WO2005036129A2 - Electrophoretic media - Google Patents
Electrophoretic media Download PDFInfo
- Publication number
- WO2005036129A2 WO2005036129A2 PCT/US2004/033188 US2004033188W WO2005036129A2 WO 2005036129 A2 WO2005036129 A2 WO 2005036129A2 US 2004033188 W US2004033188 W US 2004033188W WO 2005036129 A2 WO2005036129 A2 WO 2005036129A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- particle
- electrophoretic
- particles
- polymer
- monomer
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C25—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES; APPARATUS THEREFOR
- C25B—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES FOR THE PRODUCTION OF COMPOUNDS OR NON-METALS; APPARATUS THEREFOR
- C25B7/00—Electrophoretic production of compounds or non-metals
-
- G—PHYSICS
- G02—OPTICS
- G02F—OPTICAL DEVICES OR ARRANGEMENTS FOR THE CONTROL OF LIGHT BY MODIFICATION OF THE OPTICAL PROPERTIES OF THE MEDIA OF THE ELEMENTS INVOLVED THEREIN; NON-LINEAR OPTICS; FREQUENCY-CHANGING OF LIGHT; OPTICAL LOGIC ELEMENTS; OPTICAL ANALOGUE/DIGITAL CONVERTERS
- G02F1/00—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics
- G02F1/01—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics for the control of the intensity, phase, polarisation or colour
- G02F1/165—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics for the control of the intensity, phase, polarisation or colour based on translational movement of particles in a fluid under the influence of an applied field
- G02F1/166—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics for the control of the intensity, phase, polarisation or colour based on translational movement of particles in a fluid under the influence of an applied field characterised by the electro-optical or magneto-optical effect
- G02F1/167—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics for the control of the intensity, phase, polarisation or colour based on translational movement of particles in a fluid under the influence of an applied field characterised by the electro-optical or magneto-optical effect by electrophoresis
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09B—ORGANIC DYES OR CLOSELY-RELATED COMPOUNDS FOR PRODUCING DYES, e.g. PIGMENTS; MORDANTS; LAKES
- C09B67/00—Influencing the physical, e.g. the dyeing or printing properties of dyestuffs without chemical reactions, e.g. by treating with solvents grinding or grinding assistants, coating of pigments or dyes; Process features in the making of dyestuff preparations; Dyestuff preparations of a special physical nature, e.g. tablets, films
- C09B67/0001—Post-treatment of organic pigments or dyes
- C09B67/0004—Coated particulate pigments or dyes
- C09B67/0008—Coated particulate pigments or dyes with organic coatings
- C09B67/0013—Coated particulate pigments or dyes with organic coatings with polymeric coatings
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N27/00—Investigating or analysing materials by the use of electric, electrochemical, or magnetic means
- G01N27/26—Investigating or analysing materials by the use of electric, electrochemical, or magnetic means by investigating electrochemical variables; by using electrolysis or electrophoresis
- G01N27/416—Systems
- G01N27/447—Systems using electrophoresis
- G01N27/44704—Details; Accessories
- G01N27/44747—Composition of gel or of carrier mixture
-
- G—PHYSICS
- G02—OPTICS
- G02F—OPTICAL DEVICES OR ARRANGEMENTS FOR THE CONTROL OF LIGHT BY MODIFICATION OF THE OPTICAL PROPERTIES OF THE MEDIA OF THE ELEMENTS INVOLVED THEREIN; NON-LINEAR OPTICS; FREQUENCY-CHANGING OF LIGHT; OPTICAL LOGIC ELEMENTS; OPTICAL ANALOGUE/DIGITAL CONVERTERS
- G02F2202/00—Materials and properties
- G02F2202/02—Materials and properties organic material
- G02F2202/022—Materials and properties organic material polymeric
Definitions
- the present invention relates to electrophoretic media.
- the present media are especially, although not exclusively, intended for use in encapsulated and microcell electrophoretic displays.
- the invention also relates to electrophoretic particles for use in such media, and to displays incorporating such media.
- Certain aspects of the present invention extend to electro-optic displays other than electrophoretic displays.
- the electrophoretic particles of the present invention are modified with polymers.
- the electro-optic displays of the present invention use an electro-optic medium having a voltage threshold.
- the electro-optic medium [0002] In the displays of the present invention, the electro-optic medium
- solid electro-optic displays when a non-electrophoretic electro-optic medium will typically be a solid (such displays may hereinafter for convenience be referred to as “solid electro-optic displays"), in the sense that the electro-optic medium has solid external surfaces, although the medium may, and often does, have internal liquid- or gas-filled spaces, and to methods for assembling displays using such an electro-optic medium.
- solid electro-optic displays includes encapsulated electrophoretic displays, encapsulated liquid crystal displays, and other types of displays discussed below.
- optical property is typically color perceptible to the human eye, it may be another optical property, such as optical transmission, reflectance, luminescence or, in the case of displays intended for machine reading, pseudo-color in the sense of a change in reflectance of electromagnetic wavelengths outside the visible range.
- gray state is used herein in its conventional meaning in the imaging art to refer to a state intermediate two extreme optical states of a pixel, and does not necessarily imply a black-white transition between these two extreme states.
- extreme states are white and deep blue, so that an intermediate "gray state” would actually be pale blue. Indeed, as already mentioned the transition between the two extreme states may not be a color change at all.
- bistable and “bistability” are used herein in their conventional meaning in the art to refer to displays comprising display elements having first and second display states differing in at least one optical property, and such that after any given element has been driven, by means of an addressing pulse of finite duration, to assume either its first or second display state, after the addressing pulse has terminated, that state will persist for at least several times, for example at least four times, the minimum duration of the addressing pulse required to change the state of the display element. It is shown in published U.S. Patent Application No.
- electro-optic displays are known.
- One type of electro- optic display is a rotating bichromal member type as described, for example, in U.S. Patents Nos. 5,808,783; 5,777,782; 5,760,761; 6,054,071 6,055,091; 6,097,531; 6,128,124; 6,137,467; and 6,147,791 (although this type of display is often referred to as a "rotating bichromal ball" display, the term "rotating bichromal member" is preferred as more accurate since in some of the patents mentioned above the rotating members are not spherical).
- Such a display uses a large number of small bodies (typically spherical or cylindrical) which have two or more sections with differing optical characteristics, and an internal dipole. These bodies are suspended within liquid-filled vacuoles within a matrix, the vacuoles being filled with liquid so that the bodies are free to rotate. The appearance of the display is changed to applying an electric field thereto, thus rotating the bodies to various positions and varying which of the sections of the bodies is seen through a viewing surface.
- This type of electro- optic medium is typically bistable.
- electro-optic display uses an electrochromic medium, for example an electrochromic medium in the form of a nanochromic film comprising an electrode formed at least in part from a semi-conducting metal oxide and a plurality of dye molecules capable of reversible color change attached to the electrode; see, for example O'Regan, B., et al., Nature 1991, 353, 737; and Wood, D., Information Display, 18(3), 24 (March 2002). See also Bach, U., et al., Adv. Mater., 2002, 14(11), 845. Nanochromic films of this type are also described, for example, in U.S. Patent No. 6,301,038, International Application Publication No. WO 01/27690, and in U.S. Patent Application 2003/0214695. This type of medium is also typically bistable.
- an electrochromic medium for example an electrochromic medium in the form of a nanochromic film comprising an electrode formed at least in part from a semi-conducting metal oxide and a plurality of
- Electrophoretic displays can have attributes of good brightness and contrast, wide viewing angles, state bistability, and low power consumption when compared with liquid crystal displays. Nevertheless, problems with the long-term image quality of these displays have prevented their widespread usage. For example, particles that make up electrophoretic displays tend to settle, resulting in inadequate service-life for these displays. [0009] Numerous patents and applications assigned to or in the names of the
- encapsulated electrophoretic media comprise numerous small capsules, each of which itself comprises an internal phase containing electrophoretically-mobile particles suspended in a liquid suspending medium, and a capsule wall surrounding the internal phase.
- the capsules are themselves held within a polymeric binder to form a coherent layer positioned between two electrodes.
- Encapsulated media of this type are described, for example, in U.S. Patents Nos.
- Known electrophoretic media both encapsulated and unencapsulated, can be divided into two main types, referred to hereinafter for convenience as “single particle” and “dual particle” respectively.
- a single particle medium has only a single type of electrophoretic particle suspended in a suspending medium, at least one optical characteristic of which differs from that of the particles. (In referring to a single type of particle, we do not imply that all particles of the type are absolutely identical. For example, provided that all particles of the type possess a charge of the same polarity, considerable variation in parameters such as particle color, size and electrophoretic mobility can be tolerated without affecting the utility of the medium.
- two particles of different color may be mixed in a single capsule, together with a single pigment (or multiple pigments) of opposite charge, to provide, by appropriate choice of the colors of these pigments, colors of any desired intermediate shade in either or both of the optical states.
- a medium When such a medium is placed between a pair of electrodes, at least one of which is transparent, depending upon the relative potentials of the two electrodes, the medium can display the optical characteristic of the particles (when the particles are adjacent the electrode closer to the observer, hereinafter called the "front” electrode) or the optical characteristic of the suspending medium (when the particles are adjacent the electrode remote from the observer, hereinafter called the "rear” electrode (so that the particles are hidden by the suspending medium).
- a dual particle medium has two different types of particles differing in at least one optical characteristic and a suspending fluid which may be uncolored or colored, but which is typically uncolored.
- the two types of particles differ in electrophoretic mobility; this difference in mobility may be in polarity (this type may hereinafter be referred to as an "opposite charge dual particle" medium) and/or magnitude.
- the medium can display the optical characteristic of either set of particles, although the exact manner in which this is achieved differs depending upon whether the difference in mobility is in polarity or only in magnitude. For ease of illustration, consider an electrophoretic medium in which one type of particles is black and the other type white.
- the two types of particles differ in polarity (if, for example, the black particles are positively charged and the white particles negatively charged), the particles will be attracted to the two different electrodes, so that if, for example, the front electrode is negative relative to the rear electrode, the black particles will be attracted to the front electrode and the white particles to the rear electrode, so that the medium will appear black to the observer. Conversely, if the front electrode is positive relative to the rear electrode, the white particles will be attracted to the front electrode and the black particles to the rear electrode, so that the medium will appear white to the observer.
- both types of particles have charges of the same polarity, but differ in electrophoretic mobility (this type of medium may hereinafter to referred to as a "same polarity dual particle" medium), both types of particles will be attracted to the same electrode, but one type will reach the electrode before the other, so that the type facing the observer differs depending upon the electrode to which the particles are attracted. For example suppose the previous illustration is modified so that both the black and white particles are positively charged, but the black particles have the higher electrophoretic mobility.
- both the black and white particles will be attracted to the front electrode, but the black particles, because of their higher mobility will reach it first, so that a layer of black particles will coat the front electrode and the medium will appear black to the observer.
- both the black and white particles will be attracted to the rear electrode, but the black particles, because of their higher mobility will reach it first, so that a layer of black particles will coat the rear electrode, leaving a layer of white particles remote from the rear electrode and facing the observer, so that the medium will appear white to the observer: note that this type of dual particle medium requires that the suspending fluid be sufficiently transparent to allow the layer of white particles remote from the rear electrode to be readily visible to the observer.
- the suspending fluid in such a display is not colored at all, but some color may be incorporated for the purpose of correcting any undesirable tint in the white particles seen therethrough, or to produce a desirable shade of color in the gray state.
- Both single and dual particle electrophoretic displays may be capable of intermediate gray states having optical characteristics intermediate the two extreme optical states already described.
- Some of the aforementioned patents and published applications I disclose encapsulated electrophoretic media having three or more different types of particles within each capsule. For purposes of the present application, such multi- particle media are regarded as sub-species of dual particle media.
- the walls surrounding the discrete microcapsules in an encapsulated electrophoretic medium could be replaced by a continuous phase, thus producing a so-called polymer-dispersed electrophoretic display, in which the electrophoretic medium comprises a plurality of discrete droplets of an electrophoretic fluid and a continuous phase of a polymeric material, and that the discrete droplets of electrophoretic fluid within such a polymer-dispersed electrophoretic display may be regarded as capsules or microcapsules even though no discrete capsule membrane is associated with each individual droplet; see for example, the aforementioned 2002/0131147.
- microcell electrophoretic display A related type of electrophoretic display is a so-called "microcell electrophoretic display".
- the charged particles and the suspending fluid are not encapsulated within microcapsules but instead are retained within a plurality of cavities formed within a carrier medium, typically a polymeric film. See, for example, International Application Publication No. WO 02/01281, and published US Application No. 2002/0075556, both assigned to Sipix Imaging, Inc.
- electrophoretic media are often opaque (since, for example, in many electrophoretic media, the particles substantially block transmission of visible light through the display) and operate in a reflective mode
- many electrophoretic displays can be made to operate in a so-called "shutter mode" in which one display state is substantially opaque and one is light-transmissive.
- Shutter mode in which one display state is substantially opaque and one is light-transmissive.
- Dielectrophoretic displays which are similar to electrophoretic displays but rely upon variations in electric field strength, can operate in a similar mode; see U.S. Patent No. 4,418,346.
- An encapsulated or microcell electrophoretic display typically does not suffer from the clustering and settling failure mode of traditional electrophoretic devices and provides further advantages, such as the ability to print or coat the display on a wide variety of flexible and rigid substrates.
- printing is intended to include all forms of printing and coating, including, but without limitation: pre-metered coatings such as patch die coating, slot or extrusion coating, slide or cascade coating, curtain coating; roll coating such as knife over roll coating, forward and reverse roll coating; gravure coating; dip coating; spray coating; meniscus coating; spin coating; brush coating; air knife coating; silk screen printing processes; electrostatic printing processes; thermal printing processes; ink jet printing processes; and other similar techniques.
- pre-metered coatings such as patch die coating, slot or extrusion coating, slide or cascade coating, curtain coating
- roll coating such as knife over roll coating, forward and reverse roll coating
- gravure coating dip coating
- spray coating meniscus coating
- spin coating spin coating
- brush coating air knife coating
- silk screen printing processes electrostatic printing processes; thermal
- This application also describes various improvements in such polymer-coated particles, including controlling the amount of polymer deposited on the particle, the structure of the polymer, techniques for forming the polymeric coating on the electrophoretic particles, and techniques for pretreatment of the electrophoretic particles before the formation of polymer coatings thereon.
- the application also describes a process for producing such a polymer-coated pigment particle, this process comprising: (a) reacting the pigment particle with a reagent having a functional group capable of reacting with, and bonding to, the particle, and also having a polymerizable or polymerization-initiating group, thereby causing the functional group to react with the particle surface and attach the polymerizable group thereto; and (b) reacting the product of step (a) with at least one monomer or oligomer under conditions effective to cause reaction between the polymerizable or polymerization-initiating group on the particle and the at least one monomer or oligomer, thereby causing the formation of polymer bonded to the pigment particle.
- the aforementioned 2002/0180687 describes an electrophoretic medium comprising a plurality of particles suspended in a hydrocarbon suspending fluid, the particles being capable of moving through the fluid upon application of an electric field to the medium, the fluid having dissolved or dispersed therein a polyisobutylene having a viscosity average molecular weight in the range of about 400,000 to 1,200,000 g/mole, the polyisobutylene comprising from about 0.25 to about 2.5 per cent by weight of the suspending fluid.
- an electrophoretic medium comprising a plurality of particles suspended in a suspending fluid, the particles being capable of moving through the fluid upon application of an electric field to the medium, the fluid having dissolved or dispersed therein a polymer having an intrinsic viscosity of ⁇ in the suspending fluid and being substantially free from ionic or ionizable groups in the suspending fluid, the polymer being present in the suspending fluid in a concentration of from about 0.5 [ ⁇ ] "1 to about 2.0 [ ⁇ ] "1 .
- the presence of the polyisobutylene (PIB) or other polymer in the suspending fluid substantially increases the bistability of the display.
- electrophoretic displays require only low electrical power to switch from one state to another.
- the aforementioned 2002/0180687 describes electrophoretic media which achieve good image stability by incorporation of a high molecular weight polymer, for example PUB, that has good solubility in the suspending medium (typically an aliphatic hydrocarbon such as Isopar G) but which is not absorbed on the electrophoretic particles.
- a high molecular weight polymer typically an aliphatic hydrocarbon such as Isopar G
- the suspending medium typically an aliphatic hydrocarbon such as Isopar G
- the present invention is in no way limited by this belief to induce a weak flocculation of the pigments by a mechanism known in the colloid science art as "depletion flocculation".
- Polymers other than PLB can be used for the same purpose.
- a second polymer that has been shown to be useful for this purpose is Kraton G, a block copolymer comprising a polystyrene block and a hydrogenated polyisobutylene block, that forms aggregate structures in the suspending medium.
- the aggregates are the species that induce the depletion flocculation, rather than the monomeric block copolymer itself.
- the switching speed of the display will be reduced by this approach to image stability. Furthermore, since the depletion flocculation mechanism is only active at concentrations of polymer above the overlap concentration (which can be operationally defined as the concentration of polymer that causes the viscosity of the medium to increase by a factor of two), all polymers that act by this mechanism can be expected to produce a similar diminution of the switching speed. In practice, the switching speed is reduced by approximately a factor of two to three when enough polymer is used to give adequate image stability.
- PUB and other polymers improve image stability by manipulating the colloidal stability of the pigment particles.
- the preferred polymer coated particles described in the aforementioned WO 02/093246 are colloidally stable in the suspending medium because of a polymer shell of (typically) poly(lauryl methacrylate) that is grown on the surface of the particles during their preparation.
- composition of the polymer shell By appropriate manipulation of the composition of the polymer shell, it is possible in principle to make particles with the same degree of colloidal stability (and hence displays with the same image stability) as that afforded by PIB, Kraton, and other polymers dispersed in the suspending medium but without requiring addenda like PIB in the suspending medium.
- Such displays should be substantially faster than displays that contain PIB, or equivalently, should operate at equivalent speed at lower applied voltage.
- this invention seeks to provide approaches to providing electrophoretic particles with modified polymer shells that allow the production of fast, image-stable displays.
- this invention seeks to provide an improved form of the two-step process for preparing such polymer-coated electrophoretic particles described in Paragraph 21 above.
- titania or a similar metal oxide pigment
- the silica-coated titania is treated with a silane containing an ethylenic group.
- the resultant silane-treated titania may then be reacted with a variety of unsaturated monomers, for example, 2- ethylhexyl acrylate or lauryl methacrylate, in the presence of a free-radical polymerization initiator, to form the desired polymer-coated titania.
- Carbon black is treated with a diazotizing agent containing an ethylenic group, for example, the reaction product of 4-vinylaniline and nitrous acid, to attach ethylenic groups to the carbon black surface, and thereafter may be reacted with a variety of unsaturated monomers in substantially the same way as described for titania.
- a diazotizing agent containing an ethylenic group for example, the reaction product of 4-vinylaniline and nitrous acid
- the final polymerization steps are conducted in toluene, primarily because this is a solvent known in the polymer industry to have good properties for use in such free radical polymerizations.
- this is a solvent known in the polymer industry to have good properties for use in such free radical polymerizations.
- its use as a solvent in processes for preparing polymer-coated electrophoretic pigment particles is markedly inconvenient.
- the suspending fluid used in electrophoretic displays is an aliphatic hydrocarbon (alone or in combination with a halocarbon).
- the polymer- coated pigment particles will eventually be dispersed in an aliphatic hydrocarbon, and it is necessary to avoid contaminating this aliphatic hydrocarbon with toluene (since the behavior of electrophoretic media tends in certain cases to be highly sensitive to small changes in the composition of the suspending fluid), after the polymerization in toluene is finished and the polymer-coated pigment separated from the toluene, it is necessary to remove all traces of the toluene before the polymer-coated pigment particles are suspended in the final suspending fluid.
- washing, centrifuging and drying steps are labor intensive and costly. Further expense is incurred by the need to re-disperse the dried pigment in the final suspending fluid. Furthermore, because of the presence of the toluene and THF, the washing, centrifuging and drying steps tend to be hazardous and commercial scale production of the polymer-coated pigment requires the use of explosion-proof ovens, mixers and centrifuges, and explosion-proof electrical control panels, which substantially increases the costs of the production equipment. Also, operator exposure to vapors during processing can be significant despite the use of protective devices or exposure prevention methods. Finally, the drying step may be detrimental to the performance of the pigment in the final electrophoretic medium. It is therefore desirable to find an alternative solvent in which the polymerization reaction can be carried out, and if possible to eliminate the need for drying and re-dispersion of the dried pigment.
- this invention seeks to provide electro-optic displays which are capable of being driven in a simplified manner. Whether a display is reflective or transmissive, and whether or not the electro-optic medium used is bistable, to obtain a high-resolution display, individual pixels of a display must be addressable without interference from adjacent pixels.
- One way to achieve this objective is to provide an array of non-linear elements, such as transistors or diodes, with at least one non-linear element is associated with each pixel, to produce an "active matrix" display.
- An addressing or pixel electrode which addresses one pixel, is connected to an appropriate voltage source through the associated non-linear element.
- the pixel electrode is connected to the drain of the transistor, and this arrangement will be assumed in the following description, although it is essentially arbitrary and the pixel electrode could be connected to the source of the transistor.
- the pixels are arranged in a two-dimensional array of rows and columns, such that any specific pixel is uniquely defined by the intersection of one specified row and one specified column.
- the sources of all the transistors in each column are connected to a single column electrode, while the gates of all the transistors in each row are connected to a single row electrode; again the assignment of sources to rows and gates to columns is conventional but essentially arbitrary, and could be reversed if desired.
- the row electrodes are connected to a row driver, which essentially ensures that at any given moment only one row is selected, i.e., that there is applied to the selected row electrode a voltage such as to ensure that all the transistors in the selected row are conductive, while there is applied to all other rows a voltage such as to ensure that all the transistors in these non-selected rows remain non-conductive.
- the column electrodes are connected to column drivers, which place upon the various column electrodes voltages selected to drive the pixels in the selected row to their desired optical states.
- a transistor includes a gate electrode, an insulating dielectric layer, a semiconductor layer and source and drain electrodes.
- Application of a voltage to the gate electrode provides an electric field across the dielectric layer, which dramatically increases the source-to-drain conductivity of the semiconductor layer. This change permits electrical conduction between the source and the drain electrodes.
- the gate electrode, the source electrode, and the drain electrode are patterned.
- the semiconductor layer is also patterned in order to minimize stray conduction (i.e., cross-talk) between neighboring circuit elements.
- Liquid crystal displays commonly employ amorphous silicon (“a-Si”) thin-film transistors (“TFT's”) as switching devices for display pixels. Such TFT's typically have a bottom-gate configuration. Within one pixel, a thin-film capacitor typically holds a charge transferred by the switching TFT. Electrophoretic displays can use similar TFT's with capacitors, although the function of the capacitors differs somewhat from those in liquid crystal displays; see WO 00/67327, and the aforementioned 2002/0106847 and 2002/0060321. Thin-film transistors can be fabricated to provide high performance. Fabrication processes, however, can result in significant cost.
- a-Si amorphous silicon
- TFT's thin-film transistors
- pixel electrodes are charged via the TFT's during a line address time.
- a TFT is switched to a conducting state by changing an applied gate voltage.
- a gate voltage is switched to a "high" state to switch the TFT into a conducting state.
- the pixel electrode typically exhibits a voltage shift when the select line voltage is changed to bring the TFT channel into depletion. The pixel electrode voltage shift occurs because of the capacitance between the pixel electrode and the TFT gate electrode.
- This voltage shift is often referred to as "gate feedthrough”.
- Gate feedthrough can compensated by shifting the top plane voltage
- Variations in C gp are caused, for example, by misalignment between the two conductive layers used to form the gate and the source-drain levels of the TFT; variations in the gate dielectric thickness; and variations in the line etch, i.e., line width errors.
- Some tolerance for mis-registered conductive layers can be obtained by utilizing a gate electrode that completely overlaps the drain electrode. This technique, however, can cause a large gate-pixel capacitance. A large gate-pixel capacitance is undesirable because it can create a need for a large compensation in one of the select line voltage levels. Moreover, existing addressing structures can produce unintended bias voltages, for example, due to pixel-to-pixel variations in gate-pixel capacitance. Such voltages can produce a detrimental effect on certain electro-optic media, particularly when present for extended periods of time. [0039] The foregoing problems render designing a bistable electro-optic display using a electro-optic medium without a voltage threshold a difficult task.
- this invention provides an electrophoretic medium comprising an electrically charged particle suspended in a suspending fluid, the particle having a polymeric shell having repeating units derived from at least one monomer the homopolymer of which is incompatible with the suspending fluid.
- This aspect of the present invention may hereinafter for convenience be called the "incompatible monomer medium".
- the polymeric shell desirably further comprises repeating units derived from at least one monomer the homopolymer of which is compatible with the suspending fluid.
- the monomer or monomers forming the compatible homopolymer may comprise from about 15 to about 99 per cent, and preferably about 50 to about 99 per cent, by weight of the polymer shell.
- the suspending fluid is typically a hydrocarbon, although a mixture of a hydrocarbon and another compatible solvent, such as a halocarbon, may be used. Alternatively, a silicone fluid or a fluorocarbon can be used as the suspending fluid.
- the monomer forming the incompatible homopolymer (this monomer may hereinafter for convenience be called the "incompatible monomer”).
- a particular monomer can be considered an incompatible monomer depends upon the specific suspending fluid being used, and that a specific monomer may be an incompatible monomer in one suspending fluid and a compatible monomer in a different suspending fluid.
- a specific monomer may be an incompatible monomer in one suspending fluid and a compatible monomer in a different suspending fluid.
- lauryl methacrylate is a compatible monomer in aliphatic hydrocarbon suspending fluids, but would normally be an incompatible monomer in a silicone suspending fluid.
- the incompatible monomer may be any one or more of acrylates and methacrylates formed from alcohols containing not more than about eight carbon atoms, said alcohols optionally containing hydroxyl or halogen or other polar substituents, such as carboxyl groups, cyano groups, ketone or aldehyde groupings; acrylamides and methacrylamides; N,N-dialkylacrylamides; N-vinylpyrrolidone; styrene and derivatives thereof; vinyl esters; and vinyl halides.
- acrylates and methacrylates formed from alcohols containing not more than about eight carbon atoms, said alcohols optionally containing hydroxyl or halogen or other polar substituents, such as carboxyl groups, cyano groups, ketone or aldehyde groupings; acrylamides and methacrylamides; N,N-dialkylacrylamides; N-vinylpyrrolidone; styrene and derivatives thereof;
- incompatible monomers include methyl methacrylate, ethyl methacrylate, butyl methacrylate, isobutyl methacrylate, t-butyl methacrylate, octyl methacrylate, 2-ethylhexyl methacrylate, 2-hydroxyethyl methacrylate, acrylamide, acrylic acid, acrylonitrile, methyl vinyl ketone, methacrylamide, N-vinylpyrrolidone, styrene, vinyl acetate, vinyl chloride, and vinylidene chloride.
- incompatible monomers include fluorine-containing esters of acrylic acid and methacrylic acid, such as trifluoroethyl methacrylate, hexafluorobutyl acrylate, or other kinds of fluorinated monomers, such as pentafluorostyrene or other polyfluoroaromatic molecules containing a polymerizable functional group.
- fluorine-containing esters of acrylic acid and methacrylic acid such as trifluoroethyl methacrylate, hexafluorobutyl acrylate, or other kinds of fluorinated monomers, such as pentafluorostyrene or other polyfluoroaromatic molecules containing a polymerizable functional group.
- fluorinated monomers such as pentafluorostyrene or other polyfluoroaromatic molecules containing a polymerizable functional group.
- Other classes of incompatible monomers in hydrocarbon media include silicone-containing molecules that include polymerizable vinyl groups in their structure.
- the compatible monomer comprises lauryl methacrylate and the incompatible monomer comprises any one or more of trifluoroethyl methacrylate, hexafluorobutyl acrylate, styrene, t-butyl methacrylate and N-vinylpyrrolidone.
- the compatible monomer may comprise a majority of silicone groups, and the incompatible monomer may comprise any of the monomers listed above except those comprising a majority of silicone functionality.
- the incompatible monomer medium may further comprise a second type of electrically charged particle having at least one optical characteristic differing from that of the other (first) electrically charged particle or particles, the second type of electrically charged particle having a polymeric shell.
- the first electrically charged particle comprises titania and the second type of electrically charged particle comprises carbon black or copper chromite.
- This invention also provides an electrophoretic particle for use in the incompatible monomer medium of the present invention.
- this electrophoretic particle (hereinafter for convenience called “the incompatible monomer particle”) comprises a pigment particle having a polymeric shell having repeating units derived from at least one monomer the homopolymer of which is incompatible with n-hexane.
- hydrocarbon suspending fluids typically used in such media comprise various mixtures of low molecular weight aliphatic hydrocarbons, and for the avoidance of ambiguity, n-hexane may be used as a single test compound for testing compatibility with such hydrocarbon mixtures.
- the polymeric shell may further comprise repeating units derived from at least one monomer the homopolymer of which is compatible with n-hexane.
- the compatible monomer may comprises from about 15 to about 99 per cent, and preferably about 50 to about 99 per cent, by weight of the polymer shell.
- the incompatible monomer may comprise any one or more of those listed in Paragraph 56 above.
- the compatible monomer comprises lauryl methacrylate and the incompatible monomer comprises any one or more or styrene, t-butyl methacrylate and N-vinylpyrrolidone.
- the pigment used to form the particle of the present invention may be, for example, any one or more of titania, carbon black and copper chromite.
- this invention provides similar electrophoretic particles for use in fluorinated and silicone-based suspending fluids.
- this invention provides an electrophoretic particle comprising a pigment particle having a polymeric shell having repeating units derived from at least one monomer the homopolymer of which is incompatible with perfluorodecalin.
- This invention also provides an electrophoretic particle comprising a pigment particle having a polymeric shell having repeating units derived from at least one monomer the homopolymer of which is incompatible with polydimethylsiloxane 200, viscosity 0.65 centistokes.
- this invention provides an electrophoretic medium comprising: a suspending fluid; a first type of electrically charged particle suspended in the suspending fluid, the first type of particle having a first optical characteristic and a polymeric shell; and second type of particle having a second optical characteristic differing from the first optical characteristic, and a polymeric shell; wherein the polymeric shells are arranged such that homoaggregation of the first and second types of particles is thermodynamically favored over heteroaggregation.
- This medium may hereinafter for convenience be called the
- homoaggregation medium of the present invention. It will be understood that there is a considerable overlap between the dual particle incompatible monomer media of the present invention and the homoaggregation media, in the sense that many media can satisfy the definitions of both simultaneously.
- the polymeric shells of the first and second types of particles may each comprise repeating units derived from at least one monomer the homopolymer of which is incompatible with the suspending fluid.
- Each polymeric shell may further comprise repeating units derived from at least one monomer the homopolymer of which is compatible with the suspending fluid.
- the compatible monomer may comprise from about 15 to about 99 per cent, and preferably about 50 to about 99 per cent, by weight of the polymer shell.
- the suspending fluid may have a dielectric constant less than about 5, and may comprise a hydrocarbon, preferably an aliphatic hydrocarbon. Alternatively, the suspending fluid may comprise an aryl-alkane or dodecylbenzene.
- the incompatible monomer may comprise any one of more of those listed in Paragraph 46 above.
- the compatible monomer comprises lauryl methacrylate and the incompatible monomer comprises any one or more or styrene, t-butyl methacrylate and N-vinylpyrrolidone.
- the homoaggregation medium of the invention may have an operating voltage threshold.
- the medium may be encapsulated, i.e., the suspending fluid and the particles may be retained within a plurality of capsules or cells.
- the present invention also provides an electrophoretic display comprising either type of electrophoretic medium of the invention and at least one electrode disposed adjacent the electrophoretic medium and arranged to apply an electric field thereto.
- This invention also provides an active matrix electro-optic display comprising a layer of electro-optic medium; and a plurality of pixel electrodes disposed adjacent the layer of electro-optic medium and arranged to apply an electric field thereto, wherein the electro-optic medium exhibits a voltage threshold.
- This electro-optic display may hereinafter for convenience be called the "voltage threshold display" of the present invention.
- a capacitor may be associated with each pixel electrode.
- the electro-optic medium may comprise a plurality of charged particles suspended in a suspending fluid and capable of moving therethrough on application of an electric field to the electro-optic medium. These charged particles may have polymeric shells having repeating units derived from at least one monomer the homopolymer of which is incompatible with the suspending fluid.
- the electro-optic medium may also be a homoaggregation medium of the present invention.
- this invention provides a process for producing a polymer- coated pigment particle, this process comprising: (a) reacting the pigment particle with a reagent having a functional group capable of reacting with, and bonding to, the particle, and also having a polymerizable or polymerization-initiating group, thereby causing the functional group to react with the particle surface and attach the polymerizable group thereto; and (b) reacting the product of step (a) with at least one monomer or oligomer under conditions effective to cause reaction between the polymerizable or polymerization-initiating group on the particle and the at least one monomer or oligomer, thereby causing the formation of polymer bonded to the pigment particle, wherein step (b) is carried out in an aliphatic hydrocarbon.
- This process may hereinafter for convenience be called the "aliphatic polymerization process" of the present invention.
- Figure 1 is a reaction diagram showing schematically the process used to produce an incompatible monomer electrophoretic particle of the present invention.
- Figure 2 is a graph showing the variation of the dynamic range of a homoaggregation electrophoretic medium of the present invention as a function of the applied voltage for various pulse lengths, as described in the Examples below.
- the present invention relates to electrophoretic media using particles having polymeric shells, electrophoretic particles for use in such media, electrophoretic displays containing such media or similar electro-optic media, and processes for the preparation of the aforementioned polymeric shells.
- the polymer shell present on the electrophoretic particles of the present invention may not completely cover the surface of the particle in the sense that no additional polymer could be formed on the polymer surface; indeed, as discussed below, it is often found that a second polymerization step will often form additional polymer on a particle's surface. Accordingly, the use of the term "polymer shell" herein does not imply a polymer coating precluding the possibility of forming additional polymer on a particle by a further polymerization step.
- the preferred class of functional groups for bonding to many ceramic oxide pigments, but especially silica- and/or alumina-coated titania and similar silica- coated pigments, are silane coupling groups, especially trialkoxy silane coupling groups.
- silyl meth(acrylates) contain charge- control groups which impart to the final electrophoretic particle the ability to acquire a desired charge in the presence of a suitable charging agent or agents.
- the reagent of Formula (II) is used when a positively charging particle is desired, while the reagent of Formula (I) gives negatively charging particles.
- Both reagents of course, comprise a polymerizable vinyl grouping that allows grafting of polymeric radicals generated in solution.
- the suspending fluid in an electrophoretic medium is typically a liquid with a low dielectric constant, such as a hydrocarbon, halocarbon (especially fluorocarbon), or silicone.
- a hydrocarbon especially fluorocarbon
- silicone especially fluorocarbon
- the incompatible monomer media of the present invention may use any of these types of suspending fluid.
- the invention will be primarily described below with reference to media containing hydrocarbon suspending fluids, but modification of the media to use other types of suspending fluid will readily be apparent to those skilled in colloid chemistry in such suspending fluids.
- the polymer shell itself typically comprises a major proportion of hydrocarbon chains (i.e., chains forming a homopolymer that is highly compatible with the suspending medium); except for groups provided for charging purposes and for purposes of adjusting compatibility with the suspending fluid, as discussed below, large numbers of strongly polar or ionic groups are undesirable.
- the medium in which the particles are to be used comprises an aliphatic hydrocarbon suspending fluid (as is commonly the case)
- Substantially longer side chains may be advantageous; for example, lauryl (C 12 ) side chains.
- the side chains may themselves be branched; for example, each side chain could be a branched alkyl group, such as a 2-ethylhexyl group. It is believed (although the invention is in no way limited by this belief) that, because of the high affinity of hydrocarbon chains for the hydrocarbon-based suspending fluid, the branches of the polymer spread out from one another in a brush or tree-like structure through a large volume of liquid, thus preventing close association with other similar particles and causing the particles to be colloidally stable in the suspending fluid.
- the denser the particle the lower the optimum proportion of polymer by weight of the particle, and the more finely divided the particle (the smaller the particle size), the higher the optimum proportion of polymer.
- the aforementioned WO 02/093246 states that the particles should be coated with at least 2 and desirably at least 4, per cent by weight of polymer, and that, in most cases, the optimum proportion of polymer will range from 4 to 15 per cent by weight of the particle, and typically from 6 to 15 per cent by weight, and most desirably from 8 to 12 per cent by weight.
- the aforementioned WO 02/093246 states that the preferred range of polymer is from 8 to 12 per cent by weight of the titania.
- the amount of polymer is in terms of the surface density of polymer (i.e., the weight of polymer per unit area of particle surface, for example milligrams of polymer per square meter of particles surface.
- T the surface density of polymer
- W the weight of polymer per gram of sample (obtained from thermogravimetric analysis)
- p the density of the base particle
- D is the diameter of the base particle.
- a useful range of surface density has been found to be 2 to 40 mg/g, with a preferred range being 14 to 24 mg/g and a particularly preferred range being 18 to 22 mg/g.
- the polymer-coated particles of the present invention may also be useful in applications other than electrophoretic displays.
- the controlled affimty for hydrocarbon materials provided by the polymer coating on the present pigments should render the pigments advantageous for use in polymeric and rubber matrices, in which the pigments should be more readily dispersible than similar but uncoated pigments.
- the flexibility in the chemical nature of the polymer coating allows the coating to be "tuned" for controlled dispersibility in any specific matrix.
- the present pigments may be used as dispersible pigments or reactive extrusion compounds.
- the polymer coating on the particles may be useful in improving the mechanical properties of such pigment/polymer or rubber blends by reducing the tendency for such blends to shear or fracture at the interface between the particles and the matrix material.
- the polymer-coated particles are produced by a process which produces the polymer-coated particles in admixture with "free" polymer not attached to the particles (as discussed above), it will, in many cases, not be necessary to separate the coated particles from the free polymer before dispersing the particles in the polymeric or rubber matrix, since the free polymer will disperse harmlessly in the matrix.
- the choice of suspending fluid may be based on concerns of chemical inertness, density matching to the electrophoretic particle, or chemical compatibility with both the electrophoretic particle and capsule or microcell wall (in the case of encapsulated electrophoretic displays).
- the viscosity of the fluid should be low when movement of the particles is desired.
- the refractive index of the suspending fluid may also be substantially matched to that of the particles. As used herein, the refractive index of a suspending fluid "is substantially matched" to that of a particle if the difference between their respective refractive indices is between about zero and about 0.3, and is preferably between about 0.05 and about 0.2.
- Titania particles are typically used as scattering particles to produce a white state in dual particle electrophoretic displays, and this material has a high refractive index (ca. 2.7), so that a low refractive index in the suspending medium is desirable.
- Useful organic solvents include, but are not limited to, epoxides, such as decane epoxide and dodecane epoxide; vinyl ethers, such as cyclohexyl vinyl ether and Decave (Registered Trade Mark of international Flavors & Fragrances, Inc., New York, NY); and aromatic hydrocarbons, such as toluene and other alkyl benzene derivatives such as dodecylbenzene and naphthalene and alkyl naphthalene derivatives.
- epoxides such as decane epoxide and dodecane epoxide
- vinyl ethers such as cyclohexyl vinyl ether and Decave (Registered Trade Mark of international Flavors & Fragrances, Inc., New York, NY)
- aromatic hydrocarbons such as toluene and other alkyl benzene derivatives such as dodecylbenzene and na
- Useful halogenated organic solvents include, but are not limited to, tetrafluorodibromoethylene, tetrachloroethylene, trifluorochloroethylene, 1,2,4- trichlorobenzene and carbon tetrachloride. These materials have high densities.
- Useful hydrocarbons include, but are not limited to, dodecane, tetradecane, the aliphatic hydrocarbons in the Isopar (Registered Trade Mark) series (Exxon, Houston, TX), Norpar (Registered Trade Mark) (a series of normal paraffinic liquids), Shell-Sol (Registered Trade Mark) (Shell, Houston, TX), and Sol-Trol (Registered Trade Mark) (Shell), naphtha, and other petroleum solvents. These materials usually have low densities.
- silicone oils include, but are not limited to, octamethyl cyclosiloxane and higher molecular weight cyclic siloxanes, poly(methyl phenyl siloxane), hexamethyldisiloxane, and polydimethylsiloxane. These materials usually have low densities.
- Useful low molecular weight halogen-containing polymers include, but are not limited to, poly(chlorotrifluoroethylene) polymer (Halogenated Hydrocarbon Inc., River Edge, NJ), Galden (Registered Trade Mark) (a perfluorinated ether from Ausimont, Morristown, NJ), or Krytox (Registered Trade Mark) from du Pont (Wilmington, DE) Many of the above materials are available in a range of viscosities, densities, and boiling points. [0074] Electrophoretic particles, media and displays with controlled particle/suspending fluid compatibility
- WO 02/093426 only a single monomer, and a single polymerization step, are required to provide a sterically stabilizing polymer shell on a colloidally stable, functional pigment.
- the monomer used to form such polymeric shell is typically lauryl methacrylate, although other monomers are also used in the aforementioned WO
- polymer compatibility with the suspending fluid means that the polymer (when detached from the associated base electrophoretic particle) is soluble in the solvent. Whether a polymer is soluble can usually be determined by simple visual inspection; solutions are generally optically clear, whereas non-solutions (mixtures or dispersions) are opaque or have two obvious phases.
- compatibility is not a binary phenomenon, that is to say, a polymer is not necessarily totally compatible or totally incompatible with a given suspending fluid. Instead, there is a range of compatibilities, depending on the relative strength of interaction between the fluid and the polymer segments (approximately, the monomers of which the polymer is constituted) and that between the polymer segments themselves.
- polymer-fluid interactions are energetically more favorable than polymer-polymer interactions.
- the polymer coil in solution will be extended relative to that of the polymer in a polymer melt. This extension can be measured using capillary viscometry or liglit scattering techniques. For example, if the intrinsic viscosity, [ ⁇ ], of a series of samples of the polymer having different molecular weights (M) is measured, it is usually found that there is a power-law relationship between the two quantities:
- the exponent ⁇ ranges between 0.5 and about 0.8 (for flexible polymers), depending on the solvent quality, with larger values representing better solvents.
- ⁇ 0.5
- the fluid is called a "theta solvent” and the conformation of the polymer is similar to that in the polymer melt (i.e., surrounded only by other polymer segments), so that in effect the polymer has neutral compatibility with the fluid. Under these conditions the conformation of the polymer chain is that of a random walk.
- the Huggins coefficient can be used as an indication of the solvent quality (i.e., of polymer compatibility with the solvent).
- the Huggins constant is the term [ ⁇ ] k' in the following expression for the relative viscosity of a dilute solution of a polymer:
- ⁇ s is the viscosity of the solvent
- [ ⁇ ] is the intrinsic viscosity
- c is the concentration of the polymer in the fluid.
- k' is in the range 0.30 to 0.40, whereas for less compatible fluids, k' is larger (0.50 to 0.80).
- the degree of swelling (the ratio of the volume of a polymer sample in the presence of fluid to that of the dry polymer) can be used as a measure of the degree of compatibility of the fluid and polymer in this regime. The greater the degree of swelling, the greater the compatibility.
- Making the polymer shell less compatible with the suspending fluid can be achieved in several ways. Firstly, the polymer shell may have repeating units derived from at least one monomer the homopolymer of which is incompatible with the suspending fluid, in accordance with the incompatible monomer electrophoretic medium aspect of the present invention.
- such a polymer shell will also include a compatible monomer, that is to say a monomer the homopolymer of which is compatible with the suspending fluid, and the compatibility of the polymer shell with the suspending fluid can be adjusted by varying the ratio of the two monomers.
- a polymer shell including both an incompatible and a compatible monomer can be a random copolymer shell formed by using a mixture of polymerizable monomers in the last polymerization step (which may be the sole polymerization step).
- a range of colloidal stabilities can be built into the particle depending on the ratio of the monomers in the shell.
- different monomer species can impart different degrees of incompatibility
- by changing the incompatible monomer the degree of incompatibility can be further modified.
- acrylate esters with short side chains containing not more than about eight carbon atoms e.g., butyl methacrylate
- lauryl methacrylate yields electrophoretic particles with excellent colloidal stability in Isopar G.
- a copolymer of lauryl methacrylate and butyl methacrylate will give a polymer shell marginally compatible with Isopar G, and a marginally colloidally stable particle, at some mole ratio of butyl methacrylate to laurel methacrylate in the polymerization mixture.
- a second method of modifying the polymer shell around the electrophoretic particles is by a second stage of polymerization.
- the surface functionality provided by the reagents of Formulae (I) and (II) above is not completely consumed during a single graft polymerization step.
- thermogravimetric analysis of the pigment isolated from the reaction mixture after heating for 16 hours shows an increase in the amount of bound polymer (as determined by thermogravimetric analysis (TGA): after first polymerization: 7.3%; after second polymerization: 8.9% and 9.9% in two different runs).
- TGA thermogravimetric analysis
- the monomer used in the second polymerization can be different from that used in the first, thus making it possible to incorporate polymer chains constructed from different monomers in the final polymer shell. Furthermore, by modifying the conditions of the first polymerization step, the initial grafting density of chains can be adjusted, as well as the availability (and accessibility) of the surface vinyl functionality for the second stage polymerization. Thus, this double polymerization method affords a second degree of flexibility in the construction of polymer stabilized electrophoretic particles.
- incompatible monomers include acrylate esters with alkyl groups that vary in chain length and branching structure but typically have eight or fewer carbon atoms (e.g.
- acrylamides such as
- a polymer shell formed entirely from compatible monomers provides steric stability to the electrophoretic particle because it is thermodynamically favorable for the polymer chains forming the shell to be surrounded by suspending fluid rather than by other polymer segments.
- a polymer shell formed entirely from compatible monomers for example a pure poly(lauryl methacrylate) polymer shell dispersed in an aliphatic hydrocarbon, is highly swollen.
- One further advantage of preferred embodiments of the present invention is the introduction of a threshold for the switching of the electrophoretic medium. If the attraction of the electrophoretic particles for each other, and hence their tendency to remain in stable homoaggregates, is sufficiently strong, it will require an appreciable field to pull the particles out of the homoaggregate so that they can migrate in the field.
- the existence of an appreciable threshold voltage for switching of the electrophoretic medium has a number of advantages. If the threshold is sufficiently large, it can enable the use of passive matrix addressing of a high resolution display. Smaller thresholds may be useful for reducing the sensitivity of an active matrix display to parasitic voltages and inter-pixel voltage leakage.
- the present invention also provides an additional approach to improving image stability in a dual particle electrophoretic medium.
- a dual particle electrophoretic medium it is necessary that both types of electrophoretic particles contribute to the overall image stability. It is not sufficient that one type of particle be "image stable" (that is, remain in a homoaggregate in the location to which it has been switched) if the second type of particle is free to settle or diffuse through the suspending fluid. If such is the case, one extreme optical state of the medium might show reasonably good image stability, but the other would display poor stability. Also if the particles can diffuse, poor image stability of one particle could easily contribute to optical degradation of the second extreme optical state as well. It is therefore desirable that both types of particles have steric stabilities that are appropriately adjusted to give good image stability.
- the degree to which the depletion mechanism applies to the two types of particles is different depending on the particle size and particle geometry, and is not independently adjustable, since only one polymer is added to the suspending fluid and used to impart image stability to both types of particles.
- the colloidal stability of each type of particle can be adjusted independently.
- adding a polymer to the suspending fluid in accordance with the depletion flocculation approach may make the two separate types of particles colloidally unstable with respect to each other as well as to themselves, thus encouraging heteroaggregation of the two types of particles.
- Heteroaggregation may cause a number of undesirable effects in a dual particle electrophoretic display, including slower response time and the requirement for the use of higher applied voltages (since a higher field strength will be required to separate the heteroaggregate) as well as poor optical states (since charged heteroaggregates may not be completely separated by the applied field if there is a large majority of particles of one type therein).
- these problems are eliminated, or at least reduced, by providing a dual particle electrophoretic medium in which homoaggregation of the two types of particles is thermodynamically favored over heteroaggregation.
- This can be achieved by providing the two types of particles with polymer shells that having their compatibility with the suspending fluid adjusted to favor homoaggregation, but also adjusting the compatibility of the two types of polymer shells to disfavor heteroaggregation.
- This is relatively easy to achieve by using different incompatible monomers in the polymer shells of the two types of particles; typically, the same compatible monomer can be used for the polymer shells on both types of particles. If the two incompatible monomers are not mutually compatible, then the tendency of the two types of particles to heteroaggregate will be minimal, and only homoaggregation encouraged, thus providing more rapid response time and purer optical states of the electrophoretic medium.
- the present invention is not restricted to the use of aliphatic hydrocarbons as suspending fluids.
- Fluoro- and halocarbon oils, silicone oils, aralkyl solvents (e.g., toluene, dodecylbenzene and the like) or mixtures thereof may be useful in this invention, since they allow further modification of the properties of the suspending fluid with respect to solvation of polymer shells, as described above.
- the present invention can be applied to all types of electrophoretic media, but may be especially useful in encapsulated electrophoretic media, whether using preformed capsules or microcells. This invention can also be applied to displays with all types of switching geometries, including both forward and back, as well as side-to-side and shutter mode switching.
- FIG. 1 of the accompanying drawings shows schematically a preferred process, as used in the Examples below, for preparing electrophoretic particles in accordance with the present invention, this process being essentially similar to that described in the aforementioned WO 02/093246.
- a base pigment particle 700 is coated with silica to produce a silicated pigment 702; this step is fully described in the aforementioned WO 02/093246.
- the silicated pigment 702 is treated with a bifunctional reagent having one functional group which reacts with the silica surface, and a second, charge control group, thus producing a surface functionalized pigment 704 bearing charge control groups on its surface.
- the bifunctional reagent also provides a site for the formation of polymer on the pigment particle.
- the surface functionalized pigment 704 is contacted with one or more monomers or oligomers under conditions effective to cause formation of polymer attached to the charge control groups, thereby producing a polymer-coated functional pigment 706, which is used in the electrophoretic medium.
- Particles were prepared with lauryl methacrylate homopolymer shells as a control experiment and are referred to by the letters J and D for white particles and black particles respectively. Modifications of the polymer shell are described as follows: for particles made using a random copolymer, one-stage polymerization, the comonomer is identified by the letter codes indicated in Table I below in parentheses after the appropriate pigment indicator together with a number indicating the mole fraction of the comonomer used in the polymerization reaction. The remainder in each case was lauryl methacrylate.
- the notation J(BMA15) indicates a white pigment made from a polymerization mixture comprising 15 mole % t-butyl methacrylate and 85 mole % lauryl methacrylate.
- a two-stage polymerization is indicated by a plus sign; in general, the mole ratio of polymers for these particles is not so well known, so that no indication is given about composition.
- J(+St) indicates a white pigment made by a two-stage polymerization.
- the starting material was the control pigment in this case, with 100 mole percent lauryl methacrylate used in the first stage polymerization; the second stage polymerization was carried out using only styrene as the polymerizable monomer. [0095] Table 1
- a single-neck, 250 mL round bottomed flask was equipped with a magnetic stir bar, reflux condenser and an argon/or nitrogen inlet and placed in a silicon oil bath.
- To the flask was added 60 g of silane-coated pigment which was pulverized into a fine powder using a mortar and pestle, followed by 60 mL of lauryl methacrylate (LMA, Aldrich) and 60 mL of toluene.
- LMA lauryl methacrylate
- the reaction mixture was then stirred rapidly and the flask purged with argon or nitrogen for one hour. During this time the silicon bath was heated to 50°C.
- a single-neck, 250 mL round bottomed flask was equipped with a magnetic stir bar, reflux condenser and an argon/or nitrogen inlet and placed in a silicon oil bath.
- To the flask were added 60 ml of toluene, 60g of titania (Z6030 coated Dupont R960), lauryl methacrylate (LMA) and a second monomer, such as styrene, t-butyl methacrylate, l-vmyl-2-pyrrolidinone, hexafluorobutyl acrylate and methacrylate, N-isopropylacrylamide or acrylonitrile (Aldrich), the amounts of LMA and second monomer depending on the desired monomer ratio.
- titania Z6030 coated Dupont R960
- LMA lauryl methacrylate
- a second monomer such as styrene, t-butyl methacrylate, l-vmyl-2
- the ratios of LMA/second monomer were usually 95/5, 85/15 and 75/25.
- the reaction mixture was then stirred rapidly and the flask purged with argon or nitrogen for one hour. During this time the silicon bath was heated to 50°C. During the purge, 0.6 g of AIBN was dissolved or partially dissolved in 13 mL of toluene. At the end of the one hour purge the ATBN/toluene solution was added quickly with a glass pipette. The reaction vessel was sealed, heated to 65°C and allowed to stir overnight. To the viscous reaction mixture was added 100 mL of ethyl acetate and the resultant mixture was allowed to stir for another 10 minutes.
- the mixture was poured into plastic bottles and centrifuged for 15-20 minutes at 3600 rpm and decanted. Fresh ethyl acetate was added to the centrifuged pigment, and the resultant mixture stirred with a stainless steel spatula and sonicated for 10 minutes. The pigment was washed twice more with ethyl acetate, centrifuged and decanted. The pigment was allowed to air dry overnight followed by drying under high vacuum for 24 hours. The free polymer in bulk solution was precipitated in methanol and dried under vacuum. The molecular weight of free polymer in the solution was determined by GPC. The polymer bound on the pigment was measured by TGA.
- a single-neck, 250 mL round bottomed flask was equipped with a magnetic stir bar, reflux condenser and an argon/or nitrogen inlet and placed in a silicon oil bath.
- To the flask were added 60 mL of toluene, 60 g of titania (Z6030 coated Dupont R960), 51 mL of lauryl methacrylate (LMA) and 3.3 mL of l-vinyl-2- pyrrolidinone.
- LMA lauryl methacrylate
- the mole ratio of LMA/l-vinyl-2-pyrrolidinone is usually 85/15.
- the reaction mixture was then stirred rapidly and the flask purged with argon or nitrogen for one hour. During this time the silicon bath was heated to 50°C. During the purge,
- AIBN 0.6 g was dissolved or partially dissolved in 13 mL of toluene. At the end of the one hour purge the AEBN/toluene solution was added quickly with a glass pipette.
- the reaction vessel was sealed, heated to 65 °C and allowed to stir overnight.
- To the viscous reaction mixture was added 100 mL of ethyl acetate and the resultant mixture was allowed to stir for another 10 minutes.
- the mixture was poured into plastic bottles and centrifuged for 15-20 minutes at 3600 rpm and decanted.
- Fresh ethyl acetate was added to the centrifuged pigment, and the resultant mixture was stirred with a stainless steel spatula and sonicated for 10 minutes.
- the pigment was washed twice more with ethyl acetate, centrifuged and decanted.
- the pigment was allowed to air dry overnight followed by drying under high vacuum for 24 hours.
- the free polymer in bulk solution was precipitated in methanol and dried under vacuum.
- the molecular weight of free polymer in the solution was determined by GPC.
- the polymer bound on the pigment was measured by TGA.
- a single-neck, 250 mL round bottomed flask was equipped with a magnetic stir bar, reflux condenser and an argon/or nitrogen inlet and placed in a silicon oil bath.
- To the flask was added 60 g of silane coated pigment which was pulverized into a fine powder using a mortar and pestle, followed by 60 mL of lauryl methacrylate and 60 mL of toluene.
- the reaction mixture was then stirred rapidly and the flask purged with argon or nitrogen for one hour. During this time the silicon bath was heated to 50°C.
- 0.6 g of AIBN was dissolved in 13 mL of toluene.
- the ATBN/toluene solution was added quickly with a glass pipette.
- the reaction vessel was sealed, heated to 65 °C and allowed to stir overnight.
- To the viscous reaction mixture was added 100 mL of ethyl acetate and the resultant mixture was allowed to stir for another 10 minutes.
- the mixture was poured into plastic bottles and centrifuged for 15-20 minutes at 3600 rpm and decanted. Fresh ethyl acetate was added to the centrifuged pigment, and the resultant mixture stirred with a stainless steel spatula. The pigment was washed twice more with ethyl acetate following the above procedure.
- the pigment was allowed to air dry overnight followed by drying under high vacuum for 24 hours.
- the free polymer in bulk solution was precipitated in methanol and dried under vacuum.
- the molecular weight of free polymer in the solution was determined by GPC.
- the polymer bound on the pigment was measured by TGA.
- the pigment was washed twice more with ethyl acetate following the above procedure.
- the pigment was allowed to air dry overnight followed by drying under high vacuum for 24 hours.
- the free polymer in bulk solution was precipitated in methanol and dried under vacuum.
- the molecular weight of free polymer in the solution was determined by GPC.
- the polymer bound on the pigment was measured by TGA.
- a single-neck, 250 mL round bottomed flask was equipped with a magnetic stir bar, reflux condenser and an argon inlet and placed in a silicon oil bath.
- To the flask was added 60 g of copper chromite (CuCr 2 O 4 , Shepherd Black 1G) coated with the silane of Formula (I) above, which had pulverized into a fine powder using a mortar and pestle, followed by 1.2 mL of styrene, 57 mL of lauryl methacrylate and 60 mL of toluene.
- the reaction mixture was then stirred rapidly and the flask purged with argon or nitrogen for one hour and the silicon bath was heated to 50°C.
- the pigment was washed twice more with ethyl acetate, centrifuged and decanted. The pigment was allowed to air dry overnight followed by drying under high vacuum for 24 hours. The free polymer in bulk solution was precipitated in methanol and dried under vacuum. The molecular weight of free polymer in the solution was determined by GPC. The polymer bound on the pigment was measured by TGA.
- a control internal phase was formulated from (a) 85 g of a stock solution of the J pigment containing 60 % by weight of LMA-coated titania in Isopar
- Span 85 (a non-ionic surfactant).
- An internal phase was formulated from (a) 40 g of a stock solution of a modified J pigment containing 60 % by weight of an LMA/TBMA-coated titania
- An internal phase was formulated from (a) 40 g of a stock solution of the same J pigment as in the control internal phase, this stock solution containing 60 % by weight of an LMA-coated titania in Isopar G; (b) 20 g of a stock solution of the D pigment containing 60 % by weight of LMA/St-coated copper chromite (85/15 or 95/5 monomer ratio) in Isopar G; (c) 5.04 g of a stock solution containing 10% by weight of Solsperse 17000 in Isopar G; (d) 14.60 g of Isopar G; and (e) 0.36 g Span 85 (a non-ionic surfactant).
- microcapsules were mixed into a slurry with a polyurethane binder and coated by a roll-to-roll process at a dry coating weight of 18 g m "2 on to the surface of a 7 mil (177 ⁇ m) poly(ethylene terephthalate) (PET) film carrying an indium tin oxide (ITO) layer on one surface, the microcapsules being deposited on the ITO-covered surface, substantially as described in Paragraphs [0075] and [0076] of the aforementioned 2002/0180687.
- PET poly(ethylene terephthalate)
- the capsule-bearing film was then formed into a front plane laminate by laminating it to a layer of a polyurethane lamination adhesive carried on a release sheet, this lamination being effected at 65 psig (0.51 mPa) at a speed of 6 inches/min (2.5 mm/sec) using a Western Magnum twin roll Laminator with both rolls held at 120°C.
- pieces of the resultant front plane laminate has their release sheets removed and were then laminated at 75 °C to a 5 cm by 5 cm PET film covered with a carbon black layer, which served as the rear electrode of the single pixel display.
- the control pixel shows relatively good black state stability, but rather poor white state stability (similar displays using carbon black instead of copper chromite as the black pigment and without PTB in the suspending fluid show much worse state stability, of the order of 10-15L* in both white and dark states).
- Any of the electrophoretic pigments of the invention results in an improvement in the image stability relative to the control, either in white state or black state or in both. Only in one case is the image stability of a pixel of the invention less good than the control (J(TBMA15)/D(St5)), and here the difference is probably within experimental pixel- to-pixel variation.
- the best overall image stability is afforded by the display made with white pigment synthesized using the two-stage method, with styrene as the second monomer.
- the incompatible monomer, particles, media and displays, and the homoaggregation medium, of the present invention provide good image stability and has a number of advantages over similar media containing polymer additives in the suspending fluid, especially since the present invention does not sacrifice response time to achieve good image stability.
- This advantage in response time can be used to operating a display at lower voltage.
- the displays reported in Table 2 switch almost to saturation in 300 msec at 7.5V, and achieve optimal contrast ratio at 10V and 250-300 msec. At 15 volts, switching times of around 100 msec can be achieved.
- the graft polymerization steps described in the aforementioned WO 02/093246 and above can be carried out in aliphatic hydrocarbons, especially isoparaffin solvents; a preferred solvent for this piupose is Isopar G, sold by Exxon Mobil Co ⁇ oration, Houston TX. Since such aliphatic hydrocarbons are the same materials typically used as the suspending fluids of electrophoretic media, the coated pigment can remain in essentially the same environment from the graft polymerization step until its inco ⁇ oration into the final display.
- the aliphatic hydrocarbon solvent may contain any one or more of excess polymerization initiator, excess monomer or oligomer, and free polymer not attached to pigment particles. These materials need to be substantially removed from the pigment particles before the pigment particles are inco ⁇ orated into the final electrophoretic medium, since the materials may adversely affect the electro-optic properties and/or operating lifetime of the medium. Accordingly, it will typically still be necessary to separate the polymer- coated pigment from the graft polymerization solvent, for example by centrifugation, and to wash the pigment particles to remove the last traces of the aforementioned materials.
- the suspending fluid in the electrophoretic medium is normally an aliphatic hydrocarbon, alone or in combination with a halocarbon.
- the suspending fluid is to be a mixture of an aliphatic hydrocarbon and a halocarbon
- a dual particle electrophoretic medium for example, a medium containing white titania particles and carbon black particles
- the process of the present invention can be applied to the two types of particles separately.
- the present invention may also be practiced by first blending the two types of pigment after providing polymerizable groups thereon, and then carrying out the graft polymerization step on this pigment mixture.
- the two pigments travel together through the remaining steps required to form the electrophoretic medium.
- the mixed polymer-coated pigment resulting from this process can be regarded as a stock solution of pigment that may require little or no further processing after washing to remove residual impurities. This variant of the present process thus further reduces the number of discrete steps which need to be carried out to convert raw pigments to the final electrophoretic medium.
- the process of the present invention decreases the safety hazards associated with the prior art processes, since the aliphatic hydrocarbon solvents used in the present process are less hazardous than toluene and THF.
- the present process reduces solvent use, and also reduces the number of steps in the overall process for producing electrophoretic media, since it eliminates the previous drying and pigment re-dispersion steps, thus decreasing dispersion processing time and possibly increasing the overall quality of the final dispersion of pigments in aliphatic hydrocarbon solvent.
- Another aspect of the present invention relates to controlling the relative sizes and masses of particles in an electrophoretic medium in order to reduce the tendency for the electrophoretic particles to agglomerate.
- the electrophoretic medium contains at least one type of particle whose average diameter is between about 0.25 and about 4 times the average diameter of a second type of particle bearing an opposite charge; desirably, the ratio of average particle mass between the two types of particles is between about 0.25 and about 4.00.
- the electrophoretic medium contains at least two types of particles bearing charges of opposite polarity that from a distance apart of gap (G) under an applied electric field of magnitude F are able to achieve velocities VI and V2 respectively such that: Vl + V2 > f(F, G) where f is a function such that when the inequality if satisfied, particles agglomeration is reduced or eliminated.
- Voltage threshold display
- this invention provides an active matrix electro- optic display which uses an electro-optic medium with a voltage threshold.
- the use of this type of electro-optic medium reduces design constraints on the backplane of the electro-optic display.
- strong voltage threshold is used to mean that the medium does not switch at all for voltages less than the threshold, no matter how long the pulse. At voltages above the threshold, the medium switches normally.
- the use of such a medium alleviates the problem of data spikes caused by coupling of the data lines to the pixel electrodes (see Paragraphs 42-52 above); if the amplitude of the voltage spikes to the pixel is less than 5V, such a strong voltage threshold medium will not respond.
- a pixel storage capacitor used in such a display need only be sized to keep the voltage spikes experienced by the electro-optic medium below the voltage threshold, which could reduce the size of the storage capacitor substantially, as compared with capacitors required in prior art electro-optic displays in which the electro-optic medium displays no voltage threshold.
- the present invention is not confined to the use of electro-optic media exhibiting strong voltage thresholds, but extends to the use of electro-optic media having only "weak voltage thresholds", a term which is used herein to refer to media which responds to voltages below a threshold applied for very long periods, but do not respond to such voltages applied over a short time of interest in the operation of an electro-optic display.
- the period of interest could be a single scan frame (typically about 20 msec), or an entire image update (typically about 1000 msec).
- the present invention may make use of any of the types of electro- optic media discussed above provided that the electro-optic media display strong or weak voltage thresholds.
- Incompatible monomer electrophoretic media having a voltage threshold, as described above, are generally preferred.
- This invention allows relaxation of the design rules for a active matrix array, typically a thin film transistor (TFT) array. More specifically, this invention allows the use of a smaller storage capacitor per unit of pixel electrode area, thus making it feasible to produce a TFT backplane using lower-resolution technologies such as printing. In addition, by reducing the size of the storage capacitor, the size of the transistor can also be reduced. Decreasing the size of these two components would result in a substantial decrease in line capacitance, and thus power consumption, for the backplane. Thus, electro-optic displays of the present invention would have improved performance over conventional designs using prior art amo ⁇ hous silicon construction.
Abstract
Description
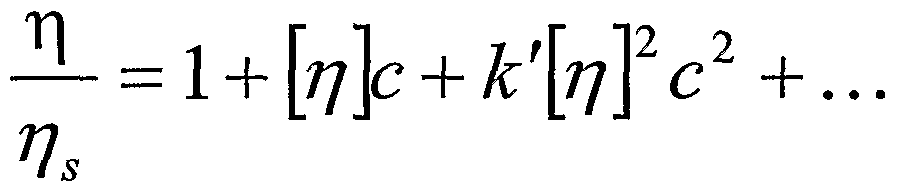



Claims
Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020107008236A KR101060980B1 (en) | 2003-10-08 | 2004-10-08 | Electrophoretic media |
| KR1020117012487A KR101197503B1 (en) | 2003-10-08 | 2004-10-08 | Electrophoretic media |
| KR1020137009666A KR20130048276A (en) | 2003-10-08 | 2004-10-08 | Electrophoretic media |
| KR1020067006846A KR101090039B1 (en) | 2003-10-08 | 2004-10-08 | Electrophoretic media |
| KR1020107021762A KR101307798B1 (en) | 2003-10-08 | 2004-10-08 | Electrophoretic media |
| JP2006534365A JP4848280B2 (en) | 2003-10-08 | 2004-10-08 | Electrophoretic medium |
| KR1020147021306A KR20140101879A (en) | 2003-10-08 | 2004-10-08 | Electrophoretic media |
| EP04794510A EP1671172A4 (en) | 2003-10-08 | 2004-10-08 | Electrophoretic media |
| HK07103167.4A HK1097602A1 (en) | 2003-10-08 | 2007-03-23 | Electrophoretic media |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US48148603P | 2003-10-08 | 2003-10-08 | |
| US60/481,486 | 2003-10-08 | ||
| US48157203P | 2003-10-28 | 2003-10-28 | |
| US60/481,572 | 2003-10-28 | ||
| US48157403P | 2003-10-29 | 2003-10-29 | |
| US60/481,574 | 2003-10-29 | ||
| US10/708,130 | 2004-02-09 | ||
| US10/708,130 US7002728B2 (en) | 1997-08-28 | 2004-02-09 | Electrophoretic particles, and processes for the production thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2005036129A2 true WO2005036129A2 (en) | 2005-04-21 |
| WO2005036129A3 WO2005036129A3 (en) | 2005-09-29 |
Family
ID=34437749
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2004/033188 WO2005036129A2 (en) | 2003-10-08 | 2004-10-08 | Electrophoretic media |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP1671172A4 (en) |
| JP (2) | JP2007328372A (en) |
| KR (3) | KR101090039B1 (en) |
| HK (1) | HK1132334A1 (en) |
| WO (1) | WO2005036129A2 (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2041739A2 (en) * | 2006-07-11 | 2009-04-01 | E Ink Corporation | Electrophoretic medium with improved image stability |
| EP2488593A1 (en) * | 2009-10-16 | 2012-08-22 | Hewlett-Packard Development Company, L.P. | Electronic inks |
| WO2013170933A1 (en) | 2012-05-14 | 2013-11-21 | Merck Patent Gmbh | Particles for electrophoretic displays |
| WO2013170937A1 (en) | 2012-05-14 | 2013-11-21 | Merck Patent Gmbh | Particles for electrophoretic displays |
| WO2013170936A1 (en) | 2012-05-14 | 2013-11-21 | Merck Patent Gmbh | Particles for electrophoretic displays |
| WO2013170938A1 (en) | 2012-05-14 | 2013-11-21 | Merck Patent Gmbh | Particles for electrophoretic displays |
| WO2013170932A1 (en) | 2012-05-14 | 2013-11-21 | Merck Patent Gmbh | Particles for electrophoretic displays |
| US8654070B2 (en) | 2009-08-21 | 2014-02-18 | Fuji Xerox Co., Ltd. | Electrophoretic particles, electrophoretic particle dispersion, display medium and display device |
| US8786936B2 (en) | 2011-12-29 | 2014-07-22 | Noroo Holdings Co., Ltd. | Ink compositions, methods of preparing ink compositions and display panels including ink compositions |
| US8961831B2 (en) | 2011-05-31 | 2015-02-24 | E Ink California, Llc | Silane-containing pigment particles for electrophoretic display |
| US9494808B2 (en) | 2012-05-14 | 2016-11-15 | Merck Patent Gmbh | Particles for electrophoretic displays |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI409305B (en) * | 2009-06-16 | 2013-09-21 | E Ink Corp | Electrophoretic particles |
| KR102577803B1 (en) | 2016-03-30 | 2023-09-13 | 주식회사 나노브릭 | Microcapsule Comprising Nanoparticle and Manufacturing Method Thereof |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4023968A (en) * | 1972-10-25 | 1977-05-17 | Xerox Corporation | Photoelectrophoretic color imaging process in which back migration is eliminated |
| US5932633A (en) * | 1997-08-22 | 1999-08-03 | Copytele, Inc. | Method for making polymers-coated pigment particles using initiator-treated pigments |
| US6445489B1 (en) * | 1998-03-18 | 2002-09-03 | E Ink Corporation | Electrophoretic displays and systems for addressing such displays |
| AU6744401A (en) * | 2000-05-31 | 2001-12-11 | Creavis Gesellschaft Fur Technologie Und Innovation Mbh | Polymerically micro-encapsulated pigments |
| JP2002244160A (en) * | 2001-02-16 | 2002-08-28 | Mitsubishi Paper Mills Ltd | Image display medium |
| EP1393122B1 (en) * | 2001-05-15 | 2018-03-28 | E Ink Corporation | Electrophoretic particles |
| WO2004099862A2 (en) * | 2003-05-02 | 2004-11-18 | E Ink Corporation | Electrophoretic displays |
-
2004
- 2004-10-08 EP EP04794510A patent/EP1671172A4/en not_active Ceased
- 2004-10-08 KR KR1020067006846A patent/KR101090039B1/en active IP Right Grant
- 2004-10-08 WO PCT/US2004/033188 patent/WO2005036129A2/en active Application Filing
- 2004-10-08 KR KR1020137009666A patent/KR20130048276A/en not_active IP Right Cessation
- 2004-10-08 KR KR1020147021306A patent/KR20140101879A/en not_active Application Discontinuation
-
2007
- 2007-08-31 JP JP2007226540A patent/JP2007328372A/en active Pending
-
2009
- 2009-09-16 HK HK09108482.9A patent/HK1132334A1/en unknown
-
2013
- 2013-01-28 JP JP2013013181A patent/JP2013140368A/en not_active Withdrawn
Non-Patent Citations (1)
| Title |
|---|
| See references of EP1671172A4 * |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2041739A4 (en) * | 2006-07-11 | 2009-11-04 | E Ink Corp | Electrophoretic medium with improved image stability |
| EP2041739A2 (en) * | 2006-07-11 | 2009-04-01 | E Ink Corporation | Electrophoretic medium with improved image stability |
| US8654070B2 (en) | 2009-08-21 | 2014-02-18 | Fuji Xerox Co., Ltd. | Electrophoretic particles, electrophoretic particle dispersion, display medium and display device |
| EP2488593A4 (en) * | 2009-10-16 | 2013-12-11 | Hewlett Packard Development Co | Electronic inks |
| EP2488593A1 (en) * | 2009-10-16 | 2012-08-22 | Hewlett-Packard Development Company, L.P. | Electronic inks |
| CN102666742A (en) * | 2009-10-16 | 2012-09-12 | 惠普发展公司,有限责任合伙企业 | Electronic inks |
| US8961831B2 (en) | 2011-05-31 | 2015-02-24 | E Ink California, Llc | Silane-containing pigment particles for electrophoretic display |
| US8786936B2 (en) | 2011-12-29 | 2014-07-22 | Noroo Holdings Co., Ltd. | Ink compositions, methods of preparing ink compositions and display panels including ink compositions |
| WO2013170933A1 (en) | 2012-05-14 | 2013-11-21 | Merck Patent Gmbh | Particles for electrophoretic displays |
| WO2013170932A1 (en) | 2012-05-14 | 2013-11-21 | Merck Patent Gmbh | Particles for electrophoretic displays |
| WO2013170938A1 (en) | 2012-05-14 | 2013-11-21 | Merck Patent Gmbh | Particles for electrophoretic displays |
| WO2013170936A1 (en) | 2012-05-14 | 2013-11-21 | Merck Patent Gmbh | Particles for electrophoretic displays |
| WO2013170937A1 (en) | 2012-05-14 | 2013-11-21 | Merck Patent Gmbh | Particles for electrophoretic displays |
| US9494808B2 (en) | 2012-05-14 | 2016-11-15 | Merck Patent Gmbh | Particles for electrophoretic displays |
| US9588357B2 (en) | 2012-05-14 | 2017-03-07 | Merck Patent Gmbh | Particles for electrophoretic displays |
| US9594260B2 (en) | 2012-05-14 | 2017-03-14 | Merck Patent Gmbh | Particles for electrophoretic displays |
| US9645416B2 (en) | 2012-05-14 | 2017-05-09 | Merck Patent Gmbh | Particles for electrophoretic displays |
| US9651846B2 (en) | 2012-05-14 | 2017-05-16 | Merck Patent Gmbh | Particles for electrophoretic displays |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20140101879A (en) | 2014-08-20 |
| WO2005036129A3 (en) | 2005-09-29 |
| KR101090039B1 (en) | 2011-12-07 |
| JP2013140368A (en) | 2013-07-18 |
| HK1132334A1 (en) | 2010-02-19 |
| EP1671172A2 (en) | 2006-06-21 |
| JP2007328372A (en) | 2007-12-20 |
| EP1671172A4 (en) | 2007-08-22 |
| KR20070028287A (en) | 2007-03-12 |
| KR20130048276A (en) | 2013-05-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5250580B2 (en) | Electrophoretic medium | |
| US20230005439A1 (en) | Colored electrophoretic displays | |
| US7230750B2 (en) | Electrophoretic media and processes for the production thereof | |
| US20100148385A1 (en) | Electrophoretic media and processes for the production thereof | |
| JP2013140368A (en) | Electrophoretic medium | |
| US10214647B2 (en) | Electrophoretic particles and processes for the production thereof | |
| US9759976B2 (en) | Electrophoretic display medium and preparing method thereof | |
| EP3888079A1 (en) | Electro-optic displays and driving methods | |
| JP7416827B2 (en) | colored electrophoretic display | |
| TW202340834A (en) | Electrophoretic media comprising electrophoretic particles and a combination of charge control agents |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200480036461.9 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006534365 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020067006846 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004794510 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004794510 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020067006846 Country of ref document: KR |