US11053243B2 - Inhibitors of hepatitis C virus replication - Google Patents
Inhibitors of hepatitis C virus replication Download PDFInfo
- Publication number
- US11053243B2 US11053243B2 US16/829,878 US202016829878A US11053243B2 US 11053243 B2 US11053243 B2 US 11053243B2 US 202016829878 A US202016829878 A US 202016829878A US 11053243 B2 US11053243 B2 US 11053243B2
- Authority
- US
- United States
- Prior art keywords
- group
- alkyl
- independently chosen
- mmol
- hydrogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 *.B.CC.CC.CC.C[2H]CC.C[2H]CC Chemical compound *.B.CC.CC.CC.C[2H]CC.C[2H]CC 0.000 description 132
- UORVGPXVDQYIDP-UHFFFAOYSA-N B Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 14
- FLTXVDVFNYXZFL-UHFFFAOYSA-N C1=CC2=C(C=C1)[Y]C=C2 Chemical compound C1=CC2=C(C=C1)[Y]C=C2 FLTXVDVFNYXZFL-UHFFFAOYSA-N 0.000 description 6
- PMEIKISOJWPZTB-UHFFFAOYSA-N CC(C)C1=CN=C(C(C)C)N1C.CC(C)C1=CN=C(C(C)C)O1.CC(C)C1=CN=C(C(C)C)S1.CC(C)C1=NN=C(C(C)C)O1.[H]N1C(C(C)C)=CN=C1C(C)C.[H]N1C(C(C)C)=NN=C1C(C)C Chemical compound CC(C)C1=CN=C(C(C)C)N1C.CC(C)C1=CN=C(C(C)C)O1.CC(C)C1=CN=C(C(C)C)S1.CC(C)C1=NN=C(C(C)C)O1.[H]N1C(C(C)C)=CN=C1C(C)C.[H]N1C(C(C)C)=NN=C1C(C)C PMEIKISOJWPZTB-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N C1=CC=CC=C1 Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 2
- BMUOLAFYQZTBAY-IMSUZSGZSA-N CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=CC1.CC(C)(C)OC(=O)N1CCC[C@H]1C=O.[H]C(=O)C([H])=O Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=CC1.CC(C)(C)OC(=O)N1CCC[C@H]1C=O.[H]C(=O)C([H])=O BMUOLAFYQZTBAY-IMSUZSGZSA-N 0.000 description 2
- CWSKOUALMLKXCD-UHFFFAOYSA-N CC(C)C1=CN=C(C(C)C)O1.[H]N1C(C(C)C)=CN=C1C(C)C Chemical compound CC(C)C1=CN=C(C(C)C)O1.[H]N1C(C(C)C)=CN=C1C(C)C CWSKOUALMLKXCD-UHFFFAOYSA-N 0.000 description 2
- UTFKXZGSBDLZRM-RKKUYNIKSA-N CC.CC.CC.CC(=O)N1CCCC1C1=NC=C(C2=CC=C(C3=CC4=C(C=CC(C5=CN=C(C6CCCN6C(C)=O)N5)=C4)[Y]3)C=C2)N1 Chemical compound CC.CC.CC.CC(=O)N1CCCC1C1=NC=C(C2=CC=C(C3=CC4=C(C=CC(C5=CN=C(C6CCCN6C(C)=O)N5)=C4)[Y]3)C=C2)N1 UTFKXZGSBDLZRM-RKKUYNIKSA-N 0.000 description 2
- MNYXEWSCEQOTLK-UHFFFAOYSA-N COC(=O)NC(C(C)C)C(C)C Chemical compound COC(=O)NC(C(C)C)C(C)C MNYXEWSCEQOTLK-UHFFFAOYSA-N 0.000 description 2
- WIPLWFMRCCXWSS-VEVNFNBLSA-N COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC3=C(C=C2)C2=C(C#N)C4=C(C=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=C4)N2CO3)N1)C(C)C Chemical compound COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC3=C(C=C2)C2=C(C#N)C4=C(C=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=C4)N2CO3)N1)C(C)C WIPLWFMRCCXWSS-VEVNFNBLSA-N 0.000 description 2
- YAQMGTJFTAQUPJ-VOIOCNMVSA-N COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3OC(C4=CC(F)=C(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)C=C4)=CC3=C2)N1)C(C)C Chemical compound COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3OC(C4=CC(F)=C(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)C=C4)=CC3=C2)N1)C(C)C YAQMGTJFTAQUPJ-VOIOCNMVSA-N 0.000 description 2
- RXRPSYRAOINUIJ-HKBQPEDESA-N O=C(NC1=CC=C(C2=CC3=CC(C4=CC=C5OCCOC5=C4)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(NC1=CC=C(C2=CC3=CC(C4=CC=C5OCCOC5=C4)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 RXRPSYRAOINUIJ-HKBQPEDESA-N 0.000 description 2
- CLMPERJUIIUMPX-XIFFEERXSA-N O=C(NC1=CC=C(C2=CC3=CC(C4=CC=CC5=CC=CN=C54)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(NC1=CC=C(C2=CC3=CC(C4=CC=CC5=CC=CN=C54)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 CLMPERJUIIUMPX-XIFFEERXSA-N 0.000 description 2
- TUXYYCPOAZENOT-NDEPHWFRSA-N O=C(NC1=CC=C(C2=CC3=CC(C4=CC=CO4)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(NC1=CC=C(C2=CC3=CC(C4=CC=CO4)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 TUXYYCPOAZENOT-NDEPHWFRSA-N 0.000 description 2
- IWVOMLNCHXCGPQ-ZPGRZCPFSA-N O=C(NC1=CC=C(C2=CC3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(NC1=CC=C(C2=CC3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 IWVOMLNCHXCGPQ-ZPGRZCPFSA-N 0.000 description 2
- MEUVAJXZPZEGQD-UHFFFAOYSA-N *.B.C Chemical compound *.B.C MEUVAJXZPZEGQD-UHFFFAOYSA-N 0.000 description 1
- KCKAAHOAQRGRKR-LKJYFVHXSA-N B=NS.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC(Br)=C(Br)N1.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=CN1 Chemical compound B=NS.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC(Br)=C(Br)N1.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=CN1 KCKAAHOAQRGRKR-LKJYFVHXSA-N 0.000 description 1
- KCJIDCMHONFGRC-HNABKQRDSA-N B=NS.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC(Br)=CC1.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC(C2=CC=C3OC(C4=CC=C([N+](=O)[O-])C=C4)=CC3=C2)=CC1.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC(C2=CC=C3OC(C4=CC=C([N+](=O)[O-])C=C4)=CC3=C2)=CC1.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=CC1.CC(C)(C)OC(=O)N1CCC[C@H]1C=O.CC1(C)OB(C2=CC=C3OC(C4=CC=C([N+](=O)[O-])C=C4)=CC3=C2)OC1(C)C.N.O.[H]C(=O)C([H])=O Chemical compound B=NS.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC(Br)=CC1.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC(C2=CC=C3OC(C4=CC=C([N+](=O)[O-])C=C4)=CC3=C2)=CC1.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC(C2=CC=C3OC(C4=CC=C([N+](=O)[O-])C=C4)=CC3=C2)=CC1.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=CC1.CC(C)(C)OC(=O)N1CCC[C@H]1C=O.CC1(C)OB(C2=CC=C3OC(C4=CC=C([N+](=O)[O-])C=C4)=CC3=C2)OC1(C)C.N.O.[H]C(=O)C([H])=O KCJIDCMHONFGRC-HNABKQRDSA-N 0.000 description 1
- XIWHVJLNYLINJF-XCPONSOVSA-N B=NS.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC(Br)=CC1.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=CC1 Chemical compound B=NS.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC(Br)=CC1.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=CC1 XIWHVJLNYLINJF-XCPONSOVSA-N 0.000 description 1
- VVRXHBNDGLTTLS-GOHZVWLPSA-N B=NS.CC(C)(C)OC(=O)N[C@@H](C(=O)C1CCCN1C(=O)NC1=CC=C2CC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1.C[C@@H](C(=O)C1CCCN1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](C)C4=CC=CC=C4)C=C3)=C(Br)C2=C1)C1=CC=CC=C1 Chemical compound B=NS.CC(C)(C)OC(=O)N[C@@H](C(=O)C1CCCN1C(=O)NC1=CC=C2CC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1.C[C@@H](C(=O)C1CCCN1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](C)C4=CC=CC=C4)C=C3)=C(Br)C2=C1)C1=CC=CC=C1 VVRXHBNDGLTTLS-GOHZVWLPSA-N 0.000 description 1
- QCGOTQJDFLQNKZ-UHFFFAOYSA-N B=NS.NC1=CC=C(Br)N=C1Br.NC1=CC=CN=C1 Chemical compound B=NS.NC1=CC=C(Br)N=C1Br.NC1=CC=CN=C1 QCGOTQJDFLQNKZ-UHFFFAOYSA-N 0.000 description 1
- HMPDIRMQCBWSIW-FWOHUZISSA-N B=NS.O=C(CC1=CC2=C(C=C1)NC(C1=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C1)=C2)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(CC1=CC2=C(C=C1)NC(C1=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C1)=C2Br)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound B=NS.O=C(CC1=CC2=C(C=C1)NC(C1=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C1)=C2)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(CC1=CC2=C(C=C1)NC(C1=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C1)=C2Br)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 HMPDIRMQCBWSIW-FWOHUZISSA-N 0.000 description 1
- AERDUUFAGYHXGH-UHFFFAOYSA-N BrC1=CC=C(C2=CC3=CC(Br)=CC=C3O2)C=C1.CC1(C)OB(B2OC(C)(C)C(C)(C)O2)OC1(C)C.CC1(C)OB(C2=CC=C(C3=CC4=CC(B5OC(C)(C)C(C)(C)O5)=CC=C4O3)C=C2)OC1(C)C Chemical compound BrC1=CC=C(C2=CC3=CC(Br)=CC=C3O2)C=C1.CC1(C)OB(B2OC(C)(C)C(C)(C)O2)OC1(C)C.CC1(C)OB(C2=CC=C(C3=CC4=CC(B5OC(C)(C)C(C)(C)O5)=CC=C4O3)C=C2)OC1(C)C AERDUUFAGYHXGH-UHFFFAOYSA-N 0.000 description 1
- QJAPPZFQUVPRDL-UHFFFAOYSA-N BrC1=CC=C(C2=CC3=CC(Br)=CC=C3O2)C=C1.CCOC(=O)C(Br)C1=CC=C(Br)C=C1.[H]C(=O)C1=CC(Br)=CC=C1O Chemical compound BrC1=CC=C(C2=CC3=CC(Br)=CC=C3O2)C=C1.CCOC(=O)C(Br)C1=CC=C(Br)C=C1.[H]C(=O)C1=CC(Br)=CC=C1O QJAPPZFQUVPRDL-UHFFFAOYSA-N 0.000 description 1
- QPRMXTXSBHMGLB-UHFFFAOYSA-N BrC1=CC=C(C2=NC3=CC(Br)=CC=C3O2)C=C1.CC1(C)OB(B2OC(C)(C)C(C)(C)O2)OC1(C)C.CC1(C)OB(C2=CC=C(C3=NC4=CC(B5OC(C)(C)C(C)(C)O5)=CC=C4O3)C=C2)OC1(C)C Chemical compound BrC1=CC=C(C2=NC3=CC(Br)=CC=C3O2)C=C1.CC1(C)OB(B2OC(C)(C)C(C)(C)O2)OC1(C)C.CC1(C)OB(C2=CC=C(C3=NC4=CC(B5OC(C)(C)C(C)(C)O5)=CC=C4O3)C=C2)OC1(C)C QPRMXTXSBHMGLB-UHFFFAOYSA-N 0.000 description 1
- MEZUZWLCRMKMIN-UHFFFAOYSA-N BrC1=CC=C(C2=NC3=CC(Br)=CC=C3O2)C=C1.NC1=CC(Br)=CC=C1O.O=C(O)C1=CC=C(Br)C=C1 Chemical compound BrC1=CC=C(C2=NC3=CC(Br)=CC=C3O2)C=C1.NC1=CC(Br)=CC=C1O.O=C(O)C1=CC=C(Br)C=C1 MEZUZWLCRMKMIN-UHFFFAOYSA-N 0.000 description 1
- IEONUJMLRPLKHY-UHFFFAOYSA-N BrC1=CC=C2OC=CC2=C1.OB(O)C1=CC2=CC(Br)=CC=C2O1 Chemical compound BrC1=CC=C2OC=CC2=C1.OB(O)C1=CC2=CC(Br)=CC=C2O1 IEONUJMLRPLKHY-UHFFFAOYSA-N 0.000 description 1
- YGAALZGRUQGHGB-UHFFFAOYSA-N BrCBr.FC1=C2C3=C(C=C(Br)C=C3)OCN2C2=CC=C(Br)C=C21.OC1=C(C2=C(F)C3=CC(Br)=CC=C3N2)C=CC(Br)=C1 Chemical compound BrCBr.FC1=C2C3=C(C=C(Br)C=C3)OCN2C2=CC=C(Br)C=C21.OC1=C(C2=C(F)C3=CC(Br)=CC=C3N2)C=CC(Br)=C1 YGAALZGRUQGHGB-UHFFFAOYSA-N 0.000 description 1
- XDORUQXPFBRJJA-UHFFFAOYSA-L Br[Cu]Br.CC(C)(C)[N+](=O)[O-].NC1=NC2=C(C=CC([N+](=O)[O-])=C2)O1.O=[N+]([O-])C1=CC2=C(C=C1)OC(Br)=N2 Chemical compound Br[Cu]Br.CC(C)(C)[N+](=O)[O-].NC1=NC2=C(C=CC([N+](=O)[O-])=C2)O1.O=[N+]([O-])C1=CC2=C(C=C1)OC(Br)=N2 XDORUQXPFBRJJA-UHFFFAOYSA-L 0.000 description 1
- YDICYGNJTVXHNZ-UHFFFAOYSA-K Br[In](Br)Br.CC(=O)CC1=CC=C(C#CC2=CC=C([N+](=O)[O-])C=C2N)C=C1.CC(=O)NC1=CC=C(/C2=C/C3=CC=C([N+](=O)[O-])C=C3N2)C=C1 Chemical compound Br[In](Br)Br.CC(=O)CC1=CC=C(C#CC2=CC=C([N+](=O)[O-])C=C2N)C=C1.CC(=O)NC1=CC=C(/C2=C/C3=CC=C([N+](=O)[O-])C=C3N2)C=C1 YDICYGNJTVXHNZ-UHFFFAOYSA-K 0.000 description 1
- QPTFSRLJKIACCB-UHFFFAOYSA-N Brc(cc1)ccc1-c1cc2cc(Br)ccc2[o]1 Chemical compound Brc(cc1)ccc1-c1cc2cc(Br)ccc2[o]1 QPTFSRLJKIACCB-UHFFFAOYSA-N 0.000 description 1
- AYOVPQORFBWFNO-UHFFFAOYSA-N Brc1ccc2[o]ccc2c1 Chemical compound Brc1ccc2[o]ccc2c1 AYOVPQORFBWFNO-UHFFFAOYSA-N 0.000 description 1
- WCHPZFFHNUCJNC-OVNKVOPASA-N C#CC1=CC=C(Br)C=C1.CON(C)C(=O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1.O=C(C#CC1=CC=C(Br)C=C1)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1 Chemical compound C#CC1=CC=C(Br)C=C1.CON(C)C(=O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1.O=C(C#CC1=CC=C(Br)C=C1)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1 WCHPZFFHNUCJNC-OVNKVOPASA-N 0.000 description 1
- YSPQVGOVEQYYAL-UHFFFAOYSA-N C#CC1=CC=C(CC(C)=O)C=C1.C#CC1=CC=C(N)C=C1.CC(=O)Cl Chemical compound C#CC1=CC=C(CC(C)=O)C=C1.C#CC1=CC=C(N)C=C1.CC(=O)Cl YSPQVGOVEQYYAL-UHFFFAOYSA-N 0.000 description 1
- XYYWZNGJHLPQAR-UHFFFAOYSA-N C#CC1=CC=C(CC(C)=O)C=C1.CC(=O)CC1=CC=C(C#CC2=CC=C([N+](=O)[O-])C=C2N)C=C1.NC1=CC([N+](=O)[O-])=CC=C1I Chemical compound C#CC1=CC=C(CC(C)=O)C=C1.CC(=O)CC1=CC=C(C#CC2=CC=C([N+](=O)[O-])C=C2N)C=C1.NC1=CC([N+](=O)[O-])=CC=C1I XYYWZNGJHLPQAR-UHFFFAOYSA-N 0.000 description 1
- LOSIIJAASZAMIX-UHFFFAOYSA-N C#CC1=CC=C(N)C=C1.C#CC1=CC=C(NC(C)=O)C=C1.CCN(CC)CC Chemical compound C#CC1=CC=C(N)C=C1.C#CC1=CC=C(NC(C)=O)C=C1.CCN(CC)CC LOSIIJAASZAMIX-UHFFFAOYSA-N 0.000 description 1
- JUUXNSQPWBSQLE-UHFFFAOYSA-N C#CC1=CC=C(NC(C)=O)C=C1.CC(=O)NC1=CC=C(C#CC2=CC([N+](=O)[O-])=CN=C2N)C=C1.NC1=NC=C([N+](=O)[O-])C=C1I Chemical compound C#CC1=CC=C(NC(C)=O)C=C1.CC(=O)NC1=CC=C(C#CC2=CC([N+](=O)[O-])=CN=C2N)C=C1.NC1=NC=C([N+](=O)[O-])C=C1I JUUXNSQPWBSQLE-UHFFFAOYSA-N 0.000 description 1
- LNQXDEMPFVSASY-UHFFFAOYSA-N C#CC1=CC=C(NC(C)=O)C=C1.CC(=O)NC1=CC=C(C#CC2=NC(Br)=CC=C2N)C=C1.NC1=CC=C(Br)N=C1Br Chemical compound C#CC1=CC=C(NC(C)=O)C=C1.CC(=O)NC1=CC=C(C#CC2=NC(Br)=CC=C2N)C=C1.NC1=CC=C(Br)N=C1Br LNQXDEMPFVSASY-UHFFFAOYSA-N 0.000 description 1
- UFDCWRUNXXVIJU-UHFFFAOYSA-N C#CC1=CC=CC(N)=C1.C#CC1=CC=CC(NC(C)=O)=C1.CC(=O)Cl.CCN(CC)CC.ClCCl Chemical compound C#CC1=CC=CC(N)=C1.C#CC1=CC=CC(NC(C)=O)=C1.CC(=O)Cl.CCN(CC)CC.ClCCl UFDCWRUNXXVIJU-UHFFFAOYSA-N 0.000 description 1
- LQJXIFOSNMDYPJ-UHFFFAOYSA-L C#CC1=CC=CC(NC(C)=O)=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.CC(=O)NC1=CC=CC(C#CC2=CC=C([N+](=O)[O-])C=C2N)=C1.Cl[Pd]Cl.NC1=CC([N+](=O)[O-])=CC=C1I Chemical compound C#CC1=CC=CC(NC(C)=O)=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.CC(=O)NC1=CC=CC(C#CC2=CC=C([N+](=O)[O-])C=C2N)=C1.Cl[Pd]Cl.NC1=CC([N+](=O)[O-])=CC=C1I LQJXIFOSNMDYPJ-UHFFFAOYSA-L 0.000 description 1
- ZWOIVVJASOYKRK-WWNYPKSESA-N C.C.CCCC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(C)C)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C.C.CCCC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(C)C)C=C3)=CC2=C1)C1=CC=CC=C1 ZWOIVVJASOYKRK-WWNYPKSESA-N 0.000 description 1
- ZIMLTTPFQHXUEK-FCJHUROFSA-N C.C.CCOC(=O)N[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C.C.CCOC(=O)N[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1 ZIMLTTPFQHXUEK-FCJHUROFSA-N 0.000 description 1
- GCDMHBGGMJCRFM-ONQOKFCBSA-N C.C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=C/C=C2\NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)[C@H](NC)C5=CC=CC=C5)N4)C=C3)=C\C2=C\1)C1=CC=CC=C1 Chemical compound C.C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=C/C=C2\NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)[C@H](NC)C5=CC=CC=C5)N4)C=C3)=C\C2=C\1)C1=CC=CC=C1 GCDMHBGGMJCRFM-ONQOKFCBSA-N 0.000 description 1
- GCDMHBGGMJCRFM-UQOUECHSSA-N C.C.CN[C@H](C(=O)N1CCC[C@H]1C(=O)CC1=C/C=C2\NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)[C@@H](NC)C5=CC=CC=C5)N4)C=C3)=C\C2=C\1)C1=CC=CC=C1 Chemical compound C.C.CN[C@H](C(=O)N1CCC[C@H]1C(=O)CC1=C/C=C2\NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)[C@@H](NC)C5=CC=CC=C5)N4)C=C3)=C\C2=C\1)C1=CC=CC=C1 GCDMHBGGMJCRFM-UQOUECHSSA-N 0.000 description 1
- LZLAGXNWNAKTLA-UHFFFAOYSA-K C.CC(=O)C1=C(O)C=C(Br)C=C1.CC(=O)OC1=CC=CC(Br)=C1.Cl[Al](Cl)Cl Chemical compound C.CC(=O)C1=C(O)C=C(Br)C=C1.CC(=O)OC1=CC=CC(Br)=C1.Cl[Al](Cl)Cl LZLAGXNWNAKTLA-UHFFFAOYSA-K 0.000 description 1
- VUXGUSWLFPKAHE-YLHZWKMJSA-N C.CC(=O)CC1=CC=C(C2=CC3=CC(C4=NN=C([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)O4)=CC=C3N2C(=O)OC(C)(C)C)C=C1.CC(=O)NC1=CC=C(Br)C=C1.CC(C)(C)OC(=O)N1C(B(O)O)=CC2=CC(C3=NN=C([C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)O3)=CC=C21 Chemical compound C.CC(=O)CC1=CC=C(C2=CC3=CC(C4=NN=C([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)O4)=CC=C3N2C(=O)OC(C)(C)C)C=C1.CC(=O)NC1=CC=C(Br)C=C1.CC(C)(C)OC(=O)N1C(B(O)O)=CC2=CC(C3=NN=C([C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)O3)=CC=C21 VUXGUSWLFPKAHE-YLHZWKMJSA-N 0.000 description 1
- PMLMYSQKJNDNRL-UHFFFAOYSA-N C.CC(=O)CC1=CC=C2NC3=C(CCCC4=CC(NC(C)=O)=CC=C43)C2=C1.CC(=O)NC1=CC=C(CN)C=C1.CC(=O)NC1=CC=C2C(=O)CCCCC2=C1 Chemical compound C.CC(=O)CC1=CC=C2NC3=C(CCCC4=CC(NC(C)=O)=CC=C43)C2=C1.CC(=O)NC1=CC=C(CN)C=C1.CC(=O)NC1=CC=C2C(=O)CCCCC2=C1 PMLMYSQKJNDNRL-UHFFFAOYSA-N 0.000 description 1
- CZZGGAXEOOMVTL-KBGXOJLMSA-N C.CC(=O)NC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)O)C4=CC=CC=C4)=CC=C3N2)C=C1 Chemical compound C.CC(=O)NC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)O)C4=CC=CC=C4)=CC=C3N2)C=C1 CZZGGAXEOOMVTL-KBGXOJLMSA-N 0.000 description 1
- BYDIPXXQFOZYQI-LQHJRYMCSA-N C.CC(=O)NCC#CC1=CC2=CC(CC(=O)[C@@H]3CCCN3C(=O)[C@H](C)C3=CC=CC=C3)=CC=C2N1.CC(=O)NCC#CC1=CC2=CC(N)=CC=C2N1 Chemical compound C.CC(=O)NCC#CC1=CC2=CC(CC(=O)[C@@H]3CCCN3C(=O)[C@H](C)C3=CC=CC=C3)=CC=C2N1.CC(=O)NCC#CC1=CC2=CC(N)=CC=C2N1 BYDIPXXQFOZYQI-LQHJRYMCSA-N 0.000 description 1
- XAYKDTWITADBNP-UHFFFAOYSA-M C.CC(=O)O[K].CC1=CC=C2C(=C1)C(F)=C1C3=C(C=C(C)C=C3)OCN21.FC1=C2C3=C(C=C(Br)C=C3)OCN2C2=CC=C(Br)C=C21 Chemical compound C.CC(=O)O[K].CC1=CC=C2C(=C1)C(F)=C1C3=C(C=C(C)C=C3)OCN21.FC1=C2C3=C(C=C(Br)C=C3)OCN2C2=CC=C(Br)C=C21 XAYKDTWITADBNP-UHFFFAOYSA-M 0.000 description 1
- XWRZPRNULOQSOT-XONKGLPHSA-N C.CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=C(F)C2=C1)C1=CC=CC=C1 Chemical compound C.CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=C(F)C2=C1)C1=CC=CC=C1 XWRZPRNULOQSOT-XONKGLPHSA-N 0.000 description 1
- KUAXFOUZWXTLSS-GAGCMDAMSA-N C.CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=CC(CC=O)=CC=C3N2)C=C1)C1=CC=CC=C1 Chemical compound C.CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=CC(CC=O)=CC=C3N2)C=C1)C1=CC=CC=C1 KUAXFOUZWXTLSS-GAGCMDAMSA-N 0.000 description 1
- CHGVZSOKCZCAGS-FTKQXCDWSA-M C.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3C(=C2)C(F)=C2C4=C(C=C(C5=CN=C([C@@H]6CCCN6C(=O)OC(C)(C)C)N5)C=C4)OCN32)N1.CC1=CC=C2C(=C1)C(F)=C1C3=C(C=C(C)C=C3)OCN21.O=COO[Na].[NaH] Chemical compound C.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3C(=C2)C(F)=C2C4=C(C=C(C5=CN=C([C@@H]6CCCN6C(=O)OC(C)(C)C)N5)C=C4)OCN32)N1.CC1=CC=C2C(=C1)C(F)=C1C3=C(C=C(C)C=C3)OCN21.O=COO[Na].[NaH] CHGVZSOKCZCAGS-FTKQXCDWSA-M 0.000 description 1
- KLJOEKMIHVNQKZ-UHFFFAOYSA-N C.CC(C)(C)OC(=O)NC1=CC2=C(C=C1)N(C(=O)OC(C)(C)C)C(C1=CC3=CC([N+](=O)[O-])=CC=C3N1)=C2.CC(C)(C)OC(=O)NC1=CC=C2CC(OBO)=CC2=C1.O=[N+]([O-])C1=CC=C2NC(Br)=CC2=C1 Chemical compound C.CC(C)(C)OC(=O)NC1=CC2=C(C=C1)N(C(=O)OC(C)(C)C)C(C1=CC3=CC([N+](=O)[O-])=CC=C3N1)=C2.CC(C)(C)OC(=O)NC1=CC=C2CC(OBO)=CC2=C1.O=[N+]([O-])C1=CC=C2NC(Br)=CC2=C1 KLJOEKMIHVNQKZ-UHFFFAOYSA-N 0.000 description 1
- KPOWVYITBUJHJK-VJJXCXNXSA-N C.CC(C)CCN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(C)CCC(C)C)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C.CC(C)CCN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(C)CCC(C)C)C=C3)=CC2=C1)C1=CC=CC=C1 KPOWVYITBUJHJK-VJJXCXNXSA-N 0.000 description 1
- XFYRASNZRIXTEN-CEOSRBJVSA-N C.CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(C)C)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C.CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(C)C)C=C3)=CC2=C1)C1=CC=CC=C1 XFYRASNZRIXTEN-CEOSRBJVSA-N 0.000 description 1
- VBKNRGFYFQXUHC-RCLAWPGESA-N C.CCOC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5)N4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C.CCOC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5)N4)C=C3)=CC2=C1)C1=CC=CC=C1 VBKNRGFYFQXUHC-RCLAWPGESA-N 0.000 description 1
- ZFHDXCIIIGDJNV-FTVHOMRLSA-N C.CCOC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)[C@@H](C5=CC=CC=C5)N(C)C)N4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C.CCOC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)[C@@H](C5=CC=CC=C5)N(C)C)N4)C=C3)=CC2=C1)C1=CC=CC=C1 ZFHDXCIIIGDJNV-FTVHOMRLSA-N 0.000 description 1
- LZFDLZKRBKEPQM-UHFFFAOYSA-N C.CCOC(=O)N1C(=O)CC2=CC([N+](=O)[O-])=CC=C21.CCOC(=O)OC1CC2=CC([N+](=O)[O-])=CC=C2N1C(=O)OCC.O=C1CC2=CC([N+](=O)[O-])=CC=C2N1 Chemical compound C.CCOC(=O)N1C(=O)CC2=CC([N+](=O)[O-])=CC=C21.CCOC(=O)OC1CC2=CC([N+](=O)[O-])=CC=C2N1C(=O)OCC.O=C1CC2=CC([N+](=O)[O-])=CC=C2N1 LZFDLZKRBKEPQM-UHFFFAOYSA-N 0.000 description 1
- ZXRAVANDEDCHAZ-KIKKKRBJSA-N C.CC[C@H](NC(=O)OC)C(=O)N1CCC[C@H]1C1=NC=C(C2=CC3=C(C=C2)C2CC4=C(C=CC(C5=CN=C(C6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=C4)N2C(=O)N3)N1 Chemical compound C.CC[C@H](NC(=O)OC)C(=O)N1CCC[C@H]1C1=NC=C(C2=CC3=C(C=C2)C2CC4=C(C=CC(C5=CN=C(C6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=C4)N2C(=O)N3)N1 ZXRAVANDEDCHAZ-KIKKKRBJSA-N 0.000 description 1
- MRTYEHIEAKKKAQ-SOCYXBEYSA-N C.CC[C@H](NC(=O)OC)C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(CC(=O)[C@@H]5CCCN5C(=O)[C@@H](CC(=O)OC)C(C)C)=CC=C4N3)C=C2)N1 Chemical compound C.CC[C@H](NC(=O)OC)C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(CC(=O)[C@@H]5CCCN5C(=O)[C@@H](CC(=O)OC)C(C)C)=CC=C4N3)C=C2)N1 MRTYEHIEAKKKAQ-SOCYXBEYSA-N 0.000 description 1
- LZPUDCBHFHDGCP-UMBBOSETSA-N C.CNC(C(=O)N1CCC[C@H]1C(=O)CC1=CC2=C(C=C1)OC(C1=CC=C(N)C=C1)=C2)C1=CC=CC=C1 Chemical compound C.CNC(C(=O)N1CCC[C@H]1C(=O)CC1=CC2=C(C=C1)OC(C1=CC=C(N)C=C1)=C2)C1=CC=CC=C1 LZPUDCBHFHDGCP-UMBBOSETSA-N 0.000 description 1
- GDQKZFSNDOVAEJ-RBIIXHJOSA-N C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(CC(=O)[C@@H]4CCCN4C(=O)C4=NC=CO4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(CC(=O)[C@@H]4CCCN4C(=O)C4=NC=CO4)C=C3)=CC2=C1)C1=CC=CC=C1 GDQKZFSNDOVAEJ-RBIIXHJOSA-N 0.000 description 1
- NCXNVCAIBYBQHA-BWCHFONSSA-N C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(CC(=O)[C@@H]4CCCN4C(=O)CC4CC4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(CC(=O)[C@@H]4CCCN4C(=O)CC4CC4)C=C3)=CC2=C1)C1=CC=CC=C1 NCXNVCAIBYBQHA-BWCHFONSSA-N 0.000 description 1
- QBYTXJSFYMOLOH-VQIDDLENSA-N C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(CC(=O)[C@@H]4CCCN4C(=O)[C@@H]4CCN(CC5=CC=CC=C5)C4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(CC(=O)[C@@H]4CCCN4C(=O)[C@@H]4CCN(CC5=CC=CC=C5)C4)C=C3)=CC2=C1)C1=CC=CC=C1 QBYTXJSFYMOLOH-VQIDDLENSA-N 0.000 description 1
- HROOLOMSUBNFKZ-FUYWROQRSA-N C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(CC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C(C)C)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(CC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C(C)C)C=C3)=CC2=C1)C1=CC=CC=C1 HROOLOMSUBNFKZ-FUYWROQRSA-N 0.000 description 1
- BLKZKTGYUIGJKL-MMIFJLSRSA-N C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(CC(=O)[C@@H]4CCCN4C(=O)[C@H]4CCCO4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(CC(=O)[C@@H]4CCCN4C(=O)[C@H]4CCCO4)C=C3)=CC2=C1)C1=CC=CC=C1 BLKZKTGYUIGJKL-MMIFJLSRSA-N 0.000 description 1
- QBYTXJSFYMOLOH-IMOBTDPZSA-N C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(CC(=O)[C@@H]4CCCN4C(=O)[C@H]4CCN(CC5=CC=CC=C5)C4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(CC(=O)[C@@H]4CCCN4C(=O)[C@H]4CCN(CC5=CC=CC=C5)C4)C=C3)=CC2=C1)C1=CC=CC=C1 QBYTXJSFYMOLOH-IMOBTDPZSA-N 0.000 description 1
- CHCGHTMKIRSAQG-DGOIJWGOSA-N C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)C4CCCC4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)C4CCCC4)C=C3)=CC2=C1)C1=CC=CC=C1 CHCGHTMKIRSAQG-DGOIJWGOSA-N 0.000 description 1
- ZEICALRDALQVAY-JIQMVMJFSA-N C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1 ZEICALRDALQVAY-JIQMVMJFSA-N 0.000 description 1
- SCNVGNNWFGDMOO-ROYBKCIRSA-N C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1 SCNVGNNWFGDMOO-ROYBKCIRSA-N 0.000 description 1
- FMJSKHSYDFCACW-IWNCSLTCSA-N C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](CC(C)C)N(C)C)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](CC(C)C)N(C)C)C=C3)=CC2=C1)C1=CC=CC=C1 FMJSKHSYDFCACW-IWNCSLTCSA-N 0.000 description 1
- KPHVPDPJHIKNNV-BNXFJJORSA-N C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1 KPHVPDPJHIKNNV-BNXFJJORSA-N 0.000 description 1
- LGRMISNRVNIZQB-OWOVFVHBSA-N C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@H](C)NC(=O)OC(C)(C)C)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@H](C)NC(=O)OC(C)(C)C)C=C3)=CC2=C1)C1=CC=CC=C1 LGRMISNRVNIZQB-OWOVFVHBSA-N 0.000 description 1
- CBZWFRKORUEQAX-OEPJFREBSA-N C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@H](NC(=O)OC(C)(C)C)C(C)C)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@H](NC(=O)OC(C)(C)C)C(C)C)C=C3)=CC2=C1)C1=CC=CC=C1 CBZWFRKORUEQAX-OEPJFREBSA-N 0.000 description 1
- QCPAWLJAWDYYLC-VYVVXECBSA-N C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(C)=O)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(C)=O)C=C3)=CC2=C1)C1=CC=CC=C1 QCPAWLJAWDYYLC-VYVVXECBSA-N 0.000 description 1
- YQACGKMJSJQUHG-LTMUXFAYSA-N C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(C)C)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(C)C)C=C3)=CC2=C1)C1=CC=CC=C1 YQACGKMJSJQUHG-LTMUXFAYSA-N 0.000 description 1
- IAEAGWKYLHQHSL-WDPAVZSUSA-N C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)C)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)C)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1 IAEAGWKYLHQHSL-WDPAVZSUSA-N 0.000 description 1
- BIZDPHTUZRBTBW-VEERUVTPSA-N C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(C)=O)=CC=C3O2)C=C1)C1=CC=CC=C1 Chemical compound C.CN[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(C)=O)=CC=C3O2)C=C1)C1=CC=CC=C1 BIZDPHTUZRBTBW-VEERUVTPSA-N 0.000 description 1
- YTEAJHLQYKHFCD-PWPHXGLMSA-N C.CN[C@H](CC(C)C)C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](CC(C)C)N(C)C)C=C3)=CC2=C1 Chemical compound C.CN[C@H](CC(C)C)C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](CC(C)C)N(C)C)C=C3)=CC2=C1 YTEAJHLQYKHFCD-PWPHXGLMSA-N 0.000 description 1
- KXOHOYADGDIKOO-UHFFFAOYSA-N C.COC(=O)C1=CC=C2NC=CC2=C1.O=C(O)C1=CC=C2NC=CC2=C1 Chemical compound C.COC(=O)C1=CC=C2NC=CC2=C1.O=C(O)C1=CC=C2NC=CC2=C1 KXOHOYADGDIKOO-UHFFFAOYSA-N 0.000 description 1
- RVSKJIYVJAFDTN-ZDOSEXAVSA-N C.COC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=C(C=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)O)C4=CC=CC=C4)=C3)N2)C=C1)C1=CC=CC=C1 Chemical compound C.COC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=C(C=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)O)C4=CC=CC=C4)=C3)N2)C=C1)C1=CC=CC=C1 RVSKJIYVJAFDTN-ZDOSEXAVSA-N 0.000 description 1
- DTZDFKJCFKNZRR-UTXOEKMMSA-N C.COC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)O)C4=CC=CC=C4)=CC=C3O2)C=C1)C1=CC=CC=C1 Chemical compound C.COC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)O)C4=CC=CC=C4)=CC=C3O2)C=C1)C1=CC=CC=C1 DTZDFKJCFKNZRR-UTXOEKMMSA-N 0.000 description 1
- NKAOLNGHXJRMKP-JNHLRUBLSA-N C.COC(=O)C[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3C(=C2)C(F)=C2C4=C(C=C(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)C=C4)OCN32)N1)C(C)C.FC1=C2C3=C(C=C(C4=CN=C([C@@H]5CCCN5)N4)C=C3)OCN2C2=CC=C(C3=CN=C([C@@H]4CCCN4)N3)C=C21 Chemical compound C.COC(=O)C[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3C(=C2)C(F)=C2C4=C(C=C(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)C=C4)OCN32)N1)C(C)C.FC1=C2C3=C(C=C(C4=CN=C([C@@H]5CCCN5)N4)C=C3)OCN2C2=CC=C(C3=CN=C([C@@H]4CCCN4)N3)C=C21 NKAOLNGHXJRMKP-JNHLRUBLSA-N 0.000 description 1
- ZDOQXKSNKPFUCU-UHFFFAOYSA-N C.NC1=CC2=C(C=C1)N/C(C1=CC3=CC([N+](=O)[O-])=CC=C3N1)=C\2.NC1=CC=C2NC(/C3=C/C4=C(C=CC(N)=C4)N3)=CC2=C1.[HH] Chemical compound C.NC1=CC2=C(C=C1)N/C(C1=CC3=CC([N+](=O)[O-])=CC=C3N1)=C\2.NC1=CC=C2NC(/C3=C/C4=C(C=CC(N)=C4)N3)=CC2=C1.[HH] ZDOQXKSNKPFUCU-UHFFFAOYSA-N 0.000 description 1
- TVXPVZUCJXQJBT-HCNRMFOCSA-N C.NC1=CC=C(/C2=C/C3=CC=C([N+](=O)[O-])C=C3N2)C=C1.O=C(NC1=CC=C(/C2=C/C3=CC=C([N+](=O)[O-])C=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(O)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound C.NC1=CC=C(/C2=C/C3=CC=C([N+](=O)[O-])C=C3N2)C=C1.O=C(NC1=CC=C(/C2=C/C3=CC=C([N+](=O)[O-])C=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(O)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 TVXPVZUCJXQJBT-HCNRMFOCSA-N 0.000 description 1
- GOBKUVNEDZWJAY-MMHRBMGHSA-N C.NC1=CC=C2/C=C(/C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)NC2=C1.O=C(NC1=CC=C(/C2=C/C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(O)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound C.NC1=CC=C2/C=C(/C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)NC2=C1.O=C(NC1=CC=C(/C2=C/C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(O)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 GOBKUVNEDZWJAY-MMHRBMGHSA-N 0.000 description 1
- LZBSYBLUOHPKAD-ZCQSKNOZSA-N C.NC1=CC=C2C(=C1)CCCC1=C2NC2=CC=C(N)C=C21.O=C(CC1=CC=C2NC3=C(CCCC4=CC(NC(=O)[C@@H]5CCCN5C(=O)CC5=CC=CC=C5)=CC=C43)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(O)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound C.NC1=CC=C2C(=C1)CCCC1=C2NC2=CC=C(N)C=C21.O=C(CC1=CC=C2NC3=C(CCCC4=CC(NC(=O)[C@@H]5CCCN5C(=O)CC5=CC=CC=C5)=CC=C43)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(O)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 LZBSYBLUOHPKAD-ZCQSKNOZSA-N 0.000 description 1
- JLJLZZDGZAFAJK-YVRGNLFHSA-N C.O=C(CC1=CN=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=C(Cl)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(CC1=CN=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=CC2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound C.O=C(CC1=CN=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=C(Cl)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(CC1=CN=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=CC2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 JLJLZZDGZAFAJK-YVRGNLFHSA-N 0.000 description 1
- VZYMDAOJYAQDKM-GJQHYITLSA-N C.O=C(CC1=NC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=C(Cl)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(CC1=NC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=CC2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound C.O=C(CC1=NC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=C(Cl)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(CC1=NC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=CC2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 VZYMDAOJYAQDKM-GJQHYITLSA-N 0.000 description 1
- MDQKCTCKSUFRND-UHFFFAOYSA-N C.OC1=C(C2=C(F)C3=CC(Br)=CC=C3N2)C=CC(Br)=C1.OC1=C(C2=CC3=CC(Br)=CC=C3N2)C=CC(Br)=C1 Chemical compound C.OC1=C(C2=C(F)C3=CC(Br)=CC=C3N2)C=CC(Br)=C1.OC1=C(C2=CC3=CC(Br)=CC=C3N2)C=CC(Br)=C1 MDQKCTCKSUFRND-UHFFFAOYSA-N 0.000 description 1
- TUCUJLRDNWYRRM-FLPKAINGSA-N C/C(=N/NC1=CC=C(Br)C=C1)C1=CC=C([N+](=O)[O-])C=C1.CC(=O)C1=CC=C([N+](=O)[O-])C=C1.NNC1=CC=C(Br)C=C1 Chemical compound C/C(=N/NC1=CC=C(Br)C=C1)C1=CC=C([N+](=O)[O-])C=C1.CC(=O)C1=CC=C([N+](=O)[O-])C=C1.NNC1=CC=C(Br)C=C1 TUCUJLRDNWYRRM-FLPKAINGSA-N 0.000 description 1
- FEQDPXQNJHBSNT-HYMQDMCPSA-N C/C(=N/NC1=CC=C(Br)C=C1)C1=CC=C([N+](=O)[O-])C=C1.O=[N+]([O-])C1=CC=C(C2=CC3=CC(Br)=CC=C3N2)C=C1 Chemical compound C/C(=N/NC1=CC=C(Br)C=C1)C1=CC=C([N+](=O)[O-])C=C1.O=[N+]([O-])C1=CC=C(C2=CC3=CC(Br)=CC=C3N2)C=C1 FEQDPXQNJHBSNT-HYMQDMCPSA-N 0.000 description 1
- XTNYBJXXXGAXGC-SRZXOJDVSA-N C1=CC(C2=CC3=CC(C4=CN=C(C5CCCN5)N4)=CC=C3O2)=CC=C1C1=CN=C([C@@H]2CCCN2)N1.CC(C)(C)OC(=O)N1CCCC1C1=NC=C(C2=CC=C3OC(C4=CC=C(C5=CN=C(C6CCCC6)N5)C=C4)=CC3=C2)N1 Chemical compound C1=CC(C2=CC3=CC(C4=CN=C(C5CCCN5)N4)=CC=C3O2)=CC=C1C1=CN=C([C@@H]2CCCN2)N1.CC(C)(C)OC(=O)N1CCCC1C1=NC=C(C2=CC=C3OC(C4=CC=C(C5=CN=C(C6CCCC6)N5)C=C4)=CC3=C2)N1 XTNYBJXXXGAXGC-SRZXOJDVSA-N 0.000 description 1
- UPGUQHBVUDTNGY-QGRPHFIJSA-N C1=CC(C2=CC3=CC(C4=CN=C(C5CCCN5)N4)=CC=C3O2)=CC=C1C1=CN=C([C@@H]2CCCN2)N1.COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=CC=C4O3)C=C2)N1)C(C)C.COC(=O)N[C@H](C(=O)O)C(C)C Chemical compound C1=CC(C2=CC3=CC(C4=CN=C(C5CCCN5)N4)=CC=C3O2)=CC=C1C1=CN=C([C@@H]2CCCN2)N1.COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=CC=C4O3)C=C2)N1)C(C)C.COC(=O)N[C@H](C(=O)O)C(C)C UPGUQHBVUDTNGY-QGRPHFIJSA-N 0.000 description 1
- YZYUVHKQQSFXIK-CGVQXEDRSA-N C1=CC(C2=NC3=CC(C4=CN=C(C5CCCN5)N4)=CC=C3O2)=CC=C1C1=CN=C([C@@H]2CCCN2)N1.CC(C)(C)OC(=O)N1CCCC1C1=NC=C(C2=CC=C3OC(C4=CC=C(C5=CN=C(C6CCCC6)N5)C=C4)=NC3=C2)N1 Chemical compound C1=CC(C2=NC3=CC(C4=CN=C(C5CCCN5)N4)=CC=C3O2)=CC=C1C1=CN=C([C@@H]2CCCN2)N1.CC(C)(C)OC(=O)N1CCCC1C1=NC=C(C2=CC=C3OC(C4=CC=C(C5=CN=C(C6CCCC6)N5)C=C4)=NC3=C2)N1 YZYUVHKQQSFXIK-CGVQXEDRSA-N 0.000 description 1
- PLDAPAMYGOPXOR-UKIIZZSZSA-N C1=CC(C2=NC3=CC(C4=CN=C(C5CCCN5)N4)=CC=C3O2)=CC=C1C1=CN=C([C@@H]2CCCN2)N1.C[C@@H](C(=O)N1CCCC1C1=NC=C(C2=CC=C3OC(C4=CC=C(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](C)C6=CC=CC=C6)N5)C=C4)=NC3=C2)N1)C1=CC=CC=C1.O=BP Chemical compound C1=CC(C2=NC3=CC(C4=CN=C(C5CCCN5)N4)=CC=C3O2)=CC=C1C1=CN=C([C@@H]2CCCN2)N1.C[C@@H](C(=O)N1CCCC1C1=NC=C(C2=CC=C3OC(C4=CC=C(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](C)C6=CC=CC=C6)N5)C=C4)=NC3=C2)N1)C1=CC=CC=C1.O=BP PLDAPAMYGOPXOR-UKIIZZSZSA-N 0.000 description 1
- WUMCCMJJCKBLIO-UHFFFAOYSA-N C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.CC(=O)NCC#CC1=CC2=CC([N+](=O)[O-])=CC=C2N1C(C)=O.CCOC(=O)N1C(OS(=O)(=O)C(F)(F)F)=CC2=CC([N+](=O)[O-])=CC=C21.[Pd] Chemical compound C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.CC(=O)NCC#CC1=CC2=CC([N+](=O)[O-])=CC=C2N1C(C)=O.CCOC(=O)N1C(OS(=O)(=O)C(F)(F)F)=CC2=CC([N+](=O)[O-])=CC=C21.[Pd] WUMCCMJJCKBLIO-UHFFFAOYSA-N 0.000 description 1
- KPDKNBPSXIGOMQ-PFMLNZFDSA-N C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.CC1=CC=C2C(=C1)C=C(B(O)O)N2C(=O)OC(C)(C)C.CC1=CC=C2C(=C1)C=C(C1=CC=C3NC([C@@H]4CCCN4C)=NC3=C1)N2C(=O)OC(C)(C)C.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NC2=CC(Br)=CC=C2N1.[Pd] Chemical compound C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.CC1=CC=C2C(=C1)C=C(B(O)O)N2C(=O)OC(C)(C)C.CC1=CC=C2C(=C1)C=C(C1=CC=C3NC([C@@H]4CCCN4C)=NC3=C1)N2C(=O)OC(C)(C)C.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NC2=CC(Br)=CC=C2N1.[Pd] KPDKNBPSXIGOMQ-PFMLNZFDSA-N 0.000 description 1
- UTFXPMGWWGHRNG-UHFFFAOYSA-N C1=CNC=N1.N#CBr.N=C(N1C=CN=C1)N1C=CN=C1 Chemical compound C1=CNC=N1.N#CBr.N=C(N1C=CN=C1)N1C=CN=C1 UTFXPMGWWGHRNG-UHFFFAOYSA-N 0.000 description 1
- ONRGHMYXRXHDTK-OHFKZJQBSA-M C1CCOC1.N=C(N)C1=CC=C(Br)C=C1.O=C(CBr)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=CN=C(C2=CC=C(Br)C=C2)N1.O=COO[Na] Chemical compound C1CCOC1.N=C(N)C1=CC=C(Br)C=C1.O=C(CBr)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=CN=C(C2=CC=C(Br)C=C2)N1.O=COO[Na] ONRGHMYXRXHDTK-OHFKZJQBSA-M 0.000 description 1
- YKDYWYYKQLTTMW-BCRBLDSWSA-N CC(=O)C1=C(C2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C2)NC2=CC=C(CC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C21 Chemical compound CC(=O)C1=C(C2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C2)NC2=CC=C(CC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C21 YKDYWYYKQLTTMW-BCRBLDSWSA-N 0.000 description 1
- XASHQGPXPFYWBR-UHFFFAOYSA-N CC(=O)C1=CC=C([N+](=O)[O-])C=C1.[H]C(=O)C(=O)C1=CC=C([N+](=O)[O-])C=C1 Chemical compound CC(=O)C1=CC=C([N+](=O)[O-])C=C1.[H]C(=O)C(=O)C1=CC=C([N+](=O)[O-])C=C1 XASHQGPXPFYWBR-UHFFFAOYSA-N 0.000 description 1
- BGNNNPBRHCWLAC-UHFFFAOYSA-N CC(=O)CC1=CC(C(C)=O)=CC=C1.CC(=O)CC1=CC2=C(C=C1)N/C(C1=CC=CC(NC(C)=O)=C1)=C\2.CC(=O)CC1=CC=C(NN)C=C1.Cl Chemical compound CC(=O)CC1=CC(C(C)=O)=CC=C1.CC(=O)CC1=CC2=C(C=C1)N/C(C1=CC=CC(NC(C)=O)=C1)=C\2.CC(=O)CC1=CC=C(NN)C=C1.Cl BGNNNPBRHCWLAC-UHFFFAOYSA-N 0.000 description 1
- HTEPRZOFFDMQCC-UHFFFAOYSA-N CC(=O)CC1=CC2=C(C=C1)N/C(C1=CC=CC(NC(C)=O)=C1)=C\2.Cl.NC1=CC(/C2=C/C3=C(C=CC(N)=C3)N2)=CC=C1 Chemical compound CC(=O)CC1=CC2=C(C=C1)N/C(C1=CC=CC(NC(C)=O)=C1)=C\2.Cl.NC1=CC(/C2=C/C3=C(C=CC(N)=C3)N2)=CC=C1 HTEPRZOFFDMQCC-UHFFFAOYSA-N 0.000 description 1
- CNUQDKSMKSDHCP-NEMIEIFKSA-N CC(=O)CC1=CC=C(/C(C)=N/NC2=CC=C(CC(C)=O)N=C2)C=C1.CC(=O)CC1=CC=C2NC(C3=CC=C(NC(C)=O)C=C3)=CC2=N1 Chemical compound CC(=O)CC1=CC=C(/C(C)=N/NC2=CC=C(CC(C)=O)N=C2)C=C1.CC(=O)CC1=CC=C2NC(C3=CC=C(NC(C)=O)C=C3)=CC2=N1 CNUQDKSMKSDHCP-NEMIEIFKSA-N 0.000 description 1
- FBLXTSILDDTWEP-DXISODENSA-N CC(=O)CC1=CC=C(C2=CC3=CC(C4=NN=C([C@@H]5CCCN5)O4)=CC=C3N2)C=C1.CC(=O)CC1=CC=C(C2=CC3=CC(C4=NN=C([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)O4)=CC=C3N2C(=O)OC(C)(C)C)C=C1 Chemical compound CC(=O)CC1=CC=C(C2=CC3=CC(C4=NN=C([C@@H]5CCCN5)O4)=CC=C3N2)C=C1.CC(=O)CC1=CC=C(C2=CC3=CC(C4=NN=C([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)O4)=CC=C3N2C(=O)OC(C)(C)C)C=C1 FBLXTSILDDTWEP-DXISODENSA-N 0.000 description 1
- FPUFCYCYOZJGOM-QWLMYISHSA-N CC(=O)CC1=CC=C(C2=CC3=CC(C4=NN=C([C@@H]5CCCN5)O4)=CC=C3N2)C=C1.CC(=O)CC1=CC=C(C2=CC3=CC(C4=NN=C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)O4)=CC=C3N2)C=C1 Chemical compound CC(=O)CC1=CC=C(C2=CC3=CC(C4=NN=C([C@@H]5CCCN5)O4)=CC=C3N2)C=C1.CC(=O)CC1=CC=C(C2=CC3=CC(C4=NN=C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)O4)=CC=C3N2)C=C1 FPUFCYCYOZJGOM-QWLMYISHSA-N 0.000 description 1
- PHPACZRORYWYFG-IOWSJCHKSA-N CC(=O)CC1=CC=C(C2=CC3=CC(C4=NN=C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)O4)=CC=C3N2)C=C1 Chemical compound CC(=O)CC1=CC=C(C2=CC3=CC(C4=NN=C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)O4)=CC=C3N2)C=C1 PHPACZRORYWYFG-IOWSJCHKSA-N 0.000 description 1
- TXFASYLMKHXSME-MHZLTWQESA-N CC(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=CC2=N1 Chemical compound CC(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=CC2=N1 TXFASYLMKHXSME-MHZLTWQESA-N 0.000 description 1
- DWENAVMSWBNMEY-USTLVJCJSA-N CC(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=CC2=N1.CC(=O)Cl.NC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=CC2=N1 Chemical compound CC(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=CC2=N1.CC(=O)Cl.NC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=CC2=N1 DWENAVMSWBNMEY-USTLVJCJSA-N 0.000 description 1
- SHOBNFAYZQRTFC-UHFFFAOYSA-N CC(=O)CC1=CC=C2NC(C3=CC=C(NC(C)=O)C=C3)=CC2=N1.Cl.NC1=CC=C(C2=CC3=NC(N)=CC=C3N2)C=C1 Chemical compound CC(=O)CC1=CC=C2NC(C3=CC=C(NC(C)=O)C=C3)=CC2=N1.Cl.NC1=CC=C(C2=CC3=NC(N)=CC=C3N2)C=C1 SHOBNFAYZQRTFC-UHFFFAOYSA-N 0.000 description 1
- WOHFPQSZFMOHOO-UHFFFAOYSA-N CC(=O)CC1=CC=C2NC3=C(CCCC4=CC(NC(C)=O)=CC=C43)C2=C1.Cl.NC1=CC=C2C(=C1)CCCC1=C2NC2=CC=C(N)C=C21 Chemical compound CC(=O)CC1=CC=C2NC3=C(CCCC4=CC(NC(C)=O)=CC=C43)C2=C1.Cl.NC1=CC=C2C(=C1)CCCC1=C2NC2=CC=C(N)C=C21 WOHFPQSZFMOHOO-UHFFFAOYSA-N 0.000 description 1
- ASCHHGQBUVDSKJ-QCENPCRXSA-N CC(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=CC2=C1 Chemical compound CC(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=CC2=C1 ASCHHGQBUVDSKJ-QCENPCRXSA-N 0.000 description 1
- FJMHGKZGHFWYMZ-UHFFFAOYSA-N CC(=O)Cl.CC(=O)OC1=CC=CC(Br)=C1.CCN(CC)CC.OC1=CC=CC(Br)=C1 Chemical compound CC(=O)Cl.CC(=O)OC1=CC=CC(Br)=C1.CCN(CC)CC.OC1=CC=CC(Br)=C1 FJMHGKZGHFWYMZ-UHFFFAOYSA-N 0.000 description 1
- HPGVBJIJWRIROF-PUBVIUOISA-N CC(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=CC2=C1 Chemical compound CC(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=CC2=C1 HPGVBJIJWRIROF-PUBVIUOISA-N 0.000 description 1
- HPGVBJIJWRIROF-GRGGVALCSA-N CC(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=CC2=C1 Chemical compound CC(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=CC2=C1 HPGVBJIJWRIROF-GRGGVALCSA-N 0.000 description 1
- FLSHBTBOXUFCER-PFDXXMEMSA-N CC(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=CC2=C1 Chemical compound CC(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=CC2=C1 FLSHBTBOXUFCER-PFDXXMEMSA-N 0.000 description 1
- PDBWPJGYEHCXKF-DTKZEWQESA-N CC(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=C(Br)C3=C(C=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(C)C)=C3)O2)C=C1 Chemical compound CC(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=C(Br)C3=C(C=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(C)C)=C3)O2)C=C1 PDBWPJGYEHCXKF-DTKZEWQESA-N 0.000 description 1
- UREBPGPHNIFBHN-QOVAEWSBSA-N CC(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)=CC=C3N2)C=C1 Chemical compound CC(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)=CC=C3N2)C=C1 UREBPGPHNIFBHN-QOVAEWSBSA-N 0.000 description 1
- RXAWAMPDDLOFSN-ASHMCCQMSA-N CC(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](C)C4=CC=CC=C4)=CC=C3N2)C=C1.CC(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](C)C4=CC=CC=C4)=CC=C3N2)C=C1 Chemical compound CC(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](C)C4=CC=CC=C4)=CC=C3N2)C=C1.CC(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](C)C4=CC=CC=C4)=CC=C3N2)C=C1 RXAWAMPDDLOFSN-ASHMCCQMSA-N 0.000 description 1
- FQQZNVPHTWQTRK-RDKKHIPBSA-N CC(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)=CC=C3N2)C=C1 Chemical compound CC(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)=CC=C3N2)C=C1 FQQZNVPHTWQTRK-RDKKHIPBSA-N 0.000 description 1
- SEKZAWKNXFGORG-KBGXGDJFSA-N CC(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=C/C(CC(=O)[C@@H]5CCCN5C(=O)[C@H](CC(=O)OC(C)(C)C)C5=CC=CC=C5)=C\C=C\4N3)C=C2)N1 Chemical compound CC(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=C/C(CC(=O)[C@@H]5CCCN5C(=O)[C@H](CC(=O)OC(C)(C)C)C5=CC=CC=C5)=C\C=C\4N3)C=C2)N1 SEKZAWKNXFGORG-KBGXGDJFSA-N 0.000 description 1
- HRLVUSPIPXBKKU-KBGXGDJFSA-N CC(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(CC(=O)[C@@H]5CCCN5C(=O)[C@H](CC(=O)OC(C)C)C5=CC=CC=C5)=CC=C4N3)C=C2)N1 Chemical compound CC(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(CC(=O)[C@@H]5CCCN5C(=O)[C@H](CC(=O)OC(C)C)C5=CC=CC=C5)=CC=C4N3)C=C2)N1 HRLVUSPIPXBKKU-KBGXGDJFSA-N 0.000 description 1
- WVTWTVHFYIUXLZ-UHFFFAOYSA-N CC(=O)NC1=CC(C2=CC3=CC=C(N)C=C3N2)=CC=C1.CC(=O)NC1=CC(C2=CC3=CC=C([N+](=O)[O-])C=C3N2)=CC=C1.N=NN=Cl.NC1=CC=C2C=C(C3=CC=CC(N)=C3)NC2=C1 Chemical compound CC(=O)NC1=CC(C2=CC3=CC=C(N)C=C3N2)=CC=C1.CC(=O)NC1=CC(C2=CC3=CC=C([N+](=O)[O-])C=C3N2)=CC=C1.N=NN=Cl.NC1=CC=C2C=C(C3=CC=CC(N)=C3)NC2=C1 WVTWTVHFYIUXLZ-UHFFFAOYSA-N 0.000 description 1
- IFNANOSZERJWNG-VEWWMAHHSA-N CC(=O)NC1=CC=C(/C(C)=N/CC2=CC=C(NC(C)=O)N=C2)C=C1.CC(=O)NC1=CC=C(C(C)=O)C=N1.CC(=O)NC1=CC=C(CN)C=N1.CCN(CC)CC Chemical compound CC(=O)NC1=CC=C(/C(C)=N/CC2=CC=C(NC(C)=O)N=C2)C=C1.CC(=O)NC1=CC=C(C(C)=O)C=N1.CC(=O)NC1=CC=C(CN)C=N1.CCN(CC)CC IFNANOSZERJWNG-VEWWMAHHSA-N 0.000 description 1
- KZONPYYGDCAQSF-UHFFFAOYSA-N CC(=O)NC1=CC=C(/C2=C/C3=CC=C([N+](=O)[O-])C=C3N2)C=C1.Cl.NC1=CC=C(/C2=C/C3=CC=C([N+](=O)[O-])C=C3N2)C=C1 Chemical compound CC(=O)NC1=CC=C(/C2=C/C3=CC=C([N+](=O)[O-])C=C3N2)C=C1.Cl.NC1=CC=C(/C2=C/C3=CC=C([N+](=O)[O-])C=C3N2)C=C1 KZONPYYGDCAQSF-UHFFFAOYSA-N 0.000 description 1
- PCFZTLDDMSLLFR-UHFFFAOYSA-N CC(=O)NC1=CC=C(C#CC2=CC([N+](=O)[O-])=CN=C2N)C=C1.CC(=O)NC1=CC=C(C2=CC3=CC([N+](=O)[O-])=CN=C3N2)C=C1 Chemical compound CC(=O)NC1=CC=C(C#CC2=CC([N+](=O)[O-])=CN=C2N)C=C1.CC(=O)NC1=CC=C(C2=CC3=CC([N+](=O)[O-])=CN=C3N2)C=C1 PCFZTLDDMSLLFR-UHFFFAOYSA-N 0.000 description 1
- ZQSJWPGREYYKPD-UHFFFAOYSA-N CC(=O)NC1=CC=C(C#CC2=NC(Br)=CC=C2N)C=C1.CC(=O)NC1=CC=C(C2=CC3=NC(Br)=CC=C3N2)C=C1 Chemical compound CC(=O)NC1=CC=C(C#CC2=NC(Br)=CC=C2N)C=C1.CC(=O)NC1=CC=C(C2=CC3=NC(Br)=CC=C3N2)C=C1 ZQSJWPGREYYKPD-UHFFFAOYSA-N 0.000 description 1
- JYCUDBXFGFUPPC-FREGXXQWSA-N CC(=O)NC1=CC=C(C2=C(Br)C3=C(C=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(C)C)=C3)O2)C=C1 Chemical compound CC(=O)NC1=CC=C(C2=C(Br)C3=C(C=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(C)C)=C3)O2)C=C1 JYCUDBXFGFUPPC-FREGXXQWSA-N 0.000 description 1
- LRXKADGASNGKEW-NSJVFKKDSA-N CC(=O)NC1=CC=C(C2=C(C#N)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)=CC=C3N2)C=C1 Chemical compound CC(=O)NC1=CC=C(C2=C(C#N)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)=CC=C3N2)C=C1 LRXKADGASNGKEW-NSJVFKKDSA-N 0.000 description 1
- LGDGNRQHHPSFDX-IUDBTDONSA-N CC(=O)NC1=CC=C(C2=C(Cl)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)=CC=C3N2)C=C1 Chemical compound CC(=O)NC1=CC=C(C2=C(Cl)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)=CC=C3N2)C=C1 LGDGNRQHHPSFDX-IUDBTDONSA-N 0.000 description 1
- XWJZHANNOYRHHM-BVZFJXPGSA-N CC(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](C)CC(=O)OC(C)(C)C)=CC=C3N2)C=C1 Chemical compound CC(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](C)CC(=O)OC(C)(C)C)=CC=C3N2)C=C1 XWJZHANNOYRHHM-BVZFJXPGSA-N 0.000 description 1
- FPJSLQNWBABAFH-JTSJOTPCSA-N CC(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(C)C)=CC=C3N2)C=C1 Chemical compound CC(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(C)C)=CC=C3N2)C=C1 FPJSLQNWBABAFH-JTSJOTPCSA-N 0.000 description 1
- CRKJPYHQVQHCKA-RYCFQHDISA-N CC(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N4CCCC4)=CC=C3N2)C=C1 Chemical compound CC(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N4CCCC4)=CC=C3N2)C=C1 CRKJPYHQVQHCKA-RYCFQHDISA-N 0.000 description 1
- SMMUGNGCHOWYAA-OFSOJUDTSA-N CC(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)=CC=C3N2)C=C1 Chemical compound CC(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)=CC=C3N2)C=C1 SMMUGNGCHOWYAA-OFSOJUDTSA-N 0.000 description 1
- REKXWSLHYPFLBA-CUBQBAPOSA-N CC(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)CC(C)C)=CC=C3N2)C=C1 Chemical compound CC(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)CC(C)C)=CC=C3N2)C=C1 REKXWSLHYPFLBA-CUBQBAPOSA-N 0.000 description 1
- RDZXDIXDZOGROS-XZWHSSHBSA-N CC(=O)NC1=CC=C(C2=CC3=CC(C4=NN=C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)O4)=CC=C3N2)C=C1 Chemical compound CC(=O)NC1=CC=C(C2=CC3=CC(C4=NN=C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)O4)=CC=C3N2)C=C1 RDZXDIXDZOGROS-XZWHSSHBSA-N 0.000 description 1
- ANKVNISGFWOZDL-ACVCQEAVSA-N CC(=O)NC1=CC=C(C2=CC3=CC(CC(=O)C4CCCN4C(=O)[C@H](C)C4=CC=CC=C4)=CC=C3O2)C=C1 Chemical compound CC(=O)NC1=CC=C(C2=CC3=CC(CC(=O)C4CCCN4C(=O)[C@H](C)C4=CC=CC=C4)=CC=C3O2)C=C1 ANKVNISGFWOZDL-ACVCQEAVSA-N 0.000 description 1
- BXEMEFDOTFICIV-RLRXPSAQSA-N CC(=O)NC1=CC=C(C2=CC3=CC(CC(=O)C4CCCN4C(=O)[C@H](C)C4=CC=CC=C4)=CC=C3O2)C=C1.CC(=O)OC(C)=O.C[C@@H](C(=O)N1CCCC1C(=O)CC1=CC=C2OC(C3=CC=C(N)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound CC(=O)NC1=CC=C(C2=CC3=CC(CC(=O)C4CCCN4C(=O)[C@H](C)C4=CC=CC=C4)=CC=C3O2)C=C1.CC(=O)OC(C)=O.C[C@@H](C(=O)N1CCCC1C(=O)CC1=CC=C2OC(C3=CC=C(N)C=C3)=CC2=C1)C1=CC=CC=C1 BXEMEFDOTFICIV-RLRXPSAQSA-N 0.000 description 1
- QMHWZNJDWXNWKO-CQTOTRCISA-N CC(=O)NC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N4CCCC4)=CC=C3N2)C=C1 Chemical compound CC(=O)NC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N4CCCC4)=CC=C3N2)C=C1 QMHWZNJDWXNWKO-CQTOTRCISA-N 0.000 description 1
- JMXOYXBISSFMKK-UZNNEEJFSA-N CC(=O)NC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N4CCCCC4)=CC=C3N2)C=C1 Chemical compound CC(=O)NC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N4CCCCC4)=CC=C3N2)C=C1 JMXOYXBISSFMKK-UZNNEEJFSA-N 0.000 description 1
- XCSDJVDRCYJXMG-CQTOTRCISA-N CC(=O)NC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N4CCOCC4)=CC=C3N2)C=C1 Chemical compound CC(=O)NC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N4CCOCC4)=CC=C3N2)C=C1 XCSDJVDRCYJXMG-CQTOTRCISA-N 0.000 description 1
- SIZJBAPBBOWPHH-PPTMTGTBSA-N CC(=O)NC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)C)C4=CC=CC=C4)=CC=C3N2)C=C1 Chemical compound CC(=O)NC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)C)C4=CC=CC=C4)=CC=C3N2)C=C1 SIZJBAPBBOWPHH-PPTMTGTBSA-N 0.000 description 1
- IOEKBYZYGANGEH-UHFFFAOYSA-N CC(=O)NC1=CC=C(C2=CC3=CC([N+](=O)[O-])=CN=C3N2)C=C1.Cl.NC1=CC=C(C2=CC3=CC([N+](=O)[O-])=CN=C3N2)C=C1 Chemical compound CC(=O)NC1=CC=C(C2=CC3=CC([N+](=O)[O-])=CN=C3N2)C=C1.Cl.NC1=CC=C(C2=CC3=CC([N+](=O)[O-])=CN=C3N2)C=C1 IOEKBYZYGANGEH-UHFFFAOYSA-N 0.000 description 1
- LIEQVRUHXGWKEH-UHFFFAOYSA-N CC(=O)NC1=CC=C(C2=CC3=NC(Br)=CC=C3N2)C=C1.N=NN=Cl.NC1=CC=C(C2=CC3=NC(Br)=CC=C3N2)C=C1 Chemical compound CC(=O)NC1=CC=C(C2=CC3=NC(Br)=CC=C3N2)C=C1.N=NN=Cl.NC1=CC=C(C2=CC3=NC(Br)=CC=C3N2)C=C1 LIEQVRUHXGWKEH-UHFFFAOYSA-N 0.000 description 1
- BCJLRNYIXQUDJK-UHFFFAOYSA-N CC(=O)NC1=CC=C(CN)C=N1.CC(=O)NC1=CC=C(N)C=N1 Chemical compound CC(=O)NC1=CC=C(CN)C=N1.CC(=O)NC1=CC=C(N)C=N1 BCJLRNYIXQUDJK-UHFFFAOYSA-N 0.000 description 1
- QWBGBONCFSORJB-UHFFFAOYSA-N CC(=O)NC1=CC=C([N+](=O)[O-])C=N1.CC(=O)OC(C)=O.NC1=CC=C([N+](=O)[O-])C=N1 Chemical compound CC(=O)NC1=CC=C([N+](=O)[O-])C=N1.CC(=O)OC(C)=O.NC1=CC=C([N+](=O)[O-])C=N1 QWBGBONCFSORJB-UHFFFAOYSA-N 0.000 description 1
- JZJWTMSIERJBBX-UHFFFAOYSA-N CC(=O)NC1=CC=CC(CCCCC(=O)O)=C1.O=C(O)CCCC(=O)C1=CC([N+](=O)[O-])=CC=C1 Chemical compound CC(=O)NC1=CC=CC(CCCCC(=O)O)=C1.O=C(O)CCCC(=O)C1=CC([N+](=O)[O-])=CC=C1 JZJWTMSIERJBBX-UHFFFAOYSA-N 0.000 description 1
- SZXOGZKYVNLZNJ-BCHFMIIMSA-N CC(=O)NCC#CC1=CC2=CC(CC(=O)[C@@H]3CCCN3C(=O)[C@H](C)C3=CC=CC=C3)=CC=C2N1 Chemical compound CC(=O)NCC#CC1=CC2=CC(CC(=O)[C@@H]3CCCN3C(=O)[C@H](C)C3=CC=CC=C3)=CC=C2N1 SZXOGZKYVNLZNJ-BCHFMIIMSA-N 0.000 description 1
- GQHUEATZTDYVPK-UHFFFAOYSA-M CC(=O)NCC#CC1=CC2=CC(N)=CC=C2N1.CC(=O)NCC#CC1=CC2=CC(N)=CC=C2N1C(C)=O.O=COO[K].[KH] Chemical compound CC(=O)NCC#CC1=CC2=CC(N)=CC=C2N1.CC(=O)NCC#CC1=CC2=CC(N)=CC=C2N1C(C)=O.O=COO[K].[KH] GQHUEATZTDYVPK-UHFFFAOYSA-M 0.000 description 1
- ABODDSHZGBKOOQ-UHFFFAOYSA-N CC(=O)NCC#CC1=CC2=CC(N)=CC=C2N1C(C)=O.CC(=O)NCC#CC1=CC2=CC([N+](=O)[O-])=CC=C2N1C(C)=O Chemical compound CC(=O)NCC#CC1=CC2=CC(N)=CC=C2N1C(C)=O.CC(=O)NCC#CC1=CC2=CC([N+](=O)[O-])=CC=C2N1C(C)=O ABODDSHZGBKOOQ-UHFFFAOYSA-N 0.000 description 1
- OOXFNHGWHYAHFH-UHFFFAOYSA-N CC(=O)O.CC(C)(C)OC(=O)NC1=CC=C(C(=O)CC2=C([N+](=O)[O-])C=CC=C2N)C=C1.NC1=CC=C(C2=NC3=C([N+](=O)[O-])C=CC=C3N2)C=C1 Chemical compound CC(=O)O.CC(C)(C)OC(=O)NC1=CC=C(C(=O)CC2=C([N+](=O)[O-])C=CC=C2N)C=C1.NC1=CC=C(C2=NC3=C([N+](=O)[O-])C=CC=C3N2)C=C1 OOXFNHGWHYAHFH-UHFFFAOYSA-N 0.000 description 1
- WEKQYECZNQKOQK-ZXNOKPTOSA-N CC(=O)O.N.O=C(CNC(=O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1)C1=CC=C(Br)C=C1.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NC=C(C2=CC=C(Br)C=C2)N1 Chemical compound CC(=O)O.N.O=C(CNC(=O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1)C1=CC=C(Br)C=C1.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NC=C(C2=CC=C(Br)C=C2)N1 WEKQYECZNQKOQK-ZXNOKPTOSA-N 0.000 description 1
- IEGLUIJIYQXQTE-YDCMEUSMSA-N CC(=O)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(CBr)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound CC(=O)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(CBr)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 IEGLUIJIYQXQTE-YDCMEUSMSA-N 0.000 description 1
- MQNPVOGILJTEBI-DMGMNCNXSA-N CC(=O)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(O)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound CC(=O)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(O)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 MQNPVOGILJTEBI-DMGMNCNXSA-N 0.000 description 1
- RIEJPUOOXWIPGN-DAGGQHOASA-N CC(C)(C)C(=O)C1=C(C2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C2)NC2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C21.CC(C)(C)C(=O)Cl.O=C(NC1=CC=C(C2=C(Br)C3=CC(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound CC(C)(C)C(=O)C1=C(C2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C2)NC2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C21.CC(C)(C)C(=O)Cl.O=C(NC1=CC=C(C2=C(Br)C3=CC(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 RIEJPUOOXWIPGN-DAGGQHOASA-N 0.000 description 1
- GAAFHALSCLOMFD-UHFFFAOYSA-N CC(C)(C)C(=O)Cl.CC1=C(CC(=O)C(C)(C)C)C=NC(NC(=O)C(C)(C)C)=C1.CC1=C(N)C=NC(N)=C1.CCN(CC)CC Chemical compound CC(C)(C)C(=O)Cl.CC1=C(CC(=O)C(C)(C)C)C=NC(NC(=O)C(C)(C)C)=C1.CC1=C(N)C=NC(N)=C1.CCN(CC)CC GAAFHALSCLOMFD-UHFFFAOYSA-N 0.000 description 1
- ACFMXPACVGYKPX-UHFFFAOYSA-N CC(C)(C)C(=O)Cl.CCN(CC)CC.CON(C)C(=O)C1=CC=C(N)C=C1.CON(C)C(=O)C1=CC=C(NC(=O)C(C)(C)C)C=C1 Chemical compound CC(C)(C)C(=O)Cl.CCN(CC)CC.CON(C)C(=O)C1=CC=C(N)C=C1.CON(C)C(=O)C1=CC=C(NC(=O)C(C)(C)C)C=C1 ACFMXPACVGYKPX-UHFFFAOYSA-N 0.000 description 1
- WQLXOUJQNOSZCH-UHFFFAOYSA-N CC(C)(C)C(=O)NC1=CC=C(C(=O)CC2=C(NC(=O)C(C)(C)C)C=NC(NC(=O)C(C)(C)C)=C2)C=C1.CC1=C(CC(=O)C(C)(C)C)C=NC(NC(=O)C(C)(C)C)=C1.CON(C)C(=O)C1=CC=C(NC(=O)C(C)(C)C)C=C1.[Li]CCCC Chemical compound CC(C)(C)C(=O)NC1=CC=C(C(=O)CC2=C(NC(=O)C(C)(C)C)C=NC(NC(=O)C(C)(C)C)=C2)C=C1.CC1=C(CC(=O)C(C)(C)C)C=NC(NC(=O)C(C)(C)C)=C1.CON(C)C(=O)C1=CC=C(NC(=O)C(C)(C)C)C=C1.[Li]CCCC WQLXOUJQNOSZCH-UHFFFAOYSA-N 0.000 description 1
- OMEOUSCVVNCEEM-UHFFFAOYSA-N CC(C)(C)C(=O)NC1=CC=C(C(=O)CC2=C(NC(=O)C(C)(C)C)C=NC(NC(=O)C(C)(C)C)=C2)C=C1.NC1=CC=C(C2=CC3=CC(N)=NC=C3N2)C=C1 Chemical compound CC(C)(C)C(=O)NC1=CC=C(C(=O)CC2=C(NC(=O)C(C)(C)C)C=NC(NC(=O)C(C)(C)C)=C2)C=C1.NC1=CC=C(C2=CC3=CC(N)=NC=C3N2)C=C1 OMEOUSCVVNCEEM-UHFFFAOYSA-N 0.000 description 1
- ASIZAGPTVDYEFV-CRCGPULASA-N CC(C)(C)CC(=O)C[C@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](NC(=O)CC(C)(C)C)C4=CC=CC=C4)=CC=C3N2)C=C1)C1=CC=CC=C1 Chemical compound CC(C)(C)CC(=O)C[C@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](NC(=O)CC(C)(C)C)C4=CC=CC=C4)=CC=C3N2)C=C1)C1=CC=CC=C1 ASIZAGPTVDYEFV-CRCGPULASA-N 0.000 description 1
- BKNXBDKBWKUZRJ-CDZUIXILSA-N CC(C)(C)CC(=O)C[C@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C2C(=C1)CCC1=C2NC2=CC=CC=C21)C1=CC=CC=C1 Chemical compound CC(C)(C)CC(=O)C[C@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C2C(=C1)CCC1=C2NC2=CC=CC=C21)C1=CC=CC=C1 BKNXBDKBWKUZRJ-CDZUIXILSA-N 0.000 description 1
- WOYNEPLAOITVRZ-FQGHPVNASA-M CC(C)(C)OC(=O)CC1=CC=C2C(=C1)/C=C(/B(O)O)N2C(=O)OC(C)(C)C.CC(C)(C)OC(=O)CC1=CC=C2C(=C1)/C=C(/C1=CC=C(C3=NN=C([C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)O3)C=C1)N2C(=O)OC(C)(C)C.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NN=C(C2=CC=C(I)C=C2)O1.O=COO[Na].[NaH] Chemical compound CC(C)(C)OC(=O)CC1=CC=C2C(=C1)/C=C(/B(O)O)N2C(=O)OC(C)(C)C.CC(C)(C)OC(=O)CC1=CC=C2C(=C1)/C=C(/C1=CC=C(C3=NN=C([C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)O3)C=C1)N2C(=O)OC(C)(C)C.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NN=C(C2=CC=C(I)C=C2)O1.O=COO[Na].[NaH] WOYNEPLAOITVRZ-FQGHPVNASA-M 0.000 description 1
- GDRQPDLYOIEBAG-NUWOOLOVSA-M CC(C)(C)OC(=O)CC1=CC=C2C(=C1)C=C(B(O)O)N2C(=O)OC(C)(C)C.CC(C)(C)OC(=O)CC1=CC=C2C(=C1)C=C(C1=CC=C(C3=CN=C([C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)N3)C=C1)N2C(=O)OC(C)(C)C.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NC=C(C2=CC=C(Br)C=C2)N1.O=COO[Na].[NaH] Chemical compound CC(C)(C)OC(=O)CC1=CC=C2C(=C1)C=C(B(O)O)N2C(=O)OC(C)(C)C.CC(C)(C)OC(=O)CC1=CC=C2C(=C1)C=C(C1=CC=C(C3=CN=C([C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)N3)C=C1)N2C(=O)OC(C)(C)C.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NC=C(C2=CC=C(Br)C=C2)N1.O=COO[Na].[NaH] GDRQPDLYOIEBAG-NUWOOLOVSA-M 0.000 description 1
- FKDJVVPNHCCTNG-JKOLFZLPSA-N CC(C)(C)OC(=O)CC1=CC=C2C(=C1)C=C(C1=CC=C(C3=CN=C([C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)N3)C=C1)N2C(=O)OC(C)(C)C.Cl.NC1=CC=C2NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)N4)C=C3)=CC2=C1 Chemical compound CC(C)(C)OC(=O)CC1=CC=C2C(=C1)C=C(C1=CC=C(C3=CN=C([C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)N3)C=C1)N2C(=O)OC(C)(C)C.Cl.NC1=CC=C2NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)N4)C=C3)=CC2=C1 FKDJVVPNHCCTNG-JKOLFZLPSA-N 0.000 description 1
- XGAPYWGBZIHDLQ-IZQZQZLFSA-N CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=C/C=C2\NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)CC5=CC=CC=C5)N4)C=C3)=C\C2=C\1)C1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=C/C=C2\NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)CC5=CC=CC=C5)N4)C=C3)=C\C2=C\1)C1=CC=CC=C1 XGAPYWGBZIHDLQ-IZQZQZLFSA-N 0.000 description 1
- NDBXTVCSGRVPHU-AGQPMONNSA-N CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC2=C(C=C1)NC(C1=CC=C(NC(=O)[C@@H]3CCCN3C(=O)[C@H](CC(=O)OC(C)(C)C)C3=CC=CC=C3)C=C1)=C2C#N)C1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC2=C(C=C1)NC(C1=CC=C(NC(=O)[C@@H]3CCCN3C(=O)[C@H](CC(=O)OC(C)(C)C)C3=CC=CC=C3)C=C1)=C2C#N)C1=CC=CC=C1 NDBXTVCSGRVPHU-AGQPMONNSA-N 0.000 description 1
- IUZHTOMPPKXZIL-IXLHQTAOSA-N CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC2=C(C=C1)NC(C1=CC=C(NC(=O)[C@@H]3CCCN3C(=O)[C@H](CC(=O)OC(C)(C)C)C3=CC=CC=C3)C=C1)=C2C(=O)O)C1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC2=C(C=C1)NC(C1=CC=C(NC(=O)[C@@H]3CCCN3C(=O)[C@H](CC(=O)OC(C)(C)C)C3=CC=CC=C3)C=C1)=C2C(=O)O)C1=CC=CC=C1 IUZHTOMPPKXZIL-IXLHQTAOSA-N 0.000 description 1
- CAQCNKMIPQCZSW-OSYFPSQLSA-N CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)C4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)C4)C=C3)=CC2=C1)C1=CC=CC=C1 CAQCNKMIPQCZSW-OSYFPSQLSA-N 0.000 description 1
- RTSHZOWLKULICZ-CPBHLAHYSA-N CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=CNC=N4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=CNC=N4)C=C3)=CC2=C1)C1=CC=CC=C1 RTSHZOWLKULICZ-CPBHLAHYSA-N 0.000 description 1
- XJEHTLZXXOPLDO-MVSFAKPFSA-N CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(N4CCCC4=O)C=C3)=C(F)C2=C1)C1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(N4CCCC4=O)C=C3)=C(F)C2=C1)C1=CC=CC=C1 XJEHTLZXXOPLDO-MVSFAKPFSA-N 0.000 description 1
- GAUMTUGHPAPHIB-IEQLCYGESA-N CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=C(F)C=C4)C=C3)=CC2=C1)C1=CC=C(F)C=C1 Chemical compound CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=C(F)C=C4)C=C3)=CC2=C1)C1=CC=C(F)C=C1 GAUMTUGHPAPHIB-IEQLCYGESA-N 0.000 description 1
- ASCKZYJQXTZVQI-NIPTWYEISA-N CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=C(C3=CC=NC=C3)C2=C1)C1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=C(C3=CC=NC=C3)C2=C1)C1=CC=CC=C1 ASCKZYJQXTZVQI-NIPTWYEISA-N 0.000 description 1
- YJYDXGGQEUQONG-QAUMXGQASA-N CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC3=C(CCCC4=CC(NC(=O)[C@@H]5CCCN5C(=O)[C@H](CC(=O)OC(C)(C)C)C5=CC=CC=C5)=CC=C43)C2=C1)C1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC3=C(CCCC4=CC(NC(=O)[C@@H]5CCCN5C(=O)[C@H](CC(=O)OC(C)(C)C)C5=CC=CC=C5)=CC=C43)C2=C1)C1=CC=CC=C1 YJYDXGGQEUQONG-QAUMXGQASA-N 0.000 description 1
- KGMCTDHYLRBBNA-IASBXJNJSA-N CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=C(Br)C2=C1)C1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=C(Br)C2=C1)C1=CC=CC=C1 KGMCTDHYLRBBNA-IASBXJNJSA-N 0.000 description 1
- KNRGFJZGRUGKDB-PGBQSJHVSA-N CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1 KNRGFJZGRUGKDB-PGBQSJHVSA-N 0.000 description 1
- WYHACQZEWLSSSL-DGPALRBDSA-N CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C2C(=C1)CCC1=C2NC2=CC=CC=C21)C1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C2C(=C1)CCC1=C2NC2=CC=CC=C21)C1=CC=CC=C1 WYHACQZEWLSSSL-DGPALRBDSA-N 0.000 description 1
- GTBPQOBBHPTSLS-VRFVMXNLSA-N CC(C)(C)OC(=O)C[C@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=C(Br)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)=CC=C3O2)C=C1)C1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)C[C@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=C(Br)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)=CC=C3O2)C=C1)C1=CC=CC=C1 GTBPQOBBHPTSLS-VRFVMXNLSA-N 0.000 description 1
- VOQHSUSUXPNXHE-FNXJSGONSA-N CC(C)(C)OC(=O)C[C@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)=CC=C3N2)C=C1)C1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)C[C@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)=CC=C3N2)C=C1)C1=CC=CC=C1 VOQHSUSUXPNXHE-FNXJSGONSA-N 0.000 description 1
- UFQJFHHAAINBBH-FIBWVYCGSA-N CC(C)(C)OC(=O)C[C@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=C(C=CC(C#N)=C3)N2)C=C1)C1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)C[C@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=C(C=CC(C#N)=C3)N2)C=C1)C1=CC=CC=C1 UFQJFHHAAINBBH-FIBWVYCGSA-N 0.000 description 1
- FWOQADHNPVAHTI-AKCYGPDLSA-N CC(C)(C)OC(=O)C[C@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)=CC=C3O2)C=C1)C1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)C[C@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)=CC=C3O2)C=C1)C1=CC=CC=C1 FWOQADHNPVAHTI-AKCYGPDLSA-N 0.000 description 1
- JXRYEAGVLOZRHE-FIBWVYCGSA-N CC(C)(C)OC(=O)C[C@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C2C(=C1)CCC1=C2NC2=CC=C(Br)C=C21)C1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)C[C@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C2C(=C1)CCC1=C2NC2=CC=C(Br)C=C21)C1=CC=CC=C1 JXRYEAGVLOZRHE-FIBWVYCGSA-N 0.000 description 1
- IHFJMEVYIJWFLO-LUEJCCLMSA-N CC(C)(C)OC(=O)C[C@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C2C(=C1)CCC1=C2NC2=CC=C(CC(=O)[C@@H]3CCCN3C(=O)[C@@H](NC(=O)OC(C)(C)C)C3=CC=CC=C3)C=C21)C1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)C[C@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C2C(=C1)CCC1=C2NC2=CC=C(CC(=O)[C@@H]3CCCN3C(=O)[C@@H](NC(=O)OC(C)(C)C)C3=CC=CC=C3)C=C21)C1=CC=CC=C1 IHFJMEVYIJWFLO-LUEJCCLMSA-N 0.000 description 1
- BKQJANGXJXDXBE-WNZKHSNXSA-N CC(C)(C)OC(=O)N(C(=O)OC(C)(C)C)C1=CC=C2CC(C3=CC=C(C4=CC([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)=NN4)C=C3)=CC2=C1.CC1=CC=C2C(=C1)C=C(B(O)O)N2C(=O)OC(C)(C)C.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NNC(C2=CC=C(Br)C=C2)=C1 Chemical compound CC(C)(C)OC(=O)N(C(=O)OC(C)(C)C)C1=CC=C2CC(C3=CC=C(C4=CC([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)=NN4)C=C3)=CC2=C1.CC1=CC=C2C(=C1)C=C(B(O)O)N2C(=O)OC(C)(C)C.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NNC(C2=CC=C(Br)C=C2)=C1 BKQJANGXJXDXBE-WNZKHSNXSA-N 0.000 description 1
- ZQBXCCNJAOBHLJ-ROPRQOIVSA-N CC(C)(C)OC(=O)N(C(=O)OC(C)(C)C)C1=CC=C2CC(C3=CC=C(C4=CC([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)=NN4)C=C3)=CC2=C1.NC1=CC=C2CC(C3=CC=C(C4=CC([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)=NN4)C=C3)=CC2=C1 Chemical compound CC(C)(C)OC(=O)N(C(=O)OC(C)(C)C)C1=CC=C2CC(C3=CC=C(C4=CC([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)=NN4)C=C3)=CC2=C1.NC1=CC=C2CC(C3=CC=C(C4=CC([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)=NN4)C=C3)=CC2=C1 ZQBXCCNJAOBHLJ-ROPRQOIVSA-N 0.000 description 1
- NWBRCLNPTPTJQA-DPMVGXLCSA-N CC(C)(C)OC(=O)N1C(B(O)O)=CC2=CC(C3=NN=C([C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)O3)=CC=C21.CC(C)(C)OC(=O)N1C=CC2=CC(C3=NN=C([C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)O3)=CC=C21 Chemical compound CC(C)(C)OC(=O)N1C(B(O)O)=CC2=CC(C3=NN=C([C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)O3)=CC=C21.CC(C)(C)OC(=O)N1C=CC2=CC(C3=NN=C([C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)O3)=CC=C21 NWBRCLNPTPTJQA-DPMVGXLCSA-N 0.000 description 1
- JFHSRYYLURHLFY-SIQLWQOGSA-N CC(C)(C)OC(=O)N1C=CC2=CC(C3=NN=C([C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)O3)=CC=C21.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NN=C(C2=CC=C3NC=CC3=C2)O1 Chemical compound CC(C)(C)OC(=O)N1C=CC2=CC(C3=NN=C([C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)O3)=CC=C21.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NN=C(C2=CC=C3NC=CC3=C2)O1 JFHSRYYLURHLFY-SIQLWQOGSA-N 0.000 description 1
- VBMLZVIDJHLRFQ-ZQVNUCEZSA-N CC(C)(C)OC(=O)N1CCCC1C(=O)CC1=CC=C2NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)N4)C=C3)=CC2=C1.O=C(CC1=CC=C2NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5)N4)C=C3)=CC2=C1)[C@@H]1CCCN1 Chemical compound CC(C)(C)OC(=O)N1CCCC1C(=O)CC1=CC=C2NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)N4)C=C3)=CC2=C1.O=C(CC1=CC=C2NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5)N4)C=C3)=CC2=C1)[C@@H]1CCCN1 VBMLZVIDJHLRFQ-ZQVNUCEZSA-N 0.000 description 1
- PRAYNNILKWRXEZ-XCDFWKGXSA-M CC(C)(C)OC(=O)N1CCCC1C1=NC=C(C2=CC=C3OC(C4=CC=C(C5=CN=C(C6CCCC6)N5)C=C4)=CC3=C2)N1.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC(Br)=CN1.CC1(C)OB(C2=CC=C(C3=CC4=CC(B5OC(C)(C)C(C)(C)O5)=CC=C4O3)C=C2)OC1(C)C.O=COO[Na].[NaH] Chemical compound CC(C)(C)OC(=O)N1CCCC1C1=NC=C(C2=CC=C3OC(C4=CC=C(C5=CN=C(C6CCCC6)N5)C=C4)=CC3=C2)N1.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC(Br)=CN1.CC1(C)OB(C2=CC=C(C3=CC4=CC(B5OC(C)(C)C(C)(C)O5)=CC=C4O3)C=C2)OC1(C)C.O=COO[Na].[NaH] PRAYNNILKWRXEZ-XCDFWKGXSA-M 0.000 description 1
- YKDZIDAYPJPRIZ-XCDFWKGXSA-M CC(C)(C)OC(=O)N1CCCC1C1=NC=C(C2=CC=C3OC(C4=CC=C(C5=CN=C(C6CCCC6)N5)C=C4)=NC3=C2)N1.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=C(Br)N1.CC1(C)OB(C2=CC=C(C3=NC4=CC(B5OC(C)(C)C(C)(C)O5)=CC=C4O3)C=C2)OC1(C)C.O=COO[Na].[NaH] Chemical compound CC(C)(C)OC(=O)N1CCCC1C1=NC=C(C2=CC=C3OC(C4=CC=C(C5=CN=C(C6CCCC6)N5)C=C4)=NC3=C2)N1.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=C(Br)N1.CC1(C)OB(C2=CC=C(C3=NC4=CC(B5OC(C)(C)C(C)(C)O5)=CC=C4O3)C=C2)OC1(C)C.O=COO[Na].[NaH] YKDZIDAYPJPRIZ-XCDFWKGXSA-M 0.000 description 1
- IFDWUTIJBQHZRT-RPEJEGKGSA-M CC(C)(C)OC(=O)N1CCCC1C1=NC=C(C2=CC=C3OC(C4=CC=C(C5=CNC([C@@H]6CCCN6C(=O)OC(C)(C)C)=N5)C(F)=C4)=CC3=C2)N1.CC1(C)OB(C2=CC=C3OC(C4=CC=C(B5OC(C)(C)C(C)(C)O5)C(F)=C4)=CC3=C2)OC1(C)C.O=COO[Na].[NaH] Chemical compound CC(C)(C)OC(=O)N1CCCC1C1=NC=C(C2=CC=C3OC(C4=CC=C(C5=CNC([C@@H]6CCCN6C(=O)OC(C)(C)C)=N5)C(F)=C4)=CC3=C2)N1.CC1(C)OB(C2=CC=C3OC(C4=CC=C(B5OC(C)(C)C(C)(C)O5)C(F)=C4)=CC3=C2)OC1(C)C.O=COO[Na].[NaH] IFDWUTIJBQHZRT-RPEJEGKGSA-M 0.000 description 1
- VAJKMFBMBCIZOA-HXUJKBDWSA-N CC(C)(C)OC(=O)N1CCCC1C1=NC=C(C2=CC=C3OC(C4=CC=C(C5=CNC([C@@H]6CCCN6C(=O)OC(C)(C)C)=N5)C(F)=C4)=CC3=C2)N1.FC1=CC(C2=CC3=CC(C4=CN=C(C5CCCN5)N4)=CC=C3O2)=CC=C1C1=CNC([C@@H]2CCCN2)=N1 Chemical compound CC(C)(C)OC(=O)N1CCCC1C1=NC=C(C2=CC=C3OC(C4=CC=C(C5=CNC([C@@H]6CCCN6C(=O)OC(C)(C)C)=N5)C(F)=C4)=CC3=C2)N1.FC1=CC(C2=CC3=CC(C4=CN=C(C5CCCN5)N4)=CC=C3O2)=CC=C1C1=CNC([C@@H]2CCCN2)=N1 VAJKMFBMBCIZOA-HXUJKBDWSA-N 0.000 description 1
- ISNWMULVSXGXMQ-JQRFZINPSA-N CC(C)(C)OC(=O)N1CCC[C@@H]1C(=O)O.CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)CC1=CC=C(C2=NC3=CC(CC(=O)[C@@H]4CCCN4C(=O)OC(C)(C)C)=CC=C3O2)C=C1.NC1=CC=C(C2=NC3=CC(N)=CC=C3O2)C=C1 Chemical compound CC(C)(C)OC(=O)N1CCC[C@@H]1C(=O)O.CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)CC1=CC=C(C2=NC3=CC(CC(=O)[C@@H]4CCCN4C(=O)OC(C)(C)C)=CC=C3O2)C=C1.NC1=CC=C(C2=NC3=CC(N)=CC=C3O2)C=C1 ISNWMULVSXGXMQ-JQRFZINPSA-N 0.000 description 1
- QWZFQBXFOHUNLJ-PBZDVIRISA-N CC(C)(C)OC(=O)N1CCC[C@@H]1C(=O)O.CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=NC3=C/C([N+](=O)[O-])=C\C=C\3N2)C=C1.NC1=CC=C(C2=NC3=CC([N+](=O)[O-])=CC=C3N2)C=C1 Chemical compound CC(C)(C)OC(=O)N1CCC[C@@H]1C(=O)O.CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=NC3=C/C([N+](=O)[O-])=C\C=C\3N2)C=C1.NC1=CC=C(C2=NC3=CC([N+](=O)[O-])=CC=C3N2)C=C1 QWZFQBXFOHUNLJ-PBZDVIRISA-N 0.000 description 1
- GJCLJJLHWIGKLE-QQKCEYOOSA-N CC(C)(C)OC(=O)N1CCC[C@@H]1C(=O)O.CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)NC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)C4=CC=CN=C4)C=C3)=NC2=C1.NC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)C4=CC=CN=C4)C=C3)=NC2=C1 Chemical compound CC(C)(C)OC(=O)N1CCC[C@@H]1C(=O)O.CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)NC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)C4=CC=CN=C4)C=C3)=NC2=C1.NC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)C4=CC=CN=C4)C=C3)=NC2=C1 GJCLJJLHWIGKLE-QQKCEYOOSA-N 0.000 description 1
- FXBQFWIVAIXFDY-NCBSXXLDSA-N CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)CC1=CC=C(C2=NC3=C(N)C=CC=C3N2)C=C1.CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)CC1=CC=C(C2=NC3=C([N+](=O)[O-])C=CC=C3N2)C=C1 Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)CC1=CC=C(C2=NC3=C(N)C=CC=C3N2)C=C1.CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)CC1=CC=C(C2=NC3=C([N+](=O)[O-])C=CC=C3N2)C=C1 FXBQFWIVAIXFDY-NCBSXXLDSA-N 0.000 description 1
- BRJVBTZAVNFVSR-PIWSJUAYSA-N CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)CC1=CC=C(C2=NC3=C(N)C=CC=C3N2)C=C1.CC1=CC=C(S(=O)(=O)Cl)C=C1.CC1=CC=C(S(=O)(=O)NC2=C3N=C(C4=CC=C(NC(=O)[C@@H]5CCCN5C(=O)OC(C)(C)C)C=C4)NC3=CC=C2)C=C1 Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)CC1=CC=C(C2=NC3=C(N)C=CC=C3N2)C=C1.CC1=CC=C(S(=O)(=O)Cl)C=C1.CC1=CC=C(S(=O)(=O)NC2=C3N=C(C4=CC=C(NC(=O)[C@@H]5CCCN5C(=O)OC(C)(C)C)C=C4)NC3=CC=C2)C=C1 BRJVBTZAVNFVSR-PIWSJUAYSA-N 0.000 description 1
- AWBFRDYEXPQEBF-FERBBOLQSA-N CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)CC1=CC=C(C2=NC3=C([N+](=O)[O-])C=CC=C3N2)C=C1.NC1=CC=C(C2=NC3=C([N+](=O)[O-])C=CC=C3N2)C=C1 Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)CC1=CC=C(C2=NC3=C([N+](=O)[O-])C=CC=C3N2)C=C1.NC1=CC=C(C2=NC3=C([N+](=O)[O-])C=CC=C3N2)C=C1 AWBFRDYEXPQEBF-FERBBOLQSA-N 0.000 description 1
- DBGJMKXSLOOHCI-NJUMWCQESA-N CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)CC1=CC=C(C2=NC3=CC(CC(=O)[C@@H]4CCCN4C(=O)OC(C)(C)C)=CC=C3O2)C=C1.O=C(CC1=CC=C(C2=NC3=CC(CC(=O)[C@@H]4CCCN4)=CC=C3O2)C=C1)[C@@H]1CCCN1 Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)CC1=CC=C(C2=NC3=CC(CC(=O)[C@@H]4CCCN4C(=O)OC(C)(C)C)=CC=C3O2)C=C1.O=C(CC1=CC=C(C2=NC3=CC(CC(=O)[C@@H]4CCCN4)=CC=C3O2)C=C1)[C@@H]1CCCN1 DBGJMKXSLOOHCI-NJUMWCQESA-N 0.000 description 1
- DRUHUDPVBMBGLE-LUDRZRIFSA-N CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C4NC([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)=NC4=C3)=CC2=C1.CC(C)(C)OC=O.CC1=CC=C2C(=C1)C=C(C1=CC=C3NC([C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)=NC3=C1)N2C(=O)OC(C)(C)C.CC1=CC=C2NC(B(O)O)=CC2=C1.NC1=C(CC(=O)[C@@H]2CCCN2C(=O)OCC2=CC=CC=C2)C=C(Br)C=C1.NC1=C(N)C=C(Br)C=C1.NC1=CC=C2NC(C3=CC=C4NC([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)=NC4=C3)=CC2=C1.O=C(O)C1CCCN1C(=O)OCC1=CC=CC=C1.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NC2=CC(Br)=CC=C2N1.[Pd] Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C4NC([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)=NC4=C3)=CC2=C1.CC(C)(C)OC=O.CC1=CC=C2C(=C1)C=C(C1=CC=C3NC([C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)=NC3=C1)N2C(=O)OC(C)(C)C.CC1=CC=C2NC(B(O)O)=CC2=C1.NC1=C(CC(=O)[C@@H]2CCCN2C(=O)OCC2=CC=CC=C2)C=C(Br)C=C1.NC1=C(N)C=C(Br)C=C1.NC1=CC=C2NC(C3=CC=C4NC([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)=NC4=C3)=CC2=C1.O=C(O)C1CCCN1C(=O)OCC1=CC=CC=C1.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NC2=CC(Br)=CC=C2N1.[Pd] DRUHUDPVBMBGLE-LUDRZRIFSA-N 0.000 description 1
- MIJCOMZDCIEQGR-MSIAAWJQSA-N CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C4NC([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)=NC4=C3)=CC2=C1.O=C(CC1=CC=C2NC(C3=CC=C4NC([C@@H]5CCCN5)=NC4=C3)=CC2=C1)[C@@H]1CCCN1 Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C4NC([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)=NC4=C3)=CC2=C1.O=C(CC1=CC=C2NC(C3=CC=C4NC([C@@H]5CCCN5)=NC4=C3)=CC2=C1)[C@@H]1CCCN1 MIJCOMZDCIEQGR-MSIAAWJQSA-N 0.000 description 1
- ODQTUOOZEKHUFF-KHXHURTPSA-N CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)OC(C)(C)C)C=C3)=CC2=C1.CCl.O=C(CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4)C=C3)=CC2=C1)[C@@H]1CCCN1.O=C(CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CCC4=CC=CC=C4)C=C3)=CC2=C1)[C@@H]1CCCN1C(=O)CCC1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)OC(C)(C)C)C=C3)=CC2=C1.CCl.O=C(CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4)C=C3)=CC2=C1)[C@@H]1CCCN1.O=C(CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CCC4=CC=CC=C4)C=C3)=CC2=C1)[C@@H]1CCCN1C(=O)CCC1=CC=CC=C1 ODQTUOOZEKHUFF-KHXHURTPSA-N 0.000 description 1
- CTLCDPLSQRUKIB-WMXJXTQLSA-N CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)OC(C)(C)C)C=C3)=CC2=C1.NC1=CC=C(C2=CC3=CC(N)=CC=C3O2)C=C1 Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)OC(C)(C)C)C=C3)=CC2=C1.NC1=CC=C(C2=CC3=CC(N)=CC=C3O2)C=C1 CTLCDPLSQRUKIB-WMXJXTQLSA-N 0.000 description 1
- FFXGBBMLHKIIRI-JHUGDEDASA-N CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=CC(C4=CN=C([C@@H]5CCCN5C(=O)OC(C)(C)C)N4)=CC=C3O2)C=C1.O=C(NC1=CC=C(C2=CC3=CC(C4=CN=C([C@@H]5CCCN5)N4)=CC=C3O2)C=C1)[C@@H]1CCCN1 Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=CC(C4=CN=C([C@@H]5CCCN5C(=O)OC(C)(C)C)N4)=CC=C3O2)C=C1.O=C(NC1=CC=C(C2=CC3=CC(C4=CN=C([C@@H]5CCCN5)N4)=CC=C3O2)C=C1)[C@@H]1CCCN1 FFXGBBMLHKIIRI-JHUGDEDASA-N 0.000 description 1
- AVDUGNNRMLNDRM-JLKOHUAHSA-N CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=NC3=CC([N+](=O)[O-])=CC=C3N2)C=C1.Cl.O=C(NC1=CC=C(/C2=N/C3=CC([N+](=O)[O-])=CC=C3N2)C=C1)[C@@H]1CCCN1 Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=NC3=CC([N+](=O)[O-])=CC=C3N2)C=C1.Cl.O=C(NC1=CC=C(/C2=N/C3=CC([N+](=O)[O-])=CC=C3N2)C=C1)[C@@H]1CCCN1 AVDUGNNRMLNDRM-JLKOHUAHSA-N 0.000 description 1
- STVOMVUVANWVPC-YOCDZIIGSA-N CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)NC1=CC=C2CC(C3=CC=C(C4=CC([C@@H]5CCCN5)=NN4)C=C3)=CC2=C1.CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)NC1=CC=C2CC(C3=CC=C(C4=CC([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)=NN4)C=C3)=CC2=C1.S Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)NC1=CC=C2CC(C3=CC=C(C4=CC([C@@H]5CCCN5)=NN4)C=C3)=CC2=C1.CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)NC1=CC=C2CC(C3=CC=C(C4=CC([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)=NN4)C=C3)=CC2=C1.S STVOMVUVANWVPC-YOCDZIIGSA-N 0.000 description 1
- XDPQILYWIJSYSD-ZZJLZXDWSA-N CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)NC1=CC=C2CC(C3=CC=C(C4=CC([C@@H]5CCCN5)=NN4)C=C3)=CC2=C1.O=C(NC1=CC=C2CC(C3=CC=C(C4=CC([C@@H]5CCCN5)=NN4)C=C3)=CC2=C1)[C@@H]1CCCN1 Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)NC1=CC=C2CC(C3=CC=C(C4=CC([C@@H]5CCCN5)=NN4)C=C3)=CC2=C1.O=C(NC1=CC=C2CC(C3=CC=C(C4=CC([C@@H]5CCCN5)=NN4)C=C3)=CC2=C1)[C@@H]1CCCN1 XDPQILYWIJSYSD-ZZJLZXDWSA-N 0.000 description 1
- ZFLBROKEFVFPDR-LWCRKSNFSA-N CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)NC1=CC=C2CC(C3=CC=C(C4=CC([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)=NN4)C=C3)=CC2=C1.CN.NC1=CC=C2CC(C3=CC=C(C4=CC([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)=NN4)C=C3)=CC2=C1 Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)NC1=CC=C2CC(C3=CC=C(C4=CC([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)=NN4)C=C3)=CC2=C1.CN.NC1=CC=C2CC(C3=CC=C(C4=CC([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)=NN4)C=C3)=CC2=C1 ZFLBROKEFVFPDR-LWCRKSNFSA-N 0.000 description 1
- KTZHWJZXNNLLAF-PJVXWQQVSA-N CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)NC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)C4=CC=CN=C4)C=C3)=NC2=C1.O=C(NC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)C4=CC=CN=C4)C=C3)=NC2=C1)[C@@H]1CCCN1 Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)NC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)C4=CC=CN=C4)C=C3)=NC2=C1.O=C(NC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)C4=CC=CN=C4)C=C3)=NC2=C1)[C@@H]1CCCN1 KTZHWJZXNNLLAF-PJVXWQQVSA-N 0.000 description 1
- PDDCKVYTJUOORZ-YMHVJCOXSA-N CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC(Br)=C(Br)N1.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=C(Br)N1.O=S(=O)([Na])O[Na] Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC(Br)=C(Br)N1.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=C(Br)N1.O=S(=O)([Na])O[Na] PDDCKVYTJUOORZ-YMHVJCOXSA-N 0.000 description 1
- CWFNKXCDKPOMRT-TXRYEIFUSA-M CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=C(Br)N1.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3OC(C4=CC=C([N+](=O)[O-])C=C4)=CC3=C2)N1.CC1(C)OB(C2=CC=C3OC(C4=CC=C([N+](=O)[O-])C=C4)=CC3=C2)OC1(C)C.O=COO[Na].[NaH] Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=C(Br)N1.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3OC(C4=CC=C([N+](=O)[O-])C=C4)=CC3=C2)N1.CC1(C)OB(C2=CC=C3OC(C4=CC=C([N+](=O)[O-])C=C4)=CC3=C2)OC1(C)C.O=COO[Na].[NaH] CWFNKXCDKPOMRT-TXRYEIFUSA-M 0.000 description 1
- OBBMPBRBSJBJLE-LIDTYJJYSA-N CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3C(=C2)C(F)=C2C4=C(C=C(C5=CN=C([C@@H]6CCCN6C(=O)OC(C)(C)C)N5)C=C4)OCN32)N1.Cl.FC1=C2C3=C(C=C(C4=CN=C([C@@H]5CCCN5)N4)C=C3)OCN2C2=CC=C(C3=CN=C([C@@H]4CCCN4)N3)C=C21 Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3C(=C2)C(F)=C2C4=C(C=C(C5=CN=C([C@@H]6CCCN6C(=O)OC(C)(C)C)N5)C=C4)OCN32)N1.Cl.FC1=C2C3=C(C=C(C4=CN=C([C@@H]5CCCN5)N4)C=C3)OCN2C2=CC=C(C3=CN=C([C@@H]4CCCN4)N3)C=C21 OBBMPBRBSJBJLE-LIDTYJJYSA-N 0.000 description 1
- ZVCUTGFXFGWZCS-HRQBZJEISA-N CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3OC(C4=CC=C(N)C=C4)=CC3=C2)N1.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3OC(C4=CC=C([N+](=O)[O-])C=C4)=CC3=C2)N1.[HH] Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3OC(C4=CC=C(N)C=C4)=CC3=C2)N1.CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3OC(C4=CC=C([N+](=O)[O-])C=C4)=CC3=C2)N1.[HH] ZVCUTGFXFGWZCS-HRQBZJEISA-N 0.000 description 1
- KAWDSIBHMVOPMB-IWXSALQQSA-N CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3OC(C4=CC=C(NC(=O)[C@@H]5CCCN5)C=C4)=CC3=C2)N1.CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=CC(C4=CN=C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)N4)=CC=C3O2)C=C1)C1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3OC(C4=CC=C(NC(=O)[C@@H]5CCCN5)C=C4)=CC3=C2)N1.CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=CC(C4=CN=C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)N4)=CC=C3O2)C=C1)C1=CC=CC=C1 KAWDSIBHMVOPMB-IWXSALQQSA-N 0.000 description 1
- WTINZEXNOCIMRZ-GRHHLOCNSA-N CC(C)(C)OC(=O)N1CCC[C@H]1CO.[H]C(=O)[C@@H]1CCCN1C(=O)OC(C)(C)C Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1CO.[H]C(=O)[C@@H]1CCCN1C(=O)OC(C)(C)C WTINZEXNOCIMRZ-GRHHLOCNSA-N 0.000 description 1
- WUNPTYXVJPDTLG-UHFFFAOYSA-N CC(C)(C)OC(=O)NC1=CC2=C(C=C1)N(C(=O)OC(C)(C)C)C(C1=CC3=CC([N+](=O)[O-])=CC=C3N1)=C2.CClO.NC1=CC2=C(C=C1)N/C(C1=CC3=CC([N+](=O)[O-])=CC=C3N1)=C\2 Chemical compound CC(C)(C)OC(=O)NC1=CC2=C(C=C1)N(C(=O)OC(C)(C)C)C(C1=CC3=CC([N+](=O)[O-])=CC=C3N1)=C2.CClO.NC1=CC2=C(C=C1)N/C(C1=CC3=CC([N+](=O)[O-])=CC=C3N1)=C\2 WUNPTYXVJPDTLG-UHFFFAOYSA-N 0.000 description 1
- HNSNPJPTDMHUOP-UHFFFAOYSA-M CC(C)(C)OC(=O)NC1=CC=C(B(O)O)C=C1.O=COO[Na].[H]C(=O)C1=C(Br)N(C(=O)OC(C)(C)C)C2=C1C=C([N+](=O)[O-])C=C2.[H]C(=O)C1=C(C2=CC=C(NC(=O)OC(C)(C)C)C=C2)N(C(=O)OC(C)(C)C)C2=C1C=C([N+](=O)[O-])C=C2.[NaH] Chemical compound CC(C)(C)OC(=O)NC1=CC=C(B(O)O)C=C1.O=COO[Na].[H]C(=O)C1=C(Br)N(C(=O)OC(C)(C)C)C2=C1C=C([N+](=O)[O-])C=C2.[H]C(=O)C1=C(C2=CC=C(NC(=O)OC(C)(C)C)C=C2)N(C(=O)OC(C)(C)C)C2=C1C=C([N+](=O)[O-])C=C2.[NaH] HNSNPJPTDMHUOP-UHFFFAOYSA-M 0.000 description 1
- GJAWIKNJCKRLMI-UHFFFAOYSA-N CC(C)(C)OC(=O)NC1=CC=C(C2=C(C(=O)NC3CC3)C3=C(C=CC(N)=C3)N2C(=O)OC(C)(C)C)C=C1.CC(C)(C)OC(=O)NC1=CC=C(C2=C(C(=O)NC3CC3)C3=C(C=CC([N+](=O)[O-])=C3)N2C(=O)OC(C)(C)C)C=C1.[HH] Chemical compound CC(C)(C)OC(=O)NC1=CC=C(C2=C(C(=O)NC3CC3)C3=C(C=CC(N)=C3)N2C(=O)OC(C)(C)C)C=C1.CC(C)(C)OC(=O)NC1=CC=C(C2=C(C(=O)NC3CC3)C3=C(C=CC([N+](=O)[O-])=C3)N2C(=O)OC(C)(C)C)C=C1.[HH] GJAWIKNJCKRLMI-UHFFFAOYSA-N 0.000 description 1
- AFQFJNPBIFKAHG-UHFFFAOYSA-N CC(C)(C)OC(=O)NC1=CC=C(C2=C(C(=O)NC3CC3)C3=C(C=CC(N)=C3)N2C(=O)OC(C)(C)C)C=C1.NC1=CC=C(C2=C(C(=O)NC3CC3)C3=C(C=CC(N)=C3)N2)C=C1 Chemical compound CC(C)(C)OC(=O)NC1=CC=C(C2=C(C(=O)NC3CC3)C3=C(C=CC(N)=C3)N2C(=O)OC(C)(C)C)C=C1.NC1=CC=C(C2=C(C(=O)NC3CC3)C3=C(C=CC(N)=C3)N2)C=C1 AFQFJNPBIFKAHG-UHFFFAOYSA-N 0.000 description 1
- XQWDFPRRTAPEFI-UHFFFAOYSA-N CC(C)(C)OC(=O)NC1=CC=C(C2=C(C(=O)NC3CC3)C3=C(C=CC([N+](=O)[O-])=C3)N2C(=O)OC(C)(C)C)C=C1.CC(C)(C)OC(=O)NC1=CC=C(C2=C(C(=O)O)C3=C(C=CC([N+](=O)[O-])=C3)N2C(=O)OC(C)(C)C)C=C1.NC1CC1 Chemical compound CC(C)(C)OC(=O)NC1=CC=C(C2=C(C(=O)NC3CC3)C3=C(C=CC([N+](=O)[O-])=C3)N2C(=O)OC(C)(C)C)C=C1.CC(C)(C)OC(=O)NC1=CC=C(C2=C(C(=O)O)C3=C(C=CC([N+](=O)[O-])=C3)N2C(=O)OC(C)(C)C)C=C1.NC1CC1 XQWDFPRRTAPEFI-UHFFFAOYSA-N 0.000 description 1
- RSTITACSAZNBDH-UHFFFAOYSA-N CC(C)(C)OC(=O)NC1=CC=C(C2=C(C(=O)O)C3=C(C=CC([N+](=O)[O-])=C3)N2C(=O)OC(C)(C)C)C=C1.[H]C(=O)C1=C(C2=CC=C(NC(=O)OC(C)(C)C)C=C2)N(C(=O)OC(C)(C)C)C2=C1C=C([N+](=O)[O-])C=C2 Chemical compound CC(C)(C)OC(=O)NC1=CC=C(C2=C(C(=O)O)C3=C(C=CC([N+](=O)[O-])=C3)N2C(=O)OC(C)(C)C)C=C1.[H]C(=O)C1=C(C2=CC=C(NC(=O)OC(C)(C)C)C=C2)N(C(=O)OC(C)(C)C)C2=C1C=C([N+](=O)[O-])C=C2 RSTITACSAZNBDH-UHFFFAOYSA-N 0.000 description 1
- JPDOGHHDCVQUMC-WPWPIJCXSA-N CC(C)(C)OC(=O)N[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=NC2=C1)C1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)N[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=NC2=C1)C1=CC=CC=C1 JPDOGHHDCVQUMC-WPWPIJCXSA-N 0.000 description 1
- JPDOGHHDCVQUMC-ZSMOYPKZSA-N CC(C)(C)OC(=O)N[C@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=NC2=C1)C1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)N[C@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=NC2=C1)C1=CC=CC=C1 JPDOGHHDCVQUMC-ZSMOYPKZSA-N 0.000 description 1
- FGSZHAWOAJVUSN-BDYUSTAISA-N CC(C)(C)OC(=O)N[C@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=NC(Br)=CC=C3N2)C=C1)C1=CC=CC=C1 Chemical compound CC(C)(C)OC(=O)N[C@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=NC(Br)=CC=C3N2)C=C1)C1=CC=CC=C1 FGSZHAWOAJVUSN-BDYUSTAISA-N 0.000 description 1
- NASVXXXLNAYRLS-UHFFFAOYSA-N CC(C)(C)OC(=O)OC(=O)OC(C)(C)C.[H]C(=O)C1=C(Br)N(C(=O)OC(C)(C)C)C2=C1C=C([N+](=O)[O-])C=C2.[H]C(=O)C1=C(Br)NC2=C1C=C([N+](=O)[O-])C=C2 Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C.[H]C(=O)C1=C(Br)N(C(=O)OC(C)(C)C)C2=C1C=C([N+](=O)[O-])C=C2.[H]C(=O)C1=C(Br)NC2=C1C=C([N+](=O)[O-])C=C2 NASVXXXLNAYRLS-UHFFFAOYSA-N 0.000 description 1
- MUSCXGSEFIKSKV-DQEYMECFSA-N CC(C)(C)OC(N(CCC1)[C@@H]1C(Nc(cc1)ccc1-c1nc2cc(NC([C@H](CCC3)N3C(OC(C)(C)C)=O)=O)ccc2[o]1)=O)=O Chemical compound CC(C)(C)OC(N(CCC1)[C@@H]1C(Nc(cc1)ccc1-c1nc2cc(NC([C@H](CCC3)N3C(OC(C)(C)C)=O)=O)ccc2[o]1)=O)=O MUSCXGSEFIKSKV-DQEYMECFSA-N 0.000 description 1
- KRFASGQNBIEBNB-ZSXSBBPPSA-N CC(C)(C)OC(N(CCC1)[C@@H]1c1nc(-c(ccc(-c2cc3cc(-c4cnc(C(CCC5)N5C(OC(C)(C)C)=O)[nH]4)ccc3[o]2)c2)c2F)c[nH]1)=O Chemical compound CC(C)(C)OC(N(CCC1)[C@@H]1c1nc(-c(ccc(-c2cc3cc(-c4cnc(C(CCC5)N5C(OC(C)(C)C)=O)[nH]4)ccc3[o]2)c2)c2F)c[nH]1)=O KRFASGQNBIEBNB-ZSXSBBPPSA-N 0.000 description 1
- ZQEBQGAAWMOMAI-SSDOTTSWSA-N CC(C)(C)OC(N(CCC1)[C@H]1C(O)=O)=O Chemical compound CC(C)(C)OC(N(CCC1)[C@H]1C(O)=O)=O ZQEBQGAAWMOMAI-SSDOTTSWSA-N 0.000 description 1
- DIQKICKSJXHDFL-YZBUDRBASA-N CC(C)(C)OC(N[C@@H](C(N(CCC1)[C@@H]1C(Nc(cc1)cc2c1[nH]c(-c(cc1)ccc1-c1cnc([C@H](CCC3)N3C(OCc3ccccc3)=O)[nH]1)c2)=O)=O)c1ccccc1)=O Chemical compound CC(C)(C)OC(N[C@@H](C(N(CCC1)[C@@H]1C(Nc(cc1)cc2c1[nH]c(-c(cc1)ccc1-c1cnc([C@H](CCC3)N3C(OCc3ccccc3)=O)[nH]1)c2)=O)=O)c1ccccc1)=O DIQKICKSJXHDFL-YZBUDRBASA-N 0.000 description 1
- VLTILDQPMYQBPI-RRPNLBNLSA-N CC(C)(C)OC(N[C@@H](C(N(CCC1)[C@@H]1C(Nc(cc1)cc2c1[nH]c(C#CCNC(C)=O)c2)=O)=O)c1ccccc1)=O Chemical compound CC(C)(C)OC(N[C@@H](C(N(CCC1)[C@@H]1C(Nc(cc1)cc2c1[nH]c(C#CCNC(C)=O)c2)=O)=O)c1ccccc1)=O VLTILDQPMYQBPI-RRPNLBNLSA-N 0.000 description 1
- NRKMCRIVYQUTLY-JPYDVTDNSA-N CC(C)(C)OC(N[C@@H](C(N(CCC1)[C@@H]1C(Nc(cc1)ccc1-c1cc2cc(NC([C@H](CCC3)N3C([C@@H](c3ccccc3)NC)=O)=O)ccc2[nH]1)=O)=O)c1ccccc1)=O Chemical compound CC(C)(C)OC(N[C@@H](C(N(CCC1)[C@@H]1C(Nc(cc1)ccc1-c1cc2cc(NC([C@H](CCC3)N3C([C@@H](c3ccccc3)NC)=O)=O)ccc2[nH]1)=O)=O)c1ccccc1)=O NRKMCRIVYQUTLY-JPYDVTDNSA-N 0.000 description 1
- NJQLGSSKALZLQU-JPYDVTDNSA-N CC(C)(C)OC(N[C@@H](C(N(CCC1)[C@@H]1C(Nc(cc1)ccc1-c1nc2cc(NC([C@H](CCC3)N3C([C@@H](c3ccccc3)NC(OC(C)(C)C)=O)=O)=O)ccc2[nH]1)=O)=O)c1ccccc1)=O Chemical compound CC(C)(C)OC(N[C@@H](C(N(CCC1)[C@@H]1C(Nc(cc1)ccc1-c1nc2cc(NC([C@H](CCC3)N3C([C@@H](c3ccccc3)NC(OC(C)(C)C)=O)=O)=O)ccc2[nH]1)=O)=O)c1ccccc1)=O NJQLGSSKALZLQU-JPYDVTDNSA-N 0.000 description 1
- ZXZCPOSQEZRHHF-MXGQMUSSSA-N CC(C)(C)OC(N[C@H](C(N(CCC1)[C@@H]1C(Nc1ccc2[nH]c(-c(cc3)c(CC4)cc3NC([C@H](CCC3)N3C([C@H](c3ccccc3)NC(OC(C)(C)C)=O)=O)=O)c4c2c1)=O)=O)c1ccccc1)=O Chemical compound CC(C)(C)OC(N[C@H](C(N(CCC1)[C@@H]1C(Nc1ccc2[nH]c(-c(cc3)c(CC4)cc3NC([C@H](CCC3)N3C([C@H](c3ccccc3)NC(OC(C)(C)C)=O)=O)=O)c4c2c1)=O)=O)c1ccccc1)=O ZXZCPOSQEZRHHF-MXGQMUSSSA-N 0.000 description 1
- UBSWKSRUZBLBRG-UHFFFAOYSA-N CC(C)(C)OC=O.CC1=CC=C2C(=C1)/C=C(C1=N/C3=C(C=CC([N+](=O)[O-])=C3)O/1)\N2C(=O)OC(C)(C)C.CC1=CC=C2NC(C)=CC2=C1.O=[N+]([O-])C1=CC2=C(C=C1)OC(Br)=N2.[Pd] Chemical compound CC(C)(C)OC=O.CC1=CC=C2C(=C1)/C=C(C1=N/C3=C(C=CC([N+](=O)[O-])=C3)O/1)\N2C(=O)OC(C)(C)C.CC1=CC=C2NC(C)=CC2=C1.O=[N+]([O-])C1=CC2=C(C=C1)OC(Br)=N2.[Pd] UBSWKSRUZBLBRG-UHFFFAOYSA-N 0.000 description 1
- KEYKPTXSYMZBQJ-DEWSGHJGSA-N CC(C)CC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)CC(C)C)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound CC(C)CC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)CC(C)C)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1 KEYKPTXSYMZBQJ-DEWSGHJGSA-N 0.000 description 1
- VIFFZNFZPWJPKL-AVEBSUOGSA-N CC(C)CCN(C)[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(C)CCC(C)C)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound CC(C)CCN(C)[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(C)CCC(C)C)C=C3)=CC2=C1)C1=CC=CC=C1 VIFFZNFZPWJPKL-AVEBSUOGSA-N 0.000 description 1
- DOQJQWHULLRQQY-FTJBHMTQSA-N CC(C)C[C@H](C(N(CCC1)[C@@H]1C(Nc(cc1)cc2c1[nH]c(-c(cc1)ccc1NC(C)=O)c2F)=O)=O)NC(OC(C)(C)C)=O Chemical compound CC(C)C[C@H](C(N(CCC1)[C@@H]1C(Nc(cc1)cc2c1[nH]c(-c(cc1)ccc1NC(C)=O)c2F)=O)=O)NC(OC(C)(C)C)=O DOQJQWHULLRQQY-FTJBHMTQSA-N 0.000 description 1
- JFQVYOBGWKJUFK-QHYNXHOBSA-N CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=C/C=C2\NC(C3=CC=C(C4=CN(C)C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)=N4)C=C3)=C\C2=C\1)C1=CC=CC=C1 Chemical compound CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=C/C=C2\NC(C3=CC=C(C4=CN(C)C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)=N4)C=C3)=C\C2=C\1)C1=CC=CC=C1 JFQVYOBGWKJUFK-QHYNXHOBSA-N 0.000 description 1
- OJCMAHYTMCMWPO-XUTPHSDNSA-N CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=C/C=C2\NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)N4)C=C3)=C\C2=C\1)C1=CC=CC=C1 Chemical compound CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=C/C=C2\NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)N4)C=C3)=C\C2=C\1)C1=CC=CC=C1 OJCMAHYTMCMWPO-XUTPHSDNSA-N 0.000 description 1
- XVRBRRLRHFSCDY-IZQZQZLFSA-N CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)N4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)N4)C=C3)=CC2=C1)C1=CC=CC=C1 XVRBRRLRHFSCDY-IZQZQZLFSA-N 0.000 description 1
- JDZBDZGGQMFNBI-IASBXJNJSA-N CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)C)C4=CC=CC=C4)C=C3)=C(Br)C2=C1)C1=CC=CC=C1 Chemical compound CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)C)C4=CC=CC=C4)C=C3)=C(Br)C2=C1)C1=CC=CC=C1 JDZBDZGGQMFNBI-IASBXJNJSA-N 0.000 description 1
- AIIIYNPPYKUBEF-PGBQSJHVSA-N CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)C)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)C)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1 AIIIYNPPYKUBEF-PGBQSJHVSA-N 0.000 description 1
- CMABHOLWDOKBMG-ZMAQEQNXSA-N CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)OC(C)(C)C)=CC=C3N2)C=C1)C1=CC=CC=C1 Chemical compound CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)OC(C)(C)C)=CC=C3N2)C=C1)C1=CC=CC=C1 CMABHOLWDOKBMG-ZMAQEQNXSA-N 0.000 description 1
- YHGJHXVKHIGPSI-ZJBJNFCASA-N CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(C)C)=CC=C3N2)C=C1)C1=CC=CC=C1 Chemical compound CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=C(F)C3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(C)C)=CC=C3N2)C=C1)C1=CC=CC=C1 YHGJHXVKHIGPSI-ZJBJNFCASA-N 0.000 description 1
- BMYDTTVVAKHBDV-CHFQRSEYSA-N CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=CC(C4=CN=C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)N4)=CC=C3O2)C=C1)C1=CC=CC=C1 Chemical compound CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=CC(C4=CN=C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)N4)=CC=C3O2)C=C1)C1=CC=CC=C1 BMYDTTVVAKHBDV-CHFQRSEYSA-N 0.000 description 1
- QXMUHZZXNWHMJW-KHTUJTCYSA-N CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=CC(C4=CNC([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)=N4)=CC=C3N2)C=C1)C1=CC=CC=C1 Chemical compound CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=CC(C4=CNC([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)=N4)=CC=C3N2)C=C1)C1=CC=CC=C1 QXMUHZZXNWHMJW-KHTUJTCYSA-N 0.000 description 1
- OEIHQFHQXZZLDQ-UHSQPCAPSA-N CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C1=NC(C2=CC=C(C3=C(Cl)C4=CC=CC=C4N3)C=C2)=CN1)C1=CC=CC=C1 Chemical compound CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C1=NC(C2=CC=C(C3=C(Cl)C4=CC=CC=C4N3)C=C2)=CN1)C1=CC=CC=C1 OEIHQFHQXZZLDQ-UHSQPCAPSA-N 0.000 description 1
- KBPYNRMZVUZKPX-CPBHLAHYSA-N CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC=CC=C4N3)C=C2)C1)C1=CC=CC=C1 Chemical compound CC(C)OC(=O)C[C@@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC=CC=C4N3)C=C2)C1)C1=CC=CC=C1 KBPYNRMZVUZKPX-CPBHLAHYSA-N 0.000 description 1
- HVOQEGFVKCKPGQ-OTEAIWORSA-N CC(C)OC(=O)Cl.CCN(CC)CC.NNC(=O)C1=CC=C(I)C=C1.O=C(NCC(=O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1)C1=CC=C(I)C=C1.O=C(O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1 Chemical compound CC(C)OC(=O)Cl.CCN(CC)CC.NNC(=O)C1=CC=C(I)C=C1.O=C(NCC(=O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1)C1=CC=C(I)C=C1.O=C(O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1 HVOQEGFVKCKPGQ-OTEAIWORSA-N 0.000 description 1
- SBYDPGBEALHJSC-SDWMJEROSA-N CC(C)OC(=O)N[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=CC([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)=NC4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound CC(C)OC(=O)N[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=CC([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)=NC4)C=C3)=CC2=C1)C1=CC=CC=C1 SBYDPGBEALHJSC-SDWMJEROSA-N 0.000 description 1
- WTXNQVJZFSZTNW-OQWQQYAOSA-N CC(C)OC(=O)N[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=CC([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)=NC4)C=C3)=CC2=C1)C1=CC=CC=C1.O=C(NC1=CC=C2CC(C3=CC=C(C4=CC([C@@H]5CCCN5)=NN4)C=C3)=CC2=C1)[C@@H]1CCCN1.S.S Chemical compound CC(C)OC(=O)N[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=CC([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)=NC4)C=C3)=CC2=C1)C1=CC=CC=C1.O=C(NC1=CC=C2CC(C3=CC=C(C4=CC([C@@H]5CCCN5)=NN4)C=C3)=CC2=C1)[C@@H]1CCCN1.S.S WTXNQVJZFSZTNW-OQWQQYAOSA-N 0.000 description 1
- QUQSMIQMYKKIJI-TWOGUARRSA-N CC(C)OC(=O)N[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)S4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound CC(C)OC(=O)N[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)S4)C=C3)=CC2=C1)C1=CC=CC=C1 QUQSMIQMYKKIJI-TWOGUARRSA-N 0.000 description 1
- XYRSZKQLFHZNHB-UJAHLCQWSA-N CC(C)OC(=O)N[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)S4)C=C3)=CC2=C1)C1=CC=CC=C1.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NC=C(C2=CC=C(Br)C=C2)S1 Chemical compound CC(C)OC(=O)N[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)S4)C=C3)=CC2=C1)C1=CC=CC=C1.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NC=C(C2=CC=C(Br)C=C2)S1 XYRSZKQLFHZNHB-UJAHLCQWSA-N 0.000 description 1
- PSSYQXBJPBVDAS-PVIZNTCDSA-N CC(C)OC(=O)N[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=NC=C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)N4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound CC(C)OC(=O)N[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=NC=C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)N4)C=C3)=CC2=C1)C1=CC=CC=C1 PSSYQXBJPBVDAS-PVIZNTCDSA-N 0.000 description 1
- LDEHSOZAERGFPZ-JNYAWNMCSA-N CC(C)OC(=O)N[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=NC=C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)N4)C=C3)=CC2=C1)C1=CC=CC=C1.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NC=C(C2=CC=C(Br)C=C2)C1 Chemical compound CC(C)OC(=O)N[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=NC=C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)C)C5=CC=CC=C5)N4)C=C3)=CC2=C1)C1=CC=CC=C1.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NC=C(C2=CC=C(Br)C=C2)C1 LDEHSOZAERGFPZ-JNYAWNMCSA-N 0.000 description 1
- HWAHNUAREDQLNZ-TZRMAZRKSA-N CC(C)OC(N[C@@H](C(N(CCC1)[C@@H]1C(Nc(cc1)ccc1-c([o]c(c1c2)ccc2NC([C@H](CCC2)N2C([C@@H](c2ccccc2)NC(OC(C)C)=O)=O)=O)c1Br)=O)=O)c1ccccc1)=O Chemical compound CC(C)OC(N[C@@H](C(N(CCC1)[C@@H]1C(Nc(cc1)ccc1-c([o]c(c1c2)ccc2NC([C@H](CCC2)N2C([C@@H](c2ccccc2)NC(OC(C)C)=O)=O)=O)c1Br)=O)=O)c1ccccc1)=O HWAHNUAREDQLNZ-TZRMAZRKSA-N 0.000 description 1
- NHCVUABPNLYZTG-LCSJGJLOSA-N CC(C)[C@@H](CC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C(C)C)C=C3)=CC2=C1 Chemical compound CC(C)[C@@H](CC(=O)OC(C)(C)C)C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C(C)C)C=C3)=CC2=C1 NHCVUABPNLYZTG-LCSJGJLOSA-N 0.000 description 1
- GLPNFIRIHKRHAF-IBGZPJMESA-N CC(Nc(cc1)ccc1-c1cc(cc(cc2)-c3nnc([C@H]4NCCC4)[o]3)c2[nH]1)=O Chemical compound CC(Nc(cc1)ccc1-c1cc(cc(cc2)-c3nnc([C@H]4NCCC4)[o]3)c2[nH]1)=O GLPNFIRIHKRHAF-IBGZPJMESA-N 0.000 description 1
- DWVOQRHCLCMQAQ-IOWSJCHKSA-N CC(Nc(cc1)ccc1-c1cc(cc(cc2)NC([C@H](CCC3)N3C([C@@H](c3ccccc3)N3CCCC3)=O)=O)c2[nH]1)=O Chemical compound CC(Nc(cc1)ccc1-c1cc(cc(cc2)NC([C@H](CCC3)N3C([C@@H](c3ccccc3)N3CCCC3)=O)=O)c2[nH]1)=O DWVOQRHCLCMQAQ-IOWSJCHKSA-N 0.000 description 1
- JNMZXCHPFVBSLB-UHFFFAOYSA-N CC(Nc(cc1)ccc1-c1cc(ccc([N+]([O-])=O)c2)c2[nH]1)=O Chemical compound CC(Nc(cc1)ccc1-c1cc(ccc([N+]([O-])=O)c2)c2[nH]1)=O JNMZXCHPFVBSLB-UHFFFAOYSA-N 0.000 description 1
- FGONUNMOVIMTIJ-UHFFFAOYSA-N CC.CC.CC.CC.CC(C)C1=CC=C2C(=C1)[W]C1=C2OC2=C1C=C(C(C)C)C=C2.CC(C)C1=CC=C2C(=C1)[W]C1=C2OC2=C1N=C(C(C)C)C=C2 Chemical compound CC.CC.CC.CC.CC(C)C1=CC=C2C(=C1)[W]C1=C2OC2=C1C=C(C(C)C)C=C2.CC(C)C1=CC=C2C(=C1)[W]C1=C2OC2=C1N=C(C(C)C)C=C2 FGONUNMOVIMTIJ-UHFFFAOYSA-N 0.000 description 1
- SPOOMHDTHVLOES-HUMMXKGPSA-N CC.CC.CC.CCC1=NC=C(C2=CC=C(C3=CC4=C(C=CC(C5=CN=C(CC)N5)=C4)[Y]3)C=C2)N1 Chemical compound CC.CC.CC.CCC1=NC=C(C2=CC=C(C3=CC4=C(C=CC(C5=CN=C(CC)N5)=C4)[Y]3)C=C2)N1 SPOOMHDTHVLOES-HUMMXKGPSA-N 0.000 description 1
- KNKOTVVVXOTUFB-UHFFFAOYSA-N CC1(C)OB(B2OC(C)(C)C(C)(C)O2)OC1(C)C.CC1(C)OB(C2=CC=C3OC(C4=CC=C(B5OC(C)(C)C(C)(C)O5)C(F)=C4)=CC3=C2)OC1(C)C.FC1=CC(C2=CC3=CC(Br)=CC=C3O2)=CC=C1Br Chemical compound CC1(C)OB(B2OC(C)(C)C(C)(C)O2)OC1(C)C.CC1(C)OB(C2=CC=C3OC(C4=CC=C(B5OC(C)(C)C(C)(C)O5)C(F)=C4)=CC3=C2)OC1(C)C.FC1=CC(C2=CC3=CC(Br)=CC=C3O2)=CC=C1Br KNKOTVVVXOTUFB-UHFFFAOYSA-N 0.000 description 1
- ZXPUQFCTUHHGKP-UHFFFAOYSA-N CC1(C)OB(B2OC(C)(C)C(C)(C)O2)OC1(C)C.CC1(C)OB(C2=CC=C3OC(C4=CC=C([N+](=O)[O-])C=C4)=CC3=C2)OC1(C)C.O=[N+]([O-])C1=CC=C(C2=CC3=CC(Br)=CC=C3O2)C=C1 Chemical compound CC1(C)OB(B2OC(C)(C)C(C)(C)O2)OC1(C)C.CC1(C)OB(C2=CC=C3OC(C4=CC=C([N+](=O)[O-])C=C4)=CC3=C2)OC1(C)C.O=[N+]([O-])C1=CC=C(C2=CC3=CC(Br)=CC=C3O2)C=C1 ZXPUQFCTUHHGKP-UHFFFAOYSA-N 0.000 description 1
- GTRPMZVGRCUVNH-YTTGMZPUSA-N CC1=C(C2=CC=C3NC(C4=CC=C(NC(=O)[C@@H]5CCCN5C(=O)CC5=CC=CC=C5)C=C4)=CC3=C2)C=NC(N(C)C)=C1 Chemical compound CC1=C(C2=CC=C3NC(C4=CC=C(NC(=O)[C@@H]5CCCN5C(=O)CC5=CC=CC=C5)C=C4)=CC3=C2)C=NC(N(C)C)=C1 GTRPMZVGRCUVNH-YTTGMZPUSA-N 0.000 description 1
- XOILKHLLGBKNEU-UHFFFAOYSA-N CC1=C(N)C=NC(N)=C1.CC1=C([N+](=O)[O-])C=NC(N)=C1 Chemical compound CC1=C(N)C=NC(N)=C1.CC1=C([N+](=O)[O-])C=NC(N)=C1 XOILKHLLGBKNEU-UHFFFAOYSA-N 0.000 description 1
- ZLHQZSZRBVEPIO-VPKLXUFRSA-N CC1=CC=C(S(=O)(=O)C[C@@H](C(=O)N2CCC[C@H]2C(=O)CC2=CC=C3NC(C4=CC=C(NC(=O)[C@@H]5CCCN5C(=O)[C@H](CS(=O)(=O)C5=CC=C(C)C=C5)C5=CC=CC=C5)C=C4)=CC3=C2)C2=CC=CC=C2)C=C1 Chemical compound CC1=CC=C(S(=O)(=O)C[C@@H](C(=O)N2CCC[C@H]2C(=O)CC2=CC=C3NC(C4=CC=C(NC(=O)[C@@H]5CCCN5C(=O)[C@H](CS(=O)(=O)C5=CC=C(C)C=C5)C5=CC=CC=C5)C=C4)=CC3=C2)C2=CC=CC=C2)C=C1 ZLHQZSZRBVEPIO-VPKLXUFRSA-N 0.000 description 1
- UZGYQVJAUKZKEM-LJAQVGFWSA-N CC1=CC=C(S(=O)(=O)NC2=C3N=C(C4=CC=C(CC(=O)[C@@H]5CCCN5C(=O)C5=CC=CC=C5)C=C4)NC3=CC=C2)C=C1 Chemical compound CC1=CC=C(S(=O)(=O)NC2=C3N=C(C4=CC=C(CC(=O)[C@@H]5CCCN5C(=O)C5=CC=CC=C5)C=C4)NC3=CC=C2)C=C1 UZGYQVJAUKZKEM-LJAQVGFWSA-N 0.000 description 1
- LSEOEMLIOVQNEP-BFMMZNOMSA-N CC1=CC=C(S(=O)(=O)NC2=C3N=C(C4=CC=C(NC(=O)[C@@H]5CCCN5C(=O)OC(C)(C)C)C=C4)NC3=CC=C2)C=C1.CCCCOC(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=NC3=C(NS(=O)(=O)C4=CC=C(C)C=C4)C=CC=C3N2)C=C1 Chemical compound CC1=CC=C(S(=O)(=O)NC2=C3N=C(C4=CC=C(NC(=O)[C@@H]5CCCN5C(=O)OC(C)(C)C)C=C4)NC3=CC=C2)C=C1.CCCCOC(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=NC3=C(NS(=O)(=O)C4=CC=C(C)C=C4)C=CC=C3N2)C=C1 LSEOEMLIOVQNEP-BFMMZNOMSA-N 0.000 description 1
- MBGDAFQPOLBKLP-YIWHZXCRSA-N CC1=CC=C2C(=C1)/C=C(/C1=CC=C(C3=NN=C([C@H]4CCCN4C(=O)OCC4=CC=CC=C4)O3)C=C1)N2C(=O)OC(C)(C)C.CCN(CC)[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2N/C(C3=CC=C(C4=NN=C([C@@H]5CCCN5C(=O)[C@@H](C5=CC=CC=C5)N(CC)CC)O4)C=C3)=C\C2=C1)C1=CC=CC=C1 Chemical compound CC1=CC=C2C(=C1)/C=C(/C1=CC=C(C3=NN=C([C@H]4CCCN4C(=O)OCC4=CC=CC=C4)O3)C=C1)N2C(=O)OC(C)(C)C.CCN(CC)[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2N/C(C3=CC=C(C4=NN=C([C@@H]5CCCN5C(=O)[C@@H](C5=CC=CC=C5)N(CC)CC)O4)C=C3)=C\C2=C1)C1=CC=CC=C1 MBGDAFQPOLBKLP-YIWHZXCRSA-N 0.000 description 1
- GIRARJPVZXCQIA-UHFFFAOYSA-N CC1=CC=C2C(=C1)/C=C(C1=N/C3=C(C=CC([N+](=O)[O-])=C3)O/1)\N2C(=O)OC(C)(C)C.NC1=CC=C2NC(/C3=N/C4=C(C=CC(N)=C4)O3)=C\C2=C1 Chemical compound CC1=CC=C2C(=C1)/C=C(C1=N/C3=C(C=CC([N+](=O)[O-])=C3)O/1)\N2C(=O)OC(C)(C)C.NC1=CC=C2NC(/C3=N/C4=C(C=CC(N)=C4)O3)=C\C2=C1 GIRARJPVZXCQIA-UHFFFAOYSA-N 0.000 description 1
- FFFXNIXCWJNXBO-UHFFFAOYSA-N CC1=CC=C2C(=C1)C=C(B(O)O)N2C(=O)OC(C)(C)C.CC1=CC=C2C(=C1)C=C(C1=NC=C(N)C=N1)N2C(=O)OC(C)(C)C.NC1=CN=C(Cl)N=C1 Chemical compound CC1=CC=C2C(=C1)C=C(B(O)O)N2C(=O)OC(C)(C)C.CC1=CC=C2C(=C1)C=C(C1=NC=C(N)C=N1)N2C(=O)OC(C)(C)C.NC1=CN=C(Cl)N=C1 FFFXNIXCWJNXBO-UHFFFAOYSA-N 0.000 description 1
- JYBSNKUPNIBZPI-UHFFFAOYSA-N CC1=CC=C2C(=C1)C=C(B(O)O)N2C(=O)OC(C)(C)C.CC1=CC=C2C(=C1)C=CN2C(=O)OC(C)(C)C Chemical compound CC1=CC=C2C(=C1)C=C(B(O)O)N2C(=O)OC(C)(C)C.CC1=CC=C2C(=C1)C=CN2C(=O)OC(C)(C)C JYBSNKUPNIBZPI-UHFFFAOYSA-N 0.000 description 1
- RNVBSQIUOYGITN-UCBFPMGHSA-N CC1=CC=C2C(=C1)C=C(C1=CC=C3NC([C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)=NC3=C1)N2C(=O)OC(C)(C)C.NC1=CC=C2NC(C3=CC=C4NC([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)=NC4=C3)=CC2=C1 Chemical compound CC1=CC=C2C(=C1)C=C(C1=CC=C3NC([C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)=NC3=C1)N2C(=O)OC(C)(C)C.NC1=CC=C2NC(C3=CC=C4NC([C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)=NC4=C3)=CC2=C1 RNVBSQIUOYGITN-UCBFPMGHSA-N 0.000 description 1
- YVYDSQULDNWUOL-UHFFFAOYSA-N CC1=CC=C2C(=C1)C=C(C1=NC=C(N)C=N1)N2C(=O)OC(C)(C)C.NC1=CC=C2NC(C3=NC=C(N)C=N3)=CC2=C1 Chemical compound CC1=CC=C2C(=C1)C=C(C1=NC=C(N)C=N1)N2C(=O)OC(C)(C)C.NC1=CC=C2NC(C3=NC=C(N)C=N3)=CC2=C1 YVYDSQULDNWUOL-UHFFFAOYSA-N 0.000 description 1
- UDYRKFUFTOEYIZ-UHFFFAOYSA-N CC1=CC=C2C(=C1)C=CN2C(=O)OC(C)(C)C.NC1=CC=C2NC=CC2=C1 Chemical compound CC1=CC=C2C(=C1)C=CN2C(=O)OC(C)(C)C.NC1=CC=C2NC=CC2=C1 UDYRKFUFTOEYIZ-UHFFFAOYSA-N 0.000 description 1
- SCPFWHXSURCJBR-RFYKNWBHSA-N CC1=CC=CC(C2=C(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)NC3=CC=C(CC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C32)=C1 Chemical compound CC1=CC=CC(C2=C(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)NC3=CC=C(CC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C32)=C1 SCPFWHXSURCJBR-RFYKNWBHSA-N 0.000 description 1
- JGWBUDVMXSUZCU-RFYKNWBHSA-N CC1=CC=CC=C1C1=C(C2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)[C@H](CC(=O)OC(C)(C)C)C3=CC=CC=C3)C=C2)NC2=CC=C(CC(=O)[C@@H]3CCCN3C(=O)[C@H](CC(=O)OC(C)(C)C)C3=CC=CC=C3)C=C21 Chemical compound CC1=CC=CC=C1C1=C(C2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)[C@H](CC(=O)OC(C)(C)C)C3=CC=CC=C3)C=C2)NC2=CC=C(CC(=O)[C@@H]3CCCN3C(=O)[C@H](CC(=O)OC(C)(C)C)C3=CC=CC=C3)C=C21 JGWBUDVMXSUZCU-RFYKNWBHSA-N 0.000 description 1
- JTLAMUADTRXLSC-URTBVJOTSA-N CC=O.NC1=C(CC(=O)[C@@H]2CCCN2C(=O)OCC2=CC=CC=C2)C=C(Br)C=C1.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NC2=CC(Br)=CC=C2N1 Chemical compound CC=O.NC1=C(CC(=O)[C@@H]2CCCN2C(=O)OCC2=CC=CC=C2)C=C(Br)C=C1.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NC2=CC(Br)=CC=C2N1 JTLAMUADTRXLSC-URTBVJOTSA-N 0.000 description 1
- LDLUWPBAIXXEIX-UWXQCODUSA-N CCC1=C(C2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C2)NC2=C1C=C(CC(=O)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1)C=C2 Chemical compound CCC1=C(C2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C2)NC2=C1C=C(CC(=O)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1)C=C2 LDLUWPBAIXXEIX-UWXQCODUSA-N 0.000 description 1
- HPWUPTUELAPWDY-XOJMQWKJSA-N CCCN(CC)[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(CC)CCC)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound CCCN(CC)[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(CC)CCC)C=C3)=CC2=C1)C1=CC=CC=C1 HPWUPTUELAPWDY-XOJMQWKJSA-N 0.000 description 1
- GJLHDIBKFBTXTL-WDEONCGHSA-N CCN(CC)C(C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=NN=C([C@@H]5CCCN5C(=O)[C@@H](C5=CC=CC=C5)N(CC)CC)O4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound CCN(CC)C(C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=NN=C([C@@H]5CCCN5C(=O)[C@@H](C5=CC=CC=C5)N(CC)CC)O4)C=C3)=CC2=C1)C1=CC=CC=C1 GJLHDIBKFBTXTL-WDEONCGHSA-N 0.000 description 1
- JYFDHWMZZZXHIO-PVIZNTCDSA-N CCN(CC)[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(CC)CC)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound CCN(CC)[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(CC)CC)C=C3)=CC2=C1)C1=CC=CC=C1 JYFDHWMZZZXHIO-PVIZNTCDSA-N 0.000 description 1
- XJOPUKDZSZMMOZ-IHMCGGPQSA-N CCN(CC)[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(CC)CC)C=C3)=C(Br)C2=C1)C1=CC=CC=C1 Chemical compound CCN(CC)[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(CC)CC)C=C3)=C(Br)C2=C1)C1=CC=CC=C1 XJOPUKDZSZMMOZ-IHMCGGPQSA-N 0.000 description 1
- KFKYLTMEROKJSN-JXKOAFQRSA-N CCN(CC)[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(CC)CC)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound CCN(CC)[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(CC)CC)C=C3)=CC2=C1)C1=CC=CC=C1 KFKYLTMEROKJSN-JXKOAFQRSA-N 0.000 description 1
- RXDHPARMXABLAX-RYCFQHDISA-N CCN(CC)[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(C)=O)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound CCN(CC)[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(C)=O)C=C3)=CC2=C1)C1=CC=CC=C1 RXDHPARMXABLAX-RYCFQHDISA-N 0.000 description 1
- ZXSFEXZUXZACHP-KIVZLOICSA-N CCN(CC)[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(C)=O)=CC=C3O2)C=C1)C1=CC=CC=C1 Chemical compound CCN(CC)[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(C)=O)=CC=C3O2)C=C1)C1=CC=CC=C1 ZXSFEXZUXZACHP-KIVZLOICSA-N 0.000 description 1
- DGBILIJUKKPTAN-UNWJUUMBSA-N CCN(CC)[C@@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](C6=CC=CC=C6)N(CC)CC)N5)=CC=C4O3)C=C2)N1)C1=CC=CC=C1 Chemical compound CCN(CC)[C@@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](C6=CC=CC=C6)N(CC)CC)N5)=CC=C4O3)C=C2)N1)C1=CC=CC=C1 DGBILIJUKKPTAN-UNWJUUMBSA-N 0.000 description 1
- HKKYCBGKXPTMPI-IOWSJCHKSA-N CCN(CC)[C@@H](C(=O)N1CCC[C@H]1C1=NN=C(C2=CC=C3NC(C4=CC=C(NC(C)=O)C=C4)=CC3=C2)O1)C1=CC=CC=C1 Chemical compound CCN(CC)[C@@H](C(=O)N1CCC[C@H]1C1=NN=C(C2=CC=C3NC(C4=CC=C(NC(C)=O)C=C4)=CC3=C2)O1)C1=CC=CC=C1 HKKYCBGKXPTMPI-IOWSJCHKSA-N 0.000 description 1
- WTZRWZGZYIWWSF-IYTFMMMVSA-N CCN(CC)[C@@H](C(N(CCC1)[C@@H]1C(Nc1ccc2[nH]c(-c(cc3)ccc3-c3nnc([C@H](CCC4)N4C([C@@H](c4ccccc4)N(CC)CC)=O)[o]3)cc2c1)=O)=O)c1ccccc1 Chemical compound CCN(CC)[C@@H](C(N(CCC1)[C@@H]1C(Nc1ccc2[nH]c(-c(cc3)ccc3-c3nnc([C@H](CCC4)N4C([C@@H](c4ccccc4)N(CC)CC)=O)[o]3)cc2c1)=O)=O)c1ccccc1 WTZRWZGZYIWWSF-IYTFMMMVSA-N 0.000 description 1
- KJTXBNTWNNWXDF-GRGGVALCSA-N CCN1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)=CC=C3N2)C=C1 Chemical compound CCN1CCC[C@H]1C(=O)NC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)=CC=C3N2)C=C1 KJTXBNTWNNWXDF-GRGGVALCSA-N 0.000 description 1
- XDSUAWIYUBHULI-UHFFFAOYSA-N CCOC(=O)C(Br)C1=CC=C(Br)C=C1.CCOC(=O)CC1=CC=C(Br)C=C1 Chemical compound CCOC(=O)C(Br)C1=CC=C(Br)C=C1.CCOC(=O)CC1=CC=C(Br)C=C1 XDSUAWIYUBHULI-UHFFFAOYSA-N 0.000 description 1
- WZFWFQVBRHFZFK-ISAZSYCMSA-N CCOC(=O)Cl.NC(=O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1.O=C(O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1 Chemical compound CCOC(=O)Cl.NC(=O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1.O=C(O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1 WZFWFQVBRHFZFK-ISAZSYCMSA-N 0.000 description 1
- JZRQZOJSLMRLBO-RXTYAQLLSA-N CCOC(=O)Cl.O=C(CBr)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1.O=C(O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1 Chemical compound CCOC(=O)Cl.O=C(CBr)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1.O=C(O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1 JZRQZOJSLMRLBO-RXTYAQLLSA-N 0.000 description 1
- PPRAWWBOUOBAON-UHFFFAOYSA-N CCOC(C(c(cc1)ccc1Br)Br)=O Chemical compound CCOC(C(c(cc1)ccc1Br)Br)=O PPRAWWBOUOBAON-UHFFFAOYSA-N 0.000 description 1
- SEONCFTTXIAERK-ZNODAVFFSA-N CC[C@@H](C)[C@H](NC(=O)OC)C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)[C@H](C)CC)N5)=CC=C4O3)C=C2)N1 Chemical compound CC[C@@H](C)[C@H](NC(=O)OC)C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)[C@H](C)CC)N5)=CC=C4O3)C=C2)N1 SEONCFTTXIAERK-ZNODAVFFSA-N 0.000 description 1
- HITORTFSSWAUOL-IEQLCYGESA-N CN(C)C(=O)C1=C(C2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)[C@H](CC(=O)OC(C)(C)C)C3=CC=CC=C3)C=C2)NC2=C1C=C(CC(=O)[C@@H]1CCCN1C(=O)[C@H](CC(=O)OC(C)(C)C)C1=CC=CC=C1)C=C2 Chemical compound CN(C)C(=O)C1=C(C2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)[C@H](CC(=O)OC(C)(C)C)C3=CC=CC=C3)C=C2)NC2=C1C=C(CC(=O)[C@@H]1CCCN1C(=O)[C@H](CC(=O)OC(C)(C)C)C1=CC=CC=C1)C=C2 HITORTFSSWAUOL-IEQLCYGESA-N 0.000 description 1
- WQBDOGHBBDFKLO-IRWHZUTBSA-N CN(C)[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(C)C)C=C3)=C(C#N)C2=C1)C1=CC=CC=C1 Chemical compound CN(C)[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(C)C)C=C3)=C(C#N)C2=C1)C1=CC=CC=C1 WQBDOGHBBDFKLO-IRWHZUTBSA-N 0.000 description 1
- VCPAAFBIACMREY-BMPBIMJCSA-N CN(C)[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(C)C)C=C3)=C(Br)C2=C1)C1=CC=CC=C1 Chemical compound CN(C)[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N(C)C)C=C3)=C(Br)C2=C1)C1=CC=CC=C1 VCPAAFBIACMREY-BMPBIMJCSA-N 0.000 description 1
- QUBSOKKEAIZSAQ-WPWPIJCXSA-N CN(C)[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C2C(=C1)CCC1=C2NC2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)[C@@H](C3=CC=CC=C3)N(C)C)C=C21)C1=CC=CC=C1 Chemical compound CN(C)[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C2C(=C1)CCC1=C2NC2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)[C@@H](C3=CC=CC=C3)N(C)C)C=C21)C1=CC=CC=C1 QUBSOKKEAIZSAQ-WPWPIJCXSA-N 0.000 description 1
- ONQDRMAAHQRIAT-ZTJXFJJRSA-N CN(C)[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C2C(=C1)CCCC1=C2NC2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)[C@@H](C3=CC=CC=C3)N(C)C)C=C21)C1=CC=CC=C1 Chemical compound CN(C)[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C2C(=C1)CCCC1=C2NC2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)[C@@H](C3=CC=CC=C3)N(C)C)C=C21)C1=CC=CC=C1 ONQDRMAAHQRIAT-ZTJXFJJRSA-N 0.000 description 1
- MBZRUNVAHSJBGN-SKQWTEPWSA-N CN(C)[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](NC(=O)NC4CCCC4)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound CN(C)[C@@H](C(=O)N1CCC[C@H]1C(=O)NC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](NC(=O)NC4CCCC4)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1 MBZRUNVAHSJBGN-SKQWTEPWSA-N 0.000 description 1
- ZZOCVTWRZHHVLX-IBSCDHMXSA-N CN(C)[C@@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(CC(=O)[C@@H]5CCCN5C(=O)[C@H](CC(=O)OC(C)(C)C)C5=CC=CC=C5)=CC=C4N3)C=C2)N1)C1=CC=CC=C1 Chemical compound CN(C)[C@@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(CC(=O)[C@@H]5CCCN5C(=O)[C@H](CC(=O)OC(C)(C)C)C5=CC=CC=C5)=CC=C4N3)C=C2)N1)C1=CC=CC=C1 ZZOCVTWRZHHVLX-IBSCDHMXSA-N 0.000 description 1
- SGOZUKMOSLYYJZ-IOWSJCHKSA-N CN(C)[C@@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC=CC=C4N3)C=C2)C1)C1=CC=CC=C1 Chemical compound CN(C)[C@@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC=CC=C4N3)C=C2)C1)C1=CC=CC=C1 SGOZUKMOSLYYJZ-IOWSJCHKSA-N 0.000 description 1
- LNZWRJCTPSPEFH-IGYGKHONSA-N CNC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)=CC=C3N2)C=C1 Chemical compound CNC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)=CC=C3N2)C=C1 LNZWRJCTPSPEFH-IGYGKHONSA-N 0.000 description 1
- DMCPIDAHPCTCER-UHFFFAOYSA-N CNOC.CON(C)C(=O)C1=CC=C([N+](=O)[O-])C=C1.O=C(Cl)C1=CC=C([N+](=O)[O-])C=C1.O=C(O)C1=CC=C([N+](=O)[O-])C=C1.O=S(Cl)Cl Chemical compound CNOC.CON(C)C(=O)C1=CC=C([N+](=O)[O-])C=C1.O=C(Cl)C1=CC=C([N+](=O)[O-])C=C1.O=C(O)C1=CC=C([N+](=O)[O-])C=C1.O=S(Cl)Cl DMCPIDAHPCTCER-UHFFFAOYSA-N 0.000 description 1
- IQMBBQUIPXHICQ-RRHZFJGQSA-N COC(=O)C1=C(C2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)[C@H](CC(=O)OC(C)(C)C)C3=CC=CC=C3)C=C2)NC2=C1C=C(CC(=O)[C@@H]1CCCN1C(=O)[C@H](CC(=O)OC(C)(C)C)C1=CC=CC=C1)C=C2 Chemical compound COC(=O)C1=C(C2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)[C@H](CC(=O)OC(C)(C)C)C3=CC=CC=C3)C=C2)NC2=C1C=C(CC(=O)[C@@H]1CCCN1C(=O)[C@H](CC(=O)OC(C)(C)C)C1=CC=CC=C1)C=C2 IQMBBQUIPXHICQ-RRHZFJGQSA-N 0.000 description 1
- QALMYIPEHRKWJC-UHFFFAOYSA-N COC(=O)C1=CC=C2NC=CC2=C1.NNC(=O)C1=CC=C2NC=CC2=C1 Chemical compound COC(=O)C1=CC=C2NC=CC2=C1.NNC(=O)C1=CC=C2NC=CC2=C1 QALMYIPEHRKWJC-UHFFFAOYSA-N 0.000 description 1
- MDKLZZMVLKDCGW-WESAGZJESA-N COC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC(C)=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound COC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC(C)=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1 MDKLZZMVLKDCGW-WESAGZJESA-N 0.000 description 1
- ODVVRBUOCCHOGX-PASMDFPHSA-N COC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC)C4=CC=CC=C4)C=C3)=C(Br)C2=C1)C1=CC=CC=C1 Chemical compound COC(=O)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)OC)C4=CC=CC=C4)C=C3)=C(Br)C2=C1)C1=CC=CC=C1 ODVVRBUOCCHOGX-PASMDFPHSA-N 0.000 description 1
- RZZJGCVXVYXXBB-ROMUGYOQSA-N COC(=O)C[C@H](C(=O)N1CCC[C@@H]1C1=NC=C(C2=CC3=C(C=C2)C2=CC4=C(C=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=C4)N2/C(C)=N\3)C1)C(C)C Chemical compound COC(=O)C[C@H](C(=O)N1CCC[C@@H]1C1=NC=C(C2=CC3=C(C=C2)C2=CC4=C(C=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=C4)N2/C(C)=N\3)C1)C(C)C RZZJGCVXVYXXBB-ROMUGYOQSA-N 0.000 description 1
- MZOASIGQSZLYSK-IKFSTVPESA-N COC(=O)C[C@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](CC(=O)OC)C(C)C)C=C3)=C(F)C2=C1)C(C)C Chemical compound COC(=O)C[C@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](CC(=O)OC)C(C)C)C=C3)=C(F)C2=C1)C(C)C MZOASIGQSZLYSK-IKFSTVPESA-N 0.000 description 1
- SVEWRQIFBXQJCB-KDCBYZHASA-N COC(=O)C[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3C(=C2)C(F)=C2C4=C(C=C(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)C=C4)OCN32)N1)C(C)C Chemical compound COC(=O)C[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3C(=C2)C(F)=C2C4=C(C=C(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)C=C4)OCN32)N1)C(C)C SVEWRQIFBXQJCB-KDCBYZHASA-N 0.000 description 1
- JAGLLEOEGWYGIL-IRWHZUTBSA-N COC(=O)N[C@@H](C(=O)N1CCC[C@H]1C1=NC(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](NC(=O)OC)C6=CC=CC=C6)N5)=CC=C4O3)C(C#N)=C2)=CN1)C1=CC=CC=C1 Chemical compound COC(=O)N[C@@H](C(=O)N1CCC[C@H]1C1=NC(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](NC(=O)OC)C6=CC=CC=C6)N5)=CC=C4O3)C(C#N)=C2)=CN1)C1=CC=CC=C1 JAGLLEOEGWYGIL-IRWHZUTBSA-N 0.000 description 1
- WXNMHZQLPWAGHG-WZIOGPPVSA-N COC(=O)N[C@@H](C(=O)N1CCC[C@H]1C1=NC(C2=CC=C3OC(C4=CC=C(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](NC(=O)OC)C6=CC=CC=C6)N5)C=C4)=CC3=C2)=CN1)C1=CC=CC=C1 Chemical compound COC(=O)N[C@@H](C(=O)N1CCC[C@H]1C1=NC(C2=CC=C3OC(C4=CC=C(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](NC(=O)OC)C6=CC=CC=C6)N5)C=C4)=CC3=C2)=CN1)C1=CC=CC=C1 WXNMHZQLPWAGHG-WZIOGPPVSA-N 0.000 description 1
- LLRDEYHDYZKVNM-DUBDIQTKSA-N COC(=O)N[C@@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC3=C(C=C2)C2=CC4=C(C=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](NC(=O)OC)C6=CC=CC=C6)N5)=C4)N2CO3)C1)C1=CC=CC=C1 Chemical compound COC(=O)N[C@@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC3=C(C=C2)C2=CC4=C(C=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](NC(=O)OC)C6=CC=CC=C6)N5)=C4)N2CO3)C1)C1=CC=CC=C1 LLRDEYHDYZKVNM-DUBDIQTKSA-N 0.000 description 1
- QKFUCROVTLGTAV-BSTNIHDZSA-N COC(=O)N[C@@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](NC(=O)OC)C(C)C)N5)=CC=C4O3)C=C2)N1)C(C)C Chemical compound COC(=O)N[C@@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](NC(=O)OC)C(C)C)N5)=CC=C4O3)C=C2)N1)C(C)C QKFUCROVTLGTAV-BSTNIHDZSA-N 0.000 description 1
- HLGAOTIJCJSNDS-WZIOGPPVSA-N COC(=O)N[C@@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](NC(=O)OC)C6=CC=CC=C6)N5)=CC=C4O3)C(F)=C2)N1)C1=CC=CC=C1 Chemical compound COC(=O)N[C@@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](NC(=O)OC)C6=CC=CC=C6)N5)=CC=C4O3)C(F)=C2)N1)C1=CC=CC=C1 HLGAOTIJCJSNDS-WZIOGPPVSA-N 0.000 description 1
- GBWCBDMHXUDBNJ-BSTNIHDZSA-N COC(=O)N[C@@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](NC(=O)OC)C6CC6)N5)=CC=C4O3)C=C2)N1)C1CC1 Chemical compound COC(=O)N[C@@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](NC(=O)OC)C6CC6)N5)=CC=C4O3)C=C2)N1)C1CC1 GBWCBDMHXUDBNJ-BSTNIHDZSA-N 0.000 description 1
- KIMPUELINZLBDS-VLRMEJBNSA-N COC(=O)N[C@@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=NC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](NC(=O)OC)C6CC6)N5)=CC=C4O3)C=C2)N1)C1CC1 Chemical compound COC(=O)N[C@@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=NC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](NC(=O)OC)C6CC6)N5)=CC=C4O3)C=C2)N1)C1CC1 KIMPUELINZLBDS-VLRMEJBNSA-N 0.000 description 1
- VPWBDPXQEHHMTD-YMMTXDPESA-N COC(=O)N[C@@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3OC(C4=CC=C(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](NC(=O)OC)C6=CC=CC=C6)N5)C=C4OC)=CC3=C2)N1)C1=CC=CC=C1 Chemical compound COC(=O)N[C@@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3OC(C4=CC=C(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](NC(=O)OC)C6=CC=CC=C6)N5)C=C4OC)=CC3=C2)N1)C1=CC=CC=C1 VPWBDPXQEHHMTD-YMMTXDPESA-N 0.000 description 1
- DTADOBLRZIBGQB-VOIOCNMVSA-N COC(=O)N[C@@H](CC(C)C)C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](CC(C)C)NC(=O)OC)N5)=CC=C4O3)C=C2)N1 Chemical compound COC(=O)N[C@@H](CC(C)C)C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](CC(C)C)NC(=O)OC)N5)=CC=C4O3)C=C2)N1 DTADOBLRZIBGQB-VOIOCNMVSA-N 0.000 description 1
- WRDIJVCDYBFIFC-KRCBVYEFSA-N COC(=O)N[C@@H](CO)C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](CO)NC(=O)OC)N5)=CC=C4O3)C=C2)N1 Chemical compound COC(=O)N[C@@H](CO)C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](CO)NC(=O)OC)N5)=CC=C4O3)C=C2)N1 WRDIJVCDYBFIFC-KRCBVYEFSA-N 0.000 description 1
- PHYFQJITYCNHHA-USOXYGONSA-N COC(=O)N[C@H](C(=O)N1CCCC1C1=NC=C(C2=CC3=C(C=C2)C2=CC4=C(C=CC(C5=NC=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=C4)N2C2(CCCCC2)O3)N1)C(C)C Chemical compound COC(=O)N[C@H](C(=O)N1CCCC1C1=NC=C(C2=CC3=C(C=C2)C2=CC4=C(C=CC(C5=NC=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=C4)N2C2(CCCCC2)O3)N1)C(C)C PHYFQJITYCNHHA-USOXYGONSA-N 0.000 description 1
- BVAZQCUMNICBAQ-HMHFXUTRSA-N COC(=O)N[C@H](C(=O)N1CCCC1C1=NC=C(C2=CC3=C(C=C2)N2C(=C3)C3=C(C=C(C4=CN=C([C@@H]5CCCN5C(=O)[C@@H](NC(=O)OC)C(C)C)N4)C=C3)OC2C2=CC=CC=C2)N1)C(C)C Chemical compound COC(=O)N[C@H](C(=O)N1CCCC1C1=NC=C(C2=CC3=C(C=C2)N2C(=C3)C3=C(C=C(C4=CN=C([C@@H]5CCCN5C(=O)[C@@H](NC(=O)OC)C(C)C)N4)C=C3)OC2C2=CC=CC=C2)N1)C(C)C BVAZQCUMNICBAQ-HMHFXUTRSA-N 0.000 description 1
- GQHVXHLZNIEEIY-SEXBWERRSA-N COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=CC=C4O3)C(C)=C2)=CN1)C(C)C Chemical compound COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=CC=C4O3)C(C)=C2)=CN1)C(C)C GQHVXHLZNIEEIY-SEXBWERRSA-N 0.000 description 1
- QUBWLYZGYLGLNH-VOIOCNMVSA-N COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=CC=C4O3)C(Cl)=C2)=CN1)C(C)C Chemical compound COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=CC=C4O3)C(Cl)=C2)=CN1)C(C)C QUBWLYZGYLGLNH-VOIOCNMVSA-N 0.000 description 1
- RULQIEFQZDOGCH-UUPVYZINSA-N COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](NC(=O)OC)C6=CC=CC=C6)N5)=CC=C4O3)C(F)=C2)=CN1)C(C)C Chemical compound COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](NC(=O)OC)C6=CC=CC=C6)N5)=CC=C4O3)C(F)=C2)=CN1)C(C)C RULQIEFQZDOGCH-UUPVYZINSA-N 0.000 description 1
- NJYCFUSPDBLKOO-LGPNGYOUSA-N COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC3=C(C(F)=C2)C2=C(F)C4=C(C=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=C4)N2CO3)N1)C(C)C Chemical compound COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC3=C(C(F)=C2)C2=C(F)C4=C(C=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=C4)N2CO3)N1)C(C)C NJYCFUSPDBLKOO-LGPNGYOUSA-N 0.000 description 1
- ROFYVXJQBKKPKR-JOJDORDVSA-N COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC3=C(C=C2)/C2=C/C4=C(/C=C/C(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=C/4)N2CCO3)N1)C(C)C Chemical compound COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC3=C(C=C2)/C2=C/C4=C(/C=C/C(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=C/4)N2CCO3)N1)C(C)C ROFYVXJQBKKPKR-JOJDORDVSA-N 0.000 description 1
- CUSFCDUPQQIFHS-BXZZQGSBSA-N COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC3=C(C=C2)C2=C(F)C4=C(C=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=C4)N2C(C)O3)C1)C(C)C Chemical compound COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC3=C(C=C2)C2=C(F)C4=C(C=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=C4)N2C(C)O3)C1)C(C)C CUSFCDUPQQIFHS-BXZZQGSBSA-N 0.000 description 1
- OGTAGEXNXIXYBO-MJYVRLNOSA-N COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC3=C(C=C2)C2=CC4=C(C=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=C4)N2/C=N\3)C1)C(C)C Chemical compound COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC3=C(C=C2)C2=CC4=C(C=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=C4)N2/C=N\3)C1)C(C)C OGTAGEXNXIXYBO-MJYVRLNOSA-N 0.000 description 1
- LIIXZJQJJDKAHS-KEAHXZLPSA-N COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC3=C(C=C2)C2=CC4=C(C=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=C4)N2C(C)(C)O3)C1)C(C)C Chemical compound COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC3=C(C=C2)C2=CC4=C(C=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=C4)N2C(C)(C)O3)C1)C(C)C LIIXZJQJJDKAHS-KEAHXZLPSA-N 0.000 description 1
- SYNNTEWUERIPID-MHAZMVRYSA-N COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC3=C(C=C2)N2CCCOC4=C(C=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=C4)C2=C3)N1)C(C)C Chemical compound COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC3=C(C=C2)N2CCCOC4=C(C=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=C4)C2=C3)N1)C(C)C SYNNTEWUERIPID-MHAZMVRYSA-N 0.000 description 1
- SKAGXYRPCVJDJE-BSTNIHDZSA-N COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=C/C(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)(C)C)N5)=C\C=C\4O3)C=C2)N1)C(C)(C)C Chemical compound COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=C/C(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)(C)C)N5)=C\C=C\4O3)C=C2)N1)C(C)(C)C SKAGXYRPCVJDJE-BSTNIHDZSA-N 0.000 description 1
- VEUGILRHMIFCOF-VOIOCNMVSA-N COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=CC=C4O3)C(F)=C2)N1)C(C)C Chemical compound COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=CC=C4O3)C(F)=C2)N1)C(C)C VEUGILRHMIFCOF-VOIOCNMVSA-N 0.000 description 1
- QKFUCROVTLGTAV-VOIOCNMVSA-N COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=CC=C4O3)C=C2)N1)C(C)C Chemical compound COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=CC=C4O3)C=C2)N1)C(C)C QKFUCROVTLGTAV-VOIOCNMVSA-N 0.000 description 1
- GBWCBDMHXUDBNJ-VOIOCNMVSA-N COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C6CC6)N5)=CC=C4O3)C=C2)N1)C1CC1 Chemical compound COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C6CC6)N5)=CC=C4O3)C=C2)N1)C1CC1 GBWCBDMHXUDBNJ-VOIOCNMVSA-N 0.000 description 1
- AOIDZCUUAJLTSB-KEAHXZLPSA-N COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C6CCOCC6)N5)=CC=C4O3)C(F)=C2)N1)C1CCOCC1 Chemical compound COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C6CCOCC6)N5)=CC=C4O3)C(F)=C2)N1)C1CCOCC1 AOIDZCUUAJLTSB-KEAHXZLPSA-N 0.000 description 1
- HFKSTVFFCHBKPA-ZATYIABESA-N COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)[C@@H](C)O)N5)=CC=C4O3)C=C2)N1)[C@@H](C)O Chemical compound COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=CC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)[C@@H](C)O)N5)=CC=C4O3)C=C2)N1)[C@@H](C)O HFKSTVFFCHBKPA-ZATYIABESA-N 0.000 description 1
- MRFREUHASWLNKQ-MKJGCKHTSA-N COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=NC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=CC=C4O3)C=C2)N1)C(C)C Chemical compound COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=NC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)=CC=C4O3)C=C2)N1)C(C)C MRFREUHASWLNKQ-MKJGCKHTSA-N 0.000 description 1
- ITQJIOAXPJDUNX-VOIOCNMVSA-N COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3C(=C2)C(F)=C2C4=C(C=C(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)C=C4)OCN32)N1)C(C)C Chemical compound COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3C(=C2)C(F)=C2C4=C(C=C(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)C=C4)OCN32)N1)C(C)C ITQJIOAXPJDUNX-VOIOCNMVSA-N 0.000 description 1
- UKOFBQYBTQKOPR-SEXBWERRSA-N COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3C(=C2)C=C2C4=C(C=C(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)C=C4)OCN32)N1)C(C)C Chemical compound COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3C(=C2)C=C2C4=C(C=C(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)C=C4)OCN32)N1)C(C)C UKOFBQYBTQKOPR-SEXBWERRSA-N 0.000 description 1
- FQGJNOBXKDKRTK-LGPNGYOUSA-N COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3OC(C4=C(F)C=C(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)C=C4F)=CC3=C2)N1)C(C)C Chemical compound COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3OC(C4=C(F)C=C(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)C=C4F)=CC3=C2)N1)C(C)C FQGJNOBXKDKRTK-LGPNGYOUSA-N 0.000 description 1
- FQOJOLFHYBYUET-RFDUZHNLSA-N COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3OC(C4=CC(F)=C(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)C=C4)=CC3=C2)N1)C(C)C.FC1=CC(C2=CC3=CC(C4=CN=C([C@@H]5CCCN5)N4)=CC=C3O2)=CC=C1C1=CNC([C@@H]2CCCN2)=N1 Chemical compound COC(=O)N[C@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C3OC(C4=CC(F)=C(C5=CN=C([C@@H]6CCCN6C(=O)[C@@H](NC(=O)OC)C(C)C)N5)C=C4)=CC3=C2)N1)C(C)C.FC1=CC(C2=CC3=CC(C4=CN=C([C@@H]5CCCN5)N4)=CC=C3O2)=CC=C1C1=CNC([C@@H]2CCCN2)=N1 FQOJOLFHYBYUET-RFDUZHNLSA-N 0.000 description 1
- RXEWMHYHCSQODW-DNMDQSTESA-N COC(N[C@@H](C(N(CCC1)[C@@H]1C(Nc(cc1)ccc1-c1cc2cc(NC([C@H](CCC3)N3C([C@@H](c3ccccc3)NC(O)=O)=O)=O)ccc2[nH]1)=O)=O)c1ccccc1)=O Chemical compound COC(N[C@@H](C(N(CCC1)[C@@H]1C(Nc(cc1)ccc1-c1cc2cc(NC([C@H](CCC3)N3C([C@@H](c3ccccc3)NC(O)=O)=O)=O)ccc2[nH]1)=O)=O)c1ccccc1)=O RXEWMHYHCSQODW-DNMDQSTESA-N 0.000 description 1
- HCALHIUDJRJMIL-AVXXXJLRSA-N COC1=CC=C(C2=C(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](C)C4=CC=CC=C4)C=C3)NC3=CC=C(CC(=O)N4CCCC4C(=O)[C@H](C)C4=CC=CC=C4)C=C32)C=C1 Chemical compound COC1=CC=C(C2=C(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](C)C4=CC=CC=C4)C=C3)NC3=CC=C(CC(=O)N4CCCC4C(=O)[C@H](C)C4=CC=CC=C4)C=C32)C=C1 HCALHIUDJRJMIL-AVXXXJLRSA-N 0.000 description 1
- XXTGXFXMTDIACZ-OBQUVBTESA-N COC1=CC=C(C2=C(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](C)C4=CC=CC=C4)C=C3)NC3=CC=C(CC(=O)N4CCCC4C(=O)[C@H](C)C4=CC=CC=C4)C=C32)C=C1.C[C@@H](C(=O)C1CCCN1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](C)C4=CC=CC=C4)C=C3)=C(Br)C2=C1)C1=CC=CC=C1 Chemical compound COC1=CC=C(C2=C(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](C)C4=CC=CC=C4)C=C3)NC3=CC=C(CC(=O)N4CCCC4C(=O)[C@H](C)C4=CC=CC=C4)C=C32)C=C1.C[C@@H](C(=O)C1CCCN1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](C)C4=CC=CC=C4)C=C3)=C(Br)C2=C1)C1=CC=CC=C1 XXTGXFXMTDIACZ-OBQUVBTESA-N 0.000 description 1
- NBDSTCGDBQWBJG-COCZKOEFSA-N COC1=CC=CC(C2=C(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)C=C3)NC3=C2C=C(CC(=O)[C@@H]2CCCN2C(=O)OCC2=CC=CC=C2)C=C3)=C1 Chemical compound COC1=CC=CC(C2=C(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)C=C3)NC3=C2C=C(CC(=O)[C@@H]2CCCN2C(=O)OCC2=CC=CC=C2)C=C3)=C1 NBDSTCGDBQWBJG-COCZKOEFSA-N 0.000 description 1
- KBDJMKFYSIVJBD-WXKUUGAPSA-N C[C@@H](C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@H](C)N(C)C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=CC2=C1)N(C)C(=O)[C@H](CC(=O)OC(C)(C)C)C1=CC=CC=C1 Chemical compound C[C@@H](C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@H](C)N(C)C(=O)[C@H](CC(=O)OC(C)(C)C)C4=CC=CC=C4)C=C3)=CC2=C1)N(C)C(=O)[C@H](CC(=O)OC(C)(C)C)C1=CC=CC=C1 KBDJMKFYSIVJBD-WXKUUGAPSA-N 0.000 description 1
- NOHSDGVYQQDULL-GXVYXIKFSA-N C[C@@H](C(=O)N1CCCC1C(=O)CC1=CC=C2OC(C3=CC=C(N)C=C3)=CC2=C1)C1=CC=CC=C1.C[C@@H](C(=O)N1CCCC1C(=O)CC1=CC=C2OC(C3=CC=C([N+](=O)[O-])C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C[C@@H](C(=O)N1CCCC1C(=O)CC1=CC=C2OC(C3=CC=C(N)C=C3)=CC2=C1)C1=CC=CC=C1.C[C@@H](C(=O)N1CCCC1C(=O)CC1=CC=C2OC(C3=CC=C([N+](=O)[O-])C=C3)=CC2=C1)C1=CC=CC=C1 NOHSDGVYQQDULL-GXVYXIKFSA-N 0.000 description 1
- LQLDGLQSHSGJRS-IBQOLQPWSA-N C[C@@H](C(=O)N1CCCC1C(=O)CC1=CC=C2OC(C3=CC=C([N+](=O)[O-])C=C3)=CC2=C1)C1=CC=CC=C1.C[C@@H](C(=O)N1CCCC1C(=O)O)C1=CC=CC=C1.NC1=CC=C2OC(C3=CC=C([N+](=O)[O-])C=C3)=CC2=C1 Chemical compound C[C@@H](C(=O)N1CCCC1C(=O)CC1=CC=C2OC(C3=CC=C([N+](=O)[O-])C=C3)=CC2=C1)C1=CC=CC=C1.C[C@@H](C(=O)N1CCCC1C(=O)O)C1=CC=CC=C1.NC1=CC=C2OC(C3=CC=C([N+](=O)[O-])C=C3)=CC2=C1 LQLDGLQSHSGJRS-IBQOLQPWSA-N 0.000 description 1
- ROPRLDOQXCEGNL-UOAMMSJBSA-N C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=C/C=C2\NC(/C3=N/C4=C(C=CC(NC(=O)[C@@H](C5=CC=CC=C5)N(C)C)=C4)O3)=C\C2=C\1)C1=CC=CC=C1.C[C@@H](C(=O)N1CCC[C@H]1C(=O)O)C1=CC=CC=C1.NC1=CC=C2NC(/C3=N/C4=C(C=CC(N)=C4)O3)=C\C2=C1 Chemical compound C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=C/C=C2\NC(/C3=N/C4=C(C=CC(NC(=O)[C@@H](C5=CC=CC=C5)N(C)C)=C4)O3)=C\C2=C\1)C1=CC=CC=C1.C[C@@H](C(=O)N1CCC[C@H]1C(=O)O)C1=CC=CC=C1.NC1=CC=C2NC(/C3=N/C4=C(C=CC(N)=C4)O3)=C\C2=C1 ROPRLDOQXCEGNL-UOAMMSJBSA-N 0.000 description 1
- XOPOQKKEIMSISB-ANQLHOQHSA-N C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC2=C(C=C1)NC(C1=CC3=C(C=CC(CC(=O)C4CCCN4C(=O)[C@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)=C3)N1)=C2)C1=CC=CC=C1 Chemical compound C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC2=C(C=C1)NC(C1=CC3=C(C=CC(CC(=O)C4CCCN4C(=O)[C@H](NC(=O)OC(C)(C)C)C4=CC=CC=C4)=C3)N1)=C2)C1=CC=CC=C1 XOPOQKKEIMSISB-ANQLHOQHSA-N 0.000 description 1
- ROXKDFNRDLKORA-JWRNMLTDSA-N C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC2=C(C=C1)NC(C1=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C1)=C2)C1=CC=CC=C1 Chemical compound C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC2=C(C=C1)NC(C1=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C1)=C2)C1=CC=CC=C1 ROXKDFNRDLKORA-JWRNMLTDSA-N 0.000 description 1
- GWOPIMJLGUMNSJ-WJWVSBNJSA-N C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC2=C(C=C1)NC(C1=CC=C(NC(=O)[C@@H]3CCCN3C(=O)[C@H](NC(=O)OC(C)(C)C)C3=CC=CC=C3)C=C1)=C2C(=O)NC1CC1)C1=CC=CC=C1 Chemical compound C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC2=C(C=C1)NC(C1=CC=C(NC(=O)[C@@H]3CCCN3C(=O)[C@H](NC(=O)OC(C)(C)C)C3=CC=CC=C3)C=C1)=C2C(=O)NC1CC1)C1=CC=CC=C1 GWOPIMJLGUMNSJ-WJWVSBNJSA-N 0.000 description 1
- OVDLFABUOKXZNP-AEOLMSHQSA-N C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)(C)C)C5=CC=CC=C5)N4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(C4=CN=C([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)(C)C)C5=CC=CC=C5)N4)C=C3)=CC2=C1)C1=CC=CC=C1 OVDLFABUOKXZNP-AEOLMSHQSA-N 0.000 description 1
- JWDVLOOEENPCIH-XOMHETQGSA-N C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(N)C=C3)=CC2=C1)C1=CC=CC=C1.C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1.O=C(O)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(N)C=C3)=CC2=C1)C1=CC=CC=C1.C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1.O=C(O)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 JWDVLOOEENPCIH-XOMHETQGSA-N 0.000 description 1
- FZIRSRNOBYJYPX-KXVUMMDDSA-N C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(N)C=C3)=CC2=C1)C1=CC=CC=C1.C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C([N+](=O)[O-])C=C3)=CC2=C1)C1=CC=CC=C1.[HH] Chemical compound C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(N)C=C3)=CC2=C1)C1=CC=CC=C1.C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C([N+](=O)[O-])C=C3)=CC2=C1)C1=CC=CC=C1.[HH] FZIRSRNOBYJYPX-KXVUMMDDSA-N 0.000 description 1
- RAXSDCKCHVCPOE-OFDVDALDSA-N C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C([N+](=O)[O-])C=C3)=CC2=C1)C1=CC=CC=C1.C[C@@H](C(=O)N1CCC[C@H]1C(=O)O)C1=CC=CC=C1.NC1=CC=C2NC(C3=CC=C([N+](=O)[O-])C=C3)=CC2=C1 Chemical compound C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C([N+](=O)[O-])C=C3)=CC2=C1)C1=CC=CC=C1.C[C@@H](C(=O)N1CCC[C@H]1C(=O)O)C1=CC=CC=C1.NC1=CC=C2NC(C3=CC=C([N+](=O)[O-])C=C3)=CC2=C1 RAXSDCKCHVCPOE-OFDVDALDSA-N 0.000 description 1
- RCECMUJBNUNICI-PQYSHTRLSA-N C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C4NC([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)(C)C)C5=CC=CC=C5)=NC4=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C4NC([C@@H]5CCCN5C(=O)[C@H](NC(=O)OC(C)(C)C)C5=CC=CC=C5)=NC4=C3)=CC2=C1)C1=CC=CC=C1 RCECMUJBNUNICI-PQYSHTRLSA-N 0.000 description 1
- NTFMZPBWTNUTDG-GJHSCVLRSA-N C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=NC4=C(C=CC(NC(=O)[C@@H](C5=CC=CC=C5)N(C)C)=C4)O3)=CC2=C1)C1=CC=CC=C1 Chemical compound C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=NC4=C(C=CC(NC(=O)[C@@H](C5=CC=CC=C5)N(C)C)=C4)O3)=CC2=C1)C1=CC=CC=C1 NTFMZPBWTNUTDG-GJHSCVLRSA-N 0.000 description 1
- ADDAPLJFIRNCRD-UYPYDWMASA-N C[C@@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=NC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](C)C6=CC=CC=C6)N5)=CC=C4O3)C=C2)N1)C1=CC=CC=C1 Chemical compound C[C@@H](C(=O)N1CCC[C@H]1C1=NC=C(C2=CC=C(C3=NC4=CC(C5=CN=C([C@@H]6CCCN6C(=O)[C@H](C)C6=CC=CC=C6)N5)=CC=C4O3)C=C2)N1)C1=CC=CC=C1 ADDAPLJFIRNCRD-UYPYDWMASA-N 0.000 description 1
- QLLRDEGLIDOZOL-SHTLUSFCSA-N C[Pd].NC1=CN=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=CC2=C1.O=C(NC1=CC=C(C2=CC3=CC([N+](=O)[O-])=CN=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound C[Pd].NC1=CN=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=CC2=C1.O=C(NC1=CC=C(C2=CC3=CC([N+](=O)[O-])=CN=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 QLLRDEGLIDOZOL-SHTLUSFCSA-N 0.000 description 1
- YSDBFOVQHQHNAJ-NDEPHWFRSA-N Cc(cc1)ccc1S(Nc1cccc2c1nc(-c(cc1)ccc1NC([C@H](CCC1)N1C(c1ccccc1)=O)=O)[nH]2)(=O)=O Chemical compound Cc(cc1)ccc1S(Nc1cccc2c1nc(-c(cc1)ccc1NC([C@H](CCC1)N1C(c1ccccc1)=O)=O)[nH]2)(=O)=O YSDBFOVQHQHNAJ-NDEPHWFRSA-N 0.000 description 1
- MNPFFHYDYMGTQU-UHFFFAOYSA-N Cl.N#CC1=CC=C(Br)C=C1.N=C(N)C1=CC=C(Br)C=C1 Chemical compound Cl.N#CC1=CC=C(Br)C=C1.N=C(N)C1=CC=C(Br)C=C1 MNPFFHYDYMGTQU-UHFFFAOYSA-N 0.000 description 1
- SZDJBBINZBSSFR-LTVBCNPBSA-N Cl.O=C(CC1=CC=C(/C2=N/C3=CC([N+](=O)[O-])=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)C1=CC=CN=C1.O=C(NC1=CC=C(/C2=N/C3=CC([N+](=O)[O-])=CC=C3N2)C=C1)[C@@H]1CCCN1.O=C(O)C1=CC=CN=C1 Chemical compound Cl.O=C(CC1=CC=C(/C2=N/C3=CC([N+](=O)[O-])=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)C1=CC=CN=C1.O=C(NC1=CC=C(/C2=N/C3=CC([N+](=O)[O-])=CC=C3N2)C=C1)[C@@H]1CCCN1.O=C(O)C1=CC=CN=C1 SZDJBBINZBSSFR-LTVBCNPBSA-N 0.000 description 1
- COKNCNYBWBHWHY-UHFFFAOYSA-L Cl[Sn]Cl.NC1=CC=C(C2=CN3C=C(CC(=O)OCC4=CC=CC=C4)C=NC3=N2)C=C1.O=C(CC1=CN2C=C(C3=CC=C([N+](=O)[O-])C=C3)N=C2N=C1)OCC1=CC=CC=C1 Chemical compound Cl[Sn]Cl.NC1=CC=C(C2=CN3C=C(CC(=O)OCC4=CC=CC=C4)C=NC3=N2)C=C1.O=C(CC1=CN2C=C(C3=CC=C([N+](=O)[O-])C=C3)N=C2N=C1)OCC1=CC=CC=C1 COKNCNYBWBHWHY-UHFFFAOYSA-L 0.000 description 1
- WIKUISIHYPIISQ-QJOPACOMSA-L Cl[Sn]Cl.NC1=CC=C(C2=NC3=CN(CC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=CN3C2=O)C=C1.O=C(CN1C=CN2C(=O)C(C3=CC=C([N+](=O)[O-])C=C3)=NC2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound Cl[Sn]Cl.NC1=CC=C(C2=NC3=CN(CC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=CN3C2=O)C=C1.O=C(CN1C=CN2C(=O)C(C3=CC=C([N+](=O)[O-])C=C3)=NC2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 WIKUISIHYPIISQ-QJOPACOMSA-L 0.000 description 1
- MNNIBHHUGMIDAX-UHFFFAOYSA-N FC1=C(Br)C=CC(I)=C1.FC1=CC(C2=CC3=CC(Br)=CC=C3O2)=CC=C1Br.OB(O)C1=CC2=CC(Br)=CC=C2O1 Chemical compound FC1=C(Br)C=CC(I)=C1.FC1=CC(C2=CC3=CC(Br)=CC=C3O2)=CC=C1Br.OB(O)C1=CC2=CC(Br)=CC=C2O1 MNNIBHHUGMIDAX-UHFFFAOYSA-N 0.000 description 1
- OYZAGXZXAAFGRB-KEKNWZKVSA-N Fc(cc(cc1)-c2cc(cc(cc3)-c4cnc(C5NCCC5)[nH]4)c3[o]2)c1-c1c[nH]c([C@H]2NCCC2)n1 Chemical compound Fc(cc(cc1)-c2cc(cc(cc3)-c4cnc(C5NCCC5)[nH]4)c3[o]2)c1-c1c[nH]c([C@H]2NCCC2)n1 OYZAGXZXAAFGRB-KEKNWZKVSA-N 0.000 description 1
- LVZPPUQAWKTGJE-BCRBLDSWSA-N N#CC1=C(C2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C2)NC2=C1C=C(CC(=O)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1)C=C2 Chemical compound N#CC1=C(C2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C2)NC2=C1C=C(CC(=O)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1)C=C2 LVZPPUQAWKTGJE-BCRBLDSWSA-N 0.000 description 1
- ZKQDQBMEPRACNK-NLOJXZQTSA-N N#CC1=C(C2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C2)NC2=CC=C(CC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C21.O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=C(Br)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound N#CC1=C(C2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C2)NC2=CC=C(CC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C21.O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=C(Br)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 ZKQDQBMEPRACNK-NLOJXZQTSA-N 0.000 description 1
- KXHHBRUGFOXJKJ-UHFFFAOYSA-N N=C(N1C=CN=C1)N1C=CN=C1.NC1=C(O)C=CC([N+](=O)[O-])=C1.NC1=NC2=C(C=CC([N+](=O)[O-])=C2)O1 Chemical compound N=C(N1C=CN=C1)N1C=CN=C1.NC1=C(O)C=CC([N+](=O)[O-])=C1.NC1=NC2=C(C=CC([N+](=O)[O-])=C2)O1 KXHHBRUGFOXJKJ-UHFFFAOYSA-N 0.000 description 1
- TVBDMMTUFHDWNQ-QHUNOZLZSA-N NC(=O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1.NC(=S)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1 Chemical compound NC(=O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1.NC(=S)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1 TVBDMMTUFHDWNQ-QHUNOZLZSA-N 0.000 description 1
- RBTSFTRDYVWZPG-UCNZBORXSA-N NC(=S)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1.O=C(CBr)C1=CC=C(Br)C=C1.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NC=C(C2=CC=C(Br)C=C2)S1 Chemical compound NC(=S)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1.O=C(CBr)C1=CC=C(Br)C=C1.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NC=C(C2=CC=C(Br)C=C2)S1 RBTSFTRDYVWZPG-UCNZBORXSA-N 0.000 description 1
- JODFDXUBCBQKNC-UHFFFAOYSA-N NC(c(cc1)ccc1Br)=N Chemical compound NC(c(cc1)ccc1Br)=N JODFDXUBCBQKNC-UHFFFAOYSA-N 0.000 description 1
- CCWHXMMQBMBIEY-UHFFFAOYSA-N NC1=CC=C(C(=O)O)C=C1.NC1=CC=C(C2=NC3=CC(N)=CC=C3O2)C=C1.NC1=CC=C(O)C(N)=C1 Chemical compound NC1=CC=C(C(=O)O)C=C1.NC1=CC=C(C2=NC3=CC(N)=CC=C3O2)C=C1.NC1=CC=C(O)C(N)=C1 CCWHXMMQBMBIEY-UHFFFAOYSA-N 0.000 description 1
- RQHOLUBQVJUVRL-UHFFFAOYSA-N NC1=CC=C(C(=O)O)C=C1.NC1=CC=C(C2=NC3=CC([N+](=O)[O-])=CC=C3N2)C=C1.NC1=CC=C([N+](=O)[O-])C=C1N Chemical compound NC1=CC=C(C(=O)O)C=C1.NC1=CC=C(C2=NC3=CC([N+](=O)[O-])=CC=C3N2)C=C1.NC1=CC=C([N+](=O)[O-])C=C1N RQHOLUBQVJUVRL-UHFFFAOYSA-N 0.000 description 1
- CJCHLGXIBHCCHR-NWJQWMSWSA-N NC1=CC=C(C2=CC3=CC(Br)=CC=C3N2)C=C1.O=C(NC1=CC=C(C2=CC3=CC(Br)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(O)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound NC1=CC=C(C2=CC3=CC(Br)=CC=C3N2)C=C1.O=C(NC1=CC=C(C2=CC3=CC(Br)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(O)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 CJCHLGXIBHCCHR-NWJQWMSWSA-N 0.000 description 1
- AIWOINDANZDRIC-UHFFFAOYSA-N NC1=CC=C(C2=CC3=CC(N)=CC=C3O2)C=C1.O=[N+]([O-])C1=CC=C(C2=CC3=CC([N+](=O)[O-])=CC=C3O2)C=C1 Chemical compound NC1=CC=C(C2=CC3=CC(N)=CC=C3O2)C=C1.O=[N+]([O-])C1=CC=C(C2=CC3=CC([N+](=O)[O-])=CC=C3O2)C=C1 AIWOINDANZDRIC-UHFFFAOYSA-N 0.000 description 1
- PPJCPDJNUROLHY-AIHXPYQNSA-N NC1=CC=C(C2=CC3=CC(N)=NC=C3N2)C=C1.O=C(CC1=NC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=CC2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound NC1=CC=C(C2=CC3=CC(N)=NC=C3N2)C=C1.O=C(CC1=NC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=CC2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 PPJCPDJNUROLHY-AIHXPYQNSA-N 0.000 description 1
- VUTRGNWQOQPNMO-UHFFFAOYSA-N NC1=CC=C(C2=CN3C=C(N)C=CC3=N2)C=C1.O=[N+]([O-])C1=CC=C(C2=CN3C=C([N+](=O)[O-])C=CC3=N2)C=C1.[HH] Chemical compound NC1=CC=C(C2=CN3C=C(N)C=CC3=N2)C=C1.O=[N+]([O-])C1=CC=C(C2=CN3C=C([N+](=O)[O-])C=CC3=N2)C=C1.[HH] VUTRGNWQOQPNMO-UHFFFAOYSA-N 0.000 description 1
- HTVUCXDZTMFSKM-YEDBJMKGSA-N NC1=CC=C(C2=CN3C=C(N)C=NC3=N2)C=C1.O=C(CC1=CN2C=C(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)N=C2N=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(O)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound NC1=CC=C(C2=CN3C=C(N)C=NC3=N2)C=C1.O=C(CC1=CN2C=C(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)N=C2N=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(O)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 HTVUCXDZTMFSKM-YEDBJMKGSA-N 0.000 description 1
- QTDWEYXICZQXJF-TVYLAEMUSA-N NC1=CC=C2/C=C(/C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)NC2=C1.O=C(NC1=CC=C(/C2=C/C3=CC=C([N+](=O)[O-])C=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound NC1=CC=C2/C=C(/C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)NC2=C1.O=C(NC1=CC=C(/C2=C/C3=CC=C([N+](=O)[O-])C=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 QTDWEYXICZQXJF-TVYLAEMUSA-N 0.000 description 1
- QITITUZHEZMFFW-RANJIIOLSA-N NC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)C4=CC=CN=C4)C=C3)=NC2=C1.O=C(CC1=CC=C(/C2=N/C3=CC([N+](=O)[O-])=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)C1=CC=CN=C1 Chemical compound NC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)C4=CC=CN=C4)C=C3)=NC2=C1.O=C(CC1=CC=C(/C2=N/C3=CC([N+](=O)[O-])=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)C1=CC=CN=C1 QITITUZHEZMFFW-RANJIIOLSA-N 0.000 description 1
- PVSPEZUAJMSRIP-UHFFFAOYSA-N NC1=CC=C2NC(C3=CC=C([N+](=O)[O-])C=C3)=CC2=C1.O=[N+]([O-])C1=CC=C(C2=CC3=CC(Br)=CC=C3N2)C=C1 Chemical compound NC1=CC=C2NC(C3=CC=C([N+](=O)[O-])C=C3)=CC2=C1.O=[N+]([O-])C1=CC=C(C2=CC3=CC(Br)=CC=C3N2)C=C1 PVSPEZUAJMSRIP-UHFFFAOYSA-N 0.000 description 1
- HWJAYIWJESVXLB-UHFFFAOYSA-N NC1=CC=C2NC=CC2=C1.O=[N+]([O-])C1=CC=C2NC=CC2=C1 Chemical compound NC1=CC=C2NC=CC2=C1.O=[N+]([O-])C1=CC=C2NC=CC2=C1 HWJAYIWJESVXLB-UHFFFAOYSA-N 0.000 description 1
- ZNUUGJZRUVYVAC-UHFFFAOYSA-N NC1=CC=C2OC(C3=CC=C([N+](=O)[O-])C=C3)=CC2=C1.O=[N+]([O-])C1=CC=C(C2=CC3=CC(Br)=CC=C3O2)C=C1 Chemical compound NC1=CC=C2OC(C3=CC=C([N+](=O)[O-])C=C3)=CC2=C1.O=[N+]([O-])C1=CC=C(C2=CC3=CC(Br)=CC=C3O2)C=C1 ZNUUGJZRUVYVAC-UHFFFAOYSA-N 0.000 description 1
- FOOIJUZEWZFFED-UHFFFAOYSA-N NC1=CN=C(Cl)N=C1.O=[N+]([O-])C1=CN=C(Cl)N=C1 Chemical compound NC1=CN=C(Cl)N=C1.O=[N+]([O-])C1=CN=C(Cl)N=C1 FOOIJUZEWZFFED-UHFFFAOYSA-N 0.000 description 1
- AXYGFQCFZHYPBL-UHFFFAOYSA-N NC1=CN=C(N)N=C1.NC1=NC=C(NC(=O)OCC2=CC=CC=C2)C=N1.NC1=NC=C(NOO)C=N1.O=C(Cl)OCC1=CC=CC=C1.[HH] Chemical compound NC1=CN=C(N)N=C1.NC1=NC=C(NC(=O)OCC2=CC=CC=C2)C=N1.NC1=NC=C(NOO)C=N1.O=C(Cl)OCC1=CC=CC=C1.[HH] AXYGFQCFZHYPBL-UHFFFAOYSA-N 0.000 description 1
- CPHORTPCOVRGDE-UHFFFAOYSA-N NC1=CN=CC=N1.O=C1C(C2=CC=C([N+](=O)[O-])C=C2)=NC2=CNC=CN12.[H]C(=O)C(=O)C1=CC=C([N+](=O)[O-])C=C1 Chemical compound NC1=CN=CC=N1.O=C1C(C2=CC=C([N+](=O)[O-])C=C2)=NC2=CNC=CN12.[H]C(=O)C(=O)C1=CC=C([N+](=O)[O-])C=C1 CPHORTPCOVRGDE-UHFFFAOYSA-N 0.000 description 1
- GRGPUTIYFRJVIG-UHFFFAOYSA-N NC1=NC=C(NC(=O)OCC2=CC=CC=C2)C=N1.O=C(CBr)C1=CC=C([N+](=O)[O-])C=C1.O=C(CC1=CN2C=C(C3=CC=C([N+](=O)[O-])C=C3)N=C2N=C1)OCC1=CC=CC=C1 Chemical compound NC1=NC=C(NC(=O)OCC2=CC=CC=C2)C=N1.O=C(CBr)C1=CC=C([N+](=O)[O-])C=C1.O=C(CC1=CN2C=C(C3=CC=C([N+](=O)[O-])C=C3)N=C2N=C1)OCC1=CC=CC=C1 GRGPUTIYFRJVIG-UHFFFAOYSA-N 0.000 description 1
- DFYOIYQFOHUFJC-UHFFFAOYSA-N NC1=NC=C([N+](=O)[O-])C=C1.NC1=NC=C([N+](=O)[O-])C=C1I Chemical compound NC1=NC=C([N+](=O)[O-])C=C1.NC1=NC=C([N+](=O)[O-])C=C1I DFYOIYQFOHUFJC-UHFFFAOYSA-N 0.000 description 1
- VVPADUMIBRNIDJ-UHFFFAOYSA-N NC1=NC=C([N+](=O)[O-])C=C1.O=C(CBr)C1=CC=C([N+](=O)[O-])C=C1.O=[N+]([O-])C1=CC=C(C2=CN3C=C([N+](=O)[O-])C=CC3=N2)C=C1 Chemical compound NC1=NC=C([N+](=O)[O-])C=C1.O=C(CBr)C1=CC=C([N+](=O)[O-])C=C1.O=[N+]([O-])C1=CC=C(C2=CN3C=C([N+](=O)[O-])C=CC3=N2)C=C1 VVPADUMIBRNIDJ-UHFFFAOYSA-N 0.000 description 1
- TZMVXFCKNLVKJX-UBTXZFNXSA-N NNC(=O)C1=CC=C2NC=CC2=C1.O=C(NCC(=O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1)C1=CC=C2NC=CC2=C1.O=C(O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1 Chemical compound NNC(=O)C1=CC=C2NC=CC2=C1.O=C(NCC(=O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1)C1=CC=C2NC=CC2=C1.O=C(O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1 TZMVXFCKNLVKJX-UBTXZFNXSA-N 0.000 description 1
- WIHHVKUARKTSBU-UHFFFAOYSA-N Nc(c(N)c1)ccc1Br Chemical compound Nc(c(N)c1)ccc1Br WIHHVKUARKTSBU-UHFFFAOYSA-N 0.000 description 1
- JNLSZEUFWIOIRS-UHFFFAOYSA-N Nc(cc1)ccc1-c([nH]c1c2)cc1ccc2[N+]([O-])=O Chemical compound Nc(cc1)ccc1-c([nH]c1c2)cc1ccc2[N+]([O-])=O JNLSZEUFWIOIRS-UHFFFAOYSA-N 0.000 description 1
- UMGYJGHIMRFYSP-UHFFFAOYSA-N Nc(cc1)ccc1-c1nc2cc(N)ccc2[o]1 Chemical compound Nc(cc1)ccc1-c1nc2cc(N)ccc2[o]1 UMGYJGHIMRFYSP-UHFFFAOYSA-N 0.000 description 1
- PNPRHZLVVXNMFI-VWLOTQADSA-N Nc1ccc(cc(-c(cc2)ccc2NC([C@H](CCC2)N2C(Cc2ccccc2)=O)=O)[nH]2)c2c1 Chemical compound Nc1ccc(cc(-c(cc2)ccc2NC([C@H](CCC2)N2C(Cc2ccccc2)=O)=O)[nH]2)c2c1 PNPRHZLVVXNMFI-VWLOTQADSA-N 0.000 description 1
- ISFVLOJMNHTOLX-VPRRGWHLSA-N O=C(C#CC1=CC=C(Br)C=C1)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NNC(C2=CC=C(Br)C=C2)=C1 Chemical compound O=C(C#CC1=CC=C(Br)C=C1)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NNC(C2=CC=C(Br)C=C2)=C1 ISFVLOJMNHTOLX-VPRRGWHLSA-N 0.000 description 1
- CCKQHBHYTYRUJH-YATWDLPUSA-N O=C(C1=CC=CC=C1)C1=C(C2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C2)NC2=CC=C(CC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C21 Chemical compound O=C(C1=CC=CC=C1)C1=C(C2=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C2)NC2=CC=C(CC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C21 CCKQHBHYTYRUJH-YATWDLPUSA-N 0.000 description 1
- YUBLWGOGUZATLJ-XSDXBZSRSA-N O=C(CBr)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(CN1C=CN2C(=O)C(C3=CC=C([N+](=O)[O-])C=C3)=NC2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C1C(C2=CC=C([N+](=O)[O-])C=C2)=NC2=CNC=CN12 Chemical compound O=C(CBr)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(CN1C=CN2C(=O)C(C3=CC=C([N+](=O)[O-])C=C3)=NC2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C1C(C2=CC=C([N+](=O)[O-])C=C2)=NC2=CNC=CN12 YUBLWGOGUZATLJ-XSDXBZSRSA-N 0.000 description 1
- RVGOTPRWPLGRNL-LBPRGKRZSA-N O=C(CBr)[C@H](CCC1)N1C(Cc1ccccc1)=O Chemical compound O=C(CBr)[C@H](CCC1)N1C(Cc1ccccc1)=O RVGOTPRWPLGRNL-LBPRGKRZSA-N 0.000 description 1
- HGLLXVCVSATZFC-UWXQCODUSA-N O=C(CC1=CC2=C(C=C1)NC(C1=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C1)=C2C1CC1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(CC1=CC2=C(C=C1)NC(C1=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C1)=C2C1CC1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 HGLLXVCVSATZFC-UWXQCODUSA-N 0.000 description 1
- JEKHJVWYYJQWQL-PXLJZGITSA-N O=C(CC1=CC2=C(C=C1)NC(C1=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C1)=C2F)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(CC1=CC2=C(C=C1)NC(C1=CC=C(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)C=C1)=C2F)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 JEKHJVWYYJQWQL-PXLJZGITSA-N 0.000 description 1
- HPSDWGWPFBIQKT-BCRBLDSWSA-N O=C(CC1=CC2=C(C=C1)NC(C1=CC=CC(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)=C1)=C2)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(CC1=CC2=C(C=C1)NC(C1=CC=CC(NC(=O)[C@@H]3CCCN3C(=O)CC3=CC=CC=C3)=C1)=C2)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 HPSDWGWPFBIQKT-BCRBLDSWSA-N 0.000 description 1
- DQMGTOOWUUSUFN-ZPGRZCPFSA-N O=C(CC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)=CC=C3O2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(CC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)=CC=C3O2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 DQMGTOOWUUSUFN-ZPGRZCPFSA-N 0.000 description 1
- LSMCSYMQJSPGQX-ANFUHZJESA-N O=C(CC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H]4CCCO4)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)[C@@H]1CCCO1 Chemical compound O=C(CC1=CC=C(C2=CC3=CC(CC(=O)[C@@H]4CCCN4C(=O)[C@@H]4CCCO4)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)[C@@H]1CCCO1 LSMCSYMQJSPGQX-ANFUHZJESA-N 0.000 description 1
- JCKCSQRUPUXAME-RANVIWFRSA-N O=C(CC1=CC=C(C2=NC3=CC(CC(=O)[C@@H]4CCCN4)=CC=C3O2)C=C1)[C@@H]1CCCN1.O=C(CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CCC4=CC=CC=C4)C=C3)=NC2=C1)[C@@H]1CCCN1C(=O)CCC1=CC=CC=C1.O=C(O)CCC1=CC=CC=C1 Chemical compound O=C(CC1=CC=C(C2=NC3=CC(CC(=O)[C@@H]4CCCN4)=CC=C3O2)C=C1)[C@@H]1CCCN1.O=C(CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CCC4=CC=CC=C4)C=C3)=NC2=C1)[C@@H]1CCCN1C(=O)CCC1=CC=CC=C1.O=C(O)CCC1=CC=CC=C1 JCKCSQRUPUXAME-RANVIWFRSA-N 0.000 description 1
- FZGNWOJOJJIYPB-HEVIKAOCSA-N O=C(CC1=CC=C(C2=NC3=CC(CC(=O)[C@@H]4CCCN4C(=O)C4=CC=CC=C4)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)C1=CC=CC=C1 Chemical compound O=C(CC1=CC=C(C2=NC3=CC(CC(=O)[C@@H]4CCCN4C(=O)C4=CC=CC=C4)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)C1=CC=CC=C1 FZGNWOJOJJIYPB-HEVIKAOCSA-N 0.000 description 1
- KKBIUZOUXMYDDE-CONSDPRKSA-N O=C(CC1=CC=C(C2=NC3=CC(CC(=O)[C@@H]4CCCN4C(=O)C4CCC4)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)C1=CC=CN=C1 Chemical compound O=C(CC1=CC=C(C2=NC3=CC(CC(=O)[C@@H]4CCCN4C(=O)C4CCC4)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)C1=CC=CN=C1 KKBIUZOUXMYDDE-CONSDPRKSA-N 0.000 description 1
- YZKHWBFYGKZWHY-ZPGRZCPFSA-N O=C(CC1=CC=C(C2=NC3=CC(CC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(CC1=CC=C(C2=NC3=CC(CC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 YZKHWBFYGKZWHY-ZPGRZCPFSA-N 0.000 description 1
- UWZOQWSMKSVJAW-UWXQCODUSA-N O=C(CC1=CC=C(C2=NC3=CC(CC(=O)[C@@H]4CCCN4C(=O)CCC4=CC=CC=C4)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CCC1=CC=CC=C1 Chemical compound O=C(CC1=CC=C(C2=NC3=CC(CC(=O)[C@@H]4CCCN4C(=O)CCC4=CC=CC=C4)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CCC1=CC=CC=C1 UWZOQWSMKSVJAW-UWXQCODUSA-N 0.000 description 1
- XIMHHHZNZIOZNO-ZAQUEYBZSA-N O=C(CC1=CC=C2NC(/C3=C/C4=C(C=CC(CC(=O)[C@@H]5CCCN5C(=O)CC5=CC=CC=C5)=C4)N3)=CC2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(CC1=CC=C2NC(/C3=C/C4=C(C=CC(CC(=O)[C@@H]5CCCN5C(=O)CC5=CC=CC=C5)=C4)N3)=CC2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 XIMHHHZNZIOZNO-ZAQUEYBZSA-N 0.000 description 1
- AZXAQKQGPFDQLV-QXPTYHDKSA-N O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)C(CC4=CC=CC=C4)C4=CC=CC=C4)C=C3)=CC2=C1)[C@@H]1CCCN1C(=O)C(CC1=CC=CC=C1)C1=CC=CC=C1 Chemical compound O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)C(CC4=CC=CC=C4)C4=CC=CC=C4)C=C3)=CC2=C1)[C@@H]1CCCN1C(=O)C(CC1=CC=CC=C1)C1=CC=CC=C1 AZXAQKQGPFDQLV-QXPTYHDKSA-N 0.000 description 1
- ZZIAJXUHJMRJKK-YATWDLPUSA-N O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=C(C3=CC=CC=C3)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=C(C3=CC=CC=C3)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 ZZIAJXUHJMRJKK-YATWDLPUSA-N 0.000 description 1
- XNZCNXNUSPCQTR-ZAQUEYBZSA-N O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=C(C3=CC=NC=C3)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=C(C3=CC=NC=C3)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 XNZCNXNUSPCQTR-ZAQUEYBZSA-N 0.000 description 1
- LZWIMKPVNVFYTF-PXLJZGITSA-N O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=C(Cl)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=C(Cl)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 LZWIMKPVNVFYTF-PXLJZGITSA-N 0.000 description 1
- YRUSOMVMERNAHQ-PXLJZGITSA-N O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=C(I)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=C(I)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 YRUSOMVMERNAHQ-PXLJZGITSA-N 0.000 description 1
- JJXXFEOCKOXOAB-ZPGRZCPFSA-N O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=CC2=N1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=CC2=N1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 JJXXFEOCKOXOAB-ZPGRZCPFSA-N 0.000 description 1
- ZFTAUPUEAAQGPS-BCRBLDSWSA-N O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CCC4=CC=CC=C4)C=C3)=NC2=C1)[C@@H]1CCCN1C(=O)CCC1=CC=CC=C1 Chemical compound O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CCC4=CC=CC=C4)C=C3)=NC2=C1)[C@@H]1CCCN1C(=O)CCC1=CC=CC=C1 ZFTAUPUEAAQGPS-BCRBLDSWSA-N 0.000 description 1
- VSCXHMNHSLHTEJ-YATWDLPUSA-N O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)C=C3)=C(C3=CC=CC=C3)C2=C1)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1 Chemical compound O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)C=C3)=C(C3=CC=CC=C3)C2=C1)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1 VSCXHMNHSLHTEJ-YATWDLPUSA-N 0.000 description 1
- RRHFTGKVNGFMGY-ZPGRZCPFSA-N O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)C=C3)=CC2=N1)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1 Chemical compound O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)OCC4=CC=CC=C4)C=C3)=CC2=N1)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1 RRHFTGKVNGFMGY-ZPGRZCPFSA-N 0.000 description 1
- PBSIRENNSDZYBQ-STDQWCJXSA-N O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N4C=CN=C4)C=C3)=CC2=C1)[C@@H]1CCCN1C(=O)[C@@H](C1=CC=CC=C1)N1C=CN=C1 Chemical compound O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N4C=CN=C4)C=C3)=CC2=C1)[C@@H]1CCCN1C(=O)[C@@H](C1=CC=CC=C1)N1C=CN=C1 PBSIRENNSDZYBQ-STDQWCJXSA-N 0.000 description 1
- KAPDREYWCVQYKS-PVIZNTCDSA-N O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N4CCCC4)C=C3)=CC2=C1)[C@@H]1CCCN1C(=O)[C@@H](C1=CC=CC=C1)N1CCCC1 Chemical compound O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N4CCCC4)C=C3)=CC2=C1)[C@@H]1CCCN1C(=O)[C@@H](C1=CC=CC=C1)N1CCCC1 KAPDREYWCVQYKS-PVIZNTCDSA-N 0.000 description 1
- PVXOUKRZSFCHQV-SDWMJEROSA-N O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N4CCCCC4)C=C3)=CC2=C1)[C@@H]1CCCN1C(=O)[C@@H](C1=CC=CC=C1)N1CCCCC1 Chemical compound O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N4CCCCC4)C=C3)=CC2=C1)[C@@H]1CCCN1C(=O)[C@@H](C1=CC=CC=C1)N1CCCCC1 PVXOUKRZSFCHQV-SDWMJEROSA-N 0.000 description 1
- ZSYCYTKPZBSYQD-PVIZNTCDSA-N O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N4CCOCC4)C=C3)=CC2=C1)[C@@H]1CCCN1C(=O)[C@@H](C1=CC=CC=C1)N1CCOCC1 Chemical compound O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@@H](C4=CC=CC=C4)N4CCOCC4)C=C3)=CC2=C1)[C@@H]1CCCN1C(=O)[C@@H](C1=CC=CC=C1)N1CCOCC1 ZSYCYTKPZBSYQD-PVIZNTCDSA-N 0.000 description 1
- NFZXFJCCBCZFFL-WKEQUWASSA-N O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](O)C4=CC=CC=C4)C=C3)=NC2=C1)[C@@H]1CCCN1C(=O)[C@H](O)C1=CC=CC=C1 Chemical compound O=C(CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](O)C4=CC=CC=C4)C=C3)=NC2=C1)[C@@H]1CCCN1C(=O)[C@H](O)C1=CC=CC=C1 NFZXFJCCBCZFFL-WKEQUWASSA-N 0.000 description 1
- ZTXWTTHGCPIMIJ-BCRBLDSWSA-N O=C(CC1=CC=C2NC(C3=CC=CC(NC(=O)[C@@H]4CCCN4C(=O)CCC4=CC=CC=C4)=C3)=NC2=C1)[C@@H]1CCCN1C(=O)CCC1=CC=CC=C1 Chemical compound O=C(CC1=CC=C2NC(C3=CC=CC(NC(=O)[C@@H]4CCCN4C(=O)CCC4=CC=CC=C4)=C3)=NC2=C1)[C@@H]1CCCN1C(=O)CCC1=CC=CC=C1 ZTXWTTHGCPIMIJ-BCRBLDSWSA-N 0.000 description 1
- DJLDGUPONJFURN-HEVIKAOCSA-N O=C(CC1=CC=C2NC(C3=NC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=N3)=CC2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(CC1=CC=C2NC(C3=NC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=N3)=CC2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 DJLDGUPONJFURN-HEVIKAOCSA-N 0.000 description 1
- LXPBUPNCQHWDES-UWXQCODUSA-N O=C(CC1=CC=C2NC3=C(CCC4=CC(NC(=O)[C@@H]5CCCN5C(=O)CC5=CC=CC=C5)=CC=C43)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(CC1=CC=C2NC3=C(CCC4=CC(NC(=O)[C@@H]5CCCN5C(=O)CC5=CC=CC=C5)=CC=C43)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 LXPBUPNCQHWDES-UWXQCODUSA-N 0.000 description 1
- UWFWMKXBCFHNPL-UWXQCODUSA-N O=C(CC1=CC=C2NC3=C(CCC4=CC(NC(=O)[C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)=CC=C43)C2=C1)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1 Chemical compound O=C(CC1=CC=C2NC3=C(CCC4=CC(NC(=O)[C@@H]5CCCN5C(=O)OCC5=CC=CC=C5)=CC=C43)C2=C1)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1 UWFWMKXBCFHNPL-UWXQCODUSA-N 0.000 description 1
- QVGJFAYUXCXUAI-GVDXJTKQSA-N O=C(CC1=CC=C2NC3C4=CC=C(NC(=O)[C@@H]5CCCN5C(=O)CC5=CC=CC=C5)C=C4CCCC3C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(CC1=CC=C2NC3C4=CC=C(NC(=O)[C@@H]5CCCN5C(=O)CC5=CC=CC=C5)C=C4CCCC3C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 QVGJFAYUXCXUAI-GVDXJTKQSA-N 0.000 description 1
- WTNQPOFISJCPEY-HEVIKAOCSA-N O=C(CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=C(Br)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=C(Br)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 WTNQPOFISJCPEY-HEVIKAOCSA-N 0.000 description 1
- QXBREQLTVPEMGX-BCRBLDSWSA-N O=C(CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CCC4=CC=CC=C4)C=C3)=CC2=C1)[C@@H]1CCCN1C(=O)CCC1=CC=CC=C1 Chemical compound O=C(CC1=CC=C2OC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CCC4=CC=CC=C4)C=C3)=CC2=C1)[C@@H]1CCCN1C(=O)CCC1=CC=CC=C1 QXBREQLTVPEMGX-BCRBLDSWSA-N 0.000 description 1
- DRUHLUBOMFMCJB-ACHIHNKUSA-N O=C(CC1=CN2C(=NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=C2Cl)N=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(CC1=CN2C(=NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=C2Cl)N=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 DRUHLUBOMFMCJB-ACHIHNKUSA-N 0.000 description 1
- NIPNUYGXDQLLCP-LQJZCPKCSA-N O=C(CC1=CN=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=C(Cl)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(CC1=CN=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=C(Cl)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 NIPNUYGXDQLLCP-LQJZCPKCSA-N 0.000 description 1
- AJDJBIDLCGHZOR-HEVIKAOCSA-N O=C(CC1=NC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=C(Cl)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(CC1=NC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=C(Cl)C2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 AJDJBIDLCGHZOR-HEVIKAOCSA-N 0.000 description 1
- NEKVBGPKZBYZHI-DEWSGHJGSA-N O=C(CC1CC1)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)CC4CC4)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1 Chemical compound O=C(CC1CC1)C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)CC4CC4)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1 NEKVBGPKZBYZHI-DEWSGHJGSA-N 0.000 description 1
- BQOAQIUIYCIDPV-ZPGRZCPFSA-N O=C(CCc1ccccc1)N(CCC1)[C@@H]1C(Nc(cc1)ccc1-c1cc(cc(cc2)NC([C@H](CCC3)N3C(CCc3ccccc3)=O)=O)c2[o]1)=O Chemical compound O=C(CCc1ccccc1)N(CCC1)[C@@H]1C(Nc(cc1)ccc1-c1cc(cc(cc2)NC([C@H](CCC3)N3C(CCc3ccccc3)=O)=O)c2[o]1)=O BQOAQIUIYCIDPV-ZPGRZCPFSA-N 0.000 description 1
- KWBATSCWSCTZTB-ACHIHNKUSA-N O=C(CN1C=CN2C(=O)C(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=NC2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(CN1C=CN2C(=O)C(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)CC4=CC=CC=C4)C=C3)=NC2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 KWBATSCWSCTZTB-ACHIHNKUSA-N 0.000 description 1
- ANSSSHMSDVDQEV-VQGCAXMISA-N O=C(C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)C4CCCCC4)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1)C1CCCCC1 Chemical compound O=C(C[C@@H](C(=O)N1CCC[C@H]1C(=O)CC1=CC=C2NC(C3=CC=C(NC(=O)[C@@H]4CCCN4C(=O)[C@H](CC(=O)C4CCCCC4)C4=CC=CC=C4)C=C3)=CC2=C1)C1=CC=CC=C1)C1CCCCC1 ANSSSHMSDVDQEV-VQGCAXMISA-N 0.000 description 1
- ZRXNYUSWXMOMEL-LQJZCPKCSA-N O=C(Cc1ccccc1)N(CCC1)[C@@H]1C(Nc(cc1)cc2c1[nH]c(-c(nc1)ncc1NC([C@H](CCC1)N1C(Cc1ccccc1)=O)=O)c2)=O Chemical compound O=C(Cc1ccccc1)N(CCC1)[C@@H]1C(Nc(cc1)cc2c1[nH]c(-c(nc1)ncc1NC([C@H](CCC1)N1C(Cc1ccccc1)=O)=O)c2)=O ZRXNYUSWXMOMEL-LQJZCPKCSA-N 0.000 description 1
- VSJMXKARWCURCB-HEVIKAOCSA-N O=C(Cc1ccccc1)N(CCC1)[C@@H]1C(Nc(cc1)ccc1-c([nH]c(c1c2)ccc2NC([C@H](CCC2)N2C(Cc2ccccc2)=O)=O)c1Cl)=O Chemical compound O=C(Cc1ccccc1)N(CCC1)[C@@H]1C(Nc(cc1)ccc1-c([nH]c(c1c2)ccc2NC([C@H](CCC2)N2C(Cc2ccccc2)=O)=O)c1Cl)=O VSJMXKARWCURCB-HEVIKAOCSA-N 0.000 description 1
- LRJOOGKZTCGUBF-VWLOTQADSA-N O=C(Cc1ccccc1)N(CCC1)[C@@H]1C(Nc(cc1)ccc1-c1cc2cc(Br)ccc2[nH]1)=O Chemical compound O=C(Cc1ccccc1)N(CCC1)[C@@H]1C(Nc(cc1)ccc1-c1cc2cc(Br)ccc2[nH]1)=O LRJOOGKZTCGUBF-VWLOTQADSA-N 0.000 description 1
- GTVSOUBTGBTHIV-NBXBJIKISA-N O=C(NC1=CC=C(C2=CC3=CC(Br)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(NC1=CC=C(C2=CC3=CC(C4=CC=CO4)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.OB(O)C1=CC=CO1 Chemical compound O=C(NC1=CC=C(C2=CC3=CC(Br)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.O=C(NC1=CC=C(C2=CC3=CC(C4=CC=CO4)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1.OB(O)C1=CC=CO1 GTVSOUBTGBTHIV-NBXBJIKISA-N 0.000 description 1
- QOKNAUSMLWZFSJ-DHUJRADRSA-N O=C(NC1=CC=C(C2=CC3=CC(C4=CN(CC5=CC=CC=C5)N=C4)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(NC1=CC=C(C2=CC3=CC(C4=CN(CC5=CC=CC=C5)N=C4)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 QOKNAUSMLWZFSJ-DHUJRADRSA-N 0.000 description 1
- YNTFVQBCUSLODA-YTTGMZPUSA-N O=C(NC1=CC=C(C2=CC3=CC(C4=CSC5=C4C=CC=C5)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(NC1=CC=C(C2=CC3=CC(C4=CSC5=C4C=CC=C5)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 YNTFVQBCUSLODA-YTTGMZPUSA-N 0.000 description 1
- QPMQOADNXAJGQR-LJAQVGFWSA-N O=C(NC1=CC=C(C2=CC3=CC(C4=CSC=C4)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(NC1=CC=C(C2=CC3=CC(C4=CSC=C4)=CC=C3N2)C=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 QPMQOADNXAJGQR-LJAQVGFWSA-N 0.000 description 1
- RVXKTENPJVVMRQ-SANMLTNESA-N O=C(NC1=CC=C2C(=C1)CCC1=C2NC2=CC=C(Br)C=C21)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(NC1=CC=C2C(=C1)CCC1=C2NC2=CC=C(Br)C=C21)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 RVXKTENPJVVMRQ-SANMLTNESA-N 0.000 description 1
- LBCQZEFISAQHMB-XDSPJLLDSA-N O=C(NC1=CC=C2C=C(C3=CC(NC(=O)[C@H]4CCCN4C(=O)CC4=CC=CC=C4)=CC=C3)NC2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 Chemical compound O=C(NC1=CC=C2C=C(C3=CC(NC(=O)[C@H]4CCCN4C(=O)CC4=CC=CC=C4)=CC=C3)NC2=C1)[C@@H]1CCCN1C(=O)CC1=CC=CC=C1 LBCQZEFISAQHMB-XDSPJLLDSA-N 0.000 description 1
- KXVMIJQFXPEEMO-NRYUZRROSA-N O=C(NCC(=O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1)C1=CC=C(I)C=C1.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NN=C(C2=CC=C(I)C=C2)O1 Chemical compound O=C(NCC(=O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1)C1=CC=C(I)C=C1.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NN=C(C2=CC=C(I)C=C2)O1 KXVMIJQFXPEEMO-NRYUZRROSA-N 0.000 description 1
- IIHFUWZJCOVZIJ-VPRRGWHLSA-N O=C(NCC(=O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1)C1=CC=C2NC=CC2=C1.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NN=C(C2=CC=C3NC=CC3=C2)O1 Chemical compound O=C(NCC(=O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1)C1=CC=C2NC=CC2=C1.O=C(OCC1=CC=CC=C1)N1CCC[C@H]1C1=NN=C(C2=CC=C3NC=CC3=C2)O1 IIHFUWZJCOVZIJ-VPRRGWHLSA-N 0.000 description 1
- WQUZFEXRJSDOQC-UHFFFAOYSA-N O=C(O)CCCC(=O)C1=CC([N+](=O)[O-])=CC=C1.O=C(O)CCCC(=O)C1=CC=CC=C1 Chemical compound O=C(O)CCCC(=O)C1=CC([N+](=O)[O-])=CC=C1.O=C(O)CCCC(=O)C1=CC=CC=C1 WQUZFEXRJSDOQC-UHFFFAOYSA-N 0.000 description 1
- GASAYYFVHNKRBT-XZVVQQHRSA-N O=C1CC2=C(C=CC([N+](=O)[O-])=C2)N1.O=P(Br)(Br)Br.[2H]CF.[H]C(=O)C1=C(Br)NC2=C1C=C([N+](=O)[O-])C=C2 Chemical compound O=C1CC2=C(C=CC([N+](=O)[O-])=C2)N1.O=P(Br)(Br)Br.[2H]CF.[H]C(=O)C1=C(Br)NC2=C1C=C([N+](=O)[O-])C=C2 GASAYYFVHNKRBT-XZVVQQHRSA-N 0.000 description 1
- UEDGZCUMJAPXOM-UHFFFAOYSA-N O=C1CC2=CC([N+](=O)[O-])=CC=C2N1.O=P(Br)(Br)Br.O=[N+]([O-])C1=CC=C2NC(Br)=CC2=C1 Chemical compound O=C1CC2=CC([N+](=O)[O-])=CC=C2N1.O=P(Br)(Br)Br.O=[N+]([O-])C1=CC=C2NC(Br)=CC2=C1 UEDGZCUMJAPXOM-UHFFFAOYSA-N 0.000 description 1
- JDJFXWMXNXXTGX-UHFFFAOYSA-N O=CC1=CC(Br)=CC=C1O.O=CC1=CC(Br)=CC=C1OCC1=CC=C([N+](=O)[O-])C=C1.O=[N+]([O-])C1=CC=C(CBr)C=C1 Chemical compound O=CC1=CC(Br)=CC=C1O.O=CC1=CC(Br)=CC=C1OCC1=CC=C([N+](=O)[O-])C=C1.O=[N+]([O-])C1=CC=C(CBr)C=C1 JDJFXWMXNXXTGX-UHFFFAOYSA-N 0.000 description 1
- SSRLSSOLWKAJAT-XZVVQQHRSA-N O=CC1=CC(Br)=CC=C1OCC1=CC=C([N+](=O)[O-])C=C1.O=[N+]([O-])C1=CC=C(C2=CC3=CC(Br)=CC=C3O2)C=C1.[2H]B[U] Chemical compound O=CC1=CC(Br)=CC=C1OCC1=CC=C([N+](=O)[O-])C=C1.O=[N+]([O-])C1=CC=C(C2=CC3=CC(Br)=CC=C3O2)C=C1.[2H]B[U] SSRLSSOLWKAJAT-XZVVQQHRSA-N 0.000 description 1
- ZJUUSOIODLFLCA-UHFFFAOYSA-N O=[N+]([O-])C1=CC=C(C2=CC3=CC([N+](=O)[O-])=CC=C3O2)C=C1.[H]C(=O)C1=CC([N+](=O)[O-])=CC=C1OCC1=CC=C([N+](=O)[O-])C=C1 Chemical compound O=[N+]([O-])C1=CC=C(C2=CC3=CC([N+](=O)[O-])=CC=C3O2)C=C1.[H]C(=O)C1=CC([N+](=O)[O-])=CC=C1OCC1=CC=C([N+](=O)[O-])C=C1 ZJUUSOIODLFLCA-UHFFFAOYSA-N 0.000 description 1
- HQMBWRQPVIXJIU-UHFFFAOYSA-N OB(c1cc2cc(Br)ccc2[o]1)O Chemical compound OB(c1cc2cc(Br)ccc2[o]1)O HQMBWRQPVIXJIU-UHFFFAOYSA-N 0.000 description 1
- PZJSZBJLOWMDRG-UHFFFAOYSA-N OB(c1ccc[o]1)O Chemical compound OB(c1ccc[o]1)O PZJSZBJLOWMDRG-UHFFFAOYSA-N 0.000 description 1
- SHKWSBAVRQZYLE-UHFFFAOYSA-N OC(CCCC(c1ccccc1)=O)=O Chemical compound OC(CCCC(c1ccccc1)=O)=O SHKWSBAVRQZYLE-UHFFFAOYSA-N 0.000 description 1
- UBQCWSGRNIOFFC-NSHDSACASA-N OC([C@H](CCC1)N1C(Cc1ccccc1)=O)=O Chemical compound OC([C@H](CCC1)N1C(Cc1ccccc1)=O)=O UBQCWSGRNIOFFC-NSHDSACASA-N 0.000 description 1
- ONQWDIQOSJSULT-UHFFFAOYSA-N OONC1=CC=C(CBr)C=C1.[H]C(=O)C1=CC([N+](=O)[O-])=CC=C1O.[H]C(=O)C1=CC([N+](=O)[O-])=CC=C1OCC1=CC=C([N+](=O)[O-])C=C1 Chemical compound OONC1=CC=C(CBr)C=C1.[H]C(=O)C1=CC([N+](=O)[O-])=CC=C1O.[H]C(=O)C1=CC([N+](=O)[O-])=CC=C1OCC1=CC=C([N+](=O)[O-])C=C1 ONQWDIQOSJSULT-UHFFFAOYSA-N 0.000 description 1
- MKKSTJKBKNCMRV-UHFFFAOYSA-N Oc(c(C=O)c1)ccc1Br Chemical compound Oc(c(C=O)c1)ccc1Br MKKSTJKBKNCMRV-UHFFFAOYSA-N 0.000 description 1
- LNWWQYYLZVZXKS-QYKNYGDISA-N [2H]C1CCCN1C(C)=O Chemical compound [2H]C1CCCN1C(C)=O LNWWQYYLZVZXKS-QYKNYGDISA-N 0.000 description 1
- MBUPVGIGAMCMBT-UHFFFAOYSA-N [O-][N+](c(cc1)ccc1C(CBr)=O)=O Chemical compound [O-][N+](c(cc1)ccc1C(CBr)=O)=O MBUPVGIGAMCMBT-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/4025—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil not condensed and containing further heterocyclic rings, e.g. cromakalim
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4178—1,3-Diazoles not condensed 1,3-diazoles and containing further heterocyclic rings, e.g. pilocarpine, nitrofurantoin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4184—1,3-Diazoles condensed with carbocyclic rings, e.g. benzimidazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
- A61K31/423—Oxazoles condensed with carbocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4245—Oxadiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/427—Thiazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/475—Quinolines; Isoquinolines having an indole ring, e.g. yohimbine, reserpine, strychnine, vinblastine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4985—Pyrazines or piperazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/536—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines ortho- or peri-condensed with carbocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5386—1,4-Oxazines, e.g. morpholine spiro-condensed or forming part of bridged ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/553—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having at least one nitrogen and one oxygen as ring hetero atoms, e.g. loxapine, staurosporine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D407/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00
- C07D407/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Definitions
- the present disclosure relates to antiviral compounds that are useful as inhibitors of hepatitis C virus (HCV) replication.
- the compounds are expected to act on HCV NS5A (non-structural 5A) protein.
- Compositions comprising such compounds, the use of such compounds for treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection, methods for inhibiting the function of the NS5A non-structural protein, and methods for inhibiting HCV viral replication and/or viral production are also provided.
- HCV infection is a major health problem that leads to chronic liver disease, such as cirrhosis and hepatocellular carcinoma, in a substantial number of infected individuals.
- Current treatments for HCV infection include immunotherapy with recombinant interferon- ⁇ alone or in combination with the nucleoside-analog ribavirin.
- RNA-dependent RNA polymerase RNA-dependent RNA polymerase
- HCV NS5A non-structural protein which is described, for example, in Seng-Lai Tan & Michael G. Katze, How Hepatitis C Virus Counteracts the Interferon Response: The Jury Is Still Out on NS 5 A, 284 V IROLOGY 1-12 (2001); and in Kyu-Jin Park et al., Hepatitis C Virus NS 5 A Protein Modulates c - Jun N - terminal Kinase through Interaction with Tumor Necrosis Factor Receptor - associated Factor 2, 278(33) J. B IO . C HEM . 30711 (2003).
- a non-structural protein, NS5A is an essential component for viral replication and assembly.
- the present disclosure relates to novel compounds of formula (I) and/or pharmaceutically acceptable salts, hydrates, solvates, prodrugs or isomers thereof. These compounds are useful, either as compounds or their pharmaceutically acceptable salts (when appropriate), in the inhibition of HCV (hepatitis C virus) NS5A (non-structural 5A) protein, the prevention or treatment of one or more of the symptoms of HCV infection, the inhibition of HCV viral replication and/or HCV viral production, and/or as pharmaceutical composition ingredients.
- HCV hepatitis C virus
- NS5A non-structural 5A
- these compounds which includes reference to hydrates and solvates of such compounds, and their salts may be the primary active therapeutic agent, and, when appropriate, may be combined with other therapeutic agents including but not limited to other HCV antivirals, anti-infectives, immunomodulators, antibiotics or vaccines.
- R 1 and R 2 may be taken together with
- each D is a group independently chosen from the group consisting of:
- each G is independently chosen from the group consisting of:
- the present invention also includes pharmaceutical compositions containing a compound of the present invention and methods of preparing such pharmaceutical compositions.
- the present invention further includes methods of treating or reducing the likelihood or severity of HCV infection, methods for inhibiting the function of the NS5A protein, and methods for inhibiting HCV viral replication and/or viral production.
- the present invention includes compounds of formula (I) above, and pharmaceutically acceptable salts thereof.
- the compounds of formula (I) are HCV NS5A inhibitors.
- a first embodiment of the invention relates to compounds having structural formula (I):
- R 1 and R 2 may be taken together with
- each E is a group independently chosen from the group consisting of:
- each G is independently chosen from the group consisting of:
- each R 1 is independently chosen from the group consisting of hydrogen, halogen, —OR 3a , —CN, —C(O)R 3 , —CO 2 R 3a , —C(O)N(R 3a ) 2 , —SR 3a , —S(O)R 3a , —S(O 2 )R 3a , —(CH 2 ) 0-6 N(R 3a ) 2 , —N(R 3a )SO 2 R 3a , —N(R 3a )CO 2 R 3a , —N(R 3a )C(O)R 3 , —N(R 3a )COR 3a , —N(R 3a )C(O)N(R 3a ), C 1-6 alkyl, C 3-8 carbocycle containing from 0 to 3 heteroatoms chosen from N, O and S, and phenyl, and the C 1-6 alkyl, C 3-8 carbocycle and phen
- R′ is substituted by from 0 to 3 additional R′, which are as provided above.
- R 1 is substituted by from 0 to 3 additional R 1 , which are as provided above.
- R 1 is substituted by from 0 to 3 additional R 1 , which are as provided above.
- each R 1 is chosen from the group consisting of hydrogen, halogen, —CN and C 1-6 alkyl.
- each R 1 is chosen from the group consisting of hydrogen, fluorine and —CN.
- each R 2 is independently chosen from the group consisting of hydrogen, halogen, —OR 4a , —CN, —CO 2 R 4a , —C(O)N(R 4a ) 2 , —N(R 4a ) 2 , —N(R 4a )CO 2 R 4a , —SR 4a , —S(O)R 4a , —S(O 2 )R 4a , —N(R 4a )SO 2 R 4a , —N(R 4a )CO 2 R 4a , —N(R 4a )CO 2 R 4a , —C ⁇ C—, phenyl, pyridinyl, pyrazinyl, pyrimidyl, 1,2,4-triazinyl, pyridazinyl, thiazyl and 9-membered bicyclic ring systems that contain from 1 to 3 heteroatoms independently chosen from the group consisting of N, O and S,
- each R 2 is independently chosen from the group consisting of fluorine, chlorine, —OH, —CH 3 , —OCH 3 and —CN.
- all other groups are as provided in the general formula above and/or in the first or second embodiments.
- W is chosen from the group consisting of —(CH 2 ) 1-3 —, —(CH 2 ) 0-2 NH(CH 2 ) 0-2 —, —(CH 2 ) 0-2 N(C 1-6 alkyl)(CH 2 ) 0-2 —, —(CH 2 ) 0-2 O(CH 2 ) 0-2 — and —(CH 2 ) 0-2 C(O)(CH 2 ) 0-2 —, where W is substituted by from 0 to 4 R w , where each R w is independently selected from C 1-6 alkyl and C 3-8 cycloalkyl; and V is chosen from the group consisting of —C(O)— and —CH 2 —, and where V is —CH 2 —, V is substituted by from 0 to 2 R v , where each R v is independently selected from the group consisting of C 1-6 alkyl and C 3-8 cycloalkyl.
- W is chosen from the group consisting of —CH 2 —, —NH—, —N(C 1-6 alkyl)-, —C(O)—, —CH 2 NH—, —CH 2 N(C 1-6 alkyl)-, —CH 2 CH 2 —, —C(O)CH 2 —, —CH 2 C(O)—, —CH 2 O—, —CH 2 CH 2 CH 2 —, —C(O)CH 2 CH 2 —, —CH 2 C(O)CH 2 —, —CH 2 OCH 2 —, —CH 2 CH 2 C(O)—, —CH 2 CH 2 O—, —CH 2 CH 2 NH—, —CH 2 CH 2 N(C 1-6 alkyl)-, —CH 2 NHCH 2 —, —CH 2 N(C 1-6 alkyl)CH 2 —, —NHCH 2 CH 2 —, and ——CH 2 CH 2 NH—, —CH 2 CH 2
- each D is independently chosen from the group consisting of a single bond, —C(O)N(R 5 )—, —NR 5 C(O)—,
- R 5 is independently chosen from the group consisting of hydrogen, halogen —OR 6 , —CN, —CO 2 R 6 , —C(O)N(R 6 ) 2 , —N(R 6 ) 2 , —N(R 6 )COR 6 , —SR 6 , —S(O)R 6 , —S(O 2 )R 6 , —N(R 6 )SO 2 R 6 , —NCO 2 R 6 , —NC(O)N(R 6 ) 2 , C 1-6 alkyl substituted by from 0 to 3 substituents R 6 and C 3-8 cycloalkyl substituted by from 0 to 3 substituents R 6 , and each R 6 is independently chosen from the group consisting of hydrogen, C 1-6 alkyl and C 3-8 cycloalkyl.
- each D is independently chosen from the group consisting of hydrogen, C 1-6 alkyl and C 3-8 cycloalkyl.
- each D is independently chosen
- each E is independently chosen from the group consisting of a single bond, —CH 2 NHC(O)—, —CH 2 N(CH 3 )C(O)—, —C(CH 3 )HNHC(O)—, —C(CH 3 )HN(CH 3 )C(O)—, —C(CH 3 ) 2 NHC(O)—, —C(CH 3 ) 2 N(CH 3 )C(O)—, —CH 2 NHC(O)O—, —CH 2 N(CH 3 )C(O)O—, —C(CH 3 )HNHC(O)O—, —C(CH 3 )HN(CH 3 )C(O)O—, —C(CH 3 ) 2 NHC(O)O—, —C(CH 3 ) 2 NHC(O)O—, —C(CH 3 ) 2 NHC(O)O—, —C(CH 3 ) 2 NHC(O)O—,
- each E is independently chosen from the group consisting of a single bond
- adjacent D and E groups each may be selected to be a single bond.
- D and E are combined to be one single bond, and all other groups are as provided in the general formula above and/or in the first, second, third and fourth embodiments. That is, where D is a single bond and the adjacent E is a single bond,
- each G is independently chosen from the group consisting of:
- G is independently chosen from the group consisting of C 1-4 alkyl having 1 to 2 substituents R 11 , wherein each R 11 is independently chosen from the group consisting of —OH, —NH 2 , —NCH 3 H, —N(CH 3 ) 2 , —N(CH 2 CH 3 ) 2 , —C(O)OCH 3 , cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, pyranyl, pyrrolidinyl, piperidinyl, oxacyclopentyl, oxacyclohexyl, phenyl, pyridinyl, pyrimidinyl and pyrrolyl.
- G is chosen such that stable compounds result.
- all other groups are as provided in the general formula above and/or in the first through sixth embodiments.
- each R 1 is independently chosen from the group consisting of hydrogen, halogen, —OR 3a , —CN, —C(O)R 3 , —CO 2 R 3a , —C(O)N(R 3a ) 2 , —SR 3a , —S(O)R 3a , —S(O 2 )R 3a , —(CH 2 ) 0-6 N(R 3a ) 2 , —N(R 3a )SO 2 R 3a , —N(Z 3a )CO 2 R 3a , —N(Z 3a )C(O)R 3 , —N(R 3a )COR 3a , —N(R 3a )C(O)N(R 3a ), C 1-6 alkyl, C 3-8 carbocycle containing from 0 to 3 heteroatoms chosen from N, O and S, and phenyl, and the C 1-6 alkyl, C 3-8 carbocycle and phen
- each R 3a is independently chosen from the group consisting of hydrogen, C 1-6 alkyl and C 3-8 cycloalkyl;
- —C ⁇ C— phenyl, pyridinyl, pyrazinyl, pyrimidyl, 1,2,4-triazinyl, pyridazinyl, thiazyl and 9-membered bicyclic ring systems that contain from 1 to 3 heteroatoms independently chosen from the group consisting of N, O and S,
- each R 2 is independently chosen from the group consisting of hydrogen, halogen, —OR 4a , —CN, —CO 2 R 4a , —C(O)N(R 4a ) 2 , —N(R 4a ) 2 , —N(R 4a )CO 2 R 4a , —SR 4a , —S(O)R 4a , —S(O 2 )R 4a , —N(R 4a )SO 2 R 4a , —N(R 4a )CO 2 R 4a , —N(R 4a )C(O)N(R 4a ), C 1-6 alkyl substituted by from 0 to 4 R 4 and C 3-8 cycloalkyl substituted by from 0 to 4 R 4 ,
- each R 4 is independently chosen from the group consisting of hydrogen, —OH, C 1-6 alkyl and C 3-8 cycloalkyl, and
- each R 4a is independently chosen from the group consisting of hydrogen, C 1-6 alkyl and C 3-8 cycloalkyl;
- each D is independently chosen from the group consisting of a single bond, —C(O)N(R 5 )—, —NR 5 C(O)—,
- each G is independently chosen from the group consisting of
- aryl ring systems G′ chosen from the group consisting of: phenyl, pyridinyl and 9-membered bicyclic ring systems containing from 0 to 2 heteroatoms independently chosen from the group consisting of N and O.
- all other groups are as provided in the general formula above.
- R 1 is substituted by from 0 to 3 additional R 1 ;
- each E is independently chosen from the group consisting of a single bond
- the compound having structural formula (Ia) is a compound having structural formula (Ib):
- Y is selected from the group consisting of O and NR 1 .
- the compound having structural formula (Ia) is a compound having structural formula (Ib):
- V is —CH 2 —
- W is —(CH 2 ) 0-2 O(CH 2 ) 0-2 —
- R 1 is fluorine
- both instances of G are
- W is chosen from the group consisting of —(CH 2 ) 1-3 —, —(CH 2 ) 0-2 NH(CH 2 ) 0-2 —, —(CH 2 ) 0-2 N(C 1-6 alkyl)(CH 2 ) 0-2 —, —(CH 2 ) 0-2 O(CH 2 ) 0-2 — and —(CH 2 ) 0-2 C(O)(CH 2 ) 0-2 —, where W is substituted by from 0 to 4 R w , where each R w is independently selected from C 1-6 alkyl and C 3-8 cycloalkyl; and
- V is chosen from the group consisting of —C(O)— and —CH 2 —, and where V is —CH 2 —, V is substituted by from 0 to 2 R v , where each R v is independently selected from the group consisting of C 1-6 alkyl and C 3-8 cycloalkyl;
- each R 2 is independently chosen from the group consisting of hydrogen, halogen, —OR 4a , —CN, —CO 2 R 4a , —C(O)N(R 4a ) 2 , —N(R 4a ) 2 , —N(R 4a )CO 2 R 4a , —SR 4a , —S(O)R 4a , —S(O 2 )R 4a , —N(R 4a )SO 2 R 4a , —N(R 4a )CO 2 R 4a , —N(R 4a )C(O)N(R 4a ), C 1-6 alkyl substituted by from 0 to 4 R 4 and C 3-8 cycloalkyl substituted by from 0 to 4 R 4 ,
- aryl ring systems G′ chosen from the group consisting of: phenyl, pyridinyl and 9-membered bicyclic ring systems containing from 0 to 2 heteroatoms independently chosen from the group consisting of N and O.
- all other groups are as provided in the general formula above.
- the compound of the invention is selected from the exemplary species depicted in Examples 1 through 215 shown below, or pharmaceutically acceptable salts thereof.
- composition comprising an effective amount of a compound of formula (I) and a pharmaceutically acceptable carrier.
- HCV antiviral agent is an antiviral selected from the group consisting of HCV protease inhibitors and HCV NSSB polymerase inhibitors.
- HCV antiviral agent is an antiviral selected from the group consisting of HCV protease inhibitors and HCV NSSB polymerase inhibitors.
- HCV antiviral agent is an antiviral selected from the group consisting of HCV protease inhibitors and HCV NSSB polymerase inhibitors.
- a method of inhibiting HCV NS5A activity in a subject in need thereof which comprises administering to the subject the pharmaceutical composition of (a), (b), or (c) or the combination of (d) or (e).
- the present invention also includes a compound of the present invention for use (i) in, (ii) as a medicament for, or (iii) in the preparation of a medicament for: (a) inhibiting HCV NS5A activity, or (b) treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection, or (c) inhibiting HCV viral replication and/or HCV viral production in a cell-based system, or (d) use in medicine.
- the compounds of the present invention can optionally be employed in combination with one or more second therapeutic agents selected from HCV antiviral agents, anti-infective agents, and immunomodulators.
- Additional embodiments of the invention include the pharmaceutical compositions, combinations and methods set forth in (a)-(o) above and the uses set forth in the preceding paragraph, wherein the compound of the present invention employed therein is a compound of one of the embodiments, aspects, classes, sub-classes, or features of the compounds described above.
- the compound may optionally be used in the form of a pharmaceutically acceptable salt, or may be present in the form of a solvate or hydrate as appropriate.
- alkyl refers to any linear or branched chain alkyl group having a number of carbon atoms in the specified range.
- C 1-6 alkyl refers to all of the hexyl alkyl and pentyl alkyl isomers as well as n-, iso-, sec- and tert-butyl, n- and isopropyl, ethyl and methyl.
- C 1-4 alkyl refers to n-, iso-, sec- and tert-butyl, n- and isopropyl, ethyl and methyl.
- halogenated refers to a group or molecule in which a hydrogen atom has been replaced by a halogen.
- haloalkyl refers to a halogenated alkyl group.
- halogen refers to atoms of fluorine, chlorine, bromine and iodine (alternatively referred to as fluoro, chloro, bromo, and iodo), preferably fluorine.
- Aryl ring systems may include, where appropriate, an indication of the variable to which a particular ring atom is attached. Unless otherwise indicated, substituents to the aryl ring systems can be attached to any ring atom, provided that such attachment results in formation of a stable ring system.
- carrier refers to (i) a C 5 to C 7 monocyclic, saturated or unsaturated ring, or (ii) a C 8 to C 10 bicyclic saturated or unsaturated ring system. Each ring in (ii) is either independent of, or fused to, the other ring, and each ring is saturated or unsaturated. Carbocycle groups may be substituted as indicated. When the carbocycles contain one or more heteroatoms independently chosen from N, O and S, the carbocycles may also be referred to as “heterocycles,” as defined below.
- heterocycle broadly refers to (i) a stable 5- to 7-membered, saturated or unsaturated monocyclic ring, or (ii) a stable 8- to 10-membered bicyclic ring system, wherein each ring in (ii) is independent of, or fused to, the other ring or rings and each ring is saturated or unsaturated, and the monocyclic ring or bicyclic ring system contains one or more heteroatoms (e.g., from 1 to 6 heteroatoms, or from 1 to 4 heteroatoms) independently selected from N, O and S and a balance of carbon atoms (the monocyclic ring typically contains at least one carbon atom and the bicyclic ring systems typically contain at least two carbon atoms); and wherein any one or more of the nitrogen and sulfur heteroatoms is optionally oxidized, and any one or more of the nitrogen heteroatoms is optionally quatern
- heteroaryl ring system refers to aryl ring systems, as defined above, that include from 1 to 4 heteroatoms (non-carbon atoms) that are independently chosen from N, O and S.
- heteroaromatic rings include pyridyl, pyrrolyl, pyrazinyl, pyrimidinyl, pyridazinyl, thienyl (or thiophenyl), thiazolyl, furanyl, imidazolyl, pyrazolyl, triazolyl, tetrazolyl, oxazolyl, isooxazolyl, oxadiazolyl, thiazolyl, isothiazolyl, and thiadiazolyl.
- bicyclic heterocycles include benzotriazolyl, indolyl, isoindolyl, indazolyl, indolinyl, isoindolinyl, quinoxalinyl, quinazolinyl, cinnolinyl, chromanyl, isochromanyl, tetrahydroquinolinyl, quinolinyl, tetrahydroisoquinolinyl, isoquinolinyl, 2,3-dihydrobenzofuranyl, 2,3-dihydrobenzo-1,4-dioxinyl and benzo-1,3-dioxolyl.
- alkyl, cycloalkyl, and aryl groups are not substituted. If substituted, preferred substituents are selected from the group that includes, but is not limited to, halo, C 1 -C 20 alkyl, —CF 3 , —NH 2 , —N(C 1 -C 6 alkyl) 2 , —NO 2 , oxo, —CN, —N 3 , —OH, —O(C 1 -C 6 alkyl), C 3 -C 10 cycloalkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, (C 0 -C 6 alkyl) S(O) 0-2 —, aryl-S(O) 0-2 —, (C 0 -C 6 alkyl)S(O) 0-2 (C 0 -C 6 alkyl)C(O)NH
- heteroaryl ring described as containing from “0 to 3 heteroatoms” means the ring can contain 0, 1, 2, or 3 heteroatoms. It is also to be understood that any range cited herein includes within its scope all of the sub-ranges within that range. The oxidized forms of the heteroatoms N and S are also included within the scope of the present invention.
- any variable for example, R 1 or R 3
- its definition on each occurrence is independent of its definition at every other occurrence.
- combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
- a “stable” compound is a compound that can be prepared and isolated and that has a structure and properties that remain or can be caused to remain essentially unchanged for a period of time sufficient to allow use of the compound for the purposes described herein (e.g., therapeutic or prophylactic administration to a subject).
- Isotopically-enriched compounds within generic formula (I) can be prepared without undue experimentation by conventional techniques well known to those skilled in the art or by processes analogous to those described in the Schemes and Examples herein using appropriate isotopically-enriched reagents and/or intermediates.
- certain of the compounds of the present invention can have asymmetric centers and can occur as mixtures of stereoisomers, or as individual diastereomers, or enantiomers. All isomeric forms of these compounds, whether isolated or in mixtures, are within the scope of the present invention.
- a reference to a compound of formula (I) is a reference to the compound per se, or to any one of its tautomers per se, or to mixtures of two or more tautomers.
- the compounds of the present invention may be administered in the form of pharmaceutically acceptable salts.
- pharmaceutically acceptable salt refers to a salt that possesses the effectiveness of the parent compound and that is not biologically or otherwise undesirable (e.g., is neither toxic nor otherwise deleterious to the recipient thereof).
- Suitable salts include acid addition salts that may, for example, be formed by mixing a solution of the compound of the present invention with a solution of a pharmaceutically acceptable acid such as hydrochloric acid, sulfuric acid, acetic acid, trifluoroacetic acid, or benzoic acid.
- administration and variants thereof (e.g., “administering” a compound) in reference to a compound of the invention mean providing the compound or a prodrug of the compound to the individual in need of treatment.
- administration and its variants are each understood to include concurrent and sequential provision of the compound or salt (or hydrate) and other agents.
- composition is intended to encompass a product comprising the specified ingredients, as well as any product that results, directly or indirectly, from combining the specified ingredients.
- pharmaceutically acceptable is meant that the ingredients of the pharmaceutical composition must be compatible with each other and not deleterious to the recipient thereof.
- subject refers to an animal, preferably a mammal, most preferably a human, who has been the object of treatment, observation or experiment.
- the term also includes herein the amount of active compound sufficient to inhibit HCV NS5A and thereby elicit the response being sought (i.e., an “inhibition effective amount”).
- an “inhibition effective amount” When the active compound (i.e., active ingredient) is administered as the salt, references to the amount of active ingredient are to the free acid or free base form of the compound.
- the compounds of this invention are useful in the preparation and execution of screening assays for antiviral compounds.
- the compounds of this invention are useful for identifying resistant HCV replicon cell lines harboring mutations within NS5A, which are excellent screening tools for more powerful antiviral compounds.
- the compounds of this invention are useful in establishing or determining the binding site of other antivirals to the HCV replicase.
- the compounds of the present invention can be administered by any means that produces contact of the active agent with the agent's site of action. They can be administered by one or more conventional means available for use in conjunction with pharmaceuticals, either as individual therapeutic agents or in a combination of therapeutic agents. They can be administered alone, but typically are administered with a pharmaceutical carrier selected on the basis of the chosen route of administration and standard pharmaceutical practice.
- the compounds of the invention can, for example, be administered by one or more of the following: orally, parenterally (including subcutaneous injections, intravenous, intramuscular, intrasternal injection or infusion techniques), by inhalation (such as in a spray form), or rectally, in the form of a unit dosage of a pharmaceutical composition containing an effective amount of the compound and conventional non-toxic pharmaceutically-acceptable carriers, adjuvants and vehicles.
- Liquid preparations suitable for oral administration e.g., suspensions, syrups, elixirs and the like
- Solid preparations suitable for oral administration can be prepared according to techniques known in the art and can employ such solid excipients as starches, sugars, kaolin, lubricants, binders, disintegrating agents and the like.
- Parenteral compositions can be prepared according to techniques known in the art and typically employ sterile water as a carrier and optionally other ingredients, such as solubility aids.
- injectable solutions can be prepared according to methods known in the art wherein the carrier comprises a saline solution, a glucose solution or a solution containing a mixture of saline and glucose. Further description of methods suitable for use in preparing pharmaceutical compositions of the present invention and of ingredients suitable for use in said compositions is provided in Remington's Pharmaceutical Sciences, 18 th edition (ed. A. R. Gennaro, Mack Publishing Co., 1990).
- the compounds of this invention can be administered orally in a dosage range of 0.001 to 1000 mg/kg of mammal (e.g., human) body weight per day in a single dose or in divided doses.
- mammal e.g., human
- One dosage range is 0.01 to 500 mg/kg body weight per day orally in a single dose or in divided doses.
- Another dosage range is 0.1 to 100 mg/kg body weight per day orally in single or divided doses.
- the compositions can be provided in the form of tablets or capsules containing 1.0 to 500 mg of the active ingredient, particularly 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, and 500 mg of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated.
- the present invention also relates to a method of inhibiting HCV replicon activity, inhibiting HCV viral replication and/or HCV viral production, treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection with a compound of the present invention in combination with one or more therapeutic agents and a pharmaceutical composition comprising a compound of the present invention and one or more therapeutic agents selected from the group consisting of a HCV antiviral agent, an immunomodulator, and an anti-infective agent.
- Such therapeutic agents active against HCV include, but are not limited to, ribavirin, levovirin, viramidine, thymosin alpha-1, R7025 (an enhanced interferon (Roche)), interferon- ⁇ , interferon- ⁇ , pegylated interferon- ⁇ (peginterferon- ⁇ ), a combination of interferon- ⁇ and ribavirin, a combination of peginterferon- ⁇ and ribavirin, a combination of interferon- ⁇ and levovirin, and a combination of peginterferon- ⁇ and levovirin.
- the combination of peginterferon- ⁇ and ribaviron represents the current Standard of Care for HCV treatment.
- Interferon- ⁇ includes, but is not limited to, recombinant interferon- ⁇ 2a (such as R OFERON interferon), pegylated interferon- ⁇ 2a (P EGASYS ), interferon- ⁇ 2b (such as I NTRON -A interferon), pegylated interferon- ⁇ 2b (P EG I NTRON ), a recombinant consensus interferon (such as interferon alphacon-1), albuferon (interferon- ⁇ bound to human serum albumin (Human Genome Sciences)), and a purified interferon- ⁇ product.
- interferon- ⁇ 2a such as R OFERON interferon
- P EGASYS pegylated interferon- ⁇ 2a
- interferon- ⁇ 2b such as I NTRON -A interferon
- P EG I NTRON pegylated interferon- ⁇ 2b
- a recombinant consensus interferon such as interferon alpha
- Amgen's recombinant consensus interferon has the brand name I NFERGEN .
- Levovirin is the L-enantiomer of ribavirin which has shown immunomodulatory activity similar to ribavirin.
- Viramidine represents an analog of ribavirin disclosed in International Patent Application Publication WO 01/60379.
- the individual components of the combination can be administered separately at different times during the course of therapy or concurrently in divided or single combination forms.
- Ribavirin, levovirin, and viramidine may exert their anti-HCV effects by modulating intracellular pools of guanine nucleotides via inhibition of the intracellular enzyme inosine monophosphate dehydrogenase (IMPDH).
- IMPDH inosine monophosphate dehydrogenase
- Ribavirin is readily phosphorylated intracellularly and the monophosphate derivative is an inhibitor of IMPDH.
- inhibition of IMPDH represents another useful target for the discovery of inhibitors of HCV replication.
- the compounds of the present invention may also be administered in combination with an inhibitor of IMPDH, such as those disclosed in International Patent Application Publications WO 97/41211, WO 01/00622 and WO 00/25780; or mycophenolate mofetil. See Anthony C. Allison and Elsie M. Eugui, Immunosuppressive and Other Anti - Rheumetic Activities of Mychophenolate Mofetil, 44 (S UPPL .) A GENTS A CTION 165 (1993).
- an inhibitor of IMPDH such as those disclosed in International Patent Application Publications WO 97/41211, WO 01/00622 and WO 00/25780; or mycophenolate mofetil. See Anthony C. Allison and Elsie M. Eugui, Immunosuppressive and Other Anti - Rheumetic Activities of Mychophenolate Mofetil, 44 (S UPPL .) A GENTS A CTION 165 (1993).
- the compounds of the present invention may also be administered in combination with the antiviral agent polymerase inhibitor R7128 (Roche).
- the compounds of the present invention may also be combined for the treatment of HCV infection with antiviral 2′-C-branched ribonucleosides disclosed in Rogers E. Harry-O'Kuru et al., A Short, Flexible Route to 2′- C - Branched Ribonucleosides, 62 J. O RG . C HEM . 1754-59 (1997); Michael S. Wolfe and Rogers E. Harry-O'Kuru, A Consise Synthesis of 2′- C - Methylribonucleosides, 36 T ET . L ETT . 7611-14 (1995); U.S. Pat. No.
- Such 2′-C-branched ribonucleosides include, but are not limited to, 2′-C-methyl-cytidine, 2′-C-methyl-uridine, 2′-C-methyl-adenosine, 2′-C-methyl-guanosine, and 9-(2-C-methyl- ⁇ -D-ribofuranosyl)-2,6-diaminopurine, and the corresponding amino acid ester of the ribose C-2′, C-3′, and C-5′ hydroxyls and the corresponding optionally substituted cyclic 1,3-propanediol esters of the 5′-phosphate derivatives.
- the compounds of the present invention may also be administered in combination with an agent that is an inhibitor of HCV NS3 serine protease.
- HCV NS3 serine protease is an essential viral enzyme and has been described to be an excellent target for inhibition of HCV replication.
- Exemplary substrate and non-substrate based inhibitors of HCV NS3 protease inhibitors are disclosed in International Patent Application Publications WO 98/22496, WO 98/46630, WO 99/07733, WO 99/07734, WO 99/38888, WO 99/50230, WO 99/64442, WO 00/09543, WO 00/59929, WO 02/48116, WO 02/48172, WO 2008/057208 and WO 2008/057209, in British Patent No. GB 2 337 262, and in U.S. Pat. Nos.
- the compounds of the present invention may also be administered in combination with an agent that is an inhibitor of HCV NSSB polymerase.
- HCV NSSB polymerase inhibitors that may be used as combination therapy include, but are not limited to, those disclosed in International Patent Application Publications WO 02/057287, WO 02/057425, WO 03/068244, WO 2004/000858, WO 04/003138 and WO 2004/007512; U.S. Pat. Nos. 6,777,392, 7,105,499, 7,125,855, 7,202,224 and U.S. Patent Application Publications US 2004/0067901 and US 2004/0110717; the content of each is incorporated herein by reference in its entirety.
- Other such HCV polymerase inhibitors include, but are not limited to, valopicitabine (NM-283; Idenix) and 2′-F-2′-beta-methylcytidine (see also WO 2005/003147).
- the compounds of the present invention may also be combined for the treatment of HCV infection with non-nucleoside inhibitors of HCV polymerase such as those disclosed in U.S. Patent Application Publications US 2006/0100262 and US 2009-0048239; International Patent Application Publications WO 01/77091, WO 01/47883, WO 02/04425, WO 02/06246, WO 02/20497, WO 2005/016927 (in particular JTK003), WO 2004/041201, WO 2006/066079, WO 2006/066080, WO 2008/075103, WO 2009/010783 and WO 2009/010785; the content of each is incorporated herein by reference in its entirety.
- non-nucleoside HCV NSSB polymerase inhibitors that are used in combination with the present HCV NS5A inhibitors are selected from the following compounds: 14-cyclohexyl-6-[2-(dimethylamino)ethyl]-7-oxo-5,6,7,8-tetrahydroindolo[2,1-a][2,5]benzodiazocine-11-carboxylic acid; 14-cyclohexyl-6-(2-morpholin-4-ylethyl)-5,6,7,8-tetrahydroindolo[2,1-a][2,5]benzodiazocine-11-carboxylic acid; 14-cyclohexyl-6-[2-(dimethylamino)ethyl]-3-methoxy-5,6,7,8-tetrahydroindolo[2,1-a][2,5]benzodiazocine-11-carboxylic acid; 14-cyclohexyl-3
- HCV replicons and NS5A inhibitory activity of the present compounds may be tested using assays known in the art.
- HCV inhibitors such as those described in the Examples herein have activities in genotype 1b, 2a and 1a replicon assays of from about 1 pM to about 1 ⁇ M.
- the assay is performed by incubating a replicon harboring cell-line in the presence of inhibitor for a set period of time and measuring the effect of the inhibitor on HCV replicon replication either directly by quantifying replicon RNA level, or indirectly by measuring enzymatic activity of a co-encoded reporter enzyme such as luciferase or ⁇ -lactamase.
- a co-encoded reporter enzyme such as luciferase or ⁇ -lactamase.
- the effective inhibitory concentration of the inhibitor (EC 50 or EC 90 ) is determined. See Jan M. Vrolijk et al., A replicons - based bioassay for the measurement of interferons in patients with chronic hepatitis C, 110 J. V IROLOGICAL M ETHODS 201 (2003). Such assays may also be run in an automated format for high through-put screening. See Paul Zuck et al., A cell - based ⁇ - lactamase reporter gene assay for the identification of inhibitors of hepatitis C virus replication, 334 A NALYTICAL B IOCHEMISTRY 344 (2004).
- the synthesis of analogs containing the 4-azaindole core can be accomplished starting from a suitably protected 2-amino-5-nitropyridine 2, which can then be reduced by catalytic hydrogenation in order to convert the resulting free amino group to its hydrazine by the action of NaNO 2 and SnCl 2 .
- the resulting pyridylhydrazine can be condensed with a ketone then subjected to Fisher indole cyclization conditions to afford the indole 6.
- Acidic deprotection of the acetyl groups can be accomplished by using a strong acid to liberate the diamine, which can be selectively coupled on the more reactive aniline nitrogen using standard coupling agents, such as HATU.
- the aminopyridine group can then be acylated with a reagent, such as acetyl chloride or a carboxylic acid, in the presence of an amide bond-forming reagent.
- 2-Bromo-3-aminopyridines can be coupled to a terminally substituted alkyne using standard Sonagashira coupling procedures to give intermediates 5, which can undergo TFAA-mediated cyclization to provide the 4-azaindole compounds 6.
- Protecting groups can be removed with a strong acid, such as aqueous HCl, and the resulting amine can be acylated using an appropriately substituted carboxylic acid and an amide bond forming reagent, such as HATU.
- scaffolds B containing a 6-azaindole core can be accomplished by the metallation of the 4-methylpyridine analog 2 with a strong base, such as BuLi, and quenching the resulting anion with the acylating agent, such as 3.
- Intermediate 4 can be globally deprotected by the action of a strong acid, such as HBr, to give the diamino azaindole structure 5.
- Both amino groups can be acylated using an appropriately substituted carboxylic acid and an amide bond forming reagent, such as HATU.
- Compounds 6 can be further functionalized at the C-3 indole position with electrophiles, such as NCS.
- Iodo aminopyridines 2 can be coupled to a terminally substituted alkyne using standard Sonagashira coupling procedures to give intermediates 5, which can undergo a base-mediated cyclization using a reagent, such as KOtBu, to provide the 7-azaindole compounds 4.
- Protecting groups can be removed with a strong acid, such as aqueous HCl, and the resulting amine can be acylated using an appropriately substituted carboxylic acid and an amide bond forming reagent, such as HATU.
- Compounds 6 can then be reduced using hydrogen and a catalyst then coupled a second time with a carboxylic acid and HATU to provide 8.
- Treatment of 8 with an electrophilic agent, such as NCS provides the desired compounds.
- Compounds in the D series can be synthesized by reacting dicarbonyl intermediate 2 with a 2-aminopyrimidine derivative in the presence of a Lewis acid, such as boron trifluoride etherate.
- a Lewis acid such as boron trifluoride etherate.
- the resulting heterocycle can be alkylated with a bromoketone analog of an amino acid, such as proline, in the presence of a tertiary amine base.
- the nitro group can be reduced, and the resulting aniline can be acylated using an appropriately substituted carboxylic acid and an amide bond forming reagent, such as HATU, to give the final products.
- Scaffold E-1 can be prepared by the condensing a benzoic acid derivative, such as 1, with a phenylenediamine counterpart 2 in the presence of a dehydrating agent, such as polyphosphoric acid.
- the resulting aniline can be acylated using an appropriately substituted carboxylic acid, such as N-Boc-L-proline, and an amide bond-forming reagent, such as HATU, and can then be subjected to acidic conditions to remove the Boc group.
- Compounds 4 can be coupled again with an appropriately substituted carboxylic acid and an amide bond-forming reagent, such as HATU.
- the nitro group in 5 can be reduced under catalytic hydrogenating conditions, and the resulting aniline can be further coupled with various amines to give the target compounds.
- Scaffold E-2 can be prepared by the reacted with a benzoic acid derivative with a phenylenediamine analog and an amide bond-forming reagent, such as HATU, to give amides 3, which can be cyclodehydrated by heating with a reagent, such as HOAc.
- the resulting aniline can be acylated using an appropriately substituted carboxylic acid and an amide bond-forming reagent, such as HATU, to give intermediates 5.
- the nitro group can be reduced under catalytic hydrogenating conditions, and the resulting aniline can be sulfonylated with an appropriately substituted sulfonyl chloride and a tertiary amine base to give the targets.
- Scaffold F can be prepared by the condensing a benzoic acid derivative, such as 1, with an amino phenol counterpart 2 in the presence of a dehydrating agent, such as polyphosphoric acid.
- the resulting aniline can be acylated using an appropriately substituted carboxylic acid, such as N-Boc-L-proline, and an amide bond-forming reagent, such as HATU, and can then be subjected to acidic conditions to remove the Boc group.
- Compounds 4 can be coupled again with an appropriately substituted carboxylic acid and an amide bond-forming reagent, such as HATU.
- the synthesis can be modified by reacting an appropriately substituted bromo salicylaldehyde with a benzyl halide, such as 4-nitrobenzyl bromide, in the presence of a tertiary amine base to give ethers 2.
- a benzyl halide such as 4-nitrobenzyl bromide
- the benzylic ethers can be treated with a base, such as DBU, and heated to elevated temperatures to effect cyclization to the benzofurans 3.
- the aryl bromide can be converted to the aryl amine by reaction with LHMDS and a palladium catalyst to provide 4, which can be coupled to an appropriately substituted carboxylic acid to give 5.
- the nitro group in 5 can be reduced under catalytic hydrogenating conditions, and the resulting aniline can be coupled with a second carboxylic acid analog to give the target compounds G-2.
- Appropriately substituted aminopyrimidines can be cyclodehydrated after acylation with an appropriately substituted ketone, such as 4′-nitro-2-bromobenzophenone, by heating in a solvent, such as MeOH, and an acid source, such as HBr.
- the resulting heterocyclic nitro compound can be converted to the aromatic amine by reduction with a reagent, such as SnCl 2 .
- the final compounds H can be obtained by reacting 4 with an appropriately substituted carboxylic acid and an amide bond-forming reagent such as HATU.
- Compounds in scheme I can be prepared by reacting the appropriately substituted aminopyridine with 4′-nitro-2-bromobenzophenone by heating in a solvent, such as acetone, then effecting a cyclodehydration reaction using methanol and an acid source, such as HBr.
- a solvent such as acetone
- the resulting heterocyclic nitro compound 3 can be converted to the aromatic amine by reduction with a reagent such as SnCl 2 .
- the final compounds can be obtained by reacting 4 with an appropriately substituted carboxylic acid and an amide bond-forming reagent, such as HATU.
- Compounds in scheme J can be prepared by coupling indole boronic acids with an appropriately substituted 2-bromoindole, such as 2, under standard Suzuki conditions.
- the protecting groups can be removed with HCl, and the nitro group in 4 can be reduced under catalytic hydrogenation conditions.
- the penultimate diamine can be coupled with an appropriately substituted carboxylic acid and an amide bond-forming reagent, such as BOP, reagent to give compounds with the targeted central scaffold.
- the synthesis of compounds with the indole core scaffold K can be prepared using standard Fisher indole synthesis protocol starting for an aryl hydrazine and a ketone such as 2. Conversion of the aryl bromide to the aryl amine 4 could be effected by the Pd-catalyzed reaction with LHMDS. The nitro group could be reduced and the diamine can be coupled with an appropriately substituted carboxylic acid and an amide bond-forming reagent, such as HATU, to give compounds with the targeted scaffold.
- indoles K can be prepared starting from a suitably protected and substituted aminoindole 3.
- Lithiation and quenching with a boronate ester affords key intermediate 4, which can be coupled to an appropriately substituted aryl or heteroaryl halide to provide targets 5.
- the Boc groups can be removed with acid, and the resulting aniline can be coupled with an appropriately substituted carboxylic acid and an amide bond-forming reagent, such as HATU.
- the nitro group in 7 can be reduced and coupled in a second amide coupling reaction to give the desired compounds.
- Tetracyclic indole scaffold L can be prepared as outline in the scheme above. Cyclization of a carboxylic acid derivative 2 with PPA can provide the ketones 3, which can participate in a Fischer indole reaction with an appropriately substituted phenylhydrazine to give 4. The acetamide groups can be deprotected under acidic conditions and the resulting aryl amines can be coupled with an appropriately substituted carboxylic acid and an amide bond-forming reagent, such as HATU, to give compounds with the targeted scaffold.
- an amide bond-forming reagent such as HATU
- Scaffold M-1 compounds can be prepared by coupling proline 2 to amino ketone 1 using standard amide bond-forming procedures to provide 3, which can be cyclized upon heating with ammonium acetate at elevated temperatures.
- Intermediate 4 can be coupled to indole boronic acids, such as using standard Suzuki-type conditions.
- the Boc groups can be removed with acid, and the resulting aniline can be coupled with an appropriately substituted carboxylic acid and an amide bond-forming reagent, such as HATU.
- the pyrrolidine protecting group can be removed under hydrogenating conditions, and the resulting amine can coupled in a second amide coupling reaction to give the desired compounds.
- Scaffold M-2 compounds can be prepared by reacting proline 1 with an anion of 4-ethynylbenzene to give intermediate ketone 2, which can be cyclized with hydrazine.
- Intermediate 3 can be coupled to indole boronic acids, such as using standard Suzuki-type conditions.
- the Boc groups can be removed with acid and the resulting aniline can be coupled with an appropriately substituted carboxylic acid, and an amide bond-forming reagent such as HATU.
- the pyrrolidine protecting group can be removed under hydrogenating conditions, and the resulting amine can coupled in a second amide coupling reaction to give the desired compounds.
- Thiazole analogs of scaffold M can be prepared from the cyclocondensation reaction of Z-proline thioamide 2 with an alpha-bromoacetophenone. Products 3 can be processed to the final compounds using methodology similar to that described in scheme M-2.
- Imidazole analogs of scaffold M can be prepared from the cyclocondensation reaction of Z-proline bromomethyl ketone 1 with an aromatic amidine derivative. Products 3 can be processed to the final compounds using methodology similar to that described in scheme M-2.
- Isomeric imidazoles can be prepared starting from a protected amino acid aldehyde, such as 1, and glyoxal in the presence of ammonia. Halogenation of the resulting imidazole 2 with NBS can be followed by a Pd-catalyzed cross coupling reaction with a functionalized indole boronate ester, such as 4. Deprotection, reduction and coupling with an appropriately substituted carboxylic acid and an amide bond-forming reagent, such as HATU, can provide intermediate compounds 8. A second deprotection/amide-coupling procedure can provide the targeted M-5 scaffold.
- a protected amino acid aldehyde such as 1, and glyoxal in the presence of ammonia.
- Halogenation of the resulting imidazole 2 with NBS can be followed by a Pd-catalyzed cross coupling reaction with a functionalized indole boronate ester, such as 4.
- Oxadiazole compounds can be prepared starting from indole hydrazide 2 and coupling to an amino acid, such as Z-proline. Cyclodehydration of intermediate 3 can be effected with a reagent, such as TPP/iodine, to give the desired oxadiazole, which can be protected on the indole nitrogen with Boc anhydride. Introduction of the boronic acid functional group activates compound 6 for coupling with a substituted aryl halide 7 to give intermediate 8. Removal of the cbz and Boc groups afford the penultimate structure 10, which can be coupled with an appropriately substituted carboxylic acid and an amide bond-forming reagent to give the targets M-6.
- a reagent such as TPP/iodine
- Oxadiazole analogs of scaffold M can be prepared by cyclocondensation reactions of diacylhydrazines 2. Coupling to heterocyclic boronic acids using methodology similar to that described in scheme M-1 can provide the targeted compounds.
- Double imidazole containing benzofuran compounds can be prepared starting from a protected amino acid aldehyde, such as 2, and glyoxal in the presence of ammonia. Halogenation of the resulting imidazole 3 with NBS can ultimately provide intermediate 5, which can be coupled to a functionalized boronate ester, such as 11, to provide 12. Deprotection and coupling with an appropriately substituted carboxylic acid and an amide bond-forming reagent, such as HATU, can provide the targeted M-8 scaffold.
- benzofurans can be realized starting from benzofuran 1, which can be converted to boronate ester 2, which can then coupled to an appropriately substituted aryl halide to afford 5.
- Intermediate 5 can subsequently be converted to a functionalized boronate ester and converted to the final products in a manner similar to that described in Scheme M-8.
- Benzoxazoles 3 can be prepared starting from a suitable substituted benzoic acid and an aminophenol, such as 2, in the presence of polyphosphoric acid. Such products can be converted to the corresponding boronate esters using standard procedures. Intermediates 4 can subsequently be coupled to a heterocyclic halide in the presence of a Pd(II) catalyst to provide compounds 5. Deprotection and coupling with an appropriately substituted carboxylic acid and an amide bond-forming reagent, such as HATU, can provide the targeted M-10 scaffold.
- an appropriately substituted carboxylic acid and an amide bond-forming reagent such as HATU
- the compounds in scheme N-1 can be prepared by heating hydrazines 1 with ketones 2 in a microwave reactor in a polar aprotic solvent, such as NMP.
- the indole acetamides 3 can be deprotected with strong acid, such as HCl.
- the resulting aryl amines can be coupled with an appropriately substituted carboxylic acid, and an amide bond-forming reagent, such as HATU, to give compounds of the targeted scaffold.
- Scaffold O can be prepared by reacting a protected proline compound (such as Cbz) with a phenylenediamine analog and an amide bond-forming reagent, such as HATU, to give amides 3, which can be cyclodehydrated by heating with a reagent, such as HOAc.
- a protected proline compound such as Cbz
- a phenylenediamine analog such as HATU
- amides 3 can be cyclodehydrated by heating with a reagent, such as HOAc.
- the resulting benzimidazole can be coupled to an indole boronic acid derivative using standard Suzuki conditions to provide 5.
- Removal of the Boc groups with acid provides 7, which can be acylated using an appropriately substituted carboxylic acid, such as Boc-L-proline, and an amide bond-forming reagent, such as HATU, to give intermediates 7.
- the Cbz group can be reduced under catalytic hydrogenating conditions, and the Boc group can be de
- Heterocycles can be fluorinated at C-3 by the action of electrophilic fluorinating agents, such as S ELECTFLUOR , to provide targets P-1.
- electrophilic fluorinating agents such as S ELECTFLUOR
- C-3 halogenated compounds can be converted to the corresponding cyano analogs by cyanating agents, such as CuCN.
- the compounds in scheme P-4 can be functionalized by the acylating indoles with Grignard reagents and zinc chloride.
- the compounds in scheme P-5 can be functionalized by deprotonating the indoles with a base such as ethylmagnesium bromide and treating the resulting intermediate with chlorosulfonyl isocyanate.
- a base such as ethylmagnesium bromide
- the indoles 3 can be prepared using Vilsmeier-Haack conditions, which can subsequently be protected and coupled under Suzuki conditions to give intermediates 5.
- the aldehydes can be oxidized using standard methodology for carboxylic acid formation.
- Amide coupling of the aniline from scheme K-1 with an appropriately substituted carboxylic acid and a coupling agent can provide intermediates 2, which can then be subjected to Pd-catalyzed cross-coupling reactions to provide the final targets R.
- Compounds in scheme S can be prepared by coupling indole boronic acids with an appropriately substituted 2-bromobenzoxazoles, such as 5, under standard Suzuki conditions.
- the nitro group in 6 can be reduced under catalytic hydrogenation conditions and the protecting groups can be removed with HCl.
- the penultimate diamine can be coupled with an appropriately substituted carboxylic acid and an amide bond-forming reagent, such as BOP, reagent to give compounds with the targeted central scaffold.
- Compounds in scheme T can be prepared by starting from a suitably substituted phenol 3 and a hydrazine reagent, such as 4, using established Fisher indole conditions.
- the indole 3 position can then be functionalized or the indole NH can be cyclized onto the C-2 aromatic ring using standard conditions to give tetracycles 8, which can subsequently be converted to the corresponding boronate esters using standard procedures.
- Intermediates 9 can then be coupled to a heterocyclic halide in the presence of a Pd(II) catalyst to provide compounds 10.
- Deprotection and coupling with an appropriately substituted carboxylic acid and an amide bond-forming reagent, such as HATU can provide the targeted T scaffold.
- step 4 The product from step 4 was pivaloylated using conditions described in step 2.
- step 7 The product from step 7 was coupled to 2 equivalents of N-phenylacetyl-L-proline using 2 equivalents of HATU and DIEA in a manner similar to that shown in Example 1.
- Arylglyoxal hydrate (5 g, 27.8 mmol) was added in one portion over a slurry of the heterocyclic amine (2.773 g, 29.2 mmol) in methylene chloride (10 ml). The resulting suspension was treated with 1 drop of freshly distilled BF 3 .Et 2 O and stirred until most of the amine was consumed. The reaction products were isolated as hydrates by filtration of the thick, intensely colored reaction mixture. The residue obtained by concentration is allowed to cool, filtered with suction, washed twice with diethyl ether and dried under reduced pressure to give the desired product (4 g). MS (ESI) m/e (M+H + ): 256.
- N-protected proline (10 g, 42.8 mmol) in dry ether (60 ml) and THF (60 ml) was stirred under argon at ⁇ 25° C.
- TEA (42.8 mol, 4.08 ml)
- ethyl chloroformate (42.8 mmol, 2.6 4.14 ml) were added to this solution.
- the solution was stirred for further 30 minutes, the temperature then allowed to reach ⁇ 10° C., and the diazomethane solution in ether (2-3 equivalents) was added drop wise.
- the suspension was stirred for an additional 3 hours and allowed to reach ambient temperature.
- the triethylamine hydrochloride was then filtered off, and the filtrate was evaporated to half of its original volume.
- step 3 The product from step 3 (0.370 g, 1.052 mmol), pyridine-3-carboxylic acid (0.157 g, 1.27 mmol), HATU (1.2 g, 3.18 mmol) and DIPEA (0.814 g, 6.36 mmol) were taken in DMF (10 mL) and stirred for overnight at RT. DMF was removed under reduced pressure, and the residue was extracted with DCM/water. The organic layer was washed with brine, dried (NaSO 4 ), concentrated and purified by column (DCM:MeOH/100:1) to afford 300 mg of desired compound.
- step 4 The product from step 4 (0.300 g, 0.657 mmol) was taken in MeOH (10 mL) and Pd/C (0.07 g) was added under N 2 . The reaction was stirred for overnight at RT under H 2 . The Pd/C was filtered through CELITE, and the filtrate was concentrated under reduced pressure to afford 234 mg of desired compound.
- step 5 The product from step 5 (0.370 g, crude), N-Boc-proline (0.157 g, 1.27 mmol), HATU (1.2 g, 3.18 mmol) and DIPEA (0.814 g, 6.36 mmol) were taken in DMF (10 mL) and stirred for overnight at RT. DMF was removed under reduced pressure, and the residue was extracted with DCM/water. The organic layer was washed with brine, dried (NaSO 4 ), concentrated and purified by column (DCM:MeOH/100:1) to afford 300 mg of targeted compound.
- step 7 The product from step 7 (0.100 g, 0.191 mmol), cyclobutanecarboxylic acid (0.018 mg, 0.183 mmol), HATU (0.116 g, 0.305 mmol) and DIPEA (0.059 g, 0.416 mmol) were taken in DMF (5 mL) and stirred for overnight at RT. DMF was removed under reduced pressure, and the residue was extracted with DCM/water. The organic layer was washed with brine, dried (NaSO 4 ) and concentrated. The residue was purified by HPLC purification to afford 12 mg of the final product.
- step 4 The product from step 4 (0.70 g, 1.13 mmol) was stirred in MeOH/HCl (20 mL) for 1 hour. The solvent was removed at high vacuum to afford the desired proline compound, which was used directly to the next step without further purification.
- 3-Phenylpropanoic acid (0.428 g, 2.85 mmol) were taken in DCM (30 mL) was reacted with the proline compound (0.500 g, 0.96 mmol) and DIPEA (0.9 g, 7.1 mmol). The reaction was stirred at RT for 5 minutes, and then HATU (1.0 g, 2.85 mmol) was added. The reaction was stirred overnight and poured into brine and extracted with EtOAc.
- step 2 The product from step 2 (600 mg, 1.3 mmol) was added into HCl (30 ml, 3M in MeOH). Then the mixture stirred at RT for 2-3 hours. When reaction was complete, the mixture was concentrated to give the crude product (400 mg). MS (ESI) m/e (M+H + ): 293.
- step 3 The product from step 3 (400 mg, 1.36 mmol) was dissolved in EtOAc and treated with Pd/C (100 mg, 20%). Then the mixture was stirred at RT overnight under H 2 atmosphere. When the reaction was complete, the Pd/C was filtered off, and the resulting solution was concentrated to give the crude product (300 mg). MS (ESI) m/e (M+H + ): 263.
- step 6 The product of step 6 above (0.22 mmol), N-phenylacetyl-L-proline (51 mg, 0.22 mmol), DIPEA (100 mg), and DMF (3 mL) was added HATU (84 mg, 0.22 mmol), and the mixture was stirred at RT overnight. The mixture was purified by RPLC to afford the product. MS (ESI) m/e (M+H + ): 656.
- HATU (20 g, 52.3 mmol) was added to a heterogeneous mixture of the amino ketone (12 g, 48.5 mmol) and L-Cbz-Pro (12.4 g, 50 mmol) in MeCN (156 mL). The mixture was cooled in an ice-water bath, and immediately afterward DIPEA (27 mL, 155 mmol) was added dropwise. After the addition of the base, the cooling bath was removed, and the reaction mixture was stirred for an additional 50 minutes. The volatile component was removed, and water (125 mL) was added to the resulting crude solid and stirred for about 1 hour. The off-white solid was filtered and washed with copious water, and dried in vacuo to provide the desired compound as a white solid (20.68 g). MS (ESI) m/e (M+H + ): 446.
- step 5 The product from step 5 (100 mg, 0.15 mmol) was dissolved in MeOH and treated with 20 mg of 20% Pd(OH) 2 then hydrogenated at 45 psi for 4 hours. The catalyst was removed by filtration through CELITE, and the filtrate was evaporated to leave the desired product. MS (ESI) m/e (M+H + ): 541.
- NBS (838.4 mg, 4.71 mmol) was added in batches over 15 minutes to a cooled (ice/water) CH 2 Cl 2 (20 mL) solution of imidazole (1.06 g, 4.50 mmol). The reaction mixture was stirred for 75 minutes and concentrated. The crude material was purified by RPLC to separate the mono bromide from its dibromo analog and the starting material. The HPLC elute was neutralized with excess NH 3 /MeOH, and the volatile component was removed in vacuo. The residue was partitioned between CH 2 Cl 2 and water, and the aqueous layer was extracted with water. The combined organic phase was dried (MgSO 4 ), filtered, and concentrated to provide compound as a white solid (374 mg).
- step 1 To a mixture of the benzofuran from Example 19, step 1 (15 g, 0.05 mol), bis(pinacolato)diboron (25.4 g, 0.1 mol), Pd(dppf)Cl 2 (1 g), KOAc (0.1 mol) in dioxane (500 mL) was stirred at reflux under N 2 atmosphere for 2 hours. Concentration of the reaction mixture left a residue that was chromatographed to give the desired compound (12 g).
- step 7 The product from step 7 (0.15 mmol) was dissolved in MeOH and treated with 20 mg of 20% Pd(OH) 2 then hydrogenated at 45 psi for 4 hours.
- the catalyst was removed by filtration through CELITE, and the filtrate was evaporated then dissolved in 1 mL of DCM then treated with 1 mL of TFA. After stirring for 2 hours, the mixture was evaporated and the residue was used directly in the next reaction without further purification.
- step 3 The product from step 3 (500 mg, 0.9 mmol) was stirred in MeOH/HCl (20 mL) for 16 hours. The solvent was removed under reduced pressure and the residue was dried at high vacuum. MS (ESI) m/e (M+H + ): 452.
- step 5 The product from step 5 (290 mg, 0.45 mmol) was dissolved in 5 mL of acetic acid and HBr (1 mL) was added. The reaction mixture was heated to 70-80° C. and stirred for 4 hours. The mixture was cooled to RT and concentrated in vacuo. The residue was extracted with EtOAc (2 ⁇ ), washed with aq NaHCO 3 and water (30 mL) and brine (30 mL), dried over anhydrous sodium sulfate. Evaporation of the solvent afforded the desired compound as brown solid (160 mg). MS (ESI) m/e (M+H + ): 415.
- Example 158 tert-butyl ⁇ (1R)-2-[(2S)-2-( ⁇ 4-[5-( ⁇ [(2S)-1- ⁇ (2R)-2-[(tert-butoxycarbonyl)amino]-2-phenylacetyl ⁇ pyrrolidin-2-yl]carbonyl ⁇ amino)-3-(cyclopropylcarbamoyl)-1H-indol-2-yl]phenyl ⁇ carbamoyl)pyrrolidin-1-yl]-2-oxo-1-phenylethyl ⁇ carbamate
- Example 159 tert-butyl ⁇ (1R)-2-[(2S)-2-( ⁇ 4-[5-( ⁇ [(2S)-1- ⁇ (2R)-2-[(tert-butoxy-carbonyl)amino]-2-phenylacetyl ⁇ pyrrolidin-2-yl]carbonyl ⁇ amino)-3-(4-methoxyphenyl)-1H-indol-2-yl]phenyl ⁇ carbamoyl)pyrrolidin-1-yl]-2-oxo-1-phenylethyl ⁇ carbamate
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description

and/or a pharmaceutically acceptable salt thereof, wherein:
is chosen from the group consisting of 9-membered bicyclic aryl ring systems that contain from 0 to 4 heteroatoms independently chosen from the group consisting of N, O and S, and that are substituted on C or N atoms by u substituents
-
- each R1 is independently chosen from the group consisting of hydrogen, halogen, —OR3a, —CN, —(CH2)0-6C(O)R3, —CO2R3a, —C(O)N(R3a)2, —SR3a, —S(O)R3a, —S(O2)R3a, —(CH2)0-6N(R3a)2, —N(R3a)SO2R3a, —N(R3a)CO2R3a, —N(R3a)C(O)R3, —N(R3a)COR3a, —N(R3a)C(O)N(R3a), C1-6alkyl, C3-8carbocycle containing from 0 to 3 heteroatoms chosen from N, O and S, and phenyl, and the C1-6alkyl, C3-8carbocycle and phenyl are substituted by from 0 to 3 substitutents independently chosen from the group consisting of hydrogen, halogen, —OR3a, —CN, —CO2R3a, —C(O)N(R3a)2, —N(R3a)2, —N(R3a)CO2R3a, —SR3a, —S(O)R3a, —S(O2)R3a, —N(R3a)SO2R3a, —N(R3a)CO2R3a, —N(R3a)C(O)N(R3a), C1-6alkyl, —O—C1-6alkyl, —S—C1-6alkyl, and C3-8cycloalkyl,
- u is from 0 to 4,
- each R3 is independently chosen from the group consisting of hydrogen, C1-6alkyl, —OH, —O—C1-6alkyl and C3-8cycloalkyl, and
- each R3a is independently chosen from the group consisting of hydrogen, C1-6alkyl and C3-8cycloalkyl;
is a group chosen from the group consisting of
-
- (a) —C≡C— and
- (b) aryl ring systems B′ chosen from the group consisting of:
- (i) 5- to 7-membered monocyclic ring systems and
- (ii) 8- to 10-membered bicyclic ring systems,
- and the aryl ring systems B′ containing from 0 to 4 heteroatoms independently chosen from the group consisting of N, O and S, and substituted on C or N atoms by v substituents R2,
- each R2 is independently chosen from the group consisting of hydrogen, halogen, —OR4a, —CN, —CO2R4a, —C(O)R4a, —C(O)N(R4a)2, —N(R4a)2, —N(R4a)COR4, —N(R4a)CO2R4a, —N(R4a)C(O)N(R4a), —N(R4a)SO2R4a, —SR4a, —S(O)R4a, —S(O2)R4a, C1-6alkyl substituted by from 0 to 4 R4 and C3-8cycloalkyl substituted by from 0 to 4 R4,
- v is from 0 to 4,
- each R4 is independently chosen from the group consisting of hydrogen, —OH, C1-6alkyl and C3-8cycloalkyl;
- each R4a is independently chosen from the group consisting of hydrogen, C1-6alkyl and C3-8cycloalkyl;
and
to form a 5- to 9-membered carbocyclic ring containing 1 or 2 heteroatoms independently chosen from the group consisting of N, O and S;
-
- (a) a single bond,
- (b) —C(O)N(R5)—,
- (c) —N(R5)C(O)—, and
- (d) a 5- or 6-membered aryl ring system D′ containing from 0 to 4 heteroatoms independently chosen from the group consisting of N, O and S, and substituted on C or N atoms by from 0 to 2 substituents R5,
- each R5 is independently chosen from the group consisting of hydrogen, halogen, —OR6, —CN, —CO2R6, —C(O)N(R6)2, —N(R6)2, —N(R6)COR6, —SR6, —S(O)R6, —S(O2)R6, —N(R6)SO2R6, —NCO2R6, —NC(O)N(R6)2, C1-6alkyl substituted by from 0 to 3 R6 and C3-8cycloalkyl substituted by from 0 to 3 R6, and
- each R6 is independently chosen from the group consisting of hydrogen, C1-6alkyl and C3-8cycloalkyl;
-
- (a) a single bond,
- (b) —(C(R7)2)0-2NR7C(O)O04—, and
- (c) a pyrrolidinyl derivative chosen from the group consisting of:

-
-
- I is a bivalent group chosen from —C(O)—, —CO2— and —C(O)N(R7)—,
- J is a fused ring system chosen from the group consisting of 3- to 7-membered carbocycles and 5- or 6-membered aryl rings containing from 0 to 4 heteroatoms independently chosen from the group consisting of N, O and S, and substituted on C or N atoms by substituents R9,
- each R8a is independently chosen from the group consisting of hydrogen, halogen, —OH, —OC1-6alkyl and C1-6alkyl, or two R8a may be taken together to form oxo,
- each R8b is independently chosen from the group consisting of hydrogen, halogen, —OH, —OC1-6alkyl and C1-6alkyl, or two R8b may be taken together to form oxo,
- each R8c is independently chosen from the group consisting of hydrogen and C1-6alkyl,
- or any two groups selected from R8a, R8b and R8c may be taken together to form a spiro-bicyclic or bridged bicyclic ring;
- each R9 is independently chosen from the group consisting of hydrogen, halogen, C1-6alkyl, —O—C1-6alkyl, —S—C1-6alkyl, —NH—C1-6alkyl and —NHC(O)—C1-6alkyl,
- each R7 is independently chosen from the group consisting of hydrogen, C1-6alkyl and phenyl, and the C1-6alkyl and phenyl are substituted by from 0 to 3 substitutents independently chosen from the group consisting of hydrogen, halogen, C1-6alkyl, —O—C1-6alkyl and —S—C1-6alkyl; and
-
-
- (a) hydrogen,
- (b) —OR10a,
- (c) —CN,
- (d) —CO2R10a,
- (e))—C(O)N(R10)2,
- (f) —SR10a,
- (g) —S(O)R10a,
- (h) —S(O2) R10a,
- (i) —N(R10)2,
- (j) —N(R10)SO2R10a,
- (k) —NCO2R10a,
- (l) —NC(O)N(R10)2,
- (m) C1-6alkyl having 0 to 4 substituents R11,
- each R11 is independently chosen from the group consisting of:
- (i) —OH,
- (ii) —N(R10)2,
- (iii) ═NR10,
- (iv) —O—C1-6alkyl,
- (v) —C(O)R10,
- (vi) —S—C1-6alkyl,
- (vii) —SO2—C1-6alkyl,
- (viii) 3- to 8-membered carbocycles containing from 0 to 3 heteroatoms independently chosen from the group consisting of N, O and S, and having from 0 to 3 substitutents R12 on N or C atoms, and each R12 is independently selected from the group consisting of hydrogen, halogen, C1-6alkyl having from 0 to 3 substituents chosen from R10, —O—C1-6alkyl, —S—C1-6alkyl, —OR10a, —CN, —C(O)R, —CO2R10a, —C(O) N(R10)2, —SR10a, —S(O)R10a, —S(O2)R10a, —N(R10)SO2R10a, —NCO2R10a, —NC(O)N(R10)2 and —N(R10)2, or two R12 are taken together to form oxo, and
- (ix) 5- or 6-membered aryl containing from 0 to 3 heteroatoms independently chosen from the group consisting of N, O and S, and having from 0 to 3 substitutents R13 on N or C atoms, and each R13 is independently selected from the group consisting of hydrogen, halogen, C1-6alkyl, —O—C1-6alkyl and 3- to 8-membered carbocycles containing from 0 to 3 heteroatoms independently chosen from the group consisting of N, O and S,
- each R11 is independently chosen from the group consisting of:
- (n) 3- to 8-membered carbocycles containing from 0 to 3 heteroatoms independently chosen from the group consisting of N, O and S, and having from 0 to 3 substitutents R10 on N or C atoms; and
- (o) aryl ring systems G′ chosen from the group consisting of:
- (i) 5- to 7-membered monocyclic ring systems and
- (ii) 8- to 10-membered bicyclic ring systems,
- and the aryl ring systems G′ containing from 0 to 4 heteroatoms independently chosen from the group consisting of N, O and S, and substituted on C or N atoms by 0 to 3 substitutents R10;
- each R10 is independently chosen from the group consisting of
- (i) hydrogen,
- (ii) —CN,
- (iii) C1-6alkyl,
- (iv) —O—C0-6alkyl,
- (v) —S—C0-6alkyl,
- (vi) C1-6alkyl-O—R14,
- (vii) —C(O)R14,
- (viii) —CO2R14,
- (ix) —SO2R14,
- (x) —N(R14)2,
- (xi) —N(R14)SO2R14,
- (xii) —NCO2R14,
- (xiii) —NC(O)N(R14)2, and
- (xiv) 3- to 8-membered carbocycles containing from 0 to 3 heteroatoms independently chosen from the group consisting of N, O and S,
- or two R10 may be taken together to form oxo;
- each R10a is independently chosen from the group consisting of
- (i) hydrogen,
- (ii) —CN,
- (iii) C1-6alkyl,
- (iv) C1-6alkyl-O—R14,
- (v) —C(O)R14,
- (vi) —CO2R14,
- (vii) —SO2R14,
- (x) —N(R14)2,
- (xi) —N(R14)SO2R14,
- (xii) —NCO2R14,
- (xiii) —NC(O)N(R14)2, and
- (xiv) 3- to 8-membered carbocycles containing from 0 to 3 heteroatoms independently chosen from the group consisting of N, O and S,
- and two R10 or R10a groups can be taken together with the N to which they are attached to form a ring, which may be substituted by from 0 to 3 substituents R14, and
- each R14 is independently chosen from the group consisting of hydrogen, C1-6alkyl, C3-8cycloalkyl, —(CH2)0-3C3-8cycloalkyl and phenyl.

and/or a pharmaceutically acceptable salt thereof, wherein:
is chosen from the group consisting of 9-membered bicyclic aryl ring systems that contain from 0 to 4 heteroatoms independently chosen from the group consisting of N, O and S, and that are substituted on C or N atoms by u substituents R′,
-
- each R1 is independently chosen from the group consisting of hydrogen, halogen, —OR3a, —CN, —C(O)R3, —CO2R3a, —C(O)N(R3a)2, —SR3a, —S(O)R3a, —S(O2)R3a, —(CH2)0-6N(R3a)2, —N(R3a)SO2R3a, —N(R3a)CO2R3a, —N(R3a)C(O)R3, —N(R3a)COR3a, —N(R3a)C(O)N(R3a), C1-6alkyl, C3-8carbocycle containing from 0 to 3 heteroatoms chosen from N, O and S, and phenyl, and the C1-6alkyl, C3-8carbocycle and phenyl are substituted by from 0 to 3 substitutents independently chosen from the group consisting of hydrogen, halogen, —OR3a, —CN, —CO2R3a, —C(O)N(R3a)2, —N(R3a)2, —N(R3a)CO2R3a, —SR3a, —S(O)R3a, —S(O2)R3a, —N(R3a)SO2R3a, —N(R3a)CO2R3a, —N(R3a)C(O)N(R3a), C1-6alkyl, —O—C1-6alkyl and —S—C1-6alkyl,
- u is from 0 to 4,
- each R3 is independently chosen from the group consisting of hydrogen, C1-6alkyl, —OH, —O—C1-6alkyl and C3-8cycloalkyl, and
- each R3a is independently chosen from the group consisting of hydrogen, C1-6alkyl and C3-8cycloalkyl;
is a group chosen from the group consisting of
-
- (a) —C≡C— and
- (b) aryl ring systems B′ chosen from the group consisting of:
- (i) 5- to 7-membered monocyclic ring systems and
- (ii) 8- to 10-membered bicyclic ring systems,
- and the aryl ring systems B′ containing from 0 to 4 heteroatoms independently chosen from the group consisting of N, O and S, and substituted on C or N atoms by v substituents R2,
- each R2 is independently chosen from the group consisting of hydrogen, halogen, —OR4a, —CN, —CO2R4a, —C(O)N(R4a)2, —N(R4a)2, —N(R4a)COR4, —N(R4a)CO2R4a, —N(R4a)C(O)N(R4a), —N(R4a)SO2R4a, —SR4a, —S(O)R4a, —S(O2)R4a, C1-6 alkyl substituted by from 0 to 4 R4 and C3-8cycloalkyl substituted by from 0 to 4 R4,
- v is from 0 to 4,
- each R4 is independently chosen from the group consisting of hydrogen, —OH, C1-6alkyl and C3-8cycloalkyl;
- each R4a is independently chosen from the group consisting of hydrogen, C1-6alkyl and C3-8cycloalkyl;
to form a 5- to 9-membered carbocyclic ring containing 1 or 2 heteroatoms independently chosen from the group consisting of N, O and S;
-
- (a) a single bond,
- (b) —C(O)N(R5)—,
- (c) —N(R5)C(O)—, and
- (d) a 5- or 6-membered aryl ring system D′ containing from 0 to 4 heteroatoms independently chosen from the group consisting of N, O and S, and substituted on C or N atoms by from 0 to 2 substituents R5,
- each R5 is independently chosen from the group consisting of hydrogen, halogen, —OR6, —CN, —CO2R6, —C(O)N(R6)2, —N(R6)2, —N(R6)COR6, —SR6, —S(O)R6, —S(O2)R6, —N(R6)SO2R6, —NCO2R6, —NC(O)N(R6)2, C1-6alkyl substituted by from 0 to 3 R6 and C3-8cycloalkyl substituted by from 0 to 3 R6, and
- each R6 is independently chosen from the group consisting of hydrogen, C1-6alkyl and C3-8cycloalkyl;
-
- (a) a single bond,
- (b) —(C(R7)2)0-2NR7C(O)O0-1—, and
- (c) a pyrrolidinyl derivative chosen from the group consisting of:

-
-
- I is a bivalent group chosen from —C(O)—, —CO2— and —C(O)N(R7)—,
- J is a fused ring system chosen from the group consisting of 3- to 7-membered carbocycles and 5- or 6-membered aryl rings containing from 0 to 4 heteroatoms independently chosen from the group consisting of N, O and S, and substituted on C or N atoms by substituents R9,
- each R8a is independently chosen from the group consisting of hydrogen, halogen, —OH, —OC1-6alkyl and C1-6alkyl, or two R8a may be taken together to form oxo,
- each R8b is independently chosen from the group consisting of hydrogen, halogen, —OH, —OC1-6alkyl and C1-6alkyl, or two R8b may be taken together to form oxo,
- each R8c is independently chosen from the group consisting of hydrogen and C1-6alkyl,
- or any two groups selected from R8a, R8b and R8c may be taken together to form a spiro-bicyclic or bridged bicyclic ring;
- each R9 is independently chosen from the group consisting of hydrogen, halogen, C1-6alkyl, —O—C1-6alkyl, —S—C1-6alkyl, —NH—C1-6alkyl and —NHC(O)—C1-6alkyl,
- each R7 is independently chosen from the group consisting of hydrogen, C1-6alkyl and phenyl, and the C1-6alkyl and phenyl are substituted by from 0 to 3 substitutents independently chosen from the group consisting of hydrogen, halogen, C1-6alkyl, —O—C1-6alkyl and —S—C1-6alkyl; and
-
-
- (a) hydrogen,
- (b) —OR10a,
- (c) —CN,
- (d) —CO2R10a,
- (e) —C(O)N(R10)2,
- (f) —SR10a,
- (g) —S(O)R10a,
- (h) —S(O2)R10a,
- (i) —N(R10)2,
- (j) —N(R10)SO2R10a,
- (k) —NCO2R10a,
- (l) —NC(O)N(R10)2,
- (m) C1-6alkyl having 0 to 4 substituents R11,
- each R11 is independently chosen from the group consisting of:
- (i) —OH,
- (ii) —N(R10)2,
- (iii) ═NR10,
- (iv) —O—C1-6alkyl,
- (v) —C(O)R10,
- (vi) —S—C1-6alkyl,
- (vii) —SO2—C1-6alkyl,
- (viii) 3- to 8-membered carbocycles containing from 0 to 3 heteroatoms independently chosen from the group consisting of N, O and S, and having from 0 to 3 substitutents R12 on N or C atoms, and each R12 is independently selected from the group consisting of hydrogen, halogen, C1-6alkyl having from 0 to 3 substituents chosen from R10, —O—C1-6alkyl, —S—C1-6alkyl, —OR10a, —CN, —C(O)R10, —CO2R10a, —C(O)N(R10)2, —SR10a, —S(O)R10a, —S(O2)R10a, N(R10)SO2R10a, —NCO2R10a, —NC(O)N(R10)2 and —N(R10)2, or two R12 are taken together to form oxo, and
- (ix) 5- or 6-membered aryl containing from 0 to 3 heteroatoms independently chosen from the group consisting of N, O and S, and having from 0 to 3 substitutents R13 on N or C atoms, and each R13 is independently selected from the group consisting of hydrogen, halogen, C1-6alkyl, —O—C1-6alkyl and 3- to 8-membered carbocycles containing from 0 to 3 heteroatoms independently chosen from the group consisting of N, O and S,
- (n) 3- to 8-membered carbocycles containing from 0 to 3 heteroatoms independently chosen from the group consisting of N, O and S, and having from 0 to 3 substitutents R10 on N or C atoms; and
- (o) aryl ring systems G′ chosen from the group consisting of:
- (i) 5- to 7-membered monocyclic ring systems and
- (ii) 8- to 10-membered bicyclic ring systems,
- and the aryl ring systems G′ containing from 0 to 4 heteroatoms independently chosen from the group consisting of N, O and S, and substituted on C or N atoms by 0 to 3 substitutents R10;
- each R10 is independently chosen from the group consisting of
- (i) hydrogen,
- (ii) —CN,
- (iii) C1-6alkyl,
- (iv) —O—O0-6alkyl,
- (v) —S—O0-6alkyl,
- (vi)
- (vii) —C(O)R14,
- (viii) —CO2R14,
- (ix) —SO2R14,
- (x) —N(R14)2,
- (xi) —N(R14)SO2R14,
- (xii) —NCO2R14,
- (xiii) —NC(O)N(R14)2, and
- (xiv) 3- to 8-membered carbocycles containing from 0 to 3 heteroatoms independently chosen from the group consisting of N, O and S,
- or two R10 may be taken together to form oxo;
- each R10a is independently chosen from the group consisting of
- (i) hydrogen,
- (ii) —CN,
- (iii) C1-6alkyl,
- (iv) C1-6alkyl-O—R14,
- (v) —C(O)R14,
- (vi) —CO2R14,
- (vii) —SO2R14,
- (x) —N(R14)2,
- (xi) —N(R14)SO2R14,
- (xii) —NCO2R14,
- (xiii) —NC(O)N(R14)2, and
- (xiv) 3- to 8-membered carbocycles containing from 0 to 3 heteroatoms independently chosen from the group consisting of N, O and S,
- and two R10 or R10a groups can be taken together with the N to which they are attached to form a ring, which may be substituted by from 0 to 3 substituents R14, and
- each R14 is independently chosen from the group consisting of hydrogen, C1-6alkyl, C3-8cycloalkyl, —(CH2)0-3C3-8cycloalkyl and phenyl. In this embodiment, all other groups are as provided in the general formula above.
- each R11 is independently chosen from the group consisting of:
is chosen from the group consisting of

where each X is independently chosen from the group consisting of CR1 and N,
is chosen from the group consisting of

each R1 is independently chosen from the group consisting of hydrogen, halogen, —OR3a, —CN, —C(O)R3, —CO2R3a, —C(O)N(R3a)2, —SR3a, —S(O)R3a, —S(O2)R3a, —(CH2)0-6N(R3a)2, —N(R3a)SO2R3a, —N(R3a)CO2R3a, —N(R3a)C(O)R3, —N(R3a)COR3a, —N(R3a)C(O)N(R3a), C1-6alkyl, C3-8carbocycle containing from 0 to 3 heteroatoms chosen from N, O and S, and phenyl, and the C1-6alkyl, C3-8carbocycle and phenyl are substituted by from 0 to 3 substitutents independently chosen from the group consisting of hydrogen, halogen, —OR3a, —CN, —CO2R3a, —C(O)N(R3a)2, —N(R3a)2, —N(R3a)CO2R3a, —SR3a, —S(O)R3a, —S(O2)R3a, —N(R3a)SO2R3a, —N(R3a)CO2R3, —N(R3a)C(O)N(R3a), C1-6alkyl, —O—C1-6alkyl and —S—C1-6alkyl, each R3 is independently chosen from the group consisting of hydrogen, C1-6alkyl, —OH, —O—C1-6alkyl and C3-8cycloalkyl, and each R3a is independently chosen from the group consisting of hydrogen, C1-6alkyl and C3-8cycloalkyl. In all aspects of this embodiment, all other groups are as provided in the general formula above or in the first embodiment above.
is chosen from the group consisting of

where
is substituted by from 0 to 3 additional R′, which are as provided above.
is chosen from the group consisting of
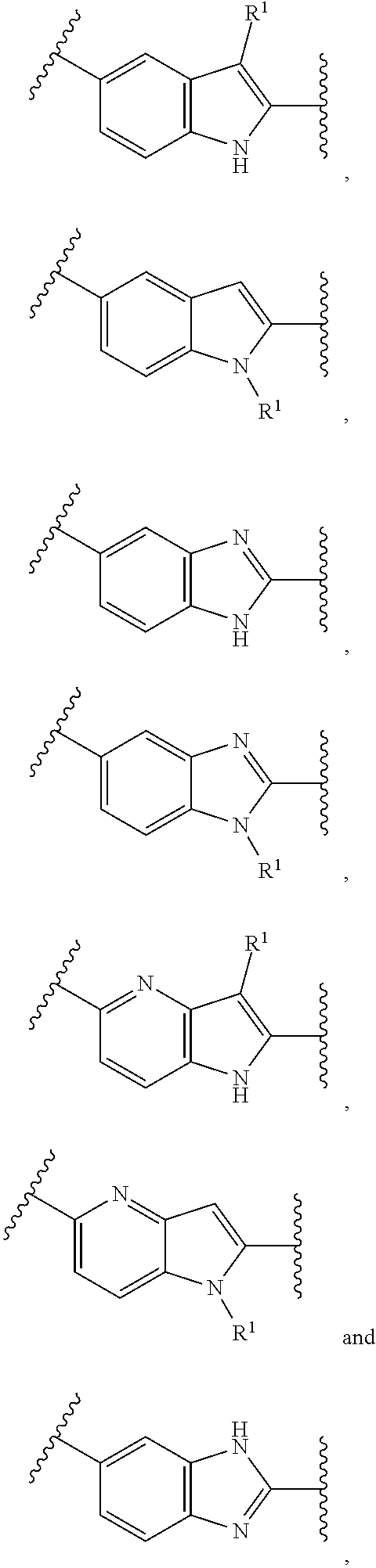
where
is substituted by from 0 to 3 additional R1, which are as provided above. In preferred instances of this aspect,
is

where
is substituted by from 0 to 3 additional R1, which are as provided above.
is chosen from the group consisting of

where
is substituted by from 0 to 3 additional R1, which are as provided above. In preferred instances of this aspect,
is

where
is substituted by from 0 to 3 additional R1, which are as provided above.
is chosen from the group consisting of —C≡C—, phenyl, pyridinyl, pyrazinyl, pyrimidyl, 1,2,4-triazinyl, pyridazinyl, thiazyl and 9-membered bicyclic ring systems that contain from 1 to 3 heteroatoms independently chosen from the group consisting of N, O and S, v is from 0 to 4, each R2 is independently chosen from the group consisting of hydrogen, halogen, —OR4a, —CN, —CO2R4a, —C(O)N(R4a)2, —N(R4a)2, —N(R4a)CO2R4a, —SR4a, —S(O)R4a, —S(O2)R4a, —N(R4a)SO2R4a, —N(R4a)CO2R4a, —N(R4a)C(O)N(R4a), C1-6alkyl substituted by from 0 to 4 R4 and C3-8cycloalkyl substituted by from 0 to 4 R4, each R4 is independently chosen from the group consisting of hydrogen, —OH, C1-6alkyl and C3-8cycloalkyl, and each R4a is independently chosen from the group consisting of hydrogen, C1-6alkyl and C3-8cycloalkyl. In particular aspects of this embodiment,
is phenyl, v is from 0 to 2, and each R2 is independently chosen from the group consisting of fluorine, chlorine, —OH, —CH3, —OCH3 and —CN. In all aspects of this embodiment, all other groups are as provided in the general formula above and/or in the first or second embodiments.
taken together with one substituent R1 and one substituent R2, are represented by a group chosen from the group consisting of:

where W is chosen from the group consisting of —(CH2)1-3—, —(CH2)0-2NH(CH2)0-2—, —(CH2)0-2N(C1-6alkyl)(CH2)0-2—, —(CH2)0-2O(CH2)0-2— and —(CH2)0-2C(O)(CH2)0-2—, where W is substituted by from 0 to 4 Rw, where each Rw is independently selected from C1-6alkyl and C3-8cycloalkyl; and V is chosen from the group consisting of —C(O)— and —CH2—, and where V is —CH2—, V is substituted by from 0 to 2 Rv, where each Rv is independently selected from the group consisting of C1-6alkyl and C3-8cycloalkyl. In a first aspect of this embodiment,
taken together with one substituent R1 and one substituent R2, are represented by a group chosen from the group consisting of

In particular instances of this embodiment, and of the first aspect of this embodiment, W is chosen from the group consisting of —CH2—, —NH—, —N(C1-6alkyl)-, —C(O)—, —CH2NH—, —CH2N(C1-6alkyl)-, —CH2CH2—, —C(O)CH2—, —CH2C(O)—, —CH2O—, —CH2CH2CH2—, —C(O)CH2CH2—, —CH2C(O)CH2—, —CH2OCH2—, —CH2CH2C(O)—, —CH2CH2O—, —CH2CH2NH—, —CH2CH2N(C1-6alkyl)-, —CH2NHCH2—, —CH2N(C1-6alkyl)CH2—, —NHCH2CH2—, and —N(C1-6alkyl)CH2CH2—. In all aspects of this embodiment, all other groups are as provided in the general formula above.

where R5 is independently chosen from the group consisting of hydrogen, halogen —OR6, —CN, —CO2R6, —C(O)N(R6)2, —N(R6)2, —N(R6)COR6, —SR6, —S(O)R6, —S(O2)R6, —N(R6)SO2R6, —NCO2R6, —NC(O)N(R6)2, C1-6alkyl substituted by from 0 to 3 substituents R6 and C3-8cycloalkyl substituted by from 0 to 3 substituents R6, and each R6 is independently chosen from the group consisting of hydrogen, C1-6alkyl and C3-8cycloalkyl. In particular aspects of this embodiment, each D is independently chosen from the group consisting of

In this embodiment, all other groups are as provided in the general formula above and/or in the first through fourth embodiments.

where one of R8a and R8b is —OH or fluorine. In a first aspect of this embodiment, each E is independently chosen from the group consisting of a single bond,

where one of R8a and R8b is —OH or fluorine. In all aspects of this embodiment, all other groups are as provided in the general formula above and/or in the first through fifth embodiments.
is connected directly to G by one single bond.
-
- (i) 5- to 7-membered monocyclic ring systems and
- (ii) 8- to 10-membered bicyclic ring systems,
- and the aryl ring systems G′ containing from 0 to 4 heteroatoms independently chosen from the group consisting of N, O and S, and substituted on C or N atoms by 0 to 3 substitutents R10. In all aspects of the seventh embodiment, G is chosen such that stable compounds result. In all aspects of this seventh embodiment, all other groups are as provided in the general formula above and/or in the first through sixth embodiments.
-
- each R11 is independently chosen from the group consisting of —OH, —NH2, —NCH3H, —N(CH3)2, —N(CH2CH3)2, ═NH, ═NCH3, —C(O)H, —C(O)OH, —C(O)CH3, —C(O)OCH3, —NHC(O)H, —NHC(O)OH, —NHC(O)CH3, —NHC(O)OCH3, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, pyranyl, pyrrolidinyl, piperidinyl, oxacyclopentyl, and oxacyclohexyl, phenyl, pyridinyl, pyrimidinyl and pyrrolyl, where
- the cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, pyranyl, pyrrolidinyl, piperidinyl, oxacyclopentyl and oxacyclohexyl are substituted by from 0 to 2 substitutents R12 on N or C atoms, and each R12 is independently selected from the group consisting of hydrogen, halogen, carboxy, C1-6alkyl, —O—C1-6alkyl and —S—C1-6alkyl; and
- the phenyl, pyridinyl, pyrimidinyl and pyrrolyl are substituted by from 0 to 3 substitutents R13 on N or C atoms, and each R13 is independently selected from the group consisting of hydrogen, halogen, C1-6alkyl and 3- to 8-membered cycloalkyl containing from 0 to 3 heteroatoms independently chosen from the group consisting of N, O and S,
is chosen from the group consisting of

where
is chosen from the group consisting of

is chosen from the group consisting of —C≡C—, phenyl, pyridinyl, pyrazinyl, pyrimidyl, 1,2,4-triazinyl, pyridazinyl, thiazyl and 9-membered bicyclic ring systems that contain from 1 to 3 heteroatoms independently chosen from the group consisting of N, O and S,

where
-
- R5 is independently chosen from the group consisting of hydrogen, halogen —OR6, —CN, —CO2R6, —C(O)N(R6)2, —N(R6)2, —N(R6)COR6, —SR6, —S(O)R6, —S(O2)R6, —N(R6)SO2R6, —NCO2R6, —NC(O)N(R6)2, C1-6alkyl substituted by from 0 to 3 substituents R6 and C3-8cycloalkyl substituted by from 0 to 3 substituents R6, and
- each R6 is independently chosen from the group consisting of hydrogen, C1-6alkyl and C3-8cycloalkyl;

-
- each R11 is independently chosen from the group consisting of —OH, —NH2, —NCH3H, —N(CH3)2, —N(CH2CH3)2, ═NH, ═NCH3, —C(O)H, —C(O)OH, —C(O)CH3, —C(O)OCH3, —NHC(O)H, —NHC(O)OH, —NHC(O)CH3, —NHC(O)OCH3, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, pyranyl, pyrrolidinyl, piperidinyl, oxacyclopentyl, and oxacyclohexyl, phenyl, pyridinyl, pyrimidinyl and pyrrolyl, where
- the cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, pyranyl, pyrrolidinyl, piperidinyl, oxacyclopentyl and oxacyclohexyl are substituted by from 0 to 2 substitutents R12 on N or C atoms, and each R12 is independently selected from the group consisting of hydrogen, halogen, carboxy, C1-6 alkyl, —O—C1-6alkyl and —S—C1-6alkyl; and
- the phenyl, pyridinyl, pyrimidinyl and pyrrolyl are substituted by from 0 to 3 substitutents R13 on N or C atoms, and each R13 is independently selected from the group consisting of hydrogen, halogen, C1-6alkyl and 3- to 8-membered cycloalkyl containing from 0 to 3 heteroatoms independently chosen from the group consisting of N, O and S,
is chosen from the group consisting of

where
is substituted by from 0 to 3 additional R1;
is phenyl; v is from 0 to 2; each R2 is independently chosen from the group consisting of fluorine, chlorine, —OH, —CH3, —OCH3 and —CN; each D is independently chosen from the group consisting of

each E is independently chosen from the group consisting of a single bond,

where one of R8a and R8b is —OH or fluorine;

or a pharmaceutically acceptable salt thereof, wherein
is substituted by u substituents R1, and Y is selected from the group consisting of 0 and NR1. In all aspects of this embodiment, all other groups are as provided in the general formula above or in any one of the first through tenth embodiments.

or a pharmaceutically acceptable salt thereof, wherein
is substituted by u substituents R1, and Y is selected from the group consisting of O and NR1. In particular aspects of this embodiment,
is substituted by u substituents R1, Y is O, and both instances of G are

In all aspects of this embodiment, all other groups are as provided in the general formula above or in the eleventh embodiment.

or a pharmaceutically acceptable salt thereof, wherein said
is substituted by u substituents R1, and said
and said
taken together with one substituent R1 and one substituent R2, are represented by a group chosen from the group consisting of

In particular aspects of this embodiment,
and said
taken together with one substituent R1 and one substituent R2, are represented by

wherein V is —CH2—, W is —(CH2)0-2O(CH2)0-2—, R1 is fluorine, and both instances of G are

In all aspects of this embodiment, all other groups are as provided in the general formula above or in the eleventh embodiment.
taken together with one substituent R1 and one substituent R2, are represented by a group chosen from the group consisting of:

where
-
- each R1 is independently chosen from the group consisting of hydrogen, halogen, —OR3, —CN, —C(O)R3, —CO2R3, —C(O)N(R3a)2, —SR3, —S(O)R3, —S(O2)R3, —N(R3a)2, —(CH2)0-6N(R3a)2, —N(R3a)SO2R3, —N(R3a)CO2R3, —N(R3a)COR3, —N(R3a)C(O)N(R3a), C1-6alkyl, C3-8carbocycle containing from 0 to 3 heteroatoms chosen from N, O and S, and phenyl, and the C1-6alkyl, C3-8carbocycle and phenyl are substituted by from 0 to 3 substitutents independently chosen from the group consisting of hydrogen, halogen, —OR3a, —CN, —CO2R3a, —C(O)N(R3a)2, —N(R3a)2, —N(R3a)CO2R3a, —SR3a, —S(O)R3a, —S(O2)R3a, —N(R3a)SO2R3a, —N(R3a)CO2R3a, —N(R3a)C(O)N(R3a), C1-6alkyl, —O—C1-6alkyl and —S—C1-6alkyl,
- each R3 is independently chosen from the group consisting of hydrogen, C1-6alkyl, —OH, —O—C1-6alkyl and C3-8cycloalkyl, and
- each R3a is independently chosen from the group consisting of hydrogen, C1-6alkyl and C3-8cycloalkyl;
-
- each R4 is independently chosen from the group consisting of hydrogen, —OH, C1-6alkyl and C3-8cycloalkyl, and
- each R4a is independently chosen from the group consisting of hydrogen, C1-6alkyl and C3-8cycloalkyl;

where
-
- R5 is independently chosen from the group consisting of hydrogen, halogen —OR6, —CN, —CO2R6, —C(O)N(R6)2, —N(R6)2, —N(R6)COR6, —SR6, —S(O)R6, —S(O2)R6, —N(R6)SO2R6, —NCO2R6, —NC(O)N(R6)2, C1-6alkyl substituted by from 0 to 3 substituents R6 and C3-8cycloalkyl substituted by from 0 to 3 substituents R6, and
- each R6 is independently chosen from the group consisting of hydrogen, C1-6alkyl and C3-8cycloalkyl;

where one of R8a and R8b is —OH or fluorine;
-
- each R11 is independently chosen from the group consisting of —OH, —NH2, —NCH3H, —N(CH3)2, —N(CH2CH3)2, ═NH, ═NCH3, —C(O)H, —C(O)OH, —C(O)CH3, —C(O)OCH3, —NHC(O)H, —NHC(O)OH, —NHC(O)CH3, —NHC(O)OCH3, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, pyranyl, pyrrolidinyl, piperidinyl, oxacyclopentyl, and oxacyclohexyl, phenyl, pyridinyl, pyrimidinyl and pyrrolyl, where
- the cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, pyranyl, pyrrolidinyl, piperidinyl, oxacyclopentyl and oxacyclohexyl are substituted by from 0 to 2 substitutents R12 on N or C atoms, and each R12 is independently selected from the group consisting of hydrogen, halogen, carboxy, C1-6alkyl, —O—C1-6alkyl and S—C1-6alkyl; and
- the phenyl, pyridinyl, pyrimidinyl and pyrrolyl are substituted by from 0 to 3 substitutents R13 on N or C atoms, and each R13 is independently selected from the group consisting of hydrogen, halogen, C1-6alkyl and 3- to 8-membered cycloalkyl containing from 0 to 3 heteroatoms independently chosen from the group consisting of N, O and S,
D, E, G, R1, R2, u, v, R3, R3a, R4, R4a, R5, R6, R7, I, J, R8a, R8b, R8c, R9, R10, R10a, R11, R12, R13, R14, W, Rw, V, and Rv, are selected independently from each other.
-
- (d) A pharmaceutical combination that is (i) a compound of formula (I) and (ii) a second therapeutic agent selected from the group consisting of HCV antiviral agents, immunomodulators, and anti-infective agents; wherein the compound of formula (I) and the second therapeutic agent are each employed in an amount that renders the combination effective for inhibiting HCV NS5A activity, or for treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection, or for inhibiting HCV viral replication and/or HCV viral production in a cell-based system.
-
- (n) A method of treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection in a subject in need thereof, which comprises administering to the subject the pharmaceutical composition of (a), (b), or (c) or the combination of (d) or (e).
- (o) A method of inhibiting HCV viral replication and/or HCV viral production in a cell-based system, which comprises administering to the subject the pharmaceutical composition of (a), (b), or (c) or the combination of (d) or (e).























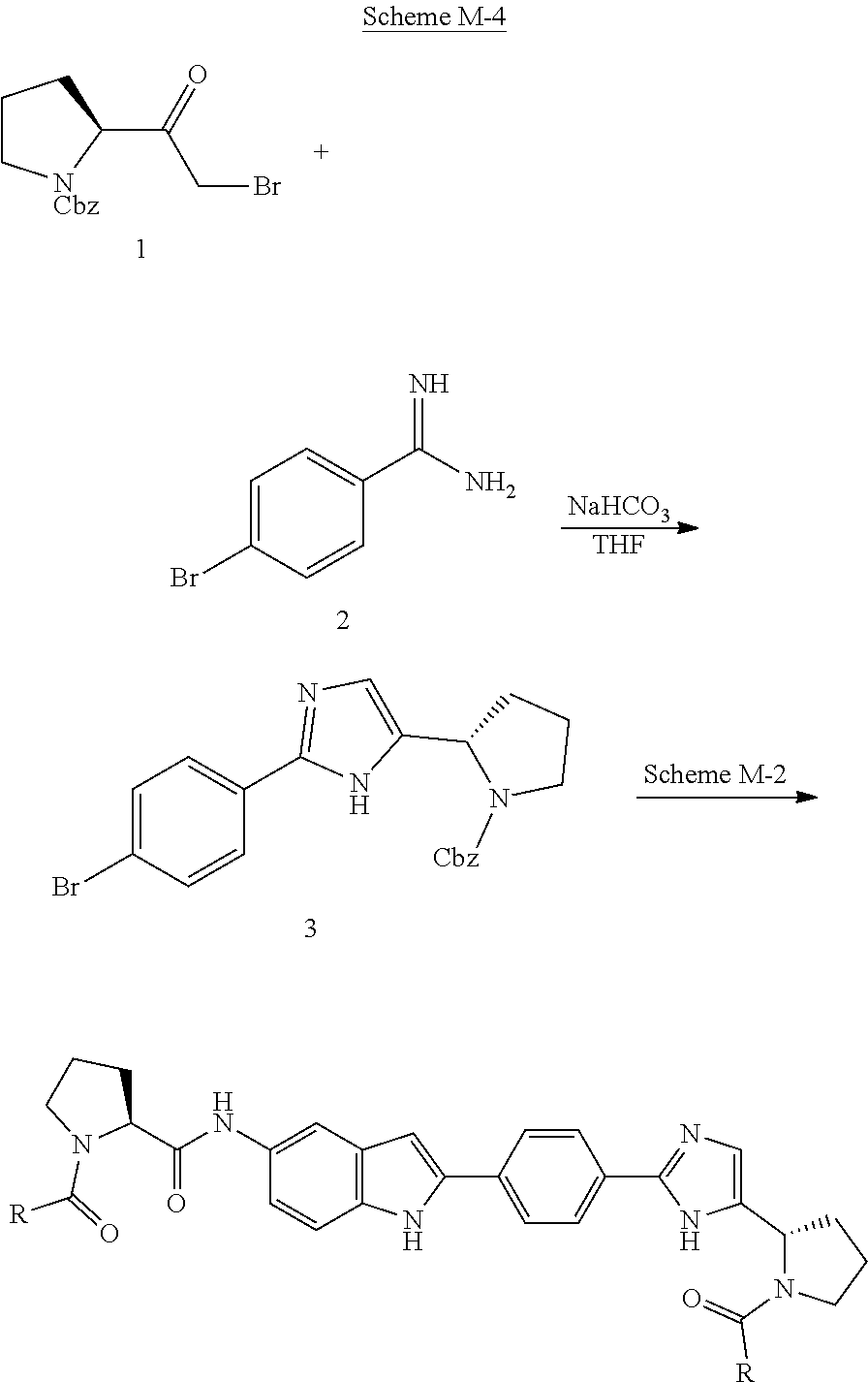














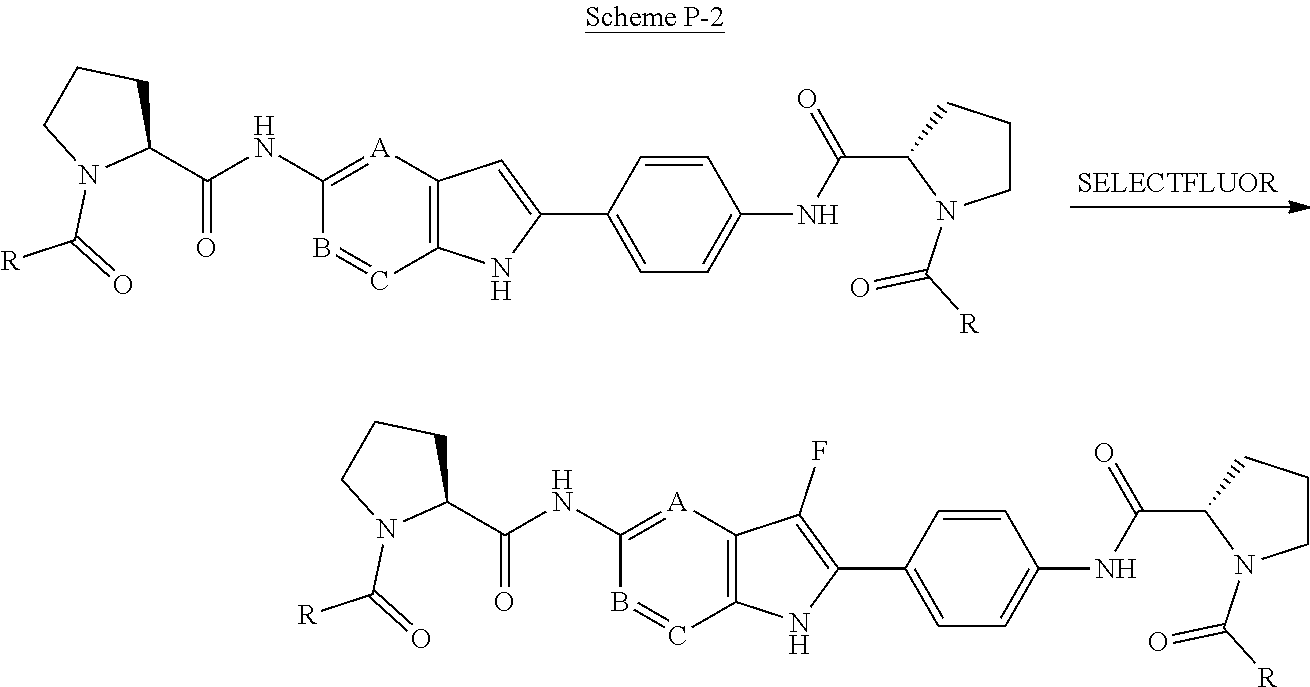








-
- Ac2O Acetic anhydride
- B(OiPr)3, (iPrO)3B Triisopropyl borate
- B(OMe)3 Trimethyl borate
- BF3 Boron trifluoride
- BOC, Boc, boc tert-Butyloxycarbonyl
- BOP Benzotriazole-1-yl-oxy-tris-(dimethylamino)-phosphonium hexafluorophosphate
- BrCN Cyanogen bromide
- BuLi, n-BuLi Butyl lithium
- CBZ, Cbz, cbz Benzyloxycarbonyl
- CDCl3 Deuterio-trichloromethane
- CH3CN, MeCN Acetonitrile
- Cs2CO3 Cesium carbonate
- CuBr2 Copper(II) bromide
- CuCN Copper(I) cyanide
- CuI Copper iodide
- DBU 1,8-Diazabicyclo[5.4.0]undec-7-ene
- DCE Dichloroethane
- DCM, CH2Cl2 Dichloromethane
- DIPEA, DIEA Diisopropylethylamine
- DMAP 4-Dimethylamino pyridine
- DMF Dimethylformamide
- DMSO Dimethyl sulfoxide
- DPPF, Dppf, dppf 1,1′-bis(Diphenylphosphino)ferrocene
- EDC, EDCI N-β-Dimethylaminopropyl)-N′-ethylcarbodiimide
- Et2O Diethyl ether
- Et3N, TEA Triethylamine
- EtMgBr Bromo(ethyl)magnesium or ethyl magnesium bromide
- EtOAc Ethyl acetate
- EtOH Ethanol
- FeCl3 Ferric chloride or Iron(III) chloride
- H2 Hydrogen or hydrogen atmosphere
- H2O Water
- H2SO4 Sulfuric acid
- HATU O-(7-Azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- HBr Hydrobromic acid
- HCl Hydrochloric acid
- HNO3 Nitric Acid
- HOAc, HAc Acetic acid
- HOBT, HOBt 1-Hydroxy benzotriazole
- HPLC High performance liquid chromatography
- InBr3 Indium tribromide
- iPr2NH Diisopropylamine
- K2CO3 Potassium carbonate
- KI Potassium iodide
- KIO3 Potassium iodate
- KOAc, AcOK Potassium acetate
- KOH Potassium hydroxide
- LDA Lithium diisopropylamide
- LHMDS, LiHMDS Lithium hexamethyldisilamide
- MeMgBr Bromo(methyl)magnesium or methyl magnesium bromide
- MeOD Methan(2H)ol
- MeOH, CH3OH Methanol
- MgSO4 Magnesium sulfate
- MOC, Moc Methoxy carbonyl
- MS Mass spectroscopy
- N2 Nitrogen or nitrogen atmosphere
- Na2CO3 Sodium carbonate
- Na2SO4 Sodium sulfate (anhydrous)
- NaClO2 Sodium perchlorate
- NaH2PO4 Dihydrogen sodium phosphate
- NaHCO3 Sodium hydrogen carbonate (sodium bicarbonate)
- NaNO2 Sodium nitrite
- NaOH Sodium hydroxide
- NBS N-bromosuccinimide
- NCS N-chlorosuccinimide
- NH4OAc Ammonium acetate
- NMM N-methylmorpholine
- NMR, 1H-NMR Proton nuclear magnetic resonance spectroscopy
- NXS N-halosuccinimide
- P2O5, P4O10 Phosphorus pentoxide
- Pd Palladium
- Pd(dppf)Cl2 Dichloro(1,1′-bis(Diphenylphosphino)ferrocene) palladium(II)
- Pd(II) Palladium(II)
- Pd(PPh3)2Cl2, PdCl2(PPh3)2 Dichlorobis(triphenylphosphine)palladium(II)
- Pd(PPh3)4 Tetrakis(triphenylphosphine)palladium(0)
- Pd/C, Pd—C Palladium on carbon
- Pd2(dba)3 Tris(dibenzylidene acetone)dipalladium(0)
- PdCl2 Palladium(II) chloride
- PE Petroleum ether
- Phg Phenylglycine
- PhCH3, PhMe Toluene
- Piv Pivaloyl
- PivCl Pivaloyl chloride
- POBr3 Phosphorus oxybromide
- PPA Polyphosphoric acid
- PPH3, TPP Triphenylphosphine
- Pro Proline
- Proc iso-Propylcarbamate
- PtBu3 Tri-tert-butyl phosphine
- Py Pyradine
- PyBOP (Benzotriazole-1-yl-oxy)-tripyrrolidinophosphonium hexafluorophosphate
- RPLC Reverse phase liquid chromatography
- RT, rt, r.t. Room temperature, approximately 25° C.
- SiO2 Silica or silica gel
- SnCl2 Stannous chloride or Tin(II) chloride
- SOCl2 Thionyl chloride
- STP Standard temperature and pressure
- t-BuLi tert-Butyl lithium
- t-BuNO2 tert-Butyl nitrate
- t-BuOH tert-Butanol
- t-BuOK, KOt-Bu Potassium tert-butoxide
- TFA Trifluoroacetic acid
- TFAA Trifluoroacetic anhydride
- THF Tetrahydrofuran
- TLC Thin layer chromatography
- ZnCl2 Zinc chloride

Step 1








| Example | Structure | MW | Name |
| 2 |
 |
654.776 | (2S)-1-(phenylacetyl)- N-{2-[4-({[(2S)-1- (phenylacetyl)pyrrolidin-2- yl]carbonyl}amino)phenyl]- 1H-pyrrolo[3,2-b]pyridin-5- yl}pyrrolidine-2-carboxamide |
| 3 |
 |
686.774 | benzyl (2S)-2-[(2-{4-[({(2S)- 1-[(benzyloxy)carbonyl] pyrrolidin- 2-yl}carbonyl)amino]phenyl}- 1H-pyrrolo[3,2-b]pyridin-5- yl)carbamoyl]pyrrolidine-1- carboxylate |

Step 1







Step 1










Step 1









Step 1




Step 4




Step 1








| Example | Structure | MW | Name |
| 9 |
 |
626.721 | (2S)-1-(phenylcarbonyl)-N-{4- [5-({[(2S)-1- (phenylcarbonyl)pyrrolidin-2- yl]carbonyl}amino)-1H- benzimidazol-2- yl]phenyl}pyrrolidine-2- carboxamide |
| 10 |
 |
682.83 | (2S)-1-[(4Z,5Z)-4- ethylidenehept-5-enoyl]-N-{2- [4-({[(2S)-1-(3- phenylpropanoyl)pyrrolidin-2- yl]carbonyl}amino)phenyl]-1H- benzimidazol-5-yl}pyrrolidine- 2-carboxamide |
| 11 |
 |
682.83 | (2S)-1-[(4Z,5Z)-4- ethylidenehept-5-enoyl]-N-{2- [3-({[(2S)-1-(3- phenylpropanoyl)pyrrolidin-2- yl]carbonyl}amino)phenyl]-1H- benzimidazol-5-yl}pyrrolidine- 2-carboxamide |
| 12 |
 |
686.774 | (2S)-1-[(2R)-2-hydroxy-2- phenylacetyl]-N-(4-{5-[({(2S)-1- [(2R)-2-hydroxy-2- phenylacetyl]pyrrolidin-2- yl}carbonyl)amino]-1H- benzimidazol-2- yl}phenyl)pyrrolidine-2- carboxamide |
| 13 |
 |
654.776 | (2S)-1-(phenylacetyl)-N-{4-[5- ({[(2S)-1-(phenylacetyl) pyrrolidin-2-yl]carbonyl} amino)-1H-benzimidazol-2- yl]phenyl}pyrrolidine-2- carboxamide |
| 14 |
 |
885.041 | tert-butyl {(1S)-2-[(2S)-2-({4- [5-({[(2S)-1-{(2S)-2-[(tert- butoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2- yl]carbonyl}amino)-1H- benzimidazol-2-yl]phenyl} carbamoyl)pyrrolidin-1-yl]-2- oxo-1-phenylethyl}carbamate |
| 15 |
 |
885.041 | tert-butyl {(1R)-2-[(2S)-2-({4- [5-({[(2S)-1-{(2R)-2-[(tert- butoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2- yl]carbonyl}amino)-1H- benzimidazol-2-yl]phenyl} carbamoyl)pyrrolidin-1-yl]-2- oxo-1-phenylethyl}carbamate |

Step 1







Step 1





Step 1





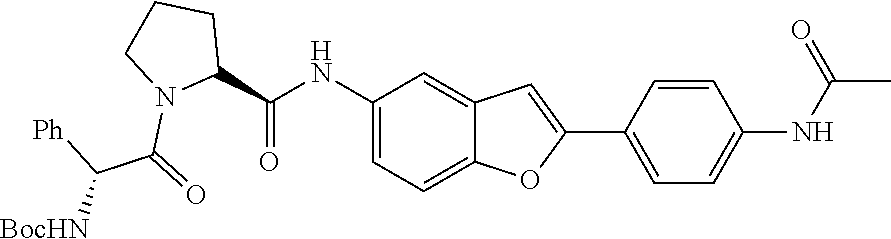
Step 1



Step 3



| Example | Structure | MW | Name |
| 20 |
 |
654.773 | (2S)-1-(phenylacetyl)-N- {4-[5- ({[(2S)-1-(phenylacetyl) pyrrolidin-2-yl]carbonyl} amino)- 1-benzofuran-2-yl]phenyl} pyrrolidine-2-carboxamide |
| 21 |
 |
885.039 | tert-butyl {(1S)-2-[(2S)-2- ({4-[5- ({[(2S)-1-{(2S)-2-[tert- butoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2- yl]carbonyl}amino)-1- benzofuran-2-yl]phenyl} carbamoyl)pyrrolidin-1-yl]-2- oxo-1-phenylethyl} carbamate |
| 22 |
 |
885.039 | tert-butyl {(1R)-2-[(2S)-2-({4-[5- ({[(2S)-1-{(2R)-2-[(tert- butoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2-yl] carbonyl}amino)-1- benzofuran- 2-yl]phenyl}carbamoyl) pyrrolidin-1-yl]-2-oxo-1- phenylethyl}carbamate |
| 23 |
 |
856.984 | propan-2-yl [(1R)-2-oxo-1- phenyl-2-{(2S)-2-[(4-{5-[({(2S)- 1-[(2R)-2-phenyl-2-{[(propan-2- yloxy)carbonyl]amino}acetyl] pyrrolidin-2-yl}carbonyl)amino]- 1-benzofuran-2-yl}phenyl) carbamoyl]pyrrolidin-1- yl}ethyl]carbamate |
| 24 |
 |
800.876 | methyl {(1R)-2-[(2S)-2-({4-[5- ({[(2S)-1-{(2R)-2- [(methoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2- yl]carbonyl}amino)-1- benzofuran-2-yl]phenyl} carbamoyl)pyrrolidin-1-yl]-2- oxo-1-phenylethyl}carbamate |
| 25 |
 |
740.91 | (2S)-1-[(2R)-2-(dimethylamino)- 2-phenylacetyl]-N-(4-{5-[({(2S)- 1-[(2R)-2-(dimethylamino)-2- phenylacetyl]pyrrolidin-2-yl} carbonyl)amino]-1-benzofuran-2- yl}phenyl)pyrrolidine-2- carboxamide |
| 26 |
 |
797.019 | (2S)-1-[(2R)-2-(diethylamino)-2- phenylacetyl]-N-(4-{5-[({(2S)-1- [(2R)-2-(diethylamino)-2- phenylacetyl]pyrrolidin-2-yl} carbonyl)amino]-1-benzofuran-2- yl}phenyl)pyrrolidine-2- carboxamide |
| 27 |
 |
798.947 | propan-2-yl [(1R)-2-{(2S)-2-[(4- {5-[({(2S)-1-[(2R)-2- (dimethylamino)-2- phenylacetyl]pyrrolidin-2-yl} carbonyl)amino]-1-benzofuran-2- yl}phenyl)carbamoyl]pyrrolidin- l-yl}-2-oxo-1- phenylethyl]carbamate |
| 28 |
 |
482.587 | N-[2-(4-aminophenyl)-1- benzofuran-5-yl]-1-[(2R)-2- (dimethylamino)-2- phenylacetyl]-L-prolinamide |
| 29 |
 |
853.127 | (2S)-1-{(2R)-2-[methyl(3- methylbutyl)amino]-2- phenylacetyl}-N-{4-[5-({[(2S)- 1-{(2R)-2-[methyl(3- methylbutyl)amino]-2- phenylacetyl}pyrrolidin-2- yl]carbonyl}amino)-1- benzofuran-2-yl]phenyl} pyrrolidine-2-carboxamide |
| 30 |
 |
552.679 | N-{2-[4-(acetylamino)pheny]]- 1-benzofuran-5-yl}-1-[(2R)-2- (diethylamino)-2-phenylacetyl]- L-prolinamide |
| 31 |
 |
649.797 | (2S)-1-acetyl-N-(2-{4-[({(2S)-1- [(2R)-2-(diethylamino)-2- phenylacetyl]pyrrolidin-2- yl}carbonyl)amino]phenyl}-1- benzofuran-5-yl)pyrrolidine-2- carboxamide |
| 32 |
 |
693.807 | tert-butyl {(1R)-2-[(2S)-2-({4-[5- ({[(2S)-1-acetylpyrrolidin-2- yl]carbonyl}amino)-1- benzofuran-2-yl]phenyl} carbamoyl)pyrrolidin-1-yl]-2- oxo-1-phenylethyl}carbamate |
| 33 |
 |
621.743 | (2S)-1-acetyl-N-(2-{4-[({(2S)-1- [(2R)-2-(dimethylamino)-2- phenylacetyl]pyrrolidin-2- yl}carbonyl)amino]phenyl}-1- benzofuran-5-yl)pyrrolidine-2- carboxamide |
| 34 |
 |
596.689 | N-{4-[5-(acetylamino)-1- benzofuran-2-yl]phenyl}-1- {{2R)-2-[{tert- butoxycarbonyl)amino]-2- phenylacelyl}-L-prolinamide |
| 35 |
 |
814.903 | methyl {(1R)-2-[(2S)-2-({4-[5- ({[(2S)-1-{(2R)-2- [(methoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2- yl]carbonyl}amino)-7-methyl-1- benzofuran-2-yl]phenyl} carbamoyl)pyrrolidin-1-yl]-2- oxo-1-pheylethyl}carbamate |
| 36 |
 |
825.073 | (2S)-1-{(2R)-2-[ethyl(propyl) amino]-2-phenylacetyl}-N-{4-[5- ({[(2S)-1-{(2R)-2-[ethyl(propyl) amino]-2-phenylacetyl} pyrrolidin-2-yl]carbonyl}amino)- 1-benzofuran-2-yl]phenyl} pyrrolidine-2-carboxamide |

Step 1






Step 1




Step 1







Step 1





Step 5


Step 1







| Example | Structure | MW | Name |
| 43 |
 |
884.054 | tert-butyl {(1R)-2-[(2S)-2-({4-[5- ({[(2S)-1-{(2R)-2-[(tert- butoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2- yl]carbonyl}amino)-1H- indol-2- yl]phenyl}carbamoyl)pyrrolidin- 1-yl]-2-oxo-1- phenylethyl}carbamate |
| 44 |
 |
799.891 | methyl {(1R)-2-[(2S)-2-({4-[5- ({[(2S)-1-{(2R)-2- [(methoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2- yl]carbonyl}amino)-1H-indol-2- yl]phenyl}carbamoyl)pyrrolidin- 1-yl]-2-oxo-1- phenylethyl}carbamate |
| 45 |
 |
792.002 | (2S)-1-[(2R)-2-phenyl-2- (pyrrolidin-1-yl)acetyl]-N-(4-{5- [({(2S)-1-[(2R)-2-phenyl-2- (pyrrolidin-1-yl)acetyl] pyrrolidin-2-yl}carbonyl)amino]- 1H-indol-2-yl}phenyl) pyrrolidine-2-carboxamide |
| 46 |
 |
848.023 | (2S)-1-{(2R)-2- [(cyclopropylacetyl)amino]-2- phenylacetyl}-N-{4-[5-({[(2S)-1- {(2R)-2-[(cyclopropylacetyl) amino]-2-phenylacetyl} pyrrolidin-2-yl]carbonyl}amino)- 1H-indol-2-yl]phenyl} pyrrolidine-2-carboxamide |
| 47 |
 |
852.055 | (2S)-1-{(2R)-2-[(3- methylbutanoyl)amino]-2- phenylacetyl}-N-{4-[5-({[(2S)-1- {{2R)-2-[(3-methylbutanoyl) amino]-2-phenylacetyl} pyrrolidin-2-yl]carbonyl}amino)- 1H-indol-2-yl]phenyl} pyrrolidine-2-carboxamide |
| 48 |
 |
824.001 | (2S)-1-[(2R)-2-(morpholin-4-yl)- 2-phenylacetyl]-N-(4-{5-[({(2S)- 1-[(2R)-2-(morpholin-4-yl)-2- phenylacetyl]|pyrrolidin-2- yl}carbonyl)amino]-1H-indol-2- yl}phenyl)pyrrolidine-2- carboxamide |
| 49 |
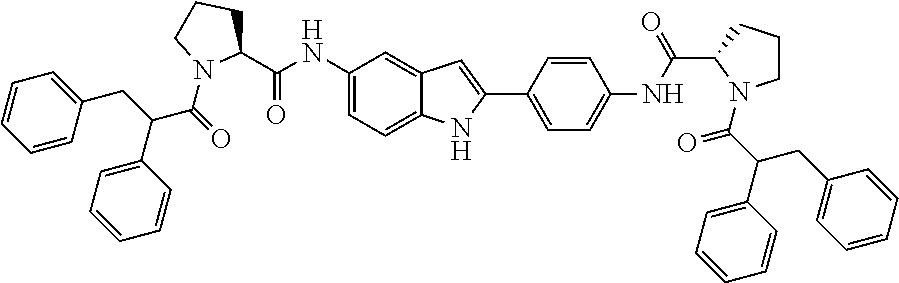 |
834.04 | (2S)-1-(2,3-diphenylpropanoyl)- N-{4-[5-({[(2S)-1-(2,3- diphenylpropanoyl)pyrrolidin-2- yl]carbonyl}amino)-1H-indol-2- yl]phenyl}pyrrolidine-2- carboxamide |
| 50 |
 |
992.195 | (2S)-1-[(2R)-2-{[(4- methylphenyl)sulfonyl]amino}-2- phenylacetyl]-N-(4-{5-[({(2S)-1- [(2R)-2-{[(4-methylphenyl) sulfonyl]amino}-2-phenylacetyl] pyrrolidin-2-yl}carbonyl)amino]- 1H-indol-2-yl}phenyl) pyrrolidine-2-carboxamide |
| 51 |
 |
904.132 | (2S)-1-{(2R)-2- [(cyclohexylcarbonyl)amino]-2- phenylacetyl}-N-{4-(5-({[(2S)-1- {(2R)-2-[(cyclohexylcarbonyl) amino]-2-phenylacetyl} pyrrolidin-2-yl]carbonyl}amino)- 1H-indol-2-yl]phenyl} pyrrolidinc-2-carboxamide |
| 52 |
 |
553.623 | methyl {(1R)-2-[(2S)-2-({2-[4- (acetylamino)phenyl]-indol- 5-yl}carbamoyl)pyrrolidin-1-yl]- 2-oxo-1-phenylethyl}carbamate |
| 53 |
 |
595.704 | tert-butyl {(1R)-2-[(2S)-2-({4-[5- (acetylamino)-1H-indol-2- yl]phenyl}carbamoyl)pyrrolidin- 1-yl]-2-oxo-1- phenylethyl}carbamate |
| 54 |
 |
565.678 | N-{2-[4-(acetylamino)phenyl]- 1H-indol-5-yl}-1-[(2R)-2- (morpholin-4-yl)-2- phenylacetyl]-L-prolinamide |
| 55 |
 |
581.677 | propan-2-yl{(1R)-2-[(2S)-2-({2- [4-(acetylamino)phenyl]-1H- indol-5-yl}carbamoyl)pyrrolidin- 1-yl]-2-oxo-1- phenylethyl}carbamate |
| 56 |
 |
817.953 | (2S)-1{(2R)-2- [(cyclopentylcarbamoyl)amino]- 2-phenylacetyl}-N-(4-{5-[({(2S)- 1-[(2R)-2-(dimethylamino)-2- phenylacetyl)pyrrolidin-2- yl}carbonyl)amino]-1H-indol-2- yl}phenyl)pyrrolidine-2- carboxamide |
| 57 |
 |
816.019 | tert-butyl {(2R)-1-[(2S)-2-({4-[5- ({[(2S)-1-{(2R)-2-[(tert- butoxycarbonyl)amino]-3- methylbutanoyl}pyrrolidin-2- yl]carbonyl}amino)-1H-indol-2- yl]phenyl}carbamoyl)pyrrolidin- 1-yl]-3-methyl-1-oxobutan-2- yl}carbamate |
| 58 |
 |
549.679 | N-{2-[4-(acetylamino)phenyl]- 1H-indol-5-yl}-1[(2R)-2-phenyl- 2-(pyrrolidin-1-yl)acetyl]-L- prolinamide |
| 59 |
 |
699.945 | (2S)-1-[(2R)-2-(dimethylamino)- 4-methylpentanoyl]-N-(4-{5- [({(2S)-1-[(2R)-2- (dimethylamino)-4- methylpentanoyl]pyrrolidin-2- yl}carbonyl)amino]-1H-indol-2- yl}phenyl)pyrrolidine-2- carboxamide (non-preferred name) |
| 60 |
 |
820.056 | (2S)-1-[(2R)-2phenyl-2- (piperidin-1-yl)acetyl]-N-(4-{5- [({(2S)-1-[(2R)-2-phenyl-2- (piperidin-1-yl)acetyl]pyrrolidin- 2-yl}carbonyl)amino]-1H-indol- 2-yl}phenyl)pyrrolidine-2- carboxamide |
| 61 |
 |
785.914 | (2S)-1-[(2R)-2-(1H-imidazol-1- yl)-2-phenylacetyl]-N-(4-{5- [({(2S)-1-[(2R)-2-(1H-imidazol- 1-yl)-2-phenylacetyl]pyrrolidin- 2-yl}carbonyl)amino]-1H-indol- 2-yl}phenyl)pyrrolidine-2- carboxamide |
| 62 |
 |
796.034 | (2S)-1-[(2R)-2-(diethylamino)-2- phenylacetyl]-N-(4-{5-[({(2S)-1- [(2R)-2-(diethylamino)-2- phenylacetyl]pyrrolidin-2- yl}carbonyl)amino]-1H-indol-2- yl}phenyl)pyrrolidine-2- carboxamide |
| 63 |
 |
692.822 | tert-butyl {(1S)-2-((2S)-2-({4-[5- ({[(2S)-1-acetylpyrrolidin-2- yl]carbonyl}amino)-1H-indol-2- yl]phenyl}carbamoyl)pyrrolidin- 1-yl]-2-oxo-1- phenyylethyl}carbamate |
| 64 |
 |
692.822 | tert-butyl {(1R)-2-[(2S)-2-({4-[5- ({[(2S)-1-acetylpyrrolidin-2- yl]carbonyl}amino)-1H-indol-2- yl]phenyl}carbamoyl)pyrrolidin- 1-yl]-2-oxo-1- phenylethyl}carbamate |
| 65 |
 |
692.866 | tert-butyl {(1R)-2-oxo-1-phenyl- 2-[(2S)-2-({2-[4-({[(2S)-1- (propan-2-yl)pyrrolidin-2- yl]carbonyl}amino)phenyl]-1H- indol-5-yl}carbamoyl)pyrrolidin- 1-yl]ethyl}carbamate |
| 66 |
 |
523.64 | N-{2-[4-(acetylamino)phenyl]- 1H-indol-5-yl}-1-[(2R)-2- (dimethylamino)-2- phenylacetyl]-L-prolinamide |
| 67 |
 |
563.706 | N-{2-[4-(acetylamino)phenyl]- 1H-indol-5-yl}-1-[(2R)-2-phenyl- 2-(piperidin-1-yl)acetyl]-L- prolinamide |
| 68 |
 |
811.99 | tert-butyl [(1R)-2-{(2S)2-[(2-{4- [({(2S)-1-[(2R)-2- (dimethylainino)-2- phenylacetyl]pyrrolidin-2- yl}carbonyl)amino]phenyl}-1H- indol-5-yl)carbamoyl]pyrrolidin- 1-yl}-2-oxo-1- phenylethyl]carbamate |
| 69 |
 |
811.99 | tert-butyl [(1R)-2-{(2S)-2-[(4-{5- [({(2S)-1-[(2R)-2- (dimethylamino)-2-phenylacetyl] pyrrolidin-2-yl}carbonyl)amino]- 1H-indol-2-yl}phenyl) carbamoyl]pyrrolidin-1-yl}-2- oxo-1-phenylethyl]carbamate |
| 70 |
 |
810.017 | (2S)-1-[(2R)-2-(dimethylamino)- 2-phenylacetyl]-N-{4-[5-({[(2S)- 1-{(2R)-2-[(3,3- dimethylbutanoyl)amino]-2- phenylacetyl}pyrrolidin-2- yl]carbonyl}amino)-1H-indol-2- yl]phenyl}pyrrolidine-2- carboxamide |
| 71 |
 |
660.823 | (2S)-1-(cyclopropylacetyl)-N-(4- {5-[({(2S)-1-[(2R)-2- (dimethylamino)-2- phenylacetyl]pyrrolidin-2- yl}carbonyl)amino]-1H-indol-2- yl}phenyl)pyrrolidine-2- carboxamide |
| 72 |
 |
676.823 | (2S)-1-[(2R)-2-(dimethylamino)- 2-phenylacetyl]-N-(2-{4-[({(2S)- 1-[(2R)-tetrahydrofuran-2- ylcarbonyl]pyrrolidin-2- yl}carbonyl)amino]phenyl}-1H- indol-5-y])pyrrolidine-2- carboxamide |
| 73 |
 |
920.035 | tert-butyl [(1R)-2-{(2S)-2-[(4-{5- [({(2S)-1-[(2R)-2-[(tert- butoxycarbonyl)amino]-2-(4- fluorophenyl)acetyl]pyrrolidin-2- yl}carbonyl)amino]-1H-indol-2- yl}phenyl)carbamoyl]pyrrolidin- 1-yl}-1-(4-fluorophenyl)-2- oxoethyl]carbamate |
| 74 |
 |
852.142 | (2S)-1-{(2R)-2-[methyl(3- methylbutyl)amino]-2- phenylacetyl}-N-{4-[5-({[(2S)-1- {(2R)-2-[methyl(3-methylbutyl) amino]-2-phenylacetyl} pyrrolidin-2-yl]carbonyl}amino)- 1H-indol-2-yl]phenyl} pyrrolidine-2-carboxamide |
| 75 |
 |
613.72 | (2S)-1-[(2S)-tetrahydrofuran-2- ylcarbonyl]-N-(4-{5-[({(2S)-1- [(2S)-tetrahydrofuran-2- ylcarbonyl]pyrrolidin-2- yl}carbonyl)amino)-1H-indol-2- yl}phenyl)pyrrolidine-2- carboxamide |
| 76 |
 |
777.972 | N-(tert-butoxycarbonyl)-D-valyl- N-{4-[5-({1-[(R)-2- (dimethylamino)-2- phenylacetyl]-L-prolyl}amino)- 1H-indol-2-yl]phenyl}-L- prolinamide |
| 77 |
 |
673.778 | (2S)-1-[(2R)-2-(dimethylamino)- 2-phenylacetyl]-N-{2-[4-({[(2S)- 1-(1,3-oxazol-2- ylcarbonyl)pyrrolidin-2- yl]carbonyl}amino)phenyl]-1H- indol-5-yl}pyrrolidinc-2- carboxamide |
| 78 |
 |
860.032 | tert-butyl [(1R)-2-{[(2S)-1-({4- [5-({(2S)-2-[{(2R)-2-[(tert- butoxycarbonyl)amino]-2- phenylacetyl}(methyl)amino] propanoyl}amino)-1H-indol-2- yl]phenyl}amino)-1-oxopropan- 2-yl)(methyl)amino}-2-oxo-1- phenylethyl]carbamate |
| 79 |
 |
567.694 | tert-butyl {(1R)-2-[(2S)-2-({2-[4- (methylamino)phenyl]-1H-indol- 5-yl}carbamoyl)pyrrolidin-1-yl]- 2-oxo-1-phenylethy}carbamate |
| 80 |
 |
696.857 | (2S)-1-[(2R)-2-(dimethylamino)- 2-phenylacetyl]-N-{2-[4-({[(2S)- 1-(phenylacetyl)pyrrolidin-2- yl]carbonyl}amino)phenyl]-1H- indol-5-yl}pyrrolidine-2- carboxamide |
| 81 |
 |
765.964 | (2S)-1-{[(3R)-1-benzylpyrrolidin- 3-yl]carbonyl}-N-(4-{5-[({(2S)- 1-[(2R)-2-(dimethylainino)-2- phenylacetyl]pyrrolidin-2- yl}carbonyl)amino]-1H-indol-2- yl}phenyl)pyrrolidine-2- carboxamide |
| 82 |
 |
765.964 | (2S)-1-{[(3S)-1-benzylpyrrolidin- 3-yl]carbonyl}-N-(4-{5-[({(2S)- 1-[(2R)-2-(dimethylamino)-2- phenylacetyl]pyrrolidin-2- yl}carbonyl)amino]-1H-indol-2- yl}phenyl)pyrrolidine-2- carboxainide |
| 83 |
 |
719.935 | N,N-dimethyl-D-leucyl-N-{4-[5- ({1-[(2R)-2-(dimethylamino)-2- phenylacetyl]-L-prolyl}amino)- 1H-indol-2-yl]phenyl}-L- prolinamide |
| 84 |
 |
577.733 | N-(2-{4-[(cyclopentylcarbonyl) amino]phenyl}-1H-indol-5-yl)-1- [(2R)-2-(dimethylamino)-2- phenylacetyl]-L-prolinamide |
| 85 |
 |
652.8 | tert-butyl [(2S)-1-({4-[5-({1- [(2R)-2-(dimethylamino)-2- phenylacetyl]-L-prolyl}amino)- 1H-indol-2-yl]phenyl}amino)-1- oxopropan-2-yl]carbamate |
| 86 |
 |
714.872 | tert-butyl [(1S)-2-({4-[5-({1- [(2R)-2-(dimethylamino)-2- phenylacetyl]-L-prolyl}amino)- 1H-indol-2-yl]phenyl}amino)-2- oxo-1-phenylethyl]carbamate |
| 87 |
 |
680.855 | tert-butyl [(2R)-1-({4-[5-({1- [(2R)-2-(dimethylamino)-2- phenylacetyl]-L-prolyl}amino)- 1H-indol-2-yl]phenyl}amino)-3- methyl-1-oxobutan-2- yl]carbamate |

Step 1






| Example | Structure | MW | Name |
| 89 |
 |
679.826 | (2S,2′S)-N,N′-6,11-dihydro-5H- benzo[a]carbazole-3,8-diylbis[1- (phenylacetyl)pyrrolidine-2- carboxamide] |
| 90 |
 |
711.825 | dibenzyl (2S,2′S)-2,2′-(6,11- dihydro-5H-benzo[a]carbazole- 3,8-diyldicarbamoyl) dipyrrolidine-1-carboxylate |
| 91 |
 |
528.453 | N-(8-bromo-6,11-dihydro-5H- benzo[a]carbazol-3-yl)-1- (phenylacetyl)-L-prolinamide |
| 92 |
 |
910.092 | di-tert-butyl (6,11-dihydro-5H- benzo[a]carbazole-3,8-diylbis {carbamoyl(2S)pyrrolidine-2,1- diyl[(1S)-2-oxo-1-phenylethane- 2,1-diyl]})biscarbamate |
| 93 |
 |
643.586 | tert-butyl [(1S)-2-{(2S)-2-[(8- bromo-6,11-dihydro-5H- benzo[a]carbazol-3-yl) carbamoyl]pyrrolidin-1-yl}-2- oxo-1-phenylethyl]carbamate |
| 94 |
 |
765.942 | (2S,2′S)-N,N′-6,11-dihydro-5H- benzo[a]carbazole-3,8-diylbis{1- [(2R)-2-(dimethylamino)-2- phenylacetyl]pyrrolidine-2- carboxamide} |
| 95 |
 |
779.968 | (2S,2′S)-N,N′-5,6,7,12- tetrahydrobenzo[6,7]cyclohepta[1,2- b]indole-3,9-diylbis{1- [(2R)-2-(dimethylamino)-2- phenylacetyl]pyrrolidine-2- carboxamide} |
| 96 |
 |
562.718 | N-(6,11-dihydro-5H- benzo[a]carbazol-3-yl)-1-{(2S)- 2-[(3,3-dimethylbutanoyl)amino]- 2-phenylacetyl}-L-prolinamide |
| 97 |
 |
564.69 | tert-butyl {(1R)-2-[(2S)-2-(6,11- dihydro-5H-benzo[a]carbazol-3- ylcarbamoyl)pyrrolidin-1-yl]-2- oxo-1-phenylethyl}carbamate |
| 98 |
 |
924.119 | di-tert-butyl (5,6,7,12- tetrahydrobenzo[6,7]cyclohepta[1,2- b]indole-3,9-diylbis {carbamoyl(2S)pyrrolidine-2,1- diyl[(1R)-2-oxo-1-phenylethane- 2,1-diyl]})biscarbamate |

Step 1



Step 3




| Example | Structure | MW | Name |
| 100 |
 |
807.958 | benzyl (2S)-2-[5-(4-{5-[(1- {(2R)-2-[(tert-butoxycarbonyl) amino]-2-phenylacetyl}-L- prolyl)amino]-1H-indol-2- yl}phenyl)-1H-imidazol-2- yl]pyrrolidine-1-carboxylate |
| 101 |
 |
762.963 | 1-[(2R)-2-(dimethylamino)-2- phenylacetyl]-N-{2-[4-(2-{(2S)- 1-[(2R)-2-(dimethylamino)-2- phenylacetyl]pyrrolidin-2-yl}- 1H-imidazol-5-yl)phenyl]-1H- indol-5-yl}-L-prolinamide |
| 102 |
 |
604.715 | tert-butyl {(1R)-2-[(2S)-2-({2- [4-(1H-imidazol-4-yl)phenyl]- 1H-indol-5-yl}carbamoyl) pyrrolidin-1-yl]-2-oxo-1- phenylethyl}carbamate |
| 103 |
 |
762.963 | 1-[(2S)-2-(dimethylamino)-2- phenylacetyl]-N-{2-[4-(2-{(2S)- 1-[(2S)-2-(dimethylamino)-2- phenylacetyl]pyrrolidin-2-yl}- 1H-imidazol-5-yl)phenyl]-1H- indol-5-yl}-L-prolinamide |
| 104 |
 |
791.958 | tert-butyl {(1R)-2-oxo-1-phenyl- 2-[(2S)-2-{[2-(4-{2-[(2S)-1- (phenylacetyl)pyrrolidin-2-yl]- 1H-imidazol-5-yl}phenyl)-1H- indol-5-yl]carbamoyl}pyrrolidin- 1-yl]ethyl}carbamate |
| 105 |
 |
715.86 | tert-butyl{(1R)-2-[(2S)-2-{[2-(4- {2-[(2S)-1-acetylpyrrolidin-2- yl]-1H-imidazol-5-yl}phenyl)- 1H-indol-5-yl]carbamoyl} pyrrolidin-1-yl]-2-oxo-1- phenylethyl}carbamate |
| 106 |
 |
793.931 | benzyl (2S)-2-(5-{4-[5-({1- [(2R)-2-phenyl-2-{[(propan-2- yloxy)carbonyl]amino}acetyl]-L- prolyl}amino)-1H-indol-2- yl]phenyl}-1H-imidazol-2- yl)pyrrolidine-1-carboxylate |
| 107 |
 |
835.027 | tert-butyl {(1R)-2-[(2S)-2-({2- [4-(2-{(2S)-1-[(2R)-2- (dimethylamino)-2- phenylacetyl]pyrrolidin-2-yl}- 1H-imidazol-5-yl)phenyl]-1H- indol-5-yl}carbamoyl)pyrrolidin- 1-yl]-2-oxo-1- phenylethyl}carbamate |
| 108 |
 |
893.064 | propan-2-yl [(1R)-2-{(2S)-2-[1- methyl-4-(4-{5-[({(2S)-1-[(2R)- 2-phenyl-2-{[(propan-2- yloxy)carbonyl]amino}acetyl] pyrrolidin-2-yl}carbonyl)amino]- 1H-indol-2-yl}phenyl)-1H- imidazol-2-yl]pyrrolidin-1-yl}-2- oxo-1-phenylethyl]carbamate |
| 109 |
 |
701.833 | propan-2-yl {(1R)-2-[(2S)-2-{[2- (4-{2-[(2S)-1-acetylpyrrolidin-2- yl]-1H-imidazol-5-yl}phenyl)- 1H-indol-5-yl]carbamoyl} pyrrolidin-1-yl]-2-oxo-1- phenylethyl}carbamate |
| 110 |
 |
659.795 | propan-2-yl {(1R)-2-oxo-1- phenyl-2-[(2S)-2-{[2-(4-{2- [(2S)-pyrrolidin-2-yl]-1H- imidazol-5-yl}phenyl)-1H-indol- 5-yl]carbamoyl}pyrrolidin-1- yl]ethyl}carbamate |
| 111 |
 |
547.663 | propan-2-yl {(1R)-2-[(2S)-2-{5- [4-(1H-indol-2-yl)phenyl]-1H- imidazol-2-yl}pyrrolidin-1-yl]-2- oxo-1-phenylethyl}carbamate |
| 112 |
 |
489.626 | (2R)-2-(dimethylamino)-1-[(2S)- 2-{5-[4-(1H-indol-2-yl)phenyl]- 1H-imidazol-2-yl}pyrrolidin-1- yl]-2-phenylethanone |
| 113 |
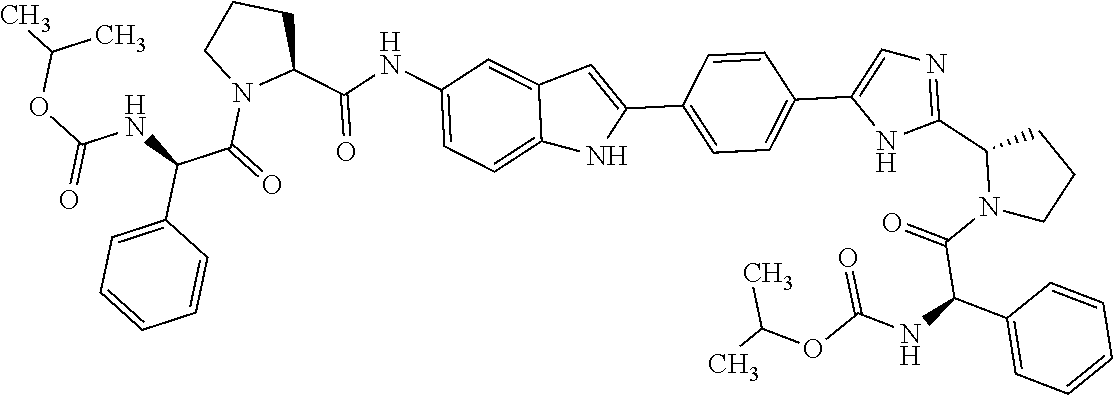 |
879.037 | propan-2-yl [(1R)-2-oxo-1- phenyl-2-{(2S)-2-[5-(4-{5- [({(2S)-1-[(2R)-2-phenyl-2- {[(propan-2-yloxy)carbonyl] amino}acetyl]pyrrolidin-2- yl}carbonyl)amino]-1H-indol-2- yl}phenyl)-1H-imidazol-2-yl] pyrrolidin-1-yl}ethyl]carbamate |
| 114 |
 |
821 | propan-2-yl {(1R)-2-[(2S)-2-({2- [4-(2-{(2S)-1-[(2R)-2- (dimethylamino)-2-phenylacetyl] pyrrolidin-2-yl}-1H-imidazol-5- yl)phenyl]-1H-indol-5- yl}carbamoyl)pyrrolidin-1-yl]-2- oxo-1-phenylethyl}carbamate |
| 115 |
 |
582.108 | propan-2-yl {(1R)-2-[(2S)-2-{4- [4-(3-chloro-1H-indol-2- yl)phenyl]-1H-imidazol-2- yl}pyrrolidin-1-yl]-2-oxo-1- phenylethyl}carbamate |
| 116 |
 |
754.894 | N-(methoxycarbonyl)-L-valyl-N- {2-[4-(2-{(2S)-1-[N- (methoxycarbonyl)-L- valyl]pyrrolidin-2-yl}-1H- imidazol-5-yl)phenyl]-1H-indol- 5-yl}-L-prolinamide |

Step 1









Step 1





Step 1


To a solution of Cbz-Pro-OH (2.9986 g, 12.0 mmol) in THF (100 mL) was added TEA (1.7 mL, 12.2 mmol). The solution was cooled to −25° C. and ethyl chloroformate (1.6 mL, 12.3 mmol) was added. The resulting solution was stirred at RT for 1 hour. The precipitate was removed by filtration, and the filtrate was used in next step without purification. A solution of 0.5 M diazomethane was added to the above reaction mixture. The sample was stirred at −10° C. for 1 hour. The reaction mixture was concentrated to one half of its original volume and washed once with saturated NaHCO3 (50 mL). The organic layer was dried over MgSO4 and filtered. The crude material was adsorbed onto silica gel and purified by flash chromatography (40 g SiO2, 0-50% ethyl acetate in hexanes) to give the diazoketone (2.29 g). 1H NMR (CDCl3) δ: 7.32 (m, 5H) 5.13 (m, 2H), 4.61 (m, 1H), 3.81, 4.03, 4.17 (s, AB quartet, 2H, J=4.0 Hz), 3.58 (m, 2H), 1.88-2.09, 2.17-2.38 (2, br m, 4H).



Step 1

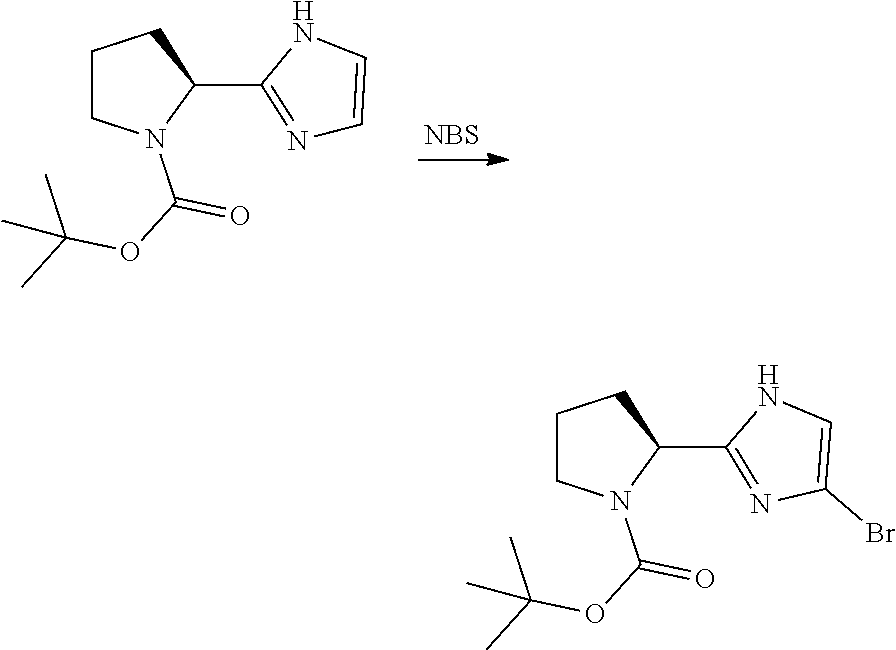





Step 7


Step 1










Step 1



Step 3

Step 1








Step 1





Step 5


Step 1









| Example | Structure | MW | Name |
| 128 |
 |
688.233 | (2S)-N-{4-[3-chloro-5-({[(2S)-1- (phenylacetyl)pyrrolidin-2- yl]carbonyl}amino)-1H-indol-2- yl]phenyl}-1-(phenylacetyl) pyrrolidine-2-carboxamide |
| 129 |
 |
671.778 | (2S)-N-{4-[3-fluoro-5-({[(2S)-1- (phenylacetyl)pyrrolidin-2- yl]carbonyl}amino)-1H-indol-2- yl]phenyl}-1-(phenylacetyl) pyrrolidine-2-carboxamide |
| 130 |
 |
902.044 | tert-butyl {(1S)-2-[(2S)-2-({4-[5- ({[(2S)-1-{(2S)-2-[(tert- butoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2- yl]carbonyl}amino)-3-fluoro-1H- indol-2-yl]phenyl}carbamoyl) pyrrolidin-1-yl]-2-oxo-1- phenylethyl}carbamate |
| 131 |
 |
630.15 | tert-butyl {(1S)-2-[(2S)-2-({2-[4- (acetylamino)phenyl]-3-chloro- 1H-indol-5-yl}carbamoyl) pyrrolidin-1-yl]-2-oxo-1- phenylethyl}carbamate |
| 132 |
 |
779.684 | (2S)-N-{4-[3-iodo-5-({[(2S)-1- (phenylacetyl)pyrrolidin-2-yl] carbonyl}amino)-1H-indol-2-yl] phenyl}-1-(phenylacetyl) pyrrolidine-2-carboxamide |
| 133 |
 |
710.813 | tert-butyl {(1S)-2-[(2S)-2-({2-[4- ({[(2S)-1-acetylpyrrolidin-2- yl]carbonyl}amino)phenyl]-3- fluoro-1H-indol-5- yl}carbamoyl)pyrrolidin-1-yl]-2- oxo-1-phenylethyl}carbamate |
| 134 |
 |
898.1 | (2S)-1-{(2S)-2-[(3,3- dimethylbutanoyl)amino]-2- phenylacetyl}-N-{4-[5-({[(2S)-1- {(2S)-2-[(3,3-dimethylbutanoyl) amino]-2-phenylacetyl} pyrrolidin-2-yl]carbonyl}amino)- 3-fluoro-1H-indol-2-yl]phenyl} pyrrolidine-2-carboxamide |
| 135 |
 |
902.044 | tert-butyl {(1R)-2-[(2S)-2-({4- [5-({[(2S)-1-{(2R)-2-[(tert- butoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2- yl]carbonyl}amino)-3-fluoro-1H- indol-2-yl]phenyl}carbamoyl) pyrrolidin-1-yl]-2-oxo-1- phenylethyl}carbamate |
| 136 |
 |
613.695 | tert-butyl {(1R)-2-[(2S)-2-({2- [4-(acetylamino)phenyl]-3- fluoro-1H-indol-5- yl}carbamoyl)pyrrolidin-1-yl]-2- oxo-1-phenylethyl}carbamate |
| 137 |
 |
710.813 | tert-butyl {(1R)-2-[(2S)-2-({2- [4-({[(2S)-1-acetylpyrrolidin-2- yl]carbonyl}amino)phenyl]-3- fluoro-1H-indol-5- yl}carbamoyl)pyrrolidin-1-yl]-2- oxo-1-phenylethyl}carbamate |
| 138 |
 |
639.733 | tert-butyl {(1R)-2-[(2S)-2-({3- fluoro-2-[4-(2-oxopyrrolidin-1- yl)phenyl]-1H-indol-5- yl}carbamoyl)pyrrolidin-1-yl]-2- oxo-1-phenylethyl}carbamate |
| 139 |
 |
567.669 | N-{2-[4-(acetylamino)phenyl]-3- fluoro-1H-indol-5-yl}-1-[(2R)-2- phenyl-2-(pyrrolidin-1- yl)acetyl]-L-prolinamide |
| 140 |
 |
551.623 | N-(tert-butoxycarbonyl)-D- alanyl-N-{2-[4- (acetylamino)phenyl]-3-fluoro- 1H-indol-5-yl}-L-prolinamide |
| 141 |
 |
541.631 | N-{2-[4-(acetylamino)phenyl]-3- fluoro-1H-indol-5-yl}-1-[(2R)-2- (dimethylamino)-2- phenylacetyl]-L-prolinamide |
| 142 |
 |
593.705 | N-(tert-butoxycarbonyl)-D- leucyl-N-{2-[4- (acetylamino)phenyl]-3-fluoro- 1H-indol-5-yl}-L-prolinamide |
| 143 |
 |
815.953 | propan-2-yl [(1R)-2-{(2S)-2-[(4- {5-[({(2S)-1-[(2R)-2- (dimethylamino)-2- phenylacetyl]pyrrolidin-2-yl} carbonyl)amino]-3-fluoro-1H- indol-2-yl}phenyl)carbamoyl] pyrrolidin-1-yl}-2-oxo-1- phenylethyl]carbamate) |
| 144 |
 |
754.866 | tert-butyl (2S)-2-[(3-fluoro-2-{4- [({(2S)-1-[(2R)-2-phenyl-2- {[(propan-2-yloxy)carbonyl] amino}acetyl]pyrrolidin-2- yl}carbonyl)amino]phenyl}-1H- indol-5-yl)carbamoyl] pyrrolidine-1-carboxylate |
| 145 |
 |
935.88 | propan-2-yl [(1R)-2-{(2S)-2-[(4- {3-bromo-5-[({(2S)-1-[(2R)-2- phenyl-2-{[(propan-2- yloxy)carbonyl]amino}acetyl] pyrrolidin-2-yl}carbonyl)amino]- 1-benzofuran-2-yl}phenyl) carbamoyl] pyrrolidin-1-yl}-2- oxo-1-phenylethyl]carbamate |
| 146 |
 |
819.806 | (2S)-N-(4-{3-bromo-5-[({(2S)-1- [(2R)-2-(dimethylamino)-2- phenylacetyl]pyrrolidin-2- yl}carbonyl)amino]-1- benzofuran-2-yl}phenyl)-1- [(2R)-2-(dimethylamino)-2- phenylacetyl]pyrrolidine-2- carboxamide |
| 147 |
 |
879.772 | methyl {(1R)-2-[(2S)-2-({4-[3- bromo-5-({[(2S)-1-{(2R)-2- [(methoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2- yl]carbonyl}amino)-1- benzofuran-2-yl]phenyl} carbamoyl)pyrrolidin-1-yl]-2- oxo-1-phenylethyl}carbamate |
| 148 |
 |
749.847 | methyl {(2S)-1-[(2S)-2-({4-[3- fluoro-5-({[(2S)-1-{(2S)-2- [(methoxycarbonyl)amino]-3- methylbutanoyl}pyrrolidin-2- yl]carbonyl}amino)-1H-indol-2- yl]phenyl}carbamoyl)pyrrolidin- 1-yl]-3-methyl-1-oxobutan-2- yl}carbamate |
| 149 |
 |
603.521 | N-{2-[4-(acetylamino)phenyl]-3- bromo-1-benzofuran-5-yl}-1- [(2R)-2-(dimethylamino)-2- phenylacetyl]-L-prolinamide |
| 150 |
 |
875.915 | (2S)-N-(4-{3-bromo-5-[({(2S)-1- [(2R)-2-(diethylamino)-2- phenylacetyl]pyrrolidin-2-yl} carbonyl)amino]-1-benzofuran- 2-yl}phenyl)-1-[(2R)-2- (diethylamino)-2-phenylacetyl] pyrrolidine-2-carboxamide |
| 151 |
 |
700.639 | (2S)-1-acetyl-N-(4-{3-bromo-5- [({(2S)-1-[(2R)-2- (dimethylamino)-2-phenylacetyl] pyrrolidin-2-yl}carbonyl)amino]- 1-benzofuran-2-yl}phenyl) pyrrolidine-2-carboxamide |
| 152 |
 |
963.935 | tert-butyl {(1S)-2-[(2S)-2-({4-[3- bromo-5-({[(2S)-1-{(2S)-2- [(tert-butoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2- yl]carbonyl}amino)-1- benzofuran-2-yl]phenyl} carbamoyl)pyrrolidin-1-yl]-2- oxo-1-phenylethyl}carbamate) |
| 153 |
 |
733.669 | (2S)-N-{4-[3-bromo-5-({[(2S)-1- (phenylacetyl)pyrrolidin-2-yl] carbonyl}amino)-1-benzofuran- 2-yl]phenyl}-1-(phenylacetyl) pyrrolidine-2-carboxamide |
| 154 |
 |
963.935 | tert-butyl {(1R)-2-[(2S)-2-({4- [3-bromo-5-({[(2S)-1-{(2R)-2- [(tert-butoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2- yl]carbonyl}amino)-1- benzofuran-2-yl]phenyl} carbamoyl)pyrrolidin-1-yl]-2- oxo-1-phenylethyl}carbamate |




Step 1




Step 4





Step 1


| Example | Structure | MW | Name |
| 160 |
 |
729.887 | (2S)-1-(phenylacetyl)-N-{4-[3- phenyl-5-({[(2S)-1- (phenylacetyl)pyrrolidin-2- yl]carbonyl}amino)-1H-indol-2- yl]phenyl}pyrrolidine-2- carboxamide |
| 161 |
 |
681.842 | (2S)-N-{4-[3-ethyl-5-({[(2S)-1- (phenylacetyl)pyrrolidin-2- yl]carbonyl}amino)-1H-indol-2- yl]phenyl}-1-(phenylacetyl) pyrrolidine-2-carboxamide |
| 162 |
 |
695.826 | (2S)-N-{4-[3-acetyl-5-({[(2S)-1- (phenylacetyl)pyrrolidin-2- yl]carbonyl}amino)-1H-indol-2- yl]phenyl}-1-(phenylacetyl) pyrrolidine-2-carboxamide |
| 163 |
 |
757.897 | (2S)-1-(phenylacetyl)-N-{4-[5- ({[(2S)-1-(phenylacetyl) pyrrolidin-2-yl]carbonyl}amino)- 3-(phenylcarbonyl)-1H-indol-2- yl]phenyl}pyrrolidine-2- carboxamide |
| 164 |
 |
730.874 | (2S)-1-(phenylacetyl)-N-{2-[4- ({[(2S)-1-(phenylacetyl) pyrrolidin-2-yl]carbonyl} amino)phenyl]-3-(pyridin-4-yl)- 1H-indol-5-yl}pyrrolidine-2- carboxamide |
| 165 |
 |
761.886 | benzyl (2S)-2-[(4-{5-[({(2S)-1- [(benzyloxy)carbonyl]pyrrolidin- 2-yl}carbonyl)amino]-3-phenyl- 1H-indol-2-yl}phenyl) carbamoyl]pyrrolidine-1- carboxylate |
| 166 |
 |
678.798 | (2S)-N-{4-[3-cyano-5-({[(2S)-1- (phenylacetyl)pyrrolidin-2- yl]carbonyl}amino)-1H-indol-2- yl]phenyl}-1-(phenylacetyl) pyrrolidine-2-carboxamide |
| 167 |
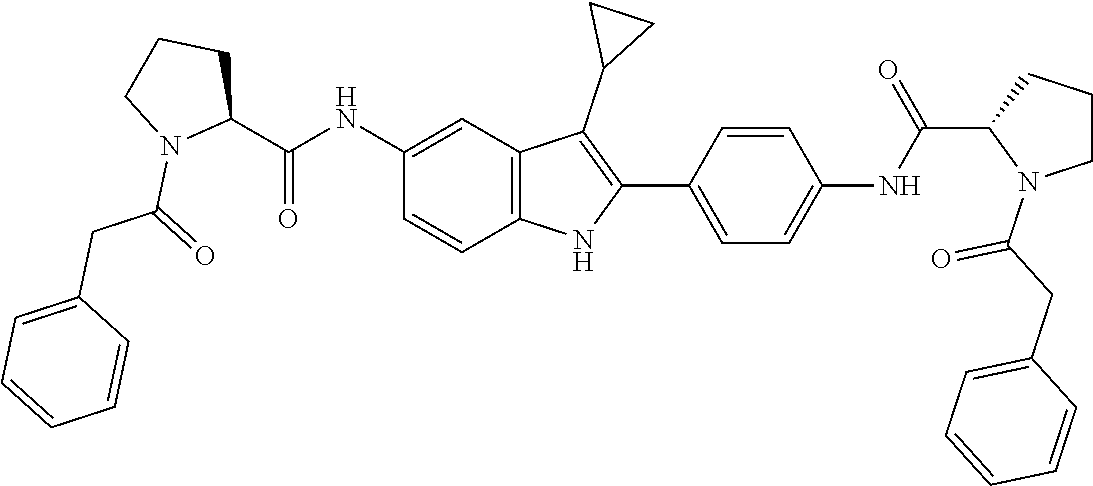 |
693.853 | (2S)-N-{4-[3-cyclopropyl-5- ({[(2S)-1-(phenylacetyl) pyrrolidin-2-yl]carbonyl}amino)- 1H-indol-2-yl]phenyl}-1- (phenylacetyl)pyrrolidine-2- carboxamide |
| 168 |
 |
791.912 | benzyl (2S)-2-[(4-{5-[({(2S)-1- [(benzyloxy)carbonyl]pyrrolidin- 2-yl}carbonyl)amino]-3-(3- methoxyphenyl)-1H-indol-2- yl}phenyl)carbamoyl]pyrrolidine- 1-carboxylate |
| 169 |
 |
974.18 | tert-butyl {(1R)-2-[(2S)-2-({4- [5-({[(2S)-1-{(2R)-2-[(tert- butoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2- yl]carbonyl}amino)-3-(3- methylphenyl)-1H-indol-2- yl]phenyl}carbamoyl)pyrrolidin- 1-yl]-2-oxo-1- phenylethyl}carbamate |
| 170 |
 |
974.18 | tert-butyl {(1R)-2-[(2S)-2-({4- [5-({[(2S)-1-{(2R)-2-[(tert- butoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2- yl]carbonyl}amino)-3-(2- methylphenyl)-1H-indol-2- yl]phenyl}carbamoyl)pyrrolidin- 1-yl]-2-oxo-1- phenylethyl}carbamate |
| 171 |
 |
961.14 | tert-butyl {(1R)-2-[(2S)-2-({2- [4-({[(2S)-1-{(2R)-2-[(tert- butoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2- yl]carbonyl}amino)phenyl]-3- (pyridin-4-yl)-1H-indol-5- yl}carbamoyl)pyrrolidin-1-yl]-2- oxo-1-phenylethyl}carbamate |
| 172 |
 |
909.064 | tert-butyl {(1R)-2-[(2S)-2-({4- [5-({[(2S)-1-{(2R)-2-[(tert- butoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2- yl]carbonyl}amino)-3-cyano-1H- indol-2-yl]phenyl}carbamoyl) pyrrolidin-1-yl]-2-oxo-1- phenylethyl}carbamate |
| 173 |
 |
764.936 | (2S)-N-(4-{3-cyano-5-[({(2S)-1- [(2R)-2-(dimethylamino)-2- phenylacetyl]pyrrolidin-2- yl}carbonyl)amino]-1H-indol-2- yl}phenyl)-1-[(2R)-2- (dimethylamino)-2-phenylacetyl] pyrrolidine-2-carboxamide |
| 174 |
 |
620.714 | tert-butyl {(1R)-2-[(2S)-2-({2- [4-(acetylamino)phenyl]-3- cyano-1H-indol-5- yl}carbamoyl)pyrrolidin-1-yl]-2- oxo-1-phenylethyl}carbamate |
| 175 |
 |
955.133 | tert-butyl {(1R)-2-[(2S)-2-({4- [5-({[(2S)-1-{(2R)-2-[(tert- butoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2- yl]carbonyl}amino)-3- (dimethylcarbamoyl)-1H-indol- 2-yl]phenyl}carbamoyl) pyrrolidin-1-yl]-2-oxo-1- phenylethyl}carbamate |
| 176 |
 |
942.091 | methyl 5-({[(2S)-1-{(2R)-2- [(tert-butoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2- yl]carbonyl}amino)-2-[4-({[(2S)- 1-{(2R)-2-[(tert- butoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2- yl]carbonyl}amino)phenyl]-1H- indole-3-carboxylate |
| 177 |
 |
928.064 | 5-({[(2S)-1-{(2R)-2-[(tert- butoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2- yl]carbonyl}amino)-2-[4-({[(2S)- 1-{(2R)-2-[(tert-butoxycarbonyl) amino]-2-phenylacetyl} pyrrolidin-2-yl]carbonyl}amino) phenyl]-1H-indole-3-carboxylic acid |

Step 1


Step 2




Step 1


| Example | Structure | MW | Name |
| 180 |
 |
557.701 | N-(4-{5-[6-(dimethylamino)-4- methylpyridin-3-yl]-1H-indol-2- yl}phenyl)-1-(phenylacetyl)-L- prolinamide |
| 181 |
 |
555.704 | N-{4-[5-(1-benzothiophen-3-yl)- 1H-indol-2-yl]phenyl}-1- (phenylacetyl)-L-prolinamide |
| 182 |
 |
579.708 | N-{4-[5-(1-benzyl-1H-pyrazol-4- yl)-1H-indol-2-yl]phenyl}-1- (phenylacetyl)-L-prolinamide |
| 183 |
 |
557.655 | N-{4-[5-(2,3-dihydro-1,4- benzodioxin-6-yl)-1H-indol-2- yl]phenyl}-1-(phenylacetyl)-L- prolinamide |
| 184 |
 |
550.666 | 1-(phenylacetyl)-N-{4-[5- (quinolin-8-yl)-1H-indol-2- yl]phenyl}-L-prolinamide |
| 185 |
 |
505.644 | 1-(phenylacetyl)-N-{4-[5- (thiophen-3-yl)-1H-indol-2- yl]phenyl}-L-prolinamide |
| 186 |
 |
563.662 | tert-butyl {(1S)-2-[(2S)-2-{[4-(5- cyano-1H-indol-2-yl)phenyl] carbamoyl}pyrrolidin-1-yl]-2- oxo-1-phenylethyl}carbamate |
| 187 |
 |
879.037 | propan-2-yl [(1R)-2-oxo-1- phenyl-2-{(2S)-2-[4-(2-{4- [({(2S)-1-[(2R)-2-phenyl-2- {[(propan-2-yloxy)carbonyl] amino}acetyl]pyrrolidin-2- yl}carbonyl)amino]phenyl}-1H- indol-5-yl)-1H-imidazol-2-yl] pyrrolidin-1-yl}ethyl]carbamate |
| 188 |
 |
606.687 | propan-2-yl {(1R)-2-[(2S)-2-(5- {2-[4-(acetylamino)phenyl]-1H- indol-5-yl}-1,3,4-oxadiazol-2- yl)pyrrolidin-1-yl]-2-oxo-1- phenylethyl}carbamate |
| 189 |
 |
576.704 | N-{4-[5-(5-{(2S)-1-[(2R)-2- (diethylamino)-2- phenylacetyl]pyrrolidin-2-yl}- 1,3,4-oxadiazol-2-yl)-1H-indol- 2-yl]phenyl}acetamide |
| 189a |
 |
806.9 | methyl [(2S)-1-{(2S)-2-[5-(10- {2-[(2S)-1-{(2S)-2- [(methoxycarbonyl)amino]-3- methylbutanoyl}pyrrolidin-2-yl]- 1H-imidazol-5-yl}indolo[1,2-c] [1,3]benzoxazin-3-yl)-1H- imidazol-2-yl]pyrrolidin-1-yl}-3- methyl-1-oxobutan-2- yl]carbamate |
| 189b |
 |
824.9 | methyl [(2S)-1-{(2S)-2-[5-(12- fluoro-10-{2-[(2S)-1-{(2S)-2- [(methoxycarbonyl)amino]-3- methylbutanoyl}pyrrolidin-2-yl]- 1H-imidazol-5-yl}indolo[1,2- c]][1,3]benzoxazin-3-yl)-1H- imidazol-2-yl]pyrrolidin-1-yl}-3- methyl-1-oxobutan-2- yl]carbamate |

Step 1










Step 1






Step 6

Step 1





Step 1



Step 3


Step 5





| Ex- | ||||
| ample | Structure | 1H NMR | M + 1 | Name |
| 193 |
 |
(MeOD) δ: 7.2-8.1 (m, 20 H), 5.2-5.6 (m, 4 H), 3.9- 4.2 (m, 2 H), 3.1 (m, 2 H), 2.6 (m, 8 H), 1.9-2.5 (m, 8 H), 1.0-1.5 (m, 12 H). | 844 | (2R)-2-(diethylamino)-1- [(2S)-2-(5-{4-[5-(2-{(2S)- 1-[(2R)-2-(diethylamino)- 2-phenylacetyl]pyrrolidin- 2-yl}-1H-imidazol-5-yl)-1- benzofuran-2-yl]phenyl}- 1H-imidazol-2-yl) pyrrolidin-1-yl]-2- phenylethanone |
| 194 |
 |
(MeOD) δ: 8.0-8.1 (m, 3 H), 7.7-7.9 (m, 6 H), 7.4 (d, J = 2.4 Hz, 1 H), 5.3 (d, J = 5.6 Hz, 2 H), 4.1 (m, 2 H), 3.9 (m, 4 H), 3.7 (m, 6 H), 2.5-2.6 (m, 2 H), 2.1- 2.3 (m, 6 H), 1.1-1.2 (m, 2 H), 0.4-0.6 (m, 9 H). | 775 | Methyl {(1S)-1- cyclopropyl-2-[(2S)-2-{5- [4-(5-{2-[(2S)-1-{(2S)-2- cyclopropyl-2- [(methoxycarbonyl)amino] acetyl}pyrrolidin-2-yl]-1H- imidazol-5-yl}-1- benzofuran-2-yl)phenyl]- 1H-imidazol-2- yl}pyrrolidin-1-yl]-2- oxoethyl}carbamate |
| 195 |
 |
(MeOD) δ: 7.7-8.1 (m, 9 H), 7.4(m, 16 H), 5.3-5.4 (m, 2 H), 3.5-4.1 (m, 12 H), 2.6 (d, J = 4.8 Hz, 26 H), 2.2 (d, J = 4.8 Hz, 6 H), 1.1-1.2 (m, 2 H), 0.4- 0.7 (m, 8 H). | 775 | Methyl {(1R)-1- cyclopropyl-2-[(2S)-2-{5- [4-(5-{2-[(2S)-1-{(2R)- 2-cyclopropyl-2- [(methoxycarbonyl)amino] acetyl}pyrrolidin-2-yl]-1H- imidazol-5-yl}-1- benzofuran-2-yl)phenyl]- 1H-imidazol- 2-yl}pyrrolidin-1- yl]-2-oxoethyl}carbamate |
| 196 |
 |
(MeOD) δ: 7.8-7.9 (m, 5 H), 7.1-7.6 (m, 5 H), 5.6- 5.7 (m, 1 H), 5.2 (d, J = 4.8 Hz, 1 H), 4.0-4.2 (m, 3 H), 3.6-3.8 (m, 8 H), 2.0-2.5 (m, 10 H), 1.6 (d, J = 4.8 Hz, 1 H), 1.3 (m, 1 H), 0.8-1.1 (m, 11 H), 0.4 (m, 26 H). | 780 | Methyl {(2R)-1-[(2S)-2- {5-[2-(4-{2-[(2S)-1- {(2R)-2- [(methoxycarbonyl) amino]-3-methylbutanoyl} pyrrolidin-2-yl]-1H- imidazol-5-yl}phenyl)-1- benzofuran-5-yl]-1H- imidazol-2-yl}pyrrolidin- 1-yl]-3-methyl-1- oxobutan-2-yl}carbamate |
| 197 |
 |
(MeOD), δ 8.10(d, J = 4 Hz, 2 H), 8.01(s, 1 H), 7.94(s, 1 H) 7.87(d, J = 2 Hz, 2 H), 7.84(m, 1 H), 7.73(d, J = 4 Hz, 1 H), 7.44(m, 1 H), 5.25(m, 2 H), 4.33(m, 2 H), 4.16(m, 2 H), 3.89(m, 2 H), 3.67(s, 6 H), 2.58(m, 2 H), 2.20(m, 6 H), 0.97(m, 18 H) | 806 | Methyl {(2S)-1-[(2S)-2- {5-[4-(5-{2-[(2S)-1-{(2S)- 2-[(methoxycarbonyl) amino]-3,3- dimethylbutanoyl} pyrrolidin-2-yl]-1H- imidazol-5-yl}-1- benzofuran-2-yl) phenyl]-1H-imidazol-2- yl}pyrrolidin-1-yl]-3,3- dimethyl-1-oxobutan-2- |
| yl}carbamate | ||||
| 198 |
 |
(MeOD) δ: 7.6-8.1 (m, 9 H), 7.3-7.5 (m, 11 H), 7.2 (m, 1 H), 5.4-5.5 (m, 2 H), 5.3 (m, 2 H), 4.1 (d, J = 4.8 Hz, 2 H), 3.7 (d, J = 2.4 Hz, 6 H), 1.9-2.4 (m, 8 H) | 848 | Methyl {(1R)-2-[(2S)-2- {5-[4-(5-{2-[(2S)-1- {(2R)-2- [(methoxycarbonyl) amino]-2- phenylacetyl}pyrrolidin- 2-yl]-1H-imidazol-4- yl}-1-benzofuran-2- yl)phenyl]-1H-imidazol- 2-yl}pyrrolidin-1-yl]-2- |
| oxo-1-phenylethyl} | ||||
| carbamate | ||||
| 199 |
 |
(MeOD) δ 8.08(d, J = 4 Hz, 2 H), 8.04(s, 1 H), 7.89(d, 3 H), 7.78(m, 1 H), 7.77 (m, 2 H), 7.42(m, 1 H), 5.34(t, J = 4 Hz, 2 H), 4.42(d, J = 4 Hz, 2 H), 4.10(s, 2 H), 3.82(m, 2 H), 3.79(m, 3 H), 3.64(s, 3 H), 2.57(m, 2 H), 2.20(m, 6 H), 1.88(m, 2 H), 1.48(m, 2 | 806 | Methyl {(2S,3R)-1- [(2S)-2-(5-{2-[4-(2- {(2S)-1-[N- (methoxycarbonyl)-L- alloisoleucyl]pyrrolidin- 2-yl}-1H-imidazol-5-yl) phenyl]-1-benzofuran-5- yl}-1H-imidazol-2- yl)pyrrolidin-1-yl]-3- methyl-1-oxopentan-2- yl}carbamate |
| H), 1.32(m, 2 H), | ||||
| 0.97(m, 12 H). | ||||
| 200 |
 |
(MeOD) δ: 7.6-8.1 (m, 9 H), 7.4 (m, 6 H), 5.4 (m, 2 H), 4.6 (m, 2 H), 3.5-4.1 (m, 13 H), 2.5-2.7 (m, 6 H), 2.3 (m, 5 H). | 755 | Methyl {(2S)-3-hydroxy- 1-[(2S)-2-{5-[4-(5-{2- [(2S)-1-{(2S)-3-hydroxy- 2-[(methoxycarbonyl) amino]propanoyl} pyrrolidin-2-yl]-1H- imidazol-5-yl}-1- benzofuran-2-yl)phenyl]- 1H-imidazol-2-yl} pyrrolidin-1-yl]-1- oxopropan-2-yl}carbamate |
| 201 |
 |
(MeOD), δ 8.04(d, J = 4 Hz, 2 H) 7.97(s, 1H), 7.84(d, J = 2 Hz, 2 H), 7.81(m, 1 H), 7.76(m, 1 H), 7.64(d, J = 4 Hz, 2 H), 7.38(m, 1 H), 5.29(m, 2 H), 4.49(m, 2 H), 4.15(m, 2 H), 3.97(m, 1 H), 3.92(m, 1 H), 3.66(m, 6 H), 2.63(m, 1 H), 2.60(m, 1 H), 2.54(m, 2 H), 2.16- | 783 | Methyl {(2S,3R)-3- hydroxy-1-[(2S)-2-{5-[4- (5-{2-[(2S)-1-{(2S,3R)- 3-hydroxy-2- [(methoxycarbonyl) amino]butanoyl} pyrrolidin-2-yl]- 1H-imidazol-5-yl}-1- benzofuran-2-yl)phenyl]- 1H-imidazol-2-yl} pyrrolidin-1-yl]-1- |
| 2.22(m, 6 H), 1.16 (d, J = | oxobutan-2-yl}carbamate | |||
| 2 Hz, 6 H). | ||||
| 202 |
 |
(MeOD), δ 8.05(d, J = 4 Hz, 2 H), 7.97(s, 1 H), 7.84(d, 1 H), 7.81(d, J = 2 Hz, 2 H), 7.76(m, 1 H), 7.64(d, J = 4 Hz, 2 H), 7.39(m, 1 H), 5.25(m, 2 H), 4.45(m, 2 H), 4.03(m, 2 H), 3.84(m, 2 H), 3.85(m, 2 H), 3.64(s, 6 H), 2.55(m, 2 H), 2.22(m, 6 H), 1.70(m, 2 H), | 806 | Methyl {(2S)-1- [(2S)-2-{5-[4-(5- {2-[(2S)-1-{(2S)-2- [(methoxycarbonyl) amino]-4- methylpentanoyl} pyrrolidin-2-yl]-1H- imidazol-5-yl}-1- benzofuran-2-yl) phenyl]-1H-imidazol- 2-yl}pyrrolidin-1- |
| 1.51(m, 4 H), 0.98 (m, 12 | yl]-4-methyl-1- | |||
| H) | oxopentan-2- | |||
| yl}carbamate | ||||
Step 1






| Ex- | |||
| ample | Structure | M + 1 | Name |
| 204 |
 |
866 | Methyl {(1R)-2-[(2S)-2-{5-[3-fluoro-4-(5-{2- [(2S)-1-{(2R)-2-[(methoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2-yl]-1H-imidazol-5- yl}-1-benzofuran-2-yl)phenyl]-1H-imidazol- 2-yl}pyrrolidin-1-yl]-2-oxo-1- phenylethyl}carbamate |
| 205 |
 |
797 | Methyl {(2S)-1-[(2S)-2-{5-[3-fluoro-4-(5-{2- [(2S)-1-{(2S)-2-[(methoxycarbonyl)amino]-3- methylbutanoyl}pyrrolidin-2-yl]-1H- imidazol-5-yl}-1-benzofuran-2-yl)phenyl]- 1H-imidazol-2-yl}pyrrolidin-1-yl]-3-methyl- 1-oxobutan-2-yl}carbamate |
| 206 |
 |
816 | Methyl {(2S)-1-[(2S)-2-{5-[2-(2,6-difluoro-4- {2-[(2S)-1-{(2S)-2-[(methoxycarbonyl)amino]- 3-methylbutanoyl}pyrrolidin-2-yl]- 1H-imidazol-5-yl}phenyl)-1-benzofuran-5- yl]-1H-imidazol-2-yl}pyrrolidin-1-yl]-3- methyl-1-oxobutan-2-yl}carbamate |
| 207 |
 |
878 | Methyl {(1R)-2-[(2S)-2-{5-[3-methoxy-4-(5- {2-[(2S)-1-{(2R)-2-[(methoxycarbonyl)amino]- 2-phenylacetyl}pyrrolidin-2-yl]-1H- imidazol-5-yl}-1-benzofuran-2-yl)phenyl]- 1H-imidazol-2-yl}pyrrolidin-1-yl]-2-oxo-1- phenylethyl}carbamate |
| 208 |
 |
814 | Methyl {(2S)-1-[(2S)-2-{5-[2-(2-chloro-4-{2- [(2S)-1-{(2S)-2-[(methoxycarbonyl)amino]-3- methylbutanoyl}pyrrolidin-2-yl]-1H- imidazol-4-yl}phenyl)-1-benzofuran-5-yl]- 1H-imidazol-2-yl}pyrrolidin-1-yl]-3-methyl- 1-oxobutan-2-yl}carbamate |
| 209 |
 |
873 | Methyl {(1R)-2-[(2S)-2-{4-[3-cyano-4-(5-{2- [(2S)-1-{(2R)-2-[(methoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2-yl]-1H-imidazol-5- yl}-1-benzofuran-2-yl)phenyl]-1H-imidazol- 2-yl}pyrrolidin-1-yl]-2-oxo-1- phenylethyl}carbamate |
| 210 |
 |
794 | Methyl {(2S)-1-[(2S)-2-{5-[2-(4-{2-[(2S)-1- {(2S)-2-[(methoxycarbonyl)amino]-3- methylbutanoyl}pyrrolidin-2-yl]-1H- imidazol-4-yl}-2-methylphenyl)-1- benzofuran-5-yl]-1H-imidazol-2- yl}pyrrolidin-1-yl]-3-methyl-1-oxobutan-2- yl}carbamate |
| 211 |
 |
Methyl [(1S)-2-[(2S)-2-(5-{3-fluoro-4-[5-(2- {(2S)-1-[(2S)-2-[(methoxycarbonyl)amino]-2- (tetrahydro-2H-pyran-4-yl)acetyl]pyrrolidin- 2-yl}-1H-imidazol-5-yl)-1-benzofuran-2- yl]phenyl}-1H-imidazol-2-yl)pyrrolidin-1-yl]- 2-oxo-1-(tetrahydro-2H-pyran-4- yl)ethyl]carbamate | |
| 212 |
 |
832 | Methyl {(2S)-1-[(2S)-2-{4-[3-fluoro-4-(5-{2- [(2S)-1-{(2R)-2-[(methoxycarbonyl)amino]-2- phenylacetyl}pyrrolidin-2-yl]-1H-imidazol-5- yl}-1-benzofuran-2-yl)phenyl]-1H-imidazol- 2-yl}pyrrolidin-1-yl]-3-methyl-1-oxobutan-2- yl}carbamate |

Step 1





| Example | Structure | 1H NMR | M + 1 | Name |
| 214 |
 |
(MeOD) δ: 8.4 (d, J = 8.4 Hz, 2 H), 8.1 (s, 1 H), 7.9 (m, 3 H), 7.8 (m, 2 H), 7.7 (m, 1 H), 5.3 (m, 2 H), 4.2 (m, 2 H), 4.1-4.0 (m, 2 H), 3.9-3.8 (m, 2 H), 3.6 (s, 2 H), 2.6 (m, 2 H), 2.3 (m, 2 H), 2.2 (m, 4 H), 2.0 (m, 2 H), 0.9 (m, 12 H). | 848 | Methyl {(2S)-1-[(2S)-2- {5-[2-(4-{2-[(2S)-1-{(2S)- 2-[(methoxycarbonyl) amino]-3- methylbutanoyl} pyrrolidin-2-yl]-1H- imidazol-5-yl}phenyl)- 1,3-benzoxazol-5-yl]-1H- imidazol-2-yl}pyrrolidin- 1-yl]-3-methyl-1- oxobutan-2-yl}carbamate |
| 215 |
 |
(MeOD) δ: 8.4 (d, J = 6.8 Hz, 2 H), 8.2-8.1 (s, 1 H), 8.0 (m, 3 H), 7.9-7.8 (m, 3 H), 5.3 (m, 2 H), 4.1-4.0 (m, 2 H), 4.0 (m, 2 H), 3.9-3.8 (m 2 H), 3.7 (m, 6 H), 2.6 (m, 2 H), 2.4-2.1 (m, 6 H), 1.3- 1.1 (m, 2 H), 0.7-0.6 (m, 3 H), 0.6-0.5 (m, 3 H), 0.4 (m, 2 H). | 776 | Methyl {(1R)-1- cyclopropyl-2-[(2S)-2-{5- [4-(5-{2-[(2S)-1-{(2R)-2- cyclopropyl-2- [(methoxycarbonyl) amino]acetyl} pyrrolidin- 2-yl]-1H-imidazol-5-yl}- 1,3-benzoxazol-2- yl)phenyl]-1H-imidazol- 2-yl}pyrrolidin-1-yl]-2- oxoethyl}carbamate |
| Example | Structure | M + 1 | Name |
| 216 |
 |
875 | dimethyl (indolo[1,2- c][1,3]benzoxazine-3,10-diylbis{1H- imidazole-5,2-diyl(2S)pyrrolidine- 2,1-diyl[(1R)-2-oxo-1-phenylethane- 2,1-diyl]})biscarbamate |
| 217 |
 |
821 | methyl [(2S)-1-{(2S)-2-[5-(11-{2- [(2S)-1-{(2S)-2-[(methoxycarbonyl) amino]-3-methylbutanoyl}pyrrolidin- 2-yl]-1H-imidazol-5-yl}-6,7- dihydroindolo[1,2-d][1,4] benzoxazepin-3-yl)-1H-imidazol-2- yl]pyrrolidin-1-yl}-3-methyl-1- oxobutan-2-yl]carbamate |
| 218 |
 |
835 | methyl [(2S)-1-{(2S)-2-[5-(3-{2- [(2S)-1-{(2S)-2-[(methoxycarbonyl) amino]-3-methylbutanoyl}pyrrolidin- 2-yl]-1H-imidazol-5-yl}-6,6- dimethylindolo[1,2-c][1,3] benzoxazin-10-yl)-1H-imidazol-2- yl]pyrrolidin-1-yl}-3-methyl-1- oxobutan-2-yl]carbamate |
| 219 |
 |
839 | methyl [(2S)-1-{(2S)-2-[5-(12-fluoro- 10-{2-[(2S)-1-{(2S)-2- [(methoxycarbonyl)amino]-3- [(methylbutanoyl}pyrrolidin-2-yl]-1H- imidazol-5-yl}-6-methylindolo[1,2- c][1,2]benzoxazin-3-yl)-1H-imidazol- 2-yl]pyrrolidin-1-yl}-3-methyl-1- oxobutan-2-yl]carbamate |
| 220 |
 |
804 | methyl [(2S)-1-{(2S)-2-[5-(3-{2- [(2S)-1-{(2S)-2-[(methoxycarbonyl) amino]-3-methylbutanoyl}pyrrolidin- 2-yl]-1H-imidazol-5-yl}indolo[1,2- c]quinazolin-10-yl)-1H-imidazol-2- yl]pyrrolidin-1-yl}-3-methyl-1- oxobutan-2-yl]carbamate |
| 221 |
 |
835 | methyl [(2S)-1-{(2S)-2-[5-(12-{2- [(2S)-1-{(2S)-2-[(methoxycarbonyl) amino]-3-methylbutanoyl}pyrrolidin- 2-yl]-1H-imidazol-5-yl}-7,8-dihydro- 6H-indolo[1,2-e][1,5]benzoxazocin- 3-yl)-1H-imidazol-2-yl]pyrrolidin-1- yl}-3-methyl-1-oxobutan-2- yl]carbamate |
| 222 |
 |
820 | methyl [(2S)-1-{(2S)-2-[5-(3-{2- [(2S)-1-{(2S)-2-[(methoxycarbonyl) amino]-3-methylbutanoyl}pyrrolidin- 2-yl]-1H-imidazol-5-yl}-6-oxo-5,6- dihydroindolo[1,2-c]quinazolin-10- yl)-1H-imidazol-2-yl]pyrrolidin-1- yl}-3-methyl-1-oxobutan-2- yl]carbamate |
| 223 |
 |
883 | methyl [(2S)-1-{(2S)-2-[5-(10-{2- [(2S)-1-{(2S)-2-[(methoxycarbonyl) amino]-3-methylbutanoyl}pyrrolidin- 2-yl]-1H-imidazol-5-yl}-6- phenylindolo[1,2-c][1,3]benzoxazin- 3-yl)-1H-imidazol-2-yl]pyrrolidin-1- yl}-3-methyl-1-oxobutan-2- yl]carbamate |
| 224 |
 |
818 | methyl [(2S)-1-{(2S)-2-[5-(3-{2- [(2S)-1-{(2S)-2-[(methoxycarbonyl) amino]-3-methylbutanoyl}pyrrolidin- 2-yl]-1H-imidazol-5-yl}-6- methylindolo[1,2-c]quinazolin-10- yl)-1H-imidazol-2-yl]pyrrolidin-1- yl}-3-methyl-1-oxobutan-2- yl]carbamate |
| 225 |
 |
875.1 | methyl [(2S)-1-{(2S)-2-[5-(10′-{2- [(2S)-1-{(2S)-2-[(methoxycarbonyl) amino]-3-methylbutanoyl}pyrrolidin- 2-yl]-1H-imidazol-5-yl}spiro [cyclohexane-1,6′-indolo[1,2- c][1,3]benzoxazin]-3′-yl)-1H- imidazol-2-yl]pyrrolidin-1-yl}-3- methyl-1-oxobutan-2-yl]carbamate |
| 226 |
 |
842 | methyl [(2S)-1-{(2S)-2-[5-(1,12- difluoro-10-{2-[(2S)-1-{(2S)-2- [(methoxycarbonyl)amino]-3- methylbutanoyl}pyrrolidin-2-yl]-1H- imidazol-5-yl}indolo[1,2- c][1,3]benzoxazin-3-yl)-1H-imidazol- 2-yl]pyrrolidin-1-yl}-3-methyl-1- oxobutan-2-yl]carbamate |
| 227 |
 |
832 | methyl [(2S)-1-{(2S)-2-[5-(12-cyano- 10-{2-[(2S)-1-{(2S)-2- [(methoxycarbonyl)amino]-3- methylbutanoyl}pyrrolidin-2-yl]-1H- imidazol-5-yl}indolo[1,2- c][1,3]benzoxazin-3-yl)-1H-imidazol- 2-yl]pyrrolidin-1-yl}-3-methyl-1- oxobutan-2-yl]carbamate |
| Activity Table |
| Example | EC50 (nM) | ||
| 2 | 9 | ||
| 6 | 200 | ||
| 14 | 10 | ||
| 15 | 0.045 | ||
| 19 | 25 | ||
| 26 | 0.063 | ||
| 30 | 26 | ||
| 39 | 0.24 | ||
| 40 | 0.026 | ||
| 41 | 0.05 | ||
| 42 | 14 | ||
| 45 | 0.02 | ||
| 49 | 0.072 | ||
| 58 | 0.97 | ||
| 60 | 0.13 | ||
| 62 | 0.067 | ||
| 72 | 0.17 | ||
| 94 | 0.006 | ||
| 95 | 0.01 | ||
| 96 | 0.015 | ||
| 99 | 0.038 | ||
| 100 | 0.031 | ||
| 101 | 0.5 | ||
| 102 | 8.3 | ||
| 103 | 5.7 | ||
| 105 | 0.08 | ||
| 107 | 0.04 | ||
| 116 | 0.065 | ||
| 119 | 0.013 | ||
| 125 | 0.016 | ||
| 129 | 0.7 | ||
| 130 | 0.05 | ||
| 131 | 17 | ||
| 137 | 0.009 | ||
| 138 | 8.5 | ||
| 144 | 0.036 | ||
| 155 | 0.9 | ||
| 158 | 0.5 | ||
| 159 | 0.002 | ||
| 169 | 0.004 | ||
| 178 | 317 | ||
| 186 | 0.015 | ||
| 189a | 0.15 | ||
| 189b | 0.001 | ||
| 190 | 0.067 | ||
| 191 | 0.02 | ||
| 192 | 0.002 | ||
| 193 | 0.05 | ||
| 203 | 0.004 | ||
| 213 | 0.009 | ||
Claims (12)





Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US16/829,878 US11053243B2 (en) | 2009-03-27 | 2020-03-25 | Inhibitors of hepatitis C virus replication |
| US17/324,799 US20220356182A1 (en) | 2009-03-27 | 2021-05-19 | Inhibitors of hepatitis c virus replication |
Applications Claiming Priority (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US16395809P | 2009-03-27 | 2009-03-27 | |
| US24731809P | 2009-09-30 | 2009-09-30 | |
| PCT/US2010/028653 WO2010111483A1 (en) | 2009-03-27 | 2010-03-25 | Inhibitors of hepatitis c virus replication |
| US201113260684A | 2011-12-19 | 2011-12-19 | |
| US14/473,117 US9090661B2 (en) | 2009-03-27 | 2014-08-29 | Inhibitors of hepatitis C virus replication |
| US14/711,345 US20150246917A1 (en) | 2009-03-27 | 2015-05-13 | Inhibitors of hepatitis c virus replication |
| US15/272,669 US20170008906A1 (en) | 2009-03-27 | 2016-09-22 | Inhibitors of hepatitis c virus replication |
| US15/800,172 US20190127365A1 (en) | 2017-11-01 | 2017-11-01 | Inhibitors of hepatitis c virus replication |
| US16/829,878 US11053243B2 (en) | 2009-03-27 | 2020-03-25 | Inhibitors of hepatitis C virus replication |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/800,172 Continuation US20190127365A1 (en) | 2009-03-27 | 2017-11-01 | Inhibitors of hepatitis c virus replication |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US17/324,799 Continuation US20220356182A1 (en) | 2009-03-27 | 2021-05-19 | Inhibitors of hepatitis c virus replication |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| US20200262836A1 US20200262836A1 (en) | 2020-08-20 |
| US11053243B2 true US11053243B2 (en) | 2021-07-06 |
Family
ID=66245222
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/800,172 Abandoned US20190127365A1 (en) | 2009-03-27 | 2017-11-01 | Inhibitors of hepatitis c virus replication |
| US16/829,878 Active US11053243B2 (en) | 2009-03-27 | 2020-03-25 | Inhibitors of hepatitis C virus replication |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/800,172 Abandoned US20190127365A1 (en) | 2009-03-27 | 2017-11-01 | Inhibitors of hepatitis c virus replication |
Country Status (1)
| Country | Link |
|---|---|
| US (2) | US20190127365A1 (en) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HUE066335T2 (en) | 2018-10-05 | 2024-07-28 | Annapurna Bio Inc | Compounds and compositions for treating conditions associated with apj receptor activity |
Citations (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH10101591A (en) | 1996-09-27 | 1998-04-21 | Eisai Co Ltd | Preventing and therapeutic agent for viral infectious disease |
| WO2003010140A2 (en) | 2001-07-25 | 2003-02-06 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis c virus polymerase inhibitors with heterobicyclic structure |
| US20060019974A1 (en) | 2000-09-20 | 2006-01-26 | Werner Mederski | 4-Amino-quinazolines |
| EP1719773A1 (en) | 2004-02-24 | 2006-11-08 | Japan Tobacco, Inc. | Fused heterotetracyclic compounds and use tehreof as hcv polymerase inhibitor |
| US20060258682A1 (en) | 2005-05-16 | 2006-11-16 | Yun Liao | Benzofuran compounds |
| US7153848B2 (en) | 2004-08-09 | 2006-12-26 | Bristol-Myers Squibb Company | Inhibitors of HCV replication |
| WO2007009120A2 (en) | 2005-07-14 | 2007-01-18 | Irm Llc | Heterotetracyclic compounds as tpo mimetics |
| US20070049593A1 (en) | 2004-02-24 | 2007-03-01 | Japan Tobacco Inc. | Tetracyclic fused heterocyclic compound and use thereof as HCV polymerase inhibitor |
| US20070110708A1 (en) | 2003-07-10 | 2007-05-17 | Miller David J | Silicon compounds and their use |
| WO2007084413A2 (en) | 2004-07-14 | 2007-07-26 | Ptc Therapeutics, Inc. | Methods for treating hepatitis c |
| US20070185175A1 (en) | 2005-08-01 | 2007-08-09 | Bristol-Myers Squibb Company | Benzothiazole and azabenzothiazole compounds useful as kinase inhibitors |
| US20080299075A1 (en) | 2006-08-11 | 2008-12-04 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| CN101336248A (en) | 2005-12-16 | 2008-12-31 | 国家健康医学研究所 | Novel compounds that interact with PEA-15 |
| US20090004111A1 (en) | 2005-02-28 | 2009-01-01 | The Rockefeller University | Structure of the Hepatitis C Ns5a Protein |
| US20090042860A1 (en) | 2007-08-09 | 2009-02-12 | Bristol-Myers Squibb Company | Compounds for the Treatment of Hepatitis C |
| WO2009023179A2 (en) | 2007-08-10 | 2009-02-19 | Genelabs Technologies, Inc. | Nitrogen containing bicyclic chemical entities for treating viral infections |
| WO2009102325A1 (en) | 2008-02-13 | 2009-08-20 | Bristol-Myers Squibb Company | Imidazolyl biphenyl imidazoles as hepatitis c virus inhibitors |
| US7662809B2 (en) | 2004-10-26 | 2010-02-16 | Istituto Di Richerche Di Biologia Molecolare P Angeletti Spa | Tetracyclic indole derivatives as antiviral agents |
| WO2010041687A1 (en) | 2008-10-09 | 2010-04-15 | コニカミノルタホールディングス株式会社 | Organic photoelectric conversion element, solar cell, and optical sensor array |
| WO2010065674A1 (en) | 2008-12-03 | 2010-06-10 | Presidio Pharmaceuticals, Inc. | Inhibitors of hcv ns5a |
| WO2010111483A1 (en) | 2009-03-27 | 2010-09-30 | Merck Sharp & Dohme Corp. | Inhibitors of hepatitis c virus replication |
| US20100316607A1 (en) | 2009-06-16 | 2010-12-16 | Yat Sun Or | Hepatitis c virus inhibitors |
| US20110104109A1 (en) | 2005-07-13 | 2011-05-05 | Frank Bennett | Tetracyclic indole derivatives and their use for treating or preventing viral infections |
| US20110130361A1 (en) | 2007-08-09 | 2011-06-02 | Jonathan Grimm | Silicon derivatives as histone deacetylase inhibitors |
| US7973040B2 (en) | 2008-07-22 | 2011-07-05 | Merck Sharp & Dohme Corp. | Macrocyclic quinoxaline compounds as HCV NS3 protease inhibitors |
| US20110223134A1 (en) | 2010-03-09 | 2011-09-15 | Anilkumar Gopinadhan Nair | Fused tricyclic silyl compounds and methods of use thereof for the treatment of viral diseases |
| US20110224211A1 (en) | 2008-11-28 | 2011-09-15 | Frank Ulrich Schmitz | Anti-Viral Compounds, Compositions, And Methods Of Use |
| US20120040962A1 (en) | 2009-03-27 | 2012-02-16 | Presidio Pharmaceuticals, Inc. | Fused ring inhibitors of hepatitis c |
| WO2012041014A1 (en) | 2010-09-29 | 2012-04-05 | Merck Sharp & Dohme Corp. | Tetracyclic indole derivatives for treating hepatitis c virus infection |
| WO2012040923A1 (en) | 2010-09-29 | 2012-04-05 | Merck Sharp & Dohme Corp. | Tetracyclic indole derivatives and methods of use thereof for the treatment of viral diseases |
| US20120083448A1 (en) | 2009-03-27 | 2012-04-05 | Van Andel Research Institute | Parathyroid Hormone Peptides And Parathyroid Hormone-Related Protein Peptides And Methods Of Use |
| WO2012050850A1 (en) | 2010-09-29 | 2012-04-19 | Merck Sharp & Dohme Corp. | Polycyclic heterocycle derivatives and methods of use thereof for the treatment of viral diseases |
| US8377980B2 (en) | 2009-12-16 | 2013-02-19 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US20130156731A1 (en) | 2009-12-22 | 2013-06-20 | Kevin X. Chen | Fused tricyclic compounds and methods of use thereof for the treatment of viral diseas |
| US20130164258A1 (en) | 2009-05-29 | 2013-06-27 | Kevin X. Chen | Antiviral Compounds Composed of Three Aligned Aryl Moieties to Treat Diseases such as Hepatitis C |
| US20140170111A1 (en) | 2010-09-29 | 2014-06-19 | Merck Sharp & Dohmn Corp. | Fused tetracycle derivatives and methods of use thereof for the treatment of viral diseases |
| US20140199264A1 (en) | 2011-03-17 | 2014-07-17 | Craig A. Coburn | Tetracyclic xanthene derivatives and methods of use thereof for the treatment of viral diseases |
| US20140377223A1 (en) | 2010-07-26 | 2014-12-25 | Joseph A. Kozlowski | Substituted biphenylene compounds and methods of use thereof for the treatment of viral diseases |
-
2017
- 2017-11-01 US US15/800,172 patent/US20190127365A1/en not_active Abandoned
-
2020
- 2020-03-25 US US16/829,878 patent/US11053243B2/en active Active
Patent Citations (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH10101591A (en) | 1996-09-27 | 1998-04-21 | Eisai Co Ltd | Preventing and therapeutic agent for viral infectious disease |
| US20060019974A1 (en) | 2000-09-20 | 2006-01-26 | Werner Mederski | 4-Amino-quinazolines |
| WO2003010140A2 (en) | 2001-07-25 | 2003-02-06 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis c virus polymerase inhibitors with heterobicyclic structure |
| US20070110708A1 (en) | 2003-07-10 | 2007-05-17 | Miller David J | Silicon compounds and their use |
| US20070049593A1 (en) | 2004-02-24 | 2007-03-01 | Japan Tobacco Inc. | Tetracyclic fused heterocyclic compound and use thereof as HCV polymerase inhibitor |
| EP1719773A1 (en) | 2004-02-24 | 2006-11-08 | Japan Tobacco, Inc. | Fused heterotetracyclic compounds and use tehreof as hcv polymerase inhibitor |
| WO2007084413A2 (en) | 2004-07-14 | 2007-07-26 | Ptc Therapeutics, Inc. | Methods for treating hepatitis c |
| US7153848B2 (en) | 2004-08-09 | 2006-12-26 | Bristol-Myers Squibb Company | Inhibitors of HCV replication |
| US7662809B2 (en) | 2004-10-26 | 2010-02-16 | Istituto Di Richerche Di Biologia Molecolare P Angeletti Spa | Tetracyclic indole derivatives as antiviral agents |
| US20090004111A1 (en) | 2005-02-28 | 2009-01-01 | The Rockefeller University | Structure of the Hepatitis C Ns5a Protein |
| US20060258682A1 (en) | 2005-05-16 | 2006-11-16 | Yun Liao | Benzofuran compounds |
| US20110104109A1 (en) | 2005-07-13 | 2011-05-05 | Frank Bennett | Tetracyclic indole derivatives and their use for treating or preventing viral infections |
| WO2007009120A2 (en) | 2005-07-14 | 2007-01-18 | Irm Llc | Heterotetracyclic compounds as tpo mimetics |
| US20070185175A1 (en) | 2005-08-01 | 2007-08-09 | Bristol-Myers Squibb Company | Benzothiazole and azabenzothiazole compounds useful as kinase inhibitors |
| CN101336248A (en) | 2005-12-16 | 2008-12-31 | 国家健康医学研究所 | Novel compounds that interact with PEA-15 |
| US20080299075A1 (en) | 2006-08-11 | 2008-12-04 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| US20090042860A1 (en) | 2007-08-09 | 2009-02-12 | Bristol-Myers Squibb Company | Compounds for the Treatment of Hepatitis C |
| US20110130361A1 (en) | 2007-08-09 | 2011-06-02 | Jonathan Grimm | Silicon derivatives as histone deacetylase inhibitors |
| WO2009023179A2 (en) | 2007-08-10 | 2009-02-19 | Genelabs Technologies, Inc. | Nitrogen containing bicyclic chemical entities for treating viral infections |
| WO2009102325A1 (en) | 2008-02-13 | 2009-08-20 | Bristol-Myers Squibb Company | Imidazolyl biphenyl imidazoles as hepatitis c virus inhibitors |
| US8080654B2 (en) | 2008-07-22 | 2011-12-20 | Insituto di Ricerche di Biologia Molecolare P. Angeletti SpA | Macrocyclic quinoxaline compounds as HCV NS3 protease inhibitors |
| US7973040B2 (en) | 2008-07-22 | 2011-07-05 | Merck Sharp & Dohme Corp. | Macrocyclic quinoxaline compounds as HCV NS3 protease inhibitors |
| WO2010041687A1 (en) | 2008-10-09 | 2010-04-15 | コニカミノルタホールディングス株式会社 | Organic photoelectric conversion element, solar cell, and optical sensor array |
| US20110224211A1 (en) | 2008-11-28 | 2011-09-15 | Frank Ulrich Schmitz | Anti-Viral Compounds, Compositions, And Methods Of Use |
| WO2010065674A1 (en) | 2008-12-03 | 2010-06-10 | Presidio Pharmaceuticals, Inc. | Inhibitors of hcv ns5a |
| WO2010111483A1 (en) | 2009-03-27 | 2010-09-30 | Merck Sharp & Dohme Corp. | Inhibitors of hepatitis c virus replication |
| US8871759B2 (en) | 2009-03-27 | 2014-10-28 | Merck Sharp & Dohme Corp. | Inhibitors of hepatitis C virus replication |
| US20120040962A1 (en) | 2009-03-27 | 2012-02-16 | Presidio Pharmaceuticals, Inc. | Fused ring inhibitors of hepatitis c |
| US9090661B2 (en) | 2009-03-27 | 2015-07-28 | Merck Sharp & Dohme Corp. | Inhibitors of hepatitis C virus replication |
| US20120083448A1 (en) | 2009-03-27 | 2012-04-05 | Van Andel Research Institute | Parathyroid Hormone Peptides And Parathyroid Hormone-Related Protein Peptides And Methods Of Use |
| US20130164258A1 (en) | 2009-05-29 | 2013-06-27 | Kevin X. Chen | Antiviral Compounds Composed of Three Aligned Aryl Moieties to Treat Diseases such as Hepatitis C |
| US20100316607A1 (en) | 2009-06-16 | 2010-12-16 | Yat Sun Or | Hepatitis c virus inhibitors |
| US8377980B2 (en) | 2009-12-16 | 2013-02-19 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US20130156731A1 (en) | 2009-12-22 | 2013-06-20 | Kevin X. Chen | Fused tricyclic compounds and methods of use thereof for the treatment of viral diseas |
| US20110223134A1 (en) | 2010-03-09 | 2011-09-15 | Anilkumar Gopinadhan Nair | Fused tricyclic silyl compounds and methods of use thereof for the treatment of viral diseases |
| US20140377223A1 (en) | 2010-07-26 | 2014-12-25 | Joseph A. Kozlowski | Substituted biphenylene compounds and methods of use thereof for the treatment of viral diseases |
| WO2012040923A1 (en) | 2010-09-29 | 2012-04-05 | Merck Sharp & Dohme Corp. | Tetracyclic indole derivatives and methods of use thereof for the treatment of viral diseases |
| US20140170111A1 (en) | 2010-09-29 | 2014-06-19 | Merck Sharp & Dohmn Corp. | Fused tetracycle derivatives and methods of use thereof for the treatment of viral diseases |
| US20130280214A1 (en) | 2010-09-29 | 2013-10-24 | Merck Sharp & Dohme Corp. | Polycyclic heterocycle derivatives and methods of use thereof for the treatment of viral diseases |
| WO2012050850A1 (en) | 2010-09-29 | 2012-04-19 | Merck Sharp & Dohme Corp. | Polycyclic heterocycle derivatives and methods of use thereof for the treatment of viral diseases |
| WO2012041014A1 (en) | 2010-09-29 | 2012-04-05 | Merck Sharp & Dohme Corp. | Tetracyclic indole derivatives for treating hepatitis c virus infection |
| US20140199264A1 (en) | 2011-03-17 | 2014-07-17 | Craig A. Coburn | Tetracyclic xanthene derivatives and methods of use thereof for the treatment of viral diseases |
Non-Patent Citations (12)
| Title |
|---|
| Alper, P.B., et al., "Discovery and biological evaluation of benzo[a]carbazole-based small molecule agonists of the trombopioetin (Tpo) receptor", Bioorganic and Medicinal Chem. Lett., 2008, vol. 18, pp. 5255-5258. |
| Caplus Accession No. 1980:471599. |
| Caplus Accession No. 2009:295362 (JP2009-054809). |
| CAR RN 1025830-17-4, STN Entry, Jun. 5, 2008. |
| CN1474815—English Abstract—Corresponding to US Application No. US2006/0019974. |
| Marsilje, T.H., et al., "Optimization of small molecule agonists of the thrombopoietin (Tpo) receptor derived from a benzo[a]carbazole hit scaffold", Bioorganic and Medicinal Chem. Lett., 2008, vol. 18, pp. 5255-5258. |
| U.S. Appl. No. 13/260,684, filed Dec. 19, 2011. |
| U.S. Appl. No. 14/473,117, filed Aug. 29, 2014. |
| U.S. Appl. No. 14/711,345, filed May 13, 2015. |
| U.S. Appl. No. 15/272,699, filed Sep. 22, 2016. |
| U.S. Appl. No. 15/800,172, filed Nov. 1, 2017. |
| Wilson, et al., "Tunable DNA Photocleavage by an Acridine-Imidazole Conjugate", Inorganic Chemistry, 2005, vol. 44, No. 18, pp. 6159-6173. |
Also Published As
| Publication number | Publication date |
|---|---|
| US20190127365A1 (en) | 2019-05-02 |
| US20200262836A1 (en) | 2020-08-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9090661B2 (en) | Inhibitors of hepatitis C virus replication | |
| EP1718608B1 (en) | Viral polymerase inhibitors | |
| MX2011010113A (en) | Method for preparing cha-type molecular sieves using novel structure directing agents. | |
| US11053243B2 (en) | Inhibitors of hepatitis C virus replication | |
| US20220356182A1 (en) | Inhibitors of hepatitis c virus replication | |
| AU2014200550A1 (en) | Inhibitors of hepatitis C virus replication | |
| HK1189577A (en) | Viral polymerase inhibitors | |
| HK1189889A (en) | Viral polymerase inhibitors | |
| HK1189576A (en) | Viral polymerase inhibitors | |
| HK1141021A (en) | Viral polymerase inhibitors | |
| HK1181390A (en) | Viral polymerase inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| FEPP | Fee payment procedure |
Free format text: ENTITY STATUS SET TO UNDISCOUNTED (ORIGINAL EVENT CODE: BIG.); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY |
|
| AS | Assignment |
Owner name: MERCK SHARP & DOHME CORP, NEW JERSEY Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:COBURN, CRAIG A.;LUDMERER, STEVEN W.;LIU, KUN;SIGNING DATES FROM 20110607 TO 20110714;REEL/FRAME:053160/0742 Owner name: MERCK SHARP & DOHME CORP, NEW JERSEY Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:WUXI APPTEC (SHANGHAI) CO., LTD.;REEL/FRAME:053160/0985 Effective date: 20110609 Owner name: WUXI APPTEC (SHANGHAI) CO., LTD., CHINA Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:WU, HAO;SOLL, RICHARD;ZHONG, BIN;AND OTHERS;REEL/FRAME:053161/0537 Effective date: 20170425 |
|
| STPP | Information on status: patent application and granting procedure in general |
Free format text: AWAITING TC RESP., ISSUE FEE NOT PAID |
|
| STPP | Information on status: patent application and granting procedure in general |
Free format text: NOTICE OF ALLOWANCE MAILED -- APPLICATION RECEIVED IN OFFICE OF PUBLICATIONS |
|
| STPP | Information on status: patent application and granting procedure in general |
Free format text: PUBLICATIONS -- ISSUE FEE PAYMENT RECEIVED |
|
| STPP | Information on status: patent application and granting procedure in general |
Free format text: PUBLICATIONS -- ISSUE FEE PAYMENT VERIFIED |
|
| STCF | Information on status: patent grant |
Free format text: PATENTED CASE |
|
| AS | Assignment |
Owner name: MERCK SHARP & DOHME LLC, NEW JERSEY Free format text: MERGER;ASSIGNOR:MERCK SHARP & DOHME CORP.;REEL/FRAME:061102/0145 Effective date: 20220407 |
|
| MAFP | Maintenance fee payment |
Free format text: PAYMENT OF MAINTENANCE FEE, 4TH YEAR, LARGE ENTITY (ORIGINAL EVENT CODE: M1551); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY Year of fee payment: 4 |