TITLE OF THE INVENTION
PROCESS TO CHIRAL BETA-AML O ACID DERIVATIVES
FIELD OF THE INVENTION
The present invention also relates to a process for the efficient preparation of enantiomerically enriched beta-amino acid derivatives which are useful in the synthesis of biologically active molecules. More particularly, the present invention relates to a process for the preparation of enantiomerically enriched beta-amino acid amide inhibitors of dipeptidyl peptidase-TN which are useful for the treatment of Type 2 diabetes.
BACKGROUND OF THE INVENTION
The present invention provides an efficient process for the preparation of chiral inhibitors of dipeptidyl peptidase-IV of general structural formula I,
or a pharmaceutically acceptable salt thereof; having the (R)-configuration at the stereogenic center marked with an *.
The present invention also provides intermediates useful in the disclosed process.
The present invention also provides an efficient process for the preparation of an enantiomerically enriched beta-amino acid derivative of structural formula 1_:
(1 ) or an amine salt thereof; having the indicated stereochemical configuration at the stereogenic center marked with an ***; wherein
R2 is Cι _8 alkyl, C5-7 cycloalkyl, aryl, heteroaryl, aryl-Cι_2 alkyl, or
heteroaryl-Ci-2 alkyl, wherein aryl and heteroaryl are unsubstituted or substituted with one to three substituents independently selected from Ci_4 alkyl, halogen, Ci .4 alkoxy, and trifluoromethyl; R3 is OR4 SR4, or NR4R5 ; R4 and R5 are each independently hydrogen, Ci_8 alkyl, aryl, or aryl~Cι _2 alkyl; or R4 and R5 together with the nitrogen atom to which they are attached form a 4- to 7-membered heterocyclic ring system optionally containing an additional heteroatom selected from O, S, and NC1 -4 alkyl.
SUMMARY OF THE INVENTION This invention is concerned with a process for preparing enantiomerically enriched compounds of structural formula I:
or a pharmaceutically acceptable salt thereof; having the (R)-configuration at the stereogenic center marked with an *; wherein
Ar is phenyl which is unsubstituted or substituted with one to five substituents independently selected from the group consisting of fluorine, trifluoromethyl, and trifluoromethoxy; and Rl is hydrogen or Ci_4 alkyl unsubstituted or substituted with one to five fluorines.
Another aspect of the present invention provides intermediate compounds of structural formulae II and IV which are useful for the preparation of compounds of structural formula I.
The products of the present process are disclosed in WO 03/004498 (published 16 January 2003) as potent inhibitors of dipeptidyl peptidase-IV which are useful for the treatment of Type 2 diabetes. The present invention is also concerned with a process for the preparation of enantiomerically-enriched beta-amino acid derivatives of structural formula 1. The process involves the elaboration of pure Z-enamines from beta-ketoesters and amides using (S)- phenylglycine amide and their hydrogenation with very high diastereoselectivities using heterogeneous catalysis. Hydrogenolytic cleavage of the (S)-phenylglycine amide affords the corresponding enantiomerically enriched beta-amino acid esters or amides.
DETAILED DESCRIPTION OF THE INVENTION
The present invention provides an efficient process for the preparation of enantiomerically enriched beta-amino acid amide derivatives of structural formula I:
or a pharmaceutically acceptable salt thereof; having the (R)-configuration at the stereogenic center marked with an *; wherein
Ar is phenyl which is unsubstituted or substituted with one to five substituents independently selected from the group consisting of fluorine, trifluoromethyl, and trifluoromethoxy; and Rl is hydrogen or Ci _4 alkyl unsubstituted or substituted with one to five fluorines; comprising the steps of:
(a) producing a compound of structural formula II:
by treating a compound of structural formula III:
with (S)-phenylglycine amide in the presence of an acid in a suitable organic solvent; (b) producing a compound of structural formula IN:
by hydrogenating a compound of structural formula II:
in the presence of a catalyst in a suitable organic solvent; and (c) hydrogenolyzing a compound of structural formula TV:
in the presence of a catalyst in a suitable organic solvent to afford a compound of structural formula I or a pharmaceutically acceptable salt thereof.
The first step in the process of the present invention entails the preparation of an enamine amide of structural formula II containing the (S)-phenylglycine amide (PGA) chiral auxiliary:
This is accomplished by treating a beta-ketoamide of formula HI:
with (S)-phenylglycine amide in the presence of acid in a suitable organic solvent. Embodiments of acids which can be employed to generate compounds of formula LT include formic acid, acetic acid, trifluoroacetic acid, methanesulfonic acid, trifluoromethanesulfonic acid, -toluenesulfonic acid, and anhydrous hydrogen chloride. Suitable solvents for this step of the process include methanol, ethanol, TPA, 2,2,2-trifluoroethanol, IP Ac, and mixtures thereof.
The second step in the process of the present invention concerns a diastereoselective hydrogenation of the enamine carbon-carbon double bond in the chiral substrate of formula II to afford protected chiral amines of formula IN having the (R)- configuration at the newly generated stereogenic center marked with an **. The hydrogenation proceeds with high diastereoselectivity (in excess of 90% de) by using platinum oxide (Ptθ2)
(Adam's catalyst) particularly when the catalyst is washed with acetic acid. Other catalysts that can be used include Pt/C, Pt/Al2θ3, Pd/C, and P0VAI2O3. The asymmetric hydrogenation is carried in a suitable organic solvent, such as tetrahydrofuran, a lower alkanol, for example, methanol, ethanol, TPA, and mixtures thereof, at a hydrogen gas pressure of about atmospheric pressure to about 200 psig.
The final step in the process of the present invention is removal of the (S)-PGA chiral auxiliary using hydrogenolytic conditions in the presence of a palladium catalyst. The hydrogenolysis can be effected with hydrogen gas or by using transfer hydrogenation conditions where hydrogen is generated in situ. Palladium catalysts that can be employed for the cleavage of the chiral auxiliary include Pd/C, Pd(OH)2/C, and Pd/Al2θ3. A preferred palladium catalyst is 20% Pd(OH)2/C. Transfer hydrogenation reagents as sources of hydrogen gas include
cyclohexene, cyclohexadiene, formic acid, ammonium formate, tetramethylammonium formate, sodium formate, potassium formate, and isopropyl alcohol. The hydrogenolysis reaction is performed in a suitable organic solvent or aqueous organic solvent, such as THF, methanol, ethanol, IP A, 2,2,2-trifluoroethanol, IP Ac, and mixtures thereof. The organic solvent may be admixed with acetic acid.
Finally, the diastereoselective hydrogenation and debenzylation can be performed in one pot by adding the palladium catalyst after the enamine hydrogenation with platinum oxide or compatible catalyst described above.
Another aspect of the process of the present invention comprises the following novel compounds of structural formula II which are intermediates in the preparation of the compounds of structural formula I:
wherein
Ar is phenyl which is unsubstituted or substituted with one to five substituents independently selected from the group consisting of fluorine, trifluoromethyl, and trifluoromethoxy; and R is hydrogen or Cι _4 alkyl unsubstituted or substituted with one to five fluorines. In one embodiment of the novel intermediates of structural formula II, Ar is 2,5-difluorophenyl or 2,4,5-trifluorophenyl and R is trifluoromethyl.
Yet a further aspect of the process of this invention comprises the following diastereomerically enriched compounds of structural formula IN which are intermediates in the preparation of the compounds of structural formula I:
having the (R)-configuration at the stereogenic center marked with an **; wherein
Ar is phenyl which is unsubstituted or substituted with one to five substituents independently selected from the group consisting of fluorine, trifluoromethyl, and trifluoromethoxy; and Rl is hydrogen or Ci .4 alkyl unsubstituted or substituted with one to five fluorines. In one embodiment of the novel intermediates of structural formula IN, Ar is 2,5-difluorophenyl or 2,4,5-trifluorophenyl and Rl is trifluoromethyl.
In one embodiment of this aspect of the present invention, the S,R-diastereomer of structural formula IV is present in a diastereomeric excess of at least 90% over the S,S- diastereomer. In a class of this embodiment the S,R-diastereomer of structural formula IV is present in a diastereomeric excess of at least 95% over the S,5-diastereomer.
Another aspect of the present invention provides an efficient process for the preparation of enantiomerically enriched beta-amino acid derivatives of structural formula 1_:
(1) or an amine salt thereof; having the indicated sterochernical configuration at the stereogenic center marked with an ***; wherein
R2 is Cι _8 alkyl, C5_7 cycloalkyl, aryl, heteroaryl, aryl-Cι _2 alkyl, or heteroaryl-Cι _2 alkyl, wherein aryl and heteroaryl are unsubstituted or substituted with one to three substituents independently selected from Ci .4 alkyl, halogen, Cι_4 alkoxy, and trifluoromethyl;
R3 is OR4, SR4, or NR4R5;
R4 and R5 are each independently hydrogen, Ci _8 alkyl, aryl, or aryl-Ci _2 alkyl; or R4 and R5 together with the nitrogen atom to which they are attached form a 4- to 7-membered heterocyclic ring system optionally containing an additional heteroatom selected from O, S, and NC1 -4 alkyl; comprising the steps of:
(a) producing a compound of structural formula 2:
(2)
by treating a compound of structural formula 3 :
(3) with (S)-phenylglycine amide in the presence of an acid in a suitable organic solvent; (b) producing a compound of structural formula 4:
(4)
itural formula 2:
(2)
in the presence of a catalyst in a suitable organic solvent; and (c) hydrogenolyzing a compound of structural formula 4:
(4)
in the presence of a catalyst in a suitable organic solvent to afford a compound of structural formula 1 or a salt thereof.
The first step in the process of the present invention entails the preparation of an enamine ester or amide of structural formula 2 containing the (S)-phenylglycine amide (PGA) chiral auxiliary:
(2)
This is accomplished by treating a beta-ketoester or amide of formula 3:
O O
R2^^R3 (3) with (S)-phenylglycine amide in the presence of acid in a suitable organic solvent. Embodiments of acids which can be employed to generate compounds of formula 2 include formic acid, acetic acid, trifluoroacetic acid, methanesulfonic acid, trifluoromethanesulfonic acid, p-toluenesulfonic acid, and anhydrous hydrogen chloride. A preferred acid is acetic acid which can be used in catalytic amounts. Suitable solvents for this step of the process include methanol, ethanol, isopropanol (IP A), 2,2,2-trifluoroethanol, isopropyl acetate, and mixtures thereof. Preferred solvents are methanol and isopropanol.
The second step in the process to compounds of formula 1 concerns a diastereoselective hydrogenation of the enamine carbon-carbon double bond in the chiral substrate of formula 2 to afford protected chiral amines of formula 4 having the indicated stereochemical configuration at the newly generated stereogenic center marked with an ***. The hydrogenation proceeds with high diastereoselectivity (in excess of 90% de) by using platinum oxide (Ptθ2) (Adam's catalyst) particularly when the catalyst is washed with acetic acid. The asymmetric hydrogenation is carried in a suitable organic solvent, such as tetrahydrofuran, a lower alkanol, for example, methanol, ethanol, TPA, and mixtures thereof, at a hydrogen gas pressure of about atmospheric pressure to about 200 psig. A preferred organic solvent is tetrahydrofuran.
The final step in the process of the present invention is removal of the (S)-PGA chiral auxiliary using hydrogenolytic conditions in the presence of a palladium catalyst. The hydrogenolysis can be effected with hydrogen gas or by using transfer hydrogenation conditions where hydrogen is generated in situ. Palladium catalysts that can be employed for the cleavage
of the chiral auxiliary include Pd/C, Pd(OH)2/C, and P IAI2O3. A preferred palladium catalyst is 20% Pd(OH)2/C. Transfer hydrogenation reagents as sources of hydrogen gas include cyclohexene, cyclohexadiene, formic acid, ammonium formate, tetramethylammonium formate, sodium formate, potassium formate, and isopropyl alcohol. The hydrogenolysis reaction is performed in a suitable organic solvent or aqueous organic solvent, such as THF, methanol, ethanol, IP A, 2,2,2-trifluoroethanol, IP Ac, and mixtures thereof. The organic solvent may be admixed with acetic acid.
Finally, the diastereoselective hydrogenation and debenzylation can be performed in one pot by adding the palladium catalyst after the enamine hydrogenation with platinum oxide or compatible catalyst described above.
In one embodiment of this aspect of the present invention, R3 is 2,4,5- trifluorobenzyl and R2 is Ci_4 alkoxy. In a class of this embodiment, R2 is methoxy.
Throughout the instant application, the following terms have the indicated meanings: The term "% enantiomeric excess" (abbreviated "ee") shall mean the % major enantiomer less the % minor enantiomer. Thus, an 80% enantiomeric excess corresponds to formation of 90% of one enantiomer and 10% of the other. The term "enantiomeric excess" is synonymous with the term "optical purity."
The term "enantiomerically enriched" shall mean that a compound of structural formula I is obtained by the process of the present invention with an enantiomeric excess of the desired (R)-enantiomer greater than 70% over the (S)-enantiomer. In one embodiment a compound of formula I having the (R)-configuration is obtained with an ee greater than 80%. In a class of this embodiment the (R)-enantiomer is obtained with an ee greater than 90%. In a subclass of this class the (R)-enantiomer is obtained with an ee greater than 95%. The term "% diastereomeric excess" (abbreviated "de") shall mean the % major diastereomer less the % minor diastereomer. Thus, an 80% diastereomeric excess corresponds to formation of 90% of one diastereomer and 10% of the other.
The term "enantioselective" shall mean a reaction in which one enantiomer is produced (or destroyed) more rapidly than the other, resulting in the predominance of the favored enantiomer in the mixture of products.
The term "diastereoselective" shall mean a reaction in which one diastereomer is produced (or destroyed) more rapidly than the other, resulting in the predominance of the favored diastereomer in the mixture of products.
Representative experimental procedures utilizing the novel process are detailed below. The following Examples are provided for the purpose of illustration only, but doing so is
not intended to limit the process of the present invention to the specific conditions for making these particular compounds.
Abbreviations: AcOH is acetic acid; TPA is isopropyl alcohol; IP Ac is isopropyl acetate; THF is tetrahydrofuran; TFE is 2,2,2-trifluoroethanol; DCM is dichloromethane; DMSO is dimethylsulfoxide; MeOH is methanol.
EXAMPLE 1
(2R -4-oxo-4-r3-(trifluoromethyl -5.6-dihvdror 2.41triazolor4.3-α1pyrazin-7(8H)-yll-l-(2.4.5- trifluorophenyl)butan-2-amine (2-6)
I. Preparation of 3-(trifluoromethyl -5,6,7,8-tetrahydror 2.41triazolor4,3-αlpyrazine, hydrochloride salt (1-4)
Scheme 1
O H π
1. CF3COOEt, CΗ3CN NΗ2NΗ2 »_- F3cΛN-NγCH*cl
H
2. CICOCH2CI, NaOH O
1 -1
1 -2
Step A: Preparation of bishydrazide (1-1)
Hydrazine (20.1 g, 35 wt% in water, 0.22 mol) was mixed with 310 mL of acetonitrile. 31.5 g of ethyl trifluoroacetate (0.22 mol) was added over 60 min. The internal temperature was increased to 25 °C from 14 °C. The resulting solution was aged at 22 - 25 °C for 60 min. The solution was cooled to 7 °C. 17.9 g of 50 wt% aqueous NaOH (0.22 mol) and 25.3 g of chloroacetyl chloride (0.22 mol) were added simultaneously over 130 min at a temperature below 16 °C. When the reaction was complete, the mixture was vacuum distilled to remove water and ethanol at 27 ~ 30 °C and under 26 ~ 27 in Hg vacuum. During the distillation, 720 mL of acetonitrile was added slowly to maintain constant volume (approximately 500 mL). The slurry was filtered to remove sodium chloride. The cake was rinsed with about 100 mL of acetonitrile. Removal of the solvent afforded bis-hydrazide 1Λ (43.2 g, 96.5% yield, 94.4 area% pure by HPLC assay). iH-NMR (400 MHz, DMSO-^6): δ 4.2 (s, 2H), 10.7 (s, 1H), and 11.6 (s, 1H) ppm. 13C-NMR (100 MHz, DMSO-^6): δ 41.0, 116.1 (q, J = 362 Hz), 155.8 (q, J = 50 Hz), and 165.4 ppm.
Step B: Preparation of 5-(trifluoromethyl)-2-(chloromethyl -l,3,4-oxadiazole (1-2)
Bishydrazide 1Λ from Step A (43.2 g, 0.21 mol) in ACN (82 mL) was cooled to 5 °C. Phosphorus oxychloride (32.2 g, 0.21 mol) was added, maintaining the temperature below 10 °C. The mixture was heated to 80 °C and aged at this temperature for 24 h until HPLC showed less than 2 area% of IΛ . In a separate vessel, 260 mL of IP Ac and 250 mL of water were mixed and cooled to 0 °C. The reaction slurry was charged to the quench keeping the internal temperature below 10 °C. After the addition, the mixture was agitated vigorously for 30 min, the temperature was increased to room temperature and the aqueous layer was cut. The organic layer was then washed with 215 mL of water, 215 mL of 5 wt% aqueous sodium bicarbonate and finally 215 mL of 20 wt% aqueous brine solution. HPLC assay yield after work up was 86-92%. Volatiles were removed by distillation at 75-80 mm Hg, 55 °C to afford an oil which could be used directly in Step C without further purification. Otherwise the product can be purified by distillation to afford J 2 in 70-80% yield. iH-NMR (400 MHz, CDC13): δ 4.8 (s, 2H) ppm.
13C-NMR (100 MHz, CDC13): 5 32.1, 115.8 (q, J = 337 Hz), 156.2 (q, J = 50 Hz), and 164.4 ppm.
Step C: Preparation of N-r(2Z)-piperazin-2-ylidene1trifluoroacetohydrazide (1-3)
To a solution of ethylenediamine (33.1 g, 0.55 mol) in methanol (150 mL) cooled at -20 °C was added distilled oxadiazole 1^2 from Step B (29.8 g, 0.16 mol) while keeping the
internal temperature at -20 °C. After the addition was complete, the resulting slurry was aged at -20 °C for 1 h. Ethanol (225 mL) was then charged and the slurry slowly warmed to -5 °C. After 60 min at -5 °C, the slurry was filtered and washed with ethanol (60 mL) at -5 °C. Amidine Lθ was obtained as a white solid in 72% yield (24.4 g, 99.5 area wt% pure by HPLC). iH-NMR (400 MHz, DMSO- 6): δ 2.9 (t, 2H), 3.2 (t, 2H), 3.6 (s, 2H), and 8.3 (b, IH) ppm.
13C-NMR (100 MHz, OMSO-dβ): δ 40.8, 42.0, 43.3, 119.3 (q, J = 350 Hz), 154.2, and 156.2 (q, J = 38 Hz) ppm.
Step D: Preparation of 3-(trifluoromethyl)-5,6 ,8-tetrahydror 2,41triazolor4,3- lpyrazine. hydrochloride salt (1-4)
A suspension of amidine 1^3 (27.3 g, 0.13 mol) in 110 mL of methanol was warmed to 55 °C. 37% Hydrochloric acid (11.2 mL, 0.14 mol) was added over 15 min at this temperature. During the addition, all solids dissolved resulting in a clear solution. The reaction was aged for 30 min. The solution was cooled down to 20 °C and aged at this temperature until a seed bed formed (10 min to 1 h). 300 mL of MTBE was charged at 20 °C over 1 h. The resulting slurry was cooled to 2 °C, aged for 30 min and filtered. Solids were washed with 50 mL of ethanohMTBE (1:3) and dried under vacuum at 45 °C. Yield of triazole L was 26.7 g (99.5 area wt% pure by HPLC). iH-NMR (400 MHz, OMSO-dβ): δ 3.6 (t, 2H), 4.4 (t, 2H), 4.6 (s, 2H), and 10.6 (b, 2H) ppm; 13C-NMR (100 MHz, DMSO-iό): δ: 39.4, 39.6, 41.0, 118.6 (q, J = 325 Hz), 142.9 (q, J = 50 Hz), and 148.8 ppm.
Scheme 2
Step A: Preparation of 5-ri-hydroxy-2-(2A5-trifluorophenyl)ethylidenel-2,2- dimethyl-1 ,3-dioxane-4,6-dione (2-2)
2,4,5-Trifluorophenylacetic acid (2-1) (11.4 g, 60 mmol) was dissolved in THF (60 mL) and l,l'-carbonyldiimidazole (10.7 g, 66 mmol) was added over 5 min. The mixture was warmed to 51 °C, Meldrum's acid (9.51 g, 66 mmol) was added, and the mixture was aged for 3 h. The reaction mixture was diluted with IP Ac (60 mL) and water (60 mL), and the pH
was adjusted to 2.4 with concentrated hydrochloric acid (11.5 mL). The aqueous layer was separated, and the organic layer was washed at 36 °C with 0.1 N HCI (60 mL). The organic layer was concentrated, flushed with IP Ac, and the residue was slurried in 2:1 heptane/TPAc (70 mL). The mixture was cooled over an ice-bath, then filtered, rinsing the solids with 2:1 heptane/TPAc. After drying, the Meldrum's acid adduct 2^2 was obtained as a solid (15.1 g).
Ste B: Preparation of 4-oxo-4-[3-(trifluoromethyl)-5,6-dihvdrori.2,41triazolor4,3- α1pyrazin-7(8Hy vn-l-(2,4,5- trifluorophenyl butan-2-one (2-3) The Meldrum's acid adduct 2/2 from Step A (22.1 g, 70 mmol) and the triazole hydrochloride L (16.0 g, 70 mmol) were slurried in IP Ac (220 mL) and NN- diisopropylethylamine (12.8 mL) was added. After aging for 3.5 h at 85 °C, water (175 mL) was added and the mixture was transferred to a separatory funnel with a 40-mL rinse with JPAc. The aqueous layer was separated and the organic layer was washed with water (100 mL). The organic layer was partially concentrated under diminished pressure to give a solution of ketoamide 2^3 (65 g) in L Ac. n-Ηeptane (30 mL) was added at room temperature, followed by seed crystals of ketoamide. More heptane (20 mL) was added dropwise, and the mixture was stirred overnight. Then more heptane (50 mL) was added slowly and after aging for 2 h, the solids were filtered and washed with 2.2:1 heptane/TPAc (30 mL). After drying, the ketoamide 2-3 was obtained in 92% yield (26.3 g).
Step C: Preparation of (25)-2-({(lZ)-3-oxo-l-(2A5- trifluorobenzviy3-F3-
(trifluoromethyl)-5 ,6-dihydror 1 ,2.41triazolo r4,3-αlpyrazin-7(8H)-yl1prop- 1 - enyl}amino)-2-phenylethanamide (2-4)
Ketoamide 2 S (98.4 wt%, 711 g, 1.72 mol) and (S)-phenylglycine amide (98.4 wt%, 276 g, 1.81 mol) were added to 2.8 L TPA, warmed to 40 °C and AcOΗ (49 mL) was added. The temperature rose initially to 48 °C after 15 min and came down to 40 °C in 1 h. After aging for 5 h, 0.4% seed was added and the mixture was aged 1 h to afford a slurry. This mixture was distilled at constant volume (39 °C, 98 Torr) with 2.0 L IPA flush over 2.5 h. The mixture was aged for 1 h and 2.8 L heptane was added over 3 h. The mixture was aged 3.5 h at 40 °C and cooled down slowly to room temperature over 5.5 h. After 21 h total reaction time, the slurry was rapidly filtered and rinsed with 800 mL 1 : 1 IP A/heptane. The solid was dried for 24 h under Ν2 to afford 972 g of PGA-enamine 2Λ (86.6 wt%, 98.7 area% purity, 91% yield) as an TPA solvate.
Step D: Preparation of (2S)-2-(((lR -3-oxo-l-(2.4,5-trifluorobenzylV3-r3-
(trifluoromethyl")-5.6-dihvdror 2.41triazolor4,3-αlpyrazin-7(8H)- yllpropyl I amino)-2-phenylethanamide (2-5)
Intermediate A (20.0 g) and PtO2 (0.500 g) were suspended in 80 mL TΗF and 20 mL MeOΗ (22 °C) in a 300 mL stirred autoclave. The stirred solution was cooled to 15 °C and placed under hydrogen pressure (90 psig). After 30 min, the solution was warmed to 22 °C over 30 min and aged for 26 h. The autoclave was vented to atmospheric pressure and the reaction solution was filtered through Solka floe. The filter cake was rinsed with methanol (2 x 20 mL). The filtrate and washings were combined and carried on directly to the final debenzylation Step E. Compound 2^ was obtained in 90% assay yield (about 17.8 g) and 96.4% diastereomeric excess (de).
The PtO2 catalyst was prepared as follows: PtO2 (36.7 g) was suspended in acetic acid (130 mL). The stirred slurry was aged at room temperature for 2 h and then filtered. The filter cake was rinsed with acetic acid (4 x 25 mL) and then dried in vacuo at 50 °C for 24 h. The PtO2 was isolated with 97% recovery (35.4 g).
Step E: Preparation of (2R -4-oxo-4-r3-(trifluoromethyl -5.6-dihvdrori.2.41triazolor4.3- αlpyrazin-7(8H)-yl"l-l-(2 ,4,5- trifluorophenyl)butan-2-amine (2-6) The crude product from Step D (41.5 g, 76.3 mmol) and 20% Pd(OΗ)2/C (12.4 g, 30 wt%) was slurried in 1 : 1 THF/MeOH (124 mL) and water (41.5 mL) and formic acid (41.5 mL) were added. The reaction mixture was heated at 60 °C for 3 h. After cooling, the reaction was treated with Solka floe (10 g) and filtered through Solka floe (20 g), rinsing with MeOH (200 mL). The filtrate contained 29.4 g (94.5% assay yield) of 2-6 with an optical purity of 97% ee.
Alternative Step E:
The crude product from Step D (82.5 g, 153 mmol) was dissolved in 1:1 THF/MeOH (200 mL) and finely pulverized 20% Pd(OH)2/C (24.8 g, 30 wt%) was added. The mixture was added to a stainless-steel autoclave. THF/MeOH (130 mL) was used to rinse the mixture into the vessel, then acetic acid (21.9 mL) and water (82.5 mL) were added. The mixture was heated at 50 °C for 10 h at 40 psig hydrogen. After cooling to room temperature and venting to ambient pressure, the autoclave was emptied with MeOH rinse (2 x 100 mL). The batch was treated with Solka floe (11 g) and filtered through Solka floe (42 g). The filter pad was washed with MeOH (200 mL). The filtrate contained 57.9 g of 2^6 with an optical purity of 96% ee.
The optical purity of 2^6 was further enhanced in the following manner. The solution from the hydrogenation reaction (18 g in 180 mL solvent) was concentrated and switched to methyl t-butyl ether (MTBE) (45 mL). Into this solution was added aqueous H3PO4 solution (0.5 M, 95 mL). After separation of the layers, 3N ΝaOH (35 mL) was added to the water layer, which was then extracted with MTBE (180 mL + 100 mL). The MTBE solution was concentrated and solvent switched to hot toluene (180 mL, about 75 °C). The hot toluene solution was then allowed to cool to 0 °C slowly (5 - 10 h). The crystals were isolated by filtration (13 g, yield 72%, 98 - 99% ee); m.p. 114.1 - 115.7 °C. IH ΝMR (300 MHz, CD3CΝ): δ 7.26 (m), 7.08 (m), 4.90 (s), 4.89 (s), 4.14 (m), 3.95 (m), 3.40 (m), 2.68 (m), 2.49 (m), 1.40 (bs).
Compound 2^6 exists as amide bond rotamers. Unless indicated, the major and minor rotamers are grouped together since the carbon- 13 signals are not well resolved:
13C NMR (CD3CN): δ 171.8, 157.4 (ddd , JCF = 242.4, 9.2, 2.5 Hz), 152.2 (major), 151.8
(minor), 149.3 (ddd; JCF = 246.7, 14.2, 12.9 Hz), 147.4 (ddd, JCF = 241.2, 12.3, 3.7 Hz), 144.2 (q, JCF = 38.8 Hz), 124.6 (ddd , JCF = 18.5, 5.9, 4.0 Hz), 120.4 (dd , JCF = 19.1, 6.2 Hz), 119.8
(q, JCF = 268.9 Hz), 106.2 (dd , JCF = 29.5, 20.9 Hz), 50.1, 44.8, 44.3 (minor), 43.2 (minor),
42.4, 41.6 (minor), 41.4, 39.6, 38.5 (minor), 36.9.
The following high-performance liquid chromatographic (HPLC) conditions were used to determine percent conversion to product: Waters Symmetry Shield RP8, 4.6 x 250 mm, 5 μm, 25 °C, 210 nm detection, 5 μL injection, 1 mL/min, 15 min run time, isocratic at 45% acetonitrile/55% 10 mM pH 6.8 phosphate buffer.
Retention times:
Compound 2-6: 4.30 min
Compound 2^5 (S,R-diastereomer): 7.73 min Compound 2-5 ( S-diastereomer): 7.11 min
The following high-performance liquid chromatographic (HPLC) conditions were used to determine optical purity (percentage ee):
Chiralcel AD-H, 5 μm, 4.6 x 250 mm, 5 μm, 35 °C, 268 nm detection, 10 μL injection, 0.7 mL/min, 25 min run time, isocratic at 40% 0.1% diethylamine in hexane/60% 0.1% diethylamine in ethanol. Retention times:
Compound 2^6 (R-enantiomer): 18.4 min
Compound 2^6 (S-enantiomer): 15.4 min
Examples of Z-PGA-Enamines:
EXAMPLE 2 Methyl (2Z)-3-{r(lS)-2-amino-2-oxo-l-phenylethyllarninolbut-2-enoate
Methyl acetoacetate (2.0 g, 17.2 mmol) and (S)-PGA (2.58 g, 17.2 mmol) were dissolved in MeOH (200 mL) at 40 °C and acetic acid (0.5 mL, 8.6 mmol) was added. Solids formed after 1 h at 40 °C. The mixture was cooled to 4 °C and filtered. The solids were rinsed with cold MeOH and dried to afford the enamine (3.3 g). [α]D 25 = -92° (c 0.44, DCM); mp = 159 °C.
1H-NMR (DMSO-d6, 400 MHz): δ 9.46 (d, J = 8 Hz, IH), 7.70 (br, IH), 7.37 (m, 4H), 7.28 (m, 2H), 5.19 (d, J = 8 Hz, IH), 4.44 (s, IH), 3.52 (s, 3H), 1.73 (s, 3H).
13C-NMR (DMSO-de, 100 MHz): δ 171.2, 169.5, 160.1, 139.8, 128.7, 127.8, 126.4, 83.1, 59.0, 49.6, 19.4
EXAMPLE 3 Methyl (2Z)-3-( r(lS)-2-amino-2-oxo-l-phenylethyl1amino }-4-methylpent-2-enoate
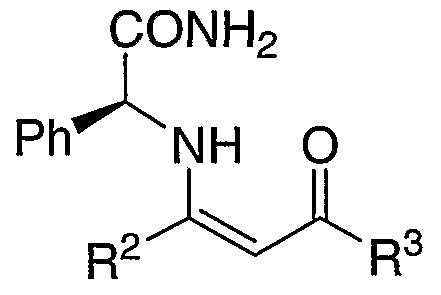
CONH2 JL pιr *NH o iPr 0Me
Methyl isobutyryl acetate (7.59 g, 50.0 mmol) and (S)-PGA (7.51 g, 50.0 mmol) were mixed in IPA (40 mL) at 40 °C and acetic acid (1.43 mL, 25 mmol) was added. A slurry formed quickly. The mixture was stirred overnight at 40 °C, cooled to 3 °C and was then filtered. The solids were rinsed with IPA and dried to afford the enamine as a white solid (11.6 g). [ ]D 25 = -57° (c 0.58, DCM); mp = 170 °C.
1H-NMR (DMSO-de, 400 MHz): δ 9.55 (d, /= 8 Hz, IH), 7.76 (s, IH), 7.40 (m, 4H), 7.30 (m, 2H), 5.29 (d, J = 8 Hz, IH), 4.48 (s, IH), 3.54 (s, 3H), 2.44 (m, IH), 1.08 (d, /= 7 Hz, 3H), 0.78
(d, /= 7 Hz, 3H).
13C-NMR (DMSO-d6, 100 MHz): δ 171.5, 170.5, 170.2, 140.6, 129.0, 128.1, 126.6, 79.2, 58.5,
49.9, 29.2, 22.4, 21.1.
EXAMPLE 4 Methyl (2Z)-3-{r(lS)-2-amino-2-oxo-l-phenylethyl1amino|-4-phenylbut-2-enoate
Methyl 3-oxo-4-phenylbutanoate (1.76 g, 9.16 mmol) and (S)-PGA (1.38 g, 9.16 mmol) were mixed in isopropanol (IPA) (18 mL) and acetic acid (0.26 mL, 4.6 mmol) and stirred 18 h over a 50 °C bath. The resulting slurry was cooled and filtered. The solids were rinsed with cold MeOH and dried to afford the enamine (2.20 g). [α
Dj
25 = -63° (c 0.40, DCM); mp = 169 °C. 1H-NMR (DMSO-de, 400 MHz): δ 9.46 (d, / = 8 Hz, IH), 7.71 (s, IH), 7.34 (d, 4H), 7.26 (m, 4H), 7.20 (m, IH), 7.08 (d, 2H), 5.10 (d, J - 8 Hz, IH), 4.30 (s, IH), 3.50 (s, 3H), 3.32 (q, 2H).
13C-NMR (DMSO-de, 100 MHz): δ 170.8, 169.4, 161.7, 139.9, 136.3, 128.7, 128.6, 128.6, 127.8, 126.7, 126.3, 84.6, 58.6, 49.7, 37.9
EXAMPLE 5
(2S)-2- 1 F( 1Z)- 1 -benzyl-3 -oxo-3-piperidin- 1 -ylprop- 1 -en- 1 -yll amino } -2-phenylacetamide
4-Oxo-l-phenyl-4-piperidin-l-ylbutan-2-one (13.8 g, 56.0 mmol) and (S)-PGA (8.42 g, 56.0 mmol) were stirred in MeOH (70 mL) with acetic acid (1.6 mL, 28 mmol) 3 days at
40 °C. The mixture was concentrated and the residue was purified by flash chromatography (80-
100% EtOAc/ hexane) and then was crystallized (EtOAc/ hexane) to afford the enamine (7.4 g).
[α]D 25 = -86° (c 0.44, MeOH); mp = 155 °C.
1H-NMR (DMSO-d6, 400 MHz): δ 10.23 (d, J = 9 Hz, IH), 7.60 (br, IH), 7.29 (m, 9H), 7.19 (m, IH), 7.10 (br, IH), 4.99 (d, J = 9 Hz, IH), 4.81 (s, IH), 3.32-3.35 (m, 6H), 2.49 (m, 2H), 1.41
(m, 4H).
13C-NMR (DMSO-d6, 100 MHz): δ 171.3, 168.3, 158.2, 140.3, 137.3, 128.4, 128.3, 127.5,
126.5, 126.4, 85.1, 58.9, 38.6, 25.8, 24.3.
EXAMPLE 6 (22D-3-I r(15)-2-amino-2-oxo-l-phenylethyllaminol-4-phenylbut-2-enamide
3-Oxo-4-phenylbutanamide (1.85 g, 10.4 mmol) and (S)-PGA (1.56 g, 10.4 mmol) were stirred in MeOH (18 mL) with acetic acid (0.30 mL, 5.2 mmol) 4 days at 40 °C. The mixture was diluted with methyl tert-butyl ether (MTBE), washed with saturated sodium bicarbonate, dried (MgSO4) and concentrated. The residue was purified by flash chromatography (50-100% EtOAc/hexane) and then crystallized (EtOAc/ hexane) to afford the enamine (0.63 g). [α]D 25 = +9° (c 0.18, DCM); mp = 188 °C.
1H-NMR (DMSO-d6, 400 MHz): δ 9.89 (d, /= 8.8 Hz, IH), 7.61 (s, IH), 7.33 (m, 4H), 7.28 (d,
/ = 7 Hz, 3 H), 7.21 (m, IH), 7.15 (d, 7 Hz, 2H), 7.10 (s, IH), 6.6 (br, IH), 6.1 (br, IH), 5.01 (d,
J= 8.8 Hz, IH), 4.37 (s, IH), 3.27 (q, 2H).
13C-NMR (DMSO-d6, 100 MHz): δ 171.7, 171.4, 157.2, 140.5, 137.2, 128.5, 128.4, 127.4, 126.4, 126.4, 89.4, 58.9, 38.1.
EXAMPLE 7 Methyl (2Z)-3-{ r(lS)-2-arnino-2-oxo-l-phenylethyl1amino 1-3-phenylacrylate
Methyl 3-oxo-3-ρhenylρropanoate (20.2 g, 114 mmol) and (S)-PGA (17.0 g, 114 mmol) were added to MeOH (60 mL) with acetic acid (3.25 mL, 57 mmol) and stirred 4 days over a 40 °C bath. The mixture was concentrated and the residue was purified by flash chromatography (40-60% EtOAc/ hexane) to afford an oil, which was crystallized in 20% . EtOAc/ hexane (250 mL) to afford after drying the enamine (19.7 g). [α]D 25 = -68° (c 0.49, DCM); mp = 142 °C.
1H-NMR (DMSO-d6, 400 MHz): δ 9.46 (d, J = 9 Hz, IH), 7.65 (s, IH), 7.39 (m, IH), 7.33 (m, 2H), 7.24 (m, 4H), 7.11 (m, 4H), 4.92 (d, J= 9 Hz, IH), 4.51 (s, IH), 3.61 (s, 3H).
13C-NMR (DMSO-d6, 100 MHz): δ 171.0, 169.0, 162.5, 140.0, 135.5, 129.2, 128.2, 128.2, 127.3, 127.2, 126.1, 86.4, 59.9, 49.8.
EXAMPLE 8 (2S)-2- { [Y 12D-3 -oxo- 1 -phenyl-3 -piperidin- 1 - ylprop- 1 -en- 1 -yll amino } -2-phenylacetamide
3-Oxo-l-phenyl-3-piperidin-l-ylpropan-l-one (10.1 g, 43.7 mmol) and (S)-PGA (6.6 g, 44 mmol) in IPA (40 mL) and acetic acid (1.9 mL, 33 mmol) were stirred 4 days at 40 °C. The mixture was concentrated and purified by flash chromatography and crystallization (EtOAc/ hexane) to afford the enamine (6.9 g).
[α]D 25 = +86° (c 0.26, DCM); mp = 126 °C.
1H-NMR (DMSO-de, 400 MHz): δ 10.17 (d, /= 9 Hz, IH), 7.56 (s, IH), 7.07-7.37 (m, 11H),
4.84 (s, IH), 4.80 (d, J = 9 Hz, IH), 3.41 (br, 4H), 1.56 (br, 2H), 1.33 (br, 4H).
13C-NMR (DMSO-d6, 100 MHz): δ 171.5, 167.7, 160.0, 140.5, 137.0, 128.8, 128.2, 128.1, 127.5, 127.2, 126.4, 87.4, 60.4, 25.8, 24.3
EXAMPLE 9 (2Z)-3-(r(lS)-2-arnino-2-oxo-l-phenylethyl1aminol-3-phenylacrylamide
3-Oxo-3-ρhenylproρanamide (6.53 g, 40.0 mmol) and (S)-PGA (6.01 g, 40.0 mmol) in IPA (35 mL) and acetic acid (1.2 mL, 20 mmol) were stirred 4 days at 40 °C. The mixture was concentrated and the residue was dissolved in EtOAc (150 mL). The undissolved solids were filtered off and the filtrate was concentrated. Purification by flash chromatography (0-10% MeOH/ EtOAc) followed by crystallization afforded the enamine (5.47 g) as a white solid.
[α]D 25 = +18° (c 0.33, DCM); mp = 111-130 °C.
1H-NMR (DMSO-de, 400 MHz): δ 9.74 (d, J= 10 Hz, IH), 7.58 (s, IH), 7.33 (m, 3H), 7.22 (m, 3H), 7.16 (m, 2H), 7.08 (m, 3H), 6.9 (br, IH), 6.3 (br, IH), 4.79 (d, J = 10 Hz, IH), 4.60 (s, IH). 13C-NMR (DMSO-d6, 100 MHz): δ 172.0, 171.5, 159.2, 140.9, 137.1, 129.2, 128.6, 128.5, 127.8, 127.6, 126.8, 92.8, 60.8.
EXAMPLE 10 Methyl (2Z)-3- ( \( lS)-2-amino-2-oxo- 1 -phenylethyll amino I -3-(4-methoxyphenyl)acrylate
Methyl 3-(4-methoxyphenyl)-3-oxopropanoate (10.41 g, 50.0 mmol) and (S)- PGA (7.51 g, 50.0 mmol) in IPA (40 mL) and acetic acid (1.43 mL, 25 mmol) were stirred 5 days at 40 °C. The mixture was concentrated and EtOAc (50 mL) was added. The undissolved solids were filtered off and the filtrate was concentrated. The residue was purified by flash chromatography (30-100% EtOAc/ hexane) to afford the enamine as an amorphous solid, which was heated and stirred in MTBE (150 mL) to afford a slurry. The solids were filtered and dried to afford the enamine (5.96 g).
[α]D 2S = -31° (c 0.56, DCM); mp = 93 °C (broad).
1H-NMR (DMSO-d6, 400 MHz): δ 9.41 (d, J = 8.9 Hz, IH), 7.64 (s, IH), 7.26 (m, 3H), 7.16 (m, 3H), 7.05 (d, J = 8.5 Hz, 2H), 6.89 (d, / = 8.6 Hz, 2H), 4.95 (d, / = 8.9 Hz, IH), 4.50 (s, IH), 3.75 (s, 3H), 3.60 (s, 3H). 13C-NMR (DMSO-d6) 100 MHz): δ 171.2, 169.1, 162.6, 160.0, 140.1, 128.8, 128.3, 127.9, 127.4, 126.2, 113.7, 86.3, 60.1, 55.2, 49.9.
EXAMPLE 11
Methyl (2Z)-3-{ r(lS)-2-amino-2-oxo-l-phenylethyllamino)-3-r4- (trifluoromethyl)phenyll acrylate
Methyl 3-oxo-3-[4-(trifluoromethyl)phenyl]propanoate (5.18 g, 20.0 mmol) and (S)-PGA (3.00 g, 20.0 mmol) in IPA (25 mL) and acetic acid (1.2 mL, 20 mmol) were stirred 2 days at 40 °C. The mixture was cooled to RT, n-heptane (10 mL) was added to precipitate the unreacted PGA and filtered. The filtrate was concentrated and the residue was purified by flash chromatography (50-70% EtOAc/ hexane) to afford the enamine as an amorphous solid (6.2 g) which contained a small amount of the E-enamine isomer. The solids were crystallized from MTBE to afford the pure Z-enamine (4.18 g) as a white solid. [α]
D 25 = -79° (c 0.57, DCM); mp = 91 °C (broad).
1H-NMR (DMSO-d6, 400 MHz): δ 9.43 (d, J = 8.6 Hz, 1 H), 7.70 (d, / = 8 Hz, 2 H), 7.60 (s, IH), 7.30 (d, J= 8 Hz, 2H), 7.24 (~t, 4 H), 7.10 (d, J= 6.5 Hz, 2 H), 4.86 (d, J= 8.6 Hz, IH), 4.57 (s, IH), 3.62 (s, 3H).
13C-NMR (DMSO-de, 100 MHz): δ 170.8, 168.9, 160.9, 139.8, 139.6, 128.4, 128.3, 127.6, 126.2, 125.2, 87.3, 59.9, 50.1.
Examples of Hydrogenated PGA- Amines:
General Procedure: A pressure reaction vessel was charged with the PGA-enamine, PtO2 and THF and the resulting slurry was cooled to 0 °C. The vessel was pressure-purged with nitrogen (3 x 40 psig) and hydrogen (2 x 40 psig) and then pressurized with hydrogen (90 psig). The reaction mixture was agitated for 30 min and then allowed to warm to room temperature over 15 min. After stirring for the prescribed amount of time, the vessel was carefully vented to atmospheric pressure and the resulting slurry was filtered through Solka floe. The filter cake was rinsed three times with THF and the filtrates were combined and concentrated. The residue was purified by flash chromatography (EtOAc/ hexane) and the product was crystallized (EtOAc/ hexane) when possible.
EXAMPLE 12 Methyl (3R)-3- j r(lS)-2-aπuno-2-oxo-l-phenylethyl1amino|butanoate
Flash chromatography (90% EtOAc/ hexane) afforded the ester.
1H-NMR (DMSO-d
6, 400 MHz): δ 7.57 (bs, IH), 7.45 (m, 2H), 7.39 (m, 2H), 7.33 (m, IH), 7.20 (bs, IH), 4.31 (bd, J = 4 Hz, IH), 3.67 (s, 3H), 2.99 (m, IH), 2.59 (dd, J = 15, 7 Hz, 2H), 2.38 (dd, J = 15, 7Hz, 2H), 1.11 (d, J= 6 Hz, 3H).
13C-NMR (DMSO-d6, 100 MHz): δ 174.1, 172.2, 140.6, 128.1, 127.3, 127.2, 63.0, 51.2 47.9 41.3, 19.8.
EXAMPLE 13 Methyl (3S)-3- { f( lS)-2-amino-2-oxo- 1 -phenylethynamino } -4-methylpentanoate
Flash chromatography (70-85% EtOAc/ hexane) afforded pure amine.
1H-NMR (DMSO-d6, 400 MHz): δ 7.42 (bs, IH), 7.37 (m, 2H), 7.31 (m, 2H), 7.24 (m, IH), 7.12
(bs, IH), 4.20 (d, J= 6 Hz, IH), 3.59 (s, 3H), 2.68 (m, IH), 2.33 (m, 2H), 1.77 (m, IH), 0.83 (d,
J = 7 Hz, 3H), 0.76 (d, J = 7 Hz, 3H).
13C-NMR (DMSO-de, 100 MHz): δ. 174.1, 172.8, 140.7, 128.0, 127.4, 127.1, 63.2, 57.4, 51.3, 29.7, 18.3, 17.4.
EXAMPLE 14 Methyl (3RV3-I r(lS)-2-amino-2-oxo-l-phenylethyl1amino }-4-phenylbutanoate
Flash chromatography (80-100% EtOAc/ hexane) afforded the amine. This was crystallized from EtOAc/ hexane.
1H-NMR (DMSO-d6, 400 MHz): δ 7.45 (s, IH), 7.24 (m, 8H), 7.11 (m, 3H), 4.31 (d, J= 6.5 Hz,
IH), 3.55 (s, 3H), 2.97 (m, IH), 2.80 (dd, J = 6.0, 13.3 Hz, 1 H), 2.58 (dd, / = 7.0 13.3 Hz, IH),
2.37-2.46 (m, 2H), 2.29 (dd, /= 5.8, 15.2 Hz, IH). 13C-NMR (DMSO-d6, 100 MHz): δ 173.8, 172.1, 140.2, 138.7, 129.2, 128.2, 128.0, 127.2,
127.2, 126.1, 62.8, 54.0, 51.4, 38.5.
EXAMPLE 15
(2SV2- 1 f( 1R)- 1 -benzyl-3 -oxo-3-piperidin- 1 -ylpropyll amino ) -2-phenylacetamide
Flash chromatography (5% MeOH/ EtOAc) afforded the amine as an oil which was crystallized from 1:1 EtOAc/ hexane to afford the product as a white solid. 1H-NMR (DMSO-d6, 400 MHz): δ 7.65 (s, IH), 7.1-7.3 (m, 11H), 4.33 (d, J = 4.6 Hz, IH), 3.39 (m, 2H), 3.20 (m, 2H), 2.98 (m, IH), 2.81 (dd, J= 5.7, 13.2 Hz, IH), 2.59 (dd, /= 6.9, 13.2 Hz, IH), 2.3-2.5 (m, 2H), 2.22 (dd, 7= 5.5, 15.2 Hz, IH), 1.52 (m, 2H), 1.36 (m, 4H). 13C-NMR (DMSO-dg, 100 MHz): δ 174.1, 169.1, 140.5, 139.2, 129.3, 128.2, 128.0, 127.3, 127.1, 126.0, 62.9, 54.2, 45.9, 41.8, 37.0, 26.0, 25.2, 23.9.
EXAMPLE 16 (3R)-3- { f( lS)-2-amino-2-oxo- 1 -phenylethyll amino ) -4-phenylbutanamide
Stirring the residue in 1:1 EtOAc/ hexane followed by filtration and drying afforded the amine as a white solid.
1H-NMR (DMSO-d6, 400 MHz): δ 7.64 (s, IH), 7.46 (s, IH), 7.25 (m, 8H), 7.11 (m, 3H), 6.83
(s, IH), 4.36 (s, IH), 2.97 (br, IH), 2.80 (dd, / = 5.5, 13.2 Hz, IH), 2.57 (dd, J = 7.0, 13.2 Hz,
IH), 2.28 (br, IH), 2.10 (m, 2H).
13C-NMR (DMSO-d6, 100 MHz): δ 174.1, 173.2, 140.5, 139.0, 129.3, 128.2, 128.0, 127.3, 127.2, 126.0, 62.8, 53.9, 39.4.
EXAMPLE 17
Methyl (3S)-3- { f( lS)-2-amino-2-oxo- 1 -phenylethyll amino 1 -3-phenylpropanoate
Flash chromatography (75% EtOAc/ hexane) afforded the pure amine.
1H-NMR (DMSO-de, 400 MHz): δ 7.59 (d, /= 2.4 Hz, IH), 7.2-7.3 (m, 11H), 3.98 (q, J = 1.1
Hz, IH), 3.72 (d, J = 5.3 Hz, IH), 3.56 (s, 3H), 3.14 (m, IH), 2.86 (dd, J = 8.1, 15.4 Hz, IH),
2.58 (dd, J= 6.4, 15.4 Hz, IH).
13C-NMR (DMSO-d6, 100 MHz): δ 173.9, 171.6, 142.4, 140.1, 128.5, 128.2, 127.4, 127.4,
127.3, 127.2, 63.5, 57.6, 51.4, 42.3.
EXAMPLE 18 (2S)-2-r(3-oxo-l-phenyl-3-piperidin-l-ylpropyl)amino1-2-phenylacetamide
Flash chromatography (EtOAc/ hexane) afforded the amine as a white solid foam . 1H-NMR (DMSO-d6, 400 MHz): δ 7.99 (s, IH), 7:36-7.19 (m, 11H), 4.03 (m, IH), 3.70 (d, /= 5 Hz, IH), 3.46 (m, IH), 3.35 (m, IH), 3.30 (m, 3H), 3.01 (bt, IH), 2.81 (dd, /= 8.6, 15 Hz, IH), 2.52 (dd, 4.9, 15 Hz, IH), 1.52 (m, 2H), 1.39 (4 H). 13C-NMR (DMSO-d6, 100 MHz): δ 174.1, 168.6, 143.2, 140.5, 128.3, 128.1, 127.2, 127.2, 127.0, 63.6, 57.8, 46.0, 42.0, 40.7, 25.8, 25.2, 24.0.
EXAMPLE 19
3- { K lS)-2-amino-2-oxo- 1 -phenylethyll amino I -3-phenylpropanamide
Suspension of the residue in EtOAc/ hexane and filtration afforded the product as a white solid. 1H-NMR (DMSO-d6) 400 MHz): δ 7.74 (s, IH), 7.42 (s, IH), 7.32 (m, 4H), 7.24 (m, 6H), 7.16 (s, IH), 6.83 (s, IH), 3.97 (bq, IH), 3.74 (d, J = 5 Hz, IH), 3.05 (bt, IH), 2.52 (dd, J= 9.1, 15 Hz, IH), 2.30 (dd, J = 5.1, 15 Hz, IH). 13C-NMR (DMSO-d6, 100 MHz): δ 174.0, 172.5, 143.2, 140.4, 128.3, 128.1, 127.2, 127.1, 127.1, 63.3, 57.5, 43.7.
EXAMPLE 20 Methyl 3-{r(lS)-2-amino-2-oxo-l-phenylethyllanιino)-3-(4-methoxyphenyl)proρanoate
Crystallization from EtOAc/ hexane afforded the amine as a white solid.
1H-NMR (DMSO-d6, 400 MHz): δ 7.55 (s, IH), 7.23 (m, 8H), 6.89 (d, 7 = 8.6 Hz, 2H), 3.92 (br- q, IH), 3.74 (s, 3H), 3.72 (IH), 3.55 (s, 3H), 3.06 (br-t, IH), 2.84 (dd, 7= 8, 15 Hz, IH), 2.54
(dd, 7= 6.5, 15 Hz, IH).
13C-NMR (DMSO-d6, 100 MHz): δ 173.8, 171.5, 158.4, 140.1, 134.1, 128.2, 128.1, 127.2,
127.1, 113.7, 63.3, 56.8, 55.0, 51.2, 42.4.
EXAMPLE 21 Methyl 3-1 r(lS)-2-arrj no-2-oxo-l-phenylethyllamino)-3-L4-(trifluoromethyl)phenyllpropanoate
Flash chromatography (80% EtOAc/ hexane) afforded the amine. 1H-NMR (CD3OD, 400 MHz): δ 7.66 (d, 7 = 8 Hz, 2H), 7.54 (d, 7 = 8 Hz, 2H), 7.27 (m, 5H),
4.25 (m, IH), 3.88 (s, IH), 3.68 (s, 3H), 2.89 (dd, 7 = 9.0, 16.0 Hz, IH), 2.70 (dd, 7= 5.6, 16.0
Hz, IH).
13C-NMR (CD3OD, 100 MHz): δ 178.0, 173.6, 148.0, 140.7, 129.8, 129.3, 129.2, 128.7, 126.8,
126.8, 65.6, 59.1, 52.4, 43.0 Examples of Deprotected Amines:
General Procedure: A pressure reaction vessel was charged with PGA-amine, acetic acid (2.5 molar equiv), water, THF, and 30% Pd(OH)2 (20 wt% based on PGA-amine) and warmed to 50 °C. The vessel was pressure-purged with nitrogen (3 x 40 psig) and pressurized with hydrogen (40 psig) and the reaction solution was stirred for 12 h. After cooling to room temperature, the vessel was vented and the solution was filtered through Solka floe. The filter cake was then
washed with THF. The filtrates were combined and the solvent was removed under reduced pressure to yield a mixture of the desired beta-amino acid derivative as the acetate salt and 2- phenylacetamide byproduct. The assay yield of the amine salt was determined by 1H-NMR using added dichloromethane as an internal reference. The stereochemical assignment was made by measuring the optical rotation of the crude product as its HCI salt and comparing its sign with what is reported in the literature or of an authentic reference sample. The HCI salt was prepared by addition of HCI/ ether and evaporation to dryness.
EXAMPLE 22 Methyl (3R)-3-aminobutanoate acetate
1H-NMR (CD3OD, 400 MHz): δ 3.73 (s, 3H), 3.65 (m, IH), 2.70 (m, 2H), 1.95 (s, 3H), 1.34 (d, 7= 6.6 Hz, 3H).
13C-NMR (CD3OD, 100 MHz): δ 178.4, 172.5, 52.8, 45.6, 39.1, 23.0, 18.9.δ Observed (HCI salt): [α]D 27= negative (water) (Thus, stereochemistry is R as shown).
Literature for (R)-enantiomer (HCI salt): [α]D 28 = -37.0 (c 0.73, water) [7. Org. Chem. 1992, 57, 2396].
EXAMPLE 23 Methyl (3S)-3-amino-4-methylpentanoate acetate
1H-NMR (CD3OD, 400 MHz): δ 3.74 (s, 3H), 3.38 (m, IH), 2.76 (dd, 7 = 4.0, 17.3 Hz, IH), 2.59 (dd, 7= 8.7, 17.3 Hz, IH), 1.96 (m, IH), 1.93 (s, 3H), 1.01 (~t, 6H). 13C-NMR (CD3OD, 100 MHz): δl79.3, 173.4, 55.2, 53.1, 35.5, 32.3, 23.7, 19.0, 18.5. Observed (HCI salt): [α]o25 = negative (MeOH) (Thus, stereochemistry is S as drawn)
Literature for the (R)-enantiomer (HCI salt): [α]D 25 = +28.2 (c 0.48, MeOH) [Tetrahedron 1995, 51, 12337].
EXAMPLE 24 Methyl (3R)-3-amino-4-phenylbutanoate acetate
1H-NMR (CD3OD, 400 MHz): δ 7.3 (m, 5H), 3.71 (m, IH), 3.69 (s, 3H), 2.64 (dd, 7= 4.8, 17.0 Hz, IH), 2.54 (dd, 7= 7.8, 17.0 Hz, IH), 1.94 (s, 3H).
13C-NMR (CD3OD, 100 MHz): δl72.9, 137.5, 130.6, 130.2, 128.6, 52.7, 51.0, 40.9, 37.9, 23.1. Observed (HCI salt): [α]D 25 = negative (MeOH) (Thus, stereochemistry is R as drawn). Literature for the (R)-enantiomer (HCI salt): [α]p= -9.2 (c 1.5, MeOH) [Biosci. Biotech. Biochem., 1996, 60, 916].
EXAMPLE 25 f( 1R)- 1 -benzyl-3 -oxo-3 -piperidin- 1 -ylpropyll amine acetate
1H-NMR (CD3OD, 400 MHz): δ 7.3 (m, 5H), 3.78 (m, IH), 3.52 (m, 2H), 3.32 (m, 2H), 3.08
(dd, 7= 6.3, 13.7 Hz, IH), 2.96 (dd, 7= 8.5, 13.7 Hz, IH), 2.64 (m, 2H), 1.96 (s, 3H), 1.63 (m,
2H), 1.51 (m, 4H). 13C-NMR (CD3OD, 100 MHz): δl77.9, 169.8, 137.3, 130.6, 130.2, 128.6, 51.5, 49.2, 47.6, 44.0,
39.6, 34.6, 27.4, 26.7, 25.4, 22.8.
Observed (HCI salt):
negative (MeOH) (Thus, stereochemistry is R as shown).
An authentic sample of (S)-enantiomer (HCI salt) was prepared from Boc-L-β- homophenylalanine via EDC coupling with piperidine, followed by HCI deprotection: [OJD 20 = +37 (c 1.4, MeOH).
EXAMPLE 26
(3R)-3-amino-4-phenylbutanamide acetate
1H-NMR (CD
3OD, 400 MHz): δ 7.3 (m, 5H), 3.75 (m, IH), 3.07 (dd, 7 = 6.1, 13.7 Hz, IH), 2.90 (dd, 7= 8.6, 13.7 Hz, IH), 2.55 (m, 2H), 1.96 (s, 3H).
13C-NMR (CD3OD, 100 MHz): δl78.4, 175.2, 136.9, 130.6, 130.2, 128.6, 51.4, 39.7, 36.4, 23.0. Observed (HCI salt): [α]D 20= negative (MeOH) (Thus, stereochemistry is R as shown).
An authentic sample of (S)-enantiomer (HCI salt) was prepared from Boc-L-β- homophenylalanine via EDC coupling with ammonia, followed by HCI deprotection: [α]D 2°: +11 (c 1.0, MeOH).
EXAMPLE 27
Step A: Preparation of Compound 3-2
A 2-liter 3-necked round-bottomed flask equipped with overhead stirrer, thermocouple, nitrogen inlet, and a condenser was charged with β-keto ester 3 (35 g), (S)- phenylglycine amide (21.35 g), methanol (700 mL) and acetic acid (44.78 mL). An exotherm of about 4 °C was observed during the acid addition. The mixture was heated to reflux. After 9 h, the mixture was removed from heating and allowed to cool to room temperature. When the temperature reached 40 °C, the product appeared as white solids. At 35 °C, the mixture turned into slurry. The mixture was stored at 4 °C. Concentration of the Z isomer in the supernatant was 7.08 mg/mL after 1 h and 3.19 mg/mL after overnight at 4 °C. Solids were filtered at 4 °C, washed with 140 mL MeOH and dried. The isolated solids were the Z isomer and have an S- configuration at the stereogenic center.
Step B: Preparation of Compound 3-3
The PGA enamine 3^2 (60 g), Ptθ2 (6 g), acetic acid (45.4 mL), and THF (1.2 L) were charged into a flask. The mixture was hydrogenated for 15 h at 90 psi and room temperature. The catalysts were filtered through solka floe and rinsed with about 500 mL THF. Water (300 mL) was added and then the pH adjusted to about 7 with 50wt% NaOH over 15 min.
An exotherm of 9 °C was observed. The layers were then separated, and the organic layer was washed with water (150 mL). The organic layer was solvent switched at 45 °C to toluene (210 mL). The mixture was transferred to a round bottom flask equipped with mechanical stirrer. The product precipitated out as the solution cooled. The mixture was heated to 63 °C to redissolve the product, then cooled at 20 °C/h. It was seeded at 57 °C. After room temperature was reached, the mixture was cooled to 0 °C within 30 min and stored in a cold room overnight. The solids were filtered and washed with 60 mL toluene and dried under nitrogen sweep. Yield 50.25 g with 100 % diastereomeric excess HPLC method: Column YMC Pro C18 (250 x 4.6 mm)
Detector: 210 nm Mobile phase A Acetonitrile Mobile phase B 10 mM K2HPO4 pH 6.6 with H3PO4 Flow: 1.25 mL / min Run Time: 15 min
Gradient Ramp from 50 % A to 60% A over 6 mins. Hold for 9 min. Typical Retention Times: Undesired isomer: 6.3 min Desired isomer: 6.5 min PGA enamine 3-2: 8.1 min