WO2006004658A2 - Furanopyrimidines - Google Patents
Furanopyrimidines Download PDFInfo
- Publication number
- WO2006004658A2 WO2006004658A2 PCT/US2005/022727 US2005022727W WO2006004658A2 WO 2006004658 A2 WO2006004658 A2 WO 2006004658A2 US 2005022727 W US2005022727 W US 2005022727W WO 2006004658 A2 WO2006004658 A2 WO 2006004658A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- unsubstituted
- phenyl
- pyrimidin
- furo
- Prior art date
Links
- JUQAECQBUNODQP-UHFFFAOYSA-N furo[3,2-d]pyrimidine Chemical class C1=NC=C2OC=CC2=N1 JUQAECQBUNODQP-UHFFFAOYSA-N 0.000 title claims abstract description 38
- 150000001875 compounds Chemical class 0.000 claims abstract description 288
- 238000000034 method Methods 0.000 claims abstract description 275
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 66
- 241000124008 Mammalia Species 0.000 claims abstract description 57
- 239000003814 drug Substances 0.000 claims abstract description 44
- 150000003839 salts Chemical class 0.000 claims abstract description 42
- 201000010099 disease Diseases 0.000 claims abstract description 37
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 29
- 229940002612 prodrug Drugs 0.000 claims abstract description 27
- 239000000651 prodrug Substances 0.000 claims abstract description 27
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 25
- 238000004519 manufacturing process Methods 0.000 claims abstract description 22
- 239000012453 solvate Substances 0.000 claims abstract description 21
- 201000011510 cancer Diseases 0.000 claims abstract description 17
- 201000004681 Psoriasis Diseases 0.000 claims abstract description 11
- 208000026935 allergic disease Diseases 0.000 claims abstract description 10
- 208000006673 asthma Diseases 0.000 claims abstract description 10
- 201000004624 Dermatitis Diseases 0.000 claims abstract description 9
- 206010063837 Reperfusion injury Diseases 0.000 claims abstract description 9
- 210000000056 organ Anatomy 0.000 claims abstract description 9
- 206010061218 Inflammation Diseases 0.000 claims abstract description 8
- 208000022559 Inflammatory bowel disease Diseases 0.000 claims abstract description 8
- 230000004054 inflammatory process Effects 0.000 claims abstract description 8
- 208000023328 Basedow disease Diseases 0.000 claims abstract description 7
- 206010009944 Colon cancer Diseases 0.000 claims abstract description 7
- 206010018364 Glomerulonephritis Diseases 0.000 claims abstract description 7
- 208000021386 Sjogren Syndrome Diseases 0.000 claims abstract description 7
- 208000006011 Stroke Diseases 0.000 claims abstract description 7
- 208000010668 atopic eczema Diseases 0.000 claims abstract description 7
- 208000037906 ischaemic injury Diseases 0.000 claims abstract description 7
- 230000000302 ischemic effect Effects 0.000 claims abstract description 7
- 206010025135 lupus erythematosus Diseases 0.000 claims abstract description 7
- 201000006417 multiple sclerosis Diseases 0.000 claims abstract description 7
- 208000010125 myocardial infarction Diseases 0.000 claims abstract description 7
- 208000026872 Addison Disease Diseases 0.000 claims abstract description 6
- 208000035285 Allergic Seasonal Rhinitis Diseases 0.000 claims abstract description 6
- 208000011231 Crohn disease Diseases 0.000 claims abstract description 6
- 208000003807 Graves Disease Diseases 0.000 claims abstract description 6
- 208000030836 Hashimoto thyroiditis Diseases 0.000 claims abstract description 6
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 claims abstract description 6
- 206010003246 arthritis Diseases 0.000 claims abstract description 6
- 208000025302 chronic primary adrenal insufficiency Diseases 0.000 claims abstract description 6
- 208000008732 thymoma Diseases 0.000 claims abstract description 6
- 208000005057 thyrotoxicosis Diseases 0.000 claims abstract description 6
- 230000007896 negative regulation of T cell activation Effects 0.000 claims abstract description 4
- 125000000217 alkyl group Chemical group 0.000 claims description 169
- -1 3-methoxy-4-(2-(l- piperidinyl)ethoxy)phenyl Chemical group 0.000 claims description 157
- 229910052739 hydrogen Inorganic materials 0.000 claims description 131
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 128
- OKKJLVBELUTLKV-UHFFFAOYSA-N methanol Substances OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 124
- 125000001072 heteroaryl group Chemical group 0.000 claims description 77
- 125000004404 heteroalkyl group Chemical group 0.000 claims description 64
- 229910052794 bromium Inorganic materials 0.000 claims description 58
- 125000000843 phenylene group Chemical group C1(=C(C=CC=C1)*)* 0.000 claims description 56
- 229910052801 chlorine Inorganic materials 0.000 claims description 53
- 229910052731 fluorine Inorganic materials 0.000 claims description 53
- 238000011282 treatment Methods 0.000 claims description 52
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 50
- 125000005549 heteroarylene group Chemical group 0.000 claims description 48
- 125000003118 aryl group Chemical group 0.000 claims description 47
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 47
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 44
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 42
- 229910052760 oxygen Inorganic materials 0.000 claims description 42
- 125000005842 heteroatom Chemical group 0.000 claims description 35
- 125000004527 pyrimidin-4-yl group Chemical group N1=CN=C(C=C1)* 0.000 claims description 35
- 229920006395 saturated elastomer Polymers 0.000 claims description 35
- 125000003545 alkoxy group Chemical group 0.000 claims description 33
- 239000003795 chemical substances by application Substances 0.000 claims description 33
- 229910052740 iodine Inorganic materials 0.000 claims description 33
- 125000004943 pyrimidin-6-yl group Chemical group N1=CN=CC=C1* 0.000 claims description 33
- 208000035475 disorder Diseases 0.000 claims description 27
- 125000001424 substituent group Chemical group 0.000 claims description 25
- 125000003342 alkenyl group Chemical group 0.000 claims description 24
- 239000003937 drug carrier Substances 0.000 claims description 23
- 150000002148 esters Chemical class 0.000 claims description 21
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 claims description 20
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 claims description 20
- 229910052717 sulfur Inorganic materials 0.000 claims description 20
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 claims description 19
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 claims description 18
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 17
- 239000001257 hydrogen Substances 0.000 claims description 17
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 17
- 230000001404 mediated effect Effects 0.000 claims description 17
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 17
- 229910052763 palladium Inorganic materials 0.000 claims description 17
- 125000001624 naphthyl group Chemical group 0.000 claims description 16
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 15
- 230000001363 autoimmune Effects 0.000 claims description 15
- 239000001301 oxygen Substances 0.000 claims description 15
- 239000003054 catalyst Substances 0.000 claims description 14
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 13
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 13
- 239000011593 sulfur Substances 0.000 claims description 13
- 125000000304 alkynyl group Chemical group 0.000 claims description 12
- 230000001028 anti-proliverative effect Effects 0.000 claims description 12
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 11
- JPIPZNJBXFDXHH-UHFFFAOYSA-N furo[2,3-d]pyrimidin-4-amine Chemical compound NC1=NC=NC2=C1C=CO2 JPIPZNJBXFDXHH-UHFFFAOYSA-N 0.000 claims description 11
- 125000001188 haloalkyl group Chemical group 0.000 claims description 11
- 230000002062 proliferating effect Effects 0.000 claims description 11
- 206010012438 Dermatitis atopic Diseases 0.000 claims description 10
- 201000008937 atopic dermatitis Diseases 0.000 claims description 10
- 125000002619 bicyclic group Chemical group 0.000 claims description 10
- 208000010247 contact dermatitis Diseases 0.000 claims description 10
- 201000005962 mycosis fungoides Diseases 0.000 claims description 10
- 206010006187 Breast cancer Diseases 0.000 claims description 9
- 230000006044 T cell activation Effects 0.000 claims description 9
- 239000003085 diluting agent Substances 0.000 claims description 9
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 9
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 8
- ZJFJRESZOCQFTB-LBPRGKRZSA-N 6-iodo-n-[[(2s)-oxolan-2-yl]methyl]-5-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C=12C(C=3C=CC=CC=3)=C(I)OC2=NC=NC=1NC[C@@H]1CCCO1 ZJFJRESZOCQFTB-LBPRGKRZSA-N 0.000 claims description 8
- 208000026310 Breast neoplasm Diseases 0.000 claims description 8
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 claims description 8
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 claims description 8
- 238000002054 transplantation Methods 0.000 claims description 8
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 7
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 claims description 7
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 7
- 208000020816 lung neoplasm Diseases 0.000 claims description 7
- 230000009885 systemic effect Effects 0.000 claims description 7
- 208000015943 Coeliac disease Diseases 0.000 claims description 6
- 241001465754 Metazoa Species 0.000 claims description 6
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 claims description 6
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 claims description 6
- 206010039085 Rhinitis allergic Diseases 0.000 claims description 6
- 230000001154 acute effect Effects 0.000 claims description 6
- 201000010105 allergic rhinitis Diseases 0.000 claims description 6
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 claims description 6
- 229910052736 halogen Inorganic materials 0.000 claims description 6
- 125000002883 imidazolyl group Chemical group 0.000 claims description 6
- 125000002757 morpholinyl group Chemical group 0.000 claims description 6
- 201000004384 Alopecia Diseases 0.000 claims description 5
- 208000009137 Behcet syndrome Diseases 0.000 claims description 5
- 206010072578 Chronic actinic dermatitis Diseases 0.000 claims description 5
- 206010009900 Colitis ulcerative Diseases 0.000 claims description 5
- 206010012442 Dermatitis contact Diseases 0.000 claims description 5
- 208000000185 Localized scleroderma Diseases 0.000 claims description 5
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 5
- 206010027982 Morphoea Diseases 0.000 claims description 5
- 208000031845 Pernicious anaemia Diseases 0.000 claims description 5
- 208000020424 Polyglandular disease Diseases 0.000 claims description 5
- 208000006311 Pyoderma Diseases 0.000 claims description 5
- 206010053613 Type IV hypersensitivity reaction Diseases 0.000 claims description 5
- 201000006704 Ulcerative Colitis Diseases 0.000 claims description 5
- 206010047642 Vitiligo Diseases 0.000 claims description 5
- 231100000360 alopecia Toxicity 0.000 claims description 5
- 208000004631 alopecia areata Diseases 0.000 claims description 5
- 201000001981 dermatomyositis Diseases 0.000 claims description 5
- 150000002367 halogens Chemical group 0.000 claims description 5
- 201000005202 lung cancer Diseases 0.000 claims description 5
- XNFHHOXCDUAYSR-SFHVURJKSA-N n-[[(2s)-oxolan-2-yl]methyl]-5,6-diphenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C([C@H]1OCCC1)NC(C1=2)=NC=NC=2OC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 XNFHHOXCDUAYSR-SFHVURJKSA-N 0.000 claims description 5
- 125000000160 oxazolidinyl group Chemical group 0.000 claims description 5
- 208000017983 photosensitivity disease Diseases 0.000 claims description 5
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 5
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 5
- 206010040400 serum sickness Diseases 0.000 claims description 5
- 125000004568 thiomorpholinyl group Chemical group 0.000 claims description 5
- 230000024664 tolerance induction Effects 0.000 claims description 5
- 230000005951 type IV hypersensitivity Effects 0.000 claims description 5
- 208000027930 type IV hypersensitivity disease Diseases 0.000 claims description 5
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 claims description 4
- PTILEOLOGGMFCS-UHFFFAOYSA-N 5,6-diphenyl-N-(2-piperazin-1-ylethyl)furo[2,3-d]pyrimidin-4-amine Chemical compound N=1C=NC=2OC(C=3C=CC=CC=3)=C(C=3C=CC=CC=3)C=2C=1NCCN1CCNCC1 PTILEOLOGGMFCS-UHFFFAOYSA-N 0.000 claims description 4
- FAKTYOGWTZOPIV-UHFFFAOYSA-N 5,6-diphenyl-n-(2-piperidin-1-ylethyl)furo[2,3-d]pyrimidin-4-amine Chemical compound N=1C=NC=2OC(C=3C=CC=CC=3)=C(C=3C=CC=CC=3)C=2C=1NCCN1CCCCC1 FAKTYOGWTZOPIV-UHFFFAOYSA-N 0.000 claims description 4
- RQXFTLMATSXLJT-UHFFFAOYSA-N 5,6-diphenyl-n-(2-pyrrolidin-1-ylethyl)furo[2,3-d]pyrimidin-4-amine Chemical compound N=1C=NC=2OC(C=3C=CC=CC=3)=C(C=3C=CC=CC=3)C=2C=1NCCN1CCCC1 RQXFTLMATSXLJT-UHFFFAOYSA-N 0.000 claims description 4
- QGULPYCGVIMWAU-UHFFFAOYSA-N 5,6-diphenyl-n-(2-thiomorpholin-4-ylethyl)furo[2,3-d]pyrimidin-4-amine Chemical compound N=1C=NC=2OC(C=3C=CC=CC=3)=C(C=3C=CC=CC=3)C=2C=1NCCN1CCSCC1 QGULPYCGVIMWAU-UHFFFAOYSA-N 0.000 claims description 4
- UIDHEXVJFRAWPL-UHFFFAOYSA-N 5-(3-methylphenyl)-n-(oxolan-2-ylmethyl)-6-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound CC1=CC=CC(C=2C3=C(NCC4OCCC4)N=CN=C3OC=2C=2C=CC=CC=2)=C1 UIDHEXVJFRAWPL-UHFFFAOYSA-N 0.000 claims description 4
- GXQQJBDHBPHSQQ-UHFFFAOYSA-N 5-(4-fluorophenyl)-n-(oxolan-2-ylmethyl)-6-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1=CC(F)=CC=C1C1=C(C=2C=CC=CC=2)OC2=NC=NC(NCC3OCCC3)=C12 GXQQJBDHBPHSQQ-UHFFFAOYSA-N 0.000 claims description 4
- MNSCWVYSADQZIV-UHFFFAOYSA-N 6-(4-aminophenyl)-n-(oxolan-2-ylmethyl)-5-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1=CC(N)=CC=C1C1=C(C=2C=CC=CC=2)C2=C(NCC3OCCC3)N=CN=C2O1 MNSCWVYSADQZIV-UHFFFAOYSA-N 0.000 claims description 4
- 208000035895 Guillain-Barré syndrome Diseases 0.000 claims description 4
- 206010049567 Miller Fisher syndrome Diseases 0.000 claims description 4
- 206010033128 Ovarian cancer Diseases 0.000 claims description 4
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 4
- 201000001263 Psoriatic Arthritis Diseases 0.000 claims description 4
- 208000036824 Psoriatic arthropathy Diseases 0.000 claims description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical group C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 4
- UPDCYTYMODWOMS-GOSISDBHSA-N n-[2-[(3r)-3-methylpiperazin-1-yl]ethyl]-5,6-diphenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1CN[C@H](C)CN1CCNC1=NC=NC2=C1C(C=1C=CC=CC=1)=C(C=1C=CC=CC=1)O2 UPDCYTYMODWOMS-GOSISDBHSA-N 0.000 claims description 4
- 201000008482 osteoarthritis Diseases 0.000 claims description 4
- 125000004193 piperazinyl group Chemical group 0.000 claims description 4
- 125000003386 piperidinyl group Chemical group 0.000 claims description 4
- 125000004076 pyridyl group Chemical group 0.000 claims description 4
- VNDVKIBKCWVFSW-UHFFFAOYSA-N 4-methoxy-5,6-diphenylfuro[2,3-d]pyrimidine Chemical compound C1=2C(OC)=NC=NC=2OC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 VNDVKIBKCWVFSW-UHFFFAOYSA-N 0.000 claims description 3
- DDMNFHVXAXKMSR-UHFFFAOYSA-N 5-(2-fluorophenyl)-n-(oxolan-2-ylmethyl)-6-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound FC1=CC=CC=C1C1=C(C=2C=CC=CC=2)OC2=NC=NC(NCC3OCCC3)=C12 DDMNFHVXAXKMSR-UHFFFAOYSA-N 0.000 claims description 3
- WVAPAQAYMQEANW-UHFFFAOYSA-N 5-(3-fluorophenyl)-n-(oxolan-2-ylmethyl)-6-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound FC1=CC=CC(C=2C3=C(NCC4OCCC4)N=CN=C3OC=2C=2C=CC=CC=2)=C1 WVAPAQAYMQEANW-UHFFFAOYSA-N 0.000 claims description 3
- QOIJEZMCZDOSJP-UHFFFAOYSA-N 5-(furan-2-yl)-n-(oxolan-2-ylmethyl)-6-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1CCOC1CNC(C1=2)=NC=NC=2OC(C=2C=CC=CC=2)=C1C1=CC=CO1 QOIJEZMCZDOSJP-UHFFFAOYSA-N 0.000 claims description 3
- GYPFRIODXXMFDZ-UHFFFAOYSA-N 5-(furan-3-yl)-n-(oxolan-2-ylmethyl)-6-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1CCOC1CNC(C1=2)=NC=NC=2OC(C=2C=CC=CC=2)=C1C=1C=COC=1 GYPFRIODXXMFDZ-UHFFFAOYSA-N 0.000 claims description 3
- RJBFZKLPTPYYSG-UHFFFAOYSA-N 6-[3-methoxy-4-(2-piperidin-1-ylethoxy)phenyl]-n-[2-(4-methylpiperazin-1-yl)ethyl]-5-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound COC1=CC(C2=C(C3=C(NCCN4CCN(C)CC4)N=CN=C3O2)C=2C=CC=CC=2)=CC=C1OCCN1CCCCC1 RJBFZKLPTPYYSG-UHFFFAOYSA-N 0.000 claims description 3
- QEQZGCQCFZQHFZ-UHFFFAOYSA-N 6-[4-(2-methoxyethoxy)phenyl]-5-phenyl-n-(2-piperazin-1-ylethyl)furo[2,3-d]pyrimidin-4-amine Chemical compound C1=CC(OCCOC)=CC=C1C1=C(C=2C=CC=CC=2)C2=C(NCCN3CCNCC3)N=CN=C2O1 QEQZGCQCFZQHFZ-UHFFFAOYSA-N 0.000 claims description 3
- GTSNNTDAKWQGOW-UHFFFAOYSA-N 6-[4-(2-morpholin-4-ylethoxy)phenyl]-n-(oxolan-2-ylmethyl)-5-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C=1C=C(C2=C(C3=C(NCC4OCCC4)N=CN=C3O2)C=2C=CC=CC=2)C=CC=1OCCN1CCOCC1 GTSNNTDAKWQGOW-UHFFFAOYSA-N 0.000 claims description 3
- BZMLKWKLFHIRRE-UHFFFAOYSA-N 6-[4-[2-(dimethylamino)ethoxy]phenyl]-5-phenyl-n-(thiolan-2-ylmethyl)furo[2,3-d]pyrimidin-4-amine Chemical compound C1=CC(OCCN(C)C)=CC=C1C1=C(C=2C=CC=CC=2)C2=C(NCC3SCCC3)N=CN=C2O1 BZMLKWKLFHIRRE-UHFFFAOYSA-N 0.000 claims description 3
- RGNHZRDNMFWDEF-UHFFFAOYSA-N 6-iodo-5-phenyl-n-propan-2-ylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1=2C(NC(C)C)=NC=NC=2OC(I)=C1C1=CC=CC=C1 RGNHZRDNMFWDEF-UHFFFAOYSA-N 0.000 claims description 3
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 claims description 3
- 208000008839 Kidney Neoplasms Diseases 0.000 claims description 3
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 claims description 3
- 206010060862 Prostate cancer Diseases 0.000 claims description 3
- 208000004403 Prostatic Hyperplasia Diseases 0.000 claims description 3
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 3
- 206010038389 Renal cancer Diseases 0.000 claims description 3
- JFDZBHWFFUWGJE-UHFFFAOYSA-N benzenecarbonitrile Natural products N#CC1=CC=CC=C1 JFDZBHWFFUWGJE-UHFFFAOYSA-N 0.000 claims description 3
- 125000005605 benzo group Chemical group 0.000 claims description 3
- 125000004276 dioxalanyl group Chemical group 0.000 claims description 3
- 125000000532 dioxanyl group Chemical group 0.000 claims description 3
- 125000005883 dithianyl group Chemical group 0.000 claims description 3
- 125000005411 dithiolanyl group Chemical group S1SC(CC1)* 0.000 claims description 3
- 125000000031 ethylamino group Chemical group [H]C([H])([H])C([H])([H])N([H])[*] 0.000 claims description 3
- 125000002541 furyl group Chemical group 0.000 claims description 3
- 125000001041 indolyl group Chemical group 0.000 claims description 3
- 201000010982 kidney cancer Diseases 0.000 claims description 3
- 201000007270 liver cancer Diseases 0.000 claims description 3
- 208000014018 liver neoplasm Diseases 0.000 claims description 3
- GFNFHSYNRPAKIP-UHFFFAOYSA-N n-(furan-2-ylmethyl)-4-[4-(oxolan-2-ylmethylamino)-5-phenylfuro[2,3-d]pyrimidin-6-yl]benzamide Chemical compound C=1C=C(C2=C(C3=C(NCC4OCCC4)N=CN=C3O2)C=2C=CC=CC=2)C=CC=1C(=O)NCC1=CC=CO1 GFNFHSYNRPAKIP-UHFFFAOYSA-N 0.000 claims description 3
- FDXKHHVUXUOKBD-UHFFFAOYSA-N n-(oxolan-2-ylmethyl)-5-phenyl-6-thiophen-2-ylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1CCOC1CNC(C=1C=2C=3C=CC=CC=3)=NC=NC=1OC=2C1=CC=CS1 FDXKHHVUXUOKBD-UHFFFAOYSA-N 0.000 claims description 3
- PDTKVFPWKDFRBL-UHFFFAOYSA-N n-(oxolan-2-ylmethyl)-6-phenyl-5-thiophen-2-ylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1CCOC1CNC(C1=2)=NC=NC=2OC(C=2C=CC=CC=2)=C1C1=CC=CS1 PDTKVFPWKDFRBL-UHFFFAOYSA-N 0.000 claims description 3
- ADYMYIXTFYSCCR-UHFFFAOYSA-N n-[2-(2,5-diazabicyclo[2.2.1]heptan-2-yl)ethyl]-5,6-diphenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1C(NC2)CC2N1CCNC(C1=2)=NC=NC=2OC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 ADYMYIXTFYSCCR-UHFFFAOYSA-N 0.000 claims description 3
- OPVOZSXJPFYFPL-UHFFFAOYSA-N n-[2-(4-methylpiperazin-1-yl)ethyl]-5,6-diphenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1CN(C)CCN1CCNC1=NC=NC2=C1C(C=1C=CC=CC=1)=C(C=1C=CC=CC=1)O2 OPVOZSXJPFYFPL-UHFFFAOYSA-N 0.000 claims description 3
- UCKQNNIWWBGUGY-SFHVURJKSA-N n-[[(2s)-oxolan-2-yl]methyl]-5,6-diphenylthieno[2,3-d]pyrimidin-4-amine Chemical compound C([C@H]1OCCC1)NC(C1=2)=NC=NC=2SC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 UCKQNNIWWBGUGY-SFHVURJKSA-N 0.000 claims description 3
- JOEDUVRAZBNLQG-UHFFFAOYSA-N n-cyclopropyl-5-phenyl-6-[4-(2-pyrrolidin-1-ylethoxy)phenyl]furo[2,3-d]pyrimidin-4-amine Chemical compound C=1C=C(C2=C(C3=C(NC4CC4)N=CN=C3O2)C=2C=CC=CC=2)C=CC=1OCCN1CCCC1 JOEDUVRAZBNLQG-UHFFFAOYSA-N 0.000 claims description 3
- 230000025020 negative regulation of T cell proliferation Effects 0.000 claims description 3
- 125000005880 oxathiolanyl group Chemical group 0.000 claims description 3
- 125000003566 oxetanyl group Chemical group 0.000 claims description 3
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 claims description 3
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 claims description 3
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 claims description 3
- 125000003507 tetrahydrothiofenyl group Chemical group 0.000 claims description 3
- 125000001544 thienyl group Chemical group 0.000 claims description 3
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 claims description 3
- 125000005871 1,3-benzodioxolyl group Chemical group 0.000 claims description 2
- FZZMTSNZRBFGGU-UHFFFAOYSA-N 2-chloro-7-fluoroquinazolin-4-amine Chemical compound FC1=CC=C2C(N)=NC(Cl)=NC2=C1 FZZMTSNZRBFGGU-UHFFFAOYSA-N 0.000 claims description 2
- MGADZUXDNSDTHW-UHFFFAOYSA-N 2H-pyran Chemical compound C1OC=CC=C1 MGADZUXDNSDTHW-UHFFFAOYSA-N 0.000 claims description 2
- NWHMTXLYOPSDLN-UHFFFAOYSA-N 4-[4-(oxolan-2-ylmethylamino)-5-phenylfuro[2,3-d]pyrimidin-6-yl]phenol Chemical compound C1=CC(O)=CC=C1C1=C(C=2C=CC=CC=2)C2=C(NCC3OCCC3)N=CN=C2O1 NWHMTXLYOPSDLN-UHFFFAOYSA-N 0.000 claims description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N Benzoic acid Natural products OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 claims description 2
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 claims description 2
- KQGXYVJZOMKLSA-UHFFFAOYSA-N furo[2,3-d]pyrimidine Chemical compound N1=CN=C2OC=CC2=C1 KQGXYVJZOMKLSA-UHFFFAOYSA-N 0.000 claims description 2
- 230000002401 inhibitory effect Effects 0.000 claims description 2
- QVRFVZHTCBJPMH-UHFFFAOYSA-N n,n-dimethyl-4-[4-(oxolan-2-ylmethylamino)-5-phenylfuro[2,3-d]pyrimidin-6-yl]benzenesulfonamide Chemical compound C1=CC(S(=O)(=O)N(C)C)=CC=C1C1=C(C=2C=CC=CC=2)C2=C(NCC3OCCC3)N=CN=C2O1 QVRFVZHTCBJPMH-UHFFFAOYSA-N 0.000 claims description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Chemical group COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 2
- 125000003944 tolyl group Chemical group 0.000 claims description 2
- OYRRZWATULMEPF-UHFFFAOYSA-N pyrimidin-4-amine Chemical compound NC1=CC=NC=N1 OYRRZWATULMEPF-UHFFFAOYSA-N 0.000 claims 5
- FACSDOITOHWERB-DEOSSOPVSA-N (4-ethylpiperazin-1-yl)-[4-[4-[[(2s)-oxolan-2-yl]methylamino]-5-phenylfuro[2,3-d]pyrimidin-6-yl]phenyl]methanone Chemical compound C1CN(CC)CCN1C(=O)C1=CC=C(C2=C(C3=C(NC[C@H]4OCCC4)N=CN=C3O2)C=2C=CC=CC=2)C=C1 FACSDOITOHWERB-DEOSSOPVSA-N 0.000 claims 1
- OBPZOBAQDUCRNE-UHFFFAOYSA-N (4-methylpiperazin-1-yl)-[4-[4-(oxolan-2-ylmethylamino)-5-phenylfuro[2,3-d]pyrimidin-6-yl]phenyl]methanone Chemical compound C1CN(C)CCN1C(=O)C1=CC=C(C2=C(C3=C(NCC4OCCC4)N=CN=C3O2)C=2C=CC=CC=2)C=C1 OBPZOBAQDUCRNE-UHFFFAOYSA-N 0.000 claims 1
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 claims 1
- CGXJGFSMZJCZRC-UHFFFAOYSA-N 1-[4-[4-(oxolan-2-ylmethylamino)-5-phenylfuro[2,3-d]pyrimidin-6-yl]phenyl]sulfonylpiperidin-4-ol Chemical compound C1CC(O)CCN1S(=O)(=O)C1=CC=C(C2=C(C3=C(NCC4OCCC4)N=CN=C3O2)C=2C=CC=CC=2)C=C1 CGXJGFSMZJCZRC-UHFFFAOYSA-N 0.000 claims 1
- CBFONFACFPVNPN-SFHVURJKSA-N 1-[[(2S)-oxolan-2-yl]methyl]-5-phenyl-6-pyridin-4-yl-2H-furo[2,3-d]pyrimidine Chemical compound C1(=CC=CC=C1)C1=C(OC=2N(CN=CC21)C[C@H]2OCCC2)C2=CC=NC=C2 CBFONFACFPVNPN-SFHVURJKSA-N 0.000 claims 1
- BZCKYNDJKUOOEU-UHFFFAOYSA-N 2,6-dimethyl-4-[5-phenyl-4-(2-piperazin-1-ylethylamino)furo[2,3-d]pyrimidin-6-yl]phenol Chemical compound CC1=C(O)C(C)=CC(C2=C(C3=C(NCCN4CCNCC4)N=CN=C3O2)C=2C=CC=CC=2)=C1 BZCKYNDJKUOOEU-UHFFFAOYSA-N 0.000 claims 1
- MRYAWEMZSXNYRM-UHFFFAOYSA-N 4-[2-[(5,6-diphenylfuro[2,3-d]pyrimidin-4-yl)amino]ethyl]piperazine-1-carboxylic acid Chemical compound C1CN(CCN1CCNC2=C3C(=C(OC3=NC=N2)C4=CC=CC=C4)C5=CC=CC=C5)C(=O)O MRYAWEMZSXNYRM-UHFFFAOYSA-N 0.000 claims 1
- XJCXOYSIUXFKHV-UHFFFAOYSA-N 4-[2-[(6-iodo-5-phenylfuro[2,3-d]pyrimidin-4-yl)amino]ethyl]piperazine-1-carboxylic acid Chemical compound C1CN(CCN1CCNC2=C3C(=C(OC3=NC=N2)I)C4=CC=CC=C4)C(=O)O XJCXOYSIUXFKHV-UHFFFAOYSA-N 0.000 claims 1
- XBGQCQRFGDCNQX-UHFFFAOYSA-N 4-[4-(oxolan-2-ylmethylamino)-5-phenylfuro[2,3-d]pyrimidin-6-yl]benzenesulfonamide Chemical compound C1=CC(S(=O)(=O)N)=CC=C1C1=C(C=2C=CC=CC=2)C2=C(NCC3OCCC3)N=CN=C2O1 XBGQCQRFGDCNQX-UHFFFAOYSA-N 0.000 claims 1
- AALKBDSPCFAJAV-QHCPKHFHSA-N 4-[4-[[(2s)-oxolan-2-yl]methylamino]-5-phenylfuro[2,3-d]pyrimidin-6-yl]-n-(pyridin-3-ylmethyl)benzenesulfonamide Chemical compound C=1C=C(C2=C(C3=C(NC[C@H]4OCCC4)N=CN=C3O2)C=2C=CC=CC=2)C=CC=1S(=O)(=O)NCC1=CC=CN=C1 AALKBDSPCFAJAV-QHCPKHFHSA-N 0.000 claims 1
- FFNQKJOPGRJEPP-IBGZPJMESA-N 4-[4-[[(2s)-oxolan-2-yl]methylamino]-5-phenylfuro[2,3-d]pyrimidin-6-yl]benzaldehyde Chemical compound C1=CC(C=O)=CC=C1C1=C(C=2C=CC=CC=2)C2=C(NC[C@H]3OCCC3)N=CN=C2O1 FFNQKJOPGRJEPP-IBGZPJMESA-N 0.000 claims 1
- OTWMQQYPLBAFNA-UHFFFAOYSA-N 4-[5-phenyl-4-(2-piperazin-1-ylethylamino)furo[2,3-d]pyrimidin-6-yl]-n-propylbenzamide Chemical compound C1=CC(C(=O)NCCC)=CC=C1C1=C(C=2C=CC=CC=2)C2=C(NCCN3CCNCC3)N=CN=C2O1 OTWMQQYPLBAFNA-UHFFFAOYSA-N 0.000 claims 1
- IJQSFGDQONQYPG-UHFFFAOYSA-N 5,6-diphenyl-n-(3-piperazin-1-ylpropyl)furo[2,3-d]pyrimidin-4-amine Chemical compound C1CNCCN1CCCNC(C1=2)=NC=NC=2OC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 IJQSFGDQONQYPG-UHFFFAOYSA-N 0.000 claims 1
- JPDAVAORUSDQDZ-UHFFFAOYSA-N 5-(3-methoxyphenyl)-n-(oxolan-2-ylmethyl)-6-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound COC1=CC=CC(C=2C3=C(NCC4OCCC4)N=CN=C3OC=2C=2C=CC=CC=2)=C1 JPDAVAORUSDQDZ-UHFFFAOYSA-N 0.000 claims 1
- DNYZVJZWTNWOBG-UHFFFAOYSA-N 5-(4-methoxyphenyl)-n-(oxolan-2-ylmethyl)-6-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1=CC(OC)=CC=C1C1=C(C=2C=CC=CC=2)OC2=NC=NC(NCC3OCCC3)=C12 DNYZVJZWTNWOBG-UHFFFAOYSA-N 0.000 claims 1
- KLVVYLBLOHGSLQ-UHFFFAOYSA-N 5-(4-methylphenyl)-n-(oxolan-2-ylmethyl)-6-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1=CC(C)=CC=C1C1=C(C=2C=CC=CC=2)OC2=NC=NC(NCC3OCCC3)=C12 KLVVYLBLOHGSLQ-UHFFFAOYSA-N 0.000 claims 1
- CYXPHNZJEDAZDD-UHFFFAOYSA-N 5-phenyl-n-(2-phenylethyl)-6-[4-(2-pyrrolidin-1-ylethoxy)phenyl]furo[2,3-d]pyrimidin-4-amine Chemical compound N=1C=NC=2OC(C=3C=CC(OCCN4CCCC4)=CC=3)=C(C=3C=CC=CC=3)C=2C=1NCCC1=CC=CC=C1 CYXPHNZJEDAZDD-UHFFFAOYSA-N 0.000 claims 1
- RHNKKXOGNZLIOK-UHFFFAOYSA-N 5-phenyl-n-(2-piperazin-1-ylethyl)-6-(3,4,5-trimethoxyphenyl)furo[2,3-d]pyrimidin-4-amine Chemical compound COC1=C(OC)C(OC)=CC(C2=C(C3=C(NCCN4CCNCC4)N=CN=C3O2)C=2C=CC=CC=2)=C1 RHNKKXOGNZLIOK-UHFFFAOYSA-N 0.000 claims 1
- DMZTWOCJNVKCBW-UHFFFAOYSA-N 5-phenyl-n-(2-piperazin-1-ylethyl)-6-[4-(2-pyrrolidin-1-ylethoxy)phenyl]furo[2,3-d]pyrimidin-4-amine Chemical compound N=1C=NC=2OC(C=3C=CC(OCCN4CCCC4)=CC=3)=C(C=3C=CC=CC=3)C=2C=1NCCN1CCNCC1 DMZTWOCJNVKCBW-UHFFFAOYSA-N 0.000 claims 1
- FWFIEBNPAPTVOD-UHFFFAOYSA-N 6-(1,3-benzodioxol-5-yl)-5-phenyl-n-(2-piperazin-1-ylethyl)furo[2,3-d]pyrimidin-4-amine Chemical compound N=1C=NC=2OC(C=3C=C4OCOC4=CC=3)=C(C=3C=CC=CC=3)C=2C=1NCCN1CCNCC1 FWFIEBNPAPTVOD-UHFFFAOYSA-N 0.000 claims 1
- NFVPGAUFFDSMQM-UHFFFAOYSA-N 6-(1-benzothiophen-2-yl)-5-phenyl-n-(2-piperazin-1-ylethyl)furo[2,3-d]pyrimidin-4-amine Chemical compound N=1C=NC=2OC(C=3SC4=CC=CC=C4C=3)=C(C=3C=CC=CC=3)C=2C=1NCCN1CCNCC1 NFVPGAUFFDSMQM-UHFFFAOYSA-N 0.000 claims 1
- KWQULIGAIVHPLJ-UHFFFAOYSA-N 6-(1-methylindol-5-yl)-5-phenyl-n-(2-piperazin-1-ylethyl)furo[2,3-d]pyrimidin-4-amine Chemical compound C=1C=C2N(C)C=CC2=CC=1C(=C(C1=2)C=3C=CC=CC=3)OC1=NC=NC=2NCCN1CCNCC1 KWQULIGAIVHPLJ-UHFFFAOYSA-N 0.000 claims 1
- ALRNLOOWLXLZQS-UHFFFAOYSA-N 6-(1h-indol-5-yl)-5-phenyl-n-(2-piperazin-1-ylethyl)furo[2,3-d]pyrimidin-4-amine Chemical compound N=1C=NC=2OC(C=3C=C4C=CNC4=CC=3)=C(C=3C=CC=CC=3)C=2C=1NCCN1CCNCC1 ALRNLOOWLXLZQS-UHFFFAOYSA-N 0.000 claims 1
- NJKQVNYTHDQZHF-INIZCTEOSA-N 6-(2-fluorophenyl)-n-[[(2s)-oxolan-2-yl]methyl]-5-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound FC1=CC=CC=C1C1=C(C=2C=CC=CC=2)C2=C(NC[C@H]3OCCC3)N=CN=C2O1 NJKQVNYTHDQZHF-INIZCTEOSA-N 0.000 claims 1
- IWTUSXPLUPCWTH-SFHVURJKSA-N 6-(3-aminophenyl)-n-[[(2s)-oxolan-2-yl]methyl]-5-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound NC1=CC=CC(C2=C(C3=C(NC[C@H]4OCCC4)N=CN=C3O2)C=2C=CC=CC=2)=C1 IWTUSXPLUPCWTH-SFHVURJKSA-N 0.000 claims 1
- YHTSUIXFAAQWLU-UHFFFAOYSA-N 6-(4-morpholin-4-ylphenyl)-5-phenyl-n-(2-piperazin-1-ylethyl)furo[2,3-d]pyrimidin-4-amine Chemical compound N=1C=NC=2OC(C=3C=CC(=CC=3)N3CCOCC3)=C(C=3C=CC=CC=3)C=2C=1NCCN1CCNCC1 YHTSUIXFAAQWLU-UHFFFAOYSA-N 0.000 claims 1
- JNTUHTLJAZVPBH-UHFFFAOYSA-N 6-[3-fluoro-4-(2-piperidin-1-ylethoxy)phenyl]-5-phenyl-n-propan-2-ylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1=2C(NC(C)C)=NC=NC=2OC(C=2C=C(F)C(OCCN3CCCCC3)=CC=2)=C1C1=CC=CC=C1 JNTUHTLJAZVPBH-UHFFFAOYSA-N 0.000 claims 1
- ZVCHQVMTVGGKOB-QHCPKHFHSA-N 6-[3-fluoro-4-(2-piperidin-1-ylethoxy)phenyl]-n-[[(2s)-oxolan-2-yl]methyl]-5-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound FC1=CC(C2=C(C3=C(NC[C@H]4OCCC4)N=CN=C3O2)C=2C=CC=CC=2)=CC=C1OCCN1CCCCC1 ZVCHQVMTVGGKOB-QHCPKHFHSA-N 0.000 claims 1
- QXJMSSPFEBATGE-UHFFFAOYSA-N 6-[3-fluoro-4-(2-pyrrolidin-1-ylethoxy)phenyl]-n-(2-methylpropyl)-5-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1=2C(NCC(C)C)=NC=NC=2OC(C=2C=C(F)C(OCCN3CCCC3)=CC=2)=C1C1=CC=CC=C1 QXJMSSPFEBATGE-UHFFFAOYSA-N 0.000 claims 1
- PQXQJSMNKNNINY-UHFFFAOYSA-N 6-[3-methoxy-4-(2-piperidin-1-ylethoxy)phenyl]-5-phenyl-n-(2-piperazin-1-ylethyl)furo[2,3-d]pyrimidin-4-amine Chemical compound COC1=CC(C2=C(C3=C(NCCN4CCNCC4)N=CN=C3O2)C=2C=CC=CC=2)=CC=C1OCCN1CCCCC1 PQXQJSMNKNNINY-UHFFFAOYSA-N 0.000 claims 1
- JLURGTUCNPVZCE-UHFFFAOYSA-N 6-[3-methoxy-4-(2-piperidin-1-ylethoxy)phenyl]-n-(2-methylpropyl)-5-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound COC1=CC(C2=C(C3=C(NCC(C)C)N=CN=C3O2)C=2C=CC=CC=2)=CC=C1OCCN1CCCCC1 JLURGTUCNPVZCE-UHFFFAOYSA-N 0.000 claims 1
- IOSKHROQBBQEOB-DEOSSOPVSA-N 6-[3-methoxy-4-(2-piperidin-1-ylethoxy)phenyl]-n-[[(2s)-oxolan-2-yl]methyl]-5-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound COC1=CC(C2=C(C3=C(NC[C@H]4OCCC4)N=CN=C3O2)C=2C=CC=CC=2)=CC=C1OCCN1CCCCC1 IOSKHROQBBQEOB-DEOSSOPVSA-N 0.000 claims 1
- NYRAJYGKYFZUAV-UHFFFAOYSA-N 6-[4-(4-methylpiperazin-1-yl)sulfonylphenyl]-5-phenyl-n-(2-piperazin-1-ylethyl)furo[2,3-d]pyrimidin-4-amine Chemical compound C1CN(C)CCN1S(=O)(=O)C1=CC=C(C2=C(C3=C(NCCN4CCNCC4)N=CN=C3O2)C=2C=CC=CC=2)C=C1 NYRAJYGKYFZUAV-UHFFFAOYSA-N 0.000 claims 1
- VEGBQINRHSCNRW-UHFFFAOYSA-N 6-[4-(dimethylamino)phenyl]-n-(oxolan-2-ylmethyl)-5-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1=CC(N(C)C)=CC=C1C1=C(C=2C=CC=CC=2)C2=C(NCC3OCCC3)N=CN=C2O1 VEGBQINRHSCNRW-UHFFFAOYSA-N 0.000 claims 1
- CKICOBOTESTQPH-UHFFFAOYSA-N 6-[4-[2-(diethylamino)ethoxy]-3-fluorophenyl]-5-phenyl-n-propan-2-ylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1=C(F)C(OCCN(CC)CC)=CC=C1C1=C(C=2C=CC=CC=2)C2=C(NC(C)C)N=CN=C2O1 CKICOBOTESTQPH-UHFFFAOYSA-N 0.000 claims 1
- UWUUCEBUCLHRDX-UHFFFAOYSA-N 6-[4-[3-(dimethylamino)propoxy]phenyl]-n-(1,3-dithiolan-2-ylmethyl)-5-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1=CC(OCCCN(C)C)=CC=C1C1=C(C=2C=CC=CC=2)C2=C(NCC3SCCS3)N=CN=C2O1 UWUUCEBUCLHRDX-UHFFFAOYSA-N 0.000 claims 1
- 239000005711 Benzoic acid Substances 0.000 claims 1
- FKEXPTQQRQJYBU-UHFFFAOYSA-N [1-[2-[(5,6-diphenylfuro[2,3-d]pyrimidin-4-yl)amino]ethyl]piperidin-4-yl]methanol Chemical compound C1CC(CO)CCN1CCNC1=NC=NC2=C1C(C=1C=CC=CC=1)=C(C=1C=CC=CC=1)O2 FKEXPTQQRQJYBU-UHFFFAOYSA-N 0.000 claims 1
- PGLYGLNQRSYVMW-IBGZPJMESA-N [3-[4-[[(2s)-oxolan-2-yl]methylamino]-5-phenylfuro[2,3-d]pyrimidin-6-yl]phenyl]methanol Chemical compound OCC1=CC=CC(C2=C(C3=C(NC[C@H]4OCCC4)N=CN=C3O2)C=2C=CC=CC=2)=C1 PGLYGLNQRSYVMW-IBGZPJMESA-N 0.000 claims 1
- 235000010233 benzoic acid Nutrition 0.000 claims 1
- 125000000896 monocarboxylic acid group Chemical group 0.000 claims 1
- CJBDQHGOKGXJEK-UHFFFAOYSA-N morpholin-4-yl-[4-[5-phenyl-4-(2-piperazin-1-ylethylamino)furo[2,3-d]pyrimidin-6-yl]phenyl]methanone Chemical compound C=1C=C(C2=C(C3=C(NCCN4CCNCC4)N=CN=C3O2)C=2C=CC=CC=2)C=CC=1C(=O)N1CCOCC1 CJBDQHGOKGXJEK-UHFFFAOYSA-N 0.000 claims 1
- XRUXFZAYYMQZMD-UHFFFAOYSA-N n,n-dimethyl-4-[5-phenyl-4-(2-piperazin-1-ylethylamino)furo[2,3-d]pyrimidin-6-yl]benzamide Chemical compound C1=CC(C(=O)N(C)C)=CC=C1C1=C(C=2C=CC=CC=2)C2=C(NCCN3CCNCC3)N=CN=C2O1 XRUXFZAYYMQZMD-UHFFFAOYSA-N 0.000 claims 1
- YSXNEICOJOYOEN-UHFFFAOYSA-N n,n-dimethyl-4-[5-phenyl-4-(2-piperazin-1-ylethylamino)furo[2,3-d]pyrimidin-6-yl]benzenesulfonamide Chemical compound C1=CC(S(=O)(=O)N(C)C)=CC=C1C1=C(C=2C=CC=CC=2)C2=C(NCCN3CCNCC3)N=CN=C2O1 YSXNEICOJOYOEN-UHFFFAOYSA-N 0.000 claims 1
- OLMIXYVQLHKCGW-UHFFFAOYSA-N n,n-dimethyl-4-[5-phenyl-4-(propan-2-ylamino)furo[2,3-d]pyrimidin-6-yl]benzenesulfonamide Chemical compound C1=2C(NC(C)C)=NC=NC=2OC(C=2C=CC(=CC=2)S(=O)(=O)N(C)C)=C1C1=CC=CC=C1 OLMIXYVQLHKCGW-UHFFFAOYSA-N 0.000 claims 1
- SHILCNKNFJKBEB-UHFFFAOYSA-N n-(1,3-dithiolan-2-ylmethyl)-5,6-diphenylfuro[2,3-d]pyrimidin-4-amine Chemical compound S1CCSC1CNC(C1=2)=NC=NC=2OC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 SHILCNKNFJKBEB-UHFFFAOYSA-N 0.000 claims 1
- HAZMKBZPNIPURP-UHFFFAOYSA-N n-(2,2-dimethylpropyl)-6-[3-fluoro-4-(2-pyrrolidin-1-ylethoxy)phenyl]-5-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1=2C(NCC(C)(C)C)=NC=NC=2OC(C=2C=C(F)C(OCCN3CCCC3)=CC=2)=C1C1=CC=CC=C1 HAZMKBZPNIPURP-UHFFFAOYSA-N 0.000 claims 1
- GYSLCLHVXAXEAR-UHFFFAOYSA-N n-(2-methoxyethyl)-4-[4-(oxolan-2-ylmethylamino)-5-phenylfuro[2,3-d]pyrimidin-6-yl]benzamide Chemical compound C1=CC(C(=O)NCCOC)=CC=C1C1=C(C=2C=CC=CC=2)C2=C(NCC3OCCC3)N=CN=C2O1 GYSLCLHVXAXEAR-UHFFFAOYSA-N 0.000 claims 1
- VDLGOWNFTBEOFX-UHFFFAOYSA-N n-(2-methoxyethyl)-4-[4-(oxolan-2-ylmethylamino)-5-phenylfuro[2,3-d]pyrimidin-6-yl]benzenesulfonamide Chemical compound C1=CC(S(=O)(=O)NCCOC)=CC=C1C1=C(C=2C=CC=CC=2)C2=C(NCC3OCCC3)N=CN=C2O1 VDLGOWNFTBEOFX-UHFFFAOYSA-N 0.000 claims 1
- HCVIUTOAKUKQLC-UHFFFAOYSA-N n-(2-morpholin-4-ylethyl)-5,6-diphenylfuro[2,3-d]pyrimidin-4-amine Chemical compound N=1C=NC=2OC(C=3C=CC=CC=3)=C(C=3C=CC=CC=3)C=2C=1NCCN1CCOCC1 HCVIUTOAKUKQLC-UHFFFAOYSA-N 0.000 claims 1
- HJQYPVYMZZRXMW-DEOSSOPVSA-N n-(3-morpholin-4-ylpropyl)-4-[4-[[(2s)-oxolan-2-yl]methylamino]-5-phenylfuro[2,3-d]pyrimidin-6-yl]benzenesulfonamide Chemical compound C=1C=C(C2=C(C3=C(NC[C@H]4OCCC4)N=CN=C3O2)C=2C=CC=CC=2)C=CC=1S(=O)(=O)NCCCN1CCOCC1 HJQYPVYMZZRXMW-DEOSSOPVSA-N 0.000 claims 1
- VYHACVWBBXBMFI-UHFFFAOYSA-N n-(oxolan-2-ylmethyl)-5-phenyl-6-[4-(2-pyridin-4-ylethoxy)phenyl]furo[2,3-d]pyrimidin-4-amine Chemical compound C=1C=C(C2=C(C3=C(NCC4OCCC4)N=CN=C3O2)C=2C=CC=CC=2)C=CC=1OCCC1=CC=NC=C1 VYHACVWBBXBMFI-UHFFFAOYSA-N 0.000 claims 1
- MIRHGZUEFRUEME-UHFFFAOYSA-N n-(oxolan-2-ylmethyl)-6-phenyl-5-thiophen-3-ylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1CCOC1CNC(C1=2)=NC=NC=2OC(C=2C=CC=CC=2)=C1C=1C=CSC=1 MIRHGZUEFRUEME-UHFFFAOYSA-N 0.000 claims 1
- DOKBHQJYGNRCOM-UHFFFAOYSA-N n-[2-(1h-imidazol-5-yl)ethyl]-6-[4-(4-methylpiperazin-1-yl)sulfonylphenyl]-5-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1CN(C)CCN1S(=O)(=O)C1=CC=C(C2=C(C3=C(NCCC=4NC=NC=4)N=CN=C3O2)C=2C=CC=CC=2)C=C1 DOKBHQJYGNRCOM-UHFFFAOYSA-N 0.000 claims 1
- GROXVTPJRLIMAW-UHFFFAOYSA-N n-[2-(4-benzylpiperazin-1-yl)ethyl]-5-(3-fluorophenyl)furo[2,3-d]pyrimidin-4-amine Chemical compound FC1=CC=CC(C=2C3=C(NCCN4CCN(CC=5C=CC=CC=5)CC4)N=CN=C3OC=2)=C1 GROXVTPJRLIMAW-UHFFFAOYSA-N 0.000 claims 1
- ZQDWQWNSBFXMDC-UHFFFAOYSA-N n-[2-(4-ethylpiperazin-1-yl)ethyl]-5,6-diphenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1CN(CC)CCN1CCNC1=NC=NC2=C1C(C=1C=CC=CC=1)=C(C=1C=CC=CC=1)O2 ZQDWQWNSBFXMDC-UHFFFAOYSA-N 0.000 claims 1
- ICZSQBHHATZYNY-UHFFFAOYSA-N n-[2-(4-ethylpiperazin-1-yl)ethyl]-5-phenyl-6-[4-(2-pyrrolidin-1-ylethoxy)phenyl]furo[2,3-d]pyrimidin-4-amine Chemical compound C1CN(CC)CCN1CCNC1=NC=NC2=C1C(C=1C=CC=CC=1)=C(C=1C=CC(OCCN3CCCC3)=CC=1)O2 ICZSQBHHATZYNY-UHFFFAOYSA-N 0.000 claims 1
- YSWLNXFQMMFDFG-DEOSSOPVSA-N n-[[(2s)-oxolan-2-yl]methyl]-5-phenyl-6-[4-(2-pyrrolidin-1-ylethoxy)phenyl]furo[2,3-d]pyrimidin-4-amine Chemical compound C=1C=C(C2=C(C3=C(NC[C@H]4OCCC4)N=CN=C3O2)C=2C=CC=CC=2)C=CC=1OCCN1CCCC1 YSWLNXFQMMFDFG-DEOSSOPVSA-N 0.000 claims 1
- WAVDWYGTDOEJFD-ZDUSSCGKSA-N n-[[(2s)-oxolan-2-yl]methyl]-5-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C([C@H]1OCCC1)NC(C1=2)=NC=NC=2OC=C1C1=CC=CC=C1 WAVDWYGTDOEJFD-ZDUSSCGKSA-N 0.000 claims 1
- VFAGEZQRPABLEY-IBGZPJMESA-N n-methyl-4-[4-[[(2s)-oxolan-2-yl]methylamino]-5-phenylfuro[2,3-d]pyrimidin-6-yl]benzamide Chemical compound C1=CC(C(=O)NC)=CC=C1C1=C(C=2C=CC=CC=2)C2=C(NC[C@H]3OCCC3)N=CN=C2O1 VFAGEZQRPABLEY-IBGZPJMESA-N 0.000 claims 1
- SHCRXFQGLRUXPI-UHFFFAOYSA-N tert-butyl 4-[2-[[6-[4-(methylcarbamoyl)phenyl]-5-phenylfuro[2,3-d]pyrimidin-4-yl]amino]ethyl]piperazine-1-carboxylate Chemical compound C1=CC(C(=O)NC)=CC=C1C1=C(C=2C=CC=CC=2)C2=C(NCCN3CCN(CC3)C(=O)OC(C)(C)C)N=CN=C2O1 SHCRXFQGLRUXPI-UHFFFAOYSA-N 0.000 claims 1
- 208000023275 Autoimmune disease Diseases 0.000 abstract description 6
- 230000035755 proliferation Effects 0.000 abstract description 5
- 206010020751 Hypersensitivity Diseases 0.000 abstract description 4
- 230000009610 hypersensitivity Effects 0.000 abstract description 2
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 186
- 230000015572 biosynthetic process Effects 0.000 description 148
- 238000003786 synthesis reaction Methods 0.000 description 144
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 104
- 239000000203 mixture Substances 0.000 description 92
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 71
- 238000006243 chemical reaction Methods 0.000 description 68
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 62
- 238000005160 1H NMR spectroscopy Methods 0.000 description 60
- 238000006069 Suzuki reaction reaction Methods 0.000 description 58
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 54
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 52
- 229910001868 water Inorganic materials 0.000 description 51
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 50
- 239000002904 solvent Substances 0.000 description 47
- 235000019439 ethyl acetate Nutrition 0.000 description 43
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 43
- 239000007787 solid Substances 0.000 description 35
- 239000000243 solution Substances 0.000 description 34
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 32
- 238000010992 reflux Methods 0.000 description 32
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 31
- 230000029936 alkylation Effects 0.000 description 28
- 238000005804 alkylation reaction Methods 0.000 description 28
- 230000000694 effects Effects 0.000 description 28
- 210000004027 cell Anatomy 0.000 description 26
- 239000011541 reaction mixture Substances 0.000 description 26
- 238000002360 preparation method Methods 0.000 description 25
- 230000002829 reductive effect Effects 0.000 description 25
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 24
- 239000002585 base Substances 0.000 description 23
- 238000007429 general method Methods 0.000 description 23
- 239000003921 oil Substances 0.000 description 23
- 235000019198 oils Nutrition 0.000 description 23
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 22
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 22
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 21
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 21
- 238000003818 flash chromatography Methods 0.000 description 21
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 20
- 239000012044 organic layer Substances 0.000 description 20
- 238000003756 stirring Methods 0.000 description 20
- 239000000047 product Substances 0.000 description 19
- 108090000623 proteins and genes Proteins 0.000 description 19
- 239000000377 silicon dioxide Substances 0.000 description 19
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 18
- 210000001744 T-lymphocyte Anatomy 0.000 description 18
- 238000003556 assay Methods 0.000 description 18
- 229940093499 ethyl acetate Drugs 0.000 description 18
- 150000003254 radicals Chemical class 0.000 description 18
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 17
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 17
- 229910000027 potassium carbonate Inorganic materials 0.000 description 17
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 16
- 150000001412 amines Chemical class 0.000 description 16
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 16
- 229910052757 nitrogen Inorganic materials 0.000 description 15
- 239000012038 nucleophile Substances 0.000 description 15
- 108091000080 Phosphotransferase Proteins 0.000 description 14
- 229910052681 coesite Inorganic materials 0.000 description 14
- 229910052906 cristobalite Inorganic materials 0.000 description 14
- 239000000543 intermediate Substances 0.000 description 14
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 14
- 102000020233 phosphotransferase Human genes 0.000 description 14
- 239000007858 starting material Substances 0.000 description 14
- 229910052682 stishovite Inorganic materials 0.000 description 14
- 229910052905 tridymite Inorganic materials 0.000 description 14
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 13
- 238000001816 cooling Methods 0.000 description 13
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 12
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 12
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 12
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 239000012267 brine Substances 0.000 description 12
- 125000000623 heterocyclic group Chemical group 0.000 description 12
- 239000003112 inhibitor Substances 0.000 description 12
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 12
- 108010087686 src-Family Kinases Proteins 0.000 description 12
- 102000009076 src-Family Kinases Human genes 0.000 description 12
- 239000000758 substrate Substances 0.000 description 12
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 11
- 230000005764 inhibitory process Effects 0.000 description 11
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 11
- 125000004433 nitrogen atom Chemical group N* 0.000 description 11
- 238000000746 purification Methods 0.000 description 11
- 238000010898 silica gel chromatography Methods 0.000 description 11
- 229910052938 sodium sulfate Inorganic materials 0.000 description 11
- NVSLDBRVXOXWQH-UHFFFAOYSA-N 1h-furo[3,2-d]pyrimidin-2-one Chemical class N1C(=O)N=CC2=C1C=CO2 NVSLDBRVXOXWQH-UHFFFAOYSA-N 0.000 description 10
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 10
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Substances IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 description 10
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 10
- 239000003480 eluent Substances 0.000 description 10
- 238000009472 formulation Methods 0.000 description 10
- 239000000843 powder Substances 0.000 description 10
- 125000006239 protecting group Chemical group 0.000 description 10
- 239000000725 suspension Substances 0.000 description 10
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 9
- 125000004432 carbon atom Chemical group C* 0.000 description 9
- 239000003153 chemical reaction reagent Substances 0.000 description 9
- 125000005843 halogen group Chemical group 0.000 description 9
- 239000007924 injection Substances 0.000 description 9
- 238000002347 injection Methods 0.000 description 9
- 230000008569 process Effects 0.000 description 9
- 150000003230 pyrimidines Chemical class 0.000 description 9
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 8
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 8
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 8
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 8
- 239000007788 liquid Substances 0.000 description 8
- 239000000741 silica gel Substances 0.000 description 8
- 229910002027 silica gel Inorganic materials 0.000 description 8
- 229960001866 silicon dioxide Drugs 0.000 description 8
- 239000011734 sodium Substances 0.000 description 8
- 235000011152 sodium sulphate Nutrition 0.000 description 8
- ZKHQWZAMYRWXGA-UHFFFAOYSA-N Adenosine triphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O ZKHQWZAMYRWXGA-UHFFFAOYSA-N 0.000 description 7
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 7
- 229940024606 amino acid Drugs 0.000 description 7
- 235000001014 amino acid Nutrition 0.000 description 7
- 239000002552 dosage form Substances 0.000 description 7
- 229940043355 kinase inhibitor Drugs 0.000 description 7
- 125000002911 monocyclic heterocycle group Chemical group 0.000 description 7
- 239000012071 phase Substances 0.000 description 7
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 7
- 238000002953 preparative HPLC Methods 0.000 description 7
- 230000004044 response Effects 0.000 description 7
- 230000001225 therapeutic effect Effects 0.000 description 7
- 210000001519 tissue Anatomy 0.000 description 7
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 7
- BQOIOXRQQPRNCO-UHFFFAOYSA-N (2-chloro-2-nitroethenyl)benzene Chemical compound [O-][N+](=O)C(Cl)=CC1=CC=CC=C1 BQOIOXRQQPRNCO-UHFFFAOYSA-N 0.000 description 6
- JIZBWFUVJZONQI-UHFFFAOYSA-N 4-chloro-5-phenyl-6-[4-(2-pyrrolidin-1-ylethoxy)phenyl]furo[2,3-d]pyrimidine Chemical compound C1=2C(Cl)=NC=NC=2OC(C=2C=CC(OCCN3CCCC3)=CC=2)=C1C1=CC=CC=C1 JIZBWFUVJZONQI-UHFFFAOYSA-N 0.000 description 6
- AEOZQAQEYHKPNA-UHFFFAOYSA-N 5-phenyl-3h-furo[2,3-d]pyrimidin-4-one Chemical compound C1=2C(=O)NC=NC=2OC=C1C1=CC=CC=C1 AEOZQAQEYHKPNA-UHFFFAOYSA-N 0.000 description 6
- QWVIMXNDWYRPLC-UHFFFAOYSA-N 5-phenyl-6-[4-(2-pyrrolidin-1-ylethoxy)phenyl]-3h-furo[2,3-d]pyrimidin-4-one Chemical compound C1=2C(=O)NC=NC=2OC(C=2C=CC(OCCN3CCCC3)=CC=2)=C1C1=CC=CC=C1 QWVIMXNDWYRPLC-UHFFFAOYSA-N 0.000 description 6
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 6
- 102000004127 Cytokines Human genes 0.000 description 6
- 108090000695 Cytokines Proteins 0.000 description 6
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 6
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 6
- 102000009490 IgG Receptors Human genes 0.000 description 6
- 108010073807 IgG Receptors Proteins 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- 239000004480 active ingredient Substances 0.000 description 6
- 125000003277 amino group Chemical group 0.000 description 6
- 229910052786 argon Inorganic materials 0.000 description 6
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 6
- 238000007796 conventional method Methods 0.000 description 6
- 125000004122 cyclic group Chemical group 0.000 description 6
- XHFGWHUWQXTGAT-UHFFFAOYSA-N dimethylamine hydrochloride Natural products CNC(C)C XHFGWHUWQXTGAT-UHFFFAOYSA-N 0.000 description 6
- IQDGSYLLQPDQDV-UHFFFAOYSA-N dimethylazanium;chloride Chemical compound Cl.CNC IQDGSYLLQPDQDV-UHFFFAOYSA-N 0.000 description 6
- 238000001914 filtration Methods 0.000 description 6
- 235000019253 formic acid Nutrition 0.000 description 6
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 6
- 210000000440 neutrophil Anatomy 0.000 description 6
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 6
- 239000003960 organic solvent Substances 0.000 description 6
- 125000004430 oxygen atom Chemical group O* 0.000 description 6
- 239000000546 pharmaceutical excipient Substances 0.000 description 6
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 6
- 238000012746 preparative thin layer chromatography Methods 0.000 description 6
- 125000004434 sulfur atom Chemical group 0.000 description 6
- 239000003826 tablet Substances 0.000 description 6
- DUFGYCAXVIUXIP-UHFFFAOYSA-N 4,6-dihydroxypyrimidine Chemical compound OC1=CC(O)=NC=N1 DUFGYCAXVIUXIP-UHFFFAOYSA-N 0.000 description 5
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 5
- SMEGBGKFGNNZTD-UHFFFAOYSA-N 6-iodo-5-phenyl-3h-furo[2,3-d]pyrimidin-4-one Chemical compound IC=1OC=2N=CNC(=O)C=2C=1C1=CC=CC=C1 SMEGBGKFGNNZTD-UHFFFAOYSA-N 0.000 description 5
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 5
- 102000004190 Enzymes Human genes 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 5
- 102000036243 Lymphocyte Specific Protein Tyrosine Kinase p56(lck) Human genes 0.000 description 5
- 108010002481 Lymphocyte Specific Protein Tyrosine Kinase p56(lck) Proteins 0.000 description 5
- 206010025323 Lymphomas Diseases 0.000 description 5
- 108091008874 T cell receptors Proteins 0.000 description 5
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 5
- 150000001299 aldehydes Chemical class 0.000 description 5
- 150000001413 amino acids Chemical class 0.000 description 5
- 210000000481 breast Anatomy 0.000 description 5
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 5
- 239000000872 buffer Substances 0.000 description 5
- 229910052799 carbon Inorganic materials 0.000 description 5
- 229910002092 carbon dioxide Inorganic materials 0.000 description 5
- 239000013058 crude material Substances 0.000 description 5
- 238000001514 detection method Methods 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 229940088598 enzyme Drugs 0.000 description 5
- JPTYCVXLSHMYJJ-UHFFFAOYSA-N furo[2,3-d]pyrimidin-2-amine Chemical class NC1=NC=C2C=COC2=N1 JPTYCVXLSHMYJJ-UHFFFAOYSA-N 0.000 description 5
- 210000005104 human peripheral blood lymphocyte Anatomy 0.000 description 5
- 239000005457 ice water Substances 0.000 description 5
- 208000032839 leukemia Diseases 0.000 description 5
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 5
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 5
- 208000024891 symptom Diseases 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 4
- NYDZWXUOHCWHGO-UHFFFAOYSA-N 2-chlorofuro[3,2-d]pyrimidine Chemical class ClC1=NC=C2OC=CC2=N1 NYDZWXUOHCWHGO-UHFFFAOYSA-N 0.000 description 4
- YMONVEHDBWPGOO-UHFFFAOYSA-N 4-bromo-5,6-diphenylfuro[2,3-d]pyrimidine Chemical compound C1=2C(Br)=NC=NC=2OC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 YMONVEHDBWPGOO-UHFFFAOYSA-N 0.000 description 4
- IXKZXFQULLZVQJ-UHFFFAOYSA-N 4-chloro-5-phenylfuro[2,3-d]pyrimidine Chemical compound C1=2C(Cl)=NC=NC=2OC=C1C1=CC=CC=C1 IXKZXFQULLZVQJ-UHFFFAOYSA-N 0.000 description 4
- URGCTJQEEYOJMX-UHFFFAOYSA-N 5,6-diphenyl-n-(thiolan-2-ylmethyl)furo[2,3-d]pyrimidin-4-amine Chemical compound C1CCSC1CNC(C1=2)=NC=NC=2OC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 URGCTJQEEYOJMX-UHFFFAOYSA-N 0.000 description 4
- PVHTVSJKENCGIS-UHFFFAOYSA-N 6-bromo-5-phenyl-3h-furo[2,3-d]pyrimidin-4-one Chemical compound BrC=1OC=2N=CNC(=O)C=2C=1C1=CC=CC=C1 PVHTVSJKENCGIS-UHFFFAOYSA-N 0.000 description 4
- 230000004544 DNA amplification Effects 0.000 description 4
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 4
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 4
- 230000004913 activation Effects 0.000 description 4
- 239000000654 additive Substances 0.000 description 4
- 150000005005 aminopyrimidines Chemical class 0.000 description 4
- 239000008346 aqueous phase Substances 0.000 description 4
- 150000001503 aryl iodides Chemical class 0.000 description 4
- 230000033228 biological regulation Effects 0.000 description 4
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 4
- DNPRVXJGNANVCZ-UHFFFAOYSA-N bromo(nitro)methane Chemical compound [O-][N+](=O)CBr DNPRVXJGNANVCZ-UHFFFAOYSA-N 0.000 description 4
- 229910000024 caesium carbonate Inorganic materials 0.000 description 4
- 239000002775 capsule Substances 0.000 description 4
- 230000003197 catalytic effect Effects 0.000 description 4
- 238000006555 catalytic reaction Methods 0.000 description 4
- HVWJATXZZQPVEL-UHFFFAOYSA-N furo[3,2-d]pyrimidin-4-amine Chemical compound NC1=NC=NC2=C1OC=C2 HVWJATXZZQPVEL-UHFFFAOYSA-N 0.000 description 4
- 230000014509 gene expression Effects 0.000 description 4
- 239000008187 granular material Substances 0.000 description 4
- 230000012010 growth Effects 0.000 description 4
- 238000002868 homogeneous time resolved fluorescence Methods 0.000 description 4
- 230000000670 limiting effect Effects 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 238000002156 mixing Methods 0.000 description 4
- 150000007524 organic acids Chemical class 0.000 description 4
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 4
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 4
- IPNPIHIZVLFAFP-UHFFFAOYSA-N phosphorus tribromide Chemical compound BrP(Br)Br IPNPIHIZVLFAFP-UHFFFAOYSA-N 0.000 description 4
- 230000026731 phosphorylation Effects 0.000 description 4
- 238000006366 phosphorylation reaction Methods 0.000 description 4
- NROKBHXJSPEDAR-UHFFFAOYSA-M potassium fluoride Chemical compound [F-].[K+] NROKBHXJSPEDAR-UHFFFAOYSA-M 0.000 description 4
- 238000011321 prophylaxis Methods 0.000 description 4
- 235000018102 proteins Nutrition 0.000 description 4
- 102000004169 proteins and genes Human genes 0.000 description 4
- 238000010926 purge Methods 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 238000006722 reduction reaction Methods 0.000 description 4
- 230000019491 signal transduction Effects 0.000 description 4
- 230000011664 signaling Effects 0.000 description 4
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 241000894007 species Species 0.000 description 4
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 4
- 230000009466 transformation Effects 0.000 description 4
- 238000001665 trituration Methods 0.000 description 4
- 239000008096 xylene Substances 0.000 description 4
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 3
- IFIDFYCBEGWGHX-UHFFFAOYSA-N (4-ethylpiperazin-1-yl)-(4-iodophenyl)methanone Chemical compound C1CN(CC)CCN1C(=O)C1=CC=C(I)C=C1 IFIDFYCBEGWGHX-UHFFFAOYSA-N 0.000 description 3
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 3
- QDXWHXMQRNDTKQ-UHFFFAOYSA-N 2-(4-bromo-2-fluorophenoxy)-n,n-diethylethanamine Chemical compound CCN(CC)CCOC1=CC=C(Br)C=C1F QDXWHXMQRNDTKQ-UHFFFAOYSA-N 0.000 description 3
- QNGBQONBZBPFQI-UHFFFAOYSA-N 2-amino-4-phenylfuran-3-carbonitrile Chemical compound N#CC1=C(N)OC=C1C1=CC=CC=C1 QNGBQONBZBPFQI-UHFFFAOYSA-N 0.000 description 3
- WCFLBCQUEZKJJD-UHFFFAOYSA-N 2-aminofuran-3-carbonitrile Chemical class NC=1OC=CC=1C#N WCFLBCQUEZKJJD-UHFFFAOYSA-N 0.000 description 3
- DEQMPWMWIVEOSX-UHFFFAOYSA-N 2-chlorofuro[2,3-d]pyrimidine Chemical class ClC1=NC=C2C=COC2=N1 DEQMPWMWIVEOSX-UHFFFAOYSA-N 0.000 description 3
- 238000010600 3H thymidine incorporation assay Methods 0.000 description 3
- GBRNPUKAAYSNOS-UHFFFAOYSA-N 3h-furo[2,3-d]pyrimidin-4-one Chemical compound O=C1NC=NC2=C1C=CO2 GBRNPUKAAYSNOS-UHFFFAOYSA-N 0.000 description 3
- IZHAGYQFIQOHTB-UHFFFAOYSA-N 4-chloro-6-iodo-5-phenylfuro[2,3-d]pyrimidine Chemical compound C1=2C(Cl)=NC=NC=2OC(I)=C1C1=CC=CC=C1 IZHAGYQFIQOHTB-UHFFFAOYSA-N 0.000 description 3
- PEFVPWSISZURQJ-UHFFFAOYSA-N 5,6-diphenyl-3h-furo[2,3-d]pyrimidin-4-one Chemical compound C1=2C(=O)NC=NC=2OC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 PEFVPWSISZURQJ-UHFFFAOYSA-N 0.000 description 3
- APAOWMPUWGJZFS-UHFFFAOYSA-N 5,6-diphenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1=2C(N)=NC=NC=2OC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 APAOWMPUWGJZFS-UHFFFAOYSA-N 0.000 description 3
- XFJBGINZIMNZBW-CRAIPNDOSA-N 5-chloro-2-[4-[(1r,2s)-2-[2-(5-methylsulfonylpyridin-2-yl)oxyethyl]cyclopropyl]piperidin-1-yl]pyrimidine Chemical compound N1=CC(S(=O)(=O)C)=CC=C1OCC[C@H]1[C@@H](C2CCN(CC2)C=2N=CC(Cl)=CN=2)C1 XFJBGINZIMNZBW-CRAIPNDOSA-N 0.000 description 3
- GHCATONDICLHRB-UHFFFAOYSA-N 5-phenyl-n-propan-2-ylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1=2C(NC(C)C)=NC=NC=2OC=C1C1=CC=CC=C1 GHCATONDICLHRB-UHFFFAOYSA-N 0.000 description 3
- QXOGBLCWQACHGH-UHFFFAOYSA-N 6-bromo-5-phenyl-n-propan-2-ylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1=2C(NC(C)C)=NC=NC=2OC(Br)=C1C1=CC=CC=C1 QXOGBLCWQACHGH-UHFFFAOYSA-N 0.000 description 3
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 3
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 3
- 229940126657 Compound 17 Drugs 0.000 description 3
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 3
- 108010052343 Gastrins Proteins 0.000 description 3
- 208000017604 Hodgkin disease Diseases 0.000 description 3
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 3
- 102000009438 IgE Receptors Human genes 0.000 description 3
- 108010073816 IgE Receptors Proteins 0.000 description 3
- 102000000588 Interleukin-2 Human genes 0.000 description 3
- 108010002350 Interleukin-2 Proteins 0.000 description 3
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 3
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 3
- 239000007832 Na2SO4 Substances 0.000 description 3
- 229910020667 PBr3 Inorganic materials 0.000 description 3
- 229910019213 POCl3 Inorganic materials 0.000 description 3
- 229920001213 Polysorbate 20 Polymers 0.000 description 3
- 102000001253 Protein Kinase Human genes 0.000 description 3
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 3
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 3
- 125000002252 acyl group Chemical group 0.000 description 3
- 239000002671 adjuvant Substances 0.000 description 3
- 239000000443 aerosol Substances 0.000 description 3
- 125000003282 alkyl amino group Chemical group 0.000 description 3
- 229940045714 alkyl sulfonate alkylating agent Drugs 0.000 description 3
- 150000008052 alkyl sulfonates Chemical class 0.000 description 3
- 229940100198 alkylating agent Drugs 0.000 description 3
- 239000002168 alkylating agent Substances 0.000 description 3
- 229910021529 ammonia Inorganic materials 0.000 description 3
- 230000003321 amplification Effects 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 238000010171 animal model Methods 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 229940125773 compound 10 Drugs 0.000 description 3
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 3
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 238000013461 design Methods 0.000 description 3
- POCFBDFTJMJWLG-UHFFFAOYSA-N dihydrosinapic acid methyl ester Natural products COC(=O)CCC1=CC(OC)=C(O)C(OC)=C1 POCFBDFTJMJWLG-UHFFFAOYSA-N 0.000 description 3
- 238000001704 evaporation Methods 0.000 description 3
- 230000008020 evaporation Effects 0.000 description 3
- 125000001153 fluoro group Chemical group F* 0.000 description 3
- 150000004820 halides Chemical class 0.000 description 3
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 3
- 238000007327 hydrogenolysis reaction Methods 0.000 description 3
- YLMAHDNUQAMNNX-UHFFFAOYSA-N imatinib methanesulfonate Chemical compound CS(O)(=O)=O.C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 YLMAHDNUQAMNNX-UHFFFAOYSA-N 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 3
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 3
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 3
- 210000002540 macrophage Anatomy 0.000 description 3
- 235000019341 magnesium sulphate Nutrition 0.000 description 3
- CUONGYYJJVDODC-UHFFFAOYSA-N malononitrile Chemical compound N#CCC#N CUONGYYJJVDODC-UHFFFAOYSA-N 0.000 description 3
- 201000001441 melanoma Diseases 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- 239000003607 modifier Substances 0.000 description 3
- 210000001616 monocyte Anatomy 0.000 description 3
- 230000035772 mutation Effects 0.000 description 3
- 238000003199 nucleic acid amplification method Methods 0.000 description 3
- 125000002971 oxazolyl group Chemical group 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 3
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 3
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 3
- 235000011056 potassium acetate Nutrition 0.000 description 3
- 239000011698 potassium fluoride Substances 0.000 description 3
- 108090000765 processed proteins & peptides Proteins 0.000 description 3
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 3
- 230000000069 prophylactic effect Effects 0.000 description 3
- 108060006633 protein kinase Proteins 0.000 description 3
- 125000003373 pyrazinyl group Chemical group 0.000 description 3
- 125000003226 pyrazolyl group Chemical group 0.000 description 3
- 239000005297 pyrex Substances 0.000 description 3
- 230000001105 regulatory effect Effects 0.000 description 3
- 230000002441 reversible effect Effects 0.000 description 3
- 238000006884 silylation reaction Methods 0.000 description 3
- 239000007921 spray Substances 0.000 description 3
- 239000003381 stabilizer Substances 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- 239000000829 suppository Substances 0.000 description 3
- 239000004094 surface-active agent Substances 0.000 description 3
- 230000004083 survival effect Effects 0.000 description 3
- 239000000375 suspending agent Substances 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- ASHOGAMAPGRXHX-UHFFFAOYSA-N tert-butyl n-(2,5-dichloropentyl)carbamate Chemical compound CC(C)(C)OC(=O)NCC(Cl)CCCCl ASHOGAMAPGRXHX-UHFFFAOYSA-N 0.000 description 3
- 229940124597 therapeutic agent Drugs 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- 125000003396 thiol group Chemical group [H]S* 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- 150000003738 xylenes Chemical class 0.000 description 3
- VHAQBOUVXRDEQM-UHFFFAOYSA-N (1-oxothiolan-2-yl)methylazanium;2,2,2-trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F.[NH3+]CC1CCCS1=O VHAQBOUVXRDEQM-UHFFFAOYSA-N 0.000 description 2
- WWTBZEKOSBFBEM-SPWPXUSOSA-N (2s)-2-[[2-benzyl-3-[hydroxy-[(1r)-2-phenyl-1-(phenylmethoxycarbonylamino)ethyl]phosphoryl]propanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound N([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)O)C(=O)C(CP(O)(=O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1C=CC=CC=1)CC1=CC=CC=C1 WWTBZEKOSBFBEM-SPWPXUSOSA-N 0.000 description 2
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 2
- HUWSZNZAROKDRZ-RRLWZMAJSA-N (3r,4r)-3-azaniumyl-5-[[(2s,3r)-1-[(2s)-2,3-dicarboxypyrrolidin-1-yl]-3-methyl-1-oxopentan-2-yl]amino]-5-oxo-4-sulfanylpentane-1-sulfonate Chemical compound OS(=O)(=O)CC[C@@H](N)[C@@H](S)C(=O)N[C@@H]([C@H](C)CC)C(=O)N1CCC(C(O)=O)[C@H]1C(O)=O HUWSZNZAROKDRZ-RRLWZMAJSA-N 0.000 description 2
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 2
- DHBXNPKRAUYBTH-UHFFFAOYSA-N 1,1-ethanedithiol Chemical compound CC(S)S DHBXNPKRAUYBTH-UHFFFAOYSA-N 0.000 description 2
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 2
- WGCYRFWNGRMRJA-UHFFFAOYSA-N 1-ethylpiperazine Chemical compound CCN1CCNCC1 WGCYRFWNGRMRJA-UHFFFAOYSA-N 0.000 description 2
- YIYBRXKMQFDHSM-UHFFFAOYSA-N 2,2'-Dihydroxybenzophenone Chemical compound OC1=CC=CC=C1C(=O)C1=CC=CC=C1O YIYBRXKMQFDHSM-UHFFFAOYSA-N 0.000 description 2
- HJKLEAOXCZIMPI-UHFFFAOYSA-N 2,2-diethoxyethanamine Chemical compound CCOC(CN)OCC HJKLEAOXCZIMPI-UHFFFAOYSA-N 0.000 description 2
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 2
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 2
- MCSXGCZMEPXKIW-UHFFFAOYSA-N 3-hydroxy-4-[(4-methyl-2-nitrophenyl)diazenyl]-N-(3-nitrophenyl)naphthalene-2-carboxamide Chemical compound Cc1ccc(N=Nc2c(O)c(cc3ccccc23)C(=O)Nc2cccc(c2)[N+]([O-])=O)c(c1)[N+]([O-])=O MCSXGCZMEPXKIW-UHFFFAOYSA-N 0.000 description 2
- PNGBKTYRDRRBHW-UHFFFAOYSA-N 5-phenyl-n-(2-piperidin-4-ylethyl)-6-[4-(2-pyrrolidin-1-ylethoxy)phenyl]furo[2,3-d]pyrimidin-4-amine Chemical compound N=1C=NC=2OC(C=3C=CC(OCCN4CCCC4)=CC=3)=C(C=3C=CC=CC=3)C=2C=1NCCC1CCNCC1 PNGBKTYRDRRBHW-UHFFFAOYSA-N 0.000 description 2
- SSVDSZIWJBBCNN-UHFFFAOYSA-N 5-phenyl-n-propan-2-yl-6-[4-(2-pyrrolidin-1-ylethoxy)phenyl]furo[2,3-d]pyrimidin-4-amine Chemical compound C1=2C(NC(C)C)=NC=NC=2OC(C=2C=CC(OCCN3CCCC3)=CC=2)=C1C1=CC=CC=C1 SSVDSZIWJBBCNN-UHFFFAOYSA-N 0.000 description 2
- RJLNLVYAPQUXHT-UHFFFAOYSA-N 6-(4-methylsulfonylphenyl)-n-(oxolan-2-ylmethyl)-5-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1=CC(S(=O)(=O)C)=CC=C1C1=C(C=2C=CC=CC=2)C2=C(NCC3OCCC3)N=CN=C2O1 RJLNLVYAPQUXHT-UHFFFAOYSA-N 0.000 description 2
- JYAWXCKBAGNTPD-UHFFFAOYSA-N 6-bromo-5-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1=2C(N)=NC=NC=2OC(Br)=C1C1=CC=CC=C1 JYAWXCKBAGNTPD-UHFFFAOYSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical class C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 2
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 2
- KCBAMQOKOLXLOX-BSZYMOERSA-N CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O Chemical compound CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O KCBAMQOKOLXLOX-BSZYMOERSA-N 0.000 description 2
- 229940126639 Compound 33 Drugs 0.000 description 2
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 2
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 2
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 2
- 102000018898 GTPase-Activating Proteins Human genes 0.000 description 2
- 108091006094 GTPase-accelerating proteins Proteins 0.000 description 2
- 102400000921 Gastrin Human genes 0.000 description 2
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 description 2
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 2
- 102000000589 Interleukin-1 Human genes 0.000 description 2
- 108010002352 Interleukin-1 Proteins 0.000 description 2
- 102000004890 Interleukin-8 Human genes 0.000 description 2
- 108090001007 Interleukin-8 Proteins 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- JLTDJTHDQAWBAV-UHFFFAOYSA-N N,N-dimethylaniline Chemical compound CN(C)C1=CC=CC=C1 JLTDJTHDQAWBAV-UHFFFAOYSA-N 0.000 description 2
- 102000048850 Neoplasm Genes Human genes 0.000 description 2
- 108700019961 Neoplasm Genes Proteins 0.000 description 2
- 235000019502 Orange oil Nutrition 0.000 description 2
- 208000002193 Pain Diseases 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- WHBMMWSBFZVSSR-UHFFFAOYSA-N R3HBA Natural products CC(O)CC(O)=O WHBMMWSBFZVSSR-UHFFFAOYSA-N 0.000 description 2
- 206010039491 Sarcoma Diseases 0.000 description 2
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 2
- PNUZDKCDAWUEGK-CYZMBNFOSA-N Sitafloxacin Chemical compound C([C@H]1N)N(C=2C(=C3C(C(C(C(O)=O)=CN3[C@H]3[C@H](C3)F)=O)=CC=2F)Cl)CC11CC1 PNUZDKCDAWUEGK-CYZMBNFOSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- 229940123237 Taxane Drugs 0.000 description 2
- 208000024313 Testicular Neoplasms Diseases 0.000 description 2
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 2
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 2
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 2
- 229940122803 Vinca alkaloid Drugs 0.000 description 2
- YNOGYQAEJGADFJ-YFKPBYRVSA-N [(2s)-oxolan-2-yl]methanamine Chemical compound NC[C@@H]1CCCO1 YNOGYQAEJGADFJ-YFKPBYRVSA-N 0.000 description 2
- CUQZAOZUFZWFPM-UHFFFAOYSA-N [4-(2-pyrrolidin-1-ylethoxy)phenyl]boronic acid Chemical compound C1=CC(B(O)O)=CC=C1OCCN1CCCC1 CUQZAOZUFZWFPM-UHFFFAOYSA-N 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 2
- 230000000996 additive effect Effects 0.000 description 2
- 150000004703 alkoxides Chemical group 0.000 description 2
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 2
- 201000009961 allergic asthma Diseases 0.000 description 2
- 230000000735 allogeneic effect Effects 0.000 description 2
- 150000001408 amides Chemical group 0.000 description 2
- 239000000908 ammonium hydroxide Substances 0.000 description 2
- 230000003110 anti-inflammatory effect Effects 0.000 description 2
- 239000000427 antigen Substances 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- 239000012223 aqueous fraction Substances 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 238000010533 azeotropic distillation Methods 0.000 description 2
- 210000003651 basophil Anatomy 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 2
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- 230000031709 bromination Effects 0.000 description 2
- 238000005893 bromination reaction Methods 0.000 description 2
- 150000001649 bromium compounds Chemical class 0.000 description 2
- 125000001246 bromo group Chemical group Br* 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 238000000423 cell based assay Methods 0.000 description 2
- 230000010261 cell growth Effects 0.000 description 2
- 238000001516 cell proliferation assay Methods 0.000 description 2
- AOXOCDRNSPFDPE-UKEONUMOSA-N chembl413654 Chemical compound C([C@H](C(=O)NCC(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](C)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]1N(CCC1)C(=O)CNC(=O)[C@@H](N)CCC(O)=O)C1=CC=C(O)C=C1 AOXOCDRNSPFDPE-UKEONUMOSA-N 0.000 description 2
- 229960004630 chlorambucil Drugs 0.000 description 2
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 208000029742 colonic neoplasm Diseases 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 229940126208 compound 22 Drugs 0.000 description 2
- 229940125833 compound 23 Drugs 0.000 description 2
- 229940126214 compound 3 Drugs 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 238000006880 cross-coupling reaction Methods 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 125000004093 cyano group Chemical group *C#N 0.000 description 2
- 229960003901 dacarbazine Drugs 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 2
- 125000004982 dihaloalkyl group Chemical group 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- 230000001747 exhibiting effect Effects 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 150000004665 fatty acids Chemical class 0.000 description 2
- 229960002949 fluorouracil Drugs 0.000 description 2
- 150000002224 folic acids Chemical class 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 239000003365 glass fiber Substances 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- 208000014829 head and neck neoplasm Diseases 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 210000003630 histaminocyte Anatomy 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 230000036571 hydration Effects 0.000 description 2
- 238000006703 hydration reaction Methods 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 150000003949 imides Chemical group 0.000 description 2
- 150000002466 imines Chemical group 0.000 description 2
- 210000002865 immune cell Anatomy 0.000 description 2
- 230000028993 immune response Effects 0.000 description 2
- 239000007943 implant Substances 0.000 description 2
- 238000011065 in-situ storage Methods 0.000 description 2
- 230000001965 increasing effect Effects 0.000 description 2
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 2
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- XKTZWUACRZHVAN-VADRZIEHSA-N interleukin-8 Chemical compound C([C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@@H](NC(C)=O)CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N[C@@H](CCSC)C(=O)N1[C@H](CCC1)C(=O)N1[C@H](CCC1)C(=O)N[C@@H](C)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC=1C=CC(O)=CC=1)C(=O)N[C@H](CO)C(=O)N1[C@H](CCC1)C(N)=O)C1=CC=CC=C1 XKTZWUACRZHVAN-VADRZIEHSA-N 0.000 description 2
- 229940096397 interleukin-8 Drugs 0.000 description 2
- 230000031146 intracellular signal transduction Effects 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 150000004694 iodide salts Chemical class 0.000 description 2
- 125000002346 iodo group Chemical group I* 0.000 description 2
- 208000028867 ischemia Diseases 0.000 description 2
- QWTDNUCVQCZILF-UHFFFAOYSA-N isopentane Chemical compound CCC(C)C QWTDNUCVQCZILF-UHFFFAOYSA-N 0.000 description 2
- 125000005956 isoquinolyl group Chemical group 0.000 description 2
- 125000000842 isoxazolyl group Chemical group 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- 238000000021 kinase assay Methods 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 2
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 239000002207 metabolite Substances 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 229960004857 mitomycin Drugs 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- HHQJWDKIRXRTLS-UHFFFAOYSA-N n'-bromobutanediamide Chemical compound NC(=O)CCC(=O)NBr HHQJWDKIRXRTLS-UHFFFAOYSA-N 0.000 description 2
- BBODICHFZAMCQW-UHFFFAOYSA-N n-[2-(2,6-dimethylmorpholin-4-yl)ethyl]-5,6-diphenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1C(C)OC(C)CN1CCNC1=NC=NC2=C1C(C=1C=CC=CC=1)=C(C=1C=CC=CC=1)O2 BBODICHFZAMCQW-UHFFFAOYSA-N 0.000 description 2
- XWQHDEYBBSQMBV-UHFFFAOYSA-N n-[4-[4-(oxolan-2-ylmethylamino)-5-phenylfuro[2,3-d]pyrimidin-6-yl]phenyl]acetamide Chemical compound C1=CC(NC(=O)C)=CC=C1C1=C(C=2C=CC=CC=2)C2=C(NCC3OCCC3)N=CN=C2O1 XWQHDEYBBSQMBV-UHFFFAOYSA-N 0.000 description 2
- 150000002825 nitriles Chemical group 0.000 description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 2
- 230000000269 nucleophilic effect Effects 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 239000010502 orange oil Substances 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 230000002018 overexpression Effects 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 125000004043 oxo group Chemical group O=* 0.000 description 2
- FDMKROMOVVBUIJ-UHFFFAOYSA-N oxolan-2-ylmethyl 4-methylbenzenesulfonate Chemical compound C1=CC(C)=CC=C1S(=O)(=O)OCC1OCCC1 FDMKROMOVVBUIJ-UHFFFAOYSA-N 0.000 description 2
- 239000003002 pH adjusting agent Substances 0.000 description 2
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 125000006684 polyhaloalkyl group Polymers 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 235000003270 potassium fluoride Nutrition 0.000 description 2
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 2
- 229960004618 prednisone Drugs 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 230000009696 proliferative response Effects 0.000 description 2
- 239000003909 protein kinase inhibitor Substances 0.000 description 2
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 2
- 150000003212 purines Chemical class 0.000 description 2
- 125000002098 pyridazinyl group Chemical group 0.000 description 2
- 125000000168 pyrrolyl group Chemical group 0.000 description 2
- 125000005493 quinolyl group Chemical group 0.000 description 2
- 210000000664 rectum Anatomy 0.000 description 2
- 230000019254 respiratory burst Effects 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 230000028327 secretion Effects 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- ZGHLCBJZQLNUAZ-UHFFFAOYSA-N sodium sulfide nonahydrate Chemical compound O.O.O.O.O.O.O.O.O.[Na+].[Na+].[S-2] ZGHLCBJZQLNUAZ-UHFFFAOYSA-N 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 230000000638 stimulation Effects 0.000 description 2
- 239000012258 stirred mixture Substances 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 150000003457 sulfones Chemical class 0.000 description 2
- 150000003462 sulfoxides Chemical class 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 231100001274 therapeutic index Toxicity 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- 150000003573 thiols Chemical class 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 238000011200 topical administration Methods 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 125000001425 triazolyl group Chemical group 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- NHDIQVFFNDKAQU-UHFFFAOYSA-N tripropan-2-yl borate Chemical compound CC(C)OB(OC(C)C)OC(C)C NHDIQVFFNDKAQU-UHFFFAOYSA-N 0.000 description 2
- 229910052722 tritium Inorganic materials 0.000 description 2
- 230000004614 tumor growth Effects 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- VCGRFBXVSFAGGA-UHFFFAOYSA-N (1,1-dioxo-1,4-thiazinan-4-yl)-[6-[[3-(4-fluorophenyl)-5-methyl-1,2-oxazol-4-yl]methoxy]pyridin-3-yl]methanone Chemical compound CC=1ON=C(C=2C=CC(F)=CC=2)C=1COC(N=C1)=CC=C1C(=O)N1CCS(=O)(=O)CC1 VCGRFBXVSFAGGA-UHFFFAOYSA-N 0.000 description 1
- WBTBMWXOILXBJI-UHFFFAOYSA-N (1,1-dioxothiolan-2-yl)methanamine;2,2,2-trifluoroacetic acid Chemical compound [O-]C(=O)C(F)(F)F.[NH3+]CC1CCCS1(=O)=O WBTBMWXOILXBJI-UHFFFAOYSA-N 0.000 description 1
- ASGMFNBUXDJWJJ-JLCFBVMHSA-N (1R,3R)-3-[[3-bromo-1-[4-(5-methyl-1,3,4-thiadiazol-2-yl)phenyl]pyrazolo[3,4-d]pyrimidin-6-yl]amino]-N,1-dimethylcyclopentane-1-carboxamide Chemical compound BrC1=NN(C2=NC(=NC=C21)N[C@H]1C[C@@](CC1)(C(=O)NC)C)C1=CC=C(C=C1)C=1SC(=NN=1)C ASGMFNBUXDJWJJ-JLCFBVMHSA-N 0.000 description 1
- UAOUIVVJBYDFKD-XKCDOFEDSA-N (1R,9R,10S,11R,12R,15S,18S,21R)-10,11,21-trihydroxy-8,8-dimethyl-14-methylidene-4-(prop-2-enylamino)-20-oxa-5-thia-3-azahexacyclo[9.7.2.112,15.01,9.02,6.012,18]henicosa-2(6),3-dien-13-one Chemical compound C([C@@H]1[C@@H](O)[C@@]23C(C1=C)=O)C[C@H]2[C@]12C(N=C(NCC=C)S4)=C4CC(C)(C)[C@H]1[C@H](O)[C@]3(O)OC2 UAOUIVVJBYDFKD-XKCDOFEDSA-N 0.000 description 1
- QGVLYPPODPLXMB-UBTYZVCOSA-N (1aR,1bS,4aR,7aS,7bS,8R,9R,9aS)-4a,7b,9,9a-tetrahydroxy-3-(hydroxymethyl)-1,1,6,8-tetramethyl-1,1a,1b,4,4a,7a,7b,8,9,9a-decahydro-5H-cyclopropa[3,4]benzo[1,2-e]azulen-5-one Chemical compound C1=C(CO)C[C@]2(O)C(=O)C(C)=C[C@H]2[C@@]2(O)[C@H](C)[C@@H](O)[C@@]3(O)C(C)(C)[C@H]3[C@@H]21 QGVLYPPODPLXMB-UBTYZVCOSA-N 0.000 description 1
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 1
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 1
- IUSARDYWEPUTPN-OZBXUNDUSA-N (2r)-n-[(2s,3r)-4-[[(4s)-6-(2,2-dimethylpropyl)spiro[3,4-dihydropyrano[2,3-b]pyridine-2,1'-cyclobutane]-4-yl]amino]-3-hydroxy-1-[3-(1,3-thiazol-2-yl)phenyl]butan-2-yl]-2-methoxypropanamide Chemical compound C([C@H](NC(=O)[C@@H](C)OC)[C@H](O)CN[C@@H]1C2=CC(CC(C)(C)C)=CN=C2OC2(CCC2)C1)C(C=1)=CC=CC=1C1=NC=CS1 IUSARDYWEPUTPN-OZBXUNDUSA-N 0.000 description 1
- YJLIKUSWRSEPSM-WGQQHEPDSA-N (2r,3r,4s,5r)-2-[6-amino-8-[(4-phenylphenyl)methylamino]purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound C=1C=C(C=2C=CC=CC=2)C=CC=1CNC1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O YJLIKUSWRSEPSM-WGQQHEPDSA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- STBLNCCBQMHSRC-BATDWUPUSA-N (2s)-n-[(3s,4s)-5-acetyl-7-cyano-4-methyl-1-[(2-methylnaphthalen-1-yl)methyl]-2-oxo-3,4-dihydro-1,5-benzodiazepin-3-yl]-2-(methylamino)propanamide Chemical compound O=C1[C@@H](NC(=O)[C@H](C)NC)[C@H](C)N(C(C)=O)C2=CC(C#N)=CC=C2N1CC1=C(C)C=CC2=CC=CC=C12 STBLNCCBQMHSRC-BATDWUPUSA-N 0.000 description 1
- UDQTXCHQKHIQMH-KYGLGHNPSA-N (3ar,5s,6s,7r,7ar)-5-(difluoromethyl)-2-(ethylamino)-5,6,7,7a-tetrahydro-3ah-pyrano[3,2-d][1,3]thiazole-6,7-diol Chemical compound S1C(NCC)=N[C@H]2[C@@H]1O[C@H](C(F)F)[C@@H](O)[C@@H]2O UDQTXCHQKHIQMH-KYGLGHNPSA-N 0.000 description 1
- OOKAZRDERJMRCJ-KOUAFAAESA-N (3r)-7-[(1s,2s,4ar,6s,8s)-2,6-dimethyl-8-[(2s)-2-methylbutanoyl]oxy-1,2,4a,5,6,7,8,8a-octahydronaphthalen-1-yl]-3-hydroxy-5-oxoheptanoic acid Chemical compound C1=C[C@H](C)[C@H](CCC(=O)C[C@@H](O)CC(O)=O)C2[C@@H](OC(=O)[C@@H](C)CC)C[C@@H](C)C[C@@H]21 OOKAZRDERJMRCJ-KOUAFAAESA-N 0.000 description 1
- MPDDTAJMJCESGV-CTUHWIOQSA-M (3r,5r)-7-[2-(4-fluorophenyl)-5-[methyl-[(1r)-1-phenylethyl]carbamoyl]-4-propan-2-ylpyrazol-3-yl]-3,5-dihydroxyheptanoate Chemical compound C1([C@@H](C)N(C)C(=O)C2=NN(C(CC[C@@H](O)C[C@@H](O)CC([O-])=O)=C2C(C)C)C=2C=CC(F)=CC=2)=CC=CC=C1 MPDDTAJMJCESGV-CTUHWIOQSA-M 0.000 description 1
- MAYZWDRUFKUGGP-VIFPVBQESA-N (3s)-1-[5-tert-butyl-3-[(1-methyltetrazol-5-yl)methyl]triazolo[4,5-d]pyrimidin-7-yl]pyrrolidin-3-ol Chemical compound CN1N=NN=C1CN1C2=NC(C(C)(C)C)=NC(N3C[C@@H](O)CC3)=C2N=N1 MAYZWDRUFKUGGP-VIFPVBQESA-N 0.000 description 1
- VUEGYUOUAAVYAS-JGGQBBKZSA-N (6ar,9s,10ar)-9-(dimethylsulfamoylamino)-7-methyl-6,6a,8,9,10,10a-hexahydro-4h-indolo[4,3-fg]quinoline Chemical compound C1=CC([C@H]2C[C@@H](CN(C)[C@@H]2C2)NS(=O)(=O)N(C)C)=C3C2=CNC3=C1 VUEGYUOUAAVYAS-JGGQBBKZSA-N 0.000 description 1
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 description 1
- ZGYIXVSQHOKQRZ-COIATFDQSA-N (e)-n-[4-[3-chloro-4-(pyridin-2-ylmethoxy)anilino]-3-cyano-7-[(3s)-oxolan-3-yl]oxyquinolin-6-yl]-4-(dimethylamino)but-2-enamide Chemical compound N#CC1=CN=C2C=C(O[C@@H]3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC(C=C1Cl)=CC=C1OCC1=CC=CC=N1 ZGYIXVSQHOKQRZ-COIATFDQSA-N 0.000 description 1
- MOWXJLUYGFNTAL-DEOSSOPVSA-N (s)-[2-chloro-4-fluoro-5-(7-morpholin-4-ylquinazolin-4-yl)phenyl]-(6-methoxypyridazin-3-yl)methanol Chemical compound N1=NC(OC)=CC=C1[C@@H](O)C1=CC(C=2C3=CC=C(C=C3N=CN=2)N2CCOCC2)=C(F)C=C1Cl MOWXJLUYGFNTAL-DEOSSOPVSA-N 0.000 description 1
- TUCIOBMMDDOEMM-ZSOIEALJSA-N (z)-n-benzyl-2-cyano-3-(3,4-dihydroxyphenyl)prop-2-enamide Chemical compound C1=C(O)C(O)=CC=C1\C=C(\C#N)C(=O)NCC1=CC=CC=C1 TUCIOBMMDDOEMM-ZSOIEALJSA-N 0.000 description 1
- 0 *c(cc1)ccc1-c1c(C23C=CC=CC2C3)c(c(NCCCCCO)ncn2)c2[o]1 Chemical compound *c(cc1)ccc1-c1c(C23C=CC=CC2C3)c(c(NCCCCCO)ncn2)c2[o]1 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- 125000004504 1,2,4-oxadiazolyl group Chemical group 0.000 description 1
- 125000004514 1,2,4-thiadiazolyl group Chemical group 0.000 description 1
- 125000004506 1,2,5-oxadiazolyl group Chemical group 0.000 description 1
- 125000004517 1,2,5-thiadiazolyl group Chemical group 0.000 description 1
- 125000001781 1,3,4-oxadiazolyl group Chemical group 0.000 description 1
- 125000004520 1,3,4-thiadiazolyl group Chemical group 0.000 description 1
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical compound C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 description 1
- APWRZPQBPCAXFP-UHFFFAOYSA-N 1-(1-oxo-2H-isoquinolin-5-yl)-5-(trifluoromethyl)-N-[2-(trifluoromethyl)pyridin-4-yl]pyrazole-4-carboxamide Chemical compound O=C1NC=CC2=C(C=CC=C12)N1N=CC(=C1C(F)(F)F)C(=O)NC1=CC(=NC=C1)C(F)(F)F APWRZPQBPCAXFP-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- WZZBNLYBHUDSHF-DHLKQENFSA-N 1-[(3s,4s)-4-[8-(2-chloro-4-pyrimidin-2-yloxyphenyl)-7-fluoro-2-methylimidazo[4,5-c]quinolin-1-yl]-3-fluoropiperidin-1-yl]-2-hydroxyethanone Chemical compound CC1=NC2=CN=C3C=C(F)C(C=4C(=CC(OC=5N=CC=CN=5)=CC=4)Cl)=CC3=C2N1[C@H]1CCN(C(=O)CO)C[C@@H]1F WZZBNLYBHUDSHF-DHLKQENFSA-N 0.000 description 1
- ABDDQTDRAHXHOC-QMMMGPOBSA-N 1-[(7s)-5,7-dihydro-4h-thieno[2,3-c]pyran-7-yl]-n-methylmethanamine Chemical compound CNC[C@@H]1OCCC2=C1SC=C2 ABDDQTDRAHXHOC-QMMMGPOBSA-N 0.000 description 1
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 1
- 102100025573 1-alkyl-2-acetylglycerophosphocholine esterase Human genes 0.000 description 1
- QXOGPTXQGKQSJT-UHFFFAOYSA-N 1-amino-4-[4-(3,4-dimethylphenyl)sulfanylanilino]-9,10-dioxoanthracene-2-sulfonic acid Chemical compound Cc1ccc(Sc2ccc(Nc3cc(c(N)c4C(=O)c5ccccc5C(=O)c34)S(O)(=O)=O)cc2)cc1C QXOGPTXQGKQSJT-UHFFFAOYSA-N 0.000 description 1
- QXQAPNSHUJORMC-UHFFFAOYSA-N 1-chloro-4-propylbenzene Chemical compound CCCC1=CC=C(Cl)C=C1 QXQAPNSHUJORMC-UHFFFAOYSA-N 0.000 description 1
- VFWCMGCRMGJXDK-UHFFFAOYSA-N 1-chlorobutane Chemical compound CCCCCl VFWCMGCRMGJXDK-UHFFFAOYSA-N 0.000 description 1
- VUQPJRPDRDVQMN-UHFFFAOYSA-N 1-chlorooctadecane Chemical class CCCCCCCCCCCCCCCCCCCl VUQPJRPDRDVQMN-UHFFFAOYSA-N 0.000 description 1
- 229940044613 1-propanol Drugs 0.000 description 1
- VOXZDWNPVJITMN-ZBRFXRBCSA-N 17β-estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 VOXZDWNPVJITMN-ZBRFXRBCSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- FBCBUVHNVPZUTE-UHFFFAOYSA-N 1h-furo[3,2-d]pyrimidin-4-one Chemical compound O=C1NC=NC2=C1OC=C2 FBCBUVHNVPZUTE-UHFFFAOYSA-N 0.000 description 1
- QUKPALAWEPMWOS-UHFFFAOYSA-N 1h-pyrazolo[3,4-d]pyrimidine Chemical class C1=NC=C2C=NNC2=N1 QUKPALAWEPMWOS-UHFFFAOYSA-N 0.000 description 1
- UTQNKKSJPHTPBS-UHFFFAOYSA-N 2,2,2-trichloroethanone Chemical group ClC(Cl)(Cl)[C]=O UTQNKKSJPHTPBS-UHFFFAOYSA-N 0.000 description 1
- HRSUAAJEGDFWMV-UHFFFAOYSA-N 2,5-diazabicyclo[2.2.1]heptane;hydrobromide Chemical compound Br.C1NC2CNC1C2 HRSUAAJEGDFWMV-UHFFFAOYSA-N 0.000 description 1
- ATGSLQBVSZNJMT-UHFFFAOYSA-N 2,5-dichloropentan-1-amine;hydrochloride Chemical compound Cl.NCC(Cl)CCCCl ATGSLQBVSZNJMT-UHFFFAOYSA-N 0.000 description 1
- WGFNXGPBPIJYLI-UHFFFAOYSA-N 2,6-difluoro-3-[(3-fluorophenyl)sulfonylamino]-n-(3-methoxy-1h-pyrazolo[3,4-b]pyridin-5-yl)benzamide Chemical compound C1=C2C(OC)=NNC2=NC=C1NC(=O)C(C=1F)=C(F)C=CC=1NS(=O)(=O)C1=CC=CC(F)=C1 WGFNXGPBPIJYLI-UHFFFAOYSA-N 0.000 description 1
- FQMZXMVHHKXGTM-UHFFFAOYSA-N 2-(1-adamantyl)-n-[2-[2-(2-hydroxyethylamino)ethylamino]quinolin-5-yl]acetamide Chemical compound C1C(C2)CC(C3)CC2CC13CC(=O)NC1=CC=CC2=NC(NCCNCCO)=CC=C21 FQMZXMVHHKXGTM-UHFFFAOYSA-N 0.000 description 1
- YFTHTJAPODJVSL-UHFFFAOYSA-N 2-(1-benzothiophen-5-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(SC=C2)C2=C1 YFTHTJAPODJVSL-UHFFFAOYSA-N 0.000 description 1
- QJEBJKXTNSYBGE-UHFFFAOYSA-N 2-(2-heptadecyl-4,5-dihydroimidazol-1-yl)ethanol Chemical compound CCCCCCCCCCCCCCCCCC1=NCCN1CCO QJEBJKXTNSYBGE-UHFFFAOYSA-N 0.000 description 1
- SHUQIGHJQMJUHB-UHFFFAOYSA-N 2-(4-ethylpiperazin-1-yl)ethanamine Chemical compound CCN1CCN(CCN)CC1 SHUQIGHJQMJUHB-UHFFFAOYSA-N 0.000 description 1
- CKIIJIDEWWXQEA-UHFFFAOYSA-N 2-(bromomethyl)-1,3-dioxolane Chemical compound BrCC1OCCO1 CKIIJIDEWWXQEA-UHFFFAOYSA-N 0.000 description 1
- VOHILFSOWRNVJJ-UHFFFAOYSA-N 2-(bromomethyl)oxolane Chemical compound BrCC1CCCO1 VOHILFSOWRNVJJ-UHFFFAOYSA-N 0.000 description 1
- VTYRPALGSNDUQQ-UHFFFAOYSA-N 2-(dimethylamino)acetyl chloride Chemical compound CN(C)CC(Cl)=O VTYRPALGSNDUQQ-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 1
- WSOHMJMYFBEAFR-UHFFFAOYSA-N 2-[4-(4-chloro-5-phenylfuro[2,3-d]pyrimidin-6-yl)phenoxy]-n,n-dimethylethanamine Chemical compound C1=CC(OCCN(C)C)=CC=C1C1=C(C=2C=CC=CC=2)C2=C(Cl)N=CN=C2O1 WSOHMJMYFBEAFR-UHFFFAOYSA-N 0.000 description 1
- PYRKKGOKRMZEIT-UHFFFAOYSA-N 2-[6-(2-cyclopropylethoxy)-9-(2-hydroxy-2-methylpropyl)-1h-phenanthro[9,10-d]imidazol-2-yl]-5-fluorobenzene-1,3-dicarbonitrile Chemical compound C1=C2C3=CC(CC(C)(O)C)=CC=C3C=3NC(C=4C(=CC(F)=CC=4C#N)C#N)=NC=3C2=CC=C1OCCC1CC1 PYRKKGOKRMZEIT-UHFFFAOYSA-N 0.000 description 1
- FMKGJQHNYMWDFJ-CVEARBPZSA-N 2-[[4-(2,2-difluoropropoxy)pyrimidin-5-yl]methylamino]-4-[[(1R,4S)-4-hydroxy-3,3-dimethylcyclohexyl]amino]pyrimidine-5-carbonitrile Chemical compound FC(COC1=NC=NC=C1CNC1=NC=C(C(=N1)N[C@H]1CC([C@H](CC1)O)(C)C)C#N)(C)F FMKGJQHNYMWDFJ-CVEARBPZSA-N 0.000 description 1
- VOXBZHOHGGBLCQ-UHFFFAOYSA-N 2-amino-3,7-dihydropurine-6-thione;hydrate Chemical compound O.N1C(N)=NC(=S)C2=C1N=CN2.N1C(N)=NC(=S)C2=C1N=CN2 VOXBZHOHGGBLCQ-UHFFFAOYSA-N 0.000 description 1
- BPFMLOQVCKVCAT-UHFFFAOYSA-N 2-amino-4,5-diphenylfuran-3-carbonitrile Chemical compound N#CC1=C(N)OC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 BPFMLOQVCKVCAT-UHFFFAOYSA-N 0.000 description 1
- UWTDTJWGYWUILP-UHFFFAOYSA-N 2-amino-5-phenylfuran-3-carbonitrile Chemical compound N#CC1=C(N)OC(C=2C=CC=CC=2)=C1 UWTDTJWGYWUILP-UHFFFAOYSA-N 0.000 description 1
- AIQYELGLPVYNCY-UHFFFAOYSA-N 2-anilinoimidazo[4,5-h]isoquinolin-9-one Chemical class N=1C2=C3C(=O)N=CC=C3C=CC2=NC=1NC1=CC=CC=C1 AIQYELGLPVYNCY-UHFFFAOYSA-N 0.000 description 1
- YEIJOVUUFJDUTH-UHFFFAOYSA-N 2-bromo-1-(4-hydroxyphenyl)-2-phenylethanone Chemical compound C1=CC(O)=CC=C1C(=O)C(Br)C1=CC=CC=C1 YEIJOVUUFJDUTH-UHFFFAOYSA-N 0.000 description 1
- YRUBIFAMCRFPPC-UHFFFAOYSA-N 2-chloro-7-fluoro-1h-quinazolin-4-one Chemical compound N1C(Cl)=NC(=O)C=2C1=CC(F)=CC=2 YRUBIFAMCRFPPC-UHFFFAOYSA-N 0.000 description 1
- VKPPFDPXZWFDFA-UHFFFAOYSA-N 2-chloroethanamine Chemical class NCCCl VKPPFDPXZWFDFA-UHFFFAOYSA-N 0.000 description 1
- RAGSWDIQBBZLLL-UHFFFAOYSA-N 2-chloroethyl(diethyl)azanium;chloride Chemical compound Cl.CCN(CC)CCCl RAGSWDIQBBZLLL-UHFFFAOYSA-N 0.000 description 1
- TWJNQYPJQDRXPH-UHFFFAOYSA-N 2-cyanobenzohydrazide Chemical compound NNC(=O)C1=CC=CC=C1C#N TWJNQYPJQDRXPH-UHFFFAOYSA-N 0.000 description 1
- LQLJZSJKRYTKTP-UHFFFAOYSA-N 2-dimethylaminoethyl chloride hydrochloride Chemical compound Cl.CN(C)CCCl LQLJZSJKRYTKTP-UHFFFAOYSA-N 0.000 description 1
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- NEAQRZUHTPSBBM-UHFFFAOYSA-N 2-hydroxy-3,3-dimethyl-7-nitro-4h-isoquinolin-1-one Chemical compound C1=C([N+]([O-])=O)C=C2C(=O)N(O)C(C)(C)CC2=C1 NEAQRZUHTPSBBM-UHFFFAOYSA-N 0.000 description 1
- ZWVHTXAYIKBMEE-UHFFFAOYSA-N 2-hydroxyacetophenone Chemical compound OCC(=O)C1=CC=CC=C1 ZWVHTXAYIKBMEE-UHFFFAOYSA-N 0.000 description 1
- JXPDNDHCMMOJPC-UHFFFAOYSA-N 2-hydroxybutanedinitrile Chemical compound N#CC(O)CC#N JXPDNDHCMMOJPC-UHFFFAOYSA-N 0.000 description 1
- JSBBMMHTNIUVTC-UHFFFAOYSA-N 2-hydroxyethanone Chemical class OC[C]=O JSBBMMHTNIUVTC-UHFFFAOYSA-N 0.000 description 1
- WMPPDTMATNBGJN-UHFFFAOYSA-N 2-phenylethylbromide Chemical class BrCCC1=CC=CC=C1 WMPPDTMATNBGJN-UHFFFAOYSA-N 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 description 1
- HCDMJFOHIXMBOV-UHFFFAOYSA-N 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-4,7-dihydropyrrolo[4,5]pyrido[1,2-d]pyrimidin-2-one Chemical compound C=1C2=C3N(CC)C(=O)N(C=4C(=C(OC)C=C(OC)C=4F)F)CC3=CN=C2NC=1CN1CCOCC1 HCDMJFOHIXMBOV-UHFFFAOYSA-N 0.000 description 1
- BYHQTRFJOGIQAO-GOSISDBHSA-N 3-(4-bromophenyl)-8-[(2R)-2-hydroxypropyl]-1-[(3-methoxyphenyl)methyl]-1,3,8-triazaspiro[4.5]decan-2-one Chemical compound C[C@H](CN1CCC2(CC1)CN(C(=O)N2CC3=CC(=CC=C3)OC)C4=CC=C(C=C4)Br)O BYHQTRFJOGIQAO-GOSISDBHSA-N 0.000 description 1
- WNEODWDFDXWOLU-QHCPKHFHSA-N 3-[3-(hydroxymethyl)-4-[1-methyl-5-[[5-[(2s)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-oxopyridin-3-yl]pyridin-2-yl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one Chemical compound C([C@@H](N(CC1)C=2C=NC(NC=3C(N(C)C=C(C=3)C=3C(=C(N4C(C5=CC=6CC(C)(C)CC=6N5CC4)=O)N=CC=3)CO)=O)=CC=2)C)N1C1COC1 WNEODWDFDXWOLU-QHCPKHFHSA-N 0.000 description 1
- SRVXSISGYBMIHR-UHFFFAOYSA-N 3-[3-[3-(2-amino-2-oxoethyl)phenyl]-5-chlorophenyl]-3-(5-methyl-1,3-thiazol-2-yl)propanoic acid Chemical compound S1C(C)=CN=C1C(CC(O)=O)C1=CC(Cl)=CC(C=2C=C(CC(N)=O)C=CC=2)=C1 SRVXSISGYBMIHR-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- XKTYXVDYIKIYJP-UHFFFAOYSA-N 3h-dioxole Chemical compound C1OOC=C1 XKTYXVDYIKIYJP-UHFFFAOYSA-N 0.000 description 1
- YELMWJNXDALKFE-UHFFFAOYSA-N 3h-imidazo[4,5-f]quinoxaline Chemical class N1=CC=NC2=C(NC=N3)C3=CC=C21 YELMWJNXDALKFE-UHFFFAOYSA-N 0.000 description 1
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 1
- TXFPEBPIARQUIG-UHFFFAOYSA-N 4'-hydroxyacetophenone Chemical compound CC(=O)C1=CC=C(O)C=C1 TXFPEBPIARQUIG-UHFFFAOYSA-N 0.000 description 1
- WYFCZWSWFGJODV-MIANJLSGSA-N 4-[[(1s)-2-[(e)-3-[3-chloro-2-fluoro-6-(tetrazol-1-yl)phenyl]prop-2-enoyl]-5-(4-methyl-2-oxopiperazin-1-yl)-3,4-dihydro-1h-isoquinoline-1-carbonyl]amino]benzoic acid Chemical compound O=C1CN(C)CCN1C1=CC=CC2=C1CCN(C(=O)\C=C\C=1C(=CC=C(Cl)C=1F)N1N=NN=C1)[C@@H]2C(=O)NC1=CC=C(C(O)=O)C=C1 WYFCZWSWFGJODV-MIANJLSGSA-N 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- KVCQTKNUUQOELD-UHFFFAOYSA-N 4-amino-n-[1-(3-chloro-2-fluoroanilino)-6-methylisoquinolin-5-yl]thieno[3,2-d]pyrimidine-7-carboxamide Chemical compound N=1C=CC2=C(NC(=O)C=3C4=NC=NC(N)=C4SC=3)C(C)=CC=C2C=1NC1=CC=CC(Cl)=C1F KVCQTKNUUQOELD-UHFFFAOYSA-N 0.000 description 1
- RYVOZMPTISNBDB-UHFFFAOYSA-N 4-bromo-2-fluorophenol Chemical compound OC1=CC=C(Br)C=C1F RYVOZMPTISNBDB-UHFFFAOYSA-N 0.000 description 1
- FPEHSPQTNJXFEZ-UHFFFAOYSA-N 4-bromofuro[3,2-d]pyrimidine Chemical compound BrC1=NC=NC2=C1OC=C2 FPEHSPQTNJXFEZ-UHFFFAOYSA-N 0.000 description 1
- DZCDERJEEHCRHG-UHFFFAOYSA-N 4-chloro-5,6-diphenylfuro[2,3-d]pyrimidine Chemical compound C1=2C(Cl)=NC=NC=2OC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 DZCDERJEEHCRHG-UHFFFAOYSA-N 0.000 description 1
- HRBMECZYDBKLBF-UHFFFAOYSA-N 4-chloro-6-iodofuro[3,2-d]pyrimidine Chemical class ClC1=NC=NC2=C1OC(I)=C2 HRBMECZYDBKLBF-UHFFFAOYSA-N 0.000 description 1
- WJFATWIQZZNARY-UHFFFAOYSA-N 4-chloro-6-phenylthieno[2,3-d]pyrimidine Chemical compound C=1C=2C(Cl)=NC=NC=2SC=1C1=CC=CC=C1 WJFATWIQZZNARY-UHFFFAOYSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- HVCNXQOWACZAFN-UHFFFAOYSA-N 4-ethylmorpholine Chemical compound CCN1CCOCC1 HVCNXQOWACZAFN-UHFFFAOYSA-N 0.000 description 1
- 125000005274 4-hydroxybenzoic acid group Chemical group 0.000 description 1
- POXFXTSTVWDWIR-UHFFFAOYSA-N 4-iodobenzenesulfonyl chloride Chemical compound ClS(=O)(=O)C1=CC=C(I)C=C1 POXFXTSTVWDWIR-UHFFFAOYSA-N 0.000 description 1
- NJAKCIUOTIPYED-UHFFFAOYSA-N 4-iodobenzoyl chloride Chemical compound ClC(=O)C1=CC=C(I)C=C1 NJAKCIUOTIPYED-UHFFFAOYSA-N 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- DDFHBQSCUXNBSA-UHFFFAOYSA-N 5-(5-carboxythiophen-2-yl)thiophene-2-carboxylic acid Chemical compound S1C(C(=O)O)=CC=C1C1=CC=C(C(O)=O)S1 DDFHBQSCUXNBSA-UHFFFAOYSA-N 0.000 description 1
- IRPVABHDSJVBNZ-RTHVDDQRSA-N 5-[1-(cyclopropylmethyl)-5-[(1R,5S)-3-(oxetan-3-yl)-3-azabicyclo[3.1.0]hexan-6-yl]pyrazol-3-yl]-3-(trifluoromethyl)pyridin-2-amine Chemical compound C1=C(C(F)(F)F)C(N)=NC=C1C1=NN(CC2CC2)C(C2[C@@H]3CN(C[C@@H]32)C2COC2)=C1 IRPVABHDSJVBNZ-RTHVDDQRSA-N 0.000 description 1
- IDPUKCWIGUEADI-UHFFFAOYSA-N 5-[bis(2-chloroethyl)amino]uracil Chemical compound ClCCN(CCCl)C1=CNC(=O)NC1=O IDPUKCWIGUEADI-UHFFFAOYSA-N 0.000 description 1
- KJIBNAKAPXNJDX-UHFFFAOYSA-N 5-bromo-6-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound BrC=1C=2C(N)=NC=NC=2OC=1C1=CC=CC=C1 KJIBNAKAPXNJDX-UHFFFAOYSA-N 0.000 description 1
- GYCPLYCTMDTEPU-UHFFFAOYSA-N 5-bromopyrimidine Chemical compound BrC1=CN=CN=C1 GYCPLYCTMDTEPU-UHFFFAOYSA-N 0.000 description 1
- QZEZNWUIKTXXHQ-UHFFFAOYSA-N 5-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1=2C(N)=NC=NC=2OC=C1C1=CC=CC=C1 QZEZNWUIKTXXHQ-UHFFFAOYSA-N 0.000 description 1
- KCBWAFJCKVKYHO-UHFFFAOYSA-N 6-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-1-[[4-[1-propan-2-yl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methyl]pyrazolo[3,4-d]pyrimidine Chemical compound C1(CC1)C1=NC=NC(=C1C1=NC=C2C(=N1)N(N=C2)CC1=CC=C(C=C1)C=1N(C=C(N=1)C(F)(F)F)C(C)C)OC KCBWAFJCKVKYHO-UHFFFAOYSA-N 0.000 description 1
- WYWHKKSPHMUBEB-UHFFFAOYSA-N 6-Mercaptoguanine Natural products N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 1
- MQLTWEIFQLVJRT-UHFFFAOYSA-N 6-bromo-n-(oxolan-2-ylmethyl)-5-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C=12C(C=3C=CC=CC=3)=C(Br)OC2=NC=NC=1NCC1CCCO1 MQLTWEIFQLVJRT-UHFFFAOYSA-N 0.000 description 1
- AYMCPPFMMNEQDF-UHFFFAOYSA-N 6-iodo-1h-pyrimidin-2-one Chemical class IC=1C=CNC(=O)N=1 AYMCPPFMMNEQDF-UHFFFAOYSA-N 0.000 description 1
- KLUPXPVKDBPOMJ-UHFFFAOYSA-N 6-phenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C=1C=2C(N)=NC=NC=2OC=1C1=CC=CC=C1 KLUPXPVKDBPOMJ-UHFFFAOYSA-N 0.000 description 1
- WDYVUKGVKRZQNM-UHFFFAOYSA-N 6-phosphonohexylphosphonic acid Chemical compound OP(O)(=O)CCCCCCP(O)(O)=O WDYVUKGVKRZQNM-UHFFFAOYSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- CYJRNFFLTBEQSQ-UHFFFAOYSA-N 8-(3-methyl-1-benzothiophen-5-yl)-N-(4-methylsulfonylpyridin-3-yl)quinoxalin-6-amine Chemical compound CS(=O)(=O)C1=C(C=NC=C1)NC=1C=C2N=CC=NC2=C(C=1)C=1C=CC2=C(C(=CS2)C)C=1 CYJRNFFLTBEQSQ-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- ZKHQWZAMYRWXGA-KQYNXXCUSA-J ATP(4-) Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O)[C@@H](O)[C@H]1O ZKHQWZAMYRWXGA-KQYNXXCUSA-J 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 102100036409 Activated CDC42 kinase 1 Human genes 0.000 description 1
- 206010001052 Acute respiratory distress syndrome Diseases 0.000 description 1
- 208000036832 Adenocarcinoma of ovary Diseases 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- YCIPQJTZJGUXND-UHFFFAOYSA-N Aglaia odorata Alkaloid Natural products C1=CC(OC)=CC=C1C1(C(C=2C(=O)N3CCCC3=NC=22)C=3C=CC=CC=3)C2(O)C2=C(OC)C=C(OC)C=C2O1 YCIPQJTZJGUXND-UHFFFAOYSA-N 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 1
- 229910000013 Ammonium bicarbonate Inorganic materials 0.000 description 1
- 206010002383 Angina Pectoris Diseases 0.000 description 1
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 208000031104 Arterial Occlusive disease Diseases 0.000 description 1
- 108010024976 Asparaginase Proteins 0.000 description 1
- 208000031212 Autoimmune polyendocrinopathy Diseases 0.000 description 1
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 1
- 229910015845 BBr3 Inorganic materials 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- KHBQMWCZKVMBLN-UHFFFAOYSA-N Benzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=CC=C1 KHBQMWCZKVMBLN-UHFFFAOYSA-N 0.000 description 1
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- 208000006386 Bone Resorption Diseases 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- 201000006474 Brain Ischemia Diseases 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- JQUCWIWWWKZNCS-LESHARBVSA-N C(C1=CC=CC=C1)(=O)NC=1SC[C@H]2[C@@](N1)(CO[C@H](C2)C)C=2SC=C(N2)NC(=O)C2=NC=C(C=C2)OC(F)F Chemical compound C(C1=CC=CC=C1)(=O)NC=1SC[C@H]2[C@@](N1)(CO[C@H](C2)C)C=2SC=C(N2)NC(=O)C2=NC=C(C=C2)OC(F)F JQUCWIWWWKZNCS-LESHARBVSA-N 0.000 description 1
- ZOOHPRAHPAXTTJ-UHFFFAOYSA-N CC(C)NS(c(cc1)ccc1-c([o]c1ncnc(NCC2OCCC2)c11)c1-c1ccccc1)(=O)=O Chemical compound CC(C)NS(c(cc1)ccc1-c([o]c1ncnc(NCC2OCCC2)c11)c1-c1ccccc1)(=O)=O ZOOHPRAHPAXTTJ-UHFFFAOYSA-N 0.000 description 1
- IPWKHHSGDUIRAH-UHFFFAOYSA-N CC1(C)OB(B2OC(C)(C)C(C)(C)O2)OC1(C)C Chemical compound CC1(C)OB(B2OC(C)(C)C(C)(C)O2)OC1(C)C IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 1
- QQMGTMLRTXAZQO-UHFFFAOYSA-N CC1(CNc2c(c(-c3ccccc3)c(-c3ccccc3)[o]3)c3ncn2)OCCO1 Chemical compound CC1(CNc2c(c(-c3ccccc3)c(-c3ccccc3)[o]3)c3ncn2)OCCO1 QQMGTMLRTXAZQO-UHFFFAOYSA-N 0.000 description 1
- YWVPNRNZLVQXHB-UHFFFAOYSA-N CC=S(c1ccc(COC)cc1)=O Chemical compound CC=S(c1ccc(COC)cc1)=O YWVPNRNZLVQXHB-UHFFFAOYSA-N 0.000 description 1
- KDXMDMRSLUFCJY-UHFFFAOYSA-N CCC1SC(CNc2c(c(-c3ccccc3)c(-c3ccccc3)[o]3)c3ncn2)SC1 Chemical compound CCC1SC(CNc2c(c(-c3ccccc3)c(-c3ccccc3)[o]3)c3ncn2)SC1 KDXMDMRSLUFCJY-UHFFFAOYSA-N 0.000 description 1
- WTFPPCPYLZHSQU-UHFFFAOYSA-N CCN(CC1)CCN1C(c1ccc(B2OC(C)(C)C(C)(C)O2)cc1)=O Chemical compound CCN(CC1)CCN1C(c1ccc(B2OC(C)(C)C(C)(C)O2)cc1)=O WTFPPCPYLZHSQU-UHFFFAOYSA-N 0.000 description 1
- BQXUPNKLZNSUMC-YUQWMIPFSA-N CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 Chemical compound CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 BQXUPNKLZNSUMC-YUQWMIPFSA-N 0.000 description 1
- IEOPZUMPHCZMCS-UHFFFAOYSA-N COCC1OCCC1 Chemical compound COCC1OCCC1 IEOPZUMPHCZMCS-UHFFFAOYSA-N 0.000 description 1
- HACIXUFIWAKFLS-UHFFFAOYSA-N COCC1OCCCC1 Chemical compound COCC1OCCCC1 HACIXUFIWAKFLS-UHFFFAOYSA-N 0.000 description 1
- HJJVHNKOXFYNTJ-UHFFFAOYSA-N COCc1cnccc1 Chemical compound COCc1cnccc1 HJJVHNKOXFYNTJ-UHFFFAOYSA-N 0.000 description 1
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical group C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 1
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical group NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 201000009030 Carcinoma Diseases 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 1
- 102000011632 Caseins Human genes 0.000 description 1
- 108010076119 Caseins Proteins 0.000 description 1
- NQUVCRCCRXRJCK-UHFFFAOYSA-N Cc(cc1)ccc1C(Cl)=O Chemical compound Cc(cc1)ccc1C(Cl)=O NQUVCRCCRXRJCK-UHFFFAOYSA-N 0.000 description 1
- 206010008120 Cerebral ischaemia Diseases 0.000 description 1
- 229920002101 Chitin Polymers 0.000 description 1
- 229920001661 Chitosan Polymers 0.000 description 1
- JWBOIMRXGHLCPP-UHFFFAOYSA-N Chloditan Chemical compound C=1C=CC=C(Cl)C=1C(C(Cl)Cl)C1=CC=C(Cl)C=C1 JWBOIMRXGHLCPP-UHFFFAOYSA-N 0.000 description 1
- 208000006332 Choriocarcinoma Diseases 0.000 description 1
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 229940127007 Compound 39 Drugs 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- HTJDQJBWANPRPF-UHFFFAOYSA-N Cyclopropylamine Chemical compound NC1CC1 HTJDQJBWANPRPF-UHFFFAOYSA-N 0.000 description 1
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 102000003915 DNA Topoisomerases Human genes 0.000 description 1
- 108090000323 DNA Topoisomerases Proteins 0.000 description 1
- 108010092160 Dactinomycin Proteins 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 238000012286 ELISA Assay Methods 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- UOACKFBJUYNSLK-XRKIENNPSA-N Estradiol Cypionate Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H](C4=CC=C(O)C=C4CC3)CC[C@@]21C)C(=O)CCC1CCCC1 UOACKFBJUYNSLK-XRKIENNPSA-N 0.000 description 1
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 1
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 1
- 102000013446 GTP Phosphohydrolases Human genes 0.000 description 1
- 108091006109 GTPases Proteins 0.000 description 1
- DSLZVSRJTYRBFB-UHFFFAOYSA-N Galactaric acid Natural products OC(=O)C(O)C(O)C(O)C(O)C(O)=O DSLZVSRJTYRBFB-UHFFFAOYSA-N 0.000 description 1
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 208000015023 Graves' disease Diseases 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 229910004373 HOAc Inorganic materials 0.000 description 1
- 108010093488 His-His-His-His-His-His Proteins 0.000 description 1
- 101000928956 Homo sapiens Activated CDC42 kinase 1 Proteins 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- VSNHCAURESNICA-UHFFFAOYSA-N Hydroxyurea Chemical compound NC(=O)NO VSNHCAURESNICA-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102000004889 Interleukin-6 Human genes 0.000 description 1
- 108090001005 Interleukin-6 Proteins 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 1
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 1
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 108010000817 Leuprolide Proteins 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 1
- 241000544038 Luronium Species 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 102000029749 Microtubule Human genes 0.000 description 1
- 108091022875 Microtubule Proteins 0.000 description 1
- 229930192392 Mitomycin Natural products 0.000 description 1
- 208000034578 Multiple myelomas Diseases 0.000 description 1
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 1
- 208000014767 Myeloproliferative disease Diseases 0.000 description 1
- TUNFSRHWOTWDNC-UHFFFAOYSA-N Myristic acid Natural products CCCCCCCCCCCCCC(O)=O TUNFSRHWOTWDNC-UHFFFAOYSA-N 0.000 description 1
- 235000021360 Myristic acid Nutrition 0.000 description 1
- MGWBBZIEPDTTFE-UHFFFAOYSA-N N#Cc(cc1)ccc1-c([o]c1ncnc(NCC2OCCC2)c11)c1-c1ccccc1 Chemical compound N#Cc(cc1)ccc1-c([o]c1ncnc(NCC2OCCC2)c11)c1-c1ccccc1 MGWBBZIEPDTTFE-UHFFFAOYSA-N 0.000 description 1
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- AMQJEAYHLZJPGS-UHFFFAOYSA-N N-Pentanol Chemical compound CCCCCO AMQJEAYHLZJPGS-UHFFFAOYSA-N 0.000 description 1
- AYCPARAPKDAOEN-LJQANCHMSA-N N-[(1S)-2-(dimethylamino)-1-phenylethyl]-6,6-dimethyl-3-[(2-methyl-4-thieno[3,2-d]pyrimidinyl)amino]-1,4-dihydropyrrolo[3,4-c]pyrazole-5-carboxamide Chemical compound C1([C@H](NC(=O)N2C(C=3NN=C(NC=4C=5SC=CC=5N=C(C)N=4)C=3C2)(C)C)CN(C)C)=CC=CC=C1 AYCPARAPKDAOEN-LJQANCHMSA-N 0.000 description 1
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- 150000001204 N-oxides Chemical group 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- YRUNEMUFLAPIPR-QFIPXVFZSA-N O=C(c(cc1)ccc1-c([o]c1c2c(NC[C@H]3OCCC3)ncn1)c2-c1ccccc1)N1CCOCC1 Chemical compound O=C(c(cc1)ccc1-c([o]c1c2c(NC[C@H]3OCCC3)ncn1)c2-c1ccccc1)N1CCOCC1 YRUNEMUFLAPIPR-QFIPXVFZSA-N 0.000 description 1
- IDRGFNPZDVBSSE-UHFFFAOYSA-N OCCN1CCN(CC1)c1ccc(Nc2ncc3cccc(-c4cccc(NC(=O)C=C)c4)c3n2)c(F)c1F Chemical compound OCCN1CCN(CC1)c1ccc(Nc2ncc3cccc(-c4cccc(NC(=O)C=C)c4)c3n2)c(F)c1F IDRGFNPZDVBSSE-UHFFFAOYSA-N 0.000 description 1
- 102000043276 Oncogene Human genes 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 208000001132 Osteoporosis Diseases 0.000 description 1
- 206010061328 Ovarian epithelial cancer Diseases 0.000 description 1
- 208000030831 Peripheral arterial occlusive disease Diseases 0.000 description 1
- 206010035226 Plasma cell myeloma Diseases 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- HFVNWDWLWUCIHC-GUPDPFMOSA-N Prednimustine Chemical compound O=C([C@@]1(O)CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)[C@@H](O)C[C@@]21C)COC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 HFVNWDWLWUCIHC-GUPDPFMOSA-N 0.000 description 1
- 101710156978 Ras-like protein Proteins 0.000 description 1
- 208000015634 Rectal Neoplasms Diseases 0.000 description 1
- 208000013616 Respiratory Distress Syndrome Diseases 0.000 description 1
- 108091008118 SFKs Proteins 0.000 description 1
- 102000038012 SFKs Human genes 0.000 description 1
- 102000000395 SH3 domains Human genes 0.000 description 1
- 108050008861 SH3 domains Proteins 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-N Salicylic acid Natural products OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 1
- 206010041067 Small cell lung cancer Diseases 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- 230000006052 T cell proliferation Effects 0.000 description 1
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 1
- 206010057644 Testis cancer Diseases 0.000 description 1
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 1
- 208000007536 Thrombosis Diseases 0.000 description 1
- 101710183280 Topoisomerase Proteins 0.000 description 1
- 241000159243 Toxicodendron radicans Species 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 102000004243 Tubulin Human genes 0.000 description 1
- 108090000704 Tubulin Proteins 0.000 description 1
- 102000044209 Tumor Suppressor Genes Human genes 0.000 description 1
- 108700025716 Tumor Suppressor Genes Proteins 0.000 description 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 1
- 206010053648 Vascular occlusion Diseases 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- LXRZVMYMQHNYJB-UNXOBOICSA-N [(1R,2S,4R)-4-[[5-[4-[(1R)-7-chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methylthiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxycyclopentyl]methyl sulfamate Chemical compound CC1=C(C=C(S1)C(=O)C1=C(N[C@H]2C[C@H](O)[C@@H](COS(N)(=O)=O)C2)N=CN=C1)[C@@H]1NCCC2=C1C=C(Cl)C=C2 LXRZVMYMQHNYJB-UNXOBOICSA-N 0.000 description 1
- SPXSEZMVRJLHQG-XMMPIXPASA-N [(2R)-1-[[4-[(3-phenylmethoxyphenoxy)methyl]phenyl]methyl]pyrrolidin-2-yl]methanol Chemical compound C(C1=CC=CC=C1)OC=1C=C(OCC2=CC=C(CN3[C@H](CCC3)CO)C=C2)C=CC=1 SPXSEZMVRJLHQG-XMMPIXPASA-N 0.000 description 1
- IOSLINNLJFQMFF-XMMPIXPASA-N [(2R)-1-[[4-[[3-[(4-fluorophenyl)methylsulfanyl]phenoxy]methyl]phenyl]methyl]pyrrolidin-2-yl]methanol Chemical compound FC1=CC=C(CSC=2C=C(OCC3=CC=C(CN4[C@H](CCC4)CO)C=C3)C=CC=2)C=C1 IOSLINNLJFQMFF-XMMPIXPASA-N 0.000 description 1
- PSLUFJFHTBIXMW-WYEYVKMPSA-N [(3r,4ar,5s,6s,6as,10s,10ar,10bs)-3-ethenyl-10,10b-dihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-6-(2-pyridin-2-ylethylcarbamoyloxy)-5,6,6a,8,9,10-hexahydro-2h-benzo[f]chromen-5-yl] acetate Chemical compound O([C@@H]1[C@@H]([C@]2(O[C@](C)(CC(=O)[C@]2(O)[C@@]2(C)[C@@H](O)CCC(C)(C)[C@@H]21)C=C)C)OC(=O)C)C(=O)NCCC1=CC=CC=N1 PSLUFJFHTBIXMW-WYEYVKMPSA-N 0.000 description 1
- MUUXBTFQEXVEEI-UHFFFAOYSA-N [2-(dimethyl-$l^{3}-silanyl)phenyl]-dimethylsilicon Chemical compound C[Si](C)C1=CC=CC=C1[Si](C)C MUUXBTFQEXVEEI-UHFFFAOYSA-N 0.000 description 1
- SMNRFWMNPDABKZ-WVALLCKVSA-N [[(2R,3S,4R,5S)-5-(2,6-dioxo-3H-pyridin-3-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [[[(2R,3S,4S,5R,6R)-4-fluoro-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl] hydrogen phosphate Chemical compound OC[C@H]1O[C@H](OP(O)(=O)OP(O)(=O)OP(O)(=O)OP(O)(=O)OC[C@H]2O[C@H]([C@H](O)[C@@H]2O)C2C=CC(=O)NC2=O)[C@H](O)[C@@H](F)[C@@H]1O SMNRFWMNPDABKZ-WVALLCKVSA-N 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- 229940022663 acetate Drugs 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 238000005903 acid hydrolysis reaction Methods 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 230000010398 acute inflammatory response Effects 0.000 description 1
- 208000005298 acute pain Diseases 0.000 description 1
- 125000004442 acylamino group Chemical group 0.000 description 1
- 210000004100 adrenal gland Anatomy 0.000 description 1
- 230000001780 adrenocortical effect Effects 0.000 description 1
- 201000000028 adult respiratory distress syndrome Diseases 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 238000001042 affinity chromatography Methods 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 239000003570 air Substances 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- IAJILQKETJEXLJ-RSJOWCBRSA-N aldehydo-D-galacturonic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-RSJOWCBRSA-N 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000003973 alkyl amines Chemical group 0.000 description 1
- 125000005197 alkyl carbonyloxy alkyl group Chemical group 0.000 description 1
- 125000005107 alkyl diaryl silyl group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- 208000028004 allergic respiratory disease Diseases 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 108010004469 allophycocyanin Proteins 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 150000001414 amino alcohols Chemical class 0.000 description 1
- ROBVIMPUHSLWNV-UHFFFAOYSA-N aminoglutethimide Chemical compound C=1C=C(N)C=CC=1C1(CC)CCC(=O)NC1=O ROBVIMPUHSLWNV-UHFFFAOYSA-N 0.000 description 1
- 229960003437 aminoglutethimide Drugs 0.000 description 1
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 1
- 239000001099 ammonium carbonate Substances 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 229960002932 anastrozole Drugs 0.000 description 1
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000005911 anti-cytotoxic effect Effects 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 229940045687 antimetabolites folic acid analogs Drugs 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 239000003972 antineoplastic antibiotic Substances 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 229940045988 antineoplastic drug protein kinase inhibitors Drugs 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 239000003886 aromatase inhibitor Substances 0.000 description 1
- 229940046844 aromatase inhibitors Drugs 0.000 description 1
- 150000004982 aromatic amines Chemical group 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 208000021328 arterial occlusion Diseases 0.000 description 1
- 150000005840 aryl radicals Chemical class 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- 239000000305 astragalus gummifer gum Substances 0.000 description 1
- METKIMKYRPQLGS-UHFFFAOYSA-N atenolol Chemical compound CC(C)NCC(O)COC1=CC=C(CC(N)=O)C=C1 METKIMKYRPQLGS-UHFFFAOYSA-N 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 201000004995 autoimmune glomerulonephritis Diseases 0.000 description 1
- 230000035578 autophosphorylation Effects 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 1
- 229960002170 azathioprine Drugs 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- FHCIILYMWWRNIZ-UHFFFAOYSA-N benzhydryl(chloro)silane Chemical compound C=1C=CC=CC=1C([SiH2]Cl)C1=CC=CC=C1 FHCIILYMWWRNIZ-UHFFFAOYSA-N 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000004618 benzofuryl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000005874 benzothiadiazolyl group Chemical group 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000001743 benzylic group Chemical group 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 239000004621 biodegradable polymer Substances 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000008512 biological response Effects 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- TXFLGZOGNOOEFZ-UHFFFAOYSA-N bis(2-chloroethyl)amine Chemical compound ClCCNCCCl TXFLGZOGNOOEFZ-UHFFFAOYSA-N 0.000 description 1
- 201000011263 bladder neck cancer Diseases 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 230000010072 bone remodeling Effects 0.000 description 1
- 230000024279 bone resorption Effects 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 230000006931 brain damage Effects 0.000 description 1
- 231100000874 brain damage Toxicity 0.000 description 1
- 208000029028 brain injury Diseases 0.000 description 1
- 201000008274 breast adenocarcinoma Diseases 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- PVAGAWKDXRTWDA-UHFFFAOYSA-N butyl n-(2,5-dichloropentyl)carbamate Chemical compound CCCCOC(=O)NCC(Cl)CCCCl PVAGAWKDXRTWDA-UHFFFAOYSA-N 0.000 description 1
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000003710 calcium ionophore Substances 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 208000029499 cancer-related condition Diseases 0.000 description 1
- 229960004117 capecitabine Drugs 0.000 description 1
- 239000007894 caplet Substances 0.000 description 1
- 235000013877 carbamide Nutrition 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- AEIMYPLMACPBOD-UHFFFAOYSA-N carbonocyanidic acid;nickel Chemical compound [Ni].OC(=O)C#N AEIMYPLMACPBOD-UHFFFAOYSA-N 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 150000001244 carboxylic acid anhydrides Chemical class 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 239000003183 carcinogenic agent Substances 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- 239000005018 casein Substances 0.000 description 1
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 1
- 235000021240 caseins Nutrition 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 230000020411 cell activation Effects 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 230000011748 cell maturation Effects 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 206010008118 cerebral infarction Diseases 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- KWYZNESIGBQHJK-UHFFFAOYSA-N chloro-dimethyl-phenylsilane Chemical compound C[Si](C)(Cl)C1=CC=CC=C1 KWYZNESIGBQHJK-UHFFFAOYSA-N 0.000 description 1
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 1
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 1
- 230000002759 chromosomal effect Effects 0.000 description 1
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 1
- 208000022371 chronic pain syndrome Diseases 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 230000000536 complexating effect Effects 0.000 description 1
- 229940125797 compound 12 Drugs 0.000 description 1
- 229940126543 compound 14 Drugs 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 229940125878 compound 36 Drugs 0.000 description 1
- 229940125807 compound 37 Drugs 0.000 description 1
- 229940127573 compound 38 Drugs 0.000 description 1
- 229940126540 compound 41 Drugs 0.000 description 1
- 229940125936 compound 42 Drugs 0.000 description 1
- 229940127271 compound 49 Drugs 0.000 description 1
- 229940127113 compound 57 Drugs 0.000 description 1
- 229940125900 compound 59 Drugs 0.000 description 1
- 235000008504 concentrate Nutrition 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- RWGFKTVRMDUZSP-UHFFFAOYSA-N cumene Chemical compound CC(C)C1=CC=CC=C1 RWGFKTVRMDUZSP-UHFFFAOYSA-N 0.000 description 1
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 description 1
- WZHCOOQXZCIUNC-UHFFFAOYSA-N cyclandelate Chemical compound C1C(C)(C)CC(C)CC1OC(=O)C(O)C1=CC=CC=C1 WZHCOOQXZCIUNC-UHFFFAOYSA-N 0.000 description 1
- 150000004292 cyclic ethers Chemical class 0.000 description 1
- 125000003678 cyclohexadienyl group Chemical group C1(=CC=CCC1)* 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- GCUVBACNBHGZRS-UHFFFAOYSA-N cyclopenta-1,3-diene cyclopenta-2,4-dien-1-yl(diphenyl)phosphane iron(2+) Chemical compound [Fe++].c1cc[cH-]c1.c1cc[c-](c1)P(c1ccccc1)c1ccccc1 GCUVBACNBHGZRS-UHFFFAOYSA-N 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 229960002433 cysteine Drugs 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- GVJHHUAWPYXKBD-UHFFFAOYSA-N d-alpha-tocopherol Natural products OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 1
- 229960000640 dactinomycin Drugs 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 150000008050 dialkyl sulfates Chemical class 0.000 description 1
- 125000005265 dialkylamine group Chemical group 0.000 description 1
- 125000005105 dialkylarylsilyl group Chemical group 0.000 description 1
- 125000005266 diarylamine group Chemical group 0.000 description 1
- ZOCHARZZJNPSEU-UHFFFAOYSA-N diboron Chemical compound B#B ZOCHARZZJNPSEU-UHFFFAOYSA-N 0.000 description 1
- 125000000950 dibromo group Chemical group Br* 0.000 description 1
- 125000006003 dichloroethyl group Chemical group 0.000 description 1
- 125000004774 dichlorofluoromethyl group Chemical group FC(Cl)(Cl)* 0.000 description 1
- WGLUMOCWFMKWIL-UHFFFAOYSA-N dichloromethane;methanol Chemical compound OC.ClCCl WGLUMOCWFMKWIL-UHFFFAOYSA-N 0.000 description 1
- 125000004772 dichloromethyl group Chemical group [H]C(Cl)(Cl)* 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 125000001664 diethylamino group Chemical group [H]C([H])([H])C([H])([H])N(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- RGLYKWWBQGJZGM-ISLYRVAYSA-N diethylstilbestrol Chemical compound C=1C=C(O)C=CC=1C(/CC)=C(\CC)C1=CC=C(O)C=C1 RGLYKWWBQGJZGM-ISLYRVAYSA-N 0.000 description 1
- 229960000452 diethylstilbestrol Drugs 0.000 description 1
- 125000006001 difluoroethyl group Chemical group 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- 125000004598 dihydrobenzofuryl group Chemical group O1C(CC2=C1C=CC=C2)* 0.000 description 1
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 description 1
- 125000005043 dihydropyranyl group Chemical group O1C(CCC=C1)* 0.000 description 1
- 125000005056 dihydrothiazolyl group Chemical group S1C(NC=C1)* 0.000 description 1
- 125000005057 dihydrothienyl group Chemical group S1C(CC=C1)* 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- AFABGHUZZDYHJO-UHFFFAOYSA-N dimethyl butane Natural products CCCC(C)C AFABGHUZZDYHJO-UHFFFAOYSA-N 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 125000000597 dioxinyl group Chemical group 0.000 description 1
- GAFRWLVTHPVQGK-UHFFFAOYSA-N dipentyl sulfate Chemical class CCCCCOS(=O)(=O)OCCCCC GAFRWLVTHPVQGK-UHFFFAOYSA-N 0.000 description 1
- ZZVUWRFHKOJYTH-UHFFFAOYSA-N diphenhydramine Chemical group C=1C=CC=CC=1C(OCCN(C)C)C1=CC=CC=C1 ZZVUWRFHKOJYTH-UHFFFAOYSA-N 0.000 description 1
- 208000037765 diseases and disorders Diseases 0.000 description 1
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical group [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000003828 downregulation Effects 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 230000036267 drug metabolism Effects 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 229960001484 edetic acid Drugs 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 150000002081 enamines Chemical group 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940095399 enema Drugs 0.000 description 1
- 238000007824 enzymatic assay Methods 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 238000011067 equilibration Methods 0.000 description 1
- 229960001433 erlotinib Drugs 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 229960005309 estradiol Drugs 0.000 description 1
- 229930182833 estradiol Natural products 0.000 description 1
- 229960005416 estradiol cypionate Drugs 0.000 description 1
- 229960001842 estramustine Drugs 0.000 description 1
- FRPJXPJMRWBBIH-RBRWEJTLSA-N estramustine Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 FRPJXPJMRWBBIH-RBRWEJTLSA-N 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- VFRSADQPWYCXDG-LEUCUCNGSA-N ethyl (2s,5s)-5-methylpyrrolidine-2-carboxylate;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CCOC(=O)[C@@H]1CC[C@H](C)N1 VFRSADQPWYCXDG-LEUCUCNGSA-N 0.000 description 1
- CJAONIOAQZUHPN-KKLWWLSJSA-N ethyl 12-[[2-[(2r,3r)-3-[2-[(12-ethoxy-12-oxododecyl)-methylamino]-2-oxoethoxy]butan-2-yl]oxyacetyl]-methylamino]dodecanoate Chemical compound CCOC(=O)CCCCCCCCCCCN(C)C(=O)CO[C@H](C)[C@@H](C)OCC(=O)N(C)CCCCCCCCCCCC(=O)OCC CJAONIOAQZUHPN-KKLWWLSJSA-N 0.000 description 1
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical class CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 238000013265 extended release Methods 0.000 description 1
- 239000012894 fetal calf serum Substances 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 125000004785 fluoromethoxy group Chemical group [H]C([H])(F)O* 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 1
- 229960002074 flutamide Drugs 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- UTVVREMVDJTZAC-UHFFFAOYSA-N furan-2-amine Chemical compound NC1=CC=CO1 UTVVREMVDJTZAC-UHFFFAOYSA-N 0.000 description 1
- 150000002240 furans Chemical class 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- DSLZVSRJTYRBFB-DUHBMQHGSA-N galactaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)C(O)=O DSLZVSRJTYRBFB-DUHBMQHGSA-N 0.000 description 1
- 229940083124 ganglion-blocking antiadrenergic secondary and tertiary amines Drugs 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000007897 gelcap Substances 0.000 description 1
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 1
- 229960005277 gemcitabine Drugs 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 230000004077 genetic alteration Effects 0.000 description 1
- 231100000118 genetic alteration Toxicity 0.000 description 1
- 238000009650 gentamicin protection assay Methods 0.000 description 1
- 229940080856 gleevec Drugs 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 208000024908 graft versus host disease Diseases 0.000 description 1
- ZRALSGWEFCBTJO-UHFFFAOYSA-N guanidine group Chemical group NC(=N)N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 1
- 125000004438 haloalkoxy group Chemical group 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 230000026030 halogenation Effects 0.000 description 1
- 238000005658 halogenation reaction Methods 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 125000006343 heptafluoro propyl group Chemical group 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid group Chemical group C(CCCCCC)(=O)O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- WGBBUURBHXLGFM-UHFFFAOYSA-N hexan-2-amine Chemical compound CCCCC(C)N WGBBUURBHXLGFM-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid group Chemical group C(CCCCC)(=O)O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229960001340 histamine Drugs 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 150000007857 hydrazones Chemical group 0.000 description 1
- 235000011167 hydrochloric acid Nutrition 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 1
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 229960001330 hydroxycarbamide Drugs 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- 230000003463 hyperproliferative effect Effects 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 229960002411 imatinib Drugs 0.000 description 1
- 229960003685 imatinib mesylate Drugs 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000010874 in vitro model Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 1
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 239000003049 inorganic solvent Substances 0.000 description 1
- 229910001867 inorganic solvent Inorganic materials 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 229940100601 interleukin-6 Drugs 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000007914 intraventricular administration Methods 0.000 description 1
- 230000009545 invasion Effects 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 229940084651 iressa Drugs 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000005929 isobutyloxycarbonyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])OC(*)=O 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- FZWBNHMXJMCXLU-BLAUPYHCSA-N isomaltotriose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O)O1 FZWBNHMXJMCXLU-BLAUPYHCSA-N 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 231100000518 lethal Toxicity 0.000 description 1
- 230000001665 lethal effect Effects 0.000 description 1
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 description 1
- 229960004338 leuprorelin Drugs 0.000 description 1
- 239000000865 liniment Substances 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 239000008297 liquid dosage form Substances 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- UBJFKNSINUCEAL-UHFFFAOYSA-N lithium;2-methylpropane Chemical compound [Li+].C[C-](C)C UBJFKNSINUCEAL-UHFFFAOYSA-N 0.000 description 1
- RENRQMCACQEWFC-UGKGYDQZSA-N lnp023 Chemical compound C1([C@H]2N(CC=3C=4C=CNC=4C(C)=CC=3OC)CC[C@@H](C2)OCC)=CC=C(C(O)=O)C=C1 RENRQMCACQEWFC-UGKGYDQZSA-N 0.000 description 1
- 229960002247 lomustine Drugs 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 125000005439 maleimidyl group Chemical group C1(C=CC(N1*)=O)=O 0.000 description 1
- 208000026037 malignant tumor of neck Diseases 0.000 description 1
- 239000000845 maltitol Substances 0.000 description 1
- 235000010449 maltitol Nutrition 0.000 description 1
- VQHSOMBJVWLPSR-WUJBLJFYSA-N maltitol Chemical compound OC[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O VQHSOMBJVWLPSR-WUJBLJFYSA-N 0.000 description 1
- 229940035436 maltitol Drugs 0.000 description 1
- 210000005075 mammary gland Anatomy 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 229960004961 mechlorethamine Drugs 0.000 description 1
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical compound ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 1
- RQZAXGRLVPAYTJ-GQFGMJRRSA-N megestrol acetate Chemical compound C1=C(C)C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 RQZAXGRLVPAYTJ-GQFGMJRRSA-N 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 230000037353 metabolic pathway Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229910000000 metal hydroxide Inorganic materials 0.000 description 1
- 150000004692 metal hydroxides Chemical class 0.000 description 1
- 238000006263 metalation reaction Methods 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- WSFSSNUMVMOOMR-BJUDXGSMSA-N methanone Chemical compound O=[11CH2] WSFSSNUMVMOOMR-BJUDXGSMSA-N 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 239000000693 micelle Substances 0.000 description 1
- 210000004688 microtubule Anatomy 0.000 description 1
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 description 1
- 230000005787 mitochondrial ATP synthesis coupled electron transport Effects 0.000 description 1
- 229960000350 mitotane Drugs 0.000 description 1
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- 238000007799 mixed lymphocyte reaction assay Methods 0.000 description 1
- 125000006682 monohaloalkyl group Chemical group 0.000 description 1
- HDZGCSFEDULWCS-UHFFFAOYSA-N monomethylhydrazine Chemical class CNN HDZGCSFEDULWCS-UHFFFAOYSA-N 0.000 description 1
- 206010028417 myasthenia gravis Diseases 0.000 description 1
- 125000001421 myristyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- CZHAWVSETSZRJW-UHFFFAOYSA-N n-(2,2-diethoxyethyl)-5,6-diphenylfuro[2,3-d]pyrimidin-4-amine Chemical compound C1=2C(NCC(OCC)OCC)=NC=NC=2OC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 CZHAWVSETSZRJW-UHFFFAOYSA-N 0.000 description 1
- DJRALJBJFAVLGR-UHFFFAOYSA-N n-[2-(1-benzylpiperidin-4-yl)ethyl]-5-phenyl-6-[4-(2-pyrrolidin-1-ylethoxy)phenyl]furo[2,3-d]pyrimidin-4-amine Chemical compound N=1C=NC=2OC(C=3C=CC(OCCN4CCCC4)=CC=3)=C(C=3C=CC=CC=3)C=2C=1NCCC(CC1)CCN1CC1=CC=CC=C1 DJRALJBJFAVLGR-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- QVBJLEMLXWUHHE-UHFFFAOYSA-N n-methyl-1,3-dithiolan-2-amine Chemical compound CNC1SCCS1 QVBJLEMLXWUHHE-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 208000015122 neurodegenerative disease Diseases 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 239000012454 non-polar solvent Substances 0.000 description 1
- 108091008046 non-receptor tyrosine kinases Proteins 0.000 description 1
- 102000037979 non-receptor tyrosine kinases Human genes 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 150000007523 nucleic acids Chemical class 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 239000012053 oil suspension Substances 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- PIDFDZJZLOTZTM-KHVQSSSXSA-N ombitasvir Chemical compound COC(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@H]1C(=O)NC1=CC=C([C@H]2N([C@@H](CC2)C=2C=CC(NC(=O)[C@H]3N(CCC3)C(=O)[C@@H](NC(=O)OC)C(C)C)=CC=2)C=2C=CC(=CC=2)C(C)(C)C)C=C1 PIDFDZJZLOTZTM-KHVQSSSXSA-N 0.000 description 1
- 230000000771 oncological effect Effects 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 208000002865 osteopetrosis Diseases 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 208000013371 ovarian adenocarcinoma Diseases 0.000 description 1
- 201000006588 ovary adenocarcinoma Diseases 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 230000036542 oxidative stress Effects 0.000 description 1
- 150000002923 oximes Chemical group 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 125000005010 perfluoroalkyl group Chemical group 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 125000001792 phenanthrenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3C=CC12)* 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- HXITXNWTGFUOAU-UHFFFAOYSA-N phenylboronic acid Chemical class OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- 125000003170 phenylsulfonyl group Chemical group C1(=CC=CC=C1)S(=O)(=O)* 0.000 description 1
- QGVLYPPODPLXMB-QXYKVGAMSA-N phorbol Natural products C[C@@H]1[C@@H](O)[C@]2(O)[C@H]([C@H]3C=C(CO)C[C@@]4(O)[C@H](C=C(C)C4=O)[C@@]13O)C2(C)C QGVLYPPODPLXMB-QXYKVGAMSA-N 0.000 description 1
- XYFCBTPGUUZFHI-UHFFFAOYSA-O phosphonium Chemical compound [PH4+] XYFCBTPGUUZFHI-UHFFFAOYSA-O 0.000 description 1
- 230000000865 phosphorylative effect Effects 0.000 description 1
- DCWXELXMIBXGTH-UHFFFAOYSA-N phosphotyrosine Chemical compound OC(=O)C(N)CC1=CC=C(OP(O)(O)=O)C=C1 DCWXELXMIBXGTH-UHFFFAOYSA-N 0.000 description 1
- 125000005545 phthalimidyl group Chemical group 0.000 description 1
- 125000000612 phthaloyl group Chemical group C(C=1C(C(=O)*)=CC=CC1)(=O)* 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- NUKCGLDCWQXYOQ-UHFFFAOYSA-N piposulfan Chemical compound CS(=O)(=O)OCCC(=O)N1CCN(C(=O)CCOS(C)(=O)=O)CC1 NUKCGLDCWQXYOQ-UHFFFAOYSA-N 0.000 description 1
- 229950001100 piposulfan Drugs 0.000 description 1
- 150000003057 platinum Chemical class 0.000 description 1
- 229960003171 plicamycin Drugs 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 229960004694 prednimustine Drugs 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- VTGOHKSTWXHQJK-UHFFFAOYSA-N pyrimidin-2-ol Chemical class OC1=NC=CC=N1 VTGOHKSTWXHQJK-UHFFFAOYSA-N 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000001422 pyrrolinyl group Chemical group 0.000 description 1
- 150000004943 pyrrolo[2,3-d]pyrimidines Chemical class 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 108010014186 ras Proteins Proteins 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 239000011535 reaction buffer Substances 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 206010038038 rectal cancer Diseases 0.000 description 1
- 239000006215 rectal suppository Substances 0.000 description 1
- 201000001275 rectum cancer Diseases 0.000 description 1
- 238000006268 reductive amination reaction Methods 0.000 description 1
- 230000012121 regulation of immune response Effects 0.000 description 1
- 230000008844 regulatory mechanism Effects 0.000 description 1
- 230000010410 reperfusion Effects 0.000 description 1
- 201000004335 respiratory allergy Diseases 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- XIIOFHFUYBLOLW-UHFFFAOYSA-N selpercatinib Chemical compound OC(COC=1C=C(C=2N(C=1)N=CC=2C#N)C=1C=NC(=CC=1)N1CC2N(C(C1)C2)CC=1C=NC(=CC=1)OC)(C)C XIIOFHFUYBLOLW-UHFFFAOYSA-N 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 208000002491 severe combined immunodeficiency Diseases 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- XGVXKJKTISMIOW-ZDUSSCGKSA-N simurosertib Chemical compound N1N=CC(C=2SC=3C(=O)NC(=NC=3C=2)[C@H]2N3CCC(CC3)C2)=C1C XGVXKJKTISMIOW-ZDUSSCGKSA-N 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 208000000587 small cell lung carcinoma Diseases 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 125000005346 substituted cycloalkyl group Chemical group 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 125000001174 sulfone group Chemical group 0.000 description 1
- 150000003461 sulfonyl halides Chemical group 0.000 description 1
- 125000003375 sulfoxide group Chemical group 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- 229940120982 tarceva Drugs 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 229960001674 tegafur Drugs 0.000 description 1
- WFWLQNSHRPWKFK-ZCFIWIBFSA-N tegafur Chemical compound O=C1NC(=O)C(F)=CN1[C@@H]1OCCC1 WFWLQNSHRPWKFK-ZCFIWIBFSA-N 0.000 description 1
- 229960004964 temozolomide Drugs 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- ZHTIJMDZNNCDOT-UHFFFAOYSA-N tert-butyl 2-amino-3-chloropropanoate Chemical compound CC(C)(C)OC(=O)C(N)CCl ZHTIJMDZNNCDOT-UHFFFAOYSA-N 0.000 description 1
- QSYTWBKZNNEKPN-UHFFFAOYSA-N tert-butyl 4-(2-aminoethyl)piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCN(CCN)CC1 QSYTWBKZNNEKPN-UHFFFAOYSA-N 0.000 description 1
- IYWPWSDVMPWRKT-UHFFFAOYSA-N tert-butyl 4-[2-[(5,6-diphenylfuro[2,3-d]pyrimidin-4-yl)amino]ethyl]piperazine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCN1CCNC1=NC=NC2=C1C(C=1C=CC=CC=1)=C(C=1C=CC=CC=1)O2 IYWPWSDVMPWRKT-UHFFFAOYSA-N 0.000 description 1
- ACXBBJXSBKBJMY-UHFFFAOYSA-N tert-butyl 4-[2-[[6-[4-(2-methoxyethoxy)phenyl]-5-phenylfuro[2,3-d]pyrimidin-4-yl]amino]ethyl]piperazine-1-carboxylate Chemical compound C1=CC(OCCOC)=CC=C1C1=C(C=2C=CC=CC=2)C2=C(NCCN3CCN(CC3)C(=O)OC(C)(C)C)N=CN=C2O1 ACXBBJXSBKBJMY-UHFFFAOYSA-N 0.000 description 1
- NXTJUGBDLXQVFR-UHFFFAOYSA-N tert-butyl n-(thiolan-2-ylmethyl)carbamate Chemical compound CC(C)(C)OC(=O)NCC1CCCS1 NXTJUGBDLXQVFR-UHFFFAOYSA-N 0.000 description 1
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 1
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 230000002381 testicular Effects 0.000 description 1
- 201000003120 testicular cancer Diseases 0.000 description 1
- 125000004192 tetrahydrofuran-2-yl group Chemical group [H]C1([H])OC([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- RAOIDOHSFRTOEL-UHFFFAOYSA-N tetrahydrothiophene Chemical class C1CCSC1 RAOIDOHSFRTOEL-UHFFFAOYSA-N 0.000 description 1
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 125000000101 thioether group Chemical group 0.000 description 1
- 150000003568 thioethers Chemical group 0.000 description 1
- RBECMGYCTFQOTG-UHFFFAOYSA-N thiolan-2-ylmethanamine;2,2,2-trifluoroacetic acid Chemical compound [NH3+]CC1CCCS1.[O-]C(=O)C(F)(F)F RBECMGYCTFQOTG-UHFFFAOYSA-N 0.000 description 1
- 229960001196 thiotepa Drugs 0.000 description 1
- 229940104230 thymidine Drugs 0.000 description 1
- 229960003087 tioguanine Drugs 0.000 description 1
- 239000003104 tissue culture media Substances 0.000 description 1
- 235000010384 tocopherol Nutrition 0.000 description 1
- 229960001295 tocopherol Drugs 0.000 description 1
- 229930003799 tocopherol Natural products 0.000 description 1
- 239000011732 tocopherol Substances 0.000 description 1
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 1
- 229960000303 topotecan Drugs 0.000 description 1
- 125000005490 tosylate group Chemical group 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- 125000004665 trialkylsilyl group Chemical group 0.000 description 1
- 125000005106 triarylsilyl group Chemical group 0.000 description 1
- 150000004654 triazenes Chemical class 0.000 description 1
- 150000003918 triazines Chemical class 0.000 description 1
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 1
- 150000008648 triflates Chemical class 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- 125000000025 triisopropylsilyl group Chemical group C(C)(C)[Si](C(C)C)(C(C)C)* 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- TUCIOBMMDDOEMM-RIYZIHGNSA-N tyrphostin B42 Chemical compound C1=C(O)C(O)=CC=C1\C=C(/C#N)C(=O)NCC1=CC=CC=C1 TUCIOBMMDDOEMM-RIYZIHGNSA-N 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 229940035893 uracil Drugs 0.000 description 1
- 229960001055 uracil mustard Drugs 0.000 description 1
- 150000003672 ureas Chemical group 0.000 description 1
- 210000003932 urinary bladder Anatomy 0.000 description 1
- 229910052720 vanadium Inorganic materials 0.000 description 1
- 208000021331 vascular occlusion disease Diseases 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 description 1
- 229960004355 vindesine Drugs 0.000 description 1
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- GVJHHUAWPYXKBD-IEOSBIPESA-N α-tocopherol Chemical compound OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/14—Drugs for dermatological disorders for baldness or alopecia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/38—Drugs for disorders of the endocrine system of the suprarenal hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
Definitions
- the present invention generally relates to furanopyrimidine compounds, pharmaceutical formulations containing the compounds, methods of treatment using the compounds, and methods of preparing medicaments comprising the compounds.
- T cells play a pivotal role in the regulation of immune responses and are important for establishing immunity to pathogens.
- T cells are often activated during inflammatory autoimmune diseases, such as rheumatoid arthritis, inflammatory bowel disease, type I diabetes, multiple sclerosis, Sjogren's disease, myasthenia gravis, psoriasis, and lupus. T cell activation is also an important component of organ transplantation rejection, allergic reactions, and asthma.
- T cells are activated by specific antigens through T cell receptors (TCR) which are expressed on the cell surface.
- TCR T cell receptors
- This activation triggers a series of intracellular signaling cascades mediated by enzymes expressed within the cell (Kane, LP et al. Current Opinion in Immunol. 200, 12, 242). These cascades lead to gene regulation events that result in the production of cytokines, including interleukin-2 (IL-2).
- IL-2 interleukin-2
- EL-2 is a critical cytokine in T cell activation, leading to proliferation and amplification of specific immune responses.
- Kinase enzymes have been shown to be important in the intracellular signal transduction.
- Src-family of protein tyrosine kinases which includes, for example: Lck, Fyn(B), Fyn(T), Lyn, Src, Yes, Hck, Fgr and BIk (for review see: Bolen, JB, and Brugge, JS Annu. Rev. Immunol 1997, 15, 371).
- PTK's protein tyrosine kinases
- Src-family kinases are also important for signaling downstream of other immune cell receptors. Fyn, like Lck, is involved in TCR signaling in T cells (Appleby, MW et al. Cell 1992, 70, 751). Hck and Fgr are involved in Fc ⁇ receptor signaling leading to neutrophil activation (Vicentini, L. et al. J. Immunol. 2002, 168, 6446). Lyn and Src also participate in Fc ⁇ receptor signaling leading to release of histamine and other allergic mediators (Turner, H. and Kinet, J-P Nature 1999, 402, B24). These findings suggest that Src family kinase inhibitors may be useful in treating allergic diseases and asthma.
- Src kinases have also been found to be activated in tumors including sarcoma, melanoma, breast, and colon cancers suggesting that Src kinase inhibitors may be useful anti ⁇ cancer agents (Abram, CL and Courtneidge, SA Exp. Cell Res. 2000, 254, 1). Src kinase inhibitors have also been reported to be effective in an animal model of cerebral ischemia (R. Paul et al. Nature Medicine 2001, 7, 222), suggesting that Src kinase inhibitors may be effective at limiting brain damage following stroke.
- Cancer is the second leading cause of death in the United States (Boring, et al., CA Cancer J. Clin., 43:7, 1993), and features uncontrolled cellular growth, which results either in local invasion of normal tissue or systemic spread (metastasis) of the abnormal growth. Cancer is caused by inherited or acquired mutations in cancer genes, which have normal cellular functions and which induce or otherwise contribute to cancer once mutated or expressed at an abnormal level. Certain well-studied tumors carry several different independently mutated genes, including activated oncogenes and inactivated tumor suppressor genes.
- Gene amplification involves a chromosomal region bearing specific genes undergoing a relative increase in DNA copy number, thereby increasing the copies of any genes that are present. In general, gene amplification results in increased levels of transcription and translation, producing higher amounts of the corresponding gene mRNA and protein. Amplification of genes causes deleterious effects, which contribute to cancer formation and proliferation (Lengauer et al. Nature, 396:643-649, 1999). Gene amplification has been established as an important genetic alteration in solid tumors (Knuutila et al., Am. J. Pathol., 152(5): 1107-23, 1998; Knuutila et al., Cancer Genet. Cytogenet, 100(l):25-30, 1998).
- Another trait of tumor cells is the over-expression or differential expression of whole collections of genes.
- pre-cancerous or cancerous cells, and tissues where both amplification of a gene and over-expression of the gene product occur, then that gene and its product present both a diagnostic target as well as a therapeutic opportunity for intervention.
- the amplified cancer genes encode an enzyme, such as a kinase, and the discovery and characterization of inhibitors of the enzymatic activity of this gene product will be a promising avenue that leads to novel therapeutics for cancer treatment.
- ACKl is a gene that is frequently amplified and over-expressed in primary human tumors (U.S. Patent Publication No. 20030175763).
- ACKl kinase activity is regulated in the context of cell attachment and detachment, and certain cancer cells depend on ACKl 's kinase activity for adhesion, anchorage independent growth and survival. Down regulation of ACKl kinase activity or ACKl expression levels can result in reduced tumor growth in animal models. Accordingly, Ack is a target believed to be useful in the regulation of cancer.
- the ACKl gene encodes an intracellular, non-receptor tyrosine kinase that binds cdc42Hs in its GTP-bound form and inhibits both the intrinsic and GTPase-activating protein (GAP)-stimulated GTPase activity of p21cdc42, a Ras-like protein involved in cell growth (Manser et al., Nature 363(6427):364-367, 1993). This binding is mediated by a unique polypeptide of 47 amino acids C-terminal to an SH3 domain. ACKl gene contains a tyrosine kinase domain and is reported to possess tyrosine kinase activity.
- GAP GTPase-activating protein
- the protein may be involved in a regulatory mechanism that sustains the GTP-bound active form of cdc42Hs and which is directly linked to a tyrosine phosphorylation signal transduction pathway.
- Src family kinase or ACK-I disclosing various chemical compounds, including 2-phenylamino-imidazo [4,5- h]isoquinolin-9-ones (Snow, RJ et al. J. Med. Chem. 2002, 45, 3394), pyrazolo [3,4- d]pyrimidines (Burchat, AF et al. Bioorganic and Med. Chem. Letters 2002, 12, 1987 and Hanke, JH et al. J.
- PCT publication WO 03/018589 discloses various 4-(substituted amino)- furanopyrimidines useful in the treatment of cardiovascular diseases caused by ischemia such as angina, pectoris, peripheral and arterial occlusion diseases, thrombosis, vascular occlusions, myocardial infarction, and reperfusion diseases.
- cardiovascular diseases caused by ischemia such as angina, pectoris, peripheral and arterial occlusion diseases, thrombosis, vascular occlusions, myocardial infarction, and reperfusion diseases.
- the publication also suggests that the disclosed compounds may be useful for the treatment of acute or chronic pain and neurodegenerative diseases.
- the publication does not disclose any 4-(heterocycloalkylamino)furanopyrimidines, and it makes no mention of using 4-(substitutedamino)furanopyrimidines to treat proliferative disease.
- the present invention relates to compounds represented by general Formula I:
- the compounds of Formula I are capable of modulating protein tyrosine kinase enzymes of the Src family, such as Lck, as well as other protein kinase enzymes such as ACK-I and Jak-3. Accordingly, these compounds are useful in the treatment, including preventative, prophylactic and therapeutic treatment, of protein tyrosine kinase-associated disorders, including but not limited to, immunologic and oncologic disorders.
- Protein tyrosine kinase-associated disorders are disorders which result from aberrant tyrosine kinase activity, and/or which are alleviated by the regulation, and inhibition in particular, of one or more of these kinase enzymes.
- Lck inhibitors are of value in the treatment of a number of such disorders (for example, the treatment of autoimmune diseases), as Lck inhibition blocks T cell activation. It is believed that the compounds of Formula I modulate T cell activation by way of inhibition of one or more of the multiple protein tyrosine kinases involved in early signal transduction steps leading to T cell activation, for example, by way of inhibition of Lck kinase.
- the compounds of Formula I are useful for the treatment of T cell mediated diseases, including inhibition of T cell activation and proliferation.
- the invention provides compounds, which selectively block T cell activation and proliferation. Further, the compounds may block the activation of endothelial cell protein tyrosine kinase by oxidative stress thereby limiting surface expression of adhesion molecules that induce neutrophil binding, and they also can inhibit protein tyrosine kinase necessary for neutrophil activation. The compounds would be useful, therefore, in the treatment of ischemia and reperfusion injury.
- methods for the treatment of protein tyrosine kinase-associated disorders are provided. The method comprises administering to a subject at least one compound of Formula I in an amount effective to treat the disorder.
- compositions comprising a compound of Formula I and a pharmaceutically acceptable carrier.
- a composition can be administered to the subject, such as a human, for the purpose of treating the disorder.
- Other therapeutic agents such as those described below may be employed in combination with the inventive compounds, such as in a combined composition, in the present methods.
- such other therapeutic agent(s) may be administered prior to, simultaneously with, or following the administration of the compound(s) of the present invention.
- the compound(s) of the present invention may be used in treating related conditions including, without limitation, arthritis (such as rheumatoid arthritis, psoriatic arthritis or osteoarthritis); transplant (such as organ transplant, acute transplant or heterograft or homograft (such as is employed in burn treatment)) rejection; protection from ischemic or reperfusion injury such as ischemic or reperfusion injury incurred during organ transplantation, myocardial infarction, stroke or other causes; transplantation tolerance induction; multiple sclerosis; inflammatory bowel disease, including ulcerative colitis and Crohn's disease; lupus (systemic lupus erythematosis); graft vs.
- arthritis such as rheumatoid arthritis, psoriatic arthritis or osteoarthritis
- transplant such as organ transplant, acute transplant or heterograft or homograft (such as is employed in burn treatment)
- protection from ischemic or reperfusion injury such as ischemic or reperfusion injury incurred during organ transplantation, my
- T -cell mediated hypersensitivity diseases including contact hypersensitivity, delayed-type hypersensitivity, and gluten-sensitive enteropathy (Celiac disease); Type 1 diabetes; psoriasis; contact dermatitis (including that due to poison ivy); Hashimoto's thyroiditis; Sjogren's syndrome; Autoimmune Hyperthyroidism, such as Graves' Disease; Addison's disease (autoimmune disease of the adrenal glands); Autoimmune polyglandular disease (also known as autoimmune polyglandular syndrome); autoimmune alopecia; pernicious anemia; vitiligo; autoimmune hypopituatarism; Guillain-Barre syndrome; other autoimmune diseases; cancers where Lck or other Src-family kinases such as Src are activated or overexpressed, such as colon carcinoma and thymoma, or cancers where Src- family kinase activity facilitates tumor growth or survival; glomerulonephritis, serum sickness; utic
- the present invention also provides methods for treating the aforementioned disorders such as atopic dermatitis by administration of a therapeutically effective amount of a compound of the present invention, which is an inhibitor of protein tyrosine kinase, to a patient suffering from dermatitis and potentially in need of such treatment.
- the compounds of the invention are also capable of modulating other kinase enzymes, such as ACK-I. Modulating ACK-I can be useful for treating various ACK-I- mediated poliferative diseases, such as cancer and cancer-related conditions. Accordingly, this is one route by which the compounds can be useful for treating cancer.
- Src-family kinases other than Lck such as Hck and Fgr, are important in the Fc ⁇ receptor induced respiratory burst of neutrophils as well as the Fc ⁇ receptor responses of monocytes and macrophages.
- the compounds of the present invention may inhibit the Fc ⁇ induced respiratory burst response in neutrophils, and may also inhibit the Fc ⁇ dependent production of TNF ⁇ .
- the ability to inhibit Fc ⁇ receptor dependent neutrophil, monocyte and macrophage responses would result in additional anti-inflammatory activity for the present compounds in addition to their effects on T cells. This activity would be especially of value, for example, in the treatment of inflammatory diseases, such as arthritis or inflammatory bowel disease.
- the present compounds may also be of value for the treatment of autoimmune glomerulonephritis and other instances of glomerulonephritis induced by deposition of immune complexes in the kidney that trigger Fc ⁇ receptor responses and which can lead to kidney damage.
- certain Src family kinases such as Lyn and Fyn(B) may be important in the Fc ⁇ receptor induced degranulation of mast cells and basophils that plays an important role in asthma, allergic rhinitis, and other allergic disease.
- Fc ⁇ receptors are stimulated by IgE-antigen complexes.
- the compounds of the present invention may inhibit the Fc ⁇ induced degranulation responses.
- the ability to inhibit Fc ⁇ receptor dependent mast cell and basophil responses may result in additional anti-inflammatory activity for the present compounds beyond their effect on T cells.
- the combined activity of the present compounds towards monocytes, macrophages, T cells, etc. may prove to be a valuable tool in the treatment of any of the aforementioned disorders.
- the compounds are useful for the treatment of the aforementioned exemplary disorders irrespective of their etiology, whether or not associated with PTK.
- the present invention provides a compound of Formula I
- X is -NR 2 R 3 , -OR 2 or -SR 2 ;
- Y is hydrogen, halogen, haloalkyl, CN, substituted or unsubstituted cycloalkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted heteroalkyl, substituted or unsubstituted phenyl, substituted or unsubstituted naphthyl, (substituted or unsubstituted phenylene)-OR 4 , (substituted or unsubstituted phenylene)-alkyl-OR 4 , (substituted or unsubstituted phenylene)-R 4 , (substituted or unsubstituted phenylene)-alkyl-R 4 , substituted or unsubstituted aralkyl, substituted or unsubstituted saturated or partially unsaturated heterocycloalkyl, substituted or unsubstituted heteroaryl, (substituted or unsubstitute
- Z is O or S(O) P , wherein p is 0,1 or 2;
- R 1 is substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted heterocycloalkyl, or substituted or unsubstituted heteroaryl, wherein the substituents are selected from F, Cl, Br, I, substituted or unsubstituted Ci -6 alkyl, or substituted or unsubstituted Q- ⁇ alkoxy, wherein the Ci- ⁇ alkyl and Ci_ 6 alkoxy substituents are selected from F, Cl, Br, or I;
- R la is H, F, Cl, Br, I, CF 3 , C 1-6 alkyl, C,. 6 haloalkyl or C 1-6 alkoxy;
- R 2 is substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted aralkyl, substituted or unsubstituted (heteroaryl)alkyl substituted or unsubstituted heteroalkyl, substituted or unsubstituted (cycloalkyl)heteroalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted (heterocycloalkyl)alkyl, substituted or unsubstituted (heterocycloalkyl)heteroalkyl, or substituted or unsubstituted fused bicyclic (arylheterocycloalkyl)alkyl, wherein the heteroalkyl moiety has 1, 2 or 3 heteroatoms independently selected from N, O, or S;
- R 3 is H, CF 3 , or Ci -6 alkyl
- R 4 is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted alkoxyl, substituted or unsubstituted heteroalkyl having 1, 2 or 3 heteroatoms independently selected from N, O, or S; substituted or unsubstituted aralkyl, substituted or unsubstituted (heteroaryl)alkyl or substituted or unsubstituted (heterocycloalkyl)alkyl; and provided that
- R 2 is substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted (heteroaryl)alkyl or substituted or unsubstituted aralkyl, and Z is O, then Y is substituted or unsubstituted naphthyl, (substituted or unsubstituted phenylene)-OR 4 or (substituted or unsubstituted 5- or 6-member heteroarylene)-OR 4 wherein R 4 is substituted or unsubstituted (heteroaryl)alkyl or substituted or unsubstituted (heterocycloalkyl)alkyl; and
- X is -NR 2 R 3 .
- X is -OR 2 .
- X is or -SR 2 .
- X is tetrahydro-2-furanylmethylamino, 2-(l-piperazinyl)ethylamino, 2-(4- raorpholinyl)ethylamino, pyrrolidinylethylamino, piperidinylethylamino, N-boc- piperazinylethylamino, N-ethyl-piperazinylethylamino, 1 -methyl-2-pyrrolidinylmethylamino or 1 -ethyl-2-pyrrolidinylmethylamino.
- Y is substituted or unsubstituted phenyl, substituted or unsubstituted naphthyl, (substituted or unsubstituted phenylene)-OR 4 , (substituted or unsubstituted phenylene)-alkyl- OR 4 , (substituted or unsubstituted phenylene)-R 4 , (substituted or unsubstituted phenylene)-alkyl-R 4 , substituted or unsubstituted aralkyl, substituted or unsubstituted saturated or partially unsaturated heterocycloalkyl, substituted or unsubstituted heteroaryl, (substituted or unsubstituted 5- or 6-member heteroarylene)-OR 4 , (substituted or unsubstituted 5- or 6-member heteroarylene)-alkyl-OR 4 , (substituted or unsubstituted 5- or 6-
- Y is substituted or unsubstituted phenyl, (substituted or unsubstituted phenylene)-OR 4 , (substituted or unsubstituted phenylene)-alkyl-OR 4 , (substituted or unsubstituted phenylene)-R 4 , (substituted or unsubstituted phenylene)-alkyl-R 4 , substituted or unsubstituted aralkyl, substituted or unsubstituted saturated heterocycloalkyl, substituted or unsubstituted heteroaryl, (substituted or unsubstituted 5- or 6-member heteroarylene)-OR 4 , (substituted or unsubstituted 5- or 6-member heteroarylene)-alkyl-OR 4 , (substituted or unsubstituted 5- or 6-member heteroarylene)-alkyl-OR 4 , (substituted
- Y is 3-methoxy-4-(2-(l-piperidinyl)ethoxy)phenyl, 3-fluoro-4-(2- (diethy lamino)ethoxy)pheny 1, 4-(COOH)pheny 1, 3 -fluoro-4-(2-( 1 -piperidiny l)ethoxy)pheny 1, 4-(CHO)phenyl, 4-((4-methyl-l-piperizinyl)methyl)phenyl, indolyl, 1-methyl-indolyl, 4- methyl-3,4-dihydro-2H-l,4-benzoxazinyl, benzofuranyl, 4-(N 5 N- dimethylsulfonamidyl)phenyl, (3,4,5-trimethyloxy)phenyl, (3,5-dimethyl-4-hydroxy)phenyl, 3 -fluoro-4-(2-(diethylamino)ethoxy)pheny
- Z is S(O) P , wherein p is 0,1 or 2.
- R 1 is substituted or unsubstituted aryl, substituted or unsubstituted heterocycloalkyl, or substituted or unsubstituted heteroaryl, wherein the substituents are selected from F, Cl, Br, I, substituted or unsubstituted C ⁇ 6 alkyl, or substituted or unsubstituted Q -6 alkoxy, wherein the Q -6 alkyl and Q. 6 alkoxy substituents are selected from F, Cl, Br, or I.
- R 1 is substituted or unsubstituted phenyl, wherein the substituents are selected from F, Cl, Br, I, substituted or unsubstituted Ci -6 alkyl, or substituted or unsubstituted C 1-6 alkoxy, wherein the Q -6 alkyl and Ci -6 alkoxy substituents are selected from F, Cl, Br, or I.
- R la is H, F, Cl, Br, I, CF 3 , Ci -4 alkyl or Ci 4 alkoxy.
- R la is H.
- R 2 is substituted or unsubstituted heteroalkyl, substituted or unsubstituted
- heterocycloalkyl heteroalkyl, or substituted or unsubstituted fused bicyclic
- heteroalkyl (arylheterocycloalkyl)alkyl, wherein the heteroalkyl moiety has 1, 2 or 3 heteroatoms independently selected from N, O, or S.
- R 2 is substituted or unsubstituted heteroalkyl, substituted or unsubstituted
- heteroalkyl (arylheterocycloalkyl)alkyl, wherein the heteroalkyl moiety has 1, 2 or 3 heteroatoms independently selected from N, O, or S.
- R 2 is substituted or unsubstituted (heterocycloalkyl)Ci-8alkyl, wherein the heterocycloalkyl is piperidine, piperazine, pyrrolidine, tetrahydrofuran, pyran or morpholine.
- R 3 is H or Ci -6 alkyl.
- R 4 is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted alkoxyl, substituted or unsubstituted heteroalkyl having 1, 2 or 3 heteroatoms independently selected from N, O, or S; substituted or unsubstituted aralkyl, substituted or unsubstituted (heteroaryl)alkyl or substituted or unsubstituted (heterocycloalkyl)alkyl.
- X is -NHR 2 ;
- Y is substituted or unsubstituted phenyl, (substituted or unsubstituted phenylene)-OR 4 , (substituted or unsubstituted phenylene)-alkyl-OR 4 , (substituted or unsubstituted phenylene)-R 4 , (substituted or unsubstituted phenylene)-alkyl-R 4 or substituted or unsubstituted heteroaryl;
- Z is O
- R 1 is substituted or unsubstituted C 6 -ioaryl, substituted or unsubstituted heterocycloalkyl, or substituted or unsubstituted heteroaryl, wherein the substituents are selected from F, Cl, Br, I, substituted or unsubstituted C 1-6 alkyl, or substituted or unsubstituted Ci -6 alkoxy, wherein the Ci -6 alkyl and C] -6 alkoxy substituents are selected from F, Cl, Br, or I;
- R la is H
- R 2 is substituted or unsubstituted (heterocycloalkyl)alkyl, substituted or unsubstituted (heterocycloalkyl)heteroalkyl, or substituted or unsubstituted fused bicyclic (arylheterocycloalkyl)alkyl, wherein the heteroalkyl moiety has 1, 2 or 3 heteroatoms independently selected from N, O, or S; and
- R 4 is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted alkoxyl, substituted or unsubstituted heteroalkyl having 1, 2 or 3 heteroatoms independently selected from N, O, or S; substituted or unsubstituted aralkyl, substituted or unsubstituted (heteroaryl)alkyl or substituted or unsubstituted (heterocycloalkyl)alkyl.
- Y is hydrogen, halogen, haloalkyl, CN, substituted or unsubstituted cycloalkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted heteroalkyl, substituted or unsubstituted phenyl, substituted or unsubstituted naphthyl, (substituted or unsubstituted phenylene)-OR 4 , (substituted or unsubstituted phenylene)-alkyl-OR 4 , (substituted or unsubstituted phenylene)-R 4 , (substituted or unsubstituted phenylene)-alkyl-R 4 , substituted or unsubstituted aralkyl, substituted or unsubstituted saturated or partially unsaturated heterocycloalkyl, substituted or unsubstituted heteroaryl, (substituted or unsubstitute
- R 1 is substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted heterocycloalkyl, or substituted or unsubstituted heteroaryl, wherein the substituents are selected from F, Cl, Br, I, substituted or unsubstituted C 1 ⁇ alkyl, or substituted or unsubstituted Q -6 alkoxy, wherein the C 1 ⁇ alkyl and Ci -6 alkoxy substituents are selected from F, Cl, Br, or I;
- R 2 is substituted or unsubstituted (heterocycloalkytyCi-galkyl
- R 4 is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted alkoxy 1, substituted or unsubstituted heteroalkyl having 1, 2 or 3 heteroatoms independently selected from N, O, or S; substituted or unsubstituted aralkyl, substituted or unsubstituted (heteroaryl)alkyl or substituted or unsubstituted (heterocycloalkyl)alkyl; provided that
- a further embodiment is a compound of Formula III
- X is -OR 2 , -SR 2 , or -NHR 2 ;
- Y is substituted or unsubstituted cycloalkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted phenyl, substituted or unsubstituted naphthyl, (substituted or unsubstituted phenylene)-OR 4 , substituted or unsubstituted aralkyl, substituted or unsubstituted saturated or unsaturated heterocycloalkyl, substituted or unsubstituted heteroaryl, or (substituted or unsubstituted 5- or 6-member heteroarylene)-OR 4 ;
- R 1 is substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted heterocycloalkyl, or substituted or unsubstituted heteroaryl, wherein the substituents are selected from F, Cl, Br, I, substituted or unsubstituted Ci -6 alkyl, or substituted or unsubstituted Q -6 alkoxy, wherein the C]. 6 alkyl and Ci- 6 alkoxy substituents are selected from F, Cl, Br, or I;
- R la is H, F, Cl, Br, I, CF 3 , C 1-6 alkyl or C 1-6 alkoxy;
- R 2 is substituted or unsubstituted heteroalkyl, substituted or unsubstituted (cycloalkyl)heteroalkyl substituted or unsubstituted (heterocycloalkyl)alkyl, substituted or unsubstituted (heterocycloalkyl)heteroalkyl, or substituted or unsubstituted fused bicyclic (arylheterocycloakyl)alkyl, wherein the heteroalkyl moiety has 1 or 2 heteroatoms independently selected from N, O, or S; and
- R 4 is substituted or unsubstituted alkyl, substituted or unsubstituted heteroalkyl having 1 or 2 heteroatoms independently selected from N, O, or S; substituted or unsubstituted aralkyl; substituted or unsubstituted (heteroaryl)alkyl or substituted or unsubstituted (heterocycloalkyl)alkyl.
- R la is H.
- R 2 is substituted or unsubstituted (heterocycloalkyl)alkyl, wherein the heterocycloalkyl group of the (heterocycloalkyl)alkyl is a saturated ring.
- X is NHR 2 .
- R 2 is substituted or unsubstituted (heterocycloalkyl)alkyl or substituted or unsubstituted (arylheterocycloalkyl)alkyl, wherein the heterocycloalkyl group of the (heterocycloalkyl)alkyl and the arylheterocycloalkyl group of the (arylheterocycloalkyl)alkyl is tetrahydrofuranyl, dioxalanyl, dithiolanyl, dioxanyl, oxathiolanyl, oxetanyl, oxazolidinyl, dithianyl, tetrahydrothiophenyl, hexahydropyranyl, hexahydrothiopyranyl, piperazinyl, pyrrolidinyl, morpholinyl, thiomorpholinyl, 1,3
- R 1 is substituted or unsubstituted aryl, wherein the substituents are selected from F, Cl, Br, I, substituted or unsubstituted C 1 ⁇ alkyl, or substituted or unsubstituted Cj -6 alkoxy, wherein the C 1-6 alkyl and C 1 ⁇ alkoxy substituents are selected from F, Cl, Br, or I.
- Y is unsubstituted cycloalkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted phenyl, substituted or unsubstituted aralkyl, substituted or unsubstituted bicyclic heteroaryl, or (substituted or unsubstituted 5- or 6-member heteroarylene)-OR 4 .
- Y is unsubstituted cycloalkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted phenyl, substituted or unsubstituted benzyl, (substituted or unsubstituted phenylene)-OR 4 , or (substituted or unsubstituted 5- or 6-member heteroarylene)-OR 4 .
- Y is unsubstituted cycloalkyl, or (unsubstituted phenylene)-OR 4 .
- R 4 is substituted or unsubstituted (heterocycloalkyl)alkyl, or substituted or unsubstituted (heteroaryl)alkyl, wherein the heterocycloalkyl moiety or heteroaryl moiety is selected from substituted or unsubstituted pyridinyl, substituted or unsubstituted piperidinyl, substituted or unsubstituted morpholinyl, substituted or unsubstituted thiomorpholinyl, substituted or unsubstituted imidazolyl, substituted or unsubstituted hexahydropyranyl, substituted or unsubstituted oxazolidinyl, or substituted or unsubstituted pyrrolidinyl.
- R 4 is C 1-6 alkyl or Ci -6 heteroalkyl, each independently substituted with one, two or three substituents selected from F, Cl, Br, I, CN, NO 2 , oxo, NH 2 , NR 5 R 6 , C(O)NR 5 R 6 , COOR 7 , OH, OR 7 , or S(O) q R 7 , wherein R 5 and R 7 are independently unsubstituted Ci -6 alkyl, aryl, aralkyl, heterocycloalkyl, heteroaryl, (heterocycloalkyl)alkyl, or (heteroaryl)alkyl; R 6 is H or unsubstituted Ci -6 alkyl, aryl, aralkyl, heterocycloalkyl, heteroaryl, (heterocycloalkyl)alkyl, or (heteroaryl)alkyl; or R 5
- R 2 is substituted or unsubstituted (heterocycloalkyl)methyl, the heterocycloalkyl ring of the (heterocycloalkyl)methyl being a 5- or 6-member saturated ring including at least one member selected from sulfur, oxygen, sulf ⁇ nyl, or sulfonyl.
- R 1 is substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted heterocycloalkyl, or substituted or unsubstituted heteroaryl, wherein the substituents are selected from F, Cl, Br, I, substituted or unsubstituted Ci -6 alkyl, or substituted or unsubstituted Ci -6 alkoxy, wherein the Ci -6 alkyl and Ci -6 alkoxy substituents are selected from F, Cl, Br, or I.
- Y is phenyl, (substituted or unsubstituted phenylene)-OR , or (substituted or unsubstituted 5- or 6-member heteroarylene)-OR 4 .
- Still another embodiment provides a compound of formula V
- Y is substituted or unsubstituted cycloalkyl, substituted or unsubstituted phenyl, substituted or unsubstituted naphthyl, (substituted or unsubstituted phenylene)-OR 4 , substituted or unsubstituted aralkyl, substituted or unsubstituted saturated or unsaturated heterocycloalkyl, substituted or unsubstituted heteroaryl, or (substituted or unsubstituted 5- or 6-member heteroarylene)-OR 4 ;
- R is H, F, Cl, Br, I, substituted or unsubstituted C 1-6 alkyl or substituted or unsubstituted C ]-6 alkoxy, wherein the substituents are selected from F, Cl, Br, or I;
- R 2 is substituted or unsubstituted (heterocycloalkyl)methyl, the heterocycloalkyl ring of the (heterocycloalkyl)methyl being a 5- or 6-member saturated ring including at least one member selected from sulfur, oxygen, sulfinyl, or sulfonyl; and
- R 4 is substituted or unsubstituted alkyl; substituted or unsubstituted heteroalkyl having 1 or 2 heteroatoms independently selected from N, O, or S; substituted or unsubstituted aralkyl; substituted or unsubstituted heterocycloalkyl; or substituted or unsubstituted heteroaryl.
- Y is unsubstituted cycloalkyl, unsubstituted phenyl, (substituted or unsubstituted phenylene)-OR 4 , substituted or unsubstituted aralkyl, or (substituted or unsubstituted 5- or 6-member heteroarylene)-OR 4 .
- the compounds of Formulae I-V, and stereoisomers, solvates, tautomers, pharmaceutically acceptable salts and derivatives, and prodrugs of these compounds are useful for treating subjects, such as humans, with various conditions and/or disease states, as previously described.
- the invention provides pharmaceutical compositions comprising one or more of the compounds of Formula I, II, III,
- IV, or V which includes compounds according to any of the various embodiments above, and a pharmaceutically acceptable carrier or diluent.
- the compounds of Formulae I-V, or pharmaceutical composition comprising such compound(s), may be administered in an effective amount to the subject to modulate one or more targets in the subject thereby treating the target-mediated disease or condition.
- another embodiment of the invention relates to a method of treating inflammation in a mammal, the method comprising administering to the mammal a therapeutically effective amount of a compound according to any one of the above embodiments.
- Another embodiment of the invention relates to a method of inhibiting T cell activation in a mammal, the method comprising administering to the mammal a therapeutically effective amount of a compound according to any one of the above embodiments.
- Another embodiment of the invention relates to a method of treating arthritis, rheumatoid arthritis, psoriatic arthritis, or osteoarthritis in a mammal, the method comprising administering to the mammal a therapeutically effective amount of a compound according to any one of the above embodiments.
- Another embodiment of the invention relates to a method of treating organ transplant, acute transplant or heterograft or homograft rejection, or transplantation tolerance induction in a mammal, the method comprising administering to the mammal a therapeutically effective amount of a compound according to any one of the above embodiments.
- Another embodiment of the invention relates to a method of treating ischemic or reperfusion injury, myocardial infarction, or stroke in a mammal, the method comprising administering to the mammal a therapeutically effective amount of a compound according to any one of the above embodiments.
- Another embodiment of the invention relates to a method of treating multiple sclerosis, inflammatory bowel disease, including ulcerative colitis, Crohn's disease, lupus, contact hypersensitivity, delayed-type hypersensitivity, and gluten-sensitive enteropathy, type
- Barre syndrome glomerulonephritis, serum sickness, uticaria, allergic diseases, asthma, hayfever, allergic rhinitis, scleracielma, mycosis fungoides, dermatomyositis, alopecia areata, chronic actinic dermatitis, eczema, Behcet's disease, Pustulosis palmoplanteris, Pyoderma gangrenum, Sezary's syndrome, atopic dermatitis, systemic schlerosis, morphea or atopic dermatitis in a mammal, the method comprising administering to the mammal a therapeutically-effective amount of a compound according to any one of the above embodiments.
- Another embodiment of the invention relates to a method of treating colon carcinoma or thymoma in a mammal, the method comprising administering to the mammal a therapeutically-effective amount of a compound according to any one of the above embodiments.
- Another embodiment of the invention relates to a method of treating a proliferative disease in a mammal, the method comprising administering to the mammal a therapeutically effective amount of a compound according to any one of the above embodiments.
- Another embodiment of the invention relates to the method of treating a proliferative disease in a mammal, the method further comprising administering to the mammal a therapeutically effective amount of a second antiproliferative agent with the compound, which was administered to the mammal.
- the proliferative disease is cancer.
- the proliferative disease is breast cancer, lung cancer, liver cancer, kidney cancer, ovarian cancer, prostate cancer, psoriasis, prostatic hyperplasia, or a benign tumor.
- Another embodiment of the invention relates to a method for treating a tyrosine kinase-mediated disorder in a mammal, comprising administering to the mammal a therapeutically effective amount of a compound according to any one of the above embodiments.
- the tyrosine kinase is Lck or ACK-I.
- Various other embodiments of the invention relate to the manufacture of a medicament for the purposes of administering the compound of Formula I, II, III, PV, or V,, or pharmaceutical composition comprising same, to the mammal for treatment thereof, as described herein.
- the invention relates to the manufacture of a medicament comprising a compound according to any one of the above embodiments.
- Another embodiment of the invention relates to a method of manufacturing a medicament for the treatment of a tyrosine kinase-mediated disease, the method comprising combining a compound according to any one of the above embodiments with a pharmaceutical carrier to form the medicament.
- Another embodiment of the invention relates to a method of manufacturing a medicament for the treatment of inflammation, the method comprising combining a compound according to any one of the above embodiments with a pharmaceutical carrier to form the medicament.
- Another embodiment of the invention relates to a method of manufacturing a medicament for the inhibition of T cell activation and proliferation, the method comprising combining a compound according to any one of the above embodiments with a pharmaceutical carrier to form the medicament.
- Another embodiment of the invention relates to the manufacture of a medicament for the treatment of arthritis, rheumatoid arthritis, psoriatic arthritis, or osteoarthritis in a mammal comprising a therapeutically-effective amount of a compound according to any one of the above embodiments.
- Another embodiment of the invention relates to a method of manufacturing a medicament for the treatment of organ transplant, acute transplant or heterograft or homograft rejection, or transplantation tolerance induction in a mammal, the method comprising combining a compound according to any one of the above embodiments with a pharmaceutical carrier to form the medicament.
- Another embodiment of the invention relates to a method of manufacturing a medicament for the treatment of ischemic or reperfusion injury, myocardial infarction, or stroke in a mammal, the method comprising combining a compound according to any one of the above embodiments with a pharmaceutical carrier to form the medicament.
- Another embodiment of the invention relates to a method of manufacturing a medicament for the treatment of multiple sclerosis, inflammatory bowel disease, including ulcerative colitis, Crohn's disease, lupus, contact hypersensitivity, delayed-type hypersensitivity, and gluten-sensitive enteropathy, type 1 diabetes, psoriasis, contact dermatitis, Hashimoto's thyroiditis, Sjogren's syndrome, autoimmune hyperthyroidism, Addison's disease, autoimmune polyglandular disease, autoimmune alopecia, pernicious anemia, vitiligo, autoimmune hypopituatarism, Guillain-Barre syndrome, glomerulonephritis, serum sickness, uticaria, allergic diseases, asthma, hayfever, allergic rhinitis, scleracielma, mycosis fungoides, dermatomyositis, alopecia areata, chronic actinic dermatitis, eczema, Behcet's disease,
- Another embodiment of the invention relates to a method of manufacturing a medicament for the treatment of colon carcinoma or thymoma in a mammal, the method comprising combining a compound according to any one of the above embodiments with a pharmaceutical carrier to form the medicament.
- Another embodiment of the invention relates to a method of making a compound as described herein, comprising the step of reacting a 2-chloro-furanopyrimidine compound (A) having the structure
- BSA Bovine Serum Albumin
- DMSO Dimethylsulfoxide dppf: 1 , 1 ' (diphenylphosphino)ferrocene
- FCS Fetal Calf Serum g: Gram(s) h: Hour(s)
- HBTU O-Benzotriazol- 1 -y 1- ⁇ , ⁇ , ⁇ ' ,N ' -tetramethy luronium hexafluorophosphate
- IC 50 value The concentration of an inhibitor that causes a 50 % reduction in a measured activity.
- LiHMDS Lithium bis(trimethylsilyl)amide
- NBS N-Bromo succinimide
- Ni-NTA Nickel-nitriloacetic acid
- NMP N-methylpyrrolidone rt: Room temperature
- references to a certain element such as hydrogen or H is meant to include all isotopes of that element.
- an R group is defined to include hydrogen or H, it also includes deuterium and tritium.
- Compounds comprising radioisotopes such as tritium, C 1 , P 32 and S 35 are thus within the scope of the invention. Procedures for inserting such labels into the compounds of the invention will be readily apparent to those skilled in the art based on the disclosure herein.
- substituted refers to a group, such as those defined below, in which one or more bonds to a hydrogen atom contained therein are replaced by a bond to non-hydrogen or non-carbon atoms such as, but not limited to, a halogen atom such as F, Cl, Br, and I; an oxygen atom in groups such as hydroxyl groups, alkoxy groups, aryloxy groups, and ester groups; a sulfur atom in groups such as thiol groups, alkyl and aryl sulfide groups, sulfoxide groups, sulfone groups, and sulfonyl groups such as sulfonyl halides and sulfonomides; a nitrogen atom in groups such as amines, amides, alkylamines, dialkylamines, arylamines, alkylarylamines, diarylamines, N-oxides, ureas, imines, imides
- Substituted alkyl groups and also substituted cycloalkyl groups and others also include groups in which one or more bonds to a carbon(s) or hydrogen(s) atom is replaced by a bond to a heteroatom such as oxygen in carboxylic acid, ester and carbamate groups; and nitrogen in groups such as imines, oximes, hydrazones, and nitriles.
- Substituents including alkyl and ring groups, may be either monovalent or polyvalent depending on the context of their usage. For example, if description contained the group R'-R ⁇ R 3 and R 2 was defined as C ⁇ alkyl, then the R 2 alkyl would be considered polyvalent because it must be bonded to at least R 1 and R 3 . Alternatively, if R 1 was defined as Ci. 6 alkyl, then the R 1 alkyl would be monovalent (excepting any further substitution language).
- unsubstituted as used herein with reference to a group, means that the group does not have one or more bonds to a hydrogen or carbon atom contained therein replaced by a bond to non-hydrogen or non-carbon atom, as described above.
- alkyl as used herein either alone or within other terms such as “haloalkyl”, “alkylamino” and “cycloalkyl”, refers to linear, branched or cyclic radicals having one to about twelve carbon atoms. "Cycloalkyl” is also used exclusively herein to refer specifically to fully or partially saturated cyclic alkyl radicals.
- alkyl radicals include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-b ⁇ tyl, pentyl, isoamyl, hexyl, cyclopropyl, cyclopentyl, cyclohexyl and the like.
- C a - b alkyl refers to an alkyl group comprising from a to b carbon atoms in a branched, cyclical or linear relationship or any combination of the three.
- the alkyl groups described in this section may also contain double or triple bonds.
- Examples of Cj-galkyl include, but are not limited to the following:
- aralkyl refers to linear or branched aryl-containing radicals each having alkyl portions of one to about ten carbon atoms. Examples of such radicals include benzyl, 2-phenyl-propane, and the like.
- Halogen and "halo” as used herein, refers to a halogen atoms selected from F, Cl, Br and I.
- haloalkyl refers to radicals wherein any one or more of the alkyl carbon atoms is substituted with halo as defined above. Specifically embraced are monohaloalkyl, dihaloalkyl and polyhaloalkyl radicals including perhaloalkyl.
- a monohaloalkyl radical for one example, may have either an iodo, bromo, chloro or fluoro atom within the radical.
- Dihalo and polyhaloalkyl radicals may have two or more of the same halo atoms or a combination of different halo radicals.
- haloalkyl radicals include fluoromethyl, difluoromethyl, trifluoromethyl, chloromethyl, dichloromethyl, trichloromethyl, pentafluoroethyl, heptafluoropropyl, difluorochloromethyl, dichlorofluoromethyl, difluoroethyl, difluoropropyl, dichloroethyl and dichloropropyl.
- Perfluoroalkyl means alkyl radicals having all hydrogen atoms replaced with fluoro atoms. Examples include trifluoromethyl and pentafluoroethyl.
- C a . b haloalkyl refers to an alkyl group, as described above, wherein any number— at least one—of the hydrogen atoms attached to the alkyl chain are replaced by F, Cl, Br or I.
- haloalkyl includes, without limitation, trifluoromethyl, pentafluoroethyl and the like.
- heteroalkyl refers to an alkyl having one or more of the carbon atoms replaced by a heteroatom, selected from nitrogen, oxygen and sulfur.
- a heteroalkyl would include an ether or a thioether chain, or an alkoxide moiety, wherein the heteroatom is in the linear region of the moeity.
- the term also includes moieties where the heteroatom is in a branched region.
- the term includes 2-amino-n- hexane or 5-hydroxy-pentane.
- hydroxyalkyl refers to linear or branched alkyl radicals having one to about ten carbon atoms any one of which may be substituted with one or more hydroxyl radicals. Examples of such radicals include hydroxymethyl, hydroxy ethyl, hydroxypropyl, hydroxyburyl and hydroxyhexyl.
- alkoxy refers to linear or branched oxy-containing radicals each having alkyl portions of one to about ten carbon atoms. Examples of such radicals include methoxy, ethoxy, propoxy, butoxy and tert-butoxy. Alkoxy radicals may be further substituted with one or more halo atoms, such as fluoro, chloro or bromo, to provide
- haloalkoxy radicals examples include fluoromethoxy, chloromethoxy, trifluoromethoxy, trifluoroethoxy, fluoroethoxy and fluoropropoxy.
- sulfonyl refers respectively to divalent radicals -SO 2 -.
- aryl refers to a carbocyclic aromatic system containing one, two or three rings wherein such rings may be attached together in a fused manner.
- aryl includes, without limitation, aromatic radicals such as phenyl, naphthyl, indenyl, tetrahydronaphthyl, and indanyl.
- the "aryl” group may have 1 to 3 substituents such as alkyl, hydroxyl, halo, haloalkyl, nitro, cyano, alkoxy and alkylamino.
- Aryl also includes the moiety wherein the aromatic carbocycle is fused with a
- C 3 .6cycloalkyl bridge wherein the bridge optionally includes 1, 2 or 3 heteroatoms selected from N, O and S.
- the bridge optionally includes 1, 2 or 3 heteroatoms selected from N, O and S.
- phenyl substituted with -0-CH 2 -O- forms the aryl benzodioxolyl substituent.
- heterocyclic refers to saturated, partially saturated and unsaturated (aromatic) heteroatom-containing ring radicals, where the heteroatoms may be selected from nitrogen, sulfur and oxygen. Accordingly “heterocyclic” includes both
- heterocycloalkyl and “heteroaryl” radicles.
- heterocycloalkyl refers to saturated and partially saturated (or partially unsaturated) heteroatom-containing ring radicals, where the heteroatoms may be selected from nitrogen, sulfur and oxygen. It does not include rings containing -O-O-,-O-S- or -S-S- portions.
- Said "heterocycloalkyl” group may have 1 to 3 substituents such as hydroxyl, Boc, halo, haloalkyl, cyano, lower alkyl, oxo, alkoxy, amino and alkylamino.
- saturated heterocycloalkyl radicals include saturated 3 to 6-membered heteromonocyclic groups containing 1 to 4 nitrogen atoms [e.g. pyrrolidinyl, imidazolidinyl, piperidinyl, pyrrolinyl, piperazinyl]; saturated 3 to 6-membered heteromonocyclic group containing 1 to 2 oxygen atoms and 1 to 3 nitrogen atoms [e.g. morpholinyl]; saturated 3 to 6- membered heteromonocyclic group containing 1 to 2 sulfur atoms and 1 to 3 nitrogen atoms [e.g., thiazolidinyl].
- nitrogen atoms e.g. pyrrolidinyl, imidazolidinyl, piperidinyl, pyrrolinyl, piperazinyl
- saturated 3 to 6-membered heteromonocyclic group containing 1 to 2 oxygen atoms and 1 to 3 nitrogen atoms e.g. morpholinyl
- heteroaryl refers fully to unsaturated heteroatom- containing ring radicals, where the heteroatoms may be selected from nitrogen, sulfur and oxygen.
- heteroaryl radicals include unsaturated 5 to 6 membered heteromonocyclyl group containing 1 to 4 nitrogen atoms, for example, pyrrolyl, imidazolyl, pyrazolyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, pyrimidyl, pyrazinyl, pyridazinyl, triazolyl [e.g., 4H-l,2,4-triazolyl, lH-l,2,3-triazolyl, 2H-l,2,3-triazolyl]; unsaturated 5- to 6-membered heteromonocyclic group containing an oxygen atom, for example, pyranyl, 2-furyl, 3-furyl, etc.; unsaturated 5 to 6-membered heteromonocyclic group containing a sulfur atom, for example, 2-thienyl, 3-thienyl, etc.; unsaturated 5- to 6-membered heteromonocyclic group containing 1 to 2
- heteroaryl also embraces radicals where heterocyclic radicals are fused/condensed with aryl radicals (also referred to herein as “arylheterocycloalkyl”): unsaturated condensed heterocyclic group containing 1 to 5 nitrogen atoms, for example, indolyl, isoindolyl, indolizinyl, benzimidazolyl, quinolyl, isoquinolyl, indazolyl, benzotriazolyl, tetrazolopyridazinyl [e.g., tetrazolo [l,5-b]pyridazinyl]; unsaturated condensed heterocyclic group containing 1 to 2 oxygen atoms and 1 to 3 nitrogen atoms [e.g.
- Exemplary heterocyclic radicals include five to ten membered fused or unfused radicals.
- heteroaryl radicals include quinolyl, isoquinolyl, imidazolyl, pyridyl, thienyl, thiazolyl, oxazolyl, furyl, and pyrazinyl.
- Other exemplary heteroaryl radicals are 5- or 6-membered heteroaryl, containing one or two heteroatoms selected from sulfur, nitrogen and oxygen, selected from thienyl, furyl, pyrrolyl, indazolyl, pyrazolyl, oxazolyl, triazolyl, imidazolyl, pyrazolyl, isoxazolyl, isothiazolyl, pyridyl, piperidinyl and pyrazinyl.
- saturated or unsaturated means a substitutent that is completely saturated, completely unsaturated, or has any degree of unsaturation in between.
- saturated or unsaturated 6-membered ring carbocycle would include phenyl, cyclohexyl, cyclohexenyl and cyclohexadienyl.
- salt refers to a salt form of a free base compound of the present invention, as appreciated by persons of ordinary skill in the art. Salts may be prepared by conventional means, known to those skilled in the art.
- pharmaceutically- acceptable when used in reference to a salt, refers to salt forms of a given compound, which are within governmental regulatory safety guidelines for ingestion and/or administration to a subject.
- pharmaceutically-acceptable salts embraces salts commonly used to form alkali metal salts and to form addition salts of free acids or free bases. The nature of the salt is not critical, provided that it is pharmaceutically-acceptable.
- Suitable pharmaceutically-acceptable acid addition salts of compounds of Formulae I-V may be prepared from an inorganic acid or from an organic acid.
- inorganic acids are hydrochloric, hydrobromic, hydroiodic, nitric, carbonic, sulfuric and phosphoric acid.
- organic acids may be selected from aliphatic, cycloaliphatic, aromatic, arylaliphatic, heterocyclic, carboxylic and sulfonic classes of organic acids, example of which are formic, acetic, adipic, butyric, propionic, succinic, glycolic, gluconic, lactic, malic, tartaric, citric, ascorbic, glucuronic, maleic, fumaric, pyruvic, aspartic, glutamic, benzoic, anthranilic, mesylic, 4-hydroxybenzoic, phenylacetic, mandelic, embonic (pamoic), methanesulfonic, ethanesulfonic, ethanedisulfonic, benzenesulfonic, pantothenic, 2- hydroxyethanesulfonic, toluenesulfonic, sulfanilic, cyclohexylaminosulfonic, camphoric, camphors
- I-V include metallic salts, such as salts made from aluminum, calcium, lithium, magnesium, potassium, sodium and zinc, or salts made from organic bases including primary, secondary and tertiary amines, substituted amines including cyclic amines, such as caffeine, arginine, diethylamine, N-ethyl piperidine, aistidine, glucamine, isopropylamine, lysine, morpholine,
- the basic nitrogen-containing groups of compounds of Formulae I-V can be quaternized with such agents as lower alkyl halides including, without limitation, methyl, ethyl, propyl, and butyl chloride, bromides and iodides; dialkyl sulfates including dimethyl, diethyl, dibutyl, and diamyl sulfates, long chain halides such as decyl, lauryl, myristyl and stearyl chlorides, bromides and iodides, aralkyl halides like benzyl and phenethyl bromides, and others. Water or oil-soluble or dispersible products may be obtained by quaternizing such basic nitrogen groups in compounds of Formulae I-V.
- lower alkyl halides including, without limitation, methyl, ethyl, propyl, and butyl chloride, bromides and iodides
- dialkyl sulfates including dimethyl, die
- “Pharmaceutically acceptable” when used with reference to a derivative is consistent in meaning with reference to a salt, and refers to a derivative that is pharmacologically safe for consumption, generally as determined by a governmental or authorized regulatory body.
- leaving group refers to groups readily displaceable by a nucleophile, such as an amine, a thiol or an alcohol nucleophile. Such leaving groups are well known in the art. Examples of such leaving groups include, but are not limited to,
- N-hydroxysuccinimide N-hydroxybenzotriazole, halides, triflates, tosylates and the like.
- Protecting group refers to groups well known in the art which are used to prevent selected reactive groups, such as carboxy, amino, hydroxy, mercapto and the like, from undergoing undesired reactions, such as nucleophilic, electrophilic, oxidation, reduction and the like.
- Protecting groups are indicated herein where appropriate. Examples of amino protecting groups include, but are not limited to, aralkyl, substituted aralkyl, cycloalkenylalkyl and substituted cycloalkenyl alkyl, allyl, substituted allyl, acyl, alkoxycarbonyl, aralkoxycarbonyl, silyl and the like.
- aralkyl examples include, but are not limited to, benzyl, ortho-methylbenzyl, trityl and benzhydryl, which can be optionally substituted with halogen, alkyl, alkoxy, hydroxy, nitro, acylamino, acyl and the like, and salts, such as phosphonium and ammonium salts.
- aryl groups include phenyl, naphthyl, indanyl, anthracenyl, 9-(9-phenylfluorenyl), phenanthrenyl, durenyl and the like.
- cycloalkenylalkyl or substituted cycloalkylenylalkyl radicals include, but are not limited to, cyclohexenyl methyl and the like.
- Suitable acyl, alkoxycarbonyl and aralkoxycarbonyl groups include benzyloxycarbonyl, t-butoxycarbonyl, iso-butoxycarbonyl, benzoyl, substituted benzoyl, butyryl, acetyl, tri- fluoroacetyl, tri-chloro acetyl, phthaloyl and the like.
- a mixture of protecting groups can be used to protect the same amino group, such as a primary amino group can be protected by both an aralkyl group and an aralkoxycarbonyl group.
- Amino protecting groups can also form a heterocyclic ring with the nitrogen to which they are attached, for example, l,2-bis(methylene)benzene, phthalimidyl, succinimidyl, maleimidyl and the like and where these heterocyclic groups can further include adjoining aryl and cycloalkyl rings.
- the heterocyclic groups can be mono-, di- or tri-substituted, such as nitrophthalimidyl.
- Amino groups may also be protected against undesired reactions, such as oxidation, through the formation of an addition salt, such as hydrochloride, toluenesulfonic acid, trifluoroacetic acid and the like.
- an addition salt such as hydrochloride, toluenesulfonic acid, trifluoroacetic acid and the like.
- Many of the amino protecting groups, including aralkyl groups for example, are also suitable for protecting carboxy, hydroxy and mercapto groups.
- Alkyl groups are also suitable groups for protecting hydroxy and mercapto groups, such as tert-butyl.
- Silyl protecting groups are groups containing silicon atoms which are optionally substituted by one or more alkyl, aryl and aralkyl groups.
- Suitable silyl protecting groups include, but are not limited to, trimethylsilyl, triethylsilyl, tri-isopropylsilyl, tert- butyldimethylsilyl, dimethy lphenylsilyl, 1 ,2-bis(dimethy lsilyl)benzene, l,2-bis(dimethylsilyl)ethane and diphenylmethylsilyl.
- Silylation of an amino groups provide mono- or di-silylamino groups. Silylation of aminoalcohol compounds can lead to a N 5 N, O- tri-silyl derivative.
- silyl function from a silyl ether function is readily accomplished by treatment with, for example, a metal hydroxide or ammonium fluoride reagent, either as a discrete reaction step or in situ during a reaction with the alcohol group.
- Suitable silylating agents are, for example, trimethylsilyl chloride, tert-butyl-dimethylsilyl chloride, phenyldimethylsilyl chloride, diphenylmethyl silyl chloride or their combination products with imidazole or DMF.
- Methods for silylation of amines and removal of silyl protecting groups are well known to those skilled in the art.
- Methods of preparation of these amine derivatives from corresponding amino acids, amino acid amides or amino acid esters are also well known to those skilled in the art of organic chemistry including amino acid/amino acid ester or aminoalcohol chemistry.
- Protecting groups are removed under conditions which will not affect the remaining portion of the molecule. These methods are well known in the art and include acid hydrolysis, hydrogenolysis and the like.
- One method involves removal of a protecting group, such as removal of a benzyloxycarbonyl group by hydrogenolysis utilizing palladium on carbon in a suitable solvent system such as an alcohol, acetic acid, and the like or mixtures thereof.
- a t-butoxycarbonyl protecting group can be removed utilizing an inorganic or organic acid, such as HCl or trifluoroacetic acid, in a suitable solvent system, such as dioxane or methylene chloride.
- the resulting amino salt can readily be neutralized to yield the free amine.
- Carboxy protecting group such as methyl, ethyl, benzyl, tert-butyl, 4- methoxyphenylmethyl and the like, can be removed under hydrolysis and hydrogenolysis conditions well known to those skilled in the art.
- Prodrugs of the compounds of this invention are also contemplated by this invention.
- a "prodrug” is a compound, which when administered to the body of a subject (such as a mammal), breaks down in the subject's metabolic pathway to provide an active compound of Formula I, II, III, IV, or V. More specifically, a prodrug is an active or inactive "masked" compound that is modified chemically through in vivo physiological action, such as hydrolysis, metabolism and the like, into a compound of this invention following administration of the prodrug to a subject or patient.
- prodrugs are well known by those skilled in the art.
- a prodrug is a masked carboxylic acid group.
- a masked carboxylate anion include a variety of esters, such as alkyl (for example, methyl, ethyl), cycloalkyl (for example, cyclohexyl), aralkyl (for example, benzyl, p-methoxybenzyl), and alkylcarbonyloxyalkyl (for example, pivaloyloxymethyl).
- esters such as alkyl (for example, methyl, ethyl), cycloalkyl (for example, cyclohexyl), aralkyl (for example, benzyl, p-methoxybenzyl), and alkylcarbonyloxyalkyl (for example, pivaloyloxymethyl).
- Amines have been masked as arylcarbonyloxymethyl substituted derivatives which are cleaved by esterases in vivo releasing the free drug and formaldehy
- stereoisomers includes enantiomers, diastereomers, atropisomers and geometric isomers. Stereoisomers generally possess different chemical properties and/or biological activity, as appreciated by those skilled in the art. For example, one stereoisomer may be more active and/or may exhibit beneficial effects in comparison to other stereoisomer(s) or when separated from the other stereoisomer(s). However, it is well within the skill of the ordinary artisan to separate, and/or to selectively prepare said stereoisomers. Accordingly, “stereoisomers” of the present invention necessarily include mixtures of stereoisomers, including racemic mixtures, individual stereoisomers, and optically active forms.
- solvate when used with reference to a compound refers to a compound, which is associated with one or more molecules of a solvent, such as an organic solvent, inorganic solvent, aqueous solvent or mixtures thereof.
- a solvent such as an organic solvent, inorganic solvent, aqueous solvent or mixtures thereof.
- the compounds of Formula Formulae I-V may also be solvated, especially hydrated. Hydration may occur during manufacturing of the compounds or compositions comprising the compounds, or the hydration may occur over time due to the hygroscopic nature of the compounds.
- Compounds of the invention may exist as organic solvates as well, including DMF, ether, and alcohol solvates among others. The identification and preparation of any particular solvate is within the skill of the ordinary artisan of synthetic organic or medicinal chemistry.
- Cytokine refers to a secreted protein that affects the functions of other cells, particularly as it relates to the modulation of interactions between cells of the immune system or cells involved in the inflammatory response.
- cytokines include but are not limited to interleukin 1 (IL-I), such as IL-IB, interleukin 6 (IL- 6), interleukin 8 (IL-8) and TNF, such as TNF- ⁇ (tumor necrosis factor- ⁇ ).
- treatment includes therapeutic treatment as well as prophylactic treatment (either preventing the onset of disorders altogether or delaying the onset of a preclinically evident stage of disorders in individuals).
- terapéuticaally-effective is intended to qualify the amount of each compound of Formula I, II, III, IV, or V, which will achieve the goal of treatment, for example, improvement in disorder severity and the frequency of incidence over treatment of each agent by itself, while avoiding adverse side effects typically associated with alternative therapies.
- Lck- or ACK-I -mediated disease or disease state refers to all disease states wherein Lck and/or ACK-I plays a role, either directly as Lck and/or ACK-I itself, or by Lck and/or ACK-I inducing another cytokine or disease-causing agent to be released.
- the specification and claims contain listing of species using the language “selected from . . . and . . .” and “is . . . or . . .” (sometimes referred to as Markush groups). When this language is used in this application, unless otherwise stated it is meant to include the group as a whole, or any single members thereof, or any subgroups thereof. The use of this language is merely for shorthand purposes and is not meant in any way to limit the removal of individual elements or subgroups from the genus.
- Compounds of Formulae I-V can be synthesized according to one or more of the following schematic procedures and specific methods wherein the substituents are as defined for Formulae I-V, above, except where further noted.
- the procedures and methods as shown relate to preparation of compounds having unspecified stereochemistry. However, such procedures and methods are generally applicable to those compounds of a specific stereochemistry, e.g., where the stereochemistry about a group is (S) or (R).
- the compounds having one stereochemistry e.g., (R)
- nitrogen atoms exhibiting less than full valency are intended to bear a number of hydrogen atoms that would satisfy full valency for an amine (i.e., "-N(H)- " or "-NH 2 ").
- oxygen atoms depicted in the structures herein and exhibiting less than full valency are to be interpreted as hydroxyl groups (i.e., "-OH").
- Scheme 1 describes a general method for preparing R 1 and R la substituted furano- pyrimidinones, which can be converted into the corresponding furano-pyrimidines.
- An optionally substituted 2-hydroxyethanone 1 can be reacted with an optionally substituted malanonitrile under basic conditions in the presence of a solvent to afford the amino-furan 2.
- Compound 2 can then be reacted with acetic anhydride in formic acid to produce furo[2,3- d]pyrimidin-4(3H)-one 3.
- desired R 1 groups such as aryl R 1 groups, and R la groups can be built into the furanopyrimidine core simultaneously.
- the specific methods below exemplify the synthesis of one possible compound 3 which can be made by this route.
- Step Two Over an ice water bath and into a 500 mL round bottom flask was placed acetic anhydride (100 mL), and formic acid (50 mL). The reaction was allowed to stir 20 minutes before adding 2-amino-4-phenylfuran-3-carbonitrile (step l, 10 g, 54.3 mmol), and stirred for another 20 minutes at 0 0 C. The reaction flask was allowed to reach RT, then equipped with a reflux condenser, and heated at reflux for 24 hours.
- reaction mixture was concentrated under reduced pressure. Water (200 mL) was added over an ice bath, and the mixture was extracted with ethyl acetate (3 x 200 mL). The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure.
- Scheme 2 describes an alternative general method for preparing R 1 and R la substituted furano-pyrimidinones 3, and chloro furanopyrimidines 9, as intermediates which can then be converted into the corresponding desired furano-pyrimidines.
- An optionally substituted aldehyde 4, such as benzaldehyde, can be reacted with bromonitromethane under basic conditions in the presence of a suitable solvent to afford the chloro-nitro olefin 6.
- Compound 6 can then be reacted with dihydroxypyrimidine in basic conditions to produce furo[2,3-d]pyrimidin-4(3H)-one 3.
- Compound 3 can then be treated with an iodide source, such as N-iodosuccinimide, in a suitable solvent under heat to afford the iodo-compound 8.
- the iodo intermediate 8 can be reacted with a chloride source, such as phosphorus oxychloride, to afford the corresponding intermediate 9.
- desired R 1 groups such as aryl R 1 groups, and R la groups can be built into the furanopyrimidine core simultaneously.
- Step 1 Synthesis of (2-Chloro-2-nitro-vinyl)-benzene
- Step 2 Synthesis of 5-Phenyl-3H-furo[2,3-d]pyrimidin-4-one (10) [0153] The title compound 10 was prepared by the method of Dauzonne, D.; Adam-Launay, A. Tetrahedron 1992, 48, 3069-3080.
- Step 3 Synthesis of 6-Iodo-5-phenyl-3H-furo[2,3-d]pyrimidin-4-one (11)
- Compound 10 1.0 g, 4.7 mmol was dissolved in dichloroethane (50 mL) and CH 3 CN (50 mL) and NIS (1.7 g, 7.1 mmol) was added. The flask was equipped with a reflux condenser and the mixture was heated at reflux for 24 h. After cooling to RT and concentration, the crude reaction mixture was taken up in EtOAc and water. K 2 CO 3 was added to raise the pH of the aqueous phase. After removal of the organic phase, the aqueous phase was extracted several times with EtOAc. The organics were then dried over Na 2 SC ⁇ and concentrated to a brown oil. Flash chromatography (SiO 2 , gradient eluent: 25% EtOAc/Hexanes to 100% EtOAc) afforded 11.
- Step 4 Synthesis of 4-Chloro-6-iodo-5-phenyl-furo[2,3-d]pyrimidine (12) [0157]
- Compound 11 (0.8 g, 2.35 mmol) was refluxed in phosphorous oxychloride (12 mL) for 90 minutes. The reaction mixture was concentrated and diluted with ice-cold water, extracted with dichloromethane. The organic layer was washed with water and dried over MgS ⁇ 4 . The organic solvent upon concentration provided a pale solid, which on trituration with MeOH provided the title compound 12.
- Compound 8 can then be reacted with a suitable chloride source, such as POCI 3 under heat to produce chloro-furo[2,3-d]pyrimidines 13.
- Compound 14 can be reacted with a bromide source, such as N-bromosuccinimide (commonly referred to as NBS) to afford the 6-bromo substituted furano-pyrimidine intermediate 15.
- NBS N-bromosuccinimide
- a desired Y-substituted boronic ester 16 can be used in a Suzuki-type reaction in the presence of a suitable palladium catalyst, under basic conditions to afford the desired 4-amino-5,6-disubstituted furano-pyrimidines 17.
- the Suzuki reaction conditions may vary.
- any of a variety of palladium catalysts may be used, and the reaction may require heat depending upon the particular Y substrate, as appreciated by those skilled in the art.
- Y is an aromatic moiety, such as phenyl
- the reaction may be complete in a short period of time with heat.
- the boronic ester need not be a cyclic boronate as shown, but may be any suitable desired boronic acid having the general formula (RO) 2 B-Y.
- desired X groups such as amino X groups
- Y groups such as aryl Y groups
- Step 1 Preparation of 4-chloro-5-phenylfuro[2,3-d]pyrimidine (18)
- Step 2 Preparation of N-isopropyl-5-phenylfuro[2,3-d]pyrimidin-4-amine (19) [0166] Into a 50 mL round bottom flask was placed 4-chloro-5-phenylfuro[2,3- d]pyrimidine, isopropanol (25 mL), and isopropylamine (1.5 g , 25 mmol). The flask was equipped with a magnetic stir bar, a reflux condenser, and argon balloon and the reaction was brought to reflux with stirring for 20 hours.
- Step 4 Preparation of N-isopropyl-5-phenyl-6-(4-(2-(pyrrolidin-l- yl)ethoxy)phenyl)furo[2,3-d]pyrimidin-4-amine (21)
- An optionally substituted iodo-pyrimidinone 8 can be reacted with a desired Y-substituted boronic ester 13 in a Suzuki-type reaction in the presence of a suitable palladium catalyst and under basic conditions to afford 6-substituted furano-pyrimidinones 22.
- the boronic species may be either a boronic acid or a boronic ester as desired.
- the boronic ester need not be a cyclic boronate as shown, but may be any suitable desired boronic ester having the general formula (RO) 2 B-Y.
- the palladium catalyst may vary, and the reaction may require heat depending upon the particular Y substrate, as appreciated by those skilled in the art. For example, where Y is an aromatic moiety, such as phenyl, the reaction may be complete in a short period of time with heat.
- Compound 22 can then be reacted with a suitable chloride source, such as POCl 3 under heat to produce chloro-furo[2,3-d]pyrimidines 23.
- Step 1 Synthesis of 5-phenyl-6-(4-(2-(pyrrolidin-l-yl)ethoxy)phenyl)furo[2,3- d]pyrimidin-4(3H)-one (26)
- Step 2 Synthesis of 4-chloro-5-phenyl-6-(4-(2-(pyrrolidin-l- yl)ethoxy)phenyl)furo[2,3-d]pyrimidine (27)
- Step 3 Synthesis of N-(2-(4-ethylpiperazin ⁇ l-yl)ethyl)-5-phenyl-6-(4-(2-(pyrrolidin-)
- Step 1 Synthesis of 5-phenyl-N-(2-(4-piperidinyl)ethyl)-6-(4-((2-(l - pyrrolidinyl)ethyl)oxy)phenyl)furo[2,3-d]pyrimidin-4-amine (30) [0189] N-(2-(l-benzylpiperidin-4-yl)ethyl)-5-phenyl-6-(4-(2-(pyrrolidin-l- yl)ethoxy)phenyl)furo[2,3-d]pyrimidin-4-amine 29 (Prepared in a manner analogous to Steps 1-3 of Example 5; 20 mg, 0.033 mmol), ammonium bicarbonate ( ⁇ 30 mg, 0.5 mmol) and 10% Pd/C ( ⁇ 5mg) were taken up in MeOH (3-5 mL).
- Scheme 5 describes a general method for preparing 4-substituted-iodo furanopyrimidines 31 as intermediates, which can then be converted into the corresponding desired Y substituted furano-pyrimidinones 32.
- X nucleophile
- the 4-X substituted furanopyrimidine 31 can be reacted with a Y- substituted boronic ester 13 in a Suzuki-type reaction in the presence of a suitable palladium catalyst and under basic conditions to afford 4-X-6-Y substituted furano-pyrimidinones 32.
- the boronic ester need not be a cyclic boronate as shown, but may be any suitable desired boronic ester having the general formula (RO) 2 B-Y.
- the palladium catalyst may vary, and the reaction may require heat depending upon the particular Y substrate, as appreciated by those skilled in the art.
- Y is an aromatic moiety, such as phenyl
- the reaction may be complete in a short period of time with heat, hi this fashion, desired X groups, such as amino X groups, and Y groups such as aryl Y groups, can be installed into the furanopyrimidine core.
- desired X groups such as amino X groups
- Y groups such as aryl Y groups
- Step 1 Synthesis of 6-iodo-N-isopropyl-5-phenylfuro[2,3-d]pyrimidin-4-amine (34) [0195] A solution of 9 (190 mg , 0.533 mmol), isopropylamine (0.45 mL, 5.33 mmol), and DIEA (0.14 mL, 0.799 mmol) in IPA (4 mL) was heated at 80 0 C for 5h, then cooled and allowed to stand at 0 0 C over night. Filtration and washing with MeOH afforded the title compound 33.
- Step 2 Synthesis of 4-(4-(isopropylamino)-5-phenylfuror23-d]pyrimidin-6-vi)-N,N- dimethylbenzenesulfonamide (34)
- Step 1 Synthesis of 6-(4-((2-(methyloxy)ethyl)oxy)phenyl)-5-phenyl-N-(2-(l- piperaziny l)ethy l)furo [2,3 -d] pyrimidin-4-amine (36)
- Step 1 Synthesis of 6-(3 -(methy loxy)-4-((2-( 1 -piperidinyl)ethyl)oxy)pheny l)-N-(2- (4-methyl-l-piperazinyl)ethyl)-5-phenylfuro[2,3-d]pyrimidin-4-amine (38) [0203] Compound 37 (Prepared in a manner analogous to Steps 1-2 of Example 7; 50 mg, 0.089 mmol) and NaBH(OAc) 3 (40 mg, 0.539) were combined in CH 3 CN (5.5 mL). After stirring at rt for 10 min.
- Step 1 Synthesis of tert-butyl 4-(2-(6-(4-((4-methy lpiperazin- 1 -yl)methy l)phenyl)-
- Step 2 Synthesis of 6-(4-((4-methyl-l-piperazinyl)methyl)phenyl)-5-phenyl-N-(2-
- Step 1 Synthesis of 6-(4-((4-methyl-l-piperazinyl)methyl)phenyl)-5-phenyl-N-
- Compound 42 was prepared from Compound 10 by a method analogous to steps 1-3 of Example 3 (Method B), and converted to the title compound 43 by reductive amination in a manner analogous to the procedure described Step 2 of Example 10. Mass for C29H33N5O 2 found: 484 (M+H 4 ).
- An optionally substituted pyrimidinone 3 can be reacted with a bromide source, such as N-bromosuccinimide (commonly referred to as NBS) to afford the 6-bromo substituted furano-pyrimidine intermediate 44.
- a bromide source such as N-bromosuccinimide (commonly referred to as NBS)
- a desired Y-substituted boronic ester 13 can be reacted with compound 44 in a Suzuki-type reaction in the presence of a suitable palladium catalyst and under basic conditions to afford 6-substituted furano- pyrimidinones 22.
- the boronic ester need not be a cyclic boronate as shown, but may be any suitable desired boronic ester having the general formula (RO) 2 B-Y.
- the palladium catalyst may vary, and the reaction may require heat depending upon the particular Y substrate, as appreciated by those skilled in the art. For example, where Y is an aromatic moiety, such as phenyl, the reaction may be complete in a short period of time with heat.
- Compound 22 can then be reacted with a suitable chloride source, such as POCl 3 under heat to produce chloro- furo[2,3-d]pyrimidines 23.
- Heat may or may not be required to effect the transformation depending upon the particular substrates involved.
- desired X groups such as amino X groups
- Y groups such as aryl Y groups
- the specific methods below exemplify the synthesis of compound 17 which can be made by this general method.
- Step 2 Preparation of 5-phenyl-6-(4-(2-(pyrrolidin-l -yl)ethoxy)phenyl)furo[2,3- d]pyrimidin-4(3H)-one (46)
- Step 3 Preparation of 4-chloro-5-phenyl-6-(4-(2-(pyrrolidin- 1 - yl)ethoxy)phenyl)furo[2,3-d]pyrimidine (47)
- the reaction was brought to reflux with some of the solvent having been distilled into the dean-stark trap.
- the reaction was allowed to cool to room temperature and phosphorous oxychloride (300 mg, 1.96 mmol) was added dropwise.
- the reaction was heated at reflux for
- Step 4 Preparation of N-cyclopropyl-5-phenyl-6-(4-(2-(pyrrolidin-l- y l)ethoxy)pheny l)furo [2,3 -d] pyrimidin-4-amine (48)
- Scheme 7 describes a general method for preparing 4-alkoxy-5,6-disubstituted furanopyrimidines 50 (R 2 is alkyl) directly from the 4-chloro-substituted furano-pyrimidine intermediate 49.
- Compound 49 can be treated with ammonia, such as ammonium in a suitable alkoxide-based solvent, such as methanol, to afford the desired 4-alkoxy-furano-pyrimidine 50.
- desired R 1 , R la , and Y groups such as aryl Y groups, can remain constant on the furanopyrimidine core while the 4-position can then be functionalized as desired.
- the specific methods below exemplify the synthesis of compound 50 which can be made by this general method.
- the palladium catalyst may vary, and the reaction may require heat depending upon the particular R 4 substitution(s), as appreciated by those skilled in the art.
- a more conventional method involving metalation chemistry halogen-metal exchange
- Such intermediates 55 may be utilized as desired Y substitutions when synthesizing the targeted furano-pyrimidine compounds. The specific methods below exemplify the synthesis of compound 55 which can be made by this general method.
- Step 1 Synthesis of 2-(4-bromo-2-fluorophenoxy)-N,N-diethylethanamine (56)
- 4-Bromo-2-fluorophenol 1.0 g, 5.24 mmol
- 2-chloro-N,N-diethylethanamine HCL (0.90 mg, 5.24 mmol)
- Cs 2 CO 3 8 g, 25 mmol
- DMF 20 mL
- H 2 O was added and the mixture was extracted with EtOAc, then washed with 2N NaOH and brine.
- the organic layers were combined, dried over Na 2 SO 4 and concentrated to a brown oil.
- Step 2 Synthesis of N,N-diethyl-2-(2-fluoro-4-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)phenoxy)ethanamine (57)
- Compound 56 500 nig, 1.72 mmol
- 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)-l,3,2-dioxaborolane 526 mg, 2.07 mmol
- KOAc (507 mg, 5.17 mmol)
- Pd(dppf) 2 Cl 2 38 mg, 0.052 mmol
- Step 1 Synthesis of l-(4-iodophenylsulfonyl)-4-methylpiperazine (58)
- Step 2 Synthesis of l-methyl-4-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)phenylsulfonyl)piperazine (59)
- Step 1 Preparation of (4-ethylpiperazin-l-yl)(4-iodophenyl)methanone (63)
- Mo a 100 mL round bottom flask was placed 4-iodobenzoyl chloride (2.67 g, 0.01 mmol) and dichloromethane (25 mL). The flask was placed in an ice water bath. Then N- ethylpiperazine (2.6 mL, 0.021 mmol) was added dropwise with stirring. The reaction was allowed to stir for 10 days at room temperature.
- Step 2 Preparation of (4-ethylpiperazin-l-yl)(4-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)phenyl)methanone (64)
- furanopyrimidines may generally be prepared using an appropriately substituted 2-amino-3-cyano-furan. Reaction of the furan with formic acid and heat, e.g., at reflux, provides the 4-hydroxy furanopyrimidine. Conversion of the hydroxyl to bromine may be accomplished using a brominating agent such as PBr 3 . The resulting 4- bromofuranopyrimidine may be reacted with a suitable amine, thiol or alcohol in the presence of base to give the product furanopyrimidine compounds.
- Exemplary bases that may be used include DIEA, other tertiary amines, sodium hydride and potassium carbonate.
- Suitable solvents include, e.g., 1-butanol.
- a hydroxyphenyl ketone may be reacted with an alkylating agent (e.g. R 4 -I) in the presence of a suitable base, such as potassium or cesium carbonate, and solvent (e.g., DMF) to give the O-alkylated product.
- a suitable base such as potassium or cesium carbonate
- solvent e.g., DMF
- the benzylic position may be brominated with bromine in dioxane or another suitable agent.
- the resulting product is cyclized to the furan by reaction with malonitrile and base (e.g. triethylamine) in a suitable solvent such as DMF.
- the 2-amino-3-cyanofurans may be converted to furanopyrimidines as shown in Scheme 9.
- the hydroxyphenyl ketone may be protected as the methyl ether until after the formation of the furanopyrimidine.
- the methyl ether may then be deprotected with a suitable agent (e.g. BBr 3 ) and alkylated with another group, such as chloroethylamines of various types.
- Scheme 11 shows an alternative route to furanopyrimidines of the invention.
- Heating bromonitromethane with a benzaldehyde in the presence of KF and the hydrochloride salt of a base such as dimethylamine provides the desired chloro-nitro-vinyl compound.
- Any suitable solvent such as xylenes and the like may be used.
- Subsequent reaction with the dihydroxypyrimidine in the presence of base and a polar solvent e.g. ethanol
- a suitable dehydrating reagent such as POCI 3
- the chloride may be displaced with an R 2 amine to provide the precursor for Suzuki coupling at the 6-position as described below.
- Furanopyrimidines of the invention may also be prepared by Suzuki coupling of aryl and heteroaryl groups with the furanopyrimidine core as shown in Scheme 12.
- the trisubstituted furan may be heated with formamide to give the 4-amine-6-aryl- furanopyrimidine.
- Bromination at position 5 with a suitable reagent e.g., bromine or N- bromosuccinimide
- bisprotection of the amino group e.g., Boc 2 O, DMAP
- the 5-iodo analog may be similarly prepared with NIS as described above.
- Panel B of Scheme 12 shows a related route to Suzuki coupling of aryl and heteroaryl groups at position 6 (Y) of the furanopyrimidine. Formation of the furanopyrimidine and bromination as before gives the 6-bromo compound.
- the R 2 moiety may be installed at this point, followed by the Pd(O) catalyzed cross-coupling reaction, or, as shown in Panel C, the Suzuki coupling may be carried out first followed by alkylation of the 4-amino group. It will be understood that many variations of Pd(O) catalyzed cross-coupling reactions may also be employed to provide compounds of the invention.
- All process steps described herein can be carried out under known reaction conditions, such as under those specifically mentioned, in the absence of or usually in the presence of solvents or diluents, which can be inert to the reagents used and able to dissolve these, in the absence or presence of catalysts, condensing agents or neutralizing agents, for example ion exchangers, typically cation exchangers, for example in the H + form, depending on the type of reaction and/or reactants at reduced, normal, or elevated temperature, for example in the range from about -100 0 C to about 190 0 C, for example from about -80 0 C to about 150 0 C, or, for another example, at about -80 to about 60 0 C, at RT, at about -20 to about 40 0 C or at the boiling point of the solvent used, under atmospheric pressure or in a closed vessel, where appropriate under pressure, and/or in an inert atmosphere, for example, under argon or nitrogen.
- solvents or diluents which can
- Salts may be present in all starting compounds and transients, if these contain salt- forming groups. Salts may also be present during the reaction of such compounds, provided the reaction is not thereby disturbed.
- the solvents from which those can be selected which are suitable for the reaction in question include, for example, water, esters, typically lower alkyl-lower alkanoates, e.g EtOAc, ethers, typically aliphatic ethers, e.g. Et 2 O, or cyclic ethers, e.g.
- liquid aromatic hydrocarbons typically benzene or toluene, alcohols, typically MeOH, EtOH, IPA or 1- propanol, nitriles, typically AcCN, halogenated hydrocarbons, typically CH 2 Cl 2 , acid amides, typically DMF, bases, typically heterocyclic nitrogen bases, e.g. pyridine, carboxylic acids, typically lower alkanecarboxylic acids, e.g. HOAc, carboxylic acid anhydrides, typically lower alkane acid anhydrides, e.g.
- acetic anhydride cyclic, linear, or branched hydrocarbons, typically cyclohexane, hexane, or isopentane, or mixtures of these solvents, e.g. aqueous solutions, unless otherwise stated in the description of the process.
- the invention relates also to those forms of the process in which one starts from a compound obtainable at any stage as a transient species and carries out the missing steps, or breaks off the process at any stage, or forms a starting material under the reaction conditions, or uses said starting material in the form of a reactive derivative or salt, or produces a compound obtainable by means of the process according to the invention and processes the said compound in situ.
- the compounds of Formulae I-V, including their salts, are also obtainable in the form of hydrates, or their crystals can include for example the solvent used for crystallization
- New starting materials and/or intermediates, as well as processes for the preparation thereof, are likewise the subject of this invention.
- such starting materials are used and reaction conditions so selected as to enable the desired compounds to be obtained.
- Step 2 4-Bromo-5,6-diphenyl-furo[2,3-d]pyrimidine (69)
- Step 3 5,6-Diphenyl-4-[(S)-(tetrahydrofuran-2-yl)-methyl]amino furo[2,3-d]pyrimidine (70)
- Step 1 BenzyI-4-methoxyphenyl ketone (74)
- Step 2 2-Ainino -5-(4-methoxy)-phenyl-4-phenyI furan-3-carbonitriIe
- Step 4 4-Chloro-6-(4-methoxy-phenyI)-5-phenyI-furo[2,3-d]pyrimidine
- Step 6 4- ⁇ 5-Phenyl-4-[(S)-(tetrahydro-furan-2-yImethyl)-amino]- furo[2,3-d]pyrimidin-6-yl ⁇ -phenol (80)
- Step 7 ⁇ 6-[4-(2-Diraethylaraino-ethoxy)-phenyl]-5-phenyl-furo[2,3- d]pyrimidin-4-yl ⁇ -(tetrahydro-furan-2-(S)-yI-methyl)-ainine (81) [0303] A mixture of 80 (80 mg, 0.21 mmol), Cs 2 CO 3 (0.12 g, 0.36 mmol) and N 3 N- dimethylaminoethyl chloride hydrochloride (59.4 mg, 0.41 mmol) in DMF (2 mL) was heated to 8O 0 C overnight.
- Compound 106 was prepared from 80 and Boc-aminoethylchloride by a method analogous to that for 81. Compound 106 was subjected to 20% TFA/DCM, stirred at rt for 2 h, then purified by silica gel chromatography to obtain 10. MS: 431.3 (M+l).
- Step 1 4-(2,2-Diethoxyethyl)amino-5,6-diphenyl furo[2,3-d]pyrimidine
- Step 2 4-[(l,3-Dithiolan-2-yl)-methyl]amino-S,6-diphenyl furo[2,3- d]pyrimidine (110)
- Method 2 via preparation of l,3-dithiolan-2-yl-methylamine A mixture of aminoacetaldehyde diethyl acetal (5.5 mL, 0.038 mol), ethanedithiol (3.2 mL, 0.038 mol) and p-toluenesulfonic acid (7.9 g, 0.042 mol)) in 50 mL toluene was heated to reflux for 6 h.
- Step 1 4- ⁇ 4-[([l,3]Dithiolan-2-ylmethyl)-amino]-5-phenyl-furo[2,3- d]pyrimidin-6-yl ⁇ -phenoI (119)
- Step 2 6- ⁇ 4-[2-(N,N-dimethylamino)ethoxy] ⁇ phenyl-5-phenyl-4-[(l,3- dithiolan-2-yl)-methyl] amino furo[2,3-d]pyrimidine (120)
- a mixture of 119 (20 mg, 0.047 mmol), cesium carbonate (0.12 g, 0.36 mmol) and N,N-dimethylaminoethyl chloride hydrochloride (14 mg, 0.097 mmol) in DMF (1 mL) was heated to 8O 0 C overnight. After cooling, the reaction mixture was concentrated, and the residue was purified with silica gel chromatography, to give 120 (12 mg).
- Example 95 Synthesis of 6- ⁇ 4-[substituted] ⁇ phenyl-5-phenyl-4-[(l,3-dithiolan- 2-yl)-methyl] amino furo[2,3-d]pyrimidine [0346]
- the following compounds 126-135 were prepared in analogy to 110.
- Example 96 Synthesis of [2-(2,5-Diaza-bicyclo[2.2.1]hept-2-yl)-ethyl]-(5,6- diphenyl-furo[2,3-d]pyrimidin-4-yl)-amine (136)
- Example 105 Synthesis of (5,6-Diphenyl-furo[2,3-d]pyrimidin-4-yl)-(2- thiomorpholin-4-yl-ethyl)-amine (145)
- Example 108 Synthesis of (5,6-Diphenyl-furo[2,3-d]pyrimidin-4-yl)-(2- piperidin-l-yl-ethyl)-amine (148)
- the title compound was prepared by the method of Dauzonne, D.; Adam-Launay, A. Tetrahedron 1992, 48, 3069-3080.
- 4,6-Dihydroxypyrimidine 161 (3.3 g, 29.4 mmol) and 160 (5.3 g, 26.7 mmol) were combined in EtOH (absolute, 110 mL) and the mixture was heated at 60°C for 10 min to dissolve 161.
- DBU (8.06 mL, 53.9 mmol) was then added drop-wise. After addition of DBU the deep green-brown solution was heated at reflux for 3 h then at 60 0 C over night. The deep red solution was then cooled to room temperature and concentrated to a thick red oil.
- Step 4 4-Chloro-6-iodo-5-phenyI-furo[2,3-d]pyrimidine (164)
- Step 5 4-[(S)-(tetrahydrofuran-2-yl)-methyl]amino-6-iodo-5-phenyl- furo[2,3-d]pyrimidine (165) [0395] A solution of 164 (550 mg , 1.54 mmol), (S)-tetrahydrofuran-2-yl-methylamine (190 ⁇ L, 1.85 mmol), and DIEA (401 ⁇ L, 2.3 mmol) in DMF (4 mL) was heated to 100 0 C for 1 h. The reaction mixture was diluted with water, the precipitated solid was filtered and dried. MS: 422 (M+l).
- Example 118 Synthesis of [6-(6-Methoxy-pyridin-3-yI)-5-phenyl-furo[2,3- d] pyrimidin-4-yI]-((5)-tetrahydro-furan-2-ylmethyl)-amine (167) [0401] Prepared by Suzuki coupling (general procedure A): LC/MS: 403 (MH + ), 1 H NMR (DMSO): 8.35 (1 H, s), 8.2 (1 H, s), 7.7 (1 H, dd), 7.55 (5 H, m), 6.8 (1 H, d), 5.1 (1 H, t), 3.8 (4H, m), 3.5 (4 H, m), 1.9-1.6 (3H, m), 1.4 (IH, m).
- Step 4 4-(2-(S)-Tetrahydrofuranyl)methylamino-5-phenyl-6-br ⁇ )inofuro-
- Example 122 Synthesis of 4-Di-tert-butyloxycarbonylamino-5-bromo-6- phenylfuro-[2,3-d]pyrimidine (269)
- Example 144 Synthesis of 4- ⁇ 5-Phenyl-4-[(tetrahydro-furan-2-yImethyI)- amino]-furo[2,3-d]pyrimidin-6-yl ⁇ -benzenesuIfonamide (187)
- Example 150 Synthesis of N-(2-Methoxy-ethyl)-4- ⁇ 5-phenyl-4-[(tetrahydro- furan-2-ylmethyl)-amino]-furo[2,3-d]pyrimidin-6-yI ⁇ -benzamide (193)
- Example 170 Synthesis of ⁇ 5-Phenyl-6-[4-(2-piperidin-l-yI-ethoxy)-phenyl]- furo[2,3-d]pyrimidin-4-yl ⁇ (tetrahydro-furan-2-ylmethyl)-amine (220)
- Step 1 (2,5-Dichloro-pentyl)-carbamic acid tert-butyl ester (221)
- 2,5-Dichloroamylamine hydrochloride (5.0 g, 26 mmol) was stirred in MeOH (250 mL) and K 2 CO 3 (7.60 g, 55 mmol) was added, followed by di-'butyl dicarbonate (7.10 g, 32.5 mmol). The mixture was stirred at room temperature for 18 hours before the MeOH was evaporated. The residue was partitioned between diethyl ether (200 mL) and water (100 mL) and separated.
- Step 2 (Tetrahydro-thiophen-2-ylmethyl)-carbamic acid tert-butyl ester
- Step 3 C-(Tetrahydro-thiophen-2-yl)-methyIamine (223)
- Step 4 1(R/S), 2(RIS)- (l-Oxo-tetrahydro-thiophen-2-yImethyl)- carbamic acid tert-butyl ester (224); l(l? ⁇ S),2(,S'//.)-(l-Oxo-tetrahydro-thiophen-2- ylmethyl)-carbamic acid tert-butyl ester (225); and (l,l-Dioxo-tetrahydro-l ⁇ ⁇ -thiophen- 2-ylmethyl)-carbamic acid tert-butyl ester (226) [0524] A solution of 2-[(N- t butoxycarbonlylamino)methyl]-tetrahydrothiophene 222 (250 mg, 1.15 mmol) in DCM (15 mL) was stirred at rt and m-CPBA (500 mg, 2.9 mmol) was added.
- the mixture was stirred for 18 h, when TLC indicated the consumption of starting material and the presence of three new compounds.
- the mixture was diluted with DCM (30 mL) and washed with sat. NaHCO 3 (2 x 10 mL), water (10 mL) and brine (10 mL).
- the solution was evaporated onto SiO 2 and purified by column chromatography, eluting with 3:1 to 1:1 cyclohexane/acetone and finally acetone to afford the sulfone 226 (98 mg, 0.39 mmol, 34%), the anti sulfoxide 225 (89 mg, 0.38 mmol, 33%) and the syn sulfoxide 224 (43 mg, 0.18 mmol, 16 %).
- Step 5A 1(R/S), 2(R/S)-C-(l-Oxo-tetrahydro-thiophen-2-yl)-methylamine (227)
- Step 5B 1(R/S), 2(S/R)-C-(l-Oxo-tetrahydro-thiophen-2-yl)-methylamine
- Step 5C C-(l,l-Dioxo-tetrahydro-l ⁇ 6 -thiophen-2-yl)-methylamine (229)
- Example 172 Synthesis of (5,6-Diphenyl-furo[2,3-d]pyrimidin-4-yl)- (tetrahydro-thiophen-2-ylmethyl)-amine (230)
- Example 175 Synthesis of ⁇ 6-[4-(2-Dimethylamino-ethoxy)-phenyl]-5-phenyl- furo[2,3-d]pyrimidin-4-yl ⁇ -(l,l-dioxo-tetrahydro-l ⁇ 6 -thiophen-2-ylmethyl)-amine (234) [0539] Prepared by the method of 233 using (1,1 -Dioxotetrahydrothiophen-2- yl)methylammonium trifluoroacetate (229).
- Example 187 Synthesis of ⁇ 6-[4-(2-Dimethylamino-ethoxy)-phenyl]-5-phenyl- furo[2,3-d]pyrimidin-4-yl ⁇ -(l(R/S),2(S/R) -l-oxo-tetrahydro-thiophen-2-ylmethyl)-amine
- Example 104 Synthesis of ⁇ 6-[4-(2-Dimethylamino-ethoxy)-phenyl]-5-phenyl- furo[2,3-d]pyrimidin-4-yl ⁇ -(l(R/S), 2(R/S)-l-oxo-tetrahydro-thiophen-2-ylmethyl)-amine (236)
- Method A Samples were run on an HP-1100 system with an HP Zorbax SB-C 8 (5 ⁇ ) reverse phase column (4.6 x 50mm) run at 3O 0 C with a flow rate of 0.75 mL/min.
- the mobile phase used solvent A (H 2 OA).1% AcOH) and solvent B (CH 3 CN/0.1% AcOH) with a 10 min gradient from 10% to 90% CH 3 CN. The gradient was followed by a 1 min return to 10%
- Method B Samples were run on an HP-1100 system with an HP Zorbax SB-C 8 (5 ⁇ ) reverse phase column (4.6 x 50mm) run at 3O 0 C with a flow rate of 1.5 mL/min.
- the mobile phase used solvent A (H 2 OA).1% AcOH) and solvent B (CH 3 CN/0.1% AcOH) with a 5 min gradient from 10% to 90% CH 3 CN. The gradient was followed by a 0.5 min return to 10% CH 3 CN and a 1.5 min flush.
- the following assays can be employed to determine the degree of activity of a compound as a protein kinase inhibitor.
- Compounds described herein have been tested in one or more of these assays, and have shown activity.
- Representative compounds of the invention were tested and found to exhibit IC 50 values of at least ⁇ 10 ⁇ M in any one of the described assays, thereby demonstrating and confirming the utility of the compounds of the invention as protein kinase inhibitors and in the prophylaxis and treatment of immune diseases, hyperproliferative disorders, etc.
- LCK-Homogeneous Time Resolved Fluorescent (HTRF) Kinase Assay begins with LCK in the presence of ATP phosphorylating the biotinylated peptide Gastrin. The reaction incubates for 90 min. To quench the assay detection reagents are added which both stop the reaction by diluting out the enzyme and chelating the metals due to the presence of EDTA. Once the detection reagents are added the assay incubates for 30 min to allow for equilibration of the detection reagents.
- the LCK HTRF assay is comprised of 10 ⁇ L of compound in 100% DMSO, 15 ⁇ L of ATP and biotinylated Gastrin, and 15 ⁇ L of LCK KD GST (225-509) for a final volume of 40 ⁇ L.
- the final concentration of gastrin is 1.2 ⁇ M.
- Buffer conditions are as follows: 5OmM HEPES pH 7.5, 5OmM NaCl, 2OmM MgCl, 5mM MnCl, 2mM DTT, 0.05% BSA.
- the assay is quenched and stopped with 160 ⁇ L of a detection reagent.
- Detection reagents are as follows: Buffer made of 5OmM Tris, pH 7.5, 10OmM NaCl, 3mM EDTA, 0.05% BSA, 0.1% Tween20. Added to this buffer prior to reading is Steptavidin allophycocyanin (SA-APC) at a final concentration in the assay of 0.0004 mg/mL, and europilated anti-phosphotyrosine Ab (Eu-anti-PY) at a final concentration of 0.025nM.
- SA-APC Steptavidin allophycocyanin
- Eu-anti-PY europilated anti-phosphotyrosine Ab
- the assay plate is read in either a Discovery or a RubyStar.
- the eu-anti-PY is excited at 320 nm and emits at 615 nm to excite the SA-APC which in turn emits at 655 nm.
- the ratio of SA-APC at 655 nm (excited due to close proximity to the Eu-anti-PY because of phosphorylation of the peptide) to free Eu-anti-PY at 615 nm will give substrate phosphorylation.
- Assays for other kinases are done in a similar way as described above, varying the concentrations of enzyme, peptide substrate, and ATP added to the reaction, depending on the specific activity of the kinase and measured Km's for the substrates.
- Exemplary compounds 3-12 and 19-104 exhibited an average IC 5O value of 25//M or less in a human HTRF assay, for the inhibition of the Lck kinase enzyme. Many of exemplary compounds exhibited activity in the human HTFR assay for the inhibition of the Lck kinase enzyme.
- the purpose of this assay is to test the potency of T cell activation inhibitors in an in vitro model of allogeneic T cell stimulation.
- Human peripheral blood lymphocytes hPBL; 2xlO 5 /well
- mitomycin C-treated B lymphoblastoid cells JY cell line; lxlO 5 /well
- JY cell line lxlO 5 /well
- the proliferative response of the hPBL is measured by 3 H-thymidine incorporation overnight between days 5 and 6 after initiation of culture.
- Cells are harvested onto glass fiber filters and 3 H-thymidine incorporation into DNA is analyzed by liquid scintillation counter.
- the purpose of this assay is to test the general anti-proliferative/cytotoxic effect of compounds on the Jurkat human T cell line.
- Jurkat cells (lxlO 5 /well) are plated in 96-well flat-bottom tissue culture plates with or without compound dilutions and cultured for 72 h at 37 0 C in 5% CO 2 .
- Viable cell number is determined during the last 4 h of culture by adding 10 ⁇ L/well WST-I dye.
- WST-I dye conversion relies on active mitochondrial electron transport for reduction of the tetrazolium dye. The dye conversion is read by OD at 450-600 nm.
- T cell receptor TCR
- CD3 T cell receptor
- CD28 signaling pathway inhibitors T cell receptor (TCR; CD3) and CD28 signaling pathway inhibitors in human T cells.
- T cells are purified from human peripheral blood lymphocytes (hPBL) and pre-incubated with or without compound prior to stimulation with a combination of an anti-CD3 and an anti-CD28 antibody in 96-well tissue culture plates (IxIO 5 T cells/well). Cells are cultured for -20 h at 37 0 C in 5% CO 2 , then secreted BL-2 in the supernatants is quantified by cytokine ELISA (Pierce/Endogen).
- the cells remaining in the wells are then pulsed with 3 H ⁇ thymidine overnight to assess the T cell proliferative response.
- Cells are harvested onto glass fiber filters and 3 H-thymidine incorporation into DNA is analyzed by liquid scintillation counter.
- phorbol myristic acid (PMA) and calcium ionophore can be used in combination to induce IL- 2 secretion from purified T cells.
- Potential inhibitor compounds can be tested for inhibition of this response as described above for anti-CD3 and -CD28 antibodies.
- IC 50 values of compounds of Formulae I-V may be assessed as follows.
- the ACKl kinase assay utilizes a protein expressed in baculovirus infected Hi-5 cells (a fusion of an N- terminal (His)6 Tag with amino acids 117 to 489 of ACKl) purified by affinity chromatography on a Ni-NTA column.
- the substrate for the reaction is ACKl itself (autophosphorylation) and poly-Glutamic acid-Tyrosine (PGT (4:1), Sigma catalog #PO275).
- PGT poly-Glutamic acid-Tyrosine
- reaction buffer (10 mM Hepes, pH 7.6; 20 mM MgCl 2 ; 75 mM NaCl, 0.125% TWEEN20 (polyoxyethylene sorbitan monolaurate); 1 mM DTT) with 5 ⁇ M ATP are added to each well.
- Test compounds are added in 10 ⁇ L DMSO, and the reaction is started by addition of 10 ⁇ L kinase in assay buffer. The reaction proceeds 2 h at room temperature.
- the ACKl cell based assay is designed to find inhibitors of ACKl kinase activity which would be prime candidates for the development of anticancer drugs.
- the assay is based on the dependence of certain transformed cell lines (e.g. C8 cells, a Ras and ElA transformed fibroblast line) on ACKl for survival under low serum conditions, whereas other cell lines (e.g. HeLa) do not. This dependency was confirmed utilizing ACKl specific siRNAs.
- test (C8) and control (HeLa) cell lines are seeded in 96 well tissue culture plates (BD Falcon) at a density of 2 to 4 x 10 4 in DMEM/F12 (C8) or DMEM (HeLa) with 0.125% FCS in the presence of ACKl inhibitors (final DMSO concentration is 0.5%, all tissue culture media are from Cellgro). After 20 to 24 h incubation at 37°C and 5% CO 2 , cell viability is determined using the Cytotox One kit (Promega) according to the manufacturer's instructions.
- the compounds of the present invention may be administered by several different modes, including without limitation, oral, parental, by spray inhalation, rectal, or topical, as discussed herein.
- parenteral includes subcutaneous, intravenous, intramuscular, intrasternal, infusion techniques or intraperitoneal administration.
- Treatment of diseases and disorders herein is intended to also include therapeutic administration of a compound of the invention (or a pharmaceutical salt, derivative or prodrug thereof) or a pharmaceutical composition containing said compound to a subject (i.e., an animal, for example a mammal, such as a human) believed to be in need of preventative treatment, such as, for example, pain, inflammation and the like.
- a subject i.e., an animal, for example a mammal, such as a human
- Treatment also encompasses administration of the compound or pharmaceutical composition to subjects not having been diagnosed as having a need thereof, i.e., prophylactic administration to the subject.
- the subject is initially diagnosed by a licensed physician and/or authorized medical practitioner, and a regimen for prophylactic and/or therapeutic treatment via administration of the compound(s) or compositions of the invention is suggested, recommended or prescribed.
- Treating means an alleviation, in whole or in part, of symptoms associated with a disorder or disease, or halt of further progression or worsening of those symptoms, or prevention or prophylaxis of the disease or disorder.
- an "effective amount” or “therapeutically effective amount” of a compound of the invention refers to an amount of the compound that alleviates, in whole or in part, symptoms associated with a disorder or disease, or halts of further progression or worsening of those symptoms, or prevents or provides prophylaxis for the disease or disorder.
- successful treatment may include a reduction in tumor adhesion and anchorage; an alleviation of symptoms related to a cancerous growth or tumor, or proliferation of diseased tissue; a halting in the progression of a disease such as cancer or in the growth of cancerous cells.
- a pharmaceutical composition comprising a compound of this invention in combination with a pharmaceutically acceptable carrier.
- Acceptable pharmaceutical carriers generally include diluents, excipients, adjuvants and the like as described herein.
- a pharmaceutical composition of the invention may comprise an effective amount of a compound of the invention or an effective dosage amount of a compound of the invention.
- An effective dosage amount of a compound of the invention includes an amount less than, equal to, or greater than an effective amount of the compound.
- a pharmaceutical composition in which two or more unit dosages, such as in tablets, capsules and the like, are required to administer an effective amount of the compound or alternatively, a multi-dose pharmaceutical composition, such as powders, liquids and the like, in which an effective amount of the compound may be administered by administering a portion of the composition.
- the pharmaceutical compositions may generally be prepared by mixing one or more compounds of Formulae I-V including stereoisomersor tautomers, solvates, pharmaceutically acceptable salts, derivatives or prodrugs thereof, with pharmaceutically acceptable carriers, excipients, binders, adjuvants, diluents and the like, to form a desired administrable formulation to treat or ameliorate a variety of disorders related to the activity of Lck, particularly inflammation, or related to the activity ACK-I, particularly cancer.
- Pharmaceutical compositions can be manufactured by methods well known in the art such as conventional granulating, mixing, dissolving, encapsulating, lyophilizing, emulsifying or levigating processes, among others.
- compositions can be in the form of, for example, granules, powders, tablets, capsules, syrup, suppositories, injections, emulsions, elixirs, suspensions or solutions.
- the instant compositions can be formulated for various routes of administration, for example, by oral administration, by transmucosal administration, by rectal administration, or subcutaneous administration as well as intrathecal, intravenous, intramuscular, intraperitoneal, intranasal, intraocular or intraventricular injection.
- the compound or compounds of the instant invention can also be administered in a local rather than a systemic fashion, such as injection as a sustained release formulation.
- powders, suspensions, granules, tablets, pills, capsules, gelcaps, and caplets are acceptable as solid dosage forms. These can be prepared, for example, by mixing one or more compounds of the instant invention, or stereoisomers, solvates, prodrugs, pharmaceutically acceptable salts or tautomers thereof, with at least one additive or excipient such as a starch or other additive and tableted, encapsulated or made into other desirable forms for conventional administration.
- Suitable additives or excipients are sucrose, lactose, cellulose sugar, mannitol, maltitol, dextran, sorbitol, starch, agar, alginates, chitins, chitosans, pectins, tragacanth gum, gum arabic, gelatins, collagens, casein, albumin, synthetic or semi-synthetic polymers or glycerides, methyl cellulose, hydroxypropylmethyl-cellulose, and/or polyvinylpyrrolidone.
- oral dosage forms can contain other ingredients to aid in administration, such as an inactive diluent, or lubricants such as magnesium stearate, or preservatives such as paraben or sorbic acid, or anti-oxidants such as ascorbic acid, tocopherol or cysteine, a disintegrating agent, binders, thickeners, buffers, sweeteners, flavoring agents or perfuming agents. Additionally, dyestuffs or pigments may be added for identification. Tablets and pills may be further treated with suitable coating materials known in the art.
- Liquid dosage forms for oral administration may be in the form of pharmaceutically acceptable emulsions, syrups, elixirs, suspensions, slurries and solutions, which may contain an inactive diluent, such as water.
- Pharmaceutical formulations may be prepared as liquid suspensions or solutions using a sterile liquid, such as, but not limited to, an oil, water, an alcohol, and combinations of these.
- Pharmaceutically suitable surfactants, suspending agents, emulsifying agents, and the like may be added for oral or parenteral administration.
- the pharmaceutical formulations may be a spray or aerosol containing an appropriate solvent and optionally other compounds such as, but not limited to, stabilizers, antimicrobial agents, antioxidants, pH modifiers, surfactants, bioavailability modifiers and combinations of these.
- a propellant for an aerosol formulation may include compressed air, nitrogen, carbon dioxide, or a hydrocarbon based low boiling solvent.
- the compound or compounds of the instant invention are conveniently delivered in the form of an aerosol spray presentation from a nebulizer or the like.
- Injectable dosage forms for parenteral administration generally include aqueous suspensions or oil suspensions, which may be prepared using a suitable dispersant or wetting agent and a suspending agent. Injectable forms may be in solution phase or a powder suitable for reconstitution as a solution. Both are prepared with a solvent or diluent. Acceptable solvents or vehicles include sterilized water, Ringer's solution, or an isotonic aqueous saline solution. Alternatively, sterile oils may be employed as solvents or suspending agents. Typically, the oil or fatty acid is non-volatile, including natural or synthetic oils, fatty acids, mono-, di- or tri-glycerides.
- the formulations may optionally contain stabilizers, pH modifiers, surfactants, bioavailability modifiers and combinations of these.
- the compounds may be formulated for parenteral administration by injection such as by bolus injection or continuous infusion.
- a unit dosage form for injection may be in ampoules or in multi-dose containers.
- the pharmaceutical formulations may be in the form of a suppository, an ointment, an enema, a tablet or a cream for release of compound in the intestines, sigmoid flexure and/or rectum.
- Rectal suppositories are prepared by mixing one or more compounds of the instant invention, or pharmaceutically acceptable salts or tautomers of the compound, with acceptable vehicles, for example, cocoa butter or polyethylene glycol, which is solid phase at room temperature but liquid phase at those temperatures suitable to release a drug inside the body, such as in the rectum.
- acceptable vehicles for example, cocoa butter or polyethylene glycol, which is solid phase at room temperature but liquid phase at those temperatures suitable to release a drug inside the body, such as in the rectum.
- acceptable vehicles for example, cocoa butter or polyethylene glycol, which is solid phase at room temperature but liquid phase at those temperatures suitable to release a drug inside the body, such as in the rectum.
- Various other agents and additives may be used in
- the formulations of the invention may be designed to be short-acting, fast-releasing, long-acting, and sustained-releasing as described below.
- the pharmaceutical formulations may also be formulated for controlled release or for slow release.
- the instant compositions may also comprise, for example, micelles or liposomes, or some other encapsulated form, or may be administered in an extended release form to provide a prolonged storage and/or delivery effect. Therefore, the pharmaceutical formulations may be compressed into pellets or cylinders and implanted intramuscularly or subcutaneously as depot injections or as implants such as stents. Such implants may employ known inert materials such as silicones and biodegradable polymers.
- Specific dosages may be adjusted depending on conditions of disease, the age, body weight, general health conditions, sex, and diet of the subject, dose intervals, administration routes, excretion rate, and combinations of drugs. Any of the above dosage forms containing effective amounts are well within the bounds of routine experimentation and therefore, well within the scope of the instant invention.
- a therapeutically effective dose may vary depending upon the route of administration and dosage form.
- the compound or compounds of the instant invention are selected to provide a formulation that exhibits a high therapeutic index.
- the therapeutic index is the dose ratio between toxic and therapeutic effects which can be expressed as the ratio between LD 50 and ED 50 .
- the LD 50 is the dose lethal to 50% of the population and the ED 50 is the dose therapeutically effective in 50% of the population.
- the LD 50 and EDs 0 are determined by standard pharmaceutical procedures in animal cell cultures or experimental animals.
- the dosage regimen for treating Lck-mediated diseases and other diseases listed above with the compounds of this invention and/or compositions of this invention is based on a variety of factors, including the type of disease, the age, weight, sex, medical condition of the patient, the severity of the condition, the route of administration, and the particular compound employed. Thus, the dosage regimen may vary widely, but can be determined routinely using standard methods. Dosage levels of the order from about 0.01 mg to 30 mg per kilogram of body weight per day, for example from about 0.1 mg to 10 mg/kg, or from about 0.25 mg to 1 mg/kg are useful for all methods of use disclosed herein.
- the pharmaceutical composition may be in the form of, for example, a capsule, a tablet, a suspension, or liquid.
- the pharmaceutical composition can be made in the form of a dosage unit containing a given amount of the active ingredient.
- these may contain an amount of active ingredient from about 1 to 2000 mg, for example from about 1 to 500 mg, or from about 5 to 150 mg.
- a suitable daily dose for a human or other mammal may vary widely depending on the condition of the patient and other factors, but, once again, can be determined using routine methods.
- the active ingredient may also be administered by injection as a composition with suitable carriers including saline, dextrose, or water.
- suitable carriers including saline, dextrose, or water.
- the daily parenteral dosage regimen will be from about 0.1 to about 30 mg/kg of total body weight, such as from about 0.1 to about 10 mg/kg, or from about 0.25 mg to 1 mg/kg.
- Formulations suitable for topical administration include liquid or semi-liquid preparations suitable for penetration through the skin (e.g., liniments, lotions, ointments, creams, or pastes) and drops suitable for administration to the eye, ear, or nose.
- liquid or semi-liquid preparations suitable for penetration through the skin e.g., liniments, lotions, ointments, creams, or pastes
- drops suitable for administration to the eye, ear, or nose e.g., liniments, lotions, ointments, creams, or pastes
- a suitable topical dose of active ingredient of a compound of the invention is 0.1 mg to 150 mg administered one to four, for example one or two times daily.
- the active ingredient may comprise from 0.001% to 10% w/w, e.g., from 1% to 2% by weight of the formulation, although it may comprise as much as 10% w/w, but typically not more than 5% w/w.
- the concentration is from 0.1% to 1% of the formulation.
- compositions may be subjected to conventional pharmaceutical operations such as sterilization and/or may contain conventional adjuvants, such as preservatives, stabilizers, wetting agents, emulsifiers, buffers etc.
- conventional adjuvants such as preservatives, stabilizers, wetting agents, emulsifiers, buffers etc.
- the pharmaceutically active compounds of this invention can be processed in accordance with conventional methods of pharmacy to produce medicinal agents for administration to patients, including humans and other mammals.
- the compounds of the present invention can be administered as the sole active pharmaceutical agent, they can also be used in combination with one or more compounds of the invention or with one or more other agents.
- the therapeutic agents can be formulated and given to the subject as a single composition or the combination of therapeutic agents can be formulated and given to the subject as separate compositions that are given at the same time or different times.
- Treatment may also include administering the pharmaceutical formulations of the present invention in combination with other therapies.
- the compounds and pharmaceutical formulations of the present invention may be administered before, during, or after surgical procedure and/or radiation therapy.
- the compounds of the invention can also be administered in conjunction with other anti-proliferative agents including those used in antisense and gene therapy.
- alkylating agents a group of highly reactive chemotherapeutics that form covalent linkages with nucleophilic centers (e.g., hydroxyl and carboxyl).
- the alkylating agents can be divided into five groups: nitrogen mustards, ethylenimines, alkylsulfonates, triazenes, and nitrosureas.
- the nitrogen mustards are frequently useful in, for example, the treatment of chronic lymphocytic leukemia, Hodgkin's disease, malignant lymphoma, small cell lung cancer and breast and testicular cancer.
- Exemplary nitrogen mustards include chlorambucil, cyclophosphamide, ifosfamide, mechlorethamine, melphalan and uracil mustard.
- the ethylenimines the most common of which is thiotepa, may be useful in bladder tumors and in breast and ovarian adenocarcinomas.
- the alkyl sulfonates are useful in the treatment of chronic myelogenous leukemia and other myeloproliferative disorders.
- Exemplary alkyl sulfonates include busulfan and piposulfan.
- the triazines which include, e.g., dacarbazine, are useful in the treatment of malignant melanomas and sarcomas.
- Temozolomide an analog of dacarbazine, may also be used in the methods and compositions of the present invention.
- the nitrosureas are especially useful against brain tumors, but also are effective for, e.g., multiple myeloma, malignant melanoma, and lymphoma.
- Exemplary nitrosureas include carmustine and lomustine.
- Another category of antiproliferative agents suitable for use in the present invention is the antimetabolites, structural analogs of normally occurring metabolites that interfere with normal nucleic acid biosynthesis.
- This category of agents may be subdivided into the folic acid analogs, purine analogs and pyrimidine analogs based on the function of the metabolite with which the agent interferes.
- the most common folic acid analog is methotrexate, useful in the treatment of choriocarcinoma, leukemias, neoplasms and psoriasis.
- the purine analogs such as mercaptopurine, thioguanine and azathioprine, may be useful in leukemias.
- pyrimidine analogs are useful in the treatment of, for example, leukemia and carcinomas of the gastrointestinal tract, mammary gland, and bladder.
- exemplary pyrimidine analogs include fluorouracil (5-FU), UFT (uracil and ftorafur), capecitabine, gemcitabine and cytarabine.
- the vinca alkaloids natural product-based agents that exert their cytotoxicity by binding to tubulin, represent another category of antiproliferative agents suitable for use in the present invention.
- the vinca alkaloids are useful in, for example, the treatment of lymphomas, leukemias, and lung, breast, testicular, bladder and head and neck cancers.
- Exemplary agents include vinblastine, vincristine, vinorelbine and vindesine.
- the taxanes, agents which promote microtubule assembly, and the podophyllotoxins, agents which inhibit topoisomerases represent related categories of antiproliferative agents that may be useful in the methods and compositions of the present invention.
- Exemplary taxanes include paclitaxol and docetaxol, which are useful in breast and lung cancers, among others.
- Exemplary podophyllotoxins include etoposide (useful in, for example, lymphoma and Hodgkin's disease), teniposide, ironotecan (useful in, for example, colon, rectal and lung cancer) and topotecan, the latter two of which act via inhibition of topoisomerase I.
- Antineoplastic antibiotics represent another category of antiproliferative agents useful in the methods and compositions of the present invention. These agents exert their effects by binding to or complexing with DNA.
- Exemplary agents include daunorubicin, doxorubicin, epirubicin, mitoxantrone, mitomycin, dactinomycin, plicamycin, and bleomycin.
- the antibiotics are useful in a diverse range of disorders, including Hodgkin's disease, leukemia, lymphoma, and lung cancer.
- the methods and compositions of the present invention may comprise other antiproliferative agents, including the platinum complexes (e.g., cisplatin and carboplatin, which are especially useful in the treatment of lung, head and neck, ovarian and breast cancer); enzymes (e.g., L-asparaginase); hormone-related therapy hormone (e.g., tamoxifen, leuprolide, flutamide, megesterol acetate, diethylstilbestrol, prednisone and estradiol cypionate); hydroxyurea; methylhydrazine derivatives such as procarbazine; adrenocortical suppressants, e.g., mitotane, aminoglutethimide; aromatase inhibitors (e.g., anastrozole); and biologic response modifiers (e.g., interferon- A).
- platinum complexes e.g., cisplatin and carboplatin, which are especially useful in the
- compositions of the present invention may comprise antiproliferative agents that result from the combination of two or more agents including, for example, prednimustine (a conjugate of prednisone and chlorambucil) and estramustine (a conjugate of nornitrogen mustard and estradiol).
- prednimustine a conjugate of prednisone and chlorambucil
- estramustine a conjugate of nornitrogen mustard and estradiol
- kinase inhibitors contemplated for use include, without limitation, tyrphostin AG490 (2-cyano-3-(3,4-dihydroxyphenyl)-N-(benzyl)-2-propenamide), Iressa (ZDl 839; Astra Zeneca); Gleevec (STI-571 or imatinib mesylate; Novartis); SU5416 (Pharmacia Corp./Sugen); and Tarceva (OSI-774; Roche/Genentech/OSI Pharmaceuticals).
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Immunology (AREA)
- Diabetes (AREA)
- Pulmonology (AREA)
- Dermatology (AREA)
- Neurology (AREA)
- Rheumatology (AREA)
- Urology & Nephrology (AREA)
- Physical Education & Sports Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Vascular Medicine (AREA)
- Cardiology (AREA)
- Obesity (AREA)
- Biomedical Technology (AREA)
- Heart & Thoracic Surgery (AREA)
- Neurosurgery (AREA)
- Emergency Medicine (AREA)
- Pain & Pain Management (AREA)
- Transplantation (AREA)
- Otolaryngology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description





















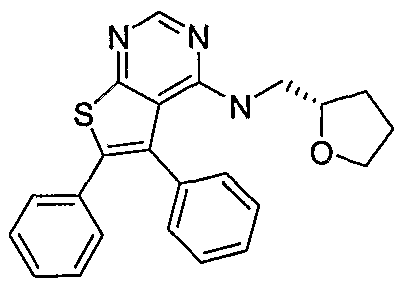



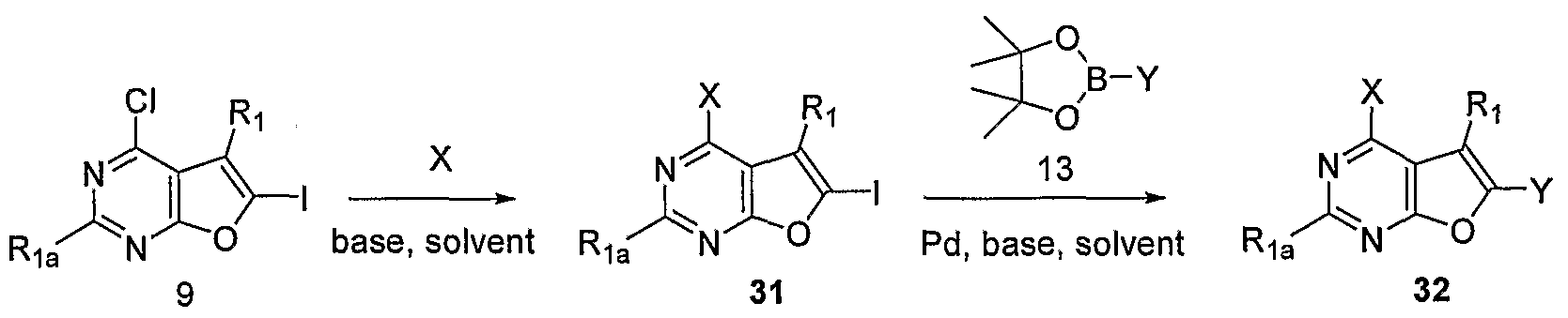
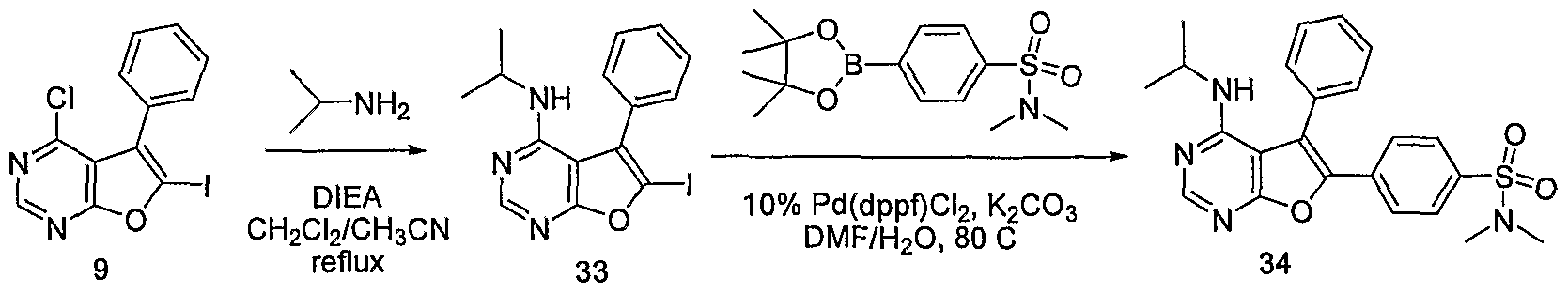


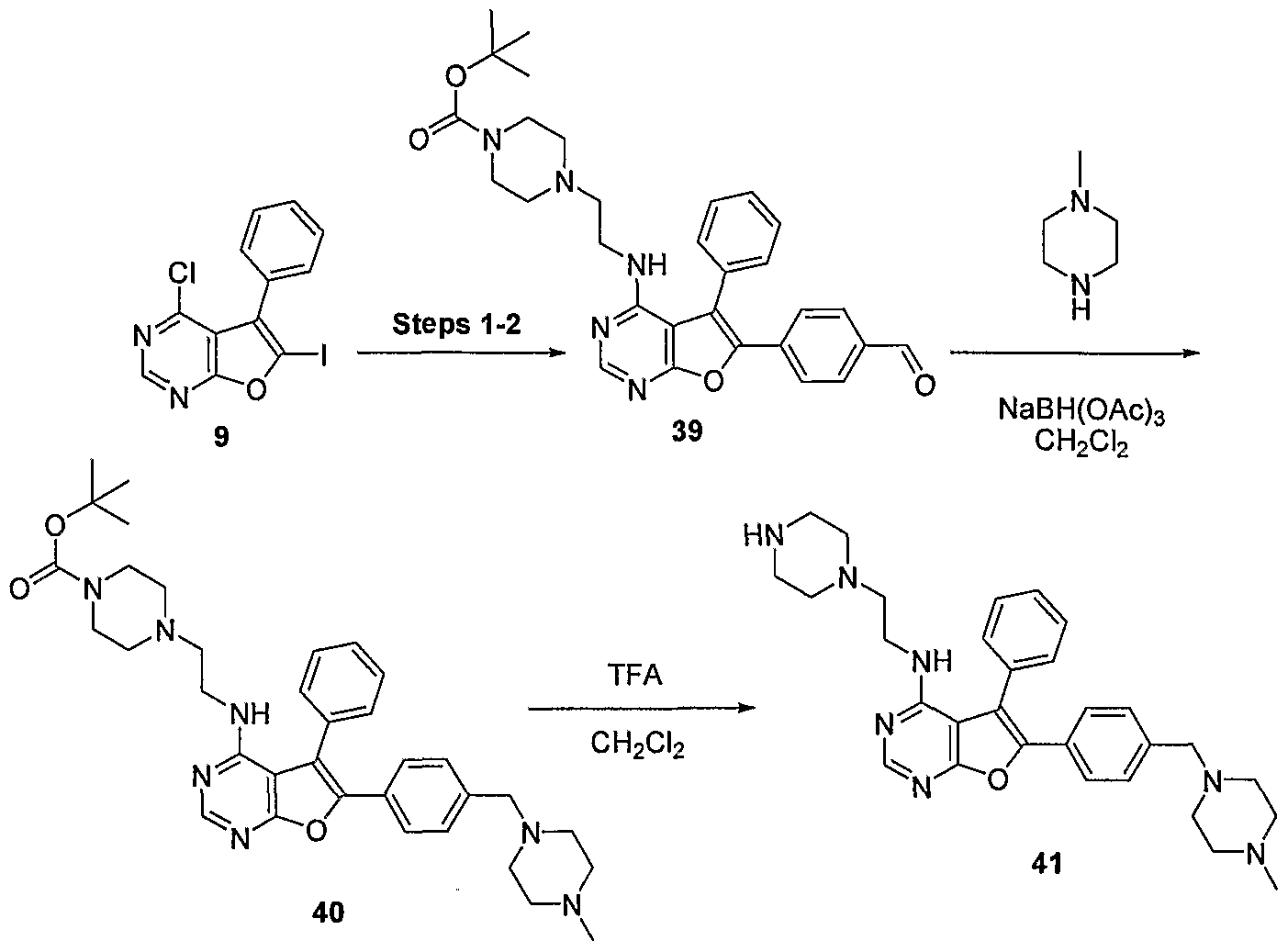







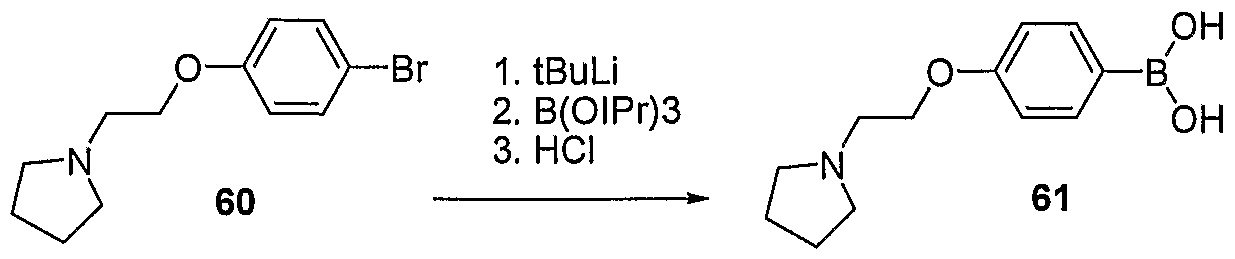










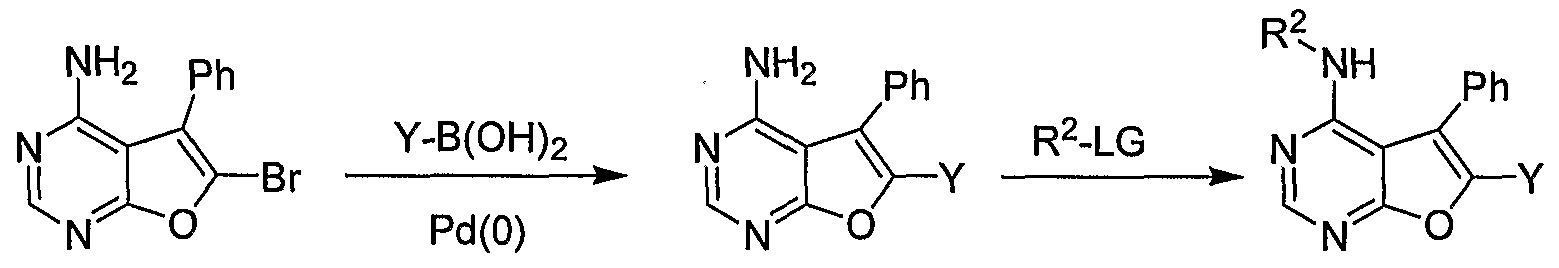
























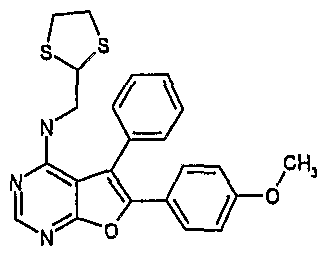






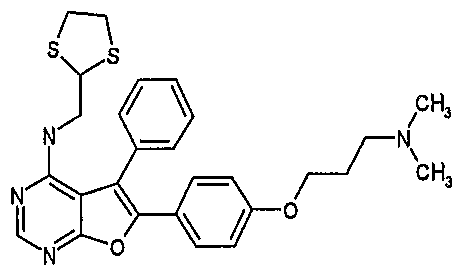









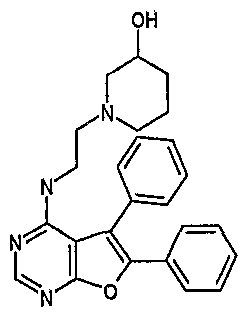




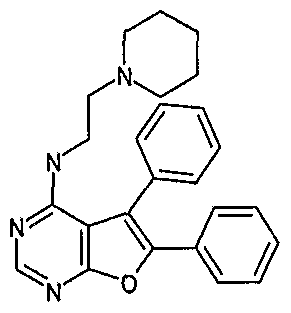













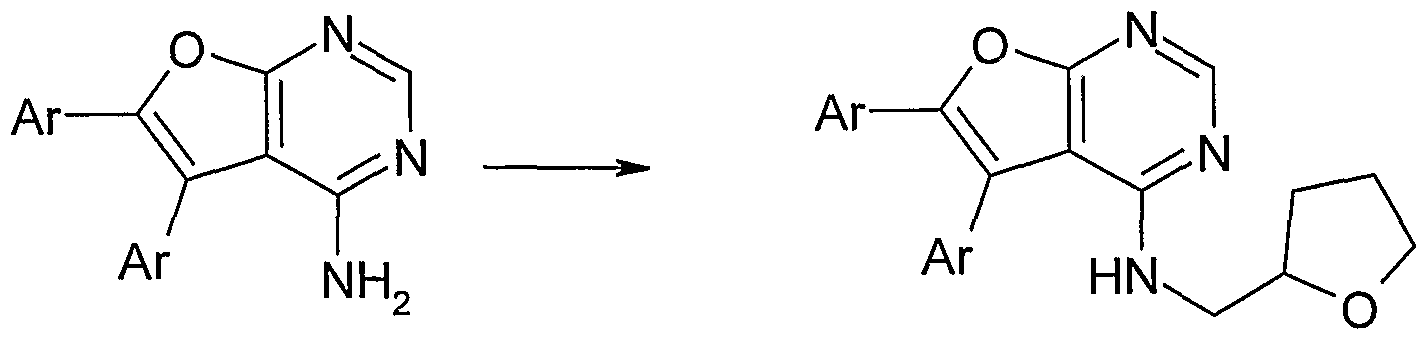









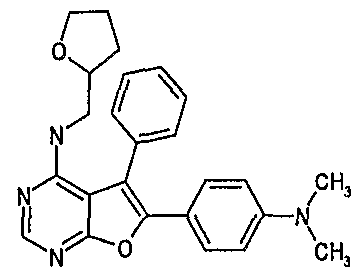

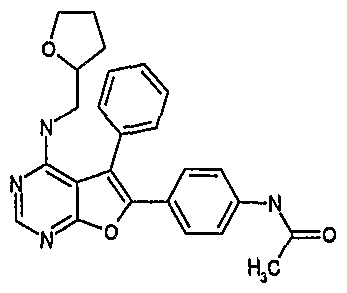




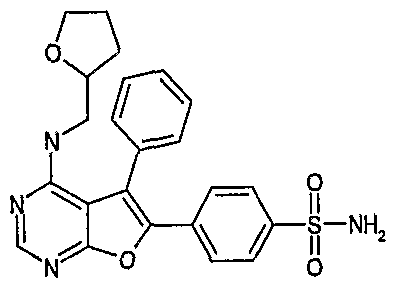





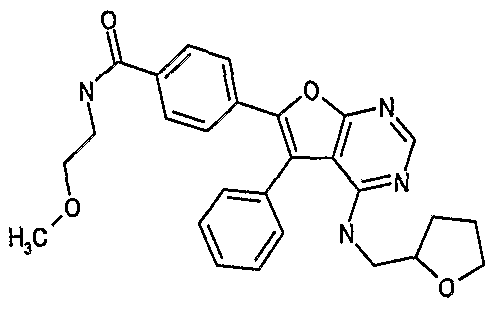


















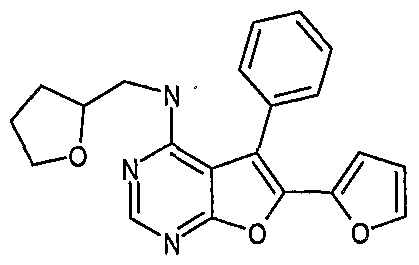



Claims


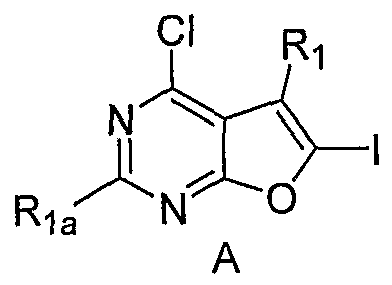









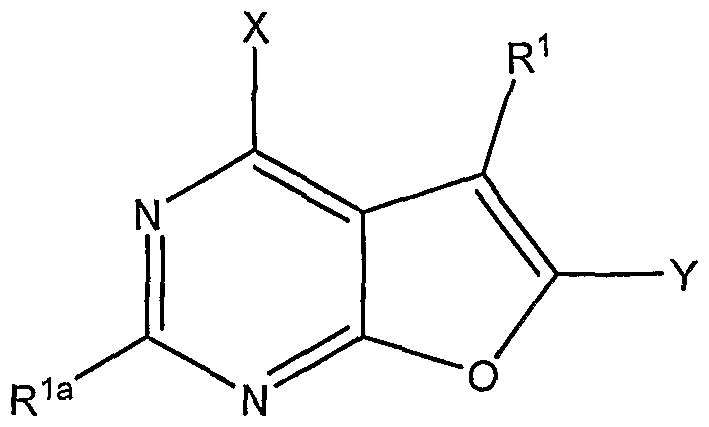
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2005260077A AU2005260077A1 (en) | 2004-06-29 | 2005-06-29 | Furanopyrimidines |
| EP05763716A EP1768986A2 (en) | 2004-06-29 | 2005-06-29 | Furanopyrimidines |
| MXPA06015223A MXPA06015223A (en) | 2004-06-29 | 2005-06-29 | Furanopyrimidines. |
| CA002571857A CA2571857A1 (en) | 2004-06-29 | 2005-06-29 | Furanopyrimidines |
| JP2007519333A JP2008505084A (en) | 2004-06-29 | 2005-06-29 | Furanopyrimidine |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US58389804P | 2004-06-29 | 2004-06-29 | |
| US60/583,898 | 2004-06-29 | ||
| US65994705P | 2005-03-08 | 2005-03-08 | |
| US60/659,947 | 2005-03-08 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2006004658A2 true WO2006004658A2 (en) | 2006-01-12 |
| WO2006004658A3 WO2006004658A3 (en) | 2006-04-20 |
Family
ID=35344939
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2005/022727 WO2006004658A2 (en) | 2004-06-29 | 2005-06-29 | Furanopyrimidines |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US7776867B2 (en) |
| EP (1) | EP1768986A2 (en) |
| JP (1) | JP2008505084A (en) |
| AU (1) | AU2005260077A1 (en) |
| CA (1) | CA2571857A1 (en) |
| MX (1) | MXPA06015223A (en) |
| WO (1) | WO2006004658A2 (en) |
Cited By (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006130160A3 (en) * | 2004-07-23 | 2007-02-22 | Amgen Inc | Furanopyridine derivatives as ack1 and lck modulators |
| WO2007079862A1 (en) * | 2005-12-21 | 2007-07-19 | Bayer Healthcare Ag | Novel, acyclic substituted furopyrimidine derivatives and use thereof for treating cardiovascular diseases |
| DE102007019690A1 (en) | 2007-04-26 | 2008-10-30 | Bayer Healthcare Ag | Use of cyclic substituted furopyrimidine derivatives for the treatment of pulmonary arterial hypertension |
| DE102007019691A1 (en) | 2007-04-26 | 2008-10-30 | Bayer Healthcare Ag | Use of acyclically substituted furopyrimidine derivatives for the treatment of pulmonary arterial hypertension |
| DE102007027799A1 (en) | 2007-06-16 | 2008-12-18 | Bayer Healthcare Ag | Substituted furopyrimidines and their use |
| DE102007027800A1 (en) | 2007-06-16 | 2008-12-18 | Bayer Healthcare Ag | Substituted bicyclic heteroaryl compounds and their use |
| DE102007054786A1 (en) | 2007-11-16 | 2009-05-20 | Bayer Healthcare Ag | Trisubstituted Furopyrimidines and their Use |
| JP2009523711A (en) * | 2005-12-21 | 2009-06-25 | バイエル・ヘルスケア・アクチェンゲゼルシャフト | Novel cyclic substituted furopyrimidine derivatives and their use to treat cardiovascular disease |
| WO2009139886A2 (en) * | 2008-05-14 | 2009-11-19 | University Of Pittsburgh-Of The Commonwealth System Of Higher Education | Targeting an hiv-1 nef-host cell kinase complex |
| US7776867B2 (en) | 2004-06-29 | 2010-08-17 | Amgen Inc. | Furanopyrimidines |
| WO2011055115A1 (en) | 2009-11-04 | 2011-05-12 | Almac Discovery Limited | Akt / pkb inhibitors |
| CN102260270A (en) * | 2010-05-28 | 2011-11-30 | 中国科学院上海药物研究所 | N-(2-methylfuran[2,3-d]pyrimidine-4-yl)acrylamide, its preparation method and application |
| US8138194B2 (en) | 2008-04-30 | 2012-03-20 | National Health Research Institutes | Fused bicyclic pyrimidine compounds as aurora kinase inhibitors |
| CN103087077A (en) * | 2011-11-03 | 2013-05-08 | 上海希迈医药科技有限公司 | Thienopyrimidine and furopyrimidine derivative, its preparation method and application in medicines |
| WO2013140189A1 (en) | 2012-03-23 | 2013-09-26 | Almac Discovery Limited | 6- (4- (1 -amino- 3 -hydroxycyclobutyl) phenyl) - 5 - phenyl (furo, thieno or pyrrolo) [2, 3-d] pyrimidin- 4 - one derivatives for the treatment of cancer |
| CN103360407A (en) * | 2012-04-10 | 2013-10-23 | 上海希迈医药科技有限公司 | Thiophene miazines derivate as well as preparation method and medical application thereof |
| CN103920056A (en) * | 2014-05-04 | 2014-07-16 | 张翠荣 | Aerosol inhalation liquid for preventing postoperative tracheospasm and preparation method thereof |
| CN103948827A (en) * | 2014-05-04 | 2014-07-30 | 李晓燕 | Medicinal composition for aerosol inhalation in preoperative period |
| US9006252B2 (en) | 2008-09-26 | 2015-04-14 | National Health Research Institutes | Fused multicyclic compounds as protein kinase inhibitors |
| CN106946896A (en) * | 2017-03-30 | 2017-07-14 | 成都知普莱生物医药科技有限公司 | The furans simultaneously amine derivative of [2,3 d] pyrimidine 4 |
| US10278972B2 (en) | 2013-12-23 | 2019-05-07 | Les Laboratoires Servier | Thienopyrimidine derivatives, a process for their preparation and pharmaceutical compositions containing them |
| CN114126620A (en) * | 2019-05-13 | 2022-03-01 | 传达治疗有限公司 | FGFR inhibitors and methods of use thereof |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6555543B2 (en) | 2000-08-04 | 2003-04-29 | Dmi Biosciences, Inc. | Method of using diketopiperazines and composition containing them |
| KR20150080004A (en) | 2003-05-15 | 2015-07-08 | 앰피오 파마슈티컬스 인코퍼레이티드 | Treatment of t-cell mediated diseases |
| WO2008070908A1 (en) * | 2006-12-11 | 2008-06-19 | Bionomics Limited | Chemical compounds and processes |
| JP5856843B2 (en) | 2008-05-27 | 2016-02-10 | アンピオ ファーマシューティカルズ,インコーポレイテッド | Pharmaceutical composition using diketopiperazine |
| GB201105659D0 (en) | 2011-04-01 | 2011-05-18 | Xention Ltd | Compounds |
| WO2013055734A1 (en) | 2011-10-10 | 2013-04-18 | Ampio Pharmaceuticals, Inc. | Treatment of degenerative joint disease |
| MY172699A (en) | 2011-10-10 | 2019-12-10 | Ampio Pharmaceuticals Inc | Implantable medical devices with increased immune tolerance, and methods for making and implanting |
| MX355446B (en) * | 2011-10-28 | 2018-04-18 | Ampio Pharmaceuticals Inc | Treatment of rhinitis. |
| EP2968315B1 (en) | 2013-03-15 | 2020-06-03 | Ampio Pharmaceuticals, Inc. | Compositions for the mobilization, homing, expansion and differentiation of stem cells and methods of using the same |
| WO2016028790A1 (en) | 2014-08-18 | 2016-02-25 | Ampio Pharmaceuticals, Inc. | Treatment of joint conditions |
| US11389512B2 (en) | 2015-06-22 | 2022-07-19 | Ampio Pharmaceuticals, Inc. | Use of low molecular weight fractions of human serum albumin in treating diseases |
| FR3046792B1 (en) * | 2016-01-19 | 2018-02-02 | Les Laboratoires Servier | NOVEL AMMONIUM DERIVATIVES, PROCESS FOR THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002105081A (en) * | 2000-07-28 | 2002-04-10 | Nikken Chem Co Ltd | Bicyclic compound of thiophene |
| WO2003018589A1 (en) * | 2001-08-22 | 2003-03-06 | Bayer Healthcare Ag | Novel 4-aminofuropyrimidines and the use thereof |
| WO2003022852A2 (en) * | 2001-09-11 | 2003-03-20 | Smithkline Beecham Corporation | Furo-and thienopyrimidine derivatives as angiogenesis inhibitors |
| WO2003080064A1 (en) * | 2002-03-21 | 2003-10-02 | Abbott Laboratories | Kinase inhibitors |
| US20030225098A1 (en) * | 2002-03-21 | 2003-12-04 | Hirst Gavin C. | Kinase inhibitors |
| WO2004111057A1 (en) * | 2003-06-11 | 2004-12-23 | Xention Discovery Limited | Thienopyrimidine derivatives as potassium channel inhibitors |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5679683A (en) * | 1994-01-25 | 1997-10-21 | Warner-Lambert Company | Tricyclic compounds capable of inhibiting tyrosine kinases of the epidermal growth factor receptor family |
| AU2096895A (en) * | 1994-03-07 | 1995-09-25 | Sugen, Incorporated | Receptor tyrosine kinase inhibitors for inhibiting cell proliferative disorders and compositions thereof |
| GB0412986D0 (en) * | 2004-06-10 | 2004-07-14 | Xention Discovery Ltd | Compounds |
| US7776867B2 (en) | 2004-06-29 | 2010-08-17 | Amgen Inc. | Furanopyrimidines |
-
2005
- 2005-06-29 US US11/169,312 patent/US7776867B2/en active Active
- 2005-06-29 MX MXPA06015223A patent/MXPA06015223A/en not_active Application Discontinuation
- 2005-06-29 WO PCT/US2005/022727 patent/WO2006004658A2/en active Application Filing
- 2005-06-29 JP JP2007519333A patent/JP2008505084A/en not_active Withdrawn
- 2005-06-29 AU AU2005260077A patent/AU2005260077A1/en not_active Abandoned
- 2005-06-29 CA CA002571857A patent/CA2571857A1/en not_active Abandoned
- 2005-06-29 EP EP05763716A patent/EP1768986A2/en not_active Withdrawn
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002105081A (en) * | 2000-07-28 | 2002-04-10 | Nikken Chem Co Ltd | Bicyclic compound of thiophene |
| WO2003018589A1 (en) * | 2001-08-22 | 2003-03-06 | Bayer Healthcare Ag | Novel 4-aminofuropyrimidines and the use thereof |
| WO2003022852A2 (en) * | 2001-09-11 | 2003-03-20 | Smithkline Beecham Corporation | Furo-and thienopyrimidine derivatives as angiogenesis inhibitors |
| WO2003080064A1 (en) * | 2002-03-21 | 2003-10-02 | Abbott Laboratories | Kinase inhibitors |
| US20030225098A1 (en) * | 2002-03-21 | 2003-12-04 | Hirst Gavin C. | Kinase inhibitors |
| WO2004111057A1 (en) * | 2003-06-11 | 2004-12-23 | Xention Discovery Limited | Thienopyrimidine derivatives as potassium channel inhibitors |
Non-Patent Citations (2)
| Title |
|---|
| MASAKU FUJITA ET AL: "Design ,Synthesis and Bioactives of Novel Diarylthiophenes: Inhibitors of Tumor Necrosis Factor-alpha(TNF-alpha) Production" BIOORGANIC CHEMISTRY, vol. 10, 2002, pages 3113-3122, XP002355371 * |
| PATENT ABSTRACTS OF JAPAN vol. 2002, no. 08, 5 August 2002 (2002-08-05) -& JP 2002 105081 A (NIKKEN CHEM CO LTD), 10 April 2002 (2002-04-10) * |
Cited By (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7776867B2 (en) | 2004-06-29 | 2010-08-17 | Amgen Inc. | Furanopyrimidines |
| US7674907B2 (en) | 2004-07-23 | 2010-03-09 | Amgen Inc. | Furanopyridine derivatives and methods of use |
| WO2006130160A3 (en) * | 2004-07-23 | 2007-02-22 | Amgen Inc | Furanopyridine derivatives as ack1 and lck modulators |
| US8183246B2 (en) | 2005-12-21 | 2012-05-22 | Bayer Intellectual Property Gmbh | Acyclically substituted furopyrimidine derivatives and use thereof |
| JP2009520713A (en) * | 2005-12-21 | 2009-05-28 | バイエル・ヘルスケア・アクチェンゲゼルシャフト | Novel acyclic substituted furopyrimidine derivatives and their use to treat cardiovascular disease |
| WO2007079862A1 (en) * | 2005-12-21 | 2007-07-19 | Bayer Healthcare Ag | Novel, acyclic substituted furopyrimidine derivatives and use thereof for treating cardiovascular diseases |
| US8324222B2 (en) | 2005-12-21 | 2012-12-04 | Bayer Intellectual Property Gmbh | Cyclically substituted furopyrimidine derivatives and use thereof |
| JP2009523711A (en) * | 2005-12-21 | 2009-06-25 | バイエル・ヘルスケア・アクチェンゲゼルシャフト | Novel cyclic substituted furopyrimidine derivatives and their use to treat cardiovascular disease |
| DE102007019690A1 (en) | 2007-04-26 | 2008-10-30 | Bayer Healthcare Ag | Use of cyclic substituted furopyrimidine derivatives for the treatment of pulmonary arterial hypertension |
| WO2008131859A3 (en) * | 2007-04-26 | 2009-07-16 | Bayer Schering Pharma Ag | Use of cyclically substituted furopyrimidine derivatives for treating pulmonary arterial hypertonia |
| WO2008131858A3 (en) * | 2007-04-26 | 2009-07-16 | Bayer Schering Pharma Ag | Use of acyclically substituted furopyrimidine derivatives for treating pulmonary arterial hypertonia |
| DE102007019691A1 (en) | 2007-04-26 | 2008-10-30 | Bayer Healthcare Ag | Use of acyclically substituted furopyrimidine derivatives for the treatment of pulmonary arterial hypertension |
| WO2008131858A2 (en) * | 2007-04-26 | 2008-11-06 | Bayer Schering Pharma Aktiengesellschaft | Use of acyclically substituted furopyrimidine derivatives for treating pulmonary arterial hypertonia |
| WO2008131859A2 (en) * | 2007-04-26 | 2008-11-06 | Bayer Schering Pharma Aktiengesellschaft | Use of cyclically substituted furopyrimidine derivatives for treating pulmonary arterial hypertonia |
| DE102007027800A1 (en) | 2007-06-16 | 2008-12-18 | Bayer Healthcare Ag | Substituted bicyclic heteroaryl compounds and their use |
| DE102007027799A1 (en) | 2007-06-16 | 2008-12-18 | Bayer Healthcare Ag | Substituted furopyrimidines and their use |
| DE102007054786A1 (en) | 2007-11-16 | 2009-05-20 | Bayer Healthcare Ag | Trisubstituted Furopyrimidines and their Use |
| US8138194B2 (en) | 2008-04-30 | 2012-03-20 | National Health Research Institutes | Fused bicyclic pyrimidine compounds as aurora kinase inhibitors |
| WO2009139886A2 (en) * | 2008-05-14 | 2009-11-19 | University Of Pittsburgh-Of The Commonwealth System Of Higher Education | Targeting an hiv-1 nef-host cell kinase complex |
| US8541415B2 (en) | 2008-05-14 | 2013-09-24 | University of Pittsburgh—of the Commonwealth System of Higher Education | Targeting an HIV-1 nef-host cell kinase complex |
| WO2009139886A3 (en) * | 2008-05-14 | 2010-03-11 | University Of Pittsburgh-Of The Commonwealth System Of Higher Education | Targeting an hiv-1 nef-host cell kinase complex |
| US9006252B2 (en) | 2008-09-26 | 2015-04-14 | National Health Research Institutes | Fused multicyclic compounds as protein kinase inhibitors |
| WO2011055115A1 (en) | 2009-11-04 | 2011-05-12 | Almac Discovery Limited | Akt / pkb inhibitors |
| CN102260270A (en) * | 2010-05-28 | 2011-11-30 | 中国科学院上海药物研究所 | N-(2-methylfuran[2,3-d]pyrimidine-4-yl)acrylamide, its preparation method and application |
| CN103087077A (en) * | 2011-11-03 | 2013-05-08 | 上海希迈医药科技有限公司 | Thienopyrimidine and furopyrimidine derivative, its preparation method and application in medicines |
| WO2013064068A1 (en) * | 2011-11-03 | 2013-05-10 | 上海希迈医药科技有限公司 | Thienopyrimidine and furopyrimidine derivatives, preparation method thereof and medical use thereof |
| CN103087077B (en) * | 2011-11-03 | 2016-05-18 | 上海希迈医药科技有限公司 | Thienopyrimidine and furans miazines derivative, its preparation method and in application pharmaceutically |
| WO2013140189A1 (en) | 2012-03-23 | 2013-09-26 | Almac Discovery Limited | 6- (4- (1 -amino- 3 -hydroxycyclobutyl) phenyl) - 5 - phenyl (furo, thieno or pyrrolo) [2, 3-d] pyrimidin- 4 - one derivatives for the treatment of cancer |
| CN103360407B (en) * | 2012-04-10 | 2016-06-22 | 上海希迈医药科技有限公司 | A kind of Thienopyrimidine analog derivative, its preparation method and in application pharmaceutically |
| CN103360407A (en) * | 2012-04-10 | 2013-10-23 | 上海希迈医药科技有限公司 | Thiophene miazines derivate as well as preparation method and medical application thereof |
| US10278972B2 (en) | 2013-12-23 | 2019-05-07 | Les Laboratoires Servier | Thienopyrimidine derivatives, a process for their preparation and pharmaceutical compositions containing them |
| CN103948827A (en) * | 2014-05-04 | 2014-07-30 | 李晓燕 | Medicinal composition for aerosol inhalation in preoperative period |
| CN103920056A (en) * | 2014-05-04 | 2014-07-16 | 张翠荣 | Aerosol inhalation liquid for preventing postoperative tracheospasm and preparation method thereof |
| CN103920056B (en) * | 2014-05-04 | 2017-02-08 | 李晓东 | Aerosol inhalation liquid for preventing postoperative tracheospasm and preparation method thereof |
| CN106946896A (en) * | 2017-03-30 | 2017-07-14 | 成都知普莱生物医药科技有限公司 | The furans simultaneously amine derivative of [2,3 d] pyrimidine 4 |
| CN106946896B (en) * | 2017-03-30 | 2019-05-21 | 成都知普莱生物医药科技有限公司 | Furans simultaneously [2,3-d] pyrimidine -4- amine derivative |
| CN114126620A (en) * | 2019-05-13 | 2022-03-01 | 传达治疗有限公司 | FGFR inhibitors and methods of use thereof |
| EP3968999A4 (en) * | 2019-05-13 | 2022-08-03 | Relay Therapeutics, Inc. | Fgfr inhibitors and methods of use thereof |
| US11780845B2 (en) | 2019-05-13 | 2023-10-10 | Relay Therapeutics, Inc. | FGFR inhibitors and methods of use thereof |
| EP4306529A3 (en) * | 2019-05-13 | 2024-04-10 | Relay Therapeutics, Inc. | Fgfr inhibitors and methods of use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| MXPA06015223A (en) | 2007-11-09 |
| JP2008505084A (en) | 2008-02-21 |
| AU2005260077A1 (en) | 2006-01-12 |
| EP1768986A2 (en) | 2007-04-04 |
| US20060040961A1 (en) | 2006-02-23 |
| US7776867B2 (en) | 2010-08-17 |
| CA2571857A1 (en) | 2006-01-12 |
| WO2006004658A3 (en) | 2006-04-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7776867B2 (en) | Furanopyrimidines | |
| US7674907B2 (en) | Furanopyridine derivatives and methods of use | |
| US7763624B2 (en) | Substituted pyrazolo[3,4-d]pyrimidines as ACK-1 and LCK inhibitors | |
| AU747920B2 (en) | Bicyclic pyridine and pyrimidine derivatives as neuropeptide Y receptor antagonists | |
| EP2872511B1 (en) | Imidazopyrazine derivatives as modulators of tnf activity | |
| JP6615752B2 (en) | Substituted nicotinimide inhibitors of BTK and their preparation and use in the treatment of cancer, inflammation and autoimmune diseases | |
| AU2008273017B2 (en) | Heterocyclic compounds useful as Raf kinase inhibitors | |
| EP2654750B1 (en) | Novel fused pyridine compounds as casein kinase inhibitors | |
| AU763758B2 (en) | Novel compounds | |
| WO2007126128A1 (en) | Dihydropyrazolopyrimidinone derivative | |
| CA2775942A1 (en) | Pi3k (delta) selective inhibitors | |
| WO2006004703A2 (en) | PYRROLO[2,3-d]PYRIMIDINES THAT MODULATE ACK1 AND LCK ACTIVITY | |
| EP2382207A2 (en) | Pi3k/mtor kinase inhibitors | |
| EP3999498A1 (en) | Inhibitors of cyclin-dependent kinases | |
| WO2014100540A1 (en) | Pyrazole substituted imidazopyrazines as casein kinase 1 d/e inhibitors | |
| EP1664053A1 (en) | Substituted heterocyclic compounds and methods of use | |
| MX2007000941A (en) | Furanopyridine derivatives as ack1 and lck modulators | |
| MX2010011136A (en) | PYRROLO[2,3-d]PYRIMIDIN-2-YL-AMINE DERIVATIVES AS PKC-THETA INHIBITORS. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2006/015223 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2571857 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007519333 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005260077 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005763716 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2005260077 Country of ref document: AU Date of ref document: 20050629 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005260077 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005763716 Country of ref document: EP |