WO2010125985A1 - Fused heterocyclic compound and use thereof - Google Patents
Fused heterocyclic compound and use thereof Download PDFInfo
- Publication number
- WO2010125985A1 WO2010125985A1 PCT/JP2010/057315 JP2010057315W WO2010125985A1 WO 2010125985 A1 WO2010125985 A1 WO 2010125985A1 JP 2010057315 W JP2010057315 W JP 2010057315W WO 2010125985 A1 WO2010125985 A1 WO 2010125985A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- active compound
- present active
- optionally substituted
- Prior art date
Links
- 0 C*CCC=*=CC(*=*)=*[C@](C*1)*C1=C1C(N)=C(C)*C(*)=C1* Chemical compound C*CCC=*=CC(*=*)=*[C@](C*1)*C1=C1C(N)=C(C)*C(*)=C1* 0.000 description 2
- CUHQLPHUMBQYKD-UHFFFAOYSA-N CC(C)(C)Oc(cncc1)c1-c([n]1C)nc2c1ncc(C(F)(F)F)c2 Chemical compound CC(C)(C)Oc(cncc1)c1-c([n]1C)nc2c1ncc(C(F)(F)F)c2 CUHQLPHUMBQYKD-UHFFFAOYSA-N 0.000 description 1
- PTJSTYOBOBYDBU-UHFFFAOYSA-N CC(C)Sc1cnccc1-c1nc(cc(C(F)(F)F)cc2)c2[n]1C Chemical compound CC(C)Sc1cnccc1-c1nc(cc(C(F)(F)F)cc2)c2[n]1C PTJSTYOBOBYDBU-UHFFFAOYSA-N 0.000 description 1
- JINSFRWPSHPFCJ-UHFFFAOYSA-N CCC(C)Nc(cc(C(F)(F)F)cc1)c1[N+]([O-])=O Chemical compound CCC(C)Nc(cc(C(F)(F)F)cc1)c1[N+]([O-])=O JINSFRWPSHPFCJ-UHFFFAOYSA-N 0.000 description 1
- NTMKEWGYBJTROE-UHFFFAOYSA-N CCS(c1c(C2(C)N(C)c(ncc(C(F)(F)F)c3)c3N2)ccnc1)=O Chemical compound CCS(c1c(C2(C)N(C)c(ncc(C(F)(F)F)c3)c3N2)ccnc1)=O NTMKEWGYBJTROE-UHFFFAOYSA-N 0.000 description 1
- SSLCVGXWZASELR-UHFFFAOYSA-N CCSC1=CNCC=C1c1nc(cc(C(F)(F)F)cc2)c2[n]1C1CC1 Chemical compound CCSC1=CNCC=C1c1nc(cc(C(F)(F)F)cc2)c2[n]1C1CC1 SSLCVGXWZASELR-UHFFFAOYSA-N 0.000 description 1
- JIQKVJVNTXWZSM-UHFFFAOYSA-N CCc(cncc1)c1-c1nc(cc(C(F)(F)F)cc2)c2[n]1C Chemical compound CCc(cncc1)c1-c1nc(cc(C(F)(F)F)cc2)c2[n]1C JIQKVJVNTXWZSM-UHFFFAOYSA-N 0.000 description 1
- ZQRLTRNZBVMDHJ-UHFFFAOYSA-N C[n]1c(-c(ccnc2)c2F)nc(C2)c1NCC2C(F)(F)F Chemical compound C[n]1c(-c(ccnc2)c2F)nc(C2)c1NCC2C(F)(F)F ZQRLTRNZBVMDHJ-UHFFFAOYSA-N 0.000 description 1
- ZMQDOGMUIVOAGG-UHFFFAOYSA-N C[n]1c(-c(ccnc2)c2S)nc2c1ccc(C(F)(F)F)c2 Chemical compound C[n]1c(-c(ccnc2)c2S)nc2c1ccc(C(F)(F)F)c2 ZMQDOGMUIVOAGG-UHFFFAOYSA-N 0.000 description 1
- HQQIRCJNPZJHCL-UHFFFAOYSA-N C[n]1c(-c(ccnc2)c2SC(C(F)F)(F)F)nc2c1ncc(C(F)(F)F)c2 Chemical compound C[n]1c(-c(ccnc2)c2SC(C(F)F)(F)F)nc2c1ncc(C(F)(F)F)c2 HQQIRCJNPZJHCL-UHFFFAOYSA-N 0.000 description 1
- YVINVSSITICRKK-UHFFFAOYSA-N C[n]1c(-c(ccnc2)c2SCC#C)nc2c1CCC(C(F)(F)F)=C2 Chemical compound C[n]1c(-c(ccnc2)c2SCC#C)nc2c1CCC(C(F)(F)F)=C2 YVINVSSITICRKK-UHFFFAOYSA-N 0.000 description 1
- OJKREQWXXWSZRS-UHFFFAOYSA-N C[n]1c(-c(ccnc2)c2SCC(OC)=O)nc2c1ccc(C(F)(F)F)c2 Chemical compound C[n]1c(-c(ccnc2)c2SCC(OC)=O)nc2c1ccc(C(F)(F)F)c2 OJKREQWXXWSZRS-UHFFFAOYSA-N 0.000 description 1
- NLJPDYHXPXYJBI-UHFFFAOYSA-N C[n]1c(-c2ccncc2)nc2c1ncc(C(F)(F)F)c2 Chemical compound C[n]1c(-c2ccncc2)nc2c1ncc(C(F)(F)F)c2 NLJPDYHXPXYJBI-UHFFFAOYSA-N 0.000 description 1
- PAFXUTUHKWRTGF-UHFFFAOYSA-N C[n]1c(-c2ccncc2C#C)nc2cc(C(F)(F)F)cnc12 Chemical compound C[n]1c(-c2ccncc2C#C)nc2cc(C(F)(F)F)cnc12 PAFXUTUHKWRTGF-UHFFFAOYSA-N 0.000 description 1
- FETIXELIHFNUFY-UHFFFAOYSA-N C[n]1c(-c2ccncc2SC(C(F)F)(F)F)nc2c1ccc(C(F)(F)F)c2 Chemical compound C[n]1c(-c2ccncc2SC(C(F)F)(F)F)nc2c1ccc(C(F)(F)F)c2 FETIXELIHFNUFY-UHFFFAOYSA-N 0.000 description 1
- HERXWVZDPRGMGG-UHFFFAOYSA-N C[n]1c(-c2ccncc2SC)nc2cc(C(F)(F)F)cnc12 Chemical compound C[n]1c(-c2ccncc2SC)nc2cc(C(F)(F)F)cnc12 HERXWVZDPRGMGG-UHFFFAOYSA-N 0.000 description 1
- AYIPDGKEGWDLRR-UHFFFAOYSA-N C[n]1c(-c2ccncc2SC2CCCC2)nc2c1NCC(C(F)(F)F)=C2 Chemical compound C[n]1c(-c2ccncc2SC2CCCC2)nc2c1NCC(C(F)(F)F)=C2 AYIPDGKEGWDLRR-UHFFFAOYSA-N 0.000 description 1
- PDDFTWJRVMJHFA-UHFFFAOYSA-N Cc(cncc1)c1-c([n]1C)nc2c1ncc(C(F)(F)F)c2 Chemical compound Cc(cncc1)c1-c([n]1C)nc2c1ncc(C(F)(F)F)c2 PDDFTWJRVMJHFA-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01N—PRESERVATION OF BODIES OF HUMANS OR ANIMALS OR PLANTS OR PARTS THEREOF; BIOCIDES, e.g. AS DISINFECTANTS, AS PESTICIDES OR AS HERBICIDES; PEST REPELLANTS OR ATTRACTANTS; PLANT GROWTH REGULATORS
- A01N43/00—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds
- A01N43/48—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds having rings with two nitrogen atoms as the only ring hetero atoms
- A01N43/50—1,3-Diazoles; Hydrogenated 1,3-diazoles
- A01N43/52—1,3-Diazoles; Hydrogenated 1,3-diazoles condensed with carbocyclic rings, e.g. benzimidazoles
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01N—PRESERVATION OF BODIES OF HUMANS OR ANIMALS OR PLANTS OR PARTS THEREOF; BIOCIDES, e.g. AS DISINFECTANTS, AS PESTICIDES OR AS HERBICIDES; PEST REPELLANTS OR ATTRACTANTS; PLANT GROWTH REGULATORS
- A01N43/00—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds
- A01N43/90—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds having two or more relevant hetero rings, condensed among themselves or with a common carbocyclic ring system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/14—Ectoparasiticides, e.g. scabicides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Definitions
- the present invention relates to a fused heterocyclic compound and its use for control of noxious arthropods
- the present invention is to provide a compound having an excellent controlling effect on noxious arthropods .
- the present inventors have intensively studied to solve the above-described problem and resultantly found that a fused heterocyclic compound of the following formula (1) has an excellent controlling effect on noxious arthropods, leading to completion of the present invention .
- the present invention provides the following .
- R 1 , R 2 , R 3 and R 4 are the same or different and represent a Cl-C ⁇ chain hydrocarbon group optionally substituted with one or more members selected from the group X, a C3-C6 alicyclic hydrocarbon group optionally substituted with one or more members selected from the group X, a phenyl group optionally substituted with one or more members selected from the group Y, a benzyl group optionally substituted with one or more members selected from the group Y, a 5-membered heterocyclic group or ⁇ -membered heterocyclic group optionally substituted with one or more members selected from the group Y, -OR 8 , -NR 8 R 9 , -NR 8 C(O)R 9 , -S(O) m R 8 , -SO 2 Cl, - SO 2 NR 8 R 9 , -CO 2 R 10 , -CONR 8 R 9 , -CONR 10 NR 11 R 12 , a cyano group, a nitro
- R 5 represents a C1-C4 chain hydrocarbon group optionally substituted with one or more halogen atoms, a cyclopropyl group optionally substituted with one or more halogen atoms, a cyclopropylmethyl group optionally substituted with one or more halogen atoms, -OR 13 , - NR 13 R 14 or a cyano group,
- R 6 and R 7 are the same or different and represent a C1-C4 chain hydrocarbon group substituted with one or more halogen atoms, -OR 15 , -S(O) 1n R 15 , a halogen atom or a hydrogen atom (here, either R 6 or R 7 represents a Cl- C4 chain hydrocarbon group substituted with one or more halogen atoms, -OR 15 , -S(O) m R 15 .), alternatively R 6 and R 7 may be bonded to form a 5-membered ring or 6-membered ring substituted with one or more halogen atoms together with carbon atoms to which R 6 and R 7 are connected,
- R 8 and R 9 are the same or different and represent a Cl-C ⁇ chain hydrocarbon group optionally substituted with one or more members selected from the group X, a C3-C6 alicyclic hydrocarbon group optionally substituted with one or more members selected from the group X, a phenyl group optionally substituted with one or more members selected from the group Y, a benzyl group optionally substituted with one or more members selected from the group Y, a 5-membered heterocyclic group or 6- membered heterocyclic group optionally substituted with one or more members selected from the group Y, or a hydrogen atom (concerning -S(O) m R 8 , m represents the same meaning as described above, but when m represents 1 or 2, R 8 does not represent a hydrogen atom.), R 10 represents a C1-C4 alkyl group optionally substituted with one or more halogen atoms, or a hydrogen atom,
- R 11 and R 12 are the same or different and represents a C1-C4 alkyl group optionally substituted with one or more halogen atoms, a C2-C5 alkoxycarbonyl group or a hydrogen atom,
- R 13 and R 14 are the same or different and represents a C1-C3 chain hydrocarbon group optionally substituted with one or more halogen atoms, a cyclopropyl group optionally substituted with one or more halogen atoms, or a hydrogen atom,
- R 15 represents a C1-C4 chain hydrocarbon group substituted with one or more halogen atoms, and n represents 0 or 1.
- Group X a group consisting of a C1-C4 alkoxy group optionally substituted with one or more halogen atoms, - CO 2 R 16 (here, R 16 represents a C1-C4 alkyl group optionally substituted by a halogen. ) , a cyano group and a halogen atom.
- Group Y a group consisting of a C1-C4 alkyl group optionally substituted with one or more halogen atoms, a C1-C4 alkoxy group optionally substituted with one or more halogen atoms, a cyano group, a nitro group and a halogen atom (hereinafter, referred to as present active compound) .
- R 1 , R 2 , R 3 and R 4 are the same or different and represent a Cl-C ⁇ chain hydrocarbon group optionally substituted with one or more members selected from the group X, a C3-C6 alicyclic hydrocarbon group optionally substituted with one or more members selected from the group X, a phenyl group optionally substituted with one or more members selected from the group Y, a benzyl group optionally substituted with one or more members selected from the group Y, a 5-membered heterocyclic group or ⁇ -membered heterocyclic group optionally substituted with one or more members selected from the group Y, -OR 8 , -NR 8 R 9 , -NR 8 C(O)R 9 , -S(O) m R 8 , -CO 2 R 10 , - CONR 8 R 9 , -CONR 10 NR 11 R 12 , a cyano group, a nitro group, a halogen atom or a hydrogen atom,
- R 5 represents a C1-C4 chain hydrocarbon group optionally substituted with one or more halogen atoms, a cyclopropyl group optionally substituted with one or more halogen atoms, a cycl ⁇ propylmethyl group optionally substituted with one or more halogen atoms, -OR 13 , - NR 13 R 14 or a cyano group,
- R 6 and R 7 are the same or different and represent a C1-C4 chain hydrocarbon group substituted with one or more halogen atoms, -OR 15 , -S(O) m R 15 , a halogen atom or a hydrogen atom (here, either R 6 or R 7 represents a Cl- C4 chain hydrocarbon group substituted with one or more halogen atoms, -OR 15 , -S(O) m R 15 .), alternatively R 6 and R 7 may be bonded to form a 5-membered ring or ⁇ -membered ring substituted with one or more halogen atoms together with carbon atoms to which R 6 and R 7 are connected,
- R 8 and R 9 are the same or different and represent a C1-C6 chain hydrocarbon group optionally substituted with one or more members selected from the group X, a
- C3-C6 alicyclic hydrocarbon group optionally substituted with one or more members selected from the group X, a phenyl group optionally substituted with one or more members selected from the group Y, a benzyl group optionally substituted with one or more members selected from the group Y, a 5-membered heterocyclic group or 6- membered heterocyclic group optionally substituted with one or more members selected from the group Y, or a hydrogen atom (concerning -S(O) m R 8 , m represents the same meaning as described above, but when m represents 1 or 2, R 8 does not represent a hydrogen atom.) ,
- R 10 represents a C1-C4 alkyl group optionally substituted with one or more halogen atoms, or a hydrogen atom
- R 11 and R 12 are the same or different and represents a C1-C4 alkyl group optionally substituted with one or more halogen atoms, a C2-C5 alkoxycarbonyl group or a hydrogen atom
- R 13 and R 14 are the same or different and represents a C1-C3 chain hydrocarbon group optionally substituted with one or more halogen atoms, a cyclopropyl group optionally substituted with one or more halogen atoms, or a hydrogen atom,
- R 15 represents a C1-C4 chain hydrocarbon group substituted with one or more halogen atoms, and n represents 0 or 1.
- Group X a group consisting of a C1-C4 alkoxy group optionally substituted with one or more halogen atoms, - CO 2 R 16 (here, R 16 represents a C1-C4 alkyl group optionally substituted by a halogen.) / a cyano group and a halogen atom.
- Group Y a group consisting of a C1-C4 alkyl group optionally substituted with one or more halogen atoms, a C1-C4 alkoxy group optionally substituted with one or more halogen atoms, a cyano group, a nitro group and a halogen atom
- R 3 is a C3-C6 alicyclic hydrocarbon group optionally substituted with one or more members selected from the group X, a phenyl group optionally substituted with one or more members selected from the group Y, a benzyl group optionally substituted with one or more members selected from the group Y, or a 5-membered heterocyclic group or 6-membered heterocyclic group optionally substituted with one or more members selected from the group Y.
- R 3 is a C1-C6 chain hydrocarbon group optionally substituted with one or more members selected from the group X, - OR 8 , -NR 8 R 9 , -NR 8 C(O)R 9 , -S(O) m R 8 , -CO 2 R 10 , -CONR 8 R 9 , - CONR 10 NR 11 R 12 , a cyano group, a nitro group, a halogen atom or a hydrogen atom, and R 8 and R 9 are the same or different and represent a C1-C6 chain hydrocarbon group optionally substituted with one or more members selected from the group X, or a hydrogen atom (here, R 8 is not a hydrogen atom when m in -S(O) m R 8 represents 1 or 2.) .
- R 3 is a Cl-C ⁇ chain hydrocarbon group optionally substituted with one or more members selected from the group X, - OR 8 , -NR 8 R 9 , -S(O) m R 8 , a halogen atom or a hydrogen atom
- R 8 and R 9 are the same or different and represent a Cl-C ⁇ chain hydrocarbon group optionally substituted with one or more members selected from the group X, or a hydrogen atom (here, R 8 is not a hydrogen atom when m in -S(O) m R 8 represents 1 or 2.)
- a noxious arthropod controlling composition which comprises the fused heterocyclic compound as described in any one of [1] to [9], [1-2], [6-2], [6-3], [9-2] or [9-3], and an inert carrier.
- a method of controlling a noxious arthropod which comprises applying an effective amount of the fused heterocyclic compound as described in any one of [1] to [9], [1-2], [6-2], [6-3], [9-2] or [9-3] to a noxious arthropod or to a habitat of a noxious arthropod (hereinafter, referred to as composition of the present invention) .
- halogen atom means a fluorine atom, chlorine atom, bromine atom and iodine atom.
- C1-C6 chain hydrocarbon group optionally substituted with one or more members selected from the group X represented by R 1 , R 2 , R 3 or R 4 include a Cl-C ⁇ alkyl group optionally substituted with one or more members selected from the group X such as a methyl group, ethyl group, propyl group, isopropyl group, butyl group, isobutyl group, sec-butyl group, tert-butyl group, pentyl group, hexyl group, methoxymethyl group, ethoxymethyl group, propoxymethyl group, isopropoxymethyl group, 2-methoxyethyl group, 2- ethoxyethyl group, 2-propoxyethyl group, 2- isopropoxyefhyl group, methoxycarbonylmethyl group, ethoxycarbonylmethyl group, 2-methoxycarbonylethyl group, 2-ethoxycarbonylethyl group, cyanomethyl group, dicyan
- C3-C6 alicyclic hydrocarbon group optionally substituted with one or more members selected from the group X" represented by R 1 , R 2 , R 3 or R 4 include a cyclopropyl group, cyclobutyl group, cyclopentyl group and cyclohexyl group.
- phenyl group optionally substituted with one or more members selected from the group Y" represented by R 1 , R 2 , R 3 or R 4 include a phenyl group, 2-chlorophenyl group, 3-chlorophenyl group, 4- chlorophenyl group, 2-methylphenyl group, 3-methylphenyl group, 4-methylphenyl group, 2-methoxyphenyl group, 3- methoxyphenyl group, 4-methoxyphenyl group, 2- (trifluoromethyl) phenyl group, 3- (trifluoromethyl) phenyl group, 4- (trifluoromethyl) phenyl group, 2-nitrophenyl group, 3-nitrophenyl group, 4-nitrophenyl group, 2- cyanophenyl group, 3-cyanophenyl group and 4-cyanophenyl group.
- Examples of "benzyl group optionally substituted with one or more members selected from the group Y" represented by R 1 , R 2 , R 3 , R 4 , R 8 or R 9 include a benzyl group, 2-chlorobenzyl group, 3-chlorobenzyl group, A- chlorobenzyl group, 2-methylbenzyl group, 3-methylbenzyl group, 4-methylbenzyl group, 2-methoxybenzyl group, 3- methoxybenzyl group and 4-methoxybenzyl group.
- Heterocyclic group in "5-membered heterocyclic group or 6-membered heterocyclic group optionally substituted with one or more members selected from the group Y" represented by R 1 , R 2 , R 3 or R 4 means a residual group of a heterocyclic compound, and examples of "5-membered heterocyclic group or 6-membered heterocyclic group optionally substituted with one or more members selected from the group Y" include a 5- membered saturated heterocyclic group such as a pyrrolidin-1-yl group and tetrahydrofuran-2-yl group; a 6-membered saturated heterocyclic group such as a piperidyl group, morpholyl group, thiomorpholyl group and 4-methylpiperazin-l-yl group; a 5-membered aromatic heterocyclic group such as a pyrazol-1-yl group, 3- chloropyrazol-1-yl group, 3-bromopyrazol-l-yl group, 3- nitropyr
- C1-C4 chain hydrocarbon group optionally substituted with one or more halogen atoms represented by R 5 include a C1-C4 alkyl group optionally substituted with one or more halogen atoms such as a methyl group, ethyl group, propyl group, isopropyl group, butyl group, isobutyl group, sec-butyl group, tert-butyl group, trifluoromethyl group, 2,2,2- trifluoroethyl group and pentafluoroethyl group; a C2-C4 alkenyl group optionally substituted with one or more halogen atoms such as a vinyl group, allyl group, and 3 , 3-difluoroallyl group; and a C2-C4 alkynyl group optionally substituted with one or more halogen atoms such as a propargyl group, and 4 , 4 , 4-trifluoro-2-butynyl group.
- C1-C4 chain hydrocarbon group substituted with one or more halogen atoms represented by R 6 or R 7 include a fluoromethyl group, difluoromethyl group, trifluoromethyl group, 2 , 2 , 2-trifluoroethyl group, pentafluoroethyl group, heptafluoropropyl group, 1,1,1,3,3, 3-hexafluoroisopropyl group, heptafluoroisopropyl group, nonafluorobutyl group, nonafluoroisobutyl group, nonafluoro-sec-butyl group and nonafluoro-tert-butyl group.
- Examples of ⁇ 5-membered ring or ⁇ -membered ring substituted with one or more halogen atoms" obtained by bonding of R 6 and R 7 , together with carbon atoms to which R 6 and R 7 are connected, include rings of the following formulae (a) to (j) (here, A 6 represents a carbon atom to which R 6 is connected, and A 7 represents a carbon atom to which R 7 is connected) .
- C1-C6 chain hydrocarbon group optionally substituted with one or more members selected from the group X include a Cl- C ⁇ alkyl group optionally substituted with one or more members selected from the group X such as a methyl group, ethyl group, propyl group, isopropyl group, butyl group, isobutyl group, sec-butyl group, tert-butyl group, 1-methylbutyl group, 2-methylbutyl group, 3- methylbutyl group, 1-ethylpropyl group, 1,2- dimethylpropyl group, 2 , 2-dimethylpropyl group, pentyl group, 1, 2-dimethylbutyl group, 2 , 2-dimethylbutyl group, 1-methylpentyl group, 2-methylpentyl group, 3- methylpentyl group, 4-methylpentyl group, hexyl group, fluoromethyl group, difluoromethyl group, trifluoromethyl
- C3-C6 alicyclic hydrocarbon group optionally substituted with one or more members selected from the group X" represented by R 8 or R 9 include a cyclopropyl group, cyclobutyl group, cyclopentyl group, cyclohexyl group and 2-cyclohexenyl group.
- phenyl group optionally substituted with one or more members selected from the group Y" represented by R 8 and R 9 include a 2-chlorophenyl group, 3-chlorophenyl group, 4-chlorophenyl group, 2- methylphenyl group, 3-methylphenyl group, 4-methylphenyl group, 2-methoxyphenyl group, 3-methoxyphenyl group, 4- methoxyphenyl group, 2- ( trifluoromethyl) phenyl group, 3- (trifluoromethyl) phenyl group, 4- (trifluoromethyl ) phenyl group, 2-cyanophenyl group, 3-cyanophenyl group, 4- cyanophenyl group, 2-nitrophenyl group, 3-nitrophenyl group and 4-nitrophenyl group.
- Examples of "5-membered heterocyclic group or 6- membered heterocyclic group optionally substituted with one or more members selected from the group Y" represented by R 8 or R 9 include a 5-membered aromatic heterocyclic group such as a 2-thienyl group and 3- thienyl group; and a ⁇ -membered aromatic heterocyclic group such as a 2-pyridyl group, 3-pyridyl group, 4- pyridyl group, 2-pyrimidinyl group and 4-pyrimidinyl group .
- C1-C4 alkyl group optionally substituted with one or more halogen atoms represented by R 10 include a methyl group, ethyl group, propyl group, isopropyl group, butyl group, isobutyl group, sec-butyl group and tert-butyl group, trifluoromethyl group and pentafluoroethyl group.
- Examples of ⁇ Cl-C4 alkyl group optionally substituted with one or more halogen atoms" represented by R 11 or R 12 include a methyl group, ethyl group, propyl group, isopropyl group, butyl group, isobutyl group, sec-butyl group, tert-butyl group, trifluoromethyl group, 2 , 2 , 2-trifluoroethyl group and pentafluoroethyl group.
- C2-C5 alkoxycarbonyl group represented by R 11 or R 12 include a methoxycarbonyl group, ethoxycarbonyl group, propoxycarbonyl group, isopropoxycarbonyl group, butoxycarbonyl' group, isobutoxycarbonyl group, sec-butoxyarbonyl group and tert-butoxycarbonyl group.
- C1-C3 chain hydrocarbon group optionally substituted with one or more halogen atoms represented by R 13 or R 14 include a C1-C3 alkyl group optionally substituted with one or more halogen atoms such as a methyl group, ethyl group, propyl group, isopropyl group, fluoromethyl group, difluoromethyl group, trifluoromethyl group, 2 , 2, 2-trifluoroethyl group and pentafluoroethyl group; a C2-C3 alkenyl group optionally substituted with one or more halogen atoms such as a vinyl group, 2-propenyl group, 3, 3-difluoro-2- propenyl group and 3 , 3-dichloro-2-propenyl group; and a C2-C3 alkynyl group optionally substituted with one or more halogen atoms such as an ethynyl group, and propargyl group.
- halogen atoms
- C1-C4 chain hydrocarbon group substituted with one or more halogen atoms represented by R 15 include a C1-C4 alkyl group substituted with one or more halogen atoms such as a fluoromethyl group, difluoromethyl group, trifluoromethyl group, 2,2,2- trifluoroethyl group, pentafluoroethyl group, heptafluoropropyl group, 1, 1, 1, 3, 3, 3-hexafluoroisopropyl group, heptafluoroisopropyl group, nonafluorobutyl group, nonafluoroisobutyl group, nonafluoro-sec-butyl group and nonafluoro-tert-butyl group; a C2-C4 alkenyl group substituted with one or more halogen atoms such as a 3 , 3-difluoroallyl group and pentafluoroallyl group; and a C2-C4 alkynyl
- C1-C4 alkyl group optionally substituted with one or more halogen atoms represented by R 16 include a methyl group, ethyl group, propyl group, isopropyl group, butyl group, isobutyl group, sec-butyl group, tert-butyl group, trifluoromethyl group, 2 , 2 , 2-trifluoroethyl group and pentafluoroethyl group and the like.
- Examples of the present active compound include the following .
- R 3 represents a C3-C6 alicyclic hydrocarbon group optionally substituted with one or more members selected from the group X, a phenyl group optionally substituted with one or more members selected from the group Y, a benzyl group optionally substituted with one or more members selected from the group Y or a 5-membered heterocyclic group or ⁇ -membered heterocyclic group optionally substituted with one or more members selected from the group Y;
- R 3 represents a C1-C6 chain hydrocarbon group optionally substituted with one or more members selected from the group X, -OR 8 , -NR 8 R 9 , -NR 8 C(O)R 9 , -S(O) m R 8 , -CO 2 R 10 , - CONR 8 R 9 , -CONR 10 NR 11 R 12 , a cyano group, a nitro group, a halogen atom or a hydrogen atom, and R 8 and R 9 are the same or different and represent a C1-C6 chain hydrocarbon group optionally substituted with one or more members selected from the group X, or a hydrogen atom (here, when m in -S(O) m R 8 represents 1 or 2, R 8 does not represent a hydrogen atom. ) ;
- R 3 represents a C1-C6 chain hydrocarbon group optionally substituted with one or more members selected from the group X, -OR 8 , -NR 8 R 9 , -S(O) m R 8 , a halogen atom or a hydrogen atom
- R 8 and R 9 are the same or different and represent a Cl-C ⁇ chain hydrocarbon group optionally substituted with one or more members selected from the group X, or a hydrogen atom (here, when m in -S(O) m R 8 represents 1 or 2, R 8 does not represent a hydrogen atom. ) ;
- the present active compound of formula (1) in which R 6 represents a fluoromethyl group The present active compound of formula (1) in which R 6 represents a difluoromethyl group;
- R 6 represents -OR 15 and R 15 represents a C1-C4 alkyl group substituted with one or more halogen atoms;
- R 7 represents -OR 15 and R 15 represents a C1-C4 alkyl group substituted with one or more halogen atoms;
- R 6 represents -OR 15 and R 15 represents a fluoromethyl group, difluoromethyl group or trifluoromethyl group;
- R 6 represents a hydrogen atom or halogen atom and R 7 represents a C1-C4 chain hydrocarbon group substituted with one or more halogen atoms;
- R 6 represents a hydrogen atom or halogen atom
- R 7 represents -OR 15
- R 15 represents a C1-C4 chain hydrocarbon group substituted with one or more halogen atoms ;
- R 6 represents a fluoromethyl group, difluoromethyl group or trifluoromethyl group and R 7 represents a hydrogen atom or halogen atom;
- R 6 represents a fluoromethyl group, difluoromethyl group or trifluoromethyl group and R 7 represents a hydrogen atom;
- R 6 represents a -OR 15
- R 15 represents a fluoromethyl group, difluoromethyl group or trifluoromethyl group
- R 7 represents a hydrogen atom or halogen atom
- R 6 represents a hydrogen atom or halogen atom and R 7 represents a fluoromethyl group, difluoromethyl group or trifluoromethyl group;
- R 6 represents a hydrogen atom and R 7 represents a fluoromethyl group, difluoromethyl group or trifluoromethyl group;
- R 6 represents a hydrogen atom or halogen atom
- R 7 represents -OR 15 and R 15 represents a fluoromethyl group, difluoromethyl group or trifluoromethyl group
- R 6 represents a hydrogen atom
- R 7 represents -OR 15
- R 15 represents a fluoromethyl group, difluoromethyl group or trifluoromethyl group
- R 3 represents -OR 8 and R 8 represents a C1-C3 alkyl group optionally substituted with one or more halogen atoms;
- R 3 represents -OR 8 and R 8 represents a methyl group, ethyl group, propyl group, isopropyl group, fluoromethyl group, difluoromethyl group, trifluoromethyl group, 2- fluoroethyl group, 2 , 2-difluoroethyl group, 2,2,2- trifluoroethyl group or pentafluoroethyl group;
- R 3 represents -OR 8 and R 8 represents a methyl group, ethyl group, propyl group, isopropyl group, 2- fluoroethyl group, 2 , 2-difluoroethyl group or 2,2,2- trifluoroethyl group;
- R 1 , R 2 and R 4 represent a hydrogen atom
- R 3 represents - OR 8
- R 8 represents a C1-C3 alkyl group optionally substituted with one or more halogen atoms
- R 1 , R 2 and R 4 represent hydrogen atoms
- R 3 represents -OR 8 and R 8 represents a methyl group, ethyl group, propyl group, isopropyl group, fluoromethyl group, difluoromethyl group, trifluoromethyl group, 2- fluoroethyl group, 2 , 2-difluoroethyl group, 2,2,2- trifluoroethyl group or pentafluoroethyl group;
- R 1 , R 2 and R 4 represent hydrogen atoms
- R 3 represents -0R ⁇
- R 8 represents a methyl group, ethyl group, propyl • group, isopropyl group, 2-fluoroethyl group, 2,2- difluoroethyl group or 2, 2, 2-trifluoroethyl group;
- R 3 represents -S(O) m R 8 and R 8 represents a C1-C3 alkyl group
- R 1 , R 2 and R 4 represent hydrogen atoms, R 3 represents - S(O) n R 8 and R 8 represents a C1-C3 alkyl group;
- the present active compound can be produced, for example, by the following (Production Method 1) to (Production Method 12) and (Production Method 21) to (Production Method 36) .
- (Production Method 1) to (Production Method 12) and (Production Method 21) to (Production Method 36) .
- a compound (4) in which n is 0 can be produced by reacting a compound (2) and a compound (3) in the presence of a base.
- solvent to be used in the reaction examples include ethers such as tetrahydrofuran (hereinafter, referred to as THF in some cases), ethylene glycol dimethyl ether, 1,4-dioxane and the like, aliphatic hydrocarbons such as hexane, heptane, octane and the like, aromatic hydrocarbons such as toluene, xylene and the like, halogenated hydrocarbons such as chlorobenzene and the like, esters such as ethyl acetate, butyl acetate and the like, nitriles such as acetonitrile and the like, acid amides such as N, N-dimethylformamide (hereinafter, referred to as DMF in some cases) and the like, sulfoxides such as dimethyl sulfoxide (hereinafter, referred to as DMSO in some cases) and the like, nitrogen-containing aromatic compounds such as pyridine, quinoline and the like,
- the reaction temperature of the reaction is usually in the range of 30 to 200°C, and the reaction time thereof is usually in the range of 0.1 to 24 hours.
- the compound (4) can be isolated by subjecting post treatment operations such as water being poured into the reaction mixture, the mixture being extracted with an organic solvent, then the organic layer being dried and concentrated.
- the isolated compound (4) can also be further purified by chromatography, recrystallization and the like.
- a compound (4) in which n is 0 can be produced by reacting a compound (2) and a compound (5) .
- the reaction is carried out usually in the presence of a solvent.
- solvent to be used in the reaction examples include ethers such as THF, ethylene glycol dimethyl ether, 1,4-dioxane and the like, aliphatic hydrocarbons such as hexane, heptane, octane and the like, aromatic hydrocarbons such as toluene, xylene and the like, halogenated hydrocarbons such as chlorobenzene and the like, esters such as ethyl acetate, butyl acetate and the like, nitriles such as acetonitrile and the like, acid amides such as DMF and the like, sulfoxides such as dimethyl sulfoxide and the like, nitrogen-containing aromatic compounds such as pyridine, quinoline and the like, and mixtures thereof.
- ethers such as THF, ethylene glycol dimethyl ether, 1,4-dioxane and the like
- aliphatic hydrocarbons such as hexane, heptane
- reaction can also be carried out with addition of a base if necessary.
- the base to be used in the reaction includes hydrogen carbonates such as sodium hydrogen carbonate, potassium hydrogen carbonate and the like, carbonates such as sodium carbonate, potassium carbonate and the like, sulfites such as sodium sulfite, potassium sulfite and the like, hydrogen sulfates such as sodium hydrogen sulfate, potassium hydrogen sulfate and the like, and mixtures thereof.
- the compound (5) is used usually in a proportion of 1 to 3 mol with respect to 1 mol of the compound (2) .
- the base is used usually in a proportion of 1 to 5 mol with respect to 1 mol of the compound (2).
- the reaction temperature of the reaction is usually in the range of 30 to 200°C, and the reaction time thereof is usually in the range of 0.1 to 24 hours.
- the compound (4) can be isolated by subjecting post treatment operations such as water being poured into the reaction mixture, the mixture being extracted with an organic solvent, then the organic layer being dried and concentrated.
- the isolated compound (4) can also be further purified by chromatography, recrystallization and the like.
- a compound (4) in which n is 0 can be produced by reacting a compound (2) and a compound (6) in the presence of a dehydrocondensing agent .
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , A 1 and A 2 represent the same meaning as described above.
- the reaction is carried out usually in the presence of a solvent.
- solvent to be used in the reaction examples include ethers such as THF, ethylene glycol dimethyl ether, 1,4-dioxane and the like, aliphatic hydrocarbons such as hexane, heptane, octane and the like, aromatic hydrocarbons such as toluene, xylene and the like, halogenated hydrocarbons such as chlorobenzene and the like, esters such as ethyl acetate, butyl acetate and the like, nitriles such as acetonitrile and the like, acid amides such as DMF and the like, sulfoxides such as DMSO and the like, nitrogen-containing aromatic compounds such as pyridine, quinoline and the like, and mixtures thereof.
- ethers such as THF, ethylene glycol dimethyl ether, 1,4-dioxane and the like
- aliphatic hydrocarbons such as hexane, heptane, oc
- the dehydrocondensing agent to be used in the reaction includes carbodiimides such as l-ethyl-3- (3- dimethylaminopropyl) carbodiimide hydrochloride (hereinafter, referred to as WSC in some cases), 1,3- dicyclohexylcarbodiimide and the like.
- WSC l-ethyl-3- (3- dimethylaminopropyl) carbodiimide hydrochloride
- 1,3- dicyclohexylcarbodiimide 1,3- dicyclohexylcarbodiimide and the like.
- the reaction may be performed in the presence of 1- hydroxybenzotriazole .
- the dehydrocondensing agent is used usually in a proportion of 1 to 5 mol
- 1- hydroxybenzotriazole is used usually in a proportion of 0.01 to 0.1 mol.
- the reaction temperature of the reaction is usually in the range of 30 to 200 0 C, and the reaction time thereof is usually in the range of 0.1 to 24 hours.
- the compound (4) can be isolated by subjecting post treatment operations such as water being poured into the reaction mixture, the mixture being extracted with an organic solvent, then the organic layer being dried and concentrated.
- the isolated compound (4) can also be further purified by chromatography, recrystallization and the like.
- a compound (4) in which n is 0 can be produced by reacting a compound (7) in the presence of a dehydrating agent.
- the reaction is carried out usually in the presence of a solvent, and it may also be permissible to use a dehydrating agent in solvent quantity.
- a solvent such as THF, ethylene glycol dimethyl ether, 1,4-dioxane and the like, aliphatic hydrocarbons such as hexane, heptane, octane and the like, aromatic hydrocarbons such as toluene, xylene and the like, halogenated hydrocarbons chlorobenzene and the like, esters such as ethyl acetate, butyl acetate and the like, nitriles such as acetonitrile and the like, acid amides such as DMF and the like, sulfoxides such as DMSO and the like, and mixtures thereof.
- the dehydrating agent to be used in the reaction includes sulfonic acids such as p-toluenesulfonic acid and the like, phosphorus oxy chloride, acetic anhydride, trifluoroacetic anhydride and the like.
- the dehydrating agent is used usually in a proportion of 0.1 to excess amount with respect to 1 mol of the compound (7) .
- the reaction temperature of the reaction is usually in the range of 30 to 200 0 C, and the reaction time thereof is usually in the range of 0.1 to 24 hours.
- the compound (4) can be isolated by subjecting post treatment operations such as the reaction mixture being extracted with an organic solvent, the organic layer being dried and concentrated. The isolated compound (4) can also be further purified by chromatography, recrystallization and the like.
- a compound (4-a) can be produced by reacting a compound (8) and a compound (9) in the presence of a base.
- R 1 , R 2 , R 4 , R 5 , R 6 , R 7 , A 1 and A 2 represent the same meaning as described above, R 31 represents a chlorine atom or a fluorine atom, and
- R 32 represents a Cl-C ⁇ chain hydrocarbon group optionally substituted with one or more members selected from the group X, a C3-C6 alicyclic hydrocarbon group optionally substituted with one or more members selected from the group X, a phenyl group optionally substituted with one or more members selected from the group Y, a benzyl group optionally substituted with one or more members selected from the group Y, a 5-membered heterocyclic " group or ⁇ -membered heterocyclic group optionally substituted with one or more members selected from the group Y . ]
- the reaction is carried out usually in the presence of a solvent, and it may also be permissible to use the compound (9) in solvent quantity.
- a solvent such as THF, ethylene glycol dimethyl ether, 1,4-dioxane and the like, aromatic hydrocarbons such as toluene, xylene and the like, nitriles such as acetonitrile and the like, acid amides such as DMF and the like, sulfoxides such as DMSO and the like, and mixtures thereof.
- Examples of the base to be used in the reaction include alkali metal hydrides such as sodium hydride and the like.
- the compound (9) is used usually in a proportion of 1 to excess amount and the base is used usually in a proportion of 1 to 10 mol with respect to 1 mol of the compound (8) .
- the reaction temperature of the reaction is usually in the range of 0 to 150°C, and the reaction time thereof is usually in the range of 0.1 to 24 hours.
- the compound (4-a) can be isolated by subjecting post treatment operations such as the reaction mixture being extracted with an organic solvent, the organic layer being dried and concentrated.
- the isolated compound (4-a) can also be further purified by chromatography, recrystallization and the like.
- a compound (4-b) can be produced by reacting a compound (8) and a compound (10) in the presence of a base.
- R 1 , R 2 , R 4 , R 5 , R 6 , R 7 , R 31 , R 32 , A 1 and A 2 represent the same meaning as described above.
- the reaction is carried out usually in the presence of a solvent.
- a solvent examples include ethers such as THF, ethylene glycol dimethyl ether, 1,4-dioxane and the like, aromatic hydrocarbons such as toluene, xylene and the like, nitriles such as acetonitrile and the like, acid amides such as DMF and the like, sulfoxides such as DMSO and the like, and mixtures thereof.
- Examples of the base to be used in the reaction include alkali metal hydrides such as sodium hydride and the like.
- the compound (10) is used usually in a proportion of 1 to 10 mol and the base is used usually in a proportion of 1 to 10 mol with respect to 1 mol of the compound (8) .
- the reaction temperature of the reaction is usually in the range of 0 to 150 0 C, and the reaction time thereof is usually in the range of 0.5 to 24 hours.
- the compound (4-b) can be isolated by subjecting post treatment operations such as the reaction mixture being extracted with an organic solvent, the organic layer being dried and concentrated.
- the isolated compound (4-b) can also be further purified by chromatography, recrystallization and the like.
- a compound (4-c) can be produced by reacting the compound (4-b) in the presence of an oxidation agent.
- the reaction is carried out usually in the presence of a solvent.
- solvent to be used in the reaction examples include aliphatic halogenated hydrocarbons such as dichloromethane, chloroform and the like, alcohols such as methanol, ethanol and the like, acetic acid, water and mixtures thereof.
- Examples of the oxidation agent to be used in the reaction include sodium periodate.
- the oxidation agent is used usually in a proportion of 1 to 3 mo1 with respect to 1 raol of the compound (4- b) .
- the reaction temperature of the reaction is usually in the range of -20 to 80°C, and the reaction time thereof is usually in the range of 0.1 to 12 hours.
- the compound (4-c) can be isolated by subjecting post treatment operations such as the reaction mixture being extracted with an organic solvent, the organic layer being washed with an aqueous solution of a reducing agent (for example, sodium sulfite, sodium thiosulfate) and an aqueous solution of a base (for example, sodium hydrogen carbonate) if necessary, then being dried and concentrated.
- a reducing agent for example, sodium sulfite, sodium thiosulfate
- a base for example, sodium hydrogen carbonate
- a compound (4-d) can be produced by reacting the compound (4-b) in the presence of an oxidation agent.
- R 1 , R 2 , R 4 , R 5 , R 6 , R 7 , R 32 , A 1 and A 2 represent the same meaning as described above.
- the reaction is carried out usually in the presence of a solvent.
- Examples of the solvent to be used in the reaction include aliphatic halogenated hydrocarbons such as dichloromethane, chloroform and the like, acetic acid, water and mixtures thereof.
- Examples of the oxidation agent to be used in the reaction include carboxylic acid peroxides such as 3- chloroperbenzoic acid and the like.
- the oxidation agent is used usually in a proportion of 2 to 4 mol with respect to 1 mol of the compound (4- b) .
- the reaction temperature of the reaction is usually in the range of -20 to 3O 0 C, and the reaction time thereof is usually in the range of 0.1 to 12 hours.
- the compound (4-d) can be isolated by subjecting post treatment operations such as the reaction mixture being extracted with an organic solvent, the organic layer being washed with an aqueous solution of a reducing agent (for example, sodium sulfite, sodium thiosulfate) and an aqueous solution of a base (for example, sodium hydrogen carbonate) if necessary, then being dried and concentrated.
- a reducing agent for example, sodium sulfite, sodium thiosulfate
- a base for example, sodium hydrogen carbonate
- a compound (4-e) can be produced by reacting a compound (11) with an acid anhydride represented by (12) or an acid chloride represented by (13) .
- R 1 , R 2 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , A 1 and A 2 represent the same meaning as described above.
- reaction is carried out usually in the presence of a solvent, and it may also be permissible to use the compound (12) in solvent quantity.
- Examples of the solvent to be used in the reaction include ethers such as THF, ethylene glycol dimethyl ether, 1,4-dioxane and the like, aromatic hydrocarbons such as toluene, xylene and the like, nitriles such as acetonitrile and the like, acid amides such as DMF and the like, sulfoxides such as DMSO and the like, nitrogen-containing aromatic compounds such as pyridine, quinoline and the like, and mixtures thereof.
- the reaction can also be carried out with addition of a base if necessary.
- Examples of the base to be used in the reaction include alkali metal hydrides such as sodium hydride and the like, carbonates such as potassium carbonate and the like, tertiary amines such as triethylamine, diisopropylethylamine and the like, and nitrogen- containing aromatic compounds such as pyridine, 4- dimethylaminopyridine and the like.
- the compound (12) or the compound (13) is used usually in a proportion of 1 to 10 mol and the base is used usually in a proportion of 1 to 10 mol with respect to 1 mol of the compound (11) .
- the reaction temperature of the reaction is usually in the range of 0 to 120°C, and the reaction time thereof is usually in the range of 0.1 to 24 hours.
- the compound (4-e) can be isolated by subjecting post treatment operations such as the reaction mixture being extracted with an organic solvent, the organic layer being dried and concentrated.
- the isolated compound (4-e) can also be further purified by chromatography, recrystallization and the like.
- a compound (4-f) can be produced by reacting a compound (14) with a boronic acid compound represented by (15) or a tin compound represented by (16) in the presence of a palladium compound.
- R 1 , R 2 , R 4 , R 5 , R 6 , R 7 , A 1 and A 2 represent the same meaning as described above,
- L represents a bromine or iodine atom
- R 3 x represents a phenyl group optionally substituted with one or more members selected from the group Y, a 5- membered aromatic heterocyclic group or a ⁇ -membered aromatic heterocyclic group optionally substituted with one or more members selected from the group Y (here, limited to aromatic heterocyclic groups bonding to a pyridine ring on its carbon atom) .
- the reaction is carried out usually in the presence of a solvent.
- solvent to be used in the reaction examples include ethers such as THF, ethylene glycol dimethyl ether, 1,4-dioxane and the like, alcohols such as methanol, ethanol and the like, aliphatic hydrocarbons such as hexane, heptane, octane and the like, aromatic hydrocarbons such as toluene, xylene and the like, acid amides such as DMF and the like, water and mixtures thereof, and the like.
- ethers such as THF, ethylene glycol dimethyl ether, 1,4-dioxane and the like
- alcohols such as methanol, ethanol and the like
- aliphatic hydrocarbons such as hexane, heptane, octane and the like
- aromatic hydrocarbons such as toluene, xylene and the like
- acid amides such as DMF and the like, water and mixtures thereof, and the like.
- the palladium compound to be used in the reaction includes palladium acetate, tetrakistriphenylphosphinepalladium, ⁇ 1,1'- bis (diphenylphosphino) ferrocene Jdichloropalladium methylene chloride complex, and bis (triphenylphosphine) palladium dichloride, and the like.
- the compound (15) or the compound (16) is used usually in a proportion of 0.5 to 5 mol and the
- the reaction can also be carried out in the presence of a base (inorganic salts such as sodium acetate, potassium acetate, potassium carbonate, tripotassium phosphate, sodium hydrogen carbonate and the like are mentioned) and / or a phase transfer catalyst (quaternary ammonium salts such as tetrabutyl ammonium bromide, benzyl triethyl ammonium bromide and the like are mentioned), if necessary.
- a base inorganic salts such as sodium acetate, potassium acetate, potassium carbonate, tripotassium phosphate, sodium hydrogen carbonate and the like are mentioned
- a phase transfer catalyst quaternary ammonium salts such as tetrabutyl ammonium bromide, benzyl triethyl ammonium bromide and the like are mentioned
- the reaction temperature of' the reaction is usually in the range of 50 to 120°C, and the reaction time thereof is usually in the range of 0.5 to 24 hours.
- the compound (4-f) can be isolated by subjecting post treatment operations such as the reaction mixture being extracted with an organic solvent, the organic layer being dried and concentrated.
- the isolated compound (4-f) can also be further purified by chromatography, recrystallization and the like.
- a compound (4-g) can be produced by reacting a compound (8) and a compound (17) in the presence of a base.
- R 1 , R 2 , R 4 , R 5 , R 6 , R 7 , R 31 , A 1 and A 2 represent the same meaning as described above,
- R 3 y represents a 5-membered heterocyclic group or a ⁇ -membered heterocyclic group optionally substituted with one or more members selected from the group Y (here, limited to heterocyclic groups bonding to a pyridine ring on its nitrogen atom) . ]
- the reaction is carried out usually in the presence of a solvent.
- Examples of the solvent to be used in the reaction include ethers such as THF, ethylene glycol dimethyl ether, 1,4-dioxane and the like, aromatic hydrocarbons such as toluene, xylene and the like, nitriles such as acetonitrile and the like, acid amides such as DMF and the like, sulfoxides such as DMSO and the like, and mixtures thereof.
- Examples of the base to be used in the reaction include alkali metal hydrides such as sodium hydride and the like, carbonates such as potassium carbonate and the like.
- the compound (17) is used usually in a proportion of 1 to 10 mol and the base is used usually in the range of 1 to 10 mol with respect to 1 mol of the compound (8) .
- the reaction temperature of the reaction is usually in the range of 0 to 100°C, and the reaction time thereof is usually in the range of 0.1 to 24 hours.
- the compound (4-g) can be isolated by subjecting post treatment operations such as the reaction mixture being extracted with an organic solvent, the organic layer being dried and concentrated.
- the isolated compound (4-g) can also be further purified by chromatography, recrystallization and the like.
- a compound (18) in which n is 1 can be produced by reacting a compound (4) in the presence of an oxidation agent.
- Examples of the solvent to be used in the reaction include aliphatic halogenated hydrocarbons such as dichloromethane, chloroform and the like, acetic acid, water and mixtures thereof.
- Examples of the oxidation agent to be used in the reaction include carboxylic acid peroxides such as 3- chloroperbenzoic acid and the like, and hydrogen peroxide water and the like. The oxidation agent is used usually in a proportion of 1 to 3 mol with respect to 1 mol of the compound (4) .
- the reaction temperature of the reaction is usually in the range of 40 to 100 0 C, and the reaction time thereof is usually in the range of 0.1 to 24 hours.
- the compound (18) can be isolated by subjecting post treatment operations such as the reaction mixture being extracted with an organic solvent, the organic layer being washed with an aqueous solution of a reducing agent (for example, sodium sulfite, sodium thiosulfate) and an aqueous solution of a base (for example, sodium hydrogen carbonate) if necessary, then being dried and concentrated.
- a reducing agent for example, sodium sulfite, sodium thiosulfate
- a base for example, sodium hydrogen carbonate
- the compound (4-h) can be produced by reacting a compound (21) and a compound (10) in the presence of a base.
- R 1 , R 2 , R 3 , R 5 , R 6 , R 7 , R 31 , R 32 , A 1 and A 2 represent the same meaning as described above.
- the reaction is carried out usually in the presence of a solvent.
- solvent to be used in the reaction examples include ethers such as THF, ethylene glycol dimethyl ether, 1,4-dioxane and the like, aromatic hydrocarbons such as toluene, xylene and the like, nitriles such as acetonitrile and the like, acid amides such as DMF and the like, sulfoxides such as DMSO and the like, and mixtures thereof.
- ethers such as THF, ethylene glycol dimethyl ether, 1,4-dioxane and the like
- aromatic hydrocarbons such as toluene, xylene and the like
- nitriles such as acetonitrile and the like
- acid amides such as DMF and the like
- sulfoxides such as DMSO and the like
- Examples of the base to be used in the reaction include alkali metal hydrides such as sodium hydride and the like, etc.
- the compound (10) is used usually in a proportion of 1 to 10 mol and the base is used usually in a proportion of 1 to 10 mol with respect to 1 mol of the compound (21) .
- the reaction temperature of the reaction is usually in the range of 0 to 100°C, and the reaction time is usually in the range of 0.1 to 24 hours.
- the compound (4-h) can be isolated by subjecting post treatment operations such as the reaction mixture being extracted with an organic solvent, the organic layer being dried and concentrated.
- the isolated compound (4-h) can also be further purified by chromatography, recrystallization and the like.
- the compound (4-j) can be produced by reacting the compound (4-h) and an oxidation agent .
- the reaction is carried out usually in the presence of a solvent.
- Examples of the solvent to be used in the reaction include aliphatic halogenated hydrocarbons such as dichloromethane , chloroform and the like, alcohols such as methanol, ethanol and the like, ethers such as T ⁇ F, ethylene glycol dimethyl ether, 1,4-dioxane and the like; acetic acid, water, and mixtures thereof.
- Examples of the oxidation agent to be used in the reaction include sodium periodate.
- the oxidation agent is used usually in a proportion of 1 to 3 mol with respect to 1 mol of the compound (4- h) .
- the reaction temperature of the reaction is usually in the range of -20 to 8O 0 C, and the reaction time is usually in the range of 0.1 to 12 hours.
- the compound (4-j) can be isolated by subjecting post treatment operations such as the reaction mixture being extracted with an organic solvent, the organic layer being washed with an aqueous solution of a reducing agent (for example, sodium sulfite, sodium thiosulfate) and an aqueous solution of a base (for example, sodium hydrogen carbonate) if necessary, then being dried and concentrated.
- a reducing agent for example, sodium sulfite, sodium thiosulfate
- a base for example, sodium hydrogen carbonate
- the compound (4-k) can be produced by reacting the compound (4-h) and an oxidation agent .
- R 1 , R 2 , R 3 , R 5 , R 6 , R 7 , R 32 , A 1 and A 2 represent the same meaning as described above.
- the reaction is carried out usually in the presence of a solvent.
- Examples of the solvent to be used in the reaction include aliphatic halogenated hydrocarbons such as dichloromethane, chloroform and the like; acetic acid, water, and mixtures thereof.
- Examples of the oxidation agent to be used in the reaction include carboxylic acid peroxides such as 3- chloroperbenzoic acid and the like.
- the oxidation agent is used usually in a proportion of 2 to 4 mol with respect to 1 mol of the compound (4- h) .
- the reaction temperature of the reaction is usually in the range of -20 to 3O 0 C, and the reaction time is usually in the range of 0.1 to 12 hours.
- the compound (4-k) can be isolated by subjecting post treatment operations such as the reaction mixture being extracted with an organic solvent, the organic layer being washed with an aqueous solution of a reducing agent (for example, sodium sulfite, sodium thiosulfate) and an aqueous solution of a base (for example, sodium hydrogen carbonate) if necessary, then being dried and concentrated.
- a reducing agent for example, sodium sulfite, sodium thiosulfate
- a base for example, sodium hydrogen carbonate
- the compound (4-k) can be produced, for example, by reacting the compound (4-j) and an oxidation agent.
- R 1 , R 2 , R 4 , R 5 , R 6 , R 7 , R 32 , A 1 and A 2 represent the same meaning as described above.
- the reaction is carried out usually in the presence of a solvent.
- a solvent examples include aliphatic halogenated hydrocarbons such as dichloromethane, chloroform and the like; acetic acid, water, and mixtures thereof.
- Examples of the oxidation agent to be used in the reaction include carboxylic acid peroxides such as 3- chloroperbenzoic acid and the like.
- the oxidation agent is used usually in a proportion of 1 to 3 mol with respect to 1 mol of the compound (4-
- the reaction temperature of the reaction is usually in the range of -20 to 30°C, and the reaction time is usually in the range of 0.1 to 12 hours.
- the compound (4-k) can be isolated by subjecting post treatment operations such as the reaction mixture being extracted with an organic solvent, the organic layer being washed with an aqueous solution of a reducing agent (for example, sodium sulfite, sodium thiosulfate) and an aqueous solution of a base (for example, sodium hydrogen carbonate) if necessary, then being dried and concentrated.
- a reducing agent for example, sodium sulfite, sodium thiosulfate
- a base for example, sodium hydrogen carbonate
- the compound (4-p) can be produced by reacting the compound (8) and a cyanide.
- R 1 , R 2 , R 4 , R 5 , R 6 , R 7 , R 31 , A 1 and A 2 represent the same meaning as described above.
- the reaction is carried out usually in the presence of a solvent .
- solvent to be used in the reaction examples include ethers such as THF, ethylene glycol dimethyl ether, 1,4-dioxane and the like, acid amides such as DMF, l-methyl-2-pyrrolidinone and the like, sulfoxides such as DMSO and the like, and mixtures thereof.
- Examples of the cyanide to be used in the reaction include sodium cyanide and potassium cyanide.
- the reaction is carried out, if necessary, in the presence of crown ethers such as 15-crown-5, 18-crown-6 and the like.
- the cyanide is used usually in a proportion of 1 to 3 mol and the catalyst is used usually in a proportion of 0.01 to 0.5 mol with respect to 1 mol of the compound (8) .
- the reaction temperature of the reaction is usually in the, range of 0 to 200°C, and the reaction time is usually in the range of 0.1 to 12 hours.
- the compound (4-p) can be isolated by subjecting post treatment operations such as the reaction mixture being extracted with an organic solvent, the organic layer being dried and concentrated.
- the isolated compound (4-p) can also be further purified by chromatography, recrystallization and the like.
- the compound (4-q) can be produced by subjecting the compound (4-p) to a hydrolysis reaction in the presence of a base.
- R 1 , R 2 , R 4 , R 5 , R 6 , R 7 , A 1 and A 2 represent the same meaning as described above.
- the reaction is carried out usually in the presence of a solvent.
- Examples of the solvent to be used in the reaction include alcohols such as methanol, ethanol and the like; water, and mixtures thereof.
- Examples of the base to be used in the reaction include hydroxides of alkali metals or alkaline earth metals such as sodium hydroxide, potassium hydroxide, barium hydroxide and the like.
- the base is used usually in a proportion of 1 to 3 mol with respect to 1 mol of the compound (4-p) .
- the reaction temperature of the reaction is usually in the range of 0 to 150°C, and the reaction time is usually in the range of 0.1 to 24 hours.
- the compound (4-q) can be isolated by subjecting post treatment operations such as neutralized with an acid of the reaction mixture, being filtration to obtain a solid or neutralized with an acid of the reaction mixture, • being extraction of the mixture with an organic solvent, then the organic layer being dried and concentrated.
- the isolated compound (4-q) can also be further purified by chromatography, recrystallization and the like.
- the compound (4-r) can be produced by reacting the compound (4-p) with hydrogen peroxide in the presence of a base.
- the reaction is carried out usually in the presence of a solvent.
- Examples of the solvent to be used in the reaction include sulfoxides such as DMSO and the like; water, and mixtures thereof.
- Examples of the base to be used in the reaction include hydroxides of alkali metals or alkaline earth metals such as sodium hydroxide, potassium hydroxide, calcium hydroxide and the like, and carbonates such as sodium carbonate, potassium carbonate and the like.
- Hydrogen peroxide is used usually in a proportion of 1 to 5 mol and the base is used usually in a proportion of 1 to 5 mol with respect to 1 mol of the compound (4- p) .
- the reaction temperature of the reaction is usually in the range of 0 to 100 0 C, and the reaction time is usually in the range of 0.1 to 24 hours.
- the compound (4-r) can be isolated by subjecting post treatment operations such as the reaction mixture being extracted with an organic solvent, the organic layer being dried and concentrated.
- the isolated compound (4-r) can also be further purified by chromatography, recrystallization and the like.
- the compound (4-s) can be produced by reacting a compound (22) and a compound (23) .
- R 1 , R 2 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , A 1 and A 2 represent the same meaning as described above.
- the reaction is carried out usually in the presence of a base, and carried out usually in the presence of a solvent .
- solvent to be used in the reaction examples include ethers such as THF, ethylene glycol dimethyl ether, 1,4-dioxane and the like, aromatic hydrocarbons such as toluene, xylene and the like, nitriles such as acetonitrile and the like, acid amides such as DMF and the like, sulfoxides such as DMSO and the like, nitrogen-containing aromatic compounds such as pyridine, quinoline and the like; water, and mixtures thereof.
- ethers such as THF, ethylene glycol dimethyl ether, 1,4-dioxane and the like
- aromatic hydrocarbons such as toluene, xylene and the like
- nitriles such as acetonitrile and the like
- acid amides such as DMF and the like
- sulfoxides such as DMSO and the like
- nitrogen-containing aromatic compounds such as pyridine, quinoline and the like
- water and mixtures thereof.
- Examples of the base to be used in the reaction include carbonates such as potassium carbonate and the like, tertiary amines such as triethylamine, diisopropylethylamine and the like, and nitrogen- containing aromatic compounds such as pyridine, 4- dimethylaminopyridine and the like.
- the compound (23) is used usually in a proportion of 1 mol to excess quantity and the base is used usually in a proportion of 1 mol to excess quantity with respect to 1 mol of the compound (22) .
- the reaction can also be carried out without using the above-described base.
- tertiary amines such as triethylamine, diisopropylethylamine and the like or liquid nitrogen- containing aromatic compounds such as pyridine and the like are used in excess amount as the base, the reaction can also be carried out without using the above- described solvent.
- the reaction temperature of the reaction is usually in the range of 0 to 100°C, and the reaction time is usually in the range of 0.1 to 24 hours.
- the compound (4-s) can be isolated by_ subj ecting post treatment operations such as the reaction mixture being extracted with an organic solvent, the organic layer being dried and concentrated.
- the isolated compound (4-s) can also be further purified by chromatography, recrystallization and the like.
- the compound (4-s) can be produced by reacting the compound (4-q), a compound (23) and a condensing agent.
- Examples of the solvent to be used in the reaction include ethers such as THF, ethylene glycol dimethyl ether, 1,4-dioxane and the like, aliphatic hydrocarbons such as hexane, heptane, octane and the like, aromatic hydrocarbons such as toluene, xylene and the like, halogenated hydrocarbons such as chlorobenzene and the like, esters such as ethyl acetate, butyl acetate and the like, nitriles such as acetonitrile and the like, acid amides such as DMF and the like, sulfoxides such as DMSO and the like, nitrogen-containing aromatic compounds such as pyridine, quinoline and the like, and mixtures thereof.
- the condensing agent to be used in the reaction include carbodiimides such as WSC, 1,3- dicyclohexylcarbodiimide and the like.
- the reaction is carried out, if necessary, in the presence of 1-hydroxybenzotriazole.
- the compound (23) is used usually in a proportion of 1 to 3 mol
- the condensing agent is used usually in a proportion of 1 to 5 mol
- 1-hydroxybenzotriazole is used usually in a proportion of 0.01 to 0.1 mol with respect to 1 mol of the compound (4-q) .
- the reaction temperature of the reaction is usually in the range of 0 to 100°C
- the reaction time is usually in the range of 0.1 to 24 hours.
- the compound (4-q) can be isolated by subjecting post treatment operations such as the reaction mixture being extracted with an organic solvent, the organic layer being dried and concentrated.
- the isolated compound (4-s) can also be further purified by chromatography, recrystallization and the like.
- the compound (4-t) can be produced by reacting the compound (4-q) and a compound (24) in the presence of an acid.
- R 1 , R 2 , R 4 , R 5 , R 6 , R 7 , A 1 and A 2 represents the same meaning as described above, and R 33 represents a C1-C4 alkyl group optionally substituted with one or more halogen atoms.
- reaction is carried out in the presence of a ⁇ solvent or in the absence of a solvent.
- solvent to be used in the reaction examples include ethers such as 1,4-dioxane and the like, aliphatic hydrocarbons such as hexane, heptane, octane and the like, aromatic hydrocarbons such as toluene, xylene and the like, halogenated hydrocarbons such as chlorobenzene and the like, and mixtures thereof.
- Examples of the acid to be used in the reaction include mineral acids such as hydrochloric acid, sulfuric acid and the like, and organic acids such as p- toluenesulfonic acid and the like.
- the compound (24) is used usually in a proportion of 1 mol to excess quantity and the acid is used usually 0.01 mol to 1 mol with respect to 1 mol of the compound (4-q) .
- reaction can also be carried out without using the above-described solvent.
- the reaction temperature of the reaction is usually in the range of 0 to 200°C, and the reaction time is usually in the range of 0.1 to 24 hours.
- the compound (4-t) can be isolated by subjecting post treatment operations such as the reaction mixture being extracted with an organic solvent, the organic layer being dried and concentrated.
- the isolated compound (4-t) can also be further purified by chromatography, recrystallization and the like.
- the compound (4-u) can be produced by reacting a compound (8) with a sulfurizing agent such as sodium hydrosulfide and the like.
- the reaction is carried out usually in the presence of a solvent.
- a solvent examples include ethers such as THF, ethylene glycol dimethyl ether, 1,4-dioxane and the like, aromatic hydrocarbons such as toluene, xylene and the like, nitriles such as acetonitrile and the like, acid amides such as DMF and the like, sulfoxides such as DMSO and the like, nitrogen-containing aromatic compounds such as pyridine, quinoline and the like; water, and mixtures thereof.
- ethers such as THF, ethylene glycol dimethyl ether, 1,4-dioxane and the like
- aromatic hydrocarbons such as toluene, xylene and the like
- nitriles such as acetonitrile and the like
- acid amides such as DMF and the like
- sulfoxides such as DMSO and the like
- nitrogen-containing aromatic compounds such as pyridine, quinoline and
- the sulfurizing agent such as sodium hydrosulfide and the like is used usually in a proportion of 1 mol to 3 mol with respect to 1 mol of the compound (8) .
- the reaction temperature of the reaction is usually in the range of 0 to 150°C, and the reaction time is usually in the range of 0.1 to 24 hours.
- the compound (4-u) can be isolated by subjecting post treatment operations such as neutralized with an acid of the reaction mixture, being filtration to obtain a solid or neutralized with an acid of the reaction mixture, being extraction of the mixture with an organic solvent, then the organic layer being dried and concentrated.
- the isolated compound (4-u) can also be further purified by chromatography, recrystallization and the like.
- the compound (4-w) can be produced by reacting the compound (4-u) and a compound (25) in the presence of a base.
- the reaction is carried out usually in the presence of a solvent.
- solvent to be used in the reaction examples include ethers such as THF, ethylene glycol dimethyl ether, 1,4-dioxane and the like, aromatic hydrocarbons such as toluene, xylene and the like, nitriles such as acetonitrile and the like, acid amides such as DMF and the like, sulfoxides such as DMSO and the like, and mixtures thereof.
- ethers such as THF, ethylene glycol dimethyl ether, 1,4-dioxane and the like
- aromatic hydrocarbons such as toluene, xylene and the like
- nitriles such as acetonitrile and the like
- acid amides such as DMF and the like
- sulfoxides such as DMSO and the like
- Examples of the base to be used in the reaction include hydrides of alkali metals or alkaline earth metals such as sodium hydride, potassium hydride, calcium hydride and the like, inorganic bases such as sodium carbonate, potassium carbonate and the like, or organic bases such as triethylamine, and the like.
- the base is used usually in a proportion of 1 to 3 mol with respect to 1 mol of the compound (4-u) .
- the reaction temperature of the reaction is usually in the range of 0 to 100 0 C, and the reaction time is usually in the range of 0.1 to 24 hours.
- the compound (4-w) can be isolated by subjecting post treatment operations such as the reaction mixture being extracted with an organic solvent, the organic layer being dried and concentrated.
- the isolated compound (4-w) can also be further purified by chromatography, recrystallization and the like.
- the compound (4-w) can be produced by reacting the compound (4-u) with an oxidation agent such as potassium nitrate and the like and a chlorinating agent such as trimethylsilyl chloride and the like.
- R 1 , R 2 , R 4 , R 5 , R 6 , R 7 , A 1 and A 2 represent the same meaning as described above.
- the reaction is carried out usually in the presence of a solvent.
- the solvent to be used in the reaction include halogenated hydrocarbons such as methylene chloride, chloroform and the like, aromatic halides such as chlorobenzene and the like, aromatic hydrocarbons such as toluene, xylene and the like, and mixtures thereof.
- the oxidation agent such as potassium nitrate and the like is used usually in a proportion of 1 to 5 mol and the chlorinating agent such as trimethylsilyl chloride and the like is used usually in a proportion of 5 1 to 5 mol with respect to 1 mol of the compound (4-u) .
- the reaction temperature of the reaction is usually in the range of 0 to 150 0 C, and the reaction time is usually in the range of 0.1 to 24 hours.
- the compound (4-w) 10 can be isolated by subjecting post treatment operations such as the reaction mixture being extracted with an organic solvent, the organic layer being dried and concentrated.
- the isolated compound (4-w) can also be further purified by recrystallization and the like.
- the compound (4-y) can be produced by reacting the compound (4-w) and a compound (23) .
- R 1 , R 2 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , A 1 and A 2 represent the same meaning as described above.
- the reaction is carried out usually in the presence of a solvent, and u.sually carried out in the presence of 25. a base.
- a solvent such as THF, ethylene glycol dimethyl ether, 1,4-dioxane and the like, aromatic hydrocarbons such as toluene, xylene and the like, and mixtures thereof.
- Examples of the base to be used in the reaction include inorganic bases such as sodium carbonate, potassium carbonate and the like or organic bases such as triethylamine, diisopropylethylamine and the like, nitrogen-containing aromatic compounds such as pyridine, quinoline and the like.
- the compound (23) is used usually in a proportion of 1 mol to excess quantity and the base is used usually in a proportion of 1 mol to excess quantity with respect to 1 mol of the compound (4-w) .
- reaction can also be carried out without using the above-described base.
- the reaction temperature of the reaction is usually in the range of 0 to 100°C, and the reaction time is usually in the range of 0.1 to 24 hours.
- the compound (4-y) can be isolated by subjecting post treatment operations such as the reaction mixture being extracted with an organic solvent, the organic layer being dried and concentrated.
- the isolated compound (4-y) can also be further purified by chromatography, recrystallization and the like.
- the compound (4-z) can be produced by reacting the compound (26) and hydrogen in the presence of a transition metal catalyst.
- the reaction is carried out usually under a hydrogen atmosphere and in the presence of a solvent.
- solvent to be used in the reaction examples include aliphatic hydrocarbons such as hexane, heptane, octane and the like, alcohols such as methanol, ethanol and the like, esters such as ethyl acetate, butyl acetate and the like, and mixtures thereof.
- transition metal catalyst to be used in the reaction examples include palladium catalysts such as palladium charcoal and the like.
- the transition metal catalyst is used usually in a proportion of 0.01 mol to 0.1 mol with respect to 1 mol of the compound (26) .
- the reaction temperature of the reaction is usually in the range of 0 to 100°C, and the reaction time is usually in the range of 0.1 to 24 hours.
- the compound (4-z) can be isolated by subjecting post treatment operations such as the reaction mixture being filtrated to remove the transition metal catalyst, the filtrate being extracted with an organic solvent, the organic layer being dried and concentrated.
- the isolated compound (4- z) can also be further purified by chromatography, recrystallization and the like.
- the compound (4-d) can be produced by reacting the compound (4-c) and an oxidation agent .
- the reaction is carried out usually in the presence of a solvent.
- solvent to be used in the reaction examples include aliphatic halogenated hydrocarbons such as dichloromethane, chloroform and the like; acetic acid, water, and mixtures thereof.
- Examples of the oxidation agent to be used in the reaction include carboxylic acid peroxides such as 3- chloroperbenzoic acid and the like.
- the oxidation agent is used usually in a proportion of 1 to 3 mol with respect to 1 mol of the compound (4- c) .
- the reaction temperature of the reaction is usually in the range of -20 to 30°C, and the reaction time is usually in the range of 0.1 to 12 hours.
- the compound (4-d) can be isolated by subjecting post treatment operations such as the reaction mixture being extracted with an organic solvent, the organic layer being washed with an aqueous solution of a reducing agent (for example, sodium sulfite, sodium thiosulfate) and an aqueous solution of a base (for example, sodium hydrogen carbonate) if necessary, then being dried and concentrated.
- a reducing agent for example, sodium sulfite, sodium thiosulfate
- a base for example, sodium hydrogen carbonate
- the intermediate according to the present invention can be produced, for example, by the following method. (Intermediate Production Method 1)
- a compound (2) can be produced by a method shown in the following scheme.
- a compound (M2) can be produced by reacting a compound (Ml) in the presence of a nitrating agent.
- the reaction is carried out usually in the presence of a solvent.
- a solvent examples include aliphatic halogenated hydrocarbons such as chloroform and the like, acetic acid, concentrated sulfuric acid, concentrated nitric acid, water and mixtures thereof.
- the nitrating agent to be used in the ' reaction examples include concentrated nitric acid and the like.
- the nitrating agent is used usually in a proportion of 1 to 3 mol with respect to 1 mol of the compound (Ml) .
- the reaction temperature of the reaction is usually in the range of -10 to 100 0 C, and the reaction time thereof is usually in the range of 0.1 to 24 hours.
- the compound (M2) can be isolated by subjecting post treatment operations such as the reaction mixture being poured into water, the mixture being extracted with an organic solvent, then the organic layer being dried and concentrated.
- the isolated compound (M2) can also be further purified by chromatography, recrystallization and the like.
- a compound (2) can be produced by reacting the compound (M2) and hydrogen in the presence of a hydrogenation catalyst.
- the reaction is carried out usually under a hydrogen atmosphere of 1 to 100 atm, usually in the presence of a solvent .
- solvent to be used in the reaction examples include ethers such as THF, 1,4-dioxane and the like, esters such as ethyl acetate, butyl acetate and the like, alcohols such as methanol, ethanol and the like, water and mixtures thereof.
- Examples of the hydrogenation catalyst to be used in the reaction include transition metal compounds such as palladium charcoal, palladium hydroxide, raney nickel, platinum oxide and the like.
- Hydrogen is used usually in a proportion of 3 mol and the hydrogenation catalyst is used usually in a proportion of 0.001 to 0.5 mol with respect to 1 mol of the compound (Ml) .
- reaction can also be carried out with addition of an acid (base or the like), if necessary.
- the reaction temperature of the reaction is usually in the range of -20 to 100°C, and the reaction time thereof is usually in the range of 0.1 to 24 hours.
- the compound (2) can be isolated by subjecting post treatment operations such as the reaction mixture being filtrated to remove the transition metal catalyst, the filtrate being extracted with an organic solvent, the organic layer being dried and concentrated.
- the isolated compound (2) can also be further purified by chromatography, recrystallization and the like. Next, specific examples of the present active compound are shown below.
- Me represents a methyl group
- Et represents an ethyl group
- Pr represents a propyl group
- iPr represents an isopropyl group
- tBu represents a tert-butyl group
- Ph represents a phenyl group
- 2-Py represents a 2-pyridyl group
- 3-Py represents a 3-pyridyl group
- 4-Py represents a 4- pyridyl group
- 1-Tz represents a 1, 2, 4-triazol-l-yl group
- 1-Pz represents a pyrazol-1-yl group
- CyPr represents a cyclopropyl group.
- the composition of the present invention contains the present active compound and an inert carrier-.
- the composition of the present invention is a formulation obtained by mixing the present active compound and an inert carrier such as a solid carrier, a liquid carrier and a gaseous carrier, and further adding a surfactant and other adjuvant for formulation, if necessary.
- the formulation includes, for example, an emulsion, an oil solution, a powder, a granule, a wettable powder, a flowable formulation, a microcapsule, an aerosol, a smoking agent, a poison bait, and a resin formulation.
- the present active compound is usually contained in an amount of 0.01% to 95% by weight.
- the solid carrier used for formulation includes, for example, a fine power and a granule of clays (e.g., kaolin clay, diatomite, bentonite, Fubasami clay, and acid clay) , synthetic hydrated silicon oxide, talc, ceramic, other inorganic minerals (e.g., sericite, quartz, sulfur, activated carbon, calcium carbonate, hydrated silica) or chemical fertilizers (e.g., ammonium sulfate, ammonium phosphate, ammonium nitrate, urea, and ammonium chloride) .
- clays e.g., kaolin clay, diatomite, bentonite, Fubasami clay, and acid clay
- synthetic hydrated silicon oxide talc
- ceramic other inorganic minerals
- other inorganic minerals e.g., sericite, quartz, sulfur, activated carbon, calcium carbonate, hydrated silica
- chemical fertilizers e.g., ammonium sulfate,
- the liquid carrier includes, for example, water, alcohols (e.g., methanol, ethanol, isopropyl alcohol, butanol, hexanol, benzyl alcohol, ethylene glycol, propylene glycol, phenoxyethanol) , ketones (e.g., acetone, methyl ethyl ketone, cyclohexanone) , aromatic hydrocarbons (e.g., toluene, xylene, ethylbenzene, dodecylbenzene, phenylxylylethane, methylnaphthalene) , aliphatic hydrocarbons (e.g., hexane, cyclohexane, kerosine, light oil), esters (e.g., ethyl acetate, butyl acetate, isopropyl mylistate, ethyl oleate, diisopropyl adipate, diisobut
- the gaseous carrier includes, for example, fluorocarbo ns, butane gas, liquefied petroleum gas (LPG), dimethyl eth er, and carbon dioxide.
- the surfactant includes, for example, nonionic surfactant, such as polyoxyethylene alkyl ether, polyoxyethylene alkylaryl ether, polyethyleneglycol fatty acid ester; and anionic surfactant, such as alkylsulfonic acid salts, alkylbenzenesulfonic acid salts, and alkylsurfic acid salts.
- nonionic surfactant such as polyoxyethylene alkyl ether, polyoxyethylene alkylaryl ether, polyethyleneglycol fatty acid ester
- anionic surfactant such as alkylsulfonic acid salts, alkylbenzenesulfonic acid salts, and alkylsurfic acid salts.
- the other adjuvant for formulation includes, for example, binders, dispersants, colorants and stabilizers, and specifically for example, casein, gelatin, polysaccharides (e.g., starch, gum arable, cellulose derivatives, alginic acid) , lignin derivatives, synthetic water-soluble polymers (e.g., polyvinyl alcohol, polyvinylpyrrolidone, polyacrylic acid), PAP (isopropyl acid phosphate), BHT (2,6-di-t- butyl-4-methylphenol) , BHA (a mixture of 2-t-butyl-4- methoxyphenol and 3-t-butyl-4-methoxyphenol) .
- binders e.g., dispersants, colorants and stabilizers
- casein e.g., gelatin, polysaccharides (e.g., starch, gum arable, cellulose derivatives, alginic acid) , lignin derivatives, synthetic water-soluble poly
- the method for controlling noxious arthropod of the present invention is applying an effective amount of the present active compound to a noxious arthropod directly and/or habitats of a noxious arthropod (e.g., plant, soil, indoor, and in-body of animals) .
- the present active compound is usually used as the composition of the present invention for the method for controlling a noxious arthropod of the present invention.
- the application amount is usually 1 to 10,000 g as the present active compound per 10,000 m 2 .
- the composition of the present invention is a formulation of emulsions, wettable powders or flowables, they are usually applied after a dilution with water to have an active ingredient concentration of 0.01 to 10,000 ppm.
- the composition of the present invention is a formulation of granules or powders, they are usually applied as such.
- formulations and the dilute aqueous solution of the formulation may be sprayed directly to the plant to be protected from a noxious arthropod, and may be applied to the soil to control the a noxious arthropod living in a soil.
- the resin formulations of sheets or strip form can be applied by a method such as winding around plants, stretching in the vicinity of plants and laying on the soil surface at the plant bottom.
- the application amount is usually 0.01 to 1,000 mg as the present active compound per 1 m 2 in case of application for plane surface, and 0.01 to 500 mg as the present active compound per 1 m 3 in case of application for space.
- the composition of the present invention is a formulation of emulsions, wettable powders or flowables, they are usually applied after a dilution with water to have an active ingredient concentration of 0.1 to 1,000 ppm.
- the composition of the present invention is a formulation of oil solutions, aerosols, smoking agents and poison baits, they are usually applied as such.
- the composition of the present invention can be a admixture with or together with other insecticides, acaricides, nematocides, fungicides, plant growth regulators, herbicides, and synergists.
- Organic phosphorus compounds Acephate, Aluminum phosphide, butathiofos, cadusafos, chlorethoxyfos, chlorfenvinphos , chlorpyrifos , chlorpyrifos-methyl, cyanophos : CYAP, diazinon, DCIP (dichlorodiisopropyl ether) , dichlofenthion: ECP, dichlorvos (DDVP) , dimethoate, dimethylvinphos , disulfoton, EPN, ethion, ethoprophos, etrimfos, fenthion: MPP, fenitrothion : MEP, fosthiazate, formothion, hydrogen phosphide, isofenphos, isoxathion, malathion, mesulfenfos, methidathion : DMTP, monocrotophos, naled: BRP, oxydeprof
- Carbamate compounds Alanycarb, bendiocarb, benfuracarb, BPMC, carbaryl, carbofuran, carbosulfan, cloethocarb, ethiofencarb, fenobucarb, fenothiocarb, fenoxycarb, furathiocarb, isoprocarb: MIPC, metolcarb, methomyl, methiocarb, NAC, oxamyl, pirimicarb, propoxur: PHC, XMC, thiodicarb, xylylcarb, aldicarb;
- Cartap bensultap, thiocyclam, monosultap, bisultap;
- Chlorfluazuron bistrifluron, diafenthiuron, diflubenzuron, fluazuron, flucycloxuron, flufenoxuron, hexaflumuron, lufenuron, novaluron, noviflumuron, teflubenzuron, triflumuron, triazuron;
- insecticidal active ingredients Machine oil, nicotine-sulfate; avermectin-B, bromopropylate, buprofezin, chlorphenapyr, cyantraniliprole, cyromazine, D-D (1, 3-Dichloropropene, emamectin-benzoate, fenazaquin, flupyrazofos , hydroprene, methoprene, indoxacarb, metoxadiazone, milbemycin-A, pymetrozine, pyridalyl, pyriproxyfen, spinosad,.
- R 100 represents chlorine, bromine or a trifluoromethyl group
- R 200 represents chlorine, bromine or a methyl group
- R 300 represents chlorine, bromine or a cyano group and, any compound represented by the following formula
- R 1000 represent s chlorine , bromine or iodide .
- Active ingredient s of the acardides Acequinocyl, amitraz, benzoximate, bifenaate, bromopropylate, chinomethionat , chlorobenzilate, CPCBS (chlorfenson) , clofentezine, cyflumetofen, dicofol, etoxazole, fenbutatin oxide, fenothiocarb, fenpyroximate, fluacrypyrim, fluproxyfen, hexythiazox, propargite: BPPS, polynactins, pyridaben, Pyrimidifen, tebufenpyrad, tetradifon, spirodiclofen, spiromesifen, spirotetramat, amidoflumet , cyenopyrafen .
- DCIP fosthiazate
- levamisol methyisothiocyanate
- morantel tartarate imicyafos.
- Active ingredients of the fungicides include propiconazole, prothioconazole, triadimenol, prochloraz, penconazole, tebuconazole, flusilazole, diniconazole, bromuconazole, epoxiconazole, difenoconazole, cyproconazole, metconazole, triflumizole, tetraconazole, myclobutanil, fenbuconazole, hexaconazole, fluquinconazole, triticonazole, bitertanol, imazalil, and flutriafol;
- Azole fungicidal compounds such as propiconazole, prothioconazole, triadimenol, prochloraz, penconazole, tebuconazole, flusilazole, diniconazole, bromuconazole, epoxiconazole, difenoconazo
- Cyclic amine fungicidal compouds such as fenpropimorph, tridemorph, and fenpropidin;
- Benzimidazole fungicidal compounds such as carbendezim, benomyl, thiabendazole, and thiophanate- methyl;
- 2,3,6-TBA dicamba, clopyralid, picloram, aminopyralid, quinclorac, and quinmerac.
- Triazine herbicidal compounds atrazine, ametoryn, cyanazine, simazine, propazine, simetryn, dimethametryn, prometryn, metribuzin, indaziflam, and triaziflam.
- Organic phosphorus herbicidal compounds amiprofos-methyl, butamifos, bensulide, piperophos, anilofos, glyphosate, glufosinate, and bialaphos.
- Chloroacetanilide herbicidal compounds acetochlor, alachlor, butachlor, dimethenamid, propachlor, metazachlor, metolachlor, pretilachlor , thenylchlor, and pethoxamid.
- Diphenylether herbicidal compounds acifluorfen-sodium, bifenox, oxyfluorfen, lactofen, fomesafen, chlomethoxynil, and aclonifen.
- Cyclic imide herbicidal compounds oxadiazon, cinidon-ethyl, carfentrazone-ethyl , surfentrazone, flumiclorac-pentyl, flumioxazin, pyraflufen-ethyl, oxadiargyl, pentoxazone, fluthiacet- methyl, butafenacil, benzfendizone, and saflufenacil .
- Triketone herbicidal compounds isoxaflutole, benzobicyclon, sulcotrione, mesotrione, tembotrione, and tefuryltrione .
- Aryloxyphenoxypropionic acid herbicidal compounds clodinafop-propargyl, cyhalofop-butyl , diclofop- methyl, fenoxaprop-ethyl, fluazifop-butyl, haloxyfop- methyl, and quizalofop-ethyl and metamifol.
- Trioneoxime herbicidal compounds alloxydim-sodium, sethoxydim, butroxydim, clethodim, cloproxydim, cycloxydim, tepraloxydim, tralkoxydim, and profoxydim.
- Sulfonylurea herbicidal compounds chlorsulfuron, sulfometuron-methyl, metsulfuron- methyl, chlorimuron-ethyl, tribenuron-methyl, triasulfuron, bensulfuron-methyl , thifensulfuron-methyl, pyrazosulfuron-ethyl, primi sulfuron-methyl, nicosulfuron, amidosulfuron, cinosulfuron, imazosulfuron, rimsulfuron, halosulfuron-methyl, prosulfuron, ethametsulfuron-methyl, triflusulfuron- methyl, flazasulfuron, cyclosulfamuron, flupyrsulfuron, sulfosulfuron, azimsulfuron, ethoxysulfuron, oxasulfuron, iodosulfuron-methyl-sodium, foramsulfuron
- Active ingredients of the synergists Piperonyl butoxide, sesamex, sulfoxide, N- (2- ethylhexyl) -8, 9, 10-trinorborn-5-ene-2 , 3-dicarboxyimide (MGK 264), N-declyimidazole, WARF-antiresistant , TBPT, TPP, IBP, PSCP, methyl iodide (CH3I), t-phenylbutenone, diethylmaleate, DMC, FDMC, ETP, and ETN.
- Noxious arthropod against which the present active compound has an activity include, for example, noxious insects and noxious acarines. Specific examples of these, noxious arthropods include the following.
- Planthoppers such as small brown planthopper (Laodelphax striatellus ) , brown rice planthopper (Nilaparvata lugens), and white-backed rice planthopper (Sogatella furcifera) ; leafhoppers (Deltocephalidae) such as green rice leafhopper (Nephotettix cincticeps), green rice leafhopper (Nephotettix virescens) , and tea green leafhopper (Empoasca onukii) ; aphids (Aphididae) such as cotton aphid (Aphis gossypii), green peach aphid (Myzus persicae) , cabbage aphid (Brevicoryne brassicae) , piraea aphid (Aphis spiraecola) , potato aphid (Macrosiphum euphorbiae), foxglove aphid (Aphididae) such
- Pyralid moths such as rice stem borer (Chilo suppressalis) , yellow rice borer (Tryporyza incertulas), rice leafroller (Cnaphalocrocis medinalis), cotton leafroller (Notarcha derogata) , Indian meal moth (Plodia interpunctella) , oriental corn borer (Ostrinia furnacalis) , cabbage webworm (Hellula undalis), and bluegrass webworm (Pediasia teterrellus) ; owlet moths (Noctuidae) such as common cutworm (Spodoptera litura) , beet armyworm (Spodoptera exigua) , armyworm (Pseudaletia separata), cabbage armyworm (Mamestra brassicae) , black cutworm (Agrotis ipsilon) , beet semi-looper (Plusia nigrisigna ) "
- Tricidae such as Adoxophyes spp., oriental fruit moth (Grapholita molesta) , soybean pod borer (Leguminivora glycinivorella) , azuki bean podworm (Matsumuraeses azukivora) , summer fruit tortrix (Adoxophyes orana fasciata) , smaller tea tortrix
- Thysanoptera
- Thrips such as yellow citrus thrips
- Culices such as common mosquito (Culex pipiens pallens) , Cluex tritaeniorhynchus, and Cluex quinquefasciatus ; Aedes spp . such as yellow fever mosquito (Aedes aegypti), and Asian tiger mosquito (Aedes albopictus); Anopheles spp.
- flies such as Anopheles sinensis; chironomids (Chironomidae) ; house flies (Muscidae) such as Musca domestica, and Muscina stabulans; blow flies (Calliphoridae) ; flesh flies ( Sarcophagidae) ; little house flies (Fanniidae); anthomyiid flies (Anthomyiidae) such as seedcorn fly (Delia platura) , and onion fly (Delia antiqua) ; leafminer flies (Agromyzidae) such as rice leafminer (Agromyza oryzae), little rice leafminer (Hydrellia griseola) , tomato leafminer (Liriomyza sativae) , legume leafminer (Liriomyza trifolii), and garden pea leafminer (Chromatomyia horticola) ; g
- Tephritidae such as melon fly (Dacus cucurbitae) , and Meditteranean fruit fly (Ceratitis capitata) ; Drosophilidae; humpbacked flies (Phoridae) such as Megaselia spiracularis; moth flies ( Psychodidae) such as Clogmia albipunctata; Simuliidae; Tabanidae such as horsefly (Tabanus trigonus); stable flies.
- Corn root worms such as Western corn root worm (Diabrotica virgifera virgifera) , and Sourthern corn root worm (Diabrotica undecimpunctata howardi); scarabs ( Scarabaeidae) such as cupreous chafer (Anomala cuprea) , soybean beetle (Anomala rufocuprea) , and Japanese beetle (Popillia japonica); weevils such as maize weevil (Sitophilus zeamais) , rice water weevil (Lissorhoptrus oryzophilus ) , azuki bean weevil (Callosobruchus chinensis), rice curculio (Echinocnemus squameus) , boll weevil (Anthonomus grandis) , and hunting billbug (Sphenophorus venatus); darkling beetles (Tenebrionid
- Asiatic locust (Locusta migratoria) , African mole cricket (Gryllotalpa africana) , rice grasshopper (Oxya yezoensis), rice grasshopper (Oxya japonica), Gryllidae.
- Hymenoptera
- Ants such as pharaoh ant (Monomorium pharaosis), negro ant (Formica fusca japonica), black house ant (Ochetellus glaber) , Pristomyrmex ponnes, Pheidole noda, leaf-cutting ant (Acromyrmex spp.), and fire ant (Solenopsis spp.); hornets (Vespidae); bethylid wasps (Betylidae); sawflies (Tenthredinidae) such as cabbage sawfly (Athalia rosae) , and Athalia japonica.
- Blattodea German cockroach (Blattella germanica) , smokybrown cockroach (Periplaneta fuliginosa) , American cockroach (Periplaneta americana) , Periplaneta brunnea, oriental cockroach (Blatta orientalis ) ;
- Spider mites such as two-spotted spider mite (Tetranychus urticae) , Kanzawa spider mite (Tetranychus kanzawai) , citrus red mite (Panonychus citri), European red mite (Panonychus ulmi), and
- Oligonychus spp . eriophyid mites (Eriophyidae) such as pink citrus rust mite (Aculops pelekassi), Phyllocoptruta citri, tomato rust mite (Aculops lycopersici) , purple tea mite (Calacarus carinatus) , pink tea rust mite (Acaphylla theavagran) , Eriophyes chibaensis, and apple rust mite (Aculus Louendali) ; tarosonemid mites (Tarsonemidae) such as broad mite ( Polyphagotarsonemus latus) ; false spider mites (Tenuipalpidae-) such as Brevipalpus phoenicis; Tuckerellidae; ticks (Ixodidae) such as Haemaphysalis longicornis, Haemaphysalis flava, Dermacentor taiwanicus,
- hemiptera noxious insects are mentioned as a noxious arthropod showing a high effect of the compound of the present invention.
- composition of the present invention could be used in farmlands on which "crops" shown below are cultivated.
- Agricultural crops corn, rice, wheat, barley, rye, oat, sorghum, cotton, soybean, peanut, sarrazin, sugar beet, rapeseed, sunflower, sugar cane, tobacco;
- Vegetables Solanaceae vegetables (eggplant, tomato, green pepper, hot pepper, and potato) , Cucurbitaceae vegetables (cucumber, pumpkin, zucchini, watermelon, and melon ) , Cruciferae vegetables (Japanese radish, turnip, horseradish, kohlrabi, Chinese cabbage, cabbage, brown mustard, broccoli, and cauliflower), Compositae vegetables (burdock, garland chrysanthemum, artichoke, and lettuce) , Liliaceae vegetables (Welsh onion, onion, garlic, and asparagus ) , Umbelliferae vegetables (carrot, parsley, celery, and parsnip) , Chenopodiaceae vegetables (spinach, and Swiss chard) , Labiatae vegetables (Japanese basil, mint, and basil), strawberry, sweat potato, yam, aroid;
- Fruit trees pomaceous fruits (apple, common pear, Japanese pear, Chinese quince, and quince) , stone fleshy fruits (peach, plum, nectarine, Japanese plum, cherry, apricot, and prune) , citrus plants (Satsuma mandarin, orange, lemon, lime, and grapefruit), nuts (chestnut, walnut, hazel nut, almond, pistachio, cashew nut, and macadamia nut) , berry fruits (blueberry, cranberry, blackberry, and raspberry) , grape, persimmon, olive, loquat, banana, coffee, date, coconut palm, and oil palm;
- Trees other fruit trees tea, mulberry, flowering trees (azalea, japonica, hydrangea, sasanqua, lllicium anisatum, cherry tree, tulip poplar, crepe myetle, and orange osmanthus) , street trees (ash tree, birch, dogwood, eucalyptus, ginkgo, lilac, maple tree, oak, poplar, cercis, Chinese sweet gum, plane tree, zelkova, Japanese arborvitae, fir tree, Japanese hemlock, needle juniper, pine, spruce, yew, elm, and horse-chestnut) , sweet viburnum, Podocarpus macrophyllus , Japanese cedar, Japanese cypress, croton, spindle tree, Chainese howthorn .
- flowers (rose, carnation, chrysanthemum, Eustoma grandiflorum Shinners (prairie gentian) , gypsophila, gerbera, pot marigold, salvia, petunia, verbena, tulip, aster, gentian, lily, pansy, cyclamen, orchid, lily of the valley, lavender, stock, ornamental kale, primula, poinsttia, gladiolus, cattleya, daisy, verbena, cymbidium, begonia), biofuel plants (Jatropha, curcas, safflower, Camelina alyssum, switchgrass, miscanthus, reed canary grass, Arundo donax, kenaf, cassava, willow, algae), foliage plant.
- the "crops” include genetically modified crops.
- Production Example 2 l-methyl-2- (pyridin-4-yl) -6-trifIuoromethyl-1H- benzimidazole (hereinafter, referred to as present active compound 2) was obtained according to the method described in Production Example 1, using N2-methyl-4- trifluoromethylbenzene-1, 2-diamine instead of 1,1,3,3- tetrafluoro-N-methyl-1, 3-dihydroisobenzofuran-5, 6- diamine .
- Production Example 3 l-methyl-2- (pyridin-4-yl) -5-trifIuoror ⁇ ethyl-1H- benzimidazole (hereinafter, referred to as present active compound 3) was obtained according to the method described in Production Example 1, using Nl-methyl-4- trifluoromethylbenzene-1 , 2-diamine instead of 1,1,3,3- tetrafluoro-N-methyl-1, 3-dihydroisobenzofuran-5, 6- diar ⁇ ine .
- Production Example 11 2- 3-isopropoxypyridin-4-yl ) -l-methyl-5- trifluoromethyl-lH-benzimidazole (hereinafter, referred to as present active compound 11) was obtained according to the method described in Production Example 10, using 2-propanol instead of 1-propanol.
- Production Example 13 1-methyl-2- [3- (2, 2 , 2-trifluoroethoxy) -pyridin-4-yl] - 5-trifluoromethyl-lH-benzimidazole (hereinafter, referred to as present active compound 13) was obtained according to the method described in Production Example 10, using 2 , 2 , 2-trifluoroethanol instead of 1-propanol.
- present active compound 14 2- [3- (2, 2-difluoroethoxy) -pyridin-4-yl] -l-methyl-5- trifluoromethyl-lH-benzimidazole (hereinafter, referred to as present active compound 14) was obtained according to the method described in Production Example 10, using 2, 2-difluoroethanol instead of 1-propanol.
- Production Example 17 l-methyl-2- (3-methylthiopyridin-4-yl) -5- trifluoromethyl-lH-benzimidazole (hereinafter, referred to as present active compound 17) was obtained according to the method described in Production Example 8, using methyl iuercaptan sodium salt instead of sodium methoxide .
- Production Example 21 2- (3-ethanesulfinylpyridin-4-yl) -l-methyl-5- trifluoromethyl-lH-benzimidazole (hereinafter, referred to as present active compound 21) was obtained according to the method described in Production Example 18, using 2- ( 3-ethylthiopyridin-4-yl) -l-methyl-5-trifluoromethyl- lH-benzimidazole instead of l-methyl-2- ( 3- methylthiopyridin-4-yl ) -5-trifIuoromethyl-1H- benzimidazole.
- Production Example 26 2- (2-fluoropyridin-4-yl) -l-methyl-5-trifluoromethyl- lH-benzimidazole (hereinafter, referred to as present active compound 26) was obtained according to the method described in Production Example 4, using 2- fluoropyridine-4-carbaldehyde instead of 3- fluoropyridine-4-carbaldehyde .
- Production Example 27 2- (2-chloropyridin-4-yl) -l-methyl-5-trifluoromethyl- lH-benzimidazole (hereinafter, referred to as present active compound 27) was obtained according to the method described in Production Example 4, using 2- chloropyridine-4-carbaldehyde instead of 3- fluoropyridine-4-carbaldehyde .
- Production Example 28 l-cyclopropyl-2- ( 3-fluoropyridin-4-yl ) -5- trifluoromethyl-lH-benzimidazole (hereinafter, referred to as present active compound 28) was obtained according to the method described in Production Example 6, using N-l-cyclopropyl-4-trifluoromethylbenzene-1 , 2 -diamine instead of N-l-methyl-4-trifluoromethylbenzene-1, 2- diamine and using 3-fluoroisonicotinic acid instead of 3-methylisonicotinic acid.
- Production Example 29 l-cyclopropyl-2- ( 3-methylthiopyridine-4-yl) -5- trifluoromethyl-lH-benzimidazole (hereinafter, referred to as present active compound 29) was obtained according to the method described in Production Example 4, using l-cyclopropyl-2- (3-fluoropyridin-4-yl) -5- trifluoromethyl-lH-benzimidazole instead of 2- (3- fluoropyridin-4-yl) -l-methyl-5-trifIuoromethyl-1H- benzimidazole and using methyl mercaptan sodium salt instead of sodium methoxide.
- Production Example 32 l-cyclopropyl-2- ( 3-ethylthiopyridin-4-yl) -5- trifluoromethyl-lH-benzimidazole (hereinafter, referred to as present active compound 32) was obtained according to the method described in Production Example 4, using l-cyclopropyl-2- ( 3-fluoropyridin-4-yl ) -5- trifluoromethyl-lH-benzimidazole instead of 2- (3- fluoropyridin-4-yl) -l-methyl-5-trifIuoromethyl-1H- benzimidazole and using ethyl mercaptan sodium salt instead of sodium methoxide.
- Production Example 35 3-methyl-2- (pyridin-4-yl) -6-trifluoromethyl-3H- imidazo [ 4 , 5-b] pyridine (hereinafter, referred to as present active compound 35) was -obtained according to the method described in Production Example 1, using N2- methyl-5-trifluoromethylpyridine-2, 3-diamine instead of 1,1,3, 3-tetrafluoro-N-methyl-1 , 3-dihydroisobenzofuran- 5, ⁇ -diamine .
- Production Example 36 A mixture of 5.51 g of N2-methyl-5- trifIuoromethylpyridine2 , 3-diamine, 6.10 g of 3- fluoroisonicotinic acid, 8.28 g of WSC and 58 ml of pyridine was stirred for 3 hours while heating at 12O 0 C. After cooling down to room temperature, water was poured, and the mixture was extracted three times with ethyl acetate. The combined organic layers were dried over magnesium sulfate, then, concentrated under reduced pressure.
- Production Example 39 2- (3-ethoxypyridin-4-yl ) -3-methyl-6-trifluoromethyl- 3H-imidazo [4, 5-b] pyridine (hereinafter, referred to as present active compound 39) was obtained according to the method described in Production Example 8, using 2- ( 3-fluoropyridin-4-yl) -3-methyl-6-trifluoromethyl-3H- imidazo [4, 5-b] pyridine instead of 2- ( 3-fluoropyridin-4- yl) -l-methyl-5-trifluoromethyl-lH-benzimidazole and using sodium ethoxide instead of sodium methoxide.
- 3-methyl-2- (3-propoxypyridin-4-yl) -6- trifluoroxnethyl-3H-imidazo [ 4 , 5-b] pyridine (hereinafter, referred to as present active compound 40) was obtained according to the method described in Production Example 10, using 2- (3-fluoropyridin-4-yl) -3-methyl- ⁇ - trifluoromethyl-3H-imidazo [4, 5-b]pyridine instead of 2- (3-fluoropyridin-4-yl) -l-methyl-5-trifIuoromethyl-1H- benzimidazole.
- Production Example 41 2- (3-isopropoxypyridin-4-yl) -3-methyl-6- trifluoromethyl-3H-imidazo [4, 5-b] pyridine (hereinafter, referred to as present active compound 41) was obtained according to the method described in Production Example 10, using 2- (3-fluoropyridin-4-yl ) -3-methyl- ⁇ - trifluoromethyl-3H-imidazo [4, 5-b] pyridine instead of 2- ( 3-fluoropyridin-4-yl) -l-methyl-5-trifIuoromethyl-1H- benzimidazole and using 2-propanol instead of 1- propanol .
- present active compound 43 (hereinafter, referred to as present active compound 43) was obtained according to the method described in Production Example 10, using 2- (3-fluoropyridin-4-yl) -3- methyl- 6-trifluoromethyl-3H-imidazo [ 4 , 5-b] pyridine instead of 2- ( 3-fluoropyridin-4-yl) -1 -methyl- 5- trifluoromethyl-lH-benzimidazole and using 2,2,2- trifluoroethanol instead of 1-propanol.
- Production Example 48 3-methyl-2- ( 3-methylthiopyridin-4-yl ) -6- trifluoromethyl-3H-imidazo [4, 5-b] pyridine (hereinafter, referred to as present active compound 48) was obtained according to the method described in Production Example 8, using 2- ( 3-fluoropyridin-4-yl) -3-methyl-6- trifluoromethyl-3H-imidazo [ 4 , 5-b] pyridine instead of 2- ( 3-fluoropyridin-4-yl) -l-methyl-5-trifIuoromethyl-1H- benzimidazole and using methyl mercaptan sodium salt instead of sodium methoxide.
- 3-methyl-2- ( l-oxypyridin-4-yl) -6-trifluoromethyl-3H- imidazo [4 , 5-b] pyridine (hereinafter, referred to as present active compound 55) was obtained according to the method described in Production Example 24, using 3- methyl-2- (pyridin-4-yl) -6-trifluoromethyl-3H- imidazo [ 4 , 5-b] pyridine instead of l-methyl-2- (pyridin-4- yl) -5-trifluoromethyl-lH-benzimidazole .
- present active compound 101 (hereinafter, referred to as present active compound 101) .
- present active compound 102 (hereinafter, referred to as present active compound 102) was obtained according to the method described in Production Example 101, using 0.09 g of 2-methyl-2- propanethiol instead of 2-propanethiol .
- Present active compound 102 was obtained according to the method described in Production Example 101, using 0.09 g of 2-methyl-2- propanethiol instead of 2-propanethiol .
- present active compound 103 (hereinafter, referred to as present active compound 103) was obtained according to the method described in Production Example 101, using 0.08 g of 1-propanethiol instead of 2-propanethiol .
- Present active compound 103
- present active compound (hereinafter, referred to as present active compound
- present active compound 105 (hereinafter, referred to as present active compound 105) was obtained according to the method described in Production Example 101, using 0.09 g of 1-butanethiol instead of 2-propanethiol .
- Present active compound 105) was obtained according to the method described in Production Example 101, using 0.09 g of 1-butanethiol instead of 2-propanethiol .
- present active compound (hereinafter, referred to as present active compound
- present active compound 107 (hereinafter, referred to as present active compound 107) .
- present active compound 108 (hereinafter, referred to as present active compound 108) was obtained according to the method described in Production Example 107, using 0.09 g of 1-butanethiol instead of 2-methyl-2-propanethiol .
- present active compound 110 (hereinafter, referred to as present active compound 110) .
- present active compound (hereinafter, referred to as present active compound
- present active compound 112 (hereinafter, referred to as present active compound 112) was obtained according to the method described in Production Example 104, using 0.07 g of 2-methyl-l- propanol instead of 2- (methylthio) ethanol .
- Present active compound 112 was obtained according to the method described in Production Example 104, using 0.07 g of 2-methyl-l- propanol instead of 2- (methylthio) ethanol .
- present active compound 113 (hereinafter, referred to as present active compound 113) .
- present active compound 114 (hereinafter, referred to as present active compound 114) .
- present active compound 115 (hereinafter, referred to as present active compound 115) .
- present active compound 116 (hereinafter, referred to as present active compound 116) .
- present active compound 116 (hereinafter, referred to as present active compound 116) .
- the present active compound 116 (0.15 g) was suspended in 2 ml of THF, and 0.1 g of a 40% methylamine aqueous solution was added, and the mixture was stirred for 2 hours at room temperature.
- the reaction mixture was concentrated under reduced pressure, and the resultant residue was subjected to silica gel column chromatography, to obtain 95 mg of a compound of formula (117) :
- present active compound 117 (hereinafter, referred to as present active compound 117) .
- present active compound 118 (hereinafter, referred to as present active compound 118) .
- present active compound (hereinafter, referred to as present active compound
- the present active compound 115 (0.15 g) was suspended in 1 ml of THF, and 0.05 g of 60% sodium hydride (oily) was added under ice cool, and the mixture was stirred for 10 minutes. To the mixture was added 0.63 g of S- (trifluoromethyl) dibenzothiophenium trifluoromethane sulfonate and 1 ml of THF, and the mixture was stirred over night and day at room temperature. Into the reaction mixture was poured water and the mixture was extracted with chloroform, then, the organic layer was washed sequentially with a saturated sodium hydrogen carbonate aqueous solution and water, dried over sodium sulfate, then, concentrated under reduced pressure. The residue was subjected to silica gel preparative thin layer chromatography, to obtain 69 mg of a compound of formula (121) :
- present active compound 121 (hereinafter, referred to as present active compound 121) .
- the present active compound 120 (0.15 g) was suspended in 2 ml of THF, and 0.03 g of 60% sodium hydride (oily) was added, and the mixture was stirred for 15 minutes. To this mixture was added 0.27 g of S- (trifluoromethyl) dibenzothiophenium-3-sulfonate, and the mixture was stirred for 8 hours at room temperature. Into the reaction mixture was poured water and the mixture was extracted with chloroform, then, the organic layer was washed sequentially with a saturated sodium hydrogen carbonate aqueous solution and water, dried over sodium sulfate, then, concentrated under reduced pressure. The residue was subjected to silica gel preparative thin layer chromatography, to obtain 96 mg of a compound of formula (122) :
- present active compound 122 (hereinafter, referred to as present active compound 122) .
- the present active compound 115 (0.15 g) was suspended in 2 ml of DMF, and 0.04 g of 60% sodium hydride (oily) was added, and the mixture was stirred for 5 minutes at about 5O 0 C To this mixture was added 0.18 g of 1 , 1-difluoro-2-iodoethane, and the mixture was stirred for 3 hours.
- the reaction mixture was allowed to cool to room temperature, water was poured and the mixture was extracted with t-butyl methyl ether, then, the organic layer was washed sequentially with a saturated sodium hydrogen carbonate aqueous solution and a saturated sodium chloride aqueous solution, dried over sodium sulfate, then, concentrated under reduced pressure. The residue was subjected to silica gel column chromatography, to obtain 0.10 g of a compound of formula (123) :
- present active compound 123 (hereinafter, referred to as present active compound 123) .
- present active compound 124 (hereinafter, referred to as present active compound 124) was obtained according to the method described in Production Example 123, using 0.15 g of the present active compound 120 instead of the present active compound 115.
- the present active compound 118 (0.15 g) was suspended in 2 ml of methanol, and 0.5 ml of a 2N sodium hydroxide aqueous solution and 0.3 ml of a 30% hydrogen peroxide aqueous solution were added, and the mixture was stirred for 1 hour at room temperature.
- Into the reaction mixture was poured water, and the mixture was extracted with chloroform, then, the organic layer was washed with a saturated sodium thiosulfate aqueous solution, dried over sodium sulfate, then, concentrated under reduced pressure. The residue was subjected to silica gel preparative thin layer chromatography, to obtain 29 mg of a compound of formula (125) :
- present active compound 125 (hereinafter, referred to as present active compound 125) .
- the present active compound 115 (0.20 g) was suspended in 3 ml of DMF, and 0.05 g of 60% sodium hydride (oily) was added under water cool, and the mixture was stirred for 5 minutes at room temperature. To the mixture was added 0.16 g of 1-bromo-l, 1, 2, 2- tetrafluoroethane, and the mixture was stirred over night and day at room temperature. Into the reaction mixture was poured water under ice cool and the mixture was extracted with t-butyl methyl ether. The organic layer was washed sequentially with water and a saturated sodium chloride aqueous solution, dried over sodium sulfate, then, concentrated under reduced pressure. The resultant solid was washed with t-butyl methyl ether, to obtain 0.16 g of a compound of formula (126) :
- present active compound 126 (hereinafter, referred to as present active compound 126) .
- present active compound (hereinafter, referred to as present active compound
- present active compound (hereinafter, referred to as present active compound
- the present active compound 120 (0.20 g) was suspended in 3 ml of DMF, and 0.05 g of 60% sodium hydride (oily) was added under water cool and the mixture was stirred- for 10 minutes at room temperature To the mixture was added 0.16 g of l-bromo-2- fluoroethane, and the mixture was stirred for 8 hours at room temperature, and allowed to stand overnight at room temperature. Into the reaction mixture was poured water under ice cool, and the generated crystal was filtrated, then, washed with water and dried under reduced pressure. The resultant crystal was subjected to silica gel preparative thin layer chromatography, to obtain 0.12 g of a compound of formula (129) :
- present active compound 129 (hereinafter, referred to as present active compound 129) and 50 ing of a compound of formula (130) :
- present active compound 130 (hereinafter, referred to as present active compound 130) .
- present active compound 130 present active compound 129
- the present active compound 116 (0.16 g) was suspended in 3 ml of THF, and 0.2 ml of a 50% dimethylamine aqueous solution was added, and the mixture was stirred for 2 hours at room temperature.
- the reaction mixture was concentrated under reduced pressure, and the resultant residue was subjected to silica gel preparative thin layer chromatography, to obtain 73 mg of a compound of formula (131) :
- present active compound 131 (hereinafter, referred to as present active compound 131) .
- present active compound 132 (hereinafter, referred to as present active compound 132) .
- present active compound (hereinafter, referred to as present active compound
- present active compound (hereinafter, referred to as present active compound
- present active compound 135) (hereinafter, referred to as present active compound 135) was obtained according to the method described in Production Example 126, using 0.16 g of 3-bromo-l- propyne instead of 1-bromo-l, 1 , 2 , 2-tetrafluoroethane .
- present active compound 136 (hereinafter, referred to as present active compound 136) was obtained according to the method described in Production Example 126, using 0.20 g of the present active compound 120 instead of the present active compound 115 and using 0.16 g of 3-bromo-l-propyne instead of 1-bromo-l, 1, 2, 2-tetrafluoroethane .
- Present active compound 136
Abstract
Description













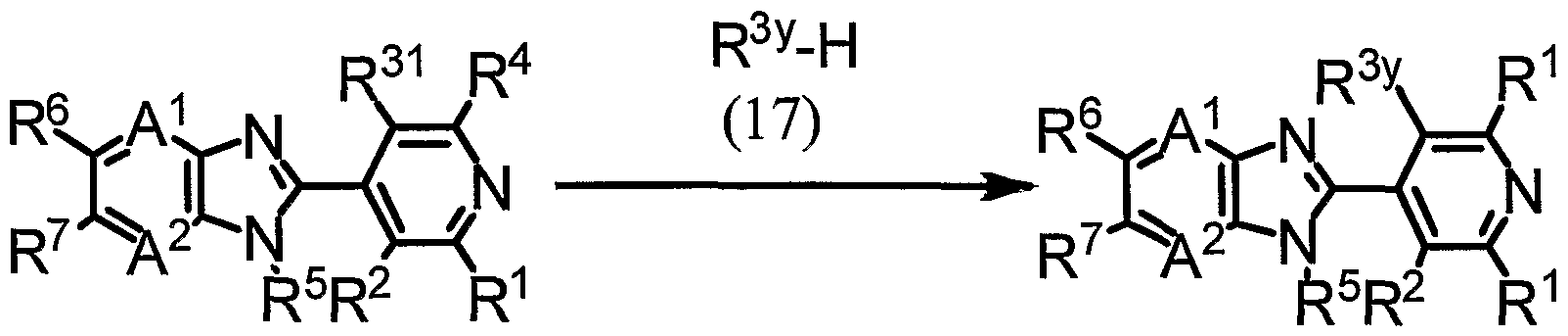







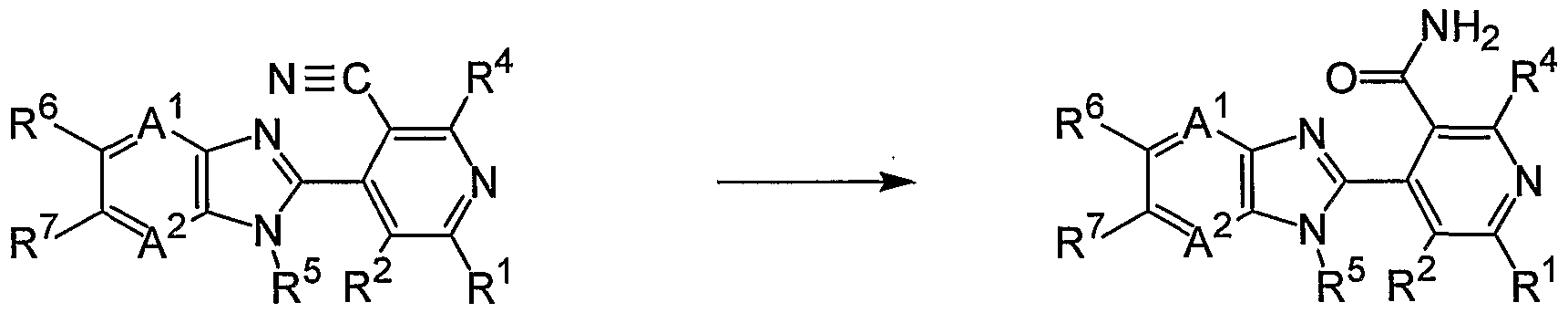




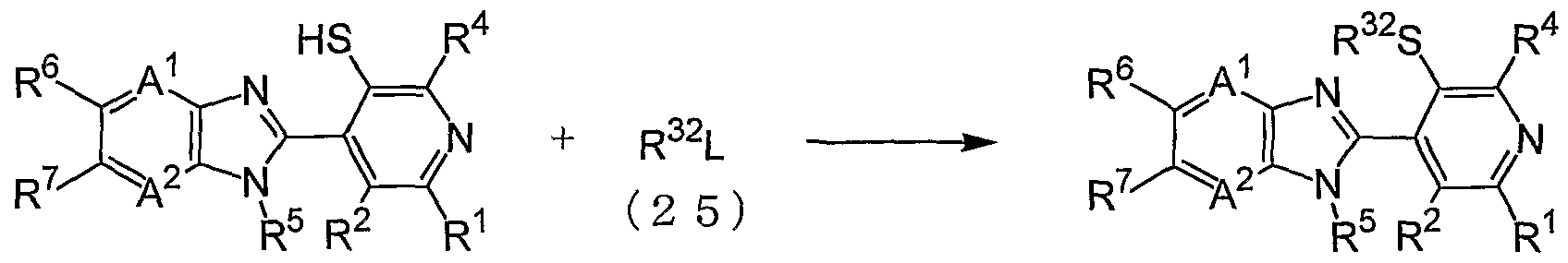















































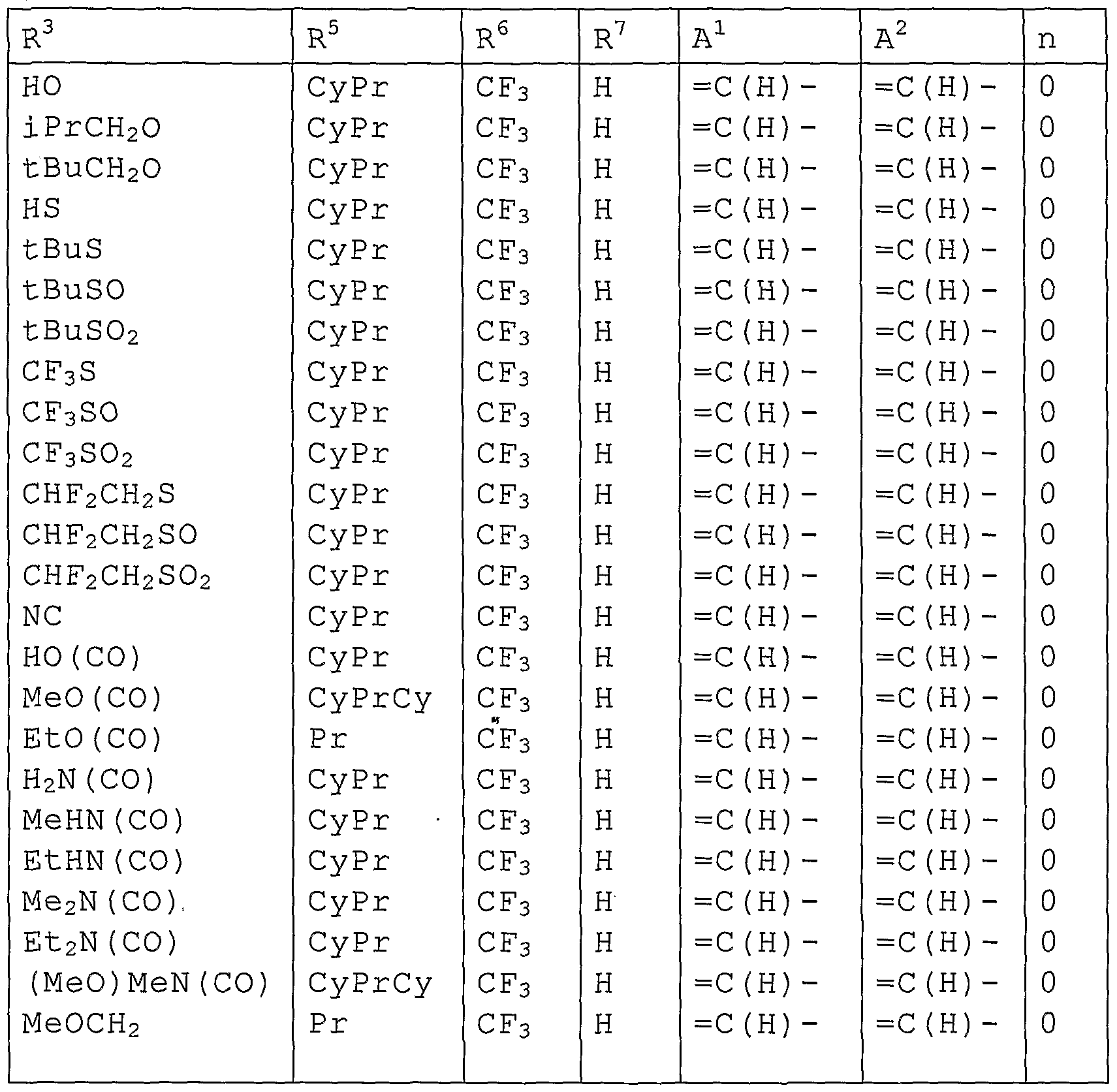








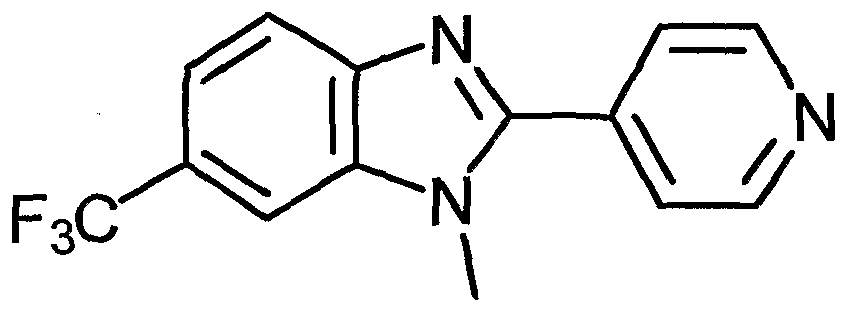













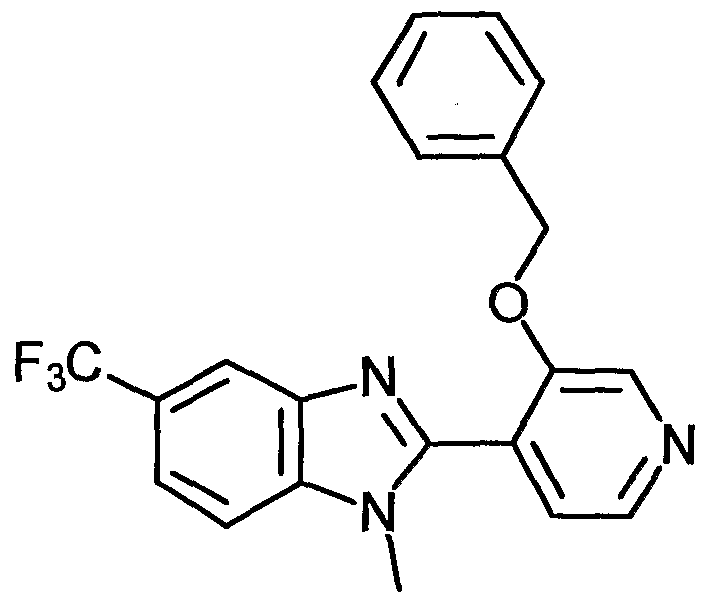




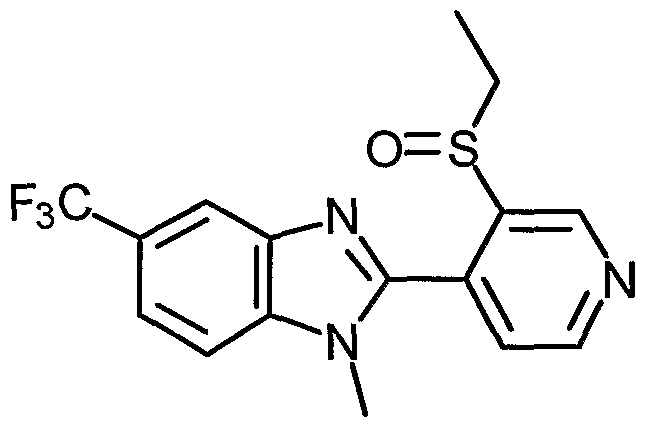





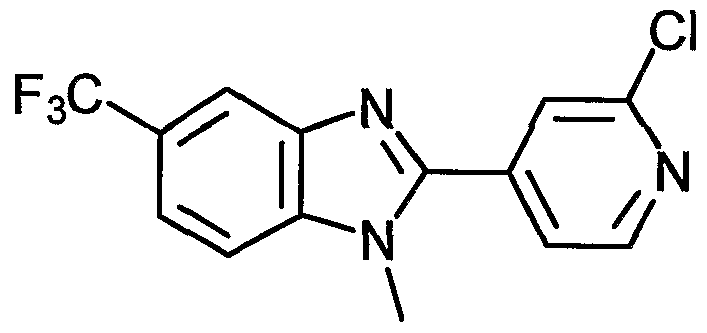











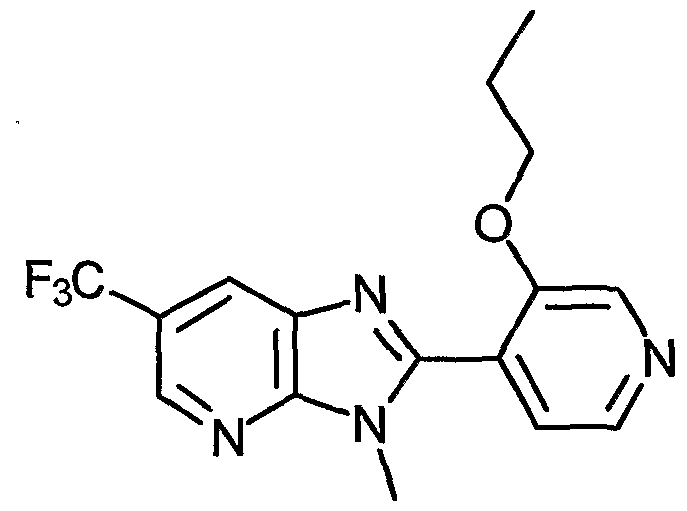


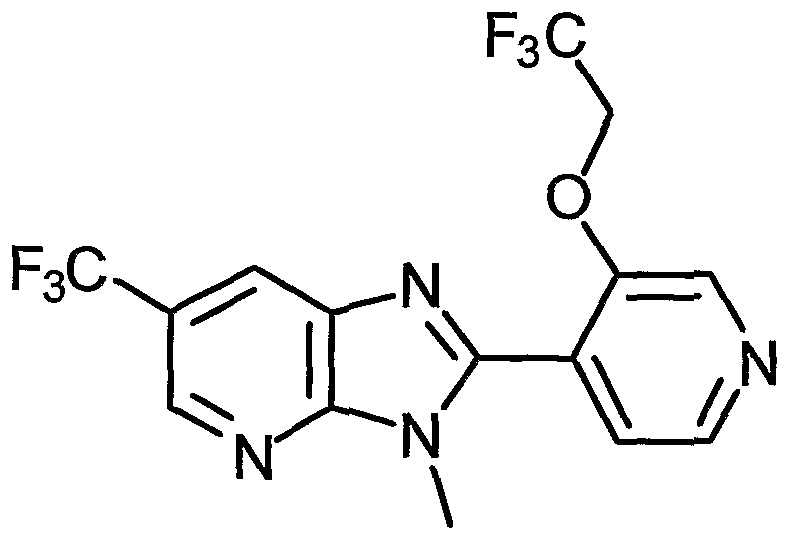






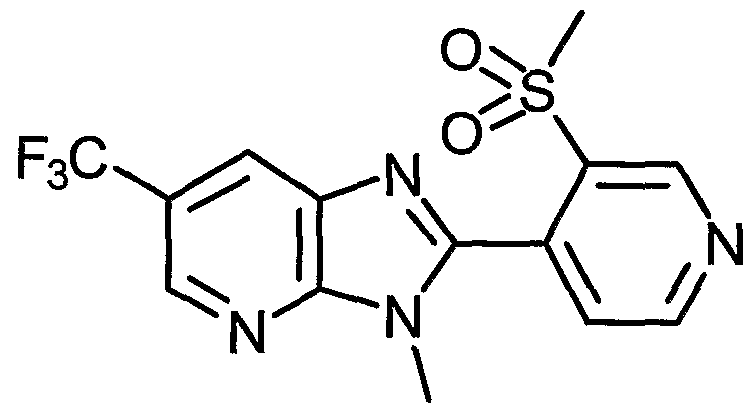



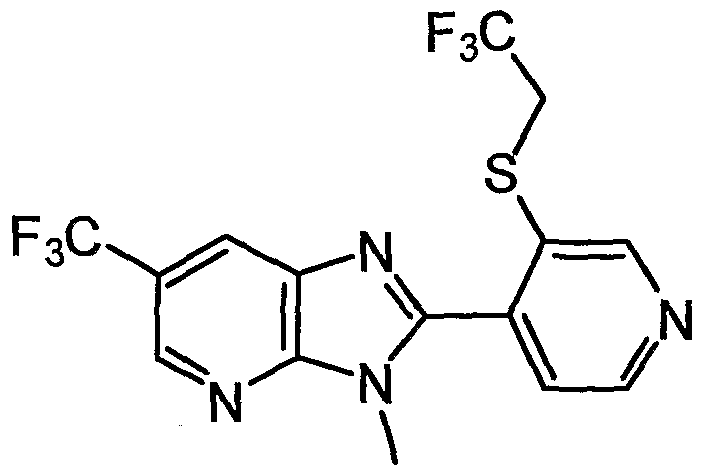



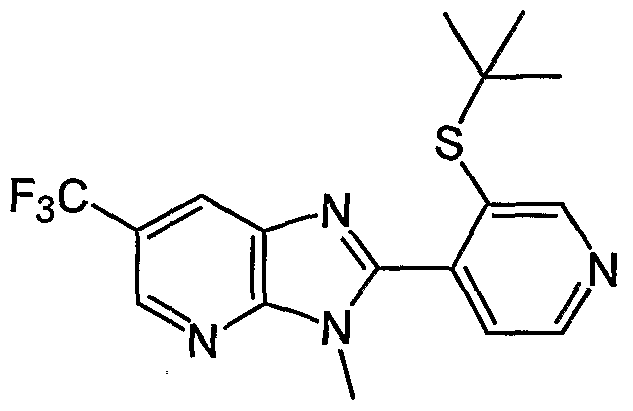
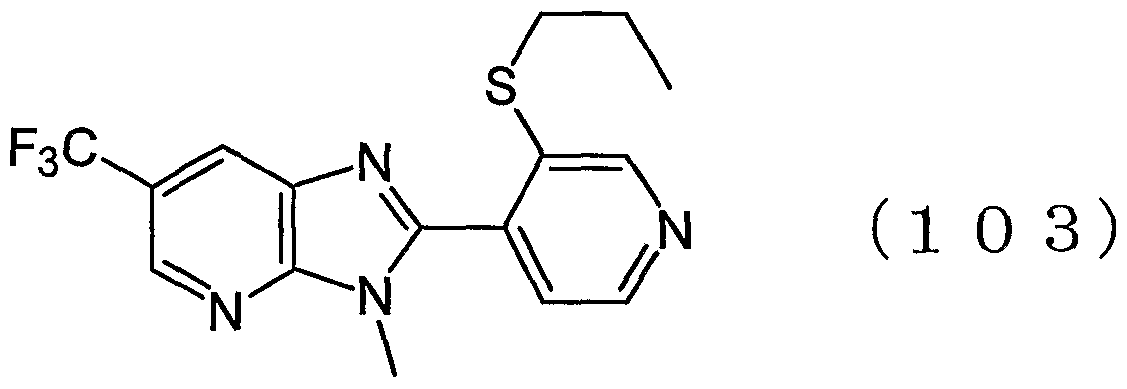




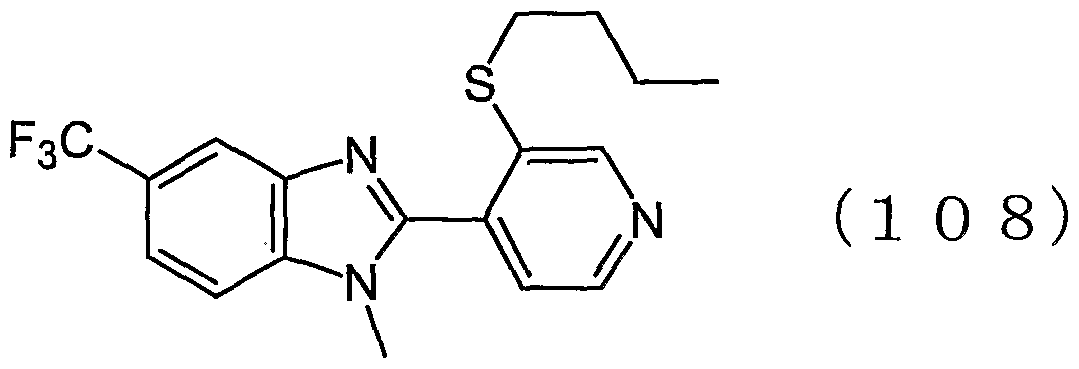




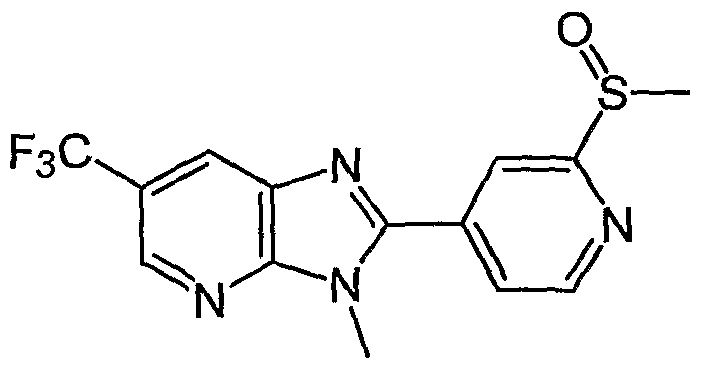




























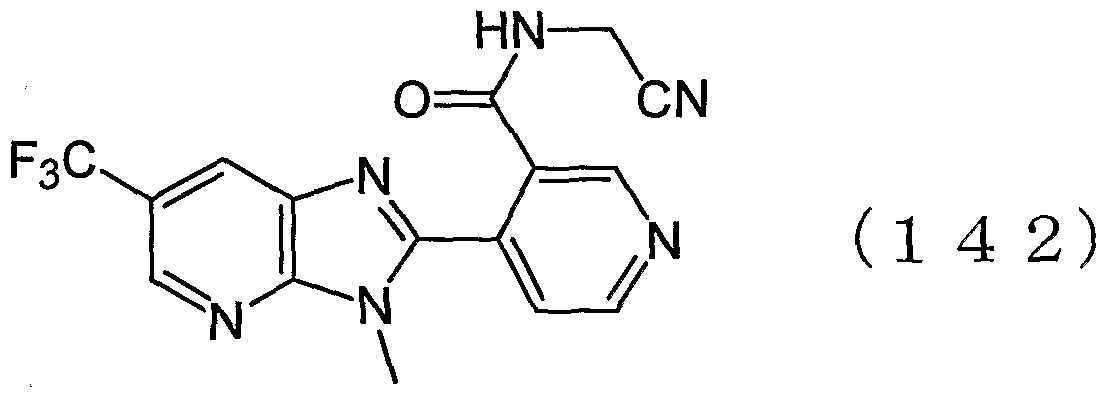




















































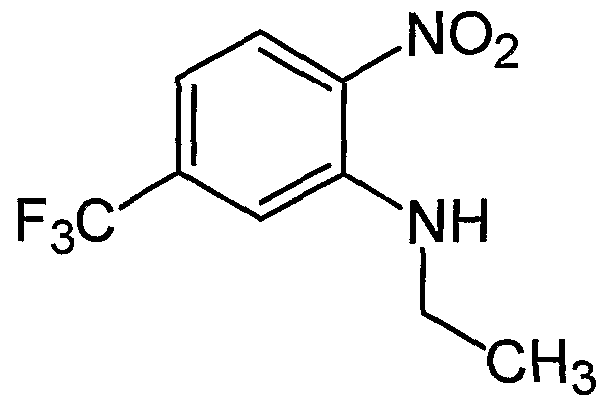













Claims

Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201080018451.8A CN102414195B (en) | 2009-04-28 | 2010-04-20 | Fused heterocyclic compound and use thereof |
| BRPI1015315A BRPI1015315B1 (en) | 2009-04-28 | 2010-04-20 | fused heterocyclic compound, its use, composition and method of controlling a harmful arthropod |
| EP10769684.1A EP2424856B1 (en) | 2009-04-28 | 2010-04-20 | Fused heterocyclic compound and use thereof |
| US13/259,058 US8273764B2 (en) | 2009-04-28 | 2010-04-20 | Fused heterocyclic compounds and use thereof |
| US13/417,156 US8461181B2 (en) | 2009-04-28 | 2012-03-09 | Fused heterocyclic compound and use thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009108926 | 2009-04-28 | ||
| JP2009-108926 | 2009-04-28 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/259,058 A-371-Of-International US8273764B2 (en) | 2009-04-28 | 2010-04-20 | Fused heterocyclic compounds and use thereof |
| US13/417,156 Division US8461181B2 (en) | 2009-04-28 | 2012-03-09 | Fused heterocyclic compound and use thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010125985A1 true WO2010125985A1 (en) | 2010-11-04 |
Family
ID=43032136
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/057315 WO2010125985A1 (en) | 2009-04-28 | 2010-04-20 | Fused heterocyclic compound and use thereof |
Country Status (7)
| Country | Link |
|---|---|
| US (2) | US8273764B2 (en) |
| EP (1) | EP2424856B1 (en) |
| JP (1) | JP5671835B2 (en) |
| CN (1) | CN102414195B (en) |
| AR (1) | AR076415A1 (en) |
| BR (1) | BRPI1015315B1 (en) |
| WO (1) | WO2010125985A1 (en) |
Cited By (95)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011045415A3 (en) * | 2009-10-15 | 2011-10-06 | Guerbet | Imaging agents and their use for the diagnostic in vivo of neurodegenerative diseases, notably alzheimer's disease and derivative diseases |
| WO2012057266A1 (en) * | 2010-10-27 | 2012-05-03 | 住友化学株式会社 | Pest control composition and method for controlling pests |
| WO2012057265A1 (en) * | 2010-10-27 | 2012-05-03 | 住友化学株式会社 | Pest control composition and method for controlling pests |
| WO2012057268A1 (en) * | 2010-10-27 | 2012-05-03 | 住友化学株式会社 | Pest control composition and method for controlling pests |
| WO2012057273A1 (en) * | 2010-10-27 | 2012-05-03 | 住友化学株式会社 | Harmful animal control composition and method for controlling harmful animals |
| WO2012057264A1 (en) * | 2010-10-27 | 2012-05-03 | 住友化学株式会社 | Pest control composition and method for controlling pests |
| WO2012057260A1 (en) * | 2010-10-27 | 2012-05-03 | 住友化学株式会社 | Pest control composition and method for controlling pests |
| WO2012057269A1 (en) * | 2010-10-27 | 2012-05-03 | 住友化学株式会社 | Harmful animal control composition and method for controlling harmful animals |
| WO2012087630A1 (en) | 2010-12-20 | 2012-06-28 | E.I. Du Pont De Nemours And Company | Pyridine and pyrimidine compounds for controlling invertebrate |
| WO2014132972A1 (en) | 2013-02-28 | 2014-09-04 | 住友化学株式会社 | Fused heterocyclic compound and pest control applications thereof |
| WO2015002211A1 (en) | 2013-07-01 | 2015-01-08 | 住友化学株式会社 | Fused heterocyclic compound and pest control use thereof |
| WO2015000715A1 (en) | 2013-07-02 | 2015-01-08 | Syngenta Participations Ag | Pesticidally active bi- or tricyclic heterocycles with sulfur containing substituents |
| EP2873668A1 (en) | 2013-11-13 | 2015-05-20 | Syngenta Participations AG. | Pesticidally active bicyclic heterocycles with sulphur containing substituents |
| EP2878199A1 (en) | 2013-11-27 | 2015-06-03 | Syngenta Participations AG. | Method of protecting a plant propagation material |
| WO2015091945A1 (en) | 2013-12-20 | 2015-06-25 | Syngenta Participations Ag | Pesticidally active substituted 5,5-bicyclic heterocycles with sulphur containing substituents |
| CN104968663A (en) * | 2013-02-06 | 2015-10-07 | 住友化学株式会社 | Condensed heterocyclic compound |
| EP2862853A4 (en) * | 2012-06-18 | 2015-11-25 | Sumitomo Chemical Co | Fused heterocyclic compound |
| EP2865671A4 (en) * | 2012-06-22 | 2015-11-25 | Sumitomo Chemical Co | Fused heterocyclic compound |
| WO2016023954A2 (en) | 2014-08-12 | 2016-02-18 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulphur containing substituents |
| US9278983B2 (en) | 2010-12-24 | 2016-03-08 | Sumitomo Chemical Company, Limited | Fused heterocyclic compound and use thereof for pest control |
| WO2016091731A1 (en) | 2014-12-11 | 2016-06-16 | Syngenta Participations Ag | Pesticidally active tetracyclic derivatives with sulfur containing substituents |
| WO2016096584A1 (en) * | 2014-12-17 | 2016-06-23 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulphur containing substituents |
| WO2016124563A1 (en) | 2015-02-05 | 2016-08-11 | Bayer Cropscience Aktiengesellschsaft | 2-(het)aryl-substituted condensed bicyclic heterocycle derivatives as pest control agents |
| WO2016124557A1 (en) | 2015-02-05 | 2016-08-11 | Bayer Cropscience Aktiengesellschaft | 2-(het)aryl-substituted condensed bicyclic heterocycle derivatives as pest control agents |
| WO2016162318A1 (en) | 2015-04-08 | 2016-10-13 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents and intermediate product |
| WO2016169886A1 (en) | 2015-04-24 | 2016-10-27 | Syngenta Crop Protection Ag | Pesticidally active polycyclic derivatives with sulfur substituted five-membered ring heterocyles |
| WO2016169882A1 (en) | 2015-04-24 | 2016-10-27 | Syngenta Participations Ag | Pesticidally active polycyclic derivatives with sulfur substituted five membered ring heterocyles |
| WO2017001311A1 (en) | 2015-07-01 | 2017-01-05 | Syngenta Participations Ag | Pesticidally active tetracyclic derivatives with sulfur containing substituents |
| WO2017001314A1 (en) | 2015-07-01 | 2017-01-05 | Syngenta Participations Ag | Pesticidally active polycyclic derivatives with sulfur containing substituents |
| US9549559B2 (en) | 2013-01-31 | 2017-01-24 | Sumitomo Chemical Company, Limited | Composition and method for controlling pests |
| WO2017050751A1 (en) | 2015-09-25 | 2017-03-30 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulphur containing substituents |
| WO2017055147A1 (en) | 2015-09-28 | 2017-04-06 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulphur containing substituents |
| WO2017072039A1 (en) | 2015-10-26 | 2017-05-04 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents |
| WO2017089190A1 (en) | 2015-11-23 | 2017-06-01 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulphur and cyclopropyl containing substituents |
| US9668482B1 (en) | 2014-03-07 | 2017-06-06 | Sumitomo Chemical Company, Limited | Fused heterocyclic compound and pest control application thereof |
| WO2017093180A1 (en) | 2015-12-01 | 2017-06-08 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents |
| US9723835B2 (en) | 2013-01-31 | 2017-08-08 | Sumitomo Chemical Company, Limited | Pest control composition and method for controlling pest |
| WO2017144341A1 (en) | 2016-02-23 | 2017-08-31 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents |
| WO2017174449A1 (en) | 2016-04-07 | 2017-10-12 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulphur containing substituents |
| WO2017174414A1 (en) | 2016-04-05 | 2017-10-12 | Bayer Cropscience Aktiengesellschaft | Naphthaline-derivatives as pest control agents |
| KR20170117079A (en) | 2015-02-12 | 2017-10-20 | 닛산 가가쿠 고교 가부시키 가이샤 | Condensed heterocyclic compound and noxious organism control agent |
| EP3241830A1 (en) | 2016-05-04 | 2017-11-08 | Bayer CropScience Aktiengesellschaft | Condensed bicyclic heterocyclic derivatives as pesticides |
| US9854805B2 (en) | 2013-01-31 | 2018-01-02 | Sumitomo Chemical Company, Limited | Composition and method for controlling pests |
| WO2018015289A1 (en) | 2016-07-19 | 2018-01-25 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents |
| US9883675B2 (en) | 2013-01-31 | 2018-02-06 | Sumitomo Chemical Company, Limited | Method for controlling pests |
| WO2018033455A1 (en) | 2016-08-15 | 2018-02-22 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents |
| WO2018041729A2 (en) | 2016-09-01 | 2018-03-08 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulphur containing substituents |
| WO2018050825A1 (en) | 2016-09-19 | 2018-03-22 | Bayer Cropscience Aktiengesellschaft | Pyrazolo [1,5-a]pyridine derivatives and their use as pesticides |
| EP3305786A2 (en) | 2018-01-22 | 2018-04-11 | Bayer CropScience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pesticides |
| WO2018065292A1 (en) | 2016-10-06 | 2018-04-12 | Bayer Cropscience Aktiengesellschaft | 2-(het)aryl-substituted condensed bicyclic heterocycle derivatives as pest control agents |
| WO2018065288A1 (en) | 2016-10-07 | 2018-04-12 | Bayer Cropscience Aktiengesellschaft | 2-[2-phenyl-1-(sulfonyl-methyl)-vinyl]-imidazo-[4,5-b] pyridine derivatives and related compounds as pesticides in plant protection |
| WO2018077565A1 (en) | 2016-10-27 | 2018-05-03 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulphur and hydroxylamine substituents |
| US9974307B2 (en) | 2013-01-31 | 2018-05-22 | Sumitomo Chemical Company, Limited | Composition and method for controlling pests |
| WO2018091389A1 (en) | 2016-11-17 | 2018-05-24 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulphur containing substituents |
| WO2018095953A1 (en) | 2016-11-23 | 2018-05-31 | Bayer Cropscience Aktiengesellschaft | 2-[3-(alkylsulfonyl)-2h-indazol-2-yl]-3h-imidazo[4,5-b]pyridine derivatives and similar compounds as pesticides |
| WO2018099812A1 (en) | 2016-12-01 | 2018-06-07 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulfur containing substituents |
| WO2018108726A1 (en) | 2016-12-15 | 2018-06-21 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulfur containing substituents |
| WO2018130443A1 (en) | 2017-01-10 | 2018-07-19 | Bayer Aktiengesellschaft | Heterocyclene derivatives as pest control agents |
| WO2018130437A1 (en) | 2017-01-10 | 2018-07-19 | Bayer Aktiengesellschaft | Heterocyclene derivatives as pest control agents |
| WO2018138050A1 (en) | 2017-01-26 | 2018-08-02 | Bayer Aktiengesellschaft | Condensed bicyclic heterocyclene derivatives as pest control agents |
| US10042251B2 (en) | 2016-09-30 | 2018-08-07 | Rohm And Haas Electronic Materials Llc | Zwitterionic photo-destroyable quenchers |
| WO2018141954A1 (en) | 2017-02-06 | 2018-08-09 | Bayer Aktiengesellschaft | Aryl or heteroaryl-substituted imidazo pyridine derivatives and their use as pesticides |
| WO2018141955A1 (en) | 2017-02-06 | 2018-08-09 | Bayer Aktiengesellschaft | Process for preparing halogenated imidazopyridine derivatives |
| WO2018153778A1 (en) | 2017-02-21 | 2018-08-30 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulfur containing substituents |
| KR20180116303A (en) | 2016-03-10 | 2018-10-24 | 닛산 가가쿠 가부시키가이샤 | Condensed heterocyclic compounds and pesticides |
| WO2018197257A1 (en) | 2017-04-24 | 2018-11-01 | Bayer Aktiengesellschaft | Condensed bicyclic heterocyclic-compound derivatives as pest control agents |
| WO2018197315A1 (en) | 2017-04-25 | 2018-11-01 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulfur containing substituents |
| WO2018202501A1 (en) | 2017-05-02 | 2018-11-08 | Bayer Aktiengesellschaft | 2-(het)aryl-substituted condensed bicyclic heterocyclic derivatives as pest control agents |
| WO2018202494A1 (en) | 2017-05-02 | 2018-11-08 | Bayer Aktiengesellschaft | 2-(het)aryl-substituted condensed bicyclic heterocyclic derivatives as pest control agents |
| WO2018215304A1 (en) | 2017-05-22 | 2018-11-29 | Syngenta Participations Ag | Tetracyclic pyridazine sulphur containing compounds and their use as pesticides |
| WO2019008115A1 (en) | 2017-07-07 | 2019-01-10 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulfur containing substituents |
| WO2019008072A1 (en) | 2017-07-05 | 2019-01-10 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulfur containing substituents |
| WO2019038195A1 (en) | 2017-08-22 | 2019-02-28 | Bayer Aktiengesellschaft | Heterocyclene derivatives as pest control agents |
| WO2019068572A1 (en) | 2017-10-04 | 2019-04-11 | Bayer Aktiengesellschaft | Derivatives of heterocyclic compounds as pest control agents |
| US10375962B2 (en) | 2016-01-11 | 2019-08-13 | Bayer Cropscience Aktiengesellschaft | Heterocycle derivatives as pesticides |
| WO2019162174A1 (en) | 2018-02-21 | 2019-08-29 | Bayer Aktiengesellschaft | Condensed bicyclic heterocyclic derivatives as pest control agents |
| WO2019175046A1 (en) | 2018-03-12 | 2019-09-19 | Bayer Aktiengesellschaft | Condensed bicyclic heterocyclic derivatives as pest control agents |
| WO2019201921A1 (en) | 2018-04-20 | 2019-10-24 | Bayer Aktiengesellschaft | Heterocyclene derivatives as pest control agents |
| WO2019229089A1 (en) | 2018-05-31 | 2019-12-05 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulfur containing substituents |
| WO2019234160A1 (en) | 2018-06-06 | 2019-12-12 | Syngenta Crop Protection Ag | Pesticidally active heterocyclic derivatives with sulfur containing substituents |
| WO2020002082A1 (en) * | 2018-06-26 | 2020-01-02 | Bayer Aktiengesellschaft | Heterocyclene derivatives as pest control agents |
| WO2020053282A1 (en) | 2018-09-13 | 2020-03-19 | Bayer Aktiengesellschaft | Heterocyclene derivatives as pest control agents |
| WO2020084075A1 (en) | 2018-10-24 | 2020-04-30 | Syngenta Crop Protection Ag | Pesticidally active heterocyclic derivatives with sulfoximine containing substituents |
| US10667517B2 (en) | 2015-08-07 | 2020-06-02 | Bayer Cropscience Aktiengesellschaft | 2-(Het)aryl-substituted fused heterocycle derivatives as pesticides |
| WO2020141135A1 (en) | 2018-12-31 | 2020-07-09 | Syngenta Crop Protection Ag | Pesticidally active heterocyclic derivatives with sulfur containing substituents |
| US10745398B2 (en) | 2015-09-28 | 2020-08-18 | Bayer Cropscience Aktiengesellschaft | 2-(het)aryl-substituted fused heterocycle derivatives as pesticides |
| WO2020173860A1 (en) | 2019-02-26 | 2020-09-03 | Bayer Aktiengesellschaft | Fused bicyclic heterocycle derivatives as pesticides |
| WO2020173861A1 (en) | 2019-02-26 | 2020-09-03 | Bayer Aktiengesellschaft | Condensed bicyclic heterocyclic derivatives as pest control agents |
| WO2020250183A1 (en) | 2019-06-13 | 2020-12-17 | Pi Industries Ltd. | Fused heterocyclic compounds and their use as pest control agents |
| WO2021053110A1 (en) | 2019-09-20 | 2021-03-25 | Syngenta Crop Protection Ag | Pesticidally active heterocyclic derivatives with sulfur and sulfoximine containing substituents |
| WO2021213978A1 (en) | 2020-04-21 | 2021-10-28 | Bayer Aktiengesellschaft | 2-(het)aryl-substituted condensed heterocyclic derivatives as pest control agents |
| WO2021219810A1 (en) | 2020-04-30 | 2021-11-04 | Syngenta Crop Protection Ag | Pesticidally active heterocyclic derivatives with sulfur containing substituents |
| WO2022002818A1 (en) | 2020-07-02 | 2022-01-06 | Bayer Aktiengesellschaft | Heterocyclene derivatives as pest control agents |
| US11229208B2 (en) | 2013-07-02 | 2022-01-25 | Syngenta Participations Ag | Pesticidally active bi- or tricyclic heterocycles with sulfur containing substituents |
| WO2022238391A1 (en) | 2021-05-12 | 2022-11-17 | Bayer Aktiengesellschaft | 2-(het)aryl-substituted condensed heterocycle derivatives as pest control agents |
Families Citing this family (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI589570B (en) | 2011-08-04 | 2017-07-01 | 住友化學股份有限公司 | Fused heterocyclic compound and use thereof for pest control |
| JP6217630B2 (en) * | 2012-05-30 | 2017-10-25 | 住友化学株式会社 | Fused heterocyclic compounds |
| EP2857396B1 (en) * | 2012-05-31 | 2018-11-21 | Sumitomo Chemical Co., Ltd | Fused heterocyclic compounds for pest control |
| WO2013187426A1 (en) * | 2012-06-15 | 2013-12-19 | 住友化学株式会社 | Pest control method for harmful arthropods |
| AU2013366867B2 (en) | 2012-12-27 | 2017-06-01 | Sumitomo Chemical Company, Limited | Fused oxazole compounds and use thereof for pest control |
| WO2014119494A1 (en) * | 2013-01-31 | 2014-08-07 | 住友化学株式会社 | Pest control composition and method for controlling pest |
| UY35421A (en) | 2013-03-15 | 2014-10-31 | Nihon Nohyaku Co Ltd | CONDENSED HETEROCYCLIC COMPOUND OR ITS SALT, AGRICULTURAL OR HERITAGE INSECTICIDE THAT INCLUDES THE COMPOSITE AND METHOD OF USE OF THE INSECTICIDE |
| TWI696612B (en) | 2015-01-29 | 2020-06-21 | 日商日本農藥股份有限公司 | Condensed heterocyclic compound having a cycloalkylpyridyl group or a salt thereof, agricultural and horticultural insecticide containing the compound, and method of using the same |
| ES2737404T3 (en) * | 2015-04-09 | 2020-01-14 | Fmc Corp | Bicyclic Pyrazole Pesticides |
| BR112018001413A2 (en) | 2015-07-24 | 2018-09-11 | Syngenta Participations Ag | pesticide-active heterocyclic derivatives with sulfur-containing substituents |
| CA3000803C (en) | 2015-10-13 | 2020-03-24 | Nihon Nohyaku Co., Ltd. | Oxime group-containing condensed heterocyclic compound or salt thereof, agricultural and horticultural insecticide comprising the compound, and method for using the insecticide |
| KR102070746B1 (en) | 2015-12-01 | 2020-01-29 | 니혼노야쿠가부시키가이샤 | 3H-pyrrolopyridine compound or N-oxide thereof, or salts thereof and agrohorticultural insecticide containing the compound and method of using the same |
| AU2017221988B2 (en) | 2016-02-26 | 2019-02-14 | Nihon Nohyaku Co., Ltd. | Benzoxazole compound or salt thereof, agricultural and horticultural insecticide comprising the compound, and method for using the insecticide |
| CA3013708C (en) | 2016-02-26 | 2021-04-13 | Nihon Nohyaku Co., Ltd. | Heterocycle-bound condensed heterocyclic compound or salt thereof, agricultural and horticultural insecticide comprising the compound, and method for using the insecticide |
| US10377754B2 (en) | 2016-09-01 | 2019-08-13 | Nihon Nohyaku Co., Ltd. | Hydrazonyl group-containing condensed heterocyclic compound or salt thereof, agricultural and horticultural insecticide comprising the compound, and method for using the insecticide |
| BR112019006217B1 (en) | 2016-10-13 | 2023-12-26 | Nihon Nohyaku Co., Ltd | CONDENSED HETEROCYCLIC COMPOUND AND ITS USE, METHOD FOR USE OF AN AGRICULTURAL AND HORTICULTURAL INSECTICIDE, METHOD FOR CONTROLLING AGRICULTURAL AND HORTICULTURAL PESTS, AND COMPOSITION FOR CONTROLLING ECTOPARASITES AND ITS USE |
| US11124507B2 (en) | 2016-10-13 | 2021-09-21 | Nihon Nohyaku Co., Ltd. | 1H-pyrrolopyridine compound, N-oxide thereof or salt thereof, agricultural and horticultural insecticide comprising the compound, and method for using the insecticide |
| ES2943264T3 (en) | 2016-11-01 | 2023-06-12 | Nihon Nohyaku Co Ltd | Quinoline compound having an oxime group, N-oxide or salt thereof, agricultural and horticultural insecticide comprising the same and method for its use |
| BR112019008952A2 (en) | 2016-11-01 | 2019-07-09 | Nihon Nohyaku Co Ltd | n-alkylsulfonia indoline compound, agricultural and horticultural insecticide comprising the compound and method for using the insecticide |
| BR112019013266B1 (en) | 2016-12-27 | 2022-10-11 | Nihon Nohyaku Co., Ltd | COMPOUND OF 4H-PYRROLOPYRIDINE OR THE SALT THEREOF, USE THEREOF, METHODS FOR USE OF AN AGRICULTURAL AND HORTICULTURAL INSECTICIDE AND FOR CONTROL OF AGRICULTURAL AND HORTICULTURAL PESTS, COMPOSITION COMPRISING SUCH COMPOUND AND ITS USE |
| US10856548B2 (en) | 2016-12-27 | 2020-12-08 | Nihon Nohyaku Co., Ltd. | Oxime group-containing condensed heterocyclic compound or salt thereof, agricultural and horticultural insecticide comprising the compound or the salt, and method for using the insecticide |
| CN110582498B (en) | 2017-04-27 | 2022-06-24 | 日本农药株式会社 | Condensed heterocyclic compound or salt thereof, agricultural and horticultural insecticide containing the same, and method of using the same |
| CN113906024B (en) | 2019-05-27 | 2023-10-20 | 日本农药株式会社 | Condensed heterocyclic compound having nitrogen atom in crosslinking part, salt thereof, agricultural and horticultural insecticide containing the same, and method for using the same |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3300505A (en) * | 1964-12-07 | 1967-01-24 | Ciba Geigy Corp | Ether-2-r-substituted benzimidazoles and derivatives and acid addition salts thereof |
| ES380609A1 (en) * | 1969-06-16 | 1973-04-01 | Lilly Co Eli | NEW 4-SUBSTITUTED AMINO-5,7-DINITRO-2-(alpha,alpha-DIFLUOROALKYL)-BENZIMIDAZOLE COMPOUNDS HAVING INSECTICIDAL ACTIVITY, AND PROCESS FOR PREPARING SAME |
-
2010
- 2010-04-20 US US13/259,058 patent/US8273764B2/en active Active
- 2010-04-20 WO PCT/JP2010/057315 patent/WO2010125985A1/en active Application Filing
- 2010-04-20 EP EP10769684.1A patent/EP2424856B1/en active Active
- 2010-04-20 BR BRPI1015315A patent/BRPI1015315B1/en active IP Right Grant
- 2010-04-20 CN CN201080018451.8A patent/CN102414195B/en active Active
- 2010-04-26 AR ARP100101384A patent/AR076415A1/en unknown
- 2010-04-27 JP JP2010101856A patent/JP5671835B2/en active Active
-
2012
- 2012-03-09 US US13/417,156 patent/US8461181B2/en active Active
Non-Patent Citations (4)
| Title |
|---|
| CHEM.PHARM.BULL., vol. 30, no. 8, 1982, pages 2996 - 3004, XP002170422 * |
| CHEMICAL ABSTRACTS, vol. 87, Columbus, Ohio, US; abstract no. 151604, XP008157444 * |
| HAKAN GOKER, ET AL.: "Synthesis and potent antifungal activity against Candida species of some novel 1H-benzimidazoles", J.HETEROCYCL.CHEM., vol. 46, no. 5, 2009, pages 936 - 948, XP002596734 * |
| See also references of EP2424856A4 * |
Cited By (132)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011045415A3 (en) * | 2009-10-15 | 2011-10-06 | Guerbet | Imaging agents and their use for the diagnostic in vivo of neurodegenerative diseases, notably alzheimer's disease and derivative diseases |
| WO2012057266A1 (en) * | 2010-10-27 | 2012-05-03 | 住友化学株式会社 | Pest control composition and method for controlling pests |
| WO2012057265A1 (en) * | 2010-10-27 | 2012-05-03 | 住友化学株式会社 | Pest control composition and method for controlling pests |
| WO2012057268A1 (en) * | 2010-10-27 | 2012-05-03 | 住友化学株式会社 | Pest control composition and method for controlling pests |
| WO2012057273A1 (en) * | 2010-10-27 | 2012-05-03 | 住友化学株式会社 | Harmful animal control composition and method for controlling harmful animals |
| WO2012057264A1 (en) * | 2010-10-27 | 2012-05-03 | 住友化学株式会社 | Pest control composition and method for controlling pests |
| WO2012057260A1 (en) * | 2010-10-27 | 2012-05-03 | 住友化学株式会社 | Pest control composition and method for controlling pests |
| WO2012057269A1 (en) * | 2010-10-27 | 2012-05-03 | 住友化学株式会社 | Harmful animal control composition and method for controlling harmful animals |
| WO2012087630A1 (en) | 2010-12-20 | 2012-06-28 | E.I. Du Pont De Nemours And Company | Pyridine and pyrimidine compounds for controlling invertebrate |
| US9278983B2 (en) | 2010-12-24 | 2016-03-08 | Sumitomo Chemical Company, Limited | Fused heterocyclic compound and use thereof for pest control |
| EP2862853A4 (en) * | 2012-06-18 | 2015-11-25 | Sumitomo Chemical Co | Fused heterocyclic compound |
| EP2865671A4 (en) * | 2012-06-22 | 2015-11-25 | Sumitomo Chemical Co | Fused heterocyclic compound |
| US9883675B2 (en) | 2013-01-31 | 2018-02-06 | Sumitomo Chemical Company, Limited | Method for controlling pests |
| US9974307B2 (en) | 2013-01-31 | 2018-05-22 | Sumitomo Chemical Company, Limited | Composition and method for controlling pests |
| US9854805B2 (en) | 2013-01-31 | 2018-01-02 | Sumitomo Chemical Company, Limited | Composition and method for controlling pests |
| US9723835B2 (en) | 2013-01-31 | 2017-08-08 | Sumitomo Chemical Company, Limited | Pest control composition and method for controlling pest |
| US9549559B2 (en) | 2013-01-31 | 2017-01-24 | Sumitomo Chemical Company, Limited | Composition and method for controlling pests |
| CN104968663A (en) * | 2013-02-06 | 2015-10-07 | 住友化学株式会社 | Condensed heterocyclic compound |
| CN104968663B (en) * | 2013-02-06 | 2017-05-17 | 住友化学株式会社 | Condensed heterocyclic compound |
| WO2014132972A1 (en) | 2013-02-28 | 2014-09-04 | 住友化学株式会社 | Fused heterocyclic compound and pest control applications thereof |
| WO2015002211A1 (en) | 2013-07-01 | 2015-01-08 | 住友化学株式会社 | Fused heterocyclic compound and pest control use thereof |
| WO2015000715A1 (en) | 2013-07-02 | 2015-01-08 | Syngenta Participations Ag | Pesticidally active bi- or tricyclic heterocycles with sulfur containing substituents |
| EP3778598A2 (en) | 2013-07-02 | 2021-02-17 | Syngenta Participations Ag | Pesticidally active bi- or tricyclic heterocycles with sulfur containing substituents |
| US11229208B2 (en) | 2013-07-02 | 2022-01-25 | Syngenta Participations Ag | Pesticidally active bi- or tricyclic heterocycles with sulfur containing substituents |
| CN105722839A (en) * | 2013-11-13 | 2016-06-29 | 先正达参股股份有限公司 | Pesticidally active bicyclic heterocycles with sulphur containing substituents |
| EP2873668A1 (en) | 2013-11-13 | 2015-05-20 | Syngenta Participations AG. | Pesticidally active bicyclic heterocycles with sulphur containing substituents |
| WO2015071180A1 (en) | 2013-11-13 | 2015-05-21 | Syngenta Participations Ag | Pesticidally active bicyclic heterocycles with sulphur containing substituents |
| US9522913B2 (en) | 2013-11-13 | 2016-12-20 | Syngenta Participations Ag | Pesticidally active bicyclic heterocycles with sulphur containing substituents |
| EP2878199A1 (en) | 2013-11-27 | 2015-06-03 | Syngenta Participations AG. | Method of protecting a plant propagation material |
| WO2015078812A1 (en) | 2013-11-27 | 2015-06-04 | Syngenta Participations Ag | Method of protecting a plant propagation material |
| WO2015091945A1 (en) | 2013-12-20 | 2015-06-25 | Syngenta Participations Ag | Pesticidally active substituted 5,5-bicyclic heterocycles with sulphur containing substituents |
| US9668482B1 (en) | 2014-03-07 | 2017-06-06 | Sumitomo Chemical Company, Limited | Fused heterocyclic compound and pest control application thereof |
| WO2016023954A2 (en) | 2014-08-12 | 2016-02-18 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulphur containing substituents |
| WO2016091731A1 (en) | 2014-12-11 | 2016-06-16 | Syngenta Participations Ag | Pesticidally active tetracyclic derivatives with sulfur containing substituents |
| RU2765282C2 (en) * | 2014-12-17 | 2022-01-28 | Зингента Партисипейшнс Аг | Pesticide-active heterocyclic derivatives with sulphur-containing substituents |
| WO2016096584A1 (en) * | 2014-12-17 | 2016-06-23 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulphur containing substituents |
| US10323030B2 (en) | 2014-12-17 | 2019-06-18 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulphur containing substituents |
| US10087192B2 (en) | 2015-02-05 | 2018-10-02 | Bayer Cropscience Aktiengesellschaft | 2-(het)aryl-substituted fused bicyclic heterocycle derivatives as pesticides |
| WO2016124563A1 (en) | 2015-02-05 | 2016-08-11 | Bayer Cropscience Aktiengesellschsaft | 2-(het)aryl-substituted condensed bicyclic heterocycle derivatives as pest control agents |
| WO2016124557A1 (en) | 2015-02-05 | 2016-08-11 | Bayer Cropscience Aktiengesellschaft | 2-(het)aryl-substituted condensed bicyclic heterocycle derivatives as pest control agents |
| US10654845B2 (en) | 2015-02-05 | 2020-05-19 | Bayer Cropscience Aktiengesellschaft | 2-(het)aryl-substituted fused bicyclic heterocycle derivatives as pesticides |
| US10882869B2 (en) | 2015-02-12 | 2021-01-05 | Nissan Chemical Corporation | Condensed heterocyclic compounds and pesticides |
| US10464950B2 (en) | 2015-02-12 | 2019-11-05 | Nissan Chemical Corporation | Condensed heterocyclic compounds and pesticides |
| KR20170117079A (en) | 2015-02-12 | 2017-10-20 | 닛산 가가쿠 고교 가부시키 가이샤 | Condensed heterocyclic compound and noxious organism control agent |
| WO2016162318A1 (en) | 2015-04-08 | 2016-10-13 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents and intermediate product |
| US10188108B2 (en) | 2015-04-08 | 2019-01-29 | Bayer Cropscience Aktiengesellschaft | Fused bicyclic heterocycle derivatives as pesticides |
| EP3656770A2 (en) | 2015-04-24 | 2020-05-27 | Syngenta Participations AG | Pesticidally active polycyclic derivatives with sulfur substituted five-membered ring heterocycles |
| WO2016169886A1 (en) | 2015-04-24 | 2016-10-27 | Syngenta Crop Protection Ag | Pesticidally active polycyclic derivatives with sulfur substituted five-membered ring heterocyles |
| WO2016169882A1 (en) | 2015-04-24 | 2016-10-27 | Syngenta Participations Ag | Pesticidally active polycyclic derivatives with sulfur substituted five membered ring heterocyles |
| WO2017001314A1 (en) | 2015-07-01 | 2017-01-05 | Syngenta Participations Ag | Pesticidally active polycyclic derivatives with sulfur containing substituents |
| WO2017001311A1 (en) | 2015-07-01 | 2017-01-05 | Syngenta Participations Ag | Pesticidally active tetracyclic derivatives with sulfur containing substituents |
| US10667517B2 (en) | 2015-08-07 | 2020-06-02 | Bayer Cropscience Aktiengesellschaft | 2-(Het)aryl-substituted fused heterocycle derivatives as pesticides |
| EP3896065A1 (en) | 2015-08-07 | 2021-10-20 | Bayer CropScience Aktiengesellschaft | 2-(het)aryl-substituted condensed heterocycle derivatives as pesticides |
| EP3896066A2 (en) | 2015-08-07 | 2021-10-20 | Bayer CropScience Aktiengesellschaft | 2-(het)aryl-substituted condensed heterocycle derivatives as pesticides |
| US10986840B2 (en) | 2015-08-07 | 2021-04-27 | Bayer Cropscience Aktiengesellschaft | 2-(het)aryl-substituted fused heterocycle derivatives as pesticides |
| WO2017050751A1 (en) | 2015-09-25 | 2017-03-30 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulphur containing substituents |
| US10745398B2 (en) | 2015-09-28 | 2020-08-18 | Bayer Cropscience Aktiengesellschaft | 2-(het)aryl-substituted fused heterocycle derivatives as pesticides |
| WO2017055147A1 (en) | 2015-09-28 | 2017-04-06 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulphur containing substituents |
| US10550116B2 (en) | 2015-10-26 | 2020-02-04 | Bayer Cropscience Aktiengesellschaft | Fused bicyclic heterocycle derivatives as pesticides |
| WO2017072039A1 (en) | 2015-10-26 | 2017-05-04 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents |
| WO2017089190A1 (en) | 2015-11-23 | 2017-06-01 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulphur and cyclopropyl containing substituents |
| WO2017093180A1 (en) | 2015-12-01 | 2017-06-08 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents |
| US10375962B2 (en) | 2016-01-11 | 2019-08-13 | Bayer Cropscience Aktiengesellschaft | Heterocycle derivatives as pesticides |
| WO2017144341A1 (en) | 2016-02-23 | 2017-08-31 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents |
| US10759815B2 (en) | 2016-03-10 | 2020-09-01 | Nissan Chemical Corporation | Condensed heterocyclic compounds and pesticides |
| US10640518B2 (en) | 2016-03-10 | 2020-05-05 | Nissan Chemical Corporation | Condensed heterocyclic compounds and pesticides |
| US10544163B2 (en) | 2016-03-10 | 2020-01-28 | Nissan Chemical Corporation | Condensed heterocyclic compounds and pesticides |
| KR20180116303A (en) | 2016-03-10 | 2018-10-24 | 닛산 가가쿠 가부시키가이샤 | Condensed heterocyclic compounds and pesticides |
| WO2017174414A1 (en) | 2016-04-05 | 2017-10-12 | Bayer Cropscience Aktiengesellschaft | Naphthaline-derivatives as pest control agents |
| WO2017174449A1 (en) | 2016-04-07 | 2017-10-12 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulphur containing substituents |
| EP3241830A1 (en) | 2016-05-04 | 2017-11-08 | Bayer CropScience Aktiengesellschaft | Condensed bicyclic heterocyclic derivatives as pesticides |
| WO2018015289A1 (en) | 2016-07-19 | 2018-01-25 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents |
| US10561145B2 (en) | 2016-07-19 | 2020-02-18 | Bayer Cropscience Aktiengesellschaft | Fused bicyclic heterocycle derivatives as pesticides |
| WO2018033455A1 (en) | 2016-08-15 | 2018-02-22 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents |
| US10660334B2 (en) | 2016-08-15 | 2020-05-26 | Bayer Cropscience Aktiengesellschaft | Fused bicyclic heterocycle derivatives as pesticides |
| WO2018041729A2 (en) | 2016-09-01 | 2018-03-08 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulphur containing substituents |
| WO2018050825A1 (en) | 2016-09-19 | 2018-03-22 | Bayer Cropscience Aktiengesellschaft | Pyrazolo [1,5-a]pyridine derivatives and their use as pesticides |
| US10611779B2 (en) | 2016-09-19 | 2020-04-07 | Bayer Cropscience Aktiengesellschaft | Fused bicyclic heterocycle derivatives as pesticides |
| US10042251B2 (en) | 2016-09-30 | 2018-08-07 | Rohm And Haas Electronic Materials Llc | Zwitterionic photo-destroyable quenchers |
| US11339155B2 (en) | 2016-10-06 | 2022-05-24 | Bayer Cropscience Aktiengesellschaft | 2-(het)aryl-substituted fused bicyclic heterocycle derivatives as pesticides |
| WO2018065292A1 (en) | 2016-10-06 | 2018-04-12 | Bayer Cropscience Aktiengesellschaft | 2-(het)aryl-substituted condensed bicyclic heterocycle derivatives as pest control agents |
| WO2018065288A1 (en) | 2016-10-07 | 2018-04-12 | Bayer Cropscience Aktiengesellschaft | 2-[2-phenyl-1-(sulfonyl-methyl)-vinyl]-imidazo-[4,5-b] pyridine derivatives and related compounds as pesticides in plant protection |
| WO2018077565A1 (en) | 2016-10-27 | 2018-05-03 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulphur and hydroxylamine substituents |
| WO2018091389A1 (en) | 2016-11-17 | 2018-05-24 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulphur containing substituents |
| US10765116B2 (en) | 2016-11-23 | 2020-09-08 | Bayer Cropscience Aktiengesellschaft | 2-[3-(alkylsulfonyl)-2H-indazol-2-yl]-3H-imidazo[4,5-B]pyridine derivatives and similar compounds as pesticides |
| WO2018095953A1 (en) | 2016-11-23 | 2018-05-31 | Bayer Cropscience Aktiengesellschaft | 2-[3-(alkylsulfonyl)-2h-indazol-2-yl]-3h-imidazo[4,5-b]pyridine derivatives and similar compounds as pesticides |
| WO2018099812A1 (en) | 2016-12-01 | 2018-06-07 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulfur containing substituents |
| WO2018108726A1 (en) | 2016-12-15 | 2018-06-21 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulfur containing substituents |
| WO2018130443A1 (en) | 2017-01-10 | 2018-07-19 | Bayer Aktiengesellschaft | Heterocyclene derivatives as pest control agents |
| WO2018130437A1 (en) | 2017-01-10 | 2018-07-19 | Bayer Aktiengesellschaft | Heterocyclene derivatives as pest control agents |
| US11058115B2 (en) | 2017-01-10 | 2021-07-13 | Bayer Aktiengesellschaft | Heterocycle derivatives as pesticides |
| US11083199B2 (en) | 2017-01-10 | 2021-08-10 | Bayer Aktiengesellschaft | Heterocycle derivatives as pesticides |
| WO2018138050A1 (en) | 2017-01-26 | 2018-08-02 | Bayer Aktiengesellschaft | Condensed bicyclic heterocyclene derivatives as pest control agents |
| US11072609B2 (en) | 2017-02-06 | 2021-07-27 | Bayer Aktiengesellschaft | Method for preparing halogenated imidazopyridine derivatives |
| WO2018141954A1 (en) | 2017-02-06 | 2018-08-09 | Bayer Aktiengesellschaft | Aryl or heteroaryl-substituted imidazo pyridine derivatives and their use as pesticides |
| US10772332B2 (en) | 2017-02-06 | 2020-09-15 | Bayer Aktiengesellschaft | 2-(het)aryl-substituted fused heterocycle derivatives as pesticides |
| WO2018141955A1 (en) | 2017-02-06 | 2018-08-09 | Bayer Aktiengesellschaft | Process for preparing halogenated imidazopyridine derivatives |
| WO2018153778A1 (en) | 2017-02-21 | 2018-08-30 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulfur containing substituents |
| WO2018197257A1 (en) | 2017-04-24 | 2018-11-01 | Bayer Aktiengesellschaft | Condensed bicyclic heterocyclic-compound derivatives as pest control agents |
| WO2018197315A1 (en) | 2017-04-25 | 2018-11-01 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulfur containing substituents |
| US11505553B2 (en) | 2017-05-02 | 2022-11-22 | Bayer Aktiengesellschaft | 2-(Het)aryl-substituted fused heterocycle derivatives as pesticides |
| WO2018202501A1 (en) | 2017-05-02 | 2018-11-08 | Bayer Aktiengesellschaft | 2-(het)aryl-substituted condensed bicyclic heterocyclic derivatives as pest control agents |
| WO2018202494A1 (en) | 2017-05-02 | 2018-11-08 | Bayer Aktiengesellschaft | 2-(het)aryl-substituted condensed bicyclic heterocyclic derivatives as pest control agents |
| US11089783B2 (en) | 2017-05-02 | 2021-08-17 | Bayer Aktiengesellschaft | 2-(het)aryl-substituted fused heterocycle derivatives as pesticides |
| WO2018215304A1 (en) | 2017-05-22 | 2018-11-29 | Syngenta Participations Ag | Tetracyclic pyridazine sulphur containing compounds and their use as pesticides |
| WO2019008072A1 (en) | 2017-07-05 | 2019-01-10 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulfur containing substituents |
| WO2019008115A1 (en) | 2017-07-07 | 2019-01-10 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulfur containing substituents |
| WO2019038195A1 (en) | 2017-08-22 | 2019-02-28 | Bayer Aktiengesellschaft | Heterocyclene derivatives as pest control agents |
| US11655258B2 (en) | 2017-08-22 | 2023-05-23 | Bayer Aktiengesellschaft | Heterocycle derivatives as pesticides |
| WO2019068572A1 (en) | 2017-10-04 | 2019-04-11 | Bayer Aktiengesellschaft | Derivatives of heterocyclic compounds as pest control agents |
| US10981935B2 (en) | 2017-10-04 | 2021-04-20 | Bayer Aktiengesellschaft | Heterocycle derivatives as pesticides |
| EP3305786A2 (en) | 2018-01-22 | 2018-04-11 | Bayer CropScience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pesticides |
| WO2019162174A1 (en) | 2018-02-21 | 2019-08-29 | Bayer Aktiengesellschaft | Condensed bicyclic heterocyclic derivatives as pest control agents |
| US11019821B2 (en) | 2018-02-21 | 2021-06-01 | Bayer Aktiengesellschaft | Fused bicyclic heterocycle derivatives as pesticides |
| WO2019175046A1 (en) | 2018-03-12 | 2019-09-19 | Bayer Aktiengesellschaft | Condensed bicyclic heterocyclic derivatives as pest control agents |
| WO2019175045A1 (en) | 2018-03-12 | 2019-09-19 | Bayer Aktiengesellschaft | Condensed bicyclic heterocyclic derivatives as pest control agents |
| WO2019201921A1 (en) | 2018-04-20 | 2019-10-24 | Bayer Aktiengesellschaft | Heterocyclene derivatives as pest control agents |
| WO2019229089A1 (en) | 2018-05-31 | 2019-12-05 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulfur containing substituents |
| WO2019234160A1 (en) | 2018-06-06 | 2019-12-12 | Syngenta Crop Protection Ag | Pesticidally active heterocyclic derivatives with sulfur containing substituents |
| US11926623B2 (en) | 2018-06-26 | 2024-03-12 | Bayer Aktiengesellschaft | Heterocycle derivatives as pesticides |
| WO2020002082A1 (en) * | 2018-06-26 | 2020-01-02 | Bayer Aktiengesellschaft | Heterocyclene derivatives as pest control agents |
| WO2020053282A1 (en) | 2018-09-13 | 2020-03-19 | Bayer Aktiengesellschaft | Heterocyclene derivatives as pest control agents |
| WO2020084075A1 (en) | 2018-10-24 | 2020-04-30 | Syngenta Crop Protection Ag | Pesticidally active heterocyclic derivatives with sulfoximine containing substituents |
| WO2020141135A1 (en) | 2018-12-31 | 2020-07-09 | Syngenta Crop Protection Ag | Pesticidally active heterocyclic derivatives with sulfur containing substituents |
| WO2020173860A1 (en) | 2019-02-26 | 2020-09-03 | Bayer Aktiengesellschaft | Fused bicyclic heterocycle derivatives as pesticides |
| WO2020173861A1 (en) | 2019-02-26 | 2020-09-03 | Bayer Aktiengesellschaft | Condensed bicyclic heterocyclic derivatives as pest control agents |
| WO2020250183A1 (en) | 2019-06-13 | 2020-12-17 | Pi Industries Ltd. | Fused heterocyclic compounds and their use as pest control agents |
| WO2021053110A1 (en) | 2019-09-20 | 2021-03-25 | Syngenta Crop Protection Ag | Pesticidally active heterocyclic derivatives with sulfur and sulfoximine containing substituents |
| WO2021213978A1 (en) | 2020-04-21 | 2021-10-28 | Bayer Aktiengesellschaft | 2-(het)aryl-substituted condensed heterocyclic derivatives as pest control agents |
| WO2021219810A1 (en) | 2020-04-30 | 2021-11-04 | Syngenta Crop Protection Ag | Pesticidally active heterocyclic derivatives with sulfur containing substituents |
| WO2022002818A1 (en) | 2020-07-02 | 2022-01-06 | Bayer Aktiengesellschaft | Heterocyclene derivatives as pest control agents |
| WO2022238391A1 (en) | 2021-05-12 | 2022-11-17 | Bayer Aktiengesellschaft | 2-(het)aryl-substituted condensed heterocycle derivatives as pest control agents |
Also Published As
| Publication number | Publication date |
|---|---|
| US20120015975A1 (en) | 2012-01-19 |
| CN102414195B (en) | 2014-11-12 |
| EP2424856A1 (en) | 2012-03-07 |
| EP2424856A4 (en) | 2012-12-12 |
| BRPI1015315B1 (en) | 2020-02-04 |
| AR076415A1 (en) | 2011-06-08 |
| US8273764B2 (en) | 2012-09-25 |
| EP2424856B1 (en) | 2014-12-31 |
| JP5671835B2 (en) | 2015-02-18 |
| CN102414195A (en) | 2012-04-11 |
| US8461181B2 (en) | 2013-06-11 |
| BRPI1015315A2 (en) | 2018-02-20 |
| US20120178779A1 (en) | 2012-07-12 |
| JP2010275301A (en) | 2010-12-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2424856B1 (en) | Fused heterocyclic compound and use thereof | |
| US8742112B2 (en) | Heterocyclic compound and its use for control of an arthropod pest | |
| US9403771B2 (en) | Fused heterocyclic compound | |
| EP2646433B1 (en) | Pyrimidine compound and use for pest control thereof | |
| EP3018130B1 (en) | Fused heterocyclic compound and pest control use thereof | |
| US8796306B2 (en) | Noxious arthropod controlling composition and heterocyclic compound | |
| US20120041009A1 (en) | Pyrimidine compound and its use in pest control | |
| US20150336881A1 (en) | Amide compound and use thereof for pest control | |
| WO2011049150A1 (en) | Pyrone compound and its use for pest control | |
| WO2010119878A1 (en) | Pyrimidine compound and its use for pest control | |
| WO2010119879A1 (en) | Pyrimidine compound and its use for pest control |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080018451.8 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10769684 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13259058 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 8638/CHENP/2011 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010769684 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: PI1015315 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: PI1015315 Country of ref document: BR Kind code of ref document: A2 Effective date: 20111026 |