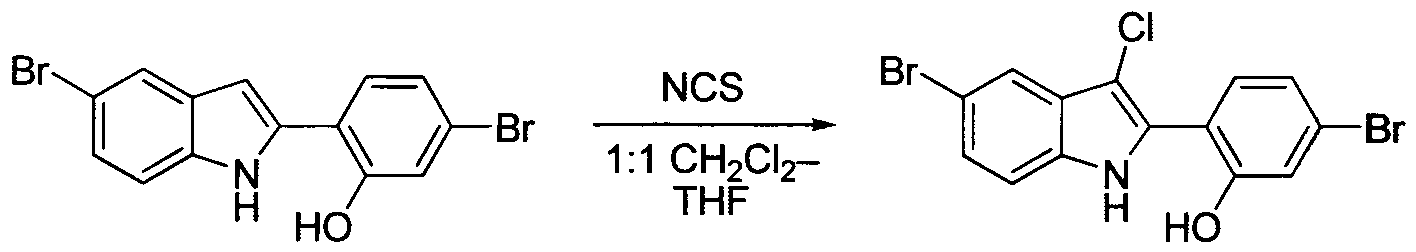
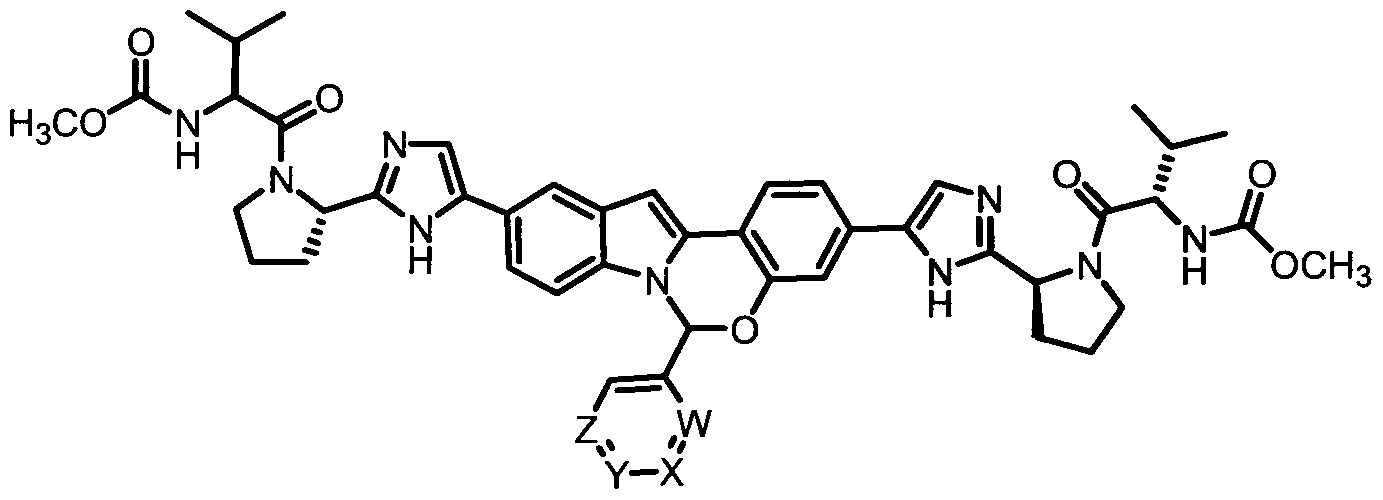
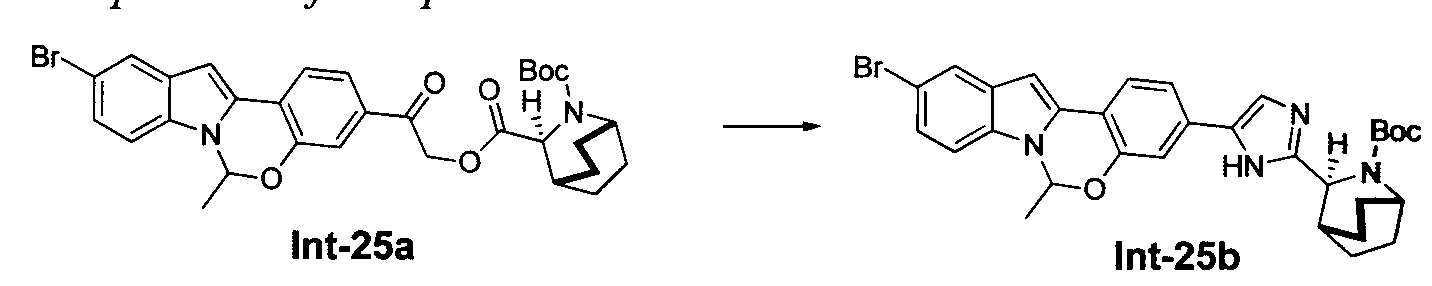
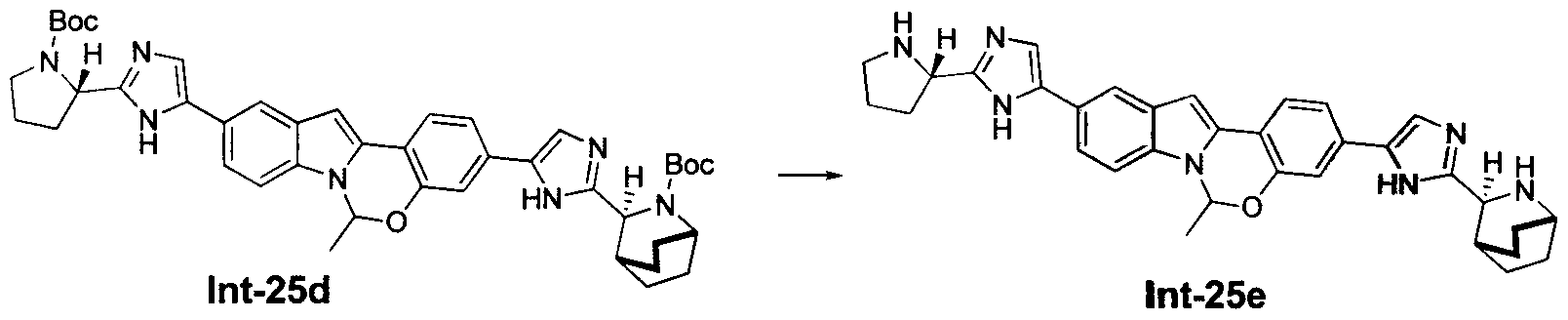
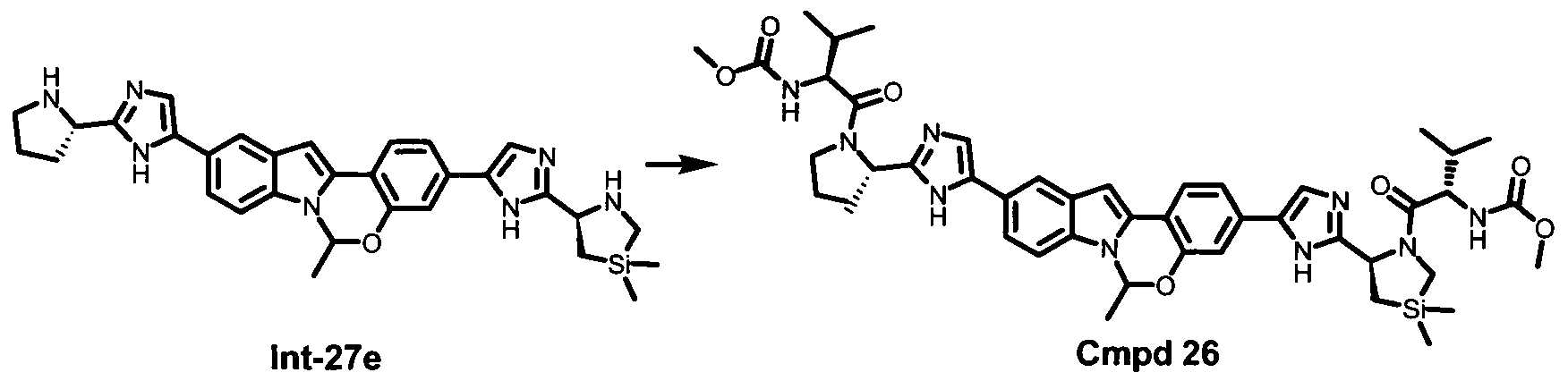
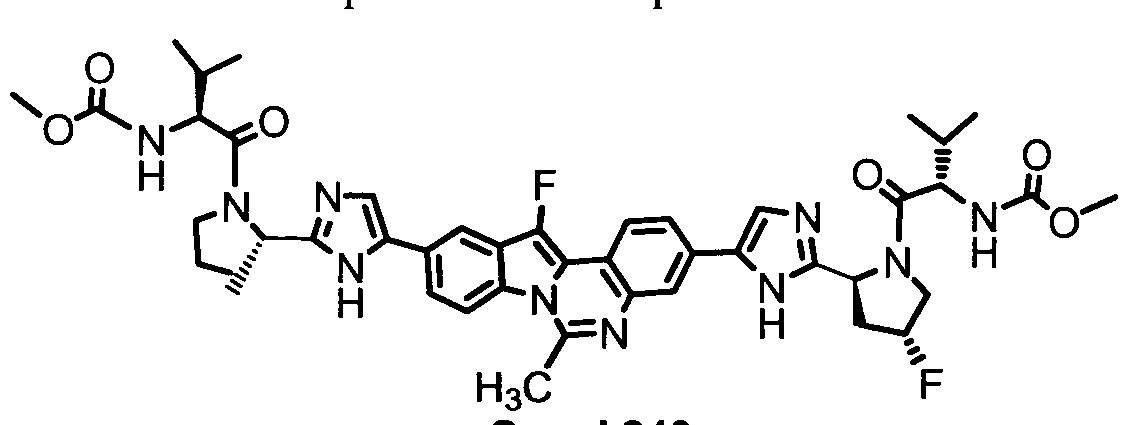
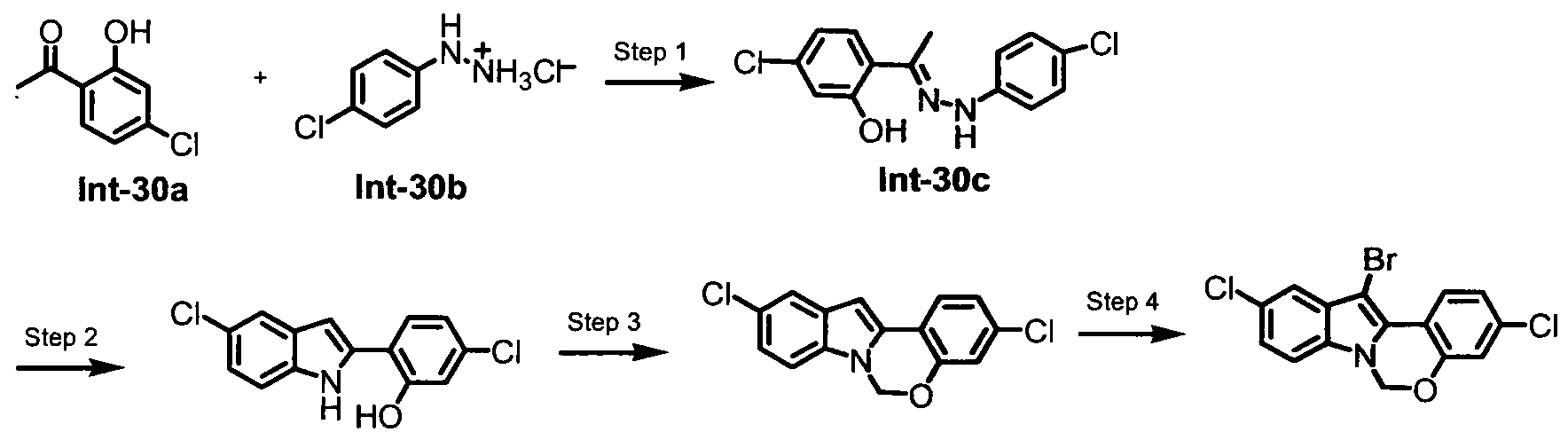
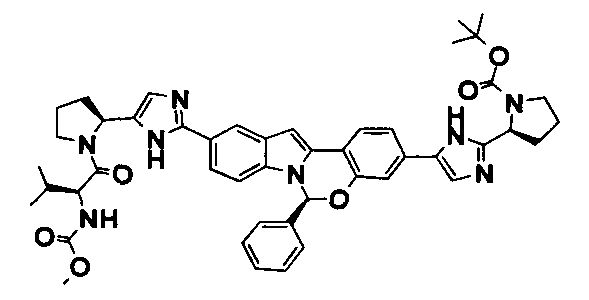
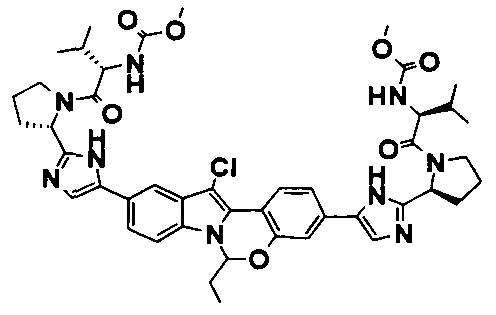
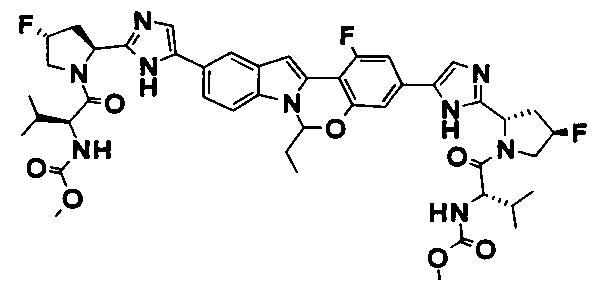
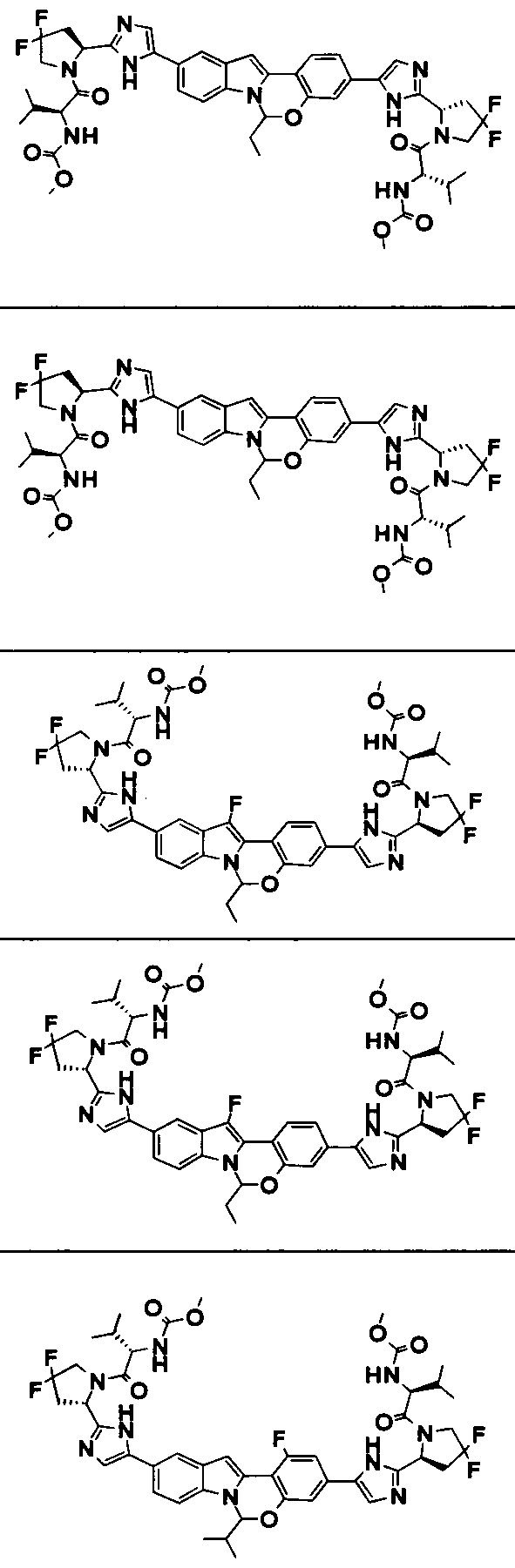
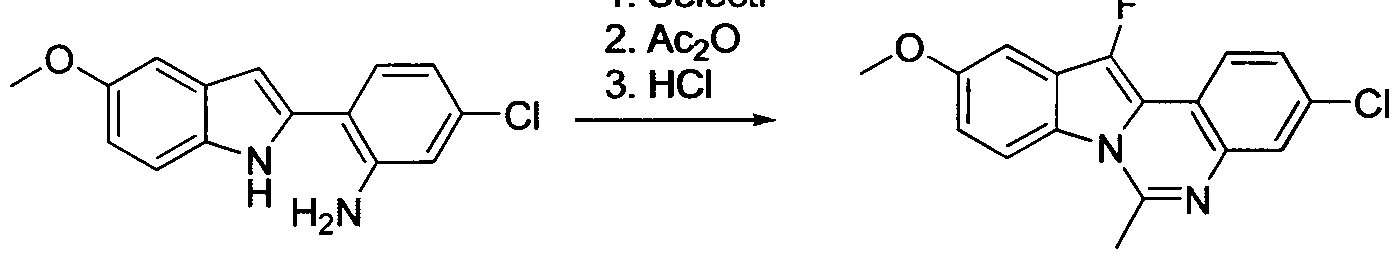TETRACYCLIC INDOLE DERIVATIVES FOR TREATING
HEPATITIS C VIRUS INFECTION
FIELD OF THE INVENTION
The present invention relates to novel Tetracyclic Indole Derivatives, compositions comprising at least one Tetracyclic Indole Derivative, and methods of using the Tetracyclic Indole Derivatives for treating or preventing HCV infection in a patient.
BACKGROUND OF THE INVENTION
Hepatitis C virus (HCV) is a major human pathogen. A substantial fraction of these HCV-infected individuals develop serious progressive liver disease, including cirrhosis and hepatocellular carcinoma, which are often fatal. HCV is a (+)- sense single-stranded enveloped RNA virus that has been implicated as the major causative agent in non-A, non-B hepatitis (NANBH), particularly in blood-associated NANBH (BB-NANBH) (see, International Publication No. WO 89/04669 and European Patent Publication No. EP 381 216). NANBH is to be distinguished from other types of viral-induced liver disease, such as hepatitis A virus (HAV), hepatitis B virus (HBV), delta hepatitis virus (HDV), cytomegalovirus (CMV) and Epstein-Barr virus (EBV), as well as from other forms of liver disease such as alcoholism and primary biliar cirrhosis.
It is well-established that persistent infection of HCV is related to chronic hepatitis, and as such, inhibition of HCV replication is a viable strategy for the prevention of hepatocellular carcinoma. Current therapies for HCV infection include a-interferon monotherapy and combination therapy comprising a-interferon and ribavirin. These therapies have been shown to be effective in some patients with chronic HCV infection, but suffer from poor efficacy and unfavorable side-effects and there are currently efforts directed to the discovery of HCV replication inhibitors that are useful for the treatment and prevention of HCV related disorders.
Current research efforts directed toward the treatment of HCV includes the use of antisense oligonucleotides, free bile acids (such as ursodeoxycholic acid and chenodeoxycholic acid) and conjugated bile acids (such as tauroursodeoxycholic
acid). Phosphonoformic acid esters have also been proposed as potentially useful for the treatment of various viral infections, including HCV. Vaccine development, however, has been hampered by the high degree of viral strain heterogeneity and immune evasion and the lack of protection against reinfection, even with the same inoculum.
In light of these treatment hurdles, the development of small-molecule inhibitors directed against specific viral targets has become a major focus of anti- HCV research. The determination of crystal structures for NS3 protease, NS3 RNA helicase, NS5A, and NS5B polymerase, with and without bound ligands, has provided important structural insights useful for the rational design of specific inhibitors.
Recent attention has been focused toward the identification of inhibitors of HCV NS5A. HCV NS5A is a 447 amino acid phosphoprotein which lacks a defined enzymatic function. It runs as 56kd and 58kd bands on gels depending on phosphorylation state (Tanji, et al. J. Virol. 69:3980-3986 (1995)). HCV NS5A resides in replication complex and may be responsible for the switch from replication of RNA to production of infectious virus (Huang, Y, et al, Virology 364:1-9 (2007)).
Multicyclic HCV NS5A inhibitors have been reported. See U.S. Patent Publication Nos. US20080311075, US20080044379, US20080050336, US20080044380, US20090202483 and US2009020478. HCV NS5A inhibitors having fused tricyclic moieties are disclosed in International Patent Publication Nos. WO 10/065681, WO 10/065668, and WO 10/065674.
Other HCV NS5A inhibitors and their use for reducing viral load in HCV infected humans have been described in U.S. Patent Publication No.
US2006027651 1.
SUMMARY OF THE INVENTION
In one aspect, the present invention provides Compounds of Formula
(I)
or a pharmaceutically acceptable salt thereof, wherein:
A and A' are each independently a 5 or 6-membered monocyclic heterocycloalkyl, wherein said 5 or 6-membered monocyclic heterocycloalkyl group can be optionally fused to an aryl group; and wherein said 5 or 6-membered monocyclic heterocycloalkyl group can be optionally and independently substituted on one or more ring carbon atoms with R13, such that any two R13 groups on the same ring, together with the carbon atoms to which they are attached, can join to form a fused, bridged or spirocyclic 3 to 6-membered cycloalkyl group or a fused, bridged or spirocyclic 4 to 6-membered heterocycloalkyl group, wherein said 5 or 6-membered monocyclic heterocycloalkyl contains from 1 to 2 ring heteroatoms, each
independently selected from N(R4), S, O and Si(RI6)2;
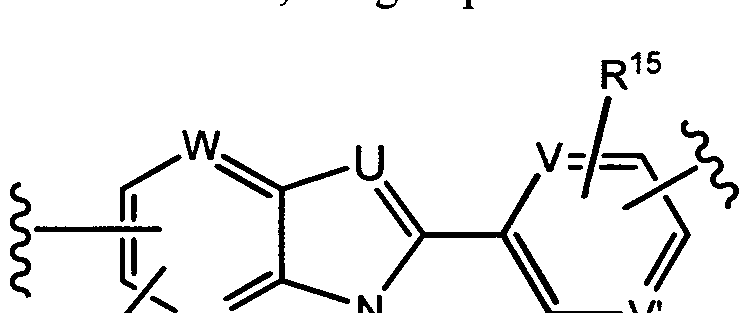
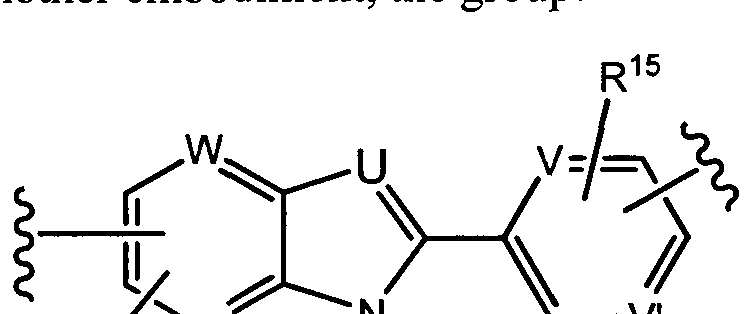
G is selected from -C(R3)2-0-, -C(R3)2-N(R5)-, -C(0)-0-, -C(0 N(R5)-, -C(0)-C(R3)2-, -C(R3)2-C(0 , -C(= R5)-N(R5)-, ^C(R3)2-S02-, -S02-C(R3)2-, -S02N(R5)-, -C(R3)2-C(R3)2-, -C(R1 )=C(R14)- and -C(R14)=N-;
U is selected from N and C(R2);
V and V are each independently selected from N and C(R15);
W and W are each independently selected from N and C(R');
X and X' are each independently selected from N and C(R10);
Y and Y' are each independently selected from N and C(R10);
R1 is selected from H, Ci-C6 alkyl, 3 to 6-membered cycloalkyl, halo, - OH, -0-(Ci-C6 alkyl), Ci-C6 haloalkyl and -0-(d-C6 haloalkyl);
each occurrence of R2 is independently selected from H, Ci-C6 alkyl, 3 to 6 membered cycloalkyl, -0-(Ci-C6 alkyl), Ci-C6 haloalkyl -0-(Ci-C6 haloalkyl); halo, -OH, aryl, and heteroaryl;
each occurrence of R3 is independently selected from H, Ci-C6 alkyl, Ci-C6 haloalkyl, -(C C6 alkylene)-0-(Ci-C6 alkyl), -(Ci-C6 alkylene)-0-(3 to 6 membered cycloalkyl), 3 to 6-membered cycloalkyl, 4 to 6-membered
heterocycloalkyl, aryl, 5 or 6-membered monocyclic heteroaryl, 9 or 10-membered bicyclic heteroaryl and benzyl, wherein said aryl group, said 5 or 6-membered monocyclic heteroaryl group, said 9 or 10-membered bicyclic heteroaryl group or the phenyl moiety of said benzyl group can be optionally substituted with up to 3 groups, which can be the same or different, and are selected from Ci-C6 alkyl, C]-C6 haloalkyl, -0-(Ci-C6 alkyl), -0(Ci-C6 haloalkyl), halo, -(Ci-C6 alkylene)-0-(Ci-C6 alkyl) and - CN and wherein two R3 groups attached to the same carbon atom, together with the common carbon atom to which they are attached, can join to form a carbonyl group, a 3 to 6-membered spirocyclic cycloalkyl group or a 3 to 6-membered spirocyclic heterocycloalkyl group;
each occurrence of R4 is independently selected from -[C(R7)2]qN(R6)2, -C(0)Rn, -C(0)-[C(R7)2]qN(R6)2, -C(0)-[C(R7)2]q-Rn, -C(0)-[C(R7)2]qN(R6)C(0)- R11, -C(0)[C(R7)2]qN(R6)S02-Rn, -C(0)-[C(R7)2]qN(R6)C(0)0-Rn, -C(O)- [C(R7)2]qC(0)0-R1 1 and -alkylene-N(R6)-[C(R7)2]q-N(R6)-C(0)0-R' 1 ;
each occurrence of R5 is independently selected from H, Ci-C6 alkyl, - (Ci-C6 alkylene)-0-(Ci-C alkyl), 3 to 6-membered cycloalkyl, 4 to 6-membered heterocycloalkyl, aryl, 5 or 6-membered monocyclic heteroaryl and benzyl, wherein said aryl group, said 5 or 6-membered monocyclic heteroaryl group or the phenyl moiety of said benzyl group can be optionally substituted with up to 3 groups, which can be the same or different, and are selected from Ci-C6 alkyl, Ci-C6 haloalkyl, -O- (Ci-C6 alkyl), -0-(d-C6 haloalkyl), halo, -(Ci-Ce alkylene)-0-(d-C6 alkyl) and - CN;
each occurrence of R6 is independently selected from H, Ci-C6 alkyl, 3 to 6-membered cycloalkyl, 4 to 6-membered heterocycloalkyl, aryl and 5 or 6- membered monocyclic heteroaryl, wherein said 3 to 6-membered cycloalkyl group, said 4 to 6-membered heterocycloalkyl group, said aryl group and said 5 or 6- membered monocyclic heteroaryl group can be optionally and independently substituted with up to two R groups, and wherein two R groups that are attached to the same nitrogen atom, together with the common nitrogen atom to which they are attached, can join to form a 4 to 6-membered heterocycloalkyl group;
each occurrence of R7 is independently selected from H, Ci-C6 alkyl, Ci-C6 haloalkyl, -alkylene-0-(Ci-C6 alkyl), 3 to 6-membered cycloalkyl, 4 to 6- membered heterocycloalkyl, aryl and 5 or 6-membered monocyclic heteroaryl, wherein said 3 to 6-membered cycloalkyl group, said 4 to 6-membered
heterocycloalkyl group, said aryl group and said 5 or 6-membered monocyclic heteroaryl group can be optionally substituted with up to three R groups;
each occurrence of R8 is independently selected from H, d-C6 alkyl, halo, -d-C6 haloalkyl, C,-C6 hydroxy alkyl, -OH, -C(0)NH-(d-C6 alkyl), -C(0)N(Ci- C6 alkyl)2, -0-(Ci-C6 alkyl), -NH2, -NH(d-C6 alkyl), -N(Ci-C6 alkyl)2 and -NHC(O)- (d-C6 alkyl);
each occurrence of R9 is independently selected from H, d-C6 alkyl, Ci-C6 haloalkyl, 3 to 6-membered cycloalkyl, 4 to 6-membered heterocycloalkyl, aryl and 5 or 6-membered monocyclic heteroaryl;
each occurrence of R10 is independently selected from H, Ci-C6 alkyl, Ci-C6 haloalkyl, halo, -OH, -0-(d-C6 alkyl) and -CN;
each occurrence of R1 1 is independently selected from H, Ci-C6 alkyl, C)-C6 haloalkyl, Ci-C6 hydroxyalkyl, 3 to 6-membered cycloalkyl and 4 to 6- membered heterocycloalkyl;
each occurrence of R is independently selected from Ci-C6 alkyl, Ci- C6 haloalkyl, 3 to 6-membered cycloalkyl, 4 to 6-membered heterocycloalkyl, aryl and 5 or 6-membered monocyclic heteroaryl;
each occurrence of R13 is independently selected from H, halo, Ci-C6 alkyl, d-C6 haloalkyl, 3 to 6-membered cycloalkyl, 4 to 6-membered
heterocycloalkyl, -CN, -OR9, -N(R9)2, -C(0)R12, -C(0)OR9, -C(0)N(R9)2, - NHC(0)R12, -NHC(0)NHR9, -NHC(0)OR9, -OC(0)R12, -SR9 and -S(0)2R12, wherein two R12 groups together with the carbon atom(s) to which they are attached, can optionally join to form a 3 to 6-membered cycloalkyl group or 4 to 6-membered heterocycloalkyl group;
each occurrence of R14 is independently selected from H, halo, Ci-C6 alkyl, -(Ci-C6 alkylene)-0-(Ci-C6 alkyl), 3 to 6-membered cycloalkyl, Ci-C6
haloalkyl, aryl, 5 or 6-membered monocyclic heteroaryl and benzyl, wherein said aryl group, said 5 or 6-membered monocyclic heteroaryl group or the phenyl moiety of said benzyl group can be optionally substituted with up to 3 groups, which can be the same or different, and are selected from halo, -CN, d-C6 alkyl, d-C6 haloalkyl, -O- (Ci-C6 alkyl), -(Ci-C6 alkylene)-0-(d-C6 alkyl) and -0-(d-C6 haloalkyl);
each occurrence of R15 is independently selected from H, Ci-C alkyl, 3 to 6-membered cycloalkyl, halo, -OH, -0-(Ci-C6 alkyl), Ci-C6 haloalkyl and -O- (Ci-C6 haloalkyl);
each occurrence of R16 is independently selected from H, halo, Ci-C6 alkyl and 3 to 6-membered cycloalkyl, wherein two R16 groups that are attached to a common silicon atom can join to form a -(CH2)4- or a -(CH2)5- group; and
each occurrence of q is independently an integer ranging from 0 to 4, provided that the compound of formula (I) is other than:
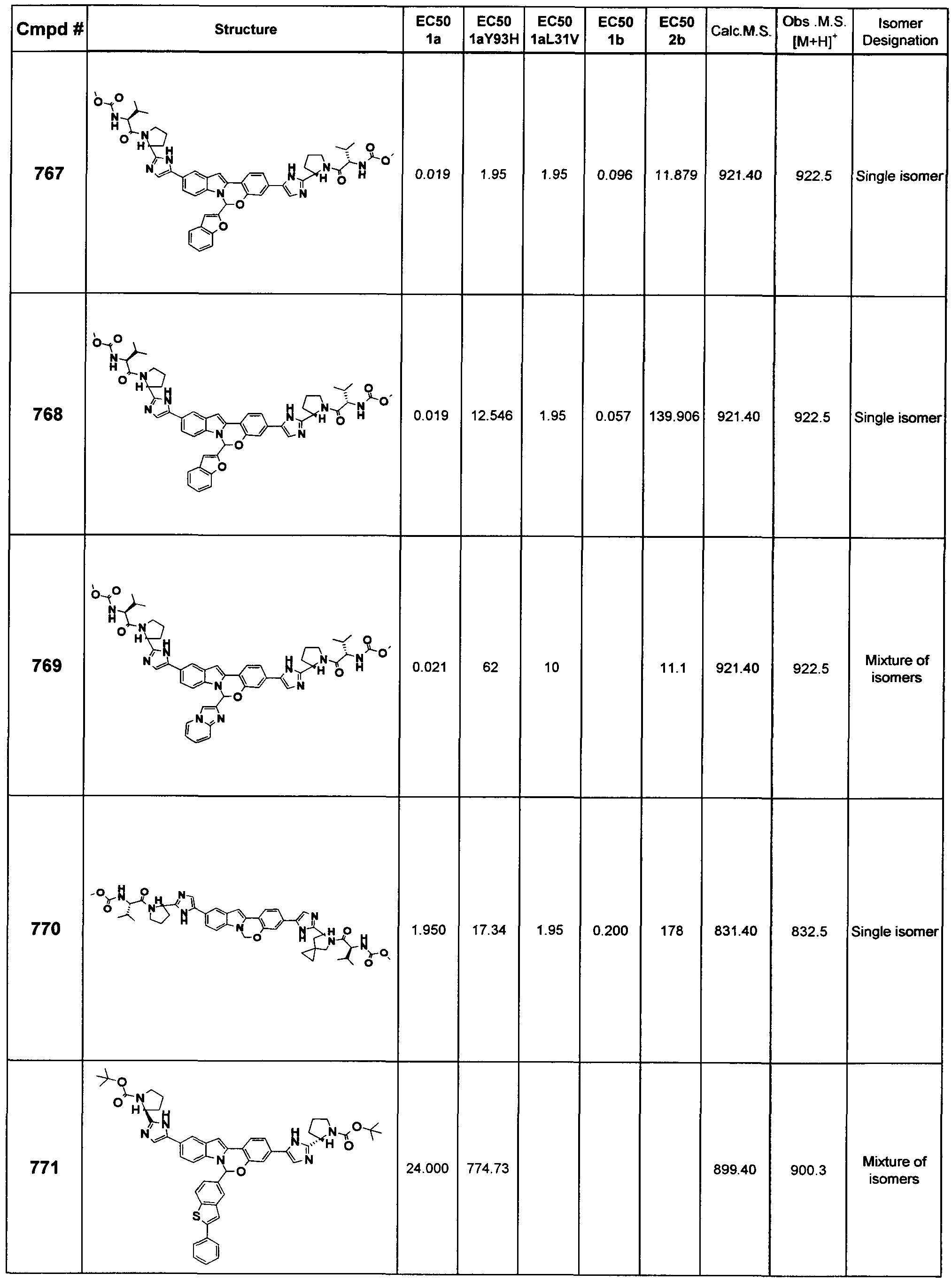
The Compounds of Formula (I) (also referred to herein as the
"Tetracyclic Indole Derivatives") and pharmaceutically acceptable salts thereof can be useful, for example, for inhibiting HCV viral replication or replicon activity, and for treating or preventing HCV infection in a patient. Without being bound by any specific theory, it is believed that the Tetracyclic Indole Derivatives inhibit HCV viral replication by inhibiting HCV NS5A.
Accordingly, the present invention provides methods for treating or preventing HCV infection in a patient, comprising administering to the patient an effective amount of at least one Tetracyclic Indole Derivative.
The details of the invention are set forth in the accompanying detailed description below.
Although any methods and materials similar to those described herein can be used in the practice or testing of the present invention, illustrative methods and materials are now described. Other embodiments, aspects and features of the present invention are either further described in or will be apparent from the ensuing description, examples and appended claims.
DETAILED DESCRIPTION OF THE INVENTION
The present invention relates to novel Tetracyclic Indole Derivatives, compositions comprising at least one Tetracyclic Indole Derivative, and methods of using the Tetracyclic Indole Derivatives for treating or preventing HCV infection in a patient.
Definitions and Abbreviations
The terms used herein have their ordinary meaning and the meaning of such terms is independent at each occurrence thereof. That notwithstanding and except where stated otherwise, the following definitions apply throughout the specification and claims. Chemical names, common names, and chemical structures
may be used interchangeably to describe the same structure. If a chemical compound is referred to using both a chemical structure and a chemical name and an ambiguity exists between the structure and the name, the structure predominates. These definitions apply regardless of whether a term is used by itself or in combination with other terms, unless otherwise indicated. Hence, the definition of "alkyl" applies to "alkyl" as well as the "alkyl" portions of "hydroxyalkyl," "haloalkyl," "-O-alkyl," etc...
As used herein, and throughout this disclosure, the following terms, unless otherwise indicated, shall be understood to have the following meanings:
A "patient" is a human or non-human mammal. In one embodiment, a patient is a human. In another embodiment, a patient is a chimpanzee.
The term "effective amount" as used herein, refers to an amount of Tetracyclic Indole Derivative and/or an additional therapeutic agent, or a composition thereof that is effective in producing the desired therapeutic, ameliorative, inhibitory or preventative effect when administered to a patient suffering from a viral infection or virus-related disorder. In the combination therapies of the present invention, an effective amount can refer to each individual agent or to the combination as a whole, wherein the amounts of all agents administered are together effective, but wherein the component agent of the combination may not be present individually in an effective amount.
The term "preventing," as used herein with respect to an HCV viral infection or HCV-virus related disorder, refers to reducing the likelihood of HCV infection.
The term "alkyl," as used herein, refers to an aliphatic hydrocarbon group having one of its hydrogen atoms replaced with a bond. An alkyl group may be straight or branched and contain from about 1 to about 20 carbon atoms. In one embodiment, an alkyl group contains from about 1 to about 12 carbon atoms. In different embodiments, an alkyl group contains from 1 to 6 carbon atoms (C]-C6 alkyl) or from about 1 to about 4 carbon atoms (C1-C4 alkyl). Non-limiting examples of alkyl groups include methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, isobutyl, tert-butyl, n-pentyl, neopentyl, isopentyl, n-hexyl, isohexyl and neohexyl. An alkyl group may be unsubstituted or substituted by one or more substituents which may be the same or different, each substituent being independently selected from the group consisting of halo, alkenyl, alkynyl, aryl, cycloalkyl, cyano, hydroxy, -O-alkyl, -O-aryl, -alkylene-O-alkyl, alkylthio, -NH2, -NH(alkyl), -N(alkyl)2, -NH(cycloalkyl),
-0-C(0)-alkyl, -0-C(0)-aryl, -0-C(0)-cycloalkyl, -C(0)OH and -C(0)O-alkyl. In one embodiment, an alkyl group is linear. In another embodiment, an alkyl group is branched. Unless otherwise indicated, an alkyl group is unsubstituted.
The term "alkenyl," as used herein, refers to an aliphatic hydrocarbon group containing at least one carbon-carbon double bond and having one of its hydrogen atoms replaced with a bond. An alkenyl group may be straight or branched and contain from about 2 to about 15 carbon atoms. In one embodiment, an alkenyl group contains from about 2 to about 12 carbon atoms. In another embodiment, an alkenyl group contains from about 2 to about 6 carbon atoms. Non-limiting examples of alkenyl groups include ethenyl, propenyl, n-butenyl, 3-methylbut-2-enyl, n- pentenyl, octenyl and decenyl. An alkenyl group may be unsubstituted or substituted by one or more substituents which may be the same or different, each substituent being independently selected from the group consisting of halo, alkenyl, alkynyl, aryl, cycloalkyl, cyano, hydroxy, -O-alkyl, -O-aryl, -alkylene-O-alkyl, alkylthio, -NH2, - NH(alkyl), -N(alkyl)2, -NH(cycloalkyl), -0-C(0)-alkyl, -0-C(0)-aryl, -O-C(O)- cycloalkyl, -C(0)OH and -C(0)0-alkyl. The term "C2-C6 alkenyl" refers to an alkenyl group having from 2 to 6 carbon atoms. Unless otherwise indicated, an alkenyl group is unsubstituted.
The term "alkynyl," as used herein, refers to an aliphatic hydrocarbon group containing at least one carbon-carbon triple bond and having one of its hydrogen atoms replaced with a bond. An alkynyl group may be straight or branched and contain from about 2 to about 15 carbon atoms. In one embodiment, an alkynyl group contains from about 2 to about 12 carbon atoms. In another embodiment, an alkynyl group contains from about 2 to about 6 carbon atoms. Non-limiting examples of alkynyl groups include ethynyl, propynyl, 2-butynyl and 3-methylbutynyl. An alkynyl group may be unsubstituted or substituted by one or more substituents which may be the same or different, each substituent being independently selected from the group consisting of halo, alkenyl, alkynyl, aryl, cycloalkyl, cyano, hydroxy, -O-alkyl, -O-aryl, -alkylene-O-alkyl, alkylthio, -NH2, -NH(alkyl), -N(alkyl)2, -NH(cycloalkyl), -0-C(0)-alkyl, -0-C(0)-aryl, -0-C(0)-cycloalkyl, -C(0)OH and -C(0)0-alkyl. The term "C2-C6 alkynyl" refers to an alkynyl group having from 2 to 6 carbon atoms. Unless otherwise indicated, an alkynyl group is unsubstituted.
The term "alkylene," as used herein, refers to an alkyl group, as defined above, wherein one of the alkyl group's hydrogen atoms has been replaced
with a bond. Non-limiting examples of alkylene groups include -CH2-, -CH2CH2-, - CH2CH2CH2-, -CH2CH2CH2CH2-, -CH(CH3)CH2CH2-, -CH(CH3)- and - CH2CH(CH3)CH2-. In one embodiment, an alkylene group has from 1 to about 6 carbon atoms. In another embodiment, an alkylene group is branched. In another embodiment, an alkylene group is linear. In one embodiment, an alkylene group is - CH2-. The term "Ci-C6 alkylene" refers to an alkylene group having from 1 to 6 carbon atoms.
The term "aryl," as used herein, refers to an aromatic monocyclic or multicyclic ring system comprising from about 6 to about 14 carbon atoms. In one embodiment, an aryl group contains from about 6 to about 10 carbon atoms. An aryl group can be optionally substituted with one or more "ring system substituents" which may be the same or different, and are as defined herein below. In one embodiment, an aryl group can be optionally fused to a cycloalkyl or cycloalkanoyl group. Non- limiting examples of aryl groups include phenyl and naphthyl. In one embodiment, an aryl group is phenyl. Unless otherwise indicated, an aryl group is unsubstituted.
The term "arylene," as used herein, refers to a bivalent group derived from an aryl group, as defined above, by removal of a hydrogen atom from a ring carbon of an aryl group. An arylene group can be derived from a monocyclic or multicyclic ring system comprising from about 6 to about 14 carbon atoms. In one embodiment, an arylene group contains from about 6 to about 10 carbon atoms. In another embodiment, an arylene group is a naphthylene group. In another embodiment, an arylene group is a phenylene group. An arylene group can be optionally substituted with one or more "ring system substituents" which may be the same or different, and are as defined herein below. An arylene group is divalent and either available bond on an arylene group can connect to either group flanking the arylene group. For example, the group "A-arylene-B," wherein the arylene group is:
is understood to represent both:
In one embodiment, an arylene group can be optionally fused to a cycloalkyl or cycloalkanoyl group. Non-limiting examples of arylene groups include phenylene and naphthalene. In one embodiment, an arylene group is unsubstituted. In another embodiment, an arylene group is:
Unless otherwise indicated, an arylene group is unsubstituted.
The term "cycloalkyl," as used herein, refers to a non-aromatic mono- or multicyclic ring system comprising from about 3 to about 10 ring carbon atoms. In one embodiment, a cycloalkyl contains from about 5 to about 10 ring carbon atoms. In another embodiment, a cycloalkyl contains from about 3 to about 7 ring atoms. In another embodiment, a cycloalkyl contains from about 5 to about 6 ring atoms. The term "cycloalkyl" also encompasses a cycloalkyl group, as defined above, which is fused to an aryl (e.g., benzene) or heteroaryl ring. Non-limiting examples of monocyclic cycloalkyls include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl. Non-limiting examples of multicyclic cycloalkyls include 1-decalinyl, norbornyl and adamantyl. A cycloalkyl group can be optionally substituted with one or more "ring system substituents" which may be the same or different, and are as defined herein below. In one embodiment, a cycloalkyl group is unsubstituted. The term "3 to 6-membered cycloalkyl" refers to a cycloalkyl group having from 3 to 6 ring carbon atoms. Unless otherwise indicated, a cycloalkyl group is unsubstituted. A ring carbon atom of a cycloalkyl group may be functionalized as a carbonyl group. An illustrative example of such a cycloalkyl group (also referred to herein as a "cycloalkanoyl" group) includes, but is not limited to, cyclobutanoyl:

The term "cycloalkenyl," as used herein, refers to a non-aromatic mono- or multicyclic ring system comprising from about 4 to about 10 ring carbon atoms and containing at least one endocyclic double bond. In one embodiment, a cycloalkenyl contains from about 4 to about 7 ring carbon atoms. In another embodiment, a cycloalkenyl contains 5 or 6 ring atoms. Non-limiting examples of monocyclic cycloalkenyls include cyclopentenyl, cyclohexenyl, cyclohepta-l,3-dienyl, and the like. A cycloalkenyl group can be optionally substituted with one or more "ring system substituents" which may be the same or different, and are as defined herein below. A ring carbon atom of a cycloalkyl group may be functionalized as a carbonyl group. In one embodiment, a cycloalkenyl group is cyclopentenyl. In another embodiment, a cycloalkenyl group is cyclohexenyl. The term "4 to 6- membered cycloalkenyl" refers to a cycloalkenyl group having from 4 to 6 ring carbon atoms. Unless otherwise indicated, a cycloalkenyl group is unsubstituted.
The term "halo," as used herein, means -F, -CI, -Br or -I.
The term "haloalkyl," as used herein, refers to an alkyl group as defined above, wherein one or more of the alkyl group's hydrogen atoms has been replaced with a halogen. In one embodiment, a haloalkyl group has from 1 to 6 carbon atoms. In another embodiment, a haloalkyl group is substituted with from 1 to 3 F atoms. Non-limiting examples of haloalkyl groups include -CH2F, -CHF2, -CF3, - CH2C1 and -CC13. The term "Ci-C6 haloalkyl" refers to a haloalkyl group having from 1 to 6 carbon atoms.
The term "hydroxyalkyl," as used herein, refers to an alkyl group as defined above, wherein one or more of the alkyl group's hydrogen atoms has been replaced with an -OH group. In one embodiment, a hydroxyalkyl group has from 1 to 6 carbon atoms. Non-limiting examples of hydroxyalkyl groups include -CH2OH, - CH2CH2OH, -CH2CH2CH2OH and -CH2CH(OH)CH3. The term "CrC6
hydroxyalkyl" refers to a hydroxyalkyl group having from 1 to 6 carbon atoms.
The term "heteroaryl," as used herein, refers to an aromatic monocyclic or multicyclic ring system comprising about 5 to about 14 ring atoms, wherein from 1 to 4 of the ring atoms is independently O, N or S and the remaining ring atoms are carbon atoms. In one embodiment, a heteroaryl group has 5 to 10 ring atoms. In another embodiment, a heteroaryl group is monocyclic and has 5 or 6 ring atoms. In another embodiment, a heteroaryl group is bicyclic and had 9 or 10 ring atoms. A heteroaryl group can be optionally substituted by one or more "ring system
substituents" which may be the same or different, and are as defined herein below. A heteroaryl group is joined via a ring carbon atom, and any nitrogen atom of a heteroaryl can be optionally oxidized to the corresponding N-oxide. The term
"heteroaryl" also encompasses a heteroaryl group, as defined above, which is fused to a benzene ring. Non-limiting examples of heteroaryls include pyridyl, pyrazinyl, furanyl, thienyl, pyrimidinyl, pyridone (including N-substituted pyridones), isoxazolyl, isothiazolyl, oxazolyl, oxadiazolyl, thiazolyl, pyrazolyl, furazanyl, pyrrolyl, triazolyl, 1,2,4-thiadiazolyl, pyrazinyl, pyridazinyl, quinoxalinyl, phthalazinyl, oxindolyl, imidazo[l,2-a]pyridinyl, imidazo[2,l-b]thiazolyl, benzofurazanyl, indolyl, azaindolyl, benzimidazolyl, benzothienyl, quinolinyl, imidazolyl, benzimidazolyl, thienopyridyl, quinazolinyl, thienopyrimidyl, pyrrolopyridyl, imidazopyridyl, isoquinolinyl, benzoazaindolyl, 1,2,4-triazinyl, benzothiazolyl and the like, and all isomeric forms thereof. The term "heteroaryl" also refers to partially saturated heteroaryl moieties such as, for example, tetrahydroisoquinolyl, tetrahydroquinolyl and the like. In one embodiment, a heteroaryl group is a 5 or 6-membered monocyclic heteroaryl. In another embodiment, a heteroaryl group is a 6-membered monocyclic heteroaryl. In another embodiment, a heteroaryl group is a 5-membered monocyclic heteroaryl. In one embodiment, a heteroaryl group is a 9 or 10-membered monocyclic heteroaryl. In another embodiment, a heteroaryl group is a 9-membered monocyclic heteroaryl. Unless otherwise indicated, a heteroaryl group is unsubstituted.
The term "heteroarylene," as used herein, refers to a bivalent group derived from an heteroaryl group, as defined above, by removal of a hydrogen atom from a ring carbon or ring heteroatom of a heteroaryl group. A heteroarylene group can be derived from a monocyclic or multicyclic ring system comprising about 5 to about 14 ring atoms, wherein from 1 to 4 of the ring atoms are each independently O, N or S and the remaining ring atoms are carbon atoms. A heteroarylene group can be optionally substituted by one or more "ring system substituents" which may be the same or different, and are as defined herein below. A heteroarylene group is joined via a ring carbon atom or by a nitrogen atom with an open valence, and any nitrogen atom of a heteroarylene can be optionally oxidized to the corresponding N-oxide. The term "heteroarylene" also encompasses a heteroarylene group, as defined above, which is fused to a benzene ring. Non-limiting examples of heteroarylenes include pyridylene, pyrazinylene, furanylene, thienylene, pyrimidinylene, pyridonylene (including those derived from N-substituted pyridonyls), isoxazolylene,
isothiazolylene, oxazolylene, oxadiazolylene, thiazolylene, pyrazolylene,
thiophenylene, furazanylene, pyrrolylene, triazolylene, 1 ,2,4-thiadiazolylene, pyrazinylene, pyridazinylene, quinoxalinylene, phthalazinylene, oxindolylene, imidazo[l,2-a]pyridinylene, imidazo[2,l-b]thiazolylene, benzofurazanylene, indolylene, azaindolylene, benzimidazolylene, benzothienylene, quinolinylene, imidazolylene, benzimidazolylene, thienopyridylene, quinazolinylene,
thienopyrimidylene, pyrrolopyridylene, imidazopyridylene, isoquinolinylene, benzoazaindolylene, 1,2,4-triazinylene, benzothiazolylene and the like, and all isomeric forms thereof. The term "heteroarylene" also refers to partially saturated heteroarylene moieties such as, for example, tetrahydroisoquinolylene,
tetrahydroquinolylene, and the like. A heteroarylene group is divalent and either available bond on a heteroarylene ring can connect to either group flanking the heteroarylene group. For example, the group "A-heteroarylene-B," wherein the heteroarylene group is:
is understood to represent both:
In one embodiment, a heteroarylene group is a monocyclic heteroarylene group or a bicyclic heteroarylene group. In another embodiment, a heteroarylene group is a monocyclic heteroarylene group. In another embodiment, a heteroarylene group is a bicyclic heteroarylene group. In still another embodiment, a heteroarylene group has from about 5 to about 10 ring atoms. In another embodiment, a heteroarylene group is monocyclic and has 5 or 6 ring atoms. In another
embodiment, a heteroarylene group is bicyclic and has 9 or 10 ring atoms. In another embodiment, a heteroarylene group is a 5-membered monocyclic heteroarylene. In another embodiment, a heteroarylene group is a 6-membered monocyclic
heteroarylene. In another embodiment, a bicyclic heteroarylene group comprises a 5 or 6-membered monocyclic heteroarylene group fused to a benzene ring. Unless otherwise indicated, a heteroarylene group is unsubstituted.
The term "heterocycloalkyl," as used herein, refers to a non-aromatic saturated monocyclic or multicyclic ring system comprising 3 to about 1 1 ring atoms, wherein from 1 to 4 of the ring atoms are independently O, S, N or Si, and the remainder of the ring atoms are carbon atoms. A heterocycloalkyl group can be joined via a ring carbon, ring silicon atom or ring nitrogen atom. In one embodiment, a heterocycloalkyl group is monocyclic and has from about 3 to about 7 ring atoms. In another embodiment, a heterocycloalkyl group is monocyclic has from about 4 to about 7 ring atoms. In another embodiment, a heterocycloalkyl group is bicyclic and has from about 7 to about 1 1 ring atoms. In still another embodiment, a
heterocycloalkyl group is monocyclic and has 5 or 6 ring atoms. In one embodiment, a heterocycloalkyl group is monocyclic. In another embodiment, a heterocycloalkyl group is bicyclic. There are no adjacent oxygen and/or sulfur atoms present in the ring system. Any -NH group in a heterocycloalkyl ring may exist protected such as, for example, as an -N(BOC), -N(Cbz), -N(Tos) group and the like; such protected heterocycloalkyl groups are considered part of this invention. The term
"heterocycloalkyl" also encompasses a heterocycloalkyl group, as defined above, which is fused to an aryl (e.g., benzene) or heteroaryl ring. A heterocycloalkyl group can be optionally substituted by one or more "ring system substituents" which may be the same or different, and are as defined herein below. The nitrogen or sulfur atom of the heterocycloalkyl can be optionally oxidized to the corresponding N-oxide, S-oxide or S,S-dioxide. Non-limiting examples of monocyclic heterocycloalkyl rings include oxetanyl, piperidyl, pyrrolidinyl, piperazinyl, morpholinyl, thiomorpholinyl, thiazolidinyl, 1 ,4-dioxanyl, tetrahydrofuranyl, tetrahydrothiophenyl, delta-lactam, delta-lactone, silacyclopentane, silapyrrolidine and the like, and all isomers thereof. Non-limiting illustrative examples of a silyl-containing heterocycloalkyl group include:
A ring carbon atom of a heterocycloalkyl group may be functionalized as a carbonyl group. An illustrative example of such a heterocycloalkyl group is:
In one embodiment, a heterocycloalkyl group is a 5-membered monocyclic heterocycloalkyl. In another embodiment, a heterocycloalkyl group is a 6-membered monocyclic heterocycloalkyl. The term "3 to 6-membered monocyclic cycloalkyl" refers to a monocyclic heterocycloalkyl group having from 3 to 6 ring atoms. The term "4 to 6-membered monocyclic cycloalkyl" refers to a monocyclic heterocycloalkyl group having from 4 to 6 ring atoms. The term "7 to 11-membered bicyclic heterocycloalkyl" refers to a bicyclic heterocycloalkyl group having from 7 to 1 1 ring atoms. Unless otherwise indicated, an heterocycloalkyl group is unsubstituted.
The term "heterocycloalkenyl," as used herein, refers to a heterocycloalkyl group, as defined above, wherein the heterocycloalkyl group contains from 4 to 10 ring atoms, and at least one endocyclic carbon-carbon or carbon-nitrogen double bond. A heterocycloalkenyl group can be joined via a ring carbon or ring nitrogen atom. In one embodiment, a heterocycloalkenyl group has from 4 to 6 ring atoms. In another embodiment, a heterocycloalkenyl group is monocyclic and has 5 or 6 ring atoms. In another embodiment, a heterocycloalkenyl group is bicyclic. A heterocycloalkenyl group can optionally substituted by one or more ring system substituents, wherein "ring system substituent" is as defined above.
The nitrogen or sulfur atom of the heterocycloalkenyl can be optionally oxidized to the corresponding N-oxide, S-oxide or S,S-dioxide. Non-limiting examples of heterocycloalkenyl groups include 1,2,3,4- tetrahydropyridinyl, 1 ,2-dihydropyridinyl, 1 ,4-dihydropyridinyl, 1,2,3,6-tetrahydropyridinyl, 1,4,5,6-tetrahydropyrimidinyl, 2- pyrrolinyl, 3-pyrrolinyl, 2-imidazolinyl, 2-pyrazolinyl, dihydroimidazolyl,
dihydrooxazolyl, dihydrooxadiazolyl, dihydrothiazolyl, 3,4-dihydro-2H-pyranyl, dihydrofuranyl, fluoro-substituted dihydrofuranyl, 7-oxabicyclo[2.2.1]heptenyl, dihydrothiophenyl, dihydrothiopyranyl, and the like and the like. A ring carbon atom of a heterocycloalkenyl group may be functionalized as a carbonyl group. In one embodiment, a heterocycloalkenyl group is a 5-membered heterocycloalkenyl. In another embodiment, a heterocycloalkenyl group is a 6-membered heterocycloalkenyl. The term "4 to 6-membered heterocycloalkenyl" refers to a heterocycloalkenyl group having from 4 to 6 ring atoms. Unless otherwise indicated, a heterocycloalkenyl group is unsubstituted.
The term "ring system substituent," as used herein, refers to a substituent group attached to an aromatic or non-aromatic ring system which, for example, replaces an available hydrogen on the ring system. Ring system substituents may be the same or different, each being independently selected from the group consisting of alkyl, alkenyl, alkynyl, aryl, heteroaryl, -alkylene-ar l, -arylene-alkyl, - alkylene-heteroaryl, -alkenylene-heteroaryl, -alkynylene-heteroaryl, -OH,
hydroxyalkyl, haloalkyl, -O-alkyl, -O-haloalkyl, -alkylene-O-alkyl, -O-aryl, -O- alkylene-aryl, acyl, -C(0)-aryl, halo, -N02, -CN, -SF5, -C(0)OH, -C(0)0-alkyl, - C(0)0-aryl, -C(0)0-alkylene-aryl, -S(0)-alkyl, -S(0)2-alkyl, -S(0)-aryl, -S(0)2-aryl, -S(0)-heteroaryl, -S(0)2-heteroaryl, -S-alkyl, -S-aryl, -S-heteroar l, -S-alkylene-aryl, -S-alkylene-heteroaryl, -S(0)2-alkylene-aryl, -S(0)2-alkylene-heteroaryl, -Si(alkyl)2, - Si(aryl)2, -Si(heteroaryl)2, -Si(alkyl)(aryl), -Si(alkyl)(cycloalkyl), - Si(alkyl)(heteroaryl), cycloalkyl, heterocycloalkyl, -0-C(0)-alkyl, -0-C(0)-aryl, -O- C(0)-cycloalkyl, -C(=N-CN)-NH2, -C(=NH)-NH2, -C(=NH)-NH(alkyl), -N(Y])(Y2), - alkylene-N(Y])(Y2), -C(0)N(Yi)(Y2) and -S(O)2N(Y (Y2), wherein Y] and Y2 can be the same or different and are independently selected from the group consisting of hydrogen, alkyl, aryl, cycloalkyl, and -alkylene-aryl. "Ring system substituent" may also mean a single moiety which simultaneously replaces two available hydrogens on two adjacent carbon atoms (one H on each carbon) on a ring system. Examples of
such moiety are methylenedioxy, ethylenedioxy, -C(CH3)2- and the like which form moieties such as, for example:
The term "silylalkyl," as used herein, refers to an alkyl group as defined above, wherein one or more of the alkyl group's hydrogen atoms has been replaced with a -Si(Rx)3 group, wherein each occurrence of Rx is independently Cj-C6 alkyl, phenyl or a 3 to 6-membered cycloalkyl group. In one embodiment, a silylalkyl group has from 1 to 6 carbon atoms. In another embodiment, a silyl alkyl group contains a -Si(CH3)3 moiety. Non-limiting examples of silylalkyl groups include -CH2-Si(CH3)3 and -CH2CH2-Si(CH3)3.
The term "substituted" means that one or more hydrogens on the designated atom is replaced with a selection from the indicated group, provided that the designated atom's normal valency under the existing circumstances is not exceeded, and that the substitution results in a stable compound. Combinations of substituents and/or variables are permissible only if such combinations result in stable compounds. By "stable compound' or "stable structure" is meant a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent.
The term "in substantially purified form," as used herein, refers to the physical state of a compound after the compound is isolated from a synthetic process (e.g., from a reaction mixture), a natural source, or a combination thereof. The term "in substantially purified form," also refers to the physical state of a compound after the compound is obtained from a purification process or processes described herein or well-known to the skilled artisan (e.g., chromatography, recrystallization and the like), in sufficient purity to be characterizable by standard analytical techniques described herein or well-known to the skilled artisan.
It should also be noted that any carbon as well as heteroatom with unsatisfied valences in the text, schemes, examples and tables herein is assumed to have the sufficient number of hydrogen atom(s) to satisfy the valences.
When a functional group in a compound is termed "protected", this means that the group is in modified form to preclude undesired side reactions at the
protected site when the compound is subjected to a reaction. Suitable protecting groups will be recognized by those with ordinary skill in the art as well as by reference to standard textbooks such as, for example, T. W. Greene et al, Protective Groups in Organic Synthesis (1991), Wiley, New York.
When any substituent or variable {e.g., alkyl, R6, Ra, etc.) occurs more than one time in any constituent or in Formula (I), its definition on each occurrence is independent of its definition at every other occurrence, unless otherwise indicated.
As used herein, the term "composition" is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
Prodrugs and solvates of the compounds of the invention are also contemplated herein. A discussion of prodrugs is provided in T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems (1987) 14 of the A.C.S. Symposium Series, and in Bioreversible Carriers in Drug Design, (1987) Edward B. Roche, ed., American Pharmaceutical Association and Pergamon Press. The term "prodrug" means a compound {e.g., a drug precursor) that is transformed in vivo to provide a Tetracyclic Indole Derivative or a pharmaceutically acceptable salt or solvate of the compound. The transformation may occur by various mechanisms {e.g., by metabolic or chemical processes), such as, for example, through hydrolysis in blood.
For example, if a Tetracyclic Indole Derivative or a pharmaceutically acceptable salt, hydrate or solvate of the compound contains a carboxylic acid functional group, a prodrug can comprise an ester formed by the replacement of the hydrogen atom of the acid group with a group such as, for example, (Ci-C8)alkyl, (C2-Ci2)alkanoyloxymethyl, l-(alkanoyloxy)ethyl having from 4 to 9 carbon atoms, 1 -methyl- l-(alkanoyloxy)-ethyl having from 5 to 10 carbon atoms,
alkoxycarbonyloxymethyl having from 3 to 6 carbon atoms, 1- (alkoxycarbonyloxy)ethyl having from 4 to 6 carbon atoms, 1 -methyl- 1- (alkoxycarbonyloxy)ethyl having from 5 to 8 carbon atoms, N- (alkoxycarbonyl)aminomethyl having from 3 to 9 carbon atoms, 1-(N- (alkoxycarbonyl)amino)ethyl having from 4 to 10 carbon atoms, 3-phthalidyl, 4- crotonolactonyl, gamma-butyrolacton-4-yl, di-N,N-(Ci-C2)alkylamino(C2-C3)alkyl (such as β-dimethylaminoethyl), carbamoyl-(Ci-C2)alkyl, N,N-di (Ci-
C2)alkylcarbamoyl-(Ci-C2)alkyl and piperidino-, pyrrolidino- or morpholino(C2- C3)alkyl, and the like.
Similarly, if a Tetracyclic Indole Derivative contains an alcohol functional group, a prodrug can be formed by the replacement of the hydrogen atom of the alcohol group with a group such as, for example, (C]-C6)alkanoyloxymethyl, 1- ((C i -C6)alkanoy loxy)ethy 1, 1 -methyl- 1 -((C i -C6)alkanoyloxy)ethyl, (C i - C6)alkoxycarbonyloxymethyl, N-(Ci-C6)alkoxycarbonylaminomethyl, succinoyl, (Ci- C6)alkanoyl, a-amino(Ci-C4)alkyl, a-amino(Ci-C4)alkylene-aryl, arylacyl and a- aminoacyl, or α-aminoacyl-a-aminoacyl, where each a-aminoacyl group is independently selected from the naturally occurring L-amino acids, -P(0)(OH)2, - P(0)(0(Ci-C6)alkyl)2 or glycosyl (the radical resulting from the removal of a hydroxyl group of the hemiacetal form of a carbohydrate), and the like.
If a Tetracyclic Indole Derivative incorporates an amine functional group, a prodrug can be formed by the replacement of a hydrogen atom in the amine group with a group such as, for example, R-carbonyl-, RO-carbonyl-, NRR'-carbonyl- wherein R and R' are each independently (Ci-C^alkyl, (C3-C7) cycloalkyl, benzyl, a natural a-aminoacyl, -QOH^C OY1 wherein Y1 is H, (CrC6)alkyl or benzyl, - C(OY2)Y3 wherein Y2 is (C1-C4) alkyl and Y3 is (d-C6)alkyl; carboxy (C C6)alkyl; amino(C C4)alkyl or mono-N- or di-N,N-(Ci-C6)alkylaminoalkyl; -C(Y4)Y5 wherein Y4 is H or methyl and Y5 is mono-N- or di-N,N-(C1-C6)alkylamino morpholino; piperidin-l-yl or pyrrolidin-l-yl, and the like.
Pharmaceutically acceptable esters of the present compounds include the following groups: (1) carboxylic acid esters obtained by esterification of the hydroxy group of a hydroxyl compound, in which the non-carbonyl moiety of the carboxylic acid portion of the ester grouping is selected from straight or branched chain alkyl (e.g., methyl, ethyl, n-propyl, isopropyl, t-butyl, sec-butyl or n-butyl), alkoxyalkyl (e.g., methoxymethyl), aralkyl (e.g., benzyl), aryloxyalkyl (for example, phenoxymethyl), aryl (e.g., phenyl optionally substituted with, for example, halogen, Ci-4alkyl, -0-(Ci-4alkyl) or amino); (2) sulfonate esters, such as alkyl- or
aralkylsulfonyl (for example, methanesulfonyl); (3) amino acid esters (e.g., L-valyl or L-isoleucyl); (4) phosphonate esters and (5) mono-, di- or triphosphate esters. The phosphate esters may be further esterified by, for example, a C1-20 alcohol or reactive derivative thereof, or by a 2,3-di (C6-24)acyl glycerol.
One or more compounds of the invention may exist in unsolvated as well as solvated forms with pharmaceutically acceptable solvents such as water, ethanol, and the like, and it is intended that the invention embrace both solvated and unsolvated forms. "Solvate" means a physical association of a compound of this invention with one or more solvent molecules. This physical association involves varying degrees of ionic and covalent bonding, including hydrogen bonding. In certain instances the solvate will be capable of isolation, for example when one or more solvent molecules are incorporated in the crystal lattice of the crystalline solid. "Solvate" encompasses both solution-phase and isolatable solvates. Non-limiting examples of solvates include ethanolates, methanolates, and the like. A "hydrate" is a solvate wherein the solvent molecule is water.
One or more compounds of the invention may optionally be converted to a solvate. Preparation of solvates is generally known. Thus, for example, M. Caira et al, J. Pharmaceutical Sci., 93(3), 601-611 (2004) describe the preparation of the solvates of the antifungal fluconazole in ethyl acetate as well as from water. Similar preparations of solvates, hemisolvate, hydrates and the like are described by E. C. van Tonder et al, AAPS PharmSciTechours. , 5(1), article 12 (2004); and A. L. Bingham et al, Chem. Commun., 603-604 (2001). A typical, non-limiting, process involves dissolving the inventive compound in desired amounts of the desired solvent (organic or water or mixtures thereof) at a higher than room temperature, and cooling the solution at a rate sufficient to form crystals which are then isolated by standard methods. Analytical techniques such as, for example IR spectroscopy, show the presence of the solvent (or water) in the crystals as a solvate (or hydrate).
The Tetracyclic Indole Derivatives can form salts which are also within the scope of this invention. Reference to a Tetracyclic Indole Derivative herein is understood to include reference to salts thereof, unless otherwise indicated. The term "salt(s)", as employed herein, denotes acidic salts formed with inorganic and/or organic acids, as well as basic salts formed with inorganic and/or organic bases. In addition, when a Tetracyclic Indole Derivative contains both a basic moiety, such as, but not limited to a pyridine or imidazole, and an acidic moiety, such as, but not limited to a carboxylic acid, zwitterions ("inner salts") may be formed and are included within the term "salt(s)" as used herein. In one embodiment, the salt is a pharmaceutically acceptable {i.e., non-toxic, physiologically acceptable) salt. In another embodiment, the salt is other than a pharmaceutically acceptable salt. Salts of
the Compounds of Formula (I) may be formed, for example, by reacting a Tetracyclic Indole Derivative with an amount of acid or base, such as an equivalent amount, in a medium such as one in which the salt precipitates or in an aqueous medium followed by lyophilization.
Exemplary acid addition salts include acetates, ascorbates, benzoates, benzenesulfonates, bisulfates, borates, butyrates, citrates, camphorates,
camphorsulfonates, fumarates, hydrochlorides, hydrobromides, hydroiodides, lactates, maleates, methanesulfonates, naphthalenesulfonates, nitrates, oxalates, phosphates, propionates, salicylates, succinates, sulfates, tartarates, thiocyanates,
toluenesulfonates (also known as tosylates) and the like. Additionally, acids which are generally considered suitable for the formation of pharmaceutically useful salts from basic pharmaceutical compounds are discussed, for example, by P. Stahl et al, Camille G. (eds.) Handbook of Pharmaceutical Salts. Properties, Selection and Use. (2002) Zurich: Wiley-VCH; S. Berge et al, Journal of Pharmaceutical Sciences (1977) 66(1) 1-19; P. Gould, International J. of Pharmaceutics (1986) 33 201-217; Anderson et al, The Practice of Medicinal Chemistry (1996), Academic Press, New York; and in The Orange Book (Food & Drug Administration, Washington, D.C. on their website). These disclosures are incorporated herein by reference thereto.
Exemplary basic salts include ammonium salts, alkali metal salts such as sodium, lithium, and potassium salts, alkaline earth metal salts such as calcium and magnesium salts, salts with organic bases (for example, organic amines) such as dicyclohexylamine, t-butyl amine, choline, and salts with amino acids such as arginine, lysine and the like. Basic nitrogen-containing groups may be quarternized with agents such as lower alkyl halides {e.g., methyl, ethyl, and butyl chlorides, bromides and iodides), dialkyl sulfates {e.g., dimethyl, diethyl, and dibutyl sulfates), long chain halides {e.g., decyl, lauryl, and stearyl chlorides, bromides and iodides), aralkyl halides {e.g., benzyl and phenethyl bromides), and others.
All such acid salts and base salts are intended to be pharmaceutically acceptable salts within the scope of the invention and all acid and base salts are considered equivalent to the free forms of the corresponding compounds for purposes of the invention.
Diastereomeric mixtures can be separated into their individual diastereomers on the basis of their physical chemical differences by methods well- known to those skilled in the art, such as, for example, by chromatography and/or
fractional crystallization. Enantiomers can be separated by converting the
enantiomeric mixture into a diastereomeric mixture by reaction with an appropriate optically active compound (e.g., chiral auxiliary such as a chiral alcohol or Mosher's acid chloride), separating the diastereomers and converting (e.g., hydrolyzing) the individual diastereomers to the corresponding pure enantiomers. Sterochemically pure compounds may also be prepared by using chiral starting materials or by employing salt resolution techniques. Also, some of the Tetracyclic Indole
Derivatives may be atropisomers (e.g., substituted biaryls) and are considered as part of this invention. Enantiomers can also be directly separated using chiral
chromatographic techniques.
It is also possible that the Tetracyclic Indole Derivatives may exist in different tautomeric forms, and all such forms are embraced within the scope of the invention. For example, all keto-enol and imine-enamine forms of the compounds are included in the invention.
All stereoisomers (for example, geometric isomers, optical isomers and the like) of the present compounds (including those of the salts, solvates, hydrates, esters and prodrugs of the compounds as well as the salts, solvates and esters of the prodrugs), such as those which may exist due to asymmetric carbons on various substituents, including enantiomeric forms (which may exist even in the absence of asymmetric carbons), rotameric forms, atropisomers, and diastereomeric forms, are contemplated within the scope of this invention. If a Tetracyclic Indole Derivative incorporates a double bond or a fused ring, both the cis- and trans-forms, as well as mixtures, are embraced within the scope of the invention.
Individual stereoisomers of the compounds of the invention may, for example, be substantially free of other isomers, or may be admixed, for example, as racemates or with all other, or other selected, stereoisomers. The chiral centers of the present invention can have the S or R configuration as defined by the TUPAC 1974 Recommendations. The use of the terms "salt", "solvate", "ester", "prodrug" and the like, is intended to apply equally to the salt, solvate, ester and prodrug of enantiomers, stereoisomers, rotamers, tautomers, positional isomers, racemates or prodrugs of the inventive compounds.
In the Compounds of Formula (I), the atoms may exhibit their natural isotopic abundances, or one or more of the atoms may be artificially enriched in a particular isotope having the same atomic number, but an atomic mass or mass
number different from the atomic mass or mass number predominantly found in nature. The present invention is meant to include all suitable isotopic variations of the compounds of generic Formula I. For example, different isotopic forms of hydrogen (H) include protium (]H) and deuterium (2H). Protium is the predominant hydrogen isotope found in nature. Enriching for deuterium may afford certain therapeutic advantages, such as increasing in vivo half-life or reducing dosage requirements, or may provide a compound useful as a standard for characterization of biological samples. Isotopically-enriched Compounds of Formula (I) can be prepared without undue experimentation by conventional techniques well known to those skilled in the art or by processes analogous to those described in the Schemes and Examples herein using appropriate isotopically-enriched reagents and/or intermediates. In one embodiment, a Compound of Formula (I) has one or more of its hydrogen atoms replaced with deuterium.
Polymorphic forms of the Tetracyclic Indole Derivatives, and of the salts, solvates, hydrates, esters and prodrugs of the Tetracyclic Indole Derivatives, are intended to be included in the present invention.
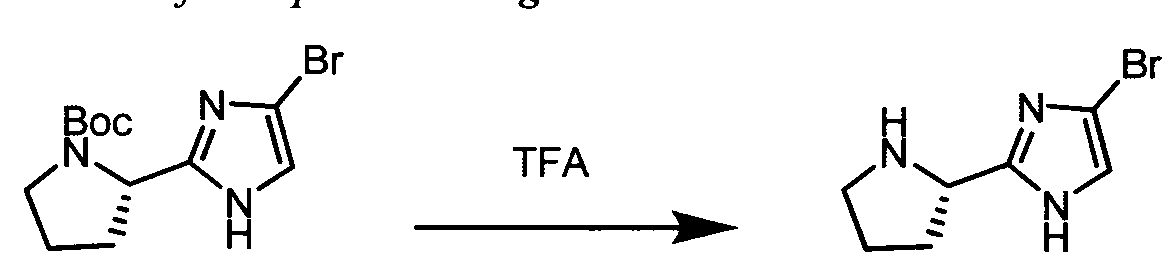
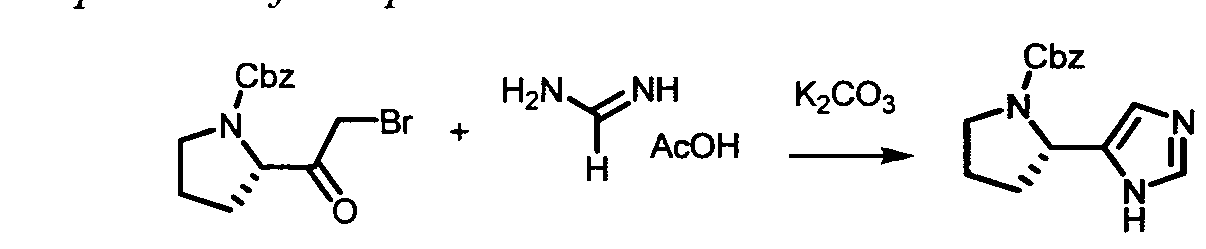
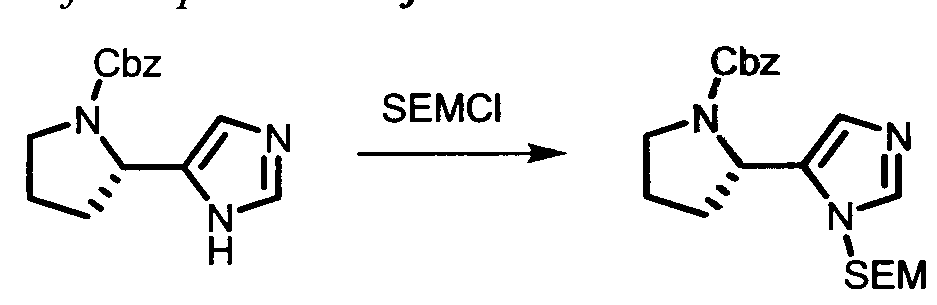
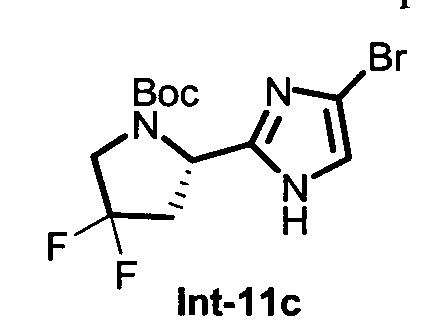
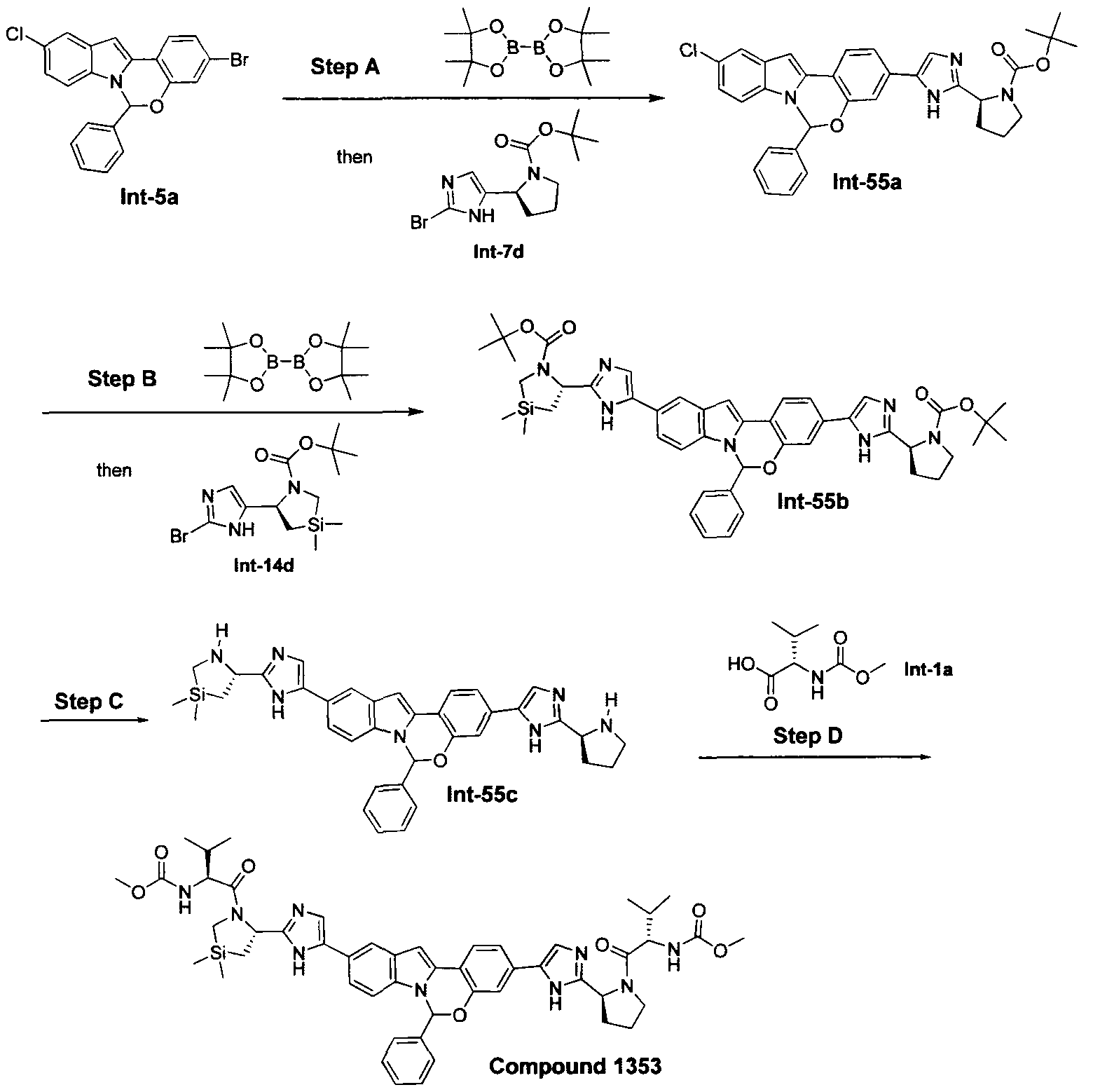
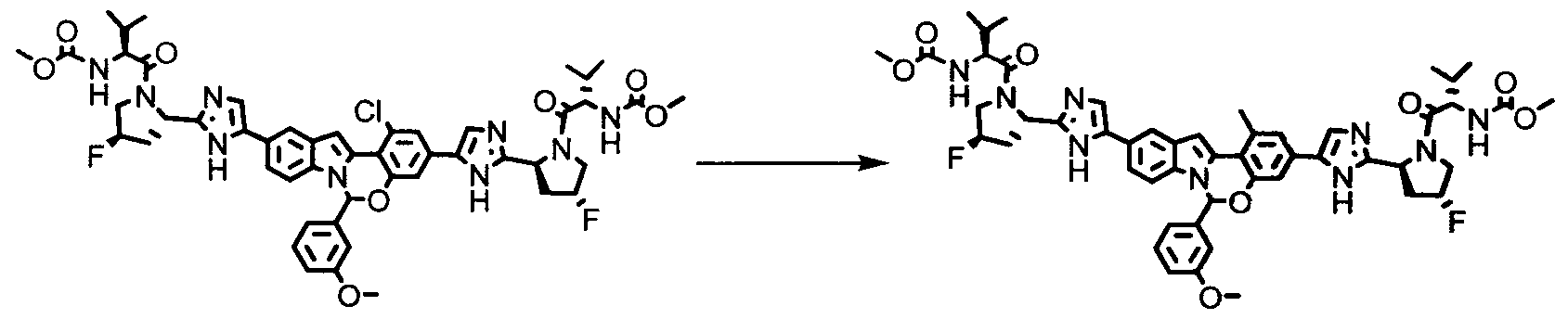
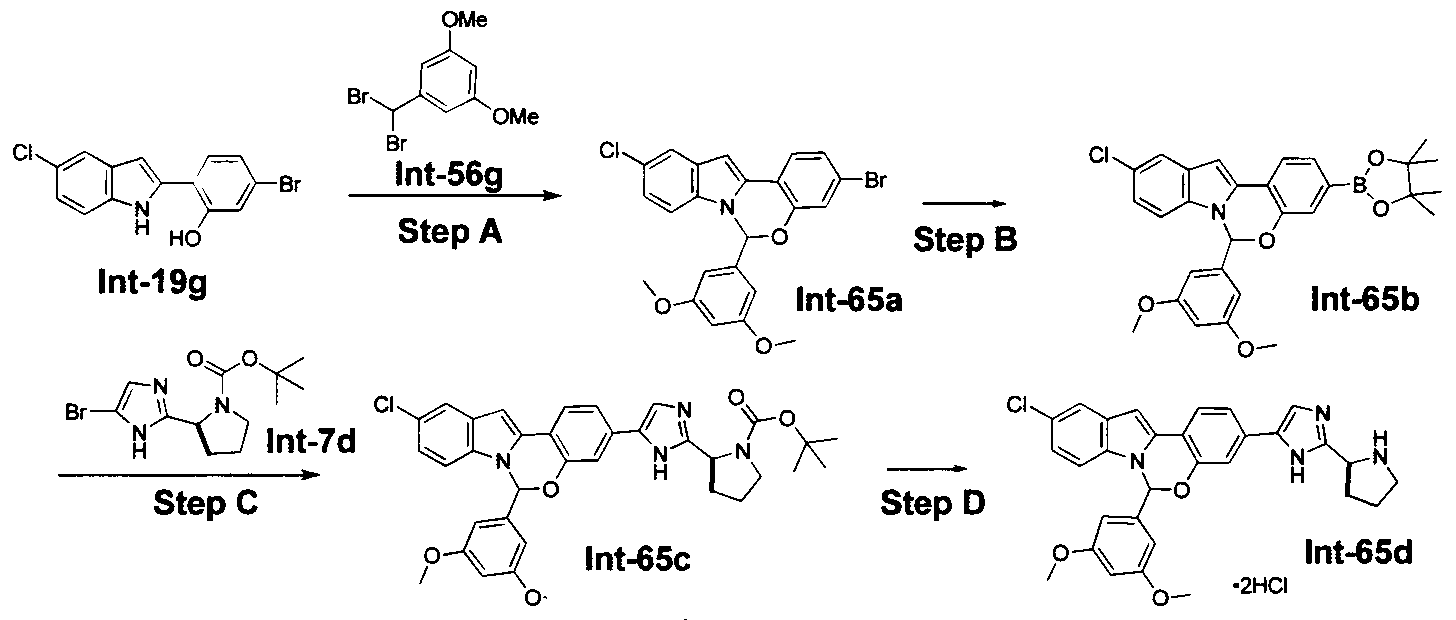
The following abbreviations are used below and have the following meanings: Ac is acyl; AcCl is acetyl chloride; AcOH or HOAc is acetic acid; Amphos is (4-(N^V)-dimethylaminophenyl)-di-tertbutylphosphine; Aq is aqueous; BF3 »OEt2 is boron trifluoride etherate; BOC or Boc is tert-butyloxycarbonyl; Boc20 is Boc anhydride; Boc-Pro-OH is Boc protected proline; L-Boc-Val-OH is Boc protected L- valine; BOP is Benzotriazole-l-yl-oxy-tris-(dimethylamino)-phosphonium
hexafluorophosphate; n-BuLi is n-butyllithium; CBZ or Cbz is carbobenzoxy; DCM is dichloromethane; DDQ is 2,3-dichloro-5,6-dicyano-l,4-benzoquinone; Dess-Martin reagent is ,l,l-Triacetoxy-l,l-dihydro-l,2-benziodoxol-3(lH)-one; DIPEA is diisopropylethylamine; DME is dimethoxyethane; DMF is N,N-dimethylformamide; dppf is diphenylphosphinoferrocene; DMSO is dimethylsulfoxide; EtMgBr is ethylmagnesium bromide; EtOAc is ethyl acetate; Et20 is diethyl ether; Et3N or NEt3 is triethylamine; HATU is 0-(7-azabenzotriazol-l-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate; HPLC is high performance liquid chromatography; HRMS is high resolution mass spectrometry; KOAc is potassium acetate; LCMS is liquid chromatography/mass spectrometry; LiHMDS is lithium hexamethyldisilazide;
LRMS is low resolution mass spectrometry; Mel is iodomethane; MeOH is methanol; NBS is N-bromosuccinimide; N¾OAc is ammonium acetate; NMM is N-
methylmorpholine; Pd/C is palladium on carbon; Pd(PPh3)4 is tetrakis
(triphenylphosphine)palladium(O); PdCl2(dppf)2 is [1,1 -Bis(diphenylphosphino) ferrocene]dichloro palladium(II); PdCl2(dppf)2 «CH2Cl2 is [1,1 - Bis(diphenylphosphino)ferrocene] dichloro palladium(II) complex with
dichloromethane; pinacol2B2 is bis(pinacolato)diboron; PPTS is pyridinium p-toluene sulfonate; RPLC is reverse-phase liquid chromatography; Select-F is 1-Chloromethyl- 4-Fluoro-l, 4-Diazoniabicyclo[2.2.2]Octane Bis-(Tetrafluoroborate); SEM-Cl is 2- (trimethylsilyl)ethoxymethyl chloride; TBAF is tetrabutylammonium fluoride;
TBDMSCl is tert-butyldimethylsilyl chloride; TFA is trifluoroacetic acid; Tf20 is triflic anhydride; THF is tetrahydrofuran; TLC is thin-layer chromatography; and TosCl is p-toluenesulfonyl chloride.
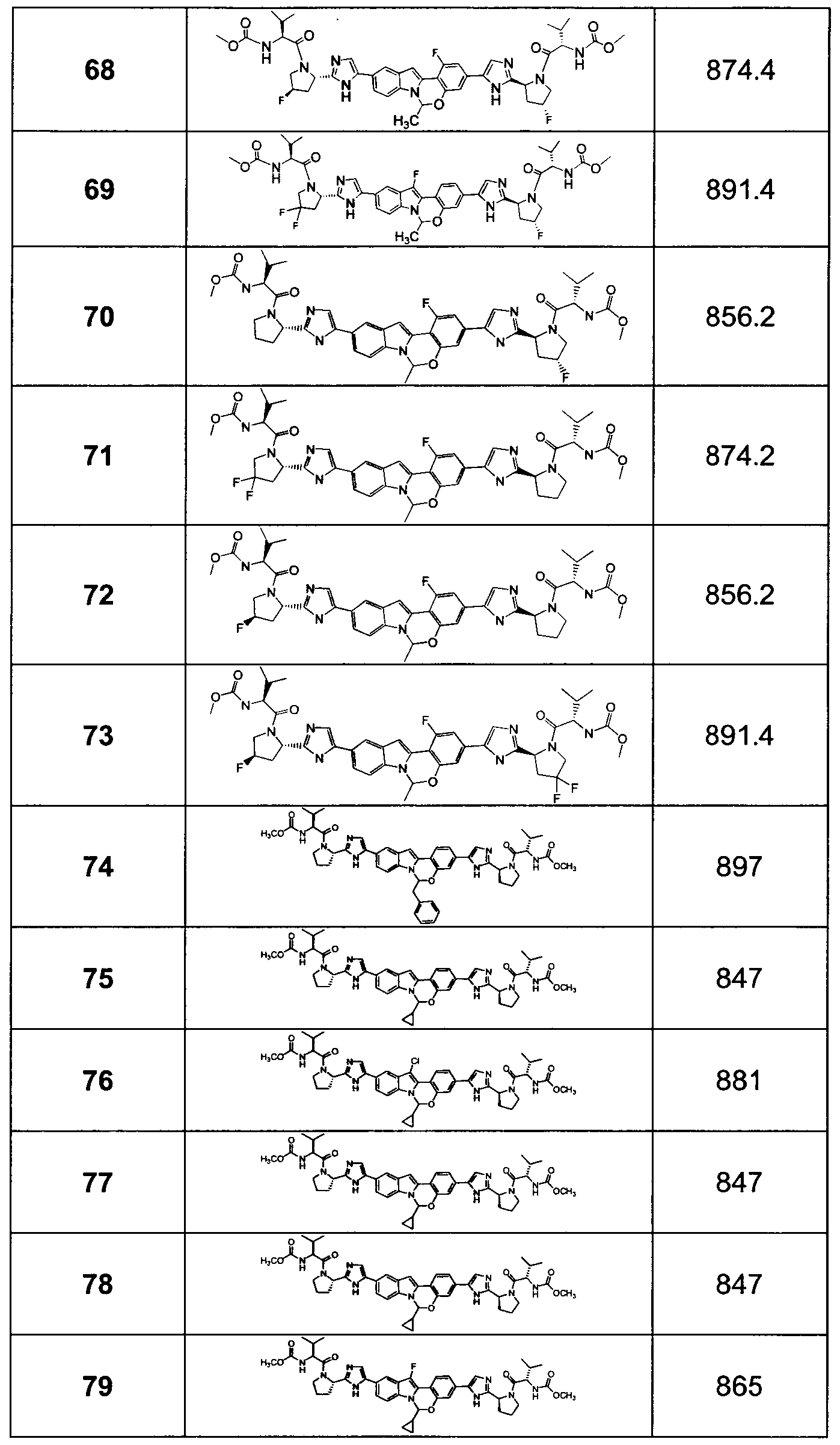
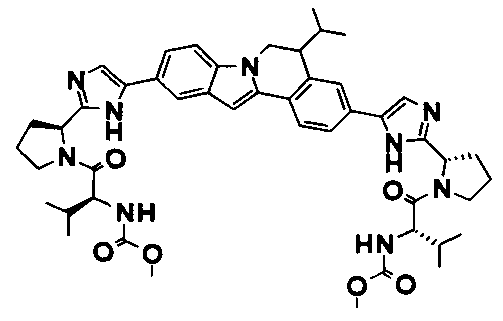
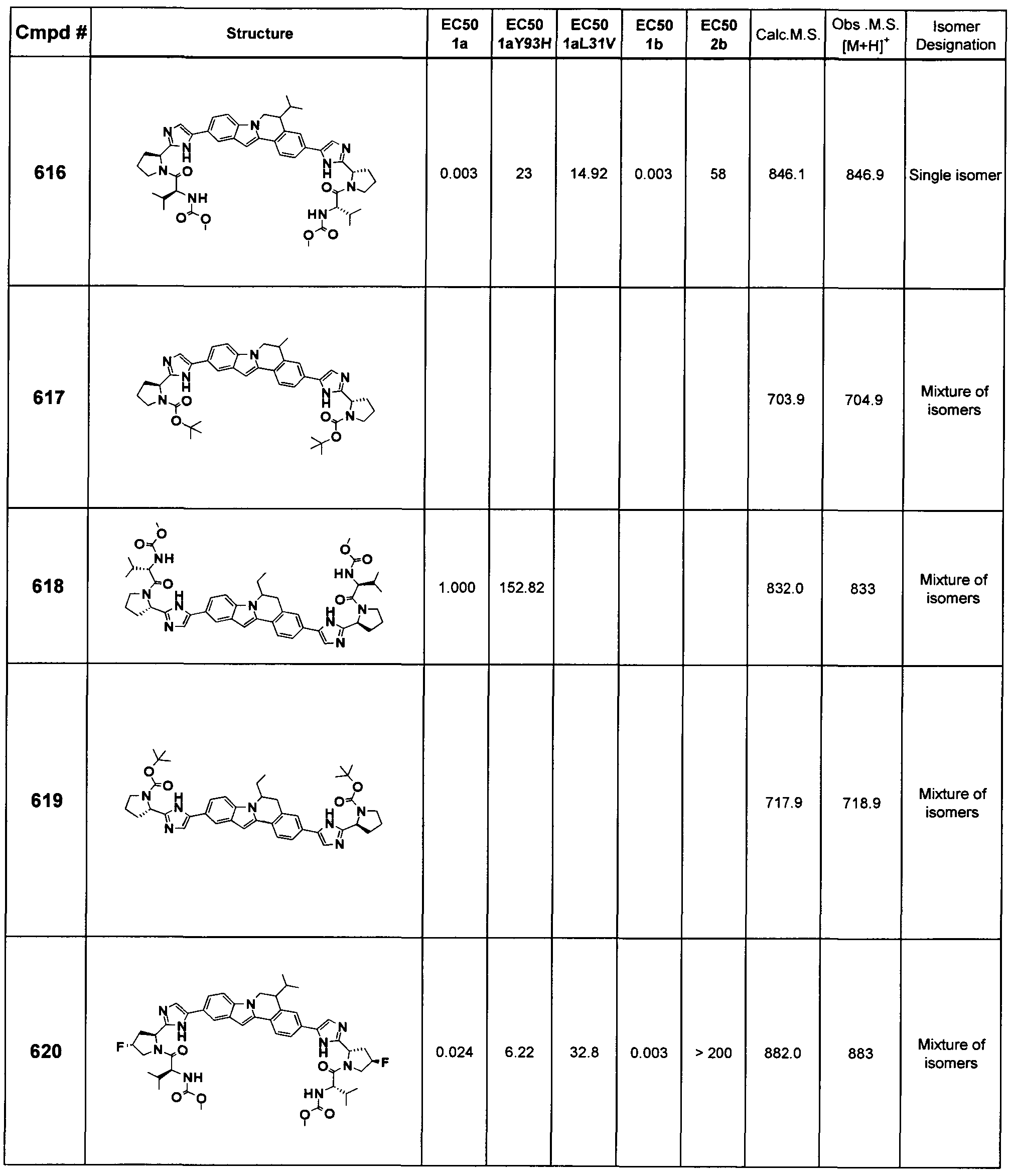
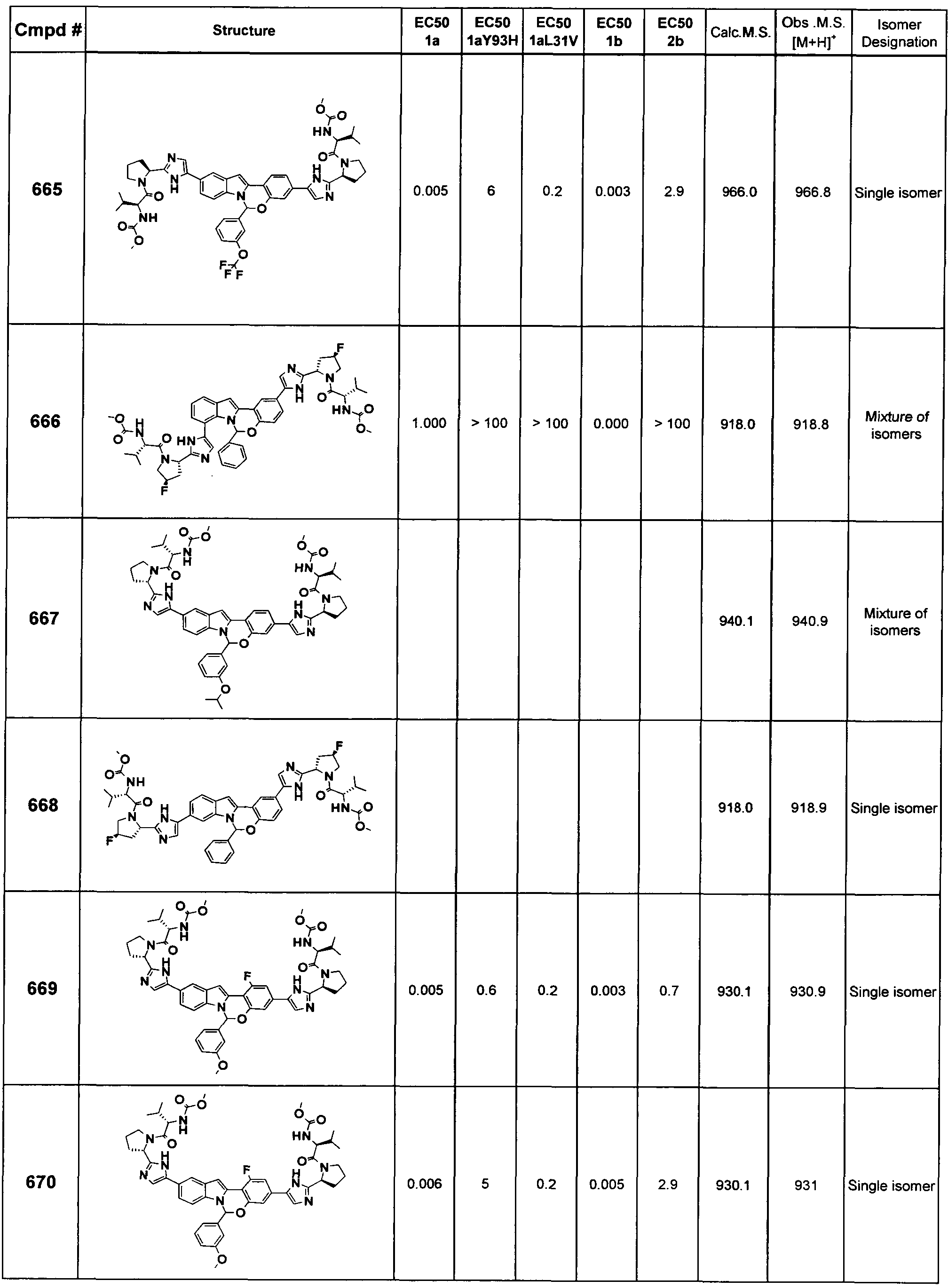
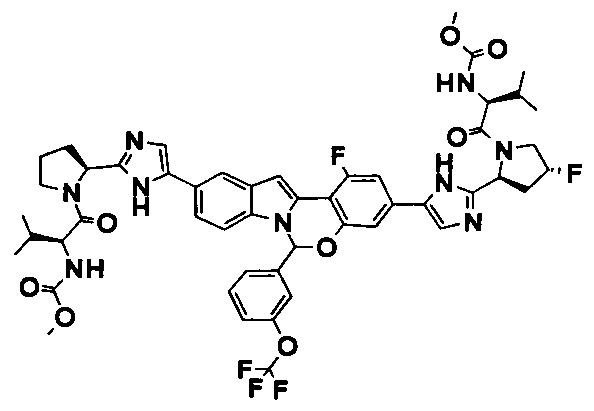
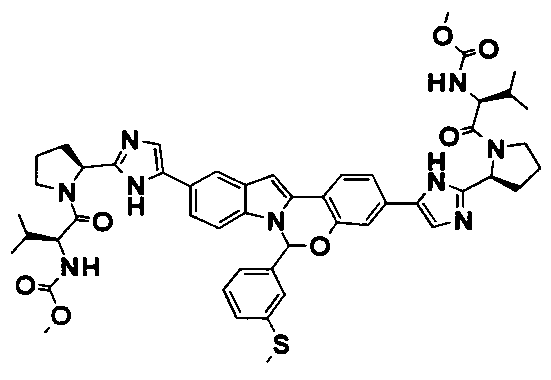
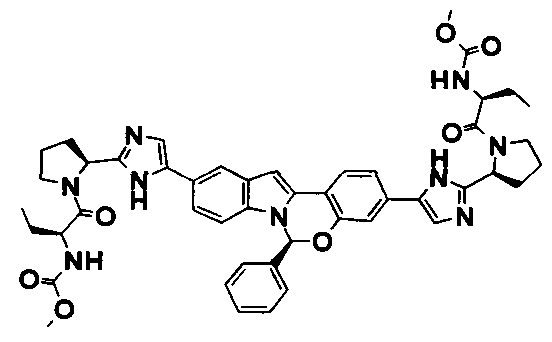
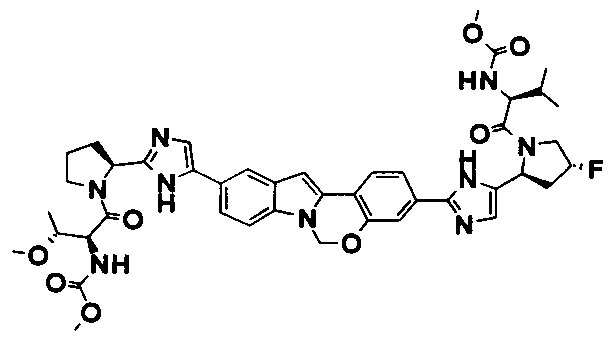
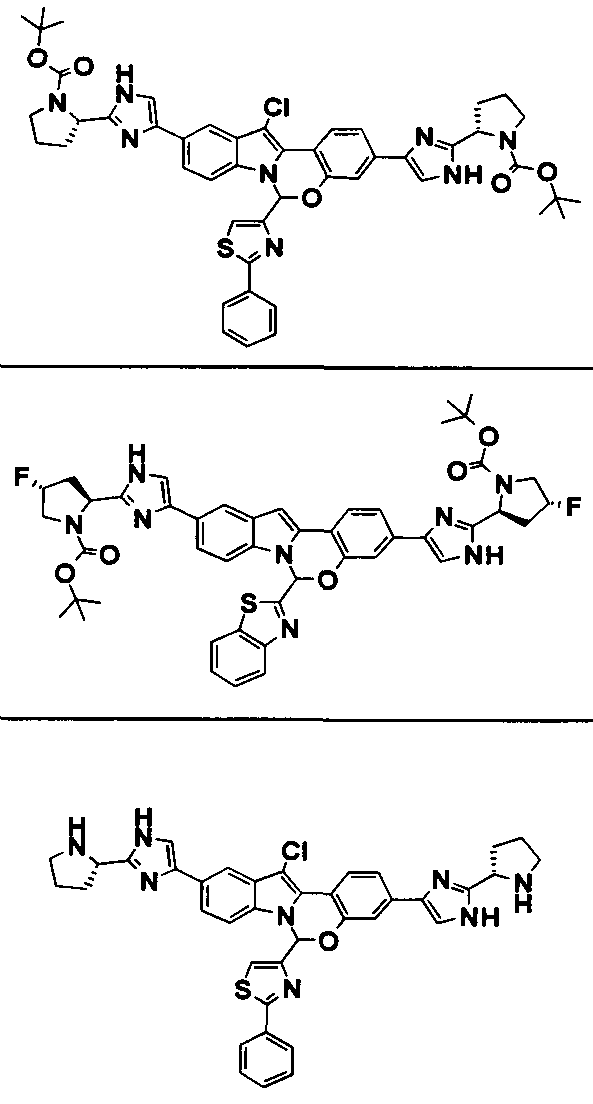
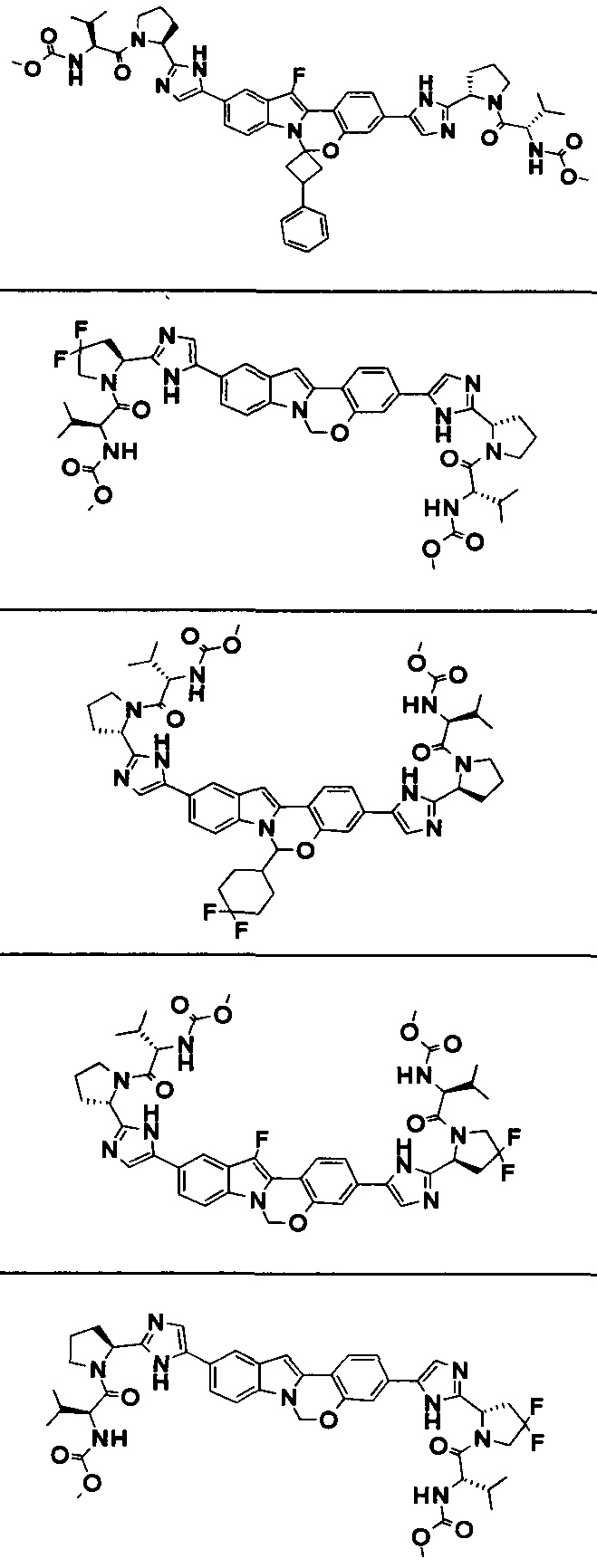
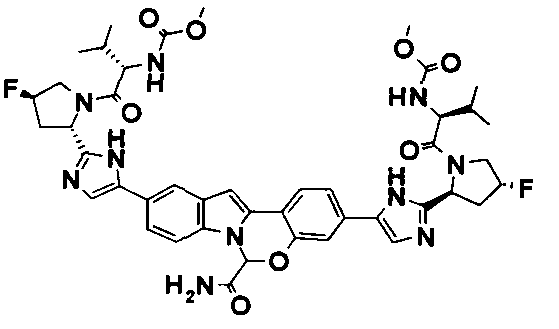
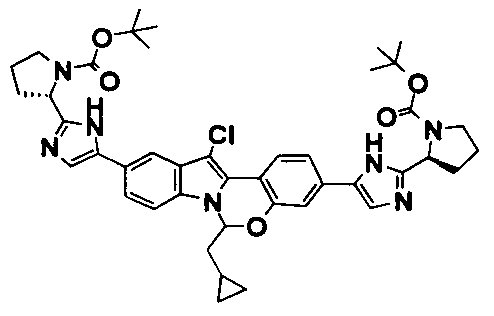
The Compounds of Formula (I)
present invention provides Tetracyclic Indole Derivatives of Formula (I):
and pharmaceutically acceptable salts thereof, wherein A, A', G, R
1, U, V, V, W, W, X, X', Y and Y' are defined above for the Compounds of Formula (I).
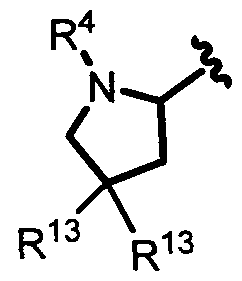
In one embodiment, A and A' are each a 5-membered heterocycloalkyl group.
In another embodiment, A and A' are each a 6-membered heterocycloalkyl group.
In another embodiment, A and A' are each independently selected from:
n another embodiment, A and A' are each independently selected from:
In another embodiment, A and A' are each independently:
In another embodiment A and A' are each independently:
wherein each occurrence of R13 is independently H, CH3, or F.
In one embodiment, each occurrence of R4 is independently -C(O)- [C(R7)2]qN(R6)C(0)0-Rn.
In another embodiment, each occurrence of R
4 is independently:
, wherein R
b is H, alkyl, haloalkyl, 3 to 6- membered cycloalkyl, 4 to 6-membered heterocycloalkyl, aryl or heteroaryl and R
a is alkyl, haloalkyl, silylalkyl, 3 to 6-membered cycloalkyl or 4 to 6-membered heterocycloalkyl, aryl or heteroaryl.
In another embodiment, each occurrence of R
4 is independently:
R
a is H, methyl, ethyl, propyl, isopropyl, t-butyl, cyclopropyl, -CH
2CH
2Si(CH
3)
3, -CH
2CH
2CF
3, pyranyl, benzyl or phenyl, and R
b is methyl, ethyl or isopropyl.
In still another embodiment, each occurrence of R4 is independently - C(0)CH(alkyl)-NHC(0)Oalkyl.
In another embodi nce of R
4 is independently:
In one embodiment, A and A' are each independently selected from:
and R
4 is:
, wherein R
b is H, alkyl, haloalkyl, 3 to 6- membered cycloalkyl, 4 to 6-membered heterocycloalkyl, aryl or heteroaryl and R
a is alkyl, haloalkyl, silylalkyl, 3 to 6-membered cycloalkyl or 4 to 6-membered heterocycloalkyl, aryl or heteroaryl.
In another embodiment, A and A' are each independently selected from:
and R
4 is:
, wherein R
a is H, methyl, ethyl, propyl, isopropyl, t-butyl, cyclopropyl, -CH
2CH
2Si(CH
3)3, -CH
2CH
2CF
3, pyranyl, benzyl or phenyl, and R
1 is methyl, ethyl or isopropyl.
In another embodiment, A and A' are each independently selected from:
In another embodiment A and A' are each independently selected from:
and R
4 is:
In yet another embodiment, A and A' are each:
, wherein each occurrence of R is independently H,
CH3, or F;
In one embodiment, G is -C(R3)2-0-.
In another embodiment, G is -C(R14)=N-
In another embodiment, G is -C(R3)2-C(R3)2- or -C(R14)=C(R14)-.
In still another embodiment, G is -C(R3)2-C(R3)2- or -C(R14)=C(R14)-.
In one embodiment, G is -C(R3)2-0- and each occurrence of R3 is independently selected from H, Ci-C6 alkyl, 3 to 6-membered cycloalkyl, 4 to 6- membered heterocycloalkyl, aryl and 5 or 6-membered monocyclic heteroaryl, wherein said 5 or 6-membered monocyclic heteroaryl group and said phenyl groups can be optionally substituted with up to 2 groups, which can be the same or different, and are selected from halo, -CN, Ci-C6 alkyl, Ci-C6 haloalkyl, -O-Ct-Ce alkyl, -(C\- C6 alkylene)-0-Ci-C6 alkyl and -0-Ci-C6 haloalkyl.
In another embodiment, G is -C(R3)2-0-, wherein one occurrence of R3 is H, and the other occurrence of R3 is selected from Ci-C6 alkyl, cycloalkyl and phenyl, wherein said phenyl group can be optionally substituted with up to 2 groups, which can be the same or different, and are selected from halo, -CN, Ci-C6 alkyl, Ci- C6 haloalkyl, -0-Ci-C6 alkyl, -(Ci-Qs alkylene)-0-C C6 alkyl and -0-Ci-C6 haloalkyl.
In one embodiment, G is -C(R3)2-0- and each occurrence of R3 is independently selected from H, methyl, ethyl, isopropyl, cyclopropyl, - methylcyclopropyl, methylenecyclopropyl, phenyl, pyridyl, and pyrimidyl wherein said phenyl, pyridyl and pyrimidinyl group can be optionally substituted with up to 2 groups, which can be the same or different, and are selected from F, CI, -CN, CH3, CF3, OCF3 and OCH2CH2OCH3.
3 3
In one embodiment, G is -CH(R )-0-, wherein R is selected from Ci- C6 alkyl, phenyl, 5 or 6-membered monocyclic heteroaryl and 9 or 10-membered
bicyclic heteroaryl, wherein said phenyl group, said 5 or 6-membered monocyclic heteroaryl group and said 9 or 10-membered bicyclic heteroaryl group can be optionally substituted with a Ci-C6 alkyl group.
In another embodiment, G is -CH(R3)-0-, wherein R3 is selected from methyl, phenyl, 5-methyl-thiophen-2-yl and benzothiophen-2-yl.
In another embodiment, G is -C(R3)2-0-, wherein one occurrence of
3 3
R is H, and the other occurrence of R is selected from phenyl, methyl, thiophenyl or benzothiophenyl, wherein said benzothiophenyl can be optionally substituted witha Ci-C6 alkyl, cycloalkyl and phenyl, wherein said phenyl group can be optionally substituted with up to 2 groups, which can be the same or different, and are selected from halo, -CN, d-C6 alkyl, Ci-C6 haloalkyl, -0-CrC6 alkyl, -(Ci-Q alkylene)-0- d-C6 alkyl and -0-d-C6 haloalkyl.
In one embodiment, G is -C(R3)2-0- and each occurrence of R3 is independently selected from H, methyl, ethyl, isopropyl, cyclopropyl, Γ- methylcyclopropyl, methylenecyclopropyl, phenyl, pyridyl, and pyrimidyl wherein said phenyl, pyridyl and pyrimidinyl group can be optionally substituted with up to 2 groups, which can be the same or different, and are selected from F, CI, -CN, CH3, CF3, OCF3 and OCH2CH2OCH3.
In another embodiment, G is -C(R3)2-0-, wherein one occurrence of
3 3
R is H, and the other occurrence of R is selected from methyl, ethyl, isopropyl, cyclopropyl, Γ-methylcyclopropyl, methylenecyclopropyl, phenyl, pyridyl, and pyrimidyl wherein said phenyl, pyridyl and pyrimidinyl group can be optionally substituted with up to 2 groups, which can be the same or different, and are selected from F, CI, -CN, CH3, CF3, OCF3 and OCH2CH2OCH3.
In one embodiment, G is -C(R3)2-0-, wherein both R3 groups, together with the common carbon atom to which they are attached, join to form a carbonyl group, a 3 to 6-membered spirocyclic cycloalkyl group or a 3 to 6-membered spirocyclic heterocycloalkyl group.
In one embodiment, G is -C(R,4)=N- wherein R14 is selected from H, Ci-C6 alkyl, cycloalkyl and phenyl, wherein said phenyl group can be optionally substituted with up to 2 groups, which can be the same or different, and are selected from halo, -CN, d-C6 alkyl, d-C6 haloalkyl, -0-d-C6 alkyl, -(Ci-C6 alkylene)-0- Ci-C6 alkyl and -0-Ci-C6 haloalkyl.
In one embodiment, each occurrence of R3 is independently selected from H, methyl, ethyl, isopropyl, cyclopropyl, Γ-methylcyclopropyl,
methylenecyclopropyl, phenyl, pyridyl, and pyrimidyl wherein said phenyl, pyridyl and pyrimidinyl group can be optionally substituted with up to 2 groups, which can be the same or different, and are selected from F, CI, -CN, CH3, CF3, OCF3 and
OCH2CH2OCH3.
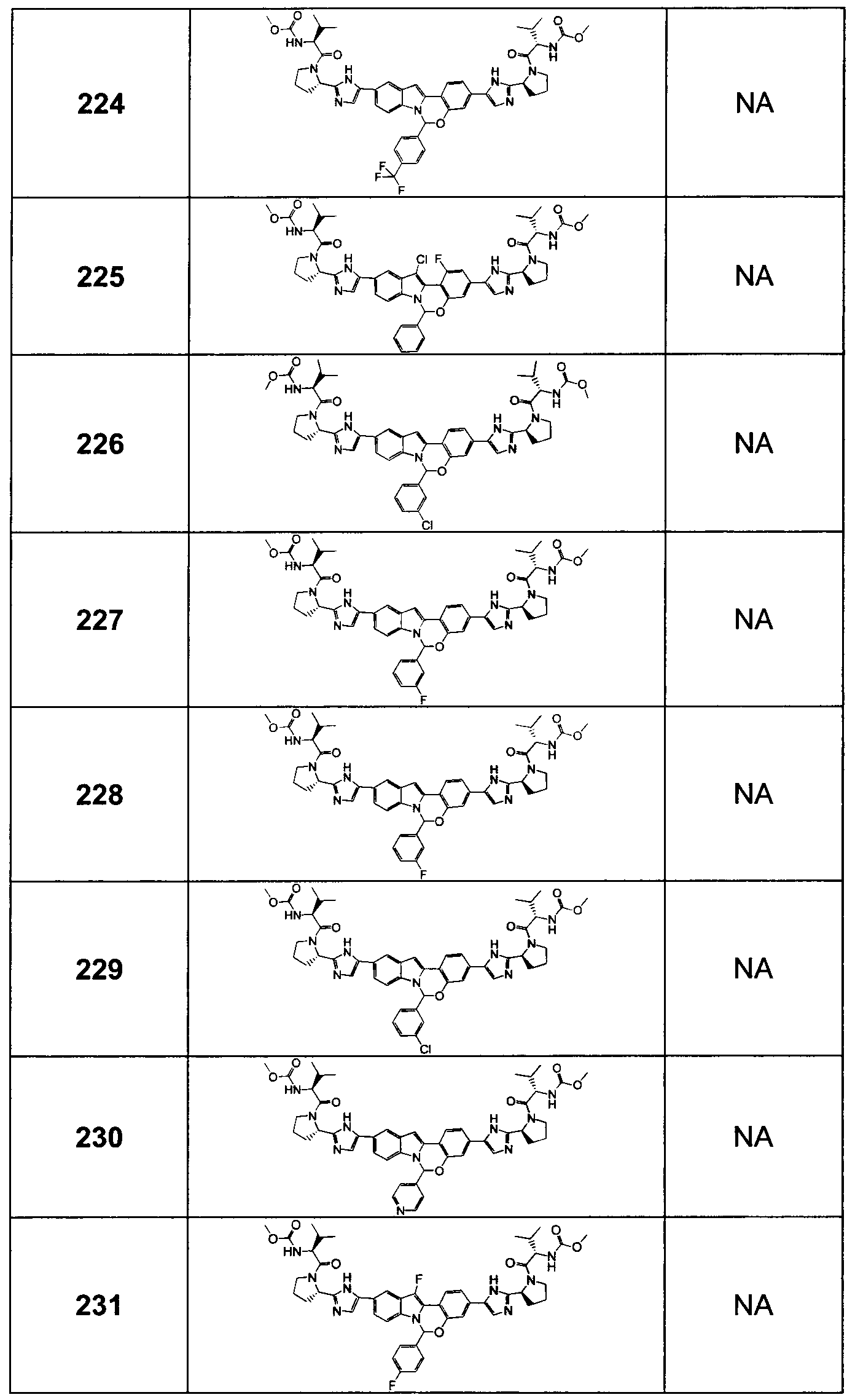
In another embodiment, two R3 groups on the same carbon atom, together with the common carbon atom to which they are attached, join to form a carbonyl group, a 3 to 6-membered spirocyclic cycloalkyl group or a 3 to 6-membered spirocyclic heterocycloalkyl group.
In one embodiment, U is C(R2).
In another embodiment, U is CH.
In another embodiment, U is CF.
In one embodiment, V is C(R15).
In another embodiment, V is CH.
In another embodiment, V is CF.
In another embodiment, V is N.
In one embodiment, V is C(R15).
In another embodiment, V is CH.
In another embodiment, V is CF.
In another embodiment, V is N.
In still another embodiment, V and V are each CH.
In one embodiment, W is C(R15).
In another embodiment, W is CH.
In another embodiment, W is CF.
In another embodiment, W is N.
In one embodiment, W ' is C(R15).
In another embodiment, W ' is CH.
In another embodiment, W ' is CF.
In another embodiment, W is N.
In still another embodiment, W and W are each CH.
In a further embodiment, V, V W and W are each CH.
In one embodiment, R1 is absent.
In another embodiment, R1 is F.
In one embodiment, each occurrence of R3 is independently selected from H, methyl, ethyl, isopropyl, cyclopropyl, l'-methylcyclopropyl,
methylenecyclopropyl, phenyl, pyridyl, and pyrimidyl wherein said phenyl, pyridyl and pyrimidinyl group can be optionally substituted with up to 2 groups, which can be the same or different, and are selected from F, CI, -CN, CH3, CF3, OCF3 and
OCH2CH2OCH3.
In another embodiment, two R3 groups on the same carbon atom, together with the common carbon atom to which they are attached, join to form a carbonyl group, a 3 to 6-membered spirocyclic cycloalkyl group or a 3 to 6-membered spirocyclic heterocycloalkyl group.
In one embodiment, each occurrence of R10 is independently H or F.
In another embodiment, each occurrence of R10 is H.
In one embodiment, the group:
has the structure:
In another embodiment, the group:
In another embodiment the group:
has the structure:
In still another embodiment, the rou :
In one embodiment, variables A, A', G, R1, U, V, V, W, W, X, X', Y and Y' for the Compounds of Formula (I) are selected independently of each other.
In another embodiment, the Compounds of Formula (I) are in substantially purified form.
embodiment, the Compounds of Formula (I) have the formula
(la)
and pharmaceutically acceptable salts thereof, wherein:
A and A' are each independently a 5-membered monocyclic heterocycloalkyl, wherein said 5-membered monocyclic heterocycloalkyl group can be optionally and independently substituted on one or more ring carbon atoms with R13, such that any two R13 groups on the same ring, together with the carbon atoms to which they are attached, can join to form a fused, bridged or spirocyclic 3 to 6- membered cycloalkyl group or a fused, bridged or spirocyclic 4 to 6-membered heterocycloalkyl group, wherein said 5-membered monocyclic heterocycloalkyl contains from 1 to 2 ring heteroatoms, each independently selected from N(R4) and Si(R16)2;
G is selected from -C(R3)2-, -C(R3)2-0-, -C(R14)=N-, -C(R3>2-C(R3)2- and -C(R14)=C(R14)-;
V and V are each independently selected from N and C(R15);
R1 represents an optional ring substituent on the phenyl ring to which R1 is attached, wherein said substituent is selected from C\-C alkyl and halo;
each occurrence of R2 is independently selected from H, C -C alkyl, 3 to 6 membered cycloalkyl, -0-(Ci-C6 alkyl), C C6 haloalkyl -0-(Ci-C6 haloalkyl); halo, -OH, aryl, and heteroaryl
each occurrence of R3 is independently selected from H, Ci-C alkyl, - (Ci-C6 alkylene)-0-(Ci-C6 alkyl), 3 to 6-membered cycloalkyl, Cj-C6 haloalkyl, aryl, 5 or 6-membered monocyclic heteroaryl and benzyl, wherein said aryl group, said 5 or 6-membered monocyclic heteroaryl group or the phenyl group of said benzyl group can be optionally substituted with up to 3 groups, which can be the same or different, and are selected from halo, -CN, Ci-C6 alkyl, Cj-C6 haloalkyl, -0-Ci-C6 alkyl, -(Ci-C6 alkylene)-0-C]-C6 alkyl and -0-(Ci-C6 haloalkyl);
each occurrence of R4 is independently -C(0)-[C(R7)2]N(R6)C(0)0-
R11;
each occurrence of R6 is independently selected from H and CrC6 alkyl;
each occurrence of R7 is independently selected from Ci-C6 alkyl, Ci- C6 haloalkyl, 3 to 6-membered cycloalkyl, 4 to 6-membered heterocycloalkyl, aryl and 5 or 6-membered monocyclic heteroaryl, wherein said 3 to 6-membered cycloalkyl group, said 4 to 6-membered heterocycloalkyl group, said aryl group and said 5 or 6-membered monocyclic heteroaryl group can be optionally and
independently substituted with up to three R8 groups;
each occurrence of R8 is independently selected from H, Ci-C6 alkyl, halo, -d-C6 haloalkyl, Ci-C6 hydroxyalkyl, -OH, -C(0)NH-(Ci-C6 alkyl), -C(0)N(Ci- C6 alkyl)2, -0-(C C6 alkyl), -NH2, -NH(Ci-C6 alkyl), -N(d-C6 alkyl)2 and -NHC(O)- (Ci-Ce alkyl);
each occurrence of R10 is independently selected from H and halo; each occurrence of R1 1 is independently Ci-C6 alkyl;
each occurrence of R13 is independently selected from H and halo, wherein two R13 groups, together with the carbon atom(s) to which they are attached, can optionally join to form a 3 to 6-membered cycloalkyl group or 4 to 6-membered heterocycloalkyl group;
each occurrence of R14 is independently selected from H, halo, CrC6 alkyl, -(Ci-C6 alkylene)-0-Ci-C6 alkyl, 3 to 6-membered cycloalkyl, Ci-C6 haloalkyl, aryl, 5 or 6-membered monocyclic heteroaryl and benzyl, wherein said aryl group, said 5 or 6-membered monocyclic heteroaryl group or the phenyl group of said benzyl group can be optionally substituted with up to 3 groups, which can be the same or different, and are selected from halo, -CN, Ci-C6 alkyl, Ci-C6 haloalkyl, -0-Cj-C6 alkyl, -(C]-C6 alkylene)-0-Ci-C6 alkyl and -0-Ci-C6 haloalkyl;
each occurrence of R15 is independently selected from H and halo; and each occurrence of R16 is independently selected from Ci-C6 alkyl.
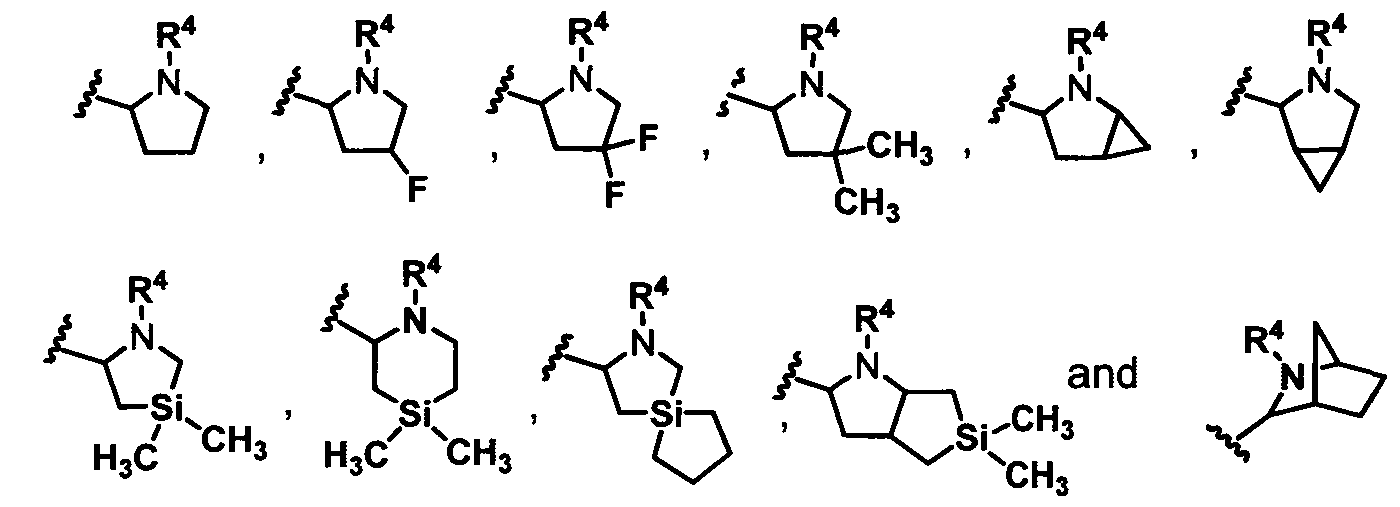
In one embodiment, for the Compounds of Formula (la), A and A' are each a 5-membered monocyclic heteroaryl group.
In another embodiment, for the Compounds of Formula (la), A and A' are each a 6-membered monocyclic heteroaryl group.
In another embodiment, for the Compounds of Formula (la), A and A' are each independently selected from:
In still another embodiment, for the Compounds of Formula (la), A and A' are each inde endently selected from:
In another embodiment, for the Compounds of Formula (la), A and A' are each inde endently selected from:
In one embodiment, for the Compounds of Formula (la), A and A' are each:
R" R 13
wherein each occurrence of Z is independently -Si(R13)2-, -C(R13)2- or -S-,
and each occurrence of R13 is independently H, Me, F or two R13 groups together with
Z, can combine to form a spirocyclic 3 to 6-membered cycloalkyl group or a spirocyclic 3 to 6-membered silyl-containing heterocycloalkyl group.
In another embodiment, for the Compounds of Formula (la), A and A' are each independently:
In another embodiment, for the Compounds of Formula (la), A and A' are each independently:
wherein each occurrence of R13 is independently H, CH3, or F.
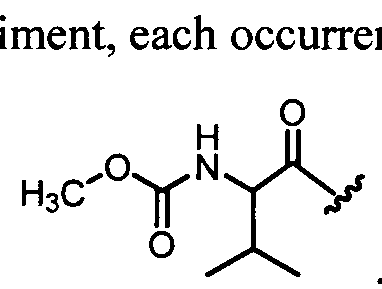
In one embodiment, for the Compounds of Formula (la), each occurrence of R4 is independently -C(0)C(R7)2NHC(0)0-Rn or -C(0)C(R7)2N(R6)2.
In another embodiment, for the Compounds of Formula (la), each occurrence of R4 is independently -C(0)-[C(R7)2]qN(R6)C(0)0-Rn.
In another embodiment, for the Compounds of Formula (la), each occurrence of R4 is independently -C(0)CH(alkyl)-NHC(0)Oalkyl,
C(0)CH(cycloalkyl)-NHC(0)Oalkyl, C(0)CH(heterocycloalkyl)-NHC(0)Oalkyl, C(0)CH(aryl)-NHC(0)Oalkyl or C(0)CH(aryl)-N(alkyl)2.
In another embodiment, for the Compounds of Formula (la), each occurrence of
4 is independently:
, wherein R
b is H, alkyl, haloalkyl, 3 to 6- membered cycloalkyl, 4 to 6-membered heterocycloalkyl, aryl or heteroaryl and R
a is alkyl, haloalkyl, silylalkyl, 3 to 6-membered cycloalkyl or 4 to 6-membered heterocycloalkyl, aryl or heteroaryl.
In another embodiment, for the Compounds of Formula (la), each occurrence of
4 is independently:
, wherein R is H, methyl, ethyl, propyl, isopropyl, t-butyl, cyclopropyl, -CH
2CH
2Si(CH
3)3, -CH
2CH
2CF
3, pyranyl, benzyl or phenyl, and R
b is methyl, ethyl or isopropyl.
In still another embodiment, for the Compounds of Formula (la), each occurrence of R4 is independently -C(0)CH(alkyl)-NHC(0)Oalkyl.
In another embodiment, for the Compounds of Formula (la), each occurrence of R
4 is independently:
In one embodiment, for the Compounds of Formula (la), A and A' are each independently selected from:
and R
4 is:
, wherein R
1 is H, alkyl, haloalkyl, 3 to 6- membered cycloalkyl, 4 to 6-membered heterocycloalkyl, aryl or heteroaryl and R
a is alkyl, haloalkyl, silylalkyl, 3 to 6-membered cycloalkyl or 4 to 6-membered heterocycloalkyl, aryl or heteroaryl.
In another embodiment, for the Compounds of Formula (la), A and A' are each independently selected from:
and R
4 is:
, wherein R
a is H, methyl, ethyl, propyl, isopropyl, t-butyl, cyclopropyl, -CH
2CH
2Si(CH
3)
3, -CH
2CH
2CF
3, pyranyl, benzyl or phenyl, and R
1 is methyl, ethyl or isopropyl.
In another embodiment, for the Compounds of Formula (la), A and A' are each independently selected from:
and R4 is:
» S
In another embodiment, for the Compounds of Formula (la), A and A' are each independently selected from:
In yet another embodiment, for the Compounds of Formula (la), A and
R , wherein each occurrence of R is independently H,
CH3, or F;
In one embodiment, for the Compounds of Formula (la), G is -C(R )2-
0-.
In another embodiment, for the Compounds of Formula (la), G is ·
C(R14)=N-
In another embodiment, for the Compounds of Formula (la), G is -
C(R3)2-C(R3)2-, -C(R14)=C(R14)-.
In still another embodiment, for the Compounds of Formula (la), G is - C(R3)2-C(R3)2-, -C(R14)=C(R14)-.
In one embodiment, for the Compounds of Formula (la), G is -C(R3)2- O- and each occurrence of R3 is independently selected from H, Ci-C6 alkyl, cycloalkyl and phenyl, wherein said phenyl group can be optionally substituted with
up to 2 groups, which can be the same or different, and are selected from halo, -CN, Ci-Ce alkyl, Ci-C6 haloalkyl, -0-C C6 alkyl, -(Ci-C6 alkylene)-0-CrC6 alkyl and - 0-C]-C6 haloalkyl.
In another embodiment, for the Compounds of Formula (la), G is - C(R3)2-0-, wherein one occurrence of R is H, and the other occurrence of R is selected from Ci-C6 alkyl, cycloalkyl and phenyl, wherein said phenyl group can be optionally substituted with up to 2 groups, which can be the same or different, and are selected from halo, -CN, Ci-C6 alkyl, Ci-C6 haloalkyl, -0-d-C6 alkyl, -(Cj-C6 alkylene)-0-C1-C6 alkyl and -0-C]-C6 haloalkyl.
In one embodiment, for the Compounds of Formula (la), G is -C(R3)2- O- and each occurrence of R3 is independently selected from H, methyl, ethyl, isopropyl, cyclopropyl, Γ-methylcyclopropyl, methylenecyclopropyl, phenyl, pyridyl, and pyrimidyl wherein said phenyl, pyridyl and pyrimidinyl group can be optionally substituted with up to 2 groups, which can be the same or different, and are selected from F, CI, -CN, CH3, CF3, OCF3 and OCH2CH2OCH3.
In another embodiment, for the Compounds of Formula (la), G is - C(R3)2-0-, wherein one occurrence of R is H, and the other occurrence of R is selected from methyl, ethyl, isopropyl, cyclopropyl, l'-methylcyclopropyl, methylenecyclopropyl, phenyl, pyridyl, and pyrimidyl wherein said phenyl, pyridyl and pyrimidinyl group can be optionally substituted with up to 2 groups, which can be the same or different, and are selected from F, CI, -CN, CH3, CF3, OCF3 and
OCH2CH2OCH3.
In one embodiment, for the Compounds of Formula (la), G is - C(RI4)=N-; wherein R14 is selected from H, C]-C6 alkyl, cycloalkyl and phenyl, wherein said phenyl group can be optionally substituted with up to 2 groups, which can be the same or different, and are selected from halo, -CN, Ci-C6 alkyl, C]-C6 haloalkyl, -0-C]-C6 alkyl, -(Ci-C6 alkylene)-0-CrC6 alkyl and -0-C C6 haloalkyl.
In one embodiment, for the Compounds of Formula (la), U is C(R ).
In another embodiment, for the Compounds of Formula (la), U is CH.
In another embodiment, for the Compounds of Formula (la), U is CF.
In one embodiment, for the Compounds of Formula (la), V is C(R15).
In another embodiment, for the Compounds of Formula (la), V is CH.
In another embodiment, for the Compounds of Formula (la), V is N.
In one embodiment, for the Compounds of Formula (la), V is C(R15).
In another embodiment, for the Compounds of Formula (la), V is CH. In another embodiment, for the Compounds of Formula (la), V is N. In still another embodiment, for the Compounds of Formula (la), V and V are each CH.
In one embodiment, for the Compounds of Formula (la), R is absent. In another embodiment, for the Compounds of Formula (la), R1 is F. In one embodiment, each occurrence of R3 is independently selected from H, methyl, ethyl, isopropyl, cyclopropyl, l'-methylcyclopropyl,
methylenecyclopropyl, phenyl, pyridyl, and pyrimidyl wherein said phenyl, pyridyl and pyrimidinyl group can be optionally substituted with up to 2 groups, which can be the same or different, and are selected from F, CI, -CN, CH3, CF3, OCF3 and
OCH2CH2OCH3.
In one embodiment, for the Compounds of Formula (la), each occurrence of R10 is independently H or F.
In another embodiment, for the Compounds of Formula (la), each occurrence of R10 is H.
In one embodiment for the Compounds of Formula (la), the group:
has the structure:
In another embodiment for the Compounds of Formula (la), the group:
In another embodiment for the Compounds of Formula (la), the group:
has the structure:
In one embodiment, variables A, A', G, R1, R2, R10, R15, U, V and V for the Compounds of Formula (la) are selected independently of each other.
In another embodiment, the Compounds of Formula (la) are in substantially purified form.
In one embodiment, the Compounds of Formula (I) have the formula
(lb):
each occurrence of R is independently selected from H, C]-C6 alkyl, Ci-C6 haloalkyl, 3 to 6 membered cycloalkyl, 4 to 6-membered heterocycioalkyl, aryl,
5 or 6-membered monocyclic heteroaryl, 9 or 10-membered bicyclic heteroaryl, -O- (C]-C6 alkyl), Ci-C6 haloalkylene -0-(Ci-C6 haloalkyl); -(Ci-C6 alkylene)C(=0)NH- alkyl, -(C!-C6 alkylene)aryl, and -(Ci-C6 alkylene)heteroaryl, wherein said aryl group, said 5 or 6-membered monocyclic heteroaryl group, said 9 or 10-membered bicyclic heteroaryl group or the phenyl group of said benzyl group can be optionally substituted with up to 3 groups, which can be the same or different, and are selected from halo, -CN, Ci-C6 alkyl, C C6 haloalkyl, -0-CrC6 alkyl, -(Ci-C6 alkylene)-0- Ci-C6 alkyl and -0-(C C6 haloalkyl);
each occurrence of R4 is independently selected from -C(0)0-(C1-C6 alkyl), -C(0)-CH(R7)N(R6)2 and -C(0)-CH(R7)C(0)0-Rn;
each occurrence of R6 is independently H or Ci-C6 alkyl; each occurrence of R7 is independently selected from Ci-C6 alkyl, phenyl, 4 to 6-membered heterocycloalkyl and 3 to 6 membered cycloalkyl;
each occurrence of R11 is independently C]-C6 alkyl;
each occurrence of R13a is independently H, Me or F; or two R13a groups that are attached to the same carbon atom, together with the common carbon atom to which they are attached, combine to form a spirocyclic 3 to 6 membered cycloalkyl group;
each occurrence of R13b is independently H, or one or both R13b groups and an R13a group that are attached to same ring, together with the ring carbon atoms to which they are attached, can combine to form a fused 3 to 6 membered cycloalkyl group; and
R15 represents up to 2 substituents, each independently selected from H, halo, Ci-C6 alkyl, Ci-C6 haloalkyl, 3 to 6 membered cycloalkyl, 4 to 6-membered heterocycloalkyl, aryl, 5 or 6-membered monocyclic heteroaryl, benzyl, -0-(CrC6 alkyl), Ci-C6 haloalkylene -0-(d-C6 haloalkyl) -(Ci-C6alkylene)C(=0)NH-alkyl, - (Ci-C6 alkylene)aryl, and -(d-Ce alkylene^eteroaryl, wherein said aryl group, said 5 or 6-membered monocyclic heteroaryl group or the phenyl group of said benzyl group can be optionally substituted with up to 3 groups, which can be the same or different, and are selected from halo, -CN, C]-C6 alkyl, C]-C6 haloalkyl, -0-C]-C6 alkyl, -(Ci-C6 alkylene)-0-C,-C6 alkyl and -0-(Ci-C6 haloalkyl).
In one embodiment, for the Compounds of Formula (lb), R2 is H.
In another embodiment, for the Compounds of Formula (lb), R2 is F.
In one embodiment, for the Compounds of Formula (lb), one occurrence of R 3 is H and the other occurrence of R 3 is selected from H, methyl, ethyl, isopropyl, cyclopropyl, 1 '-methylcyclopropyl, methylenecyclopropyl, phenyl, pyridyl, and pyrimidyl wherein said phenyl, pyridyl and pyrimidinyl group can be optionally substituted with up to 2 groups, which can be the same or different, and are selected from F, CI, -CN, CH3, CF3, OCF3, OCH2CH2OCH3.
In another embodiment, for the Compounds of Formula (lb), two R3 groups that are attached to the same carbon atom, together with the common carbon atom that they are attached to, join to form a carbonyl group, a 3 to 6-membered spirocyclic cycloalkyl group or a 3 to 6-membered spirocyclic heterocycloalkyl group.
In one embodiment, for the Compounds of Formula (lb), each occurrence of R3 is C]-C6 alkyl.
In another embodiment, for the Compounds of Formula (lb), one occurrence of R3 is H.
In another embodiment, for the Compounds of Formula (lb), one occurrence of R3 is H and the other occurrence of R3 is methyl, phenyl, 5 or 6- membered monocyclic heteroaryl or 9-membered bicyclic heteroaryl.
In still another embodiment, for the Compounds of Formula (lb), one occurrence of R3 is H and the other occurrence of R3 is phenyl, methyl,
In one embodiment, for the Compounds of Formula (lb), each occurrence of R4 is -C(0)CH(R7)NHC(0)ORn.
In another embodiment, for the Compounds of Formula (lb), each occurrence of R4 is -C(0)CH(R7)NHC(0)OR' 1 and each occurrence of R11 is methyl.
In another embodiment, for the Compounds of Formula (lb), each occurrence of R4 is -C(0)CH(R7)NHC(0)ORn; each occurrence of R7 is isopropyl, benzyl, cyclopropyl or tetrahyropyranyl; and each occurrence of R11 is methyl.
In still another embodiment, for the Compounds of Formula (lb), each occurrence of R4 is -C(0)CH(R7)NHC(0)OR11 ; each occurrence of R7 is isopropyl or tetrahyropyranyl; and each occurrence of R11 is methyl.
In another embodiment, for the Compounds of Formula (lb), each occurrence of R4 is -C(0)CH(R7)NHC(0)ORn; each occurrence of R7 is isopropyl; and each occurrence of R11 is methyl.
In yetanother embodiment, for the Compounds of Formula (lb), each occurrence of R4 is -C(0)CH(R7)NHC(0)ORn; each occurrence of R7 is
tetrahyropyranyl; and each occurrence of R11 is methyl.
In one embodiment, for the Compounds of Formula (lb), each occurrence of R13a is independently H, or F.
In another embodiment, for the Compounds of Formula (lb), two R13a groups that are attached to the same carbon atom, together with the common carbon atom to which they are attached, combine to form a spirocyclic 3 to 6 membered cycloalkyl group.
In another embodiment, for the Compounds of Formula (lb), two R13a groups that are attached to the same carbon atom, together with the common carbon atom to which they are attached, combine to form a spirocyclic cyclopropyl group.
In one embodiment, for the Compounds of Formula (lb), each occurrence of R13b is H.
In another embodiment, for the Compounds of Formula (lb), one or both RI3b groups and an R13a group that are attached to same ring, together with the ring carbon atoms to which they are attached, can combine to form a fused 3 to 6 membered cycloalkyl group.
In another embodiment, for the Compounds of Formula (lb), one or both R13b groups and an R13a group that are attached to same ring, together with the ring carbon atoms to which they are attached, can combine to form a fused 3 to 6 membered cyclopropyl group.
In one embodiment, for the Compounds of Formula (lb), each occurrence of R15 is independently selected from H and F.
In another embodiment, for the Compounds of Formula (lb), each occurrence of R15 is H.
In one embodiment, for the Compounds of Formula (lb), each occurrence of R2, R13 and R15 is independently selected from H and F.
In another embodiment, for the Compounds of Formula (lb), each occurrence of R22,, RR1133 , and R15 is independently selected from H and F and one occurrence of R3 is H.
In one embodiment, variables R2, R3, R13 and R15 for the Compounds of Formula (lb) are selected independently of each other.
In another embodiment, the Compounds of Formula (lb) are in substantially purified form. embodiment, the Compounds of Formula (I) have the formula
(Ic):
(Ic)
and pharmaceutically acceptable salts thereof, wherein:
Ry is isopropyl or tetrahydropyranyl;
Rz is isopropyl or tetrahydropyranyl;
R is H or halo;
R3 is selected from 3 to 6-membered cycloalkyl or phenyl, wherein said phenyl group can be optionally substituted with up to 2 groups, which can be the same or different, and are selected from halo, -CN, Ci-C6 alkyl, Cj-C6 haloalkyl, -O- Ci-C6 alkyl, -(Ci-C6 alkylene)-0-Ci-C6 alkyl and -0-C C6 haloalkyland
each occurrence of R13 is independently selected from H and halo; and each occurrence of R15 is independently selected from H and halo.
In one embodiment, for the compounds of formula (Ic), R3 is phenyl, wherein said phenyl group can be optionally substituted with up to 2 groups, which can be the same or different, and are selected from F, CI, -CN, CH3, CF3, OCF3, OCH2CH2OCH3.
In another embodiment, for the compounds of formula (Ic), R3 is cyclopropyl.
In another embodiment, for the compounds of formula (Ic), R and R are each independently H or F.
In another embodiment, for the compounds of formula (Ic), each occurrence of R13 is independently H or F;
In one embodiment, for the compounds of formula (Ic), R3 is phenyl; each occurrence of R13 is independently H or F; and R2 and R15 are each
independently H or F, wherein said phenyl group can be optionally substituted with up to 2 groups, which can be the same or different, and are selected from F, CI, -CN, CH3, CF3, OCF3, OCH2CH2OCH3.
In another embodiment, for the compounds of formula (Ic), R3 is cyclopropyl; each occurrence of R13 is independently H or F; and R2 and R15 are each independently H or F.
In one embodiment, the Compounds of Formula (I) have the formula
(Id):
(Id)
or a pharmaceutically acceptable salt thereof,
wherein:
R30 is Ci-C6 alkyl, aryl, 5 or 6-membered monocyclic heteroaryl or 9- membered bicyclic heteroaryl;
Rw is H, or Rw and Rx, together with the ring carbon atoms to which they are attached, combine to form a fused 3 to 6-membered cycloalkyl group;
Rx is H or F, or Rw and Rx, together with the ring carbon atoms to which they are attached, combine to form a fused 3 to 6-membered cycloalkyl group;
Ry is H, or Ry and Rz, together with the ring carbon atoms to which they are attached, combine to form a fused 3 to 6-membered cycloalkyl group; and
Rz is H or F, or Ry and Rz, together with the ring carbon atoms to which they are attached, combine to form a fused 3 to 6-membered cycloalkyl group.
In one embodiment, for the compounds of formula (Id), R is phenyl, methyl,
In another embodiment, for the compounds of formula (Id), Rw and Rx, together with the ring carbon atoms to which they are attached, combine to form a fused cyclopropyl group.
In another embodiment, for the compounds of formula (Id), Ry and Rz, together with the ring carbon atoms to which they are attached, combine to form a fused cyclopropyl group.
In still another embodiment, for the compounds of formula (Id), Ry and Rz, together with the ring carbon atoms to which they are attached, combine to form a fused cyclopropyl group and Rw and Rx, together with the ring carbon atoms to which they are attached, combine to form a fused cyclopropyl group.
In another embodiment, for the compounds of formula (Id), Rw, Rx, and Ry are each H and Rz is F.
In another embodiment, for the compounds of formula (Id), Rw and Rx, together with the ring carbon atoms to which they are attached, combine to form a fused cyclopropyl group; Ry is H and Rz is F.
In one embodiment, for the compounds of formula (Id), variables R30, Rw, Rx, Ry, and Rz are selected independently of each other.
In another embodiment, the Compounds of Formula (Ic) are in substantially purified form.
Other embodiments of the present invention include the following:
(a) A pharmaceutical composition comprising an effective amount of a Compound of Formula (I) or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
(b) The pharmaceutical composition of (a), further comprising a second therapeutic agent selected from the group consisting of HCV antiviral agents, immunomodulators, and anti-infective agents.
(c) The pharmaceutical composition of (b), wherein the HCV antiviral agent is an antiviral selected from the group consisting of HCV protease inhibitors and HCV NS5B polymerase inhibitors.
(d) A pharmaceutical combination that is (i) a Compound of Formula (I) and (ii) a second therapeutic agent selected from the group consisting of HCV antiviral agents, immunomodulators, and anti-infective agents; wherein the Compound of Formula (I) and the second therapeutic agent are each employed in an amount that renders the combination effective for inhibiting HCV replication, or for treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection.
(e) The combination of (d), wherein the HCV antiviral agent is an antiviral selected from the group consisting of HCV protease inhibitors and HCV NS5B polymerase inhibitors.
(f) A method of inhibiting HCV replication in a subject in need thereof which comprises administering to the subject an effective amount of a Compound of Formula (I).
(g) A method of treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection in a subject in need thereof which comprises administering to the subject an effective amount of a Compound of Formula (I).
(h) The method of (g), wherein the Compound of Formula (I) is administered in combination with an effective amount of at least one second therapeutic agent selected from the group consisting of HCV antiviral agents, immunomodulators, and anti-infective agents.
(i) The method of (h), wherein the HCV antiviral agent is an antiviral selected from the group consisting of HCV protease inhibitors and HCV NS5B polymerase inhibitors.
(j) A method of inhibiting HCV replication in a subject in need thereof which comprises administering to the subject the pharmaceutical composition of (a), (b) or (c) or the combination of (d) or (e).
(k) A method of treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection in a subject in need thereof which comprises administering to the subject the pharmaceutical composition of (a), (b) or (c) or the combination of (d) or (e).
The present invention also includes a compound of the present invention for use (i) in, (ii) as a medicament for, or (iii) in the preparation of a medicament for: (a) medicine; (b) inhibiting HCV replication or (c) treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection. In these uses, the compounds of the present invention can optionally be employed in combination with one or more second therapeutic agents selected from HCV antiviral agents, anti-infective agents, and immunomodulators.
The present invention also includes the use of a compound of the present invention for (i) inhibiting HCV replication or (ii) treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection.
Additional embodiments of the invention include the pharmaceutical compositions, combinations and methods set forth in (a)-(k) above and the uses set forth in the preceding paragraph, wherein the compound of the present invention employed therein is a compound of one of the embodiments, aspects, classes, subclasses, or features of the compounds described above. In all of these embodiments, the compound may optionally be used in the form of a pharmaceutically acceptable salt or hydrate as appropriate.
It is further to be understood that the embodiments of compositions and methods provided as (a) through (k) above are understood to include all embodiments of the compounds, including such embodiments as result from combinations of embodiments.
The Compounds of Formula (I) may be referred to herein by chemical structure and/or by chemical name. In the instance that both the structure and the name of a Compound of Formula (I) are provided and a discrepancy is found to exist between the chemical structure and the corresponding chemical name, it is understood that the chemical structure will predominate.
Non-limiting examples of the Compounds of Formula (I) include (i) compounds 1-1542, as set forth in Tables 1 and 2 in the Examples Section below.
Methods For Making the Compounds of Formula (T)
The Compounds of Formula (I) may be prepared from known or readily prepared starting materials, following methods known to one skilled in the art of organic synthesis. Methods useful for making the Compounds of Formula (I) are set forth in the Examples below and generalized in Schemes 1-5 below. Alternative synthetic pathways and analogous structures will be apparent to those skilled in the art of organic synthesis.
Scheme 1 shows methods useful for making the Compounds of formula G8, which correspond to the Compounds of Formula (I), wherein B is phenyl and the group -U-V-W- is -C(R2)=CH-N-.
Scheme 1
G8
Wherein Q and Q' are each independently halo, hydroxy, or a protected hydroxy such as methoxy or benzyloxy; M, M', M" are each independently halo, hydroxy, or a protected hydroxy, triflate, boronic acid or boronic ester; K represents a group that can form a bond to the indole nitrogen. One skilled in the art of organic synthesis will recognize that when G is single or multiatom bridge, K should contain all the atoms of the bridge and a reactive group capable of forming a bond to nitrogen of the indole. Examples of reactive groups capable of forming a bond to nitrogen are
well known to one skilled in the art of organic synthesis and non-limiting examples include an alkyl halide, vinyl halide, aldehyde group or a vicinal dihalide. Z represents an appropriate aryl coupling partner which will be well known to one skilled in the art of organic chemistry. An example of aryl coupling partners include but are not limited to halide and triflate when the other partner is an arylboron or arylstannane derivative.
Tetracyclic Compounds of formula G8 can be prepared from suitably substituted indole derivatives of formula G6. An indole derivative of formula G6 is cyclized to provide tetracyclic Compounds of formula G7. Indole derivatives of formula G6 may be obtained commercially or prepared by using methods known to those skilled in the art of organic synthesis. In an illustrative example, the
Compounds of formula G6 can be made via dehydration of a hydrazide of formula Gl with a ketone of formula G2 to provide hydrazones of formula G3, which can then be cyclized in the presence of a strong acid such as PPA or a Lewis acid such as aluminum chloride, to provide the hydroxyl-substituted indole Compounds of formula G4. A Compound of formula G4 can then be reacted with an aldehyde of formula R3- CHO to provide the cyclized Compounds of formula G8, wherein G is -CHR3-0-.
Compounds of formula G7 can be made, for example, via the arylation of the 2-position of an indole of formula G5 with a coupling partner of formula G6. A Compound of formula G7 can then be cyclized by reacting Y and K' to provide the Compounds of formula G8. It will be obvious to one skilled in the art of organic synthesis that the Compounds of formulas G4 and G7 may undergo further functional group manipulations prior to cyclization as necessary in order to provide the scope of the Compounds of Formula (I).
Scheme 2 shows a method useful for making the Compounds of formula G12, which correspond to the Compounds of Formula (I), wherein B is phenyl; X and X' are each CH; Y and Y' are each N; and the group -U-V-W- is - C(R2)=CH-N-.
Scheme 2
Wherein D and D' are each independently C(R13)2, N(R4), S, O or Si(R16) 2; M and M' are each independently halo, triflate, boronic acid or boronic ester; PG is a protecting group, such as Boc or 4-methoxybenzyl; R4 is -C(0)Rn, -C(O)- [C(R7)2]qN(R6)2, -C(0)-[C(R7)2]q-Rn, -C(0)-[C(R7)2]qN(R6)C(0)-Rn, - C(0)[C(R7)2]qN(R6)S02-Rn, -C(0)-[C(R7)2]qN(R6)C(0)0-Rn or -C(O)- [C(R7)2]qC(0)0-Rn; and G, R1, R2 and R15 are defined above for the Compounds of Formula (I).
Scheme 3 shows a method useful for making the Compounds of formula G16, which correspond to the Compounds of Formula (I), wherein B is phenyl; X and X' are each CH; Y and Y' are each N; and the group -U-V-W- is - N=CH-N-.
Scheme 3
G16 G15
Wherein Z and Z' are each independently C(R
I3)
2, N(R
4), S, O or Si(R
16)
2; M and M' are each independently halo, triflate, boronic acid or boronic ester; X is halo; R
4 is -C(0)R
n, -C(0)-[C(R
7)
2]
qN(R
6)
2, -C(0)-[C(R
7)
2]
q-R
n, -C(0)-
-C(0)[C(R
7)
2]
qN(R
6)S0
2-R
1 -C(0)-[C(R
7)
2]
qN(R
6)C(0)0- R
11 or -C(0)-[C(R
7)
2]
qC(0)0-R
n; K, Q and Q' are defined above in connection with Scheme 1 ; and G, R
2 and R
15 are defined above for the Compounds of Formula (I).
A 2-amino aniline derivative of formula G12 can be reacted with an acyl halide of formula G13 to provide the 2-substituted benzimidazole Compounds of formula G14. The Compounds of formula G14.can be cyclized and derivatized to provide Compounds of formula G15, using at methods analogous to those described in Scheme 1 for the conversion of G6 to G8. A Compound of formula G15 can then be carried forth to the Compounds of formula G16 using methods analogous to those described in Scheme 2.
Scheme 4 shows a method useful for making the Compounds of formula G20, which correspond to the Compounds of Formula (I), wherein B is pyridyl; X and X' are each CH; Y and Y' are each N; and the group -U-V-W- is - C(R2)=CH-N-.
Scheme 4
G20
Wherein Z and Z' are each independently C(R13)2, N(R4), S, O or Si(R16) 2; M and M' are each independently halo, triflate, boronic acid or boronic ester; R4 is -C(0)Rn, -C(0)-[C(R7)2]qN(R6)2, -C(0)-[C(R7)2]q-Rn, -C(O)- [C(R7)2]qN(R6)C(0)-R' \ -C(0)[C(R7)2]qN(R6)S02-R1 -C(0)-[C(R7)2]qN(R6)C(0)0- R1 1 or -C(0)-[C(R7)2]qC(0)0-Ru; and G, R1 and R2 are defined above for the
Compounds of Formula (I).
A pyridyl hydrazone of formula G17 can be converted to the tetracyclic Compounds of formula G19 using methods analogous to those described in Scheme 1 for the conversion of G3 to G8. A Compound of formula G19 can then
be carried forth to the Compounds of G20 using methods analogous to those described in Scheme 2.
Scheme 5 shows methods useful for making the Compounds of formula G24, which are useful intermediates for making the Compounds of Formula (I) wherein X and X' are each CH and Y and Y' are each N.
Scheme 5
G23
Wherein Z or Z' is C(R13)2, N(R4), S, O or Si(R16)2; X is halo or inflate; and PG is a amino protecting group, such as Boc or 4-methoxybenzyl..
An appropriately functionalized aldehyde of formula G21 can be reacted with glyoxal and ammonia to provide a substituted imidazole of formula G22. A Compound of formula G22 can subsequently be selectively mono-halogenated to provide a mono-halogenated imidazole Compound of formula G24. Alternatively, a Compound of formula G24 can subsequently be di-halogenated to provide a
Compound of formula G23, which is then selectively reduced to provide a mono- halogenated imidazole Compound of formula G24.
In some of the Compounds of Formula (I) contemplated in Schemes 1- 5, amino acids (such as, but not limited to proline, 4-(R)-fluoroproline, 4-(S)- fluoroproline, 4,4-difluoroproline, 4,4-dimethylsilylproline, aza-bicyclo[2.2.1]heptane carboxylic acid, aza-bicyclo[2.2.2]octane carboxylic acid, (S)-2-piperidine carboxylic acid, valine, alanine, norvaline, etc.) are incorporated as part of the structures.
Methods have been described in the organic chemistry literature as well as in
Banchard US 2009/0068140 (Published March 9th 2009) for the preparation of such amino acid-derived intermediates.
One skilled in the art of organic synthesis will recognize that the synthesis of fused tetracyclic cores contained in Compounds of Formula (I) may require protection of certain functional groups (i.e., derivatization for the purpose of
chemical compatibility with a particular reaction condition). Suitable protecting groups for the various functional groups of these Compounds and methods for their installation and removal are well known in the art of organic chemistry. A summary of many of these methods can be found in Greene et ah, Protective Groups in Organic Synthesis, Wiley-Interscience, New York, (1999).
One skilled in the art of organic synthesis will also recognize that one route for the synthesis of the fused tetracyclic cores of the Compounds of Formula (I) may be more desirable depending on the choice of appendage substituents.
Additionally, one skilled in the art will recognize that in some cases the order of reactions may differ from that presented herein to avoid functional group
incompatibilities and thus adjust the synthetic route accordingly.
One skilled in the art of organic synthesis will recognize that the synthesis of certain fused tetracyclic cores of the Compounds of Formula (I) require the construction of an amide bond. Methods useful for making such amide bonds, include but are not limited to, the use of a reactive carboxy derivative {e.g., an acid halide, or ester at elevated temperatures) or the use of an acid with a coupling reagent {e.g., HOBt, EDCI, DCC, HATU, PyBrop) with an amine.
The preparation of multicyclic intermediates useful for making the fused tetracyclic ring systems of the Compounds of Formula (I) have been described in the literature and in compendia such as "Comprehensive Heterocyclic Chemistry" editions I, II and III, published by Elsevier and edited by A.R. Katritzky & R. JK Taylor. Manipulation of the required substitution patterns have also been described in the available chemical literature as summarized in compendia such as
"Comprehensive Organic Chemistry" published by Elsevier and edited by DH R. Barton and W. D. Ollis; "Comprehensive Organic Functional Group Transformations" edited by edited by A.R. Katritzky & R. JK Taylor and "Comprehensive Organic Transformation" published by Wily-CVH and edited by R. C. Larock.
The Compounds Formula (I) may contain one or more silicon atoms. The Compounds contemplated in this invention in general can be prepared using the carba-analog methodology unless otherwise noted. A recent review of the synthesis of silicon containing Compounds can be found in "Silicon Chemistry: from Atom to Extended Systems", Ed P. Jutzi & U. Schubet; ISBN 978-3-527-30647-3.
Preparation of silyl containing amino acids has been described. See Bolm et al, Angew. Chem. Int Ed., 39:2289 (2000). Descriptions of improved cellular update
( Giralt, J. Am. Chem. Soc, 128:8479 (2006)) and reduced metabolic processing of silyl containing Compounds have been described ( Johansson et ah, Drug Metabolism & Disposition, 38:73 (2009)).
The starting materials used and the intermediates prepared using the methods set forth in Schemes 1-5 may be isolated and purified if desired using conventional techniques, including but not limited to filtration, distillation, crystallization, chromatography and alike. Such materials can be characterized using conventional means, including physical constants and spectral data.
Uses of the Tetracyclic Indole Derivatives
The Tetracyclic Indole Derivatives are useful in human and veterinary medicine for treating or preventing a viral infection in a patient. In one embodiment, the Tetracyclic Indole Derivatives can be inhibitors of viral replication. In another embodiment, the Tetracyclic Indole Derivatives can be inhibitors of HCV replication. Accordingly, the Tetracyclic Indole Derivatives are useful for treating viral infections, such as HCV. In accordance with the invention, the Tetracyclic Indole Derivatives can be administered to a patient in need of treatment or prevention of a viral infection.
Accordingly, in one embodiment, the invention provides methods for treating a viral infection in a patient comprising administering to the patient an effective amount of at least one Tetracyclic Indole Derivative or a pharmaceutically acceptable salt thereof.
Treatment or Prevention of a Flaviviridae Virus
The Tetracyclic Indole Derivatives can be useful for treating or preventing a viral infection caused by the Flaviviridae family of viruses.
Examples of Flaviviridae infections that can be treated or prevented using the present methods include but are not limited to, dengue fever, Japanese encephalitis, Kyasanur Forest disease, Murray Valley encephalitis, St. Louis encephalitis, Tick-borne encephalitis, West Nile encephalitis, yellow fever and Hepatitis C Virus (HCV) infection.
In one embodiment, the Flaviviridae infection being treated is hepatitis C virus infection.
Treatment or Prevention of HCV Infection
The Tetracyclic Indole Derivatives are useful in the inhibition of HCV (e.g., HCV NS5A), the treatment of HCV infection and/or reduction of the likelihood or severity of symptoms of HCV infection and the inhibition of HCV viral replication and/or HCV viral production in a cell-based system. For example, the Tetracyclic Indole Derivatives are useful in treating infection by HCV after suspected past exposure to HCV by such means as blood transfusion, exchange of body fluids, bites, accidental needle stick, or exposure to patient blood during surgery or other medical procedures.
In one embodiment, the hepatitis C infection is acute hepatitis C. In another embodiment, the hepatitis C infection is chronic hepatitis C.
Accordingly, in one embodiment, the invention provides methods for treating HCV infection in a patient, the methods comprising administering to the patient an effective amount of at least one Tetracyclic Indole Derivative or a pharmaceutically acceptable salt thereof. In a specific embodiment, the amount administered is effective to treat or prevent infection by HCV in the patient. In another specific embodiment, the amount administered is effective to inhibit HCV viral replication and/or viral production in the patient.
The Tetracyclic Indole Derivatives are also useful in the preparation and execution of screening assays for antiviral compounds. For example the
Tetracyclic Indole Derivatives are useful for identifying resistant HCV replicon cell lines harboring mutations within NS5A, which are excellent screening tools for more powerful antiviral compounds. Furthermore, the Tetracyclic Indole Derivatives are useful in establishing or determining the binding site of other antivirals to the HCV replicase.
The compositions and combinations of the present invention can be useful for treating a patient suffering from infection related to any HCV genotype. HCV types and subtypes may differ in their antigenicity, level of viremia, severity of disease produced, and response to interferon therapy as described in Holland et al, Pathology, 30(21:192-195 (1998). The nomenclature set forth in Simmonds et al, J Gen Virol, 74(Ptl l):2391-2399 (1993) is widely used and classifies isolates into six major genotypes, 1 through 6, with two or more related subtypes, e.g., la and lb. Additional genotypes 7-10 and 1 1 have been proposed, however the phylogenetic basis on which this classification is based has been questioned, and thus types 7, 8, 9 and 11 isolates have been reassigned as type 6, and type 10 isolates as type 3 (see
Lamballerie et al, J Gen Virol, 78(Ptl):45-51 (1997)). The major genotypes have been defined as having sequence similarities of between 55 and 72% (mean 64.5%), and subtypes within types as having 75%-86% similarity (mean 80%) when sequenced in the NS-5 region (see Simmonds et al, J Gen Virol, 75(Pt 5^:1053-1061 (1994)).
Combination Therapy
In another embodiment, the present methods for treating or preventing HCV infection can further comprise the administration of one or more additional therapeutic agents which are not Tetracyclic Indole Derivatives.
In one embodiment, the additional therapeutic agent is an antiviral agent.
In another embodiment, the additional therapeutic agent is an immunomodulatory agent, such as an immunosuppressive agent.
Accordingly, in one embodiment, the present invention provides methods for treating a viral infection in a patient, the method comprising
administering to the patient: (i) at least one Tetracyclic Indole Derivative, or a pharmaceutically acceptable salt thereof, and (ii) at least one additional therapeutic agent that is other than a Tetracyclic Indole Derivative, wherein the amounts administered are together effective to treat or prevent a viral infection.
When administering a combination therapy of the invention to a patient, therapeutic agents in the combination, or a pharmaceutical composition or
compositions comprising therapeutic agents, may be administered in any order such as, for example, sequentially, concurrently, together, simultaneously and the like. The amounts of the various actives in such combination therapy may be different amounts (different dosage amounts) or same amounts (same dosage amounts). Thus, for non- limiting illustration purposes, a Tetracyclic Indole Derivative and an additional therapeutic agent may be present in fixed amounts (dosage amounts) in a single dosage unit {e.g., a capsule, a tablet and the like).
In one embodiment, the at least one Tetracyclic Indole Derivative is administered during a time when the additional therapeutic agent(s) exert their prophylactic or therapeutic effect, or vice versa.
In another embodiment, the at least one Tetracyclic Indole Derivative and the additional therapeutic agent(s) are administered in doses commonly employed when such agents are used as monotherapy for treating a viral infection.
In another embodiment, the at least one Tetracyclic Indole Derivative and the additional therapeutic agent(s) are administered in doses lower than the doses commonly employed when such agents are used as monotherapy for treating a viral infection.
In still another embodiment, the at least one Tetracyclic Indole Derivative and the additional therapeutic agent(s) act synergistically and are administered in doses lower than the doses commonly employed when such agents are used as monotherapy for treating a viral infection.
In one embodiment, the at least one Tetracyclic Indole Derivative and the additional therapeutic agent(s) are present in the same composition. In one embodiment, this composition is suitable for oral administration. In another embodiment, this composition is suitable for intravenous administration. In another embodiment, this composition is suitable for subcutaneous administration. In still another embodiment, this composition is suitable for parenteral administration.
Viral infections and virus-related disorders that can be treated or prevented using the combination therapy methods of the present invention include, but are not limited to, those listed above.
In one embodiment, the viral infection is HCV infection.
The at least one Tetracyclic Indole Derivative and the additional therapeutic agent(s) can act additively or synergistically. A synergistic combination may allow the use of lower dosages of one or more agents and/or less frequent administration of one or more agents of a combination therapy. A lower dosage or less frequent administration of one or more agents may lower toxicity of therapy without reducing the efficacy of therapy.
In one embodiment, the administration of at least one Tetracyclic Indole Derivative and the additional therapeutic agent(s) may inhibit the resistance of a viral infection to these agents.
Non-limiting examples of additional therapeutic agents useful in the present compositions and methods include an interferon, an immunomodulator, a viral replication inhibitor, an antisense agent, a therapeutic vaccine, a viral polymerase inhibitor, a nucleoside inhibitor, a viral protease inhibitor, a viral helicase inhibitor, a
virion production inhibitor, a viral entry inhibitor, a viral assembly inhibitor, an antibody therapy (monoclonal or polyclonal), and any agent useful for treating an R A-dependent polymerase-related disorder.
In one embodiment, the additional therapeutic agent is a viral protease inhibitor.
In another embodiment, the additional therapeutic agent is a viral replication inhibitor.
In another embodiment, the additional therapeutic agent is an HCV NS3 protease inhibitor.
In still another embodiment, the additional therapeutic agent is an HCV NS5B polymerase inhibitor.
In another embodiment, the additional therapeutic agent is a nucleoside inhibitor.
In another embodiment, the additional therapeutic agent is an interferon.
In yet another embodiment, the additional therapeutic agent is an HCV replicase inhibitor.
In another embodiment, the additional therapeutic agent is an antisense agent.
In another embodiment, the additional therapeutic agent is a therapeutic vaccine.
In a further embodiment, the additional therapeutic agent is a virion production inhibitor.
In another embodiment, the additional therapeutic agent is an antibody therapy.
In another embodiment, the additional therapeutic agent is an HCV
NS2 inhibitor.
In still another embodiment, the additional therapeutic agent is an HCV NS4A inhibitor.
In another embodiment, the additional therapeutic agent is an HCV NS4B inhibitor.
In another embodiment, the additional therapeutic agent is an HCV NS5A inhibitor
In yet another embodiment, the additional therapeutic agent is an HCV NS3 helicase inhibitor.
In another embodiment, the additional therapeutic agent is an HCV IRES inhibitor.
In another embodiment, the additional therapeutic agent is an HCV p7 inhibitor.
In a further embodiment, the additional therapeutic agent is an HCV entry inhibitor.
In another embodiment, the additional therapeutic agent is an HCV assembly inhibitor.
In one embodiment, the additional therapeutic agents comprise a viral protease inhibitor and a viral polymerase inhibitor.
In still another embodiment, the additional therapeutic agents comprise a viral protease inhibitor and an immunomodulatory agent.
In yet another embodiment, the additional therapeutic agents comprise a polymerase inhibitor and an immunomodulatory agent.
In another embodiment, the additional therapeutic agents comprise a viral protease inhibitor and a nucleoside.
In another embodiment, the additional therapeutic agents comprise an immunomodulatory agent and a nucleoside.
In one embodiment, the additional therapeutic agents comprise an HCV protease inhibitor and an HCV polymerase inhibitor.
In another embodiment, the additional therapeutic agents comprise a nucleoside and an HCV NS5A inhibitor.
In another embodiment, the additional therapeutic agents comprise a viral protease inhibitor, an immunomodulatory agent and a nucleoside.
In a further embodiment, the additional therapeutic agents comprise a viral protease inhibitor, a viral polymerase inhibitor and an immunomodulatory agent.
In another embodiment, the additional therapeutic agent is ribavirin.
HCV polymerase inhibitors useful in the present compositions and methods include, but are not limited to, VP- 19744 (Wyeth/ViroPharma), PSI-7851 (Pharmasset), RG7128 (Roche/Pharmasset), PSI-938 (Pharmasset), PSI-7977
(Pharmasset), PF-868554/filibuvir (Pfizer), VCH-759 (ViroChem Pharma), HCV-796 (Wyeth/ViroPharma), IDX-184 (Idenix), IDX-375 (Idenix), NM-283
(Idenix/Novartis), R-1626 (Roche), MK-0608 (Isis/Merck), I X-8014 (Inhibitex), ΓΝΧ-8018 (Inhibitex), ΓΝΧ-189 (Inhibitex), GS 9190 (Gilead), A-848837 (Abbott), ABT-333 (Abbott), ABT-072 (Abbott), A-837093 (Abbott), BI-207127 (Boehringer- Ingelheim), BILB-1941 (Boehringer-Ingelheim), MK-3281 (Merck), VCH222 (ViroChem), VCH916 (ViroChem), VCH716(ViroChem), GSK-71185 (Glaxo SmithKline), ANA598 (Anadys), GSK-625433 (Glaxo SmithKline), XTL-2125 (XTL Biopharmaceuticals), and those disclosed in Ni et al., Current Opinion in Drug Discovery and Development, 7(4):446 (2004); Tan et al., Nature Reviews,1:867 (2002); and Beaulieu et al., Current Opinion in Investigational Drugs, 5:838 (2004).
Other HCV polymerase inhibitors useful in the present compositions and methods include, but are not limited to, those disclosed in International
Publication Nos. WO 08/082484, WO 08/082488, WO 08/083351, WO 08/136815, WO 09/0321 16, WO 09/032123, WO 09/032124 and WO 09/032125.
Interferons useful in the present compositions and methods include, but are not limited to, interferon alfa-2a, interferon alfa-2b, interferon alfacon-1 and PEG- interferon alpha conjugates. "PEG-interferon alpha conjugates" are interferon alpha molecules covalently attached to a PEG molecule. Illustrative PEG-interferon alpha conjugates include interferon alpha-2a (Roferon™, Hoffman La-Roche, Nutley, New Jersey) in the form of pegylated interferon alpha-2a (e.g., as sold under the trade name Pegasys™), interferon alpha-2b (Intron™, from Schering-Plough Corporation) in the form of pegylated interferon alpha-2b (e.g., as sold under the trade name PEG- Intron™from Schering-Plough Corporation), interferon alpha-2b-XL (e.g., as sold under the trade name PEG-Intron™), interferon alpha-2c (Berofor Alpha™,
Boehringer Ingelheim, Ingelheim, Germany), PEG-interferon lambda (Bristol-Myers Squibb and ZymoGenetics), interferon alfa-2b alpha fusion polypeptides, interferon fused with the human blood protein albumin (Albuferon™, Human Genome
Sciences), Omega Interferon (Intarcia), Locteron controlled release interferon (Biolex/OctoPlus), Biomed-510 (omega interferon), Peg-IL-29 (ZymoGenetics), Locteron CR (Octoplus), IFN-a-2b-XL (Flamel Technologies), and consensus interferon as defined by determination of a consensus sequence of naturally occurring interferon alphas (Infergen™, Amgen, Thousand Oaks, California).
Antibody therapy agents useful in the present compositions and methods include, but are not limited to, antibodies specific to IL-10 (such as those disclosed in US Patent Publication No. US2005/0101770, humanized 12G8, a
humanized monoclonal antibody against human IL-10, plasmids containing the nucleic acids encoding the humanized 12G8 light and heavy chains were deposited with the American Type Culture Collection (ATCC) as deposit numbers PTA-5923 and PTA-5922, respectively), and the like).
Examples of viral protease inhbitors useful in the present compositions and methods include, but are not limited to, an HCV protease inhibitor.
HCV protease inhibitors useful in the present compositions and methods include, but are not limited to, those disclosed in U.S. Patent Nos. 7,494,988, 7,485,625, 7,449,447, 7,442,695, 7,425,576, 7,342,041, 7,253,160, 7,244,721, 7,205,330, 7,192,957, 7,186,747, 7,173,057, 7,169,760, 7,012,066, 6,914,122, 6,911,428, 6,894,072, 6,846,802, 6,838,475, 6,800,434, 6,767,991, 5,017,380, 4,933,443, 4,812,561 and 4,634,697; U.S. Patent Publication Nos. US20020068702, US20020160962, US20050119168, US20050176648, US20050209164,
US20050249702 and US20070042968; and International Publication Nos. WO 03/006490, WO 03/087092, WO 04/092161 and WO 08/124148.
Additional HCV protease inhibitors useful in the present compositions and methods include, but are not limited to, SCH503034 (Boceprevir, Schering- Plough), SCH900518 (Schering-Plough), VX-950 (Telaprevir, Vertex), VX-500 (Vertex), VX-813 (Vertex), VBY-376 (Virobay), MK-7009 (Merck), MK-5172 (Merck), BI-201335 (Boehringer Ingelheim), TMC-435 (Medivir/Tibotec), ABT-450 (Abbott), TMC-435350 (Medivir), ITMN-191/R7227 (InterMune/Roche), EA-058 (Abbott/Enanta), EA-063 (Abbott/Enanta), GS-9132 (Gilead/Achillion), ACH-1095 (Gilead/Achillon), IDX-136 (Idenix), IDX-316 (Idenix), ITMN-8356 (InterMune), ITMN-8347 (InterMune), ITMN-8096 (InterMune), ITMN-7587 (InterMune), BMS- 650032 (Bristol-Myers Squibb), VX-985 (Vertex) and PHX1766 (Phenomix).
Further examples of HCV protease inhbitors useful in the present compositions and methods include, but are not limited to, those disclosed in Landro et al, Biochemistry, 36(31 :9340-9348 (1997); Ingallinella et al, Biochemistry,
37(25):8906-8914 (1998); Llinas-Brunet et al, Bioorg Med Chem Lett, 8(13):1713- 1718 (1998); Martin et al, Biochemistry, 37(33):! 1459-11468 (1998); Dimasi et al, J Virol, 71(10):7461-7469 (1997); Martin et al, Protein Eng, 10(5):607-614 (1997); Elzouki et al, J Hepat, 27(l):42-48 (1997); BioWorld Today, 9(217):4 (November 10, 1998); U.S. Patent Publication Nos. US2005/0249702 and US 2007/0274951; and
International Publication Nos. WO 98/14181, WO 98/17679, WO 98/17679, WO 98/22496 and WO 99/07734 and WO 05/087731.
Further examples of HCV protease inhibitors useful in the present compositions and methods include, but are not limited to, the following compounds:
Viral replication inhibitors useful in the present compositions and methods include, but are not limited to, HCV replicase inhibitors, IRES inhibitors, NS4A inhibitors, NS3 helicase inhibitors, NS5A inhibitors, NS5B inhibitors, ribavirin, AZD-2836 (Astra Zeneca), BMS-790052 (Bristol-Myers Squibb, see Gao et al, Nature, 465:96-100 (2010)), viramidine, A-831 (Arrow Therapeutics); an antisense agent or a therapeutic vaccine.
HCV NS4A inhibitors useful in the useful in the present compositions and methods include, but are not limited to, those disclosed in U.S. Patent Nos.
7,476,686 and 7,273,885; U.S. Patent Publication No. US20090022688; and
International Publication Nos. WO 2006/019831 and WO 2006/019832. Additional HCV NS4A inhibitors useful in the useful in the present compositions and methods include, but are not limited to, AZD2836 (Astra Zeneca) and ACH-806 (Achillon Pharmaceuticals, New Haven, CT).
HCV replicase inhibitors useful in the useful in the present compositions and methods include, but are not limited to, those disclosed in U.S. Patent Publication No. US20090081636.
Therapeutic vaccines useful in the present compositions and methods include, but are not limited to, IC41 (Intercell Novartis), CSL123 (Chiron/CSL), GI 5005 (Globeimmune), TG-4040 (Transgene), GNI-103 (GENimmune), Hepavaxx C (ViRex Medical), ChronVac-C (Inovio/Tripep), PeviPROTM (Pevion Biotect), HCV/MF59 (Chiron Novartis) and Civacir (NABI).
Examples of further additional therapeutic agents that may be useful in the present compositions and methods include, but are not limited to, Ritonavir (Abbott), TT033 (Benitec/Tacere Bio/Pfizer), Sirna-034 (Sirna Therapeutics), GNI- 104 (GENimmune), GI-5005 (Globeimmune), IDX-102 (Idenix), Levovirin™ (ICN Pharmaceuticals, Costa Mesa, California); Humax (Genmab), ITX-2155
(Ithrex/Novartis), PRO 206 (Progenies), HepaCide-I (NanoVirocides), MX3235 (Migenix), SCY-635 (Scynexis); PE02003002 (Kemin Pharma), Lenocta (VioQuest Pharmaceuticals), IET - Interferon Enhancing Therapy (Transition Therapeutics), Zadaxin (SciClone Pharma), VP 50406™ (Viropharma, Incorporated, Exton, Pennsylvania); Taribavirin (Valeant Pharmaceuticals); Nitazoxanide (Romark); Debio 025 (Debiopharm); GS-9450 (Gilead); PF-4878691 (Pfizer); ANA773 (Anadys); SCV-07 (SciClone Pharmaceuticals); NIM-881 (Novartis); ISIS 14803™ (ISIS Pharmaceuticals, Carlsbad, California); Heptazyme™ (Ribozyme Pharmaceuticals, Boulder, Colorado); Thymosin™ (SciClone Pharmaceuticals, San Mateo, California); Maxamine™ (Maxim Pharmaceuticals, San Diego, California); NKB-122 (JenKen Bioscience Inc., North Carolina); Alinia (Romark Laboratories), INFORM- 1 (a combination of R7128 and ITMN-191); and mycophenolate mofetil (Hoffman- LaRoche, Nutley, New Jersey).
The doses and dosage regimen of the other agents used in the combination therapies of the present invention for the treatment or prevention of HCV infection can be determined by the attending clinician, taking into consideration the approved doses and dosage regimen in the package insert; the age, sex and general health of the patient; and the type and severity of the viral infection or related disease or disorder. When administered in combination, the Tetracyclic Indole Derivative(s) and the other agent(s) can be administered simultaneously (i.e., in the same composition or in separate compositions one right after the other) or sequentially. This particularly useful when the components of the combination are given on different dosing schedules, e.g., one component is administered once daily and another component is administered every six hours, or when the preferred
pharmaceutical compositions are different, e.g., one is a tablet and one is a capsule. A kit comprising the separate dosage forms is therefore advantageous.
Generally, a total daily dosage of the at least one Tetracyclic Indole Derivative(s) alone, or when administered as combination therapy, can range from about 1 to about 2500 mg per day, although variations will necessarily occur depending on the target of therapy, the patient and the route of administration. In one embodiment, the dosage is from about 10 to about 1000 mg/day, administered in a single dose or in 2-4 divided doses. In another embodiment, the dosage is from about 1 to about 500 mg/day, administered in a single dose or in 2-4 divided doses. In still another embodiment, the dosage is from about 1 to about 100 mg/day, administered in a single dose or in 2-4 divided doses. In yet another embodiment, the dosage is from about 1 to about 50 mg/day, administered in a single dose or in 2-4 divided doses. In another embodiment, the dosage is from about 500 to about 1500 mg/day,
administered in a single dose or in 2-4 divided doses. In still another embodiment, the dosage is from about 500 to about 1000 mg/day, administered in a single dose or in 2- 4 divided doses. In yet another embodiment, the dosage is from about 100 to about 500 mg/day, administered in a single dose or in 2-4 divided doses.
In one embodiment, when the additional therapeutic agent is INTRON- A interferon alpha 2b (commercially available from Schering-Plough Corp.), this agent is administered by subcutaneous injection at 3MIU(12 mcg)/0.5mL/TIW for 24 weeks or 48 weeks for first time treatment.
In another embodiment, when the additional therapeutic agent is PEG- INTRON interferon alpha 2b pegylated (commercially available from Schering-
Plough Corp.), this agent is administered by subcutaneous injection at 1.5
mcg/kg/week, within a range of 40 to 150 meg/week, for at least 24 weeks.
In another embodiment, when the additional therapeutic agent is ROFERON A interferon alpha 2a (commercially available from Hoffmann-La Roche), this agent is administered by subcutaneous or intramuscular injection at 3MIU(1 1.1 mcg/mL)/TIW for at least 48 to 52 weeks, or alternatively 6MIU/TIW for 12 weeks followed by 3MIU/TIW for 36 weeks.
In still another embodiment, when the additional therapeutic agent is PEGASUS interferon alpha 2a pegylated (commercially available from Hoffmann-La Roche), this agent is administered by subcutaneous injection at 180 mcg/lmL or 180 mcg/0.5mL, once a week for at least 24 weeks.
In yet another embodiment, when the additional therapeutic agent is INFERGEN interferon alphacon-1 (commercially available from Amgen), this agent is administered by subcutaneous injection at 9 mcg/TIW is 24 weeks for first time treatment and up to 15 mcg/TIW for 24 weeks for non-responsive or relapse treatment.
In a further embodiment, when the additional therapeutic agent is Ribavirin (commercially available as REBETOL ribavirin from Schering-Plough or COPEGUS ribavirin from Hoffmann-La Roche), this agent is administered at a daily dosage of from about 600 to about 1400 mg/day for at least 24 weeks.
In one embodiment, one or more compounds of the present invention are administered with one or more additional therapeutic agents selected from: an interferon, an immunomodulator, a viral replication inhibitor, an antisense agent, a therapeutic vaccine, a viral polymerase inhibitor, a nucleoside inhibitor, a viral protease inhibitor, a viral helicase inhibitor, a viral polymerase inhibitor a virion production inhibitor, a viral entry inhibitor, a viral assembly inhibitor, an antibody therapy (monoclonal or polyclonal), and any agent useful for treating an RNA- dependent polymerase-related disorder.
In another embodiment, one or more compounds of the present invention are administered with one or more additional therapeutic agents selected from an HCV protease inhibitor, an HCV polymerase inhibitor, an HCV replication inhibitor, a nucleoside, an interferon, a pegylated interferon and ribavirin. The combination therapies can include any combination of these additional therapeutic agents.
In another embodiment, one or more compounds of the present invention are administered with one additional therapeutic agent selected from an HCV protease inhibitor, an interferon, a pegylated interferon and ribavirin.
In still another embodiment, one or more compounds of the present invention are administered with two additional therapeutic agents selected from an HCV protease inhibitor, an HCV replication inhibitor, a nucleoside, an interferon, a pegylated interferon and ribavirin.
In another embodiment, one or more compounds of the present invention are administered with an HCV protease inhibitor and ribavirin. In another specific embodiment, one or more compounds of the present invention are administered with a pegylated interferon and ribavirin.
In another embodiment, one or more compounds of the present invention are administered with three additional therapeutic agents selected from an HCV protease inhibitor, an HCV replication inhibitor, a nucleoside, an interferon, a pegylated interferon and ribavirin.
In one embodiment, one or more compounds of the present invention are administered with one or more additional therapeutic agents selected from an HCV polymerase inhibitor, a viral protease inhibitor, an interferon, and a viral replication inhibitor. In another embodiment, one or more compounds of the present invention are administered with one or more additional therapeutic agents selected from an HCV polymerase inhibitor, a viral protease inhibitor, an interferon, and a viral replication inhibitor. In another embodiment, one or more compounds of the present invention are administered with one or more additional therapeutic agents selected from an HCV polymerase inhibitor, a viral protease inhibitor, an interferon, and ribavirin.
In one embodiment, one or more compounds of the present invention are administered with one additional therapeutic agent selected from an HCV polymerase inhibitor, a viral protease inhibitor, an interferon, and a viral replication inhibitor. In another embodiment, one or more compounds of the present invention are administered with ribavirin.
In one embodiment, one or more compounds of the present invention are administered with two additional therapeutic agents selected from an HCV polymerase inhibitor, a viral protease inhibitor, an interferon, and a viral replication inhibitor.
In another embodiment, one or more compounds of the present invention are administered with ribavirin, interferon and another therapeutic agent.
In another embodiment, one or more compounds of the present invention are administered with ribavirin, interferon and another therapeutic agent, wherein the additional therapeutic agent is selected from an HCV polymerase inhibitor, a viral protease inhibitor, and a viral replication inhibitor.
In still another embodiment, one or more compounds of the present invention are administered with ribavirin, interferon and a viral protease inhibitor.
In another embodiment, one or more compounds of the present invention are administered with ribavirin, interferon and an HCV protease inhibitor.
In another embodiment, one or more compounds of the present invention are administered with ribavirin, interferon and boceprevir or telaprevir.
In a further embodiment, one or more compounds of the present invention are administered with ribavirin, interferon and an HCV polymerase inhibitor.
In another embodiment, one or more compounds of the present invention are administered with pegylated-interferon alpha and ribavirin.
In one embodiment, one or more compounds of the present invention are administered with from one to three additional therapeutic agents, wherein the additional therapeutic agents are each independently selected from HCV protease inhibitors, HCV NS5A inhibitors and HCV NS5B polymerase inhibitors.
In one embodiment, one or more compounds of the present invention are administered with MK-5172.
In another embodiment, one or more compounds of the present invention are administered with MK-7009.
In another embodiment, one or more compounds of the present invention are administered with boceprevir.
In still another embodiment, one or more compounds of the present invention are administered with telaprevir.
In another embodiment, one or more compounds of the present invention are administered with PSI-938.
In another embodiment, one or more compounds of the present invention are administered with PSI-7977.
In yet another embodiment, one or more compounds of the present invention are administered with RG-7128.
In one embodiment, one or more compounds of the present invention are administered with (i) a compound selected from PSI-7977, PSI-938 RG-7128; and (ii) a compound selected from boceprevir, telaprevir, MK-7009 and MK-5172.
In another embodiment, one or more compounds of the present invention are administered with PSI-7977 and MK-5172.
Compositions and Administration
Due to their activity, the Tetracyclic Indole Derivatives are useful in veterinary and human medicine. As described above, the Tetracyclic Indole
Derivatives are useful for treating or preventing HCV infection in a patient in need thereof.
When administered to a patient, the Tetracyclic Indole Derivatives can be administered as a component of a composition that comprises a pharmaceutically acceptable carrier or vehicle. The present invention provides pharmaceutical compositions comprising an effective amount of at least one Tetracyclic Indole Derivative and a pharmaceutically acceptable carrier. In the pharmaceutical compositions and methods of the present invention, the active ingredients will typically be administered in admixture with suitable carrier materials suitably selected with respect to the intended form of administration, i.e., oral tablets, capsules (either solid-filled, semi-solid filled or liquid filled), powders for constitution, oral gels, elixirs, dispersible granules, syrups, suspensions, and the like, and consistent with conventional pharmaceutical practices. For example, for oral administration in the form of tablets or capsules, the active drug component may be combined with any oral non-toxic pharmaceutically acceptable inert carrier, such as lactose, starch, sucrose, cellulose, magnesium stearate, dicalcium phosphate, calcium sulfate, talc, mannitol, ethyl alcohol (liquid forms) and the like. Solid form preparations include powders, tablets, dispersible granules, capsules, cachets and suppositories. Powders and tablets may be comprised of from about 0.5 to about 95 percent inventive composition. Tablets, powders, cachets and capsules can be used as solid dosage forms suitable for oral administration.
Moreover, when desired or needed, suitable binders, lubricants, disintegrating agents and coloring agents may also be incorporated in the mixture.
Suitable binders include starch, gelatin, natural sugars, corn sweeteners, natural and synthetic gums such as acacia, sodium alginate, carboxymethylcellulose, polyethylene glycol and waxes. Among the lubricants there may be mentioned for use in these dosage forms, boric acid, sodium benzoate, sodium acetate, sodium chloride, and the like. Disintegrants include starch, methylcellulose, guar gum, and the like.
Sweetening and flavoring agents and preservatives may also be included where appropriate.
Liquid form preparations include solutions, suspensions and emulsions and may include water or water-propylene glycol solutions for parenteral injection.
Liquid form preparations may also include solutions for intranasal administration.
Aerosol preparations suitable for inhalation may include solutions and solids in powder form, which may be in combination with a pharmaceutically acceptable carrier, such as an inert compressed gas.
Also included are solid form preparations which are intended to be converted, shortly before use, to liquid form preparations for either oral or parenteral administration. Such liquid forms include solutions, suspensions and emulsions.
For preparing suppositories, a low melting wax such as a mixture of fatty acid glycerides or cocoa butter is first melted, and the active ingredient is dispersed homogeneously therein as by stirring. The molten homogeneous mixture is then poured into convenient sized molds, allowed to cool and thereby solidify.
Additionally, the compositions of the present invention may be formulated in sustained release form to provide the rate controlled release of any one or more of the components or active ingredients to optimize therapeutic effects, i.e., antiviral activity and the like. Suitable dosage forms for sustained release include layered tablets containing layers of varying disintegration rates or controlled release polymeric matrices impregnated with the active components and shaped in tablet form or capsules containing such impregnated or encapsulated porous polymeric matrices.
In one embodiment, the one or more Tetracyclic Indole Derivatives are administered orally.
In another embodiment, the one or more Tetracyclic Indole Derivatives are administered intravenously.
In another embodiment, the one or more Tetracyclic Indole Derivatives are administered topically.
In still another embodiment, the one or more Tetracyclic Indole Derivatives are administered sublingually.
In one embodiment, a pharmaceutical preparation comprising at least one Tetracyclic Indole Derivative is in unit dosage form. In such form, the preparation is subdivided into unit doses containing effective amounts of the active components.
Compositions can be prepared according to conventional mixing, granulating or coating methods, respectively, and the present compositions can contain, in one embodiment, from about 0.1% to about 99% of the Tetracyclic Indole Derivative(s) by weight or volume. In various embodiments, the present
compositions can contain, in one embodiment, from about 1% to about 70% or from about 5% to about 60% of the Tetracyclic Indole Derivative(s) by weight or volume.
The quantity of Tetracyclic Indole Derivative in a unit dose of preparation may be varied or adjusted from about 1 mg to about 2500 mg. In various embodiments, the quantity is from about 10 mg to about 1000 mg, 1 mg to about 500 mg, 1 mg to about 100 mg, and 1 mg to about 100 mg.
For convenience, the total daily dosage may be divided and administered in portions during the day if desired. In one embodiment, the daily dosage is administered in one portion. In another embodiment, the total daily dosage is administered in two divided doses over a 24 hour period. In another embodiment, the total daily dosage is administered in three divided doses over a 24 hour period. In still another embodiment, the total daily dosage is administered in four divided doses over a 24 hour period.
The amount and frequency of administration of the Tetracyclic Indole Derivatives will be regulated according to the judgment of the attending clinician considering such factors as age, condition and size of the patient as well as severity of the symptoms being treated. Generally, a total daily dosage of the Tetracyclic Indole Derivatives range from about 0.1 to about 2000 mg per day, although variations will necessarily occur depending on the target of therapy, the patient and the route of administration. In one embodiment, the dosage is from about 1 to about 200 mg/day, administered in a single dose or in 2-4 divided doses. In another embodiment, the dosage is from about 10 to about 2000 mg/day, administered in a single dose or in 2-4 divided doses. In another embodiment, the dosage is from about 100 to about 2000 mg/day, administered in a single dose or in 2-4 divided doses. In still another
embodiment, the dosage is from about 500 to about 2000 mg/day, administered in a single dose or in 2-4 divided doses.
The compositions of the invention can further comprise one or more additional therapeutic agents, selected from those listed above herein. Accordingly, in one embodiment, the present invention provides compositions comprising: (i) at least one Tetracyclic Indole Derivative or a pharmaceutically acceptable salt thereof; (ii) one or more additional therapeutic agents that are not a Tetracyclic Indole Derivative; and (iii) a pharmaceutically acceptable carrier, wherein the amounts in the
composition are together effective to treat HCV infection.
In one embodiment, the present invention provides compositions comprising a Compound of Formula (I) and a pharmaceutically acceptable carrier.
In another embodiment, the present invention provides compositions comprising a Compound of Formula (I), a pharmaceutically acceptable carrier, and a second therapeutic agent selected from the group consisting of HCV antiviral agents, immunomodulators, and anti-infective agents.
In another embodiment, the present invention provides compositions comprising a Compound of Formula (I), a pharmaceutically acceptable carrier, and wto additional therapeutic agents, each of which are independently selected from the group consisting of HCV antiviral agents, immunomodulators, and anti-infective agents.
Kits
In one aspect, the present invention provides a kit comprising a therapeutically effective amount of at least one Tetracyclic Indole Derivative, or a pharmaceutically acceptable salt, solvate, ester or prodrug of said compound and a pharmaceutically acceptable carrier, vehicle or diluent.
In another aspect the present invention provides a kit comprising an amount of at least one Tetracyclic Indole Derivative, or a pharmaceutically acceptable salt, solvate, ester or prodrug of said compound and an amount of at least one additional therapeutic agent listed above, wherein the amounts of the two or more active ingredients result in a desired therapeutic effect. In one embodiment, the one or more Tetracyclic Indole Derivatives and the one or more additional therapeutic agents are provided in the same container. In one embodiment, the one or more
Tetracyclic Indole Derivatives and the one or more additional therapeutic agents are provided in separate containers.
EXAMPLES
General Methods
Solvents, reagents, and intermediates that are commercially available were used as received. Reagents and intermediates that are not commercially available were prepared in the manner as described below. Ή NMR spectra when reported, were obtained on either a Varian VNMR System 400 (400 MHz) or a Bruker Avance 500 (500 MHz) and resonances are reported as ppm downfield from Me4Si with number of protons, multiplicities, and coupling constants in Hertz indicated parenthetically. Where LC/MS data are presented, analyses was performed using an Agilent 6110A MSD or an Applied Biosystems API- 100 mass spectrometer and Shimadzu SCL-IOA LC column: Alltech platinum C18 column, 3 micron, 33 mm x 7mm ID; typical gradient flow: 0 minutes - 10% CH3CN, 5 minutes - 95% CH3CN, 5-7 minutes - 95% CH3CN, 7 minutes - stop. The retention time and observed parent ion are given. Chromatography was performed using partially automated systems manufactured by Gilson, ISCO or Biotage. Unless otherwise indicated, chromatography was performed using a gradient elution of hexanes/ethyl acetate, from 100% hexanes to 100% ethyl acetate.
EXAMPLE 1
Preparation of Compound Int-la
Int-la
To a solution of L-valine (10.0 g, 85.3 mmol) in 1M aqueous NaOH solution (86 mL) at room temperature was added solid sodium carbonate (4.60 g, 43.4 mmol). The reaction mixture was cooled to 0 °C (ice bath) and then methyl
chloroformate (7.20 mL, 93.6 mmol) was added dropwise over 20 minutes. The reaction mixture was then allowed to warm to room temperature, and allowed to stir at room temperature for an additional 4 hours. The reaction mixture was then diluted with diethyl
ether ( 100 mL), the resulting solution was cooled to at 0 °C, and then concentrated hydrochloric acid (18 mL, 216 mmol) was added slowly. The reaction was extracted with EtOAc (3 x 100 mL) and the combined organics were dried over MgSC^, filtered and concentrated in vacuo to provide Compound Int-la (13.5 g, 90%), which was used without further purification.
The following intermediates can be prepared by the reaction of L-valine or L- threonine with isopropyl chloroformate, 2-methoxyethyl chloroformate or with 1- methylcyclopropyl hydroxysuccinimide respectively as above.
Int-lb Int-lc
Int-ld Int-le
EXAMPLE 2
Preparation of Intermediate Compound Int-2a
Int-2a
To a solution of D-phenylglycine (10.0 g, 66.1 mmol) and NaOH (21.2 g, 265 mmol) in water (60 mL) at 0 °C was added methyl chloroformate (10.2 mL, 133 mmol) dropwise over 20 minutes. The resulting mixture was allowed to stir at 0 °C for 1 hour, then was acidified using concentrated hydrochloric acid (25 mL, 300 mmol). The acidic solution was extracted with EtOAc (3 x 100 mL) and the combined organics were
dried over MgS04, filtered and concentrated in vacuo to provide Compound Int-2a (12.6 g, 91%), which was used without further purification.
The following intermediates can be prepared by the reaction of glycine, L-Alanine and 4- F phenylglycine respectively with methyl chloroformate (Aldrich Inc.) using the method described above:
Int- 2b Int-2c Int-2d
EXAMPLE 3
Preparation of Intermediate Compound Int-3a
Int-3a
A solution of D-phenylglycine (20.0 g, 132 mmol), 37% aqueous formaldehyde (66 mL, 814 mmol) and 5 % Pd on carbon (8.0 g, mmol) in a mixture of methanol (80 mL) and 1 N HCl (60 mL) was placed on a hydrogenation shaker and shook under an atmosphere of 35-40 psi hydrogen for 4 hours. The reaction was then flushed with nitrogen, filtered through a Celite pad and concentrated in vacuo to provide
Compound Int-3a (29.7 g, quant.) as a white solid, which was used without further purification.
lnt-3b lnt-3c
To a solution of (R)-2-amino-2-(4-fluorophenyl)acetic acid (Int 3b) in MeOH (20 mL) at 0 °C was added sodium cyanoborohydride portionwise over ~ 20 minutes. The resulting mixture was allowed to stir for 10 minutes and then acetaldehyde was added dropwise via syringe over -10 minutes. The resulting solution was allowed to stir for 1 hour at 0 °C, and then allowed to warm to room temperature. After 12h, LC-MS indicated disappearance of Int-3b, and the mixture was recooled to 0 °C, carefully treated with water (3 mL) followed by addition of cone HCl over -40 minutes (pH -2.0). The cooling bath was removed and the mixture was allowed to stand for about 15 hours. The precipitate was collected by filtration to provide Int-3c.
Intermediate Int-3d can be prepared using the procedure above from R- Phenyl glycine.
Phenylglycine lnt-3d
EXAMPLE 4
Preparation of Intermediate Compound Int-4f
lnt-4d |nt-4e lnt-4f
Step A - Preparation of Compound Int-4b
To a solution of methyl 2-(benzyloxycarbonylamino)-2- (dimethoxyphosphoryl) acetate (10.0 g, 30.2 mmol, made as decribed in Hamada et al, Organic Letters; English, 20:4664-4667 (2009)) in THF (100 mL) at -20 °C was added tetramethylguanidine (4.20 mL, 33.2 mmol). The reaction mixture was allowed to stir at -20 °C for 1 hour then dihydro-2H-pyran-4(3H)-one (4a) was added (3.1 mL, 33.2 mmol) in THF (5 mL) and the reaction mixture was warmed to room temperature and allowed to stir for about 15 hours. EtOAc (200 mL) was added and the organic mixture was washed with water (3 χ 50 mL) and brine (50 mL). The organic layers were combined and dried with Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using flash chromatography on an ISCO 330 g Redi-Sep column using 0 35%
EtOAc/hexanes as the eluent to provide Compound Int-4b as a white solid (615 mg, 45%). !H NMR (CDC13) δ 7.40 7.30 (m, 5H), 6.00 (br s, 1H), 5.12 (s, 2H), 3.80 3.65 (m, 7H), 2.92 (m, 2H), 2.52 2.48 (m, 2H).
Step B - Preparation of Compound Int-4c
To a solution of Int-4b (2.43 g, 7.96 mmol) in methanol (160 mL) previously purged with N2 was added (-)-l,2-Bis((25,,51S)-2,5-dimethylphospholano)
ethane (cyclooctadiene)rhodium(I) tetrafluoroborate (487 mg, 0.880 mmol) under N2. The mixture was shaken in a Parr shaker apparatus for 18 hours at 50 psi of H2. After evacuating the hydrogen, the suspension was filtered and the filtrate was concentrated in vacuo to provide Compound Int-4c as a white solid (1.30 g, 53%). Ή NMR (CDC13) δ 7.40 7.30 (m, 5H), 5.32 (br s, 1H), 5.12 (s, 2H), 4.40 4.30 (m, 1H), 4.00 3.95 (m, 2H), 3.75 (s, 3H), 3.40 3.25 (m, 2H), 2.10 1.95 (m, 1H), 1.50 1.45 (m, 4H).
Step C - Preparation of Compound Int-4d
To a suspension of 50% palladium on carbon (10% wet, 200 mg) in absolute ethanol (20 mL) under nitrogen was added Int-4c (1.06 g, 3.45 mmol). With stirring, the solution was placed in vacuo for 30 seconds and then was opened to a hydrogen gas balloon for 2 hours. After evacuating the hydrogen, the suspension was filtered through a Celite pad and the pad was washed with ethanol (2 χ 20 mL). The filtrate was concentrated in vacuo to provide Compound Int-4d as a colorless oil (585 mg, 98%). !H NMR (CDC13) δ 4.06 3.96 (m, 2H), 3.73 (s, 3H), 3.48 3.28 (m, 3H), 1.92 1.78 (m, 1H), 1.61 1.47 (m, 6H).
Step D - Preparation of Compound Int-4e
To a solution of Compound Int-4d (585 mg, 3.37 mmol) and triethylamine (0.710 mL, 5.09 mmol) in CH2C12 (6 mL) was added methyl chloroformate (0.290 mL, 3.76 mmol). The reaction was allowed to stir at room temperature for about 15 hours, then water (15 mL) was added and the aqueous mixture was extracted with CH2C12 (3 x 20 mL). The combined organic extracts were dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using flash
chromatography on an ISCO 24 g Redi-Sep column using 0 3% MeOH/CH2Cl2 as the eluent to provide Compound Int-4e as a colorless oil (600 mg, 77%). Ή NMR (CDC13) 8 5.27 5.18 (m, 1H), 4.38 4.28 (m, 1H), 4.06 3.96 (m, 2H), 3.75 (s, 3H), 3.69 (s, 3H), 3.39 3.30 (m, 2H), 2.09 1.94 (m, 1H), 1.59 1.48 (m, 4H).
Step E - Preparation of Compound Int-4f
To a solution of Compound Int-4e (600 mg, 2.59 mmol) in THF (5 mL) was added lithium hydroxide monohydrate (218 mg, 5.19 mmol) in water (5 mL). The reaction was allowed to stir at room temperature for 2 hours then was concentrated in vacuo to half of its original volume. The concentrated mixture was then acidified with 6N HC1 and extracted with EtOAc (7 * 50 mL). The combined organic extracts were dried over Na2S04, filtered and concentrated in vacuo to provide Compound Int-4f as an off-white solid (485 mg, 86%). Ή NMR (CD3OD) δ 4.09 4.07 (m, 1H), 3.96 3.92 (m, 2H), 3.65 (s, 3H), 3.40 3.34 (m, 2H), 2.10 1.99 (m, 1H), 1.56 1.47 (m, 4H).
EXAMPLE 5
Preparation of Intermediate Compound Int-5f
lnt-5a lnt-5b lnt-5c
CI(CO)OCH3
lnt-5d int-5e lnt-5f
Step A - Preparation of Compound Int-Sa
To a solution of methyl 2-(benzyloxycarbonylamino)-2- (dimethoxyphosphoryl) acetate (1.50 g, 4.52 mmol) in THF (5 mL) at -20 °C was added tetramethylguanidine (625 μί, 4.98 mmol). The reaction mixture was allowed to stir at - 20 °C for 1 hour then tert-butyl 4-oxopiperidine-l-carboxylate was added (992 mg, 4.97 mmol) in THF (2 mL) and the reaction mixture was warmed to room temperature and allowed to stir for about 15 hours. EtOAc (90 mL) was added and the organic mixture was washed with water (3 χ 20 mL) and brine (25 mL). The combined organic extracts
were dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using flash chromatography on an ISCO 40 g Redi-Sep column using 0 35% EtOAc/hexanes as the eluent to provide Compound Int-5a as a white semi-solid (1.1 g, 61%). Ή NMR (CDC13) δ 7.40 7.30 (m, 5H), 6.02 (br s, 1H), 5.12 (s, 2H), 3.80 3.40 (m, 7H), 2.90 2.80 (m, 2H), 2.45 2.35 (m, 2H), 1.45 (s, 9H).
Step B - Preparation of Compound IntSb
To a solution of Int-5a (1.30 g, 3.21 mmol) in methanol (90 mL) previously purged with N2 was added (-)-l ,2-Bis((2S,5S)-2,5-dimethylphospholano) ethane(cyclooctadiene)rhodium(I) tetrafluoroborate (197 mg, 0.354 mmol) under N2. The mixture was then shaken in a Parr shaker apparatus for 18 hours at 50 psi of H2. After evacuating the hydrogen, the suspension was filtered and the filtrate was concentrated in vacuo to provide Compound Int-5b as colorless oil (1.00 g, 77%). Ή NMR (CDCI3) δ 7.40 7.30 (m, 5H), 5.35 5.25 (m, 1H), 5.10 (s, 2H), 4.40 4.35 (m, 1H), 4.20 4.10 (m, 2H), 3.70 (s, 3H), 2.70 2.55 (m, 2H), 2.00 1.90 (m, 1H), 1.65 1.40 (m, 11H), 1.30 1.20 (m, 2H).
Step C - Preparation of Compound Int-Sc
To a solution of 50% palladium on carbon (10% wet, 250 mg) in absolute ethanol (20 mL) under nitrogen was added Int-5b (1.00 g, 2.46 mmol). The reaction was evacuated, then put under an H2 atmosphere using a hydrogen-filled balloon and allowed to stir for 2 hours. The hydrogen was evacuated and the resulting suspension was filtered through a Celite pad and the pad washed with ethanol (2 χ 20 mL). The filtrate and ethanol washings were combined and concentrated in vacuo to provide Compound Int-5c as a colorless oil (670 mg, quant.). lH NMR (CDC13) δ 4.21 4.08 (m, 2H), 3.73 (s, 3H), 3.31 (d, J= 6.0 Hz, 1H), 2.75 2.57 (m, 2H), 1.84 1.70 (m, 1H), 1.68 1.56 (m, 1H), 1.45 (s, 9H), 1.45 1.20 (m, 5H).
Step D - Preparation of Compound Int-Sd
To a solution of Compound Int-5c (670 mg, 2.46 mmol) and triethylamine (0.520 mL, 3.73 mmol) in CH2C12 (10 mL) was added methyl chloroformate (0.210 mL,
2.72 mmol). The reaction mixture was allowed to stir at room temperature for about 15 hours. Water (20 mL) was added and the aqueous mixture was extracted with CH2CI2 (2 x 15 mL). The combined organic extracts were dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using flash chromatography on an ISCO 24 g Redi-Sep column using 0 3% MeOH/CH2Cl2 as the eluent to provide Compound Int-5d as an off-white solid (515 mg, 63%). Ή NMR (CDCI3) δ 5.26 5.17 (m, 1H), 4.38 4.30 (m, 1H), 4.20 4.07 (m, 2H), 3.75 (s, 3H), 3.68 (s, 3H), 2.71 2.57 (m, 2H), 2.00 1.85 (m, 1H), 1.87 1.48 (m, 2H), 1.44 (s, 9H), 1.35 1.18 (m, 2H).
Step E - Preparation of Compound JntSe
Compound Int-5d (300 mg, 0.908 mmol) was dissolved in a mixture of TFA (2 mL) and CH2C12 (10 mL) and the solution was allowed to stir at room
temperature for 1 hour, then was concentrated in vacuo. To the resulting residue was added triethylamine (0.760 mL, 5.45 mmol) in CH2CI2 (10 mL), then acetic anhydride (0.086 mL, 0.915 mmol). The reaction was allowed to stir at room temperature for about 15 hours then concentrated in vacuo. The residue obtained was purified using flash chromatography on an ISCO 12 g Redi-Sep column using 0 4% ΜεΟΗ/0¾02 as the eluent to provide Compound Int-5e as colorless oil (247 mg, 99%). *H NMR (CDC13) δ 5.27 5.21 (m, 1H), 4.73 4.62 (m, 1H), 4.42 4.32 (m, 1H), 3.69 (s, 3H), 3.18 (s, 3H), 3.18 3.09 (m, 1H), 3.07 2.95 (m ,1H), 2.55 2.41 (m, 1H), 2.07 (s, 3H), 1.78 1.49 (m, 3H), 1.38 1.21 (m, 2H).
Step F - Preparation of Compound Int-Sf
To a solution of Compound Int-5e (247 mg, 2.59 mmol) in THF (3 mL) was added lithium hydroxide monohydrate (77 mg, 1.83 mmol) in water (3 mL). The reaction mixture was allowed to stir at room temperature for about 15 hours then concentrated in vacuo to 50% of its original volume. The concentrated solution was then acidified with IN HC1 to pH 4 and extracted with EtOAc (7 x 15 mL). The combined organic extracts were dried over Na2S04, filtered and concentrated in vacuo to provide Compound Int-5f as an off-white solid (106 mg, 45%). Ή NMR (CD3OD) δ 5.52 5.43
(m, 1H), 4.71 4.62 (m, 1H), 4.44 4.31 (m, IH), 3.91 3.81 (M, IH), 3.70 (s, 3H), 3.12 2.99 (m, IH), 2.58 2.46 (m, IH), 2.10 (m, 4H), 1.86 1.54 (m, 2H), 1.50 1.21 (m, 3H).
EXAMPLE 6
Preparation of Intermediate Compound Int-6f
Int-6c
OH
LiOH-H20
}f9
09 N b
Boc
Int-6f
- Preparation of Compound Int-6c
A stirred mixture of D-(+)-a-methylbenzyl amine Int-6a (50.0 g, 0.412 mol), ethyl glyoxylate (81.5 mL, 50% in toluene, 0.412 mol) and PPTS (0.50 g, 2.00 mmol) in benzene (600 mL) was heated to reflux in a Dean-Stark apparatus and allowed to remain at reflux until no further water (~8 mL) azeotroped from the reaction (~ 4 hours). The resulting mixture was concentrated in vacuo to provide Compound Int-6b,
which was used without further purification: lH NMR (300 MHz, CDC13) δ 7.72 (s, 1H), 7.36 7.24 (m, 5H), 4.61 (q, J= 6.9 Hz, 1H), 4.35 (q, J= 7.2 Hz, 2H), 1.62 (d, J= 6.6 Hz, 3H), 1.34 (t, J = 7.2 Hz, 3H).
To a stirred solution of crude Int-6b in methylene chloride (600 mL) at - 78 °C were added the following in 10 minutesute intervals: TFA (31.0 mL, 0.416 mol), boron trifluoride etherate (51.3 mL, 0.416 mol) and freshly distilled cyclopentadiene (32.7 g, 0.494 mol). After less than 2 minutes following the addition of cyclopentadiene, the reaction mixture formed a thick brown mass, which was allowed to stir for 6 hours at -78 °C. The reaction mixture was then allowed to warm to room temperature on its own and stir for an additional 15 hours. The resulting dark brown reaction mixture was quenched with sat. aq. Na2CC>3 (~ 900 mL) and allowed to stir for 30 minutes. The resultant suspension was filtered through a pad of Celite® and the filtrate was extracted with methylene chloride (3 χ 100 mL). The combined organic extracts were washed with sat. aq. NaCl (2 χ 75 mL), dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using flash column chromatography (silica; 8 x 18 cm, 10% to 25% ethyl acetate/hexanes as the eluent) to provide endo Int-6c (10.9 g, 9%) as a brown oil: Ή NMR (300 MHz, CDC13) δ 7.34 7.19 (m, 5H), 6.00 5.95 (m, 1H), 4.18 (q, J= 7.1 Hz, 3H), 3.47 (s, 1H), 3.03 (s, 1H), 2.97 (q, J= 6.5 Hz, 1H), 2.41 (s, 1H), 1.86 (d, J= 8.2 Hz, 1H), 1.26 (t, J= 6.6 Hz, 3H), 1.17 (t, J= 6.6 Hz, 3H). Exo Int-6c (84.3 g, 74%) was also collected as a brown oil: Ή NMR (300 MHz, CDC13) δ 7.34 7.19 (m, 5H), 6.36 6.33 (m, 1H), 6.22 6.18 (m, 1H), 4.37 (s, 1H), 3.87 (q, J= 6.8 Hz, 2H), 3.10 (q, J= 6.5 Hz, 1H), 2.96 (s, 1H), 2.27 (s, 1H), 2.20 (d, J= 8.4 Hz, 1H), 1.48 (d, J= 6.5 Hz, 3H), 1.01 (d, J = 7.0 Hz, 3H), 1.00 (m, 1H).
Step B- Representative Example for the Preparation of Compound \nt-6d
A mixture of ejco-Int-6c (15.8 g, 0.582 mol) and 10% Pd/C (4.07 g, 50% wet) in a 1:2 mixture of EtOH/EtOAc (150 mL) was shaken for 23 hours in a Parr hydrogenation apparatus under an atmosphere of H2 (50 psi). The reaction mixture was then filtered through Celite® and the filtrate was concentrated in vacuo. Ή NMR analysis of the residue (10.8 g) showed some aromatic resonances present. Repetition of the hydrogenation procedure using 10% Pd/C (2.0 g) provided Int-6d (10.0 g, quant.) as
a brown oil, which was used without further purification. Ή NMR (300 MHz, CDC ) δ 4.18 (q, J= 7.2 Hz, 3H), 3.54 (s, 1H), 3.32 (s, 1H), 2.62 (s, 1H), 2.23 (s, 1H), 1.64 1.39 (m, 5H), 1.31 1.20 (m, 4H).
Step C - Preparation of Compound \at-6e
To a solution of Int-6d (36.6 g, 0.236 mol) and sat. aq. Na2C03 (300 mL) in THF (600 mL) at 0 °C was added di- rt-butyl dicarbonate (59.0 g, 0.270 mol). The resulting reaction was allowed to slowly warm to room temperature with stirring over 6 hours, then was allowed to stir at room temperature for an additional 68 hours. The reaction mixture was diluted with EtOAc (250 mL) and water (250 mL) and the aqueous layer was extracted with EtOAc (2 χ 200 mL). The combined organic extracts were washed with sat. aq. NaCl (2 χ 75 mL), dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using flash column chromatography (silica; 16 x 10 cm) using 10 20% ethyl acetate/hexanes as the eluent to provide Compound Int-6e (49.0 g, 84%) as a pale yellow oil: Ή NMR (300 MHz, CDCI3) δ 4.35 (s, 0.6H), 4.22 4.10 (m, 2.4H), 3.81 (s, 0.45H), 3.71 (s, 0.55H), 2.66 (s, 1H), 1.96 1.90 (m, 1H), 1.76 1.50 (m, 3H), 1.55 1.45 (m, 5H), 1.39 (s, 5H), 1.30 1.23 (m, 4H).
Step D- Preparation of Compound 2.2.1 Bicyclic Acid Intermediate lnt-6f
To a stirred mixture of Int-6e (49.0 g, 0.182 mmol) in 1:1 THF/water (600 mL) was added LiOH»H20 (15.3 g, 0.364 mol). The reaction mixture was heated to 60 °C and allowed to stir at this temperature for 47 hours. The reaction mixture was then cooled to room temperature, concentrated in vacuo, and the residue obtained was diluted with CH2CI2 (200 mL) then acidified with 2N HC1 to pH ~ 4. The acidic solution was extracted with CH2CI2 (4 x 100 mL) and the combined organic extracts were washed with sat. aq. NaCl (25 mL), dried over Na2S04, filtered and concentrated in vacuo to provide Compound Int-6f, (1R, 3S, 4S)-N-Boc-2-azabicyclo[2.2.1]heptane-3-carboxylic acid (41.2 g, 93%) as an off white solid, which was used without further purification: Ή NMR (400 MHz, DMSO- 6) δ 12.44 (s, 1H), 4.13 (s, 0.56H), 4.06 (s, 0.47H), 3.61 (d, J= 4.0 Hz, 1H), 2.59 (s, 1H), 1.75 1.45 (m, 5H), 1.39 (s, 4H), 1.32 (s, 5H), 1.23 (t, J= 8.4 Hz, 1H); Optical Rotation: [a]D 25 -169.0° (c = 1.1 , CHC13).
EXAMPLE 7
Preparation of Intermediate Compound Int-7h
lnt-7h
- Preparation of Compound Int-7b
lnt-7a lnt-7b
A 2 L, 3-necked round bottomed flask equipped with an overhead stirrer and a N2 inlet was charged with a solution of oxalyl chloride (130 mL, 0.26 mol) in dichloromethane (250 mL). The solution was cooled to -78 °C, and a solution of DMSO (20 mL, 0.28 mol) in dichloromethane (30 mL) was added dropwise. After 30 minutes, a solution of (S)-N-Boc-prolinol, Int-7a (40 g, 0.20 mol) in dichloromethane (200 mL) was added dropwise. After 30 minutes, triethylamine (140 mL, 1.00 mol) was added to the solution, and the flask was transferred to an ice/water bath and allowed to stir for another 30 minutes. The reaction mixture was diluted with dichloromethane (200 mL) and washed successively with H20, 1M HC1, saturated NaHC03, and brine. The organic layer was dried over Na2S04, filtered, and concentrated in vacuo to provide crude (5)-2- formyl-pyrrolidine-l-carboxylic acid ter/-butyl ester, Int-7b (40 g) as oil, which was used without further purification.
Step B - Preparation of Compound Int-7c
lnt-7b lnt-7c
To (5)-Boc-prolinal, Int-7b (crude, 80g, 0.4 mol) was added a solution of ammonia in MeOH (prepared from 150 mL of 7 N ammonia/MeOH and 200 mL MeOH, 1.05 mol, 260 mol%). An exotherm was noted with the internal temperature rising to ~ 30 °C. The solution was allowed to stir for 0.5 hours at room temperature, then glyoxal (76 g, 0.52 mol, 130 mole%) was added over 5 minutes in portions, with the internal temperature rising to ~ 60 °C and then returning to room temperature after 1 hour. The reaction was allowed to stir for an additional 15 hours and the reaction mixture was concentrated in vacuo. The resulting residue was diluted with dichloromethane (1 L) and water (0.5 L) were added and the organic phase was washed with water (0.25 L), dried over MgSC>4, filtered and concentrated in vacuo. The residue obtained was slurried with warm ethyl acetate ( ~ 100 mL) and Hexane (100 mL), then was cooled and filtered. The solid obtained was washed with 30%ethyl acetate/Hexane to provide Compound Int-7c (66.2g, 70% yield).
- Preparation of Compound Int-7d
lnt-7c lnt-7d
N-Bromosuccinimide (838.4 mg, 4.71 mmol) was added in portions over 15 minutes to a cooled (ice/water) CH2CI2 (20 mL) solution of imidazole Int-7c (1.06 g, 4.50 mmol). The reaction mixture was allowed to stir for 75 minutes and concentrated in
vacuo to oil. The residue obtained was purified using silica-gel RPLC (Acetonitrile/ water/ 0.1% TFA) to separate the mono bromide from its dibromo analog (over bromination) and the starting material. The RPLC elute was neutralized with excess Ν¾/ΜεΟΗ, and the volatile component was removed in vacuo. The residue obtained was partitioned between CH2CI2 and water, and the aqueous layer was extracted with water. The combined organic phase was dried (MgSC ), filtered, and concentrated in vacuo to provide Compound Int-7d as a white solid (374 mg). Ή NMR (DMSO) δ: 12.12 (br s, IH), 7.10 (m, IH), 4.70 (m, IH), 3.31 (m, IH; overlapped with water signal), 2.25-1.73 (m, 4H), 1.39/1.17 (s, 3.8H + 5.2H).
Step D - Alternative Synthesis of Int-7d
lnt-7b lnt-7e
To a suspension of Int-7b (140 g, 0.59 mol) in THF (2000 mL) was added N-bromosuccinimide (200 g, 1.1 mol). The mixture was allowed to stir at room temperature under N
2 gas for about 15 hours. The solvent was then removed in vacuo, and the residue obtained was purified using silica-gel chromatography (ethyl acetate eluent) to provide 230 g of the desired dibromo Compound Int-7e. MS (ESI) m/e (M+H
+): 396.
lnt-7e lnt-7d
To a suspension of Int-7e (230 g, 0.58 mol) in EtOH H20 (1: 1 ratio, 3000 mL) was added Na2S03 (733 g, 5.8 mol). The resulting mixture was allowed to stir at mild reflux for about 15 hours. After cooling to room temperature, the mixture was extracted with dichloromethane twice and the combined organic layer was concentrated
in vacuo to a semi-solid. The residue obtained was purified using chromatography on silica gel to provide the desired Compound Int-7d. MS (ESI) m/e (M+H+): 317.
- Preparation of Compound Int-7f
lnt-7e lnt-7f
Compound Int-7e (2.63 g, 5.0 mmol) was dissolved in THF (30 mL) and cooled to - 78 °C, n-BuLi (1M in hexane, 2.2 mL, 5.5 mmol) was added and the reaction was allowed to stir for 20 minutes. N-fluorodibenzenesulfonamide (1.6 mL, 5.0 mmol) was added at -78 °C and the reaction mixture was allowed to warm slowly to room temperature again. The reaction was quenched with aq. NH4CI then partitioned between water and ethyl acetate. The organic layer was dried over Na2S04 and concentrated in vacuo. The residue obtained was purified using flash column chromatography (Gradient Ethyl acetaterpetroleum ether from 0-20% Ethyl acetate) to provide Compound Int-7f. (63 % yield). MS (ESI) m/z (M+H)+: 464, 466. 19 F NMR = - 151.8 ppm .
Step F - Preparation of Compound Int-7g
lnt-7d lnt-7g
Intermediate 7d (2.51 g, 7.94 mmol, 1.0 eq) was dissolved in 20 mL of CH2CI2 and to the resulting solution was added trifluoroacetic acid (5 mL). The reaction mixture was allowed to stir for about 15 hours at room temperature under N2, and the reaction was diluted with hexanes (15 mL) and concentrated in vacuo to provide a yellow oil. CH2CI2 and toluene were added and the solution was re-concentrated in vacuo. This step was repeated until excess TFA was removed, giving a solid which was dried in vacuo for 1 hour to provide 3.5 g of solid Int-7g. MS (ESI) m/z (M+H)+:217/ 218.1.
- Preparation of Compound Int-7h
InMa lnt-7g lnt-7h
Int-7g (3.01g, 6.78 mmol, 1.0 eq) and Int-la (1.202 g, 6.86 mmol, 1.01 eq) were added to a 250 mL round-bottomed flask equipped with a stir bar. DMF was added, and the flask was connected to a vacuum line. The flask was cycled between vacuum and N2 twice, then cooled in an ice-methanol bath for 10 minutes. HATU (2.75 g, 7.23 mmol, 1.07 eq) was added, followed by diisopropylethyl amine (2.80 mL). The reaction mixture was allowed to stir at -15 °C for 20 minutes. Additional diisopropylethyl amine (2.0 mL) was added. The reaction mixture was allowed to stir for 40 minutes, then quenched with water (1.5 mL). The resulting solution was diluted with EtOAc (100 mL) and Et20 (100 mL), then washed with water (6 x 15 mL) and brine (2 x 25 mL). The organic layer was dried with MgS04, filtered, and concentrated in vacuo yielding 2.23 g of a clear oil. The residue obtained was purified via chromatography using an 80 g Isco Gold Si02 cartridge with a 0.5%-2.5% MeOH/ CH2C12 gradient as the mobile phase. The major peak was collected to provide 1.28 g Int-7h as a white foam. This material was further purified via sgc on an 80 g Isco Gold Si02 cartridge using a 45%-65% gradient of (5% methanol in EtOAc)/hexanes. Triethylamine 1% by volume was added to the MeOH/EtOAc solution. The fractions were assayed via TLC using Hanessian's stain. (See Example 13 below for more information on Hanessian's stain.) The major peak was collected as product to provide 1.18 g of Int-7h as a white foam. MS (ESI) m/z (M+H)+:373.1.
EXAMPLE 7B
Preparation of Intermediate Compound Int-7i
N-Moc-(S)-tetrahydropyranyl glycine (InMf) (252 mg, 1.160 mmol), Int-7g (354 mg, 1.225 mmol), DMF (6 mL), and DIPEA (0.7 mL, 4.01 mmol) were added to a 40 mL screw cap vial equipped with a stir bar. The reaction mixture was placed under a blanket of N2 and the vial was capped. The vial was cooled in an ice- methanol bath for 10 minutes. HATU (445 mg, 1.215 mmol) was added, and the reaction mixture was left stirring at -15 °C. After 3 hours, the bath temp was 10 °C. The reaction mixture was diluted with ethyl acetate and aqueous ammonium chloride. The layers were separated. The organic layer was washed with water and brine, gravity filtered, dried with MgS04, and filtered again. The solvent was evaporated under reduced pressure on the rotovap to provide a clear oil-(458 mg). The crude product was purified via flash silica gel column chromatography on an Isco 24 g Si02 Gold cartridge, using a
MeOH(NH3)/CH2Cl2 gradient (0-5%) as the mobile phase to provide Int-7h as a clear oil.
Weight = 246 mg Took 1 H NMR and LC/MS. Obsd M+H = 415.1.
N-Moc (S)-tetrahydropyranyl glycine In f (236 mg, 1.086 mmol) and Int-lOg (333 mg, 1.085 mmol), DMF (5 mL), and DIPEA (0.6 mL, 3.44 mmol) were added to a 40 mL screw cap vial equipped with a stir bar. The reaction mixture was placed under a blanket of N2 and the vial was capped. The vial was cooled in an ice- methanol bath for 15 minutes. HATU (418 mg, 1.141 mmol) was added, and the reaction mixture was left stirring at -15 °C. After 3h, the bath temp was 10 °C. The reaction mixture was diluted with ethyl acetate and water. The layers were separated. The organic
layer was washed with water and brine, gravity filtered, dried with MgSCH, and filtered again. The solvent was evaporated under reduced pressure on the rotovap to provide a clear oil. The crude product was dissolved in methanol and left standing at room temperature over the weekend.
The reaction mixture was concentrated in vacuo. The crude product was purified via flash silica gel column chromatography on an Isco 40 g Si02 Gold cartridge. The column was initially eluted (mistakenly) with a 0%-50% EtOAc/hexanes gradient, then flushed with 5% (MeOH/(l%NHD(Aq.)))/CH2Cl2 The fractions were combined to provide 0.50 g of impure product as a clear oil.
The impure product was purified via flash silica gel column chromatography on an Isco 24 g Si02 Gold cartridge, using a 0%-5% MeOH/CH2Cl2 gradient as the mobile phase to provide Int-7i as a clear oil-(0.306g). When a sample was dissolved in deuterated methanol, a white solid formed in the flask. Took Ή NMR and LC/MS. Obsd M+H = 433.1
EXAMPLE 8
Preparation of Intermediate Compound Int-8h
lnt-8h
- Preparation of Compound Int-8b
lnt-8a lnt-8b
A solution of Int-8a (1 1.0 g, 42.6 mmol) in THF (50 mL) was cooled to 0 °C and to the cooled solution was added EtMgBr (82 mmol). After addition was complete, the cooling bath was removed and the resulting reaction was allowed to stir at room temperature for 6 hours. 3 N HC1 was then added and the reaction mixture was
extracted with ethyl acetate (2 x 50 mL). The combined organic extracts were washed with water, brine, dried over Na2S04, and concentrated in vacuo. The residue obtained was purified using flash column chromatography on silica gel to provide Compound Int- 8b (7.5 g, 50% yield).
- Preparation of Compound Int-8c
lnt-8b lnt-8c
Int-8b (7.5 g, 21.3 mmol) was dissolved in 100 mL of dichloromethane and cooled to 0 °C. TFA (100 mL) was added and the reaction was allowed to stir to room temperature over 2h. The solvent was removed and the residue obtained was redissolved in EtOAc then washed with saturated bicarbonate solution then brine. The extracts were dried over magnesium sulfate, filtered and concentrated in vacuo to provide Compound Int-8c as an oil, which was used without further purification.
Step C - Preparation
To a solution of Compound Int-8c(4.2 g, 33 mmol) in THF (30 mL) was added Et3N (4.1 g, 49 mmol) and then trityl chloride (8.7 g, 40 mmol). The mixture was allowed to stir at room temperature for 2 hours, then concentrated in vacuo. The residue obtained was purified using flash chromatography on silica gel to provide Compound Int-8d (8.7 g, 71% yield). MS (ESI) m/z (M+H)+: 370.
Step D - Preparation of Compound Int-8e
To a solution of Compound Int-8d (3.6 g, 10.0 mmol) in THF (30 mL) was added LiHMDS (11.0 mmol) and then NBS (1.8 g, 10 mmol) at 0 °C. The mixture was allowed to stir at room temperature for 2 hours and then 3 N HCl was added to the mixture and the resulting solution was extracted with ethyl acetate (2 x 25 mL). The combined organic extracts were concentrated in vacuo and the residue obtained was purified using chromatography to provide Compound Int-8e (1.98 g, 44% yield). MS (ESI) m/z (M+H)+: 478, 480.
- Preparation of Compound Int-8f
To a solution of Compound Int-8e (3.6 g, 10.0 mmol) in THF (30 mL) was added LiHMDS (11.0 mmol) and then NBS (1.8 g, 10 mmol). The mixture was allowed to stir at room temperature for 2 hours and then 3 N HCl was added to the mixture and extracted with ethyl acetate twice. The organic layer was concentrated in vacuo. The residue obtained was purified using chromatography to provide the Int-8f (1.98 g, 44% yield). MS (ESI) m/z (M+H)+: 478, 480.
Preparation of Compound Int-8g
lnt-8f lnt-8g
To a solution of Compound Int-8f (3.9 g, 10 mmol) in chloroform (30 mL) was added NBS (1.76 g, 10 mmol) and the mixture was allowed to stir at room temperature for 2 hours. The reaction mixture was then concentrated in vacuo and the residue obtained was purified using flash chromatography to provide Compound Int-8g (2.2 g, 47% yield).
- Preparation of Compound Int-Sh
lnt-8g lnt-8h
To a solution of Compound Int-8g (1.28 g, 2.7 mmol) in dichloromethane (10 mL) was added TFA (10 mL) and the mixture was allowed to stir at room
temperature for 2 hours. Then the mixture was concentrated in vacuo and used in the next reaction directly. The residue obtained was dissolved in THF (20 mL) and ΕίβΝ (5 mL) and to the resulting solution was added BOC anhydride (590 mg, 2.7 mmol). The mixture was allowed to stir at room temperature for 2 hours and concentrated in vacuo. The residue obtained was purified using chromatography to provide Compound Int-8h (600 mg, 67% yield). MS (ESI) m/z (M+H)+: 331.
EXAMPLE 9
Preparation of Intermediate Compound Int-9g
lnt-9g
Step A - Preparation of Compound Int- 9b
lnt-9a lnt-9b
To a solution of Compound Int-9a (50 g, 0.2 mol) in THF (500 mL) and Et3N (20 mL) was added dropwise isopropyl chloroformate (25 g, 0.22 mol) at ice water bath. Then the resulting solution was allowed to warm to room temperature and allowed to stir for lh. Then a solution of CH2N2 (0.22 mol) in ether was added slowly until no N2 gas evolution was noted. Acetic acid (4 mL) was added and the reaction mixture was allowed to stir for 10 minutes. NaHCC>3 solution was then added and the reaction mixture extracted three times with ethyl acetate. The organic layers were combined, dried over Na2S04, and concentrated in vacuo to provide crude product. The crude product was then purified using column chromatography on silica gel (Pet Ether: EthylAcetate = 3:1) to provide Compound Int-9b (38 g, 70% yield)
Step B - Preparation of Compound Int-9c
lnt-9b int-9c
To a solution of Int-9b (38 g, 0.14 mol) in HOAc (20 mL) was added dropwise an aqueous HBr solution (11.2 g, 0.14 mol). After 10 minutes, the mixture was
poured into an aqueous NaHCC>3 solution and extracted three times with ethyl acetate. The combined organic extracts were washed with brine, water, dried over Na2SC>4 and concentrated in vacuo to provide the product Int-9c (30 g, 68% yield).
Step C - Preparation of Compound Int-9e
lnt-9c 9d lnt-9e
To a solution of Int-9c (10 g, 32 mmol) and Compound 9d (8.4 g, 64 mmol) in DMF (70 mL) was added K2C03 (18 g, 126 mmol). The mixture was allowed to stir at 100 °C in a sealed tube for about 15 hours. The solvent was removed and the residue obtained was purified using column chromatography on silica gel
(dichloromethane: MeOH = 20:1) to provide the product Int-9e. (6 g, 59% yield).
Step D - Preparation of Compound Int-9f
lnt-9e lnt-9f
To a solution Int-9e (4 g, 14.7 mmol) in THF (40 mL) was added NaH (6.6 g, 60 % content, 16.17 mmol) at 0 °C. The mixture was allowed to stir at room temperature for 30 minutes, and then cooled to 0 °C, and SEM-C1 (2.4 g, 14.7 mmol) added dropwise. The resulting mixture was allowed to stir at 0 °C for 2 hours. The solvent was removed in vacuo and the residue obtained was purified using column chromatography on silica gel (dichloromethane: MeOH =20:1) to provide the product Int-9f. (2 g, 34 % yield).
Step E - Preparation of Compound Int-9g
To a solution of Int-9f (2 g, 5 mmol) in THF (20 mL) was added dropwise n-BuLi (2.5 mL, 6.3 mmol) at -78 °C (bath) under N2 protection. The resulting solution was allowed to stir at this temperature for 30 minutes. Then a solution of NBS (0.89 g, 5 mmol) in THF (10 mL) was added dropwise at -78 °C. The mixture was allowed to stir at -78 °C for 1 hour and then aqueous NH4CI solution was added. The organic layer was separated and concentrated off to provide a crude residue, which was purified using column chromatography on silica gel (petroleum ether :EA=3: 1 as the eluent) to provide Compound Int-9g (400 mg, 16.5% yield).
EXAMPLE 10
Preparation of Intermediate Compound Int-lOf
Step A - Preparation of Compound Int-lOb
(2S,4R)- 1 -(½ /-butoxycarbonyl)-4-fluoropyrrolidine-2-carboxy lie acid (Int-lOa, 20 g, 85.75 mmol) was dissolved in anhydrous THF and cooled to 0 °C.
BH3 THF (1M in THF, 171 mL, 171 mmol) was added via an addition funnel. The
solution was gradually warmed up to room temperature and allowed to stir at room temperature for about 15 hours. MeOH was added until no bubbles came out. The solution was concentrated in vacuo and the residue obtained was purified using flash column chromatography on silica gel (330g, 0% to 60% of EtOAc in Hexane) to provide Compound Int-lOb (15.1 g, 80.3%)
Step B - Preparation of Compound Int-lOc
To a dry 1000 mL round bottom flask was added oxalyl chloride (7.50 mL, 88.9 mmol) and dry dichloromethane (250 mL). After the solution was cooled to -78 °C, DMSO (6.80 mL, 95.8 mmol) in dichloromethane (20 mL) was added dropwise. The solution was allowed to stir at -78 °C for 30 minutes. Int-lOb (15.0 g, 68.4 mmol) in dichloromethane (50 mL) was added via syringe. After the solution was allowed to stir at - 78 °C for 30 minutes, TEA (38.1 mL, 273.6 mmol) was added. The solution was allowed to stir at -78 °C for 30 minutes and at 0 °C for one hour. The solution was diluted with dichloromethane (300 mL) and washed with water, IN HC1, sat NaHCC>3, and brine. It was dried over anhydrous Na2S04, filtered and concentrated in vacuo. The residue obtained was dried in vacuo for 1 hour to provide Compound Int-lOc which was used without further purification.
Step C - Preparation of Compound Int-lOd
To a 1000 mL round bottom flask was added Int-lOc and NH3 (7N in MeOH, 150 mL). Glyoxal (15 mL, 40% in water, 131 mmol) was added slowly. The solution was allowed to stir at room temperature for about 15 hours. Additional glyoxal (5 mL, 44 mmol) was added and the reaction was allowed to stir at room temperature for another 24 hours. The solution was concentrated in vacuo and the residue obtained was purified using flash column chromatography on silica gel (240g, 0% to 5% of MeOH in dichloromethane, with 0.1% ΝΗ3Ή20) to provide Compound Int-lOd (8.5 g, 48.7% from 2)
Step D - Preparation of Compound Int-lOe
To a 100 mL round bottom flask was added Int-lOd (8.5 g, 33.3 mmol) and CH3CN (250 mL). More CH3CN was added to form a clear solution. NBS (1 1.3 g, 63.3 mmol) was added in one portion and the solution was allowed to stir at room temperature for about 15 hours. C¾CN was removed in vacuo and dichloromethane (50 mL) was added with stirring. The solid was filtered and washed with dichloromethane twice. The filtrate was concentrated in vacuo to about 30 mL and filtered again. The filtrate was purified using flash column chromatography on silica gel (120g, 20% to 80% of EtOAc in Hexane) to provide Compound Int-lOe (11.88 g, 86.4%).
Step E - Preparation of Compound Int-lOf
To a 1000 mL round bottom flask was added Int-IOd (11.88 g, 28.76 mmol), sodium sulfite (Na2S03, 36.0 g, 288 mmol), EtOH (270 mL) and water (130 mL). The solution was allowed to stir at reflux for about 15 hours. More Na2S03 (10 g, 79 mmol) was added and the solution was allowed to stir at reflux for another 24 hours. After cooling down, the solid was filtered and washed with EtOAc three times. The filtrate was concentrated in vacuo and the residue obtained was dissolved in a mixture of EtOAc (300 mL) and water (200 mL). The organic layer was separated and washed with brine, dried over anhydrous Na2S04, filtered, and concentrated in vacuo. The residue obtained was purified using flash column chromatography on silica gel (240g, 0% to 33% of EtOAc in Hexane) to provide Compound Int-lOf (5.12 g, 53.3%).
EXAMPLE 11
Preparation of Intermediate Compound Int-llc
Step A - Preparation of Compound Int-llb
Int-lla Int-llb
The aldehyde Int-lla was prepared from the commercially available alcohol using the method described in Example 10.
A flask was charged with aldehyde Int-lla (82g, 0.35 mol) and a 2.33 N ammonia/MeOH solution was added with good stirring (600 mL, 4.0 eq., prepared from 200ml 7N ammonia/MeOH diluted with 400 mL MeOH). The reaction was then heated to 35 °C and allowed to stir at this temperature for 2 hours, after which time a solution of 40 wt% glyoxal in water (80 mL, 2.0 eq.) was added dropwise over about 15 minutes. After stirring for an additional 2 hours, a solution of 7 ammonia/MeOH (100 mL, 2.0 eq.) was added and the reaction was allowed to stir at 35 °C for 1 hour. Additional glyoxal (40 mL, 1.0 eq.) was then added dropwise over 5 minutes and the resulting reaction was allowed to stir at 35 °C for 1 hour. The reaction mixture was then allowed to cool room temperature and stir for about 15 hours. Additional 7N ammonia/MeOH (50 mL, 1.0 eq.) was then added and the reaction reheated to 35 °C and allowed to stir at this temperature for 1 hour. An additional amount of glyoxal (20 mL, 0.5 eq.) was then added and the resulting reaction was allowed to stir at 35 °C for 1 hour, then the reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated in vacuo and the residue obtained was diluted with dichloromethane and water (2 L, 1: 1). The organic layer was separated, washed with 1L of water, then brine and dried (MgS04), filtered and concentrated in vacuo. The brown foam residue obtained was further purified using being passed through a short silica gel column to provide Compound Int- llb (60g, 62%).
Step B - Preparation of Compound Int-llc
Int-llc was prepared from Int-llb using the method described in
Example 10.
Intermediate Compounds Int-lld, Int-lle and Int-llf can be prepared using the methods described in Example 10 and Example 1 1.
lnt-1 1 d lnt-11e ,nt-11f
EXAMPLE 12
Preparation of Intermediate Compound Int-12i
Step A - Preparation of Compound Int-12b
lnt-12a lnt-12b
To a solution of Compound Int-12a (60 g, 0.24 mol) in dry THF (1 L) allowed to stir at -78 °C was added lithium hexamethyldisilazide (82 g, 0.49 mol, 1 M in THF). After the reaction mixture had been allowed to stir at -78 °C for 1 hour, the iodomethane (66 g, 0.46 mol) dissolved in dry THF (100 mL) was added at -78 °C and the
mixture was allowed to stir for 15 minutes at this temperature and 2 hours at 25 °C. The reaction mixture was quenched with saturated ammonium chloride solution and extracted with dichloromethane (3 x 300 mL). The combined organic phases were dried over MgS04, filtered, and concentrated in vacuo. The products were purified using flash column chromatography on silica gel to provide Compound Int-12b (18.3 g, 27% yield). Ή NMR 6: 4.38-4.34 (m, 1 H), 4.08-4.05 (m, 2 H), 2.09-2.03 (m, 1 H), 1.77-1.73 (m, 1 H), 1.35 (s, 9 H), 1.12 (t, J= 8 Hz, 3 H), 1.06 (s, 6 H).
- Preparation of Compound Int-12c
To a solution of Compound Int-12b (18.3 g, 60 mmol) in dichloromethane (150 mL) was added TFA (15 mL) and the mixture allowed to stir at room temperature for 30 minutes. The solvent was removed to provide Compound Int-12c (11.2 g, 100% yield).
Step C - Preparation of Compound Int-12d
lnt-12c lnt-12d
A suspension of L1AIH4 (16.2 g, 0.44 mol) and Compound Int-12c (11.2 g, 54.8 mmol) in THF (200 mL) was allowed to stir under reflux for 8 hours. After successive addition of 17 mL of water, 17 mL of 10% aq NaOH, and 51 mL of water, and filtration, the filtrate was concentrated in vacuo to provide Compound Int-12d (6.7 g, 94% yield).
Step D - Preparation of Compound Int-12e
lnt-12d lnt-12e
Compound Int-12D was dissolved in THF and ΕίβΝ, (Boc)20 were added.
The mixture was allowed to stir at room temperature for 2 hours and concentrated in vacuo. The residue obtained was purified using chromatography to provide Compound Int-12e (14 g, 100% yield).
Step E - Preparation of Compound Int-12f
lnt-12e lnt-12f
To a solution of Compound Int-12e (14g, 65.4 mmol) in dichloromethane was added Dess-Martin reagent (41.6 g, 98.1 mol). After stirring at room temperature for about 15 hours, the solvent was removed and the residue obtained was purified using flash column chromatography on silica gel to provide Compound Int-12f (7 g, 47% yield). Ή NMR δ: 9.40 (s, 1 H), 4.05-4.03 (m, 1 H), 3.14-3.1 1 (m, 2 H), 1.83-1.79 (m, 1 H), 1.66- 1.63 (m, 1 H), 1.36 (s, 9 H), 1.02 (s, 6 H).
Step F - Preparation of Compound Int-12g
lnt-12f lnt-12g
Glyoxal (1.75 mL of 40% in water) was added dropwise over 1 1 minutes to a solution of NH4OH (26 mL) and Compound Int-12f (6.1 g, 28.8 mmol) in methanol
and allowed to stir at room temperature for 19 hours. The volatile component was removed in vacuo and the residue obtained was purified using a flash chromatography on silica gel to provide Compound Int-12g (3 g, 39% yield).
MS (ESI) m/z (M+H)+: 266.
- Preparation of Compound Int-12h
A mixture of Compound Int-12g (2.2 g, 8.3 mmol), N-bromosuccinimide (2.66 g, 14.9 mmol) in anhydrous THF (80 mL) was heated at reflux for about 15 hours. After cooling to room temperature, the solids are removed by filtration and the filtrate was concentrated in vacuo and the residue obtained was purified using chromatography to provide Compound Int-12h (2.0 g, 57% yield). Ή NMR (J0001201 17 H10170-003-1 CDC13 varian 400 MHz) δ: 1 1.03 (s, 1 H), 4.79 (t, J= 8 Hz, 1 H), 3.25 (t, J= 12 Hz, 1 H), 2.96 (t, J= 12 Hz, 1 H), 2.58-2.53 (m, 1 H), 2.95-1.90 (m, 1 H), 1.34 (s, 9 H), 1.05 (s, 3 H), 0.99 (s, 3 H). MS (ESI) m/z (M+H)+: 422.
Preparation of Compound Int-12i
To a solution of Compound Int-12h (1.9 g, 4.5 mmol) in H20/EtOH (40 mL /20 mL) was added Na2S03 (5.6 g, 4.5 mmol) and the mixture was allowed to stir at room temperature for about 15 hours. The reaction mixture was concentrated in vacuo and the residue obtained was dissolved in ethyl acetate, washed with brine, dried over MgS04, filtered, and concentrated in vacuo. The residue obtained was purified using
chromatography on silica gel to provide Compound Int-12i (0.75 g, 48% yield). Ή NMR δ: 6.92 (s, 1 H), 4.71-4.67 (m, 1 H), 3.26-3.21 (m, 2 H), 2.01-1.96 (m, 1 H), 1.78-1.72 (m, 1 H), 1.13 (s, 9 H), 1.00 (s, 3 H).
EXAMPLE 12A
Preparation of Intermediate Compound Int-12o
0 i"Boc Step l f-Boc Step 2 H t-Boc Step 3
lnt-12j lnt-12k lnt-121 lnt-12m
lnt-12n lnt-12o
Step A
Acid Int-12j (22.7 g, 100 mmol) was dissolved in dry THF (400 ml) in a 1000 mL flask, and cooled with an ice-water bath. Borane tetrahydrofuran complex (1.0 M in THF, 200 ml, 200 mmol) was added via an additional funnel dropwise over a period of 80 minutes. After 1 hour at 0 °C the reaction was allowed to warm to room temperature and stir for about 15 hours. Methanol was then added dropwise via an additional funnel (-100 ml) and then the reaction was then concentrated in vacuo. The residue was purified on a 300 g ISCO silica column/ Combi-Flash Rf system using a gradient of 0-70% ethyl acetate in hexanes to provide alcohol Int-12k as a colorless oil (18.2 g, 85%).
Step B
Oxalyl chloride (14.08 g, 111 mmol) was dissolved in methylene chloride (340 ml) in a 1000 mL flask and cooled to -78 °C under nitrogen atmosphere. DMSO (9.33 g, 119 mmol) was added slowly via syringe over a period of 10 minutes. The resulting solution was allowed to stir at -78 °C for 45 minutes prior to the slow addition of the alcohol Int-12k (15.2 g, 85 mmol) in methylene chloride (50 ml) and stirred at -
78 °C under nitrogen for 45 minutes before addition of triethylamine (34.5 g, 341 mmol). After 40 minutes at -78 °C, then reaction was warmed to 0 °C and stirred at 0 °C for an additional 1 hour. After addition of 500 mL of methylene chloride, the organic solution was washed with water, IN HC1 solution (300 ml), and water. The organic layer was dried over sodium sulfate, concentrated in vacuo to provide aldehyde Int-121 as a colorless oil (18.14 g, -100%). This crude product was used for the next reaction without purification.
Step C
The aldehyde Int-121 (18.14 g, 86 mmol) was dissolved in methanol (37 ml) and the resulting solution was cooled with a RT water bath. A 7N ammonia solution in methanol (31.9 ml, 223 mmol) was then added dropwise via an additional funnel over a period of 15 minutes. The reaction mixture was allowed to stir at room temperature for 20 minutes before a 40% aqueous solution of glyoxal (16.2 g, 112 mmol) was added. The reaction mixture was allowed to stir at room temperature for about 15 hours and then concentrated in vacuo. The residue was purified using a 220 g ISCO silica
column/Combi-Flash Rf system (0-7% methanol in dichloromethane eluent) to provide Compound Int-12m as a slightly yellow solid (10.8 g, 51.5%).
Step D
Intermediate Int-12m (10.81 g, 43.4 mmol) was dissolved in THF (200 ml) in a 250 mL flask and NBS (15.43 g, 87 mmol) was added slowly at room temperature. The resulting solution was allowed to stir at room temperature for 4.5 hours and concentrated to semi-solid. The residue was dissolved in ethyl acetate (300 ml), washed with brine (3X100 ml), dried over sodium sulfate, and concentrated in vacuo. The crude material was purified using crystallization from dichloromethane to provide Compound Int-12n as a white solid (7.68 g, 43.5%). The mixture from mother liquid was purified using a 220 g ISCO silica column/Combi-Flash Rf system using 0-70% ethyl acetate in hexanes as the eluent to provide a second batch of Int-12n as a pale solid (7.73g, 43.8).
Step E
Intermediate Int-12n (14.4 g, 35.4 mmol) was dissolved in methanol (45 ml) and water (16 ml) and placed in a water bath. EDTA (10.34 g, 35.3 mmol) followed by 7N ammonia in methanol (20.21 ml, 141 mmol) were then added. Zinc powder (2.314 g, 45.4 mmol) was then added and the resulting solution was allowed to stir at room temperature. After 6 hours the reaction was then concentrated and the residue was redissolved with ethyl acetate (100 ml), washed with water (2x50 ml), dried over sodium sulfate, and concentrated in vacuo. The crude product was purified on a 80 g silica column with a Combi-Flash Rf system using a gradient of 0-70% ethyl acetate in hexanes to provide Int-12o as a white solid (7.56 g, 65%).
EXAMPLE 13
Preparation of Intermediate Compounds Int-13d and Int-13e
Int-13b lnt-13c lnt-13c' lnt-13d
Step A - Preparation of Compound Int - 13c
A 5 L- 3 necked round bottomed flask, equipped with a mechanical stirrer, temperature probe, addition funnel and N2 inlet, was charged with the Schollkopf chiral auxiliary-(Int-13a, 200 g, 1.09 mol, 1.0 eq), bis(chloromethyl) dimethylsilane (Int-13b, 256 g, 1.63 mol, 1.5 eq), and THF (2 L, Aldrich anhydrous). The flask was cooled in a dry ice/ 2-propanol bath until the internal temperature reached -75 °C. n-Butyllithium (Aldrich 2.5 M in hexanes , 478 mL, 1.19 mol, 1.09 eq) was added via a dropping funnel over 1 hour while maintaining the internal reaction temperature between -67 °C and - 76 °C. The resulting orange-red solution was allowed to gradually warm to room temperature for about 15 hours. The reaction mixture was then re-cooled to 0 °C and quenched with 500 mL of water. Diethyl ether (2L) was added and the layers were
separated. The aqueous layer was extracted with 1 L of diethyl ether. The combined organic extracts was washed with water and brine, dried with MgSC^, filtered, and concentrated in vacuo, giving 480 g of orange oil. This material was left in vacuo for about 15 hours to provide 420 g of oil. The crude product was split into two batches and purified via silica gel chromatography on a 1.6 kg flash column. The column was eluted with gradient of 0-4% Εί20 in hexanes. The product fractions were concentrated in vacuo at a bath temperature at or below 40 °C giving 190 grams of Int-13c-(60%yield).
Step B - Preparation of Compound Int-13d
A 5 L, 3-necked round bottomed flask equipped with a mechanical stirrer, addition funnel, temperature probe, external water bath and N2 inlet was charged with Compound Int-13c (196 g, 0.643 mol, 1.0 eq) and methanol (1.5 L). Aqueous HCl (500 mL of 10% by volume) was added at room temperature over 30 minutes, with a mild exotherm observed. The temperature increased to 37 °C then dropped back down. The reaction mixture was allowed to stir at room temperature for 3 hours and was monitored by TLC and LC/MS. The reaction mixture was then concentrated in vacuo to an oil. Additional methanol (3 x 200 mL) was added and the reaction mixture was concentrated in vacuo again. The resulting crude product was dried under house vacuum for about 15 hours. The crude product was then dissolved in CH2C12 (750 mL) and Et20 (1250 mL) and sodium iodide (96.4 g, 0.643 mol, 1.0 eq) was added. Diisopropylethylamine (336 mL, 1.929 mol, 3.0 eq) was added slowly over 25 minutes with stirring, causing the temperature to increase to 35 °C then decrease to room temperature again. The reaction mixture was allowed to stir at room temperature for 2 hours, after which time the MS of an aliquot indicated consumption of the starting material. The reaction mixture was allowed to stir for an additional 2 hours and then Boc-anhydride (281 g, 1.286 mol, 2.0 eq) was added. The reaction mixture was then allowed to stir at room temperature. After two days, the reaction mixture was diluted with EtOAc (2 L) and water (1 L), and he layers were separated. The aqueous phase was extracted with 500 mL of EtO Ac. The combined organic extracts were washed with water (500 mL) and brine (500 mL), dried with MgSC , filtered, and concentrated in vacuo to a yellow oil (380 g). The crude product was split into two 180 g portions for convenience and each portion was purified
via flash silica gel chromatography. Column conditions for a 180 g portion of crude product are as follows. The 180 gram sample of crude product was loaded onto a 191 g Si02 cartridge and purified on a 1.5 kg Si02 column. The column was eluted using a 0%- 20% EtOAc/hexanes gradient as the mobile phase to provide 52 grams of pure Int-13d and additional fractions of Int-13d that contained a small amount of a Boc-valine impurity. The impure fractions from the two columns were recombined and re-purified. After chromatography, Compound Int-13d was obtained as an oil which solidified to a white solid on standing (128 g, 65 % yield over the three steps.)
- Preparation of Compound Int-13e
lnt-13d lnt-13e
A solution of Int-13d (8.5 g, 31.1 mmol) in methanol (100 mL) and 1.0 M aqueous KOH solution (48 mL, 48 mmol) was allowed to stir at room temperature for about 15 hours. The reaction was then neutralized with 48 mL of 1.0 M aqueous HC1 solution to pH ~5, and partially concentrated in vacuo. The aqueous layer was then extracted twice with dichloromethane (2 x 100 mL). The combined organic solutions were concentrated in vacuo to provide Compound Int-13e as a gel (7.74 g, 96%).
Note: The above reactions were monitored by TLC using Hanessian's stain. To prepare the visualization stain, combine 450 mL of H20, 25 g ammonium molybdate, 5 g of eerie sulfate, and 50 mL of cone. HC1 or cone. H2SO4.
EXAMPLE 14
Preparation of Intermediate Compound Int-14d
lnt-14c lnt-14d
Step A - Preparation of Compound Int-14a
To a mixture of carboxylic acid Int-13e (20 g, 77 mmol) in THF (400 mL) at 0 °C was added 1M BH3 in THF ( 0.17 L) via addition funnel at 0 °C. The mixture was allowed to warm to room temperature and stir for about 15 hours. The reaction was carefully quenched by addition of MeOH (~ 75 mL) until bubbling ceased. The reaction mixture was concentrated in vacuo whereupon the residue obtained was partitioned between EtOAc and H2O. The layers were separated and the aqueous layer was extracted with EtOAc (2x). The organic layers were combined, washed with brine, dried (Na2S04), and concentrated in vacuo to provide Compound Int-14d (18 g, 99%) as a clear oil, which was used without further purification. MS (ESI) m/e (M+H+Na)+: 268.
Step B - Preparation of Compound Int-14b
To a dry 2-necked flask equipped with a stir bar was added oxalyl chloride (8.2 mL, 96 mmol) and CH2C12 (280 mL). The solution was cooled to -78 °C whereupon a solution of DMSO (7.4 mL, 0.10 mol) in CH2C12 (22 mL) was added and the mixture was allowed to stir for 30 minutes at -78 °C. A solution of alcohol Int-14a (18 g, 74 mmol) from Step A in CH2CI2 (60 mL) was added dropwise via addition funnel over 30 minutes. The resulting solution was allowed to stir for an additional 30 minutes at -78 °C whereupon Et3N (42 mL, 0.30 mol) was added dropwise. The mixture was allowed to stir for 30 minutes at -78 °C, warmed to 0 °C, and allowed to stir for an additional 1.5 hours. The mixture was diluted with CH2C12 (400 mL) and was transferred to a separatory funnel. The organic layer was washed with sat. aq NH4CI (2 x 100 mL) and brine (2 x 100 mL). The organic layer was dried (Na2S04), filtered, and concentrated in vacuo to provide Compound Int-14b,l 8 g (99%) as a clear oil, which was used without further purification.
Step C - Preparation of Compound Int-14c
To a round bottom flask charged with aldehyde Int-14b (18 g, 74 mmol) from Step B was added a 7N NH3 in MeOH solution (28 mL, 0.19 mol) in MeOH (37 mL) at room temperature. The mixture was allowed to stir for 30 minutes at room temperature whereupon a solution of glyoxal (14 g, 96 mmol) was added over 5 minutes. The resulting solution was allowed to stir for 12 hours at room temperature and was concentrated in vacuo. The residue obtained was purified using column chromatography using a gradient of 100% CH2Cl2to 97.5% CH2Cl2/2.5% MeOH to provide Compound Int-14c, 9.9 g (48%) as yellow oil. MS (ESI) m/e (M+H) +: 282.
Step D - Preparation of Compound Int-14d
To a solution of imidazole Int-14c (1.0 g, 3.6 mmol) from Step C in CH2C12 (5 mL) at 0 °C, was added NBS (0.44 g, 2.5 mmol) in CH2C12 (10 mL) dropwise via addition funnel. The resulting mixture was allowed to stir for 90 minutes at 0 °C whereupon the mixture was concentrated in vacuo. The crude residue obtained was partitioned between CHC13 (10 mL) and water (3 mL) and the layers were separated. The organic layer was washed with water (3 x 3 mL), dried (Na2S04), filtered, and
concentrated in vacuo. The residue obtained was purified using column chromatography (80g) using a gradient of 100% hexanesto 65% hexanes/35% EtOAc to provide
Compound Int-14d, (0.35 g, 27%) as a white solid. MS (ESI) m/e (M+H)+: 360/362.
EXAMPLE 15
Preparation of pound Int-15c
Int-15c
Step A - Preparation of Compound Int-15a
Int-15a
To a solution of dichlorozirconocene (Cp2ZrCl2) (4.2 g, 14.2 mmol) in 40 mL THF at -78 °C was added n-BuLi (1.6 M in hexane, 18 mL, 28.4 mmol). The resulting reaction was allowed to stir for 1 hour, then diphenyldiallylsilane (2 g, 14.2 mmol) in 17 mL of THF was added at -78 °C . The reaction was allowed to stir for 1 hour at -78 °C and for 18 hours at 25 °C. Iodine (9 g, 35.5 mmol) in 20 mL THF was then added at -78 °C and the mixture was allowed to stir for 1 hour. The reaction was quenched with 10% aqueous H2SO4 and the organic phase was extracted by ether. The organic solution was washed with saturated aqueous NaHCCb solution, brine solution, and dried (Na2SC>4). After filtration, the filtrate was concentrated in vacuo and the residue obtained was purified using an ISCO 120 g column (hexane) to provide
Compound Int-15a, 2.75 g (49%). Ή NMR (CDC13) δ 3.44 (dd, J= 2.2, 10.0 Hz, 2H), 3.33 (dd, J= 4.7, 10.0 Hz, 2H), 1.20 (m, 2H), 0.93 (dd, J= 5.9, 14.7 Hz, 2H), 0.63 (dd, J = 11.1, 14.2 Hz, 2H), 0.19 (s, 6H).
- Preparation of Compound Int-15b
Int-15b
To a solution of (2R)-(-)-2,5-dihydro-3,6-dimethoxy-2-isopropylpyrazine (0.61 g, 4.36 mmol) in THF (8 mL) was added n-BuLi (2.5 M in hexane, 1.8 mL, 4.58 mmol) at -78 °C. After allowed to stir for 0.3 hours, Compound Int-15a (2.75 g, 6.98 mmol) in 2 mL of THF was added and the mixture was allowed to stir at the temperature for 4 hours. The reaction was quenched by saturated aqueous NH4CI solution and the
organic layers were extracted with EtOAc. The combined organic solution was washed with brine solution, dried (Na2S04), and concentrated in vacuo. The residue obtained was purified using an ISCO 40 g column (gradient from 0% to 2.5% ether in hexane) to provide Compound Int-15b, 783 mg (44%). Ή NMR (CDC13) δ 4.05 (m, 1H), 3.96 (t, J = 3.4 Hz, 1H), 3.72 (s, 3H), 3.71 (s, 3H), 3.49 (dd, J = 2,8, 0.4 Hz, 1H), 3.26 (dd, J= 6, 9.4 Hz, 1H), 2.30 (m, 1H), 1.96 (m, 1H), 1.60 (m, 2H), 1.37 1.17 (m, 3H), 1.08 (d, J= 6.9 Hz, 3H), 0.99 0.86 (m, 2H), 0.72 (d, J= 6.6 Hz, 3H), 0.49 (dd, J= 11.0, 14.4 Hz, 1H), 0.35 (dd, J= 11.0, 14.2 Hz, 1H), 0.16 (s, 6H).
Step C - Preparation of Compound Int-15c
To a solution of Compound Int-15b (780 mg, 1.92 mmol) in MeOH (9 mL) was added 10% aqueous HC1 (3 mL) at 0 °C and the mixture was allowed to stir at 25 °C for 18 hours. The mixture was concentrated in vacuo and the residue obtained was reconcentrated in vacuo with MeOH twice. The resulting white foam was dissolved in ether (6 mL) and CH2CI2 (9 mL), and diisopropylethylamine (1 mL, 5.7 mmol) was added. After allowed to stir at 25 °C for 18 hours, di-t-butyl dicarbonate (922 mg, 4.22 mmol) was added and the resulting mixture was allowed to stir at 25 °C for 2 days. The mixture was added to cold water and the organic layers were extracted with EtOAc. The combined organic solution was washed with brine solution, dried (Na2S04), and concentrated in vacuo. Then the residue obtained was dissolved in MeOH (8 mL) and treated with aqueous 1 M KOH solution (3.3 mL, 3.3 mmol). After allowed to stir at 0 °C to 25 °C, the reaction mixture was acidified with 10% aqueous HC1 and the organic layers were extracted with CH2CI2. The combined organic solution was washed with brine solution, dried (Na2S04), and concentrated in vacuo to provide Compound Iiit-15c, which was used without further purification.
EXAMPLE 16
Preparation of Intermediate Compound Int-16e
lnt-16a lnt-16b lnt-16c
lnt-16d lnt-16e
Step A - Preparation of Compound Int-16b
To a 1000 mL flame dried flask was added 1, 1-dichlorosilolane (Int-16a, 28.09 g, 181.1 mmol), bromochloromethane (23.5 mL, 362.2 mmol), and anhydrous THF (400 mL). The solution was cooled to -70 °C, then M-BuLi (2.5M in hexane, 145 mL, 362 mmol) was added slowly over a period of 1 hour. The resulting reaction was allowed to stir at -70 to -60 °C for 20 minutes, then was allowed to warm to room temperature over 1 hour. Saturated NH4CI solution (200 mL) and Εί20 (200 mL) were then added and the organic layer was separated and the aqueous layer was extracted with Et20 (100 mL) twice. The organic layers were combined, washed with brine, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using Si02 chromatography (240 g, eluted with hexane) to provide Compound Int-16b (17.2 g, 51.9%).
Step B - Preparation of Compound Int-16c
To a 500 mL flame dried flask was added (i?)-2-isopropyl-3, 6- dimethoxy-2,5-dihydropyrazine (10.0 g, 54.3 mmol) and anhydrous THF (200 mL). The solution was cooled to -78 °C. «-BuLi (2.5M in hexane, 24.0 mL, 59.7 mmol) was added dropwise. After the solution was allowed to stir at -78 °C for 30 minutes, Compound Int-16b (in 5 mL anhydrous THF) was added dropwise. After the solution was allowed to stir at -78 °C for 1 hour, it was allowed to warm up to room temperature in two hours. Water (100 mL) and Et20 (150 mL) were added. The organic layer was
separated and the aqueous layer was extracted with Et20 (100 mL) twice. The organic layers were combined, washed with brine, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using Si02 chromatography (40 g, eluted with Et20 in Hexane: 0% to 3%) to provide Compound Int-16c (10.43 g, 58.0%).
Step C - Preparation of Compound Int-16d
To a 500 mL flask was added Compound Int-16c (11.5 g, 34.8 mmol) and MeOH (80 mL). 10% HCl (20 mL) was added. The solution was allowed to stir at room temperature for 5 hours and concentrated in vacuo. The residue obtained was dissolved in 20 mL MeOH and concentrated again to remove water and HCl. This process was repeated three times. The residue obtained was dissolved in
dichloromethane (50 mL) and Et20 (70 mL). DIPEA (15.4 mL, 86.9 mmol) and Nal (5.2 g, 34.75 mmol) were added. The solution was allowed to stir at room temperature for about 15 hours. Di-tert-butyl dicarbonate (18.9 g, 86.9 mmol) was added. The solution was allowed to stir at room temperature for 4 hours. Water (100 mL) and EtOAc (100 mL) were added. The organic layer was separated and the aqueous layer was extracted with EtOAc (100 mL) twice. The organic layers were combined and washed with brine, dried over anhydrous Na2S04, filtered, and concentrated in vacuo. The residue obtained was purified using Si02 chromatography (220g, Hexane EtOAC: 0% to 20%) to provide Compound Int-16d (7.9 g, 75.9%).
Step D - Preparation of Compound Int-16e
Compound Int-16d (7.9 g, 26.4 mmol) was dissolved in MeOH (100 mL) and cooled to 0 °C. KOH (1M in water, 39.6 mL, 39.6 mmol) was added. The solution was allowed to stir at 0 °C for 2 hours, and then at room temperature for 3 hours. HCl (2 N, 20 mL) was added, then additional HCl was added slowly to adjust the solution to pH 4. The acidified solution was concentrated in vacuo and to the residue obtained was added water (1 0 mL) and EtOAc (200 mL). The organic layer was separated and the aqueous layer was extracted with EtOAc (2 x 100 mL). The combined organic extracts were washed with brine, dried over anhydrous Na S04, filtered, and concentrated in
vacuo. The residue obtained was dried in vacuo for 48 hours to provide Compound Int- 16e (7.45 g, 99%), which was used without further purification.
EXAMPLE 17
Preparation of Intermediate Compounds Int-17c and Int-17d
— Preparation of Compound Int-17b
lnt-17a lnt-17b
To a 500 mL flask was added Int-17a (25.0 g, 130 mmol), dry dichloromethane (250 mL) and DIPEA (25.37 g, 195 mmol). The solution was cooled to 0 °C and acetyl chloride (13.27g, 169 mmol, in 30 mL dry dichloromethane) was added dropwise. The resulting reaction was allowed to stir at 0 °C for one hour and then at room temperature for about 15 hours. The solution was diluted with EtOAc and washed with water. The organic phase was dried over anhydrous Na2S04, filtered, and concentrated in vacuo. The residue obtained was purified using flash column
chromatography on silica gel (330g, 0% to 50% of EtOAc in Hexane) to provide
Compound Int-17b (22.58 g, 74.5%)
Step B - Preparation o Compound Int-17c
Int-17b Int-17c
To a 500 mL flask was added Int-17b (21.45 g, 92.05 mmol) and dry dichloromethane (200 mL). It was cooled to 0 °C and aluminum trichloride (A1C13, 36.82 g, 276.2 mmol) was added in portions. After the solution was allowed to stir at 0 °C for 30 minutes, it was concentrated in vacuo. The semi-solid residue obtained was heated at 140 °C for three hours. After it was cooled to 80 °C, water (10 mL) was added dropwise.
It was then cooled to 0 °C and EtOAc (300 mL) and water (200 mL) were added. The suspension was allowed to stir at 0 °C until the entire solid dissolved. More EtOAc was added and the organic layer was separated. The organic layer was washed with water, dried over anhydrous Na2S04, filtered, and concentrated in vacuo. The residue obtained was purified using flash column chromatography on silica gel (330g, 0% to 10% of EtOAc in Hexane)to provide Compound Int-17c (18.76 g, 87%).
Step C - Preparation of Compound Int-17d
lnt-17d
Compound Int-17d was prepared using the method described above for the synthesis of Compound Int-17c and substituting 2-bromophenol for Compound Int- 17a in Step A
EXAMPLE 18
Preparation of Intermediate Compound Int-18c
Int-18c
- Preparation of Compound Int-18b
Int-18a Int-18b
To a stirred solution of (3-methyloxetan-3-yl)methanol (Int-18a,10.0 g, 97.9 mmol) in methylene chloride (400 mL) at 0 °C, under inert atmosphere, was added silica gel (20 g). PCC (29.5 g, 137 mmol) was then added in portions over a 2 minutesute
period. The solution was allowed to slowly warm to room temperature and stirred for 6.5 hours. The reaction mixture was then filtered through a mixture of Celite:silica gel (1 : 1, 400 g total) and the Celite: Silica gel was washed with methylene chloride (4 L). The filtrate and washing were combined and concentrated in vacuo to provide 4.98 g (51%) of Int-18b as a clear solution (48.5 wt%) in methylene chloride. Ή NMR (CDC13 500 MHz): 5 9.94 (s, 1H), 4.89 4.83 (m, 2H), 4.52 4.46 (m, 2H), 1.48 (s, 3H).
Step B - Preparation of Compound Int-18c
lnt-18b lnt-18c
To a stirred solution of triphenylphosphite (5.10 mL, 19.5 mmol) in methylene chloride (9 mL) at 0 °C, under inert atmosphere, was added bromine (1.00 mL, 19.5 mmol) dropwise at 0 °C. A solution of Compound Int-18b (1.00 g, 9.99 mmol) in methylene chloride (1 mL) was then added and the resulting reaction was allowed to stir for 40 minutes at 0 °C. The reaction mixture was diluted with hexanes (10 mL) and the solution was passed through a plug of silica gel (4 g). The solids were washed with MTBE (20 mL). The filtrate and washing was combined and concentrated in vacuo to ~10 mL and purified using flash column chromatography on silica gel (methylene chloride/pentane) to provide 1.06 g (44%) of Compound Int-18c as clear colorless oil. Ή NMR (CDC13,500 MHz): δ 5.98 (s, 1H), 3.76 (d, J= 10.5 Hz, 2H), 3.65 (d, J= 10.5 Hz, 2H), 1.44 (s, 3H).
EXAMPLE 19
Preparation of Intermediate Compound Int-19e
tep A - Preparation of Compound Int-19a
lnt-17d lnt-19a
A mixture of Int-17d (4.2 g, 20 mmol) and 4-bromophenyl hydrazine hydrochloride (4.4 g, 20 mmol) in AcOH and EtOH (1:10, 100 mL) was heated to reflux and allowed to stir at this temperature for 6 hours. The reaction mixture was cooled to room temperature and concentrate in vacuo to provide Compound Int-19a as a solid, which was used without further purification (9.2 g). MS (ESI) m / e (M+lT): 383.
- Preparation of Compound Int-19b
lnt-19a lrrt-19b
A mixture of Int-19a (9.2 g) in PPA was heated to 80 °C and allowed to stir at this temperature for 2 hours. After cooling to room temperature, the reaction mixture was poured into ice water. The resulting solution was extracted with dichloromethane and the organic extract was washed with brine, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using column chromatography to provide Compound Int-19b (4.8 g). MS (ESI) m / e (M+HT): 368.
Step C - Preparation of Compound Int-19c
lnt-19b lnt-19c
To a solution of Int-19b (6 g, 16.3 mmol) in DMSO/CH3CN (1 :1, 24 mL) was added Select-F (5.8 g, 16.3 mmol) in portions. The reaction was allowed to stir for 1 hour at room temperature, then the reaction mixture was concentrated in vacuo and the residue obtained was purified using HPLC to provide Compound Int-19c as a solid (1.0 g). MS (ESI) m / e (M+Ff): 386.
- Preparation of Compound Int-19d
ini-TC lnt-19d
A suspension of Int-17c (51.6 g, 221 mmol, 1.0 eq) in 910 mL of absolute ethanol and 100 mL of glacial acetic acid was heated to 40 °C and 4-chlorophenyl hydrazine hydrochloride (41.66 g/232 mmol/1.05 eq) was added in portions, with stirring, followed by 3 Angstrom molecular sieves (23 g) and additional acetic acid (350 mL). The reaction mixture was placed under a N2 atmosphere, heated to 70 °C and allowed to stir at this temperature for 4 hours. The reaction mixture was allowed to cool to room temperature and was allowed to stand for about 15 hours, without stirring, under N2. The reaction mixture was filtered, the filtrate was concentrated in vacuo and the residue obtained was taken up in toluene (230 mL) and absolute ethanol (100 mL). The resulting solution was then concentrated in vacuo. The residue obtained was diluted with absolute ethanol (400 mL) and the resulting solution was allowed to stand in a 54 °C water bath for 45 minutes, then was allowed to cool to room temperature with stirring. The resulting precipitate was filtered and the collected solid was washed with 30 mL of absolute ethanol and 75 mL of hexanes, then dried in vacuo to provide Compound Int- 19d as an off white solid (50.2 grams (63%)). This material was used without further purification. MS (ESI) m / e (M+Ff): 357.0, 359.0.
Step E - Preparation of Compound Int-19e
lnt-19d lnt-19e
Polyphosphoric acid (111.8 g) and xylenes (260 mL) were added to a 1 liter 3-necked flask. The flask was placed in a 100 °C oil bath, connected to a N2 inlet, and equipped with a mechanical stirrer. The PPA/xylenes mixture was allowed to stir for 30 minutes to bring the internal temperature up to 100 °C. Compound Int-19d was then added in portions over 10 minutes. The reaction was placed under N2 atmosphere, capped, stirred for 30 minutes at 100 °C, and then stirred for 2.5 hours at 110 °C. The flask was lifted out of the oil bath and allowed to cool for 15 minutes. Ice (750 mL) was added in portions to the reaction mixture with stirring. After about 15 minutes, the reaction mixture was suction filtered through fiberglass filter paper in a Buchner funnel and an orange solid was collected. The collected solid was dissolved in EtOAc, and the resulting purple solution was washed with water and brine, then dried over MgSC^, filtered, and concentrated in vacuo. The residue obtained was purified using flash chromatography on a 345 g Si02 column using 5%-25% EtOAc/hexanes gradient, to provide Compound Int-19e (11.22 g) as a yellow solid (47%).
The following 2-aryl indole intermediates can be made using the method described above and substituting the appropriate reactants:
EXAMPLE 19a
Preparation of Intermediate Compound Int-19i
lnt-17d lnt-19f lnt-19g
lnt-19h lnt-19i
Step A - Preparation of Compound Int-19f
To a solution of Int-17d (14.0 g, 65.1 mmol), (4-chlorophenyl)hydrazine (23.3g, 130 mmol) in EtOH (400 mL) was added glacial acetic acid (40 mL). The reaction was heated to 90 °C and allowed to stir at this temperature for about 15 hours. The reaction mixture was cooled to room temperature, filtered, and the filtrate was concentrated in vacuo and dried in vacuo for 15 minutes. The resulting residue was diluted with dichloromethane (600 mL) and the resulting suspension was allowed to stir at room temperature for 30 minutes. The solid was removed by filtration and washed with dichloromethane five times. The filtrate was concentrated in vacuo and MeOH (100 mL) was added. The suspension was allowed to stir at room temperature for 15 minutes and filtered. The solid was dried in vacuo for two hours to provide Compound Int-19f (17.9 g, 81.0%).
Step B - Preparation of Compound Int-19g
To a 250 mL three-neck flask with a mechanic stirrer was added polyphosphoric acid (PPA, lOOg). PPA was heated to 110 °C and Int-19f (10.3 g, 30.3 mmol) was added in small portions. The reaction mixture gradually became dark green. The reaction mixture was allowed to stir at 110 °C for two hours. After cooling down, crushed ice was added slowly with stirring until the dark green color disappeared. Water was added and the suspension was transferred into a 1000 mL beak. The suspension was allowed to stir for 10 minutes and filtered. The solid was washed with water (100 mL)
three times and dried in vacuo at 60 °C for about 15 hours to provide Compound Int-19g (9.72 g, 99.4%).
Step C - Preparation of Compound Int-19h
To a 100 mL round bottom flask was added Int-19g (2.62 g, 8.12 mmol), DMSO (15 mL), and MeCN (15 mL). The solution was cooled to 0 °C and Select-F (2.3 g, 6.5 mmol) was added in three portions. The reaction was allowed to stir at 0 °C for 1.5 hours, then gradually warmed up to room temperature in one hour. The reaction mixture was then diluted with 20 mL MeOH and filtered. The filtrate was concentrated in vacuo to about 20 mL and purified using CI 8 chromatography (150g, 50% to 100% of MeCN in water, with 0.05% TFA) to provide Compound Int-19h (964 mg, 35%).
Step D - Preparation of Compound Int-19i
A solution of Int-19h (2.05 g, 6.02 mmol), DMF (120 mL), Cs2C03 (10.0 g, 31.0 mmol), and dibromoethane (5.2 mL, 60.2 mmol) was heated to 100 °C and allowed to stir at this temperature for about 15 hours. Additional dibromoethane (4.0 ml, 46 mmol) and CS2CO3 (3.0 g, 9.2 mmol) were added and the reaction was allowed to stir at 100 °C for 8 hours. The reaction mixture was cooled to room temperature and water (200 mL) and EtOAc (250 mL) were added. The organic layer was separated and the aqueous layer was extracted with EtOAc (100 mL). The organic layers were combined, washed with water (2 x 100 mL) and brine, dried over anhydrous Na2S04, filtered and concentrated in vacuo. The resulting residue was purified using flash column chromatography on silica gel (0% to 50% of EtOAc in Hexane) to provide Compound Int-19i (1.24 g, 56.2%).
EXAMPLE 20
Preparation of Compound 37
lnt-20c Compound 37
Step A - Preparation of Compound Int-20a
To a 40 mL vial was added Int-19i (329 mg, 0.898 mmol), bis(pinacolato)diboron (228 mg, 0.898 mmol), Pd(dppf)2Cl2-dichloromethane (146 mg, 0.18 mmol), and KOAc (264 mg, 2.7 mmol). The vial was degassed, refilled with N2, and capped. Dioxane was added via a syringe and the solution was allowed to stir at 90 °C for 2 hours. (25',4i?)-½rt-butyl-2-(5-bromo-lH-imidazol-2- /)-4-fluoropyrrolidine- 1 -carboxylate praline (300 mg, 0.90 mmol), Pd(dppf)2Cl2-dichloromethane (83 mg, 0.1 mmol), and K2CO3 (1M, 3.3 mL, 3.3 mmol) were added and the reaction was allowed to stir at 90 °C for 2 hours. The reaction mixture was cooled to room temperature, diluted with 5 mL EtOAc, and the aqueous layer was separated and extracted with 3 mL EtOAc. The combined organic extracts were dried over anhydrous Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using flash column
chromatography on silica gel (24 g, 15% to 70% of EtOAc in Hexane) to provide Compound Int-20a (387 mg, 79.7%).
Step B - Preparation of Compound Int-20b
To a 40 mL vial was added Int-20a (182 mg, 0.336 mmol), bis(pinacolato)diboron (89.7 mg, 0.353 mmol), Pd2(dba)3 CHCl3 (35 mg, 0.034 mmol), X-phos (32 mg, 0.067 mmol), and KOAc (98 mg, 1.0 mmol). The vial was degassed, refilled with N2, and capped. Dioxane was added via a syringe and the solution was allowed to stir at 120 °C for 2 hours. (S)- rt-butyl-2-(5-bromo-lH-imidazol-2- yl)pyrrolidine-l -carboxylate (116.9 mg, 0.37 mmol), Pd(dppf)2Cl2 dichloromethane (28 mg, 0.034 mmol), and K2CO3 (1M, 1.0 mL, 1.0 mmol) were added. The reaction was
allowed to stir at 80 °C for about 15 hours, then was cooled to room temperature. The aqueous layer were separated and extracted with 5 mL EtOAc. The organic extracts were combined and dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using flash column chromatography on silica gel (43 g, A:
dichloromethane; B: 10% MeOH in EtOAc: A/B: 0% to 80%) to provide Compound Int- 20b (191 mg, 89.9%).
Step C - Preparation of Compound Int-20c
To a 40 mL vial was added Int-20a (190 mg, 0.256 mmol), MeOH (2 mL), and HC1 (4M in dioxane, 6 mL, 24 mmol). The solution was allowed to stir at room temperature for two hours, then was concentrated in vacuo and the residue obtained was dried in vacuo for 30 minutes to provide Compound Int-20c, which was used without further purification.
Step D - Preparation of Compound 37
To a 40 mL vial was added Int-20c (~ 0.256 mmol), (S)-2- (methoxycarbonylamino)-3-methylbutanoic acid (90.0 mg, 0.512 mmol), HATU (214 mg, 0.56 mmol), and DMF (3 mL). The resulting solution was cooled to 0 °C and DIPEA (0.32 ml, 1.79 mmol) was added. The reaction was allowed to stir at 0 °C for 2 hours, then was diluted with water (0.2 mL) and the resulting solution was purified using a CI 8 column (43 g, 10% to 60%, of CH3CN in water with 0.05% TFA) to provide Compound 37 (46 mg, 21.4% from Int-20b). MS 874.4 [M+H]+
The following Compounds of the present invention were made using the method described in Example 20.
Compound Structure MS
864.3
61
[M+H]+
EXAMPLE 21
Preparation of Intermediate Compound Int-21a
lnt-19h lnt-21a
To a 20 mL microwave vial was added Int-19h (1.16 g, 3.41 mmol), anhydrous toluene (15 mL), cyclopropane-carboxaldehyde (1.28 mL, 17.1 mmol), and p- toluenesulfonyl chloride (65 mg, 0.34 mmol). The vial was capped and sealed, then placed in a microwave reactor and heated to 170 °C for three hours. The reaction mixture was cooled to room temperature and concentrated in vacuo. The residue obtained was dissolved in dichloromethane (40 mL) and filtered through a short pad of Celite. The filtrate was concentrated in vacuo and purified using flash column chromatography on silica gel (80 g, hexane) to provide Compound Int-21a (778 mg, 58.1%).
EXAMPLE 22
Preparation of Intermediate Compound Int-22c
Int-19b (5.82 g, 0.016 mmol) was dissolved in dichloromethane (50 mL) and THF (50 mL) and the mixture was allowed to stir at room temperature until all solids dissolved. The resulting solution was cooled in an ice-water bath for 30 minutes, after which NCS (2.13 g, 0.016 mmol) was added to the stirred reaction mixture in portions over -10 minutes. The reaction mixture was allowed to stir at 0 °C for 30 minutes and then at room temperature for 2 hours. The reaction mixture was concentrated in vacuo to provide a brown semi-solid, which was dissolved in dichloromethane (-300 mL). The organic solution was washed sequentially with water (1 x -200 mL), 10% (w/v) aq. sodium thiosulfate (1 x -200 mL), and brine (1 x -200 mL), then dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo. The resulting solid residue was purified using column chromatography (330 g
Teledyne-Isco RediSep® silica column, 0-30% EtOAc/hexanes over 12 column volumes at 200 mL/min) to provide 2.97 g of Int-22a (47% yield) as a brown solid. MS (ESI) m / e (M+tT): 400.
In a 20-mL microwave tube, Int-22a (1.075 g, 2.68 mmol) was dissolved in dry toluene (13 mL). Cyclopropanecarboxaldehyde (1.0 mL, 0.94 g, 13.4 mmol),/>- toluenesulfonyl chloride (51 mg, 0.27 mmol), and a magnetic stir bar were added. The tube was sealed and the reaction mixture was heated at 170 °C (microwave) with stirring for 3 hours. The reaction mixture was cooled to room temperature, the tube opened, and further aliquots of each of cyclopropanecarboxaldehyde (1.0 mL, 0.94 g, 13.4 mmol) and >-toluenesulfonyl chloride (51 mg, 0.27 mmol) were added. The tube was re-sealed and the reaction was again subjected to microwave heating at 170 °C for 4 hours, then cooled to room temperature and concentrated in vacuo to provide a brown solid residue. The brown solid residue was adsorbed onto silica gel (19 g) using EtOAc (-100 mL), followed by evaporation of the solvent, and then loaded onto a 100 g Biotage
® KP-Sil SNAP cartridge. Elution with 100% hexanes over 13 column volumes at 85 mL/min provided 600 mg of Int-22b (50% yield) as a light brown solid. MS (ESI) m / e (M+Yf): 452.
EXAMPLE 23
Preparation of Compound 23A
lnt-19b lnt-23a
A mixture of Compound Int-19b (1.1 g, 3 mmol),
(dibromomethyl)benzene (2.25 g, 9 mmol) and K2CO3 (1.2 g, 9 mmol) in 15 mL of DMF was heated to 100 °C and allowed to stir at this temperature for 3 hours. The reaction mixture was cooled to room temperature, concentrated in vacuo and the residue obtained was dissolved with dichloromethane and water. The aqueous phase was extracted with dichloromethane. The combined organic extracts were washed with brine, dried over Na2S04, filtered and concentrated in vacuo. The resulting residue was purified using flash column chromatography on silica gel to provide Compound Int-23a (380 mg, 28 %) as a white solid. Ή NMR (CDCI3): δ 7.72 (bs, 1 H), 7.44 - 7.46 (d, J= 8.4 Hz, 1 H), 7.21 - 7.28 (m, 3 H), 7.09 - 7.12 (m, 3 H), 7.04 (s, 1 H), 6.99 - 7.01 (bs, J= 6.8 Hz, 2 H), 6.78 (s, 1 H), 6.63 - 6.65 (d, J= 8.4 Hz, 1 H). MS (ESI) m/e (M+H+): 456.
- Pre aration of Compound Int-23b
lnt-23a lnt-23b
To a solution of Int-23a (456 mg, 1.0 mmol) in 1,4-dioxane was added bis pinacol borate (2.2 mmol) , Pd(dppf)Cl2 (0.04 mmol) and KOAc (4 mmol). The reaction mixture was put under N2, heated to 110°C and allowed to stir at this temperature for 3 hours. The reaction mixture was cooled to room temperature, concentrated in vacuo, and the residue obtained was purified using column chromatography on silica gel to provide Compound Int-23b (590 mg, 87 % yield). !H NMR (CDC13): δ 8.13 (s, 1 H), 7.60 (d, J= 7.6 Hz, 1 H), 7.52 (d, J= 8.0 Hz, 1H), 7.36 - 7.39 (m, 1 H), 7.14 -7.19 (m, 4 H), 6.93 - 6.95 (m, 3 H), 6.90 (s, 1 H), 1.26 - 1.29 (s, 24 H). MS (ESI) m / e (M+H+): 550.
A suspension of Int-23b (550 mg, 1.0 mmol), tert-butyl 2-(2-bromo-lH- imidazol-5-yl) pyrrolidine- 1-carboxylate (2.4 mmol), Pd(dppf) Cl2 (200 mg), Na2CC>3 (3 mmol) and in THF/H20 (10: 1, 33 mL) was allowed to stir at reflux for about 15 hours under N2. The reaction mixture was cooled to room temperature and filtered, and the filtrate was washed with water (50 mL) and extracted with EtOAc (100 mL). The organic extract was washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The resulting residue was purified using column chromatography on silica gel to provide Compound Int-23c (160 mg). MS (ESI) m / e (M+H+): 768.
- Preparation of Compound Int-23d
Int-23c (0.10 g, 0.13 mmol) was added to HCl/CH3OH (5 mL, 3M) and the resulting reaction was allowed to stir at room temperature for about 3 hours. The reaction mixture was then concentrated in vacuo to provide Compound Int-23d, which was used without further purification. MS (ESI) m / e (M+H+): 568.
- Pre aration of Compoun
To a solution of Int-23d (56.8 mg, 0.10 mmol), (S)-2-
(methoxycarbonylamino)-3-methylbutanoic acid (35.0 mg, 0.20 mmol) and DIPEA (0.8 mmol) in CH3CN (1 mL) was added BOP (98 mg, 0.22 mmol). The resulting reaction was allowed to stir at room temperature and monitored using LC/MS. After LC/MS showed the starting material to be consumed, the reaction mixture was filtered, and the filtrate was purified using HPLC to provide Compound A as a white solid. 1H NMR (MeOD): δ 7.94 (s, 1 H), 7.85 (d, J= 8.0 Hz, 1 H), 7.74 (s, 1 H), 7.63 (s, 1 H), 7.48 (s, 1 H), 7.35 - 7.37 (m, 2 H), 7.31 (s, 1 H), 7.17 - 7.18 (m, 4 H), 7.11 (s, 1 H), 6.96 - 6.98 (d, J = 7.6 Hz, 2 H), 5.09 - 5.17 (m, 2 H), 4.13 (t, J= 8.0 Hz, 2 H), 3.99 (bs, 2 H), 3.78 (bs, 2 H), 3.56 (s, 6 H), 2.44 - 2.47 (m, 2 H), 1.92 - 2.19 (m, 8 H), 0.77 - 0.85 (m, 12 H). MS (ESI) m / e (M+H+): 882.
The diastereomers were separated on a chiral SFC column:
Isomer A: Ή NMR (MeOD): δ 8.08 (s, 1H), 7.91 - 7.93 (m, 1 H), 7.72 (s, 1 H), 7.56 (s, 1 H), 7.24 - 7.43 (m, 7 H), 7.19 (s, 1 H), 7.03 - 7.05 (m, 2 H), 5.16 - 5.24 (m, 2 H), 3.81 - 4.21 (m, 6 H), 3.62 (s, 6 H), 2.52 - 2.54 (m, 2 H), 2.00 - 2.25 (m, 8 H), 0.84 - 0.91 (m, 12 H). MS (ESI) m/z (M+H)+: 882.
Isomer B: !H NMR (MeOD): δ 7.90 (s, 1 H), 7.81 - 7.83 (m, 1 H), 7.72 (s, 1 H), 7.62 (s, 1 H), 7.45 (s, 1 H), 7.14 - 7.33 (m, 6 H), 7.09 (s, 1 H), 6.93 - 6.95 (m, 2 H), 5.06 - 5.14 (m, 2 H), 3.71 - 4.11 (m, 6 H), 3.52 (s, 6 H), 2.41 - 2.44 (m, 2 H), 1.90 - 2.15 (m, 8 H), 0.74 - 0.86 (m, 12 H). MS (ESI) m/z (M+H)+: 882.
The Compounds of the present invention depicted in the table below were made using the method described in Example 23 and substituting the appropriate dibromotoluene derivative in Step A
Cpd W X Y Z MS
95 CH CH C(OCH3) CH 912
99 CH CH CF CH 900
916,
103 CH CH CC1 CH
918
104 CH CH C(CF3) CH 950
108 CH N CH CH 883
109 N CH CH CH 883
110 CH CF CH CH 900
916,
117 CH CC1 CH CH
918
121 CH CH N CH 883
125 CH CH C(OCF3) CH 966
126 CH CF CF CH 918
129 CH CH C(CH3) CH 896
916,
130 CC1 CH CH CH
918
133 CF CF CH CH 918
136 CH CH CCN CH 907
143 CH CF CH CF 918
145 CH C(OCH3) CH CH 912
149 CF CH CH CH 900
150 CH C(CF3) CH CH 950
153 CF CH CH C(OCH3) 930
EXAMPLE 24
Preparation of Compound 16
16
- Preparation of Compound Int-24b
lnt-24a lnt-24b
To a solution of Compound Int-24a(1.48 g, 3.76 mmol) in 11 mL THF at -78 °C was added n-BuLi (2.5 M in hexane, 1.66 mL, 4.14 mmol). The reaction was allowed to stir at -78 °C for 30 minutes, then 2-chloro-N-methoxy-N- methylacetamide(l .l g, 7.52 mmol) in 2 mL of THF was added at -78 °C . The reaction was allowed to stir for 1 hour at -78 °C, then was quenched with saturated aqueous
NH4CI. The resulting solution was extracted with EtOAc and the organic extract was washed with brine solution, dried (Na
2S04), filtered and concentrated in vacuo. The resulting residue was purified using an ISCO 80 g column (hexane to 50% EtOAc-hexane, gradient) to provide Compound Int-24b, 503 mg (35%). LRMS: (M+H)
+ = 390.
- Preparation of Compound Int-24d
lnt-24b lnt-6f lnt-24c
To a solution of Compound Int-24b (97 mg, 0.25 mmol) and Int- mg, 0.38 mmol) in DMF (2 mL) was added CS2CO3 (163 mg, 0.50 mmol). The resulting reaction was heated to 40 °C, allowed to stir at this temperature for 1 hour, then cooled to 25 °C. The reaction mixture was poured into ice-water and the organic phase was extracted with EtOAc. The organic extract was washed with brine, dried (Na?S04), filtered and concentrated in vacuo. The residue obtained was purified using an ISCO 24 g column (gradient from hexane to 40% EtOAc in hexane) to provide Compound Int-24c, 135 mg (91%).
To a solution of Compound Int-24c (135 mg, 0.23 mmol) in o-xylene (2 mL) was added ammonium acetate (107 mg, 1.38 mmol) and the resulting reaction was allowed to stir at 140 °C for 3 hours. After being cooled to 25 °C, the reaction mixture was added to aqueous NaHCC"3 solution and the organic layer were extracted with EtOAc. The combined organic solutions was washed with brine, dried (Na2S04), filtered and concentrated in vacuo. The residue obtained was purified using an ISCO 24 g column (gradient from hexane to 50% EtOAc in hexane) to provide Compound Int-24d, 84 mg (64%). LRMS: (M+H)+ = 575
Step D - Preparation of Compound Int-24g
24g
a solution of Compound Int-24d (81 mg, 0.14 mmo pinacolatodiborane (53 mg, 0.21 mmol), PdCl2(dppf 2 CH2CI2 complex (11.5 mg, 0.014 mmol) in 1,4-dioxane (2 mL) was added potassium acetate (41 mg, 0.42 mmol). The reaction was degassed and allowed to stir at 100 °C for 2 hours. After being cooled to 25 °C, the reaction mixture was diluted with EtOAc and filtered through a Celite pad. The filtrate was concentrated in vacuo to provide Compound Int-24f, which was combined with Int-7d (66 mg, 0.21 mmol), and PdCkidppf CH2CI2 complex (11.5 mg, 0.014 mmol) and dissolved in 1,4-dioxane (2 mL). The resulting solution was treated with aqueous 2 M Na2CC>3 solution (0.21 mL, 0.42 mmol) and the reaction mixture was degassed and allowed to stir at 100 °C for 2 hours. After being cooled to 25 °C, the reaction mixture was diluted with EtOAc and filtered through a Celite pad. The filtrate was concentrated in vacuo and the residue obtained was purified using an ISCO 24 g column (gradient from 0% to 100% EtOAc in hexane) to provide Compound Int-24g (40 mg, 39%). LRMS: (M+H)+ = 732.
- Preparation of Compound Int-24h
lnt-24g lnt-24h
To a 0 °C solution of Compound Int-24g (40 mg, 0.054 mmol) in dichloromethane (2 mL) was added TFA (0.4 mL). The reaction was allowed to stir at 0 °C for 0.5 hours and then was warmed to 25 °C and allowed to stir for 2 additional hours. The reaction mixture was concentrated in vacuo and the residue obtained was dissolved in MeOH (2 mL) followed by addition of 4N HCl in dioxane (0.3 mL). The solution was concentrated in vacuo to provide Compound Int-24h as its HCl salt (40 mg), which was used without further purification. LRMS: (M+H)+ = 532.
Preparation of Compound 16
lnt-24h lnt-1a
To a -30 °C solution of Compound Int-24h (41 mg, 0.068 mmol),
Compound Int-la (36 mg, 0.20 mmol), and diisopropylethylamine (83 ί, 0.48 mmol) in DMF (1.5 mL) was added HATU (103 mg, 0.27mmol). The mixture was allowed to stir at -30 °C to 0 °C for 1 hour and for an additional 2 hours at 0 °C. The reaction was then quenched by addition of cold water and the resulting mixture was purified using Gilson HPLC (CH3CN-H2O, 0.1% TFA) to provide Compound 16. Compound 16 was dissolved in MeOH (10 mL) and treated with 4N HCl in dioxane (0.3 mL) followed by concentration in vacuo to provide the HCl salt of Compound 16 as a ~1 : 1 mixture of diastereomers, 16 mg (28%).
The diastereomers were separated by chiral HPLC using Chiral OD (Lux Cellulose-1) Semi-prep column (20% EtOH-hexane, 0.1% DEA) to provide Compound 16A (retention time: 44 minutes), 6 mg, and Compound 37B (retention time : 66 minutes), 3 mg.
EXAMPLE 25
Preparation of Compound 17
- Preparation of Compound Int-25a
Compound Int-25a was prepared from Compound Int-24b using the method described in Example 24, Step B (100%).
— Preparation of Compound Int-25b
Compound Int-25b was prepared from Compound Int-25a using the method described in Example 24, Step C yield (45%). LRMS (M+H)+ = 589.
Step C - Preparation of Compound Int-25d
Compound Int-25d was prepared from Compound Int-25b using method described in Example 24, Step D yield (44%). LRMS: (M+H)+= 746.
- Preparation of Compound Int-25e
Compound Int-25e was prepared from Compound Int-25d using the method described in Example 24, Step Eyield (100%).
Step E - Preparation of Compound 17
Compound 17 (HCl salt) was prepared from Compound Int-25e using the method described in Example 24, Step F yield (50%).
The diastereomers were separated by chiral HPLC using Chiral Lux C-2 Semi-prep column (50% EtOH-hexane, 0.1% DEA) to provide Compound 17A (retention time: 45 minutes) and Compound 17B (retention time: 59 minutes).
EXAMPLE 26
Preparation of Compound 23
lnt-24b lnt-26a Compound Int-26a was prepared from Compound Int-24b using the method described in Example 24, step B (87%).
- Preparation of Compound Int-26b
Compound Int-26b was prepared from Compound Int-26a using the method described in Example 24, step C (72%). LRMS (M+H)+ = 585.
Step C - Preparation of Compound Int-26d
To a solution of Compound Int-26b (243 mg, 0.42 mmol), bis-pinacolato diborane (127 mg, 0.50 mmol), PdCl2(dppf)2 CH2C12 complex (34 mg, 0.042 mmol) in 1,4-dioxane (3 mL) was added potassium acetate (83 mg, 0.84 mmol). The mixture was degassed and allowed to stir at 100 °C for 2 hours. After being cooled to 25 °C, Int-7d (265 mg, 0.84 mmol), PdCl2(dppf)2 CH2C12 complex (34 mg, 0.042 mmol), and K2C03 (IN aqueous solution, 1.2 mL, 1.2 mmol) were added. The mixture was degassed and allowed to stir at 90 °C for 18 hours. After being cooled to 25 °C, the mixture was diluted with EtOAc and filtered through a Celite pad. The filtrate was concentrated in vacuo and the residue obtained was purified using Prep TLC (5% MeOH in CH2C12) to provide Int-26d, 146 mg (47%). LRMS: (M+H)+ = 742.
- Preparation of Compound Int-26e
Compound Int-26e was prepared from Int-26d using the method described in Example 24, step E (100%). LRMS: (M+H)+ = 542.6.
Preparation of Compound 23
lnt-26e 'nt-1a 23
Compound 23 (HCl salt) was prepared from Compound Int-26e using the method described in Example 24, step F (53%).
The diastereomers were separated by chiral HPLC using Chiral Lux C-2 Semi-prep column (50% EtOH-hexane, 0.1% DEA) to provide Compound 23A (retention time: 16 minutes) and Compound 23B (retention time: 27 minutes).
EXAMPLE 27
Preparation of Compound 26
26
Step A - Preparation of Compound Int-27a
lnt-24b lnt-13e lnt-27a
Compound Int-27a was prepared from Compound Int-24b using the method described in Example 24, step B (85%).
- Preparation of Compound Int-27b
Compound Int-27b was prepared from Compound Int-27a using the method described in Example 24, step C (75%). LRMS (M+H)
+ = 593.
- Preparation of Compound Int-27d
Compound Int-27d was prepared from Compound Int-27b
method described in Example 24, step D (40%). LRMS (M+H)+ = 750.
- Preparation of Compound Int-27e
Compound Int-27e was prepared from Compound Int-27d using the method described in Example 24, step E (100%). LRMS: (M+H)+= 550.
Step E - Preparation of Compound 26
Compound 26 (HC1 salt) was prepared from Compound Int-27e using the method described in Example 24, step F (50%).
The diastereomers were separated by chiral HPLC using a Chiral Lux C-2 Semi-prep column (35% EtOH-hexane, 0.1% DEA) to provide Compound 26A (retention time: 38 minutes) and Compound 26B (retention time:50 minutes).
EXAMPLE 28
Preparation of Compound 240
Cmpd 240
Step A - Preparation of Compound Int-28c
,n a lnt-28b |nt-28c
A solution of Int-28a (13.2 g, 46mM), Int-28b (9.0g, 38 mM), Pd(PPh3)4 (4.4 g, 3.8 mM), K2C03 (13. lg, 95 mmol) in 28 mL H20 and 140 mL DME was purged with nitrogen. The reaction was allowed to stir at refluxed for 3 hours. Another portion of boronic acid (0.5 equiv.), Pd(PPh3)4 (0.01 eq) were added and the reaction was allowed to stir at reflux for an additional 4 hours. The reaction mixture was diluted with EtOAc and filtered through a small Celite plug. The filtrate was concentrated in vacuo and the
residue obtained was purified using flash LC ( 0%-10% EtOAc/Hexane) to provide Compound Int-28c (14.5 g). MS (ESI) m / e (M+Na+): 425.
Step B - Preparation of Compound Int-28d
lnt-28c lnt-28d
To a suspension of Compound Int-28c (2 g, 5 raM) in CH2CI2 (8 mL), TFA (4 mL) was added dropwise and the reaction was allowed to stir at room
temperature for 14 hours. The reaction mixture was concentrated in vacuo and the resulting residue was suspended in a solvent mixture of THF (25 mL), ethanol (6 mL) and water (2.5 mL). Zn dust (3.25 g, 50 mmol) and NH4C1 (1.3 g, 25 mmol) were added and the reaction was allowed to stir at reflux for 1 hour. The reaction mixture was diluted with EtOAc and filtered through a small Celite plug. The filtrate was washed with water and brine, dried over MgS04, filtered and concentrated in vacuo to provide Compound Int-28d (1.9 g). MS (ESI) m / e (M+Ff): 273.
- Preparation of Compound Int-28e
lnt-28d lnt-28e
To a suspension of Int-28d (1 g, 3.6 mM) in DMSO (5 mL)/ Acetonitrile (5 mL) was added Select-F (1.53 g, 4.3 mmol). The reaction was allowed to stir for 30 minutes, then was diluted with EtOAc and washed with water and brine, and the organic phase was dried over MgS04, filtered and concentrated in vacuo. The residue obtained was suspended in acetic anhydride (4 mL) and allowed to stir at room temperature for 2 hours. The reaction mixture was then diluted with EtOAc and washed with NaHC03
solution and water. The organic phase was dried over MgS04, filtered and concentrated in vacuo and the residue obtained was suspended in EtOAc (10 mL). To the resulting suspension was added 4M HC1 in dioxane (4 mL) and the reaction was allowed to stir at room temperature for 2 hours. The reaction mixture was filtered and the collected solid was washed with hexane, then recrystallized from ethanol to provide Compound Int-28e (200 mg). MS (ESI) m / e (M+H+): 315.
- Preparation of Compound Int-28f
lnt-28e lnt-28f
To a 0 °C suspension of Int-28e (200 mg, 0.63 mM) in CH2C12 (5 mL) was added 1M solution of BBr3 in CH2C12 (5 mL) at 0 °C. The reaction was allowed to stir at 0 °C for 1.5 hours, then an additional 5 mL of 1M solution of BBr3 in CH2CI2 was added and the reaction was heated to 40 °C and allowed to stir at this temperature for 5 hours. The reaction mixture was then diluted with EtOAc (200 mL) and the resulting solution was washed with NaHCC>3 solution and NaOH solution. The organic layer was then washed with water and brine, dried over MgS04, filtered and concentrated in vacuo to provide a residue which was subsequently suspended in CH2CI2 (5 mL) cooled at 0 °C. To this solution was added Et3N (0.6 mL) and Tf20 (0.5 mL) and the resulting reaction was allowed to stir at 0 °C for 1.5 hours. The reaction was then diluted with
dichloromethane and quenched with 10 % citric acid. The organic layer was washed with water and brine, dried over MgS04, filtered and concentrated in vacuo. The residue obtained was suspended in dioxane (8 mL) and to the resulting solution was added bis- pinacolatodiborane (265 mg), PdCl2(dppf)2 CH2CI2 complex (26 mg) and potassium acetate (206 mg). The mixture was degassed, purged with nitrogen and allowed to stir at 100 °C for 1.6 hours. The reaction mixture was cooled to 25 °C, diluted with EtOAc and filtered through a Celite pad. The filtrate was concentrated in vacuo and the residue
obtained was purified using flash LC (0-100 % EtOAc-Hex) to provide Compound Int- 28f (100 mg).
Step - Preparation of Compound 240
Compound 240 was prepared from Compound Int-28f using the method described in Example 20.
EXAMPLE 29
Preparation of Intermediate Compound Int-29a
To a suspension of Compound Int-28d (0.51 g, 1.87 mmol) in CH2CI2 (3 mL) was added cyclopropyl anhydride (2 mL). The resulting reaction was allowed to stir at room temperature for 2 hours, then a solution of 4M HCl in dioxane ( 3 mL) was added and the reaction was allowed to stir at room temperature for 2 hours. The reaction mixture was then filtered and the collected solid was washed with hexane and dried in vacuo to provide Compound Int-29a (590 mg). MS (ESI) m / e (M+H+): 323.
EXAMPLE 30
Step A
Commercially available phenol Int-30a (125.g, 73.3 mmol), hydrazine Int-30b (13.1 g, 73.3 mmol) and methanol (200 mg) were charged to a 500 mL flask. To the suspension was added potassium acetate (14.5 g, 148 mmol) and the resulting reaction mixture was allowed to stir at reflux. After 3 hours, the reaction was cooled, and the solid collected by filtration, washed with methanol (50 ml) and water (2x50 ml), and dried in vacuo to provide hydrazone Int-30c as a slightly orange solid (18.5 g, 50%).
Step B
Int-30c (18.5 g, 62.7 mmol) and polyphosphoric acid (50 g) were added to a 250 mL flask equipped with mechanical stirrer. The mixture was allowed to stir at 120 °C for 30 minutes and cooled to room temperature. To the mixture were added ice and water. The solid was collected by filtration, washed with water (2x100 ml) and then dissolved in ethyl acetate (200 ml), and washed with water (2x200 ml) again. The solution was then dried over sodium sulfate, and concentrated in vacuo to provide Indole Int-30d as a solid (17 g, 98%).
Step C
Indole Int-30d (18.3 g, 65.8 mmol), cesium carbonate powder (356.6 g, 117 mmol), and DMSO (100 ml) were charged into a 500 mL flask. To the resulting suspension was added diiodomethane (134.4 g, 36 mmol) via a syringe. The reaction mixture was allowed to stir at room temperature for about 15 hours, treated with water
(300 ml) and filtered. The solid was purified using a 120 g silica column/Combi-Flash Rf system using a gradient of 0-20% ethyl acetate in hexanes to provide Int-30e as a white solid (8.5 g, 45%).
Step D
Int-30e (2.4 g, 8.27 mmol), NBS (1.47 g, 8.27 mmol), and THF (50 ml) were added to a 100 mL flask and stirred at room temperature. After 5h the reaction was concentrated in vacuo to a semi-solid and the residue was treated with water (100 ml), stirred at room temperature for about 15 hours, and filtered. The filter cake was washed with water (3X20 ml) and dried to provide indole Int-30f as a pale solid (2.7 g, 88%).
EXAMPLE 31
Preparation of Compound 1525 & Compound 1541
Compound
Step A
To a 35 mL microwave reaction tube were added Int-30f (500 mg, 1.36 mmol), bis(triphenylphosphine)palladium (II) dichloride (95 mg, 0.135 mmol), copper iodide (258 mg, 1.355 mmol), and DMF (10 mL). The resulting suspension was degassed and heated to 100 °C, and then Int-31a was then added in portions via a syringe. The resulting mixture was allowed to stir at 100 °C for additional 6 hours under nitrogen. After cooling, the solution was diluted with 10 mL of ethyl acetate, filtered, and concentrated in vacuo. The residue was purified using a 40 g silica column/Combi-Flask Rf system (0-15% ethyl acetate in hexanes eluent) to provide Int-31b as a wax (370 mg, 65%).
Step B
Int-31b (120 mg, 0.286 mmol), bis(pinacolato)diboron (152 mg, 0.6 mmol), potassium acetate (280 mg, 2.86 mmol), Pd2(dba)3-CHCl3 (59.1 mg, 0.057 mmol), X-PHOS (54.4 mg, 0.114 mmol), and dioxane (4 ml) were added to a 35 mL microwave reaction tube. The sealed mixture was degassed and stirred at 1 10 °C under nitrogen atmosphere for 8 hours then cooled to room temperature. To this mixture were added bromide Int-7h (246 mg, 0.658 mmol), PdCl2(dppf)-CH2Cl2 (46.7 mg, 0.0057 mmol), 1.5 M aqueous solution of sodium carbonate (1.9 ml, 2.9 mmol). The resulting mixture was degassed and stirred at 95 °C under nitrogen atmosphere for 6 hours, cooled to room temperature, concentrated, purified using Gilson reverse phase chromatography (10-80% acetonitrile in water with 0.1% TFA eluent) to provide Compound 1525 as a wax (68 mg, 20%). LC/MS anal, calcd. for: Cs^ss^Og 935.4; Found: 937.1 (M+H)+.
Step C
Compound 1525 (16 mg, 0.014 mmol) and 10% palladium on activated carbon (5 mg, 4.7 μΜ) were added to 8 mL of methanol in a 250 mL pressure vessel and the reaction was shaken at room temperature under 35 psi hydrogen atmosphere using PARR hydrogenation apparatus for 6 hours. The reaction mixture was filtered through celite, concentrated in vacuo to provide Compound 1541 as a solid (15 mg, 93%).
LC/MS anal, calcd. for: C52H6iN908 939.4; Found: 939.7 (M+H)+.
EXAMPLE 32
Preparation of Compound 752
lnt-32a Cmpd 752
A round bottom flask was charged with Int-32a (89 mg, 0.14 mmol), (R)- 2-(diethylamino)-2-phenylacetic acid hydrochloride (66 mg, 0.32 mmol), HATU (58 mg, 0.153 mmol) and 2 mL DMF provided 50 mg (34%) of the title Compound 752 using the capping procedure as in Step D of Compound 752. LC-MS (M+H) = 946.8.
EXAMPLE 33
Preparation of Compound 1359
Compound 1359
Step A
CS2CO3 (48.5g, 150 mmol)and dibromoethane (28g, 150mmol) was added to a stirred, solution of Int-19g (9.6g, 30mmol) in DMSO (100 mL) and the mixture was allowed to stir at 90 °C for for about 15 hours. The mixture was cooled, diluted with water (-200 mL). and extracted with EtOAc (3 x lOOmL). The combined organic extracts and a EtOAc washing was washed with brine (1 x 80 mL), dried over Na2S04. The dried layer was evaporated, the solid residue solid was triturated with methylene chloride, filtered to provide first crop of Int-33a as off white solid (4.33g). The filtrate was purified using column chromatography on silica gel 330g column, eluting with Hex/EtOAc (0 to 10% then 20%) to provide the 2nd crop of Int-33a as a off white solid (2.5g) yield 62.3%.
Step B
Int-33a (6.4g) was resolved on SFC (Chiral AD, 30% MeOH/AcCN (2: 1) in C02, to provide Int-33a' (~3g) and Int-33a" (~2.8g).
Step C
Int-33a" (0.51 g, 1.463 mmol), bis(pinacolato)diboron 0.446 g, 1.755 mmol), KOAc(0.431 g, 4.40 mmol) and PdCl2(dppf)2 (0.107 g, 0.146 mmol) were added into a microwave tube. After the flask was flashed with N2, dioxane (5 mL) was added.
The mixture was allowed to stir at 95 °C for 4 hours. The crude Int-33b was used in the next step without purification
Step D
Int-7d (0.51 g, 1.61 mmol), PdCl2(dppf)2 (0.107 g, 0.146 mmol) and
K2CO3 (1 N aq., 5 ml) were added to the reaction mixture of above mentioned Int-33b . The tube was sealed and degassed and heated to 100 °C for about 15 hours. After cooling, EtOAc (30mL) was added and it was extracted with Brine (30 mL). The organic layer was separated and dried and concentrated in vacuo. The crude material was purified on a ISCO column (40 g) and eluted with Hex:EtOAc 0% to 70% to provide the Int-33c (350 mg, 45%).
Step E
Int-33c (160mg, 0.317 mmol), Pd2(dba)3, (44mg, 0.048 mmol), X-phos
(45.3mg, 0.095 mmol), KOAc (93mg, 0.950 mmol), bis(pinacolato)diboron (88 mg, 0.349 mmol) and dioxane (3 mL) are added into a 25 mL sealed tube. After the tube was degassed in vacuo followed by flashing with N2 for three times. The mixture was allowed to stir at 120 °C for for about 15 hours. LC-MS indicated that the reaction was complete, the crude product Int-33d was used in the next step without further purification.
Step F
Int-33d (131 mg, 0.392mmol), PdCl2(dppf)2, (26 mg, 0.036mmol) and
1M K2CO3 (~3 mL) were added to the above mentioned mixture of Int-7e. The mixture was allowed to stir at 90 °C for 4 hours. After cooling down, the aqueous layer was separated and extracted with 10 mL EtOAc. The organic layers were combined and dried over anhydrous Na2SC"4. The solution was filtered and concentrated in vacuo. The product was purified using Si(>> chromatography (24 g, solvent A: DCM; solvent B:0-
50%) to provide Int-33e as desired product (95 mg, 37%).
Step G
Int-33e (95 mg) was allowed to stir in dioxane (10 mL). HC1 (4N in dioxane, 3 mL) was added and it was allowed to stir at room temperature for 1.5 hr. The solvent was removed and the Int-33f was isolated without further purification (95 mg, 100%).
Step H
Int-33f (50 mg, 0.075 mmol) was dissolved in DMF (1.5 mL) and cooled to 0°C. HATU(68.2 mg, 0.179 mmol) Compound 10A (34.1 mg, 0.157 mmol were added followed by addition of Hunig's base (0.062 mL, 0.45 mmol). The reaction was allowed to stir at 0°C for 45 minutes. Water was added to quench the reaction. The mixture purified using RP HPLC (AcCN/H2O,0-80%) to provide title Compound 1359 1 1 45 mg (52.4%).
EXAMPLE 34
Preparation of Compound 851
Compound 851
Step A
A mixture of Int-34a (0.3G,0.773 mmol), cesium carbonate (0.755g, 2.318mmol) and lR,3S,5R)-2-(tert-butoxycarbonyl)-2-azabicyclo[3.1.0]hexane-3- carboxylic acid (0.386g, 1.7mmol) in DMF(lOml) were combined in a microwave tube and heated at 40 °C. After 4 hrs. TLC indicated complete reaction. The reaction was diluted with EtOAc (30ml) washed with water (3x20ml), brine(lx20ml), dried (Na2S04), filtered and concentrated under reduced pressure to provide Int-34b as red oil. Int-34b was used in the next step with out additional purification.
Step B
Int-34b (0.59g, 0.766mmol), ammonium acetate (1.182g, 15.33mmol) and xylenes (15ml) were charged in a microwave tube and heated at 120 °C (oil bath) for 4 hrs. (Note: This reaction should be carried out in a fume-hood with shield protection). The reaction was cooled and then diluted with EtOAc (25ml) and water (25ml). The organic layer were washed with water (2x20ml), brine (1x20ml), dried (Na2S04), filtered and concentrated to provide crude Int-34c which was purified on a ISCO
chromatography system using 5%MeOH/ CH2CI2 .The relevant fraction were collected
and concentrated to provide Int-34c as an orange solid,
Step C
Standard capping procedure was used as for the Compound 851 from Int- 34c as above (41%).
EXAMPLE 35
Preparation of Intermediate Compound Int-35e
Step A
The indole phenol Int-19-g (10.0 g, 31.0 mmol), cesium carbonate (40.g, 123 mmol), and DMSO (77 ml) were added to a 500 mL round bottomed pressure flask equipped with a stir bar. 1, 1-Dichloropropane (10.09 g, 89 mmol) was added to the reaction mixture, N2 was blown over the reaction mixture, and the flask was capped. The flask was placed in a 90 °C oil bath which was then heated to 110 °C. After ~16 hours at 1 10 °C, the reaction mixture was allowed to cool to room temperature. Additional 1,1- dichloropropane was added (4 g, 35 mmol), the reaction mixture was blanketed with N2 and reheated to 110 °C. After 4.5 hours, the reaction was cooled to room temperature, and poured into 300 mL of water. EtOAc was added (500 mL) and the layers were separated. The aq. layer was extracted with additional EtOAc. The combined organic layer was washed with water and brine, gravity filtered and dried over MgS04. The mixture was filtered and the solvent concentrated under reduced pressure to provide 11.62 g of a tan solid. The crude product Int-35a was dissolved in CH2CI2 , Silica gel was added (62 g) and the mixture was concentrated in vacuo. The silica gel containing the crude product was dry loaded on a silica gel column (262 g) that had been packed with hexanes. The column was eluted with an EtOAc/hexanes gradient (0%-1.5%). The
first major peak was collected as product to provide Int-35a as an off white solid (3.1 1 g). LC/MS. Obsd. M+H = 361.8.
Int-35a (1.59 g, 4.38 mmol), PdCl2(dppf) (0.493 g, 0.674 mmol),
Bis(pinacolato)diboron (1.08 g, 4.26 mmol), and potassium acetate (1.49 g, 15.18 mmol) were added to a 20 mL microwave vial equipped with a stir bar. The vial was cycled between vacuum and nitrogen five times. Dioxane (16 ml) was added via syringe, and the vial was cycled between vacuum and nitrogen three more times. The vial was placed in a preheated reaction block and the reaction mixture was left stirring at 85 °C. After 2.5 hours, the reaction mixture was allowed to cool to room temperature. The reaction mixture was diluted with ethyl acetate and water and the layers separated. The organic layer was washed with water and brine, filtered through a pad of Celite, dried with MgS04, and filtered again. The solvent was evaporated under reduced pressure to provide Int-35b as a yellow oil. The crude product was further purified via silica gel column chromatography on an 80 g Isco Gold Si02 cartridge, using a MeOH/Ct^C gradient (0%-5%) as the mobile phase to provide Int-35b (1.29 g) as an off white foam. LC/MS. Obsd M+H = 410.11.
Int-35b (0.66 g, 1.611 mmol), Int-lOf (0.658 g, 1.682 mmol) and PdCl2(dppf) (0.120 g, 0.164 mmol) were added to 100 mL round bottomed flask equipped with a stir bar. The flask was capped with a septum, connected to a vacuum line via needle and tubing and cycled between vacuum and nitrogen five times. Dioxane (8 ml) was added via syringe, and the flask was cycled between vacuum and nitrogen three more times. Aqueous 2.0 M potassium carbonate (2.8 ml, 5.60 mmol) was added and the flask was cycled between vacuum and nitrogen five times and the flask was heated at 85 °C in a heating block. After 16.5 hours, the reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate and water and the layers were separated. The organic layer was washed with water and brine, gravity filtered, dried with MgS04, and filtered again. The solvent was evaporated under reduced pressure to provide Int-35c (1.16 g) as a brown foam. The crude product was purified further via flash silica gel column chromatography on an ISCO 80 g Si02 Gold cartridge, using a MeOH/CH2Cl2 (0%-5%) gradient as the mobile phase. The major peak was isolated as product to provide Int-35c (0.41 g) as a tan foam.
LC/MS- Obsd M+H = 594.2.
Step D
X-Phos (0.1 16 g, 0.243 mmol), Pd
2(dba)
3 chloroform adduct (0.110 g, 0.106 mmol), Bis-(pinacolato)diboron (0.175 g, 0.689 mmol), and potassium acetate (0.254 g, 2.59 mmol) were added to a 5 mL microwave tube equipped with a stir bar. The tube was capped and connected to a vacuum line via needle and tubing. The tube was cycled between vacuum and nitrogen five times. Dioxane (0.3 mL) was added via syringe and the tube was cycled between vacuum and nitrogen five times. After five minutes, a solution of Int-35c (0.44 g, 0.741 mmol) in 2.2 mL of dioxane was added via syringe. The tube was cycled between vacuum and nitrogen five more times and the tube was placed in a heating block at 120 °C. After 4h the reaction mixture was allowed to cool to room temperature and was used in the next step without further purification.
Step E
PdCl2(dppf) (81 mg, 0.111 mmol) and Int-7d (246 mg, 0.777 mmol) were added to a 5 mL microwave tube equipped with a stir bar. The tube was capped and connected to a vacuum line via needle and tubing. The tube was cycled between vacuum and nitrogen five times. The crude Int-35c was added via syringe to the tube containing the Suzuki reaction. The tube was cycled between vacuum and nitrogen three times. Aq. 2.0 M potassium carbonate (1.480 ml, 2.96 mmol) was added via syringe. The tube was cycled between vacuum and nitrogen three more times. The tube was placed in an 85 °C heating block and left stirring for about 15 hours. After ~16h, the reaction was cooled and the aq layer was removed via pipette. The remaining organic layer was diluted with 1.5 mL of DMF and 0.3 mL of water. The resulting material was passed through a micron syringe filter while injecting it directly onto an ISCO Gold C-18 cartridge. The cartridge had been conditioned with 15% acetonitrile in water. TFA (0.1%) was added to
each component of the mobile phase. The column was eluted with an acetonitrile/water gradient (15%-90% with and isocratic hold at 45% acetonitrile while the main peak eluted. Int-35d was obtained as an off white solid (262 mg). LC/MS Obsd M+H = 795.3.
Int-35d (257 mg, 0.323 mmol) and Methanol (15 ml) were added to a round bottomed flask equipped with a stir bar. HC1 in Dioxane (4.0 M) (5 ml, 20.00 mmol) was added, and the reaction mixture stirred at room temperature. After ~ 45 minutes the reaction mixture was concentrated in vacuo. Int-35e was obtained as a colored solid (Int-5060. LC/MS. Obsd M+H = 695.3. The product was used in the subsequent reactions without further purification.
EXAMPLE 36
Preparation of Compounds 814, 1450, and 1451
Amino acid Int-4f (44.6 mg, 0.205 mmol) and a solution containing Int- 35e (57 mg, 0.082 mmol), acetonitrile (410 μΐ), THF (410 μΐ), and DIPEA (71.6 μΐ, 0.410 mmol) were added to a 1 dram vial equipped with a stir bar. Propylphosphonic anhydride (aka T3P) (164 μΐ, 0.246 mmol) was added, and the reaction mixture was allowed to stir at room temperature. After 4 hours, the reaction mixture was diluted with EtOAc and water and the layers were separated. The organic layer was washed with water and brine, dried with MgSC>4, filtered, and concentrated to a brown oil. The aqueous layer was then basified with 2.0 M potassium carbonate and extracted with EtOAc and CH2CI2. The
combined organic layer was filtered, dried with MgS04, filtered again, and concentrated to dryness. The crude product was purified via silica gel column chromatography on an ISCO 4 g Si02 cartridge, using a MeOH/ CH2C12 gradient as the mobile phase to provide an off white solid Compound 814 LC/MS. Obsd. M+H = 894.3.
Compound 814 (isomer mixture at ethyl position) was separated on a Chiralcel OD column using 30% ethanol in hexanes as the mobile phase. Diethyl amine (0.1% by volume) was added to each component of the mobile phase. Two peaks were isolated that contained a molecular ion at 894.4 in the LC MS. LC/MS. Obsd. M+H = 895.0. Peak A = Compound 1450; Peak B = Compound 1451.
Int-35b (113 mg, 0.276 mmol), Int-7b (103 mg, 0.238 mmol), and PdCl
2(dppf) (26 mg, 0.036 mmol) were added to a 2 mL microwave vial equipped with a stir bar. Using the procedure for Example 35, Step C provided 129 mg of Int-37a as a clear oil. LC/MS. Obsd M+H = 636.1
X-Phos (30 mg, 0.063 mmol), Pd2(dba)3 (31 mg, 0.034 mmol), Bis(pinacolato)-diboron (47mg, 0.185 mmol), and potassium acetate (65 mg, 0.662 mmol) were added to a 2 mL microwave vial equipped with a stir bar. The vial was capped and connected to a vacuum line via needle and tubing. The vial was cycled between vacuum and nitrogen five times. A solution of Int-37a (125 mg, 0.197 mmol) in dioxane (800 μΐ) was added via syringe, and the vial was cycled between house vacuum and nitrogen five times. The vial was placed in a preheated reaction block and the reaction mixture was left stirring at 120 °C, for 3.5 hours. The reaction mixture was allowed to cool to room temperature and left stirring for about 15 hours at room temperature. The crude reaction mixture, which contains intermediate compound Int-37b, was used without further purification.
EXAMPLE 38
Preparation of Compound 1453
The microwave vial containing the Int-37b crude reaction mixture was charged with Int-7i (0.063 g, 0.145 mmol) and PdCl2(dppf) (17 mg, 0.023 mmol). The vial was recapped, and connected to a vacuum line via syringe needle and tubing. Using the procedure for Example 35, step C provided Compound 1453 as a tan solid-(43 mg). LC/MS. Obsd. M+H = 936.4.
EXAMPLE 39
Preparation of Compound 1452
Int-7i (0.070 g, 0.169 mmol) and PdCl2(dppf) (0.024 g, 0.033 mmol) were added to a 2 mL microwave tube equipped with a stir bar. The vial was capped and connected to a vacuum line via needle and tubing. The vial was cycled between vacuum and nitrogen five times. A solution of compound 1453 (0.135 g, 0.185 mmol) in dioxane (0.9 mL) was added via syringe into the reaction vial. Using the procedure for Example 35, step C provided Compound 1452 as a tan solid-(49 mg). LC/MS. Obsd. M+H = 936.4.
EXAMPLE 40
Preparation of Compound 751
Step A
Using the method described in Example 35, step B, Int-lOa (0.34 g, 0.83 mmol) was converted to Int-40a (0.24 g , 40% yield) as a brown solid. LC/MS Obs M+H = 720.4.
Step B
Using the method described in Example 35, step F, Int-40a (78 mg, 0.1 1 mmol) was converted to Int-40b 72 mg (99%) as a dihydrochloride salt. LC/MS Obs M+H = 521.2.
Step C
Using the method described in Example 36, step A, Int-40b (72 mg, 0.11 mmol) was treated with Int-2b (49 mg, 0.23 mmol) to provide Compound 751 (80 mg, 75% yield) as the dihydrochloride salt. LC/MS Obs M+H = 918.5.
EXAMPLE 41
Preparation of Compound 1491
Step A
In a 20-mL Biotage® microwave tube, cyclopropylacetaldehyde (2.0 g, 24 mmol) was dissolved in toluene (10 mL) to provide a milky mixture. Int-22a (1.78 g, 4.43 mmol) was added and the resulting red-brown suspension was allowed to stir at room temperature for 10 minutes. j9-Toluenesulfonyl chloride (85 mg, 0.443 mmol) and
toluene (2 mL) were added and the tube was flushed with nitrogen. The sealed reaction was heated and stirred under microwave conditions (Biotage® Initiator 8, reaction temperature = 170 °C; total heating time = 12 h), after which the reaction mixture was allowed to cool to room temperature . The reaction mixture was concentrated under reduced pressure (bath temperature -50-60 °C) and then coevaporated with EtOAc (2 x 100 mL) to provide a dark-brown semi-solid as crude product. The crude product was adsorbed onto 6.0 g silica gel and purified using flash silica gel chromatography (ISCO®; 200 g RediSep® Gold silica gel column; Eluent of 0-30% EtOAc/hexanes gradient @ 150 mL/min) to provide Int-41a as a light orange-yellow solid (710 mg, 34% yield).
Step B
In a 125-mL round-bottom flask, Int-41a (0.707 g, 1.51 mmol), bis(pinacolato)-diboron (0.806 g, 3.18 mmol), (dppf)PdCl2 «CH2Cl2 (111 mg, 0.151 mmol) and KOAc (445 mg, 4.54 mmol) were admixed. A magnetic stir bar was added, the flask was sealed and alternately evacuated and refilled with nitrogen (5x). Dry dioxane (7.5 mL) was added and the flask was immersed in a preheated 90 °C oil bath. After 1.5 hours the reaction mixture was allowed to cool to room temperature, diluted with EtOAc (-100 mL) and washed with brine (-50 mL). The organic layer was dried over anhydrous MgSC , filtered, and concentrated under reduced pressure (water bath temperature -50- 60 °C) to provide a dark brown semi-solid as crude product. The crude product was purified using flash silica gel chromatography (ISCO®; 120 g RediSep® Gold silica gel column; Eluent 0-70% EtOAc/hexanes gradient @ 85 mL/min) to provide Int-41b as a beige solid (630 mg, 74% yield).
Step C
In a 125-mL round-bottom flask, Int-41b (618 mg, 1.10 mmol), bromo- imidazole Int-7d (731 mg, 2.31 mmol), (dppf)PdCl2'CH2Cl2 (81 mg, 0.1 10 mmol) were admixed. A magnetic stir bar was added and the flask was sealed with a rubber septum. The flask was alternately evacuated and refilled with nitrogen (5x). Dioxane (1 1 mL) was added and the reaction mixture was allowed to stir at room temperature for 5 minutes after which aqueous potassium carbonate solution (5.5 mL, 1 M aqueous, 5.5 mmol) was
added. The reaction mixture was allowed to stir at 90 °C for 18 hours. The reaction mixture was allowed to cool to room temperature and was diluted with EtOAc (-100 mL). The resulting solution was poured into a separatory funnel containing EtOAc (-50 mL) and water (-50 mL). The organic layer was washed with brine (-50 mL). The organic layer was dried over anhydrous MgSC>4, filtered, and concentrated under reduced pressure to provide an orange-brown as crude product. The crude product was purified using flash silica gel chromatography (ISCO®; 80 g RediSep® Gold silica gel column; 0- 100% EtOAc-hexanes gradient @ 60 mL/min). Product-containing fractions were collected, concentrated, and re-purified using reverse-phase chromatography (Gilson®; Phenomenex Gemini 150 x 21.20 mm x 5 μιη column; Eluent of 10-70% MeCN/water (+0.1% TFA) gradient over 20 minutes) to provide Int-41c as a beige solid (467 mg, 54% yield).
Step D
In a 50-mL round-bottom flask, Int-41c (41 1 mg, 0.527 mmol) was dissolved in methanol (5.0 mL) and hydrogen chloride solution (1.5 mL, 4 M in dioxane, (1.8 g, 6 mmol) was added. The reaction mixture was allowed to stir at room temperature for 24 hours. The reaction mixture was concentrated under reduced pressure to provide Int-41d as a beige (396 mg, quantitative yield).
Step E
Int-4f (85 mg, 0.392 mmol), was weighed into a pre-tarred vial and transferred to a 50-mL round-bottom flask containing Int-41d (142 mg, 0.196 mmol) with the aid of dry DMF (4 x 500 μί). Diisopropylamine (200 μί, 148 mg, 1.15 mmol) was added by syringe. The mixture was allowed to stir at room temperature for -1 minutes, during which time all solids dissolved. The flask was cooled in an ice-water bath for -10 minutes. Solid HATU (157 mg, 0.412 mmol) was added in one portion at 0 °C but gradually allowed to warm to room temperature as the cooling bath expired. After 24 hrs methanol (2 mL), water (0.2 mL) and potassium carbonate (135 mg, 0.980 mmol) were added sequentially. The reaction mixture was extracted with EtOAc (2 x 50 mL), the combined extracts were washed with brine (-50 mL), and dried over anhydrous
MgS04. After filtration, the organic layer was concentrated under reduced pressure to provide a light brown solid as the crude product. Further purification by reverse-phase chromatography (Gilson®; Phenomenex® Gemini 150 x 21.20 mm x 5 μπι column; 10- 70% MeCN/water (+0.1% TFA) gradient over 15 minutes) provided Compound 1491 as a beige solid (164 mg, 86% yield).
EXAMPLE 42
Preparation of Compound 1490
In a 50-mL round-bottom flask, Int-41d (196 mg, 0.270 mmol) and Int-la (95 mg, 0.540 mmol) were admixed. A magnetic stir bar was added and the solids were dissolved in dry DMF (2.7 mL). Diisopropylethylamine (283 μί, 209 mg, 1.62 mmol) was added, the reaction mixture cooled to 0 °C (ice-water bath) and then stirred for 15 minutes. Solid HATU (216 mg, 0.567 mmol) was added in one portion and the reaction mixture was allowed to stir at 0 °C for 24 hours. Methanol (2 mL), water (0.2 mL) and potassium carbonate (187 mg, 1.35 mmol) were added and the reaction was allowed to stir at room temperature for 18 hours. Water (20 mL) was added and the reaction mixture was extracted with EtOAc (2 x 50 mL). The combined organic extracts were washed with brine (-50 mL), dried over anhydrous MgS04, filtered, and concentrated under reduced pressure to provide a light brown solid as crude product. The crude product was purified directly by reverse-phase chromatography (Gilson®; Phenomenex® Gemini 150 x 21.20 mm x 5 μηι column; 10-70% MeCN/water (+0.1% TFA) gradient over 15 minutes to provide Compound 1490 as a beige solid (151 mg, 63% yield).
EXAMPLE 43
Preparation of Compound 1499
1490 1499
In a 5-mL Biotage microwave tube, Compound 1490 (128 mg, 0.143 mmol), bis(pinacolato)diboron (73 mg, 0.286 mmol), Pd2(dba)3*CHCl3 (15 mg, 0.014 mmol) and X-Phos (14 mg, 0.029 mmol) were admixed. A magnetic stir bar was added and the tube was alternately evacuated and back-filled with nitrogen (5x). Dry dioxane (1.0 mL) was added and the reaction mixture immersed into a preheated 120 °C oil bath. After 2 hours the reaction mixture was diluted with EtOAc (-50 mL) and washed with brine (-25 mL). The organic layer was dried over anhydrous MgSC , filtered, and concentrated under reduced pressure to provide an orange-red as crude product. The crude product was purified using reverse-phase chromatography (Gilson®; Phenomenex13 Gemini 150 x 21.20 mm x 5 μιη column; 10-70% MeCN/water (+0.1% TFA) gradient over 15 minutes) to provide Compound 1499 as a beige solid (75 mg, 61% yield).
EXAMPLE 44
Preparation of Compound 1500
Using the method described in Example 43, Compound 1491 was transformed to Compound 1500 and the crude product was purified directly by reverse- phase chromatography (Gilson®; Phenomenex® Gemini 150 x 21.20 mm x 5 μιη column; 10-70% MeCN/water (+0.1% TFA) gradient over 15 minutes) to provide Compound 1500 as a beige solid (89 mg, 64% yield).
EXAMPLE 45
Preparation of Int-45a and Int-45b
Chiral SFC separation (Chiral AD, 30% MeOH/AcCN (2:1) in C02) of Int-23a yielded Compounds Int-45a and Int-45b.
EXAMPLE 46
Preparation of Compound 728
Step A
To a 250 mL round bottomed flask with a stir bar under N2 was added dibromoindole Int-45a (3 g, 6.6 mmol) followed by bis(pinacolato)diboron (3.7 g, 14.5
mmol), KOAc (1.9 g, 20 mmol), and PdCl2 (dppf) CH2Cl2 (1.6 g, 2.0 mmol). Dioxane (-45 mL) was added to the mixture which was degassed six times under house vacuum filling with N2 after each evacuation. The reaction flask was affixed with a reflux condenser and the mixture was heated to 90 °C. After 5 hours the mixture was deemed to be complete by LC-MS, and the crude bisboronate used as is.
To a cooled flask containing the crude bisboronate above was added bromo imidazole Int-4f (4.6 g, 14.5 mmol), PdCl2dppf CH2Cl2 (1.6 g, 1.98 mmol), and 1 M K2C03 (-20 mL). The flask was flushed with N2, capped, and heated to 95 °C. After 12 hours at 95 °C and the mixture was cooled to room temperature and the mixture diluted with EtOAc (100 mL) and water (20 mL). The layers were separated and the aqueous layer was extracted with EtOAc (3 x 75 mL). The organic layers were combined and were washed with brine (1 x 50 mL), dried (Na2S04), filtered, and concentrated under reduced pressure. The crude material was purified using RS ISCO Gold 220 gm column using a gradient of 100% CH2C12 to 92/8 % CH2Cl2/MeOH to provide 2.0 (39%) of Int-46a as a brown solid.
LC-MS M+H = 769.2.
Step B
To a solution of Int-46a (0.11 g, 0.14 mmol) in CH2C12 (1.5 mL) under N2 was added excess TFA (1 mL) and the resultant mixture stirred at room temperature for 2 hours. The reaction was concentrated in vacuo and then taken up in - 2-3 mL 4.0 M HC1 in dioxane and concentrated to dryness to yield Int-46b (75 mg, 99% yield) as the HC1 salt.
LC-MS M+= 568.2. Step C
To a solution of Int-46b (75 mg, 0.13 mmol) in 1.5 mL DMF (1.5 mL) at - 15 °C was added (5)-2-(methoxycarbonylamino)-2-(tetrahydro-2H-pyran-4-yl)acetic acid Int-4f (60 mg, 0.28 mmol) and HATU (0.105 g, 0.277 mmol). The mixture was allowed to stir for - 15 minutes whereupon DIPEA (0.17 mL, 0.925 mmol) was added. The mixture was allowed to stir at - 15 °C for 90 minutes whereupon H20 (3 mL) and EtOAc
(15 mL) were added. The organic layer was washed with H20 (3 X 3 mL), brine (3 x 3 mL), dried (Na2S04), filtered, and concentrated under reduced pressure. The crude material was purified using reverse-phase HPLC (Gilson) using a CI 8 column with a gradient: 0% ACN to 90% ACN/10% water (both with 0.1% TFA) to provide 120 mg (87%) of the title Compound 728 as a light yellow dihydrochloride salt after treatment with HC1. LC-MS (M+H) = 966.6.
EXAMPLE 47
Preparation of Compound 538
Step A
Using the procedure for the preparation of Int-46a, Int-45b (2.5 g, 5.5 mmol) was converted to 2.5 g (56%) of Int-47a as a brown solid. LC-MS M+H 768.4.
Step B
Using the procedure for the preparation of Int-46b, Int-47a (0.10 g, 0.14 mmol) was converted to 98 mg (99%) of Int-47b as the hydrochloride salt. LC-MS (M+H) = 568.3.
Step C
Using the method described in Example 46, step C, Int-47b (98 mg, 0.14 mmol) was treated with (S)-2-(methoxycarbonylamino)-2-(tetrahydro-2H-pyran-4- yl)acetic acid Int-4f (65 mg, 0.30 mmol) to provide 39 mg (26%) of Compound 538 as the dihydrochloride salt after HC1 treatment. LC-MS M+H 966.4.
EXAMPLE 48
Preparation of Compound 725
Step A
Following the procedure for Example 46, Step C, treatment of Int-47b (75 mg, 0.13 mmol) with (R)-2-(diethylamino)-2-phenylacetic acid hydrochloride Int-2c (68 mg, 0.28 mmol) provided 0.12 g (83%) of the title Compound 725. LC-MS (M+H) = 946.8.
Chiral SFC separation (Chiral AD, 30% MeOH/AcCN (2: 1) in C02) of Int-49 yielded Compounds Int-49a and Int-49b.
Optical rotation: Int-49b [alpha]D 23 -362.4°
EXAMPLE 50
Preparation of Compound 758
Step A
Using the procedure for the preparation of Int-46a, Int-49a (l .Og, 2.4 mmol) was converted to 0.73 g (49%) of Int-50a as a brown solid. LC-MS M+H 567.2.
Step B
To a round bottom flask charged with Int-50a (0.25 g, 0.44 mmol) and a stir bar was added MeOH (1 mL) to provide a yellow, heterogeneous mixture. 4 N HCl
in dioxane (~1 mL) was added dropwise and the resulting solution was allowed to stir for 2.5 hours at room temperature. The mixture was concentrated under reduced pressure to provide an orange solid. The solid was triturated with Et20 (4 x 4 mL), concentrated under reduced pressure, and placed under high vacuum to provide Int-50b (206 mg ,99%) of a light yellow solid. LC-MS M+H = 467.2. This material was taken without any further characterization or purification.
Step C
To a solution of Int-50b (0.24 g, 0.44 mmol) in DMF (2.5 mL) at -10 °C (ice/acetone) was added (S)-2-(methoxycarbonylamino)-3-methylbutanoic acid Int-2d (85 mg, 0.49 mmol), HATU (0.18 g, 0.49 mmol), followed by dropwise addition of DIPEA (0.23 mL, 1.3 mmol) to provide an orange, homogenous solution. The resulting solution was allowed to stir for 1 hour at -10 °C whereupon the reaction mixture was diluted with water (1.5 mL) and EtOAc (4 mL) and the layers were separated. The aqueous layer was extracted with EtOAc (3 x 4 mL) and the organic layers were combined. The organic layer was washed with brine (1 x 3 mL), dried (Na2SC>4), filtered, and concentrated under reduced pressure. The resulting orange/brown semisolid was placed under high vacuum to provide a yellow semisolid. The crude material was taken up in CH2C12 (2 mL) and was loaded onto a 40 g silica gold column. A gradient of 100% CH2C12 to 85% CH2C12/15% MeOH was run over roughly 35 minutes. The major fraction that was collected was concentrated under reduced pressure to provide 0.27 g (95%) of Int-50c an off-white solid. LC-MS (M+H) = 624.2.
Step D
To a 20 mL pressure tube with a stir bar was added Int-50c (0.30 g, 0.48 mmol) in dioxane (4 mL). Bis(pinacolato)diboron (0.13 g, 0.53 mmol), KOAc (0.14 g, 1.4 mmol), and Pd2(dba)3.CHCl3 (75 mg, 0.07 mmol), and X-phos (69 mg, 0.14 mmol) were added to the tube to provide a heterogeneous mixture. The reaction mixture was degassed under house vacuum and filled with N2 five times. The tube was capped, the reaction was heated to 120 °C. After 4 hours LC-MS (M+H 716.2) in dicated that the reaction was complete. To the cooled pressure tube containing the crude boronate was
added bromo imidazole Int-2a (0.18 g, 0.58 mmol), PdCl2dppfCH2Cl2 (79 mg, 0.096 mmol), and 1 M K2CO3 (1.4 mL). The tube was flushed with N2, capped, and heated to 95 °C for 12 hours. The reaction was then cooled to room temperature, diluted with EtOAc (100 mL) and water (20 mL). The layers were separated and the aqueous layer was extracted with EtOAc (3 x 75 mL). The organic layers were combined and were washed with brine (1 x 50 mL). dried (Na2SC>4), filtered, and concentrated under reduced pressure. The crude material was purified using RS ISCO Gold 40 gm column using a gradient of 100% CH2C12 to 90/10 % CH2Cl2 MeOH to provide 0.16 (37%) of Int-50d as a brown solid. LC-MS (M+H) = 825.4.
Step E
Using the procedure for the preparation of Int-3b, Int-50d (71 mg, 0.086 mmol) was converted to 71 mg (99%) of the free diamine as the hydrochloride salt. LC- MS (M+H) = 726.2.
Using the procedure in example 45, step C, The diamine intermediate Int- 50d' (71 mg, 0.086 mmol) was treated with (5)-2-(methoxycarbonylamino)-2- (tetrahydro-2H-pyran-4-yl)acetic acid Int-4f (20 mg, 0.094 mmol) to provide 70 mg (82%) of Compound 758 as the dihydrochlonde salt after HCl treatment. LC-MS (M+H) = 966.4.
Preparation of Compound 734
Using the methods described in Example 7, Steps A-E, compound Int-49b was converted into Compound 734. LC-MS (M+H) = 925.3.
EXAMPLE 51
Preparation of Compound 760
Step A
Using the method described in Example 50, Int-50a (0.40 g, 0.71 mmol) was treated with Int-7b (0.32 g, 0.85 mmol) after initial boronate formation to provide 0.27 g (44%) of Int-51a as an off-white solid. LC-MS (M+H) = 825.2.
Step B
Using the method described in Example 5o, Int-51a (86 mg, 0.10 mmol) was converted to 86 mg (99%) of Int-51b as the hydrochloride salt. LC-MS (M+H) = 725.4.
Step C
Using the procedure in example 50, Int-51b (86 mg, 0.10 mmol) was treated with (5)-2-(methoxycarbonylamino)-2-(tetrahydro-2H-pyran-4-yl)acetic acid Int-
4f (25 mg, 0.12 mmol) to provide 65 mg (62%) of Compound 760 as the dihydrochloride salt after HC1 treatment. LC-MS (M+H) = 924.5.
Preparation of Compound 731
In an analogous procedure, Int-50a was converted into Compound 731. LC-MS (M+H) = 924.5.
EXAMPLE 52
Preparation of Compound 762
'Using the method described in Example 50, Int-51b (86 mg, 0.10 mmol) was treated with (25',3R)-3-methoxy-2-(methoxycarbonylamino)butanoic acid Int-le (22 mg, 0.12 mmol) to provide 70 mg (69%) of Compound 762 as the dihydrochloride salt after HC1 treatment. LC-MS (M+H) = 899.4.
EXAMPLE 53
Preparation of Compound 732
Steps A-C; Example 50
lnt-49b 732
In an analogous procedure to Example 50 using (R)-2-(diethylamino)-2- phenylacetic acid hydrochloride Int-2c, and Int-49b was converted into Compound 732.
LC-MS (M+H) = 914.4.
EXAMPLE 54
Preparation of Compound 1178 & compound 1179
lnt-54a InMOf lnt-54b lnt-54c
1178 (Isomer A)
Step A
Int-54a (prepared from Int-19i, 800 mg, 1.87 mmol), bis(pinacolato)diboron (474 mg, 1.87 mmol), PdCl2 (dppf) 2 (273 mg, 0.37 mmol), and KOAc (549 mg, 5.6 mmol) were added into a 100 mL flask. After the flask was flashed with N2, dry dioxane (18 mL) was added and the reaction was allowed to stir at 90 °C for 2 hours. After cooling down, Int-lOf (624 mg, 1.87 mmol), PdCl2 (dppf)2 (136 mg, 0.19 mmol) and 1M ^COj solution (1M, 5.6 mL, 5.6 mmol) were added.
The mixture was allowed to stir at 90 °C for 4 hours and allowed to cool to room temperature . The aqueous layer was separated and extracted with 10 mL EtOAc. The organic layers were combined
and dried over anhydrous Na.S04, filtered and concentrated in vacuo. The product was purified using silica-gel chromatography (80 g, EluentrEtOAc in Hexane: 0% to 80%) to provide Int-54b (791 mg, 70.3%).
Step B
Int-54b (791 mg, 1.31 mmol), bis(pinacolato)diboron (333 mg, 1.31 mmol), Pd2(dba)3 (120 mg, 0.13 mmol), dicyclohexyl(2',4',6'-triisopropylbiphenyl-2-yl)phosphine (125 mg, 0.262) and KOAc (386 mg, 3.93 mmol) were added into a 100 mL flask. After the flask was flashed with N2, dioxane (13 mL) was added. The mixture was allowed to stir at 110 °C for 2 hours. After cooling down, Compound Int-lOf (438 mg, 1.31 mmol), PdCl2 (dppf)2 (96 mg, 0.13 mmol) and 1M K^COj solution (1M, 3.9 mL, 3.9 mmol) were added. The mixture was allowed to stir at 90 °C for an additional 4 hours and allowed to cool to room temperature. The aqueous layer was separated and extracted thrice with 10 mL EtOAc. The combined organic extracts, dried over anhydrous Na2S04, filtered and concentrated in vacuo. The product was purified using silica-gel chromatography (40 g, Eluent: EtOAC (10% MeOH) in CH2C12: 0% to 80% to provide Int-54c (364 mg, 33.8%).
Step C
Int-54c was charged in a 50 mL flask, MeOH (0.5 mL) was added and the reaction was allowed to stir at room temperature for 1 minutes. HC1 (4M in dioxane, 6.6 mL, 26.4 mmol) was then added and the solution was allowed to stir at room temperature. After 1 hr the solution was concentrated and the residue was dried in vacuo to provide Int-54d (364 mg, 100%) which were used in the next step without further purification.
Step D
Int-54d (364 mg, 0.443 mmol), (S)-2-(methoxycarbonylamino)-3-methylbutanoic acid (155 mg. 0.886 mmol), HATU (337 mg, 0.886 mmol), and DMF (4.5 mL) were added into a 40 mL flask. The reaction mixture was cooled to 0 °C and DIPEA (0.55 mL, 3.1 mmol) was added. After 1 hour, water (0.7 mL) and TFA (0.7 mL) were added at 0 °C. The solution was then stirred at room temperature for an additional 30 minutes, before concentration to an oil.. The
solution was purified using CI 8 column (80 g, CH3CN/water 10% to 70%, with 0.05% TFA) to provide Int-54e (312 mg, 60.5%).
Step E
Int-54e was resolved by Chiral SFC (Chiracel AS-H, 20x250 mm, Eluent: 40% MeOH (0.2% DEA)/C02, 50 mL/min) to provide isomer A (Compound 1178, 1st peak, 110 mg, 35.2%) and B (Compound 1179, 2nd peak, 108 mg, 34.6%).
The following Compounds were prepared as described above
EXAMPLE 55
Preparation of Compound 1353
Step A
Using the method described in Example 50 step A, Int-5a (1.0 g, 2.4 mmol) was converted to the boronate and was treated with Int-7d (0.92 g, 2.9 mmol) to provide 0.92 g (67%) of Int-55a as a brown solid. LC-MS (M+H) = 566.7.
Step B
Using the method described in Example 50, Int-55a (0.70 g, 0.1.2 mmol) was converted to the intermediate boronate followed by treatment with Int-7d (0.53 g, 1.5 mmol) to provide 0.25 g (25%) of Int-55b. LC-MS (M+H) = 811.6.
Step C
Using the method described in Example 50, Int-55b (0.25 g, 0.31 mmol) was converted to 0.23 g (99%) of Int-55c as the hydrochloride salt. LC-MS (M+H) = 611.8.
Step D Preparation of Compound 1353
Using the procedure Example 50 step E, Int-55c (23 mg, 0.31 mmol) was treated with (S)-2-(methoxycarbonylamino)-3-methylbutanoic acid Int-la (0.11 g, 0.65 mmol) to provide 0.19 g (66%) of Compound 1353 as the dihydrochloride salt. LC-MS (M+H) = 926.2.
EXAMPLE 56
A 500-mL round-bottom flask was charged with triphenyl phosphite (31 mL, 37 g, 120 mmol), dichloromethane (250 mL) and cooled for 15 minutes in a dry ice- acetone bath that was maintained at -50 to -60 °C. Bromine (6.2 mL, 19 g, 120 mmol) was added dropwise over 15 minutes through an addition funnel. Triethylamine (19 mL, 13 g, 132 mmol) and 3,5-dimethoxybenzaldehyde (Int-56a; 10.0 g, 60.2 mmol) were added sequentially. The reaction mixture was allowed to stir at -60 °C for 1 hour. The cold bath was removed and the reaction mixture was allowed to stir for a further 18 hours as the temperature was allowed to reach RT. The reaction mixture was concentrated by rotary evaporation under reduced pressure (water bath temperature -50-60 °C) to provide a dark brown, viscous liquid as crude product. The crude product was taken up in EtOAc (-100 mL) and filtered. The filtrate was concentrated under reduced pressure (water bath temperature -50-60 °C) to provide a dark brown, crude Int-56b as a viscous liquid. Int- 56b was loaded directly onto a pre-equilibrated 330 g RediSep® Gold silica gel column
and purified using flash chromatography (Isco®; Eluent: 0-5% EtOAc/hexanes gradient, to 5-70% EtOAc/ hexane to provide Int-56b as a white solid (12.8 g, 68% yield).
In the same manner, the following 1 , 1 -dibromo intermediates were prepared from the corresponding aldehydes.
EXAMPLE 57
Preparation of Compound 1286
Step A
Int-19g (10.0 g, 31 mmol), NCS (4.14 g, 31 mmol), dichloromethane (300 ml), and THF (300 ml) were added to a 1 L flask and the resulting mixture was allowed to stir at 0 °C for 1 hour and then at room temperature for 2 hours. The reaction mixture was then concentrated to a semi-solid and the residue was suspended in dichloromethane (150 ml) and filtered. The solid was washed with dichloromethane (2x15 ml) and dried to provide Int-57a as a solid (6.4 g, 58%).
Step B
Int-57a (1.0 g, 2.8 mmol), 3-methoxybenzaldehyde (0.572 g, 0.58 mmol), /?-toluenesulfonic acid (0.0053 g, 0.28 mmol), and o-xylene (10 ml) were added to a 35 mL pressure vessel. The resulting mixture was allowed to stir at 170 °C for about 15 hours under protection of a shield, cooled to room temperature, and purified on a 80 g
silica column/ Combi-Flash Rf system (Eluent: 0-5% ethyl acetate in hexanes eluent) to provide Int-57b as a gel (0.2 g, 15%).
Step C
Int-57b (200 mg, 0.421 mmol), bis(pinacolato)diboron (118 mg, 0.421 mmol), potassium acetate (207 mg, 2.1 mmol), PdCl2(dppf)-CH2Cl2 (34.4 mg, 0.042 mmol), and dioxane (5 ml) were added to a 35 mL microwave reaction tube. The sealed tube was degassed and stirred at 95 °C under nitrogen atmosphere for 4 hours then cooled to room temperature. To this mixture were added bromide Int-7d (160 mg, 0.505 mmol), PdCl2(dppf)-CH2Cl2 (34.4 mg, 0.042 mmol), 1.5 M aqueous solution of sodium carbonate (1.4 ml, 2.1 mmol). The resulting mixture was degassed and stirred at 95 °C under nitrogen atmosphere for 6 hours, cooled to room temperature, concentrated, purified using a 12 g silica column on Combi-Flash Rf system (0-60% ethyl acetate in hexanes eluent) to provide Int-57c as a wax (1 14 mg, 43%).
Step D
Int-57c (65 mg, 0.103 mmol), bis(pinacolato)diboron ( 57.5 mg, 0.226 mmol), potassium acetate (101 mg, 1.03 mmol), Pd2(dba)3-CHCl3 (21.3 mg, 0.02 mmol), X-PHOS (19.6 mg, 0.04 mmol), and dioxane (3 ml) were added to a 35 mL microwave reaction tube. The sealed mixture was degassed and stirred at 110 °C under nitrogen atmosphere for 8 hours then cooled to room temperature. To this mixture were added bromide 27 (85 mg, 0.258 mmol), PdCl2(dppf)-CH2Cl2 (16.8 mg, 0.02 mmol), 1.5 M aqueous solution of sodium carbonate (0.7 ml, 1.05 mmol). The resulting mixture was degassed and stirred at 95 °C under nitrogen atmosphere for 6 hours, cooled to room temperature, concentrated, purified using a 4 g silica column / Combi-Flash Rf system (0- 100% ethyl acetate in hexanes eluent) to provide Int-57d as a solid (39 mg, 47%).
Step E
Int-57d (39 mg, 0.048 mmol), TFA (1 ml) and dichloromethane (1 ml) were added to a 25 mL flask and stirred at room temperature for 4 hours and concentrated in vacuo. The residue was dissolved in methanol (2 ml), treated with 0.1 mL of 4.0 M
HC1 (0.4 mmol) solution in dioxane and concentrated again in vacuo to provide Int-57e as a white solid. This crude product was used for the next reaction without purification.
Step F
Diamine Int-57e , Valine-MOC acid Int-la (14.3 mg, 0.081 mmol), and DMF (1 ml) were added to a 50 mL flask and cooled to 0 °C. To this cooled solution was added HATU (30 mg, 0.08 mmol) and the reaction was allowed to stir at 0 °C over a period of 1 hour, quenched with water (3 drops). The reaction mixture was purified using reverse phase chromatography (0-90% acetonitrile in water with 0.1% TFA eluent) to provide Compound 1286 as a wax (13 mg, 31%). LC/MS anal, calcd. for: C51H57N9O8 923.4; Found: 924.5 (M+H)+.
EXAMPLE 58
Preparation of Compound 1198 & Compound 1199
1198 1199
Compound 1198 (20 mg, 0.020 mmol), trimethylboroxine (7.67 mg, 0.061 mmol), Pd2(dba) 3 (3.73 mg, 4.07 μπιοΐ), and dicyclohexyl(2',4',6'-triisopropylbiphenyl-2- yl)phosphine (3.88 mg, 8.14 μηιοΐ) are added into a 50 mL flask. After the flask was flashed with N2, 1,4-Dioxane (204 μΐ) and K2C03 (61.1 μΐ, 0.061 mmol) was added. The mixture was allowed to stir at 1 10 °C for 16 hours. After cooling down, the aqueous layer was separated and extracted with 5 mL EtOAc. The organic layers were combined and dried over anhydrous Na2S04. The solution was filtered and concentrated in vacuo.
The solution was concentrated and purified using S1O2 chromatography (24 g, MeOH (Eluent: 10% concentrated MeOH/NH3-H2°) in CH2CI2, 0% to 80%) to provide Compound 1199 (15 mg, 77%).
The following compound was made using the method described in the Example above:
EXAMPLE 59
Preparation of Compound 1014
Step A
In a 250-mL round-bottom flask, Int-19b (2.006 g, 5.47 mmol) was dissolved in DMSO (22 mL). Neat dibromide Int-56c (1.710 g, 6.01 mmol) was added, followed by solid cesium carbonate (5.34 g, 16.4 mmol). The reaction mixture was immersed into a preheated 90 °C oil bath and stirred for 18 hours, then allowed to cool to room temperature, and poured into water (-100 mL), whereupon a tan solid precipitated. The aqueous mixture was extracted with EtOAc (2 x 100 mL). The combined organic phases were washed with brine (-50 mL), then dried over anhydrous MgSC filtered,
and concentrated in vacuo to provide a tan-brown solid as crude product. The crude product was purified using flash silica gel chromatography (Isco®; 220 g RediSep® Gold silica gel column; Eluent: 0-30% EtOAc/hexanes gradient to provide Int-59a as a pale yellow solid (683 mg, 26% yield).
Step B
In a 20-mL Biotage® microwave tube was charged with stir basr was added Int-59a (670 mg, 1.37 mmol), bis(pinacolato)diboron (695 mg, 2.74 mmol), (dppf PdCl2,CH2Cl2 (68 mg, 0.083 mmol) and potassium acetate (403 mg, 4.11 mmol). The tube was alternately evacuated and back-filled with nitrogen 5 times. Dioxane (14 mL) was added and the tube was immersed into a preheated 90 °C oil bath. After 1.5 hours, then reaction was allowed to cool to room temperature, diluted with EtOAc (~20 mL) and filtered through a Celite® pad. The pad was rinsed with EtOAc (-50 mL) and the combined filtrate were washed with brine (-25 mL), dried over anhydrous MgSC>4, filtered, and concentrated under reduced pressure to provide an light brown solid as crude product. The crude product was purified using flash silica gel chromatography (Isco®; 40 g RediSep® Gold silica gel column; Eluent 0-50% EtOAc/hexanes gradient) to provide Int-59b as a beige solid (705 mg, 88% yield).
Step C
A 20-mL Biotage® microwave tube was charged with a stir bar, Int-59b (700 mg, 1.20 mmol), bromoimidazole Int-7d (834 mg, 2.64 mmol), and
(dppf)PdCl2 ,CH2Cl2 (49 mg, 0.060 mmol). The tube was alternately evacuated and refilled with nitrogen (5x). Dioxane (8 mL) was added and the reaction mixture was allowed to stir at room temperature for 5 minutes. Aqueous potassium carbonate solution (6 mL, 1 M aqueous, 6 mmol) was then added and the reaction was immersed into a preheated 90 °C oil bath. After 18 hours the reaction was allowed to cool to room temperature and was diluted with EtOAc (-50 mL), filtered through a polyethylene filter frit and the filtrate was washed with brine (-25 mL). The organic layer was dried over anhydrous MgS04, filtered, and concentrated under reduced pressure to provide an orange-brown solid as crude product. The crude product was purified using flash silica
gel chromatography (Isco ; 120 g RediSep Gold silica gel column; Eluent 0-10%
MeOH/CH2C12 gradient) to provide Int-59c as a beige solid (719 mg, 75% yield).
Step D
A 5-mL Biotage® microwave tube was charged with a stir bar, Int-59c (130 mg, 0.162 mmol), (dba)3Pd2 »CHCl3 (25 mg, 0.024 mmol) and X-Phos (23 mg, 0.049 mmol). The tube was sealed and alternately evacuated and back-filled with nitrogen (5x). 5-methylthienyl-2-boronate (36 mg, 0.16 mmol), dissolved in dioxane (1.6 mL), and potassium carbonate (0.8 mL, 1 M aqueous; 0.8 mmol) were added by syringe. The tube was immersed in preheated 120 °C oil bath and stirred for 4 hours. The reaction mixture was then cooled, diluted with EtOAc (-50 mL), filtered, and washed with brine (-25 mL). The organic layer was dried over anhydrous MgS04, filtered, and
concentrated under reduced pressure to provide the crude product as a golden yellow solid. Further purification by reverse-phase chromatography (Gilson®; Phenomenex® Gemini 150 x 21.20 mm x 5 μηι column; 10-70% eCN/water (+0.1% TFA) gradient over 20 minutes) to provide Int-59d as a beige solid (26 mg, 19% yield).
Step E
In a 50-mL round-bottom flask Int-59d (20 mg, 0.03 mmol) was dissolved in methanol (500 μί) and HC1 solution (60 μί, 4 M in dioxane, 0.240 mmol) was added. The clear, pale yellow tinged solution was allowed to stir at room temperature for 24 hours. The reaction mixture was concentrated in vacuo to provide Int-59e a beige solid (15.6 mg, 83% yield).
Step F
In a 50-mL round-bottom flask, Int-59e (16 mg, 0.019 mmol) and Int-la (7 mg, 0.040 mmol) were dissolved in DMF (200 μί). Diisopropylethylamine (20 xL, 15 mg, 0.118 mmol) was added and the reaction mixture was cooled in an ice-water bath for 15 minutes. Solid HATU (15 mg, 0.039 mmol) was added slowly and the reaction allowed to warm slowly to room temperature. After 3 hours the reaction was purified directly by reverse-phase chromatography (Gilson®; Phenomenex® Gemini 150 x 21.20
mm χ 5 μπι column; 10-70% MeCN/water (+0.1% TFA) gradient over minutes) to provide Compound 1014 as a beige solid (12 mg, 62% yield).
EXAMPLE 60
Preparation of Compound 1005
Step A
A 5-mL Biotage® microwave tube equipped with a magnetic stir bar was added, Int-59c (254 mg, 0.317 mmol), phenylboronic acid (77 mg, 0.634 mmol), Pd2(dba)3'CHCl3 (66 mg, 0.063 mmol) and X-Phos (61 mg, 0.127 mmol). The tube was sealed and alternately evacuated and back-filled with nitrogen (5x). Dioxane (3 mL) and potassium carbonate (0.78 mL, 1 M aqueous; 0.78 mmol) were added and the reaction immersed in a preheated 110 °C oil bat. After 22 hours the reaction was allowed to cool, diluted with EtOAc (-30 mL), and washed sequentially with water (-20 mL) and brine (-20 mL). The organic layer was dried over anhydrous MgSC^, filtered, and concentrated in vacuo to provide the crude product as a light brown solid. The crude product was purified using flash silica gel chromatography (Isco®; 40 g RediSep® Gold silica gel column; Eluent 0-10% MeOH/CH2Cl2 gradient) to provide Int-60a (134 mg, 50% yield) as a pale yellow-orange solid.
Step B
In a 125-mL round-bottom flask, Int-60a (100 mg, 0.118 mmol) was dissolved in methanol (1.2 mL). Hydrogen chloride solution (0.300 mL, 4 M in dioxane, 1.2 mmol) was added and the reaction mixture was allowed to stir at room temperature. After 17 hours the reaction mixture was concentrated under reduced pressure to provide a golden-brown solid, which was dried in a vacuum oven (house vacuum, ~60 °C) for 20 hours to provide Int-60b as a golden brown solid (99 mg, quantitative yield).
Step C
To a 50-mL flask equipped with a stir bar was added Int-60b (39 mg, 0.047 mmol) and Int-la (17 mg, 0.094 mmol) , and dry DMF (472 L).
Diisopropylethylamine (41 μί, 31 mg, 0.236 mmol) was added and the reaction mixture was cooled to 0 °C in an ice-water bath. After -15 minutes, solid HATU (40 mg, 0.104 mmol) was added and the reaction mixture stirred at 0 °C, After 2 hours the reaction was quenched by addition of water (20 mL), whereupon a beige solid precipitated. The solid was collected by vacuum filtration and washed further with water (-50 mL). The solid was dissolved in EtOAc (-100 mL) and the resulting solution was washed with brine (-25 mL). The organic layer was collected, dried over MgS04, filtered, and concentrated under reduced pressure to provide a beige crude product. Further purification by reverse phase CI 8 chromatography (Gilson®, Phenomenex® Gemini C18 5 μπι 150 x 21.20 mm column, Eluent: 10-70% MeCN/water + 0.1% TFA over 20 minutes @ 20 mL/min} afford Compound 1005 as a beige solid (28.4 mg, 63% yield).
EXAMPLE 61
Preparation of Compounds 1166, 1171 & 1173
Step A
Compound Int-61a (150 mg, 0.179 mmol, prepared by a similar route as in example 59), biphenyl-4-ylboronic acid (35.4 mg, 0.179 mmol), Pd2(dba)3 (18.5 mg, 0.018 mmol), and dicyclohexyl(2',4',6'-triisopropylbiphenyl-2-yl)phosphine (17 mg, 0.036 mmol) were added into a 40 mL flask. The flask was put in vacuo and filled with N This process was repeated once. Dioxane (1.8 mL) and K2CO3 (1M, 0.9 mL, 0.9 mmol) were added, and the sealed flask was allowed to stir at 110 °C. After 3 hours the reaction was cooled, the aqueous layer was separated and extracted with 3 mL EtOAc. The organic layers were combined and dried over anhydrous Na2S04, filtered and concentrated to provide crude product. Further purification by Silica-gel chromatography (Eluent: EtOAC (10% MeOH) in CH2C12: 0% to 80%) gave Compound lnt-61 a (137 mg, 80%).
Steps B to D were carried out using the methods described in Example 50.
The following compound was made using the method described in the Example above:
EXAMPLE 62
Preparation of Compound 1528
Step A
To a 250 mL flask were added Int-19g (5.0 g, 15.5 mmol), dibromide Int- 56h (5.8 g, 80% purity, 15.5 mmol), cesium carbonate (25.3 g, 77 mmol), and acetonitrile (50 ml) and the resulting suspension was allowed to stir at 60 °C for about 15 hours. Ethyl acetate (200 ml) was then added, and the organic layer was washed with water (2x150 ml), dried over sodium sulfate, and concentrated in vacuo. The residue was purified on a 120 g silica column/ Combi-Flash Rf system(Eluent: 0-10% ethyl acetate in hexanes) to provide Int-62a as a white solid (3.7 g, 52%).
Step B
Intermediate Int-62a (500 mg, 1.09 mmol), bis(pinacolato)diboron ( 304 mg, 1.2 mmol), potassium acetate (535 mg, 5.45 mmol), PdCl2(dppf)-CH2Cl2 (89 mg, 0.109 mmol), and dioxane (8 ml) were added to a 35 mL microwave reaction tube. The sealed mixture was degassed and stirred at 95 °C under nitrogen atmosphere for 4 hours and then cooled to room temperature. To this mixture were added bromide Int-12o (429 mg, 1.31 mmol), PdCl2(dppf)-CH2Cl2 (89 mg, 0.109 mmol), 1.5 M aqueous solution of sodium carbonate (3.6 ml, 5.4 mmol). The resulting mixture was degassed and stirred at 95 °C under nitrogen atmosphere for 6 hours, cooled to room temperature, concentrated, to provide crude product. Further purification was accomplished on by a 40 g pre-packed silica gel column/ Combi-Flash Rf system (Eluent: 0-90% ethyl acetate in hexanes) to provide Int-62b as a wax (530 mg, 78%).
Step C
Int-62b (130 mg, 0.207 mmol), bis(pinacolato)diboron ( 58 mg, 0.23 mmol), potassium acetate (102 mg, 1.04 mmol), Pd2(dba)3-CHC13 (21.5 mg, 0.02 mmol), X-PHOS (19.8 mg, 0.04 mmol), and dioxane (2 ml) were added to a 35 mL microwave reaction tube. The sealed mixture was degassed and stirred at 110 °C under nitrogen atmosphere for 8 hours then cooled to room temperature. To this mixture were added bromide Int-7b (78 mg, 0.21 mmol), PdCl2(dppf)-CH2Cl2 (14.2 mg, 0.02 mmol), 1.5 M aqueous solution of sodium carbonate (0.6 ml, 0.9 mmol). The resulting mixture was degassed and stirred at 95 °C under nitrogen atmosphere for 6 hours, cooled to room temperature, concentrated to provide crude product. Further purification on a 4 g silica gel pre-packed column/ Combi-Flash Rf system (Eluent: 0-100% ethyl acetate in hexanes) provided Int-62c as a solid (105 mg, 68%).
Step D
Int-62c (98 mg, 0.11 mmol), TFA (1 ml) and dichloromethane (1 ml) were added to a 25 mL flask. The resulting solution was allowed to stir at room temperature for 4 hours and concentrated in vacuo. The residue was dissolved in methanol (2 ml), treated with 0.1 mL of 4.0 M HC1 (0.4 mmol) solution in dioxane and concentrated again in vacuo to provide Int-62d as a solid (99 mg, 100%).
Step E
Int-62d (30 mg, 0.034 mmol), acid Int-la (6.5 mg, 0.04 mmol), and DMF (1 ml) were added to a 25 mL flask and the resulting solution was cooled to 0 °C. To this cooled solution was added HATU (13 mg, 0.034 mmol) and the reaction was allowed to stir at 0 °C, After lh , water (3 drops) was added and the reaction directly purified using reverse phase chromatography (10-80% acetonitrile in water with 0.1% TFA eluent ) to provide Compound 1528 as a white solid (5 mg, 13%). LC/MS anal, calcd. for:
C5iH56FN908 941.4; Found: 942.5 (M+H)+.
EXAMPLE 63
Preparation of Compound 1496
Step A
Into a 250-mL round-bottom flask equipped with a stir bar , dibromoindole Int-19b (4.41 g, 12.02 mmol), Int-56c (4.47 g, 14.4 mmol) were added
and dissolved in dry DMSO (50 mL). Solid cesium carbonate (20 g, 61 mmol) was added. The reaction mixture was allowed to stir at 100 °C for 14 hours. Water (-150 mL) was added to the reaction mixture, whereupon a beige solid precipitated. The suspension was extracted with EtOAc (3 x 250 mL). Combined extracts were washed with brine (-250 mL). The organic layer was dried over anhydrous MgS04, filtered, and
concentrated under reduced pressure to provide the crude product as a light orange-brown solid. The crude product was adsorbed onto silica gel (10.0 g), and then further purified using flash silica gel chromatography (Isco®; 330 g RediSep® Gold silica gel column; Eluent: 0-10% EtOAc/hexanes gradient) to provide Int-63a as a light brown solid (2.77 g, 46% yield).
Step B
A 125-mL round-bottom flask equipped with a stir bar was charged with Int-63a (1.46 g, 2.90 mmol), bis(pinacolato)diboron (1.55 g, 6.09 mmol),
(dppf)PdCl2'CH2Cl2 (106 mg, 0.145 mmol) and KOAc (854 mg, 8.71 mmol). The reaction was sealed and alternately evacuated and refilled with nitrogen (5x). Dry dioxane (19 mL) was added and the flask was immersed in a preheated 90 °C oil bath. After 1 hour the reaction mixture was allowed to cool to room temperature, diluted with EtOAc (100 mL), filtered through a polyethylene frit and washed with brine (-50 mL). The organic layer was dried over anhydrous MgSC , filtered, and concentrated to provide the crude product as a dark-brown semi-solid. The crude product was purified using flash silica gel chromatography (Isco®; 220 g RediSep® Gold silica gel column; Eluent: 0-30% EtOAc/hexanes gradient) to provide Int-63b (1.09 g, 63% yield).
Step C
A 125-mL round-bottom flask equipped with a stir bar was charged with Int-63b (707 mg, 1.184 mmol), bromoimidazole Int-7d (786 mg, 2.49 mmol),
(dppf PdCl2 »CH2Cl2 (87 mg, 0.1 18 mmol). The flask was alternately evacuated and refilled with nitrogen (5x). Dioxane (12 mL) was added and the reaction mixture was allowed to stir at room temperature for 5 minutes. Aqueous potassium carbonate solution (6 mL, 1 M aqueous, 6 mmol) was then added. The reaction mixture was allowed to stir
at 90 °C for 2.5 hours, cooled to room temperature and was diluted with EtOAc (-50 mL). The resulting solution was poured into a separatory funnel containing EtOAc (-50 mL) and water (-50 mL). The organic layer was washed with brine (-50 mL). The organic layer was dried over anhydrous MgS04, filtered, and concentrated under reduced pressure to provide the crude product as an orange-brown solid. The crude product was further purified using flash silica gel chromatography (Isco®; 120 g RediSep® Gold silica gel column; Eluent : 0-100% { 10% MeOH/EtO Ac }— hexanes gradient) to provide Int- 63c a light brown solid (644 mg, 67% yield).
Step D
In a 50-mL round-bottom flask, Int-63c (633 mg, 0.776 mmol) was dissolved in methanol (8.0 mL) and hydrogen chloride solution (2.0 mL, 4 M in dioxane, (2.4 g, 8 mmol) was added. The reaction mixture was allowed to stir at room temperature for 24 hours. The reaction mixture was concentrated under reduced pressure to provide Int-63d as a beige solid (572 mg, 97% yield).
Step E
Int-4f (57 mg, 0.263 mmol) was dissolved in dry DMF (1.3 mL). The resulting solution was added to a 50-mL round-bottom flask containing solid Int-63d
(100 mg, 0.131 mmol). N,N-Diisopropylethylamine (140 μί, 104 mg, 0.802 mmol) was added and the mixture agitated by sonication until no more solid adhered to the walls of the flask. The reaction mixture was allowed to stir at 0 °C (ice-water bath) for -15 minutes. Solid HATU (110 mg, 0.289 mmol) was added and the reaction mixture was allowed to stir at 0 °C. After 1.5 hours the reaction was diluted with methanol (1 mL), and water (-0.1 mL) and solid potassium carbonate (36 mg, 0.263 mmol) were added sequentially. After 24 hours the reaction mixture was partitioned between EtOAc (-100 mL) and brine (-25 mL). The aqueous layer was extracted with a second portion of EtOAc (-25 mL). The combined extracts were washed with brine (-25 mL), dried over anhydrous MgS04, filtered, and concentrated under reduced pressure to provide crude product as a light brown solid.. The crude product was purified directly by reverse-phase chromatography (Gilson®; Phenomenex Gemini 150 x 21.20 mm x 5 μπι column; Eluent:
0-70% MeCN/water (+0.1% TFA) gradient over 15 minutes) to provide the targeted product Compound 1496 as a white solid (84 mg, 63% yield).
EXAMPLE 64
Preparation of Compounds 1002, 1024, & 1025
Step A
A 250-mL round-bottom flask was charged with Int-19b (3 g, 8.2 mmol) and DMSO (35 mL). The 1,1-dibromide Int-56g (2.5 g, 8.1 mmol) and cesium carbonate (8.0 g, 25 mmol) were added and the mixture was heated with stirring at 90 °C for 18 hours. The reaction mixture was poured into water (-300 mL) and extracted with EtOAc (3 x 250 mL). Combined extracts were washed with brine (-250 mL). The organic layer was dried over anhydrous MgSC , filtered, and concentrated under reduced pressure to provide the crude product as a brown oil. The crude product was adsorbed onto 8.5 g silica gel and further purified using flash silica gel chromatography (Isco®; 300 g RediSep® Gold silica gel column; Eluent: 0-50% EtOAc/hexanes gradient) to provide Int-64a (751 mg, 18% yield).
Step B
In a 20-mL Biotage® microwave tube, Int-64a (276 mg, 0.535 mmol), bis(pinacolato)diboron (220 mg, 0.866 mmol), (dppf)PdCl «CH2Cl2 (34 mg, 0.042 mmol) and KOAc (122 mg, 1.24 mmol) were added. The tube was sealed and alternately evacuated and refilled with nitrogen (5x). Dry dioxane (3.5 mL) was added and the reaction mixture was allowed to stir until homogeneity was achieved (< 1 minutes). The tube was immersed in a preheated 90 °C oil bath and stirred for 45 minutes.
The reaction mixture was cooled, diluted with EtOAc (~10 mL) and filtered through a Celite® pad with washing (EtOAc). The combined filtrates were washed with brine (-25 mL), dried over anhydrous MgS04, filtered, and concentrated under reduced pressure to provide the crude product as an orange-brown solid. Further purification by flash silica gel chromatography (Isco®; 40 g ediSep® Gold silica gel column; Eluent: 0-30% EtOAc/hexanes gradient) to provide Int-64b as an off-white solid (127 mg, 39% yield).
Step C
In a 20-mL Biotage® microwave tube, Int-64b (122 mg, 0.200 mmol), bromoimidazole Int-7d (133 mg, 0.420 mmol), and (dppf)PdCl2 «CH2Cl2 (16 mg, 0.020 mmol) were mixed. The tube was sealed and alternately evacuated and back-filled with nitrogen (5x). Dioxane (2 mL) and potassium carbonate (0.60 mL, 1 M aqueous; 0.60 mmol) were added and the reaction immersed in a preheated 90 °C oil bath. After 17 hours the reaction mixture was allowed to cool, diluted with EtOAc (-100 mL) and was washed with brine (-50 mL). The organic layer was dried over anhydrous MgS04, filtered, and concentrated under reduced pressure to provide a light brown solid. The crude product was purified using flash silica gel chromatography (Isco ; 24 g RediSep Gold silica gel column; Eluent: 0-60% MeOH/CH2Cl2 gradient) to provide Int-64c as a beige solid (111 mg, 67% yield).
Step D
In a 50-mL round-bottom flask, Int-64d (101 mg, 0.122 mmol) was dissolved in methanol (2.0 mL) and HC1 solution (300 μί, 4 M in dioxane, 1.2 mmol) was added. The pale yellow solution was allowed to stir at room temperature for 23
hours, then concentrated under reduced pressure to provide Intermediate 900D (100 mg, -100% yield) as a pale yellow powder..
Step E
A 50-mL flask was charged with Int-64d (55 mg, 0.072 mmol) and Int-la (25 mg, 0.143 mmol) and dissolved in dry DMF (716 \xL). Diisopropylethylamine (61xL, 46 mg, 0.358 mmol) was added and the reaction mixture was cooled to 0 °C in an ice-water bath. After -15 minutes, solid HATU (57 mg, 0.150 mmol) was added . After 3 hours at 0 °C the reaction was quenched by addition of water (20 mL), whereupon a beige solid precipitated. The solid was collected by vacuum filtration and washed again with water (-50 mL). The solid was dissolved in EtOAc (-100 mL) and the resulting solution was washed with brine (-25 mL). The organic layer was collected, dried over MgS04, filtered, and concentrated under reduced pressure to provide the crude product. Further purification by reverse phase CI 8 chromatography (Gilson®, Phenomenex Gemini C18 5 μπι 150 x 21.20 mm column, Eluent: 10-70% MeCN/water + 0.1% TFA) provided Compound 1002 as a beige solid (26 mg, 39% yield).
Step F: Isomer separation by HPLC.
Compound 1002 (48.8 mg) was dissolved in abs. EtOH (1.0 mL) and the solution was filtered through a Whatman Puradisc 13 mm syringe filter. The sample was injected onto a Phenomenex Lux Cellulose-2 (5 μηι, 150 x 21.20 mm) semi-preparative column; detection wavelength = 350 nm. Elution with 50% EtOH/hexane @ 10 mL/min provided the first peak (eluted between t = 0.5 minutes and t = 35 minutes) which was collected and concentrated to provide Compound 1024 (15 mg) as an off-white solid. The eluent solvent polarity was increased to 60% EtOH/hexane at t = 120 minutes while maintaining a flow rate of 10 mL. The second component, (between t = 125 minutes and t = 185 minutes), was collected and concentrated to provide Compound 1025 (15 mg) as an off-white solid.
EXAMPLE 65
Preparation of Compound 1019
Step A
In a 250-mL round-bottom flask, the 2-(hydroxyphenyl)indole Int-19g (3.03 g, 9.4 mmol) and gem-dibromide Int-56g (8.7 g, 28 mmol) were mixed and dissolved in dry DMSO (94 mL). Solid cesium carbonate (21 g, 66 mmol) and a magnetic stir bar were added. The reaction mixture was allowed to stir at 100 °C for 21 hours. Water (-500 mL) was added to the reaction mixture, whereupon a beige solid precipitated. The suspension was extracted with EtOAc (3 x 250 mL) and the combined extracts were washed with brine (-250 mL). The organic layer was dried over anhydrous MgS04, filtered, and concentrated under reduced pressure to provide the crude product as
a dark orange-brown solid. The crude product was adsorbed onto silica gel (10 g), and then purified using flash silica gel chromatography (Isco®; 120 g RediSep® Gold silica gel column; Eluent 0-50% EtOAc/hexanes gradient) to provide Int-65a as a light brown solid (1.80 g, 41% yield).
Step B
In a 125-mL round-bottom flask, Int-65a (2.644 g, 6.18 mmol), bis(pinacolato)diboron (1.57 g, 6.18 mmol), (dppf)PdCl2 »CH2Cl2 (138 mg, 0.168 mmol) and KOAc (1.65 g, 16.85 mmol) were mixed. The reaction was alternately evacuated and refilled with nitrogen (5x) followed by dry dioxane (38 mL). The flask was immersed in a preheated 90 °C oil bath and the reaction mixture, was allowed to stir at 90 °C for 2 hours. The reaction mixture was allowed to cool to room temperature, diluted with EtOAc (-300 mL) and washed with brine (-200 mL). The organic layer was dried over anhydrous MgS04, filtered, and concentrated in vacuo to provide a dark yellow solid, which was purified using flash silica gel chromatography (Isco®; 120 g RediSep® Gold silica gel column; Eluent: 0-50% EtOAc hexanes gradient) to provide Int-65b as yellow solid (1.99 g, 68% yield).
Step C
In a 125-mL round-bottom flask, the boronate Int-65b (1.14 g, 2.21 mmol), bromoimidazole Int-7d (750 mg, 2.37 mmol), (dppi)PdCl2 »CH2Cl2 (90 mg, 0.110 mmol) were mixed. The flask was alternately evacuated and refilled with nitrogen (5x) and dry dioxane (15 mL) was added. The reaction mixture was allowed to stir at room temperature for 5 minutes and then aqueous potassium carbonate solution (11 mL, 1 M aqueous, 11 mmol) was added. The flask was immersed into a preheated 90 °C oil bath and stirred at 90 °C for 3 hours. The reaction mixture was allowed to cool to room temperature, diluted with EtOAc (-100 mL) and the resulting solution was filtered and washed with brine (-50 mL). The organic layer was dried over anhydrous MgS04, filtered, and concentrated ration under reduced pressure to provide an orange-brown solid. The crude product was purified using flash silica gel chromatography (Isco®; 220 g
RediSep Gold silica gel column; Eluent: 0-100% EtOAc/hexanes) to provide Int-65c as a golden yellow solid (1.06 g, 76% yield).
Step D
In a 150-mL round-bottom flask, substrate Int-65c (754 mg, 1.202 mmol) was dissolved in methanol (12 mL) and HC1 solution (3 mL, 4 M in dioxane, 12 mmol) was added. The clear, pale yellow solution was allowed to stir at room temperature for 18 hours. The reaction mixture was concentrated by rotary evaporation under reduced pressure to provide Intermediate Int-65d as a pale yellow solid (728 mg, quantitative yield).
Step E
In a 50-mL round-bottom flask, Int-65d (719 mg, 1.20 mmol) and Int-la (231 mg, 1.318 mmol) were dissolved in dry DMF (12 mL). Diisopropylethylamine (1.0 mL, 774 mg, 5.99 mmol) was added and the he reaction mixture cooled to 0 °C (ice- water bath). After 15 minutes., solid HATU (684 mg, 1.80 mmol) was added in one portion. The reaction mixture was allowed to stir at 0 °C for 1 hour. Water (-20 mL) was added and the precipitated solid was collected by vacuum filtration. The collected solid was washed with water (~5 mL) and air-dried. The crude product was subsequently purified using flash silica gel chromatography (Isco®; 40 g RediSep® Gold silica gel column; Eluent: 0-10% MeOH/CH2Cl2 gradient). All product-containing fractions were collected, concentrated, and re-purified using flash silica gel chromatography (Isco®; 80 g RediSep® Gold silica gel column; Eluent: 0-3.5% MeOH/CH2Cl2 gradient) to provide Int-65e (286 mg, 35% yield).
Step F
In a 50-mL round-bottom flask, Int-65e (285 mg, 0.417 mmol), bis(pinacolato)diboron (127 mg, 0.500 mmol), Pd2(dba)3*CHCl3 (43 mg, 0.042 mmol), X-Phos (40 mg, 0.083 mmol) and KOAc (123 mg, 1.25 mmol) were mixed. The flask was alternately evacuated and refilled with nitrogen (5x). Dioxane (3 mL) was added and the reaction was allowed to stir at 120 °C for 1.5 hours. The reaction mixture was
allowed to cool slowly to room temperature for 12 hours. The reaction mixture was diluted with EtOAc (-100 mL) and washed with brine (~50 mL). The organic layer was dried over anhydrous MgS04, filtered, and concentrated under reduced pressure to provide an orange solid as crude product. The crude product was purified using flash silica gel chromatography (Isco®; 40 g RediSep® Gold silica gel column; Eluent: 0-9% MeOH/Cf Cb gradient) to provide Int-65f as an orange-yellow foamy solid (253 mg, 78% yield).
Step G
In a 5-mL Biotage® microwave tube, Int-65f (123 mg, 0.159 mmol), bromoimidazole Int-lOf (64 mg, 0.190 mmol), (dppf)PdCl2 »CH2Cl2 (13 mg, 0.016 mmol) were mixed. The tube was alternately evacuated and refilled with nitrogen (5x), dry dioxane (1.5 mL) was added and the reaction mixture stirred at room temperature for 5 minutes. Aqueous potassium carbonate solution (0.800 mL, 1 M aqueous, 0.8 mmol) was then added. The tube was immersed into a preheated 90 °C oil bath and the reaction was allowed to stir for 16 hours. The reaction mixture was allowed to cool to room temperature, diluted with EtOAc (-50 mL), and filtered through a polyethylene filter frit. The filtrate was washed with brine (-25 mL), dried over anhydrous MgS04, filtered, and concentrated under reduced pressure to provide a light brown solid. The crude product was purified using flash silica gel chromatography (Isco®; 24 g RediSep® Gold silica gel column; Eluent: 0-9% MeOH/CH2Cl2 gradient) to provide Int-65g as a beige solid (92 mg, 64% yield).
Step H:
In a 50-mL round-bottom flask, the Int-65g (73 mg, 0.081 mmol) was dissolved in methanol (0.8 mL) and hydrogen chloride solution (200 μί, 4 M in dioxane, (240 mg, 0.800 mmol) was added. The reaction mixture was allowed to stir at room temperature for 18 hours. The reaction mixture was concentrated under reduced pressure to provide Int-65h as a beige solid (77 mg, quantitative yield).
Step I:
In a 50-mL round-bottom flask, Int-65h (73 mg, 0.080 mmol) and Int-la (17 mg, 0.096 mmol) were mixed and dry DMF (1 mL) was added.
Diisopropylethylamine (70 μί, 53 mg, 0.412 mmol) was added and the reaction cooled to 0 °C (ice-water bath). After 15 minutes, solid HATU (46 mg, 0.120 mmol) was added in one portion. The reaction mixture was allowed to stir at 0 °C for 1 hour. Water (-20 mL) was added and the precipitated solid was collected by vacuum filtration. The collected solid was washed with water (~5 mL), air-dried briefly, dissolved in DMF (-1 mL) and purified using reverse-phase C18 chromatography (Gilson®, Phenomenex® Gemini CI 8 5 μπι 150 x 21.20 mm column, Eluent: 10-70% MeCN/water + 0.1% TFA) to provide Compound 1019 (21 mg, 28% yield) as a beige solid.
EXAMPLE 66
Preparation of Compound 1033
Step A
In a 200-mL pear-shaped flask was charged with Int-19c (1.64 g, 4.26 mmol), Int-56g (2.64 g, 8.52 mmol), DMSO (17 mL) and stirred until homogeneous.
Solid cesium carbonate (10 g, 66 mmol) was added, the flask fitted with a condenser and then immersed into a preheated 100 °C oil bath. After 18 hours the reaction mixture was poured into water (-400 mL) and Extracted with EtOAc (2 x 150 mL, 1 x 300 mL). The aqueous layer was diluted with brine (-200 mL) and was extracted with EtOAc (-150 mL). The combined organic phases were washed with brine (-100 mL), dried over anhydrous MgSC>4, filtered, and concentrated under reduced pressure to provide an orange-red semi-solid. The crude product was adsorbed onto silica gel (10.0 g) and was purified using flash silica gel chromatography (Isco®; 220 g RediSep® Gold silica gel column; Eluent: 0-40% EtOAc/hexanes gradient) to provide Int-66a (1.09 g, 48% yield) as a beige solid.
Step B
In a 125-mL round-bottom flask, Int-66a (1.03 mg, 1.93 mmol), bis(pinacolato)diboron (1.08 g, 4.25 mmol), (dppf)PdCl2'CH2Cl2 (158 mg, 0.193 mmol) and KOAc (569 mg, 5.80 mmol) were mixed. The tube was sealed and alternately evacuated and refilled with nitrogen (5x) and dry dioxane (13 mL) was added. The flask was immersed in a preheated 90 °C oil bath and the reaction mixture was allowed to stir for 1 hour. The reaction mixture was allowed to cool to room temperature, diluted with EtOAc (100 mL), filtered and washed with brine (-50 mL). The organic layer was dried over anhydrous MgS04, filtered, and concentrated to provide a light brown solid. The crude product was purified using flash silica gel chromatography (Isco®; 120 g RediSep® Gold silica gel column; Eluent: 0-40% EtOAc/hexanes gradient) to provide Int-66b as a dark beige (1.00 g, 83% yield).
Step C
A 125-mL round-bottom flask was charged with intermediate Int-66b (992 mg, 1.58 mmol), bromoimidazole Int-7d (1100 mg, 3.48 mmol), and
(dppf)PdC12 CH2C12 (129 mg, 0.158 mmol). The flask was sealed and alternately evacuated and refilled with nitrogen (5x). Dioxane (11 mL) was added and the reaction mixture was allowed to stir at room temperature for 5 minutes. Aqueous potassium carbonate solution (5 mL, 1 M aqueous, 5 mmol) was added and the flask immersed into
a preheated 90 °C oil bath. After 22 hours the reaction was allowed to cool to room temperature, diluted with EtOAc (-100 mL) and the resulting solution washed with brine (-50 mL). The organic layer was dried over anhydrous MgS04, filtered, and
concentrated under reduced pressure to provide a light brown solid. The crude product was purified using flash silica gel chromatography (Isco®; 80 g RediSep® Gold silica gel column; Eluent: 0-6% MeOH/CEbCb gradient) to provide Int-66c as an orange-yellow solid (867 mg, 65% yield).
Step D
In a 100-mL round-bottom flask, Int-66c (690 mg, 0.816 mmol) was dissolved in methanol (8 mL) and hydrogen chloride solution (2 mL, 4 M in dioxane, (2.4 g, 8 mmol) was added. The reaction mixture was allowed to stir at room temperature for 12 hours. The reaction mixture was concentrated under reduced pressure to provide Int- 66d as a beige solid (648 mg, quantitative yield).
Step E
In a 50-mL round-bottom flask was charged with Int-66d (200 mg, 0.253 mmol) and Int-la (97 mg, 0.556 mmol) and dry DMF (2.5 mL). Diisopropylethylamine (265 μί, 196 mg, 1.5 mmol) was added and the reaction cooled to 0 °C (ice-water bath). After 15 minutes, solid HATU (240 mg, 0.632 mmol) was added in one portion. The reaction mixture was allowed to stir at 0 °C for 10 hours. Water (20 mL) was added to quench the reaction. The cream-colored suspension was extracted with EtOAc (2 x 50 mL) and the combined extracts were washed with brine (-25 mL). The organic layer was dried over anhydrous MgSC>4, filtered, and concentrated under reduced pressure to provide a light yellow-brown solid. The crude product was purified using reverse-phase chromatography (Gilson®; Phenomenex® Gemini 150 x 21.20 mm x 5 μπι column;
Eluent: 10-70% MeCN/water (+0.1% TFA) gradient) to provide Compound 1033 as a beige solid (79 mg, 33% yield).
EXAMPLE 67
Preparation of Compound 1038
Step A
In a 250-mL round-bottom flask was charged with Int-65f (1.51 g, 1.95 mmol), bromoimidazole Int-7d (739 mg, 2.34 mmol), (dppf)PdCl2'CH2Cl2 (143 mg, 0.195 mmol). The flask was alternately evacuated and refilled with nitrogen (5x) and dry dioxane (19 mL) was added. After 5 minutes, aqueous potassium carbonate solution (10 mL, 1 M aqueous, 10 mmol) was added and the reaction immersed into a preheated 90 °C oil bath. After 10 hours, the reaction was allowed to cool to room temperature and was diluted with EtOAc (-100 mL) and water (-50 mL). The organic layer was washed with brine (-50 mL), dried over anhydrous MgS04, filtered, and concentrated by rotary evaporation under reduced pressure to provide an orange-brown solid. The crude product was purified using flash silica gel chromatography (Isco®; 220 g RediSep® Gold silica gel column; Eluent: 0-100% EtOAc/hexanes gradient) to provide Int-67a as a golden yellow solid (1.23 g, 71% yield).
Step B
In a 50-mL round-bottom flask, Int-67a (1.222 g, 1.381 mmol) was dissolved in methanol (14 mL) and hydrogen chloride solution (3.5 mL, 4 M in dioxane, (4.20 g, 14 mmol) was added. The reaction mixture was allowed to stir at room temperature for 9 hours, and then concentrated under reduced pressure to provide Int-67b as a beige solid (1.222 g, 99% yield).
Step C
A 50-mL round-bottom flask was charged with Int-67b (155 mg, 0.73 mmol), Int-4f (45 mg, 0.208 mmol) and the solids were dissolved in a solution of diisopropylethylamine (151 μί, 1 12 mg, 0.867 mmol) in dry DMF (1.7 mL). The reaction mixture was cooled to 0 °C (ice-water bath) and stirred for 15 minutes. Solid HATU (99 mg, 0.260 mmol) was added in one portion. The reaction mixture was allowed to stir at 0 °C for 2 hours. Methanol (1 mL) and TFA (56 μί) were added sequentially at room temperature and the reaction mixture was allowed to stir at room temperature for an additional 2 hours. Water (20 mL) and aqueous sodium bicarbonate solution (~10 mL) were added, and the aqueous phase was extracted with EtOAc (2 x -50 mL). The combined organic phase was washed with brine (-25 mL), dried over anhydrous MgS04, filtered, and concentrated under reduced pressure to provide a light brown solid. The crude product was purified directly by reverse-phase chromatography (Gilson®; Phenomenex Gemini 150 x 21.20 mm x 5 μηι column; Run 1 : Eluent: 10-70% MeCN/water (+0.1% TFA) gradient; Run 2: 10-60% MeCN/water (+0.1% TFA) gradient) to provide Compound 1038 as a beige solid (80 mg, 47% yield).
EXAMPLE 68
Preparation of Compound 1048
lnt-64d 1048
A 50-mL round-bottom flask was charged with Int-64d (167 mg, 0.216 mmol), Int-4f (103 mg, 0.475 mmol) and the solids were dissolved in dry DMF (2 mL). Diisopropylethylamine (226 μί, (167 mg, 1.30 mmol) was then added to the reaction at 0 °C (ice-water bath) and stirred for 15 minutes. Solid HATU (204 mg, 0.537 mmol) was then added in one portion and the reaction was allowed to stir at 0 °C for 1 hour.
Methanol (1 mL) and trifluoroacetic acid (200 μί) were added and the reaction was allowed to stir at room temperature for 30 minutes. Water (20 mL) was added to quench the reaction. The reaction mixture was extracted with EtOAc (2 x 50 mL), the combined organic phase was washed with brine (-50 mL), dried over anhydrous MgS04, filtered, and concentrated under reduced pressure to provide a light orange-yellow solid. The crude product was purified directly by reverse-phase chromatography (Gilson®;
Phenomenex Gemini 150 x 21.20 mm x 5 μπι column; Eluent: 10-60% MeCN/water (+0.1% TFA)) to provide the Compound 1048 as a beige solid (135 mg, 61% yield)
EXAMPLE 69
Preparation of Compound 1488
Step A
A 20-mL Biotage® microwave vial was charged with Int-64b (392 mg, 0.643 mmol), bromoimidazole Int-lOf (451 mg, 1.35 mmol), and (dppf)PdCl2 »CH2Cl2
(47 mg, 0.064 mmol). The flask was alternately evacuated and refilled with nitrogen (5x) and dry dioxane (6.5 mL) was added and stirred vigorously. After 5 minutes aqueous potassium carbonate solution (3 mL, 1 M aqueous, 3 mmol) was added and the reaction immersed into a preheated 90 °C oil bath. After 18 hours the reaction was allowed to cool to room temperature, and diluted with EtOAc (-100 mL) and water was added. The reaction was extracted thrice with EtOAc (-50 mL), and the combined organic phase was washed with brine (-50 mL). The organic phase was dried over anhydrous MgSC>4, filtered, and concentrated under reduced pressure to provide an orange-brown solid. The crude product was purified using flash silica gel chromatography (Isco®; 40 g RediSep® Gold silica gel column; Eluent: 0-100% EtOAc/hexanes gradient) to provide Int-69a as a golden yellow solid (409 mg, 74% yield).
Step B
In a 50-mL round-bottom flask, the Int-69a (375 mg, 0.434 mmol) was dissolved in methanol (4.5 mL) and a hydrogen chloride solution (1.0 mL, 4 M in dioxane, (1.2 g, 4 mmol) was added. The reaction mixture was allowed to stir at room temperature for 24 hours. The reaction mixture was concentrated under reduced pressure to provide Int-69b as a beige solid (344 mg, 98% yield).
Step C
Int-4f (99 mg, 0.454 mmol) was weighed into a pre-tarred vial and transferred using DMF solvent (4 x 500 \L) to a 50-mL round-bottom flask containing Int-69b (167 mg, 0.206 mmol)). Diisopropylethylamine (220 μί, 163 mg, 1.26 mmol) was added by syringe. The mixture was allowed to stir at room temperature for -1 minutes, during which time all solids dissolved. The flask was cooled in an ice-water bath for -15 minutes and solid HATU (196 mg, 0.516 mmol) was added in one portion. After 1.5 hours at 0 °C, Methanol (1 mL) and TFA (190 μί) were added sequentially and the reaction mixture was allowed to stir for an additional 2 hours. Water (-20 mL) was added and the reaction mixture was extracted with EtOAc (2 x -50 mL). The combined organic phases was washed with brine (-50 mL), dried over anhydrous MgS04, filtered, and concentrated under reduced pressure to provide a light orange solid. The crude
product was purified directly by reverse-phase chromatography (Gilson ; Phenomenex Gemini 150 x 21.20 mm x 5 μπι column; Eluent: 0-60% MeCN/water (+0.1% TFA) gradient over 15 minutes. Major components that eluted were Compound 1488 and a TFA adduct of same. The total yield of the final product was 146 mg, 67% yield.
EXAMPLE 70:
Preparation of Compound 1492
A 50-mL round-bottom flask was charged with Int-64d (183 mg, 0.237 mmol) and ( ?)-N,N-diethylphenylglycine hydrochloride (127 mg, 0.520 mmol) and the solids were dissolved in dry DMF (2.5 mL). Diisopropylethylamine (400 μί, 296 mg, 2.29 mmol) was added, the reaction cooled to 0 °C (ice-water bath) and stirred for 15 minutes. Solid HATU (225 mg, 0.591 mmol) was added in one portion. After 1 hour methanol (1 mL) and trifluoroacetic acid (365 uL) were added and the reaction was allowed to stir at room temperature for 30 minutes. The reaction was quenched with water (20 mL) and the product was extracted into EtOAc (2 x 50 mL). The combined organic phase were washed with brine (-50 mL), dried over anhydrous MgS04, filtered, and concentrated under reduced pressure to provide a light orange-yellow solid. The crude product was purified directly by reverse-phase chromatography (Gilson®;
Phenomenex Gemini 150 x 21.20 mm x 5 μπι column; Eluent: 10-60% MeCN/water (+0.1% TFA) gradient over 15 minutes) to provide fractions containing Compound 1492 and a TFA adduct of same. Retreatment of the TFA-adduct fractions with methanol as above, provided additional amounts of the desired Compound. Total yield of Compound 1492 was 201 mg, 74% yield.
EXAMPLE 71
A 50-mL round-bottom flask was charged with Int-67b (123 mg, 0.138 mmol) and (7?)-N,N-diethylphenylglycine hydrochloride (40 mg, 0.165 mmol) and the solids were dissolved in a solution of diisopropylethylamine (240 μί, 178 mg, 1.375 mmol) in dry DMF (1.4 mL). The reaction mixture was cooled to 0 °C (ice-water bath) and stirred for 15 minutes. Solid HATU (78 mg, 0.206 mmol) was added in one portion. After 1 hour the reaction mixture was concentrated under reduced pressure to provide brown, viscous oil, which was purified using reverse-phase chromatography (Gilson®; Phenomenex® Gemini 150 x 21.20 mm x 5 μπι C-18 column; Run 1 : 450 μΕ injection; 10-70% MeCN/water (+0.1% TFA) gradient over 15 minutes. Run 2: 600 uL injection; 10-60% MeCN/water (+0.1% TFA) gradient over 20 minutes) to provide Compound 1044 as a beige solid (88 mg, 66% yield).
EXAMPLE 72
Preparation of Compound 1039
A 50-mL round-bottom flask was charged with Inter Int-67b (104 mg, 0.1 16 mmol), Int-le (27 mg, 0.140 mmol) and a solution of diisopropylethylamine (102
μί, 75 mg, 0.581 mmol) in dry DMF (1 mL). The reaction mixture was cooled to 0 °C (ice-water bath) and stirred for 15 minutes. Solid HATU (66 mg, 0.174 mmol) was added in one portion and the reaction mixture was allowed to stir for 2 hours and allowed to warm to room temperature. Methanol (1 mL) and TFA (56 μί) were added sequentially at room temperature and the reaction was allowed to stir at room temperature for 2 hours. Water (20 mL) followed by aqueous sodium bicarbonate solution (-10 mL) were then added. The reaction was extracted with EtOAc (2 x ~50 mL) and the combined extracts were washed with brine (-25 mL). The organic phase was dried over anhydrous MgSCv, filtered, and concentrated under reduced pressure to provide a light brown solid. The crude product was purified directly by reverse-phase chromatography (Gilson®; Phenomenex® Gemini 150 x 21.20 mm x 5 μπι column; Run 1 : 10-70% MeCN/water (+0.1% TFA) gradient over 20 minutes. Run 2: 10-60% MeCN/water (+0.1% TFA) gradient over 20 minutes) to provide Compound 1039 as a beige solid (65 mg, 82% yield).
EXAMPLE 73
Preparation of Compounds 959, 950, & 951
Step A
Int-22a (lg, 2.8 mmol), 2-methyl thiophenecarboxaldehyde (1.06g, 8.4 mmol) and />-tosyl chloride were dissolved in toluene (10 mL) and stirred in a pressure tube at 150 °C for 6 nr. After cooling, the crude material was purified using an ISCO silica gel column (pre-packed, 80 g) eluted with EtOAc : Hex (0% to 5%) to yield Int- 73a (500 mg, 38%).
Step B
Int-73a (0.5 g, 1.08 mmol), bis(pinacolato)diboron (0.3 g, 1.2 mmol), KOAc (316 mg, 3.2 mmol) and PdCl2(dppf)2 (88 mg g, 0.11 mmol) were added into a microwave tube. After the flask was flashed with N2, and dioxane (3 mL) was added.
The mixture was allowed to stir at 1 10 °C for 1 hour. The crude reaction containing Int- 73b was used without further purification.
Step C
To the reaction flask charged with Int-73b was added Int-lOf (430 mg, 1.3 mmol), PdCl2(dppf 2 (88 mg, 0.1 1 mmol) and K2C03 (1 N aq., 3.23 ml). The tube was sealed, degassed and stirred at 90 °C for about 15 hours. After cooling, EtOAc (lOOmL) was added , the layers separated and the organic phase washed with brine (100 mL). The organic phase dried and concentrated to provide a semi-solid. The crude product was purified on a ISCO column (pre-packed silica gel, 40 g) and eluted with Hex:EtOAc 0% to 70% gradient to provide product Int-73c 650 mg (94%).
Step D
Int-73c (640 mg, 1.0 mmol), bis(pinacolato)diboron (508 mg, 2 mmol), Pd2dba3 (155 mg, 0.15 mmol), X-Phos (143 mg, 0.3 mmol) and KOAc (491 mg, 5 mmol) were added to a 20 mL microwave tube. The tube was sealed, degassed and the reaction was allowed to stir at 1 17 °C for 8 hr. To this reaction mixture was added Int-lOf (259 mg, 0.78 mmol), PdCl2(dppf)2 ( 06 mg, 0.13 mmol) and K2C03 (1 N aq., 1.9 ml) The tube was sealed and degassed and heated to 100 °C for an additional 24 hr. After cooling, EtOAc (lOOmL) was added, the layers separated, and the organic phase washed with Brine (100 mL). The organic phase was dried and concentrated to provide a solid. The crude material was purified on an ISCO column (pre-packed silica gel, 24 g) and eluted with DCM: DCM/MeOH/NH3.MeOH (90: 10: 1) 0% to 80% to provide the product Int- 73d 130 mg (24%).
Step E
Int-73d (145 mg, 0.18 mmol) was dissolved in dioxane (2 mL) and HC1 (4N in dioxane, 0.9 mL) was added at room temperature. After 1.5 hours, the reaction was concentrated in vacuo in vacuo. The product Int-73e was isolated without further purification (123 mg, 100%).
Step F
Int-73e (123 mg, 0.18 mmol) was dissolved in DMF (5 mL) and cooled to 0 °C. HATU(154 mg, 0.41 mmol), Int-la (71.1 mg, 0.41 mmol ) were added followed by addition of Hunig's base (0.19 mL, 1.06 mmol). After 1.5 hours at 0°C, water was added to quench the reaction. The mixture was diluted with EtOAc and extracted with NaClaq. The organic pahse was dried and concentrated to affors a solid. Further purification with by silica- gel chromatography (pre-packed column, 23 g) eluted with DCM and EtOAC/MeOH/NH3.H20 (90:10: 1) 0% to 80% to gave Compound 959 103 mg (62%).
Compounds 950 & 951
The diastereomers of Compound 959 (103 mg) were separated by chiral SFC separation on a AS-H column (50% MeOH (0.2% DEA)/C02, 50 ml/min, 100 bar), to provide isomer A Compound 950 (27 mg, 35%) and isomer B Compound 951 (28 mg).
EXAMPLE 74
Preparation of Compound 1464
2-Chloro-5-dichloromethylthiophene prepared from 2-chlorothiophene- aldehyde (5 g, 13.62 mmol) and CS2CO3 (19.97 g, 61.3 mmol) was charged in a flask and dissolved in DMSO (50 mL). Int-19b (5.49 g, 27.2 mmol) was added and the reaction was heated to 100 °C. After 1 hour, the reaction was filtered and the filtrated was extracted with NaCl aq. The organic phase was dried and concentrated in vacuo to a semi-solid. The crude material was purified using flash column chromatography on silica gel (220 g column) eluted with EtOAc in Hex 0% to 5% to provide Int-74a (1.35g, 20%).
Step B
Int-74a (1.53 g, 3.09 mmol), dipinacolatoborane (1.8 g, 7.1 mmol), KOAc( 1.52 g, 15.44 mmol) and PdCl2(dppf)2 (0.504 g, 0.62 mmol) were charged into a microwave tube. After the flask was flashed with N2, dioxane (20 mL) was added. The mixture was allowed to stir at 95 °C for 4 hours. The crude reaction was diluted with EtOAc ( 100 mL) and it was extracted with NaCl aq. The organic phase was dried and concentrated in vacuo. The crude material was purified using flash column
chromatography on silica gel with EtOAc in Hex (0% to 20%) eluent to provide Int-74b (990 mg, 54%).
Step C
Int-74b (990 mg, 1.68 mmol), Int-lOf (1.35 g, 4.03 mmol), PdCl2(dppf)2
(0.274 g, 0.342 mmol) and K2CO3 (I N aq., 8.4 ml) were added to a 20 mL microwave tube. The tube was sealed, degassed with nitrogen and stirred at 100 °C for about 15 hours. After cooling, EtOAc (lOOmL) was added and the reaction was extracted with brine (100 mL). The organic phase was separated, dried and concentrated in vacuo. The crude material was purified on a ISCO silica-gel column (40 g) and with EtOAc/Hex (0% to 70%) eluent to provide product Int-74c 500 mg (33%).
Int-74c (504 mg) was subjected to SFC chiral separation on OD-H column (IPA (0.05% DEA)/C02) to provide isomers Int-74c' and Int-74c" (176 mg, 35%).
Step D
Int-74c" ((176 mg) was dissolved in dioxane (10 mL) and HC1 (4N in dioxane, 0.53 mL) was added and stirred at room temperature. After 1.5 hr. the solvent was removed in vacuo. Int-74d was isolated without further purification (167 mg, 100%).
Step E
Int-74d (diastereomer B, 167 mg, 0.21 mmol) was dissolved in DMF (3 mL) and cooled to 0 °C. HATU(169 mg, 0.44 mmol), Int-lOf (74.1 mg, 0.423 mmol were added followed by addition of Hunig's base (0.22 mL, 1.27 mmol) and the reaction was allowed to stir at 0°C. After 1.5 hours, water was added and the reaction diluted with EtOAc and extracted with NaClaq. The organic phase was dried and concentrated in vacuo to provide a solid.. Purification by silica gel chromatography (23 g) with DCM and EtOAC/MeOH/NH3 (90: 10: 1 - 0% to 100%) eluent provided the title Compound 970 (140 mg, 69.1%).
Compound 1464 (diastereomer B).
Compound 970 (60 mg, 0.063 mmol), cyclopropylboronic acid (81 mg, 0.94 mmol), Pd2dba3 (6.5 mg, 6.26 μπιοΐ), X-Phos (5.97 mg, 0.013 mmol) and K2C03 (1 N aq., 188 μΐ) were added to a 20 mL microwave tube. The tube was sealed and degassed with nitrogen. The reaction was allowed to stir at 110 °C for 5 hr. The crude material was purified on silica gel using DCM to EtOAc/MeOH/NH3.H20 (100:10: 1- 0% to 90%) eluent to provide Compound 1464 (40 mg, 62%).
EXAMPLE 75
Preparation of Compound 1459
lnt-75a
lnt-75e
1. HATU, N,N-diisopropyl,ethyl
/1.5h
2. 2MHCI in Ether(XS)
Step A
To a solution of 4-Methyl-2-thiazole-2-carboxaldehyde (2.0g, 15.73mmol) in CH2Cl2 (40 mL) at -20 c, added pyridine(0.254ml, 3.15mmol) followed by addition of PCl5(6.55g, 31.5mmol) . The mixture was allowed to stir at -20 °C for 30 minutes. NaHC03 (13.2g, lOeq.) was added as solid to the reaction mixture. After stirring for additional 30 minutes the reaction was filtered through celite and washed with 2 X 25 mL CH2C12. The filtrate was concentrated under reduced pressure to provide the crude. It was re-dissolved in CH2C12 and filtered through a pad of silica-gel. Filtrate on concentration and drying gave Int-75a as brown oil. (32%)
Step B
The dibromo indole (Int-19b, 0.5g, 1.362mmol), 2-(dichloromethyl)-4- methylthiazole(Int-75a, 0.496g, 2.72mmol) and Cesium carbonate (0.976g, 3.00mmol) were combined in acetonitrile (10ml) in a 50mL round bottomed flask equipped with condenser and heated at 55°C for 15 hrs. TLC analysis showed consumption of starting
material. The reaction was diluted with EtOAc, washed with water(3X20ml), brine (1x20ml), dried(Na2S04), filtered and concentrated under reduced pressure to provide brown semi-solid crude. It was allowed to stir with ether and filtered to provide Int-75b as yellow solid. The filterate was concentrated and purified using ISCO silica-gel column. The combined yield of 4 was 0.32g (49%).
Step C
Intermediate Int-75b (0.095g, 0.2mmol), bis(pinacolato)diboron ( 0.106g, 0.419mmol), potassium acetate( 0.117g, 1.197mmol) and PdC12(dppf).CH2C12 (0.065g, 0.08mmol) and Dioxane (2.0ml) were combined in a microwave tube and sealed and purged with nitrogen (3 x). The reaction was heated at 90°C for 2hrs. TLC showed complete reaction. The reaction mixture containing Int-75c was used without additional workup.
Step D
To the above reaction mixture (Intermediate Int-75c (0.1 14g, 0.2mmol) in the microwave tube, was added N-Bocproline imidazole bromide(Int-7d, 0.139g, 0.44mmol), PdC12(dppf).CH2C12 (0.033g, 0.04mmol) and Potassium carbonate
( 1.199ml of 1M aqueous solution, 1.199mmol) . Sealed and purged with nitrogen (3 x). The reaction was heated at 90°C for 4hrs. Reaction was worked up by diluting with EtOAc (25ml) and water (20ml). The resulting mixture was vigorously stirred for 10 minutes and then filtered through Celite. Filtrate was partitioned. The organics were washed with water (3x15ml) and brine(lxl5ml), dried ^SC ), filtered and concentrated in vacuo. The resulting crude was purified using preparatory silica gel column
chromatography, using 5%MeOH/CH2C12 to provide desired product Int-75d ( 79%).
Step E
Trifluoroacetic acid (0.25ml, 3.24mmol) was added to Intermediate Int- 75d at °C. The mixture was allowed to warm room temperature and stirred for additional 1 hour. The solvent was removed under reduced pressure. The product was treated with
0.36ml of 4MHC1 in dioxane (1.44mmol). After 10 minutes of stirring, excess acid and solvent removed and the product Int-75e was dried for about 15 hours.
Preparation of Compound 1459
To a solution of Intermediate Int-75e (0.035g, 0.045mmol) in DMF (1.4ml) was added (S)-2-(methoxycarbonylamino)-2-(tetrahydro-2H-pyran-4-yl) acetic acid (0.022g, O.lmmol), HATU(0.038g, O.lmmol). The reaction was cooled to -15°C and Hunig's base (0.051ml, 0.363mmol) was added drop-wise. The resulting mixture was allowed to stir for 1.5 hrs at -15°C. The reaction was quenched with water (20ml). The product was extracted with EtOAC (3x20ml). The organics were washed with water (3x20ml), brine (1x20ml), dried (Na^SC^), filtered and concentrated under reduced pressure to provide crude which was purified using Gilson reverse phase
chromatography using gradient elution of 0% to90% C¾CN with 0.1%TFA and water with 0.1% TFA. The desired fractions were collected and concentrated under reduced pressure and then treated with 0.3ml of 2MHC1 in ether. The solvent was removed and the sample was dried for about 15 hours to provide Compound 1459 as an orange brown solid. (32%).
EXAMPLE 76
Preparation of Intermediate Compound Int-76d
Step A - Preparation of Int-76a
To a cooled mixture of thionyl chloride (20 ml, 274 mmol) and DMF (0.7 ml) at 0 °C, benzothiophene 2-carboxaldehyde (4.7 g, 29.0 mmol) was added in 3 portionas, and stirred at 0 °C for 30 minutes and then a;lowed to warm for about 15 hours. The mixture was poured into ice and aqueous IN sodium hydrogen carbonate and then extracted with EtOAc. The combined organic solution was washed with brine and dried (Na2S04) and concentrated in vacuo to provide Int-76a (6.1 g, 28.1 mmol, 97 % yield).
Step B - Preparation of Compound Int-76b
Int-19g (4.5 g, 13.95 mmol), Int-76a (6.06 g, 27.9 mmol) , and cesium carbonate (18.18 g, 55.8 mmol) in DMSO (22 ml) was allowed to stir at 80 °C for 2 hours. The mixture was then added to cold water and the resulting solid was filtered off and washed with water to provide a 1.55 g of a solid . The filtrate was concentrated and the residue was allowed to stir with 1:1 MeOH-MC to provide crude solid material which was further purified using silica-gel Chromatography (Pre-packed Biotage column, 80g solid loading, Eluent: 1000% Hex to 15% EtOAc/Hex) to provide the desired product Int-76b (650 mg, Yield 33.8%).
Step C - Preparation of Int-76c
A mixture of Int76b(0.418 g, 0.90 mmol), bis(pinacolato)diboron (0.25 g, 0.99 mmol), KOAc (0.176 g, 1.80 mmol), and Pd(dppf)Cl2 (0.066 g, 0.09 mmol) in 1,4- Dioxane (3 ml) was degassed (by N2 flush) and heated to 100 °C. After 4h , the reaction was cooled to room temperature, and Int-lOf (329 mg, 0.99 mmol), Pd(dppf)Cl2 (66 mg, 0.09 mmol) and IN K2CO3 (1.8 ml, 1.8 mmol) were added. The mixture was degassed and heated at 100 °C for 2 hours. The mixture was cooled to room temperature, diluted in EtOAc, and filtered through celite pad. The filtrate was concentrated in vacuo and the residue was purified on an ISCO 80 g gold column (Eluent: CH2Cl2-5%MeOH/ CH2C12) to provide Int-76c (503 mg, 0.785 mmol, 88 % yield) as a pale yellow solid.
LC/MS (M+H) = 641.2.
Step D - Preparation o/Int-76d
A mixture of Int-76c (0.292 g, 0.455 mmol), bis(pinacolato)diboron (0.127 g, 0.50 mmol), KOAc (0.089 g, 0.91 mmol), X-Phos (0.043 g, 0.091 mmol), and Pd2dba3 (0.047 g, 0.046 mmol) in 1,4-Dioxane (3.5 ml) was degassed (by N2 flush) and heated to 100 °C. After 18 hours, the reaction was cooled to room temperature, the crude mixture was treated with Int-7d (160 mg, 0.51 mmol), Pd(dppf)Cl2 (34 mg, 0.046 mmol) and IN K2CO3 (0.92 ml, 0.92 mmol). The mixture was degassed and stirred at 100 °C for 6 hours, cooled to room temperature, diluted in EtOAc, and filtered through celite pad. The filtrate was concentrated in vacuo and the residue was purified on an ISCO 40 g gold column (Eluent: Hex-EtOAc 100:1 to 85: 15 gradient) to provide Int-76d (225 mg, 58 % yield) as a pale yellow solid. LC/MS (M+H) = 842.3.
EXAMPLE 77
Preparation of Compounds 792, 422 & 423
Step A
Trifluoroacetic acid (1 ml, 12.98 mmol) was added to a stirred, cooled 0 °C solution of Int-76d (0.171 g, 0.203 mmol) in CH2C12 (3 ml). After 5 minutes the reaction was allowed to warm to room temperature and stir an additional 90 minutes.. The mixture was concentrated in vacuo and the residue was dissolved in MeOH followed by treatment with 2N HC1 in ether. The methanol solution was then concentrated to dryness providing Int-77a (0.145 g, 0.203 mmol, 100 % yield) which was used without further purification.
LC/MS (M+H) = 642.3.
Step B
A round flask was charged with Int-77a (145 mg, 0.203 mmol), DMF (1.5 ml) and Int-4a (88 mg, 0.406 mmol) and cooled to -15 °C. To the reaction mixture were added N,N-diisopropylethylamine (0.248 ml, 1.42 mmol) and HATU (154 mg, 0.406 mmol). After 10 minutes and the reaction was allowed to warm to 0 °C. After 3 hours, the reaction was quenched by 0.5 mL of water and the mixture was filtered and purified
on a Gilson HPLC (Eluent: Acetonitrile/Water + 0.1% TFA) to provide Compound 792 (106 mg, 41 % yield) as a diastereomeric mixture (-1 :1).
The diastereomers of Compound 792 were separated by SFC to provide pure diastereomers Compound 422 and Compound 423.
LC/MS (M+H) = 1041.4. SFC separation condition:
Instrument: Thar 80 SFC ; Column: Chiral Cel OJ, 20μιη ,Daicel Chemical Industries, Ltd 250x30mmI.D. Mobile phase: A: Supercritical C02 , B:ETOH(contained 0.2% DEA) , A:B =45:55 at 80ml/min; Column Temp: 38 °C
EXAMPLE 78
Preparation of Intermediate Compound Int-78a
Int-78a (248 mg, 0.288 mmol, 62 % yield) was prepared from (343 mg, 0.47 mmol) using the method described in Example 50..
LC/MS (M+H) = 860.3.
EXAMPLE 79
Preparation of Compounds 791, 703 & 704
Step A
Compound Int-79a (211 mg, 0.29 mmol, 100% crude yield). was prepared from Int-78a (248 mg, 0.29 mmol) following the method described in Example 77 Step A LC/MS (M+H) = 660.3.
Step B
Compound 791 (112 mg, 0.087 mmol, 44% yield) was prepared from Int- 79a (147 mg, 0.20 mmol) using the method described in Example 77, Step B SFC separation provided the pure diastereomers Compound 703 and Compound 704 LC/MS (M+H) = 1058.2.
SFC Separation condition: (Thar 80 SFC , Chiral Pak AS, 20μπι, Daicel Chemical Industries, Ltd 250x30mmI.D.
Mobile phase: A: Supercritical C02 , B:ETOH(contained 0.2% DEA) , A:B =60:40at 80ml/min
EXAMPLE 80
Preparation of Compound 789
Compound 789 (106 mg, 0.084 mmol, 41% yield) was prepared from Int- 77a (145 mg, 0.203 mmol) using Int-la using the method described in Example 77, Step B LC/MS (M+H) = 1041.4.
Step A
n-BuLi (5.79 ml, 14.47 mmol) was added to a stirred, cooled -78 °C solution of 7-bromo-4-methyl-3,4-dihydro-2H-l,4-benzoxazine (Int-81a, 3 g, 13.15 mmol) in THF (24 ml). After stirring lh at -78 °C for 1 hour., DMF (2.037 ml, 26.3
mmol) was added dropwise and the mixture allowed to warm slowly over 2 hours to room temperature. The reaction was quenched with aqueous ammonium chloride, and the product extracted into ethyl acetate. The organic phase was washed with brine, dried (NaiSC ), filtered and under reduced pressure to provide Int-81b (2.32 g, 13.09 mmol, 100 % yield) as a green solid.
Step B
A 0.2-0.5 mL microwave tube was charged with Int-22a (lg, 2.491 mmol), Int-81b (1.12 g, 6.32 mmol), p-TsCl (0.142 g, 0.747 mmol) and Toluene (8 ml). The reaction was heated in the microwave reactor at 170 °C for 6 hours. The mixture was cooled, concentrated in vacuo and the residue was purified on an ISCO 24 g gold column (Eluent: 100% Hex to 50% EA/Hex gradient) to provide Int-81c (190 mg, 0.339 mmol, 13.61 % yield).
Step C
A mixture of Int-81c (350 mg, 0.624 mmol), bis(pinacolato)diboron (349 mg, 1.373 mmol), KOAc (245 mg, 2.497 mmol), and Pd(dppf)Cl2 (45.7 mg, 0.062 mmol) in 1 ,4-Dioxane (5 ml) was degassed (by N2 flush) and heated to 100 °C. After 18h, the reaction wascooled to room temperature, the mixture was treated with Int-lOf (438 mg, 1.310 mmol), IN K2C03 (2.5 ml, 2.5 mmol) and Pd(dppf)Cl2 (45.7 mg, 0.062 mmol). The mixture was degassed and heated to 100 °C for 18 hours. The mixture was cooled, diluted in EtOAc and filtered through celite pad, and the filtrate was concentrated in vacuo to provide a solid. The crude product was purified using flash column
chromatography on silica gel on (ISCO 40g gold, Eluent:Hex-EtOAc 100:0 to 85: 15) to provide Int-81d (233 mg, 0.256 mmol, 41.1 % yield) as a pale yellow solid. LC/MS (M+H) = 909.4.
EXAMPLE 82
Preparation of Compound 793
Step A
Compound Int-82a was prepared from Int-81d (100 mg, 0.1 1 mmol) using the method described in Example 77, Step A (86 mg, 0.1 1 mmol, 100% yield). LC/ MS (M+H) = 709.3.
Step B
Compound 793 was prepared from Int-82a (86 mg, 0.1 1 mmol) using the method described in Example 77, Step B (63 mg, 0.050 mmol, 46% yield). LC/ MS (M+H) = 1024.4.
EXAMPLE 83
Preparation of Compound 794
A mixture of X-Phos (4.27 mg, 8.95 μπιοΐ), Compound 793 (56 mg, 0.045 mmol), Pd
2dba
3 (4.63 mg, 4.47 μπιοΐ), KOAc (10.98 mg, 0.1 12 mmol) and
bis(pinacolato)diboron (17.04 mg, 0.067 mmol) in 1 ,4-Dioxane (1 ml) was degassed and heated to 100 °C for about 15 hours. The mixture was the then cooled to room temperature, filtered and the crude reaction mixture purified on a Gilson HPLC (Eluent: Acetonitrile/Water + 0.1% TFA) to provide Compound 794 (30.5 mg, 0.025 mmol, 56% yield). LC/MS (M+H) = 989.5.
EXAMPLE 84
Preparation of Compounds 1051, 1061 & 1062
Step A
A 20-mL microwave tube was charged with Int-22a (1.0 g, 2.5 mmol), 3- phenylpropanal (3.3 mL, 3.3 g, 25 mmol) and 7-toluenesulfonyl chloride (48 mg, 0.25
mmol) and toluene (8 mL). The reaction mixture was heated and stirred at 170 °C in a microwave for 12 hours. The reaction mixture was concentrated in vacuo in vacuo, and the residue adsorbed onto silica gel. Purification by by silica gel chromatography
(Eluent:0-15% EtOAc/hexanes) provided Int-84a as a yellow oil (901 mg, 70% yield)
Step B
A 20-mL microwave tube was charged with Int-8 a (901 mg, 1.74 mmol), bis(pinacolato)diboron (1.1 g, 4.4 mmol), (dppf)PdCl2 »CH2Cl2 (142 mg, 0.17 mmol) and KOAc (512 mg, 5.22 mmol). Dioxane (10 mL) was added, and the sealed reaction degassed with dry nitrogen. The reaction was allowed to stir at 90 °C for 2 hours, then allowed to cool to room temperature, and diluted with EtOAc (100 mL). The organic phase was washed sequentially with water (10 mL) and brine (10 mL). The organic phase was dried over MgSC , filtered, and concentrated in vacuo to provide a solid. The crude product was purified using flash column chromatography on silica gel (Eluent: 0- 20% EtOAc/hexanes) to provide Int-84b (1.3 g).
Step C
A 20-mL microwave tube was charged with Int-84b (572 mg, 0.93 mmol), Int-lOf (687 mg, 2.05 mmol), and (dppf)PdCl2'CH2Cl2 (38 mg, 0.047 mmol). The tube was sealed, dioxane (8 mL) was added , degassed with nitrogen, and aqueous potassium carbonate (6 mL, 1 M, 6 mmol) added. The reaction mixture was heated at 90 °C for 16 hours, cooled to room temperature and diluted with EtOAc (100 mL). The aqueous layer was extracted with EtOAc (2 x 20 mL), and the combined organic extracts were washed with brine (20 mL), dried over MgS04, filtered, and concentrated in vacuo. The crude product was purified using flash column chromatography on silica gel (Eluent: EtOAc (containing 10% MeOH): hexanes of 10:90 to 90: 10) to provide Int-84c (580 mg, 72% yield).
Step D
A 125-mL round-bottom flask was charged with Int-84c (411 mg, 0.47 mmol) and methanol (9 mL). HC1 (9.4 mL, 2 M in diethyl ether, 19 mmol) was added
and the reaction mixture was allowed to stir for about 15 hours at room temperature. The reaction mixture was concentrated in vacuo to provide Int-84d (384 mg, quantitative yield).
Step E
In a 125-mL round-bottom flask, Int-84d (382 mg, 0.52 mmol) and Int-la (228 mg, 1.3 mmol) were dissolved in DMF (7.5 mL) and diisopropylethylamine (0.63 mL, 0.47 g, 3.6 mmol) was added. The reaction mixture was cooled to 0 °C and was allowed to stir for 15 minutes. HATU (395 mg, 1.04 mmol) was added and the reaction mixture was allowed to stir at 0 °C for 30 minutes, and then at room temperature for 2.5 hours. The reaction mixture was poured into water (30 mL). The precipitate was collected by filtration, then dissolved in methylene chloride (200 mL), dried over MgS04, filtered, and concentrated in vacuo. The resulting crude product was purified using reverse-phase CI 8 chromatography (Gilson, 0-90% CH3CN (+ 0.1% TFA)— water (+ 0.1% TFA) over 15 minutes) to provide Compound 1051 as a yellow foam (199 mg, 39% yield).
Step F
Compound 1051 (247 mg, 0.251 mmol) was dissolved in methanol (13 mL) and palladium (268 mg, 10 wt% on carbon, containing 50 wt% water) was added. The reaction mixture was hydrogenated for 71 hours, at which point LC/MS analysis showed a 4:1 mixture of desired product and starting mixture. The hydrogenation was continued for a further 92 hours. The reaction mixture was filtered and the catalyst was rinsd with methanol (-100 mL). The filtrate was concentrated, adsorbed onto silica gel (15 mL), then purified using flash column chromatography on silica gel (0-10% MeOH (+1% NH4OH)/CH2Cl2) to provide Int-85e (164 mg, 69% yield).
Step G
The isomers of Int-85e were separated by HPLC. Int-85e (164 mg) was dissolved in abs. EtOH (6.0 mL) and the solution was filtered. The sample was divided into four equal portions, each of which was injected onto a Phenomenex Lux Cellulose-2
(5 μιη, 150 x 21.20 mm) semi-preparative column; detection wavelength = 350 nm. Initial elution with 25% EtOH/hexane @ 10 mL/min for 159 minutes gave Compound 1061 (tR = 83 minutes; 62 mg). The solvent polarity was increased to 35% EtOH/hexane, and further elution at 10 mL/min gave Compound 1062 (tR = 163 minutes; 72 mg).
EXAMPLE 85
Preparation of Compounds 1049, 1054, 1059 & 1060
Step A
In a 20-mL microwave tube, Int-84b (229 mg, 0.37 mmol), Int-7d (261 mg, 0.82 mmol), and (dppf)PdCl2 «CH2Cl2 (15 mg, 0.019 mmol) were combined. The tube was sealed, evacuated, and placed under nitrogen atmosphere. Dioxane (4 mL) and aqueous potassium carbonate (3 mL, 1 M, 3 mmol) was added. The reaction mixture was allowed to stir for about 15 hours at 90 °C, then allowed to cool to room temperature. The reaction mixture was diluted with EtOAc (50 mL). The aqueous layer was extracted with EtOAc (2 x 10 mL). The combined organic extracts were washed with brine (20 mL), dried over MgSOi, filtered, and concentrated in vacuo. The residue was purified
using flash column chromatography on silica gel (0-100% EtOAc(containing 10%
MeOH)— hexanes) to provide Int-85a (212 mg, 69% yield).
Step B
In a 125-mL round-bottom flask, Int-85a (456 mg, 0.55 mmol) was dissolved in methanol (11 mL). HCl (5.5 mL, 2 M in diethyl ether, 11 mmol) was added and the reaction mixture was allowed to stir for about 15 hours at room temperature. The reaction mixture was concentrated in vacuo to provide Int-85b (425 mg, quantitative yield).
Step C
In a 125-mL round-bottom flask, Int-85b (439 mg, 0.62 mmol) and Int-la (274 mg, 1.56 mmol) were dissolved in DMF (8 mL) and diisopropylethylamine (0.76 mL, 0.56 g, 4.3 mmol) was added. The reaction mixture was cooled to 0 °C and was allowed to stir for 15 minutes. HATU (475 mg, 1.24 mmol) was added and the reaction mixture was allowed to stir at 0 °C for 30 minutes, and then at room temperature for 2 hours. The reaction mixture was poured into water (30 mL). The precipitate was collected by filtration, then dissolved in EtOAc (200 mL), dried over MgS04, filtered, and concentrated in vacuo. The resulting crude product was purified using reverse-phase CI 8 chromatography (Gilson, 0-90% CH3CN (+ 0.1% TFA}— water (+ 0.1% TFA) over 15 minutes) to provide Compound 1049 as a yellow foam (362 mg, 62% yield).
Step D
Compound 1049 (362 mg, 0.383 mmol) was dissolved in methanol (20 mL) and palladium (163 mg, 10 wt% on carbon, containing 50 wt% water) was added. The reaction mixture was hydrogenated for 71 hours, at which point LCMS analysis showed only a trace amount of remaining starting material. The reaction mixture was filtered and the catalyst was rinsed with methanol (-100 mL). The filtrate was concentrated, adsorbed onto silica gel (15 mL), then purified using flash column chromatography on silica gel (0-10% MeOH (+1% NH4OH)/CH2Cl2) to provide
Compound 1054 (231 mg, 66% yield).
Step E
The isomers comprising Compound 1054 were separated by HPLC.
Compound 1054 (222 mg) was dissolved in abs. EtOH (6.0 mL) and the solution was filtered. The sample was divided into two equal portions, each of which was injected onto a Phenomenex Lux Cellulose-2 (5 μηι, 150 x 21.20 mm) semi-preparative column; detection wavelength = 350 nm. Elution with 45% EtOH/hexane (+ 0.1% diethylamine) @ 10 mL/min gave Fraction A: Compound 1059 (tR = 32 minutes, 91 mg) and Fraction B: Compound 1060 (tR = 97 minutes, 68 mg).
EXAMPLE 86
Preparation of Compound 1100
Step A
A 20-mL microwave tube was charged with Int-22a (1.0 g, 2.8 mmol), 3- (3'-methoxyphenyl)propanal (2.3 g, 14 mmol), /?-toluenesulfonyl chloride (53 mg, 0.280 mmol) and dissolved in toluene (9 mL). The tube was sealed and heated in a microwave with stirring at 170 °C. After 12 hours the reaction was concentrated partially in vacuo,
and the residue was adsorbed onto silica gel (20 mL). The crude product was purified using flash column chromatography on silica gel (Eluent: 0-10% EtOAc: hexanes) to provide Int-86a (1.39 g, 99% yield)
Step B
A20-mL microwave tube was charged with, Int-86a (1.39 g, 2.76 mmol), bis(pinacolato)diboron (772 mg, 3.04 mmol), (dppf)PdCl2'CH2Cl2 (202 mg, 0.276 mmol) and KOAc (813 mg, 8.29 mmol). The tube was sealed, evacuated, and placed under nitrogen atmosphere. Dioxane (11 mL) was added, the reaction was allowed to stir at 90 °C for 2 hours, and then allowed to cool to room temperature. EtOAc (40 mL) and water ( 40 mL) were added . The aqueous layer was extracted with EtOAc (2 x 40 mL). Combined organic layers were washed with brine (40 mL), dried over MgS04, filtered, and concentrated in vacuo. The crude product was purified using flash column chromatography on silica gel (Eluent 0-30% EtOAc/hexanes) to provide Int-86b (1.07 g, 70% yield).
Step C
A 20-mL microwave tube was charged with Int-86b (500 mg, 0.91 mmol), Int-7h (373 mg, 0.999 mmol), and (dppf)PdCl2'CH2Cl2 (67 mg, 0.091 mmol). The tube was sealed, evacuated, and placed under nitrogen atmosphere. Dioxane (9 mL) and aqueous potassium carbonate (2.7 mL, 1 M, 2.7 mmol) was added and the reaction was allowed to stir for about 15 hours at 80 °C. After cooling to room temperature, EtOAc (50 mL) and water (50 mL) were added, and the layers separated. The aqueous layer was extracted with EtOAc (2 x 10 mL), and the combined organic extracts were washed with brine (20 mL), dried over MgS04, filtered, and concentrated in vacuo. The crude product was purified using flash column chromatography on silica gel (Eluent: 0-100%
EtOAc/hexanes) to provide Int-86c (385 mg, 59% yield).
Step D
In a 20-mL microwave tube, Int-86c (380 mg, 0.530 mmol), bis(pinacolato)diboron (336 mg, 1.3 mmol), (dba)3Pd2*CHCl3 (55 mg, 0.053 mmol), X-
Phos (51 mg, 0.106 mmol) and KOAc (156 mg, 1.61 mmol) were combined. The tube was sealed, evacuated, and placed under nitrogen atmosphere. Dioxane (5.3 mL) was added and the reaction was allowed to stir at 120 °C for 1 hour, then allowed to cool to room temperature. The reaction mixture was diluted with EtOAc (20 mL) and washed sequentially with water (5 mL) and brine (5 mL). The organic layer was dried over MgSC>4, filtered, and concentrated in vacuo. The crude product was purified using flash column chromatography on silica gel (Eluent: 0-100% EtOAc/hexanes) to provide Int- 86d (382 mg, 93% yield).
Step E
In a 20-mL microwave tube, Int-86d (302 mg, 0.391 mmol), Int-7d (124 mg, 0.391 mmol), and (dppf)PdCl2 »CH2Cl2 (32 mg, 0.039 mmol) were combined. The tube was sealed, evacuated, and placed under nitrogen atmosphere. Dioxane (8 mL) and aqueous potassium carbonate (1.2 mL, 1 M, 1.2 mmol) was added. The reaction was allowed to stir for about 15 hours at 80 °C, then allowed to cool to room temperature. The reaction mixture was diluted with EtOAc (200 mL). The aqueous layer was extracted with EtOAc (2 x 10 mL). The combined organic extracts were washed with brine (50 mL), dried over MgSC , filtered, and concentrated in vacuo. The crude product was purified using flash column chromatography on silica gel (Eluent: 0-100% EtOAc (containing 10% MeOH)— hexanes) to provide Int-86e (78 mg, 22% yield).
Step F
A 125-mL round-bottom flask was charged with Int-86e (81 mg, 0.092 mmol) and methanol (2 mL). HC1 (0.84 mL, 2 M in diethyl ether, 1.7 mmol) was added and the reaction was allowed to stir for about 15 hours at room temperature. The reaction mixture was concentrated in vacuo to provide Int-86f (109 mg, quantitative yield).
Step G
A 25-mL round-bottom flask was charged with Int-le (53 mg, 0.067 mmol), Int-86f (15.5 mg, 0.081 mmol) and DMF (1 mL). Diisopropylethylamine (82 uL, 61 mg, 0.472 mmol) was added to the solution. The reaction mixture was cooled to 0 °C,
stirred for 15 minutes and HATU (395 mg, 1.04 mmol) was added. The reaction mixture was allowed to stir at 0 °C for 30 minutes, and then at room temperature for 2 hours. Additional diisopropylethylamine (20 uL, 2 eq) was added and the reaction was allowed to proceed for an additional 1 hour. The reaction mixture was diluted with EtOAc (30 mL) and poured into water (30 mL). The aqueous layer was extracted with EtOAc (2 x 10 mL). The combined organic phase were dried over MgS04, filtered, and concentrated in vacuo. The resulting crude product was purified using reverse-phase C18
chromatography (Gilson, 0-90% CH3CN (+ 0.1% TF A)— water (+ 0.1% TFA) over 15 minutes) to provide Compound 1100 as a yellow solid (31 mg, 48% yield).
A 25-mL round-bottom flask was charged with Int-86f (53 mg, 0.067 mmol), Int-4f (18 mg, 0.081 mmol), DMF (1 mL) and diisopropylethylamine (82 uL, 61 mg, 0.47 mmol). The reaction mixture was cooled to 0 °C and was allowed to stir for 15 minutes. HATU (26 mg, 0.067 mmol) was added and the reaction mixture was allowed to stir at 0 °C for 30 minutes, and then at allowed to warm to room temperature over 2 hours. Additional Int-4f (8.8 mg, 0.6 eq), HATU (5 mg, 0.2 eq) and
diisopropylethylamine (20 uL, 2 eq) were added and the reaction was allowed to proceed for an additional 1 hour. The reaction was diluted with EtOAc (30 mL) and poured into water (30 mL). The aqueous layer was extracted with EtOAc (2 x 10 mL) and the combined organic phase were dried over MgS04, filtered, and concentrated in vacuo. The crude product was purified using reverse-phase CI 8 chromatography (Gilson, 0-90% CH3CN (+ 0.1% TFA)— water (+ 0.1% TFA) over 15 minutes) to provide Compound 1099 as a yellow solid (28 mg, 43% yield).
EXAMPLE 88
Preparation of Compounds 1502 and 1505
Compound 793 (prepared as above in example 57 from Int-14d and Int-19j) (38 mg, 0.039 mmol) and Int-la (7.5 mg, 0.04 mmol) using the method described in
Example 57, step E to provide Isomeric Compounds 1502 and 1505 (19 mg, 46% yield).
EXAMPLE 89
Cell-Based HCV Replicon Assay
To measure cell-based anti-HCV activity of selected Compounds of the present invention, replicon cells were seeded at 5000 cells/well in 96-well collagen I- coated Nunc plates in the presence of the test Compound. Various concentrations of test Compound, typically in 10 serial 2-fold dilutions, were added to the assay mixture, with the starting concentration ranging from 250 μΜ to 1 μΜ. The final concentration of DMSO was 0.5%, fetal bovine serum was 5%, in the assay media. Cells were harvested on day 3 by the addition of lx cell lysis buffer (Ambion cat #8721). The replicon RNA level was measured using real time PCR (Taqman assay). The amplicon was located in 5B. The PCR primers were: 5B.2F, ATGGACAGGCGCCCTGA (SEQ ID NO. 1);
5B.2R, TTGATGGGCAGCTTGGTTTC (SEQ ID NO. 2); the probe sequence was FAM-labeled CACGCCATGCGCTGCGG (SEQ ID NO. 3). GAPDH RNA was used as endogenous control and was amplified in the same reaction as NS5B (multiplex PCR) using primers and VIC-labeled probe recommended by the manufacturer (PE Applied Biosystem). The real-time RT-PCR reactions were run on ABI PRISM 7900HT Sequence Detection System using the following program: 48 C for 30 minutes, 95°C for 10 minutes, 40 cycles of 95 C for 15 sec, 60 C for 1 minutes. The ACT values (CT5B-CTGAPDH) were
plotted against the concentration of test Compound and fitted to the sigmoid dose- response model using XLfit4 (MDL). EC50 was defined as the concentration of inhibitor necessary to achieve ACT=1 over the projected baseline; EC90 the concentration necessary to achieve ACT=3.2 over the baseline. Alternatively, to quantitate the absolute amount of replicon RNA, a standard curve was established by including serially diluted T7 transcripts of replicon RNA in the Taqman assay. All Taqman reagents were from PE Applied Biosystems. Such an assay procedure was described in detail in e.g. Malcolm et al, Antimicrobial Agents and Chemotherapy 50: 1013-1020 (2006).
HCV replicon assay ECgo data was calculated for selected Compounds of the present invention using this method and is provided in the table immediately below and in Table 2 in Example 89.
Compound la WT Ib WT la Y93H 2a WT 3a WT No. (nM) (nM) (nM) (nM) (nM)
31 0.004 0.003 15 0.05 1.2
32 0.002 0.002 3 0.007 0.3
61 0.036 0.01 1 14.6 0.27 2.7
62 0.011 0.006 8.4 0.12 3
63 0.005 0.003 1.8 0.045 2.7
64 0.013 0.008 6.2 0.083 4.8
66 0.006 0.004 27 0.06 2.7
67 0.005 0.008 29 0.1 2.5
68 0.002 0.002 0.02 0.21 1.0
69 0.001 0.001 0.6 0.11 1.8
75 0.005 0.006 28.937 0.006 0.465
76 0.010 0.005 68.123 0.018 5.367
79 0.004 0.004 28.273 0.010 1.357
84 0.012 0.007 6.086 NA 1.239
86 0.004 0.004 1.619 0.298 0.402
87 0.007 0.006 13 0.22 1.7
88 0.004 0.002 13.648 0.133 0.727
89 0.014 0.022 77.94 0.682 3.921
90 0.012 0.004 2.840 NA 2.301
91 0.003 0.004 0.401 0.306 0.354
92 0.011 0.007 6 0.46 1.2
93 0.026 0.009 14.7 0.36 3
94 0.003 0.004 0.06 0.3 0.35
95 0.02 0.01 100 0.11 0.98
99 0.004 0.007 39 0.022 0.17
103 0.015 0.016 38 0.032 0.16
104 0.005 0.003 14 0.023 0.09
108 0.014 0.015 68 0.13 5.4
109 0.015 0.016 139 0.13 0.4
110 0.006 0.006 68 0.02 0.25
117 0.004 0.003 44 0.063 0.08
121 0.027 0.021 256 0.15 3.5
125 0.020 0.016 18 0.04 0.14
126 0.003 0.005 20 0.066 >10
129 0.016 0.009 80 0.047 0.65
130 0.03 0.006 211 0.3 5.8
133 0.002 0.002 32 0.15 0.58
136 0.006 0.006 78 0.07 0.31
143 0.020 0.013 26 0.23 0.31
145 0.004 0.005 8 0.007 0.1
149 0.004 0.004 29 0.04 0.4
150 0.001 0.003 7 0.11 0.21
153 0.003 0.003 12 0.008 0.074
EXAMPLE 90
Additional Compounds of the Invention
Additional illustrative compounds of the present invention are set forthlow in Table 2. The Replicon data provided for selected compounds depicted in Table was generated using the method described in Example 89.
Table 1
ot Available
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
247 0.003 1.4 0.1 0.004 8 984.15 985.1
isomers
248 0.013 2.54 0.005 17 948.05 948.5 Mixture of isomers
Mixture of
249 0.009 0.8 0.004 4 958.11 959
isomers
250 0.013 0.3 4.05 0.003 36 966.04 966.7 Single isomer
251 0.003 1.12 0.004 25 966.04 966.7 Single isomer p
Mixture of
252 0.024 9.9 1.15 0.011 15 956.12 957
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalC.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
253 56.000 500 918.02 918.6 Single isomer
254 127.000 489 918.02 918.6 Single isomer
255 0.008 3.2 0.28 0.004 4 984.03 985 Single isomer
256 0.007 12.9 14 984.03 984.8 Single isomer
257 0.030 0.44 4.73 0.003 110 1020.01 1021 Single isomer
J °r° A
258 0.003 3.94 0.003 46 1020.01 1021 Single isomer
0
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [ +Hf Designation
Mixture of
264 0.009 6.7 0.411 0.008 6.9 916.03 917
isomers
Mixture of
265 0.012 6.39 0.2 0.005 3.3 926.09 926.7
isomers
Mixture of
266 0.009 4.8 0.35 0.006 4 1002.17 1003
isomers
Mixture of
267 0.006 0.5 0.459 0.005 40 1038.15 1003
isomers
268 0.009 0.6 0.205 0.005 1 984.15 985.1 Single isomer
269 0.004 10.86 0.3 0.004 16.8 984.15 985.1 Single isomer ο
0
EC50 EC50 EC50 EC50 EC50 Obs .M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
276 0.045 2.1 1 1.9 0.014 98.3 1037.12 1019.9
7 ° O -
277 940.12 941 Single isomer
O
278 940.12 941 Single isomer
279 1001.14 1002 Single isomer
280 1001.14 002 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs .M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [Μ+ΗΓ Designation
281 0.003 58.4 0.7 0.006 13.7 956.12 956.8 Single isomer
282 956.12 956.8 Single isomer
283 958.11 958.9 Single isomer
284 0.026 24.3 0.007 9.4 958.11 959 Single isomer
285 0.015 3.4 0.005 47.7 994.09 995 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+H]* Designation
J. °r°
296 983.15 984 Single isomer o
HN
J. °r ό
297 944.09 945 Single isomer
0
298 944.09 944.9 Single isomer
299 0.009 40.3 0.004 14.4 954.15 954.9 Single isomer
300 0.014 5.1 0.7 0.004 12.4 954.15 955 Single isomer
EC50 EC50 ECSO EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
306 0.007 4.8 0.3 0.006 6.6 984.18 985 Single isomer
307 1020.16 1021 Single isomer
308 1020.16 1021 Single isomer
309 0.029 22.9 0.015 > 100 1037.12 1038 Single isomer
310 0.143 13.3 19.1 0.018 > 100 1037.12 1038 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M S Isomer
Cmpd # Structure Calc .S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
317 0.00817 1.184 0.195 6.912 988.14 988.9 Single isomer
318 0.015 0.5 0.3209 13.88 916.03 916.8 Single isomer
319 0.004 41.02 0.3329 9 916.03 916.8 Single isomer
Mixture of
320 0.01 128 0.1398 1.207 87.61 994.09 995
isomers
Mixture of
321 0.00982 2.132 0.195 2.146 944.09 945.1
isomers
Mixture of
322 0.0162 0.4183 0.1426 1.192 942.07 943.1
isomers
EC50 ECS0 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
323 0.01563 0.02 2.674 68.32 980.07 981
isomers
324 0.02436 6.242 1.81 25.43 948.05 948.9 Single isomer
325 1 100 100 100 948.05 948.9 Single isomer
Mixture of
326 0.02664 2.77 49.99 976.10 977
isomers
Mixture of
327 0.01 2.3 0.1873 1.328 946.12 947
isomers
Mixture of
328 0.01029 0.04 2.205 73 1008.12 1009.1
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc. .S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
329 0.0056 1.517 1.656 18.49 984.03 985
isomers
Mixture of
330 0.003 1.486 0.2152 7.645 1050.21 1051.1
isomers
Mixture of
331 0.004 7.523 0.2071 4.54 928.13 928.8
isomers
332 0.01225 43.76 0.4942 17.35 972.14 973 Single isomer
333 0.01234 1.19 0.195 5.178 972.14 973 Single isomer
334 0.005 52.66 0.2183 12.86 926.09 926.8 Single isomer
ECSO EC50 EC50 EC50 EC50 ObS M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
340 1 100 100 100 1019.13 1020.2 Single isomer
341 0.01328 54.63 0.2997 11.13 940.12 941.2 Single isomer
342 0.01308 2.627 0.2881 2.418 940.12 941.2 Single isomer
Mixture of
343 0.02634 0.07918 3.088 61.91 1020.13 1021
isomers
344 15.9 469.9 724.87 725.5 Single isomer
o ό ,
345 0.01349 8.592 1.789 31.37 926.09 926.6 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
351 0.02156 0.4577 0.1775 9.245 930.06 931.1
isomers
Mixture of
352 0.00976 0.04956 0.5672 9.561 978.05 979.2
353 8.626 366 824.99 825.9 Single isomer
354 1.179 678.1 824.99 825.9 Single isomer
355 12.16 110.2 724.87 725.8 Single isomer
356 0.01505 3.088 0.2035 2.379 926.09 927 Single isomer o
EC50 EC50 EC50 EC50 EC50 Obs .M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [ +ΗΓ Designation
357 0.0162 20.67 0.3557 2.697 942.07 943 Single isomer
358 0.02086 1.514 0.03596 0.5689 942.07 943 Single isomer
359 0.02836 0.5365 4.157 76.52 1024.12 1025.2 Single isomer
360 0.01321 0.06267 2.885 11 1.6 1024.12 1025.2 Single isomer
361 0.01345 20.71 0.572 33.65 998.18 999.1 Single isomer
362 0.02602 2.432 0.07 4.6 998.18 999.1 Single isomer
EC50 ECS0 EC50 ECS0 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
363 0.018 43.3 0.4796 10.63 954.15 955 Single isomer
364 0.028 48.67 0.6579 10.78 954.15 955 Single isomer
365 0.00804 0.04587 2.188 68.84 1008.12 1009.1 Single isomer
366 0.0389 0.2949 2.96 79.37 1008.12 1009.1 Single isomer
Mixture of
367 0.01121 5.385 0.2396 7.747 954.15 955.2
Mixture of
368 V 0 H 0.02062 7.579 0.363 32.25 972.14 973
isomers
ECSO EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
369 0.02 15.21 0.112 3.65 928.07 929.2 Single isomer
Mixture of
370 0.01857 7.238 0.028 0.55 946.06 946.7
isomers
Mixture of
371 0.01427 2.28 0.2345 11.7 944.09 944.6
Mixture of
372 0.02522 1.79 0.055 946.06 947.8
isomers
Mixture of
373 0.01613 5.214 0.2444 5.925 972.14 973
isomers
Mixture of
374 0.0105 8.422 0.3063 8.9 974.11 975
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
375 0.09597 1.528 6.8 198 934.02 935
isomers
Mixture of
376 0.00973 2.25 0.035 11.21 972.14 973
isomers
Mixture of
377 0.00736 0.025 1.792 59.8 1008.12 1009
isomers
378 0.023 0.43 2.018 73.85 994.09 995 Single isomer
379 0.005 0.0098 2.687 71.51 994.09 995 Single isomer
380 0.01344 3.12 0.1024 0.4612 928.07 929 Single isomer p
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+H Designation
416 990.13 990.8 Single isomer
417 990.13 990.8 Single isomer
418 1050.21 1051 Single isomer
419 1050.21 1051 Single isomer
420 930.06 931 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
421 930.06 931 Single isomer
422 1040.19 1041 Single isomer
423 1040.19 1041 Single isomer
424 0.00389 27 0.6629 6.776 972.14 973.1 Single isomer
425 0.01474 1.047 0.1416 16.74 972.14 973.1 Single isomer
426 0.00649 0.2636 10.16 977.05 978 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
434 0.011 4 1 0.001 10 865.40 867.2 Single isomer
435 0.002 0.5 2 0.001 32 865.40 866.5 Single isomer
436 0.002 30 1 0.001 5 865.40 866.5 Single isomer
437 0.003 6 1 0.001 2 865.40 866.5 Single isomer
Mixture of
438 0.008 45.6 10.38 0.003 37.4 879.40 880.5
isomers
439 0.014 540.8 7.4 0.002 6.1 879.40 880.7 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
440 0.002 24.2 4.05 0.001 52.7 879.40 880.8 Single isomer
441 0.010 14.2 0.020 22.9 911.40 912.7 Single isomer
442 0.010 18 0.020 151.8 911.40 .7 Single isomer
ixture of
443 0.019 1.95 42.864 0.008 883.40 884.8
isomers
Mixture of
444 1.320 14.582 2.81266 0.792 6.101 792.40 793.4
isomers
Mixture of
445 > 10 > 000 855.866 > 2 682.615 825.40 826.4
isomers
EC50 EC50 EC50 EC50 EC50 Obs .M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
446 1.000 4.26 1.95 0.200 2 906.40 907.4
isomers
ixture of
447 1.950 1.95 1.95 0.200 2 939.40 940.4
isomers
Mixture of
448 2.830 1000 212.36 2.400 1000 925.40 926.5
isomers
Mixture of
449 > 2000 > 5000 592.30 593.3
isomers
Mixture of
450 365.116 5099 811.40 812.3
Mixture of
451 131.553 670.918 788.30 789.7
isomers
N
EC50 EC50 EC50 EC50 EC50 Obs .M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
458 188.892 227.88 788.40 789.7
isomers
Mixture of
459 0.246 19.21 18.4 0.027 200 968.50 968.8
Mixture of
460 512.000 > 20000 838.40 839.4
isomers
Mixture of
461 0.097 > 200 14 0.151 99.105 964.50 965.5
isomers
Mixture of
462 0.018 > 200 88.9 0.022 76.8 882.40 883.4
isomers
>-0 N-t°
EC50 EC50 EC50 EC50 EC50 Obs .M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+H]* Designation
Mixture of
463 0.024 > 200 0.4 0.014 41 952.50 953.5
isomers
Mixture of
464 485.000 2619 881.50 882.1
isomers
Mixture of
465 0.073 > 200 0.27 0.033 0.2 970.50 971.1
isomers
Mixture of
466 0.005 0.2 3.2 0.002 49 942.40 943.4
V 0 Λ Mixture of
467 >1 >100 >100 0.130 >100 995.50 996.5
isomers
ECSO EC50 EC50 EC50 EC50 ObS M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
480 1227.6 691.30 692.3 Single isomer
481 0.124 62.1 109.9 0.012 110 805.40 806.4 Single isomer p
Mixture of
482 0.028 0.382 0.014 2.7 738.40 739.3
483 ° v ° 609.000 >1000 721.40 722.3 Single isomer
484 0.026 11.9 17.3 0.019 > 100 835.40 836.4 Single isomer
Mixture of
485 0.012 22.58 0.23 4.35 936.40 937.4
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
486 934.400 >1000 822.40 823.4
isomers
N
487 605.000 >1000 806.40 808.3 Single isomer
Mixture of
488 0.011 114.2 1.62 35.93 931.40 933.2
489 0.860 115.2 236.2 >1000 835.40 836.3 Single isomer
490 0.090 51 65 234 920.40 921.3 Single isomer
491 0.010 9.02 0.42 10.9 924.40 925.3 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs .M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
0
508 > 1 > 100 > 100 0.154 > 100 747.39 748.2 Single isomer
NH
509 0.070 > 100 > 100 0.026 > 100 793.35 794.3 Single isomer
510 0.015 > 100 25 0.017 99.5 820.37 821.4 Single isomer
511 0.020 110 50 0.015 69.3 845.33 846.3 Single isomer
512 0.043 109.9 44.71 0.020 110 859.35 860.4 Single isomer
513 0.140 109.9 109.9 0.030 110 859.35 860.4 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+H]* Designation
Mixture of
519 0.038 > 100 8.7 0.034 > 100 821.39 822.4
isomers
520 0.165 1.6 0.082 > 100 807.37 808.4 Single isomer
521 0.044 > 100 4.6 0.020 > 100 835.40 836.4 Single isomer
522 0.054 > 100 24.1 0.022 > 100 833.42 834.4 Single isomer
523 0.085 > 100 27.9 0.040 > 100 817.39 818.3 Single isomer
EC50 EC50 CS0 EC50 EC50 Obs M.S.
Cmpd # E Isomer
Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
524 820.40 NA Single isomer
525 25.000 1000 592 1000 923.45 924.2 Single isomer
526 1.680 377 195 823.40 824.3 Single isomer
527 0.050 13 0.7 15 949.34 950.2 Single isomer
528 0.420 143 108 >1000 880.42 881.7 Single isomer
EC50 EC50 ECSO EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
534 0.013 12 9.11 0.005 9 901.5 901.8
isomers
535 0.020 27.82 1.1 0.002 111 859.4 859.9 Single isomer
536 890.530 10000 767.4 768.4 Single isomer
537 0.013 109.9 18 914 915.5 Single isomer
538 0.006 0.9 0.020 0.2 965.4 966.4 Single isomer
Q O
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
539 0.180 >100 93 >1 824.4 825.4 Single isomer
540 0.500 >100 71 >1 824.4 825.2 Single isomer
541 478.000 >1000 >1000 360.000 763.40 765.0 Single isomer
Mixture of
542 0.003 19.19 1.815 0.002 44.5 868.4 869.6
isomers
ECSO EC50 EC50 ECSO EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+H]* Designation
Mixture of
543 0.005 12.18 4.748 0.004 3 834.0 834.7
544 0.005 2.26 1.724 0.002 1.1 888.1 889 Single isomer
Mixture of
545 0.004 4.69 0.5041 0.002 1 908.5 909.6
isomers
Mixture of
546 0.006 19.64 0.6745 0.003 1.3 874.1 875
Mixture of
547 0.005 16.84 0.619 0.002 8.7 898.4 899.4
isomers
ECSO EC50 ECSO EC50 EC50 ObS M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
548 0.013 31.05 2.474 0.003 > 100 823.9 824.8
Mixture of
549 0.003 1.97 0.9988 0.003 1.3 864.0 865
Mixture of
550 0.009 66.79 1.92 0 890.1 891
isomers
1 C ίΐ ' Λ Mixture of
55 0 7.42 6.055 0 > 100 818.0 818.8
isomers
Mixture of
552 0.003 2.15 1.334 0 3.1 852.0 852.7
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
563 0.001 12.15 1.339 0.001 4.5 866.0 866.8 Mixture of isomers
564 0.005 48.97 11.08 0.002 17.9 864.0 865 Mixture of isomers
Mixture of
565 0.002 0.93 1.06 0.001 61.1 918.0 919
566 0.022 > 100 33.97 0.006 > 100 858.0 858.7 Single isomer
xture of
567 0.033 > 100 12.3 0.003 > 100 902.0 903 Mi
isomers
EC50 EC50 EC50 EC50 EC50 Obs .M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
568 0.011 0.22 11.63 0.001 43.7 902.0 903
isomers
Mixture of
569 0.023 > 100 11.76 0.010 60.5 846.0 846.8
Mixture of
570 0.012 0.2 12.86 0.001 76 888.0 888.8
isomers
571 0.001 4.02 0.3006 0.001 3.7 882.0 882.6 Single isomer
572 0.003 22.65 1.359 0.001 2.6 864.0 865 Single isomer
573 882.0 883 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
574 0.010 > 100 43.2 0.001 336.4 793.9 794.5 Single isomer
575 NVjJ c 0.019 0.81 19.86 0.002 44 884.0 884.6 Single isomer
576 0.011 0.41 45.42 0.001 90.8 870.0 870.7 Single isomer
577 0.004 5.64 1.526 0.001 104.4 938.0 938.6 Single isomer
578 0.002 1.09 5.191 0.001 102.9 923.9 925 Single isomer
579 0.011 26.56 2.158 0.009 918.0 919 Single isomer
Cmpd # EC50 EC50 EC50 EC50 ECSO Obs .M.S. Isomer
Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
580 0.005 60.12 5.531 0.002 2.2 846.0 846.8 Single isomer
581 0.020 0.39 11.49 0.001 70.5 902.0 902.8 Single isomer
582 0.013 1.11 83.06 0.002 93.9 888.0 889 Single isomer
583 864.0 865 Single isomer
584 900.0 900.8 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
585 0.014 0.2 23.5 0.007 64 888.0 888.8 Single isomer
586 0.002 4.2 1.4 0.001 41.7 906.0 906.7 Single isomer
587 0.001 14.82 2.6 0.001 95.4 906.0 906.7 Single isomer
588 0.002 7.68 1 0.001 41.4 923.9 924.7 Single isomer
589 0.001 14.23 2.5 0.001 158.6 923.9 924.7 Single isomer
590 0.003 24 1.7 0.001 59.4 938.0 938.7 Single isomer
591 0.002 43 1.6 0.001 159.4 938.0 938.7 Single isomer
ECS0 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+H]* Designation
592 0.007 53 4 0.001 151.3 923.9 924 Single isomer
593 0.003 59.04 9.4 0.001 209.9 923.9 925 Single isomer
594 0.002 9.6 1.3 0.001 3.1 920.0 921 Single isomer
595 0.001 0.9 0.001 5.4 920.0 920.8 Single isomer
596 0.005 9.18 0.001 134.8 936.0 936.7 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Ca!c M S.
1a 1aY93H 1aL31V 1b 2b [M+HJ* Designation
604 0.014 0.008 852.0 852.8 Single isomer
605 0.003 21.58 1.9 0.002 2 848.0 848.8 Single isomer
606 0.002 15.22 2.1 0.001 2 834.0 835 Single isomer
607 0.001 16.54 1.7 0.002 7 848.0 849 Single isomer
608 0.002 4.62 3 0.002 12 834.0 834.8 Single isomer
609 0.002 24.89 8.6 0.003 71 852 0 852.8 Single isomer
Mixture of
610 0.009 35 1.4 0.003 3.9 900.0 900.9
isomers
P
EC50 EC50 EC50 EC50 EC50 Obs .M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+H]+ Designation
611 1.950 27.45 1.95 0.200 12 884.0 885 Mixture of isomers
612 1.950 241.5 7.47 0.200 200 846.1 846.9 Mixture of isomers
613 0.023 359 10 13.2 884.0 885 Single isomer
614 ¾! n¾ 0.004 12 10 9.8 884.0 885 Single isomer
615 0.016 190 9.11 0.003 46 846 1 846.9 Single isomer
mer
Cmpd # Structure EC alcM.S
1a50 1a EYC9530H 1 EaLC3510V EC 1b50 EC 2b50 Obs M.S. Iso
C .
[M+Hf Designation
671 0.016 0.6 2.4 0.005 52.5 1002.0 1002 Single isomer o
672 0.006 0.22 2.1 0.005 26.8 1002.0 1003 Single isomer o
Mixture of
673 0.00685 1.4 0.1408 948.0 948.9
isomers
Mixture of
674 0.01579 7.862 0.1396 930.1 931
Mixture of
675 0.01313 5.192 0.1423 980.2 981.1
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+H]* Designation
686 0.00799 0.01904 1.958 1002.0 1003 Single isomer
687 0.02801 0.197 2.78 1002.0 1003 Single isomer
688 0.00919 15.96 0.9876 984.0 984.8 Single isomer
689 0.00254 1.935 2.455 984.0 984.8 Single isomer
690 0.00529 0.6467 0.9878 1002.0 1003 Single isomer
EC50 EC50 EC50 EC50 EC50 Qbs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+H]* Designation
691 0.00274 5.758 1.067 1002.0 1003 Single isomer
692 0.01126 0.3635 0.7222 1002.0 1003 Single isomer
693 0.00297 1.13 0.5323 1002.0 1003 Single isomer
694 0.00191 1.68 0.4014 928.1 929.9 Single isomer
695 0.00523 1.498 0.2222 928.1 929.1 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc .S.
-la 1aY93H 1aL31V 1b 2b [M+H]* Designation
708 902.10 903.4 Single isomer
Mixture of
709 0.008 91 1 0.005 9 860.00 861.3
isomers
Mixture of
710 0.050 > 1000 21.6615 0.016 890.515 977.42 978.7
Mixture of
711 0.019 5.02934 1.9555 0.002 25.9449 909.45 910.7
J °r° A
Mixture of
712 0.019 2.93552 1.9555 0.007 22.3421 909.45 651.8
isomers
ECSO EC50 EC50 EC50 EC50 Obs .M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [ +Hf Designation
713 0.019 11.559 3.693 0.002 37.251 849.40 850.7 Single isomer
714 925.43 925.8 Single isomer
715 0.002 18.59 9.11 0.002 85.53 925.43 925.8 Single isomer
716 0.016 > 50 27.49 0.006 15.61 909.45 910.8 Single isomer
717 0.005 6.23 981.72 0.005 130.23 909.45 910.8 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc .S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
724 945.51 946.3 Single isomer
725 949.39 950.6 Single isomer
726 803.36 803.9 Single isomer
727 825.36 826.7 Single isomer
728 965.44 966.7 Single isomer
Mixture of
729 0.013 47 12.2 0.009 12 915.41 916.4
isomers
EC50 ECSO ECSO EC50 EC50 Obs .M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
730 0.016 > 100 1.6 0.009 0.254 853.39 855.4
Mixture of
731 0.013 4.287 0.2 0.012 1.031 923.43 924.5
Mixture of
732 0.029 55.72 0.39 0.019 2.354 913.46 914.4
Mixture of
733 0.371 > 100 44.8 0.035 32.027 766.36 767.3
Mixture of
734 0.008 0.71 0.19 0.009 0.9 923.43 924.3
isomers
ECSO EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
735 0.008 19.11 0.008 1.911 913.41 914.4 Single isomer
736 0.005 49 1.7 0.006 41.4 933.45 934.7 Single isomer
Mixture of
737 0.018 11 0.2 0.010 913.46 916.4
isomers
738 965.44 967.5 Single isomer
739 909.45 9 1.4 Single isomer
740 ¾ H°¾ 0.007 32 1 0.004 16.8 909.45 911.4 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs .M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
745 0.014 3.18 0.8 0.010 5 847.40 848.5 Single isomer
746 0.082 3.45 0.4 0.038 5.9 889.41 890.5 Single isomer
747 0.014 41.65 9 0.010 < 100 803.38 804.4 Single isomer
748 0.022 92.12 2.3 0.017 16 837.38 838.4 Single isomer
Mixture of
749 0.020 20.05 0.3 0.007 6.1 897.42 899.3
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
750 0.030 6.3 0.007 38.1 831.41 832.3 Single isomer
Mixture of
751 0.055 3.5 0.3 0.044 1.6 917.44 918.5
752 0.227 > 100 1.6 0.038 0.6 869.47 870.3 Single isomer
753 0.021 1.5 0.013 4.5 837.43 839.3 Single isomer
754 0.026 5.3 1.4 0.750 15 847.40 848.3 Single isomer
Mixture of
755 0.200 >100 0.85 0.120 0.52 897.51 899.5
°T j isomers
O
ECS0 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+H]+ Designation
756 >1 >100 51.6 >1 >100 763.35 764.3 Single isomer
757 0.009 16.2 0.85 0.410 33 853.39 854.4 Single isomer
758 0.036 1.3 0.12 0.120 0.36 923.43 925.5 Single isomer
759 0.015 13.5 0.33 0.090 1.5 897.42 899.4 Single isomer
760 0.028 3.3 0.076 0.021 2 923.43 924.5 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
772 > 2000 > 20000 995.40 996.3
isomers
Mixture of
773 > 2000 > 20000 935.40 936.3
Mixture of
774 > 2000 > 20000 953.40 954.3
isomers
775 165.000 > 20000 751.30 752.3
Mixture of
776 0.036 1.8 3.8 0.005 46.59 1109.40 1110.3
isomers o.
Mixture of
777 0.007 0.6 0.8 0.003 28.18 1067.40 1069.0
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+H]* Designation
Mixture of
795 1.69 1005.40 1006.4
isomers
Mixture of
796 _ -o >1 >100 >100 857.40 859.4
isomers
Mixture of
797 0.023 2.234 0.26 987.40 988.4
isomers
Mixture of
798 >1 >100 >100 839.40 840.4
Mixture of
799 0.018 13.2 2.1 0.006 380 971.40 972.6
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
806 0.021 11 16.84 0.016 12 883.40 884.4 Mixture of isomers
807 0.065 2.76 39 0.012 < 100 857.40 858.4 Mixture of isomers
808 0.009 11.82 10.9 0.009 92 839.40 840.4 Mixture of isomers
809 26.800 314.1 794.40 795.4 Mixture of isomers o ^.o
810 .036 14.18 58.24 246.6 849.40 850.4 Mixture of isomers
O NH < , 0
811 0.011 17.84 35.03 169.9 865.40 866.5 Mixture of isomers
EC50 EC50 EC50 EC50 EC50 Obs .M.S. Isomer
Cmpd # Structure Calc.M S.
1a 1aY93H 1aL31V 1b 2b [M+H]* Designation
Mixture of
812 0.045 3.576 5.465 10.04 893.40 894.4
isomers
Mixture of
813 1.160 11.25 794.40 795.3
Mixture of
814 0.035 1.72 4.1 0.21 893.40 894.3
isomers
Mixture of
815 0.020 3.76 36.2 150.3 849.40 850.3
Mixture of
816 0.042 2.57 12.3 42.46 867.40 868.2
817 0.001 0.4 2 0.001 5.7 896 02 897.0 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc M.S
1a 1aY93H 1aL31V 1b 2b [M+H]* Designation
°r
°NH
830 0.015 6.149 1 0.013 0.881 902.39 902.7 Single isomer
Mixture of
831 > 1000 1480 884.32 884.6
isomers
Mixture of
832 518.300 1953 860.33 860.6
isomers
Mixture of
833 > 1000 3481 684.22 684.6
Mixture of
834 > 1000 7347 660.22 660.7
isomers
Mixture of
835 0.008 0.15 2.7 0.007 35 974.37 975.2
isomers
EC50 ECSO EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
853 0.005 1.4 3.7 0.001 18 910.04 911.0
854 0.010 1 2.6 0.020 7.7 922.05 923.1
isomers
Mixture of
855 0.010 0.5 1.2 0.020 8.3 954.10 955.1
Mixture of
856 > 0.2 > 100 63 > 0.2 369 839.99 841.0
isomers
Mixture of
857 0.002 619.1 0.79 0.002 9.4 940.10 941.1
858 > 2 < 100 > 102 > 0.2 > 2000 735.91 736 9 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
864 0.002 5.6 1 0.002 2.2 890.11 891.1 Mixture of isomers
865 0.005 31 4.2 0.001 13 940.10 941.1 Single isomer
866 0.003 4 1 0.001 42 841.92 842.9 Single isomer
Mixture of
867 0.002 19.4 1.6 0.001 4 924.07 925.1
isomers
868 0.002 6.3 1.4 0.000 79 859.91 860.9 Single isomer
869 0.002 3.3 2 0.002 68 841.92 842.9 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
876 > 2 > 200 200 0.033 200 780.94 781.9 Single isomer
877 0.354 > 200 200 0.123 200 839.96 841.0 Single isomer
878 > 2 > 200 200 0.070 200 760.86 761.9 Single isomer
879 > 2 > 200 200 0.037 200 788.87 789.9 Single isomer
9 °
N N HTV-ZN
880 0^NH Ο Ν 0.316 > 200 200 0.028 200 819.97 821.0 Single isomer
881 0 777 > 200 200 0.030 200 720.84 721.8 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
895 0.014 7 6.5 0.013 96 791.92 792.9 Single isomer
896 0.013 32.89 10.1 0.007 1.166 819.97 821.0 Single isomer
897 218.024 2408.33 834.04 835.0 Single isomer
898 32.000 401 782.95 784.0 Single isomer
899 0.004 43 3 0.002 51.1 819.97 821.0 Single isomer
900 0.233 > 100 93 0.070 > 100 924.08 925.1 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc .S.
1a 1aY93H 1aL31V 1b 2b [M+H]+ Designation
918 0.131 109.9 109.9 0.019 110 914.00 915.0 Single isomer
919 0.300 > 100 > 100 0.088 789.94 790.9 Single isomer
920 > 2 109.9 109.9 0.368 110 804.01 805.0 Single isomer
9 °
921 0.136 109.9 109.9 0.016 110 855.96 857.0 Single isomer
9 °
922 0.154 109.9 65.72 0.034 110 89500 896.0 Single isomer
9 °
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
929 860.03 861.0
isomers
Mixture of
930 876.03 877.0
isomers
Mixture of
931 > 10 > 1000 > 1000 > 2 > 1000 734.86 735.9
Mixture of
932 0.362 > 1000 524 0.205 > 1000 862.99 863.8
Mixture of
933 > 10 > 1000 > 1000 > 2 > 1000 770.84 771.7
Mixture of
934 > 10 465.092 > 1000 0.100 > 1000 884.95 885.9
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
983 0.026 0.49 8.1 0.001 67 966.40 966.0
Mixture of
984 0.006 29.88 0.7 0.002 57 930.40 930.0
isomers
Mixture of
985 0.003 8 1 0.002 15 896.40 896.5
Mixture of
986 0.003 20.8 0.27 0.001 6.2 946.40 946.2
isomers
Mixture of
987 0.004 8.8 0.8 0.004 1.9 912.40 912.4
Mixture of
988 > 10 275.009 35.1221 > 2 156.107 802.40 802.4
isomers
CI
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
989 16.000 95.7794 77.0776 > 2 845.40 845.4
Mixture of
990 143.510 1000 544.35 23.770 456 834.40 834.4
isomers
Mixture of
991 98.590 1000 412.46 21.370 374 860.40 860.4
isomers
Mixture of
992 349.000 > 5000 645.30 645.3
isomers
Mixture of
993 3.000 > 5000 660.30 660.3
isomers
Mixture of
994 1.950 10.44 1.95 0.200 2 974.50 974 7
isomers
EC50 EC50 EC50 EC50 ECSO Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+H]* Designation
Mixture of
995 0.010 >100 10 24.8 948.40 948.3
isomers
Mixture of
996 0.007 40 10 9.8 959.50 959.4
Mixture of
997 139.157 3562.44 861.40 861.3
isomers
NH2
Mixture of
998 3.900 52.94 27 0.218 > 100 844.40 844.4
isomers
Mixture of
999 325.000 > 5000 875.40 875.4
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
1000 189.210 1230.32 828.40 828.3
isomers
Mixture of
1001 592.717 > 20000 981.50 981.4
Mixture of
1002 0.004 1.48 0.005 0.3 942.40 942.4
isomers p
Mixture of
1003 0.007 2.76 0.5 0.005 11.9 1094.50 1094.5
NVj3 Mixture of
1004 0.003 0.19 2 0.003 12 9 989.50 989.4
isomers
Ί>-ο
EC50 EC50 EC50 EC50 EC50 Obs .M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
J °r° A
C Mixture of
1005 0.003 7.89 0.3 0.004 1 958.50 958.5
isomers
Mixture of
1006 230.000 > 20000 853.40 853.3
Mixture of
1007 0.006 25.94 0.2 0.008 7.6 959.50 959.5
isomers
Mixture of
1008 0.004 52.2 0.3 0.006 16.869 967.50 967.5
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+H]+ Designation
Mixture of
1018 0.007 0.69 0.004 7 960.40 960.5
isomers
Mixture of
1019 0.006 1.17 0.497 0.006 0.512 960.40 960.5
Mixture of
1020 1.111 403.99 885.40 885.4
isomers p
Mixture of
1021 2.045 520.8 903.40 903.5
Mixture of
1022 7.490 1228.75 785.40 785.4
Mixture of
1023 2.836 700.87 803.40 803.4
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [ +Hf Designation
1024 0.003 25.32 0.2 0.003 4.1 942.40 942.5 Single isomer
1025 0.003 0.68 0.31 0.002 0.2 942.20 942.2 Single isomer
Mixture of
1026 16.000 443 893.40 893.5
isomers
O
Mixture of
1027 693.30 693.3
isomers
O
Mixture of
1028 > 1 > 100 > 100 > 1 > 100 843.40 843.5
isomers
Mixture of
1029 0.004 3.17 1 0.008 3.8 1007.40 1007.2
isomers
ECS0 EC50 EC50 EC50 EC50 Obs M S. Isomer
Cmpd # Structure Calc M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
1030 > 1 > 100 > 100 > 1 > 100 842.40 842.5
Mixture of
1031 199.700 606 846.40 846.4
Mixture of
1032 1.000 > 100 > 100 1.000 > 100 844.40 844.5
Mixture of
1033 0.006 2 0.2 0.005 0.7 960.40 960.5
isomers o-
1034 0.030 82 13 > 1 51.11 844.40 844.5 Single isomer
1035 0.247 11 0.066 6.15 844.40 844.4 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc .S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
1036 219.000 628 874.40 874.5
isomers
Mixture of
1037 0.010 3.2 0.005 10.4 988.50 988.5
Mixture of
1038 0.024 0.4 0.009 0.2 984.50 984.5
isomers
Mixture of
1039 0.009 12.1 0.007 0.6 958.40 958.5
Mixture of
1040 942.40 942.2
isomers ρ
EC50 EC50 EC50 EC50 EC50 Obs M S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
1041 >100 39 74 884.40 884.2
Mixture of
1042 0.020 58 0.96 19.80. 976.40 976.2
Mixture of
1043 0.022 72.2 0.28 994.40 994.3
isomers p
Mixture of
1044 0.024 3.5 <0.195 <0.195 974.50 974.2
isomers o-
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+H]* Designation
Mixture of
1045 >1 >100 7.2 >100 914.40 914.4
isomers
OH
330.000 325 864.40 864.3 Mixture of
1046 isomers
1047 319.000 527 780.40 780.3 Mixture of isomers
Mixture of
1048 0.030 0.43 0.03 0.13 1026.50 1026.6
isomers p
J. °r° ό
Mixture of
1049 0.004 10.5 1.1 0.001 21 944.50 944.5 isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
1050 0.039 < 100 12 0.003 1 13 910.50 910.4
Mixture of
1051 0.030 0.4 9 0.001 129 980.50 980.4
NYY TI N Γ Mixture of
1052 O NH o 0.007 48 5.5 0.001 85 876.50 876.8
isomers
Mixture of
1053 0.005 37.5 1.1 0.001 19 896.40 896.0
isomers
EC50 EC50 EC50 EC50 EC50 Obs .M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
1058 0.001 10.73 0.771 0.001 10 862.50 862.5
1059 0.003 27 1 0.001 2 910.50 910.5 Single isomer
1060 0.002 1.5 0 0.001 2 910.50 910.5 Single isomer
1061 > 0.2 0.8 25 0.001 15 946.40 946.5 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
1072 7.301 642.739 896.40 896.3
O NH s °T Mixture of
1073 0.006 19 9.11 0.005 9 987.50 987.4
isomers
Mixture of
1074 267.000 777.33 854.40 854.3
isomers q
Mixture of
1075 0.008 36.28 148.08 0.003 > 5000 968.50 968.4
isomers
Mixture of
1076 27.260 45.888 898.20 898.2
isomers
Mixture of
1077 0.628 > 200 200 > 2 200 955.50 955.3
isomers
EC50 EC50 EC50 EC50 ECSO Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
1078 455.503 1819.04 B4B.40 848.3
Mixture of
1079 156.944 831.55 911.40 911.4
isomers q
Mixture of
1080 0.061 > 200 10.5 0.009 14 953.50 953.4
isomers
Mixture of
1081 0.010 87.6 4.6 0.015 2.1 981.50 981.2
Mixture of
1082 33.936 30.1 868.40 868.1
isomers
Mixture of
1083 1.000 > 100 > 100 1.000 > 100 806.40 806.4
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+H]* Designation
Mixture of
1084 1.000 > 100 97 0.188 97.3 882.40 882.5
Mixture of
1085 1.000 > 100 > 100 0.159 > 100 806.40 806.7
Mixture of
1086 0.074 > 100 45.5 0.010 22.24 882.40 882.7
Mixture of
1087 0.478 > 100 > 100 0.430 > 100 841.40 841.3
> ° N Mixture of
1088 0.417 > 100 94 0 237 > 100 916.40 916.4
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
1100 0.010 19.2 1.56 0.006 3.51 956.50 956.5
isomers
1101 0.002 1.26 7.719 0.001 50.2 881.40 882.4 Single isomer
1102 0.002 1.95 19.68 0.002 > 100 873.40 874.4 Single isomer
1103 0.007 17.1 1.542 0.006 23.4 899.40 900.4 Single isomer
1104 0.004 2.26 0.588 0.003 23.7 899.40 900.4 Single isomer
1105 0.003 2.97 3.046 0.005 17 855.40 856.4 Single isomer
ECS0 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+H]* Designation
1106 0.002 1.95 0.637 0.001 31 855.40 856.4 Single isomer
Mixture of
1107 0.005 1.95 1.591 0.001 18.3 923.50 924.5
isomers
Mixture of
1108 0.003 1.95 1.521 0.001 19.3 923.50 924.5
1109 0.020 43 0.3 0.001 52 899.40 900.3 Single isomer
Mixture of
1110 0.001 1.1 0.9 0.001 9.4 909.40 910.4
isomers
EC50 EC50 EC50 EC50 EC50 Obs .M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
1111 0.002 0.8 1 0.001 9.7 963.50 964.7
/"" >-0 H l F Mixture of
1112 0.003 4.108 0.001 8.5 883.40 884.4
isomers
J A
Mixture of
1113 0.003 11 1.2 0.001 12 941.40 942.8
isomers F
1114 0.002 1.2 0.4 0.001 76 859.40 860.4 Single isomer
1115 0.064 92.5 64 0.004 64 863.40 863.7 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc .S.
1a 1aY93H 1aL31V 1b 2b [M+H]* Designation
J °r° A
1116 0.010 0.8 2.4 0.020 20.9 863.40 863.7 Single isomer
1117 0.023 19.7 4.8 0.001 7 891.40 892.7 Single isomer
1118 0.003 0.5 2 0.001 23 891.40 892.7 Single isomer
1119 0.011 8.8 10 0.001 31 877.40 877.7 Single isomer
1120 0.003 0.5 4.8 0.001 21 877.40 877.7 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs .M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
1126 0.018 46.74 26.5 0.001 142 873.50 874.5
isomers
1127 0.003 12 12 0.001 49 881.40 882.5 Single isomer
Mixture of
1128 0.002 0.8 0 0.001 9 917.40 918.5
1129 0.003 3 2 0.002 14 869.40 870.3 Single isomer
1130 0.001 2 1 0.001 37 869.40 870.3 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+H]* Designation
1131 0.003 29 2 0.002 5 847.40 848.4 Single isomer
1132 0.003 3 1 0.002 3 847.40 848.4 Single isomer
1133 0.004 27 1 0.002 16 883.40 884.4 Single isomer
1134 0.001 1 1 0.001 17 883.40 884.4 Single isomer
1135 0.003 17 0 0.001 26 917.40 918.4 Single isomer
1136 0.002 1 0 0.001 11 917.40 918.4 Single isomer
1137 0.002 19.1 4.23 0.001 40.6 857.40 858.5 Single isomer
9X°
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
1138 0.003 18.7 0.26 0.004 2 958.50 959.2
Mixture of
1139 0.010 1 1.1 0.020 1.1 957.50 958.5
Mixture of
1140 1.100 2 1.1 0.020 1.1 921.50 922 5
1141 o NH < °γΝ 0.010 2 13 0.020 12.4 869.40 870.5 Single isomer t
1142 1.100 11.2 0.020 45.1 869.40 870.5 Single isomer
9 0
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
1143 0.010 > 100 7.7 0.020 10.2 963.50 964 5 Single isomer
1144 0.010 2 1.1 0.020 26.5 963.50 964.5 Single isomer
Mixture of
1145 0.019 5.764 2.511 0.054 457.395 897.40 898.4
Mixture of
1146 0.019 4.6 2.4 0.070 318.37 895.40 8964
isomers
Mixture of
1147 0.019 1.9555 1.9555 0.002 1.9555 1033.50 1034.4
isomers
Mixture of
1148 0.019 1.9555 5.39248 0.002 67.2647 981.40 9824
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
1155 0.030 16.31 3.4 0.023 11.75 959.40 960.4
isomers
Mixture of
1156 0.016 26.75 2.4 0.011 4.4 997.50 998.4
Mixture of
1157 0.007 0.27 6.6 0.004 32 987.50 988.5
isomers
Mixture of
1158 0.032 15.22 1 0.021 4.92 989.50 990.5
Mixture of
1159 0.005 0.2 4.6 0.005 20.1 1012.40 1013.3
isomers
Cmpd # Structure EC50 EC50 EC50 EC50 EC50 Obs M.S.
Calc.M.S. Isomer 1a 1aY93H 1aL31V 1b 2b [ +Hf Designation
Mixture of
1160 922.301 724.584 897.40 898.3
Mixture of
1161 _ "° 8.766 5099 973.40 974.3
isomers
Mixture of
1162 0.006 9.111 9.111 0.005 14.818 1087.50 1088.3
isomers
1163 0.004 58.732 9.111 0.004 9.111 957.50 958.5 Single isomer
1164 0.006 9.111 9.111 0.005 9.111 957.50 958.5 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [ +H]* Designation
Mixture of
1165 0.005 0.13 889.65 0.003 99.34 1023.50 1024.4
Mixture of
1166 0.005 0.09 9.11 0.003 25.34 1069.50 1070.4
_)-o isomers
1167 0.015 0.2 685.23 0.002 > 5000 969.40 970.3 Single isomer
1168 0.005 0.19 0.9 0.006 8.6 1087.50 1088.4 Single isomer
1169 0.049 0.64 9 0.002 49.8 993.40 994.5 Single isomer
EC50 EC50 EC50 EC50 EC50 ObS M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [ +Hf Designation
1170 0.002 0.2 2.3 0.003 25.3 993.40 994.5 Single isomer
1171 0.015 0.67 5.5 0.003 51 1069.50 1070.4 Single isomer
1172 0.025 0.76 10.7 0.005 37.6 1087.50 1088.4 Single isomer
1173 0.002 0.19 0.6 0.003 11.5 1087.50 1088.4 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+H]+ Designation
1174 0.016 0.38 6.5 0.004 61 1029.40 1030.3 Single isomer
1175 0.022 0.25 6.9 0.002 36.5 1047.40 1048.3 Single isomer
Mixture of
1176 0.004 0.2 21.3 0.002 90 901.40 902.4
Mixture of
1177 0.065 0.3 23.8 > 200 935.40 936.3
isomers
1178 0.007 0.4 5.1 0.002 28.8 935.40 936.1 Single isomer
ECSO EC50 ECSO EC50 EC50 Obs .M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
1179 0.002 0.1 4 0.002 27.9 935.40 936.1 Single isomer
1180 0.002 5.4 0.004 39.9 1012.40 1013.0 Single isomer
1181 0.003 2.71 0.005 20.8 1012.40 1013.9 Single isomer
1182 0.003 1.245 0.004 27.4 1042.40 1043.0 Single isomer
Mixture of
1183 0.250 >100 >100 973.40 974.3
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+H]* Designation
Mixture of
1184 0.011 0.2 28.29 0.009 53 899.40 900.5
isomers
1185 0.045 0.59 39 0.005 61.2 901.40 902.4 Single isomer
1186 0.008 0.19 22 0.005 61 901.40 902.4 Single isomer
1187 0.164 3.5 16 0.043 > 100 1252.50 1253.8 Single isomer
1188 0.014 0.2 49 0.005 41 887.40 888.4 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc .S.
1a 1aY93H 1aL31V 1b 2b [M+H]* Designation
1189 0.006 0.2 28 0.004 82 887.40 888.4 Single isomer
1190 0.220 > 100 71 0.006 74.4 899.40 900.4 Single isomer
1191 0.008 0.3 17.9 0.005 56 899.40 900.4 Single isomer
1192 0.007 2.365 0.005 48 1011.40 1012.4 Single isomer
1193 0.037 0.9 11.514 0.004 60 1011.40 1012.4 Single isomer
1194 533.000 943 803.30 804.4 Mixture of isomers
% r
EC50 EC50 EC50 ECSO EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
1195 0.135 1 68.9 0.010 > 100 917.40 918.4
Mixture of
1196 0.150 8.2 95 >100 883.40 884.4
Mixture of
1197 608.000 >1000 867.30 868.3
isomers p
Mixture of
1198 0.058 0.75 4.7 95 981.40 982.2
Mixture of
1199 0.130 0.92 2.9 85 961.43 962.4
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
1210 > 0.2 46.31 24 0.098 374 696.30 698.2 Single isomer
Mixture of
1211 1.100 2 1.1 0.020 5 893.40 894.4
Mixture of
1212 1.100 2 1.1 0.020 44.2 905.40 906.5
1213 1.100 2 1.1 0.020 3.1 893.40 894.4 Single isomer
1214 0.010 134 123.5 0.760 982.1 697.30 697.2 Single isomer
1215 0.010 8.48 1.1 0 020 82.3 853.40 854.3 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
1222 4.725 352.297 50.7592 0.301 > 1000 877.40 878.0 Single isomer
1223 502.760 1000 1000 > 100 1000 691.40 692.3 Single isomer
1224 180.440 1000 1000 15.920 510 1002.50 1004.0 Single isomer
Mixture of
1225 645.63 1000 > 100 1000 835.30 836.3
Mixture of
1226 129.300 > 5000 635.20 636.2
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
1227 > 2 > 5000 3844 4709 991.40 992.0 Single isomer
1228 > 2 > 5000 4660 > 5000 1059.40 1061.0 Single isomer
Mixture of
1229 8.600 1254 792.30 793.2
isomers
Mixture of
1230 > 2 1827 404 948.6 951.30 952.1
isomers
1231 501.958 5099 702.30 703.3 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc .S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
1232 0.025 > 10 9.11 0.020 9.11 1173.60 1174.5 Single isomer
Mixture of
1233 0.059 29.393 9.111 0.029 9.111 1173.60 1175.4
isomers
Mixture of
1234 > 2 381.08 156.186 0.075 211.543 824.40 825.3
isomers
Mixture of
1235 0.270 155.87 65.774 0.025 81.879 824.40 825.4
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+H]+ Designation
1236 0.037 49 11 0.019 28 1058.50 1060.3 Single isomer
1237 Cry rsH° H 0.203 13 83 0.008 219 897.40 898.3 Single isomer
1238 > 1 > 50 76.7 0.036 987.75 748.40 749.3 Single isomer
1239 0.112 > 50 > 5000 0 004 164.09 766.40 767.2 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
1240 0.606 > 50 > 5000 0.039 745.66 762.40 763.2 Single isomer
1241 0.071 > 50 > 5000 0.014 325.09 763.30 764.2 Single isomer
Mixture of
1242 0.510 > 50 9.11 0.024 1248.86 788.30 789.2
Mixture of
1243 0.320 > 50 9.11 0.021 1564.83 85940 860.2
1244 H2NV O N 0.366 > 50 > 5000 0.025 1457.91 747.40 748.3 Single isomer
° HN-V
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+H]* Designation
1245 0.266 > 50 > 5000 0.019 614.1 777.40 778.3 Single isomer
LN iiYtvry N
Mixture of
1246 0.023 > 50 9.11 0.004 103.13 853.40 854.3
isomers
Mixture of
1247 > 2 > 50 612.51 0.012 217.17 853.40 854.4
1248 > 1 > 50 9.11 0.053 281.25 754.30 755.3 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+H]* Designation
1249 0.045 > 200 43.2 0.004 57.81 1078.50 1060.4 Single isomer
1250 2.633 > 20000 > 200 0.192 > 200 937.40 938.5 Single isomer
1251 0.037 > 200 10.1 0.004 > 200 1010.50 1012.4 Single isomer
1252 0.019 > 200 17.8 0.004 > 200 1010.50 1011.5 Single isomer
- H
1253 3.974 359.7 > 200 > 2 > 200 758.40 759.3 Single isomer
EC50 EC50 EC50 EC50 EC50 Obs .M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
1254 4.765 1234.4 > 200 0.177 > 200 978.50 979.4 Single isomer
1255 0.043 46.8 200 0.022 > 200 910.50 911.2 Single isomer
1256 0.100 > 200 > 200 0.014 > 200 910.50 911.1 Single isomer
1257 0.020 > 200 200 0.092 > 200 1020.50 1023.0 Single isomer
1258 0.041 > 200 56.12 0.028 12.5 1093.50 1094.9 Single isomer
1259 0.064 > 200 20 0.005 12.7 1025.50 1026.0 Single isomer
EC50 EC50 EC50 EC50 EC50 ObS M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+H]* Designation
1271 0.094 50.63 25.7 0.009 58.827 957.50 958.5 Single isomer
1272 0.005 1.826 0.004 5.138 993.40 994.4 Single isomer
1273 0.002 3 0.789 0.003 27.939 847.40 849.3 Single isomer
1274 0.056 72 7.5 0.013 29.2 1104.60 1105.4 Single isomer
1275 0.044 99 0.9 0.013 15.1 1024.50 1026.4 Single isomer
EC50 EC50 EC50 EC50 EC50 ObS M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
1300 f° Ho > 688.40 688.8
isomers
Mixture of
1301 0.080 0.14 0.19 11 983.40 983.6
isomers
Mixture of
1302 0.060 1 0.02 0.3 965.40 965.6
Ογ Η isomers
Mixture of
1303 0.060 1.8 2.3 1.3 887.40 887.8
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc .S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
1304 0.020 0.18 0.7 2.7 905.40 905.7
Mixture of
1305 0.020 1 0.2 1.7 887.40 887.8
Mixture of
1306 0.010 0.4 2.1 13 863.40 863.8
isomers
Mixture of
1307 0.010 7 1.6 11 845.40 845.8
isomers
Mixture of
1308 0.004 20 1.11 0.003 855.95 857.0
isomers
\
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
■J- °Γ° ° „
Mixture of
1309 0.002 4.03 0.1997 0.001 24.3 936.012 937.0
isomers
Mixture of
1310 0.001 0.46 0.019 0.001 12.3 936.012 937.0
Mixture of
1311 0.001 0.2 0.6 0.001 9 918.022 919.0
isomers
Mixture of
1312 0.004 5.4 0.7 0.001 42 918.022 919.0
Mixture of
1313 0.016 7.6 3.8 0.001 10 910.042 911.0
isomers
ECSO EC50 EC50 EC50 EC50 Obs .M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
1314 0.001 0.3 1.1 0.001 31 910.042 911.0
Mixture of
1315 0.009 7.7 3 0.001 6 924.069 925.1
Mixture of
1316 0.002 0.3 1 0.001 30 924.069 925.1
Mixture of
1317 0.020 < 100 9.94 0.002 100.2 932.218 933.2
Mixture of
1318 0.010 59.96 3.6 0.020 19.9 924.069 925.1
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
1319 0.010 2 1.1 0.020 26.1 924.069 925.1
isomers
Mixture of
1320 0.018 105 10 12 890.136 891.1
Mixture of
1321 0.014 208 10 72.7 892.152 893.2
Mixture of
1322 0.019 1 3.6 0.003 89 1000.48 1001.5
isomers p
Mixture of
1323 0.005 0.47 9.6 0.004 9.7 970.457 971.5
isomers
CI
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+H]* Designation
Mixture of
1324 0.009 0.43 10.2 0.007 26.3 1016.1 1017.1
isomers
Mixture of
1325 0.003 0.19 1.1 0.003 8.1 1021.12 1022.1
J °r° A
Mixture of
1326 0.006 0.38 9.11 0.004 25.14 1019.15 1020.2
isomers
Mixture of
1327 0.007 2.51 9.11 0.002 17.64 1042.14 1043.1
isomers
Mixture of
1328 0.007 0.36 88.43 0.006 180.29 1046.13 1047.1
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure CalcM.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
1335 0.017 > 100 18.1 0.008 1.248 976.077 977.1
isomers
„ NH
Mixture of
1336 0.032 15.67 5.4 0.008 2.38 1051.15 1052.2
1337 0.010 0.5 19 0.007 167 859.913 860.9 Single isomer
1338 0.009 53 1.7 0.008 26 821.941 822.9 Single isomer
1339 0.040 52.8 4.88 0.017 110 843.951 845.0 Single isomer
CN HN Y
EC50 Obs .M.S. Isomer
Cmpd # Structure EC50 EC50 EC50 EC50 CalC.M.S.
1a 1aY93H 1aL31V 1b 2b [M+H]+ Designation
Mixture of
1346 > 2 109.9 48.617 0.126 110 851.971 853.0
isomers
Mixture of
1347 > 1 > 100 > 100 0.238 > 100 851.971 853.0
isomers
1348 0.008 > 100 6.1 0.010 55.2 821.941 822.9 Single isomer
Mixture of
1349 -°-£> ° HOTN 0.691 > 100 90.1 0.107 > 100 923.05 924.1
isomers p r-VirNH Q HJNL-O "f
1350 0.051 > 100 > 100 0.015 > 100 805.898 806.9 Single isomer
1351 0.018 78.9 4.1 0.006 54.2 869.986 871.0 Single isomer
EC50 EC50 EC50 EC50 ECSO Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
1352 0.017 > 100 21.3 0.023 25.9 845.926 846.9 Single isomer
1353 0.004 13.96 1.16 0.006 53.67 926.17 927.2 Mixture of isomers
1354 0.051 > 200 31.6 0.010 > 200 919.997 921.0 Single isomer
1355 > 2 > 200 200 > 2 > 200 805.892 806.9 Single isomer
1356 > 2 > 200 > 200 > 2 > 200 769.911 770.9 Single isomer
Mixture of
1357 0.178 12.01 71.7 0.004 > 200 868.005 869.0
Mixture of
1358 0.20 1.14 1.54 0.16 80.00 922.04 923.30
isomers
EC50 EC50 EC50 EC50 EC50 Obs M.S. Isomer
Cmpd # Structure Calc.M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
1366 0.00981 0.567 1.38 0.00765 32.2 966.16 967.0 Single isomer
1367 0.00734 23.7 0.397 0.00568 4.85 984.03 985.0 Single isomer
1368 0.00574 15.3 0.297 0.0105 10.8 930.06 931.0 Single isomer
1369 0.0449 0.0706 7.05 0.0124 63.3 1018.16 1019.0 Single isomer
1370 0.133 49.2 0.52 0.0896 64.3 1054.15 1055.0 Single isomer o-
Mixture of
1371 0.0282 16.4 0.358 0.00711 7 1038.24 1039.0
isomers
EC50 EC50 EC50 EC50 EC50 Obs .M.S. Isomer
Cmpd # Structure Calc M.S.
1a 1aY93H 1aL31V 1b 2b [M+Hf Designation
Mixture of
1378 0.00577 2.87 0.0649 0.00497 3.3 946.06 947.0
Mixture of
1379 0.00754 25.3 0.211 0.00462 8.25 946.06 947.0
isomers
Mixture of
1380 0.00556 0.798 0.0467 0.00628 0.874 948.05 949.0
isomers
Mixture of
1381 0.0283 44.6 0.519 0.00798 11 948.05 949.0
isomers
Mixture of
1382 0.00634 2.27 0.106 0.00699 0.147 930.06 931.0
Mixture of
1383 0.0205 56.4 0.419 0.0139 61.8 930.06 931.0
isomers o-
The present invention is not to be limited by the specific embodiments disclosed in the examples that are intended as illustrations of a few aspects of the invention and any embodiments that are functionally equivalent are within the scope of this invention. Indeed, various modifications of the invention in addition to those shown and described herein will become apparent to those skilled in the art and are intended to fall within the scope of the appended claims.
A number of references have been cited herein, the entire disclosures of which are incorporated herein by reference.