WO2011154331A1 - Polymers for delivery of nucleic acids - Google Patents
Polymers for delivery of nucleic acids Download PDFInfo
- Publication number
- WO2011154331A1 WO2011154331A1 PCT/EP2011/059239 EP2011059239W WO2011154331A1 WO 2011154331 A1 WO2011154331 A1 WO 2011154331A1 EP 2011059239 W EP2011059239 W EP 2011059239W WO 2011154331 A1 WO2011154331 A1 WO 2011154331A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polymer
- acid
- fmoc
- resin
- group
- Prior art date
Links
- 0 CC(CN)C(*)N**C(O)=O Chemical compound CC(CN)C(*)N**C(O)=O 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/02—Polyamines
- C08G73/028—Polyamidoamines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/0008—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'non-active' part of the composition delivered, e.g. wherein such 'non-active' part is not delivered simultaneously with the 'active' part of the composition
- A61K48/0025—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'non-active' part of the composition delivered, e.g. wherein such 'non-active' part is not delivered simultaneously with the 'active' part of the composition wherein the non-active part clearly interacts with the delivered nucleic acid
- A61K48/0041—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'non-active' part of the composition delivered, e.g. wherein such 'non-active' part is not delivered simultaneously with the 'active' part of the composition wherein the non-active part clearly interacts with the delivered nucleic acid the non-active part being polymeric
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G69/00—Macromolecular compounds obtained by reactions forming a carboxylic amide link in the main chain of the macromolecule
- C08G69/02—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids
- C08G69/08—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids derived from amino-carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G69/00—Macromolecular compounds obtained by reactions forming a carboxylic amide link in the main chain of the macromolecule
- C08G69/02—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids
- C08G69/26—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids derived from polyamines and polycarboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/111—General methods applicable to biologically active non-coding nucleic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/14—Type of nucleic acid interfering N.A.
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/35—Nature of the modification
- C12N2310/351—Conjugate
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2320/00—Applications; Uses
- C12N2320/30—Special therapeutic applications
- C12N2320/32—Special delivery means, e.g. tissue-specific
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Genetics & Genomics (AREA)
- Biomedical Technology (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Polymers & Plastics (AREA)
- Biotechnology (AREA)
- Molecular Biology (AREA)
- General Engineering & Computer Science (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biochemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
- Veterinary Medicine (AREA)
- Plant Pathology (AREA)
- Public Health (AREA)
- Microbiology (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
Abstract
Polyamide polymers containing defined oligo (alkylene amino) acids, methods for their production, their use as a delivery reagent for nucleic acids, pharmaceutical compositions containing said polyamide polymers, and uses thereof are provided.
Description
POLYMERS FOR DELIVERY OF NUCLEIC ACIDS
The present invention relates to polyamide polymers (PAA) containing defined oligo (alkylene amino) acids (OAA), methods for their production, their use as a delivery reagent for nucleic acids, pharmaceutical compositions containing said polyamide polymers, and uses thereof. In the recent years the application of nucleic acids has become a powerful approach in molecular medicine and diagnostics. Of particular interest is the specific targeting and delivery of double stranded RNA molecules (dsRNA) to and into target tissues and target cells. Double- stranded ribonucleic acid (dsRNA) molecules have been shown to block gene expression in a highly conserved regulatory mechanism known as RNA interference (RNAi). However the in vivo application requires the nucleic acids specifically and efficiently interacting with target cells. The efficient in vivo delivery of functional therapeutic or diagnostic agents to a target tissue or cell still remains one of the biggest obstacles in drug development. Specific targeting and delivery of nucleic acids to and into target tissues and target cells is a major bottleneck, which has not satisfactorily been solved by current technologies. Most so far described nucleic acid delivery entities consist not of one defined molecule but rather are a cocktail of molecules or particles. However, for therapeutic applications, homogenous defined entities are desired.
The nucleic acid payloads must be attached covalently or non-covalently with good stability to assure specific targeting and avoid systemic nonspecific release of the payload. The carrier protects the nucleic acids from degradation in the extracellular environment. However, to enable entry into the cell, the payload is ideally released at or within target cells. To combine good stability within the circulation with effective release at the target is a major bottleneck in conjugate development. In addition the carrier has to be thus designed that it does not cause immunogenicity and is not toxic when accumulating in the body. One approach is to attach the nucleic acids covalently or non-covalently to a polymeric carrier. For example polyamine structures have been widely used as artificial non-viral carrier systems. A major drawback of the polyamine carriers known so far is that they are random
polymerization derived macromolecules and thus poorly defined, heterogeneous molecules in terms of their molecular weight and isomer distribution. The exact composition of these macromolecular polymeric systems is unknown and hence selective modification is impossible and structure activity relationships are difficult to predict. Targeted nucleic acids frequently accumulate in endosomes from which they need to escape to be active. However, effective non-toxic, non-immunogenic endosome escape mechanisms for targeted dsRNAs still have to be found.
Hartmann et al. (e.g. Hartmann L, Hafele S, Peschka-Suss R, Antonietti M, Borner HG. Tailor-Made Poly(amidoamine)s for Controlled Complexation and Condensation of DNA. Chemistry 2008;14(7):2025-2033.) describe linear PEG-polyamidoamines for controlled complexation and condensation of DNA, which are composed of propyleneamine subunits and sometimes combined with spermine. These structures are assembled using commercially available PEG (2.7 kDa, -63 repating units), diamino-N-methyl-dipropylamine, spermine succinic acid anhydride, and lysine. Hartmann et al describe the successful synthesis optimization using a double condensation strategy and show successful complexation of DNA at high N/P ratios. However, these polymers are not suitable for successful siRNA/Oligo/pDNA delivery in vivo. None of the described structures contain lipophilic modifications or thiols for polyplex stabilization. Incorporation of a spacer peptide sequence is described but without any relevance to either polymer properties or delivery. Polymers were not optimized for pDNA binding or lytic activity. Polymer design is restricted by the boundaries of the synthesis and by the limited scope of commercial available amine building blocks. Furthermore the use of tri/tetramines results in a decreased charge density further weakening the nucleic acid complexation. Taken together all described sequences apparently possess no delivery potential. Substances were not tested for lytic activity but with regard to the published cytotoxicity results lytic activity is not very likely.
Wang et al (e.g. Wang XL, Ramusovic S, Nguyen T, Lu ZR, Novel polymerizable surfactants with pH-sensitive amphiphilicity and cell menrane disruption for efficient siRNA delivery, Bioconjugate Chemistry, 2007, 18, 2169-2177) specifically aimed at the development of amphiphilic cationic lipids with classical Y-shape. The substances are characterized by a maximum of 5 charges per molecule and a rather high HLB value. The synthetic route used does not allow the incorporation of additional amines. The structure is therefore not a polymer and could be described as a dipeptide and fatty acid modified oligoamine. Polymer design is
restricted by the narrow boundaries of the synthesis and by the limited scope of commercial available amine building blocks. None of the described structures contain more than one amine building block and the synthesis is limited to commercially available amines. The synthesized structures are exclusively branched structures and have more similarity to PAMAM dendrons than to classical linear polyamidoamines as the amine building block is connected via an alkyl bond to the peptide domain. The polymer design is restricted by the boundaries of the synthesis and by the limited scope of commercial available amine building blocks. Furthermore the use of a single amine building block results in a decreased charge density compared to classical PAAs. Complex, multimodal, programmable delivery polymers can't be synthesized by this strategy as its inherently restricted by its inability to incorporate more than one amino acid unit. Structures were tested for lytic activity and siRNA delivery but show only moderate efficiency. DNA delivery results were not reported. Targeting/shielding can only be incorporated via post- pegylation approaches making the system less flexible and difficult to control.
These drawbacks of prior art are overcome by the polymers of the present invention, providing defined polymeric systems which can be used as effective carriers for the delivery of nucleic acids, with the ability to deliver the nucleic acids into the cytosol across cellular membranes, for example by delivery into and release from endosomal compartments. The polymers of the invention are pH specifically protonated and thus act as pH responsive lytic polymers.
Solid-phase synthesis of linear PEG-polyamidoamines was first reported by Hartmann et al. using a alternating condensation approach resulting in defined, linear, propylenimine based polyamidoamines with high purity. To generate a oligoethyleneimine based library the corresponding boc-protected oligoethylenimine building blocks were synthesized. Application of the published synthesis protocols on synthesis of the novel polymers comprising oligo(alkyleneamine) of this invention was not succesful as the reaction tended to generate cross- linked fragments on the resin. Therefore a new solid phase synthesis strategy based on fmoc/tBu was developed. An object of the present invention is to provide a synthetically defined nucleic acid delivery carrier of low toxicity and high safety based on oligo(alkyleneamine) units. The carrier, when used to administer a nucleic acid such as a siRNA into an animal-derived cell or organism, is capable of delivering efficiently the nucleic acids into the cells while protecting it from being
degraded. In one preferred embodiment, the carrier is (totally or partly) prepared by iterative solid-phase synthesis. The nucleic acid delivery formulation is prepared by either mixing the nucleic acid delivery carrier with a nucleic acid or covalently conjugating the nucleic acid to the carrier, or a combination of both. A polymer comprising oligo(alkyleneamino) acid units is provided, wherein the units have the general structure 1 :

Z is chosen from
either an alkyl group with the general structure L , wherein the number c of methylene (CH2) groups is 0-8;

an amino group with the general structure , wherein the number n of methylene groups is 1-7;
or an aromatic group with the structure
 , or
, or
R is either a methylene group (CH2) or a carbonylgroup (C=0); and the number a of alkyleneamino ((CH2)b NH) groups is 1-7, wherein the number b of methylene (CH2) groups is 2-7 .
The oligo (alkyleneamino) acid units of the polymer are covalently linked via the terminal carboxygroup of a first oligo (alkyleneamino) acid unit and a terminal amino group of a second oligo(alkyleneamino) acid unit. Thus, the polymer comprises oligo (alkyleneamino) acid units covalently linked to each other via an amide bond, and is terminated by a free carboxyl group (-
COOH) at the one end and terminated by a free amine group (-NH2) at the other end. Hence the polymer has a C (carboxy) terminus and an N (amino) terminus.
Said polymer comprises 2-60 oligo(alkyleneamino) acid units having the general structure 1, preferably 2-10 oligo(alkyleneamino) acid units having the general structure 1, most preferably 2-5 oligo(alkyleneamino) acid units having the general structure 1. The polymer can optionally have the same oligo(alkyleneamino)acid units recurring or may also have a combination of varying oligo(alkyleneamino)acid units recurring.
In other embodiments said polymer comprises identical oligo(alkyleneamino)acid units and at least one amino acid recurring, in yet other embodiments said polymer comprises a combination of varying oligo(alkyleneamino)acid units and at least one amino acid recurring.
Below preferred embodiments of the oligo(alkyleneamino)acid units of general structure 1 are described.
As stated above, substituent Z of the oligo(alkyleneamino) acid units are selected from an alkyl group, an amino group or an aromatic group. Below preferred but not limiting embodiments are described wherein Z is an alkyl group.
In a preferred embodiment, Z is an alkyl group wherein the number c of methylene (CH2) groups is 0-8; R is either a methylene group or a carbonyl group; and the number a of alkyleneamino ((CH2)b NH) groups is 1-7, wherein the number b of methylene (CH2) groups is -7. Accordingly said oligo(alkyleneamino) acid units have the general structure 2:

Said polymer comprises 2-60 oligo(alkyleneamino) acid units having the general structure 2, preferably 2-10 oligo(alkyleneamino) acid units having the general structure 2, most preferably 2-5 oligo(alkyleneamino) acid units having the general structure 2. The polymer can optionally have the same oligo(alkyleneamino)acid units recurring or may also have a combination of varying oligo(alkyleneamino)acid units recurring.
In a preferred embodiment, Z is an alkyl group wherein the number c of methylene (CH2) groups is 0-8, R is a carbonylgroup, and the number a of alkyleneamino ((CH2)b NH) groups is 1-7, wherein the number b of methylene (CH2) groups is 2-7 . Accordingly said oligo(alkyleneamino) acid units have the general structure 3:

As illustrated above in the general structure 3, said oligo(alkyleneamino) acid units preferably comprise of 2 - 7, most preferably 3 or 4, alkyleneamine monomers wherein one alkyleneamine monomer is covalently linked to an aliphatic dicarboxylic acid. Preferably said alkyleneamine monomer is selected from the group: Ethyleneamine, propyleneamine, butyleneamine, pentylenamine and hexyleneamine. Preferably said dicarboxylic acid is an aliphatic dicarboxylic acid selected from the group oxalic acid, malonic acid, succinic acid, glutaric acid, adipic acid, pimelic acid, suberic acid, azelaic acid and sebacic acid. Said polymer comprises 2-60 oligo(alkyleneamino) acid units having the general structure 3, preferably 2-10 oligo(alkyleneamino) acid units having the general structure 3, most preferably 2-5 oligo(alkyleneamino) acid units having the general structure 3. The polymer can optionally have the same oligo(alkyleneamino)acid units recurring or may also have a combination of varying oligo(alkyleneamino)acid units recurring. In a preferred embodiment, Z is an alkyl group wherein the number c of methylene (CH2) groups is 0-8, R is a carbonylgroup, and the number a of alkyleneamino ((CH2)b NH) groups is 1-7, wherein the number b of methylene (CH2) groups is 2 . Accordingly in this embodiment said oligo(alkyleneamino) acid units are oligo (ethyleneamino) acid units, said units having the general structure 4:

, wherein a: 2-7, c: 0-8.
GENERAL STRUCTURE 4
Said polymer comprises 2-60 oligo(ethyleneamino) acid units having the general structure 4, preferably 2-10 oligo(ethylenamino) acid units having the general structure 4, most preferably 2- 5 oligo(ethylenamino) acid units having the general structure 4.
As illustrated above in the general structure 4, said oligo(ethyleneamino) acid units preferably comprise of 2 to 7 ethyleneamine monomers wherein one ethyleneamine monomer is covalently linked to a dicarboxylic acid. Preferably said dicarboxylic acid is an aliphatic dicarboxylic acid selected from the group oxalic acid, malonic acid, succinic acid, glutaric acid, adipic acid, pimelic acid, suberic acid, azelaic acid and sebacic acid. In a preferred embodiment, Z is an alkyl group wherein the number c of methylene (CH2) groups is 1, R is a carbonylgroup, and the number a of alkyleneamino ((CH2)b NH) groups is 4, wherein the number b of methylene (CH2) groups is 2. Hence the oligo(ethylenamino) acid units of this embodiment are tetraethylenepentamine succinic acid (Stp) units. Preferably the polymer comprises 2-5 of these (Stp) units. In another embodiment, Z is an alkyl group wherein the number c of methylene (CH2) groups is 0-8; R is a methylene group; and the number a of alkyleneamino ((CH2)b NH) groups is 2-7, wherein the number b of methylene (CH2) groups is 2-6. Accordingly said oligo(alkyleneamino) acid units have the general structure 5:
ΗΟ¾^Ν{¾Ν¾Η
H GENERAL STRUCTURE 5 a: 2-7; b: 2-6, c: 0-8
Said polymer comprises 2-60 oligo(alkyleneamino) acid units having the general structure 5, preferably 2-10 oligo(alkyleneamino) acid units having the general structure 5, most preferably 2-5 oligo(alkyleneamino) acid units having the general structure 5. The polymer can optionally have the same oligo(alkyleneamino)acid units recurring or may also have a combination of varying oligo(alkyleneamino)acid units recurring.
As illustrated above in the general structure 5, said oligo(alkyleneamino) acid units preferably comprise of 2 - 7 alkyleneamine monomers wherein one alkyleneamine monomer is
covalently linked to a monocarboxylic acid. Preferably said alkyleneamine monomer is selected from the group: Ethyleneamine, propyleneamine, butyleneamine, pentylenamine and
hexyleneamine. Preferably said carboxylic acid is an aliphatic carboxylic acid selected from the group acetic acid, propionic acid, butanoic acid, pentanoic acid, hexanoic acid, heptanoic acid, octanoic acid and nonaoic acid.
In another embodiment, Z is an alkyl group wherein the number c of methylene (CH2) groups is 0-8; R is a methylene group; and the number a of alkyleneamino ((CH2)b NH) groups is 2-7, wherein the number b of methylene (CH2) groups is 2. Accordingly said
oligo(alkyleneamino) acid units are oligo (ethyleneamino) acid units with the general structure 6

wherein a: 2-7, c: 0-8. GENERAL STRUCTURE 6
Said polymer comprises 2-60 oligo(ethyleneamino) acid units having the general structure 6, preferably 2-10 oligo(ethylenamino) acid units having the general structure 6, most preferably 2-5 oligo(ethylenamino) acid units having the general structure 6.
As illustrated above in the general structure 6, said oligo(ethyleneamino) acid units preferably comprise of 2 to 7 ethyleneamine monomers wherein one ethyleneamine monomer is covalently linked to a carboxylic acid. Preferably said carboxylic acid is an aliphatic carboxylic acid selected from the group acetic acid, propionic acid, butanoic acid, pentanoic acid, hexanoic acid, heptanoic acid, octanoic acid and nonaoic acid.
As stated above, substituent Z of the oligo(alkyleneamino) acid units are selected from an alkyl group, an amino group or an aromatic group. Below preferred but not limiting
 embodiments are described wherein Z is an amino group of the general structure , wherein the number n of methylene groups is 1-7. Preferably, R is a carbonyl group. In one preferred option, R is a carbonyl group and n is 1 or 2.
In one embodiment said oligo(alkyleneamino) acid polymers has the formula
embodiments are described wherein Z is an amino group of the general structure , wherein the number n of methylene groups is 1-7. Preferably, R is a carbonyl group. In one preferred option, R is a carbonyl group and n is 1 or 2.
In one embodiment said oligo(alkyleneamino) acid polymers has the formula

The oligo(alkyleneamino) acid polymers and derivatives described above are preferably synthesized by solid phase synthesis. The oligo(alkyleneamino) acid polymers provided herein are defined monodisperse molecules of low toxicity with the ability to noncovalently bind nucleic acids. Nucleic acids bear anionic groups and can form an ionic bond with the polymers of this invention. The protonation of the alkyleneamine subunits of the polymers is pH dependent, therefore the payload is released specifically from the endosome. Hence in a preferred embodiment the oligo(alkyleneamino) acid polymers comprise a nucleic acid complexed or covalently conjugated to the polymer. Therefore the oligo(alkyleneamino) acid polymers of the invention are potent carrier molecules useful for the delivery of nucleic acids. In addition, the oligo(alkyleneamino) acid polymers can be designed according to the different requirements for the delivery of various nucleic acid species and are thus useful for the delivery of such different nucleic acid species like DNA, RNA, siRNA, LNA, PNA and others.
To meet the certain needs required for the successful in vivo delivery of nucleic acids, the oligo(alkyleneamino) acid polymers of the invention can be further modified. Targeted nucleic acids frequently accumulate in endosomes from which they need to escape to be active. It is known that the delivery of the nucleic acid into the cytoplasm can be facilitated by the addition of hydrophobic structures to the carrier molecule, as these hydrophobic structures increase the ability of the carrier to lyse lipid membranes. Due to the acidic environment of the endosomal/lysosomal compartments it is crucial for a nucleic acid delivery carrier to display a low buffering capacity at physiological pH values and a high buffering capacity at endosomal/lysosomal pH, thus facilitating selective endosomal/lysosomal membrane disruption. In one preferred embodiment of the invention the oligo(alkyleneamino) acid polymers additionally comprise a hydrophobic domain, which provides a pH-responsive lytic activity.
The hydrophobic domain comprises either multiple hydrophobic amino acid residues, such as leucine, valine, isoleucine, tyrosine or phenylalanine, or at least one fatty acid, such as, for example, myristic acid, palmitic acid, arachidic acid or other fatty acids. Preferred fatty acids
include but are not limited to butyric acid, caprylic acid, myristic acid, oleic acid, linolic acid, arachidic acid, stearic acid, lauric acid and palmitic acid. Additionally, linker groups, between the hydrophobic domain and the oligo(alkyleneamino) acid polymer are also part of the invention. Preferably such linker groups are amino acids, most preferably lysine (for stable linkage) or cysteine (for bioreducible linkage). The use of naturally occurring amino acids or fatty acids is preferable to other hydrophobic moieties because they are relatively non- immunogenic and non-toxic. In addition, other hydrophobic domains such as hydrocarbon chains (saturated or unsaturated long chain aliphatic alcohols or thiols), stabilizing siRNA complexes and supporting endosome escape can be covalently linked to the polymers of the invention. The hydrophobic domain described above is preferably covalently linked to the oligo(alkyleneamino) acid polymers of the invention.
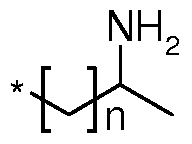
In one embodiment, said hydrophobic domain is linked to the amine group at the N- terminus of the oligo(alkyleneamino) acid polymer, resulting in an i-shape as illustrated in figure 1 b. In other embodiments, said hydrophobic domain is connected to the N- terminus of the oligo(alkyleneamino) acid polymer through a linker group. Said linker group is covalently linked to the amine group at the N-terminus of the polymer and comprises preferably at least one amino acid. Preferably said at least one amino acid is selected from the group of: one lysine, two lysines covalently linked to each other via an amide bond, a cysteine and a lysine covalently linked to each other via an amide bond, and two cysteines covalently linked to each other via an amide bond.
In one embodiment the hydrophobic domain consists of two fatty acids covalently linked to a lysine residue via an amide bond using both amino functions. The lysine residue is either covalently connected to the N-terminus of the oligo(alkyleneamino) acid polymer via its carboxyl group, or covalently linked to the N-terminus of another aminoacid which is then connected to the N-terminus of the oligo(alkyleneamino) acid polymer via its carboxyl group. This second amino acid of the linker is preferably a lysine or a cysteine, as stated above. Said cysteine can be part of a crosslinking domain as stated below (see also Figure 1 d).
Other linker groups envisaged are amino acids with thiol groups such as cysteine. In this embodiment the hydrophobic domain is covalently linked to cysteine residues at the N-terminus of the oligo(alkyleneamino) acid polymer via a disulfide linkage.
In other embodiments said hydrophobic domain is covalently linked to two oligo(alkyleneamino) acid polymer chains of the invention, resulting in a t-shape as illustrated in figure 1 e. Preferably, said hydrophobic domain is connected to the oligo(alkyleneamino) acid polymers through a linker group. Said linker group is covalently linked to the two oligo(alkyleneamino) acid polymers via the amine group at the N-terminus or the carboxygroup at the C-terminus and comprises preferably at least one amino acid. Preferably said at least one amino acid is selected from the group of: one lysine, two lysines covalently linked to each other via an amide bond, a cysteine and a lysine covalently linked to each other via an amide bond, and two cysteines covalently linked to each other via an amide bond.
In one embodiment, said linker group is lysine, which is covalently linked to the amine group at the N-terminus of a first oligo(alkyleneamino) acid polymer chain and covalently linked to the carboxygroup of the C-terminus of a second oligo(alkyleneamino) acid polymer chain, and which is covalently linked via its second amine group with one fatty acid. In other embodiments, the second amine group is covalently linked to the carboxygroup of a lysine group, which is covalently linked to two fatty acids via an amide bond using both amino functions.
Apart from the t-shaped polymers described above, other branched oligo(alkyleneamino) acid polymers are also part of this invention.
A key requirement for efficient transport using synthetic nucleic acid delivery systems is their ability to compact nucleic acids and the formation of stable polyplexes suitable for the efficient delivery to and into the cell. The nucleic acid compacting ability of polymers is strongly influenced by charge density, molecular weight and hydrophobic interactions of the polymers with the nucleic acid. Hence, structural modifications of the oligo(alkyleneamino) acid polymers that enhance compaction of the nucleic acid payload and formation of stable polyplexes are part of the invention. In one preferred embodiment, said polymer described above additionally comprises at least one head group which increases binding to the nucleic acid, formulation stability or interaction with lipid membranes.
The head group is preferably covalently linked to the N- or C-terminus of the oligo(alkyleneamino) acid polymers, preferably via a peptide bond. In one preferred embodiment the headgroup preferably comprises a polar positively charged head group, a coupling or crosslinking domain or a polar positively charged head group in the vicinity of a coupling or crosslinking domain.
Said coupling and crosslinking domain facilitates cross-linking of the oligo(alkyleneamino) acid polymers (dimerisation or polymerisation) and the coupling onto macromolecules such as nucleic acids. Thus the coupling or crosslinking domain increases self- stabilisation of oligo(alkyleneamino) acid polymers. The dimerisation/polymerisation of polymer molecules increases the positive charge and the number of aliphatic chains per molecule, which positively influence the transfection efficiency as well as the noncovalent binding of nucleic acids to the polymers. The resulting crosslinked polyplexes (i.e. crosslinked oligo(alkyleneamino) acid polymers) disintegrate after transport into the cell due to the bioreversible nature of the crosslinking. In addition, it is possible to covalently link the nucleic acid to the carrier polymer via the coupling and crosslinking domain.
Said coupling and crosslinking domain preferably comprises a functional group which facilitates bioreversible ligation, and comprises inter alia a cysteine (for disulfide bond formation), tyrosine trimers or oligomers (for noncovalent aromatic stabilization), ureido- pyrimidinones (for noncovalent hydrogen bonding), aldehydes (for imine, oxime or hydrazone formation) or azide (for click chemistry with alkyne groups). In one embodiment, the oligo(alkyleneamino) acid polymer has a coupling domain comprising at least one, preferably two or three cysteine groups (Figure 1 c to Figure 1 f). Preferably the oligo(alkyleneamino) acid polymer comprises two cysteine groups covalently linked via a peptide bond to each terminus of the polymer. In one embodiment, the oligo(alkyleneamino) acid polymer additionally comprises a hydrophobic domain which is covalently linked to one of the terminal cysteine groups, which results in the i-shape topology (Figure 1 d). As stated above, the hydrophobic group is either covalently linked via a peptide bond to the cysteine group directly or via a lysine group that is covalently linked to the cysteine and the hydrophobic group. In another embodiment, a polymer molecule with t-shape conformation is envisaged, comprising two oligo(alkyleneamino) acid polymer chains to each of which a cysteine group is covalently linked to one terminus, said oligo(alkyleneamino) acid polymers being connected via a central linker group, and a lipophilic domain which is covalently linked to the central linker group (Figure 1 e). Preferably said linker
group is at least one amino acid, most preferably one lysine or two lysines covalently linked to each other via a peptide bond. In another embodiment, the oligo(alkyleneamino) acid polymer has a coupling domain comprising at least one, tyrosine trimer, or a tyrosine oligomer comprising 3 to 50, preferably 3 to 10, most preferably 3-5 tyrosines, instead of cysteine groups. The tyrosine groups are linked to said polymers as outlined for cysteine above.
In another embodiment, the oligo(alkyleneamino) acid polymer has a coupling domain comprising at least one, preferably two or three ureido-pyrimidinones instead of cysteine groups. Preferably said ureido-pyrimidinones is 2-(6-Isocyanahexylaminocarbonylamino)-6-methyl- 4(1 H) pyrimidinone (ICH-CAMP). The ureido-pyrimidinones are linked to said polymers as outlined for cysteine above.
In another embodiment, the oligo(alkyleneamino) acid polymer has a coupling domain comprising aldehydes. Preferred aldehydes used herein are aliphatic aldehydes such as propionaldehyde linked at the omega carbon position to the polymer.
In another embodiment, the oligo(alkyleneamino) acid polymer has a coupling domain comprising azides which can be used for coupling with alkynes by click reaction.
In other embodiments, said head group comprises a polar head group. Said polar head group facilitates nucleic acid/formulation stability or lipid membrane interaction and preferably comprises a thiol group, such as cysteine and is preferably flanked by at least one polar positively charged amino acid, such as lysine, arginine or histidin. In one preferred embodiment, said oligo(alkyleneamino) acid polymer comprises a polar headgroup and a hydrophobic domain. In another preferred option, said oligo(alkyleneamino) acid polymer comprises a crosslinking domain and a hydrophobic domain. Said domains are covalently linked to the oligo(alkyleneamino) acid polymer, either directly via the amine group or via a linker group. Said domains can be thus linked to the oligo(alkyleneamino) acid polymer to form a chain, an i- shaped polymer or a t-shaped polymer. In addition, other configurations (branched polymers) are envisaged. One non-limiting example is shown in figure lh. In another embodiment a branched polymer with four or five arms is envisaged, as shown in figure 36.
Further, in one embodiment the oligo(alkyleneamino) acid polymers additionally comprise agents that facilitate other functions in the eukaryotic cell, e.g. receptor recognition (targeting), internalization, release, nucleus localization and systemic in vivo stabilization. Such
agent preferably comprises a targeting ligand, which recognizes tissue specific cell-surface structures, such as receptors. Most preferably said targeting ligand comprises a targeting peptide, a targeting antibody, a protein, a small molecule or a carbohydrate ligand. In one embodiment said targeting ligand is selected from the group of transferrin receptor binding molecules such as transferrin or phage-derived synthetic peptides such as B6, epidermal growth factor (EGF) receptor binding molecules such as EGF or phage derived synthetic peptide GE11, folic acid, cyclic RDG peptide (for binding integrin) and peptide CMP (for binding the hepatocyte growth factor c-Met). In a preferred embodiment said targeting ligand is CMP.
In another embodiment, said agents that facilitate other functions in the eukaryotic cell are selected from agents that facilitate endosomal escape. Preferred therein is the use of endosome-buffering imidazoles such as histidine for enabling endosomal escape. Therefore one or more imidazoles are incorporated in the oligo(alkyleneamino) acid polymer backbone, preferably in form of the amino acid histidine via amide bond formation like in standard peptide synthesis. In another preferred embodiment, non-amino acid forms of imidazoles are incorporated, such as derivatives of 3-aminopropyl-imidazole via amide bond formation. One non-limiting example of a 3-aminopropyl-imidazole derivative is 3-(imidazole)propane amino- Ν,Ν-diacetic acid.
Other agents that facilitate endosomal escape are endosomolytic peptides such as mellitin, Inf7 and cell penetrating peptides such as the TAT peptide or oligoarginine peptide. Thus, in another embodiment said polymer comprises an agent that facilitates endosomal escape chosen from the group of imidazoles and endosomolytic peptides.
In one embodiment said polymer comprises a targeting ligand chosen from the group of a peptide sequence, a vitamin, a proteins, an antibody, or a synthetic chemical receptor-binding ligand. Non-limiting examples of targeting proteins useful herein are EGF and Transferrin. A preferred vitamin is Folic Acid (FolA). Preferred synthetic chemical receptor-binding ligands are anisamide, clenbuterol and iloprost.
In one preferred embodiment, said polymer described above additionally comprises a shielding moiety. Said shielding moiety is useful for in vivo stabilization of the oligo(alkyleneamino) acid polymer/ nucleic acid polyplex while circulating in the blood and to reduce undesired interactions or agglomeration oligo(alkyleneamino) acid polymer/ nucleic acid polyplex and preferably comprises monodisperse or polydisperse polyethylene glycol (PEG).
In one preferred embodiment, said polymer comprises a targeting ligand attached to a monodisperse PEG which is attached to the polymer (Figure 1 g). In one preferred embodiment, a bifunctionally modified PEG chain is attached via a carboxy group to the amino group of a targeting ligand, and the oligo(alkyleneamino) acid polymer is attached to the amino-group of the PEG chain.
For shielding/stabilization purposes a PEG-chain (preferably monodisperse) of a Mw > 1 kDa can be attached at any position of the oligo(alkyleneamino) acid polymer using the epsilon- amino function of lysine or the amino function at the N-terminus, or the carboxy function at the C-terminus. In other embodiments, the oligo(alkyleneamino) acid polymers are combined with lipids, including also PEG-lipids. In another embodiment the oligo(alkyleneamino) acid polymers are combined with transfection reagents. Non-limiting examples of transfection reagents are lipoplexes, liposomes and cationic lipids.
In one preferred embodiment use of the oligo(alkyleneamino) acid polymers and its derivatives for the delivery of nucleic acids are envisaged. Particularly of interest herein is the delivery of siRNA into its target cell. The siRNA can be modified or conjugated to contain functional units such as endosomolytic peptides (such as Inf7) or targeting ligands (such as FolA). Other therapeutic nucleic acids of interest are antisense molecules, microRNAs, microRNA blocking antagomirs, apoptosis-inducing RNAs, immunostimulatory RNAs, messenger RNAs, or DNA molecules encoding gene expression units.
Another embodiment provides a pharmaceutical composition comprising the oligo(alkyleneamino) acid polymers and a nucleic acid, and may further comprise an agent that is complexed to the polymer. In particular, said pharmaceutical composition comprises a oligo(alkyleneamino) acid polymers and an siRNA molecule. These pharmaceutical compositions are particularly useful in the inhibition of the expression of a target gene in a cell, a tissue or an organism. The pharmaceutical composition comprising the oligo(alkyleneamino) acid polymers of the invention may also comprise (a) pharmaceutically acceptable carrier(s), diluent(s) and/or excipient(s). Yet a further embodiment provides a method of treating a mammal, comprising administering the oligo(alkyleneamino) acid polymers complexed with a nucleic acid to a mammal.
Definitions:
The term "nucleic acid" as used herein means an oligomer or polymer composed of nucleotides, e.g., deoxyribonucleotides or ribonucleotides, or compounds produced synthetically (e.g., PNA as described in U.S. Pat. No. 5,948,902 and the references cited therein) which can hybridize with naturally occurring nucleic acids in a sequence specific manner analogous to that of two naturally occurring nucleic acids, e.g., can participate in Watson-Crick base pairing interactions. Non-naturally occurring nucleic acids are oligomers or polymers which contain nucleobase sequences which do not occur in nature, or species which contain functional equivalents of naturally occurring nucleobases, sugars, or inter-sugar linkages, like peptide nucleic acids (PNA), threose nucleic acids (TNA), locked nucleic acids (LNA), or glycerol nucleic acids (GNA). This term includes oligomers that contain the naturally occurring nucleic acid nucleobases adenine (A), guanine (G), thymine (T), cytosine (C) and uracil (U), as well as oligomers that contain base analogs or modified nucleobases. Nucleic acids can derive from a variety of natural sources such as viral, bacterial and eukaryotic DNAs and RNAs. Other nucleic acids can be derived from synthetic sources, and include any of the multiple oligonucleotides that are being manufactured for use as research reagents, diagnostic agents or potential and definite therapeutic agents. The term includes oligomers comprising of a single strand nucleic acid or a double strand nucleic acid.
The term "siRNA" as used herein refers to double- stranded RNA molecules (dsRNA) capable of blocking gene expression in a highly conserved regulatory mechanism known as RNA interference (RNAi). The term "double-stranded RNA", "dsRNA molecule", or "dsRNA", as used herein, refers to a ribonucleic acid molecule, or complex of ribonucleic acid molecules, having a duplex structure comprising two anti-parallel and substantially complementary nucleic acid strands. The dsRNAs may comprise naturally occurring nucleotides or at least one modified nucleotide, such as a 2'-0-methyl modified nucleotide, a nucleotide comprising a 5'- phosphorothioate group, and a terminal nucleotide linked to a cholesteryl derivative or dodecanoic acid bisdecylamide group. 2' modified nucleotides may have the additional advantage that certain immunostimulatory factors or cytokines are suppressed when the inventive dsRNA molecules are employed in vivo, for example in a medical setting. Alternatively and non-limiting, the modified nucleotide may be chosen from the group of: a 2'- deoxy-2'-fluoro modified nucleotide, a 2'-deoxy-modified nucleotide, a locked nucleotide, an abasic nucleotide, 2'-amino-modified nucleotide, 2'-alkyl-modified nucleotide, morpholino
nucleotide, a phosphoramidate, and a non-natural base comprising nucleotide. The two strands forming the duplex structure may be different portions of one larger RNA molecule, or they may be separate RNA molecules. Where the two strands are part of one larger molecule, and therefore are connected by an uninterrupted chain of nucleotides between the 3 '-end of one strand and the 5' end of the respective other strand forming the duplex structure, the connecting RNA chain is referred to as a "hairpin loop". The RNA strands may have the same or a different number of nucleotides. In addition to the duplex structure, a dsRNA may comprise one or more nucleotide overhangs. The nucleotides in said "overhangs" may comprise between 0 and 5 nucleotides, whereby "0" means no additional nucleotide(s) that form(s) an "overhang" and whereas "5" means five additional nucleotides on the individual strands of the dsRNA duplex. These optional "overhangs" are located in the 3' end of the individual strands. As will be detailed below, also dsRNA molecules which comprise only an "overhang" in one the two strands may be useful and even advantageous in context of this invention. The "overhang" comprises preferably between 0 and 2 nucleotides. Most preferably 2 "dT" (deoxythymidine) nucleotides are found at the 3' end of both strands of the dsRNA. Also 2 "U"(uracil) nucleotides can be used as overhangs at the 3' end of both strands of the dsRNA. Accordingly, a "nucleotide overhang" refers to the unpaired nucleotide or nucleotides that protrude from the duplex structure of a dsRNA when a 3'-end of one strand of the dsRNA extends beyond the 5'-end of the other strand, or vice versa. For example the antisense strand comprises 23 nucleotides and the sense strand comprises 21 nucleotides, forming a 2 nucleotide overhang at the 3' end of the antisense strand. Preferably, the 2 nucleotide overhang is fully complementary to the mRNA of the target gene. "Blunt" or "blunt end" means that there are no unpaired nucleotides at that end of the dsRNA, i.e., no nucleotide overhang. A "blunt ended" dsRNA is a dsRNA that is double- stranded over its entire length, i.e., no nucleotide overhang at either end of the molecule. The term "antisense strand" refers to the strand of a dsRNA which includes a region that is substantially complementary to a target sequence. As used herein, the term "region of complementarity" refers to the region on the antisense strand that is substantially complementary to a sequence, for example a target sequence. Where the region of complementarity is not fully complementary to the target sequence, the mismatches are most tolerated outside nucleotides 2-7 of the 5' terminus of the antisense strand
The term "sense strand," as used herein, refers to the strand of a dsRNA that includes a region that is substantially complementary to a region of the antisense strand. "Substantially
complementary" means preferably at least 85% of the overlapping nucleotides in sense and antisense strand are complementary.
As used herein, the term "fatty acid" includes both saturated, i.e. an alkane chain as known in the art, having no double bonds between carbons of the chain and having the maximum number of hydrogen atoms, and unsaturated, i.e. an alkene or alkyne chain, having at least one double or alternatively triple bond between carbons of the chain, respectively, and further terminating the chain in a carboxylic acid as is commonly known in the art, wherein the hydrocarbon chain is not less then four carbon atoms.
The process of delivering a nucleic acid to a cell has been commonly termed transfection or the process of transfecting. The term transfecting as used herein refers to the introduction of a nucleic acid or other biologically active compound from outside a cell to inside cell such the nucleic acid has biologically activity. The nucleic acid may be used for research purposes or to produce a change in a cell that can be therapeutic. The delivery of a nucleic acid can lead to modification of the genetic material present in the target cell. A transfection reagent or delivery vehicle is a compound or compounds that bind(s) to or complex(es) with oligonucleotides and nucleic acids, and mediates their entry into cells.
In vitro transfection reagents, or delivery vehicles, are compounds or compositions of compounds that bind to or complex with nucleic acids and mediate their entry into cells. Examples of transfection reagents include, but are not limited to, protein and polymer complexes (polyplexes), lipids and liposomes (lipoplexes), combinations of polymers and lipids (lipopolyplexes), calcium phosphate precipitates, and dendrimers. Typically, the transfection reagent has a component with a net positive charge that binds to the oligonucleotide's or nucleic acid's negative charge. Cationic transfection agents may also condense large nucleic acids. Transfection agents may also be used to associate functional groups with a nucleic acid. Functional groups include cell targeting signals, nuclear localization signals, compounds that enhance release of contents from endosomes or other intracellular vesicles (such as membrane active compounds), and other compounds that alter the behavior or interactions of the compound or complex to which they are attached (interaction modifiers).
As used herein, a "pharmaceutical composition" comprises a pharmacologically effective amount of a nucleic acid complexed with the oligo(alkyleneamino) acid polymers of the
invention and a pharmaceutically acceptable carrier. However, such a "pharmaceutical composition" may also comprise individual strands of such a nucleic acid molecule. As used herein, "pharmacologically effective amount," "therapeutically effective amount" or simply "effective amount" refers to that amount of a nucleic acid effective to produce the intended pharmacological, therapeutic or preventive result.
The term "pharmaceutically acceptable carrier" refers to a carrier for administration of a therapeutic agent. Such carriers include, but are not limited to, saline, buffered saline, dextrose, water, glycerol, ethanol, and combinations thereof. The term specifically excludes cell culture medium. For drugs administered orally, pharmaceutically acceptable carriers include, but are not limited to pharmaceutically acceptable excipients such as inert diluents, disintegrating agents, binding agents, lubricating agents, sweetening agents, flavoring agents, coloring agents and preservatives as known to persons skilled in the art.
It is in particular envisaged that the pharmaceutically acceptable carrier allows for the systemic administration of the nucleic acids complexed with the oligo(alkyleneamino) acid polymers of this invention. Whereas also the enteric administration is envisaged the parenteral administration and also transdermal or transmucosal (e.g. insufflation, buccal, vaginal, anal) administration as well was inhalation of the drug are feasible ways of administering to a patient in need of medical intervention the compounds of this invention. When parenteral administration is employed, this can comprise the direct injection of the compounds of this invention into the diseased tissue or at least in close proximity. However, also intravenous, intraarterial, subcutaneous, intramuscular, intraperitoneal, intradermal, intrathecal and other administrations of the compounds of this invention are within the skill of the artisan, for example the attending physician.
For intramuscular, subcutaneous and intravenous use, the pharmaceutical compositions of the invention will generally be provided in sterile aqueous solutions or suspensions, buffered to an appropriate pH and isotonicity. In a preferred embodiment, the carrier consists exclusively of an aqueous buffer. In this context, "exclusively" means no auxiliary agents or encapsulating substances are present which might affect or mediate uptake of nucleic acids in the cells. Aqueous suspensions according to the invention may include suspending agents such as cellulose derivatives, sodium alginate, polyvinyl-pyrrolidone and gum tragacanth, and a wetting agent such as lecithin. Suitable preservatives for aqueous suspensions include ethyl and n-propyl p-hydroxybenzoate.
The term "oligo(alkyleneamino) acid polymer" as used herein includes all variations and derivatives of polymers formed by oligo (alkylenamino) acid units of the general structure

Z is chosen from either an alkyl group with the general structure L J , wherein c
NH2 is 0-8; or an amino group with the eneral structure , wherein n is 1-7; or an aromatic
group with the structure
 , or either CH2 or C=0; and a is 1-7 and b is 2-7. The oligo (alkyleneamino) acid units of the polymer are covalently linked via the terminal carboxygroup of a first oligo (alkyleneamino) acid unit and a terminal aminegroup of a second oligo(alkyleneamino) acid unit. Thus, the polymer comprises oligo (alkyleneamino) acid units covalently linked to each other via an amide bond, and is terminated by a free carboxyl group (- COOH) at the one end and terminated by a free amine group (-NH2) at the other end. Hence the polymer has a C (carboxy) terminus and an N (amino) terminus.
, or either CH2 or C=0; and a is 1-7 and b is 2-7. The oligo (alkyleneamino) acid units of the polymer are covalently linked via the terminal carboxygroup of a first oligo (alkyleneamino) acid unit and a terminal aminegroup of a second oligo(alkyleneamino) acid unit. Thus, the polymer comprises oligo (alkyleneamino) acid units covalently linked to each other via an amide bond, and is terminated by a free carboxyl group (- COOH) at the one end and terminated by a free amine group (-NH2) at the other end. Hence the polymer has a C (carboxy) terminus and an N (amino) terminus.
Said polymer comprises 2-60 oligo(alkyleneamino) acid units having the general structure 1, preferably 2-10 oligo(alkyleneamino) acid units having the general structure 1, most preferably 2-5 oligo(alkyleneamino) acid units having the general structure 1. The polymer can optionally have the same oligo(alkyleneamino)acid units recurring or may also have a combination of varying oligo(alkyleneamino)acid units recurring.
In other embodiments said polymer comprises identical oligo(alkyleneamino)acid units and at least one amino acid recurring, in yet other embodiments said polymer comprises a combination of varying oligo(alkyleneamino)acid units and at least one amino acid recurring.
A oligo(alkyleneamino) acid polymer can comprise more than one chain. Where the two oligo(alkyleneamino) acid polymer chains are connected covalently by means other than an uninterrupted chain of oligo(alkyleneamino) acid units, the connecting structure is referred to as a "linker". Mostly the linker is at least one amino acid. Through introduction of a linker into the oligo(alkyleneamino) acid polymer, branching of the oligo(alkyleneamino) acid polymer is possible. One example of a branched oligo(alkyleneamino) acid polymer is a t-shape, wherein two oligo(alkyleneamino) acid polymer chains are connected through an amino acid, to which a hydrophobic domain is covalently linked via another linker amino acid group. In addition to the t-shape, other branched oligo(alkyleneamino) acid polymers are envisaged with the oligo(alkyleneamino) acid polymer comprising 1-8 amino acids as branching points, preferably lysines and cysteines. To these branching points within the oligo(alkyleneamino) acid polymer, more oligo(alkyleneamino) acid polymer chains are attached either on solid-phase or in solution, resulting in a multiple branched oligo(alkyleneamino) acid polymer.
If not stated otherwise all domains are connected by peptide bonds, linking the carboxy term of the domain covalently to the oligo(alkyleneamino) acid polymer. The peptide bond is the preferred option but can be substituted by bond types stable to the conditions of peptide synthesis either on solid-phase or in solution e.g. disulfide bond, Huisgen cycloaddition product: 1,2,3- triazole.
The term "head group" as used herein refers to any functional group covalently linked to the C or N-terminus of the oligo(alkyleneamino) acid polymer, preferably at the N-terminus, which increases binding to the nucleic acid, formulation stability or interaction with lipid membranes. The head group can either be a polar positively charged head group, a coupling or crosslinking domain or a polar positively charged head group in the vicinity of a coupling or crosslinking domain. Therefore the term "polar positively charged head group" refers to any polar positively charged functional group covalently linked to the C- or N-terminus of the oligo(alkyleneamino) acid polymer. Non-limiting examples of polar positively charged functional groups are thiol groups, such as cysteine, and polar positively charged amino acids like lysine, arginine or histidine. The term "coupling or crosslinking domain" refers to any functional group covalently linked to the C- or N-terminus of the oligo(alkyleneamino) acid
polymer which enables crosslinking of the oligo(alkyleneamino) acid polymer (dimerisation or polymerisation). Said crosslinking is preferably bioreversible, therefore preferred functional groups of a crosslinking domain are disulfide-forming cysteine, oxime- or imine-forming aldehydes, or hydrazone-forming hydrazines. Said crosslinking can also be noncovalent, for example hydrogen bonding by ureido-pyrimidinones, or aromatic stabilization by tyrosine trimers.
The term "shielding moiety" as used herein refers to any moities that can be attached to the oligo(alkyleneamino) acid polymer resulting in a macromolecular structure to reduce unspecific interactions, uptake and agglomeration tendencies. For example a PEG-chain (preferably monodisperse) of a Mw > 1 kDa can be attached at any position of the polymer sequence using the gamma-amino function of lysine or the N-terminus and provide effiecient shielding of the oligo(alkyleneamino) acid polymer.
The term "hydrophobic domain" as used herein refers to highly hydrophobic functional groups covalently attached of the oligo(alkyleneamino) acid polymer, preferably to the N- terminus. Highly hydrophobic groups include but are not limited to amino acids or fatty acids. Preferably two fatty acids or a stretch of hydrophobic amino acids are linked to the amino functions of a N-terminal lysine residue or the epsilon amino function of a branching lysine.
Short description of the figures:
Figure 1 - Topological structures of the polymer (a) chain b) - d) i- shape e) t- shape f) branched polymer g) shielded and targeted polymer h) four arm polymer.
Figure 2 - Representative DNA gel-shift assay. Comparison of the gel retardation of DNA of the different PAAs. All polymers were tested at a w/w of 10. Polymers showing no complete retardation at that concentration are shown in increasing concentrations. Picture 1 - 4 dimerizing i-shape family (67, 68, 69, 70); Picture 5 - 7 crosslinking i-shape family (45, 46, 51) 7 w/w 5,10,20; Picture 8 - 11 t-shape family (74, 49, 78, 82); 12 - 14 crosslinking chains family (72, 76, 80) w/w 5, 10.
Figure 3 - Representative RNA gel-shift assay. Comparison of the gel retardation of siRNA of the different PAA families. Picture 1 - 4 dimerizing i-shape family (69, 70, 72, 71) N/P 12, 20; Picture 5 - 8 crosslinking i-shape family (50, 45, 46, 51) N/P 12, 20; Picture 9 - 12 t-shape family (49, 76, 80, 84) N/P 6, 12.
Figure 4 - Dynamic light scattering. Particle formation study of in vivo applied polymeric carrier systems. N/P ratio: molar ratio of protonable polymer nitrogens to DNA phosphates.
Figure 5 - Erythrocyte leakage assay. Analysis of the lytic activity of the polymers at a concentration of 5 μΜ by erythrocyte leakage assay at different pHs. Figure 6 - Cell viability assay and Luciferase reporter gene expression. Numbers in brackets depict ID numbers of polymers tested, (a) Reporter gene expression and metabolic activity of cells 24 h after transfection with pCMVLuc (Plank C, Zatloukal K, Cotten M, Mechtler K, Wagner E. Bioconjug Chem. 1992, 3:533-9.) using i-shape PAA carrier systems, (b) Reporter gene expression and metabolic activity of cells 24h after transfection with pCMVLuc using t- shape PAA carrier systems (c) Reporter gene expression and metabolic activity of cells 24 h after transfection with pCMVLuc using non hydrophobical modified t-shape PAA carrier systems.
Figure 7 - Cell viability assay and Luciferase reporter gene silencing, (a) i- shapes. Comparison of different FA modifications (Polymer ID 45: C-Stp3-C-K-MyrA2, Polymer ID 46: C-Stp3-C-K- 01eA2, Polymer ID 51: C-Stp3-C-K) - "Luc siRNA": SEQ ID No 1/2 , "Mut siRNA": SEQ ID No. 3/4 (b) t-shapes. Comparison one FA vs. 2FA (Polymer ID 58: C-Stp2- K(01eA)-Stp2-C, Polymer ID 49: C-Stp2-K-(K-01eA2)- Stp2-C) "Luc siRNA": SEQ ID No 1/2 , "Mut siRNA": SEQ ID No. 3/4 (c) Dimer-forming PAAs. (Polymer ID 72: C-Stp2-K- 01eA2, Polymer ID 71: C-K-Stp2-K-01eA2), "Luc siRNA": SEQ ID No 1/2 , "Mut siRNA": SEQ ID No. 3/4 (d) TShape, Comparison on two different cell lines (Polymer ID 49: C-Stp2-K- (K-01eA2)-Stp2-C), "Luc siRNA": SEQ ID No 5/6 , "Mut siRNA": SEQ ID No. 7/8 (e) siRNA Knockdown comparison of two cross-linking i-shape homologues (Stp-backbone Polymer ID 46: C-Stp3-C-K-01eA2 vs. Gtp-backbone Polymer ID 213: C-Gtp3-C-K-01eA2). "Luc siRNA": SEQ ID No 5/6 , "Mut siRNA": SEQ ID No. 7/8. Figure 8 - Luciferase reporter gene expression of targeted polymers/cell viability (a) Reporter gene expression 24 h after transfection with targeted and shielded carrier systems. HUH7 cells were transfected using 200 ng pCMVLuc (2 μg/mL DNA) plasmid. Polyplexes were prepared at different w/w ratios and compared to standard LPEI polyplexes. Luciferase reporter gene expression is presented as mean value + SD of quintuplicates. (b) Metabolic activity of transfected cells 24 h after transfection with targeted and shielded carrier systems. HUH7 cells were transfected using 200 ng pCMVLuc (2 μg/mL DNA) plasmid.
Polyplexes were prepared at different w/w ratios and compared to standard LPEI polyplexes. Metabolic activity was measured by MTT assay and is presented as mean value + SD of quintuplicates.
Figure 9 - In vivo luciferase reporter gene expression in different organs (He, heart; Lu, lung; Li, liver; Sp, spleen; Ki, kidney; Tu, tumor; Mu, muscle; Va, isolated blood vessel ) after systemic application. Systemic gene transfer in tumor bearing mice was carried out using polyplexes containing 50 μg pEGFPLuc DNA (Clontech Laboratories, Mountain View, CA) per 20 g body weight at a concentration of 200 μg/mL DNA in HBG and a polymer/DNA w/w ratio of 10. Results for polymers 46 (Figure 9 a) , 49 (Figure 9 b), 82 (Figure c) are shown. Figure 10 - Cell viability assay and Luciferase reporter gene expression in Neuro2A GFPLuc cells with Ptp-containing i- shapes, (a) Polymer ID 364: C-K-Ptp2- K- 01eA2, Polymer ID 365: C-Ptp3-C-K-01eA2 (b) Polymer ID 367: C-Gtt2-Ptp-C-K- 01eA2, Polymer ID 368: C-Stp-Gtt-Ptp-C-K-01eA2, Polymer ID 370: C-Gtp-Ptp2-C-K- 01eA2.N/P ratios are as indicated. "GFP" siRNA: SEQ ID No 5/6 , "control" siRNA: SEQ ID No. 7/8. Figure 11 - Cell viability assay and Luciferase reporter gene expression in Neuro2AGFPLuc cells with optimised siRNA carriers, (a) Polymer ID 230 (b) Polymers ID 199, ID 386, ID 277 (c) Polymers ID280, ID278, ID 352 (d) Polymer ID279. N/P ratios are as indicated. "GFP" siRNA: SEQ ID No 5/6 , "control" siRNA: SEQ ID No. 7/8.
Figure 12 - Luciferase gene expression and cell viability after transfection of (a,b) Neuro2A or (c,d) HUH7 cells. Transfections (a,c) and viability assays (b,d) were performed as described for "Luciferase reporter gene expression" and "Cell viability assay (MTT Assay)". Cells were transfected with polymers ID23: K-Stp5-K; ID286: A- K-(K-(Stp)2)2; ID287: A-K- (K-(Stp2)2)2; ID288: A-K-(K-(Stp3)2)2; ID289: A-K-(K- (Stp4)2)2.
Figure 13 - Luciferase gene expression after transfection of cells using transferrin receptor targeting. Neuro2A-cells were transfected with B6-PEG24-K-(Stp4-C)2, #203 (targeting transferrin receptor) or B6mod-PEG24-K-(Stp4-C)2, #204 (modified non targeting sequence) in different N/P ratios as indicated and treated with chloroquine lh after transfection.
Figure 14 - Luciferase gene expression after transfection of cells using ανβ3 integrin receptor targeting. Dul45-cells were transfected with cRGDfK-A-PEG24-K-(Stp4-C)2, #391
(targeting for α νβ3 integrin receptor) or A-PEG24-K-(Stp4-C)2, #188 (non targeted sequence) in different N/P ratios as indicated and treated with chloroquine lh after transfection.
Figure 15 - Luciferase gene expression after transfection of cells using folic acid receptor targeting. IGROV-cells were transfected with FolA-PEG24-K-(Stp4-C)2, #356 (targeting for folic acid receptor) or A-PEG24-K-(Stp4-C)2, #188 (non targeted sequence) in different N/P ratios as indicated and either not treated or treated with chloroquine lh after transfection.
Figure 16 a - Cell viability assay and luciferase reporter gene silencing in KB-eGFPLuc cells with FolA-siRNA /siRNA at indicated % ratios using Polymer ID 233 as carrier system. As control FolA-siRNA was transfected without polymer. "GFP" siRNA: SEQ ID No 5/6 , "siControl" siRNA: SEQ ID No. 7/8.
Figure 16 b - Transfection via folate receptor targeting using ligand bound PAA and endosomolytic siRNA. Transfection of KB-EGFPLuc cells with functional polymer ID 356 in combination with Inf7 -GFP- siRNA (a), functional polymer ID 356 in combination with GFP- siRNA (b), control polymer ID 188 without ligand in combination with Inf7-GFP-siRNA (c), control polymer ID 420 with serines instead of cysteines in combination with Inf7-GFP-siRNA (d) or functional polymer ID 356 in combination with Inf7-Control-siRNA (e) at indicated N/P ratios. Transfections were performed using 250ng siRNA.
Figure 17 - Erythrocyte leakage assay. Analysis of the lytic activity of free Inf7 and Inf7 covalently attached to siRNA at a concentration of 5 μΜ at different pHs. Figure 18 - Cell viability assay and Luciferase reporter gene silencing, (a) Reporter gene expression 48 h after transfection of endosomolytic siRNA with i-shape or t-shape carrier system. Neuro2A-eGFPLuc cells were transfected using 500 ng siGFP/ siGFP-Inf7/ siGFP- DMMAnMel (5 μg/mL siRNA). Polyplexes were prepared at different N/P ratios as indicated using i-shape (#46) and t-shape (#49) structures. Luciferase reporter gene expression is presented as relative mean value + SD of triplicates. Expression level of HBG treated cells was set to 100%. (b) Metabolic activity of transfected cells 48 h after transfection of endosomolytic siRNA with i-shape or t-shape carrier system. Neuro2A-eGFPLuc cells were transfected using 500 ng siGFP/ siGFP-Inf7/ siGFP-DMMAnMel (5 μg/mL siRNA). Polyplexes were prepared at different N/P ratios as indicated using i-shape (#46) and t-shape (#49) structures. Metabolic
activity was measured by MTT assay and is presented as rel. mean value + SD of triplicates. Metabolic activity of HBG treated cells was set to 100%.
Figure 19 - Cell viability assay and Luciferase reporter gene silencing. Comparison of knockdown efficiency of siRNA and endosomolytic siRNA. Neuro2A eGFPLuc cells were treated with siGFP and siCtrl mixted with polymer #76 or siGFP-Inf7 and siCtrl-Inf7 mixed with polymer #76 in different N/P ratios as indicated.
Figure 20 - Transfection via transferrin receptor targeting using ligand bound PAA and endosmolytic siRNA. (a) Transfection of DU145-eGFPLuc cells with functional polymer #203 containing functional targeting ligand. Comparison of transfection efficiency with endosomolytic or nonmodified siRNA (b) Transfection of DU145-eGFPLuc cells with functional polymer #204 containing nonfunctional targeting ligand. Comparison of transfection efficiency with endosomolytic or nonmodified siRNA (c) Transfection of DU145- eGFPLuc cells with functional polymer #188 without targeting ligand. Comparison of transfection efficiency with endosomolytic or nonmodified siRNA. Figure 21 - Microscopic pictures of stained tumor slices. Slices are prepared as described out of subcutaneous KB tumors from NMRI mice (A) or subcutaneous N2A tumors from A/J mice (B-D). Animals were treated with one intravenous injection of either polymer # 356 in combination with EG5-siRNA (A), polymer #233 in combination with EG5-siRNA-Inf7 (B), polymer #49 in combination with EG5-siRNA-Inf7 (C) or polymer #49 in combination with EG5-siRNA. Arrows mark mitotic figures ("Asters") due to silencing of EG5.
Figure 22 - Luciferase reporter gene silencing after transfection with a T-shape polymers containing a Y3-motif instead of fatty acids. Neuro2A-eGFPLuc cells were either transfected with C-Stp3-K(K-(Y3)2)-Stp3-C ID 304 or K(Stp3-Y3-C)2), ID 331. "GFP siRNA": SEQ ID No 5/6 , "Control siRNA": SEQ ID No. 7/8. Figure 23 - Luciferase reporter gene silencing after transfection with a T-shape polymer containing Y3-motifes instead of cysteines. Neuro2A-eGFPLuc cells were transfected with Y3- Stp2-K(K-01eA2)-Stp2-Y3, ID 332. "GFP siRNA": SEQ ID No 5/6, "Control siRNA": SEQ ID No. 7/8.
Figure 24 - Luciferase reporter gene silencing after transfection with a T-shape polymer containing ICH-CAMP motifes instead of cysteines. Neuro2A-eGFPLuc cells were
transfected with (01eA2-K)K(K(Stp2-ICH-CAMP)2)), ID 354. "GFP siRNA": SEQ ID No 5/6 , "Control siRNA": SEQ ID No. 7/8.
Figure 25 - Luciferase reporter gene silencing after transfection with a T-shape polymer containing ICH-CAMP and Y3 motifes. Neuro2A-eGFPLuc cells were transfected with ((Y3)2-K)K(K(Stp2-ICH-CAMP)2)), ID 355. "GFP siRNA": SEQ ID No 5/6 , "Control siRNA": SEQ ID No. 7/8.
Figure 26 - Luciferase gene expression and cell viability after transfection of Neuro2A.
Transfections and viability assays were performed as described for "Luciferase reporter gene expression" and "Cell viability assay (MTT Assay)". Cells were transfected with polymers ID402: A-K-(K-(Stp4-C)2)2.
Figure 27 - Luciferase gene expression and cell viability after transfection of Neuro2A.
Transfections and viability assays were performed as described for "Luciferase reporter gene expression" and "Cell viability assay (MTT Assay)". Cells were transfected with polymers ID421: A-K-(K-(Stp5-C)2)2. Figure 28 - Luciferase gene expression and cell viability after transfection of Neuro2A.
Transfections and viability assays were performed as described for "Luciferase reporter gene expression" and "Cell viability assay (MTT Assay)". Cells were transfected with polymers ID424: A-Stp3-K-(K-(Stp3-C)2)2; ID425: A-Stp4-K-(K-(Stp4-C)2)2.
Figure 29 - Luciferase reporter gene silencing. Neuro2A-eGFPLuc cells were transfected with polymer ID392: A- K-(K-(Stp3-C)2)2 at indicated ratios. Black bars: transfection using GFP- siRNA SEQ ID No 5/6, grey bars: transfection using Control-siRNA SEQ ID No. 7/8.
Figure 30 - Luciferase reporter gene silencing. Neuro2A-eGFPLuc cells were transfected with polymer ID414: A-Stp2-K-(K-(Stp2-C)2)2 or ID415: A-C-Stp2-K-(K-(Stp2-C)2)2.at indicated ratios. Black bars: transfection using GFP-siRNA SEQ ID No 5/6, grey bars: transfection using Control-siRNA SEQ ID No. 7/8.
Figure 31 - Luciferase gene expression after transfection of cells using HGF receptor targeting. PC3-cells were transfected with CMP-PEG24-K-(Stp4-C)2, ID443 (targeting HGF receptor c-Met) in different N/P ratios as indicated or A-PEG24-K-(Stp4-C)2, ID 188 (non targeted sequence) in a N/P ratio of 6 and treated with chloroquine lh after transfection.
Figure 32 - Luciferase gene expression after transfection of cells using transferrin receptor targeting and histidine modified polymer. DU145-cells were transfected with "B6-PEG24-K- (Stp4-C)2-HIS", ID 441 (targeting transferrin receptor, containing histidines in polymeric backbone), or "B6-PEG24-K-(Stp4-C)2", ID 203 (targeting transferrin receptor, no histidines in polymeric backbone) in a N/P ratio of 6. A subset of cells was treated with chloroquine lh after transfection as indicated.
Figure 33 - Luciferase gene expression after transfection of cells using HGF receptor targeting and histidine modified polymer. PC3-cells were transfected with "CMP-PEG24-K- (Stp4-C)2-HIS", ID 442 (targeting HGF receptor c-Met, containing histidines in polymeric backbone) or "CMP-PEG24-K-(Stp4-C)2", ID 443 (targeting HGF receptor c-Met, no histidines in polymeric backbone) in different N/P ratios as indicated. A subset of cells was treated with chloroquine lh after transfection as indicated.
Figure 34 - Luciferase gene expression after transfection of cells using transferrin receptor targeting and histidine modified polymer. DU145-cells were transfected with "CMP-PEG24- K-(Stp4-C)2-HIS", ID 442 (targeting HGF receptor c-Met, containing histidines in polymeric backbone) in different N/P ratios as indicated. A subset of cells was treated with chloroquine lh after transfection as indicated.
Figure 35 - Reporter gene silencing via targeted siRNA. KB-EGFPLuc cells were treated with FolA-PEG24-triazol-s-s-siPvNA (using FolA for folate receptor targeting) mixed in different ratios with unmodified siRNA at indicated amounts (% of modified siRNA). For transfection, polyplexes were formed with polymer ID386 at N/P ratio 20.
Figure 36 - Topological structures of four and five arm polymers
Examples:
Abbreviations A/J mice Strain A of the Jackson Laboratory
Bisboc bis-tert-butoxycarbonyl
Bistfa bis-trifluoroacetyl
Boc tert-butoxycarbonyl
Boc-Cys(trt)-OH Na-Boc-S-trityl-L-cysteine, N-(tert-Butoxycarbonyl)-S-trityl-L- cysteine
C, Cys cysteine
CMP c-Met binding peptide
cRGDfK See RGD (real structure: cyclicRGD, f: (D)-Phenylalanine, K:
Lysine)
CuBr Copper(I) bromide
DAPI 4' ,6-Diamidin-2' -phenylindoldihydrochlorid
DBU l,8-diazabicyclo[5.4.0]undec-7-ene
DCM Dichloromethane
dde-Lys(fmoc)OH Na-(4-4-Dimethyl-2,6-dioxocyclohex-l-ylidene)ethyl-N-e-(9- fuorenylmethyloxycarbonyl)-L-lysine
DIPEA Di-isopropylethyleneamine
DMF Dimethylformamide
DMMAnMel 2,3-Dimethylmaleic anhydride modified mellitin
EG5 Kinesin 11 gene, kinesin spindle protein (KSP) gene
EtOAc Ethyl acetate
FA fatty acid
Fmoc (9-Fluorenylmethoxycarbonyloxy)
Fmoc-Gtp-OH l-(9-Fluorenylmethoxycarbonylamino)-3,6,9-tris(tert- butoxycarbonyl)- 13-oxo-3,6,9, 12-tetraazaheptadecan-17-oic acid
Fmoc-Gtt-OH l-(9-Fluorenylmethoxycarbonylamino)-3,6,-bis(tert- butoxycarbonyl)- 10-oxo-3,6,9-triazatetradecan- 14-oic acid
Fmoc-His(Trt)-OH Fmoc-Nim-trityl-L-histidine
Fmoc-Lys(ivDDE)-OH Fmoc-Ne-l-(4,4-dimethyl-2,6-dioxocyclohex-l-ylidene)-3- methylbutyl-L-lysine
Fmoc-Osu N-(9-Fluorenylmethoxycarbonyloxy) succinimide
Fmoc-Stp(boc3)-OH l-(9-Fluorenylmethoxycarbonylamino)-3,6,9-tris(tert- butoxycarbonyl)- 13-oxo-3,6,9, 12-tetraazahexadecan-16-oic acid FolA folic acid
Gtp Glutaryl tetraethylenepentamine
Gtt Glutaryl triethylenetetramine
HATU 2-(lH-7-Azabenzotriazol-l-yl)-l,l,3,3-tetramethyl uronium HFIP
Hexafluoro-2-propanol
HOAt (l-Hydroxy-7-azabenzotriazole)
HOBt Hydroxybenzotriazole hexafluorophosphate
ICH-CAMP 2-(6-Isocyanahexylaminocarbonylamino)-6-methyl-
4(lH)pyrimidinone
INF7 Influenza peptide
K, Lys lysine
KB cells Human cervix carcinoma cell line
LinA Linoleic acid
MTBE Methyl tert-butyl ether
NMRI mice Rj:NMRI-nu (nu/nu) (Naval Medical Research Institute)
OAA oligo (alkylene amino) acids
OleA Oleic acid
PAA polyamide polymers
PEG24 poly(ethylene glycol) containing 24 ethylenoxide monomers
Ptp ortho-Phthaloyl-tetraethylenpentamine
Pybop (Benzotriazol- 1 -yloxy)tripyrrolidinophosphonium
hexafluorophosphate
Pybop/HOBt PyBOP®/l-Hydroxybenzotriazole
RBF Round-bottom flask
RGD avB3 integrin targeting molecule (R: arginine, G: Glycine, D:
Aspartic acid)
SEC size-exclusion chromatography
Stp Succinyl tetraethylenepentamine (l-Amino-13-oxo-3,6,9,12- tetraazahexadecan-16-oic acid)
TBTA Tris [( 1 -benzyl- 1 H- 1 ,2,3-triazol-4-yl)methyl] amine
TEPA Tetraethylenepentamine
TETA Triethylenetetramine
TFA Trifluoroacetic acid
TFA/TIS Trifluoroacetic acid/ Triisopropylsilane
THF Tetrahydrofuran
Trisboc tris-tert-butoxycarbonyl
Y Tyrosine
Y3 Oligotyrosine consisting of 3 monomers
Synthesis of bistfa-trisboc-Tetraethylenepentamine (bistfa-trisboc-TEPA) using tetraethylenepentamine pentahydrochloride
10 g (26.9 mmol) tetraethylenepentamine pentahydrochloride were weighed in a 1 1 round bottom flask and dissolved in 250 ml dichloromethane (DCM) /methanol (2: 1). 18.75 ml (134.5 mmol) triethylamine was added and after stirring for 2 - 12 h the mixture was cooled down to 0 °C. 6.72 ml (56.5 mmol) trifluoroacetic ethyl ester were diluted in dichloromethane (40 ml) and added dropwise over 2 h at 0 °C. The round bottom flask was allowed to warm to room temperature after 2 h and was stirred for another hour. Di-t-butyl dicarbonate (23.4 g, 107.6 mmol) was dissolved in 40 ml dichloromethane (DCM) and added dropwise over one hour. Afterwards 15 ml of triethylamine (107.5 mmol) were added and the mixture was stirred over night. The organic phase was reduced to approximately 150 ml and washed three times with saturated sodium bicarbonate, then three times with 5 % sodium citrate solution and finally three times with water. The organic phase was dried over sodium sulfate and the solvent was evaporated to a yellowish viscous, waxy solid. The residue was recrystallized: Therefore it was dissolved in the minimal amount of dichloromethane (37 ml) which was heated to reflux. Then slowly n-hexane (65 ml) was added to the boiling dichloromethane till clouding could be observed at the drop-in point. The crystallization solution was stored over night at 4 °C. The microcrystalline residue was filtered, washed with cooled n-hexane and dried, yielding 12.48 g (68.0 %) of bistfa-trisboc-tetraethylenepentamine.
1H-Nuclear Magnetic Resonance (NMR) spectrum (500 MHz, CDC13, 24.1 °C): δ = 1.39-1.48 (m, 27 H, OC(CH3)3), 3.20-3.55 (m, 16 H, CH2), 7.93 (d, J = 46.5, 0.15 H, NH), 8.21 (d, J = 41.3, 0.35 H, NH) ppm.
Mass spectrometry analysis (MS), Electrospray injection (ESI); m/z ( ) = 699.3527 [M+NH4]+ (100), 682.3268 [M+l]+ (22)
Synthesis of bistfa-trisboc-Tetraethylenepentamine using technical grade (85%) tetraethylenepentamine
12 g (53,8 mmol) of tetraethylenepentamine were weighed in a 1 L round bottom flask and dissolved in 500 ml dichloromethane. The mixture was cooled down to 0 °C. Trifluoroacetic ethyl ester (13.45 ml, 16.05 g, 56.5 mmol) was diluted in 220 ml dichloromethane, transferred into a dropping funnel and added dropwise to the cooled mixture in the round bottom flask over 2.5 h. After complete addition of the trifluoroacetic ethyl ester the reaction was stirred for an additional hour at RT. Di-t-butyl dicarbonate (47 g, 215.3 mmol) was dissolved in 80 ml dichloromethane and added dropwise over one hour. Afterwards 30 ml (215 mmol) of triethylamine were added and the mixture was stirred over night. The organic phase was reduced to approximately 200 ml and washed three times with saturated sodium bicarbonate, then three times with 5 % sodium citrate solution and finally three times with water. The organic phase was dried over sodium sulfate and the solvent was evaporated to a yellowish viscous, waxy solid. The oily residue was recrystallised: Therefore it was solved in the minimal amount of boiling dichloromethane (60 ml). Then slowly n-hexane (140 ml) was added to the boiling dichloromethane till clouding was observed at the drop-in site. The crystallisation solution was stored over night in a refrigerator at 4 °C. The microcrystalline residue was filtered, washed with cooled n-hexane and dried.
Yield = 17.2 g (47%)
1H NMR (500 MHz, CDC13, 24.1 °C): δ = 1.39-1.48 (m, 27 H, OC(CH3)3), 3.20-3.55 (m, 16 H, CH2), 7.93 (d, J = 46.5, 0.15 H, NH), 8.21 (d, J = 41.3, 0.35 H, NH) ppm. MS (ESI); m/z (%) = 699.3527 [M+NH4]+ (100), 682.3268 [M+l]+ (22)
Synthesis of trisboc-Tetraethylenepentamine
10 g bistfa-trisboc-Tetraethylenepentamine were suspended in 75 ml ethanol. 100 ml of 3 M aqueous sodium hydroxide were slowly added via a dropping funnel under stirring. After a reaction time of 6-20 hours the alcohol was evaporated and the aqueous phase was extracted with 3 x 100 ml dichloromethane. The pooled organic phases were dried over sodium sulfate. After evaporation of the solvent and HV-treatment trisboc-tetraethylenepentamine was isolated in a
99.8 % yield as viscous oil which solidified slowly after addition of crystallization seeds and storage at 4 °C.
1H NMR (400 MHz, CDC13, 50.0 °C): δ = 1.40-1.50 (m, 27 H, OC(CH3)3), 2.36-2.58 (bs, 4 H, NH2), 2.79-2.96 (bt, J = 5.1, 4 H, CH2), 3.21 - 3.41 (m, 12 H, CH2) ppm. MS (ESI); m/z (%) = 245.6837 [M+2H]+ (100), 490.3610 [M+H]+ (22)
Synthesis of bistfa-bisboc-triethylenetetramine (bistfa-bisboc-TETA)
Triethylenetetramine (TETA, 2.0 g, 13.7 mmol, 2.05 ml) was dissolved in 27 ml dichloromethane. A solution of trifluoroacetic ethyl ester (27) (4.09 g, 28.8 mmol, 3.43 ml) in 57 ml dichloromethane (99.5 %) was added dropwise at 0 °C and stirred for 1 h at 0°C. Triethylamine (2.91 g, 28.8 mmol, 4.00 ml,) was added and the reaction was brought to RT, followed by dropwise addition of a solution of di-t-butyl dicarbonate (8.9838 g, 41.2 mmol) in 21 ml dichloromethane. The reaction was stirred overnight and washed 3 x 5% NaHC03 solution, 3 x 5% citric acid solution and 3 x water, dried over sodium sulfate, filtered and concentrated. The resulting white solid was recrystallized from dichloromethane/hexanes, yielding bistfa-bisboc-triethylenetetramine as a white solid.
Bistfa-bisboc-triethylenetetramine: 2.6745 g (36%).
1H NMR (500 MHz, CDC13, 24.1 °C): δ = 1.40 (s, 9 H, OC(CH3)3), 1.46 (s, 9 H, OC(CH3)3), 3.24-3.63 (m, 12 H, CH2), 7.83 (sbr, 0.30 H, NH), 9.11 (sbr, 0.79 H, NH) ppm.
MS (ESI); m/z (%) = 556.2572 [M+NH4]+ (100), 539.2312 [M+l]+ (11) Synthesis of bisboc-Triethylenetetramine
0.4882 g (0.907 mmol) of bistfa-bisboc-triethylenetetramine were suspended in 5 ml ethanol. While stirring 5 ml of aqueous 3 M NaOH were added dropwise over 10 min. After 16 h the solution was concentrated to approx. 5 ml and extracted with dichloromethane. The combined organic phase was dried over sodium sulfate and filtrated. The organic phase was evaporated yielding a crystalline solid which was further dried under high vacuum. bisboc-Triethylenetetramine: 0.315 g (36%)
1H NMR (400 MHz, CDC13, 19.1 °C): δ = 1.24 (sbr, 4 H, NH2), 1.45 (s, 18 H, OC(CH3)3), 2.812 (t, J = 6.2 Hz, 4 H, CH2), 3.25 (t, J = 6.2 Hz, 4 H, CH2), 3.32 (t, J = 7.5 Hz, 4 H, CH2) ppm.
13C NMR (125 MHz, CDC13, 21.3 °C): δ = 28.8 (q, 6 C, C(CH3)3), 40.9 (t, 1 C, CH2), 41.3 (t, 1 C, CH2), 45.9 (t, 1 C, CH2), 46.2 (t, 1 C, CH2), 51.0 (t, 1 C, CH2), 51.6 (t, 1 C, CH2), 80.1 (q, 1 C, C(CH3)3), 80.2 (q, 1 C, C(CH3)3), 156.0 (s, 2 C, C=0) ppm.
Synthesis of Fmoc-Stp-OH
4.0 g of trisboc-tetraethylenepentamine (8.2 mmol) were dissolved in 16.5 ml of tetrahydrofuran and cooled to -75 °C. 0.514 g (0.5 mmol) of succinic anhydride were dissolved in 110 ml tetrahydrofuran and added dropwise over the course of 1 h. This was repeated once. The reaction was stirred for an additional hour at -75 °C and then for 1 h at RT. 4.19 ml hiinig's base (DIPEA, mmol) were added to the RBF and the reaction mixture cooled to 0 °C. 4.128 g Fmoc-Osu (mmol) were dissolved in a mixture of acetonitril/ tetrahydrofuran (25 ml/ 12 ml). This solution was added dropwise to the reaction mixture and stirred over night. The solution was concentrated to approximately 100 ml, mixed with 100 ml of dichloromethane and was washed 5 x with 0.1 M sodium citrate buffer (pH 5.2). The organic phase was dried, concentrated and the resulting foamy, off-white product further purified by dry column vacuum chromatography using a n-heptane/ethyl acetate gradient to elute fmoc-byproducts, followed by a ethyl acetate /methanol gradient. Fmoc-Stp-OH: 2,67 g (40%), bristle foamy, off-white solid.
1H-NMR (400 MHz, CDC13, 19.1 °C): δ =
MS (ESI); m/z (%) =812.4419 [M+H]+ (42), 829.4682 [M+NH4]+ (100), 834.4237 [M+Na]+
Synthesis of Fmoc-Gtt-OH
Trisboc-TETA (8.65 g, 25 mmol) was dissolved in acetonitrile: tetrahydrofuran (200:50 ml) at room temperature and cooled to -70 °C, then dropwise glutaric anhydride solution (3.15 g, 27.6 mmol in 50 ml acetonitrile) was added for 20 min to obtain a solid mass. The reaction mixture
was kept at same temp for 10 min then warmed up to room temperature and stirred for further 60 min. Hiinig's base (8.34 g, 75 mmol) was added, the reaction mixture was cooled to 0 °C and stirred for further 15 min. Fmoc-OSu (12.60 g, 37.5 mmol in 250 ml acetonitrile) was added dropwise (1 h) and stirred for 20 min at the same temperature followed by warm up to room temperature and stirring it for 12 h. The solvent was removed under reduced pressure then dichloromethane (700 ml) was added and the organic phase was washed with sodium citrate hydrochloride solution (0.1 M, pH 5.5) five times, dried with sodium sulfate and evaporated to obtain a solid mass which was purified by dry column vacuum chromatography (Ethyl Acetate:Methanol; 9.5:0.5 to 7:3 v/v)
Yield: 7.35g, 42%
1H NMR (400 MHz, CDC13): δ 7.75 (d, 2H, J=8Hz), 7.56 (d, 2H, J=8Hz), 7.38 (t, 2H, J=8Hz), 7.28 (t, 2H, J=8Hz), 4.40 (m, 2H), 4.20 (m, 1H), 3.32-3.41 (m, 12H), 2.37-2.40 (m, 2H), 2.26- 2.29 (m, 2H), 1.91-1.98 (m, 2H), 1.45 (s, 27H).
13C NMR (100 MHz, CDC13): δ 173 (C=0), 171 (C=0),143, 141, 128, 127, 125, 119 (Ar-C- Fmoc), 80 (CH-Fmoc), 60 (OCH2-Fmoc), 47 (CH2-TETA), 28 (CH3-Tert-but), 21, 20 14 (CH2- Glu)
High Resolution Mass Spectroscopy (HRMS), (EI): Calculated for C36H5oN409, 682.3577, found: 681.3494, [M-l].
Synthesis of Fmoc-Gtp-OH Trisboc-TEPA (9.8g, 20 mmol) was dissolved in tetrahydrofuran (250 ml) at room temperature and cooled to -70 °C, then glutaric anhydride solution (2.52 g, 24 mmol in 40 ml tetrahydrofuran) was added dropwise and the resulting suspension was stirred at -70 °C for 30 min, then brought to room temperature and stirred for further 60 min. Hiinig's base (7.8 g, 60 mmol) was added and the solution was cooled to 0 °C. Fmoc-OSu (10.08 g, 30 mmol in 250 ml acetonitrile) was added dropwise (1 h), stirred for 20 min and was brought to room temperature, followed by stirring it for 12 h. The solvent was removed under reduced pressure followed by addition of dichloromethane (500 ml). The organic phase was washed with sodium citrate hydrochloride
solution (0.1 M, pH 5.5) five times, dried with sodium sulfate and was evaporated to obtain a semi solid crude mass. The crude product was purified by dry column vacuum chromatography (ethyl acetate: methanol; 10:0 to 7:3 v/v) Yield: 7.42g, 45%
1H NMR (400 MHz, CDC13): δ 7.73 (d, 2H, J=8Hz), 7.55 (d, 2H, J=8Hz), 7.34 (t, 2H, J=8Hz), 7.26 (t, 2H, J=8Hz), 4.37 (m, 2H), 4.17 (m, 1H), 3.30-3.43 (m, 16H), 2.35-2.38 (m, 2H), 2.20- 2.24 (m, 2H), 1.91-1.95 (m, 2H), 1.43 (s, 27H).
13 C NMR (100 MHz, CDC13): δ 172 (C=0, Glu), 171 (C=0, Glu),144, 141, 128, 127, 125, 119 (Ar-C-Fmoc), 80 (CH-Fmoc), 60 (OCH2-Fmoc), 47, 45 (CH2-TEPA), 35 (CH2-Glu), 33 (CH2- TEPA), 28 (CH3-Ter-but), 21, 20 (CH2-Glu), 14 (CH2-Glu)
HRMS(EI): Calculated for C43H63N5011, 825.4524, found: 824.4431, [M-l].
Solid-phase protocols
General Polyamidoamine synthesis protocol


Synthesis of Peptide precursors for targeting domain
Peptides were assembled in a fully automatic fashion using by fmoc/tBu chemistry on an Applied Biosystems 431 A Peptide Synthesizer by employing the Applied Biosystems Small Scale FastMoc protocols.
General procedure: Manual synthesis of OAA-chains:
An amount of resin corresponding to 25-50 μιηοΐ of loaded amino acid was weighed into a syringe reactor and swelled for 30 min in an appropriate solvent. The general reaction cycle is depicted in the table above. Briefly, each cycle began with fmoc-removal by treatment with 20% piperidine in DMF followed by DMF washing steps. Coupling was done using a mixture of protected OAA unit/Pybop/HOBt/DIPEA (4/4/4/8 equivalents (eq)) for 30 min or until complete conversion was indicated by Kaisertest.
General procedure: Synthesis of i-shapes terminated by one fatty acid (FA): exemplified
 After swelling of fmoc-K(boc)-Wang resin (0.05-0.20 mmol) in DMF and cleavage of the fmoc protecting group, 4 equivalents of a solution of fmoc-Stp-OH in DMF, 8 eq Hiinigs base (DIPEA) and 4 eq PyBOP/HOBt were added to the resin and the vessel was agitated until Kaisertest indicated complete conversion (30 min). The reaction solvent was drained and the resin was washed five times with DMF. To cap residual, unreacted primary amino groups before introduction of the fatty acid the resin was acetylated using 5 equivalents of acetic anhydride and
10 equivalents of DIPEA in dichloromethane (DCM), before the subsequent removal of the fmoc protecting group. The resin was washed three times with DMF followed by three DCM washes after removal of the fmoc protecting group. Five equivalents of fatty acid were dissolved in DCM while 5 equivalents of PyBOP/HOBt and 10 equivalents of DIPEA dissolved in the smallest possible amount of DMF were added to the resin and the mixture was agitated until Kaisertest indicated complete conversion (normally 30 min). After completion of the reaction the resin was washed and dried for 12 h over KOH in vacuo. The PAA was cleaved from the resin by suspending it in a solution of TFA/H20 (95:5, v/v) for 3 h. The cleavage solution was collected by filtration and the resin washed twice with TFA and once with DCM. The collected solutions were concentrated under reduced pressure. The residue was dissolved in 5 % acetic acid and lyophilised.
After swelling of fmoc-K(boc)-Wang resin (0.05-0.20 mmol) in DMF and cleavage of the fmoc protecting group, 4 equivalents of a solution of fmoc-Stp-OH in DMF, 8 eq Hiinigs base (DIPEA) and 4 eq PyBOP/HOBt were added to the resin and the vessel was agitated until Kaisertest indicated complete conversion (30 min). The reaction solvent was drained and the resin was washed five times with DMF. To cap residual, unreacted primary amino groups before introduction of the fatty acid the resin was acetylated using 5 equivalents of acetic anhydride and
10 equivalents of DIPEA in dichloromethane (DCM), before the subsequent removal of the fmoc protecting group. The resin was washed three times with DMF followed by three DCM washes after removal of the fmoc protecting group. Five equivalents of fatty acid were dissolved in DCM while 5 equivalents of PyBOP/HOBt and 10 equivalents of DIPEA dissolved in the smallest possible amount of DMF were added to the resin and the mixture was agitated until Kaisertest indicated complete conversion (normally 30 min). After completion of the reaction the resin was washed and dried for 12 h over KOH in vacuo. The PAA was cleaved from the resin by suspending it in a solution of TFA/H20 (95:5, v/v) for 3 h. The cleavage solution was collected by filtration and the resin washed twice with TFA and once with DCM. The collected solutions were concentrated under reduced pressure. The residue was dissolved in 5 % acetic acid and lyophilised.
General Procedure: Synthesis of i-shapes terminated by 2 fatty acids: exemplified by K- Stp K-FAz
After swelling of fmoc-Lys(boc)-Wang resin (0.05-0.20 mmol) in DMF and cleavage of the fmoc protecting group, four equivalents of a solution of fmoc-Stp-OH in DMF, DIPEA (8 eq) and PyBOP/HOBt (4 eq) were added to the resin and the vessel was agitated until Kaisertest indicated complete conversion (30 min). The reaction solvent was drained and the resin was washed five times with DMF. To covalently attach two fatty acids to the N-terminus of the PAA, fmoc-Lys(fmoc)-OH was incorporated before the coupling of the fatty acid. To cap residual, unreacted primary amino groups before introduction of the fatty acid the resin was acetylated using 5 equivalents of acetic anhydride and 10 equivalents of DIPEA in DMF, before subsequent removal of the fmoc protecting group. The resin was washed three times with DMF followed by three DCM washes after removal of the fmoc protecting group. Ten equivalents of the fatty acid were dissolved in DCM while 10 equivalents of PyBOP/ HOBt and 20 equivalents of DIPEA in the smallest possible amount of DMF were added to the resin and the mixture was agitated until Kaisertest did indicate complete conversion (normally 30 min). After completion of the reaction the resin was washed and dried for 12 h over KOH in vacuo. The PAA was cleaved from the resin by suspending it in a solution of TFA/H20 (95:5, v/v) for 3 h. The cleavage solution was collected by filtration and the resin washed twice with TFA and once with DCM. The collected solutions were concentrated under reduced pressure. The residue was dissolved in 5 % acetic acid and lyophilised.
General Procedure: Synthesis of i-shapes with a single coupling domain: exemplified by

For PAAs containing a C-terminal cysteine fmoc-Cys(trt)-Wang resin was used. Other steps of the synthesis were performed as described in General Procedure: synthesis of i-shapes terminated by 2 fatty acids. For cleavage the resin was suspended in a solution of TFA/TIS/H20 (95:2.5:2.5, v/v) and agitated for 3 h. The cleavage solution was drained and collected. The resin was washed twice with TFA and twice with DCM. The collected solutions were concentrated under reduced pressure to approximately 1 ml. The concentrated solution was dropped slowly in a 1: 1 mixture (40 ml) of cooled (0 °C) MTBE and n-hexane. The resulting precipitate was centrifuged at 4 °C for 10 min (3000 rpm). The solvent was decanted and the pellet was washed twice with ice-cold MTBE. The resulting pellet was dissolved in 5 % acetic acid and lyophilised.
General Procedure: Synthesis of i-shapes with two coupling domains: exemplified by C- StprC-K-FAz
After swelling 0.035 mmol of a fmoc-Cys(trt)-Wang resin in DMF and cleavage of the fmoc protecting group, four equivalents of a solution of fmoc-Stp-fmoc in DMF, DIPEA (8 eq) and Pybop/HOBt (4 eq) were added to the resin and the vessel was agitated until Kaisertest indicated complete conversion (normally 30 min). The reaction solvent was drained and the resin was washed five times with DMF. This cycle was repeated twice. Afterwards the amino acid fmoc-Cys(trt)-OH was coupled. Then, in order to covalently attach two fatty acids to the linear PAA, fmoc-Lys(fmoc)-OH was incorporated N-terminal before the coupling of the fatty acid. To cap unreacted primary amino groups, the resin was acetylated using 5 equivalents of acetic anhydride and 10 equivalents of DIPEA in DMF, before the subsequent removal of the fmoc protecting group. To covalently attach the fatty acid, the solvent was changed to DCM after fmoc cleavage. The resin was washed three times with DMF and DCM after removal of the fmoc protecting group. 10 equivalents of the fatty acid were dissolved in DCM, 20 equivalents of DIPEA and 20 equivalents of Pybop/ HOBt in DMF were added to the resin and the mixture was agitated for 30 min. After completion of the reaction the resin was washed and dried over KOH in vacuo. For cleavage the resin was suspended in a solution of TFA/TIS/H20 (95:2.5:2.5, v:v:v) and agitated for 3 h. The cleavage solution was drained and collected. The resin was washed twice with TFA and once with DCM. The collected solutions were concentrated under reduced pressure to approximately 1 ml. The concentrated solution was dropped slowly in a 1: 1 mixture
for 10 min (2000-3000 rpm). The solvents were decanted and the pellet was washed twice with ice-cold MTBE. The resulting pellet was dissolved in 5 % acetic acid and lyophilised.
General Procedure: Synthesis of T-shapes with one FA: exemplified by C-Stpj_-K(FA)-
StplzC After swelling 0.05-0.20 mmol of fmoc-Cys(trt)-Wang resin in DMF for 30 min the fmoc-protection group was cleaved by double treatment with 20% piperidine in DMF. After washing the resin, four equivalents (related to resin loading) of fmoc-Stp-OH, DIPEA (8 eq) and Pybop/ HOBt (4 eq) were added for 30 min. The reaction solvent was drained and the resin was washed five times with DMF. Reaction progress was monitored by Kaisertest. To introduce a branching point dde-Lys(fmoc)-OH was used in the next coupling step. Dde-Lys(fmoc)-OH (4 eq) solved in DMF, DIPEA (8 eq) and Pybop/ HOBt (4 eq) solved in DMF were added and the synthesis vessel was agitated for 30 min. After a negative Kaiser test, the resin was washed with DMF. To cap unreacted primary amino groups, the resin was acetylated using 5 equivalents of acetic anhydride and 10 equivalents of DIPEA in DMF, before the subsequent removal of the fmoc protecting group. To couple the fatty acid, the solvent was changed to DCM after fmoc- cleavage. Therefore the resin was washed three times with DMF and DCM after removal of the fmoc-protecting group. 5 equivalents of the fatty acid solved in DCM, 10 equivalents of DIPEA and 5 equivalents of Pybop/ HOBt were added to the resin for 30 min. After completion of the reaction the resin was washed five times with DCM and three times with DMF. The dde- protecting group was cleaved with 2 % hydrazine monohydrate in DMF (v/v) (5-10 times for 5 min) till no significant A300 was measurable in the deprotection mixture. In-between the deprotection-steps the resin was washed twice with DMF. Fmoc-Stp-OH dissolved in DMF, DIPEA (8 eq) and Pybop/ HOBt (4 eq) were added for 30 min. After a successful reaction the resin was treated twice with 20 % piperidine in DMF. After washing the resin, boc-Cys(trt)-OH (4 eq) solved in DMF, DIPEA (8 eq) and Pybop/ HOBt (4 eq) were added and the vessel agitated for 30 min. Afterwards the resin was washed and dried over KOH in vacuo. For cleavage the resin was suspended in a solution of TFA/TIS/H20 (95:2.5:2.5, v/v/v) and agitated for 3 h. The cleavage solution was drained and collected. The resin was washed twice with TFA and once with DCM. The collected solutions were concentrated under reduced pressure to approximately 1 ml. The concentrated solution was dropped slowly in a 1: 1 mixture (40 ml) of cooled (0 °C) MTBE and n-hexane. The resulting precipitate was centrifuged at 4 °C for 10 min (2000-
3000 rpm). The solvents were decanted and the pellet was washed twice with ice-cold MTBE. The resulting pellet was dissolved in 5 % acetic acid and lyophilised.
General Procedure: Synthesis of T-shapes with two FAs: exemplified by C-Stpj_-K(K- FAZ -Stp,-C After swelling 0.05-0.20 mmol of fmoc-Cys(trt)-Wang resin in DMF for 30 min the fmoc-protection group was cleaved by double treatment with 20 % piperidine in DMF. After washing the resin, four equivalents (related to resin loading) of fmoc-Stp-OH, DIPEA (8 eq) and Pybop/ HOBt (4 eq) were added for 30 min. The reaction solvent was drained and the resin was washed five times with DMF. Reaction progress was monitored by Kaiser test. To introduce a branching point dde-Lys(fmoc)-OH was used in the next coupling step. Dde-Lys(fmoc)-OH (4 eq) solved in DMF, DIPEA (8 eq) and Pybop/ HOBt (4 eq) solved in DMF were added and the synthesis vessel was agitated for 30 min. After a negative Kaiser test, the resin was washed with DMF. After treatment with 20% piperidine in DMF and washing the resin with DMF, fmoc- Lys(fmoc)-OH (4 eq), DIPEA (8 eq) and Pybop/ HOBt (4 eq) was added. In order to cap unreacted primary amino groups, the resin was acetylated using 5 equivalents of acetic anhydride and 10 equivalents of DIPEA, before the subsequent removal of the fmoc protecting group. To couple the fatty acid, the solvent was changed to DCM after fmoc-cleavage. Therefore the resin was washed three times with DMF and DCM after removal of the fmoc-protecting group. 10 equivalents of the fatty acid solved in DCM, 20 equivalents of DIPEA and 10 equivalents of Pybop/ HOBt were added to the resin for 30 min. After completion of the reaction the resin was washed five times with DCM and three times with DMF. The dde-protecting group was cleaved with 2 % hydrazine monohydrate in DMF (v/v) (5-10 times for 5 min) till no significant A30o was measurable in the deprotection mixture. In-between the deprotection-steps the resin was washed twice with DMF. fmoc-Stp-OH solved in DMF, DIPEA (8 eq) and Pybop/ HOBt (4 eq) were added for 30 min. After successful reaction the resin was treated twice with 20 % piperidine in DMF. After washing the resin, boc-Cys(trt)-OH (4 eq) solved in DMF, DIPEA (8 eq) and Pybop/ HOBt (4 eq) were added for 30 min. Afterwards the resin was washed and dried over KOH in vacuo. For cleavage the resin was suspended in a solution of TFA/TIS/H20 (95/2.5/2.5, v/v/v) and agitated for 3 h. The cleavage solution was drained and collected. The resin was washed twice with TFA and once with DCM. The collected solutions were concentrated under reduced pressure to approximately 1 ml. The concentrated solution was dropped slowly in a 1: 1 mixture (40 ml) of cooled (0 °C) MTBE and n-hexane. The resulting precipitate was centrifuged
at 4 °C for 10 min (2000-3000 rpm). The solvents were decanted and the pellet was washed twice with ice-cold MTBE. The resulting pellet was dissolved in 5 % acetic acid and lyophilised.
General Procedure: Synthesis of T-Shapes containing shielding and targeting domains: exemplified by HO-IVNOPTYGYWHY -PEG^-K-iStp^-C)?: After swelling 20 μηιοΐ of resin-bound HO-IVNQPTYGYWHY-NH2 (GE 11 ) in DMF for
30 min four equivalents of a solution of fmoc-PEG24-OH in DCM/DMF(1: 1, v/v), DIPEA (8 eq) and PyBOP/HOBt (4 eq) were added to the resin and the vessel was agitated until Kaisertest indicated complete conversion (60 min). The reaction solvent was drained and the resin was washed three times with DCM followed by three DMF washs. After removal of the fmoc protective group by treatment with 20% piperidine in DMF fmoc-Lys(fmoc)-OH was introduced as a branching point using standard coupling conditions (AA/PyBOP/HOBt/DIPEA, 4/4/4/8 eq in DMF). After succesfull deprotection 8 equivalents of fmoc-Stp-OH, 8 equivalents of PyBOP/ HOBt and 16 equivalents of DIPEA in the smallest possible amount of DMF were added to the resin and the mixture was agitated until Kaisertest did indicate complete conversion (normally 30 min). The fmoc group was removed and the whole cycle repeated 4 times exchanging Stp by fmoc-C(Trt)-OH in the last step. After completion of the reaction the resin was washed and dried for 12 h over KOH in vacuo. The PAA was cleaved from the resin by suspending it in a solution of TFA/H20/TIS/EDT (92.5:2.5:2.5:2.5, v/v) for 1 h. The cleavage solution was collected by filtration and the resin washed twice with TFA and once with DCM. The concentrated solution was dropped slowly in a 1: 1 mixture (40 ml) of cooled (0 °C) MTBE and n-hexane. The resulting precipitate was centrifuged at 4 °C for 10 min (2000-3000 rpm). The solvents were decanted and the pellet was washed twice with ice-cold MTBE. The resulting pellet was dissolved in 5 % acetic acid and lyophilised.
General cleavage procedure: The resin was transferred into a syringe reactor of appropriate size and treated with 10 ml/g(resin) of a TFA/Water/TIS (95:2.5:2.5) mixture for 3 h. The resin was filtered off and washed twice using pure TFA followed by two DCM washes. The combined filtrates were concentrated in a rotovap and either precipitated by dropwise addition into ice-cold MTBE (50 ml MTBE/1 ml TFA) or other suitable mixtures. If precipitation wasn't possible the TFA was further concentrated to a glassy film and washed 3x with ice-cold MTBE. The precipitate/film was dissolved in 5% acetic acid, snap-frozen and lyophilized to obtain the crude peptide. Above
described are general methods which can be applied for the synthesis of many different variations of oligo(alkyleneamino) acid polymers comprising oligo(alkyleneamino) acid subunits of the general structure 1. Non-limiting examples of oligo(alkyleneamino) acid polymer derivatives are listed in the table below: Table 1: Examples of Oligo(alkyleneamino) acid polymer derivatives. Abbreviations:
K= Lysine, C= Cysteine, Stp = Succinyl tetraethylenepentamine , 01eA= Oleic acid, MyrA=Myristic Acid, CapA=Caprylic acid, ButA= Butyric acid, AraA= Arachidic acid, SteA= Stearic acid, prot a = protonable amines per molecule. Sequences from C- to N-terminus, as assembled on the solid support.




Calculation of the polymer/nucleic acid ratio for polyplex formation
Defined charge ratios of cationic polymer/anionic nucleic acid were tested. The charge ratio can be defined either as N/P ratio or as w/w ratio. The N/P ratio is the molar ratio of protonable polymer nitrogens to DNA phosphates. The w/w ratio is the polymer/DNA weight/weight ratio. The polymer/DNA weight/weight (w/w) ratios can be calculated from the corresponding N/P ratio by the formula: w/w ratio = (N/P ratio) x molecular weight of the polymer / (number of protonable polymer groups x molecular weight of nucleotide monomer). The average molecular weight of nucleotide monomers is 330 for DNA and 348 for RNA.
DNA polyplex formation
Polyplex formulations for DNA delivery were prepared as follows: 200 ng of DNA and the calculated amount of PAA were diluted in separate tubes in Hepes Buffered Glucose pH 7.4 (HBG). The polycation solution was added to the nucleic acid, rapidly mixed by pipetting up and down and incubated for 30-40 min at RT in order to form the polyplexes necessary for transfection and gel- shift experiments.
siRNA polyplex formation
Non-covalent polyplex formulations for siRNA delivery were prepared as follows: 500 ng of siRNA and the calculated amount of PAA were diluted in separate tubes in HBG. Then the polycation solution was added to the nucleic acid, mixed by pipetting and incubated for 30-40 min at RT in order to form the polyplexes necessary for transfection and gel-shift experiments. DNA gel- shift assay
A 1% agarose gel was prepared by dissolving 1.2 g agarose (Sigma- Aldrich, Taufkirchen, Germany) in 120 ml TBE buffer and heating the mixture to 100 °C. After cooling down to approximately 50 °C, 120 μΐ Gel-Red were added and the gel was poured in the casting unit. Polyplex- samples containing 100 ng DNA, PAA, HBG-buffer and loading buffer were placed into the pockets after an incubation time of 30 min at RT. Electrophoresis was performed at 120 V for 80 min. Results of a representative DNA gel shift are shown in figure 2. siRNA gel-shift assay
A 2.5% agarose gel was prepared by dissolving 3.0 g agarose (Sigma-Aldrich, Taufkirchen, Germany) in 120 ml TBE buffer and heating the mixture to 100 °C. After cooling down to approximately 50 °C, 120 μΐ^ Gel-Red () were added and the gel was poured in the casting unit. Polyplex- samples containing 500 ng siRNA, PAA, HBG-buffer and loading buffer were placed into the pockets after an incubation time of 30 min at RT. Electrophoresis was performed at 120 V for 40 min. Results of a representative RNA gel shift are shown in figure 3.
Erythrocyte leakage assay Freshly collected, citrate buffered murine blood was washed by four centrifugation cycles, each in phosphate-buffered saline (PBS) at 2000 rpm (600-800 g) at 4 °C for 10 min. The erythrocytes in the pellet were counted. The pellet was then diluted with different PBS buffers (pH 7.4, 6.5 and 5.5) to 5 x 107 erythrocytes/ml. The suspension was always freshly prepared and used within 24 h. 75 μΐ of the PAA solutions prepared at different concentration and different pH-values were mixed with 75 μΐ erythrocyte suspension in a 96-well plate (NUNC, V- bottom, Denmark). After incubating the plates under constant shaking at 37 °C for 60 min, intact blood cells and cell debris was removed by centrifugation (4 °C, 600-800 g (2000 rpm), 10 min). 80 μΐ of the supernatant was transferred to a new 96-well plate (TPP 96F, Trasadingen, Switzerland). Haemoglobin absorption was determined at 405 nm using a microplate reader (Spectrafluor Plus, Tecan Autstria GmbH, Grodig, Austria). PBS-buffers with pH- values of 7.4,
6.5 and 5.5 were used as negative control, 1 % TritonX-100 in PBS as positive control. Haemolysis was defined as percent ((ODPAA - ODbuffer)/(ODTriton-X100 - ODbuffer))*100. Results are shown in figure 5.
Cell viability assay (MTT Assay) The metabolic activity of the cells was determined using a methylthiazole tetrazolium
(MTT) assay as follows: 10 μΐ^ per 100 μΐ^ of medium of a 5 mg/ml solution of MTT in sterile PBS-buffer was added to each well of the 96-well plate. After incubation for 1-2 h at 37 °C the medium was removed and the cells were frozen at -80 °C for at least 1 h. 200 μΐ DMSO were added and the samples were incubated under constant shaking at 37 °C for 30 min to dissolve the crystals completely. The optical absorbance was measured at 590 nm with a reference wavelength of 630 nm using a microplate reader (Spectrafluor Plus,Tecan Autstria GmbH, Grodig, Austria). The cell viability was defined as percent: (ODPAA treated cells/ODBuffer treated cells)* 100. Results are shown in figures 6 - 8 and 10.
Luciferase reporter gene silencing
All experiments were performed in stably transfected Neuro2A-eGFPLuc cells. Cells were seeded in 96-well plates (TPP, Trasadingen, Switzerland) using 5000 cells per well 24 h prior to transfection. Transfection complexes containing siRNA were then added to cells in 100 μΐ culture medium containing 10% serum, 100 U/ml penicillin and 100 μg/ml streptomycin (final siRNA-concentration 367 nmol/1). 48 h after initial transfection medium was removed and cells were lysed in 50 μΐ 0.5X Promega cell lysis solution to measure the gene expression as described below. Transfections were performed in parallel using a specific target siRNA and a non-specific control siRNA to distinguish between specific gene silencing and unspecific knockdown of protein expression due to carrier toxicity. Qualitative information on the toxicity of the conjugates was obtained by diminution in luciferase expression upon delivery of the non-specific control siRNA compared to the luciferase expression from the same number of untreated control cells. Results are shown in figures 6-8. The used vector pEGFPLuc DNA was obtained from Clontech Laboratories, Mountain View, CA
Sequences of siRNAs employed in this study:
Luciferase-siRNA GL3 luciferase duplex:
5'-CUUACGCUGAGUACUUCGAdTdT-3' (sense) [Seq ID. No 1]
5'-UCGAAGUACUCAGCGUAAGdTdT-3' (antisense) [Seq ID. No 2]
control-siRNA (non-targeting control duplex):
5'-AUGUAUUGGCCUGUAUUAGUU-3' (sense) [Seq ID. No 3]
5'-CUAAUACAGGCCAAUACAUUU-3' (antisense) [Seq ID. No 4]
EGFPLuc-siRNA duplex:
5 '-AuAucAuGGccGAcAAGcAdTsdT-3 ' (sense) [Seq ID. No 5]
5'-UGCUUGUCGGCcAUGAuAUdTsdT-3' (antisense) [Seq ID. No 6]
Control siRNA duplex:
5'-AuGuAuuGGccuGuAuuAGdTsdT-3' (sense) [Seq ID. No 7]
5 '-CuAAuAcAGGCcAAuAcAUdTsdT-3 '(antisense) [Seq ID. No 8]
Luciferase reporter gene expression
Cells were plated in 96 well plates at a density of 10.000 cells per well 24 h prior to transfection. The polyplexes formed using 200 ng of pDNA/well were added to the cells in 100 μΐ culture medium containing 10% serum, 100 U/ml penicillin and 100 μg/ml streptomycin. LPEI polyplexes (Zou SM, Erbacher P, Remy JS, Behr JP. J Gene Med. 2000 Mar-Apr;2(2): 128- 34) formed in HBG using 200 ng of pDNA and 160 ng of linear polyethylenimine, (LPEI, molecular weight 22 kDa) /well were used for comparison. 24 h after initial transfection medium was removed and cells were lysed in 50 μΐ 0.5X Promega cell lysis solution to measure the gene expression. Luciferase activity was measured using a Lumat LB9507 instrument (Berthold, Bad Wildbad, Germany). Luciferase light units were recorded from an 20 μΐ aliquot of the cell lysate with 10 s integration time after automatic injection of freshly prepared luciferin using the luciferase assay system (Promega, Mannheim, Germany). Transfection efficiency was evaluated as relative light units (RLU) per number of seeded cells. Two ng of recombinant luciferase (Promega, Mannheim, Germany) corresponded to 10 light units. Results are shown in figures 6- 8 and 10.
Dynamic light scattering
Particle size of siRNA and DNA formulations was measured by laser-light scattering using a Zetasizer Nano ZS (Malvern Instruments, Worcestershire, U.K.). Therefore polyplexes were formed for 30-40 min at RT, containing 10 μg nucleic acid and the PAA at a molar ratio of PAA nitrogen to DNA phosphate of 6, 12 or 20 (N/P = 6, 12, 20). The polyplexes were diluted to 500 μΐ with HBG or water before measurement. Results are shown in figure 4.
In vivo biocompatibility of polymers in mice
Polymers were administered by tail vein injection in 200 μΐ^ HBG (20 mM Hepes buffered 5% glucose, pH 7.1) into nude mice. In every case two doses were tested that correspond to amounts present in 50 μg siRNA polyplexes per 20 g mouse at a protonable nitrogen: phosphate charge ratio (N/P) of either 6 or 12. No visible toxic effects were observed, immediately or after 3 hours. All animals survived the observation period of one day.
Tested polymers:
Polymer ID Sequence
45 C-Stp3-C-K-MyrA2
46 C-Stp3-C-K-01eA2
51 C-Stp3-C-K
26 K-Stp5-K-MyrA2
27 K-Stp5-K-01eA2
23 K-Stp5-K
49 C-Stp2-K-(K-01eA2)-Stp2-C
69 C-Stp2-K-MyrA2
70 C-K-Stp2-K-MyrA2
72 C-Stp2-K-01eA2
71 C-K-Stp2-K-01eA2
In vivo luciferase reporter gene expression after systemic application
Female 8-week-old A/J mice were inoculated subcutaneously in the flank with 1 million Neuro2a cells. Experiments started when tumors reached a weight of 100-750 mg. Systemic gene transfer in tumor bearing mice ( 4-5 mice per group) was carried out using polyplexes containing 50 μg pEGFPLuc DNA (Clontech Laboratories, Mountain View, CA) per 20 g body weight at a concentration of 200 μg/ml DNA in HBG and a polymer/DNA w/w ratio of 10. Polyplexes were applied into the tail vein and animals were sacrificed 24 h after application. Tissues as indicated were dissected and homogenized in cell culture lysis reagent (Promega, Mannheim, Germany) using an IKA-Ultra-Turrax homogeniser and subsequently centrifuged at 4000 rpm at 4 °C for 20 minutes to separate insoluble cell components. Luciferase activity was determined in the supernatant using a luminometer as described above. The results (luciferase gene expression per organs: He, heart; Lu, lung; Li, liver; Sp, spleen; Ki, kidney; Tu, tumor; Mu, muscle; Va, isolated blood vessel) are shown in figure 9. Results for polymers 46 , 49, 82 are shown.
Synthesis of Fmoc-Ptp-OH

2.45g (5mmol, 1 eq) of trisboc-tetraethylenepentamine was dissolved in THF (25 ml) at room temperature and cooled to -70 °C, followed by dropwise addition of a phthalic anhydride solution (0.814g, 6mmol, 1.1 eq) in acetonitrile:THF (2: 10 ml) over 30 minutes. The reaction mixture was stirred at that temperature for 60 minutes, then brought to room temperature and stirred for additional60 minutes. DIPEA (1.95g, 15mmol, 3 eq) was added to the reaction mixture which was cooled to 0 °C. Fmoc-OSu (2.52g, 7.5mmol, 1.5eq) in acetonitrile:THF (25: 10 ml) was added dropwise over one hour. The reaction mixture was stirred for additional 20 min at 0 °C, was brought to room temperature and stirred over night. The solvent was evaporated under reduced pressure and the resulting residue dissolved in 70 ml dichloromethane (DCM). The organic phase was washed 5 x with 0.1 M sodium citrate buffer (pH 5.2). The organic phase was dried over NaHC03, concentrated and purified by dry column vacuum chromatography (DCVC) using an n-Heptane/EtOAc gradient to elute fmoc-byproducts, followed by a EtOAc/MeOH gradient to isolate the product.
Yield: l.OOg, 24%
1H NMR (400 MHz, CDC13): δ 7.92 (bs, 1H, NH), 7.67 (d, 2H, J=8Hz, ArH-Fmoc), 7.51 (d, 2H, J=8Hz, ArH-Fmoc), 7.37 (s, 4H, ArH-Ph), 7.29 (t, 2H, J=8Hz, ArH-Fmoc), 7.20 (t, 2H, J=8Hz, ArH-Fmoc), 4.29 (m, 2H, CH2-Fmoc), 4.11 (m, 1H, CH-Fmoc), 3.25-3.50 (m, 16H, 8xCH2- Tepa), 1.27-1.36 (m, 27H, CH3-Ter-but).
13C NMR (100 MHz, CDC13): δ 171, 156, 155, 144, 141,128, 127, 125, 120, 80, 67, 60, 47, 46, 28, 20, 14
HRMS (ESI): Calculated for C46H61N5011, 859.4367, found: 858.4266, [M- H].
Synthesis of Ptp-containing polymers
Ptp-containing polymers were synthesized as described by the "General Procedure: Synthesis of i-shapes with two coupling domains."
Table 2: Examples of ortho-Phthaloyl-tetraethylenpentamine polymer derivatives. Sequences (from C- to N-terminus, as assembled on the solid support). Abbreviations: K= Lysine, C= Cysteine, Stp = Succinyl tetraethylenepentamine, Ptp = ortho-Phthaloyl- tetraethylenpentamine, Gtp = Glutaryl tetraethylenepentamine, Gtt = Glutaryl triethylenetetramine, 01eA= Oleic acid.

Neuro2AGFPLuc cells with Ptp-containing i- shapes (protocols for both assays see above).
Optimised siRNA carriers (i-shaped, branched, double T-shape)
Table 3: Examples of Optimised siRNA carriers. Sequences (from C- to N-terminus, as assembled on the solid support). Abbreviations: K= Lysine, C= Cysteine, Stp = Succinyl tetraethylenepentamine, Ptp = ortho-Phthaloyl-tetraethylenpentamine, Gtp = Glutaryl tetraethylenepentamine, Gtt = Glutaryl triethylenetetramine, OleA= Oleic acid, LinA= Linoleic Acid.
ID Mol Weight Formula Sequence Type
230 1676,40 C84H158N1g012S2 C-Gtp-Gtt-Stp-C-K-LinA2 I- shape
199 2211,94 CgsHigiNsvOigSs K-C-Stp2-K-(Stp2-C)2 branched
386 2877,95 Ci23H266N5o02lS3 C-Stp3-K(Stp3-C)2 branched
277 3679,05 C159H344N65026S3 C-Stp4-K-(Stp4-C)2 branched
C-K-(K-(LinA)2)-Stp2-K-(K-
280 2329,43 C126H23oN2o015S2 T- shape
(LinA)2)-C
C-K-(K-(LinA)2)-Stp3-K-(K-
278 2600,79 C138H255N25017S2 T- shape
(LinA)2)-C
C-K-(K-(LinA)2)-Stp4-K-(K-
352 2872,15 C15oH28oN3o019S2 T- shape
(LinA)2)-C
A-K-(K-(LinA)2)-Stp3-K-(K-
279 2536,66 C138H255N25017 T- shape
(LinA)2)-A
Synthesis of i-shape polymer #230
i-shape polymer #230 with the sequence C-Gtp-Gtt-Stp-C-K-LinA2 was synthesized as described by the "General Procedure: Synthesis of i-shapes with two coupling domains." General Procedure: Synthesis of three-branch polymers exemplified by C-(Stp3)-K-(Stp3-
After swelling 0.20 mmol of 2 Chloro Trityl Chloride resin in DCM for 30 min and DMF for 10 min, 0,7 equivalents of fmoc-Cys-(trt) and DIPEA (10 eq) were added for 60 min. 0,8 mL of methanol pro gram of resin were then added and after 10 min the resin was washed five times with DMF and five times with DCM. Reaction progress was monitored by Kaiser test. Fmoc- protection group was cleaved by double treatment with 20% piperidine in DMF. After washing the resin, four equivalents of fmoc-Stp-OH, DIPEA (8 eq) and Pybop/ HOBt (4 eq) were added for 45 min. The reaction solvent was drained and the resin was washed five times with DMF and five times with DCM. Reaction progress was monitored by Kaisertest. The resin was treated twice with 20 % piperidine in DMF in order to deprotect the fmoc group. The whole process was repeated two times. Fmoc-Lys(fmoc)-OH was used in the next coupling step. Fmoc-Lys(fmoc)-
OH (4 eq) DIPEA (8 eq) and Pybop/ HOBt (4 eq) were added for 45 min. The reaction solvent was drained and the resin was washed five times with DMF and five times with DCM. Reaction progress was monitored by Kaiser test. The resin was treated twice with 20 % piperidine in DMF in order to deprotect the fmoc group. After deprotection of the lysine 3 Stp units were again coupled, as described above, using AA/HOBt/PyBop/DIPEA in a ratio of 8/8/8/16 equivalents. Boc-Cys-(trt) was the last aminoacid to be added. Boc-Cys(trt)-OH (8 eq) solved in DMF, DIPEA (16 eq) and Pybop/ HOBt (8 eq) solved in DMF were added and the synthesis vessel was agitated for 45 min. After a negative Kaiser test, the resin was washed with DMF. Afterwards the resin was washed with DCM and dried over KOH in vacuo. For cleavage the resin was suspended in a solution of TFA/TIS/H20 (95:2.5:2.5, v/v/v) and agitated for 1 h. The cleavage solution was drained and collected. The resin was washed twice with TFA and once with DCM. The collected solutions were concentrated under reduced pressure to approximately 3 ml. The concentrated solution was dropped slowly in a 1: 1 mixture (40 ml) of cooled (0 °C) MTBE. The resulting precipitate was centrifuged at 4 °C for 10 min (3000 rpm). The solvents were decanted and the pellet was washed twice with ice-cold MTBE. The resulting pellet was dissolved in water and lyophilized.
General Procedure: Synthesis of double T-shapes with four FA: exemplified by C-K(K- LinA9)-Stp3- K(K-LinA9)-C
After swelling 0.20 mmol of 2 Chloro Trityl Chloride resin in DCM for 30 min and DMF for 10 min, 0,7 equivalents of fmoc-Cys-(trt) and DIPEA (10 eq) were added for 60 min. 0,8 mL of methanol pro gram of resin were then added and after 10 min the resin was washed five times with DMF and five times with DCM. Reaction progress was monitored by Kaiser test. Fmoc- protection group was cleaved by double treatment with 20% piperidine in DMF. fmoc-Lys(dde)- OH was used in the next coupling step. Dde-Lys(fmoc)-OH (4 eq) (related to resin loading) solved in DMF, DIPEA (8 eq) and Pybop/ HOBt (4 eq) solved in DMF were added and the synthesis vessel was agitated for 45 min. After a negative Kaiser test, the resin was washed with DMF. After a successful reaction the resin was treated twice with 20 % piperidine in DMF in order to deprotect the fmoc group. After washing the resin, four equivalents of fmoc-Stp-OH, DIPEA (8 eq) and Pybop/ HOBt (4 eq) were added for 45 min. The reaction solvent was drained and the resin was washed five times with DMF and five times with DCM. Reaction progress was monitored by Kaiser test. The resin was treated twice with 20 % piperidine in DMF in order to deprotect the fmoc group. The whole process was repeated two times. After, fmoc-Lys(dde)-OH
was used in the next coupling step, as described above. Boc-Cys-(trt) was the following aminoacid to be added. Boc-Cys(trt)-OH (4 eq) solved in DMF, DIPEA (8 eq) and Pybop/ HOBt (4 eq) solved in DMF were added and the synthesis vessel was agitated for 45 min. After a negative Kaiser test, the resin was washed with DMF. Then, the Dde groups from the lysines were removed. The dde protecting group was cleaved with 2 % hydrazine monohydrate in DMF (v/v) (20-30 times for 5 min) till no significant A300 was measurable in the deprotection mixture. In-between the deprotection- steps the resin was washed twice with DMF. Then 8 equivalents of Fmoc-Lys(Fmoc) solved in DMF, DIPEA (16 eq) and Pybop/ HOBt (8 eq) were added to the resin for 45 min. After a negative Kaiser test, the resin was washed with DMF. After a successful reaction the resin was treated twice with 20 % piperidine in DMF in order to deprotect the fmoc group. Last step was the coupling of Linoleic Acid (LinA). 20 equivalents of the fatty acid solved in DCM, 40 equivalents of DIPEA and 20 equivalents of Pybop/ HOBt were added to the resin for 45 min. After completion of the reaction the resin was washed five times with DCM and three times with DMF. Afterwards the resin was washed with DCM and dried over KOH in vacuo. For cleavage the resin was suspended in a solution of TFA/TIS/H20 (95:2.5:2.5, v/v/v) and agitated for 1 h. The cleavage solution was drained and collected. The resin was washed twice with TFA and once with DCM. The collected solutions were concentrated under reduced pressure to approximately 3 ml. The concentrated solution was dropped slowly in a 1: 1 mixture (40 ml) of cooled (0 °C) n- hexane. The resulting precipitate was centrifuged at 4 °C for 10 min (3000 rpm). The solvents were decanted and the pellet was washed twice with ice-cold n-hexane. The resulting pellet was dissolved in 50 % acetonitrile and lyophilized.
Figure 11 shows the Cell viability assay and luciferase reporter gene silencing in Neuro2AGFPLuc cells with (a) Polymer ID 230 (b) Polymers ID 199, ID 386, ID 277 (c) Polymers ID280, ID278, ID 352 (d) Polymer ID279 (protocols for both assays see above).
General Procedure: Synthesis of four-arm polymers exemplified by A-K-fK-fStp^)?)?
After swelling 0.2 mmol of fmoc- Ala-Wang resin Low Load (Novabiochem) in DMF and cleavage of the fmoc protecting group by double treatment with 20% piperidine in DMF, a solution of fmoc-Lys(fmoc)-OH (0.4 eq), PyBOP/HOBt (0.4 eq) and DIPEA (0.8 eq) in DMF was added to the resin and the vessel was agitated for 1 h. The unreacted amino groups were subsequently acetylated using 20 equivalents of acetic anhydride and 40 equivalents of DIPEA in
DMF before removal of the fmoc protecting group. The second coupling was performed by adding a solution of fmoc-Lys(fmoc)-OH (4 eq), DIPEA (8 eq) and Pybop/ HOBt (4 eq) in DMF and incubation for 1 h. Reaction progress was monitored by Kaiser test. After completion of the reaction the resin was washed five times with DMF and five times with DCM and dried for 12 h in vacuo. The loading of the resin was determined by spectrophotometric fmoc-quantification using 20% piperidine in DMF.
The preloaded resin (0.02-0.04 mmol according to fmoc loading) was swollen over night in DMF. After the swelling 1% Triton X-100 was added to the DMF for use, and all subsequent steps including washing, coupling and fmoc-deprotection were performed using this mixture. Fmoc protecting groups were cleaved by sixfold treatment (each 5 min) with 2% piperidine, 2% diazabicyclo-undecene (DBU) in DMF/Triton. After washing the resin, a solution of fmoc-Stp- OH (4 eq), DIPEA (8 eq) and Pybop/ HOBt (4 eq) in DMF/Triton was added for 90 min. The reaction solvent was drained and the resin was washed five times with DMF. Reaction progress was monitored by Kaiser test. The whole process was repeated three times. The resin was treated six-times (each 5 min) with 2% piperidine, 2% l,8-diazabicyclo[5.4.0]undec-7-ene (DBU; Sigma- Aldrich) in DMF/Triton in order to deprotect the fmoc group. The resin was washed five times with DMF and five times with DCM and dried over KOH in vacuo. For cleavage the resin was suspended in a solution of TFA/H20 (95:5, v/v) and agitated for 3 h. The cleavage solution was drained and collected. The resin was washed twice with TFA and twice with DCM. The collected solutions were concentrated under reduced pressure to minimum volume. The residue was washed twice with ice-cold MTBE, then dissolved in water and lyophilized. The lyophilized product was purified on an Akta Basic HPLC system using the cation exchange column Resource S 1 ml (GE Healthcare). The buffers used were A: lOmM HC1 and B: lOmM HC1, 3M NaCl. The step gradient was started with 5% B to 35% B in 10 min, isocratic elution (2 ml/min) for 15 min and continued to 100% B in 10 min. The product fractions were pooled, dialyzed in water for 24 h (Spectra/Por® Dialysis Membrane, MWCO 1000 Da) and freeze dried.
Table 4. Sequences of four-arm polymers exemplified by A-K-(K-(Stp.j)2)2. Sequences (from C- to N-terminus, as assembled on the solid support). Abbreviations: K= Lysine, Stp = Succinyl tetraethylenepentamine, A= Alanine.
ID Mol Weight Formula Sequence Type
286 1559,05 C69H143N27013 A-K-(K-(Stp)2)2 branched
287 2644,48 A-K-(K-(Stp2)2)2 branched
288 3729,92
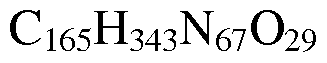 A-K-(K-(Stp3)2)2 branched
A-K-(K-(Stp3)2)2 branched
289 4815,36 C213H443N87O37 A-K-(K-(Stp4)2)2 branched
Figure 12 shows the Cell viability assay and luciferase reporter gene silencing after transfection of (a,b) Neuro2A or (c,d) HUH7 cells. Transfections (a,c) and viability assays (b,d) were performed as described for "Luciferase reporter gene expression" and "Cell viability assay (MTT Assay)". Cells were transfected with polymers ID23: K-Stp5-K; ID286: A-K-(K-(Stp)2)2; ID287: A-K-(K-(Stp2)2)2; ID288: A-K-(K-(Stp3)2)2; ID289: A-K-(K-(Stp4)2)2.
Targeting Ligand - Polymer Conjugates
The targeting ligands B6 (for binding the transferrin receptor, HO-KRPGKAKHG), non- functional control sequences (B6mod, Ala), folic acid (FolA), cyclic RGD (for integrin binding) and CMP (for binding the hepatocyte growth factor receptor c-Met, HO-KHHHIHDHRSLSK) were synthesized as PEG-polymer conjugate as described in the following and were demonstrated as functional in receptor-targeted DNA delivery. Synthesis of HO-KRPGKAKHG-PEG94-K-(Stp4-C)9.:
HO-KRPGKAKHG-PEG24-K-(Stp4-C)2 ("B6-PEG24-K-(Stp4-C)2") was synthesized according to
"General Procedure: Synthesis of T-Shapes containing shielding and targeting domains: exemplified by HO-IVNQPTYGYWHY-PEG24-K-(Stp4-C)2".
Synthesis of HO-GRPGGAGHG-PEG94-K-(StP4-C)9:
HO-GRPGGAGHG-PEG24-K-(Stp4-C)2 ("B6mod-PEG24-K-(Stp4-C)2") was synthesized according to "General Procedure: Synthesis of T-Shapes containing shielding and targeting domains: exemplified by HO-IVNQPTYGYWHY-PEG24-K-(Stp4-C)2".
Synthesis of HO- A- PEG24-K-(Stp4-C)2:
HO-A-PEG24-K-(Stp4-C)2 was synthesized according to "General Procedure: Synthesis of T- Shapes containing shielding and targeting domains: exemplified by HO-IVNQPTYGYWHY-

Synthesis of HO-K(PEG94-K-(StP4-C)9)-HHHIHDHRSLSK ("CMP-PEG94-K-(StP4-C)9") At first, resin bound K(ivDDE)-HHHIHDHRSLSK-Boc (protected CMP) was synthesized by standard peptide synthesis. After swelling 20 μιηοΐ of resin bound K(ivDDE)-
HHHIHDHRSLSK-Boc (CMP) in DMF for 30 min the ivDDE protecting group was removed with 2% hydrazine monohydrate in DMF (v/v) (5-10 times for 5 min) until no significant A30o was measurable in the deprotection mixture. In between the deprotection- steps the resin was washed twice with DMF. Subsequent synthesis steps were accomplished according to„General Procedure: Synthesis of T-Shapes containing shielding and targeting domains: examplified by HO-IVNQPTYGYWHY-PEG24-K-(STP4-C)2".
Synthesis of T-Shapes containing shielding and targeting domains, with N-terminal targeting domain, exemplified by FolA-PEG^-K-fStp^-C)?:
After swelling 20 μιηοΐ of 2-Chloro Trityl Chloride resin with resin-bound Cys(Trt)-NH2 in DMF for 30 min four equivalents of a solution of fmoc-Stp-OH in the smallest possible amount of DMF, DIPEA (8 eq) and PyBOP/HOBt (4 eq) were added to the resin and the vessel was agitated until Kaisertest indicated complete conversion (60 min). The reaction solvent was drained and the resin was washed three times with DCM followed by three DMF washs. After removal of the fmoc protective group by treatment with 20% piperidine in DMF this cycle was repeated 3 times. After last fmoc removing step HO-Lys(fmoc)-dde was introduced as a branching point using standard coupling conditions (AA/PyB OP/HOB t/DIPEA, 4/4/4/8 eq in DMF). After successful deprotection 4 equivalents of fmoc-Stp-OH, 4 equivalents of PyBOP/ HOBt and 8 equivalents of DIPEA in the smallest possible amount of DMF were added to the resin and the mixture was agitated until Kaisertest did indicate complete conversion (normally 30 min). The fmoc group was removed and the whole cycle repeated 4 times exchanging Stp by Boc-Cys(trt)-OH in the last cycle. After finishing the backbone HO-C(trt)-Stp4-K(dde)-Stp4- C(trt)-Boc the dde protective group was removed with 2 % hydrazine monohydrate in DMF (v/v) (5-10 times for 5 min) till no significant A300 was measurable in the deprotection mixture. In- between the deprotection-steps the resin was washed twice with DMF. Fmoc-PEG24-OH dissolved in DCM/DMF (l: l,v/v), DIPEA (8 eq) and Pybop/ HOBt (4 eq) were added to the resin and the vessel was agitated until Kaisertest indicated complete conversion (60 min). fmoc cleavage 4 equivalents of fmoc-Glu(OH)-OtBu in DCM/DMF (1: 1, v/v) were added, using DIPEA (8 eq) and Pybop/ HOBt (4 eq). As last step N10-(Trifuoroacetyl)pteroic acid was coupled under standard coupling conditions (AA/PyB OP/HOB t/DIPEA, 4/4/4/8 eq in DMF). After washing the resin 5 times with DMF the TFA group was removed by incubating the resin with ammonium solution/DMF (1: 1, v/v) for 2h. After completion of the reaction the resin was washed and dried for 12 h over KOH in vacuo. The PAA was cleaved from the resin by
suspending it in a solution of TFA/H20/TIS/EDT (92.5:2.5:2.5:2.5, v/v) for 1 h. The cleavage solution was collected by filtration and the resin washed twice with TFA and once with DCM. The concentrated solution was dropped slowly in a 1 : 1 mixture (40 ml) of cooled (0 °C) MTBE and n-hexane. The resulting precipitate was centrifuged at 4 °C for 10 min (2000-3000 rpm). The solvents were decanted and the pellet was washed twice with ice-cold MTBE. The resulting pellet was dissolved in 50% acetonitrile and lyophilised.
General Procedure: Synthesis of T-Shapes containing shielding and targeting domains, coupling of ligand in solution: cRGDfK-A-PEG24-K-(Stp4-C) :
After swelling 0.20 mmol of 2-Chlorotrityl Chloride Resin resin in DCM for 30 min and DMF for 10 min, 0,7 equivalents of fmoc-Ala-OH and DIPEA (10 eq) were added for 60 min. 0,8 mL of methanol pro gram of resin were then added and after 10 min the resin was washed five times with DMF and five times with DCM. Reaction progress was monitored by Kaisertest. After removal of the fmoc protective group by treatment with 20% piperidine in DMF four equivalents of a solution of fmoc-PEG24-OH in DCM/DMF(1: 1, v/v), DIPEA (8 eq) and PyBOP/HOBt (4 eq) were added to the resin and the vessel was agitated until Kaisertest indicated complete conversion (60 min). The reaction solvent was drained and the resin was washed three times with DCM followed by three DMF washs. After removal of the fmoc protective group by treatment with 20% piperidine in DMF fmoc-Lys(fmoc)-OH was introduced as a branching point using standard coupling conditions (AA/PyB OP/HOB t/DIPEA, 4/4/4/8 eq in DMF). After succesfull deprotection 8 equivalents of fmoc-Stp-OH, 8 equivalents of PyBOP/ HOBt and 16 equivalents of DIPEA in the smallest possible amount of DMF were added to the resin and the mixture was agitated until Kaisertest did indicate complete conversion 15 (normally 30 min). The fmoc group was removed and the whole cycle repeated 4 times, exchanging Stp by boc-C(Trt)-OH in the last step. After completion of the reaction the resin was washed and dried for 12 h over KOH in vacuo. The PAA was cleaved from the resin by suspending it in a solution of 10% HFIP in DMF for 5 min 5 times. The cleavage solution was collected by filtration. The solvent was evaporated by vacuum speed.
1 eq of the polymer was dissolved in 500 μΐ^ of dry DMF, in the presence of HATU (1.2 eq) and HOAt (1.2 eq), when the mixture was dissolved, DIEA (5 eq) is added, and the mixture is pre- activated at room temperature for 4.5 h. Next, 1 eqivalent of cyclo(-R(Pbf)GD(OtBu)fK-), the protected form of cyclo(RGDfK) (Haubner R, Gratias R, Diefenbach B, Goodman SL, Jonczyk A, and Kessler H., J. Am. Chem. Soc, 1996, 118, 7461-7472) was added. After 24h of reaction
time, DMF was evaporated the solid was re-dissolved in NaHC03, the product was extracted with AcOEt, and washed with NaCl and finally the solvent was evaporated. The compound was treated with TFA/H20/TIS (90:2.5:2.5) for 2 h in order to deprotect the side chains. Then, it was precipitated with cold Et20, centrifuged and decanted. The solid was re-dissolved in water and lyophilized.
Luciferase reporter gene expression using chloroquine for endosomal escape
Transfection was performed as described for "luciferase reporter gene expression" with following modification: lh after incubation of cells with polyplexes, medium was removed and exchanged against medium containing ΙΟΟμΜ chloroquine (Cotten M, Langle-Rouault F, Kirlappos H, Wagner E, Mechtler K, Zenke M, Beug H, Birnstiel, ML, Proc. Natl. Acad. Sci. U.S.A 1990, 87, 4033-4037). After additional 4 h incubation the medium was removed again and exchanged against medium without chloroquine. Results are shown in Figures 13-15.
Ligand-PEG-siRNA conjugates
General procedure: Synthesis of pegylated targeting domain with C-terminal ligand, exemplified by HO-KRPGKAKHG-PEG24-Cvs:
After swelling 20 μπιοΐ of resin-bound HO-KRPGKAKHG-NH2 (B6) (2-Chloro Trityl Chloride resin) in DMF for 30 min four equivalents of a solution of fmoc-PEG24-OH in DCM/DMF (1: 1, v/v), DIPEA (8 eq) and PyBOP/HOBt (4 eq) were added to the resin and the vessel was agitated until Kaisertest indicated complete conversion (60 min). The reaction solvent was drained and the resin was washed three times with DCM followed by three DMF washes. After removal of the fmoc protective group by treatment with 20% piperidine in DMF, 4 eq boc-Cys(trt)-OH was added with 4 eq PyBop/HOBt and 8 eq DIPEA in DCM/DMF (1: 1, v/v). After completion of the reaction the resin was washed and dried for 12 h over KOH in vacuo. The PAA was cleaved from the resin by suspending it in a solution of TFA/H20/TIS/EDT (92.5:2.5:2.5:2.5, v/v) for 1 h. The cleavage solution was collected by filtration and the resin washed twice with TFA and once with DCM. Solvent was evaporated. The peptidic structure was diluted in a buffer containing 20mM Hepes pH 6,5, 150mM NaCl, 30% (v/v) acetonitrile and purified by size exclusion chromatography.
General procedure: Synthesis of pegylated targeting domain with N-terminal ligand, exemplified by Cys-PEG24-FolicAcid:
After swelling 20 μηιοΐ of resin-bound Cys(trt)-NH2 (2-Chloro Trityl Chloride resin) in DMF for 30 min four equivalents of a solution of fmoc-PEG24-OH in DCM/DMF(1: 1, v/v), DIPEA (8 eq) and PyBOP/HOBt (4 eq) were added to the resin and the vessel was agitated until Kaiser test indicated complete conversion (60 min). The reaction solvent was drained and the resin was washed three times with DCM followed by three DMF washes. After removal of the fmoc protective group by treatment with 20% piperidine in DMF 4 eq fmoc-Glu(OH)-OtBu was added with 4 eq PyBop/HOBt and 8 eq DIPEA in DCM/DMF (1: 1, v/v). After removal of the fmoc protective group N10-(Trifuoroacetyl)pteroic acid was coupled as last step under standard coupling conditions (AA/PyBOP/HOBt/DIPEA, 4/4/4/8 eq in DMF). After washing the resin 5 times with DMF the TFA group was removed by incubating the resin with ammonium solution/DMF (1: 1, v/v) for 2h. After completion of the reaction the resin was washed and dried for 12 h over KOH in vacuo. The PAA was cleaved from the resin by suspending it in a solution of TFA/H20/TIS/EDT (92.5:2.5:2.5:2.5, v/v) for 2 h. The cleavage solution was collected by filtration and the resin washed twice with TFA and once with DCM. Solvent was evaporated. The peptidic structure was diuted in a buffer containing 20mM Hepes pH 6,5, 150mM NaCl, 30% (v/v) acetonitrile and purified by size exclusion chromatography.
Synthesis of targeted siRNA; exemplified by FolA-PEG24-Cys-s-s-siRNA
TNB-s-s-C6-siRNA was incubated 60min with 2 eq Cys-PEG24-FolicAcid diluted in 500μί 20mM Hepes pH 6.5, 30% (v/v) acetonitrile. Purification of the resulting FolA-PEG24-Cys-s-s- siRNA was performed on a lmL ResourceQ column connected to an Aktabasic system. For that purpose the reaction solution was diluted 1:2 with buffer A (20mM Hepes pH 6.5, 30% (v/v) acetonitrile) and loaded onto the column using the same buffer A containing 20mM NaCl. After unbound material was washed away with buffer A containing 200mM NaCl. The product was eluted setting a gradient of lOmM NaCl/min and a flow of lmL/min. Resulting fractions were analyzed on a 2,5% agarose gel. Samples containing gel-retarded siRNA compared to unmodified control were pooled.
Synthesis of HO-KRPGKAKHG-PEG24-Cvs-s-s-siRNA
Synthesis was performed by the general procedure starting by incubation of TNB-s-s-C6-siRNA with 2 eq HO-KRPGKAKHG-PEG24-Cys. Figure 16 shows the Cell viability assay and luciferase reporter gene silencing in KB-eGFPLuc cells with FolA-siRNA /siRNA at indicated % ratios using Polymer ID 233 as carrier system. As
control FolA-siRNA was transfected without polymer. "GFP" siRNA: SEQ ID No 5/6 ,
"siControl" siRNA: SEQ ID No. 7/8.
Table 5 - Exemplified sequences with enhanced with receptor targeting moiety Sequences (from C- to N-terminus, as assembled on the solid support). Abbreviations: K = Lysine, Stp = Succinyltetraethylenepentamine, A = Alanine, C = Cysteine, PEG24 = polyethylene glycol with 24 subunits, CMP = peptide for HGF receptor targeting, B6 = peptide for transferrin receptor targeting, FolA = folic acid.
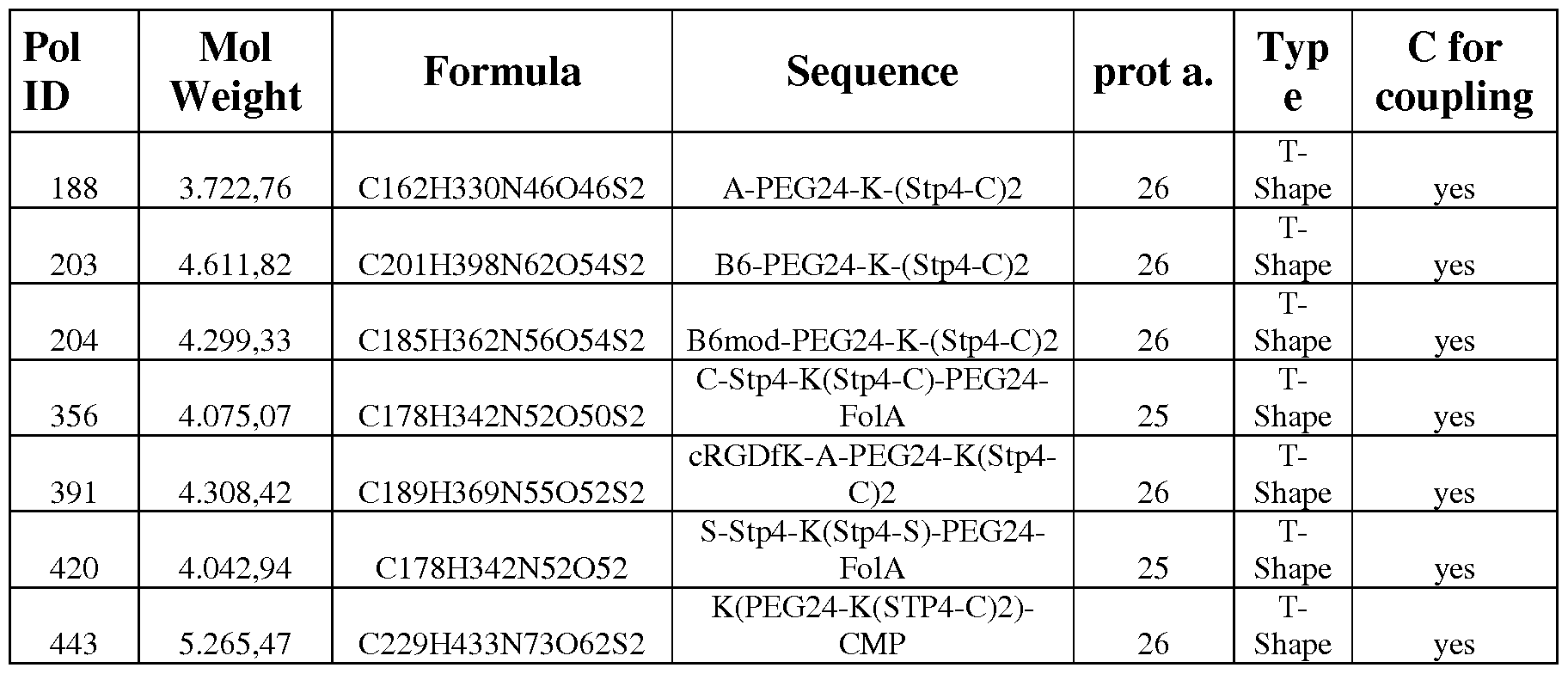
Endosomolytic peptide-siRNA Conjugates
Deprotection and activation of thiol modified siRNA
370nmol of siRNA containing a C6-s-s-C6 modification at the 5' end of the sense strand was diluted in 200μ1 water and incubated for 30min with 10 eq tris(2-carboxyethyl)phosphine (TCEP) to cleave the disulfide bridge. The resulting HS-C6-siRNA was purified by ion exchange chromatography using a lmL ResourceQ column connected to an Akta basic system. The mixture was loaded onto the column with buffer A containing 20mM Hepes, lOmM NaCl, pH 6,5, 30% (v/v) acetonitrile. After washing away unbound material with same buffer containing 200mM NaCl the modified siRNA was eluted using a buffer containing 500mM NaCl.
After deprotection the thiol group was activated using 10 eq of 2,2'-Dinitro-5,5'-dithio-dibenzoic acid (DTNB) diluted in 200μL· 20mM Hepes pH 8,0. The siRNA solution was added dropwise to avoid dimerization. After 30min incubation the mixture was diluted 1:2 with buffer containing 20mM Hepes, 30% (v/v) acetonitrile. The TNB-s-s-C6-siRNA was purified as explained for the deprotection.
Synthesis of an endosomolytic active siRNA; exemplified by siRNA-Inf7
TNB-s-s-C6-siRNA was incubated 60min with 1,5 eq
H2N-GLFEAIEGFIENGWEGMIDGWYGC-Amid (Inf7; Plank C, Oberhauser B, Mechtler K, Koch C, Wagner E.J Biol Chem. 1994; 269: 12918-24) diluted in 500μΕ 20mM Hepes pH 8,5, 30% (v/v) acetonitrile. Purification of the resulting Inf7-s-s-siRNA was performed on a lmL ResourceQ column connected to an Akta basic system. For that purpose the reaction solution was diluted 1:2 with buffer A (20mM Hepes pH 6,5, 30% (v/v) acetonitrile) and loaded onto the column using the same buffer A containing 200mM NaCl. After unbound material was washed away the product was eluted setting a gradient of lOmM NaCl/min and a flow of lmL/min. Resulting fractions were analysed on a 2,5% agarose gel. Samples containing gel-retarded siRNA compared to unmodified control were pooled.
Synthesis of siRNA-DMMAnMel
The synthesis of siRNA-DMMAnMel was modified according to the synthesis of siRNA-Inf7. In a preincubation step mellitin-peptide (H2N-CIGAVLKVLTTGLPALISWIKRKRQQ-OH, all-D- form) was modified by 2,3-dimethylmaleic anhydride (DMMAn) as described in (Meyer M, Dohmen C, Philipp A, Kiener D, Maiwald G, Scheu C, Ogris M, Wagner E. Mol Pharm. 2009; 6: 752-62). Thus mellitin was diluted in a buffer containing 250mM HEPPS pH 8,5, 30% (v/v) acetonitrile, mixed with 12 eq DMMAn in acetonitrile and incubated for 30min at 25°C. After that 60 eq. L-lysine were added and incubated for another 30min at 25°C. This mixture was used without purification to be coupled to siRNA as described for siRNA-Inf7 using same buffers at pH 8,5.
Purification of the resulting siRNA- s-s-DMMAnMel was performed on a lmL ResourceQ column connected to a Akta basic system. For that purpose the reaction solution was diluted 1:2 with buffer A (20mM Hepes pH 8,5, 30% (v/v) acetonitrile) and loaded onto the column using the same buffer A containing 200mM NaCl. After unbound material was washed away the product was eluted setting a gradient of lOmM NaCl/min and a flow of lmL/min. Resulting fractions were analysed on a 2,5% agarose gel. Samples containing gel-retarded siRNA compared to unmodified control were pooled.
Figure 17 shows the results of the erythrocyte leakage assay and Figures 18 and 19 show the results of Cell viability assay and Luciferase reporter gene silencing.
Combination of targeting polymer with endosomolytic ΙηΠ-siRNA conjugate
Luciferase reporter gene silencing with targeted polymers
Transfection procedure was performed as described for„Luciferase reporter gene silencing" with the following modification: the polyplexes were left for 30min on the cells. After that medium was removed and exchanged against fresh medium.
Results are shown in Figure 20.
Silencing of EG5 gene in tumors after systemic application of polymer/EG5-siRNA in vivo
EG5 siRNA duplex:
5'-ucGAGAAucuAAAcuAAcudTsdT-3' (sense) [Seq ID. No 9] 5'-AGUuAGUUuAGAUUCUCGAdTsdT-3' (antisense) [Seq ID. No 10]
Female 6-8-week-old A/J mice (for N2A cell experiments) or 6-8-week-old NMRI mice (for KB cell experiments) were inoculated subcutaneously in the flank with 1 million Neuro2a cells or 5 million KB cells. Experiments started when tumors reached a weight of 100-750 mg. Systemic gene transfer in tumor bearing mice (2 mice per group N2A experiments, 3 mice per group KB experiments) was carried out using polyplexes containing 50 μg EG5 siRNA per 20 g body weight (2,5mg/kg) at a concentration of 200 μg/ml siRNA in HBG and a polymer/siRNA N/P of 12 (EG5 siRNA) or 16 (Inf7-EG5 siRNA). Polyplexes were applied into the tail vein and animals were sacrificed 24 h after application. Tissues were dissected, transferred into
Cryomolds 10mm x 10mm x 5mm (Sakura Finetek GmbH, Staufen, Germany) and embedded in O.C.T. compound (Sakura Finetek GmbH, Staufen, Germany). Different tissues (He, heart; Lu, lung; Li, liver; Sp, spleen; Ki, kidney; Tu, tumor;) were cut into 5μιη fine sections using an LEICA CM 3050s cryostat (LEICA, Wetzlar, Germany) and stained with a 1: 1000 diluted 4', 6- Diamidin-2'-phenylindoldihydrochlorid (DAPI, Roche Diagnostics, Mannheim, Germany) solution for 5 min at room temperature. Tissue slices were covered with cover slips and analysed using a Zeiss Axiovert 200 microscope and Zeiss Axio Cam camera (Carl Zeiss GmbH, Leipzig Germany). Mitotic arrest of tumor cells (visible as aster formation) as a consequence of EG5 silencing could only be observed in tumor samples treated with EG5 siRNA pictures are shown in Figure 21. Other tissues or animals treated with control siRNA did not show aster formation.
Oligotyrosine-containing stabilizing polymers
For polyplex stabilization by noncovalent hydrophobic π-stacking, oligomeric tyrosines (preferred form: trimers) were introduced into the polymers (instead of the either cysteines or fatty acids). These oligotyrosine containing polymers were able to bind nucleic acids effectively. Oligotyrosines in combination with either cysteines or fatty acids were functional in efficient siRNA transfection.
Loading of a 2-chlorotrityl chloride resin with fmoc-Tyr(tBu)-OH
A 2-chlorotrityl chloride resin (1.6 mmol/g) was loaded with fmoc-Tyr(tBu)-OH. After swelling in anhydrous DCM for 10 min, the amino acid fmoc-Tyr(tBu)-OH (0.625 eq) and DIPEA (1.3 eq) solved in anhydrous DCM were added for 1 h. To cap the residual reactive chloride groups, a mixture of DCM/MeOH/DIPEA (80/15/5) was added twice for 10 min. The loading of the resin was determined by measuring the absorption at 301 nm, after adding 1 ml 20 % piperidine in DMF to a small part of the resin for 1 h. The fmoc-protection group of the fmoc-Tyr(tBu)- chlorotrityl resin was cleaved by double treatment with 20 % piperidine in DMF. Afterwards the resin was washed with DMF, DCM and n-hexane and dried in vacuo.
Synthesis of Tyr(tBu)3-chlorotrityl resin:
After swelling of the preloaded Tyr(tBu)-chlorotrityl resin, four equivalents (related to resin loading) of fmoc-Tyr(tBu)-OH, DIPEA (8 eq) and Pybop/ HOBt (4 eq) were added for 35 min. The reaction solvent was drained and the resin was washed four times with DCM and once with DMF. Reaction progress was monitored by Kaiser test. After a negative Kaiser test, the resin was treated with 20 % piperidine in DMF by double treatment. Fmoc-Tyr(tBu)-OH was attached again as described above.
After negative Kaiser test, the fmoc-protection group was cleaved as described above. The resin was washed with DMF and DCM and then dried in vacuo resulting in a Tyr(tBu)3-chlorotrityl resin.
Synthesis of fmoc-Tyr(tBu)3-OH:
The synthesis of fmoc-Tyr(tBu)3-OH was modified according to "Synthesis of Tyr(tBu)3- chlorotrityl resin" as followed: After the attachment of the third fmoc-Tyr(tBu)-OH the resin was washed with DMF and DCM and dried in vaccuo. The fmoc-Tyr(tBu)3-OH was cleaved from the resin with acetic acid/trifluoroethanol/DCM (1/2/7) five times for 20 min (5 ml) till no absorption was observed on a TLC. The cleavage solutions were pooled and evaporated. To
remove the acetic acid, the residue was solved in chloroform and washed five times with sodium citrate buffer (0.1 M, pH 5.5). The organic phase was dried over sodium sulphate and then dried in vacuo. The resulting protected fmoc-Tyr(tBu)3-OH was used as building block for further syntheses. The building block was characterized with NMR and mass spectra.
Synthesis of boc-Tyr(tBu)3-OH
The synthesis of boc-Tyr(tBu)3-OH was modified according to "Synthesis of fmoc-Tyr(tBu)3- OH". As followed: After attachment of the third fmoc-Tyr(tBu)-OH, the fmoc-protection group was cleaved of and 8 equivalents di-ie/t-butyl dicarbonate (Boc20) and DIPEA (16 eq) were added for 45 min to the resin. The resulting boc-Tyr(tBu)3-OH was cleaved from the resin and purified as described above.
General Procedure: Synthesis of T-shaped oligotyrosines: exemplified by Y3-Stp2-K(K- FA2)-Stp2-Y3
The synthesis of Y3-Stp2-K(K-FA2)-Stp2-Y3 was performed according to "General Procedure: Synthesis of T-shapes with two FAs: exemplified by C-Stpl-K(KFA2)-Stpl-C" with following modification:After swelling 0.03 mmol of Tyr(tBu)3-chlorotrityl resin in DMF over night, four equivalents (related to resin loading) of fmoc-Stp-OH, DIPEA (8 eq) and Pybop/ HOBt (4 eq) were added for 35 min. After removing the protection group this cycles was repeated once.
Instead of the attachment of a final cysteine, boc-Tyr(tBu)3-OH (2 eq) solved in DCM, DIPEA (4 eq) and Pybop/ HOBt (2 eq) were added for 60 min. Afterwards the resin was washed and dried over KOH in vacuo. For cleavage the resin was suspended in a solution of TFA/TIS/H20 (95/2.5/2.5, v/v/v) and agitated for 2 h. The cleavage solution was drained and collected. The resin was washed twice with TFA and once with DCM. The collected solutions were evaporated under reduced pressure and the residue was washed 3 times with cooled n-hexane. The residue was solved in 10 mM HC1 in water with 30 % acetonitrile and purified via SEC (G10, 10 mM HC1 + 30 % ACN). The fractions were lyophilised after SEC and characterized via NMR and mass. General Procedure: Synthesis of T-shaped oligotyrosines: C-Stp3-K(K-(Y3)2)-Stp3-C
The synthesis of C-Stp3-K(K-(Y3)2)-Stp3-C was performed according to "General Procedure: Synthesis of T-shapes with two FAs: exemplified by C-Stpl-K(KFA2)-Stpl-C" with following modification:The attachment of Stp and the removal of its fmoc protective group were always
performed 3 times. Instead of the coupling of the fatty acids, 4 equivalents of the boc-Tyr(tBu)3- OH solved in DCM, 8 equivalents of DIPEA and 4 equivalents of Pybop/ HOBt were added to the resin for 55 min. After precipitation in cooled n-hexane, the residue was solved in 10 mM HC1 in water with 30 % acetonitrile and purified via SEC (G10, 10 mM HC1 + 30 % ACN). The fractions were lyophilised after SEC and characterized via NMR and mass.
General Procedure: Synthesis of two-arm oligotyrosines: K(Stp3-Y3-C)2
After swelling 0.03-0.06 mmol of a Rink amide resin in DMF over night and cleavage of the fmoc protecting group, four equivalents of a solution of fmoc-Lys(fmoc)-OH (4 eq) in DMF, DIPEA (8 eq) and Pybop/HOBt (4 eq) were added to the resin and the vessel was agitated until Kaiser test indicated complete conversion (normally 35 min). The reaction solvent was drained and the resin was washed five times with DMF and DCM. After fmoc-deprotection, eight equivalents fmoc-Stp-OH in DMF, DIPEA (16 eq) and Pybop/HOBt (8 eq) were added to the resin and the vessel was agitated until Kaiser test indicated complete conversion. The reaction solvent was drained and the resin was washed five times with DMF and DCM. This cycle was repeated twice. Afterwards fmoc-Tyr(tBu)3-OH (4 eq), DIPEA (8 eq) and Pybop/HOBt (4 eq) were added to the resin and the vessel was agitated for 60 min. After complete coupling, the fmoc-protection group was removed with 20 % piperidine in DMF and boc-Cys(trt)-OH (8 eq.) was coupled. After completion of the reaction the resin was washed and dried over KOH in vacuo. For cleavage the resin was suspended in a solution of TFA/TIS/H20 (95/2.5/2.5, v/v/v) and agitated for 2 h. The cleavage solution was drained and collected. The resin was washed twice with TFA and once with DCM. The collected solutions were evaporated under reduced pressure and the residue was washed 3 times with cooled n-hexane. The residue was solved in 10 mM HC1 in water with 30 % acetonitrile and purified via SEC (G10, 10 mM HC1 + 30 % ACN). The fractions were lyophilised after SEC and characterized via NMR and mass.
Ureido-pyrimidinone-containing stabilizing polymers
For polyplex stabilization via noncolvalent hydrogen-bonding, ureido-pyrimidinones were used instead of the crosslinking cysteine groups. The ureido-pyrimidinones are fully compatible with solid phase peptide synthesis (Dankers, Meijer: Convenient Solid-Phase Synthesis of Ureido- Pyrimidinone Modified Peptides, Eur. J. Org. Chem. 2007) and are attached at the N-terminus of the sequences.

• W Hydrogen bond formation between two ICH-CAMP groups
Ureido-pyrimidinone synthesis
The ureido-pyrimidinone 2-(6-Isocyanahexylaminocarbonylamino)-6-methyl-4(7H) pyrimidinone (ICH-CAMP) was synthesised as described by Dankers et al.
Loading of a 2-chlorotrityl chloride resin with dde-Lys(fmoc)-OH
The loading of the resin was performed according to "Loading of a 2-chlorotrityl chloride resin with fmoc-Tyr(tBu)-OH" using dde-Lys(fmoc)-OH instead of fmoc-Tyr(tBu)-OH.
General Procedure: Synthesis of T-shaped ureido-pyrimidinone polymers examplified by (QleA2-K)K(K(Stp2-ICH-CAMP)2)
After swelling 0.03-0.08 mmol of dde-Lys-chlorotrityl resin in DMF over night, four equivalents (related to resin loading) of fmoc-Lys(fmoc)-OH, DIPEA (8 eq) and Pybop/ HOBt (4 eq) were added for 35 min. The reaction solvent was drained and the resin was washed five times with DCM/DMF. In order to cap unreacted primary amino groups, the resin was acetylated using 10 equivalents of acetic anhydride and 20 equivalents of DIPEA, before the subsequent removal of the fmoc protecting group. To couple the fatty acid, the solvent was changed to DCM after fmoc- cleavage. Therefore the resin was washed three times with DMF and DCM after removal of the fmoc-protecting group. 10 equivalents of the fatty acid solved in DCM, 20 equivalents of DIPEA and 10 equivalents of Pybop/ HOBt were added to the resin for 35 min. After completion of the reaction the resin was washed five times with DCM and three times with DMF. The dde-
protecting group was cleaved with 2 % hydrazine monohydrate in DMF (v/v) (5-15 times for 5 min) till no significant A30o was measurable in the deprotection mixture. To introduce another branching point fmoc-Lys(fmoc)-OH was used in the next coupling step. fmoc-Lys(fmoc)-OH (4 eq) solved in DMF, DIPEA (8 eq) and Pybop/ HOBt (4 eq) solved in DMF were added and the synthesis vessel was agitated for 40 min. After a negative Kaiser test, the resin was washed with DMF. After treatment with 20 % piperidine in DMF and washing the resin with DMF and DCM, fmoc-Stp-OH (6 eq), DIPEA (12 eq) and Pybop/ HOBt (6 eq) was added. After monitoring the reaction progress by Kaiser test, the fmoc -protection group was cleaved with 20 % piperidine in DMF. The deprotection solution was drained and the resin was washed five times with DMF and once with DCM. This cycle was repeated once. The ureido-pyrimidinone ICH-CAMP (10 eq) was solved in DMF and added, after filtration, to the resin for 50 h. After a negative Kaiser test, the resin was washed 7 times with DMF, twice with DCM and n-hexane and dried in vacuo. For cleavage the resin was suspended in a solution of TFA/TIS/H20 (95/2.5/2.5, v/v/v) and agitated for 2 h. The cleavage solution was drained and collected. The resin was washed twice with TFA and once with DCM. The collected solutions were evaporated under reduced pressure and the residue was washed 3 times with cooled n-hexane. The residue was solved in 10 mM HCl in water with 30 % acetonitrile and purified via SEC (G10, 10 mM HCl + 30 % ACN). The fractions were lyophilised after SEC and characterized via NMR and mass.
General Procedure: Synthesis of T-shaped polymers containing stabilizing ureido- pyrimidinones and oligotyrosines exemplified by ((Y3)2-K)K(K(Stp2-ICH-CAMP)2). The synthesis was performed according to "General Procedure: Synthesis of T-shaped ureido- pyrimidinone polymers examplified by (01eA2-K)K(K(Stp2-ICH-CAMP)2)" with following modification: Instead of the fatty acid 3.5 equivalents of boc-Tyr(tBu)3-OH solved in DCM, 7 equivalents of DIPEA and 3.5 equivalents of Pybop/ HOBt were added to the resin for 45 min.
Figures 21 to 25 show Luciferase reporter gene silencing after transfection with T- shape polymers containing a Y3 motif or a ICH-CAMP motif respectively instead of fatty acids. Table 6: Examples of polymers containing stabilizing ureido-pyrimidinones or
oligotyrosines. Sequences (from C- to N-terminus, as assembled on the solid support).
Abbreviations: K = lysine, Y = tyrosine, Stp = Succinyltetraethylenepentamine, OleA = olic acid, C = Cysteine, ICH-CAMP = ureido-pyrimidinones.

General Procedure: Synthesis of four-arm polymers with four coupling domains exemplified by A-K-(K-(Stp4-C)z)2
After swelling 0.2 mmol of fmoc- Ala-Wang resin Low Load (Novabiochem) in DMF and cleavage of the fmoc protecting group by double treatment with 20% piperidine in DMF, a solution of fmoc-Lys(fmoc)-OH (0.4 eq), PyBOP/HOBt (0.4 eq) and DIPEA (0.8 eq) in DMF was added to the resin and the vessel was agitated for 1 h. The unreacted amino groups were subsequently acetylated using 20 equivalents of acetic anhydride and 40 equivalents of DIPEA in DMF before removal of the fmoc protecting group. The second coupling was performed by adding a solution of fmoc-Lys(fmoc)-OH (4 eq), DIPEA (8 eq) and Pybop/ HOBt (4 eq) in DMF
and incubation for 1 h. Reaction progress was monitored by Kaiser test. After completion of the reaction the resin was washed five times with DMF and five times with DCM and dried for 12 h in vacuo. The loading of the resin was determined by spectrophotometric fmoc-quantification using 20% piperidine in DMF. The preloaded resin (0.02-0.04 mmol according to fmoc loading) was swollen over night in DMF. After the swelling 1% Triton X-100 was added to the DMF for use, and all subsequent steps including washing, coupling and fmoc-deprotection were performed using this mixture. Fmoc protecting groups were cleaved by sixfold treatment (each 5 min) with 2% piperidine, 2% l,8-diazabicyclo[5.4.0]undec-7-ene (DBU; Sigma-Aldrich) in DMF/Triton. After washing the resin, a solution of fmoc-Stp-OH (4 eq), DIPEA (8 eq) and Pybop/ HOBt (4 eq) in DMF/Triton was added for 90 min. The reaction solvent was drained and the resin was washed five times with DMF. Reaction progress was monitored by Kaiser test. The whole process was repeated three times. After fmoc deprotection a solution of fmoc-Cys(trt)-OH (4 eq), DIPEA (8 eq) and Pybop/HOBt (4 eq) in DMF/Triton was added for 90 min.
The resin was treated six-times (each 5 min) with 2% piperidine, 2% 1,8- diazabicyclo[5.4.0]undec-7-ene (DBU; Sigma-Aldrich) in DMF/Triton in order to deprotect the fmoc group. The resin was washed five times with DMF and five times with DCM and dried over KOH in vacuo. For cleavage the resin was suspended in a solution of TFA/TIS/H20
(95:2.5:2.5, v/v/v) and agitated for 2 h. The cleavage solution was drained and collected. The resin was washed twice with TFA and twice with DCM. The collected solutions were
concentrated under reduced pressure to approximately 2 ml. The concentrated solution was dropped slowly in a 1: 1 mixture (40 ml) of cooled (0 °C) MTBE and n-hexane. The resulting precipitate was centrifuged at 4 °C for 5 min (3000 rpm).The residue was washed once with ice- cold MTBE. The pellet was then dissolved in lOmM HC1 containing 30% acetonitrile and purified by size exclusion chromatography (column material: G10 GE Healthcare, volume 47 ml, flow rate 2 ml/min). The buffer used was 10 mM HC1 containing 30% acetonitrile. The product fractions were pooled and freeze dried.
General Procedure: Synthesis of five-arm polymers with four coupling domains exemplified by A-Stp4-K-(K-(Stp4-C)2>? After swelling 0.1 mmol of fmoc-Ala-Wang resin Low Load (Novabiochem) in DMF and cleavage of the fmoc protecting group by double treatment with 20% piperidine in DMF, a
solution of fmoc-Stp-OH (0.5 eq), PyBOP/HOBt (0.5 eq) and DIPEA (1.0 eq) in DMF was added to the resin and the vessel was agitated for 1 h. The unreacted amino groups were subsequently acetylated using 20 equivalents of acetic anhydride and 40 equivalents of DIPEA in DMF. Reaction progress was monitored by Kaiser test. After completion of the reaction the resin was washed five times with DMF and five times with DCM and dried for 12 h in vacuo. The loading of the resin was determined by spectrophotometric fmoc-quantification using 20% piperidine in DMF. The preloaded resin (0.01 mmol) was swollen in DMF for 1 h. After cleavage of the fmoc protecting group by double treatment with 20% piperidine in DMF, a solution of fmoc-Stp-OH (4 eq), PyBOP/HOBt (4 eq) and DIPEA (8 eq) in DMF was added to the resin and incubated for 1 h. In the next two coupling steps fmoc-Lys(fmoc)-OH was used as described above, 4 eq for the first coupling and 8 eq for the second. After removal of the fmoc protection group with 20% piperidine in DMF the synthesis was carried on according to
"General Procedure: Synthesis of four-arm polymers with four coupling domains exemplified by A-K-(K-(Stp4-C)2)2" using 16 eq building block 16 eq HOBt/PyBop and 32 eq DIPEA in each coupling step.
General Procedure: Synthesis of a five-arm polymer with five coupling domains: A-C-
Stp2-K-(K-(StP7-C)9.)7
The synthesis was performed according to the "General Procedure: Synthesis of five-arm polymers with four coupling domains exemplified by A-Stp4-K-(K-(Stp4-C)2)2" with following modifications:
Instead of fmoc-Stp-OH fmoc-Cys(trt)-OH (0.5 eq.) was added in the first coupling step. After acetylation of free amino groups and determination of resin loading by spectrophotometric fmoc- quantification using 20% piperidine in DMF, the synthesis was carried on as described repeating each fmoc-Stp-OH coupling step once.


Figures 26 to 30 show Luciferase reporter gene silencing after transfection with four and five arm polymers.
Polymers containing histidines for increased buffering capacity at endosomal pH, improved proton-sponge effect and endosomal escape
Endosomal respectively lysosomal escape after internalization of nucleic acid carriers is crucial for the efficient delivery of the cargo to the target site. Besides the incorporation of hydrophobic domains for an improved lytic activity of the transfecting agent, an increased buffering capacity at endosomal/lysosomal pH can facilitate selective endosomal/lysosomal membrane disruption and subsequent nucleic acid delivery to the cytoplasm. Exceeding buffering of the acidic conditions within endo- or lysosomes leads to a reactionally augmented active transport of protons and corresponding chloride counter ions into the membrane vesicles. By implication of the resulting increased osmotic pressure water flows into the compartments and causes swelling and rupture of the endosomal/lysosomal membranes. By this means an exceeding buffering capacity at endosomal/lysosomal pH can cause selective burst of endo- and lysosomes. This so
called "proton sponge effect" is well-known and reported for certain established transfecting agents and accounts for their endosomal escape and efficient nucleic acid delivery.
Imidazole containing structures with pka values between 5 and 7 provide remarkable properties capable of augmenting the buffering capacity at endosomal/lysosomal pH. Native histidine with a pka value of around 6 provides ideal properties for this purpose and is convenient for its use in the described solid-phase synthesis. Therefore in one embodiment of the invention the oligo(alkyleneamino) acid polymers comprise numerous histidines within the sequence in order to increase the buffering capacity at endosomal/lysosomal pH and to achieve selective endosomal/lysosomal membrane disruption. Targeting Ligand - Polymer Conjugates compromising histidines for enhanced endosomal escape
Since the polymer conjugates with T-Shapes containing shielding and targeting domains have been demonstrated to be functional in receptor targeted DNA delivery together with the addition of endosome disruptive chloroquine, but lack sufficient endosomal escape performance by itself, corresponding T-Shapes were synthesized incorporating numerous histidines for an improved endosomal escape performance without addition of endosome disruptive agents. The targeting ligands B6 (for binding the transferrin receptor, HO-KRPGKAKHG), non-functional control conjugate (Ala) and CMP (for binding the hepatocyte growth factor receptor c-Met, HO- KHHHIHDHRSLSK) were synthesized as PEG-Polymer conjugates incorporating eleven histidines in the oligo(alkyleneamino) acid polymer backbone as described in the following. Luciferase reporter gene expression was investigated with and without use of chloroquine - according to "luciferase reporter gene expression" and "luciferase reporter gene expression using chloroquine for endosomal escape" and compared to the results of corresponding T-Shapes lacking histidines in the polymeric backbone. Results are shown in Figures 32- 34. General Procedure: Synthesis of T-Shapes containing histidines for augmented buffering capacity at endosomal/lysosomal pH: examplified by H0-KRPGKAKHG-PEG?4-HK(H-(STP- H C 9 ("B6-PEG24-K-(Stp4-C 2-HIS" :
After swelling 20 μηιοΐ of resin bound HO- KRPGKAKHG- NH2 (B6) in DMF for 30 min a solution of Fmoc-PEG24-OH (4 eq), DIPEA (8 eq), PyBOP (4 eq) and HOBt (4 eq) in
DCM/DMF (1: 1, v/v) were added to the resin and the vessel was agitated until Kaiser test
indicated complete conversion (60 min). The reaction solvent was drained and the resin was washed three times with DCM followed by three DMF washes. After removal of the Fmoc protective group by treatment with 20% piperidine in DMF Fmoc-His(Trt)-OH was introduced using standard coupling conditions ( AA/PyB op/HOBt/DIPEA, 4/4/4/8 eq in DMF) followed by Fmoc deprotection with 20% piperidine in DMF. Fmoc-Lys(Fmoc)-OH was introduced as a branching point using standard coupling conditions (AA/PyBop/HOBt/DIPEA, 4/4/4/8 eq in DMF). After Fmoc deprotection a solution of 8 equivalents Fmoc-His(Trt)-OH, 8 equivalents of PyBop/HOBt and 16 equivalents of DIPEA in DCM/DMF (1: 1) was added to the resin and the mixture was agitated until Kaisertest indicated complete conversion (60 min). After Fmoc deprotection a solution of 8 equivalents Fmoc-stp-OH, 8 equivalents of PyBop/HOBt and 16 equivalents of DIPEA in DMF was added to the resin and the mixture was agitated until Kaiser test indicated complete conversion (normally 30 min). After Fmoc deprotection, the coupling and deprotection cycles of Fmoc-His(Trt)-OH and Fmoc-Stp-OH were alternately repeated three times. Subsequently Fmoc-His(Trt)-OH was coupled once more under the mentioned conditions followed by Fmoc deprotection. Finally a solution of 8 equivalents Boc-Cys(Trt)-OH, 8 equivalents of PyBop/HOBt and 16 equivalents of DIPEA in DCM/DMF (1: 1) was added to the resin and the mixture was agitated until Kaiser test indicated complete conversion (60 min). The resin was washed and dried for 12 h over KOH in vacuo. The PAA was cleaved from the resin by suspending it in a solution of TFA/H20/TIS/EDT (92.5:2.5:2.5:2.5, v/v) for 1 h. The cleavage solution was collected by filtration and the resin washed twice with TFA and once with DCM. The solution was concentrated and dropped slowly in a 1: 1 mixture (40 ml) of cooled (0° C) MTBE and n-hexane. The resulting precipitate was centrifuged at 4° C for 10 min (2000-3000 rpm). The solvents were decanted and the pellet was washed twice with ice-cold MTBE. The resulting pellet was dissolved in 30% acetonitrile in water and lyophilized. Synthesis of HO-KrPEG94-HK(H-(Stp-H)4-C)zl-HHHIHDHRSLSK ("CMP-PEG24-K-(Stp4- Q2-HIS")
After swelling 20 μπιοΐ of resin bound K(ivDDE) -HHHIHDHRS LS K-B oc (CMP) in DMF for 30 min the ivDDE protecting group was removed with 2% hydrazine monohydrate in DMF (v/v) (5-10 times for 5 min) until no significant A300 was measurable in the deprotection mixture. In between the deprotection-steps the resin was washed twice with DMF. Subsequent synthesis steps were accomplished according to„Synthesis of T-Shapes containing histidines for
augmented buffering capacity at endosomal/lysomal pH: examplified by HO-KRPGKAKHG- PEG24-HK((H-STP)4-H-C)2".
Synthesis of HO-A-PEG^-HKiH-iStp-HWQ? ("A-PEG24-K-(Stp4-C)2-HIS")
HO-A-PEG24-HK(H-(Stp-H)4-C)2 was synthesized according to„Synthesis of T-Shapes containing histidines for augmented buffering capacity at endosomal/lysomal pH: examplified by HO-KRPGKAKHG-PEG24-HK((H-STP)4-H-C)2" with following modification: Resin bound Ala was used instead of HO- KRPGKAKHG .
Table 8- Exemplified sequences with enhanced endosomolytic activity due to histidine incorporation Sequences (from C- to N-terminus, as assembled on the solid support). Abbreviations: K = Lysine, H = Histidine, Stp = Succinyltetraethylenepentamine, A = Alanine, C = Cysteine, PEG24 = polyethylene glycol with 24 subunits, CMP = peptide for HGF receptor targeting, B6 = peptide for transferrin receptor targeting.

Figures 31 to 34 show Luciferase reporter gene expression after transfection with polymers with enhanced endosomolytic activity due to histidine incorporation.
Synthesis of Azido-PEG^-FolA:
Azido-PEG24-FolA was synthesized according to "General procedure: Synthesis of pegylated targeting domain with N-terminal ligand, exemplified by Cys-PEG24-FolicAcid" with following modification: Instead of the resin-bound Cys(trt)-NH2, (S)-5-Azido-2-(Fmoc-amino)pentanoic acid was used. For cleavage a solution of TFA/H20/TIS (95:2.5:2.5) for lh was used. No SEC purification was performed.
Synthesis of Azido-PEG^-NH?:
Azido-PEG24-NH2 was synthesized according to "Synthesis of Azido-PEG24-Folic Acid" stopping the synthesis after deprotection of the PEG building block.
Synthesis of targeted siRNA using azide-alkyne cycloaddition; exemplified by Folic
Acid-PEG24-triazol-s-s-siRNA:
74 nmol siRNA (modified with a Hexynyl-ss-C6-linker at the 5' end of its sense strand) in ΙΟΟμί H20 were mixed with 296 nmol Azido-PEG24-FolA in 300μΕ DMSO/tertiary Butanol (3: 1 ; v/v) and 60 μΙ, TBTA/CuBr solution (0,1M TBTA/Ο,ΙΜ CuBr 2: 1 (v/v) each in DMSO/tertiary Butanol (3: 1 (v/v)) . After 3h at 37°C under constant shaking, the mixture was filled up with 1.5mL buffer A (20mM Hepes, pH 6.5, 30% acetonitrile) and centrifuged for 5min at 13000rpm. The targeted siRNA in the supernatant was purified using a lmL ResourceQ column connected to an Akta basic system. For that purpose the supernatant was loaded onto the column using the same buffer A containing 20mM NaCl. After unbound material was washed away with buffer A containing 200mM NaCl, the product was eluted setting a gradient of lOmM NaCl/min and a flow of lmL/min. Resulting fractions were analyzed on a 2.5% agarose gel. Samples containing gel-retarded siRNA compared to unmodified control were pooled.
Synthesis of pegylated siRNA using azide-alkyne cycloaddition:
Pegylated siRNA was synthesised according to "Synthesis of targeted siRNA using azide-alkyne cycloaddition; exemplified by FolA-PEG24-triazol-s-s- siRNA" using Azido-PEG24 instead of Azido-PEG24-FolA.
Figure 35 shows Luciferase reporter gene silencing after transfection with FolA-PEG24-triazol- s-s-siRNA
Claims
Claims
1. A polymer formed by 2-60 oligo(alkyleneamino) acid units of the general structure

Z is chosen from either an alkyl group with the general structure L , wherein c
NH2 is 0-8; or an amino group with the eneral structure , wherein n is 1-7; or an aromatic
group with the structure
 either CH2 or C=0; and a is 1-7 and b is 2-7.
either CH2 or C=0; and a is 1-7 and b is 2-7.
The polymer of claim 1, formed by 2-10 oligo(alkyleneamino) acid units, preferably by 2-5 oligo(alkyleneamino) acid units.
The polymer of any of claims 1 to 2 wherein Z is an alkyl group with the general structure , wherein c is 0-8.
4. The polymer of any of claims 1 to 3, wherein R is C=0.
5. The polymer of any of claims 1 to 4, wherein b is 2 .
6. The polymer of any of claims 1 to 5, further comprising a hydrophobic domain
covalently linked to the polymer.
7. The polymer of claim 6, wherein the hydrophobic domain comprises 1-10 fatty acids.
8. The polymer of any of claims 6 to 7, wherein the hydrophobic domain comprises 2 fatty acids.
9. The polymer of any of claims 6 to 8, wherein the fatty acid is chosen from the group of butyric acid, caprylic acid, myristic acid, oleic acid, arachidic acid, stearic acid, lauric acid and palmitic acid.
10. The polymer of any of claims 6 to 9, wherein the hydrophobic domain is covalently linked to an amine group at the N-terminus of the polymer.
11. The polymer of any of claims 6 to 10, wherein the hydrophobic domain is covalently linked to an amine group at the N-terminus of the polymer via a linker moiety, wherein the linker moiety is an amino acid.
12. The polymer of any of claims 1 to 5, wherein the polymer is branched.
13. The branched polymer of claim 12, wherein two polymer chains according to claims 1 to 5 are connected via a linker moiety, wherein the linker moiety is an amino acid.
14. The branched polymer of claim 12, further comprising a hydrophobic domain
according to claims 7-9 covalently linked to the amine group of the linker moiety.
15. The polymer of any of claims 1 tol4, further comprising a headgroup covalently linked to one terminus of the polymer.
16. The polymer of claim 15, wherein the headgroup is covalently linked to the carboxyl group at the C-terminus.
17. The polymer of any of claims 15 or 16 wherein the headgroup is a polar positively charged head group.
18. The polymer of any of claims 15 to 17 wherein the polar positively charged head group is chosen from cysteine, lysine, arginine, histidine or a combination thereof.
19. The polymer of any of claims 15 to 18 wherein the headgroup is a coupling or
crosslinking domain.
20. The polymer of claim 19 wherein the coupling or crosslinking domain is selected from at least one of the group of a cysteine group, an imine group, a hydrazone group, a tyrosine trimer, a tyrosine oligomer or a ureido-pyrimidinone group
21. The polymer of any of claims 1 to 20, further comprising a targeting ligand, chosen from the group of a peptide, a protein, an antibody, a vitamin, or a synthetic chemical receptor-binding ligand.
22. The polymer of any of claims 1 to 21, further comprising a ligand for mediating
endosomal escape selected from the group of imidazoles or an endosomolytic peptide
23. The polymer of any of claims 1 to 22, further comprising a shielding moiety.
24. The polymer of any of claims 1 to 23, wherein the polymer is complexed with a nucleic acid.
25. The polymer of claim 24, wherein the nucleic acid is a siRNA.
26. Use of the polymer of any of claims 1 to 25 for the delivery of nucleic acids.
27. A composition comprising the polymer of any of claims 1 to 23 and a nucleic acid.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP10165502.5 | 2010-06-10 | ||
| EP10165502A EP2395041A1 (en) | 2010-06-10 | 2010-06-10 | Polymers for delivery of nucleic acids |
| EP10191030.5 | 2010-11-12 | ||
| EP10191030 | 2010-11-12 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011154331A1 true WO2011154331A1 (en) | 2011-12-15 |
Family
ID=44303305
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2011/059239 WO2011154331A1 (en) | 2010-06-10 | 2011-06-06 | Polymers for delivery of nucleic acids |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2011154331A1 (en) |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8426554B2 (en) | 2010-12-29 | 2013-04-23 | Arrowhead Madison Inc. | In vivo polynucleotide delivery conjugates having enzyme sensitive linkages |
| WO2014207231A1 (en) | 2013-06-28 | 2014-12-31 | Ethris Gmbh | Compositions for introducing rna into cells |
| CN104356231A (en) * | 2014-11-10 | 2015-02-18 | 北海开元生物科技有限公司 | Method for effectively removing human immune globulin polymer |
| WO2015128030A1 (en) | 2014-02-26 | 2015-09-03 | Ethris Gmbh | Compositions for gastrointestinal administration of rna |
| WO2016028149A1 (en) * | 2014-08-20 | 2016-02-25 | Tu Eindhoven | Ureidopyrimidone supramolecular complexes for compound delivery into cells |
| EP3034539A1 (en) | 2014-12-19 | 2016-06-22 | Ethris GmbH | Compositions for introducing nucleic acid into cells |
| CN106362163A (en) * | 2016-08-26 | 2017-02-01 | 上海交通大学 | Nucleic acid delivery nano systems having double-targeting function |
| US9809632B2 (en) | 2013-10-23 | 2017-11-07 | University Of Washington Through Its Center For Commercialization | Universal protein tag for double stranded nucleic acid delivery |
| WO2019128611A1 (en) * | 2017-12-29 | 2019-07-04 | Suzhou Ribo Life Science Co., Ltd. | Conjugates and preparation and use thereof |
| US10543232B2 (en) | 2014-05-14 | 2020-01-28 | Targimmune Therapeutics Ag | Polyplex of double-stranded RNA and polymeric conjugate |
| WO2021037205A1 (en) * | 2019-08-29 | 2021-03-04 | 苏州瑞博生物技术股份有限公司 | Compound and drug conjugate, and preparation method and use thereof |
| WO2022216977A1 (en) * | 2021-04-07 | 2022-10-13 | Batelle Memorial Institute | Rapid design, build, test, and learn technologies for identifying and using non-viral carriers |
| US11492620B2 (en) | 2017-12-01 | 2022-11-08 | Suzhou Ribo Life Science Co., Ltd. | Double-stranded oligonucleotide, composition and conjugate comprising double-stranded oligonucleotide, preparation method thereof and use thereof |
| RU2795400C2 (en) * | 2017-12-29 | 2023-05-03 | Сучжоу Рибо Лайф Сайенс Ко., Лтд | Conjugates and their obtaining and application |
| US11660347B2 (en) | 2017-12-01 | 2023-05-30 | Suzhou Ribo Life Science Co., Ltd. | Nucleic acid, composition and conjugate containing same, preparation method, and use thereof |
| WO2023239922A1 (en) * | 2022-06-09 | 2023-12-14 | Battelle Memorial Institute | Non-viral delivery compositions and screening methods |
| US11896674B2 (en) | 2018-09-30 | 2024-02-13 | Suzhou Ribo Life Science Co., Ltd. | SiRNA conjugate, preparation method therefor and use thereof |
| US11918600B2 (en) | 2018-08-21 | 2024-03-05 | Suzhou Ribo Life Science Co., Ltd. | Nucleic acid, pharmaceutical composition and conjugate containing nucleic acid, and use thereof |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1509967A (en) * | 1974-07-19 | 1978-05-10 | Basf Ag | Crosslinked polyamidoamine condensation products for papermaking |
| US5948902A (en) | 1997-11-20 | 1999-09-07 | South Alabama Medical Science Foundation | Antisense oligonucleotides to human serine/threonine protein phosphatase genes |
| EP1400551A1 (en) * | 1998-08-28 | 2004-03-24 | Gryphon Sciences | Polyamide chains of precise length and their conjugates with proteins |
| US20040198947A1 (en) * | 2003-04-01 | 2004-10-07 | Hercules Incorporated | Synthesis of high solids resins from amine terminated polyamides |
-
2011
- 2011-06-06 WO PCT/EP2011/059239 patent/WO2011154331A1/en active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1509967A (en) * | 1974-07-19 | 1978-05-10 | Basf Ag | Crosslinked polyamidoamine condensation products for papermaking |
| US5948902A (en) | 1997-11-20 | 1999-09-07 | South Alabama Medical Science Foundation | Antisense oligonucleotides to human serine/threonine protein phosphatase genes |
| EP1400551A1 (en) * | 1998-08-28 | 2004-03-24 | Gryphon Sciences | Polyamide chains of precise length and their conjugates with proteins |
| US20040198947A1 (en) * | 2003-04-01 | 2004-10-07 | Hercules Incorporated | Synthesis of high solids resins from amine terminated polyamides |
Non-Patent Citations (13)
| Title |
|---|
| C LOCHER: "Enhancement of a human immunodeficiency virus env DNA vaccine using a novel polycationic nanoparticle formulation", IMMUNOLOGY LETTERS, vol. 90, no. 2-3, 15 December 2003 (2003-12-15), pages 67 - 70, XP055003774, ISSN: 0165-2478, DOI: 10.1016/j.imlet.2003.02.001 * |
| COTTEN M, LANGLE-ROUAULT F, KIRLAPPOS H, WAGNER E, MECHTLER K, ZENKE M, BEUG H, BIRNSTIEL, ML, PROC. NATL. ACAD. SCI. U.S.A, vol. 87, 1990, pages 4033 - 4037 |
| DANKERS, MEIJER: "Convenient Solid-Phase Synthesis of Ureido-Pyrimidinone Modified Peptides", EUR. J. ORG. CHEM., 2007 |
| HARTMANN L, HAFELE S, PESCHKA-SUSS R, ANTONIETTI M, BORNER HG: "Tailor-Made Poly(amidoamine)s for Controlled Complexation and Condensation of DNA", CHEMISTRY, vol. 14, no. 7, 2008, pages 2025 - 2033 |
| HAUBNER R, GRATIAS R, DIEFENBACH B, GOODMAN SL, JONCZYK A, KESSLER H., J. AM. CHEM. SOC., vol. 118, 1996, pages 7461 - 7472 |
| LAURA HARTMANN, STEFANIE HÄFELE, REGINE PESCHKA-SÜSS, MARKUS ANTONIETTI, HANS G. BÖRNER: "Tailor-Made Poly(amidoamine)s for Controlled Complexation and Condensation of DNA", CHEM. EUR. J., vol. 14, no. 7, 7 February 2008 (2008-02-07), pages 2025 - 2033, XP002595989, DOI: 10.1002/chem.200701223 * |
| MEYER M, DOHMEN C, PHILIPP A, KIENER D, MAIWALD G, SCHEU C, OGRIS M, WAGNER E., MOL PHARM., vol. 6, 2009, pages 752 - 62 |
| PAOLO FERRUTI ET AL: "POLY(AMIDO-AMINE)S: BIOMEDICAL APPLICATIONS", MACROMOLECULAR: RAPID COMMUNICATIONS, WILEY VCH VERLAG, WEINHEIM, DE LNKD- DOI:10.1002/1521-3927(20020401)23:5/6<,332::AID-MARC332>,3.0.CO,2-I, vol. 23, 1 January 2002 (2002-01-01), pages 332 - 355, XP008077050, ISSN: 1022-1336 * |
| PLANK C, OBERHAUSER B, MECHTLER K, KOCH C, WAGNER E., J BIOL CHEM., vol. 269, 1994, pages 12918 - 24 |
| PLANK C, ZATLOUKAL K, COTTEN M, MECHTLER K, WAGNER E, BIOCONJUG CHEM., vol. 3, 1992, pages 533 - 9 |
| SABRINA CARROCCIO, CONCETTO PUGLISI, GIORGIO MONTAUDO: "MALDI Investigation of the Photooxidation of Nylon-66", MACROMOLECULES, vol. 37, no. 16, 14 July 2004 (2004-07-14), pages 6037 - 6049, XP002595990, DOI: 10.1021/ma049521n * |
| WANG XL, RAMUSOVIC S, NGUYEN T, LU ZR: "Novel polymerizable surfactants with pH-sensitive amphiphilicity and cell menrane disruption for efficient siRNA delivery", BIOCONJUGATE CHEMISTRY, vol. 18, 2007, pages 2169 - 2177 |
| ZOU SM, ERBACHER P, REMY JS, BEHR JP, J GENE MED., vol. 2, no. 2, March 2000 (2000-03-01), pages 128 - 34 |
Cited By (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9198947B2 (en) | 2010-12-29 | 2015-12-01 | Hoffmann-La Roche Inc. | Small molecule conjugates for intracellular delivery of nucleic acids |
| US8426554B2 (en) | 2010-12-29 | 2013-04-23 | Arrowhead Madison Inc. | In vivo polynucleotide delivery conjugates having enzyme sensitive linkages |
| US9301990B2 (en) | 2010-12-29 | 2016-04-05 | Hoffmann-La Roche, Inc. | Small molecule conjugates for intracellular delivery of biologically active compounds |
| US9968647B2 (en) | 2010-12-29 | 2018-05-15 | Hoffmann-La Roche Inc. | Small molecule conjugates for intracellular delivery of biologically active compounds |
| WO2014207231A1 (en) | 2013-06-28 | 2014-12-31 | Ethris Gmbh | Compositions for introducing rna into cells |
| EA036400B1 (en) * | 2013-06-28 | 2020-11-06 | Этрис Гмбх | Compositions for introducing rna into cells |
| US9809632B2 (en) | 2013-10-23 | 2017-11-07 | University Of Washington Through Its Center For Commercialization | Universal protein tag for double stranded nucleic acid delivery |
| WO2015128030A1 (en) | 2014-02-26 | 2015-09-03 | Ethris Gmbh | Compositions for gastrointestinal administration of rna |
| US10543232B2 (en) | 2014-05-14 | 2020-01-28 | Targimmune Therapeutics Ag | Polyplex of double-stranded RNA and polymeric conjugate |
| US11298376B2 (en) | 2014-05-14 | 2022-04-12 | Targimmune Therapeutics Ag | Method of treating cancer |
| WO2016028149A1 (en) * | 2014-08-20 | 2016-02-25 | Tu Eindhoven | Ureidopyrimidone supramolecular complexes for compound delivery into cells |
| CN104356231A (en) * | 2014-11-10 | 2015-02-18 | 北海开元生物科技有限公司 | Method for effectively removing human immune globulin polymer |
| EP3034539A1 (en) | 2014-12-19 | 2016-06-22 | Ethris GmbH | Compositions for introducing nucleic acid into cells |
| CN106362163A (en) * | 2016-08-26 | 2017-02-01 | 上海交通大学 | Nucleic acid delivery nano systems having double-targeting function |
| US11660347B2 (en) | 2017-12-01 | 2023-05-30 | Suzhou Ribo Life Science Co., Ltd. | Nucleic acid, composition and conjugate containing same, preparation method, and use thereof |
| US11492620B2 (en) | 2017-12-01 | 2022-11-08 | Suzhou Ribo Life Science Co., Ltd. | Double-stranded oligonucleotide, composition and conjugate comprising double-stranded oligonucleotide, preparation method thereof and use thereof |
| US11633482B2 (en) | 2017-12-29 | 2023-04-25 | Suzhou Ribo Life Science Co., Ltd. | Conjugates and preparation and use thereof |
| RU2795400C2 (en) * | 2017-12-29 | 2023-05-03 | Сучжоу Рибо Лайф Сайенс Ко., Лтд | Conjugates and their obtaining and application |
| WO2019128611A1 (en) * | 2017-12-29 | 2019-07-04 | Suzhou Ribo Life Science Co., Ltd. | Conjugates and preparation and use thereof |
| US11918600B2 (en) | 2018-08-21 | 2024-03-05 | Suzhou Ribo Life Science Co., Ltd. | Nucleic acid, pharmaceutical composition and conjugate containing nucleic acid, and use thereof |
| US11896674B2 (en) | 2018-09-30 | 2024-02-13 | Suzhou Ribo Life Science Co., Ltd. | SiRNA conjugate, preparation method therefor and use thereof |
| CN113891892A (en) * | 2019-08-29 | 2022-01-04 | 苏州瑞博生物技术股份有限公司 | Compound and drug conjugate, preparation method and application thereof |
| WO2021037205A1 (en) * | 2019-08-29 | 2021-03-04 | 苏州瑞博生物技术股份有限公司 | Compound and drug conjugate, and preparation method and use thereof |
| CN113891892B (en) * | 2019-08-29 | 2024-01-30 | 苏州瑞博生物技术股份有限公司 | Compounds and drug conjugates, methods of preparation and uses thereof |
| WO2022216977A1 (en) * | 2021-04-07 | 2022-10-13 | Batelle Memorial Institute | Rapid design, build, test, and learn technologies for identifying and using non-viral carriers |
| WO2023239922A1 (en) * | 2022-06-09 | 2023-12-14 | Battelle Memorial Institute | Non-viral delivery compositions and screening methods |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2011154331A1 (en) | Polymers for delivery of nucleic acids | |
| Kim et al. | Arginine-conjugated polypropylenimine dendrimer as a non-toxic and efficient gene delivery carrier | |
| Li et al. | Copolymer of poly (ethylene glycol) and poly (L-lysine) grafting polyethylenimine through a reducible disulfide linkage for siRNA delivery | |
| JP5481614B2 (en) | Block copolymer introduced with phenylboronic acid group and use thereof | |
| US8153110B2 (en) | Environment-responding siRNA carrier using disulfide-cross-linked polymeric micelle | |
| Lin et al. | Structural contributions of blocked or grafted poly (2-dimethylaminoethyl methacrylate) on PEGylated polycaprolactone nanoparticles in siRNA delivery | |
| AU2014306021B2 (en) | Polyconjugates for delivery of RNAi triggers to tumor cells in vivo | |
| Cheng et al. | The effect of guanidinylation of PEGylated poly (2-aminoethyl methacrylate) on the systemic delivery of siRNA | |
| US20140140929A1 (en) | Chemically modified cell-penetrating peptides for improved delivery of gene modulating compounds | |
| Chang et al. | Development of lysine–histidine dendron modified chitosan for improving transfection efficiency in HEK293 cells | |
| AU2014300980A1 (en) | Compositions for introducing RNA into cells | |
| Qin et al. | Development of cholesteryl peptide micelles for siRNA delivery | |
| Pan et al. | Short multi-armed polylysine-graft-polyamidoamine copolymer as efficient gene vectors | |
| Zavradashvili et al. | Library of cationic polymers composed of polyamines and arginine as gene transfection agents | |
| US20230313181A1 (en) | Histidine-lysine polymers and methods for delivering mrna using the same | |
| Begum et al. | Bombesin/oligoarginine fusion peptides for gastrin releasing peptide receptor (GRPR) targeted gene delivery | |
| Han et al. | A comprehensive review on histone-mediated transfection for gene therapy | |
| Yang et al. | Synthetic helical polypeptide as a gene transfection enhancer | |
| JP2002526456A (en) | Cationic compounds and their use as macromolecular carriers | |
| WO2021134023A2 (en) | Compositions and methods for nucleic acid delivery | |
| KR20220110174A (en) | Tumor-Targeting Polypeptide Nanoparticle Delivery Systems for Nucleic Acid Therapeutics | |
| WO2019046937A1 (en) | Tri-modal nucleic acid delivery systems | |
| Grijalvo et al. | Synthesis and in vitro inhibition properties of oligonucleotide conjugates carrying amphipathic proline-rich peptide derivatives of the sweet arrow peptide (SAP) | |
| EP2395041A1 (en) | Polymers for delivery of nucleic acids | |
| Sun et al. | New amphiphilic N-phosphoryl oligopeptides designed for gene delivery |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11723464 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11723464 Country of ref document: EP Kind code of ref document: A1 |