WO2008006432A1 - Use of ampk-activating imidazole derivatives, preparation process therefor and pharmaceutical compositions comprising them - Google Patents
Use of ampk-activating imidazole derivatives, preparation process therefor and pharmaceutical compositions comprising them Download PDFInfo
- Publication number
- WO2008006432A1 WO2008006432A1 PCT/EP2007/005164 EP2007005164W WO2008006432A1 WO 2008006432 A1 WO2008006432 A1 WO 2008006432A1 EP 2007005164 W EP2007005164 W EP 2007005164W WO 2008006432 A1 WO2008006432 A1 WO 2008006432A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- imidazole
- hydroxy
- carboxamide
- benzyl
- alkyl
- Prior art date
Links
- 238000002360 preparation method Methods 0.000 title claims abstract description 38
- 150000002460 imidazoles Chemical class 0.000 title claims abstract description 25
- 239000008194 pharmaceutical composition Substances 0.000 title claims abstract description 13
- 229940079865 intestinal antiinfectives imidazole derivative Drugs 0.000 title abstract description 9
- 150000001875 compounds Chemical class 0.000 claims abstract description 182
- 238000000034 method Methods 0.000 claims abstract description 34
- 230000008569 process Effects 0.000 claims abstract description 22
- 206010012601 diabetes mellitus Diseases 0.000 claims abstract description 20
- 239000003814 drug Substances 0.000 claims abstract description 9
- 208000001072 type 2 diabetes mellitus Diseases 0.000 claims abstract description 9
- 230000007170 pathology Effects 0.000 claims abstract description 8
- 208000008589 Obesity Diseases 0.000 claims abstract description 6
- 235000020824 obesity Nutrition 0.000 claims abstract description 6
- 239000012190 activator Substances 0.000 claims abstract description 5
- 206010022489 Insulin Resistance Diseases 0.000 claims abstract description 4
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 79
- 125000003118 aryl group Chemical group 0.000 claims description 73
- 125000000217 alkyl group Chemical group 0.000 claims description 72
- 229910052739 hydrogen Inorganic materials 0.000 claims description 53
- 125000003545 alkoxy group Chemical group 0.000 claims description 51
- 239000001257 hydrogen Substances 0.000 claims description 51
- -1 amino, hydroxyl Chemical group 0.000 claims description 44
- 102000014156 AMP-Activated Protein Kinases Human genes 0.000 claims description 43
- 108010011376 AMP-Activated Protein Kinases Proteins 0.000 claims description 43
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 40
- 150000001412 amines Chemical class 0.000 claims description 38
- 239000007787 solid Substances 0.000 claims description 34
- 150000003839 salts Chemical class 0.000 claims description 30
- 229910052736 halogen Inorganic materials 0.000 claims description 29
- 150000002367 halogens Chemical class 0.000 claims description 29
- 239000002253 acid Substances 0.000 claims description 27
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 27
- 125000005842 heteroatom Chemical group 0.000 claims description 27
- 150000002431 hydrogen Chemical class 0.000 claims description 27
- 125000004414 alkyl thio group Chemical group 0.000 claims description 22
- 239000002585 base Substances 0.000 claims description 22
- 125000001951 carbamoylamino group Chemical group C(N)(=O)N* 0.000 claims description 22
- 238000006243 chemical reaction Methods 0.000 claims description 22
- 239000000651 prodrug Substances 0.000 claims description 22
- 229940002612 prodrug Drugs 0.000 claims description 22
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 20
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 18
- 125000004448 alkyl carbonyl group Chemical group 0.000 claims description 16
- 125000001072 heteroaryl group Chemical group 0.000 claims description 16
- 230000003287 optical effect Effects 0.000 claims description 16
- 150000003568 thioethers Chemical class 0.000 claims description 16
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 15
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 15
- 150000004677 hydrates Chemical class 0.000 claims description 15
- 229910052760 oxygen Inorganic materials 0.000 claims description 14
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 13
- 125000002252 acyl group Chemical group 0.000 claims description 13
- 125000004104 aryloxy group Chemical group 0.000 claims description 13
- 125000003282 alkyl amino group Chemical group 0.000 claims description 12
- 230000000694 effects Effects 0.000 claims description 12
- ORTFAQDWJHRMNX-UHFFFAOYSA-N hydroxidooxidocarbon(.) Chemical group O[C]=O ORTFAQDWJHRMNX-UHFFFAOYSA-N 0.000 claims description 12
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 12
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 12
- 239000012453 solvate Substances 0.000 claims description 12
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 11
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 10
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 10
- 239000001301 oxygen Substances 0.000 claims description 10
- GKASDNZWUGIAMG-UHFFFAOYSA-N triethyl orthoformate Chemical compound CCOC(OCC)OCC GKASDNZWUGIAMG-UHFFFAOYSA-N 0.000 claims description 10
- KDBUUJSOUXNTOE-UHFFFAOYSA-N 2-bromopropanediamide Chemical compound NC(=O)C(Br)C(N)=O KDBUUJSOUXNTOE-UHFFFAOYSA-N 0.000 claims description 9
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 9
- 229910052757 nitrogen Inorganic materials 0.000 claims description 9
- 229910052717 sulfur Inorganic materials 0.000 claims description 9
- 239000011593 sulfur Substances 0.000 claims description 9
- 230000009471 action Effects 0.000 claims description 8
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 8
- WBJINCZRORDGAQ-UHFFFAOYSA-N formic acid ethyl ester Natural products CCOC=O WBJINCZRORDGAQ-UHFFFAOYSA-N 0.000 claims description 8
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims description 7
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 claims description 7
- 239000003153 chemical reaction reagent Substances 0.000 claims description 7
- 125000005915 C6-C14 aryl group Chemical group 0.000 claims description 6
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 claims description 5
- 239000007788 liquid Substances 0.000 claims description 5
- 125000006239 protecting group Chemical group 0.000 claims description 5
- JINBYESILADKFW-UHFFFAOYSA-N aminomalonic acid Chemical compound OC(=O)C(N)C(O)=O JINBYESILADKFW-UHFFFAOYSA-N 0.000 claims description 4
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 4
- 125000005914 C6-C14 aryloxy group Chemical group 0.000 claims description 3
- 125000000738 acetamido group Chemical group [H]C([H])([H])C(=O)N([H])[*] 0.000 claims description 3
- 229910021529 ammonia Inorganic materials 0.000 claims description 3
- 239000007822 coupling agent Substances 0.000 claims description 3
- 201000001119 neuropathy Diseases 0.000 claims description 3
- 230000007823 neuropathy Effects 0.000 claims description 3
- 208000033808 peripheral neuropathy Diseases 0.000 claims description 3
- 239000003981 vehicle Substances 0.000 claims description 3
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 claims description 2
- 125000006376 (C3-C10) cycloalkyl group Chemical group 0.000 claims description 2
- 208000032928 Dyslipidaemia Diseases 0.000 claims description 2
- 208000017442 Retinal disease Diseases 0.000 claims description 2
- 206010038923 Retinopathy Diseases 0.000 claims description 2
- 208000037849 arterial hypertension Diseases 0.000 claims description 2
- 210000004204 blood vessel Anatomy 0.000 claims description 2
- 230000000747 cardiac effect Effects 0.000 claims description 2
- 125000005843 halogen group Chemical group 0.000 claims description 2
- 210000003734 kidney Anatomy 0.000 claims description 2
- 210000005036 nerve Anatomy 0.000 claims description 2
- 239000002798 polar solvent Substances 0.000 claims description 2
- 238000010561 standard procedure Methods 0.000 claims description 2
- LVEGZFXIIKKYTA-UHFFFAOYSA-N 2-iminopropanedioic acid Chemical compound OC(=O)C(=N)C(O)=O LVEGZFXIIKKYTA-UHFFFAOYSA-N 0.000 claims 3
- PYOKUURKVVELLB-UHFFFAOYSA-N trimethyl orthoformate Chemical compound COC(OC)OC PYOKUURKVVELLB-UHFFFAOYSA-N 0.000 claims 2
- 125000006313 (C5-C8) alkyl group Chemical group 0.000 claims 1
- 125000001088 1-naphthoyl group Chemical group C1(=CC=CC2=CC=CC=C12)C(=O)* 0.000 claims 1
- QSFMCUMGHDQZGU-UHFFFAOYSA-N 5-hydroxy-3-[[4-[(4-methoxyphenyl)carbamoylamino]phenyl]methyl]imidazole-4-carboxamide Chemical compound C1=CC(OC)=CC=C1NC(=O)NC(C=C1)=CC=C1CN1C(C(N)=O)=C(O)N=C1 QSFMCUMGHDQZGU-UHFFFAOYSA-N 0.000 claims 1
- 102100036009 5'-AMP-activated protein kinase catalytic subunit alpha-2 Human genes 0.000 abstract 1
- 101000783681 Homo sapiens 5'-AMP-activated protein kinase catalytic subunit alpha-2 Proteins 0.000 abstract 1
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 167
- 238000005160 1H NMR spectroscopy Methods 0.000 description 54
- WRIRWRKPLXCTFD-UHFFFAOYSA-N malonamide Chemical class NC(=O)CC(N)=O WRIRWRKPLXCTFD-UHFFFAOYSA-N 0.000 description 34
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 29
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 27
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 27
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 26
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 26
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 23
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 21
- 239000000203 mixture Substances 0.000 description 19
- 239000002609 medium Substances 0.000 description 17
- 239000002904 solvent Substances 0.000 description 17
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 15
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 15
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 15
- GFQBSQXXHYLABK-UHFFFAOYSA-N 2-aminopropanediamide Chemical class NC(=O)C(N)C(N)=O GFQBSQXXHYLABK-UHFFFAOYSA-N 0.000 description 14
- RTRQQBHATOEIAF-UUOKFMHZSA-N acadesine Chemical compound NC1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 RTRQQBHATOEIAF-UUOKFMHZSA-N 0.000 description 14
- 238000001035 drying Methods 0.000 description 14
- 238000003756 stirring Methods 0.000 description 14
- 229910052786 argon Inorganic materials 0.000 description 13
- 239000002244 precipitate Substances 0.000 description 13
- 230000004913 activation Effects 0.000 description 12
- 239000012429 reaction media Substances 0.000 description 11
- 235000014113 dietary fatty acids Nutrition 0.000 description 10
- 239000000194 fatty acid Substances 0.000 description 10
- 229930195729 fatty acid Natural products 0.000 description 10
- 150000004665 fatty acids Chemical class 0.000 description 10
- 0 *[n]1c(OI)c(C(N)=O)nc1 Chemical compound *[n]1c(OI)c(C(N)=O)nc1 0.000 description 9
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 238000010992 reflux Methods 0.000 description 9
- 239000000243 solution Substances 0.000 description 9
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 8
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 8
- 230000003647 oxidation Effects 0.000 description 8
- 238000007254 oxidation reaction Methods 0.000 description 8
- 230000009467 reduction Effects 0.000 description 8
- 238000006722 reduction reaction Methods 0.000 description 8
- RAXXELZNTBOGNW-UHFFFAOYSA-N 1H-imidazole Chemical class C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 7
- ZBNZAJFNDPPMDT-UHFFFAOYSA-N 1h-imidazole-5-carboxamide Chemical compound NC(=O)C1=CNC=N1 ZBNZAJFNDPPMDT-UHFFFAOYSA-N 0.000 description 7
- RTRQQBHATOEIAF-UHFFFAOYSA-N AICA riboside Natural products NC1=C(C(=O)N)N=CN1C1C(O)C(O)C(CO)O1 RTRQQBHATOEIAF-UHFFFAOYSA-N 0.000 description 7
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 7
- 125000004432 carbon atom Chemical group C* 0.000 description 7
- 239000008103 glucose Substances 0.000 description 7
- 229960001031 glucose Drugs 0.000 description 7
- 210000004185 liver Anatomy 0.000 description 7
- 238000004519 manufacturing process Methods 0.000 description 7
- 210000002027 skeletal muscle Anatomy 0.000 description 7
- IMLAIXAZMVDRGA-UHFFFAOYSA-N 2-phenoxyethanamine Chemical compound NCCOC1=CC=CC=C1 IMLAIXAZMVDRGA-UHFFFAOYSA-N 0.000 description 6
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 6
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 6
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 6
- 230000037396 body weight Effects 0.000 description 6
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 230000007423 decrease Effects 0.000 description 6
- 239000003921 oil Substances 0.000 description 6
- 235000019198 oils Nutrition 0.000 description 6
- 230000026731 phosphorylation Effects 0.000 description 6
- 238000006366 phosphorylation reaction Methods 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- 102000004190 Enzymes Human genes 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- 238000005481 NMR spectroscopy Methods 0.000 description 5
- 235000011054 acetic acid Nutrition 0.000 description 5
- 150000007513 acids Chemical class 0.000 description 5
- 239000011149 active material Substances 0.000 description 5
- 239000001768 carboxy methyl cellulose Substances 0.000 description 5
- 235000001727 glucose Nutrition 0.000 description 5
- 238000001727 in vivo Methods 0.000 description 5
- 230000005764 inhibitory process Effects 0.000 description 5
- 230000002503 metabolic effect Effects 0.000 description 5
- 230000004060 metabolic process Effects 0.000 description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 238000003786 synthesis reaction Methods 0.000 description 5
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 5
- 229940086542 triethylamine Drugs 0.000 description 5
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 4
- 102000000452 Acetyl-CoA carboxylase Human genes 0.000 description 4
- 108010016219 Acetyl-CoA carboxylase Proteins 0.000 description 4
- 108010018763 Biotin carboxylase Proteins 0.000 description 4
- 102000004877 Insulin Human genes 0.000 description 4
- 108090001061 Insulin Proteins 0.000 description 4
- 208000001145 Metabolic Syndrome Diseases 0.000 description 4
- 241000699670 Mus sp. Species 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- 241000700159 Rattus Species 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 201000000690 abdominal obesity-metabolic syndrome Diseases 0.000 description 4
- 150000001408 amides Chemical class 0.000 description 4
- 239000012267 brine Substances 0.000 description 4
- 230000003197 catalytic effect Effects 0.000 description 4
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 4
- 238000004090 dissolution Methods 0.000 description 4
- 125000004494 ethyl ester group Chemical group 0.000 description 4
- 238000001704 evaporation Methods 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 239000000499 gel Substances 0.000 description 4
- 230000014101 glucose homeostasis Effects 0.000 description 4
- 230000006377 glucose transport Effects 0.000 description 4
- 230000006872 improvement Effects 0.000 description 4
- 229910052500 inorganic mineral Inorganic materials 0.000 description 4
- 229940125396 insulin Drugs 0.000 description 4
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 4
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin Chemical compound CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 4
- 229960003105 metformin Drugs 0.000 description 4
- 235000010755 mineral Nutrition 0.000 description 4
- 239000011707 mineral Substances 0.000 description 4
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 4
- 230000000638 stimulation Effects 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 4
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 3
- YVFCKAVZCQKJFC-UHFFFAOYSA-N 5-hydroxy-3-[[4-(naphthalene-1-carbonylamino)phenyl]methyl]imidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC(C=C1)=CC=C1NC(=O)C1=CC=CC2=CC=CC=C12 YVFCKAVZCQKJFC-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- 102000011690 Adiponectin Human genes 0.000 description 3
- 108010076365 Adiponectin Proteins 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 3
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 208000031773 Insulin resistance syndrome Diseases 0.000 description 3
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 3
- 230000003213 activating effect Effects 0.000 description 3
- 239000011575 calcium Substances 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 3
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 3
- 230000001413 cellular effect Effects 0.000 description 3
- 235000012000 cholesterol Nutrition 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 230000008878 coupling Effects 0.000 description 3
- 238000010168 coupling process Methods 0.000 description 3
- 238000005859 coupling reaction Methods 0.000 description 3
- 229960001270 d- tartaric acid Drugs 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- YCWBWMZDUHRBGG-UHFFFAOYSA-N ethyl 5-hydroxy-1-(2-phenoxyethyl)imidazole-4-carboxylate Chemical compound OC1=C(C(=O)OCC)N=CN1CCOC1=CC=CC=C1 YCWBWMZDUHRBGG-UHFFFAOYSA-N 0.000 description 3
- 229940093915 gynecological organic acid Drugs 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- 229910052749 magnesium Inorganic materials 0.000 description 3
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 3
- 230000007246 mechanism Effects 0.000 description 3
- 238000002844 melting Methods 0.000 description 3
- 230000008018 melting Effects 0.000 description 3
- 125000002950 monocyclic group Chemical group 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 235000005985 organic acids Nutrition 0.000 description 3
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 description 3
- 230000002265 prevention Effects 0.000 description 3
- SSOLNOMRVKKSON-UHFFFAOYSA-N proguanil Chemical compound CC(C)\N=C(/N)N=C(N)NC1=CC=C(Cl)C=C1 SSOLNOMRVKKSON-UHFFFAOYSA-N 0.000 description 3
- 230000035484 reaction time Effects 0.000 description 3
- 235000010413 sodium alginate Nutrition 0.000 description 3
- 239000000661 sodium alginate Substances 0.000 description 3
- 229940005550 sodium alginate Drugs 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- IJXJGQCXFSSHNL-MRVPVSSYSA-N (2s)-2-amino-2-phenylethanol Chemical compound OC[C@@H](N)C1=CC=CC=C1 IJXJGQCXFSSHNL-MRVPVSSYSA-N 0.000 description 2
- JXSPKRUNMHMICQ-UHFFFAOYSA-N 1-(2-bromoethoxy)-4-fluorobenzene Chemical compound FC1=CC=C(OCCBr)C=C1 JXSPKRUNMHMICQ-UHFFFAOYSA-N 0.000 description 2
- YBCACHFIFLJIBZ-UHFFFAOYSA-N 1-[2-(4-fluorophenoxy)ethyl]-5-hydroxyimidazole-4-carboxamide Chemical compound OC1=C(C(=O)N)N=CN1CCOC1=CC=C(F)C=C1 YBCACHFIFLJIBZ-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- WXGXQQVRTZNAMF-UHFFFAOYSA-N 2-[(4-fluorophenyl)methylamino]propanediamide Chemical compound NC(=O)C(C(N)=O)NCC1=CC=C(F)C=C1 WXGXQQVRTZNAMF-UHFFFAOYSA-N 0.000 description 2
- WNZQDUSMALZDQF-UHFFFAOYSA-N 2-benzofuran-1(3H)-one Chemical compound C1=CC=C2C(=O)OCC2=C1 WNZQDUSMALZDQF-UHFFFAOYSA-N 0.000 description 2
- RFUVZXXHTCYBRQ-UHFFFAOYSA-N 3-[(4-acetamidophenyl)methyl]-5-hydroxyimidazole-4-carboxamide Chemical compound C1=CC(NC(=O)C)=CC=C1CN1C(C(N)=O)=C(O)N=C1 RFUVZXXHTCYBRQ-UHFFFAOYSA-N 0.000 description 2
- LZTYBPWCJFKEAQ-UHFFFAOYSA-N 3-[(4-aminophenyl)methyl]-5-[tert-butyl(dimethyl)silyl]oxyimidazole-4-carboxamide Chemical compound NC(=O)C1=C(O[Si](C)(C)C(C)(C)C)N=CN1CC1=CC=C(N)C=C1 LZTYBPWCJFKEAQ-UHFFFAOYSA-N 0.000 description 2
- RKKDOZQERXWUOA-UHFFFAOYSA-N 3-[(4-chlorophenyl)methyl]-5-hydroxyimidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC1=CC=C(Cl)C=C1 RKKDOZQERXWUOA-UHFFFAOYSA-N 0.000 description 2
- VBDUJUUKWQGJBX-UHFFFAOYSA-N 3-[(4-fluorophenyl)methyl]-5-hydroxyimidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC1=CC=C(F)C=C1 VBDUJUUKWQGJBX-UHFFFAOYSA-N 0.000 description 2
- IYOZDZYQRFQEJL-UHFFFAOYSA-N 3-[[4-(2,3-dihydro-1h-inden-5-ylcarbamoylamino)phenyl]methyl]-5-hydroxyimidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC(C=C1)=CC=C1NC(=O)NC1=CC=C(CCC2)C2=C1 IYOZDZYQRFQEJL-UHFFFAOYSA-N 0.000 description 2
- BODWFTNLXPPCGI-UHFFFAOYSA-N 3-[[4-(benzylcarbamoylamino)phenyl]methyl]-5-hydroxyimidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC(C=C1)=CC=C1NC(=O)NCC1=CC=CC=C1 BODWFTNLXPPCGI-UHFFFAOYSA-N 0.000 description 2
- ZIOFDPLRKUTPDI-UHFFFAOYSA-N 3-[[4-[(2-fluorobenzoyl)amino]phenyl]methyl]-5-hydroxyimidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC(C=C1)=CC=C1NC(=O)C1=CC=CC=C1F ZIOFDPLRKUTPDI-UHFFFAOYSA-N 0.000 description 2
- CHVQCFQUMKSLPG-UHFFFAOYSA-N 3-[[4-[(4-chlorobenzoyl)amino]phenyl]methyl]-5-hydroxyimidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC(C=C1)=CC=C1NC(=O)C1=CC=C(Cl)C=C1 CHVQCFQUMKSLPG-UHFFFAOYSA-N 0.000 description 2
- GDWCDXPQPPLNKA-UHFFFAOYSA-N 3-[[4-[(4-fluorobenzoyl)amino]phenyl]methyl]-5-hydroxyimidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC(C=C1)=CC=C1NC(=O)C1=CC=C(F)C=C1 GDWCDXPQPPLNKA-UHFFFAOYSA-N 0.000 description 2
- YURKSASXBUTRFK-UHFFFAOYSA-N 4-[[4-[(5-carbamoyl-4-hydroxyimidazol-1-yl)methyl]phenyl]carbamoylamino]benzoic acid Chemical compound NC(=O)C1=C(O)N=CN1CC(C=C1)=CC=C1NC(=O)NC1=CC=C(C(O)=O)C=C1 YURKSASXBUTRFK-UHFFFAOYSA-N 0.000 description 2
- MPLLLQUZNJSVTK-UHFFFAOYSA-N 5-[3-[4-[2-(4-fluorophenyl)ethoxy]phenyl]propyl]furan-2-carboxylic acid Chemical compound O1C(C(=O)O)=CC=C1CCCC(C=C1)=CC=C1OCCC1=CC=C(F)C=C1 MPLLLQUZNJSVTK-UHFFFAOYSA-N 0.000 description 2
- HQLBLKJBVKUWLU-UHFFFAOYSA-N 5-hydroxy-1-(2-phenoxyethyl)imidazole-4-carboxamide Chemical compound OC1=C(C(=O)N)N=CN1CCOC1=CC=CC=C1 HQLBLKJBVKUWLU-UHFFFAOYSA-N 0.000 description 2
- DIHOQLFJHGTMBD-UHFFFAOYSA-N 5-hydroxy-3-(2-phenoxyethyl)imidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CCOC1=CC=CC=C1 DIHOQLFJHGTMBD-UHFFFAOYSA-N 0.000 description 2
- AKCWLEUBZPTWCV-UHFFFAOYSA-N 5-hydroxy-3-[(4-nitrophenyl)methyl]imidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC1=CC=C([N+]([O-])=O)C=C1 AKCWLEUBZPTWCV-UHFFFAOYSA-N 0.000 description 2
- VUSCITBBOVNZIU-UHFFFAOYSA-N 5-hydroxy-3-[[4-(phenylcarbamoylamino)phenyl]methyl]imidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC(C=C1)=CC=C1NC(=O)NC1=CC=CC=C1 VUSCITBBOVNZIU-UHFFFAOYSA-N 0.000 description 2
- NOTIAMDYUHBRJZ-UHFFFAOYSA-N 5-hydroxy-3-[[4-[(3-methoxybenzoyl)amino]phenyl]methyl]imidazole-4-carboxamide Chemical compound COC1=CC=CC(C(=O)NC=2C=CC(CN3C(=C(O)N=C3)C(N)=O)=CC=2)=C1 NOTIAMDYUHBRJZ-UHFFFAOYSA-N 0.000 description 2
- HEFKRWSKBHEJLA-UHFFFAOYSA-N 5-hydroxy-3-phenylimidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1C1=CC=CC=C1 HEFKRWSKBHEJLA-UHFFFAOYSA-N 0.000 description 2
- SLAHUKJCEGMKEL-UHFFFAOYSA-N 5-methoxy-3-[(4-nitrophenyl)methyl]imidazole-4-carboxamide Chemical compound NC(=O)C1=C(OC)N=CN1CC1=CC=C([N+]([O-])=O)C=C1 SLAHUKJCEGMKEL-UHFFFAOYSA-N 0.000 description 2
- WFXKCCVYVIECNU-UHFFFAOYSA-N 5-methoxy-3-[[4-(naphthalene-1-carbonylamino)phenyl]methyl]imidazole-4-carboxamide Chemical compound NC(=O)C1=C(OC)N=CN1CC(C=C1)=CC=C1NC(=O)C1=CC=CC2=CC=CC=C12 WFXKCCVYVIECNU-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- 239000005995 Aluminium silicate Substances 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 2
- 239000001856 Ethyl cellulose Substances 0.000 description 2
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 102000003638 Glucose-6-Phosphatase Human genes 0.000 description 2
- 108010086800 Glucose-6-Phosphatase Proteins 0.000 description 2
- 229920002907 Guar gum Polymers 0.000 description 2
- 108010086524 Hepatocyte Nuclear Factor 4 Proteins 0.000 description 2
- 102000015902 Hepatocyte nuclear factor 4-alpha Human genes 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- 102000004286 Hydroxymethylglutaryl CoA Reductases Human genes 0.000 description 2
- 108090000895 Hydroxymethylglutaryl CoA Reductases Proteins 0.000 description 2
- UQSXHKLRYXJYBZ-UHFFFAOYSA-N Iron oxide Chemical compound [Fe]=O UQSXHKLRYXJYBZ-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 102000016267 Leptin Human genes 0.000 description 2
- 108010092277 Leptin Proteins 0.000 description 2
- 108010081805 Malonyl-CoA decarboxylase Proteins 0.000 description 2
- 102100029461 Malonyl-CoA decarboxylase, mitochondrial Human genes 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 108091000080 Phosphotransferase Proteins 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- LOUPRKONTZGTKE-WZBLMQSHSA-N Quinine Chemical compound C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@@H]2[C@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-WZBLMQSHSA-N 0.000 description 2
- 241000283984 Rodentia Species 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- 230000001154 acute effect Effects 0.000 description 2
- 150000001263 acyl chlorides Chemical class 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 210000001789 adipocyte Anatomy 0.000 description 2
- 230000001476 alcoholic effect Effects 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 239000000783 alginic acid Substances 0.000 description 2
- 229960001126 alginic acid Drugs 0.000 description 2
- 150000004781 alginic acids Chemical class 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 235000012211 aluminium silicate Nutrition 0.000 description 2
- PZZYQPZGQPZBDN-UHFFFAOYSA-N aluminium silicate Chemical compound O=[Al]O[Si](=O)O[Al]=O PZZYQPZGQPZBDN-UHFFFAOYSA-N 0.000 description 2
- 229910000323 aluminium silicate Inorganic materials 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 125000002619 bicyclic group Chemical group 0.000 description 2
- 238000004166 bioassay Methods 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 230000036772 blood pressure Effects 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 239000001110 calcium chloride Substances 0.000 description 2
- 229910001628 calcium chloride Inorganic materials 0.000 description 2
- 235000013877 carbamide Nutrition 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 210000004027 cell Anatomy 0.000 description 2
- 230000019522 cellular metabolic process Effects 0.000 description 2
- 235000010980 cellulose Nutrition 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 229960004926 chlorobutanol Drugs 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- 235000015165 citric acid Nutrition 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 230000008602 contraction Effects 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- MKRTXPORKIRPDG-UHFFFAOYSA-N diphenylphosphoryl azide Chemical compound C=1C=CC=CC=1P(=O)(N=[N+]=[N-])C1=CC=CC=C1 MKRTXPORKIRPDG-UHFFFAOYSA-N 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- PVTYJHAPUVNPAH-UHFFFAOYSA-N ethyl 1-[2-(4-cyanophenoxy)ethyl]-5-hydroxyimidazole-4-carboxylate Chemical compound OC1=C(C(=O)OCC)N=CN1CCOC1=CC=C(C#N)C=C1 PVTYJHAPUVNPAH-UHFFFAOYSA-N 0.000 description 2
- PWLRASXCGHGROK-UHFFFAOYSA-N ethyl 1-[2-(4-fluorophenoxy)ethyl]-5-hydroxyimidazole-4-carboxylate Chemical compound OC1=C(C(=O)OCC)N=CN1CCOC1=CC=C(F)C=C1 PWLRASXCGHGROK-UHFFFAOYSA-N 0.000 description 2
- DHMRVVNEGWPPOE-UHFFFAOYSA-N ethyl 2-[[4-[(5-carbamoyl-4-hydroxyimidazol-1-yl)methyl]phenyl]carbamoylamino]acetate Chemical compound C1=CC(NC(=O)NCC(=O)OCC)=CC=C1CN1C(C(N)=O)=C(O)N=C1 DHMRVVNEGWPPOE-UHFFFAOYSA-N 0.000 description 2
- COMMVKWVFGHLMO-UHFFFAOYSA-N ethyl 2-[[4-[(5-carbamoyl-4-hydroxyimidazol-1-yl)methyl]phenyl]carbamoylamino]benzoate Chemical compound CCOC(=O)C1=CC=CC=C1NC(=O)NC(C=C1)=CC=C1CN1C(C(N)=O)=C(O)N=C1 COMMVKWVFGHLMO-UHFFFAOYSA-N 0.000 description 2
- BKDDOQDARTWKKM-UHFFFAOYSA-N ethyl 3-[[4-[(5-carbamoyl-4-hydroxyimidazol-1-yl)methyl]phenyl]carbamoylamino]benzoate Chemical compound CCOC(=O)C1=CC=CC(NC(=O)NC=2C=CC(CN3C(=C(O)N=C3)C(N)=O)=CC=2)=C1 BKDDOQDARTWKKM-UHFFFAOYSA-N 0.000 description 2
- YPKNBPQPSQJZKR-UHFFFAOYSA-N ethyl 4-[[4-[(5-carbamoyl-4-hydroxyimidazol-1-yl)methyl]phenyl]carbamoylamino]benzoate Chemical compound C1=CC(C(=O)OCC)=CC=C1NC(=O)NC(C=C1)=CC=C1CN1C(C(N)=O)=C(O)N=C1 YPKNBPQPSQJZKR-UHFFFAOYSA-N 0.000 description 2
- 235000019325 ethyl cellulose Nutrition 0.000 description 2
- 229920001249 ethyl cellulose Polymers 0.000 description 2
- 229940052308 general anesthetics halogenated hydrocarbons Drugs 0.000 description 2
- 230000004153 glucose metabolism Effects 0.000 description 2
- 239000000665 guar gum Substances 0.000 description 2
- 235000010417 guar gum Nutrition 0.000 description 2
- 229960002154 guar gum Drugs 0.000 description 2
- 150000008282 halocarbons Chemical class 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 238000005984 hydrogenation reaction Methods 0.000 description 2
- 239000012442 inert solvent Substances 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 2
- TWBYWOBDOCUKOW-UHFFFAOYSA-N isonicotinic acid Chemical compound OC(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 229960001375 lactose Drugs 0.000 description 2
- NRYBAZVQPHGZNS-ZSOCWYAHSA-N leptin Chemical compound O=C([C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](N)CC(C)C)CCSC)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](CS)C(O)=O NRYBAZVQPHGZNS-ZSOCWYAHSA-N 0.000 description 2
- 229940039781 leptin Drugs 0.000 description 2
- FEWJPZIEWOKRBE-LWMBPPNESA-N levotartaric acid Chemical compound OC(=O)[C@@H](O)[C@H](O)C(O)=O FEWJPZIEWOKRBE-LWMBPPNESA-N 0.000 description 2
- 230000007774 longterm Effects 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 239000001630 malic acid Substances 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 229960001855 mannitol Drugs 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- SMRYWTZXDXXQKC-UHFFFAOYSA-N methyl 4-(2-aminoethoxy)benzoate Chemical compound COC(=O)C1=CC=C(OCCN)C=C1 SMRYWTZXDXXQKC-UHFFFAOYSA-N 0.000 description 2
- VOSTYMNOYWRWRC-UHFFFAOYSA-N methyl 4-[2-(4-carbamoyl-5-hydroxyimidazol-1-yl)ethoxy]benzoate Chemical compound C1=CC(C(=O)OC)=CC=C1OCCN1C(O)=C(C(N)=O)N=C1 VOSTYMNOYWRWRC-UHFFFAOYSA-N 0.000 description 2
- 229920000609 methyl cellulose Polymers 0.000 description 2
- 239000001923 methylcellulose Substances 0.000 description 2
- 235000010981 methylcellulose Nutrition 0.000 description 2
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 2
- 239000008108 microcrystalline cellulose Substances 0.000 description 2
- 229940016286 microcrystalline cellulose Drugs 0.000 description 2
- 210000003205 muscle Anatomy 0.000 description 2
- ODUCDPQEXGNKDN-UHFFFAOYSA-N nitroxyl Chemical compound O=N ODUCDPQEXGNKDN-UHFFFAOYSA-N 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 239000003208 petroleum Substances 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 239000000825 pharmaceutical preparation Substances 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 102000020233 phosphotransferase Human genes 0.000 description 2
- WLJVNTCWHIRURA-UHFFFAOYSA-N pimelic acid Chemical compound OC(=O)CCCCCC(O)=O WLJVNTCWHIRURA-UHFFFAOYSA-N 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 150000003254 radicals Chemical class 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- FZHAPNGMFPVSLP-UHFFFAOYSA-N silanamine Chemical compound [SiH3]N FZHAPNGMFPVSLP-UHFFFAOYSA-N 0.000 description 2
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 2
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 229940032147 starch Drugs 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 229960004793 sucrose Drugs 0.000 description 2
- 239000011975 tartaric acid Substances 0.000 description 2
- 229960001367 tartaric acid Drugs 0.000 description 2
- 239000002562 thickening agent Substances 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 150000003672 ureas Chemical class 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- 238000011683 zucker rat (lean) Methods 0.000 description 2
- 238000011684 zucker rat (obese) Methods 0.000 description 2
- DTGKSKDOIYIVQL-WEDXCCLWSA-N (+)-borneol Chemical compound C1C[C@@]2(C)[C@@H](O)C[C@@H]1C2(C)C DTGKSKDOIYIVQL-WEDXCCLWSA-N 0.000 description 1
- KPWKPGFLZGMMFX-VHSXEESVSA-N (-)-camphanic acid Chemical compound C1C[C@]2(C(O)=O)OC(=O)[C@@]1(C)C2(C)C KPWKPGFLZGMMFX-VHSXEESVSA-N 0.000 description 1
- KWGRBVOPPLSCSI-WPRPVWTQSA-N (-)-ephedrine Chemical compound CN[C@@H](C)[C@H](O)C1=CC=CC=C1 KWGRBVOPPLSCSI-WPRPVWTQSA-N 0.000 description 1
- REPVLJRCJUVQFA-UHFFFAOYSA-N (-)-isopinocampheol Natural products C1C(O)C(C)C2C(C)(C)C1C2 REPVLJRCJUVQFA-UHFFFAOYSA-N 0.000 description 1
- FMCGSUUBYTWNDP-ONGXEEELSA-N (1R,2S)-2-(dimethylamino)-1-phenyl-1-propanol Chemical compound CN(C)[C@@H](C)[C@H](O)C1=CC=CC=C1 FMCGSUUBYTWNDP-ONGXEEELSA-N 0.000 description 1
- RQEUFEKYXDPUSK-ZETCQYMHSA-N (1S)-1-phenylethanamine Chemical compound C[C@H](N)C1=CC=CC=C1 RQEUFEKYXDPUSK-ZETCQYMHSA-N 0.000 description 1
- ZICDZTXDTPZBKH-OAHLLOKOSA-N (1r)-2-(4-methylphenyl)-1-phenylethanamine Chemical compound C1=CC(C)=CC=C1C[C@@H](N)C1=CC=CC=C1 ZICDZTXDTPZBKH-OAHLLOKOSA-N 0.000 description 1
- KPWKPGFLZGMMFX-ZJUUUORDSA-N (1s,4r)-1,7,7-trimethyl-2-oxo-3-oxabicyclo[2.2.1]heptane-4-carboxylic acid Chemical compound C1C[C@@]2(C(O)=O)OC(=O)[C@]1(C)C2(C)C KPWKPGFLZGMMFX-ZJUUUORDSA-N 0.000 description 1
- DFQWOAPAERBEID-NSHDSACASA-N (2s)-1-phenylmethoxybutan-2-amine Chemical compound CC[C@H](N)COCC1=CC=CC=C1 DFQWOAPAERBEID-NSHDSACASA-N 0.000 description 1
- TXMNBKSKRZKHEV-KRWDZBQOSA-N (2s)-3-[2-(4-cyanophenoxy)ethylamino]-3-oxo-2-(phenylmethoxycarbonylamino)propanoic acid Chemical compound N([C@H](C(=O)O)C(=O)NCCOC=1C=CC(=CC=1)C#N)C(=O)OCC1=CC=CC=C1 TXMNBKSKRZKHEV-KRWDZBQOSA-N 0.000 description 1
- QGUVABOEOQGGNM-INIZCTEOSA-N (2s)-3-[2-(4-fluorophenoxy)ethylamino]-3-oxo-2-(phenylmethoxycarbonylamino)propanoic acid Chemical compound N([C@H](C(=O)O)C(=O)NCCOC=1C=CC(F)=CC=1)C(=O)OCC1=CC=CC=C1 QGUVABOEOQGGNM-INIZCTEOSA-N 0.000 description 1
- RJWIAUHQNPUHNB-KRWDZBQOSA-N (2s)-3-[2-(4-methoxycarbonylphenoxy)ethylamino]-3-oxo-2-(phenylmethoxycarbonylamino)propanoic acid Chemical compound C1=CC(C(=O)OC)=CC=C1OCCNC(=O)[C@@H](C(O)=O)NC(=O)OCC1=CC=CC=C1 RJWIAUHQNPUHNB-KRWDZBQOSA-N 0.000 description 1
- IIFVWLUQBAIPMJ-UHFFFAOYSA-N (4-fluorophenyl)methanamine Chemical compound NCC1=CC=C(F)C=C1 IIFVWLUQBAIPMJ-UHFFFAOYSA-N 0.000 description 1
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 description 1
- IWYDHOAUDWTVEP-SSDOTTSWSA-N (R)-mandelic acid Chemical compound OC(=O)[C@H](O)C1=CC=CC=C1 IWYDHOAUDWTVEP-SSDOTTSWSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- ADFXKUOMJKEIND-UHFFFAOYSA-N 1,3-dicyclohexylurea Chemical compound C1CCCCC1NC(=O)NC1CCCCC1 ADFXKUOMJKEIND-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- GOFIUEUUROFVMA-UHFFFAOYSA-N 1-(1h-indol-5-yl)ethanone Chemical compound CC(=O)C1=CC=C2NC=CC2=C1 GOFIUEUUROFVMA-UHFFFAOYSA-N 0.000 description 1
- LYMPMKYTOXEEKQ-UHFFFAOYSA-N 1-[2-(4-cyanophenoxy)ethyl]-5-hydroxyimidazole-4-carboxamide Chemical compound OC1=C(C(=O)N)N=CN1CCOC1=CC=C(C#N)C=C1 LYMPMKYTOXEEKQ-UHFFFAOYSA-N 0.000 description 1
- UEZADNGATFAIBL-UHFFFAOYSA-N 1-[4-[(4-hydroxyimidazol-1-yl)methyl]phenyl]-3-naphthalen-1-ylurea Chemical compound C1=NC(O)=CN1CC(C=C1)=CC=C1NC(=O)NC1=CC=CC2=CC=CC=C12 UEZADNGATFAIBL-UHFFFAOYSA-N 0.000 description 1
- SXPWYSNMKUDXOU-UHFFFAOYSA-N 2,2,3,4-tetramethyl-5-phenyl-1,3-oxazolidine Chemical compound O1C(C)(C)N(C)C(C)C1C1=CC=CC=C1 SXPWYSNMKUDXOU-UHFFFAOYSA-N 0.000 description 1
- HTMIRIIZZGJJBK-UHFFFAOYSA-N 2-(4-fluorophenoxy)ethanamine Chemical compound NCCOC1=CC=C(F)C=C1 HTMIRIIZZGJJBK-UHFFFAOYSA-N 0.000 description 1
- OXQGTIUCKGYOAA-UHFFFAOYSA-N 2-Ethylbutanoic acid Chemical compound CCC(CC)C(O)=O OXQGTIUCKGYOAA-UHFFFAOYSA-N 0.000 description 1
- IOIGGGYXFDEEMX-UHFFFAOYSA-N 2-[(4-chlorophenyl)methylamino]propanediamide Chemical compound NC(=O)C(C(N)=O)NCC1=CC=C(Cl)C=C1 IOIGGGYXFDEEMX-UHFFFAOYSA-N 0.000 description 1
- ITGQYOBJQTWWHB-UHFFFAOYSA-N 2-[2-(4-cyanophenoxy)ethylamino]propanediamide Chemical compound NC(=O)C(C(N)=O)NCCOC1=CC=C(C#N)C=C1 ITGQYOBJQTWWHB-UHFFFAOYSA-N 0.000 description 1
- VVNAGOXDBCDUPI-UHFFFAOYSA-N 2-[2-(4-fluorophenoxy)ethyl]isoindole-1,3-dione Chemical compound C1=CC(F)=CC=C1OCCN1C(=O)C2=CC=CC=C2C1=O VVNAGOXDBCDUPI-UHFFFAOYSA-N 0.000 description 1
- NTVIENJYWNRRIP-UHFFFAOYSA-N 2-[2-(4-fluorophenoxy)ethylamino]propanediamide Chemical compound NC(=O)C(C(N)=O)NCCOC1=CC=C(F)C=C1 NTVIENJYWNRRIP-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- PJBFHGHMMOGYNF-UHFFFAOYSA-N 2-amino-3-ethoxy-3-oxopropanoic acid;hydrochloride Chemical compound Cl.CCOC(=O)C(N)C(O)=O PJBFHGHMMOGYNF-UHFFFAOYSA-N 0.000 description 1
- SGFXYZGBSOTQOK-UHFFFAOYSA-N 2-amino-n'-(2-phenoxyethyl)propanediamide Chemical compound NC(=O)C(N)C(=O)NCCOC1=CC=CC=C1 SGFXYZGBSOTQOK-UHFFFAOYSA-N 0.000 description 1
- OUKDSFPJPWQCCG-UHFFFAOYSA-N 2-amino-n'-[2-(4-cyanophenoxy)ethyl]propanediamide Chemical compound NC(=O)C(N)C(=O)NCCOC1=CC=C(C#N)C=C1 OUKDSFPJPWQCCG-UHFFFAOYSA-N 0.000 description 1
- VOMPFMBTZUWROY-UHFFFAOYSA-N 2-amino-n'-[2-(4-fluorophenoxy)ethyl]propanediamide Chemical compound NC(=O)C(N)C(=O)NCCOC1=CC=C(F)C=C1 VOMPFMBTZUWROY-UHFFFAOYSA-N 0.000 description 1
- VTLFFDBQXSVCBV-UHFFFAOYSA-N 2-anilinopropanediamide Chemical compound NC(=O)C(C(N)=O)NC1=CC=CC=C1 VTLFFDBQXSVCBV-UHFFFAOYSA-N 0.000 description 1
- 125000001216 2-naphthoyl group Chemical group C1=C(C=CC2=CC=CC=C12)C(=O)* 0.000 description 1
- FDHIEBLLIJSUME-UHFFFAOYSA-N 3-[(4-aminophenyl)methyl]-5-hydroxyimidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC1=CC=C(N)C=C1 FDHIEBLLIJSUME-UHFFFAOYSA-N 0.000 description 1
- DPNORYDXBOIHIX-UHFFFAOYSA-N 3-[(4-benzamidophenyl)methyl]-5-hydroxyimidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC(C=C1)=CC=C1NC(=O)C1=CC=CC=C1 DPNORYDXBOIHIX-UHFFFAOYSA-N 0.000 description 1
- UDGQQXXGPDBJRQ-UHFFFAOYSA-N 3-[(4-nitrophenyl)methyl]-5-phenylmethoxyimidazole-4-carboxamide Chemical compound N1=CN(CC=2C=CC(=CC=2)[N+]([O-])=O)C(C(=O)N)=C1OCC1=CC=CC=C1 UDGQQXXGPDBJRQ-UHFFFAOYSA-N 0.000 description 1
- PKLBPJAJONBGBT-UHFFFAOYSA-N 3-[2-(4-cyanophenoxy)ethyl]-5-hydroxyimidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CCOC1=CC=C(C#N)C=C1 PKLBPJAJONBGBT-UHFFFAOYSA-N 0.000 description 1
- WTSZKLYYBXRWLE-UHFFFAOYSA-N 3-[2-(4-fluorophenoxy)ethyl]-5-hydroxyimidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CCOC1=CC=C(F)C=C1 WTSZKLYYBXRWLE-UHFFFAOYSA-N 0.000 description 1
- MLFAPFALCMVRJJ-UHFFFAOYSA-N 3-[[4-(3,3-dimethylbutanoylamino)phenyl]methyl]-5-hydroxyimidazole-4-carboxamide Chemical compound C1=CC(NC(=O)CC(C)(C)C)=CC=C1CN1C(C(N)=O)=C(O)N=C1 MLFAPFALCMVRJJ-UHFFFAOYSA-N 0.000 description 1
- AXSJMQYBKADWPE-UHFFFAOYSA-N 3-[[4-(3-cyclopentylpropanoylamino)phenyl]methyl]-5-hydroxyimidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC(C=C1)=CC=C1NC(=O)CCC1CCCC1 AXSJMQYBKADWPE-UHFFFAOYSA-N 0.000 description 1
- OFUACMQZHBNPLK-UHFFFAOYSA-N 3-[[4-(carbamoylamino)phenyl]methyl]-5-hydroxyimidazole-4-carboxamide Chemical compound C1=CC(NC(=O)N)=CC=C1CN1C(C(N)=O)=C(O)N=C1 OFUACMQZHBNPLK-UHFFFAOYSA-N 0.000 description 1
- HATNBAPKFYRDJM-UHFFFAOYSA-N 3-[[4-(cyclohexanecarbonylamino)phenyl]methyl]-5-hydroxyimidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC(C=C1)=CC=C1NC(=O)C1CCCCC1 HATNBAPKFYRDJM-UHFFFAOYSA-N 0.000 description 1
- SFHSDGPRUWFYSS-UHFFFAOYSA-N 3-[[4-(cyclopentylcarbamoylamino)phenyl]methyl]-5-hydroxyimidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC(C=C1)=CC=C1NC(=O)NC1CCCC1 SFHSDGPRUWFYSS-UHFFFAOYSA-N 0.000 description 1
- LABDLKVRSOVUQT-UHFFFAOYSA-N 3-[[4-(hexanoylamino)phenyl]methyl]-5-hydroxyimidazole-4-carboxamide Chemical compound C1=CC(NC(=O)CCCCC)=CC=C1CN1C(C(N)=O)=C(O)N=C1 LABDLKVRSOVUQT-UHFFFAOYSA-N 0.000 description 1
- WLNYFQIXZNOUNG-UHFFFAOYSA-N 3-[[4-[(2-chlorophenyl)carbamoylamino]phenyl]methyl]-5-hydroxyimidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC(C=C1)=CC=C1NC(=O)NC1=CC=CC=C1Cl WLNYFQIXZNOUNG-UHFFFAOYSA-N 0.000 description 1
- BTMIXHMGKFFLLK-UHFFFAOYSA-N 3-[[4-[(4-chlorophenyl)carbamoylamino]phenyl]methyl]-5-hydroxyimidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC(C=C1)=CC=C1NC(=O)NC1=CC=C(Cl)C=C1 BTMIXHMGKFFLLK-UHFFFAOYSA-N 0.000 description 1
- JXOYEPFVERCJHS-UHFFFAOYSA-N 3-[[4-[(4-ethylphenyl)carbamoylamino]phenyl]methyl]-5-hydroxyimidazole-4-carboxamide Chemical compound C1=CC(CC)=CC=C1NC(=O)NC(C=C1)=CC=C1CN1C(C(N)=O)=C(O)N=C1 JXOYEPFVERCJHS-UHFFFAOYSA-N 0.000 description 1
- HUFGOOOAVVXAIN-UHFFFAOYSA-N 3-[[4-[(5-carbamoyl-4-hydroxyimidazol-1-yl)methyl]phenyl]carbamoylamino]benzoic acid Chemical compound NC(=O)C1=C(O)N=CN1CC(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(O)=O)=C1 HUFGOOOAVVXAIN-UHFFFAOYSA-N 0.000 description 1
- NOAXUUMAJUOHGW-UHFFFAOYSA-N 3-[[4-[[2-(4-chlorophenyl)acetyl]amino]phenyl]methyl]-5-hydroxyimidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC(C=C1)=CC=C1NC(=O)CC1=CC=C(Cl)C=C1 NOAXUUMAJUOHGW-UHFFFAOYSA-N 0.000 description 1
- FIOLYQVLESMAMM-UHFFFAOYSA-N 3-[[4-[[2-(furan-2-yl)acetyl]amino]phenyl]methyl]-5-hydroxyimidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC(C=C1)=CC=C1NC(=O)CC1=CC=CO1 FIOLYQVLESMAMM-UHFFFAOYSA-N 0.000 description 1
- AGMZSYQMSHMXLT-UHFFFAOYSA-N 3-aminobutan-1-ol Chemical compound CC(N)CCO AGMZSYQMSHMXLT-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- RUQIUASLAXJZIE-UHFFFAOYSA-N 3-methoxybenzoyl chloride Chemical compound COC1=CC=CC(C(Cl)=O)=C1 RUQIUASLAXJZIE-UHFFFAOYSA-N 0.000 description 1
- VVUJBFUHEWGKAZ-UHFFFAOYSA-N 4-(2-aminoethoxy)benzonitrile Chemical compound NCCOC1=CC=C(C#N)C=C1 VVUJBFUHEWGKAZ-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- INTCGJHAECYOBW-UHFFFAOYSA-N 4-(dimethylamino)-3-methyl-1,2-diphenylbutan-2-ol Chemical compound C=1C=CC=CC=1C(O)(C(CN(C)C)C)CC1=CC=CC=C1 INTCGJHAECYOBW-UHFFFAOYSA-N 0.000 description 1
- KZELLPISMCSTBM-UHFFFAOYSA-N 4-[2-(1,3-dioxoisoindol-2-yl)ethoxy]benzonitrile Chemical compound O=C1C2=CC=CC=C2C(=O)N1CCOC1=CC=C(C#N)C=C1 KZELLPISMCSTBM-UHFFFAOYSA-N 0.000 description 1
- 125000001255 4-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1F 0.000 description 1
- 125000005274 4-hydroxybenzoic acid group Chemical class 0.000 description 1
- DXUGGXVUMIKPQU-UHFFFAOYSA-N 5-hydroxy-1-[2-(4-methoxycarbonylphenoxy)ethyl]imidazole-4-carboxylic acid Chemical compound COC(=O)c1ccc(OCCn2cnc(C(O)=O)c2O)cc1 DXUGGXVUMIKPQU-UHFFFAOYSA-N 0.000 description 1
- WIIXHCVGKKSRAE-UHFFFAOYSA-N 5-hydroxy-3-[[4-(2-phenylbutanoylamino)phenyl]methyl]imidazole-4-carboxamide Chemical compound C=1C=CC=CC=1C(CC)C(=O)NC(C=C1)=CC=C1CN1C=NC(O)=C1C(N)=O WIIXHCVGKKSRAE-UHFFFAOYSA-N 0.000 description 1
- MYQDVMHHXYRVEN-UHFFFAOYSA-N 5-hydroxy-3-[[4-(naphthalen-2-ylcarbamoylamino)phenyl]methyl]imidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC(C=C1)=CC=C1NC(=O)NC1=CC=C(C=CC=C2)C2=C1 MYQDVMHHXYRVEN-UHFFFAOYSA-N 0.000 description 1
- MJEQJACECOJXMW-UHFFFAOYSA-N 5-hydroxy-3-[[4-(naphthalene-2-carbonylamino)phenyl]methyl]imidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC(C=C1)=CC=C1NC(=O)C1=CC=C(C=CC=C2)C2=C1 MJEQJACECOJXMW-UHFFFAOYSA-N 0.000 description 1
- DKOPDBGEBMOPNF-UHFFFAOYSA-N 5-hydroxy-3-[[4-[(2-methoxyphenyl)carbamoylamino]phenyl]methyl]imidazole-4-carboxamide Chemical compound COC1=CC=CC=C1NC(=O)NC(C=C1)=CC=C1CN1C(C(N)=O)=C(O)N=C1 DKOPDBGEBMOPNF-UHFFFAOYSA-N 0.000 description 1
- BIAFLIADQLSCMR-UHFFFAOYSA-N 5-hydroxy-3-[[4-[(4-methoxybenzoyl)amino]phenyl]methyl]imidazole-4-carboxamide Chemical compound C1=CC(OC)=CC=C1C(=O)NC(C=C1)=CC=C1CN1C(C(N)=O)=C(O)N=C1 BIAFLIADQLSCMR-UHFFFAOYSA-N 0.000 description 1
- PYMCUIJTHUYGFD-UHFFFAOYSA-N 5-hydroxy-3-[[4-[(4-methylbenzoyl)amino]phenyl]methyl]imidazole-4-carboxamide Chemical compound C1=CC(C)=CC=C1C(=O)NC(C=C1)=CC=C1CN1C(C(N)=O)=C(O)N=C1 PYMCUIJTHUYGFD-UHFFFAOYSA-N 0.000 description 1
- MNXHKIBYHMWADU-UHFFFAOYSA-N 5-hydroxy-3-[[4-[[2-(4-nitrophenyl)acetyl]amino]phenyl]methyl]imidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC(C=C1)=CC=C1NC(=O)CC1=CC=C([N+]([O-])=O)C=C1 MNXHKIBYHMWADU-UHFFFAOYSA-N 0.000 description 1
- FBUGESRPPXJMBJ-UHFFFAOYSA-N 5-hydroxy-3-[[4-[[2-(trifluoromethyl)phenyl]carbamoylamino]phenyl]methyl]imidazole-4-carboxamide Chemical compound NC(=O)C1=C(O)N=CN1CC(C=C1)=CC=C1NC(=O)NC1=CC=CC=C1C(F)(F)F FBUGESRPPXJMBJ-UHFFFAOYSA-N 0.000 description 1
- ROFZRKMOZRZSNN-UHFFFAOYSA-N 5-methoxy-1-methylimidazole-4-carboxamide Chemical compound COC1=C(C(N)=O)N=CN1C ROFZRKMOZRZSNN-UHFFFAOYSA-N 0.000 description 1
- NOTGFIUVDGNKRI-UUOKFMHZSA-N AICA ribonucleotide Chemical compound NC1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(O)=O)O1 NOTGFIUVDGNKRI-UUOKFMHZSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N Acrylic acid Chemical compound OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- 229920002126 Acrylic acid copolymer Polymers 0.000 description 1
- 102000057234 Acyl transferases Human genes 0.000 description 1
- 108700016155 Acyl transferases Proteins 0.000 description 1
- WSVLPVUVIUVCRA-KPKNDVKVSA-N Alpha-lactose monohydrate Chemical compound O.O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O WSVLPVUVIUVCRA-KPKNDVKVSA-N 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N Benzoic acid Natural products OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 101001005186 Bos taurus Hormone-sensitive lipase Proteins 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 235000001258 Cinchona calisaya Nutrition 0.000 description 1
- 244000223760 Cinnamomum zeylanicum Species 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 208000002249 Diabetes Complications Diseases 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 208000017701 Endocrine disease Diseases 0.000 description 1
- 229920003134 Eudragit® polymer Polymers 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 101710091951 Glycerol-3-phosphate acyltransferase Proteins 0.000 description 1
- 102000008100 Human Serum Albumin Human genes 0.000 description 1
- 108091006905 Human Serum Albumin Proteins 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- 206010021143 Hypoxia Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 229920002774 Maltodextrin Polymers 0.000 description 1
- 239000005913 Maltodextrin Substances 0.000 description 1
- 235000006679 Mentha X verticillata Nutrition 0.000 description 1
- 235000002899 Mentha suaveolens Nutrition 0.000 description 1
- 235000001636 Mentha x rotundifolia Nutrition 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 1
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 1
- 229920000881 Modified starch Polymers 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 1
- BXDZOYLPNAIDOC-UHFFFAOYSA-N N-[5-[(5-tert-butyl-1,3-oxazol-2-yl)methylsulfanyl]-1,3-thiazol-2-yl]-1-[2-[2-[2-[2-[2-[[2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindol-4-yl]amino]ethoxy]ethoxy]ethoxy]ethylamino]-2-oxoethyl]piperidine-4-carboxamide Chemical compound CC(C)(C)c1cnc(CSc2cnc(NC(=O)C3CCN(CC(=O)NCCOCCOCCOCCNc4cccc5C(=O)N(C6CCC(=O)NC6=O)C(=O)c45)CC3)s2)o1 BXDZOYLPNAIDOC-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 229910020700 Na3VO4 Inorganic materials 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- 108090000472 Phosphoenolpyruvate carboxykinase (ATP) Proteins 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 208000006268 Sarcoma 180 Diseases 0.000 description 1
- 244000000231 Sesamum indicum Species 0.000 description 1
- 235000003434 Sesamum indicum Nutrition 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- ABBQHOQBGMUPJH-UHFFFAOYSA-M Sodium salicylate Chemical compound [Na+].OC1=CC=CC=C1C([O-])=O ABBQHOQBGMUPJH-UHFFFAOYSA-M 0.000 description 1
- 229920002125 Sokalan® Polymers 0.000 description 1
- 102000000019 Sterol Esterase Human genes 0.000 description 1
- 108010055297 Sterol Esterase Proteins 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 244000299461 Theobroma cacao Species 0.000 description 1
- 235000009470 Theobroma cacao Nutrition 0.000 description 1
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 125000005041 acyloxyalkyl group Chemical group 0.000 description 1
- 230000006978 adaptation Effects 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 125000003342 alkenyl group Chemical group 0.000 description 1
- 125000005205 alkoxycarbonyloxyalkyl group Chemical group 0.000 description 1
- 230000003281 allosteric effect Effects 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 239000004411 aluminium Substances 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 150000001414 amino alcohols Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 125000005428 anthryl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C3C(*)=C([H])C([H])=C([H])C3=C([H])C2=C1[H] 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000003243 anti-lipolytic effect Effects 0.000 description 1
- 229940125708 antidiabetic agent Drugs 0.000 description 1
- 239000003472 antidiabetic agent Substances 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- 150000005347 biaryls Chemical class 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 125000004057 biotinyl group Chemical group [H]N1C(=O)N([H])[C@]2([H])[C@@]([H])(SC([H])([H])[C@]12[H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C(*)=O 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 229940116229 borneol Drugs 0.000 description 1
- CKDOCTFBFTVPSN-UHFFFAOYSA-N borneol Natural products C1CC2(C)C(C)CC1C2(C)C CKDOCTFBFTVPSN-UHFFFAOYSA-N 0.000 description 1
- RRKTZKIUPZVBMF-IBTVXLQLSA-N brucine Chemical compound O([C@@H]1[C@H]([C@H]2C3)[C@@H]4N(C(C1)=O)C=1C=C(C(=CC=11)OC)OC)CC=C2CN2[C@@H]3[C@]41CC2 RRKTZKIUPZVBMF-IBTVXLQLSA-N 0.000 description 1
- RRKTZKIUPZVBMF-UHFFFAOYSA-N brucine Natural products C1=2C=C(OC)C(OC)=CC=2N(C(C2)=O)C3C(C4C5)C2OCC=C4CN2C5C31CC2 RRKTZKIUPZVBMF-UHFFFAOYSA-N 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 229960001631 carbomer Drugs 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 1
- 150000001733 carboxylic acid esters Chemical class 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 238000010531 catalytic reduction reaction Methods 0.000 description 1
- 239000003610 charcoal Substances 0.000 description 1
- KXZJHVJKXJLBKO-UHFFFAOYSA-N chembl1408157 Chemical compound N=1C2=CC=CC=C2C(C(=O)O)=CC=1C1=CC=C(O)C=C1 KXZJHVJKXJLBKO-UHFFFAOYSA-N 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- LOUPRKONTZGTKE-UHFFFAOYSA-N cinchonine Natural products C1C(C(C2)C=C)CCN2C1C(O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-UHFFFAOYSA-N 0.000 description 1
- 235000017803 cinnamon Nutrition 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 229960000913 crospovidone Drugs 0.000 description 1
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- KWGRBVOPPLSCSI-UHFFFAOYSA-N d-ephedrine Natural products CNC(C)C(O)C1=CC=CC=C1 KWGRBVOPPLSCSI-UHFFFAOYSA-N 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 229950006137 dexfosfoserine Drugs 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 125000004663 dialkyl amino group Chemical group 0.000 description 1
- ZWWWLCMDTZFSOO-UHFFFAOYSA-N diethoxyphosphorylformonitrile Chemical compound CCOP(=O)(C#N)OCC ZWWWLCMDTZFSOO-UHFFFAOYSA-N 0.000 description 1
- HRTDMVVQLLUHMP-UHFFFAOYSA-N diethyl 2-methoxy-2-(methylideneamino)propanedioate Chemical compound CCOC(=O)C(OC)(N=C)C(=O)OCC HRTDMVVQLLUHMP-UHFFFAOYSA-N 0.000 description 1
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical compound [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- DTGKSKDOIYIVQL-UHFFFAOYSA-N dl-isoborneol Natural products C1CC2(C)C(O)CC1C2(C)C DTGKSKDOIYIVQL-UHFFFAOYSA-N 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 230000002124 endocrine Effects 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- AFAXGSQYZLGZPG-UHFFFAOYSA-N ethanedisulfonic acid Chemical compound OS(=O)(=O)CCS(O)(=O)=O AFAXGSQYZLGZPG-UHFFFAOYSA-N 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- ZVLHUOGPYYNFIA-UHFFFAOYSA-N ethyl 1-benzyl-5-methoxyimidazole-4-carboxylate Chemical compound COC1=C(C(=O)OCC)N=CN1CC1=CC=CC=C1 ZVLHUOGPYYNFIA-UHFFFAOYSA-N 0.000 description 1
- VQSVJNWJPIVFFE-UHFFFAOYSA-N ethyl 4-[(5-carbamoyl-4-hydroxyimidazol-1-yl)methyl]benzoate Chemical compound C1=CC(C(=O)OCC)=CC=C1CN1C(C(N)=O)=C(O)N=C1 VQSVJNWJPIVFFE-UHFFFAOYSA-N 0.000 description 1
- JEUGZMVDNAEZLB-UHFFFAOYSA-N ethyl 4-[[(1,3-diamino-1,3-dioxopropan-2-yl)amino]methyl]benzoate Chemical compound CCOC(=O)C1=CC=C(CNC(C(N)=O)C(N)=O)C=C1 JEUGZMVDNAEZLB-UHFFFAOYSA-N 0.000 description 1
- LVOIKKRXQOVEPO-UHFFFAOYSA-N ethyl 5-hydroxy-1-[2-(4-methoxycarbonylphenoxy)ethyl]imidazole-4-carboxylate Chemical compound OC1=C(C(=O)OCC)N=CN1CCOC1=CC=C(C(=O)OC)C=C1 LVOIKKRXQOVEPO-UHFFFAOYSA-N 0.000 description 1
- 229960004667 ethyl cellulose Drugs 0.000 description 1
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 1
- QUSNBJAOOMFDIB-UHFFFAOYSA-O ethylaminium Chemical compound CC[NH3+] QUSNBJAOOMFDIB-UHFFFAOYSA-O 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 230000004136 fatty acid synthesis Effects 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 230000008570 general process Effects 0.000 description 1
- 230000001890 gluconeogenic effect Effects 0.000 description 1
- 239000000174 gluconic acid Substances 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 230000009229 glucose formation Effects 0.000 description 1
- 230000004190 glucose uptake Effects 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 125000000268 heptanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 description 1
- 125000003104 hexanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000013632 homeostatic process Effects 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 238000007327 hydrogenolysis reaction Methods 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 229920003063 hydroxymethyl cellulose Polymers 0.000 description 1
- 229940031574 hydroxymethyl cellulose Drugs 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- 229940031704 hydroxypropyl methylcellulose phthalate Drugs 0.000 description 1
- 229920003132 hydroxypropyl methylcellulose phthalate Polymers 0.000 description 1
- 229940071676 hydroxypropylcellulose Drugs 0.000 description 1
- 201000001421 hyperglycemia Diseases 0.000 description 1
- 230000007954 hypoxia Effects 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- JEIPFZHSYJVQDO-UHFFFAOYSA-N iron(III) oxide Inorganic materials O=[Fe]O[Fe]=O JEIPFZHSYJVQDO-UHFFFAOYSA-N 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 229960001021 lactose monohydrate Drugs 0.000 description 1
- 230000003520 lipogenic effect Effects 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 229960003511 macrogol Drugs 0.000 description 1
- 235000001055 magnesium Nutrition 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 229940091250 magnesium supplement Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 229940035034 maltodextrin Drugs 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N mandelic acid Chemical compound OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 201000000083 maturity-onset diabetes of the young type 1 Diseases 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 208000030159 metabolic disease Diseases 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- DEPYTKJPEVOSST-UHFFFAOYSA-N methyl 4-[2-(1,3-dihydroisoindol-2-yl)ethoxy]benzoate Chemical compound C1=CC(C(=O)OC)=CC=C1OCCN1CC2=CC=CC=C2C1 DEPYTKJPEVOSST-UHFFFAOYSA-N 0.000 description 1
- VNIILKHYZDHDLS-UHFFFAOYSA-N methyl 4-[2-(5-carbamoyl-4-hydroxyimidazol-1-yl)ethoxy]benzoate Chemical compound C1=CC(C(=O)OC)=CC=C1OCCN1C(C(N)=O)=C(O)N=C1 VNIILKHYZDHDLS-UHFFFAOYSA-N 0.000 description 1
- SSEJZOMGFHLEAJ-UHFFFAOYSA-N methyl 4-[2-[(2,3-diamino-3-oxopropanoyl)amino]ethoxy]benzoate Chemical compound COC(=O)C1=CC=C(OCCNC(=O)C(N)C(N)=O)C=C1 SSEJZOMGFHLEAJ-UHFFFAOYSA-N 0.000 description 1
- DOPRIRFMNIINFP-UHFFFAOYSA-N methyl 4-hydroxy-1H-imidazole-5-carboxylate Chemical class COC(=O)C=1N=CNC=1O DOPRIRFMNIINFP-UHFFFAOYSA-N 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- LNOPIUAQISRISI-UHFFFAOYSA-N n'-hydroxy-2-propan-2-ylsulfonylethanimidamide Chemical compound CC(C)S(=O)(=O)CC(N)=NO LNOPIUAQISRISI-UHFFFAOYSA-N 0.000 description 1
- YKYONYBAUNKHLG-UHFFFAOYSA-N n-Propyl acetate Natural products CCCOC(C)=O YKYONYBAUNKHLG-UHFFFAOYSA-N 0.000 description 1
- YZMHQCWXYHARLS-UHFFFAOYSA-N naphthalene-1,2-disulfonic acid Chemical compound C1=CC=CC2=C(S(O)(=O)=O)C(S(=O)(=O)O)=CC=C21 YZMHQCWXYHARLS-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 229920001206 natural gum Polymers 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 229960003512 nicotinic acid Drugs 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 150000002828 nitro derivatives Chemical class 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 231100000956 nontoxicity Toxicity 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 125000002801 octanoyl group Chemical group C(CCCCCCC)(=O)* 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 150000002940 palladium Chemical class 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 230000006611 pharmacological activation Effects 0.000 description 1
- 125000005561 phenanthryl group Chemical group 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- DGTNSSLYPYDJGL-UHFFFAOYSA-N phenyl isocyanate Chemical compound O=C=NC1=CC=CC=C1 DGTNSSLYPYDJGL-UHFFFAOYSA-N 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 239000010773 plant oil Substances 0.000 description 1
- 210000002381 plasma Anatomy 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 235000013809 polyvinylpolypyrrolidone Nutrition 0.000 description 1
- 229920000523 polyvinylpolypyrrolidone Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- FYRHIOVKTDQVFC-UHFFFAOYSA-M potassium phthalimide Chemical compound [K+].C1=CC=C2C(=O)[N-]C(=O)C2=C1 FYRHIOVKTDQVFC-UHFFFAOYSA-M 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229940090181 propyl acetate Drugs 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 235000018102 proteins Nutrition 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 229960000948 quinine Drugs 0.000 description 1
- 239000004172 quinoline yellow Substances 0.000 description 1
- IZMJMCDDWKSTTK-UHFFFAOYSA-N quinoline yellow Chemical compound C1=CC=CC2=NC(C3C(C4=CC=CC=C4C3=O)=O)=CC=C21 IZMJMCDDWKSTTK-UHFFFAOYSA-N 0.000 description 1
- 229940051201 quinoline yellow Drugs 0.000 description 1
- 235000012752 quinoline yellow Nutrition 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 108091006091 regulatory enzymes Proteins 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 125000003607 serino group Chemical group [H]N([H])[C@]([H])(C(=O)[*])C(O[H])([H])[H] 0.000 description 1
- AWUCVROLDVIAJX-GSVOUGTGSA-N sn-glycerol 3-phosphate Chemical compound OC[C@@H](O)COP(O)(O)=O AWUCVROLDVIAJX-GSVOUGTGSA-N 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 229960004025 sodium salicylate Drugs 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 229940071117 starch glycolate Drugs 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 125000000446 sulfanediyl group Chemical group *S* 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000004149 thio group Chemical group *S* 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 229910052723 transition metal Inorganic materials 0.000 description 1
- 150000003624 transition metals Chemical class 0.000 description 1
- UFTFJSFQGQCHQW-UHFFFAOYSA-N triformin Chemical compound O=COCC(OC=O)COC=O UFTFJSFQGQCHQW-UHFFFAOYSA-N 0.000 description 1
- IHIXIJGXTJIKRB-UHFFFAOYSA-N trisodium vanadate Chemical compound [Na+].[Na+].[Na+].[O-][V]([O-])([O-])=O IHIXIJGXTJIKRB-UHFFFAOYSA-N 0.000 description 1
- DCXXMTOCNZCJGO-UHFFFAOYSA-N tristearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCCCCCCCCCC)COC(=O)CCCCCCCCCCCCCCCCC DCXXMTOCNZCJGO-UHFFFAOYSA-N 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 125000003774 valeryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/90—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
Definitions
- the present invention relates to the use of imidazole derivatives as AMP-acti- vated protein kinase (AMPK) activators, for the prevention of or treating pathologies, such as diabetes, metabolic syndrome and obesity.
- AMPK AMP-acti- vated protein kinase
- the invention also relates to processes for the preparation of the said derivatives, to pharmaceutical compositions com- prising them and to the use of these derivatives for the preparation of medicaments.
- AMPK is a sensor and a regulator of the homeostasis of cellular energy " (Hardie D. G. and Hawley S. A., "AMP-activated protein kinase: the energy charge hypothesis revisited", Bioassays, 23, (2001), 1112; Kemp B. E. et al., "AMP-activated protein kinase, super metabolic regulator", Biochem. Soc. Transactions, 31, (2003), 162). Allosteric activation of this kinase originating from an increase in the level of AMP takes place under conditions of cellular energy depletion.
- AMPK activation results in phosphorylation of the serine/threonine residues of the target enzymes, which leads to an adaptation of the cellular metabolism towards lower energy states.
- the most marked effect of the changes induced by AMPK activation is inhibition of the process of ATP consumption and activation of ATP generation, the consequence being regeneration of the stock of ATP.
- Examples of AMPK substrates include acetyl- CoA-carboxylase (ACC) and HMG-CoA-reductase (Carting D. et al., "A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis", FEBS Letters, 223, (1987), 217).
- Phosphorylation leads to a reduction in fatty acid synthesis (consumption of ATP) and simultaneously to an increase in fatty acid oxidation (generation of ATP). Phosphorylation, and the inhibition of HMG-CoA reductase resulting therefrom, leads to a decrease in cholesterol synthesis.
- Other AMPK substrates include hormone-sensitive lipase (Garton A. J. et al., "Phosphorylation of bovine hormone-sensitive lipase by the AMP-activated protein kinase. A possible antilipolytic mechanism", Eur. J.
- AMPK adipocyte hormone that stimulates stimulation of AMPK and, consequently, leads to an increase in fatty acid oxidation in skeletal muscle (Mino- koshi Y. et al., "Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase", Nature, 415, (2002), 339). It has also been shown that adiponectin, another adipocyte hormone that results in an improvement in carbohydrate and fat metabolism, stimulates AMPK in the liver and skeletal muscle (Yamauchi T.
- AMPK activation appears to be independent of the increase in the cellular levels of AMP, and is thought to be due rather to a phosphorylation by one or more kinases that have not been identified to date.
- AMPK activation beneficial effects of in vivo AMPK activation can be envisaged.
- a reduction in the expression of the glucogenesis enzymes should result in a decrease in glucose production in the liver and an improvement in glucose homeostasis, whereas the inhibition and/or reduction of expression of the key enzymes of fat metabolism would lead to a reduction in fatty acid and cholesterol synthesis and to an increase in fatty acid oxidation.
- the present invention relates to AMPK-activating imidazole derivatives that can be used in the treatment of diabetes and related pathologies.
- the invention relates firstly to the use of the imidazole derivatives of the general formula (1) below:
- A represents -NH- or -O-;
- R 1 is chosen from:
- heteroaryl group of each of these groups itself being optionally substituted by one or more groups Y, which may be identical or different, it being understood that the said heteroaryl group may contain one or more heteroatoms chosen from nitrogen, oxygen and sulfur;
- R 2 is chosen from:
- C 3 -Cio)cycloalkyl optionally substituted by one or more groups, which may be identical or different, chosen from cycloalkyl, alkoxy, carboxyl and alkylcarbonyl;
- R 3 represents hydrogen or linear or branched (Ci-C 8 )alkyl, optionally substituted by one or more groups, which may be identical or different, chosen from cycloalkyl, alkoxy, carboxyl and alkylcarbonyl;
- Y is chosen from hydrogen, amino, nitro, hydroxyl, cyano, thio, halogen, (Ci-C 8 )- alkyl, (Ci-C 8 )alkoxy, (d-C 8 )alkoxycarbonyl, (Ci-C 8 )alkylthio, (Ci-C 8 )alkylamino, (C 6 -Ci 4 )aryl, (C 6 -Ci 4 )aryloxy and (C 6 -Ci 4 )aryl(Ci-C 8 )alkoxy,
- T is chosen from:
- heteroaryl group of each of these groups itself possibly being substituted by one or more groups independently chosen from halogen, (CrCsJalkoxy, (C 3 -C 8 )cycloalkyl(Ci-Cs)alkyl, (Ci-C 8 )alkoxycarbonyl, hydroxycarbonyl and (Ci-C 8 )alkylthio; it being understood that the said heteroaryl group may contain one or more heteroatoms chosen from nitrogen, oxygen and sulfur.
- the invention also relates to the use of the possible geometrical and/or optical isomers, epimers and tautomeric forms, possible oxidised forms, especially amine oxides, thioethers and hydrates of the compounds of the formula (1) defined above, and also to the use of the possible salts thereof with a pharmaceutically acceptable acid or base, or the pharmaceutically acceptable prodrugs of these compounds.
- the dashed line indicates the presence of a double bond between one of the two nitrogen atoms and the carbon atom bearing the hydrogen atom.
- the compounds of the formula (1) can thus be represented either by the formula (1i) or by the formula (1 2 ):
- (Ci-C 8 )alkyl denotes a linear or branched alkyl radical containing from 1 to 8 carbon atoms.
- CrC 8 alkyl radicals mention may be made especially of methyl, ethyl, propyl, isopropyl, butyl, sec-butyl, tert- butyl, pentyl, hexyl and octyl radicals;
- (Ci-C 8 )acyl denotes a group of the formula in which R' represents hydrogen or a linear or branched hydrocarbon-based radical containing from 1 to 7 carbon atoms.
- acyl radicals containing from 1 to 8 carbon atoms include formyl, acetyl, propionyl, butanoyl, pentanoyl, hexanoyl, heptanoyl, octanoyl, isopropionyl, isobutanoyl and 2,2-dimethylacetyl radicals;
- alkoxy refers to the term "alkyl-oxy";
- halogen(s) refers in a non-limiting manner to fluorine, chlorine or bromine
- (C 6 -Ci4)aryl refers to a monocyclic or polycyclic aromatic group containing from 6 to 14 carbon atoms, at least one of the rings having a system of con- jugated ⁇ electrons, and includes biaryls that may be optionally substituted, as indicated hereinabove for the aryls. Mention will be made in particular of biphenyl, phenyl, naphthyl, anthryl, phenanthryl, indanyl and tetralyl radicals;
- hetero(C 6 -Ci4)aryl refers to a 6- to 14-membered monocyclic or polycyclic aromatic heterocycle containing from 1 to 4 heteroatoms, the other atoms being carbon atoms.
- heteroatoms that will be mentioned in particular are oxygen, sulfur and nitrogen.
- heteroaryl radicals that will be mentioned more particularly are furyl, thienyl, pyridyl, pyrrolyl, pyrimidyl, pyrazinyl, oxazolyl, oxadiazolyl, isoxazolyl, quinolyl and thiazolyl radicals;
- (C 3 -Cio)cycloalkyl refers to a saturated hydrocarbon-based ring and comprises monocyclic, bicyclic or polycyclic radicals containing from 3 to 10 carbon atoms. Mention will be made in a non-limiting manner of cyclopropyl, cyclobutyl, cyclo- pentyl and cyclohexyl radicals.
- the bases that can be used for the formation of salts of compounds of the formula (1) are organic or mineral bases.
- the resulting salts are, for example, the salts formed with metals, and especially alkali metals, alkaline-earth metals and transition metals (such as sodium, potassium, calcium, magnesium or aluminium) or with bases, for instance ammonia or secondary or tertiary amines (such as diethylamine, triethyl- amine, piperidine, piperazine or morpholine) or with basic amino acids, or with osamines (such as meglumine) or with amino alcohols (such as 3-aminobutanol and 2-aminoetha- nol).
- alkali metals alkaline-earth metals and transition metals (such as sodium, potassium, calcium, magnesium or aluminium)
- bases for instance ammonia or secondary or tertiary amines (such as diethylamine, triethyl- amine, piperidine, piperazine
- the acids that can be used for the formation of salts of compounds of the formula (1) are mineral or organic acids.
- mineral acids that will be mentioned, by way of example and in a non-limiting manner, are the following mineral acids: sulfuric acid, nitric acid, hydrochloric acid, hydrobromic acid, phosphoric acid, sulfamic acid.
- organic acids that will be mentioned, by way of example and in a non-limiting manner, are the following organic acids: formic acid, acetic acid, propionic acid, pivalic acid, diethylacetic acid, malonic acid, succinic acid, pimelic acid, fumaric acid, maleic acid, lactic acid, tartaric acid, malic acid, citric acid, gluconic acid, ascorbic acid, nicotinic acid, isonicotinic acid, methanesulfonic acid, ethanesulfonic acid, ethanedisulfonic acid, 2-hydroxyethanesulfonic acid, benzenesulfonic acid, para-toluenesulfonic acid, naph- thalenemonosulfonic acid, naphthalenedisulfonic acid, laurylsulfuric acid.
- formic acid acetic acid, propionic acid, pivalic acid, diethylacetic acid, malonic acid, succinic acid,
- the invention also relates to the chiral salts used for the separation of race- mates.
- the following chiral acids are used: (+)-D-di-O-benzoyl- tartaric acid, (-)-L-di-O-benzoyltartaric acid, (-)-L-di-O,O'-p-toluyl- ⁇ _-tartaric acid, (+)-D-di- O,O'-p-toluyl-L-tartaric acid, (R)-(+)-ma ⁇ c acid, fSH-)-malic acid, (+)-camphanic acid, (-)- camphanic acid, f?-(-)-1 ,1'-binaphthalene-2,2'-diylhydrogenophosphonic acid, ⁇ -camphoric acid, (-)-camphoric acid, CS > )-(+)-2-phenylpropionic acid, (f?H + )-2
- the chiral acid is preferentially chosen from (-)-di-O,O'-p-toluyl-L-tartaric acid, (+)-D-di-O,O'-p-toluyl-D-tartaric acid, (R>(-)- 1 . 1 l -binaphthalene-2,2'-diyl hydrogen phosphate, L-tartaric acid and D-tartaric acid, or a mixture of two or more thereof.
- Chiral amines may also optionally be used, for example quinine, brucine, (S)-1- (benzyloxymethyl)propylamine (III), (-)-ephedrine, (4S,5R)-(+)-1 ,2,2,3,4-tetramethyl-5- phenyl-1 ,3-oxazolidine, (R)-1-phenyl-2-p-tolylethylamine, (S)-phenylglycinol, (-)-N- methylephedrine, (+)-(2S,3R)-4-dimethylamino-3-methyl-1 ,2-diphenyl-2-butanol, (S)- phenylglycinol or (S)- ⁇ -methylbenzylamine, or a mixture of two or more thereof.
- quinine brucine
- (-)-ephedrine (4S,5R)-(
- the compounds of the formula (1) above also comprise the prodrugs of these compounds.
- prodrugs means compounds which, once administered into a biological system, are transformed into compounds of the formula (1) (biologically active compound), chemically and/or biologically, via spontaneous chemical reaction(s), via chemical reaction(s) catalysed with one or more enzymes and/or via metabolic chemical reaction (s).
- the conventional "prodrugs” are formed using groups linked to a functional group, such as hydroxyl, thio, carboxyl, alkylcarbonyl, amino, alkylamino or dialkylamino, associated with the AMPK activator, and which become separated in vivo.
- Non-limiting examples of conventional "prodrugs” include carboxylic esters, in which the ester group is alkyl, aryl, aralkyl, acyloxyalkyl or alkoxy- carbonyloxyalkyl, and also the esters of hydroxyl, thio and amine groups, in which the attached group is an acyl, an alkoxycarbonyl, an aminocarbonyl, a phosphate or a sulfate.
- the prodrugs should undergo at least one chemical transformation to produce the compound that is biologically active, or alternatively should be a precursor of the biologi- cally active compound. In certain cases, the prodrugs is biologically active, but generally, however, less than the compound itself, and is useful for improving the efficacy or the non-toxicity by means of improving the oral bioavailability, the pharmacodynamic half-life and the like.
- patent US-4 140 788 describes imidazole derivatives used in the treatment of "Sarcoma 180" tumour and corresponding to the general formula (1) in which A represents -NH-, and R 1 and R 3 represent, independently of each other, hydrogen, (C 1 - C 3 )alkyl, (C 3 -C 5 )alkenyl, or benzyl optionally substituted by (Ci-C 3 )alkoxy.
- the present invention also relates to the novel imidazole derivatives of the general formula (1'):
- A represents -NH- or -O-;
- R' 1 is chosen from:
- Cs-C ⁇ JalkyI optionally substituted by one or more groups, which may be identical or different, chosen from cycloalkyl, alkoxy, carboxyl and alkylcarbonyl;
- heteroaryl group of each of these groups itself being optionally substituted by one or more groups Y 1 , which may be identical or different, it being understood that the said heteroaryl group may contain one or more heteroatoms chosen from nitrogen, oxygen and sulfur; * R' 2 is chosen from:
- - linear or branched (Ci-C 8 )alkyl optionally substituted by one or more groups, which may be identical or different, chosen from cycloalkyl, alkoxy, carboxyl and alkylcarbonyl;
- - (C 3 -C 10 )cycloalkyl optionally substituted by one or more groups, which may be identical or different, chosen from cycloalkyl, alkoxy, carboxyl and alkylcarbonyl;
- (C 2 -C 14 )acyl optionally substituted by one or more groups independently chosen from amino, hydroxyl, cyano, thio, halogen, (Ci-C 8 )alkyl, (Ci-C ⁇ )alkoxy, (Ci-C 8 )alkylthio, (Ci-C 8 )alkylamino, (C 6 -Ci 4 )aryl, (C 6 -Ci 4 )aryloxy, (C 6 -Ci 4 )- aryl(C r C 8 )alkoxy and Y'; and
- R 3 represents hydrogen or linear or branched (Ci-C 8 )alkyl, optionally substituted by one or more groups, which may be identical or different, chosen from cycloalkyl, alkoxy, carboxyl and alkylcarbonyl;
- Y is chosen from hydrogen, amino, nitro, hydroxyl, cyano, thio, halogen, (Ci-C 8 )alkyl, (C r C 8 )alkoxy, (Ci-C 8 )alkoxycarbonyl, (Ci-C 8 )alkylthio, (Ci-C 8 )alkylamino,
- T is chosen from: - optionally substituted linear or branched (CrCsJalkyl;
- (Ci-C 8 )alkyl the heteroaryl group of each of these groups itself possibly being substituted by one or more groups independently chosen from halogen, (C 1 -C 8 )alkoxy, (C 3 -C 8 )cycloalky!(Ci-C 8 )alkyl, (Ci-C 8 )alkoxycarbonyl, hydroxycarbonyl and (C-i-C8)alkylthio; it being understood that the said heteroaryl group may contain one or more heteroatoms chosen from nitrogen, oxygen and sulfur; with the restriction that if R' 2 represents hydrogen, then R' 1 cannot represent benzyl or benzyl substituted in position 4 with nitro or methoxy, and the possible geometrical and/or optical isomers, epimers and tautomeric forms, possible oxidised forms, especially amine oxide, thioethers and hydrates thereof, and also the possible addition salts thereof with a pharmaceutically acceptable acid or base, or alternatively the pharmaceutically acceptable prodrugs of these compounds.
- the term "compound(s) of the formula (1)” means “compound(s) of the formula (1) or of the formula (V)".
- the compounds of the formula (1) for which A represents -NH-, and more preferably the compounds of the formula (1) for which A represents -NH- and R 3 represents hydrogen are preferred.
- the compounds of the formula (1) for which A represents -O- are preferred.
- a first preferred subgroup according to the invention corresponds to the compounds of the formula (1') in which A represents -NH- and R' 3 represents hydrogen, the said subgroup being represented by formula (1A):
- R' 1 and R' 2 are as defined above for the compounds of the formula (1'), with the restriction that if R' 2 represents hydrogen, then R' 1 cannot represent benzyl or benzyl substituted in position 4 with nitro or methoxy, the possible geometrical and/or optical isomers, epimers and tautomeric forms thereof, possible oxidised forms, especially amine oxide or thioethers, and the solvates and hydrates of these compounds; and also the possible salts thereof with a pharmaceutically acceptable acid or base, or alternatively the pharmaceutically acceptable prodrugs of these compounds.
- a second preferred subgroup corresponds to the compounds of the formula (1') according to the invention, in which -A- is -O-, and represented by formula (1B):
- R' 1 and R' 2 are as defined above, and R l3B represents (Ci-C 8 )alkyl, with the restriction that if R' 2 represents hydrogen, then R' 1 cannot represent benzyl or benzyl substituted in position 4 with nitro or methoxy, the possible geometrical and/or optical isomers, epimers and tautomeric forms thereof, possible oxidised forms, especially amine oxide or thioethers, and the solvates and hydrates of these compounds; and also the possible salts thereof with a pharmaceutically acceptable acid or base, or alternatively the pharmaceutically acceptable prodrugs of these compounds.
- a third preferred subgroup corresponds to the compounds of the formula (1'), according to the invention, represented by formula (1C):
- R' 1 is as defined above and R l2C is chosen from hydrogen, (Ci-C ⁇ )alkyl and (C 2 -C 14 )acyl, with the restriction that if R' 2 represents hydrogen, then R' 1 cannot represent benzyl or benzyl substituted in position 4 with nitro or methoxy, the possible geometrical and/or optical isomers, epimers and tautomeric forms thereof, possible oxidised forms, especially amine oxide or thioethers, and the solvates and hydrates of these compounds; and also the possible salts thereof with a pharmaceutically acceptable acid or base, or alternatively the pharmaceutically acceptable prodrugs of these compounds.
- R l2C is chosen from hydrogen, (Ci-C ⁇ )alkyl and (C 2 -C 14 )acyl, with the restriction that if R' 2 represents hydrogen, then R' 1 cannot represent benzyl or benzyl substituted in position 4 with nitro or methoxy, the possible geometrical and/or optical isomers, epimers and
- R' 2C represents hydrogen, (Ci-C 8 B ))aallkkyyll oorr (((C 2 -Ci 4 )acyl and Y' is as de- fined above for the compounds of the formula (1'), with the restriction that if R' 2 represents hydrogen, then Y' cannot represent hydrogen, nitro or methoxy, the possible geometrical and/or optical isomers, epimers and tautomeric forms thereof, possible oxidised forms, especially amine oxide or thioethers, and the solvates and hydrates of these compounds, and also the possible salts thereof with a pharmaceutically acceptable acid or base, or alternatively the pharmaceutically acceptable prodrugs of these compounds.
- a more particularly preferred subgroup among the compounds of the formula (1Ca) is defined by the compounds represented by formula (1Caa):
- R >2C represents hydrogen, (Ci-C 8 )alkyl or (C 2 -Ci 4 )acyl and T is as defined above for the compounds of the formula (1'), the possible geometrical and/or optical isomers, epimers and tautomeric forms thereof, possible oxidised forms, especially amine oxide or thioethers, and the solvates and hydrates of these compounds; and also the possible salts thereof with a pharmaceutically acceptable acid or base, or alternatively the pharmaceutically acceptable prodrugs of these compounds.
- R' 2C represents hydrogen, (C- ⁇ -C ⁇ )alkyl or (C ⁇ -C-uJacyl and T' is as defined above for the compounds of the formula (1'), the possible geometrical and/or optical isomers, epimers and tautomeric forms thereof, possible oxidised forms, especially amine oxide or thioethers, and the solvates and hydrates of these compounds; and also the possible salts thereof with a pharmaceutically acceptable acid or base, or alternatively the pharmaceutically acceptable prodrugs of these compounds.
- the preferred compounds of the formula (1) are chosen from: 01) ethyl 5-hydroxy-1-(2-phenoxyethyl)-1H-imidazole-4-carboxylate;
- the present invention also relates to a process for the preparation of these compounds of the formula (1) or (1'), characterised in that a compound of the formula (4a) or (4b):
- R 1 and R 3 are as defined above, is used in a cyclisation reaction, the said cyclisation reaction being performed according to known methods, for example with ethyl orthoformate [HC(OC2H 5 ) 3 ], to give the imidazoles of the formula (1OH): corresponding to the compounds of the formula (1), in which R 2 represents hy- drogen, in which imidazoles of the formula (1OH) the hydroxyl group may be etherified, according to standard techniques known to those skilled in the art, for example using a reagent of the formula R-X, in which R has the same definition as R 2 defined above, with the exception of hydrogen, and X represents a halogen atom, to give the compounds of the formula (1R): the set of compounds of the formulae (1OH) and (1R) forming the set of compounds of the formula (1).
- the starting reagent (4) is either commercially available or obtained via standard chemical synthetic routes known to those skilled in the art.
- G represents a protecting group chosen from Z (benzyloxycarbonyl), BOC (tert- butoxycarbonyl) and Fmoc (fluorenemethoxycarbonyl).
- the starting malonate (O) was prepared according to the method described in J. Org. Chem., 54(6), (1989), 1364-70.
- the compounds of the formulae (1A R ) and (1 A O H) form the set of compounds of the formula (1A) defined above and are obtained in four steps from the malonate (O):
- the preferred agents are, for example, dicyclohexylcarbodiimide (DCC), diethyl cyanophosphate (DEPC), carbonyldiimidazole, diphenylphosphorylazide (DPPA), or more particularly the dicyclohexylcarbodiimide/1-hydroxybenzotriazole (HBTO) couple, or the diethyl azo- dicarboxylate/triphenylphosphine couple or the dicyclohexylcarbodiimide/1-hydroxy- benzotriazole couple.
- the dicyclohexylcarbodiimide/1-hydroxybenzotriazole couple is more particularly preferred.
- the reaction is preferably performed in the presence of an inert solvent, such as aromatic hydrocarbons, for example benzene, toluene or xylene; a halogenated hydrocarbon and in particular aliphatic halogenated hydrocarbons, for in- stance methylene chloride, dichloroethane or chloroform; esters, for instance ethyl acetate or propyl acetate, ethers, such as diethyl ether, tetrahydrofuran and dioxane; amides, such as dimethylformamide, dimethylacetamide or hexamethylphosphorotriamide; and nitriles, such as acetonitrile.
- aromatic hydrocarbons for example benzene, toluene or xylene
- a halogenated hydrocarbon and in particular aliphatic halogenated hydrocarbons for in- stance methylene chloride, dichloroethane or chloroform
- esters for instance
- Ethers such as tetrahydrofuran, halogenated hydro- carbons, such as methylene chloride, amides, such as dimethylformamide and esters, such as ethyl acetate are more particularly preferred.
- the reaction can take place over a wide temperature range, in particular between -10 0 C and 50 0 C and preferably between - 1O 0 C and room temperature.
- the reaction time varies according to many factors and in particular the reaction temperature and the reagents used. It is preferably between 30 minutes and 24 hours;
- the compound of the formula (3A) is obtained via the action of ammonia on compound (2A), in a solvent, such as an alcohol, for example methanol or ethanol, at a temperature of between O 0 C and 30 0 C and preferably between 1O 0 C and room temperature.
- a solvent such as an alcohol, for example methanol or ethanol
- the reaction time varies according to many factors and in particular the reaction temperature and the reagents used. It is preferably between 30 minutes and 24 hours;
- the compound of the formula (4A) is obtained by deprotection ("cleavage") of the protecting group G, for example via hydrogenolysis according to known techniques, for example as described in Greene et al., "Protective Groups in Organic Synthesis", 2 nd ed., John Wiley & Sons, New York, 1999, 17-292; - the cyclisation of (4A) to (1A O H) is performed according to Tarumi et al.
- the amino malonate (2B) is either obtained from commercial sources or prepared according to the literature (for example according to EP 811 602).
- Compound (3B) is obtained from (2B) according to Lim et al. (J. Org. Chem., 50(25), (1985), 5111-5115).
- the cyclisation reaction of (3B) with (5) to give (1B O H) is also performed according to Lim et al. (ibid.) (the acetonitrile or methanol can advantageously be replaced with ethyl acetate), and the reaction time is preferably between 2 hours and 24 hours.
- Compound (1 B O H) is generally obtained in the form of the ammonium salt (1B 0 ) with the amine (5):
- (1 B 0 ) By treating an aqueous solution of (1Bo) with an acid, and more particularly hydrochloric acid, (1 BOH) is obtained, and is then converted into (1 BR) according to Atsumi et al. (US 4 140788), in which R' has the same definition as R' 2 ' with the exception of hydrogen, the set of compounds of the formulae (1 BOH) and (1 BR) forming the set of compounds of the formula (1 B).
- the coupling of R' 1 NH 2 with (3C) is preferably performed in an anhydrous alcoholic solvent (for example methanol or ethanol), in the presence of a base, such as triethylamine or pyridine, for a time preferably of between 2 hours and 8 hours, and at a temperature of between room temperature and the reflux temperature of the solvent.
- a base such as triethylamine or pyridine
- the cyclisation of (3C) is performed with an excess of ethyl orthoformate, for example between 4 and 10 equivalents relative to (3C), in an anhydrous alcoholic solvent, such as those defined above, for a time preferably of between 2 hours and 8 hours and at a temperature of between room temperature and the reflux temperature of the solvent.
- the set of compounds of the formulae (1 CQ H ) and (1 CR) forms the set of com- pounds of the formula (1C).
- the compounds of the formulae (1Caa) and (1Cb) defined above can also be prepared according to the following reaction scheme:
- compound (2C) is reacted with TBDMS in a solvent, such as dimethylformamide, toluene or acetone, in the presence of imidazole (1 eq.), at a temperature of between 20 0 C and 80 0 C, for a time of between 2 hours and 8 hours.
- a solvent such as dimethylformamide, toluene or acetone
- Compound (4C) is placed in a solvent, such as anhydrous tetrahydrofuran, dimethylformamide, toluene or the like. It is then treated, according to the known techniques of organic chemistry, with an acyl chloride, at a temperature preferably of between 20 0 C and the reflux temperature of the solvent, and for a time preferably of be- tween 2 hours and 72 hours. Compound (5Ca) is thus obtained, and is not isolated.
- a solvent such as anhydrous tetrahydrofuran, dimethylformamide, toluene or the like. It is then treated, according to the known techniques of organic chemistry, with an acyl chloride, at a temperature preferably of between 20 0 C and the reflux temperature of the solvent, and for a time preferably of be- tween 2 hours and 72 hours.
- Cleavage of the silyl ether is then performed by treating compound (5Ca) in acidic medium according to the known techniques of organic chemistry, to give compound (5CaoH), which is then optionally treated with a compound of the formula R'-X, as indicated previously, to give compound (5Ca R ).
- the set of compounds of the formulae (5CaoH) and (5Ca R ) forms the set of compounds of the formula (5Ca).
- the present invention also relates to pharmaceutical preparations comprising at least one compound of the formula (I) and/or a pharmaceutically acceptable salt thereof.
- the present invention also relates to pharmaceutical compositions comprising a pharmaceutically effective amount of at least one compound of the formula (I) and/or a salt thereof, in combination with one or more pharmaceutically acceptable vehicles.
- compositions of the invention comprise formulations, such as granules, powders, tablets, gel capsules, syrups, emulsions and suspensions, and also the forms used for non-oral administration, for example injections, sprays, suppositories and the like.
- the pharmaceutical forms can be prepared via known conventional techniques.
- the preparation of a solid pharmaceutical form for oral administration can be performed, for example, according to the following process: an excipient (for example lactose, sucrose, starch, mannitol and the like), a disintegrant (for example calcium carbonate, calcium carboxymethylcellulose, alginic acid, sodium carboxymethylcellulose, colloidal silicon dioxide, sodium croscarmellose, crospovidone, guar gum, magnesium aluminium silicate, microcrystalline cellulose, cellulose powder, pregelatinised starch, sodium alginate, starch glycolate and the like), a binder (for example ⁇ -starch, gum ara- bic, carboxymethylcellulose, polyvinylpyrrolidone, hydroxypropylcellulose, alginic acid, carbomer, dextrin, ethylcellulose, sodium alginate, maltodextrin, liquid glucose, magnesium aluminium silicate, hydroxyethylcellulose, methylcellulose, guar gum
- the tab- lets can be coated via the known techniques, in order to mask the taste (for example with cocoa powder, mint, borneol, cinnamon powder, etc.) or to allow enteric dissolution or to allow sustained release of the active principles.
- the coating products that can be used are, for example, ethylcellulose, hydroxymethylcellulose, polyoxyethylene glycol, cellulose acetophthalate, hydroxypropylmethylcellulose phthalate and Eudragit ® (methacrylic acid/acrylic acid copolymer) or Opadry ® (hydroxypropylmethylcellulose + macrogol + titanium oxide + lactose monohydrate).
- Pharmaceutically acceptable colorants can also be added (for example yellow iron oxide, red iron oxide, quinoline yellow lake, etc.).
- compositions such as tablets, powders, sachets and gel capsules can be used for oral administration.
- the liquid pharmaceutical forms for oral administration comprise solutions, sus- pensions and emulsions.
- the aqueous solutions can be obtained by dissolving the active principles in water, followed by addition of flavourings, colorants, stabilisers and thickeners, if necessary. In order to improve the solubility, it is possible to add ethanol, propylene glycol or other pharmaceutically acceptable non-aqueous solvents.
- the aqueous suspensions for oral use can be obtained by dispersing the finely divided active principles in water with a viscous product, such as natural or synthetic gums, resins, methylcellulose or sodium carboxymethylcellulose.
- the pharmaceutical forms for injection can be obtained, for example, according to the following process: the active principle(s) is (are) dissolved, suspended or emulsi- fied either in an aqueous medium (for example distilled water, physiological saline, Ringer's solution, etc.) or in an oily medium (for example a plant oil, such as olive oil, sesame seed oil, cottonseed oil, corn oil, etc.
- an aqueous medium for example distilled water, physiological saline, Ringer's solution, etc.
- an oily medium for example a plant oil, such as olive oil, sesame seed oil, cottonseed oil, corn oil, etc.
- a dispersant for example Tween 80, HCO 60 (Nikko Chemicals), polyethylene glycol, carboxymethylcellulose, sodium alginate, etc.
- a preserving agent for example methyl para-hydroxy- benzoate, propyl para-hydroxybenzoate, benzyl alcohol, chlorobutanol, phenol, etc.
- an isotonic agent for example sodium chloride, glycerol, sorbitol, glucose, etc.
- a solubilising agent for example sodium salicylate, sodium acetate, etc.
- a stabiliser for example human serum albumin
- a pharmaceutical form for external use can be obtained from a solid, semi-solid or liquid composition comprising the active principle(s).
- the active principle(s) alone or mixed with excipients (for example lactose, man- nitol, starch, microcrystalline cellulose, sucrose, etc.) and a thickener (for example natural gums, cellulose derivatives, acrylic polymers, etc.) are treated so as to transform them into powder.
- excipients for example lactose, man- nitol, starch, microcrystalline cellulose, sucrose, etc.
- a thickener for example natural gums, cellulose derivatives, acrylic polymers, etc.
- the liquid pharmaceutical compositions are prepared in substantially the same manner as the forms for injection as indicated above.
- the semi-solid pharmaceutical forms are preferentially in the form of an aqueous or oily gel or in the form of an ointment.
- compositions may optionally comprise a pH regulator (for example carbonic acid, phosphoric acid, citric acid, hydrochloric acid, sodium hydroxide, etc.) and a preserving agent (for example para-hydroxybenzoic acid esters, chlorobutanol, benz- alkonium chloride, etc.), and also other additives.
- a pH regulator for example carbonic acid, phosphoric acid, citric acid, hydrochloric acid, sodium hydroxide, etc.
- a preserving agent for example para-hydroxybenzoic acid esters, chlorobutanol, benz- alkonium chloride, etc.
- Insulin resistance is characterised by a reduction in the action of insulin (cf. Presse Medicate, 26(14), (1997), 671-677) and is involved in many pathological conditions, such as diabetes and more particularly non-insulin-dependent diabetes (type Il diabetes or NIDDM), dyslipidaemia, obesity, arterial hypertension, and also certain cardiac, microvascular and macrovascular complications, for instance atherosclerosis, retinopathy and neuropathy.
- type Il diabetes or NIDDM non-insulin-dependent diabetes
- dyslipidaemia for instance atherosclerosis, retinopathy and neuropathy.
- atherosclerosis retinopathy and neuropathy
- the aim of the present invention is to propose a pharmaceutical composition comprising at least one compound of the formula (1) or (T) for significantly improving the condition of a diabetic patient.
- the pharmaceutical compositions of the invention especially have hypoglycae- miant activity.
- the compounds of the formula (1) or (T) are therefore useful in the treatment of pathologies associated with hyperglycaemia.
- the effective doses and posologies for administration of the com- pounds of the invention intended for the prevention of and treating a disorder or condition caused by or associated with modulation of AMPK activity, depends on a great many factors, for example on the nature of the compound, the size of the patient, the desired aim of the treatment, the nature of the pathology to be treated, the specific pharmaceutical composition used, and the observations and conclusions of the treating physician.
- a suitable posology of the compounds is between about 0.1 mg/kg and about 100 mg/kg of body weight per day, preferably between about 0.5 mg/kg and about
- suitable dosages of the compounds of the formula (1) or (1 ') will be between about 1-10 mg and 1000-10 000 mg per day, preferably between about 5-50 mg and 500-5000 mg per day and preferentially between 10-100 mg and 100-1000 mg per day of active material comprising a compound of the formula (1) according to the invention.
- dosage ranges represent the total amounts of active material per day for a given patient.
- the number of administrations per day for which a dose is administered may vary within wide proportions as a function of pharmacokinetic and pharmacological factors, such as the half-life of the active material, which reflects its rate of catabolism and of clearance, and also the minimum and optimum levels of the said active material reached in blood plasma or other body fluids of patients and which are required for therapeutic efficacy.
- NMR The NMR spectra were obtained using a Br ⁇ ker Advanced DPX 200 MHz NMR spectrometer or a Br ⁇ ker Advanced DPX 500 MHz NMR spectrometer. Mass: The masses were determined by HPLC coupled to an Agilent 1100 Series mass detector.
- THF Tetrahydrofuran
- TBDMS terf-Butyldimethylsilyl; s: singlet; d: doublet; t: triplet; q: quartet; o: octet; m: complex peak.
- the medium When hot, the medium becomes pale yellow and clear, and then opacifies again (about 30 minutes after the start of refluxing) and becomes white.
- the insoluble matter (phthalide hydrazide) is filtered off and washed with water.
- the malonate monoester (O) (7.5 g), the amine (5A)-1 (4.14 g) in ethanolic solution (30%) and hydroxybenzotriazole (HOBT) (3.97 g) in 70 ml of THF are placed in a 500 ml three-necked flask. After complete dissolution, the whole is cooled to -7°C and DCC (5.5 g) diluted in 30 ml of THF is then added dropwise over 30 minutes.
- the mixture is allowed to warm to room temperature and is then left stirring for 19 hours. At room temperature, the medium becomes cloudy (white precipitate).
- the precipitate (dicyclohexyl urea) is filtered off and concentrated to dryness. 15.77 g of a beige-coloured solid are obtained.
- the amide (2A)-2 (6 g; 15 mmol) is dissolved in methanol (60 ml) in a 250 ml round-bottomed flask.
- the amine (3A)-2 (3 g; 7.7 mmol) is dissolved in methanol (170 ml) in a 500 ml conical flask.
- stirring is stopped to flush the medium with argon, and the contents of the conical flask are then filtered through a Clarcel filter, and the methanol is concentrated.
- Example 1 1 -[2-(4-fluorophenoxy)ethyl]-5-hydroxy-1 H-imidazole-4-carbox- amide
- the reagents (1 eq. of 2-amino-N-[2-(4-fluorophenoxy)ethyl]malonamide 3 per
- Reaction medium temperature 130 0 C
- the amine (18.1 g; 103.3 mmol) in 100 ml of acetonitrile is added dropwise over 5 hours.
- the acid form (formula (1 B O H)) is freed by adding 4.83 ml (1 eq) of 1N HCI in a flask containing 2 g of salt (1Bo) described above in 20 ml of water, followed by maintaining the stirring overnight at room temperature.
- Example 8 ethyl 5-hydroxy-1- ⁇ 2-[-4(methoxycarbonyl)phenoxy]ethyl ⁇ -1H- imidazole-4-carboxylate Characteristics of the salt 2-(4-methoxycarbonylphenoxy)ethylammonium 5- ethoxycarbonyl-3-[2-(4-methoxycarbonylphenoxy)ethyl]-3/-/-imidazol-4-ate:
- a solution of bromine in acetic acid (78 g, i.e. 25 ml in 200 ml) is added dropwise over 5 hours to 50 g of malonamide (0.49 mol) dissolved in 300 ml of acetic acid at 60 0 C, with stirring and while maintaining the temperature at 6O 0 C.
- the medium decol- orises instantaneously. It turns yellow after 2 hours 30 minutes of addition.
- 90.94 g of a pale pink solid are obtained. This solid is triturated from 95% ethanol and then suction-filtered to give 79.24 g of 2-bromo- malonamide.
- the mixture is soluble while hot (yellow solution).
- reaction medium is stirred for 12 hours at room temperature and is then poured into brine and extracted with ether. After drying and evaporation, 1.88 g of an orange solid are isolated and recrystallised from 95% ethanol to give 0.672 g of an orange solid.
- Example 21 5-hvdroxv-3-r4-(3-methoxybenzovlamino)benzvl1-3H-imida- zole-4-carboxamide 1 g of silyl amine 3C (2.88 mmol) is placed in a round-bottomed flask under an atmosphere of nitrogen containing 15 ml of dry THF. 0.349 g (0.481 ml) of triethylamine (3.46 mmol, 1.2 eq.) is added, followed by addition of 0.492 g of 3-methoxybenzoyl chloride (0.1 eq.) dissolved in 5 ml of dry THF. The reaction medium is stirred for 72 hours at room temperature.
- the imidazole compounds (ICao t -O below are prepared according to a similar process (purity greater than 90% determined by HPLC-mass).
- Example 22 3-(4-acetylaminobenzyl)-5-hydroxy-3H-imidazole-4-carboxamide
- Example 23 3-(4-benzoylaminobenzyl)-5-hydroxy-3H-imidazole-4-carbox- amide
- Example 27 5-hydroxy-3-[4-(4-fluorobenzoylamino)benzyl]-3H-imidazole-4- carboxamide
- Example 28 5-hydroxy-3- ⁇ 4-[(naphthalene-1-carbonyl)amino]benzyl ⁇ -3H- imidazole-4-carboxamide
- Example 32 3-(4-hexanoylaminobenzyl)-5-hydroxy-3H-imidazole-4-carbox- amide
- Example 33 5-hydroxy-3-[4-(2-fluorobenzoy lamino)benzyl]-3H-imidazole-4- carboxamide
- Example 37 5-hydroxy-3- ⁇ 4-[2-(4-nitrophenyl)acetylamino]benzyl ⁇ -3H-imid- azole-4-carboxamide
- Example 38 5-hydroxy-3-[4-(2-phenylbutyrylamino)benzyl]-3H-imidazole-4- carboxamide
- Example 41 5-hydroxy-3-[4-(3-phenylureido)benzyl]-3H-imidazole-4- carboxamide 0.5 g of silyl amine 3C (1.44 mmol) is placed in a round-bottomed flask under a nitrogen atmosphere containing 10 ml of dry THF 1 followed by addition of 0.172 g (1.44 mmol; 1 eq.) of phenyl isocyanate in a single portion.
- a precipitate forms after 5 hours 30 minutes of stirring at room temperature.
- the reaction medium is stirred for 29 hours. 10 ml of water and 1 ml of 1 N HCI are then added. After stirring for a further 48 hours at room temperature, the solvent is evaporated off and the residue is taken up in a water-ice mixture. The precipitate is washed with water and then with petroleum ether.
- Example 42 5-hydroxy-3-[4-ureidobenzyl]-3H-imidazole-4-carboxamide 0.187 g of sodium cyanide (24.7 mmol; 2 eq.) dissolved in 5 ml of water is added to a round-bottomed flask containing 10 ml of water and 0.5 g (14.4 mmol) of 3-(4-amino- benzyl)-5-(tert-butyldimethylsilanyloxy)-3H-imidazole-4-carboxamide, followed by drop- wise addition of 10 ml of acetic acid over 5 minutes. The medium turns yellow. After stirring for 24 hours at room temperature, the solvent is evaporated off. 30 ml of water and 1 ml of 1 N HCI are added to the residue and the resulting mixture is stirred for a further 24 hours at room temperature.
- Example 46 3-r4-(3-cvclopentvlureido)benzvn-5-hydroxv-3H-imidazole-4- carboxamide
- Example 47 5-hydroxy-3-[4-(3-naphthalen-1-ylureido)benzyl]-3H-imidazole-
- Example 51 3-[4-(3-benzylureido)benzyl]-5-hydroxy-3H-imidazole-4- carboxamide
- Example 52 5-hydroxy-3-[4-(3-mefa-tolylureido)benzyl]-3H-imidazole-4- carboxamide
- Example 54 3-f4-r3-(2-fluorobenzvl)ureido]benzvl)-5-hvdroxy-3H-imida- zole-4-carboxamide
- Example 55 5-hydroxy-3- ⁇ 4-[3-(2-methoxyphenvl)ureido1benzvl>-3H-imida- zole-4-carboxamide
- Example 56 5-hvdroxv-3-f4-f3-f4-ethvlphenyl)ureido1ben2vl>-3H-imidazole- 4-carboxamide
- Example 57 5-hvdroxv-3-(4-[3-(3-methvlsulfanylphenvl)ureido1benzyl>-3H- imidazole-4-carboxamide
- Example 61 5-hydroxy-3- ⁇ 4-[3-(2-chlorophenyl)ureido]benzyl ⁇ -3H-imida- zole-4-carboxamide
- Example 62 5-hydroxy-3- ⁇ 4-[3-(2-trifluoromethylphenyl)ureido]-benzyl ⁇ -3H- imidazole-4-carboxamide
- Example 65 ethyl 3- ⁇ 3-[4-(5-carbamoyl-4-hydroxyimidazol-1-ylmethyl)- phenyl]ureido ⁇ benzoate
- Example 66 ethyl 4- ⁇ 3-[4-(5-carbamoyl-4-hydroxyimidazol-1-ylmethyl)- phenyl]ureido ⁇ benzoate
- Example 67 4- ⁇ 3-[4-(5-carbamoyl-4-hydroxyimidazol-1-ylmethyl)- phenyl]ureido ⁇ benzoic acid
- Example 69 ethyl ⁇ 3-[4-(5-carbamoyl-4-hydroxyimidazol-1-ylmethyl)- phenyl]ureido ⁇ acetate
- Example 70 3- ⁇ 3-[4-(5-carbamoyl-4-hydroxyimidazol-1 -ylmethy I)- phenyQureido ⁇ benzoic acid
- AMPK 25 mil of AMPK are incubated in the presence of different concentrations of AMP or of products, for 30 minutes at 30 0 C, in a final volume of 30 ⁇ l comprising 50 mM
- the phosphorylation of the peptide AMARAA is then measured according to a Delfia protocol (Perkin-Elmer), using a europium-labelled anti-phospho-serine specific antibody.
- the AMPK used in this test is a partially purified protein from rat liver.
- the percentage of activation is calculated relative to the basal activity (100%) obtained in the absence of AMP.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Diabetes (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Child & Adolescent Psychology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Ophthalmology & Optometry (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Urology & Nephrology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description



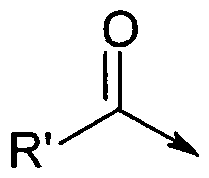




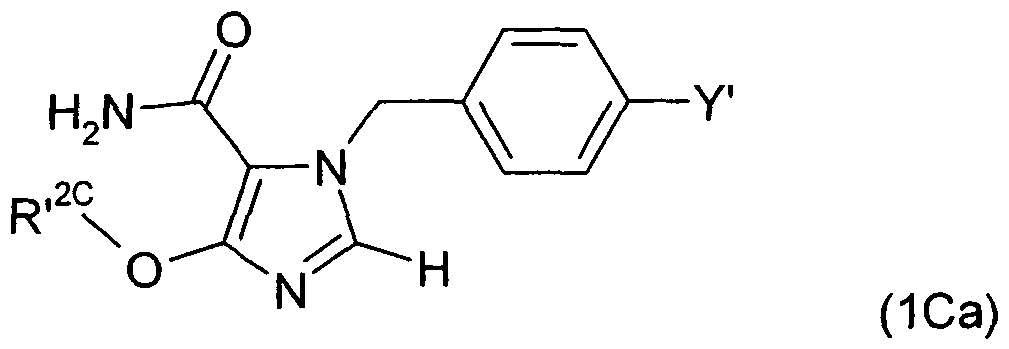














Claims










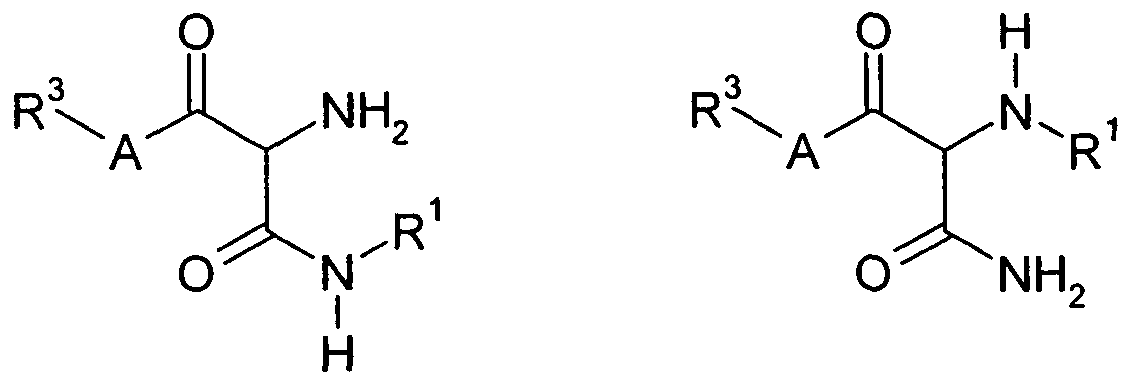



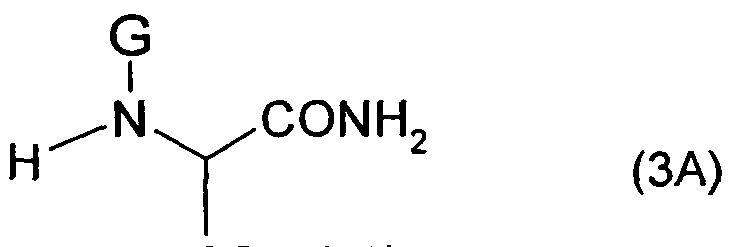


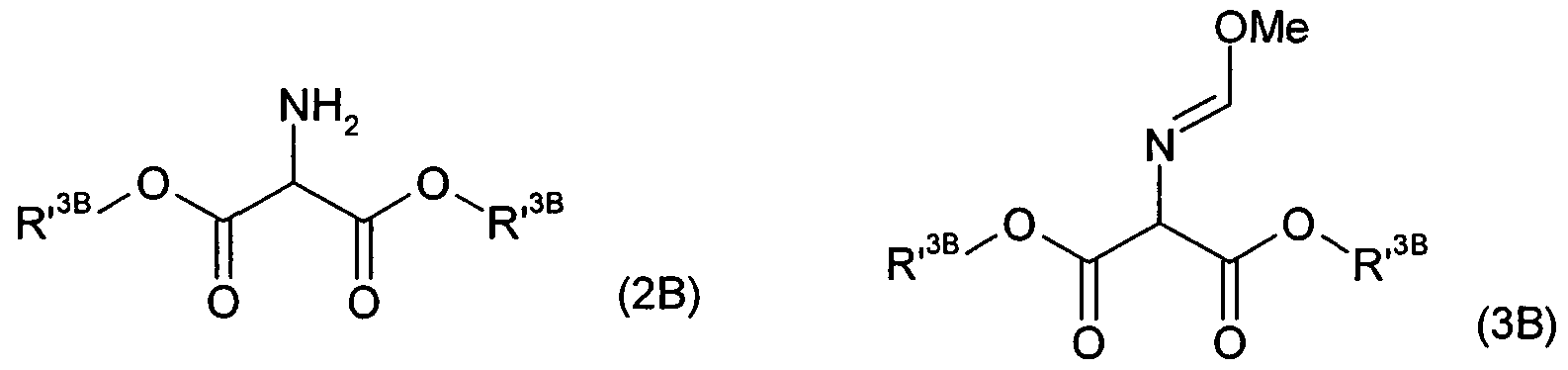





Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2007272082A AU2007272082B2 (en) | 2006-07-13 | 2007-06-12 | Use of AMPK-activating imidazole derivatives, preparation process therefor and pharmaceutical compositions comprising them |
| JP2009518732A JP5237941B2 (en) | 2006-07-13 | 2007-06-12 | Use of AMPK-activated imidazole derivatives, process for their preparation and pharmaceutical compositions containing them |
| US12/373,310 US8329738B2 (en) | 2006-07-13 | 2007-06-12 | Use of AMPK-activating imidazole derivatives, preparation process therefor and pharmaceutical compositions comprising them |
| DK07725977.8T DK2040702T3 (en) | 2006-07-13 | 2007-06-12 | Use of AMPK-activating imidazole derivatives, processes for their preparation and pharmaceutical preparations comprising them |
| AT07725977T ATE517621T1 (en) | 2006-07-13 | 2007-06-12 | USE OF AMPK-ACTIVATED IMIDAZOLE DERIVATIVES, PRODUCTION PROCESS THEREOF AND PHARMACEUTICAL COMPOSITIONS CONTAINING SAME |
| CA2657335A CA2657335C (en) | 2006-07-13 | 2007-06-12 | Use of ampk-activating imidazole derivatives, preparation process therefor and pharmaceutical compositions comprising them |
| EP07725977A EP2040702B1 (en) | 2006-07-13 | 2007-06-12 | Use of ampk-activating imidazole derivatives, preparation process therefor and pharmaceutical compositions comprising them |
| PL07725977T PL2040702T3 (en) | 2006-07-13 | 2007-06-12 | Use of ampk-activating imidazole derivatives, preparation process therefor and pharmaceutical compositions comprising them |
| IL196280A IL196280A (en) | 2006-07-13 | 2008-12-30 | Ampk-activating imidazole derivatives, processes for their preparation, uses thereof and pharmaceutical compositions comprising them |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR06/06415 | 2006-07-13 | ||
| FR0606415A FR2903695B1 (en) | 2006-07-13 | 2006-07-13 | USE OF AMPAP ACTIVATOR IMIDAZOLE DERIVATIVES, PROCESS FOR PREPARING THEM AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008006432A1 true WO2008006432A1 (en) | 2008-01-17 |
Family
ID=37696444
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2007/005164 WO2008006432A1 (en) | 2006-07-13 | 2007-06-12 | Use of ampk-activating imidazole derivatives, preparation process therefor and pharmaceutical compositions comprising them |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US8329738B2 (en) |
| EP (1) | EP2040702B1 (en) |
| JP (1) | JP5237941B2 (en) |
| AR (1) | AR061994A1 (en) |
| AT (1) | ATE517621T1 (en) |
| AU (1) | AU2007272082B2 (en) |
| CA (1) | CA2657335C (en) |
| DK (1) | DK2040702T3 (en) |
| ES (1) | ES2367852T3 (en) |
| FR (1) | FR2903695B1 (en) |
| IL (1) | IL196280A (en) |
| PL (1) | PL2040702T3 (en) |
| PT (1) | PT2040702E (en) |
| WO (1) | WO2008006432A1 (en) |
Cited By (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| FR2933694A1 (en) * | 2008-07-10 | 2010-01-15 | Oreal | USE OF PHENOXYALKYLAMINES IN COSMETIC AND / OR DERMATOLOGICAL COMPOSITIONS. |
| WO2010047982A1 (en) | 2008-10-22 | 2010-04-29 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| WO2010051206A1 (en) | 2008-10-31 | 2010-05-06 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| WO2011078370A1 (en) * | 2009-12-25 | 2011-06-30 | 杏林製薬株式会社 | Novel parabanic acid derivative and drug having the same as active ingredient |
| JP2011524384A (en) * | 2008-06-16 | 2011-09-01 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツング | Quinoxalinedione derivatives |
| WO2011106273A1 (en) | 2010-02-25 | 2011-09-01 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011121109A1 (en) * | 2010-04-02 | 2011-10-06 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and compositions comprising ampk activator (metformin/troglitazone) for the treatment of myotonic dystrophy type 1 (dm1) |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| JP2012503661A (en) * | 2008-09-26 | 2012-02-09 | メルク・シャープ・エンド・ドーム・コーポレイション | Novel cyclic benzimidazole derivatives useful as antidiabetic agents |
| WO2012026495A1 (en) * | 2010-08-25 | 2012-03-01 | 杏林製薬株式会社 | Novel hydantoin derivative and medicinal agent comprising same as active ingredient |
| WO2012116145A1 (en) | 2011-02-25 | 2012-08-30 | Merck Sharp & Dohme Corp. | Novel cyclic azabenzimidazole derivatives useful as anti-diabetic agents |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2014031465A1 (en) | 2012-08-22 | 2014-02-27 | Merck Sharp & Dohme Corp. | Novel azabenzimidazole tetrahydropyran derivatives |
| WO2014139388A1 (en) | 2013-03-14 | 2014-09-18 | Merck Sharp & Dohme Corp. | Novel indole derivatives useful as anti-diabetic agents |
| US8889730B2 (en) | 2012-04-10 | 2014-11-18 | Pfizer Inc. | Indole and indazole compounds that activate AMPK |
| US9290517B2 (en) | 2012-08-22 | 2016-03-22 | Merck Sharp & Dohme Corp. | Azabenzimidazole hexahydrofuro[3,2-b]furan derivatives |
| EP2946780A4 (en) * | 2013-01-15 | 2016-06-08 | Fujifilm Corp | Tablet containing 5-hydroxy-1h-imidazole-4-carboxamide |
| US9394285B2 (en) | 2013-03-15 | 2016-07-19 | Pfizer Inc. | Indole and indazole compounds that activate AMPK |
| US9527839B2 (en) | 2012-08-22 | 2016-12-27 | Merck Sharp & Dohme Corp. | Benzimidazole tetrahydropyran derivatives |
| US9540364B2 (en) | 2012-08-22 | 2017-01-10 | Merck Sharp & Dohme Corp. | Benzimidazole tetrahydrofuran derivatives |
| US9556193B2 (en) | 2012-08-22 | 2017-01-31 | Merck Shapr & Dohme Corp. | Benzimidazole hexahydrofuro[3,2-b]furan derivatives |
| US9868733B2 (en) | 2012-08-22 | 2018-01-16 | Merck Sharp & Dohme Corp. | Azabenzimidazole tetrahydrofuran derivatives |
| EP3608312A4 (en) * | 2017-04-07 | 2020-10-28 | Xiamen Vivohealths Technology Co., Ltd. | Substituted imidazole salt compounds, preparation method therefor, pharmaceutical composition thereof, and applications thereof |
| US11279702B2 (en) | 2020-05-19 | 2022-03-22 | Kallyope, Inc. | AMPK activators |
| US11407768B2 (en) | 2020-06-26 | 2022-08-09 | Kallyope, Inc. | AMPK activators |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2013118021A (en) * | 2010-09-20 | 2014-10-27 | Кареус Терапьютикс, Са | METHODS AND COMPOSITIONS FOR TREATMENT OF DIABETES MELLITUS AND DYSLIPIDEMIA |
| US9005909B2 (en) | 2011-01-06 | 2015-04-14 | Rigel Pharmaceuticals, Inc. | Whole blood assay for measuring AMPK activation |
| PL223830B1 (en) | 2012-04-03 | 2016-11-30 | Univ Jagiellonski | Derivatives of aromatic imidazolidinones and their use |
| WO2015103480A1 (en) | 2014-01-02 | 2015-07-09 | Massachusetts Eye & Ear Infirmary | Treating ocular neovascularization |
| EP3355876A2 (en) | 2015-09-30 | 2018-08-08 | Instituto de Medicina Molecular | Methods for attenuating parasite virulence |
| CN116322663A (en) | 2020-09-30 | 2023-06-23 | 比奥维拉迪维治疗股份有限公司 | AMPK activators and methods of use thereof |
| WO2022106892A1 (en) | 2020-11-17 | 2022-05-27 | Instituto De Medicina Molecular | Anti-malarial compounds |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4140788A (en) | 1976-11-10 | 1979-02-20 | Sumitomo Chemical Company, Limited | N-Substituted imidazolecarboxamide derivatives |
| JPS5748971A (en) | 1980-09-08 | 1982-03-20 | Sumitomo Chem Co Ltd | Novel alkylimidazole derivative |
| JPS5748971B2 (en) | 1976-12-24 | 1982-10-19 | ||
| WO2002009726A1 (en) | 2000-07-31 | 2002-02-07 | Trustees Of Boston University | Methods of treating conditions associated with insulin resistance with aicar, (5-amino-4-imidazole carboxamide riboside) and related compounds |
| US6482844B1 (en) | 2000-04-07 | 2002-11-19 | Neurogen Corporation | 1-benzylimidazole derivatives |
| DE10150172A1 (en) | 2001-10-11 | 2003-04-30 | Morphochem Ag | New 5-amino-4-phenyl-1H-imidazole derivatives useful as protein tyrosine phosphatase 1B inhibitors, for treating e.g. diabetes and obesity |
| WO2004043957A1 (en) | 2002-11-05 | 2004-05-27 | Les Laboratoires Servier | Imidazopyridine derivatives, preparation method and pharmaceutical compositions containing same |
| US20050038068A1 (en) | 2003-05-16 | 2005-02-17 | Iyengar Rajesh R. | Thienopyridones as AMPK activators for the treatment of diabetes and obesity |
| WO2005028464A1 (en) | 2003-09-18 | 2005-03-31 | Kao Corporation | Endurance improving agent |
| JP2005225804A (en) | 2004-02-13 | 2005-08-25 | Kao Corp | Ampk activator |
-
2006
- 2006-07-13 FR FR0606415A patent/FR2903695B1/en active Active
-
2007
- 2007-06-12 US US12/373,310 patent/US8329738B2/en active Active
- 2007-06-12 JP JP2009518732A patent/JP5237941B2/en active Active
- 2007-06-12 AU AU2007272082A patent/AU2007272082B2/en active Active
- 2007-06-12 PT PT07725977T patent/PT2040702E/en unknown
- 2007-06-12 EP EP07725977A patent/EP2040702B1/en active Active
- 2007-06-12 PL PL07725977T patent/PL2040702T3/en unknown
- 2007-06-12 ES ES07725977T patent/ES2367852T3/en active Active
- 2007-06-12 WO PCT/EP2007/005164 patent/WO2008006432A1/en active Application Filing
- 2007-06-12 CA CA2657335A patent/CA2657335C/en active Active
- 2007-06-12 DK DK07725977.8T patent/DK2040702T3/en active
- 2007-06-12 AT AT07725977T patent/ATE517621T1/en active
- 2007-07-13 AR ARP070103120A patent/AR061994A1/en unknown
-
2008
- 2008-12-30 IL IL196280A patent/IL196280A/en active IP Right Grant
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4140788A (en) | 1976-11-10 | 1979-02-20 | Sumitomo Chemical Company, Limited | N-Substituted imidazolecarboxamide derivatives |
| JPS5748971B2 (en) | 1976-12-24 | 1982-10-19 | ||
| JPS5748971A (en) | 1980-09-08 | 1982-03-20 | Sumitomo Chem Co Ltd | Novel alkylimidazole derivative |
| US6482844B1 (en) | 2000-04-07 | 2002-11-19 | Neurogen Corporation | 1-benzylimidazole derivatives |
| WO2002009726A1 (en) | 2000-07-31 | 2002-02-07 | Trustees Of Boston University | Methods of treating conditions associated with insulin resistance with aicar, (5-amino-4-imidazole carboxamide riboside) and related compounds |
| DE10150172A1 (en) | 2001-10-11 | 2003-04-30 | Morphochem Ag | New 5-amino-4-phenyl-1H-imidazole derivatives useful as protein tyrosine phosphatase 1B inhibitors, for treating e.g. diabetes and obesity |
| WO2004043957A1 (en) | 2002-11-05 | 2004-05-27 | Les Laboratoires Servier | Imidazopyridine derivatives, preparation method and pharmaceutical compositions containing same |
| US20050038068A1 (en) | 2003-05-16 | 2005-02-17 | Iyengar Rajesh R. | Thienopyridones as AMPK activators for the treatment of diabetes and obesity |
| WO2005028464A1 (en) | 2003-09-18 | 2005-03-31 | Kao Corporation | Endurance improving agent |
| JP2005225804A (en) | 2004-02-13 | 2005-08-25 | Kao Corp | Ampk activator |
Non-Patent Citations (42)
| Title |
|---|
| BENJAMIN B. LIM: "Reagents for bioorganic synthesis", J. ORG. CHEM, vol. 50, 1985, pages 5111 - 5115, XP002419940 * |
| BENJAMIN B.: "Lim published syntheses of other imidazole derivatives", J. ORG. CHEM., vol. 50, 1985, pages 5111 - 5115 |
| BERGERON R. ET AL.: "Effect of 5-aminoimidazole-4-carboxamide-1 [3-D-ribofuranoside infusion on in vivo glucose metabolism in lean and obese Zucker rats", DIABETES, vol. 50, 2001, pages 1076 |
| BERGERON R. ET AL.: "Effect of 5-aminoimidazote-4-carboxamide-1 [3-D-ribofuranoside infusion on in vivo glucose metabolism in lean and obese Zucker rats", DIABETES, vol. 50, 2001, pages 1076 |
| BUHL E. S. ET AL.: "Long-term AICAR administration reduces metabolic disturbances and lowers blood pressure in rats displaying feature of the insulin resistance syndrome", DIABETES, vol. 51, 2002, pages 2199 |
| CARLING D. ET AL.: "A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis", FEBS LETTERS, vol. 223, 1987, pages 217 |
| CORTON J. M. ET AL.: "5-Aminoimidazole-4-carboxamide ribonucleoside, a specific method for activating AMP-activated protein kinase in intact cells", EUR. J. BIOCHEM., vol. 229, 1995, pages 558 |
| CUR. OPIN. INVESTIG. DRUGS, vol. 7, 2006, pages 324 - 337 |
| DATABASE CA [online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; FRANCHETTI, PALMARISA ET AL: "Ribose-modified Mizoribine Analogs: Synthesis and Biological Evaluation", XP002420468, retrieved from STN Database accession no. 2005:1327555 * |
| DIABETES, vol. 37, 1988, pages 1595 - 1607 |
| FRANCHETTI, P. ET AL., NUCLEOSIDES, NUCLEOTIDES & NUCLEIC ACIDS, vol. 24, no. 10-12, 2005, pages 2023 - 2027 |
| GARTON A. J. ET AL.: "Phosphorylation of bovine hormone-sensitive lipase by the AMP-activated protein kinase. A possible antilipolytic mechanism", EUR. J. BIOCHEM., vol. 179, 1989, pages 249 |
| GREENE ET AL.: "Protective Groups in Organic Synthesis", 1999, JOHN WILEY & SONS, pages: 17 - 292 |
| HALSETH A. E. ET AL.: "Acute and chronic treatment of ob/ob and db/db mice with AICAR decreases blood glucose concentrations", BIOCHEM. BIOPHYS. RES. COMM., vol. 294, 2002, pages 798 |
| HARDIE D. G.; HAWLEY S. A.: "AMP-activated protein kinase: the energy charge hypothesis revisited", BIOASSAYS, vol. 23, 2001, pages 1112 |
| HORM. RES., vol. 38, 1992, pages 28 - 32 |
| HOSMANE R. S.; B. B. LIM, TET. LETT., vol. 26, no. 16, 1985, pages 1915 - 1918 |
| J. HET. CHEM., vol. 20, no. 4, 1983, pages 875 - 885 |
| J. ORG. CHEM., vol. 50, no. 25, 1985, pages 5111 - 5115 |
| JOURNAL OF DIABETES AND ITS COMPLICATIONS, vol. 12, 1998, pages 110 - 119 |
| KEMP B. E. ET AL.: "AMP-activated protein kinase, super metabolic regulator", BIOCHEM. SOC. TRANSACTIONS, vol. 31, 2003, pages 162 |
| LECLERC I. ET AL.: "Hepatocyte nuclear factor-4a involved in type-1 maturity-onset diabetes of the young is a novel target of AMP-activated protein kinase", DIABETES, vol. 50, 2001, pages 1515 |
| LOCHHEAD P. A. ET AL.: "5-aminoimidazole-4-carboxamide riboside mimics the effects of insulin on the expression of the 2 key gluconeogenic genes PEPCK and glucose-6-phosphatase", DIABETES, vol. 49, 2000, pages 896 |
| MARVIN E. LEVIN ET AL., UNCOMPLICATED GUIDE TO DIABETES COMPLICATIONS, 2002 |
| MINO- KOSHI Y. ET AL.: "Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase", NATURE, vol. 415, 2002, pages 339 |
| MU J. ET AL.: "A role for AMP-activated protein kinase in contraction and hypoxia-regulated glucose transport in skeletal muscle", MOLECULAR CELL, vol. 7, 2001, pages 1085 |
| MUOIO D. M. ET AL.: "AMP-activated kinase reciprocally regulates triacylglycerol synthesis and fatty acid oxidation in liver and muscle: evidence that sn-glycerol-3-phosphate acyltransferase is a novel target", BIOCHEM. J., vol. 338, 1999, pages 783 |
| MUSI N. ET AL.: "Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes", DIABETES, vol. 51, 2002, pages 2074 |
| MUSI N.; GOOD- YEAR L. J.: "Targeting the AMP-activated protein kinase for the treatment of type 2 diabetes", CURRENT DRUG TARGETS-IMMUNE, ENDOCRINE AND METABOLIC DISORDERS, vol. 2, 2002, pages 119 |
| NUCLEOSIDES, NUCLEOTIDES & NUCLEIC ACIDS , 24(10-12), 2023-2027 CODEN: NNNAFY; ISSN: 1525-7770, 2005 * |
| PRESSE MÉDICALE, vol. 26, no. 14, 1997, pages 671 - 677 |
| SAHA A. K. ET AL.: "Activation of malonyl-CoA decarboxylase in rat skeletal muscle by contraction and the AMP-activated protein kinase activator 5-aminoimidazo)e-4-carboxamide-1-P-D-ribofuranoside", J. BIOI. CHEM., vol. 275, 2000, pages 24279 |
| SLEBOCKA-TILK ET AL., J. ORG. CHEM., vol. 50, 1985, pages 4638 |
| SONG S. M. ET AL.: "5-Aminoimidazole-4-carboxamide ribonucleoside treatment improves glucose homeostasis in insulin-resistant diabeted (ob/db) mice", DIABETOLOGIA, vol. 56, 2002 |
| SONG S. M. ET AL.: "5-Aminoimidazole-4-carboxamide ribonucleoside treatment improves glucose homeostasis in insulin-resistant diabeted (ob/ob) mice", DIABETOLOGIA, vol. 45, 2002, pages 56 |
| TARUMI ET AL., J. HET. CHEM., vol. 21, no. 3, 1984, pages 849 |
| TARUMI Y. ET AL., J. HET. CHEM., vol. 21, no. 3, 1984, pages 849 - 854 |
| TARUMI, Y. ET AL.: "published syntheses of other imidazole derivatives", J. HETEROCYCL. CHEM., vol. 21, no. 2, 1984, pages 529 - 37 |
| TARUMI, Y. ET AL: "Studies on imidazole derivatives and related compounds. 3. Regioselective glycosylation of 4-carbamoylimidazolium-5-olate", JOURNAL OF HETEROCYCLIC CHEMISTRY , 21(2), 529-37 CODEN: JHTCAD; ISSN: 0022-152X, 1984, XP002419939 * |
| TOMAS E. ET AL.: "Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: Acetyl-CoA carboxylase inhibition and AMP-activated protein kinase activation", PNAS, vol. 99, 2002, pages 16309 |
| YAMAUCHI T. ET AL.: "Adiponectin stimulates glucose utilisation and fatty acid oxidation by activating AMP-activated protein kinase", NATURE MEDICINE, vol. 8, 2002, pages 1288 |
| ZHOU G. ET AL.: "Role of AMP-activated protein kinase in mechanism of metformin action", J. CLIN. INVEST., vol. 108, 2001, pages 1167 |
Cited By (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| JP2011524384A (en) * | 2008-06-16 | 2011-09-01 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツング | Quinoxalinedione derivatives |
| FR2933694A1 (en) * | 2008-07-10 | 2010-01-15 | Oreal | USE OF PHENOXYALKYLAMINES IN COSMETIC AND / OR DERMATOLOGICAL COMPOSITIONS. |
| JP2012503661A (en) * | 2008-09-26 | 2012-02-09 | メルク・シャープ・エンド・ドーム・コーポレイション | Novel cyclic benzimidazole derivatives useful as antidiabetic agents |
| US8394969B2 (en) | 2008-09-26 | 2013-03-12 | Merck Sharp & Dohme Corp. | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| US8410284B2 (en) | 2008-10-22 | 2013-04-02 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| WO2010047982A1 (en) | 2008-10-22 | 2010-04-29 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| WO2010051206A1 (en) | 2008-10-31 | 2010-05-06 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| US8329914B2 (en) | 2008-10-31 | 2012-12-11 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| WO2011078370A1 (en) * | 2009-12-25 | 2011-06-30 | 杏林製薬株式会社 | Novel parabanic acid derivative and drug having the same as active ingredient |
| WO2011106273A1 (en) | 2010-02-25 | 2011-09-01 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| US8895596B2 (en) | 2010-02-25 | 2014-11-25 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011121109A1 (en) * | 2010-04-02 | 2011-10-06 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and compositions comprising ampk activator (metformin/troglitazone) for the treatment of myotonic dystrophy type 1 (dm1) |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012026495A1 (en) * | 2010-08-25 | 2012-03-01 | 杏林製薬株式会社 | Novel hydantoin derivative and medicinal agent comprising same as active ingredient |
| WO2012116145A1 (en) | 2011-02-25 | 2012-08-30 | Merck Sharp & Dohme Corp. | Novel cyclic azabenzimidazole derivatives useful as anti-diabetic agents |
| EP3243385A1 (en) | 2011-02-25 | 2017-11-15 | Merck Sharp & Dohme Corp. | Novel cyclic azabenzimidazole derivatives useful as anti-diabetic agents |
| US8796258B2 (en) | 2011-02-25 | 2014-08-05 | Merck Sharp & Dohme Corp. | Cyclic azabenzimidazole derivatives useful as anti-diabetic agents |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8889730B2 (en) | 2012-04-10 | 2014-11-18 | Pfizer Inc. | Indole and indazole compounds that activate AMPK |
| US9290517B2 (en) | 2012-08-22 | 2016-03-22 | Merck Sharp & Dohme Corp. | Azabenzimidazole hexahydrofuro[3,2-b]furan derivatives |
| US9868733B2 (en) | 2012-08-22 | 2018-01-16 | Merck Sharp & Dohme Corp. | Azabenzimidazole tetrahydrofuran derivatives |
| WO2014031465A1 (en) | 2012-08-22 | 2014-02-27 | Merck Sharp & Dohme Corp. | Novel azabenzimidazole tetrahydropyran derivatives |
| US9382243B2 (en) | 2012-08-22 | 2016-07-05 | Merck Sharp & Dohme Corp. | Azabenzimidazole tetrahydropyran derivatives |
| US9556193B2 (en) | 2012-08-22 | 2017-01-31 | Merck Shapr & Dohme Corp. | Benzimidazole hexahydrofuro[3,2-b]furan derivatives |
| US9527839B2 (en) | 2012-08-22 | 2016-12-27 | Merck Sharp & Dohme Corp. | Benzimidazole tetrahydropyran derivatives |
| US9540364B2 (en) | 2012-08-22 | 2017-01-10 | Merck Sharp & Dohme Corp. | Benzimidazole tetrahydrofuran derivatives |
| EP2946780A4 (en) * | 2013-01-15 | 2016-06-08 | Fujifilm Corp | Tablet containing 5-hydroxy-1h-imidazole-4-carboxamide |
| US9650375B2 (en) | 2013-03-14 | 2017-05-16 | Merck Sharp & Dohme Corp. | Indole derivatives useful as anti-diabetic agents |
| WO2014139388A1 (en) | 2013-03-14 | 2014-09-18 | Merck Sharp & Dohme Corp. | Novel indole derivatives useful as anti-diabetic agents |
| US9394285B2 (en) | 2013-03-15 | 2016-07-19 | Pfizer Inc. | Indole and indazole compounds that activate AMPK |
| EP3608312A4 (en) * | 2017-04-07 | 2020-10-28 | Xiamen Vivohealths Technology Co., Ltd. | Substituted imidazole salt compounds, preparation method therefor, pharmaceutical composition thereof, and applications thereof |
| US11584722B2 (en) | 2017-04-07 | 2023-02-21 | Xiamen Vivohealths Technology Co., Ltd. | Substituted imidazole salt compounds, preparation method thereof, pharmaceutical composition thereof and application thereof |
| US11279702B2 (en) | 2020-05-19 | 2022-03-22 | Kallyope, Inc. | AMPK activators |
| US11851429B2 (en) | 2020-05-19 | 2023-12-26 | Kallyope, Inc. | AMPK activators |
| US11407768B2 (en) | 2020-06-26 | 2022-08-09 | Kallyope, Inc. | AMPK activators |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2040702B1 (en) | 2011-07-27 |
| AR061994A1 (en) | 2008-08-10 |
| AU2007272082A1 (en) | 2008-01-17 |
| FR2903695B1 (en) | 2008-10-24 |
| FR2903695A1 (en) | 2008-01-18 |
| CA2657335C (en) | 2015-01-27 |
| PT2040702E (en) | 2011-11-04 |
| EP2040702A1 (en) | 2009-04-01 |
| ES2367852T3 (en) | 2011-11-10 |
| DK2040702T3 (en) | 2011-09-05 |
| AU2007272082B2 (en) | 2012-05-24 |
| JP5237941B2 (en) | 2013-07-17 |
| IL196280A (en) | 2013-02-28 |
| IL196280A0 (en) | 2009-09-22 |
| PL2040702T3 (en) | 2011-11-30 |
| ATE517621T1 (en) | 2011-08-15 |
| US8329738B2 (en) | 2012-12-11 |
| CA2657335A1 (en) | 2008-01-17 |
| US20090253764A1 (en) | 2009-10-08 |
| JP2009542734A (en) | 2009-12-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2657335C (en) | Use of ampk-activating imidazole derivatives, preparation process therefor and pharmaceutical compositions comprising them | |
| RU2416409C2 (en) | Application of thienopyridone derivatives as ampa-activators and pharmaceutic compositions, containing them | |
| US7534800B2 (en) | 7-azaindoles as inhibitors of c-Jun N-terminal kinases for the treatment of neurodegenerative disorders | |
| RU2247121C2 (en) | 1-(para-thienylbenzyl)-imidazoles as agonists of angiotensin-(1-7) receptors and pharmaceutical composition containing thereof | |
| US8299102B2 (en) | Heteroarylcyclopropanecarboxamides and their use as pharmaceuticals | |
| RU2377238C2 (en) | Imidazole derivatives active to cb1 receptor | |
| EA017756B1 (en) | Thienopyridone derivatives as amp-activated protein kinase (ampk) activators | |
| JP2010500999A (en) | Imidazoleamine as an inhibitor of beta-secretase | |
| US20110105567A1 (en) | Pyrrole Compounds Having Sphingosine-1-Phosphate Receptor Agonist Or Antagonist Biological Activity | |
| JP2011503042A (en) | p38 MAP kinase inhibitor | |
| HUT64040A (en) | Process for producing imidazolylmethyl-substituted phenylacetamide derivatives and pharmaceutical compositions comprising same | |
| JPH05213939A (en) | Substituted imidazolyl-propenic acid derivative | |
| PT2240482E (en) | Cyclic azaindole-3-carboxamides, their preparation and their use as pharmaceuticals | |
| US20040259860A1 (en) | Substituted piperidines and pyrrolidines as calcium sensing receptor modulators and method | |
| US6462063B1 (en) | C-proteinase inhibitors | |
| JP2003206230A (en) | Cyanoheterocyclic derivative or its salt | |
| WO2003090732A1 (en) | Lxr modulators for the treatment of cardiovascular diseases | |
| PL198827B1 (en) | N-ARYLSULFONYL AMINO ACID OMEGA AMIDES, method of making them, pharmaceutical agent and their application | |
| US6956039B2 (en) | Nitrogenous heterocylic compounds | |
| US20200165233A1 (en) | Ldha activity inhibitors | |
| CZ51693A3 (en) | Propenoyl-imidazole derivatives | |
| US5112841A (en) | Imidazole derivatives and antiepileptics comprising said imidazole derivatives as effective ingredients | |
| WO2003082263A1 (en) | Sulfamic acids as inhibitors of human cytoplasmic protein tyrosine phosphatases | |
| EP3553061A1 (en) | New inhibitors of bone resorption | |
| EP0502110B1 (en) | (hetero) 4-arylmethoxy phenyl diazole derivatives, method for preparing same, and their therapeutical applications |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07725977 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007725977 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 196280 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2657335 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12373310 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009518732 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007272082 Country of ref document: AU |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |
|
| ENP | Entry into the national phase |
Ref document number: 2007272082 Country of ref document: AU Date of ref document: 20070612 Kind code of ref document: A |