WO2005097755A2 - Imidazolderivate als hemmer von tyrosinkinase - Google Patents
Imidazolderivate als hemmer von tyrosinkinase Download PDFInfo
- Publication number
- WO2005097755A2 WO2005097755A2 PCT/EP2005/002746 EP2005002746W WO2005097755A2 WO 2005097755 A2 WO2005097755 A2 WO 2005097755A2 EP 2005002746 W EP2005002746 W EP 2005002746W WO 2005097755 A2 WO2005097755 A2 WO 2005097755A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compounds
- phenyl
- cancer
- amino
- Prior art date
Links
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 title abstract description 3
- 239000005483 tyrosine kinase inhibitor Substances 0.000 title abstract description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N 1H-imidazole Chemical class C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 218
- 238000011282 treatment Methods 0.000 claims abstract description 77
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 66
- 108010077182 raf Kinases Proteins 0.000 claims abstract description 27
- 102000009929 raf Kinases Human genes 0.000 claims abstract description 27
- 102100022014 Angiopoietin-1 receptor Human genes 0.000 claims abstract description 16
- 101000753291 Homo sapiens Angiopoietin-1 receptor Proteins 0.000 claims abstract description 16
- -1 COOA Chemical group 0.000 claims description 107
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 81
- 201000010099 disease Diseases 0.000 claims description 78
- 150000003839 salts Chemical class 0.000 claims description 72
- 239000000203 mixture Substances 0.000 claims description 54
- 239000012453 solvate Substances 0.000 claims description 36
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 claims description 34
- 239000003814 drug Substances 0.000 claims description 32
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 claims description 31
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 claims description 31
- 238000000034 method Methods 0.000 claims description 30
- 201000011510 cancer Diseases 0.000 claims description 28
- 230000000694 effects Effects 0.000 claims description 28
- 230000033115 angiogenesis Effects 0.000 claims description 25
- 239000003112 inhibitor Substances 0.000 claims description 24
- 239000002253 acid Substances 0.000 claims description 22
- 230000019491 signal transduction Effects 0.000 claims description 21
- 238000004519 manufacturing process Methods 0.000 claims description 18
- 241000251730 Chondrichthyes Species 0.000 claims description 17
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 claims description 17
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 17
- 108091000080 Phosphotransferase Proteins 0.000 claims description 16
- 102000020233 phosphotransferase Human genes 0.000 claims description 16
- 238000002360 preparation method Methods 0.000 claims description 16
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 claims description 15
- 108090000623 proteins and genes Proteins 0.000 claims description 15
- 102100033479 RAF proto-oncogene serine/threonine-protein kinase Human genes 0.000 claims description 14
- 101710141955 RAF proto-oncogene serine/threonine-protein kinase Proteins 0.000 claims description 14
- 108091008605 VEGF receptors Proteins 0.000 claims description 14
- 230000005764 inhibitory process Effects 0.000 claims description 14
- 239000000460 chlorine Substances 0.000 claims description 13
- 229910052801 chlorine Inorganic materials 0.000 claims description 13
- 102000004169 proteins and genes Human genes 0.000 claims description 13
- 201000004681 Psoriasis Diseases 0.000 claims description 11
- 206010012689 Diabetic retinopathy Diseases 0.000 claims description 10
- 125000000217 alkyl group Chemical group 0.000 claims description 10
- 125000004432 carbon atom Chemical group C* 0.000 claims description 10
- 206010006187 Breast cancer Diseases 0.000 claims description 9
- 208000026310 Breast neoplasm Diseases 0.000 claims description 9
- 230000003463 hyperproliferative effect Effects 0.000 claims description 9
- 208000027866 inflammatory disease Diseases 0.000 claims description 9
- 206010061218 Inflammation Diseases 0.000 claims description 8
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 8
- 230000004054 inflammatory process Effects 0.000 claims description 8
- 208000032839 leukemia Diseases 0.000 claims description 8
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 8
- 230000001404 mediated effect Effects 0.000 claims description 8
- 201000002528 pancreatic cancer Diseases 0.000 claims description 8
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 8
- 230000007170 pathology Effects 0.000 claims description 8
- 230000008569 process Effects 0.000 claims description 8
- 206010009944 Colon cancer Diseases 0.000 claims description 7
- 239000004202 carbamide Substances 0.000 claims description 7
- 208000002780 macular degeneration Diseases 0.000 claims description 7
- 125000004434 sulfur atom Chemical group 0.000 claims description 7
- KKVYYGGCHJGEFJ-UHFFFAOYSA-N 1-n-(4-chlorophenyl)-6-methyl-5-n-[3-(7h-purin-6-yl)pyridin-2-yl]isoquinoline-1,5-diamine Chemical compound N=1C=CC2=C(NC=3C(=CC=CN=3)C=3C=4N=CNC=4N=CN=3)C(C)=CC=C2C=1NC1=CC=C(Cl)C=C1 KKVYYGGCHJGEFJ-UHFFFAOYSA-N 0.000 claims description 6
- 208000023275 Autoimmune disease Diseases 0.000 claims description 6
- 101100381978 Mus musculus Braf gene Proteins 0.000 claims description 6
- 206010064930 age-related macular degeneration Diseases 0.000 claims description 6
- 230000001028 anti-proliverative effect Effects 0.000 claims description 6
- 210000000988 bone and bone Anatomy 0.000 claims description 6
- 210000004556 brain Anatomy 0.000 claims description 6
- 239000003795 chemical substances by application Substances 0.000 claims description 6
- 208000030533 eye disease Diseases 0.000 claims description 6
- 208000014829 head and neck neoplasm Diseases 0.000 claims description 6
- 125000000623 heterocyclic group Chemical group 0.000 claims description 6
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 6
- 125000004193 piperazinyl group Chemical group 0.000 claims description 6
- 230000000644 propagated effect Effects 0.000 claims description 6
- 230000004276 retinal vascularization Effects 0.000 claims description 6
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 6
- 101150019464 ARAF gene Proteins 0.000 claims description 5
- 208000010507 Adenocarcinoma of Lung Diseases 0.000 claims description 5
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 5
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 5
- 206010003246 arthritis Diseases 0.000 claims description 5
- 239000002834 estrogen receptor modulator Substances 0.000 claims description 5
- 206010017758 gastric cancer Diseases 0.000 claims description 5
- 208000005017 glioblastoma Diseases 0.000 claims description 5
- 201000005249 lung adenocarcinoma Diseases 0.000 claims description 5
- 125000002757 morpholinyl group Chemical group 0.000 claims description 5
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 5
- 201000008482 osteoarthritis Diseases 0.000 claims description 5
- 125000003386 piperidinyl group Chemical group 0.000 claims description 5
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 5
- 102000027483 retinoid hormone receptors Human genes 0.000 claims description 5
- 108091008679 retinoid hormone receptors Proteins 0.000 claims description 5
- 208000000587 small cell lung carcinoma Diseases 0.000 claims description 5
- JISRJXWZXHFTJF-UHFFFAOYSA-N 5-amino-1-[4-[[2-methoxy-5-(trifluoromethyl)phenyl]carbamoylamino]phenyl]imidazole-4-carboxamide Chemical compound COC1=CC=C(C(F)(F)F)C=C1NC(=O)NC1=CC=C(N2C(=C(C(N)=O)N=C2)N)C=C1 JISRJXWZXHFTJF-UHFFFAOYSA-N 0.000 claims description 4
- 206010012442 Dermatitis contact Diseases 0.000 claims description 4
- 108091008794 FGF receptors Proteins 0.000 claims description 4
- 101000692455 Homo sapiens Platelet-derived growth factor receptor beta Proteins 0.000 claims description 4
- 206010020751 Hypersensitivity Diseases 0.000 claims description 4
- 208000029462 Immunodeficiency disease Diseases 0.000 claims description 4
- 102100026547 Platelet-derived growth factor receptor beta Human genes 0.000 claims description 4
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 4
- 230000033228 biological regulation Effects 0.000 claims description 4
- 201000008275 breast carcinoma Diseases 0.000 claims description 4
- 208000010247 contact dermatitis Diseases 0.000 claims description 4
- 102000052178 fibroblast growth factor receptor activity proteins Human genes 0.000 claims description 4
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 4
- 208000026278 immune system disease Diseases 0.000 claims description 4
- 201000005202 lung cancer Diseases 0.000 claims description 4
- 208000020816 lung neoplasm Diseases 0.000 claims description 4
- 201000008968 osteosarcoma Diseases 0.000 claims description 4
- 238000001959 radiotherapy Methods 0.000 claims description 4
- 208000007442 rickets Diseases 0.000 claims description 4
- 239000000849 selective androgen receptor modulator Substances 0.000 claims description 4
- 210000002784 stomach Anatomy 0.000 claims description 4
- 201000011549 stomach cancer Diseases 0.000 claims description 4
- YSDAXATUNMEKLG-UHFFFAOYSA-N 5-amino-1-[4-[[2-fluoro-5-(trifluoromethyl)phenyl]carbamoyl]phenyl]imidazole-4-carboxamide Chemical compound NC1=C(C(=O)N)N=CN1C1=CC=C(C(=O)NC=2C(=CC=C(C=2)C(F)(F)F)F)C=C1 YSDAXATUNMEKLG-UHFFFAOYSA-N 0.000 claims description 3
- IUKSOEWBILXDEE-UHFFFAOYSA-N 5-amino-1-[4-[[4-fluoro-3-(trifluoromethyl)phenyl]carbamoylamino]phenyl]imidazole-4-carboxamide Chemical compound NC1=C(C(=O)N)N=CN1C(C=C1)=CC=C1NC(=O)NC1=CC=C(F)C(C(F)(F)F)=C1 IUKSOEWBILXDEE-UHFFFAOYSA-N 0.000 claims description 3
- 206010000830 Acute leukaemia Diseases 0.000 claims description 3
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 claims description 3
- 208000003174 Brain Neoplasms Diseases 0.000 claims description 3
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 claims description 3
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims description 3
- 201000009273 Endometriosis Diseases 0.000 claims description 3
- 208000008839 Kidney Neoplasms Diseases 0.000 claims description 3
- 206010025323 Lymphomas Diseases 0.000 claims description 3
- 206010030155 Oesophageal carcinoma Diseases 0.000 claims description 3
- 208000004403 Prostatic Hyperplasia Diseases 0.000 claims description 3
- 206010038389 Renal cancer Diseases 0.000 claims description 3
- 208000024770 Thyroid neoplasm Diseases 0.000 claims description 3
- 125000003342 alkenyl group Chemical group 0.000 claims description 3
- 230000029936 alkylation Effects 0.000 claims description 3
- 238000005804 alkylation reaction Methods 0.000 claims description 3
- 125000000304 alkynyl group Chemical group 0.000 claims description 3
- 239000004037 angiogenesis inhibitor Substances 0.000 claims description 3
- 229940121369 angiogenesis inhibitor Drugs 0.000 claims description 3
- 210000004369 blood Anatomy 0.000 claims description 3
- 239000008280 blood Substances 0.000 claims description 3
- 239000000969 carrier Substances 0.000 claims description 3
- AOGYCOYQMAVAFD-UHFFFAOYSA-N chlorocarbonic acid Chemical class OC(Cl)=O AOGYCOYQMAVAFD-UHFFFAOYSA-N 0.000 claims description 3
- 230000001684 chronic effect Effects 0.000 claims description 3
- 208000024207 chronic leukemia Diseases 0.000 claims description 3
- 229940127089 cytotoxic agent Drugs 0.000 claims description 3
- 239000002254 cytotoxic agent Substances 0.000 claims description 3
- 231100000599 cytotoxic agent Toxicity 0.000 claims description 3
- 239000004030 hiv protease inhibitor Substances 0.000 claims description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 3
- 210000000987 immune system Anatomy 0.000 claims description 3
- 201000010982 kidney cancer Diseases 0.000 claims description 3
- 210000000867 larynx Anatomy 0.000 claims description 3
- 210000004185 liver Anatomy 0.000 claims description 3
- 201000007270 liver cancer Diseases 0.000 claims description 3
- 208000014018 liver neoplasm Diseases 0.000 claims description 3
- 210000004072 lung Anatomy 0.000 claims description 3
- 210000004324 lymphatic system Anatomy 0.000 claims description 3
- 208000026037 malignant tumor of neck Diseases 0.000 claims description 3
- 210000001616 monocyte Anatomy 0.000 claims description 3
- 239000003419 rna directed dna polymerase inhibitor Substances 0.000 claims description 3
- 229920006395 saturated elastomer Polymers 0.000 claims description 3
- 230000037390 scarring Effects 0.000 claims description 3
- 201000002510 thyroid cancer Diseases 0.000 claims description 3
- 210000001685 thyroid gland Anatomy 0.000 claims description 3
- 239000003558 transferase inhibitor Substances 0.000 claims description 3
- RXSJISQBNSJHAK-UHFFFAOYSA-N 1-[4-(5-amino-4-cyanoimidazol-1-yl)phenyl]-3-[2-fluoro-5-(trifluoromethyl)phenyl]urea Chemical compound NC1=C(C#N)N=CN1C(C=C1)=CC=C1NC(=O)NC1=CC(C(F)(F)F)=CC=C1F RXSJISQBNSJHAK-UHFFFAOYSA-N 0.000 claims description 2
- 125000003635 2-dimethylaminoethoxy group Chemical group [H]C([H])([H])N(C([H])([H])[H])C([H])([H])C([H])([H])O* 0.000 claims description 2
- RBZMPHOZJYRRMS-UHFFFAOYSA-N 5-amino-1-[4-[[3-(trifluoromethyl)phenyl]carbamoylamino]phenyl]imidazole-4-carboxamide Chemical compound NC1=C(C(=O)N)N=CN1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1 RBZMPHOZJYRRMS-UHFFFAOYSA-N 0.000 claims description 2
- KNTXUYQBZXBXRI-UHFFFAOYSA-N 5-amino-1-[4-[[3-fluoro-5-(trifluoromethyl)phenyl]carbamoylamino]phenyl]imidazole-4-carboxamide Chemical compound NC1=C(C(=O)N)N=CN1C(C=C1)=CC=C1NC(=O)NC1=CC(F)=CC(C(F)(F)F)=C1 KNTXUYQBZXBXRI-UHFFFAOYSA-N 0.000 claims description 2
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 claims description 2
- 206010005003 Bladder cancer Diseases 0.000 claims description 2
- 208000000461 Esophageal Neoplasms Diseases 0.000 claims description 2
- 102000009465 Growth Factor Receptors Human genes 0.000 claims description 2
- 108010009202 Growth Factor Receptors Proteins 0.000 claims description 2
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 claims description 2
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 claims description 2
- AKZWRTCWNXHHFR-PDIZUQLASA-N [(3S)-oxolan-3-yl] N-[(2S,3S)-4-[(5S)-5-benzyl-3-[(2R)-2-carbamoyloxy-2,3-dihydro-1H-inden-1-yl]-4-oxo-3H-pyrrol-5-yl]-3-hydroxy-1-phenylbutan-2-yl]carbamate Chemical compound NC(=O)O[C@@H]1Cc2ccccc2C1C1C=N[C@](C[C@H](O)[C@H](Cc2ccccc2)NC(=O)O[C@H]2CCOC2)(Cc2ccccc2)C1=O AKZWRTCWNXHHFR-PDIZUQLASA-N 0.000 claims description 2
- 230000001154 acute effect Effects 0.000 claims description 2
- 210000003679 cervix uteri Anatomy 0.000 claims description 2
- 201000004101 esophageal cancer Diseases 0.000 claims description 2
- 210000003238 esophagus Anatomy 0.000 claims description 2
- 210000003128 head Anatomy 0.000 claims description 2
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 claims description 2
- 210000000936 intestine Anatomy 0.000 claims description 2
- 210000003734 kidney Anatomy 0.000 claims description 2
- 125000001844 prenyl group Chemical group [H]C([*])([H])C([H])=C(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 2
- 210000002307 prostate Anatomy 0.000 claims description 2
- 206010041823 squamous cell carcinoma Diseases 0.000 claims description 2
- 208000017572 squamous cell neoplasm Diseases 0.000 claims description 2
- 210000003932 urinary bladder Anatomy 0.000 claims description 2
- 201000005112 urinary bladder cancer Diseases 0.000 claims description 2
- ZBNZAJFNDPPMDT-UHFFFAOYSA-N 1h-imidazole-5-carboxamide Chemical compound NC(=O)C1=CNC=N1 ZBNZAJFNDPPMDT-UHFFFAOYSA-N 0.000 claims 2
- 229940123468 Transferase inhibitor Drugs 0.000 claims 2
- 125000000896 monocarboxylic acid group Chemical group 0.000 claims 2
- 229940075993 receptor modulator Drugs 0.000 claims 2
- BNPZIJUBJBORDD-UHFFFAOYSA-N 5-(dimethylamino)-1-[4-[[2-fluoro-5-(trifluoromethyl)phenyl]carbamoylamino]phenyl]imidazole-4-carboxamide Chemical compound CN(C)C1=C(C(N)=O)N=CN1C(C=C1)=CC=C1NC(=O)NC1=CC(C(F)(F)F)=CC=C1F BNPZIJUBJBORDD-UHFFFAOYSA-N 0.000 claims 1
- ADDBCEUOQLJGAF-UHFFFAOYSA-N 5-amino-1-[4-[(3-methylphenyl)carbamoylamino]phenyl]imidazole-4-carboxamide Chemical compound CC1=CC=CC(NC(=O)NC=2C=CC(=CC=2)N2C(=C(C(N)=O)N=C2)N)=C1 ADDBCEUOQLJGAF-UHFFFAOYSA-N 0.000 claims 1
- AZIMSEDYULKZFG-UHFFFAOYSA-N 5-amino-1-[4-[(4-chloro-2-methoxy-5-methylphenyl)carbamoylamino]phenyl]imidazole-4-carboxamide Chemical compound COC1=CC(Cl)=C(C)C=C1NC(=O)NC1=CC=C(N2C(=C(C(N)=O)N=C2)N)C=C1 AZIMSEDYULKZFG-UHFFFAOYSA-N 0.000 claims 1
- SMDJJUSJKQASMR-UHFFFAOYSA-N 5-amino-1-[4-[[2-(2-morpholin-4-ylethoxy)-5-(trifluoromethyl)phenyl]carbamoylamino]phenyl]imidazole-4-carboxamide Chemical compound NC1=C(C(=O)N)N=CN1C(C=C1)=CC=C1NC(=O)NC1=CC(C(F)(F)F)=CC=C1OCCN1CCOCC1 SMDJJUSJKQASMR-UHFFFAOYSA-N 0.000 claims 1
- PDLDJUUGTXZNPH-UHFFFAOYSA-N 5-amino-1-[4-[[2-fluoro-5-(trifluoromethyl)phenyl]carbamoylamino]phenyl]-2-methylimidazole-4-carboxamide Chemical compound CC1=NC(C(N)=O)=C(N)N1C(C=C1)=CC=C1NC(=O)NC1=CC(C(F)(F)F)=CC=C1F PDLDJUUGTXZNPH-UHFFFAOYSA-N 0.000 claims 1
- MAGVBSGEHJKHAF-UHFFFAOYSA-N 5-amino-1-[4-[[4-chloro-3-(trifluoromethyl)phenyl]carbamoylamino]phenyl]imidazole-4-carboxamide Chemical compound NC1=C(C(=O)N)N=CN1C(C=C1)=CC=C1NC(=O)NC1=CC=C(Cl)C(C(F)(F)F)=C1 MAGVBSGEHJKHAF-UHFFFAOYSA-N 0.000 claims 1
- NMVVHECYRKUHER-UHFFFAOYSA-N 5-amino-2-ethyl-1-[4-[[2-fluoro-5-(trifluoromethyl)phenyl]carbamoylamino]phenyl]imidazole-4-carboxamide Chemical compound CCC1=NC(C(N)=O)=C(N)N1C(C=C1)=CC=C1NC(=O)NC1=CC(C(F)(F)F)=CC=C1F NMVVHECYRKUHER-UHFFFAOYSA-N 0.000 claims 1
- 102100032187 Androgen receptor Human genes 0.000 claims 1
- 229940122440 HIV protease inhibitor Drugs 0.000 claims 1
- 102000004286 Hydroxymethylglutaryl CoA Reductases Human genes 0.000 claims 1
- 108090000895 Hydroxymethylglutaryl CoA Reductases Proteins 0.000 claims 1
- 208000028018 Lymphocytic leukaemia Diseases 0.000 claims 1
- 108010080146 androgen receptors Proteins 0.000 claims 1
- 210000003739 neck Anatomy 0.000 claims 1
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 claims 1
- 210000004027 cell Anatomy 0.000 description 52
- 235000002639 sodium chloride Nutrition 0.000 description 49
- 108010053099 Vascular Endothelial Growth Factor Receptor-2 Proteins 0.000 description 31
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 30
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 30
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 29
- 239000004480 active ingredient Substances 0.000 description 27
- 238000000668 atmospheric pressure chemical ionisation mass spectrometry Methods 0.000 description 26
- 239000000243 solution Substances 0.000 description 25
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 20
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 18
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 18
- 238000003556 assay Methods 0.000 description 17
- 108010014186 ras Proteins Proteins 0.000 description 17
- 102000016914 ras Proteins Human genes 0.000 description 17
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 16
- 108700020796 Oncogene Proteins 0.000 description 16
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 15
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 15
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 15
- 239000002585 base Substances 0.000 description 15
- 230000012010 growth Effects 0.000 description 15
- 230000002265 prevention Effects 0.000 description 15
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 14
- 238000006243 chemical reaction Methods 0.000 description 14
- 238000009472 formulation Methods 0.000 description 14
- 230000002401 inhibitory effect Effects 0.000 description 14
- 239000008194 pharmaceutical composition Substances 0.000 description 14
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 13
- 239000003826 tablet Substances 0.000 description 13
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 12
- 239000000843 powder Substances 0.000 description 12
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 12
- 102000005962 receptors Human genes 0.000 description 12
- 108020003175 receptors Proteins 0.000 description 11
- 239000000758 substrate Substances 0.000 description 11
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 11
- 102000004190 Enzymes Human genes 0.000 description 10
- 108090000790 Enzymes Proteins 0.000 description 10
- 102000009484 Vascular Endothelial Growth Factor Receptors Human genes 0.000 description 10
- 230000026731 phosphorylation Effects 0.000 description 10
- 238000006366 phosphorylation reaction Methods 0.000 description 10
- 235000018102 proteins Nutrition 0.000 description 10
- 238000012360 testing method Methods 0.000 description 10
- DLFVBJFMPXGRIB-UHFFFAOYSA-N thioacetamide Natural products CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 10
- 201000009030 Carcinoma Diseases 0.000 description 9
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 9
- 235000013877 carbamide Nutrition 0.000 description 9
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 9
- 239000003102 growth factor Substances 0.000 description 9
- BJHCYTJNPVGSBZ-YXSASFKJSA-N 1-[4-[6-amino-5-[(Z)-methoxyiminomethyl]pyrimidin-4-yl]oxy-2-chlorophenyl]-3-ethylurea Chemical compound CCNC(=O)Nc1ccc(Oc2ncnc(N)c2\C=N/OC)cc1Cl BJHCYTJNPVGSBZ-YXSASFKJSA-N 0.000 description 8
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 8
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 8
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 8
- 230000001086 cytosolic effect Effects 0.000 description 8
- 229940079593 drug Drugs 0.000 description 8
- 230000014509 gene expression Effects 0.000 description 8
- 239000012442 inert solvent Substances 0.000 description 8
- 239000011734 sodium Substances 0.000 description 8
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 8
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 7
- 230000004913 activation Effects 0.000 description 7
- 230000001419 dependent effect Effects 0.000 description 7
- 239000007788 liquid Substances 0.000 description 7
- 229910052757 nitrogen Inorganic materials 0.000 description 7
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 7
- 239000002904 solvent Substances 0.000 description 7
- 230000004614 tumor growth Effects 0.000 description 7
- 150000003672 ureas Chemical class 0.000 description 7
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 7
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- 101100381481 Caenorhabditis elegans baz-2 gene Proteins 0.000 description 6
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 6
- 241000124008 Mammalia Species 0.000 description 6
- 241001465754 Metazoa Species 0.000 description 6
- 229910019142 PO4 Inorganic materials 0.000 description 6
- 102000001253 Protein Kinase Human genes 0.000 description 6
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 6
- 101100372762 Rattus norvegicus Flt1 gene Proteins 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 6
- 239000012911 assay medium Substances 0.000 description 6
- 239000011230 binding agent Substances 0.000 description 6
- 239000002775 capsule Substances 0.000 description 6
- 239000003446 ligand Substances 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 239000003226 mitogen Substances 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- 239000003208 petroleum Substances 0.000 description 6
- YBYRMVIVWMBXKQ-UHFFFAOYSA-N phenylmethanesulfonyl fluoride Chemical compound FS(=O)(=O)CC1=CC=CC=C1 YBYRMVIVWMBXKQ-UHFFFAOYSA-N 0.000 description 6
- 239000010452 phosphate Substances 0.000 description 6
- 108060006633 protein kinase Proteins 0.000 description 6
- 239000007787 solid Substances 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 210000001519 tissue Anatomy 0.000 description 6
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 5
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 5
- 101001004391 Drosophila melanogaster Protein jim lovell Proteins 0.000 description 5
- 108090000379 Fibroblast growth factor 2 Proteins 0.000 description 5
- 241000282412 Homo Species 0.000 description 5
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 5
- 102000043276 Oncogene Human genes 0.000 description 5
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 5
- 239000007983 Tris buffer Substances 0.000 description 5
- 239000013543 active substance Substances 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 229910052791 calcium Inorganic materials 0.000 description 5
- 239000011575 calcium Substances 0.000 description 5
- 230000010261 cell growth Effects 0.000 description 5
- 239000006071 cream Substances 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 239000012458 free base Substances 0.000 description 5
- 230000006870 function Effects 0.000 description 5
- 235000011187 glycerol Nutrition 0.000 description 5
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 5
- 239000007924 injection Substances 0.000 description 5
- 238000002347 injection Methods 0.000 description 5
- 239000010410 layer Substances 0.000 description 5
- 239000000314 lubricant Substances 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 5
- 239000003921 oil Substances 0.000 description 5
- 229920000642 polymer Polymers 0.000 description 5
- 229910052700 potassium Inorganic materials 0.000 description 5
- 239000011591 potassium Substances 0.000 description 5
- 150000003254 radicals Chemical group 0.000 description 5
- 229910052708 sodium Inorganic materials 0.000 description 5
- 235000015424 sodium Nutrition 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- 210000003556 vascular endothelial cell Anatomy 0.000 description 5
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical group COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 4
- 102100034594 Angiopoietin-1 Human genes 0.000 description 4
- 208000005623 Carcinogenesis Diseases 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 4
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 4
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 4
- 101000753253 Homo sapiens Tyrosine-protein kinase receptor Tie-1 Proteins 0.000 description 4
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 4
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 4
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 4
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 4
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 4
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 4
- 206010029113 Neovascularisation Diseases 0.000 description 4
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 4
- 208000034038 Pathologic Neovascularization Diseases 0.000 description 4
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 4
- 102000052575 Proto-Oncogene Human genes 0.000 description 4
- 108700020978 Proto-Oncogene Proteins 0.000 description 4
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 4
- 229920002472 Starch Polymers 0.000 description 4
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 4
- 102100022007 Tyrosine-protein kinase receptor Tie-1 Human genes 0.000 description 4
- 239000008186 active pharmaceutical agent Substances 0.000 description 4
- 150000001340 alkali metals Chemical class 0.000 description 4
- 150000001342 alkaline earth metals Chemical class 0.000 description 4
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 4
- 210000004204 blood vessel Anatomy 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 230000036952 cancer formation Effects 0.000 description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 4
- 231100000504 carcinogenesis Toxicity 0.000 description 4
- 230000004663 cell proliferation Effects 0.000 description 4
- 230000001413 cellular effect Effects 0.000 description 4
- 239000007884 disintegrant Substances 0.000 description 4
- 210000002889 endothelial cell Anatomy 0.000 description 4
- 239000008103 glucose Substances 0.000 description 4
- 239000008187 granular material Substances 0.000 description 4
- 238000000338 in vitro Methods 0.000 description 4
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 4
- 230000003834 intracellular effect Effects 0.000 description 4
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 4
- 229940043355 kinase inhibitor Drugs 0.000 description 4
- 230000035800 maturation Effects 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- 239000012528 membrane Substances 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- 235000001968 nicotinic acid Nutrition 0.000 description 4
- 239000011664 nicotinic acid Substances 0.000 description 4
- LQNUZADURLCDLV-UHFFFAOYSA-N nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC=C1 LQNUZADURLCDLV-UHFFFAOYSA-N 0.000 description 4
- 231100000252 nontoxic Toxicity 0.000 description 4
- 230000003000 nontoxic effect Effects 0.000 description 4
- 150000007530 organic bases Chemical class 0.000 description 4
- 230000037361 pathway Effects 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 4
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 4
- 230000035755 proliferation Effects 0.000 description 4
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 4
- 238000011321 prophylaxis Methods 0.000 description 4
- 230000009822 protein phosphorylation Effects 0.000 description 4
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 4
- 230000002285 radioactive effect Effects 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- 230000001105 regulatory effect Effects 0.000 description 4
- 238000012552 review Methods 0.000 description 4
- 230000008054 signal transmission Effects 0.000 description 4
- 235000019698 starch Nutrition 0.000 description 4
- 239000008107 starch Substances 0.000 description 4
- 239000000454 talc Substances 0.000 description 4
- 229910052623 talc Inorganic materials 0.000 description 4
- 235000012222 talc Nutrition 0.000 description 4
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- CDTVHWZDLMFHAW-UHFFFAOYSA-N 5-amino-1-[4-[[2-fluoro-5-(trifluoromethyl)benzoyl]amino]phenyl]imidazole-4-carboxamide Chemical compound NC1=C(C(=O)N)N=CN1C(C=C1)=CC=C1NC(=O)C1=CC(C(F)(F)F)=CC=C1F CDTVHWZDLMFHAW-UHFFFAOYSA-N 0.000 description 3
- SVGGJEGMNWIXOC-UHFFFAOYSA-N 5-amino-1-[4-[[2-fluoro-5-(trifluoromethyl)phenyl]carbamoylamino]phenyl]imidazole-4-carboxamide Chemical compound NC1=C(C(=O)N)N=CN1C(C=C1)=CC=C1NC(=O)NC1=CC(C(F)(F)F)=CC=C1F SVGGJEGMNWIXOC-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 3
- ZKHQWZAMYRWXGA-UHFFFAOYSA-N Adenosine triphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O ZKHQWZAMYRWXGA-UHFFFAOYSA-N 0.000 description 3
- 108010039627 Aprotinin Proteins 0.000 description 3
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 3
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 3
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 3
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 3
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 3
- GDBQQVLCIARPGH-UHFFFAOYSA-N Leupeptin Natural products CC(C)CC(NC(C)=O)C(=O)NC(CC(C)C)C(=O)NC(C=O)CCCN=C(N)N GDBQQVLCIARPGH-UHFFFAOYSA-N 0.000 description 3
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 3
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 3
- 241000699666 Mus <mouse, genus> Species 0.000 description 3
- JLTDJTHDQAWBAV-UHFFFAOYSA-N N,N-dimethylaniline Chemical compound CN(C)C1=CC=CC=C1 JLTDJTHDQAWBAV-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- 241000288906 Primates Species 0.000 description 3
- 102000009516 Protein Serine-Threonine Kinases Human genes 0.000 description 3
- 108010009341 Protein Serine-Threonine Kinases Proteins 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 3
- 229920004890 Triton X-100 Polymers 0.000 description 3
- 239000013504 Triton X-100 Substances 0.000 description 3
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 3
- 102000016549 Vascular Endothelial Growth Factor Receptor-2 Human genes 0.000 description 3
- 108010053100 Vascular Endothelial Growth Factor Receptor-3 Proteins 0.000 description 3
- 241000700605 Viruses Species 0.000 description 3
- AXIWSBMKUQLHRL-UHFFFAOYSA-N [2-fluoro-5-(trifluoromethyl)phenyl]urea Chemical compound NC(=O)NC1=CC(C(F)(F)F)=CC=C1F AXIWSBMKUQLHRL-UHFFFAOYSA-N 0.000 description 3
- 239000003513 alkali Substances 0.000 description 3
- 229910052783 alkali metal Inorganic materials 0.000 description 3
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 3
- 229960004405 aprotinin Drugs 0.000 description 3
- 229940098773 bovine serum albumin Drugs 0.000 description 3
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 229910002091 carbon monoxide Inorganic materials 0.000 description 3
- 238000004113 cell culture Methods 0.000 description 3
- 230000030833 cell death Effects 0.000 description 3
- 238000000576 coating method Methods 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 230000018109 developmental process Effects 0.000 description 3
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 3
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 3
- 208000035475 disorder Diseases 0.000 description 3
- 239000012154 double-distilled water Substances 0.000 description 3
- 239000000975 dye Substances 0.000 description 3
- 239000012636 effector Substances 0.000 description 3
- 239000006260 foam Substances 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 3
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 3
- 230000001771 impaired effect Effects 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- ZPNFWUPYTFPOJU-LPYSRVMUSA-N iniprol Chemical compound C([C@H]1C(=O)NCC(=O)NCC(=O)N[C@H]2CSSC[C@H]3C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@H](C(N[C@H](C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC=4C=CC=CC=4)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC=4C=CC=CC=4)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC2=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]2N(CCC2)C(=O)[C@@H](N)CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N2[C@@H](CCC2)C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N2[C@@H](CCC2)C(=O)N3)C(=O)NCC(=O)NCC(=O)N[C@@H](C)C(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H](C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@H](C(=O)N1)C(C)C)[C@@H](C)O)[C@@H](C)CC)=O)[C@@H](C)CC)C1=CC=C(O)C=C1 ZPNFWUPYTFPOJU-LPYSRVMUSA-N 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- GDBQQVLCIARPGH-ULQDDVLXSA-N leupeptin Chemical compound CC(C)C[C@H](NC(C)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C=O)CCCN=C(N)N GDBQQVLCIARPGH-ULQDDVLXSA-N 0.000 description 3
- 108010052968 leupeptin Proteins 0.000 description 3
- 239000012139 lysis buffer Substances 0.000 description 3
- 229910052749 magnesium Inorganic materials 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 230000007246 mechanism Effects 0.000 description 3
- 230000005012 migration Effects 0.000 description 3
- 238000013508 migration Methods 0.000 description 3
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 3
- 230000035772 mutation Effects 0.000 description 3
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 3
- 235000019198 oils Nutrition 0.000 description 3
- 239000002674 ointment Substances 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 239000006072 paste Substances 0.000 description 3
- 108010091212 pepstatin Proteins 0.000 description 3
- 229950000964 pepstatin Drugs 0.000 description 3
- FAXGPCHRFPCXOO-LXTPJMTPSA-N pepstatin A Chemical compound OC(=O)C[C@H](O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)C[C@H](O)[C@H](CC(C)C)NC(=O)[C@H](C(C)C)NC(=O)[C@H](C(C)C)NC(=O)CC(C)C FAXGPCHRFPCXOO-LXTPJMTPSA-N 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 238000002953 preparative HPLC Methods 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- 230000035484 reaction time Effects 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 208000037803 restenosis Diseases 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 235000011121 sodium hydroxide Nutrition 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 230000000638 stimulation Effects 0.000 description 3
- 235000000346 sugar Nutrition 0.000 description 3
- 239000000829 suppository Substances 0.000 description 3
- 239000006188 syrup Substances 0.000 description 3
- 235000020357 syrup Nutrition 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 230000000699 topical effect Effects 0.000 description 3
- 238000012546 transfer Methods 0.000 description 3
- 210000004881 tumor cell Anatomy 0.000 description 3
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 3
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 3
- 230000002792 vascular Effects 0.000 description 3
- 239000011534 wash buffer Substances 0.000 description 3
- 239000001993 wax Substances 0.000 description 3
- NXLNNXIXOYSCMB-UHFFFAOYSA-N (4-nitrophenyl) carbonochloridate Chemical compound [O-][N+](=O)C1=CC=C(OC(Cl)=O)C=C1 NXLNNXIXOYSCMB-UHFFFAOYSA-N 0.000 description 2
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- JRWAUKYINYWSTA-UHFFFAOYSA-N 2-amino-2-cyanoacetamide Chemical compound N#CC(N)C(N)=O JRWAUKYINYWSTA-UHFFFAOYSA-N 0.000 description 2
- ZNQVEEAIQZEUHB-UHFFFAOYSA-N 2-ethoxyethanol Chemical compound CCOCCO ZNQVEEAIQZEUHB-UHFFFAOYSA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- 108700028369 Alleles Proteins 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- 108010048154 Angiopoietin-1 Proteins 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 241000416162 Astragalus gummifer Species 0.000 description 2
- 201000001320 Atherosclerosis Diseases 0.000 description 2
- 208000009137 Behcet syndrome Diseases 0.000 description 2
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 2
- 201000004569 Blindness Diseases 0.000 description 2
- 208000006386 Bone Resorption Diseases 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 2
- QGJOPFRUJISHPQ-UHFFFAOYSA-N Carbon disulfide Chemical compound S=C=S QGJOPFRUJISHPQ-UHFFFAOYSA-N 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 2
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 2
- 101000827763 Drosophila melanogaster Fibroblast growth factor receptor homolog 1 Proteins 0.000 description 2
- 206010016654 Fibrosis Diseases 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 102000013446 GTP Phosphohydrolases Human genes 0.000 description 2
- 101710113436 GTPase KRas Proteins 0.000 description 2
- 108091006109 GTPases Proteins 0.000 description 2
- 108010070675 Glutathione transferase Proteins 0.000 description 2
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 2
- 102100029100 Hematopoietic prostaglandin D synthase Human genes 0.000 description 2
- 101000924552 Homo sapiens Angiopoietin-1 Proteins 0.000 description 2
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 2
- 108060003951 Immunoglobulin Proteins 0.000 description 2
- 102000004877 Insulin Human genes 0.000 description 2
- 108090001061 Insulin Proteins 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- 206010023825 Laryngeal cancer Diseases 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 108090000744 Mitogen-Activated Protein Kinase Kinases Proteins 0.000 description 2
- 102000004232 Mitogen-Activated Protein Kinase Kinases Human genes 0.000 description 2
- 102100023482 Mitogen-activated protein kinase 14 Human genes 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 241001529936 Murinae Species 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 2
- 229910002651 NO3 Inorganic materials 0.000 description 2
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 2
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 2
- 102000003992 Peroxidases Human genes 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 108700019535 Phosphoprotein Phosphatases Proteins 0.000 description 2
- 102000045595 Phosphoprotein Phosphatases Human genes 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 108090000315 Protein Kinase C Proteins 0.000 description 2
- 102000003923 Protein Kinase C Human genes 0.000 description 2
- 101150040459 RAS gene Proteins 0.000 description 2
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- 241000906446 Theraps Species 0.000 description 2
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 2
- 239000004473 Threonine Substances 0.000 description 2
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 2
- 229920001615 Tragacanth Polymers 0.000 description 2
- XSTXAVWGXDQKEL-UHFFFAOYSA-N Trichloroethylene Chemical group ClC=C(Cl)Cl XSTXAVWGXDQKEL-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 2
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 2
- 108010034265 Vascular Endothelial Growth Factor Receptors Proteins 0.000 description 2
- 102100033179 Vascular endothelial growth factor receptor 3 Human genes 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 239000008272 agar Substances 0.000 description 2
- 235000010419 agar Nutrition 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 229940072056 alginate Drugs 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 150000001350 alkyl halides Chemical class 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 230000002491 angiogenic effect Effects 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- 208000006673 asthma Diseases 0.000 description 2
- 230000035578 autophosphorylation Effects 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 239000000440 bentonite Substances 0.000 description 2
- 235000012216 bentonite Nutrition 0.000 description 2
- 229910000278 bentonite Inorganic materials 0.000 description 2
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 2
- 229940077388 benzenesulfonate Drugs 0.000 description 2
- ZLLVUAAESHIVAZ-UHFFFAOYSA-N benzo[g]isoquinoline-5,10-dione Chemical compound N1=CC=C2C(=O)C3=CC=CC=C3C(=O)C2=C1 ZLLVUAAESHIVAZ-UHFFFAOYSA-N 0.000 description 2
- 229940050390 benzoate Drugs 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 229920002988 biodegradable polymer Polymers 0.000 description 2
- 239000004621 biodegradable polymer Substances 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 230000024279 bone resorption Effects 0.000 description 2
- 229910052792 caesium Inorganic materials 0.000 description 2
- TVFDJXOCXUVLDH-UHFFFAOYSA-N caesium atom Chemical compound [Cs] TVFDJXOCXUVLDH-UHFFFAOYSA-N 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 2
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 2
- 150000001735 carboxylic acids Chemical class 0.000 description 2
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 2
- 230000003915 cell function Effects 0.000 description 2
- 230000006037 cell lysis Effects 0.000 description 2
- 230000009134 cell regulation Effects 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- 150000008280 chlorinated hydrocarbons Chemical class 0.000 description 2
- VDANGULDQQJODZ-UHFFFAOYSA-N chloroprocaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1Cl VDANGULDQQJODZ-UHFFFAOYSA-N 0.000 description 2
- 229960002023 chloroprocaine Drugs 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 2
- 229960001231 choline Drugs 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 210000001072 colon Anatomy 0.000 description 2
- 239000000470 constituent Substances 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- UQHKFADEQIVWID-UHFFFAOYSA-N cytokinin Natural products C1=NC=2C(NCC=C(CO)C)=NC=NC=2N1C1CC(O)C(CO)O1 UQHKFADEQIVWID-UHFFFAOYSA-N 0.000 description 2
- 239000004062 cytokinin Substances 0.000 description 2
- 231100000433 cytotoxic Toxicity 0.000 description 2
- 230000001472 cytotoxic effect Effects 0.000 description 2
- 229960000975 daunorubicin Drugs 0.000 description 2
- 206010012601 diabetes mellitus Diseases 0.000 description 2
- 238000000502 dialysis Methods 0.000 description 2
- 230000004069 differentiation Effects 0.000 description 2
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 2
- 239000013024 dilution buffer Substances 0.000 description 2
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 2
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 238000000132 electrospray ionisation Methods 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 230000003511 endothelial effect Effects 0.000 description 2
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- LYCAIKOWRPUZTN-UHFFFAOYSA-N ethylene glycol Natural products OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 2
- 239000013604 expression vector Substances 0.000 description 2
- 239000003889 eye drop Substances 0.000 description 2
- 229940012356 eye drops Drugs 0.000 description 2
- 238000010265 fast atom bombardment Methods 0.000 description 2
- 230000001605 fetal effect Effects 0.000 description 2
- 238000003818 flash chromatography Methods 0.000 description 2
- 238000002875 fluorescence polarization Methods 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 108020001507 fusion proteins Proteins 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 229940050410 gluconate Drugs 0.000 description 2
- 238000005469 granulation Methods 0.000 description 2
- 230000003179 granulation Effects 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 2
- 102000018358 immunoglobulin Human genes 0.000 description 2
- 230000001976 improved effect Effects 0.000 description 2
- 238000011065 in-situ storage Methods 0.000 description 2
- 238000010348 incorporation Methods 0.000 description 2
- 229940125396 insulin Drugs 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 230000000302 ischemic effect Effects 0.000 description 2
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 2
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 238000000021 kinase assay Methods 0.000 description 2
- GXESHMAMLJKROZ-IAPPQJPRSA-N lasofoxifene Chemical compound C1([C@@H]2[C@@H](C3=CC=C(C=C3CC2)O)C=2C=CC(OCCN3CCCC3)=CC=2)=CC=CC=C1 GXESHMAMLJKROZ-IAPPQJPRSA-N 0.000 description 2
- 229960002367 lasofoxifene Drugs 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 239000007937 lozenge Substances 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 2
- 238000004949 mass spectrometry Methods 0.000 description 2
- 229960003194 meglumine Drugs 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 150000002739 metals Chemical class 0.000 description 2
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical compound COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 2
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 2
- 238000000520 microinjection Methods 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 230000002297 mitogenic effect Effects 0.000 description 2
- 230000008747 mitogenic response Effects 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 125000004312 morpholin-2-yl group Chemical group [H]N1C([H])([H])C([H])([H])OC([H])(*)C1([H])[H] 0.000 description 2
- 125000004572 morpholin-3-yl group Chemical group N1C(COCC1)* 0.000 description 2
- 150000002825 nitriles Chemical class 0.000 description 2
- 150000002828 nitro derivatives Chemical class 0.000 description 2
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 description 2
- 102000039446 nucleic acids Human genes 0.000 description 2
- 108020004707 nucleic acids Proteins 0.000 description 2
- 150000007523 nucleic acids Chemical class 0.000 description 2
- 238000011580 nude mouse model Methods 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 2
- 230000035699 permeability Effects 0.000 description 2
- 108040007629 peroxidase activity proteins Proteins 0.000 description 2
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 2
- 230000000704 physical effect Effects 0.000 description 2
- 125000004483 piperidin-3-yl group Chemical group N1CC(CCC1)* 0.000 description 2
- 125000004482 piperidin-4-yl group Chemical group N1CCC(CC1)* 0.000 description 2
- 239000002798 polar solvent Substances 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 229920002643 polyglutamic acid Polymers 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 229920001592 potato starch Polymers 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 238000003825 pressing Methods 0.000 description 2
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 2
- 229960004919 procaine Drugs 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 2
- 229960004622 raloxifene Drugs 0.000 description 2
- 108700042226 ras Genes Proteins 0.000 description 2
- 239000002994 raw material Substances 0.000 description 2
- 239000011535 reaction buffer Substances 0.000 description 2
- 239000002464 receptor antagonist Substances 0.000 description 2
- 229940044551 receptor antagonist Drugs 0.000 description 2
- 238000007634 remodeling Methods 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 210000001525 retina Anatomy 0.000 description 2
- 238000012216 screening Methods 0.000 description 2
- 229940095743 selective estrogen receptor modulator Drugs 0.000 description 2
- 239000000333 selective estrogen receptor modulator Substances 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- FQENQNTWSFEDLI-UHFFFAOYSA-J sodium diphosphate Chemical compound [Na+].[Na+].[Na+].[Na+].[O-]P([O-])(=O)OP([O-])([O-])=O FQENQNTWSFEDLI-UHFFFAOYSA-J 0.000 description 2
- 229940048086 sodium pyrophosphate Drugs 0.000 description 2
- PFNFFQXMRSDOHW-UHFFFAOYSA-N spermine Chemical compound NCCCNCCCCNCCCN PFNFFQXMRSDOHW-UHFFFAOYSA-N 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 239000012089 stop solution Substances 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 150000003462 sulfoxides Chemical class 0.000 description 2
- 230000002195 synergetic effect Effects 0.000 description 2
- 229940095064 tartrate Drugs 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 235000019818 tetrasodium diphosphate Nutrition 0.000 description 2
- 239000001577 tetrasodium phosphonato phosphate Substances 0.000 description 2
- YAPQBXQYLJRXSA-UHFFFAOYSA-N theobromine Chemical compound CN1C(=O)NC(=O)C2=C1N=CN2C YAPQBXQYLJRXSA-UHFFFAOYSA-N 0.000 description 2
- 229940104230 thymidine Drugs 0.000 description 2
- 238000002877 time resolved fluorescence resonance energy transfer Methods 0.000 description 2
- 230000000451 tissue damage Effects 0.000 description 2
- 231100000827 tissue damage Toxicity 0.000 description 2
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 2
- 235000010487 tragacanth Nutrition 0.000 description 2
- 239000000196 tragacanth Substances 0.000 description 2
- 229940116362 tragacanth Drugs 0.000 description 2
- 238000010361 transduction Methods 0.000 description 2
- 230000026683 transduction Effects 0.000 description 2
- 238000002054 transplantation Methods 0.000 description 2
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 description 2
- GKASDNZWUGIAMG-UHFFFAOYSA-N triethyl orthoformate Chemical compound CCOC(OCC)OCC GKASDNZWUGIAMG-UHFFFAOYSA-N 0.000 description 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 229960000281 trometamol Drugs 0.000 description 2
- 230000005747 tumor angiogenesis Effects 0.000 description 2
- 210000003606 umbilical vein Anatomy 0.000 description 2
- 239000002691 unilamellar liposome Substances 0.000 description 2
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 2
- 230000006459 vascular development Effects 0.000 description 2
- 208000019553 vascular disease Diseases 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- 239000008096 xylene Substances 0.000 description 2
- BMKDZUISNHGIBY-ZETCQYMHSA-N (+)-dexrazoxane Chemical compound C([C@H](C)N1CC(=O)NC(=O)C1)N1CC(=O)NC(=O)C1 BMKDZUISNHGIBY-ZETCQYMHSA-N 0.000 description 1
- DNXHEGUUPJUMQT-UHFFFAOYSA-N (+)-estrone Natural products OC1=CC=C2C3CCC(C)(C(CC4)=O)C4C3CCC2=C1 DNXHEGUUPJUMQT-UHFFFAOYSA-N 0.000 description 1
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical compound CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- JKFZMIQMKFWJAY-RQJQXFIZSA-N (1r,3s,5z)-5-[(2e)-2-[(3as,7as)-1-[(2r)-6-hydroxy-6-methylhept-4-yn-2-yl]-7a-methyl-3a,5,6,7-tetrahydro-3h-inden-4-ylidene]ethylidene]-4-methylidenecyclohexane-1,3-diol Chemical compound C1(/[C@@H]2CC=C([C@]2(CCC1)C)[C@@H](CC#CC(C)(C)O)C)=C\C=C1\C[C@@H](O)C[C@H](O)C1=C JKFZMIQMKFWJAY-RQJQXFIZSA-N 0.000 description 1
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 1
- ZUQBAQVRAURMCL-DOMZBBRYSA-N (2s)-2-[[4-[2-[(6r)-2-amino-4-oxo-5,6,7,8-tetrahydro-1h-pyrido[2,3-d]pyrimidin-6-yl]ethyl]benzoyl]amino]pentanedioic acid Chemical compound C([C@@H]1CC=2C(=O)N=C(NC=2NC1)N)CC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 ZUQBAQVRAURMCL-DOMZBBRYSA-N 0.000 description 1
- XSAKVDNHFRWJKS-IIZANFQQSA-N (2s)-n-benzyl-1-[(2s)-1-[(2s)-2-[[(2s)-2-[[(2s)-2-(dimethylamino)-3-methylbutanoyl]amino]-3-methylbutanoyl]-methylamino]-3-methylbutanoyl]pyrrolidine-2-carbonyl]pyrrolidine-2-carboxamide Chemical compound CC(C)[C@H](N(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H](C(C)C)C(=O)N1CCC[C@H]1C(=O)N1[C@H](C(=O)NCC=2C=CC=CC=2)CCC1 XSAKVDNHFRWJKS-IIZANFQQSA-N 0.000 description 1
- PSVUJBVBCOISSP-SPFKKGSWSA-N (2s,3r,4s,5s,6r)-2-bis(2-chloroethylamino)phosphoryloxy-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound OC[C@H]1O[C@@H](OP(=O)(NCCCl)NCCCl)[C@H](O)[C@@H](O)[C@@H]1O PSVUJBVBCOISSP-SPFKKGSWSA-N 0.000 description 1
- ZKSNZYLCOXUJIR-VOKUKXJJSA-N (5s,5ar,8ar,9r)-5-[[(2r,4ar,6r,7r,8r,8as)-7-(dimethylamino)-8-hydroxy-2-methyl-4,4a,6,7,8,8a-hexahydropyrano[3,2-d][1,3]dioxin-6-yl]oxy]-9-(4-hydroxy-3,5-dimethoxyphenyl)-5a,6,8a,9-tetrahydro-5h-[2]benzofuro[6,5-f][1,3]benzodioxol-8-one Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)N(C)C)[C@@H]3[C@@H]2C(OC3)=O)=C1 ZKSNZYLCOXUJIR-VOKUKXJJSA-N 0.000 description 1
- DLROLUIVVKTFPW-LVEBQJTPSA-N (5s,5as,8ar,9r)-9-(4-hydroxy-3,5-dimethoxyphenyl)-5-(4-nitroanilino)-5a,6,8a,9-tetrahydro-5h-[2]benzofuro[5,6-f][1,3]benzodioxol-8-one Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](NC=3C=CC(=CC=3)[N+]([O-])=O)[C@@H]3[C@@H]2C(OC3)=O)=C1 DLROLUIVVKTFPW-LVEBQJTPSA-N 0.000 description 1
- WTSKMKRYHATLLL-UHFFFAOYSA-N (6-benzoyloxy-3-cyanopyridin-2-yl) 3-[3-(ethoxymethyl)-5-fluoro-2,6-dioxopyrimidine-1-carbonyl]benzoate Chemical compound O=C1N(COCC)C=C(F)C(=O)N1C(=O)C1=CC=CC(C(=O)OC=2C(=CC=C(OC(=O)C=3C=CC=CC=3)N=2)C#N)=C1 WTSKMKRYHATLLL-UHFFFAOYSA-N 0.000 description 1
- BSRQHWFOFMAZRL-BODGVHBXSA-N (7s,9s)-7-[(2r,4s,5s,6s)-5-[(2s,4s,5s,6s)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-4-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-8,10-dihydro-7h-tetracene-5,12-dione;hydron;chloride Chemical compound Cl.C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@H]1[C@@H](O)C[C@H](O[C@@H]2C3=C(O)C=4C(=O)C5=CC=CC=C5C(=O)C=4C(O)=C3C[C@](O)(C2)C(=O)CO)O[C@H]1C BSRQHWFOFMAZRL-BODGVHBXSA-N 0.000 description 1
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- LKJPYSCBVHEWIU-KRWDZBQOSA-N (R)-bicalutamide Chemical compound C([C@@](O)(C)C(=O)NC=1C=C(C(C#N)=CC=1)C(F)(F)F)S(=O)(=O)C1=CC=C(F)C=C1 LKJPYSCBVHEWIU-KRWDZBQOSA-N 0.000 description 1
- ZGNLFUXWZJGETL-YUSKDDKASA-N (Z)-[(2S)-2-amino-2-carboxyethyl]-hydroxyimino-oxidoazanium Chemical compound N[C@@H](C\[N+]([O-])=N\O)C(O)=O ZGNLFUXWZJGETL-YUSKDDKASA-N 0.000 description 1
- 0 *c(c(*)c1*)c(*)c(*)c1N Chemical compound *c(c(*)c1*)c(*)c(*)c1N 0.000 description 1
- 125000005919 1,2,2-trimethylpropyl group Chemical group 0.000 description 1
- 125000005940 1,4-dioxanyl group Chemical group 0.000 description 1
- HJTAZXHBEBIQQX-UHFFFAOYSA-N 1,5-bis(chloromethyl)naphthalene Chemical compound C1=CC=C2C(CCl)=CC=CC2=C1CCl HJTAZXHBEBIQQX-UHFFFAOYSA-N 0.000 description 1
- RVNVDKZBWXYULM-UHFFFAOYSA-N 1-(4-aminophenyl)-3-[2-methoxy-5-(trifluoromethyl)phenyl]urea Chemical compound COC1=CC=C(C(F)(F)F)C=C1NC(=O)NC1=CC=C(N)C=C1 RVNVDKZBWXYULM-UHFFFAOYSA-N 0.000 description 1
- MZNMZWZGUGFQJP-UHFFFAOYSA-N 1-[11-(dodecylamino)-10-hydroxyundecyl]-3,7-dimethylpurine-2,6-dione Chemical compound O=C1N(CCCCCCCCCC(O)CNCCCCCCCCCCCC)C(=O)N(C)C2=C1N(C)C=N2 MZNMZWZGUGFQJP-UHFFFAOYSA-N 0.000 description 1
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 1
- VUQPJRPDRDVQMN-UHFFFAOYSA-N 1-chlorooctadecane Chemical compound CCCCCCCCCCCCCCCCCCCl VUQPJRPDRDVQMN-UHFFFAOYSA-N 0.000 description 1
- 125000006218 1-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- PZNPLUBHRSSFHT-RRHRGVEJSA-N 1-hexadecanoyl-2-octadecanoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCCCC(=O)O[C@@H](COP([O-])(=O)OCC[N+](C)(C)C)COC(=O)CCCCCCCCCCCCCCC PZNPLUBHRSSFHT-RRHRGVEJSA-N 0.000 description 1
- 125000006039 1-hexenyl group Chemical group 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- 125000006023 1-pentenyl group Chemical group 0.000 description 1
- 125000006017 1-propenyl group Chemical group 0.000 description 1
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 1
- DEPDDPLQZYCHOH-UHFFFAOYSA-N 1h-imidazol-2-amine Chemical class NC1=NC=CN1 DEPDDPLQZYCHOH-UHFFFAOYSA-N 0.000 description 1
- ROZCIVXTLACYNY-UHFFFAOYSA-N 2,3,4,5,6-pentafluoro-n-(3-fluoro-4-methoxyphenyl)benzenesulfonamide Chemical compound C1=C(F)C(OC)=CC=C1NS(=O)(=O)C1=C(F)C(F)=C(F)C(F)=C1F ROZCIVXTLACYNY-UHFFFAOYSA-N 0.000 description 1
- HCSBTDBGTNZOAB-UHFFFAOYSA-N 2,3-dinitrobenzoic acid Chemical compound OC(=O)C1=CC=CC([N+]([O-])=O)=C1[N+]([O-])=O HCSBTDBGTNZOAB-UHFFFAOYSA-N 0.000 description 1
- 125000003870 2-(1-piperidinyl)ethoxy group Chemical group [*]OC([H])([H])C([H])([H])N1C([H])([H])C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- XWNJMSJGJFSGRY-UHFFFAOYSA-N 2-(benzylamino)-3,7-dihydropurin-6-one Chemical compound N1C=2N=CNC=2C(=O)N=C1NCC1=CC=CC=C1 XWNJMSJGJFSGRY-UHFFFAOYSA-N 0.000 description 1
- FBRJYBGLCHWYOE-UHFFFAOYSA-N 2-(trifluoromethyl)benzoic acid Chemical compound OC(=O)C1=CC=CC=C1C(F)(F)F FBRJYBGLCHWYOE-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- ZKKBWNOSVZIFNJ-UHFFFAOYSA-N 2-amino-3,7-dihydropurin-6-one;diphosphono hydrogen phosphate Chemical compound O=C1NC(N)=NC2=C1NC=N2.OP(O)(=O)OP(O)(=O)OP(O)(O)=O ZKKBWNOSVZIFNJ-UHFFFAOYSA-N 0.000 description 1
- JWDLEVZYUTZUBA-UHFFFAOYSA-N 2-amino-3,7-dihydropurin-6-one;phosphono dihydrogen phosphate Chemical compound OP(O)(=O)OP(O)(O)=O.O=C1NC(N)=NC2=C1NC=N2 JWDLEVZYUTZUBA-UHFFFAOYSA-N 0.000 description 1
- GOJUJUVQIVIZAV-UHFFFAOYSA-N 2-amino-4,6-dichloropyrimidine-5-carbaldehyde Chemical group NC1=NC(Cl)=C(C=O)C(Cl)=N1 GOJUJUVQIVIZAV-UHFFFAOYSA-N 0.000 description 1
- 125000000022 2-aminoethyl group Chemical group [H]C([*])([H])C([H])([H])N([H])[H] 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- 229940013085 2-diethylaminoethanol Drugs 0.000 description 1
- 125000006176 2-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- NJRXVEJTAYWCQJ-UHFFFAOYSA-L 2-mercaptosuccinate Chemical compound OC(=O)CC([S-])C([O-])=O NJRXVEJTAYWCQJ-UHFFFAOYSA-L 0.000 description 1
- RKUSRLUGUVDNKP-UHFFFAOYSA-N 2-methoxy-5-(trifluoromethyl)aniline Chemical compound COC1=CC=C(C(F)(F)F)C=C1N RKUSRLUGUVDNKP-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-M 2-methylbenzenesulfonate Chemical compound CC1=CC=CC=C1S([O-])(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- OPJKGTJXHVPYIM-UHFFFAOYSA-N 2-methylprop-2-enamide;phenol Chemical compound CC(=C)C(N)=O.OC1=CC=CC=C1 OPJKGTJXHVPYIM-UHFFFAOYSA-N 0.000 description 1
- 229940080296 2-naphthalenesulfonate Drugs 0.000 description 1
- WMPPDTMATNBGJN-UHFFFAOYSA-N 2-phenylethylbromide Chemical compound BrCCC1=CC=CC=C1 WMPPDTMATNBGJN-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000004485 2-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])([H])C1([H])* 0.000 description 1
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- FFRFGVHNKJYNOV-DOVUUNBWSA-N 3',4'-Anhydrovinblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C=C(C2)CC)N2CCC2=C1NC1=CC=CC=C21 FFRFGVHNKJYNOV-DOVUUNBWSA-N 0.000 description 1
- CURYRIVJTBNEGU-UHFFFAOYSA-L 3-bromo-1-[12-(3-bromopropanoyl)-3,12-diaza-6,9-diazoniadispiro[5.2.5^{9}.2^{6}]hexadecan-3-yl]propan-1-one;dichloride Chemical compound [Cl-].[Cl-].C1CN(C(=O)CCBr)CC[N+]21CC[N+]1(CCN(CC1)C(=O)CCBr)CC2 CURYRIVJTBNEGU-UHFFFAOYSA-L 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000004575 3-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001397 3-pyrrolyl group Chemical group [H]N1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- SFTRDTVZSPPXNS-UHFFFAOYSA-N 4-[N-(2,4-dinitroanilino)-C-phenylcarbonimidoyl]cyclohexa-2,4-diene-1,1-diol Chemical compound [N+](=O)([O-])C1=C(C=CC(=C1)[N+](=O)[O-])NN=C(C1=CCC(C=C1)(O)O)C1=CC=CC=C1 SFTRDTVZSPPXNS-UHFFFAOYSA-N 0.000 description 1
- OZBUFFXESDBEHG-FXILSDISSA-N 4-[[(2e,4e,6e,8e)-3,7-dimethyl-9-(2,6,6-trimethylcyclohexen-1-yl)nona-2,4,6,8-tetraenoyl]amino]benzoic acid Chemical compound C=1C=C(C(O)=O)C=CC=1NC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C OZBUFFXESDBEHG-FXILSDISSA-N 0.000 description 1
- TVZGACDUOSZQKY-LBPRGKRZSA-N 4-aminofolic acid Chemical compound C1=NC2=NC(N)=NC(N)=C2N=C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 TVZGACDUOSZQKY-LBPRGKRZSA-N 0.000 description 1
- HVCNXQOWACZAFN-UHFFFAOYSA-N 4-ethylmorpholine Chemical compound CCN1CCOCC1 HVCNXQOWACZAFN-UHFFFAOYSA-N 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- KDDQRKBRJSGMQE-UHFFFAOYSA-N 4-thiazolyl Chemical compound [C]1=CSC=N1 KDDQRKBRJSGMQE-UHFFFAOYSA-N 0.000 description 1
- IQWWLZSDQDCAAZ-UHFFFAOYSA-N 5-amino-1-[4-[(2-fluoro-5-methylphenyl)carbamoylamino]phenyl]imidazole-4-carboxamide Chemical compound CC1=CC=C(F)C(NC(=O)NC=2C=CC(=CC=2)N2C(=C(C(N)=O)N=C2)N)=C1 IQWWLZSDQDCAAZ-UHFFFAOYSA-N 0.000 description 1
- CDAKEFCFURDSPN-UHFFFAOYSA-N 5-amino-1-[4-[(3-tert-butylphenyl)carbamoylamino]phenyl]imidazole-4-carboxamide Chemical compound CC(C)(C)C1=CC=CC(NC(=O)NC=2C=CC(=CC=2)N2C(=C(C(N)=O)N=C2)N)=C1 CDAKEFCFURDSPN-UHFFFAOYSA-N 0.000 description 1
- JUQPYRUQAKOWRR-UHFFFAOYSA-N 5-amino-1-[4-[[3-(trifluoromethoxy)phenyl]carbamoylamino]phenyl]imidazole-4-carboxamide Chemical compound NC1=C(C(=O)N)N=CN1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(OC(F)(F)F)=C1 JUQPYRUQAKOWRR-UHFFFAOYSA-N 0.000 description 1
- XAUDJQYHKZQPEU-KVQBGUIXSA-N 5-aza-2'-deoxycytidine Chemical compound O=C1N=C(N)N=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 XAUDJQYHKZQPEU-KVQBGUIXSA-N 0.000 description 1
- 125000004539 5-benzimidazolyl group Chemical group N1=CNC2=C1C=CC(=C2)* 0.000 description 1
- CWDWFSXUQODZGW-UHFFFAOYSA-N 5-thiazolyl Chemical group [C]1=CN=CS1 CWDWFSXUQODZGW-UHFFFAOYSA-N 0.000 description 1
- KAEVHZSIYLATMK-UHFFFAOYSA-N 6-n-[bis(aziridin-1-yl)phosphoryl]-2-n,2-n,7-trimethylpurine-2,6-diamine Chemical compound C=12N(C)C=NC2=NC(N(C)C)=NC=1NP(=O)(N1CC1)N1CC1 KAEVHZSIYLATMK-UHFFFAOYSA-N 0.000 description 1
- RGVRUQHYQSORBY-UHFFFAOYSA-N 7-(4-amino-5-hydroxy-6-methyloxan-2-yl)oxy-6,9,11-trihydroxy-9-(2-hydroxyethyl)-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione Chemical compound C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(CCO)CC1OC1CC(N)C(O)C(C)O1 RGVRUQHYQSORBY-UHFFFAOYSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- KABRXLINDSPGDF-UHFFFAOYSA-N 7-bromoisoquinoline Chemical compound C1=CN=CC2=CC(Br)=CC=C21 KABRXLINDSPGDF-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- SHGAZHPCJJPHSC-ZVCIMWCZSA-N 9-cis-retinoic acid Chemical compound OC(=O)/C=C(\C)/C=C/C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-ZVCIMWCZSA-N 0.000 description 1
- ZKHQWZAMYRWXGA-KQYNXXCUSA-J ATP(4-) Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O)[C@@H](O)[C@H]1O ZKHQWZAMYRWXGA-KQYNXXCUSA-J 0.000 description 1
- 208000003200 Adenoma Diseases 0.000 description 1
- OGSPWJRAVKPPFI-UHFFFAOYSA-N Alendronic Acid Chemical compound NCCCC(O)(P(O)(O)=O)P(O)(O)=O OGSPWJRAVKPPFI-UHFFFAOYSA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 102100038778 Amphiregulin Human genes 0.000 description 1
- 108010033760 Amphiregulin Proteins 0.000 description 1
- 102100034608 Angiopoietin-2 Human genes 0.000 description 1
- 108010048036 Angiopoietin-2 Proteins 0.000 description 1
- 108020004491 Antisense DNA Proteins 0.000 description 1
- 108020005544 Antisense RNA Proteins 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 206010003210 Arteriosclerosis Diseases 0.000 description 1
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 1
- 208000027496 Behcet disease Diseases 0.000 description 1
- GUBGYTABKSRVRQ-DCSYEGIMSA-N Beta-Lactose Chemical compound OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-DCSYEGIMSA-N 0.000 description 1
- 101800001382 Betacellulin Proteins 0.000 description 1
- KWIUHFFTVRNATP-UHFFFAOYSA-N Betaine Natural products C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 1
- 229940122361 Bisphosphonate Drugs 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 208000020084 Bone disease Diseases 0.000 description 1
- 241000283725 Bos Species 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 206010048962 Brain oedema Diseases 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- CUWBAOQFLODAEN-UHFFFAOYSA-N C1=C2C=CC(=O)CC2=CC2=C1OCO2 Chemical compound C1=C2C=CC(=O)CC2=CC2=C1OCO2 CUWBAOQFLODAEN-UHFFFAOYSA-N 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- AOCCBINRVIKJHY-UHFFFAOYSA-N Carmofur Chemical compound CCCCCCNC(=O)N1C=C(F)C(=O)NC1=O AOCCBINRVIKJHY-UHFFFAOYSA-N 0.000 description 1
- 102000004171 Cathepsin K Human genes 0.000 description 1
- 108090000625 Cathepsin K Proteins 0.000 description 1
- ZEOWTGPWHLSLOG-UHFFFAOYSA-N Cc1ccc(cc1-c1ccc2c(n[nH]c2c1)-c1cnn(c1)C1CC1)C(=O)Nc1cccc(c1)C(F)(F)F Chemical compound Cc1ccc(cc1-c1ccc2c(n[nH]c2c1)-c1cnn(c1)C1CC1)C(=O)Nc1cccc(c1)C(F)(F)F ZEOWTGPWHLSLOG-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 208000005590 Choroidal Neovascularization Diseases 0.000 description 1
- 206010060823 Choroidal neovascularisation Diseases 0.000 description 1
- 206010009900 Colitis ulcerative Diseases 0.000 description 1
- 206010048832 Colon adenoma Diseases 0.000 description 1
- 108010071942 Colony-Stimulating Factors Proteins 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 229930188224 Cryptophycin Natural products 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- CYESCLHCWJKRKM-UHFFFAOYSA-N DCPU Natural products NC(=O)NC1=CC=C(Cl)C(Cl)=C1 CYESCLHCWJKRKM-UHFFFAOYSA-N 0.000 description 1
- 230000006820 DNA synthesis Effects 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- QXNVGIXVLWOKEQ-UHFFFAOYSA-N Disodium Chemical compound [Na][Na] QXNVGIXVLWOKEQ-UHFFFAOYSA-N 0.000 description 1
- SNRUBQQJIBEYMU-UHFFFAOYSA-N Dodecane Natural products CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 1
- LQKSHSFQQRCAFW-UHFFFAOYSA-N Dolastatin 15 Natural products COC1=CC(=O)N(C(=O)C(OC(=O)C2N(CCC2)C(=O)C2N(CCC2)C(=O)C(C(C)C)N(C)C(=O)C(NC(=O)C(C(C)C)N(C)C)C(C)C)C(C)C)C1CC1=CC=CC=C1 LQKSHSFQQRCAFW-UHFFFAOYSA-N 0.000 description 1
- ZQZFYGIXNQKOAV-OCEACIFDSA-N Droloxifene Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=C(O)C=CC=1)\C1=CC=C(OCCN(C)C)C=C1 ZQZFYGIXNQKOAV-OCEACIFDSA-N 0.000 description 1
- 102000012545 EGF-like domains Human genes 0.000 description 1
- 108050002150 EGF-like domains Proteins 0.000 description 1
- 238000012286 ELISA Assay Methods 0.000 description 1
- 102100039578 ETS translocation variant 4 Human genes 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- SAMRUMKYXPVKPA-VFKOLLTISA-N Enocitabine Chemical compound O=C1N=C(NC(=O)CCCCCCCCCCCCCCCCCCCCC)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 SAMRUMKYXPVKPA-VFKOLLTISA-N 0.000 description 1
- 101800003838 Epidermal growth factor Proteins 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 241000206602 Eukaryota Species 0.000 description 1
- CWYNVVGOOAEACU-UHFFFAOYSA-N Fe2+ Chemical compound [Fe+2] CWYNVVGOOAEACU-UHFFFAOYSA-N 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 102000003974 Fibroblast growth factor 2 Human genes 0.000 description 1
- 102100023593 Fibroblast growth factor receptor 1 Human genes 0.000 description 1
- 101710182386 Fibroblast growth factor receptor 1 Proteins 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- VWUXBMIQPBEWFH-WCCTWKNTSA-N Fulvestrant Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3[C@H](CCCCCCCCCS(=O)CCCC(F)(F)C(F)(F)F)CC2=C1 VWUXBMIQPBEWFH-WCCTWKNTSA-N 0.000 description 1
- 102000034286 G proteins Human genes 0.000 description 1
- 108091006027 G proteins Proteins 0.000 description 1
- 101150066516 GST gene Proteins 0.000 description 1
- 206010018364 Glomerulonephritis Diseases 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 108010024636 Glutathione Proteins 0.000 description 1
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 239000012981 Hank's balanced salt solution Substances 0.000 description 1
- 101800001649 Heparin-binding EGF-like growth factor Proteins 0.000 description 1
- 102400001369 Heparin-binding EGF-like growth factor Human genes 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 108010033040 Histones Proteins 0.000 description 1
- 101000813747 Homo sapiens ETS translocation variant 4 Proteins 0.000 description 1
- 101001005128 Homo sapiens LIM domain kinase 1 Proteins 0.000 description 1
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 1
- 101000808011 Homo sapiens Vascular endothelial growth factor A Proteins 0.000 description 1
- 101000851007 Homo sapiens Vascular endothelial growth factor receptor 2 Proteins 0.000 description 1
- 241000725303 Human immunodeficiency virus Species 0.000 description 1
- 206010021143 Hypoxia Diseases 0.000 description 1
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 1
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 1
- JJKOTMDDZAJTGQ-DQSJHHFOSA-N Idoxifene Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN2CCCC2)=CC=1)/C1=CC=C(I)C=C1 JJKOTMDDZAJTGQ-DQSJHHFOSA-N 0.000 description 1
- 206010061598 Immunodeficiency Diseases 0.000 description 1
- 102000000646 Interleukin-3 Human genes 0.000 description 1
- 108010002386 Interleukin-3 Proteins 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- 108010044467 Isoenzymes Proteins 0.000 description 1
- SHGAZHPCJJPHSC-NUEINMDLSA-N Isotretinoin Chemical compound OC(=O)C=C(C)/C=C/C=C(C)C=CC1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-NUEINMDLSA-N 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- MLFKVJCWGUZWNV-UHFFFAOYSA-N L-alanosine Natural products OC(=O)C(N)CN(O)N=O MLFKVJCWGUZWNV-UHFFFAOYSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- 108010043135 L-methionine gamma-lyase Proteins 0.000 description 1
- 125000002842 L-seryl group Chemical group O=C([*])[C@](N([H])[H])([H])C([H])([H])O[H] 0.000 description 1
- 102100026023 LIM domain kinase 1 Human genes 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- NNJVILVZKWQKPM-UHFFFAOYSA-N Lidocaine Chemical compound CCN(CC)CC(=O)NC1=C(C)C=CC=C1C NNJVILVZKWQKPM-UHFFFAOYSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- 208000030289 Lymphoproliferative disease Diseases 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 206010064912 Malignant transformation Diseases 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- WAEMQWOKJMHJLA-UHFFFAOYSA-N Manganese(2+) Chemical compound [Mn+2] WAEMQWOKJMHJLA-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 229920000715 Mucilage Polymers 0.000 description 1
- 101100372766 Mus musculus Kdr gene Proteins 0.000 description 1
- 101100268066 Mus musculus Zap70 gene Proteins 0.000 description 1
- 102000006386 Myelin Proteins Human genes 0.000 description 1
- 108010083674 Myelin Proteins Proteins 0.000 description 1
- ONXPDKGXOOORHB-BYPYZUCNSA-N N(5)-methyl-L-glutamine Chemical compound CNC(=O)CC[C@H](N)C(O)=O ONXPDKGXOOORHB-BYPYZUCNSA-N 0.000 description 1
- KWIUHFFTVRNATP-UHFFFAOYSA-O N,N,N-trimethylglycinium Chemical compound C[N+](C)(C)CC(O)=O KWIUHFFTVRNATP-UHFFFAOYSA-O 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- QIAFMBKCNZACKA-UHFFFAOYSA-N N-benzoylglycine Chemical compound OC(=O)CNC(=O)C1=CC=CC=C1 QIAFMBKCNZACKA-UHFFFAOYSA-N 0.000 description 1
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- 206010028851 Necrosis Diseases 0.000 description 1
- 102400000058 Neuregulin-1 Human genes 0.000 description 1
- 108090000556 Neuregulin-1 Proteins 0.000 description 1
- 229920000305 Nylon 6,10 Polymers 0.000 description 1
- DKMYEBKKKCBLST-UHFFFAOYSA-N O=C1C2=C(O)C=CC(O)=C2N2N=C(CNCCO)C3=C2C1=CC=C3 Chemical compound O=C1C2=C(O)C=CC(O)=C2N2N=C(CNCCO)C3=C2C1=CC=C3 DKMYEBKKKCBLST-UHFFFAOYSA-N 0.000 description 1
- REYJJPSVUYRZGE-UHFFFAOYSA-N Octadecylamine Chemical compound CCCCCCCCCCCCCCCCCCN REYJJPSVUYRZGE-UHFFFAOYSA-N 0.000 description 1
- 108091034117 Oligonucleotide Proteins 0.000 description 1
- 108010038807 Oligopeptides Proteins 0.000 description 1
- 102000015636 Oligopeptides Human genes 0.000 description 1
- 208000005072 Oncogenic osteomalacia Diseases 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 208000010191 Osteitis Deformans Diseases 0.000 description 1
- 208000001132 Osteoporosis Diseases 0.000 description 1
- YIKSCQDJHCMVMK-UHFFFAOYSA-N Oxamide Chemical compound NC(=O)C(N)=O YIKSCQDJHCMVMK-UHFFFAOYSA-N 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 208000027868 Paget disease Diseases 0.000 description 1
- 208000031481 Pathologic Constriction Diseases 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- KMSKQZKKOZQFFG-HSUXVGOQSA-N Pirarubicin Chemical compound O([C@H]1[C@@H](N)C[C@@H](O[C@H]1C)O[C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1CCCCO1 KMSKQZKKOZQFFG-HSUXVGOQSA-N 0.000 description 1
- 108010020346 Polyglutamic Acid Proteins 0.000 description 1
- 108010039918 Polylysine Proteins 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- HRHKSTOGXBBQCB-UHFFFAOYSA-N Porfiromycine Chemical compound O=C1C(N)=C(C)C(=O)C2=C1C(COC(N)=O)C1(OC)C3N(C)C3CN12 HRHKSTOGXBBQCB-UHFFFAOYSA-N 0.000 description 1
- HFVNWDWLWUCIHC-GUPDPFMOSA-N Prednimustine Chemical compound O=C([C@@]1(O)CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)[C@@H](O)C[C@@]21C)COC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 HFVNWDWLWUCIHC-GUPDPFMOSA-N 0.000 description 1
- 102100033237 Pro-epidermal growth factor Human genes 0.000 description 1
- 102100029837 Probetacellulin Human genes 0.000 description 1
- AHHFEZNOXOZZQA-ZEBDFXRSSA-N Ranimustine Chemical compound CO[C@H]1O[C@H](CNC(=O)N(CCCl)N=O)[C@@H](O)[C@H](O)[C@H]1O AHHFEZNOXOZZQA-ZEBDFXRSSA-N 0.000 description 1
- 241000713810 Rat sarcoma virus Species 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 101000852966 Rattus norvegicus Interleukin-1 receptor-like 1 Proteins 0.000 description 1
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 1
- 101710100969 Receptor tyrosine-protein kinase erbB-3 Proteins 0.000 description 1
- 102100029986 Receptor tyrosine-protein kinase erbB-3 Human genes 0.000 description 1
- 102100029981 Receptor tyrosine-protein kinase erbB-4 Human genes 0.000 description 1
- 101710100963 Receptor tyrosine-protein kinase erbB-4 Proteins 0.000 description 1
- 229940123934 Reductase inhibitor Drugs 0.000 description 1
- 206010063837 Reperfusion injury Diseases 0.000 description 1
- 206010039085 Rhinitis allergic Diseases 0.000 description 1
- OWPCHSCAPHNHAV-UHFFFAOYSA-N Rhizoxin Natural products C1C(O)C2(C)OC2C=CC(C)C(OC(=O)C2)CC2CC2OC2C(=O)OC1C(C)C(OC)C(C)=CC=CC(C)=CC1=COC(C)=N1 OWPCHSCAPHNHAV-UHFFFAOYSA-N 0.000 description 1
- IIDJRNMFWXDHID-UHFFFAOYSA-N Risedronic acid Chemical compound OP(=O)(O)C(P(O)(O)=O)(O)CC1=CC=CN=C1 IIDJRNMFWXDHID-UHFFFAOYSA-N 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- 206010041067 Small cell lung cancer Diseases 0.000 description 1
- OCOKWVBYZHBHLU-UHFFFAOYSA-N Sobuzoxane Chemical compound C1C(=O)N(COC(=O)OCC(C)C)C(=O)CN1CCN1CC(=O)N(COC(=O)OCC(C)C)C(=O)C1 OCOKWVBYZHBHLU-UHFFFAOYSA-N 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- BCKXLBQYZLBQEK-KVVVOXFISA-M Sodium oleate Chemical compound [Na+].CCCCCCCC\C=C/CCCCCCCC([O-])=O BCKXLBQYZLBQEK-KVVVOXFISA-M 0.000 description 1
- 241000256251 Spodoptera frugiperda Species 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 102000005450 TIE receptors Human genes 0.000 description 1
- 108010006830 TIE receptors Proteins 0.000 description 1
- NAVMQTYZDKMPEU-UHFFFAOYSA-N Targretin Chemical compound CC1=CC(C(CCC2(C)C)(C)C)=C2C=C1C(=C)C1=CC=C(C(O)=O)C=C1 NAVMQTYZDKMPEU-UHFFFAOYSA-N 0.000 description 1
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 1
- 208000007536 Thrombosis Diseases 0.000 description 1
- IVTVGDXNLFLDRM-HNNXBMFYSA-N Tomudex Chemical compound C=1C=C2NC(C)=NC(=O)C2=CC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)S1 IVTVGDXNLFLDRM-HNNXBMFYSA-N 0.000 description 1
- 206010052779 Transplant rejections Diseases 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 1
- 102000044209 Tumor Suppressor Genes Human genes 0.000 description 1
- 108700025716 Tumor Suppressor Genes Proteins 0.000 description 1
- 201000006704 Ulcerative Colitis Diseases 0.000 description 1
- 102000016663 Vascular Endothelial Growth Factor Receptor-3 Human genes 0.000 description 1
- 208000032594 Vascular Remodeling Diseases 0.000 description 1
- 206010058990 Venous occlusion Diseases 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- XMYKNCNAZKMVQN-NYYWCZLTSA-N [(e)-(3-aminopyridin-2-yl)methylideneamino]thiourea Chemical compound NC(=S)N\N=C\C1=NC=CC=C1N XMYKNCNAZKMVQN-NYYWCZLTSA-N 0.000 description 1
- XSMVECZRZBFTIZ-UHFFFAOYSA-M [2-(aminomethyl)cyclobutyl]methanamine;2-oxidopropanoate;platinum(4+) Chemical compound [Pt+4].CC([O-])C([O-])=O.NCC1CCC1CN XSMVECZRZBFTIZ-UHFFFAOYSA-M 0.000 description 1
- CJJFXIYSNIDTMY-UHFFFAOYSA-N [3-(trifluoromethoxy)phenyl]urea Chemical compound NC(=O)NC1=CC=CC(OC(F)(F)F)=C1 CJJFXIYSNIDTMY-UHFFFAOYSA-N 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- QCLRUNIZLOTOPA-UHFFFAOYSA-N [Pt].CC1=NC=CC(Cl)=C1Cl Chemical compound [Pt].CC1=NC=CC(Cl)=C1Cl QCLRUNIZLOTOPA-UHFFFAOYSA-N 0.000 description 1
- UVIQSJCZCSLXRZ-UBUQANBQSA-N abiraterone acetate Chemical compound C([C@@H]1[C@]2(C)CC[C@@H]3[C@@]4(C)CC[C@@H](CC4=CC[C@H]31)OC(=O)C)C=C2C1=CC=CN=C1 UVIQSJCZCSLXRZ-UBUQANBQSA-N 0.000 description 1
- 229960004103 abiraterone acetate Drugs 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 239000003655 absorption accelerator Substances 0.000 description 1
- IPBVNPXQWQGGJP-UHFFFAOYSA-N acetic acid phenyl ester Natural products CC(=O)OC1=CC=CC=C1 IPBVNPXQWQGGJP-UHFFFAOYSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 230000001464 adherent effect Effects 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 229950005033 alanosine Drugs 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 229940062527 alendronate Drugs 0.000 description 1
- 229960001445 alitretinoin Drugs 0.000 description 1
- 229910001854 alkali hydroxide Inorganic materials 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 229910000272 alkali metal oxide Inorganic materials 0.000 description 1
- 150000008051 alkyl sulfates Chemical class 0.000 description 1
- 125000005278 alkyl sulfonyloxy group Chemical group 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 1
- 201000010105 allergic rhinitis Diseases 0.000 description 1
- AEMOLEFTQBMNLQ-BKBMJHBISA-N alpha-D-galacturonic acid Chemical compound O[C@H]1O[C@H](C(O)=O)[C@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-BKBMJHBISA-N 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 229960000473 altretamine Drugs 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 229960003896 aminopterin Drugs 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- VJZITPJGSQKZMX-XDPRQOKASA-N amrubicin Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC=C4C(=O)C=3C(O)=C21)(N)C(=O)C)[C@H]1C[C@H](O)[C@H](O)CO1 VJZITPJGSQKZMX-XDPRQOKASA-N 0.000 description 1
- 229960002550 amrubicin Drugs 0.000 description 1
- 239000003098 androgen Substances 0.000 description 1
- 229940030486 androgens Drugs 0.000 description 1
- 239000002870 angiogenesis inducing agent Substances 0.000 description 1
- 238000002399 angioplasty Methods 0.000 description 1
- 229950001104 anhydrovinblastine Drugs 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- CIDNKDMVSINJCG-GKXONYSUSA-N annamycin Chemical compound I[C@@H]1[C@H](O)[C@@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(=O)CO)C1 CIDNKDMVSINJCG-GKXONYSUSA-N 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 230000003288 anthiarrhythmic effect Effects 0.000 description 1
- 230000001772 anti-angiogenic effect Effects 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 230000000123 anti-resoprtive effect Effects 0.000 description 1
- 239000003416 antiarrhythmic agent Substances 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000003816 antisense DNA Substances 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229960003121 arginine Drugs 0.000 description 1
- GOLCXWYRSKYTSP-UHFFFAOYSA-N arsenic trioxide Inorganic materials O1[As]2O[As]1O2 GOLCXWYRSKYTSP-UHFFFAOYSA-N 0.000 description 1
- 208000011775 arteriosclerosis disease Diseases 0.000 description 1
- 239000008122 artificial sweetener Substances 0.000 description 1
- 235000021311 artificial sweeteners Nutrition 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 125000005279 aryl sulfonyloxy group Chemical group 0.000 description 1
- MCGDSOGUHLTADD-UHFFFAOYSA-N arzoxifene Chemical compound C1=CC(OC)=CC=C1C1=C(OC=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 MCGDSOGUHLTADD-UHFFFAOYSA-N 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- TWHSQQYCDVSBRK-UHFFFAOYSA-N asulacrine Chemical compound C12=CC=CC(C)=C2N=C2C(C(=O)NC)=CC=CC2=C1NC1=CC=C(NS(C)(=O)=O)C=C1OC TWHSQQYCDVSBRK-UHFFFAOYSA-N 0.000 description 1
- 229950011088 asulacrine Drugs 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 108010044540 auristatin Proteins 0.000 description 1
- 230000003305 autocrine Effects 0.000 description 1
- KLNFSAOEKUDMFA-UHFFFAOYSA-N azanide;2-hydroxyacetic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OCC(O)=O KLNFSAOEKUDMFA-UHFFFAOYSA-N 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- 239000000022 bacteriostatic agent Substances 0.000 description 1
- 230000003385 bacteriostatic effect Effects 0.000 description 1
- RQPZNWPYLFFXCP-UHFFFAOYSA-L barium dihydroxide Chemical compound [OH-].[OH-].[Ba+2] RQPZNWPYLFFXCP-UHFFFAOYSA-L 0.000 description 1
- 229910001863 barium hydroxide Inorganic materials 0.000 description 1
- 150000007514 bases Chemical class 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- 125000004604 benzisothiazolyl group Chemical group S1N=C(C2=C1C=CC=C2)* 0.000 description 1
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 description 1
- 229940073608 benzyl chloride Drugs 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 229960002938 bexarotene Drugs 0.000 description 1
- 229960000997 bicalutamide Drugs 0.000 description 1
- 230000008238 biochemical pathway Effects 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 229950008548 bisantrene Drugs 0.000 description 1
- 150000004663 bisphosphonates Chemical class 0.000 description 1
- 229920001400 block copolymer Polymers 0.000 description 1
- 230000017531 blood circulation Effects 0.000 description 1
- UBJAHGAUPNGZFF-XOVTVWCYSA-N bms-184476 Chemical compound O([C@H]1[C@@H]2[C@]3(OC(C)=O)CO[C@@H]3C[C@@H]([C@]2(C(=O)[C@H](OC(C)=O)C2=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)C=3C=CC=CC=3)C=3C=CC=CC=3)C[C@]1(O)C2(C)C)C)OCSC)C(=O)C1=CC=CC=C1 UBJAHGAUPNGZFF-XOVTVWCYSA-N 0.000 description 1
- GMJWGJSDPOAZTP-MIDYMNAOSA-N bms-188797 Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](OC(C)=O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)C=4C=CC=CC=4)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)OC)C(=O)C1=CC=CC=C1 GMJWGJSDPOAZTP-MIDYMNAOSA-N 0.000 description 1
- 239000012888 bovine serum Substances 0.000 description 1
- 208000006752 brain edema Diseases 0.000 description 1
- 210000005013 brain tissue Anatomy 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 1
- 239000000920 calcium hydroxide Substances 0.000 description 1
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 150000003857 carboxamides Chemical class 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 229940105329 carboxymethylcellulose Drugs 0.000 description 1
- 238000013130 cardiovascular surgery Methods 0.000 description 1
- 229960003261 carmofur Drugs 0.000 description 1
- 230000021164 cell adhesion Effects 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 230000012292 cell migration Effects 0.000 description 1
- 230000036755 cellular response Effects 0.000 description 1
- 230000005754 cellular signaling Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 108010046713 cemadotin Proteins 0.000 description 1
- 229950009017 cemadotin Drugs 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 230000002490 cerebral effect Effects 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- ROWSTIYZUWEOMM-UHFFFAOYSA-N chembl488755 Chemical compound C12=CC=CC=C2C(=O)C2=C1C1=CC=C(O)C=C1N=C2NCCN(C)C ROWSTIYZUWEOMM-UHFFFAOYSA-N 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- KVSASDOGYIBWTA-UHFFFAOYSA-N chloro benzoate Chemical compound ClOC(=O)C1=CC=CC=C1 KVSASDOGYIBWTA-UHFFFAOYSA-N 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 208000037976 chronic inflammation Diseases 0.000 description 1
- 230000006020 chronic inflammation Effects 0.000 description 1
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 1
- 230000007882 cirrhosis Effects 0.000 description 1
- 208000019425 cirrhosis of liver Diseases 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 230000004186 co-expression Effects 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 230000024203 complement activation Effects 0.000 description 1
- 239000003184 complementary RNA Substances 0.000 description 1
- 229940035811 conjugated estrogen Drugs 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 238000010411 cooking Methods 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- POADTFBBIXOWFJ-VWLOTQADSA-N cositecan Chemical compound C1=CC=C2C(CC[Si](C)(C)C)=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 POADTFBBIXOWFJ-VWLOTQADSA-N 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 108010006226 cryptophycin Proteins 0.000 description 1
- PSNOPSMXOBPNNV-VVCTWANISA-N cryptophycin 1 Chemical compound C1=C(Cl)C(OC)=CC=C1C[C@@H]1C(=O)NC[C@@H](C)C(=O)O[C@@H](CC(C)C)C(=O)O[C@H]([C@H](C)[C@@H]2[C@H](O2)C=2C=CC=CC=2)C/C=C/C(=O)N1 PSNOPSMXOBPNNV-VVCTWANISA-N 0.000 description 1
- PSNOPSMXOBPNNV-UHFFFAOYSA-N cryptophycin-327 Natural products C1=C(Cl)C(OC)=CC=C1CC1C(=O)NCC(C)C(=O)OC(CC(C)C)C(=O)OC(C(C)C2C(O2)C=2C=CC=CC=2)CC=CC(=O)N1 PSNOPSMXOBPNNV-UHFFFAOYSA-N 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- 230000001351 cycling effect Effects 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- BALGDZWGNCXXES-UHFFFAOYSA-N cyclopentane;propanoic acid Chemical compound CCC(O)=O.C1CCCC1 BALGDZWGNCXXES-UHFFFAOYSA-N 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 229960003603 decitabine Drugs 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 230000030609 dephosphorylation Effects 0.000 description 1
- 238000006209 dephosphorylation reaction Methods 0.000 description 1
- 230000003831 deregulation Effects 0.000 description 1
- 229960000605 dexrazoxane Drugs 0.000 description 1
- 150000001982 diacylglycerols Chemical class 0.000 description 1
- 238000002405 diagnostic procedure Methods 0.000 description 1
- 150000004985 diamines Chemical class 0.000 description 1
- 229950007457 dibrospidium chloride Drugs 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- AZFLJNIPTRTECV-FUMNGEBKSA-N dienogest Chemical compound C1CC(=O)C=C2CC[C@@H]([C@H]3[C@@](C)([C@](CC3)(O)CC#N)CC3)C3=C21 AZFLJNIPTRTECV-FUMNGEBKSA-N 0.000 description 1
- 229940043237 diethanolamine Drugs 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 125000001664 diethylamino group Chemical group [H]C([H])([H])C([H])([H])N(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 150000004683 dihydrates Chemical class 0.000 description 1
- 125000004598 dihydrobenzofuryl group Chemical group O1C(CC2=C1C=CC=C2)* 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 229950009278 dimesna Drugs 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- GAFRWLVTHPVQGK-UHFFFAOYSA-N dipentyl sulfate Chemical compound CCCCCOS(=O)(=O)OCCCCC GAFRWLVTHPVQGK-UHFFFAOYSA-N 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 230000006806 disease prevention Effects 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- KQYGMURBTJPBPQ-UHFFFAOYSA-L disodium;2-(2-sulfonatoethyldisulfanyl)ethanesulfonate Chemical compound [Na+].[Na+].[O-]S(=O)(=O)CCSSCCS([O-])(=O)=O KQYGMURBTJPBPQ-UHFFFAOYSA-L 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- AMRJKAQTDDKMCE-UHFFFAOYSA-N dolastatin Chemical compound CC(C)C(N(C)C)C(=O)NC(C(C)C)C(=O)N(C)C(C(C)C)C(OC)CC(=O)N1CCCC1C(OC)C(C)C(=O)NC(C=1SC=CN=1)CC1=CC=CC=C1 AMRJKAQTDDKMCE-UHFFFAOYSA-N 0.000 description 1
- 229930188854 dolastatin Natural products 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- ZWAOHEXOSAUJHY-ZIYNGMLESA-N doxifluridine Chemical compound O[C@@H]1[C@H](O)[C@@H](C)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ZWAOHEXOSAUJHY-ZIYNGMLESA-N 0.000 description 1
- 229950005454 doxifluridine Drugs 0.000 description 1
- 229950004203 droloxifene Drugs 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 229950005450 emitefur Drugs 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000026721 endothelial cell chemotaxis Effects 0.000 description 1
- 210000003038 endothelium Anatomy 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 229950011487 enocitabine Drugs 0.000 description 1
- 238000006911 enzymatic reaction Methods 0.000 description 1
- 229940116977 epidermal growth factor Drugs 0.000 description 1
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 1
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 1
- 210000002615 epidermis Anatomy 0.000 description 1
- 230000008472 epithelial growth Effects 0.000 description 1
- 210000000981 epithelium Anatomy 0.000 description 1
- 229960001842 estramustine Drugs 0.000 description 1
- FRPJXPJMRWBBIH-RBRWEJTLSA-N estramustine Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 FRPJXPJMRWBBIH-RBRWEJTLSA-N 0.000 description 1
- 229940011871 estrogen Drugs 0.000 description 1
- 239000000262 estrogen Substances 0.000 description 1
- JKKFKPJIXZFSSB-CBZIJGRNSA-N estrone 3-sulfate Chemical compound OS(=O)(=O)OC1=CC=C2[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CCC2=C1 JKKFKPJIXZFSSB-CBZIJGRNSA-N 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- JEFPWOBULVSOTM-PPHPATTJSA-N ethyl n-[(2s)-5-amino-2-methyl-3-phenyl-1,2-dihydropyrido[3,4-b]pyrazin-7-yl]carbamate;2-hydroxyethanesulfonic acid Chemical compound OCCS(O)(=O)=O.C=1([C@H](C)NC=2C=C(N=C(N)C=2N=1)NC(=O)OCC)C1=CC=CC=C1 JEFPWOBULVSOTM-PPHPATTJSA-N 0.000 description 1
- 229940012017 ethylenediamine Drugs 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 229960000752 etoposide phosphate Drugs 0.000 description 1
- LIQODXNTTZAGID-OCBXBXKTSA-N etoposide phosphate Chemical compound COC1=C(OP(O)(O)=O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 LIQODXNTTZAGID-OCBXBXKTSA-N 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 230000004438 eyesight Effects 0.000 description 1
- 230000009647 facial growth Effects 0.000 description 1
- 230000004761 fibrosis Effects 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 238000005429 filling process Methods 0.000 description 1
- DBEPLOCGEIEOCV-WSBQPABSSA-N finasteride Chemical compound N([C@@H]1CC2)C(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](C(=O)NC(C)(C)C)[C@@]2(C)CC1 DBEPLOCGEIEOCV-WSBQPABSSA-N 0.000 description 1
- 229960004039 finasteride Drugs 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 108091071773 flk family Proteins 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 1
- 229960002074 flutamide Drugs 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 229960004783 fotemustine Drugs 0.000 description 1
- YAKWPXVTIGTRJH-UHFFFAOYSA-N fotemustine Chemical compound CCOP(=O)(OCC)C(C)NC(=O)N(CCCl)N=O YAKWPXVTIGTRJH-UHFFFAOYSA-N 0.000 description 1
- 229960002258 fulvestrant Drugs 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 229940083124 ganglion-blocking antiadrenergic secondary and tertiary amines Drugs 0.000 description 1
- 208000010749 gastric carcinoma Diseases 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 238000003209 gene knockout Methods 0.000 description 1
- 102000034356 gene-regulatory proteins Human genes 0.000 description 1
- 108091006104 gene-regulatory proteins Proteins 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 239000003365 glass fiber Substances 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- 229950011595 glufosfamide Drugs 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 229940049906 glutamate Drugs 0.000 description 1
- 229960002989 glutamic acid Drugs 0.000 description 1
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 1
- 210000004524 haematopoietic cell Anatomy 0.000 description 1
- 230000035876 healing Effects 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- 239000000833 heterodimer Substances 0.000 description 1
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 1
- NAQMVNRVTILPCV-UHFFFAOYSA-N hexane-1,6-diamine Chemical compound NCCCCCCN NAQMVNRVTILPCV-UHFFFAOYSA-N 0.000 description 1
- 238000000589 high-performance liquid chromatography-mass spectrometry Methods 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 238000001794 hormone therapy Methods 0.000 description 1
- 102000055590 human KDR Human genes 0.000 description 1
- 102000058223 human VEGFA Human genes 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 1
- 229910000043 hydrogen iodide Inorganic materials 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 239000008309 hydrophilic cream Substances 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 230000000521 hyperimmunizing effect Effects 0.000 description 1
- 230000001146 hypoxic effect Effects 0.000 description 1
- 229960000908 idarubicin Drugs 0.000 description 1
- 229950002248 idoxifene Drugs 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 150000002460 imidazoles Chemical class 0.000 description 1
- JBFYUZGYRGXSFL-UHFFFAOYSA-N imidazolide Chemical compound C1=C[N-]C=N1 JBFYUZGYRGXSFL-UHFFFAOYSA-N 0.000 description 1
- 208000003669 immune deficiency disease Diseases 0.000 description 1
- 229940127121 immunoconjugate Drugs 0.000 description 1
- 230000007813 immunodeficiency Effects 0.000 description 1
- DBIGHPPNXATHOF-UHFFFAOYSA-N improsulfan Chemical compound CS(=O)(=O)OCCCNCCCOS(C)(=O)=O DBIGHPPNXATHOF-UHFFFAOYSA-N 0.000 description 1
- 229950008097 improsulfan Drugs 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 210000004969 inflammatory cell Anatomy 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 102000006495 integrins Human genes 0.000 description 1
- 108010044426 integrins Proteins 0.000 description 1
- 239000000138 intercalating agent Substances 0.000 description 1
- 229940076264 interleukin-3 Drugs 0.000 description 1
- 102000010681 interleukin-8 receptors Human genes 0.000 description 1
- 108010038415 interleukin-8 receptors Proteins 0.000 description 1
- 229940079865 intestinal antiinfectives imidazole derivative Drugs 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- 229950005254 irofulven Drugs 0.000 description 1
- NICJCIQSJJKZAH-AWEZNQCLSA-N irofulven Chemical compound O=C([C@@]1(O)C)C2=CC(C)=C(CO)C2=C(C)C21CC2 NICJCIQSJJKZAH-AWEZNQCLSA-N 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 229960005280 isotretinoin Drugs 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-N lactobionic acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-N 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 201000005264 laryngeal carcinoma Diseases 0.000 description 1
- 206010023841 laryngeal neoplasm Diseases 0.000 description 1
- 201000004962 larynx cancer Diseases 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- UGFHIPBXIWJXNA-UHFFFAOYSA-N liarozole Chemical compound ClC1=CC=CC(C(C=2C=C3NC=NC3=CC=2)N2C=NC=C2)=C1 UGFHIPBXIWJXNA-UHFFFAOYSA-N 0.000 description 1
- 229950007056 liarozole Drugs 0.000 description 1
- 229960004194 lidocaine Drugs 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 229950008991 lobaplatin Drugs 0.000 description 1
- 229950000909 lometrexol Drugs 0.000 description 1
- 229960003538 lonidamine Drugs 0.000 description 1
- WDRYRZXSPDWGEB-UHFFFAOYSA-N lonidamine Chemical compound C12=CC=CC=C2C(C(=O)O)=NN1CC1=CC=C(Cl)C=C1Cl WDRYRZXSPDWGEB-UHFFFAOYSA-N 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 201000005296 lung carcinoma Diseases 0.000 description 1
- RVFGKBWWUQOIOU-NDEPHWFRSA-N lurtotecan Chemical compound O=C([C@]1(O)CC)OCC(C(N2CC3=4)=O)=C1C=C2C3=NC1=CC=2OCCOC=2C=C1C=4CN1CCN(C)CC1 RVFGKBWWUQOIOU-NDEPHWFRSA-N 0.000 description 1
- 229950002654 lurtotecan Drugs 0.000 description 1
- 230000001926 lymphatic effect Effects 0.000 description 1
- 229960003646 lysine Drugs 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 230000036212 malign transformation Effects 0.000 description 1
- 210000001161 mammalian embryo Anatomy 0.000 description 1
- 208000027202 mammary Paget disease Diseases 0.000 description 1
- MMIPFLVOWGHZQD-UHFFFAOYSA-N manganese(3+) Chemical compound [Mn+3] MMIPFLVOWGHZQD-UHFFFAOYSA-N 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- PSGAAPLEWMOORI-PEINSRQWSA-N medroxyprogesterone acetate Chemical compound C([C@@]12C)CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2CC[C@]2(C)[C@@](OC(C)=O)(C(C)=O)CC[C@H]21 PSGAAPLEWMOORI-PEINSRQWSA-N 0.000 description 1
- 239000001525 mentha piperita l. herb oil Substances 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 208000030159 metabolic disease Diseases 0.000 description 1
- 125000005341 metaphosphate group Chemical group 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- 230000006510 metastatic growth Effects 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- 229940095102 methyl benzoate Drugs 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 1
- 239000003094 microcapsule Substances 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 229950010913 mitolactol Drugs 0.000 description 1
- VFKZTMPDYBFSTM-GUCUJZIJSA-N mitolactol Chemical compound BrC[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-GUCUJZIJSA-N 0.000 description 1
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 1
- 150000004682 monohydrates Chemical class 0.000 description 1
- 230000006740 morphological transformation Effects 0.000 description 1
- 239000002324 mouth wash Substances 0.000 description 1
- 210000003097 mucus Anatomy 0.000 description 1
- 125000001421 myristyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- ACTNHJDHMQSOGL-UHFFFAOYSA-N n',n'-dibenzylethane-1,2-diamine Chemical compound C=1C=CC=CC=1CN(CCN)CC1=CC=CC=C1 ACTNHJDHMQSOGL-UHFFFAOYSA-N 0.000 description 1
- UDGSVBYJWHOHNN-UHFFFAOYSA-N n',n'-diethylethane-1,2-diamine Chemical group CCN(CC)CCN UDGSVBYJWHOHNN-UHFFFAOYSA-N 0.000 description 1
- BXQCAFCKILLBJO-UHFFFAOYSA-N n-(4-aminophenyl)-2-fluoro-5-(trifluoromethyl)benzamide Chemical compound C1=CC(N)=CC=C1NC(=O)C1=CC(C(F)(F)F)=CC=C1F BXQCAFCKILLBJO-UHFFFAOYSA-N 0.000 description 1
- NJSMWLQOCQIOPE-OCHFTUDZSA-N n-[(e)-[10-[(e)-(4,5-dihydro-1h-imidazol-2-ylhydrazinylidene)methyl]anthracen-9-yl]methylideneamino]-4,5-dihydro-1h-imidazol-2-amine Chemical compound N1CCN=C1N\N=C\C(C1=CC=CC=C11)=C(C=CC=C2)C2=C1\C=N\NC1=NCCN1 NJSMWLQOCQIOPE-OCHFTUDZSA-N 0.000 description 1
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-M naphthalene-2-sulfonate Chemical compound C1=CC=CC2=CC(S(=O)(=O)[O-])=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-M 0.000 description 1
- 239000007923 nasal drop Substances 0.000 description 1
- 229940100662 nasal drops Drugs 0.000 description 1
- 229940097496 nasal spray Drugs 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 235000021096 natural sweeteners Nutrition 0.000 description 1
- 230000017074 necrotic cell death Effects 0.000 description 1
- 229950007221 nedaplatin Drugs 0.000 description 1
- 230000017066 negative regulation of growth Effects 0.000 description 1
- 229960000801 nelarabine Drugs 0.000 description 1
- IXOXBSCIXZEQEQ-UHTZMRCNSA-N nelarabine Chemical compound C1=NC=2C(OC)=NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1O IXOXBSCIXZEQEQ-UHTZMRCNSA-N 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 210000002569 neuron Anatomy 0.000 description 1
- XWXYUMMDTVBTOU-UHFFFAOYSA-N nilutamide Chemical compound O=C1C(C)(C)NC(=O)N1C1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 XWXYUMMDTVBTOU-UHFFFAOYSA-N 0.000 description 1
- 229960002653 nilutamide Drugs 0.000 description 1
- 229960001420 nimustine Drugs 0.000 description 1
- VFEDRRNHLBGPNN-UHFFFAOYSA-N nimustine Chemical compound CC1=NC=C(CNC(=O)N(CCCl)N=O)C(N)=N1 VFEDRRNHLBGPNN-UHFFFAOYSA-N 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 229950000891 nolatrexed Drugs 0.000 description 1
- XHWRWCSCBDLOLM-UHFFFAOYSA-N nolatrexed Chemical compound CC1=CC=C2NC(N)=NC(=O)C2=C1SC1=CC=NC=C1 XHWRWCSCBDLOLM-UHFFFAOYSA-N 0.000 description 1
- 102000037979 non-receptor tyrosine kinases Human genes 0.000 description 1
- 108091008046 non-receptor tyrosine kinases Proteins 0.000 description 1
- 230000000263 nonmitogenic effect Effects 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-M octanoate Chemical compound CCCCCCCC([O-])=O WWZKQHOCKIZLMA-UHFFFAOYSA-M 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 230000005853 oncogenic activation Effects 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 210000002997 osteoclast Anatomy 0.000 description 1
- 230000001599 osteoclastic effect Effects 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 229960001756 oxaliplatin Drugs 0.000 description 1
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 125000001312 palmitoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 230000003076 paracrine Effects 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000011236 particulate material Substances 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- QOFFJEBXNKRSPX-ZDUSSCGKSA-N pemetrexed Chemical compound C1=N[C]2NC(N)=NC(=O)C2=C1CCC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 QOFFJEBXNKRSPX-ZDUSSCGKSA-N 0.000 description 1
- 229960005079 pemetrexed Drugs 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 125000004817 pentamethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[*:1] 0.000 description 1
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 229960002340 pentostatin Drugs 0.000 description 1
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 1
- 235000019477 peppermint oil Nutrition 0.000 description 1
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical compound [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 235000019271 petrolatum Nutrition 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- PUXKSJCSTXMIKR-UHFFFAOYSA-N phenyl 2,2-dimethylpropanoate Chemical compound CC(C)(C)C(=O)OC1=CC=CC=C1 PUXKSJCSTXMIKR-UHFFFAOYSA-N 0.000 description 1
- DYUMLJSJISTVPV-UHFFFAOYSA-N phenyl propanoate Chemical compound CCC(=O)OC1=CC=CC=C1 DYUMLJSJISTVPV-UHFFFAOYSA-N 0.000 description 1
- 229940049953 phenylacetate Drugs 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 150000008105 phosphatidylcholines Chemical class 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- LFGREXWGYUGZLY-UHFFFAOYSA-N phosphoryl Chemical group [P]=O LFGREXWGYUGZLY-UHFFFAOYSA-N 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 229960001221 pirarubicin Drugs 0.000 description 1
- 229950010765 pivalate Drugs 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 210000002826 placenta Anatomy 0.000 description 1
- 229940096701 plain lipid modifying drug hmg coa reductase inhibitors Drugs 0.000 description 1
- HRGDZIGMBDGFTC-UHFFFAOYSA-N platinum(2+) Chemical compound [Pt+2] HRGDZIGMBDGFTC-UHFFFAOYSA-N 0.000 description 1
- 229950008499 plitidepsin Drugs 0.000 description 1
- UUSZLLQJYRSZIS-LXNNNBEUSA-N plitidepsin Chemical compound CN([C@H](CC(C)C)C(=O)N[C@@H]1C(=O)N[C@@H]([C@H](CC(=O)O[C@H](C(=O)[C@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N2CCC[C@H]2C(=O)N(C)[C@@H](CC=2C=CC(OC)=CC=2)C(=O)O[C@@H]1C)C(C)C)O)[C@@H](C)CC)C(=O)[C@@H]1CCCN1C(=O)C(C)=O UUSZLLQJYRSZIS-LXNNNBEUSA-N 0.000 description 1
- 108010049948 plitidepsin Proteins 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 229920001610 polycaprolactone Polymers 0.000 description 1
- 229920002721 polycyanoacrylate Polymers 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- 229920000656 polylysine Polymers 0.000 description 1
- 229920006324 polyoxymethylene Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 230000008092 positive effect Effects 0.000 description 1
- RPDAUEIUDPHABB-UHFFFAOYSA-N potassium ethoxide Chemical compound [K+].CC[O-] RPDAUEIUDPHABB-UHFFFAOYSA-N 0.000 description 1
- 229960004694 prednimustine Drugs 0.000 description 1
- 229940063238 premarin Drugs 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- WGYKZJWCGVVSQN-UHFFFAOYSA-N propylamine Chemical compound CCCN WGYKZJWCGVVSQN-UHFFFAOYSA-N 0.000 description 1
- 239000011241 protective layer Substances 0.000 description 1
- 238000001742 protein purification Methods 0.000 description 1
- 229940126409 proton pump inhibitor Drugs 0.000 description 1
- 239000000612 proton pump inhibitor Substances 0.000 description 1
- 229950007401 pumitepa Drugs 0.000 description 1
- 239000012264 purified product Substances 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 229960004432 raltitrexed Drugs 0.000 description 1
- 229960002185 ranimustine Drugs 0.000 description 1
- 230000036647 reaction Effects 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 230000002207 retinal effect Effects 0.000 description 1
- 229940100552 retinamide Drugs 0.000 description 1
- OWPCHSCAPHNHAV-LMONGJCWSA-N rhizoxin Chemical compound C/C([C@H](OC)[C@@H](C)[C@@H]1C[C@H](O)[C@]2(C)O[C@@H]2/C=C/[C@@H](C)[C@]2([H])OC(=O)C[C@@](C2)(C[C@@H]2O[C@H]2C(=O)O1)[H])=C\C=C\C(\C)=C\C1=COC(C)=N1 OWPCHSCAPHNHAV-LMONGJCWSA-N 0.000 description 1
- 229940089617 risedronate Drugs 0.000 description 1
- 238000011808 rodent model Methods 0.000 description 1
- VHXNKPBCCMUMSW-FQEVSTJZSA-N rubitecan Chemical compound C1=CC([N+]([O-])=O)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VHXNKPBCCMUMSW-FQEVSTJZSA-N 0.000 description 1
- 229950009213 rubitecan Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- MOODSJOROWROTO-UHFFFAOYSA-N salicylsulfuric acid Chemical compound OC(=O)C1=CC=CC=C1OS(O)(=O)=O MOODSJOROWROTO-UHFFFAOYSA-N 0.000 description 1
- 229960005399 satraplatin Drugs 0.000 description 1
- 190014017285 satraplatin Chemical compound 0.000 description 1
- 238000003345 scintillation counting Methods 0.000 description 1
- 238000002821 scintillation proximity assay Methods 0.000 description 1
- 125000003548 sec-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 210000000329 smooth muscle myocyte Anatomy 0.000 description 1
- 229950010372 sobuzoxane Drugs 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- RYYKJJJTJZKILX-UHFFFAOYSA-M sodium octadecanoate Chemical compound [Na+].CCCCCCCCCCCCCCCCCC([O-])=O RYYKJJJTJZKILX-UHFFFAOYSA-M 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- RCOSUMRTSQULBK-UHFFFAOYSA-N sodium;propan-1-olate Chemical compound [Na+].CCC[O-] RCOSUMRTSQULBK-UHFFFAOYSA-N 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 239000008347 soybean phospholipid Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 229940063675 spermine Drugs 0.000 description 1
- 108010087686 src-Family Kinases Proteins 0.000 description 1
- 102000009076 src-Family Kinases Human genes 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 230000036262 stenosis Effects 0.000 description 1
- 208000037804 stenosis Diseases 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 201000000498 stomach carcinoma Diseases 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 125000003107 substituted aryl group Chemical group 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 229940086735 succinate Drugs 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 229940071103 sulfosalicylate Drugs 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 229960005566 swainsonine Drugs 0.000 description 1
- FXUAIOOAOAVCGD-FKSUSPILSA-N swainsonine Chemical compound C1CC[C@H](O)[C@H]2[C@H](O)[C@H](O)CN21 FXUAIOOAOAVCGD-FKSUSPILSA-N 0.000 description 1
- FXUAIOOAOAVCGD-UHFFFAOYSA-N swainsonine Natural products C1CCC(O)C2C(O)C(O)CN21 FXUAIOOAOAVCGD-UHFFFAOYSA-N 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- 229960003102 tasonermin Drugs 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 229960001674 tegafur Drugs 0.000 description 1
- WFWLQNSHRPWKFK-ZCFIWIBFSA-N tegafur Chemical compound O=C1NC(=O)C(F)=CN1[C@@H]1OCCC1 WFWLQNSHRPWKFK-ZCFIWIBFSA-N 0.000 description 1
- 229960004964 temozolomide Drugs 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- LFKDJXLFVYVEFG-UHFFFAOYSA-N tert-butyl carbamate Chemical compound CC(C)(C)OC(N)=O LFKDJXLFVYVEFG-UHFFFAOYSA-N 0.000 description 1
- NBRKLOOSMBRFMH-UHFFFAOYSA-N tert-butyl chloride Chemical compound CC(C)(C)Cl NBRKLOOSMBRFMH-UHFFFAOYSA-N 0.000 description 1
- KCZFBLNQOSFGSH-UHFFFAOYSA-N tert-butyl n-(2-aminophenyl)carbamate Chemical compound CC(C)(C)OC(=O)NC1=CC=CC=C1N KCZFBLNQOSFGSH-UHFFFAOYSA-N 0.000 description 1
- WIVYTYZCVWHWSH-UHFFFAOYSA-N tert-butyl n-(4-aminophenyl)carbamate Chemical compound CC(C)(C)OC(=O)NC1=CC=C(N)C=C1 WIVYTYZCVWHWSH-UHFFFAOYSA-N 0.000 description 1
- 229960004559 theobromine Drugs 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 238000011287 therapeutic dose Methods 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 229960003723 tiazofurine Drugs 0.000 description 1
- FVRDYQYEVDDKCR-DBRKOABJSA-N tiazofurine Chemical compound NC(=O)C1=CSC([C@H]2[C@@H]([C@H](O)[C@@H](CO)O2)O)=N1 FVRDYQYEVDDKCR-DBRKOABJSA-N 0.000 description 1
- 230000017423 tissue regeneration Effects 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 229940100611 topical cream Drugs 0.000 description 1
- 229940100615 topical ointment Drugs 0.000 description 1
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 1
- 229960000303 topotecan Drugs 0.000 description 1
- XFCLJVABOIYOMF-QPLCGJKRSA-N toremifene Chemical compound C1=CC(OCCN(C)C)=CC=C1C(\C=1C=CC=CC=1)=C(\CCCl)C1=CC=CC=C1 XFCLJVABOIYOMF-QPLCGJKRSA-N 0.000 description 1
- 229960005026 toremifene Drugs 0.000 description 1
- PKVRCIRHQMSYJX-AIFWHQITSA-N trabectedin Chemical compound C([C@@]1(C(OC2)=O)NCCC3=C1C=C(C(=C3)O)OC)S[C@@H]1C3=C(OC(C)=O)C(C)=C4OCOC4=C3[C@H]2N2[C@@H](O)[C@H](CC=3C4=C(O)C(OC)=C(C)C=3)N(C)[C@H]4[C@@H]21 PKVRCIRHQMSYJX-AIFWHQITSA-N 0.000 description 1
- 229960000977 trabectedin Drugs 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 102000027257 transmembrane receptors Human genes 0.000 description 1
- 108091008578 transmembrane receptors Proteins 0.000 description 1
- 229960000575 trastuzumab Drugs 0.000 description 1
- 229960001727 tretinoin Drugs 0.000 description 1
- 229960005526 triapine Drugs 0.000 description 1
- WTVXIBRMWGUIMI-UHFFFAOYSA-N trifluoro($l^{1}-oxidanylsulfonyl)methane Chemical group [O]S(=O)(=O)C(F)(F)F WTVXIBRMWGUIMI-UHFFFAOYSA-N 0.000 description 1
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 1
- 229960001099 trimetrexate Drugs 0.000 description 1
- NOYPYLRCIDNJJB-UHFFFAOYSA-N trimetrexate Chemical compound COC1=C(OC)C(OC)=CC(NCC=2C(=C3C(N)=NC(N)=NC3=CC=2)C)=C1 NOYPYLRCIDNJJB-UHFFFAOYSA-N 0.000 description 1
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- UMKFEPPTGMDVMI-UHFFFAOYSA-N trofosfamide Chemical compound ClCCN(CCCl)P1(=O)OCCCN1CCCl UMKFEPPTGMDVMI-UHFFFAOYSA-N 0.000 description 1
- 229960000875 trofosfamide Drugs 0.000 description 1
- 229950010147 troxacitabine Drugs 0.000 description 1
- RXRGZNYSEHTMHC-BQBZGAKWSA-N troxacitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1O[C@@H](CO)OC1 RXRGZNYSEHTMHC-BQBZGAKWSA-N 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- 230000005748 tumor development Effects 0.000 description 1
- 102000003390 tumor necrosis factor Human genes 0.000 description 1
- 229940046728 tumor necrosis factor alpha inhibitor Drugs 0.000 description 1
- 239000002451 tumor necrosis factor inhibitor Substances 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- ZOCKGBMQLCSHFP-KQRAQHLDSA-N valrubicin Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC(OC)=C4C(=O)C=3C(O)=C21)(O)C(=O)COC(=O)CCCC)[C@H]1C[C@H](NC(=O)C(F)(F)F)[C@H](O)[C@H](C)O1 ZOCKGBMQLCSHFP-KQRAQHLDSA-N 0.000 description 1
- 229960000653 valrubicin Drugs 0.000 description 1
- 230000006444 vascular growth Effects 0.000 description 1
- 230000004862 vasculogenesis Effects 0.000 description 1
- 239000002525 vasculotropin inhibitor Substances 0.000 description 1
- 230000035899 viability Effects 0.000 description 1
- 229960005212 vindesine sulfate Drugs 0.000 description 1
- NMDYYWFGPIMTKO-HBVLKOHWSA-N vinflunine Chemical compound C([C@@](C1=C(C2=CC=CC=C2N1)C1)(C2=C(OC)C=C3N(C)[C@@H]4[C@@]5(C3=C2)CCN2CC=C[C@]([C@@H]52)([C@H]([C@]4(O)C(=O)OC)OC(C)=O)CC)C(=O)OC)[C@H]2C[C@@H](C(C)(F)F)CN1C2 NMDYYWFGPIMTKO-HBVLKOHWSA-N 0.000 description 1
- 229960000922 vinflunine Drugs 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
- 230000029663 wound healing Effects 0.000 description 1
- 239000000230 xanthan gum Substances 0.000 description 1
- 235000010493 xanthan gum Nutrition 0.000 description 1
- 229920001285 xanthan gum Polymers 0.000 description 1
- 229940082509 xanthan gum Drugs 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
- FBTUMDXHSRTGRV-ALTNURHMSA-N zorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(\C)=N\NC(=O)C=1C=CC=CC=1)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 FBTUMDXHSRTGRV-ALTNURHMSA-N 0.000 description 1
- 229960000641 zorubicin Drugs 0.000 description 1
- 229930195724 β-lactose Natural products 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/90—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/88—Nitrogen atoms, e.g. allantoin
Definitions
- the object of the invention was to find new compounds with valuable properties, in particular those which can be used for the production of medicaments.
- the present invention relates to compounds and the use of compounds in which the inhibition, regulation and / or modulation of the signal transduction of kinases, in particular the tyrosine kinases and / or serine / threonine kinases plays a role, furthermore pharmaceutical compositions which contain these compounds, and the use of the compounds for the treatment of kinase-related diseases.
- the present invention relates to compounds of the formula I which inhibit, regulate and / or modulate the signal transduction of the tyrosine kinases, compositions which contain these compounds and methods for their use for the treatment of tyrosine kinase-related diseases and conditions such as angiogenesis, cancer, tumorigenesis, Growth and spread, arteriosclerosis, eye diseases, such as age-related macular degeneration, choroidal neovascularization and diabetic retinopathy, inflammatory diseases, arthritis, thrombosis, fibrosis, glomerulonephritis, neurodegeneration, psoriasis, restenosis, wound healing of the immune system, transplantation and metabolic disorders , also autoimmune diseases, cirrhosis, diabetes and diseases of the blood vessels, including instability and permeability (permeability) and the like in mammals.
- diseases and conditions such as angiogenesis, cancer, tumorigenesis, Growth and spread, arteriosclerosis, eye diseases, such as age-related macular degeneration, choroidal ne
- Tyrosine kinases are a class of enzymes with at least 400 members that catalyze the transfer of the terminal phosphate of adenosine triphosphate (gamma-phosphate) to tyrosine residues on protein substrates. It is believed that the tyrosine 5 kinases play an important role in signal transduction in various cell functions via substrate phosphorylation. Although the exact mechanisms of signal transduction are still unclear, it has been shown that tyrosine kinases are important factors in the
- the tyrosine kinases can be divided into receptor tyrosine kinases and cytosolic tyrosine kinases.
- the receptor tyrosine kinases have an extracellular part, a transmembrane part and an intra-
- the receptor tyrosine kinases consist of a large number of transmembrane receptors with different biological effectiveness. Around 20 different subfamilies of receptor tyrosine kinases have been identified.
- a tyrosine kinase subfamily called the HER subfamily consists of EGFR, HER2, HER3 and HER4.
- the ligands of this receptor subfamily include the epithelial growth factor, TGF- ⁇ , amphiregulin, HB-EGF, betacellulin and heregulin.
- the insulin subfamily which includes INS-R, IGF-IR and IR-R, is another subfamily of these receptor tyrosine kinases.
- the PDGF subfamily includes the PDGF- ⁇ and -R receptor, CSFIR, c- kit and Q FLK-II.
- There is also the FLK family which consists of the kinase insert domain receptor (KDR), the fetal liver kinase-1 (FLK-1), the fetal liver kinase-4 (FLK-4) and the fms tyrosine kinase-1 (fit-1 ) consists.
- KDR kinase insert domain receptor
- FLK-1 the fetal liver kinase-1
- FLK-4 fetal liver kinase-4
- fit-1 fms tyrosine kinase-1
- the RTKs (receptor tyrosine kinases) also include TIE2 and its ligands angiopoietin 1 and 2. More and more 5 homologs of these ligands have now been found, the effect of which has not yet been clearly demonstrated.
- TIE1 is known as a homologue of TIE2.
- the TIE RTKs are selectively expressed on endothelial cells and are used in processes of angiogenesis and maturation
- ⁇ c Vascularization can be a valuable target for active ingredients.
- the cytosolic tyrosine kinases also consist of a large number of subfamilies, including Src, Frk, Btk, Csk, Abi, Zap70, Fes / Fps, Fak, Jak, Ack, and LIMK. Each of these subfamilies is further divided into 5 different receptors.
- the Src subfamily is one of the largest subfamilies. It includes Src, Yes, Fyn, Lyn, Lck, BIk, Hck, Fgr and Yrk.
- the Src enzyme subfamily has been linked to oncogenesis.
- Both the receptor tyrosine kinases and the cytosolic tyrosine kinases are involved in cell signaling pathways that lead to various conditions, including cancer, psoriasis and hyper-immune reactions.
- Various receptor tyrosine kinases and the growth factors that bind them have been suggested to play a role in angiogenesis, although some may promote angiogenesis indirectly (Mustonen and Alitalo, J. CellBiol. 129: 895-898, 1995).
- One of these receptor tyrosine kinases is fetal liver kinase 1, also called FLK-1.
- the human analogue of FLK-1 is the kinase insert domain-containing receptor KDR, which is also known as vascular endothelial cell growth factor receptor 2 or VEGFR-2, because it binds VEGF with high affinity.
- VEGFR-2 vascular endothelial cell growth factor receptor 2
- VEGF and KDR represent a ligand-receptor pair that play an essential role in the proliferation of vascular endothelial cells and
- vasculogenesis Formation and sprouting of the blood vessels, which are called vasculogenesis or angiogenesis, plays.
- VEGF vascular endothelial growth factor
- the KDR induces the mitogenic function of VEGF, while Flt-1 appears to modulate non-mitogenic functions, such as those related to cell adhesion. Inhibition of the KDR therefore modulates the level of mitogenic VEGF activity.
- VEGF receptor antagonists Two PTK (protein tyrosine kinase) receptors for VEGFR have been identified
- VEGFR-1 (Flt-1); VEGRF-2 (Flk-1 or KDR) and VEGFR-3 (Flt-4). VEGFR-2 is of particular interest.
- Solid tumors can therefore be treated with tyrosine kinase inhibitors ⁇ 5 , since these tumors depend on angiogenesis for the formation of the blood vessels required to support their growth.
- These solid tumors include monocyte leukemia, brain, urogenital, lymphatic, gastric, larynx, and lung cancer, including lung adenocarcinoma and small cell lung cancer.
- lung cancer including lung adenocarcinoma and small cell lung cancer.
- carcinomas in which overexpression or activation of Raf-activating oncogenes eg K-ras, erb-B
- These cancers include pancreatic and breast cancer. Inhibitors of these tyrosine kinases are therefore suitable for the prevention and treatment of prol iterative diseases which are caused by these enzymes.
- VEGF vascular endothelial growth factor
- VEGF mRNA and protein levels in the eye are increased due to conditions such as retinal venous occlusion in the primate and reduced pO 2 levels in the mouse, which lead to neovascularization.
- Intraocularly injected monoclonal anti-VEGF antibodies, or VEGF receptor immunoconjugates inhibit both in primate and in Rodent model the neovascularization in the eye.
- VEGF vascular endothelial growth factor
- Anti-VEGF monoclonal antibodies inhibit the growth of human tumors in the nude mouse. Although the same tumor cells continue to express VEGF in culture, the antibodies do not decrease their cell division rate. This is how it comes from tumors
- VEGF not as an autocrine mitogenic factor. VEGF therefore contributes to tumor growth in vivo by promoting angiogenesis through its paracrine vascular endothelial cell chemotaxis and mitogenesis activity.
- monoclonal antibodies also inhibit the growth of typically less vascularized human colon carcinomas in 0 thymus-less mice and reduce the number of tumors arising from inoculated cells.
- VEGF-binding construct of Flk-1, Flt-1 the mouse KDR receptor homolog shortened to remove the cytoplasmic tyrosine kinase domains, but 5 while maintaining a membrane anchor, virtually stops the growth of a transplantable glioblastoma in the mouse, presumably due to the dominant-negative mechanism of heterodimer formation with trans-Q membranous endothelial cell VEGF receptors.
- Embryo stem cells which usually grow in the form of solid tumors in the nude mouse, do not form any detectable tumors when all VEGF alleles are knocked out.
- KDR or Flt-1 are involved in 5 pathological angiogenesis, and these receptors are useful for the treatment of diseases in which angiogenesis is a part overall pathology, e.g. inflammation, diabetic retinal vascularization and various forms of cancer, since it is known that tumor growth is dependent on angiogenesis (Weidner et al., N. Engl. J. Med., 324, pp. 1- 8, 1991). 5
- Angiopoietin 1 (Ang1), a ligand for the endothelium-specific receptor tyrosine kinase TIE-2, is a new angiogenic factor (Davis et al, Cell, 1996, 87: 1161-1169; Partanen et al, Mol.
- TIE tyrosine kinase with Ig and EGF homology domains. TIE is used to identify a class of receptor tyrosine kinases that are unique to
- TIE receptor kinases are typically characterized by the presence of an EGF-like domain and an immunoglobulin (IG) -like domain, which consists of extracellular folding units that are stabilized by disulfide bridges between the chains 0 (Partanen et al Curr. Topics Microbiol. Immunol ., 1999, 237: 159-172).
- IG immunoglobulin
- Ang1 and its receptor TIE-2 act during the later stages of vascular development, 5 i.e.
- VEGFR-2 block the phosphorylation of tyrosine residues and so on serve to interrupt the initiation of angiogenesis. It can therefore be assumed that the inhibition of TIE-2 and / or VEGFR-2 should prevent tumor angiogenesis and serve to slow down or completely eliminate tumor growth. Accordingly, treatment for cancer and other diseases associated with inappropriate angiogenesis could be provided.
- the present invention is directed to methods for regulating, modulating or inhibiting TIE-2 for the prevention and / or treatment of diseases in connection with unregulated or impaired TIE-2 activity.
- the compounds of the formula I can also be used in the treatment of certain forms of cancer.
- the compounds of formula I may be used to provide additive or synergistic effects in certain existing cancer chemotherapies, and / or can be used to restore effectiveness of certain of the existing cancer chemotherapies and radiotherapies.
- the compounds of the formula I can be used to isolate and to study the activity or expression of TIE-2. They are also particularly suitable for use in 5 diagnostic procedures for diseases in connection with unregulated or impaired TIE-2 activity.
- the present invention is further directed to methods for 0 the regulation, modulation or inhibition of VEGFR-2 for the prevention / or treatment of diseases and in connection with unregulated or disturbed VEGFR-2 activity.
- the present invention further relates to the compounds of formula 5 as inhibitors of Raf kinases.
- Protein phosphorylation is a fundamental process for the regulation of cell functions. The coordinated action of both protein kinases and phosphatases controls the levels of phosphorylation and consequently the activity of specific target proteins.
- Role of protein phosphorylation is in signal transduction when extracellular signals are amplified and by a cascade of protein
- Phosphorylation and dephosphorylation events e.g. B. are propagated in the p21 ras / raf way.
- the p21 ras gene was discovered as an oncogene of the Harvey and Kirsten rat sarcoma viruses (H-Ras and K-Ras, respectively).
- H-Ras and K-Ras characteristic mutations in the cellular Ras gene (c-Ras) have been associated with many different types of cancer.
- c-Ras characteristic mutations in the cellular Ras gene
- These mutant alleles that make Ras constitutively active have been shown to transform cells, such as the murine cell line NIH 3T3, in culture.
- the p21 ras oncogene is an important contributing factor in the development and progression of solid human carcinomas and is mutated in 30% of all human carcinomas (Bolton et al. (1994) Ann. Rep. Med. Chem., 29, 165-74; Bos. (1989) Cancer Res., 49, 4682-9).
- the Ras protein is a key element of the signal transduction cascade, which is controlled by growth factor receptors in almost all tissues (Avruch et al. (1994) Trends Biochem. Sei., 19, 279-83) ,
- Ras is a guanine nucleotide binding protein, and the cycling between a GTP-linked activated and a GDP-linked quiescent form is strictly controlled by Ras endogenous GTPase activity and other regulatory proteins.
- the Ras gene product binds to guanine triphosphate (GTP) and guanine diphosphate (GDP) and hydrolyzes GTP to GDP. Ras is active in the GTP-bound state.
- the endogenous GTPase activity is reduced in the Ras mutants in cancer cells. weakens, and consequently the protein emits constitutive growth signals to "downstream" effectors such as the Raf kinase enzyme. This leads to cancerous growth of the cells carrying these mutants (Magnuson et al.
- the Ras proto-oncogene requires a functionally intact C-Raf-1 proto-oncogene in order to generate growth and differentiation signals in higher eukaryotes by receptor and non-receptor tyrosine kinases to transduce.
- Ras Activated Ras is necessary for the activation of the C-Raf-1 proto-oncogene, but the biochemical steps by which Ras activates the Raf-1 protein (Ser / Thr) kinase have now been well characterized. It has been shown to inhibit the effect of active Ras
- Raf kinase signaling pathway Inhibition of the Raf kinase signaling pathway by administration of deactivating antibodies against Raf kinase or by co-expression of dominant negative Raf kinase or dominant negative MEK (MAPKK), the substrate of Raf kinase, leads to the reversion of transformed cells to the normal growth phenotype, see: Daum et al. (1994) Trends Biochem. Sci., 19, 474-80; Fridman et al. (1994) J Biol. Chem., 269, 30105-8. Kolch et al. (1991) Nature, 349, 426-28) and for discussion Weinstein-Oppenheimer et al. Pharm. & Therap. (2000), 88, 229-279.
- Raf kinase by Antisense-Oligodesoxynucleot.de
- inhibition of Raf kinase has been associated in vitro and in vivo with the inhibition of growth in a number of different human tumor types (Monia et al., Nat. Med. 1996, 2, 668-75).
- Raf-serine and threonine-specific protein kinases are cytosolic enzymes that stimulate cell growth in a number of different cell systems (Rapp, U.R., et al. (1988) in The Oncogene Handbook; T.
- Raf-1 is found in all organs and in all cell lines that have been examined
- ⁇ p- and A- and B-Raf are expressed in urogenital and brain tissues, respectively (Storm, S.M. (1990) Oncogene 5: 345-351).
- Raf genes are proto-oncogenes: they can initiate the malignant transformation of cells if they are expressed in specifically modified forms. Genetic changes that lead to oncogenic activation produce a constitutively active protein kinase by removal or interference with an N-terminal negative regulatory domain of the protein (Heidecker, G., et al. (1990) Mol. Cell. Biol. 10: 2503-2512 ; Rapp, UR, et 5 al. (1987) in Oncogenes and Cancer; SA Aaronson, J. Bishop, T. Sugimura, M. Terada, K. Toyoshima and PK Vogt (ed.) Japan Scientific Press, Tokyo).
- Raf-1 is an intracellular activator of cell growth.
- Raf-1 protein serine kinase is a candidate for the "downstream" effector of mitogen signal transduction because Raf oncogenes face growth arrest resulting from blockade of cellular Ras activity due to a cellular mutation (Ras revertant cells) or micro-injection of anti-Ras antibodies results (Rapp, UR, et al. (1988) in The Oncogene Handbook, T. Curran, EP Reddy and A. Skalka (ed.), Elsevier Science Publishers; Netherlands, S. 213-253; Smith, MR, et al. (1986) Nature (London) 320: 540-543).
- the C-Raf function is required for transformation by a number of different membrane-bound oncogenes and for growth stimulation by mitogens contained in sera (Smith, M.R., et al. (1986) Nature (London) 320: 540-543).
- Raf-1 protein serine kinase activity is regulated by mitogens via phosphorylation (Morrison, DK, et al. (1989) Cell 58: 648-657), which also effects the subcellular distribution (Olah, Z., et al. (1991) Exp. Brain Res. 84: 403; Rapp, UR, et al. (1988) Cold Spring Harbor Sym. Quant. Biol. 53: 173-184.
- Raf-1 activating growth factors include those from platelets growth factor (PDGF) (Morrison, DK, et al. (1988) Proc. Natl. Acad. Sci. USA 85: 8855-8859), the colony-stimulating factor (Baccarini, M “et al. (1990) EMBO J 9: 3649-3657), insulin (Blackshear, PJ, et al. (1990) J. Biol. Chem. 265: 12115-12118), epidermal growth factor (EGF) (Morrison, RK, et al. (1988) Proc. Natl. Acad. Sci. USA 85: 8855-8859), Inter! Eukin-2 (Turner, BC, et al.
- Raf-1 protein serine kinase translocates into the perinuclear region and the nucleus (Olah, Z., et al. (1991) Exp. Brain Res. 84: 403; Rapp, UR, et al. (1988) Cold Spring Habor Sym. Quant. Biol. 53: 173-184).
- Cells containing activated Raf are changed in their gene expression pattern (Heidecker, G., et al. (1989) in Genes and signal transduction in multistage carcinogenesis, N. Colburn (ed.), Marcel Dekker, Inc., New York, Pp.
- Raf-oncogenes activate transcription from Ap-1 / PEA3-dependent promotors in transient transfeetion assays (Jamal, S., et al. (1990) Science 344: 463-466; Kaibuchi, K., et al. (1989) J. Biol. Chem. 264: 20855-20858; Wasylyk, C, et al. (1989) Mol. Cell. Biol. 9: 2247-2250).
- Raf-1 protein phosphorylation may be a consequence of a kinase cascade that is amplified by autophosphorylation, or may be entirely caused by autophosphorylation that is by binding a putative activation ligand to the Raf-1 regulator domain, analogous to PKC activation Diacylglycerol is initiated (Nishizuka, Y. (1986) Science 233: 305-312).
- Proteins occur predominantly with serine, threonine or tyrosine residues, and protein kinases were therefore selected according to their specificity for the location of the phosphoryl. H. of serine / threonine kinases and tyrosine kinases. Because phosphorylation is such a common process in
- the compounds according to the invention are inhibitors of the enzyme Raf kinase. Since the enzyme is a "downstream" effector of p21 ras , the inhibitors in pharmaceutical compositions for human or veterinary use are found to be useful when inhibiting the Raf kinase pathway, for example in the treatment of tumors and / or by Raf kinase mediated cancerous cell growth is indicated.
- the compounds are particularly useful in the treatment of solid carcinomas in humans and animals, e.g. B. Murine cancer, since the progression of these cancers is dependent on the Ras protein signal
- the compound of the invention or a pharmaceutically acceptable salt thereof is used for the treatment of
- cancer including solid cancers, such as cancers (e.g., the lungs, pancreas, thyroid, bladder, or colon), myeloid disorders (e.g. myeloid
- Ap-leukemia or adenomas (e.g. villous colon adenoma), pathological angiogenesis and metastatic cell migration.
- the compounds are also useful in the treatment of complement activation-dependent chronic inflammation (Niculescu et al. (2002) Immunol. Res., 24: 191-199) and immunodeficiency induced by HIV-1 (Human Immunodeficiency Virus 0 type 1) (Popik et al. (1998) J Virol, 72: 6406-6413).
- the 5 compounds according to the invention can interact with signaling pathways, in particular with the signaling pathways described herein and preferably the Raf kinase signaling pathway.
- the compounds according to the invention preferably exhibit an advantageous biological activity in enzyme-based assays, for example assays Q as described herein, is readily detectable.
- the compounds according to the invention preferably show and bring about an inhibitory effect which is usually documented by IC 50 values in a suitable range, preferably in the micromolar range and more preferably in the nanomolar range 5. As discussed herein, these signaling pathways are different
- the present invention therefore relates to the invention
- Preferred objects of the invention are therefore compounds according to the invention as promoters or inhibitors, preferably as inhibitors of the Raf kinase pathway.
- a preferred subject of the invention is therefore compounds according to the invention as promoters or inhibitors, preferably as inhibitors of Raf kinase.
- a more preferred subject of the invention are compounds according to the invention as promoters or inhibitors, preferably as inhibitors of one or more Raf kinases, selected from the group consisting of A-Raf, B-Raf and C-Raf-1.
- a particularly preferred subject of the invention are compounds according to the invention as promoters or inhibitors, preferably as inhibitors of C-Raf-1.
- the present invention furthermore relates to the use of one or more compounds according to the invention in the treatment and / or prophylaxis of diseases, preferably the diseases described here, which are caused, mediated and / or propagated by Raf kinases and in particular diseases which are caused by Raf - Kinases selected from the group consisting of A-Raf, B-Raf and C-Raf-1 are caused, mediated and / or propagated.
- the diseases discussed here are usually divided into two groups, hyperproliferative and non-hyperproliferative diseases.
- psoriasis, arthritis, inflammation, endometriosis, scarring, benign prostatic hyperplasia, immunological diseases, autoimmune diseases and immune deficiency diseases are not considered cancerous diseases, of which arthritis, inflammation, immunological diseases, autoimmune diseases and immunodeficiency diseases are usually considered non-hyperproliferative diseases.
- cancerous diseases to consider all of which are commonly considered hyperproliferative diseases.
- cancerous cell growth and in particular cancerous cell growth mediated by Raf kinase, is a disease that is a goal of the present
- a c represents invention.
- the present invention therefore relates to compounds according to the invention as medicaments and / or active pharmaceutical ingredients in the treatment and / or prophylaxis of the diseases mentioned and the use of compounds according to the invention for the preparation of a pharmaceutical for the treatment and / or prophylaxis of the diseases mentioned and also a process for Treatment of said diseases comprising the administration of one or more compounds according to the invention to a patient in need of such an administration. 5
- the compounds according to the invention have an in vivo antiproliferative effect in a xenograft tumor model.
- the compounds of the invention are administered to Q a patient with a hyperproliferative disease, e.g.
- the present compounds are useful for prophylactic or therapeutic purposes.
- the term "treat” is used to refer to both the Disease prevention as well as the treatment of pre-existing ailments are used.
- the prevention of proliferation is achieved by administration of the compounds according to the invention before the development of the evident disease, e.g. B. for the prevention of tumor growth, 5 prevention of metastatic growth, the reduction of restenoses associated with cardiovascular surgery, etc. achieved.
- the compounds are used to treat persistent diseases by stabilizing or improving clinical
- the host or patient can belong to any mammalian species, e.g. B. a primate species, especially humans; Rodents, including AC mice, rats and hamsters; Rabbits; Horses, cattle, dogs, cats, etc. Animal models are of interest for experimental studies, providing a model for treating a human disease.
- the susceptibility of a particular cell to treatment with the compounds according to the invention can be determined by testing in vitro. Typically, a culture of the cell is combined with a compound of the invention at various concentrations for a period of time sufficient to enable the active agents to induce cell death or to inhibit migration, usually between about an hour and a week. Cultured cells from a biopsy sample can be used for in vitro testing. The 0 remaining after the treatment viable cells are then counted.
- the dose varies depending on the specific compound used, the specific disease, patient status, etc. Typically, a therapeutic dose is sufficient to significantly reduce the unwanted cell population in the target tissue while maintaining the patient's viability.
- the treatment will generally continue until there is a substantial reduction, e.g. B. at least about 50% reduction in cell load and can be continued until essentially no more unwanted cells are detected in the body.
- Suitable models or model systems have been developed by various scientists to identify a signal transmission path and to demonstrate interactions between different signal transmission paths, e.g. Cell culture models (e.g. Khwaja et al.,
- EMBO, 1997, 16, 2783-93 models of transgenic animals (e.g. White et al., Oncogene, 2001, 20, 7064-7072).
- interacting compounds can be used to modulate the signal (e.g. Stephens et al., Biochemical J., 2000, 351, 95-105).
- the compounds according to the invention can also be used as reagents for testing kinase-dependent signal transmission paths in animals and / or cell culture models or in the clinical diseases mentioned in this application.
- Measuring kinase activity is a technique well known to those skilled in the art.
- Generic test systems for determining kinase activity with substrates e.g. Histone (e.g. Alessi et al., FEBS Lett. 1996, 399, 3, pages 333-338) or the basic myelin protein are described in the literature (e.g. Campos-Gonzalez, R. and Glenney, Jr., JR 1992, J. Biol. Chem. 267, page 14535).
- phospho-AK specific phospho-antibodies
- the phospho-AK only binds the phosphorylated substrate. This binding can be detected with a second peroxidase-conjugated anti-sheep antibody by chemiluminescence (Ross et al., 2002, Biochem. J., immediately before publication, manuscript BJ20020786).
- the sufferings of interest include, but are not limited to, the following sufferings.
- the compounds of the invention are useful in the treatment of a number of different conditions in which proliferation and / or migration of smooth muscle cells and / or inflammatory cells is present in the intimal layer of a vessel, resulting in reduced blood flow to this vessel, e.g. B. in neointimal occlusive lesions.
- Occlusive graft vascular diseases of interest include atherosclerosis, coronary vascular disease after transplantation, venous graft stenosis, peri-anastomotic prosthetic restenosis, restenosis after angioplasty or stent placement and the like.
- the compounds according to the invention are also suitable as p38 kinase inhibitors.
- WO 200104115 is known for aryl- and heteroaryl-substituted ureas, which act as cytokinin inhibitors for the treatment of inflammatory and
- aryl- and heteroaryl-substituted ureas which can be used as cytokinin inhibitors for the treatment of osteoarthritis or ulcerative colitis are described in WO 200055139.
- Heterocyclic-substituted ureas for the treatment of inflammatory diseases are described in WO 00/43384.
- EP 0 286 979 discloses aryl- and heteroaryl-substituted ureas which can be used as antiarrhythmics.
- WO 200006550 describes phenyl-substituted ureas as lipid peroxidase inhibitors. Madsen et al.
- substituted aryl and heteroaryl compounds for the treatment, inter alia, in WO 03/000245.
- allergic rhinitis, atherosclerosis, asthma or cancer Aryl- and heteroaryl-substituted urea derivatives are described in WO 00/76515 as IL-8 receptor antagonists.
- the invention relates to compounds of the formula I.
- R 4 , R 5 each independently of one another H, A, OH, OA, alkenyl, alkynyl, NO 2 , NH 2) NHA, NA 2) shark, CN, COOH, COOA, -OHet, -O-alkylene-Het, - O-alkylene-NR 10 R 11 or CONR 10 R 11 , two adjacent radicals selected from R 1 , R 2 , R 3 , R 4 , R 5 together also -O-CH 2 -CH 2 -, -O-CH 2 -O- or -O-CH 2 -CH 2 -O-,
- R 6 , R 7 each independently of one another H, A, shark, OH, OA or CN, R 8 CN, COOH, COOA, CONH2, CONHA or CONA 2 , R 9 H or A,
- a alkyl having 1 to 10 C atoms, where 1-7 H atoms can also be replaced by F and / or chlorine,
- Hai F. CI. Br or l mean as well as their pharmaceutically usable derivatives, solvates, salts and stereoisomers, including their mixtures in all ratios.
- the invention also relates to the optically active forms 5 (stereoisomers), the enantiomers, the racemates, the diastereomers and the hydrates and solvates of these compounds.
- Solvates of the compounds are understood to mean the addition of inert solvent molecules to the compounds, which are mutually exclusive due to their mutual nature
- Solvates are e.g. Mono- or dihydrates or alcoholates.
- compositions are understood to mean, for example, the 5 salts of the compounds according to the invention and so-called prodrug compounds.
- Prodrug derivatives are understood with z. B. alkyl or acyl groups, sugars or oligopeptides modified compounds of formula I, which are quickly cleaved in the organism to the active 0 compounds of the invention.
- This also includes biodegradable polymer derivatives of the compounds according to the invention, as described, for. B. in Int. J. Pharm. 115, 61-67 (1995).
- an effective amount means the amount of a drug or active pharmaceutical ingredient that elicits a biological or medical response in a tissue, system, animal or human that is sought or sought by, for example, a researcher or medical professional .
- therapeutically effective amount means an amount which, compared to a subject who has not received this amount, does the following:
- terapéuticaally effective amount also includes
- the invention also relates to the use of mixtures of the compounds of the formula I, e.g. Mixtures of two diastereomers e.g. in a ratio of 1: 1, 1: 2, 1: 3, 1: 4, 1: 5, 1: 10, 1: 100 or 1: 1000. These are particularly preferably mixtures of stereoisomeric compounds.
- the compounds of the invention can also exist in various polymorphic forms, e.g. as amorphous and crystalline polymorphic forms. All polymorphic forms of the compounds according to the invention belong to the scope of the invention and are a further aspect of the invention.
- the invention relates to the compounds of formula I and their salts and a process for the preparation of compounds of formula I according to claims 1-10 and their pharmaceutically usable derivatives, salts, solvates and stereoisomers, characterized in that
- R 6 , R 7 , R 8 , R 9 , R 10 and R 11 have the meanings given in claim 1,
- R 1 , R 2 , R 3 , R 4 and R 5 have the meanings given in claim 1, are reacted with a chloroformate derivative to give an intermediate carbamate derivative which is then reacted with a compound of the formula II,
- R - 8 8 CN COOA, CONH 2 , CONHA or CONA 2 ,
- R 10 , R 11 H mean a compound of formula V.
- R 8 is CN, COOA, CONH 2 , CONHA or CONA 2 ,
- R has the meaning given in claim 1 and A 'denotes alkyl having 1, 2, 3, 4, 5 or 6 carbon atoms,
- R 1 , R 2 , R 3 , R 4 and R 5 have the meanings given in claim 1, and L is Cl, Br, I or a free or reactively modified OH group, implements,
- A is preferably methyl, furthermore ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl or tert-butyl, furthermore also pentyl, 1-, 2- or 3-methylbutyl, 1, 1-, 1, 2- or 2,2-dimethylpropyl, 1-ethylpropyl, hexyl, 1-, 2-, 3- or 4-M ethyl pentyl, 1,1-, 1, 2-, 1,3-, 2,2-, 2,3 - or 3,3-dimethylbutyl, 1- or 2-ethylbutyl, 1-ethyl-1-methylpropyl, 1-ethyl-2-methylpropyl, 1,1,2- or 1,2,2-trimethylpropyl, further preferred eg Trifluoromethyl.
- A also means cycloalkyl. Cycloalkyl preferably means cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or cycloheptyl.
- a ' is preferably methyl, ethyl, propyl or butyl.
- Alkylene is preferably unbranched and preferably means methylene,
- Ethylene, propylene, butylene or pentylene Ethylene, propylene, butylene or pentylene.
- R 1 , R 2 , R 3 , R 4 , R 5 each independently of one another, denote H, A, OH, OA, NO 2 , NH 2) NHA, NA 2 , shark, CN,
- R 1 , R 2 , R 3 , R 4 , R 5 each independently of one another, denote H, A, OH, OA, shark, O-alkylene-het or -O-alkylene-NR 10 R 11 .
- R 1 , R 2 , R 3 , R 4 , R 5 each independently, preferably, for example, H; A, such as methyl or ethyl; OH, OA, such as
- R ⁇ and R 7 are preferably H.
- R 8 is preferably CONH 2 or CN.
- R 10 and R 11 are preferably H.
- Het means, for example, 2- or 3-furyl, 2- or 3-thienyl, 1-, 2- or 3-pyrrolyl, 1-, 2, 4- or 5-imidazolyl, 1-, 3-, 4 - or 5-pyrazolyl, 2-, 4- or 5-oxazolyl, 3-, 4- or 5-isoxazolyl, 2-, 4- or 5-thiazolyl, 3-, 4- or 5-isothiazolyl, 2-, 3rd - or 4-pyridyl, 2-, 4-, 5- or 6- 5 pyrimidinyl, further preferably 1, 2,3-triazol-1-, -4- or -5-yl, 1, 2,4-triazol- 1-, -3- or 5-yl, 1- or 5-tetrazolyl, 1, 2,3-oxadiazol-4- or -5-yl, 1, 2,4-oxadiazol-3- or -5-yl, 1, 3,4-thiadiazol-2- or -5-yl, 1, 2,4-thiadiazol-3- or -5-yl, 1, 2,4-
- heterocyclic radicals can also be partially or completely hydrogenated.
- Het can, for. B. also mean 2,3-dihydro-2-, -3-, -4- or -5-furyl, 5 2,5-dihydro-2-, -3-, -4- or 5-furyl, tetrahydro- 2- or -3-furyl, 1, 3-dioxolan-4-yl, tetrahydro-2- or -3-thienyl, 2,3-dihydro-1-, -2-, -3-, -4- or -5-pyrrolyl, 2,5-dihydro-1-, -2-, -3-, -4- or -5-pyrrolyl, 1-, 2- or 3-pyrrolidinyl, tetrahydro-1-, - 2- or -4-imidazolyl, 2,3-dihydro-1-, -2-, -3-, -4- or -5-pyrazolyl, tetrahydro-1-,
- Dihydro-2H-1,5-benzodioxepin-6- or -7-yl further preferably 2,3-dihydro-benzofuranyl or 2,3-dihydro-2-oxo-furanyl.
- Het means a mononuclear saturated heterocycle with 1 to 3 N-, O- and / or S-
- the mononuclear saturated heterocycle here means particularly preferably piperidinyl, pyrrolidinyl, morpholinyl or piperazinyl.
- Het very particularly preferably denotes 1-, 2-, 3- or 4-piperidinyl or 2-, 3- or 4-morpholinyl, it being possible for Het to be substituted by COOA.
- Shark is preferably F, Cl or Br, but also I, particularly preferably F or Cl.
- Alkenyl is preferably vinyl, 1- or 2-propenyl, 1-butenyl, isobutenyl, sec-butenyl, further preferred is 1-pentenyl, isopentyl or 1-hexenyl.
- Alkynyl is preferably ethynyl, propyn-1-yl, furthermore butyn-1, butyn-2-yl, pentyn-1, pentyn-2 or pentyn-3-yl.
- the invention relates in particular to those compounds of the formula I in which at least one of the radicals mentioned has one of the preferred meanings indicated above.
- Some preferred groups of compounds can be expressed by the following sub-formulas Ia to Ih, which correspond to the formula I and in which the radicals not specified have the meaning given for the formula I, but in which
- R 1 , R 2 , R 3 , R 4 , R 5 each independently of one another H, A, OH, OA, NO 2 , NH 2 , NHA, NA 2 , shark, CN, -OHet, -O-alkylene- Het or -O-alkylene-NR 8 ° oR9 mean;
- Ic Het denotes a mononuclear saturated heterocycle having 1 to 3 N, O and / or S atoms, which is unsubstituted or can simply be substituted by COOA;
- NH, R 1 , R 2 , R 3 , R 4 , R 5 each independently NH 2 , NHA, NA 2 , shark, CN, -OHet, -O-alkylene-het or - O-alkylene-NR 1'0 u rR .1'1 ', R 6 , R 7 H, R 8 CONH 2 or CN, Het unsubstituted.es or piperidinyl, pyrrolidinyl, morpholinyl or piperazinyl substituted by COOA;
- R 1 , R 2 , R 3 , R 4 , R 5 each independently of one another H, A, OH, OA, shark, O-alkylene-het or -O-alkylene -NR 10 R 11 , R 6 , R 7 H, R 8 CONH 2 or CN, R 9 H or A, R 10 , R 11 each independently of one another H or A, Het unsubstituted or simply substituted by COOA, piperidinyl, pyrrolidinyl, morpholinyl or piperazinyl, 5 A alkyl having 1 to 10 C atoms, 1-7 H atoms also being represented by F and / or can be replaced by chlorine, mean shark F, Cl, Br or I,
- the starting materials can also be formed in situ, 5 so that they are not isolated from the reaction mixture, but instead are immediately reacted further to give the compounds of the formula I.
- the compounds of the formula II and those of the formula III are generally known. 5
- the reaction is usually carried out in an inert solvent, in the presence of an organic base such as triethylamine, dimethylaniline, pyridine or quinoline.
- an organic base such as triethylamine, dimethylaniline, pyridine or quinoline.
- the reaction time is between a few minutes and 14 days, the reaction temperature is between about 0 ° and 150 °, normally between 15 ° and 90 °, particularly preferably between 15 and 30 ° C.
- Suitable inert solvents are e.g. Hydrocarbons such as hexane, petroleum ether, benzene, toluene or xylene; chlorinated hydrocarbons such as trichlorethylene, 1,2-dichloroethane, carbon tetrachloride, chloroform or dichloromethane; Alcohols such as methanol, ethanol, isopropanol, n-propanol, n-butanol or tert-butanol; Ethers such as diethyl ether, diisopropyl ether, A g tetrahydrofuran (THF) or dioxane; Glycol ethers such as ethylene glycol monomethyl or monoethyl ether (methyl glycol or ethyl glycol), ethylene glycol dimethyl ether (diglyme); Ketones such as acetone or butanone; Amides such as acetamide, dimethylacetamide or dimethylformamide (
- reaction is usually carried out in an inert solvent, in the presence of an acid-binding agent, preferably an alkali metal or alkaline earth metal hydroxide, carbonate or bicarbonate or one
- an acid-binding agent preferably an alkali metal or alkaline earth metal hydroxide, carbonate or bicarbonate or one
- 35 other salt of a weak acid of the alkali or alkaline earth metals preferably potassium, sodium, calcium or cesium.
- an organic base such as triethylamine, dimethylaniline, N, N'-
- the reaction time is between a few minutes and 14 days
- the reaction temperature is between about 0 ° and 150 °, normally between 20 ° and 130 °, particularly preferably between 60 and 90 °.
- Suitable inert solvents are e.g. Hydrocarbons such as hexane, petroleum ether, benzene, toluene or xylene; chlorinated hydrocarbons such as trichlorethylene, 1,2-dichloroethane, carbon tetrachloride, chloroform or dichloromethane; Alcohols such as methanol, ethanol, isopropanol, n-propanol, n-butanol or tert-butanol; Ethers such as diethyl ether, diisopropyl ether, tetrahydrofuran (THF) or dioxane; Glycol ethers such as ethylene glycol monomethyl or monoethyl ether (methyl glycol or ethyl glycol), ethylene glycol dimethyl ether (diglyme); Ketones such as acetone or butanone; Amides such as acetamide, dimethylacetamide or dimethylformamide (DMF);
- the starting compounds of the formula IV are generally known.
- R 8 CN, COOA, CONH 2 , CONHA or CONA 2 ,
- R 10 , R 11 H can further preferably be obtained by combining compounds of the formula V with compounds of the formula VI and compounds of the
- the reaction is usually carried out in an inert solvent as indicated above.
- the response time depends on the one used Conditions between a few minutes and 14 days, the reaction temperature between about 0 ° and 150 °, normally between 20 ° and 130 °, particularly preferably between 60 and 90 °.
- X is absent and X 'is NH, can further preferably be obtained by reacting compounds of the formula II with compounds of the formula VIII.
- the compounds of the formula VIII are generally known.
- L is preferably Cl, Br, I or a free or a reactively modified OH group such as e.g. an activated ester, an imidazolide or alkylsulfonyloxy with 1-6 C atoms (preferably methylsulfonyloxy or trifluoromethylsulfonyloxy) or arylsulfonyloxy with 6-10 C atoms (preferably phenyl- or p-tolylsulfonyloxy).
- an activated ester an imidazolide or alkylsulfonyloxy with 1-6 C atoms (preferably methylsulfonyloxy or trifluoromethylsulfonyloxy) or arylsulfonyloxy with 6-10 C atoms (preferably phenyl- or p-tolylsulfonyloxy).
- the reaction is usually carried out in an inert solvent
- an acid-binding agent preferably an organic base such as DIPEA, triethylamine, dimethylaniline, pyridine or quinoline or an excess of the carboxy component of the formula VIII.
- alkali or alkaline earth metal hydroxide carbonate or bicarbonate or another salt of a weak acid Alkali or alkaline earth metals, preferably potassium, sodium,
- reaction time is between a few minutes and 14 days, the reaction temperature between about
- the compounds according to the invention mentioned can be used in their final non-salt form.
- the present invention also encompasses the use of these compounds in the form of their pharmaceutically acceptable salts, which can be derived from various organic and inorganic acids and bases by methods known in the art.
- Most of the pharmaceutically acceptable salt forms of the compounds of formula I are prepared conventionally. If the compound of the formula I contains a carboxylic acid group, one of its suitable salts can be formed by reacting the compound with a suitable base to give the corresponding base addition salt.
- Such bases are, for example, alkali metal hydroxides, including potassium hydroxide, sodium hydroxide and lithium hydroxide; Alkaline earth metal hydroxides such as barium hydroxide and calcium hydroxide; Alkali metal alcoholates, for example potassium ethanolate and sodium propanolate; as well as various organic bases such as piperidine, diethanolamine and N-methylglutamine.
- alkali metal hydroxides including potassium hydroxide, sodium hydroxide and lithium hydroxide
- Alkaline earth metal hydroxides such as barium hydroxide and calcium hydroxide
- Alkali metal alcoholates for example potassium ethanolate and sodium propanolate
- various organic bases such as piperidine, diethanolamine and N-methylglutamine.
- acid addition salts can be formed by Compounds with pharmaceutically acceptable organic and inorganic acids, e.g.
- hydrogen halides such as hydrogen chloride, hydrogen bromide or hydrogen iodide, other mineral acids and their corresponding salts such as sulfate, nitrate or phosphate and the like 5 as well as alkyl and monoarylsulfonates such as ethanesulfonate, toluenesulfonate and benzenesulfonate, and other organic acids and their corresponding salts such as acetate, trifluoroacetate, tartrate, maleate, succinate, citrate, benzoate, salicylate, ascorbate and the like. accordingly
- pharmaceutically acceptable acid addition salts of the compounds of formula I include the following: acetate, adipate, alginate, arginate, aspartate, benzoate, benzenesulfonate (besylate), bisulfate, bisulfite, bromide, butyrate, camphorate, camphor sulfonate, caprylate, chloride,
- the base salts of the 0 compounds according to the invention include aluminum, ammonium, calcium, copper, iron (III), iron (II), lithium, magnesium, manganese (III), manganese (II), Potassium, sodium and zinc salts, but this is not intended to be a limitation.
- Preferred among the salts mentioned above are ammonium; the alkali metal salts sodium and potassium, and the alkaline earth metal salts
- Amines, cyclic amines and basic ion exchange resins e.g.
- Hydrabamine isopropylamine, lidocaine, lysine, meglumine, N-methyl-D-glucamine, morpholine, piperazine, piperidine, polyamine resins, procain, purines,
- Contain groups can be with agents such as (C 1 -C 4 ) alkyl halides, for example methyl, ethyl, isopropyl and tert-butyl chloride, bromide and iodide;
- Di (-C 4 ) alkyl sulfates such as dimethyl, diethyl and diamyl sulfate; (C 10 -
- Ci 8 alkyl halides, for example decyl, dodecyl, lauryl, myristyl and
- the above-mentioned pharmaceutical salts which are preferred include acetate, trifluoroacetate, besylate, citrate, fumarate, gluconate, hemisuccinate, hippurate, hydrochloride, hydrobromide, isethionate, mandelate, meglumine, nitrate, oleate, phosphonate, pivalate, sodium phosphate, stearate, Sulfate, sulfosalicylate, tartrate, thiomalate, tosylate and tromethamine, but this is not intended to be a limitation.
- the acid addition salts of basic compounds of formula I are prepared in that the free base form with a sufficient Contact the amount of the desired acid, whereby the salt is prepared in the usual way.
- the free base can be regenerated in a conventional manner by contacting the salt form with a base and isolating the free base.
- the free base forms differ in a sense from their corresponding salt forms in terms of certain physical properties such as solubility in polar solvents; in the context of the invention, however, the salts otherwise correspond to their respective free base forms.
- the pharmaceutically acceptable base addition salts of the compounds of the formula I are formed with metals or amines such as alkali metals and alkaline earth metals or organic amines.
- metals are sodium, potassium, magnesium and calcium.
- Preferred organic amines are N.N'-dibenzylethylenediamine, chloroprocaine, choline, diethanolamine, ethylenediamine, N-methyl-D-glucamine and procaine.
- the base addition salts of acidic compounds according to the invention are prepared by contacting the free acid form with a sufficient amount of the desired base, whereby the salt is prepared in the usual way.
- the free acid can be regenerated in a conventional manner by contacting the salt form with an acid and isolating the free acid.
- the free acid forms differ in a sense from their corresponding salt forms in terms of certain physical properties such as solubility in polar solvents; in the context of the invention, however, the salts otherwise correspond to their respective free acid forms.
- a compound according to the invention contains more than one group which can form such pharmaceutically acceptable salts, this includes
- Invention also multiple salts.
- Typical multiple salt forms include, for example, bitartrate, diacetate, difumarate, dimeglumine, Diphosphate, disodium and trihydrochloride, but this is not intended to be a limitation.
- the expression 5 “pharmaceutically acceptable salt” in the present context is to be understood as an active substance which contains a compound of the formula I in the form of one of its salts, in particular when this salt form is the active substance compared to the free form of the active substance or
- the pharmaceutically acceptable salt form of the active ingredient can only give this active ingredient a desired pharmacokinetic property
- the invention further relates to medicaments containing at least 0 a compound of the formula I and / or its pharmaceutically usable derivatives, solvates and stereoisomers, including their mixtures in all ratios, and, if appropriate, carriers and / or auxiliaries.
- Pharmaceutical formulations can be administered in the form of dose units containing a predetermined amount of active ingredient per dose unit.
- Such a unit can contain, for example, 0.5 mg to 1 g, preferably 1 mg to 700 mg, particularly preferably 5 mg to 100 mg Q of a compound according to the invention, depending on the condition of the disease treated, the route of administration and the age, weight and condition of the patient , or pharmaceutical formulations can be presented in the form of dose units containing a predetermined amount of active ingredient per dose unit.
- Preferred dosage unit formulations are those that have a daily dose or partial dose as indicated above, or a corresponding fraction thereof Active ingredient included.
- such pharmaceutical formulations can be produced using one of the methods generally known in the pharmaceutical field.
- compositions can be administered for administration by any suitable route, for example by oral (including buccal or sublingual), rectal, nasal, topical (including buccal, sublingual or transdermal),
- vaginal or parenteral including subcutaneous, intramuscular, intravenous, or intradermal) routes.
- Such formulations can be prepared by any method known in the pharmaceutical art, for example by the
- ⁇ g of active ingredient is brought together with the carrier (s) or auxiliary (s).
- compositions adapted for oral administration can be used as separate units, e.g. Capsules or tablets; powder
- the active ingredient component can be combined with an oral, non-toxic and pharmaceutically acceptable inert carrier, such as Q, for example ethanol, glycerol, water and others. Powders are made by crushing the compound to an appropriate fine size and mixing it with a similarly crushed pharmaceutical carrier such as an edible carbohydrate such as starch or mannitol. A flavor, preservative, dispersant and color may also be present.
- an oral, non-toxic and pharmaceutically acceptable inert carrier such as Q, for example ethanol, glycerol, water and others.
- Powders are made by crushing the compound to an appropriate fine size and mixing it with a similarly crushed pharmaceutical carrier such as an edible carbohydrate such as starch or mannitol.
- a flavor, preservative, dispersant and color may also be present.
- Capsules are made by making a powder mixture as described above and filling shaped gelatin shells with it.
- Lubricants and lubricants such as highly disperse silica, talc, magnesium stearate, calcium stearate or polyethylene glycol in solid form can be added to the powder mixture before the filling process.
- suitable binding agents can also be incorporated into the mixture.
- suitable binders include starch, gelatin, natural sugars such as glucose or beta-lactose, sweeteners from corn, natural and synthetic gums such as acacia, tragacanth or sodium alginate, carboxymethyl cellulose, polyethylene glycol, waxes, etc.
- Lubricants include sodium oleate, sodium stearate, magnesium stearate, sodium benzoate, sodium acetate, sodium chloride, etc.
- Disintegrants include, but are not limited to, starch, methyl cellulose, agar, bentonite, xanthan gum, and the like.
- the tablets are formulated by, for example, producing a powder mixture, granulating or pressing them dry, adding a lubricant and a disintegrant and compressing the whole thing into tablets.
- a powder mixture is produced by the compound, which has been comminuted in a suitable manner, with a diluent or a base, as described above, and optionally with a binder, such as, for example, carboxymethyl cellulose, an alginate, gelatin or polyvinylpyrrolidone, a solution-reducing agent, such as, for example, paraffin Absorption accelerator, such as a quaternary salt and / or an absorbent, such as bentonite, kaolin or dicalcium phosphate, is mixed.
- a binder such as, for example, carboxymethyl cellulose, an alginate, gelatin or polyvinylpyrrolidone
- a solution-reducing agent such as, for example, paraffin Absorption accelerator, such as a quaternary salt and / or an absorb
- the powder mixture can be granulated by mixing it with a binder such as syrup, starch paste, Acadia mucilage or solutions made from cellulose or polymer. materials are wetted and pressed through a sieve.
- a binder such as syrup, starch paste, Acadia mucilage or solutions made from cellulose or polymer. materials are wetted and pressed through a sieve.
- the powder mixture can be run through a tabletting machine, resulting in irregularly shaped lumps which are broken up into granules.
- the granules can be greased by adding 5 stearic acid, a stearate salt, talc or mineral oil to prevent sticking to the tablet molds. The greased mixture is then compressed into tablets.
- the compounds of the invention can also be used with a free-flowing inert
- a clear or opaque protective layer consisting of a shellac seal, a layer of sugar or polymer
- a 5 material and a gloss layer of wax may be present. Dyes can be added to these coatings in order to be able to differentiate between different dosage units.
- Oral liquids such as solution, syrups and elixirs, can be prepared in the form of dosage units so that a given quantity contains a given amount of the compound.
- Syrups can be made by dissolving the compound in an aqueous solution with a suitable taste, while elixirs are made using a non-toxic alcoholic vehicle.
- Suspensions can be formulated by dispersing the compound in a non-toxic vehicle.
- Solubilizers and emulsifiers such as, for example, ethoxylated isostearyl alcohols and polyoxyethylene sorbitol ethers, Q preservatives, flavor additives, such as, for example, peppermint oil or natural sweeteners or saccharin or other artificial sweeteners and the like. can also be added.
- Dosage unit formulations for oral administration can be any suitable dosage unit formulations for oral administration.
- the formulation can also be prepared so that the release is prolonged or delayed is, such as by coating or embedding particulate material in polymers, wax, etc.
- the compounds of the formula I and salts, solvates and physiologically functional derivatives thereof can also be in the form of liposome delivery systems, such as administer small unilamellar vesicles, large unilamellar vesicles and multilamellar vesicles.
- Liposomes can be made from various phospholipids, e.g. Cholesterol, stearylamine or phosphatidylcholines.
- the compounds of the formula I and the salts, solvates and physiologically functional derivatives thereof can also be supplied using monoclonal antibodies as individual carriers to which the compound molecules are coupled.
- the compounds can also be coupled with soluble polymers as targeted drug carriers.
- Such polymers can include polyvinylpyrrolidone, pyran copolymer, polyhydroxypropyl methacrylamide phenol, polyhydroxyethylaspartamide phenol or polyethylene oxide polylysine substituted with palmitoyl residues.
- the compounds can be linked to a class of biodegradable polymers suitable for achieving controlled release of a drug, e.g.
- Polylactic acid Polyepsilon-caprolactone, polyhydroxybutyric acid, polyorthoesters, polyacetals, polydihydroxy-pyrans, polycyanoacrylates and cross-linked or amphipatic block copolymers of hydrogels.
- Formulations can be given as stand-alone patches for prolonged, close contact with the recipient's epidermis.
- the active ingredient can be supplied from the patch by means of iontophoresis, as generally described in Pharmaceutical Research, 3 (6), 318 (1986).
- Pharmaceutical compounds adapted for topical administration can be formulated as ointments, creams, suspensions, lotions, powders, solutions, pastes, gels, sprays, aerosols or oils.
- the formulations are preferably applied as a topical ointment or cream.
- the active ingredient can be either paraffinic or water-miscible
- cream base can be used.
- the active ingredient can be formulated into a cream with an oil-in-water cream base or a water-in-oil base.
- compositions adapted for topical application to the eye include eye drops, the active ingredient being dissolved or suspended in a suitable carrier, in particular an aqueous solvent.
- suitable carrier in particular an aqueous solvent.
- Pharmaceutical formulations adapted for topical application in the mouth include lozenges, lozenges and mouthwashes.
- compositions 5 adapted for rectal administration can be administered in the form of suppositories or enemas.
- compositions adapted for nasal administration in which the carrier substance is a solid, contain a coarse Q powder with a particle size, for example in the range of 20-500 micrometers, which is administered in the manner in which snuff is taken up, ie by Quick inhalation via the nasal passages from a container with the powder held close to the nose.
- Suitable formulations for administration as a nasal spray or 5 Nasal drops with a liquid as a carrier substance comprise active ingredient solutions in water or oil.
- compositions adapted for administration by inhalation comprise fine particulate dusts or mists which can be generated by means of various types of pressurized metering dispensers with aerosols, nebulizers or insufflators.
- compositions adapted for vaginal administration can be administered as pessaries, tampons, creams, gels, pastes, foams or spray formulations.
- a [- Pharmaceutical formulations adapted for parenteral administration include aqueous and non-aqueous sterile injection solutions which contain antioxidants, buffers, bacteriostatics and solutes, by which the formulation is rendered isotonic with the blood of the recipient to be treated; and aqueous and non-aqueous sterile suspensions, which may contain suspending agents and thickeners.
- the formulations can be presented in single-dose or multi-dose containers, for example sealed ampoules and vials, and stored in a freeze-dried (lyophilized) state, so that only the addition of the sterile carrier liquid, for example water for injections, is required immediately before use.
- Injection solutions and suspensions prepared according to the recipe can be made from sterile powders, granules and tablets. It is understood that the formulations may contain, in addition to the above-mentioned components, other means customary in the art with regard to the respective type of formulation; for example, formulations suitable for oral administration may contain 5 flavorings.
- a therapeutically effective amount of a compound of formula I depends on a number of factors, including, for example, the age and weight of the animal, the exact condition of the disease that requires treatment, and its severity, the nature of the formulation
- neoplastic growth e.g. Colon or breast cancer
- the actual daily amount would usually be between 70 and 700 mg
- a r- this amount can be given as a single dose per day or more usually in a series of divided doses (such as two, three, four, five or six) per day so that the total daily dose is the same.
- An effective amount of a salt or solvate or a physiologically functional derivative thereof can be determined as a proportion of the effective amount of the compound according to the invention. It is believed that similar dosages are suitable for the treatment of the other conditions mentioned above.
- the invention further relates to medicaments containing at least one compound of the formula I and / or their pharmaceutically usable derivatives, solvates and stereoisomers, including their mixtures in all ratios, and at least one further active pharmaceutical ingredient.
- the invention also relates to a set (kit) consisting of separate packs of (a) an effective amount of a compound of the formula I and / or its pharmaceutically usable derivatives, solvates and stereo ⁇
- 35 isomers, including their mixtures in all proportions, and (b) an effective amount of another drug ingredient.
- the set contains suitable containers, such as boxes or cartons, individual bottles, bags or ampoules.
- suitable containers such as boxes or cartons, individual bottles, bags or ampoules.
- the set can e.g. separate
- Derivatives, solvates and stereoisomers, including their mixtures in all proportions, and an effective amount of another active pharmaceutical ingredient are dissolved or in lyophilized form.
- the present compounds are suitable as pharmaceutical active substances for mammals, in particular for humans, in the treatment of tyrosine kinase-related diseases.
- diseases include tumor cell proliferation, pathological neovascularization (or angiogenesis) that promotes the growth of solid tumors, neovascularization (diabetic retinopathy, age-related macular degeneration and the like) and inflammation (psoriasis, rheumatoid arthritis and the like).
- the present invention encompasses the use of the compounds of
- Preferred carcinomas for the treatment come from the group of brain carcinoma, urogenital tract carcinoma, carcinoma of the lymphatic system, gastric carcinoma, larynx carcinoma and lung carcinoma.
- Another group of preferred forms of cancer are monocyte leukemia, lung adenocarcinoma, small cell lung carcinoma, pancreatic cancer, glioblastoma and breast carcinoma.
- the use of the compounds according to the invention and / or their physiologically acceptable salts and solvates for the manufacture of a medicament for the treatment or prevention of a disease in which angiogenesis 5 is involved is also included.
- One such disease in which angiogenesis is involved is an eye disorder such as retinal vascularization, diabetic retinopathy, age-related macular degeneration and the like.
- Such inflammatory diseases include, for example, rheumatoid arthritis, psoriasis, contact dermatitis, late-type hypersensitivity reaction and the like.
- the compounds of the formula I and / or their physiologically acceptable salts and solvates for the preparation of a medicament for the treatment or prevention of a tyrosine kinase-related disease or a tyrosine kinase-related illness in a mammal, whereby this method involves a sick mammal such treatment requires a therapeutically effective amount of a compound of the invention.
- the therapeutic amount depends on the respective disease and can be determined by the person skilled in the art without any great effort.
- the present invention also encompasses the use of compounds 0 of the formula I and / or their physiologically acceptable salts and solvates for the production of a medicament for the treatment or prevention of retinal vascularization.
- Methods for the treatment or prevention of eye diseases such as diabetic retinopathy and age-related macular degeneration are also part of the invention.
- tyrosine kinase-related diseases or conditions refers to pathological conditions which are dependent on the activity of one or more tyrosine kinases.
- the tyrosine kinases are either directly or indirectly on the signal transduction pathways of various cell activ
- tyrosine kinase activity 10 activities, including proliferation, adhesion and migration as well as differentiation.
- Diseases associated with tyrosine kinase activity include tumor cell proliferation, pathological neovascularization that promotes the growth of solid tumors, neovascularization in the
- a ⁇ eye diabetic retinopathy, age-related macular degeneration and the like
- inflammation psoriasis, rheumatoid arthritis and the like.
- the compounds of formula I can be administered to patients for the treatment of cancer.
- the present compounds inhibit tumor angiogenesis and thus influence the growth of tumors (J. Rak et al. Cancer Research, 55: 4575-4580, 1995).
- the angiogenesis-inhibiting properties of the present compounds of the formula I 5 are also suitable for the treatment of certain forms of blindness which are associated with retinal vascularization.
- the compounds of the formula I are also suitable for the treatment of certain bone pathologies such as osteosarcoma, osteoarthritis and Q rickets, which is also known under the name of oncogenic osteomalacia (Hasegawa et al., Skeletal Radiol. 28, pp. 41-45, 1999; Gerber et al., Nature Medicine, Vol.
- the present compounds are also suitable for treatment and prevention of suffering associated with Bone resorption is related, such as osteoporosis and Paget's disease.
- the compounds can also be used to reduce or prevent tissue damage that occurs after cerebral ischemic events such as stroke (Drug News Perspect 11: 265-270 (1998); J. Clin. Invest. 104: 1613-1620 (1999)).
- the invention thus relates to the use of compounds of the formula I and their pharmaceutically usable derivatives, solvates and stereoisomers, including their mixtures in all A c ratios, for the manufacture of a medicament for the treatment of diseases in which the inhibition, regulation and / or modulation of signal transduction by kinases plays a role.
- Kinases are preferably selected from the group of 0 tyrosine kinases and Raf kinases.
- the tyrosine kinases are preferably TIE-2, VEGFR, PDGFR, FGFR and / or FLT / KDR. 5 Preferred is the use of compounds of formula I, as well as their pharmaceutically usable derivatives, solvates and stereoisomers, including their mixtures in all ratios, Q for the preparation of a medicament for the treatment of diseases which are influenced by the compounds according to claim 1 by inhibiting the tyrosine kinases become.
- the disease is a solid tumor.
- the solid tumor is preferably selected from the group of tumors of the squamous epithelium, the blisters, the stomach, the kidneys, the head and neck, the esophagus, the cervix, the thyroid, the intestine, the liver, the brain, the prostate, the Urogenital tract, lymphatic system, stomach, larynx and / or lungs.
- the solid tumor is also preferably selected from the group of lung adenocarcinoma, small cell lung carcinoma, pancreatic cancer, glioblastoma, colon carcinoma and breast carcinoma.
- a tumor of the blood and immune system preferably for the treatment of a tumor selected from the group of acute myelotic leukemia, chronic myelotic leukemia, acute lymphatic leukemia and / or chronic lymphatic leukemia.
- the invention further relates to the use of
- the disease is preferably an eye disease.
- the invention further relates to the use for the treatment of retinal vascularization, diabetic retinopathy, and age-related
- the inflammatory disease is preferably selected from the group rheumatoid arthritis, psoriasis, contact dermatitis and late-type hypersensitivity reaction.
- the invention further relates to the use of the compounds according to the invention for the treatment of bone pathologies, the bone pathology coming from the group of osteosarcoma, osteoarthritis and rickets.
- the compounds of formula I are suitable for the manufacture of a medicament for the treatment of diseases which are caused, mediated and / or propagated by Raf kinases, the Raf kinase
- a j - is selected from the group consisting of A-Raf, B-Raf and Raf-1.
- Preferred is the use for the treatment of diseases, preferably from the group of hyperproliferative and non-hyperproliferative diseases. These are cancerous or non-cancerous 0 diseases.
- the non-cancerous diseases are selected from the group consisting of psoriasis, arthritis, inflammation, endometriosis, scarring, benign prostatic hyperplasia, immunological diseases, 5 autoimmune diseases and immunodeficiency diseases.
- the cancerous diseases are selected from the group consisting of brain cancer, lung cancer, squamous cell, bladder, 0 gastric cancer, pancreatic cancer, liver cancer, renal cancer, colorectal cancer, breast cancer, head cancer, neck cancer, oesophageal cancer, gynecological cancer, thyroid cancer, lymphoma, chronic leukemia and acute leukemia. 5
- the compounds of the formula I can also be used together with other well-known therapeutic agents which, owing to their particular suitability for the Treated ailments are selected to be administered.
- combinations that contain antiresorptive bisphosphonates, such as alendronate and risedronate, integrin blockers (as defined below), such as ⁇ v? 3 antagonists would be beneficial
- Conjugated estrogens such as Prempro®, Premarin® and Endometrion® used in hormone therapy; contain selective estrogen receptor modulators (SERMs) such as raloxifene, droloxifene, CP-336,156 (Pfizer) and lasofoxifene, cathepsin K inhibitors and ATP proton pump inhibitors.
- SERMs selective estrogen receptor modulators
- the present compounds are also suitable for combination with known anti-cancer agents.
- known anti-cancer agents include the following: estrogen receptor modulators, androgen receptor modulators, retinoid receptor modulators, cytotoxics, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiproliferative, antiprolife
- AC agents prenyl protein transferase inhibitors, HMG-CoA reductase inhibitors, HIV protease inhibitors, reverse transcriptase inhibitors and other angiogenesis inhibitors.
- the present compounds are particularly suitable for joint use with radiotherapy.
- the synergistic effects of inhibiting VEGF in combination with 0 radiotherapy have been described in the specialist field (see WO 00/61186).
- Estrogen receptor modulators refers to compounds which interfere with or inhibit the binding of estrogen to the receptor, regardless of how this is done.
- the estrogen receptor modulators include, for example, tamoxifen, raloxifene, idoxifene, LY353381, LY 117081, toremifene, fulvestrant, 4- [7- (2,2-dimethyl-1-oxopropoxy-4-methyl-2- [4- [2- (1 - piperidinyl) ethoxy] phenyl] -2H-1-0 benzopyran-3-yl ] phenyl-2,2-dimethylpropanoate, 4,4-dihydroxybenzophenone-2,4-dinitrophenylhydrazone, and SH646, which, however, is not intended to be a limitation.
- “Androgen receptor modulators” refers to compounds which interfere with the binding of androgens to the receptor or inhibit them,
- Androgen receptor modulators include, for example, finasteride and others 5 ⁇ -reductase inhibitor, nilutamide, flutamide, bicalutamide, liarozole and abiraterone acetate.
- Retinoid receptor modulators refers to compounds that interfere with or inhibit the binding of retinoids to the receptor, regardless of how this is done.
- retinoid receptor modulators include, for example, bexarotene, tretinoin, 13-cis-retinoic acid, 9-cis Retinoic acid, ⁇ -difluoromethylornithine, ILX23-7553, trans-N- (4'-hydroxyphenyl) retinamide and N-4-carboxyphenylretinamide.
- Cytotoxics refers to compounds that are primarily affected by cell function leading to cell death or inhibit or interfere with cell myosis, including alkylating agents, tumor necrosis factors, intercalating agents, microtubulin inhibitors and topoisomerase inhibitors.
- Cytotoxic agents include, for example tirapazimine, sertenef, cachectin, ifosfamide, tasonermin, lonidamine, carboplatin, altretamine, prednimustine, Dibromdulcit, ranimustine, fotemustine, nedaplatin, oxaliplatin, temozolomide, heptaplatin, estramustine, improsulfan tosylate, trofosfamide, nimustine, Dibrospidium- chloride, Pumitepa, lobaplatin, satraplatin, profiromycin, cisplatin, irofulven, dexifosfamide, cis-amine dichloro (2-methylpyridine) platinum, benzylguanine, glufosfamide, GPX100, (trans, trans, trans) -bis-mu- (hexane-1, 6 -diamine) -mu-
- MEN 10755 and 4-desmethoxy-3-desamino-3-aziridinyl-4-methylsulfonyl-daunorubicin see WO 00/50032, which, however, is not intended to be a restriction.
- microtubulin inhibitors include, for example, paclitaxel, vindesine sulfate, S ' ⁇ ' - Dideshydro ⁇ '- deoxy- ⁇ '-norvincaleukoblastin, docetaxol, rhizoxin, dolastatin, mivobulin isethionate, auristatin, cemadotin, RPR109881, BMS184476, vinflunine, cryptophycin, 2,3,4,5,6-pentafluoro-N- (3-fluoro-4-methoxyphenyl) benzenesulfonamide, anhydrovinblastine, N, N-dimethyl-L-valyl-L-valyl-N -methyl-L-valyl-L-prolyl-L-prolin-t-butylamide, TDX258 and BMS188797.
- paclitaxel vindesine sulfate
- 5 topoisomerase inhibitors are, for example, topotecan, hycaptamine, irinotecan, rubitecan, 6-ethoxypropionyl-3 ', 4'-O-exo-benzylidene-chartreusin, 9-methoxy-N, N-dimethyl-5-nitropyrazolo [3,4 , 5-kl] acridin-2- (6H) propanamine, 1-amino-9-ethyl-5-fluoro-2,3-dihydro-9-hydroxy-4-methyl-
- antiproliferative agents include antisense RNA and DNA oligonucleotides such as G3139, ODN698, RVASKRAS, GEM231 and 0 INX3001, as well as antimetabolites such as enocitabine, Carmofur, Tegafur, pentostatin, doxifluridine, trimetrexate, flitarabinabin, fludarabin, and fludarabin -ocfosfate, fosteabin sodium hydrate, raltitrexed, paltitrexid, emitefur, tiazofurin, decitabine, nolatrexed, pemetrexed, nelzarabine, 2-deoxy-2'-methylididine, 2'-fluoromethylene-2 , -deoxycytidine, N- [5- ( 3- 5 dihydrobenzofuryl) sulfonyl] -N '- (3,4-dichlorophenyl) urea
- antiproliferative agents also include monoclonal antibodies against growth factors other than those already mentioned under the “angiogenesis inhibitors”, such as trastuzumab, and tumor suppressor genes, such as p53, which can be released via recombinant virus-mediated gene transfer (see, for example, US Pat. No. 6,069,134 ).
- the invention further relates to the use of the compounds of the formula I for the manufacture of a medicament for the treatment of diseases, the disease being characterized by disturbed angiogenesis.
- the disease is preferably cancer.
- the disturbed angiogenesis preferably results from an impaired VEGFR-1, VEGFR-2 and / or VEGFR-3 activity. It is therefore particularly preferred to use the compounds according to the invention for the manufacture of a medicament for inhibiting VEGFR-2 activity.
- VEGF receptor kinase activity is determined by incorporating radioactively labeled phosphate in 4: 1 polyglutamic acid / tyrosine substrate (pEY).
- pEY polyglutamic acid / tyrosine substrate
- the phosphorylated pEY product is held on a filter membrane and the incorporation of the radioactively labeled phosphate is quantified by scintillation counting.
- the intracellular tyrosine kinase domains of human KDR (Terman, BI et al. Oncogene (1991) Vol. 6, pp. 1677-1683.) And Flt-1 (Shibuya, M. et al. Oncogene (1990) Vol. 5 , Pp. 519-524) were cloned as glutathione-S-transferase (GST) gene fusion proteins. This was done by cloning the cytoplasmic domain of the KDR kinase as a read-frame fusion at the carboxy terminus of the GST gene. The soluble recombinant GST-kinase domain fusion proteins were found in Spodoptera frugiperda (Sf21) insect cells (Invitrogen)
- Enzyme dilution buffer 10 50 mM Tris, pH 7.4, 0.1 M NaCl, 1 mM DTT, 10% glycerin, 100 mg / ml BSA.
- the supernatant was then passed through a glutathione-Sepharose acid (Pharmacia) equilibrated with lysis buffer 0 and washed with 5 volumes of the same buffer and then 5 volumes of washing buffer.
- the recombinant GST-KDR protein was eluted with wash buffer / 10 mM reduced glutathione (Sigma) and counteracted
- Method B VEGF receptor kinase assay 1. Add 5 ⁇ ⁇ inhibitor or control in 50% DMSO to the assay. 2. Add 35 ⁇ l reaction mixture containing 5 // I 10 * reaction buffer, 5 l 25 mM ATP / 10 Ci [ 33 P] ATP (Amersham) and 5 ⁇ ⁇ 10 * substrate. 5 3. Start the reaction by adding 10 ⁇ ⁇ KDR (25 nM) in enzyme dilution buffer. 4. Mix and incubate for 15 minutes at room temperature. 5. Stop the reaction by adding 50 ⁇ ⁇ stop solution. Incubate at 6.degree. C. for 15 minutes. 7. Transfer 90 ⁇ l aliquot to filter plate. 8.
- VEGF vascular endothelial growth factor
- bFGF basic fibroblast growth factor
- endothelial Growth Medium EGM; Clonetics
- NUNCLON 96-well polystyrene tissue culture plates (NUNC # 167008).
- Dulbecco modified Eagle medium with 1 g / ml glucose (DMEM with low glucose content; Mediatech) plus 10% (v / v) fetal
- VEGF 165 500 ng / ml; R&D Systems
- bFGF 10 ng / ml; R&D Systems
- HUVEC single cell layers kept in EGM are harvested by trypsin treatment and inoculated in a density of 4000 cells per 100 ⁇ ⁇ assay medium per well in 96-well plates.
- the growth of the cells is stopped for 24 hours at 37 ° C. in a humid atmosphere containing 5% CO 2 .
- Procedure 2 The growth stop medium is replaced with 100 ⁇ ⁇ assay medium, which is either the constituent (0.25% [v / v] DMSO) or the desired one
- the cells are added by adding 10 ⁇ l assay medium, 10 * VEGF solution or 10 * bFGF solution
- the cells are then incubated at 37 ° C / 5% CO 2 .
- the medium is suctioned off and the cells are washed twice with washing medium (400 ⁇ l / well, then 200 ⁇ l / well).
- the washed, adherent cells are then solubilized by adding cell lysis solution (100 ⁇ l / well) and heating to 37 ° C. for 30 minutes.
- the cell lysates are transferred to 7 ml glass scintillation tubes containing 150 ul water.
- the scintillation cocktail (5 ml / tube) is added and the radioactivity associated with the cells is determined by liquid scintillation spectroscopy.
- the compounds of the formula I are VEGF inhibitors and are therefore suitable for inhibiting angiogenesis, such as in the treatment of eye diseases, for example diabetic retinopathy, and for the treatment of carcinomas, for example solid tumors.
- the present compounds inhibit VEGF-stimulated mitogenesis of cultured human vascular endothelial cells with HK50 values of 0.01-5.0 ⁇ M.
- These compounds are compared to related tyrosine kinases (e.g. FGFR1 and Src family; the relationship between Src kinases and VEGFR kinases see also Eliceiri et al., Molecular Cell, Vol. 4, pp. 915-924, December 1999) also selectively.
- the 77 £ -2 tests can be carried out, for example, analogously to the methods given in WO 02/44156.
- the assay determines the inhibitory activity of the substances to be tested in the phosphorylation of the substrate poly (Glu, Tyr) by Tie-2-kinase in the presence of radioactive 33 P-ATP.
- the phosphorylated substrate binds to the surface of a "flashplate" microtiter plate during the incubation period. After removing the reaction mixture, it is washed several times and then the radioactivity is measured on the surface of the microtiter plate. An inhibiting effect of the substances to be measured results in a lower radioactivity compared to an undisturbed enzymatic reaction.
- customary work-up means: if necessary, water is added, if necessary, depending on the constitution of the end product, the pH is between 2 and 10, , extracted with ethyl acetate or dichloromethane, separated, the organic phase dried over sodium sulfate, evaporated and purified by chromatography on 5 silica gel and / or by crystallization. Rf values on silica gel; Mobile solvent: ethyl acetate / methanol 9: 1.
- Mass spectrometry (MS): El (electron impact ionization) M + FAB (Fast Atom Bombardment) (M + H) + Q ESI (Electrospray ionization) (M + H) + APCI-MS (atmospheric pressure chemical ionization - mass spectrometry) ( M + H) + .
- Chlorine-1-methyl-pyridinium iodide combined and heated at 50 ° C for 2 h.
- the desired fractions are pooled, concentrated and freeze-dried.
- Example A Injection glasses
- a solution of 100 g of an active ingredient of the formula I and 5 g of disodium hydrogen phosphate is adjusted to pH 6.5 in 3 l of double-distilled water with 2N hydrochloric acid, sterile filtered, filled into injection glasses, lyophilized under sterile conditions and sealed sterile. Each injection jar contains 5 mg of active ingredient.
- a mixture of 20 g of an active ingredient of the formula I is melted with 100 g of soy lecithin and 1400 g of cocoa butter, poured into molds and allowed to cool. Each suppository contains 20 mg of active ingredient.
- a solution is prepared from 1 g of an active ingredient of the formula I, 9.38 g of NaH 2 PO 4 .2H 2 O, 28.48 g of Na 2 HPO. 12H 2 O and 0.1 g of benzalkonium chloride in 940 ml double distilled water. It is adjusted to pH 6.8, made up to 1 l and sterilized by irradiation. This solution can be used in the form of eye drops.
- Example D ointment
- Example E tablets
- a mixture of 1 kg of active ingredient of the formula I, 4 kg of lactose, 1, 2 kg of potato starch, 0.2 kg of talc and 0.1 kg of magnesium stearate is more common
- each tablet contains 10 mg of active ingredient.
- Example F coated tablets
- Example E tablets are pressed, which are then coated in a conventional manner with a coating of sucrose, potato starch, talc, tragacanth and dye.
- Example G capsules
- each capsule contains 20 mg of the active ingredient.
- a solution of 1 kg of active ingredient of the formula I in 60 l of double-distilled water is sterile filtered, filled into ampoules, lyophilized under sterile conditions and sealed sterile. Each ampoule contains 10 mg of active ingredient.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Physical Education & Sports Medicine (AREA)
- Immunology (AREA)
- Rheumatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Dermatology (AREA)
- Reproductive Health (AREA)
- Hematology (AREA)
- Pain & Pain Management (AREA)
- Gynecology & Obstetrics (AREA)
- Pregnancy & Childbirth (AREA)
- Ophthalmology & Optometry (AREA)
- Endocrinology (AREA)
- Urology & Nephrology (AREA)
- Oncology (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description








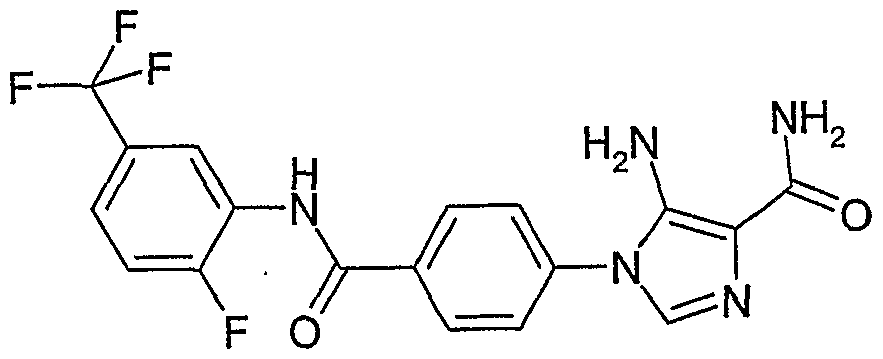

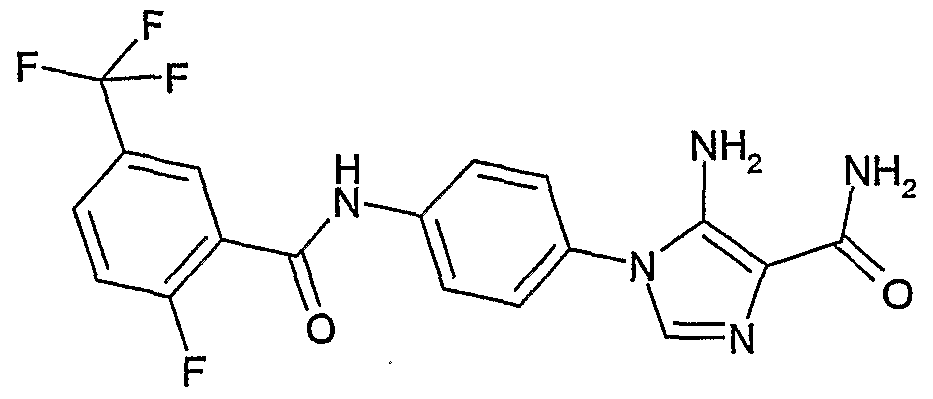
Claims






Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP05716076A EP1761503A2 (de) | 2004-03-29 | 2005-03-15 | Imidazolderivate als hemmer von tyrosinkinase |
| CA002561585A CA2561585A1 (en) | 2004-03-29 | 2005-03-15 | Imidazol derivatives |
| JP2007505422A JP2007530609A (ja) | 2004-03-29 | 2005-03-15 | イミダゾール誘導体 |
| AU2005231907A AU2005231907A1 (en) | 2004-03-29 | 2005-03-15 | Imidazol derivatives as tyrosine kinase inhibitors |
| US10/593,295 US20070225347A1 (en) | 2004-03-29 | 2005-03-15 | Imidazole Derivatives |
| BRPI0508881-0A BRPI0508881A (pt) | 2004-03-29 | 2005-03-15 | derivados de imidazol |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102004015099.0 | 2004-03-29 | ||
| DE102004015099A DE102004015099A1 (de) | 2004-03-29 | 2004-03-29 | Imidazolderivate |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2005097755A2 true WO2005097755A2 (de) | 2005-10-20 |
| WO2005097755A3 WO2005097755A3 (de) | 2006-03-09 |
Family
ID=34981611
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2005/002746 WO2005097755A2 (de) | 2004-03-29 | 2005-03-15 | Imidazolderivate als hemmer von tyrosinkinase |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US20070225347A1 (de) |
| EP (1) | EP1761503A2 (de) |
| JP (1) | JP2007530609A (de) |
| KR (1) | KR20060132965A (de) |
| CN (1) | CN1938282A (de) |
| AR (1) | AR048327A1 (de) |
| AU (1) | AU2005231907A1 (de) |
| BR (1) | BRPI0508881A (de) |
| CA (1) | CA2561585A1 (de) |
| DE (1) | DE102004015099A1 (de) |
| RU (1) | RU2006138150A (de) |
| WO (1) | WO2005097755A2 (de) |
| ZA (1) | ZA200608998B (de) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006076592A1 (en) * | 2005-01-14 | 2006-07-20 | Cgi Pharmaceuticals, Inc. | 1,3 substituted diaryl ureas as modulators of kinase activity |
| WO2007115670A1 (en) * | 2006-04-12 | 2007-10-18 | Merck Patent Gmbh | N-oxides of heterocyclic substituted bisarylureas for treating kinase-mediated diseases |
| US7777040B2 (en) | 2005-05-03 | 2010-08-17 | Cgi Pharmaceuticals, Inc. | Certain substituted ureas, as modulators of kinase activity |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2729012A1 (en) | 2008-06-27 | 2009-12-30 | Amgen Inc. | Ang-2 inhibition to treat multiple sclerosis |
| JP2011057661A (ja) * | 2009-08-14 | 2011-03-24 | Bayer Cropscience Ag | 殺虫性カルボキサミド類 |
| CN103910714A (zh) * | 2013-01-09 | 2014-07-09 | 天津泰瑞倍药研科技有限公司 | 氟代环丁烷基咪唑类化合物 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996040673A1 (en) * | 1995-06-07 | 1996-12-19 | Sugen, Inc. | Novel urea- and thiourea-type compounds |
| WO2003072541A2 (en) * | 2001-09-27 | 2003-09-04 | Smithkline Beecham Corporation | Chemical compounds |
-
2004
- 2004-03-29 DE DE102004015099A patent/DE102004015099A1/de not_active Withdrawn
-
2005
- 2005-03-15 AU AU2005231907A patent/AU2005231907A1/en not_active Abandoned
- 2005-03-15 KR KR1020067020089A patent/KR20060132965A/ko not_active Application Discontinuation
- 2005-03-15 EP EP05716076A patent/EP1761503A2/de not_active Withdrawn
- 2005-03-15 JP JP2007505422A patent/JP2007530609A/ja active Pending
- 2005-03-15 US US10/593,295 patent/US20070225347A1/en not_active Abandoned
- 2005-03-15 WO PCT/EP2005/002746 patent/WO2005097755A2/de active Application Filing
- 2005-03-15 CN CNA200580010619XA patent/CN1938282A/zh active Pending
- 2005-03-15 RU RU2006138150/04A patent/RU2006138150A/ru not_active Application Discontinuation
- 2005-03-15 CA CA002561585A patent/CA2561585A1/en not_active Abandoned
- 2005-03-15 BR BRPI0508881-0A patent/BRPI0508881A/pt not_active IP Right Cessation
- 2005-03-23 AR ARP050101136A patent/AR048327A1/es unknown
-
2006
- 2006-10-30 ZA ZA200608998A patent/ZA200608998B/xx unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996040673A1 (en) * | 1995-06-07 | 1996-12-19 | Sugen, Inc. | Novel urea- and thiourea-type compounds |
| WO2003072541A2 (en) * | 2001-09-27 | 2003-09-04 | Smithkline Beecham Corporation | Chemical compounds |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006076592A1 (en) * | 2005-01-14 | 2006-07-20 | Cgi Pharmaceuticals, Inc. | 1,3 substituted diaryl ureas as modulators of kinase activity |
| US7625931B2 (en) | 2005-01-14 | 2009-12-01 | Cgi Pharmaceuticals, Inc. | Certain substituted diphenyl ureas, as modulators of kinase activity |
| US7777040B2 (en) | 2005-05-03 | 2010-08-17 | Cgi Pharmaceuticals, Inc. | Certain substituted ureas, as modulators of kinase activity |
| WO2007115670A1 (en) * | 2006-04-12 | 2007-10-18 | Merck Patent Gmbh | N-oxides of heterocyclic substituted bisarylureas for treating kinase-mediated diseases |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2007530609A (ja) | 2007-11-01 |
| KR20060132965A (ko) | 2006-12-22 |
| CN1938282A (zh) | 2007-03-28 |
| WO2005097755A3 (de) | 2006-03-09 |
| ZA200608998B (en) | 2008-07-30 |
| US20070225347A1 (en) | 2007-09-27 |
| AU2005231907A1 (en) | 2005-10-20 |
| CA2561585A1 (en) | 2005-10-20 |
| EP1761503A2 (de) | 2007-03-14 |
| DE102004015099A1 (de) | 2005-10-20 |
| RU2006138150A (ru) | 2008-05-10 |
| AR048327A1 (es) | 2006-04-19 |
| BRPI0508881A (pt) | 2007-09-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1866302B1 (de) | Pyrazolderivate | |
| EP1799679B1 (de) | Als kinaseinhibitoren geeignete derivate des n,n'-diphenylharnstoffs | |
| EP1912998B1 (de) | Pyrazolderivate mit tyrosinkinase aktivität | |
| EP1809628B1 (de) | Phenylharnstoffderivate als hemmstoffe von tyrosinkinasen zur behandlung von tumorerkrankungen | |
| EP1809630B1 (de) | 4-amino-5-oxo-8-phenyl-5h-pyrido-[2,3-d]-pyrimidin-derivate als inhibitoren der tyrosinkinasen und der raf-kinasen zur behandlung von tumoren | |
| EP1682548B1 (de) | Pyridopyrimidinone zur behandlung von krebserkrankungen | |
| EP1675849A1 (de) | Benzimidazolylderivate | |
| EP1718614A1 (de) | Pyridinamid-derivate als kinase-inhibitoren | |
| WO2006094600A1 (de) | Substituierte tetrahydro-pyrrolo-chinolinderivate als kinasemodulatoren, speziell der tyrosin- und raf-kinasen | |
| EP1664039B1 (de) | 1,3-benzoxazolylderivate als kinase-inhibitoren | |
| WO2006108482A1 (de) | Purinderivate als inhibitoren von rezeptor-tyrosinkinase-aktivität | |
| EP1809629B1 (de) | Zur Behandlungen von Tumoren, Augenkrankheiten, Entzündungen sowie Erkrankungen des Immunsystems geeignete Derivate des N,N'-Diphenylharnstoffs | |
| WO2005019216A1 (de) | Aminobenzimidazolderivate | |
| EP1663988A1 (de) | Benzyl-benzimidazolylderivate | |
| EP1473293A1 (de) | Chromenonderivate | |
| EP1737858A1 (de) | Sulfonamide | |
| WO2005097755A2 (de) | Imidazolderivate als hemmer von tyrosinkinase | |
| WO2005085220A1 (de) | Verwendung von thiadiazolharnstoffderivaten |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2005716076 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2398/KOLNP/2006 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12006501731 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2006/010968 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2561585 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020067020089 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200580010619.X Country of ref document: CN Ref document number: 2007505422 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005231907 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006/08998 Country of ref document: ZA Ref document number: 200608998 Country of ref document: ZA Ref document number: 2006138150 Country of ref document: RU |
|
| ENP | Entry into the national phase |
Ref document number: 2005231907 Country of ref document: AU Date of ref document: 20050315 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005231907 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020067020089 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10593295 Country of ref document: US Ref document number: 2007225347 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005716076 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0508881 Country of ref document: BR |
|
| WWP | Wipo information: published in national office |
Ref document number: 10593295 Country of ref document: US |