WO2006037574A1 - Improved synthesis scheme for lacosamide - Google Patents
Improved synthesis scheme for lacosamide Download PDFInfo
- Publication number
- WO2006037574A1 WO2006037574A1 PCT/EP2005/010603 EP2005010603W WO2006037574A1 WO 2006037574 A1 WO2006037574 A1 WO 2006037574A1 EP 2005010603 W EP2005010603 W EP 2005010603W WO 2006037574 A1 WO2006037574 A1 WO 2006037574A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- anyone
- methylation
- lacosamide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/02—Preparation of carboxylic acid amides from carboxylic acids or from esters, anhydrides, or halides thereof by reaction with ammonia or amines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C273/00—Preparation of urea or its derivatives, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups
- C07C273/18—Preparation of urea or its derivatives, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups of substituted ureas
- C07C273/1809—Preparation of urea or its derivatives, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups of substituted ureas with formation of the N-C(O)-N moiety
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C273/00—Preparation of urea or its derivatives, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups
- C07C273/18—Preparation of urea or its derivatives, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups of substituted ureas
- C07C273/1854—Preparation of urea or its derivatives, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups of substituted ureas by reactions not involving the formation of the N-C(O)-N- moiety
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C275/00—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups
- C07C275/04—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to acyclic carbon atoms
- C07C275/06—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to acyclic carbon atoms of an acyclic and saturated carbon skeleton
- C07C275/10—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to acyclic carbon atoms of an acyclic and saturated carbon skeleton being further substituted by singly-bound oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C275/00—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups
- C07C275/04—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to acyclic carbon atoms
- C07C275/06—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to acyclic carbon atoms of an acyclic and saturated carbon skeleton
- C07C275/16—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to acyclic carbon atoms of an acyclic and saturated carbon skeleton being further substituted by carboxyl groups
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
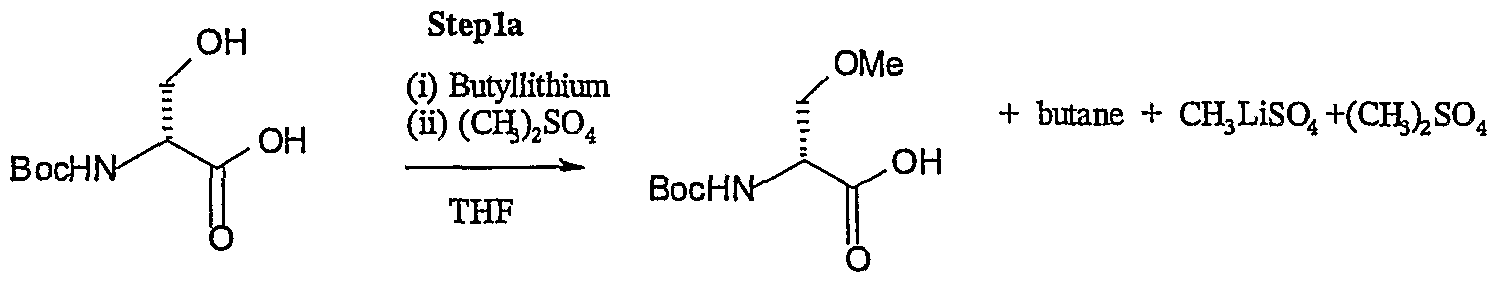
- the present invention provides an improved lacosamide synthesis route, wherein the O-methylation method is selective for the alcoholic hydroxy group of the N-protected D-serine. Accordingly, compared to the unspecific methylation suggested in scheme 1 of US 6,048,899, which also leads to the esterification of the carboxylic group, the present invention results in a shortened, more effective synthesis, wherein the subsequent step of hydrolysing the methyl ester group of an intermediate is avoided.
- the present invention relates to an improved method of producing (R)-2-acetamido-N-benzyl-3-methoxypropionamide comprising the O-methylation of a compound of formula I
- Rx is an N-protecting group
- one-step reaction means that when transforming a compound of formula I to a compound of formula Il no significant amount (i.e. an amount of 5 Mol% or more) of ester of the carboxyl group is formed that needs to be hydrolysed in a separate step. Usually, even less than 1 Mol% of ester is formed which is then removed during the further processing to lacosamide as described further below without the need for any additional hydrolysis step.
- the inventive O-methylation can be achieved by adding to a compound of formula I 1 such as e.g. to N-Boc-D-serine, a methylation agent in the presence of an organo metal compound, preferably an organo lithium compound. Suitable methylation agents are e.g.
- the organo lithium compound is preferably an alkyl lithium compound, such as butyllithium, methyllithium or hexyllithium or an aryllithium compound, such as phenyl lithium. More preferably the organo lithium compound is t-butyllithium or n-butyllithium and particularly preferred n-butyllithium.
- organo metal compounds comprising a metal-carbon binding
- organo zinc compounds including organo zinc halides
- organo aluminium compounds including organo aluminium halides
- organo tin compounds including organo tin halides or/and organo magnesium compounds including organo magnesium halides
- halides include Cl, Br or/and I.
- the organo moiety may be aryl or alkyl.
- Grignard compounds Alkyl-Mg-Y, or Aryl-Mg-Y, wherein Y is Cl, Br or I.
- THF/2-methoxyethyl ether mixtures, diethoxymethane or, preferably, THF may be used as solvent.
- the reaction is usually allowed to proceed for at least 5 hours at 0-10 0 C, and preferably for 7-24 hours at 0-10 0 C, most preferably for 9-18 hours at 0-5 0 C. Also, the reaction may be performed at higher or lower temperatures such as any temperature between -10 and +25°C if the reaction time is adapted accordingly.
- N-protecting group Rx of the compound of formula I is N-Boc a typical reaction can be illustrated by the following scheme (step 1-A)
- the yield of the methylation according to the method of the present invention using an organo metal compound may be at least 85%, preferably at least 90%.
- the amount of ester impurity after methylation using an organo metal or preferably an organo lithium compound, in particular after step 1a is significantly below 1 Mol%, preferably below 0,1 Mol% and is regularly below the limit of detection.
- PTC phase transfer catalysis
- phase transfer catalyst e.g. a quarternary ammonium, phosphonium or sulfonium salt, such as e.g. a tetraalkylammonium halide, is used as phase transfer catalyst.
- Suitable catalysts and PTC reagents can be purchased from many vendors, e.g. from Sigma-Aldrich or Hawks Chemical.
- one embodiment of the present invention relates to a method of producing lacosamide, characterized in that a compound of formula I is O- methylated to a compound of formula Il by performing the reaction as a phase transfer catalysis.
- this method comprises the addition of a methylation reagent, such as dimethylsulfate, methyl iodide or trimethyl phosphate to a phase transfer reaction system comprising the compound of formula I, an aqueous phase, an organic phase and a phase transfer catalyst.
- a methylation reagent such as dimethylsulfate, methyl iodide or trimethyl phosphate
- the methylation agent is selected from dimethylsulfate, methyl iodide or trimethyl phosphate, wherein dimethylsulfate is particularly preferred;
- the first (aqueous) phase is an alkaline aqueous solution, such as aqueous sodium hydroxide, aqueous lithium hydroxide, aqueous potassium hydroxide, aqueous sodium carbonate or aqueous potassium carbonate, wherein aqueous sodium hydroxide is particularly preferred;
- the second (organic) phase is selected from toluene, hexane, methylene chloride or methyl f-butyl ether, with toluene being particularly preferred and
- phase transfer catalyst is an ammonium or phosphonium salt of formula IV 1 a sulfonium salt of formula V or a pyridinium salt of formula Vl
- R, R', R" and R'" are independently selectable alkyl, aryl or aralkyl groups, Q is a nitrogen or phosphorus and X is a halide, acetate, p-toluenesulfonate, trifluoromethanesulfonate, hexafluoroantimonate, hydroxide, perchlorate, hydrogensulfate, thiocyanate or tetrafluoroborate.
- phase transfer catalysts examples include tetraethylammonium p- toluenesulfonate, tetrapropylammonium trifluoromethanesulfonate, tetraphenylphosphonium hexafluoroantimonate, cetylpyridinium bromide, triphenylmethyl triphenylphosponium chloride, benzyltriethylammonium chloride, benzyltrimethylammonium chloride, benzyltriphenylphosphonium chloride, benzytributylammonium chloride, butyltriethylammonium bromide, butyltriphenylphosphonium bromide, cetyltri methyl ammonium bromide, cetyltri methyl ammonium chloride, ethyltriphenylphosphonium bromide, ethyltriphenylphosphonium iodide, methyltrioctylammoni
- aqueous alkali is provided as a 5 to 50% w/w solution and in an amount of 1.1 to 10 molar equivalents with respect to the compound of formula I
- the amount of organic solvent with respect to the compound of formula I is preferably between 3-20 volumes, in particular 3-20 I/kg compound of formula I
- the amount of phase transfer catalyst is between 0.01 to 0.1 molar equivalents of the compound of formula I
- aryl refers to an aromatic group, substituted with one ore more substituents or unsubstituted, which contains from 6 up to 18 ring carbon atoms and up to a total of 25 carbon atoms and includes polynuclear aromatics.
- aryl groups may be monocyclic, bicyclic, tricyclic or polycyclic and may be fused rings.
- a polynuclear aromatic compound as used herein, is meant to encompass bicyclic fused aromatic ring systems containing from 10-18 ring carbon atoms and up to a total of 25 carbon atoms.
- one to six carbon atoms may be replaced by a heteroatom, such as oxygen, sulfur or/and nitrogen.
- Aryl comprises unsubstituted phenyl; unsubstituted naphtyl; phenyl or napthyl substituted with one or more substituents selected from e.g. hydroxy, carboxy, halogen, nitro, C1-6 alkyl, C1-6 alkoxy, amino; substituted or unsubstituted heteroaryls such as pyrroyl; thienyl, indolyl, etc.
- "aryl” is preferably chosen from unsubstituted phenyl or substituted phenyl, e.g. 2,6- difluorophenyl, p-nitrophenyl or p-toluyl. Unsubstituted phenyl is particularly preferred.
- alkyl comprises branched or linear saturated hydrocarbon chains.
- alkyl is a branched or linear hydrocarbon with up to 20 carbon atoms, more preferably with up to 6 carbon atoms; most preferably with up to 4 carbon atoms.
- the hydrocarbon may be substituted with one ore more substituents or unsubstituted.
- Preferred examples of “alkyl” are cetyl, octyl, heptyl, pentyl, butyl, propyl, ethyl and methyl.
- aralkyl means a group aryl-alkyl wherein “aryl” and “alkyl” are as defined above.
- aralkyl is benzyl.
- substitution refers to substitution of a H atom by e.g. hydroxy, carboxy, halogen, nitro, C1-6 alkyl, C1-6 alkoxy, amino.
- the PTC reaction is usually allowed to proceed at 0-10 0 C for at least 30 minutes, e.g. for 0.5 to 24 hours, preferably for at least 45 minutes and even more preferred for at least 1 hour.
- the yield of the PTC of the present invention may thus be at least 85%, preferably at least 90%, even more preferably at least 91 %, 92%, 93%, 94%, 95% or 96%.
- N-Boc-D-serine can also be produced by reacting D- serine with di-t-butyl dicarbonate to N-Boc-D-serine in a phase transfer catalysis reaction using essentially the conditions (e.g. choice and concentration/amount of alkali, solvent, PCT-catalysts, temperature, reaction time etc) as described above, except that di-t-butyl dicarbonate is used as the reagent instead of a methylation agent.
- N-protecting group of the compound of formula I is Boc (t- Butoxycarbonyl)
- the amount of ester impurity after methylation by PTC, in particular after step 1-B is well below 1 Mol%, preferably below 0,1 Mol%, and usually below the limit of detection.
- the process of the current invention may further comprise the step of processing the compound of formula Il to a compound of formula III (Step 2)
- the benzyl amide formation can be performed by adding to a compound of formula Il an amount of benzylamine in the presence of
- a base such as triethylamine, diisopropylethylamine, 1 ,8-diazabicyclo [5.4.0]undec-7-ene, potassium bicarbonate or a morpholine derivative, preferably 4-methylmorpholine and
- an activator of the carboxyl group such as a carbodiimide or an alkyl chloroformate, preferably isobutyl chloroformate.
- the yield of the benzylamid formation under the conditions of e.g. Example 3 was typically between 90% and 99%.
- the yield of the benzylamid formation of the present invention may be in the range of at least 85% up to 99,9%, preferably in the range of at least 90% up to 99% product.
- Suitable protecting groups in the method according to the present invention are e.g. t-butoxycarbonyl (Boc) or carbobenzoxy (Cbz), with the Boc group being particularly preferred.
- the protecting group Rx can be cleaved off to obtain (R)-2-amino-N-benzyl-3-methoxypropionamide by appropriate measures known from the art.
- the protecting group Rx is a carbobenzoxy group it may be cleaved off with H2, Pd/C as described in US 6,048,899.
- the protecting group is a Boc group this group may be conveniently removed with an acid, such as hydrochloric acid, e.g. at room temperature (step 3).
- step 3 typically yielded product in an amount ranging from 95% to 100%.
- the amine formation step in the method of the present invention may yield at least 85%, preferably at least 90%, more preferably at least 95% product.
- (R)-2-amino-N-benzyl-3-methoxypropionamide can then be transformed to lacosamide by N-acetylation using acetic anhydride (step 4)
- step 4 typically yielded between 81% and 95%.
- the yield of the acetyiation step in the method of the present invention may thus be in the range of at least 70% up to 99%, preferably in the range of at least 80% up to 95%.
- One embodiment of the present invention is thus the production of lacosamide comprising a step of N-acetylation of (R)-2-amino-N-benzyl-3- methoxypropionamide with acetic anhydride in the absence of a base, in particular in the absence of pyridine.
- the advantage of a base-free reaction is that toxic bases, such as pyridine, can be excluded.
- lacosamide can be isolated from the reaction mixture of step 4 with improved purity by crystallisation in appropriate solvents, such as ethyl acetate.
- lacosamide was obtained under the specific conditions of Examples 1 to 4 in a yield of typically 63%-70% (using butyllithium in step 1- A) or 66% to 75% (using PTC in step 1-B).
- lacosamide may be obtained by the method of the present invention in a total yield ranging from at least 50% up to 90%, preferably from at least 60% up to 80%. If an organo metal compound is employed the total yield of lacosamide may be more preferably in the range of at least 60% up to 70%, most preferably in the range of at least 63% up to 70%. If a PTC is employed, the total yield of lacosamide may be more preferably in the range of at least 60% up to 75%, most preferably at least 66% up to 75%.
- a compound of formula Il from a compound of formula I is comprised by the method of lacosamide synthesis as described above.
- Subject of the present invention is thus a method for production of a compound of formula (II) from a compound of formula (I) by O-methylation as described above essentially in the abscence of methyl ester formation or significant racemisation.
- D-serine derivatives or L-serine derivatives or mixtures of D- and L-serine derivatives in any ratio may be used in the method of the present invention.
- the method of the present invention for production of a compound of formula (II) or/and of lacosamide leads to an improved yield and an improved enantiomeric purity of the product.
- the invention also relates to important intermediates of the current process.
- the most important intermediate (R)-2-N-Boc-amino-3-methoxypropanoic acid (C-936) results from the improved O-methylation step according to the present invention (see figure 1).
- the compound can be easily isolated from the reaction mixture as the free acid or by forming a salt, such as e.g. a cyclohexylammonium salt.
- a salt such as e.g. a cyclohexylammonium salt.
- Suitable C-936 salts are also regarded to be part of the invention.
- Another aspect of the invention relates to the use of C-936 or C-937 or any salt thereof as an adduct or intermediate in a method of producing (R)-2- acetamido-N-benzyl-3-methoxypropionamide (lacosamide). Yet another aspect of the invention pertains to a method of producing a pharmaceutical formulation by the subsequent steps of
- Another aspect of the present invention is a method of producing a compound of formula VIII comprising the O-methylation of a compound of formula VII,
- R 4 is H, an N-protecting group, or/and a group having 0-30 C atoms
- R 1 , R 2 , and R 3 are independently selected from H and groups having 0-30 C atoms, characterized in that the O-methylation in being carried out in a one-step reaction and wherein the compound of formula VIII is obtained in the same configuration as the compound VII, of at least 88%, preferably of at least 90 %, more preferably of at least 95%, 96%, 97%, 98% or 99% enantiomeric purity.
- R 1 , R 2 , and R 3 are independently hydrogen, -OH, -SH, -NH 2 , -NO 2 , -CN, -COOH, -SOH, -SO 2 H, -SO 3 H, halogen, -OR 10 , -SR 10 , -NR 10 R 11 , -SOR 10 , -SO 2 R 10 , -SO 3 R 10 , substituted or unsubstituted alkyl as definied above, substituted or unsubstituted C 2 -C 6 -alkenyl, substituted or unsubstituted C 2 -C 6 -alkynyl, -(CO)-R 10 , -(CO)-O-R 10 , -0-(CO)-R 10 , substituted or unsubstituted aryl as defined above, substituted or unsubstituted C 3 -C 13 - hetaryl having 1-3 heteroatoms independently selected
- R 1 is H
- R 2 is H or/and R 3 is H. It is most preferred that R 1 is H, R 2 is H and R 3 is H.
- R 4 is selected from R 1 and N-protecting groups. More preferably, R 4 is the N-protecting group Rx as described above.
- R 1 being H
- R 2 being H
- R 3 being H
- R 4 being Rx as described above.
- R 4 , R 1 , R 2 , R 3 , the groups R 10 and R 11 are independently hydrogen, substituted or unsubstituted alkyl as defined above, substituted or unsubstituted C 2 -C 6 -alkenyl, substituted or unsubstituted C 2 -C 6 -alkynyl, substituted or unsubstituted aryl as defined above, substituted or unsubstituted C 3 -Ci 3 -hetaryl having 1-3 heteroatoms independently selected from N, S, O, substituted or unsubstituted aralkyl as defined above, substituted or unsubstituted C 7 -Ci 5 -alkaryl, substituted or unsubstituted C 4 - Cu-hetaralkyl having 1-3 heteroatoms independently selected from N, S, O; substituted or unsubstituted C 4 -Ci 4 -alkhetaryl having 1-3 heteroatoms independently selected from N
- substitution in the groups R 4 , R 1 , R 2 , R 3 , R 10 and R 11 refers to substitution by one or more substituents as defined above, e.g. by hydroxy, carboxy, halogen, nitro, C1-C6 alkyl, C1-C6 alkoxy, amino, etc.
- the same configuration of the compound VIII with reference to compound VII means that essentially no racemisation takes place, or compound VIII is obtained in the same configuration as compound VII with an enantiomeric purity as defined above. If compound VII is in the R configuration, compound VIII is also in the R configuration. If compound VII is in the S configuration, compound VIII is also in the S configuration.
- compound VII is in the R configuration.
- the parameter of enantiomeric purity can be applied mutatis mutandis to enantiomer mixtures.
- compound VII is a mixture of the R and S configuration
- compound VIII is essentially the same mixture of the R and S configuration, i.e. the ratio of the R and S configuration remains essentially unaltered, or an enantiomeric ratio as follows is obtained.
- the obtained enantiomeric ratio of compound VIII may be at least 88%, preferably at least 90 %, more preferably in at least 95, 96, 97, 98 or 99% of the enantiomeric ratio of compound VII.
- Th ⁇ reaction schema of compound VII to compound VlII is a generalization of the O-methylation of the present invention of the compound of formula I to produce a compound of formula Il as described above. If the compound VII is in the R configuration, R 1 is H, R 2 is H, R 3 is H and R 4 is Rx, the compound VIII corresponds to the compound of formula Il and may be used for the production of lacosamide, e.g. by the reaction steps as described above. Starting from the compound Il or VII, lacosamide may be produced by any suitable method to introduce the N-benzylamide group and the N-acetyl group. Therefore, in a particular preferred embodiment, the compound VII is in the R configuration and R 4 is Rx. It is most preferred that R 4 is Rx, R 1 is H, R 2 is H, and R 3 is H and the compound VII is in the R configuration.
- the inventive O-methylation of compound VII can be achieved by adding to a compound of formula VII a methylation agent in the presence of an organo metal compound, in particular an organo lithium compound, as defined above. Suitable methylation agents are defined above.
- the selective O-methylation of the alcoholic group may be performed by phase transfer catalysis as defined above.
- Specific embodiments of the O-methylation of the compound of formula VII correspond to the specific embodiments of the production method of lacosamide comprising the O-methylation of the compound of formula I as described above, in particular specific embodiments relating to the phase transfer catalysis, phase transfer catalysts, in particular as defined in formula IV, V, and Vl, the phase transfer reaction system and its components, the organo metal compound, reaction conditions during phase transfer catalysis or in the presence of the organo metal compound, further reaction steps and reaction conditions leading to lacosamide including N-benzylamide formation, N-deprotection and N-acetylation, etc.
- the yield of the methylation of compound VII by the method of the present invention may be at least 85%, preferably at least 90% when using an organo metal compound.
- the yield of methylation of compound VII may be at least 85%, preferably at least 90%, even more preferably at least 91%, 92%, 93%, 94%, 95 % or 96%.
- Example 1 Production of (R)-2-N-Boc-amino-3-methoxypropanoic acid (C-936) using butyllithium (step 1a)
- Example 2 Production of (R)-2-N-Boc-amino-3-methoxypropanoic acid (C-936) using PTC (step 1b)
- a suspension of N-Boc-D-serine (22g, 0.107 mol) and tetrabutylammonium bromide (1.3g, 0.004mol) in toluene (110ml) was cooled to ⁇ 10 ° C.
- reaction mixture is warmed to room temperature over 30 minutes and aged for a further 30 minutes.
- the mixture is then washed with water (44ml),
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Life Sciences & Earth Sciences (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Pharmacology & Pharmacy (AREA)
- Neurosurgery (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pain & Pain Management (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description



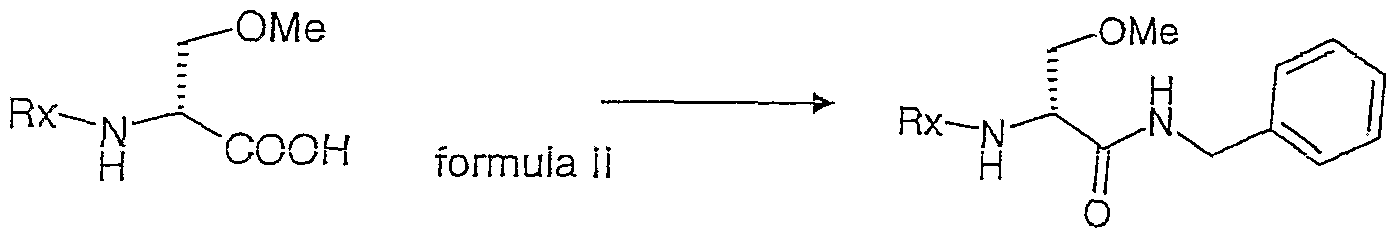




Claims






Priority Applications (20)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EA200700746A EA012588B1 (en) | 2004-10-02 | 2005-09-30 | Improved synthesis scheme for lacosamide |
| SI200530992T SI1799635T1 (en) | 2004-10-02 | 2005-09-30 | Improved synthesis scheme for lacosamide |
| KR1020077002247A KR101246169B1 (en) | 2004-10-02 | 2005-09-30 | Improved synthesis scheme for lacosamide |
| EP05794396A EP1799635B1 (en) | 2004-10-02 | 2005-09-30 | Improved synthesis scheme for lacosamide |
| CA2570598A CA2570598C (en) | 2004-10-02 | 2005-09-30 | Improved synthesis scheme for lacosamide |
| DK05794396.1T DK1799635T3 (en) | 2004-10-02 | 2005-09-30 | Improved lacosamide synthesis scheme |
| CN2005800241755A CN1989102B (en) | 2004-10-02 | 2005-09-30 | Improved synthetic method of lacosamide |
| HR20100239T HRP20100239T1 (en) | 2004-10-02 | 2005-09-30 | Improved synthesis scheme for lacosamide |
| AU2005291456A AU2005291456B2 (en) | 2004-10-02 | 2005-09-30 | Improved synthesis scheme for lacosamide |
| JP2007533955A JP5128281B2 (en) | 2004-10-02 | 2005-09-30 | Improved synthetic scheme for lacosamide |
| AT05794396T ATE457975T1 (en) | 2004-10-02 | 2005-09-30 | IMPROVED SYNTHESIS SCHEME FOR LACOSAMIDE |
| US11/664,316 US7884134B2 (en) | 2004-10-02 | 2005-09-30 | Synthesis scheme for lacosamide |
| NZ552076A NZ552076A (en) | 2004-10-02 | 2005-09-30 | Improved synthesis scheme for lacosamide |
| DE602005019437T DE602005019437D1 (en) | 2004-10-02 | 2005-09-30 | |
| BRPI0516735A BRPI0516735B8 (en) | 2004-10-02 | 2005-09-30 | methods for producing (r)-2-acetamido-n-benzyl-3-methoxypropionamide (lacosamide) and a pharmaceutical formulation, and use of (r)-n-benzyl-2-nboc-amino-3-methoxypropionamide (c -937) |
| MX2007001253A MX2007001253A (en) | 2004-10-02 | 2005-09-30 | Improved synthesis scheme for lacosamide. |
| PL05794396T PL1799635T3 (en) | 2004-10-02 | 2005-09-30 | Improved synthesis scheme for lacosamide |
| IL180479A IL180479A (en) | 2004-10-02 | 2007-01-01 | Synthesis scheme for lacosamide |
| NO20072250A NO336765B1 (en) | 2004-10-02 | 2007-04-30 | Process for the preparation of (R) -2-acetamido-N-benzyl-3-methoxypropionamide (lacosamide). |
| US12/961,705 US8809585B2 (en) | 2004-10-02 | 2010-12-07 | Synthesis scheme for lacosamide |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP04023556.6 | 2004-10-02 | ||
| EP04023556A EP1642889A1 (en) | 2004-10-02 | 2004-10-02 | Improved synthesis scheme for lacosamide |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/664,316 A-371-Of-International US7884134B2 (en) | 2004-10-02 | 2005-09-30 | Synthesis scheme for lacosamide |
| US12/961,705 Division US8809585B2 (en) | 2004-10-02 | 2010-12-07 | Synthesis scheme for lacosamide |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006037574A1 true WO2006037574A1 (en) | 2006-04-13 |
Family
ID=34926827
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2005/010603 Ceased WO2006037574A1 (en) | 2004-10-02 | 2005-09-30 | Improved synthesis scheme for lacosamide |
Country Status (24)
| Country | Link |
|---|---|
| US (2) | US7884134B2 (en) |
| EP (2) | EP1642889A1 (en) |
| JP (1) | JP5128281B2 (en) |
| KR (1) | KR101246169B1 (en) |
| CN (2) | CN1989102B (en) |
| AT (1) | ATE457975T1 (en) |
| AU (1) | AU2005291456B2 (en) |
| BR (1) | BRPI0516735B8 (en) |
| CA (1) | CA2570598C (en) |
| CY (1) | CY1109994T1 (en) |
| DE (1) | DE602005019437D1 (en) |
| DK (1) | DK1799635T3 (en) |
| EA (1) | EA012588B1 (en) |
| ES (1) | ES2341470T3 (en) |
| HR (1) | HRP20100239T1 (en) |
| IL (1) | IL180479A (en) |
| MX (1) | MX2007001253A (en) |
| NO (1) | NO336765B1 (en) |
| NZ (1) | NZ552076A (en) |
| PL (1) | PL1799635T3 (en) |
| SI (1) | SI1799635T1 (en) |
| UA (1) | UA95600C2 (en) |
| WO (1) | WO2006037574A1 (en) |
| ZA (1) | ZA200610000B (en) |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2067765A2 (en) | 2007-12-04 | 2009-06-10 | Ranbaxy Laboratories Limited | Intermediate compounds and their use in preparation of lacosamide |
| DE102008059155A1 (en) | 2008-11-27 | 2010-06-02 | Ratiopharm Gmbh | Dry processing and new forms of lacosamide |
| EP2297092A1 (en) | 2008-05-28 | 2011-03-23 | Pliva Hrvatska D.O.O. | Polymorphic and amorphous forms of lacosamide and amorphous compositions |
| WO2011039781A1 (en) * | 2009-09-25 | 2011-04-07 | Cadila Healthcare Limited | Processes for the preparation of lacosamide and intermediates thereof |
| WO2011061610A2 (en) | 2009-11-19 | 2011-05-26 | Ranbaxy Laboratories Limited | Processes for preparation of polymorphic forms of lacosamide |
| WO2011092559A1 (en) | 2010-01-29 | 2011-08-04 | Archimica Srl | Process for the synthesis of lacosamide |
| WO2011092672A2 (en) | 2010-01-29 | 2011-08-04 | Ranbaxy Laboratories Limited | Processes for reducing impurities in lacosamide |
| WO2011095995A1 (en) | 2010-02-08 | 2011-08-11 | Natco Pharma Limited | A process for the preparation of lacosamide |
| WO2011099033A1 (en) * | 2010-02-09 | 2011-08-18 | Msn Laboratories Limited | Process for preparing (r)-2-acetamido-n-benzyl-3-methoxy-propionamide |
| EP2399901A1 (en) | 2010-06-23 | 2011-12-28 | Archimica GmbH | Intermediate for producing lacosamide and a process for its preparation and conversion to lacosamide |
| WO2012041986A1 (en) | 2010-10-01 | 2012-04-05 | Ucb Pharma Gmbh | Process for the preparation of amino acid derivatives |
| EP2444390A1 (en) | 2010-10-19 | 2012-04-25 | Archimica GmbH | Process for producing Lacosamide |
| WO2012065891A1 (en) | 2010-11-17 | 2012-05-24 | Ucb Pharma Gmbh | Process for preparing lacosamide |
| WO2013030654A1 (en) | 2011-08-29 | 2013-03-07 | Signa S.A. De C.V. | Processes for the preparation of (r)-2-acetamido-n-benzyl-3-methoxypropionamide and intermediates thereof |
| US8440861B2 (en) | 2009-08-06 | 2013-05-14 | Medichem, S.A. | Solid forms of an N-(phenylmethyl)propanamide derivative and processes of preparation |
| US8748660B2 (en) | 2012-07-09 | 2014-06-10 | Council Of Scientific & Industrial Research | Process for the synthesis of antiepileptic drug lacosamide |
| US8829226B2 (en) | 2010-02-06 | 2014-09-09 | Zhejiang Jiuzhou Pharmaceutical Co., Ltd | Lacosamide intermediate compound, preparation method thereof and use thereof |
| WO2014155264A1 (en) | 2013-03-25 | 2014-10-02 | Jubilant Life Sciences Limited | Process for the preparation of lacosamide using novel intermediates |
| WO2015068977A1 (en) | 2013-11-08 | 2015-05-14 | 에스티팜 주식회사 | Method for preparing lacosamide |
| US9095557B2 (en) | 2006-06-15 | 2015-08-04 | Ucb Pharma Gmbh | Anticonvulsant combination therapy |
| EP2990399A1 (en) | 2014-08-28 | 2016-03-02 | Rao, Davuluri Ramamohan | Improved process for the preparation of lacosamide and its novel intermediate |
| US9308183B2 (en) | 2006-06-30 | 2016-04-12 | Ucb Pharma Gmbh | Therapy for hyperexcitability disorders |
| EP3659997A1 (en) | 2015-11-13 | 2020-06-03 | API Corporation | Method for producing lacosamide and intermediate thereof |
Families Citing this family (49)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE60120104T2 (en) | 2001-03-20 | 2006-09-21 | Schwarz Pharma Ag | New use of peptide compounds in the treatment of non-neuropathic inflammatory pain |
| DK1243263T3 (en) * | 2001-03-21 | 2003-03-17 | Sanol Arznei Schwarz Gmbh | Hitherto unknown use of a class of peptide compounds to treat allodynia or other various types of chronic or phantom pain |
| JP4664924B2 (en) * | 2003-12-02 | 2011-04-06 | ウーツェーベー ファルマ ゲーエムベーハー | Novel use of peptide compounds for the treatment of central neuropathic pain |
| EP1579858A1 (en) * | 2004-03-26 | 2005-09-28 | Schwarz Pharma Ag | Novel use of peptide compounds for treating pain in painful diabetic neuropathy |
| US20100256179A1 (en) * | 2004-03-26 | 2010-10-07 | Ucb Pharma Gmbh | Combination therapy for pain in painful diabetic neuropathy |
| US20070042969A1 (en) * | 2004-03-26 | 2007-02-22 | Srz Properties, Inc. | Combination therapy for pain in painful diabetic neuropathy |
| AU2005232395B2 (en) * | 2004-04-16 | 2010-09-09 | Schwarz Pharma Ag | Use of peptidic compounds for the prophylaxis and treatment of chronic headache |
| EP1604655A1 (en) * | 2004-06-09 | 2005-12-14 | Schwarz Pharma Ag | Novel use of peptide compounds for treating pain in trigeminal neuralgia |
| EA014055B1 (en) * | 2004-08-27 | 2010-08-30 | Шварц Фарма Аг | Use of peptide compounds for treating bone cancer pain, chemotherapy-and nucleoside-induced pain |
| EP1642889A1 (en) * | 2004-10-02 | 2006-04-05 | Schwarz Pharma Ag | Improved synthesis scheme for lacosamide |
| US20060252749A1 (en) * | 2005-01-28 | 2006-11-09 | Srz Properties, Inc. | Lacosamide for add-on therapy of psychosis |
| US20070043120A1 (en) * | 2005-08-18 | 2007-02-22 | Bettina Beyreuther | Therapeutic combination for painful medical conditions |
| EP1754476A1 (en) * | 2005-08-18 | 2007-02-21 | Schwarz Pharma Ag | Lacosamide (SPM 927) for treating myalgia, e.g. fibromyalgia |
| ATE439193T1 (en) | 2005-11-24 | 2009-08-15 | S D Warren Company D B A | COATING SYSTEM WITH FLOWABLE COATING MATERIAL FOR SMOOTH OR STRUCTURED SURFACES |
| EP1873527A1 (en) * | 2006-06-30 | 2008-01-02 | Schwarz Pharma Ag | Method for identifying CRMP modulators |
| WO2009053070A1 (en) * | 2007-10-23 | 2009-04-30 | Schwarz Pharma Ag | Compounds for treating demyelination conditions |
| HRP20130456T1 (en) * | 2008-11-07 | 2013-06-30 | Ucb Pharma Gmbh | Novel process for the preparation of amino acid derivatives |
| CN101591300B (en) * | 2009-02-19 | 2011-05-04 | 成都伊诺达博医药科技有限公司 | Novel method for synthesizing lacosamide |
| WO2010107993A2 (en) | 2009-03-18 | 2010-09-23 | Pliva Hrvatska D.O.O. | Process for preparing (r)-7v-benzyl-2- (benyloxycarbonylamino)-s-methoxypropionamide |
| CN102020589B (en) * | 2009-09-19 | 2013-11-06 | 浙江九洲药业股份有限公司 | Tert-butyl carbamate derivative and preparation method and application thereof |
| CN102146048B (en) * | 2010-02-06 | 2013-11-13 | 浙江九洲药业股份有限公司 | Lacosamide intermediate compound and preparation method and application thereof |
| WO2011130615A2 (en) * | 2010-04-15 | 2011-10-20 | Dr. Reddy's Laboratories Ltd. | Preparation of lacosamide |
| US20130123537A1 (en) * | 2010-05-17 | 2013-05-16 | K a s s Narayan Garimella | Process for the preparation of lacosamide |
| EP2582833A1 (en) | 2010-06-15 | 2013-04-24 | Medichem, S.A. | Enzymatic resolution of racemic (2r,s)-2-(acetylamino)-3-methoxy-n-(phenylmethyl)propanamide |
| WO2012001710A1 (en) * | 2010-07-02 | 2012-01-05 | Sun Pharmaceutical Industries Ltd | An improved process for the preparation of lacosamide |
| US8957252B2 (en) | 2010-07-27 | 2015-02-17 | Indoco Remedies Limited | Process for preparation of lacosamide and some N-benzyl-propanamide intermediate derivatives |
| GB201020026D0 (en) | 2010-11-25 | 2011-01-12 | Cambrex Karlskoga Ab | New process |
| EP2468261A1 (en) | 2010-12-02 | 2012-06-27 | UCB Pharma GmbH | Formulation of lacosamide |
| CN102320993B (en) * | 2011-07-13 | 2013-11-27 | 石家庄四药有限公司 | Method for preparing (R)-2- Boc-amidogen-3-methoxypropionic acid |
| WO2013024383A1 (en) * | 2011-08-12 | 2013-02-21 | Alembic Pharmaceuticals Limited | An improved process for the preparation of lacosamide |
| WO2013072933A2 (en) * | 2011-10-03 | 2013-05-23 | Glenmark Generics Limited | Process for preparation of 2-acetamido-n-benzyl-3-methoxypropionamide |
| WO2013072936A2 (en) | 2011-11-10 | 2013-05-23 | Ramamohan Rao Davuluri | A novel process for the preparation of (r)-n-benzyl-2 acetamido-3-methoxypropionamide |
| CN103930397B (en) | 2011-11-15 | 2015-10-21 | 塞诺菲-安万特德国有限公司 | Process for the production of N-substituted 2-(acetylamino)-N'-benzyl-3-methoxypropionamides |
| CN102816083B (en) * | 2012-07-30 | 2015-09-23 | 永光制药有限公司 | The preparation method of scheme for lacosamide |
| GB201219627D0 (en) | 2012-11-01 | 2012-12-12 | Cambrex Karlskoga Ab | New process |
| CN103342655A (en) * | 2013-07-02 | 2013-10-09 | 扬州大学 | New method for synthesizing substituted amide by using substituted ethanedione dianiline Schiff base |
| WO2016039393A1 (en) * | 2014-09-10 | 2016-03-17 | 株式会社エーピーアイ コーポレーション | Production method for amino acid derivative |
| CN104761465B (en) * | 2015-03-18 | 2017-05-31 | 四川同晟生物医药有限公司 | A kind of preparation method of scheme for lacosamide |
| CN106699595B (en) * | 2015-07-21 | 2019-04-12 | 上海医药集团股份有限公司 | A kind of scheme for lacosamide preparation method |
| CN106699605B (en) * | 2015-07-21 | 2019-08-20 | 上海医药集团股份有限公司 | A kind of methylation method of scheme for lacosamide intermediate |
| IN2015CH05001A (en) | 2015-09-18 | 2015-10-16 | Divis Lab Ltd | |
| MA43532A (en) | 2015-12-30 | 2018-11-07 | Adamas Pharmaceuticals Inc | METHODS AND COMPOSITIONS FOR THE TREATMENT OF DISORDERS RELATED TO EPILEPTIC SEIZURES |
| US9718765B1 (en) | 2016-06-21 | 2017-08-01 | Sci Pharmtech, Inc. | Process for preparation of optically pure N-substituted-3-methoxypropionic acid derivatives |
| BR112018015215A2 (en) * | 2016-09-28 | 2018-12-11 | Unichem Laboratories Limited | process for the preparation of lacosamide compound of the formula and prepared lacosamide purification process |
| CN106811492B (en) * | 2017-01-18 | 2019-11-01 | 长兴制药股份有限公司 | A kind of preparation method of scheme for lacosamide |
| KR20190081386A (en) | 2017-12-29 | 2019-07-09 | 강원대학교산학협력단 | Composition for preventing the ischemia stroke disease containing rufinamide |
| KR20190081385A (en) | 2017-12-29 | 2019-07-09 | 강원대학교산학협력단 | Composition for preventing the ischemia stroke disease containing oxcarbazepine |
| CN110320291A (en) * | 2019-06-21 | 2019-10-11 | 山东省药学科学院 | A kind of method of HPLC standard measure detection lacosamide injection liquid hold-up |
| JP2022072636A (en) * | 2020-10-30 | 2022-05-17 | 住友化学株式会社 | Method for producing amide compound |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH1129544A (en) * | 1997-07-09 | 1999-02-02 | Tama Kagaku Kogyo Kk | Production of n-alkoxycarbonylated, n-alkenyloxycarbonylated or n-arylalkoxycarbonylated amino acids |
| US6048899A (en) * | 1997-03-17 | 2000-04-11 | Research Corporation Tech., Inc. | Anticonvulsant enantiomeric amino acid derivatives |
| WO2004014895A1 (en) * | 2002-08-05 | 2004-02-19 | Eli Lilly And Company | Piperazine substituted aryl benzodiazepines |
Family Cites Families (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5129456A (en) * | 1974-08-30 | 1976-03-12 | Tanabe Seiyaku Co | Kogakukatsusei oo benjiruserinjudotai no seiho |
| JPS62132849A (en) * | 1985-12-04 | 1987-06-16 | Toray Ind Inc | Production of d-or l-n-t-butoxycarbonyl-o-benzylserine |
| US5585358A (en) * | 1993-07-06 | 1996-12-17 | Yissum Research Development Corporation Of The Hebrew University Of Jerusalem | Derivatives of valproic acid amides and 2-valproenoic acid amides, method of making and use thereof as anticonvulsant agents |
| WO1997033861A1 (en) * | 1996-03-15 | 1997-09-18 | Research Corporation Technologies, Inc. | Anticonvulsant enantiomeric amino acid derivatives |
| IL154470A0 (en) * | 2000-08-17 | 2003-09-17 | Teva Pharma | Use of derivatives of valproic acid amides and 2-valproenic acid amides for the treatment or prevention of pain and/or headache disorders |
| DE60120104T2 (en) * | 2001-03-20 | 2006-09-21 | Schwarz Pharma Ag | New use of peptide compounds in the treatment of non-neuropathic inflammatory pain |
| DK1243263T3 (en) * | 2001-03-21 | 2003-03-17 | Sanol Arznei Schwarz Gmbh | Hitherto unknown use of a class of peptide compounds to treat allodynia or other various types of chronic or phantom pain |
| JP4664924B2 (en) * | 2003-12-02 | 2011-04-06 | ウーツェーベー ファルマ ゲーエムベーハー | Novel use of peptide compounds for the treatment of central neuropathic pain |
| US20060009384A1 (en) * | 2003-12-05 | 2006-01-12 | David Rudd | Novel use of peptide compounds for treating status epilepticus or related conditions |
| US20100256179A1 (en) * | 2004-03-26 | 2010-10-07 | Ucb Pharma Gmbh | Combination therapy for pain in painful diabetic neuropathy |
| US20070042969A1 (en) * | 2004-03-26 | 2007-02-22 | Srz Properties, Inc. | Combination therapy for pain in painful diabetic neuropathy |
| EP1579858A1 (en) * | 2004-03-26 | 2005-09-28 | Schwarz Pharma Ag | Novel use of peptide compounds for treating pain in painful diabetic neuropathy |
| AU2005232395B2 (en) * | 2004-04-16 | 2010-09-09 | Schwarz Pharma Ag | Use of peptidic compounds for the prophylaxis and treatment of chronic headache |
| EP1604654A1 (en) * | 2004-05-18 | 2005-12-14 | Schwarz Pharma Ag | Novel use of peptide compounds for treating dyskinesia |
| EP1604656A1 (en) * | 2004-06-09 | 2005-12-14 | Schwarz Pharma Ag | Novel use of peptide compounds for treating amyotrophic lateral sclerosis (ALS) |
| EP1604655A1 (en) * | 2004-06-09 | 2005-12-14 | Schwarz Pharma Ag | Novel use of peptide compounds for treating pain in trigeminal neuralgia |
| US7427601B2 (en) * | 2004-06-24 | 2008-09-23 | Schwarz Pharma Ag | Method for treating tremor |
| EA014055B1 (en) * | 2004-08-27 | 2010-08-30 | Шварц Фарма Аг | Use of peptide compounds for treating bone cancer pain, chemotherapy-and nucleoside-induced pain |
| EP1642889A1 (en) * | 2004-10-02 | 2006-04-05 | Schwarz Pharma Ag | Improved synthesis scheme for lacosamide |
| US20060252749A1 (en) * | 2005-01-28 | 2006-11-09 | Srz Properties, Inc. | Lacosamide for add-on therapy of psychosis |
| US20070048372A1 (en) * | 2005-08-18 | 2007-03-01 | Srz Properties, Inc. | Method for treating non-inflammatory osteoarthritic pain |
| US20070043120A1 (en) * | 2005-08-18 | 2007-02-22 | Bettina Beyreuther | Therapeutic combination for painful medical conditions |
| EP1754476A1 (en) * | 2005-08-18 | 2007-02-21 | Schwarz Pharma Ag | Lacosamide (SPM 927) for treating myalgia, e.g. fibromyalgia |
| EP1873527A1 (en) * | 2006-06-30 | 2008-01-02 | Schwarz Pharma Ag | Method for identifying CRMP modulators |
| CN101466390B (en) * | 2006-06-15 | 2014-03-12 | 优时比制药有限公司 | Peptide compounds for the treatment of refractory status epilepticus |
| EA019757B1 (en) * | 2006-06-15 | 2014-06-30 | ЮСиБи ФАРМА ГМБХ | Pharmaceutical composition with synergistic anticonvulsant effect |
| WO2009053070A1 (en) * | 2007-10-23 | 2009-04-30 | Schwarz Pharma Ag | Compounds for treating demyelination conditions |
| EP2468261A1 (en) * | 2010-12-02 | 2012-06-27 | UCB Pharma GmbH | Formulation of lacosamide |
| BR112013013525A2 (en) * | 2010-12-02 | 2016-10-18 | Ucb Pharma Gmbh | lacosamide solid controlled release formulation for oral administration, solid pharmaceutical composition, and method for the prevention, alleviation and / or treatment of a central nervous system disease |
-
2004
- 2004-10-02 EP EP04023556A patent/EP1642889A1/en not_active Withdrawn
-
2005
- 2005-09-30 CN CN2005800241755A patent/CN1989102B/en not_active Expired - Fee Related
- 2005-09-30 HR HR20100239T patent/HRP20100239T1/en unknown
- 2005-09-30 SI SI200530992T patent/SI1799635T1/en unknown
- 2005-09-30 JP JP2007533955A patent/JP5128281B2/en not_active Expired - Lifetime
- 2005-09-30 BR BRPI0516735A patent/BRPI0516735B8/en not_active IP Right Cessation
- 2005-09-30 DK DK05794396.1T patent/DK1799635T3/en active
- 2005-09-30 DE DE602005019437T patent/DE602005019437D1/de not_active Expired - Lifetime
- 2005-09-30 NZ NZ552076A patent/NZ552076A/en not_active IP Right Cessation
- 2005-09-30 UA UAA200704328A patent/UA95600C2/en unknown
- 2005-09-30 MX MX2007001253A patent/MX2007001253A/en active IP Right Grant
- 2005-09-30 US US11/664,316 patent/US7884134B2/en not_active Expired - Fee Related
- 2005-09-30 WO PCT/EP2005/010603 patent/WO2006037574A1/en not_active Ceased
- 2005-09-30 KR KR1020077002247A patent/KR101246169B1/en not_active Expired - Fee Related
- 2005-09-30 CN CN2010101835217A patent/CN101928230B/en not_active Expired - Fee Related
- 2005-09-30 EP EP05794396A patent/EP1799635B1/en not_active Expired - Lifetime
- 2005-09-30 CA CA2570598A patent/CA2570598C/en not_active Expired - Fee Related
- 2005-09-30 PL PL05794396T patent/PL1799635T3/en unknown
- 2005-09-30 AU AU2005291456A patent/AU2005291456B2/en not_active Ceased
- 2005-09-30 EA EA200700746A patent/EA012588B1/en not_active IP Right Cessation
- 2005-09-30 AT AT05794396T patent/ATE457975T1/en active
- 2005-09-30 ES ES05794396T patent/ES2341470T3/en not_active Expired - Lifetime
-
2006
- 2006-11-30 ZA ZA200610000A patent/ZA200610000B/en unknown
-
2007
- 2007-01-01 IL IL180479A patent/IL180479A/en active IP Right Grant
- 2007-04-30 NO NO20072250A patent/NO336765B1/en not_active IP Right Cessation
-
2010
- 2010-04-30 CY CY20101100383T patent/CY1109994T1/en unknown
- 2010-12-07 US US12/961,705 patent/US8809585B2/en active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6048899A (en) * | 1997-03-17 | 2000-04-11 | Research Corporation Tech., Inc. | Anticonvulsant enantiomeric amino acid derivatives |
| JPH1129544A (en) * | 1997-07-09 | 1999-02-02 | Tama Kagaku Kogyo Kk | Production of n-alkoxycarbonylated, n-alkenyloxycarbonylated or n-arylalkoxycarbonylated amino acids |
| WO2004014895A1 (en) * | 2002-08-05 | 2004-02-19 | Eli Lilly And Company | Piperazine substituted aryl benzodiazepines |
Non-Patent Citations (6)
| Title |
|---|
| ANDURKAR, SHRIDHAR V. ET AL: "Synthesis and anticonvulsant activities of (R)-(O)-methylserine derivatives", TETRAHEDRON: ASYMMETRY, 9(21), 3841-3854 CODEN: TASYE3; ISSN: 0957-4166, 1998, XP002317153 * |
| BARLOS, K. ET AL: "Convenient synthesis of N-trityl-O-alkyl-L-hydroxyamino acids and derivatives. Application to the synthesis of related peptides", TETRAHEDRON, 39(3), 475 -8 CODEN: TETRAB; ISSN: 0040-4020, 1983, XP008056137 * |
| CHEN, FRANCIS M. F. ET AL: "Simple preparations of N-(tert-butyloxycarbonyl)-O-methyl-L-serine and N-(tert-butyloxycarbonyl)-O-methyl-L-threonine by direct methylation", JOURNAL OF ORGANIC CHEMISTRY, 44(13), 2299-300 CODEN: JOCEAH; ISSN: 0022-3263, 1979, XP002317152 * |
| DATABASE CA [online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; NOBESHIMA, HIROFUMI: "Preparation of N-alkoxycarbonylated, N-alkenyloxycarbonylated, and N-arylalkoxycarbonylated amino acids or peptides", XP002317154, retrieved from STN Database accession no. 1999:78500 * |
| MEDERSKI W W K R ET AL: "Chlorothiophenecarboxamides as P1 surrogates of inhibitors of blood coagulation factor Xa", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, OXFORD, GB, vol. 14, no. 23, 6 December 2004 (2004-12-06), pages 5817 - 5822, XP004611126, ISSN: 0960-894X * |
| VARGA, JANOS R. ET AL: "Ring-formation by methylation of phenylserine derivatives", ACTA CHIMICA HUNGARICA , 120(4), 247 -9 CODEN: ACHUDC; ISSN: 0231-3146, 1985, XP008056138 * |
Cited By (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9095557B2 (en) | 2006-06-15 | 2015-08-04 | Ucb Pharma Gmbh | Anticonvulsant combination therapy |
| US9446011B2 (en) | 2006-06-15 | 2016-09-20 | Ucb Pharma Gmbh | Anticonvulsant combination therapy |
| US9308183B2 (en) | 2006-06-30 | 2016-04-12 | Ucb Pharma Gmbh | Therapy for hyperexcitability disorders |
| US8378142B2 (en) | 2007-12-04 | 2013-02-19 | Ranbaxy Laboratories Limited | Intermediate compounds and their use in preparation of lacosamide |
| EP2067765A3 (en) * | 2007-12-04 | 2010-09-08 | Ranbaxy Laboratories Limited | Intermediate compounds and their use in preparation of lacosamide |
| US8093426B2 (en) | 2007-12-04 | 2012-01-10 | Ranbaxy Laboratories Limited | Intermediate compounds and their use in preparation of lacosamide |
| EP2067765A2 (en) | 2007-12-04 | 2009-06-10 | Ranbaxy Laboratories Limited | Intermediate compounds and their use in preparation of lacosamide |
| EP2297092A1 (en) | 2008-05-28 | 2011-03-23 | Pliva Hrvatska D.O.O. | Polymorphic and amorphous forms of lacosamide and amorphous compositions |
| DE102008059155A1 (en) | 2008-11-27 | 2010-06-02 | Ratiopharm Gmbh | Dry processing and new forms of lacosamide |
| US8440861B2 (en) | 2009-08-06 | 2013-05-14 | Medichem, S.A. | Solid forms of an N-(phenylmethyl)propanamide derivative and processes of preparation |
| US8946477B2 (en) | 2009-08-06 | 2015-02-03 | Medichem, S.A. | Solid forms of an N-(phenylmethyl) propanamide derivative and processes of preparation |
| US8853439B2 (en) | 2009-09-25 | 2014-10-07 | Cadila Healthcare Limited | Processes for the preparation of lacosamide and intermediates thereof |
| WO2011039781A1 (en) * | 2009-09-25 | 2011-04-07 | Cadila Healthcare Limited | Processes for the preparation of lacosamide and intermediates thereof |
| US9242926B2 (en) | 2009-09-25 | 2016-01-26 | Cadila Healthcare Limited | Processes for the preparation of lacosamide and intermediates thereof |
| WO2011061610A2 (en) | 2009-11-19 | 2011-05-26 | Ranbaxy Laboratories Limited | Processes for preparation of polymorphic forms of lacosamide |
| US8853453B2 (en) | 2010-01-29 | 2014-10-07 | Ranbaxy Laboratories Limited | Processes for reducing impurities in lacosamide |
| WO2011092672A2 (en) | 2010-01-29 | 2011-08-04 | Ranbaxy Laboratories Limited | Processes for reducing impurities in lacosamide |
| WO2011092559A1 (en) | 2010-01-29 | 2011-08-04 | Archimica Srl | Process for the synthesis of lacosamide |
| US8796488B2 (en) | 2010-01-29 | 2014-08-05 | Euticals S.P.A. | Process for the preparation of lacosamide |
| US8829226B2 (en) | 2010-02-06 | 2014-09-09 | Zhejiang Jiuzhou Pharmaceutical Co., Ltd | Lacosamide intermediate compound, preparation method thereof and use thereof |
| WO2011095995A1 (en) | 2010-02-08 | 2011-08-11 | Natco Pharma Limited | A process for the preparation of lacosamide |
| US8759582B2 (en) | 2010-02-08 | 2014-06-24 | Natco Pharma Limited | Process for the preparation of lacosamide |
| EP2534127A4 (en) * | 2010-02-09 | 2013-11-27 | Msn Lab Ltd | PROCESS FOR THE PREPARATION OF (R) -2-ACETAMIDO-N-BENZYL-3-METHOXYPROPIONAMIDE |
| US8907132B2 (en) | 2010-02-09 | 2014-12-09 | Msn Laboratories Private Limited | Process for preparing (R)-2-acetamido-N-benzyl-3-methoxy-propionamide |
| WO2011099033A1 (en) * | 2010-02-09 | 2011-08-18 | Msn Laboratories Limited | Process for preparing (r)-2-acetamido-n-benzyl-3-methoxy-propionamide |
| EP2534127A1 (en) | 2010-02-09 | 2012-12-19 | MSN Laboratories Limited | Process for preparing (r)-2-acetamido-n-benzyl-3-methoxy-propionamide |
| EP2399901A1 (en) | 2010-06-23 | 2011-12-28 | Archimica GmbH | Intermediate for producing lacosamide and a process for its preparation and conversion to lacosamide |
| WO2012041986A1 (en) | 2010-10-01 | 2012-04-05 | Ucb Pharma Gmbh | Process for the preparation of amino acid derivatives |
| US8969620B2 (en) | 2010-10-01 | 2015-03-03 | Ucb Pharma Gmbh | Process for the preparation of amino acid derivatives |
| EP2444390A1 (en) | 2010-10-19 | 2012-04-25 | Archimica GmbH | Process for producing Lacosamide |
| US8598386B2 (en) | 2010-10-19 | 2013-12-03 | Euticals Gmbh | Process for producing lacosamide |
| EP2487152A1 (en) | 2010-11-17 | 2012-08-15 | UCB Pharma GmbH | Process for the preparation of Lacosamide including resolution of O-methyl-DL-serine |
| WO2012065891A1 (en) | 2010-11-17 | 2012-05-24 | Ucb Pharma Gmbh | Process for preparing lacosamide |
| WO2013030654A1 (en) | 2011-08-29 | 2013-03-07 | Signa S.A. De C.V. | Processes for the preparation of (r)-2-acetamido-n-benzyl-3-methoxypropionamide and intermediates thereof |
| US9133101B2 (en) | 2011-08-29 | 2015-09-15 | Signa S.A. De C.V. | Processes for the preparation of (R)-2-acetamido-N-benzyl-3-methoxypropionamide and intermediates thereof |
| US8748660B2 (en) | 2012-07-09 | 2014-06-10 | Council Of Scientific & Industrial Research | Process for the synthesis of antiepileptic drug lacosamide |
| WO2014155264A1 (en) | 2013-03-25 | 2014-10-02 | Jubilant Life Sciences Limited | Process for the preparation of lacosamide using novel intermediates |
| US9790170B2 (en) | 2013-11-08 | 2017-10-17 | St Pharm Co., Ltd. | Method for preparing lacosamide |
| WO2015068977A1 (en) | 2013-11-08 | 2015-05-14 | 에스티팜 주식회사 | Method for preparing lacosamide |
| EP2990399A1 (en) | 2014-08-28 | 2016-03-02 | Rao, Davuluri Ramamohan | Improved process for the preparation of lacosamide and its novel intermediate |
| EP3659997A1 (en) | 2015-11-13 | 2020-06-03 | API Corporation | Method for producing lacosamide and intermediate thereof |
| US10975117B2 (en) | 2015-11-13 | 2021-04-13 | Api Corporation | Method for producing lacosamide and intermediate thereof |
| US11623943B2 (en) | 2015-11-13 | 2023-04-11 | Api Corporation | Method for producing lacosamide and intermediate thereof |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1799635B1 (en) | Improved synthesis scheme for lacosamide | |
| US20130041180A1 (en) | Process for preparing (r)-2-acetamido-n-benzyl-3-methoxy-propionamide | |
| KR101036536B1 (en) | Synthesis of (S)-(+)-3- (aminomethyl) -5-methyl hexanoic acid | |
| WO2024092892A1 (en) | Edoxaban intermediate and preparation method therefor | |
| WO2010021093A1 (en) | Asymmetric organic catalyst | |
| US6646150B1 (en) | Processes for producing (aminomethyl)trifluorocarbinol derivatives | |
| JP2004526735A (en) | Method for producing benazepril | |
| US7122696B2 (en) | Processes for preparation of N-protected-β-amino alcohols and N-protected-β-amino epoxides | |
| EP2736875B1 (en) | Chemical process for opening ring compounds | |
| EP1344763A1 (en) | Process for producing optically active 3-halogenocarboxylic acid esters and 3-azide-carboxylic acid esters | |
| JP2007063267A (en) | Method for producing optically active diphenylalanine compound | |
| WO1994011336A1 (en) | Improved process for the synthesis of substituted 1,2,3-aminodiols |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KP KR KZ LC LK LR LS LT LU LV LY MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU LV MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 200610000 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3624/KOLNP/2006 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2570598 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12006502511 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 552076 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 180479 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200580024175.5 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020077002247 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2007/001253 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007533955 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005291456 Country of ref document: AU |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2005291456 Country of ref document: AU Date of ref document: 20050930 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005291456 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005794396 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: a200704328 Country of ref document: UA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200700746 Country of ref document: EA |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005794396 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11664316 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 11664316 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: PI0516735 Country of ref document: BR |