WO2004106348A1 - アミノメチル置換フルオロチアゾロベンゾイミダゾール誘導体 - Google Patents
アミノメチル置換フルオロチアゾロベンゾイミダゾール誘導体 Download PDFInfo
- Publication number
- WO2004106348A1 WO2004106348A1 PCT/JP2004/007655 JP2004007655W WO2004106348A1 WO 2004106348 A1 WO2004106348 A1 WO 2004106348A1 JP 2004007655 W JP2004007655 W JP 2004007655W WO 2004106348 A1 WO2004106348 A1 WO 2004106348A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- acid
- added
- mixture
- optionally substituted
- compound
- Prior art date
Links
- -1 Aminomethyl-substituted fluorothiazolobenzimidazole Chemical class 0.000 title claims abstract description 35
- 150000003839 salts Chemical class 0.000 claims abstract description 28
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 22
- 239000003814 drug Substances 0.000 claims abstract description 14
- 125000002947 alkylene group Chemical group 0.000 claims abstract description 12
- 125000000623 heterocyclic group Chemical group 0.000 claims abstract description 12
- 229910052717 sulfur Inorganic materials 0.000 claims abstract description 7
- 125000003342 alkenyl group Chemical group 0.000 claims abstract description 6
- 125000000304 alkynyl group Chemical group 0.000 claims abstract description 6
- 239000001301 oxygen Substances 0.000 claims abstract description 6
- 229910052760 oxygen Inorganic materials 0.000 claims abstract description 6
- 125000005346 substituted cycloalkyl group Chemical group 0.000 claims abstract description 6
- 101100490437 Mus musculus Acvrl1 gene Proteins 0.000 claims abstract description 5
- 239000011593 sulfur Substances 0.000 claims abstract description 5
- 125000004093 cyano group Chemical group *C#N 0.000 claims abstract description 4
- 125000005842 heteroatom Chemical group 0.000 claims description 15
- 208000004296 neuralgia Diseases 0.000 claims description 15
- 208000021722 neuropathic pain Diseases 0.000 claims description 15
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 12
- 239000004480 active ingredient Substances 0.000 claims description 8
- 208000002193 Pain Diseases 0.000 claims description 7
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 7
- 229940124597 therapeutic agent Drugs 0.000 claims description 7
- 230000036407 pain Effects 0.000 claims description 6
- 125000004043 oxo group Chemical group O=* 0.000 claims description 5
- 125000005843 halogen group Chemical group 0.000 claims description 4
- 239000008194 pharmaceutical composition Substances 0.000 claims description 4
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical compound C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 claims description 3
- 150000002926 oxygen Chemical class 0.000 claims description 3
- XIZCDQOKKYYCRH-UHFFFAOYSA-N 1h-benzimidazole-2-carboxamide Chemical compound C1=CC=C2NC(C(=O)N)=NC2=C1 XIZCDQOKKYYCRH-UHFFFAOYSA-N 0.000 claims description 2
- JUGHOUIZKUEEBN-UHFFFAOYSA-N [1,3]thiazolo[3,2-a]benzimidazole-2-carboxamide Chemical compound S1C(=CN2C1=NC1=C2C=CC=C1)C(=O)N JUGHOUIZKUEEBN-UHFFFAOYSA-N 0.000 claims description 2
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 claims description 2
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 2
- 150000003463 sulfur Chemical class 0.000 claims description 2
- 201000001119 neuropathy Diseases 0.000 claims 1
- 230000007823 neuropathy Effects 0.000 claims 1
- 208000033808 peripheral neuropathy Diseases 0.000 claims 1
- 230000001568 sexual effect Effects 0.000 claims 1
- 230000000694 effects Effects 0.000 abstract description 21
- 108010010914 Metabotropic glutamate receptors Proteins 0.000 abstract description 10
- 102000016193 Metabotropic glutamate receptors Human genes 0.000 abstract description 10
- 229910052739 hydrogen Inorganic materials 0.000 abstract description 10
- 239000001257 hydrogen Substances 0.000 abstract description 10
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 abstract description 7
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 abstract description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 abstract description 3
- 150000002431 hydrogen Chemical class 0.000 abstract description 2
- 125000001188 haloalkyl group Chemical group 0.000 abstract 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 129
- 150000001875 compounds Chemical class 0.000 description 110
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 107
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 87
- 239000000203 mixture Substances 0.000 description 76
- 239000000243 solution Substances 0.000 description 70
- 230000002829 reductive effect Effects 0.000 description 56
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 51
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 48
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 45
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 40
- 238000001914 filtration Methods 0.000 description 36
- 238000010438 heat treatment Methods 0.000 description 34
- 238000004519 manufacturing process Methods 0.000 description 34
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 33
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 33
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 29
- 238000006243 chemical reaction Methods 0.000 description 29
- 239000013078 crystal Substances 0.000 description 27
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 26
- 238000005160 1H NMR spectroscopy Methods 0.000 description 25
- 238000012360 testing method Methods 0.000 description 22
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 21
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 20
- 239000002253 acid Substances 0.000 description 19
- 239000002904 solvent Substances 0.000 description 19
- 235000011054 acetic acid Nutrition 0.000 description 16
- 238000000034 method Methods 0.000 description 16
- QGJOPFRUJISHPQ-UHFFFAOYSA-N Carbon disulfide Chemical compound S=C=S QGJOPFRUJISHPQ-UHFFFAOYSA-N 0.000 description 15
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 15
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 14
- 239000000725 suspension Substances 0.000 description 14
- 239000012046 mixed solvent Substances 0.000 description 13
- 238000001816 cooling Methods 0.000 description 12
- 239000003921 oil Substances 0.000 description 12
- 235000019198 oils Nutrition 0.000 description 12
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 11
- 239000007864 aqueous solution Substances 0.000 description 11
- 239000011541 reaction mixture Substances 0.000 description 11
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 10
- 241000700159 Rattus Species 0.000 description 10
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 10
- 125000003277 amino group Chemical group 0.000 description 10
- 238000002156 mixing Methods 0.000 description 10
- AMLBAOPYPUXEQF-UHFFFAOYSA-N 1-(2,6-difluorophenyl)-1,3-dihydro-[1,3]thiazolo[3,4-a]benzimidazole Chemical class FC1=CC=CC(F)=C1C1N2C3=CC=CC=C3N=C2CS1 AMLBAOPYPUXEQF-UHFFFAOYSA-N 0.000 description 9
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 9
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 9
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 230000002378 acidificating effect Effects 0.000 description 9
- 235000011114 ammonium hydroxide Nutrition 0.000 description 9
- 239000003795 chemical substances by application Substances 0.000 description 9
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 9
- 239000012442 inert solvent Substances 0.000 description 9
- 108090000623 proteins and genes Proteins 0.000 description 9
- 102000005962 receptors Human genes 0.000 description 9
- 108020003175 receptors Proteins 0.000 description 9
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 8
- 239000002244 precipitate Substances 0.000 description 8
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 7
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 7
- 238000004440 column chromatography Methods 0.000 description 7
- 230000007886 mutagenicity Effects 0.000 description 7
- 231100000299 mutagenicity Toxicity 0.000 description 7
- 239000012044 organic layer Substances 0.000 description 7
- 230000008569 process Effects 0.000 description 7
- 230000004044 response Effects 0.000 description 7
- 229920006395 saturated elastomer Polymers 0.000 description 7
- 239000012279 sodium borohydride Substances 0.000 description 7
- 229910000033 sodium borohydride Inorganic materials 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- 125000001424 substituent group Chemical group 0.000 description 7
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 6
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 6
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 6
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 6
- 208000000114 Pain Threshold Diseases 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- 239000005557 antagonist Substances 0.000 description 6
- 239000012298 atmosphere Substances 0.000 description 6
- 239000003480 eluent Substances 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- 238000010265 fast atom bombardment Methods 0.000 description 6
- 238000006460 hydrolysis reaction Methods 0.000 description 6
- 239000005457 ice water Substances 0.000 description 6
- 230000037040 pain threshold Effects 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- 239000011734 sodium Substances 0.000 description 6
- 229910052708 sodium Inorganic materials 0.000 description 6
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 5
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 5
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 5
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 5
- 230000004913 activation Effects 0.000 description 5
- 206010008118 cerebral infarction Diseases 0.000 description 5
- 208000026106 cerebrovascular disease Diseases 0.000 description 5
- 206010012601 diabetes mellitus Diseases 0.000 description 5
- 229910052731 fluorine Inorganic materials 0.000 description 5
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 5
- 230000002503 metabolic effect Effects 0.000 description 5
- 238000007254 oxidation reaction Methods 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 239000012264 purified product Substances 0.000 description 5
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 5
- 235000017557 sodium bicarbonate Nutrition 0.000 description 5
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 5
- 238000003756 stirring Methods 0.000 description 5
- SVUOLADPCWQTTE-UHFFFAOYSA-N 1h-1,2-benzodiazepine Chemical compound N1N=CC=CC2=CC=CC=C12 SVUOLADPCWQTTE-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 4
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 4
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 4
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 4
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N N-phenyl amine Natural products NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 4
- 206010029174 Nerve compression Diseases 0.000 description 4
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 4
- 230000009471 action Effects 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- 229940049706 benzodiazepine Drugs 0.000 description 4
- MTZQAGJQAFMTAQ-UHFFFAOYSA-N benzoic acid ethyl ester Natural products CCOC(=O)C1=CC=CC=C1 MTZQAGJQAFMTAQ-UHFFFAOYSA-N 0.000 description 4
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 4
- 229910052794 bromium Inorganic materials 0.000 description 4
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 4
- 239000003638 chemical reducing agent Substances 0.000 description 4
- 238000006297 dehydration reaction Methods 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 206010015037 epilepsy Diseases 0.000 description 4
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 4
- 125000001153 fluoro group Chemical group F* 0.000 description 4
- 230000007062 hydrolysis Effects 0.000 description 4
- 239000003701 inert diluent Substances 0.000 description 4
- 239000008101 lactose Substances 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 210000002569 neuron Anatomy 0.000 description 4
- 125000004430 oxygen atom Chemical group O* 0.000 description 4
- LVTJOONKWUXEFR-FZRMHRINSA-N protoneodioscin Natural products O(C[C@@H](CC[C@]1(O)[C@H](C)[C@@H]2[C@]3(C)[C@H]([C@H]4[C@@H]([C@]5(C)C(=CC4)C[C@@H](O[C@@H]4[C@H](O[C@H]6[C@@H](O)[C@@H](O)[C@@H](O)[C@H](C)O6)[C@@H](O)[C@H](O[C@H]6[C@@H](O)[C@@H](O)[C@@H](O)[C@H](C)O6)[C@H](CO)O4)CC5)CC3)C[C@@H]2O1)C)[C@H]1[C@H](O)[C@H](O)[C@H](O)[C@@H](CO)O1 LVTJOONKWUXEFR-FZRMHRINSA-N 0.000 description 4
- 238000006722 reduction reaction Methods 0.000 description 4
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 4
- 125000004434 sulfur atom Chemical group 0.000 description 4
- AKEJUJNQAAGONA-UHFFFAOYSA-N sulfur trioxide Chemical compound O=S(=O)=O AKEJUJNQAAGONA-UHFFFAOYSA-N 0.000 description 4
- 239000006228 supernatant Substances 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- 229920001817 Agar Polymers 0.000 description 3
- 241000894006 Bacteria Species 0.000 description 3
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 3
- 102000018899 Glutamate Receptors Human genes 0.000 description 3
- 108010027915 Glutamate Receptors Proteins 0.000 description 3
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- 208000007271 Substance Withdrawal Syndrome Diseases 0.000 description 3
- 229930006000 Sucrose Natural products 0.000 description 3
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 3
- 206010048010 Withdrawal syndrome Diseases 0.000 description 3
- 125000000738 acetamido group Chemical group [H]C([H])([H])C(=O)N([H])[*] 0.000 description 3
- 150000008065 acid anhydrides Chemical class 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 239000008272 agar Substances 0.000 description 3
- 125000004202 aminomethyl group Chemical group [H]N([H])C([H])([H])* 0.000 description 3
- 239000012300 argon atmosphere Substances 0.000 description 3
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 3
- 230000027455 binding Effects 0.000 description 3
- 210000004556 brain Anatomy 0.000 description 3
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 3
- 125000004432 carbon atom Chemical group C* 0.000 description 3
- 238000010531 catalytic reduction reaction Methods 0.000 description 3
- 210000004027 cell Anatomy 0.000 description 3
- 230000030833 cell death Effects 0.000 description 3
- 235000015165 citric acid Nutrition 0.000 description 3
- 238000002425 crystallisation Methods 0.000 description 3
- 230000008025 crystallization Effects 0.000 description 3
- 125000000753 cycloalkyl group Chemical group 0.000 description 3
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 239000001530 fumaric acid Substances 0.000 description 3
- 229930195712 glutamate Natural products 0.000 description 3
- 239000004220 glutamic acid Substances 0.000 description 3
- 235000013922 glutamic acid Nutrition 0.000 description 3
- 229910052736 halogen Inorganic materials 0.000 description 3
- 150000002367 halogens Chemical class 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 229910017604 nitric acid Inorganic materials 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 230000003647 oxidation Effects 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 description 3
- 229940002612 prodrug Drugs 0.000 description 3
- 239000000651 prodrug Substances 0.000 description 3
- 235000018102 proteins Nutrition 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 239000008247 solid mixture Substances 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 239000005720 sucrose Substances 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 2
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 2
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical class C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 2
- CWVVNPJEMXUWBE-UHFFFAOYSA-N 1h-benzimidazole-2,4-dicarboxylic acid Chemical compound C1=CC=C2NC(C(=O)O)=NC2=C1C(O)=O CWVVNPJEMXUWBE-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- 206010058019 Cancer Pain Diseases 0.000 description 2
- 206010007269 Carcinogenicity Diseases 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- 229940126062 Compound A Drugs 0.000 description 2
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 2
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 2
- 102000004310 Ion Channels Human genes 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 2
- 108010065028 Metabotropic Glutamate 5 Receptor Proteins 0.000 description 2
- 102100036837 Metabotropic glutamate receptor 2 Human genes 0.000 description 2
- 102100038357 Metabotropic glutamate receptor 5 Human genes 0.000 description 2
- 102100038300 Metabotropic glutamate receptor 6 Human genes 0.000 description 2
- 102100037636 Metabotropic glutamate receptor 8 Human genes 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 208000019695 Migraine disease Diseases 0.000 description 2
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 206010036376 Postherpetic Neuralgia Diseases 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- MMWCIQZXVOZEGG-HOZKJCLWSA-N [(1S,2R,3S,4S,5R,6S)-2,3,5-trihydroxy-4,6-diphosphonooxycyclohexyl] dihydrogen phosphate Chemical compound O[C@H]1[C@@H](O)[C@H](OP(O)(O)=O)[C@@H](OP(O)(O)=O)[C@H](O)[C@H]1OP(O)(O)=O MMWCIQZXVOZEGG-HOZKJCLWSA-N 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 239000002671 adjuvant Substances 0.000 description 2
- 235000010419 agar Nutrition 0.000 description 2
- 239000000556 agonist Substances 0.000 description 2
- 230000001476 alcoholic effect Effects 0.000 description 2
- 238000007112 amidation reaction Methods 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 150000001448 anilines Chemical class 0.000 description 2
- 150000001450 anions Chemical class 0.000 description 2
- 235000003704 aspartic acid Nutrition 0.000 description 2
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 231100000260 carcinogenicity Toxicity 0.000 description 2
- 230000007670 carcinogenicity Effects 0.000 description 2
- 150000001768 cations Chemical class 0.000 description 2
- 210000001638 cerebellum Anatomy 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 239000012024 dehydrating agents Substances 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 2
- 238000000132 electrospray ionisation Methods 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- PQJJJMRNHATNKG-UHFFFAOYSA-N ethyl bromoacetate Chemical compound CCOC(=O)CBr PQJJJMRNHATNKG-UHFFFAOYSA-N 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 238000004992 fast atom bombardment mass spectroscopy Methods 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- WBJINCZRORDGAQ-UHFFFAOYSA-N formic acid ethyl ester Natural products CCOC=O WBJINCZRORDGAQ-UHFFFAOYSA-N 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 231100000025 genetic toxicology Toxicity 0.000 description 2
- 230000001738 genotoxic effect Effects 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 2
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 108010038421 metabotropic glutamate receptor 2 Proteins 0.000 description 2
- 108010038450 metabotropic glutamate receptor 6 Proteins 0.000 description 2
- 108010038448 metabotropic glutamate receptor 8 Proteins 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 206010027599 migraine Diseases 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- LZGUHMNOBNWABZ-UHFFFAOYSA-N n-nitro-n-phenylnitramide Chemical class [O-][N+](=O)N([N+]([O-])=O)C1=CC=CC=C1 LZGUHMNOBNWABZ-UHFFFAOYSA-N 0.000 description 2
- 210000001640 nerve ending Anatomy 0.000 description 2
- 230000000802 nitrating effect Effects 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- 239000008188 pellet Substances 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 150000003905 phosphatidylinositols Chemical class 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- FGIUAXJPYTZDNR-UHFFFAOYSA-N potassium nitrate Chemical compound [K+].[O-][N+]([O-])=O FGIUAXJPYTZDNR-UHFFFAOYSA-N 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 230000000069 prophylactic effect Effects 0.000 description 2
- 239000002464 receptor antagonist Substances 0.000 description 2
- 229940044551 receptor antagonist Drugs 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 238000006268 reductive amination reaction Methods 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 238000007363 ring formation reaction Methods 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- 239000012064 sodium phosphate buffer Substances 0.000 description 2
- 210000001032 spinal nerve Anatomy 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- 238000010998 test method Methods 0.000 description 2
- SFLXUZPXEWWQNH-UHFFFAOYSA-K tetrabutylazanium;tribromide Chemical compound [Br-].[Br-].[Br-].CCCC[N+](CCCC)(CCCC)CCCC.CCCC[N+](CCCC)(CCCC)CCCC.CCCC[N+](CCCC)(CCCC)CCCC SFLXUZPXEWWQNH-UHFFFAOYSA-K 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- VXUYXOFXAQZZMF-UHFFFAOYSA-N titanium(IV) isopropoxide Chemical compound CC(C)O[Ti](OC(C)C)(OC(C)C)OC(C)C VXUYXOFXAQZZMF-UHFFFAOYSA-N 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 229920002554 vinyl polymer Polymers 0.000 description 2
- 238000010792 warming Methods 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- TXUICONDJPYNPY-UHFFFAOYSA-N (1,10,13-trimethyl-3-oxo-4,5,6,7,8,9,11,12,14,15,16,17-dodecahydrocyclopenta[a]phenanthren-17-yl) heptanoate Chemical compound C1CC2CC(=O)C=C(C)C2(C)C2C1C1CCC(OC(=O)CCCCCC)C1(C)CC2 TXUICONDJPYNPY-UHFFFAOYSA-N 0.000 description 1
- 125000003161 (C1-C6) alkylene group Chemical group 0.000 description 1
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- IMLSAISZLJGWPP-UHFFFAOYSA-N 1,3-dithiolane Chemical compound C1CSCS1 IMLSAISZLJGWPP-UHFFFAOYSA-N 0.000 description 1
- OGYGFUAIIOPWQD-UHFFFAOYSA-N 1,3-thiazolidine Chemical compound C1CSCN1 OGYGFUAIIOPWQD-UHFFFAOYSA-N 0.000 description 1
- LOZWAPSEEHRYPG-UHFFFAOYSA-N 1,4-dithiane Chemical compound C1CSCCS1 LOZWAPSEEHRYPG-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- LSTPKMWNRWCNLS-UHFFFAOYSA-N 1-amino-2,3-dihydroindene-1,5-dicarboxylic acid Chemical compound OC(=O)C1=CC=C2C(N)(C(O)=O)CCC2=C1 LSTPKMWNRWCNLS-UHFFFAOYSA-N 0.000 description 1
- PLRACCBDVIHHLZ-UHFFFAOYSA-N 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine Chemical compound C1N(C)CCC(C=2C=CC=CC=2)=C1 PLRACCBDVIHHLZ-UHFFFAOYSA-N 0.000 description 1
- WIIFZYZMLRVMIP-UHFFFAOYSA-N 2,3-dihydro-1h-benzimidazole-5-carboxylic acid Chemical compound OC(=O)C1=CC=C2NCNC2=C1 WIIFZYZMLRVMIP-UHFFFAOYSA-N 0.000 description 1
- VZSRBBMJRBPUNF-UHFFFAOYSA-N 2-(2,3-dihydro-1H-inden-2-ylamino)-N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]pyrimidine-5-carboxamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C(=O)NCCC(N1CC2=C(CC1)NN=N2)=O VZSRBBMJRBPUNF-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- HOOWCUZPEFNHDT-UHFFFAOYSA-N 2-amino-2-(3,5-dihydroxyphenyl)acetic acid Chemical compound OC(=O)C(N)C1=CC(O)=CC(O)=C1 HOOWCUZPEFNHDT-UHFFFAOYSA-N 0.000 description 1
- GSFNQBFZFXUTBN-UHFFFAOYSA-N 2-chlorothiophene Chemical compound ClC1=CC=CS1 GSFNQBFZFXUTBN-UHFFFAOYSA-N 0.000 description 1
- QGCDCQXHCJKJHS-UHFFFAOYSA-N 2-hydroxypropanoate;morpholin-4-ium Chemical compound CC(O)C([O-])=O.C1COCC[NH2+]1 QGCDCQXHCJKJHS-UHFFFAOYSA-N 0.000 description 1
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- JHUUPUMBZGWODW-UHFFFAOYSA-N 3,6-dihydro-1,2-dioxine Chemical compound C1OOCC=C1 JHUUPUMBZGWODW-UHFFFAOYSA-N 0.000 description 1
- GJKMGBBFFJQKLO-UHFFFAOYSA-N 3,6-dihydro-2h-imidazo[4,5-g][1,3]benzothiazole Chemical group C1=C2NC=NC2=C2SCNC2=C1 GJKMGBBFFJQKLO-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- COYPLDIXZODDDL-UHFFFAOYSA-N 3h-benzimidazole-5-carboxylic acid Chemical compound OC(=O)C1=CC=C2N=CNC2=C1 COYPLDIXZODDDL-UHFFFAOYSA-N 0.000 description 1
- HCCNHYWZYYIOFM-UHFFFAOYSA-N 3h-benzo[e]benzimidazole Chemical compound C1=CC=C2C(N=CN3)=C3C=CC2=C1 HCCNHYWZYYIOFM-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 238000010953 Ames test Methods 0.000 description 1
- 231100000039 Ames test Toxicity 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 208000019901 Anxiety disease Diseases 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 1
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 description 1
- BHPQYMZQTOCNFJ-UHFFFAOYSA-N Calcium cation Chemical compound [Ca+2] BHPQYMZQTOCNFJ-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- PTHCMJGKKRQCBF-UHFFFAOYSA-N Cellulose, microcrystalline Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC)C(CO)O1 PTHCMJGKKRQCBF-UHFFFAOYSA-N 0.000 description 1
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 208000000094 Chronic Pain Diseases 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- GSNUFIFRDBKVIE-UHFFFAOYSA-N DMF Natural products CC1=CC=C(C)O1 GSNUFIFRDBKVIE-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 208000032131 Diabetic Neuropathies Diseases 0.000 description 1
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 1
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical class OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- 108091006027 G proteins Proteins 0.000 description 1
- 102000030782 GTP binding Human genes 0.000 description 1
- 108091000058 GTP-Binding Proteins 0.000 description 1
- 206010064571 Gene mutation Diseases 0.000 description 1
- 235000003332 Ilex aquifolium Nutrition 0.000 description 1
- 241000209027 Ilex aquifolium Species 0.000 description 1
- WRYCSMQKUKOKBP-UHFFFAOYSA-N Imidazolidine Chemical compound C1CNCN1 WRYCSMQKUKOKBP-UHFFFAOYSA-N 0.000 description 1
- 206010061216 Infarction Diseases 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 102100036834 Metabotropic glutamate receptor 1 Human genes 0.000 description 1
- 102100038352 Metabotropic glutamate receptor 3 Human genes 0.000 description 1
- 102100038354 Metabotropic glutamate receptor 4 Human genes 0.000 description 1
- 102100038294 Metabotropic glutamate receptor 7 Human genes 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- MKYBYDHXWVHEJW-UHFFFAOYSA-N N-[1-oxo-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propan-2-yl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(C(C)NC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 MKYBYDHXWVHEJW-UHFFFAOYSA-N 0.000 description 1
- NIPNSKYNPDTRPC-UHFFFAOYSA-N N-[2-oxo-2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(CNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 NIPNSKYNPDTRPC-UHFFFAOYSA-N 0.000 description 1
- AFCARXCZXQIEQB-UHFFFAOYSA-N N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(CCNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 AFCARXCZXQIEQB-UHFFFAOYSA-N 0.000 description 1
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 208000004550 Postoperative Pain Diseases 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 241000293869 Salmonella enterica subsp. enterica serovar Typhimurium Species 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 241000256011 Sphingidae Species 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 238000006859 Swern oxidation reaction Methods 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 229910021626 Tin(II) chloride Inorganic materials 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 102000014384 Type C Phospholipases Human genes 0.000 description 1
- 108010079194 Type C Phospholipases Proteins 0.000 description 1
- JAWMENYCRQKKJY-UHFFFAOYSA-N [3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-ylmethyl)-1-oxa-2,8-diazaspiro[4.5]dec-2-en-8-yl]-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]methanone Chemical compound N1N=NC=2CN(CCC=21)CC1=NOC2(C1)CCN(CC2)C(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F JAWMENYCRQKKJY-UHFFFAOYSA-N 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000001785 acacia senegal l. willd gum Substances 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 239000003377 acid catalyst Substances 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 239000005456 alcohol based solvent Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 125000004450 alkenylene group Chemical group 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- WMGSQTMJHBYJMQ-UHFFFAOYSA-N aluminum;magnesium;silicate Chemical compound [Mg+2].[Al+3].[O-][Si]([O-])([O-])[O-] WMGSQTMJHBYJMQ-UHFFFAOYSA-N 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 230000003042 antagnostic effect Effects 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- QOJJMZDSPHXWMB-UHFFFAOYSA-N azanium;4-amino-2-fluoro-5-nitrobenzoate Chemical compound [NH4+].NC1=CC(F)=C(C([O-])=O)C=C1[N+]([O-])=O QOJJMZDSPHXWMB-UHFFFAOYSA-N 0.000 description 1
- ZSIQJIWKELUFRJ-UHFFFAOYSA-N azepane Chemical compound C1CCCNCC1 ZSIQJIWKELUFRJ-UHFFFAOYSA-N 0.000 description 1
- 239000003899 bactericide agent Substances 0.000 description 1
- MYONAGGJKCJOBT-UHFFFAOYSA-N benzimidazol-2-one Chemical compound C1=CC=CC2=NC(=O)N=C21 MYONAGGJKCJOBT-UHFFFAOYSA-N 0.000 description 1
- 125000003310 benzodiazepinyl group Chemical group N1N=C(C=CC2=C1C=CC=C2)* 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- OUGIDAPQYNCXRA-UHFFFAOYSA-N beta-naphthoflavone Chemical compound O1C2=CC=C3C=CC=CC3=C2C(=O)C=C1C1=CC=CC=C1 OUGIDAPQYNCXRA-UHFFFAOYSA-N 0.000 description 1
- MJKDQDZHUIFGLN-UHFFFAOYSA-N bis(1H-imidazol-2-yl)diazene Chemical class C1=CNC(N=NC=2NC=CN=2)=N1 MJKDQDZHUIFGLN-UHFFFAOYSA-N 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 230000003491 cAMP production Effects 0.000 description 1
- 229910001424 calcium ion Inorganic materials 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- QGJOPFRUJISHPQ-NJFSPNSNSA-N carbon disulfide-14c Chemical compound S=[14C]=S QGJOPFRUJISHPQ-NJFSPNSNSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 230000002490 cerebral effect Effects 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000007810 chemical reaction solvent Substances 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- KRVSOGSZCMJSLX-UHFFFAOYSA-L chromic acid Substances O[Cr](O)(=O)=O KRVSOGSZCMJSLX-UHFFFAOYSA-L 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 210000003792 cranial nerve Anatomy 0.000 description 1
- 239000010779 crude oil Substances 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 125000001162 cycloheptenyl group Chemical group C1(=CCCCCC1)* 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 238000005947 deacylation reaction Methods 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- VAYGXNSJCAHWJZ-UHFFFAOYSA-N dimethyl sulfate Chemical compound COS(=O)(=O)OC VAYGXNSJCAHWJZ-UHFFFAOYSA-N 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 239000000386 donor Substances 0.000 description 1
- 229960003638 dopamine Drugs 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 238000010931 ester hydrolysis Methods 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- SPGMDXDPJKEDGC-UHFFFAOYSA-N ethyl 4-amino-3-fluorobenzoate Chemical compound CCOC(=O)C1=CC=C(N)C(F)=C1 SPGMDXDPJKEDGC-UHFFFAOYSA-N 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- VGGRCVDNFAQIKO-UHFFFAOYSA-N formic anhydride Chemical compound O=COC=O VGGRCVDNFAQIKO-UHFFFAOYSA-N 0.000 description 1
- 239000003205 fragrance Substances 0.000 description 1
- AWJWCTOOIBYHON-UHFFFAOYSA-N furo[3,4-b]pyrazine-5,7-dione Chemical compound C1=CN=C2C(=O)OC(=O)C2=N1 AWJWCTOOIBYHON-UHFFFAOYSA-N 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 235000001727 glucose Nutrition 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 230000002140 halogenating effect Effects 0.000 description 1
- 230000026030 halogenation Effects 0.000 description 1
- 238000005658 halogenation reaction Methods 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 1
- 239000000852 hydrogen donor Substances 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 229920003132 hydroxypropyl methylcellulose phthalate Polymers 0.000 description 1
- 229940031704 hydroxypropyl methylcellulose phthalate Drugs 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 230000007574 infarction Effects 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 239000012155 injection solvent Substances 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 239000002198 insoluble material Substances 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 238000007914 intraventricular administration Methods 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 238000006317 isomerization reaction Methods 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 210000003292 kidney cell Anatomy 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 210000003141 lower extremity Anatomy 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 108010038445 metabotropic glutamate receptor 3 Proteins 0.000 description 1
- 108010038422 metabotropic glutamate receptor 4 Proteins 0.000 description 1
- 108010038449 metabotropic glutamate receptor 7 Proteins 0.000 description 1
- 108010014719 metabotropic glutamate receptor type 1 Proteins 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- QRUYXKCCDNOAAX-UHFFFAOYSA-N n,2,2-trimethylpropan-1-amine;hydrochloride Chemical compound Cl.CNCC(C)(C)C QRUYXKCCDNOAAX-UHFFFAOYSA-N 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- 230000016273 neuron death Effects 0.000 description 1
- 239000002858 neurotransmitter agent Substances 0.000 description 1
- 230000003957 neurotransmitter release Effects 0.000 description 1
- 238000006396 nitration reaction Methods 0.000 description 1
- JCZMXVGQBBATMY-UHFFFAOYSA-N nitro acetate Chemical compound CC(=O)O[N+]([O-])=O JCZMXVGQBBATMY-UHFFFAOYSA-N 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 230000003040 nociceptive effect Effects 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 230000001473 noxious effect Effects 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- AHHWIHXENZJRFG-UHFFFAOYSA-N oxetane Chemical compound C1COC1 AHHWIHXENZJRFG-UHFFFAOYSA-N 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- DDBREPKUVSBGFI-UHFFFAOYSA-N phenobarbital Chemical compound C=1C=CC=CC=1C1(CC)C(=O)NC(=O)NC1=O DDBREPKUVSBGFI-UHFFFAOYSA-N 0.000 description 1
- 229960002695 phenobarbital Drugs 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-N picric acid Chemical compound OC1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-N 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 229940068968 polysorbate 80 Drugs 0.000 description 1
- 210000004044 posterior horn cell Anatomy 0.000 description 1
- 230000002980 postoperative effect Effects 0.000 description 1
- 230000001242 postsynaptic effect Effects 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- RPDAUEIUDPHABB-UHFFFAOYSA-N potassium ethoxide Chemical compound [K+].CC[O-] RPDAUEIUDPHABB-UHFFFAOYSA-N 0.000 description 1
- 239000004323 potassium nitrate Substances 0.000 description 1
- 235000010333 potassium nitrate Nutrition 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 238000011533 pre-incubation Methods 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 238000002731 protein assay Methods 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000007115 recruitment Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 210000001525 retina Anatomy 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 239000013049 sediment Substances 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- JVBXVOWTABLYPX-UHFFFAOYSA-L sodium dithionite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])=O JVBXVOWTABLYPX-UHFFFAOYSA-L 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- GYBMSOFSBPZKCX-UHFFFAOYSA-N sodium;ethanol;ethanolate Chemical compound [Na+].CCO.CC[O-] GYBMSOFSBPZKCX-UHFFFAOYSA-N 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 238000003797 solvolysis reaction Methods 0.000 description 1
- 230000009870 specific binding Effects 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000001119 stannous chloride Substances 0.000 description 1
- 235000011150 stannous chloride Nutrition 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 238000009495 sugar coating Methods 0.000 description 1
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- DZLFLBLQUQXARW-UHFFFAOYSA-N tetrabutylammonium Chemical compound CCCC[N+](CCCC)(CCCC)CCCC DZLFLBLQUQXARW-UHFFFAOYSA-N 0.000 description 1
- 229960003495 thiamine Drugs 0.000 description 1
- 239000011721 thiamine Substances 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 210000000427 trigeminal ganglion Anatomy 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 230000007306 turnover Effects 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
Definitions
- the present invention relates to a novel aminomethyl-substituted rotiazolobenzimidazole derivative or a salt thereof, which is useful as a medicament.
- Glutamate acts as a neurotransmitter in the mammalian central nervous system
- Glutamate receptors are currently classified into two broad categories based on various pharmacological and physiological studies. One is a receptor with a built-in ion channel, and the other is a metabotropic (metatropic) receptor (Hollmann M. and Heinemann S., Annu. Rev. Neurosc, 17 (1994) 31). -108).
- mGluR metapotropic dalmycin receptor
- mGluR6 does not exist in the brain but only on the retina ( Nakanishi S., Neuron 13 (1995) 1031-1037). MGluR is more selective than glutamate receptors with built-in ion channels. Compounds have been reported (Hayashi Y. et al., Br. J. Pharmacol. 107 (1992) 539-543; Hayashi Y. et al., J. Neurosci. 14 (1995) 3370-3377). Through studies using compounds, the association between mGluR and various disease states has been reported in (1) to (4) below.
- Epilepsy is induced by administration of the mGluR agonist (1S, 3R) —1-aminocyclopentane-1,3-dicarbonic acid (hereinafter referred to as (1S, 3R) -ACPD) (Tizzano JP) et a, Neurosci. Lett., 162 (1993) 12-16; McDonald JW et al., J. Neurosci., 13 (1993) 4445-4455).
- (1S, 3R) -ACPD mGluR agonist of mGluR2
- mGluRI antagonists suppress the leakage of proteins out of dural vessels due to trigeminal ganglion electrical stimulation (WO0V32632). That is, the above reports show that compounds acting on mGluFM are useful for controlling epilepsy, pain, nerve cell death, benzodiazepine withdrawal syndrome, and migraine.
- the mGluFTI antagonist since the efficacy of the mGluRI antagonist has been confirmed in a rat cerebral infarction model, the mGluFTI antagonist is considered to be useful as a prophylactic / therapeutic agent for cerebral infarction (Patent Document 1).
- Patent Document 2 As compounds having an mGluRI antagonistic activity, thiazolobenzimidazole derivatives are disclosed in Patent Documents 1, 2, 3, and Patent Document 4 described above, and Patent Document 4 discloses benzene of thiazo benzobenzimidazole. A compound in which the ring is substituted by bromine and an amino group is disclosed (Example 90). However, the thiazolobenzimidazole derivative of the present invention in which the benzene ring is substituted with a fluorine atom and an aminomethyl group has no specific disclosure in these documents.
- Patent Document 2 reports that a thiazolobenzimidazole derivative in which an amino group is substituted on the benzene ring of the thiazolobenzimidazole ring has a therapeutic effect on neuropathic pain by oral administration.
- Patent Document 2 PCT international publication pamphlet WO O 1/0 8 7 0 5
- An object of the present invention is to provide a novel clinically useful aminomethyl-substituted fluorothiazo oral benzoimidazole derivative and a salt thereof as a metabolic daltime receptor antagonist having excellent oral activity.
- Patent Document 2 has oral activity, it has been confirmed by our research that it has a gene mutation inducing property. It is considered that this gene mutagenicity is expressed due to structural characteristics having an aniline amino group, and a compound having an aniline amino group may have carcinogenicity even if it has oral activity. It has the disadvantage that it cannot be used clinically as a pharmaceutical.
- the present inventors have conducted intensive studies to achieve the above object, and found that the benzene ring of the thiazolobenzoimidazole derivative was substituted with a fluorine atom and substituted aminomethyl, and did not have an aniline-like amino group as a substituent.
- Thiazolobenzimidazole derivatives particularly the aminomethyl-substituted thiazolobenzimidazole derivatives in which the benzene ring has a fluorine atom and an oxygen-containing heterocyclic ring, are known compounds in which a fluorine atom is not substituted L, Patent Documents 1, 3, and 4 Compared to the thiazolobenzimidazole derivative described in (1), it has better activity on metabotropic glumemate receptor, has no risk of carcinogenicity, and has excellent oral activity, especially neuropathic pain due to nerve compression In contrast, the present inventors have found an unexpected effect having excellent oral activity, and have completed the present invention.
- the present invention provides an aminomethyl-substituted rotiazo lipstick represented by the following general formula (I):
- the present invention relates to an azoimidazole derivative or a salt thereof, and a medicament containing them as an active ingredient. Specifically, it is as shown below.
- Alk2 lower alkylene optionally substituted with a oxo group
- n 0 or 1
- X bond, 0, S, or NR 5
- R 3 H, lower alkyl, halogeno lower alkyl, lower alkenyl, lower alkynyl, optionally substituted cycloalkyl, cyano, or saturated heterocyclic ring
- R 2 , R 4 , R 5 , R 6 and R 7 the same or different, H or lower alkyl
- R 1 is oxetane or dioxolane
- R 2 is H
- R 3 is neopentyl
- X is a bond
- R 4 is methyl
- Alk 1 is methylene
- n is 0.
- a pharmaceutical composition comprising, as an active ingredient, the aminomethyl-substituted fluorothiazolobenzimidazole derivative or the pharmaceutically acceptable salt thereof according to (1) to (3).
- a therapeutic agent for neuropathic pain comprising the aminomethyl-substituted fluorothiazolobenzimidazole derivative or the pharmaceutically acceptable salt thereof according to any one of (1) to (3) as an active ingredient.
- a therapeutic agent for neuropathic pain caused by nerve compression comprising the aminomethyl-substituted flurothiazolobene zoimidazole derivative or a pharmaceutically acceptable salt thereof according to any one of (1) to (3) above, More preferably, the neuropathic pain comprising an aminomethyl-substituted flurothiazolobenzimidazole derivative or a pharmaceutically acceptable salt thereof according to any one of (1) to (3) having no gene mutagenicity as an active ingredient. It is a therapeutic agent.
- the present invention further relates to an mGluRI receptor antagonist comprising, as an active ingredient, the aminomethyl-substituted fluorinated rothiazo benzoimidazole derivative or a salt thereof according to any one of the above (1) to (3).
- “Lower alkyl” is C 1-6 alkyl, preferably straight-chain or branched C 1-4 alkyl such as methyl, ethyl, propyl, isopropyl, t-butyl, and more preferably C 1- alkyl. 3 alkyl.
- “Lower alkylene J is C 1-6 alkylene, preferably straight-chain or branched C 1-4 alkylene such as methylene, ethylene, methylmethylene, trimethylene, propylene, ethylethylene, tetrabutylene and the like. Is C 1-3 alkylene.
- the lower alkylene substituted with a oxo group means a group in which any carbon atom of the above lower alkylene is substituted with an oxo group, and is preferably —CH 2 —C (0) —, 1 C (0 ) One CH 2 -,-CH 2 -C (0) one CH 2- , one (CH 2 ) 2 -C (0) one, — C (0) one (CH 2 ) 2- .
- the "lower alkenyl J be a C 2-6 alkenyl, preferably vinyl, pro Beniru, C 2 linear or branched, such as cycloheptenyl 4 alkenyl and more preferably C 2 - a 3 alkenyl.
- lower alkynyl are C 2-6 alkynyl, preferably Echiniru, flop port pinyl, straight or branched C 2-4 alkynyl more preferably C 2 _ 3 alkynyl, such heptynyl.
- Halogen means a halogen atom, for example, a fluorine, chlorine, bromine, or iodine atom.
- octogeno lower alkyl refers to a group in which any one or more hydrogen atoms of the lower alkyl are substituted by the above-mentioned halogen atom, and trifluoylmethyl is preferable.
- Cycloalkyl means a 3- to 8-membered cycloalkyl, preferably cyclopropyl, cyclopentyl, cyclohexyl, cycloheptyl and the like.
- “Saturated heterocycle-!” Means a 3- to 8-membered saturated heterocycle containing 1 to 4 heteroatoms selected from a nitrogen atom, an oxygen atom or a sulfur atom, and includes pyrrolidine, piperidine, Piperazine, homopirazine, imidazolidine, morpholine, lactate morpholine, lactate xylane, lactate xetane, chetan, tetrahydrofuran, tetrahydropyran, [1,3] dilute xolan, [4] dilute xane, tetra Lahydrothifenphen, [1,4] dithian, hexahydroazepine, hexahydro-pyrro [2,11c] [1,4] -year-old xazine, and the like.
- Oxygen-containing saturated hetero ring means a saturated hetero ring that always contains an oxygen atom as a hetero atom in the ring among the above-mentioned saturated hetero rings. That is, it means a 3- to 8-membered saturated hetero ring which may contain one or two nitrogen or sulfur atoms in addition to one to three oxygen atoms. Preferably, it is a 4- to 6-membered saturated heterocycle, and more preferably, xetane, tetrahydrofuran, 1,3-dioxolan, tetrahydropyran, and morpholine.
- the “sulfur-containing saturated hetero ring” means a saturated hetero ring containing a sulfur atom as a hetero atom in the ring among the above-mentioned saturated hetero rings. That is, it means a 3- to 8-membered saturated heterocyclic ring which may contain 1 or 2 nitrogen or oxygen atoms in addition to 1 to 3 sulfur atoms. Preferred are 4- to 6-membered saturated heterocycles, and more preferred are chetan, 1,3-dithiolane, tetrahydrothienefen, thiazolidine, and thiaminemorpholine. 1 to 3 carbon atoms or heteroatoms on the ring may be substituted oxygen-containing saturated heterocycles, sulfur-containing saturated heterocycles which may be substituted, or cycloalkyl which may be substituted. May have a substituent.
- the substituent means a usual substituent commonly used in the art for the group to be substituted, and most preferred substituents include halogen, cyano, halogen lower alkyl, lower alkyl, OH, lower alkyl 10-, Oxo, lower alkyl-1 C (0) -1, carboxyl, lower alkyl— 0—C (0) 1, lower alkyl-1 O—lower alkyl—, nitro, even if substituted with 1 or 2 lower alkyls Good amino and the like.
- the compound of the present invention has an optical isomer (optically active substance, diastereomer, etc.) depending on the type of the group.
- the compounds of the present invention include compounds having an amide bond and a double bond, and also include tautomers and geometric isomers.
- the present invention includes a separated form or a mixture of these isomers.
- Salts with acids include mineral acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid, phosphoric acid, etc .; formic acid, acetic acid, propionic acid, oxalic acid, malonic acid, succinic acid, Examples thereof include acid addition salts with organic acids such as fumaric acid, maleic acid, lactic acid, malic acid, citric acid, tartaric acid, carbonic acid, picric acid, methanesulfonic acid, ethanesulfonic acid, and glutamic acid.
- mineral acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid, phosphoric acid, etc .
- formic acid acetic acid, propionic acid, oxalic acid, malonic acid, succinic acid
- organic acids such as fumaric acid, maleic acid, lactic acid, malic acid, citric acid, tartaric acid, carbonic acid, pic
- salts with bases include inorganic bases such as sodium, potassium, magnesium, calcium, and aluminum; organic bases such as methylamine, ethylamine, medalmine, and ethanolamine; and basic amino acids such as lysine, arginine, and ordinine. Salt and ammonium salt.
- the compound of the present invention can form a hydrate, a solvate with ethanol or the like, or a polymorph.
- the compound of the present invention also includes a pharmacologically acceptable prodrug.
- the group forming a pharmacologically acceptable prodrug of the compound include the group described in Prog. Med. 5: 2157-2161 (1985) and the group described in Hirokawa Shoten 1990 Vol.
- the prodrug include mono OC (O) —optionally substituted lower alkylene—C (O) 0 R (R represents H or lower alkyl, the same applies hereinafter), —OC (0) monosubstituted Lower alkenylene mono C (O) OR, -OC (O) monosubstituted aryl, mono C (O) mono lower alkylene-0-lower alkylene-C (0) OR, — OC (O) one C (O) R, one OC (O) - optionally substituted lower alkyl, one OS 0 2 - optionally substituted lower alkylene - C (0) 0R, one 0-phthalidyl, 5-methyl-1, 3-dioxolen-2-one-4-y
- the compound (1) of the present invention can be produced by the following production method.
- DM F dimethylformamide
- THF tetrahydrofuran
- Production method 1 Production of 4-amino-5-promo 2-furosyl-3-nitrobenzoic acid derivative Nitration
- R 8 represents hydrogen or lower alkyl, R 9 ttC Br, I.
- Production method 1 is a method for producing a dinitroaniline derivative from an aniline derivative.
- Step 1 is the halogenation of the benzene ring. That is, (II) is converted to a solvent such as carbon tetrachloride, chloroform, methylene chloride, 4-dioxane, DMF, DMSO, methanol, acetic acid, bromine, hydrogen bromide, tetrabutylammonium tribromide, etc. (III) can be produced by reacting with a halogenating agent such as an ammonium complex of N, bromosuccinimide, chlorine, or N-chlorosuccinimide under ice-cooling to heating conditions.
- a halogenating agent such as an ammonium complex of N, bromosuccinimide, chlorine, or N-chlorosuccinimide under ice-cooling to heating conditions.
- Step 2 is the acylation of the amino group. That is, (III) is used in an inert solvent such as THF, black porphyrum, dimethyl ether, DMF, acetate nitrile, or an active solvent such as pyridine, acid octride, acid anhydride, active (IV) can be produced by reacting with an acylating agent such as an ester under low-to-warming conditions.
- an inert solvent such as THF, black porphyrum, dimethyl ether, DMF, acetate nitrile, or an active solvent such as pyridine, acid octride, acid anhydride, active (IV)
- an acylating agent such as an ester under low-to-warming conditions.
- Step 3 is the conversion of the benzene ring to a disulfide. That is, (IV) is reacted in a solvent such as sulfuric acid or acetic acid, with a nitrating agent such as nitric acid, potassium nitrate, or acetyl nitrate, or with nitric acid as a solvent under low to room temperature conditions and, if necessary, under heating conditions. By doing this, (V) can be manufactured. In addition, in an inert solvent such as toluene, acetonitrile, THF, or sulfolane, a nitrating agent such as dinitronium tetrachloride is used at low to room temperature conditions, if necessary. (V) can also be produced by reacting under heating conditions.
- a solvent such as sulfuric acid or acetic acid
- a nitrating agent such as nitric acid, potassium nitrate, or acetyl nitrate
- Step 4 is a deacylation reaction by hydrolysis. That is, (VI-a) can be produced by subjecting (V) to a hydrolysis reaction under acidic or alkaline conditions.
- acidic conditions use sulfuric acid, hydrochloric acid, acetic acid, etc.
- alkaline conditions use sodium hydroxide, potassium hydroxide, potassium carbonate, etc., methanol, ethanol, TH
- the reaction is carried out from room temperature to heating in a solvent such as F, acetate and water, a mixed solvent thereof, or no solvent.
- Production method 2 Production of 4-amino-2-fluoro-5-dibenzobenzoic acid derivative
- Production method 2 is a method for producing aminobenzene derivatives from full-year-old robenzene derivatives by the ipso reaction. That is, by reacting (VII) with an alcohol-based solvent such as ethanol and isopropanol, an amine donor such as ammonium chloride or ammonia in a solvent such as DMF, DMSO and water under heating conditions from room temperature. ) Can be manufactured.
- Production method 3 Production of thioxobenzimidazole ring
- Production method 2 is a method for producing a thixobenzimidazole ring from a dinitroaniline derivative.
- Step (2) is a reduction reaction. That is, (VI) is subjected to a catalytic reduction reaction using a metal hornworm medium such as palladium in an inert solvent such as methanol, ethanol, or DMF, in a hydrogen atmosphere, or in the presence of a hydrogen donor such as ammonium formate.
- a metal hornworm medium such as palladium in an inert solvent such as methanol, ethanol, or DMF
- a hydrogen donor such as ammonium formate.
- R 9 is hydrogen
- the reaction is carried out using iron, stannous chloride or the like under acidic conditions such as acetic acid, hydrochloric acid, ammonium chloride or the like.
- (VIII) can also be produced by reacting with a reducing agent such as sodium hydrosulfite in a mixed solvent such as methanol, ethanol, and THF under heating conditions from room temperature.
- Step 2 is a cyclization reaction of a thioxobenzimidazole ring. That is, (VIII) can be prepared by reacting (VIII) with an inert solvent such as methanol, ethanol, or DMF in the presence or absence of a base such as triethylamine, carbon disulfide, 1,1'-thiamine carbonyldiimidazole, titani carbonate, etc. (IX) can be produced by reacting
- step 2 can be continued.
- Yes Production method 4: Production of thiazolobenzimidazole ring
- Production method 4 is a method for producing a thiazolobenzimidazole ring from a thioxobenzimidazole ring.
- Step 1 is an alkylation reaction. That is, (IX) and a-octaacetic acid derivative were treated with an alcoholic solvent such as ethanol or methanol, or an inert solvent such as DMF, THF, or acetonitrile in sodium methoxide or sodium methoxide.
- the reaction of (X) by heating at room temperature to heating in the presence or absence of a base such as trioxide, hydroxyl sodium, potassium hydroxide, sodium hydride, potassium carbonate, and sodium hydrogen carbonate. Can be manufactured.
- Step 2 is a cyclization reaction. That is, sodium (ethoxide) and potassium ethoxide are mixed with (X) and a formylating agent such as formate or formic anhydride in an alcoholic solvent such as ethanol or methanol, or in an inert solvent such as DMF, HF, or acetonitrile.
- (XI) can be produced by reacting from room temperature to heating under basic conditions such as sodium hydroxide, sodium hydride, pyridine and triethylamine. This (XI) may take the equilibrium state of Equation 1 in the solution state. (Equation 1)
- Step 3 is an isomerization reaction of a dihydrothiazolobenzimidazole ring. That is, after (XI) is prepared in the form of a solution or suspension in the form of an equilibrium mixture represented by the formula 1, this is treated under acidic conditions such as hydrochloric acid, sulfuric acid, acetic acid, and trifluroacetic acid to precipitate (XII). Can be.
- acidic conditions such as hydrochloric acid, sulfuric acid, acetic acid, and trifluroacetic acid to precipitate (XII).
- Step 4 is a dehydration reaction from a dihydrothiazolobenzoimidazole ring to a thiazolobenzoimidazole ring. That is, (XII) can be produced by reacting (XII) with an acid such as concentrated sulfuric acid, acetic acid, or trifluroacetic acid from room temperature under heating conditions. In addition, in an inert solvent such as toluene or benzene, under acidic conditions such as p-toluenesulfonic acid or camphorsulfonic acid, or in the presence of a dehydrating agent such as molecular sieves if necessary, or Dean-Stark dehydration. (XIII) can also be produced by reacting from room temperature to heating under dehydration reaction conditions in an apparatus or the like. Process 5: Reduction of carboxylic acid
- Production method 5 is an ordinary method for producing an alcohol compound by reduction of a carboxylic acid.
- Process 6 is the hydrolysis of an ester. That is, (XV) can be produced by subjecting (XIV) to a hydrolysis reaction under acidic conditions or alkaline conditions.
- acidic conditions use sulfuric acid, hydrochloric acid, acetic acid, etc.
- alkaline conditions use sodium hydroxide, potassium hydroxide, potassium carbonate, etc., to obtain methanol, ethanol, THF, acetate nitrile,
- the reaction is carried out in a solvent such as water, a mixed solvent thereof, or no solvent at room temperature to heating.
- Production Method 7 Amidation k 2 ) n _ X -R
- Production method 7 is a usual amidation reaction of a carboxylic acid. That is, (XV) is converted into dicyclohexyl carbodiimide, 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide, diphenylphosphoryl triazide, in an inert solvent such as DMF, THF, 1,2-dichloroethane, and chloroform.
- (XVI) can be produced by activating with a condensing agent such as 1,1'-carboxyl 1H- ⁇ T midazole or 1-hydroxybenzotriazole, and reacting the activated form with the corresponding amine.
- Production method 8 is an oxidation reaction of an alcohol to an aldehyde. That is, (XVI) can be produced by reacting (XVI) with sulfur trioxide / pyridine complex under ice-cooling to room temperature conditions using DMSO as a solvent. (XV II) can also be produced by ordinary oxidation reactions such as SWERN oxidation, chromic acid oxidation, and permanganic acid oxidation. Process 9: Reductive amination — X— R 3
- (XV II) to (I) are ordinary reductive amination reactions. That is, (XV II) is converted into a solvent such as methylene chloride, 12-dichloroethane, chloromethium, THF, methanol, or ethanol, and if necessary, an acid catalyst such as acetic acid or hydrochloric acid, or titanium tetraisopropoxide. In the presence of a corresponding amine with a reducing agent such as sodium triacetoxyborohydride, sodium cyanoborohydride, sodium borohydride, etc. Thus, (I) can be manufactured.
- a solvent such as methylene chloride, 12-dichloroethane, chloromethium, THF, methanol, or ethanol
- an acid catalyst such as acetic acid or hydrochloric acid, or titanium tetraisopropoxide.
- a corresponding amine with a reducing agent such as sodium triacetoxyborohydride, sodium cyanoborohydride, sodium boro
- (I) is obtained by reacting (XVII) with the corresponding amine in an inert solvent such as toluene or benzene, if necessary, using a dehydrating agent such as molecular sheep or a dehydration reaction using a Dean Stark dehydrator.
- the imine is reacted under a warming condition from room temperature, and treated with a reducing agent such as sodium borohydride in a solvent such as methanol or ethanol, or in a mixed solvent with the above reaction solvent.
- a reducing agent such as sodium borohydride
- solvent such as methanol or ethanol
- (I) can be produced by using a metal catalyst such as palladium under ordinary catalytic reduction conditions, specifically, a hydrogen atmosphere.
- the compound of the present invention produced in this manner is isolated or purified as free or as a salt thereof.
- Isolation and purification are performed by applying ordinary chemical operations such as extraction, concentration, evaporation, crystallization, filtration, recrystallization, and various types of chromatography.
- Various isomers can be separated by selecting an appropriate starting compound or by utilizing a difference in physical properties between the isomers.
- the optical isomers can be stereochemically selected by selecting appropriate raw materials or by a racemic resolution method of a racemic compound (for example, a method of optically resolving a diastereomer salt with a general optically active base). It can lead to a pure isomer.
- a preparation containing one or more of the compound of the present invention or a salt thereof as an active ingredient is prepared using a carrier, an excipient, and other additives which are usually used for preparation of a preparation.
- the carrier or excipient for the preparation may be solid or liquid, for example, lactose, magnesium stearate, starch, talc, gelatin, agar, pectin, arabic gum, leabe oil, sesame oil, cocoa butter, ethylene Glycols and the like are also commonly used.
- Administration may be in the form of tablets, pills, capsules, granules, powders, liquids, etc., oral administration, intravenous injection, intramuscular injection, parenteral administration, suppositories, transdermal, etc. Good.
- the dose is determined as appropriate depending on the individual case, taking into account the symptoms, age, sex, etc. of the administration subject, but it is usually 1 to 100 mg / day, preferably 50 to 100 mg / day for an adult. 2 Oral dose in the range of OO mg once or several times a day or iv in a dose of 1 to 50 mg per adult per day, once to several times a day Or continuous intravenous administration for 1 hour to 24 hours per day.
- the one or more active substances include at least one inert diluent, such as lactose, mannitol, glucose, hydroxypropyl cellulose, microcrystalline cellulose, starch, polyvinyl. It is mixed with pyrrolidone and magnesium aluminate metasilicate.
- the composition may contain additives other than inert diluents, such as lubricants such as magnesium stearate, disintegrants such as starch, calcium cellulose dalcholate, and stable agents such as lactose. It may contain a solubilizing agent such as an agent, dartamic acid or aspartic acid. If necessary, tablets or pills may be coated with sugar coating such as sucrose, gelatin, hydroxypropylcellulose, hydroxypropylmethylcellulose phthalate or the like, or with a film of gastric or enteric substance.
- lubricants such as magnesium stearate
- disintegrants such as starch, calcium cellulose dalcholate
- stable agents such as lactose.
- solubilizing agent such as an agent, dartamic acid or aspartic acid.
- tablets or pills may be coated with sugar coating such as sucrose, gelatin, hydroxypropylcellulose, hydroxypropylmethylcellulose phthalate or the like, or with a film of gastric or enter
- Liquid compositions for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups, elixirs and the like, and commonly used inert diluents such as purified Contains water and ethanol.
- the composition may contain, in addition to the inert diluent, adjuvants such as wetting agents and suspending agents, sweetening agents, flavoring agents, fragrances, and preservatives.
- Injections for parenteral administration include sterile aqueous or non-aqueous solutions, suspensions, and emulsions.
- Aqueous solutions and suspensions include, for example, distilled water for injection and physiological saline.
- water-insoluble solutions and suspensions examples include propylene glycol, polyethylene glycol, vegetable oils such as crude oil, alcohols such as ethanol, and polysorbate 80.
- Such compositions may also contain adjuvants such as preserving, wetting, emulsifying, dispersing, and stabilizing agents (eg, lactose) and solubilizing agents (eg, glutamic acid, aspartic acid). May be included. These are sterilized by, for example, filtration through a pateria retaining filter, blending of a bactericide or irradiation. In addition, these can be used by producing a sterile solid composition and dissolving it in sterile water or a sterile injection solvent before use.
- MS mass spectrometry (FAB +: cation fast atom bombardment mass spectrometry, FAB-: anion fast atom bombardment mass spectrometry, ES +: cation electrospray ionization, ES-: anion electrospray ionization.
- M molecular weight
- the crystallized residue was recrystallized from hexane (50 ml) and ethyl acetate (10 ml), filtered and washed with a mixed solvent of hexane and ethyl acetate (mixing ratio: 10: 1). The obtained crystals were dried under reduced pressure while heating at 50 ° C to give the title compound (10.1 g).
- Acetyl chloride (104.4 ml) was added to a THF (800 ml) solution of the compound (128.27 g) of Reference Example 1 under ice-cooling. And stirred at room temperature for 24 hours. After completion, the reaction solution was concentrated under reduced pressure, and extracted with ethyl acetate. The organic layer was washed with a saturated aqueous solution of sodium hydrogen carbonate and saturated saline, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. Ethyl acetate (200 ml) was added to the crystallized residue, and the mixture was dissolved by heating under reflux, and then hexane (250 ml) was added for crystallization.
- the precipitated crystals were collected by filtration and washed with a mixed solvent of hexane and ethyl acetate (mixing ratio: 3: 1). The obtained crystals were dried under reduced pressure at 50 ° C. while heating to obtain the title compound (140.5 g).
- the compound of Reference Example 3 (88.4 g) was dissolved in a mixed solvent of concentrated hydrochloric acid and acetic acid (mixing ratio 1: 1, 1 L), followed by stirring in a 100 ° C. oil bath for 24 hours. After completion, the reaction solution was ice-cooled and poured into ice water (4 L). The precipitated crystals were collected by filtration, washed with water, and dried under reduced pressure at 70 ° C. while heating to obtain the title compound (65.2 g).
- the compound of Reference Example 8 (15.5 g) was dissolved in concentrated sulfuric acid (50 ml), and the mixture was stirred in an oil bath at 45 ° C for 4 hours. After completion, the reaction solution was ice-cooled and then dispersed in ice-cooled ethyl acetate (400 ml). The precipitated crystals were collected by filtration, washed with ethyl acetate, and dried under reduced pressure while heating at 60 ° C. to give the title compound (14.5 g).
- the compound of Reference Example 9 (14.5 g) was suspended in acetonitrile (150 ml), and 1,1′-carboxydiimidazole (17.4 g) was added at room temperature under an argon atmosphere. After the addition, the reaction mixture was stirred at room temperature for 3 hours. Subsequently, the reaction mixture was ice-cooled, sodium borohydride (4.05 g) /0.1% aqueous sodium hydroxide solution (20 ml) was added, and the mixture was stirred for 5 hours while cooling on ice, and further heated to room temperature and stirred for 4 hours. did. After completion, concentrated hydrochloric acid (25.3 g) and water (230 ml) were added to the reaction suspension,
- the compound of Reference Example 10 (7.95 g) was suspended in a 40% aqueous solution of acetonitrile (100 ml), added to a 1 M aqueous sodium hydroxide solution (32 ml) under ice cooling, and stirred at room temperature for 4 hours. After completion, a 1 M aqueous hydrochloric acid solution (32 ml) was added to the reaction solution, and the mixture was cooled on ice, and the precipitated crystals were collected by filtration. The collected residue was washed with water, and dried under reduced pressure while heating at 60 ° C to obtain the title compound (6.49 g).
- the reaction solution was extracted with chloroform, and the organic layer was washed with water and saturated saline, dried over anhydrous sodium sulfate, and concentrated under reduced pressure.
- the crystallized residue was recrystallized from ethanol and ethyl acetate, and the obtained crystals were collected by filtration.
- the crystals were washed with a mixed solvent of ethanol and ethyl acetate (mixing ratio: 1.3), and dried under reduced pressure at 60 ° C under heating to obtain the title compound (3.65 g).
- 2,4-Difluoro-5-dibenzobenzoic acid (26.29 g) was dissolved in a concentrated aqueous ammonia solution (500 ml), and the reaction mixture was stirred at room temperature for 4 hours. After completion, the mixture was concentrated under reduced pressure. Ethanol was added to the residue, and the mixture was concentrated again under reduced pressure. The residue was washed with water and a small amount of ethanol, and dried under reduced pressure to obtain the title compound (26.38 g).
- Reference Example 18 Water (30 ml) and a 1 M aqueous solution of sodium hydroxide (26.1 ml) were added to a suspension of the compound of Example 8 (6.40 g) in acetonitrile (50 ml). The mixture was stirred at room temperature for 6 hours. After completion, a 1 M aqueous hydrochloric acid solution (26.1 ml) was added to the reaction mixture, and the mixture was stirred at room temperature for 30 minutes. The insolubles were collected by filtration, washed with water and acetate nitrile, and dried under reduced pressure to obtain the title compound (5.69 g).
- Acetic anhydride (81.4 ml) was added to a solution of ethyl 4-amino-3-fluorobenzoate (31.88 g) in pyridine (300 ml) and the mixture was stirred for 72 hours. Water (30 ml) was added thereto, and the mixture was further stirred at room temperature for 30 minutes. After completion, the mixture was concentrated under reduced pressure. The residue was partitioned between ethyl acetate and water. The aqueous layer was separated and extracted with ethyl acetate.
- Triethylamine (3.36 ml) was added to the mixture, and the mixture was further stirred for 1 hour in a 40 ° C oil bath. After completion, the reaction mixture was concentrated under reduced pressure. The residue was diluted with water, and then adjusted to acidic with 1 M aqueous hydrochloric acid. The precipitate was collected by filtration, washed with water, and dried under reduced pressure to give the title compound (4.93 g).
- Acetic acid (206I), 3-aminosixetane hydrochloride (267 mg), and triethylamine (301 1) were added to a dichloroethane solution (15 ml) of the compound of Reference Example 12 (250 mg), and the mixture was stirred at room temperature for 2 hours. . Subsequently, sodium triacetoxyborohydride (458 mg) was added, and the mixture was stirred at room temperature for 14 hours. Further, triacetoxy sodium borohydride (458 mg) was added, and the mixture was stirred at room temperature for 4.5 hours. After completion of the reaction, a 1 M aqueous solution of sodium hydroxide was added to the reaction solution, followed by extraction with a 5% methanol / chloroform solution and concentration under reduced pressure.
- Acetic acid (123 1) and (1,3-dioxolan-12-yl) methylamine (122iUl) were added to a dichloroethane solution (10 ml) of the compound of Reference Example 12 (150 mg), and stirred at room temperature for 2.5 hours. did. Subsequently, sodium triethoxy borohydride (274 mg) was added, and the mixture was stirred at room temperature for 14 hours. Further, sodium triacetoxyborohydride (274 mg) was added, and the mixture was stirred at room temperature for 4.5 hours. After completion, a 1 M aqueous sodium hydroxide solution was added to the reaction solution, and the mixture was extracted with a 5% methanol / chloroform solution and concentrated under reduced pressure.
- the effect of the compound of the present invention on mGluRI was confirmed according to the method described in Patent Document 3.
- the effect of the compound of the present invention is that of 6-amino N-cyclohexyl N, 3-dimethylthiazolo [3,2-a] benzimidazo mono-l-2-loxaboxamide, which has a selective and potent action on mGluRI.
- Tritium label (specific activity: 81 Ci / mmol Five
- Rats (Wistar, male, 9-12 weeks old) were decapitated and the cerebellum was removed. The weight was measured and homogenized with 7-10 volumes of 0.32 M sucrose solution. After centrifugation at 900 xg for 15 minutes, the supernatant was stored in another container (on ice). The sediment was homogenized again with the same amount of 0.32 M sucrose solution as in the first run, and centrifuged at 900 X g for 15 minutes. The supernatant obtained at this time and the supernatant obtained above were combined and subjected to eccentricity at 15,000 X g for 20 minutes. The pellet was homogenized with 5 mM Tris-HCI, pH 7.4, and centrifuged at 15,000 xg for 15 minutes.
- the pellet was homogenized with 50 mM Tris-HCU pH 7.4 and centrifuged at 15,000 xg for 15 minutes.
- the precipitate was homogenized with an appropriate amount of 50 mM Tris-HCl, pH 7.4, aliquoted and stored at -80 ° C.
- Test Example 2 Suppressive effect on neuropathic pain
- the experiment was performed by modifying a part of the report (Pharmacol Biochem Behav 39, 541-544, 1991). Intraperitoneal administration of 200 mg / kg STZ to 4-week-old ICR mice. Two weeks after administration, a pre-test of the tail pinch test was performed in the afternoon, and only animals with a reaction latency of 3 seconds or less were subjected to the next day's experiment.
- Control compounds A, B, and C were selected as compounds having a substituted aminomethyl group on the benzene ring of thiazolobenzoimidazole.
- control compounds A and B were orally administered at 100 mg / kg and evaluated in the same manner.
- mice not loaded with STZ show an average response latency of 6-7 seconds in this test.
- the STZ-loaded mice used in this study were those with a response latency of 3 seconds or less, where a clear decrease in pain threshold was observed.
- Control Compound C Compound disclosed in Patent Document 4 and Example 113 The above test confirmed that the compound of the present invention is a compound that specifically binds to mGIuR1.
- the compound of the present invention showed an average response latency difference of 2 seconds or more in all cases of oral administration at 10 mg / kg, confirming that it has a therapeutic effect on neuropathic pain due to diabetes.
- control compounds A and B had an average response latency difference of less than about ⁇ seconds when administered orally at 100 mg / kg.
- the compound of the present invention is a compound which is at least 10 times more orally active than the control compounds A and B and is useful as an oral preparation.
- control compound C showed a response latency difference of 2 seconds or more.
- the left lumbar nerve (L5 and L6) was ligated with a silk thread under pentovalpital anesthesia.
- the drug was orally administered at 10 mg / kg, and 30 minutes later, a von Frey hair (VFH) test was performed to determine the pain threshold for mechanical nociceptive stimulus.
- the measurements were performed on the left and right hind limbs.
- the pain threshold of the sham operation rat was 17-20 g (log (g): 1.23-1.30) on average, with no difference between left and right.
- the L5 / L6 spinal nerve ligation rat operation Decreased pain thresholds for mechanical noxious stimuli were found in the lateral paws.
- the significance test was performed using the Dunnett method between the control group and the drug administration group on each of the right and left feet.
- Control compound D Patent document 2, compound A
- the compound of the present invention shown in Table 3 is about 8 to 50 times or more that of control compound C which showed a response latency difference of 2 seconds or more in an STZ-induced diabetes model. The effect was shown.
- the compound of the present invention showed 4- to 30-fold the action of control compound D having an aniline-like amino group, which was confirmed in Patent Document 2 to have an action of lowering the pain threshold due to nerve compression.
- the gene mutagenicity of the compound of the present invention was confirmed by a reverse mutation test using bacteria.
- test method was performed by the preincubation method in the presence and absence of a metabolic activation system in accordance with the guidelines for testing genotoxicity of drugs (Pharmaceutical Affairs Council No. 1604, January 1, 2001).
- test strain was performed by the preincubation method in the presence and absence of a metabolic activation system in accordance with the guidelines for testing genotoxicity of drugs (Pharmaceutical Affairs Council No. 1604, January 1, 2001).
- test tube To the test tube, add 0.1 mL of 0.1 M sodium phosphate buffer (pH 7.4), 0.5 mL of the test bacterium suspension cultured overnight, and 0.1 mL of the test substance solution, and shake at 37 for 20 minutes (60 reciprocations / minute). ), 2 mL of soft agar kept at about 45 ° C was added, spread on a minimal glucose agar plate (plate), and cultured at 37 ° C for about 48 hours. In the case of the metabolic activation test, the same operation was performed by adding the same amount of S9Mix instead of 0.1 M sodium phosphate buffer.
- S9 Mix used for metabolic activation test was S-9 / cofactor A set (9000Xg supernatant of rat liver homogenate induced with drug metabolizing enzyme with phenobarbital and 5,6-benzoflavone) and Cofactor, Ames test, Oriental yeast
- the amount of S9 in S9Mix was 0.1 mL / mL.
- the solvent used was dimethylsulphate.
- the compounds of Example 1 and Example 2 did not have gene mutagenicity.
- the control compound D having an aniline-like amino group had mutagenicity. From these results, the compound of the present invention has no anilinic amino group and therefore does not have gene mutagenicity, and has excellent oral activity that cannot be predicted from Patent Documents 1 to 4, especially suppression of neuropathic pain due to nerve compression. The effect was confirmed. The invention's effect
- the compound of the present invention is a compound having a strong action on the metabolic daltamet receptor and excellent in activity of Jiangro, and can be used for oral administration.
- the compound of the present invention is a disease in which mGluRI receptor is considered to be involved, for example, Epilepsy, pain, suppression of neuronal cell death, benzodiazepine withdrawal syndrome, Parkinson's disease, migraine, anxiety disorder, cerebral infarction (preferably an agent for preventing the development of infarct lesions administered during acute phase of cerebral infarction) or neuropathic pain (preferably Is useful as a prophylactic / therapeutic agent for diabetic neuropathic pain, postherpetic neuralgia, cancer pain, and postoperative pain.
- mGluRI receptor is considered to be involved, for example, Epilepsy, pain, suppression of neuronal cell death, benzodiazepine withdrawal syndrome, Parkinson's disease, migraine, anxiety disorder, cerebral infarction (preferably an agent for preventing the development of infarct lesions administered during acute phase of cerebral infarction) or neuropathic pain (preferably Is useful as a prophylactic / therapeutic agent for diabetic neuropathic pain, postherpetic neuralgia,
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Neurosurgery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Neurology (AREA)
- Pharmacology & Pharmacy (AREA)
- Biomedical Technology (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pain & Pain Management (AREA)
- Heart & Thoracic Surgery (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Cardiology (AREA)
- Psychology (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Description








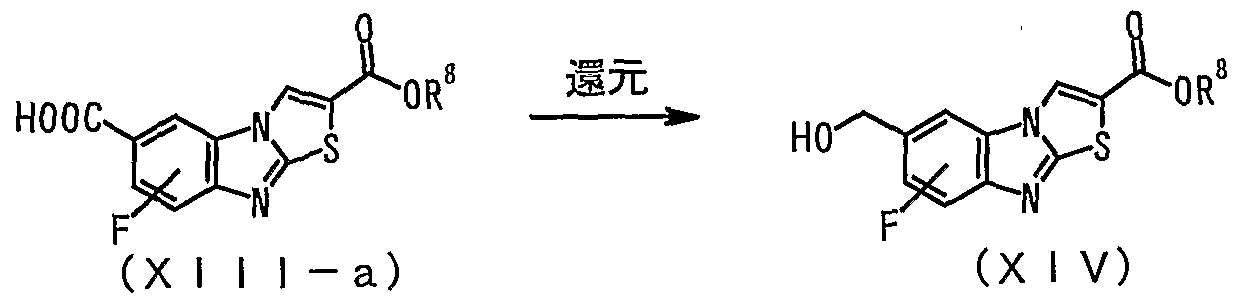










Claims

Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005506550A JPWO2004106348A1 (ja) | 2003-05-28 | 2004-05-27 | アミノメチル置換フルオロチアゾロベンゾイミダゾール誘導体 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003150430 | 2003-05-28 | ||
| JP2003-150430 | 2003-05-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004106348A1 true WO2004106348A1 (ja) | 2004-12-09 |
Family
ID=33487182
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2004/007655 WO2004106348A1 (ja) | 2003-05-28 | 2004-05-27 | アミノメチル置換フルオロチアゾロベンゾイミダゾール誘導体 |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JPWO2004106348A1 (ja) |
| WO (1) | WO2004106348A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009501146A (ja) * | 2005-06-23 | 2009-01-15 | アレイ バイオファーマ、インコーポレイテッド | ベンズイミダゾール化合物の調製方法 |
| US8329914B2 (en) | 2008-10-31 | 2012-12-11 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| US8563746B2 (en) | 2008-10-29 | 2013-10-22 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999044639A1 (fr) * | 1998-03-03 | 1999-09-10 | Yamanouchi Pharmaceutical Co., Ltd. | Medicaments contre l'infarcissement du cerveau |
| JP2000351782A (ja) * | 1999-04-06 | 2000-12-19 | Yamanouchi Pharmaceut Co Ltd | メタボトロピックグルタメート受容体作用薬及び新規チアゾロベンゾイミダゾール誘導体 |
| WO2001008705A1 (fr) * | 1999-08-02 | 2001-02-08 | Yamanouchi Pharmaceutical Co., Ltd. | Remedes contre les douleurs neurogenes |
| WO2003078441A1 (fr) * | 2002-03-20 | 2003-09-25 | Yamanouchi Pharmaceutical Co., Ltd. | Derive de thiazolobenzimidazole substitue par aminomethyle |
-
2004
- 2004-05-27 WO PCT/JP2004/007655 patent/WO2004106348A1/ja active Application Filing
- 2004-05-27 JP JP2005506550A patent/JPWO2004106348A1/ja not_active Withdrawn
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999044639A1 (fr) * | 1998-03-03 | 1999-09-10 | Yamanouchi Pharmaceutical Co., Ltd. | Medicaments contre l'infarcissement du cerveau |
| JP2000351782A (ja) * | 1999-04-06 | 2000-12-19 | Yamanouchi Pharmaceut Co Ltd | メタボトロピックグルタメート受容体作用薬及び新規チアゾロベンゾイミダゾール誘導体 |
| WO2001008705A1 (fr) * | 1999-08-02 | 2001-02-08 | Yamanouchi Pharmaceutical Co., Ltd. | Remedes contre les douleurs neurogenes |
| WO2003078441A1 (fr) * | 2002-03-20 | 2003-09-25 | Yamanouchi Pharmaceutical Co., Ltd. | Derive de thiazolobenzimidazole substitue par aminomethyle |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009501146A (ja) * | 2005-06-23 | 2009-01-15 | アレイ バイオファーマ、インコーポレイテッド | ベンズイミダゾール化合物の調製方法 |
| EP1904481A4 (en) * | 2005-06-23 | 2010-11-10 | Array Biopharma Inc | PROCESS FOR THE PREPARATION OF BENZIMIDAZOL COMPOUNDS |
| US8039637B2 (en) | 2005-06-23 | 2011-10-18 | Array Biopharma Inc. | Process for preparing benzimidazole compounds |
| US8383832B2 (en) | 2005-06-23 | 2013-02-26 | Array Biopharma Inc. | Process for preparing benzimidazole compounds |
| US8501956B2 (en) | 2005-06-23 | 2013-08-06 | Array Biopharma Inc. | Benzimidazole compounds |
| US9024040B2 (en) | 2005-06-23 | 2015-05-05 | Array Biopharma Inc. | Processes for preparing benzimidazole compounds |
| US8563746B2 (en) | 2008-10-29 | 2013-10-22 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| US8329914B2 (en) | 2008-10-31 | 2012-12-11 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2004106348A1 (ja) | 2006-07-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8153658B2 (en) | Piperidine derivative or salt thereof | |
| TWI417100B (zh) | 二唑衍生物及其作為代謝型麩胺酸受體增效劑-842之用途 | |
| US8637510B2 (en) | Morpholinothiazoles as alpha 7 positive allosteric modulators | |
| KR101521405B1 (ko) | 베타-락타민 페닐알라닌, 시스테인 그리고 세린 바소프레신 길항물질 | |
| TWI382027B (zh) | 代謝型麩胺酸受體異唑配位體及其作為增效劑之用途-286 | |
| JP5466292B2 (ja) | イソオキサゾール−ピリジン誘導体 | |
| DE60132618T2 (de) | 4,6-diphenylpyridinderivate als entzündungshemmende mittel | |
| WO2004110986A1 (ja) | ベンズアミド誘導体又はその塩 | |
| JP6975515B2 (ja) | Trpa1モデュレーターとしてのスルホニルシクロアルキルカルボキサミド化合物 | |
| JP2009509921A (ja) | 置換イソインドロン類及び代謝調節型グルタミン酸受容体増強剤としてのその使用 | |
| TW201514157A (zh) | 環丙胺化合物及其用途 | |
| JP5478714B2 (ja) | イソオキサゾール誘導体 | |
| JPWO2007105753A1 (ja) | トリアゾール誘導体またはその塩 | |
| WO2011021678A1 (ja) | 縮合複素環化合物 | |
| JP7195436B2 (ja) | バニン阻害剤としての複素芳香族化合物 | |
| WO2019205983A1 (zh) | 氧杂螺环类化合物及其制备方法和用途 | |
| JP2012509352A (ja) | Bace1のキナゾリン阻害剤および使用方法 | |
| TW201522330A (zh) | 2-醯胺噻唑衍生物或其鹽 | |
| JP2003012653A (ja) | キナゾリン誘導体 | |
| JP2022531144A (ja) | 神経疾患又は精神疾患を治療するためのKv3カリウムチャネル活性化薬としてのN-((ヘテロアリール)メチル)-1-トシル-1H-ピラゾール-3-カルボキサミド誘導体 | |
| JP2015531392A (ja) | Mglur5受容体活性のモジュレーターとしてのエチニル誘導体 | |
| TW200811137A (en) | mGluR5 modulators II | |
| KR20130122531A (ko) | Mglur5 양성 알로스테릭 조절물로서 치환된 6메틸니코틴아미드 | |
| EP1167369A1 (en) | Novel thiazolobenzimidazole derivatives | |
| FR2930249A1 (fr) | Nouveaux derives de 3-aminoalkyl-1,3-dihydro-2h-indol-2-one, leur preparation et leur application en therapeutique. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| DPEN | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed from 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2005506550 Country of ref document: JP |
|
| 122 | Ep: pct application non-entry in european phase |