WO2011072241A1 - 4 -oxo- ih -quinoline- 3 - carboxamides as modulators of atp -binding cassette transporters - Google Patents
4 -oxo- ih -quinoline- 3 - carboxamides as modulators of atp -binding cassette transporters Download PDFInfo
- Publication number
- WO2011072241A1 WO2011072241A1 PCT/US2010/059920 US2010059920W WO2011072241A1 WO 2011072241 A1 WO2011072241 A1 WO 2011072241A1 US 2010059920 W US2010059920 W US 2010059920W WO 2011072241 A1 WO2011072241 A1 WO 2011072241A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- oxo
- carboxamide
- phenyl
- hydroxy
- quinoline
- Prior art date
Links
- 102000005416 ATP-Binding Cassette Transporters Human genes 0.000 title abstract description 38
- 108010006533 ATP-Binding Cassette Transporters Proteins 0.000 title abstract description 38
- 239000000203 mixture Substances 0.000 claims abstract description 307
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 95
- 201000010099 disease Diseases 0.000 claims abstract description 77
- 238000000034 method Methods 0.000 claims abstract description 53
- 239000012634 fragment Substances 0.000 claims abstract description 14
- CVAWACBLSANHSQ-UHFFFAOYSA-N 4-oxo-1h-quinoline-3-carboxamide Chemical class C1=CC=CC2=C(O)C(C(=O)N)=CN=C21 CVAWACBLSANHSQ-UHFFFAOYSA-N 0.000 claims abstract description 4
- -1 4-oxo- lH-quinolin-3-yl Chemical group 0.000 claims description 175
- 150000001875 compounds Chemical class 0.000 claims description 165
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 105
- 239000002253 acid Substances 0.000 claims description 80
- 230000000694 effects Effects 0.000 claims description 64
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 56
- 230000002829 reductive effect Effects 0.000 claims description 48
- 239000003795 chemical substances by application Substances 0.000 claims description 39
- 150000003857 carboxamides Chemical class 0.000 claims description 37
- 201000003883 Cystic fibrosis Diseases 0.000 claims description 32
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 30
- 230000035772 mutation Effects 0.000 claims description 30
- 150000003839 salts Chemical class 0.000 claims description 27
- 230000007812 deficiency Effects 0.000 claims description 25
- 208000006673 asthma Diseases 0.000 claims description 24
- 102100026383 Vasopressin-neurophysin 2-copeptin Human genes 0.000 claims description 23
- 201000010064 diabetes insipidus Diseases 0.000 claims description 23
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 23
- 201000009266 primary ciliary dyskinesia Diseases 0.000 claims description 21
- 206010006451 bronchitis Diseases 0.000 claims description 20
- 108090000623 proteins and genes Proteins 0.000 claims description 20
- 102100025027 E3 ubiquitin-protein ligase TRIM69 Human genes 0.000 claims description 18
- 101000830203 Homo sapiens E3 ubiquitin-protein ligase TRIM69 Proteins 0.000 claims description 18
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 18
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 claims description 17
- 208000025678 Ciliary Motility disease Diseases 0.000 claims description 17
- 206010033645 Pancreatitis Diseases 0.000 claims description 17
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 15
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 claims description 13
- 239000012472 biological sample Substances 0.000 claims description 12
- 208000011580 syndromic disease Diseases 0.000 claims description 12
- 206010006474 Bronchopulmonary aspergillosis allergic Diseases 0.000 claims description 11
- 208000003556 Dry Eye Syndromes Diseases 0.000 claims description 11
- 206010014561 Emphysema Diseases 0.000 claims description 11
- 208000006778 allergic bronchopulmonary aspergillosis Diseases 0.000 claims description 11
- 230000007547 defect Effects 0.000 claims description 11
- 206010006458 Bronchitis chronic Diseases 0.000 claims description 10
- 206010010356 Congenital anomaly Diseases 0.000 claims description 10
- 208000015439 Lysosomal storage disease Diseases 0.000 claims description 10
- 208000007451 chronic bronchitis Diseases 0.000 claims description 10
- 206010012601 diabetes mellitus Diseases 0.000 claims description 10
- HWYHDWGGACRVEH-UHFFFAOYSA-N n-methyl-n-(4-pyrrolidin-1-ylbut-2-ynyl)acetamide Chemical compound CC(=O)N(C)CC#CCN1CCCC1 HWYHDWGGACRVEH-UHFFFAOYSA-N 0.000 claims description 10
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 9
- 238000012545 processing Methods 0.000 claims description 9
- BLTDCIWCFCUQCB-UHFFFAOYSA-N quinoline-3-carboxamide Chemical compound C1=CC=CC2=CC(C(=O)N)=CN=C21 BLTDCIWCFCUQCB-UHFFFAOYSA-N 0.000 claims description 9
- 230000000306 recurrent effect Effects 0.000 claims description 9
- 206010010774 Constipation Diseases 0.000 claims description 8
- 206010067265 Heterotaxia Diseases 0.000 claims description 8
- 201000000101 Hyperekplexia Diseases 0.000 claims description 8
- 206010058271 Hyperexplexia Diseases 0.000 claims description 8
- 208000031733 Situs inversus totalis Diseases 0.000 claims description 8
- 208000021386 Sjogren Syndrome Diseases 0.000 claims description 8
- 230000002146 bilateral effect Effects 0.000 claims description 8
- 208000008797 situs inversus Diseases 0.000 claims description 8
- 210000001177 vas deferen Anatomy 0.000 claims description 8
- 102100034452 Alternative prion protein Human genes 0.000 claims description 7
- 208000019693 Lung disease Diseases 0.000 claims description 7
- 208000035467 Pancreatic insufficiency Diseases 0.000 claims description 7
- 108091000054 Prion Proteins 0.000 claims description 7
- 150000002632 lipids Chemical class 0.000 claims description 7
- 208000024827 Alzheimer disease Diseases 0.000 claims description 6
- 102100032187 Androgen receptor Human genes 0.000 claims description 6
- 206010013883 Dwarfism Diseases 0.000 claims description 6
- 208000024720 Fabry Disease Diseases 0.000 claims description 6
- 201000011240 Frontotemporal dementia Diseases 0.000 claims description 6
- 206010019860 Hereditary angioedema Diseases 0.000 claims description 6
- 101000775732 Homo sapiens Androgen receptor Proteins 0.000 claims description 6
- 208000000563 Hyperlipoproteinemia Type II Diseases 0.000 claims description 6
- 208000000038 Hypoparathyroidism Diseases 0.000 claims description 6
- 208000022559 Inflammatory bowel disease Diseases 0.000 claims description 6
- 102100024640 Low-density lipoprotein receptor Human genes 0.000 claims description 6
- 208000007466 Male Infertility Diseases 0.000 claims description 6
- 208000002678 Mucopolysaccharidoses Diseases 0.000 claims description 6
- 208000012902 Nervous system disease Diseases 0.000 claims description 6
- 208000025966 Neurological disease Diseases 0.000 claims description 6
- 206010031243 Osteogenesis imperfecta Diseases 0.000 claims description 6
- 208000018737 Parkinson disease Diseases 0.000 claims description 6
- 208000000609 Pick Disease of the Brain Diseases 0.000 claims description 6
- 201000005660 Protein C Deficiency Diseases 0.000 claims description 6
- 206010045261 Type IIa hyperlipidaemia Diseases 0.000 claims description 6
- 208000006269 X-Linked Bulbo-Spinal Atrophy Diseases 0.000 claims description 6
- 239000003937 drug carrier Substances 0.000 claims description 6
- 201000001386 familial hypercholesterolemia Diseases 0.000 claims description 6
- 208000013746 hereditary thrombophilia due to congenital protein C deficiency Diseases 0.000 claims description 6
- 238000001727 in vivo Methods 0.000 claims description 6
- 201000001441 melanoma Diseases 0.000 claims description 6
- 206010028093 mucopolysaccharidosis Diseases 0.000 claims description 6
- 230000004770 neurodegeneration Effects 0.000 claims description 6
- 208000015122 neurodegenerative disease Diseases 0.000 claims description 6
- 206010003591 Ataxia Diseases 0.000 claims description 5
- 206010062264 Congenital hyperthyroidism Diseases 0.000 claims description 5
- 206010013774 Dry eye Diseases 0.000 claims description 5
- 206010051125 Hypofibrinogenaemia Diseases 0.000 claims description 5
- 206010068871 Myotonic dystrophy Diseases 0.000 claims description 5
- 208000001132 Osteoporosis Diseases 0.000 claims description 5
- SMWDFEZZVXVKRB-UHFFFAOYSA-N anhydrous quinoline Natural products N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 claims description 5
- 230000007613 environmental effect Effects 0.000 claims description 5
- 238000000338 in vitro Methods 0.000 claims description 5
- 230000020978 protein processing Effects 0.000 claims description 5
- 210000001215 vagina Anatomy 0.000 claims description 5
- 208000009575 Angelman syndrome Diseases 0.000 claims description 4
- 206010002961 Aplasia Diseases 0.000 claims description 4
- 208000000668 Chronic Pancreatitis Diseases 0.000 claims description 4
- 208000024940 Dent disease Diseases 0.000 claims description 4
- 208000003892 Kartagener syndrome Diseases 0.000 claims description 4
- 208000029725 Metabolic bone disease Diseases 0.000 claims description 4
- 208000010316 Myotonia congenita Diseases 0.000 claims description 4
- 206010028980 Neoplasm Diseases 0.000 claims description 4
- 206010049088 Osteopenia Diseases 0.000 claims description 4
- 206010033647 Pancreatitis acute Diseases 0.000 claims description 4
- 206010033649 Pancreatitis chronic Diseases 0.000 claims description 4
- 208000025237 Polyendocrinopathy Diseases 0.000 claims description 4
- 208000024777 Prion disease Diseases 0.000 claims description 4
- 208000035954 Thomsen and Becker disease Diseases 0.000 claims description 4
- 201000003229 acute pancreatitis Diseases 0.000 claims description 4
- 239000002671 adjuvant Substances 0.000 claims description 4
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 claims description 4
- 239000003242 anti bacterial agent Substances 0.000 claims description 4
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 claims description 4
- 201000001883 cholelithiasis Diseases 0.000 claims description 4
- 230000001886 ciliary effect Effects 0.000 claims description 4
- 206010015037 epilepsy Diseases 0.000 claims description 4
- 208000001130 gallstones Diseases 0.000 claims description 4
- 208000021302 gastroesophageal reflux disease Diseases 0.000 claims description 4
- 235000016709 nutrition Nutrition 0.000 claims description 4
- 108010040003 polyglutamine Proteins 0.000 claims description 4
- 229920000155 polyglutamine Polymers 0.000 claims description 4
- 201000002212 progressive supranuclear palsy Diseases 0.000 claims description 4
- 201000009890 sinusitis Diseases 0.000 claims description 4
- 239000000779 smoke Substances 0.000 claims description 4
- 208000012904 Bartter disease Diseases 0.000 claims description 3
- 208000010062 Bartter syndrome Diseases 0.000 claims description 3
- 206010008609 Cholangitis sclerosing Diseases 0.000 claims description 3
- 208000019683 Gorham-Stout disease Diseases 0.000 claims description 3
- 208000033981 Hereditary haemochromatosis Diseases 0.000 claims description 3
- 208000008955 Mucolipidoses Diseases 0.000 claims description 3
- 206010072928 Mucolipidosis type II Diseases 0.000 claims description 3
- 208000004622 abetalipoproteinemia Diseases 0.000 claims description 3
- 239000002260 anti-inflammatory agent Substances 0.000 claims description 3
- 229940121363 anti-inflammatory agent Drugs 0.000 claims description 3
- 206010003246 arthritis Diseases 0.000 claims description 3
- 208000027157 chronic rhinosinusitis Diseases 0.000 claims description 3
- 230000002496 gastric effect Effects 0.000 claims description 3
- 208000019423 liver disease Diseases 0.000 claims description 3
- 230000036210 malignancy Effects 0.000 claims description 3
- 208000020460 mucolipidosis II alpha/beta Diseases 0.000 claims description 3
- 201000000742 primary sclerosing cholangitis Diseases 0.000 claims description 3
- 208000010157 sclerosing cholangitis Diseases 0.000 claims description 3
- 208000014644 Brain disease Diseases 0.000 claims description 2
- 208000031976 Channelopathies Diseases 0.000 claims description 2
- 208000032274 Encephalopathy Diseases 0.000 claims description 2
- 208000007984 Female Infertility Diseases 0.000 claims description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 claims description 2
- 206010021928 Infertility female Diseases 0.000 claims description 2
- YNPNZTXNASCQKK-UHFFFAOYSA-N Phenanthrene Natural products C1=CC=C2C3=CC=CC=C3C=CC2=C1 YNPNZTXNASCQKK-UHFFFAOYSA-N 0.000 claims description 2
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 claims description 2
- 229960005475 antiinfective agent Drugs 0.000 claims description 2
- 239000004599 antimicrobial Substances 0.000 claims description 2
- 230000003115 biocidal effect Effects 0.000 claims description 2
- 239000003172 expectorant agent Substances 0.000 claims description 2
- 229940066491 mucolytics Drugs 0.000 claims description 2
- 210000004291 uterus Anatomy 0.000 claims description 2
- 102000027257 transmembrane receptors Human genes 0.000 claims 4
- 108091008578 transmembrane receptors Proteins 0.000 claims 4
- 239000008194 pharmaceutical composition Substances 0.000 claims 3
- RUZLIIJDZBWWSA-INIZCTEOSA-N methyl 2-[[(1s)-1-(7-methyl-2-morpholin-4-yl-4-oxopyrido[1,2-a]pyrimidin-9-yl)ethyl]amino]benzoate Chemical group COC(=O)C1=CC=CC=C1N[C@@H](C)C1=CC(C)=CN2C(=O)C=C(N3CCOCC3)N=C12 RUZLIIJDZBWWSA-INIZCTEOSA-N 0.000 claims 2
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 claims 1
- FONBNPKVUAKNKL-UHFFFAOYSA-N 5-hydroxy-n-(3-hydroxy-4-propan-2-ylphenyl)-4-oxo-1h-quinoline-3-carboxamide Chemical compound C1=C(O)C(C(C)C)=CC=C1NC(=O)C1=CNC2=CC=CC(O)=C2C1=O FONBNPKVUAKNKL-UHFFFAOYSA-N 0.000 claims 1
- BNERSGDXTXPIBE-UHFFFAOYSA-N 6-fluoro-n-[3-hydroxy-4-(trifluoromethyl)phenyl]-4-oxo-1h-quinoline-3-carboxamide Chemical compound C1=C(C(F)(F)F)C(O)=CC(NC(=O)C=2C(C3=CC(F)=CC=C3NC=2)=O)=C1 BNERSGDXTXPIBE-UHFFFAOYSA-N 0.000 claims 1
- DCJKUTWCEOPLQN-UHFFFAOYSA-N 8-cyano-n-(2,4-ditert-butyl-5-hydroxyphenyl)-4-oxo-1h-quinoline-3-carboxamide Chemical compound C1=C(O)C(C(C)(C)C)=CC(C(C)(C)C)=C1NC(=O)C1=CNC2=C(C#N)C=CC=C2C1=O DCJKUTWCEOPLQN-UHFFFAOYSA-N 0.000 claims 1
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 claims 1
- 206010020850 Hyperthyroidism Diseases 0.000 claims 1
- 208000034189 Sclerosis Diseases 0.000 claims 1
- 230000020764 fibrinolysis Effects 0.000 claims 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims 1
- BQPJRGXMKCQXSZ-UHFFFAOYSA-N n-(4-cyclohexyl-3-hydroxyphenyl)-4-oxo-1h-quinoline-3-carboxamide Chemical compound OC1=CC(NC(=O)C=2C(C3=CC=CC=C3NC=2)=O)=CC=C1C1CCCCC1 BQPJRGXMKCQXSZ-UHFFFAOYSA-N 0.000 claims 1
- KBWIYIDPGFVKKU-UHFFFAOYSA-N n-(4-cyclohexyl-3-hydroxyphenyl)-6-fluoro-4-oxo-1h-quinoline-3-carboxamide Chemical compound OC1=CC(NC(=O)C=2C(C3=CC(F)=CC=C3NC=2)=O)=CC=C1C1CCCCC1 KBWIYIDPGFVKKU-UHFFFAOYSA-N 0.000 claims 1
- XJLSEXAGTJCILF-UHFFFAOYSA-N nipecotic acid Chemical compound OC(=O)C1CCCNC1 XJLSEXAGTJCILF-UHFFFAOYSA-N 0.000 claims 1
- 102000012605 Cystic Fibrosis Transmembrane Conductance Regulator Human genes 0.000 abstract description 78
- 108010079245 Cystic Fibrosis Transmembrane Conductance Regulator Proteins 0.000 abstract description 78
- 230000001404 mediated effect Effects 0.000 abstract description 8
- 108010078791 Carrier Proteins Proteins 0.000 abstract description 6
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 167
- 239000000243 solution Substances 0.000 description 152
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 130
- 229910001868 water Inorganic materials 0.000 description 121
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 120
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 116
- 125000001931 aliphatic group Chemical group 0.000 description 101
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 100
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 96
- 229910052739 hydrogen Inorganic materials 0.000 description 90
- 239000001257 hydrogen Substances 0.000 description 90
- 235000019439 ethyl acetate Nutrition 0.000 description 78
- 239000012044 organic layer Substances 0.000 description 75
- 238000005481 NMR spectroscopy Methods 0.000 description 74
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 71
- 239000011734 sodium Substances 0.000 description 69
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 68
- 238000004440 column chromatography Methods 0.000 description 62
- 239000012267 brine Substances 0.000 description 61
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 61
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 57
- 239000000706 filtrate Substances 0.000 description 57
- 125000005843 halogen group Chemical group 0.000 description 55
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 52
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 52
- 239000007787 solid Substances 0.000 description 51
- 125000001424 substituent group Chemical group 0.000 description 50
- 150000002431 hydrogen Chemical group 0.000 description 49
- 150000002148 esters Chemical class 0.000 description 45
- 229910052760 oxygen Inorganic materials 0.000 description 45
- 238000010992 reflux Methods 0.000 description 45
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 44
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 43
- 229910052717 sulfur Chemical group 0.000 description 42
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 41
- 230000015572 biosynthetic process Effects 0.000 description 40
- 238000003786 synthesis reaction Methods 0.000 description 39
- 238000001914 filtration Methods 0.000 description 37
- 239000011541 reaction mixture Substances 0.000 description 37
- 238000004128 high performance liquid chromatography Methods 0.000 description 36
- 229910052757 nitrogen Inorganic materials 0.000 description 36
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 33
- 150000001412 amines Chemical class 0.000 description 33
- 125000003118 aryl group Chemical group 0.000 description 33
- 125000005842 heteroatom Chemical group 0.000 description 33
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 33
- 239000000725 suspension Substances 0.000 description 33
- 239000002243 precursor Substances 0.000 description 32
- 125000004494 ethyl ester group Chemical group 0.000 description 31
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 30
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 30
- 229920006395 saturated elastomer Polymers 0.000 description 30
- 239000002904 solvent Substances 0.000 description 30
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 29
- 239000005457 ice water Substances 0.000 description 29
- 235000002639 sodium chloride Nutrition 0.000 description 29
- 239000011593 sulfur Chemical group 0.000 description 29
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 28
- 238000007792 addition Methods 0.000 description 28
- 239000001301 oxygen Chemical group 0.000 description 28
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 25
- 125000000623 heterocyclic group Chemical group 0.000 description 25
- 238000000746 purification Methods 0.000 description 25
- 239000003208 petroleum Substances 0.000 description 24
- FGIUAXJPYTZDNR-UHFFFAOYSA-N potassium nitrate Inorganic materials [K+].[O-][N+]([O-])=O FGIUAXJPYTZDNR-UHFFFAOYSA-N 0.000 description 24
- 239000002244 precipitate Substances 0.000 description 24
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 23
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 23
- 230000032258 transport Effects 0.000 description 23
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 22
- 239000000741 silica gel Substances 0.000 description 20
- 229910002027 silica gel Inorganic materials 0.000 description 20
- 210000004027 cell Anatomy 0.000 description 19
- 239000003921 oil Substances 0.000 description 19
- 235000019198 oils Nutrition 0.000 description 19
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 19
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 18
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 18
- 238000006243 chemical reaction Methods 0.000 description 18
- 238000003756 stirring Methods 0.000 description 18
- 239000010410 layer Substances 0.000 description 17
- 125000002950 monocyclic group Chemical group 0.000 description 17
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 16
- 208000035475 disorder Diseases 0.000 description 16
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 16
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 15
- 238000001816 cooling Methods 0.000 description 15
- 125000004093 cyano group Chemical group *C#N 0.000 description 15
- 239000000284 extract Substances 0.000 description 15
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 14
- 125000000217 alkyl group Chemical group 0.000 description 14
- 125000004429 atom Chemical group 0.000 description 14
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical compound NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 13
- 206010012735 Diarrhoea Diseases 0.000 description 13
- JNCMHMUGTWEVOZ-UHFFFAOYSA-N F[CH]F Chemical compound F[CH]F JNCMHMUGTWEVOZ-UHFFFAOYSA-N 0.000 description 13
- 239000003054 catalyst Substances 0.000 description 13
- 239000003814 drug Substances 0.000 description 13
- 125000001072 heteroaryl group Chemical group 0.000 description 13
- 239000012528 membrane Substances 0.000 description 13
- 108091006146 Channels Proteins 0.000 description 12
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 12
- 238000000576 coating method Methods 0.000 description 12
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 12
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 11
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 11
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 11
- 229960000583 acetic acid Drugs 0.000 description 11
- 150000001450 anions Chemical class 0.000 description 11
- 210000000170 cell membrane Anatomy 0.000 description 11
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 11
- 241000124008 Mammalia Species 0.000 description 10
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 10
- 125000002619 bicyclic group Chemical group 0.000 description 10
- 230000002950 deficient Effects 0.000 description 10
- 210000003097 mucus Anatomy 0.000 description 10
- 235000018102 proteins Nutrition 0.000 description 10
- 102000004169 proteins and genes Human genes 0.000 description 10
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 9
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N DMSO Substances CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 9
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 9
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 9
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 9
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 9
- 235000019441 ethanol Nutrition 0.000 description 9
- CXHHBNMLPJOKQD-UHFFFAOYSA-M methyl carbonate Chemical compound COC([O-])=O CXHHBNMLPJOKQD-UHFFFAOYSA-M 0.000 description 9
- 229910000027 potassium carbonate Inorganic materials 0.000 description 9
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 9
- 125000004076 pyridyl group Chemical group 0.000 description 9
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 9
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 8
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 8
- 241001465754 Metazoa Species 0.000 description 8
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 8
- 239000008346 aqueous phase Substances 0.000 description 8
- 239000012043 crude product Substances 0.000 description 8
- 125000000753 cycloalkyl group Chemical group 0.000 description 8
- 239000012530 fluid Substances 0.000 description 8
- 235000010333 potassium nitrate Nutrition 0.000 description 8
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 8
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 7
- 241001553178 Arachis glabrata Species 0.000 description 7
- 239000005711 Benzoic acid Substances 0.000 description 7
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 7
- 229910052799 carbon Inorganic materials 0.000 description 7
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 7
- 125000001309 chloro group Chemical group Cl* 0.000 description 7
- 230000006870 function Effects 0.000 description 7
- 230000001771 impaired effect Effects 0.000 description 7
- 239000007788 liquid Substances 0.000 description 7
- 229920001223 polyethylene glycol Polymers 0.000 description 7
- 239000000843 powder Substances 0.000 description 7
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 7
- 230000028327 secretion Effects 0.000 description 7
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 6
- DTYLXDLAOLOTKT-UHFFFAOYSA-N 1,4-dihydroquinoline-3-carboxylic acid Chemical compound C1=CC=C2CC(C(=O)O)=CNC2=C1 DTYLXDLAOLOTKT-UHFFFAOYSA-N 0.000 description 6
- VGUWZCUCNQXGBU-UHFFFAOYSA-N 3-[(4-methylpiperazin-1-yl)methyl]-5-nitro-1h-indole Chemical compound C1CN(C)CCN1CC1=CNC2=CC=C([N+]([O-])=O)C=C12 VGUWZCUCNQXGBU-UHFFFAOYSA-N 0.000 description 6
- JVVRCYWZTJLJSG-UHFFFAOYSA-N 4-dimethylaminophenol Chemical compound CN(C)C1=CC=C(O)C=C1 JVVRCYWZTJLJSG-UHFFFAOYSA-N 0.000 description 6
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 6
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-dimethylaminopyridine Substances CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 6
- RHMPLDJJXGPMEX-UHFFFAOYSA-N 4-fluorophenol Chemical compound OC1=CC=C(F)C=C1 RHMPLDJJXGPMEX-UHFFFAOYSA-N 0.000 description 6
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 6
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 6
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- 238000010521 absorption reaction Methods 0.000 description 6
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 6
- 125000001118 alkylidene group Chemical group 0.000 description 6
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 6
- 230000034994 death Effects 0.000 description 6
- 231100000517 death Toxicity 0.000 description 6
- 229960004132 diethyl ether Drugs 0.000 description 6
- 125000004786 difluoromethoxy group Chemical group [H]C(F)(F)O* 0.000 description 6
- 239000002552 dosage form Substances 0.000 description 6
- 229940079593 drug Drugs 0.000 description 6
- 238000009472 formulation Methods 0.000 description 6
- 230000001965 increasing effect Effects 0.000 description 6
- 125000001041 indolyl group Chemical group 0.000 description 6
- 208000015181 infectious disease Diseases 0.000 description 6
- 229910052740 iodine Inorganic materials 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 6
- 229910017604 nitric acid Inorganic materials 0.000 description 6
- 239000000546 pharmaceutical excipient Substances 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- 238000002360 preparation method Methods 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- 229910052979 sodium sulfide Inorganic materials 0.000 description 6
- GRVFOGOEDUUMBP-UHFFFAOYSA-N sodium sulfide (anhydrous) Chemical compound [Na+].[Na+].[S-2] GRVFOGOEDUUMBP-UHFFFAOYSA-N 0.000 description 6
- 208000024891 symptom Diseases 0.000 description 6
- 238000002560 therapeutic procedure Methods 0.000 description 6
- PSWCIARYGITEOY-UHFFFAOYSA-N 6-nitro-1h-indole Chemical compound [O-][N+](=O)C1=CC=C2C=CNC2=C1 PSWCIARYGITEOY-UHFFFAOYSA-N 0.000 description 5
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 5
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 5
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 5
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 5
- 108010067035 Pancrelipase Proteins 0.000 description 5
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 5
- ZTHYODDOHIVTJV-UHFFFAOYSA-N Propyl gallate Chemical compound CCCOC(=O)C1=CC(O)=C(O)C(O)=C1 ZTHYODDOHIVTJV-UHFFFAOYSA-N 0.000 description 5
- 229920002472 Starch Polymers 0.000 description 5
- 239000002775 capsule Substances 0.000 description 5
- 238000004587 chromatography analysis Methods 0.000 description 5
- 239000011248 coating agent Substances 0.000 description 5
- 125000004122 cyclic group Chemical group 0.000 description 5
- 239000012065 filter cake Substances 0.000 description 5
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 5
- 150000002500 ions Chemical class 0.000 description 5
- 239000008101 lactose Substances 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 5
- 231100000252 nontoxic Toxicity 0.000 description 5
- 230000003000 nontoxic effect Effects 0.000 description 5
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 5
- 239000006187 pill Substances 0.000 description 5
- 229920000642 polymer Polymers 0.000 description 5
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 5
- 230000003248 secreting effect Effects 0.000 description 5
- 229910052708 sodium Inorganic materials 0.000 description 5
- 235000017557 sodium bicarbonate Nutrition 0.000 description 5
- 239000007909 solid dosage form Substances 0.000 description 5
- 235000019698 starch Nutrition 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- 125000001113 thiadiazolyl group Chemical group 0.000 description 5
- 210000001519 tissue Anatomy 0.000 description 5
- 239000003981 vehicle Substances 0.000 description 5
- 239000001993 wax Substances 0.000 description 5
- WJUGEERCZOQFJC-UHFFFAOYSA-N 1-nitro-4-(2-nitropropyl)benzene Chemical compound [O-][N+](=O)C(C)CC1=CC=C([N+]([O-])=O)C=C1 WJUGEERCZOQFJC-UHFFFAOYSA-N 0.000 description 4
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 4
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 4
- 108091006515 Anion channels Proteins 0.000 description 4
- 102000037829 Anion channels Human genes 0.000 description 4
- 239000004215 Carbon black (E152) Substances 0.000 description 4
- 208000010693 Charcot-Marie-Tooth Disease Diseases 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 4
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 4
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 4
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 4
- 229910002651 NO3 Inorganic materials 0.000 description 4
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 4
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 4
- YIKSCQDJHCMVMK-UHFFFAOYSA-N Oxamide Chemical compound NC(=O)C(N)=O YIKSCQDJHCMVMK-UHFFFAOYSA-N 0.000 description 4
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 4
- 239000002202 Polyethylene glycol Substances 0.000 description 4
- 229930006000 Sucrose Natural products 0.000 description 4
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 4
- 239000012346 acetyl chloride Substances 0.000 description 4
- 230000001154 acute effect Effects 0.000 description 4
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 4
- 150000001408 amides Chemical class 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 235000010233 benzoic acid Nutrition 0.000 description 4
- 230000033228 biological regulation Effects 0.000 description 4
- 125000001246 bromo group Chemical group Br* 0.000 description 4
- 150000001721 carbon Chemical group 0.000 description 4
- 230000001684 chronic effect Effects 0.000 description 4
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 4
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 4
- CJYQQUPRURWLOW-YDLUHMIOSA-M dmsc Chemical compound [Na+].OP(=O)=O.OP(=O)=O.OP(=O)=O.[O-]P(=O)=O.O=C1C2=C(O)C=CC=C2[C@H](C)[C@@H]2C1=C(O)[C@]1(O)C(=O)C(C(N)=O)=C(O)[C@@H](N(C)C)[C@@H]1[C@H]2O CJYQQUPRURWLOW-YDLUHMIOSA-M 0.000 description 4
- 210000000981 epithelium Anatomy 0.000 description 4
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 4
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical class CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 4
- 238000001704 evaporation Methods 0.000 description 4
- 230000008020 evaporation Effects 0.000 description 4
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 4
- 238000010438 heat treatment Methods 0.000 description 4
- 229930195733 hydrocarbon Natural products 0.000 description 4
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 4
- 239000003701 inert diluent Substances 0.000 description 4
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 4
- 239000000314 lubricant Substances 0.000 description 4
- 210000004072 lung Anatomy 0.000 description 4
- GPSDUZXPYCFOSQ-UHFFFAOYSA-N m-toluic acid Chemical compound CC1=CC=CC(C(O)=O)=C1 GPSDUZXPYCFOSQ-UHFFFAOYSA-N 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 4
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 4
- PXHVJJICTQNCMI-UHFFFAOYSA-N nickel Substances [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 4
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 4
- 125000004433 nitrogen atom Chemical group N* 0.000 description 4
- 229910052698 phosphorus Inorganic materials 0.000 description 4
- 239000011574 phosphorus Substances 0.000 description 4
- ODLMAHJVESYWTB-UHFFFAOYSA-N propylbenzene Chemical compound CCCC1=CC=CC=C1 ODLMAHJVESYWTB-UHFFFAOYSA-N 0.000 description 4
- 125000006239 protecting group Chemical group 0.000 description 4
- 125000003373 pyrazinyl group Chemical group 0.000 description 4
- 230000000241 respiratory effect Effects 0.000 description 4
- 208000023504 respiratory system disease Diseases 0.000 description 4
- 239000000377 silicon dioxide Substances 0.000 description 4
- 229910000029 sodium carbonate Inorganic materials 0.000 description 4
- 239000005720 sucrose Substances 0.000 description 4
- 235000011149 sulphuric acid Nutrition 0.000 description 4
- 229940124597 therapeutic agent Drugs 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- 229920002554 vinyl polymer Polymers 0.000 description 4
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 description 3
- GLEFBYIVFCNNOK-UHFFFAOYSA-N 2-(6-nitro-1h-indol-3-yl)acetonitrile Chemical compound [O-][N+](=O)C1=CC=C2C(CC#N)=CNC2=C1 GLEFBYIVFCNNOK-UHFFFAOYSA-N 0.000 description 3
- 125000004861 4-isopropyl phenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- KFPFJUDNNBTXBL-UHFFFAOYSA-N 5-ethyl-6-nitro-1h-indole Chemical compound C1=C([N+]([O-])=O)C(CC)=CC2=C1NC=C2 KFPFJUDNNBTXBL-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- ZKHQWZAMYRWXGA-UHFFFAOYSA-N Adenosine triphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O ZKHQWZAMYRWXGA-UHFFFAOYSA-N 0.000 description 3
- 101150029409 CFTR gene Proteins 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- 206010061218 Inflammation Diseases 0.000 description 3
- 102000004310 Ion Channels Human genes 0.000 description 3
- 108090000862 Ion Channels Proteins 0.000 description 3
- ZSXGLVDWWRXATF-UHFFFAOYSA-N N,N-dimethylformamide dimethyl acetal Chemical compound COC(OC)N(C)C ZSXGLVDWWRXATF-UHFFFAOYSA-N 0.000 description 3
- 102000007399 Nuclear hormone receptor Human genes 0.000 description 3
- 108020005497 Nuclear hormone receptor Proteins 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- NFHFRUOZVGFOOS-UHFFFAOYSA-N Pd(PPh3)4 Substances [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 3
- 102000035195 Peptidases Human genes 0.000 description 3
- 108091005804 Peptidases Proteins 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- 239000004365 Protease Substances 0.000 description 3
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 3
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 3
- 206010047924 Wheezing Diseases 0.000 description 3
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 3
- 230000002159 abnormal effect Effects 0.000 description 3
- 238000009825 accumulation Methods 0.000 description 3
- 235000010443 alginic acid Nutrition 0.000 description 3
- 229920000615 alginic acid Polymers 0.000 description 3
- 125000003545 alkoxy group Chemical group 0.000 description 3
- 229910052786 argon Inorganic materials 0.000 description 3
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 3
- 125000003710 aryl alkyl group Chemical group 0.000 description 3
- 230000001580 bacterial effect Effects 0.000 description 3
- 230000009286 beneficial effect Effects 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 125000005605 benzo group Chemical group 0.000 description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical compound BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 3
- 239000006172 buffering agent Substances 0.000 description 3
- 125000002837 carbocyclic group Chemical group 0.000 description 3
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- 230000018044 dehydration Effects 0.000 description 3
- 238000006297 dehydration reaction Methods 0.000 description 3
- 230000003111 delayed effect Effects 0.000 description 3
- 238000012217 deletion Methods 0.000 description 3
- 230000037430 deletion Effects 0.000 description 3
- 235000014113 dietary fatty acids Nutrition 0.000 description 3
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 206010013781 dry mouth Diseases 0.000 description 3
- 239000003995 emulsifying agent Substances 0.000 description 3
- 229940088598 enzyme Drugs 0.000 description 3
- 239000012259 ether extract Substances 0.000 description 3
- 125000000816 ethylene group Chemical group [H]C([H])([*:1])C([H])([H])[*:2] 0.000 description 3
- 230000001747 exhibiting effect Effects 0.000 description 3
- 239000000194 fatty acid Substances 0.000 description 3
- 229930195729 fatty acid Natural products 0.000 description 3
- 239000000945 filler Substances 0.000 description 3
- 238000003818 flash chromatography Methods 0.000 description 3
- 230000004907 flux Effects 0.000 description 3
- 125000004005 formimidoyl group Chemical group [H]\N=C(/[H])* 0.000 description 3
- 210000001035 gastrointestinal tract Anatomy 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 150000002430 hydrocarbons Chemical group 0.000 description 3
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 3
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical compound O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 description 3
- 208000013403 hyperactivity Diseases 0.000 description 3
- 230000004054 inflammatory process Effects 0.000 description 3
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 3
- 230000033001 locomotion Effects 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 230000007257 malfunction Effects 0.000 description 3
- 125000006217 methyl sulfide group Chemical group [H]C([H])([H])S* 0.000 description 3
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 238000006396 nitration reaction Methods 0.000 description 3
- 239000012074 organic phase Substances 0.000 description 3
- 235000021317 phosphate Nutrition 0.000 description 3
- 125000005936 piperidyl group Chemical group 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- 239000011698 potassium fluoride Substances 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 230000000750 progressive effect Effects 0.000 description 3
- 125000003226 pyrazolyl group Chemical group 0.000 description 3
- 238000001953 recrystallisation Methods 0.000 description 3
- 229910052710 silicon Inorganic materials 0.000 description 3
- 239000010703 silicon Substances 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 229910000104 sodium hydride Inorganic materials 0.000 description 3
- 239000008247 solid mixture Substances 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 239000000829 suppository Substances 0.000 description 3
- FQFILJKFZCVHNH-UHFFFAOYSA-N tert-butyl n-[3-[(5-bromo-2-chloropyrimidin-4-yl)amino]propyl]carbamate Chemical compound CC(C)(C)OC(=O)NCCCNC1=NC(Cl)=NC=C1Br FQFILJKFZCVHNH-UHFFFAOYSA-N 0.000 description 3
- 125000000437 thiazol-2-yl group Chemical group [H]C1=C([H])N=C(*)S1 0.000 description 3
- KJAMZCVTJDTESW-UHFFFAOYSA-N tiracizine Chemical compound C1CC2=CC=CC=C2N(C(=O)CN(C)C)C2=CC(NC(=O)OCC)=CC=C21 KJAMZCVTJDTESW-UHFFFAOYSA-N 0.000 description 3
- 229960000707 tobramycin Drugs 0.000 description 3
- NLVFBUXFDBBNBW-PBSUHMDJSA-N tobramycin Chemical compound N[C@@H]1C[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N NLVFBUXFDBBNBW-PBSUHMDJSA-N 0.000 description 3
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 3
- 125000004306 triazinyl group Chemical group 0.000 description 3
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 3
- 230000003612 virological effect Effects 0.000 description 3
- 238000010792 warming Methods 0.000 description 3
- 239000000080 wetting agent Substances 0.000 description 3
- 229910052725 zinc Inorganic materials 0.000 description 3
- 239000011701 zinc Substances 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 2
- QNBJYUUUYZVIJP-UHFFFAOYSA-N 2,4-dichloroquinoline Chemical compound C1=CC=CC2=NC(Cl)=CC(Cl)=C21 QNBJYUUUYZVIJP-UHFFFAOYSA-N 0.000 description 2
- XSZBLRGKCBUWRJ-UHFFFAOYSA-N 2,4-dimethoxyquinoline Chemical compound C1=CC=CC2=NC(OC)=CC(OC)=C21 XSZBLRGKCBUWRJ-UHFFFAOYSA-N 0.000 description 2
- MOTLBIPBNZTCMS-UHFFFAOYSA-N 2,4-dimethoxyquinoline-3-carboxylic acid Chemical compound C1=CC=C2C(OC)=C(C(O)=O)C(OC)=NC2=C1 MOTLBIPBNZTCMS-UHFFFAOYSA-N 0.000 description 2
- KLIDCXVFHGNTTM-UHFFFAOYSA-N 2,6-dimethoxyphenol Chemical group COC1=CC=CC(OC)=C1O KLIDCXVFHGNTTM-UHFFFAOYSA-N 0.000 description 2
- 125000004198 2-fluorophenyl group Chemical group [H]C1=C([H])C(F)=C(*)C([H])=C1[H] 0.000 description 2
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 2
- OAIIKIWHCVTWKK-UHFFFAOYSA-N 2-methyl-1h-indol-6-amine Chemical compound C1=C(N)C=C2NC(C)=CC2=C1 OAIIKIWHCVTWKK-UHFFFAOYSA-N 0.000 description 2
- OOUGLTULBSNHNF-UHFFFAOYSA-N 3-[5-(2-fluorophenyl)-1,2,4-oxadiazol-3-yl]benzoic acid Chemical compound OC(=O)C1=CC=CC(C=2N=C(ON=2)C=2C(=CC=CC=2)F)=C1 OOUGLTULBSNHNF-UHFFFAOYSA-N 0.000 description 2
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 2
- 125000004207 3-methoxyphenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(OC([H])([H])[H])=C1[H] 0.000 description 2
- YJXYKDQEOIPVBD-UHFFFAOYSA-N 3-nitro-4-propylaniline Chemical compound CCCC1=CC=C(N)C=C1[N+]([O-])=O YJXYKDQEOIPVBD-UHFFFAOYSA-N 0.000 description 2
- CURBJZDRYTXCIC-UHFFFAOYSA-N 3-nitro-n-(propylideneamino)aniline Chemical compound CCC=NNC1=CC=CC([N+]([O-])=O)=C1 CURBJZDRYTXCIC-UHFFFAOYSA-N 0.000 description 2
- HMOBRUPKXAKFHS-UHFFFAOYSA-N 4,4-dimethyl-1,3-dihydroquinolin-2-one Chemical compound C1=CC=C2C(C)(C)CC(=O)NC2=C1 HMOBRUPKXAKFHS-UHFFFAOYSA-N 0.000 description 2
- ASTIKEBMMAIZAS-UHFFFAOYSA-N 4-(2-ethoxyphenyl)-3-nitroaniline Chemical compound CCOC1=CC=CC=C1C1=CC=C(N)C=C1[N+]([O-])=O ASTIKEBMMAIZAS-UHFFFAOYSA-N 0.000 description 2
- UDSLQZULGNKWBB-UHFFFAOYSA-N 4-bromophenol Chemical compound OC1=CC=C(Br)C=C1.OC1=CC=C(Br)C=C1 UDSLQZULGNKWBB-UHFFFAOYSA-N 0.000 description 2
- WRTDVWHCSGVIOT-UHFFFAOYSA-N 5-amino-2-tert-butylphenol Chemical compound CC(C)(C)C1=CC=C(N)C=C1O WRTDVWHCSGVIOT-UHFFFAOYSA-N 0.000 description 2
- WNEGVTLAQRMHSQ-UHFFFAOYSA-N 5-amino-4-fluoro-2-(1-methylcyclohexyl)phenol Chemical compound C=1C(F)=C(N)C=C(O)C=1C1(C)CCCCC1 WNEGVTLAQRMHSQ-UHFFFAOYSA-N 0.000 description 2
- SBVQALZKTIHCSD-UHFFFAOYSA-N 5-bromo-1h-indol-6-amine Chemical compound C1=C(Br)C(N)=CC2=C1C=CN2 SBVQALZKTIHCSD-UHFFFAOYSA-N 0.000 description 2
- QEDCHCLHHGGYBT-UHFFFAOYSA-N 5-bromo-2,3-dihydro-1h-indole Chemical compound BrC1=CC=C2NCCC2=C1 QEDCHCLHHGGYBT-UHFFFAOYSA-N 0.000 description 2
- QZRTXAUAKONXQB-UHFFFAOYSA-N 5-ethyl-1h-indol-6-amine Chemical compound C1=C(N)C(CC)=CC2=C1NC=C2 QZRTXAUAKONXQB-UHFFFAOYSA-N 0.000 description 2
- DWLKQZKMPDGRNB-UHFFFAOYSA-N 5-ethyl-2,3-dihydro-1h-indole Chemical compound CCC1=CC=C2NCCC2=C1 DWLKQZKMPDGRNB-UHFFFAOYSA-N 0.000 description 2
- OWUNYWQSRORGBQ-UHFFFAOYSA-N 5-fluoro-1h-indol-6-amine Chemical compound C1=C(F)C(N)=CC2=C1C=CN2 OWUNYWQSRORGBQ-UHFFFAOYSA-N 0.000 description 2
- 208000030507 AIDS Diseases 0.000 description 2
- ZKHQWZAMYRWXGA-KQYNXXCUSA-J ATP(4-) Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O)[C@@H](O)[C@H]1O ZKHQWZAMYRWXGA-KQYNXXCUSA-J 0.000 description 2
- 108091006112 ATPases Proteins 0.000 description 2
- 102000057290 Adenosine Triphosphatases Human genes 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- WZPBZJONDBGPKJ-UHFFFAOYSA-N Antibiotic SQ 26917 Natural products O=C1N(S(O)(=O)=O)C(C)C1NC(=O)C(=NOC(C)(C)C(O)=O)C1=CSC(N)=N1 WZPBZJONDBGPKJ-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 208000035143 Bacterial infection Diseases 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 102000011045 Chloride Channels Human genes 0.000 description 2
- 108010062745 Chloride Channels Proteins 0.000 description 2
- 241000711573 Coronaviridae Species 0.000 description 2
- 208000020406 Creutzfeldt Jacob disease Diseases 0.000 description 2
- 208000003407 Creutzfeldt-Jakob Syndrome Diseases 0.000 description 2
- 208000010859 Creutzfeldt-Jakob disease Diseases 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- XRHVZWWRFMCBAZ-UHFFFAOYSA-L Endothal-disodium Chemical compound [Na+].[Na+].C1CC2C(C([O-])=O)C(C(=O)[O-])C1O2 XRHVZWWRFMCBAZ-UHFFFAOYSA-L 0.000 description 2
- 102000003837 Epithelial Sodium Channels Human genes 0.000 description 2
- 108090000140 Epithelial Sodium Channels Proteins 0.000 description 2
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 2
- 239000005977 Ethylene Substances 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 108010024636 Glutathione Proteins 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 208000018565 Hemochromatosis Diseases 0.000 description 2
- 101000632319 Homo sapiens Septin-7 Proteins 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- 206010021143 Hypoxia Diseases 0.000 description 2
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 2
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 2
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 2
- 108010021466 Mutant Proteins Proteins 0.000 description 2
- 102000008300 Mutant Proteins Human genes 0.000 description 2
- 229910020889 NaBH3 Inorganic materials 0.000 description 2
- NBBJYMSMWIIQGU-UHFFFAOYSA-N Propionic aldehyde Chemical compound CCC=O NBBJYMSMWIIQGU-UHFFFAOYSA-N 0.000 description 2
- 206010057190 Respiratory tract infections Diseases 0.000 description 2
- 229910006074 SO2NH2 Inorganic materials 0.000 description 2
- 229910006124 SOCl2 Inorganic materials 0.000 description 2
- 102100027981 Septin-7 Human genes 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- 102000003673 Symporters Human genes 0.000 description 2
- 108090000088 Symporters Proteins 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 208000005946 Xerostomia Diseases 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- 235000010419 agar Nutrition 0.000 description 2
- 125000003342 alkenyl group Chemical group 0.000 description 2
- 230000000172 allergic effect Effects 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 description 2
- MDFFNEOEWAXZRQ-UHFFFAOYSA-N aminyl Chemical compound [NH2] MDFFNEOEWAXZRQ-UHFFFAOYSA-N 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 125000005160 aryl oxy alkyl group Chemical group 0.000 description 2
- 208000010668 atopic eczema Diseases 0.000 description 2
- 230000003190 augmentative effect Effects 0.000 description 2
- 125000003725 azepanyl group Chemical group 0.000 description 2
- WZPBZJONDBGPKJ-VEHQQRBSSA-N aztreonam Chemical compound O=C1N(S([O-])(=O)=O)[C@@H](C)[C@@H]1NC(=O)C(=N/OC(C)(C)C(O)=O)\C1=CSC([NH3+])=N1 WZPBZJONDBGPKJ-VEHQQRBSSA-N 0.000 description 2
- 229960003644 aztreonam Drugs 0.000 description 2
- 208000022362 bacterial infectious disease Diseases 0.000 description 2
- SESFRYSPDFLNCH-UHFFFAOYSA-N benzyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OCC1=CC=CC=C1 SESFRYSPDFLNCH-UHFFFAOYSA-N 0.000 description 2
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 2
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 229920002988 biodegradable polymer Polymers 0.000 description 2
- 239000004621 biodegradable polymer Substances 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- MYSWGUAQZAJSOK-UHFFFAOYSA-N ciprofloxacin Chemical compound C12=CC(N3CCNCC3)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 MYSWGUAQZAJSOK-UHFFFAOYSA-N 0.000 description 2
- 229940110456 cocoa butter Drugs 0.000 description 2
- 235000019868 cocoa butter Nutrition 0.000 description 2
- 230000001276 controlling effect Effects 0.000 description 2
- 235000012343 cottonseed oil Nutrition 0.000 description 2
- 239000010779 crude oil Substances 0.000 description 2
- NLUNLVTVUDIHFE-UHFFFAOYSA-N cyclooctylcyclooctane Chemical group C1CCCCCCC1C1CCCCCCC1 NLUNLVTVUDIHFE-UHFFFAOYSA-N 0.000 description 2
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 239000002274 desiccant Substances 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- USIUVYZYUHIAEV-UHFFFAOYSA-N diphenyl ether Chemical compound C=1C=CC=CC=1OC1=CC=CC=C1 USIUVYZYUHIAEV-UHFFFAOYSA-N 0.000 description 2
- 238000007598 dipping method Methods 0.000 description 2
- 239000008298 dragée Substances 0.000 description 2
- 239000006196 drop Substances 0.000 description 2
- 238000002651 drug therapy Methods 0.000 description 2
- 239000003792 electrolyte Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 239000002702 enteric coating Substances 0.000 description 2
- 238000009505 enteric coating Methods 0.000 description 2
- SRCZQMGIVIYBBJ-UHFFFAOYSA-N ethoxyethane;ethyl acetate Chemical compound CCOCC.CCOC(C)=O SRCZQMGIVIYBBJ-UHFFFAOYSA-N 0.000 description 2
- URJOBQJGQMZSTN-UHFFFAOYSA-N ethyl 6-amino-1h-indole-7-carboxylate Chemical compound CCOC(=O)C1=C(N)C=CC2=C1NC=C2 URJOBQJGQMZSTN-UHFFFAOYSA-N 0.000 description 2
- RIFGWPKJUGCATF-UHFFFAOYSA-N ethyl chloroformate Chemical compound CCOC(Cl)=O RIFGWPKJUGCATF-UHFFFAOYSA-N 0.000 description 2
- MMXKVMNBHPAILY-UHFFFAOYSA-N ethyl laurate Chemical compound CCCCCCCCCCCC(=O)OCC MMXKVMNBHPAILY-UHFFFAOYSA-N 0.000 description 2
- 230000029142 excretion Effects 0.000 description 2
- 150000004665 fatty acids Chemical class 0.000 description 2
- 210000003608 fece Anatomy 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 238000002866 fluorescence resonance energy transfer Methods 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 210000004907 gland Anatomy 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 229960003180 glutathione Drugs 0.000 description 2
- 150000002334 glycols Chemical class 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 2
- 125000004475 heteroaralkyl group Chemical group 0.000 description 2
- 230000001744 histochemical effect Effects 0.000 description 2
- BKOYKMLGFFASBG-UHFFFAOYSA-N hydron;(3-nitrophenyl)hydrazine;chloride Chemical compound Cl.NNC1=CC=CC([N+]([O-])=O)=C1 BKOYKMLGFFASBG-UHFFFAOYSA-N 0.000 description 2
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 2
- 230000035874 hyperreactivity Effects 0.000 description 2
- 239000007972 injectable composition Substances 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N iron Substances [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- 206010023332 keratitis Diseases 0.000 description 2
- 201000010666 keratoconjunctivitis Diseases 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 239000008297 liquid dosage form Substances 0.000 description 2
- 239000012280 lithium aluminium hydride Substances 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 201000005296 lung carcinoma Diseases 0.000 description 2
- 230000002132 lysosomal effect Effects 0.000 description 2
- 125000000040 m-tolyl group Chemical group [H]C1=C([H])C(*)=C([H])C(=C1[H])C([H])([H])[H] 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 229960001855 mannitol Drugs 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 238000002779 membrane potential assay Methods 0.000 description 2
- 239000002207 metabolite Substances 0.000 description 2
- TWXDDNPPQUTEOV-FVGYRXGTSA-N methamphetamine hydrochloride Chemical compound Cl.CN[C@@H](C)CC1=CC=CC=C1 TWXDDNPPQUTEOV-FVGYRXGTSA-N 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- XMJHPCRAQCTCFT-UHFFFAOYSA-N methyl chloroformate Chemical compound COC(Cl)=O XMJHPCRAQCTCFT-UHFFFAOYSA-N 0.000 description 2
- 150000004702 methyl esters Chemical class 0.000 description 2
- 239000004530 micro-emulsion Substances 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- CQDGTJPVBWZJAZ-UHFFFAOYSA-N monoethyl carbonate Chemical compound CCOC(O)=O CQDGTJPVBWZJAZ-UHFFFAOYSA-N 0.000 description 2
- 230000000420 mucociliary effect Effects 0.000 description 2
- 239000012299 nitrogen atmosphere Substances 0.000 description 2
- 239000000346 nonvolatile oil Substances 0.000 description 2
- ZWLPBLYKEWSWPD-UHFFFAOYSA-N o-toluic acid Chemical compound CC1=CC=CC=C1C(O)=O ZWLPBLYKEWSWPD-UHFFFAOYSA-N 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 239000004006 olive oil Substances 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- FWFGVMYFCODZRD-UHFFFAOYSA-N oxidanium;hydrogen sulfate Chemical compound O.OS(O)(=O)=O FWFGVMYFCODZRD-UHFFFAOYSA-N 0.000 description 2
- 125000003854 p-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Cl 0.000 description 2
- 229940045258 pancrelipase Drugs 0.000 description 2
- AQIXEPGDORPWBJ-UHFFFAOYSA-N pentan-3-ol Chemical compound CCC(O)CC AQIXEPGDORPWBJ-UHFFFAOYSA-N 0.000 description 2
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- 239000002304 perfume Substances 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 150000003904 phospholipids Chemical class 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 235000015320 potassium carbonate Nutrition 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 230000000069 prophylactic effect Effects 0.000 description 2
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 2
- 125000000714 pyrimidinyl group Chemical group 0.000 description 2
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 2
- 238000010791 quenching Methods 0.000 description 2
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 2
- 150000003254 radicals Chemical class 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 208000020029 respiratory tract infectious disease Diseases 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 125000006413 ring segment Chemical group 0.000 description 2
- XMVJITFPVVRMHC-UHFFFAOYSA-N roxarsone Chemical compound OC1=CC=C([As](O)(O)=O)C=C1[N+]([O-])=O XMVJITFPVVRMHC-UHFFFAOYSA-N 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- 201000009881 secretory diarrhea Diseases 0.000 description 2
- 239000008159 sesame oil Substances 0.000 description 2
- 235000011803 sesame oil Nutrition 0.000 description 2
- BNRNXUUZRGQAQC-UHFFFAOYSA-N sildenafil Chemical compound CCCC1=NN(C)C(C(N2)=O)=C1N=C2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(C)CC1 BNRNXUUZRGQAQC-UHFFFAOYSA-N 0.000 description 2
- 239000012279 sodium borohydride Substances 0.000 description 2
- 229910000033 sodium borohydride Inorganic materials 0.000 description 2
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- 210000001138 tear Anatomy 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- NBHLCQIUMVKKSV-UHFFFAOYSA-N tert-butyl n-(3-amino-4-ethylphenyl)-n-methylcarbamate Chemical compound CCC1=CC=C(N(C)C(=O)OC(C)(C)C)C=C1N NBHLCQIUMVKKSV-UHFFFAOYSA-N 0.000 description 2
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 125000000335 thiazolyl group Chemical group 0.000 description 2
- 230000008719 thickening Effects 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- 125000001425 triazolyl group Chemical group 0.000 description 2
- 239000002753 trypsin inhibitor Substances 0.000 description 2
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical compound CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- XJRIDJAGAYGJCK-UHFFFAOYSA-N (1-acetyl-5-bromoindol-3-yl) acetate Chemical compound C1=C(Br)C=C2C(OC(=O)C)=CN(C(C)=O)C2=C1 XJRIDJAGAYGJCK-UHFFFAOYSA-N 0.000 description 1
- MRZZXDUZVJNFQL-UHFFFAOYSA-N (2,4-ditert-butylphenyl) methyl carbonate Chemical compound COC(=O)OC1=CC=C(C(C)(C)C)C=C1C(C)(C)C MRZZXDUZVJNFQL-UHFFFAOYSA-N 0.000 description 1
- DGFCTCGCMKEILT-UHFFFAOYSA-N (2-ethoxyphenyl)boronic acid Chemical compound CCOC1=CC=CC=C1B(O)O DGFCTCGCMKEILT-UHFFFAOYSA-N 0.000 description 1
- AHMPDVNAZQIQFR-UHFFFAOYSA-N (2-tert-butyl-4-fluoro-6-nitrophenyl) methyl carbonate Chemical compound COC(=O)OC1=C([N+]([O-])=O)C=C(F)C=C1C(C)(C)C AHMPDVNAZQIQFR-UHFFFAOYSA-N 0.000 description 1
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 1
- QSIRXSYRKZHJHX-TWXHAJHVSA-N (2s)-6-amino-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-6-amino-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-6-amino-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-6-amino-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2,6-diaminohexanoyl]amino]-4-methylpen Chemical compound NCCCC[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(O)=O QSIRXSYRKZHJHX-TWXHAJHVSA-N 0.000 description 1
- PLHVDNXEWINUIK-UHFFFAOYSA-N (3-formamidophenyl) acetate Chemical compound CC(=O)OC1=CC=CC(NC=O)=C1 PLHVDNXEWINUIK-UHFFFAOYSA-N 0.000 description 1
- FZYLFMAKCKNQRT-UHFFFAOYSA-N (3-nitro-4-propylphenyl)carbamic acid Chemical compound CCCC1=CC=C(NC(O)=O)C=C1[N+]([O-])=O FZYLFMAKCKNQRT-UHFFFAOYSA-N 0.000 description 1
- 125000003161 (C1-C6) alkylene group Chemical group 0.000 description 1
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- WDCYWAQPCXBPJA-UHFFFAOYSA-N 1,3-dinitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC([N+]([O-])=O)=C1 WDCYWAQPCXBPJA-UHFFFAOYSA-N 0.000 description 1
- KDDHVPXDYDGORO-UHFFFAOYSA-N 1-(benzenesulfonyl)-5-ethyl-2,3-dihydroindole Chemical compound C1CC2=CC(CC)=CC=C2N1S(=O)(=O)C1=CC=CC=C1 KDDHVPXDYDGORO-UHFFFAOYSA-N 0.000 description 1
- VTBOTOBFGSVRMA-UHFFFAOYSA-N 1-Methylcyclohexanol Chemical compound CC1(O)CCCCC1 VTBOTOBFGSVRMA-UHFFFAOYSA-N 0.000 description 1
- DQFQCHIDRBIESA-UHFFFAOYSA-N 1-benzazepine Chemical compound N1C=CC=CC2=CC=CC=C12 DQFQCHIDRBIESA-UHFFFAOYSA-N 0.000 description 1
- NJZQOCCEDXRQJM-UHFFFAOYSA-N 1-benzylindole Chemical compound C1=CC2=CC=CC=C2N1CC1=CC=CC=C1 NJZQOCCEDXRQJM-UHFFFAOYSA-N 0.000 description 1
- HLVFKOKELQSXIQ-UHFFFAOYSA-N 1-bromo-2-methylpropane Chemical compound CC(C)CBr HLVFKOKELQSXIQ-UHFFFAOYSA-N 0.000 description 1
- BTQZKHUEUDPRST-UHFFFAOYSA-N 1-fluoro-3-methylbenzene Chemical compound CC1=CC=CC(F)=C1 BTQZKHUEUDPRST-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- CHHHXKFHOYLYRE-UHFFFAOYSA-M 2,4-Hexadienoic acid, potassium salt (1:1), (2E,4E)- Chemical compound [K+].CC=CC=CC([O-])=O CHHHXKFHOYLYRE-UHFFFAOYSA-M 0.000 description 1
- 125000004201 2,4-dichlorophenyl group Chemical group [H]C1=C([H])C(*)=C(Cl)C([H])=C1Cl 0.000 description 1
- NUMXHEUHHRTBQT-AATRIKPKSA-N 2,4-dimethoxy-1-[(e)-2-nitroethenyl]benzene Chemical compound COC1=CC=C(\C=C\[N+]([O-])=O)C(OC)=C1 NUMXHEUHHRTBQT-AATRIKPKSA-N 0.000 description 1
- KYUUZCUXYXNPFO-UHFFFAOYSA-N 2,4-dinitro-1-(trifluoromethoxy)benzene Chemical compound [O-][N+](=O)C1=CC=C(OC(F)(F)F)C([N+]([O-])=O)=C1 KYUUZCUXYXNPFO-UHFFFAOYSA-N 0.000 description 1
- ZLMNRPCLXQDADC-UHFFFAOYSA-N 2,4-dinitrobenzoic acid Chemical compound OC(=O)C1=CC=C([N+]([O-])=O)C=C1[N+]([O-])=O.OC(=O)C1=CC=C([N+]([O-])=O)C=C1[N+]([O-])=O ZLMNRPCLXQDADC-UHFFFAOYSA-N 0.000 description 1
- PBOPJYORIDJAFE-UHFFFAOYSA-N 2,4-dinitrobromobenzene Chemical compound [O-][N+](=O)C1=CC=C(Br)C([N+]([O-])=O)=C1 PBOPJYORIDJAFE-UHFFFAOYSA-N 0.000 description 1
- 125000001917 2,4-dinitrophenyl group Chemical group [H]C1=C([H])C(=C([H])C(=C1*)[N+]([O-])=O)[N+]([O-])=O 0.000 description 1
- DCHWFDIPDWETDQ-UHFFFAOYSA-N 2-(2-adamantyl)-4-methyl-5-nitrophenol Chemical compound C1=C([N+]([O-])=O)C(C)=CC(C2C3CC4CC(C3)CC2C4)=C1O DCHWFDIPDWETDQ-UHFFFAOYSA-N 0.000 description 1
- PTCBIMMBZIKAKX-UHFFFAOYSA-N 2-(6-nitro-1h-indol-3-yl)ethylcarbamic acid Chemical compound [O-][N+](=O)C1=CC=C2C(CCNC(=O)O)=CNC2=C1 PTCBIMMBZIKAKX-UHFFFAOYSA-N 0.000 description 1
- ZOZVLTMZUMMWED-UHFFFAOYSA-N 2-[3-amino-4-[1-(2-methoxyethoxy)-2-methylpropan-2-yl]phenyl]acetamide Chemical compound COCCOCC(C)(C)C1=CC=C(CC(N)=O)C=C1N ZOZVLTMZUMMWED-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- 125000004174 2-benzimidazolyl group Chemical group [H]N1C(*)=NC2=C([H])C([H])=C([H])C([H])=C12 0.000 description 1
- 125000006276 2-bromophenyl group Chemical group [H]C1=C([H])C(Br)=C(*)C([H])=C1[H] 0.000 description 1
- ZVSCENGNXWPDPL-UHFFFAOYSA-N 2-methyl-2-phenylpropan-1-ol Chemical compound OCC(C)(C)C1=CC=CC=C1 ZVSCENGNXWPDPL-UHFFFAOYSA-N 0.000 description 1
- YOETUEMZNOLGDB-UHFFFAOYSA-N 2-methylpropyl carbonochloridate Chemical compound CC(C)COC(Cl)=O YOETUEMZNOLGDB-UHFFFAOYSA-N 0.000 description 1
- 229940080296 2-naphthalenesulfonate Drugs 0.000 description 1
- FCERNOPRZOOFSL-UHFFFAOYSA-N 2-tert-butyl-4-fluorophenol Chemical compound CC(C)(C)C1=CC(F)=CC=C1O FCERNOPRZOOFSL-UHFFFAOYSA-N 0.000 description 1
- UAPURNYDDXMJCS-UHFFFAOYSA-N 2-tert-butyl-5-nitrophenol Chemical compound CC(C)(C)C1=CC=C([N+]([O-])=O)C=C1O UAPURNYDDXMJCS-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- ZYOATBKPEFZNGP-UHFFFAOYSA-N 3,5-diamino-6-chloro-n-[n'-[4-[4-(2,3-dihydroxypropoxy)phenyl]butyl]carbamimidoyl]pyrazine-2-carboxamide;methanesulfonic acid Chemical compound CS(O)(=O)=O.N1=C(Cl)C(N)=NC(N)=C1C(=O)NC(=N)NCCCCC1=CC=C(OCC(O)CO)C=C1 ZYOATBKPEFZNGP-UHFFFAOYSA-N 0.000 description 1
- 125000004211 3,5-difluorophenyl group Chemical group [H]C1=C(F)C([H])=C(*)C([H])=C1F 0.000 description 1
- MTJGVAJYTOXFJH-UHFFFAOYSA-N 3-aminonaphthalene-1,5-disulfonic acid Chemical compound C1=CC=C(S(O)(=O)=O)C2=CC(N)=CC(S(O)(=O)=O)=C21 MTJGVAJYTOXFJH-UHFFFAOYSA-N 0.000 description 1
- CWLKGDAVCFYWJK-UHFFFAOYSA-N 3-aminophenol Chemical compound NC1=CC=CC(O)=C1 CWLKGDAVCFYWJK-UHFFFAOYSA-N 0.000 description 1
- 229940018563 3-aminophenol Drugs 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- 125000004179 3-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(Cl)=C1[H] 0.000 description 1
- ZRPLANDPDWYOMZ-UHFFFAOYSA-N 3-cyclopentylpropionic acid Chemical compound OC(=O)CCC1CCCC1 ZRPLANDPDWYOMZ-UHFFFAOYSA-N 0.000 description 1
- XKIRHOWVQWCYBT-UHFFFAOYSA-N 3-ethylpentan-3-ol Chemical compound CCC(O)(CC)CC XKIRHOWVQWCYBT-UHFFFAOYSA-N 0.000 description 1
- 125000004180 3-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(F)=C1[H] 0.000 description 1
- WAJVIALFRQVUJA-UHFFFAOYSA-N 3-methyl-1-nitroindole Chemical compound C1=CC=C2C(C)=CN([N+]([O-])=O)C2=C1 WAJVIALFRQVUJA-UHFFFAOYSA-N 0.000 description 1
- MRBCJYBSVUJBOX-UHFFFAOYSA-N 3-methyl-1h-indol-6-amine Chemical compound NC1=CC=C2C(C)=CNC2=C1 MRBCJYBSVUJBOX-UHFFFAOYSA-N 0.000 description 1
- FXOBRUMCGPOJLL-UHFFFAOYSA-N 3-methyl-6-nitro-1h-indole Chemical compound [O-][N+](=O)C1=CC=C2C(C)=CNC2=C1 FXOBRUMCGPOJLL-UHFFFAOYSA-N 0.000 description 1
- RINIQJLZLHTWKN-UHFFFAOYSA-N 3-methyl-n-phenylbut-2-enamide Chemical compound CC(C)=CC(=O)NC1=CC=CC=C1 RINIQJLZLHTWKN-UHFFFAOYSA-N 0.000 description 1
- XJCVRTZCHMZPBD-UHFFFAOYSA-N 3-nitroaniline Chemical compound NC1=CC=CC([N+]([O-])=O)=C1 XJCVRTZCHMZPBD-UHFFFAOYSA-N 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- 125000004800 4-bromophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Br 0.000 description 1
- WXNZTHHGJRFXKQ-UHFFFAOYSA-N 4-chlorophenol Chemical compound OC1=CC=C(Cl)C=C1 WXNZTHHGJRFXKQ-UHFFFAOYSA-N 0.000 description 1
- 125000001255 4-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1F 0.000 description 1
- HDHQZCHIXUUSMK-UHFFFAOYSA-N 4-hydroxy-2-quinolone Chemical compound C1=CC=C2C(O)=CC(=O)NC2=C1 HDHQZCHIXUUSMK-UHFFFAOYSA-N 0.000 description 1
- 125000004203 4-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 1
- UZOFELREXGAFOI-UHFFFAOYSA-N 4-methylpiperidine Chemical compound CC1CCNCC1 UZOFELREXGAFOI-UHFFFAOYSA-N 0.000 description 1
- SSQKYXXODSFITD-UHFFFAOYSA-N 4-nitro-1h-indole-2-carbonitrile Chemical compound [O-][N+](=O)C1=CC=CC2=C1C=C(C#N)N2 SSQKYXXODSFITD-UHFFFAOYSA-N 0.000 description 1
- AKZHSEGCFVRYDR-UHFFFAOYSA-N 4-nitro-1h-indole-2-carboxamide Chemical compound C1=CC=C2NC(C(=O)N)=CC2=C1[N+]([O-])=O AKZHSEGCFVRYDR-UHFFFAOYSA-N 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- VJYNRXFXHKIGLT-UHFFFAOYSA-N 5-[3-(1,3-diethyl-4,6-dioxo-2-sulfanylidene-1,3-diazinan-5-yl)prop-2-enylidene]-1,3-diethyl-2-sulfanylidene-1,3-diazinane-4,6-dione Chemical compound O=C1N(CC)C(=S)N(CC)C(=O)C1C=CC=C1C(=O)N(CC)C(=S)N(CC)C1=O VJYNRXFXHKIGLT-UHFFFAOYSA-N 0.000 description 1
- PPXAIJGGCHQXBC-UHFFFAOYSA-N 5-bromo-6-nitro-1h-indole Chemical compound C1=C(Br)C([N+](=O)[O-])=CC2=C1C=CN2 PPXAIJGGCHQXBC-UHFFFAOYSA-N 0.000 description 1
- YABGMLXPMPPWKX-UHFFFAOYSA-N 5-bromo-6-nitro-2,3-dihydro-1h-indole Chemical compound C1=C(Br)C([N+](=O)[O-])=CC2=C1CCN2 YABGMLXPMPPWKX-UHFFFAOYSA-N 0.000 description 1
- 125000006163 5-membered heteroaryl group Chemical group 0.000 description 1
- SESSPRSOEAVOMC-UHFFFAOYSA-N 5-methyl-2,4-dinitrobenzoic acid Chemical compound CC1=CC(C(O)=O)=C([N+]([O-])=O)C=C1[N+]([O-])=O SESSPRSOEAVOMC-UHFFFAOYSA-N 0.000 description 1
- JIXJNWVLWQQNDB-UHFFFAOYSA-N 6-nitro-1h-indole-2-carboxylic acid Chemical compound C1=C([N+]([O-])=O)C=C2NC(C(=O)O)=CC2=C1 JIXJNWVLWQQNDB-UHFFFAOYSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- SDDSJMXGJNWMJY-BRHAQHMBSA-N 7-[(2r,4ar,5r,7ar)-2-[(3s)-1,1-difluoro-3-methylpentyl]-2-hydroxy-6-oxo-3,4,4a,5,7,7a-hexahydrocyclopenta[b]pyran-5-yl]heptanoic acid Chemical compound O1[C@](C(F)(F)C[C@@H](C)CC)(O)CC[C@@H]2[C@@H](CCCCCCC(O)=O)C(=O)C[C@H]21 SDDSJMXGJNWMJY-BRHAQHMBSA-N 0.000 description 1
- WSWMGHRLUYADNA-UHFFFAOYSA-N 7-nitro-1,2,3,4-tetrahydroquinoline Chemical compound C1CCNC2=CC([N+](=O)[O-])=CC=C21 WSWMGHRLUYADNA-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 102000041092 ABC transporter family Human genes 0.000 description 1
- 108091060858 ABC transporter family Proteins 0.000 description 1
- 208000010444 Acidosis Diseases 0.000 description 1
- 206010027654 Allergic conditions Diseases 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 1
- 101710081722 Antitrypsin Proteins 0.000 description 1
- 235000003276 Apios tuberosa Nutrition 0.000 description 1
- 102000010637 Aquaporins Human genes 0.000 description 1
- 108010063290 Aquaporins Proteins 0.000 description 1
- 244000105624 Arachis hypogaea Species 0.000 description 1
- 235000010777 Arachis hypogaea Nutrition 0.000 description 1
- 235000010744 Arachis villosulicarpa Nutrition 0.000 description 1
- 206010003557 Asthma exercise induced Diseases 0.000 description 1
- 102000014461 Ataxins Human genes 0.000 description 1
- 108010078286 Ataxins Proteins 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 208000020084 Bone disease Diseases 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 206010006448 Bronchiolitis Diseases 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- PCTVZYCIOLEJIU-UHFFFAOYSA-N C(C)OC1=C(C=CC=C1)C1=C(C=C(C=C1)NC(O)=O)N Chemical compound C(C)OC1=C(C=CC=C1)C1=C(C=C(C=C1)NC(O)=O)N PCTVZYCIOLEJIU-UHFFFAOYSA-N 0.000 description 1
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical group C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 description 1
- 101100075829 Caenorhabditis elegans mab-3 gene Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 101000654354 Canis lupus familiaris Sodium channel protein type 10 subunit alpha Proteins 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 102100035673 Centrosomal protein of 290 kDa Human genes 0.000 description 1
- 206010008025 Cerebellar ataxia Diseases 0.000 description 1
- VGCXGMAHQTYDJK-UHFFFAOYSA-N Chloroacetyl chloride Chemical compound ClCC(Cl)=O VGCXGMAHQTYDJK-UHFFFAOYSA-N 0.000 description 1
- 206010008631 Cholera Diseases 0.000 description 1
- 101150065749 Churc1 gene Proteins 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 241000223935 Cryptosporidium Species 0.000 description 1
- 101710190437 Cytotoxin 3 Proteins 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- 241001366015 Desis Species 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- RPNUMPOLZDHAAY-UHFFFAOYSA-N Diethylenetriamine Chemical compound NCCNCCN RPNUMPOLZDHAAY-UHFFFAOYSA-N 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- 208000000059 Dyspnea Diseases 0.000 description 1
- 206010013975 Dyspnoeas Diseases 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- XXRCUYVCPSWGCC-UHFFFAOYSA-N Ethyl pyruvate Chemical compound CCOC(=O)C(C)=O XXRCUYVCPSWGCC-UHFFFAOYSA-N 0.000 description 1
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 1
- 208000004657 Exercise-Induced Asthma Diseases 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 108010049003 Fibrinogen Proteins 0.000 description 1
- 102000008946 Fibrinogen Human genes 0.000 description 1
- 239000004606 Fillers/Extenders Substances 0.000 description 1
- 206010016818 Fluorosis Diseases 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 102000002464 Galactosidases Human genes 0.000 description 1
- 108010093031 Galactosidases Proteins 0.000 description 1
- 241000206672 Gelidium Species 0.000 description 1
- 206010064571 Gene mutation Diseases 0.000 description 1
- 241000224467 Giardia intestinalis Species 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 102100020948 Growth hormone receptor Human genes 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- 229910004373 HOAc Inorganic materials 0.000 description 1
- 208000028782 Hereditary disease Diseases 0.000 description 1
- 239000004705 High-molecular-weight polyethylene Substances 0.000 description 1
- 101000715664 Homo sapiens Centrosomal protein of 290 kDa Proteins 0.000 description 1
- 101000907783 Homo sapiens Cystic fibrosis transmembrane conductance regulator Proteins 0.000 description 1
- 108091006905 Human Serum Albumin Proteins 0.000 description 1
- 102000008100 Human Serum Albumin Human genes 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 1
- 208000026350 Inborn Genetic disease Diseases 0.000 description 1
- 108010001127 Insulin Receptor Proteins 0.000 description 1
- 102100036721 Insulin receptor Human genes 0.000 description 1
- 102100027612 Kallikrein-11 Human genes 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 229910010084 LiAlH4 Inorganic materials 0.000 description 1
- 208000026709 Liddle syndrome Diseases 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 101150068888 MET3 gene Proteins 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 208000002720 Malnutrition Diseases 0.000 description 1
- 240000003183 Manihot esculenta Species 0.000 description 1
- 235000016735 Manihot esculenta subsp esculenta Nutrition 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- 102000003939 Membrane transport proteins Human genes 0.000 description 1
- 108090000301 Membrane transport proteins Proteins 0.000 description 1
- 108700011325 Modifier Genes Proteins 0.000 description 1
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Natural products C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 1
- 102000014842 Multidrug resistance proteins Human genes 0.000 description 1
- 108050005144 Multidrug resistance proteins Proteins 0.000 description 1
- XCUAIINAJCDIPM-XVFCMESISA-N N(4)-hydroxycytidine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=NO)C=C1 XCUAIINAJCDIPM-XVFCMESISA-N 0.000 description 1
- GXCLVBGFBYZDAG-UHFFFAOYSA-N N-[2-(1H-indol-3-yl)ethyl]-N-methylprop-2-en-1-amine Chemical compound CN(CCC1=CNC2=C1C=CC=C2)CC=C GXCLVBGFBYZDAG-UHFFFAOYSA-N 0.000 description 1
- 229910017912 NH2OH Inorganic materials 0.000 description 1
- 101100022915 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) cys-11 gene Proteins 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- PCKPVGOLPKLUHR-UHFFFAOYSA-N OH-Indolxyl Natural products C1=CC=C2C(O)=CNC2=C1 PCKPVGOLPKLUHR-UHFFFAOYSA-N 0.000 description 1
- HSVIINWFPKEOEQ-NSHDSACASA-N Ofloxacin methyl ester Chemical compound C([C@H](C)N1C=C(C(C(=C11)C=C2F)=O)C(=O)OC)OC1=C2N1CCN(C)CC1 HSVIINWFPKEOEQ-NSHDSACASA-N 0.000 description 1
- 240000007817 Olea europaea Species 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- 102100036893 Parathyroid hormone Human genes 0.000 description 1
- 101150034459 Parpbp gene Proteins 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 102100035917 Peripheral myelin protein 22 Human genes 0.000 description 1
- 101710199257 Peripheral myelin protein 22 Proteins 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- QSXMZJGGEWYVCN-UHFFFAOYSA-N Pirbuterol acetate Chemical compound CC(O)=O.CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=N1 QSXMZJGGEWYVCN-UHFFFAOYSA-N 0.000 description 1
- 229920002732 Polyanhydride Polymers 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 208000004880 Polyuria Diseases 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 108010050254 Presenilins Proteins 0.000 description 1
- 102000015499 Presenilins Human genes 0.000 description 1
- 108010050808 Procollagen Proteins 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 102000007327 Protamines Human genes 0.000 description 1
- 108010007568 Protamines Proteins 0.000 description 1
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 1
- 239000007868 Raney catalyst Substances 0.000 description 1
- 229910000564 Raney nickel Inorganic materials 0.000 description 1
- 108700005075 Regulator Genes Proteins 0.000 description 1
- 208000037656 Respiratory Sounds Diseases 0.000 description 1
- 235000004443 Ricinus communis Nutrition 0.000 description 1
- 241000702670 Rotavirus Species 0.000 description 1
- 241000607142 Salmonella Species 0.000 description 1
- 101100512790 Schizosaccharomyces pombe (strain 972 / ATCC 24843) mei3 gene Proteins 0.000 description 1
- 101100022918 Schizosaccharomyces pombe (strain 972 / ATCC 24843) sua1 gene Proteins 0.000 description 1
- 108010000303 Secretory Proteinase Inhibitory Proteins Proteins 0.000 description 1
- 102000002255 Secretory Proteinase Inhibitory Proteins Human genes 0.000 description 1
- 238000012300 Sequence Analysis Methods 0.000 description 1
- GIIZNNXWQWCKIB-UHFFFAOYSA-N Serevent Chemical compound C1=C(O)C(CO)=CC(C(O)CNCCCCCCOCCCCC=2C=CC=CC=2)=C1 GIIZNNXWQWCKIB-UHFFFAOYSA-N 0.000 description 1
- 206010041235 Snoring Diseases 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 235000002595 Solanum tuberosum Nutrition 0.000 description 1
- 244000061456 Solanum tuberosum Species 0.000 description 1
- 108010068542 Somatotropin Receptors Proteins 0.000 description 1
- 208000009415 Spinocerebellar Ataxias Diseases 0.000 description 1
- SSZBUIDZHHWXNJ-UHFFFAOYSA-N Stearinsaeure-hexadecylester Natural products CCCCCCCCCCCCCCCCCC(=O)OCCCCCCCCCCCCCCCC SSZBUIDZHHWXNJ-UHFFFAOYSA-N 0.000 description 1
- 102000018075 Subfamily B ATP Binding Cassette Transporter Human genes 0.000 description 1
- 108010091105 Subfamily B ATP Binding Cassette Transporter Proteins 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- 201000009594 Systemic Scleroderma Diseases 0.000 description 1
- 206010042953 Systemic sclerosis Diseases 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 229910021626 Tin(II) chloride Inorganic materials 0.000 description 1
- 241000009298 Trigla lyra Species 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 101710152431 Trypsin-like protease Proteins 0.000 description 1
- 102000003425 Tyrosinase Human genes 0.000 description 1
- 108060008724 Tyrosinase Proteins 0.000 description 1
- 108090000643 Vasopressin Receptors Proteins 0.000 description 1
- 102000004136 Vasopressin Receptors Human genes 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 239000001089 [(2R)-oxolan-2-yl]methanol Substances 0.000 description 1
- LXPZOELMLMOFST-UHFFFAOYSA-N [2-(2-adamantyl)-4-methyl-5-nitrophenyl] ethyl carbonate Chemical compound CCOC(=O)OC1=CC([N+]([O-])=O)=C(C)C=C1C1C(C2)CC3CC2CC1C3 LXPZOELMLMOFST-UHFFFAOYSA-N 0.000 description 1
- VUZBUFLXXRLXNM-UHFFFAOYSA-N [2-(2-adamantyl)-4-methylphenyl] ethyl carbonate Chemical compound CCOC(=O)OC1=CC=C(C)C=C1C1C(C2)CC3CC2CC1C3 VUZBUFLXXRLXNM-UHFFFAOYSA-N 0.000 description 1
- ZXQYGBMAQZUVMI-QQDHXZELSA-N [cyano-(3-phenoxyphenyl)methyl] (1r,3r)-3-[(z)-2-chloro-3,3,3-trifluoroprop-1-enyl]-2,2-dimethylcyclopropane-1-carboxylate Chemical compound CC1(C)[C@@H](\C=C(/Cl)C(F)(F)F)[C@H]1C(=O)OC(C#N)C1=CC=CC(OC=2C=CC=CC=2)=C1 ZXQYGBMAQZUVMI-QQDHXZELSA-N 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 239000003655 absorption accelerator Substances 0.000 description 1
- ORWKVZNEPHTCQE-UHFFFAOYSA-N acetic formic anhydride Chemical compound CC(=O)OC=O ORWKVZNEPHTCQE-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000007950 acidosis Effects 0.000 description 1
- 208000026545 acidosis disease Diseases 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 230000009056 active transport Effects 0.000 description 1
- 201000009840 acute diarrhea Diseases 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- NDAUXUAQIAJITI-UHFFFAOYSA-N albuterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-UHFFFAOYSA-N 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 201000009961 allergic asthma Diseases 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- HUMHYXGDUOGHTG-HEZXSMHISA-N alpha-D-GalpNAc-(1->3)-[alpha-L-Fucp-(1->2)]-D-Galp Chemical compound O[C@H]1[C@H](O)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](O[C@@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](O)[C@@H](CO)OC1O HUMHYXGDUOGHTG-HEZXSMHISA-N 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- LKCWBDHBTVXHDL-RMDFUYIESA-N amikacin Chemical compound O([C@@H]1[C@@H](N)C[C@H]([C@@H]([C@H]1O)O[C@@H]1[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O1)O)NC(=O)[C@@H](O)CCN)[C@H]1O[C@H](CN)[C@@H](O)[C@H](O)[C@H]1O LKCWBDHBTVXHDL-RMDFUYIESA-N 0.000 description 1
- 229960004821 amikacin Drugs 0.000 description 1
- XSDQTOBWRPYKKA-UHFFFAOYSA-N amiloride Chemical compound NC(=N)NC(=O)C1=NC(Cl)=C(N)N=C1N XSDQTOBWRPYKKA-UHFFFAOYSA-N 0.000 description 1
- 229960002576 amiloride Drugs 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- BIVUUOPIAYRCAP-UHFFFAOYSA-N aminoazanium;chloride Chemical class Cl.NN BIVUUOPIAYRCAP-UHFFFAOYSA-N 0.000 description 1
- 235000011114 ammonium hydroxide Nutrition 0.000 description 1
- 150000001448 anilines Chemical class 0.000 description 1
- 125000002490 anilino group Chemical group [H]N(*)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000001475 anti-trypsic effect Effects 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 125000005228 aryl sulfonate group Chemical group 0.000 description 1
- 125000005362 aryl sulfone group Chemical group 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 229960003995 ataluren Drugs 0.000 description 1
- 201000004562 autosomal dominant cerebellar ataxia Diseases 0.000 description 1
- 229960004099 azithromycin Drugs 0.000 description 1
- MQTOSJVFKKJCRP-BICOPXKESA-N azithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)N(C)C[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 MQTOSJVFKKJCRP-BICOPXKESA-N 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- CSKNSYBAZOQPLR-UHFFFAOYSA-N benzenesulfonyl chloride Chemical compound ClS(=O)(=O)C1=CC=CC=C1 CSKNSYBAZOQPLR-UHFFFAOYSA-N 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- MTZQAGJQAFMTAQ-UHFFFAOYSA-N benzoic acid ethyl ester Natural products CCOC(=O)C1=CC=CC=C1 MTZQAGJQAFMTAQ-UHFFFAOYSA-N 0.000 description 1
- 125000004190 benzothiazol-2-yl group Chemical group [H]C1=C([H])C([H])=C2N=C(*)SC2=C1[H] 0.000 description 1
- 229960002903 benzyl benzoate Drugs 0.000 description 1
- 102000007478 beta-N-Acetylhexosaminidases Human genes 0.000 description 1
- 108010085377 beta-N-Acetylhexosaminidases Proteins 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 230000036760 body temperature Effects 0.000 description 1
- 229910000085 borane Inorganic materials 0.000 description 1
- MCQRPQCQMGVWIQ-UHFFFAOYSA-N boron;methylsulfanylmethane Chemical compound [B].CSC MCQRPQCQMGVWIQ-UHFFFAOYSA-N 0.000 description 1
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 1
- RDHPKYGYEGBMSE-UHFFFAOYSA-N bromoethane Chemical compound CCBr RDHPKYGYEGBMSE-UHFFFAOYSA-N 0.000 description 1
- 239000004044 bronchoconstricting agent Substances 0.000 description 1
- 230000003435 bronchoconstrictive effect Effects 0.000 description 1
- 230000003182 bronchodilatating effect Effects 0.000 description 1
- DQXBYHZEEUGOBF-UHFFFAOYSA-N but-3-enoic acid;ethene Chemical compound C=C.OC(=O)CC=C DQXBYHZEEUGOBF-UHFFFAOYSA-N 0.000 description 1
- ZTQSAGDEMFDKMZ-UHFFFAOYSA-N butyric aldehyde Natural products CCCC=O ZTQSAGDEMFDKMZ-UHFFFAOYSA-N 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- FATUQANACHZLRT-KMRXSBRUSA-L calcium glucoheptonate Chemical compound [Ca+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O FATUQANACHZLRT-KMRXSBRUSA-L 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- XASIMHXSUQUHLV-UHFFFAOYSA-N camostat Chemical compound C1=CC(CC(=O)OCC(=O)N(C)C)=CC=C1OC(=O)C1=CC=C(N=C(N)N)C=C1 XASIMHXSUQUHLV-UHFFFAOYSA-N 0.000 description 1
- 229960000772 camostat Drugs 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 238000000423 cell based assay Methods 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 229910052729 chemical element Inorganic materials 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 150000001805 chlorine compounds Chemical class 0.000 description 1
- FZFAMSAMCHXGEF-UHFFFAOYSA-N chloro formate Chemical compound ClOC=O FZFAMSAMCHXGEF-UHFFFAOYSA-N 0.000 description 1
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- 208000019902 chronic diarrheal disease Diseases 0.000 description 1
- 239000003541 chymotrypsin inhibitor Substances 0.000 description 1
- 235000019504 cigarettes Nutrition 0.000 description 1
- 229960003405 ciprofloxacin Drugs 0.000 description 1
- TXXHDPDFNKHHGW-CCAGOZQPSA-N cis,cis-muconic acid Chemical compound OC(=O)\C=C/C=C\C(O)=O TXXHDPDFNKHHGW-CCAGOZQPSA-N 0.000 description 1
- 239000004927 clay Substances 0.000 description 1
- 229950005980 cobiprostone Drugs 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 238000010835 comparative analysis Methods 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 239000012050 conventional carrier Substances 0.000 description 1
- 229910000366 copper(II) sulfate Inorganic materials 0.000 description 1
- 210000000399 corneal endothelial cell Anatomy 0.000 description 1
- 239000003246 corticosteroid Substances 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 229940092125 creon Drugs 0.000 description 1
- LDHQCZJRKDOVOX-NSCUHMNNSA-N crotonic acid Chemical compound C\C=C\C(O)=O LDHQCZJRKDOVOX-NSCUHMNNSA-N 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000005520 cutting process Methods 0.000 description 1
- 125000000392 cycloalkenyl group Chemical group 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 230000007123 defense Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 108700041286 delta Proteins 0.000 description 1
- 208000004042 dental fluorosis Diseases 0.000 description 1
- CARVNSROHCBVAO-BUGJESOBSA-N depelestat Chemical compound O=C([C@H](C(C)C)NC(=O)CNC(=O)[C@@H]1CSSC[C@@H](C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N2CCC[C@H]2C(=O)N[C@H](C(N[C@H](C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N2CCC[C@H]2C(=O)N[C@@H]2C(=O)N[C@H](C(=O)N[C@@H](C)C(=O)N[C@@H](CC=3C=CC=CC=3)C(=O)N[C@@H](CC=3C=CC=CC=3)C(=O)N3CCC[C@H]3C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=3C4=CC=CC=C4NC=3)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=3C=CC=CC=3)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@H](C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@H]3CSSC[C@H](NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=4C=CC(O)=CC=4)NC(=O)[C@H](CC=4C=CC=CC=4)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC(N)=O)NC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC=4C=CC(O)=CC=4)NC(=O)[C@H]4N(CCC4)C(=O)[C@H](CC=4C=CC=CC=4)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC3=O)C(C)C)CSSC2)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N1)C(C)C)[C@@H](C)CC)C(C)C)=O)[C@@H](C)CC)NC(=O)[C@H](C)NC(=O)[C@@H](N)CCC(O)=O)N1CCC[C@H]1C(O)=O CARVNSROHCBVAO-BUGJESOBSA-N 0.000 description 1
- 108010077021 depelestat Proteins 0.000 description 1
- 229950003912 depelestat Drugs 0.000 description 1
- 201000001981 dermatomyositis Diseases 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 230000000741 diarrhetic effect Effects 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- PYPCDUKQEIPHAF-UHFFFAOYSA-N diethyl 2-(anilinomethylidene)propanedioate Chemical compound CCOC(=O)C(C(=O)OCC)=CNC1=CC=CC=C1 PYPCDUKQEIPHAF-UHFFFAOYSA-N 0.000 description 1
- LTMHNWPUDSTBKD-UHFFFAOYSA-N diethyl 2-(ethoxymethylidene)propanedioate Chemical compound CCOC=C(C(=O)OCC)C(=O)OCC LTMHNWPUDSTBKD-UHFFFAOYSA-N 0.000 description 1
- NFWNKZGKJXKUKP-UHFFFAOYSA-N diethyl 2-[[(3-oxocyclohexen-1-yl)amino]methylidene]propanedioate Chemical compound CCOC(=O)C(C(=O)OCC)=CNC1=CC(=O)CCC1 NFWNKZGKJXKUKP-UHFFFAOYSA-N 0.000 description 1
- 230000001079 digestive effect Effects 0.000 description 1
- GXGAKHNRMVGRPK-UHFFFAOYSA-N dimagnesium;dioxido-bis[[oxido(oxo)silyl]oxy]silane Chemical compound [Mg+2].[Mg+2].[O-][Si](=O)O[Si]([O-])([O-])O[Si]([O-])=O GXGAKHNRMVGRPK-UHFFFAOYSA-N 0.000 description 1
- 150000002009 diols Chemical class 0.000 description 1
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 1
- 229910000396 dipotassium phosphate Inorganic materials 0.000 description 1
- 235000019797 dipotassium phosphate Nutrition 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- XNFVGEUMTFIVHQ-UHFFFAOYSA-N disodium;sulfide;hydrate Chemical compound O.[Na+].[Na+].[S-2] XNFVGEUMTFIVHQ-UHFFFAOYSA-N 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 230000035619 diuresis Effects 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- 108010067396 dornase alfa Proteins 0.000 description 1
- 239000003596 drug target Substances 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 239000003221 ear drop Substances 0.000 description 1
- 229940047652 ear drops Drugs 0.000 description 1
- 238000003372 electrophysiological method Methods 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 239000002713 epithelial sodium channel blocking agent Substances 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- DQYBDCGIPTYXML-UHFFFAOYSA-N ethoxyethane;hydrate Chemical compound O.CCOCC DQYBDCGIPTYXML-UHFFFAOYSA-N 0.000 description 1
- 125000005448 ethoxyethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- MQSRIZPXSLOTND-UHFFFAOYSA-N ethyl 2,4-dimethoxyquinoline-3-carboxylate Chemical compound C1=CC=CC2=C(OC)C(C(=O)OCC)=C(OC)N=C21 MQSRIZPXSLOTND-UHFFFAOYSA-N 0.000 description 1
- IKGAXAXURUGYGO-UHFFFAOYSA-N ethyl 2-[2-(dimethylamino)ethenyl]-3,5-dinitrobenzoate Chemical compound CCOC(=O)C1=CC([N+]([O-])=O)=CC([N+]([O-])=O)=C1C=CN(C)C IKGAXAXURUGYGO-UHFFFAOYSA-N 0.000 description 1
- DJFQGBZJRMAHMS-UHFFFAOYSA-N ethyl 2-methyl-3,5-dinitrobenzoate Chemical compound CCOC(=O)C1=CC([N+]([O-])=O)=CC([N+]([O-])=O)=C1C DJFQGBZJRMAHMS-UHFFFAOYSA-N 0.000 description 1
- IOLXKMCXWZWJDI-UHFFFAOYSA-N ethyl 3-[2-(dimethylamino)ethenyl]-2,6-dinitrobenzoate Chemical compound CCOC(=O)C1=C([N+]([O-])=O)C=CC(C=CN(C)C)=C1[N+]([O-])=O IOLXKMCXWZWJDI-UHFFFAOYSA-N 0.000 description 1
- LXUABEANNNKOLF-UHFFFAOYSA-N ethyl 4-nitro-1h-indole-2-carboxylate Chemical compound C1=CC=C2NC(C(=O)OCC)=CC2=C1[N+]([O-])=O LXUABEANNNKOLF-UHFFFAOYSA-N 0.000 description 1
- YBEOYBKKSWUSBR-UHFFFAOYSA-N ethyl 4-oxo-1h-quinoline-3-carboxylate Chemical compound C1=CC=C2C(=O)C(C(=O)OCC)=CNC2=C1 YBEOYBKKSWUSBR-UHFFFAOYSA-N 0.000 description 1
- IUNWYBCKKXSNTK-UHFFFAOYSA-N ethyl 5-methyl-2,4-dinitrobenzoate Chemical compound CCOC(=O)C1=CC(C)=C([N+]([O-])=O)C=C1[N+]([O-])=O IUNWYBCKKXSNTK-UHFFFAOYSA-N 0.000 description 1
- SOMZLSPJSSDTAP-UHFFFAOYSA-N ethyl 6-nitro-1h-indole-2-carboxylate Chemical compound C1=C([N+]([O-])=O)C=C2NC(C(=O)OCC)=CC2=C1 SOMZLSPJSSDTAP-UHFFFAOYSA-N 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- MFFXVVHUKRKXCI-UHFFFAOYSA-N ethyl iodoacetate Chemical compound CCOC(=O)CI MFFXVVHUKRKXCI-UHFFFAOYSA-N 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 239000005038 ethylene vinyl acetate Substances 0.000 description 1
- UKFXDFUAPNAMPJ-UHFFFAOYSA-N ethylmalonic acid Chemical compound CCC(C(O)=O)C(O)=O UKFXDFUAPNAMPJ-UHFFFAOYSA-N 0.000 description 1
- 230000005713 exacerbation Effects 0.000 description 1
- 208000024695 exercise-induced bronchoconstriction Diseases 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 101150023575 fer3 gene Proteins 0.000 description 1
- 230000035558 fertility Effects 0.000 description 1
- 229940012952 fibrinogen Drugs 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- VUWZPRWSIVNGKG-UHFFFAOYSA-N fluoromethane Chemical compound F[CH2] VUWZPRWSIVNGKG-UHFFFAOYSA-N 0.000 description 1
- 125000001207 fluorophenyl group Chemical group 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- FATAVLOOLIRUNA-UHFFFAOYSA-N formylmethyl Chemical group [CH2]C=O FATAVLOOLIRUNA-UHFFFAOYSA-N 0.000 description 1
- 229960000308 fosfomycin Drugs 0.000 description 1
- YMDXZJFXQJVXBF-STHAYSLISA-N fosfomycin Chemical compound C[C@@H]1O[C@@H]1P(O)(O)=O YMDXZJFXQJVXBF-STHAYSLISA-N 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 208000016361 genetic disease Diseases 0.000 description 1
- TZBJGXHYKVUXJN-UHFFFAOYSA-N genistein Natural products C1=CC(O)=CC=C1C1=COC2=CC(O)=CC(O)=C2C1=O TZBJGXHYKVUXJN-UHFFFAOYSA-N 0.000 description 1
- 229940085435 giardia lamblia Drugs 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 235000001727 glucose Nutrition 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- 229960002449 glycine Drugs 0.000 description 1
- 125000003630 glycyl group Chemical group [H]N([H])C([H])([H])C(*)=O 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 125000004438 haloalkoxy group Chemical group 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- XLYOFNOQVPJJNP-ZSJDYOACSA-N heavy water Substances [2H]O[2H] XLYOFNOQVPJJNP-ZSJDYOACSA-N 0.000 description 1
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 102000056427 human CFTR Human genes 0.000 description 1
- 239000003906 humectant Substances 0.000 description 1
- 230000036571 hydration Effects 0.000 description 1
- 238000006703 hydration reaction Methods 0.000 description 1
- YPGCWEMNNLXISK-UHFFFAOYSA-N hydratropic acid Chemical compound OC(=O)C(C)C1=CC=CC=C1 YPGCWEMNNLXISK-UHFFFAOYSA-N 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- WVKIFIROCHIWAY-UHFFFAOYSA-N hydron;2-(methylamino)acetic acid;chloride Chemical compound Cl.CNCC(O)=O WVKIFIROCHIWAY-UHFFFAOYSA-N 0.000 description 1
- FUKUFMFMCZIRNT-UHFFFAOYSA-N hydron;methanol;chloride Chemical compound Cl.OC FUKUFMFMCZIRNT-UHFFFAOYSA-N 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- XXSMGPRMXLTPCZ-UHFFFAOYSA-N hydroxychloroquine Chemical compound ClC1=CC=C2C(NC(C)CCCN(CCO)CC)=CC=NC2=C1 XXSMGPRMXLTPCZ-UHFFFAOYSA-N 0.000 description 1
- 229960004171 hydroxychloroquine Drugs 0.000 description 1
- 125000004464 hydroxyphenyl group Chemical group 0.000 description 1
- 230000001077 hypotensive effect Effects 0.000 description 1
- 230000007954 hypoxia Effects 0.000 description 1
- 230000001146 hypoxic effect Effects 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 229910052738 indium Inorganic materials 0.000 description 1
- JYGFTBXVXVMTGB-UHFFFAOYSA-N indolin-2-one Chemical compound C1=CC=C2NC(=O)CC2=C1 JYGFTBXVXVMTGB-UHFFFAOYSA-N 0.000 description 1
- LPAGFVYQRIESJQ-UHFFFAOYSA-N indoline Chemical compound C1=CC=C2NCCC2=C1 LPAGFVYQRIESJQ-UHFFFAOYSA-N 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 239000012678 infectious agent Substances 0.000 description 1
- 208000000509 infertility Diseases 0.000 description 1
- 230000036512 infertility Effects 0.000 description 1
- 231100000535 infertility Toxicity 0.000 description 1
- 208000021267 infertility disease Diseases 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 229940102213 injectable suspension Drugs 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 208000003243 intestinal obstruction Diseases 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- CPJRRXSHAYUTGL-UHFFFAOYSA-N isopentenyl alcohol Chemical compound CC(=C)CCO CPJRRXSHAYUTGL-UHFFFAOYSA-N 0.000 description 1
- 125000000555 isopropenyl group Chemical group [H]\C([H])=C(\*)C([H])([H])[H] 0.000 description 1
- FMKOJHQHASLBPH-UHFFFAOYSA-N isopropyl iodide Chemical compound CC(C)I FMKOJHQHASLBPH-UHFFFAOYSA-N 0.000 description 1
- 150000002540 isothiocyanates Chemical class 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-N lactobionic acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-N 0.000 description 1
- 229940070765 laurate Drugs 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 239000003591 leukocyte elastase inhibitor Substances 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 230000004199 lung function Effects 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 229910000386 magnesium trisilicate Inorganic materials 0.000 description 1
- 235000019793 magnesium trisilicate Nutrition 0.000 description 1
- 229940099273 magnesium trisilicate Drugs 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 230000001071 malnutrition Effects 0.000 description 1
- 235000000824 malnutrition Nutrition 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- QLNWXBAGRTUKKI-UHFFFAOYSA-N metacetamol Chemical compound CC(=O)NC1=CC=CC(O)=C1 QLNWXBAGRTUKKI-UHFFFAOYSA-N 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- AWUPLMYXZJKHEG-UHFFFAOYSA-N methyl 2-chloro-2,2-difluoroacetate Chemical compound COC(=O)C(F)(F)Cl AWUPLMYXZJKHEG-UHFFFAOYSA-N 0.000 description 1
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical group COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 1
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 1
- UFEJKYYYVXYMMS-UHFFFAOYSA-N methylcarbamic acid Chemical compound CNC(O)=O UFEJKYYYVXYMMS-UHFFFAOYSA-N 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 230000003274 myotonic effect Effects 0.000 description 1
- LNQQUYVEYRCVQF-UHFFFAOYSA-N n-(3-amino-4-methylphenyl)-4-oxo-1h-quinoline-3-carboxamide Chemical compound C1=C(N)C(C)=CC=C1NC(=O)C1=CNC2=CC=CC=C2C1=O LNQQUYVEYRCVQF-UHFFFAOYSA-N 0.000 description 1
- GQHSRKKKIUETPF-UHFFFAOYSA-N n-(4,4-dimethyl-2,3-dihydrochromen-7-yl)acetamide Chemical compound O1CCC(C)(C)C=2C1=CC(NC(=O)C)=CC=2 GQHSRKKKIUETPF-UHFFFAOYSA-N 0.000 description 1
- UOBXADQDGUWLQN-UHFFFAOYSA-N n-[3-(3-methylbut-3-enoxy)phenyl]acetamide Chemical compound CC(=C)CCOC1=CC=CC(NC(C)=O)=C1 UOBXADQDGUWLQN-UHFFFAOYSA-N 0.000 description 1
- ZVSJFPOKRSIYSH-UHFFFAOYSA-N n-butyl-3-nitroaniline Chemical compound CCCCNC1=CC=CC([N+]([O-])=O)=C1 ZVSJFPOKRSIYSH-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-M naphthalene-2-sulfonate Chemical compound C1=CC=CC2=CC(S(=O)(=O)[O-])=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-M 0.000 description 1
- 229940097496 nasal spray Drugs 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- LQNUZADURLCDLV-UHFFFAOYSA-N nitrobenzene Substances [O-][N+](=O)C1=CC=CC=C1 LQNUZADURLCDLV-UHFFFAOYSA-N 0.000 description 1
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 1
- 230000008518 non respiratory effect Effects 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- 239000012038 nucleophile Substances 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 208000015380 nutritional deficiency disease Diseases 0.000 description 1
- 208000007892 occupational asthma Diseases 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 229940041678 oral spray Drugs 0.000 description 1
- 239000000668 oral spray Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- WCPAKWJPBJAGKN-UHFFFAOYSA-N oxadiazole Chemical compound C1=CON=N1 WCPAKWJPBJAGKN-UHFFFAOYSA-N 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 125000000636 p-nitrophenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)[N+]([O-])=O 0.000 description 1
- 125000001037 p-tolyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical compound [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 1
- 239000002831 pharmacologic agent Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N phenylalanine group Chemical group N[C@@H](CC1=CC=CC=C1)C(=O)O COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 230000035479 physiological effects, processes and functions Effects 0.000 description 1
- 229940075930 picrate Drugs 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-M picrate anion Chemical compound [O-]C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-M 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229960004994 pirbuterol acetate Drugs 0.000 description 1
- 229950010765 pivalate Drugs 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 229920001200 poly(ethylene-vinyl acetate) Polymers 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 239000004632 polycaprolactone Substances 0.000 description 1
- 229920001610 polycaprolactone Polymers 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- 229920000137 polyphosphoric acid Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 229920001021 polysulfide Polymers 0.000 description 1
- 239000005077 polysulfide Substances 0.000 description 1
- 150000008117 polysulfides Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 235000011181 potassium carbonates Nutrition 0.000 description 1
- NNFCIKHAZHQZJG-UHFFFAOYSA-N potassium cyanide Chemical compound [K+].N#[C-] NNFCIKHAZHQZJG-UHFFFAOYSA-N 0.000 description 1
- WFIZEGIEIOHZCP-UHFFFAOYSA-M potassium formate Chemical compound [K+].[O-]C=O WFIZEGIEIOHZCP-UHFFFAOYSA-M 0.000 description 1
- 239000004323 potassium nitrate Substances 0.000 description 1
- 235000010241 potassium sorbate Nutrition 0.000 description 1
- 239000004302 potassium sorbate Substances 0.000 description 1
- 229940069338 potassium sorbate Drugs 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 108010091624 preproparathormone Proteins 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 229950008679 protamine sulfate Drugs 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 238000001799 protein solubilization Methods 0.000 description 1
- 230000007925 protein solubilization Effects 0.000 description 1
- 208000007153 proteostasis deficiencies Diseases 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 229940107568 pulmozyme Drugs 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 201000010727 rectal prolapse Diseases 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 239000011369 resultant mixture Substances 0.000 description 1
- 239000003340 retarding agent Substances 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 102200073741 rs121909602 Human genes 0.000 description 1
- 229960002052 salbutamol Drugs 0.000 description 1
- 210000003296 saliva Anatomy 0.000 description 1
- 229960004017 salmeterol Drugs 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- DCKVNWZUADLDEH-UHFFFAOYSA-N sec-butyl acetate Chemical compound CCC(C)OC(C)=O DCKVNWZUADLDEH-UHFFFAOYSA-N 0.000 description 1
- 230000000580 secretagogue effect Effects 0.000 description 1
- 210000000582 semen Anatomy 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 229960003310 sildenafil Drugs 0.000 description 1
- 150000004760 silicates Chemical class 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 229960000802 sinapultide Drugs 0.000 description 1
- 108010081062 sinapultide Proteins 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- AWUCVROLDVIAJX-GSVOUGTGSA-N sn-glycerol 3-phosphate Chemical compound OC[C@@H](O)COP(O)(O)=O AWUCVROLDVIAJX-GSVOUGTGSA-N 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 229940125794 sodium channel blocker Drugs 0.000 description 1
- 239000003195 sodium channel blocking agent Substances 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- VWDWKYIASSYTQR-UHFFFAOYSA-N sodium nitrate Inorganic materials [Na+].[O-][N+]([O-])=O VWDWKYIASSYTQR-UHFFFAOYSA-N 0.000 description 1
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 1
- 239000008279 sol Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- ZRRGOUHITGRLBA-UHFFFAOYSA-N stattic Chemical compound [O-][N+](=O)C1=CC=C2C=CS(=O)(=O)C2=C1 ZRRGOUHITGRLBA-UHFFFAOYSA-N 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 239000003206 sterilizing agent Substances 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 210000004243 sweat Anatomy 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- IDMWHCGVDQMWTG-UHFFFAOYSA-N tert-butyl n-(3-nitro-4-propylphenyl)carbamate Chemical compound CCCC1=CC=C(NC(=O)OC(C)(C)C)C=C1[N+]([O-])=O IDMWHCGVDQMWTG-UHFFFAOYSA-N 0.000 description 1
- KEVMXKJNAMYXDE-UHFFFAOYSA-N tert-butyl n-methyl-n-(3-nitro-4-propylphenyl)carbamate Chemical compound CCCC1=CC=C(N(C)C(=O)OC(C)(C)C)C=C1[N+]([O-])=O KEVMXKJNAMYXDE-UHFFFAOYSA-N 0.000 description 1
- ZGNPLWZYVAFUNZ-UHFFFAOYSA-N tert-butylphosphane Chemical compound CC(C)(C)P ZGNPLWZYVAFUNZ-UHFFFAOYSA-N 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000006223 tetrahydrofuranylmethyl group Chemical group 0.000 description 1
- BSYVTEYKTMYBMK-UHFFFAOYSA-N tetrahydrofurfuryl alcohol Chemical compound OCC1CCCO1 BSYVTEYKTMYBMK-UHFFFAOYSA-N 0.000 description 1
- PASYJVRFGUDDEW-WMUGRWSXSA-J tetrasodium;[[(2r,3s,5r)-5-(4-amino-2-oxopyrimidin-1-yl)-3-hydroxyoxolan-2-yl]methoxy-oxidophosphoryl] [[[(2r,3s,4r,5r)-5-(2,4-dioxopyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-oxidophosphoryl]oxy-oxidophosphoryl] phosphate Chemical compound [Na+].[Na+].[Na+].[Na+].O=C1N=C(N)C=CN1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])(=O)OP([O-])(=O)OC[C@@H]2[C@H]([C@@H](O)[C@@H](O2)N2C(NC(=O)C=C2)=O)O)[C@@H](O)C1 PASYJVRFGUDDEW-WMUGRWSXSA-J 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- AXZWODMDQAVCJE-UHFFFAOYSA-L tin(II) chloride (anhydrous) Chemical compound [Cl-].[Cl-].[Sn+2] AXZWODMDQAVCJE-UHFFFAOYSA-L 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- CWMFRHBXRUITQE-UHFFFAOYSA-N trimethylsilylacetylene Chemical group C[Si](C)(C)C#C CWMFRHBXRUITQE-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 229940054369 ultrase Drugs 0.000 description 1
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical compound CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical class CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 210000002268 wool Anatomy 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C215/00—Compounds containing amino and hydroxy groups bound to the same carbon skeleton
- C07C215/74—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups and amino groups bound to carbon atoms of six-membered aromatic rings of the same carbon skeleton
- C07C215/76—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups and amino groups bound to carbon atoms of six-membered aromatic rings of the same carbon skeleton of the same non-condensed six-membered aromatic ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/10—Laxatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/04—Artificial tears; Irrigation solutions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
- A61P5/16—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4 for decreasing, blocking or antagonising the activity of the thyroid hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/18—Drugs for disorders of the endocrine system of the parathyroid hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/12—Antidiuretics, e.g. drugs for diabetes insipidus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/08—Indoles; Hydrogenated indoles with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/14—Radicals substituted by nitrogen atoms, not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/18—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/30—Indoles; Hydrogenated indoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to carbon atoms of the hetero ring
- C07D209/42—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/80—[b, c]- or [b, d]-condensed
- C07D209/94—[b, c]- or [b, d]-condensed containing carbocyclic rings other than six-membered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/06—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom containing only hydrogen and carbon atoms in addition to the ring nitrogen atom
- C07D213/16—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom containing only hydrogen and carbon atoms in addition to the ring nitrogen atom containing only one pyridine ring
- C07D213/20—Quaternary compounds thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/73—Unsubstituted amino or imino radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/20—Oxygen atoms
- C07D215/22—Oxygen atoms attached in position 2 or 4
- C07D215/227—Oxygen atoms attached in position 2 or 4 only one oxygen atom which is attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/38—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/48—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
- C07D215/54—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen attached in position 3
- C07D215/56—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen attached in position 3 with oxygen atoms in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/02—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with only hydrogen atoms or radicals containing only carbon and hydrogen atoms, directly attached to carbon atoms of the nitrogen-containing ring; Alkylene-bis-isoquinolines
- C07D217/06—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with only hydrogen atoms or radicals containing only carbon and hydrogen atoms, directly attached to carbon atoms of the nitrogen-containing ring; Alkylene-bis-isoquinolines with the ring nitrogen atom acylated by carboxylic or carbonic acids, or with sulfur or nitrogen analogues thereof, e.g. carbamates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D223/00—Heterocyclic compounds containing seven-membered rings having one nitrogen atom as the only ring hetero atom
- C07D223/14—Heterocyclic compounds containing seven-membered rings having one nitrogen atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D223/16—Benzazepines; Hydrogenated benzazepines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D265/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom and one oxygen atom as the only ring hetero atoms
- C07D265/28—1,4-Oxazines; Hydrogenated 1,4-oxazines
- C07D265/34—1,4-Oxazines; Hydrogenated 1,4-oxazines condensed with carbocyclic rings
- C07D265/36—1,4-Oxazines; Hydrogenated 1,4-oxazines condensed with carbocyclic rings condensed with one six-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D279/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom and one sulfur atom as the only ring hetero atoms
- C07D279/10—1,4-Thiazines; Hydrogenated 1,4-thiazines
- C07D279/14—1,4-Thiazines; Hydrogenated 1,4-thiazines condensed with carbocyclic rings or ring systems
- C07D279/16—1,4-Thiazines; Hydrogenated 1,4-thiazines condensed with carbocyclic rings or ring systems condensed with one six-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D311/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings
- C07D311/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D311/04—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring
- C07D311/58—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring other than with oxygen or sulphur atoms in position 2 or 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/10—Spiro-condensed systems
Definitions
- the present invention relates to modulators of ATP-Bmding Cassette (“ABC”) transporters or fragments thereof, including cystic fibrosis transmembrane conductance regulator (“CFTR”), compositions thereof, and methods therewith.
- the present invention also relates to methods of treating ABC transporter mediated diseases using such modulators,
- ABC transporters are a family of membrane transporter proteins that regulate the transport of a wide variety of pharmacological agents, potentially toxic drugs, and xenobioiics, as well as anions, ABC transporters are homologous membrane proteins mat bind and use cellular adenosine triphosphate (ATI 5 ) for their specific activities. Some of these transporters were discovered as multidrug resistance proteins (like the MDR1-P glycoprotein, or tire multidrug resistance protein, MRP! ), defending malignant cancer cells against chemotherapeutic agents. To date, 48 ABC Transporters have been identified and grouped into 7 families based on their sequence identity and function.
- ABC transporters regulate a variety of important physiological roles within the body and provide defense against harmful environmental compounds. Because of this, they represent important potential drug targets for the treatment of diseases associated with defects in the transporter, prevention of drug transport out of the target cell, and intervention in other diseases in which modulation of ABC transporter activity may be beneficial.
- CFTR cAMP/ATP-mediated anion channel
- CFTR is expressed In a variety of cells types, including absorptive and secretory epithelia cells, where It regulates anion flux across the membrane, as well as the activity of other ion channels and proteins. In epithelia ceils, normal functioning of CFTR is critical for the maintenance of electrolyte transport throughout the body, including respiratory and digestive tissue.
- CFTR is composed of approximately 1480 amino acids that encode a protein made up of a tandem repeat of transmembrane domains, each containing six transmembrane helices and a nucleotide binding domain. Be two transmembrane domains are linked by a large, polar, regulatory (R)-domain with multiple phosphorylation sites that regulate channel activity and cellular trafficking.
- CFTR cystic fibrosis
- a defect in this gene causes mutations in CFTR resulting in cystic fibrosis ("CF"), the most common fatal genetic diseas in humans. Cystic fibrosis affects approximately one in every 2,500 infants in the United States. Within the general United Stales population, up to 10 million people carry a single copy of the defective gene without apparent ill effects, in contrast, individuals with two copies of the CF associated gene suffer from the debilitating and fatal effects of CF, including chronic lung disease.
- Ri I7H-CFTR and G551D-CFTR other disease causing mutations in CFTR that result in defective trafficking, synthesis, and or channel gating could be up- or down-regulated to alter anion secretion and modify disease progression and/or severity.
- CFTR transports a variety of molecules in addition to anions
- this role represents one element in an important mechanism of transporting ions and water across the epithelium.
- the other elements include the epithelia! Na ⁇ channel, ENaC, Na + /2C!YK + co-transporter, a + -K + - ATPase pump and the basolaterai membrane K' channels, that are responsible for the uptake of chloride into the cell.
- Mutations in CFTR that are associated with moderate CFTR dysfunction are also evident in patients with conditions that share certain disease manifestations with CF but do not meet the diagnostic criteria for CF. These include congenital bilateral absence of die vas deferens, idiopathic chronic pancreatitis, chronic bronchitis, and chronic rhinosinusitis. Other diseases in which mutant CFTR is believed to be a risk factor along with modifier genes
- environmental factors include primary sclerosing cholangitis, allergic bronchopulmonary aspergillosis, and asthma,
- Cigarette smoke, hypoxia, and environmental factors that induce hypoxic signaling have also been demonstrated to impair CFT function and may contribute to certain forms of respiratory disease, such as chronic bronchitis. Diseases that ma be due to defective CFTR function but do not meet the diagnostic criteria for CF are characterized as CFTR- related diseases.
- CFTR in addition to cystic fibrosis, modulation of CFTR activity may be beneficial for other diseases not directly caused by mutations in CFTR, such as secretory diseases and other protein folding diseases mediated by CFTR.
- CFTR regulates chloride and bicarbonate flux across the epithe!ia of many cells to control fluid movement, protein solubilization, mucus viscosity, and enzyme activity. Defects in CFTR can cause blockage of the airway or ducts in many organs, including the liver and pancreas.
- Potentiators are compounds that enhance the gating activity of CFTR present in the cell membrane. Any disease which involves thickening of the mucus, impaired fluid regulation, impaired mucus clearance, or blocked ducts leading to inflammation and tissue destruction could be a candidate for potentiators,
- COPD chronic obstructive pulmonary disease
- asthma smoke induced COPD
- chronic bronchitis chronic bronchitis
- rhinosinusitis constipation
- dry eye disease dry eye disease
- Sjogren's Syndrome gastroesophageal reflux disease
- gallstones rectal prolapse
- inflammatory bowel disease characterized by airflow limitation that is progressive and not fully reversible.
- the airflow limitation is due to mucus hypersecretion, emphysema, and bronchiolitis.
- Activators of mutant or wild-type CFTR offer a potential treatment of mucus hypersecretion and impaired mucociliary clearance that is common in COPD.
- CFTR modulators may prevent or slow the parenchimal destruction of the airway that characterizes emphysema and reduce or reverse the increase in mucus secreting cell number and size that underiyses mucus hypersecretion in airway diseases. Dry eye disease is characterized by a decrease in tear- aqueous production and abnormal tear film lipid, protein and mucin profiles.
- Sjogrens's syndrome is an autoimmune disease in which the immune system attacks moisture-producing glands throughout the body, including the eye, mouth, skin, respiratory tissue, liver, vagina, and gut. Symptoms, include, dry eye, mouth, and vagina, as well as lung disease.
- the disease is also associated with rheumatoid arthritis, systemic lupus, systemic sclerosis, and polymypositis/dermatomyositis. Defective protein trafficking is believed to cause the disease, for which treatment options are limited.
- Modulators of CFTR activity may hydrate the various organs afflicted by the disease and may help to alleviate the associated symptoms.
- Individuals with cystic fibrosis have recurrent episodes of intestinal obstruction and higher incidences of rectal polapse, gallstones, gastroesophageal reflux disease, GI malignancies, and inflammatory bowel disease, indicating that CFTR function may play an important role in preventing such diseases.
- the diseases associated with the first class of ER malfunction are cystic fibrosis (due to misfolded AF5Q8-CFTR as discussed above), hereditary emphysema (due to a 1 -antitrypsin; non Piz variants), hereditar hemochromatosis, coaguiation-fibrinolysis deficiencies, such as protein C deficiency, Type 1 hereditary angioedema, lipid processing deficiencies, such as familial hypercholesterolemia. Type 1 chylor eronernia,
- abetalipoproteineniia lysosomal storage diseases, such as I-celi disease/pseudo-Hurler, Mucopolysaccharidoses (due to lysosomal processing enzymes), Sandhof/Tay-Sachs (due to ⁇ - hexosaminidase), Crigler-Najjar type II (due to UDP-glucuronyl-sialyc-transferase), polyendocrinopathy/hyperinsulemia, Diabetes mellitus (due to insulin receptor), Laron
- hypoparathyroidism due to preproparathyroid hormone
- melanoma due to tyrosinase
- the diseases associated with the latter class of ER malfunction are Glycanosis CDG type 1, hereditary emphysema (due to al- Antitrypsin (PiZ variant), congenital hyperthyroidism, osteogenesis imperfecta (due to Type I, ⁇ , IV procollagen), hereditary hypofibrinogenemia (due to fibrinogen), ACT deficiency (due to al-antichymotrypsin), Diabetes insipidus (DI), neurophysea!
- neprogenic DI due to aquaporin II
- Charcot-Marie Tooth syndrome due to peripheral myelin protein 22
- Perlizaeus- Merzbacher disease neurodegenerative diseases such as Alzheimer's disease (due to ⁇ and presenilins), Parkinson's disease, amyotrophic lateral sclerosis, progressive supranuclear palsy, Pick's disease, several poiyglutamine neurological disorders such as Huntington's, spinocerebullar ataxia type I, spinal and bulbar muscular atrophy, dentatorubal pallidoluysian, and myotonic dystrophy, as well as spongiform encephalopathies, such as hereditary
- Creutzfeldt- Jakob disease due to prion protein processing defect
- Fabry disease due to lysosomal a ⁇ galactosidase A
- Straussler-Scheinker syndrome due to Prp processing defect
- infertility pancreatitis pancreatic insufficiency
- osteoporosis osteopenia, Gorham's
- PCD Primary Ciliary Dyskinesia
- PCD with situs inversus also known as Kartagener syndrome
- PCD without situs inversus and ciliary aplasia and liver disease.
- CFTR- opathies disease phenotypes associated with cystic fibrosis transmembrane regulator gene mutations
- CFTR modulators may be beneficial for the treatment of secretory diarrheas, in which epithelial water transport is dramatically increased as a result of secretagogue activated chloride transport.
- the mechanism involves elevation of cAMP and stimulation of CFTR.
- Diarrhea is both a significant factor in malnutrition and the leading cause of death (5,000,000 deaths/year) in children less than five years old.
- Diarrhea in bam animals and pets such as cows, pigs and horses, sheep, goats, cats and dogs, also known as scours, is a major cause of death in these animals. Diarrhea can result from any major transition, such as weaning or physical movement, as well as in response to a variety of bacterial or viral infections and generally occurs within the first few hours of the animal's life.
- ETEC enterotoxogenic E.coli
- Common viral causes of diarrhea include rotavirus and coronavirus.
- Oilier infectious agents include Cryptosporidium, giardia lamblia, and salmonella, among others.
- Symptoms of rotavirai infection include excretion of watery feces, dehydration and weakness. Coronavirus causes a more severe illness in the newborn animals, and has a higher mortality rate than rotavirai infection. Often, however, a young animal may be infected with more than one virus or with a combination of viral and bacterial microorganisms at one time. This dramatically increases the severity of the disease.
- R 1 , R 2 , R 3 , R 4 , R 3 , R 6 , R', and Ar l are described generally and in classes and subclasses below.
- compositions are useful for treating or lessening the severity of a variety of diseases, disorders, or conditions, including, but not limited to, cystic fibrosis, Hereditary emphysema, Hereditar hemochromatosis, Coagulation-Fibrinolysis deficiencies, such as Protein C deficiency, Type 1 hereditary angioedema. Lipid processing deficiencies, such as Familial hypercholesterolemia.
- Type 1 chylomicronemia Abetalipoproteinemia, Lysosomal storage diseases, such as I-ceS1 disease Pseudo-Hurler, Mucopolysaccharidoses, Sandhof/Tay-Saehs, Crig!er-Najjar type ⁇ , Poiyendocrinopathy/Hyperinsulemia, Diabetes mellitus, Laron dwarfism, Myleoperoxidase deficiency, Primary hypoparathyroidism, Melanoma, Glycanosis CDG type 1, Hereditary emphysema. Congenital hyperthyroidism, Osteogenesis imperfecta, Hereditary
- hypofibrinogenemia ACT deficiency
- Diabetes insipidus DI
- Neurophyseal DI Neprogenic DI
- Charcot-Marie Tooth syndrome Perlizaeus-Merzbacher disease
- neurodegenerative diseases such as AlzheimeR's disease, Parkinson's disease, Amyotrophic lateral sclerosis, Progressive supranuclear plasy, Pick's disease, several polyglutamine neurological disorders asuch as Huntington, Spinocerebuliar ataxia type I, Spinal and bulbar muscular atrophy, Dentatorubai pallidoluysian, and Myotonic dystrophy, as well as Spongiform
- encephalopathies such as Hereditary Creutzfe!dt- Jakob disease, Fabry disease, Straussler- Scheinker syndrome, COPD, dry-eye disease, and Sjogren's disease.
- the present i vention relates to compounds of formula I useful as modulators of ABC transporter activity:
- AT is a 5-6 membered aromatic monocyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, wherein said ring is optionally fused to a 5-12 membered monocyclic or bicyclic, aromatic, partially unsaturated, or saturated ring, wherem each ring contains 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, wherein Ar ! has m substituents, each independently selected from -WR W ;
- W is a bond or is an optionally substituted Q-Cg alkylidene chain wherein up to two methylene units of W are optionally and independently replaced by -CO-, -CS-, -COCO-, - CONR'-, -CONR'NR'-, -CO2-, -OCO-, -NR'COr, -0-, -NR'CONR'-, -OCONR'-, -NR.' R.', - NR'NR'CO-, -NR'CO-, -S-, -SO, -SO?.-, -NR'-, -S0 2 NR'-, NR'S0 2 -, or -NR'80 2 NR' ⁇ ;
- R w is independently R', halo, N0 2 , CN, CF 3 , or OCF3;
- n 0-5;
- each of R ! , R 2 , R 3 , R 4 , and R 5 is indendently ⁇ X-R ;
- X is a bond or is an optionally substituted C C g alkylidene chain wherein up to two methylene units of X are optionally and independently replaced by -CO-, -CS-, -COCO-, - CONR'-, -CONR'NR'-, -CO2-, -OCO-, -NR'COr, -0-, -NR'CONR'-, -OCONR'-, -NR'NR', - NR'NR'CO-, -NR'CO-, -S-, -SO, -SQr, -NR'-, -SO2NR'-, NR'SQr, or -NR'SCfeNR * -;
- R x is independently R', halo, N0 2 , CN, CF 3 , or OCF3;
- R 6 is hydrogen, CF3, -OR', -SR', or an optionally substituted Ci-e aliphatic group
- R 7 is hydrogen or a C;- 6 aliphatic group optionally substituted with -X-R ;
- R' is independently selected from hydrogen or an optionally substituted group selected from a Cj Cs aliphatic group, a 3-8-membered saturated, partially unsaturated, or fully unsaturated monocyclic ring having 0-3 heteroatoms independently selected from nitrogen, oxygen, or sulfur, or an 8-12 membered saturated, partially unsaturated, or fully unsaturated bicyclic ring system having 0-5 heteroatoms independently selected from nitrogen, oxygen, or
- R' 8544833.1 sulfur; or two occurrences of R' are taken together with the atoro(s) to which they are bound to form an optionally substituted 3-12 membered saturated, partially unsaturated, or fully unsaturated monocyclic or bicyclic ring having 0-4 heieroatoms independently selected from nitrogen, oxygen, or sulfur.
- Ar ⁇ is a 5-6 membered aromatic monocyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, wherein said ring is optionally fused to a 5-12 membered monocyclic or bicyclic, aromatic, partially unsaturated, or saturated ring, wherein each ring contains 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, wherein Ar 1 has m substituents each independently selected from -WR W ;
- W is a bond or is an optionally substituted C 1 -C6 alkylidene chain wherein up to two methylene units of W are optionally and independently replaced by -CO-, -CS-, -COCO-, - CONR'-, -CONR'NR'-, -CO2-, -OCO-, -NR'CCfe-, -0-, -NR'CONR'-, -OCONR'-, -NR'NR', - NR'NR'CO-, -NR'CO-, -S-, -SO, -SO2-, -NR'-, -80 2 NR' ⁇ , NR'S0 2 -, -NR'SCfcNR'-;
- R w is independently R', halo, N0 2 , CN, CF 3 , or OCF3;
- n 0-5;
- each of R 5 , R 2 , R 3 , R 4 , and R 5 is independentlyTMX-R X ;
- X is a bond or is an optionally substituted C ⁇ -Cs alkylidene chain wherein up to two methylene units of X are optionally and independently replaced by -CO-, -CS-, -COCO-, - CONR'-, -CONR'NR'-, -CO2-, -OCO-, -NR'COr, -0-, -NR'CONR'-, -OCONR'-, -NR'NR', - NR'NR'CO-, -NR'CO-, -S-, -SO, -SO2-, -NR'-, -80 2 NR ⁇ NR'S0 2 -, or -NR'S0 2 NR'-;
- R x is independently R', halo, N0 2 , CN, CF 3 , or OCF 3 ;
- R 6 is hydrogen, CF 3 , -OR', -SR', or an optionally substituted C1-C8 aliphatic group
- R 7 is hydrogen or a C1-C6 aliphatic group optionally substituted with -X-R ;
- R' is independently selected from hydrogen or an optionally su stituted group selected from a Q.Cs aliphatic group, a 3-8-membered saturated, partially unsaturated, or fully unsaturated monocyclic ring having 0-3 heteroatoms independently selected from nitrogen, oxygen, or sulfur, or an 8- 12 membered saturated, partially unsaturated, or fully unsaturated
- bicyclic ring system having 0-5 heteroatoms independently selected from nitrogen, oxygen, or sulfur; or two occurrences of R * are taken together with the atom(s) to which they are bound to form an optionally substituted 3-12 membered saturated, partially unsaturated, or fully unsaturated monocyclic or bicyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur;
- Ar ! is not phenyl, 2- meihoxyphenyl, 4-meihoxyphenyl, 2-methyiphenyi, 2,6-dichlorophenyI, 2,4-dichiorophenyl, 2- bromophenyl, 4-bron ophenyl, 4-hydroxyphenyl, 2,4-dinitrophenyl, 3,5 ⁇ dicarboxyiic acid phenyl, 2,4-dimethy .phenyl, 2,6-dimethylphenyl, 2-ethylphenyi, 3-nitro-4-methyIphenyl, 3- carboxylic-acid phenyl, 2-fiuorophenyl, 3-fiuorophenyi, 3-trifluoromethyIphenyl, 3- ethoxyphenyi, 4-chlorophenyi, 3-methoxyphenyl, 4-dimethyl
- Ar J is not 4-chiorophenyl, 4-bromophenyl, 4-nitrophenyl, 4- earboethoxyphenyl, 6-ethoxy-benzothiazol-2-yl, 6-carboethoxy-benzothiazol-2-yl, 6-halo- benzothiazo!-2-yl, 6-nitro-benzothiazol-2-yl, or 6-thiocyano-benzothiazoi-2-yl.
- Ar* is not 4-substituted phenyl wherein said substituent is -S0 2 NHR xx , wherein R xx is 2-pyridinyl, 4-methyi-2-pyrimidinyl, 3,4-dimethyl-5-isoxazoiyi;
- Ar* is not thiazol-2-yl, IH- l,2,4-triazoI-3-y!, or lH-l,3,4-triazol-2-yl;
- Ar 5 is not 5-methyl-l ,2-oxazol-3-yl, thiazol-2-yi, 4-fluorophenyl, pyrimidin-2-yl, 1- methyl-l ,2-(lH)-pyrazol-5-yl, pyridine-2-yL phenyl, N-methyl-imidazol-2-yl, imidazoi-2-yl, 5- methyl-imidazol-2-yl, l,3-oxazol-2-yl, or l,3,5-(IH)-triazo!-2-yl:
- Ar 1 is not pyrimidin-2-yl, 4,6-dimethyl-pyrimidin-2-yl, 4-methoxy-6-methyi- 1 ,3,5-triazin-2-yS; 5-bromo-pyridin-2-yi, pyridin-2-y], or 3,5-dichloro-pyridin-2-yl;
- Ar 1 is not an optionally substituted ring selected from thiazol-2-yl, pyridyl, phenyl, thiadiazolyl, benzothiazol-2-yl, or indazolyl;
- Ar 1 is not optionally substituted tetrazolyl
- R 2 , R 4 , R 5 , R 6 , and R 7 each is hydrogen, and R 1 and R 3 both are
- Ar 1 is not 4,5-dihydro-l, 3-thiazol-2-yi, thiazol-2-yi, or [3,5-bis(trifluororaethyi)-i/f-pyrazol- 1 -yljphenyl;
- R 1 , R 4 , R 5 , R 6 , and R 7 each is hydrogen, and Ar 1 is thiazol-2-yl, then neither R 2 nor R J is isopropyl, chloro, or CF 3 ;
- Ar 1 is 4-methoxy phenyl, 4-trifluororaethyIphenyl, 2-fluorophenyl, phenyl, or 3-chlorophenyl, then:
- R ! , R 2 , R 4 , R 5 , R 6 , and R 7 each is hydrogen, then R" is not methoxy; or b) when R 1 , R 3 , R 4 , R 5 , R 6 , and R' each is hydrogen, then R' 1 is not chloro; or c) when R ! , R 2 , R 3 , R 5 , R 6 , and R' each is hydrogen, then R 4 is not methoxy; or d) when when R ! , R J , R 4 , R 6 , and R 7 each is hydrogen, and R 5 is ethyl, then R 2 is not chloro;
- Arl is not a phenyl substituted with -0 € ⁇ 2 03 ⁇ 4 ⁇ 1 ⁇ , -OCH 2 CH 2 (2-trifluorometliyl-phenyI), - OCH 2 CH 2 -(6,7-dimethoxy-l,2,3,4-tetrahydroisoquinolin-2- l), or substituted l f-pyrazoi-3-yl; and
- ABS-transporter as used herein means an ABC-transporter protein or a fragment thereof comprising at least one binding domain, wherein said protein or fragment, thereof is present in vivo or in vitro.
- binding domain as used herein means a domain on the ABC-transporter that can bind to a modulator. See, e.g., Hwang, T. C. et l, L Gen. Physiol. (1998): 1 1 1(3), 477-90.
- CFTR cystic fibrosis transmembrane conductance regulator or a mutation thereof capable of regulator activity, including, but not limited to, AF508 CFTR and G551D CFTR (see, e.g., http://www.genet.sickkids.on.ca/cftr/, for CFTR mutations).
- modulating means increasing or decreasing by a measurable amount.
- compounds of the invention may optionally be substituted with one or more substituents, such as are illustrated generally above, or as exemplified by
- a stable compound or chemically feasible compound is one that is not substantially altered when kept at a temperature of 40°C or less, in the absence of moisture or other chemically reactive conditions, for at least a week.
- aliphatic or "aliphatic group”, as used herein, means a straight- chain (i.e., unbranched) or branched, substituted or unsubstituted hydrocarbon chain that is completely saturated or that contains one or more units of unsaturation, or a monocyclic hydrocarbon or bicyclic hydrocarbon that is completely saturated or that contains one or more units of unsaturation, but which is not aromatic (also referred to herein as “carbocyc!e” "cycloaiiphatic” or “cycloalkyl”), mat has a single point of attachment to the rest of the molecule.
- aliphatic groups contain 1-20 aliphatic carbon atoms.
- aliphatic groups contain 1-10 aliphatic carbon atoms. In other embodiments, aliphatic groups contain 1-8 aliphatic caibon atoms, in still other embodiments, aliphatic groups contain 1-6 aliphatic carbon atoms, and in yet other embodiments aliphatic groups contain 1-4 aliphatic carbon atoms.
- cycloaiiphatic refers to a monocyclic Cs-Cg hydrocarbon or bicyclic or tricyclic Cg-C ⁇ hydrocarbon that is completely saturated or that contains one or more units of unsaturation, but which is not aromatic, that has a single point of attachment to the rest of the molecule wherein any individual ring in said bicyclic ring system has 3-7 members.
- Suitable aliphatic groups include, but are not limited to, linear or branched, substituted or unsubstituted alkyl, alkenyl, alkynyi groups and hybrids thereof such as (cycloalkyl)alkyl,
- Suitable cycloaiiphatic groups include cycloalkyl.
- bicyclic cycloalkyl e.g., deealin
- bridged bicycloaikyl such as norbomyl or [2.2.2]bicyek>- octyl
- bridged tricyclic such as adamantyl
- heteroaliphatic means aliphatic groups wherein one or two carbon atoms are independently replaced by one or more of oxygen, sulfur, nitrogen, phosphorus, or silicon. Heteroaliphatic groups may be substituted or unsubstiiuied, branched or unbranched, cyclic or acyclic, and include “heterocycle”, “heierocyelyl”,
- heterocycloaliphatic'' or “heterocyclic” groups.
- heterocyclic as used herein means non-aromatic, monocyclic, bicyclic, or tricyclic ring systems in which one or more ring members is an independently selected heteroatom.
- the "heterocycle”, “heterocyclyl”, “heterocycloaliphatic”, or “heterocyclic” group has three to fourteen ring members in which one or more ring members is a heteroatom independently selected from oxygen, sulfur, nitrogen, or phosphorus, and each ring in the system contains 3 to 7 ring members.
- heteroatom means one or more of oxygen, sulfur, nitrogen, phosphorus, or silicon (including, any oxidized fomi of nitrogen, sulfur, phosphorus, or silicon; the quatemszed form of any basic nitrogen or; a substitutable nitrogen of a heterocyclic ring, for example N (as in 3,4-dihydro-2H-pyrrolyl), NH (as in pyrrolidinyl) or N * (as in N- substituted pyrrolidinyl)).
- alkoxy refers to an alkyi group, as previously defined, attached to the principal carbon chain through an oxygen (“alkoxy”) or sulfur (“thioaikyl”) atom.
- haloaliphatic and haloalkoxy means aliphatic or alkoxy, as the case may be, substituted with one or more halo atoms.
- halogen or “halo” means F, CI, Br, or L Examples of haloaliphatic incude -CHF 2 , -C3 ⁇ 4F, -CF 3 , -CF 2 -, or perhaloalkyl, such as, -CF 2 CF 3 .
- aryl used alone or as pari of a larger moiety as in “aralkyl”, “aralkoxy”, or “aryloxyalkyl”, refers to monocyclic, bicyclic, and tricyclic ring systems having a total of five to fourteen ring members, wherein at least one ring in the system is aromatic and wherein each ring in the system contains 3 to 7 ring members.
- aryl may be used interchangeably with the term “aryl ring”.
- aryl also refers to heteroaryl ring systems as defined hereinbelow.
- heteroaryl refers to monocyclic, bicyciic, and tricyclic ring systems having a total of five to fourteen ring members, wherein at least one ring in the system is aromatic, at least one ring in the system contains one or more heteroatoms, and wherein each ring in the system contains 3 to 7 ring members.
- heteroaryl may be used interchangeably with the term “heteroaryl ring” or the term “heteroaromatic".
- An aryl (including aralkyl, aralkoxy, aryloxyalkyl and the like) or heteroaryl (including heteroaralkyl and heteroaryiaikoxy and the like) group may contain one or more substituents.
- Optional substituents on the aliphatic group of R° are selected from N3 ⁇ 4, (Ci_ 4 aliphatic) 2 , halo, C s , 4 aliphatic, OH, N0 2 , CN, C0 2 0(haloCi_4 aliphatic), or haloCi ⁇ aliphatic, wherein each of the foregoing Ct ⁇ aliphatic groups of R° is unsubstituted.
- Optional substituents on the aliphatic group of R* are selected from T3 ⁇ 4, NH(Ci-4 aliphatic), aliphatic ) 2 , halo, Ct-4 aliphatic, OH, 0(C aliphatic), N0 2 , CN,
- substituents on the aliphatic group or the phenyl ring of R + are selected from ⁇ 2 , NH(Cj-4 aliphatic), N(C[. 4 aiiphatic) 2 , halo, C M aliphatic, OH, 0(C t-4 aliphatic), N0 2 , CN, C0 2 H, C0 2 (C M aliphatic), 0(haSo C t .4 aliphatic), or haSo(Ci-4 aliphatic), wherein each of the foregoing €i. 4 aliphatic groups of R + is unsubstituted.
- alkylidene chain refers to a straight or branched carbon chain mat may be fully saturated or have one or more units of unsaturation and has two points of attachment to the rest of the molecule.
- spirocycloalkylidene refers to a carbocyclic l ing that may be fully saturated or have one or more units of unsaturation and has two points of attachment from the same ring carbon atom to the rest of the molecule,
- two independent occurrences of R° are taken together together with the atom ⁇ s) to which each variable is bound to form a 3-8-membered cycloalkyl, heterocyclyl, aryl, or heteroary! ring having 0-3 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- Exemplar)-' rings that are formed when two independent occurrences of R° (or R ⁇ , or any other variable similarly defined herein) are taken together with the atom(s) to which each variable is bound include, but are not limited to the following: a) two independent occurrences of R° (or R + , or any other variable similarly defined herein) that are bound to the same atom and are taken together with that atom to form a ring, for example, N(R°) 2 , where both occurrences of R° are taken together with the nitrogen atom to form a piperidin-l-yl, piperazin- -yl, or morpholin-4-yl group; and b) two independent occurrences of R° (or R ⁇ , or any other variable similarly defined herein) that are bound to different atoms and are taken
- a substituent bond in, e.g., a bicycHc ring system, as shown below, means that the substituent can be attached to any substitutahle ring atom on either ring of the bicyclic ring system:
- structures depicted herein are also meant to include all isomeric (e.g., enantiomeric, diastereomeric, and geometric (or conformational)) forms of the structure; for example, the R and S configurations for each asymmetric center, (Z) and (E) double bond isomers, and (Z) and (E) conformational isomers. Therefore, single isomeric (e.g., enantiomeric, diastereomeric, and geometric (or conformational)) forms of the structure; for example, the R and S configurations for each asymmetric center, (Z) and (E) double bond isomers, and (Z) and (E) conformational isomers. Therefore, single isomeric (e.g., enantiomeric, diastereomeric, and geometric (or conformational)) forms of the structure; for example, the R and S configurations for each asymmetric center, (Z) and (E) double bond isomers, and (Z) and (E) conformational isomers
- structures depicted herein are also meant to include compounds that differ only in the presence of one or more isotopically enriched atoms.
- compounds having the present structures except for the replacement of hydrogen by deuterium or tritium, or tlie replacement of a carbon by a l 3 C- or , C-enriched carbon are within
- Such compounds are useful, for example, as analytical tools or probes in biological assays.
- Ar 5 is selected from:
- a i and A 2 together, is an 8-14 aromatic, hicyclie or tricyclic aryl ring, wherein each ring contains 0-4 heteroaioms independently selected from nitrogen, oxygen, or sulfur.
- At is an optionally substituted 6 mernbered aromatic ring having 0-4 heteroaioms, wherein said heteroatom is nitrogen.
- a t is an optionally substituted phenyl.
- is an optionally substituted pyridyl, pyrimidinyl, pyrazinyl or triazinyl.
- As is an optionally substituted pyrazinyl or triazinyl.
- a ⁇ is an optionally substituted pyridyl.
- At is an optionally substituted 5-membered aromatic ring having 0-3 heteroaioms, wherein said heteroatom is niirogen, oxygen, or sulfur.
- Aj is an optionally substituted 5-membered aromatic ring having 1-2 nitrogen atoms.
- a ⁇ is an optionally substituted 5-membered aromatic ring oilier than thiazolyl.
- a 2 is an optionally substituted 6 mernbered aromatic ring having 0-4 heteroaioms, wherein said heteroatom is niirogen.
- a 2 is an optionally substituted phenyl.
- a 2 is an optionally substituted pyridyl, pyrimidinyl, pyrazinyl, or triazinyl.
- a 2 is an optionally substituted 5-membered aromatic ring having 0-3 heteroaioms, wherein said heieroaiom is nitrogen, oxygen, or sulfur. In some embodiments, A 2 is an optionally substituted 5-membered aromatic ring having 1-2 nitrogen atoms. In certain embodiments, A 2 is an optionally substituted pyrro!yl.
- a 2 is an optionally substituted 5-7 mernbered saturated or unsatusaied heterocyclic ring having 1-3 heteroaioms independently selected from niirogen,
- exemplary such rings include piperidyl, piperazyl, morphoiinyl, thiomorpholinyl, pyrrolidinyl, tetrahydrofuranyl, etc.
- a 2 is an optionally substituted 5- 10 membered saturated or unsaturated carbocyclic ring, in one embodiment, A 2 is an optionally substituted 5-10 membered saturated carbocyclic ring.
- Exemplary such rings include cyclohexyl, cyc!opentyl, etc.
- ring A 2 is selected from:
- W is a bond or is an optionally substituted
- alkyliderse chain wherein one or two methylene units are optionally and independently replaced by O, NR', S, SO, S0 2 , or COO, CO, S0 2 NR', NR'S0 2 , C(0)NR ⁇ NR'C(O), OC(O), OC(0)NR ⁇ and R w is R' or halo.
- each occurrence of WR W is independently -C1 -C3 a!kyl, C1 -C3 perhaloalkyl, -0(C1-C3alkyl), -CF 3 , -OCF3, - SCFj, -F, -CI, -Br, or -COOR', -COR' , -0(C3 ⁇ 4) 2 N(R')(R'), -0(CH 2 )N(R')(R'), - CON(R')(R'), -(C3 ⁇ 4) 2 QK ⁇ -(CH 2 )OR ⁇ optionally substituted monocyclic or bicyclic aromatic ring, optionally substituted arylsulfone, optionally substituted 5-membered heteroaryl ring, - N(R')iR'), -(CH ? .) 2 N ⁇ R')(R') 5 or -(CH 2 ) (R')(R 5 ).
- m is 0. Or, m is 1, Or, m is 2. In some embodiments, m is 3. In yet other embodiments, m is 4.
- R 5 is X-R x .
- R 5 is hydrogen.
- R 5 is an optionally substituted Cj-g aliphatic group.
- R 5 is optionally substituted CM aliphatic.
- R 5 is benzyl.
- R 6 is hydrogen. Or, R 6 is an optionally substituted Cj.g aliphatic group. In some embodiments, R 6 is optionally substituted C aliphatic. In certain other embodiments, R 6 is -(O-CM aliphatic) or -(S-CM aliphatic). Preferably, R 6 is -OMe or - SMe. In certain other embodiments, 6 is CF 3 .
- R 1 , R 2 , R 3 , and R 4 are
- R 6 and R 7 are both simultaneously hydrogen.
- R ⁇ R 2 , R 3 , R 4 , and R 5 are simultaneously hydrogen.
- R l , R 2 , R ⁇ R 4 , R 5 and R 6 are simultaneously hydrogen,
- R 2 is X-R x , wherein X is - S0 2 R'-, and R x is R': i.e., R 2 is -S0 2 N(R') 2 .
- the two R' therein taken together form an optionally substituted 5-7 membered ring with 0-3 additional heteroatoms selected from nitrogen, oxygen, or sulfur.
- R 1 , R ⁇ R 4 , R 5 and R 6 are simultaneously hydrogen, and R 2 is S0 2 N(R 5 ) 2 .
- X is a bond or is an optionally substituted Q
- each occurrence of XR X is independently -C h lky!, -0(Cs- 3 alkyl), -CF 3 , -OCF 3 ⁇ 4 , -SCF 3 , -F, -CI, -Br, OH, -COOR ⁇ -COR', -0(CH 2 )2N(R , )(R'), -0(CH 2 )N(R , )(R') !
- R 7 is hydrogen, in certain other embodiment, R 7 is €5.4 straight or branched aliphatic,
- R w is selected from halo, cyano, CF 3 , CHF 2 , OCHF 2 , Me, Et, CH(Me) 2 , CHMeEt, n-propyl, t-butyl, OMe, QEt, OPh, O-fluorophenyl, O- difluorophenyl, O-methoxyphenyl, O-tolyl, O-benzyl, SMe, SCF 3 , SCHF 2( SEt, C3 ⁇ 4CN, NH 2 , NHMe, N(Me) 2 , NHEt, N(Ei) 2 , C(0)CH 3 , C(0)Ph, C(0)N3 ⁇ 4, SPh, S0 2 -(amino-pyridyl), SO2NH2, S0 2 Ph, SQ 2 NHPh, SC -N-morpholino, SCfe-N-pyrrolidyl, N-pyrrolyl
- R' is hydrogen
- R' is a C1-C8 aliphatic group, optionally substituted with up to 3 substituenis selected from halo, CN, CF3, CHF 2 , OCF3, or OCHF 2 , wherein up to two methylene units of said C1-C8 aliphatic is optionally replaced with -CO-, -CONH(C1 -C4 alky!)-, -CO2-, -OCO-, -N(C1-C4 alkyl)CO , -0-, -N(C1-C4 alkyl)CON(Cl-C4 alky])-, -OCON(Cl-C4 alkyl)-, -N(C1-C4 alkyl)CO-, -S-, -N(C1-C4 alky!)-, -S(3 ⁇ 4 (C1-C4 alkyl , N(C1-C4 alkyl)SO , or -N(C1
- R' is a 3-8 membered saturated, partially unsaturated, or fully unsaturated monocyclic ring having 0-3 heteroatoms independently selected from
- R" is optionally substituted with up to 3 substituents selected from halo, CN, CF3, CHF 2 , OCF 3 , OCHF 2 , or C1-C6 alkyl, wherein up to two methylene units of said C1-C6 alkyl is optionally replaced with -CO-, -CONH(Cl-C4 alkyl)-, -CO 2 -, -OCO-, -N(Cl-C alkyI)CO , -0-, ⁇ N(C1-C4 aikyl)CON(Cl -C4 alky!)-, -OCON(Cl- C4 alkyl)-, -N(C1-C4 alkyl)CO-, -S-, -N(C1-C4 alkyl)-, -S0 2 N(C1 ⁇ C4 alkyl)-, N(C1-C4 alkyl)
- R' is an 8-12 membered saturated, partially unsaturated, or fully unsaturated bicyclic ring system having 0-5 heteroatoms independently selected from nitrogen, oxygen, or sulfur; wherein R' is optionally substituted with up to 3 substituents selected from halo, CN, CF 3 , CHF 2 , OCF3, OCHF2, or C1-C6 alkyl, wherein up to two methylene units of said C1-C6 alkyi is optionally replaced with -CO-, -CONH(Cl-C4 alkyl)-, -CO2-, -OCO-, -N(C1-C4 alkyl)C0 2 -, -0-, -N(C1-C4 alkyl)CON(Cl-C4 alkyl)-, -OCGN ⁇ Cl ⁇ C4 alkyi)-, -N(C1-C4 alkyl)CO-, -S-, -N(C1 -C
- two occurrences of R' are taken together with the atom(s) to which they are bound to form an optionally substituted 3-12 membered saturated, partially unsaturated, or fully unsaturated monocyclic or bicyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, wherein R' is optionally substituted with up to 3 substituents selected from halo, CN, CF 3 , CHF 2 , OCF 3 , OCHF 2 , or C1-C6 alkyl, wherein up to two methylene units of said C1-C6 alkyl is optionally replaced with -CO-, - CONH(Cl-C4 alkyi)-, -CO , -OCO-, -N(C1-C4 alkyl)C0 2 -, -0-, -N(C1-C4 alkyl)CON(Cl- C4 alkyl)-, -OCON(Cl-C4 alkyl)
- the present invention provides compounds of formula IIA or formula IBB:
- the present invention provides compounds of formula ⁇ , formula HIB, formula IIIC, formula HID, or formula HIE:
- each of Xi, X 2 , X 3 , X4, and X5 is independently selected from CH or N; and Xe is O, S, or NR' .
- compounds of formula IIIA, formula ⁇ , formula IIIC, formula MID, OF formula ⁇ have y occurrences of substituent X-R x , wherein y is 0-4. Or, y is 1. Or, y is 2.
- [ ⁇ 89] is selected from halo, cyano, Q3 ⁇ 4, CHF 2 , OCHF 2 , Me, Et, CH(Me>2, CHMeEt, n-propyl, t-butyl, OMe, OEt, OPh, O-fluorophenyl, O- difluorophenyl.
- X and R x taken together, is Me, Et, halo, CN, CF 3 , OH, OMe, QEt, S0 2 N(Me)(fluorophenyl), SO -(4-methyl ⁇ piperidin ⁇ l ⁇ yl, or 80 2 -N-pyrrolidinyi. f3 ⁇ 49I] According to another embodiment the present invention provides compounds of formula TV A,
- compounds of formula IVA, formula IVB, and formula IVC have y occurrences of substituent X-R x , wherein y is 0-4, Or, y is 1. Or, y is 2.
- the present invention provides compounds of formula IVA, formula IVB, and formula IVC, wherein X is a bond and R x is hydrogen.
- the present invention provides compounds of formula formula IVB, and formula IVC, wherein ring A 2 is an optionally substituted, saturated, unsaturated, or aromatic seven membered ring with 0-3 heteroatoms selected from O, S, or N.
- exemplary rings include azepanyl, 5,5-dimeihyl azepanyl, etc.
- the present invention provides compounds of formula IVB and IVC, wherein ring A 2 is an optionally substituted, saturated, unsaturated, or aromatic six membered ring with 0-3 heteroatoms selected from O, S, or N.
- exemplary rings include piperidinyl, 4,4-dimethylpiperidinyl, etc.
- the present invention provides compounds of formula IVB and IVC, wherein ring A 2 is an optionally substituted, saturated, unsaturated, or aromatic five membered ring with 0-3 heteroatoms selected from O, S, or N.
- the present invention provides compounds of formula IVB and IVC, wherein ring A 2 is an optionally substituted five membered ring with one nitrogen atom, e.g., pyrrolyl or pyrrolidinyL
- each of WR* 2 and WR W4 is independently selected from hydrogen, CN, CF3, halo, C1-C6 straight or branched alkyl, 3-12 membered cycloaliphatic, phenyl, C5-C10 heteroaryi or C3-C7 heterocyclic, wherein said heteroaryl or heterocyclic has up to 3 heteroatoms selected from O, S, or N, wherein said WR* 2 and WR W4 is independently and
- - 3 - 44833.1 optionally substituted with up to three substituents selected from -OR', -CF3, -OCF 3 , SR ⁇ S(0)R', SQ 2 R ⁇ -SCF 3 , halo, C , -COOR ⁇ -COR', -0(CH 2 ) 2 N(R')(R'), -0(CH 2 )N(R')(R'), - CON(R')(R'), ⁇ iCH 2 ) 2 OR', -(C3 ⁇ 4)GR ⁇ CH 2 CN, optionally substituted phenyl or phenoxy, - N(R')(R'), -NR'C(0)GR ⁇ -NR'C(0)R ⁇ -(C3 ⁇ 4) 2 N(R')(R 5 ), or -(CH 2 )N(R')(R'); and
- WR WS is selected from hydrogen, -OH, NH 2> CN, CHF 2 , NHR', N(R') 2 , -NHC(0)R', -NHC(0)OR', NHSO 2 R', -OR ⁇ CH 2 OH, CH2N(R') 2 , C(0)OR ⁇ SG 2 NHR ⁇ SO ⁇ R'h, or CH 2 NHC(0)OR ⁇
- WR W4 and WR WS taken togetlier form a 5-7 membered ring containing 0-3 three heteroatoms selected from N, O, or S, wherein said ring is optionally substituted with up to three WR W substituents.
- compounds of formula VA-i have y occurrences of X-R x , wherein y is 0-4. in one embodiment, y is 0.
- the present invention provides compounds of formula VA- 1, wherein X is a bond and R x is hydrogen.
- the present invention provides compounds of formula VA- 1, wherein:
- each of WR W2 and WR W4 is independently selected from hydrogen, CN, CF 3 , halo, C1-C6 straight or branched alky!, 3-12 membered cycioaliphatic, or phenyl, wherein said WR W2 and WR W is independently and optionally substituted with up to three substituents selected from -OR', -CF 3 , -OCF 3 , -SCF 3 , halo, -COOR ⁇ -COR', -0(CH 2 ) 2 N(R')(R'), - 0(C3 ⁇ 4)N(R')(R !
- WR W5 is selected from hydrogen, -OH, NH 2 , CN, NHR', N(R') 2 , -NHC(0)R', - NHC(0)OR ⁇ NHSO 2 R', -OR', CH 2 OH, C(0)OR', SO 2 NHR', or C3 ⁇ 4 iC ⁇ 0)0-(R').
- the present invention provides compounds of formula VAIL, wherein:
- WR W2 is a pheny ring optionally substituted with up to three substituents selected from - OR', -CF 3 , -OCF3, SR ⁇ S(0)R ⁇ S0 2 R', -SCF 3 , halo, CN, -COOR', -COR', - 0(CH 2 ) 2 N(R')(R'), -0(C3 ⁇ 4)N(R')(R'), -CON(R')(R'), -(CH 2 ) 2 OR', -(CH 2 )OR', CH 2 CN, optionally substituted phenyl or phenoxy, -N(R')(R'), -NR'C(0)OR ⁇ -NR'C(0)R', - (CH 2 ) 2 N(R')(R'), or -(CH 2 )N(R')(R');
- WR W4 is C1-C6 straight or branched alkyl
- WR WS is OH.
- each of WR W2 and WR W4 is independently selected from CF 3 or halo, in one embodiment, each of WR W2 and WE? 4 is independently selected from
- each of of WR W2 and WR W is independently selected from optionally substituted n-propyl, isopropyi, n-b tyl, sec-butyf, t- butyl, l,l-dimethyl-2-hydroxyethyl, l,l-dimethyl ⁇ 2 ⁇
- each of W W2 and WR W4 is independently selected from optionally substituted 3-12 menibered cycloaliphatic.
- exemplary embodiments of such eycioaliphaiic include cyclopentyl, eyclohexyl, cycioheptyl, norbornyl, adamantyl,
- WR W2 is hydrogen and WR W4 is C1-C6 straight or branched alkyl.
- W W4 is selected from methyl, ethyl, propyl, n-butyi, sec-butyl, or i-butyL
- WR W4 is hydrogen and W ft,; is C1-C6 straight or branched alkyl.
- WR W2 is selected from methyl, ethyl, propyl, n-butyl, sec -butyl, t-butyl, or n-pentyl.
- each of WR W2 and WR 3 ⁇ 44 is CI-C6 straight or branched alkyl.
- each of WR wi and WR W4 is selected from methyl, ethyl, propyl, n-butyl, sec-butyl, t-butyl, or pentyl.
- WR Wi is selected from hydrogen, CHF 2 , NH 2 , CN, NHR ⁇ N(R') 2 , C3 ⁇ 4N(R') 2 , -NHC(0)R', -NHC(0)OR ⁇ -OR', C(0)OR', or S0 2 NHR'.
- WR WS is - OR', e.g., OH.
- WR W5 is selected from hydrogen, NH 2 , CN, CHF 2 , NH ⁇ Ci-C6 alkyl), N(C1-C6 alkylfc, -NHC(0)(C I-C6 alkyl), -CH 2 NHC(0)0(C1 ⁇ C6 alkyl), - NHC(0)0(C1-C6 alkyl), -OH, -Q(Ci-C6 alkyl), C(0)0(C1 -C6 alkyl), CH 2 Q(C1-C6 alkyl), or S0 2 N3 ⁇ 4.
- WR W5 is selected from -OH, OMe, NH 2 , -NHMe, -N(Me) 2 , -CH 2 NH 2 , CH 2 OH, NHC(0)OMe, NHC(0)OEt, CN, CHF 2 , -C3 ⁇ 4NHC(0)0(t-butyi), -O- (ethoxyethyl), -O-(hydroxyethyl), -C(0)OMe, or -SO ? N3 ⁇ 4.
- compound of formula VA-1 has one, preferably more, or more preferably all, of the following features:
- WR W2 is hydrogen
- WR W4 is C1-C6 straight or branched alkyl or monocyclic or bicyclic aliphatic; and iii) WR W5 is selected from hydrogen, CN, CHF 2 , N3 ⁇ 4, NHCC1-C6 alkyl), N(C1-C6
- compound of formula VA-1 has one, preferably more, or more preferably all, of the following features:
- WR 2 is halo, C1-C6 alky!, CF , CN, or phenyl optionally substituted with up to 3 substituents selected from C1-C4 aikyl, -0(0-C4 alkyi), or halo;
- WR W4 is CF 3 , halo, C1-C6 aikyl or C6-C10 cycloaliphatic;
- WR W3 is OH, N3 ⁇ 4, H(C1-C6 aikyl), or N(C1-C6 alkyi).
- X-R x is at the 6-posiiion of the quinolinyl ring, in certain embodiments, X-R x taken together is C1-C6 alkyi, -0-(O-C6 alkyi), or halo.
- X-R x is at the 5-position of the quinolinyl ring, in certain embodiments, X-R x taken together is -OH.
- the present invention provides compounds of formula VA-1, wherein WR W4 and W W5 taken together form a 5-7 membered ring containing 0-3 three heteroaioms selected from N, O, or S, wherein said ring is optionally substituted with up to three WR W substituents.
- WR W4 and WR 5 taken together form an optionally substituted 5-7 membered saturated, unsatiiraied, or aromatic ring containing 0 heteroatoms.
- WR W4 and WR 3 ⁇ 45 taken together form an optionally substituted 5-7 membered ring containing 1-3 heteroatoms selected from N, O, or S
- WR W4 and WR W5 taken together form an optionally substituted saturated, unsaturated, or aromatic 5-7 membered ring containing 1 nitrogen heteroatom.
- WR 4 and WR W5 taken together form an optionally substituted 5-7 membered ring containing 1 oxygen heteroatom.
- the present invention provides compounds of formula
- Y is CH 2 , C(0)0, C(O), or S(0) 2 ;
- n 0-4;
- X, R x , W, and R are as defined above.
- compounds of formula VA-2 have y occurrences of X-R , wherein y is 0-4. In one embodiment, y is 0. Or, y is 1. Or, y is 2.
- Y is C(O). in another embodiment, Y is C ⁇ 0)0. Or, Y is S(0) 2 , Or, Y s CH 2 .
- n 1 or 2. Or, m is 1. Or, rn is 0.
- W is a bond
- R w is C1-C6 aliphatic, halo, CF 3 , or phenyl optionally substituted with C1-C6 alkyi, halo, cyano, or CF 3 , wherein up to two jneihylene units of said C1-C6 aliphatic or C1-C6 alkyl is optionally replaced with -CO-, -CONR'-, -C0 2 -, -OCO-, -NR'CGr, -0-, -NR'CONR'-, -OCO R'-, -NR'CO-, -S-, -NR'-, -SO2 '-, NR'SOr, or - NR'S ⁇ 3 ⁇ 4NR'-.
- R" above is C1-C4 alkyl.
- Exemplary embodiments of WR* include methyl, ethyl, propyl, tert-butyl, or 2-ethoxyphenyl.
- R w in Y-R* is C1-C6 aliphatic optionally substituted with N(R") 2 , wherein R" is hydrogen, C1-C6 alkyl, or two R" taken together form a 5-7 membered heterocyclic ring with up to 2 additional heteroatoms selected from O, S, or NR'.
- exemplary such heterocyclic rings include pyrro!idinyt, piperidyl, morpholinyl, or thiomorphoiinyl.
- the present invention provides compounds of formula -A-3:
- n 0-4;
- n 0-4;
- R x , W, and R w are as defined above.
- compounds of formula VA-3 have y occurrences of X ⁇ R X , wherein y is 0-4. In one embodiment, y is 0. Or, y is 1. Or, y is 2,
- n is 0-2.
- m is 0-2. In one embodiment, m is 0. In one embodiment, m is 1. Or, m is 2.
- QR Q taken together is halo, CF 3 , OCF 3 , CN, C1-C6 aliphatic, 0-C1-C6 aliphatic, O-phenyl, NH(C1-C6 aliphatic), or N(C1-C6 aliphatic) 2 , wherein said aliphatic and phenyl are optionally substituted with up to three substiiuents selected from C1 -C6 alky I, 0-C1-C6 alkyi, halo, cyano, OH, or CF 3 , wherein up to two methylene units of said C1-C6 aliphatic or C1-C6 alkyi is optionally replaced with -CO-, -CONR'-, -CO 2 -, -OCO-, -NR'COr, -0-, -NR'CONR'-, -OCONR'-, -NR'CO-, -8-, -NR
- R' above is C1-C4 alkyi.
- Exemplary QR Q include methyl, isopropyi, sec-buty!, hydroxymethyl, CF 3 , NMe 2 , CN, CH 2 CN, fluoro, chloro, OEt, OMe, SMe, OCF 3 , OPh, C(0)OMe, C(0)0-iPr, S(0)Me, NHC(0)Me, or S(0) 2 Me.
- the present invention provides compounds of formula
- compounds of formula VA-4 have y occurrences of X-R , wherein y is 0-4. In one embodiment, y is 0. Or, y is 1. Or, y is 2,
- R w is C1-C12 aliphatic, C5-C10 cycloaliphatic, or C5-C7 heterocyclic ring, wherein said aliphatic, cycloaliphatic, or heterocyclic ring is optionally substituted with up to three substiiuents selected from C1-C6 alkyi, halo, cyano, oxo, OH, or CF 3 , wherein up to two methylene units of said C1-C6 aliphatic or C1-C6 alkyi is optionally- replaced with -CO-, -CONR'-, -CO2-, -OCO-, -NR'CC , -0-, -NR'CONR'-, -OCONR'-, -NR'CO-, -S-, -NR * -, -SO2NR'-, NR'SOr, or -NR'S0 2 NR'-.
- R' above is
- Exemplary R includes methyl, ethyl, n-propyl, isopropyi, n-butyl, sec-butyi, t- butyl, n-pentyl, vinyl, cyanomethyi, hydroxymethyl, hydroxyethyl, hydroxybutyl, cyclohexyl, adamantyl, or -C(CH 3 ) 2 -NHC(0)0-T, wherein T is C1-C4 alkyi, methoxyethyl, or tetrahydrofuranyl methyl.
- the present invention provides compounds of foi
- n 0-4;
- compounds of formula VA-5 have y occurrences of X-R , wherein y is 0-4, In one embodiment, y is 0. Or, y is 1. Or, y is 2,
- n is 0-2. Or, m is I, Or, m is 2,
- both R' are hydrogen.
- one R' is hydrogen and the other W is C1-C4 alkyl, e.g., methyl
- both R' are C1-C4 aikyt e.g., methyl.
- m is I or 2, and is halo, CF 3 , CN, C1-C6 aliphatic, 0-C1-C6 aliphatic, or phenyl, wherein said aliphatic and phenyl are optionally substituted with up to three substituents selected from C1-C6 alkyl, 0-CI-C6 alkyl, halo, cyano, OH, or CF 3 , wherein up to two methylene units of said CI -C6 aliphatic or C1-C6 alky!
- R' above is C1-C4 alkyl
- R w include chloro, CF 3 , OCF 3 , methyl, ethyl, n- propyl, isopropyl, n-butyl, t-butyl, methoxy, ethoxy, propyloxy, or 2-ethoxyphenyl.
- 4 S33.1 ring B is a 5-7 membered monocyclic or bicyclic, heterocyclic or heteroaryl ring optionally substituted with up to n occurrences of ⁇ Q ⁇ R Q , wherein n is 0-4, and Q and R Q are as defined above;
- compounds of formula VA-6 have y occurrences of X ⁇ R X , wherein y is 0-4. in one embodiment, y is 0, Or, y is 1. Or, y is 2,
- n is 0-2. Or, m is 0. Or m is 1.
- n is 0-2. Or, n is 0. Or, n is 1.
- ring B is a 5-7 membered monocyclic, heterocyclic ring having up to 2 heteroaioms selected from O, S, or N, optionally substituted with up to n occurrences of -Q-R Q .
- exemplary heterocyclic rings include N-morpholinyl, N-piperidinyS, 4- benzoyl-piperazin-l-y!, pyrrolidm-l-yl, or 4-methyl-piperidin ⁇ 1 -yl.
- ring B is a 5-6 membered monocyclic, heteroaryl ring having up to 2 heteroaioms selected from O, S, or N, optionally substituted with up to n occurrences of ⁇ Q-R Q .
- Exemplary such rings include benzimidazol-2-yl, 5-methyl-furan-2-yS, 2,5-dimcthyl-pyrroi-i-yI, pyridine-4-yl, indoi-5-yi, indol-2-yi, 2,4-dimethoxy-pyrimidin-5-yi, furan-2-yl, furan-3-yl, 2-acyl-thien-2-yl, benzothiophen-2-yl, 4-methyl-thien-2-yi, 5-cyano- thien-2-yl, 3-chloro-5-trifluoromethyl-pyridin-2-yl.
- one of Qs and (3 ⁇ 4 is N(WR ) and the other of Q ⁇ and Q 3 is selected from O, S, or N(WR W );
- Q 2 is C(0), C3 ⁇ 4-C(0), C(0)-CH 2 , CH 2 , CH 2 -C3 ⁇ 4, CF 2s or CF 2 -CF 2 ;
- n 0-3;
- X, W, R x , and R w are as defined above.
- compounds of formula V-B-l have y occurrences of X- , wherein y is 0-4. in one embodiment, y is 0. Or, y is h Or, y is 2.
- Q 3 is N(WR*); exemplary WR W include hydrogen, Cl-Ci aliphatic, C(0)C1-C6 aliphatic, or C(0)OC1-C6 aliphatic.
- ⁇ 3 ⁇ 4 is N(WR W )
- Q 2 is C(0), CH 2 , CH 2 -CH 2
- Qi is
- the present invention provides compounds of formula
- R is hydrogen or C 1 -C6 aliphatic
- each of R W3 is hydrogen or C1-C6 aliphatic;
- n 0-4;
- compounds of formula V-B-2 have y occurrences of X-R x » wherein y is 0-4. in one embodiment, y is 0. Or, y is 1. Or, y is 2.
- WR Wi is hydrogen, C1-C6 aliphatic, C(0)C1-C6 aliphatic, or C(0)OCl-C6 aliphatic.
- each R W3 is hydrogen, C1-C4 alkyl.
- both R W3 taken togetlier form a C3-C6 cycloaliphatic ring or 5-7 rnembered heterocyclic ring having up to two heteroatoms selected from O, S, or N, wherein said cycloaliphatic or heterocyclic ring is optionally substituted with up to three substitutents selected from WR WI .
- Exemplary such rings include cyciopropyi, cyclopentyl, optionally substituted piperidyl, etc.
- the present invention provides compounds of formula
- Q 4 is a bond, C(0), C(0)0, or S(G>>;
- R Wi is hydrogen or C1-C6 aliphatic
- n 0-4;
- X, W, R w , and R x are as defined above.
- compoimds of formula V-B-3 have y occurrences of X ⁇ R X , wherein y is 0-4. in one embodiment, y is G.
- Q 4 is C(O). Or Q 4 is C(0)0, in another embodiment, R i is CI-C6 aikyl.
- R W1 include methyl, ethyl, or t-butyl.
- the present invention provides compounds of formula
- n 0-4;
- X, x , W, and R w are as defined above,
- compounds of formula V-B-4 have y occurrences of X-R x , wherein y is 0-4. in one embodiment, y is 0, Or, y is 1. Or, y is 2.
- n is 0-2. Or, m is 0. Or, m is 1.
- said cycloaliphatic ring is a 5-menibered ring.
- said ring is a six-membered ring.
- the present invention provides compounds of formula
- ring A 2 is a phenyl or a 5-6 membered heteroaryl ring, wherein ring A 2 and the phenyl ring fused thereto together have up 4 substituents independently selected from WR W ;
- n 0-4;
- X, W, R w and R x are as defined above.
- compounds of formula V-B-5 have y occurrences of X-R x , wherein y is 0-4. In one embodiment, y is 0. Or, y is I . Or, y is 2.
- ring A 2 is an optionally substituted 5-membered ring selected from pyrroiyi, furanyl, thienyi, pyrazolyl, iraidazoly!, thiazolyl, oxazoiyi, thiadiazolyl, oxadiazo!yl, or triazolyl.
- ring A is an optionally substituted 5-membered ring selected from pyrroiyi, pyrazolyl, thiadiazolyl, imidazolyl, oxazoiyi, or triazolyl, Exemplary such rings include:
- ring A 2 is an optionally substituted 6-membered ring.
- exemplary such rings include pyridyl, pyrazinyl, or triazinyi.
- said ring is an optionally pyridyl,
- ring A 2 is phenyl
- ring A 2 is pyrroiyi, pyrazolyl, pyridyl, or thiadiazolyl
- Exemplary W in formula V-B-5 includes a bond, C(O), C(0)0 or C1-C6 alkylene.
- R in formula V-B-5 include cyano, halo, C1-C6 aliphatic, C3-C6 cycloaliphatic, aryl, 5-7 membered heterocyclic ring having up to two heteroatoms selected from O, S, or N, wherein said aliphatic, phenyl, and heterocyclic are independently and optionally substituted with up to three substituents selected from C1-C6 alkyi, 0-C1-C6 alkyi, halo, cyano, OH, or CF 3 , wherein up to two methylene units of said C1-C6 aliphatic or C1-C6 alkyi is optionally replaced with -CO-, -CONR'-, -CO2-, -OCO-, -NR'C0 2 -, -0-, - NR'CONR'-, -OCONR'-, -NR'CO-, -S-, - R'-
- the present invention provides compounds of formula V-B-
- G 4 is hydrogen, halo, CN, CF 3 , CHF 2 , CH 2 F, optionally substituted CI -C6 aliphatic, aryl-Cl-C6 alkyi, or a phenyl, wherein G 4 is optionally substituted with up to 4 WR W substituents; wherein up to two methylene units of said CI -C6 aliphatic or C1-C6 alkyi is optionally replaced with -CO-, -CONR'-, -CO2-, -OCO-, -NR'C0 2 -, -0-, -NR'CONR'-, -OCONR'-, -NR'CO-, -S-, -NR'-, -S ⁇ 1 ⁇ 4NR ⁇ NR'SO s -, or -NR'S0 2 NR'-. ;
- G 5 is hydrogen or an optionally substituted C1-C6 aliphatic
- indole ring system is further optionally substituted with up to 3 substituents independently selected from WR W .
- compounds of formula V-B-5-a have y occurrences of X ⁇ R x , wherein y is 0-4. In one embodiment, y is 0. Or, y is 1. Or, y is 2,
- G 4 is hydrogen.
- G 5 is hydrogen.
- G 4 is hydrogen
- G5 is C1-C6 aliphatic, wherein said aliphatic is optionally substituted with C1-C6 alkyi, halo, cyano, or CF 3 , and wherein up to two methylene units of said C1-C6 aliphatic or C1-C6 alkyi is optionally replaced with - CO-, -CONR'-, -CO 2 -, -OCO-, -NR'CQr, -0-, -NR'CONR'-, -OCONR'-, -NR'CO-, -S-, - NR'-, -SO2NR'-, NR'SG , or -NR'SQsNR'-.
- R' above is C1-C4 alkyi.
- G 4 is hydrogen
- G5 is cyano, methyl, ethyl, propyl, isopropyl, butyl, sec-butyl, t-butyl, cyanomethyl, methoxyethyl, CH 2 C(0)OMe, (CH ) 2 ⁇ NHC(0)0-iert-butyl, or cyclopeniy!.
- G 5 is hydrogen
- G4 is halo, C1-C6 aliphatic or phenyl, wherein said aliphatic or phenyl is optionally substituted wife C1-C6 alkyl, halo, cyano, or CF3, wherein up to two methylene units of said C1-C6 aliphatic or C1 -C6 alkyl is optionally replaced wife -CO-, -CONR'-, ⁇ CG 2 ⁇ , -OCO-, -NR'C0 2 -, -0-, -NR'CONR'-, -OCONR'-, -NR'CO-, -S-, -NR'-, -80 2 NR ⁇ NR'S0 2 ⁇ , or ⁇ NR'SQ 2 NR'-.
- R' above is C 1-C4 alkyl.
- G 5 is hydrogen
- G 4 is halo, CFj, ethoxycarbonyl, t- butyl, 2-raethoxyphenyi, 2-ethoxyphenyl, (4 ⁇ C(0)NH(CH 2 ) 2 ⁇ NMe 2 )-phenyl, 2-mefeoxy ⁇ 4- chioro-phenyl, pyridine-3-yl, 4-isopropylphenyl, 2,6-dimethoxyphenyl, sec- butylaminocarbonyl, ethyl, t-butyl, or piperidin-l-ylcarbonyl.
- G 4 and G5 are both hydrogen, and the nitrogen ring atom of said indole ring is substituted with C1-C6 aliphatic, C(0)(C1-C6 aliphatic), or benzyl, wherein said aliphatic or benzyl is optionally substituted with C1-C6 alkyl, halo, cyano, or CF 3 , wherein up to two methylene units of said C1-C6 aliphatic or CI -C6 alkyl is optionally replaced with -CO-, -CONR'-, -CO2-, -OCO-, -NR'COr, -0-, -NR'CONR'-, -OCONR'-, -NR'CO-, -S-, -NR'-, -SO 2 NR'-, NR'SOr, or -NR'S0 2 NR'-.
- R' above is C1-C4 alkyl
- G 4 and Gj are both hydrogen, and the nitrogen ring atom of said indole ring is substituted with acyl, benzyl, C(0)CH 2 N(Me)C(0)CH 2 NHMe, or ethoxycarbonyl.
- each of R ⁇ R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , and Ar ! in compounds of formula P is independently as defined above for any of the embodiments of compounds of formula I.
- the present invention provides compounds useful as intermediates in the synthesis of compounds of formula I.
- such compounds have formula A-I:
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Diabetes (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Pulmonology (AREA)
- Endocrinology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Hematology (AREA)
- Rheumatology (AREA)
- Psychology (AREA)
- Reproductive Health (AREA)
- Ophthalmology & Optometry (AREA)
- Obesity (AREA)
- Immunology (AREA)
- Emergency Medicine (AREA)
- Pain & Pain Management (AREA)
- Psychiatry (AREA)
- Hospice & Palliative Care (AREA)
- Heart & Thoracic Surgery (AREA)
- Gynecology & Obstetrics (AREA)
- Pregnancy & Childbirth (AREA)
- Cardiology (AREA)
- Otolaryngology (AREA)
- Gastroenterology & Hepatology (AREA)
Abstract
Description





























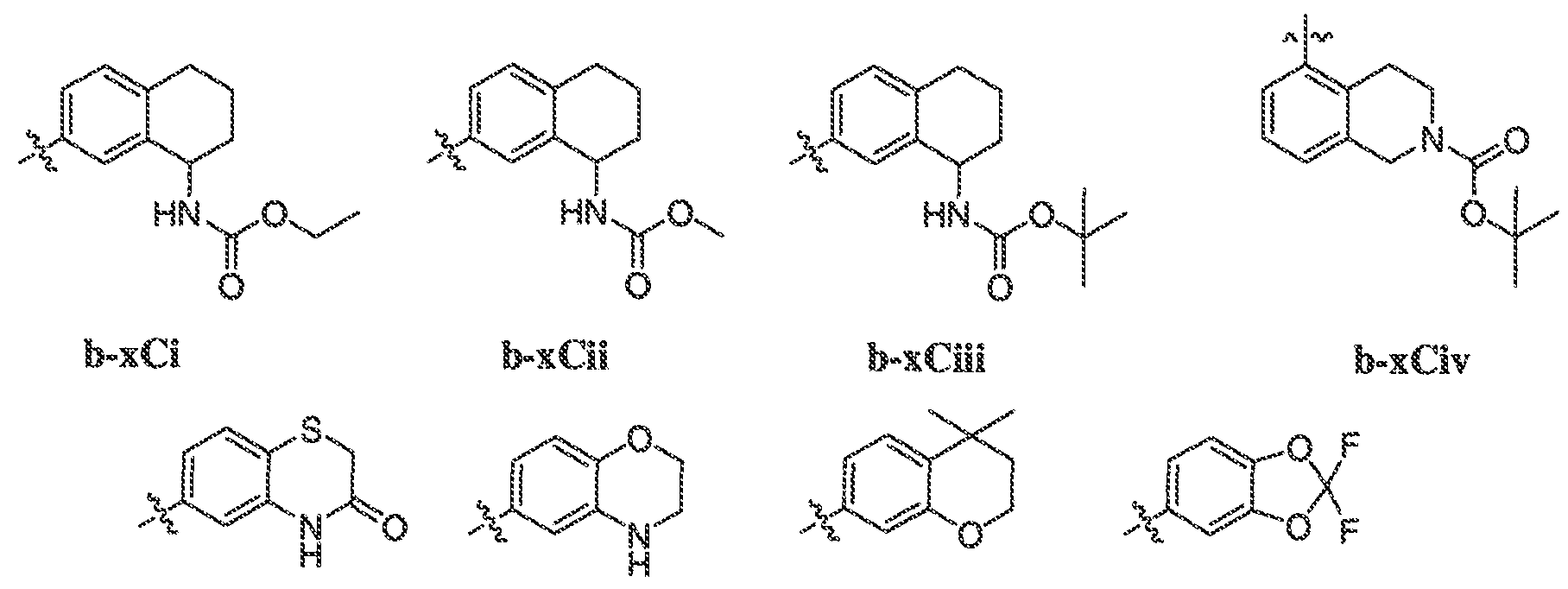
















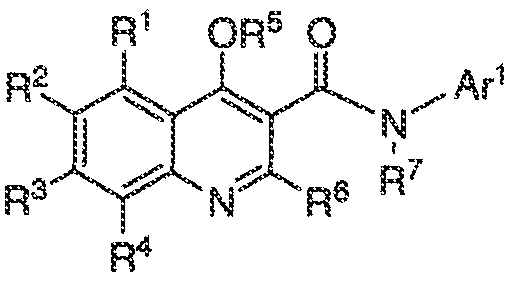










































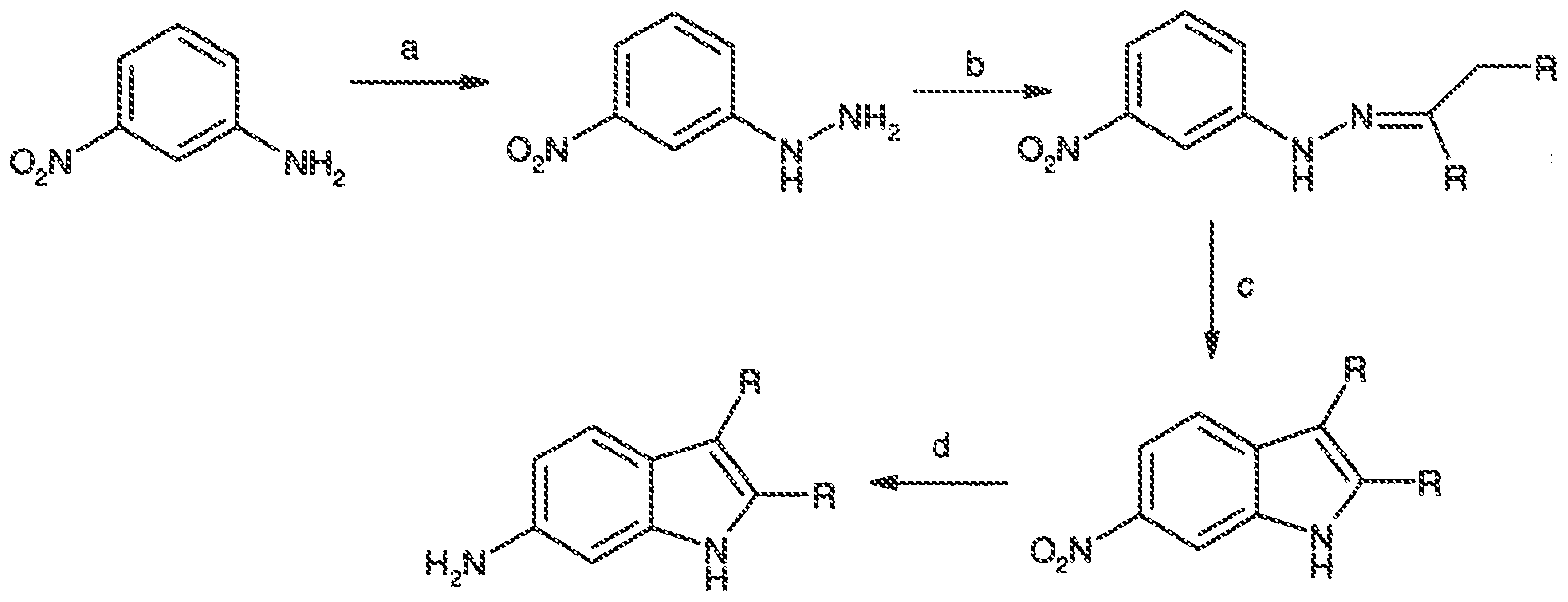









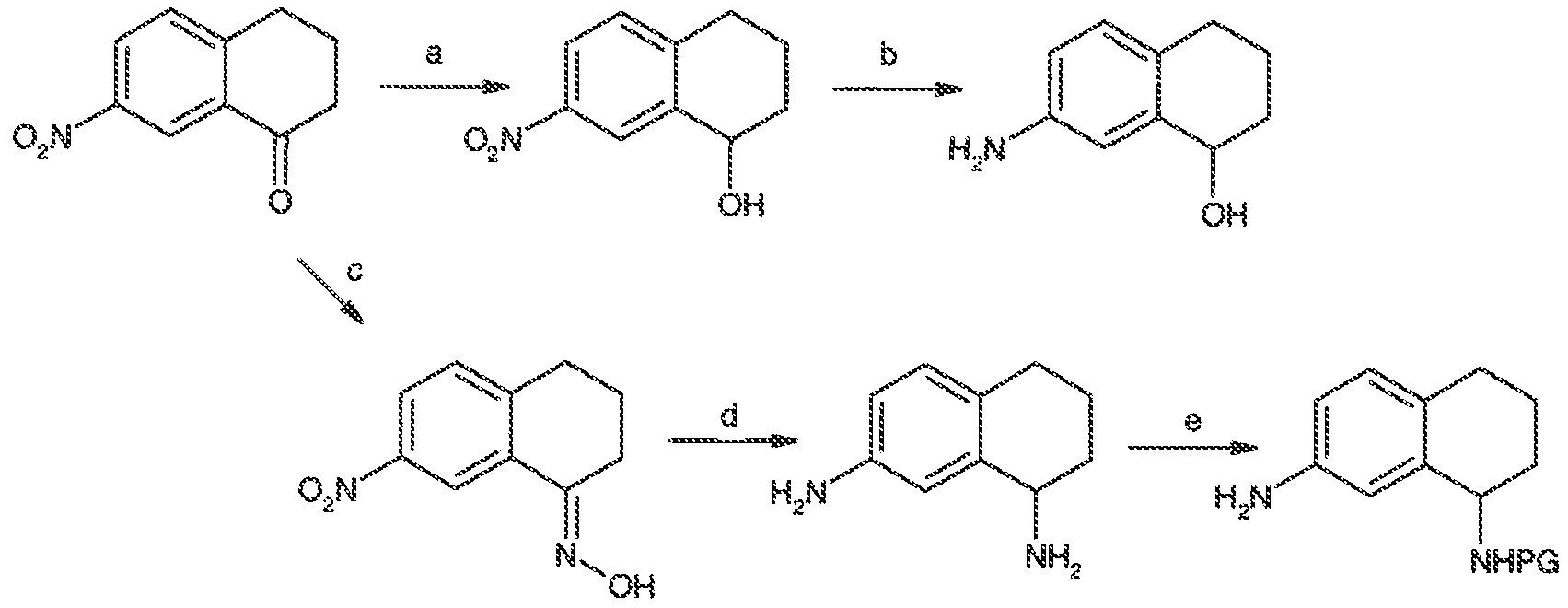






























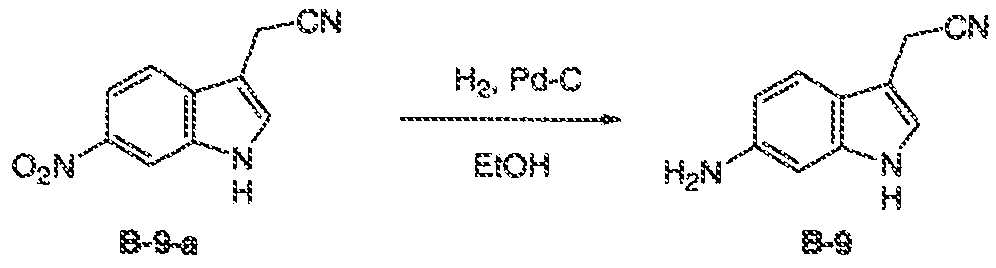


































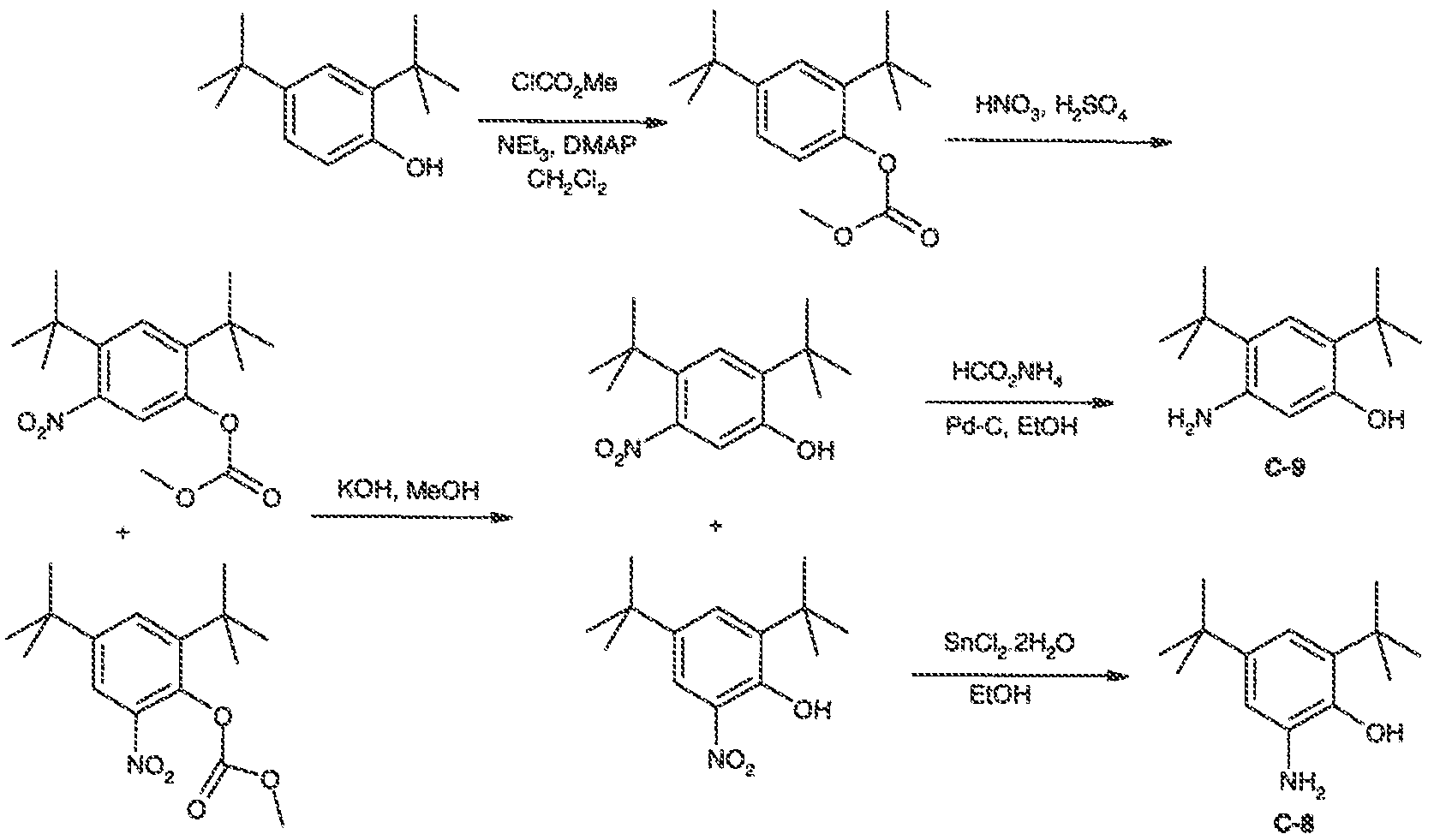


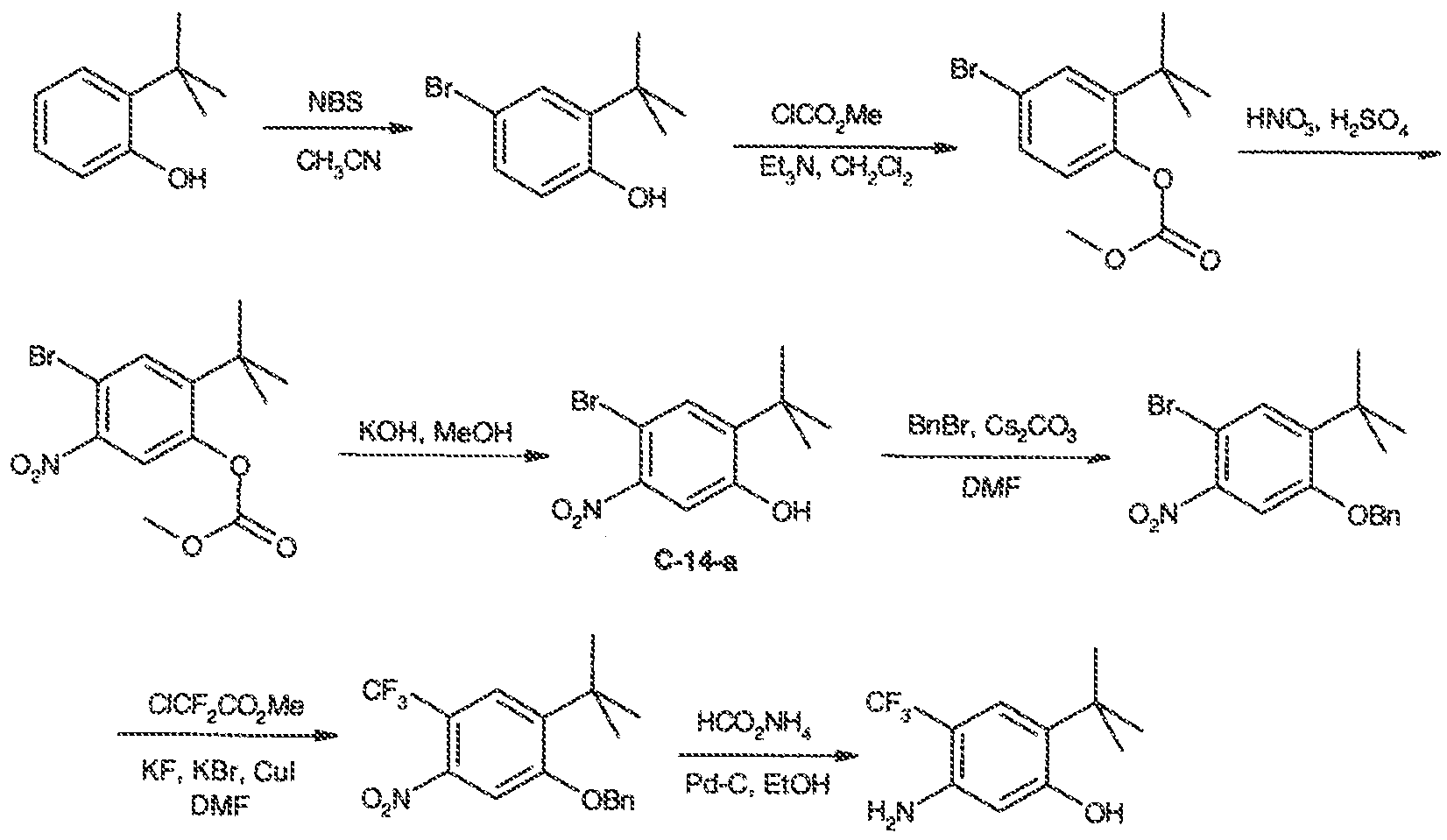






















































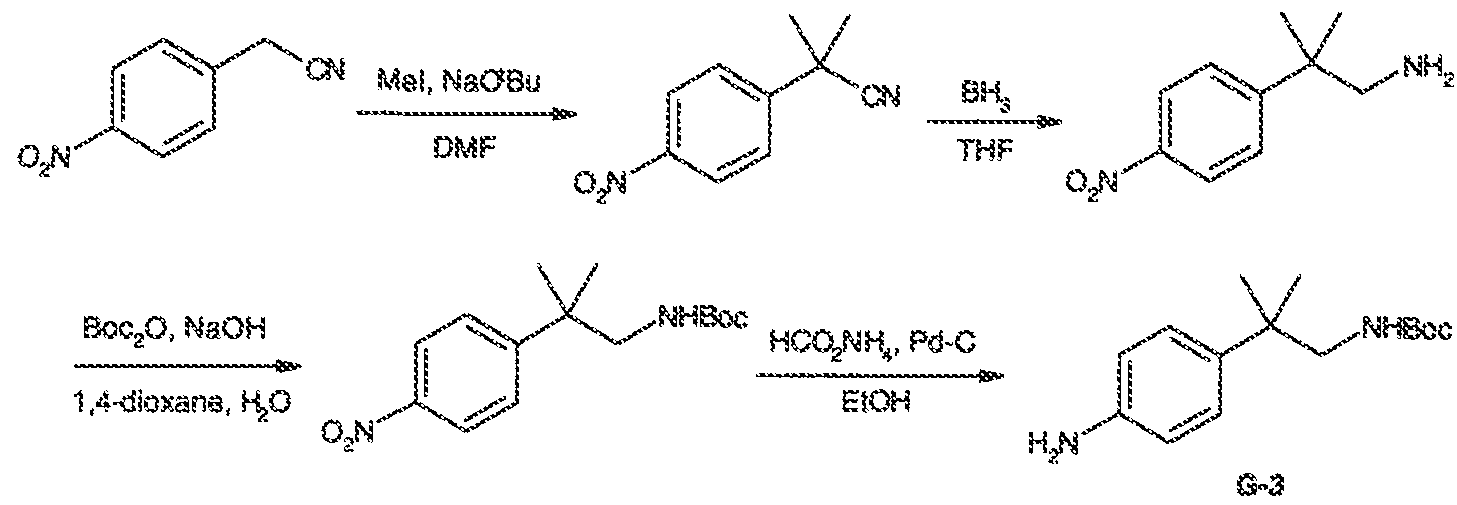
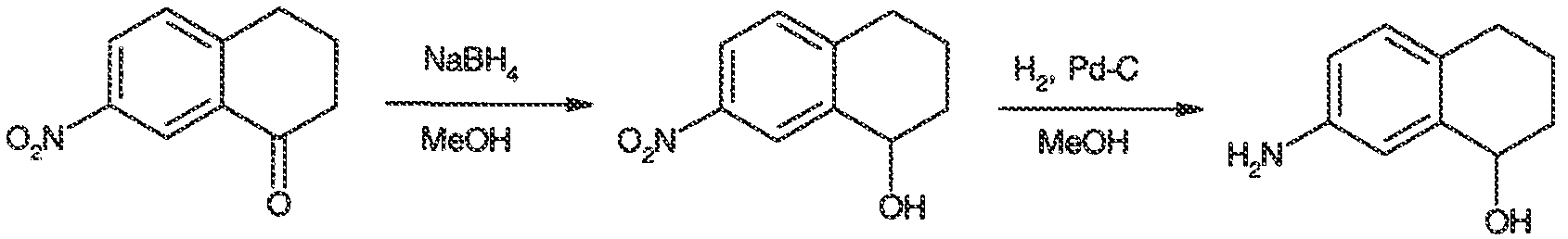































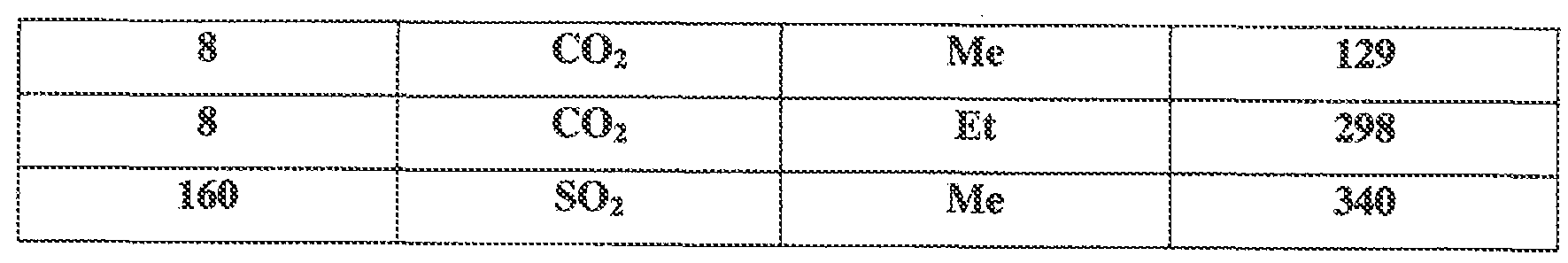



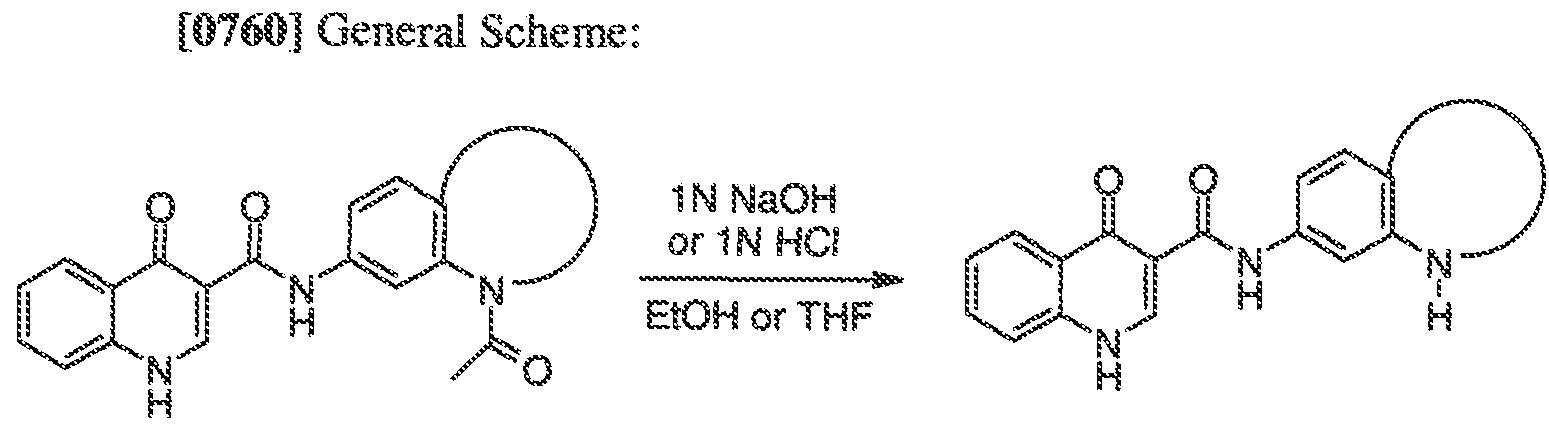






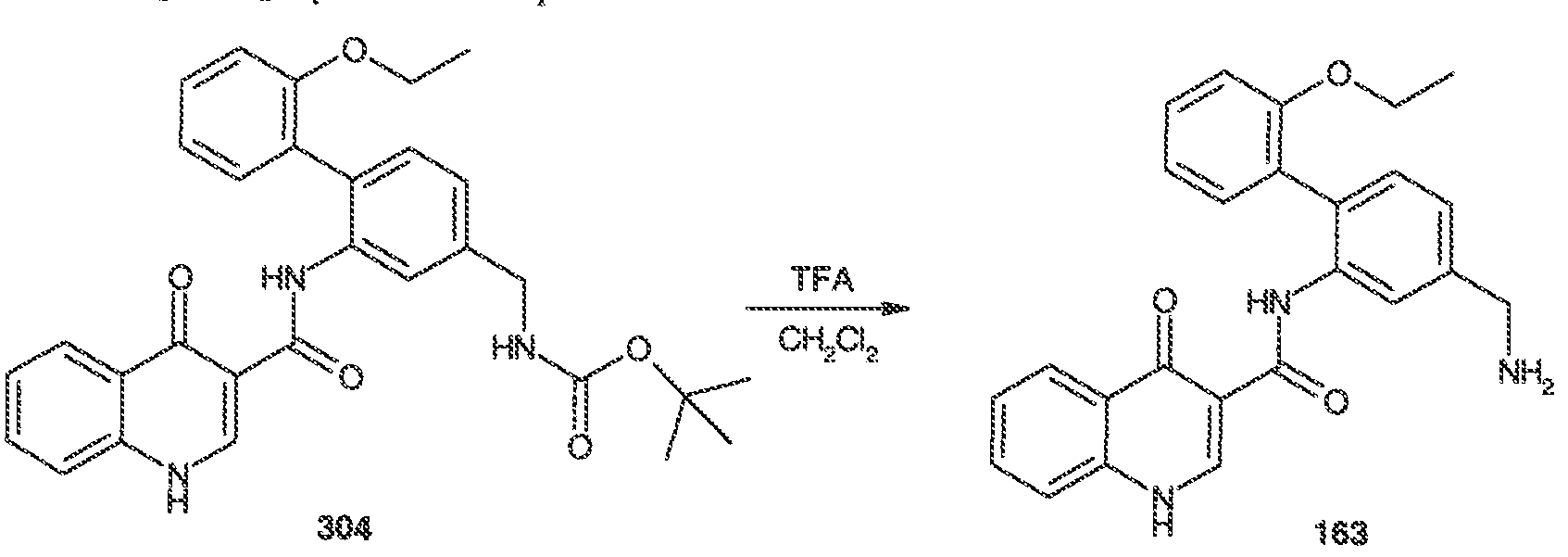




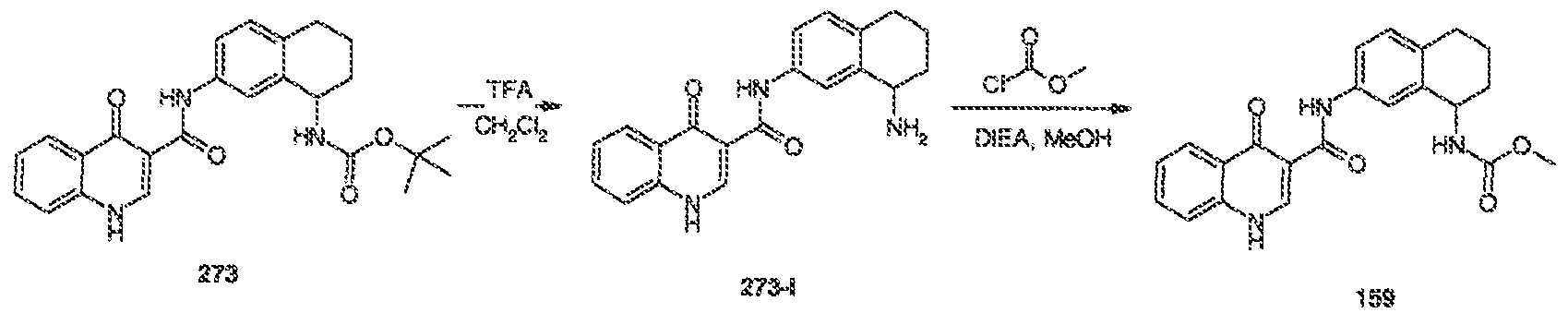















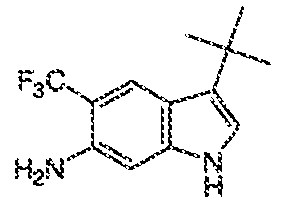

















































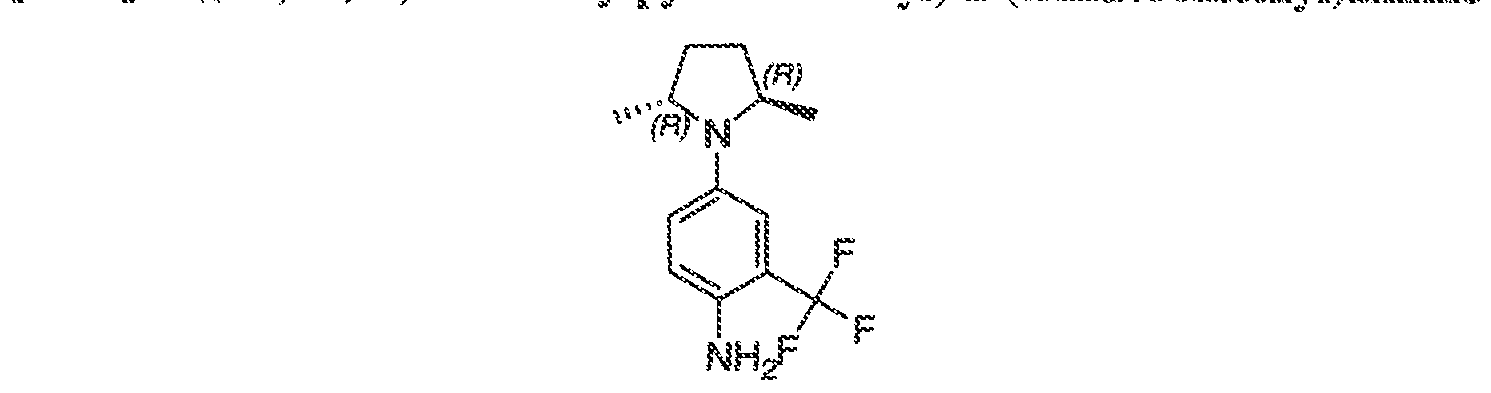
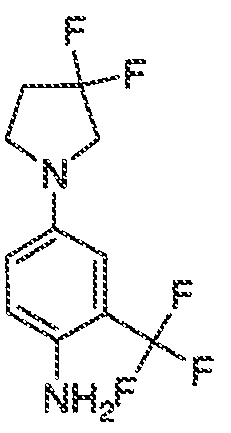




















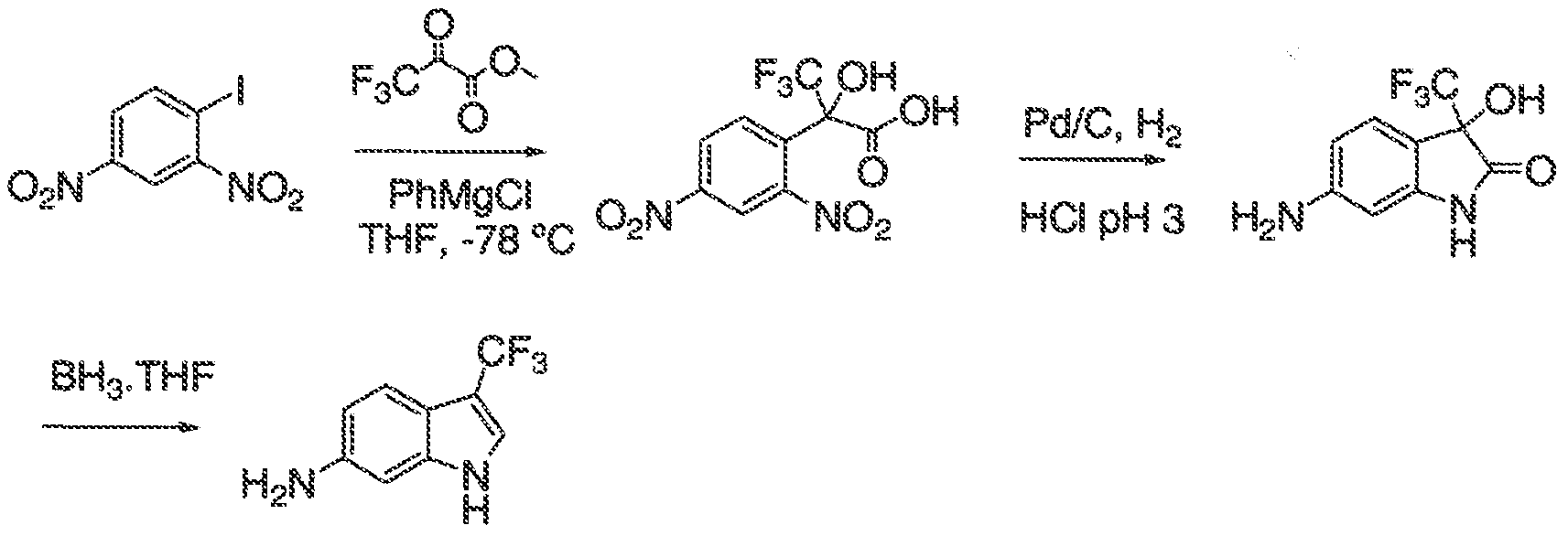







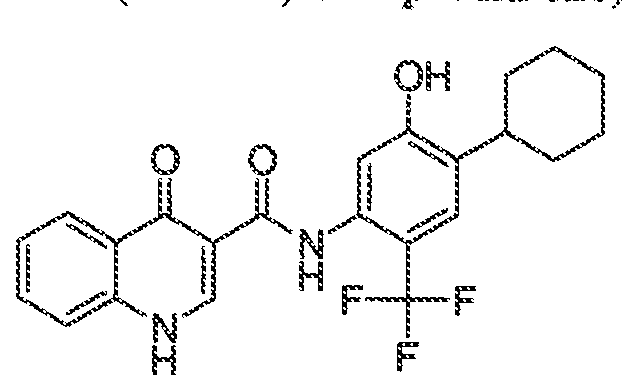






























Claims
Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| NZ600707A NZ600707A (en) | 2009-12-11 | 2010-12-10 | 4-oxo-1h-quinoline-3-carboxamides as modulators of atp-binding cassette transporters |
| CA2783314A CA2783314A1 (en) | 2009-12-11 | 2010-12-10 | 4-oxo-1h-quinoline-3-carboxamides as modulators of atp-binding cassette transporters |
| CN201080056022XA CN102652128A (en) | 2009-12-11 | 2010-12-10 | 4 -oxo- iH -quinoline- 3 - carboxamides as modulators of ATP -binding cassette transporters |
| AU2010327993A AU2010327993A1 (en) | 2009-12-11 | 2010-12-10 | 4-oxo-1H-quinoline-3-carboxamides as modulators of ATP-Binding cassette transporters |
| RU2012129206/04A RU2552353C2 (en) | 2009-12-11 | 2010-12-10 | Modulators of atp-binding transporters |
| EP10793116A EP2509954A1 (en) | 2009-12-11 | 2010-12-10 | 4 -oxo- 1h -quinoline- 3 - carboxamides as modulators of atp -binding cassette transporters |
| JP2012543310A JP2013513617A (en) | 2009-12-11 | 2010-12-10 | 4-oxo-1H-quinoline-3-carboxamide as modulator of ATP-binding cassette transporter |
| MX2012006764A MX2012006764A (en) | 2009-12-11 | 2010-12-10 | 4 -oxo- ih -quinoline- 3 - carboxamides as modulators of atp -binding cassette transporters. |
| US13/492,164 US8802700B2 (en) | 2010-12-10 | 2012-06-08 | Modulators of ATP-Binding Cassette transporters |
| IL220283A IL220283A0 (en) | 2009-12-11 | 2012-06-10 | 4-oxo-ih-quinoline-3- carboxamides, compositions comprising the same and uses thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/635,927 | 2009-12-11 | ||
| US12/635,927 US8354427B2 (en) | 2004-06-24 | 2009-12-11 | Modulators of ATP-binding cassette transporters |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/492,164 Continuation US8802700B2 (en) | 2010-12-10 | 2012-06-08 | Modulators of ATP-Binding Cassette transporters |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2011072241A1 true WO2011072241A1 (en) | 2011-06-16 |
| WO2011072241A9 WO2011072241A9 (en) | 2012-06-14 |
Family
ID=43447725
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2010/059920 WO2011072241A1 (en) | 2009-12-11 | 2010-12-10 | 4 -oxo- ih -quinoline- 3 - carboxamides as modulators of atp -binding cassette transporters |
Country Status (12)
| Country | Link |
|---|---|
| US (2) | US8354427B2 (en) |
| EP (1) | EP2509954A1 (en) |
| JP (1) | JP2013513617A (en) |
| CN (1) | CN102652128A (en) |
| AU (1) | AU2010327993A1 (en) |
| BE (1) | BE2015C040I2 (en) |
| CA (1) | CA2783314A1 (en) |
| IL (1) | IL220283A0 (en) |
| MX (1) | MX2012006764A (en) |
| NZ (1) | NZ600707A (en) |
| RU (1) | RU2552353C2 (en) |
| WO (1) | WO2011072241A1 (en) |
Cited By (57)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014114649A1 (en) * | 2013-01-22 | 2014-07-31 | Technische Universität Graz | Lipase inhibitors |
| US9150855B2 (en) | 2010-05-21 | 2015-10-06 | Universität Für Bodenkultur Wien | Methods for diagnosing bone or cardiovascular disorders |
| US9206115B2 (en) | 2010-05-21 | 2015-12-08 | Technische Universität Graz | ATGListatin and pharmaceutical composition comprising the same |
| US9254291B2 (en) | 2011-11-08 | 2016-02-09 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| WO2016069891A1 (en) | 2014-10-31 | 2016-05-06 | Abbvie Inc. | Substituted tetrahydropyrans and method of use |
| US9382208B1 (en) | 2015-01-26 | 2016-07-05 | Vanderbilt University | Negative allosteric modulators of metabotropic glutamate receptor 2 |
| WO2016193812A1 (en) | 2015-06-02 | 2016-12-08 | Abbvie S.A.R.L. | Substituted pyridines and method of use |
| US9540349B2 (en) | 2014-07-16 | 2017-01-10 | Gruenenthal Gmbh | Substituted pyrimidine compounds |
| WO2017009804A1 (en) | 2015-07-16 | 2017-01-19 | Abbvie S.Á.R.L. | Substituted tricyclics and method of use |
| WO2017053711A2 (en) | 2015-09-25 | 2017-03-30 | Concert Pharmaceuticals, Inc. | Deuterated cftr potentiators |
| WO2017060879A1 (en) | 2015-10-09 | 2017-04-13 | AbbVie S.à.r.l. | Novel compounds for treatment of cystic fibrosis |
| WO2017060874A1 (en) | 2015-10-09 | 2017-04-13 | Abbvie S.Á.R.L | N-sulfonylated pyrazolo[3,4-b]pyridin-6-carboxamides and method of use |
| WO2017060873A1 (en) | 2015-10-09 | 2017-04-13 | AbbVie S.à.r.l. | Substituted pyrazolo[3,4-b]pyridin-6-carboxylic acids and their use |
| WO2017187321A1 (en) | 2016-04-26 | 2017-11-02 | AbbVie S.à.r.l. | Modulators of cystic fibrosis transmembrane conductance regulator protein |
| WO2017208115A1 (en) | 2016-06-03 | 2017-12-07 | AbbVie S.à.r.l. | Heteroaryl substituted pyridines and methods of use |
| WO2018065962A1 (en) | 2016-10-07 | 2018-04-12 | AbbVie S.à.r.l. | Substituted pyrrolidines and their use in the treatment of cystic fiibrosis |
| WO2018065921A1 (en) | 2016-10-07 | 2018-04-12 | Abbvie S.Á.R.L. | Substituted pyrrolidines as cftr modulators |
| WO2018116185A1 (en) | 2016-12-20 | 2018-06-28 | AbbVie S.à.r.l. | Deuterated cftr modulators and methods of use |
| WO2018154493A1 (en) | 2017-02-24 | 2018-08-30 | AbbVie S.à.r.l. | Modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| US10138232B2 (en) | 2014-06-03 | 2018-11-27 | Novartis Ag | Naphthyridinedione derivatives |
| WO2019053634A1 (en) | 2017-09-14 | 2019-03-21 | AbbVie S.à.r.l. | Modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| US10391099B2 (en) | 2014-07-16 | 2019-08-27 | Gruenenthal Gmbh | Method of treating psoriatic arthritis, psoriasis, and chronic obstructive pulmonary disease with a novel 2,5-substituted pyrimidines |
| WO2019193062A1 (en) | 2018-04-03 | 2019-10-10 | Abbvie S.Á.R.L | Substituted pyrrolidines and their use |
| US10479766B2 (en) | 2011-05-18 | 2019-11-19 | Verex Pharmaceuticals (Europe) Limited | Deuterated CFTR potentiators |
| US10751363B2 (en) | 2015-03-23 | 2020-08-25 | Algipharma As | Use of aliginate oligomers and CFTR modulators in treatment of conditions associated with CFTR dysfunction |
| US10758534B2 (en) | 2014-10-06 | 2020-09-01 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| US10793547B2 (en) | 2016-12-09 | 2020-10-06 | Vertex Pharmaceuticals Incorporated | Modulator of the cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |
| EP3747882A1 (en) | 2019-06-03 | 2020-12-09 | AbbVie Overseas S.à r.l. | Prodrug modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| WO2021097054A1 (en) | 2019-11-12 | 2021-05-20 | Genzyme Corporation | 6-membered heteroarylaminosulfonamides for treating diseases and conditions mediated by deficient cftr activity |
| WO2021113809A1 (en) | 2019-12-05 | 2021-06-10 | Genzyme Corporation | Arylamides and methods of use thereof |
| WO2021113806A1 (en) | 2019-12-05 | 2021-06-10 | Genzyme Corporation | Arylamides and methods of use thereof |
| EP3664822A4 (en) * | 2017-08-04 | 2021-07-07 | Axial Therapeutics, Inc. | Inhibitors of microbially induced amyloid |
| US11066417B2 (en) | 2018-02-15 | 2021-07-20 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulators |
| US11155533B2 (en) | 2017-10-19 | 2021-10-26 | Vertex Pharmaceuticals Incorporated | Crystalline forms and compositions of CFTR modulators |
| US11179367B2 (en) | 2018-02-05 | 2021-11-23 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for treating cystic fibrosis |
| US11186566B2 (en) | 2016-09-30 | 2021-11-30 | Vertex Pharmaceuticals Incorporated | Modulator of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |
| US11253509B2 (en) | 2017-06-08 | 2022-02-22 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| WO2022150173A1 (en) | 2021-01-06 | 2022-07-14 | AbbVie Global Enterprises Ltd. | Modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| WO2022150174A1 (en) | 2021-01-06 | 2022-07-14 | AbbVie Global Enterprises Ltd. | Modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| US11413306B2 (en) | 2015-10-06 | 2022-08-16 | Algipharma As | Alginate oligomers for the treatment or prevention of microbial overgrowth in the intestinal tract |
| US11414439B2 (en) | 2018-04-13 | 2022-08-16 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |
| US11434201B2 (en) | 2017-08-02 | 2022-09-06 | Vertex Pharmaceuticals Incorporated | Processes for preparing pyrrolidine compounds |
| US11465985B2 (en) | 2017-12-08 | 2022-10-11 | Vertex Pharmaceuticals Incorporated | Processes for making modulators of cystic fibrosis transmembrane conductance regulator |
| US11517564B2 (en) | 2017-07-17 | 2022-12-06 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| US11584761B2 (en) | 2019-08-14 | 2023-02-21 | Vertex Pharmaceuticals Incorporated | Process of making CFTR modulators |
| US11591350B2 (en) | 2019-08-14 | 2023-02-28 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2023034992A1 (en) | 2021-09-03 | 2023-03-09 | Genzyme Corporation | Indole compounds and methods of use |
| WO2023034946A1 (en) | 2021-09-03 | 2023-03-09 | Genzyme Corporation | Indole compounds and uses thereof in the treatement of cystic fibrosis |
| US11708331B2 (en) | 2017-12-01 | 2023-07-25 | Vertex Pharmaceuticals Incorporated | Processes for making modulators of cystic fibrosis transmembrane conductance regulator |
| US11873300B2 (en) | 2019-08-14 | 2024-01-16 | Vertex Pharmaceuticals Incorporated | Crystalline forms of CFTR modulators |
| WO2024054851A1 (en) | 2022-09-07 | 2024-03-14 | Sionna Therapeutics | Macrocyclic compounds, compositions and methods of using thereof |
| WO2024054840A1 (en) | 2022-09-07 | 2024-03-14 | Sionna Therapeutics | Macrocyclic compounds, compositions, and methods of using thereof |
| WO2024054845A1 (en) | 2022-09-07 | 2024-03-14 | Sionna Therapeutics | Macrocycic compounds, compositions, and methods of using thereof |
| WO2024056791A1 (en) | 2022-09-15 | 2024-03-21 | Idorsia Pharmaceuticals Ltd | Combination of macrocyclic cftr modulators with cftr correctors and / or cftr potentiators |
| WO2024056798A1 (en) | 2022-09-15 | 2024-03-21 | Idorsia Pharmaceuticals Ltd | Macrocyclic cftr modulators |
| US11992553B2 (en) | 2014-08-29 | 2024-05-28 | Algipharma As | Inhalable powder formulations of alginate oligomers |
| US12122788B2 (en) | 2023-01-04 | 2024-10-22 | Vertex Pharmaceuticals Incorporated | Process of making CFTR modulators |
Families Citing this family (78)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100074949A1 (en) | 2008-08-13 | 2010-03-25 | William Rowe | Pharmaceutical composition and administration thereof |
| RU2006111093A (en) | 2003-09-06 | 2007-10-27 | Вертекс Фармасьютикалз Инкорпорейтед (Us) | MODULATORS OF ATR-BINDING CASSETTE TRANSPORTERS |
| NZ547220A (en) * | 2003-11-14 | 2009-12-24 | Vertex Pharma | Thiazoles and oxazoles useful as modulators of ATP-binding cassette transporters |
| US7977322B2 (en) | 2004-08-20 | 2011-07-12 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US20140343098A1 (en) * | 2004-06-24 | 2014-11-20 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| BR122018075478B8 (en) * | 2004-06-24 | 2023-10-31 | Vertex Pharma | atp link cassette carrier modulators |
| US8354427B2 (en) * | 2004-06-24 | 2013-01-15 | Vertex Pharmaceutical Incorporated | Modulators of ATP-binding cassette transporters |
| JP5143738B2 (en) * | 2005-08-11 | 2013-02-13 | バーテックス ファーマシューティカルズ インコーポレイテッド | Modulator of cystic fibrosis membrane conductance regulator |
| ES2439736T3 (en) | 2005-11-08 | 2014-01-24 | Vertex Pharmaceuticals Incorporated | Heterocyclic modulators of ATP binding cassette transporters |
| CA2635214A1 (en) * | 2005-12-27 | 2007-07-05 | Vertex Pharmaceuticals Incorporated | Compounds useful in cftr assays and methods therewith |
| HUE049976T2 (en) | 2005-12-28 | 2020-11-30 | Vertex Pharma | Pharmaceutical compositions of the amorphous form of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| CA2856037C (en) * | 2005-12-28 | 2017-03-07 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| US7671221B2 (en) * | 2005-12-28 | 2010-03-02 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-Binding Cassette transporters |
| RU2451018C2 (en) | 2006-04-07 | 2012-05-20 | Вертекс Фармасьютикалз Инкорпорейтед | Modulators of atp-binding cassette transporters |
| US10022352B2 (en) | 2006-04-07 | 2018-07-17 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US7645789B2 (en) | 2006-04-07 | 2010-01-12 | Vertex Pharmaceuticals Incorporated | Indole derivatives as CFTR modulators |
| US8563573B2 (en) * | 2007-11-02 | 2013-10-22 | Vertex Pharmaceuticals Incorporated | Azaindole derivatives as CFTR modulators |
| CN104447716A (en) | 2007-05-09 | 2015-03-25 | 沃泰克斯药物股份有限公司 | Modulators of CFTR |
| US20110177999A1 (en) * | 2007-08-09 | 2011-07-21 | Vertex Pharmaceuticals Incorporated | Therapeutic Combinations Useful in Treating CFTR Related Diseases |
| WO2009038913A2 (en) | 2007-08-24 | 2009-03-26 | Vertex Pharmaceuticals Incorporated | Isothiazolopyridinones useful for the treatment of (inter alia) cystic fibrosis |
| US8551534B2 (en) * | 2007-10-10 | 2013-10-08 | Parion Sciences, Inc. | Inhaled hypertonic saline delivered by a heated nasal cannula |
| CA2705562C (en) * | 2007-11-16 | 2016-05-17 | Vertex Pharmaceuticals Incorporated | Isoquinoline modulators of atp-binding cassette transporters |
| CN101910156B (en) | 2007-12-07 | 2013-12-04 | 沃泰克斯药物股份有限公司 | Solid forms of 3-(6-(1-(2,2-difluorobenzo[d][1,3] dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl) benzoic acid |
| US20100036130A1 (en) | 2007-12-07 | 2010-02-11 | Vertex Pharmaceuticals Incorporated | Processes for producing cycloalkylcarboxamido-pyridine benzoic acids |
| CA2989620C (en) * | 2007-12-07 | 2022-05-03 | Vertex Pharmaceuticals Incorporated | Processes for producing cycloalkylcarboxamido-pyridine benzoic acids |
| WO2009076141A2 (en) * | 2007-12-07 | 2009-06-18 | Vertex Pharmaceuticals Incorporated | Formulations of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cycklopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid |
| EP2271622B1 (en) | 2008-02-28 | 2017-10-04 | Vertex Pharmaceuticals Incorporated | Heteroaryl derivatives as CFTR Modulators |
| PT2615085E (en) | 2008-03-31 | 2015-10-09 | Vertex Pharma | Pyridyl derivatives as cftr modulators |
| CN102164587A (en) * | 2008-09-29 | 2011-08-24 | 沃泰克斯药物股份有限公司 | Dosage units of 3-(6-(1-(2,2-difluorobenzo [D] [1,3] dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid |
| EA018891B1 (en) | 2008-10-23 | 2013-11-29 | Вертекс Фармасьютикалз, Инкорпорейтед | Modulators of cystic fibrosis transmembrane conductance regulator |
| SG10201504084QA (en) | 2009-03-20 | 2015-06-29 | Vertex Pharma | Process for making modulators of cystic fibrosis transmembrane conductance regulator |
| EP2490687A1 (en) | 2009-10-22 | 2012-08-29 | Vertex Pharmaceuticals Incorporated | Compositions for treatment of cystic fibrosis and other chronic diseases |
| US8802868B2 (en) | 2010-03-25 | 2014-08-12 | Vertex Pharmaceuticals Incorporated | Solid forms of (R)-1(2,2-difluorobenzo[D][1,3]dioxo1-5-yl)-N-(1-(2,3-dihydroxypropyl-6-fluoro-2-(1-hydroxy-2-methylpropan2-yl)-1H-Indol-5-yl)-Cyclopropanecarboxamide |
| DK3150198T3 (en) | 2010-04-07 | 2021-11-01 | Vertex Pharma | PHARMACEUTICAL COMPOSITIONS OF 3- (6- (1- (2,2-DIFLUOROBENZO [D] [1,3] DIOXOL-5-YL) -CYCLOPROPANCARBOXAMIDO) -3-METHYLPYRIODIN-2-YL) BENZOIC ACID AND ADMINISTRATION |
| BR112012026257A2 (en) | 2010-04-07 | 2017-03-14 | Vertex Pharma | solid forms of 3- (6- (1- (2-, 2-difluorbenzo [d] [1,3] dioxol-5-yl) cyclopropanecarboxamido) -3-methylpyridin-2-yl) benzoic acid |
| MX2012012204A (en) | 2010-04-22 | 2012-12-05 | Vertex Pharma | Process of producing cycloalkylcarboxamido-indole compounds. |
| NZ603043A (en) | 2010-04-22 | 2015-02-27 | Vertex Pharma | Pharmaceutical compositions comprising cftr modulators and administrations thereof |
| EP2560649A1 (en) | 2010-04-22 | 2013-02-27 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions and administrations thereof |
| AU2011255237A1 (en) * | 2010-05-20 | 2012-11-29 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions and administrations thereof |
| US8563593B2 (en) | 2010-06-08 | 2013-10-22 | Vertex Pharmaceuticals Incorporated | Formulations of (R)-1-(2,2-difluorobenzo[D] [1,3] dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide |
| US20140113898A1 (en) * | 2010-11-08 | 2014-04-24 | Zalicus Pharmaceuticals Ltd. | Bisarylsulfone and dialkylarylsulfone compounds as calcium channel blockers |
| US8802700B2 (en) * | 2010-12-10 | 2014-08-12 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-Binding Cassette transporters |
| JP6219271B2 (en) | 2011-06-07 | 2017-10-25 | パリオン・サイエンシィズ・インコーポレーテッド | Method of treatment |
| US8945605B2 (en) | 2011-06-07 | 2015-02-03 | Parion Sciences, Inc. | Aerosol delivery systems, compositions and methods |
| AR086745A1 (en) | 2011-06-27 | 2014-01-22 | Parion Sciences Inc | 3,5-DIAMINO-6-CHLORINE-N- (N- (4- (4- (2- (HEXIL (2,3,4,5,6-PENTAHYDROXIHEXIL)) AMINO) ETOXI) PHENYL) BUTIL) CARBAMIMIDOIL) PIRAZINA -2-CARBOXAMIDE |
| CN109966264A (en) | 2012-02-27 | 2019-07-05 | 沃泰克斯药物股份有限公司 | Pharmaceutical composition and its application |
| US8674108B2 (en) | 2012-04-20 | 2014-03-18 | Vertex Pharmaceuticals Incorporated | Solid forms of N-[2,4-bis(1,1-dimethylethy)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| AU2013267504B2 (en) | 2012-05-29 | 2017-11-02 | Parion Sciences, Inc. | Dendrimer like amino amides possessing sodium channel blocker activity for the treatment of dry eye and other mucosal diseases |
| AR092857A1 (en) | 2012-07-16 | 2015-05-06 | Vertex Pharma | PHARMACEUTICAL COMPOSITIONS OF (R) -1- (2,2-DIFLUOROBENZO [D] [1,3] DIOXOL-5-IL) -N- (1- (2,3-DIHYDROXIPROPIL) -6-FLUORO-2- ( 1-HYDROXI-2-METHYLPROPAN-2-IL) -1H-INDOL-5-IL) CYCLOPROPANCARBOXAMIDE AND ADMINISTRATION OF THE SAME |
| PT2931713T (en) | 2012-12-17 | 2017-02-22 | Parion Sciences Inc | Chloro-pyrazine carboxamide derivatives useful for the treatment of diseases favoured by insufficient mucosal hydration |
| ES2674665T3 (en) | 2012-12-17 | 2018-07-03 | Parion Sciences, Inc. | 3,5-Diamino-6-Chloro-N- (N- (4-phenylbutyl) carbamimidoyl) -pyrazine-2-carboxamide compounds |
| BR112015014178A2 (en) | 2012-12-17 | 2017-07-11 | Parion Sciences Inc | 3,5-diamino-6-chloro-n- (n- (4-phenylbutyl) carbamimidoyl) pyrazine-2-carboxamide compounds |
| CN103224466A (en) * | 2013-04-15 | 2013-07-31 | 北京大学 | Compound with beta-secretase inhibition function, its preparation method and application |
| US10231932B2 (en) | 2013-11-12 | 2019-03-19 | Vertex Pharmaceuticals Incorporated | Process of preparing pharmaceutical compositions for the treatment of CFTR mediated diseases |
| ES2957761T3 (en) | 2014-04-15 | 2024-01-25 | Vertex Pharma | Pharmaceutical compositions for the treatment of diseases mediated by the cystic fibrosis transmembrane conductance regulator |
| US20200347038A1 (en) | 2014-08-29 | 2020-11-05 | Russell Dahl | Quinolines that modulate serca and their use for treating disease |
| US20170275249A1 (en) * | 2014-09-17 | 2017-09-28 | The Regents Of The University Of California | Small lipopeptidomimetic inhibitors of ghrelin o-acyl transferase |
| US9573948B2 (en) | 2014-10-06 | 2017-02-21 | Flatley Discovery Lab | Triazolopyridine compounds and methods for the treatment of cystic fibrosis |
| CN107250113B (en) | 2014-10-07 | 2019-03-29 | 弗特克斯药品有限公司 | Co-crystals of modulators of cystic fibrosis transmembrane conductance regulator |
| US20170281612A1 (en) * | 2014-10-08 | 2017-10-05 | Nivalis Therapeutics, Inc. | Methods for the Treatment of Cystic Fibrosis |
| JP6494757B2 (en) | 2014-11-18 | 2019-04-03 | バーテックス ファーマシューティカルズ インコーポレイテッドVertex Pharmaceuticals Incorporated | Process for high-throughput high performance liquid chromatography |
| EP3256116B1 (en) | 2015-02-11 | 2022-08-17 | Icahn School of Medicine at Mount Sinai | Benzenesulfonamide upregulators of npc1 for neimann-pick disease and other lysosomal storage disorders |
| CN106467541B (en) | 2015-08-18 | 2019-04-05 | 暨南大学 | Substituted quinolone analog derivative or its pharmaceutically acceptable salt or stereoisomer and its Pharmaceutical composition and application |
| HUP1600270A2 (en) | 2016-04-25 | 2017-10-30 | Druggability Tech Ip Holdco Ltd | Complexes of ivacaftor and its salts and derivatives, process for the preparation thereof and pharmaceutical compositions containing them |
| US10383865B2 (en) | 2016-04-25 | 2019-08-20 | Druggability Technologies Ip Holdco Limited | Pharmaceutical combination composition comprising complex formulations of Ivacaftor and Lumacaftor and their salts and derivatives, process for their preparation thereof and pharmaceutical compositions containing them |
| US10206915B2 (en) | 2016-04-25 | 2019-02-19 | Druggability Technologies Ip Holdco Limited | Complexes of Ivacaftor and its salts and derivatives, process for the preparation thereof and pharmaceutical compositions containing them |
| HUP1600271A2 (en) | 2016-04-25 | 2017-10-30 | Druggability Tech Ip Holdco Ltd | Pharmaceutical composition comprising complex formulations of ivacaftor and lumacaftor and their salts and derivatives, process for their preparation thereof and pharmaceutical compositions containing them |
| CN106366002B (en) * | 2016-08-30 | 2019-01-22 | 京博农化科技股份有限公司 | A kind of Boscalid intermediate 4 '-chloro- 2- aminobphenyl synthetic method |
| CA3103711A1 (en) * | 2018-07-12 | 2020-01-16 | UCB Biopharma SRL | Spirocyclic indane analogues as il-17 modulators |
| WO2020158870A1 (en) * | 2019-01-30 | 2020-08-06 | 学校法人慶應義塾 | Parkinson's disease therapeutic |
| WO2021011327A1 (en) | 2019-07-12 | 2021-01-21 | Orphomed, Inc. | Compound for treating cystic fibrosis |
| NZ788464A (en) | 2019-12-11 | 2024-07-05 | Taro Pharmaceutical Ind Ltd | Preparation of trifarotene and intermediates and polymorphs thereof |
| CN113831301B (en) * | 2020-06-08 | 2023-06-06 | 沈阳药科大学 | Benzothiazole derivative and application thereof |
| US11730729B2 (en) | 2020-07-20 | 2023-08-22 | Neurodon Corporation | Quinolines that modulate SERCA and their use for treating disease |
| CN113149881B (en) * | 2021-03-04 | 2022-07-19 | 宁波大学 | Chiral derivatization reagent and preparation method and application thereof |
| CN114085197B (en) * | 2021-12-01 | 2024-03-29 | 合肥创新医药技术有限公司 | Synthesis method of 4- (3-trifluoromethyl phenyl) -1-piperazine-ethanol, flibanserin intermediate and flibanserin |
| CN116082163B (en) * | 2023-01-06 | 2023-10-13 | 河北凡克新材料有限公司 | Preparation method of 3',4' -difluoro-2 ' -aminobiphenyl |
| CN116410180B (en) * | 2023-03-01 | 2024-09-10 | 青岛大学 | 4-Oxo quinoline-3-carboxamide derivative and application thereof |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5304121A (en) | 1990-12-28 | 1994-04-19 | Boston Scientific Corporation | Drug delivery system making use of a hydrogel polymer coating |
| US5886026A (en) | 1993-07-19 | 1999-03-23 | Angiotech Pharmaceuticals Inc. | Anti-angiogenic compositions and methods of use |
| US6099562A (en) | 1996-06-13 | 2000-08-08 | Schneider (Usa) Inc. | Drug coating with topcoat |
| WO2006002421A2 (en) * | 2004-06-24 | 2006-01-05 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| WO2009074575A2 (en) | 2007-12-10 | 2009-06-18 | Novartis Ag | Organic compounds |
Family Cites Families (128)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5874424A (en) | 1995-12-20 | 1999-02-23 | Vertex Pharmaceuticals Incorporated | Inhibitors of interleukin-1β converting enzyme |
| US3524858A (en) | 1967-05-18 | 1970-08-18 | Warner Lambert Pharmaceutical | 1,4 - dihydro-1-substituted alkyl-6,7-methylenedioxy - 4 - oxoquinoline-3-carboxylic acid |
| BE757639A (en) | 1969-10-17 | 1971-04-16 | Roussel Uclaf | NEW CEPHALOSPORIN DERIVATIVES AND PREPARATION PROCESS |
| GB1433774A (en) | 1973-02-26 | 1976-04-28 | Allen & Hanburys Ltd | Heterocyclic compounds apparatus for conveying articles |
| GB1433151A (en) | 1973-04-05 | 1976-04-22 | Allen & Hanburys Ltd | Benzo-ij-quinolizines |
| FR2281761A1 (en) | 1974-08-13 | 1976-03-12 | Roussel Uclaf | NEW DERIVATIVES OF 3-QUINOLEINE CARBOXYLIC ACID, THEIR METHOD OF PREPARATION AND THEIR APPLICATION AS A MEDICINAL PRODUCT |
| FR2324304A2 (en) | 1975-09-22 | 1977-04-15 | Roussel Uclaf | Analgesic (4)-hydroxy-quinoline-(3)-amides - prepd. from a quinoline-(3)-carboxyl chloride and an amine |
| FR2340092A2 (en) | 1976-02-09 | 1977-09-02 | Roussel Uclaf | Analgesic (N)-thiazolyl-quinoline carboxamides - prepd. from the corresp. quinoline carboxylic acid and (2)-amino-thiazole |
| FR2340735A1 (en) * | 1976-02-11 | 1977-09-09 | Roussel Uclaf | NEW DERIVATIVES OF 3-QUINOLEINE CARBOXYLIC ACID, THEIR METHOD OF PREPARATION AND THEIR APPLICATION AS A MEDICINAL PRODUCT |
| US4312870A (en) | 1979-06-21 | 1982-01-26 | Ciba-Geigy Corporation | Pyrazoloquinolines |
| HU190796B (en) | 1981-06-12 | 1986-11-28 | Roussel Uclaf,Fr | Process for producing n-dihydrothiazolyl-3-quinoline-carboxamide derivatives |
| FR2509728A1 (en) | 1981-07-17 | 1983-01-21 | Roussel Uclaf | NOVEL QUINOLINE DERIVATIVES, THEIR SALTS, PREPARATION METHOD, MEDICAMENT APPLICATION AND COMPOSITIONS COMPRISING THE SAME |
| FR2537140B1 (en) | 1982-12-07 | 1986-07-18 | Roussel Uclaf | NOVEL 4-HYDROXY-3-QUINOLEINE CARBOXAMIDE DERIVATIVES, SALTS THEREOF, PROCESS FOR THEIR PREPARATION, APPLICATION AS MEDICAMENTS, AND COMPOSITIONS CONTAINING THEM |
| SU1360584A3 (en) * | 1983-08-12 | 1987-12-15 | Варнер-Ламберт Компани (Фирма) | Method of producing naphthyrydine quinoline- or benzoxazine carbolic acids or their pharmaceutically acceptable salts of acid addition |
| US4845105A (en) | 1984-10-30 | 1989-07-04 | Roussel Uclaf | 4-OH-quinoline carboxylic acid amides having analgesic and anti-inflammatory activity |
| US4687539A (en) | 1986-10-29 | 1987-08-18 | International Business Machines Corp. | End point detection and control of laser induced dry chemical etching |
| DE3702393A1 (en) | 1987-01-28 | 1988-08-11 | Bayer Ag | 8-CYANO-1-CYCLOPROPYL-1,4-DIHYDRO-4-OXO- 3-CHINOLINE CARBONIC ACIDS, METHOD FOR THEIR PRODUCTION AND ANTIBACTERIAL AGENTS CONTAINING THEM |
| DD279887A1 (en) | 1987-07-03 | 1990-06-20 | Inst Pharmakologische Forschun | METHOD OF PREPARING D-ALPHA- (4 (1H) -1,5-NAPHTHYRIDONE-3-CARBOXAMIDO) -BENZYLPENICILLIN AND OTHER BETA LACTAMANTIBIOTICS |
| US4777252A (en) | 1987-08-13 | 1988-10-11 | E. R. Squibb & Sons, Inc. | 2-oxo-1-[[(substituted sulfonyl)amino]-carbonyl]azetidines |
| DE3827253A1 (en) | 1987-08-20 | 1989-03-02 | Sandoz Ag | Esters and amides of cyclic carboxylic acids and cyclic alcohols and amines, processes for their preparation and therapeutic compositions containing them |
| DE3731516A1 (en) | 1987-09-18 | 1989-03-30 | Bayer Ag | N-ARYL NITROGEN HETEROCYCLES |
| DE3811341A1 (en) | 1987-10-09 | 1989-04-27 | Bayer Ag | 7-POSITION C-LINKED CHINOLONIC AND 1,8-NAPHTHYRIDINE-4-ON-CARBONIC ACID AND A METHOD FOR THE PRODUCTION THEREOF |
| US4786644A (en) | 1987-11-27 | 1988-11-22 | Hoechst-Roussel Pharmaceuticals Inc. | 1-aryl-3-quinolinecarboxamide |
| DE3808118A1 (en) | 1988-03-11 | 1989-09-21 | Bayer Ag | METHOD FOR PRODUCING 1-CYCLOPROPYL-QUINOLON CARBONIC ACIDS AND THEIR DERIVATIVES |
| DE3808117A1 (en) | 1988-03-11 | 1989-09-21 | Bayer Ag | N-CYCLOPROPYLANILINE AND THE USE THEREOF IN METHOD FOR THE PRODUCTION OF 1-CYCLOPROPYL-CHINOLONIC CARBONIC ACIDS AND THEIR DERIVATIVES |
| DE3816119A1 (en) | 1988-05-11 | 1989-11-23 | Bayer Ag | 7-SUBSTITUTED CHINOLON AND NAPHTHYRIDONE CARBONIC ACID DERIVATIVES |
| DK273689A (en) | 1988-06-06 | 1989-12-07 | Sanofi Sa | 4-AMINO-3-CARBOXYQUINOLINES AND -NAPHTHYRIDINES, PROCEDURES FOR THEIR PREPARATION AND USE OF THEM IN PHARMACEUTICALS |
| DE3827221A1 (en) | 1988-08-11 | 1990-02-15 | Bayer Ag | SUBSTITUTED N-PHENYL NITROGEN OR NITROGEN-SULFUR HETEROCYCLES, METHODS AND CORRESPONDING HETEROCYCLIC PHENOL DERIVATIVES, PHENYLISO (THIO) CYANATES AND CARBAMATES AS INTERMEDIATE PRODUCTS FOR THEIR PRODUCTION, THEIR USE IN PLANTING PLANT |
| US5491139A (en) | 1988-10-24 | 1996-02-13 | The Procter & Gamble Company | Antimicrobial quinolonyl lactams |
| LU87611A1 (en) | 1989-10-20 | 1991-05-07 | Oreal | TINCTORIAL COMPOSITION FOR KERATINIC FIBERS CONTAINING OXIDATION DYE PRECURSORS AND AMINO INDOLIC COUPLERS, DYEING METHODS USING SAID COMPOSITIONS AND COMPOUNDS |
| FR2662713B1 (en) | 1990-05-29 | 1994-04-08 | Oreal | PROCESS FOR DYEING KERATINIC FIBERS WITH AN AMINOINDOLE ASSOCIATED WITH A QUINON DERIVATIVE. |
| DE4017516A1 (en) | 1990-05-31 | 1991-12-05 | Wella Ag | OXIDATION HAIR COLORING AGENT CONTAINING 3-AMINOPHENOL DERIVATIVES, METHOD FOR THE OXIDATIVE COLORING OF HAIR AND NEW 3-AMINOPHENOL DERIVATIVES |
| DE4026530A1 (en) | 1990-08-22 | 1992-02-27 | Basf Ag | SYNERGISTIC AGENTS FOR REGULATING PLANT GROWTH |
| AU8448491A (en) | 1990-09-07 | 1992-03-30 | Schering Corporation | Antiviral compounds and antihypertensive compounds |
| US5175151A (en) | 1990-09-07 | 1992-12-29 | Schering Corporation | Antiviral compounds and antihypertensive compounds |
| CA2091172C (en) | 1990-09-07 | 1997-05-20 | Adriano Afonso | Antiviral compounds and antihypertensive compounds |
| JPH04171521A (en) | 1990-11-05 | 1992-06-18 | Fujitsu Ltd | Touch panel |
| IL100917A0 (en) | 1991-02-16 | 1992-11-15 | Fisons Plc | Pyridinone and pyrimidinone derivatives,their preparation and pharmaceutical compositions containing them |
| CA2065106A1 (en) | 1991-04-04 | 1992-10-05 | Junichi Fukawa | Silver halide photographic light-sensitive material and photographic product for film-making process |
| FR2675380A1 (en) | 1991-04-18 | 1992-10-23 | Oreal | PROCESS FOR DYING KERATIN FIBERS WITH AMINOINDOLES, BASIC PH, COMPOSITIONS IMPLEMENTED AND NOVEL COMPOUNDS. |
| GB9108547D0 (en) | 1991-04-22 | 1991-06-05 | Fujisawa Pharmaceutical Co | Quinoline derivatives |
| CA2075154A1 (en) | 1991-08-06 | 1993-02-07 | Neelakantan Balasubramanian | Peptide aldehydes as antithrombotic agents |
| JPH05345780A (en) | 1991-12-24 | 1993-12-27 | Kumiai Chem Ind Co Ltd | Pyrimidine or triazine derivative and herbicide |
| DE69329856D1 (en) | 1992-05-20 | 2001-02-15 | Merck & Co Inc | ESTER DERIVATIVES OF 4-AZA STEROIDS |
| EP0641204B1 (en) | 1992-05-20 | 2000-08-16 | Merck & Co. Inc. | 17-ethers and thioethers of 4-aza-steroids |
| US5352690A (en) | 1992-07-01 | 1994-10-04 | Eli Lilly And Company | 1,2,4-trioxygenated benzene derivatives useful as leukotriene antagonists |
| AU4567193A (en) | 1992-07-10 | 1994-01-31 | Laboratoires Glaxo S.A. | Anilide derivatives |
| US5322847A (en) | 1992-11-05 | 1994-06-21 | Pfizer Inc. | Azabenzimidazoles in the treatment of asthma, arthritis and related diseases |
| WO1994014797A1 (en) | 1992-12-23 | 1994-07-07 | Smithkline Beecham Corporation | Quinoline compounds and the treatment of leucotriene related diseases therewith |
| US5750754A (en) | 1993-03-29 | 1998-05-12 | Zeneca Limited | Heterocyclic compounds |
| JP3760474B2 (en) | 1993-04-22 | 2006-03-29 | ダイキン工業株式会社 | Method and apparatus for generating electric energy, and compound having NF bond used therefor |
| IL111266A (en) | 1993-10-22 | 2002-03-10 | Zeneca Ltd | 2-HETEROARYL OR 2-ARYLPYRIDAZINO [4,5-b] QUINOLINE - 1, 10 - DIONES, THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
| EP0746561B1 (en) | 1994-02-25 | 1999-04-28 | Laboratorios Aranda, S.A. De C.V. (Mx/Mx) | Quinolonylcarboxamidocephalosporin derivatives and pharmaceutical compositions containing them |
| FR2720397B1 (en) | 1994-05-24 | 1996-08-23 | Laphal Laboratoires Sa | New oxathiolanes, process for their preparation and pharmaceutical compositions containing them. |
| ATE246677T1 (en) | 1994-05-27 | 2003-08-15 | Smithkline Beecham Farma | QUINOLINE DERIVATIVES AS TACHYKININ NK3 RECEPTOR ANTAGONISTS |
| EP0705835A1 (en) | 1994-09-01 | 1996-04-10 | Ciba-Geigy Ag | Quinoxalic-2,3-diones with an oxa or thiaheterocyclic fused ring |
| WO1996015099A1 (en) | 1994-11-09 | 1996-05-23 | Novo Nordisk A/S | Heterocyclic compounds, their preparation and use |
| GB9501567D0 (en) | 1995-01-26 | 1995-03-15 | Pharmacia Spa | Hydrosoluble 3-arylidene-2-oxindole derivatives as tyrosine kinase inhibitors |
| DE19601142A1 (en) | 1995-07-13 | 1997-01-16 | Agfa Gevaert Ag | Colour photographic process and material using masked developer - combining high colour densities with decreased fogging and sensitivity to process variations, also allowing use of developer free baths |
| EP0841929B1 (en) | 1995-08-02 | 2003-05-07 | Darwin Discovery Limited | Quinolones and their therapeutic use |
| ATE258437T1 (en) | 1995-08-02 | 2004-02-15 | Darwin Discovery Ltd | QUINOLONES AND THEIR THERAPEUTIC USE |
| DE19532235A1 (en) | 1995-08-31 | 1997-03-06 | Keppler Bernhard K Priv Doz Dr | New antibacterial contg. osteotropic mol. fragments |
| DE69623655T2 (en) | 1995-10-19 | 2003-04-24 | Takeda Chemical Industries, Ltd. | CHINOLINE DERIVATIVES AS GNRH ANTAGONISTS |
| WO1997030999A1 (en) | 1996-02-21 | 1997-08-28 | Darwin Discovery Limited | Quinolones and their therapeutic use |
| US6215016B1 (en) | 1996-03-27 | 2001-04-10 | Toray Industries, Inc. | Ketone derivatives and medical application thereof |
| DE19615262A1 (en) | 1996-04-18 | 1997-10-23 | Bayer Ag | Hetero-linked phenylglycinolamides |
| PL329922A1 (en) | 1996-05-20 | 1999-04-26 | Darwin Discovery Ltd | Quinoline carboxamides as inhibitors of the tumour necrosis factor and of phosphodiesterase |
| BR9709105A (en) | 1996-05-20 | 1999-08-03 | Darwin Discovery Ltd | Quinoline sulfonamides as tnf inhibitors and as pde-iv inhibitors |
| CA2258822A1 (en) | 1996-06-20 | 1997-12-24 | Sean Kerwin | Compounds and methods for providing pharmacologically active preparations and uses thereof |
| AU3671897A (en) | 1996-07-23 | 1998-02-10 | Neurogen Corporation | Certain amido- and amino-substituted benzylamine derivatives; a new class of neuropeptite y1 specific ligands |
| GB9717576D0 (en) | 1997-08-19 | 1997-10-22 | Xenova Ltd | Pharmaceutical compounds |
| US6069151A (en) | 1996-11-06 | 2000-05-30 | Darwin Discovery, Ltd. | Quinolines and their therapeutic use |
| DE19651099A1 (en) | 1996-12-09 | 1998-06-10 | Consortium Elektrochem Ind | Multi-component system for changing, breaking down or bleaching lignin, lignin-containing materials or similar substances as well as methods for their use |
| AU6209098A (en) | 1997-01-15 | 1998-08-07 | Novartis Ag | Herbicidal agent |
| ZA986594B (en) | 1997-07-25 | 1999-01-27 | Abbott Lab | Urokinase inhibitors |
| US6258822B1 (en) | 1997-08-06 | 2001-07-10 | Abbott Laboratories | Urokinase inhibitors |
| US6429207B1 (en) | 1997-11-21 | 2002-08-06 | Nps Pharmaceuticals, Inc. | Metabotropic glutamate receptor antagonists and their use for treating central nervous system diseases |
| TR200002616T2 (en) | 1997-12-22 | 2000-11-21 | Bayer Corporation | Inhibition of raf kinase using symmetric and asymmetrically substituted diphenyl ureas |
| DE19818614A1 (en) | 1998-04-20 | 1999-10-21 | Basf Ag | New benzamide derivatives useful as cysteine protease inhibitors for treating neurodegenerative diseases, neuronal damage, stroke, cranial trauma, Alzheimer's disease, etc. |
| WO2000006549A1 (en) | 1998-07-28 | 2000-02-10 | Nihon Nohyaku Co., Ltd. | Fused-heterocycle dicarboxylic diamide derivatives or salts thereof, herbicides and usage thereof |
| TR200100286T2 (en) | 1998-08-03 | 2001-07-23 | Applied Research Systems Ars Holding N.V. | Process for the synthesis of (1H) -Benzo [C] quinolizine-3-one derivatives. |
| FR2786483B1 (en) | 1998-12-01 | 2001-02-16 | Rhodia Chimie Sa | PROCESS FOR THE PREPARATION OF 4-HYDROXYQUINOLEINS AND / OR TAUTOMERIC FORMS |
| JP2000256358A (en) | 1999-03-10 | 2000-09-19 | Yamanouchi Pharmaceut Co Ltd | Pyrazole derivative |
| CA2371472A1 (en) | 1999-05-06 | 2000-11-16 | Neurogen Corporation | Substituted 4-oxo-quinoline-3-carboxamides: gaba brain receptor ligands |
| SE0002320D0 (en) | 1999-10-25 | 2000-06-21 | Active Biotech Ab | Malignant tumors |
| RS51019B (en) | 1999-10-25 | 2010-10-31 | Active Biotech Ab. | Drugs for the treatment of malignant tumours |
| WO2001030757A1 (en) | 1999-10-28 | 2001-05-03 | Microcide Pharmaceuticals, Inc. | Drug discharge pump inhibitors |
| WO2001034570A1 (en) | 1999-11-08 | 2001-05-17 | Sankyo Company, Limited | Nitrogenous heterocycle derivatives |
| UA75055C2 (en) | 1999-11-30 | 2006-03-15 | Пфайзер Продактс Інк. | Benzoimidazole derivatives being used as antiproliferative agent, pharmaceutical composition based thereon |
| GT200000203A (en) | 1999-12-01 | 2002-05-24 | COMPOUNDS, COMPOSITIONS AND METHODS TO STIMULATE THE GROWTH AND ELONGATION OF NEURONS. | |
| JP2001233859A (en) | 2000-02-23 | 2001-08-28 | Yamanouchi Pharmaceut Co Ltd | New method for producing anti-helicobacter pylori compound and its intermediate |
| GB0011409D0 (en) | 2000-05-11 | 2000-06-28 | Smithkline Beecham Plc | Novel compounds |
| AU2001268691A1 (en) | 2000-06-29 | 2002-01-21 | Clairol Incorporated | Iodo-containing organic couplers for use in oxidative hair dyeing |
| CN1441794A (en) | 2000-07-13 | 2003-09-10 | 武田药品工业株式会社 | Lipid-rich plague inhibitors |
| WO2002012189A1 (en) | 2000-08-09 | 2002-02-14 | Mitsubishi Pharma Corporation | Fused bicyclic amide compounds and medicinal use thereof |
| JP2002212179A (en) | 2001-01-15 | 2002-07-31 | Wakunaga Pharmaceut Co Ltd | New anilide derivative or salt thereof and medicine containing the same |
| GB2372986A (en) | 2001-01-17 | 2002-09-11 | Xenova Ltd | 2-oxo, 4-hydroxy pyrroles and quinolines |
| GB0102687D0 (en) | 2001-02-02 | 2001-03-21 | Pharmacia & Upjohn Spa | Oxazolyl-pyrazole derivatives active as kinase inhibitors,process for their preparation and pharmaceutical compositions comprising them |
| TWI243164B (en) | 2001-02-13 | 2005-11-11 | Aventis Pharma Gmbh | Acylated indanyl amines and their use as pharmaceuticals |
| DE10108271A1 (en) | 2001-02-21 | 2002-08-22 | Schering Ag | New oxo substituted quinoline, isoquinoline and phthalazine derivatives useful as GnRH antagonists in male fertility control, female infertility and contraception and tumor control |
| US6515001B2 (en) | 2001-03-05 | 2003-02-04 | Chemokine Therapeutic Corporation | IL-8 receptor ligands-drugs for inflammatory and autoimmune diseases |
| DE10110750A1 (en) | 2001-03-07 | 2002-09-12 | Bayer Ag | Novel aminodicarboxylic acid derivatives with pharmaceutical properties |
| DK1379239T3 (en) | 2001-03-29 | 2008-01-07 | Lilly Co Eli | N (2-aryl-ethyl) -benzylamines as antagonists of the HT6 receptor |
| US6878713B2 (en) | 2001-04-25 | 2005-04-12 | Wockhardt Limited | Generation triple-targeting, chiral, broad-spectrum antimicrobial 7-substituted piperidino-quinolone carboxylic acid derivatives, their preparation, compositions and use as medicaments |
| JP2002322054A (en) | 2001-04-26 | 2002-11-08 | Dai Ichi Seiyaku Co Ltd | Drug discharging pump inhibitor |
| JP2002322154A (en) | 2001-04-27 | 2002-11-08 | Dai Ichi Seiyaku Co Ltd | Antifungal compound |
| CA2448076A1 (en) | 2001-05-24 | 2002-11-28 | Masahiko Hayakawa | 3-quinoline-2-(1h)-ylideneindolin-2-one derivatives |
| CA2462442A1 (en) | 2001-10-12 | 2003-04-24 | Warner-Lambert Company Llc | Alkyne matrix metalloproteinase inhibitors |
| US7084156B2 (en) | 2001-11-27 | 2006-08-01 | Merck & Co., Inc. | 2-Aminoquinoline compounds |
| MXPA04005427A (en) | 2001-12-10 | 2005-04-19 | Amgen Inc | Vanilloid receptor ligands and their use in treatments. |
| JP2003238413A (en) | 2002-02-14 | 2003-08-27 | Kyowa Hakko Kogyo Co Ltd | Steroid sulfatase inhibitor |
| DE20221945U1 (en) | 2002-03-15 | 2009-10-15 | Wella Aktiengesellschaft | Quinolinium salt-containing colorants |
| US6930131B2 (en) | 2002-04-10 | 2005-08-16 | Wyeth | Aryl substituted 3-ethoxy phenyl trifluoromethane sulfonamides for the treatment of non-insulin dependent diabetes mellitus (NIDDM) |
| US7037913B2 (en) | 2002-05-01 | 2006-05-02 | Bristol-Myers Squibb Company | Bicyclo 4.4.0 antiviral derivatives |
| ES2319176T3 (en) | 2002-05-14 | 2009-05-05 | The Regents Of The University Of California | QUINOLONA-CARBOXILIC ACIDS SUBSTITUTED, ITS DERIVATIVES, SITE OF ACTION AND USE OF THE SAME. |
| WO2003095447A1 (en) | 2002-05-14 | 2003-11-20 | Xenova Limited | Process for the preparation of a hydrate of an anthranilic acid derivative |
| KR20050029209A (en) | 2002-07-15 | 2005-03-24 | 미리어드 제네틱스, 인크. | Compounds, compositions, and methods employing same |
| US20040033959A1 (en) | 2002-07-19 | 2004-02-19 | Boehringer Ingelheim Pharmaceuticals, Inc. | Pharmaceutical compositions for hepatitis C viral protease inhibitors |
| JP2006503008A (en) | 2002-08-13 | 2006-01-26 | ワーナー−ランバート カンパニー リミティド ライアビリティー カンパニー | 4-Hydroxyquinoline derivatives as matrix metalloproteinase inhibitors |
| TWI314041B (en) | 2002-10-21 | 2009-09-01 | Sankyo Agro Co Ltd | Quinolyl-3-carboxamide compound |
| WO2004105779A2 (en) | 2003-05-27 | 2004-12-09 | Cesare Montecucco | Green tea and oplyphenol inhibitors of bacterial proteases |
| CA2530352A1 (en) | 2003-07-24 | 2005-02-03 | Astellas Pharma Inc. | Quinolone derivative or salt thereof |
| ZA200603515B (en) | 2003-10-08 | 2007-11-28 | Vertex Pharma | Modulators of ATP-binding cassette transporters |
| US8354427B2 (en) * | 2004-06-24 | 2013-01-15 | Vertex Pharmaceutical Incorporated | Modulators of ATP-binding cassette transporters |
| US7804769B1 (en) * | 2005-12-01 | 2010-09-28 | Juniper Networks, Inc. | Non-stop forwarding in a multi-chassis router |
| WO2007067559A2 (en) | 2005-12-06 | 2007-06-14 | Regents Of The University Of Minnesota | Antibacterial agents |
| KR20080083680A (en) * | 2005-12-23 | 2008-09-18 | 스미스클라인 비참 코포레이션 | Azaindole inhibitors of aurora kinases |
| US20110257223A1 (en) * | 2008-10-23 | 2011-10-20 | Vertex Pharmaceuticals Incorporated | Modulators of Cystic Fibrosis Transmembrane Conductance Regulator |
-
2009
- 2009-12-11 US US12/635,927 patent/US8354427B2/en active Active
-
2010
- 2010-12-10 CA CA2783314A patent/CA2783314A1/en not_active Abandoned
- 2010-12-10 EP EP10793116A patent/EP2509954A1/en not_active Withdrawn
- 2010-12-10 RU RU2012129206/04A patent/RU2552353C2/en active
- 2010-12-10 WO PCT/US2010/059920 patent/WO2011072241A1/en active Application Filing
- 2010-12-10 MX MX2012006764A patent/MX2012006764A/en active IP Right Grant
- 2010-12-10 JP JP2012543310A patent/JP2013513617A/en active Pending
- 2010-12-10 CN CN201080056022XA patent/CN102652128A/en active Pending
- 2010-12-10 AU AU2010327993A patent/AU2010327993A1/en not_active Abandoned
- 2010-12-10 NZ NZ600707A patent/NZ600707A/en unknown
-
2012
- 2012-06-10 IL IL220283A patent/IL220283A0/en unknown
- 2012-11-30 US US13/690,924 patent/US8614327B2/en active Active
-
2015
- 2015-07-06 BE BE2015C040C patent/BE2015C040I2/nl unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5304121A (en) | 1990-12-28 | 1994-04-19 | Boston Scientific Corporation | Drug delivery system making use of a hydrogel polymer coating |
| US5886026A (en) | 1993-07-19 | 1999-03-23 | Angiotech Pharmaceuticals Inc. | Anti-angiogenic compositions and methods of use |
| US6099562A (en) | 1996-06-13 | 2000-08-08 | Schneider (Usa) Inc. | Drug coating with topcoat |
| WO2006002421A2 (en) * | 2004-06-24 | 2006-01-05 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| WO2009074575A2 (en) | 2007-12-10 | 2009-06-18 | Novartis Ag | Organic compounds |
Non-Patent Citations (27)
| Title |
|---|
| "Handbook of Chemistry and Physics" |
| "March's Advanced Organic Chemistry", 2001, JOHN WILEY & SONS |
| ARIDOR M ET AL., NATURE MED., vol. 5, no. 7, 1999, pages 745 - 751 |
| BROSS P. ET AL., HUMAN MUT., vol. 14, 1999, pages 186 - 198 |
| CUTTING, G. R. ET AL., NATURE, vol. 346, 1990, pages 366 - 369 |
| DEAN, M. ET AL., CELL, vol. 61, no. 863, 1990, pages 870 |
| DOHNANS ET AL., NATURE LOND., vol. 354, 1991, pages 526 - 528 |
| E. W. MARTIN: "Remington's Pharmaceutical Sciences", 1980, MACK PUBLISHING CO. |
| GONZALEZ, J. E.; K. OADES ET AL.: "Cell-based assays and instrumentation for screening ion-channel targets", DRUG DISCOV TODAY, vol. 4, no. 9, 1999, pages 431 - 439 |
| GONZALEZ, J. E.; R. Y. TSIEN: "Improved indicators of cell membrane potential that use fluorescence resonance energy transfer", CHEM BIOL, vol. 4, no. 4, 1997, pages 269 - 77 |
| GONZALEZ, J. E.; R. Y. TSIEN: "Voltage sensing by fluorescence resonance energy transfer in single cells", BIOPHYS J, vol. 69, no. 4, 1995, pages 1272 - 80 |
| GREGORY, R. J. ET AL., NATURE, vol. 347, 1990, pages 382 - 386 |
| HWANG, T. C. ET AL., J. GEN. PHYSIOL., vol. 111, no. 3, 1998, pages 477 - 90 |
| KEREM, B-S ET AL., PROC. NATL. ACAD. SCI. USA, vol. 87, 1990, pages 8447 - 8451 |
| KEREM, B-S. ET AL., SCIENCE, vol. 245, 1989, pages 1073 - 1080 |
| LEE R. CHOO-KANG; PAMELA L.: "Zeitlin. Type I, II, III, IV, and V cystic fibrosis Tansmembrane Conductance Regulator Defects and Opportunities of Therapy", CURRENT OPINION IN PULMONARY MEDICINE, vol. 6, 2000, pages 521 - 529 |
| MORELLO, JP ET AL., TIPS, vol. 21, 2000, pages 466 - 469 |
| PASYK; FOSKETT, J. CELL. BIOCHEM., vol. 270, 1995, pages 12347 - 50 |
| PAUL M. QUINTON: "Cystic fibrosis: impaired bicarbonate secretion and mucoviscidosis", LANCET, vol. 372, 2008, pages 415 - 417 |
| PEADER G. NOONE; MICHAEL R. KNOWLES: "CFTR- opathies: disease phenotypes associated with cystic fibrosis transmembrane regulator gene mutations", RESPIR. RES., vol. 2, 2001, pages 328 - 332 |
| QUINTON, P. M., FASEB J., vol. 4, 1990, pages 2709 - 2727 |
| RICH, D. P. ET AL., NATURE, vol. 347, 1990, pages 358 - 362 |
| RIORDAN, J. R. ET AL., SCIENCE, vol. 245, 1989, pages 1066 - 1073 |
| RUTISHAUSER, J. ET AL., SWISS MED WKLY, vol. 132, 2002, pages 211 - 222 |
| S. M. BERGE ET AL., J. PHARMACEUTICAL SCIENCES, vol. 66, 1977, pages 1 - 19 |
| SHASTRY, B.S. ET AL., NEUROCHEM. INTERNATIONAL, vol. 43, 2003, pages 1 - 7 |
| THOMAS SORRELL: "Organic Chemistry", 1999, UNIVERSITY SCIENCE BOOKS |
Cited By (83)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9150855B2 (en) | 2010-05-21 | 2015-10-06 | Universität Für Bodenkultur Wien | Methods for diagnosing bone or cardiovascular disorders |
| US9206115B2 (en) | 2010-05-21 | 2015-12-08 | Technische Universität Graz | ATGListatin and pharmaceutical composition comprising the same |
| US10894773B2 (en) | 2011-05-18 | 2021-01-19 | Vertex Pharmaceuticals (Europe) Limited | Deuterated CFTR potentiators |
| US10479766B2 (en) | 2011-05-18 | 2019-11-19 | Verex Pharmaceuticals (Europe) Limited | Deuterated CFTR potentiators |
| US9254291B2 (en) | 2011-11-08 | 2016-02-09 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| WO2014114649A1 (en) * | 2013-01-22 | 2014-07-31 | Technische Universität Graz | Lipase inhibitors |
| US10138232B2 (en) | 2014-06-03 | 2018-11-27 | Novartis Ag | Naphthyridinedione derivatives |
| US10189854B2 (en) | 2014-07-16 | 2019-01-29 | Gruenenthal Gmbh | Substituted pyrimidine compounds |
| US9540349B2 (en) | 2014-07-16 | 2017-01-10 | Gruenenthal Gmbh | Substituted pyrimidine compounds |
| EP3527561A1 (en) | 2014-07-16 | 2019-08-21 | Grünenthal GmbH | Novel substituted pyrimidine compounds |
| US10391099B2 (en) | 2014-07-16 | 2019-08-27 | Gruenenthal Gmbh | Method of treating psoriatic arthritis, psoriasis, and chronic obstructive pulmonary disease with a novel 2,5-substituted pyrimidines |
| US11992553B2 (en) | 2014-08-29 | 2024-05-28 | Algipharma As | Inhalable powder formulations of alginate oligomers |
| US10758534B2 (en) | 2014-10-06 | 2020-09-01 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| US11426407B2 (en) | 2014-10-06 | 2022-08-30 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2016069891A1 (en) | 2014-10-31 | 2016-05-06 | Abbvie Inc. | Substituted tetrahydropyrans and method of use |
| US9382208B1 (en) | 2015-01-26 | 2016-07-05 | Vanderbilt University | Negative allosteric modulators of metabotropic glutamate receptor 2 |
| US10751363B2 (en) | 2015-03-23 | 2020-08-25 | Algipharma As | Use of aliginate oligomers and CFTR modulators in treatment of conditions associated with CFTR dysfunction |
| WO2016193812A1 (en) | 2015-06-02 | 2016-12-08 | Abbvie S.A.R.L. | Substituted pyridines and method of use |
| WO2017009804A1 (en) | 2015-07-16 | 2017-01-19 | Abbvie S.Á.R.L. | Substituted tricyclics and method of use |
| US10759721B2 (en) | 2015-09-25 | 2020-09-01 | Vertex Pharmaceuticals (Europe) Limited | Deuterated CFTR potentiators |
| EP3352758A4 (en) * | 2015-09-25 | 2018-08-01 | Vertex Pharmaceuticals (Europe) Limited | Deuterated cftr potentiators |
| WO2017053711A2 (en) | 2015-09-25 | 2017-03-30 | Concert Pharmaceuticals, Inc. | Deuterated cftr potentiators |
| US11413306B2 (en) | 2015-10-06 | 2022-08-16 | Algipharma As | Alginate oligomers for the treatment or prevention of microbial overgrowth in the intestinal tract |
| WO2017060873A1 (en) | 2015-10-09 | 2017-04-13 | AbbVie S.à.r.l. | Substituted pyrazolo[3,4-b]pyridin-6-carboxylic acids and their use |
| WO2017060874A1 (en) | 2015-10-09 | 2017-04-13 | Abbvie S.Á.R.L | N-sulfonylated pyrazolo[3,4-b]pyridin-6-carboxamides and method of use |
| WO2017060879A1 (en) | 2015-10-09 | 2017-04-13 | AbbVie S.à.r.l. | Novel compounds for treatment of cystic fibrosis |
| US10118916B2 (en) | 2016-04-26 | 2018-11-06 | Abbvie S.Á.R.L. | Modulators of cystic fibrosis transmembrane conductance regulator protein |
| WO2017187321A1 (en) | 2016-04-26 | 2017-11-02 | AbbVie S.à.r.l. | Modulators of cystic fibrosis transmembrane conductance regulator protein |
| WO2017208115A1 (en) | 2016-06-03 | 2017-12-07 | AbbVie S.à.r.l. | Heteroaryl substituted pyridines and methods of use |
| US10604515B2 (en) | 2016-06-03 | 2020-03-31 | Abbvie S.Á.R.L. | Heteroaryl substituted pyridines and methods of use |
| US10138227B2 (en) | 2016-06-03 | 2018-11-27 | Abbvie S.Á.R.L. | Heteroaryl substituted pyridines and methods of use |
| US11186566B2 (en) | 2016-09-30 | 2021-11-30 | Vertex Pharmaceuticals Incorporated | Modulator of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |
| US9981910B2 (en) | 2016-10-07 | 2018-05-29 | Abbvie S.Á.R.L. | Substituted pyrrolidines and methods of use |
| US10399940B2 (en) | 2016-10-07 | 2019-09-03 | Abbvie S.Á.R.L. | Substituted pyrrolidines and methods of use |
| WO2018065962A1 (en) | 2016-10-07 | 2018-04-12 | AbbVie S.à.r.l. | Substituted pyrrolidines and their use in the treatment of cystic fiibrosis |
| WO2018065921A1 (en) | 2016-10-07 | 2018-04-12 | Abbvie S.Á.R.L. | Substituted pyrrolidines as cftr modulators |
| US10793547B2 (en) | 2016-12-09 | 2020-10-06 | Vertex Pharmaceuticals Incorporated | Modulator of the cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |
| US11453655B2 (en) | 2016-12-09 | 2022-09-27 | Vertex Pharmaceuticals Incorporated | Modulator of the cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |
| WO2018116185A1 (en) | 2016-12-20 | 2018-06-28 | AbbVie S.à.r.l. | Deuterated cftr modulators and methods of use |
| WO2018154493A1 (en) | 2017-02-24 | 2018-08-30 | AbbVie S.à.r.l. | Modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| WO2018154519A1 (en) | 2017-02-24 | 2018-08-30 | AbbVie S.à.r.l. | Modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| US10428017B2 (en) | 2017-02-24 | 2019-10-01 | Abbvie S.Á.R.L. | Modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| US11253509B2 (en) | 2017-06-08 | 2022-02-22 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| US11517564B2 (en) | 2017-07-17 | 2022-12-06 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| US11434201B2 (en) | 2017-08-02 | 2022-09-06 | Vertex Pharmaceuticals Incorporated | Processes for preparing pyrrolidine compounds |
| US11505528B2 (en) | 2017-08-04 | 2022-11-22 | Axial Therapeutics, Inc. | Inhibitors of microbially induced amyloid |
| EP3664822A4 (en) * | 2017-08-04 | 2021-07-07 | Axial Therapeutics, Inc. | Inhibitors of microbially induced amyloid |
| EP3736267A1 (en) | 2017-09-14 | 2020-11-11 | AbbVie Overseas S.à r.l. | Modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| EP3736270A1 (en) | 2017-09-14 | 2020-11-11 | AbbVie Overseas S.à r.l. | Modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| WO2019053634A1 (en) | 2017-09-14 | 2019-03-21 | AbbVie S.à.r.l. | Modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| US10829473B2 (en) | 2017-09-14 | 2020-11-10 | Abbvie Overseas S.À.R.L. | Modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| US10844042B2 (en) | 2017-09-14 | 2020-11-24 | Abbvie Overseas S.À.R.L. | Modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| EP3865474A1 (en) | 2017-09-14 | 2021-08-18 | AbbVie Overseas S.à r.l. | Modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| US10844041B2 (en) | 2017-09-14 | 2020-11-24 | Abbvie Overseas S.À.R.L. | Modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| US10981890B2 (en) | 2017-09-14 | 2021-04-20 | Abbvie Overseas S.À.R.L. | Modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| US10988454B2 (en) | 2017-09-14 | 2021-04-27 | Abbvie Overseas S.À.R.L. | Modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| US11155533B2 (en) | 2017-10-19 | 2021-10-26 | Vertex Pharmaceuticals Incorporated | Crystalline forms and compositions of CFTR modulators |
| US12024491B2 (en) | 2017-12-01 | 2024-07-02 | Vertex Pharmaceuticals Incorporated | Processes for making modulators of cystic fibrosis transmembrane conductance regulator |
| US11708331B2 (en) | 2017-12-01 | 2023-07-25 | Vertex Pharmaceuticals Incorporated | Processes for making modulators of cystic fibrosis transmembrane conductance regulator |
| US11465985B2 (en) | 2017-12-08 | 2022-10-11 | Vertex Pharmaceuticals Incorporated | Processes for making modulators of cystic fibrosis transmembrane conductance regulator |
| US11179367B2 (en) | 2018-02-05 | 2021-11-23 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for treating cystic fibrosis |
| US11866450B2 (en) | 2018-02-15 | 2024-01-09 | Vertex Pharmaceuticals Incorporated | Modulators of Cystic Fibrosis Transmembrane Conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulators |
| US11066417B2 (en) | 2018-02-15 | 2021-07-20 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulators |
| WO2019193062A1 (en) | 2018-04-03 | 2019-10-10 | Abbvie S.Á.R.L | Substituted pyrrolidines and their use |
| US11414439B2 (en) | 2018-04-13 | 2022-08-16 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |
| US11345691B2 (en) | 2019-06-03 | 2022-05-31 | AbbVie Global Enterprises Ltd. | Prodrug modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| EP3747882A1 (en) | 2019-06-03 | 2020-12-09 | AbbVie Overseas S.à r.l. | Prodrug modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| US11584761B2 (en) | 2019-08-14 | 2023-02-21 | Vertex Pharmaceuticals Incorporated | Process of making CFTR modulators |
| US11591350B2 (en) | 2019-08-14 | 2023-02-28 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| US11873300B2 (en) | 2019-08-14 | 2024-01-16 | Vertex Pharmaceuticals Incorporated | Crystalline forms of CFTR modulators |
| WO2021097054A1 (en) | 2019-11-12 | 2021-05-20 | Genzyme Corporation | 6-membered heteroarylaminosulfonamides for treating diseases and conditions mediated by deficient cftr activity |
| WO2021113806A1 (en) | 2019-12-05 | 2021-06-10 | Genzyme Corporation | Arylamides and methods of use thereof |
| WO2021113809A1 (en) | 2019-12-05 | 2021-06-10 | Genzyme Corporation | Arylamides and methods of use thereof |
| WO2022150173A1 (en) | 2021-01-06 | 2022-07-14 | AbbVie Global Enterprises Ltd. | Modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| WO2022150174A1 (en) | 2021-01-06 | 2022-07-14 | AbbVie Global Enterprises Ltd. | Modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| WO2023034946A1 (en) | 2021-09-03 | 2023-03-09 | Genzyme Corporation | Indole compounds and uses thereof in the treatement of cystic fibrosis |
| WO2023034992A1 (en) | 2021-09-03 | 2023-03-09 | Genzyme Corporation | Indole compounds and methods of use |
| WO2024054840A1 (en) | 2022-09-07 | 2024-03-14 | Sionna Therapeutics | Macrocyclic compounds, compositions, and methods of using thereof |
| WO2024054845A1 (en) | 2022-09-07 | 2024-03-14 | Sionna Therapeutics | Macrocycic compounds, compositions, and methods of using thereof |
| WO2024054851A1 (en) | 2022-09-07 | 2024-03-14 | Sionna Therapeutics | Macrocyclic compounds, compositions and methods of using thereof |
| WO2024056791A1 (en) | 2022-09-15 | 2024-03-21 | Idorsia Pharmaceuticals Ltd | Combination of macrocyclic cftr modulators with cftr correctors and / or cftr potentiators |
| WO2024056798A1 (en) | 2022-09-15 | 2024-03-21 | Idorsia Pharmaceuticals Ltd | Macrocyclic cftr modulators |
| US12122788B2 (en) | 2023-01-04 | 2024-10-22 | Vertex Pharmaceuticals Incorporated | Process of making CFTR modulators |
Also Published As
| Publication number | Publication date |
|---|---|
| BE2015C040I2 (en) | 2024-10-08 |
| AU2010327993A8 (en) | 2012-07-19 |
| WO2011072241A9 (en) | 2012-06-14 |
| US8354427B2 (en) | 2013-01-15 |
| MX2012006764A (en) | 2012-11-23 |
| NZ600707A (en) | 2014-09-26 |
| RU2012129206A (en) | 2014-01-20 |
| US20100184739A1 (en) | 2010-07-22 |
| US8614327B2 (en) | 2013-12-24 |
| EP2509954A1 (en) | 2012-10-17 |
| CA2783314A1 (en) | 2011-06-16 |
| AU2010327993A1 (en) | 2012-07-12 |
| RU2552353C2 (en) | 2015-06-10 |
| US20130165442A1 (en) | 2013-06-27 |
| CN102652128A (en) | 2012-08-29 |
| JP2013513617A (en) | 2013-04-22 |
| IL220283A0 (en) | 2012-07-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2509954A1 (en) | 4 -oxo- 1h -quinoline- 3 - carboxamides as modulators of atp -binding cassette transporters | |
| US8802700B2 (en) | Modulators of ATP-Binding Cassette transporters | |
| AU2010251787B2 (en) | Modulators of ATP-Binding Cassette Transporters | |
| US20140343098A1 (en) | Modulators of atp-binding cassette transporters | |
| AU2007326449A1 (en) | Carboxylic acid derivative | |
| WO2010078103A1 (en) | Modulators of cystic fibrosis transmembrane conductance regulator | |
| EP2502911B1 (en) | Modulators of ATP-binding cassette transporters | |
| AU2013204751A1 (en) | Modulators of ATP-Binding Cassette Transporters |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080056022.X Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10793116 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1294/KOLNP/2012 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 2783314 Country of ref document: CA |
|
| REEP | Request for entry into the european phase |
Ref document number: 2010793116 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010793116 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012543310 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 220283 Country of ref document: IL |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2012/006764 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010327993 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012129206 Country of ref document: RU |
|
| ENP | Entry into the national phase |
Ref document number: 2010327993 Country of ref document: AU Date of ref document: 20101210 Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112012015851 Country of ref document: BR |
|
| ENPW | Started to enter national phase and was withdrawn or failed for other reasons |
Ref document number: 112012015851 Country of ref document: BR |