WO2006125554A1 - Verwendung von cyanopyrimidinen für die behandlung von herz-kreislauf-erkrankungen - Google Patents
Verwendung von cyanopyrimidinen für die behandlung von herz-kreislauf-erkrankungen Download PDFInfo
- Publication number
- WO2006125554A1 WO2006125554A1 PCT/EP2006/004639 EP2006004639W WO2006125554A1 WO 2006125554 A1 WO2006125554 A1 WO 2006125554A1 EP 2006004639 W EP2006004639 W EP 2006004639W WO 2006125554 A1 WO2006125554 A1 WO 2006125554A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- mmol
- preparation
- compounds
- group
- Prior art date
Links
- 0 *CC(NC1=O)=NC(N(*)*)=C1C#N Chemical compound *CC(NC1=O)=NC(N(*)*)=C1C#N 0.000 description 2
- NTFCEELFQOCFJN-UHFFFAOYSA-N CCC(CC)CCC1OC(C)(C)OC1 Chemical compound CCC(CC)CCC1OC(C)(C)OC1 NTFCEELFQOCFJN-UHFFFAOYSA-N 0.000 description 1
- YDPBNKNMCBNLOY-UHFFFAOYSA-N CN(CC1)CCN1C(N=C(Cc1c[s]cc1)NC1=O)=C1C#N Chemical compound CN(CC1)CCN1C(N=C(Cc1c[s]cc1)NC1=O)=C1C#N YDPBNKNMCBNLOY-UHFFFAOYSA-N 0.000 description 1
- INGVXKAYXKBQND-UHFFFAOYSA-M COc(nc1)ccc1-c1ccccc1CC([N-]C1=O)=NC(SC)=C1C#N Chemical compound COc(nc1)ccc1-c1ccccc1CC([N-]C1=O)=NC(SC)=C1C#N INGVXKAYXKBQND-UHFFFAOYSA-M 0.000 description 1
- OTYPWODJYNSMMD-UHFFFAOYSA-N CSC(N=C(Cc(cc1)ccc1Cl)NC1=O)=C1C#N Chemical compound CSC(N=C(Cc(cc1)ccc1Cl)NC1=O)=C1C#N OTYPWODJYNSMMD-UHFFFAOYSA-N 0.000 description 1
- JZRJAARTOLAUDD-UHFFFAOYSA-N CSC(N=C(Cc(cccc1)c1F)NC1=O)=C1C#N Chemical compound CSC(N=C(Cc(cccc1)c1F)NC1=O)=C1C#N JZRJAARTOLAUDD-UHFFFAOYSA-N 0.000 description 1
- UOMGOPZFBGARRV-UHFFFAOYSA-N Cc1ccccc1CC(NC1=O)=NC(SC)=C1C#N Chemical compound Cc1ccccc1CC(NC1=O)=NC(SC)=C1C#N UOMGOPZFBGARRV-UHFFFAOYSA-N 0.000 description 1
- IDJJUOHWPMXSFS-UHFFFAOYSA-N N#CC1=C(N2CCCC2)N=C(Cc2cc(Cl)ccc2)NC1=O Chemical compound N#CC1=C(N2CCCC2)N=C(Cc2cc(Cl)ccc2)NC1=O IDJJUOHWPMXSFS-UHFFFAOYSA-N 0.000 description 1
- ILUHDQDONXZWMQ-UHFFFAOYSA-N N#CC1=C(N2CCNCC2)N=C(Cc2c[s]cc2)NC1=O Chemical compound N#CC1=C(N2CCNCC2)N=C(Cc2c[s]cc2)NC1=O ILUHDQDONXZWMQ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/513—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim having oxo groups directly attached to the heterocyclic ring, e.g. cytosine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
Definitions
- the invention relates to the use of cynoopyrimidines for the preparation of medicaments for the treatment of cardiovascular diseases.
- Inhibition of phosphodiesterases modulates levels of cyclic nucleotides 5'-3 'cyclic adenosine monophosphate (cAMP) and 5'-3' cyclic guanosine monophosphate (cGMP), respectively.
- cAMP and cGMP cyclic nucleotides
- cAMP and cGMP are important second messengers and therefore play a central role in the cellular signal transduction cascades. Both activate, inter alia, but not exclusively, again protein kinases.
- the cAMP-activated protein kinase is called protein kinase A (PKA), and the cGMP-activated protein kinase is called protein kinase G (PKG).
- PKA protein kinase A
- PKG protein kinase G
- Activated PKA or PKG can phosphorylate a variety of cellular effector proteins (e.g., ion channels, G protein-coupled receptors, structural proteins).
- the second messengers cAMP and cGMP can control the most diverse physiological processes in various organs.
- the cyclic nucleotides can also act directly on effector molecules.
- cGMP can act directly on ion channels and thereby influence the cellular ion concentration (reviewed in: Wei et al., Prog. Neurobiol., 1998, 56: 37-64).
- PDE phosphodiesterases
- the human PDE9A was cloned and sequenced in 1998.
- the amino acid identity to other PDEs is a maximum of 34% (PDE8A) and a minimum of 28% (PDE5A).
- Km value Michaelis-Menten constant
- PDE9A is highly affine for cGMP.
- PDE9A has no cGMP binding domain suggestive of allosteric enzyme regulation by cGMP.
- PDE9A has been shown to be expressed in human, inter alia, in the testes, brain, small intestine, skeletal muscle, heart, lungs, thymus and spleen. The highest expression has been found in brain, small intestine, heart and spleen (Fisher et al., J. Biol. Chem., 1998, 273 (25): 15559-15564).
- the gene for human PDE9A is located on chromosome 21q22.3 and contains 21 exons.
- 20 alternative splice variants of PDE9A have been identified (Guipponi et al., Hum.
- Mouse PDE9A was described by Soderling et al. (J. Biol. Chem., 1998, 273 (25): 15553-15558) and sequenced. Like the human form, this is highly affine for cGMP with a Km of 70 nM. In the mouse, a particularly high expression in the kidney, brain, lung and heart was found. The mouse PDE9A is also not inhibited by IBMX in concentrations below 200 ⁇ M; the IC 50 value for zaprinast is 29 ⁇ M (Soderling et al., J. Biol. Chem., 1998, 273 (19): 15553-15558). In the rat brain, it has been shown that PDE9A is strongly expressed in some brain regions.
- hippocampus hippocampus
- cortex basal ganglia
- basal forebrain Basal forebrain
- US 5,002,949 discloses cyanopyrimidinones for the inhibition of white thrombus formations.
- WO 02/06288 describes cyanopyrimidinones with mGluR antagonistic action.
- cyanopyrimidinones are disclosed for the treatment of depression and Alzheimer's disease.
- EP 130735 describes cyanopyrimidines as cardiotonic reagents.
- the present invention relates to compounds of the formula
- A is CpCg-alkyl, C 3 -Cg -cycloalkyl, tetrahydrofuryl or tetrahydropyranyl, which optionally with up to 3 radicals independently selected from the group
- R 3 and R 4 are independently hydrogen or C 1 -C 6 -alkyl
- R 3 and R 4 together with the nitrogen atom to which they are attached, denote 5- to 8-membered heterocyclyl
- B is phenyl or heteroaryl optionally substituted with up to 3 groups each independently selected from the group Ci-C ⁇ alkyl, Ci-C ⁇ alkoxy, hydroxycarbonyl which
- Ci-C ⁇ alkylaminocarbonyl C r C 6 alkoxycarbonyl, dC 6 alkylcarbonyl, CpC 6 - alkylsulfonyl, and Ci-C 6 substituted alkylthio,
- R 3 and R 4 have the meanings given above,
- a further embodiment of the invention relates to compounds of the formula (I)
- Ci-C 5 alkyl or C 3 -C 6 cycloalkyl which is optionally substituted by up to 3 radicals independently selected from the group Ci-C4 alkyl, Ci-C4-alkoxy, hydroxycarbonyl, cyano, amino, hydroxy , 4 alkoxycarbonyl, C r C 6 alkylcarbonyl, Ci-C4-alkylsulfonyl and C r C 4 -alkylthio Ci-GrAlkylamino fluorine, chlorine, bromine, Ci-C,
- C r C 4 -alkyl and CpC 4 -alkoxy optionally have a radical selected from the group consisting of hydroxyl, cyano, fluorine, chlorine, bromine, hydroxycarbonyl and a group of the formula -NR 3 R 4 ,
- R 3 and R 4 independently of one another are hydrogen or C 1 -C 4 -alkyl
- R 3 and R 4 together with the nitrogen atom to which they are attached, denote 5- to 6-membered heterocyclyl, are substituted
- B is phenyl, thienyl or pyridyl, which are optionally each independently selected from the group consisting of C 1 -C 4 -alkyl, C 1 -C 4 -alkoxy, hydroxycarbonyl, with up to 3 radicals
- C r C 4 alkyl and Ci-C 4 alkoxy optionally substituted with a radical selected from the group hydroxy, cyano, fluorine, chlorine, bromine, hydroxycarbonyl and a group of the formula -ISfR 3 R 4,
- R 3 and R 4 have the meanings given above,
- a further embodiment of the invention relates to compounds of the formula (I)
- B is phenyl or pyridyl, which is optionally selected independently of one another from the group consisting of methyl, ethyl, 2-propyl, trifluoromethyl, methoxy, ethoxy, fluorine and chlorine, with up to 3 radicals,
- a further embodiment of the invention relates to compounds of the formula (I)
- a further embodiment of the invention relates to compounds of the formula (I)
- A is 2-methylpropyl, 2-butyl, 2-pentyl or 3-pentyl and
- a further embodiment of the invention relates to compounds of the formula (I)
- B is phenyl, thienyl or pyridyl, which are optionally each independently selected from the group consisting of C 1 -C 3 -alkyl, trifluoromethyl, hydroxy, methoxy, with up to 3 radicals
- Cycs alkyl. dVCfi-alkyl, CVCs-alkyl and CrCV-alkyl are a straight-chain or branched alkyl radical having 1 to 8, preferably 1 to 6, particularly preferably 1 to 5 and 1 to 4 carbon atoms.
- Preferred examples include methyl, ethyl, n-propyl, isopropyl, 2-butyl, 2-pentyl and 3-pentyl.
- for a straight-chain or branched alkoxy radical having 1 to 6, preferably 1 to 4, particularly preferably 1 to 3 carbon atoms.
- Preferred examples include methoxy, ethoxy, n-propoxy, isopropoxy, tert-butoxy, n-pentoxy and n-hexoxy.
- CVCfi-alkoxycarbonyl represents a straight-chain or branched alkoxycarbonyl radical having 1 to 6, preferably 1 to 4 and particularly preferably 1 to 3 carbon atoms.
- Preferred examples include methoxycarbonyl, ethoxycarbonyl, n-propoxycarbonyl, isopropoxycarbonyl and tert-butoxycarbonyl.
- C 1 -C 8 -alkylamino represents a straight-chain or branched mono- or dialkylamino radical having 1 to 6, preferably 1 to 4 and particularly preferably 1 to 3, carbon atoms.
- Preferred examples include methylamino, ethylamino, n-propylamino, isopropylamino, tert-butylamino, n-pentylamino and n-hexylamino, dimethylamino, diethylamino, di-n-propylamino, diisopropylamino, di-tert-butylamino, di-n-pentylamino , Di-n-hexylamino, ethylmethylamino, isopropylmethylamino, n-butylethylamino and n-hexyl-i-pentylamino.
- CVCV alkylaminocarbonyl is a mono- or dialkylamino radical linked via a carbonyl group, where the alkyl radicals may be identical or different, are straight-chain or branched and contain in each case 1 to 6, preferably 1 to 4, and particularly preferably 1 to 3, carbon atoms.
- Preferred examples include methylaminocarbonyl, ethylaminocarbonyl, n -propylaminocarbonyl, isopropylaminocarbonyl, tert-butylaminocarbonyl, n -pentylaminocarbonyl, n-hexylaminocarbonyl, dimethylaminocarbonyl, diethylaminocarbonyl, di-n-propylaminocarbonyl, diisopropylaminocarbonyl, di-t-butylaminocarbonyl, di-n-pentylaminocarbonyl, di -n-hexylaminocarbonyl, ethylmethylaminocarbonyl, isopropylmethylaminocarbonyl, n-butylethylaminocarbonyl and n-hexyl-i-pentylaminocarbonyl. Furthermore, in the case of a dialkylamino radical, the two alky
- dVCfi-alkylcarbonyl represents a straight-chain or branched alkylcarbonyl radical having 1 to 6 and preferably 1 to 4 carbon atoms. Examples which may be mentioned are: acetyl, ethylcarbonyl, propylcarbonyl, isopropylcarbonyl, butylcarbonyl, isobutylcarbonyl, pentylcarbonyl and hexylcarbonyl. Particularly preferred are acetyl and ethylcarbonyl.
- C r Cft-alkylsulfonyl is a straight-chain or branched alkylsulfonyl group having 1 to 6, preferably 1 to 4 and particularly preferably having 1 to 3 carbon atoms.
- Preferred examples include methylsulfonyl, ethylsulfonyl, n-propylsulfonyl, isopropylsulfonyl, t-butylsulfonyl, n-pentylsulfonyl and n-hexylsulfonyl.
- C j -C f -alkylthio represents a straight-chain or branched alkylthio radical having 1 to 6, preferably 1 to 4 and particularly preferably 1 to 3 carbon atoms.
- Preferred examples include methylthio, ethylthio, n-propylthio, isopropylthio, tert-butylthio, n-pentylthio and n-hexylthio.
- Halogen is fluorine, chlorine, bromine and iodine. Preference is given to fluorine, chlorine, bromine, more preferably fluorine and chlorine.
- Heteroaryl is an aromatic, monocyclic radical having 5 to 6 ring atoms and up to 3 heteroatoms from the series S, O and / or N. Preferred are 5- to 6-membered heteroaryls having up to 2 heteroatoms. The heteroaryl group may be bonded via a carbon or nitrogen atom. Preferred examples include thienyl, furyl, pyrrolyl, thiazolyl, oxazolyl, imidazolyl, tetrazolyl, pyridyl, pyrimidinyl and pyridazinyl.
- 3- to 8-membered cycloalkyl represents saturated and partially unsaturated non-aromatic cycloalkyl radicals having 3 to 8, preferably 3 to 6 and more preferably 5 to 6 carbon atoms in the cycle.
- Preferred examples include cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl and cyclohexenyl.
- 5- to 8-membered heterocyclyl is a mono- or polycyclic, heterocyclic radical having 5 to 8 ring atoms and up to 3, preferably 2 heteroatoms or hetero groups from the series N, O, S, SO, SO 2 .
- Mono- or bicyclic heterocyclyl is preferred. Particularly preferred is monocyclic heterocyclyl.
- heteroatoms N and O are preferred.
- the heterocyclyl radicals may be saturated or partially unsaturated. Saturated heterocyclyl radicals are preferred. Particularly preferred are 5- to 7-membered heterocyclyl radicals.
- Preferred examples include oxetan-3-yl, pyrrolidin-2-yl, pyrrolidin-3-yl, pyrrolinyl, tetrahydrofuranyl, tetrahydrothienyl, pyranyl, piperidinyl, thiopyranyl, morpholinyl, perhydroazepinyl.
- radicals in the compounds according to the invention are optionally substituted, unless otherwise specified, a substitution with up to three identical or different substituents is preferred.
- Another object of the present invention relates to compounds of the formula
- D is phenyl, heteroaryl or a group of the formula
- phenyl and heteroaryl where appropriate, having up to 2 radicals, independently of one another, are selected from the group consisting of heteroaryl, halogen, C 1 -C 6 -alkyl, C 1 -C 6 -alkyl,
- Ci-C ⁇ -alkyl optionally with a group of the formula -NR 5 R 6 , in which R 5 is C r C 6 alkyl and R 6 is hydrogen or C r C 6 alkoxy (Ci-C 6 ) - alkyl, and
- Heteroaryl is optionally substituted by Ci-C ⁇ - alkoxy,
- R 1 is C 3 -C 8 cycloalkyl, C r C 6 alkyl, C 1 -C 6 alkoxy (Ci-C 6) alkyl, benzyl or a group of
- C 3 -C 8 -cycloalkyl is optionally substituted by hydroxy, CpC ⁇ -alkyl or trifluoromethyl
- benzyl is optionally substituted by C 1 -C 6 -alkoxy or halogen
- R 2 is hydrogen
- R 1 and R 2 together with the nitrogen atom to which they are attached form a 5- to 6-membered heterocyclyl, which is optionally selected from up to 2 substituents independently selected from the group CpC ⁇ -alkyl, hydroxy,
- C 1 -C 6 -alkyl is optionally substituted by hydroxy or heteroaryl
- Another embodiment of the invention relates to compounds of the formula (II) in which
- phenyl and heteroaryl optionally with up to 2 radicals independently of one another are selected from the group heteroaryl, halogen, C 1 -C 4 -alkyl, CpC 4 -
- Ci-Gj-alkyl optionally with a group of the formula -NR 3 R 4 , in which R 3 is C, -C 4 alkyl and R 4 is hydrogen or C r C 4 alkoxy (CpC 4 ) - alkyl, and
- Heteroaryl is optionally substituted by C 1 -C 4 -alkoxy
- R 1 is C 3 -C 6 cycloalkyl, C r C 4 alkyl, Ci-C 4 alkoxy (Ci-C 4) alkyl, benzyl or a group of
- C 3 -C 6 -cycloalkyl is optionally substituted by hydroxyl, C 1 -C 4 -alkyl or trifluoromethyl,
- benzyl is optionally substituted by C 1 -C 4 -alkoxy or halogen
- R 2 is hydrogen
- R 1 and R 2 together with the nitrogen atom to which they are attached, a 5- to 6-membered heterocyclyl, which is optionally substituted with up to 2 substituents independently selected from the group C r C 4 alkyl, hydroxy,
- C r C 4 -alkyl is optionally substituted by hydroxy or heteroaryl
- Another embodiment of the invention relates to compounds of the formula (II) in which A is phenyl, thienyl or a group of the formula
- phenyl and thienyl are optionally substituted by up to 2 radicals independently of one another selected from the group of pyridyl, fluorine, chlorine, bromine, C 1 -C -alkyl, C 1 -C 4 -alkoxy, trifluoromethyl, trifluoromethoxy, benzyloxy and benzyl,
- Ci-C 4 alkyl optionally having a group of the formula -NR 3 R 4 in which R 3 is C r C 4 alkyl and R 4 is hydrogen or C r C 4 alkoxy (C r C 4) - alkyl, and
- Pyridyl is optionally substituted by C 1 -C 4 -alkoxy
- R 1 is C 3 -C 6 cycloalkyl, C r C 4 alkyl, C r C 4 alkoxy (Ci-C 4) alkyl, benzyl or a group of
- C 3 -C 6 -cycloalkyl is optionally substituted by hydroxy, C 1 -C 4 -alkyl or trifluoromethyl
- benzyl is optionally substituted by C 1 -C 4 -alkoxy, fluorine, chlorine or bromine,
- R 2 is hydrogen
- R 1 and R 2 together with the nitrogen atom to which they are attached, a 5- to 6-membered heterocyclyl selected from the group pyrrolidinyl, piperidinyl,
- C 1 -C 4 -alkyl is optionally substituted by hydroxy or pyridyl
- Another embodiment of the invention relates to compounds of the formula (II) in which
- A is phenyl, thienyl or a group of the formula
- phenyl is optionally substituted by up to 2 independently selected from the group consisting of pyridyl, fluorine, chlorine, methyl, methoxy, ethoxy, trifluoromethyl, trifluoromethoxy, benzyloxy and benzyl,
- methyl is optionally substituted by a group of the formula -NR 3 R 4 , in which R 3 is methyl and R 4 is hydrogen or 2-methoxyethyl, and
- Pyridyl is optionally substituted with methoxy
- R 1 is C 3 -C 6 -cycloalkyl, methyl, ethyl, propyl, 2-methoxyethyl, benzyl or a group of
- C 3 -C 6 -cycloalkyl is optionally substituted by hydroxy, methyl or trifluoromethyl
- R 2 is hydrogen
- R 1 and R 2 together with the nitrogen atom to which they are attached, a 5- to 6-membered heterocyclyl selected from the group pyrrolidinyl, piperidinyl,
- Piperazinyl and morpholinyl which are optionally selected with up to 2 substituents independently selected from the group of methyl, ethyl, propyl, tert-butyl, hydroxy, cyano, oxo, pyridyl, benzyl, formyl, methylcarbonyl, ethylcarbonyl, propylcarbonyl and a the following groups
- OrCfi-alkyl is a straight-chain or branched alkyl radical having 1 to 6, preferably 1 to 4 carbon atoms. Preferred examples include methyl, ethyl, n-propyl, isopropyl, 2-butyl, tert-butyl, 2-pentyl, 3-pentyl and n-hexyl.
- C 1 -Cs-AIkOXV represents a straight-chain or branched alkoxy radical having 1 to 6, preferably 1 to 4, particularly preferably 1 to 3 carbon atoms.
- Preferred examples include methoxy, ethoxy, n-propoxy, isopropoxy, tert-butoxy, n-pentoxy and n-hexoxy.
- Preferred examples include methoxymethyl, 2-methoxyethyl, ethoxymethyl and 2-ethoxyethyl.
- C j -CFT-Alkylcarbonyl is a straight-chain or branched alkylcarbonyl group having 1 to 6, preferably 1 to 4 and particularly preferably 1 to 3 carbon atoms.
- Preferred examples include methylcarbonyl, ethylcarbonyl, n-propylcarbonyl, isopropylcarbonyl and tert-butylcarbonyl.
- 3- to 8-membered cycloalkyl represents saturated cycloalkyl radicals having 3 to 8, preferably 3 to 6 and more preferably 5 to 6 carbon atoms in the cycle.
- Preferred examples include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and cycloheptyl.
- Halogen is fluorine, chlorine, bromine and iodine. Preference is given to fluorine, chlorine, bromine, more preferably fluorine and chlorine.
- Heteroaryl is an aromatic, monocyclic radical having 5 to 6 ring atoms and up to 3 heteroatoms from the series S, O and / or N. Preferred are 5- to 6-membered heteroaryls having up to 2 heteroatoms. The heteroaryl group may be bonded via a carbon or nitrogen atom. Preferred examples include thienyl, furyl, pyrrolyl, thiazolyl, oxazolyl, imidazolyl, pyridyl, pyrimidinyl and pyridazinyl.
- 5- to 6-membered heterocyclyl is a monocyclic, saturated or partially unsaturated heterocyclic radical having 5 to 6 ring atoms and up to 2 heteroatoms from the series N, O, S.
- heteroatoms N and O are preferred.
- Preferred examples include pyrrolidinyl, pyrrolinyl, tetrahydrofuranyl, tetrahydrothienyl, pyranyl, thiopyranyl, piperidinyl, morpholinyl, thiomorpholinyl and piperazinyl.
- radicals in the compounds of the invention are optionally substituted, unless otherwise specified, substitution with up to three identical or different substituents is preferred.
- R 1 and R 2 have the meanings given above,
- R 1 and R 2 have the meanings given above,
- the compound of the formula (III) is known from the literature (R. Gompper, W. Toepfl, Chem. Ber. 1962, 95, 2861-2870).
- the compounds of the formulas (IV) and (VI) are commercially available, known from the literature or can be prepared in analogy to processes known from the literature (see, for example, H. Gielen, C. Alonso-Alija, M. Hendrix, U. Nie Strukturer, D. Schauß, Tetrahedron Lett., 2002, 43, 419-421).
- high-boiling, inert organic solvents are suitable which do not change under the reaction conditions. These include preferably toluene, acetonitrile, dimethylformamide, dimethyl sulfoxide or sulfolane. It is likewise possible to carry out the reaction without solvent in the melt. Particularly preferred is a reaction without solvent or in dimethylformamide, acetonitrile or toluene.
- the reaction is generally carried out in a temperature range from + 70 ° C to +200 0 C, preferably in a temperature range from +100 0 C to + 15O 0 C.
- the reaction may be at atmospheric, ER- be performed elevated or at reduced pressure (eg from 0.5 to 5 bar). Generally, one works at normal pressure.
- the compound of the formula (IV) is used here in an amount of 1 to 2 mol, preferably in an equivalent amount of 1 mol, based on 1 mol of the compound of the formula (HI).
- organic solvents which do not change under the reaction conditions. These preferably include dimethylformamide, dimethylsulfoxide or acetonitrile. It is also possible to carry out the reaction without solvent. Particularly preferred is a reaction without solvent or in acetonitrile.
- the reaction is generally carried out in a temperature range from + 5O 0 C to +150 0 C, preferably installed into a temperature range of + 7O 0 C to +100 0 C.
- the reaction can be carried out at atmospheric, elevated or reduced pressure (eg from 0.5 to 5 bar). Generally, one works at normal pressure.
- the compound of the formula (TV) is in this case used in an amount of 1 to 10 mol, preferably in an excess of 3 to 10 mol, based on 1 mol of the compound of the formula (VII).
- ком ⁇ онентs which do not change under the reaction conditions. These include preferably dimethylformamide, dimethyl sulfoxide, acetonitrile, dioxane or alcohols such as methanol, ethanol, propanol, isopropanol, n-butanol or tert-butanol. It is also possible to use mixtures of said solvents. Particularly preferred for the process step (V) + (VI) -> (II) dimethylformamide or acetonitrile and for the process step (DI) + (VI) -> (VIT) ethanol.
- the reaction is generally carried out in a temperature range of +50 0 C to +150 0 C, preferably in a temperature range of +70 0 C to +100 0 C.
- the reaction can be carried out at normal, elevated or at reduced pressure (eg 0.5 to 5 bar). Generally, one works at normal pressure.
- Suitable bases for process step (V) + (VI) ⁇ (E) or (HI) + (VI) ⁇ (VE) are preferably alkali metal carbonates such as lithium, sodium, potassium or cesium carbonate or organic amine bases such as for example, pyridine, triethylamine, ethyldiisopropylamine, N-methylmorpholine or N-methylpiperidine. Particularly preferred is potassium carbonate or triethylamine.
- the base is used here in an amount of 1.5 to 4 mol, preferably in an amount of 1.5 to 2 mol, based on 1 mol of the compound of the formula (V) or (BT).
- the connection of Formula (VI) is used in an amount of 1 to 1.5 mol, preferably in an amount of 1.2 mol, based on 1 mol of the compound of formula (V) or (III).
- Compounds of the invention are the compounds of formula (I) and (II) and their salts, solvates and solvates of the salts; the compounds of formulas (I) and (II) of the following formulas and their salts, solvates and solvates of the salts and of formulas (I) and (II), hereinafter referred to as exemplary compounds and their salts, solvates and solvates the salts, as far as the compounds encompassed by formulas (I) and (II), are not already salts, solvates and solvates of the salts.
- the compounds of the invention may exist in stereoisomeric forms (enantiomers, diastereomers).
- the invention therefore relates to the enantiomers or diastereomers and their respective mixtures. From such mixtures of enantiomers and / or diastereomers, the stereoisomerically uniform components can be isolated in a known manner.
- Physiologically acceptable salts of the compounds of formulas (I) and (II) include acid addition salts of mineral acids, carboxylic acids and sulfonic acids, e.g. Salts of hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, methanesulfonic acid, ethanesulfonic acid, toluenesulfonic acid, benzenesulfonic acid, naphthalenedisulfonic acid, acetic acid, propionic acid, lactic acid, tartaric acid, malic acid, citric acid, fumaric acid, maleic acid and benzoic acid.
- salts of hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, methanesulfonic acid, ethanesulfonic acid, toluenesulfonic acid, benzenesulfonic acid, naphthalenedisulfonic acid acetic acid, propionic acid, lactic acid, tartaric
- Physiologically acceptable salts of the compounds of formulas (I) and (II) also include salts of conventional bases such as, for example, and preferably alkali metal salts (eg, sodium and potassium salts), alkaline earth salts (eg, calcium and magnesium salts), and ammonium salts derived from ammonia or organic amines having 1 to 16 carbon atoms such as, by way of example and by way of preference, ethylamine, diethylamine, triethylamine, ethyldiisopropylamine, monoethanolamine, diethanolamine, triethanolamine, dicyclohexylamine, dimethylaminoethanol, procaine, dibenzylamine, N-methylmorpholine, dehydroabietylamine, arginine, lysine, ethylenediamine and methylpiperidine.
- alkali metal salts eg, sodium and potassium salts
- alkaline earth salts eg, calcium and magnesium salts
- solvates are those forms of the compounds which form a complex in the solid or liquid state by coordination with solvent molecules. Hydrates are a special form of solvates that coordinate with water.
- the present invention also includes prodrugs of the compounds of the invention.
- prodrugs includes compounds which may themselves be biologically active or inactive, but which are converted during their residence time in the body into compounds of the invention (for example metabolically or hydrolytically).
- the compounds of the invention show an unpredictable, valuable pharmacological and pharmacokinetic activity spectrum. They are characterized in particular by an inhibition of PDE9A.
- the compounds according to the invention are suitable for the preparation of medicaments for the treatment of cardiovascular diseases.
- the compounds according to the invention can be used alone or in combination with other medicaments for the treatment of cardiovascular diseases.
- cardiovascular diseases such as hypertension and cardiac insufficiency, acute heart failure, stable and unstable angina pectoris, peripheral and cardiovascular diseases, arrhythmias, for the treatment of thromboembolic disorders and ischaemias such as myocardial infarction, stroke, transient and ischemic attacks, peripheral circulatory disorders, prevention of restenosis such as after thrombolytic therapy, percutaneous transluminal angioplasties (PTA), percutaneous transluminal coronary angioplasty (PTCA), bypass and for the treatment of arteriosclerosis, asthmatic diseases and diseases of the genitourinary system such as prostatic hypertrophy, erectile dysfunction, female sexual Dysfunction, osteoporosis, glaucoma, pulmonary hypertension, portal hypertension, ischemic kidney disease, nephrosis, such as the nephrotic syndrome, renal stenosis, kidney insufficiency, acute renal failure, metabolic syndrome, gastroparesis, incontinence, liver
- cardiovascular diseases such as hypertension and cardiac in
- Device type MS Micromass ZQ
- Device type HPLC Waters Alliance 2795
- Eluent A 1 l of water + 0.5 ml of 50% formic acid
- eluent B 1 l of acetonitrile + 0.5 ml of 50% formic acid
- Gradient: 0.0 min 90% A flow 1 ml / min ⁇ 2.5 min 30% A, flow 2 ml / min ⁇ 3.0 min 5% A, flow 2 ml / min - »4.5 min 5% A, flow 2 ml / min ; Oven: 5O 0 C
- UV detection 210 nm.
- Device type MS Micromass ZQ
- Device type HPLC HP 1100 Series
- UV DAD Column: Phenomenex Synergi 2 ⁇ Hydro-RP Mercury 20 mm x 4 mm
- Eluent A 1 l of water + 0.5 ml of 50% formic acid
- eluent B 1 l of acetonitrile + 0.5 ml of 50% formic acid
- Gradient: 0.0 min 90% A flow 1 ml / min ⁇ 2.5 min 30% A, flow 2 ml / min ⁇ 3.0 min 5% A, flow 2 ml / min ⁇ 4.5 min 5% A, flow 2 ml / min
- Oven 50 ° C .
- UV detection 210 nm.
- Device type MS Micromass ZQ
- Device type HPLC HP 1100 Series
- UV DAD Column: Grom-Sil 120 ODS-4 HE 50 mm ⁇ 2 mm, 3.0 ⁇ m
- Eluent A water + 500 ⁇ l 50% formic acid / 1
- eluent B acetonitrile + 500 ⁇ l 50% formic acid / 1
- Oven 50 ° C .
- Flow 0.8 ml / min
- UV detection 210 nm.
- Device type MS Micromass ZQ
- Device type HPLC Waters Alliance 2790
- Eluent A water + 500 ⁇ l 50% formic acid / 1
- eluent B acetonitrile + 500 ⁇ l 50% formic acid / 1
- Oven 45 ° C .
- UV detection 210 nm.
- Device type MS Micromass ZQ
- Device type HPLC Waters Alliance 2790
- Eluent A water + 500 ⁇ l 50% formic acid / 1
- eluent B acetonitrile + 500 ⁇ l 50% formic acid / 1
- Oven 45 ° C .
- UV detection 210 nm.
- Device type MS Micromass ZQ
- Device type HPLC Waters Alliance 2795; Column: Merck Chromolith SpeedROD RP-18e 50 mm x 4.6 mm; Eluent A: water + 500 ⁇ l 50% formic acid / 1, eluent B: acetonitrile + 500 ⁇ l 50% formic acid / 1; Gradient: 0.0 min 10% B ⁇ 3.0 min 95% B ⁇ 4.0 min 95% B; Oven: 35 ° C; Flow: 0.0 min 1.0 ml / min ⁇ 3.0 min 3.0 ml / min ⁇ 4.0 min 3.0 ml / min; UV detection: 210 nm.
- Device type MS Micromass ZQ
- Device type HPLC Waters Alliance 2790; Column: Uptisphere C 18 50 mm x 2.0 mm, 3.0 ⁇ m; Eluent B: acetonitrile + 0.05% formic acid, eluent A: water + 0.05% formic acid; Gradient: 0.0 min 5% B ⁇ 2.0 min 40% B ⁇ 4.5 min 90% B ⁇ 5.5 min 90% B; Oven: 45 ° C; Flow: 0.0 min 0.75 ml / min ⁇ 4.5 min 0.75 ml / min ⁇ 5.5 min 1.25 ml / min; UV detection: 210 nm.
- amidines required for the following reactions are prepared from the corresponding nitriles or esters according to Gielen H., Alonso-Aliaja C, Hendrix M., Nieelleer U., Schauß D., Tetrahedron Lett. 43, 419-421 (2002).
- the product is separated from the precipitated solid by suction and the filter cake washed several times with methanol.
- the combined filtrates are concentrated to dryness, the residue is then slurried in dichloromethane / methanol 10: 1 and again filtered off with suction. After concentration, the filtrate gives 0.5 g (46% of theory) of the title compound.
- Example 2A Analogously to the preparation of Example 2A, 0.5 g (2.00 mmol) of 2- (3-chlorophenyl) ethanamidine hydrobromide with 0.41 g (2.00 mmol) of methyl 2-cyano-3,3-dimethylthioprop-2-enoate and 0.81 g (8.01 mmol ) Reacted triethylamine to 0.5 g (86% of theory) of the title compound.
- Example 2A Analogously to the preparation of Example 2A, 1.00 g (4.17 mmol) of 2- (3,4-dichlorophenyl) ethanediamine hydrochloride with 0.85 g (4.17 mmol) of methyl 2-cyano-3,3-dimethylthioprop-2-enoate and 1.69 g (16.7 mmol) of triethylamine to 0.6 g (44% of theory) of the title compound.
- Example 2A Analogously to the preparation of Example 2A, 100 mg (0.43 mmol) of 2- (3-fluorophenyl) ethanediamine hydrochloride are reacted with 87 mg (0.43 mmol) of methyl 2-cyano-3,3-dimethylthioprop-2-enoate and 174 mg ( 1.72 mmol) of triethylamine were reacted to give 28 mg (24% of theory) of the title compound. - -
- Example 2A Analogously to the preparation of Example 2A, 0.5 g (2.10 mmol) of 2- [3- (trifluoromethyl) phenyl] ethanamidine hydrochloride with 0.43 g (2.10 mmol) of methyl 2-cyano-3,3-dimethylthioprop-2-enoate and 0.85 g (8.38 mmol) of triethylamine to 0.4 g (59% of th.) Of the title compound.
- PDE1C Recombinant PDE1C (GenBank / EMBL Accession Number: NM_005020, Loughney et al., J. Biol. Chem., 1996 271, 796-806), PDE2A (GenBank / EMBL Accession Number: NM_002599, Rosman et al., Gene 1997, 191, 89-95 ), PDE3B (GenBank / EMBL Accession Number: NM_000922, Miki et al, Genomics 1996, 36, 476-485), PDE4B (GenBank / EMBL Accession Number: NM_002600, Obernolte et al., Gene., 1993, 129, 239-247).
- PDE5A GenBank / EMBL Accession Number: NM 001083, Loughney et al, Gene 1998, 216, 139-147
- PDE7B GenBank / EMBL Accession Number: NM_018945, Hetman et al. Proc. Natl. Acad. Be. USA 2000, 97, 472-476
- PDE8A GenBank / EMBL Accession Number: AF_056490, Fisher et al., Biochem Biophys Res. Commun. 1998 246, 570-577
- PDE9A Fisher et al., J.
- test substances are dissolved in 100% DMSO and serially diluted to determine their in vitro effect on PDE 9A.
- serial dilutions 200 ⁇ M to 1.6 ⁇ M are prepared (resulting final concentrations in the assay: 4 ⁇ M to 0.032 ⁇ M).
- 2 ⁇ L each of the diluted substance solutions are placed in the wells of microtiter plates (Isoplate, Wallac Inc., Atlanta, GA). Subsequently, 50 ⁇ L of a dilution of the PDE9A preparation described above is added.
- the dilution of the PDE9A preparation is chosen such that during the later incubation less than 70% of the substrate is reacted (typical dilution: 1: 10,000; dilution buffer: 50 mM Tris / HCl pH 7.5, 8.3 mM MgCl 2 , 1.7 mM EDTA, 0.2% BSA).
- the substrate, [8- 3 H] guanosine 3 ', 5 ( cyclic phosphate (1 ⁇ Ci / ⁇ L; Ameram Pharmacia Biotech., Piscataway, NJ) is diluted 1: 2000 with assay buffer (50 mM Tris / HCl pH 7.5, 8.3 mM MgCl 2 , 1.7 mM EDTA) to a concentration of 0.0005 ⁇ Ci / ⁇ L
- 50 ⁇ L (0.025 ⁇ Ci) of the diluted substrate the enzyme reaction is finally started
- the test mixtures are incubated for 60 min at room temperature and the reaction by 25 ⁇ l of a suspension containing 18 mg / ml of Yttrium Scintillation Proximity Beads (Amersham Pharmacia Biotech., Piscataway, Biol.) Are then added, followed by 25 ⁇ l of a PDE9A inhibitor dissolved in assay buffer (eg the inhibitor from Preparation Example 1, 10
- microtiter plates are sealed with a foil and allowed to stand at room temperature for 60 min, then the plates are measured for 30 s per well in a microbeta scintillation counter (Wallac Inc., Atlanta, GA) IC 5 o values are determined by plotting the concentration of the substance against the percent inhibition.
- PDE10A and PDEI IA are determined according to the assay protocol described above for PDE 9A with the following adaptations:
- the substrate used is [5 ', 8- 3 H] adenosine 3', 5'-cyclic phosphate (1 - 7 - ⁇ Ci / ⁇ L; Amersham Pharmacia Biotech., Piscataway, NJ).
- the addition of an inhibitor solution to stop the reaction is not necessary. Instead, following the incubation of substrate and PDE, the addition of the Yttrium Scintillation Proximity Beads is continued as described above, thereby stopping the reaction.
- PDE2A PDE5 and the protocol is additionally adapted as follows: In pdel Calmodulin 10 -7 M and CaCl 2 are added 3 mM to the reaction mixture in addition. PDE2A is stimulated in the assay by addition of cGMP 1 ⁇ M and tested with a BSA concentration of 0.01%.
- the force of contraction is detected with Statham UC2 cells, amplified and digitized via A / D converters (DAS-1802 HC, Keithley Instruments Munich) and registered in parallel on chart recorders.
- DAS-1802 HC A / D converters
- phenylephrine is added cumulatively to the bath in increasing concentration.
- the substance to be examined is examined in each subsequent passage in increasing dosages and the height of the contraction is compared with the height of the contraction achieved in the last predistortion. This is used to calculate the concentration required to reduce the level of the control value by 50% (IC 50 ).
- the standard application volume is 5 ⁇ l, the DMSO content in the bath solution corresponds to 0.1%.
- test substances are dissolved in a mixture of glycerol: water: polyethylene glycol 6: 10: 9.69 and applied in a volume of 1 ml / kg with the gavage.
- mice Male Wistar rats weighing 300-350 g (Harlan Winkelmann, Germany) were anesthetized with 1-2.5% isofluoran in a mixture of nitrous oxide / O 2 (2: 1).
- a catheter was inserted into the femoral artery, the substance was administered via a femoral vein catheter and urine collection via a bladder catheter. After the operation, 5 ml / kg of phys. NaCl were given intravenously as a bolus to equalize the fluid and the animals were infused continuously for 1 h with phys. NaCl at a rate of 100 ⁇ l / kg / min via the venous catheter. The body temperature of the animals was kept constant over a hot plate.
- test substances along with ANP were continuously infused via the venous catheter at a rate of 100 ⁇ l / kg / min.
- Urine was collected every 15 min and the volume, cGMP content (RIA), and Na + and K + concentrations (flame photometry) were measured.
- the substance to be tested is administered intravenously to animals (for example mouse, rat, dog) as a solution, and the oral administration is carried out as a solution or suspension via a gavage.
- animals for example mouse, rat, dog
- the animals are bled at fixed times, this is heparinized and then plasma is recovered therefrom by centrifugation.
- the substance is analytically quantified in the plasma via LC / MSMS.
- the pharmacokinetic parameters are calculated from the plasma concentration / time profiles determined in this way by means of a validated pharmacokinetic calculation program.
- Recombinant enzymes eg CYP 1A2, 2C8, 2C9, 2CI9, 2D6 or 3A4
- substrates generally containing fluorescein or coumarin substructures are used in the assay for the formation of fluorescent metabolites.
- a substrate concentration and 8 concentrations of the potential inhibitor are used.
- the extent of fluorescent metabolites in comparison to the control (without inhibitor) is determined by means of fluorescence readers and an IC 50 value is calculated [Anal. Biochem. 248, 188 (1997)].
- the second assay uses human liver microsomes as the enzyme source and phenacetin (CYP1A2), diclofenac (CYP2C9), dextromethorphan (CYP2D6) and CYP isoform-selective substrates Midazolam (CYP3A4) used.
- CYP1A2 phenacetin
- CYP2C9 diclofenac
- CYP2D6 dextromethorphan
- CYP3A4 CYP isoform-selective substrates Midazolam
- K 1 values are calculated from the reduction in metabolite formation compared to the control (1 substrate, 3 inhibitor concentrations).
- the hepatocytes are treated for 5 days in duplicate with different concentrations of the test substances compared with the inducers rifampicin (RIF, 50 ⁇ M), omeprazole (OME, 100 ⁇ M) and phenobarbital (PB, 2 mM).
- the final concentrations of the test substances are 0.01-10 ⁇ g / ml.
- the cell cultures show the inductive effect of the test substances on the cytochrome (CYP) P450 enzymes 1A2, 2B6, 2C19 and 3A4 by adding the substrates 7-ethoxyresorufin (CYP1A2), [ 14 C] -S-mephenytoin (CYP2B6 and 2Cl 9) and [ 14 C] -testosterone (CYP3A4) at day 8. From the thus measured enzyme activities CYP 1A2, 2B6, 2Cl 9 and 3 A4 treated cells compared to untreated cells, the inductive potential of the test substances is determined.
- CYP cytochrome
- Another object of the present invention are pharmaceutical compositions containing at least one compound of the invention and at least one or more other active ingredients, in particular for the treatment and / or prophylaxis of the aforementioned diseases.
- the compounds according to the invention can act systemically and / or locally.
- they may be applied in a suitable manner, e.g. oral, parenteral, pulmonary, nasal, sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctivae otic or as an implant or stent.
- the compounds according to the invention can be administered in suitable administration forms.
- Contain mold such as tablets (uncoated or coated tablets, for example with enteric or delayed-dissolving or insoluble coatings controlling the release of the compound of the invention), rapidly disintegrating tablets or films / wafers, films / lyophilisates, capsules (eg hard or soft gelatin capsules), dragees, granules, pellets, powders , Emulsions, suspensions, aerosols or solutions.
- Parenteral administration can be accomplished by bypassing a resorption step (e.g., intravenously, intraarterially, intracardially, intraspinal, or intralumbar) or by resorting to absorption (e.g., intramuscularly, subcutaneously, intracutaneously, percutaneously, or intraperitoneally).
- a resorption step e.g., intravenously, intraarterially, intracardially, intraspinal, or intralumbar
- absorption e.g., intramuscularly, subcutaneously, intracutaneously, percutaneously, or intraperitoneally.
- parenteral administration are suitable as application forms u.a. Injection and infusion preparations in the form of solutions, suspensions, emulsions, lyophilisates or sterile powders.
- Inhalation medicines including powder inhalers, nebulizers
- nasal drops solutions, sprays
- lingual, sublingual or buccal tablets films / wafers or capsules
- suppositories ear or eye preparations
- vaginal capsules aqueous suspensions (lotions, shake mixtures)
- lipophilic suspensions ointments
- creams transdermal therapeutic systems (such as patches)
- milk, pastes, foams, scattering powders, implants or stents for example Inhalation medicines (including powder inhalers, nebulizers), nasal drops, solutions, sprays; lingual, sublingual or buccal tablets, films / wafers or capsules, suppositories, ear or eye preparations, vaginal capsules, aqueous suspensions (lotions, shake mixtures), lipophilic suspensions, ointments, creams, transdermal therapeutic systems (such as patches), Milk, pastes, foams, scattering powders, implants or stents.
- the compounds according to the invention can be converted into the stated administration forms. This can be done in a conventional manner by mixing with inert, non-toxic, pharmaceutically suitable excipients.
- excipients for example microcrystalline cellulose, lactose, mannitol
- solvents for example liquid polyethylene glycols
- emulsifiers and dispersants or wetting agents for example sodium dodecyl sulfate, polyoxysorbitanoleate
- binders for example polyvinylpyrrolidone
- synthetic and natural polymers for example albumin
- stabilizers for example, antioxidants such as ascorbic acid
- dyes eg, inorganic pigments such as iron oxides
- compositions containing at least one compound of the invention usually together with one or more inert, non-toxic, pharmaceutically suitable excipients, and their use for the purposes mentioned above.
- parenteral administration per day amounts of about 0.001 to 10 mg / kg body weight to achieve effective results.
- the amount per day is about 0.005 to 3 mg / kg of body weight.
- the compounds according to the invention can be converted into pharmaceutical preparations as follows:
- the mixture of compound of the invention, lactose and starch is granulated with a 5% solution (m / m) of the PVP in water.
- the granules are mixed after drying with the magnesium stearate for 5 minutes.
- This mixture is compressed with a conventional tablet press (for the tablet format see above).
- a pressing force of 15 kN is used as a guideline for the compression.
- a single dose of 100 mg of the compound of the invention corresponds to 10 ml of oral suspension.
- the rhodigel is suspended in ethanol, the erf ⁇ ndungshiele connection is fed to the suspension. While stirring, the addition of water. Until the completion of the swelling of Rhodigels is stirred for about 6 h.
- the compound of the invention is suspended in the mixture of polyethylene glycol and polysorbate with stirring. The stirring is continued until complete dissolution of the compound according to the invention.
- the compound of the invention is sterile at a concentration below saturation solubility in a physiologically acceptable solvent (e.g., isotonic saline, glucose solution 5%, and / or PEG 400 solution.)
- a physiologically acceptable solvent e.g., isotonic saline, glucose solution 5%, and / or PEG 400 solution.
- the solution is sterile filtered and sterile and pyrogen-free injection wells are filled.
- the compound of the present invention is dissolved in the water with stirring together with polyethylene glycol 400.
- the solution is sterile-filtered (pore diameter 0.22 ⁇ m) and filled under raseptic conditions into heat-sterilized infusion bottles. These are closed with infusion stoppers and bristle caps.
Abstract
Die Erfindung betrifft die Verwendung von Cynaopyrimidinen zur Herstellung von Arzneimitteln zur Behandlung von Herz-Kreislauf-Erkrankungen.
Description
VERWENDUNG VON CYÄNOPYRIMIDINEN FÜR DIE BEHANDLUNG VON HERZ-KREISLAUF-ERKRANKUNGEN
Die Erfindung betrifft die Verwendung von Cynaopyrimidinen zur Herstellung von Arzneimitteln zur Behandlung von Herz-Kreislauf-Erkrankungen.
Inhibition von Phosphodiesterasen moduliert die Spiegel der zyklischen Nukleotide 5'-3' zyklisches Adenosinmonophosphat (cAMP) bzw. 5'-3' zyklisches Guanosinmonophosphat (cGMP). Diese zyklischen Nukleotide (cAMP und cGMP) sind wichtige second messenger und spielen daher eine zentrale Rolle in den zellulären Signaltransduktionskaskaden. Beide aktivieren unter anderem, aber nicht ausschließlich, jeweils wieder Proteinkinasen. Die von cAMP aktivierte Proteinkinase wird Proteinkinase A (PKA) genannt, die von cGMP aktivierte Proteinkinase wird Proteinkinase G (PKG) genannt. Aktivierte PKA bzw. PKG können wiederum eine Reihe zellulärer Effektorproteine phosphorylieren (z.B. Ionenkanäle, G-Protein-gekoppelte Rezeptoren, Strukturproteine). Auf diese Weise können die second messengers cAMP und cGMP die unterschiedlichsten physiologischen Vorgänge in den verschiedensten Organen kontrollieren. Die zyklischen Nukleotide können aber auch direkt auf Effektormoleküle wirken. So ist z.B. bekannt, dass cGMP direkt auf Ionenkanäle wirken kann und hiermit die zelluläre Ionenkonzentration beeinflussen kann (Übersicht in: Wei et al., Prog. Neurobiol., 1998, 56: 37-64). Ein Kontrollmechanismus, um die Aktivität von cAMP und cGMP und damit diese physiologischen Vorgänge wiederum zu steuern, sind die Phosphodiesterasen (PDE). PDEs hydrolysieren die zyklischen Monophosphate zu den inaktiven Monophosphaten AMP und GMP. Es sind mittlerweile mindestens 21 PDE-Gene beschrieben (Exp. Opin. Investig. Drugs 2000, 9, 1354-3784). Diese 21 PDE-Gene lassen sich aufgrund ihrer Sequenzhomologie in 1 1 PDE-Familien einteilen (Nomenklatur-Vorschlag siehe http://depts.washington.edu/pde/Nomenclature.html.). Einzelne PDE-Gene innerhalb einer Familie werden durch Buchstaben unterschieden (z.B. PDElA und PDElB). Falls noch unterschiedliche Splice-Varianten innerhalb eines Genes vorkommen, wird dies dann durch eine zusätzliche Nummerierung nach dem Buchstaben angegeben (z.B. PDElAl).
Die humane PDE9A wurde 1998 kloniert und sequenziert. Die Aminosäurenidentität zu anderen PDEs liegt bei maximal 34 % (PDE8A) und minimal 28 % (PDE5A). Mit einer Michaelis-Menten- Konstante (Km-Wert) von 170 nM ist PDE9A hochaffin für cGMP. Darüber hinaus ist PDE9A selektiv für cGMP (Km-Wert für cAMP = 230 μM). PDE9A weist keine cGMP-Bindungsdomäne auf, die auf eine allosterische Enzymregulation durch cGMP schließen ließe. In einer Northern Blot-Analyse wurde gezeigt, dass die PDE9A im Mensch unter anderem in Hoden, Gehirn, Dünndarm, Skelettmuskulatur, Herz, Lunge, Thymus und Milz exprimiert wird. Die höchste Expression wurde in Gehirn, Dünndarm, Herz und Milz gefunden (Fisher et al., J. Biol. Chem., 1998, 273 (25): 15559-15564). Das Gen für die humane PDE9A liegt auf Chromosom 21q22.3 und
enthält 21 Exons. Bislang wurden 20 alternative Spleißvarianten der PDE9A identifiziert (Guipponi et al., Hum. Geriet, 1998, 103: 386-392, Wang et al., Gene, 2003, 314: 15-27, Rentero et al., Biochem. Biophys. Res. Commun., 2003, 301: 686-692). Klassische PDE-Inhibitoren hemmen die humane PDE9A nicht. So zeigen IBMX, Dipyridamole, SKF94120, Rolipram und Vinpocetin in Konzentrationen bis 100 μM keine Inhibition am isolierten Enzym. Für Zaprinast wurde ein ICso-Wert von 35 μM nachgewiesen (Fisher et al., J. Biol. Chem., 1998, '273 (25): 15559-15564).
Die Maus-PDE9A wurde 1998 von Soderling et al. (J. Biol. Chem., 1998, 273 (25): 15553-15558) kloniert und sequenziert. Diese ist wie die humane Form hochaffin für cGMP mit einem Km von 70 nM. In der Maus wurde eine besonders hohe Expression in der Niere, Gehirn, Lunge und Herz gefunden. Auch die Maus-PDE9A wird von IBMX in Konzentrationen unter 200 μM nicht gehemmt; der IC50- Wert für Zaprinast liegt bei 29 μM (Soderling et al., J. Biol. Chem., 1998, 273 (19): 15553-15558). Im Rattengehirn wurde gezeigt, dass PDE9A in einigen Hirnregionen stark exprimiert wird. Dazu zählen der Bulbus olfactorius, Hippocampus, Cortex, Basalganglien und basales Vorderhirn (Andreeva et al., J. Neurosci., 2001, 21 (22): 9068-9076). Insbesondere Hippocampus, Cortex und basales Vorderhirn spielen eine wichtige Rolle bei Lern- und Gedächtnisvorgängen.
Wie oben bereits erwähnt, zeichnet sich PDE9A durch eine besonders hohe Affinität für cGMP aus. Deshalb ist PDE9A im Gegensatz zu PDE2A (Km = 10 μM; Martins et al., J. Biol. Chem., 1982, 257: 1973-1979), PDE5A (Km = 4 μM; Francis et al., J. Biol. Chem., 1980, 255: 620-626), PDE6A (Km = 17 μM; Gillespie and Beavo, J. Biol. Chem., 1988, 263 (17): 8133-8141) und PDEl IA (Km = 0.52 μM; Fawcett et al., Proc. Nat. Acad. Sei., 2000, 97 (7): 3702-3707) schon bei niedrigen physiologischen Konzentrationen aktiv. Im Gegensatz zu PDE2A (Murashima et al., Bio- chemistry, 1990, 29: 5285-5292) wird die katalytische Aktvität von PDE9A nicht durch cGMP gesteigert, da es keine GAF-Domäne (cGMP-Bindedomäne, über die die PDE-Aktivität allosterisch gesteigert wird) aufweist (Beavo et al., Current Opinion in Cell Biology, 2000, 12: 174-179). PDE9A-Inhibitoren können deshalb zu einer Erhöhung der basalen cGMP-Konzen- tration führen.
Aus der WO 04/1 13306 und der dort zitierten Literatur sind Cyanopyrimidine und deren Verwendung zur Herstellung von Arzneimitteln zur Behandlung von verschiedenen Krankheiten bereits bekannt.
Die US 5,002,949 offenbart Cyanopyrimidinone zur Inhibierung von weißen Thrombusformationen.
In WO 02/06288 werden Cyanopyrimidinone mit mGluR antagonistischer Wirkung beschrieben.
In WO 95/10506 offenbart Cyanopyrimidinone zur Behandlung von Depression und der AIz- heimer'schen Krankheit.
In EP 130735 werden Cyanopyrimidine als cardiotonische Reagentien beschrieben.
US 5,256,668 und WO 99/41253 offenbaren Cyanopyrimidine mit antiviraler Wirkung.
Die vorliegende Erfindung betrifft Verbindungen der Formel

in welcher
A CpCg-Alkyl, C3-Cg-Cycloalkyl, Tetrahydrofuryl oder Tetrahydropyranyl, welche ge- gebenenfalls mit bis zu 3 Resten unabhängig voneinander ausgewählt aus der Gruppe
CpCö-Alkyl, Ci-Cβ-Alkoxy, Hydroxycarbonyl, Cyano, Trifluormethyl, Trifluormethoxy,
Amino, Hydroxy, Ci-Cβ-Alkylamino, Halogen, Ci-Cβ-Alkylaminocarbonyl, Ci-Cβ-Alkoxy- carbonyl, Ci-Cβ-Alkylcarbonyl, Ci-C6-Alkylsulfonyl und Ci-C6-Alkylthio substituiert sind,
wobei Ci-C6-Alkyl, Ci-C6-Alkoxy, Ci-C6-Alkylamino, C]-C6-Alkylaminocarbonyl, Ci-Cβ- Alkoxycarbonyl, Ci-Cβ-Alkylcarbonyl, Ci-Cβ-Alkylsulfonyl und Ci-Cö-Alkylthio gegebenenfalls mit einem oder mehreren Resten ausgewählt aus der Gruppe
Hydroxy, Cyano, Halogen, Hydroxycarbonyl und einer Gruppe der Formel
-NR3R4,
wobei
R3 und R4 unabhängig voneinander Wasserstoff oder C1-C6-AIlCyI,
oder
R3 und R4 zusammen mit dem Stickstoffatom, an das sie gebunden sind, 5- bis 8- gliedriges Heterocyclyl bedeuten,
substituiert sind,
B Phenyl oder Heteroaryl, die gegebenenfalls mit bis zu 3 Resten jeweils unabhängig voneinander ausgewählt aus der Gruppe Ci-Cβ-Alkyl, Ci-Cδ-Alkoxy, Hydroxycarbonyl,
Cyano, Trifluormethyl, Trifluormethoxy, Amino, Nitro, Hydroxy, Ci-Cβ-Alkylamino,
Halogen, Ci-Cό-Alkylaminocarbonyl, CrC6-Alkoxycarbonyl, d-C6-Alkylcarbonyl, CpC6- Alkylsulfonyl und Ci-C6-Alkylthio substituiert sind,
wobei Ci-C6-Alkyl, CrC6-Alkoxy, Ci-C6-Alkylamino, Cj-Cö-Alkylaminocarbonyl, Ci-C6- Alkoxycarbonyl, Ci-Cδ-Alkylcarbonyl, CrC6-Alkylsulfonyl und Ci-C6-Alkylthio gegebenenfalls mit einem Rest ausgewählt aus der Gruppe Hydroxy, Cyano, Halogen, Hydroxycarbonyl und einer Gruppe der Formel -NR3R4,
wobei
R3 und R4 die oben angegebenen Bedeutungen aufweisen,
substituiert sind,
bedeuten, sowie deren Salze, Solvate und/oder Solvate der Salze, zur Herstellung von Arzneimitteln zur Behandlung von Herz-Kreislauf-Erkrankungen.
Eine weitere Ausführungsform der Erfindung betrifft Verbindungen der Formel (I),
in welcher
A Ci-C5-Alkyl oder C3-C6-Cycloalkyl, welche gegebenenfalls mit bis zu 3 Resten unabhängig voneinander ausgewählt aus der Gruppe Ci-C4-Alkyl, Ci-C4-Alkoxy, Hydroxycarbonyl, Cyano, Amino, Hydroxy, Ci-GrAlkylamino, Fluor, Chlor, Brom, Ci-C4-Alkoxycarbonyl, CrC6-Alkylcarbonyl, Ci-C4-Alkylsulfonyl und CrC4-Alkylthio substituiert sind,
wobei CrC4-Alkyl und CpC4-AIkOXy gegebenenfalls mit einem Rest ausgewählt aus der Gruppe Hydroxy, Cyano, Fluor, Chlor, Brom, Hydroxycarbonyl und einer Gruppe der Formel -NR3R4,
wobei
R3 und R4 unabhängig voneinander Wasserstoff oder Ci-C4-Alkyl,
oder
R3 und R4 zusammen mit dem Stickstoffatom, an das sie gebunden sind, 5- bis 6- gliedriges Heterocyclyl bedeuten,
substituiert sind,
B Phenyl, Thienyl oder Pyridyl, die gegebenenfalls mit bis zu 3 Resten jeweils unabhängig voneinander ausgewählt aus der Gruppe Ci-C4-Alkyl, Ci-C4-Alkoxy, Hydroxycarbonyl,
Cyano, Trifluormethyl, Trifluormethoxy, Amino, Hydroxy, Ci -C4-AIlCy lamino, Fluor, Chlor, Brom, Ci-C4-Alkylaminocarbonyl, Ci-C4-Alkoxycarbonyl, Ci-C4-Alkylcarbonyl,
C]-C4-Alkylsulfonyl und Ci-C4-Alkylthio substituiert sind,
wobei CrC4-Alkyl und Ci-C4-Alkoxy gegebenenfalls mit einem Rest ausgewählt aus der Gruppe Hydroxy, Cyano, Fluor, Chlor, Brom, Hydroxycarbonyl und einer Gruppe der Formel -ISfR3R4,
wobei
R3 und R4 die oben angegebenen Bedeutungen aufweisen,
substituiert sind,
bedeuten, sowie deren Salze, Solvate und/oder Solvate der Salze, zur Herstellung von Arzneimitteln zur Behandlung von Herz-Kreislauf-Erkrankungen.
Eine weitere Ausführungsform der Erfindung betrifft Verbindungen der Formel (I),
in welcher
A die oben angegebenen Bedeutungen aufweist, und
B Phenyl oder Pyridyl, die gegebenenfalls mit bis zu 3 Resten jeweils unabhängig voneinander ausgewählt aus der Gruppe Methyl, Ethyl, 2-Propyl, Trifluormethyl, Methoxy, Ethoxy, Fluor und Chlor,
wobei einer der Reste am Phenyl oder Pyridyl in ortho-Position relativ zur Anknüpfungsstelle der Aminofunktion lokalisiert ist,
substituiert sind,
bedeutet, sowie deren Salze, Solvate und/oder Solvate der Salze, zur Herstellung von Arznei- mittein zur Behandlung von Herz-Kreislauf-Erkrankungen.
Eine weitere Ausführungsform der Erfindung betrifft Verbindungen der Formel (I),
in welcher
A C3-C6-Cycloalkyl bedeutet und
B die oben angegebenen Bedeutungen aufweist,
bedeuten, sowie deren Salze, Solvate und/oder Solvate der Salze, zur Herstellung von Arzneimitteln zur Behandlung von Herz-Kreislauf-Erkrankungen.
Eine weitere Ausführungsform der Erfindung betrifft Verbindungen der Formel (I),
in welcher
A 2-Methylpropyl, 2-Butyl, 2-Pentyl oder 3-Pentyl bedeutet und
B die oben angegebenen Bedeutungen aufweist,
sowie deren Salze, Solvate und/oder Solvate der Salze, zur Herstellung von Arzneimitteln zur Behandlung von Herz-Kreislauf-Erkrankungen.
Eine weitere Ausführungsform der Erfindung betrifft Verbindungen der Formel (I),
in welcher
A C3-C5-Alkyl oder C5-C6-Cycloalkyl,
B Phenyl, Thienyl oder Pyridyl, die gegebenenfalls mit bis zu 3 Resten jeweils unabhängig voneinander ausgewählt aus der Gruppe Ci-C3-Alkyl, Trifiuormethyl, Hydroxy, Methoxy,
Ethoxy, Cyano, Dimethylamino, Diethylamino, Methoxycarbonyl, Ethoxycarbonyl, Methylcarbonyl, Ethylcarbonyl, Fluor und Chlor substituiert sind,
bedeuten, sowie deren Salze, Solvate und/oder Solvate der Salze, zur Herstellung von Arzneimitteln zur Behandlung von Herz-Kreislauf-Erkrankungen.
Im Rahmen der vorliegenden Erfindung haben die Substituenten der erfindungsgemäßen Verbindungen gemäß Formel (I), soweit nicht anders spezifiziert, die folgende Bedeutung:
CyCs-Alkyl. dVCfi-Alkyl, CVCs-Alkyl und CrCV-Alkyl stehen für einen geradkettigen oder verzweigten Alkylrest mit 1 bis 8, bevorzugt 1 bis 6, besonders bevorzugt 1 bis 5 und 1 bis 4 Kohlenstoffatomen. Bevorzugte Beispiele umfassen Methyl, Ethyl, n-Propyl, Isopropyl, 2-Butyl, 2-Pentyl und 3-Pentyl.
 für einen geradkettigen oder verzweigten Alkoxyrest mit 1 bis 6, bevorzugt 1 bis 4, besonders bevorzugt mit 1 bis 3 Kohlenstoffatomen. Bevorzugte Beispiele umfassen Methoxy, Ethoxy, n-Propoxy, Isopropoxy, tert.-Butoxy, n-Pentoxy und n-Hexoxy.
für einen geradkettigen oder verzweigten Alkoxyrest mit 1 bis 6, bevorzugt 1 bis 4, besonders bevorzugt mit 1 bis 3 Kohlenstoffatomen. Bevorzugte Beispiele umfassen Methoxy, Ethoxy, n-Propoxy, Isopropoxy, tert.-Butoxy, n-Pentoxy und n-Hexoxy.
CVCfi-Alkoxycarbonyl steht für einen geradkettigen oder verzweigten Alkoxycarbonylrest mit 1 bis 6, bevorzugt 1 bis 4 und besonders bevorzugt 1 bis 3 Kohlenstoffatomen. Bevorzugte Beispiele umfassen Methoxycarbonyl, Ethoxycarbonyl, n-Propoxycarbonyl, Isopropoxycarbonyl und tert.Butoxycarbonyl.
CrCfi-Alkylamino steht für einen geradkettigen oder verzweigten Mono- oder Dialkylaminorest mit 1 bis 6, bevorzugt 1 bis 4 und besonders bevorzugt mit 1 bis 3 Kohlenstoffatomen. Bevorzugte Beispiele umfassen Methylamino, Ethylamino, n-Propylamino, Isopropylamino, tert.-Butylamino, n- Pentylamino und n-Hexylamino, Dimethylamino, Diethylamino, Di-n-propylamino, Diisopropylamino, Di-tert.-butylamino, Di-n-pentylamino, Di-n-hexylamino, Ethylmethylamino, Iso- propylmethylamino, n-Butylethylamino und n-Hexyl-i-pentylamino.
CVCVAlkylaminocarbonyl steht für einen über eine Carbonyl-Gruppe verknüpften Mono- oder Dialkylaminorest, wobei die Alkylreste gleich oder verschieden sein können, geradkettig oder verzweigt sind und jeweils 1 bis 6, bevorzugt 1 bis 4, und besonders bevorzugt 1 bis 3 Kohlenstoffatome enthalten. Bevorzugte Beispiele umfassen Methylaminocarbonyl, Ethylaminocarbonyl, n- Propylaminocarbonyl, Isopropylaminocarbonyl, tert.Butylaminocarbonyl, n-Pentylaminocarbonyl, n- Hexylaminocarbonyl, Dimethylaminocarbonyl, Diethylaminocarbonyl, Di-n-propylaminocarbonyl, Diisopropylaminocarbonyl, Di-t-butylaminocarbonyl, Di-n-pentylaminocarbonyl, Di-n-hexylamino- carbonyl, Ethylmethylaminocarbonyl, Isopropylmethylaminocarbonyl, n-Butylethylaminocarbonyl und n-Hexyl-i-pentylaminocarbonyl. Weiterhin können im Falle eines Dialkylaminorestes die beiden Alkylreste zusammen mit dem Stickstoffatom, an das sie gebunden sind, einen 5- bis 8-gliedriges Heterocyclyl bilden.
dVCfi-Alkylcarbonyl steht für einen geradkettigen oder verzweigten Alkylcarbonylrest mit 1 bis 6 und bevorzugt 1 bis 4 Kohlenstoffatomen. Beispielsweise seien genannt: Acetyl, Ethylcarbonyl, Propylcarbonyl, Isopropylcarbonyl, Butylcarbonyl, Isobutylcarbonyl, Pentylcarbonyl und Hexylcarbonyl. Besonders bevorzugt sind Acetyl und Ethylcarbonyl.
CrCft-Alkylsulfonyl steht für einen geradkettigen oder verzweigten Alkylsulfonylrest mit 1 bis 6, bevorzugt 1 bis 4 und besonders bevorzugt mit 1 bis 3 Kohlenstoffatomen. Bevorzugte Beispiele umfassen Methylsulfonyl, Ethylsulfonyl, n-Propylsulfonyl, Isopropylsulfonyl, tert.Butylsulfonyl, n- Pentylsulfonyl und n-Hexylsulfonyl.
- -
Cj-Cfi-Alkylthio steht für einen geradkettigen oder verzweigten Alkylthiorest mit 1 bis 6, bevorzugt 1 bis 4 und besonders bevorzugt mit 1 bis 3 Kohlenstoffatomen. Bevorzugte Beispiele umfassen Methylthio, Ethylthio, n-Propylthio, Isopropylthio, tert.Butylthio, n-Pentylthio und n-Hexylthio.
Halogen steht für Fluor, Chlor, Brom und Iod. Bevorzugt sind Fluor, Chlor, Brom, besonders bevorzugt Fluor und Chlor.
Heteroaryl steht für einen aromatischen, monocyclischen Rest mit 5 bis 6 Ringatomen und bis zu 3 Heteroatomen aus der Reihe S, O und/oder N. Bevorzugt sind 5- bis 6-gliedrige Heteroaryle mit bis zu 2 Heteroatomen. Der Heteroarylrest kann über ein Kohlenstoff- oder Stickstoffatom gebunden sein. Bevorzugte Beispiele umfassen Thienyl, Furyl, Pyrrolyl, Thiazolyl, Oxazolyl, Imidazolyl, Tetra- zolyl, Pyridyl, Pyrimidinyl und Pyridazinyl.
3- bis 8-gliedriges Cycloalkyl steht für gesättigte und teilweise ungesättigte nicht-aromatische Cycloalkylreste mit 3 bis 8, bevorzugt 3 bis 6 und besonders bevorzugt 5 bis 6 Kohlenstoffatomen im Cyclus. Bevorzugte Beispiele umfassen Cyclopropyl, Cyclobutyl, Cyclopentyl, Cyclopentenyl, Cyclohexyl und Cyclohexenyl.
5- bis 8-gliedriges Heterocyclyl steht für einen mono- oder polycyclischen, heterocyclischen Rest mit 5 bis 8 Ringatomen und bis zu 3, vorzugsweise 2 Heteroatomen bzw. Heterogruppen aus der Reihe N, O, S, SO, SO2. Mono- oder bicyclisches Heterocyclyl ist bevorzugt. Besonders bevorzugt ist monocyclisches Heterocyclyl. Als Heteroatome sind N und O bevorzugt. Die Heterocyclyl- Reste können gesättigt oder teilweise ungesättigt sein. Gesättigte Heterocyclyl-Reste sind bevor- zugt. Besonders bevorzugt sind 5- bis 7-gliedrige Heterocyclylreste. Bevorzugte Beispiele umfassen Oxetan-3-yl, Pyrrolidin-2-yl, Pyrrolidin-3-yl, Pyrrolinyl, Tetrahydrofuranyl, Tetra- hydrothienyl, Pyranyl, Piperidinyl, Thiopyranyl, Morpholinyl, Perhydroazepinyl.
Wenn Reste in den erfindungsgemäßen Verbindungen gegebenenfalls substituiert sind, ist, soweit nicht anders spezifiziert, eine Substitution mit bis zu drei gleichen oder verschiedenen Substituen- ten bevorzugt.
Die Verbindungen der Formeln (I) und deren Herstellung sind aus WO 04/113306 bekannt.
Ein weiterer Gegenstand der vorliegenden Erfindung betrifft Verbindungen der Formel

in welcher
D Phenyl, Heteroaryl oder eine Gruppe der Formel

wobei Phenyl und Heteroaryl gegebenenfalls mit bis zu 2 Resten unabhängig von- einander ausgewählt aus der Gruppe Heteroaryl, Halogen, Ci-Cβ-Alkyl, Ci-Cβ-
Alkoxy, Trifluormethyl, Trifluormethoxy, Benzyloxy und Benzyl substituiert sind,
wobei Ci-Cβ-Alkyl gegebenenfalls mit einer Gruppe der Formel -NR5R6, in welcher R5 CrC6-Alkyl und R6 Wasserstoff oder CrC6-Alkoxy(Ci-C6)- alkyl bedeuten, und
Heteroaryl gegebenenfalls mit Ci-Cό-Alkoxy substituiert ist,
R1 C3-C8-Cycloalkyl, CrC6-Alkyl, C1-C6-Alkoxy(Ci-C6)alkyl, Benzyl oder eine Gruppe der
Formel

wobei C3-C8-Cycloalkyl gegebenenfalls mit Hydroxy, CpCβ-Alkyl oder Trifluormethyl,
CpCβ-Alkyl gegebenenfalls mit Heteroaryl, C3-Cg-Cycloalkyl oder Hydroxy,
und Benzyl gegebenenfalls mit Ci-Cβ-Alkoxy oder Halogen substituiert ist,
R2 Wasserstoff,
oder
- -
R1 und R2 zusammen mit dem Stickstoffatom, an dem sie gebunden sind, ein 5- bis 6- gliedriges Heterocyclyl bilden, welches gegebenenfalls mit bis zu 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe CpCβ-Alkyl, Hydroxy,
Cyano, Oxo, Heteroaryl, Benzyl, Formyl, Q-Ce-Alkylcarbonyl und einer der folgenden Gruppen

Kohlenstoffatome im Heterocyclus gebunden sind, substituiert ist,
wobei Ci-Cβ-Alkyl gegebenenfalls mit Hydroxy oder Heteroaryl substituiert ist,
bedeuten, sowie deren Salze, Solvate und/oder Solvate der Salze, zur Herstellung von Arzneimitteln zur Behandlung von Herz-Kreislauf-Erkrankungen.
Eine weitere Ausführungsform der Erfindung betrifft Verbindungen der Formel (II), in welcher
Phenyl, Heteroaryl oder eine Gruppe der Formel

wobei Phenyl und Heteroaryl gegebenenfalls mit bis zu 2 Resten unabhängig von- einander ausgewählt aus der Gruppe Heteroaryl, Halogen, Ci-C4-Alkyl, CpC4-
Alkoxy, Trifluormethyl, Trifluormethoxy, Benzyloxy und Benzyl substituiert sind,
wobei Ci-Gj-Alkyl gegebenenfalls mit einer Gruppe der Formel -NR3R4, in welcher R3 C,-C4-Alkyl und R4 Wasserstoff oder CrC4-Alkoxy(CpC4)- alkyl bedeuten, und
Heteroaryl gegebenenfalls mit Ci-C4-Alkoxy substituiert ist,
R1 C3-C6-Cycloalkyl, CrC4-Alkyl, Ci-C4-Alkoxy(Ci-C4)alkyl, Benzyl oder eine Gruppe der
Formel

wobei C3-C6-Cycloalkyl gegebenenfalls mit Hydroxy, Ci-C4-Alkyl oder Trifluor- methyl,
CrC4-Alkyl gegebenenfalls mit Heteroaryl, C3-C6-Cycloalkyl oder Hydroxy,
und Benzyl gegebenenfalls mit Ci-C4-Alkoxy oder Halogen substituiert ist,
R2 Wasserstoff,
oder
R1 und R2 zusammen mit dem Stickstoffatom, an dem sie gebunden sind, ein 5- bis 6- gliedriges Heterocyclyl bilden, welches gegebenenfalls mit bis zu 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe CrC4-Alkyl, Hydroxy,
Cyano, Oxo, Heteroaryl, Benzyl, Formyl, Ci-C4-Alkylcarbonyl und einer der folgenden Gruppen

wobei CrC4-Alkyl gegebenenfalls mit Hydroxy oder Heteroaryl substituiert ist,
bedeuten, sowie deren Salze, Solvate und/oder Solvate der Salze, zur Herstellung von Arzneimitteln zur Behandlung von Herz-Kreislauf-Erkrankungen.
Eine weitere Ausführungsform der Erfindung betrifft Verbindungen der Formel (II), in welcher
A Phenyl, Thienyl oder eine Gruppe der Formel

wobei Phenyl und Thienyl gegebenenfalls mit bis zu 2 Resten unabhängig voneinander ausgewählt aus der Gruppe Pyridyl, Fluor, Chlor, Brom, Ci-Gj-Alkyl, Ci-C4-Alkoxy, Trifluormethyl, Trifluormethoxy, Benzyloxy und Benzyl substi- tuiert sind,
wobei Ci-C4-Alkyl gegebenenfalls mit einer Gruppe der Formel -NR3R4, in welcher R3 CrC4-Alkyl und R4 Wasserstoff oder CrC4-Alkoxy(CrC4)- alkyl bedeuten, und
Pyridyl gegebenenfalls mit Ci-C4-Alkoxy substituiert ist,
R1 C3-C6-Cycloalkyl, CrC4-Alkyl, CrC4-Alkoxy(Ci-C4)alkyl, Benzyl oder eine Gruppe der
Formel

wobei C3-C6-Cycloalkyl gegebenenfalls mit Hydroxy, Ci-C4-Alkyl oder Trifluormethyl,
C)-C4-Alkyl gegebenenfalls mit Pyridyl, C3-C6-Cycloalkyl oder Hydroxy,
und Benzyl gegebenenfalls mit Ci-C4-Alkoxy, Fluor, Chlor oder Brom substituiert ist,
R2 Wasserstoff,
oder
R1 und R2 zusammen mit dem Stickstoffatom, an dem sie gebunden sind, ein 5- bis 6- gliedriges Heterocyclyl ausgewählt aus der Gruppe Pyrrolidinyl, Piperidinyl,
Piperazinyl und Morpholinyl bilden, welches gegebenenfalls mit bis zu 2 Sub- stituenten unabhängig voneinander ausgewählt aus der Gruppe Ci-C4-Alkyl,
Hydroxy, Cyano, Oxo, Heteroaryl, Benzyl, Formyl, Ci-Q-Alkylcarbonyl und einer der folgenden Gruppen

Kohlenstoffatome im Heterocyclus gebunden sind, substituiert ist,
wobei Ci-C4-Alkyl gegebenenfalls mit Hydroxy oder Pyridyl substituiert ist,
bedeuten, sowie deren Salze, Solvate und/oder Solvate der Salze, zur Herstellung von Arzneimitteln zur Behandlung von Herz-Kreislauf-Erkrankungen.
Eine weitere Ausführungsform der Erfindung betrifft Verbindungen der Formel (II), in welcher
A Phenyl, Thienyl oder eine Gruppe der Formel

wobei Phenyl gegebenenfalls mit bis zu 2 Resten unabhängig voneinander ausgewählt aus der Gruppe Pyridyl, Fluor, Chlor, Methyl, Methoxy, Ethoxy, Trifluor- methyl, Trifluormethoxy, Benzyloxy und Benzyl substituiert ist,
wobei Methyl gegebenenfalls mit einer Gruppe der Formel -NR3R4, in welcher R3 Methyl und R4 Wasserstoff oder 2-Methoxyethyl bedeuten, und
Pyridyl gegebenenfalls mit Methoxy substituiert ist,
R1 C3-C6-Cycloalkyl, Methyl, Ethyl, Propyl, 2-Methoxyethyl, Benzyl oder eine Gruppe der
Formel

wobei C3-C6-Cycloalkyl gegebenenfalls mit Hydroxy, Methyl oder Trifluormethyl,
Methyl, Ethyl, Propyl gegebenenfalls mit Pyridyl, Cyclopropyl oder Hydroxy,
und Benzyl gegebenenfalls mit Methoxy, Ethoxy, Fluor oder Chlor substituiert ist,
R2 Wasserstoff,
oder
R1 und R2 zusammen mit dem Stickstoffatom, an dem sie gebunden sind, ein 5- bis 6- gliedriges Heterocyclyl ausgewählt aus der Gruppe Pyrrolidinyl, Piperidinyl,
Piperazinyl und Morpholinyl bilden, welches gegebenenfalls mit bis zu 2 Sub- stituenten unabhängig voneinander ausgewählt aus der Gruppe Methyl, Ethyl, Propyl, tert.-Butyl, Hydroxy, Cyano, Oxo, Pyridyl, Benzyl, Formyl, Methylcarbonyl, Ethylcarbonyl, Propylcarbonyl und einer der folgenden Gruppen

Kohlenstoffatome im Heterocyclus gebunden sind, substituiert ist,
wobei Methyl, Ethyl und Propyl gegebenenfalls mit Hydroxy oder Pyridyl substituiert sind,
bedeuten, sowie deren Salze, Solvate und/oder Solvate der Salze, zur Herstellung von Arzneimitteln zur Behandlung von Herz-Kreislauf-Erkrankungen.
Im Rahmen der vorliegenden Erfindung haben die Substituenten der erfindungsgemäßen Verbindungen der Formel (IT), soweit nicht anders spezifiziert, die folgende Bedeutung:
OrCfi-Alkyl steht für einen geradkettigen oder verzweigten Alkylrest mit 1 bis 6, bevorzugt 1 bis 4 Kohlenstoffatomen. Bevorzugte Beispiele umfassen Methyl, Ethyl, n-Propyl, Isopropyl, 2-Butyl, tert.-Butyl, 2-Pentyl, 3-Pentyl und n-Hexyl.
C1-Cs-AIkOXV steht für einen geradkettigen oder verzweigten Alkoxyrest mit 1 bis 6, bevorzugt 1 bis 4, besonders bevorzugt mit 1 bis 3 Kohlenstoffatomen. Bevorzugte Beispiele umfassen Methoxy, Ethoxy, n-Propoxy, Isopropoxy, tert.-Butoxy, n-Pentoxy und n-Hexoxy.
Cj-Cft-Alkylcarbonyl steht für einen geradkettigen oder verzweigten Alkylcarbonylrest mit 1 bis 6, bevorzugt 1 bis 4 und besonders bevorzugt 1 bis 3 Kohlenstoffatomen. Bevorzugte Beispiele um- fassen Methylcarbonyl, Ethylcarbonyl, n-Propylcarbonyl, Isopropylcarbonyl und tert.-Butylcarbonyl.
3- bis 8-gliedriges Cycloalkyl steht für gesättigte Cycloalkylreste mit 3 bis 8, bevorzugt 3 bis 6 und besonders bevorzugt 5 bis 6 Kohlenstoffatomen im Cyclus. Bevorzugte Beispiele umfassen Cyclo- propyl, Cyclobutyl, Cyclopentyl, Cyclohexyl und Cycloheptyl.
Halogen steht für Fluor, Chlor, Brom und Iod. Bevorzugt sind Fluor, Chlor, Brom, besonders be- vorzugt Fluor und Chlor.
Heteroaryl steht für einen aromatischen, monocyclischen Rest mit 5 bis 6 Ringatomen und bis zu 3 Heteroatomen aus der Reihe S, O und/oder N. Bevorzugt sind 5- bis 6-gliedrige Heteroaryle mit bis zu 2 Heteroatomen. Der Heteroarylrest kann über ein Kohlenstoff- oder Stickstoffatom gebunden sein. Bevorzugte Beispiele umfassen Thienyl, Furyl, Pyrrolyl, Thiazolyl, Oxazolyl, Imidazolyl, Pyridyl, Pyrimidinyl und Pyridazinyl.
5- bis 6-gliedriges Heterocyclyl steht für einen monocyclischen, gesättigten oder partiell ungesättigten heterocyclischen Rest mit 5 bis 6 Ringatomen und bis zu 2 Heteroatomen aus der Reihe N, O, S. Als Heteroatome sind N und O bevorzugt. Bevorzugte Beispiele umfassen Pyrrolidinyl, Pyrrolinyl, Tetrahydrofuranyl, Tetrahydrothienyl, Pyranyl, Thiopyranyl, Piperidinyl, Morpholinyl, Thiomorpholinyl und Piperazinyl.
Wenn Reste in den erfindungsgemäßen Verbindungen gegebenenfalls substituiert sind, ist, soweit nicht anders spezifiziert, eine Substitution mit bis zu drei gleichen oder verschiedenen Substituenten bevorzugt.
Außerdem wurde ein Verfahren zur Herstellung der erfindungsgemäßen Verbindungen der Formel (II) gefunden, dadurch gekennzeichnet, dass man entweder
[A] eine Verbindung der Formel

zunächst mit einer Verbindung der Formel
HNR1R2 (IV),
in welcher
R1 und R2 die oben angegebenen Bedeutungen aufweisen,
bei erhöhter Temperatur in einem inerten Lösemittel oder auch in Abwesenheit eines Lösemittels in eine Verbindung der Formel

in welcher
R1 und R2 die oben angegebenen Bedeutungen aufweisen,
überführt und diese dann in einem inerten Lösemittel in Gegenwart einer Base mit einer Verbindung der Formel

X = Cl1 Br oder I
in welcher
D die oben angegebenen Bedeutungen aufweist,
umsetzt
oder in veränderter Reihenfolge der Reaktionspartner
[B] eine Verbindung der Formel (III) zunächst mit einer Verbindung der Formel (VI) in einem inerten Lösemittel in Gegenwart einer Base in eine Verbindung der Formel

in welcher
D die oben angegebenen Bedeutungen aufweist,
überführt und diese dann bei erhöhter Temperatur in einem inerten Lösemittel oder auch in Abwesenheit eines Lösemittels mit einer Verbindung der Formel (IV) umsetzt,
und die jeweils resultierenden Verbindungen der Formel (II) gegebenenfalls mit den entsprechenden (i) Lösemitteln und/oder (ii) Basen oder Säuren zu ihren Solvaten, Salzen und/oder Solvaten der Salze umsetzt.
Die Verbindung der Formel (III) ist literaturbekannt (R. Gompper, W. Toepfl, Chem. Ber. 1962, 95, 2861-2870). Die Verbindungen der Formeln (IV) und (VI) sind kommerziell erhältlich, literaturbekannt oder können in Analogie zu literaturbekannten Verfahren hergestellt werden (siehe z.B. H. Gielen, C. Alonso-Alija, M. Hendrix, U. Niewöhner, D. Schauß, Tetrahedron Lett. 2002, 43, 419-421).
Für den Verfahrensschritt (III) + (FV) -» (V) eignen sich hochsiedende, inerte organische Lösemittel, die sich unter den Reaktionsbedingungen nicht verändern. Hierzu gehören bevorzugt Toluol, Acetonitril, Dimethylformamid, Dimethylsulfoxid oder Sulfolan. Ebenso ist es möglich, die Reaktion ohne Lösemittel in der Schmelze durchzuführen. Besonders bevorzugt ist eine Reaktionsführung ohne Lösemittel oder in Dimethylformamid, Acetonitril oder Toluol.
Die Reaktion erfolgt im Allgemeinen in einem Temperaturbereich von +70°C bis +2000C, bevorzugt in einem Temperaturbereich von +1000C bis +15O0C. Die Umsetzung kann bei normalem, er-
höhtem oder bei erniedrigtem Druck durchgeführt werden (z.B. von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Die Verbindung der Formel (IV) wird hierbei in einer Menge von 1 bis 2 Mol, bevorzugt in einer äquivalenten Menge von 1 Mol, bezogen auf 1 Mol der Verbindung der Formel (HI), eingesetzt.
Für den Verfahrensschritt (VII) + (IV) -» (II) eignen sich übliche organische Lösemittel, die sich unter den Reaktionsbedingungen nicht verändern. Hierzu gehören bevorzugt Dimethylformamid, Di- methylsulfoxid oder Acetonitril. Ebenso ist es möglich, die Reaktion ohne Lösemittel durchzuführen. Besonders bevorzugt ist eine Reaktionsführung ohne Lösemittel oder in Acetonitril.
Die Reaktion erfolgt im Allgemeinen in einem Temperaturbereich von +5O0C bis +1500C, bevor- zugt in einem Temperaturbereich von +7O0C bis +1000C. Die Umsetzung kann bei normalem, erhöhtem oder bei erniedrigtem Druck durchgeführt werden (z.B. von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Die Verbindung der Formel (TV) wird hierbei in einer Menge von 1 bis 10 Mol, bevorzugt in einem Überschuss von 3 bis 10 Mol, bezogen auf 1 Mol der Verbindung der Formel (VII), einge- setzt.
Für den Verfahrensschritt (V) + (VI) -> (II) bzw. (III) + (VI) → (VII) eignen sich übliche organische Lösemittel, die sich unter den Reaktionsbedingungen nicht verändern. Hierzu gehören bevorzugt Dimethylformamid, Dimethylsulfoxid, Acetonitril, Dioxan oder Alkohole wie Methanol, Ethanol, Propanol, Isopropanol, n-Butanol oder tert.-Butanol. Ebenso ist es möglich, Gemische der genannten Lösemittel einzusetzen. Besonders bevorzugt ist für den Verfahrensschritt (V) + (VI) -> (II) Dimethylformamid oder Acetonitril und für den Verfahrensschritt (DI) + (VI) -> (VIT) Ethanol.
Die Reaktion erfolgt im Allgemeinen in einem Temperaturbereich von +500C bis +1500C, bevorzugt in einem Temperaturbereich von +700C bis +1000C. Die Umsetzung kann bei normalem, erhöhtem oder bei erniedrigtem Druck durchgeführt werden (z.B. von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Als Basen für den Verfahrensschritt (V) + (VI) → (E) bzw. (HI) + (VI) → (VE) eignen sich bevorzugt Alkalicarbonate wie Lithium-, Natrium-, Kalium- oder Cäsiumcarbonat oder organische Amin-Basen wie beispielsweise Pyridin, Triethylamin, Ethyldiisopropylamin, N-Methylmorpholin oder N-Methylpiperidin. Besonders bevorzugt ist Kaliumcarbonat oder Triethylamin.
Die Base wird hierbei in einer Menge von 1.5 bis 4 Mol, bevorzugt in einer Menge von 1.5 bis 2 Mol, bezogen auf 1 Mol der Verbindung der Formel (V) bzw. (BT), eingesetzt. Die Verbindung der
Formel (VI) wird in einer Menge von 1 bis 1.5 Mol, bevorzugt in einer Menge von 1.2 Mol, bezogen auf 1 Mol der Verbindung der Formel (V) bzw. (III), eingesetzt.
Das erfindungsgemäße Verfahren kann durch die folgenden Formelschemata beispielhaft erläutert werden:
Schema I:


X = Cl, Br
a) Ethanol, Triethylamin, 5-16 h Rückfluss; b) Acetonitril, 85-900C, 1-7 Tage.
Schema II:


a) 1. Toluol, Bortrifluorid-Etherat, RT, 30 min.; 2. Amin-Komponente R1R2NH, 15O0C, 16 h; oder: Schmelze der Ausgangsverbindungen bei 15O0C, 1-16 h; b) DMF, Triethylamin, 1000C, 16 h oder DMF, Kaliumcarbonat, 9O0C, 16 h.
Erfindungsgemäße Verbindungen sind die Verbindungen der Formel (I) und (II) und deren Salze, Solvate und Solvate der Salze; die von Formeln (I) und (II) umfassten Verbindungen der nachfolgend genannten Formeln und deren Salze, Solvate und Solvate der Salze sowie die von Formeln (I) und (II) umfassten, nachfolgend als Ausführungsbeispiele genannten Verbindungen und deren Salze, Solvate und Solvate der Salze, soweit es sich bei den von Formeln (I) und (II) umfassten, nachfolgend genannten Verbindungen nicht bereits um Salze, Solvate und Solvate der Salze handelt.
Die erfindungsgemäßen Verbindungen können in Abhängigkeit von ihrer Struktur in stereoisomeren Formen (Enantiomere, Diastereomere) existieren. Die Erfindung betrifft deshalb die En- antiomeren oder Diastereomeren und ihre jeweiligen Mischungen. Aus solchen Mischungen von Enantiomeren und/oder Diastereomeren lassen sich die stereoisomer einheitlichen Bestandteile in bekannter Weise isolieren.
Als Salze sind im Rahmen der Erfindung physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen bevorzugt.
Physiologisch unbedenkliche Salze der Verbindungen gemäß den Formeln (I) und (II) umfassen Säureadditionssalze von Mineralsäuren, Carbonsäuren und Sulfonsäuren, z.B. Salze der Chlorwasserstoffsäure, Bromwasserstoffsäure, Schwefelsäure, Phosphorsäure, Methansulfonsäure, Ethan- sulfonsäure, Toluolsulfonsäure, Benzolsulfonsäure, Naphthalindisulfonsäure, Essigsäure, Propionsäure, Milchsäure, Weinsäure, Äpfelsäure, Zitronensäure, Fumarsäure, Maleinsäure und Benzoesäure.
Physiologisch unbedenkliche Salze der Verbindungen der Formeln (I) und (II) umfassen auch Salze üblicher Basen, wie beispielhaft und vorzugsweise Alkalimetallsalze (z.B. Natrium- und Kaliumsalze), Erdalkalisalze (z.B. Calcium- und Magnesiumsalze) und Ammoniumsalze, abgeleitet von Ammoniak oder organischen Aminen mit 1 bis 16 C-Atomen, wie beispielhaft und vorzugsweise Ethylamin, Diethylamin, Triethylamin, Ethyldiisopropylamin, Monoethanolamin, Diethanolamin, Triethanolamin, Dicyclohexylamin, Dimethylaminoethanol, Prokain, Dibenzylamin, N-Methylmorpholin, Dehydroabietylamin, Arginin, Lysin, Ethylendiamin und Methylpiperidin.
- -
AIs Solvate werden im Rahmen der Erfindung solche Formen der Verbindungen bezeichnet, welche in festem oder flüssigem Zustand durch Koordination mit Lösungsmittelmolekülen einen Komplex bilden. Hydrate sind eine spezielle Form der Solvate, bei denen die Koordination mit Wasser erfolgt.
Außerdem umfasst die vorliegende Erfindung auch Prodrugs der erfindungsgemäßen Verbindungen. Der Begriff „Prodrugs" umfasst Verbindungen, welche selbst biologisch aktiv oder inaktiv sein können, jedoch während ihrer Verweilzeit im Körper zu erfindungsgemäßen Verbindungen umgesetzt werden (beispielsweise metabolisch oder hydrolytisch).
Die erfindungsgemäßen Verbindungen zeigen ein nicht vorhersehbares, wertvolles pharma- kologisches und pharmakokinetisches Wirkspektrum. Sie zeichnen sich insbesondere durch eine Inhibition von PDE9A aus.
Überraschenderweise wurde gefunden, dass die erfindungsgemäßen Verbindungen zur Herstellung von Arzneimitteln zur Behandlung von Herz-Kreislauf-Erkrankungen geeignet sind.
Die erfindungsgemäßen Verbindungen können aufgrund ihrer pharmakologischen und pharma- kokinetischen Eigenschaften allein oder in Kombination mit anderen Arzneimitteln zur Behandlung von Herz-Kreislauf-Erkrankungen eingesetzt werden.
Sie können daher in Arzneimitteln zur Behandlung von kardiovaskulären Erkrankungen wie beispielsweise zur Behandlung des Bluthochdrucks und der Herzinsuffizienz, akutem Herzversagen, stabiler und instabiler Angina pectoris, peripheren und kardialen Gefäßerkrankungen, von Arrhythmien, zur Behandlung von thromboembolischen Erkrankungen und Ischämien wie Myokardinfarkt, Hirnschlag, transistorischen und ischämischen Attacken, peripheren Durchblutungsstörungen, Verhinderung von Restenosen wie nach Thrombolysetherapien, percutan transluminalen Angioplastien (PTA), percutan transluminalen Koronarangioplastien (PTCA), Bypass sowie zur Behandlung von Arteriosklerose, asthmatischen Erkrankungen und Krankheiten des Urogenitalsystems wie beispielsweise Prostatahypertrophie, erektiler Dysfunktion, weiblicher sexueller Dysfunktion, Osteoporose, Glaukom, pulmonaler Hypertonie, portaler Hypertonie, ischämischen Nierenerkrankungen, Nephrosen, wie dem nephrotischem Syndrom, Nierenstenosen, Niereninsuffizienz, akutes Nierenversagen, metabolischem Syndrom, Gastroparese, Inkontinenz, Leberfibrose und Leberzirrhose eingesetzt werden.
Beispiele der erfindungsgemäßen Verbindungen der Formel (I) sind aus WO 04/113306 bekannt.
Beispiele der erfindungsgemäßen Verbindungen der Formel (II) sind im folgenden aufgeführt.
Abkürzungen:
DCI direkte chemische Ionisation (bei MS)
DMSO Dimethylsulfoxid d. Th. der Theorie (bei Ausbeute)
ESI Elektrospray-Ionisation (bei MS)
Fp. Schmelzpunkt
H Stunde(n)
HPLC Hochdruck-, Hochleistungsflüssigchromatographie
LC-MS Flüssigchromatographie-gekoppelte Massenspektroskopie min Minute(n)
MS Massenspektroskopie
NMR Kernresonanzspektroskopie
Rf Retentionsindex (bei DC)
RT Raumtemperatur
R. Retentionszeit (bei HPLC)
LC-MS- und HPLC-Methoden;
Methode 1 :
Instrument: Micromass Platform LCZ mit HPLC Agilent Serie 1100; Säule: Grom-Sil 120 ODS-4 HE, 50 mm x 2.0 mm, 3 μm; Eluent A: 1 1 Wasser + 1 ml 50 %-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 1 ml 50 %-ige Ameisensäure; Gradient: 0.0 min 100 % A → 0.2 min 100 % A → 2.9 min 30 % A → 3.1 min 10 % A → 4.5 min 10 % A; Ofen: 550C; Fluss: 0.8 ml/min; UV- Detektion: 208-400 nm.
Methode 2:
Instrument: Micromass Quattro LCZ mit HPLC Agilent Serie 1100; Säule: Grom-Sil 120 ODS-4 HE, 50 mm x 2.0 mm, 3 μm; Eluent A: 1 1 Wasser + 1 ml 50 %-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 1 ml 50 %-ige Ameisensäure; Gradient: 0.0 min 100 % A → 0.2 min 100 % A → 2.9 min 30 % A → 3.1 min 10 % A → 4.5 min 10% A; Ofen: 55°C; Fluss: 0.8 ml/min; UV- Detektion: 208-400 nm.
- -
Methode 3:
Instrument: Micromass Platform LCZ mit HPLC Agilent Serie 1100; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50 %-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50 %-ige Ameisensäure; Gradient: 0.0 min 90 % A, Fluss 1 ml/min → 2.5 min 30 % A, Fluss 2 ml/min → 3.0 min 5 % A, Fluss 2 ml/min → 4.5 min 5 % A, Fluss 2 ml/min; Ofen: 500C; UV-Detektion: 210 nm.
Methode 4:
Instrument: Micromass Quattro LCZ mit HPLC Agilent Serie 1100; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50 %-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50 %-ige Ameisensäure; Gradient: 0.0 min 90 % A, Fluss 1 ml/min → 2.5 min 30 % A, Fluss 2 ml/min → 3.0 min 5 % A, Fluss 2 ml/min → 4.5 min 5 % A, Fluss 2 ml/min; Ofen: 500C; UV-Detektion: 208- 400 nm.
Methode 5:
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2795; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50 %-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50 %-ige Ameisensäure; Gradient: 0.0 min 90 % A, Fluss 1 ml/min → 2.5 min 30 % A, Fluss 2 ml/min → 3.0 min 5 % A, Fluss 2 ml/min -» 4.5 min 5 % A, Fluss 2 ml/min; Ofen: 5O0C; UV-Detektion: 210 nm.
Methode 6:
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: HP 1100 Series; UV DAD; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50 %-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50 %-ige Ameisensäure; Gradient: 0.0 min 90 % A, Fluss 1 ml/min → 2.5 min 30 % A, Fluss 2 ml/min → 3.0 min 5 % A, Fluss 2 ml/min → 4.5 min 5 % A, Fluss 2 ml/min; Ofen: 500C; UV-Detektion: 210 nm.
Methode 7:
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: HP 1100 Series; UV DAD; Säule: Grom-Sil 120 ODS-4 HE 50 mm x 2 mm, 3.0 μm; Eluent A: Wasser + 500 μl 50 %-ige Ameisensäure / 1, Eluent B: Acetonitril + 500 μl 50 %-ige Ameisensäure / 1; Gradient: 0.0 min 0 % B → 2.9 min 70 % B → 3.1 min 90 % B → 4.5 min 90 % B; Ofen: 500C; Fluss: 0.8 ml/min; UV-Detektion: 210 nm.
Methode 8:
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2790; Säule: Grom-Sil 120 ODS-4 HE 50 mm x 2 mm, 3.0 μm; Eluent A: Wasser + 500 μl 50 %-ige Ameisensäure / 1, Eluent B: Acetonitril + 500 μl 50 %-ige Ameisensäure / 1; Gradient: 0.0 min 5 % B → 2.0 min 40 % B -> 4.5 min 90 % B → 5.5 min 90 % B; Fluss: 0.0 min 0.75 ml/min → 4.5 min 0.75 ml/min → 5.5 min 1.25 ml/min; Ofen: 450C; UV-Detektion: 210 nm.
Methode 9:
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2790; Säule: Grom-Sil 120 ODS-4 HE 50 mm x 2 mm, 3.0 μm; Eluent A: Wasser + 500 μl 50 %-ige Ameisensäure / 1, Eluent B: Acetonitril + 500 μl 50 %-ige Ameisensäure / 1; Gradient: 0.0 min 0 % B → 0.2 min 0 % B → 2.9 min 70 % B → 3.1 min 90 % B → 4.5 min 90 % B; Ofen: 450C; Fluss: 0.8 ml/min; UV- Detektion: 210 nm.
Methode 10:
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2795; Säule: Merck Chromolith SpeedROD RP-18e 50 mm x 4.6 mm; Eluent A: Wasser + 500 μl 50 %-ige Ameisensäure / 1, Eluent B: Acetonitril + 500 μl 50 %-ige Ameisensäure / 1; Gradient: 0.0 min 10 % B → 3.0 min 95 % B → 4.0 min 95 % B; Ofen: 35°C; Fluss: 0.0 min 1.0 ml/min → 3.0 min 3.0 ml/min → 4.0 min 3.0 ml/min; UV-Detektion: 210 nm.
Methode 1 1 :
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2790; Säule: Uptisphere C 18 50 mm x 2.0 mm, 3.0 μm; Eluent B: Acetonitril + 0.05 % Ameisensäure, Eluent A: Wasser + 0.05 % Ameisensäure; Gradient: 0.0 min 5 % B → 2.0 min 40 % B → 4.5 min 90 % B → 5.5 min 90 % B; Ofen: 45°C; Fluss: 0.0 min 0.75 ml/min → 4.5 min 0.75 ml/min → 5.5 min 1.25 ml/min; UV-Detektion: 210 nm.
Methode 12:
Instrument: HP 1100 mit DAD-Detektion; Säule: Kromasil RP-18, 60 mm x 2 mm, 3.5 μm; Eluent A: 5 ml HClO4 / 1 Wasser, Eluent B: Acetonitril; Gradient: 0 min 2% B → 0.5 min 2% B → 4.5 min 90 % B → 6.5 min 90 % B; Fluss: 0.75 ml/min; Temperatur: 300C; UV-Detektion: 210 nm.
Methode 13:
Instrument: HP 1100 mit DAD-Detektion; Säule: Kromasil RP-18, 125 mm x 4 mm, 5 μm; Eluent A: 5 ml HClO4 / 1 Wasser, Eluent B: Acetonitril; Gradient: 0 min 2 % B → 0.5 min 2 % B → 4.5 min 90 % B → 6.5 min 90% B; Fluss: 0.75 ml/min; Temperatur: 3O0C; UV-Detektion: 210 nm.
- -
Ausgangsverbindungen ;
Die für die folgenden Umsetzungen benötigten Amidine werden aus den entsprechenden Nitrilen bzw. Estern gemäß Gielen H., Alonso-Alija C, Hendrix M., Niewöhner U., Schauß D., Tetrahedron Lett. 43, 419-421 (2002) hergestellt.
eispiel IA
2-(3,4-Dichlθφhenyl)ethanamidin-Hydrochlorid

Unter einer Argonatmosphäre werden 2.88 g (54 mmol) Ammoniumchlorid in 50 ml Toluol suspendiert und auf 00C abgekühlt. Nach Zutropfen von 27 ml einer 2 M Trimethylaluminium- Lösung in Toluol wird das Gemisch auf Raumtemperatur erwärmt und 1.5 h nachgerührt. Es werden 5 g (27 mmol) 3,4-Dichlorphenylacetonitril zugegeben und der Ansatz über Nacht bei 800C gerührt. Nach Abkühlen auf O0C werden 50 ml Methanol zugetropft. Das Produkt wird vom ausgefallenen Feststoff durch Absaugen getrennt und der Filterkuchen mehrfach mit Methanol gewaschen. Die vereinigten Filtrate werden bis zur Trockene eingeengt, der Rückstand dann in Dichlormethan/Methanol 10:1 aufgeschlämmt und wiederum abgesaugt. Das Filtrat ergibt nach Einengen 6.2 g (77% d. Th.) der Titelverbindung.
MS (ESIpos): m/z = 203 [M+H]+.
Beispiel 2A
6-Methoxypyridin-3-ylboronsäure

1 g (5.32 mmol) 5-Brom-2-methoxypyridin wird in 10 ml absolutem Tetrahydrofuran gelöst und auf -780C abgekühlt. Nach Zugabe von 0.4 g (6.38 mmol) einer 1.6 M n-Butyllithium-Lösung in
Hexan entsteht eine gelbe Lösung, die 30 min bei der gegebenen Temperatur gerührt wird. Nach
Zugabe von 3 g (15.9 mmol) Triisopropylborat wird eine weitere Stunde gerührt, wobei sich die
Lösung auf -200C erwärmt. Man versetzt mit Wasser und rührt über Nacht nach. Die Rohlösung wird mit 1 N Salzsäure auf pH 5 angesäuert und zweimal mit Ethylacetat extrahiert. Die organische Phase wird über Natriumsulfat getrocknet, filtriert und eingeengt, wobei ein hellbrauner Feststoff erhalten wird, der mit Diethylether aufgeschlämmt und filtriert wird. Es werden 0.38 g (47 % d. Th.) des Produktes isoliert.
MS (ESIpos): m/z = 154 [M+H]+
1H-NMR (DMSO-d6, 300 MHz): δ = 3.83 (s, 3H), 6.76 (d, IH), 8.0 (dd, IH), 8.52 (s, IH).
Beispiel 3 A
Methyl [2-(6-methoxypyridin-3-yl)phenyl]acetat

1.35 g (5.89 mmol) Methyl (2-bromphenyl)acetat werden mit 1 g (6.55 mmol) 6-Methoxypyridin-
3-ylboronsäure und 1.98 g (13.09 mmol) Cäsiumfluorid unter Argon in 20 ml 1 ,2-Dimethoxyethan vorgelegt. Nach Zugabe von 0.22 g (0.19 mmol) Tetrakis(triphenylphosphin)palladium(0) wird die
Reaktionsmischung 4 h bei 1000C gerührt. Nach Abkühlen auf Raumtemperatur wird eine Mischung aus Ethylacetat und Wasser zugesetzt und mit Ethylacetat extrahiert. Nach Trocknen der organischen Phase über Magnesiumsulfat und Entfernen des Lösungsmittels im Vakuum wird der Rückstand säulenchromatographisch an Kieselgel gereinigt (Laufmittel: Cyclohexan/Ethylacetat 9: 1 ). Es werden 1.1 g (68% d. Th.) des Produktes erhalten.
LC-MS (Methode 5): R, = 2.1 min., MS (ESIpos): m/z = 258 [M+H]+
1H-NMR (DMSOd6, 300 MHz): δ = 3.51 (s, 3H), 3.63 (s, 2H), 3.9 (s, 3H), 6.89 (d, IH), 7.26 (m, IH), 7.38 (m, 3H), 7.63 (dd, IH), 8.08 (m, IH).
Beispiel 4A
2-[2-(6-Methoxypyridin-3-yl)phenyl]ethanimidamid-Hydrochlorid

Unter einer Argonatmosphäre werden 1.14 g (21.37 mmol) Ammoniumchlorid in 20 ml Toluol suspendiert und auf 00C abgekühlt. Nach Zutropfen von 10.7 ml einer 2 M Trimethylaluminium- Lösung in Toluol wird das Gemisch auf Raumtemperatur erwärmt und 1.5 h nachgerührt. Es werden 1.1 g (4.27 mmol) Methyl [2-(6-methoxypyridin-3-yl)phenyl]acetat zugegeben und der Ansatz zwei Tage bei 8O0C gerührt. Nach Abkühlen auf 50C werden 50 ml Methanol zugetropft. Das Produkt wird vom ausgefallenen Feststoff durch Absaugen getrennt und der Filterkuchen mehrfach mit Methanol gewaschen. Die vereinigten Filtrate werden bis zur Trockene eingeengt, der Rückstand dann in Dichlormethan/Methanol 10:1 aufgeschlämmt und wiederum abgesaugt. Das Filtrat ergibt nach Einengen 0.5 g (46% d. Th.) der Titelverbindung.
LC-MS (Methode 5): R, = 0.94 min., MS (ESIpos): m/z = 242 [M+H]+
1H-NMR (DMSO-d6, 300 MHz): δ = 3.79 (s, 2H), 3.9 (s, 3H), 6.89 (d, IH), 7.31 (m, 2H), 7.46 (m, 2H), 7.68 (dd, IH), 8.12 (m, IH), 8.72 (s, IH), 8.8 (s, 2H).
Beispiel 5A
2-[2-(6-Methoxypyridin-3-yl)benzyl]-4-(methylsulfanyl)-6-oxo-l,6-dihydropyrimidin-5-carbonitril

0.55 g (1.96 mmol) 2-[2-(6-Methoxypyridin-3-yl)phenyl]ethanimidamid-Hydrochlorid werden mit 0.4 g (1.96 mmol) Methyl 2-cyano-3,3-dimethylthioprop-2-enoat und 0.79 g (7.81 mmol) Triethyl- amin in 20 ml Dioxan gelöst und über Nacht bei 9O0C gerührt. Anschließend wird das Lösungsmittel bis auf ca. 2 ml im Vakuum entfernt und die verbleibende Lösung mit Acetonitril versetzt, wobei das Produkt ausfällt. Nach Filtration wird mit Acetonitril und Methanol gewaschen und das Produkt im Hochvakuum getrocknet. Man erhält 276 mg (38% d. Th.) der Titelverbindung.
LC-MS (Methode 5): R1 = 2.08 min., MS (ESIpos): m/z = 365 [M+H]+
1H-NMR (DMSO-d6, 300 MHz): δ = 2.31 (s, 3H), 3.9 (s, 3H), 3.98 (s, 2H), 6.89 (d, IH), 7.28 (m, IH), 7.39 (m, 3H), 7.63 (dd, IH), 8.08 (m, IH).
Beispiel 6A
2-Benzyl-4-(methylsulfanyl)-6-oxo-l,6-dihydro-5-pyrimidincarbonitril

100 mg (0.59 mmol) 2-Phenylethanamidin-Hydrochlorid werden mit 119 mg (0.59 mmol) Methyl 2-cyano-3,3-dimethylthioprop-2-enoat und 237 mg (2.34 mmol) Triethylamin in 2 ml Ethanol gelöst und 5 h bei 7O0C gerührt. Anschließend wird das Lösungsmittel im Vakuum entfernt, der
Rückstand in 50 ml Dichlormethan aufgenommen und mit 2 M Salzsäure gewaschen. Nach Trocknen der organischen Phase über Magnesiumsulfat wird das Lösungsmittel im Vakuum entfernt und der Rückstand an Kieselgel flash-chromatographiert (Laufmittel: Dichlormethan/ Methanol 200:1, 100:1). Man erhält 75 mg (50% d. Th.) der Titelverbindung.
HPLC (Methode 12): R, = 4.2 min.
MS (ESIpos): m/z = 258 [M+H]+.
Beispiel 7A
2-(3-Methylbenzyl)-4-(methylsulfanyl)-6-oxo-l,6-dihydro-5-pyrimidincarbonitril

1.45 g (9.83 mmol) 2-(3-Methylphenyl)ethanamidin-Hydrochlorid werden mit 2 g (9.83 mmol) Methyl 2-cyano-3,3-dimethylthioprop-2-enoat und 2 g (19.67 mmol) Triethylamin in 40 ml Ethanol gelöst und 5 h bei 7O0C gerührt. Anschließend wird das Lösungsmittel im Vakuum entfernt und der Rückstand mittels präparativer HPLC gereinigt. Es werden 0.4 g (15 % d. Th.) des Produkts erhalten.
LC-MS (Methode 2): R, = 2.83 min., MS (ESIpos): m/z = 272 [M+H]+
1H-NMR (DMSOd6, 300 MHz): δ = 2.25 (s, 3H), 2.50 (s, 3H), 3.91 (s, 2H), 7.19 (m, 4H).
Beispiel 8A
2-(2-Methylbenzyl)-4-(methylsulfanyl)-6-oxo-l,6-dihydro-5-pyrimidincarbonitril
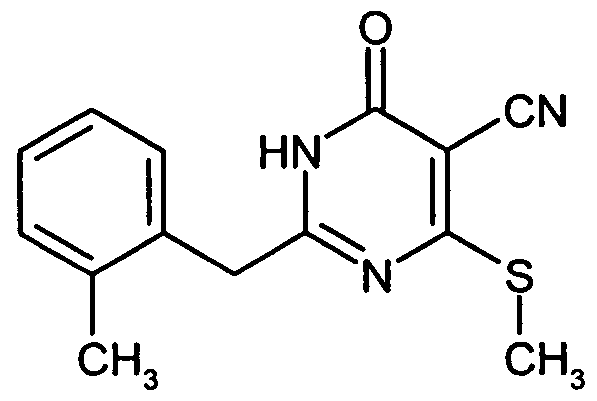
LC-MS (Methode 10): R, = 2.13 min., MS (ESIpos): m/z = 272 [M+H]+.
Beispiel 9A
2-(2-Fluorbenzyl)-4-(methylsulfanyl)-6-oxo-l,6-dihydro-5-pyrimidincarbonitril

3.5 g (18.5 mmol) 2-(2-Fluorphenyl)ethanamidin-Hydrochlorid werden mit 3.8 g (18.5 mmol) Methyl 2-cyano-3,3-dimethylthioprop-2-enoat und 7.5 g (74.2 mmol) Triethylamin in 50 ml Dioxan gelöst und über Nacht bei 900C gerührt. Nach dem Abfiltrieren der Triethylammonium- salze wird das Filtrat eingeengt und der Rückstand in Ethylacetat aufgenommen. Das Produkt wird durch Zugabe von 1 N Salzsäure und Wasser ausgefällt. Es werden 3.8 g (75 % d. Th.) der Titel- Verbindung erhalten.
LC-MS (Methode 10): R, = 2.03 min., MS (ESIpos): m/z = 276 [M+H]+
1H-NMR (DMSOd6, 300 MHz): δ = 2.31 (s, 3H), 4.06 (s, 2H), 7.19 (m, 2H), 7.41 (m, 2H).
Beispiel IQA
2-(2-Ethoxybenzyl)-4-(methylsulfanyl)-6-oxo-l,6-dihydro-5-pyrimidincarbonitril

LC-MS (Methode 3): R, = 2.32 min., MS (ESIpos): m/z = 302 [M+H]+
1H-NMR (DMSOd6, 300 MHz): δ = 1.19 (t, 3H), 2.30 (s, 3H), 3.99 (q, 2H + s, 2H), 6.93 (m, 2H), 7.27 (m, 2H).
Beispiel I IA
2-(3-Chlorbenzyl)-4-(methylsulfanyl)-6-oxo-l,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 2A werden 0.5 g (2.00 mmol) 2-(3-Chlorphenyl)ethanamidin- Hydrobromid mit 0.41 g (2.00 mmol) Methyl 2-cyano-3,3-dimethylthioprop-2-enoat und 0.81 g (8.01 mmol) Triethylamin zu 0.5 g (86 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R. = 4.4 min.
MS (DCI, NH3): m/z = 292 [M+H]+, 309 [M+NHJ*.
Beispiel 12A
2-(4-Chlorbenzyl)-4-(methylsulfanyl)-6-oxo-l,6-dihydropyrimidin-5-carbonitril

HPLC (Methode 12): R, = 4.35 min.
MS (ESIpos): m/z = 292 [M+H]+.
Beispiel 13A
2-(3 ,4-Dichlorbenzyl)-4-(methy lsulfany l)-6-oxo- 1 ,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 2A werden 1.00 g (4.17 mmol) 2-(3,4-Dichlorphenyl)ethan- amidin-Hydrochlorid mit 0.85 g (4.17 mmol) Methyl 2-cyano-3,3-dimethylthioprop-2-enoat und 1.69 g (16.7 mmol) Triethylamin zu 0.6 g (44 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 4.7 min.
MS (DCI, NH3): m/z = 343 [M+NHL,]*.
Beispiel 14A
2-(3-Fluorbenzyl)-4-(methylsulfanyl)-6-oxo-l ,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 2A werden 100 mg (0.43 mmol) 2-(3-Fluorphenyl)ethan- amidin-Hydrochlorid mit 87 mg (0.43 mmol) Methyl 2-cyano-3,3-dimethylthioprop-2-enoat und 174 mg (1.72 mmol) Triethylamin zu 28 mg (24% d. Th.) der Titelverbindung umgesetzt.
- -
HPLC (Methode 12): R, = 4.2 min.
MS (ESIpos): m/z = 276 [M+H]+.
Beispiel 15A
4-(Methylsulfanyl)-6-oxo-2-[3-(trifluormethyl)benzyl]-l,6-dihydropyrimiäin-5-carbonitril

Analog zur Darstellung von Beispiel 2A werden 0.5 g (2.10 mmol) 2-[3-(Trifluormethyl)phenyl]- ethanamidin-Hydrochlorid mit 0.43 g (2.10 mmol) Methyl 2-cyano-3,3-dimethylthioprop-2-enoat und 0.85 g (8.38 mmol) Triethylamin zu 0.4 g (59% d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 4.4 min.
MS (ESIpos): m/z = 326 [M+H]+.
Beispiel 16A
4-(Methylsulfanyl)-6-oxo-2-(3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 2A werden 3.10 g (17.6 mmol) 2-(3-Thienyl)ethanamidin- Hydrochlorid mit 3.57 g (17.6 mmol) Methyl 2-cyano-3,3-dimethylthioprop-2-enoat und 7.10 g (70.2 mmol) Triethylamin zu 2.19 g (47% d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R1 = 4.1 min.
MS (ESIpos): m/z = 263.9 [M+H]+.
Beispiel 17A
Methyl (2E/Z)-2-cyano-3-(cyclohexylamino)-3-methylthio-prop-2-enoat

0.6 g (2.9 mmol) Methyl 3,3-bis(methylthio)-2-cyanoacrylat werden mit 0.29 g (2.9 mmol) Cyclo- hexylamin in 20 ml Acetonitril 1 h bei Raumtemperatur gerührt. Die flüchtigen Bestandteile werden im Vakuum entfernt. Es werden 0.74 g (98 % d. Th.) des Produkts als gelbes Öl erhalten.
HPLC (Methode 12): R, = 4.85 min.
MS (DCI, NH3): m/z = 254.9 [M+H]+, 272 [M+NH(]+.
Beispiel 18A
Methyl (2E/Z)-2-cyano-3-(cyclopentylamino)-3-methylthio-prop-2-enoat

0.3 g (1.47 mmol) Methyl 3,3-bis(methylthio)-2-cyanoacrylat werden mit 0.13 g (1.47 mmol) Cyclopentylamin in 3 ml Acetonitril 30 min auf 700C erhitzt. Anschließend werden die flüchtigen Bestandteile im Vakuum entfernt. Man erhält 0.35 g (98 % d. Th.) des Produkts als gelbes Öl.
HPLC (Methode 12): R1 = 4.6 min.
MS (DCI, NH3): m/z = 241 [M+H]+, 258 [M+NH,]+.
Ausfuhrungsbeispiele:
Die nachfolgenden Verbindungen werden nach dem in Schema I dargestellten allgemeinen Syntheseweg hergestellt:
Schema I:


X = Cl, Br
a) Ethanol, Triethylamin, 5-16 h Rückfluss; b) Acetonitril, 85-9O0C, 1-7 Tage.
Beispiel 1
2-(3 ,4-Dichlorbenzy l)-6-oxo-4-( 1 -piperidiny I)- 1 ,6-dihydropyrimidin-5-carbonitril

100 mg (0.34 mmol) 2-(3,4-Dichlorbenzyl)-4-(methylsulfanyl)-6-oxo-l,6-dihydropyrimidin 5-carbonitril werden mit 261 mg (3.07 mmol) Piperidin 16 h bei 85°C gerührt. Nach Entfernen der flüchtigen Bestandteile im Vakuum wird der Rückstand mittels präparativer HPLC gereinigt. Es werden 42 mg (38 % d. Th.) der Titelverbindung erhalten.
HPLC (Methode 12): R, = 4.8 min.
MS (ESIpos): m/z = 363 [M+H]+
1H-NMR (DMSO-dö, 200 MHz): δ = 1.45-1.70 (m, 6H), 3.73-3.89 (m, 6H), 7.35 (dd, IH), 7.57- 7.66 (m, 2H), 11.1 (s, IH).
Beispiel 2
2-Benzy l-6-oxo-4-( 1 -piperidiny I)- 1 ,6-dihydropyrimidin-5-carbonitril

43 mg (0.17 mmol) 2-Benzyl-4-(methylsulfanyl)-6-oxo-l,6-dihydropyrimidin-5-carbonitril werden in 0.3 ml Acetonitril suspendiert und mit 42.7 mg (0.50 mmol) Piperidin 16 h bei 85°C gerührt. Im Anschluss wird das entstandene Rohprodukt mittels präparativer HPLC gereinigt. Es werden 11 mg (22 % d. Th.) der Titelverbindung erhalten.
HPLC (Methode 12): R, = 4.3 min.
MS (ESIpos): m/z = 295 [M+H]+
1H-NMR (CD3OD, 300 MHz): δ = 1.59-1.77 (m, 6H), 3.83 (s, 2H), 3.93 (t, 4H), 7.24-7.34 (m, 5H).
Beispiel 3
4-[4-(2-Hydroxyethyl)-l-piperidinyl]-2-(3-methylbenzyl)-6-oxo-l,6-dihydro-5-pyrimidincarbonitril

0.1 g (0.37 mmol) 2-(3-Methylbenzyl)-4-(methylsulfanyl)-6-oxo-l,6-dihydro-5-pyrimidincarbo- nitril werden unter Argon mit 0.142 g (1.16 mmol) 2-(4-Piperidinyl)ethan-l-ol in 3 ml Acetonitril sieben Tage auf 90°C erhitzt. Nach Abkühlen auf Raumtemperatur wird das Rohprodukt mittels
präparativer HPLC gereinigt. Es werden 0.047 g (36 % d. Th.) der Titelverbindung als farbloser Feststoff erhalten.
LC-MS (Methode 7): R, = 3.01 min., MS (ESIpos): m/z = 353 [M+H]+
1H-NMR (DMSO-d6, 300 MHz): δ = 1.05 (m, 2H), 1.36 (m, 2H), 1.69 (m, 3H), 2.25 (s, 3H), 2.89 (t, 2H), 3.42 (t, 2H), 3.68 (s, 2H), 4.51 (d, 2H), 7.18 (m, 4H).
Beispiel 4
4-(Cyclopentylamino)-2-(3-methylbenzyl)-6-oxo-l,6-dihydro-5-pyrimidincarbonitril

0.1 g (0.37 mmol) 2-(3-Methylbenzyl)-4-(methylsulfanyl)-6-oxo-l,6-dihydro-5-pyrimidincarbo- nitril werden unter Argon mit 0.31 g (3.65 mmol) Cyclopentylamin in 3 ml Acetonitril über Nacht auf 9O0C erhitzt. Nach Abkühlen auf Raumtemperatur wird das Rohprodukt mittels präparativer HPLC gereinigt. Es werden 0.03 g (26 % d. Th.) der Titelverbindung als farbloser Feststoff erhalten.
LC-MS (Methode 10): R, = 2.27 min., MS (ESIpos): m/z = 309 [M+H]+.
Beispiel 5
4-[(2S)-2-(Hydroxymethyl)-l-pyrrolidinyl]-2-(3-methylbenzyl)-6-oxo-l,6-dihydro-5-pyrimidin- carbonitril

0.1 g (0.37 mmol) 2-(3-Memylbenzyl)-4-(methylsulfanyl)-6-oxo-l,6-dihydro-5-pyrirnidincarbo- nitril werden unter Argon mit 0.11 g (1.1 mmol) (<S)-(+)-2-Pyrrolidinmethanol in 3 ml Acetonitril fünf Tage auf 900C erhitzt. Nach Abkühlen auf Raumtemperatur wird das Rohprodukt mittels präparativer HPLC gereinigt. Es werden 0.035 g (29 % d. Th.) der Titelverbindung als farbloser Feststoff erhalten.
LC-MS (Methode 7): R, = 2.91 min., MS (ESIpos): m/z = 325 [M+H]+
1H-NMR (DMSO-d6, 300 MHz): δ = 1.94 (m, 4H), 2.28 (s, 2H), 3.76 (m, 3H), 3.70 (s, 2H), 3.81 (m, 2H), 4.4 (m, IH), 7.15 (m, 4H), 12.34 (s, IH).
Beispiel 6
4-[4-(2-Hydroxyethyl)-l -piperidinyl]-2-(2-methylbenzyl)-6-oxo-l ,6-dihydro-5-pyrimidincarbonitril

0.1 g (0.37 mmol) 2-(2-Methylbenzyl)-4-(methylsulfanyl)-6-oxo-l,6-dihydro-5-pyrimidincarbo- nitril werden unter Argon mit 0.14 g (1.1 mmol) 2-(4-Piperidinyl)ethan-l-ol in 3 ml Acetonitril fünf Tage auf 9O0C erhitzt. Nach Abkühlen auf Raumtemperatur wird das Rohprodukt mittels präparativer HPLC gereinigt. Es werden 13 mg (10 % d. Th.) der Titelverbindung als farbloser Feststoff erhalten.
LC-MS (Methode 5): R, = 1.74 min., MS (ESIpos): m/z = 353 [M+H]+
1H-NMR (DMSO-de, 300 MHz): δ = 1.06 (m, 2H), 1.34 (m, 2H), 1.68 (m, 3H), 2.30 (s, 3H), 3.00 (t, 2H), 3.42 (t, 2H), 3.80 (s, 2H), 4.51 (d, 2H), 7.16 (m, 4H), 12.33 (s, IH).
Beispiel 7
2-(2-Fluorbenzyl)-4-[4-(2-hydroxyethyl)-l-piperidinyl]-6-oxo-l,6-dihydro-5-pyrimidincarbonitril

0.1 g (0.37 mmol) 2-(2-Fluorbenzyl)-4-(methylsulfanyl)-6-oxo-l,6-dihydro-5-pyrimidincarbonitril werden unter Argon mit 0.14 g (1.1 mmol) 2-(4-Piperidinyl)ethan-l-ol in 3 ml Acetonitril sechs Tage auf 9O0C erhitzt. Nach Abkühlen auf Raumtemperatur wird das Rohprodukt mittels präpara- tiver HPLC gereinigt. Es werden 31 mg (24 % d. Th.) der Titelverbindung als farbloser Feststoff erhalten.
LC-MS (Methode 2): R, = 2.94 min., MS (ESIpos): m/z = 357 [M+H]+
1H-NMR (DMSO-dö, 300 MHz): δ = 1.01 (m, 2H), 1.33 (m, 2H), 1.67 (m, 3H), 2.97 (t, 2H), 3.41 (dd, 2H), 3.87 (s, 2H), 4.32 (t, IH), 4.47 (d, 2H), 7.16 (m, 2H), 7.36 (m, 2H), 12.38 (s, IH).
Beispiel 8
2-(2-Ethoxybenzyl)-4-[4-(2-hydroxyethyl)-l-piperidinyl]-6-oxo-l,6-dihydro-5-pyrimidincarbonitril

0.1 g (0.37 mmol) 2-(2-Ethoxybenzyl)-4-(methylsulfanyl)-6-oxo-l,6-dihydro-5-pyrimidincarbo- nitril werden unter Argon mit 0.13 g (0.99 mmol) 2-(4-Piperidinyl)ethan-l-ol in 3 ml Acetonitril fünf Tage auf 900C erhitzt. Nach Abkühlen auf Raumtemperatur wird das Rohprodukt mittels prä- parativer HPLC gereinigt. Es werden 45 mg (43 % d. Th.) der Titelverbindung als farbloser Feststoff erhalten.
LC-MS (Methode 6): R1 = 2.01 min., MS (ESIpos): m/z = 383 [M+H]+
1H-NMR (DMSO-d6, 200 MHz): δ = 1.04 (m, 2H), 1.23 (t, 3H), 1.34 (m, 2H), 1.67 (m, 3H), 2.94 (t, 2H), 3.41 (t, 2H), 3.87 (s, 2H), 3.96 (q, 2H), 4.48 (d, 2H), 6.92 (m, 2H), 7.17 (m, 2H), 12.26 (s, IH).
Beispiel 9
2-(3-Chlorbenzyl)-6-oxo-4-(propylamino)-l,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 100 mg (0.34 mmol) 2-(3-Chlorbenzyl)-4-(methyl- sulfanyl)-6-oxo-l,6-dihydropyrimidin-5-carbonitril mit 203 mg (3.43 mmol) n-Propylamin zu 12 mg (12 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R1 = 4.4 min.
MS (ESIpos): m/z = 303 [M+H]+
1H-NMR (DMSOd6, 200 MHz): δ = 0.75 (t, 3H), 1.40 (m, 2H), 3.23 (m, 2H), 3.83 (s, 2H), 7.23- 7.38 (m, 4H), 7.42 (s, IH), 12.34 (s, IH).
Beispiel 10
2-(3-Chlorbenzyl)-4-(cyclopentylamino)-6-oxo-l,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 100 mg (0.34 mmol) 2-(3-Chlorbenzyl)-4-(methyl- sulfanyl)-6-oxo-l,6-dihydropyrimidin-5-carbonitril mit 292 mg (3.43 mmol) Cyclopentylamin zu 10 mg (9 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 4.6 min.
MS (ESIpos): m/z = 329 [M+H]+
1H-NMR (DMSOd6, 200 MHz): δ = 1.38-1.85 (m, 8H), 3.82 (s, 2H), 4.20-4.37 (m, IH), 7.23-7.46 (m, 4H), 7.66-7.80 (s, IH), 12.25-12.44 (s, IH).
Beispiel 11
2-(3-Chlorbenzyl)-6-oxo-4-(l-pyrrolidinyl)-l,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 100 mg (0.34 mmol) 2-(3-Chlorbenzyl)-4-(methyl- sulfanyl)-6-oxo-l,6-dihydropyrimidin-5-carbonitril mit 244 mg (3.43 mmol) Pyrrolidin zu 29 mg (27 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 4.4 min.
MS (ESIpos): m/z = 315 [M+H]+
1H-NMR (DMSOd6, 200 MHz): δ = 1.80-1.95 (m, 4H), 3.58-3.71 (m, 4H), 3.79 (s, 2H), 7.25-7.45 (m, 4H), 12.28-12.39 (s, IH).
Beispiel 12
4-(4,4-Dimethylpiperidin-l-yl)-2-(3-chlorbenzyl)-6-oxo-l,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 150 mg (0.51 mmol) 2-(3-Chlorbenzyl)-4-(methyl- sulfanyl)-6-oxo-l,6-dihydropyrimidin-5-carbonitril mit 582 mg (5.14 mmol) 4,4-Dimethylpiperidin zu 96 mg (52 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 4.8 min.
MS (ESIpos): m/z = 357 [M+H]+
1H-NMR (DMSO-d6, 200 MHz): δ = 0.96 (s, 6H), 1.30-1.41 (m, 4H), 3.75-3.87 (m, 6H, s bei 3.81), 7.24-7.45 (m, 4H), 12.39 (s, IH).
Beispiel 13
2-(3-Chlorbenzyl)-4-[(2-methoxyethyl)amino]-6-oxo-l,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 100 mg (0.34 mmol) 2-(3-Chlorbenzyl)-4-(methyl- sulfanyl)-6-oxo-l,6-dihydropyrimidin-5-carbonitril mit 257 mg (3.43 mmol) 2-Methoxyethylamin zu 61 mg (56 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R4 = 4.0 min.
MS (ESIpos): m/z = 319 [M+H]+
1H-NMR (CDCl3, 300 MHz): δ = 3.38 (s, 3H), 3.53 (t, 2H), 3.75 (q, 2H), 3.88 (s, 2H), 6.00 (t, IH), 7.25-7.31 (m, 3H), 7.38 (s, IH), 12.56 (s, IH).
Beispiel 14
2-(3-Chlorbenzyl)-4-(morpholin-4-yl)-6-oxo-l ,6-dihydropyrimidin-5-carbonitril
Analog zur Darstellung von Beispiel 1 werden 150 mg (0.51 mmol) 2-(3-Chlorbenzyl)-4-(methyl- sulfanyl)-6-oxo-l,6-dihydropyrimidin-5-carbonitril mit 448 mg (5.14 mmol) Morpholin zu 109 mg (63 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 4.0 min.
MS (ESIpos): m/z = 331 [M+H]+
1H-NMR (DMSOd6, 200 MHz): δ = 3.59-3.69 (t, 4H), 3.79-3.90 (m, 6H, s bei 3.82), 7.25-7.44 (m, 4H), 12.53 (s, IH).
Beispiel 15
2-(3-Chlorbenzyl)-4-(4-methylpiperazin-l-yl)-6-oxo-l,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 100 mg (0.34 mmol) 2-(3-Chlorbenzyl)-4-(methyl- sulfanyl)-6-oxo-l,6-dihydropyrimidin-5-carbonitril mit 343 mg (3.43 mmol) N-Methylpiperazin zu 101 mg (84 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 3.5 min.
MS (ESIpos): m/z = 344 [M+H]+
1H-NMR (DMSOd6, 200 MHz): δ = 2.18 (s, 3H), 2.35 (t, 4H), 3.79-3.88 (m, 6H, s bei 3.82), 7.25- 7.44 (m, 4H), 12.48 (s, IH).
Beispiel 16
2-(3-Chlorbenzyl)-4-[(2-methoxybenzyl)amino]-6-oxo-l ,6-dihydropyrimidin-5-carbonitril

HPLC (Methode 12): R, = 4.6 min.
MS (ESIpos): m/z = 381 [M+H]+
1H-NMR (DMSO-d6, 300 MHz): δ = 3.77 (s, 2H), 3.79 (s, 3H), 4.52 (d, 2H), 6.84 (t, IH), 6.92- 6.99 (m, 2H), 7.12 (d, IH), 7.19-7.33 (m, 4H), 8.14 (t, IH), 12.39 (s, IH).
Beispiel 17
2-(4-Chlorbenzyl)-4-(cyclobutylamino)-6-oxo-l,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 100 mg (0.34 mmol) 2-(4-Chlorbenzyl)-4-(methyl- sulfanyl)-6-oxo-l,6-dihydropyrimidin-5-carbonitril mit 244 mg (3.43 mmol) Cyclobutylamin zu 15 mg (14 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 4.5 min.
MS (ESIpos): m/z = 315 [M+H]+
1H-NMR (DMSO-d6, 300 MHz): δ = 1.50-1.67 (m, 2H), 2.03-2.15 (m, 4H), 3.79 (s, 2H), 4.37-4.51 (m, IH), 7.31-7.43 (m, 4H), 8.00 (s, IH), 12.33 (s, IH).
Beispiel 18
2-(4-Chlorbenzyl)-6-oxo-4-(l-pyrrolidinyl)-l,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 100 mg (0.34 mmol) 2-(4-Chlorbenzyl)-4-(methyl- sulfanyl)-6-oxo-l,6-dihydropyrimidin-5-carbonitril mit 244 mg (3.43 mmol) Pyrrolidin zu 45 mg (42% d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 4.4 min.
MS (ESIpos): m/z = 315 [M+H]+
1H-NMR (CD3OD, 300 MHz): δ = 1.88-2.03 (m, 4H), 3.69-3.86 (m, 6H, s bei 3.82), 7.25-7.35 (m, 4H).
Beispiel 19
4-(Cyclopentylamino)-2-(3-fluorbenzyl)-6-oxo-l ,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 100 mg (0.36 mmol) 2-(3-Fluorbenzyl)-4-(methyl- sulfanyl)-6-oxo-l,6-dihydropyrimidin-5-carbonitril mit 309 mg (3.63 mmol) Cyclopentylamin zu 12 mg (11 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 4.4 min.
MS (ESIpos): m/z = 313 [M+H]+
1H-NMR (DMSO-d6, 200 MHz): δ = 1.29-1.71 (m, 8H), 3.89 (s, 2H), 3.98-4.14 (m, IH), 7.11-7.42 (m, 4H), 7.63-7.75 (s, IH), 12.34-12.43 (s, IH).
Beispiel 20
2-(3-Fluorbenzyl)-6-oxo-4-(l-piperidinyl)-l,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 100 mg (0.36 mmol) 2-(3-Fluorbenzyl)-4-(methyl- sulfanyl)-6-oxo-l,6-dihydropyrimidin-5-carbonitril mit 309 mg (3.63 mmol) Piperidin zu 40 mg (35 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 4.3 min.
MS (ESIpos): m/z = 313 [M+H]+
1H-NMR (DMSOd6, 200 MHz): δ = 1.40-1.66 (m, 6H), 3.66-3.76 (m, 4H), 3.76 (s, 2H), 7.12-7.42 (m, 4H), 12.27-12.41 (s, IH).
Beispiel 21
6-Oxo-4-( 1 -piperidiny l)-2-[3 -(trifluormethy l)benzy I]- 1 ,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 100 mg (0.31 mmol) 4-(Methylsulfanyl)-6-oxo-2- [3-(trifluormethyl)benzyl]-l,6-dihydropyrimidin-5-carbonitril mit 262 mg (3.07 mmol) Piperidin zu 26 mg (22 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 4.7 min.
MS (ESIpos): m/z = 363 [M+H]+
1H-NMR (DMSO-d6, 200 MHz): δ = 1.43-1.70 (m, 6H), 3.71-3.83 (m, 4H), 3.93 (s, 2H), 7.51-7.78 (m, 4H), 12.42 (s, IH).
Beispiel 22
6-Oxo-4-(propylamino)-2-(3 -thieny lmethyl)- 1 ,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 100 mg (0.38 mmol) 4-(Methylsulfanyl)-6-oxo-2- (3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril mit 224 mg (3.80 mmol) n-Propylamin zu 14 mg (13 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 4.1 min.
MS (ESIpos): m/z = 275.2 [M+H]+
1H-NMR (DMSOd6, 300 MHz): δ = 0.79 (t, 3H), 1.46 (m, 2H), 3.29 (m, 2H), 3.81 (s, 2H), 7.06 (d, IH), 7.33 (s, IH), 7.49 (m, IH), 7.87 (s, IH), 12.27 (s, IH).
Beispiel 23
4-(Cyclopropylamino)-6-oxo-2-(3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 100 mg (0.38 mmol) 4-(Methylsulfanyl)-6-oxo-2- (3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril mit 217 mg (3.80 mmol) Cyclopropylamin zu 44 mg (43 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 3.8 min. MS (ESIpos): m/z = 273 [M+H]+
1H-NMR (DMSO-d6, 300 MHz): δ = 0.60-0.78 (m, 4H), 2.84-2.98 (m, IH), 3.80 (s, 2H), 7.08 (d, IH), 7.35 (s, IH), 7.49 (m, IH), 7.85-8.05 (s, IH), 12.32 (s, IH).
Beispiel 24
4-(Cyclopentylamino)-6-oxo-2-(3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 150 mg (0.57 mmol) 4-(Methylsulfanyl)-6-oxo-2- (3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril mit 485 mg (5.70 mmol) Cyclopentylamin zu 46 mg (26 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 4.4 min.
MS (ESIpos): m/z = 301.2 [M+H]+
1H-NMR (DMSOd6, 300 MHz): δ = 1.40-1.91 (m, 8H), 3.81 (s, 2H), 4.36 (m, IH), 7.07 (d, IH), 7.34 (s, IH), 7.50 (m, IH), 7.65 (s, IH), 12.28 (s, IH).
Beispiel 25
6-Oxo-4-(l-pyrrolidinyl)-2-(3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 100 mg (0.38 mmol) 4-(Methylsulfanyl)-6-oxo-2- (3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril mit 270 mg (3.80 mmol) Pyrrolidin zu 64 mg (59 % d. Th.) der Titelverbindung umgesetzt.
- -
HPLC (Methode 12): R, = 4.1 min.
MS (ESIpos): m/z = 287 [M+H]+
1H-NMR (DMSO-d6, 300 MHz): δ = 1.80-1.96 (m, 4H), 3.60-3.76 (m, 4H), 3.78 (s, 2H), 7.08 (d, IH), 7.35 (s, IH), 7.48 (m, IH), 12.27 (s, IH).
Beispiel 26
4-(4-Methylpiperidin- 1 -yl)-6-oxo-2-(3-thienylmethyl)- 1 ,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 100 mg (0.38 mmol) 4-(Methylsulfanyl)-6-oxo-2- (3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril mit 377 mg (3.80 mmol) 4-Methylpiperidin zu 65 mg (54 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 4.5 min.
MS (ESIpos): m/z = 315 [M+H]+
1H-NMR (DMSO-d6, 300 MHz): δ = 0.91 (d, 3H), 1.05-1.18 (t, 2H), 1.62-1.77 (m, 3H), 3.06 (t, 2H), 3.80 (s, 2H), 4.61 (d, 2H), 7.07 (d, IH), 7.34 (s, IH), 7.49 (m, IH), 12.32 (s, IH).
Beispiel 27
4-(4,4-Dimethy lpiperidin- 1 -y l)-6-oxo-2-(3-thienylmethy I)- 1 ,6-dihydropyrimidin-5-carbonitril

HPLC (Methode 12): R, = 4.6 min.
MS (ESIpos): m/z = 329 [M+H]+
1H-NMR (DMSO-d6, 300 MHz): δ = 0.97 (s, 6H), 1.37 (t, 4H), 3.77-3.87 (m, 6H, s bei 3.80), 7.06 (d, IH), 7.34 (s, IH), 7.49 (m, IH), 12.31 (s, IH).
Beispiel 28
4-[4-(ferf.-Butyl)piperidin-l-yl]-6-oxo-2-(3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 100 mg (0.38 mmol) 4-(Methylsulfanyl)-6-oxo-2- (3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril mit 536 mg (3.80 mmol) 4-tert.-Butyl- piperidin zu 57 mg (42 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 4.9 min.
MS (ESIpos): m/z = 357 [M+H]+
1H-NMR (DMSO-d6, 200 MHz): δ = 0.82 (s, 9H), 1.02-1.42 (m, 3H), 1.74 (d, 2H), 2.96 (t, 2H), 3.78 (s, 2H), 4.72 (d, 2H), 7.06 (d, IH), 7.33 (s, IH), 7.49 (m, IH), 12.35 (s, IH).
Beispiel 29
4-(4-Hydroxypiperidin-l-yl)-6-oxo-2-(3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 100 mg (0.38 mmol) 4-(Methylsulfanyl)-6-oxo-2- (3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril mit 384 mg (3.80 mmol) Piperidin-4-ol zu 47 mg (39 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 3.50 min.
MS (ESIpos): m/z = 317 [M+H]+
1H-NMR (DMSO-d6, 200 MHz): δ = 1.29-1.50 (m, 2H), 1.72-1.90 (m, 2H), 3.42-3.60 (m, 2H), 3.68-3.86 (m, 3H, s bei 3.79), 4.10-4.26 (m, 2H), 4.83 (d, IH, OH), 7.07 (d, IH), 7.35 (s, IH), 7.50 (m, IH), 1 1.79-12.29 (s, IH, NH).
Beispiel 30
4-[4-(2-Hydroxyethyl)piperidin-l-yl]-6-oxo-2-(3-thienylmethyl)-l,6-dihydropyrimidin-5-carbo- nitril

Analog zur Darstellung von Beispiel 1 werden 100 mg (0.38 mmol) 4-(Methylsulfanyl)-6-oxo-2- (3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril mit 491 mg (3.80 mmol) 2-(4-Piperidinyl)- ethan-1-ol zu 64 mg (49 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 3.70 min.
MS (ESIpos): m/z = 345 [M+H]+
1H-NMR (DMSO-d6, 300 MHz): δ = 1.02-1.20 (m, 2H), 1.32-1.42 (q, 2H), 1.68-1.81 (m, 3H), 3.05 (t, 2H), 3.44 (q, 2H), 3.80 (s, 2H), 4.34 (t, IH, OH), 4.62 (d, 2H), 7.06 (d, IH), 7.34 (s, IH), 7.49 (m, IH), 12.31 (s, IH5 NH).
Beispiel 31
4-[(2-Methoxyethyl)amino]-6-oxo-2-(3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 100 mg (0.38 mmol) 4-(Methylsulfanyl)-6-oxo-2- (3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril mit 285 mg (3.80 mmol) 2-Methoxyethyl- amin zu 76 mg (69 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 3.7 min.
MS (ESIpos): m/z = 291 [M+H]+
1H-NMR (DMSOd6, 300 MHz): δ = 3.20 (s, 3H), 3.35-3.42 (m, 2H), 3.47-3.56 (m, 2H), 3.82 (s, 2H), 7.07 (d, IH), 7.34 (s, IH), 7.49 (m, IH), 7.79 (s, IH), 12.33 (s, IH).
Beispiel 32
4-(l ,4-Dioxa-8-azaspiro[4.5]dec-8-yl)-6-oxo-2-(3-thienylmethyl)-l ,6-dihydropyrimidin-5-carbo- nitril

Analog zur Darstellung von Beispiel 1 werden 100 mg (0.38 mmol) 4-(Methylsulfanyl)-6-oxo-2- (3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril mit 544 mg (3.80 mmol) 1 ,4-Dioxa-8-aza- spiro[4.5]decan zu 65 mg (48 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R1 = 4.0 min.
MS (ESIpos): m/z = 359 [M+H]+
1H-NMR (DMSO-d6, 300 MHz): δ = 1.70 (t, 4H), 3.81 (s, 2H), 3.86-3.95 (m, 8H, s bei 3.92), 7.07 (d, IH), 7.34 (s, IH), 7.49 (m, IH), 12.41 (s, IH).
Beispiel 33
4-(7,l l-Dioxa-3-azaspiro[5.5]undec-3-yl)-6-oxo-2-(3-thienylmethyl)-l,6-dihydropyrimidin-5- carbonitril

Analog zur Darstellung von Beispiel 1 werden 100 mg (0.38 mmol) 4-(Methylsulfanyl)-6-oxo-2- (3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril mit 597 mg (3.80 mmol) 1 ,5-Dioxa-9-aza- spiro[5.5]undecan zu 86 mg (61 % d. Th.) der Titel Verbindung umgesetzt.
HPLC (Methode 12): R, = 4.0 min.
MS (ESIpos): m/z = 373 [M+H]+
1H-NMR (DMSOd6, 200 MHz): δ = 1.56 (m, 2H), 1.82-1.94 (m, 4H), 3.75-3.92 (m, 10H, s bei 3.80), 7.07 (d, IH), 7.35 (s, IH), 7.50 (m, IH), 12.43 (br. s, IH).
Beispiel 34
4-(4-Methylpiperazin-l-yl)-6-oxo-2-(3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril

HPLC (Methode 12): R, = 3.2 min.
MS (ESIpos): m/z = 316 [M+H]+
1H-NMR (DMSOd6, 200 MHz): δ = 2.19 (s, 3H), 2.37 (t, 4H), 3.77-3.91 (m, 6H, s bei 3.80), 7.07 (d, IH), 7.35 (s, IH), 7.50 (m, IH), 12.43 (s, IH).
Beispiel 35
6-Oxo-4-(piperazin-l-yl)-2-(3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 80 mg (0.30 mmol) 4-(Methylsulfanyl)-6-oxo-2-(3- thienylmethyl)-l,6-dihydropyrimidm-5-carbonitril mit 326 mg (3.80 mmol) Piperazin zu 45 mg (49 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 3.15 min.
MS (ESIpos): m/z = 302 [M+H]+
1H-NMR (DMSO-d6, 200 MHz): δ = 2.75 (t, 4H), 3.76-3.82 (m, 6H, s bei 3.80), 7.06 (d, IH), 7.34 (s, IH), 7.49 (m, IH).
Beispiel 36
4-(Benzylamino)-6-oxo-2-(3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 100 mg (0.38 mmol) 4-(Methylsulfanyl)-6-oxo-2- (3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril mit 407 mg (3.80 mmol) Benzylamin zu 43 mg (35 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 4.3 min.
MS (ESIpos): m/z = 323 [M+H]+
1H-NMR (DMSO-d6, 200 MHz): δ = 3.80 (s, 2H), 4.52 (d, 2H), 6.95 (d, IH), 7.16-7.32 (m, 6H), 7.45 (m, IH), 8.49 (t, IH), 12.40 (s, IH).
Beispiel 37
4-[(2-Methoxybenzyl)amino]-6-oxo-2-(3-thienylmethyl)-l ,6-dihydropyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 100 mg (0.38 mmol) 4-(Methylsulfanyl)-6-oxo-2- (3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril mit 521 mg (3.80 mmol) 2-Methoxybenzyl- amin zu 62 mg (46 % d. Th.) der Titelverbindung umgesetzt.
HPLC (Methode 12): R, = 4.4 min.
MS (ESIpos): m/z = 353 [M+H]+
1H-NMR (DMSO-(I6, 300 MHz): δ = 3.75 (s, 2H), 3.80 (s, 3H), 4.56 (d, 2H), 6.84-6.90 (m, 2H), 6.96-7.04 (m, 2H), 7.16 (s, IH), 7.23 (t, IH), 7.39 (m, IH), 8.15 (t, IH), 12.34 (s, IH).
Beispiel 38
4-[4-(2-Hydroxyethyl)piperidin-l-yl]-2-[2-(6-methoxypyridin-3-yl)benzyl]-6-oxo-l,6-dihydro- pyrimidin-5-carbonitril

Analog zur Darstellung von Beispiel 1 werden 80 mg (0.22 mmol) 2-[2-(6-Methoxypyridin-3-yl)- benzyl]-4-(methylsulfanyl)-6-oxo-l,6-dihydropyrimidin-5-carbonitril mit 85 mg (0.65 mmol) 2-(4- Piperidinyl)ethan-l-ol zu 62 mg (63 % d. Th.) der Titelverbindung umgesetzt.
Die nachfolgende Verbindung wird nach dem in Schema II dargestellten allgemeinen Syntheseweg hergestellt:
Schema II:


X = Cl, Br
a) 1. Toluol, Bortrifluorid-Etherat, RT, 30 min.; 2. Amin-Komponente R1R2NH, 1500C, 16 h; oder: Schmelze der Ausgangsverbindungen bei 15O0C, 1-16 h; b) DMF, Triethylamin, 1000C, 16 h oder DMF, Kaliumcarbonat, 900C, 16 h.
Beispiel 39
4-(Cyclohexylamino)-6-oxo-2-(3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril

69.5 mg (0.39 mmol) 2-(3-Thienyl)ethanamidin-Hydrochlorid werden mit 100 mg (0.39 mmol) Methyl (2E/Z)-2-cyano-3-(cyclohexylamino)-3-methylthio-prop-2-enoat und 159 mg (1.57 mmol) Triethylamin in 0.5 ml DMF gelöst und über Nacht bei 1000C gerührt. Der abgekühlte Ansatz wird in wenig Wasser aufgenommen und mit Dichlormethan extrahiert. Die Dichlormethan-Phase wird über Natriumsulfat getrocknet, eingeengt und der Rückstand an Kieselgel flash-chromatographiert
- -
(Laufmittel: Dichlormethan, dann Dichlormethan/ Methanol 200:1, 100:1). Es werden 23 mg (19 % d. Th.) des Produkts erhalten.
HPLC (Methode 12): R1 = 4.55 min.
MS (DCI, NH3): m/z = 315 [M+H]+
1H-NMR (DMSOd6, 400 MHz): δ = 0.98-1.43 (m, 5H), 1.53-1.74 (m, 5H), 3.78-3.94 (m, 3H, s bei 3.82), 7.06 (d, IH), 7.33 (s, IH), 7.50 (m, IH), 7.54 (m, IH), 12.28 (s, IH).
Beispiel 40
4-(4-Formy lpiperazin- 1 -y l)-6-oxo-2-(3-thieny lmethy I)- 1 ,6-dihydropyrimidin-5-carbonitril

Unter einer Argonatmosphäre werden 11.3 mg (0.17 mmol) Imidazol mit 33.6 mg (0.33 mmol) Triethylamin und 7.64 mg (0.17 mmol) Ameisensäure in 5 ml Dichlormethan vorgelegt und auf 00C gekühlt. Dann wird eine Lösung von 21.1 mg (0.17 mmol) Oxalylchlorid in Dichlormethan zugetropft und die Mischung im Anschluss 15 min lang gerührt. Es werden 50 mg (0.17 mmol) 6- Oxo-4-(piperazin-l-yl)-2-(3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril zugefügt und der Ansatz über Nacht bei RT gerührt. Dann wird mit 1 N Kaliumhydrogensulfat-Lösung gewaschen, die Dichlormethan-Phase über Natriumsulfat getrocknet, eingeengt und der Rückstand an Kieselgel flash-chromatographiert (Laufmittel: Dichlormethan/Methanol 100:1, 80:1, 60:1). Man erhält 16 mg (29 % d. Th.) der Titelverbindung.
HPLC (Methode 12): R, = 3.4 min.
MS (ESIpos): m/z = 330 [M+H]+
1H-NMR (DMSOd6, 200 MHz): δ = 3.42-3.54 (m, 4H), 3.79-3.94 (m, 6H, s bei 3.82), 7.09 (d, IH), 7.36 (s, IH), 7.51 (m, IH), 8.06 (s, IH), 12.55 (s, IH).
- -
Beispiel 41
4-(4-Acetylpiperazin-l-yl)-6-oxo-2-(3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril

50 mg (0.17 mmol) 6-Oxo-4-(piperazin-l-yl)-2-(3-thienylmethyl)-l,6-dihydropyrimidin-5-carbo- nitril werden mit 34 mg (0.33 mmol) Triethylamin in DMF gelöst und mit 14.3 mg (0.18 mmol) Acetylchlorid über Nacht bei RT gerührt. Anschließend wird der Ansatz mit Dichlormethan verdünnt und mit Wasser gewaschen. Die organische Phase wird abgetrennt, über Natriumsulfat getrocknet und der Rückstand an Kieselgel flash-chromatographiert (Laufmittel: Dichlormethan/ Methanol 100:1, 80: 1, 60:1). Man erhält 42 mg (74% d. Th.) der Titelverbindung.
HPLC (Methode 12): R, = 3.5 min.
MS (ESIpos): m/z = 344 [M+H]+
1H-NMR (DMSOd6, 300 MHz): δ = 2.02 (s, 3H), 3.50-3.61 (m, 4H), 3.80-3.95 (m, 6H, s bei 3.82), 7.08 (d, IH), 7.36 (s, IH), 7.50 (m, IH), 12.47 (s, IH).
Beispiel 42
4-(4-Ethylpiperazin-l-yl)-6-oxo-2-(3-thienylmethyl)-l,6-dihydropyrimidin-5-carbonitril

50 mg (0.17 mmol) 6-Oxo-4-(piperazin-l-yl)-2-(3-thienylmethyl)-l,6-dihydropyrimidin-5-carbo- nitril werden mit 34 mg (0.33 mmol) Triethylamin in DMF gelöst, mit 19.9 mg (0.18 mmol)
Bromethan versetzt und über Nacht bei RT gerührt. Dann wird der Ansatz mit Dichlormethan verdünnt und mit Wasser gewaschen. Die organische Phase wird abgetrennt, über Natriumsulfat getrocknet und der Rückstand an Kieselgel flash-chromatographiert (Laufmittel: Dichlormethan/ Methanol 100:1, 80:1, 60:1, 40:1). Man erhält 38 mg (70% d. Th.) der Titelverbindung.
HPLC (Methode 12): R, = 3.3 min.
MS (ESIpos): m/z = 330 [M+H]+
1H-NMR (DMSO-d6, 300 MHz): δ = 1.00 (t, 3H), 2.34 (q, 2H), 2.42 (t, 4H), 3.80 (s, 2H), 3.86 (t, 4H), 7.06 (d, IH), 7.34 (s, IH), 7.49 (m, IH), 12.39 (s, IH).
Die in der folgenden Tabelle aufgeführten Ausführungsbeispiele werden in Analogie zu den zuvor beschriebenen Beispielen hergestellt:







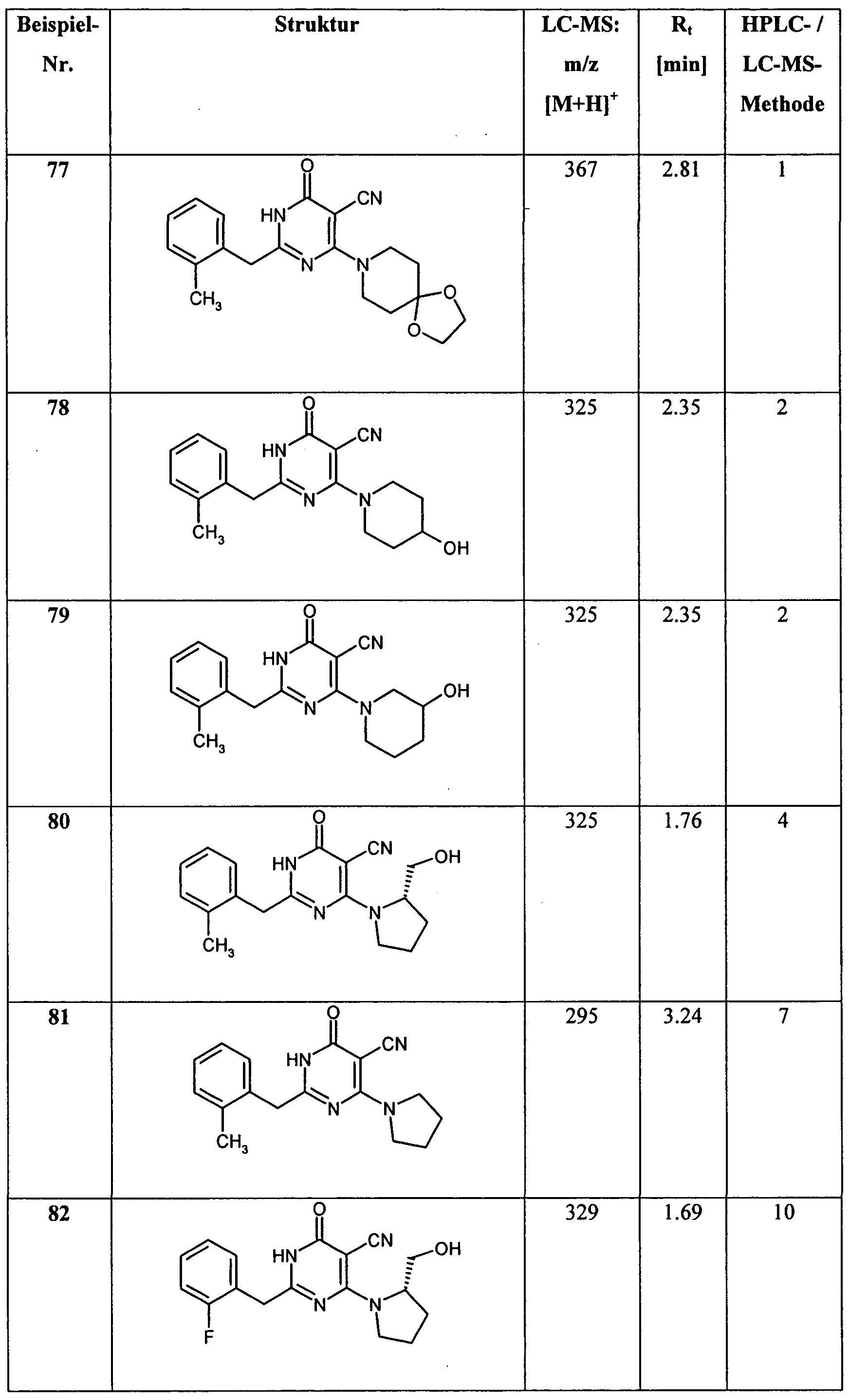

















Die in v/Yro-Wirkung der erfindungsgemäßen Verbindungen der Fromel (I) und (II) kann mit folgenden biologischen Assays gezeigt werden:
PDE-Inhibition
Rekombinante PDElC (GenBank/EMBL Accession Number: NM_005020, Loughney et al. J. Biol. Chem. 1996 271, 796-806), PDE2A (GenBank/EMBL Accession Number: NM_002599, Rosman et al. Gene 1997 191, 89-95), PDE3B (GenBank/EMBL Accession Number: NM_000922, Miki et al. Genomics 1996, 36, 476-485), PDE4B (GenBank/EMBL Accession Number: NM_002600, Obernolte et al. Gene. 1993, 129, 239-247), PDE5A (GenBank/EMBL Accession Number: NM 001083, Loughney et al. Gene 1998, 216, 139-147), PDE7B (GenBank/EMBL
Accession Number: NM_018945, Hetman et al. Proc. Natl. Acad. Sei. U.S.A. 2000, 97, 472-476), PDE8A (GenBank/EMBL Accession Number: AF_056490, Fisher et al. Biochem. Biophys. Res. Commun. 1998 246, 570-577), PDE9A (Fisher et al., J. Biol. Chem, 1998, 273 (25): 15559-15564), PDElOA (GenBank/EMBL Accession Number: NM_06661, Fujishige et al. J Biol Chem. 1999, 274, 18438-45), PDEl IA (GenBank/EMBL Accession Number: NM_016953, Fawcett et al. Proc. Natl. Acad. Sei. 2Ö00, 97, 3702-3707) wurden mit Hilfe des pFASTBAC Baculovirus-Expressi- onssystems (GibcoBRL) in Sf9-Zellen exprimiert. PDE5 wurde aus humanen Blutplättchen gereinigt: Nach Ultraschallaufschluss und Zentrifugation folgte eine Ionenaustauscherchromatographie des Überstandes über eine Mono Q 10/10 Säule (linearer NaCl Gradient, Eluation mit 0,2- 0,3 M NaCl in Puffer (20 mM Hepes pH7,2 und 2 mM MgC12)). PDE 1 (aus Rinderhirn) war von Sigma.
Die Testsubstanzen werden zur Bestimmung ihrer in vitro Wirkung an PDE 9A in 100 % DMSO aufgelöst und seriell verdünnt. Typischerweise werden Verdünnungsreihen von 200 μM bis 1.6 μM hergestellt (resultierende Endkonzentrationen im Test: 4 μM bis 0.032 μM). Jeweils 2 μL der verdünnten Substanzlösungen werden in die Vertiefungen von Mikrotiterplatten (Isoplate; Wallac Inc., Atlanta, GA) vorgelegt. Anschließend werden 50 μL einer Verdünnung des oben beschriebenen PDE9A-Präparates hinzugefügt. Die Verdünnung des PDE9A-Präparates wird so gewählt, dass während der späteren Inkubation weniger als 70% des Substrates umgesetzt wird (typische Verdünnung: 1 :10000; Verdünnungspuffer: 50 mM Tris/HCl pH 7.5, 8.3 mM MgCl2, 1.7 mM EDTA, 0.2% BSA). Das Substrat, [8-3H] guanosine 3',5(-cyclic phosphate (1 μCi/μL; Amer- sham Pharmacia Biotech., Piscataway, NJ) wird 1 :2000 mit Assaypuffer (50 mM Tris/HCl pH 7.5, 8.3 mM MgCl2, 1.7 mM EDTA) auf eine Konzentration von 0.0005 μCi/μL verdünnt. Durch Zugabe von 50 μL (0.025 μCi) des verdünnten Substrates wird die Enzymreaktion schließlich gestartet. Die Testansätze werden für 60 min bei Raumtemperatur inkubiert und die Reaktion durch Zugabe von 25 μl eines in Assaypuffer gelösten PDE9A-Inhibitors (z.B. der Inhibitor aus Herstellbeispiel 1, 10 μM Endkonzentration) gestoppt. Direkt im Anschluß werden 25 μL einer Suspension mit 18 mg/mL Yttrium Scintillation Proximity Beads (Amersham Pharmacia Biotech., Piscataway, NJ) hinzugefügt. Die Mikrotiterplatten werden mit einer Folie versiegelt und für 60 min bei Raumtemperatur stehen gelassen. Anschließend werden die Platten für 30 s pro Vertiefung in einem Microbeta-Szintillationzähler (Wallac Inc., Atlanta, GA) vermessen. IC5o-Werte werden anhand der graphischen Auftragung der Substanzkonzentration gegen die prozentuale Inhibition bestimmt.
Die in vitro Wirkung von Testsubstanzen an rekombinanter PDE3B, PDE4B, PDE7B, PDE8A,
PDE10A und PDEI lA wird nach dem oben für PDE 9A beschriebenen Testprotokoll mit folgenden Anpassungen bestimmt: Als Substrat wird [5',8-3H] adenosine 3',5'-cyclic phosphate (1
- 7 - μCi/μL; Amersham Pharmacia Biotech., Piscataway, NJ) verwendet. Die Zugabe einer Inhibitorlösung zum Stoppen der Reaktion ist nicht notwendig. Stattdessen wird im Anschluss an die Inkubation von Substrat und PDE direkt mit der Zugabe der Yttrium Scintillation Proximity Beads wie oben beschrieben fortgefahren und dadurch die Reaktion gestoppt. Für die Bestimmung einer entsprechenden Wirkung an PDEl, PDE2A und PDE5 wird das Protokoll zusätzlich wie folgt angepaßt: Bei PDEl werden zusätzlich Calmodulin 10'7 M und CaCl2 3 mM zum Reaktionsansatz gegeben. PDE2A wird im Test durch Zugabe von cGMP 1 μM stimuliert und mit einer BSA-Konzentration von 0.01% getestet. Für PDEl und PDE2A wird als Substrat [5',8-3H] adenosine 3',5'-cycIic phosphate (1 μCi/μL; Amersham Pharmacia Biotech., Piscataway, NJ), für PDE5 [8-3H] guanosine 3',5'-cyclic phosphate (1 μCi/μL; Amersham Pharmacia Biotech., Piscataway, NJ) eingesetzt.
Gefäßrelaxierende Wirkung in vitro
Kaninchen werden durch Nackenschlag betäubt und entblutet. Die Arteria Saphena wird entnommen, von anhaftendem Gewebe befreit, in 3 mm breite Ringe geteilt und einzeln unter einer Vorspannung in 5 ml-Organbäder mit 37°C warmer, carbogen-begaster Krebs-Henseleit-Lösung folgender Zusammensetzung (mM) gebracht: NaCl: 119; KCl: 4,8; CaCl2 x 2 H2O: 1; MgSO4 x 7 H2O: 1,4; KH2PO4: 1,2; NaHCO3: 25; Glucose: 10. Die Kontraktionskraft wird mit Statham UC2-Zellen erfasst, verstärkt und über A/D-Wandler (DAS-1802 HC, Keithley Instruments München) digitalisiert sowie parallel auf Linienschreiber registriert. Zur Erzeugung einer Kontraktion wird Phenylephrin dem Bad kumulativ in ansteigender Konzentration zugesetzt. Nach mehreren Kontrollzyklen wird die zu untersuchende Substanz in jedem weiteren Durchgang in jeweils steigender Dosierung untersucht und die Höhe der Kontraktion mit der Höhe der im letzten Vordurchgang erreichten Kontraktion verglichen. Daraus wird die Konzentration errechnet, die erforderlich ist, um die Höhe des Kontrollwertes um 50 % zu reduzieren (IC50). Das Standardapplikationsvolumen beträgt 5 μl, der DMSO-Anteil in der Badlösung entspricht 0, 1 %.
Die Ergebnisse sind in Tabelle 1 gezeigt:
Tabelle 1 : Gefäßrelaxierende Wirkung in vitro

Männliche Wistar Ratten mit einem Gewicht von 300-350 g (Harlan Winkelmann, Borchen, Deutschland) wurden mit 100 mg kg'1 i.p. Thiopental "Nycomed®" (Nycomed, München, Deutschland) anästhesiert. Eine Tracheotomie wurde durchgeführt und Katheter wurden zur Messung von Blutdruck und Herzfrequenz (Gould Druck Transducer und Rekorder, Modell RS 3400) in die Femoralarterie und zur Substanzapplikation in die Femoralvene eingeführt. Die Tiere wurden mit Raumluft beatmet und ihre Körpertemperatur kontrolliert. Testsubstanzen wurden oral oder intravenös appliziert.
Kaninchen-Modell
Adulte, männliche Chinchilla-Kaninchen mit einem Gewicht von 3 - 5 kg werden nach Lieferung mehrere Tage in Einzelhaltung adaptiert. Sie haben freien Zugang zu Wasser und können zwei Stunden pro Tag Futter zu sich nehmen. Die Tiere werden in einem 10/14 Stunden Tag-Nacht Rhythmus gehalten (Licht an, ab 8.00 Uhr), die Raumtemperatur beträgt 22 - 24°C.
Pro Behandlungsgruppe werden drei bis sechs Tiere verwendet und direkt vor Versuchsbeginn gewogen. Für die i.V. Gabe werden die Substanzen in Transcutol (GATTEFOSSE GmbH) gelöst und im Verhältnis 3/7 mit einer 20%igen Cremophorlösung (Cremophor (BASF), Wasser) verdünnt. Es wird ein Volumen von 0,5 ml/kg in die Ohrvene injiziert. Wasserlösliche Substanzen werden in 0,9 % Kochsalzlösung injiziert.
Für die orale Gabe werden die Testsubstanzen in einer Mischung von Glycerin: Wasser: PoIy- ethylenglycol 6:10:9.69 gelöst und in einem Volumen von 1 ml/kg mit der Schlundsonde appliziert.
Unter Ruhebedingungen ist der Kaninchenpenis in der Schamregion nicht sichtbar und von der Penishaut vollständig bedeckt. Die Erektion wird gewertet, indem man die Länge des hervortretenden Penis mit einer Schiebelehre misst. Die Messung wird 5, 10, 15, 30, 45, 60 und 120 Minuten nach Substanzgabe durchgeführt, nach oraler Gabe zusätzlich auch noch nach 3, 4, 5 und 6 Stunden. Die Tiere werden dazu jedes Mal aus dem Käfig geholt, am Nackenfell und den Hinterläufen festgehalten, auf den Rücken gedreht und gemessen. Entsprechende Lösungsmittelkontrollen werden durchgeführt, (vergleiche Literatur: E. Bischoff, K. Schneider, Int. J. of Impotence Res. 2001, 13, 230-235; E. Bischoff, U. Niewoehner, H. Haning, M. Es Sayed, T. Schenke, K. H. Schlemmer, The Journal of Urology, 2001, 165, 1316-1318; E. Bischoff, Int. J. Impotence Res. 2001, 13, 146-148).
Diurese in anästhesierten Ratten
Männliche Wistar Ratten mit einem Gewicht von 300-350 g (Harlan Winkelmann, Deutschland) wurden mit 1-2,5% Isofluoran in einem Gemisch von Lachgas / O2 (2:1) anästhesiert. Zur Messung von Blutdruck und Herzfrequenz wurde ein Katheter in die Femoralarterie eingeführt, die Substanzapplikation erfolgte über einen Femoralvenenkatheter und die Urinsammlung über einen Blasenkatheter. Nach der OP wurden 5ml/kg phys. NaCl als Bolus zum Flüssigkeitsausgleich intravenös gegeben und die Tiere für Ih mit phys. NaCl kontinuierlich mit einer Geschwindigkeit von lOOμl/kg/min über den Venekatheter infundiert. Die Körpertemperatur der Tiere wurde über eine Heizplatte konstant gehalten. Nach der ersten Stunde wurden die Testsubstanzen zusammen mit ANP kontinuierlich mit einer Geschwindigkeit von lOOμl/kg/min über den Venenkatheter infundiert. Urin wurde alle 15 min gesammelt und das Volumen, der cGMP-Gehalt (RIA), sowie die Na+ und K+ Konzentration (Flammenphotometrie) gemessen.
Bestimmung pharmakokinetischer Kenngrößen nach intravenöser und oraler Gabe
Die zu untersuchende Substanz wird Tieren (z.B. Maus, Ratte, Hund) intravenös als Lösung appliziert, die orale Applikation erfolgt als Lösung oder Suspension über eine Schlundsonde.
Nach Substanzgabe wird den Tieren zu festgelegten Zeitpunkten Blut entnommen, dieses wird heparinisiert und anschließend wird daraus durch Zentrifugation Plasma gewonnen. Die Substanz wird im Plasma über LC/MSMS analytisch quantifiziert. Aus den so ermittelten Plasmakon- zentrations-Zeit-Verläufen werden die pharmakokinetischen Kenngrößen mittels eines validierten pharmakokinetischen Rechenprogramms berechnet.
Inhibition von Cvtochrom P450-Enzvmen
Das Potential der Inhibition von P-450 Isoenzymen, die für den Metabolismus wichtig sind, wird automatisiert im 96-well Format untersucht. Hierbei werden zwei verschiedene Assays verwendet.
Bei dem auf Bildung von fluoreszierenden Metaboliten basierenden Assay werden rekombinante Enzyme (z.B. CYP 1A2, 2C8, 2C9, 2Cl 9, 2D6 oder 3 A4) und im allgemeinen Fluorescein- oder Coumarin-Teilstrukturen enthaltene Substrate eingesetzt. Es werden jeweils eine Substratkonzentration und 8 Konzentrationen des potentiellen Inhibitors verwendet. Nach Inkubation mit dem jeweiligen rekombinanten CYP Enzym wird mittels Fluoreszenzreader das Ausmaß an fluoreszierenden Metaboliten im Vergleich zur Kontrolle (ohne Inhibitor) ermittelt und ein IC50- Wert berechnet [Anal. Biochem. 248, 188 (1997)].
Beim 2. Assay werden als Enzymquelle humane Lebermikrosomen und als CYP Isoform-selektive Substrate Phenacetin (CYP1A2), Diclofenac (CYP2C9), Dextromethorphan (CYP2D6) und
Midazolam (CYP3A4) verwendet. Die Bildung des jeweiligen Metaboliten wird mittels LC- MS/MS gemessen. Unter Annahme kompetitiver Inhibition werden aus der Verminderung der Metabolitenbildung im Vergleich zur Kontrolle K1- Werte berechnet (1 Substrat-, 3 Inhibitorkonzentrationen).
Induktion von Cytochrom P450-Enzymen in humanen Leberzellkulturen
Zur Untersuchung des Nebenwirkungspotentials der erfindungsgemäßen Substanzen bezüglich einer Induktion von Cytochrom P450-Enzymen werden primäre humane Hepatozyten mit einer Zelldichte von 2,5 x 10^ Zellen zwischen zwei Schichten von Collagen in 24 well-Mikrotiter- platten bei 370C bei 5 % CO2 8 Tage kultiviert. Das Zellkulturmedium wird täglich gewechselt.
Nach 48 Stunden in Kultur werden die Hepatozyten über 5 Tage in Doppelbestimmung mit unterschiedlichen Konzentrationen der Testsubstanzen im Vergleich mit den Induktoren Rifampicin (RIF; 50 μM), Omeprazol (OME; 100 μM) und Phenobarbital (PB; 2 mM) behandelt. Die Endkonzentrationen der Testsubstanzen liegen bei 0,01 - 10 μg/ml.
Von den Zellkulturen wird der induktive Effekt der Testsubstanzen auf die Cytochrom (CYP) P450-Enzyme 1A2, 2B6, 2C19 und 3A4 durch Zugabe der Substrate 7-Ethoxyresorufin (CYP1A2), [14C]-S-Mephenytoin (CYP2B6 und 2Cl 9) und [14C]-Testosteron (CYP3A4) am Tag 8 bestimmt. Von den so gemessenen Enzymaktivitäten CYP 1A2, 2B6, 2Cl 9 und 3 A4 behandelter Zellen im Vergleich zu unbehandelten Zellen wird das induktive Potential der Testsubstanzen ermittelt.
Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, enthaltend mindestens eine erfindungsgemäße Verbindung und mindestens einen oder mehrere weitere Wirkstoffe, insbesondere zur Behandlung und/oder Prophylaxe der zuvor genannten Erkrankungen.
Die erfϊndungsgemäßen Verbindungen können systemisch und/oder lokal wirken. Zu diesem Zweck können sie auf geeignete Weise appliziert werden, wie z.B. oral, parenteral, pulmonal, nasal, sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctivae otisch oder als Implantat bzw. Stent.
Für diese Applikationswege können die erfindungsgemäßen Verbindungen in geeigneten Applikationsformen verabreicht werden.
Für die orale Applikation eignen sich nach dem Stand der Technik funktionierende schnell und/oder modifiziert die erfindungsgemäßen Verbindungen abgebende Applikationsformen, die die erfindungsgemäßen Verbindungen in kristalliner und/ oder amorphisierter und/oder gelöster
Form enthalten, wie z.B. Tabletten (nichtüberzogene oder überzogene Tabletten, beispielsweise
mit magensaftresistenten oder sich verzögert auflösenden oder unlöslichen Überzügen, die die Freisetzung der erfindungsgemäßen Verbindung kontrollieren), in der Mundhöhle schnell zerfallende Tabletten oder Filme/Oblaten, Filme/Lyophylisate, Kapseln (beispielsweise Hart- oder Weichgelatinekapseln), Dragees, Granulate, Pellets, Pulver, Emulsionen, Suspensionen, Aerosole oder Lösungen.
Die parenterale Applikation kann unter Umgehung eines Resorptionsschrittes geschehen (z.B. intravenös, intraarteriell, intrakardial, intraspinal oder intralumbal) oder unter Einschaltung einer Resorption (z.B. intramuskulär, subcutan, intracutan, percutan oder intraperitoneal). Für die parenterale Applikation eignen sich als Applikationsformen u.a. Injektions- und Infusionszu- bereitungen in Form von Lösungen, Suspensionen, Emulsionen, Lyophilisaten oder sterilen Pulvern.
Für die sonstigen Applikationswege eignen sich z.B. Inhalationsarzneiformen (u.a. Pulverinhalatoren, Nebulizer), Nasentropfen, -lösungen, -sprays; lingual, sublingual oder buccal zu applizierende Tabletten, Filme/Oblaten oder Kapsem, Suppositorien, Ohren- oder Augen- präparationen, Vaginalkapseln, wässrige Suspensionen (Lotionen, Schüttelmixturen), lipophile Suspensionen, Salben, Cremes, transdermale therapeutische Systeme (wie beispielsweise Pflaster), Milch, Pasten, Schäume, Streupuder, Implantate oder Stents.
Die erfϊndungsgemäßen Verbindungen können in die angeführten Applikationsformen überführt werden. Dies kann in an sich bekannter Weise durch Mischen mit inerten, nichttoxischen, pharmazeutisch geeigneten Hilfsstoffen geschehen. Zu diesen Hilfsstoffen zählen u.a. Trägerstoffe (beispielsweise mikrokristalline Cellulose, Laktose, Mannitol), Lösungsmittel (z.B. flüssige Polyethylenglycole), Emulgatoren und Dispergier- oder Netzmittel (beispielsweise Natrium- dodecylsulfat, Polyoxysorbitanoleat), Bindemittel (beispielsweise Polyvinylpyrrolidon), synthetische und natürliche Polymere (beispielsweise Albumin), Stabilisatoren (z.B. Antioxidantien wie beispielsweise Ascorbinsäure), Farbstoffe (z.B. anorganische Pigmente wie beispielsweise Eisenoxide) und Geschmacks- und / oder Geruchskorrigentien.
Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, die mindestens eine erfindungsgemäße Verbindung, üblicherweise zusammen mit einem oder mehreren inerten, nichttoxischen, pharmazeutisch geeigneten Hilfsstoffen enthalten, sowie deren Verwendung zu den zuvor genannten Zwecken.
Im allgemeinen hat es sich als vorteilhaft erwiesen, bei parenteraler Applikation pro Tag Mengen von etwa 0.001 bis 10 mg/kg Körpergewicht zur Erzielung wirksamer Ergebnisse zu verabreichen. Bei oraler Applikation beträgt die Menge pro Tag etwa 0.005 bis 3 mg/kg Körpergewicht.
Trotzdem kann es gegebenenfalls erforderlich sein, von den genannten Mengen abzuweichen, und zwar in Abhängigkeit von Körpergewicht, Applikationsweg, individuellem Verhalten gegenüber dem Wirkstoff, Art der Zubereitung und Zeitpunkt bzw. Intervall, zu welchem die Applikation erfolgt. So kann es in einigen Fällen ausreichend sein, mit weniger als der vorgenannten Mindest- menge auszukommen, während in anderen Fällen die genannte obere Grenze überschritten werden muss. Im Falle der Applikation größerer Mengen kann es empfehlenswert sein, diese in mehreren Einzelgaben über den Tag zu verteilen.
Die Prozentangaben in den folgenden Tests und Beispielen sind, sofern nicht anders angegeben, Gewichtsprozente; Teile sind Gewichtsteile, Lösungsmittelverhältnisse, Verdünnungsverhältnisse und Konzentrationsangaben von flüssig/flüssig-Lösungen beziehen sich jeweils auf das Volumen.
Die erfindungsgemäßen Verbindungen können folgendermaßen in pharmazeutische Zubereitungen überfuhrt werden:
- - Tablette:
Zusammenfassung:
100 mg der erfindungsgemäßen Verbindung, 50 mg Lactose (Monohydrat) 50 mg Maisstärke (nativ), 10 mg Polyvinylpyrolidon (PVP 25) (Fa. BASF, Ludwigshafen, Deutschland) und 2 mg Magnesiumstearat.
Tablettengewicht 212 mg, Durchmesser 8 mm, Wölbungsradius 12 mm.
Herstellung:
Die Mischung aus erfindungsgemäßer Verbindung, Lactose und Stärke wird mit einer 5 %igen Lösung (m/m) des PVPs in Wasser granuliert. Das Granulat wird nach dem Trocknen mit dem Magnesiumstearat 5 Minuten gemischt. Diese Mischung wird mit einer üblichen Tablettenpresse verpresst (Format der Tablette siehe oben). Als Richtwert für die Verpressung wird eine Presskraft von 15 kN verwendet.
Oral applizierbare Suspension:
Zusammensetzung:
1000 mg der erfindungsgemäßen Verbindung, 1000 mg Ethanol (96 %), 400 mg Rhodigel® (Xanthan gum der Firma FMC, Pennsylvania, USA) und 99 g Wasser.
Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 10 ml orale Suspension.
Herstellung:
Das Rhodigel wird in Ethanol suspendiert, die erfϊndungsgemäße Verbindung wird der Suspension zugeführt. Unter Rühren erfolgt die Zugabe des Wassers. Bis zum Abschluß der Quellung des Rhodigels wird ca. 6 h gerührt.
Oral applizierbare Lösung:
Zusammenfassung:
500 mg der erfindungsgemäßen Verbindung, 2,5 g Polysorbat und 97 g Polyethylenglycol 400. Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 20 g orale Lösung.
n Λ
- 94 -
Herstellung:
Die erfindungsgemäße Verbindung wird in der Mischung aus Polyethylenglycol und Polysorbat unter Rühren suspendiert. Der Rührvorgang wird bis zur vollständigen Auflösung der erfindungsgemäßen Verbindung fortgesetzt.
i.v.Lösung:
Die erfindungsgemäße Verbindung wird in einer Konzentration unterhalb der Sättigungslöslichkeit in einem physiologisch verträglichen Lösungsmittel (z.B. isotonische Kochsalzlösung, Glucoselösung 5 % und/oder PEG 400 Lösung 30 %. Die Lösung wird steril filtriert und sterile und pyrogenfreie Injektionsbehältnisse aufgefüllt.
Intravenös applizierbare Lösung:
Zusammensetzung:
1 mg der erfindungsgemäßen Verbindung, 15 g Polyethylenglykol 400 und 250 g Wasser für Injektionszwecke.
Herstellung:
Die erfindungsgemäße Verbindung wird zusammen mit Polyethylenglykol 400 in dem Wasser unter Rühren gelöst. Die Lösung wird sterilfilriert (Porendurchmesser 0,22 μm) und unter raseptischen Bedingungen in hitzesterilisierte Infusionsflaschen abgefüllt. Diese werden mit Infusionsstopfen und Bordelkappen verschlossen.
Claims
1. Verwendung von Verbindungen der Formel

in welcher
A CpCg-Alkyl, C3-Cg-Cycloalkyl, Tetrahydrofuryl oder Tetrahydropyranyl, welche gegebenenfalls mit bis zu 3 Resten unabhängig voneinander ausgewählt aus der Gruppe CpCö-AIkyl, Ci-Cβ-Alkoxy, Hydroxycarbonyl, Cyano, Trifluormethyl, Trifluormethoxy, Amino, Hydroxy, CrC6-Alkyl- amino, Halogen, Ci-Cβ-Alkylaminocarbonyl, Ci-Cβ-Alkoxycarbonyl, Cp Ce-Alkylcarbonyl, CrC6-Alkylsulfonyl und CrC6-Alkylthio substituiert sind,
wobei Ci-Cβ-Alkyl, Ci-Cβ-Alkoxy, Ci-C6-Alkylamino, Ci-Cβ-Alkylaminocarbonyl, Ci-Cβ-Alkoxycarbonyl, Q-Ce-Alkylcarbonyl, CpCδ-Alkylsulfonyl und Q- Cβ-Alkylthio gegebenenfalls mit einem oder mehreren Resten ausgewählt aus der Gruppe Hydroxy, Cyano, Halogen, Hydroxycarbonyl und einer
Gruppe der Formel -NR3R4,
wobei
R3 und R4 unabhängig voneinander Wasserstoff oder Ci-Cβ-Alkyl,
oder
R3 und R4 zusammen mit dem Stickstoffatom, an das sie gebunden sind, 5- bis 8-gliedriges Heterocyclyl bedeuten,
substituiert sind,
B Phenyl oder Heteroaryl, die gegebenenfalls mit bis zu 3 Resten jeweils unabhängig voneinander ausgewählt aus der Gruppe Ci-C6-Alkyl, Ci-C6-Alkoxy, Hydroxy- carbonyl, Cyano, Trifluormethyl, Trifluormethoxy, Amino, Nitro, Hydroxy, CrC6- - -
Alkylamino, Halogen, Ci-Cβ-Alkylaminocarbonyl, Ci-Cβ-Alkoxycarbonyl, Q-C6- Alkylcarbonyl, Q-C6-Alkylsulfonyl und Q-C6-Alkylthio substituiert sind,
wobei Ci-C6-Alkyl, Ci-C6-Alkoxy, Ci-C6-Alkylamino, Q-C6-Alkylamino- carbonyl, Ci-C6-Alkoxycarbonyl, Q-Cό-Alkylcarbonyl, Q-C6-AIlCyI- sulfonyl und-Q-C6-Alkylthio gegebenenfalls mit einem Rest ausgewählt aus der Gruppe Hydroxy, Cyano, Halogen, Hydroxycarbonyl und einer Gruppe der Formel -NR3R4,
wobei
R3 und R4 die oben angegebenen Bedeutungen aufweisen,
substituiert sind,
bedeuten, sowie deren Salze, Solvate und/oder Solvate der Salze, zur Herstellung von Arzneimitteln zur Behandlung von Herz-Kreislauf-Erkrankungen.
2. Verwendung von Verbindungen der Formel

in welcher
D Phenyl, Heteroaryl oder eine Gruppe der Formel 
wobei Phenyl und Heteroaryl gegebenenfalls mit bis zu 2 Resten unabhängig voneinander ausgewählt aus der Gruppe Heteroaryl, Halogen, Q- C6-Alkyl, Q-C6-Alkoxy, Trifluormethyl, Trifluormethoxy, Benzyloxy und Benzyl substituiert sind,
wobei Q-C6-Alkyl gegebenenfalls mit einer Gruppe der Formel -NR5R , in welcher R5 Q-C6-Alkyl und R6 Wasserstoff oder Q-C6-Alkoxy(Q-C6)- alkyl bedeuten, und - -
Heteroaryl gegebenenfalls mit CrC6-Alkoxy substituiert ist,
R1 C3-C8-Cycloalkyl, CrC6-Alkyl, C]-C6-Alkoxy(Ci-C6)alkyl, Benzyl oder eine
Gruppe der Formel 
wobei C3-Cg-Cycloalkyl gegebenenfalls mit Hydroxy, Ci-Cβ-Alkyl oder Trifluormethyl,
Ci-Cβ-Alkyl gegebenenfalls mit Heteroaryl, C3-Cg-Cycloalkyl oder Hydroxy,
und Benzyl gegebenenfalls mit Ci-Cβ-Alkoxy oder Halogen substituiert ist,
R2 Wasserstoff,
oder
R1 und R2 zusammen mit dem Stickstoffatom, an dem sie gebunden sind, ein 5- bis 6- gliedriges Heterocyclyl bilden, welches gegebenenfalls mit bis zu 2 Sub- stituenten unabhängig voneinander ausgewählt aus der Gruppe CI-CO-
Alkyl, Hydroxy, Cyano, Oxo, Heteroaryl, Benzyl, Formyl, Ci-Cβ-Alkyl- carbonyl und einer der folgenden Gruppen

wobei Ci-Cβ-Alkyl gegebenenfalls mit Hydroxy oder Heteroaryl substituiert ist,
bedeuten, sowie deren Salze, Solvate und/oder Solvate der Salze, zur Herstellung von
Arzneimitteln zur Behandlung von Herz-Kreislauf-Erkrankungen.
3. Verwendung von Verbindungen der allgemeinen Formel (I-II) gemäß Ansprüchen 1 und 2 zur Herstellung von Arzneimitteln zur Behandlung von Bluthochdruck. - -
4. Verwendung von Verbindungen der allgemeinen Formel (IS) gemäß Ansprüchen 1 und 2 zur Herstellung von Arzneimitteln zur Behandlung von stabiler und instabiler Angina Pectoris.
5. Verwendung von Verbindungen der allgemeinen Formel (IS) gemäß Ansprüchen 1 und 2 zur Herstellung von Arzneimitteln zur Behandlung von Niereninsuffizienz.
6. Verwendung von Verbindungen der allgemeinen Formel (LS) gemäß Ansprüchen 1 und 2 zur Herstellung von Arzneimitteln zur Behandlung von akutem Nierenversagen.
7. Verwendung von Verbindungen der allgemeinen Formel (IS) gemäß Ansprüchen 1 und 2 zur Herstellung von Arzneimitteln zur Behandlung von ischämischen Nierenerkrankungen.
8. Verwendung von Verbindungen der allgemeinen Formel (IS) gemäß Ansprüchen 1 und 2 zur Herstellung von Arzneimitteln zur Behandlung von Ischämien.
9. Verwendung von Verbindungen der allgemeinen Formel (IS) gemäß Ansprüchen 1 und 2 zur Herstellung von Arzneimitteln zur Behandlung von Herzinsuffizienz und akutem Herzversagen.
10. Verwendung von Verbindungen der allgemeinen Formel (I-II) gemäß Ansprüchen 1 und 2 zur Herstellung von Arzneimitteln zur Behandlung von venösen Erkrankungen.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP06742947A EP1890702A1 (de) | 2005-05-27 | 2006-05-17 | Verwendung von cyanopyrimidinen für die behandlung von herz-kreislauf-erkrankungen |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102005024494.7 | 2005-05-27 | ||
| DE102005024494A DE102005024494A1 (de) | 2005-05-27 | 2005-05-27 | Verwendung von Cyanopyrimidinen |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006125554A1 true WO2006125554A1 (de) | 2006-11-30 |
Family
ID=36888992
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2006/004639 WO2006125554A1 (de) | 2005-05-27 | 2006-05-17 | Verwendung von cyanopyrimidinen für die behandlung von herz-kreislauf-erkrankungen |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP1890702A1 (de) |
| DE (1) | DE102005024494A1 (de) |
| WO (1) | WO2006125554A1 (de) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007080382A1 (en) * | 2006-01-11 | 2007-07-19 | Astrazeneca Ab | Morpholino pyrimidine derivatives and their use in therapy |
| WO2008023180A1 (en) * | 2006-08-24 | 2008-02-28 | Astrazeneca Ab | Morpholino pyrimidine derivatives useful in the treatment of proliferative disorders |
| WO2008023159A1 (en) * | 2006-08-24 | 2008-02-28 | Astrazeneca Ab | Morpholino pyrimidine derivatives useful in the treatment of proliferative disorders |
| US8138183B2 (en) | 2007-07-09 | 2012-03-20 | Astrazeneca Ab | Morpholino pyrimidine derivatives used in diseases linked to mTOR kinase and/or PI3K |
| US8252802B2 (en) | 2010-06-11 | 2012-08-28 | Astrazeneca Ab | Chemical compounds |
| WO2015185499A1 (en) * | 2014-06-06 | 2015-12-10 | H. Lundbeck A/S | Pde9 inhibitors with 1-benzyl-2,5,6,8-tetrahydro-3-oxo-2,7-naphthyridine-4-carbonitrile backbone |
| CN109020837A (zh) * | 2018-07-27 | 2018-12-18 | 广东省石油与精细化工研究院 | 一种2-取代苯基-乙脒盐酸盐的制备方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0130735A1 (de) * | 1983-06-30 | 1985-01-09 | American Home Products Corporation | Aminopyrimidin-Derivate |
| WO2003037899A1 (en) * | 2001-11-02 | 2003-05-08 | Pfizer Limited | Pde9 inhibitors for treating cardiovascular disorders |
| WO2003041725A2 (de) * | 2001-11-15 | 2003-05-22 | Bayer Healthcare Ag | REGULATION DER cGMP-SPEZIFISCHEN PHOSPHODIESTERASE 9A |
| WO2004053495A1 (en) * | 2002-12-09 | 2004-06-24 | Bayer Healthcare Ag | Diagnostics and therapeutics for diseases associated with phosphodiesterase 9a1 (pde9a1) |
| WO2004113306A1 (de) * | 2003-06-25 | 2004-12-29 | Bayer Healthcare Ag | 6-arylamino-5-cyano-4-pyrimidinone als pde9a-inhibitoren |
-
2005
- 2005-05-27 DE DE102005024494A patent/DE102005024494A1/de not_active Withdrawn
-
2006
- 2006-05-17 WO PCT/EP2006/004639 patent/WO2006125554A1/de not_active Application Discontinuation
- 2006-05-17 EP EP06742947A patent/EP1890702A1/de not_active Withdrawn
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0130735A1 (de) * | 1983-06-30 | 1985-01-09 | American Home Products Corporation | Aminopyrimidin-Derivate |
| WO2003037899A1 (en) * | 2001-11-02 | 2003-05-08 | Pfizer Limited | Pde9 inhibitors for treating cardiovascular disorders |
| WO2003041725A2 (de) * | 2001-11-15 | 2003-05-22 | Bayer Healthcare Ag | REGULATION DER cGMP-SPEZIFISCHEN PHOSPHODIESTERASE 9A |
| WO2004053495A1 (en) * | 2002-12-09 | 2004-06-24 | Bayer Healthcare Ag | Diagnostics and therapeutics for diseases associated with phosphodiesterase 9a1 (pde9a1) |
| WO2004113306A1 (de) * | 2003-06-25 | 2004-12-29 | Bayer Healthcare Ag | 6-arylamino-5-cyano-4-pyrimidinone als pde9a-inhibitoren |
Non-Patent Citations (1)
| Title |
|---|
| BAGLI ET AL: "Chemistry and positive inotropic effect of pelrinone and related derivatives. A novel class of 2-methylpyrimidones as inotropic agents", JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY. WASHINGTON, US, vol. 31, no. 4, 1988, pages 814 - 823, XP002300134, ISSN: 0022-2623 * |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007080382A1 (en) * | 2006-01-11 | 2007-07-19 | Astrazeneca Ab | Morpholino pyrimidine derivatives and their use in therapy |
| WO2008023180A1 (en) * | 2006-08-24 | 2008-02-28 | Astrazeneca Ab | Morpholino pyrimidine derivatives useful in the treatment of proliferative disorders |
| WO2008023159A1 (en) * | 2006-08-24 | 2008-02-28 | Astrazeneca Ab | Morpholino pyrimidine derivatives useful in the treatment of proliferative disorders |
| US7750003B2 (en) | 2006-08-24 | 2010-07-06 | Astrazeneca Ab | Compounds-943 |
| US8138183B2 (en) | 2007-07-09 | 2012-03-20 | Astrazeneca Ab | Morpholino pyrimidine derivatives used in diseases linked to mTOR kinase and/or PI3K |
| US8252802B2 (en) | 2010-06-11 | 2012-08-28 | Astrazeneca Ab | Chemical compounds |
| US8552004B2 (en) | 2010-06-11 | 2013-10-08 | Astrazeneca Ab | Chemical compounds |
| US8999997B2 (en) | 2010-06-11 | 2015-04-07 | Astrazeneca Ab | Chemical compounds |
| US9155742B2 (en) | 2010-06-11 | 2015-10-13 | Astrazeneca Ab | Chemical compounds |
| US9421213B2 (en) | 2010-06-11 | 2016-08-23 | Astrazeneca Ab | Chemical compounds |
| WO2015185499A1 (en) * | 2014-06-06 | 2015-12-10 | H. Lundbeck A/S | Pde9 inhibitors with 1-benzyl-2,5,6,8-tetrahydro-3-oxo-2,7-naphthyridine-4-carbonitrile backbone |
| CN109020837A (zh) * | 2018-07-27 | 2018-12-18 | 广东省石油与精细化工研究院 | 一种2-取代苯基-乙脒盐酸盐的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| DE102005024494A1 (de) | 2006-11-30 |
| EP1890702A1 (de) | 2008-02-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1534713B1 (de) | Alkyl-substituierte pyrazolopyrimidine | |
| JP5457476B2 (ja) | 知覚力、集中力、学習力および/または記憶力の改善のために使用される6−アミノ−5−シアノ−ピリミジン−4−オン化合物 | |
| EP1902037B1 (de) | 2,4-diamino-pyrimidine als aurora inhibitoren | |
| EP2417112B1 (de) | Sulfonamid- und sulfoximin-substituierte diaryldihydropyrimidinone und ihre verwendung | |
| MX2010011591A (es) | Pirimidin-5-carboxamidas sustituidas 281. | |
| WO2004018474A1 (de) | Phenyl-substituierte pyrazolopyrimidine | |
| EP1626971B1 (de) | 6-cyclylmethyl- und 6-alkylmethyl-substituierte pyrazolopyrimidine | |
| WO2006125554A1 (de) | Verwendung von cyanopyrimidinen für die behandlung von herz-kreislauf-erkrankungen | |
| EP0531883A1 (de) | Kondensierte 5-gliedrige Heterocyclen, Verfahren zu ihrer Herstellung und diese Verbindungen enthaltende Arzneimittel | |
| EP1628980B1 (de) | 6-arylmethyl-substituierte pyrazolopyrimidine | |
| DE102006031314A1 (de) | Verwendung von 1,4-Diaryl-dihydropyrimidin-2-on-Derivaten zur Behandlung der pulmonalen arteriellen Hyptertonie | |
| EP1644339B1 (de) | 6-arylamino-5-cyano-4-pyrimidinone als pde9a-inhibitoren | |
| WO2004005291A1 (de) | Heterocyclisch substituierte imidazotriazine | |
| DE102005024493A1 (de) | Verwendung von Pyrazolopyrimidinen | |
| EP2298773B1 (de) | 6-Cyclylmethyl- und 6-Alkylmethyl-substituierte Pyrazolopyrimidine | |
| MXPA06007982A (en) | 6-amino-5-cyano-pyrimidine-4-ones used for improving perception, power of concentration, learning efficiency, and/or memory power |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006742947 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: DE |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: RU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006742947 Country of ref document: EP |