WO2004085370A1 - Verfahren der heterogen katalysierten partiellen gasphasenoxidation von acrolein zu acrylsäure - Google Patents
Verfahren der heterogen katalysierten partiellen gasphasenoxidation von acrolein zu acrylsäure Download PDFInfo
- Publication number
- WO2004085370A1 WO2004085370A1 PCT/EP2004/002934 EP2004002934W WO2004085370A1 WO 2004085370 A1 WO2004085370 A1 WO 2004085370A1 EP 2004002934 W EP2004002934 W EP 2004002934W WO 2004085370 A1 WO2004085370 A1 WO 2004085370A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- zone
- fixed bed
- catalyst bed
- temperature
- bed catalyst
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/16—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation
- C07C51/21—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation with molecular oxygen
- C07C51/25—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation with molecular oxygen of unsaturated compounds containing no six-membered aromatic ring
- C07C51/252—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation with molecular oxygen of unsaturated compounds containing no six-membered aromatic ring of propene, butenes, acrolein or methacrolein
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/42—Separation; Purification; Stabilisation; Use of additives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C57/00—Unsaturated compounds having carboxyl groups bound to acyclic carbon atoms
- C07C57/02—Unsaturated compounds having carboxyl groups bound to acyclic carbon atoms with only carbon-to-carbon double bonds as unsaturation
- C07C57/03—Monocarboxylic acids
- C07C57/04—Acrylic acid; Methacrylic acid
Definitions
- the present invention relates to a process of heterogeneously catalyzed partial gas phase oxidation of acrolein to acrylic acid, in which an acrolein, molecular oxygen and at least one inert gas reaction mixture containing the molecular oxygen and acrolein in a molar ratio O 2 : C 3 H 4 O ⁇ 0.5 contains, in a reaction stage with the proviso via a fixed bed catalyst bed whose active composition is at least one multimetal oxide containing the elements Mo and V, that
- the fixed bed catalyst bed is arranged in two spatially successive temperature zones A, B,
- both the temperature of temperature zone A and the temperature of temperature zone B is a temperature in the range from 230 to 320 ° C.
- the fixed bed catalyst bed consists of at least two spatially consecutive fixed bed catalyst bed zones, the volume-specific activity within a fixed bed catalyst bed zone being essentially constant and increasing suddenly in the direction of flow of the reaction gas mixture when moving from a fixed bed catalyst bed zone into another fixed bed catalyst bed zone,
- temperature zone A extends to a conversion of acrolein of 45 to 85 mol%
- the acrolein conversion is> 90 mol% and the selectivity of the acrylic acid formation, based on converted acrolein, is> 90 mol%
- the loading of the fixed bed catalyst bed with the acrolein contained in the reaction gas starting mixture is> 70 Nl acrolein / l fixed bed catalyst bed • h
- the difference T axA - T maxB , formed from the highest temperature T maxA that the reaction gas mixture has within temperature zone A and the highest temperature ⁇ maxB that the reaction gas mixture has within temperature zone B is> 0 ° C.
- reaction gas starting mixture contains i.a. therefore inert gas in order to keep the reaction gas outside the explosion area.
- One objective of such a heterogeneously catalyzed partial gas phase oxidation of acrolein to acrylic acid is to achieve the highest possible selectivity S M of acrylic acid (that is the number of moles of acrolein converted to acrylic acid) when the reaction gas mixture passes through the reaction stage under otherwise specified boundary conditions. based on the number of moles of acrolein reacted).
- Another objective is to achieve the highest possible conversion U AC of acrolein when the reaction gas mixture passes through the reaction stage under otherwise specified boundary conditions (this is the mole number of converted acrolein, based on the mole number of acrolein used).
- Another objective of such a heterogeneously catalyzed partial gas phase oxidation of acrolein to acrylic acid is to achieve the highest possible space-time yield (STA) of acrylic acid (in the case of a continuous procedure this is the hour and volume of the fixed bed catalyst bed used in liters) total amount of acrylic acid produced).
- STA space-time yield
- EP-A 1106598 also teaches a process of heterogeneously catalyzed partial gas phase oxidation of acrolein to acrylic acid, in which the fixed bed catalyst bed of the reaction stage consists of several spatially successive fixed bed catalyst bed zones, the volume-specific activity of which is essentially constant within a fixed bed catalyst bed zone and in the direction of flow of the reaction gas mixture increases suddenly during the transition from a fixed bed catalyst bed zone into another fixed bed catalyst bed zone and can be arranged in several temperature zones.
- the individual effects in the transition area are often neutralized and the U AC is reduced.
- the object of the present invention was therefore to provide a process for heterogeneously catalyzed partial gas phase oxidation of acrolein to acrylic acid in a multi-zone arrangement which does not have the disadvantages of the prior art processes.
- a process of heterogeneously catalyzed partial gas phase oxidation of acrolein to acrylic acid in which an acrolein, molecular oxygen and at least one inert gas reaction mixture containing the molecular oxygen and the acrolein in a molar ratio O 2 : C 3 H 4 O> 0 , 5 contains, in a reaction stage with the proviso via a fixed bed catalyst bed whose active composition is at least one multimetal oxide containing the elements Mo and V, that
- the fixed bed catalyst bed is arranged in two spatially successive temperature zones A, B,
- both the temperature of temperature zone A and the temperature of temperature zone B is a temperature in the range from 230 to 320 ° C.
- the fixed bed catalyst bed consists of at least two spatially successive fixed bed catalyst bed zones, the volume-specific activity within a fixed bed catalyst bed zone being essentially constant and increasing abruptly in the direction of flow of the reaction gas mixture when transitioning from a fixed bed catalyst bed zone into another fixed bed catalyst bed zone,
- temperature zone A extends to a conversion of acrolein of 45 to 85 mol%
- the acrolein conversion is> 90 mol% and the selectivity of the acrylic acid formation, based on converted acrolein, is> 90 mol%
- the loading of the fixed bed catalyst bed with the acrolein contained in the reaction gas starting mixture is> 70 Nl acrolein / l fixed bed catalyst bed • h
- the difference T ma A - T maxB formed from the highest temperature T maxA which the reaction gas mixture has within the temperature zone A and the highest temperature T maxB which the reaction gas mixture has within the temperature zone B is> 0 ° C.
- transition from temperature zone A to temperature zone B in the fixed bed catalyst bed does not coincide with a transition from a fixed bed catalyst bed zone to another fixed bed catalyst bed zone.
- the temperature of a temperature zone is understood to mean the temperature of the part of the fixed bed catalyst bed located in the temperature zone when the process according to the invention is carried out, but in the absence of a chemical reaction. If this temperature is not constant within the temperature zone, the term temperature of a temperature zone here means the (number) average of the temperature of the fixed bed catalyst bed along the reaction zone. It is essential that the temperature of the individual temperature zones is essentially independent of one another. Since the heterogeneously catalyzed partial gas phase oxidation of acrolein to acrylic acid is a markedly exothermic reaction, the temperature of the reaction gas mixture during the reactive passage through the fixed bed catalyst bed is generally different from the temperature of a temperature zone. It is usually above the temperature of the temperature zone and usually passes through a maximum (hotspot maximum) within a temperature zone or decreases from a maximum value.
- T maxA -T maxB in the method according to the invention will not be more than 75 ° C.
- T axA -T maxB is preferably > 3 ° C and ⁇ 60 ° C.
- T maxA -T ma B is very particularly preferably ⁇ 5 ° C. and ⁇ 40 ° C.
- T axA - T maxB required according to the invention are usually found when the process according to the invention is carried out in the case of rather low (> 70 Nl / I • h and ⁇ 150 Nl / I • h) acrolein loads on the fixed bed catalyst bed when both Temperature of reaction zone A and the temperature of reaction zone B is in the range from 230 to 320 ° C.
- reaction zone B T B
- T A the difference between the temperature of reaction zone B (T B ) and the temperature of reaction zone A (T A ), ie, T B - T A , ⁇ 0 ° C and> -20 ° C or> -10 ° C, or ⁇ 0 ° C and> -5 ° C, or often ⁇ 0 ° C and> -3 ° C.
- both the temperature of reaction zone A and the temperature of reaction zone B are in the range from 230 to 320 ° C. and T B - T A > 0 ° C. and ⁇ 40 ° C., or> 5 ° C. and ⁇ 35 ° C. or .30 ° C, or> 10 ° C and ⁇ 25 ° C or 20 ° C, or 15 ° C.
- the acrolein contamination of the fixed bed catalyst bed can thus be, for example,> 70 Nl / I • h or 90 Nl / I • h and ⁇ 300 Nl / I • h, or> 110 Nl / I • h and ⁇ 280 Nl / I • h in the process according to the invention , or> 130 Nl / I • h and ⁇ 260 Nl / I • h, or> 150 NI / I • h and ⁇ 240 NI / I • h, or> 170 Nl / I • h and ⁇ 220 NI / I • h, or 190 NI / I • h and ⁇ 200 NI / I • h.
- temperature zone A preferably extends to a conversion of acrolein of 50 to 85 mol% or 60 to 85 mol%.
- the working pressure in the method according to the invention can be both below normal pressure (e.g. up to 0.5 bar) and above normal pressure. Typically, the working pressure will be 1 to 5 bar, often 1 to 3 bar. The reaction pressure will normally not exceed 100 bar.
- the molar ratio of O 2 : acrolein in the reaction gas starting mixture must be> 0.5. It will often be 1. This ratio will usually be at values ⁇ 3. According to the invention, the molar ratio of O 2 : acrolein in the reaction gas starting mixture is frequently 1 to 2 or 1 to 1.5.
- the acrolein conversion based on the single pass in the process according to the invention can be> 92 mol%, or> 94 mol%, or> 96 mol%, or> 98 mol% and often even> 99 mol%.
- the selectivity of the acrylic acid formation will regularly be> 92 mol% or> 94 mol%, often> 95 mol% or> 96 mol% or> 97 mol%.
- Suitable catalysts for the fixed bed catalyst bed of the gas phase catalytic acrolein oxidation according to the invention are all those whose active composition is at least one Mo and V containing multimetal oxide.
- Such suitable multi-metal oxide catalysts can be found, for example, in US Pat. No. 3,775,474, US Pat. No. 3,954,555, US Pat. No. 3,893,951, and US Pat. No. 4,339,355.
- the multimetal oxide materials of EP-A 427508, DE-A 2909671, DE-C 3151805, DE-AS 2626887, DE-A 4302991, EP-A 700893, EP-A 714700 and EP are particularly suitable DE-A 19736105 and DE-A 10046928.
- EP-A 714700 and DE-A 19736105 are also suitable.
- X 1 W, Nb, Ta, Cr and / or Ce
- X 2 Cu, Ni, Co, Fe, Mn and / or Zn
- X 3 Sb and / or Bi
- X 4 one or more alkali metals
- X 5 one or more alkaline earth metals
- X 6 Si, Al, Ti and / or Zr
- n a number , which is determined by the valency and frequency of elements other than oxygen in IV,
- X 1 W, Nb, and / or Cr
- X 2 Cu, Ni, Co, and / or Fe
- X 5 Ca, Sr and / or Ba
- X 6 Si, Al, and / or Ti
- n a number that is determined by the valency and frequency of elements other than oxygen in IV.
- multimetal oxides IV which are particularly preferred according to the invention are those of the general formula V.
- Y 1 W and / or Nb
- Y 2 Cu and / or Ni
- Y 5 Ca and / or Sr
- Y 6 Si and / or Al
- n' a number which is determined by the valency and frequency of the Different oxygen elements in V is determined.
- multimetal oxide active compositions (IV) which are suitable according to the invention are known per se, e.g. available in DE-A 4335973 or in EP-A 714700.
- multimetal oxide active compositions suitable for the catalysts of the fixed bed catalyst bed can be prepared in a simple manner by producing an intimate, preferably finely divided, dry mixture composed of suitable elementary constituents according to their stoichiometry, and this calcined at temperatures from 350 to 600 ° C.
- the caicination can be carried out both under inert gas and under an oxidative atmosphere such as air (mixture of inert gas and oxygen) and under a reducing atmosphere (eg mixtures of inert gas and reducing gases such as H 2 , NH 3 , CO, methane and / or acrolein or the reducing gases mentioned are carried out by themselves).
- the calcination time can range from a few minutes to a few hours and usually decreases with temperature.
- Suitable sources for the elementary constituents of the multimetal oxide active compositions IV are those compounds which are already oxides and / or those compounds which can be converted into oxides by heating, at least in the presence of oxygen.
- the intimate mixing of the starting compounds for the production of multimetal oxide compositions IV can be carried out in dry or in wet form. If it is carried out in dry form, the starting compounds are expediently used as finely divided powders and, after mixing and optionally compacting, are subjected to the calcination. However, the intimate mixing is preferably carried out in wet form.
- the starting compounds are mixed together in the form of an aqueous solution and / or suspension.
- Particularly intimate dry mixtures are obtained in the mixing process described if only sources of the elementary constituents present in dissolved form are used. Water is preferably used as the solvent.
- the aqueous mass obtained is then dried, the drying process preferably being carried out by spray drying the aqueous mixture at exit temperatures of 100 to 150 ° C.
- the resulting multimetal oxide compositions are generally used in the fixed bed catalyst bed, not in powder form but shaped into specific catalyst geometries, it being possible for the shaping to take place before or after the final caicination.
- full catalysts can be produced from the powder form of the active composition or its uncalcined precursor composition by compression to the desired catalyst geometry (e.g. by tableting, extrusion or extrusion), where appropriate auxiliaries such as e.g. Graphite or stearic acid can be added as a lubricant and / or molding aid and reinforcing agent such as microfibers made of glass, asbestos, silicon carbide or potassium titanate.
- Suitable unsupported catalyst geometries are e.g.
- Solid cylinder or hollow cylinder with an outer diameter and a length of 2 to 10 mm.
- a wall thickness of 1 to 3 mm is appropriate.
- the full catalyst can also have a spherical geometry, the spherical diameter being 2 to 10 mm.
- the powdery active composition or its powdery, not yet calcined, precursor composition can also be shaped by application to preformed inert catalyst supports.
- the coating of the support bodies for the production of the coated catalysts is usually carried out in a suitable rotatable container, as is e.g. is known from DE-A 2909671, EP-A 293859 or from EP-A 714700.
- the powder mass to be applied is expediently moistened and dried again after application, for example by means of hot air.
- the layer thickness of the powder mass applied to the carrier body becomes expediently selected in the range from 10 to 1000 ⁇ m, preferably in the range from 50 to 500 ⁇ m and particularly preferably in the range from 150 to 250 ⁇ m.
- carrier materials Conventional porous or non-porous aluminum oxides, silicon dioxide, thorium dioxide, zirconium dioxide, silicon carbide or silicates such as magnesium or aluminum silicate can be used as carrier materials.
- the carrier bodies can have a regular or irregular shape, with regularly shaped carrier bodies with a clearly formed surface roughness, e.g. Balls or hollow cylinders with a split layer are preferred. It is suitable to use essentially non-porous, rough-surface, spherical supports made of steatite (e.g. steatite C220 from CeramTec), the diameter of which is 1 to 8 mm, preferably 4 to 5 mm.
- steatite e.g. steatite C220 from CeramTec
- cylinders as carrier bodies, the length of which is 2 to 10 mm and the outside diameter is 4 to 10 mm.
- the wall thickness is also usually 1 to 4 mm.
- Annular support bodies to be used preferably have a length of 2 to 6 mm, an outer diameter of 4 to 8 mm and a wall thickness of 1 to 2 mm. Rings of geometry 7 mm x 3 mm x 4 mm (outer diameter x length x inner diameter) are particularly suitable as carrier bodies.
- the fineness of the catalytically active oxide compositions to be applied to the surface of the carrier body is of course adapted to the desired shell thickness (cf. EP-A 714 700).
- compositions to be used for the catalysts of the fixed bed catalyst bed are also compositions of the general formula VI,
- Z 1 W, Nb, Ta, Cr and / or Ce
- z 2 Cu, Ni, Co, Fe, Mn and / or Zn
- z 3 Sb and / or Bi
- z 4 Li, Na, K, Rb, Cs and / or H
- z 5 Mg, Ca, Sr and / or Ba
- z 6 Si, Al, Ti and / or Zr
- z 7 Mo, W, V, Nb and / or Ta, preferably Mo and / or W
- starting mass 1 separately in finely divided form (starting mass 1) and then the preformed solid starting mass 1 in an aqueous solution, an aqueous suspension or in a finely divided dry mixture of sources of the elements Mo, V, Z ⁇ Z 2 , Z 3 , Z 4 , Z 5 , Z 6 , which the aforementioned elements in the Stoichiomethe D
- multimetal oxide compositions VI in which the preformed solid starting composition 1 is incorporated into an aqueous starting composition 2 at a temperature of ⁇ 70 ° C.
- a detailed description of the production of multimetal oxide materials VI catalysts contain e.g. EP-A 668104, DE-A 19736105, DE-A 10046928, DE-A 19740493 and DE-A 19528646.
- suitable multimetal oxide compositions suitable for the catalysts of the fixed bed catalyst bed are those of DE-A 19815281, in particular all exemplary embodiments from this document. With regard to the shape, what has been said above applies.
- Catalysts which are particularly suitable for the fixed bed catalyst bed of the process according to the invention are shell catalysts S1 (stoichiometry: Mo 12 V 3 W 1 ⁇ 2 Cu 2 ⁇ 4 O n ) and S7 (stoichiometry: Mo 12 V 3 W 1 ⁇ 2 Cu 1 ⁇ 6 Ni 0
- the catalysts recommended above for the reaction stage according to the invention are also suitable for the reaction stage according to the invention if everything is retained and only the carrier geometry is changed to 5 mm ⁇ 3 mm ⁇ 1.5 mm (outer diameter ⁇ length ⁇ inner diameter).
- the multimetal oxides mentioned can also be used in the form of the corresponding unsupported catalyst rings in the reaction stage according to the invention.
- the fixed bed catalyst bed consists of at least two spatially consecutive fixed bed catalyst bed zones, the volume-specific activity within a fixed bed catalyst bed zone being essentially constant and increasing suddenly in the direction of flow of the reaction gas mixture during the transition from a fixed bed catalyst bed zone into another fixed bed catalyst bed zone.
- the volume-specific (ie, the normalized to the unit of the respective bed volume) activity of a fixed bed catalyst bed zone can now be adjusted in a substantially constant manner via the fixed bed catalyst bed zone by starting from a basic quantity of shaped catalyst bodies produced uniformly (their bed corresponds to the maximum achievable volume-specific Activity) and this in the respective fixed bed catalyst bed zone is homogeneously diluted with shaped bodies (diluent shaped bodies) which are essentially inert with respect to the heterogeneously catalyzed partial gas phase oxidation.
- shaped bodies shaped bodies
- all those materials which are also suitable as support material for coated catalysts suitable according to the invention are suitable as materials for such inert shaped diluent bodies.
- Such materials come e.g. porous or non-porous aluminum oxides, silicon dioxide, thorium dioxide, zirconium dioxide, silicon carbide, silicates such as magnesium or aluminum silicate or the already mentioned steatite (e.g. steatite C-220 from CeramTec).
- the geometry of such inert shaped dilution bodies can be as desired. That means, for example, spheres, polygons, solid cylinders or rings. According to the invention, preference will be given to choosing the inert shaped diluent whose geometry corresponds to that of the shaped catalyst body to be diluted with them.
- the chemical composition of the active composition used does not change over the entire fixed bed catalyst bed.
- the active composition used for a single shaped catalyst body can be a mixture of different multimetal oxides containing the elements Mo and V, but the same mixture must then be used for all shaped catalyst bodies in the fixed bed catalyst bed.
- a volume-specific activity which increases in zones in the direction of flow of the reaction gas mixture over the fixed bed catalyst bed can thus be achieved in a simple manner for the process according to the invention, e.g. adjust by starting the bed in a first fixed bed catalyst bed zone with a high proportion of inert diluent shaped bodies based on a type of shaped catalyst bodies, and then reducing this proportion of diluent shaped bodies in zones in the flow direction.
- a zone-by-zone increase in the volume-specific activity is also possible, for example, by increasing the thickness of the layer of active mass applied to the support with the same geometry and type of active mass of a shaped shell catalyst, or zone-wise in a mixture of shell catalysts with the same geometry but with a different proportion by weight of the active mass
- the proportion of shaped catalyst bodies with a higher proportion of active mass increases.
- one can also dilute the active materials themselves for example by incorporating inert, diluting materials such as high-grade silicon dioxide into the dry mixture to be calcined from starting compounds. Different additions of thinner Material automatically leads to different activities. The more diluting material is added, the lower the resulting activity will be.
- An analogous effect can also be achieved, for example, by changing the mixing ratio in a corresponding manner in the case of mixtures of unsupported catalysts and of shell catalysts (with identical active composition). Furthermore, a variation of the volume-specific activity can be achieved through the use of catalyst geometries with different bulk densities (for example with full catalysts with identical active mass composition of the different geometries). Of course, the variants described can also be used in combination.
- mixtures of catalysts with chemically different active composition and, as a result of this different composition, different activity can also be used for the fixed bed catalyst bed.
- the composition of these mixtures can in turn be varied zone by zone and / or diluted with different amounts of inert diluent tablets.
- the fixed bed catalyst bed Before and / or after the fixed bed catalyst bed, there can only be beds consisting of inert material (e.g. only dilution tablets) (in this document they are not conceptually included in the fixed bed catalyst bed because they do not contain any tablets that have a multimetal oxide active composition).
- the shaped dilution bodies used for the inert bed can have the same geometry as the shaped catalyst bodies used in the fixed bed catalyst bed.
- the geometry of the shaped diluent bodies used for the inert bed can also differ from the aforementioned geometry of the shaped catalyst bodies (for example spherical instead of annular).
- the temperature zones A and B can also extend to the inert beds in the method according to the invention. According to the invention, both the temperature zone A and the temperature zone B advantageously do not record more than three fixed-bed catalyst bed zones (according to the invention, at least one fixed-bed catalyst bed zone is necessarily detected by both temperature zones).
- the entire fixed bed catalyst bed particularly advantageously comprises no more than five, suitably no more than four or three, fixed bed catalyst bed zones.
- the volume-specific active mass ie the weight of the multimetal oxide mass contained in the unit bed volume
- % preferably increase by at least 10% by weight (this also applies in particular to uniform shaped catalyst bodies over the entire fixed bed catalyst bed).
- this increase in the method according to the invention will not amount to more than 50% by weight, usually not more than 40% by weight.
- the difference in the volume-specific active mass of that fixed bed catalyst bed zone with the lowest volume-specific activity and that fixed bed catalyst bed zone with the highest volume-specific activity should advantageously not exceed 50% by weight, preferably not more than 40% by weight. -% and particularly preferably not more than 30 wt .-%.
- the fixed bed catalyst bed will often consist of only two fixed bed catalyst bed zones.
- the last fixed bed catalyst bed zone in the flow direction of the reaction mixture is undiluted. That is, it preferably consists exclusively of shaped catalyst bodies. If necessary, it can also consist of a bed of shaped catalyst bodies whose volume-specific activity e.g. lowered by dilution with inert material, e.g. by 10%. If the fixed bed catalyst bed consists of only two fixed bed catalyst bed zones, it is generally advantageous according to the invention (as is very general in the process according to the invention) if the fixed bed catalyst bed zone with the highest volume-specific activity extends into temperature zone A (especially if in temperature zone A and in Temperature zone B, the temperature is controlled by means of a flowing heat transfer medium which flows in countercurrent to the reaction gas mixture).
- the fixed bed catalyst bed consists of only three fixed bed catalyst bed zones, it is generally also advantageous according to the invention if the fixed bed catalyst bed zone with the highest volume-specific activity extends into the
- Temperature zone A protrudes (in particular if temperature control in temperature zone A and temperature zone B takes place by means of a flowing heat transfer medium which flows in countercurrent to the reaction gas mixture).
- the fixed bed catalyst bed consists of four fixed bed catalyst bed zones, it is generally advantageous according to the invention if the fixed bed catalyst bed zone with the second highest volume-specific activity protrudes both into temperature zone A and into temperature zone B (in particular when in temperature zone A and in the temperature zone B the temperature is controlled by means of a flowing heat transfer medium which flows in countercurrent to the reaction gas mixture).
- the volume-specific activity between two fixed bed catalyst bed zones can be differentiated experimentally in such a simple manner that under identical boundary conditions (preferably the conditions of the process envisaged) over fixed bed catalyst beds of the same length, but in each case corresponding to the composition of the respective fixed bed catalyst bed zone, and containing the same acrolein Reaction gas starting mixture is performed.
- the higher amount of acrolein converted shows the higher volume-specific activity.
- the fixed bed catalyst bed in the flow direction of the reaction gas mixture is preferably structured as follows in the process according to the invention.
- first or these two first zones is then advantageously up to the end of the length of the fixed bed catalyst bed (ie, for example over a length of 2.00 to 3.00 m, preferably 2.50 to 3.00 m ) either a bed of the shaped catalyst bodies which is only diluted to a lesser extent (than in the first or in the first two zones), or, very particularly preferably, a bed of the same shaped catalyst bodies which were also used in the first zone.
- both the shaped catalyst bodies or their carrier rings and the shaped dilution bodies in the process according to the invention essentially have the ring geometry 7 mm ⁇ 3 mm ⁇ 4 mm (outer diameter ⁇ length ⁇ inner diameter).
- shell catalyst moldings are used instead of inert shaped diluent bodies whose active mass fraction is 2 to 15% by weight points lower than the active mass fraction of the shell catalyst shaped bodies at the end of the fixed bed catalyst bed.
- a pure inert material bed the length of which, based on the length of the fixed bed catalyst bed, is expediently 5 to 20%, generally introduces the fixed bed catalyst bed in the direction of flow of the reaction gas mixture. It is normally used to temper the reaction gas mixture.
- the temperature zone A (which according to the invention advantageously also extends to the pre-filling of the inert material) now extends to 5 to 20%, often to 5 to 15% of the length of the last (most active in terms of volume) in the direction of flow of the reaction gas mixture. fixed catalyst bed zone.
- the reaction stage of the process according to the invention is carried out in a manner which is expedient in terms of application technology, in a two-zone tube bundle reactor, as described, for example, in DE-A's 19910508, 19948523, 19910506 and 19948241 is.
- a preferred variant of a two-zone tube bundle reactor which can be used according to the invention is disclosed in DE-C 2830765. But also those disclosed in DE-C 2513405, US-A 3147084, DE-A 2201528, EP-A 383224 and DE-A 2903218 Two-zone tube bundle reactors are suitable for carrying out the reaction stage of the process according to the invention.
- the fixed bed catalyst bed to be used according to the invention (possibly with upstream and / or downstream inert beds) is in the simplest manner in the metal tubes of a tube bundle reactor and two temperature-regulating media which are essentially spatially separated from one another, usually molten salts, are guided around the metal tubes.
- the pipe section over which the respective salt bath extends represents a temperature zone.
- a salt bath A means those sections of the tubes (the temperature zone A) in which the oxidative conversion of acrolein (in a single pass) is particularly high until a conversion value in the range from 45 to 85 mol% (preferably 50 to 85 mol%) is reached preferably 60 to 85 mol%) and a salt bath B flows around the section of the tubes (temperature zone B) in which the oxidative subsequent conversion of acrolein (in a single pass) until a conversion value of at least 90 mol% is reached carried out (if necessary, further temperature zones can be connected to the temperature zones A, B to be used according to the invention, which are kept at individual temperatures).
- the reaction stage of the process according to the invention does not comprise any further temperature zones. That is, the salt bath B expediently flows around the section of the tubes in which the oxidative subsequent conversion of acrolein (in a single pass) up to a conversion value of> 92 mol%, or> 94 mol% or> 96 mol% or> 98 mol% and often even> 99 mol% or more.
- the start of temperature zone B is usually behind the hotspot maximum of temperature zone A.
- the two salt baths A, B can be conducted in cocurrent or countercurrent through the space surrounding the reaction tubes relative to the flow direction of the reaction gas mixture flowing through the reaction tubes.
- a parallel flow can also be used in temperature zone A and a counterflow in temperature zone B (or vice versa).
- a transverse flow can be superimposed on the parallel flow of the molten salt within the respective temperature zone relative to the reaction tubes, so that the individual temperature zone corresponds to a tube bundle reactor as described in EP-A 700714 or EP-A 700893 and overall a longitudinal section through the contact tube bundle results in a meandering flow of the heat exchange medium.
- the contact tubes are usually made of ferritic steel and typically have a wall thickness of 1 to 3 mm. Their inner diameter is usually 20 to 30 mm, often 21 to 26 mm. Their length is expediently 3 to 4, preferably 3.5 m.
- the fixed bed catalyst bed occupies at least 60%, or at least 75%, or at least 90% of the length of the zone. The remaining length, if any, may be occupied by an inert bed.
- the number of contact tubes accommodated in the tube bundle container is at least 5000, preferably at least 10,000.
- the number of contact tubes accommodated in the reaction vessel is frequently 15,000 to 30,000.
- Tube bundle reactors with a number of contact tubes above 40,000 are rather the exception.
- the contact tubes are normally arranged homogeneously distributed within the container (preferably 6 equidistant neighboring tubes per contact tube), the distribution being expediently chosen so that the distance between the central inner axes of the closest contact tubes (the so-called contact tube division) is 35 to 45 mm ( see EP-B 468290).
- Fluid heat transfer media are particularly suitable as heat exchange medium.
- melts of salts such as potassium nitrate, potassium nitrite, sodium nitrite and / or sodium nitrate, or of low-melting metals such as sodium, mercury and alloys of various metals is particularly favorable.
- the flow rate within the two required heat exchange medium circuits is selected such that the temperature of the heat exchange medium from the entry point into the temperature zone to the exit point from the reaction zone is around 0 to 15 ° C increases. That is, the aforementioned ⁇ T can be 1 to 10 ° C, or 2 to 8 ° C or 3 to 6 ° C according to the invention.
- the entry temperature of the heat exchange medium into the temperature zone A is normally in the range from 230 to 320 ° C., preferably in the range from 250 to 300 ° C. and particularly preferably in the range from 260 to 280 ° C.
- the entry temperature of the heat exchange medium into reaction zone B is normally also in the range from 230 to 320 ° C. or 280 ° C., but at the same time normally according to the invention expediently 0 ° C to ⁇ 20 ° C or ⁇ 10 ° C, or> 0 ° C and ⁇ 5 ° C, or often> 0 ° C and ⁇ 3 ° C below the inlet temperature of the heat exchange medium entering reaction zone A.
- the entry temperature of the heat exchange medium into temperature zone B is also in the range of 230 up to 320 ° C, but at the same time normally expediently according to the invention> 0 ° C and ⁇ 40 ° C, or> 5 ° C and ⁇ 35 ° C or ⁇ 30 ° C, or> 10 ° C and ⁇ 25 ° C or ⁇ 20 ° C, or 15 ° C above the inlet temperature of the heat exchange medium entering temperature zone A.
- the two-zone tube bundle reactor type described in DE-AS 2201528 which includes the possibility of the hotter heat exchange medium of the reaction zone, can be used in particular to carry out the reaction stage of the process according to the invention B to discharge a partial amount to the reaction zone A in order to possibly heat up a reaction gas starting mixture which is too cold or a cold circulating gas.
- the tube bundle characteristic can be designed within an individual temperature zone as described in EP-A 382098.
- Acrolein which was generated by catalytic gas phase oxidation of propene, is normally used in the process according to the invention.
- the acrolein-containing reaction gases of this propene oxidation are used without intermediate purification, which is why the reaction gas starting mixture according to the invention also contains small amounts of e.g. unconverted propene or by-products of propene oxidation.
- the oxygen required for acrolein oxidation must also be added to the product gas mixture from the propene oxidation.
- two two-zone tube bundle reactors can be connected in series or fused into a four-zone tube bundle reactor, as described, for example, in WO 01/36364, propene oxidation being carried out in the first two-zone part and acrolein oxidation in the second two-zone part ,
- the propene oxidation stage can also be a separate single-zone tube bundle reactor or one which is fused in a corresponding manner with the subsequent acrolein oxidation stage.
- the length of the reaction tubes often corresponds to the sum of the lengths of the unmelted tube bundle reactors.
- the proportion of propene in the reaction gas starting mixture for the reaction stage preceding the process according to the invention can be e.g. with values of 4 to 15% by volume, often 5 to 12% by volume or 5 to 8% by volume (in each case based on the total volume).
- the process preceding the process according to the invention will be in the case of a propene: oxygen: indifferent gases (including water vapor) volume ratio in the reaction gas starting mixture of 1: (1.0 to 3.0): (5 to 25), preferably 1: (1 , 4 to 2.3): Carry out (10 to 15).
- the indifferent (inert) gas will consist of at least 20% of its volume of molecular nitrogen.
- inert diluent gases should be those that pass through the reaction step less than less than 5%, preferably less than 2%; in addition to molar nitrogen, these include, for example, gases such as propane, ethane, methane, pentane, butane, CO 2 , CO, water vapor and / or noble gases).
- the inert diluent gas in the process preceding the process according to the invention can also consist of up to 50 mol% or up to 75 mol% and more of propane. Circulating gas can also be part of the diluent gas, as it remains after the acrylic acid has been separated off from the product gas mixture of the process according to the invention.
- Suitable catalysts for such a gas phase catalytic propene oxidation are in particular those of EP-A 15565, EP-A 575897, DE-A 19746210 and DE -A 19855913.
- the inert gas to be used in the process according to the invention can be 20% by volume, or> 30% by volume, or> 40% by volume, or> 50% by volume, or 60% by volume, or consist of> 70 vol .-%, or 80 vol .-%, or 90 vol .-%, or> 95 vol .-% of molecular nitrogen.
- the inert diluent gas in the process according to the invention will expediently consist of 5 to 20% by weight of H 2 O and 70 to 90% by volume of N 2 .
- inert diluent gases should generally be those which convert to less than 5%, preferably less than 2%, in one pass
- propane, Ethane, methane, butane, pentane, CO 2 , CO, water vapor and / or noble gases are recommended.
- these gases can also be used at lower loads. It is also possible to use an inert gas consisting only of one or more of the aforementioned gases.
- the acrolein load in the process according to the invention will not exceed 600 NI / l • h.
- the acrolein content in the reaction gas starting mixture for the process according to the invention can e.g. with values of 3 to 15% by volume, often 4 to 10% by volume or 5 to 8% by volume (in each case based on the total volume).
- the process according to the invention is frequently used with an acrolein: oxygen: water vapor: inert gas - volume ratio (NI) present in the reaction gas starting mixture of 1: (1 to 3): (0 to 20): (3 to 30), preferably 1: (1 to 3): (0.5 to 10): execute (7 to 10).
- acrolein oxygen: water vapor: inert gas - volume ratio (NI) present in the reaction gas starting mixture of 1: (1 to 3): (0 to 20): (3 to 30), preferably 1: (1 to 3): (0.5 to 10): execute (7 to 10).
- the process according to the invention can also be carried out using an acrolein present in the reaction gas starting mixture: oxygen: water vapor: Other - Perform volume ratio (NI) of 1: (0.9 to 1.3): (2.5 to 3.5): (10 to 12).
- multimetal oxide compositions of DE-A 10261186 are also favorable as active compositions for the fixed bed catalyst bed of the process according to the invention.
- Inexpensive embodiments of a two-zone tube bundle reactor according to the invention for a propene partial oxidation stage preceding the reaction stage according to the invention can be as follows (the constructive detailed design can be as in utility model applications 202 19 277.6, 2002 19 278.4 and 202 19 279.2 or in PCT applications PCT / EP02 / 14187, PCT / EP02 / 14188 or PCT / EP02 / 14189):
- Contact tube material ferritic steel
- Dimensions of the contact tubes e.g. 3500 mm length; e.g. 30 mm outer diameter; e.g. 2 mm wall thickness;
- Number of contact tubes in the tube bundle e.g. 30,000, or 28,000, or 32,000, or 34,000; additionally up to 10 thermotubes (as described in EP-A 873 783 and EP-A 12 70 065), which are loaded like the contact tubes (rotating like a screw from the outside to the inside) e.g. the same length and wall thickness, but with an outer diameter of e.g. 33.4 mm and a centered thermal sleeve of e.g. 8 mm outer diameter and e.g. 1 mm wall thickness;
- Cylindrical container with an inner diameter of 6000 - 8000 mm;
- Reactor covers clad with type 1 stainless steel, 4541; Plating thickness: a few mm;
- ring-shaped tube bundle e.g. with a free central space
- Diameter of the central free space eg 1000 - 2500 mm (eg 1200 mm, or 1400 mm, or 1600 mm, or 1800 mm, or 2000 mm, or 2200 mm, or 2400 mm);
- Normally homogeneous contact tube distribution in the tube bundle (6 equidistant neighboring tubes per contact tube), arrangement in an equilateral triangle, contact tube division (distance of the central inner axes from the closest contact tubes): 35 - 45 mm, e.g. 36 mm, or 38 mm, or 40 mm, or 42 mm , or 44 mm;
- the contact tubes are sealed with their ends in contact tube plates (upper plate and lower plate e.g. with a thickness of 100-200 mm) and open at the upper end into a hood connected to the container, which has an approval for the reaction gas starting mixture;
- a e.g. Partition plate on the half contact tube length of 20 - 100 mm thick divides the reactor space symmetrically into two temperature zones A '(upper zone) and B' (lower zone); each temperature zone is divided into 2 equidistant longitudinal sections by a deflection disk;
- the deflection disk preferably has a ring geometry; the contact tubes are advantageously fastened to the separating plate in a sealing manner; they are not attached in a sealing manner to the deflection disks, so that the cross-flow velocity of the molten salt is as constant as possible within a zone;
- each zone is supplied by its own salt pump with molten salt as a heat transfer medium; the supply of molten salt is e.g. below the deflection disc and the removal is e.g. above the pulley;
- the cooled salt melt stream is divided, combined with the respective residual stream and pressed into the reactor by the respective pump into the corresponding ring channel which distributes the salt melt over the circumference of the container;
- the salt melt reaches the tube bundle through windows in the reactor jacket; the inflow is e.g. in the radial direction to the tube bundle;
- the salt melt flows in each zone according to the specification of the baffle, e.g. in the sequence
- the molten salt collects at each end of the zone in an annular channel arranged around the reactor jacket in order to be pumped in a circuit, including partial flow cooling;
- the molten salt is led from bottom to top over each temperature zone.
- the reaction gas mixture leaves the reactor upstream of the reaction stage according to the invention at a temperature a few degrees higher than the salt bath inlet temperature of this reactor.
- the reaction gas mixture is expediently cooled to 220 ° C. to 280 ° C., preferably 240 ° C. to 260 ° C., in a separate aftercooler, which is connected downstream of this reactor.
- the aftercooler is usually flanged below the lower tube plate and usually consists of tubes with ferritic steel.
- Stainless steel sheet spirals which can be partially or fully coiled, are advantageously inserted into the tubes of the aftercooler to improve the heat transfer.
- a mixture of 53% by weight of potassium nitrate, 40% by weight of sodium nitrite and 7% by weight of sodium nitrate can be used as the molten salt; both reaction zones and the aftercooler advantageously use a molten salt of the same composition; the amount of salt pumped around in the reaction zones can be approximately 10000 m 3 / h per zone.
- reaction gas starting mixture for the propene oxidation expediently flows through the reactor from top to bottom, while the differently tempered salt melts of the individual zones are expediently conveyed from bottom to top;
- Section 1 50 cm in length
- Section 3 160 cm in length
- Catalyst feed with annular (5 mm x 3 mm x 2 mm outer diameter x length x inner diameter) full catalyst according to Example 1 of DE-A 10046957 (stoichiometry: [Bi 2 W 2 O 9 x 2 WO 3 ] 0.5 [M ⁇ i 2 C ⁇ 5 t5 Fe 2 , 9 Si ⁇ , 59 Ko, o 8 ⁇ ] ⁇ ).
- the thickness of the upper and lower contact tube bottoms is often 100-200 mm, e.g. 110 mm, or 130 mm, or 150 mm, or 170 mm, or 190 mm.
- the aftercooler is omitted; instead, the contact tubes open with their lower openings into a hood connected to the container at the lower end with an outlet for the product gas mixture; the upper temperature zone is zone A and the lower temperature zone is temperature zone B.
- the outlet “aftercooler” and inlet “reactor for the reaction stage according to the invention” there is expediently a possibility of supplying compressed air.
- the contact tube and thermal tube loading (from top to bottom) can e.g. be as follows:
- Section 1 20 cm long
- Section 2 90 cm length of catalyst feed with a homogeneous mixture of 30% by weight of steatite rings of geometry 7 mm x 3 mm x 4 mm (outside diameter x length x inside diameter) and 70% by weight coated catalyst from section 4.
- Section 3 50 cm long
- Section 4 190 cm length
- the propene step contact tube and thermotube loading can also look like this:
- Section 1 50 cm in length
- Section 2 300 cm in length
- the acrol-stage contact tube and thermotube feeding can also look like this (from top to bottom):
- Section 1 20 cm length steatite rings of geometry 7 mm x 7 mm x 4 mm (outer diameter x
- Section 2 140 cm in length
- Catalyst feed with ring-shaped (7 mm x 3 mm x 4 mm outer diameter x length x inner diameter) coated catalyst according to production example 5 of DE-A 10046928 (stoichiometry: Mo 12 V 3 W 1 ⁇ 2 Cu 2 , 4 O x ).
- the unsupported catalyst from Example 1 of DE-A 10046957 can also be replaced by:
- Example 1c a catalyst according to Example 1c from EP-A 15565 or a catalyst to be produced according to this example, but which has the active composition Mo ⁇ Nie.sZnaFe ⁇ iiPo.ooes o.oeOx • 10 SiO 2 ;
- steatite from CeramTec is preferred as the carrier material (type: C220).
- the coated catalyst according to preparation example 5 of DE-A 10046928 can be replaced by:
- the fixed bed catalyst bed for the propene oxidation stage and the fixed bed catalyst bed for the acrolein oxidation stage according to the invention are expediently chosen (for example by dilution with, for example, inert material) such that the temperature difference between the hotspot maximum of the reaction gas mixture in the individual reaction zones and the respective temperature of the reaction zone generally 80 ° C does not exceed.
- this temperature difference is ⁇ 70 ° C, often it is 20 to 70 ° C, preferably this temperature difference is small.
- these fixed bed catalyst beds are selected in a manner known per se to the person skilled in the art (for example by dilution with, for example, inert material) such that the "peak-to-salt-temperature sensitivity" as defined in EP-A 1106598 ⁇ 9 ° C. , or ⁇ 7 ° C, or ⁇ 5 ° C, or ⁇ 3 ° C.
- Aftercooler and reactor for the acrolein reaction stage are connected by a connecting pipe, the length of which is less than 25 m.
- the annular shaped diluent bodies and the annular shaped catalyst bodies can also be formed by spherical shaped diluent bodies and spherical shaped catalyst bodies (each with a radius of 2 to 5 mm and with an active mass fraction of 10 to 30% by weight, often 10 up to 20% by weight).
- a reaction tube (V2A steel; 30 mm outside diameter, 2 mm wall thickness, 26 mm inside diameter, length: 350 cm) as well as a thermotube centered in the middle of the reaction tube (4 mm outside diameter) for receiving a thermocouple with which the temperature in the reaction tube is determined over its entire length is loaded from top to bottom as follows:
- Section 1 20 cm long
- Section 3 50 cm long
- Section 4 190 cm length
- Catalyst feed with ring-shaped (7 mm x 3 mm x 4 mm outer diameter x length x inner diameter) coated catalyst according to production example 5 of DE-A 10046928 (stoichiometry: Mo 12 V 3 W 1 2 Cu 2 , O x ).
- the first 175 cm are thermostatted from top to bottom by means of a salt bath A pumped in countercurrent.
- the second 175 cm are thermostatted by means of a salt bath B pumped in countercurrent.
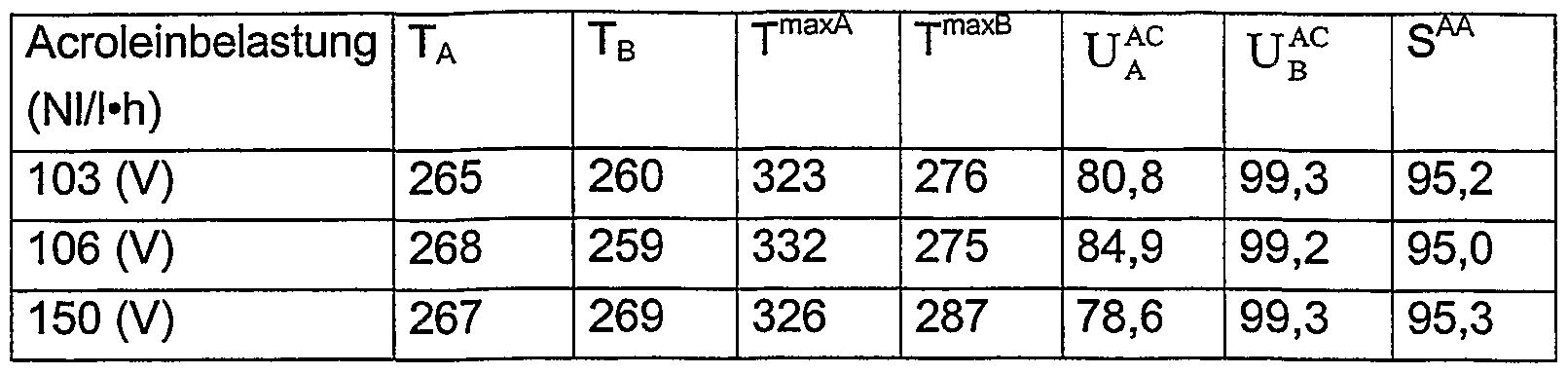
- reaction tube described above is continuously charged with a reaction gas starting mixture of the following composition, the load and the thermostatting of the reaction tube being varied:
- the reaction gas mixture flows through the reaction tube from top to bottom.
- the pressure at the inlet of the reaction tube varies between 1.6 and 2.1 bar depending on the acrolein load.
- a small sample is taken from the product gas mixture at the reaction tube outlet for gas chromatographic analysis.
- An analysis point is also located at the end of reaction zone A.
- T A , T B stand for the temperatures of the pumped salt baths in reaction zones A and B.
- U AC is the acrolein conversion at the end of reaction zone A in mol%.
- U AC is the acrolein conversion at the end of reaction zone B in mol%.
- -AA is the selectivity of acrylic acid formation in the product gas mixture based on converted acrolein in mol%.
- Section 3 65 cm instead of 50 cm.
- Section 4 175 cm instead of 190 cm.
Abstract
Description

Claims
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP04722167A EP1611079B1 (de) | 2003-03-25 | 2004-03-20 | Verfahren der heterogen katalysierten partiellen gasphasenoxidation von acrolein zu acryls ure |
| BRPI0408642-2B1A BRPI0408642B1 (pt) | 2003-03-25 | 2004-03-20 | processo para oxidar parcialmente acroleÍna a Ácido em fase gasosa. |
| JP2006504767A JP4537387B2 (ja) | 2003-03-25 | 2004-03-20 | アクロレインからアクリル酸への不均一系接触部分気相酸化法 |
| DE502004003884T DE502004003884D1 (de) | 2003-03-25 | 2004-03-20 | Verfahren der heterogen katalysierten partiellen gasphasenoxidation von acrolein zu acryls ure |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE10313214A DE10313214A1 (de) | 2003-03-25 | 2003-03-25 | Verfahren der heterogen katalysierten partiellen Gasphasenoxidation von Acrolein zu Acrylsäure |
| DE10313214.7 | 2003-03-25 | ||
| US47579203P | 2003-06-05 | 2003-06-05 | |
| US60/475,792 | 2003-06-05 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004085370A1 true WO2004085370A1 (de) | 2004-10-07 |
Family
ID=33099283
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2004/002934 WO2004085370A1 (de) | 2003-03-25 | 2004-03-20 | Verfahren der heterogen katalysierten partiellen gasphasenoxidation von acrolein zu acrylsäure |
Country Status (8)
| Country | Link |
|---|---|
| EP (1) | EP1611079B1 (de) |
| JP (1) | JP4537387B2 (de) |
| KR (1) | KR101006585B1 (de) |
| AT (1) | ATE362909T1 (de) |
| BR (1) | BRPI0408642B1 (de) |
| DE (1) | DE502004003884D1 (de) |
| ES (1) | ES2286626T3 (de) |
| WO (1) | WO2004085370A1 (de) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7884238B2 (en) | 2006-01-18 | 2011-02-08 | Basf Aktiengesellschaft | Process for the long-term operation of a heterogeneously catalyzed partial gas phase oxidation of an organic starting compound |
| DE102010048405A1 (de) | 2010-10-15 | 2011-05-19 | Basf Se | Verfahren zum Langzeitbetrieb einer heterogen katalysierten partiellen Gasphasenoxidation von Proben zu Acrolein |
| DE102013202048A1 (de) | 2013-02-07 | 2013-04-18 | Basf Se | Verfahren zur Herstellung einer katalytisch aktiven Masse, die ein Gemisch aus einem die Elemente Mo und V enthaltenden Multielementoxid und wenigstens einem Oxid des Molybdäns ist |
| US8431743B2 (en) | 2004-07-01 | 2013-04-30 | Basf Aktiengesellschaft | Preparation of acrylic acid by heterogeneously catalyzed partial gas phase oxidation of propylene |
| WO2015039982A1 (de) * | 2013-09-17 | 2015-03-26 | Basf Se | Katalysator zur herstellung einer ungesättigten carbonsäure durch gasphasenoxidation eines ungesättigten aldehyds |
| EP3770145A1 (de) | 2019-07-24 | 2021-01-27 | Basf Se | Verfahren zur kontinuierlichen herstellung von acrolein oder acrylsäure als zielprodukt aus propen |
| WO2021213823A1 (de) | 2020-04-21 | 2021-10-28 | Basf Se | Verfahren zur herstellung eines die elemente mo, w, v und cu enthaltenden katalytisch aktiven multielementoxids |
| WO2022090019A1 (de) | 2020-10-29 | 2022-05-05 | Basf Se | Verfahren zur herstellung eines schalenkatalysators |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000053559A1 (de) * | 1999-03-10 | 2000-09-14 | Basf Aktiengesellschaft | Verfahren der katalytischen gasphasenoxidation von acrolein zu acrylsäure |
| EP1055662A1 (de) * | 1999-05-27 | 2000-11-29 | Nippon Shokubai Co., Ltd | Verfahren zum Herstellen von Acrylsäure |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MY121878A (en) * | 1999-03-10 | 2006-02-28 | Basf Ag | Method for the catalytic gas-phase oxidation of propene into acrylic acid |
| JP3732080B2 (ja) * | 1999-08-31 | 2006-01-05 | 株式会社日本触媒 | 接触気相酸化反応器 |
-
2004
- 2004-03-20 DE DE502004003884T patent/DE502004003884D1/de not_active Expired - Lifetime
- 2004-03-20 JP JP2006504767A patent/JP4537387B2/ja not_active Expired - Lifetime
- 2004-03-20 BR BRPI0408642-2B1A patent/BRPI0408642B1/pt active IP Right Grant
- 2004-03-20 ES ES04722167T patent/ES2286626T3/es not_active Expired - Lifetime
- 2004-03-20 EP EP04722167A patent/EP1611079B1/de not_active Expired - Lifetime
- 2004-03-20 WO PCT/EP2004/002934 patent/WO2004085370A1/de active IP Right Grant
- 2004-03-20 AT AT04722167T patent/ATE362909T1/de not_active IP Right Cessation
- 2004-03-20 KR KR1020057017974A patent/KR101006585B1/ko active IP Right Grant
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000053559A1 (de) * | 1999-03-10 | 2000-09-14 | Basf Aktiengesellschaft | Verfahren der katalytischen gasphasenoxidation von acrolein zu acrylsäure |
| EP1055662A1 (de) * | 1999-05-27 | 2000-11-29 | Nippon Shokubai Co., Ltd | Verfahren zum Herstellen von Acrylsäure |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8431743B2 (en) | 2004-07-01 | 2013-04-30 | Basf Aktiengesellschaft | Preparation of acrylic acid by heterogeneously catalyzed partial gas phase oxidation of propylene |
| US8188310B2 (en) | 2006-01-18 | 2012-05-29 | Basf Se | Process for the long-term operation of a heterogeneously catalyzed partial gas phase oxidation of an organic starting compound |
| US7884238B2 (en) | 2006-01-18 | 2011-02-08 | Basf Aktiengesellschaft | Process for the long-term operation of a heterogeneously catalyzed partial gas phase oxidation of an organic starting compound |
| DE102010048405A1 (de) | 2010-10-15 | 2011-05-19 | Basf Se | Verfahren zum Langzeitbetrieb einer heterogen katalysierten partiellen Gasphasenoxidation von Proben zu Acrolein |
| WO2012049246A2 (de) | 2010-10-15 | 2012-04-19 | Basf Se | Verfahren zum langzeitbetrieb einer heterogen katalysierten partiellen gasphasenoxidation von propen zu acrolein |
| US8618336B2 (en) | 2010-10-15 | 2013-12-31 | Basf Se | Process for long-term operation of a heterogeneously catalyzed partial gas phase oxidation of propene to acrolein |
| US9061988B2 (en) | 2013-02-07 | 2015-06-23 | Basf Se | Process for producing a catalytically active composition being a mixture of a multielement oxide comprising the elements Mo and V and at least one oxide of molybdenum |
| DE102013202048A1 (de) | 2013-02-07 | 2013-04-18 | Basf Se | Verfahren zur Herstellung einer katalytisch aktiven Masse, die ein Gemisch aus einem die Elemente Mo und V enthaltenden Multielementoxid und wenigstens einem Oxid des Molybdäns ist |
| WO2014122043A1 (de) | 2013-02-07 | 2014-08-14 | Basf Se | Verfahren zur herstellung einer katalytisch aktiven masse, die ein gemisch aus einem die elemente mo und v enthaltenden multielementoxid und wenigstens einem oxid des molybdäns ist |
| WO2015039982A1 (de) * | 2013-09-17 | 2015-03-26 | Basf Se | Katalysator zur herstellung einer ungesättigten carbonsäure durch gasphasenoxidation eines ungesättigten aldehyds |
| US9238217B2 (en) | 2013-09-17 | 2016-01-19 | Basf Se | Catalyst for preparation of an unsaturated carboxylic acid by gas phase oxidation of an unsaturated aldehyde |
| RU2678847C2 (ru) * | 2013-09-17 | 2019-02-04 | Басф Се | Катализатор для синтеза ненасыщенной карбоновой кислоты путем газофазного окисления ненасыщенного альдегида |
| EP3770145A1 (de) | 2019-07-24 | 2021-01-27 | Basf Se | Verfahren zur kontinuierlichen herstellung von acrolein oder acrylsäure als zielprodukt aus propen |
| WO2021013640A1 (en) | 2019-07-24 | 2021-01-28 | Basf Se | A process for the continuous production of either acrolein or acrylic acid as the target product from propene |
| WO2021213823A1 (de) | 2020-04-21 | 2021-10-28 | Basf Se | Verfahren zur herstellung eines die elemente mo, w, v und cu enthaltenden katalytisch aktiven multielementoxids |
| WO2022090019A1 (de) | 2020-10-29 | 2022-05-05 | Basf Se | Verfahren zur herstellung eines schalenkatalysators |
Also Published As
| Publication number | Publication date |
|---|---|
| BRPI0408642B1 (pt) | 2013-07-02 |
| ES2286626T3 (es) | 2007-12-01 |
| JP2006523187A (ja) | 2006-10-12 |
| EP1611079B1 (de) | 2007-05-23 |
| DE502004003884D1 (de) | 2007-07-05 |
| KR101006585B1 (ko) | 2011-01-07 |
| EP1611079A1 (de) | 2006-01-04 |
| BRPI0408642A (pt) | 2006-03-28 |
| KR20050115311A (ko) | 2005-12-07 |
| ATE362909T1 (de) | 2007-06-15 |
| JP4537387B2 (ja) | 2010-09-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1159246B1 (de) | Verfahren der katalytischen gasphasenoxidation von acrolein zu acrylsäure | |
| EP1611077B1 (de) | Verfahren der heterogen katalysierten partiellen gasphasenoxidation von propen zu acrylsäure | |
| EP1979305B1 (de) | Verfahren zum langzeitbetrieb einer heterogen katalysierten partiellen gasphasenoxidation einer organischen ausgangsverbindung | |
| EP1159248B1 (de) | Verfahren der katalytischen gasphasenoxidation von propen zu acrylsäure | |
| EP1159244B1 (de) | Verfahren der katalytischen gasphasenoxidation von propen zu acrolein | |
| EP1230204B1 (de) | Verfahren der katalytischen gasphasenoxidation von propen zu acrylsäure | |
| EP1973641B1 (de) | Verfahren der heterogen katalysierten gasphasen-partialoxidation wenigstens einer organischen ausgangsverbindung | |
| EP1159247B1 (de) | Verfahren der katalytischen gasphasenoxidation von propen zu acrylsäure | |
| EP1682477B1 (de) | Verfahren zum langzeitbetrieb einer heterogen katalysierten gasphasenpartialoxidation von acrolein zu acrylsäure | |
| EP1963248B1 (de) | Verfahren der heterogen katalysierten partiellen gasphasenoxidation von propylen zu acrylsäure | |
| EP2627622B1 (de) | Verfahren zum langzeitbetrieb einer heterogen katalysierten partiellen gasphasenoxidation von propen zu acrolein | |
| DE10313208A1 (de) | Verfahren der heterogen katalysierten partiellen Gasphasenoxidation von Propen zu Acrylsäure | |
| EP1611080B1 (de) | Verfahren der heterogen katalysierten partiellen gasphasenoxidation von acrolein zu acrylsäure | |
| DE10313213A1 (de) | Verfahren der heterogen katalysierten partiellen Gasphasenoxidation von Propen zu Acrylsäure | |
| EP1611073B1 (de) | Verfahren der heterogen katalysierten partiellen gasphasenoxidation von propen zu acrolein | |
| EP1611076B1 (de) | Verfahren der heterogen katalysierten partiellen gasphasenoxidation von propen zu acrylsäure | |
| EP1611079B1 (de) | Verfahren der heterogen katalysierten partiellen gasphasenoxidation von acrolein zu acryls ure | |
| DE10313212A1 (de) | Verfahren der heterogen katalysierten partiellen Gasphasenoxidation von Propen zu Acrolein | |
| DE10313214A1 (de) | Verfahren der heterogen katalysierten partiellen Gasphasenoxidation von Acrolein zu Acrylsäure | |
| DE10313211A1 (de) | Verfahren der heterogen katalysierten partiellen Gasphasenoxidation von Acrolein zu Acrylsäure | |
| EP1613579B1 (de) | Verfahren der heterogen katalysierten partiellen gasphasenoxidation von propen zu acrolein | |
| DE10313210A1 (de) | Verfahren der heterogen katalysierten partiellen Gasphasenoxidation von Propen zu Acrolein |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2004722167 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020057017974 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006504767 Country of ref document: JP Ref document number: 20048080975 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005/08599 Country of ref document: ZA Ref document number: 200508599 Country of ref document: ZA |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020057017974 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004722167 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0408642 Country of ref document: BR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2004722167 Country of ref document: EP |