DERIVES DE THIOPHÈNE-2-CARBOXAMIDE, LEUR PREPARATION ET LEUR APPLICATION EN THERAPEUTIQUE.
La présente invention a pour objet des dérivés de 4,5-diarylthiophène-2- carboxamide, leur préparation et leur application en thérapeutique.
Des dérivés de diphénylpyrazole, présentant une affinité pour les récepteurs CBj des cannabinoïdes, ont été décrits notamment dans les brevets US 5 624 941, EP 0 576 357, EP 0 656 354 et EP 1 150 961.
Des dérivés de 5,6-diphényl-2-pyrazine-carboxamide sont décrits dans la demande internationale WO 03/051 850 comme des antagonistes des récepteurs CBj.
Des dérivés de l,2-diphényl-4-imidazole-carboxamide sont décrits dans la demande internationale WO 03/027 076 comme des agonistes des récepteurs CBj, des agonistes partiels ou des antagonistes.
Des dérivés de 4,5-diarylthiophène ayant des propriétés analgésiques sont décrits dans la demande internationale WO 91/19 708.
D'autres dérivés de 4,5-diarylthiophène sont décrits dans la demande internationale WO 2005/035 488 comme des antagonistes des récepteurs CBj.
On a maintenant trouvé des nouveaux dérivés de 4,5-diarylthiophène-2- carboxamide portant un substituant particulier sur l'un des groupes aryle qui possèdent des propriétés antagonistes des récepteurs CBj des cannabinoïdes au niveau central et au niveau périphérique. En particulier, ces nouveaux dérivés ont des propriétés antagonistes des récepteurs CBl périphériques et présentent une faible pénétration au niveau du cerveau.
Ainsi la présente invention a pour objet des composés répondant à la formule (I) :
- Rj et R2 ensemble avec l'atome d'azote auquel ils sont liés constituent :
O soit un radical hétérocyclique saturé de 5 à 7 atomes comprenant deux atomes d'azote, non substitué ou substitué par un groupe phényle, benzyle, benzodioxolyle, benzodioxolylméthyle, tétrahydrofuranylcarbonyle, -CORj j, et/ou -CH2CORJ J, le groupe phényle étant lui-même non substitué ou substitué
une ou plusieurs fois par un substituant choisi chacun indépendamment parmi un atome d'halogène, un groupe (Cj-C4)alkyle , trifluorométhyle, hydroxyle, (Cj-
C4)alcoxy et/ou cyano ; 0 soit un radical hétérocyclique saturé de 4 à 7 atomes comprenant un atome d'azote, non substitué ou substitué une ou deux fois par un substituant choisi chacun indépendamment parmi :
. un groupe cyano, -CORn, -CH2NHRj2, -(Cβ-CyXycloalkyle, -CH2COR11, -NR12Ri3, -NHCOR14, -SO2Ri4 » et/ou -SO2NR12Ri3 ;
. et/ou un groupe phényle, benzyle, pyridinyle ; lesdits groupes étant non substitués ou substitués une ou plusieurs fois par un substituant choisi chacun indépendamment parmi un atome d'halogène, un groupe (Cj-C4)alkyle, trifluorométhyle, hydroxyle, (Cj-C4)alcoxy et/ou cyano ;
. et/ou un groupe pipéridin-1-yle, pyrrolidin-1-yle, azétidin-1-yle, lesdits groupes étant non substitués ou substitués une ou plusieurs fois par un substituant choisi chacun indépendamment parmi un atome de fluor, un groupe (Cj-C4)alkyle, (Cj-C4)alcoxy, hydroxy, trifluorométhyle et/ou OCF3 ;
. et/ou un groupe, phénylamino, benzylamino, lesdits groupes étant non substitués ou substitués une ou plusieurs fois par un substituant choisi chacun indépendamment parmi un atome d'halogène, un groupe (Cj-C4)alkyle, trifluorométhyle, hydroxyle, (Cj-C4)alcoxy et/ou cyano ;
. et/ou un groupe amino(Cj-C(5)alkyle non substitué ou substitué une ou plusieurs fois par un substituant choisi chacun indépendamment parmi un atome d'halogène, un groupe hydroxyle, (Cj-C4)alcoxy, (Cβ-Cy^ycloalkyle et/ou cyano ;
. et/ou un groupe amino(C3-C7)cycloalkyle non substitué ou substitué une ou plusieurs fois par un substituant choisi chacun indépendamment parmi un atome d'halogène, un groupe hydroxyle, (Cj-C4)alkyle, (Cj-C4)alcoxy et/ou cyano, ledit groupe (Cj-C4)alkyle étant non substitué ou substitué une ou plusieurs fois par un atome de fluor ;
- R3, R4, R5, Rg, R7 et Rg représentent chacun indépendamment un atome d'hydrogène, un atome d'halogène, un groupement -CN, -S(O)nRj4, -OSO2Rj4, un groupe (Cj-Cg)alkyle et/ou un groupe (Cj-Cg)alcoxy , lesdits groupes étant non substitués ou substitués une ou plusieurs fois par un atome de fluor, à la condition que l'un des deux substituants R3, Rg représente un groupe Y-A-R9 ;
- Y représente un atome d'oxygène, un groupe -S(O)n'-, ou -OSO2 ;
- A représente un groupe alkylène en (Cj-C4) non substitué ou substitué une ou plusieurs fois par un groupe (C j-C3)alkyle et/ou par un atome de fluor ;
- R9 représente un groupe -OR19, -CN, -CH3, -CF3, -NR19R20, -CO2R19, -
CONR19R2O, -NR15COR19, -CONHNH2, -CONHOH, -CONHSO2R21 -S(O)nR21, -SO2NR19R20, -NR18SO2R21, -NR15SO2NR19R20 ;
- R10 représente un atome d'hydrogène ou un groupe (Q-C^alkyle, et de préférence un atome d'hydrogène ;
- R1 \ représente :
O un groupe
phényle, benzyle,
ou (C
1-C3)alkylène-0- (C
1-C3)alkyle, lesdits groupes étant non substitués ou substitués par un substituant choisi chacun indépendamment parmi un groupe
et/ou un groupe hydroxy et/ou par un ou plusieurs atomes de fluor ;
O et/ou un groupement -NR1^R1Y ;
- R
12 et R^ représentent chacun indépendamment un atome d'hydrogène ou un groupe
éventuellement substitué une ou plusieurs fois par un substituant choisi chacun indépendamment parmi un atome de fluor, un groupe -OH et/ou un groupe -OR^ ;
- ou R12 et R^ ensemble avec l'atome d'azote auquel ils sont liés constituent un radical hétérocyclique de 4 à 7 chaînons pouvant comporter un second hétéroatome choisi parmi un atome d'azote, d'oxygène ou de soufre ;
- n représente O, 1 ou 2 ;
- n' représente O, 1 ou 2 ;
- R^ représente un groupe (C1-C^aIkVIe non substitué ou substitué une ou plusieurs fois par un atome de fluor ;
- R15 représente un atome d'hydrogène ou un groupe (C1-C^aIkVIe ;
- R1^ et R1Y représentent chacun indépendamment : 0 un atome d'hydrogène ;
0 et/ou un groupe benzyle non substitué ou substitué une ou plusieurs fois par un substituant choisi chacun indépendamment parmi un atome d'halogène, un groupe (C
1-C4)alkyle, trifluorométhyle, hydroxyle,
et/ou cyano ;
0 et/ou un groupe
éventuellement substitué une ou plusieurs fois par un substituant choisi chacun indépendamment parmi un atome d'halogène, un groupe -OH et/ou -OR^ ;
- R18 représente un atome d'hydrogène ou un groupe (Q-C^alkyle non substitué ou substitué une ou plusieurs fois par un atome de fluor ;
- R
19 et R
20 représentent chacun indépendamment un atome d'hydrogène ou un groupe
éventuellement substitué une ou plusieurs fois par un substituant choisi chacun indépendamment parmi un atome de fluor, un groupe -OH et/ou un groupe -OR^ ;
- ou Rj 9 et R20 ensemble avec l'atome d'azote auquel ils sont liés constituent un radical hétérocyclique de 4 à 7 chaînons pouvant comporter un second hétéroatome choisi parmi un atome d'azote, d'oxygène ou de soufre ;
- R21 représente un groupe (Ci-C4)alkyle non substitué ou substitué une ou plusieurs fois par un atome de fluor ; à l'état de bases (= correspondent aux formes libres des composés) ainsi que leurs sels acceptables pharmaceutiquement ou acceptables pour la purification et/ou l'isolement desdits composés de formule (I).
Les composés de formule (I) peuvent comporter un ou plusieurs atomes de carbone asymétriques. Ils peuvent donc exister sous forme d'énantiomères ou de diastéréoisoméres. Ces énantiomères, diastéréoisomères, ainsi que leurs mélanges, y compris les mélanges racémiques font partie de l'invention.
Les composés de formule (I) peuvent exister à l'état de bases (c'est-à-dire tels quels sous leurs formes libres), de sels d'addition à des acides ou de sels d'addition à des bases. Ces sels sont avantageusement préparés avec des sels pharmaceutiquement acceptables ; les sels d'autres acides utiles, par exemple, pour la purification ou l'isolement des composés de formule (I) font également partie de l'invention.
Par groupe alkyle, on entend un radical carboné linéaire ou ramifié, tel qu'en particulier : méthyle, éthyle, propyle, isopropyle, butyle, isobutyle, tert-butyle, n-pentyle, isopentyle, n-hexyle, isohexyle. Le groupe méthyle est préféré pour un (C\- C4)alkyle et pour un (Ci-Cg)alkyle.
Par groupe aminoalkyle, on entend un groupe amino lié à un radical carboné linéaire ou ramifié, tel que par exemple : méthyle, éthyle, propyle, isopropyle, butyle, isobutyle, tert-butyle, n-pentyle, isopentyle, n-hexyle, isohexyle, heptyle, isoheptyle.
Par groupe alkylène, on entend un radical carboné bivalent linéaire tel que -(CH2)-, -(CH2)2-, -(CH2)3-, -(CH2)4-, -(CH2)5-.
Par (Cj-C4)alcoxy et (Cj-C^alcoxy , on entend respectivement un atome d'oxygène lié à un radical carboné linéaire ou ramifié de un à quatre atomes de carbones et de un à six atomes de carbones, tel que le radical méthoxy, éthoxy, propoxy, isopropoxy, butoxy, sec-butoxy, tert-butoxy, pentoxy, hexyloxy, le groupe méthoxy étant préféré.
Par atome d'halogène, on entend un atome de fluor, de chlore, de brome ou d'iode, les atomes de fluor, chlore ou brome étant préférés.
Par groupe cycloalkyle, on entend un radical carboné cyclopropyle, cyclobutyle, cyclopentyle, cyclohexyle, cycloheptyle.
Par groupe aminocycloalkyle, on entend un groupe amino lié à un radical carboné cyclique, tel que par exemple : cyclopropyle, cyclobutyle, cyclopentyle, cyclohexyle, cycloheptyle.
Par radical hétérocyclique, saturé de 4 à 7 chaînons, contenant 1 ou 2 atomes d'azote, on entend notamment des radicaux tels que azétidin-1-yle, pyrrolidin-1-yle, pipéridin-1-yle, homopipéridin-1-yle, imidazolin-1-yle, pipérazin-1-yle et 1 ,4-diazépan- 1-yle.
Selon la présente invention, on distingue :
- les composés de formule (IA) dans laquelle Y représente un atome d'oxygène ;
- les composés de formule (IB) dans laquelle Y représente un groupe -S(O)n'- ;
- les composés de formule (IC) dans laquelle Y représente un groupe -O(SO2)-;
- les autres substituants étant tels que définis pour les composés de formule (I).
Au sein des composés de formules (I), (IA), (IB) et (IC), on distingue en particulier :
- les composés dans lesquels le substituant R3 représente un groupe Y-A-R9 et le substituant Rg représente un atome d'hydrogène, un atome d'halogène, un groupement -CN, -S(O)nRj 4, -OSO2R14, un groupe (Cj-Cg)alkyle ou un groupe (Ci-Cg) alcoxy, lesdits groupes étant non substitués ou substitués par un ou plusieurs atomes de fluor ;
- et les composés dans lesquels le substituant Rg représente un groupe Y-A-R9 et le substituant R3 représente un atome d'hydrogène, un atome d'halogène, un groupement -CN, -S(O)nR^, -OSO2R14, un groupe (Ci-Cg)alkyle ou un groupe (C i"Cg) alcoxy, lesdits groupes étant non substitués ou substitués par un ou plusieurs atomes de fluor ;
- les autres substituants étant tels que définis ci-avant pour les composés de formule (I).
De préférence pour les composés dans lesquels le substituant R3 représente un groupe Y-A-R9, R4, R5, Rg, R7 et Rg représentent chacun indépendamment un atome d'hydrogène, un atome d'halogène, un groupement -CN et/ou un groupe (Ci-Cg)alkyle, ledit groupe étant non substitué ou substitué une ou plusieurs fois par un atome de fluor.
De préférence pour les composés dans lesquels le substituant Rg représente un groupe Y-A-R9, R3, R4, R5, R7 et Rg représentent chacun indépendamment un atome d'hydrogène, un atome d'halogène, un groupement -CN et/ou un groupe (Ci-Cg)alkyle, ledit groupe étant non substitué ou substitué une ou plusieurs fois par un atome de fluor.
Pour ces deux modes préférentiels, les autres substituants sont tels que définis ci- avant pour les composés de formule (I).
Pour les composés dans lesquels le substituant R3 représente un groupe Y-A-R9, le substituant R4 et/ou le substituant R5 correspondent de préférence à un atome
d'hydrogène. Plus préférentiellement, R3 est en position 4 sur le phényle. Pour cette variante des composés de formule (I) selon l'invention, on préfère de plus Rg correspondant à un atome d'hydrogène ou d'halogène tel que le chlore ou le fluor, R7 correspondant à un atome d'halogène, de préférence un atome de chlore et Rg correspondant à un atome d'hydrogène.
Selon la présente invention, on préfère les composés de formule (I) dans lesquels :
- A représente un groupe alkylène en (C1-C4) non substitué ;
- R9 représente un groupe -ORj 9, -CH3, -CF3, -NR19R2Q, -CONR19R2Q, -
NR15COR19, -S(O)nR21, ou -NR18SO2R2I ;
- et les autres substituants étant tels que définis ci-avant pour les composés de formule (I).
En particulier pour Y, on préfère un atome d'oxygène ou un atome de soufre. En particulier pour R9, on préfère un groupe -OR19, -NR19R2Q, -CONR19R20, -S(O)nR21, ou -NR18SO2R21.
Selon la présente invention, on préfère les composés de formule (I) dans lesquels :
- R1 et R2 ensemble avec l'atome d'azote auquel ils sont liés constituent un radical homopipéridin-1-yle, pipéridin-1-yle, pyrrolidin-1-yle ou azétidin-1-yle, lesdits radicaux étant substitués une ou deux fois par un substituant choisi chacun indépendamment parmi :
O un groupe cyano, -COR1 1, -NR12R13, -NHCOR1^ -CH2COR1 1, -SO2R1^ et/ou -SO2NR12R13 ;
O et/ou un groupe phényle, benzyle, pyridinyle ; lesdits groupes étant non substitués ou substitués une ou plusieurs fois par un substituant choisi chacun indépendamment parmi un atome d'halogène, un groupe (Q-C^alkyle, trifluorométhyle, hydroxyle,
et/ou cyano ;
O et/ou un groupe pipéridin-1-yle, pyrrolidin-1-yle, azétidin-1-yle, lesdits groupes étant non substitués ou substitués une ou plusieurs fois par un substituant choisi chacun indépendamment parmi un atome de fluor, un groupe (Q-C^alkyle, (C1- C4)alcoxy, hydroxy, trifluorométhyle et/ou OCF3 ;
O et/ou un groupe phénylamino, benzylamino, lesdits groupes étant non substitués ou substitués une ou plusieurs fois par un substituant choisi chacun indépendamment parmi un atome d'halogène, un groupe (Q-C^alkyle, trifluorométhyle, hydroxyle,
et/ou cyano ;
O et/ou un groupe amino (C
1-Cg)alkyle non substitué ou substitué une ou plusieurs fois par un substituant choisi chacun indépendamment parmi un atome d'halogène, un groupe hydroxyle,
et/ou cyano ;
O et/ou un groupe amino (Cβ-Cy^ycloalkyle non substitué ou substitué une ou plusieurs fois par un substituant choisi chacun indépendamment parmi un atome d'halogène, un groupe hydroxyle, (Cj-C^alkyle, (Cj-C^alcoxy et/ou cyano, ledit groupe (Ci-C4)alkyle étant non substitué ou substitué une ou plusieurs fois par un atome de fluor ;
- les autres substituants étant tels que définis pour les composés de formule (I).
En particulier, on préfère les composés de formule (I) dans lesquels :
- R1 et R2 ensemble avec l'atome d'azote auquel ils sont liés constituent un radical homopipéridin-1-yle, pipéridin-1-yle, pyrrolidin-1-yle ou azétidin-1-yle, ledit radical étant gem-disubstitué :
O le premier substituant dudit radical étant choisi parmi un groupe cyano, -COR1 1 ? -
NHCOR14, ou -SO2Ri4 ; O le second substituant dudit radical étant choisi parmi :
. NR12Ri3;
. et/ou un groupe phényle, ledit groupe étant non substitué ou substitué une ou plusieurs fois par un substituant choisi chacun indépendamment parmi un atome d'halogène, un groupe (Cj-C^alkyle, trifluorométhyle, hydroxyle, (Cj-C^alcoxy et/ou cyano ;
. et/ou un groupe pipéridin-1-yle, ledit groupe étant non substitué ou substitué une ou plusieurs fois par un substituant choisi chacun indépendamment parmi un atome de fluor, un groupe (Cj-C4)alkyle, (C^-C^alcoxy, hydroxy, trifluorométhyle et/ou OCF3 ;
. et/ou un groupe benzylamino, ledit groupe étant non substitué ou substitué une ou plusieurs fois par un substituant choisi chacun indépendamment parmi un atome d'halogène, un groupe (Cj-C4)alkyle, trifluorométhyle, hydroxyle, (C1-
C4)alcoxy et/ou cyano ;
- les autres substituants étant tels que définis pour les composés de formule (I).
Plus particulièrement, on préfère les composés de formule (I) dans lesquels R1 et R2 ensemble avec l'atome d'azote auquel ils sont liés constituent un radical pipéridin-1- yle ou azétidin-1-yle. Dans ce cas :
- le premier substituant du radical pipéridin-1-yle ou azétidin-1-yle est de préférence
-COR1 1 ;
- et le second substituant du radical pipéridin-1-yle ou azétidin-1-yle est de préférence choisi parmi -NR12R13, un groupe phényle, un groupe benzylamino ou un groupe pipéridin-1-yle, ledit groupe pipéridin-1-yle étant non substitué ou substitué une ou plusieurs fois par un substituant choisi chacun indépendamment parmi un atome de
fluor, un groupe (Cj-C^alkyle, (Cj-C^alcoxy, hydroxy, trifluorométhyle et/ou OCF3 ; - les autres substituants étant tels que définis pour les composés de formule (I).
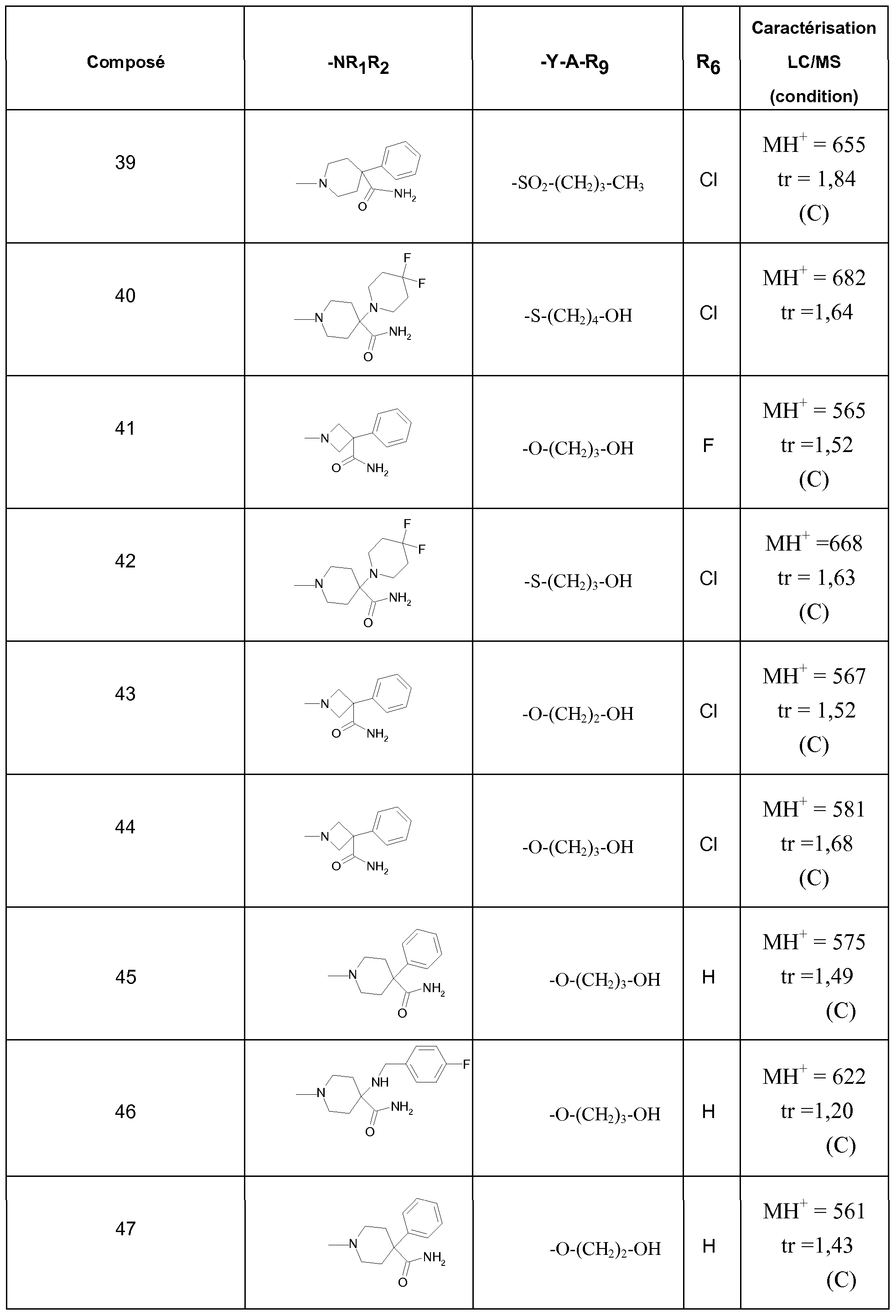
Parmi les composés selon l'invention, on peut notamment citer les composés ci- après, tels quels ainsi que leurs sels :
La présente invention a également pour objet un procédé de préparation des composés selon l'invention.
Ce procédé est caractérisé en ce que l'on traite avec une aminé de formule (III) HNRi ^2 dans laquelle R^ et R2 sont tels que définis pour (I) l'acide de formule (II) ou un dérivé fonctionnel de cet acide de formule (II) :
dans laquelle R3, R4, R5, Rg, R7, Rg et RJQ sont tels que définis pour (I) et le groupe Y-A-R9 est remplacé par un groupe Z précurseur chimique du groupe Y-A-R9
Par groupe Z, on entend des groupes chimiques qui conduisent après une ou plusieurs étapes réactionnelles connues de l'homme de l'art aux groupes Y-A-R9. Par exemple, le groupe précurseur Z correspond à Y-H, un atome d'halogène, un groupe Y- A-OH, Y-A-Cl, Y-A-CO2AIk, Y-A-S-AIk, Y-AIk, Y-A-OPr, Y-A-NHPg. Le radical AIk correspond à un radical carboné linéaire ou ramifié comprenant notamment 1 à 6 atomes de carbone, et de préférence correspond à un radical méthyle. Le groupe Pg correspond à un groupe protecteur de la fonction aminé, par exemple le tert- butyloxycarbonyl. D'autres exemples de groupes protecteurs Pg sont donnés dans «Protective Group in Organic Synthesis», Green et al, 4e édition , John Wiley & Sons, Inc., New York, 2007. Le groupe Pr correspond à un groupe protecteur de la fonction alcool ; par exemple, le tétrahydropyranyle (THP), le tétrabutyldiméthylsilyle (TBDMS) et le triméthylsilyle (TMS).
Les composés de formule (I) obtenus par les différents modes opératoires peuvent être ultérieurement séparés du milieu réactionnel et purifiés selon les méthodes classiques, par exemple par cristallisation ou chromatographie.
Eventuellement, on transforme le composé de formule (I) ainsi obtenu en un de ses sels.
Comme dérivé fonctionnel de l'acide (II) on peut utiliser le chlorure d'acide, l'anhydride, un anhydride mixte, un ester alkylique en CJ-C4 dans lequel l'alkyle est droit ou ramifié, un ester benzylique, un ester activé, par exemple l'ester de p-nitrophényle, ou l'acide libre opportunément activé, par exemple avec un agent
de couplage tel que le N,N-dicyclohexylcarbodiimide, l'hexafluorophosphate de benzotriazol-l-yloxotris(diméthylamino)-phosphonium (BOP), l'hexafluorophosphate de benzotriazol-l-yloxotris-(pyrrolidino)phosphonium (PyBOP), l'hexafluorophosphate de 2-(l-H-benzotriazol-l-yl)-l, 1, 3, 3-tétraméthyl-uronium) (HBTU) ou le O-benzotriazol 1 yl N, N, N', N', tetraméthylurronium tetrafluoroborate (TBTU).
Ainsi dans le procédé selon l'invention, on peut faire réagir le chlorure de l'acide de formule (II), obtenu par réaction du chlorure de thionyle ou du l-chloro-N,N-2- triméthyl-l-propen-1 -aminé effectué selon Chem. Comm., 1988, 475-477, à 00C sur l'acide de formule (II), avec une aminé HNRj R-2, dans un solvant inerte, tel qu'un solvant chloré (le dichlorométhane, le dichloroéthane, le chloroforme par exemple), un éther (tétrahydrofurane, dioxane par exemple), ou un amide (N,N-diméthylformamide par exemple) sous une atmosphère inerte, à une température comprise entre 00C et la température ambiante, en présence d'une aminé tertiaire telle que la triéthylamine, la N- méthylmorpholine ou la pyridine.
Une variante consiste à préparer l'anhydride mixte de l'acide de formule (II) par réaction du chloroformiate d'éthyle avec l'acide de formule (II), en présence d'une base telle que la triéthylamine, et à le faire réagir avec une aminé HNRJR^, dans un solvant tel que le dichlorométhane, sous une atmosphère inerte, à la température ambiante, en présence d'une base telle que la triéthylamine.
Les composés de formule (II) peuvent être préparés selon le schéma 1 ci-après.
Pour ce mode de préparation, le groupe précurseur Z est :
- soit présent au sein du composé (IV) ; dans ce cas R3 représente le groupe Z.
- soit présent au sein du composé (V); dans ce cas Rg représente le groupe Z.
SCHEMA 1
La préparation des composés de formule (VI) s'effectue à partir des composés de formule (IV) et (V) en présence d'une base forte telle que par exemple l'hexaméthyldisilazane de sodium (NaHMDS), Phexaméthyldisilazane de lithium (LiHMDS), le dicyclohexylamide de sodium (LiNCy2) ou le diisopropylamide de lithium (LDA).
Après réaction à l'étape (bl) avec la l,l-diméthoxy-N,N- diméthylméthanamine, on fait réagir les composés (VII) avec le trichlorure de phosphoryle suivi d'une hydrolyse en présence d'un mélange solvant-eau tel que par exemple le tétrahydofurane-eau ou le dioxane-eau.
La cyclisation par le mercaptoacétate de méthyle à l'étape (dl) est effectuée en présence d'une base tel que par exemple une aminé encombrée : le 1,8 diazabicyclo[5,4,0] undec-7-ène (DBU) ou l,3-diazabicyclo[5.4.0]undécane (DBN). Les composés de formule (IX) obtenus sont ensuite traités avec de l'hydroxyde de sodium pour conduire aux composés de formule (II).
Les composés de formule (II) et dérivés fonctionnels de l'acide (II) peuvent également être préparés selon les modes de préparations décrits dans la demande internationale WO 2005/035 488 (voir notamment page 4 ligne 31 à page 9 ligne 32).
Ces composés de formule (II) et dérivés fonctionnels de l' acides (II) conduisent ensuite en une ou plusieurs étapes aux composés de formule (I) selon l'invention.
Ainsi, selon le schéma 2 ci-après, on part de l'acide de formule (lia) dans laquelle Rg est remplacé par le groupe Z. Cet acide est traité avec une aminé de formule (III) HNR \ R-2, Ri et R2 étant tels que définis pour (I). On obtient un amide de formule (Xa).
Puis le groupe Z du composé de formule (Xa) obtenu est transformé en une ou plusieurs étapes en groupe Y-A-R9 par une des méthodes connues de l'homme de l'art pour conduire aux composés de formule (I).
SCHEMA 2
(lia) (Xa)
(I)
Ou bien selon le schéma 2' ci-après, on part de l'acide de formule (Hb) dans laquelle R3 est remplacé par le groupe Z. Cet acide est traité avec une aminé de formule (III) HNR 1R2, Ri et R2 étant tels que définis pour (I). On obtient un amide de formule (Xb).
Puis le groupe Z du composé de formule (Xb) obtenu est transformé en une ou plusieurs étapes en groupe Y-A-R9 par une des méthodes connues de l'homme de l'art pour conduire aux composés de formule (I).
SCHEMA 2'
(Mb) (Xb) (I)
A titre d'exemples, les composés de formule (I) dans laquelle Y correspond à un atome d'oxygène et R9 représente un groupe OR19, CO2R19, CONR19R20, NR19R20, NRigSθ2R2b S(O)nR2i peuvent être obtenus à partir de composés de formule (Hb)
dans laquelle R3 est remplacé par Z, et Z correspond à Y-AIk, AIk étant un radical carboné linéaire ou ramifié, de préférence un radical méthyle.
Selon le schéma 3, le composé (XI) obtenu par réaction du composé (Hb) et de l'aminé HNR4R2 est traité à l'étape (b3) avec du tribromure de bore puis le composé (XII) est traité à l'étape (c3) avec un chloroalcanol de formule Cl-A-OH ou bromoalcanol de formule Br-A-OH en milieu alcalin (ex: carbonate de potassium ou de césium) pour mener au composé de formule (I) dans laquelle R9 correspond à un radical -OH.
SCHEMA 3
Les composés de formule (I) obtenus à l'issu du schéma réactionnel 3, référencés (F), peuvent conduire à d'autres composés de formule (I).
Notamment selon le schéma 4, les composés (F) permettent de préparer les composés de formule (I) dans laquelle Y-A-R9 correspond à Y-A-S-R21. A l'étape (a4), le composé de formule (F) est traité avec du chlorure de mésyle en présence d'une aminé tel que par exemple la triéthy lamine. Puis, le composé obtenu est traité avec un alkylthiolate de sodium de formule -R2iSNa pour mener au composé de formule (I") dans laquelle R9 correspond à un radical -S-R21.
SCHEMA 4
(I")
En accord avec le schéma 5, les composés de formule (I) obtenus à l'issu du schéma réactionnel 4, référencés (I"), peuvent ensuite être traités avec de l'acide 3- chloroperoxybenzoique (MCPBA) pour conduire aux composés de formule (F") dans laquelle R9 correspond à un radical -S(O)n'-R2i avec n' égal à 1 ou 2.
SCHEMA 5
(I") (I-)
Les composés de formule (XII) peuvent conduire à d'autres composés de formule (I). Par exemple, on ajoute dans une première étape à un composé de formule (XII) du tert-butyi (3-bromopropyl)carbamate puis dans une deuxième étape du TFA pour obtenir un composé de formule (I) dans laquelle -R9 correspond à un radical -NH2.
Les composés de formule (II) permettent également de conduire à des composés de formule (I) dans laquelle Y correspond au radical divalent -S(O)n'- et -R9 représente un radical -ORi 9, -CH3 ou -CF3.
Par exemple, on part d'un composé de formule (Hb) dans laquelle Z correspond à un atome de Br.
Selon le schéma 6, le composé de formule (Hb) avec Z correspondant à Br est mis en contact avec un réactif de formule HS-A-ORj 9 et de Phydrure de sodium. On ajoute ensuite un catalyseur tel que Pd2(dba)3 et un ligand tel que le composé organophosphoré Xantphos pour arriver à un composé de formule (XIII). Ce composé de formule (XIII) correspond à un composé de formule (I) dans lequel- Y- A-R9 est -S-A-ORj 9 .
SCHEMA 6
Selon le schéma 7, le composé obtenu (XIII) via le mode opératoire du schéma 6 peut être ensuite traité avec du MCPBA pour oxyder le soufre présent sur la chaine latérale du phényle et mener à des composés de formule (I) dans laquelle -Y-A-R9 est -(SO)-A-O Rj 9 (ci-après le composé de formule (XIV) ou -SO2-A-OR19 (ci-après le composé de formule (XV).
SCHEMA 7
En remplaçant dans la séquence réactionnelle du schéma 6 le réactif HS-A-ORj 9 par un réactif de formule HS-A-CH3 ou HS-A-CF3, les composés de formules (XIIF) et (XIII") avec n" égal à O, 1 ou 2 sont préparés :
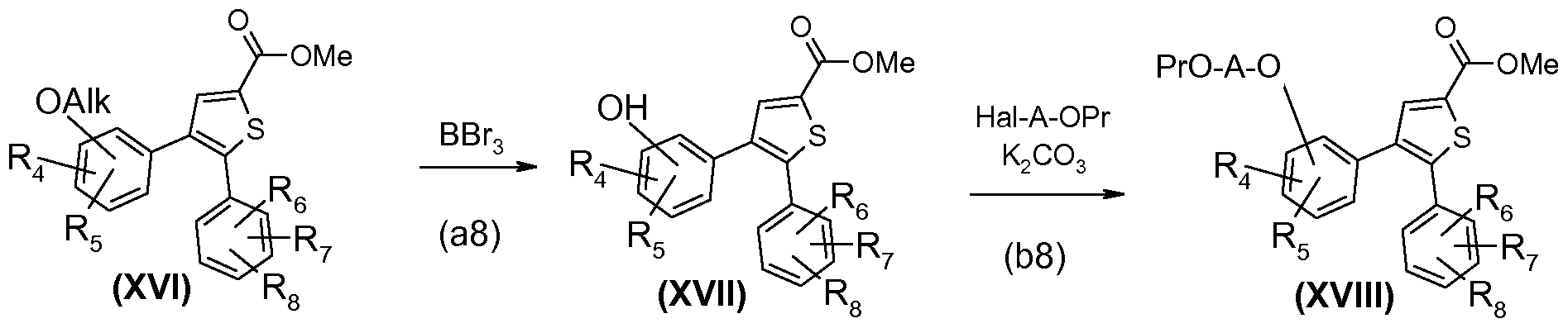
Les composés de formule (IX) où R3 ou Rg correspond à Z et Z est OAIk permettent également de conduire à des composés de formule (I) où Y-A-R9 correspond à 0-A-OH ou O-A-NHR19 selon le schéma 8 réactionnel ci-après.
Ainsi selon le schéma 8, on part du composé de formule (IX) où R3 correspond à Z et Z est OAIk, référencé ci-après (XVI).
Alternativement, on peut partir du composé de formule (IX) où Rg correspond à Z et Z est OAIk. Dans ce cas, le radical fonctionnalisé Y-A- R9 du composé final est présent sur le phényle en position 5 du thiophène.
SCHEMA 8
Selon le schéma 8, le composé de formule (XVI) est traité avec BBrç Le composé obtenu de formule (XVII) est alkylé par un dérivé halogène de formule HaI-A-OPr avec HaI égal à Br ou Cl pour obtenir un composé intermédiaire de formule (XVIII) dans laquelle Z est devenu 0-A-OPr.
Après saponification avec par exemple NaOH, l'acide de formule (XIX) est traité à l'étape (d8) avec une aminé de formule HNRjR2, en présence d'un agent de couplage
tel que par exemple TBTU. Puis, le composé obtenu est traité en milieu acide pour conduire au composé de formule (F).
A l'étape (b8), on peut remplacer HaI-A-OPr par HaI-A-NR^Pg . Dans ce cas, on obtient après saponification un composé intermédiaire de formule (XXI) qui conduit ensuite aux composés de formule (I) où Y-A-R9 correspond à O-A-NHR49.
SCHEMA 9
A l'étape (b8), on peut également remplacer HaI-A-OPr par HaI- A-NR 19R2O avec Rj 9 et R20 différents d'un atome d'hydrogène. Dans ce cas, on obtient après saponification un composé intermédiaire de formule (XXIII) qui conduit ensuite aux composés de formule (I) où Y-A-R9 correspond à 0-A-NR^R2O-
SCHEMA 10
A partir des composés de formule (XVIII), on peut de plus préparer en suivant le schéma réactionnel 11 des composés de formule (I) où Y-A-R9 correspond à O- A- SR2I.
A l'issue de l'étape (bl l), les composés intermédiaires de formule (XXV) peuvent ensuite être traités avec de l'acide 3-chloroperoxybenzoique (MCPBA) pour conduire à des composés intermédiaires (XXV) dans laquelle -R9 correspond à un radical - S(0)n'-R2i avec n' égal à 1 ou 2. Ensuite, ces composés (XXV) suivent les étapes
(ci l) et (dl l) pour mener à des composés de formule (F") où Y-A-R9 correspond à 0-A-S(O)n-R2I .
SCHEMA 11
A partir de composés de formule (XXVII) en suivant le schéma réactionnel 12, on peut préparer des composés de formule (I) où Y-A-R9 correspond à S-A-OR19. L'étape (a 12) est réalisée dans des conditions similaires à celles du schéma réactionnel 6 et l'étape (bl2) de saponification puis celle de couplage (cl 2) sont effectuées dans des conditions similaires à celles des étapes (c8) et (d8) du schéma réactionnel 8.
SCHEMA 12
Les aminés de formule (III) HNR jR-2 sont connues ou préparées par des méthodes connues, par exemple celle décrite dans J. Med. Chem., 7, 1964, 619-622.
Les composés intermédiaires de formules (XXII), (XXIII), (XXV), (XXVI), (XXVIII) et (XXIX) sont nouveaux et sont utilisés pour la préparation des composés de formule (I).
Les composés intermédiaires de formule (XXVII) correspondent aux composés de formule (IX) dans lequel R3 correspond à un atome de brome. Ces composés de formule (XXVII) sont préparés selon le schéma réactionnel 1.
La présente invention a également pour objet les composés de formules (XXX) (XXXI) et (XXXII), incluant notamment les composés de formules (XXII), (XXIII), (XXV), (XXVI), (XXVIII), et (XXIX). Ces composés sont utilisés pour la préparation des composés de formule (I) et correspondent aux formules suivantes :
dans lesquelles :
- W représente un groupe (Cj-C^alcoxy, de préférence méthoxy, un halogène, de préférence un atome de chlore ou un radical hydroxyle, - et les autres substituants sont tels que définis pour les composés de formule (I).
Lorsque W représente un groupe (Cj-C^alcoxy, on traite les composés de formule (XXX), (XXXI) ou (XXXII) avec un agent de saponification tel que par exemple NaOH puis avec une aminé de formule (III) HNR \K2 dans laquelle K\ et R2 sont tels que définis pour (I) pour préparer les composés de formule (I).
Lorsque W représente un radical hydroxyle ou un atome de chlore, on traite les composés de formule (XXX), (XXXI) ou (XXXII) avec une aminé de formule (III) HNRi ^2 dans laquelle R^ et R2 sont tels que définis pour (I) pour préparer les composés de formule (I).
Les exemples suivants décrivent la préparation de certains composés conformes à l'invention. Ces exemples ne sont pas limitatifs et ne font qu'illustrer la présente invention.
Dans les exemples, on utilise les abréviations suivantes :
AcOEt : acétate d'éthyle
ACONH4 : acétate d'ammonium
BBrβ : tribromure de bore
CH2CI2 : dichlorométhane
DBU : 1,8 diazabicyclo[ 5,4,0 ]undèc-7-ene
DIPEA : diisopropyléthylamine
DCM : dichlorométhane
DMF : N,N-diméthylformamide.
Et3N : triéthylamine
HPLC : chromatographie en phase liquide sous haute pression
LiHMDS : hexaméthyldisilazane de lithium
MeOH : méthanol
MCPBA : acide 3-chloroperoxybenzoique
MsCl : chlorure de mésyle
NaHMDS : hexaméthyldisilylane de sodium
NH3 : ammoniac
NH4OH : ammoniaque
Pd(dba)2 : bis(dibenzyllideneacétone)palladium
Pd2(dba)3 : tris(dibenzyllideneacétone)dipalladium
POCI3 : trichlorure de phosphoryle
P(tBu)3 : triterbutylphosphine
TA : température ambiante
TBTU : O-benzotriazol-1-yl-N, N, N', N'- tétraméthylurronium tetrafluoroborate
Tf : point de fusion
TFA : acide trifluoroacétique
THF : tétrahydrofurane
UPLC : chromatographie en phase liquide 'Ultra Performance'
Xantphos :
Les spectres de résonance magnétique nucléaire sont enregistrés à 250 MHz ou 400 MHz dans le DMSO-d6. Pour l'interprétation des spectres, on utilise les abréviations suivantes : s : singulet, d : doublet, t : triplet, q : quadruplet, qui : quintuplet, m : massif, si : singulet large, dd : doublet de doublet. Les composés selon l'invention sont analysés par couplage LC/UV/MS (chromatographie liquide/détection UV/spectrométrie de masse). On mesure le pic moléculaire caractéristique (MH+) et le temps de rétention (tr) en minutes (min).
Les composés sont analysés par couplage HPLC-UV-MS ou bien UPLC-UV-MS (chromatographie liquide-détection UV et détection masse). Les conditions analytiques sont les suivantes :
Conditions A (HPLC) :
On utilise une colonne: Symmetry C18 (50 x 2,1 mm ; 3,5 μm)
Eluant A : 0,005 % d'acide trifluoroacétique (TFA) dans l'eau à environ pH 3,1
Eluant B : 0,005% de TFA 0.005% dans l'acétonitrile.
Gradient:
Température colonne : 300C ; débit : 0,4 ml/minute. Détection : λ = 210 nm - 220 nm
Conditions B (HPLC) :
On utilise une colonne XT erra MS C18 ( 50 x 2,1 mm ; 3,5 μm)
Eluant A : AcONH4 1OmM à environ pH 7
Eluant B : acétonitrile
Gradient :
Température colonne : 300C ; débit : 0,4 ml/minute. Détection : λ = 220 nm
Conditions C (UPLC) :
On utilise une colonne Acquity BEH C18 ( 50 x 2,1 mm ; 1,7 μm)
Eluant A : 0,005 % de TFA dans l'eau à environ pH 3,1 / acétonitrile (97 / 3)
Eluant B : 0,035% de TFA dans l' acétonitrile.
Gradient:
Température colonne : 400C ; débit : 1 ml/minute. Détection : λ = 220 nm
Conditions de spectrométrie de masse
L'enregistrement des spectres de masse est effectué en mode electrospray (ESI) positif, afin d'observer les ions issus de la protonation de composés analysés (MH+), ou de la formation d'adduits avec d'autres cations tels que Na+, K+, etc.
PREPARATIONS
Préparation 1 : 4-[(3,3,3-trifluoropropyl)amino]pipéridine-4-carboxamide.
(i) 4-Carbamoyl-4-[(3,3,3-trifluoropropyl)amino]pipéridine-l-carboxylate de tert- butyle.
Dans 250 ml de CH2CI2 on ajoute à TA, 10 g de 4-amino-4-carbamoylpipéridine- 1-carboxylate de tert-butyle, 4,42 g de 3,3,3-trifluoro-propionaldéhyde, 17,61 g de triacétoxyborohydrure de sodium, 4,51 ml d'acide acétique puis on agite le mélange pendant 3 heures. On ajoute de l'eau et extrait le composé attendu au CH2CI2, lave la phase organique avec une solution saturée de NaHCOβ, puis avec de l'eau. Après séchage de la phase organique, fïltration et évaporation à sec, on obtient 13 g de composé attendu.
(ii) 4-[(3,3,3-trifluoropropyl)amino]pipéridine-4-carboxamide
On agite 13 g de composé obtenu en (i) dans 25 ml de MeOH, puis on ajoute 30 ml d'éther chlorhydrique (2M) et on laisse sous agitation pendant une nuit. Après fïltration et séchage on obtient 9, 57 g de composé attendu.
Préparation 2 : Chlorhydrate de 4-phénylpipéridine-4-carboxamide.
(i) Chlorhydrate de l-benzyl-4-phénylpipéridine-4-carboxamide.
Dans 77 ml d'acide sulfurique concentré, on ajoute 10 g de l-benzyl-4-phényl- pipéridine-4-carbonitrile puis chauffe à 1000C pendant une heure. Le mélange réactionnel est ensuite ajouté sur de la glace puis basifîé avec NH4OH. Le produit est extrait au CH2CI2, séché, évaporé. Après cristallisation du chlorhydrate dans l'éther, on obtient 5,7 g de produit attendu.
(ii) Chlorhydrate de 4-phénylpipéridine-4-carboxamide.
Dans 70 ml de MeOH, on ajoute 5,6 g de produit obtenu en (i), 6,7 g de cyclohexadiène, 0,5 g de palladium à 10% sur charbon et on chauffe le mélange réactionnel à reflux pendant 6 heures. On filtre le catalyseur sur de la célite et évapore à sec. Après cristallisation dans un mélange éther-éther isopropylique, on obtient 3 g de produit attendu.
Préparation 3 : Formiate de 3-phényl-azétidine-3-carboxamide.
(i) 1 -Benzyl-3-méthyl- 1 ,3-azétidinedicarboxylate.
Dans 150 ml de CH2CI2, on ajoute 10 g de chlorhydrate d'ester méthylique d'acide azétidine-3-carboxylique puis 23 ml de triéthylamine. On refroidit et ajoute 13,5 g de benzyl chloroformate. Après une nuit à TA, on lave à l'eau puis à HCl (N). Après séchage, concentration à sec et purification par chromatographie sur silice (éluant : Heptane- AcOEt (gradient de 0% à 50%)), on obtient 13, 1 g de composé attendu.
(ii) 1 -Benzyl-3-méthyl-3-phényl- 1 ,3-azétidinedicarboxylate
Dans 40 ml de toluène, on ajoute 5,11 ml de bromo-benzène puis 0,5 g de Pd(dba)2, 2,15 ml de solution à 10% dans l'hexane de triterbutylphosphine (P(tBu)3) puis 8,4 g de LiHMDS. On ajoute 11 g du composé obtenu à l'étape précédente (i) dans 10 ml de toluène en maintenant la température entre 15 et 200C. Après une nuit, on verse sur une solution saturée de NH4CI, extrait à l'éther, sèche sur MgSÛ4 et concentre à sec. Après purification par chromatographie sur silice (éluant : Heptane- AcOEt (gradient de 0% à 30%)), on obtient 3,25 g de composé attendu.
(iii) Ester benzylique d'acide 3-phényl-azétidine-l,3-dicarboxylique.
Dans 40 ml de méthanol, on ajoute 3,7 g de composé obtenu à l'étape précédente (ii) puis 3 ml de soude à 30%. On chauffe à reflux pendant 1 heure puis on concentre à sec. On reprend dans l'eau, lave à l'éther, puis acidifie avec HCl concentré. Après
extraction à l'éther, séchage, concentration à sec et cristallisation dans le pentane, on obtient 2,87 g de composé attendu.
(iv) Ester benzylique d'acide 3-carbamoyl-3-phényl-azétidine-l-carboxylique.
Dans 60 ml de CH2CI2, on ajoute 2,87 g de composé obtenu à l'étape précédente (iii), 2,4 ml de DIPEA puis 3,3 g de TBTU. On fait barboter du NH3 dans le milieu en maintenant la température à 25°C. On agite à TA pendant une nuit. On concentre à sec, reprend dans l'acétate d'éthyle, lave à l'eau puis à l'HCl (N) et finalement avec une solution aqueuse à 10% de NaHCOβ.
Après séchage, concentration à sec puis cristallisation dans l'éther, on obtient 1,51 g de composé attendu.
(v) Formiate de 3-phényl-azétidine-3-carboxamide.
Dans 30 ml d'éthanol, on ajoute 1,51 g de composé obtenu à l'étape précédente (iv), 1,57 g de formiate d'ammonium, puis 0,15 g de palladium sur charbon à 10% .On agite à TA pendant 3 heures. On filtre le catalyseur et concentre à sec. Après cristallisation dans l'acétone, on obtient 0,985 g de composé attendu.
EXEMPLES
Exemple 1 : l-({5-(2,4-dichlorophényl)-4-[4-(3-hydroxypropoxy)phényl]thién-2- yl}carbonyl)-4-phénylpipéridine-4-carboxamide.
IA) 1 -(2,4-dichlorophényl)-2-(4-méthoxyphényl)éthanone.
Dans 250 ml de THF, on ajoute 367 ml de NaHMDS (2M dans le THF) et refroidit le mélange à -700C. On ajoute alors 54 g d'acide (4- méthoxyphényl) acétique ; on laisse sous agitation pendant 2 heures puis ajoute 49 g de 2,4-dichlorobenzoate de méthyle. On laisse le mélange réactionnel revenir à 100C, verse sur 2 litres d' HCl (2N) glacé et on extrait le produit à l'éther. Après lavage avec une solution saturée de bicarbonate de sodium, puis une solution saturée de NaCl, séchage de la phase organique et évaporation, le produit cristallise dans l'heptane ; on obtient 32 g du composé attendu.
IB) (2E)l-(2,4-dichlorophényl)-3-(diméthylamino)-2-(4-méthoxyphényl)prop-2-én-
1-one.
Dans 100 ml de THF, on ajoute 15 g de composé obtenu en IA) et 33,76 ml de 1,1- diméthoxy-Λ/,Λ/-diméthylméthanamine et on chauffe à 8O0C pendant une nuit. Après évaporation à sec du mélange réactionnel, on ajoute de l'eau et extrait à l'éther. On sèche et on évapore. Après purification par chromatographie sur silice (éluant : Heptane- AcOEt (gradient de 0%à 30%)), on obtient 18 g de composé attendu.
IC) (2E)-3-chloro-3-(2,4-dichlorophényl)-2-(4-méthoxyphényl)acrylaldéhyde.
Dans 100 ml de CH2CI2, on ajoute 18 g de composé obtenu en IB), 12 ml de POCI3 et chauffe à 45 0C pendant 3 heures. Après une nuit à TA, on évapore à sec,
reprend dans du THF puis ajoute 50 ml d'eau. On extrait à l'éther, lave à l'eau puis avec une solution aqueuse saturée en bicarbonate de sodium. Après séchage sur MgSC>4 puis concentration à sec, on obtient 15,5 g de composé attendu.
1 D) 5-(2,4-dichlorophényl)-4-(4-méthoxyphényl)thiophène-2-carboxylate de méthyle.
Dans 80 ml d'acétonitrile, on mélange 11 g de composé obtenu en IC) et 7,2 ml de mercaptoacétate de méthyle. On chauffe à 600C et on ajoute du DBU. On maintient la température du milieu réactionnel à 600C et on revient à TA pendant une nuit. On évapore à sec, ajoute une solution d'HCl IN et extrait le composé à l'acétate d'éthyle. On sèche la phase organique et évapore. Après purification par chromatographie sur silice (éluant : Heptane-AcOEt (gradient de 0% à 30%)), on obtient 6,3 g du composé attendu.
IE) Acide 5-(2,4-dichlorophényl)-4-(4-méthoxyphényl)thiophène-2-carboxylique.
Dans un mélange 50-50 Dioxane-Méthanol, on ajoute 7,2 g du composé obtenu à l'étape ID) et 1,83 g de NaOH. On chauffe le mélange réactionnel à 500C pendant une nuit. On évapore à sec, ajoute de l'eau distillée et extrait les résidus organiques avec de l'éther. On acidifie la phase aqueuse avec HCl concentré puis on extrait l'acide avec de l'éther. La phase organique est séchée puis évaporée. On obtient 5,6 g du composé attendu.
1 F) 1 - { [5-(2,4-dichlorophényl)-4-(4-méthoxyphényl)thién-2-yl] carbonyl} -A- phénylpipéridine-4-carboxamide Dans 40 ml de CH2CI2, on ajoute à 2 g du composé obtenu à l'étape IE), 1,27 g de 4-phénylpipéridine-4-carboxamide (préparation 2), 2,22 ml de Et3N et 1,86 g de TBTU. Le mélange est mis sous agitation à TA pendant 3 heures. On évapore à sec, ajoute de l'eau et extrait à l'acétate d'éthyle. La phase organique est séchée puis évaporée. Après purification par chromatographie sur silice (éluant : Heptane-AcOEt (gradient de 0% à 30%)), on obtient 2,8 g du composé attendu.
1 G) 1 - { [5-(2,4-dichlorophényl)-4-(4-hydroxyphényl)thién-2-yl] carbonyl} -A- phénylpipéridine-4-carboxamide.
Dans 70 ml de CH2CI2 à la température de la glace fondante, on ajoute à 2,8 g de composé obtenu à l'étape IF), 19,8 ml de solution IM de BBrβ dans CH2CI2. On laisse remonter la température à TA tout en continuant d'agiter pendant 3 heures. On verse ensuite le milieu réactionnel sur de l'eau et on récupère la phase organique qui est ensuite séchée puis évaporée. Après cristallisation dans un mélange éther - acétate d'éthyle, on obtient 1,8 g du composé attendu.
IH) l-({5-(2,4-dichlorophényl)-4-[4-(3-hydroxypropoxy)phényl]thién-2- yl}carbonyl)-4-phénylpipéridine-4-carboxamide.
Dans 10 ml de DMF, on ajoute à 1,65 g du composé obtenu à l'étape IG), 0,57 g de K2CO3 et 0,83 g de 3-chloropropan-l-ol. Le milieu réactionnel est chauffé à 900C
pendant 3 heures. On verse alors le milieu réactionnel sur de l'eau distillée et extrait le composé attendu à l'acétate d'éthyle. On sèche et on évapore la phase organique. Après cristallisation dans l'acétate d'éthyle, on obtient 1, 2 g du composé attendu.
Exemple 2 : l-{[5-(2,4-dichlorophényl)-4-(4-{3-[(méthylsulfonyl)amino]propoxy} phényl)-thién-2-yl]carbonyl}-4-phénylpipéridine-4-carboxamide.
2A) Méthanesulfonate de 3-(4-{5-[(4-carbamoyl-4-phénylpipéridin-l-yl)carbonyl]- 2-(2,4-dichlorophényl)thién-3-yl}phénoxy)propyle.
A 1,5 g de composé obtenu à l'étape IH) mis dans 10 ml de CH2CI2, on ajoute 0,07 ml de MsCl puis 0,02 ml de EtβN. On laisse sous agitation pendant 1 heure. On évapore à sec, ajoute une solution d'HCl 0,1N et on extrait le composé attendu à l'acétate d'éthyle. Après séchage et évaporation, on obtient 0,15 g du composé attendu.
2B) l-{[5-(2,4-dichlorophényl)-4-(4-{3-[(méthylsulfonyl)amino]propoxy}phényl) thién- 2-yl]carbonyl} -4-phénylpipéridine-4-carboxamide.
Dans 5 ml de DMF, on ajoute 0,029 g de méthanesulfonamide et 0,016 g d'hydrure de sodium puis on agite pendant 5 minutes. On ajoute alors 0,14 g du composé obtenu à l'étape 2A) dissout dans 2 ml de DMF et on agite 2 heures. On verse le milieu réactionnel sur de l'eau et on extrait à l'acétate d'éthyle. On sèche et on évapore. Après purification par chromatographie sur silice (éluant : CF^C^-MeOH (gradient de 0% à 3%), on obtient 0,035 g de composé attendu.
Exemple 3 : l-({5-(2,4-dichlorophényl)-4-[4-(3-pyrrolidin-l- ylpropoxy)phényl]thién-2-yl}carbonyl)-4-phénylpipéridine-4-carboxamide.
Dans 5 ml de DMF, on ajoute 0,12 ml de pyrrolidine à 0,2 g du composé obtenu à l'étape 2A), puis on chauffe à 700C pendant 2 heures. On verse le milieu réactionnel sur de l'eau distillée et on extrait le composé à l'acétate d'éthyle. On sèche et on évapore. Après lavage à l'éther et fîltration, on obtient 0,1 g du composé attendu.
Exemple 4 : l-({4-(2,4-dichlorophényl)-5-[4-(3-hydroxypropoxy)phényl]thién-2- yl}carbonyl)-4-phénylpipéridine-4-carboxamide.
4A) l-{[4-(2,4-dichlorophényl)-5-(4-méthoxyphényl)thién-2-yl]carbonyl}-4- phénylpipéridine-4-carboxamide.
L'acide 4-(2,4-dichlorophényl)-5-(4-méthoxyphényl)thiophène-2-carboxylique est préparé suivant le mode opératoire utilisé dans les étapes IA - IE à partir d'acide (2,4- dichlorophényl)acétique et de 4-méthoxybenzoate de méthyle . Dans 100 ml de CH2CI2, on ajoute à TA 3,8 g d'acide 4-(2,4-dichlorophényl)-5-(4-méthoxyphényl) thiophène-2-carboxylique, 2,53 g de 4-phénylpipéridine-4-carboxamide (préparation 2), 0,72 ml de EtβN et 3,53 g de TBTU. Le mélange est mis sous agitation pendant une nuit. On évapore à sec, on ajoute de l'eau et on extrait le composé obtenu à l'acétate
d'éthyle. On sèche la phase organique et évapore à sec. Après cristallisation, lavage avec de l'éther éthylique, fïltration puis séchage, on obtient 5,1 g du composé attendu.
4B) l-{[4-(2,4-dichlorophényl)-5-(4-hydroxyphényl)thién-2-yl]carbonyl}-4- phénylpipéridine-4-carboxamide.
Dans 100 ml de CH2CI2 à la température de la glace fondante, on ajoute 5 g du composé issu de l'étape 4A) puis 35,37 ml de solution de IM de BBrç dans CH2CI2. On laisse la température du milieu réactionnel revenir à TA pendant la nuit. On verse ensuite le milieu réactionnel sur de l'eau distillée et extrait le composé obtenu au CH2CI2. On sèche et on évapore la phase organique. Après cristallisation dans l'éther éthylique, on obtient 3,1 g du composé attendu.
4C) 1 -( {4-(2,4-dichlorophényl)-5-[4-(3-hydroxypropoxy)phényl]thién-2- yl}carbonyl)-4-phénylpipéridine-4-carboxamide.
Dans 6 ml de DMF, on ajoute 1 g du composé obtenu à l'étape 4B), 0, 376 g de carbonate de potassium et 0,223g de 3-chloropropan-l-ol. On chauffe le milieu réactionnel à 1100C pendant 3 heures. On verse ensuite le milieu réactionnel sur de l'eau distillée et on extrait le composé obtenu à l'acétate d'éthyle. On sèche et on évapore la phase organique. Après purification par chromatographie sur silice (éluant : CH2Cl2-MeOH (gradient de 0 % à 4 %)), on obtient 0,6 g de composé attendu.
Exemple 5 : Chlorhydrate de l-({5-[4-(3-aminopropoxy)phényl]-4-(2,4- dichlorophényl)thién-2-yl}carbonyl)-4-phénylpipéridine-4-carboxamide.
5A) [3-(4- {5-[(4-Carbamoyl-4-phénylpipéridin- 1 -yl)carbonyl]-3-(2,4-dichloro phényl)thién-2-yl}phénoxy)propyl]carbamate de tert-butyle.
Dans 60 ml d'acétone, on ajoute 1 g du composé obtenu à l'étape 4B), 0,504 g de carbonate de potassium et 0,868 g de (3-bromopropyl)carbamate de tert-butyle puis on chauffe le milieu réactionnel à reflux pendant 4 heures. On filtre les insolubles et évapore à sec. Après purification par chromatographie sur silice (éluant : CH2CI2 - MeOH (gradient de 0 % à 7 %)). On obtient 0,6 g de composé attendu.
5B) Chlorhydrate de l-({5-[4-(3-aminopropoxy)phényl]-4-(2,4- dichlorophényl)thién-2-yl} carbonyl)-4-phénylpipéridine-4-carboxamide.
Dans 5 ml de CH2CI2, on ajoute 0,4g de composé obtenu en 5A), 1,28 g de TFA et on laisse sous agitation à TA pendant 1 heure. On évapore à sec, ajoute une solution de soude IN et extrait le composé au CH2CI2. La phase organique est séchée avant d'ajouter de l'éther chlorhydrique pour former le sel. Après évaporation à sec et séchage sous vide, on obtient 0,28 g de composé attendu.
Exemple 6 : l-{[4-(2,4-dichlorophényl)-5-(4-{3-[(méthylsulfonyl)amino] propoxy}phényl)thién-2-yl]carbonyl}-4-phénylpipéridine-4-carboxamide.
Dans 10 ml de CH2CI2, on ajoute 0,19 g de composé obtenu en 5B), 0,063 ml de EtβN puis 0,03 ml de MsCl et on laisse sous agitation pendant 2 heures. On évapore à
sec, ajoute une solution de soude puis extrait le composé au CH2CI2. On sèche et on évapore. Après purification par chromatographie sur silice (éluant : CH2CI2 - MeOH (gradient de 0% à 5%)), on obtient 0,085 g de composé attendu.
Exemple 7 : l-{[4-(2,4-dichlorophényl)-5-{4-[3- (méthylthio)propoxy]phényl}thién-2-yl] carbonyl}-4-phénylpipéridine-4-carboxamide.
7A) Méthanesulfonate de 3-(4-{5-[(4-carbamoyl-4-phénylpipéridin-l-yl)carbonyl]- 3-(2,4-dichlorophényl)thién-2-yl}phénoxy)propyle.
Dans 20 ml de CH2CI2 à TA, on ajoute 0,8 g de composé obtenu en 4C), 0,37 ml de EtβN, 0,12 ml de MsCl puis on laisse sous agitation pendant 1 heure. On évapore à sec, ajoute une solution d'HCl O,1N et extrait le composé à l'acétate d'éthyle. Après séchage et évaporation, on obtient 0,79 g de composé attendu.
7B) l-{[4-(2,4-dichlorophényl)-5-{4-[3-(méthylthio)propoxy]phényl}thién-2-yl] carbonyl}-4-phénylpipéridine-4-carboxamide.
Dans 4 ml de DMF, on ajoute 0,79 g de composé obtenu en 7A), 0,097 g de méthanethiolate de sodium puis on laisse sous agitation pendant 4 heures à TA. On verse ensuite le mélange réactionnel sur de l'eau distillée et extrait le composé à l'acétate d'éthyle. On sèche et on évapore. Après purification par chromatographie sur silice (éluant : CH2CI2 - MeOH (gradient de 0 % à 5 %)), on obtient 0,55 g de composé attendu.
Exemple 8 : l-{[4-(2,4-dichlorophényl)-5-{4-[3-(méthylsulfonyl)propoxy] phényl}thién-2-yl] carbonyl} -4-phénylpipéridine-4-carboxamide.
Dans 20 ml de CH2CI2 à TA, on ajoute 0,27 g de composé obtenu en 7B), 0,26 g de MCPBA à 70% puis on laisse sous agitation pendant une nuit. On ajoute alors une solution de Na2CÛ3 à 10 % et agite vigoureusement pendant 30 minutes. On sépare la phase organique, la sèche et évapore. Après purification par chromatographie sur silice (éluant : CH2CI2 - MeOH (gradient de 0 % à 4 %)), on obtient 0,07 g de composé attendu.
Exemple 9 : l-{[5-(2,4-dichlorophényl)-4-{4-[(4-hydroxybutyl)thio]phényl}thién- 2-yl]carbonyl}-4-phénylpipéridine-4-carboxamide.
9A) l-{[4-(4-bromophényl)-5-(2,4-dichlorophényl)thién-2-yl]carbonyl}-4- phénylpipéridine-4-carboxamide.
L'acide 4-(4-bromophényl)-5-(2,4-dichlorophényl)-thiophène-2-carboxylique est préparé suivant le mode opératoire utilisé dans les étapes étapes IA - IE à partir de l'acide (4-bromophényl)acétique et de 2,4-dichlorobenzoate de méthyle . Dans 100 ml de CH2CI2, on ajoute à TA 7 g d'acide 4-(4-bromophényl)-5-(2,4-dichlorophényl)- thiophène-2-carboxylique, 3,94 g de chlorhydrate de 4-phénylpipéridine-4-carboxamide (préparation 2), 6,83 ml de EtβN, 5,77 g de TBTU puis on agite le mélange pendant une
nuit. On évapore à sec, ajoute de l'eau et extrait le composé attendu à l'acétate d'éthyle. On sèche la phase organique avec MgSOφ filtre et évapore à sec. Après cristallisation, lavage avec de l'éther éthylique, fîltration et séchage, on obtient 8,41 g de composé attendu.
9B) l-{[5-(2,4-dichlorophényl)-4-{4-[(4-hydroxybutyl)thio]phényl}thién-2-yl] carbonyl}-4-phénylpipéridine-4-carboxamide.
Dans 25 ml de xylène à 00C et préalablement dégazé 15 minutes à l'argon, on ajoute 0,455 g de 4-mercaptobutan-l-ol et 0,072 g d'hydrure de sodium puis on laisse agiter 1 heure en laissant remonter à TA. On ajoute alors 1 g de composé obtenu en 9A), 0,119 g de Pd2(dba)3 et 0,087 g de xantphos. Le milieu réactionnel est ensuite chauffé à 1500C pendant la nuit. On évapore le milieu réactionnel à sec, ajoute de l'eau et de l'acétate d'éthyle, filtre l'insoluble, lave la phase organique avec de l'eau, sèche la phase organique, filtre et concentre. Après purification par chromatographie sur silice (éluant : CH2CI2 - MeOH (gradient de 0% à 3%)), on obtient 0,35 g de composé attendu.
Exemple 10 : l-{[5-(2,4-dichlorophényl)-4-{4-[(4-hydroxybutyl)sulfînyl]phényl} thién-2-yl]carbonyl}-4-phénylpipéridine-4-carboxamide.
Dans 20 ml de CH2CI2 à TA, on ajoute 0,23 g de composé obtenu en 9B), 0,109 g de MCPBA à 70 % puis on laisse sous agitation pendant une nuit. On ajoute alors une solution de Na2CÛ3 à 10% et agite ensuite pendant 15 minutes. La phase organique est séparée, séchée et évaporée. Après purification par chromatographie sur silice (éluant : CH2CI2 - MeOH (gradient de 0 % à 4 %)), on obtient 0,14 g de composé attendu.
Exemple 11 : l-{[5-(2,4-dichlorophényl)-4-{4-[(4- hydroxybutyl)sulfonyl]phényl}thién-2-yl]carbonyl}-4-phénylpipéridine-4-carboxamide.
Dans 20 ml de CH2CI2 à TA, on ajoute 0,16 g de composé obtenu à l'exemple 10 et 0,121 g de MCPBA à 70 % puis on laisse sous agitation pendant une nuit. On ajoute alors une solution de Na2CÛ3 à 10 % et agite ensuite pendant 15 minutes. On sépare la phase organique, la sèche et évapore. Après purification par chromatographie sur silice (éluant : CH2CI2 - MeOH (gradient de 0 % à 4 %)), on obtient 0,05 g de composé attendu.
Exemple 12 : Propane- 1-sulfonate de 4-{5-[(4-carbamoyl-4-phénylpipéridin-l- yl)carbonyl]-2-(2-chlorophényl)thién-3-yl}phényle.
12A) Acide 5-(2-chlorophényl)-4-(4-méthoxyphényl)thiophène-2-carboxylique.
L'acide 5-(2-chlorophényl)-4-(4-méthoxyphényl)-thiophène-2-carboxylique est préparée suivant le mode opératoire utilisé dans les étapes IA - IE à partir de l'acide (4-méthoxyphényl) acétique et de 2-chlorobenzoate de méthyle. A la dernière étape de ce mode opératoire, on ajoute 5 g de méthyl 5-(2-chlorophényl)-4-(4-méthoxyphényl)-
2-thiophènecarboxylate et 1,11 g de soude en pastille dans 30 ml de méthanol et 3 ml de dioxane. On chauffe le milieu réactionnel à 50-600C pendant 5 heures. On évapore à sec, ajoute de l'eau et extrait les impuretés à l'éther. On acidifie la phase aqueuse et extrait le composé attendu à l'éther. Après séchage et évaporation, on obtient 3,5 g de composé attendu.
12B) 1 - { [5-(2-chlorophényl)-4-(4-méthoxyphényl)thién-2-yl]carbonyl} -4- phénylpipéridine-4-carboxamide.
Dans 100 ml de CH2CI2, on ajoute à TA 1,5 g de composé obtenu en 12A), 1,047 g de chlorhydrate de 4-phénylpipéridine-4-carboxamide, 1,83 ml de EtβN, 1,56 g de TBTU puis on agite le mélange pendant une nuit. On évapore à sec, ajoute de l'eau et extrait le composé attendu à l'acétate d'éthyle. On sèche la phase organique, filtre et évapore à sec. Le composé qui a cristallisé est lavé avec de l'éther éthylique puis filtré et séché. On obtient 2 g de composé attendu.
12C) 1 - { [5-(2-chlorophényl)-4-(4-hydroxyphényl)thién-2-yl]carbonyl} -4- phénylpipéridine-4-carboxamide.
Dans 50 ml de CH2CI2 à la température de la glace fondante, on ajoute 1,7 g de composé obtenu à l'étape 12B) puis 12,8 ml de solution IM de BBrβ dans CH2CI2 et on laisse revenir la température du milieu réactionnel à TA pendant la nuit. On verse ensuite le milieu réactionnel sur de l'eau distillée et extrait le composé attendu au CH2CI2. On sèche et on évapore la phase organique. Le composé cristallise dans l'éther éthylique. On obtient 1,2 g de composé attendu.
12D) Propane- 1-sulfonate de 4-{5-[(4-carbamoyl-4-phénylpipéridin-l-yl)carbonyl] -2-(2-chlorophényl)thién-3-yl}phényle.
Dans 30 ml de CH2CI2, on ajoute 0,3 g de composé obtenu en 12C), 0,29 ml de EtβN puis 0,207 g de propane- 1-sulfonyl chloride à TA. Après 3 heures, on verse le milieu réactionnel sur de l'eau distillée et extrait le composé. Après séchage, évaporation puis lavage avec de l'éther, on obtient 0,13 g de composé attendu.
Exemple 13 : 3,3,3-trifluoropropane-l-sulfonate de 4-{5-[(4-carbamoyl-4- phénylpipéridin-l-yl)carbonyl]-2-(2-chlorophényl)thién-3-yl}phényle.
Dans 30 ml de CH2CI2, on ajoute 0,3 g de composé obtenu à l'étape 12C), 0,29 ml de EtβN, puis 0,28 g de chlorure de 3,3,3-trifluoropropane-l-sulfonyle à TA. Après 3 heures, on évapore à sec. On ajoute de l'eau et de l'acétate d'éthyle puis on agite l'ensemble. Après fïltration, rinçage à l'éther puis séchage, on obtient 0,13 g de composé attendu.
Exemple 14 : l-{[5-(2,4-dichlorophényl)-4-{4-[(4,4,4-trifluorobutyl)thio]phényl thién-2-yl]carbonyl}-4-phénylpipéridine-4-carboxamide.
Dans 25 ml de xylène à 00C et préalablement dégazé 15 minutes à l'Argon, on ajoute 0,455 g de 4,4,4-trifluorobutane-l-thiol et 0,072 g d'hydrure de sodium puis on
laisse agiter 1 heure en laissant remonter à TA. On ajoute alors 1 g de composé obtenu en 9A), 0,119 g de Pd2(dba)3 et 0,087 g de xantphos. Le milieu réactionnel est ensuite chauffé à 1500C pendant la nuit. On évapore le milieu réactionnel à sec, ajoute de l'eau et de l'acétate d'éthyle, filtre l'insoluble, lave la phase organique avec de l'eau, sèche la phase organique, filtre et concentre. Après purification par chromatographie sur silice (éluant : Cï^C^-MeOH (gradient de 0% à 4 %)), on obtient 0,16 g de composé attendu.
Exemple 15 : l-{[5-(2,4-dichlorophényl)-4-{4-[(4,4,4-trifluorobutyl)sulfonyl] phényl}thién-2-yl]carbonyl}-4-phénylpipéridine-4-carboxamide.
Dans 20 ml de CH2CI2 à TA, on ajoute 0,36 g de composé obtenu à l'exemple 14), 0,385g de MCPBA à 70 % puis on laisse sous agitation pendant une nuit. On ajoute alors une solution de Na2CÛ3 à 10 % et agite vigoureusement pendant 30 minutes. On sépare la phase organique, la sèche et l'évaporé Après purification par chromatographie sur silice (éluant : Cf^C^-MeOH (gradient de 0 % à 4 %)), on obtient 0,19 g de composé attendu.
Exemple 16 : l-({5-(2-Chloro-4-fluorophényl)-4-[4-(3-hydroxypropoxy)phényl] thién-2-yl}carbonyl)-4-phénylpipéridine-4-carboxamide.
16A) 1 -(2-chloro-4-fluorophényl)-2-(4-méthoxyphényl)éthanone.
Dans 250 ml de THF, on introduit sous azote 230 ml de solution 2M de NaHMDS dans le THF. On refroidit à -6O0C puis ajoute à cette température 30,5 g d'acide (4- méthoxyphényl)acétique dans 120 ml de THF. Après Ih 30 mn à -6O0C, on additionne 33 g de 2-chloro-4-fluorobenzoate de méthyle, agite à -6O0C pendant 45 mn puis on laisse revenir à 00C. On verse le milieu réactionnel sur 500 ml d' HCl 2N glacé, extrait à l'éther, lave à l'eau puis avec une solution aqueuse saturée en chlorure de sodium. Après séchage, concentration à sec puis cristallisation dans le pentane, on obtient 27, 4 g de composé attendu.
16B) (2E)- 1 -(2-chloro-4-fluorophényl)-3-(diméthylamino)-2-(4-méthoxyphényl) prop-2-én-l-one
Dans 100 ml de THF, on ajoute 27,4 g de composé obtenu en 16A), 35 g de 1,1- diméthoxy-Λ/,Λ/-diméthylméthanamine et on porte à reflux pendant 3 heures. Après concentration à sec puis cristallisation dans l'éther isopropylique, on obtient 24,7 g de composé attendu.
16C) (2E)-3-chloro-3-(2-chloro-4-fluorophényl)-2-(4-méthoxyphényl)acrylal- déhyde.
Dans 200 ml de CH2CI2, on ajoute 24,7 g de composé obtenu à l'étape 16B) puis 17,3 g de POCI3. On chauffe à 45°C pendant une nuit puis on concentre à sec. On reprend dans du THF puis ajoute 50 ml d'eau. On extrait à l'éther, lave à l'eau puis avec une solution aqueuse saturée en chlorure de sodium. Après séchage, concentration à sec, on obtient 24 g de composé attendu.
16D) 5-(2-chloro-4-fluorophényl)-4-(4-méthoxyphényl)thiophène-2-carboxylate de méthyle.
Dans 240 ml d'acétonitrile, on ajoute 24 g de composé obtenu en 16C) puis 16,5 ml de mercaptoacétate de méthyle. On chauffe à 6O0C et ajoute 12 ml de DBU. On maintient à 6O0C pendant 2 heures puis à TA pendant une nuit. On concentre à sec, reprend dans l'acétate d'éthyle, lave à l' HCl IN puis à l'eau. Après séchage, concentration à sec puis cristallisation dans le méthanol, on obtient 20,6 g de composé attendu.
16E) 5-(2-chloro-4-fluorophényl)-4-(4-hydroxyphényl)thiophène-2-carboxylate de méthyle.
Dans 100 ml de CH2CI2, on ajoute le composé obtenu à l'étape 16D) et on refroidit à -500C, ajoute 40 ml de solution IM de BBrβ dans CH2CI2. On laisse revenir la température à 200C puis on ajoute 20 ml de méthanol. On concentre à sec, reprend dans du CH2CI2, lave à l'eau, sèche sur MgSOφ concentre à sec. Après purification par chromatographie sur silice (heptane jusqu'à heptane / AcOEt 75/25). On obtient 4,8 g de composé attendu.
16F) 5-(2-chloro-4-fluorophényl)-4- {4-[3-(tétrahydro-2H-pyran-2-yloxy)propoxy] phényl}thiophène-2-carboxylate de méthyle.
Dans 30 ml de DMF, on ajoute 4,8 g de composé obtenu à l'étape 16E), 3,6 g de 2- (3-bromopropoxy)tétrahydro-2H-pyran, 2,2 g de carbonate de potassium puis on chauffe à 6O0C pendant 7 heures. On verse sur eau glacée, extrait à l'éther, sèche et concentre à sec. Après purification par chromatographie sur silice (heptane puis heptane / AcOEt : 90 / 10), on obtient 6,2 g de composé attendu.
16G) Acide 5-(2-chloro-4-fluorophényl)-4- {4-[3-(tétrahydro-2Η-pyran-2- yloxy)propoxy]phényl}thiophène-2-carboxylique.
Dans 30 ml de méthanol et 1 ml d'eau, on ajoute 3,1 g de composé obtenu à l'étape 16F) puis 800 mg de soude en pastille. On porte le mélange réactionnel à reflux pendant 2 heures. On concentre à sec, reprend dans 500 ml de solution tampon à pH 2, extrait à l'éther, sèche et concentre à sec. On obtient 3 g de composé attendu, cristallisé dans le pentane.
16H) l-{[5-(2-chloro-4-fluorophényl)-4-{4-[3-(tétrahydro-2H-pyran-2-yloxy) propoxy]phényl}thién-2-yl]carbonyl}-4-phénylpipéridine-4-carboxamide.
Dans 10 ml de CH2CI2, on ajoute 0,5 g de composé obtenu à l'étape 16G), 0,25 g de chlorhydrate de 4-phénylpipéridine-4-carboxamide, 0,43 ml de EtβN et 0,36 g de TBTU puis on laisse la réaction 2 heures à TA. Après concentration à sec, reprise dans un mélange eau / éther, et fïltration, on obtient 0,6 g de composé attendu.
161) l-({5-(2-chloro-4-fluorophényl)-4-[4-(3-hydroxypropoxy)phényl]thién-2- yl}carbonyl)-4-phénylpipéridine-4-carboxamide.
Dans 15 ml de méthanol, on ajoute 0,6 g de composé obtenu à l'étape 16H), 2 ml d' éther chlorhydrique 2N. Après 2 heures à TA, on filtre le composé attendu. On obtient 0,385 g de composé attendu.
Exemple 17 : Chlorhydrate de l'-({5-(2-Chloro-4-fluorophényl)-4-[4-(3- hydroxypropoxy)phényl]-2-thiényl} carbonyl)-4,4-difluoro- 1 ,4'-bipipéridine-4'- carboxamide.
17A) r-{[5-(2-chloro-4-fluorophényl)-4-{4-[3-(tétrahydro-2H-pyran-2-yloxy) propoxy]phényl}thién-2-yl]carbonyl} -4,4-difluoro- 1 ,4'-bipipéridine-4'-carboxamide.
Dans 10 ml de CΗ2CI2, on ajoute 0,5 g de composé obtenu à l'étape 16G), 0,33 g de 4,4-difluoro-[l,4']bipipéridinyl-4'-carboxamide (décrit à la préparation 5 page 23 de la demande WO 2008/068 423), 0,57 ml de EtβN et 0,36 g de TBTU. Après 2 heures à TA, concentration à sec, reprise dans un mélange eau/éther et fïltration, on obtient 0,63 g de composé attendu.
17B) Chlorhydrate de r-({5-(2-chloro-4-fluorophényl)-4-[4-(3-hydroxypropoxy) phényl]thién-2-yl} carbonyl)-4,4-difluoro- 1 ,4'-bipipéridine-4'-carboxamide.
Dans 15 ml de méthanol, on ajoute 0,63 g du composé obtenu à l'étape 17A) et 2 ml d' éther chlorhydrique 2N. Après 1 heure à TA, concentration à sec puis cristallisation dans l'AcOEt, on obtient 0, 612 g de composé attendu.
Exemple 18 : Chlorydrate de l-({5-(2-chloro-4-fluorophényl)-4-[4-(3- hydroxypropoxy)phényl]thién-2-yl}carbonyl)-4-[(3,3,3-trifluoropropyl)amino] pipéridine-4-carboxamide.
18A) l-{[5-(2-chloro-4-fluorophényl)-4-{4-[3-(tétrahydro-2H-pyran-2-yloxy) propoxy]phényl}thién-2-yl]carbonyl}-4-[(3,3,3-trifluoropropyl)amino]pipéridine-4- carboxamide.
Dans 10 ml de CΗ2CI2, on ajoute 0,5 g de composé obtenu à l'étape 16G), 0,41 g de 4-(3,3,3-trifluoro-propylamino)-pipéridine-4-carboxamide (préparation 1), 0,71 ml de EtβN et 0,36 g de TBTU. Après 3 heures à TA, on concentre à sec, reprend dans l' éther, lave à l'eau, sèche et concentre à sec. Après purification par chromatographie sur silice (gradient d'élution : heptane/ AcOEt : 90/10 jusqu'à AcOEt pur). On obtient 0,7 g de composé attendu.
18B) Chlorydrate de l-({5-(2-chloro-4-fluorophényl)-4-[4-(3-hydroxypropoxy) phényl]thién-2-yl}carbonyl)-4-[(3,3,3-trifluoropropyl)amino]pipéridine-4-carboxamide. Dans 15 ml de méthanol, on ajoute 0,7g de composé obtenu à l'étape 18A) et 3 ml d'éther chlorhydrique 2N. Après 2 heures à TA, concentration à sec, reprise dans l'éther puis fïltration on obtient 0,57 g de composé attendu.
Exemple 19 : l-{[5-(2-chloro-4-fluorophényl)-4-{4-[3-(méthylthio)propoxy] phényl}thién-2-yl]carbonyl}-4-phénylpipéridine-4-carboxamide.
19A) 5-(2-chloro-4-fluorophényl)-4-[4-(3-hydroxypropoxy)phényl]thiophène-2- carboxylate de méthyle.
Dans 40 ml de méthanol, on ajoute 3,1 g de composé obtenu à l'étape 16F) et 1 ml de résine Amberlyst 15. On porte à reflux pendant 2 heures, filtre la résine et concentre à sec. On reprend dans l'éther, lave à l'eau, sèche puis concentre à sec. Après purification par chromatographie sur silice (gradient d'élution : heptane jusqu'à heptane / AcOEt ; 60/40), on obtient 2,0 g de composé attendu.
19B) 5-(2-chloro-4-fluorophényl)-4-(4- (3-[(méthylsulfonyl)oxy]propoxy}phényl) thiophène-2-carboxylate de méthyle.
Dans 40 ml de CH2CI2, on ajoute 2 g de composé obtenu en 19A) puis 0,79 ml de Et3N. On refroidit à 00C puis ajoute 0,44 ml de MsCl. Après 15 minutes, on lave à l'eau, sèche et concentre à sec. On obtient 2,38 g de composé attendu.
19C) 5-(2-chloro-4-fluorophényl)-4- {4-[3-(méthylthio)propoxy]phényl}thiophène- 2-carboxylate de méthyle.
Dans 12 ml de DMF, on ajoute 2,38 g de composé obtenu en 19B), 0,83 g de méthanethiolate de sodium et agite à TA pendant 2 heures. On verse sur de l'eau, extrait à l'éther, lave à l'HCl, dilue à l'eau, sèche et concentre à sec. Après purification par chromatographie sur silice (gradient d'élution : heptane jusqu'à heptane / AcOEt 93/7), on obtient 1,68 g de composé attendu.
19D) Acide 5-(2-chloro-4-fluorophényl)-4- {4-[3-(méthylthio)propoxy]phényl} thiophène-2-carboxylique.
Dans 20 ml de MeOH et 1 ml d'eau, on ajoute 1,68 g de composé obtenu en 19C) puis 0,45g de NaOH. On porte à reflux pendant 2 heures, puis concentre à sec. On reprend dans l'eau, acidifie avec de HCl 2N, extrait à l'éther, sèche et concentre à sec. On obtient 1,51 g de composé attendu.
19E) l-{[5-(2-Chloro-4-fluorophényl)-4-{4-[3-(méthylthio)propoxy]phényl}thién- 2-yl] carbonyl} -4-phénylpipéridine-4-carboxamide.
Dans 10 ml de CH2CI2, on ajoute 0,5 g de composé obtenu en 19D), 0,28 g de chlorhydrate de 4-phénylpipéridine-4-carboxamide (préparation 2), 0,48 ml de EtβN et 0,405 g de TBTU. Après 2 heures à TA, on concentre à sec, reprend dans un mélange eau / éther et filtre. On obtient 0,635 g de composé attendu.
Exemple 20 : l-{[5-(2-Chloro-4-fluorophényl)-4-{4-[3- (méthylsulfonyl)propoxy]phényl}thién-2-yl]carbonyl}-4-phénylpipéridine-4- carboxamide.
Dans 10 ml de CH2CI2, on ajoute 0,43 g de composé obtenu à l'étape 19E), 0,425 g de MCPBA et on agite à TA pendant 3 heures. On lave avec une solution aqueuse de bicarbonate de sodium, sèche et concentre à sec. Après purification par chromatographie sur silice (gradient d'élution : CH2CI2 jusqu'à CH2CI2 / MeOH 98/2), on obtient 0,216 g de composé attendu.
Exemple 21 : Chlorhydrate de r-{[5-(2-chloro-4-fluorophényl)-4-{4-[3- (méthylthio)propoxy]phényl}thién-2-yl]carbonyl} -4,4-difluoro- 1 ,4'-bipipéridine-4'- carboxamide.
Dans 10 ml de CH2CI2, on ajoute 0,5 g de composé obtenu à l'étape 19D), 0,51 g de 4,4-difluoro-[l,4']bipipéridinyl-4'-carboxamide (décrit à la préparation 5 page 23 de la demande WO 2008/068 423), 0,8 ml de Et3N et 0,405 g de TBTU. Après 3 heures à TA, on concentre à sec, reprend dans l'éther, lave à l'eau, sèche et concentre à sec. Après purification par chromatographie sur silice (gradient d'élution : CH2CI2 jusqu'à CH2Cl2/MeOH 98/2), on dissout le composé obtenu dans CH2CI2, ajoute de l'éther chlorhydrique jusqu' à pH 1, puis concentre à sec. Après reprise dans l'éther et fîltration, on obtient 0,723 g de composé attendu.
Exemple 22 : Chlorhydrate de r-{[5-(2-chloro-4-fluorophényl)-4-{4-[3- (méthylsulfonyl)propoxy]phényl}thién-2-yl]carbonyl} -4,4-difluoro- 1 ,4'-bipipéridine-4'- carboxamide.
22A) Acide 5-(2-chloro-4-fluorophényl)-4- {4-[3-(méthylsulfonyl)propoxy]phényl} thiophène-2-carboxylique.
Dans 15 ml de CH2CI2, on ajoute 0,5 g de composé obtenu à l'étape 19D), 0,62 g de MCPBA puis on agite à TA pendant 4 heures. Après concentration, fîltration et rinçage à l'éther isopropylique, on obtient 0,585 g de composé attendu.
22B) Chlorhydrate de r-{[5-(2-chloro-4-fluorophényl)-4-{4-[3-(méthylsulfonyl) propoxy]phényl}thién-2-yl]carbonyl} -4,4-difluoro- 1 ,4'-bipipéridine-4'-carboxamide. Dans 10 ml de CH2CI2, on ajoute dans l'ordre 0,3 g de composé obtenu à l'étape 22A), 0,3 g de 4,4-difluoro-[l,4']bipipéridinyl-4'-carboxamide (décrit à la préparation 5 page 23 de la demande WO 2008/068 423) , 0,45 ml de Et3N, et 0,25 g de TBTU.
Après 3 heures à TA, on concentre à sec, reprend dans l' AcOEt, lave à l'eau, sèche et concentre à sec. Après purification par chromatographie sur silice (gradient d'élution : CH2CI2 jusqu'à ClH^C^/MeOH ; 98/2), on dissout le composé obtenu dans du CH2CI2, ajoute de l'éther chlorhydrique jusqu' à pH 1 puis concentre à sec. On obtient 0,28 g de composé attendu.
Exemple 23 : l-({5-(2-chlorophenyl)-4-[4-(3-hydroxypropoxy)phényl]thién-2- yl}carbonyl)-4-phénylpipéridine-4-carboxamide.
23 A) 1 -(2-chlorophényl)-2-(4-méthoxyphényl)éthanone.
Dans 250 ml de THF, on introduit sous azote 230 ml de solution 2M de NaHMDS dans le THF. On refroidit à -600C puis ajoute à cette température 30,5g d'acide (4- méthoxyphényl)acétique dans 120 ml de THF. Après Ih 30 mn à -600C, on additionne 29,8 g de 2-chlorobenzoate de méthyle, agite à -600C pendant 45 mn puis on laisse revenir à 00C. On verse le milieu réactionnel sur 500 ml d' HCl 2N glacé, extrait à l'éther, lave à l'eau puis avec une solution aqueuse saturée en chlorure de sodium. Après séchage, concentration à sec, on purifie par chromatographie sur silice (gradient : heptane jusqu'à heptane / AcOEt : 80 / 20). On obtient 7, 3 g de composé attendu.
23B) (2E)- 1 -(2-chlorophényl)-3-(diméthylamino)-2-(4-méthoxyphényl)prop-2-én- 1-one.
Dans 25 ml de THF, on ajoute 7,3 g de composé obtenu en 23A), 9,5 g de 1,1- diméthoxy-Λ/,Λ/-diméthylméthanamine et on porte à reflux pendant 3 heures. Après concentration à sec, on purifie par chromatographie sur silice (gradient : CH2CI2 jusqu'à CH2CI2 / AcOEt : 80 / 20). On obtient 9,2 g de composé attendu.
23C) (2E)-3-chloro-3-(2-chlorophényl)-2-(4-méthoxyphényl)acrylaldéhyde.
Dans 75 ml de CH2CI2, on ajoute 9,2 g de composé obtenu à l'étape 23B) puis 6,8g de POCI3. On chauffe à 45°C pendant une nuit puis on concentre à sec. On reprend dans du THF puis ajoute 20 ml d'eau. On extrait à l'éther, lave à l'eau puis avec une solution aqueuse saturée en chlorure de sodium. Après séchage, concentration à sec, on obtient 9,1 g de composé attendu.
23D) 5-(2-chlorophényl)-4-(4-méthoxyphényl)thiophène-2-carboxylate de méthyle.
Dans 90 ml d'acétonitrile, on ajoute 9,1 g de composé obtenu en 23C) puis 6,62 ml de mercaptoacétate de méthyle. On chauffe à 6O0C et ajoute 4,83 ml de DBU. On maintient à 6O0C pendant 2 heures puis à TA pendant une nuit. On concentre à sec, reprend dans l'acétate d'éthyle, lave à l' HCl IN puis à l'eau. Après séchage, concentration à sec puis cristallisation dans le méthanol, on obtient 7,8 g de composé attendu.
23E) 5-(2-chlorophényl)-4-(4-hydroxyphényl)thiophène-2-carboxylate de méthyle.
Dans 100 ml de CH2CI2, on ajoute 5g de composé obtenu à l'étape 23D) et on refroidit à -500C, ajoute 41,8 ml de solution IM de BBrç dans CH2CI2. On laisse revenir la température à 200C puis on ajoute 20 ml de méthanol. On concentre à sec, reprend dans du CH2CI2, lave à l'eau, sèche sur MgSOφ concentre à sec. Après purification par chromatographie sur silice (heptane jusqu'à heptane / AcOEt 75/25), on obtient 4,57 g de composé attendu.
23F) 5 -(2-chlorophényl)-4- {4- [3 -(tétrahydro-2H-pyran-2-yloxy)-propoxy] - phényl}thiophène-2-carboxylate de méthyle.
Dans 15 ml de DMF, on ajoute 2,3 g de composé obtenu à l'étape 23E), 1,95 g de 2-(3-bromopropoxy)tétrahydro-2H-pyran, 1,2 g de carbonate de potassium puis on chauffe à 600C pendant 7 heures. On verse sur eau glacée, extrait à l'éther, sèche et concentre à sec. Après purification par chromatographie sur silice (heptane puis heptane / AcOEt : 80 / 20), on obtient 3,1 g de composé attendu.
23G) Acide 5-(2-chlorophényl)-4- {4-[3-(tétrahydro-2H-pyran-2-yloxy)-propoxy]- phényl}thiophène-2-carboxylique.
Dans 30 ml de méthanol et 1 ml d'eau, on ajoute 3,1 g de composé obtenu à l'étape 23F) puis 800 mg de soude en pastille. On porte le mélange réactionnel à reflux pendant 2 heures. On concentre à sec, reprend dans 500 ml de solution tampon à pΗ 2, extrait à l'éther, sèche et concentre à sec. On obtient 2,8 g de composé attendu, cristallisé dans le mélange éther isopropylique/ pentane.
23Η) l-{[5-(2-chlorophényl)-4-{4-[3-(tétrahydro-2H-pyran-2-yloxy)propoxy] phényl}thién-2-yl]carbonyl}-4-phénylpipéridine-4-carboxamide.
Dans 15 ml de CΗ2CI2, on ajoute 0,5 g de composé obtenu à l'étape 23G), 0,26 g de chlorhydrate de 4-phényl-pipéridine-4-carboxamide, 0,44 ml de EtβN et 0,38 g de TBTU puis on laisse la réaction 2 heures à TA. Après concentration à sec, reprise dans un mélange eau / éther, et fîltration, on obtient 0,45 g de composé attendu.
231) l-({5-(2-chlorophényl)-4-[4-(3-hydroxypropoxy)phényl]thién-2-yl}carbonyl)- 4-phénylpipéridine-4-carboxamide.
Dans 10 ml de méthanol, on ajoute 0,45 g de composé obtenu à l'étape 23H), 2 ml d'éther chlorhydrique 2N. Après 2 heures à TA, on filtre le composé attendu. On obtient 0,26 g de composé attendu.Les tableaux 1 et 2 indiquent les structures chimiques de quelques composés selon l'invention ainsi que leurs propriétés physiques (analyse par couplage LC/UV/MS : chromatographie liquide/détection UV/spectrométrie de masse). Ces composés sont exemplifïés ci-avant ou préparés selon des modes opératoires similaires à ceux des composés exemplifïés (exemples 1 à 23).
Dans les tableaux 1 et 2, Me représente un groupe méthyle.
Les analyses effectuées par RMN pour les composés 6, 8, 13, 27, 31 et 45 sont données ci-après :
Composé 6 : RMN 1H : DMSO-d6 (250 MHz) : δ (ppm) : 1.67- 1.90 : m : 2 H ; 2.39 - 2.58 : m : 2 H ; 3.13 - 3.38 : m : 2 H ; 3.63 : q : 2 H ; 3.91 : t : 2 H ; 4.03 - 4.22 : m : 2 H ; 4.81 : t : 1 H ; 6.86 : d : 2 H ; 7.00 - 7.12 : m : 3 H ; 7.15 - 7.47 : m : 9 H ; 7.68 : d : 1 H.
Composé 8 : RMN 1H : DMSO-d6 (250 MHz) : δ (ppm) : 1.60 - 2.04 : m : 4 H ; 2.37 - 2.62 : m : 2 H ; 3.15 - 3.41 : m : 2 H ; 3.48 : q : 2 H ; 3.94 : t : 2 H ; 4.02 - 4.21 : m : 2 H ; 4.46 : t : 1 H ; 6.80 : d : 2 H ; 6.97 - 7.13 : m : 3 H ; 7.14 - 7.25 : m : 2 H ; 7.27
- 7.41 : m : 4 H ; 7.44 : s : 2 H ; 7.55 : s : 1 H ; 7.68 : s : 1 H.
Composé 13 : RMN 1H : DMSO-d6 (250 MHz) : δ (ppm) : 1.31 - 1.66 : m : 4 H ; 1.76 - 1.97 : m : 2 H ; 2.48 - 2.63 : m : 2 H ; 3.19 - 3.50 : m : 6 H ; 4.10 - 4.25 : m : 2 H ; 4.42 : t : 1 H ; 7.12 : s : 1 H ; 7.20 - 7.60 : m : 10 H ; 7.75 : d : 2 H ; 7.82 : d : 2 H.
Composé 27 : RMN 1H : DMSO-d6 (250 MHz) : δ (ppm) : 1.77 - 2.02 : m : 2 H ; 2.46 - 2.62 : m : 2 H ; 3.20 - 3.53 : m : 2 H ; 3.68 : q : 2 H ; 3.95 : t : 2 H ; 4.10 - 4.25 : m : 2 H ; 4.83 : t : 1 H ; 6.85 : d : 2 H ; 7.07 - 7.17 : m : 3 H ; 7.22 - 7.30 : m : 2 H : 7.32
- 7.47 : m : 4 H ; 7.49 : d : 2 H ; 7.61 : s : 1 H ; 7.73 : t : 1 H.
Composé 31 : RMN 1H : DMSO-d6 (400 MHz) : δ (ppm) : 1.00 : t : 3 H ; 1.71- 1.85 : m : 2 H ; 1.84 - 1.95 : m : 2 H ; 2.53 : d : 2 H ; 3.32 - 3.44 : m : 2 H ; 3.47 : t : 2 H ; 4.15 : d : 2 H ; 7.09 : s : 1 H ; 7.20-7.32 : m : 6 H ; 7.34 : t : 2 H ; 7.41 : d : 2 H ; 7.48 : dd : 1 H ; 7.52 : d : 1 H ; 7.67 : s : 1 H ; 7.73 : d : 1 H.
Composé 45 : RMN 1H : DMSO-d6 (250 MHz) : δ (ppm) : 1,70 - 1,99 : m : 4 H ; 2,45 - 2,63 : m : 2 H ; 3,31 : br : s : 2H ; 3,53 : q : 2 H ; 3,98 : t : 2 H ; 4,17 : dt : 2 H ; 4,51 : t : IH ; 6,82 : d : 2 H ; 7,06 - 7,18 ; m ; 3 H ; 7,20 - 7,65 ; m 11 H. Les composés de formule (I) possèdent une très bonne affinité in vitro (IC50 ≤ 5.10 M) pour les récepteurs aux cannabinoïdes CBj, dans les conditions expérimentales décrites par M. Rinaldi-Carmona et al. (FEBS Letters, 1994, 350, 240-244).
La nature antagoniste des composés de formule (I) a été démontrée in vitro par les résultats obtenus dans les modèles de l'inhibition de l'adénylate-cyclase comme décrits
dans M. Bouaboula et al, J. Biol. Chem., 1995, 270, 13973-13980, M. Rinaldi-Carmona et al, J. Pharmacol. Exp. Ther., 1996, 278, 871-878 et M. Bouaboula et al, J. Biol. Chem., 1997, 272, 22330-22339.
La faible pénétration des composés de formule (I) au niveau de la barrière hématoencéphalique (BHE) a été évaluée in vivo par :
- Mesure (1) : la quantification des composés de formule (I) (inchangé) dans des échantillons de cerveau de souris après une administration par voie intraveineuse
(iv, 3 mg/kg) ou orale, à l'aide de technique analytique (LC-MS/MS). quantité présente dans le cerveau
Le ratio inférieur à 0,2 traduit une faible quantité présente dans le plasma pénétration du composé au niveau du cerveau.
- Mesure (2) : la mesure de l'interaction des composés de formule (I) avec les récepteurs CBj présents dans le cerveau chez la souris à l'aide d'un test de binding ex vivo du [3HJ-CP55940 (agoniste CBj) après une administration par voie intraveineuse (10 mg/kg) comme décrit dans M. Rinaldi-Carmona et al., FEBS Letters, 1994, 350, 240-244 et M. Rinaldi-Carmona et al, Life Sciences, 1995, 56, 1941-1947, M. Rinaldi-Carmona et al, J. Pharmacol. Exp. Ther., 2004, 310, 905- 914.
Un pourcentage d'inhibition de la liaison du [3HJ-CP55940 au niveau du cerveau inférieur à 50% à 10 mg/kg traduit une faible pénétration au niveau du cerveau. De préférence, ce pourcentage est inférieur à 40% et plus préférentiellement inférieur à 30%.
- Mesure (3) : la mesure du blocage par les composés de formule (I) de l'effet hypothermique induit par un agoniste des récepteurs CBi (CP55940), après une administration par voie intraveineuse (10 mg/kg), comme décrit dans Rinaldi- Carmona M. et al, JPET 2004, 310, 905-914).
Un pourcentage de réversion de l'effet hypothermique du CP55940 inférieur ou égal à 60 % à 10 mg/kg traduit une faible pénétration au niveau du cerveau. De préférence, ce pourcentage est inférieur à 40% et plus préférentiellement inférieur à 30%.
L'interaction d'un composé de formule (I) selon l'invention avec les récepteurs CBl présents à la périphérie a été démontrée chez la souris par la mesure du blocage de l'effet inhibiteur induit par le CP55940 sur le transit gastro -intestinal (TGI), après une administration orale (po, 10 mg/kg), comme décrit dans M. Rinaldi-Carmona et al, J. Pharmacol. Exp. Ther., 2004, 310, 905-914.
Un pourcentage de réversion de l'effet hypothermique du CP55940 supérieur à 50 % à 10 mg/kg traduit un pouvoir antagoniste significatif au niveau des récepteurs CBi
présent à la périphérie. De préférence, le pourcentage de réversion est compris entre 70% et 100%.
A titre d'exemples, les mesures suivantes ont été faites pour les composés n° 8 et
45 du Tableau 3
Tableau 3
Les composés de formule (I) sont compatibles avec leur utilisation en tant que médicament.
Ainsi, selon un autre de ses aspects, l'invention a pour objet des médicaments pour la médecine humaine ou vétérinaire qui comprennent un composé de formule (I), ou un sel d'addition de ce dernier à un acide ou une base pharmaceutiquement acceptable du composé de formule (I).
Ainsi les composés selon l'invention peuvent être utilisés chez l'homme ou chez l'animal (notamment chez les mammifères incluant de façon non limitative les chiens, les chats, les chevaux, les bovins, les moutons) dans le traitement et/ou la prévention de maladies impliquant les récepteurs aux cannabinoïdes CBj.
Par exemple et de manière non limitative, les composés de formule (I) sont utiles comme médicaments psychotropes, notamment pour le traitement et/ou la prévention des désordres psychiatriques incluant l'anxiété, la dépression, les troubles de l'humeur, l'insomnie, les troubles délirants, les troubles obsessionnels, les psychoses en général, la schizophrénie, les troubles du déficit de l'attention et de l'hyperactivité (TDAH) chez les enfants hyperkinétiques ainsi que pour le traitement et/ou la prévention des troubles liés à l'utilisation de substances psychotropes, notamment dans le cas d'un abus d'une
substance et/ou de dépendance à une substance, y compris la dépendance alcoolique et la dépendance nicotinique.
Les composés de formule (I) selon l'invention peuvent être utilisés comme médicaments pour le traitement et/ou la prévention de la migraine, du stress, des maladies d'origine psychosomatique, des crises d'attaques de panique, de l'épilepsie, des troubles du mouvement, en particulier des dyskinésies ou de la maladie de Parkinson, des tremblements et de la dystonie.
Les composés de formule (I) selon l'invention peuvent également être utilisés comme médicaments dans le traitement et/ou la prévention des troubles mnésiques, des troubles cognitifs, en particulier dans le traitement et/ou la prévention des démences séniles, de la maladie d'Alzheimer, ainsi que dans le traitement et/ou la prévention des troubles de l'attention ou de la vigilance.
De plus, les composés de formule (I) peuvent être utiles comme neuroprotecteurs, dans le traitement de l'ischémie, des traumatismes crâniens et le traitement et/ou la prévention des maladies neurodégénératives aiguës ou chroniques : incluant la chorée, la chorée de Huntington, le syndrome de Tourrette.
Les composés de formule (I) selon l'invention peuvent être utilisés comme médicaments dans le traitement et/ou la prévention de la douleur dont notamment : les douleurs neuropathiques, les douleurs aiguës périphériques, les douleurs chroniques d'origine inflammatoire, les douleurs induites par un traitement anticancéreux.
Les composés de formule (I) selon l'invention peuvent être utilisés comme médicaments dans la médecine humaine ou vétérinaire dans le traitement et/ou la prévention des troubles du métabolisme, de l'appétit, de l'appétence (pour les sucres, carbohydrates, drogues, alcools ou toute substance appétissante) et/ou des conduites alimentaires, notamment pour le traitement et/ou la prévention de l'obésité, de la boulimie ainsi que pour le traitement et/ou la prévention du diabète, notamment diabète de type II ou diabète non insulinodépendant, et pour le traitement et/ou la prévention des dyslipidémies, du syndrome métabolique. Ainsi les composés de formule (I) selon l'invention sont utiles dans le traitement et/ou la prévention de l'obésité et des risques associés à l'obésité, notamment les risques cardio-vasculaires.
De plus, les composés de formule (I) selon l'invention peuvent être utilisés en tant que médicaments dans le traitement et/ou la prévention des troubles gastro-intestinaux, des troubles diarrhéiques, des ulcères, des vomissements, des troubles vésicaux et urinaires, des maladies du foie d'origine alcoolique ou non telles que la cirrhose chronique, la fïbrose, la stéatose hépatique, la stéatohépatite ; ainsi que des troubles d'origine endocrinienne, des troubles cardio-vasculaires, de l'hypotension, de l'athérosclérose, du choc hémorragique, du choc septique, de l'asthme, de la bronchite chronique, des maladies pulmonaires chroniques obstructives, du syndrome de
Raynaud, du glaucome, des troubles de la fertilité, de l'accouchement prématuré, de l'interruption de grossesse, des phénomènes inflammatoires, des maladies du système immunitaire, en particulier autoimmunes et neuroinflammatoires tel que l'arthrite rhumatoïde, l'arthrite réactionne Ile, les maladies entraînant une démyélinisation, la sclérose en plaque, des maladies infectieuses et virales telles que les encéphalites, des accidents vasculaires cérébraux ainsi qu'en tant que médicaments pour la chimiothérapie anticancéreuse, pour le traitement du syndrome de Guillain-Barré et pour le traitement et/ou la prévention des maladies des os et de l'ostéoporose.
De plus, les composés de formule (I) selon l'invention peuvent être utilisés pour leurs effets protecteurs contre la cardiotoxicité induite par les médicaments.
Selon la présente invention, les composés de formule (I) sont tout particulièrement utiles pour la préparation de médicaments utiles pour le traitement et/ou la prévention des désordres psychiatriques, en particulier la schizophrénie, les troubles de l'attention et de la vigilance, les troubles du déficit de l'attention et de l'hyperactivité (TDAH) chez les enfants hyperkinétiques; pour le traitement et/ou la prévention des déficits mnésiques et des troubles cognitifs ; de la dépendance et du sevrage à une substance, en particulier la dépendance alcoolique, la dépendance nicotinique, le sevrage alcoolique et le sevrage tabagique ; des maladies neurodégénératives aiguës ou chroniques.
Selon la présente invention, les composés de formule (I) sont également tout particulièrement utiles pour la préparation de médicaments utiles dans le traitement et/ou la prévention des troubles de l'appétit, les troubles de l'appétence, des troubles du métabolisme, l'obésité, le diabète de type II, le syndrome métabolique, la dyslipidémie, des troubles gastro-intestinaux, des phénomènes inflammatoires, des maladies du système immunitaire, des troubles psychotiques, de la dépendance alcoolique, de la dépendance nicotinique.
Selon un de ses aspects, la présente invention est relative à l'utilisation d'un composé de formule (I) et de ses sels pharmaceutiquement acceptables pour le traitement et/ou la prévention des troubles et maladies indiqués ci-dessus.
Selon un autre de ses aspects, la présente invention concerne des compositions pharmaceutiques comprenant, en tant que principe actif, un composé de formule (I) selon l'invention. Ces compositions pharmaceutiques contiennent une dose efficace d'au moins un composé selon l'invention, ou un sel pharmaceutiquement acceptable dudit composé, ainsi qu'au moins un excipient pharmaceutiquement acceptable.
Lesdits excipients sont choisis selon la forme pharmaceutique et le mode d'administration souhaité, parmi les excipients habituels qui sont connus de l'Homme du métier.
Les compositions pharmaceutiques selon la présente invention peuvent contenir à côté d'un composé de formule (I) un (ou plusieurs) autre principe actif utile dans le traitement et/ou la prévention des troubles et maladies indiquées ci-dessus.
Ainsi, la présente invention a également pour objet des compositions pharmaceutiques contenant un composé de formule (I) selon la présente invention associé à un (ou plusieurs) principe actif choisi parmi l'une des classes thérapeutiques suivantes :
- un autre antagoniste ou modulateurs allosteriques des récepteurs CBj aux cannabinoïdes ;
- un modulateur des récepteurs CE$2 aux cannabinoïdes ;
- un antagoniste des récepteurs ATi de l'angiotensine II ;
- un inhibiteur de l'enzyme de conversion ;
- un antagoniste calcique ;
- un diurétique ;
- un béta-bloquant ;
- un antihyperlipémiant ou un antihypercholestérolémiant ;
- un antidiabétique ;
- un autre agent anti-obésité ou agissant sur les troubles du métabolisme ;
- un agoniste nicotinique, un agoniste nicotinique partiel ;
- un antidépresseur, un antipsychotique, un anxiolytique ;
- un anticancéreux ou un agent antiprolifératif ;
- un antagoniste des opioïdes ; ainsi que :
- un agent améliorant la mémoire ;
- un agent utile dans le traitement de l'alcoolisme ou des symptômes de sevrage ;
- un agent utile pour traiter l'ostéoporose ;
- un anti-inflammatoire non stéroïdien ou stéroïdien ;
- un anti-infectieux ;
- un analgésique ;
- un antiasthmatique.
Selon un autre aspect de l'invention, le composé de formule (I), un de ses sels pharmaceutiquement acceptables et l'autre principe actif associé peuvent être administrés de manière simultanée, séparée ou étalée dans le temps.
On entend par «utilisation simultanée» l'administration des composés de la composition selon l'invention compris dans une seule et même forme pharmaceutique.
On entend par « utilisation séparée » l'administration, en même temps, des deux composés de la composition selon l'invention chacun compris dans une forme pharmaceutique distincte.
On entend par « utilisation étalée dans le temps » l'administration successive, du premier composé de la composition de l'invention, compris dans une forme pharmaceutique, puis du deuxième composé de la composition selon l'invention, compris dans une forme pharmaceutique distincte. Dans ce cas, le laps de temps écoulé entre l'administration du premier composé de la composition selon l'invention et l'administration du deuxième composé de la même composition selon l'invention n'excède généralement pas 24 heures.
Dans les compositions pharmaceutiques de la présente invention pour l'administration orale, sublinguale, sous-cutanée, intramusculaire, intra- veineuse, topique, locale, intratrachéale, intranasale, transdermique ou rectale, le principe actif de formule (I) ci-dessus, ou son sel éventuel, peut être administré sous forme unitaire d'administration, en mélange avec des excipients pharmaceutiques classiques, aux animaux et aux êtres humains pour la prophylaxie, le traitement et/ou la prévention des troubles ou des maladies ci-dessus.
Les formes unitaires d'administration appropriées comprennent les formes par voie orale telles que les comprimés, les gélules molles ou dures, les poudres, les granules et les solutions ou suspensions orales, les formes d'administration sublinguale, buccale, intratrachéale, intraoculaire, intranasale, par inhalation, les formes d'administration topique, transdermique, sous-cutanée, intramusculaire ou intraveineuse, les formes d'administration rectale et les implants. Pour l'application topique, on peut utiliser les composés selon l'invention dans des crèmes, gels, pommades ou lotions.
A titre d'exemple, une forme unitaire d'administration d'un composé selon l'invention sous forme de comprimé peut comprendre les composants suivants :
Composé selon l'invention 50,0 mg Mannitol 223,75 mg
Croscarmellose sodique 6,0 mg Amidon de maïs 15,0 mg Hydroxypropyl-méthylcellulose 2,25 mg Stéarate de magnésium 3,0 mg
Par voie orale, la dose de principe actif administrée par jour peut atteindre 0,01 à 100 mg/kg, en une ou plusieurs prises, préférentiellement 0,02 à 50 mg/kg.
Il peut y avoir des cas particuliers où des dosages plus élevés ou plus faibles sont appropriés ; de tels dosages ne sortent pas du cadre de l'invention. Selon la pratique habituelle, le dosage approprié à chaque patient est déterminé par le médecin selon le mode d'administration, le poids et la réponse dudit patient.
La présente invention, selon un autre de ses aspects, concerne également une méthode de traitement et/ou de prévention des pathologies ci-dessus indiquées qui
comprend l'administration, à un patient, d'une dose efficace d'un composé selon l'invention, ou un de ses sels pharmaceutiquement acceptables.