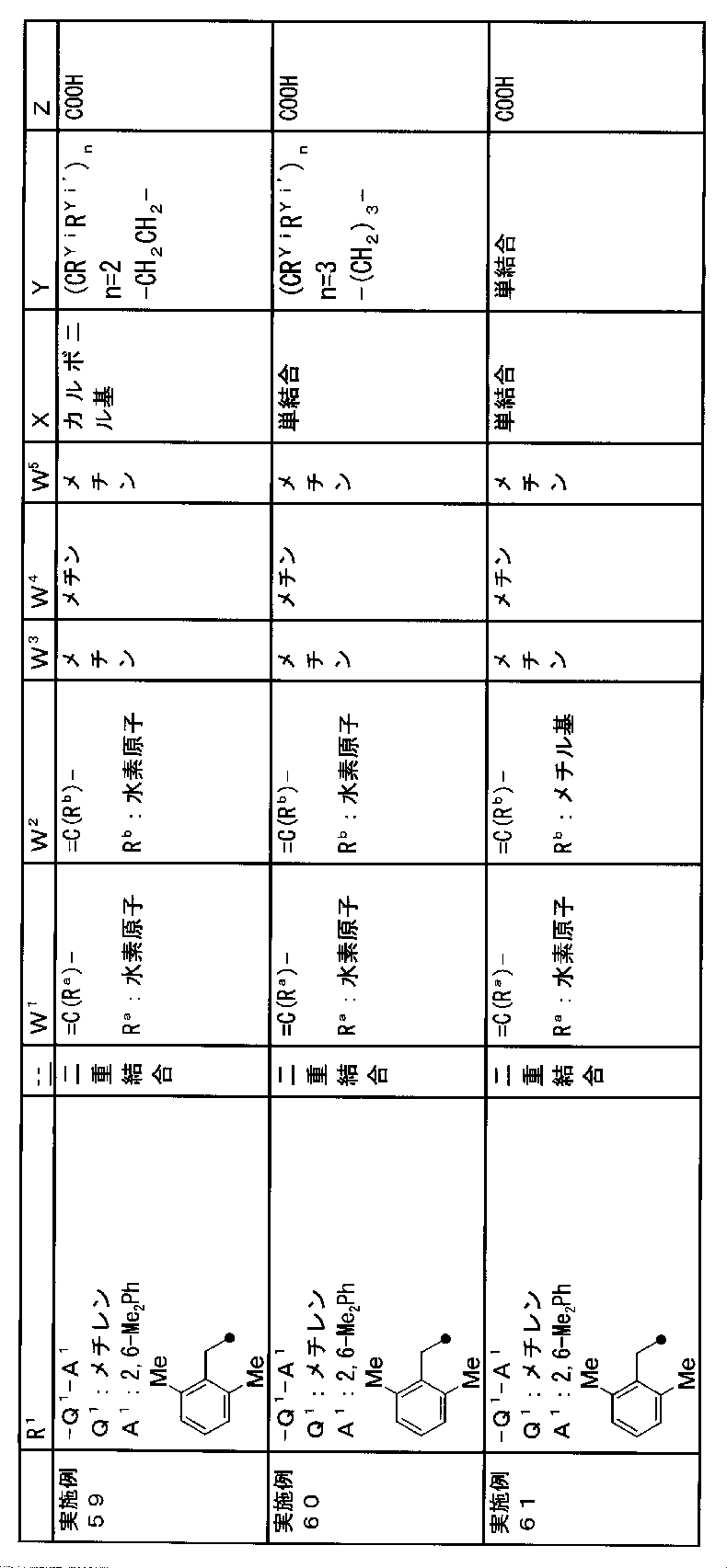
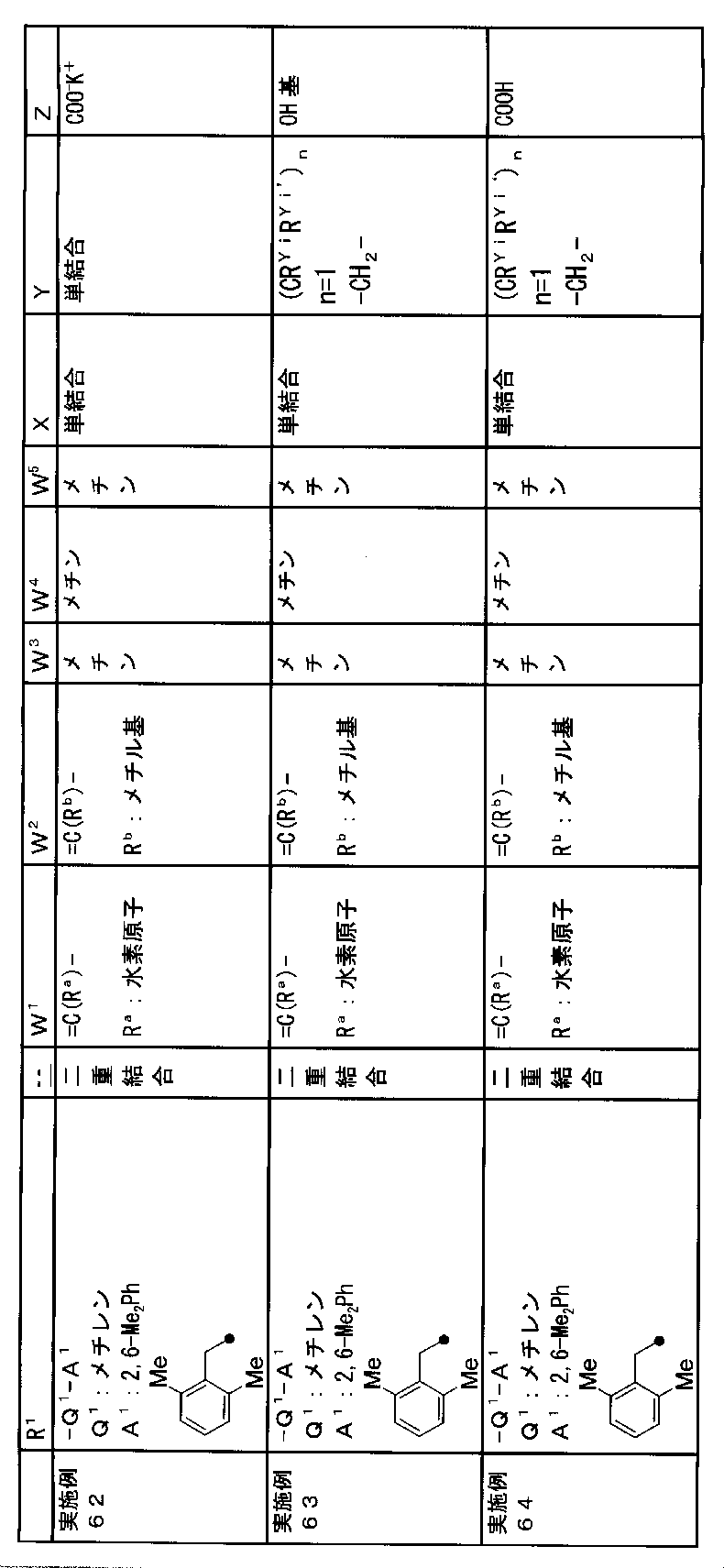


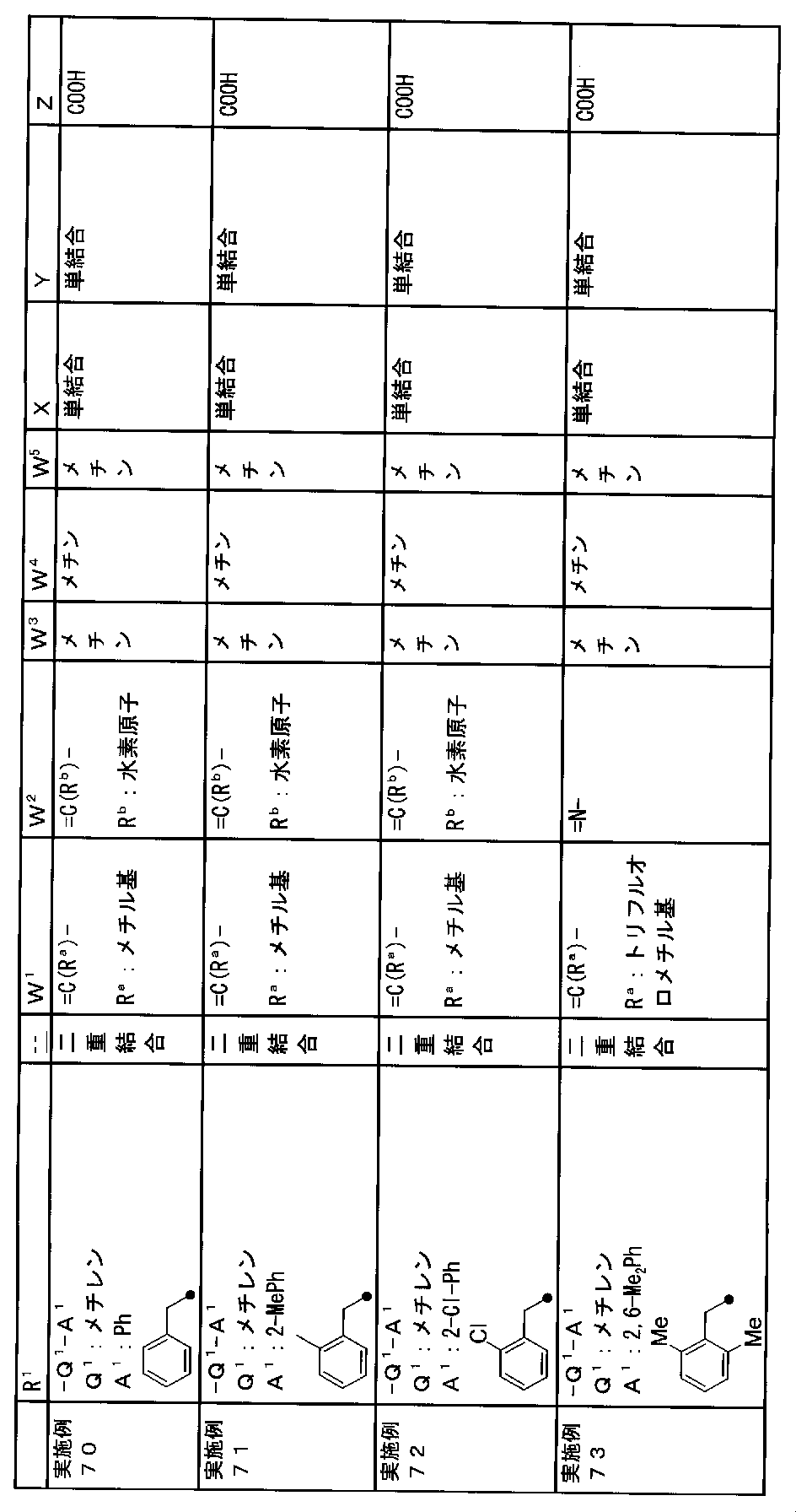


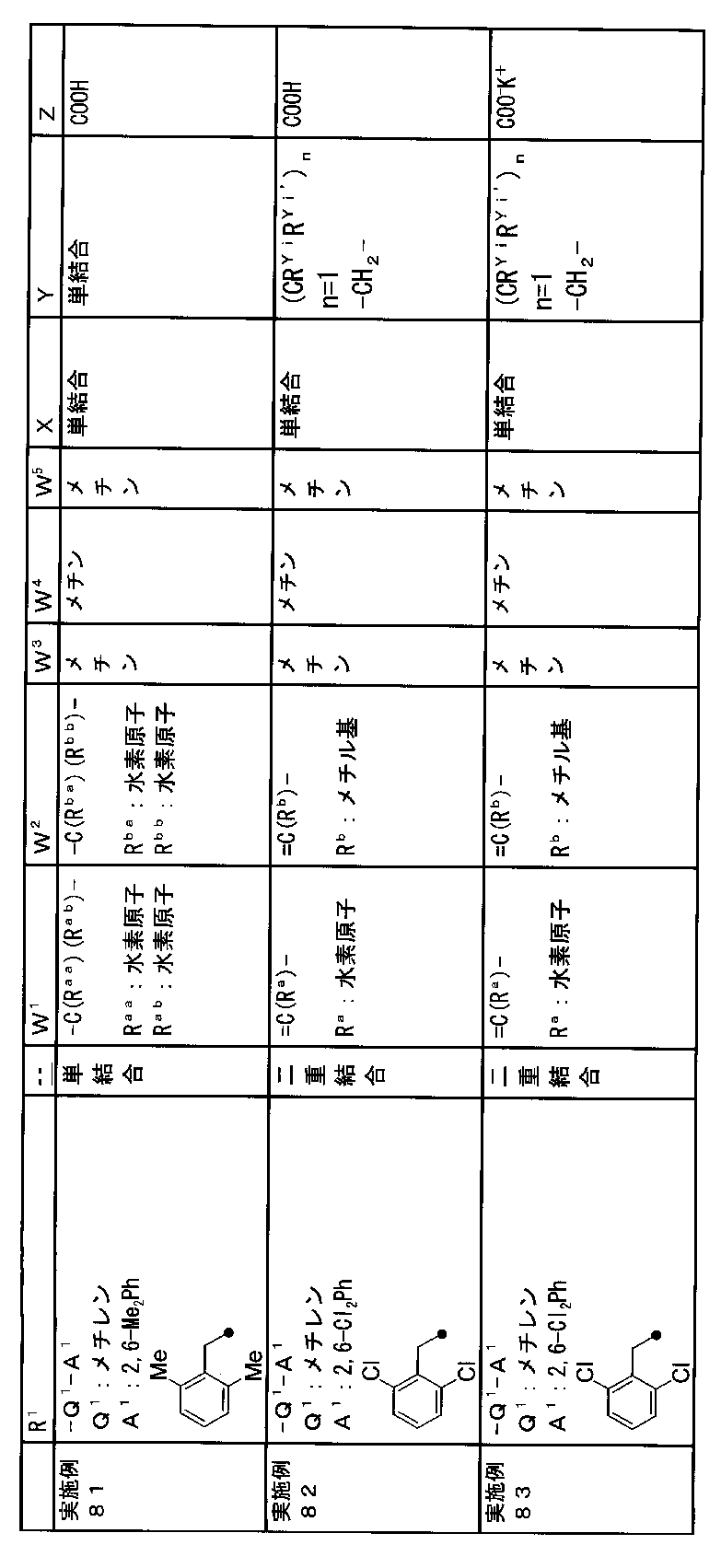








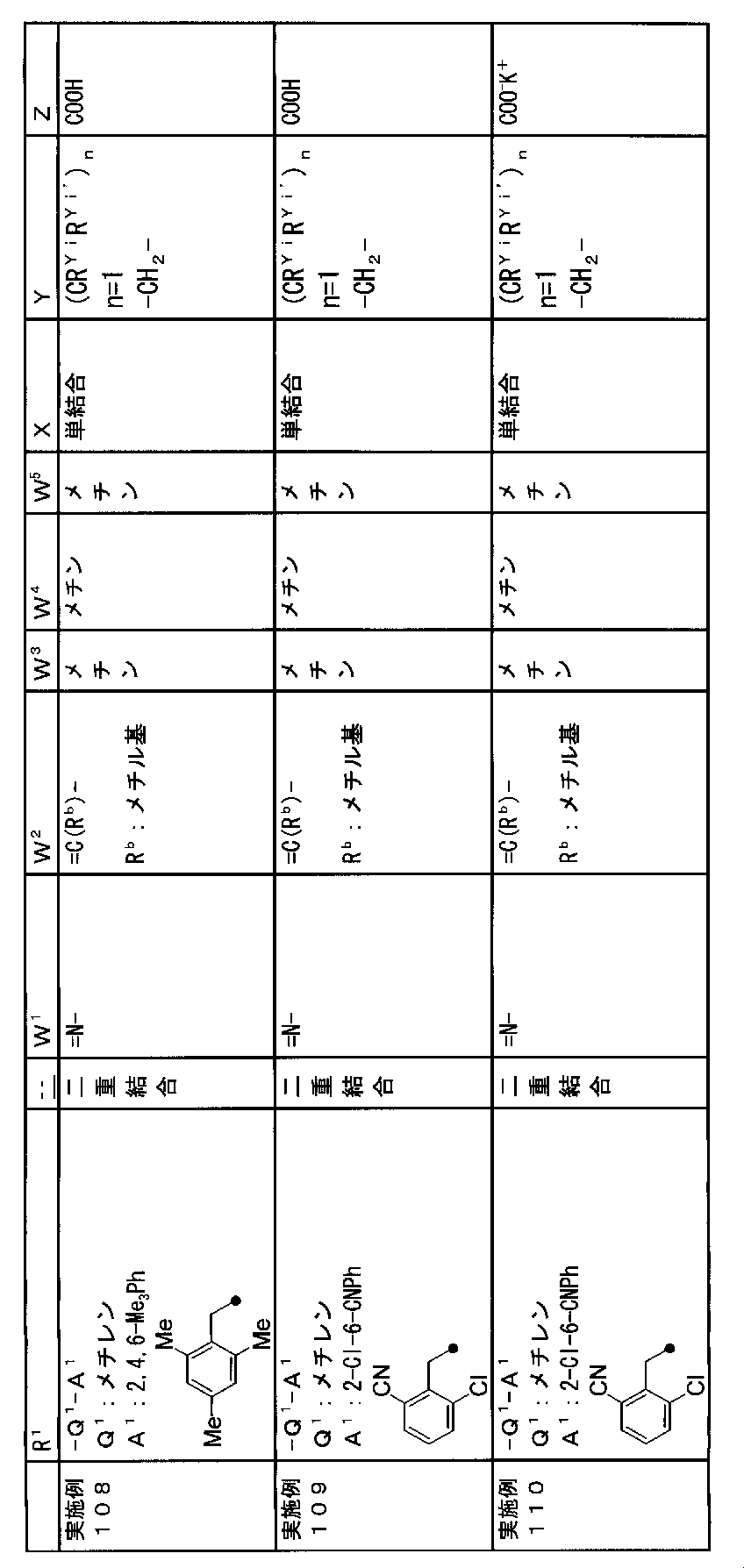







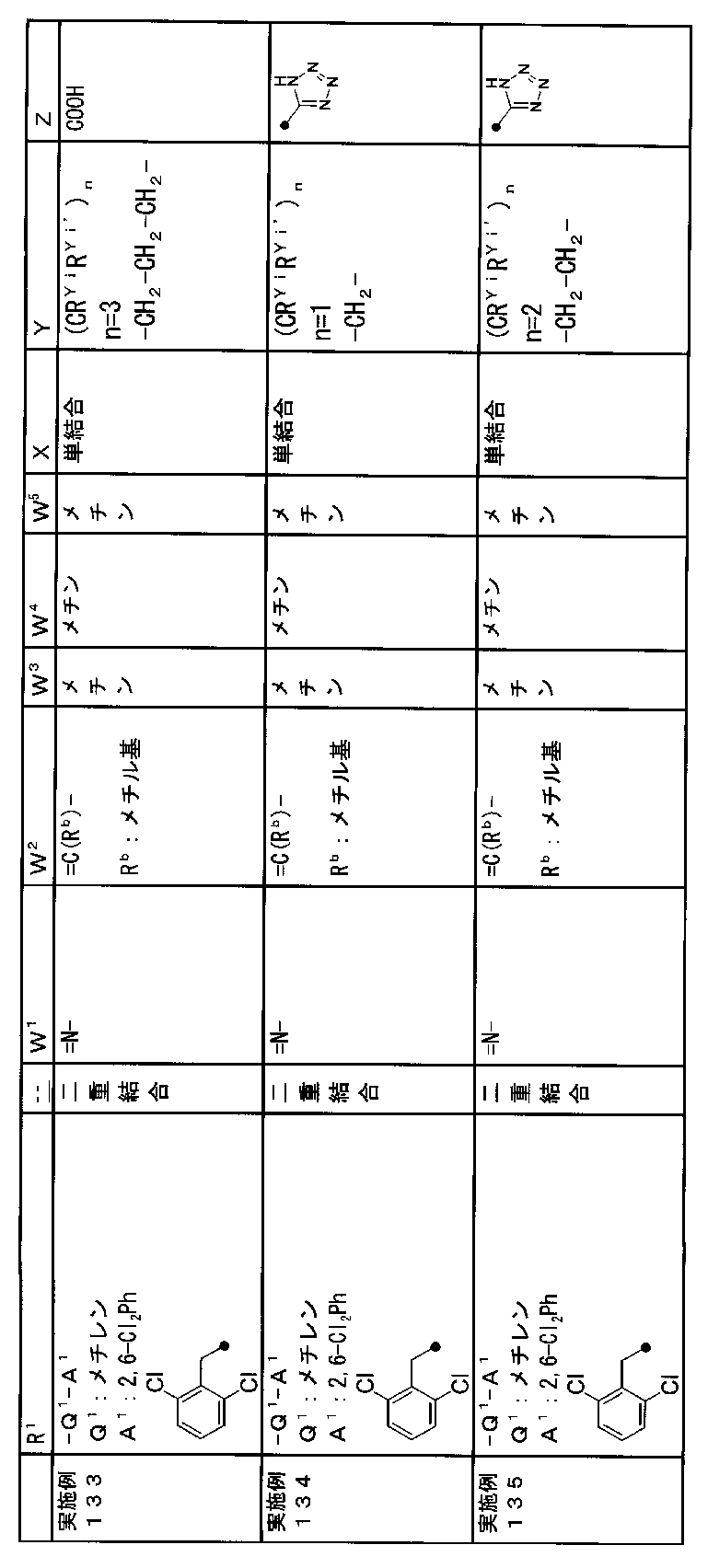
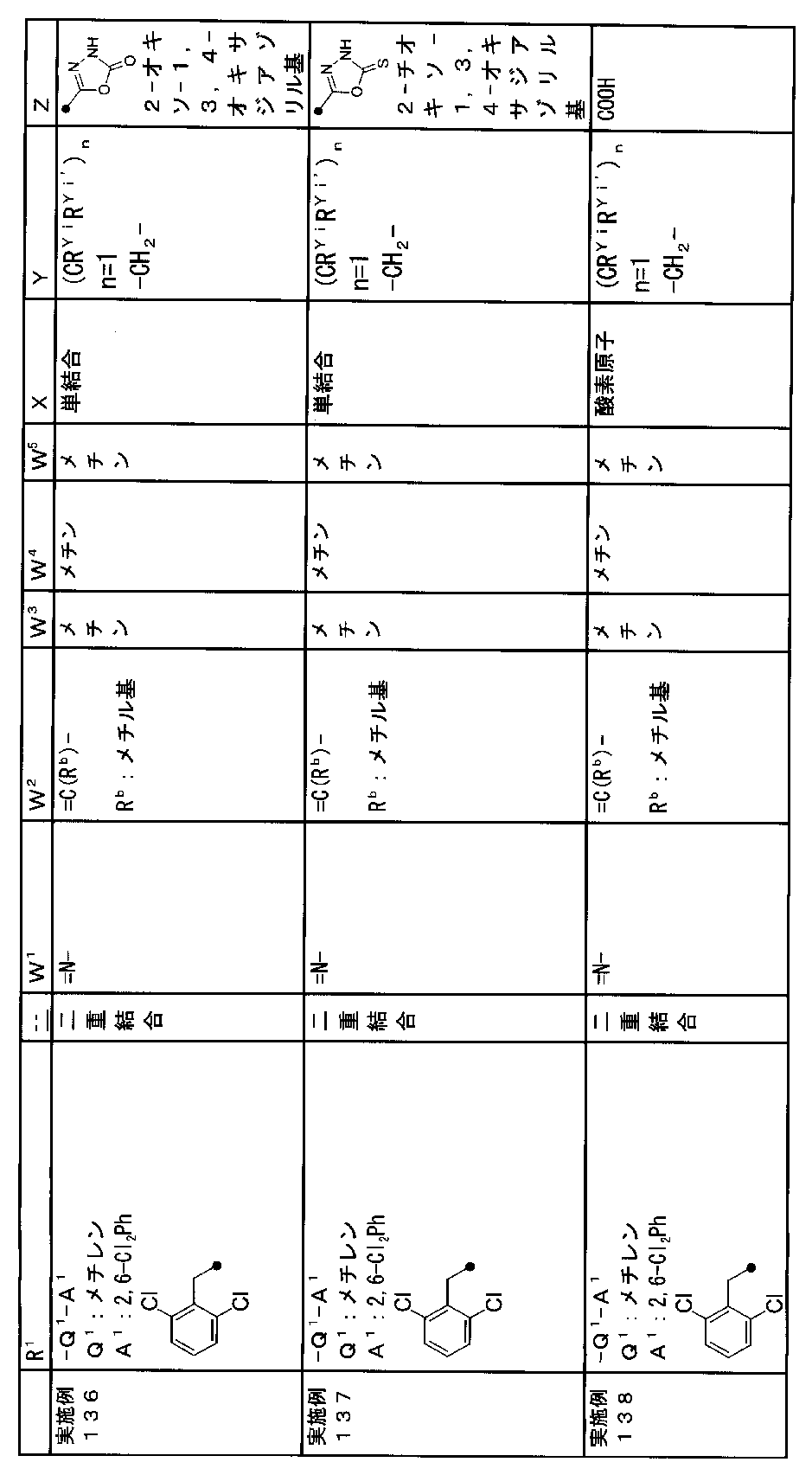
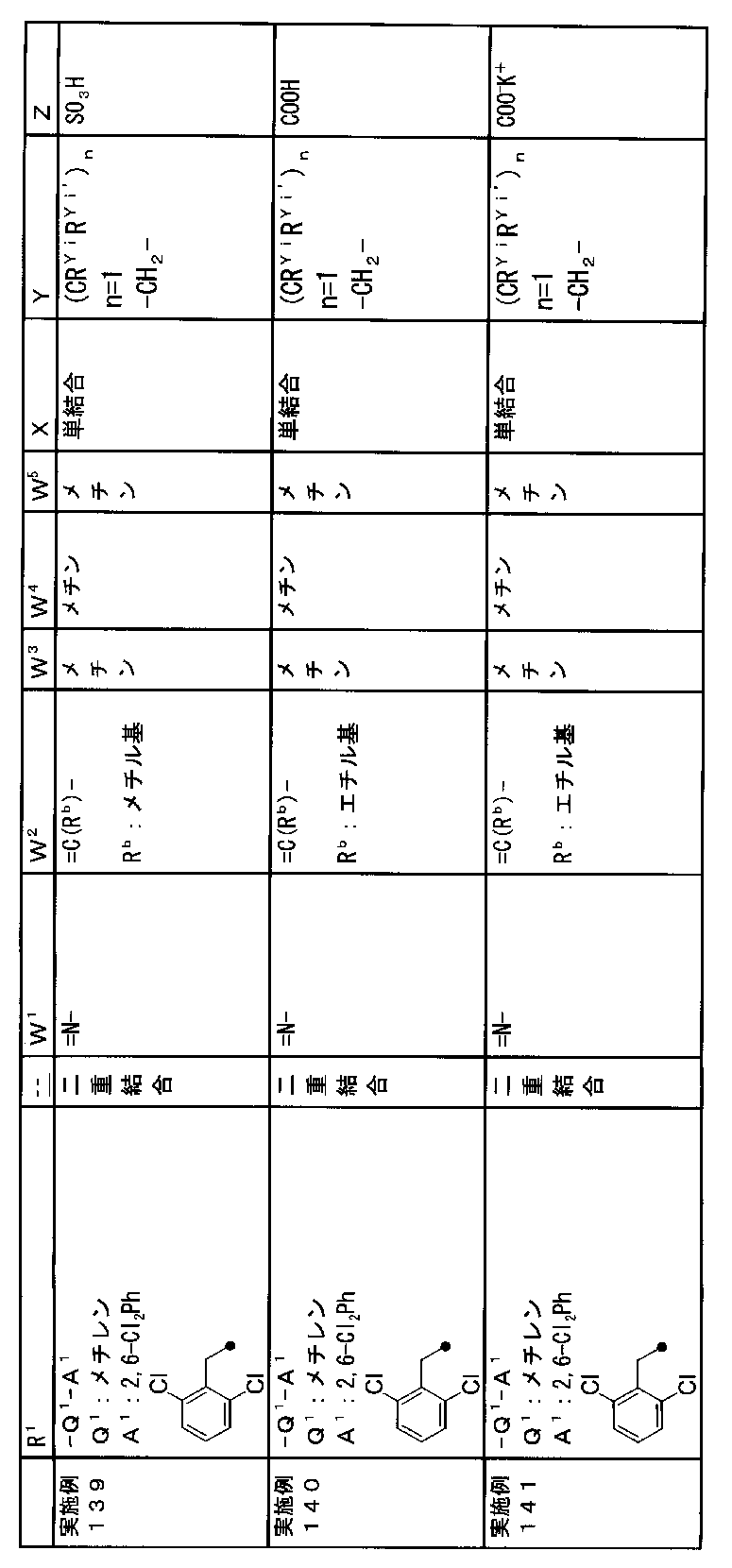


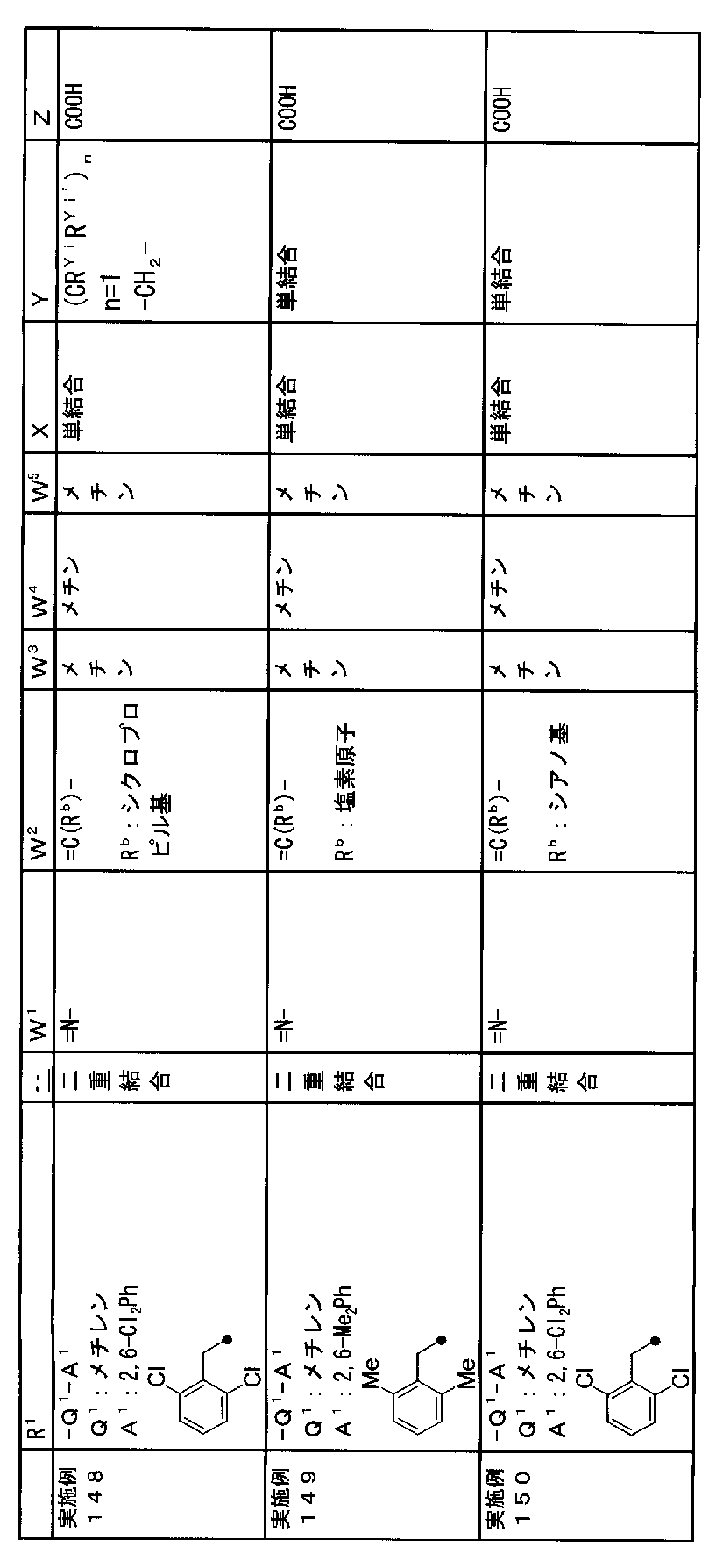




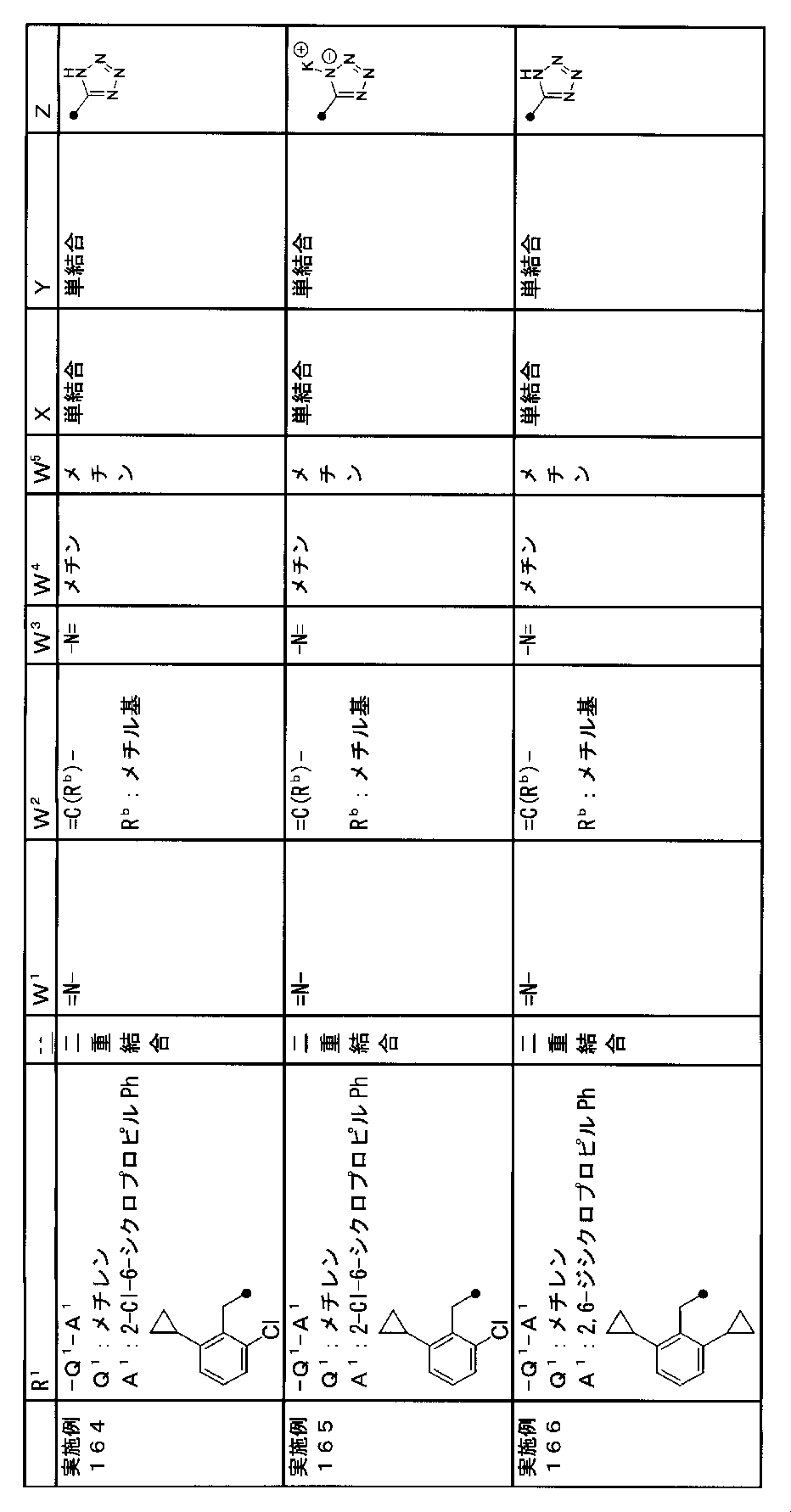
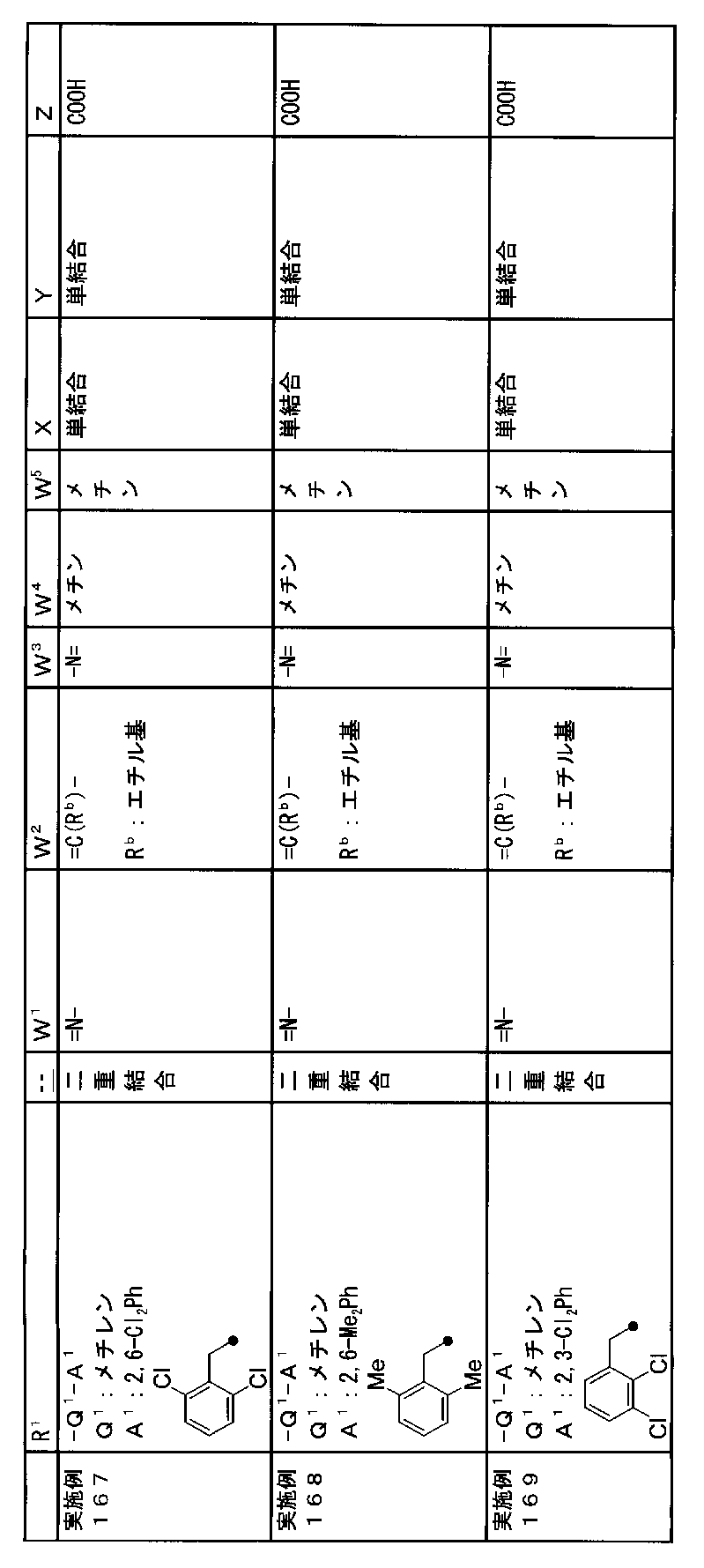

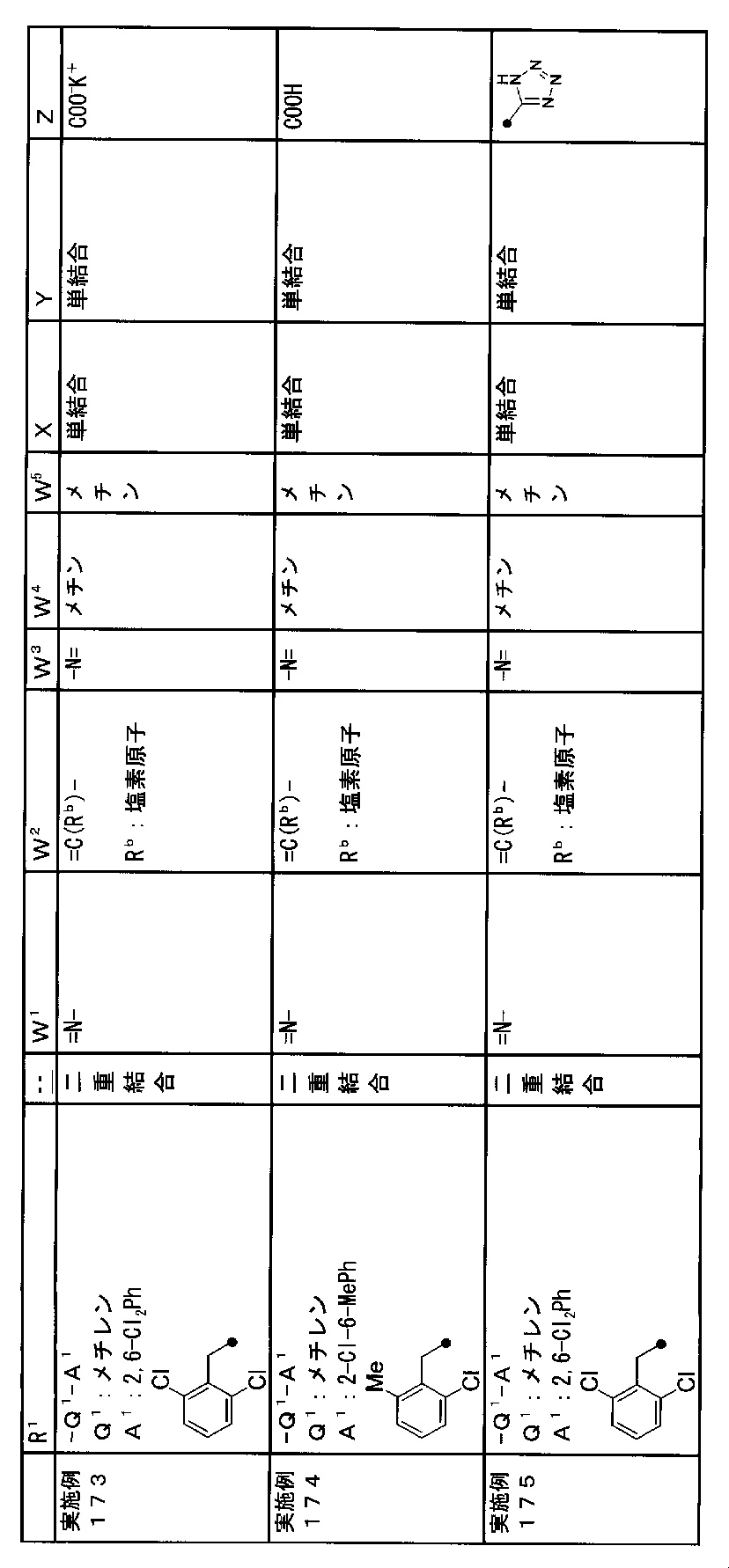


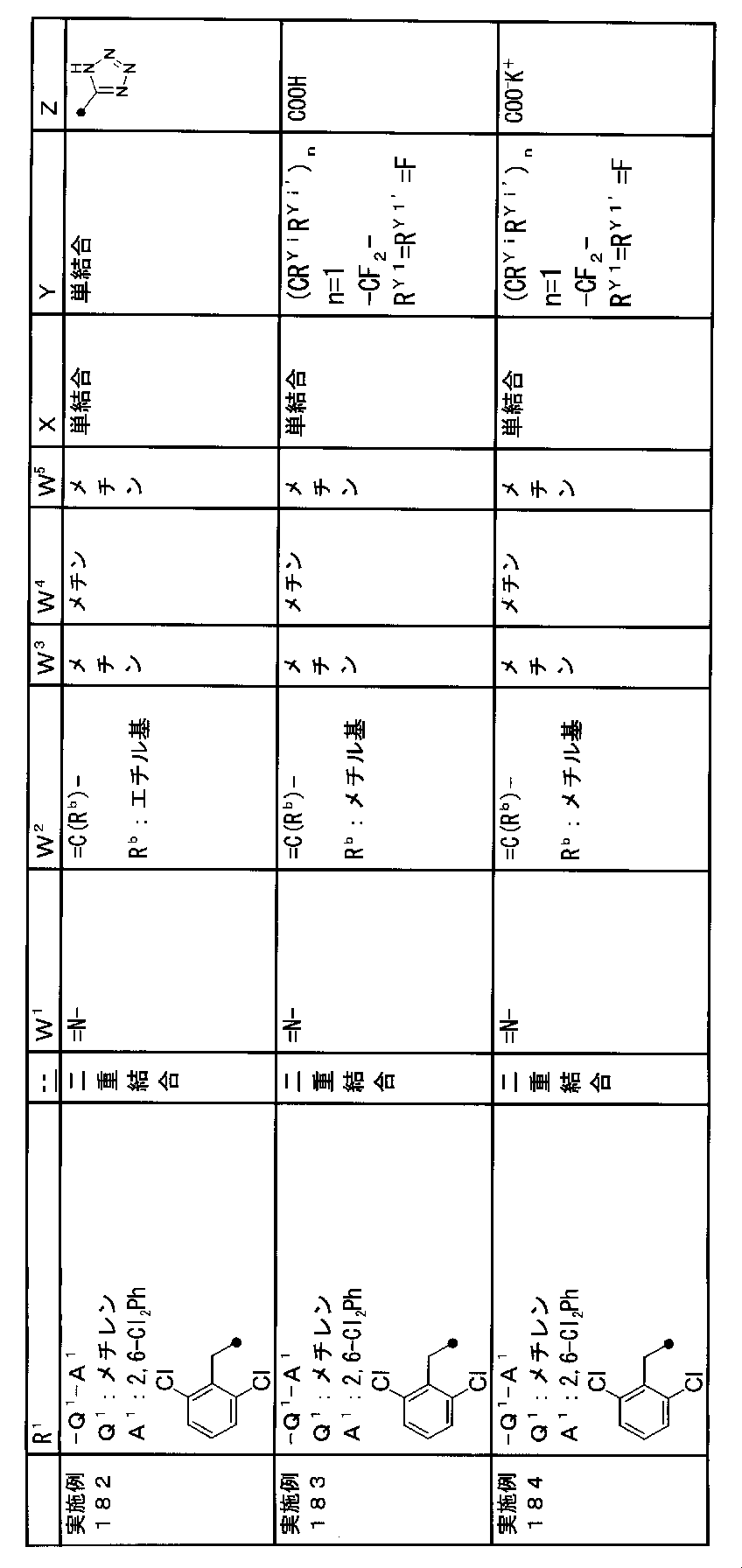

Claims (35)
- 式(I):

R1は、シクロアルキル基で置換されてもよい低級アルキル基、シクロアルキル基、ハロ低級アルキル基、ヒドロキシ低級アルキル基、低級アルコキシ低級アルキル基、低級アルコキシカルボニル基、低級アルキルスルホニル基又は一般式:-Q1-A1で表される基を表し;
Q1は、単結合又は低級アルキレン基を表し(ここで、該低級アルキレン基を構成する1若しくは2以上のメチレン基は、それぞれ独立して、カルボニル基、スルフィニル基若しくはスルホニル基でメチレン基全体が置き換えられていてもよく、及び/又は、メチレン基を構成する水素が低級アルキル基で置換されていてもよい。);
A1は、後記の<置換基群L>から選択される1乃至3個の置換基で置換されていてもよい、アリール基又はヘテロアリール基を表し(ここで、当該アリール基又はヘテロアリール基上のお互いに隣接する任意の二つの置換基が一緒になって低級アルキレンジオキシ基を形成してもよい。);
- - - - は、二重結合又は単結合を表し;
- - - - が二重結合である場合、
W1は、窒素原子又は一般式:=C(Ra)-で表される基を表し、かつ
W2は、窒素原子又は一般式:=C(Rb)-で表される基を表し;
- - - - が単結合である場合、
W1は、一般式:-C(Raa)(Rab)-で表される基又は一般式:-(C=O)-で表される基を表し、かつ
W2は、一般式:-C(Rba)(Rbb)-で表される基、一般式:-(C=O)-で表される基又は一般式:-N(Rbc)-で表される基を表し;
Ra及びRbは、それぞれ独立して、水素原子、後記の<置換基群M>から選択される置換基又は一般式:-Q2-A2で表される基を表し;
Raa及びRabは、それぞれ独立して、水素原子、後記の<置換基群N>から選択される置換基又は一般式:-Q2-A2で表される基を表すか、或いは、Raa及びRabが一緒になって低級アルキレン基を形成してもよく(ここで、該低級アルキレン基を構成する1乃至2以上のメチレン基は、それぞれ独立して、酸素原子、カルボニル基、ビニレン基若しくは一般式:-N(Rc)-で表される基でメチレン基全体が置き換えられていてもよく、及び/又は、メチレン基を構成する水素が水酸基、低級アルキル基若しくはハロゲン原子で置換されていてもよい。);
Rba及びRbbは、それぞれ独立して、水素原子、ハロゲン原子、アミノ基、低級アルキルアミノ基、ジ低級アルキルアミノ基、ヒドロキシ低級アルキルアミノ基、低級アルキルスルホニルアミノ基、低級アルコキシカルボニルアミノ基、<置換基群N>から選択される置換基又は一般式:-Q2-A2で表される基を表すか、或いは、Rba及びRbbが一緒になって低級アルキレン基を形成してもよく(ここで、該低級アルキレン基を構成する1乃至2以上のメチレン基は、それぞれ独立して、酸素原子、カルボニル基、ビニレン基若しくは一般式:-N(Rc)-で表される基でメチレン基全体が置き換えられていてもよく、及び/又は、メチレン基を構成する水素が水酸基、低級アルキル基若しくはハロゲン原子で置換されていてもよい。);
Rbcは、水素原子、低級アルキル基、シクロアルキル基、ハロ低級アルキル基、低級アルコキシカルボニル基、カルバモイル基、モノ低級アルキルカルバモイル基、ジ低級アルキルカルバモイル基及び低級アルカノイル基からなる群から選択される基又は一般式:-Q2-A2で表される基を表し;
Q2は、単結合、低級アルキレン基又は低級アルケニレン基を表し(ここで、該低級アルキレン基を構成する1若しくは2以上のメチレン基は、それぞれ独立して、酸素原子、窒素原子若しくはカルボニル基でメチレン基全体が置き換えられていてもよく、及び/又は、メチレン基を構成する水素がハロゲン原子、シアノ基、水酸基若しくは低級アルキル基で置換されていてもよい。);
A2は、<置換基群L>から選択される1乃至3個の置換基で置換されていてもよい、シクロアルキル基、脂肪族複素環基、アリール基又はヘテロアリール基を表し(ここで、当該アリール基又はヘテロアリール基上のお互いに隣接する任意の二つの置換基が一緒になって低級アルキレンジオキシ基を形成してもよい。);
W3、W4及びW5は、それぞれ独立して、窒素原子、又は、ハロゲン原子、水酸基、シアノ基、低級アルキル基、シクロアルキル基、ハロ低級アルキル基、低級アルコキシ基及びハロ低級アルコキシ基からなる群から選択される置換基を有してもよいメチン基を表し;但し、W1、W2、W3、W4及びW5のうち0個乃至4個が窒素原子であり;
Xは、単結合、酸素原子、硫黄原子、スルフィニル基、スルホニル基、カルボニル基、低級アルケニレン基、低級アルキニレン基又は一般式:-N(RX)-で表される基を表し(ここで、RXは、水素原子又は低級アルキル基である。);
Yは、単結合又は(CRYiRYi')nであり(ここで、nは、1乃至6のいずれかの整数であり、iは、1乃至nのいずれかの整数であり、(CRYiRYi')nは、n=1のとき(CRY1RY1')を表し、n=2のとき(CRY1RY1')-(CRY2RY2')を表し、n=3のとき(CRY1RY1')-(CRY2RY2')-(CRY3RY3')を表し、n=4のとき(CRY1RY1')-(CRY2RY2')-(CRY3RY3')-(CRY4RY4')を表し、n=5のとき(CRY1RY1')-(CRY2RY2')-(CRY3RY3')-(CRY4RY4')-(CRY5RY5')を表し、n=6のとき(CRY1RY1')-(CRY2RY2')-(CRY3RY3')-(CRY4RY4')-(CRY5RY5')-(CRY6RY6')を表し(ここで、RY1、RY1'、RY2、RY2'、RY3、RY3'、RY4、RY4'、RY5、RY5'、RY6及びRY6'は、それぞれ独立して、水素原子、ハロゲン原子又は<置換基群N>から選択される置換基であるか、
或いは、RX、RY1、RY1'、RY2、RY2'、RY3、RY3'、RY4、RY4'、RY5、RY5'、RY6及びRY6'に関して、以下の2個の基の組み合わせ:「RY1とRY1'」、「RY2とRY2'」、「RY3とRY3'」、「RY4とRY4'」、「RY5とRY5'」、「RY6とRY6'」、「RXとRY1」、「RXとRY2」、「RXとRY3」、「RY1とRY2」、「RY1とRY3」、「RY1とRY4」、「RY2とRY3」、「RY2とRY4」又は「RY2とRY5」は、当該組み合わせを構成する2個の基が一緒になって、低級アルキレン基を形成してもよく、ここで、該低級アルキレン基を構成する1若しくは2以上のメチレン基は、それぞれ独立して、酸素原子、カルボニル基、ビニレン基若しくは一般式:-N(Rc)-でメチレン基全体が置き換えられていてもよく、及び/又は、メチレン基を構成する水素が水酸基、低級アルキル基若しくはハロゲン原子で置換されていてもよい。);
Rcは、水素原子、低級アルキル基、ハロ低級アルキル基又は低級アルカノイル基を表し;
Zは、水酸基、COOR2、CONR3R4、SO3R2、SO3NR3R4、5-テトラゾリル基、5-オキソ-1,2,4-オキサジアゾリル基、2-オキソ-1,3,4-オキサジアゾリル基、5-イミノ-4,5-ジヒドロ-1,3,4-オキサジアゾリル基、2-チオキソ-1,3,4-オキサジアゾリル基又は5-オキソ-1,2,4-チアジアゾリル基を表し;
ここでR2、R3及びR4は、それぞれ独立して、水素原子又は低級アルキル基を表し; <置換基群L>、<置換基群M>及び<置換基群N>は、下記のように定義される。
<置換基群L>:
ハロゲン原子、水酸基、ニトロ基、シアノ基、ホルミル基、アミノ基、カルボキシル基、低級アルキル基、ハロ低級アルキル基、シクロアルキル基、低級アルコキシ基、ハロ低級アルコキシ基、ヒドロキシ低級アルキル基、低級アルコキシ低級アルキル基、低級アルコキシカルボニル基、低級アルカノイル基、低級アルキルチオ基、低級アルキルスルホニル基、低級アルキルアミノ基、ジ低級アルキルアミノ基、カルバモイル基、モノ低級アルキルカルバモイル基、ジ低級アルキルカルバモイル基、低級アルカノイルアミノ基、低級アルキルスルホニルアミノ基、低級アルコキシカルボニルアミノ基、アラルキル基、アリールオキシ基、ヘテロアリールオキシ基及び低級アルケニル基
<置換基群M>:
ハロゲン原子、水酸基、ニトロ基、シアノ基、ホルミル基、アミノ基、カルボキシル基、低級アルキル基、ハロ低級アルキル基、シクロアルキル基、低級アルコキシ基、ハロ低級アルコキシ基、ヒドロキシ低級アルキル基、低級アルコキシ低級アルキル基、低級アルコキシカルボニル基、低級アルカノイル基、低級アルキルチオ基、低級アルキルスルホニル基、低級アルキルアミノ基、ジ低級アルキルアミノ基、カルバモイル基、モノ低級アルキルカルバモイル基、ジ低級アルキルカルバモイル基、低級アルカノイルアミノ基、低級アルキルスルホニルアミノ基及び低級アルコキシカルボニルアミノ基
<置換基群N>:
水酸基、シアノ基、ホルミル基、カルボキシル基、低級アルキル基、ハロ低級アルキル基、シクロアルキル基、低級アルコキシ基、ハロ低級アルコキシ基、ヒドロキシ低級アルキル基、低級アルコキシ低級アルキル基、低級アルコキシカルボニル基、低級アルカノイル基、カルバモイル基、モノ低級アルキルカルバモイル基及びジ低級アルキルカルバモイル基]
で示される化合物又は該化合物の薬学的に許容できる塩若しくはエステル。 - 下記部分構造:

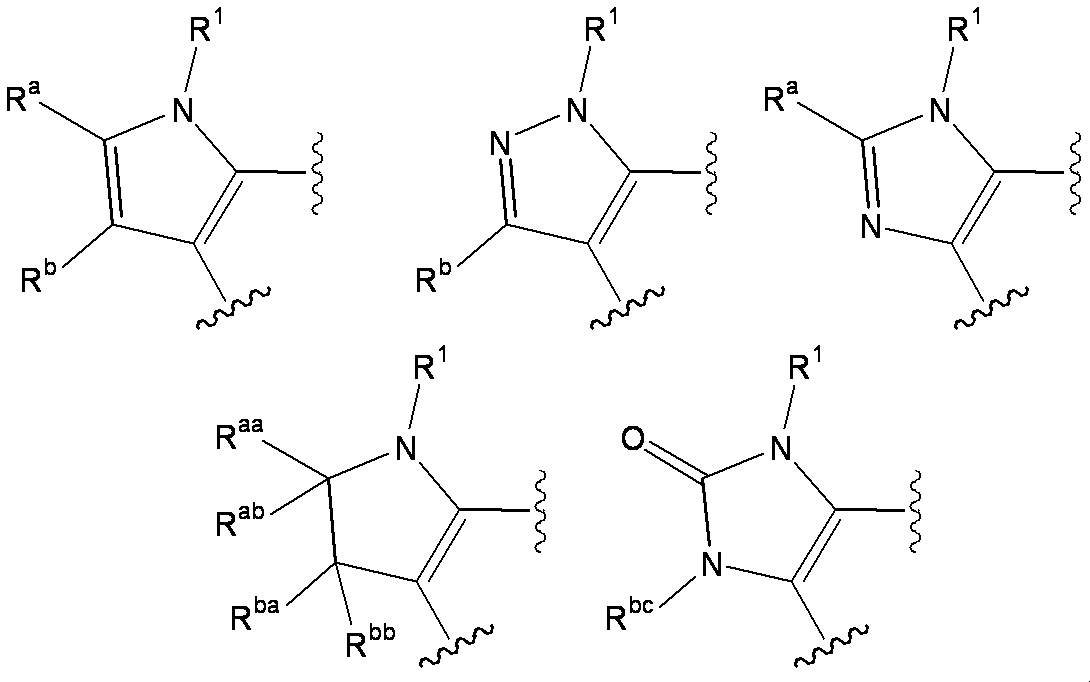
- W3、W4及びW5が、それぞれ独立して、窒素原子、又は、ハロゲン原子及び低級アルキル基からなる群より選択される置換基を有してもよいメチン基である、請求項2記載の化合物又は該化合物の薬学的に許容できる塩若しくはエステル。
- Xが、単結合、酸素原子、カルボニル基、ビニレン基又は一般式:-N(RX)-で表される基である、請求項3記載の化合物又は該化合物の薬学的に許容できる塩若しくはエステル。
- R1が、一般式-Q1-A1で表される基である、請求項4記載の化合物又は該化合物の薬学的に許容できる塩若しくはエステル。
- Q1が、単結合又は低級アルキル基で置換されてもよい低級アルキレン基である、請求項5記載の化合物又は該化合物の薬学的に許容できる塩若しくはエステル。
- Yが、単結合又は(CRYiRYi')nである(ここで、nは、1乃至6のいずれかの整数であり、iは、1乃至nのいずれかの整数であり、(CRYiRYi')nは、n=1のとき(CRY1RY1')を表し、n=2のとき(CRY1RY1')-(CRY2RY2')を表し、n=3のとき(CRY1RY1')-(CRY2RY2')-(CRY3RY3')を表し、n=4のとき(CRY1RY1')-(CRY2RY2')-(CRY3RY3')-(CRY4RY4')を表し、n=5のとき(CRY1RY1')-(CRY2RY2')-(CRY3RY3')-(CRY4RY4')-(CRY5RY5')を表し、n=6のとき(CRY1RY1')-(CRY2RY2')-(CRY3RY3')-(CRY4RY4')-(CRY5RY5')-(CRY6RY6')を表し、ここで、RY1、RY1'、RY2、RY2'、RY3、RY3'、RY4、RY4'、RY5、RY5'、RY6及びRY6'は、それぞれ独立して、水素原子、ハロゲン原子又は<置換基群N>から選択される置換基である。)、請求項6記載の化合物又は該化合物の薬学的に許容できる塩若しくはエステル。
- Q1がメチレン基である、請求項7記載の化合物又は該化合物の薬学的に許容できる塩若しくはエステル。
- A1のアリール基又はヘテロアリール基が、フェニル基、ナフチル基、ピリジル基、ピリダニジル基、ピラジニル基、ピリミジニル基、キノリル基、イソキノリル基又はベンゾチエニル基である、請求項8記載の化合物又は該化合物の薬学的に許容できる塩若しくはエステル。
- <置換基群L>が、水酸基、ハロゲン原子、シアノ基、メチル基、エチル基、シクロプロピル基、トリフルオロメチル基、ヒドロキシメチル基、メトキシ基及びトリフルオロメトキシ基からなる群である、請求項9記載の化合物又は薬学的に許容できる塩若しくはエステル。
- 以下の(a)~(s)の化合物:
(a)1-(2,6-ジメチルベンジル)-3-メチル-1H-インドール-6-カルボン酸、
(b)2-[1-(2,6-ジメチルベンジル)-3-メチル-1H-インドール-6-イル]酢酸、
(c)2-[1-(2,6-ジメチルベンジル)-3-メチル-1H-インダゾール-6-イル]酢酸、
(d)2-[3-クロロ-1-(2,6-ジメチルベンジル)-1H-インドール-6-イル]酢酸、
(e)2-[1-(2,6-ジメチルベンジル)-2,3-ジヒドロ-1H-インドール-6-イル]酢酸、
(f)(3-RS)-2-[1-(2,6-ジメチルベンジル)-2,3-ジヒドロ-3-メチル-1H-インドール-6-イル]酢酸、
(g)2-[1-(2,6-ジメチルベンジル)-2,3-ジヒドロ-3,3-ジメチル-1H-インドール-6-イル]酢酸、
(h)2-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インドール-6-イル]酢酸、
(i)2-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]酢酸、
(j)2-[1-(2-クロロ-6-メチルベンジル)-3-メチル-1H-インダゾール-6-イル]酢酸、
(k)3-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]プロピオン酸、
(l)1-(2,6-ジクロロベンジル)-3-メチル-6-(1H-テトラゾール-5-イルメチル)-1H-インダゾール、
(m)2-[1-(2,6-ジクロロベンジル)-3-エチル-1H-インダゾール-6-イル]酢酸、
(n)1-(2-クロロ-6-メチルベンジル)-3-メチル-6-(1H-テトラゾール-5-イル)-1H-ピラゾロ[4,3-b]ピリジン、
(o)1-(2-クロロ-6-シクロプロピルベンジル)-3-メチル-6-(1H-テトラゾール-5-イル)-1H-ピラゾロ[4,3-b]ピリジン、
(p)3-クロロ-1-(2,6-ジクロロベンジル)-1H-ピラゾロ[4,3-b]ピリジン-6-カルボン酸、
(q)3-クロロ-1-(2-クロロ-6-メチルベンジル)-1H-ピラゾロ[4,3-b]ピリジン-6-カルボン酸、
(r)3-クロロ-1-(2-クロロ-6-メチルベンジル)-6-(1H-テトラゾール-5-イル)-1H-ピラゾロ[4,3-b]ピリジン、又は
(s)[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]ジフルオロ酢酸
である、請求項1記載の化合物又はその薬学的に許容できる塩若しくはエステル。 - 1-(2,6-ジメチルベンジル)-3-メチル-1H-インドール-6-カルボン酸である、請求項1記載の化合物又はその薬学的に許容できる塩若しくはエステル。
- 2-[1-(2,6-ジメチルベンジル)-3-メチル-1H-インドール-6-イル]酢酸である、請求項1記載の化合物又はその薬学的に許容できる塩若しくはエステル。
- 2-[1-(2,6-ジメチルベンジル)-3-メチル-1H-インダゾール-6-イル]酢酸である、請求項1記載の化合物又はその薬学的に許容できる塩若しくはエステル。
- 2-[3-クロロ-1-(2,6-ジメチルベンジル)-1H-インドール-6-イル]酢酸である、請求項1記載の化合物又はその薬学的に許容できる塩若しくはエステル。
- 2-[1-(2,6-ジメチルベンジル)-2,3-ジヒドロ-1H-インドール-6-イル]酢酸である、請求項1記載の化合物又はその薬学的に許容できる塩若しくはエステル。
- (3-RS)-2-[1-(2,6-ジメチルベンジル)-2,3-ジヒドロ-3-メチル-1H-インドール-6-イル]酢酸である、請求項1記載の化合物又はその薬学的に許容できる塩若しくはエステル。
- 2-[1-(2,6-ジメチルベンジル)-2,3-ジヒドロ-3,3-ジメチル-1H-インドール-6-イル]酢酸である、請求項1記載の化合物又はその薬学的に許容できる塩若しくはエステル。
- 2-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インドール-6-イル]酢酸である、請求項1記載の化合物又はその薬学的に許容できる塩若しくはエステル。
- 2-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]酢酸である、請求項1記載の化合物又はその薬学的に許容できる塩若しくはエステル。
- 2-[1-(2-クロロ-6-メチルベンジル)-3-メチル-1H-インダゾール-6-イル]酢酸である、請求項1記載の化合物又はその薬学的に許容できる塩若しくはエステル。
- 3-[1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]プロピオン酸である、請求項1記載の化合物又はその薬学的に許容できる塩若しくはエステル。
- 1-(2,6-ジクロロベンジル)-3-メチル-6-(1H-テトラゾール-5-イルメチル)-1H-インダゾールである、請求項1記載の化合物又はその薬学的に許容できる塩。
- 2-[1-(2,6-ジクロロベンジル)-3-エチル-1H-インダゾール-6-イル]酢酸である、請求項1記載の化合物又はその薬学的に許容できる塩若しくはエステル。
- 1-(2-クロロ-6-メチルベンジル)-3-メチル-6-(1H-テトラゾール-5-イル)-1H-ピラゾロ[4,3-b]ピリジンである、請求項1記載の化合物又はその薬学的に許容できる塩。
- 1-(2-クロロ-6-シクロプロピルベンジル)-3-メチル-6-(1H-テトラゾール-5-イル)-1H-ピラゾロ[4,3-b]ピリジンである、請求項1記載の化合物又はその薬学的に許容できる塩。
- 3-クロロ-1-(2,6-ジクロロベンジル)-1H-ピラゾロ[4,3-b]ピリジン-6-カルボン酸である、請求項1記載の化合物又はその薬学的に許容できる塩若しくはエステル。
- 3-クロロ-1-(2-クロロ-6-メチルベンジル)-1H-ピラゾロ[4,3-b]ピリジン-6-カルボン酸である、請求項1記載の化合物又はその薬学的に許容できる塩若しくはエステル。
- 3-クロロ-1-(2-クロロ-6-メチルベンジル)-6-(1H-テトラゾール-5-イル)-1H-ピラゾロ[4,3-b]ピリジンである、請求項1記載の化合物又はその薬学的に許容できる塩。
- [1-(2,6-ジクロロベンジル)-3-メチル-1H-インダゾール-6-イル]ジフルオロ酢酸である、請求項1記載の化合物又はその薬学的に許容できる塩若しくはエステル。
- 請求項1乃至30のいずれかに記載の化合物又はその薬学的に許容できる塩若しくはエステルを含む、URAT1阻害剤。
- 請求項1乃至30のいずれかに記載の化合物又はその薬学的に許容できる塩若しくはエステルを含む、血中尿酸値低下剤。
- 請求項1乃至30のいずれかに記載の化合物又はその薬学的に許容できる塩若しくはエステルと、薬学的に許容できる担体とを含む、血中尿酸が関与する病態を治療又は予防するための医薬組成物。
- 血中尿酸が関与する病態が、高尿酸血症、痛風結節、急性通風関節炎、慢性通風関節炎、痛風腎、尿路結石、腎機能障害、冠動脈疾患及び虚血性心疾患からなる群より選択される、請求項33に記載の医薬組成物。
- 血中尿酸が関与する病態が高尿酸血症である、請求項33に記載の医薬組成物。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012554878A JP5990106B2 (ja) | 2011-01-28 | 2012-01-30 | 縮環化合物 |
| EP12739424.5A EP2669270B1 (en) | 2011-01-28 | 2012-01-30 | Indole-related compounds as urat1 inhibitors |
| DK12739424.5T DK2669270T3 (en) | 2011-01-28 | 2012-01-30 | Indole-related compounds such as URAT1 inhibitors |
| US13/982,200 US8987473B2 (en) | 2011-01-28 | 2012-01-30 | Ring-fused compound |
| US14/619,980 US9359350B2 (en) | 2011-01-28 | 2015-02-11 | Ring-fused compound |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011-016950 | 2011-01-28 | ||
| JP2011016950 | 2011-01-28 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/982,200 A-371-Of-International US8987473B2 (en) | 2011-01-28 | 2012-01-30 | Ring-fused compound |
| US14/619,980 Division US9359350B2 (en) | 2011-01-28 | 2015-02-11 | Ring-fused compound |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012102405A1 true WO2012102405A1 (ja) | 2012-08-02 |
Family
ID=46580968
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/052009 WO2012102405A1 (ja) | 2011-01-28 | 2012-01-30 | 縮環化合物 |
Country Status (5)
| Country | Link |
|---|---|
| US (2) | US8987473B2 (ja) |
| EP (1) | EP2669270B1 (ja) |
| JP (2) | JP5990106B2 (ja) |
| DK (1) | DK2669270T3 (ja) |
| WO (1) | WO2012102405A1 (ja) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102875590A (zh) * | 2012-10-22 | 2013-01-16 | 北京林业大学 | 芳香族三丁基锡衍生物的制备方法 |
| WO2013096496A3 (en) * | 2011-12-21 | 2013-08-15 | Allergan, Inc. | Compounds acting at multiple prostaglandin receptors giving a general anti-inflammatory response |
| WO2014017643A1 (ja) * | 2012-07-27 | 2014-01-30 | 佐藤製薬株式会社 | ジフルオロメチレン化合物 |
| JP2018509451A (ja) * | 2015-03-24 | 2018-04-05 | シャンハイ インリ ファーマシューティカル カンパニー リミティド | 縮合環誘導体、その製造方法、中間体、薬学的組成物及び応用 |
| WO2018199284A1 (ja) * | 2017-04-28 | 2018-11-01 | 佐藤製薬株式会社 | ジフルオロメチレン化合物の製造法 |
| US11471455B2 (en) | 2018-10-05 | 2022-10-18 | Annapurna Bio, Inc. | Compounds and compositions for treating conditions associated with APJ receptor activity |
| US11767321B2 (en) | 2020-10-05 | 2023-09-26 | Enliven Inc. | 5- and 6-azaindole compounds for inhibition of BCR-ABL tyrosine kinases |
| US11820747B2 (en) | 2021-11-02 | 2023-11-21 | Flare Therapeutics Inc. | PPARG inverse agonists and uses thereof |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BR112018010216B1 (pt) | 2015-11-20 | 2024-02-15 | Forma Therapeutics, Inc | Purinonas como inibidores da protease específica da ubiquitina 1 e composição farmacêutica compreendendo os referidos compostos |
| CN106045898B (zh) * | 2016-06-28 | 2019-05-24 | 广东东阳光药业有限公司 | 一种吲哚类化合物及其制备方法和用途 |
| CN106146533B (zh) * | 2016-07-14 | 2018-04-03 | 华润赛科药业有限责任公司 | 含硫杂环羧酸类衍生物、其制备方法和应用 |
| EP3538151B1 (en) | 2016-11-11 | 2024-06-26 | Whitehead Institute for Biomedical Research | Human plasma-like medium |
| AU2018281131B2 (en) | 2017-06-08 | 2022-01-20 | Merck Sharp & Dohme Corp. | Pyrazolopyrimidine PDE9 inhibitors |
| US20200172483A1 (en) * | 2017-06-22 | 2020-06-04 | Curadev Pharma Limited | Heterocyclic small molecule modulators of human sting |
| BR112021023562A2 (pt) | 2019-06-14 | 2022-01-04 | Janssen Pharmaceutica Nv | Carbamatos de pirazina e seus usos como moduladores do receptor de glun2b |
| MA56196A (fr) * | 2019-06-14 | 2022-04-20 | Janssen Pharmaceutica Nv | Pyrazolo-pyridine amides substitués et leur utilisation en tant que modulateurs du récepteur glun2b |
| CA3143276A1 (en) | 2019-06-14 | 2020-12-17 | Janssen Pharmaceutica Nv | Substituted heteroaromatic pyrazolo-pyridines and their use as glun2b receptor modulators |
| JP2022536773A (ja) | 2019-06-14 | 2022-08-18 | ヤンセン ファーマシューティカ エヌ.ベー. | 置換ピラゾロピラジン及びglun2b受容体調節因子としてのそれらの使用 |
| CA3143103A1 (en) | 2019-06-14 | 2020-12-17 | Janssen Pharmaceutica Nv | Pyridine carbamates and their use as glun2b receptor modulators |
| WO2023208108A1 (zh) * | 2022-04-27 | 2023-11-02 | 江苏新元素医药科技有限公司 | 可用于降尿酸的化合物 |
Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH05222000A (ja) * | 1991-10-14 | 1993-08-31 | Fujisawa Pharmaceut Co Ltd | ベンズイミダゾール誘導体およびその製造方法 |
| WO1997024334A1 (fr) * | 1995-12-28 | 1997-07-10 | Fujisawa Pharmaceutical Co., Ltd. | Derives du benzimidazole |
| WO1998005327A1 (en) * | 1996-08-05 | 1998-02-12 | Rhone-Poulenc Rorer Pharmaceuticals Inc. | Substituted aromatic compounds |
| JPH10503488A (ja) * | 1994-11-25 | 1998-03-31 | 三共株式会社 | 複素環化合物、その製造及びその治療的用途 |
| WO1998015530A1 (fr) * | 1996-10-08 | 1998-04-16 | Fujisawa Pharmaceutical Co., Ltd. | Derives d'indol |
| WO1999000372A1 (en) * | 1997-06-27 | 1999-01-07 | Fujisawa Pharmaceutical Co., Ltd. | Sulfonamide compounds and medicinal use thereof |
| WO1999000373A1 (fr) * | 1997-06-27 | 1999-01-07 | Fujisawa Pharmaceutical Co., Ltd. | Derives de benzimidazole |
| JPH11503445A (ja) * | 1995-04-10 | 1999-03-26 | 藤沢薬品工業株式会社 | cGMP−PDE阻害剤としてのインドール誘導体 |
| WO2000039099A1 (fr) * | 1998-12-24 | 2000-07-06 | Fujisawa Pharmaceutical Co., Ltd. | Derives de benzimidazole |
| JP2003513075A (ja) * | 1999-11-05 | 2003-04-08 | スミスクライン ビーチャム パブリック リミテッド カンパニー | 複合5ht1a、5ht1bおよび5ht1d受容体活性を有するイソキノリンおよびキナゾリン誘導体 |
| WO2006057460A1 (ja) * | 2004-11-29 | 2006-06-01 | Japan Tobacco Inc. | 窒素含有縮合環化合物及びその用途 |
| WO2007097197A1 (ja) * | 2006-02-20 | 2007-08-30 | Astellas Pharma Inc. | アミド誘導体またはその塩 |
| WO2007109178A2 (en) * | 2006-03-16 | 2007-09-27 | Pharmacyclics, Inc. | Indole derivatives as inhibitors of histone deacetylase |
| WO2009129335A2 (en) * | 2008-04-15 | 2009-10-22 | Pharmacyclics, Inc. | Selective inhibitors of histone deacetylase |
| WO2010107739A2 (en) * | 2009-03-18 | 2010-09-23 | The Board Of Trustees Of The Leland Stanford Junior University | Methods and compositions of treating a flaviviridae family viral infection |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0470543A1 (de) * | 1990-08-10 | 1992-02-12 | Dr. Karl Thomae GmbH | Heterocyclische Imidazole, diese Verbindungen enthaltende Arzneimittel und Verfahren zur ihrer Herstellung |
| CZ24197A3 (en) * | 1994-07-27 | 1997-08-13 | Sankyo Co | Heterocyclic compounds, usable as allosteric efectors in muscarine receptors |
| AUPO118896A0 (en) * | 1996-07-23 | 1996-08-15 | Fujisawa Pharmaceutical Co., Ltd. | New use |
| JPH10114654A (ja) * | 1996-10-09 | 1998-05-06 | Fujisawa Pharmaceut Co Ltd | 新規用途 |
| US6410584B1 (en) * | 1998-01-14 | 2002-06-25 | Cell Pathways, Inc. | Method for inhibiting neoplastic cells with indole derivatives |
| US6358992B1 (en) * | 1998-11-25 | 2002-03-19 | Cell Pathways, Inc. | Method of inhibiting neoplastic cells with indole derivatives |
| AU2002349477A1 (en) * | 2001-11-26 | 2003-06-10 | Takeda Chemical Industries, Ltd. | Bicyclic derivative, process for producing the same, and use |
| AU2003902860A0 (en) * | 2003-06-06 | 2003-06-26 | Daicel Chemical Industries, Ltd | Benzimidazole compounds |
| US20070197512A1 (en) | 2006-01-27 | 2007-08-23 | Japan Tobacco Inc. | Carboxylic Acid Compounds and Use Thereof |
| WO2007086504A1 (ja) | 2006-01-27 | 2007-08-02 | Japan Tobacco Inc. | カルボン酸化合物及びその用途 |
| JP5093096B2 (ja) | 2006-03-02 | 2012-12-05 | アステラス製薬株式会社 | 17βHSDtype5阻害剤 |
| ATE543819T1 (de) * | 2006-10-19 | 2012-02-15 | Signal Pharm Llc | Heteroarylverbindungen, zusammensetzungen daraus und behandlungsverfahren damit |
| US8501747B2 (en) * | 2007-02-13 | 2013-08-06 | Merck Sharp & Dohme Corp. | Functionally selective alpha2C adrenoreceptor agonists |
| DK2133332T3 (da) | 2007-04-11 | 2013-09-30 | Kissei Pharmaceutical | (aza)indolderivat og anvendelse deraf til medicinske formål |
| US9149463B2 (en) | 2007-09-18 | 2015-10-06 | The Board Of Trustees Of The Leland Standford Junior University | Methods and compositions of treating a Flaviviridae family viral infection |
| CN101969773B (zh) | 2008-03-13 | 2015-08-19 | 维尔斯达医疗公司 | 用于降低尿酸的化合物和方法 |
| KR101048448B1 (ko) * | 2008-11-21 | 2011-07-11 | 한국화학연구원 | 신규 피라졸로[4,3-b]피리딘 유도체 또는 이의 약학적으로허용가능한 염, 이의 제조방법 및 이를 유효성분으로 함유하는 약학적 조성물 |
| JP2011246389A (ja) * | 2010-05-26 | 2011-12-08 | Oncotherapy Science Ltd | Ttk阻害作用を有する縮環ピラゾール誘導体 |
-
2012
- 2012-01-30 DK DK12739424.5T patent/DK2669270T3/en active
- 2012-01-30 WO PCT/JP2012/052009 patent/WO2012102405A1/ja active Application Filing
- 2012-01-30 JP JP2012554878A patent/JP5990106B2/ja not_active Expired - Fee Related
- 2012-01-30 US US13/982,200 patent/US8987473B2/en not_active Expired - Fee Related
- 2012-01-30 EP EP12739424.5A patent/EP2669270B1/en not_active Not-in-force
-
2015
- 2015-02-11 US US14/619,980 patent/US9359350B2/en not_active Expired - Fee Related
-
2016
- 2016-08-12 JP JP2016158397A patent/JP6223514B2/ja not_active Expired - Fee Related
Patent Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH05222000A (ja) * | 1991-10-14 | 1993-08-31 | Fujisawa Pharmaceut Co Ltd | ベンズイミダゾール誘導体およびその製造方法 |
| JPH10503488A (ja) * | 1994-11-25 | 1998-03-31 | 三共株式会社 | 複素環化合物、その製造及びその治療的用途 |
| JPH11503445A (ja) * | 1995-04-10 | 1999-03-26 | 藤沢薬品工業株式会社 | cGMP−PDE阻害剤としてのインドール誘導体 |
| WO1997024334A1 (fr) * | 1995-12-28 | 1997-07-10 | Fujisawa Pharmaceutical Co., Ltd. | Derives du benzimidazole |
| WO1998005327A1 (en) * | 1996-08-05 | 1998-02-12 | Rhone-Poulenc Rorer Pharmaceuticals Inc. | Substituted aromatic compounds |
| WO1998015530A1 (fr) * | 1996-10-08 | 1998-04-16 | Fujisawa Pharmaceutical Co., Ltd. | Derives d'indol |
| WO1999000373A1 (fr) * | 1997-06-27 | 1999-01-07 | Fujisawa Pharmaceutical Co., Ltd. | Derives de benzimidazole |
| WO1999000372A1 (en) * | 1997-06-27 | 1999-01-07 | Fujisawa Pharmaceutical Co., Ltd. | Sulfonamide compounds and medicinal use thereof |
| WO2000039099A1 (fr) * | 1998-12-24 | 2000-07-06 | Fujisawa Pharmaceutical Co., Ltd. | Derives de benzimidazole |
| JP2003513075A (ja) * | 1999-11-05 | 2003-04-08 | スミスクライン ビーチャム パブリック リミテッド カンパニー | 複合5ht1a、5ht1bおよび5ht1d受容体活性を有するイソキノリンおよびキナゾリン誘導体 |
| WO2006057460A1 (ja) * | 2004-11-29 | 2006-06-01 | Japan Tobacco Inc. | 窒素含有縮合環化合物及びその用途 |
| WO2007097197A1 (ja) * | 2006-02-20 | 2007-08-30 | Astellas Pharma Inc. | アミド誘導体またはその塩 |
| WO2007109178A2 (en) * | 2006-03-16 | 2007-09-27 | Pharmacyclics, Inc. | Indole derivatives as inhibitors of histone deacetylase |
| WO2009129335A2 (en) * | 2008-04-15 | 2009-10-22 | Pharmacyclics, Inc. | Selective inhibitors of histone deacetylase |
| WO2010107739A2 (en) * | 2009-03-18 | 2010-09-23 | The Board Of Trustees Of The Leland Stanford Junior University | Methods and compositions of treating a flaviviridae family viral infection |
Non-Patent Citations (2)
| Title |
|---|
| HULME C. ET AL.: "Orally active indole N-Oxide PDE4 inhibitors", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 8, no. 21, 1998, pages 3053 - 3058, XP004141874 * |
| See also references of EP2669270A4 * |
Cited By (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9567328B2 (en) | 2011-12-21 | 2017-02-14 | Allergan, Inc. | Compounds acting at multiple prostaglandin receptors giving a general anti-inflammatory response |
| WO2013096496A3 (en) * | 2011-12-21 | 2013-08-15 | Allergan, Inc. | Compounds acting at multiple prostaglandin receptors giving a general anti-inflammatory response |
| US10988474B2 (en) | 2011-12-21 | 2021-04-27 | Allergan, Inc. | Compounds acting at multiple prostaglandin receptors giving a general anti-inflammatory response |
| US10392382B2 (en) | 2011-12-21 | 2019-08-27 | Allergan, Inc. | Compounds acting at multiple prostaglandin receptors giving a general anti-inflammatory response |
| EP3424904A1 (en) * | 2011-12-21 | 2019-01-09 | Allergan, Inc. | Compounds acting at multiple prostaglandin receptors giving a general anti-inflammatory response |
| US9512119B2 (en) | 2012-07-27 | 2016-12-06 | Sato Pharmaceutical Co., Ltd. | Difluoromethylene compound |
| WO2014017643A1 (ja) * | 2012-07-27 | 2014-01-30 | 佐藤製薬株式会社 | ジフルオロメチレン化合物 |
| US9650380B2 (en) | 2012-07-27 | 2017-05-16 | Sato Pharmaceutical Co., Ltd. | Difluoromethylene compound |
| JPWO2014017643A1 (ja) * | 2012-07-27 | 2016-07-11 | 佐藤製薬株式会社 | ジフルオロメチレン化合物 |
| EP3444238A3 (en) * | 2012-07-27 | 2019-05-15 | Sato Pharmaceutical Co., Ltd. | Process for preparing difluoromethylene compounds |
| JP2016169241A (ja) * | 2012-07-27 | 2016-09-23 | 佐藤製薬株式会社 | ジフルオロメチレン化合物 |
| CN102875590A (zh) * | 2012-10-22 | 2013-01-16 | 北京林业大学 | 芳香族三丁基锡衍生物的制备方法 |
| JP2018509451A (ja) * | 2015-03-24 | 2018-04-05 | シャンハイ インリ ファーマシューティカル カンパニー リミティド | 縮合環誘導体、その製造方法、中間体、薬学的組成物及び応用 |
| WO2018199284A1 (ja) * | 2017-04-28 | 2018-11-01 | 佐藤製薬株式会社 | ジフルオロメチレン化合物の製造法 |
| JPWO2018199284A1 (ja) * | 2017-04-28 | 2020-03-12 | 佐藤製薬株式会社 | ジフルオロメチレン化合物の製造法 |
| TWI774755B (zh) * | 2017-04-28 | 2022-08-21 | 日商佐藤製藥股份有限公司 | 二氟甲烯化合物之製造法 |
| JP7198201B2 (ja) | 2017-04-28 | 2022-12-28 | 佐藤製薬株式会社 | ジフルオロメチレン化合物の製造法 |
| US11471455B2 (en) | 2018-10-05 | 2022-10-18 | Annapurna Bio, Inc. | Compounds and compositions for treating conditions associated with APJ receptor activity |
| US11944622B2 (en) | 2018-10-05 | 2024-04-02 | Annapurna Bio, Inc. | Compounds and compositions for treating conditions associated with APJ receptor activity |
| US11767321B2 (en) | 2020-10-05 | 2023-09-26 | Enliven Inc. | 5- and 6-azaindole compounds for inhibition of BCR-ABL tyrosine kinases |
| US11807638B2 (en) | 2020-10-05 | 2023-11-07 | Enliven Inc. | 5- and 6-azaindole compounds for inhibition of Bcr-Abl tyrosine kinases |
| US11820747B2 (en) | 2021-11-02 | 2023-11-21 | Flare Therapeutics Inc. | PPARG inverse agonists and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| JP6223514B2 (ja) | 2017-11-01 |
| US20150203490A1 (en) | 2015-07-23 |
| US9359350B2 (en) | 2016-06-07 |
| JP5990106B2 (ja) | 2016-09-07 |
| EP2669270B1 (en) | 2018-01-03 |
| JP2017008079A (ja) | 2017-01-12 |
| US20140005221A1 (en) | 2014-01-02 |
| JPWO2012102405A1 (ja) | 2014-07-03 |
| US8987473B2 (en) | 2015-03-24 |
| DK2669270T3 (en) | 2018-02-26 |
| EP2669270A4 (en) | 2014-07-02 |
| EP2669270A1 (en) | 2013-12-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6223514B2 (ja) | 縮環化合物 | |
| JP6229020B2 (ja) | ジフルオロメチレン化合物 | |
| EP2516439B1 (en) | Diaza-spiro[5.5]undecanes as orexin receptor antagonists | |
| AU757290B2 (en) | Imidazolone anorectic agents: II. phenyl derivatives | |
| EA035185B1 (ru) | Производные спиродиамина в качестве ингибиторов альдостеронсинтазы | |
| EP3240783B1 (en) | New benzimidazole derivatives as antihistamine agents | |
| JP2023542845A (ja) | Rnaヘリカーゼdhx33を阻害する多環式化合物及びその応用 | |
| WO2008018306A1 (fr) | Dérivé quinazoline | |
| JP2905599B2 (ja) | ピリジルイミダゾール誘導体およびその製造方法 | |
| JP2023547306A (ja) | アリールアミド化合物、それを含む医薬組成物、その製造方法及びそれらの使用 | |
| DK2781508T3 (en) | NITROGEN CONCENTRATED RING COMPOUNDS FOR USE AS CRTH2 ANTAGONISTS | |
| Sharma et al. | SYNTHESIS AND BIOLOGICAL ACTIVITY OF 4'-(5-AMINO-6-CHLORO-2-SUBSTITUTED-BENZOIMIDAZOL-1-YLMETHYL)]-BIPHENYL-2-CARBOXYLIC ACID AS ANTIHYPERTENSIVE AGENTS. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12739424 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2012554878 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012739424 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13982200 Country of ref document: US |