WO2008076243A2 - Acyl bipiperidinyl compounds, compositions containing such compounds and methods of treatment - Google Patents
Acyl bipiperidinyl compounds, compositions containing such compounds and methods of treatment Download PDFInfo
- Publication number
- WO2008076243A2 WO2008076243A2 PCT/US2007/025225 US2007025225W WO2008076243A2 WO 2008076243 A2 WO2008076243 A2 WO 2008076243A2 US 2007025225 W US2007025225 W US 2007025225W WO 2008076243 A2 WO2008076243 A2 WO 2008076243A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- optionally substituted
- phenyl
- group
- bipiperidine
- Prior art date
Links
- ZJPBMHPNMMDOFN-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CCC1C(CC1)CCN1c(cc1)cc(Cl)c1N)=O Chemical compound CC(C)(C)OC(N(CC1)CCC1C(CC1)CCN1c(cc1)cc(Cl)c1N)=O ZJPBMHPNMMDOFN-UHFFFAOYSA-N 0.000 description 1
- ULHGRYYDJZNACS-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CCC1C(CC1)CCN1c(cc1)cc(Cl)c1NC(c1c[s]cn1)=O)=O Chemical compound CC(C)(C)OC(N(CC1)CCC1C(CC1)CCN1c(cc1)cc(Cl)c1NC(c1c[s]cn1)=O)=O ULHGRYYDJZNACS-UHFFFAOYSA-N 0.000 description 1
- IHQVNLNQOCIRPT-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CCC1C(CC1)CCN1c(cn1)ncc1Cl)=O Chemical compound CC(C)(C)OC(N(CC1)CCC1C(CC1)CCN1c(cn1)ncc1Cl)=O IHQVNLNQOCIRPT-UHFFFAOYSA-N 0.000 description 1
- OEHSOQBBWXZTSY-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CCC1C(CC1)CCN1c1cc(S(C)(=O)=O)ncn1)=O Chemical compound CC(C)(C)OC(N(CC1)CCC1C(CC1)CCN1c1cc(S(C)(=O)=O)ncn1)=O OEHSOQBBWXZTSY-UHFFFAOYSA-N 0.000 description 1
- GIIDYBTVCAWKOC-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CCC1C(CC1)CCN1c1cc(SC)ncn1)=O Chemical compound CC(C)(C)OC(N(CC1)CCC1C(CC1)CCN1c1cc(SC)ncn1)=O GIIDYBTVCAWKOC-UHFFFAOYSA-N 0.000 description 1
- IUTKEXNDCBCKKW-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CCC1C(CC1)CCN1c1nc(Cl)cnc1)=O Chemical compound CC(C)(C)OC(N(CC1)CCC1C(CC1)CCN1c1nc(Cl)cnc1)=O IUTKEXNDCBCKKW-UHFFFAOYSA-N 0.000 description 1
- NXNZCIXBIDICRH-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CCC1C(CC1)CCN1c1nccnc1)=O Chemical compound CC(C)(C)OC(N(CC1)CCC1C(CC1)CCN1c1nccnc1)=O NXNZCIXBIDICRH-UHFFFAOYSA-N 0.000 description 1
- NERBLCVCQKXTEP-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CCC1C1CCNCC1)=O Chemical compound CC(C)(C)OC(N(CC1)CCC1C1CCNCC1)=O NERBLCVCQKXTEP-UHFFFAOYSA-N 0.000 description 1
- LSEAAPGIZCDEEH-UHFFFAOYSA-N Clc1nc(Cl)cnc1 Chemical compound Clc1nc(Cl)cnc1 LSEAAPGIZCDEEH-UHFFFAOYSA-N 0.000 description 1
- RHJLTOVSJKKHLY-UHFFFAOYSA-N FC(c1nccc(Br)n1)(F)F Chemical compound FC(c1nccc(Br)n1)(F)F RHJLTOVSJKKHLY-UHFFFAOYSA-N 0.000 description 1
- PDCVDVCPQWFGAX-UHFFFAOYSA-N Oc1nc(C(F)(F)F)ncc1 Chemical compound Oc1nc(C(F)(F)F)ncc1 PDCVDVCPQWFGAX-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/26—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by nitrogen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
- A61P5/50—Drugs for disorders of the endocrine system of the pancreatic hormones for increasing or potentiating the activity of insulin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- the present invention relates to G-protein coupled receptor agonists.
- the present invention is directed to agonists of GPR 119 that are useful for the treatment of diabetes, especially type 2 diabetes, obesity, the metabolic syndrome and related diseases and conditions.
- Diabetes is a disease derived from multiple causative factors. It is characterized by elevated levels of plasma glucose (hyperglycemia) in the fasting state or after administration of glucose during an oral glucose tolerance test.
- type 1 diabetes or insulin-dependent diabetes mellitus (IDDM)
- IDDM insulin-dependent diabetes mellitus
- T2DM noninsul in-dependent diabetes mellitus
- insulin is still produced in the body, and patients demonstrate resistance to the effects of insulin in stimulating glucose and lipid metabolism in the main insulin-sensitive tissues, namely, muscle, liver and adipose tissue.
- T2DM noninsul in-dependent diabetes mellitus
- T2DM noninsul in-dependent diabetes mellitus
- These patients often have normal levels of insulin, and may have hyperinsulinemia (elevated plasma insulin levels), as they compensate for the reduced effectiveness of insulin by secreting increased amounts of insulin.
- Insulin resistance is not primarily caused by a diminished number of insulin receptors but rather by a post-insulin receptor binding defect that is not yet completely understood. This lack of responsiveness to insulin results in insufficient insulin-mediated activation of uptake, oxidation and storage of glucose in muscle, and inadequate insulin-mediated repression of lipolysis in adipose tissue and of glucose production and secretion in the liver.
- Persistent or uncontrolled hyperglycemia that occurs with diabetes is associated with increased and premature morbidity and mortality. Often abnormal glucose homeostasis is associated both directly and indirectly with obesity, hypertension, and alterations of the lipid, lipoprotein and apolipoprotein metabolism, as well as other metabolic and hemodynamic disease. Patients with type 2 diabetes mellitus have a significantly increased risk of macrovascular and microvascular complications, including atherosclerosis, coronary heart disease, stroke, peripheral vascular disease, hypertension, nephropathy, neuropathy, and retinopathy. Therefore, therapeutic control of glucose homeostasis, lipid metabolism, obesity, and hypertension are critically important in the clinical management and treatment of diabetes mellitus.
- a patient having metabolic syndrome is characterized as having three or more symptoms selected from the following group of five symptoms: (1) abdominal obesity; (2) hypertriglyceridemia; (3) low high-density lipoprotein cholesterol (HDL); (4) high blood pressure; and (5) elevated fasting glucose, which may be in the range characteristic of Type 2 diabetes if the patient is also diabetic.
- Each of these symptoms is defined clinically in the Third Report of the National Cholesterol Education Program Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel IH, or ATP m), National Institutes of Health, 2001, NIH Publication No. 01-3670.
- Patients with metabolic syndrome whether or not they have or develop overt diabetes mellitus, have an increased risk of developing the macrovascular and microvascular complications that occur with type 2 diabetes, such as atherosclerosis and coronary heart disease.
- Obesity a multifactorial pathophysiological state, is characterized by excessive adiposity relative to body mass.
- Obesity and being overweight increases the risk of developing conditions such as high blood pressure, type 2 diabetes, heart disease, stroke, osteoarthritis, sleep apnea, gallbladder disease and cancer of the breast, prostate and colon. Higher body weights are also associated with increases in all-cause mortality.
- the mainstay of treatment for obesity is an energy-limited diet and increased exercise.
- Orlistat which reduces intestinal fat absorption by inhibiting pancreatic lipase
- sibutramine anorectic agent sibutramine
- Reductil ® anorectic agent
- Other anorectics such as phentermine, bupropion, and diethylpropion have limited use due to side effects and, many of these drugs, like sibutramine, are schedule IV controlled substances due to the risk of addiction.
- a CB-1 antagonist, rimonabant (Accomplia) has been launched but long term durability and efficacy remains to be determined, and the drug has CNS side effects including dysphoria.
- the biguanides are a class of drugs that are widely used to treat type 2 diabetes.
- the two best known biguanides, phenformin and metformin cause some correction of hyperglycemia.
- the biguanides act primarily by inhibiting hepatic glucose production, and they also are believed to modestly improve insulin sensitivity.
- the biguanides can be used as monotherapy or in combination with other anti-diabetic drugs, such as insulin or an insulin secretogogues, without increasing the risk of hypoglycemia.
- phenformin and metformin can induce lactic acidosis and nausea/diarrhea. Metformin has a lower risk of side effects than phenformin and is widely prescribed for the treatment of Type 2 diabetes.
- the glitazones are a newer class of compounds that can ameliorate hyperglycemia and other symptoms of type 2 diabetes.
- the glitazones that are currently marketed are agonists of the peroxisome proliferator activated receptor (PPAR) gamma subtype.
- PPAR peroxisome proliferator activated receptor
- the PPAR-gamma agonists substantially increase insulin sensitivity in muscle, liver and adipose tissue in several animal models of type 2 diabetes, resulting in partial or complete correction of elevated plasma glucose levels without the occurrence of hypoglycemia.
- PPAR-gamma agonism is believed to be responsible for the improved insulin sensititization that is observed in human patients who are treated with the glitazones.
- New PPAR agonists are currently being developed. Many of the newer PPAR compounds are agonists of one or more of the PPAR alpha, gamma and delta subtypes. Compounds that are agonists of both the PPAR alpha and PPAR gamma subtypes (PPAR alpha/gamma dual agonists) are promising because they reduce hyperglycemia and also improve lipid metabolism.
- the currently marketed PPAR gamma agonists are modestly effective in reducing plasma glucose and HemoglobinAlC.
- the currently marketed compounds do not greatly improve lipid metabolism and may actually have a negative effect on the lipid profile.
- the PPAR compounds represent an important advance in diabetic therapy, but further improvements are still needed.
- Another widely used drug treatment involves the administration of insulin secretagogues, such as the sulfonylureas (e.g. tolbutamide and glipizide). These drugs increase the plasma level of insulin by stimulating the pancreatic ⁇ -cells to secrete more insulin. Insulin secretion in the pancreatic ⁇ -cell is under strict regulation by glucose and an array of metabolic, neural and hormonal signals.
- Glucose stimulates insulin production and secretion through its metabolism to generate ATP and other signaling molecules, whereas other extracellular signals act as potentiators or inhibitors of insulin secretion through GPCR's present on the plasma membrane.
- Sulfonylureas and related insulin secretagogues act by blocking the ATP-dependent K+ channel in ⁇ -cells, which causes depolarization of the cell and the opening of the voltage-dependent Ca2+ channels with stimulation of insulin release. This mechanism is non-glucose dependent, and hence insulin secretion can occur regardless of the ambient glucose levels. This can cause insulin secretion even if the glucose level is low, resulting in hypoglycemia, which can be fatal in severe cases. The administration of insulin secretagogues must therefore be carefully controlled.
- the insulin secretagogues are often used as a first-line drug treatment for Type 2 diabetes.
- GPCR G-protein coupled receptors
- GPR119 agonists may be effective anti-obesity agents.
- Synthetic GPR119 agonists also augment the release of insulin from isolated static mouse islets only under conditions of elevated glucosej and improve glucose tolerance in diabetic mice and diet-induced obese (DIO) C57/B6 mice without causing hypoglycemia. GPR119 agonists therefore have the potential to function as anti-hyperglycemic agents that produce weight loss.
- GPR119 As a potential target for the treatment of type 2 diabetes and obesity.
- the weight loss efficacy of GPR119 agonists should contribute to antihyperglycemic efficacy in diabetic and prediabetic obese subjects, and activation of GPR119 may allow for the simultaneous treatment of the common co-morbidities of obesity and impaired glucose tolerance/diabetes.
- the limited tissue distribution of GPR119 in humans suggests that there would be less chance for side effects associated with GPR119 activity in other tissues.
- GPR119 agonists may have the potential to restore or preserve islet function since GPR119 agonists reportedly increase GLP-1 levels in rodents (Chu, Z L. et al Abstract Pl-19, ENDO 2005 87 th Annual Meeting, San Diego, CA).
- GLP-1 is an incretin hormone that effects GDIS and exerts anti-apoptotic and proliferative effects on islets.
- a protective effect on islets upon GPR119 agonism would be highly advantageous, because long term diabetes therapy often leads to the gradual diminution of islet activity, such that after extended periods of treatment with multiple oral antihyperglycemic agents, it is often necessary to treat type 2 diabetic patients with daily insulin injections.
- GPR119 agonists may delay or prevent the diminution and loss of islet function in a type 2 diabetic patient.
- R 1 represents a 6-10 membered Aryl group or a 5-10 membered Heteroaryl group containing one or two rings, at least one nitrogen atom, 0-2 additional nitrogen atoms, and 0-1 oxygen or sulfur atom, said Aryl or Heteroaryl group being optionally substituted with 1-3 halo groups and 1-2 members selected from the group consisting of:
- Phenyl the phenyl portion of which being optionally substituted with 1-3 halo groups and 0-1 C 1-3 alkyl, haloalkyl, alkoxy or haloalkoxy groups, and each of said C 1-8 alkyl, C 3-6 cycloalkyl and OC 1-8 alkyl being optionally substituted with 1-3 halo groups, and 0-1 members selected from the group consisting of OH, oxo, CN, OC 1-6 alkyl, haloC 1-6 alkoxy, pyridyl, C(O)C 1-3 alkyl, phenyl, and C(O)phenyl, said phenyl, pyridyl and the phenyl portion of C(O)phenyl being optionally substituted with 1-3 halo atoms, and 1-2 C 1-6 alkyl or OC 1-6 alkyl groups.
- Alkyl as well as other groups having the prefix "alk”, such as alkoxy, and the like, means carbon chains which may be linear, branched, or cyclic, or combinations thereof, containing the indicated number of carbon atoms. If no number is specified, 1-6 carbon atoms are intended for linear and 3-7 carbon atoms for branched alkyl groups. Examples of alkyl groups include methyl, ethyl, propyl, isopropyl, butyl, sec- and tert-butyl, pentyl, hexyl, heptyl, octyl, nonyl and the like.
- Cycloalkyl is a subset of alkyl; if no number of atoms is specified, 3-7 carbon atoms are intended, forming 1-3 carbocyclic rings that are fused. "Cycloalkyl” also includes monocyclic rings fused to an aryl group in which the point of attachment is on the non-aromatic portion. Examples of cycloalkyl include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, tetrahydronaphthyl, decahydronaphthyl, indanyl and the like.
- Haloalkoxy and haloOalkyl are used interchangeably and refer to halo substituted alkyl groups linked through the oxygen atom.
- Haloalkyl and haloalkoxy include mono- substituted as well as multiple substituted alkyl and alkoxy groups, up to perhalo substituted alkyl and alkoxy.
- trifluoromethyl and trifluoromethoxy are included.
- Aryl (Ar) means phenyl or naphthyl, preferably phenyl.
- Heteroaryl (HAR) unless otherwise specified, means monocyclic aromatic ring systems containing 5-6 atoms, at least one of which is a heteroatom selected from O, S, S(O), SO 2 and N. Examples include, but are not limited to, pyrrolyl, isoxazolyl, isothiazolyl, pyrazolyl, pyridyl, oxazolyl, oxadiazolyl, thiadiazolyl, thiazolyl, imidazolyl, triazolyl, tetrazolyl, furanyl, triazinyl, thienyl, pyrimidyl, pyridazinyl, pyrazinyl and the like. Heteroaryl also includes such groups in charged form, e.g., pyridinium.
- Halogen includes fluorine, chlorine, bromine and iodine.
- R 1 represents a 6-10 membered Aryl group or a 5-10 membered Heteroaryl group containing one or two rings, at least one nitrogen atom, 0-2 additional nitrogen atoms, and 0-1 oxygen or sulfur atom, said Aryl or Heteroaryl group being optionally substituted with 1-3 halo groups and 1-2 members selected from the group consisting of:
- Phenyl a 5-10 membered Heteroaryl or a 5-10 membered Heterocyclic moiety, each being optionally substituted with 1-3 halo atoms and 1-2 C 1-3 alkyl groups;
- R 2 is selected from the group consisting of C 1-8 alkyl, C 3-6 cycloalkyl, OC 1-8 alkyl and O- Phenyl, the phenyl portion of which being optionally substituted with 1-3 halo groups and 0-1 C 1-3 alkyl, haloalkyl, alkoxy or haloalkoxy groups, and each of said C 1-8 alkyl, C 3-6 cycloalkyl and OC 1-8 alkyl being optionally substituted with 1-3 halo groups, and 0-1 members selected from the group consisting of OH, oxo, CN, OC 1-6 alkyl, haloC 1-6 alkoxy, pyridyl, C(O)C 1-3 alkyl, phenyl, and C(O)phenyl, said phenyl, pyridyl and the phenyl portion of C(O)phenyl being optionally substituted with 1-3 halo atoms, and 1-2 C 1-6
- An aspect of the invention that is of interest relates to a compound of formula I or a pharmaceutically acceptable salt or solvate thereof wherein R 1 represents a 6-10 membered Aryl group, optionally substituted as described above.
- R 1 represents a 6-10 membered Aryl group, optionally substituted as described above.
- an aspect of the invention that is of interest relates to a compound of formula I or a pharmaceutically acceptable salt or solvate thereof wherein R 1 represents phenyl optionally substituted as described above.
- R 1 represents phenyl optionally substituted as described above.
- Another aspect of the invention that is of interest relates to a compound of formula I or a pharmaceutically acceptable salt or solvate thereof wherein R 1 represents a 5-10 membered heteroaryl group, optionally substituted as described above.
- R 1 represents a 5-10 membered heteroaryl group, optionally substituted as described above.
- R 1 represents a 5-10 membered heteroaryl group, selected from the group consisting of: thiazolyl, thiadiazolyl, pyridyl, pyrimidinyl, pyrazinyl and benzopyrazinyl, said heteroaryl group being optionally substituted as described above.
- R 1 represents a 5-10 membered heteroaryl group, selected from the group consisting of: thiazolyl, thiadiazolyl, pyridyl, pyrimidinyl, pyrazinyl and benzopyrazinyl, said heteroaryl group being optionally substituted as described above.
- an aspect of the invention that is of interest relates to a compound of formula I or a pharmaceutically acceptable salt or solvate thereof wherein R 1 represents a member selected from the group consisting of phenyl, pyridyl, pyrimidinyl and pyrazinyl, said group being optionally substituted as described above.
- R 1 represents a member selected from the group consisting of phenyl, pyridyl, pyrimidinyl and pyrazinyl, said group being optionally substituted as described above.
- an aspect of the invention that is of interest relates to a compound of formula I or a pharmaceutically acceptable salt or solvate thereof wherein R 1 represents phenyl or pyrimidinyl, said group being optionally substituted as described above.
- R 1 represents phenyl or pyrimidinyl, said group being optionally substituted as described above.
- Another aspect of the invention that is of interest relates to a compound of formula I or a pharmaceutically acceptable salt wherein R 1 is described as above and is optionally substituted with 1-2 halo atoms selected from fluoro, chloro and bromo, and 1 -2 members selected from the group consisting of: (1) CN or OH;
- R e is selected from H, methyl, C 3-6 cycloalkyl, phenyl and a 5-6 membered Heteroaryl group selected from thiazole, imidazole and triazole; x) CO 2 R e , wherein R e is as defined above; xi) Heteroaryl optionally substituted with 1-2 halo or C 1-3 alkyl groups;
- Phenyl a 5-10 membered Heteroaryl or a 5-10 membered Heterocyclic moiety, each being optionally substituted with 1-3 fluorine atoms and 1-2 C 1-3 alkyl groups.
- all other variables are as originally defined.
- an aspect of the invention that is of interest relates to a compound of formula I or a pharmaceutically acceptable salt wherein R 1 is phenyl or a 5-9 membered Heteroaryl group as described above, optionally substituted with 1-2 halo atoms selected from fluoro and chloro, and 1-2 members selected from the group consisting of: (1) CN or OH;
- CH 3 , OCH 3 or cyclopropyl being optionally substituted with 1-3 halo atoms selected from fluoro and chloro and 1 member selected from the group consisting of: i) SO 2 C 1-3 alkyl, wherein the alkyl portion is optionally substituted with 1-3 fluorine atoms; ii) a 5-6 membered Heteroaryl moiety selected from trhazole, pyrazole, thiadiazole, imidazole and oxazole, said moiety being optionally substituted with 1-2 halo atoms selected from fluorine and chlorine, or CH 3 groups; iii) OCH 3 or OCF 3 ; iv) CN; v) NH 2 , NHCH 3 and N(CH 3 ) 2 the alkyl portions of which are optionally substituted with 1-3 fluorine atoms; vi) OH; vii) C(O)CH 3 the alkyl portion being optionally substituted with 1-3 fluorine
- R e is selected from H, methyl, C 3-6 cycloalkyl, phenyl and a 5-6 membered Heteroaryl group selected from thiazole, imidazole and triazole; x) CO 2 R e , wherein R e is as defined above; xi) Heteroaryl optionally substituted with 1-2 halo or C 1-3 alkyl groups; (3) S(O) x C 1-3 alkyl, and SO 2 NR d R e wherein the x, R d and R e are as defined above and the C 1-3 alkyl portion is optionally substituted with 1-3 halo groups selected from Cl and F; (4) NH 2 , NHCH 3 or N(CH 3 ) 2 , wherein the alkyl portions are optionally substituted with 1-3 fluorine atoms;
- Another aspect of the invention that is of interest relates to a compound of formula I or a pharmaceutically acceptable salt thereof wherein:
- R 2 is selected from the group consisting of C 1-7 alkyl, C 3-6 cycloalkyl, OC 1-6 alkyl and O- Phenyl, the phenyl portion of which being optionally substituted with 1-3 halo groups selected from chloro and fluoro, and 0-1 C 1-3 alkyl, haloC 1-3 alkyl, C 1-3 alkoxy or haloC 1-3 alkoxy groups, wherein the halo atoms are fluorine or chlorine, and each of said C 1-7 alkyl, C 3-6 cycloalkyl and OC 1-6 alkyl being optionally substituted with 1-3 fluoro or chloro groups, and 0-1 members selected from the group consisting of OH, oxo, CN, OC 1-3 alkyl, haloC 1-3 alkoxy, C(O)C 1-3 alkyl, phenyl, and C(O)phenyl, said phenyl and the phenyl portion of C(O)phen
- R 2 is selected from the group consisting of C 1-4 alkyl, cyclopropyl, cyclohexyl, OC 1-6 alkyl and O-Phenyl, the phenyl portion of which being optionally substituted with 1-3 halo groups selected from chloro and fluoro, and 0-1 C 1-3 alkyl, haloC 1-3 alkyl, C 1-3 alkoxy or haloC 1-3 alkoxy groups, wherein the halo atoms are fluorine or chlorine, and each of said C 1-4 alkyl, cyclopropyl, cyclohexyl and OC 1-6 alkyl being optionally substituted with 1-3 fluoro or chloro groups, and 0-1 members selected from the group consisting of OH, oxo, CN, OCH 3 , OCF 3 , C(O)CH 3 , phenyl, and C(O)phenyl, said phenyl and the phenyl portion of C(O)pheny
- An aspect of the invention that is of more interest relates to a compound of formula I or a pharmaceutically acceptable salt or solvate thereof wherein:
- R 1 represents a member selected from the group consisting of phenyl, pyridyl, pyrimidinyl and pyrazinyl; said group being optionally substituted with 1-2 halo atoms selected from fluoro, chloro and bromo, and 1-2 members selected from the group consisting of:
- R e is selected from H, methyl, C 3-6 cycloalkyl, phenyl and a 5-6 membered Heteroaryl group selected from thiazole, imidazole and triazole; x) CO 2 R e , wherein R e is as defined above; xi) Heteroaryl optionally substituted with 1-2 halo or C 1-3 alkyl groups;
- Phenyl the phenyl portion of which being optionally substituted with 1-3 halo groups selected from chloro and fluoro, and 0-1 C 1-3 alkyl, haloC 1-3 alkyl, C 1-3 alkoxy or haloC 1-3 alkoxy groups, wherein the halo atoms are fluorine or chlorine, and each of said C 1-7 alkyl, C 3-6 cycloalkyl and OC 1-6 alkyl being optionally substituted with 1-3 fluoro or chloro groups, and 0-1 members selected from the group consisting of OH, oxo, CN, OC 1-3 alkyl, haloC 1-3 alkoxy, C(O)C 1-3 alkyl, phenyl, and C(O)phenyl, said phenyl and the phenyl portion of C(O)phenyl being optionally substituted with 1-3 chlorine or fluorine atoms, and 1-2 C 1-3 alkyl or OC 1-3 alkyl groups.
- an aspect of the invention that is of increased interest relates to a compound of formula I or a pharmaceutically acceptable salt or solvate thereof wherein:
- R 1 represents a member selected from the group consisting of phenyl, pyridyl, pyrimidinyl and pyrazinyl, optionally substituted with 1-2 halo atoms selected from fluoro and chloro, and 1 -2 members selected from the group consisting of:
- R e is selected from H, methyl, C 3-6 cycloalkyl, phenyl and a 5-6 membered Heteroaryl group selected from thiazole, imidazole and triazole; x) CO 2 R e , wherein R e is as defined above; xi) Heteroaryl optionally substituted with 1-2 halo or C 1-3 alkyl groups; (3) S(O) x C 1-3 alkyl, and SO 2 NR d R e wherein the x, R d and R e are as defined above and the C 1-3 alkyl portion is optionally substituted with 1-3 halo groups selected from Cl and F; (4) NH 2 , NHCH 3 or N(CH 3 ) 2 , wherein the alkyl portions are optionally substituted with 1-3 fluorine atoms;
- R 2 is selected from the group consisting of C 1-4 alkyl, cyclopropyl, cyclohexyl, OC 1-6 alkyl and O-Phenyl, the phenyl portion of which being optionally substituted with 1-3 halo groups selected from chloro and fluoro, and 0-1 C 1-3 alkyl, haloC 1-3 alkyl, C 1-3 alkoxy or haloC 1-3 alkoxy groups, wherein the halo atoms are fluorine or chlorine, and each of said C 1-4 alkyl, cyclopropyl, cyclohexyl and OC 1-6 alkyl being optionally substituted with 1-3 fluoro or chloro groups, and 0-1 members selected from the group consisting of OH, oxo, CN, OCH 3 , OCF 3 , C(O)CH 3 , phenyl, and C(O)phenyl, said phenyl and the phenyl portion of C(O)pheny
- Compounds of the present invention are potent agonists of the GPR119 receptor.
- the compounds of the invention, and pharmaceutically acceptable salts thereof are modulators of the receptor known as GPR 119, and are therefore useful in the treatment of diseases that are modulated by GPR119 ligands and agonists. Many of these diseases are summarized below.
- the compounds of the invention may be used for the manufacture of a medicament for treating one or more of these diseases or conditions:
- non- insulin dependent diabetes mellitus type 2 diabetes
- hypercholesterolemia (6) hypertriglyceridemia (elevated levels of triglyceride-rich-lipoproteins);
- the following diseases and conditions can be treated using the compounds of formula I or a pharmaceutically acceptable salt or solvate thereof.
- the compounds may be used for manufacturing a medicament for the treatment or prevention of one or more of these diseases or conditions: (1) Type 2 diabetes, and specifically hyperglycemia;
- the compounds are agonists of the GPR119 receptor, the compounds will be useful for lowering glucose, lipids, and insulin resistance in diabetic patients and in non-diabetic patients who have impaired glucose tolerance and/or are in a pre-diabetic condition.
- the compounds are useful to ameliorate hyperinsulinemia, which often occurs in diabetic or pre-diabetic patients, by modulating the swings in the level of serum glucose that often occurs in these patients.
- the compounds are useful for treating or reducing insulin resistance.
- the compounds are useful for treating or preventing gestational diabetes.
- the compounds, compositions, and medicaments as described herein are useful for reducing the risks of adverse sequelae associated with metabolic syndrome, and in reducing the risk of developing atherosclerosis, delaying the onset of atherosclerosis, and/or reducing the risk of sequelae of atherosclerosis.
- Sequelae of atherosclerosis include angina, claudication, heart attack, stroke, and others.
- the compounds are useful to delay or for preventing vascular restenosis and diabetic retinopathy.
- the compounds of this invention are useful in improving or restoring ⁇ -cell function, so that they may be useful in treating type 1 diabetes or in delaying or preventing a patient with type 2 diabetes from needing insulin therapy.
- the compounds may be useful for reducing appetite and body weight in obese subjects and may thereforebe useful in reducing the risk of co-morbidities associated with obesity such as hypertension, atherosclerosis, diabetes, and dyslipidemia.
- the compounds are useful in treating neurological disorders such as Alzheimer's disease, multiple sclerosis, and schizophrenia.
- the compounds generally are useful for treating the following diseases and conditions:
- type 2 diabetes also known as non-insulin dependent diabetes mellitus, or T2DM
- hyperglycemia also known as non-insulin dependent diabetes mellitus, or T2DM
- hyperglycemia also known as non-insulin dependent diabetes mellitus, or T2DM
- obesity also known as non-insulin dependent diabetes mellitus, or T2DM
- hyperglycemia also known as non-insulin dependent diabetes mellitus, or T2DM
- hyperglycemia also known as non-insulin dependent diabetes mellitus, or T2DM
- hyperglycemia also known as non-insulin dependent diabetes mellitus, or T2DM
- hyperlipid disorders also known as non-insulin dependent diabetes mellitus, or T2DM
- dyslipidemia (8) hyperlipidemia, (9) hypertriglyceridemia, (10) hypercholesterolemia, (11) low HDL levels, (12) high LDL levels, (13) atherosclerosis and its sequelae, (14) vascular restenosis, (15
- One aspect of the invention provides a method for the treatment and control of mixed or diabetic dyslipidemia, hypercholesterolemia, atherosclerosis, low HDL levels, high LDL levels, hyperlipidemia, and/or hypertriglyceridemia, which comprises administering to a patient in need of such treatment a therapeutically effective amount of a compound of formula I or a pharmaceutically acceptable salt or solvate therof.
- the compound may be used alone or advantageously may be administered with a cholesterol biosynthesis inhibitor, particularly an HMG-CoA reductase inhibitor such as lovastatin, simvastatin, rosuvastatin, pravastatin, fluvastatin, atorvastatin, rivastatin, itavastatin, or ZD-4522.
- the compound may also be used advantageously in combination with other lipid lowering drugs such as cholesterol absorption inhibitors (for example stanol esters, sterol glycosides such as tiqueside, and azetidinones such as ezetimibe), ACAT inhibitors (such as avasimibe), CETP inhibitors (for example torcetrapib), niacin, bile acid sequestrants, microsomal triglyceride transport inhibitors, and bile acid reuptake inhibitors.
- cholesterol absorption inhibitors for example stanol esters, sterol glycosides such as tiqueside, and azetidinones such as ezetimibe
- ACAT inhibitors such as avasimibe
- CETP inhibitors for example torcetrapib
- niacin for example bile acid sequestrants
- microsomal triglyceride transport inhibitors microsomal triglyceride transport inhibitors
- Another aspect of the invention provides a method for the treatment and control of obesity or metabolic syndrome, which comprises administering to a patient in need of such treatment a therapeutically effective amount of a compound having formula I or a pharmaceutically acceptable salt or solvate thereof.
- the compound may be used alone or advantageously may be administered with an anti- obesity agent, particularly a lipase inhibitor such as orlistat, or a monoamine neurotransmitter uptake inhibitor such as sibutramine, phentermine and the like.
- the compound may also be used advantageously in combination with CB-1 inverse agonists or antagonists such as rimonabant.
- Any suitable route of administration may be employed for providing a mammal, especially a human, with an effective dose of a compound of the present invention.
- oral, rectal, topical, parenteral, ocular, pulmonary, nasal, and the like may be employed.
- Dosage forms include tablets, troches, dispersions, suspensions, solutions, capsules, creams, ointments, aerosols, and the like.
- compounds of Formula I or a pharmaceutically acceptable salt or solvate thereof are administered orally.
- the effective dosage of active ingredient employed may vary depending on the particular compound employed, the mode of administration, the condition being treated and the severity of the condition being treated. Such dosage may be ascertained readily by a person skilled in the art.
- a daily dosage of from about 0.1 milligram to about 100 milligram per kilogram of animal body weight, preferably given as a single daily dose or in divided doses two to six times a day, or in sustained release form.
- the total daily dosage is from about 1.0 milligrams to about 1000 milligrams.
- the total daily dose will generally be from about 1 milligram to about 350 milligrams.
- the dosage for an adult human may be as low as 0.1 mg.
- the dosage regimen may be adjusted within this range or even outside of this range to provide the optimal therapeutic response.
- Oral administration will usually be carried out using tablets or capsules.
- Examples of doses in tablets and capsules are 0.1 mg, 0.25 mg, 0.5 mg, 1 mg, 2 mg, 5 mg, 10 mg, 25 mg, 50 mg, 100 mg, 200 mg, 350 mg, 500mg, 700mg, 750mg, 800mg and 1000mg.
- Other oral forms may also have the same or similar dosages.
- compositions which comprise a compound of Formula I and a pharmaceutically acceptable carrier.
- the pharmaceutical compositions of the present invention comprise a compound of Formula I or a pharmaceutically acceptable salt or solvate as an active ingredient, as well as a pharmaceutically acceptable carrier and optionally other therapeutic ingredients.
- pharmaceutically acceptable salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic bases or acids and organic bases or acids.
- a pharmaceutical composition may also comprise a prodrug, or a pharmaceutically acceptable salt thereof, if a prodrug is administered.
- compositions are typically suitable for oral, rectal, topical, parenteral (including subcutaneous, intramuscular, and intravenous), ocular (ophthalmic), pulmonary (nasal or buccal inhalation), or nasal administration, although the most suitable route in any given case will depend on the nature and severity of the condition being treated and on the particular active ingredient selected. They may be conveniently presented in unit dosage form and prepared by any of the methods well-known in the art of pharmacy.
- the compound of Formula I, or the pharmaceutically acceptable salt or solvate thereof can be combined as the active ingredient in intimate admixture with the pharmaceutical carrier according to conventional pharmaceutical compounding techniques.
- the carrier may take a wide variety of forms depending on the form of preparation desired for administration, e.g., oral or parenteral (including intravenous),
- any of the usual pharmaceutical media may be employed, such as, for example, water, glycols, oils, alcohols, flavoring agents, preservatives, coloring agents and the like in the case of oral liquid preparations, such as, for example, suspensions, elixirs and solutions; or carriers such as starches, sugars, microcrystalline cellulose, diluents, granulating agents, lubricants, binders, disintegrating agents and the like in the case of oral solid preparations such as, for example, powders, hard and soft capsules and tablets, with the solid oral preparations being preferred over the liquid preparations.
- tablets and capsules represent the most advantageous oral dosage form.
- Solid pharmaceutical carriers are therefore typically employed.
- tablets may be coated by standard aqueous or nonaqueous techniques.
- Such compositions and preparations typically comprise at least about 0.1 percent of active compound, the remainder of the composition being the carrier.
- the percentage of active compound in these compositions may, of course, be varied and is conveniently between about 2 percent to about 60 percent of the weight of the dosage form.
- the amount of active compound in such therapeutically useful compositions is such that an effective dosage will be delivered.
- the active compound can be administered intranasally as, for example, in the form of liquid drops or a spray.
- the tablets, capsules and the like also typically contain a binder.
- suitable binders include gum tragacanth, acacia, gelatin and a synthetic or semisynthetic starch derivative, such as hydroxypropylmethylcellulose (HPMC); excipients such as dicalcium phosphate; a disintegrating agent such as corn starch, potato starch, alginic acid; a lubricant such as magnesium stearate; and in some instances, a sweetening agent such as sucrose, lactose or saccharin.
- HPMC hydroxypropylmethylcellulose
- the dosage form employed is a capsule, it may contain, in addition to the components described above, a liquid carrier such as a fatty oil.
- a liquid carrier such as a fatty oil.
- Various other materials may be present as coatings or to modify the physical form of the dosage unit. For instance, tablets may be coated with shellac, sugar or both.
- Syrups and elixirs typically contain, in addition to the active ingredient, sucrose as a sweetening agent, methyl or propylparabens as a preservative, a dye and a flavoring such as cherry or orange flavor.
- the compound of formula I or a pharmaceutically acceptable salt or solvate thereof may also be administered parenterally.
- Solutions or suspensions of these active compounds can be prepared in water, saline or another biocompatible vehicle, suitably mixed with a surfactant, buffer, and the like. Dispersions can also be prepared in glycerol, liquid polyethylene glycols and mixtures thereof in an oil. Under ordinary conditions of storage and use, these preparations can also contain a preservative to prevent the growth of microorganisms.
- the pharmaceutical forms suitable for injectable use include sterile aqueous solutions and dispersions, and sterile powders for the extemporaneous preparation of sterile injectable solutions and dispersions.
- the preparation should be prepared under sterile condituons and be fluid to the extent that easy syringability exists. It should be sufficiently stable under the conditions of manufacture and storage and preserved against the growth of microorganisms such as bacteria and fungi.
- the carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (e.g. glycerol, propylene glycol and liquid polyethylene glycol), suitable mixtures thereof, and suitable oils.
- Combination Therapy Compounds of Formula I may be used in combination with other drugs that may also be useful in the treatment or amelioration of the diseases and conditions described herein.
- Such other drugs may be administered, by a route and in an amount commonly used therefor, contemporaneously or sequentially with a compound of Formula I or a pharmaceutically acceptable salt or solvate thereof.
- a compound of Formula I or a pharmaceutically acceptable salt or solvate thereof In the treatment of patients who have type 2 diabetes, insulin resistance, obesity, metabolic syndrome, neurological disorders, and co-morbidities that accompany these diseases, more than one drug is commonly administered.
- the compounds of this invention may generally be administered to a patient who is already taking one or more other drugs for these conditions.
- a pharmaceutical composition in unit dosage form containing such other drugs and the compound of Formula I is preferred.
- the combination therapy also includes therapies in which the compound of Formula I and one or more other drugs are administered on different overlapping schedules.
- the pharmaceutical compositions of the present invention include those that contain one or more other active ingredients, in addition to a compound of Formula I.
- Examples of other active ingredients that may be administered in combination with a compound of Formula I, and either administered separately or in the same pharmaceutical composition include, but are not limited to:
- PPAR gamma agonists and partial agonists including both glitazones and non- glitazones (e.g. troglitazone, pioglitazone, englitazone, MCC-555, rosiglitazone, balaglitazone, netoglitazone, T-131, LY-300512, and LY-818;
- glitazones and non- glitazones e.g. troglitazone, pioglitazone, englitazone, MCC-555, rosiglitazone, balaglitazone, netoglitazone, T-131, LY-300512, and LY-818;
- DPP-4 dipeptidyl peptidase IV (DPP-4) inhibitors, such as sitagliptin, saxagliptin, denagliptin, SYR-322, and vildagliptin;
- sulfonylureas such as tolbutamide, glimepiride, glipizide, and related materials
- ⁇ -glucosidase inhibitors such as acarbose
- agents which improve a patient's lipid profile such as (i) HMG-CoA reductase inhibitors (lovastatin, simvastatin, rosuvastatin, pravastatin, fluvastatin, atorvastatin, rivastatin, itavastatin, ZD-4522 and other statins), (ii) bile acid sequestrants (cholestyramine, colestipol, and dialkylaminoalkyl derivatives of a cross-linked dextran), (iii) nicotinyl alcohol, nicotinic acid or a salt thereof, (iv) PPAR ⁇ agonists such as fenofibric acid derivatives (gemfibrozil, clofibrate, fenofibrate and bezafibrate), (v) cholesterol absorption inhibitors, such as for example ezetimibe, (vi) acyl CoAxholesterol acyltransferase (ACAT) a
- PPAR ⁇ / ⁇ dual agonists such as muraglitazar, tesaglitazar, farglitazar, and JT-501
- PPAR ⁇ agonists such as those disclosed in WO97/28149
- antiobesity compounds such as fenfluramine, dexfenfluramine, phentiramine, subitramine, orlistat, neuropeptide Y5 inhibitors, Mc4r agonists, cannabinoid receptor 1 (CB-1) antagonists/inverse agonists, and ⁇ 3 adrenergic receptor agonists;
- ileal bile acid transporter inhibitors agents intended for use in inflammatory conditions such as aspirin, non-steroidal anti-inflammatory drugs, glucocorticoids, azulfidine, and cyclo-oxygenase 2 selective inhibitors;
- GLP-1 analogs such as exendins, for example exenatide (Byetta), exenatide-LAR, and liraglutide
- the above combinations include combinations of a compound of the present invention not only with one other active compound, but also with two or more other active compounds.
- Non- limiting examples include combinations of compounds having Formula I or a pharmaceutically acceptable salt or solvate thereof with two or more active compounds selected from biguanides, sulfonylureas, HMG-CoA reductase inhibitors, other PPAR agonists, PTP-1B inhibitors, DPP-4 inhibitors, and anti-obesity compounds.
- Another aspect of the invention that is of interest is a pharmaceutical composition comprised of a compound of formula I or a pharmaceutically acceptable salt or solvate thereof in combination with a pharmaceutically acceptable carrier.
- Another aspect of the invention that is of interest relates to a method of treating hyperglycemia, diabetes or insulin resistance in a mammalian patient in need of such treatment which comprises administering to said patient a compound in accordance with formula I or a pharmaceutically acceptable salt or solvate thereof in an amount that is effective to treate hyperglycemia, diabetes or insulin resistance.
- another aspect of the invention that is of interest relates to a method of treating type 2 diabetes in a mammalian patient in need of such treatment comprising administering to the patient a compound in accordance with formula I or a pharmaceutically acceptable salt or solvate thereof in an amount that is effective to treat type 2 diabetes.
- Yet another aspect of the invention that is of interest relates to a method of treating non- insulin dependent diabetes mellitus in a mammalian patient in need of such treatment comprising administering to the patient a compound in accordance with formula I or a pharmaceutically acceptable salt or solvate thereof in an amount that is effective to treat non-insulin dependent diabetes mellitus.
- Yet another aspect of the invention that is of interest relates to a method of treating obesity in a mammalian patient in need of such treatment compriseing administering to said patient a compound in accordance with formula I or a pharmaceutically acceptable salt or solvate thereof in an amount that is effective to treat obesity.
- Yet another aspect of the invention that is of interest relates to a method of treating
- Syndrome X in a mammalian patient in need of such treatment, comprising administering to said patient a compound in accordance with formula I or a pharmaceutically acceptable salt or solvate thereof in an amount that is effective to treat Syndrome X.
- Yet another aspect of the invention that is of interest relates to a method of treating a lipid disorder selected from the group consisting of dyslipidemia, hyperlipidemia, hypertriglyceridemia, hypercholesterolemia, low HDL and high LDL in a mammalian patient in need of such treatment, comprising administering to said patient a compound in accordance with formula I or a pharmaceutically acceptable salt or solvate thereof in an amount that is effective to treat said lipid disorder.
- Yet another aspect of the invention that is of interest relates to a method of treating atherosclerosis in a mammalian patient in need of such treatment, comprising administering to said patient a compound in accordance with a compound in accordance with formula I or a pharmaceutically acceptable salt or solvate thereof in an amount that is effective to treat atherosclerosis.
- Yet another aspect of the invention that is of interest relates to a method of treating a condition selected from the group consisting of: (1) hyperglycemia, (2) low glucose tolerance, (3) insulin resistance, (4) obesity, (5) lipid disorders, (6) dyslipidemia, (7) hyperlipidemia, (8) hypertriglyceridemia, (9) hypercholesterolemia, (10) low HDL levels, (11) high LDL levels, (12) atherosclerosis and its sequelae, (13) vascular restenosis, (14) pancreatitis, (15) abdominal obesity, (16) neurodegenerative disease, (17) retinopathy, (18) nephropathy, (19) neuropathy, (20) Syndrome X, (21) hypertension and other conditions and disorders where insulin resistance is a component, in a mammalian patient in need of such treatment, comprising administering to the patient a compound in accordance with formula I or a pharmaceutically acceptable salt or solvate thereof in an amount that is effective to treat said condition.
- a condition selected from the group consisting of: (1) hyperglycemia
- Yet another aspect of the invention that is of interest relates to a method of delaying the onset of a condition selected from the group consisting of (1 ) hyperglycemia, (2) low glucose tolerance, (3) insulin resistance, (4) obesity, (5) lipid disorders, (6) dyslipidemia, (7) hyperlipidemia, (8) hypertriglyceridemia, (9) hypercholesterolemia, (10) low HDL levels, (11) high LDL levels, (12) atherosclerosis and its sequelae, (13) vascular restenosis, (14) pancreatitis, (15) abdominal obesity, (16) neurodegenerative disease, (17) retinopathy, (18) nephropathy, (19) neuropathy, (20) Syndrome X, (21) hypertension and other conditions and disorders where insulin resistance is a component in a mammalian patient in need thereof, comprising administering to the patient a compound in accordance with formula I or a pharmaceutically acceptable salt or solvate thereof in an amount that is effective to delay the onset of said condition.
- a condition selected from the group consisting of
- Yet another aspect of the invention that is of interest relates to a method of reducing the risk of developing a condition selected from the group consisting of (1) hyperglycemia, (2) low glucose tolerance, (3) insulin resistance, (4) obesity, (5) lipid disorders, (6) dyslipidemia, (7) hyperlipidemia, (8) hypertriglyceridemia, (9) hypercholesterolemia, (10) low HDL levels, (11) high LDL levels, (12) atherosclerosis and its sequelae, (13) vascular restenosis, (14) pancreatitis, (15) abdominal obesity, (16) neurodegenerative disease, (17) retinopathy, (18) nephropathy, (19) neuropathy, (20) Syndrome X, (21) hypertension and other conditions and disorders where insulin resistance is a component in a mammalian patient in need thereof, comprising administering to the patient a compound in accordance with formula I or a pharmaceutically acceptable salt or solvate thereof in an amount that is effective to reduce the risk of developing said condition.
- a condition selected from the group consisting of (1) hyper
- Yet another aspect of the invention that is of interest relates to a method of treating a condition selected from the group consisting of (1) hyperglycemia, (2) low glucose tolerance, (3) insulin resistance, (4) obesity, (5) lipid disorders, (6) dyslipidemia, (7) hyperlipidemia, (8) hypertriglyceridemia, (9) hypercholesterolemia, (10) low HDL levels, (11) high LDL levels, (12) atherosclerosis and its sequelae, (13) vascular restenosis, (14) pancreatitis, (15) abdominal obesity, (16) neurodegenerative disease, (17) retinopathy, (18) nephropathy, (19) neuropathy, (20) Syndrome X, (21) hypertension and other conditions and disorders where insulin resistance is a component, in a mammalian patient in need of such treatment, comprising administering to the patient a compound in accordance with formula I or a pharmaceutically acceptable salt or solvate thereof in an amount that is effective to treat said condition, and a compound selected from the group consisting of:
- insulin sensitizers selected from the group consisting of (i) PPAR agonists and (ii) biguanides;
- glucagon receptor antagonists (f) glucagon receptor antagonists; (g) GLP-1 , GLP-1 mimetics, and GLP-1 receptor agonists;
- PACAP PACAP, PACAP mimetics, and PACAP receptor 3 agonists
- HMG-CoA reductase inhibitors HMG-CoA reductase inhibitors, (ii) sequestrants, (iii) nicotinyl alcohol, nicotinic acid and salts thereof, (iv) PPAR ⁇ agonists, (v) PPAR ⁇ / ⁇ dual agonists, (vi) inhibitors of cholesterol absorption, (vii) acyl CoA:cholesterol acyltransferase inhibitors, and (viii) anti-oxidants; (k) PPAR ⁇ agonists; (1) antiobesity compounds; (m) ileal bile acid transporter inhibitors;
- anti-inflammatory agents excluding glucocorticoids
- PTP-1B protein tyrosine phosphatase-1B
- antihypertensives including those acting on the angiotensin or renin systems, such as angiotensin converting enzyme inhibitors, angiotensin II receptor antagonists or renin inhibitors, such as captopril, cilazapril, enalapril, fosinopril, lisinopril, quinapril, ramapril, zofenopril, candesartan, cilexetil, eprosartan, irbesartan, losartan, tasosartan, telmisartan, and valsartan; said compounds being administered to the patient in an amount that is effective to treat said condition.
- angiotensin converting enzyme inhibitors such as captopril, cilazapril, enalapril, fosinopril, lisinopril, quinapril, ramapril, zofenopril
- candesartan cilexe
- Yet another aspect of the invention that is of interest relates to a method of treating a condition selected from the group consisting of hypercholesterolemia, atherosclerosis, low HDL levels, high LDL levels, hyperlipidemia, hypertriglyceridemia and dyslipidemia, in a mammalian patient in need of such treatment, comprising administering to the patient a compound in accordance with formula I or a pharmaceutically acceptable salt or solvate thereof and an HMG-CoA reductase inhibitor, in amounts that are effective to treat said condition.
- another aspect of the invention that is of interest relates to a method of treating a condition selected from the group consisting of hypercholesterolemia, atherosclerosis, low HDL levels, high LDL levels, hyperlipidemia, hypertriglyceridemia and dyslipidemia, in a mammalian patient in need of such treatment, comprising administering to the patient a compound in accordance with formula I or a pharmaceutically acceptable salt or solvate thereof and an HMG-CoA reductase inhibitor in the form of a statin, said compounds being administered in amounts that are effective for treating said condition.
- Statins useful in this regard include the group consisting of lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, itavastatin, ZD-4522 and rivastatin.
- a method of reducing the risk of developing a condition selected from the group consisting of hypercholesterolemia, atherosclerosis, low HDL levels, high LDL levels, hyperlipidemia, hypertriglyceridemia and dyslipidemia, and the sequelae of such conditions comprising administering to a mammalian patient in need of such treatment a therapeutically effective amount of a compound as defined in Claim 1 and an HMG-CoA reductase inhibitor.
- another aspect of the invention that is of interest relates to a method of delaying the onset or reducing the risk of developing atherosclerosis in a human patient in need of such treatment comprising administering to the patient a compound in accordance with formula I or a pharmaceutically acceptable salt or solvate thereof and an HMG-CoA reductase inhibitor in the form of a statin, said compounds being administered in amounts that are effective for treating said condition.
- Statins useful in this regard include the group consisting of lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, itavastatin, ZD-4522 and rivastatin.
- another aspect of the invention that is of interest relates to a method of treating, delaying the onset or reducing the risk of developing atherosclerosis in a human patient in need of such treatment comprising administering to the patient a compound in accordance with formula I or a pharmaceutically acceptable salt or solvate thereof and a cholesterol absorption inhibitor, said compounds being administered in amounts that treat, delay the onset, or reduce the risk of developing atherosclerosis.
- another aspect of the invention that is of interest relates to a method of treating, delaying the onset or reducing the risk of developing atherosclerosis in a human patient in need of such treatment comprising administering to the patient a compound in accordance with formula I or a pharmaceutically acceptable salt or solvate thereof and a cholesterol absorption inhibitor, wherein the cholesterol absorption inhibitor is ezetimibe, said compounds being administered in amounts that treat, delay the onset, or reduce the risk of developing atherosclerosis.
- compositions that is comprised of: (1) a compound according to formula I or a pharmaceutically acceptable salt or solvate thereof, (2) a compound selected from the group consisting of : (a) DP-rV inhibitors;
- insulin sensitizers selected from the group consisting of (i) PPAR agonists and (ii) biguanides;
- PACAP PACAP
- PACAP mimetics PACAP receptor 3 agonists
- PACAP receptor 3 agonists PACAP receptor 3 agonists
- cholesterol lowering agents selected from the group consisting of (i) HMG-CoA reductase inhibitors, (ii) sequestrants, (iii) nicotinyl alcohol, nicotinic acid or a salt thereof, (iv) PPAR ⁇ agonists, (v) PPAR ⁇ / ⁇ dual agonists, (vi) inhibitors of cholesterol absorption, (vii) acyl CoAxholesterol acyltransferase inhibitors, and (viii) anti-oxidants;
- antihypertensives including those acting on the angiotensin or renin systems, such as angiotensin converting enzyme inhibitors, angiotensin II receptor antagonists or renin inhibitors, such as captopril, cilazapril, enalapril, fosinopril, lisinopril, quinapril, ramapril, zofenopril, candesartan, cilexetil, eprosartan, irbesartan, losartan, tasosartan, telmisartan, and valsartan; and (3) a pharmaceutically acceptable carrier
- Another aspect of the invention that is of interest relates to the use of a compound in accordance with formula I or a pharmaceutically acceptable salt or solvate thereof in the manufacture of a medicament for use in treating a disease or condition described herein.
- the compounds of the invention can be prepared using the synthetic schemes described herein.
- the following abbreviations are used in the synthetic schemes:
- Ac is acetyl [CH 3 C(O)-]; Ac 2 O is acetic anhydride; AcAc is acetyl acetonoate; Ar is Aryl; ArX is an aryl halide; 9-BBN is 9-borabicyclo[3.3.1]nonane; Bn is benzyl; BOC is tert
- R 1 - R 5 serve as placeholders.
- the ring containing H shown below denotes a heterocycle.
- E is an ester forming group, e.g., propyl, butyl, and the like.
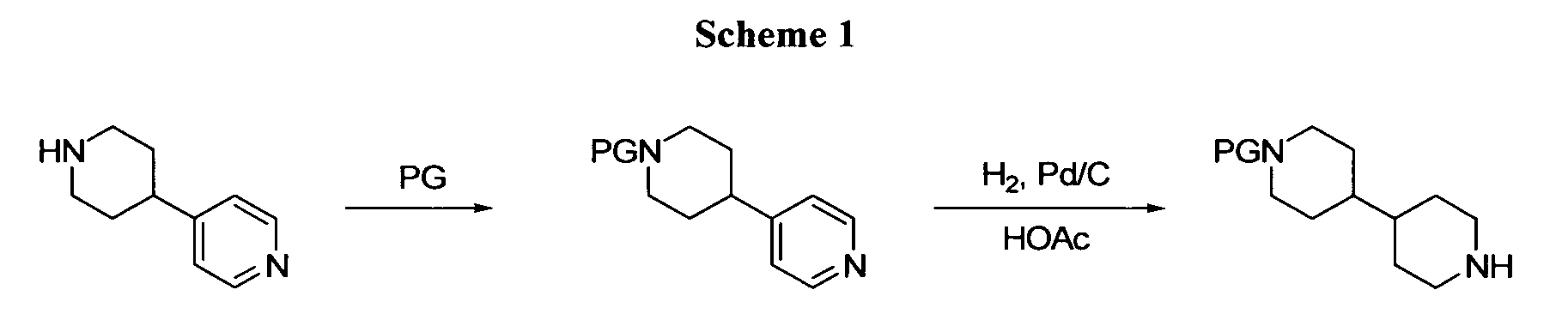
- the substituted bipiperidines of this invention can be prepared by any of several methods. The specific examples detailed below may employ some of the following general procedures. Substituted aryl and heteroaryl coupling intermediates shown in the schemes are commercially available or may be prepared from readily accessible aryl, heterocycles, or other congeners via a host of routes. Intermediates are accessible through either modification of a preformed heteroaryl scaffold or through de novo ring synthesis. Many of the piperidine intermediates required for the preparation of this invention are available commercially or through published procedures. One of the most useful means synthese utilizes a reduction of a suitable pyridine. Low pressure reductions with hydrogen and 5-10% Pd on charcoal or similar hydrogenation catalyst in acetic acid lead to the appropriate piperidine (scheme 1).
- Heterocyclic residues of either ArI or the Het fragment are obtainable by de novo ring synthesis.
- An example of de novo ring synthesis for thiazole and pyrimidine cases is shown in Scheme 4. A host of methods can be used.
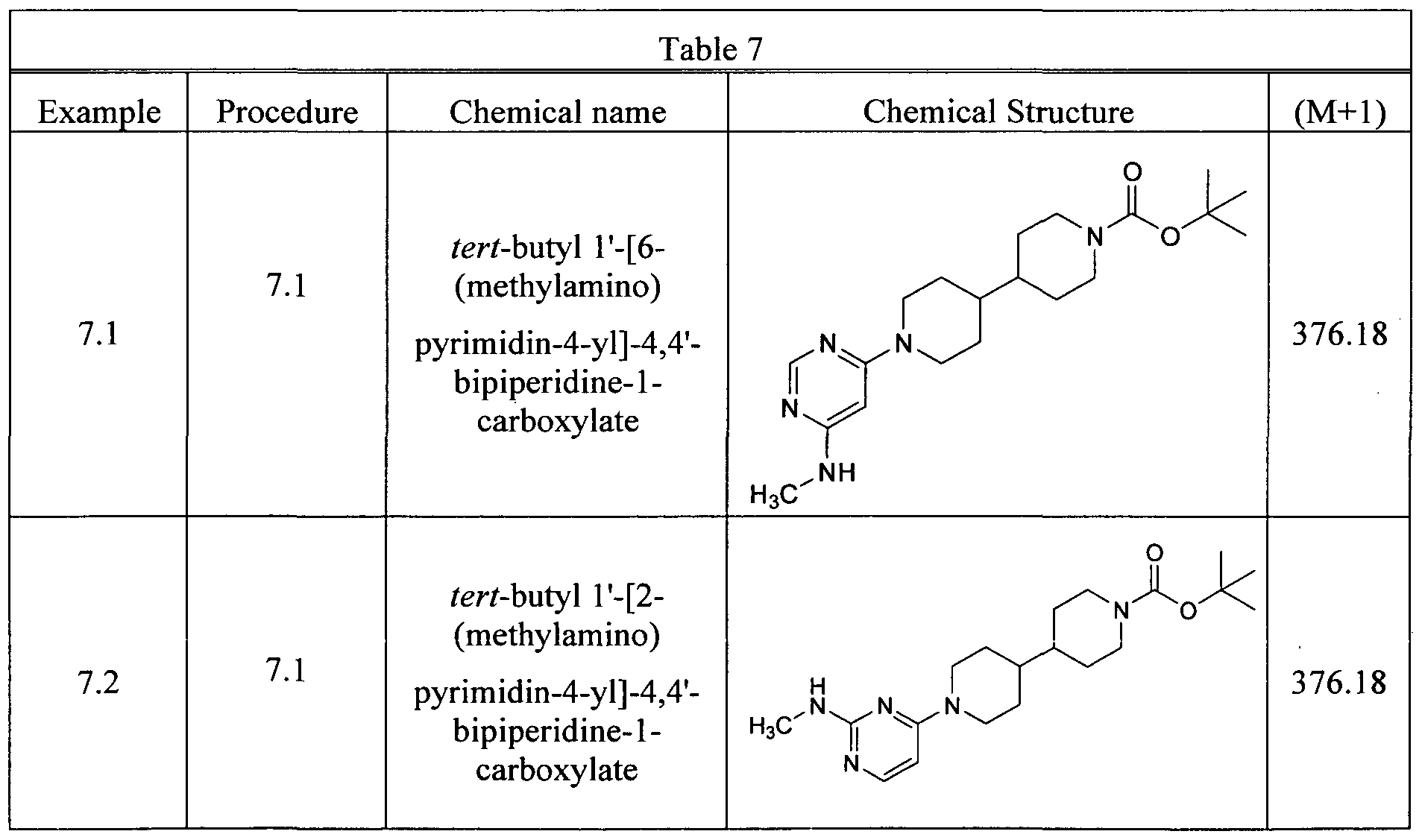
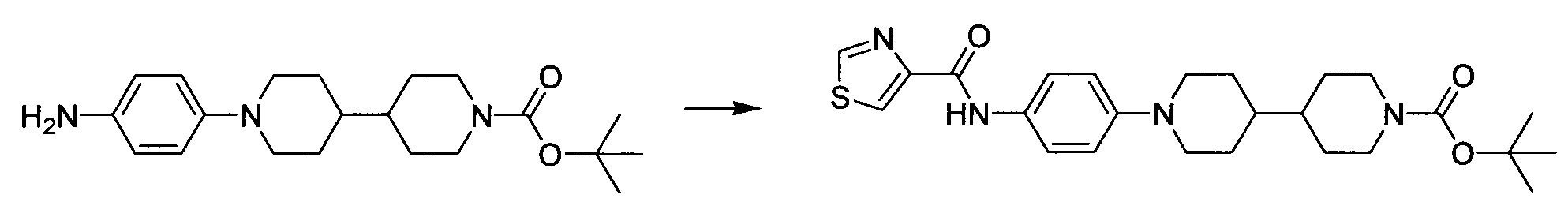
- Example 2.1 tert-butyl 1'-[4-(methylsulfonyl)phenyl]-4,4'-bipiperidine-1-carboxylate.
- the tert-butyl 4,4'-bipiperidine-1-carboxylate (2.15g, 8.0 mmol) and cesium carbonate (7.8 g, 24 mmol) were slurried in NMP (80 ml) and the 2,6-dichloropyrazine (1.25 mg, 8.4 mmol) was added. The mixture was stirred at 70 °C for 2h. The reactive mixture was cooled down to r.t. and diluted with 100 mL of ethyl acetate, washed with water 80 mL (xl) and brine 80 mL (xl). The organic phase was dried over magnesium sulfate, filtered, and concentrated on rotavapor.
- Step 1 4-bromo-2-(trifluoromethyl)pyrimidine.
- Step 2 tert-butyl 1'-[2-(trifluoromethyl)pyrimidin-4-yl]-4,4'-bipiperidine-1-carboxylate.
- Step 1 tert-butyl 1'-[4-(methylsulfonyl)phenyl]-4,4'-bipiperidine-1-carboxylate: same as Procedure 2.1.
- Step 2 tert-butyl 1'-(6-methylpyrazin-2-yl)-4,4'-bipiperidine-1-carboxylate.
- the tert-butyl 1'-[4-(methylsulfonyl)phenyl]-4,4'-bipiperidine-1-carboxylate (100 mg, 0.26 mmol) was dissolved in THF and NMP (5 mL, 1: 1) at r.t.
- the catalyst iron (III) acetylacetonate (18.4 mg, 0.052 mmol) was added under N 2 .
- Methyl magnesium bromide (3M, 0.19 mL, 0.57 mmol) was added dropwise. The reaction was stirred for more 15 min and was quenched with iced ammonia chloride solution (10 mL) and extracted with ethyl acetate (10 mL).
- Step 1 tert-butyl 1'-[4-(methylsulfonyl)phenyl]-4,4'-bipiperidine-1-carboxylate: same as Procedure 2.1.
- Step 2 tert-butyl 1'-(6-cyanopyrazin-2-yl)-4,4'-bipiperidine-1-carboxylate.
- Step 1 tert-butyl 1'-(6-chloropyrimidin-4-yl)-4,4'-bipiperidine-1-carboxylate: same as Procedure 2.1.
- Step 2 tert-butyl 1'-[6-(1H-1,2,4-triazol-1-yl)pyrimidin-4-yl]-4,4'-bipiperidine-1-carboxylate.
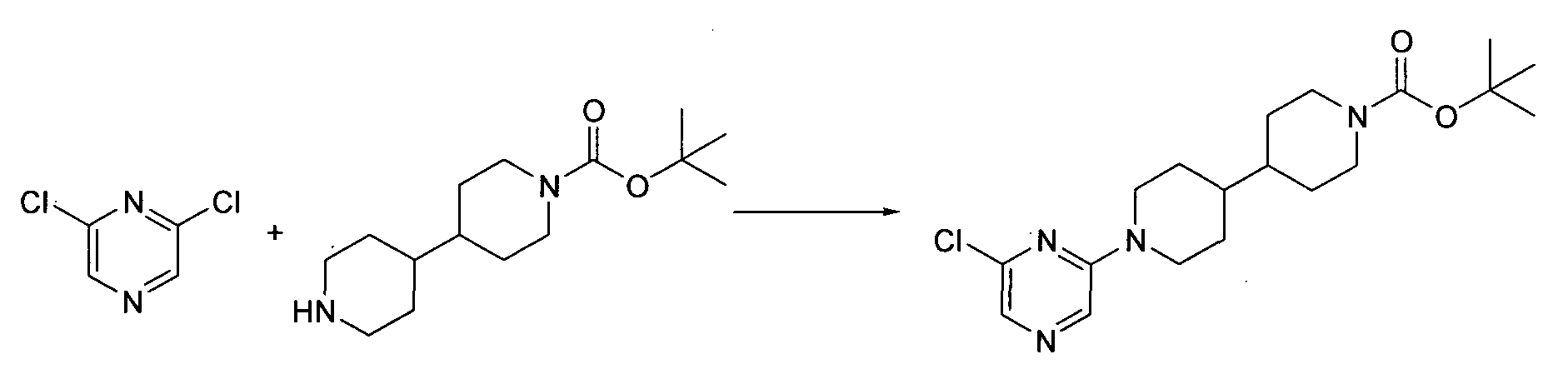
- Step 1 tert-butyl 1'-(6-chloropyrimidin-4-yl)-4,4'-bipiperidine-1-carboxylate: same as Procedure 2.1.
- Step 2 tert-butyl 1'-[6-(methylamino)pyrimidin-4-yl]-4,4'-bipiperidine-1-carboxylate.
- Step 1 tert-butyl 1'-(6-chloropyrimidin-4-yl)-4,4'-bipiperidine-1-carboxylate: same as Procedure 2.1.
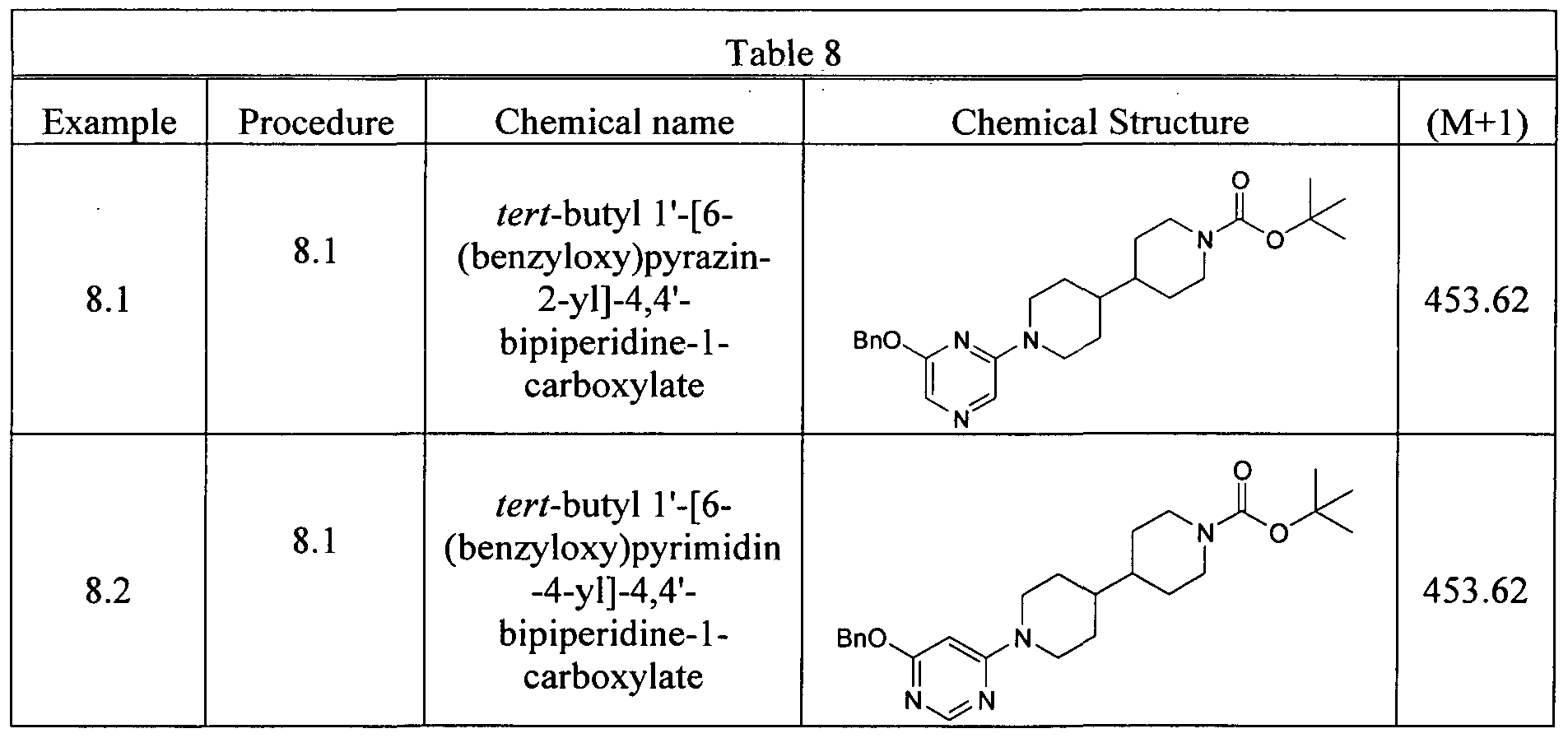
- Step 2 Preparation of tert-butyl 1'-[6-(benzyloxy)pyrimidin-4-yl]-4,4'-bipiperidine-1-carboxylate.
- tert-butyl 1'-(6-chloropyrimidin-4-yl)-4,4'-bipiperidine-1-carboxylate 100 mg, 0.26 mmol
- phenylmethanol 0.031 mL, 0.29 mmol
- sodium hydride 7 mg, 0.29 mmol
- the reaction was heated to 30 °C for 30 min.
- the reaction crude was cooled down to r.t. and quenched with ice ammonia chloride solution (10 mL) and extracted with ethyl acetate (10 mL).
- Step 1 tert-butyl 1'-(2-chloro-5-fluoropyrimidin-4-yl)-4,4'-bipiperidine-1-carboxylate: same as Procedure
- Step 2 tert-butyl 1'-(5-fluoro-2-methoxypyrimidin-4-yl)-4,4'-bipiperidine-1-carboxylate.
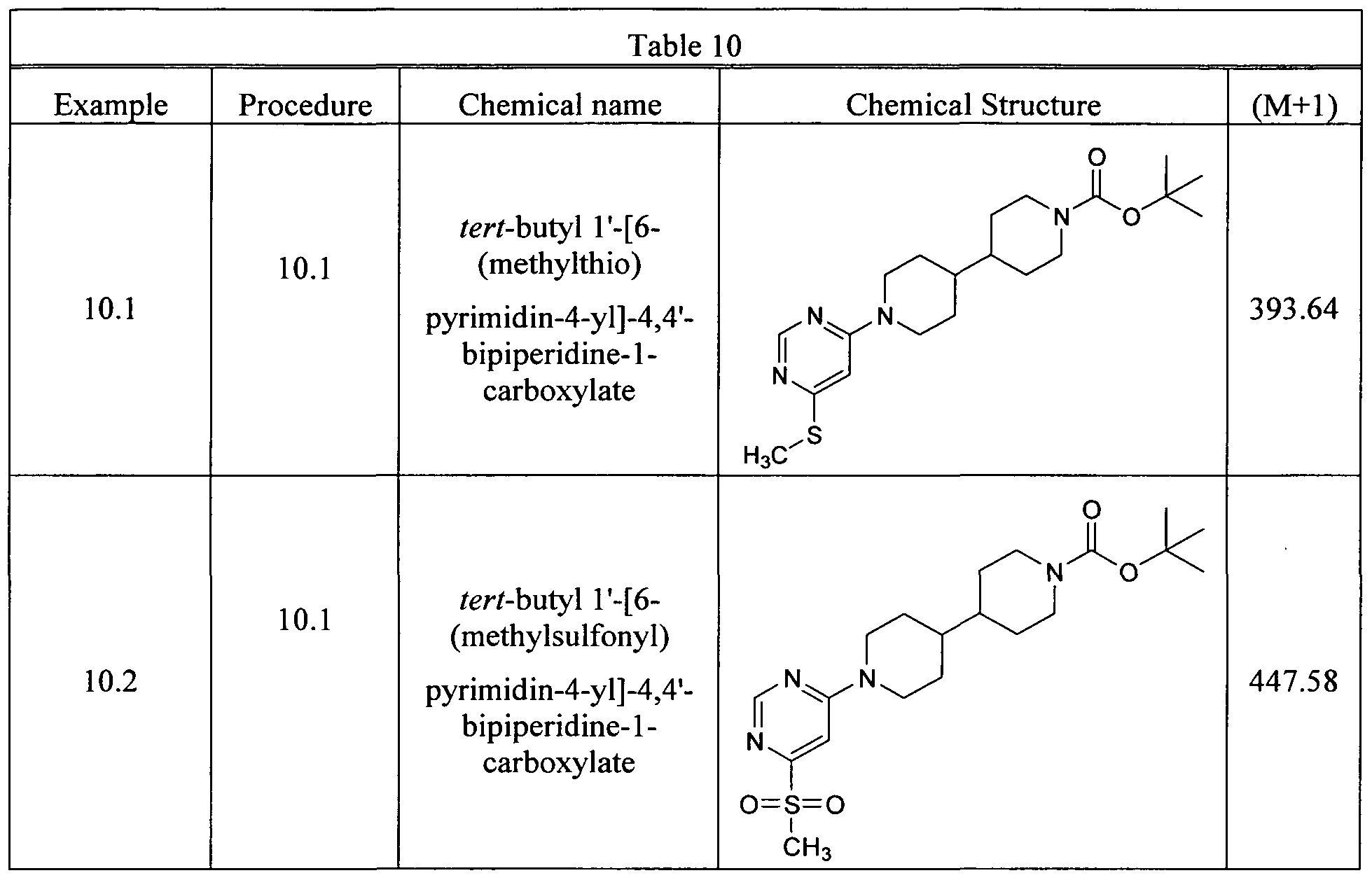
- the tert-butyl 1'-(6-chloropyrimidin-4-yl)-4,4'-bipiperidine-1-carboxylate (100 mg, 0.26 mmol) was dissolved in NMP (2.6 mL), and sodium methanethiolate (18.2 mg, 0.26 mmol) was added. The reaction was stirred at r.t. for 30 min. The reaction crude was diluted with ethyl acetate (10 mL) and was washed with water (10 mL, xl) and brine (10 mL, xl), dried over magnesium sulfate, filtered and concentrated by rotavapor. The crude mixture was purified by preparative TLC with 30% ethylacetate: hexane to yield the title compound. LC-MS: 393.64 (M+H).
- Step 3 tert-butyl 1'-[6-(methylsulfonyl)pyrimidin-4-yl]-4,4'-bipiperidine-1-carboxylate.
- tert-butyl 1'-pyrazin-2-yl-4,4'-bipiperidine-1-carboxylate (16 mg, 0.046 mmol, prepared as described for Example 8) and N-bromosuccinimide (8.2 mg, 0.046 mmol) were dissolved in DMF (0.2 ml) and the solution stirred overnight at 50 C.
- the slurry was diluted with 15% brinerwater (40 ml) and extracted with ethyl acetate (3 x 25 ml). The combined organic fraction was washed with brine, dried over magnesium sulfate, filtered, and the volatiles removed in vac.
- a suspension of 4-methylthiophenylboronic acid (240mg; 1.431 mmol) in dichloromethane (6mL) was treated with activated powdered 4A molecular sieves (1.5g), Cu(OAc) 2 (87mg; 0.477mmol), tert-butyl 4,4'-bipiperidine-1-carboxylate (128mg; 0.477mmol) and triethylamine (134 ⁇ L; 0.954mmol).
- the reaction vessel was purged with oxygen and the mixture stirred under an oxygen atmosphere at ambient temperature for 18hr.
- the now brown mixture was filtered through Celite.
- the eluant was adsorbed onto silica gel.
- the LRMS of all BOC containing compounds gave an M+l and an M-100+1 (M-BOC+1) ion.
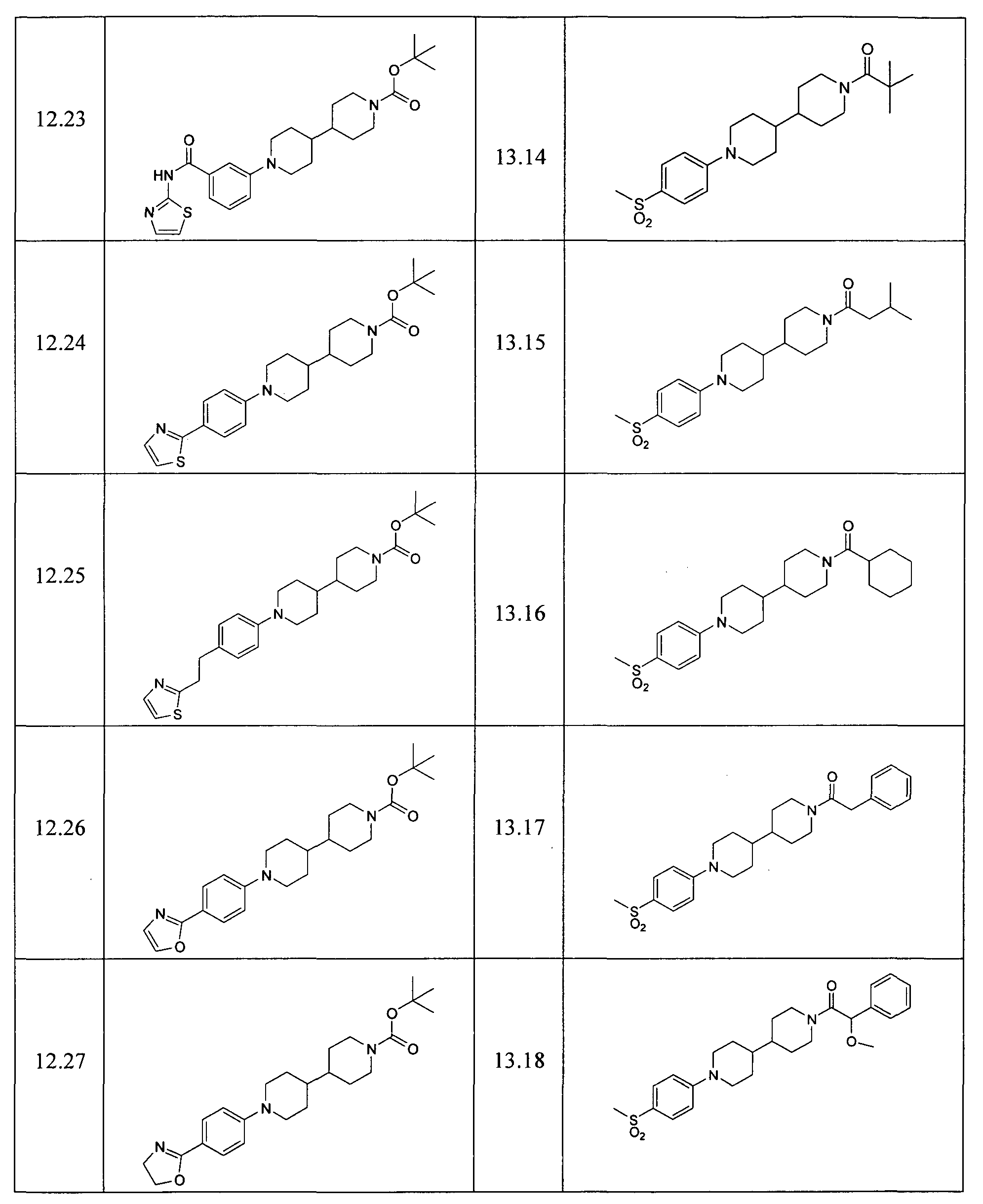
- Example 12.24 Preparation of tert-Butyl-1'-[4-(1,3-thiazol-2-yl)phenyl]-4,4'-bipiperidine-1-carboxylate Step 1: tert-Butyl-1'-[4-(aminocarbonothioyl)phenyl]-4,4'-bipiperidine-1-carboxylate
- Example 12.25 tert-butyl-1'- ⁇ 4-[2-(1,3-thiazol-2-yl)ethyl]phenyl ⁇ -4,4'-bipiperidine-1-carboxylate
- Step 1 tert-butyl-1'-[4-(3-amino-3-oxopropyl)phenyl]-4,4'-bipiperidine-1-carboxylate
- Step 3 tert-butyl- 1'- ⁇ 4-[2-( 1,3-thiazol-2-yl)ethyl]phenyl ⁇ -4,4'-bipiperidine-1-carboxylate
- Example 12.26 Preparation of tert-butyl- 1'- [4-(1,3-oxazol-2-yl)phenyl]-4,4'-bipiperidine-1-carboxylate Step 1: tert-butyl 1'-(4-iodophenyl)-4,4'-bipiperidine-1-carboxylate
- Step 2 tert-butyl-1'-[4-(1,3-oxazol-2-yl)phenyl]-4,4'-bipiperidine-1-carboxylate
- Step 1 tert-butyl 1'-(4- ⁇ [(2-hydroxyethyl)amino]carbonyl ⁇ phenyl)-4,4'-bipiperidine-1-carboxylate
- This compound was prepared using the method of General Procedure 12.5 and ethanolamine as starting material.
- Step 2 tert-butyl-1'-[4-(4,5-dihydro-1,3-oxazol-2-yl)phenyl]-4,4'-bipiperidine-1-carboxylate
- Step 2 tert-Butyl 1'-(4-aminophenyl)-4,4'-bipiperidine-1-carboxylate
- Step 4 tert-Butyl-1'- ⁇ 4-[(1,3-thiazol-2-ylcarbonyl)amino]phenyl ⁇ -4,4'-bipiperidine-1-carboxylate.
- Example 12.31 Preparation of tert-butyl-1'- ⁇ 3-chloro-4-[(1,3-thiazol-4-ylcarbonyl)amino]phenyl ⁇ -4,4'- bipiperidine-1-carboxylate Step 1: tert-Butyl 1'-(3-chloro-4-nitrophenyl)-4,4'-bipiperidine-1-carboxylate
- Step 2 tert-Butyl 1'-(3-chloro-4-aminophenyl)-4,4'-bipiperidine-1-carboxylate
- Step 3 tert-Butyl-1'- ⁇ 3-chloro-4-[(1,3-thiazol-4-ylcarbonyl)amino]phenyl ⁇ -4,4'-bipiperidine-1- carboxylate
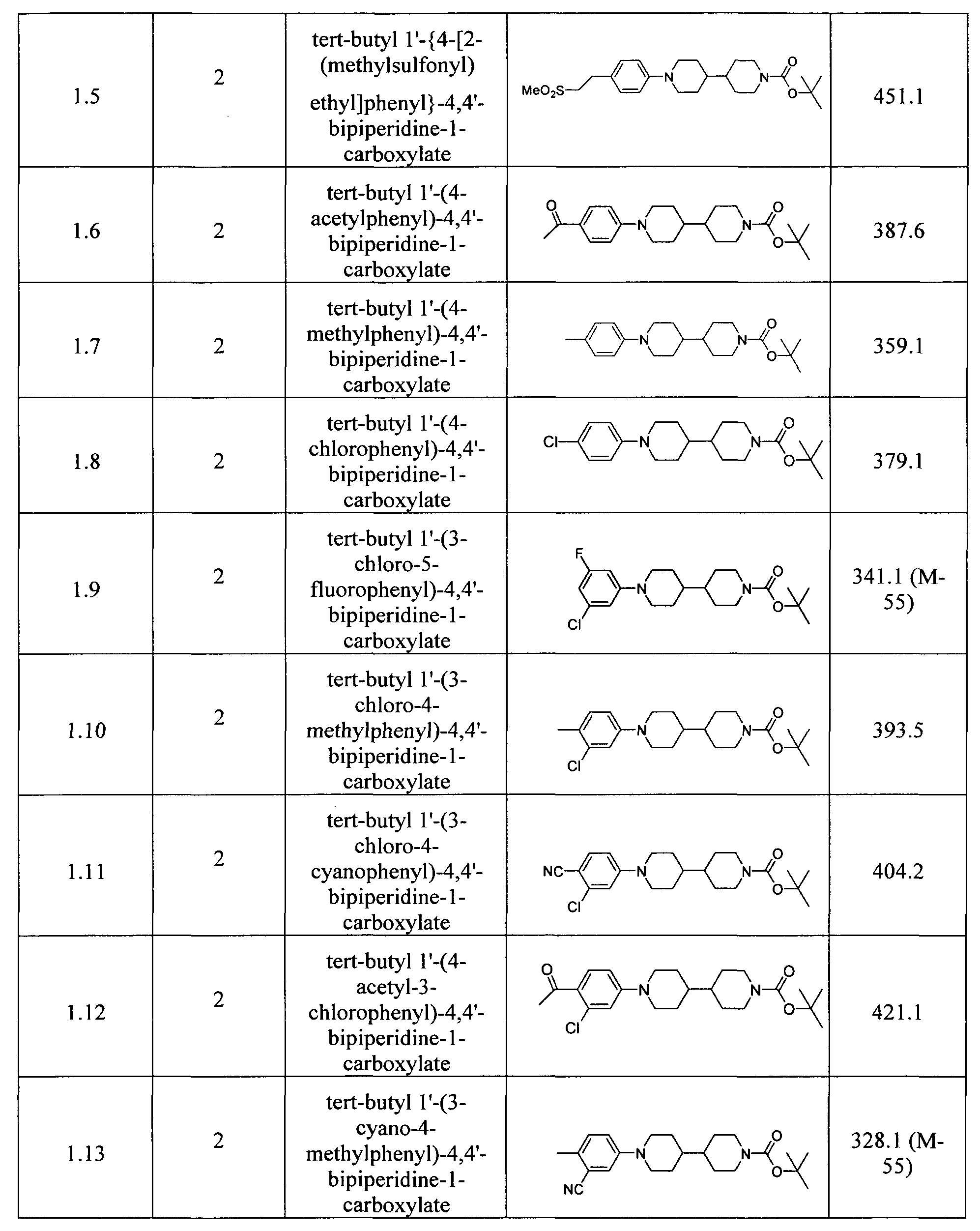
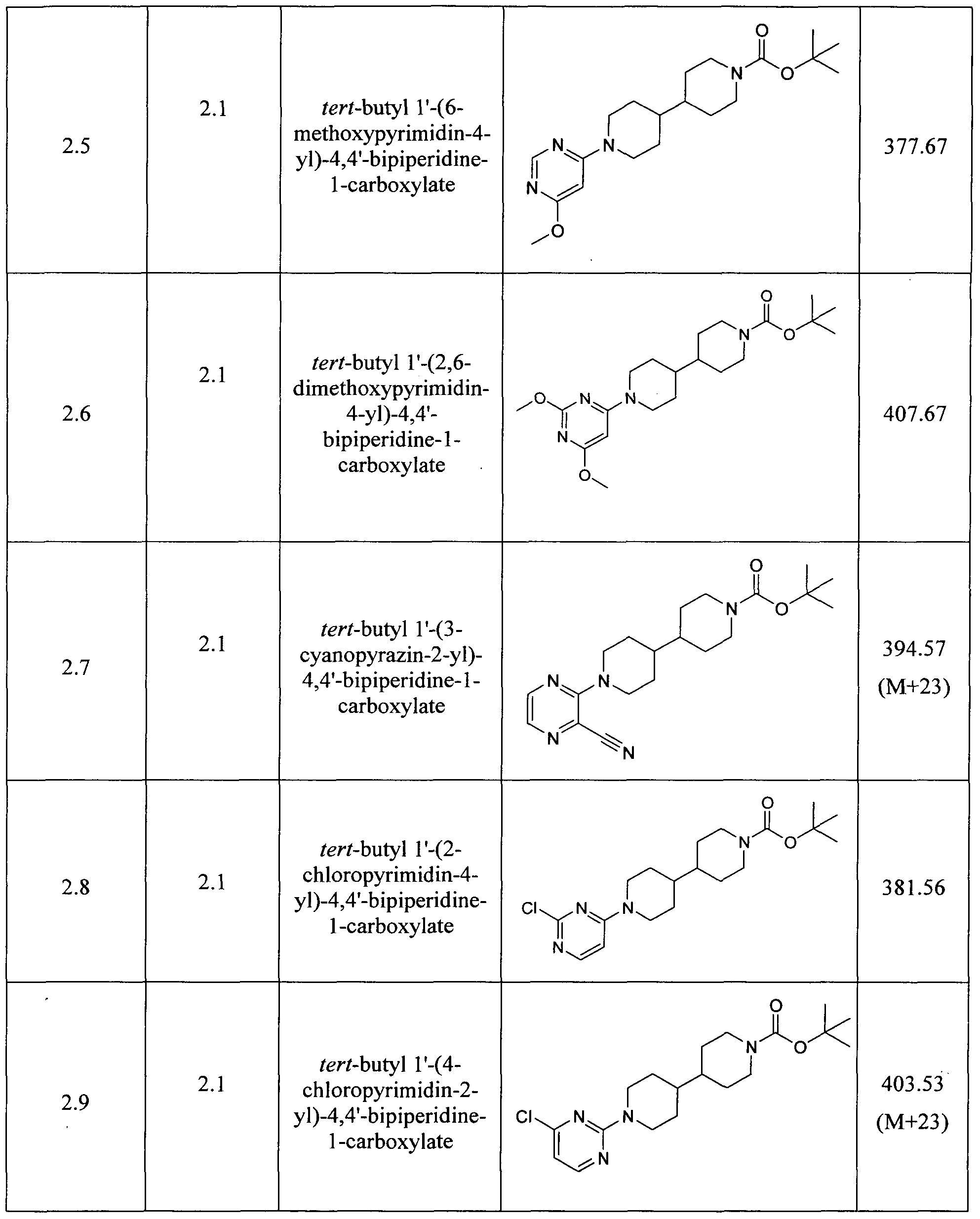
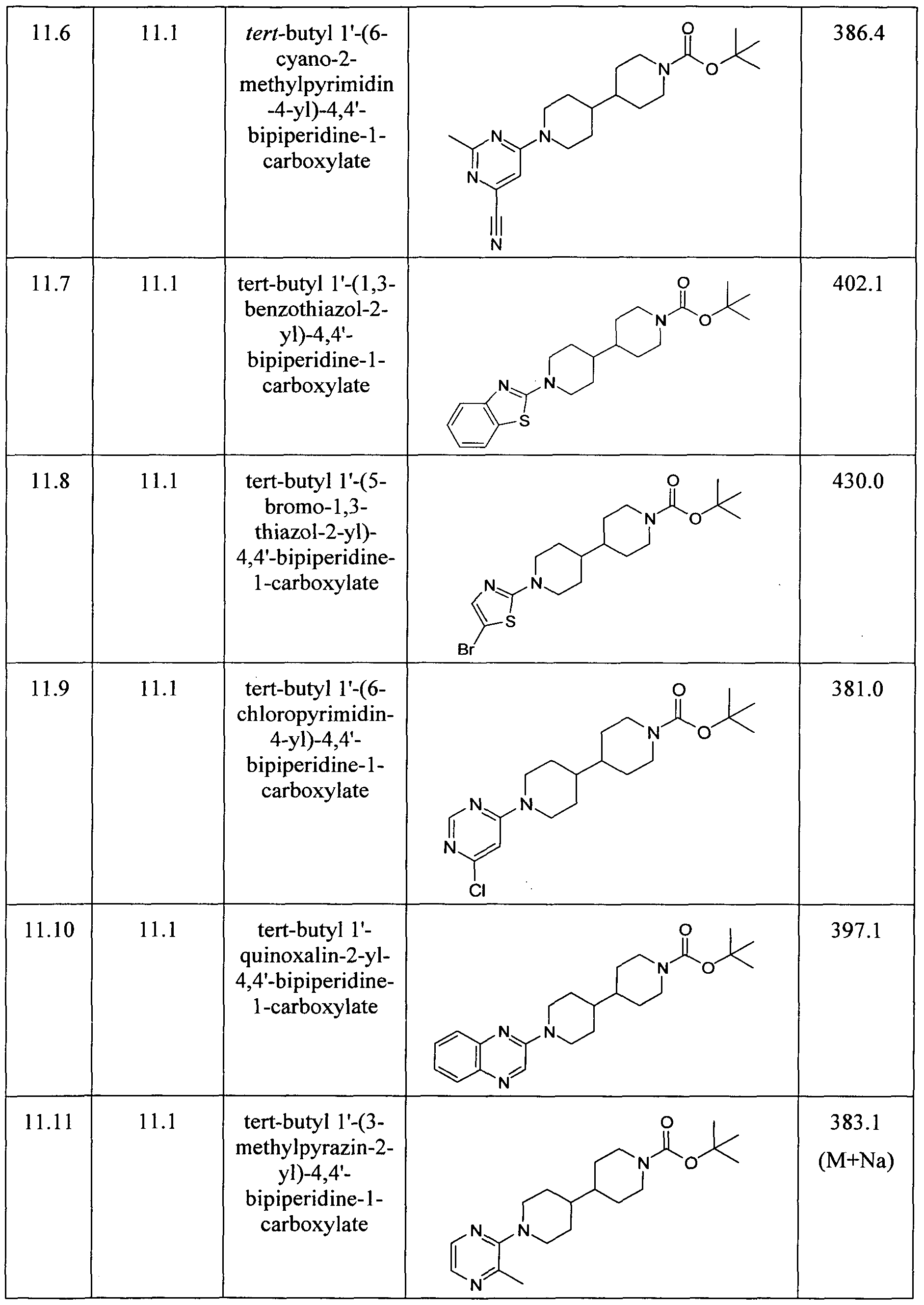
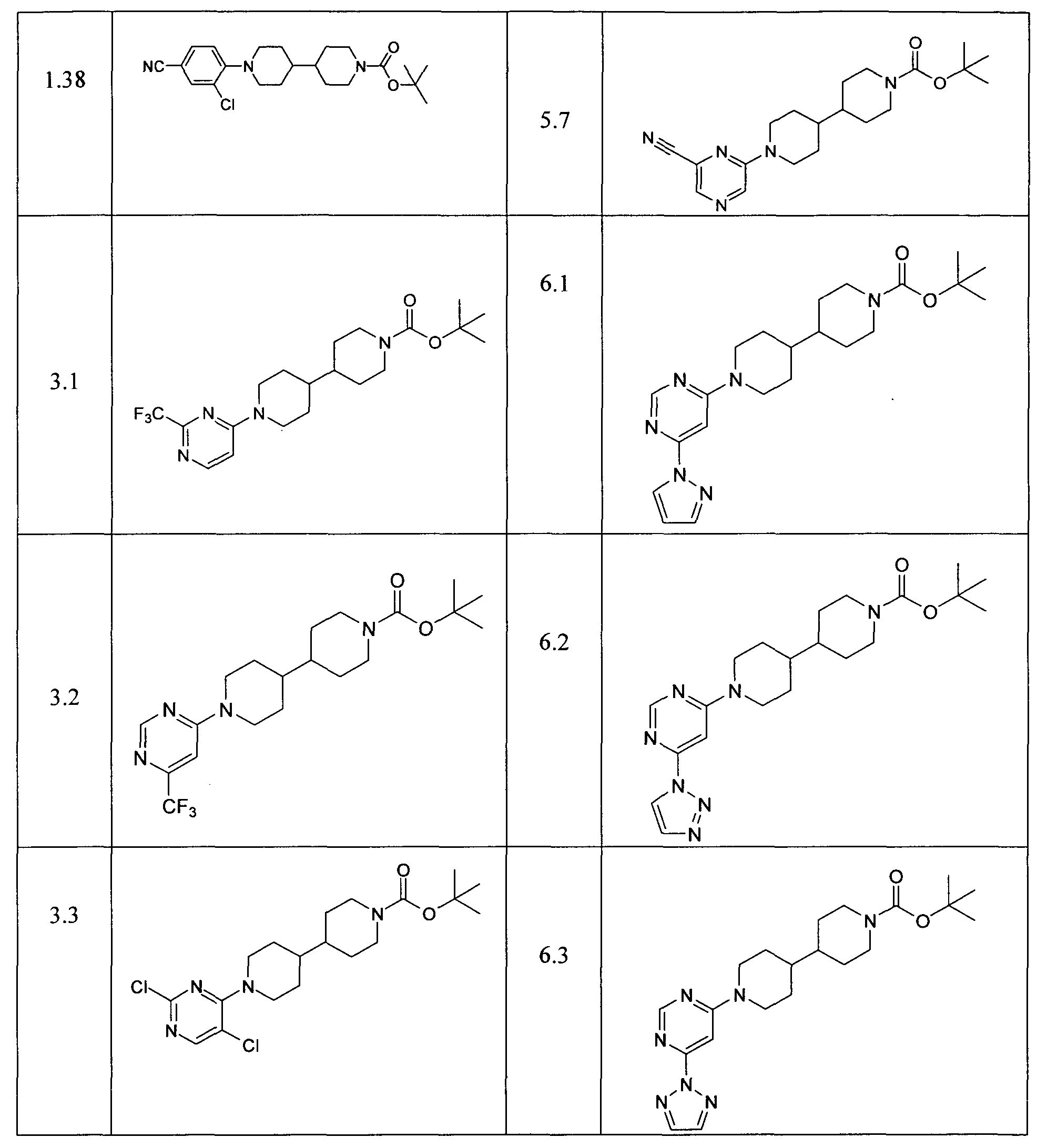
- Examples reported in Table 13 can be prepared by the procedures described above from readily available starting materials and tert-butyl 4,4'-bipiperidine-1-carboxylate.
- Step 1 tert-butyl 1'-(2-chloro-5-fluoropyrimidin-4-yl)-4,4'-bipiperidine-1-carboxylate: same as Procedure
- Step 2 tert-butyl 1'-(5-fluoro-2-methylpyrirriidin-4-yl)-4,4'-bipiperidine-1-carboxylate: same as Procedure 4.1, step 2.
- Step 1 tert-butyl 1'-(2-chloro-5-fluoropyrimidin-4-yl)-4,4'-bipiperidine-1-carboxylate: same as Procedure 14.1 above.
- Step 2 tert-butyl 1'-(5-fluoro-2-methylpyrimidin-4-yl)-4,4'-bipiperidine-1-carboxylate: same as Procedure 4..1, step 2.
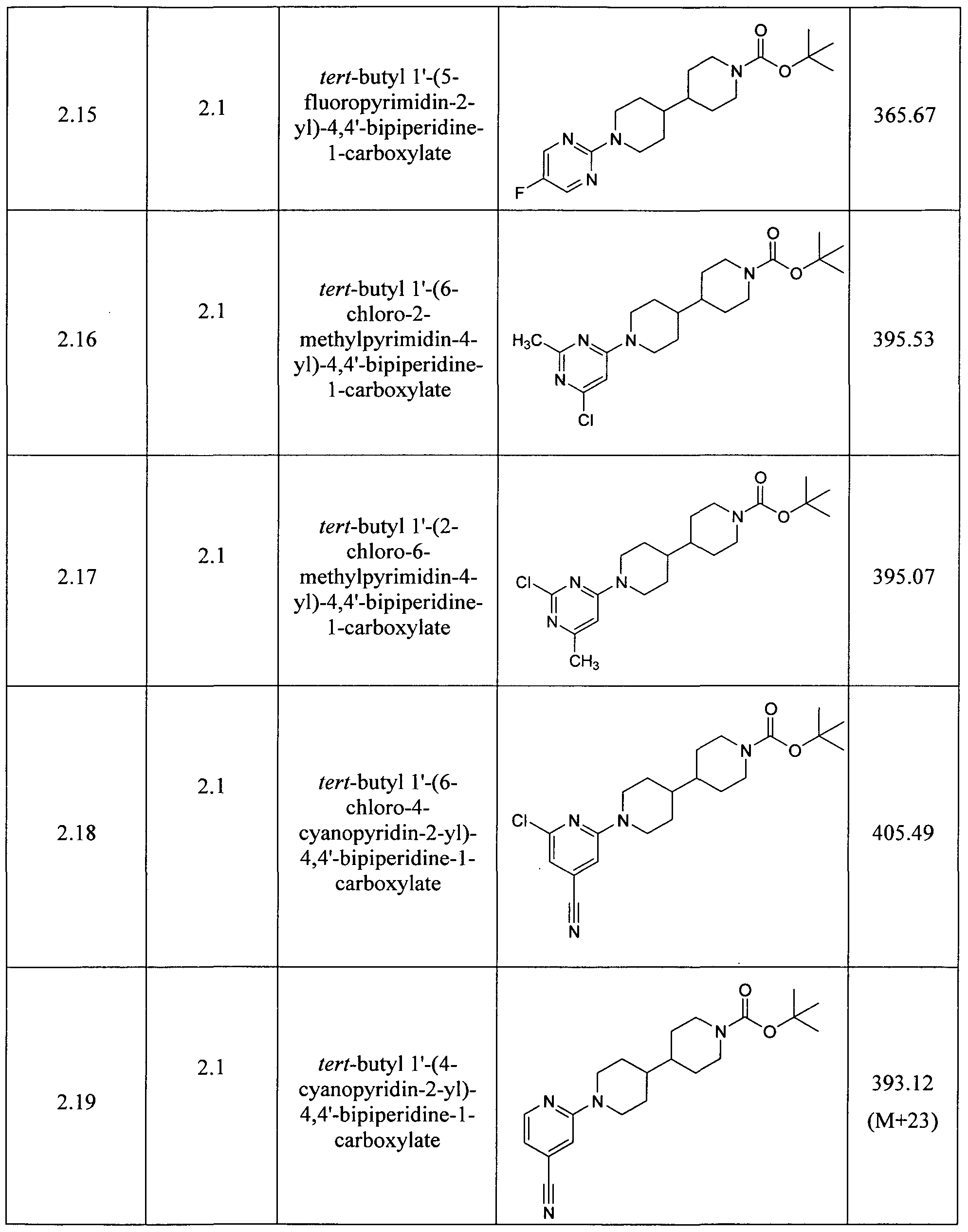
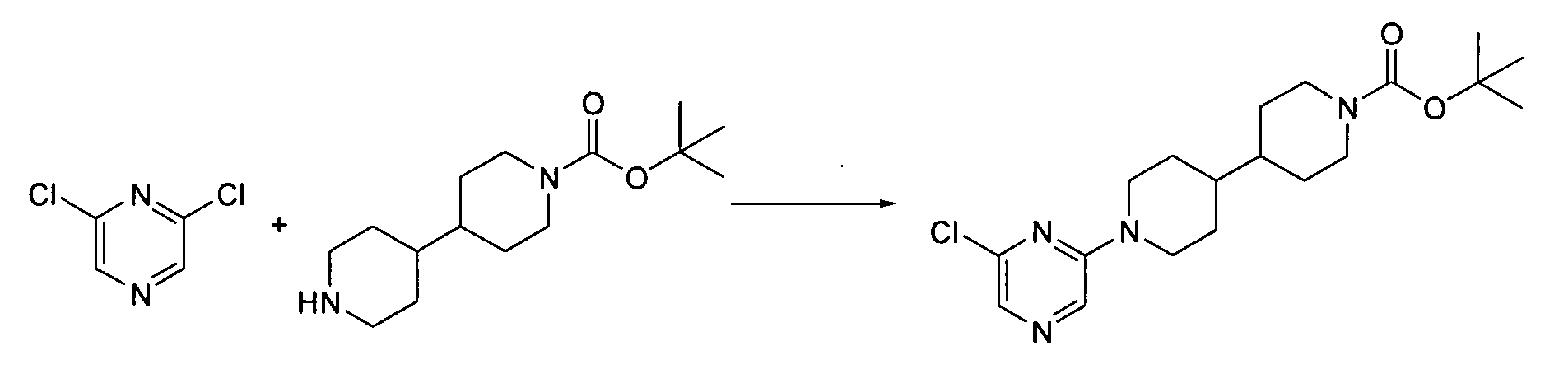
- Step 1 tert-butyl 1'-(6-chloropyrimidin-4-yl)-4,4'-bipiperidine-1-carboxylate: same as Procedure 2.1, substituting thedichloropyrimidine shown below for dichloropyrazine.
- Step 2 tert-butyl 1'-(6-cyanopyrimidin-4-yl)-4,4'-bipiperidine-1-carboxylate: same as Procedure 5.1, step
- Step 1 tert-butyl 1'-(6-chloropyrimidin-4-yl)-4,4'-bipiperidine-1-carboxylate: same as Procedure 2.1.
- Step 2 tert-butyl 1'-(6-cyanopyrimidin-4-yl)-4,4'-bipiperidine-1-carboxylate: same as Procedure 5.1, step
- CHO cell lines stably transfected with the permissive guanine nucleotide binding protein alpha 15 (G ⁇ l5) and a human SNP variant of GPR119 or murine GPR119 were maintained in DMEM media containing FBS, penicillin-streptomycin, puromycin, and G418.
- a homogenous time resolved fluorescence (HTRF) assay for GPR119 receptor activation was used to measure cAMP accumulation in transfected cells upon incubation with compounds of this invention as per the manufacturer's instructions (CisBio, Bedford, MA). Cells were incubated with compound at room temperature for 60 min, and subsequently with XL-665 and anti-cAMP cryptate for an additional 60 min.
- HTRF time resolved fluorescence
- activation of the GPR119 receptor in a cAMP HTRF assay denotes induction of about a five fold increase in intracellular cAMP concentration.
- GDIS glucose dependent insulin secretion
- Pancreatic islets of Langerhans were isolated from the pancreata of 10-12 wk-old C57BL/6 mice by collagenase digestion and discontinuous Ficoll gradient separation, a modification of the original method of Lacy and Kostianovsky (Lacy & Kostianovsky, 1967 Diabetes 16-35-39).
- the islets were cultured overnight in RPMI 1640 medium (11 mM glucose, 10% FCS) before experimental treatment.
- the acute effects of compounds of this invention on GDIS were determined by 60-min static incubation with islets in Krebs-Ringers' bicarbonate (KRB) medium.
- the KRB medium contained, in mM, 143.5 Na + , 5.8 K + , 2.5 Ca 2+ , 1.2 Mg 2+ , 124.1 Cl-, 1.2 PO 4 3- , 1.2 SO 4 2+ , 25 CO 3 2- , and 10 HEPES, pH 7.4, in addition to 2 mg/ml bovine serum albumin, and either 2 (G2) or 16 (G 16) mM glucose (pH 7.4).
- the static incubation was performed with round-bottomed 96-well plates (one islet/well with 200 ⁇ l KRB medium). The compounds were added to KRB medium just before the initiation of the 60-min incubation. Insulin concentration in aliquots of the incubation buffer was measured by the ultrasensitive rat insulin EIA kit from ALPCO Diagnostics (Windham, NH).
- EXAMPLE OF A PHARMACEUTICAL FORMULATION As a specific embodiment of an oral composition of a compound of the present invention, 50 mg of any of the examples is formulated with sufficient finely divided lactose to provide a total amount of 580 to 590 mg to fill a size O hard gelatin capsule.
Abstract
Description










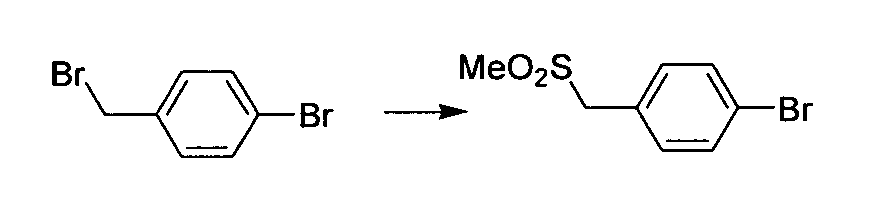





















































































Claims
















Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002672059A CA2672059A1 (en) | 2006-12-14 | 2007-12-10 | Acyl bipiperidinyl compounds, compositions containing such compounds and methods of treatment |
| EP07862712A EP2112880A4 (en) | 2006-12-14 | 2007-12-10 | Acyl bipiperidinyl compounds, compositions containing such compounds and methods of treatment |
| AU2007334519A AU2007334519A1 (en) | 2006-12-14 | 2007-12-10 | Acyl bipiperidinyl compounds, compositions containing such compounds and methods of treatment |
| US12/517,142 US8399485B2 (en) | 2006-12-14 | 2007-12-10 | Acyl bipiperidinyl compounds useful as GPR 119 agonists |
| JP2009541322A JP2010513272A (en) | 2006-12-14 | 2007-12-10 | Acylbipiperidinyl compounds, compositions containing such compounds, and therapeutic methods |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US87490706P | 2006-12-14 | 2006-12-14 | |
| US60/874,907 | 2006-12-14 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2008076243A2 true WO2008076243A2 (en) | 2008-06-26 |
| WO2008076243A3 WO2008076243A3 (en) | 2009-03-26 |
Family
ID=39536880
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/025225 WO2008076243A2 (en) | 2006-12-14 | 2007-12-10 | Acyl bipiperidinyl compounds, compositions containing such compounds and methods of treatment |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US8399485B2 (en) |
| EP (1) | EP2112880A4 (en) |
| JP (1) | JP2010513272A (en) |
| AU (1) | AU2007334519A1 (en) |
| CA (1) | CA2672059A1 (en) |
| WO (1) | WO2008076243A2 (en) |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| EP1808168B1 (en) * | 2005-01-10 | 2009-06-03 | Arena Pharmaceuticals, Inc. | Combination therapy for the treatment of diabetes and conditions related thereto and for the treatment of conditions ameliorated by increasing a blood GLP-1 level |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011113947A1 (en) | 2010-03-18 | 2011-09-22 | Boehringer Ingelheim International Gmbh | Combination of a gpr119 agonist and the dpp-iv inhibitor linagliptin for use in the treatment of diabetes and related conditions |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012046249A1 (en) | 2010-10-08 | 2012-04-12 | Cadila Healthcare Limited | Novel gpr 119 agonists |
| WO2012145361A1 (en) * | 2011-04-19 | 2012-10-26 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8883714B2 (en) | 2008-04-07 | 2014-11-11 | Arena Pharmaceuticals, Inc. | Pharmaceutical compositions comprising GPR119 agonists which act as peptide YY (PYY) secretagogues |
| WO2017049321A1 (en) | 2015-09-17 | 2017-03-23 | Miller Marvin J | Benzyl amine-containing heterocyclic compounds and compositions useful against mycobacterial infection |
| US9777018B2 (en) | 2011-06-09 | 2017-10-03 | Rhizen Pharmaceuticals Sa | Compounds as modulators of GPR-119 |
| US10894787B2 (en) | 2010-09-22 | 2021-01-19 | Arena Pharmaceuticals, Inc. | Modulators of the GPR119 receptor and the treatment of disorders related thereto |
| US11007175B2 (en) | 2015-01-06 | 2021-05-18 | Arena Pharmaceuticals, Inc. | Methods of treating conditions related to the S1P1 receptor |
| US11534424B2 (en) | 2017-02-16 | 2022-12-27 | Arena Pharmaceuticals, Inc. | Compounds and methods for treatment of primary biliary cholangitis |
| US11884626B2 (en) | 2015-06-22 | 2024-01-30 | Arena Pharmaceuticals, Inc. | Crystalline L-arginine salt of (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-1,2,3,4-tetrahydrocyclo-penta [b]indol-3-yl)acetic acid(Compound1) for use in S1P1 receptor-associated disorders |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK1599468T3 (en) | 2003-01-14 | 2008-02-04 | Arena Pharm Inc | 1,2,3-Trisubstituted aryl and heteroaryl derivatives as modulators of metabolism and the prevention and treatment of disorders associated therewith such as diabetes and hyperglycemia |
| AR045047A1 (en) * | 2003-07-11 | 2005-10-12 | Arena Pharm Inc | ARILO AND HETEROARILO DERIVATIVES TRISUSTITUIDOS AS MODULATORS OF METABOLISM AND PROFILAXIS AND TREATMENT OF DISORDERS RELATED TO THEMSELVES |
| WO2017083219A1 (en) * | 2015-11-12 | 2017-05-18 | Merck Sharp & Dohme Corp. | Bispiperidinyl derivatives as liver x receptor beta agonists, compositions, and their use |
| WO2018068297A1 (en) * | 2016-10-14 | 2018-04-19 | Merck Sharp & Dohme Corp. | N-ARYL AND N-HETEROARYL PIPERIDINE DERIVATIVES AS LIVER X RECEPTOR β AGONISTS, COMPOSITIONS, AND THEIR USE |
| WO2018068296A1 (en) | 2016-10-14 | 2018-04-19 | Merck Sharp & Dohme Corp. | PIPERIDINE DERIVATIVES AS LIVER X RECEPTOR β AGONISTS, COMPOSITIONS, AND THEIR USE |
Family Cites Families (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5242924A (en) | 1992-07-02 | 1993-09-07 | Sterling Winthrop Inc. | Tetrazolyl-(phenoxy and phenoxyalkyl)-piperidinylpyridazines as antiviral agents |
| US5364865A (en) * | 1992-12-30 | 1994-11-15 | Sterling Winthrop Inc. | Phenoxy- and phenoxyalkyl-piperidines as antiviral agents |
| AR012443A1 (en) * | 1997-04-16 | 2000-10-18 | Uriach & Cia Sa J | NEW CARBOXAMIDS AS INHIBITORS OF THE PLATELET AGGREGATION, PROCEDURE FOR THEIR PREPARATION, PHARMACEUTICAL COMPOSITIONS THAT CONTAIN THEM AND USE OF THE SAME IN THE MANUFACTURE OF MEDICINES |
| AR013693A1 (en) | 1997-10-23 | 2001-01-10 | Uriach & Cia Sa J | NEW PIPERIDINES AND PIPERAZINAS AS INHIBITORS OF THE PLAQUETARY AGREGATION |
| HUP0200119A3 (en) | 1999-02-08 | 2003-04-28 | G D Searle & Co Chicago | Sulfamato hydroxamic acid metalloprotease inhibitor |
| US6800646B1 (en) * | 1999-02-08 | 2004-10-05 | Pharmacia Corporation | Sulfamato hydroxamic acid metalloprotease inhibitor |
| US6498161B1 (en) * | 1999-04-06 | 2002-12-24 | Merck & Co., Inc. | Pyrrolidine modulators of chemokine receptor activity |
| AUPR213700A0 (en) | 2000-12-18 | 2001-01-25 | Biota Scientific Management Pty Ltd | Antiviral agents |
| AU2003225027A1 (en) | 2002-04-16 | 2003-11-03 | Merck And Co., Inc. | Combination therapy using a ppar alpha/gamma agonist |
| DK1599468T3 (en) | 2003-01-14 | 2008-02-04 | Arena Pharm Inc | 1,2,3-Trisubstituted aryl and heteroaryl derivatives as modulators of metabolism and the prevention and treatment of disorders associated therewith such as diabetes and hyperglycemia |
| CN1751038A (en) | 2003-02-24 | 2006-03-22 | 艾尼纳制药公司 | Substituted aryl and heteroaryl derivatives as modulators of glucose metabolism and the prophylaxis and treatment of disorders thereof |
| AR045047A1 (en) | 2003-07-11 | 2005-10-12 | Arena Pharm Inc | ARILO AND HETEROARILO DERIVATIVES TRISUSTITUIDOS AS MODULATORS OF METABOLISM AND PROFILAXIS AND TREATMENT OF DISORDERS RELATED TO THEMSELVES |
| EP1711491A1 (en) | 2003-12-24 | 2006-10-18 | Prosidion Limited | Heterocyclic derivatives as gpcr receptor agonists |
| TW200610761A (en) | 2004-04-23 | 2006-04-01 | Astrazeneca Ab | Chemical compounds |
| KR20070046150A (en) * | 2004-07-28 | 2007-05-02 | 아이알엠 엘엘씨 | Compounds and compositions as modulators of steroid hormone nuclear receptors |
| GB0419160D0 (en) * | 2004-08-27 | 2004-09-29 | Novartis Ag | Organic compounds |
| US20080312281A1 (en) | 2004-12-24 | 2008-12-18 | Matthew Colin Thor Fyfe | G-Protein Coupled Receptor (Gpr116) Agonists and Use Thereof for Treating Obesity and Diabetes |
| US8193359B2 (en) | 2004-12-24 | 2012-06-05 | Prosidion Limited | G-protein coupled receptor agonists |
| GB0428514D0 (en) | 2004-12-31 | 2005-02-09 | Prosidion Ltd | Compounds |
| DOP2006000008A (en) * | 2005-01-10 | 2006-08-31 | Arena Pharm Inc | COMBINED THERAPY FOR THE TREATMENT OF DIABETES AND RELATED AFFECTIONS AND FOR THE TREATMENT OF AFFECTIONS THAT IMPROVE THROUGH AN INCREASE IN THE BLOOD CONCENTRATION OF GLP-1 |
-
2007
- 2007-12-10 EP EP07862712A patent/EP2112880A4/en not_active Withdrawn
- 2007-12-10 CA CA002672059A patent/CA2672059A1/en not_active Abandoned
- 2007-12-10 US US12/517,142 patent/US8399485B2/en not_active Expired - Fee Related
- 2007-12-10 AU AU2007334519A patent/AU2007334519A1/en not_active Abandoned
- 2007-12-10 WO PCT/US2007/025225 patent/WO2008076243A2/en active Application Filing
- 2007-12-10 JP JP2009541322A patent/JP2010513272A/en not_active Withdrawn
Non-Patent Citations (1)
| Title |
|---|
| See references of EP2112880A4 * |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1808168B1 (en) * | 2005-01-10 | 2009-06-03 | Arena Pharmaceuticals, Inc. | Combination therapy for the treatment of diabetes and conditions related thereto and for the treatment of conditions ameliorated by increasing a blood GLP-1 level |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| US8883714B2 (en) | 2008-04-07 | 2014-11-11 | Arena Pharmaceuticals, Inc. | Pharmaceutical compositions comprising GPR119 agonists which act as peptide YY (PYY) secretagogues |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011113947A1 (en) | 2010-03-18 | 2011-09-22 | Boehringer Ingelheim International Gmbh | Combination of a gpr119 agonist and the dpp-iv inhibitor linagliptin for use in the treatment of diabetes and related conditions |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| US10894787B2 (en) | 2010-09-22 | 2021-01-19 | Arena Pharmaceuticals, Inc. | Modulators of the GPR119 receptor and the treatment of disorders related thereto |
| WO2012046249A1 (en) | 2010-10-08 | 2012-04-12 | Cadila Healthcare Limited | Novel gpr 119 agonists |
| WO2012145361A1 (en) * | 2011-04-19 | 2012-10-26 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| US9777018B2 (en) | 2011-06-09 | 2017-10-03 | Rhizen Pharmaceuticals Sa | Compounds as modulators of GPR-119 |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US11007175B2 (en) | 2015-01-06 | 2021-05-18 | Arena Pharmaceuticals, Inc. | Methods of treating conditions related to the S1P1 receptor |
| US11884626B2 (en) | 2015-06-22 | 2024-01-30 | Arena Pharmaceuticals, Inc. | Crystalline L-arginine salt of (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-1,2,3,4-tetrahydrocyclo-penta [b]indol-3-yl)acetic acid(Compound1) for use in S1P1 receptor-associated disorders |
| WO2017049321A1 (en) | 2015-09-17 | 2017-03-23 | Miller Marvin J | Benzyl amine-containing heterocyclic compounds and compositions useful against mycobacterial infection |
| US11534424B2 (en) | 2017-02-16 | 2022-12-27 | Arena Pharmaceuticals, Inc. | Compounds and methods for treatment of primary biliary cholangitis |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2008076243A3 (en) | 2009-03-26 |
| CA2672059A1 (en) | 2008-06-26 |
| EP2112880A4 (en) | 2011-12-21 |
| US20100075987A1 (en) | 2010-03-25 |
| AU2007334519A1 (en) | 2008-06-26 |
| US8399485B2 (en) | 2013-03-19 |
| JP2010513272A (en) | 2010-04-30 |
| EP2112880A2 (en) | 2009-11-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2112880A2 (en) | Acyl bipiperidinyl compounds, compositions containing such compounds and methods of treatment | |
| AU2007342531B2 (en) | Bipiperidinyl compounds, compositions containing such compounds and methods of treatment | |
| EP2297129B1 (en) | Substituted cyclopropyl compounds, compositions containing such compounds and methods of treatment | |
| US9365539B2 (en) | Prolylcarboxypeptidase inhibitors | |
| JP6047144B2 (en) | Substituted cyclopropyl compounds, compositions containing such compounds and methods of treatment | |
| US8921394B2 (en) | Prolylcarboxypeptidase inhibitors | |
| US20130059830A1 (en) | Novel prolylcarboxypeptidase inhibitors | |
| US8669252B2 (en) | Prolylcarboxypeptidase inhibitors | |
| US8569299B2 (en) | Prolylcarboxypeptidase inhibitors | |
| US20120041012A1 (en) | Substituted spirocyclic amines useful as antidiabetic compounds | |
| US8759539B2 (en) | Substituted bicyclic amines for the treatment of diabetes | |
| WO2014052379A1 (en) | Substituted cyclopropyl compounds | |
| EP2352373A1 (en) | Triazole beta carboline derivatives as antidiabetic compounds | |
| TW201102374A (en) | Substituted cyclopropyl compounds, compositions containing such compounds and methods of treatment |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07862712 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007334519 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12517142 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2009541322 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2672059 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007862712 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2007334519 Country of ref document: AU Date of ref document: 20071210 Kind code of ref document: A |