WO2004078163A2 - Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen - Google Patents
Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen Download PDFInfo
- Publication number
- WO2004078163A2 WO2004078163A2 PCT/US2004/006288 US2004006288W WO2004078163A2 WO 2004078163 A2 WO2004078163 A2 WO 2004078163A2 US 2004006288 W US2004006288 W US 2004006288W WO 2004078163 A2 WO2004078163 A2 WO 2004078163A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- crystal
- acid
- degrees
- ray diffraction
- diffraction pattern
- Prior art date
Links
- 239000013078 crystal Substances 0.000 title claims abstract description 1329
- 239000000203 mixture Substances 0.000 title claims description 109
- FFGPTBGBLSHEPO-UHFFFAOYSA-N carbamazepine Chemical compound C1=CC2=CC=CC=C2N(C(=O)N)C2=CC=CC=C21 FFGPTBGBLSHEPO-UHFFFAOYSA-N 0.000 title claims description 74
- 229960000623 carbamazepine Drugs 0.000 title claims description 74
- RZEKVGVHFLEQIL-UHFFFAOYSA-N celecoxib Chemical compound C1=CC(C)=CC=C1C1=CC(C(F)(F)F)=NN1C1=CC=C(S(N)(=O)=O)C=C1 RZEKVGVHFLEQIL-UHFFFAOYSA-N 0.000 title claims description 42
- 229960000590 celecoxib Drugs 0.000 title claims description 42
- 229960001165 modafinil Drugs 0.000 title claims description 41
- YFGHCGITMMYXAQ-UHFFFAOYSA-N 2-[(diphenylmethyl)sulfinyl]acetamide Chemical compound C=1C=CC=CC=1C(S(=O)CC(=O)N)C1=CC=CC=C1 YFGHCGITMMYXAQ-UHFFFAOYSA-N 0.000 title claims description 38
- JZUFKLXOESDKRF-UHFFFAOYSA-N Chlorothiazide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC2=C1NCNS2(=O)=O JZUFKLXOESDKRF-UHFFFAOYSA-N 0.000 title claims description 36
- 229960002003 hydrochlorothiazide Drugs 0.000 title claims description 36
- 229960004130 itraconazole Drugs 0.000 title claims description 35
- KVWDHTXUZHCGIO-UHFFFAOYSA-N olanzapine Chemical compound C1CN(C)CCN1C1=NC2=CC=CC=C2NC2=C1C=C(C)S2 KVWDHTXUZHCGIO-UHFFFAOYSA-N 0.000 title claims description 32
- 229960005017 olanzapine Drugs 0.000 title claims description 32
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 title claims description 29
- 229960002949 fluorouracil Drugs 0.000 title claims description 29
- SYTBZMRGLBWNTM-UHFFFAOYSA-N flurbiprofen Chemical compound FC1=CC(C(C(O)=O)C)=CC=C1C1=CC=CC=C1 SYTBZMRGLBWNTM-UHFFFAOYSA-N 0.000 title claims description 25
- 229960002390 flurbiprofen Drugs 0.000 title claims description 25
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 title claims description 18
- KJADKKWYZYXHBB-XBWDGYHZSA-N Topiramic acid Chemical compound C1O[C@@]2(COS(N)(=O)=O)OC(C)(C)O[C@H]2[C@@H]2OC(C)(C)O[C@@H]21 KJADKKWYZYXHBB-XBWDGYHZSA-N 0.000 title claims description 18
- 229960004394 topiramate Drugs 0.000 title claims description 18
- VHVPQPYKVGDNFY-DFMJLFEVSA-N 2-[(2r)-butan-2-yl]-4-[4-[4-[4-[[(2r,4s)-2-(2,4-dichlorophenyl)-2-(1,2,4-triazol-1-ylmethyl)-1,3-dioxolan-4-yl]methoxy]phenyl]piperazin-1-yl]phenyl]-1,2,4-triazol-3-one Chemical compound O=C1N([C@H](C)CC)N=CN1C1=CC=C(N2CCN(CC2)C=2C=CC(OC[C@@H]3O[C@](CN4N=CN=C4)(OC3)C=3C(=CC(Cl)=CC=3)Cl)=CC=2)C=C1 VHVPQPYKVGDNFY-DFMJLFEVSA-N 0.000 title claims description 16
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 title claims description 14
- 229960001680 ibuprofen Drugs 0.000 title claims description 14
- CXOFVDLJLONNDW-UHFFFAOYSA-N Phenytoin Chemical compound N1C(=O)NC(=O)C1(C=1C=CC=CC=1)C1=CC=CC=C1 CXOFVDLJLONNDW-UHFFFAOYSA-N 0.000 title claims description 13
- 229960002036 phenytoin Drugs 0.000 title claims description 13
- 229960005489 paracetamol Drugs 0.000 title claims description 9
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 title claims description 8
- 229960001138 acetylsalicylic acid Drugs 0.000 title claims description 8
- 239000003814 drug Substances 0.000 title description 4
- 229940079593 drug Drugs 0.000 title description 2
- -1 nitrate ester Chemical class 0.000 claims abstract description 190
- 239000002253 acid Substances 0.000 claims abstract description 91
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims abstract description 84
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 claims abstract description 84
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 70
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims abstract description 68
- 150000001732 carboxylic acid derivatives Chemical class 0.000 claims abstract description 59
- 150000003141 primary amines Chemical class 0.000 claims abstract description 57
- 150000003335 secondary amines Chemical class 0.000 claims abstract description 57
- 238000002425 crystallisation Methods 0.000 claims abstract description 56
- 230000008025 crystallization Effects 0.000 claims abstract description 53
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims abstract description 48
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims abstract description 48
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 claims abstract description 46
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 claims abstract description 46
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 claims abstract description 44
- 150000002148 esters Chemical class 0.000 claims abstract description 41
- 125000000524 functional group Chemical group 0.000 claims abstract description 40
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims abstract description 37
- 150000001408 amides Chemical class 0.000 claims abstract description 25
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims abstract description 24
- NEAQRZUHTPSBBM-UHFFFAOYSA-N 2-hydroxy-3,3-dimethyl-7-nitro-4h-isoquinolin-1-one Chemical compound C1=C([N+]([O-])=O)C=C2C(=O)N(O)C(C)(C)CC2=C1 NEAQRZUHTPSBBM-UHFFFAOYSA-N 0.000 claims abstract description 23
- XZMCDFZZKTWFGF-UHFFFAOYSA-N Cyanamide Chemical compound NC#N XZMCDFZZKTWFGF-UHFFFAOYSA-N 0.000 claims abstract description 23
- 229910002651 NO3 Inorganic materials 0.000 claims abstract description 23
- 229910019142 PO4 Inorganic materials 0.000 claims abstract description 23
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical compound OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 claims abstract description 23
- ZMZDMBWJUHKJPS-UHFFFAOYSA-M Thiocyanate anion Chemical compound [S-]C#N ZMZDMBWJUHKJPS-UHFFFAOYSA-M 0.000 claims abstract description 23
- 150000001299 aldehydes Chemical class 0.000 claims abstract description 23
- 229910021529 ammonia Inorganic materials 0.000 claims abstract description 23
- SKOLWUPSYHWYAM-UHFFFAOYSA-N carbonodithioic O,S-acid Chemical compound SC(S)=O SKOLWUPSYHWYAM-UHFFFAOYSA-N 0.000 claims abstract description 23
- 150000008282 halocarbons Chemical class 0.000 claims abstract description 23
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N hydrogen thiocyanate Natural products SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 claims abstract description 23
- 150000002466 imines Chemical class 0.000 claims abstract description 23
- 150000002576 ketones Chemical class 0.000 claims abstract description 23
- 150000002923 oximes Chemical class 0.000 claims abstract description 23
- 150000002978 peroxides Chemical class 0.000 claims abstract description 23
- 239000010452 phosphate Substances 0.000 claims abstract description 23
- 150000003512 tertiary amines Chemical class 0.000 claims abstract description 23
- 150000007970 thio esters Chemical class 0.000 claims abstract description 23
- 150000003568 thioethers Chemical class 0.000 claims abstract description 23
- 150000003573 thiols Chemical class 0.000 claims abstract description 23
- 229930192474 thiophene Natural products 0.000 claims abstract description 23
- ACVYVLVWPXVTIT-UHFFFAOYSA-N phosphinic acid Chemical compound O[PH2]=O ACVYVLVWPXVTIT-UHFFFAOYSA-N 0.000 claims abstract description 22
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 claims abstract description 21
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims abstract description 17
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 claims abstract description 13
- 150000002118 epoxides Chemical class 0.000 claims abstract 17
- 239000008186 active pharmaceutical agent Substances 0.000 claims description 452
- 230000003993 interaction Effects 0.000 claims description 230
- FTOAOBMCPZCFFF-UHFFFAOYSA-N 5,5-diethylbarbituric acid Chemical compound CCC1(CC)C(=O)NC(=O)NC1=O FTOAOBMCPZCFFF-UHFFFAOYSA-N 0.000 claims description 154
- 238000000034 method Methods 0.000 claims description 101
- 239000001257 hydrogen Substances 0.000 claims description 99
- 229910052739 hydrogen Inorganic materials 0.000 claims description 99
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 claims description 87
- 230000008569 process Effects 0.000 claims description 81
- 150000001875 compounds Chemical class 0.000 claims description 79
- 230000001965 increasing effect Effects 0.000 claims description 75
- 150000003839 salts Chemical class 0.000 claims description 58
- 239000000243 solution Substances 0.000 claims description 57
- 238000001938 differential scanning calorimetry curve Methods 0.000 claims description 56
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 51
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 49
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 claims description 47
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 claims description 46
- 238000000227 grinding Methods 0.000 claims description 46
- VFQXVTODMYMSMJ-UHFFFAOYSA-N isonicotinamide Chemical compound NC(=O)C1=CC=NC=C1 VFQXVTODMYMSMJ-UHFFFAOYSA-N 0.000 claims description 46
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 44
- 150000003140 primary amides Chemical class 0.000 claims description 43
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 claims description 43
- 229960002319 barbital Drugs 0.000 claims description 42
- 238000010438 heat treatment Methods 0.000 claims description 42
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 claims description 40
- XEZNGIUYQVAUSS-UHFFFAOYSA-N 18-crown-6 Chemical compound C1COCCOCCOCCOCCOCCO1 XEZNGIUYQVAUSS-UHFFFAOYSA-N 0.000 claims description 39
- ICSNLGPSRYBMBD-UHFFFAOYSA-N 2-aminopyridine Chemical group NC1=CC=CC=N1 ICSNLGPSRYBMBD-UHFFFAOYSA-N 0.000 claims description 39
- 229960003966 nicotinamide Drugs 0.000 claims description 39
- 235000005152 nicotinamide Nutrition 0.000 claims description 39
- 239000011570 nicotinamide Substances 0.000 claims description 39
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 claims description 38
- 238000003181 co-melting Methods 0.000 claims description 38
- 238000004090 dissolution Methods 0.000 claims description 38
- 150000003334 secondary amides Chemical class 0.000 claims description 37
- MWVTWFVJZLCBMC-UHFFFAOYSA-N 4,4'-bipyridine Chemical compound C1=NC=CC(C=2C=CN=CC=2)=C1 MWVTWFVJZLCBMC-UHFFFAOYSA-N 0.000 claims description 36
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 claims description 35
- LJXQPZWIHJMPQQ-UHFFFAOYSA-N pyrimidin-2-amine Chemical compound NC1=NC=CC=N1 LJXQPZWIHJMPQQ-UHFFFAOYSA-N 0.000 claims description 34
- ZFXYFBGIUFBOJW-UHFFFAOYSA-N theophylline Chemical compound O=C1N(C)C(=O)N(C)C2=C1NC=N2 ZFXYFBGIUFBOJW-UHFFFAOYSA-N 0.000 claims description 32
- 239000007787 solid Substances 0.000 claims description 31
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 claims description 30
- 239000012453 solvate Substances 0.000 claims description 29
- 229910052757 nitrogen Inorganic materials 0.000 claims description 27
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 claims description 26
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 claims description 26
- 229940124530 sulfonamide Drugs 0.000 claims description 26
- 150000003456 sulfonamides Chemical class 0.000 claims description 26
- 230000003247 decreasing effect Effects 0.000 claims description 24
- 239000000370 acceptor Substances 0.000 claims description 23
- 239000004202 carbamide Substances 0.000 claims description 23
- 238000001237 Raman spectrum Methods 0.000 claims description 22
- QMKYBPDZANOJGF-UHFFFAOYSA-N benzene-1,3,5-tricarboxylic acid Chemical compound OC(=O)C1=CC(C(O)=O)=CC(C(O)=O)=C1 QMKYBPDZANOJGF-UHFFFAOYSA-N 0.000 claims description 22
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical group Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 21
- 125000000664 diazo group Chemical group [N-]=[N+]=[*] 0.000 claims description 21
- 150000002825 nitriles Chemical class 0.000 claims description 21
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 claims description 20
- 231100000673 dose–response relationship Toxicity 0.000 claims description 20
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 claims description 19
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 claims description 19
- 239000001530 fumaric acid Substances 0.000 claims description 19
- 239000007788 liquid Substances 0.000 claims description 19
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 claims description 18
- 229960000278 theophylline Drugs 0.000 claims description 17
- 125000003118 aryl group Chemical group 0.000 claims description 16
- 229920000877 Melamine resin Polymers 0.000 claims description 15
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 claims description 15
- 235000011090 malic acid Nutrition 0.000 claims description 15
- JDSHMPZPIAZGSV-UHFFFAOYSA-N melamine Chemical compound NC1=NC(N)=NC(N)=N1 JDSHMPZPIAZGSV-UHFFFAOYSA-N 0.000 claims description 15
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 15
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical group Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 claims description 14
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 claims description 14
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 claims description 14
- 239000011976 maleic acid Substances 0.000 claims description 14
- VYWYYJYRVSBHJQ-UHFFFAOYSA-N 3,5-dinitrobenzoic acid Chemical compound OC(=O)C1=CC([N+]([O-])=O)=CC([N+]([O-])=O)=C1 VYWYYJYRVSBHJQ-UHFFFAOYSA-N 0.000 claims description 12
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 12
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 claims description 12
- 239000003085 diluting agent Substances 0.000 claims description 12
- 229940116298 l- malic acid Drugs 0.000 claims description 12
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 claims description 12
- 229940081974 saccharin Drugs 0.000 claims description 12
- 235000019204 saccharin Nutrition 0.000 claims description 12
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 claims description 12
- 239000001384 succinic acid Substances 0.000 claims description 12
- 229960001367 tartaric acid Drugs 0.000 claims description 12
- 239000011975 tartaric acid Substances 0.000 claims description 12
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 claims description 11
- 238000004519 manufacturing process Methods 0.000 claims description 11
- 239000007790 solid phase Substances 0.000 claims description 11
- NBDAHKQJXVLAID-UHFFFAOYSA-N 5-nitroisophthalic acid Chemical compound OC(=O)C1=CC(C(O)=O)=CC([N+]([O-])=O)=C1 NBDAHKQJXVLAID-UHFFFAOYSA-N 0.000 claims description 10
- MUIPLRMGAXZWSQ-UHFFFAOYSA-N 9-ethylpurin-6-amine Chemical compound N1=CN=C2N(CC)C=NC2=C1N MUIPLRMGAXZWSQ-UHFFFAOYSA-N 0.000 claims description 10
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 claims description 10
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 claims description 10
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 claims description 10
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 9
- DATYUTWESAKQQM-UHFFFAOYSA-N 4,7-phenanthroline Chemical compound C1=CC=C2C3=CC=CN=C3C=CC2=N1 DATYUTWESAKQQM-UHFFFAOYSA-N 0.000 claims description 8
- ALYNCZNDIQEVRV-UHFFFAOYSA-N 4-aminobenzoic acid Chemical compound NC1=CC=C(C(O)=O)C=C1 ALYNCZNDIQEVRV-UHFFFAOYSA-N 0.000 claims description 8
- WWZKQHOCKIZLMA-UHFFFAOYSA-N Caprylic acid Natural products CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 claims description 8
- FEWJPZIEWOKRBE-XIXRPRMCSA-N Mesotartaric acid Chemical compound OC(=O)[C@@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-XIXRPRMCSA-N 0.000 claims description 8
- KKEYFWRCBNTPAC-UHFFFAOYSA-N Terephthalic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C=C1 KKEYFWRCBNTPAC-UHFFFAOYSA-N 0.000 claims description 8
- 229940048879 dl tartaric acid Drugs 0.000 claims description 8
- 235000021355 Stearic acid Nutrition 0.000 claims description 7
- YRKCREAYFQTBPV-UHFFFAOYSA-N acetylacetone Natural products CC(=O)CC(C)=O YRKCREAYFQTBPV-UHFFFAOYSA-N 0.000 claims description 7
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 claims description 7
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 claims description 7
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 claims description 7
- BLNVISNJTIRAHF-UHFFFAOYSA-N 4-chlorobenzamide Chemical compound NC(=O)C1=CC=C(Cl)C=C1 BLNVISNJTIRAHF-UHFFFAOYSA-N 0.000 claims description 6
- OTLNPYWUJOZPPA-UHFFFAOYSA-N 4-nitrobenzoic acid Chemical compound OC(=O)C1=CC=C([N+]([O-])=O)C=C1 OTLNPYWUJOZPPA-UHFFFAOYSA-N 0.000 claims description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 6
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 claims description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 6
- DFPAKSUCGFBDDF-ZQBYOMGUSA-N [14c]-nicotinamide Chemical compound N[14C](=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-ZQBYOMGUSA-N 0.000 claims description 6
- IPPWILKGXFOXHO-UHFFFAOYSA-N chloranilic acid Chemical compound OC1=C(Cl)C(=O)C(O)=C(Cl)C1=O IPPWILKGXFOXHO-UHFFFAOYSA-N 0.000 claims description 6
- OZHNSXNGSKKWMC-UHFFFAOYSA-N iron;1,2,3,4,5-pentamethylcyclopentane Chemical compound [Fe].C[C]1[C](C)[C](C)[C](C)[C]1C.C[C]1[C](C)[C](C)[C](C)[C]1C OZHNSXNGSKKWMC-UHFFFAOYSA-N 0.000 claims description 6
- 235000006408 oxalic acid Nutrition 0.000 claims description 6
- MCJGNVYPOGVAJF-UHFFFAOYSA-N quinolin-8-ol Chemical compound C1=CN=C2C(O)=CC=CC2=C1 MCJGNVYPOGVAJF-UHFFFAOYSA-N 0.000 claims description 6
- 235000002906 tartaric acid Nutrition 0.000 claims description 6
- RTHCYVBBDHJXIQ-MRXNPFEDSA-N (R)-fluoxetine Chemical compound O([C@H](CCNC)C=1C=CC=CC=1)C1=CC=C(C(F)(F)F)C=C1 RTHCYVBBDHJXIQ-MRXNPFEDSA-N 0.000 claims description 5
- RTBFRGCFXZNCOE-UHFFFAOYSA-N 1-methylsulfonylpiperidin-4-one Chemical compound CS(=O)(=O)N1CCC(=O)CC1 RTBFRGCFXZNCOE-UHFFFAOYSA-N 0.000 claims description 5
- 239000005711 Benzoic acid Substances 0.000 claims description 5
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 claims description 5
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 claims description 5
- YJLYANLCNIKXMG-UHFFFAOYSA-N N-Methyldioctylamine Chemical compound CCCCCCCCN(C)CCCCCCCC YJLYANLCNIKXMG-UHFFFAOYSA-N 0.000 claims description 5
- JFCQEDHGNNZCLN-UHFFFAOYSA-N anhydrous glutaric acid Natural products OC(=O)CCCC(O)=O JFCQEDHGNNZCLN-UHFFFAOYSA-N 0.000 claims description 5
- 235000010233 benzoic acid Nutrition 0.000 claims description 5
- 229960001948 caffeine Drugs 0.000 claims description 5
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 claims description 5
- 229960002464 fluoxetine Drugs 0.000 claims description 5
- 235000001968 nicotinic acid Nutrition 0.000 claims description 5
- 229960003512 nicotinic acid Drugs 0.000 claims description 5
- 239000011664 nicotinic acid Substances 0.000 claims description 5
- 229960004889 salicylic acid Drugs 0.000 claims description 5
- 239000008117 stearic acid Substances 0.000 claims description 5
- WXTMDXOMEHJXQO-UHFFFAOYSA-N 2,5-dihydroxybenzoic acid Chemical compound OC(=O)C1=CC(O)=CC=C1O WXTMDXOMEHJXQO-UHFFFAOYSA-N 0.000 claims description 4
- NEGFNJRAUMCZMY-UHFFFAOYSA-N 3-(dimethylamino)benzoic acid Chemical compound CN(C)C1=CC=CC(C(O)=O)=C1 NEGFNJRAUMCZMY-UHFFFAOYSA-N 0.000 claims description 4
- IJFXRHURBJZNAO-UHFFFAOYSA-N 3-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=CC(O)=C1 IJFXRHURBJZNAO-UHFFFAOYSA-N 0.000 claims description 4
- IHSIWAKWCWADBV-UHFFFAOYSA-N 4-[2-[1-(2-hydroxyethyl)pyridin-4-ylidene]ethylidene]cyclohexa-2,5-dien-1-one Chemical compound C1=CN(CCO)C=CC1=CC=C1C=CC(=O)C=C1 IHSIWAKWCWADBV-UHFFFAOYSA-N 0.000 claims description 4
- YBAZINRZQSAIAY-UHFFFAOYSA-N 4-aminobenzonitrile Chemical compound NC1=CC=C(C#N)C=C1 YBAZINRZQSAIAY-UHFFFAOYSA-N 0.000 claims description 4
- AOCTXBGGCGPOKT-UHFFFAOYSA-N 5-tert-butylpyrimidine-2,4,6-triamine Chemical compound CC(C)(C)C1=C(N)N=C(N)N=C1N AOCTXBGGCGPOKT-UHFFFAOYSA-N 0.000 claims description 4
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 claims description 4
- 229920000858 Cyclodextrin Polymers 0.000 claims description 4
- 239000001116 FEMA 4028 Substances 0.000 claims description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N Formic acid Chemical compound OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 claims description 4
- HCUARRIEZVDMPT-UHFFFAOYSA-N Indole-2-carboxylic acid Chemical compound C1=CC=C2NC(C(=O)O)=CC2=C1 HCUARRIEZVDMPT-UHFFFAOYSA-N 0.000 claims description 4
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 4
- 239000007983 Tris buffer Substances 0.000 claims description 4
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 claims description 4
- 229960004050 aminobenzoic acid Drugs 0.000 claims description 4
- RWZYAGGXGHYGMB-UHFFFAOYSA-N anthranilic acid Chemical compound NC1=CC=CC=C1C(O)=O RWZYAGGXGHYGMB-UHFFFAOYSA-N 0.000 claims description 4
- WHGYBXFWUBPSRW-FOUAGVGXSA-N beta-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO WHGYBXFWUBPSRW-FOUAGVGXSA-N 0.000 claims description 4
- 235000011175 beta-cyclodextrine Nutrition 0.000 claims description 4
- 229960004853 betadex Drugs 0.000 claims description 4
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 claims description 4
- KWIUHFFTVRNATP-UHFFFAOYSA-N glycine betaine Chemical compound C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 claims description 4
- 150000002460 imidazoles Chemical class 0.000 claims description 4
- SEOVTRFCIGRIMH-UHFFFAOYSA-N indole-3-acetic acid Chemical compound C1=CC=C2C(CC(=O)O)=CNC2=C1 SEOVTRFCIGRIMH-UHFFFAOYSA-N 0.000 claims description 4
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 claims description 4
- GPSDUZXPYCFOSQ-UHFFFAOYSA-N m-toluic acid Chemical compound CC1=CC=CC(C(O)=O)=C1 GPSDUZXPYCFOSQ-UHFFFAOYSA-N 0.000 claims description 4
- 229940099690 malic acid Drugs 0.000 claims description 4
- 239000001630 malic acid Substances 0.000 claims description 4
- LVWZTYCIRDMTEY-UHFFFAOYSA-N metamizole Chemical compound O=C1C(N(CS(O)(=O)=O)C)=C(C)N(C)N1C1=CC=CC=C1 LVWZTYCIRDMTEY-UHFFFAOYSA-N 0.000 claims description 4
- IIFCLXHRIYTHPV-UHFFFAOYSA-N methyl 2,4-dihydroxybenzoate Chemical compound COC(=O)C1=CC=C(O)C=C1O IIFCLXHRIYTHPV-UHFFFAOYSA-N 0.000 claims description 4
- FBFBRAFXKGRRHI-UHFFFAOYSA-N n-(4-aminophenyl)sulfonyl-4-propan-2-yloxybenzamide Chemical compound C1=CC(OC(C)C)=CC=C1C(=O)NS(=O)(=O)C1=CC=C(N)C=C1 FBFBRAFXKGRRHI-UHFFFAOYSA-N 0.000 claims description 4
- FUZZWVXGSFPDMH-UHFFFAOYSA-N n-hexanoic acid Natural products CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 claims description 4
- BDJRBEYXGGNYIS-UHFFFAOYSA-N nonanedioic acid Chemical compound OC(=O)CCCCCCCC(O)=O BDJRBEYXGGNYIS-UHFFFAOYSA-N 0.000 claims description 4
- XNGIFLGASWRNHJ-UHFFFAOYSA-N o-dicarboxybenzene Natural products OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 claims description 4
- LPNBBFKOUUSUDB-UHFFFAOYSA-N p-toluic acid Chemical compound CC1=CC=C(C(O)=O)C=C1 LPNBBFKOUUSUDB-UHFFFAOYSA-N 0.000 claims description 4
- IZUPBVBPLAPZRR-UHFFFAOYSA-N pentachlorophenol Chemical compound OC1=C(Cl)C(Cl)=C(Cl)C(Cl)=C1Cl IZUPBVBPLAPZRR-UHFFFAOYSA-N 0.000 claims description 4
- LCPDWSOZIOUXRV-UHFFFAOYSA-N phenoxyacetic acid Chemical compound OC(=O)COC1=CC=CC=C1 LCPDWSOZIOUXRV-UHFFFAOYSA-N 0.000 claims description 4
- ZFACJPAPCXRZMQ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O.OC(=O)C1=CC=CC=C1C(O)=O ZFACJPAPCXRZMQ-UHFFFAOYSA-N 0.000 claims description 4
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Substances [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 claims description 4
- PWEBUXCTKOWPCW-UHFFFAOYSA-N squaric acid Chemical compound OC1=C(O)C(=O)C1=O PWEBUXCTKOWPCW-UHFFFAOYSA-N 0.000 claims description 4
- 229950001296 sulfaproxyline Drugs 0.000 claims description 4
- IEDVJHCEMCRBQM-UHFFFAOYSA-N trimethoprim Chemical compound COC1=C(OC)C(OC)=CC(CC=2C(=NC(N)=NC=2)N)=C1 IEDVJHCEMCRBQM-UHFFFAOYSA-N 0.000 claims description 4
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 claims description 3
- AZQWKYJCGOJGHM-UHFFFAOYSA-N 1,4-benzoquinone Chemical compound O=C1C=CC(=O)C=C1 AZQWKYJCGOJGHM-UHFFFAOYSA-N 0.000 claims description 3
- AXQJTBIZHUSQNN-UHFFFAOYSA-N 3,4-dichloro-2-nitrophenol Chemical compound OC1=CC=C(Cl)C(Cl)=C1[N+]([O-])=O AXQJTBIZHUSQNN-UHFFFAOYSA-N 0.000 claims description 3
- AWQSAIIDOMEEOD-UHFFFAOYSA-N 5,5-Dimethyl-4-(3-oxobutyl)dihydro-2(3H)-furanone Chemical compound CC(=O)CCC1CC(=O)OC1(C)C AWQSAIIDOMEEOD-UHFFFAOYSA-N 0.000 claims description 3
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 claims description 3
- WJJMNDUMQPNECX-UHFFFAOYSA-N Dipicolinic acid Natural products OC(=O)C1=CC=CC(C(O)=O)=N1 WJJMNDUMQPNECX-UHFFFAOYSA-N 0.000 claims description 3
- BLXXJMDCKKHMKV-UHFFFAOYSA-N Nabumetone Chemical compound C1=C(CCC(C)=O)C=CC2=CC(OC)=CC=C21 BLXXJMDCKKHMKV-UHFFFAOYSA-N 0.000 claims description 3
- 235000011054 acetic acid Nutrition 0.000 claims description 3
- 229960000583 acetic acid Drugs 0.000 claims description 3
- VWAIZPYLEYEEFK-UHFFFAOYSA-N adamantane-1,3,5,7-tetracarboxylic acid Chemical compound C1C(C2)(C(O)=O)CC3(C(O)=O)CC1(C(=O)O)CC2(C(O)=O)C3 VWAIZPYLEYEEFK-UHFFFAOYSA-N 0.000 claims description 3
- GONOPSZTUGRENK-UHFFFAOYSA-N benzyl(trichloro)silane Chemical compound Cl[Si](Cl)(Cl)CC1=CC=CC=C1 GONOPSZTUGRENK-UHFFFAOYSA-N 0.000 claims description 3
- 229960003237 betaine Drugs 0.000 claims description 3
- 235000013985 cinnamic acid Nutrition 0.000 claims description 3
- 229930016911 cinnamic acid Natural products 0.000 claims description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 3
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 claims description 3
- 229960004270 nabumetone Drugs 0.000 claims description 3
- 125000001624 naphthyl group Chemical group 0.000 claims description 3
- 150000004045 organic chlorine compounds Chemical group 0.000 claims description 3
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 claims description 3
- 229910052697 platinum Inorganic materials 0.000 claims description 3
- 235000019260 propionic acid Nutrition 0.000 claims description 3
- YONLFQNRGZXBBF-KBPBESRZSA-N (2s,3s)-2,3-dibenzoyloxybutanedioic acid Chemical compound O([C@H](C(=O)O)[C@H](OC(=O)C=1C=CC=CC=1)C(O)=O)C(=O)C1=CC=CC=C1 YONLFQNRGZXBBF-KBPBESRZSA-N 0.000 claims description 2
- IWYDHOAUDWTVEP-ZETCQYMHSA-N (S)-mandelic acid Chemical compound OC(=O)[C@@H](O)C1=CC=CC=C1 IWYDHOAUDWTVEP-ZETCQYMHSA-N 0.000 claims description 2
- XLYMOEINVGRTEX-ONEGZZNKSA-N (e)-4-ethoxy-4-oxobut-2-enoic acid Chemical compound CCOC(=O)\C=C\C(O)=O XLYMOEINVGRTEX-ONEGZZNKSA-N 0.000 claims description 2
- FIDRAVVQGKNYQK-UHFFFAOYSA-N 1,2,3,4-tetrahydrotriazine Chemical compound C1NNNC=C1 FIDRAVVQGKNYQK-UHFFFAOYSA-N 0.000 claims description 2
- LLBBBYLDTDJMNU-UHFFFAOYSA-N 1-(2,4-dihydroxyphenyl)propan-1-one Chemical compound CCC(=O)C1=CC=C(O)C=C1O LLBBBYLDTDJMNU-UHFFFAOYSA-N 0.000 claims description 2
- GYSCBCSGKXNZRH-UHFFFAOYSA-N 1-benzothiophene-2-carboxamide Chemical compound C1=CC=C2SC(C(=O)N)=CC2=C1 GYSCBCSGKXNZRH-UHFFFAOYSA-N 0.000 claims description 2
- MCTWTZJPVLRJOU-UHFFFAOYSA-N 1-methyl-1H-imidazole Chemical compound CN1C=CN=C1 MCTWTZJPVLRJOU-UHFFFAOYSA-N 0.000 claims description 2
- DVVGIUUJYPYENY-UHFFFAOYSA-N 1-methylpyridin-2-one Chemical compound CN1C=CC=CC1=O DVVGIUUJYPYENY-UHFFFAOYSA-N 0.000 claims description 2
- ILAOVOOZLVGAJF-UHFFFAOYSA-N 1-methylpyrrole-2-carboxylic acid Chemical compound CN1C=CC=C1C(O)=O ILAOVOOZLVGAJF-UHFFFAOYSA-N 0.000 claims description 2
- YYKBWYBUCFHYPR-UHFFFAOYSA-N 12-bromododecanoic acid Chemical compound OC(=O)CCCCCCCCCCCBr YYKBWYBUCFHYPR-UHFFFAOYSA-N 0.000 claims description 2
- KAQBNBSMMVTKRN-UHFFFAOYSA-N 2,4,6-trinitrobenzoic acid Chemical compound OC(=O)C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O KAQBNBSMMVTKRN-UHFFFAOYSA-N 0.000 claims description 2
- OVSKIKFHRZPJSS-UHFFFAOYSA-N 2,4-D Chemical compound OC(=O)COC1=CC=C(Cl)C=C1Cl OVSKIKFHRZPJSS-UHFFFAOYSA-N 0.000 claims description 2
- 239000005631 2,4-Dichlorophenoxyacetic acid Substances 0.000 claims description 2
- DWUFVOJMPJBOFS-UHFFFAOYSA-N 2,5-dihydroxy-3,6-dioxocyclohexa-1,4-diene-1,4-dicarbonitrile Chemical compound OC1=C(C#N)C(=O)C(O)=C(C#N)C1=O DWUFVOJMPJBOFS-UHFFFAOYSA-N 0.000 claims description 2
- YHBAZQDEMYQPJL-UHFFFAOYSA-N 2-[(2-aminoacetyl)amino]acetic acid;hydron;chloride Chemical compound Cl.NCC(=O)NCC(O)=O YHBAZQDEMYQPJL-UHFFFAOYSA-N 0.000 claims description 2
- SLWIPPZWFZGHEU-UHFFFAOYSA-N 2-[4-(carboxymethyl)phenyl]acetic acid Chemical compound OC(=O)CC1=CC=C(CC(O)=O)C=C1 SLWIPPZWFZGHEU-UHFFFAOYSA-N 0.000 claims description 2
- NIONDZDPPYHYKY-UHFFFAOYSA-N 2-hexenoic acid Chemical compound CCCC=CC(O)=O NIONDZDPPYHYKY-UHFFFAOYSA-N 0.000 claims description 2
- KAKASWBIQZXDDS-UHFFFAOYSA-N 2-n,4-n-bis(3-bromophenyl)-1,3,5-triazine-2,4,6-triamine Chemical compound N=1C(NC=2C=C(Br)C=CC=2)=NC(N)=NC=1NC1=CC=CC(Br)=C1 KAKASWBIQZXDDS-UHFFFAOYSA-N 0.000 claims description 2
- IWKOYSAFDXNJAE-UHFFFAOYSA-N 2-n,4-n-bis(3-iodophenyl)-1,3,5-triazine-2,4,6-triamine Chemical compound N=1C(NC=2C=C(I)C=CC=2)=NC(N)=NC=1NC1=CC=CC(I)=C1 IWKOYSAFDXNJAE-UHFFFAOYSA-N 0.000 claims description 2
- OVRPPHQYAMQXEL-UHFFFAOYSA-N 2-n,4-n-bis(4-fluorophenyl)-1,3,5-triazine-2,4,6-triamine Chemical compound N=1C(NC=2C=CC(F)=CC=2)=NC(N)=NC=1NC1=CC=C(F)C=C1 OVRPPHQYAMQXEL-UHFFFAOYSA-N 0.000 claims description 2
- AASRCBJBSGLGIM-UHFFFAOYSA-N 2-n,4-n-bis(4-tert-butylphenyl)-1,3,5-triazine-2,4,6-triamine Chemical compound C1=CC(C(C)(C)C)=CC=C1NC1=NC(N)=NC(NC=2C=CC(=CC=2)C(C)(C)C)=N1 AASRCBJBSGLGIM-UHFFFAOYSA-N 0.000 claims description 2
- DWPNRKFKRCODNR-UHFFFAOYSA-N 2-n,4-n-bis[4-(trifluoromethyl)phenyl]-1,3,5-triazine-2,4,6-triamine Chemical compound N=1C(NC=2C=CC(=CC=2)C(F)(F)F)=NC(N)=NC=1NC1=CC=C(C(F)(F)F)C=C1 DWPNRKFKRCODNR-UHFFFAOYSA-N 0.000 claims description 2
- KYDAUSBHOHGKTN-UHFFFAOYSA-N 2-n-(4-chlorophenyl)-1,3,5-triazine-2,4,6-triamine Chemical compound NC1=NC(N)=NC(NC=2C=CC(Cl)=CC=2)=N1 KYDAUSBHOHGKTN-UHFFFAOYSA-N 0.000 claims description 2
- SLAMLWHELXOEJZ-UHFFFAOYSA-N 2-nitrobenzoic acid Chemical compound OC(=O)C1=CC=CC=C1[N+]([O-])=O SLAMLWHELXOEJZ-UHFFFAOYSA-N 0.000 claims description 2
- SNYRXHULAWEECU-UHFFFAOYSA-N 3,4-dichlorophenoxyacetic acid Chemical compound OC(=O)COC1=CC=C(Cl)C(Cl)=C1 SNYRXHULAWEECU-UHFFFAOYSA-N 0.000 claims description 2
- HVFQJWGYVXKLTE-UHFFFAOYSA-N 3,5-bis(trifluoromethyl)benzoic acid Chemical compound OC(=O)C1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 HVFQJWGYVXKLTE-UHFFFAOYSA-N 0.000 claims description 2
- WAOQONBSWFLFPE-VIFPVBQESA-N 3,5-dichloro-N-[[(2S)-1-ethyl-2-pyrrolidinyl]methyl]-2-hydroxy-6-methoxybenzamide Chemical compound CCN1CCC[C@H]1CNC(=O)C1=C(O)C(Cl)=CC(Cl)=C1OC WAOQONBSWFLFPE-VIFPVBQESA-N 0.000 claims description 2
- LWFUFLREGJMOIZ-UHFFFAOYSA-N 3,5-dinitrosalicylic acid Chemical compound OC(=O)C1=CC([N+]([O-])=O)=CC([N+]([O-])=O)=C1O LWFUFLREGJMOIZ-UHFFFAOYSA-N 0.000 claims description 2
- AJHPGXZOIAYYDW-UHFFFAOYSA-N 3-(2-cyanophenyl)-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound CC(C)(C)OC(=O)NC(C(O)=O)CC1=CC=CC=C1C#N AJHPGXZOIAYYDW-UHFFFAOYSA-N 0.000 claims description 2
- UEKHVFDHZRFETL-UHFFFAOYSA-N 3-(5-methyl-1,2,4-oxadiazol-3-yl)benzaldehyde Chemical compound O1C(C)=NC(C=2C=C(C=O)C=CC=2)=N1 UEKHVFDHZRFETL-UHFFFAOYSA-N 0.000 claims description 2
- MWBWKVQOODCNCH-UHFFFAOYSA-N 3-[[2-(2,2-dimethylhydrazinyl)-1,3-thiazol-4-yl]methylsulfanyl]-n'-sulfamoylpropanimidamide Chemical compound CN(C)NC1=NC(CSCCC(N)=NS(N)(=O)=O)=CS1 MWBWKVQOODCNCH-UHFFFAOYSA-N 0.000 claims description 2
- BPYHGTCRXDWOIQ-UHFFFAOYSA-N 3-nitropyridin-2-amine Chemical compound NC1=NC=CC=C1[N+]([O-])=O BPYHGTCRXDWOIQ-UHFFFAOYSA-N 0.000 claims description 2
- VBDCTFDIXXYZOO-UHFFFAOYSA-N 4,6-dichloro-1,3-dimethylpyrazolo[3,4-d]pyrimidine Chemical compound ClC1=NC(Cl)=C2C(C)=NN(C)C2=N1 VBDCTFDIXXYZOO-UHFFFAOYSA-N 0.000 claims description 2
- LJBCIJXZALEYFR-UHFFFAOYSA-N 4-(dimethylamino)benzoic acid Chemical compound CN(C1=CC=C(C(=O)O)C=C1)C.CN(C1=CC=C(C(=O)O)C=C1)C LJBCIJXZALEYFR-UHFFFAOYSA-N 0.000 claims description 2
- FUCDWLXYNKZXHC-UHFFFAOYSA-N 4-[(4-cyanophenyl)-(1h-imidazol-2-yl)methyl]benzonitrile Chemical compound C1=CC(C#N)=CC=C1C(C=1C=CC(=CC=1)C#N)C1=NC=CN1 FUCDWLXYNKZXHC-UHFFFAOYSA-N 0.000 claims description 2
- ALYNCZNDIQEVRV-PZFLKRBQSA-N 4-amino-3,5-ditritiobenzoic acid Chemical compound [3H]c1cc(cc([3H])c1N)C(O)=O ALYNCZNDIQEVRV-PZFLKRBQSA-N 0.000 claims description 2
- SJZRECIVHVDYJC-UHFFFAOYSA-M 4-hydroxybutyrate Chemical compound OCCCC([O-])=O SJZRECIVHVDYJC-UHFFFAOYSA-M 0.000 claims description 2
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 claims description 2
- IKVPZYAOGOJTLK-UHFFFAOYSA-N 5,5-diphenyl-1,3-diazinane-2,4,6-trione Chemical compound O=C1NC(=O)NC(=O)C1(C=1C=CC=CC=1)C1=CC=CC=C1 IKVPZYAOGOJTLK-UHFFFAOYSA-N 0.000 claims description 2
- SSHFCFRJYJIJDV-UHFFFAOYSA-N 5-nitropyrimidin-2-amine Chemical compound NC1=NC=C([N+]([O-])=O)C=N1 SSHFCFRJYJIJDV-UHFFFAOYSA-N 0.000 claims description 2
- FDBDETBIRVYJIJ-UHFFFAOYSA-N 9-ethylpurine-2,6-diamine Chemical compound N1=C(N)N=C2N(CC)C=NC2=C1N FDBDETBIRVYJIJ-UHFFFAOYSA-N 0.000 claims description 2
- RMMXTBMQSGEXHJ-UHFFFAOYSA-N Aminophenazone Chemical compound O=C1C(N(C)C)=C(C)N(C)N1C1=CC=CC=C1 RMMXTBMQSGEXHJ-UHFFFAOYSA-N 0.000 claims description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 claims description 2
- GHVNFZFCNZKVNT-UHFFFAOYSA-N Decanoic acid Natural products CCCCCCCCCC(O)=O GHVNFZFCNZKVNT-UHFFFAOYSA-N 0.000 claims description 2
- 239000004471 Glycine Substances 0.000 claims description 2
- 102000004877 Insulin Human genes 0.000 claims description 2
- 108090001061 Insulin Proteins 0.000 claims description 2
- VEQPNABPJHWNSG-UHFFFAOYSA-N Nickel(2+) Chemical compound [Ni+2] VEQPNABPJHWNSG-UHFFFAOYSA-N 0.000 claims description 2
- 229910021610 Silver(III) fluoride Inorganic materials 0.000 claims description 2
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N Valeric acid Natural products CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 claims description 2
- KFVXPZFMARZYNX-UHFFFAOYSA-L [2-[3-(hydroxymethyl)pyridin-2-yl]pyridin-3-yl]methanol platinum(2+) dichloride Chemical compound [Cl-].[Cl-].[Pt+2].OCC1=CC=CN=C1C1=NC=CC=C1CO KFVXPZFMARZYNX-UHFFFAOYSA-L 0.000 claims description 2
- 239000001361 adipic acid Substances 0.000 claims description 2
- 235000011037 adipic acid Nutrition 0.000 claims description 2
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 claims description 2
- OBETXYAYXDNJHR-UHFFFAOYSA-N alpha-ethylcaproic acid Natural products CCCCC(CC)C(O)=O OBETXYAYXDNJHR-UHFFFAOYSA-N 0.000 claims description 2
- 229960000212 aminophenazone Drugs 0.000 claims description 2
- PNLLQYWFZSKTKY-UHFFFAOYSA-N butanedioic acid;piperidin-1-yl-[3-[[4-(2-propan-2-yloxyphenyl)piperazin-1-yl]methyl]phenyl]methanone Chemical compound OC(=O)CCC(O)=O.CC(C)OC1=CC=CC=C1N1CCN(CC=2C=C(C=CC=2)C(=O)N2CCCCC2)CC1.CC(C)OC1=CC=CC=C1N1CCN(CC=2C=C(C=CC=2)C(=O)N2CCCCC2)CC1 PNLLQYWFZSKTKY-UHFFFAOYSA-N 0.000 claims description 2
- LSPHULWDVZXLIL-QUBYGPBYSA-N camphoric acid Chemical compound CC1(C)[C@H](C(O)=O)CC[C@]1(C)C(O)=O LSPHULWDVZXLIL-QUBYGPBYSA-N 0.000 claims description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 2
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Natural products NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 claims description 2
- 229940104302 cytosine Drugs 0.000 claims description 2
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 claims description 2
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 claims description 2
- HOXINJBQVZWYGZ-UHFFFAOYSA-N fenbutatin oxide Chemical compound C=1C=CC=CC=1C(C)(C)C[Sn](O[Sn](CC(C)(C)C=1C=CC=CC=1)(CC(C)(C)C=1C=CC=CC=1)CC(C)(C)C=1C=CC=CC=1)(CC(C)(C)C=1C=CC=CC=1)CC(C)(C)C1=CC=CC=C1 HOXINJBQVZWYGZ-UHFFFAOYSA-N 0.000 claims description 2
- KSEBMYQBYZTDHS-HWKANZROSA-N ferulic acid Chemical compound COC1=CC(\C=C\C(O)=O)=CC=C1O KSEBMYQBYZTDHS-HWKANZROSA-N 0.000 claims description 2
- KSEBMYQBYZTDHS-UHFFFAOYSA-N ferulic acid Natural products COC1=CC(C=CC(O)=O)=CC=C1O KSEBMYQBYZTDHS-UHFFFAOYSA-N 0.000 claims description 2
- 235000019253 formic acid Nutrition 0.000 claims description 2
- XLYMOEINVGRTEX-UHFFFAOYSA-N fumaric acid monoethyl ester Natural products CCOC(=O)C=CC(O)=O XLYMOEINVGRTEX-UHFFFAOYSA-N 0.000 claims description 2
- 235000010299 hexamethylene tetramine Nutrition 0.000 claims description 2
- VKYKSIONXSXAKP-UHFFFAOYSA-N hexamethylenetetramine Chemical compound C1N(C2)CN3CN1CN2C3 VKYKSIONXSXAKP-UHFFFAOYSA-N 0.000 claims description 2
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 claims description 2
- MTNDZQHUAFNZQY-UHFFFAOYSA-N imidazoline Chemical compound C1CN=CN1 MTNDZQHUAFNZQY-UHFFFAOYSA-N 0.000 claims description 2
- 239000003617 indole-3-acetic acid Substances 0.000 claims description 2
- 229940125396 insulin Drugs 0.000 claims description 2
- ZKZFPRUSWCYSGT-UHFFFAOYSA-N mazapertine Chemical compound CC(C)OC1=CC=CC=C1N1CCN(CC=2C=C(C=CC=2)C(=O)N2CCCCC2)CC1 ZKZFPRUSWCYSGT-UHFFFAOYSA-N 0.000 claims description 2
- 229950004476 mazapertine Drugs 0.000 claims description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 2
- XLSZMDLNRCVEIJ-UHFFFAOYSA-N methylimidazole Natural products CC1=CNC=N1 XLSZMDLNRCVEIJ-UHFFFAOYSA-N 0.000 claims description 2
- DUWWHGPELOTTOE-UHFFFAOYSA-N n-(5-chloro-2,4-dimethoxyphenyl)-3-oxobutanamide Chemical compound COC1=CC(OC)=C(NC(=O)CC(C)=O)C=C1Cl DUWWHGPELOTTOE-UHFFFAOYSA-N 0.000 claims description 2
- ABMFBCRYHDZLRD-UHFFFAOYSA-N naphthalene-1,4-dicarboxylic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=C(C(O)=O)C2=C1 ABMFBCRYHDZLRD-UHFFFAOYSA-N 0.000 claims description 2
- JRNGUTKWMSBIBF-UHFFFAOYSA-N naphthalene-2,3-diol Chemical compound C1=CC=C2C=C(O)C(O)=CC2=C1 JRNGUTKWMSBIBF-UHFFFAOYSA-N 0.000 claims description 2
- GEVPUGOOGXGPIO-UHFFFAOYSA-N oxalic acid;dihydrate Chemical compound O.O.OC(=O)C(O)=O GEVPUGOOGXGPIO-UHFFFAOYSA-N 0.000 claims description 2
- UQGPCEVQKLOLLM-UHFFFAOYSA-N pentaneperoxoic acid Chemical compound CCCCC(=O)OO UQGPCEVQKLOLLM-UHFFFAOYSA-N 0.000 claims description 2
- HUPQYPMULVBQDL-UHFFFAOYSA-N pentanoic acid Chemical compound CCCCC(O)=O.CCCCC(O)=O HUPQYPMULVBQDL-UHFFFAOYSA-N 0.000 claims description 2
- 229950001518 raclopride Drugs 0.000 claims description 2
- 239000004334 sorbic acid Substances 0.000 claims description 2
- 235000010199 sorbic acid Nutrition 0.000 claims description 2
- 229940075582 sorbic acid Drugs 0.000 claims description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 claims description 2
- 229960002135 sulfadimidine Drugs 0.000 claims description 2
- ASWVTGNCAZCNNR-UHFFFAOYSA-N sulfamethazine Chemical compound CC1=CC(C)=NC(NS(=O)(=O)C=2C=CC(N)=CC=2)=N1 ASWVTGNCAZCNNR-UHFFFAOYSA-N 0.000 claims description 2
- 229910052717 sulfur Inorganic materials 0.000 claims description 2
- UVZICZIVKIMRNE-UHFFFAOYSA-N thiodiacetic acid Chemical compound OC(=O)CSCC(O)=O UVZICZIVKIMRNE-UHFFFAOYSA-N 0.000 claims description 2
- QERYCTSHXKAMIS-UHFFFAOYSA-N thiophene-2-carboxylic acid Chemical compound OC(=O)C1=CC=CS1 QERYCTSHXKAMIS-UHFFFAOYSA-N 0.000 claims description 2
- 238000002441 X-ray diffraction Methods 0.000 claims 220
- 230000007704 transition Effects 0.000 claims 22
- 125000004093 cyano group Chemical group *C#N 0.000 claims 15
- KUCOHFSKRZZVRO-UHFFFAOYSA-N terephthalaldehyde Chemical compound O=CC1=CC=C(C=O)C=C1 KUCOHFSKRZZVRO-UHFFFAOYSA-N 0.000 claims 9
- SULYEHHGGXARJS-UHFFFAOYSA-N 2',4'-dihydroxyacetophenone Chemical compound CC(=O)C1=CC=C(O)C=C1O SULYEHHGGXARJS-UHFFFAOYSA-N 0.000 claims 2
- QGBNDEUSOBSRDE-UHDJGPCESA-N (E)-3-phenylprop-2-enoic acid hydrate Chemical compound O.OC(=O)\C=C\C1=CC=CC=C1 QGBNDEUSOBSRDE-UHDJGPCESA-N 0.000 claims 1
- DYMCFZJBNPRRHQ-UHFFFAOYSA-N 2-[4-[[4-amino-6-[4-(carboxymethyl)anilino]-1,3,5-triazin-2-yl]amino]phenyl]acetic acid Chemical compound N=1C(NC=2C=CC(CC(O)=O)=CC=2)=NC(N)=NC=1NC1=CC=C(CC(O)=O)C=C1 DYMCFZJBNPRRHQ-UHFFFAOYSA-N 0.000 claims 1
- ZKIJPLJGLBNQIV-UHFFFAOYSA-N 2-n,4-n-bis(3-fluorophenyl)-1,3,5-triazine-2,4,6-triamine Chemical compound N=1C(NC=2C=C(F)C=CC=2)=NC(N)=NC=1NC1=CC=CC(F)=C1 ZKIJPLJGLBNQIV-UHFFFAOYSA-N 0.000 claims 1
- YXFLBRCWYYGDMD-UHFFFAOYSA-N 2-n,4-n-bis[3-(trifluoromethyl)phenyl]-1,3,5-triazine-2,4,6-triamine Chemical compound N=1C(NC=2C=C(C=CC=2)C(F)(F)F)=NC(N)=NC=1NC1=CC=CC(C(F)(F)F)=C1 YXFLBRCWYYGDMD-UHFFFAOYSA-N 0.000 claims 1
- AHLVXBLYNKLIGD-UHFFFAOYSA-N 2-n,4-n-diphenyl-1,3,5-triazine-2,4,6-triamine Chemical compound N=1C(NC=2C=CC=CC=2)=NC(N)=NC=1NC1=CC=CC=C1 AHLVXBLYNKLIGD-UHFFFAOYSA-N 0.000 claims 1
- 229940090248 4-hydroxybenzoic acid Drugs 0.000 claims 1
- COBZZWPZKRRPEN-UHFFFAOYSA-N 9-ethyl-3h-purin-6-one Chemical compound N1C=NC(=O)C2=C1N(CC)C=N2 COBZZWPZKRRPEN-UHFFFAOYSA-N 0.000 claims 1
- 125000003158 alcohol group Chemical group 0.000 claims 1
- 125000001033 ether group Chemical group 0.000 claims 1
- 229960002598 fumaric acid Drugs 0.000 claims 1
- 229960004275 glycolic acid Drugs 0.000 claims 1
- 238000010348 incorporation Methods 0.000 claims 1
- 229940098895 maleic acid Drugs 0.000 claims 1
- KLAKIAVEMQMVBT-UHFFFAOYSA-N p-hydroxy-phenacyl alcohol Natural products OCC(=O)C1=CC=C(O)C=C1 KLAKIAVEMQMVBT-UHFFFAOYSA-N 0.000 claims 1
- 229960005141 piperazine Drugs 0.000 claims 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 claims 1
- MCJGNVYPOGVAJF-UHFFFAOYSA-O quinolin-8-yloxidanium Chemical compound C1=C[NH+]=C2C(O)=CC=CC2=C1 MCJGNVYPOGVAJF-UHFFFAOYSA-O 0.000 claims 1
- 125000000565 sulfonamide group Chemical group 0.000 claims 1
- 229940045136 urea Drugs 0.000 claims 1
- 150000001412 amines Chemical class 0.000 abstract description 2
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 abstract 3
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 abstract 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 abstract 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 abstract 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 abstract 1
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 abstract 1
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 abstract 1
- 125000002816 methylsulfanyl group Chemical group [H]C([H])([H])S[*] 0.000 abstract 1
- 150000003457 sulfones Chemical class 0.000 abstract 1
- 125000003396 thiol group Chemical class [H]S* 0.000 abstract 1
- 235000002639 sodium chloride Nutrition 0.000 description 54
- 230000015572 biosynthetic process Effects 0.000 description 18
- 230000008901 benefit Effects 0.000 description 12
- 239000002904 solvent Substances 0.000 description 12
- 230000002829 reductive effect Effects 0.000 description 11
- 239000000126 substance Substances 0.000 description 11
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 9
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 9
- 238000009472 formulation Methods 0.000 description 9
- 239000011230 binding agent Substances 0.000 description 8
- 239000003795 chemical substances by application Substances 0.000 description 8
- 239000012458 free base Substances 0.000 description 8
- 239000003826 tablet Substances 0.000 description 8
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 7
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 7
- 229920002472 Starch Polymers 0.000 description 7
- 239000002585 base Substances 0.000 description 7
- 229910052791 calcium Inorganic materials 0.000 description 7
- 229960005069 calcium Drugs 0.000 description 7
- 239000011575 calcium Substances 0.000 description 7
- 239000008380 degradant Substances 0.000 description 7
- 239000011734 sodium Substances 0.000 description 7
- 229910052708 sodium Inorganic materials 0.000 description 7
- 235000019698 starch Nutrition 0.000 description 7
- 239000000080 wetting agent Substances 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 6
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 6
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 6
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 6
- 239000007884 disintegrant Substances 0.000 description 6
- 239000012530 fluid Substances 0.000 description 6
- 229910052744 lithium Inorganic materials 0.000 description 6
- 239000011777 magnesium Substances 0.000 description 6
- 229910052749 magnesium Inorganic materials 0.000 description 6
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 6
- 239000008108 microcrystalline cellulose Substances 0.000 description 6
- 229940016286 microcrystalline cellulose Drugs 0.000 description 6
- 150000002924 oxiranes Chemical class 0.000 description 6
- 230000036470 plasma concentration Effects 0.000 description 6
- 229910052700 potassium Inorganic materials 0.000 description 6
- 239000011591 potassium Substances 0.000 description 6
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 5
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 5
- 238000010521 absorption reaction Methods 0.000 description 5
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 5
- 235000014113 dietary fatty acids Nutrition 0.000 description 5
- 239000000194 fatty acid Substances 0.000 description 5
- 229930195729 fatty acid Natural products 0.000 description 5
- 150000002500 ions Chemical class 0.000 description 5
- 230000000670 limiting effect Effects 0.000 description 5
- 238000012216 screening Methods 0.000 description 5
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 5
- 239000000454 talc Substances 0.000 description 5
- 229910052623 talc Inorganic materials 0.000 description 5
- 229940033134 talc Drugs 0.000 description 5
- 235000012222 talc Nutrition 0.000 description 5
- 230000001225 therapeutic effect Effects 0.000 description 5
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 4
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Chemical compound CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 4
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 239000000853 adhesive Substances 0.000 description 4
- 230000001070 adhesive effect Effects 0.000 description 4
- 229920001400 block copolymer Polymers 0.000 description 4
- 235000010980 cellulose Nutrition 0.000 description 4
- 229920002678 cellulose Polymers 0.000 description 4
- 238000007906 compression Methods 0.000 description 4
- 230000006835 compression Effects 0.000 description 4
- 239000006185 dispersion Substances 0.000 description 4
- 239000002552 dosage form Substances 0.000 description 4
- 239000007789 gas Substances 0.000 description 4
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 4
- 229960001375 lactose Drugs 0.000 description 4
- 239000008101 lactose Substances 0.000 description 4
- 239000000314 lubricant Substances 0.000 description 4
- 229910052751 metal Inorganic materials 0.000 description 4
- 239000002184 metal Substances 0.000 description 4
- 230000000704 physical effect Effects 0.000 description 4
- 229920000642 polymer Polymers 0.000 description 4
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 4
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 230000004044 response Effects 0.000 description 4
- 241000894007 species Species 0.000 description 4
- 238000000859 sublimation Methods 0.000 description 4
- 229920002785 Croscarmellose sodium Polymers 0.000 description 3
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 3
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 3
- 235000010443 alginic acid Nutrition 0.000 description 3
- 229920000615 alginic acid Polymers 0.000 description 3
- 125000003277 amino group Chemical group 0.000 description 3
- 230000000181 anti-adherent effect Effects 0.000 description 3
- 239000003911 antiadherent Substances 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- 239000001913 cellulose Substances 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 238000006243 chemical reaction Methods 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 229960001681 croscarmellose sodium Drugs 0.000 description 3
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 3
- 238000010586 diagram Methods 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 230000002496 gastric effect Effects 0.000 description 3
- 150000004677 hydrates Chemical class 0.000 description 3
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 3
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 3
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 3
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 3
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 3
- 230000000968 intestinal effect Effects 0.000 description 3
- 238000003801 milling Methods 0.000 description 3
- 238000012856 packing Methods 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 238000012545 processing Methods 0.000 description 3
- 230000003381 solubilizing effect Effects 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 229940032147 starch Drugs 0.000 description 3
- 239000007916 tablet composition Substances 0.000 description 3
- 238000001757 thermogravimetry curve Methods 0.000 description 3
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- XMIIGOLPHOKFCH-UHFFFAOYSA-N 3-phenylpropionic acid Chemical compound OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 2
- WSVLPVUVIUVCRA-KPKNDVKVSA-N Alpha-lactose monohydrate Chemical compound O.O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O WSVLPVUVIUVCRA-KPKNDVKVSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical group OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- KWIUHFFTVRNATP-UHFFFAOYSA-O N,N,N-trimethylglycinium Chemical compound C[N+](C)(C)CC(O)=O KWIUHFFTVRNATP-UHFFFAOYSA-O 0.000 description 2
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- 238000001069 Raman spectroscopy Methods 0.000 description 2
- BCKXLBQYZLBQEK-KVVVOXFISA-M Sodium oleate Chemical compound [Na+].CCCCCCCC\C=C/CCCCCCCC([O-])=O BCKXLBQYZLBQEK-KVVVOXFISA-M 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- 102400000336 Thyrotropin-releasing hormone Human genes 0.000 description 2
- 101800004623 Thyrotropin-releasing hormone Proteins 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 239000000783 alginic acid Substances 0.000 description 2
- 229960001126 alginic acid Drugs 0.000 description 2
- 150000004781 alginic acids Chemical class 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 150000003927 aminopyridines Chemical class 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 239000012296 anti-solvent Substances 0.000 description 2
- YFGHCGITMMYXAQ-LJQANCHMSA-N armodafinil Chemical compound C=1C=CC=CC=1C([S@](=O)CC(=O)N)C1=CC=CC=C1 YFGHCGITMMYXAQ-LJQANCHMSA-N 0.000 description 2
- 229960004823 armodafinil Drugs 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- 235000012216 bentonite Nutrition 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 239000001569 carbon dioxide Substances 0.000 description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 239000004359 castor oil Substances 0.000 description 2
- 235000019438 castor oil Nutrition 0.000 description 2
- 238000002288 cocrystallisation Methods 0.000 description 2
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 2
- 239000002178 crystalline material Substances 0.000 description 2
- 239000000539 dimer Substances 0.000 description 2
- 239000012738 dissolution medium Substances 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 230000008030 elimination Effects 0.000 description 2
- 238000003379 elimination reaction Methods 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 229960001031 glucose Drugs 0.000 description 2
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 2
- 238000005469 granulation Methods 0.000 description 2
- 230000003179 granulation Effects 0.000 description 2
- 229910052736 halogen Inorganic materials 0.000 description 2
- 150000002367 halogens Chemical class 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 239000002608 ionic liquid Substances 0.000 description 2
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 2
- QQVIHTHCMHWDBS-UHFFFAOYSA-N isophthalic acid Chemical compound OC(=O)C1=CC=CC(C(O)=O)=C1 QQVIHTHCMHWDBS-UHFFFAOYSA-N 0.000 description 2
- 235000014655 lactic acid Nutrition 0.000 description 2
- 239000004310 lactic acid Substances 0.000 description 2
- 229960001021 lactose monohydrate Drugs 0.000 description 2
- 230000002045 lasting effect Effects 0.000 description 2
- 238000005461 lubrication Methods 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- 229920000609 methyl cellulose Polymers 0.000 description 2
- 239000001923 methylcellulose Substances 0.000 description 2
- 235000010981 methylcellulose Nutrition 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 150000004682 monohydrates Chemical class 0.000 description 2
- DNIAPMSPPWPWGF-UHFFFAOYSA-N monopropylene glycol Natural products CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 2
- TXXHDPDFNKHHGW-UHFFFAOYSA-N muconic acid Chemical group OC(=O)C=CC=CC(O)=O TXXHDPDFNKHHGW-UHFFFAOYSA-N 0.000 description 2
- 229910017604 nitric acid Inorganic materials 0.000 description 2
- 229910052755 nonmetal Inorganic materials 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 2
- 229920000053 polysorbate 80 Polymers 0.000 description 2
- 229940069328 povidone Drugs 0.000 description 2
- 235000019814 powdered cellulose Nutrition 0.000 description 2
- 229920003124 powdered cellulose Polymers 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- XNSAINXGIQZQOO-SRVKXCTJSA-N protirelin Chemical compound NC(=O)[C@@H]1CCCN1C(=O)[C@@H](NC(=O)[C@H]1NC(=O)CC1)CC1=CN=CN1 XNSAINXGIQZQOO-SRVKXCTJSA-N 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- RYYKJJJTJZKILX-UHFFFAOYSA-M sodium octadecanoate Chemical class [Na+].CCCCCCCCCCCCCCCCCC([O-])=O RYYKJJJTJZKILX-UHFFFAOYSA-M 0.000 description 2
- 239000007909 solid dosage form Substances 0.000 description 2
- 238000001179 sorption measurement Methods 0.000 description 2
- 230000003068 static effect Effects 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- YAPQBXQYLJRXSA-UHFFFAOYSA-N theobromine Chemical compound CN1C(=O)NC(=O)C2=C1N=CN2C YAPQBXQYLJRXSA-UHFFFAOYSA-N 0.000 description 2
- 238000002411 thermogravimetry Methods 0.000 description 2
- 229940034199 thyrotropin-releasing hormone Drugs 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical class OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 1
- SPFMQWBKVUQXJV-BTVCFUMJSA-N (2r,3s,4r,5r)-2,3,4,5,6-pentahydroxyhexanal;hydrate Chemical compound O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O SPFMQWBKVUQXJV-BTVCFUMJSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- METKIMKYRPQLGS-GFCCVEGCSA-N (R)-atenolol Chemical compound CC(C)NC[C@@H](O)COC1=CC=C(CC(N)=O)C=C1 METKIMKYRPQLGS-GFCCVEGCSA-N 0.000 description 1
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 1
- ICLYJLBTOGPLMC-KVVVOXFISA-N (z)-octadec-9-enoate;tris(2-hydroxyethyl)azanium Chemical compound OCCN(CCO)CCO.CCCCCCCC\C=C/CCCCCCCC(O)=O ICLYJLBTOGPLMC-KVVVOXFISA-N 0.000 description 1
- ZORQXIQZAOLNGE-UHFFFAOYSA-N 1,1-difluorocyclohexane Chemical compound FC1(F)CCCCC1 ZORQXIQZAOLNGE-UHFFFAOYSA-N 0.000 description 1
- IWUCXVSUMQZMFG-RGDLXGNYSA-N 1-[(2s,3s,4r,5s)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,2,4-triazole-3-carboxamide Chemical compound N1=C(C(=O)N)N=CN1[C@@H]1[C@@H](O)[C@@H](O)[C@H](CO)O1 IWUCXVSUMQZMFG-RGDLXGNYSA-N 0.000 description 1
- RJCLZEKYUQKDAL-WNQIDUERSA-M 1-ethyl-3-methylimidazol-3-ium;(2s)-2-hydroxypropanoate Chemical compound C[C@H](O)C([O-])=O.CCN1C=C[N+](C)=C1 RJCLZEKYUQKDAL-WNQIDUERSA-M 0.000 description 1
- AMMPLVWPWSYRDR-UHFFFAOYSA-N 1-methylbicyclo[2.2.2]oct-2-ene-4-carboxylic acid Chemical compound C1CC2(C(O)=O)CCC1(C)C=C2 AMMPLVWPWSYRDR-UHFFFAOYSA-N 0.000 description 1
- YGTUPRIZNBMOFV-UHFFFAOYSA-N 2-(4-hydroxybenzoyl)benzoic acid Chemical compound OC(=O)C1=CC=CC=C1C(=O)C1=CC=C(O)C=C1 YGTUPRIZNBMOFV-UHFFFAOYSA-N 0.000 description 1
- KUXGUCNZFCVULO-UHFFFAOYSA-N 2-(4-nonylphenoxy)ethanol Chemical compound CCCCCCCCCC1=CC=C(OCCO)C=C1 KUXGUCNZFCVULO-UHFFFAOYSA-N 0.000 description 1
- NJMKKMGDQNAJBH-UHFFFAOYSA-N 2-N,4-N-bis(2-fluorophenyl)-1,3,5-triazine-2,4,6-triamine Chemical compound FC1=C(C=CC=C1)NC1=NC(=NC(=N1)NC1=C(C=CC=C1)F)N NJMKKMGDQNAJBH-UHFFFAOYSA-N 0.000 description 1
- SBWQNDUROAZSDH-UHFFFAOYSA-N 2-N,4-N-bis[2-(trifluoromethyl)phenyl]-1,3,5-triazine-2,4,6-triamine Chemical compound FC(F)(F)C1=C(C=CC=C1)NC1=NC(=NC(=N1)NC1=C(C=CC=C1)C(F)(F)F)N SBWQNDUROAZSDH-UHFFFAOYSA-N 0.000 description 1
- CABMTIJINOIHOD-UHFFFAOYSA-N 2-[4-methyl-5-oxo-4-(propan-2-yl)-4,5-dihydro-1H-imidazol-2-yl]quinoline-3-carboxylic acid Chemical compound N1C(=O)C(C(C)C)(C)N=C1C1=NC2=CC=CC=C2C=C1C(O)=O CABMTIJINOIHOD-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- RPVDWHOGJOXVJK-UHFFFAOYSA-N 2-ethyl-3,7-dihydropurin-6-one Chemical compound O=C1NC(CC)=NC2=C1NC=N2 RPVDWHOGJOXVJK-UHFFFAOYSA-N 0.000 description 1
- UPHOPMSGKZNELG-UHFFFAOYSA-N 2-hydroxynaphthalene-1-carboxylic acid Chemical group C1=CC=C2C(C(=O)O)=C(O)C=CC2=C1 UPHOPMSGKZNELG-UHFFFAOYSA-N 0.000 description 1
- CYYPGGXVPOQRKO-UHFFFAOYSA-N 2-n,4-n-bis(4-bromophenyl)-1,3,5-triazine-2,4,6-triamine Chemical compound N=1C(NC=2C=CC(Br)=CC=2)=NC(N)=NC=1NC1=CC=C(Br)C=C1 CYYPGGXVPOQRKO-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- CDOUZKKFHVEKRI-UHFFFAOYSA-N 3-bromo-n-[(prop-2-enoylamino)methyl]propanamide Chemical compound BrCCC(=O)NCNC(=O)C=C CDOUZKKFHVEKRI-UHFFFAOYSA-N 0.000 description 1
- ZRPLANDPDWYOMZ-UHFFFAOYSA-N 3-cyclopentylpropionic acid Chemical compound OC(=O)CCC1CCCC1 ZRPLANDPDWYOMZ-UHFFFAOYSA-N 0.000 description 1
- JSTYJLJRZTZPPL-UHFFFAOYSA-N 4-nitrobenzoic acid;quinolin-8-ol Chemical compound C1=CN=C2C(O)=CC=CC2=C1.OC(=O)C1=CC=C([N+]([O-])=O)C=C1 JSTYJLJRZTZPPL-UHFFFAOYSA-N 0.000 description 1
- XZIIFPSPUDAGJM-UHFFFAOYSA-N 6-chloro-2-n,2-n-diethylpyrimidine-2,4-diamine Chemical compound CCN(CC)C1=NC(N)=CC(Cl)=N1 XZIIFPSPUDAGJM-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 229920000856 Amylose Polymers 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- PTHCMJGKKRQCBF-UHFFFAOYSA-N Cellulose, microcrystalline Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC)C(CO)O1 PTHCMJGKKRQCBF-UHFFFAOYSA-N 0.000 description 1
- 240000008886 Ceratonia siliqua Species 0.000 description 1
- 235000013912 Ceratonia siliqua Nutrition 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 241000694440 Colpidium aqueous Species 0.000 description 1
- 244000303965 Cyamopsis psoralioides Species 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Chemical group OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 229920002907 Guar gum Polymers 0.000 description 1
- SQUHHTBVTRBESD-UHFFFAOYSA-N Hexa-Ac-myo-Inositol Natural products CC(=O)OC1C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C1OC(C)=O SQUHHTBVTRBESD-UHFFFAOYSA-N 0.000 description 1
- 238000004566 IR spectroscopy Methods 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- HZQDCMWJEBCWBR-UUOKFMHZSA-N Mizoribine Chemical compound OC1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 HZQDCMWJEBCWBR-UUOKFMHZSA-N 0.000 description 1
- 229920000881 Modified starch Polymers 0.000 description 1
- TXXHDPDFNKHHGW-CCAGOZQPSA-N Muconic acid Chemical group OC(=O)\C=C/C=C\C(O)=O TXXHDPDFNKHHGW-CCAGOZQPSA-N 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 108010019160 Pancreatin Proteins 0.000 description 1
- GMZVRMREEHBGGF-UHFFFAOYSA-N Piracetam Chemical compound NC(=O)CN1CCCC1=O GMZVRMREEHBGGF-UHFFFAOYSA-N 0.000 description 1
- RVGRUAULSDPKGF-UHFFFAOYSA-N Poloxamer Chemical compound C1CO1.CC1CO1 RVGRUAULSDPKGF-UHFFFAOYSA-N 0.000 description 1
- 229920002511 Poloxamer 237 Polymers 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 229920003081 Povidone K 30 Polymers 0.000 description 1
- GOOHAUXETOMSMM-UHFFFAOYSA-N Propylene oxide Chemical group CC1CO1 GOOHAUXETOMSMM-UHFFFAOYSA-N 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 239000001744 Sodium fumarate Substances 0.000 description 1
- IYFATESGLOUGBX-YVNJGZBMSA-N Sorbitan monopalmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O IYFATESGLOUGBX-YVNJGZBMSA-N 0.000 description 1
- HVUMOYIDDBPOLL-XWVZOOPGSA-N Sorbitan monostearate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O HVUMOYIDDBPOLL-XWVZOOPGSA-N 0.000 description 1
- 235000015125 Sterculia urens Nutrition 0.000 description 1
- 240000001058 Sterculia urens Species 0.000 description 1
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 1
- 239000013504 Triton X-100 Substances 0.000 description 1
- 229920004890 Triton X-100 Polymers 0.000 description 1
- TVXBFESIOXBWNM-UHFFFAOYSA-N Xylitol Natural products OCCC(O)C(O)C(O)CCO TVXBFESIOXBWNM-UHFFFAOYSA-N 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000005215 alkyl ethers Chemical class 0.000 description 1
- 238000005275 alloying Methods 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- SNAAJJQQZSMGQD-UHFFFAOYSA-N aluminum magnesium Chemical class [Mg].[Al] SNAAJJQQZSMGQD-UHFFFAOYSA-N 0.000 description 1
- 229960004977 anhydrous lactose Drugs 0.000 description 1
- 239000003945 anionic surfactant Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 229940027983 antiseptic and disinfectant quaternary ammonium compound Drugs 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229960003121 arginine Drugs 0.000 description 1
- BHIAIPWSVYSKJS-UHFFFAOYSA-N arotinolol Chemical compound S1C(SCC(O)CNC(C)(C)C)=NC(C=2SC(=CC=2)C(N)=O)=C1 BHIAIPWSVYSKJS-UHFFFAOYSA-N 0.000 description 1
- 229950010731 arotinolol Drugs 0.000 description 1
- 229960002274 atenolol Drugs 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 229960001950 benzethonium chloride Drugs 0.000 description 1
- UREZNYTWGJKWBI-UHFFFAOYSA-M benzethonium chloride Chemical compound [Cl-].C1=CC(C(C)(C)CC(C)(C)C)=CC=C1OCCOCC[N+](C)(C)CC1=CC=CC=C1 UREZNYTWGJKWBI-UHFFFAOYSA-M 0.000 description 1
- HQYMUNCIMNFLDT-UHFFFAOYSA-N benzyl n-(2-amino-2-oxoethyl)carbamate Chemical compound NC(=O)CNC(=O)OCC1=CC=CC=C1 HQYMUNCIMNFLDT-UHFFFAOYSA-N 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 1
- 239000004327 boric acid Substances 0.000 description 1
- MSYKRHVOOPPJKU-BDAKNGLRSA-N brivaracetam Chemical compound CCC[C@H]1CN([C@@H](CC)C(N)=O)C(=O)C1 MSYKRHVOOPPJKU-BDAKNGLRSA-N 0.000 description 1
- 229960002161 brivaracetam Drugs 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- XAAHAAMILDNBPS-UHFFFAOYSA-L calcium hydrogenphosphate dihydrate Chemical compound O.O.[Ca+2].OP([O-])([O-])=O XAAHAAMILDNBPS-UHFFFAOYSA-L 0.000 description 1
- PASHVRUKOFIRIK-UHFFFAOYSA-L calcium sulfate dihydrate Chemical compound O.O.[Ca+2].[O-]S([O-])(=O)=O PASHVRUKOFIRIK-UHFFFAOYSA-L 0.000 description 1
- UBWYRXFZPXBISJ-UHFFFAOYSA-L calcium;2-hydroxypropanoate;trihydrate Chemical compound O.O.O.[Ca+2].CC(O)C([O-])=O.CC(O)C([O-])=O UBWYRXFZPXBISJ-UHFFFAOYSA-L 0.000 description 1
- ZHZFKLKREFECML-UHFFFAOYSA-L calcium;sulfate;hydrate Chemical compound O.[Ca+2].[O-]S([O-])(=O)=O ZHZFKLKREFECML-UHFFFAOYSA-L 0.000 description 1
- 239000007894 caplet Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- NWPJLRSCSQHPJV-UHFFFAOYSA-N carpipramine Chemical compound C1CN(CCCN2C3=CC=CC=C3CCC3=CC=CC=C32)CCC1(C(=O)N)N1CCCCC1 NWPJLRSCSQHPJV-UHFFFAOYSA-N 0.000 description 1
- 229960000700 carpipramine Drugs 0.000 description 1
- SRZNHPXWXCNNDU-RHBCBLIFSA-N cefotetan Chemical compound N([C@]1(OC)C(N2C(=C(CSC=3N(N=NN=3)C)CS[C@@H]21)C(O)=O)=O)C(=O)C1SC(=C(C(N)=O)C(O)=O)S1 SRZNHPXWXCNNDU-RHBCBLIFSA-N 0.000 description 1
- 229960005495 cefotetan Drugs 0.000 description 1
- SYLKGLMBLAAGSC-QLVMHMETSA-N cefsulodin Chemical compound C1=CC(C(=O)N)=CC=[N+]1CC1=C(C([O-])=O)N2C(=O)[C@@H](NC(=O)[C@@H](C=3C=CC=CC=3)S(O)(=O)=O)[C@H]2SC1 SYLKGLMBLAAGSC-QLVMHMETSA-N 0.000 description 1
- 229960003202 cefsulodin Drugs 0.000 description 1
- 235000013339 cereals Nutrition 0.000 description 1
- 229960001927 cetylpyridinium chloride Drugs 0.000 description 1
- NFCRBQADEGXVDL-UHFFFAOYSA-M cetylpyridinium chloride monohydrate Chemical compound O.[Cl-].CCCCCCCCCCCCCCCC[N+]1=CC=CC=C1 NFCRBQADEGXVDL-UHFFFAOYSA-M 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000010668 complexation reaction Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 229940099112 cornstarch Drugs 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 229960000913 crospovidone Drugs 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 230000001351 cycling effect Effects 0.000 description 1
- HXGBXQDTNZMWGS-RUZDIDTESA-N darifenacin Chemical compound C=1C=CC=CC=1C([C@H]1CN(CCC=2C=C3CCOC3=CC=2)CC1)(C(=O)N)C1=CC=CC=C1 HXGBXQDTNZMWGS-RUZDIDTESA-N 0.000 description 1
- 229960002677 darifenacin Drugs 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 238000000280 densification Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 229940096516 dextrates Drugs 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 229960000673 dextrose monohydrate Drugs 0.000 description 1
- 229940061607 dibasic sodium phosphate Drugs 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 238000000113 differential scanning calorimetry Methods 0.000 description 1
- 238000009792 diffusion process Methods 0.000 description 1
- 150000004683 dihydrates Chemical class 0.000 description 1
- FSBVERYRVPGNGG-UHFFFAOYSA-N dimagnesium dioxido-bis[[oxido(oxo)silyl]oxy]silane hydrate Chemical compound O.[Mg+2].[Mg+2].[O-][Si](=O)O[Si]([O-])([O-])O[Si]([O-])=O FSBVERYRVPGNGG-UHFFFAOYSA-N 0.000 description 1
- 235000019329 dioctyl sodium sulphosuccinate Nutrition 0.000 description 1
- MSJMDZAOKORVFC-SEPHDYHBSA-L disodium fumarate Chemical compound [Na+].[Na+].[O-]C(=O)\C=C\C([O-])=O MSJMDZAOKORVFC-SEPHDYHBSA-L 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- LLRANSBEYQZKFY-UHFFFAOYSA-N dodecanoic acid;propane-1,2-diol Chemical compound CC(O)CO.CCCCCCCCCCCC(O)=O LLRANSBEYQZKFY-UHFFFAOYSA-N 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical group CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- AFAXGSQYZLGZPG-UHFFFAOYSA-N ethanedisulfonic acid Chemical compound OS(=O)(=O)CCS(O)(=O)=O AFAXGSQYZLGZPG-UHFFFAOYSA-N 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 229940012017 ethylenediamine Drugs 0.000 description 1
- CKSJXOVLXUMMFF-UHFFFAOYSA-N exalamide Chemical compound CCCCCCOC1=CC=CC=C1C(N)=O CKSJXOVLXUMMFF-UHFFFAOYSA-N 0.000 description 1
- 229950010333 exalamide Drugs 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- WAAPEIZFCHNLKK-PELKAZGASA-N fidarestat Chemical compound C([C@@H](OC1=CC=C(F)C=C11)C(=O)N)[C@@]21NC(=O)NC2=O WAAPEIZFCHNLKK-PELKAZGASA-N 0.000 description 1
- 229950007256 fidarestat Drugs 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- SIBNYOSJIXCDRI-SECBINFHSA-N frovatriptan Chemical compound C1=C(C(N)=O)[CH]C2=C(C[C@H](NC)CC3)C3=NC2=C1 SIBNYOSJIXCDRI-SECBINFHSA-N 0.000 description 1
- 229960002284 frovatriptan Drugs 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 229940083124 ganglion-blocking antiadrenergic secondary and tertiary amines Drugs 0.000 description 1
- 238000004817 gas chromatography Methods 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000000174 gluconic acid Chemical group 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 125000003976 glyceryl group Chemical group [H]C([*])([H])C(O[H])([H])C(O[H])([H])[H] 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 239000000665 guar gum Substances 0.000 description 1
- 235000010417 guar gum Nutrition 0.000 description 1
- 229960002154 guar gum Drugs 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 229960002885 histidine Drugs 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- 230000036571 hydration Effects 0.000 description 1
- 238000006703 hydration reaction Methods 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- 239000008172 hydrogenated vegetable oil Substances 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 229960000367 inositol Drugs 0.000 description 1
- CDAISMWEOUEBRE-GPIVLXJGSA-N inositol Chemical compound O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@H](O)[C@@H]1O CDAISMWEOUEBRE-GPIVLXJGSA-N 0.000 description 1
- 238000007689 inspection Methods 0.000 description 1
- 230000031891 intestinal absorption Effects 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- VHVPQPYKVGDNFY-ZPGVKDDISA-N itraconazole Chemical compound O=C1N(C(C)CC)N=CN1C1=CC=C(N2CCN(CC2)C=2C=CC(OC[C@@H]3O[C@](CN4N=CN=C4)(OC3)C=3C(=CC(Cl)=CC=3)Cl)=CC=2)C=C1 VHVPQPYKVGDNFY-ZPGVKDDISA-N 0.000 description 1
- 125000000468 ketone group Chemical group 0.000 description 1
- HPHUVLMMVZITSG-ZCFIWIBFSA-N levetiracetam Chemical compound CC[C@H](C(N)=O)N1CCCC1=O HPHUVLMMVZITSG-ZCFIWIBFSA-N 0.000 description 1
- 229960004002 levetiracetam Drugs 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 229960003646 lysine Drugs 0.000 description 1
- 229940037627 magnesium lauryl sulfate Drugs 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 235000019793 magnesium trisilicate Nutrition 0.000 description 1
- 229940099273 magnesium trisilicate Drugs 0.000 description 1
- 229910000386 magnesium trisilicate Inorganic materials 0.000 description 1
- HBNDBUATLJAUQM-UHFFFAOYSA-L magnesium;dodecyl sulfate Chemical compound [Mg+2].CCCCCCCCCCCCOS([O-])(=O)=O.CCCCCCCCCCCCOS([O-])(=O)=O HBNDBUATLJAUQM-UHFFFAOYSA-L 0.000 description 1
- 229960001855 mannitol Drugs 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- HEBKCHPVOIAQTA-UHFFFAOYSA-N meso ribitol Natural products OCC(O)C(O)C(O)CO HEBKCHPVOIAQTA-UHFFFAOYSA-N 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- GVXHSMAJJFVLGD-UHFFFAOYSA-N methyl 5-chloro-7-(trifluoromethyl)thieno[3,2-b]pyridine-3-carboxylate Chemical compound C1=C(Cl)N=C2C(C(=O)OC)=CSC2=C1C(F)(F)F GVXHSMAJJFVLGD-UHFFFAOYSA-N 0.000 description 1
- 229950000844 mizoribine Drugs 0.000 description 1
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 1
- 229940111688 monobasic potassium phosphate Drugs 0.000 description 1
- 235000019796 monopotassium phosphate Nutrition 0.000 description 1
- 239000004570 mortar (masonry) Substances 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229940073555 nonoxynol-10 Drugs 0.000 description 1
- 229920004918 nonoxynol-9 Polymers 0.000 description 1
- 229940087419 nonoxynol-9 Drugs 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 150000007523 nucleic acids Chemical class 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- IHLAQQPQKRMGSS-UHFFFAOYSA-N oxiracetam Chemical compound NC(=O)CN1CC(O)CC1=O IHLAQQPQKRMGSS-UHFFFAOYSA-N 0.000 description 1
- 229960001227 oxiracetam Drugs 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- 229940055695 pancreatin Drugs 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- XYJRXVWERLGGKC-UHFFFAOYSA-D pentacalcium;hydroxide;triphosphate Chemical compound [OH-].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O XYJRXVWERLGGKC-UHFFFAOYSA-D 0.000 description 1
- 150000004686 pentahydrates Chemical class 0.000 description 1
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N phenylbenzene Natural products C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 229960004526 piracetam Drugs 0.000 description 1
- 229920001983 poloxamer Polymers 0.000 description 1
- 229940044519 poloxamer 188 Drugs 0.000 description 1
- 229920001993 poloxamer 188 Polymers 0.000 description 1
- 229920000193 polymethacrylate Polymers 0.000 description 1
- 235000011185 polyoxyethylene (40) stearate Nutrition 0.000 description 1
- 239000001194 polyoxyethylene (40) stearate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 229940068977 polysorbate 20 Drugs 0.000 description 1
- 229940068968 polysorbate 80 Drugs 0.000 description 1
- 235000013809 polyvinylpolypyrrolidone Nutrition 0.000 description 1
- 229920000523 polyvinylpolypyrrolidone Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- GNSKLFRGEWLPPA-UHFFFAOYSA-M potassium dihydrogen phosphate Chemical compound [K+].OP(O)([O-])=O GNSKLFRGEWLPPA-UHFFFAOYSA-M 0.000 description 1
- WSHYKIAQCMIPTB-UHFFFAOYSA-M potassium;2-oxo-3-(3-oxo-1-phenylbutyl)chromen-4-olate Chemical compound [K+].[O-]C=1C2=CC=CC=C2OC(=O)C=1C(CC(=O)C)C1=CC=CC=C1 WSHYKIAQCMIPTB-UHFFFAOYSA-M 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- 238000003908 quality control method Methods 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 238000009790 rate-determining step (RDS) Methods 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- CDAISMWEOUEBRE-UHFFFAOYSA-N scyllo-inosotol Natural products OC1C(O)C(O)C(O)C(O)C1O CDAISMWEOUEBRE-UHFFFAOYSA-N 0.000 description 1
- PNCPYILNMDWPEY-QGZVFWFLSA-N silodosin Chemical compound N([C@@H](CC=1C=C(C=2N(CCCO)CCC=2C=1)C(N)=O)C)CCOC1=CC=CC=C1OCC(F)(F)F PNCPYILNMDWPEY-QGZVFWFLSA-N 0.000 description 1
- 229960004953 silodosin Drugs 0.000 description 1
- 238000004467 single crystal X-ray diffraction Methods 0.000 description 1
- 238000005245 sintering Methods 0.000 description 1
- 238000004513 sizing Methods 0.000 description 1
- 239000010454 slate Substances 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 229940005573 sodium fumarate Drugs 0.000 description 1
- 235000019294 sodium fumarate Nutrition 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 238000010996 solid-state NMR spectroscopy Methods 0.000 description 1
- 238000007614 solvation Methods 0.000 description 1
- 229940035044 sorbitan monolaurate Drugs 0.000 description 1
- 235000011069 sorbitan monooleate Nutrition 0.000 description 1
- 239000001593 sorbitan monooleate Substances 0.000 description 1
- 229940035049 sorbitan monooleate Drugs 0.000 description 1
- 235000011071 sorbitan monopalmitate Nutrition 0.000 description 1
- 239000001570 sorbitan monopalmitate Substances 0.000 description 1
- 229940031953 sorbitan monopalmitate Drugs 0.000 description 1
- 235000011076 sorbitan monostearate Nutrition 0.000 description 1
- 239000001587 sorbitan monostearate Substances 0.000 description 1
- 229940035048 sorbitan monostearate Drugs 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 230000008022 sublimation Effects 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 229960004964 temozolomide Drugs 0.000 description 1
- FBWNMEQMRUMQSO-UHFFFAOYSA-N tergitol NP-9 Chemical compound CCCCCCCCCC1=CC=C(OCCOCCOCCOCCOCCOCCOCCOCCOCCO)C=C1 FBWNMEQMRUMQSO-UHFFFAOYSA-N 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 229960004559 theobromine Drugs 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- FVRDYQYEVDDKCR-DBRKOABJSA-N tiazofurine Chemical compound NC(=O)C1=CSC([C@H]2[C@@H]([C@H](O)[C@@H](CO)O2)O)=N1 FVRDYQYEVDDKCR-DBRKOABJSA-N 0.000 description 1
- 229960003723 tiazofurine Drugs 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 235000019731 tricalcium phosphate Nutrition 0.000 description 1
- 229940117013 triethanolamine oleate Drugs 0.000 description 1
- 150000004684 trihydrates Chemical class 0.000 description 1
- 239000013638 trimer Substances 0.000 description 1
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 229960004224 tyloxapol Drugs 0.000 description 1
- MDYZKJNTKZIUSK-UHFFFAOYSA-N tyloxapol Chemical compound O=C.C1CO1.CC(C)(C)CC(C)(C)C1=CC=C(O)C=C1 MDYZKJNTKZIUSK-UHFFFAOYSA-N 0.000 description 1
- 229920001664 tyloxapol Polymers 0.000 description 1
- 238000000870 ultraviolet spectroscopy Methods 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 238000005550 wet granulation Methods 0.000 description 1
- 239000000811 xylitol Substances 0.000 description 1
- 235000010447 xylitol Nutrition 0.000 description 1
- HEBKCHPVOIAQTA-SCDXWVJYSA-N xylitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)CO HEBKCHPVOIAQTA-SCDXWVJYSA-N 0.000 description 1
- 229960002675 xylitol Drugs 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1617—Organic compounds, e.g. phospholipids, fats
- A61K9/1623—Sugars or sugar alcohols, e.g. lactose; Derivatives thereof; Homeopathic globules
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/141—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers
- A61K9/145—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers with organic compounds
Definitions
- the present mvention relates to co-crystal API-containing compositions, pharmaceutical compositions comprising such APIs, and methods for preparing the same.
- APIs Active pharmaceutical ingredients (API or APIs (plural)) in pharmaceutical compositions can be prepared in a variety of different forms. Such APIs can be prepared so as to have a variety of different chemical forms including chemical derivatives or salts. Such APIs can also be prepared to have different physical forms. For example, the APIs may be amorphous, may have different crystalline polymorphs, or may exist in different solvation or hydration states. By varying the form of an API, it is possible to vary the physical properties thereof. For example, crystalline polymorphs typically have different solubilities from one another, such that a more thermodynamically stable polymorph is less soluble than a less thermodynamically stable polymorph. Pharmaceutical polymorphs can also differ in properties such as shelf-life, bioavailability, morphology, vapour pressure, density, colour, and compressibility. Accordingly, variation of the crystalline state of an API is one of many ways in which to modulate the physical properties thereof.
- APIs that have improved properties, in particular, as oral formulations. Specifically, it is desirable to identify improved forms of APIs that exhibit significantly improved properties including increased aqueous solubility and stability. Further, it is desirable to improve the processability, or preparation of pharmaceutical formulations. For example, needle-like crystal forms or habits of APIs can cause aggregation, even in compositions where the API is mixed with other substances, such that a non- uniform mixture is obtained. It is also desirable to increase or decrease the dissolution rate of API-containing pharmaceutical compositions in water, increase or decrease the bioavailability of orally-administered compositions, and provide a more rapid or more delayed onset to therapeutic effect.
- APIs can be obtained which improve the properties of APIs as compared to such APIs in a non-co-crystalline slate (free acid, free base, zwitter ions, salts, etc.).
- the present invention provides a co-crystal pharmaceutical composition
- a co-crystal pharmaceutical composition comprising an API compound and a co-crystal former, such that the API and co- crystal former are capable of co-crystallizing from a solid or solution phase under crystallization conditions.
- Another aspect of the present invention provides a process for the production of a pharmaceutical composition, which process comprises:
- an API which has at least one functional group selected from ether, thioether, alcohol, thiol, aldehyde, ketone, thioketone, nitrate ester, phosphate ester, thiophosphate ester, ester, thioester, sulfate ester, carboxylic acid, phosphonic acid, phosphinic acid, sulfonic acid, amide, primary amine, secondary amine, ammonia, tertiary amine, imine, thiocyanate, cyanamide, oxime, nitrile, diazo, organohalide, nitro, S-heterocyclic ring, thiophene, N-heterocyclic ring, pyrrole, O-heterocyclic ring, furan, epoxide, peroxide, hydroxamic acid, imidazole, and pyridine;
- a co-crystal former which has at least one functional group selected from ether, thioether, alcohol, thiol, aldehyde, ketone, thioketone, nitrate ester, phosphate ester, thiophosphate ester, ester, thioester, sulfate ester, carboxylic acid, phosphonic acid, phosphinic acid, sulfonic acid, amide, primary amine, secondary amine, ammonia, tertiary amine, imine, thiocyanate, cyanamide, oxime, nitrile, diazo, organohalide, nitro, S-heterocyclic ring, thiophene, N-heterocyclic ring, pyrrole, O-heterocyclic ring, furan, epoxide, peroxide, hydroxamic acid, imidazole, and pyridine;
- a further aspect of the present invention provides a process for the production of a pharmaceutical composition, which comprises:
- the present invention provides a process for the production of a pharmaceutical composition, which comprises:
- the present invention provides a process for modulating the solubility of an API, which process comprises:
- the present invention provides a process for modulating the dissolution of an API, whereby the aqueous dissolution rate or the dissolution rate in simulated gastric fluid or in simulated intestinal fluid, or in a solvent or plurality of solvents is increased or decreased, which process comprises:
- the present invention provides a process for modulating the bioavailability of an API, whereby the AUC is increased, the time to T max is reduced, the length of time the concentration of the API is above ' _ T max is increased, or C max is increased, which process comprises: (1) grinding, heating, co-subliming, co-melting, or contacting in solution the API with a co-crystal former under crystallization conditions, so as to form a co-crystal of the API and the co-crystal former; and
- the present invention provides a process for improving the linearity of a dose response of an API, which process comprises:
- the present invention provides a process for improving the stability of a pharmaceutical salt, which process comprises:
- the present invention provides a process for making co-crystals of difficult to salt or unsaltable APIs, which process comprises:
- the present invention provides a method for decreasing the hygroscopicity of an API, which method comprises: (1) grinding, heating, co-subliming, co-melting, or contacting in solution the API with a co-crystal former under crystallization conditions, so as to form a co-crystal of the API and the co-crystal former; and
- the present invention provides a process for crystallizing an amorphous compound, which process comprises:
- the present invention provides a process for reducing the form diversity of an API, which process comprises:
- the present invention provides a process for modifying the morphology of an API, which process comprises:
- the present invention provides a co-crystal composition
- a co-crystal composition comprising a co-crystal, wherein said co-crystal comprises an API compound and a co-crystal former.
- the co-crystal has an improved property as compared to the free form (including a free acid, free base, zwitter ion, hydrate, solvate, etc.) or a salt (which includes salt hydrates and solvates).
- the improved property is selected from the group consisting of: increased solubility, increased dissolution, increased bioavailability, increased dose response, decreased hygroscopicity, a crystalline form of a normally amorphous compound, a crystalline form of a difficult to salt or unsaltable compound, decreased form diversity, more desired morphology, or other property described herein.
- Figs. I A-B PXRD diffractograms of a co-crystal comprising celecoxib and nicotinamide, with the background removed and as collected, respectively.
- Fig. 2 DSC thermogram for a co-crystal comprising celecoxib and nicotinamide.
- Fig. 3 TGA thermogram for a co-crystal comprising celecoxib and nicotinamide.
- Fig. 4 Raman spectrum for a co-crystal comprising celecoxib and nicotinamide.
- Figs. 5A-B PXRD diffractograms of a co-crystal comprising celecoxib and 18-crown-6, with the background removed and as collected, respectively.
- Fig. 6 DSC thermogram for a co-crystal comprising celecoxib and 18-crown-6.
- Fig. 7 TGA thermogram for a co-crystal comprising celecoxib and 18-crown-6.
- Figs. 8A-B PXRD diffractograms of a co-crystal comprising topiramate and 18-crown-6, with the background removed and as collected, respectively.
- Fig. 9 DSC thermogram for a co-crystal comprising topiramate and 18-crown-6.
- Figs. 10A-B PXRD diffractograms of a co-crystal comprising olanzapine and nicotinamide (Form
- Fig. 11 DSC thermogram for a co-crystal comprising olanzapine and nicotinamide (Form I).
- Fig. 12 PXRD diffractogram of a co-crystal comprising olanzapine and nicotinamide (Form II).
- Figs. 13A-B PXRD diffractograms of a co-crystal comprising olanzapine and nicotinamide (Form
- Figs. 14A-D Packing diagrams and crystal structure of a co-crystal comprising olanzapine and nicotinamide (Form III).
- Fig. 15 PXRD diffractogram of a co-crystal comprising c ⁇ -itraconazole and succinic acid.
- Fig. 16 DSC thermogram for a co-crystal comprising c ⁇ -itraconazole and succinic acid.
- Fig. 17 PXRD diffractogram of a co-crystal comprising c/s-itraconazole and fumaric acid.
- Fig. 18 DSC thermogram for a co-crystal comprising CM-itraconazole and fumaric acid.
- Fig. 19 PXRD diffractogram of a co-crystal comprising czAitraconazole and L-tartaric acid.
- Fig. 20 DSC thermogram for a co-crystal comprising c ⁇ -itraconazole and L-tartaric acid.
- Fig. 21 PXRD diffractogram of a co-crystal comprising cw-itraconazole and L-malic acid.
- Fig. 22 DSC thermogram for a co-crystal comprising c/ ' s-itraconazole and L-malic acid.
- Fig. 23 PXRD diffractogram of a co-crystal comprising cw-itraconazoleHCl and DL-tartaric acid.
- Fig 24 DSC thermogram for a co-crystal comprising c ⁇ -itraconazoleHCl and DL-tartaric acid.
- Fig. 26 DSC thermogram for a co-crystal comprising modafinil and malonic acid (Form I).
- Fig. 27 Raman spectrum for a co-crystal comprising modafinil and malonic acid (Form I).
- Fig. 28 PXRD diffractogram of a co-crystal comprising modafinil and malonic acid (Form II).
- Figs. 29A-B PXRD diffractograms of a co-crystal comprising modafinil and glycolic acid, with the background removed and as collected, respectively.
- Figs. 30A-B PXRD diffractograms of a co-crystal comprising modafinil and maleic acid, with the background removed and as collected, respectively.
- Figs. 31 A-B PXRD diffractograms of a co-crystal comprising 5-fluorouracil and urea, with the background removed and as collected, respectively.
- Fig. 32 DSC thermogram for a co-crystal comprising 5-ftuorouracil and urea.
- Fig. 33 TGA thermogram for a co-crystal comprising 5-fluorouracil and urea.
- Fig. 34 Raman spectrum for a co-crystal comprising 5-fluorouracil and urea.
- Figs. 35 A-B PXRD diffractograms of a co-crystal comprising hydrochlorothiazide and nicotinic acid, with the background removed and as collected, respectively.
- Figs. 37A-B PXRD diffractograms of a co-crystal comprising hydrochlorothiazide and piperazine, with the background removed and as collected, respectively.
- Figs. 38A-B An acetaminophen 1-D polymeric chain and a co-crystal of acetaminophen and 4,4'- bipyridine, respectively.
- Figures 40C and 40D show the supramolecular entity containing the synthon and corresponding co-crystal of aspirin and 4,4'-bipyridine, respectively.
- FIGs. 41A-D Pure ibuprofen and the corresponding crystal structure are shown in Figures 41A and 4 IB, respectively.
- Figures 41C and 41D show the supramolecular entity containing the synthon and corresponding co-crystal of ibuprofen and 4,4'-bipyridine, respectively.
- FIGs. 2A-D Pure flurbiprofen and the corresponding crystal structure are shown in Figures 42A and 2B, respectively.
- Figures 42C and 42D show the supramolecular synthon and corresponding co-crystal of flurbiprofen and 4,4'-bipyridine, respectively.
- Figs. 43A-B The supramolecular entity containing the synthon and the corresponding co-crystal structure of flurbiprofen and trans- l,2-bis(4-pyridyl)ethylene, respectively.
- Figs. 44A-B The crystal structure of pure carbamazepine and the co-crystal structure of carbamazepine and -phthalaldehyde, respectively.
- Fig. 45 A packing diagram of the co-crystal structure of carbamazepine and nicotinamide.
- Fig. 46 PXRD diffractogram of a co-crystal comprising carbamazepine and nicotinamide.
- Fig. 47 DSC thermogram for a co-crystal comprising carbamazepine and nicotinamide.
- Fig. 48 A packing diagram of the co-crystal structure of carbamazepine and saccharin.
- Fig. 49 PXRD diffractogram of a co-crystal comprising carbamazepine and saccharin.
- Fig. 50 DSC thermogram for a co-crystal comprising carbamazepine and saccharin.
- Figs. 51 A-B The crystal structure of carbamazepine and the co-crystal structure of carbamazepine and 2,6-pyridinedicarboxylic acid, respectively.
- Figs. 52A-B The crystal structure of carbamazepine and the co-crystal structure of carbamazepine and 5-nitroisophthalic acid, respectively.
- Figs. 53 A-B The crystal structure of carbamazepine and the co-crystal structure of carbamazepine and 1,3,5,7-adamantanetetracarboxylic acid, respectively.
- Figs. 54A-B The crystal structure of carbamazepine and the co-crystal structure of carbamazepme and benzoquinone, respectively.
- Figs. 55A-B The crystal structure of carbamazepine and the co-crystal structure of carbamazepine and trimesic acid, respectively.
- Fig. 56 PXRD diffractogram of a co-crystal comprising carbamazepine and trimesic acid.
- Fig. 57 Dissolution profile for a co-crystal of celecoxib :nicotinamide vs. celecoxib free acid.
- Fig. 58 Dissolution profile for co-crystals of itraconazole: succinic acid, itraconazle:tartaric acid and itraconazole:malic acid vs. itraconazole free base.
- Fig. 61 Dissolution profile of several formulations of modafinil free form and modafinikmalonic acid (Form I).
- co-crystal as used herein means a crystalline material comprised of two or more unique solids at room temperature, each containing distinctive physical characteristics, such as structure, melting point and heats of fusion, with the exception that, if specifically stated, the API may be a liquid at room temperature.
- the co-crystals of the present invention comprise a co-crystal former H-bonded to an API.
- the co- crystal former may be H-bonded directly to the API or may be H-bonded to an additional molecule which is bound to the API.
- the additional molecule may be H-bonded to the API or bound ionically or covalently to the API.
- the additional molecule could also be a different API.
- Solvates of API compounds that do not further comprise a co-crystal former are not co-crystals according to the present invention.
- the co-crystals may however, include one or more solvate molecules in the crystalline lattice. That is, solvates of co-crystals, or a co-crystal further comprising a solvent or compound that is a liquid at room temperature, is included in the present invention, but crystalline material comprised of only one solid and one or more liquids (at room temperature) are not included in the present invention, with the previously noted exception of specifically stated liquid APIs.
- the co-crystals may also be a co-crystal between a co-crystal former and a salt of an API, but the API and the co-crystal former of the present invention are constructed or bonded together through hydrogen bonds.
- Other modes of molecular recognition may also be present including, pi-stacking, guest-host complexation and van der Waals interactions.
- hydrogen-bonding is the dominant interaction in the formation of the co-crystal, (and a required interaction according to the present invention) whereby a non-covalent bond is formed between a hydrogen bond donor of one of the moieties and a hydrogen bond acceptor of the other. Hydrogen bonding can result in several different intermolecular configurations.
- hydrogen bonds can result in the formation of dimers, linear chains, or cyclic structures. These configurations can further include extended (two-dimensional) hydrogen bond networks and isolated triads (Fig. 60).
- An alternative embodiment provides for a co-crystal wherein the co-crystal former is a second API. In another embodiment, the co-crystal former is not an API. In another embodiment the co-crystal comprises two co-crystal formers.
- the chemical and physical properties of an API in the form of a co-crystal may be compared to a reference compound that is the same API in a different form.
- the reference compound may be specified as a free form, or more specifically, a free acid, free base, or zwitterion; a salt, or more specifically for example, an inorganic base addition salt such as sodium, potassium, lithium, calcium, magnesium, ammonium, aluminum salts or organic base addition salts, or an inorganic acid addition salts such as HBr, HCl, sulfuric, nitric, or phosphoric acid addition salts or an organic acid addition salt such as acetic, propionic, pyruvic, malanic, succinic, malic, maleic, fumaric, tartaric, citric, benzoic, methanesulfonic, ethanesulforic, stearic or lactic acid addition salt; an anhydrate or hydrate of a free form or salt, or more specifically, for example, a hemihydrate, monohydrate, dihydrate, trihydrate, quadrahydrate, pentahydrate, sesquihydrate; or a solvate of a free form or salt.
- the reference compound for an API in salt form co-crystallized with a co-crystal former can be the API salt form.
- the reference compound for a free acid API co-crystallized with a co-crystal former can be the free acid API.
- the reference compound may also be specified as crystalline or amorphous.
- the co-crystals can include an acid addition salt or base addition salt of an API.
- Acid addition salts include, but are not limited to, inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, and phosphoric acid, and organic acids such as acetic acid, propionic acid, hexanoic acid, heptanoic acid, cyclopentanepropionic acid, glycolic acid, pyruvic acid, lactic acid, malonic acid, succinic acid, malic acid, maleic acid, fumaric acid, tartatic acid, citric acid, benzoic acid, o-(4-hydroxybenzoyl)benzoic acid, cinnamic acid, madelic acid, methanesulfonic acid, ethanesulfonic acid, 1,2-ethanedisulfonic acid, 2- hydroxyethanesulfonic acid, benzenesulfonic acid,/?-chlorobenzenes
- Base addition salts include, but are not limited to, inorganic bases such as sodium, potassium, lithium, ammonium, calcium and magnesium salts, and organic bases such as primary, secondary and tertiary amines (e.g. isopropylamine, trimethyl amine, diethyl amine, tri(iso-propyl) amine, tri(n-propyl) amine, ethanolamine, 2-dimethylaminoethanol, tromethamine, lysine, arginine, histidine, procaine, hydrabamine, choline, betaine, ethylenediamine, glucosamine, N-alkylglucamines, theobromine, purines, piperazine, piperidine, morpholine, and N-ethylpiperidine).
- inorganic bases such as sodium, potassium, lithium, ammonium, calcium and magnesium salts
- organic bases such as primary, secondary and tertiary amines (e.g. isopropylamine,
- the ratio of API to co-crystal former may be stoichiometric or non-stoichiometric according to the present invention. For example, 1 :1, 1.5:1, 1 :1.5, 2:1 and 1 :2 ratios of API:co-crystal former are acceptable.
- a co-crystal form of an API is particularly advantageous where the original API is insoluble or sparingly soluble in water.
- the co-crystal properties conferred upon the API are also useful because the bioavailability of the API can be improved and the plasma concentration and/or serum concentration of the API can be improved. This is particularly advantageous for orally-administrable formulations.
- the dose response of the API can be improved, for example by increasing the maximum attainable response and/or increasing the potency of the API by increasing the biological activity per dosing equivalent.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a co-crystal of an API and a co-crystal former, such that the API and co-crystal former are capable of co-crystallizing from a solution phase under crystallization conditions or from the solid-state, for example, through grinding, heating, or through vapor transfer (e.g., co-sublimation).
- the API has at least one functional group selected from ether, thioether, alcohol, thiol, aldehyde, ketone, thioketone, nitrate ester, phosphate ester, thiophosphate ester, ester, thioester, sulfate ester, carboxylic acid, phosphonic acid, phosphinic acid, sulfonic acid, amide, primary amine, secondary amine, ammonia, tertiary amine, imine, thiocyanate, cyanamide, oxime, nitrile, diazo, organohalide, nitro, S-heterocyclic ring, thiophene, N-heterocyclic ring, pyrrole, O-heterocyclic ring, furan, epoxide, peroxide, hydroxamic acid, imidazole, and pyridine and a co-crystal former which has at least one functional group selected from ether, thioether,
- co-crystals of the present invention are formed where the API and co-crystal former are bonded together through hydrogen bonds.
- Other non-covalent interactions including pi-stacking and van der Waals interactions, may also be present.
- the co-crystal former is selected from the co-crystal formers of Table I and Table II. In other embodiments, the co-crystal former of Table I is specified as a Class 1, Class 2, or Class 3 co-crystal former (see column labeled "class" Table I).
- the difference in pK a value of the co-crystal former and the API is less than 2. In other embodiments, the difference in pK a values of the co- crystal former and API is less than 3, less than 4, less than 5, between 2 and 3, between 3 and 4, or between 4 and 5. Table I lists multiple pK a values for co-crystal formers having multiple functionalities. It is readily apparent to one skilled in the art the particular functional group corresponding to a particular pK a value.
- the particular functional group of a co-crystal former interacting with the API is specified (see for example Table I, columns labeled “Functionality” and “Molecular Structure” and the column of Table II labeled "Co- Crystal Former Functional Group”).
- the functional group of the API interacting with the co-crystal former functional group is specified (see, for example, Tables II and III).
- the co-crystal comprises more than one co-crystal former.
- two, three, four, five, or more co-crystal formers can be incorporated in a co-crystal with an API.
- Co-crystals which comprise two or more co-crystal formers and an API are bound together via hydrogen bonds.
- incorporated co- crystal formers are hydrogen bonded to the API molecules.
- co- crystal formers are hydrogen bonded to either the API molecules or the incorporated co- crystal formers.
- co-crystal formers can be contained in a single compartment, or kit, for ease in screening an API for potential co-crystal species.
- the co-crystal kit can comprise 5, 10, 15, 20, 25, 30, 40, 50, 60, 70, 80, 90, 100, or more of the co-crystal formers in Tables I and II.
- the co-crystal formers are in solid form or in solution and in an array of individual reaction vials such that individual co-crystal formers can be tested with one or more APIs by one or more crystallization methods or multiple co-crystal formers can be easily tested against one or more compounds by one or more crystallization methods.
- the crystallization methods include, but are not limited to, melt recrystallization, grinding, milling, standing, co-crystal formation from solution by evaporation, thermally driven crystallization from solution, co-crystal formation from solution by addition of anti-solvent, co-crystal formation from solution by vapor- diffusion, co-crystal formation from solution by drown-out, co-crystal formation from solution by any combination of the above mentioned techniques, co-crystal formation by co-sublimation, co-crystal formation by sublimation using a Knudsen cell apparatus, co- crystal formation by standing the desired components of the co-crystal in the presence of solvent vapor, co-crystal formation by slurry conversion of the desired components of the co-crystal in a solvent or mixtures of solvents, or co-crystal formation by any combination of the above techniques in the presence of additives, nucleates, crystallization enhancers, precipitants, chemical stabilizers, or anti-oxidants.
- kits can be used alone or as part of larger crystallization experiments.
- kits can be constructed as single co-crystal former single well kits, single co- crystal former multi-well kits, multi-co-crystal former single well kits, or multi-co-crystal former multi-well kits.
- High-throughput crystallization e.g., the CrystalMaxTM platform
- Multi-well plates e.g., 96 wells, 384 wells, 1536 wells, etc.
- the API is selected from an API of Table IV or elsewhere herein.
- co-crystals can comprise such APIs in free form (i.e. free acid, free base, zwitter ion), salts, solvates, hydrates, or the like.
- the API can either be of the form listed in Table IV or its corresponding free form, or of another form that is not listed.
- Table IV includes the CAS number, chemical name, or a PCT or patent reference (each incorporated herein in their entireties).
- the functional group of the particular API interacting with the co-crystal former is specified.
- a specific functional group of a co-crystal former, a specific co-crystal former, or a specified functional group or a specific co-crystal former interacting with the particular API may also be specified. It is noted that for Table II, the co-crystal former, and optionally the specific functionality, and each of the listed corresponding interacting groups are included as individual species of the present invention. Thus, each specific combination of a co-crystal former and one of the interacting groups in the same row may be specified as a species of the present invention. The same is true for other combinations as discussed in the Tables and elsewhere herein.
- the co-crystal comprises an API wherein the API forms a dimeric primary amide structure via hydrogen bonds with an R 2 2 (8) motif.
- the dimeric primary amide structure further comprises one, two, three, or four hydrogen bond donors.
- the dimeric primary amide structure further comprises one or two hydrogen bond acceptors.
- the dimeric primary amide structure further comprises a combination of hydrogen bond donors and acceptors.
- the dimeric primary amide structure can further comprise one hydrogen bond donor and one hydrogen bond acceptor, one hydrogen bond donor and two hydrogen bond acceptors, two hydrogen bond donors and one hydrogen bond acceptor, two hydrogen bond donors and two hydrogen bond acceptors, or three hydrogen bond donors and one hydrogen bond acceptor.
- Two non- limiting examples of APIs which form a dimeric primary amide co-crystal structure include modafinil and carbamazepine.
- APIs which include a primary amide functional group include, but are not limited to, arotinolol, atenolol, carpipramine, cefotetan, cefsulodin, docapromine, darifenacin, exalamide, fidarestat, frovatriptan, silodosin, levetiracetam, MEN- 10700, mizoribine, oxiracetam, piracetam, protirelin, TRH, ribavirm, valrecemide, temozolomide, tiazofurin, antiPARP-2, levovirin, N- benzyloxycarbonyl glycinamide, and UCB-34714.
- each process according to the invention there is a need to contact the API with the co-crystal former. This may involve grinding or milling the two solids together or melting one or both components and allowing them to recrystallize.
- the use of a granulating liquid may improve or may impede co-crystal formation.
- Non-limiting examples of tools useful for the formation of co-crystals may include, for example, an extruder or a mortar and pestle.
- contacting the API with the co-crystal former may also involve either solubilizing the API and adding the co-crystal former, or solubilizing the co-crystal former and adding the API. Crystallization conditions are applied to the API and co-crystal former.
- This may entail altering a property of the solution, such as pH or temperature and may require concentration of the solute, usually by removal of the solvent, typically by drying the solution. Solvent removal results in the concentration of both API and co-crystal former increasing over time so as to facilitate crystallization. For example, evaporation, cooling, co-sublimation, or the addition of an antisolvent may be used to crystallize co-crystals. In another embodiment, a slurry comprising an API and a co-crystal former is used to form co-crystals. Once the solid phase comprising any crystals is formed, this may be tested as described herein.
- co-crystals on a large and/or commercial scale may be successfully completed using one or more of the processes and techniques described herein.
- crystallization of co-crystals from a solvent and grinding or milling are conceivable non-limiting processes.
- co-crystals with stoichiometries of 1:1, 2:1, or 1 :2 can be produced by adding co-crystal former in an amount that is 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 50, 75, 100 times or more than the stoichiometric amount for a given co- crystal.
- Such an excessive use of a co-crystal former to form a co-crystal can be employed in solution or when grinding an API and a co-crystal former to drive co-crystal formation.
- the present invention provides for the use of an ionic liquid as a medium for the formation of a co-crystal, and can also be used to crystallize other forms in addition to co-crystals (e.g., salts, solvates, free acid, free base, zwitterions, etc.).
- This medium is useful, for example, where the above methods do not work or are difficult or impossible to control.
- ionic liquids useful in co-crystal formation are: l-butyl-3-methylimidazolium lactate, 1-ethyl- 3-methylimidazolium lactate, and 1-butylpyridinium hexafluorophosphate.
- compositions in general are discussed in further detail below and may further comprise a pharmaceutically-acceptable diluent, excipient or carrier.
- the present invention provides a process for the production of a pharmaceutical composition, which process comprises:
- an API which has at least one functional group selected from ether, thioether, alcohol, thiol, aldehyde, ketone, thioketone, nitrate ester, phosphate ester, thiophosphate ester, ester, thioester, sulfate ester, carboxylic acid, phosphonic acid, phosphinic acid, sulfonic acid, amide, primary amine, secondary amine, ammonia, tertiary amine, imine, thiocyanate, cyanamide, oxime, nitrile diazo, organohalide, nitro, S-heterocyclic ring, thiophene, N-heterocyclic ring, pyrrole, O-heterocyclic ring, furan, epoxide, peroxide, hydroxamic acid, imidazole, and pyridine or of Table II or III;
- a co-crystal former which has at least one functional group selected from ether, thioether, alcohol, thiol, aldehyde, ketone, thioketone, nitrate ester, phosphate ester, thiophosphate ester, ester, thioester, sulfate ester, carboxylic acid, phosphonic acid, phosphinic acid, sulfonic acid, amide, primary amine, secondary amine, ammonia, tertiary amine, imine, thiocyanate, cyanamide, oxime, nitrile, diazo, organohalide, nitro, S-heterocyclic ring, thiophene, N-heterocyclic ring, pyrrole, O- heterocyclic ring, furan, epoxide, peroxide, hydroxamic acid, imidazole, and pyridine or of Table I, II, or III; (3) grinding, heating or contacting in solution the
- the present invention provides a process for the production of a pharmaceutical composition, which comprises:
- Assaying the solid phase for the presence of co-crystals of the API and the co- crystal former may be carried out by conventional methods known in the art. For example, it is convenient and routine to use powder X-ray diffraction techniques to assess the presence of co-crystals. This may be affected by comparing the spectra of the API, the crystal former and putative co-crystals in order to establish whether or not true co- crystals had been formed. Other techniques, used in an analogous fashion, include differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), solid state NMR spectroscopy, and Raman spectroscopy. Single crystal X-ray diffraction is especially useful in identifying co-crystal structures.
- DSC differential scanning calorimetry
- TGA thermogravimetric analysis
- TGA solid state NMR spectroscopy
- Raman spectroscopy Raman spectroscopy
- the present invention therefore provides a process of screening for co-crystal compounds, which comprises:
- APIs and co-crystal formers of the present invention have one or more chiral centers and may exist in a variety of stereoisomeric configurations. As a consequence of these chiral centers, several APIs and co-crystal formers of the present invention occur as racemates, mixtures of enantiomers and as individual enantiomers, as well as diastereomers and mixtures of diastereomers. All such racemates, enantiomers, and diastereomers are within the scope of the present invention including, for example, cis- and tr ⁇ s-isomers, R- and S-enantiomers, and (D)- and (L)-isomers.
- Co-crystals of the present invention can include isomeric forms of either the API or the co-crystal former or both.
- Isomeric forms of APIs and co-crystal formers include, but are not limited to, stereoisomers such as enantiomers and diastereomers.
- a co-crystal can comprise a racemic API and/or co-crystal former.
- a co-crystal can comprise an enantiomerically pure API and/or co-crystal former.
- a co-crystal can comprise an API or a co-crystal former with an enantiomeric excess of about 50 percent, 55 percent, 60 percent, 65 percent, 70 percent, 75 percent, 80 percent, 85 percent, 90 percent, 95 percent, 96 percent, 97 percent, 98 percent, 99 percent, greater than 99 percent, or any intermediate value.
- stereoisomeric APIs include modafinil, cw-itraconazole, ibuprofen, and flurbiprofen.
- stereoisomeric co-crystal formers include tartaric acid and malic acid.
- Co-crystals comprising enantiomerically pure components (e.g., API or co-crystal former) can give rise to chemical and/or physical properties which are modulated with respect to those of the corresponding co-crystal comprising a racemic component.
- the modafinil :malonic acid co-crystal from Example 10 comprises racemic modafinil.
- Enantiomerically pure R-modafinil:malonic acid can conceivably be synthesized via the same or another method of the present invention and is therefore included in the scope of the invention.
- enantiomerically pure S- modaf ⁇ nikmalonic acid can conceivably be synthesized via a method of the present invention and is therefore included in the scope of the invention.
- a co-crystal comprising an enantiomerically pure component can give rise to a modulation of, for example, activity, bioavailability, or solubility, with respect to the corresponding co-crystal comprising a racemic component.
- the co-crystal R-modafinil:malonic acid can have modulated properties as compared to the racemic modafinikmalonic acid co-crystal.
- racemic co-crystal refers to a co-crystal which is comprised of an equimolar mixture of two enantiomers of the API, the co-crystal former, or both.
- a co-crystal comprising a stereoisomeric API and a non-stereoisomeric co-crystal former is a "racemic co-crystal" when there is present an equimolar mixture of the API enantiomers.
- a co-crystal comprising a non-stereoisomeric API and a stereoisomeric co-crystal former is a "racemic co-crystal" when there is present an equimolar mixture of the co-crystal former enantiomers.
- a co-crystal comprising a stereoisomeric API and a stereoisomeric co-crystal former is a "racemic co-crystal" when there is present an equimolar mixture of the API enantiomers and of the co-crystal former enantiomers.
- enantiomerically pure co- crystal refers to a co-crystal which is comprised of a stereoisomeric API or a stereoisomeric co-crystal former or both where the enantiomeric excess of the stereoisomeric species is greater than or equal to about 90 percent ee.
- the present invention includes a pharmaceutical composition comprising a co-crystal with an enantiomerically pure API or co-crystal former wherein the bioavailability is modulated with respect to the racemic co-crystal.
- the present invention includes a pharmaceutical composition comprising a co-crystal with an enantiomerically pure API or co-crystal former wherein the activity is modulated with respect to the racemic co-crystal.
- the present invention includes a pharmaceutical composition comprising a co-crystal with an enantiomerically pure API or co-crystal former wherein the solubility is modulated with respect to the racemic co-crystal.
- enantiomerically pure includes a composition which is substantially enantiomerically pure and includes, for example, a composition with greater than or equal to about 90, 91, 92, 93, 94, 95, 96, 97, 98, or 99 percent enantiomeric excess.
- the present invention provides a process for modulating the solubility of an API, which process comprises:
- solubility of the API is modulated such that the aqueous solubility is increased.
- Solubility of APIs may be measured by any conventional means such as chromatography (e.g., HPLC) or spectroscopic determination of the amount of API in a saturated solution of the API, such as UV-spectroscopy, IR-spectroscopy, Raman spectroscopy, quantitative mass spectroscopy, or gas chromatography.
- the API may have low aqueous solubility.
- low aqueous solubility in the present application refers to a compound having a solubility in water which is less than or equal to 10 mg/mL, when measured at 37 degrees C, and preferably less than or equal to 5 mg/mL or 1 mg/mL.
- Low aqueous solubility can further be specifically defined as less than or equal to 900, 800, 700, 600, 500, 400, 300, 200 150 100, 90, 80, 70, 60, 50, 40, 30, 20 micrograms/mL, or further 10, 5 or 1 micrograms/mL, or further 900, 800, 700, 600, 500, 400, 300, 200 150, 100 90, 80, 70, 60, 50, 40, 30, 20, or 10 ng/mL, or less than 10 ng/mL when measured at 37 degrees C.
- Aqueous solubility can also be specified as less than 500, 400, 300, 200, 150, 100, 75, 50 or 25 mg/mL.
- solubility can be increased 2, 3, 4, 5, 7, 10, 15, 20, 25, 50, 75, 100, 200, 300, 500, 750, 1000, 5000, or 10,000 times by making a co-crystal of the reference form (e.g., crystalline or amorphous free acid, free base or zwitter ion, hydrate or solvate), or a salt thereof.
- aqueous solubility can be measured in simulated gastric fluid (SGF) or simulated intestinal fluid (SIF) rather than water.
- the pH of the solvent used may also be specified as 1, 1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, 1.8, 1.9, 2, 2.1, 2.2, 2.3, 2.4, 2.5, 2.6, 2.7, 2.8, 2.9, 3, 3.5, 4, 4.5, 5, 5.5, 6, 6.5, 7, 7.5, 8, 8.5, 9, 9.5, 10, 10.5, 11, 11.5, 12, 12.5, 13, 13.5, or 14 or any pH in between successive values.
- Examples of embodiments includes: co-crystal compositions with an aqueous solubility, at 37 degrees C and a pH of 7.0, that is increased at least 5 fold over the reference form, co-crystal compositions with a solubility in SGF that is increased at least 5 fold over the reference form, co-crystal compositions with a solubility in SIF that is increased at least 5 fold over the reference form.
- the dissolution profile of the API is modulated whereby the aqueous dissolution rate or the dissolution rate in simulated gastric fluid or in simulated intestinal fluid, or in a solvent or plurality of solvents is increased.
- Dissolution rate is the rate at which API solids dissolve in a dissolution medium.
- the rate-limiting step in the absorption process is often the dissolution rate. Because of a limited residence time at the absorption site, APIs that are not dissolved before they are removed from intestinal absorption site are considered useless. Therefore, the rate of dissolution has a major impact on the performance of APIs that are poorly soluble. Because of this factor, the dissolution rate of APIs in solid dosage forms is an important, routine, quality control parameter used in the API manufacturing process.
- Dissolution rate K S (C 3 -C)
- K dissolution rate constant
- S is the surface area
- C s is the apparent solubility
- C is the concentration of API in the dissolution medium.
- C 3 -C is approximately equal to C s .
- the dissolution rate of APIs may be measured by conventional means known in the art.
- the increase in the dissolution rate of a co-crystal, as compared to the reference form (e.g., free form or salt), may be specified, such as by 10, 20, 30, 40, 50, 60, 70, 80, 90, or 100%, or by 2, 3, 4, 5 ,6, 7, 8, 9, 10, 15, 20, 25, 30, 40, 50, 75, 100, 125, 150, 175, 200, 250, 300, 350, 400, 500, 1000, 10,000, or 100,000 fold greater than the reference form (e.g., free form or salt form) in the same solution.
- Conditions under which the dissolution rate is measured is the same as discussed above
- the increase in dissolution may be further specified by the time the composition remains supersaturated before reaching equilibrium solubility.
- Examples of above embodiments include: co-crystal compositions with a dissolution rate in aqueous solution, at 37 degrees C and a pH of 7.0, that is increased at least 5 fold over the reference form, co-crystal compositions with a dissolution rate in SGF that is increased at least 5 fold over the reference form, co-crystal compositions with a dissolution rate in SIF that is increased at least 5 fold over the reference form.
- the methods of the present invention are used to make a pharmaceutical API formulation with greater solubility, dissolution, and bioavailability. Bioavailability can be improved via an increase in AUC, reduced time to T ma , (the time to reach peak blood serum levels), or increased C max ,.
- the present invention can result in higher plasma concentrations of API when compared to the neutral form or salt alone (reference form).
- AUC is the area under the plot of plasma concentration of API (not logarithm of the concentration) against time after API administration. The area is conveniently determined by the "trapezoidal rule": The data points are connected by straight line segments, perpendiculars are erected from the abscissa to each data point, and the sum of the areas of the triangles and trapezoids so constructed is computed. When the last measured concentration (C Thread, at time t n ) is not zero, the AUC from t n to infinite time is estimated by C Cincinnati/k eI .
- the AUC is of particular use in estimating bioavailability of APIs, and in estimating total clearance of APIs (Cl ⁇ ).
- AUC F • D/Cl ⁇ , where F is the absolute bioavailability of the API.
- the present invention provides a process for modulating the bioavailability of an API when administered in its normal and effective dose range as a co-crystal, whereby the AUC is increased, the time to T max is reduced, or C max is increased, as compared to a reference form, which process comprises:
- Examples of the above embodiments include: co-crystal compositions with a time to T max that is reduced by at least 10% as compared to the reference form, co-crystal compositions with a time to T max that is reduced by at least 20% over the reference form, co-crystal compositions with a time to T max that is reduced by at least 40% over the reference form, co-crystal compositions with a time to T max that is reduced by at least 50% over the reference form, co-crystal compositions with a T max that is reduced by at least 60% over the reference form, co-crystal compositions with a T max that is reduced by at least 70% over the reference form, co-crystal compositions with a T ma that is reduced by at least 80% over the reference form, co-crystal compositions with a T max that is reduced by at least 90% over the reference form, co-crystal compositions with a C ma ⁇ that is increased by at least 20% over the reference form, co-crystal compositions with a C max that is increased by
- the present invention provides a process for improving the dose response of an API, which process comprises:
- Dose response is the quantitative relationship between the magnitude of response and the dose inducing the response and may be measured by conventional means known in the art.
- the curve relating effect (as the dependent variable) to dose (as the independent variable) for an API-cell system is the "dose-response curve".
- dose-response curve is the measured response to an API plotted against the dose of the API (mg/kg) given.
- the dose response curve can also be a curve of AUC against the dose of the API given.
- a co-crystal of the present mvention has an increased dose response curve or a more linear dose response curve than the corresponding reference compound.
- the present invention provides a process for improving the stability of an API (as compared to a reference form such as its free form or a salt thereof), which process comprises:
- compositions of the present invention including the API or active pharmaceutical ingredient (API) and formulations comprising the API, are suitably stable for pharmaceutical use.
- the API or formulations thereof of the present invention are stable such that when stored at 30 degrees C for 2 years, less than 0.2 % of any one degradant is formed.
- degradant refers herein to product(s) of a single type of chemical reaction. For example, if a hydrolysis event occurs that cleaves a molecule into two products, for the purpose of the present invention, it would be considered a single degradant. More preferably, when stored at 40 degrees C for 2 years, less than 0.2 % of any one degradant is formed.
- the relative humidity (RH) may be specified as ambient (RH), 75 % (RH), or as any single integer between 1 to 99 %. Difficult to Salt or Unsaltable Compounds
- the present invention provides a process for making co- crystals of unsaltable or difficult to salt APIs which process comprises:
- Difficult to salt compounds include bases with a pKa less than 3 or acids with a pKa greater than 10.
- Zwitter ions are also difficult to salt or unsaltable compounds according to the present invention.
- the present invention provides a method for decreasing the hygroscopicity of an API, which method comprises:
- An aspect of the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a co-crystal of an API that is less hygroscopic than amorphous or crystalline, free form or salt (including metal salts such as sodium, potassium, lithium, calcium, magnesium) or another reference compound.
- Hygroscopicity can be assessed by dynamic vapor sorption analysis, in which 5-50 mg of the compound is suspended from a Cahn microbalance.
- the compound being analyzed should be placed in a non- hygroscopic pan and its weight should be measured relative to an empty pan composed of identical material and having nearly identical size, shape, and weight. Ideally, platinum pans should be used.
- the pans should be suspended in a chamber through which a gas, such as air or nitrogen, having a controlled and known percent relative humidity (%RH) is flowed until eqilibrium criteria are met.
- a gas such as air or nitrogen
- Typical equilibrium criteria include weight changes of less than 0.01 % over 3 minutes at constant humidity and temperature.
- the relative humidity should be measured for samples dried under dry nitrogen to constant weight ( ⁇ 0.01 % change in 3 minutes) at 40 degrees C unless doing so would de-solvate or otherwise convert the material to an amorphous compound.
- the hygroscopicity of a dried compound can be assessed by increasing the RH from 5 to 95 % in increments of 5 % RH and then decreasing the RH from 95 to 5 % in 5 % increments to generate a moisture sorption isotherm.
- the sample weight should be allowed to equilibrate between each change in % RH. If the compound deliquesces or becomes amorphous above 75 % RH, but below 95 % RH, the experiment should be repeated with a fresh sample and the relative humidity range for the cycling should be narrowed to 5-75 % RH or 10-75 % RH, instead of 5-95 %RH.
- Hygroscopicity can be defined using various parameters. For purposes of the present invention, a non-hygroscopic molecule should not gain or lose more than 1.0 %, or more preferably, 0.5 % weight at 25 degrees C when cycled between 10 and 75 % RH (relative humidity at 25 degrees C).
- the non-hygroscopic molecule more preferably should not gain or lose more than 1.0 %, or more preferably, 0.5 % weight when cycled between 5 and 95 % RH at 25 degrees C, or more than 0.25 % of its weight between 10 and 75 % RH. Most preferably, a non-hygroscopic molecule will not gain or lose more than 0.25 % of its weight when cycled between 5 and 95 % RH.
- hygroscopicity can be defined using the parameters of Callaghan et al., "Equilibrium moisture content ofpharmaceutical excipients", in Api Dev. Ind. Pharm., Vol. 8, pp. 335-369 (1982). Callaghan et al. classified the degree of hygroscopicity into four classes.
- Class 1 Non-hygroscopic Essentially no moisture increases occur at relative humidities below 90 %.
- Class 2 Slightly hygroscopic Essentially no moisture increases occur at relative humidities below 80%.
- Class 3 Moderately hygroscopic Moisture content does not increase more than 5 % after storage for 1 week at relative humidities below 60 %.
- Class 4 Very hygroscopic Moisture content increase may occur at relative humidities as low as 40 to 50 %.
- hygroscopicity can be defined using the parameters of the European Pharmacopoeia Technical Guide (1999, p. 86) which has defined hygrospocity, based on the static method, after storage at 25 degrees C for 24 hours at 80 % RH:
- Hygroscopic Increase in mass is less than 15 percent m/m and equal to or greater than 0.2 percent m/m.
- Co-crystals of the present invention can be set forth as being in Class 1, Class 2, or Class 3, or as being Slightly hygroscopic, Hygroscopic, or Very Hygroscopic. Co- crystals of the present invention can also be set forth based on their ability to reduce hygroscopicity. Thus, preferred co-crystals of the present invention are less hygroscopic than a reference compound.
- the reference compound can be specified as the API in free form (free acid, free base, hydrate, solvate, etc.) or salt (e.g., especially metal salts such as sodium, potassium, lithium, calcium, or magnesium).
- co-crystals that do not gain or lose more than 1.0 % weight at 25 degrees C when cycled between 10 and 75 % RH, wherein the reference compound gains or loses more than 1.0 % weight under the same conditions.
- co-crystals that do not gain or lose more than 0.5 % weight at 25 degrees C when cycled between 10 and 75 % RH, wherein the reference compound gains or loses more than 0.5 % or more than 1.0 % weight under the same conditions.
- co-crystals that do not gain or lose more than 1.0 % weight at 25 degrees C when cycled between 5 and 95 % RH, wherein the reference compound gains or loses more than 1.0 % weight under the same conditions.
- co-crystals that do not gain or lose more than 0.5 % weight at 25 degrees C when cycled between 5 and 95 % RH, wherein the reference compound gains or loses more than 0.5 % or more than 1.0 % weight under the same conditions.
- co-crystals that do not gain or lose more than 0.25 % weight at 25 degrees C when cycled between 5 and 95 % RH, wherein the reference compound gains or loses more than 0.5 % or more than 1.0 % weight under the same conditions.
- co-crystals that have a hygroscopicity (according to Callaghan et al.) that is at least one class lower than the reference compound or at least two classes lower than the reference compound. Included are a Class 1 co-crystal of a Class 2 reference compound, a Class 2 co-crystal of a Class 3 reference compound, a Class 3 co-crystal of a Class 4 reference compound, a Class 1 co- crystal of a Class 3 reference compound, a Class 1 co-crystal of a Class 4 reference compound, or a Class 2 co-crystal of a Class 4 reference compound.
- co-crystals that have a hygroscopicity (according to the European Pharmacopoeia Technical Guide) that is at least one class lower than the reference compound or at least two classes lower than the reference compound.
- a slightly hygroscopic co-crystal of a hygroscopic reference compound a hygroscopic co-crystal of a very hygroscopic reference compound, a very hygroscopic co-crystal of a deliquescent reference compound, a slightly hygroscopic co-crystal of a very hygroscopic reference compound, a slightly hygroscopic co-crystal of a deliquescent reference compound, and a hygroscopic co-crystal of a deliquescent reference compound.
- the present invention provides a process for crystallizing an amorphous compound, which process comprises: (1) grinding, heating, co-subliming, co-melting, or contacting in solution the API with a co-crystal former under crystallization conditions, so as to form a co-crystal of the API and the co-crystal former; and
- An amorphous compound includes compounds that do not crystallize using routine methods in the art.
- the present invention provides a process for reducing the form diversity of an API, which process comprises:
- the number of forms of a co-crystal is compared to the number of forms of a reference compound (e.g. the free form or a salt of the API) that can be made using routine methods in the art.
- a reference compound e.g. the free form or a salt of the API
- the present invention provides a process for modifying the morphology of an API, which process comprises:
- the co-crystal comprises or consists of a co-crystal former and a pharmaceutical wherein the interaction between the two, e.g., H-bonding, occurs between a functional group of Table III of an API with a corresponding interacting group of Table III.
- the co-crystal comprises a co-crystal former of Table I or II and an API with a corresponding interacting group of Table III.
- the co-crystal comprises an API from Table IV and a co-crystal former with a functional group of Table III.
- the co-crystal is from Table I or II.
- co-crystals having an H-bond acceptor on the first molecule and an H-bond donor on the second molecule are included in the present invention.
- Table IV includes the CAS number, chemical name or a PCT or patent reference (each incorporated herein in their entireties). Thus, whether a particular API contains an H-bond donor, acceptor or both is readily apparent.
- the co-crystal former and API each have only one H- bond donor/acceptor.
- the molecular weight of the API is less than 2000, 1500, 1000, 750, 500, 350, 200, or 150 Daltons. In another embodiment, the molecular weight of the API is between 100-200, 200-300, 300-400, 400-500, 500-600, 600-700, 700-800, 800-900, 900-1000, 1000-1200, 1200-1400, 1400-1600, 1600-1800, or 1800-2000. APIs with the above molecular weights may also be specifically excluded from the present invention.
- the hydrogen bond donor moieties of a co-crystal can include, but are not limited to, any one, any two, any three, any four, or more of the following: amino-pyridine, primary amine, secondary amine, sulfonamide, primary amide, secondary amide, alcohol, and carboxylic acid.
- the hydrogen bond acceptor moieties of a co-crystal can include, but are not limited to, any one, any two, any three, any four, or more of the following: amino-pyridine, primary amine, secondary amine, sulfonamide, primary amide, secondary amide, alcohol, carboxylic acid, carbonyl, cyano, dimethoxyphenyl, sulfonyl, aromatic nitrogen (6 membered ring), ether, chloride, organochloride, bromide, organobromide, and organoiodide. Hydrogen bonds are known to form many supramolecular structures including, but not limited to, a catemer, a dimer, a trimer, a t ⁇ tramer, or a higher order structure.
- Tables V-XXI list specific hydrogen bond donor and acceptor moieties and their approximate interaction distances from the electromagnetic donor atom through the hydrogen atom to the electromagnetic acceptor atom.
- Table V lists functional groups that are known to hydrogen bond with amino-pyridines. Amino-pyridines comprise two distinct sites of hydrogen bond donation/acceptance. Both the aromatic nitrogen atom (Npy) and the amine group (NH 2 ) can participate in hydrogen bonds. The ability of a given functional group to participate in a hydrogen bond as a donor or as an acceptor or both can be determined by inspection by those skilled in the art.
- Tables V-XXI are taken from an analysis of solid-state structures as reported in the Cambridge Structural Database (CSD). These data include a number of hydrogen bonding interactions between many functional groups and their associated interaction distances.
- peptides, proteins, nucleic acids or other biological APIs are excluded from the present invention.
- all non- pharmaceutically acceptable co-crystal formers are excluded from the present invention.
- organometalic APIs are excluded from the present invention.
- a co-crystal former comprising any one or more of the functional groups of Table III may be specifically excluded from the present invention.
- any one or more of the co-crystal formers of Table I or II may be specifically excluded from the present invention. Any APIs currently known in the art may also be specifically excluded from the present invention.
- the API is not a salt, is not a non-metal salt, or is not a metal salt, e.g., sodium, potassium, lithium, calcium or magnesium.
- the API is a salt, is a non-metal salt, or is a metal salt, e.g., sodium, potassium, lithium, calcium, magnesium.
- the API does not contain a halogen. In one embodiment, the API does contain a halogen.
- any one or more of the APIs of Table IV may be specifically excluded from the present invention.
- Any APIs currently known in the art may also be specifically excluded from the present invention.
- nabumetone:2,3-naphthalenediol fluoxetine HCkbenzoic acid, fluoxetine HCLsuccinic acid, acetaminophe piperazine, acetaminophe theophylline, theophylline: salicylic acid, theophylline :p-hydiOxybenzoic acid, theophylline: sorbic acid, theophylline :l-hydroxy-2- naphthoic acid, theophylline :glycolic acid, theophylline:2,5-dihydroxybenzoic acid, theophylline xhloroacetic acid, bis(diphenylhydantoin):9-ethyladenine acetylacetone solvate, bis(diphenylhydantoin)
- a pharmaceutical composition can be formulated to contain an API in co-crystal form as micronized or nano-sized particles. More specifically, another embodiment couples the processing of a pure API to a co-crystal form with the process of making a controlled particle size for manipulation into a pharmaceutical dosage form. This embodiment combines two processing steps into a single step via techniques such as, but not limited to, grinding, alloying, or sintering (i.e., heating a powder mix). The coupling of these processes overcomes a serious limitation of having to isolate and store the bulk drug that is required for a formulation, which in some cases can be difficult to isolate (e.g., amorphous, chemically or physically unstable).
- Excipients employed in pharmaceutical compositions of the present invention can be solids, semi-solids, liquids or combinations thereof. Preferably, excipients are solids.
- Compositions of the invention containing excipients can be prepared by any known technique of pharmacy that comprises admixing an excipient with an API or therapeutic agent.
- a pharmaceutical composition of the invention contains a desired amount of API per dose unit and, if intended for oral administration, can be in the form, for example, of a tablet, a caplet, a pill, a hard or soft capsule, a lozenge, a cachet, a dispensable powder, granules, a suspension, an elixir, a dispersion, or any other form reasonably adapted for such administration.
- oral dosage forms that are discrete dose units each containing a predetermined amount of the API, such as tablets or capsules.
- APIs with an inappropriate pH for transdermal patches can be co-crystallized with an appropriate co-crystal former, thereby adjusting its pH to an appropriate level for use as a transdermal patch.
- an APIs pH level can be optimized for use in a transdermal patch via co-crystallization with an appropriate co-crystal former.
- Non-limiting examples follow of excipients that can be used to prepare pharmaceutical compositions of the invention.
- compositions of the invention optionally comprise one or more pharmaceutically acceptable carriers or diluents as excipients.
- suitable carriers or diluents illustratively include, but are not limited to, either individually or in combination, lactose, including anhydrous lactose and lactose monohydrate; starches, including directly compressible starch and hydrolyzed starches (e.g., CelutabTM and EmdexTM); mannitol; sorbitol; xylitol; dextrose (e.g., CereloseTM 2000) and dextrose monohydrate; dibasic calcium phosphate dihydrate; sucrose-based diluents; confectioner's sugar; monobasic calcium sulfate monohydrate; calcium sulfate dihydrate; granular calcium lactate trihydrate; dextrates; inositol; hydrolyzed cereal solids; amylose; celluloses including microcrystalline cellulose, food grade sources of alpha- and a
- Such carriers or diluents constitute in total about 5% to about 99%, preferably about 10% to about 85%, and more preferably about 20% to about 80%, of the total weight of the composition.
- the carrier, carriers, diluent, or diluents selected preferably exhibit suitable flow properties and, where tablets are desired, compressibility.
- Lactose, mannitol, dibasic sodium phosphate, and microcrystalline cellulose are preferred diluents. These diluents are chemically compatible with many co-crystals described herein.
- the use of extragranular microcrystalline cellulose that is, microcrystalline cellulose added to a granulated composition
- Lactose, especially lactose monohydrate is particularly preferred.
- Lactose typically provides compositions having suitable release rates of co-crystals, stability, pre- compression flowability, and/or drying properties at a relatively low diluent cost. It provides a high density substrate that aids densification during granulation (where wet granulation is employed) and therefore improves blend flow properties and tablet properties.
- compositions of the invention optionally comprise one or more pharmaceutically acceptable disintegrants as excipients, particularly for tablet formulations.
- Suitable disintegrants include, but are not limited to, either individually or in combination, starches, including sodium starch glycolate (e.g., ExplotabTM of PenWest) and pregelatinized corn starches (e.g., NationalTM 1551 of National Starch and Chemical Company, NationalTM 1550, and ColorconTM 1500), clays (e.g., VeegumTM HV of R.T.
- Vanderbilt celluloses such as purified cellulose, microcrystalline cellulose, methylcellulose, carboxymethylcellulose and sodium carboxymethylcellulose, croscarmellose sodium (e.g., Ac-Di-SolTM of FMC), alginates, crospovidone, and gums such as agar, guar, locust bean, karaya, pectin and fragacanth gums.
- Disintegrants may be added at any suitable step during the preparation of the composition, particularly prior to granulation or during a lubrication step prior to compression. Such disintegrants, if present, constitute in total about 0.2% to about 30%, preferably about 0.2% to about 10%, and more preferably about 0.2% to about 5%, of the total weight of the composition.
- Croscarmellose sodium is a preferred disintegrant for tablet or capsule disintegration, and, if present, preferably constitutes about 0.2% to about 10%, more preferably about 0.2% to about 7%, and still more preferably about 0.2% to about 5%, of the total weight of the composition. Croscarmellose sodium confers superior intragranular disintegration capabilities to granulated pharmaceutical compositions of the present invention.
- Pharmaceutical compositions of the invention optionally comprise one or more pharmaceutically acceptable binding agents or adhesives as excipients, particularly for tablet formulations. Such binding agents and adhesives preferably impart sufficient cohesion to the powder being tableted to allow for normal processing operations such as sizing, lubrication, compression and packaging, but still allow the tablet to disintegrate and the composition to be absorbed upon ingestion.
- binding agents may also prevent or inhibit crystallization or recrystallization of a co-crsytal of the present invention once the salt has been dissolved in a solution.
- Suitable binding agents and adhesives include, but are not limited to, either individually or in combination, acacia; fragacanth; sucrose; gelatin; glucose; starches such as, but not limited to, pregelatinized starches (e.g., NationalTM 1511 and NationalTM 1500); celluloses such as, but not limited to, methylcellulose and carmellose sodium (e.g., TyloseTM); alginic acid and salts of alginic acid; magnesium aluminum silicate; PEG; guar gum; polysaccharide acids; bentonites; povidone, for example povidone K-15, K-30 and K-29/32; polymethacrylates; HPMC; hydroxypropylcellulose (e.g., KlucelTM of Aqualon); and ethylcellulose (e.g., EthocelTM of the Dow Chemical
- binding agents are polymers comprising amide, ester, ether, alcohol or ketone groups and, as such, are preferably included in pharmaceutical compositions of the present invention.
- Polyvinylpyrrolidones such as povidone K-30 are especially preferred.
- Polymeric binding agents can have varying molecular weight, degrees of crosslinking, and grades of polymer.
- Polymeric binding agents can also be copolymers, such as block co-polymers that contain mixtures of ethylene oxide and propylene oxide units. Variation in these units' ratios in a given polymer affects properties and performance. Examples of block co-polymers with varying compositions of block units are Poloxamer 188 and Poloxamer 237 (BASF Corporation).
- compositions of the invention optionally comprise one or more pharmaceutically acceptable wetting agents as excipients.
- Such wetting agents are preferably selected to maintain the co-crystal in close association with water, a condition that is believed to improve bioavailability of the composition.
- Such wetting agents can also be useful in solubilizing or increasing the solubility of co-crystals.
- Non-limiting examples of surfactants that can be used as wetting agents in pharmaceutical compositions of the invention include quaternary ammonium compounds, for example benzalkonium chloride, benzethonium chloride and cetylpyridinium chloride, dioctyl sodium sulfosuccinate, polyoxyethylene alkylphenyl ethers, for example nonoxynol 9, nonoxynol 10, and degrees Ctoxynol 9, poloxamers (polyoxyethylene and polyoxypropylene block copolymers), polyoxyethylene fatty acid glycerides and oils, for example polyoxyethylene (8) caprylic/capric mono- and diglycerides (e.g., LabrasolTM of Gattefosse), polyoxyethylene (35) castor oil and polyoxyethylene (40) hydrogenated castor oil; polyoxyethylene alkyl ethers, for example polyoxyethylene (20) cetostearyl ether, polyoxyethylene fatty acid esters, for example polyoxyethylene (40) stearate, poly
- Sodium lauryl sulfate is a particularly preferred wetting agent.
- Sodium lauryl sulfate if present, constitutes about 0.25% to about 7%, more preferably about 0.4% to about 4%, and still more preferably about 0.5% to about 2%, of the total weight of the pharmaceutical composition.
- compositions of the invention optionally comprise one or more pharmaceutically acceptable lubricants (including anti-adherents and/or glidants) as excipients.
- suitable lubricants include, but are not limited to, either individually or in combination, glyceryl behapate (e.g., CompritolTM 888 of Gattefosse); stearic acid and salts thereof, including magnesium, calcium and sodium stearates; hydrogenated vegetable oils (e.g., Sterotex of Abitec); colloidal silica; talc; waxes; boric acid; sodium benzoate; sodium acetate; sodium fumarate; sodium chloride; DL-leucine; PEG (e.g., CarbowaxTM 4000 and CarbowaxTM 6000 of the Dow Chemical Company); sodium oleate; sodium lauryl sulfate; and magnesium lauryl sulfate.
- Such lubricants if present, constitute in total about 0. 1% to about 10%, preferably about
- Magnesium stearate is a preferred lubricant used, for example, to reduce friction between the equipment and granulated mixture during compression of tablet formulations.
- Suitable anti-adherents include, but are not limited to, talc, cornstarch, DL- leucine, sodium lauryl sulfate and metallic stearates.
- Talc is a preferred anti-adherent or glidant used, for example, to reduce formulation sticking to equipment surfaces and also to reduce static in the blend.
- Talc if present, constitutes about 0.1% to about 10%, more preferably about 0.25% to about 5%, and still more preferably about 0.5% to about 2%, of the total weight of the pharmaceutical composition.
- Glidants can be used to promote powder flow of a solid formulation. Suitable glidants include, but are not limited to, colloidal silicon dioxide, starch, talc, tribasic calcium phosphate, powdered cellulose and magnesium trisilicate. Colloidal silicon dioxide is particularly preferred.
- compositions of the invention can further comprise, for example, buffering agents.
- one or more effervescent agents can be used as disintegrants and/or to enhance organoleptic properties of pharmaceutical compositions of the invention.
- one or more effervescent agents are preferably present in a total amount of about 30% to about 75%, and preferably about 45% to about 70%, for example about 60%, by weight of the pharmaceutical composition.
- an effervescent agent present in a solid dosage form in an amount less than that effective to promote disintegration of the dosage form, provides improved dispersion of the API in an aqueous medium.
- the effervescent agent is effective to accelerate dispersion of the API from the dosage form in the gastrointestinal tract, thereby further enhancing absorption and rapid onset of therapeutic effect.
- an effervescent agent is preferably present in an amount of about 1% to about 20%, more preferably about 2.5% to about 15%, and still more preferably about 5% to about 10%, by weight of the pharmaceutical composition.
- an “effervescent agent” herein is an agent comprising one or more compounds which, acting together or individually, evolve a gas on contact with water.
- the gas evolved is generally oxygen or, most commonly, carbon dioxide.
- Preferred effervescent agents comprise an acid and a base that react in the presence of water to generate carbon dioxide gas.
- the base comprises an alkali metal or alkaline earth metal carbonate or bicarbonate and the acid comprises an aliphatic carboxylic acid.
- Non-limiting examples of suitable bases as components of effervescent agents useful in the invention include carbonate salts (e.g., calcium carbonate), bicarbonate salts (e.g., sodium bicarbonate), sesquicarbonate salts, and mixtures thereof. Calcium carbonate is a preferred base.
- Non-limiting examples of suitable acids as components of effervescent agents and/or solid organic acids useful in the invention include citric acid, tartaric acid (as D-, L-, or D/L-tartaric acid), malic acid (as D-, L-, or DL-malic acid), maleic acid, fumaric acid, adipic acid, succinic acid, acid anhydrides of such acids, acid salts of such acids, and mixtures thereof.
- Citric acid is a preferred acid.
- the weight ratio of the acid to the base is about 1 : 100 to about 100:1, more preferably about 1:50 to about 50:1, and still more preferably about 1 : 10 to about 10:1.
- the ratio of the acid to the base is approximately stoichiometric.
- Excipients which solubilize APIs typically have both hydrophilic and hydrophobic regions, or are preferably amphiphilic or have amphiphilic regions.
- One type of amphiphilic or partially-amphiphilic excipient comprises an amphiphilic polymer or is an amphiphilic polymer.
- a specific amphiphilic polymer is a polyalkylene glycol, which is commonly comprised of ethylene glycol and/or propylene glycol subunits.
- Such polyalkylene glycols can be esterified at their termini by a carboxylic acid, ester, acid anhyride or other suitable moiety.
- excipients examples include poloxamers (symmetric block copolymers of ethylene glycol and propylene glycol; e.g., poloxamer 237), polyalkyene glycolated esters of tocopherol (including esters formed from a di- or multi-functional carboxylic acid; e.g., d-alpha-tocopherol polyethylene glycol- 1000 succinate), and macrogolglycerides (formed by alcoholysis of an oil and esterification of a polyalkylene glycol to produce a mixture of mono-, di- and tri-glycerides and mono- and di-esters; e.g., stearoyl macrogol-32 glycerides).
- poloxamers symmetric block copolymers of ethylene glycol and propylene glycol
- polyalkyene glycolated esters of tocopherol including esters formed from a di- or multi-functional carboxylic acid; e.g., d-alpha-tocopherol
- compositions of the present invention can comprise about 10 % to about 50 %, about 25 % to about 50 %, about 30 % to about 45 %, or about 30 % to about 35 % by weight of a co-crystal; about 10 % to about 50 %, about 25 % to about 50 %, about 30 % to about 45 %, or about 30 % to about 35 % by weight of an excipient which inhibits crystallization in aqueous solution, in simulated gastric fluid, or in simulated intestinal fluid; and about 5 % to about 50 %, about 10 % to about 40 %, about 15 % to about 35 %, or about 30 % to about 35 % by weight of a binding agent.
- the weight ratio of the co-crystal to the excipient which inhibits crystallization to binding agent is about 1 to 1 to 1.
- Solid dosage forms of the invention can be prepared by any suitable process, not limited to processes described herein.
- An illustrative process comprises (a) a step of blending an API of the invention with one or more excipients to form a blend, and (b) a step of tableting or encapsulating the blend to form tablets or capsules, respectively.
- solid dosage forms are prepared by a process comprising (a) a step of blending a co-crystal of the invention with one or more excipients to form a blend, (b) a step of granulating the blend to form a granulate, and (c) a step of tableting or encapsulating the blend to form tablets or capsules respectively.
- Step (b) can be accomplished by any dry or wet granulation technique known in the art, but is preferably a dry granulation step.
- a salt of the present invention is advantageously granulated to form particles of about 1 micrometer to about 100 micrometer, about 5 micrometer to about 50 micrometer, or about 10 micrometer to about 25 micrometer.
- One or more diluents, one or more disintegrants and one or more binding agents are preferably added, for example in the blending step, a wetting agent can optionally be added, for example in the granulating step, and one or more disintegrants are preferably added after granulating but before tableting or encapsulating.
- a lubricant is preferably added before tableting. Blending and granulating can be performed independently under low or high shear.
- a process is preferably selected that forms a granulate that is uniform in API content, that readily disintegrates, that flows with sufficient ease so that weight variation can be reliably controlled during capsule filling or tableting, and that is dense enough in bulk so that a batch can be processed in the selected equipment and individual doses fit into the specified capsules or tablet dies.
- solid dosage forms are prepared by a process that includes a spray drying step, wherein an API is suspended with one or more excipients in one or more sprayable liquids, preferably a non-protic (e.g., non-aqueous or nonalcoholic) sprayable liquid, and then is rapidly spray dried over a current of warm air.
- a granulate or spray dried powder resulting from any of the above illustrative processes can be compressed or molded to prepare tablets or encapsulated to prepare capsules. Conventional tableting and encapsulation techniques known in the art can be employed. Where coated tablets are desired, conventional coating techniques are suitable.
- Excipients for tablet compositions of the invention are preferably selected to provide a disintegration time of less than about 30 minutes, preferably about 25 minutes or less, more preferably about 20 minutes or less, and still more preferably about 15 minutes or less, in a standard disintegration assay.
- compositions can be administered by controlled-, sustained-, or delayed-release means.
- Controlled-release pharmaceutical products have a common goal of improving drug therapy over that achieved by their non-controlled release counterparts.
- the use of an optimally designed controlled-release preparation in medical treatment is characterized by a minimum of drug substance being employed to cure or control the condition in a minimum amount of time.
- Controlled-release formulations include: 1) extended activity of the drug; 2) reduced dosage frequency; 3) increased patient compliance; 4) usage of less total drug; 5) reduction in local or systemic side effects; 6) minimization of drug accumulation; 7) reduction in blood level fluctuations; 8) improvement in efficacy of treatment; 9) reduction of potentiation or loss of drug activity; and 10) improvement in speed of control of diseases or conditions.
- Conventional dosage forms generally provide rapid or immediate drug release from the formulation. Depending on the pharmacology and pharmacokinetics of the drug, use of conventional dosage forms can lead to wide fluctuations in the concentrations of the drug in a patient's blood and other tissues. These fluctuations can impact a number of parameters, such as dose frequency, onset of action, duration of efficacy, maintenance of therapeutic blood levels, toxicity, side effects, and the like.
- controlled- release formulations can be used to control a drug's onset of action, duration of action, plasma levels within the therapeutic window, and peak blood levels.
- controlled- or extended-release dosage forms or formulations can be used to ensure that the maximum effectiveness of a drug is achieved while minimizing potential adverse effects and safety concerns, which can occur both from under dosing a drug (i.e., going below the minimum therapeutic levels) as well as exceeding the toxicity level for the drug.
- Controlled-release formulations are designed to initially release an amount of drug (active ingredient) that promptly produces the desired therapeutic effect, and gradually and continually release other amounts of drug to maintain this level of therapeutic or prophylactic effect over an extended period of time. In order to maintain this constant level of drug in the body, the drug must be released from the dosage form at a rate that will replace the amount of drug being metabolized and excreted from the body.
- Controlled-release of an active ingredient can be stimulated by various conditions including, but not limited to, pH, ionic strength, osmotic pressure, temperature, enzymes, water, and other physiological conditions or compounds.
- a variety of known controlled- or extended-release dosage forms, formulations, and devices can be adapted for use with the co-crystals and compositions of the mvention.
- Examples include, but are not limited to, those described in U.S. Pat. Nos.: 3,845,770; 3,916,899; 3,536,809; 3,598,123; 4,008,719; 5,674,533; 5,059,595; 5,591,767; 5,120,548; 5,073,543; 5,639,476; 5,354,556; 5,733,566; and 6,365,185 Bl; each of which is incorporated herein by reference.
- dosage forms can be used to provide slow or controlled-release of one or more active ingredients using, for example, liydroxypropylmethyl cellulose, other polymer matrices, gels, permeable membranes, osmotic systems (such as OROS® (Alza Corporation, Mountain View, Calif. USA)), multilayer coatings, microparticles, liposomes, or microspheres or a combination thereof to provide the desired release profile in varying proportions.
- ion exchange materials can be used to prepare immobilized, adsorbed co-crystals and thus effect controlled delivery of the drug. Examples of specific anion exchangers include, but are not limited to, Duolite® A568 and Duolite® AP143 (Rohm & Haas, Spring House, PA. USA).
- One embodiment of the invention encompasses a unit dosage form which comprises a pharmaceutically acceptable co-crystal, or a solvate, hydrate, dehydrate, anhydrous, or amorphous form thereof, and one or more pharmaceutically acceptable excipients or diluents, wherein the pharmaceutical composition or dosage form is formulated for controlled-release.
- Specific dosage forms utilize an osmotic drug delivery system.
- OROS® osmotic drug delivery system
- This technology can readily be adapted for the delivery of compounds and compositions of the invention.
- Various aspects of the technology are disclosed in U.S. Pat. Nos. 6,375,978 Bl; 6,368,626 Bl; 6,342,249 Bl; 6,333,050 B2; 6,287,295 Bl; 6,283,953 Bl; 6,270,787 Bl; 6,245,357 Bl; and 6,132,420; each of which is incorporated herein by reference.
- OROS® that can be used to administer compounds and compositions of the invention
- OROS® Push-PullTM Delayed Push-PullTM, Multi- Layer Push-PullTM, and Push-StickTM Systems, all of which are well known. See, e.g., http://www.alza.com.
- Additional OROS® systems that can be used for the controlled oral delivery of compounds and compositions of the invention include OROS®-CT and L- OROS®. Id.; see also, Delivery Times, vol. II, issue II (Alza Corporation).
- OROS® oral dosage forms are made by compressing a drag powder (e.g. co-crystal) into a hard tablet, coating the tablet with cellulose derivatives to form a semi-permeable membrane, and then drilling an orifice in the coating (e.g., with a laser).
- a drag powder e.g. co-crystal
- Kim, Cherng-ju, Controlled Release Dosage Form Design, 231-238 (Technomic Publishing, Lancaster, Pa.: 2000).
- the advantage of such dosage forms is that the delivery rate of the drug is not influenced by physiological or experimental conditions. Even a drug with a pH-dependent solubility can be delivered at a constant rate regardless of the pH of the delivery medium.
- OROS® drug delivery systems cannot be used to effectively deliver drugs with low water solubility. Id. at 234. Because co-crystals of this invention can be far more soluble in water than the API itself, they are well suited for osmotic-based delivery to patients. This invention does, however, encompass the incorporation of conventional crystalline API (e.g. pure API without co-crystal former), and non-salt isomers and isomeric mixtures thereof, into OROS® dosage forms.
- conventional crystalline API e.g. pure API without co-crystal former
- a specific dosage form of the invention comprises: a wall defining a cavity, the wall having an exit orifice formed or formable therein and at least a portion of the wall being semipermeable; an expandable layer located within the cavity remote from the exit orifice and in fluid communication with the semipermeable portion of the wall; a dry or substantially dry state drug layer located within the cavity adjacent to the exit orifice and in direct or indirect contacting relationship with the expandable layer; and a flow- promoting layer interposed between the inner surface of the wall and at least the external surface of the drug layer located within the cavity, wherein the drug layer comprises a co- crystal, or a solvate, hydrate, dehydrate, anhydrous, or amorphous form thereof. See U.S. Pat. No. 6,368,626, the entirety of which is incorporated herein by reference.
- Another specific dosage form of the invention comprises: a wall defining a cavity, the wall having an exit orifice formed or formable therein and at least a portion of the wall being semipermeable; an expandable layer located within the cavity remote from the exit orifice and in fluid communication with the semipermeable portion of the wall; a drug layer located within the cavity adjacent the exit orifice and in direct or indirect contacting relationship with the expandable layer; the drug layer comprising a liquid, active agent formulation absorbed in porous particles, the porous particles being adapted to resist compaction forces sufficient to form a compacted drug layer without significant exudation of the liquid, active agent formulation, the dosage form optionally having a placebo layer between the exit orifice and tlie drug layer, wherein the active agent formulation comprises a co-crystal, or a solvate, hydrate, dehydrate, anhydrous, or amorphous form thereof. See U.S. Pat. No. 6,342,249, the entirety of which is incorporated herein by reference.
- CrystalMaxTM comprises a sequence of automated, integrated high throughput robotic stations capable of rapid generation, identification and characterization of polymorphs, salts, and co-crystals of APIs and API candidates. Worksheet generation and combinatorial mixture design is carried out using proprietary design software ArchitectTM. Typically, an API or an API candidate is dispensed from an organic solvent into tubes and dried under a stream of nitrogen. Salts and/or co-crystal formers may also be dispensed and dried in the same fashion. Water and organic solvents may be combinatorially dispensed into the tubes using a multi-channel dispenser. Each tube in a 96-tube array is then sealed within 15 seconds of combinatorial dispensing to avoid solvent evaporation.
- the mixtures are then rendered supersaturated by heating to 70 degrees C for 2 hours followed by a 1 degree C/minute cooling ramp to 5 degrees C.
- Optical checks are then conducted to detect crystals and/or solid material. Once a solid has been identified in a tube, it is isolated through aspiration and drying. Raman spectra are then obtained on the solids and cluster classification of the spectral patterns is performed using proprietary software (InquireTM).
- Co-crystals may be obtained by dissolving the separate components in a solvent and adding one to the other. The co-crystal may then precipitate or crystallize as the solvent mixture is evaporated slowly. The co-crystal may also be obtained by dissolving the two components in the same solvent or a mixture of solvents.
- a co-crystal may be obtained by melting the two components together (i.e., co- melting) and allowing recrystallization to occur.
- an anti-solvent may be added to facilitate crystallization.
- a co-crystal may be obtained by melting the higher melting component on a glass slide and allowing it to recrystallize. The second component is then melted and is also allowed to recrystallize. The co-crystal may form as a separated phase/band in between the eutectic bands of the two original components.
- a co-crystal may be obtained by mixing or grinding two components together in the solid state.
- a co-crystal may be obtained by co-subliming a mixture of an API and a co- crystal former in the same sample cell as an intimate mixture either by heating, mixing or placing the mixture under vacuum.
- a co-crystal may also be obtained by co-sublimation using a Kneudsen apparatus where the API and the co-crystal former are contained in separate sample cells, connected to a single cold finger, each of the sample cells is maintained at the same or different temperatures under a vaccum atmosphere in order to co-sublime the two components onto the cold-finger forming the desired co-crystal.
- the purge gas used was dry nitrogen
- the reference material was an empty aluminum pan that was crimped
- the sample purge was 50 mL/minute.
- DSC analysis of the sample was performed by placing ⁇ 2 mg of sample in an aluminum pan with a crimped pan closure.
- the starting temperature was typically 20 degrees C with a heating rate of 10 degrees C/minute, and the ending temperature was 300 degrees C. Unless otherwise indicated, all reported transitions are as stated +/- 1.0 degrees C.
- TGA analysis of samples was performed using a Q500 Thermogravimetric Analyzer (TA Instruments, New Castle, DE, U.S.A.), which uses Advantage for QW- Series, version 1.0.0.78, Thermal Advantage Release 2.0 (2001 TA Instruments-Water LLC).
- the analysis software used was Universal Analysis 2000 for Windows 95/95/2000/NT, version 3.1E;Build 3.1.0.40 (2001 TA Instruments-Water LLC).
- the purge gas used was dry nitrogen, the balance purge was 40 mL/minute N 2 , and the sample purge was 60 mL/minute N 2 .
- TGA of the sample was performed by placing ⁇ 2 mg of sample in a platinum pan.
- the starting temperature was typically 20 degrees C with a heating rate of 10 degrees C/minute, and the ending temperature was 300 degrees C.
- a powder X-ray diffraction pattern for the samples was obtained using a D/Max Rapid, Contact (Rigaku/MSC, The Woodlands, TX, U.S.A.), which uses as its control software RINT Rapid Control software, Rigaku Rapid/XRD, version 1.0.0 (1999 Rigaku Co.).
- RINT Rapid Control software Rigaku Rapid/XRD, version 1.0.0 (1999 Rigaku Co.
- analysis software used were RINT Rapid display software, version 1.18 (Rigaku MSC), and JADE XRD Pattern Processing, versions 5.0 and 6.0 ((1995- 2002, Materials Data, Inc.).
- the acquisition parameters were as follows: source was Cu with a K line at 1.5406A; x-y stage was manual; collimator size was 0.3 or 0.8 mm; capillary tube (Charles Supper Company, Natick, MA, U.S.A.) was 0.3 mm ID; reflection mode was used; the power to the X-ray tube was 46 kV; the current to the X-ray tube was 40 mA; the omega-axis was oscillating in a range of 0-5 degrees at a speed of 1 degree/minute; the phi-axis was spinning at an angle of 360 degrees at a speed of 2 degrees/second; 0.3 or 0.8 mm collimator; the collection time was 60 minutes; the temperature was room temperature; and the heater was not used.
- the sample was presented to the X-ray source in a boron rich glass capillary.
- the analysis parameters were as follows: the integration 2-theta range was 2-40 or 60 degrees; the integration chi range was 0-360 degrees; the number of chi segments was 1; the step size used was 0.02; the integration utility was cylint; normalization was used; dark counts were 8; omega offset was 180; and chi and phi offsets were 0.
- the relative intensity of peaks in a diffractogram is not necessarily a limitation of the PXRD pattern because peak intensity can vary from sample to sample, e.g., due to crystalline impurities. Further, the angles of each peak can vary by about +/- 0.1 degrees, preferably +/-0.05. The entire pattern or most of the pattern peaks may also shift by about +/- 0.1 degree due to differences in calibration, settings, and other variations from instrument to instrument and from operator to operator. Procedure for Raman Acquisition. Filtering and Binning
- the sample was either left in the glass vial in which it was processed or an aliquot of the sample was transferred to a glass slide.
- the glass vial or slide was positioned in the sample chamber.
- the measurement was made using an AlmegaTM Dispersive Raman (AlmegaTM Dispersive Raman, Thermo-Nicolet, 5225 Verona Road, Madison, WI 53711- 4495) system fitted with a 785nm laser source.
- the sample was manually brought into focus using the microscope portion of the apparatus with a lOx power objective (unless otherwise noted), thus directing the laser onto the surface of the sample.
- the spectrum was acquired using the parameters outlined in Table XXII. (Exposure times and number of exposures may vary; changes to parameters will be indicated for each acquisition.)
- Each spectrum in a set was filtered using a matched filter of feature size 25 to remove background signals, including glass contributions and sample fluorescence. This is particularly important as large background signal or fluorescence limit the ability to accurately pick and assign peak positions in the subsequent steps of the binning process.
- Filtered spectra were binned using the peak pick and bin algorithm with the parameters given in Table XXIII.
- the sorted cluster diagrams for each sample set and the corresponding cluster assignments for each spectral file were used to identify groups of samples with similar spectra, which was used to identify samples for secondary analyses.
- Single crystal x-ray data were collected on a Bruker SMART-APEX CCD diffractometer (M. J. Zaworotko, Department of Chemistry, University of South Florida). Lattice parameters were determined from least squares analysis. Reflection data was integrated using the program SAINT. The structure was solved by direct methods and refined by full matrix least squares using the program SHELXTL (Sheldrick, G. M. SHELXTL, Release 5.03; Siemans Analytical X-ray Instruments Inc.: Madison, WI).
- the co-crystals of the present invention can be characterized, e.g., by the TGA or DSC data or by any one, any two, any three, any four, any five, any six, any seven, any eight, any nine, any ten, or any single integer number of PXRD 2-theta angle peaks or Raman shift peaks listed herein or disclosed in a figure, or by single crystal x-ray diffraction data.
- celecoxib icotinamide co-crystals were prepared. Celecoxib (100 mg, 0.26 mmol) and nicotinamide (32.0 mg, 0.26 mmol) were each dissolved in acetone (2 mL). The two solutions were mixed and the resulting mixture was allowed to evaporate slowly overnight. The precipitated solid was redissolved in acetone a second time and left to evaporate to dryness. The powder was collected and characterized. Detailed characterization of the celecoxib icotinamide co-crystal is listed in Table XXIV. Fig. 1A shows the PXRD diffractogram after subtraction of background noise. Fig. IB shows the raw PXRD data. Fig.
- FIG. 2 shows a DSC thermogram of the celecoxib :nicotinamide co- crystal.
- Fig. 3 shows a TGA thermogram of the celecoxib :nicotinamide co-crystal.
- Fig. 4 shows a Raman spectrum of the celecoxib:nicotinamide co-crystal.
- Co-crystals of topiramate and 18-crown-6 were prepared. To topiramate (100 mg, 0.29 mmol) dissolved in diethyl ether (5 mL) was added 18-crown-6 (78 mg, 0.29 mmol) in diethyl ether (5 mL). Upon addition of 18-crown-6, the solution became cloudy and was sonicated for 30 seconds. The solution was left standing for 1 hour and a colorless precipitate was observed. The precipitate was collected, washed with diethyl ether and dried to give a 1 :1 co-crystal of topiramate: 18-crown-6 as a colorless solid.
- Fig. 8 A shows the PXRD diffractogram after subtraction of background noise.
- Fig. 8B shows the raw PXRD data.
- Fig. 9 shows a DSC thermogram of the topiramate: 18-crown-6 co-crystal.
- Co-crystals of olanzapine and nicotinamide were prepared.
- a 9-block experiment was designed with 12 solvents.
- a block is a receiving plate, which can be, for example, an industry standard 24 well, 96 well, 384 well, or 1536 well format, or a custom format.
- 864 crystallization experiments with 10 co-crystal formers and 3 concentrations were carried out using the CrystalMaxTM platform.
- Form I was obtained from mixtures containing 1:1 and 1:2 molar ratios of olanzapine:nicotinamide in 1,2- dichloroethane.
- Form II was obtained from mixtures containing a 1 :2 molar ratio of olanzapine and nicotinamide in isopropyl acetate.
- PXRD and DSC characterization of the olanzapine:nicotinamide co-crystals are listed in Table XXIV.
- Fig. 10A shows the PXRD diffractogram of form I after subtraction of background noise.
- Fig. 10B shows the raw PXRD data of form I.
- Fig. 11 shows a DSC thermogram of the olanzapine :nicotinamide form I co-crystal.
- Fig. 12 shows the PXRD diffractogram of olanzapinemicotinamide form II after subtraction of background noise.
- Co-crystals of olanzapine and nicotinamide were prepared.
- Olanzapine 40 microliters of 25 mg/mL stock solution in tetrahydrofuran
- nicotinamide 37.6 microliters of 20 mg/mL stock solution in methanol
- To the solid mixture was added isopropyl acetate (100 microliters) and the vial was sealed with an aluminum cap.
- the suspension was then heated at 70 degrees C for two hours in order to dissolve all of the solid material.
- the solution was then cooled to 5 degrees C and maintained at that temperature for 24 hours.
- Fig. 13 A shows the PXRD diffractogram of form III after subtraction of background noise.
- Fig. 13B shows the raw PXRD data of form III.
- Figs. 14A-D show packing diagrams of the olanzapine:nicotinamide form III co-crystal.
- the olanzapine :nicotinamide (Form III) co-crystal is made up of a ternary system containing olanzapine, nicotinamide, water and isopropyl acetate in the unit cell.
- the co-crystal crystallizes in the monoclinic space group P2i/c and contains two olanzapine molecules, one nicotinamide molecule, 4 water molecules and one isopropyl acetate molecule in the asymmetric unit.
- the packing diagram is made up of a two-dimensional hydrogen-bonded network with the water molecules connecting the olanzapine and nicotinamide moieties.
- the packing diagram is also comprised of alternating olanzapine and nicotinamide layers connected through hydrogen bonding via the water and isopropyl acetate molecules, as shown in Figure 14B.
- the olanzapine layer propagates along the b axis at c/4 and 3c/4.
- the nicotinamide layer propagates along the b axis at c/2.
- the top of Figure 14C illustrates the nicotinamide superstructure.
- the nicotinamide molecules form dimers which hydrogen bond to chains of 4 water molecules.
- the water chains terminate with isopropyl acetate molecules on each side.
- a co-crystal of cw-itraconazole and succinic acid was prepared.
- succinic acid (16.8 mg, 0.142 mmol) in tetrahydrofuran (THF) (0.50 mL) was added cis- itraconazole (100 mg, 0.142 mmol).
- THF tetrahydrofuran
- a clear solution formed with heating (60 degrees C) and stirring. Upon cooling to room temperature (25 degrees C), crystals began to form.
- the solid was collected by filtration and washed with cold THF (2 mL).
- the white solid was air-dried and placed in a glass vial.
- the crystalline substance was found to be a succinic acid co-crystal of cz ' s-itraconazole.
- the solid was characterized by PXRD and DSC.
- Fig. 15 shows the PXRD diffractogram after subtraction of background noise.
- Fig. 16 shows a DSC thermogram
- a co-crystal of czAitraconazole and fumaric acid was prepared.
- To a blend of fumaric acid (8.40 mg, 0.072 mmol) and czAitraconazole (51.8 mg, 0.073 mmol) was added tetrahydrofuran (THF) (1.0 mL).
- THF tetrahydrofuran
- To the clear solution was added t-butyl methyl ether (1.0 mL).
- a white solid formed immediately and was collected by filtration and washed with cold t-butyl methyl ether (2 mL). The white solid was air-dried and placed in a glass vial.
- the crystalline substance was found to be a fumaric acid co-crystal of czAitraconazole.
- the solid was characterized by PXRD and DSC.
- Fig. 17 shows the PXRD diffractogram after subtraction of background noise.
- Fig. 18 shows a DSC thermogram of the co-crystal.
- a co-crystal of czAitraconazole and L-tartaric acid was prepared.
- L-tartaric acid (21.3 mg, 0.142 mmol) in tetrahydrofuran (THF) (0.50 mL) was added cis- itraconazole (100 mg, 0.142 mmol).
- THF tetrahydrofuran
- cis- itraconazole 100 mg, 0.142 mmol
- a clear solution formed with heating (60 degrees C) and stirring. Upon cooling to room temperature (25 degrees C), crystals began to form.
- the solid was collected by filtration and washed with cold THF (2 mL).
- the white solid was air-dried and placed in a glass vial.
- the crystalline substance was found to be an L- tartaric acid co-crystal of czAitraconazole.
- the solid was characterized by PXRD and DSC.
- Fig. 19 shows the PXRD diffractogram after
- a co-crystal of czAitraconazole and L-malic acid was prepared.
- L-malic acid (19.1 mg, 0.143 mmol) in tetrahydrofuran (THF) (0.50 mL) was added cis- itraconazole (100 mg, 0.142 mmol).
- THF tetrahydrofuran
- a clear solution formed with heating (60 degrees C) and stirring. Upon cooling to room temperature (25 degrees C), crystals began to form.
- the solid was collected by filtration and washed with cold THF (2 mL).
- the white solid was air-dried and placed in a glass vial.
- the crystalline substance was found to be an L- malic acid co-crystal of czAitraconazole.
- the solid was characterized by PXRD and DSC.
- Fig. 21 shows the PXRD diffractogram after subtraction of background noise.
- Fig. 22 shows a DSC thermogram of the
- a co-crystal of czAitraconazole hydrochloride and DL-tartaric acid was prepared.
- a suspension of c/Aitraconazole freebase (20.1 g, 0.0285 mol) in absolute ethanol (100 mL) was added a solution of hydrochloric acid (1.56 g, 0.0428 mol) and DL-tartaric acid (2.99 g, 0.0171mol) in absolute ethanol (100 mL).
- a clear solution formed with stirring and heating to reflux.
- the hot solution was gravity filtered and allowed to cool to room temperature (25 degrees C). Upon cooling white crystals formed.
- the solid was collected by filtration and washed with cold absolute ethanol (15 mL).
- the white solid was dried in a vacuum oven overnight at 80 degrees C.
- the crystalline substance was found to be a DL-tartaric acid co-crystal of ezAitraconazole hydrochloride.
- the solid was characterized by PXRD and DSC.
- Fig. 23 shows the PXRD diffractogram after subtraction of background noise.
- Fig. 24 shows a DSC thermogram of the co-crystal.
- Co-crystals of modafinil and malonic acid were prepared. Using a 250 mg/ml modafinil-acetic acid solution, malonic acid was dissolved on a hotplate (about 67 degrees C) at a 1 :2 modafinil to malonic acid ratio. The mixture was dried under flowing nitrogen overnight. A powdery white solid was produced. After further drying for 1 day, acetic acid was removed (as determined by TGA) and the crystal structure of the modafinikmalonic acid (Form I) co-crystal, as determined by PXRD, remained the same. The modafinil :malonic acid (Form I) co-crystal was also prepared by grinding the API and co-crystal former together.
- FIG. 26 shows a DSC thermogram of the modafinil :malonic acid Form I co-crystal.
- Fig. 27 shows the Raman spectrum of the modafinil :malonic acid Form I co-crystal.
- Fig. 27 comprises peaks, in order of decreasing intensity, of 1004, 222, 633, 265, 1032, 1183, 814, 1601, 490, 718, 767, 361, 917, 1104, 889, 412, 1225, 1251, 1398, 1442, 1731, 1298, 3065, and 2949 cm "1 .
- Single crystal data of the modafinikmalonic acid Form I co-crystal were acquired and are reported below.
- a polymorph of the modafinikmalonic acid Form I co-crystal was prepared in a vial. 11.4 mg of modafinil and 8.9 mg of malonic acid were dissolved in 2 mL of acetone. The solids dissolved at room temperature, and the vial was left open to evaporate the solvent in air. Large parallelogram shaped crystals formed on the walls and bottom of the vial.
- the PXRD diffractogram of the large crystals showed modafinil :malonic acid co-crystals Form II, a polymorphic form of modafinil :malonic acid Form I.
- Fig. 28 shows the PXRD diffractogram of the modafinil :malonic acid Form II co-crystal after subtraction of background noise.
- Co-crystals of modafinil and glycolic acid were prepared. Modafinil (1 mg, 0.0037mmol) and glycolic acid (0.30 mg, 0.0037 mmol) were dissolved in acetone (400 microliters). The solution was allowed to evaporate to dryness and the resulting solid was characterized using PXRD.
- PXRD data for the modafinil: glycolic acid co-crystal is listed in Table XXIV.
- Fig. 29A shows the PXRD diffractogram after subtraction of background noise.
- Fig. 29B shows the raw PXRD data.
- Co-crystals of modafinil and maleic acid were prepared. Using a 250 mg/ml modafinil-acetic acid solution, maleic acid was dissolved on a hotplate (about 67 degrees C) at a 2:1 modafinil to maleic ratio. The mixture was dried under flowing nitrogen overnight. A clear amorphous material remained. Solids began to grow after 2 days stored in a sealed vial at room temperature. The solid was collected and characterized as the modafinil :maleic acid co-crystal using PXRD. Fig. 30A shows the PXRD diffractogram after subtraction of background noise. Fig. 3 OB shows the raw PXRD data.
- FIG. 32 shows a DSC thermogram of the 5-fluorouracil:urea co-crystal.
- Fig. 33 shows a TGA thermogram of the 5-fluorouracil:urea co-crystal.
- Fig. 34 shows a Raman spectrum of the 5-fluorouracil:urea co-crystal. Single crystal data of the 5-fluorouracil:urea co- crystal were acquired and are reported below.
- Co-crystals of hydrochlorothiazide and nicotinic acid were prepared. Hydrochlorothiazide (12.2 mg, 0.041 mmol) and nicotinic acid (5 mg, 0.041 mmol) were dissolved in methanol (1 mL). The solution was then cooled to 5 degrees C and maintained at that temperature for 12 hours. A white solid precipitated and was collected and characterized as the hydrochlorothiazide:nicotinic acid co-crystal using PXRD.
- Fig. 35A shows the PXRD diffractogram after subtraction of background noise.
- Fig. 35B shows the raw PXRD data.
- Co-crystals of hydrochlorothiazide and 18-crown-6 were prepared. Hydrochlorothiazide (100 mg, 0.33 mmol) was dissolved in diethyl ether (15 mL) and was added to a solution of 18-crown-6 (87.2 mg, 0.33 mmol) in diethyl ether (15 mL). A white precipitate immediately began to form and was collected and characterized as the hydrochlorothiazide: 18-crown-6 co-crystal using PXRD.
- Fig. 36A shows the PXRD diffractogram after subtraction of background noise.
- Fig. 36B shows the raw PXRD data.
- Crystal packing The co-crystals contain bilayered sheets in which water molecules act as a hydrogen bonded bridge between the network bipyridine moieties and the acetaminophen. Bipyridine guests are sustained by ⁇ - ⁇ stacking interactions between two network bipyridines. The layers stack via ⁇ - ⁇ interactions between the phenyl groups of the acetaminophen moieties.
- Crystal packing The co-crystal is sustained by hydrogen bonding of adjacent phentoin molecules between the carbonyl and the amine closest to the tetrahedral carbon, and by hydrogen bonding between pyridone carbonyl functionalities and the amine not involved in phenytoin-phenytoin interactions.
- the pyridone carbonyl also hydrogen bonds with adjacent pyridone molecules forming a one-dimensional network.
- Infrared Spectroscopy (Nicolet Avatar 320 FTIR), characteristic peaks for the co-crystal were identified as: 2° amine found at 3311cm "1 , carbonyl (ketone) found at 1711cm "1 , olephin peak found at 1390cm "1 .
- Thermogravimetric Analysis (TA Instruments 2950 Hi-Resolution TGA), a 29.09% weight loss starting at 192.80 degrees C, 48.72% weight loss starting at 238.27 degrees C, and 18.38% loss starting at 260.17 degrees C followed by complete decomposition.
- the co-crystal contains the carboxylic acid-pyridine heterodimer that crystallizes in the Pbcn space group.
- the structure is an inclusion compound containing disordered solvent in the channels.
- ⁇ - ⁇ stacking of the bipyridine and phenyl groups of the aspirin and hydrophobic interactions contribute to the overall packing interactions.
- Infrared Spectroscopy (Nicolet Avatar 320 FTIR), characteristic (-COOH) peak at 1679 cm “1 was shifted up and less intense at 1694cm " , where as the lactone peak is shifted down slightly from 1750cm “1 to 1744cm “1 .
- Thermogravimetric Analysis (TA Instruments 2950 Hi-Resolution TGA), weight loss of 9% starting at 22.62 degrees C, 49.06% weight loss starting at 102.97 degrees C followed by complete decomposition starting at 209.37 degrees C.
- Crystal packing The co-crystal contains ibuprofembipyridine heterodimers, sustained by two hydrogen bonded carboxylic acidpyridine supramolecular synthons, arranged in a herringbone motif that packs in the space group P-1.
- the heterodimer is an extended version of the homodimer and packs to form a two-dimensional network sustained by ⁇ - ⁇ stacking of the bipyridine and phenyl groups of the ibuprofen and hydrophobic interactions from the ibuprofen tails.
- Thermogravimetric Analysis (TA Instruments 2950 Hi-Resolution TGA), 13.28% weight loss between room temperature and 100.02 degrees C immediately followed by complete decomposition.
- the co-crystal contains flurbiprofe bipyridine heterodimers, sustained by two hydrogen bonded carboxylic acidpyridine supramolecular synthon, arranged in a herringbone motif that packs in the space group P2j/n.
- the heterodimer is an extended version of the homodimer and packs to form a two-dimensional network sustained by ⁇ - ⁇ stacking and hydrophobic interactions of the bipyridine and phenyl groups of the flurbiprofen.
- Thermogravimetric Analysis (TA Instruments 2950 Hi-Resolution TGA), 30.93% weight loss starting at 31.13 degrees C and a 46.26% weight loss starting at 168.74 degrees C followed by complete decomposition.
- the co-crystal contains flurbiprofen: 1 ,2-bis (4-pyridyl) ethylene heterodimers, sustained by two hydrogen bonded carboxylic acid-pyridine supramolecular synthons, arranged in a herringbone motif that packs in the space group P2]/n.
- the heterodimer from 1,2-bis (4-pyridyl) ethylene further extends the homodimer relative to example 21 and packs to form a two-dimensional network sustained by ⁇ - ⁇ stacking and hydrophobic interactions of the bipyridine and phenyl groups of the flurbiprofen.
- Thermogravimetric Analysis (TA Instruments 2950 Hi-Resolution TGA), 91.79% weight loss starting at 133.18 degrees C followed by complete decomposition.
- Crystal packing The co-crystals contain hydrogen bonded carboxamide homodimers that crystallize in the space group C2/c.
- the 1° amines of the homodimer are bifurcated to the carbonyl of the/7-phthalaldehyde forming a chain with an adjacent homodimer.
- the chains pack in a crinkled tape motif sustained by ⁇ - ⁇ interactions between phenyl rings of the carbamazepine.
- Thermogravimetric Analysis (TA Instruments 2950 Hi-Resolution TGA), 17.66% weight loss starting at 30.33 degrees C then a 17.57% weight loss starting at 100.14 degrees C followed by complete decomposition.
- PXRD derived from the single crystal data, experimental (calculated): 8.5 (8.7); 10.6 (10.8); 11.9 (12.1); 14.4 (14.7) 15.1 (15.2); 18.0 (18.1); 18.5 (18.2); 19.8 (18.7); 23.7 (24.0); 24.2 (24.2); 26.4 (26.7); 27.6 (27.9); 27.8 (28.2); 28.7 (29.1); 29.3
- Crystal packing The co-crystals contain hydrogen bonded carboxamide homodimers.
- the 1° amines are bifurcated to the carbonyl of the nicotinamide on each side of the dimer.
- the 1° amines of each nicotinamide are hydrogen bonded to the carbonyl of the adjoining dimer.
- the dimers form chains with ⁇ - ⁇ interactions from the phenyl groups of the carbamazepine.
- Thermogravimetric Analysis (TA Instruments 2950 Hi-Resolution TGA), 57.94% weight loss starting at 205.43 degrees C followed by complete decomposition.
- PXRD Showed analogous peaks to the simulated PXRD derived from the single crystal data.
- carbamazepine saccharin co-crystals were also prepared via another method.
- a 12-block experiment was designed with 12 solvents.
- a block is a receiving plate, which can be an industry standard 96 well, 384 well, or 1536 well format, or a custom format.
- 1152 crystallization experiments were carried out using the CrystalMaxTM platform.
- the carbamazepine: saccharin co-crystal was obtained from a mixture of isopropyl acetate and heptane.
- the resulting co-crystal was characterized by PXRD and DSC and these data are shown in Figures 49 and 50, respectively.
- the co-crystal prepared from a mixture of isopropyl acetate and heptane may contain impurities such as carbamazepine in free form due to incomplete purification.
- Crystal packing The co-crystals contain hydrogen bonded carboxamide homodimers.
- the 2° amines of the saccharin are hydrogen bonded to the carbonyl of the carbamazepine on each side forming a tetramer.
- the crystal has a space group of P-1 with ⁇ - ⁇ interactions between the phenyl groups of the carbamazepine and the saccharin phenyl groups.
- PXRD derived from the single crystal data, experimental (calculated): 6.9 (7.0); 12.2 (12.2); 13.6 (13.8); 14.0 (14.1); 14.1 (14.4); 15.3 (15.6); 15.9 (15.9); 18.1 (18.2); 18.7 (18.8); 20.2 (20.3); 21.3 (21.5); 23.7 (23.9); 26.3 (26.4); 28.3 (28.3).
- Crystal packing Each hydrogen on the carbamazepine 1° amine is hydrogen bonded to a carbonyl group of a different 2,6-pyridinedicarboxylic acid moiety.
- the carbonyl of the carbamazepine carboxamide is hydrogen bonded to two hydroxide groups of one 2,6-pyridinedicarboxylic acid moiety.
- Crystal packing The co-crystals are sustained by hydrogen bonded carboxylic acid homodimers between the two 5-nitroisophthalic acid moieties and hydrogen bonded carboxy-amide heterodimers between the carbamazepine and 5-nitroisophthalic acid moiety. There is solvent hydrogen bonded to an additional N-H donor from the carbamazepine moiety.
- Thermogravimetric Analysis (TA Instruments 2950 Hi-Resolution TGA). 32.02% weight loss starting at 202 degrees C, a 12.12% weight loss starting at 224 degrees C and a 17.94% weight loss starting at 285 degrees C followed by complete decomposition.
- PXRD Showed analogous peaks to the simulated PXRD derived from the single crystal data.
- PXRD analysis experimental (calculated): 10.138 (10.283), 15.291 (15.607), 17.438 (17.791), 21.166 (21.685), 31.407 (31.738), 32.650 (32.729).
- Crystal packing The co-crystals form a single 3D network of four tetrahedron, linked by square planes similar to the PtS topology. The crystals are sustained by hydrogen bonding.
- Crystal packing The co-crystals contain hydrogen bonded carboxamide homodimers. Each 1° amine on the carbamazepine is bifurcated to a carbonyl group of a benzoquinone moiety. The dimers form infinite chains.
- Crystal packing The co-crystals are sustained by hydrogen bonded carboxylic acid homodimers between carbamazepine and trimesic acid moieties and hydrogen bonded carboxylic acid-amine heterodimers between two trimesic acid moieties arranged in a stacked ladder formation.
- Thermogravimetric Analysis (TA Instruments 2950 Hi-Resolution TGA). 62.83%o weight loss starting at 253 degrees C and a 30.20% weight loss starting at 278 degrees C followed by complete decomposition.
- PXRD analysis experimental 10.736, 12.087, 16.857, 24.857, 27.857.
- DSC Two endothermic transitions at about 117 and 119 degrees C and a sharp endotherm at about 130 degrees C
- TGA Decomposition beginning at about 150 degrees
- TGA Decomposition above 200 degrees C with a 25% weight loss between about 190-
- TGA Rapid decomposition beginning at about 135 degrees C and leveling off slightly after 200 degrees C
- TGA 29.09 percent weight loss starting at about 193 degrees C, 48.72 percent weight loss starting at about 238 degrees C, 18.38 percent weight loss starting at about 260 degrees C
- TGA 9 percent weight loss starting at about 23 degrees C, 49.06 percent weight loss starting at about 103 degrees C, decomposition starting at about 209 degrees C
- PXRD 16.8, 17.1, 18.1, 19.0, 20.0, 21.3, 22.7, 25.0, 26.0, 26.1, 28.2, 29.1
- TGA 30.93 percent weight loss starting at about 31 degrees C, 46.26 percent weight loss starting at about 169 degrees C
- TGA 17.66 percent weight loss starting at about 30 degrees C, 17.57 percent weight loss starting at about 100 degrees C
- TGA Decomposition beginning at about 150 degrees
- TGA 3.342 percent weight loss starting at about 67 degrees C, 55.09 percent weight loss starting at about 119 degrees C
- TGA 32.02 percent weight loss starting at about 202 degrees C, 12.12 percent weight loss starting at about 224 degrees C, 17.94 percent weight loss starting at about 285 degrees C
- TGA 9 percent weight loss starting at about 189 degrees C, 52 percent weight loss starting at about 251 degrees C, 31 percent weight loss starting at about 374 degrees C
- TGA 20.62 percent weight loss starting at about 168 degrees C, 78 percent weight loss starting at about 223 degrees C
- TGA 62.83 percent weight loss starting at about 253 degrees C, 30.20 percent weight loss starting at about 278 degrees C
- a co-crystal with a modulated dissolution profile has been prepared.
- Celecoxib nicotinamide co-crystals were prepared via methods shown in Example 1. (See Fig. 57)
- a co-crystal with a modulated dissolution profile has been prepared, cis- Itraconazole: succinic acid, cw-itraconazolei-tartaric acid and czAitraconazole:L-malic acid co-crystals were prepared via methods shown in Examples 5, 7 and 8. (See Fig. 58)
- a co-crystal of an unsaltable or difficult to salt API has been prepared.
- Celecoxib nicotinamide co-crystals were prepared via methods shown in Example 1.
- Example 34
- a co-crystal with an improved hygroscopicity profile has been prepared.
- Celecoxib nicotinamide co-crystals were prepared via methods shown in Example 1. (See Fig. 59)
- a co-crystal with reduced form diversity as compared to the API has been prepared.
- Co-crystals of carbamazepine and saccharin have been prepared via method shown in Example 25.
- the formulation of a modafinil :malonic acid form I co-crystal was completed using lactose.
- Two mixtures, one of modafinil and lactose, and the second of modafinil :malonic acid co-crystal and lactose, were ground together in a mortar an pestle.
- the mixtures targeted a 1 :1 weight ratio of modafinil to lactose.
- 901.2 mg of modafinil and 901.6 mg of lactose were ground together.
- 1221.6 mg of co-crystal and 871.4 mg of lactose were ground together.
- the resulting powders were analyzed by PXRD and DSC.
- the PXRD patterns and DSC thermograms of the mixtures showed virtually no change upon comparison with both individual components.
- the DSC of the co-crystal mixture showed only the co-crystal melting peak at 113.6 degrees C with a heat of fusion of 75.9 J/g. This heat of fusion is 59.5 % of that found for the co-crystal alone (127.5 J/g). This result is consistent with a 58.4 % weight ratio of co-crystal in the mixture.
- the DSC of the modafinil and lactose mixture had a melting point of 165.7 degrees C. This is slightly lower then the measured melting point of modafinil (168.7 degrees C).
- the heat of fusion of the mixture (59.3 J/g) is 46.9 % that of the modafinil alone (126.6 J/g), which is consistent with the estimated value of 50 %.
- Modafinil and the modafinikmalonic acid form I co-crystal were passed through a 38 micrometer sieve.
- Gelatin capsules Size 0, B&B Pharmaceuticals, Lot # 15-01202
- Dissolution studies were performed in a Vankel VK 7000 Benchsaver Dissolution Testing Apparatus with the VK750D heater/circulator set at 37 degrees C. At 0 minutes, the capsules were dropped into vessels containing 900 mL 0.01 M HCl and stirred by paddles.
- Absorbance readings were taken using a Cary 50 Spectrophotometer (wavelength set at 260nm) at the following time points: 0, 5, 10, 15, 20, 25, 30, 40, 50, and 60 minutes. The absorbance values were compared to those of standards and the modafinil concentrations of the solutions were calculated.
- Modafinil and the modafinil :malonic acid form I co-crystal were mixed with equivalent amounts of lactose (Spectrum, Lot QV0460) for approximately 5 minutes.
- Gelatin capsules Size 0, B&B Pharmaceuticals, Lot # 15-01202 were filled with 400.2 mg modafinil and lactose (approximately 200 mg modafinil), or 561.0 mg modafinil :malonic acid form I co-crystal and lactose (approximately 200 mg modafinil).
- HPMC capsules (Size 0, Shionogi, Lot # A312A6) were filled with 399.9 mg modafinil and lactose, 560.9 mg modafinikmalonic acid co-crystal and lactose, 199.9 mg modafinil, or 280.5 mg modafinil :malonic acid form I co-crystal.
- the dissolution study was carried out as described above.
- the modafinikmalonic acid form I co-crystal (from Example 10) was administered to dogs in a pharmacokinetic study.
- Particles of modafinil :malonic acid co- crystal with a median particle size of about 16 micrometers were administered in the study.
- micronized modafinil with a median particle size of about 2 micrometers was also administered in the study.
- the AUC of the modafinil :malonic acid co-crystal was determined to be 40 to 60 percent higher than that of the pure modafinil.
- Such a higher bioavailability illustrates the modulation of an important pharmacokinetic parameter due to an embodiment of the present invention.
- a compilation of important pharmacokinetic parameters measured during the animal study are included in Table XXV.
- the increased half-life and bioavailability of modafinil in the malonic acid form I co-crystal may be due to the presence of malonic acid. It is believed that the malonic acid may be inhibiting one or more pathways responsible for the metabolism or elimination of modafinil. It is noted that modafinil and malonic acid share a similar structure: each including two carbonyl or sulfonyl groups separated by a -CH 2 - and each molecule is terminated with a group that is capable of participation in a hydrogen bond with an enzyme. Such a mechanism may take place with other APIs or co-crystal formers of similar structure.
- the stability of the modafinikmalonic acid form I co-crystal was measured at various temperatures and relative humidities over a four week period. No degradation was found to occur at 20 or 0 degrees C. At 60 degrees C, about 0.14 percent degradation per day was determined based on a simple exponential model. At 80 degrees C, about 8 percent degradation per day was determined. TABLE I
Landscapes
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- General Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Biophysics (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- Molecular Biology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description






















































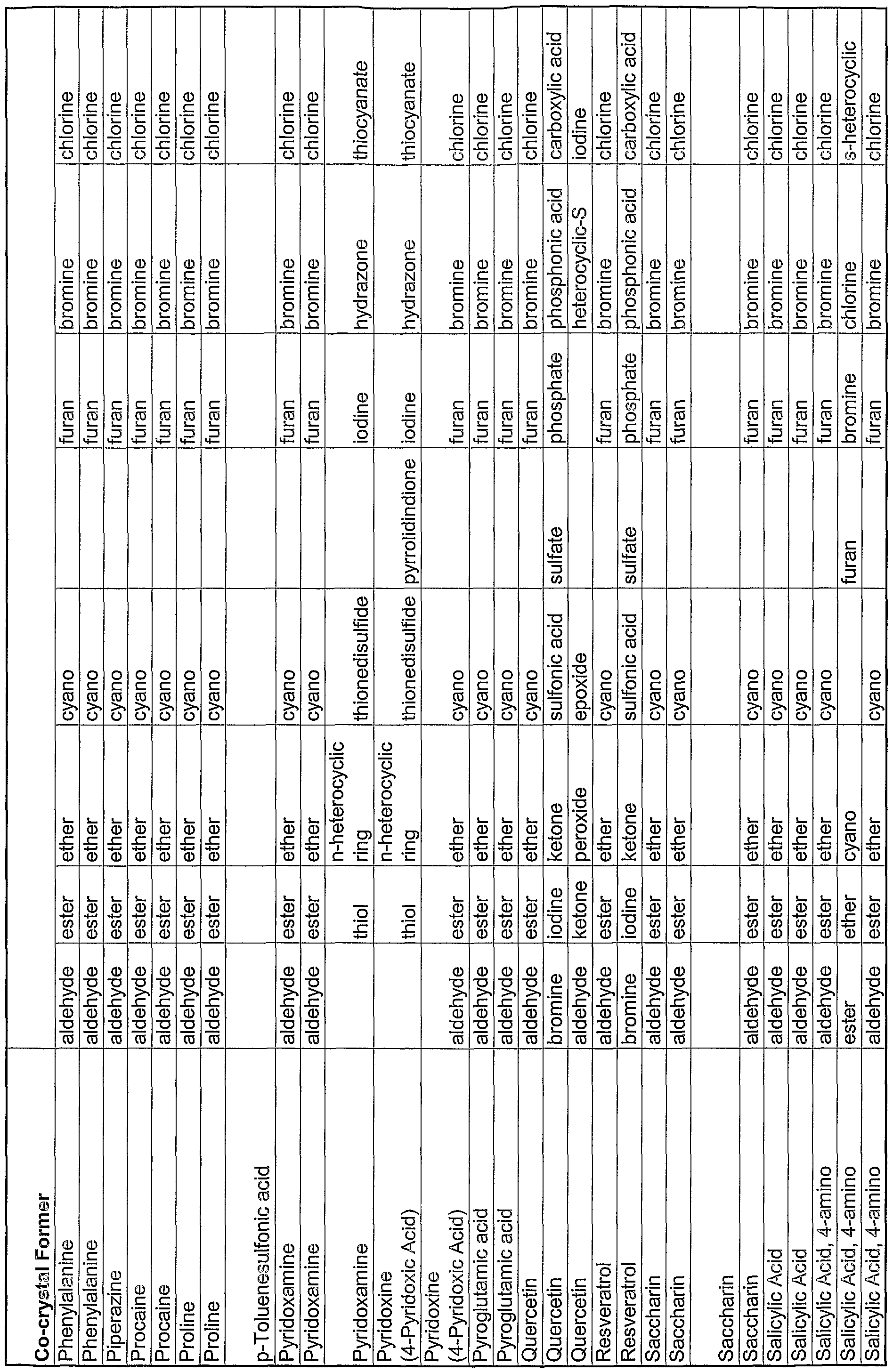








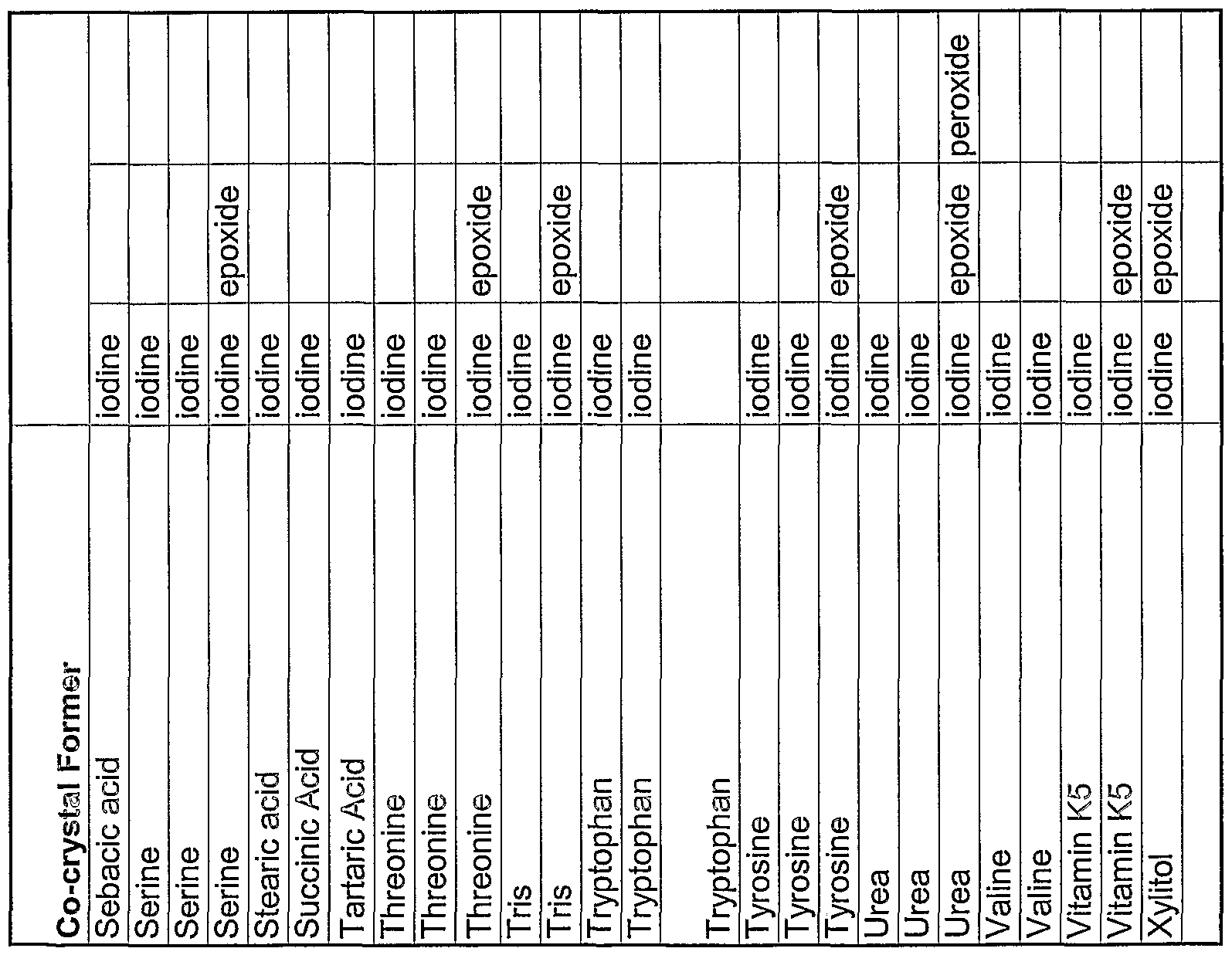



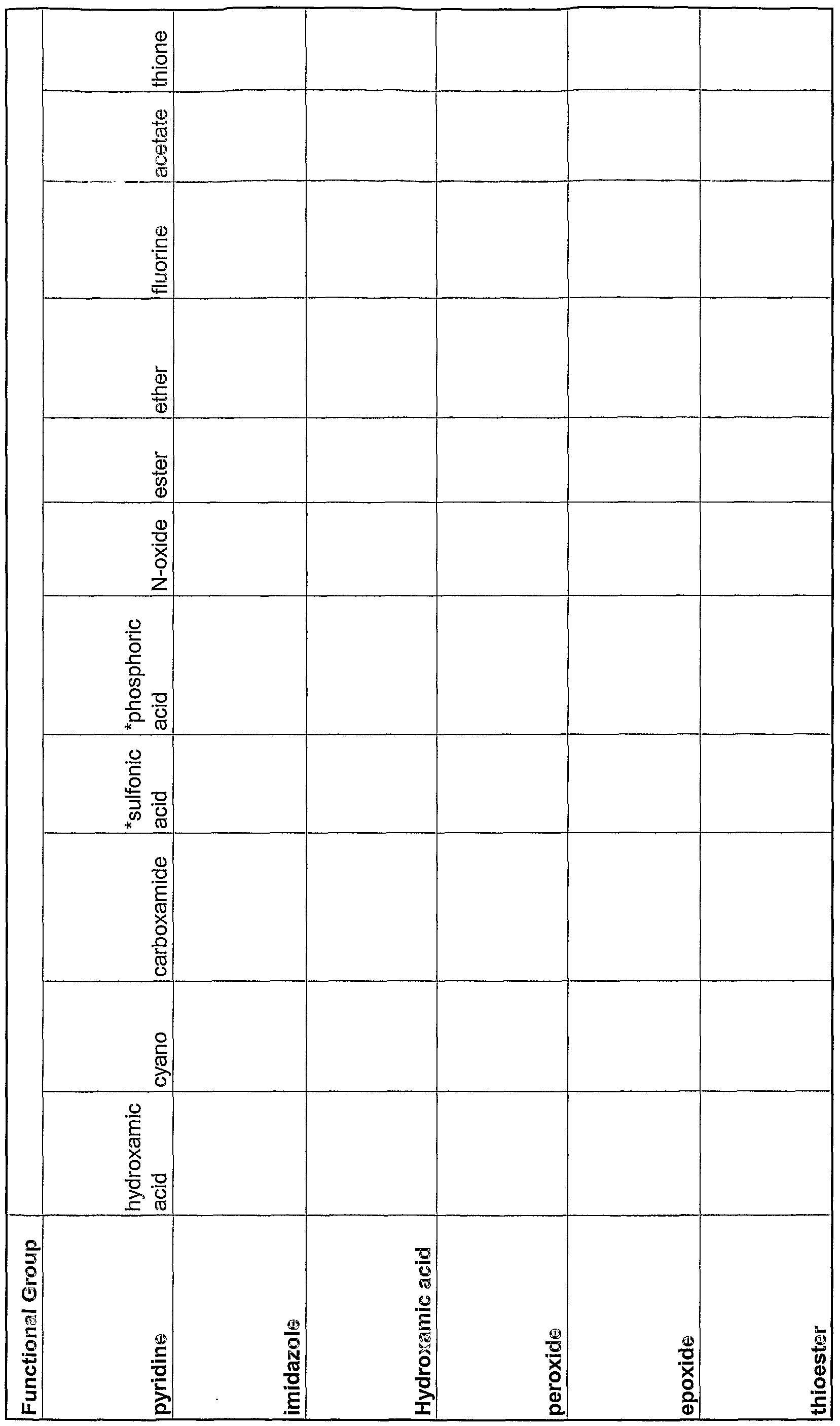















































































































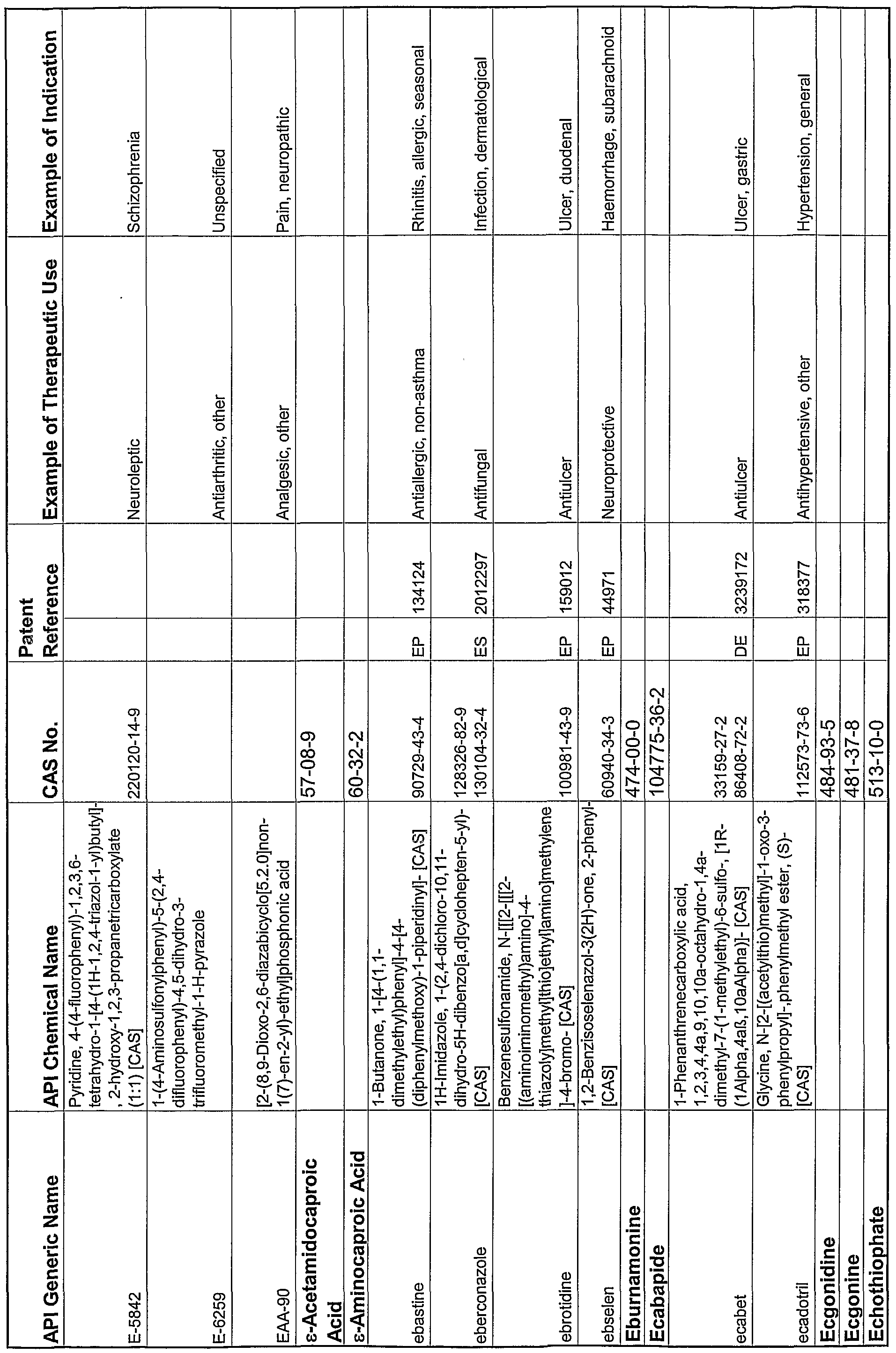


























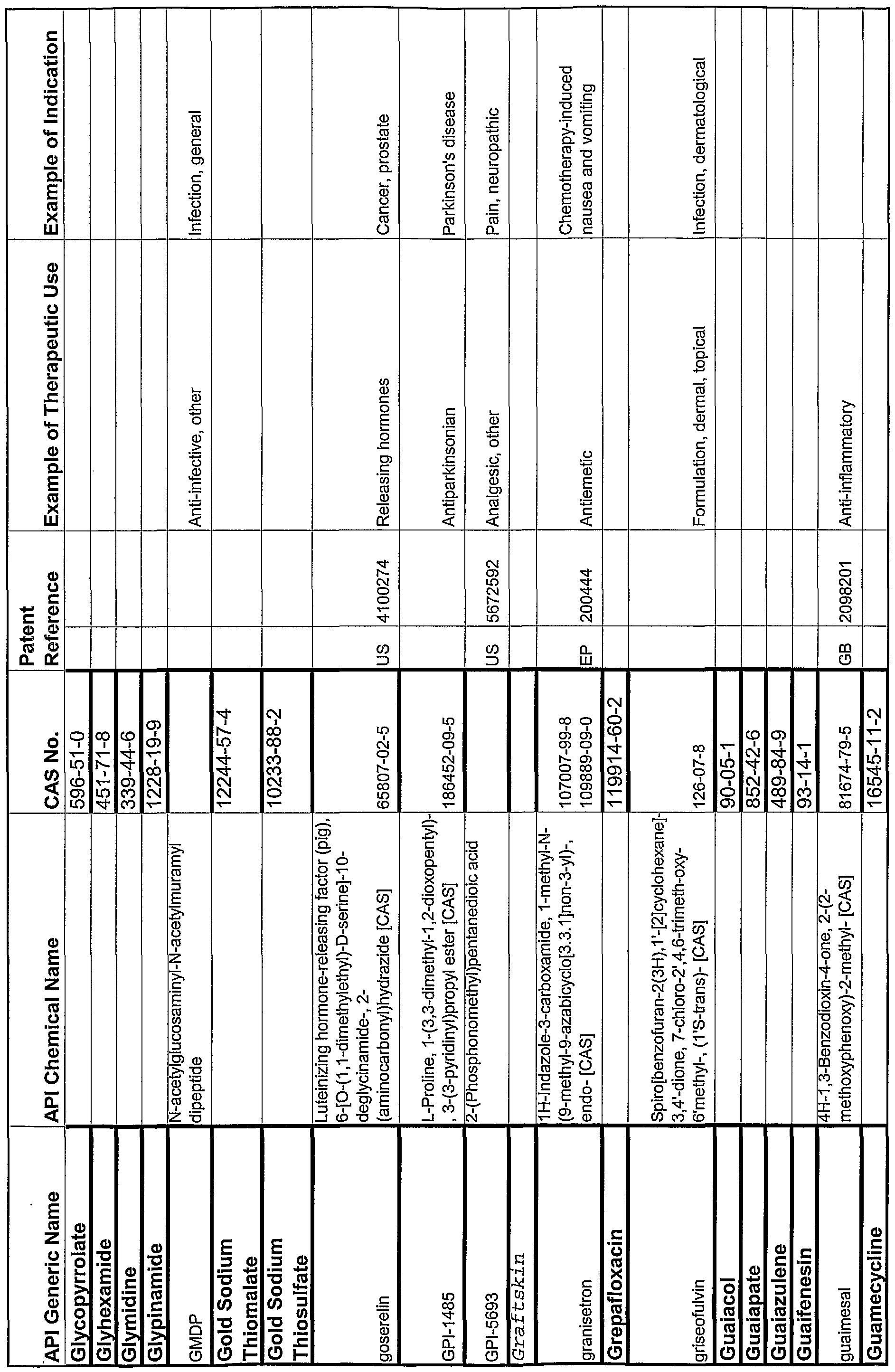



































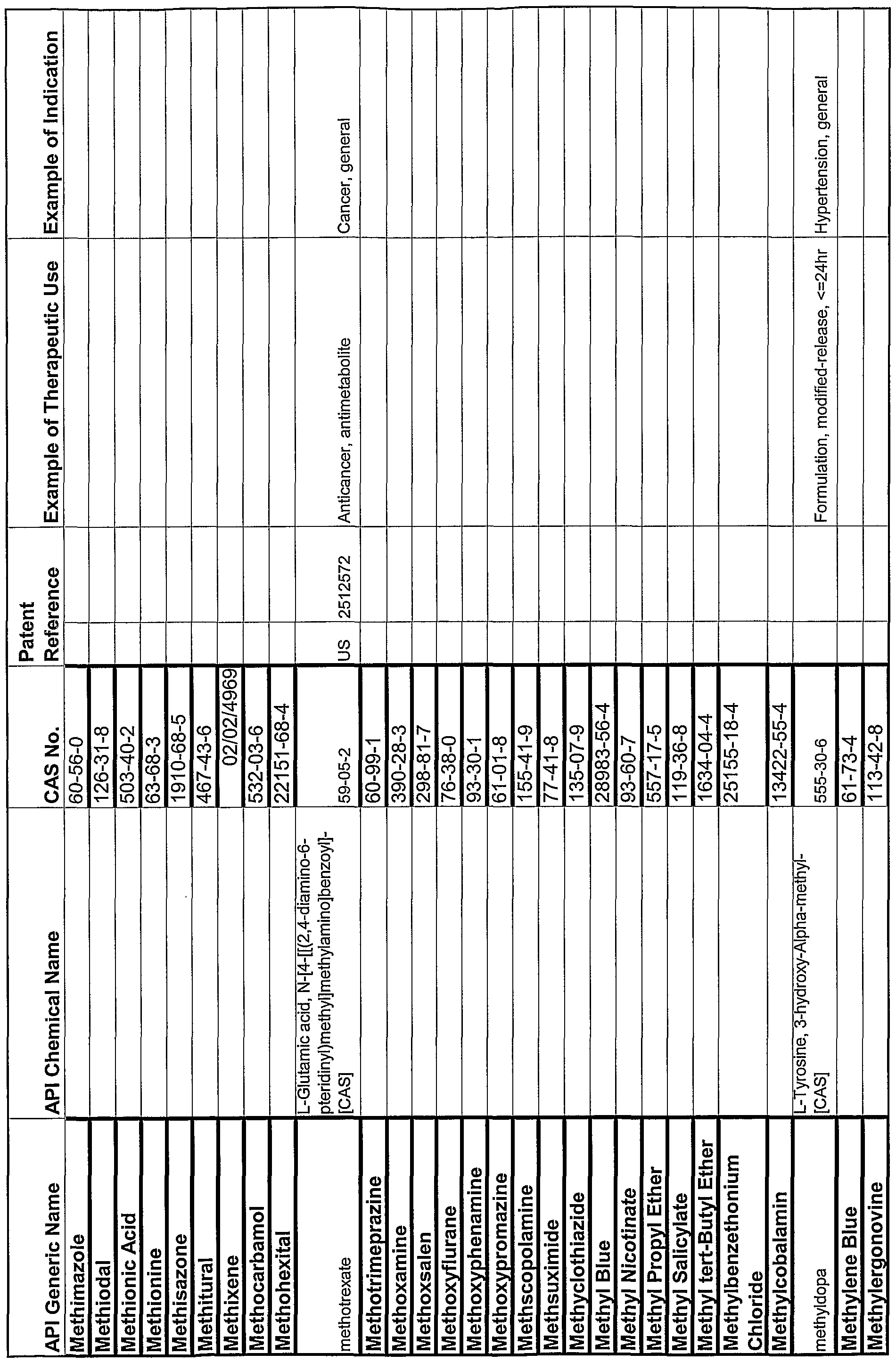





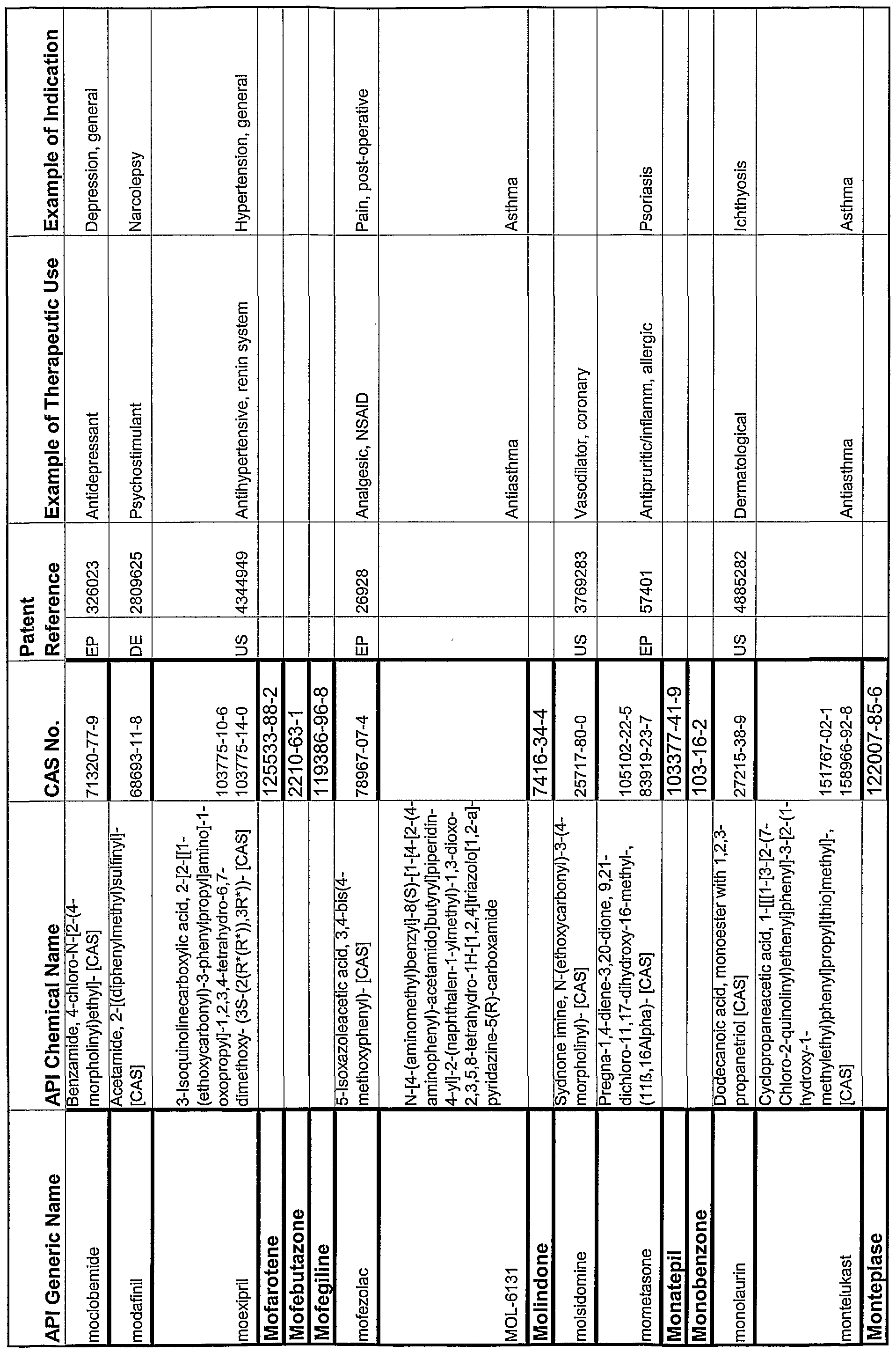




































































































Claims
Priority Applications (17)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002514733A CA2514733A1 (en) | 2003-02-28 | 2004-02-26 | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| JP2006508979A JP2007524596A (en) | 2003-02-28 | 2004-02-26 | Co-crystal pharmaceutical composition |
| EP04715190A EP1631260A2 (en) | 2003-02-28 | 2004-02-26 | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| US10/546,963 US20070059356A1 (en) | 2002-05-31 | 2004-02-26 | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| PCT/US2004/009947 WO2004089313A2 (en) | 2003-04-01 | 2004-03-31 | Novel olanzapine forms and related methods of treatment |
| US10/551,014 US20060223794A1 (en) | 2002-02-15 | 2004-03-31 | Novel olanzapine forms and related methods of treatment |
| US10/926,842 US7446107B2 (en) | 2002-02-15 | 2004-08-26 | Crystalline forms of conazoles and methods of making and using the same |
| JP2007500742A JP2007525502A (en) | 2004-02-26 | 2004-09-01 | Novel crystalline forms of conazoles and methods for their production and use |
| EP04782868A EP1718640A4 (en) | 2004-02-26 | 2004-09-01 | Novel crystalline forms of conazoles and methods of making and using the same |
| PCT/US2004/028456 WO2005092884A1 (en) | 2004-02-26 | 2004-09-01 | Novel crystalline forms of conazoles and methods of making and using the same |
| JP2006525508A JP4842819B2 (en) | 2003-09-04 | 2004-09-04 | Modafinil composition |
| EP20040783308 EP1670753A4 (en) | 2003-09-04 | 2004-09-04 | Modafinil compositions |
| IL173575A IL173575A0 (en) | 2003-09-04 | 2006-02-07 | Modafinil compositions |
| NO20060669A NO20060669L (en) | 2003-09-11 | 2006-02-10 | Modafinilsammensetninger |
| US12/234,420 US20090088443A1 (en) | 2002-02-15 | 2008-09-19 | Novel crystalline forms of conazoles and methods of making and using the same |
| IL199140A IL199140A (en) | 2003-09-04 | 2009-06-03 | Modafinil compositions |
| US12/792,415 US20100311701A1 (en) | 2002-02-15 | 2010-06-02 | Pharmaceutical Co-Crystal Compositions |
Applications Claiming Priority (26)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US45121303P | 2003-02-28 | 2003-02-28 | |
| US60/451,213 | 2003-02-28 | ||
| PCT/US2003/006662 WO2003074474A2 (en) | 2002-03-01 | 2003-03-03 | Multiple-component solid phases containing at least one active pharmaceutical ingredient |
| USPCT/US03/06662 | 2003-03-03 | ||
| US45602703P | 2003-03-18 | 2003-03-18 | |
| US60/456,027 | 2003-03-18 | ||
| US46396203P | 2003-04-18 | 2003-04-18 | |
| US60/463,962 | 2003-04-18 | ||
| US10/449,307 | 2003-05-30 | ||
| US10/449,307 US7078526B2 (en) | 2002-05-31 | 2003-05-30 | CIS-itraconazole crystalline forms and related processes, pharmaceutical compositions and methods |
| PCT/US2003/019574 WO2004000284A1 (en) | 2002-06-21 | 2003-06-20 | Pharmaceutical compositions with improved dissolution |
| USPCT/US03/19574 | 2003-06-20 | ||
| US10/601,092 US20050025791A1 (en) | 2002-06-21 | 2003-06-20 | Pharmaceutical compositions with improved dissolution |
| US10/601,092 | 2003-06-20 | ||
| US48706403P | 2003-07-11 | 2003-07-11 | |
| US60/487,064 | 2003-07-11 | ||
| USPCT/US03/27772 | 2003-09-04 | ||
| PCT/US2003/027772 WO2004078161A1 (en) | 2003-02-28 | 2003-09-04 | Pharmaceutical co-crystal compositions of drugs such as carbamazeptine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| US10/660,202 | 2003-09-11 | ||
| US10/660,202 US7927613B2 (en) | 2002-02-15 | 2003-09-11 | Pharmaceutical co-crystal compositions |
| US50820803P | 2003-10-02 | 2003-10-02 | |
| US60/508,208 | 2003-10-02 | ||
| USPCT/US03/41273 | 2003-12-24 | ||
| PCT/US2003/041273 WO2004061433A1 (en) | 2002-12-30 | 2003-12-24 | Pharmaceutical compositions with improved dissolution |
| US54275204P | 2004-02-06 | 2004-02-06 | |
| US60/542,752 | 2004-02-06 |
Related Parent Applications (6)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2003/006662 Continuation-In-Part WO2003074474A2 (en) | 2002-02-15 | 2003-03-03 | Multiple-component solid phases containing at least one active pharmaceutical ingredient |
| US10/449,307 Continuation-In-Part US7078526B2 (en) | 2002-02-15 | 2003-05-30 | CIS-itraconazole crystalline forms and related processes, pharmaceutical compositions and methods |
| US10/601,092 Continuation-In-Part US20050025791A1 (en) | 2002-02-15 | 2003-06-20 | Pharmaceutical compositions with improved dissolution |
| US10/637,829 Continuation-In-Part US20040053853A1 (en) | 2002-02-15 | 2003-08-08 | Topiramate salts and compositions comprising and methods of making and using the same |
| PCT/US2003/027772 Continuation-In-Part WO2004078161A1 (en) | 2002-02-15 | 2003-09-04 | Pharmaceutical co-crystal compositions of drugs such as carbamazeptine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| US10/660,202 Continuation-In-Part US7927613B2 (en) | 2002-02-15 | 2003-09-11 | Pharmaceutical co-crystal compositions |
Related Child Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/601,092 Continuation-In-Part US20050025791A1 (en) | 2002-02-15 | 2003-06-20 | Pharmaceutical compositions with improved dissolution |
| US10/926,842 Continuation-In-Part US7446107B2 (en) | 2002-02-15 | 2004-08-26 | Crystalline forms of conazoles and methods of making and using the same |
| US12/792,415 Continuation US20100311701A1 (en) | 2002-02-15 | 2010-06-02 | Pharmaceutical Co-Crystal Compositions |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2004078163A2 true WO2004078163A2 (en) | 2004-09-16 |
| WO2004078163A3 WO2004078163A3 (en) | 2005-01-20 |
Family
ID=56290526
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2004/006288 WO2004078163A2 (en) | 2002-02-15 | 2004-02-26 | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP1631260A2 (en) |
| JP (1) | JP2007524596A (en) |
| CA (1) | CA2514733A1 (en) |
| WO (1) | WO2004078163A2 (en) |
Cited By (395)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1511490A2 (en) * | 2002-05-31 | 2005-03-09 | Transform Pharmaceuticals, Inc. | Novel conazole crystalline forms and related processes, pharmaceutical compositions and methods |
| EP1670753A2 (en) * | 2003-09-04 | 2006-06-21 | Cephalon, Inc. | Modafinil compositions |
| WO2006103011A1 (en) * | 2005-03-30 | 2006-10-05 | Aicuris Gmbh & Co. Kg | Pharmaceutical preparation of n -[5-(aminosulfonyl)-4-methyl-1,3-thiazol-2-yl]-n-methyl-2-[4-(2-pyridinyl)phenyl] acetamide |
| WO2006138421A2 (en) * | 2005-06-15 | 2006-12-28 | Elan Pharma International Limited | Nanoparticulate azelnidipine formulations |
| WO2008054609A2 (en) * | 2006-10-04 | 2008-05-08 | The Regents Of The University Of Michigan | Dissolution and precipitation of cocrystals with ionizable components |
| WO2008063284A2 (en) * | 2006-10-10 | 2008-05-29 | Janssen Pharmaceutica Nv | Novel crystal of (s)-(+)-2-(2-chlorophenyl)-2-hydroxy-ethyl carbamate |
| EP1951393A2 (en) * | 2005-10-31 | 2008-08-06 | The Regents of the University of Michigan | Reaction co-crystallization of molecular complexes or co-crystals |
| EP1962600A2 (en) * | 2005-12-08 | 2008-09-03 | SSCI, Inc. | Metronidazole cocrystals and imipramine cocrystals |
| WO2008108639A1 (en) * | 2007-03-08 | 2008-09-12 | Avantium Holding B.V. | Co-crystalline forms of carbamazepine |
| WO2008153945A2 (en) * | 2007-06-06 | 2008-12-18 | University Of South Florida | Nutraceutical co-crystal compositions |
| US7507823B2 (en) | 2004-05-06 | 2009-03-24 | Bristol-Myers Squibb Company | Process of making aripiprazole particles |
| WO2009094155A1 (en) * | 2008-01-22 | 2009-07-30 | Thar Pharmaceuticals | In vivo studies of crystalline forms of meloxicam |
| WO2009140466A2 (en) * | 2008-05-14 | 2009-11-19 | Dr. Reddy's Laboratories Ltd. | Linezolid co-crystals |
| EP2123626A1 (en) * | 2008-05-21 | 2009-11-25 | Laboratorios del Dr. Esteve S.A. | Co-crystals of duloxetine and co-crystal formers for the treatment of pain |
| WO2009152347A2 (en) * | 2008-06-13 | 2009-12-17 | Bionevia Pharmaceuticals Inc. | Crystalline forms of zotepine hydrochloride |
| WO2010043412A1 (en) * | 2008-10-17 | 2010-04-22 | Laboratorios Del Dr. Esteve, S.A. | Co-crystals of tramadol and nsaids |
| WO2010017343A3 (en) * | 2008-08-06 | 2010-05-14 | Bionevia Pharmaceuticals, Inc. | Flupirtine hydrochloride maleic acid cocrystal |
| EP2199274A1 (en) * | 2008-12-16 | 2010-06-23 | Laboratorios Del. Dr. Esteve, S.A. | Co-crystals of tramadol and paracetamol |
| WO2010090491A2 (en) * | 2009-02-09 | 2010-08-12 | 주식회사 한독약품 | Novel salts of adefovir dipivoxil and preparation method thereof |
| US7790905B2 (en) | 2002-02-15 | 2010-09-07 | Mcneil-Ppc, Inc. | Pharmaceutical propylene glycol solvate compositions |
| WO2010115981A1 (en) | 2009-04-10 | 2010-10-14 | Novartis Ag | 7-azadispiro [3.0.4.1] decane-8-carboxamides as hepatitis c virus inhibitors |
| WO2010116248A1 (en) | 2009-04-10 | 2010-10-14 | Novartis Ag | Organic compounds and their uses |
| WO2010130796A1 (en) | 2009-05-15 | 2010-11-18 | Novartis Ag | Aryl pyridine as aldosterone synthase inhibitors |
| WO2010130773A2 (en) | 2009-05-15 | 2010-11-18 | Novartis Ag | Benzoxazolone derivatives as aldosterone symthase inhibitors |
| WO2010136493A1 (en) | 2009-05-28 | 2010-12-02 | Novartis Ag | Substituted aminopropionic derivatives as neprilysin inhibitors |
| EP2262488A1 (en) * | 2008-04-07 | 2010-12-22 | Mutual Pharmaceutical Company, Inc. | Colchicine solid complex; methods of making; and methods of use thereof |
| WO2011009943A1 (en) | 2009-07-24 | 2011-01-27 | Novartis Ag | Oxazine derivatives and their use as bace inhibitors for the treatment of neurological disorders |
| WO2011009852A2 (en) | 2009-07-21 | 2011-01-27 | Novartis Ag | Pharmaceutical compositions |
| US7883713B2 (en) | 2001-06-22 | 2011-02-08 | Aicuris Gmbh & Co. Kg | Topical application of thiazolyl amides |
| EP2281558A1 (en) * | 2009-08-06 | 2011-02-09 | Laboratorios Del. Dr. Esteve, S.A. | Pharmaceutical compounds of O-Desmethyl-Tramadol and COX-inhibitors |
| WO2011015652A1 (en) | 2009-08-07 | 2011-02-10 | Novartis Ag | 3-heteroarylmethyl-imidazo[1,2-b]pyridazin-6-yl derivatives as c-met tyrosine kinase modulators |
| WO2011018454A1 (en) | 2009-08-12 | 2011-02-17 | Novartis Ag | Heterocyclic hydrazone compounds and their uses to treat cancer and inflammation |
| WO2011020861A1 (en) | 2009-08-20 | 2011-02-24 | Novartis Ag | Heterocyclic oxime compounds |
| WO2011023677A1 (en) | 2009-08-26 | 2011-03-03 | Novartis Ag | Tetra-substituted heteroaryl compounds and their use as mdm2 and/or mdm4 modulators |
| WO2011026904A1 (en) | 2009-09-04 | 2011-03-10 | Novartis Ag | Pyrazinylpyridines useful for the treatment of proliferative diseases |
| WO2011026917A1 (en) | 2009-09-04 | 2011-03-10 | Novartis Ag | Heteroaryl compounds as kinase inhibitors |
| WO2011026911A1 (en) | 2009-09-04 | 2011-03-10 | Novartis Ag | Bipyridines useful for the treatment of proliferative diseases |
| WO2011029842A1 (en) | 2009-09-10 | 2011-03-17 | Novartis Ag | Sulfonamides as inhibitors of bcl-2 family proteins for the treatment of cancer |
| WO2011029915A1 (en) | 2009-09-10 | 2011-03-17 | Novartis Ag | Ether derivatives of bicyclic heteroaryls |
| US7927613B2 (en) | 2002-02-15 | 2011-04-19 | University Of South Florida | Pharmaceutical co-crystal compositions |
| WO2011054828A1 (en) | 2009-11-04 | 2011-05-12 | Novartis Ag | Heterocyclic sulfonamide derivatives useful as mek inhibitors |
| EP2325172A1 (en) * | 2009-11-02 | 2011-05-25 | Laboratorios Del. Dr. Esteve, S.A. | Co-crystals of celecoxib and L-proline |
| WO2011061271A1 (en) | 2009-11-20 | 2011-05-26 | Novartis Ag | Substituted carbamoylmethylamino acetic acid derivatives as novel nep inhibitors |
| WO2011061168A1 (en) | 2009-11-17 | 2011-05-26 | Novartis Ag | Aryl-pyridine derivatives as aldosterone synthase inhibitors |
| WO2011064376A1 (en) | 2009-11-30 | 2011-06-03 | Novartis Ag | Imidazole derivatives as aldosterone synthase inhibitors |
| WO2011073316A1 (en) | 2009-12-18 | 2011-06-23 | Novartis Ag | 4-aryl-butane-1,3-diamides |
| WO2011076747A1 (en) | 2009-12-21 | 2011-06-30 | Novartis Ag | Diaza-spiro[5.5]undecanes as orexin receptor antagonists |
| WO2011076786A1 (en) | 2009-12-22 | 2011-06-30 | Novartis Ag | Substituted isoquinolinones and quinazolinones |
| WO2011076744A1 (en) | 2009-12-21 | 2011-06-30 | Novartis Ag | Disubstituted heteroaryl-fused pyridines |
| WO2011080176A1 (en) | 2009-12-31 | 2011-07-07 | Novartis Ag | Pyrazine derivatives and their use in the treatment of neurological disorders |
| WO2011092290A1 (en) | 2010-02-01 | 2011-08-04 | Novartis Ag | Pyrazolo[5,1b]oxazole derivatives as crf-1 receptor antagonists |
| WO2011092293A2 (en) | 2010-02-01 | 2011-08-04 | Novartis Ag | Cyclohexyl amide derivatives as crf receptor antagonists |
| WO2011095450A1 (en) | 2010-02-02 | 2011-08-11 | Novartis Ag | Cyclohexyl amide derivatives as crf receptor antagonists |
| WO2011095452A1 (en) | 2010-02-02 | 2011-08-11 | Novartis Ag | Aryl benzylamine compounds |
| WO2011104266A1 (en) | 2010-02-25 | 2011-09-01 | Novartis Ag | Dimeric iap inhibitors |
| WO2011113894A1 (en) | 2010-03-19 | 2011-09-22 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of cf |
| WO2011131709A1 (en) | 2010-04-21 | 2011-10-27 | Novartis Ag | Furopyridine compounds and uses thereof |
| WO2011144666A1 (en) | 2010-05-20 | 2011-11-24 | Novartis Ag | 2,4-dioxo-1,4-dihydro-2h-quinazolin-3-yl-sulfonamide derivatives |
| WO2011157787A1 (en) | 2010-06-17 | 2011-12-22 | Novartis Ag | Biphenyl substituted 1,3-dihydro-benzoimidazol-2-ylideneamine derivatives |
| WO2011157793A1 (en) | 2010-06-17 | 2011-12-22 | Novartis Ag | Piperidinyl substituted 1,3-dihydro-benzoimidazol-2-ylideneamine derivatives |
| WO2011097372A3 (en) * | 2010-02-03 | 2011-12-29 | Aptuit Laurus Private Limited | Pterostilbene cocrystals |
| WO2012003189A1 (en) | 2010-06-29 | 2012-01-05 | Irm Llc | Compositions and methods for modulating the wnt signaling pathway |
| WO2012004299A1 (en) | 2010-07-06 | 2012-01-12 | Novartis Ag | Tetrahydro-pyrido-pyrimidine derivatives |
| WO2012006953A1 (en) | 2010-07-13 | 2012-01-19 | Novartis Ag | Oxazine derivatives and their use in the treatment of neurological disorders |
| WO2012007539A1 (en) | 2010-07-14 | 2012-01-19 | Novartis Ag | Ip receptor agonist heterocyclic compounds |
| WO2012010663A1 (en) | 2010-07-22 | 2012-01-26 | Novartis Ag | 2,3,5-trisubstituted thiophene compounds and uses thereof |
| WO2012017020A1 (en) | 2010-08-04 | 2012-02-09 | Novartis Ag | N-((6-amino-pyridin-3-yl)methyl)-heteroaryl-carboxamides as inhibitors of plasma kallikrein |
| WO2011158110A3 (en) * | 2010-04-28 | 2012-03-15 | Nuformix Limited | Cilostazol cocrystals and compositions |
| WO2012035158A1 (en) | 2010-09-17 | 2012-03-22 | Novartis Ag | Pyrazine derivatives as enac blockers |
| WO2012035023A1 (en) | 2010-09-13 | 2012-03-22 | Novartis Ag | Triazine-oxadiazoles |
| EP2432320A1 (en) * | 2009-05-20 | 2012-03-28 | Nutracryst Therapeutics Private Limited | Pharmaceutical co-crystals of quercetin |
| WO2012040230A1 (en) | 2010-09-20 | 2012-03-29 | Envivo Pharmaceuticals, Inc. | Imidazotriazinone compounds |
| WO2012048235A1 (en) | 2010-10-08 | 2012-04-12 | Novartis Ag | Vitamin e formulations of sulfamide ns3 inhibitors |
| WO2012055888A1 (en) | 2010-10-26 | 2012-05-03 | Novartis Ag | Diaza-spiro[5.5]undecanes useful as orexin receptor antagonists |
| US8183290B2 (en) | 2002-12-30 | 2012-05-22 | Mcneil-Ppc, Inc. | Pharmaceutically acceptable propylene glycol solvate of naproxen |
| WO2012065956A1 (en) | 2010-11-16 | 2012-05-24 | Novartis Ag | Substituted amino bisphenyl pentanoic acid derivatives as nep inhibitors |
| WO2012065953A1 (en) | 2010-11-16 | 2012-05-24 | Novartis Ag | Substituted carbamoylcycloalkyl acetic acid derivatives as nep inhibitors |
| WO2012065958A1 (en) | 2010-11-16 | 2012-05-24 | Novartis Ag | Method of treating contrast-induced nephropathy |
| WO2012080260A1 (en) | 2010-12-13 | 2012-06-21 | Novartis Ag | Dimeric iap inhibitors |
| WO2012080271A1 (en) | 2010-12-13 | 2012-06-21 | Novartis Ag | Dimeric iap inhibitors |
| WO2012087520A1 (en) | 2010-12-20 | 2012-06-28 | Irm Llc | Compositions and methods for modulating farnesoid x receptors |
| WO2012087521A1 (en) | 2010-12-20 | 2012-06-28 | Irm Llc | Compositions and methods for modulating farnesoid x receptors |
| WO2012084873A1 (en) | 2010-12-20 | 2012-06-28 | Novartis Ag | 4- (hetero) aryl - ethynyl - octahydro - indole - 1 - carboxylic acid esters |
| WO2012087519A1 (en) | 2010-12-20 | 2012-06-28 | Irm Llc | Compositions and methods for modulating fxr |
| WO2012093101A1 (en) | 2011-01-04 | 2012-07-12 | Novartis Ag | Indole compounds or analogues thereof useful for the treatment of age-related macular degeneration (amd) |
| WO2012095521A1 (en) | 2011-01-13 | 2012-07-19 | Novartis Ag | Bace-2 inhibitors for the treatment of metabolic disorders |
| WO2012095463A1 (en) | 2011-01-12 | 2012-07-19 | Novartis Ag | Oxazine derivatives and their use in the treatment of neurological disorders |
| WO2012095469A1 (en) | 2011-01-13 | 2012-07-19 | Novartis Ag | Novel heterocyclic derivatives and their use in the treatment of neurological disorders |
| WO2012098501A1 (en) * | 2011-01-21 | 2012-07-26 | Ranbaxy Laboratories Limited | Febuxostat co-crystals |
| WO2012101058A1 (en) | 2011-01-24 | 2012-08-02 | Novartis Ag | 4-tolyl-ethynyl-octahydro-indole-1-ester derivatives |
| WO2012101066A1 (en) | 2011-01-28 | 2012-08-02 | Novartis Ag | Pyridine biaryl amine compounds and their uses |
| WO2012101064A1 (en) | 2011-01-28 | 2012-08-02 | Novartis Ag | N-acyl pyrimidine biaryl compounds as protein kinase inhibitors |
| WO2012101487A1 (en) | 2010-12-22 | 2012-08-02 | Novartis Ag | Di/tri-aza-spiro-c9-c11alkanes |
| WO2012101063A1 (en) | 2011-01-28 | 2012-08-02 | Novartis Ag | N-acyl pyridine biaryl compounds and their uses |
| WO2012101065A2 (en) | 2011-01-28 | 2012-08-02 | Novartis Ag | Pyrimidine biaryl amine compounds and their uses |
| WO2012104823A2 (en) | 2011-02-04 | 2012-08-09 | Novartis Ag | Pyridopyrimidinone compounds in the treatment of neurodegenerative diseases |
| WO2012104776A1 (en) | 2011-01-31 | 2012-08-09 | Novartis Ag | Novel heterocyclic derivatives |
| WO2012107500A1 (en) | 2011-02-10 | 2012-08-16 | Novartis Ag | [1, 2, 4] triazolo [4, 3 -b] pyridazine compounds as inhibitors of the c-met tyrosine kinase |
| WO2012116217A1 (en) | 2011-02-25 | 2012-08-30 | Irm Llc | Compounds and compositions as trk inhibitors |
| US8258155B2 (en) | 2008-06-30 | 2012-09-04 | Mutual Pharmaceutical Company, Inc. | Quinine sulfate/bisulfate solid complex; methods of making; and methods of use thereof |
| WO2012117224A1 (en) | 2011-03-02 | 2012-09-07 | Sabrepharm Limited | Dipyridinium derivatives |
| WO2012120469A1 (en) | 2011-03-08 | 2012-09-13 | Novartis Ag | Fluorophenyl bicyclic heteroaryl compounds |
| WO2012131633A1 (en) | 2011-04-01 | 2012-10-04 | Novartis Ag | Pyrazolo pyrimidine derivatives |
| WO2012138648A1 (en) | 2011-04-06 | 2012-10-11 | Irm Llc | Compositions and methods for modulating lpa receptors |
| WO2012140597A1 (en) | 2011-04-14 | 2012-10-18 | Novartis Ag | Glycoside derivatives and their uses for the treatment of diabetes |
| WO2012140596A1 (en) | 2011-04-14 | 2012-10-18 | Novartis Ag | Glycoside derivatives and uses thereof |
| US8298580B2 (en) | 2006-11-17 | 2012-10-30 | Supernus Pharmaceuticals, Inc. | Sustained-release formulations of topiramate |
| WO2012146371A1 (en) * | 2011-04-28 | 2012-11-01 | Zentiva, K.S. | Pharmaceutically acceptable cocrystals of n-[2-(7-methoxy-1-naphtyl)ethyl]acetamide and methods of their preparation |
| WO2012129568A3 (en) * | 2011-03-24 | 2012-11-22 | University Of South Florida | Lithium compositions |
| WO2012156936A1 (en) | 2011-05-17 | 2012-11-22 | Novartis Ag | Substituted indole derivatives for the treatment of immunological disorders |
| WO2012164473A1 (en) | 2011-05-27 | 2012-12-06 | Novartis Ag | 3-spirocyclic piperidine derivatives as ghrelin receptor agonists |
| WO2012175520A1 (en) | 2011-06-20 | 2012-12-27 | Novartis Ag | Hydroxy substituted isoquinolinone derivatives |
| WO2012175487A1 (en) | 2011-06-20 | 2012-12-27 | Novartis Ag | Cyclohexyl isoquinolinone compounds |
| WO2013008095A1 (en) | 2011-07-08 | 2013-01-17 | Novartis Ag | Novel pyrrolo pyrimidine derivatives |
| WO2013008164A2 (en) | 2011-07-08 | 2013-01-17 | Novartis Ag | Method of treating atherosclerosis in high triglyceride subjects |
| WO2013008162A1 (en) | 2011-07-08 | 2013-01-17 | Novartis Ag | Novel trifluoromethyl-oxadiazole derivatives and their use in the treatment of disease |
| WO2013014627A1 (en) | 2011-07-27 | 2013-01-31 | Novartis Ag | Pyrazoline derivatives and their use as selective androgen receptor modulators |
| WO2013016197A1 (en) | 2011-07-22 | 2013-01-31 | Novartis Ag | Tetrahydropyrido-pyridine and tetrahydropyrido-pyrimidine compounds and use thereof as c5a receptor modulators |
| WO2013027188A1 (en) | 2011-08-25 | 2013-02-28 | Novartis Ag | 2 -amino-4 - (pyridin- 2 -yl) - 5, 6 -dihydro-4h- 1, 3 -oxazine derivatives and their use as bace-1 and/or bace - 2 inhibitors |
| WO2013033167A1 (en) | 2011-09-01 | 2013-03-07 | Irm Llc | Compounds and compositions as c-kit kinase inhibitors |
| WO2013033620A1 (en) | 2011-09-01 | 2013-03-07 | Irm Llc | Compounds and compositions as pdgfr kinase inhibitors |
| WO2013033203A1 (en) | 2011-09-01 | 2013-03-07 | Irm Llc | Compounds and compositions as c-kit kinase inhibitors |
| WO2013033070A1 (en) | 2011-09-01 | 2013-03-07 | Irm Llc | COMPOUNDS AND COMPOSITIONS AS c-KIT KINASE INHIBITORS |
| WO2013030802A1 (en) | 2011-09-01 | 2013-03-07 | Novartis Ag | Bicyclic heterocycle derivatives for the treatment of pulmonary arterial hypertension |
| WO2013033116A1 (en) | 2011-09-01 | 2013-03-07 | Irm Llc | Compounds and compositions as c-kit kinase inhibitors |
| WO2013035047A1 (en) | 2011-09-06 | 2013-03-14 | Novartis Ag | Benzothiazolone compound |
| US8399023B2 (en) | 2009-07-31 | 2013-03-19 | Thar Pharmaceuticals, Inc. | Crystallization method and bioavailability |
| WO2013038378A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | Pyridine amide derivatives |
| WO2013038362A1 (en) | 2011-09-15 | 2013-03-21 | Novartis Ag | 6 - substituted 3 - (quinolin- 6 - ylthio) - [1,2,4] triazolo [4, 3 -a] pyradines as tyrosine kinase |
| WO2013038386A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | Heterocyclic compounds for the treatment of cystic fibrosis |
| WO2013038373A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | Pyridine amide derivatives |
| WO2013038381A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | Pyridine/pyrazine amide derivatives |
| WO2013038390A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | N-substituted heterocyclyl carboxamides |
| WO2013050987A1 (en) | 2011-10-08 | 2013-04-11 | Novartis Ag | Carbamate/ urea derivatives containing piperidin and piperazin rings as h3 receptor inhibitors |
| WO2013054291A1 (en) | 2011-10-13 | 2013-04-18 | Novartis Ag | Novel oxazine derivatives and their use in the treatment of disease |
| WO2013057169A1 (en) | 2011-10-19 | 2013-04-25 | Universiteit Gent | Pharmaceutical nanosuspension |
| WO2013057711A1 (en) | 2011-10-21 | 2013-04-25 | Novartis Ag | Quinazoline derivatives as pi3k modulators |
| WO2013061305A1 (en) | 2011-10-28 | 2013-05-02 | Novartis Ag | Novel purine derivatives and their use in the treatment of disease |
| WO2013080141A1 (en) | 2011-11-29 | 2013-06-06 | Novartis Ag | Pyrazolopyrrolidine compounds |
| WO2013080120A1 (en) | 2011-11-28 | 2013-06-06 | Novartis Ag | Novel trifluoromethyl-oxadiazole derivatives and their use in the treatment of disease |
| WO2013093850A1 (en) | 2011-12-22 | 2013-06-27 | Novartis Ag | Quinoline derivatives |
| WO2013093849A1 (en) | 2011-12-22 | 2013-06-27 | Novartis Ag | Dihydro-benzo-oxazine and dihydro-pyrido-oxazine derivatives |
| WO2013105061A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | Fused dihydropyrido [2,3 -b] pyrazines as ip receptor agonists for the treatment of pulmonary arterial hypertension (pah) and related disorders |
| WO2013105058A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | 7,8- dihydropyrido [3, 4 - b] pyrazines as ip receptor agonists for the treatment of pulmonary arterial hypertension (pah) and related disorders |
| WO2013105065A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | Fused piperidines as ip receptor agonists for the treatment of pah and related disorders |
| WO2013105063A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | Fused piperidines as ip receptor agonists for the treatment of pulmonary arterial hypertension (pah) and related disorders |
| WO2013105057A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | Fused pyrroles as ip receptor agonists for the treatment of pulmonary arterial hypertension (pah) and related disorders |
| WO2013111108A1 (en) | 2012-01-27 | 2013-08-01 | Novartis Ag | 5-membered heteroarylcarboxamide derivatives as plasma kallikrein inhibitors |
| WO2013111107A1 (en) | 2012-01-27 | 2013-08-01 | Novartis Ag | Aminopyridine derivatives as plasma kallikrein inhibitors |
| US8507561B2 (en) | 2009-01-22 | 2013-08-13 | Absorption Pharmaceuticals, LLC | Desensitizing drug product |
| US8519002B2 (en) | 2008-04-07 | 2013-08-27 | Takeda Pharmaceuticals U.S.A., Inc. | Colchicine solid complex; methods of making; and methods of use thereof |
| WO2013128421A1 (en) | 2012-03-02 | 2013-09-06 | Novartis Ag | Spirohydantoin compounds and their use as selective androgen receptor modulators |
| US8555875B2 (en) | 2008-12-23 | 2013-10-15 | Map Pharmaceuticals, Inc. | Inhalation devices and related methods for administration of sedative hypnotic compounds |
| WO2013163508A1 (en) | 2012-04-27 | 2013-10-31 | Novartis Ag | Tetrahydropyran dgat1 inhibitors |
| WO2013160873A1 (en) | 2012-04-27 | 2013-10-31 | Novartis Ag | Cyclic bridgehead ether dgat1 inhibitors |
| WO2013164790A1 (en) | 2012-05-03 | 2013-11-07 | Novartis Ag | L-malate salt of 2, 7 - diaza - spiro [4.5 ] dec- 7 - yle derivatives and crystalline forms thereof as ghrelin receptor agonists |
| WO2013164802A1 (en) | 2012-05-04 | 2013-11-07 | Novartis Ag | Complement pathway modulators and uses thereof |
| WO2013171690A1 (en) | 2012-05-16 | 2013-11-21 | Novartis Ag | Monocyclic heteroaryl cycloalkyldiamine derivatives |
| WO2013175417A1 (en) | 2012-05-24 | 2013-11-28 | Novartis Ag | Pyrrolopyrrolidinone compounds |
| WO2013184766A1 (en) | 2012-06-06 | 2013-12-12 | Irm Llc | Compounds and compositions for modulating egfr activity |
| WO2013184757A1 (en) | 2012-06-06 | 2013-12-12 | Irm Llc | Compounds and compositions for modulating egfr activity |
| WO2013192345A1 (en) | 2012-06-20 | 2013-12-27 | Novartis Ag | Complement pathway modulators and uses thereof |
| WO2013189910A1 (en) * | 2012-06-22 | 2013-12-27 | Basf Se | Multicomponent crystals comprising imatinib mesilate and selected co-crystal formers |
| WO2014002051A2 (en) | 2012-06-28 | 2014-01-03 | Novartis Ag | Complement pathway modulators and uses thereof |
| WO2014002053A1 (en) | 2012-06-28 | 2014-01-03 | Novartis Ag | Pyrrolidine derivatives and their use as complement pathway modulators |
| WO2014002052A1 (en) | 2012-06-28 | 2014-01-03 | Novartis Ag | Pyrrolidine derivatives and their use as complement pathway modulators |
| WO2014002054A1 (en) | 2012-06-28 | 2014-01-03 | Novartis Ag | Pyrrolidine derivatives and their use as complement pathway modulators |
| WO2014002057A1 (en) | 2012-06-28 | 2014-01-03 | Novartis Ag | Pyrrolidine derivatives and their use as complement pathway modulators |
| WO2014002058A2 (en) | 2012-06-28 | 2014-01-03 | Novartis Ag | Complement pathway modulators and uses thereof |
| WO2014009833A2 (en) | 2012-07-12 | 2014-01-16 | Novartis Ag | Complement pathway modulators and uses thereof |
| WO2014013469A1 (en) | 2012-07-20 | 2014-01-23 | Novartis Ag | Carbamate/urea derivatives |
| US8652527B1 (en) | 2013-03-13 | 2014-02-18 | Upsher-Smith Laboratories, Inc | Extended-release topiramate capsules |
| WO2014028459A1 (en) | 2012-08-13 | 2014-02-20 | Novartis Ag | 1,4-disubstituted pyridazine analogs and methods for treating smn-deficiency-related conditions |
| WO2014027300A1 (en) | 2012-08-13 | 2014-02-20 | Novartis Ag | Bicyclic heteroaryl cycloalkyldiamine derivatives as inhibitors of spleen tyrosine kinases (syk) |
| WO2014033617A1 (en) | 2012-08-31 | 2014-03-06 | Novartis Ag | 2'-ethynyl nucleoside derivatives for treatment of viral infections |
| WO2014033630A1 (en) | 2012-08-31 | 2014-03-06 | Novartis Ag | Novel aminothiazole carboxamides as kinase inhibitors |
| WO2014033654A1 (en) | 2012-08-30 | 2014-03-06 | Novartis Ag | Salts of benzothiazolone compound as beta-2-adrenoceptor agonist |
| WO2014033631A1 (en) | 2012-08-31 | 2014-03-06 | Novartis Ag | N-(3-pyridyl) biarylamides as kinase inhibitors |
| WO2014037900A1 (en) | 2012-09-07 | 2014-03-13 | Novartis Ag | Indole carboxamide derivatives and uses thereof |
| WO2014047020A1 (en) | 2012-09-19 | 2014-03-27 | Novartis Ag | Dihydropyrrolidino-pyrimidines as kinase inhibitors |
| WO2014049540A2 (en) | 2012-09-29 | 2014-04-03 | Novartis Ag | Cyclic peptides and use as medicines |
| WO2014049514A1 (en) | 2012-09-25 | 2014-04-03 | Novartis Ag | Compounds for use in gastric complication |
| WO2014052619A1 (en) | 2012-09-27 | 2014-04-03 | Irm Llc | Piperidine derivatives and compositions as modulators of gpr119 activity |
| US8697735B2 (en) | 2008-07-25 | 2014-04-15 | Bionevia Pharmaceuticals, Inc. | Solid forms of epalrestat |
| WO2014072911A1 (en) | 2012-11-07 | 2014-05-15 | Novartis Ag | Substituted indole derivatives |
| WO2014072956A1 (en) | 2012-11-12 | 2014-05-15 | Novartis Ag | Oxazolidin-2-one-pyrimidine derivatives |
| WO2014078813A1 (en) | 2012-11-19 | 2014-05-22 | Irm Llc | Compounds and compositions for the treatment of parasitic diseases |
| WO2014078802A1 (en) | 2012-11-19 | 2014-05-22 | Irm Llc | Compounds and compositions for the treatment of parasitic diseases |
| WO2014082935A1 (en) | 2012-11-30 | 2014-06-05 | Novartis Ag | Cyclic nucleoside derivatives and uses thereof |
| WO2014091446A1 (en) | 2012-12-13 | 2014-06-19 | Novartis Ag | Pyrimido [4,5-b]quinoline-4,5 (3h,10h)-diones as nonsense mutation suppressors |
| WO2014093606A1 (en) | 2012-12-13 | 2014-06-19 | Novartis Ag | Pyridone derivatives and uses thereof in the treatment of tuberculosis |
| WO2014097148A1 (en) | 2012-12-19 | 2014-06-26 | Novartis Ag | Tricyclic compounds for inhibiting the cftr channel |
| WO2014099880A1 (en) | 2012-12-19 | 2014-06-26 | Novartis Ag | Aryl-substituted fused bicyclic pyridazine compounds |
| WO2014097151A2 (en) | 2012-12-19 | 2014-06-26 | Novartis Ag | Autotaxin inhibitors |
| WO2014097147A1 (en) | 2012-12-19 | 2014-06-26 | Novartis Ag | Tricyclic compounds as cftr inhibitors |
| WO2014115077A1 (en) | 2013-01-22 | 2014-07-31 | Novartis Ag | Substituted purinone compounds |
| WO2014115080A1 (en) | 2013-01-22 | 2014-07-31 | Novartis Ag | Pyrazolo[3,4-d]pyrimidinone compounds as inhibitors of the p53/mdm2 interaction |
| WO2014116845A1 (en) | 2013-01-23 | 2014-07-31 | Novartis Ag | Thiadiazole analogs thereof and methods for treating smn-deficiency-related-conditions |
| WO2014126979A1 (en) | 2013-02-14 | 2014-08-21 | Novartis Ag | Substituted bisphenyl butanoic phosphonic acid derivatives as nep (neutral endopeptidase) inhibitors |
| WO2014125413A1 (en) | 2013-02-13 | 2014-08-21 | Novartis Ag | Ip receptor agonist heterocyclic compounds |
| WO2014128612A1 (en) | 2013-02-20 | 2014-08-28 | Novartis Ag | Quinazolin-4-one derivatives |
| WO2014132205A1 (en) | 2013-02-28 | 2014-09-04 | Novartis Ag | Formulation comprising benzothiazolone compound |
| WO2014143638A1 (en) | 2013-03-14 | 2014-09-18 | Novartis Ag | 2-(1h-indol-4-ylmethyl)-3h-imidazo[4,5-b]pyridine-6-carbonitrile derivatives as complement factor b inhibitors useful for the treatment of ophthalmic diseases |
| WO2014151784A1 (en) | 2013-03-15 | 2014-09-25 | Irm Llc | Compounds and compositions for the treatment of parasitic diseases |
| WO2014151729A1 (en) | 2013-03-15 | 2014-09-25 | Irm Llc | Compounds and compositions for the treatment of parasitic diseases |
| WO2014151616A1 (en) | 2013-03-14 | 2014-09-25 | Novartis Ag | Biaryl amide compounds as kinase inhibitors |
| WO2014160649A1 (en) | 2013-03-29 | 2014-10-02 | Novartis Ag | Hydroxamic acid derivatives as lpxc inhibitors for the treatment of bacterial infections |
| US8859607B2 (en) | 2007-02-09 | 2014-10-14 | Basf Se | Crystalline complexes of agriculturally active organic compounds |
| WO2014167528A1 (en) | 2013-04-11 | 2014-10-16 | Novartis Ag | Spiropyrazolopyridine derivatives and uses thereof for the treatment of viral infections |
| WO2014178040A1 (en) | 2013-04-29 | 2014-11-06 | Mapi Pharma Ltd. | Co-crystals of dapagliflozin |
| WO2014184778A1 (en) | 2013-05-17 | 2014-11-20 | Novartis Ag | Pyrimidin-4-yl)oxy)-1h-indole-1-carboxamide derivatives and use thereof |
| WO2014191906A1 (en) | 2013-05-28 | 2014-12-04 | Novartis Ag | Pyrazolo-pyrrolidin-4-one derivatives as bet inhibitors and their use in the treatment of disease |
| WO2014191894A1 (en) | 2013-05-27 | 2014-12-04 | Novartis Ag | Imidazopyrrolidinone derivatives and their use in the treatment of disease |
| WO2014191896A1 (en) | 2013-05-27 | 2014-12-04 | Novartis Ag | Pyrazolopyrrolidine derivatives and their use in the treatment of disease |
| WO2014191911A1 (en) | 2013-05-28 | 2014-12-04 | Novartis Ag | Pyrazolo-pyrrolidin-4-one derivatives and their use in the treatment of disease |
| US8906948B2 (en) | 2008-09-06 | 2014-12-09 | Bionevia, LLC | Choline cocrystal of epalrestat |
| WO2015008230A1 (en) | 2013-07-18 | 2015-01-22 | Novartis Ag | Autotaxin inhibitors comprising a heteroaromatic ring-benzyl-amide-cycle core |
| WO2015008229A1 (en) | 2013-07-18 | 2015-01-22 | Novartis Ag | Autotaxin inhibitors |
| WO2015009616A1 (en) | 2013-07-15 | 2015-01-22 | Novartis Ag | Piperidinyl indole derivatives and their use as complement factor b inhibitors |
| WO2015059668A1 (en) | 2013-10-25 | 2015-04-30 | Novartis Ag | Ring-fused bicyclic pyridyl derivatives as fgfr4 inhibitors |
| WO2015066241A1 (en) | 2013-10-30 | 2015-05-07 | Novartis Ag | 2-benzyl-benzimidazole complement factor b inhibitors and uses thereof |
| WO2015066188A1 (en) | 2013-11-01 | 2015-05-07 | Novartis Ag | Aminoheteroaryl benzamides as kinase inhibitors |
| WO2015066413A1 (en) | 2013-11-01 | 2015-05-07 | Novartis Ag | Oxazolidinone hydroxamic acid compounds for the treatment of bacterial infections |
| WO2015075665A1 (en) | 2013-11-21 | 2015-05-28 | Novartis Ag | Pyrrolopyrrolone derivatives and their use as bet inhibitors |
| WO2015079417A1 (en) | 2013-11-29 | 2015-06-04 | Novartis Ag | Novel amino pyrimidine derivatives |
| WO2015095301A2 (en) | 2013-12-17 | 2015-06-25 | Irm Llc | Cytotoxic peptides and conjugates thereof |
| WO2015095477A1 (en) | 2013-12-19 | 2015-06-25 | Irm Llc | [1,2,4]triazolo[1,5-a]pyrimidine derivatives as protozoan proteasome inhibitors for the treatment of parasitic diseases such as leishmaniasis |
| WO2015092740A1 (en) | 2013-12-20 | 2015-06-25 | Novartis Ag | Heteroaryl butanoic acid derivatives as lta4h inhibitors |
| WO2015102929A1 (en) | 2013-12-30 | 2015-07-09 | Novartis Ag | Tricyclic sulfonamide derivatives |
| US9101545B2 (en) | 2013-03-15 | 2015-08-11 | Upsher-Smith Laboratories, Inc. | Extended-release topiramate capsules |
| WO2015148379A1 (en) | 2014-03-24 | 2015-10-01 | Novartis Ag | Monobactam organic compounds for the treatment of bacterial infections |
| WO2015070177A3 (en) * | 2013-11-11 | 2015-10-08 | Collaborative Medicinal Development, Llc | Metal complexes and methods of treatment |
| WO2015161052A1 (en) | 2014-04-17 | 2015-10-22 | Novartis Ag | Polycyclic herg activators |
| US9169279B2 (en) | 2009-07-31 | 2015-10-27 | Thar Pharmaceuticals, Inc. | Crystallization method and bioavailability |
| WO2015162461A1 (en) | 2014-04-24 | 2015-10-29 | Novartis Ag | Pyrazine derivatives as phosphatidylinositol 3-kinase inhibitors |
| WO2015164458A1 (en) | 2014-04-22 | 2015-10-29 | Novartis Ag | Isoxazoline hydroxamic acid derivatives as lpxc inhibitors |
| WO2015162558A1 (en) | 2014-04-24 | 2015-10-29 | Novartis Ag | Autotaxin inhibitors |
| WO2015162459A1 (en) | 2014-04-24 | 2015-10-29 | Novartis Ag | Amino pyrazine derivatives as phosphatidylinositol 3-kinase inhibitors |
| WO2015162456A1 (en) | 2014-04-24 | 2015-10-29 | Novartis Ag | Amino pyridine derivatives as phosphatidylinositol 3-kinase inhibitors |
| WO2015173659A2 (en) | 2014-05-14 | 2015-11-19 | Novartis Ag | Carboxamide derivatives |
| WO2015175487A1 (en) | 2014-05-13 | 2015-11-19 | Novartis Ag | Compounds and compositions for inducing chondrogenesis |
| WO2015173656A1 (en) | 2014-05-14 | 2015-11-19 | Novartis Ag | Carboxamide derivatives |
| WO2015175796A1 (en) | 2014-05-14 | 2015-11-19 | Novartis Ag | Carboxamide derivatives |
| WO2015177646A1 (en) | 2014-05-14 | 2015-11-26 | Novartis Ag | Carboxamide derivatives |
| WO2015181747A1 (en) | 2014-05-28 | 2015-12-03 | Novartis Ag | Novel pyrazolo pyrimidine derivatives and their use as malt1 inhibitors |
| WO2015186062A1 (en) | 2014-06-03 | 2015-12-10 | Novartis Ag | PYRIMIDO[4,5-b]QUINOLINE-4,5(3H,10H)-DIONE DERIVATIVES |
| WO2015186061A1 (en) | 2014-06-03 | 2015-12-10 | Novartis Ag | Pyridopyrimidinedione derivatives |
| WO2015186063A1 (en) | 2014-06-03 | 2015-12-10 | Novartis Ag | Naphthyridinedione derivatives |
| WO2015189791A1 (en) | 2014-06-13 | 2015-12-17 | Novartis Ag | Auristatin derivatives and conjugates thereof |
| WO2016001876A1 (en) | 2014-07-02 | 2016-01-07 | Novartis Ag | Thiophen-2-yl-pyridin-2-yl-1h-pyrazole-4-carboxylic acid derivatives and the use thereof as soluble guanylate cyclase activators |
| WO2016001878A1 (en) | 2014-07-02 | 2016-01-07 | Novartis Ag | Cyclohexen-1-yl-pyridin-2-yl-1h-pyrazole-4-carboxylic acid derivatives and the use thereof as soluble guanylate cyclase activators |
| WO2016001875A1 (en) | 2014-07-02 | 2016-01-07 | Novartis Ag | Indane and indoline derivatives and the use thereof as soluble guanylate cyclase activators |
| WO2016020836A1 (en) | 2014-08-06 | 2016-02-11 | Novartis Ag | Quinolone derivatives as antibacterials |
| CN105399665A (en) * | 2015-12-11 | 2016-03-16 | 吉林大学珠海学院 | Deferiprone pharmaceutical cocrystal with p-hydroxybenzoic acid as precursor and preparation method thereof |
| CN105399664A (en) * | 2015-12-11 | 2016-03-16 | 吉林大学珠海学院 | Deferiprone pharmaceutical cocrystal with 2,5-dihydroxybenzoic acid as precursor and preparation method thereof |
| WO2016038583A1 (en) | 2014-09-12 | 2016-03-17 | Novartis Ag | Compounds and compositions as kinase inhibitors |
| WO2016038582A1 (en) | 2014-09-12 | 2016-03-17 | Novartis Ag | Compounds and compositions as raf kinase inhibitors |
| WO2016038581A1 (en) | 2014-09-12 | 2016-03-17 | Novartis Ag | Compounds and compositions as raf kinase inhibitors |
| WO2016049266A1 (en) * | 2014-09-24 | 2016-03-31 | Pain Therapeutics, Inc. | 4:3 naltrexone: 5-methyl-2-furaldehyde cocrystal |
| CN105541701A (en) * | 2015-12-11 | 2016-05-04 | 吉林大学珠海学院 | Pharmaceutical cocrystal of deferiprone with maleic acid as precursor, and preparation method thereof |
| US9340565B2 (en) | 2010-11-24 | 2016-05-17 | Thar Pharmaceuticals, Inc. | Crystalline forms |
| WO2016079669A1 (en) | 2014-11-19 | 2016-05-26 | Novartis Ag | Labeled amino pyrimidine derivatives |
| WO2016088082A1 (en) | 2014-12-05 | 2016-06-09 | Novartis Ag | Amidomethyl-biaryl derivatives complement factor d inhibitors and uses thereof |
| WO2016097995A1 (en) | 2014-12-16 | 2016-06-23 | Novartis Ag | Isoxazole hydroxamic acid compounds as lpxc inhibitors |
| WO2016103155A1 (en) | 2014-12-23 | 2016-06-30 | Novartis Ag | Triazolopyrimidine compounds and uses thereof |
| US9388134B2 (en) | 2005-11-09 | 2016-07-12 | Novartis, Ag | Compounds containing S-N-valeryl-N-{[2′-(1H-tetrazole-5-yl)-biphenyl-4-yl]-methyl)-valine and (2R,4S)-5-biphenyl-4-yl-4-(3-carboxy-propionylamino)-2-methyl-pentanoic acid ethyl ester moieties and cations |
| EP3048100A1 (en) | 2009-05-28 | 2016-07-27 | Novartis AG | Substituted aminobutyric derivatives as neprilysin inhibitors |
| EP3064502A1 (en) | 2012-01-26 | 2016-09-07 | Novartis AG | Imidazopyrrolidinone compounds |
| WO2016151499A1 (en) | 2015-03-25 | 2016-09-29 | Novartis Ag | Formylated n-heterocyclic derivatives as fgfr4 inhibitors |
| WO2016164580A1 (en) | 2015-04-07 | 2016-10-13 | Novartis Ag | Combination of chimeric antigen receptor therapy and amino pyrimidine derivatives |
| WO2016174616A1 (en) | 2015-04-30 | 2016-11-03 | Novartis Ag | Fused tricyclic pyrazole derivatives useful for modulating farnesoid x receptors |
| WO2016203432A1 (en) | 2015-06-17 | 2016-12-22 | Novartis Ag | Antibody drug conjugates |
| US9532993B2 (en) | 2011-11-25 | 2017-01-03 | Nuformix Limited | Aprepitant L-proline solvates—compositions and cocrystals |
| CN106432136A (en) * | 2016-06-30 | 2017-02-22 | 中国药科大学 | Hydrochlorothiazide and atenolol co-amorphous system and preparation method thereof |
| WO2017053936A1 (en) | 2015-09-24 | 2017-03-30 | Pain Therapeutic, Inc. | Cocrystals of naloxone and naltrexone |
| WO2017081641A1 (en) | 2015-11-13 | 2017-05-18 | Novartis Ag | Novel pyrazolo pyrimidine derivatives |
| WO2017089985A1 (en) | 2015-11-26 | 2017-06-01 | Novartis Ag | Diamino pyridine derivatives |
| WO2017103888A1 (en) | 2015-12-18 | 2017-06-22 | Novartis Ag | Indane derivatives and the use thereof as soluble guanylate cyclase activators |
| WO2017103824A1 (en) | 2015-12-18 | 2017-06-22 | Novartis Ag | Tricyclic compounds and compositions as kinase inhibitors |
| WO2017103677A1 (en) | 2015-12-16 | 2017-06-22 | Druggability Technologies Ip Holdco Limited | Complexes of celecoxib and its salts and derivatives process for the preparation thereof and pharmaceutical compositions containing them |
| WO2017125898A1 (en) | 2016-01-21 | 2017-07-27 | Novartis Ag | Compounds and compositions for the treatment of cryptosporidiosis |
| US9725453B2 (en) | 2012-03-19 | 2017-08-08 | Ironwood Pharmaceuticals, Inc. | Imidazotriazinone compounds |
| WO2017140821A1 (en) | 2016-02-19 | 2017-08-24 | Novartis Ag | Tetracyclic pyridone compounds as antivirals |
| US9744137B2 (en) | 2006-08-31 | 2017-08-29 | Supernus Pharmaceuticals, Inc. | Topiramate compositions and methods of enhancing its bioavailability |
| WO2017149463A1 (en) | 2016-03-01 | 2017-09-08 | Novartis Ag | Cyano-substituted indole compounds and uses thereof as lsd1 inhibitors |
| WO2017153919A1 (en) | 2016-03-08 | 2017-09-14 | Novartis Ag | Tricyclic compounds useful to treat orthomyxovirus infections |
| WO2017163186A1 (en) | 2016-03-24 | 2017-09-28 | Novartis Ag | Alkynyl nucleoside analogs as inhibitors of human rhinovirus |
| WO2017175185A1 (en) | 2016-04-08 | 2017-10-12 | Novartis Ag | Heteroaryl butanoic acid derivatives as lta4h inhibitors |
| WO2017216685A1 (en) | 2016-06-16 | 2017-12-21 | Novartis Ag | Pentacyclic pyridone compounds as antivirals |
| WO2017216686A1 (en) | 2016-06-16 | 2017-12-21 | Novartis Ag | 8,9-fused 2-oxo-6,7-dihydropyrido-isoquinoline compounds as antivirals |
| WO2017221100A1 (en) | 2016-06-20 | 2017-12-28 | Novartis Ag | Imidazopyrimidine compounds useful for the treatment of cancer |
| WO2017221092A1 (en) | 2016-06-20 | 2017-12-28 | Novartis Ag | Triazolopyridine compounds and uses thereof |
| WO2018014829A1 (en) | 2016-07-20 | 2018-01-25 | Novartis Ag | Aminopyridine derivatives and their use as selective alk-2 inhibitors |
| WO2018042303A1 (en) | 2016-08-29 | 2018-03-08 | Novartis Ag | Fused tricyclic pyridazinone compounds useful to treat orthomyxovirus infections |
| WO2018042316A1 (en) | 2016-08-29 | 2018-03-08 | Novartis Ag | N-(pyridin-2-yl)pyridine-sulfonamide derivatives and their use in the treatment of disease |
| WO2018047109A1 (en) | 2016-09-09 | 2018-03-15 | Novartis Ag | Polycyclic pyridone compounds as antivirals |
| WO2018047081A1 (en) | 2016-09-09 | 2018-03-15 | Novartis Ag | Compounds and compositions as inhibitors of endosomal toll-like receptors |
| WO2018055551A1 (en) | 2016-09-23 | 2018-03-29 | Novartis Ag | Aza-indazole compounds for use in tendon and/or ligament injuries |
| WO2018055550A1 (en) | 2016-09-23 | 2018-03-29 | Novartis Ag | Indazole compounds for use in tendon and/or ligament injuries |
| WO2018060926A1 (en) | 2016-09-28 | 2018-04-05 | Novartis Ag | Beta-lactamase inhibitors |
| WO2018073753A1 (en) | 2016-10-18 | 2018-04-26 | Novartis Ag | Fused tetracyclic pyridone compounds as antivirals |
| WO2018087677A1 (en) | 2016-11-10 | 2018-05-17 | Novartis Ag | Bmp potentiators |
| WO2018141749A1 (en) | 2017-02-01 | 2018-08-09 | Medivir Ab | Therapeutic applications of malt1 inhibitors |
| WO2018172997A1 (en) | 2017-03-24 | 2018-09-27 | Novartis Ag | Isoxazole carboxamide compounds and uses thereof |
| US10093691B2 (en) | 2009-07-31 | 2018-10-09 | Grunenthal Gmbh | Crystallization method and bioavailability |
| WO2018198079A1 (en) | 2017-04-27 | 2018-11-01 | Novartis Ag | Fused indazole pyridone compounds as antivirals |
| WO2018203302A1 (en) | 2017-05-05 | 2018-11-08 | Novartis Ag | Tricyclic 2-quinolinones as antibacterials |
| WO2018234978A1 (en) | 2017-06-19 | 2018-12-27 | Novartis Ag | Substituted 5-cyanoindole compounds and uses thereof |
| US10195218B2 (en) | 2016-05-31 | 2019-02-05 | Grunenthal Gmbh | Crystallization method and bioavailability |
| WO2019087162A1 (en) | 2017-11-06 | 2019-05-09 | Novartis Ag | Polycyclic herg activators |
| WO2019087163A1 (en) | 2017-11-06 | 2019-05-09 | Novartis Ag | Polycyclic herg activators |
| WO2019097479A1 (en) | 2017-11-17 | 2019-05-23 | Novartis Ag | Novel dihydroisoxazole compounds and their use for the treatment of hepatitis b |
| WO2019102256A1 (en) | 2017-11-24 | 2019-05-31 | Novartis Ag | Pyridinone derivatives and their use as selective alk-2 inhibitors |
| WO2019123285A1 (en) | 2017-12-20 | 2019-06-27 | Novartis Ag | Fused tricyclic pyrazolo-dihydropyrazinyl-pyridone compounds as antivirals |
| WO2019124507A1 (en) | 2017-12-21 | 2019-06-27 | 杏林製薬株式会社 | Therapeutic agent for nocturnal pollakiuria |
| WO2019154416A1 (en) | 2018-02-07 | 2019-08-15 | Novartis Ag | Substituted bisphenyl butanoic ester derivatives as nep inhibitors |
| WO2019166951A1 (en) | 2018-02-28 | 2019-09-06 | Novartis Ag | Indole-2-carbonyl compounds and their use for the treatment of hepatitis b |
| WO2019166950A1 (en) | 2018-02-28 | 2019-09-06 | Novartis Ag | 10-(di(phenyl)methyl)-4-hydroxy-8,9,9a,10-tetrahydro-7h-pyrrolo[1 ',2':4,5]pyrazino[1,2-b]pyridazine-3,5-dione derivatives and related compounds as inhibitors of the orthomyxovirus replication for treating influenza |
| US10463653B2 (en) | 2014-10-03 | 2019-11-05 | Novartis Ag | Use of ring-fused bicyclic pyridyl derivatives as FGFR4 inhibitors |
| WO2019223632A1 (en) | 2018-05-22 | 2019-11-28 | Js Innomed Holdings Ltd. | Heterocyclic compounds as kinase inhibitors, compositions comprising the heterocyclic compound, and methods of use thereof |
| WO2019244049A1 (en) | 2018-06-19 | 2019-12-26 | Novartis Ag | Cyanotriazole compounds and uses thereof |
| WO2020016594A1 (en) | 2018-07-19 | 2020-01-23 | Benevolentai Bio Limited | Imidazo[1,2-b]pyridazine derivatives as trk inhibitors |
| EP3610883A1 (en) | 2015-10-21 | 2020-02-19 | Hox Therapeutics Limited | Peptides |
| WO2020035779A1 (en) | 2018-08-17 | 2020-02-20 | Novartis Ag | Urea compounds and compositions as smarca2/brm atpase inhibitors |
| WO2020039209A1 (en) | 2018-08-23 | 2020-02-27 | Benevolentai Bio Limited | Imidazo[1,2-b]pyridazines as trk inhibitors |
| WO2020053654A1 (en) | 2018-09-12 | 2020-03-19 | Novartis Ag | Antiviral pyridopyrazinedione compounds |
| WO2020058913A1 (en) | 2018-09-21 | 2020-03-26 | Novartis Ag | Isoxazole carboxamide compounds and uses thereof |
| WO2020075046A1 (en) * | 2018-10-09 | 2020-04-16 | Novartis Ag | Solid forms of n-(4-fluoro-3-(6-(3-methylpyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)phenyl)-2,4-dimethyloxazole-5-carboxamide |
| US10633344B2 (en) | 2002-03-01 | 2020-04-28 | University Of South Florida | Multiple-component solid phases containing at least one active pharmaceutical ingredient |
| WO2020128768A1 (en) | 2018-12-18 | 2020-06-25 | Novartis Ag | N-(pyridin-2-ylsulfonyl)cyclopropanecarboxamide derivatives and their use in the treatment of a cftr mediated disease |
| WO2020144604A1 (en) | 2019-01-11 | 2020-07-16 | Novartis Ag | Lta4h inhibitors for the treatment of hidradenitis suppurativa |
| WO2020150626A1 (en) | 2019-01-18 | 2020-07-23 | Biogen Ma Inc. | Imidazo[1,2-a]pyridinyl derivatives as irak4 inhibitors |
| WO2020154581A1 (en) * | 2019-01-24 | 2020-07-30 | Assia Chemical Industries Ltd | Solid state forms of fedovapagon-salicyclic acid co-crystal |
| US10766865B2 (en) | 2012-10-16 | 2020-09-08 | Sumitomo Dainippon Pharma Oncology, Inc. | PKM2 modulators and methods for their use |
| WO2020198077A1 (en) | 2019-03-22 | 2020-10-01 | Sumitomo Dainippon Pharma Oncology, Inc. | Compositions comprising pkm2 modulators and methods of treatment using the same |
| WO2020250116A1 (en) | 2019-06-10 | 2020-12-17 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of cf, copd, and bronchiectasis |
| WO2020263967A1 (en) | 2019-06-27 | 2020-12-30 | Biogen Ma Inc. | 2h-indazole derivatives and their use in the treatment of disease |
| WO2020263980A1 (en) | 2019-06-27 | 2020-12-30 | Biogen Ma Inc. | Imidazo[1,2-a]pyridinyl derivatives and their use in the treatment of disease |
| WO2021003417A1 (en) | 2019-07-03 | 2021-01-07 | Sumitomo Dainippon Pharma Oncology, Inc. | Tyrosine kinase non-receptor 1 (tnk1) inhibitors and uses thereof |
| WO2021038426A1 (en) | 2019-08-28 | 2021-03-04 | Novartis Ag | Substituted 1,3-phenyl heteroaryl derivatives and their use in the treatment of disease |
| WO2021044351A1 (en) | 2019-09-06 | 2021-03-11 | Novartis Ag | Methods of treating liver disease using lta4h inhibitors |
| CN112521292A (en) * | 2020-12-18 | 2021-03-19 | 深圳市萱嘉生物科技有限公司 | Eutectic crystal of betaine and organic acid and preparation method and application thereof |
| CN112552189A (en) * | 2020-11-10 | 2021-03-26 | 中国海洋大学 | Pharmaceutical co-crystal of amantadine hydrochloride and resveratrol and preparation method thereof |
| WO2021060949A1 (en) * | 2019-09-26 | 2021-04-01 | 대봉엘에스 주식회사 | Co-crystalline efinaconazole, and method for producing same |
| US11040038B2 (en) | 2018-07-26 | 2021-06-22 | Sumitomo Dainippon Pharma Oncology, Inc. | Methods for treating diseases associated with abnormal ACVR1 expression and ACVR1 inhibitors for use in the same |
| WO2021124172A1 (en) | 2019-12-18 | 2021-06-24 | Novartis Ag | 3-(5-methoxy-1-oxoisoindolin-2-yl)piperidine-2,6-dione derivatives and uses thereof |
| WO2021148805A1 (en) | 2020-01-22 | 2021-07-29 | Benevolentai Bio Limited | Topical pharmaceutical compositions comprising imidazo[1,2-b]pyridazine compounds |
| WO2021148807A1 (en) | 2020-01-22 | 2021-07-29 | Benevolentai Bio Limited | Pharmaceutical compositions and their uses |
| US11091489B2 (en) | 2016-06-20 | 2021-08-17 | Novartis Ag | Crystalline forms of a triazolopyrimidine compound |
| EP3884938A1 (en) | 2020-03-25 | 2021-09-29 | ArtemiFlow GmbH | 1,2,4-trioxane compounds and compositions comprising the same for use in the treatment of covid-19 |
| WO2021214073A1 (en) | 2020-04-20 | 2021-10-28 | Novartis Ag | Antiviral 1,3-di-oxo-indene compounds |
| WO2021214080A1 (en) | 2020-04-20 | 2021-10-28 | Novartis Ag | Antiviral 1,3-di-oxo-indene compounds |
| CN113582927A (en) * | 2020-04-30 | 2021-11-02 | 苏州恩华生物医药科技有限公司 | Celecoxib and pregabalin co-amorphous compound and preparation method thereof |
| WO2021255608A1 (en) | 2020-06-16 | 2021-12-23 | Novartis Ag | Methyl 2-methyl-5-oxo-1,4,5,7-tetradhydrofuro[3,4- b]pyridine-3-carboxylate compounds as cav1.2 activators |
| WO2021255607A1 (en) | 2020-06-15 | 2021-12-23 | Novartis Ag | Methyl 2-(fluoromethyl)-5-oxo-4-phenyl-4,5,6,7-tetrahydro-1h-cyclopenta[b]pyridine-3-carboxylates and methyl 2-(fluoromethyl)-5-oxo-4-phenyl-1,4,5,7-tetrahydrofuro[3,4-b]pyridine-3-carboxylates as cav1.2 activators |
| CN113845438A (en) * | 2021-10-12 | 2021-12-28 | 河北大学 | Acetaminophen-piracetam pharmaceutical co-crystal and preparation method thereof |
| CN114031515A (en) * | 2021-11-29 | 2022-02-11 | 河北大学 | Paracetamol-ibuprofen pharmaceutical co-crystal and preparation method thereof |
| WO2022058902A1 (en) | 2020-09-17 | 2022-03-24 | Novartis Ag | Compounds and compositions as sppl2a inhibitors |
| CN114644669A (en) * | 2022-03-10 | 2022-06-21 | 华润紫竹药业有限公司 | Preparation method and application of progesterone eutectic |
| WO2022140415A1 (en) | 2020-12-22 | 2022-06-30 | Biogen Ma Inc. | 2h-indazole derivatives as irak4 inhibitors and their use in the treatment of disease |
| WO2022140425A1 (en) | 2020-12-22 | 2022-06-30 | Biogen Ma Inc. | Imidazo[1,2-a]pyridine derivatives as irak4 inhibitors and their use in the treatment of disease |
| WO2022150446A1 (en) | 2021-01-07 | 2022-07-14 | Biogen Ma Inc. | Tyk2 inhibitors |
| CN114835657A (en) * | 2022-06-10 | 2022-08-02 | 大连工业大学 | Method for regulating and controlling ethenzamide-saccharin eutectic crystal polycrystalline type based on ionic liquid |
| WO2022195355A1 (en) | 2021-03-15 | 2022-09-22 | Novartis Ag | Benzisoxazole derivatives and uses thereof |
| WO2022195454A1 (en) | 2021-03-15 | 2022-09-22 | Novartis Ag | Pyrazolopyridine derivatives and uses thereof |
| WO2022201097A1 (en) | 2021-03-26 | 2022-09-29 | Novartis Ag | 1,3-substituted cyclobutyl derivatives and uses thereof |
| WO2022217248A1 (en) | 2021-04-10 | 2022-10-13 | Sunovion Pharmaceuticals Inc. | Substituted sulfonamide-chroman compounds, and pharmaceutical compositions, and methods of use thereof |
| WO2022217232A1 (en) | 2021-04-10 | 2022-10-13 | Sunovion Pharmaceuticals Inc. | Chromans and benzofurans as 5-ht1a and taar1 agonists |
| WO2022219546A1 (en) | 2021-04-16 | 2022-10-20 | Novartis Ag | Heteroaryl aminopropanol derivatives as inhibitors of lta4h |
| WO2022234287A1 (en) | 2021-05-06 | 2022-11-10 | Benevolentai Bio Limited | Imidazopyridazine derivatives useful as trk inhibitors |
| WO2022249060A1 (en) | 2021-05-26 | 2022-12-01 | Novartis Ag | Triazolo-pyrimidine analogues for treating diseases connected to the inhibiton of werner syndrome recq helicase (wrn) |
| WO2022254362A1 (en) | 2021-06-03 | 2022-12-08 | Novartis Ag | 3-(1-oxoisoindolin-2-yl)piperidine-2,6-dione derivatives and medical uses thereof |
| EP4105208A1 (en) | 2013-07-31 | 2022-12-21 | Novartis AG | 1,4-disubstituted pyridazine derivatives and their use for treating smn-deficiency-related conditions |
| WO2023275796A1 (en) | 2021-07-01 | 2023-01-05 | Novartis Ag | Heterocyclic derivatives as sphingosine-1-phosphate 3 inhibitors |
| WO2023002443A1 (en) | 2021-07-22 | 2023-01-26 | Novartis Ag | Substituted pyridone compounds useful to treat orthomyxovirus infections |
| WO2023049721A1 (en) | 2021-09-23 | 2023-03-30 | Sunovion Pharmaceuticals Inc. | Methods of treating metabolic disorders |
| CN116162053A (en) * | 2023-01-10 | 2023-05-26 | 山东省分析测试中心 | Gliclazide-piperazine eutectic crystal and preparation method thereof |
| WO2023094965A1 (en) | 2021-11-23 | 2023-06-01 | Novartis Ag | Naphthyridinone derivatives for the treatment of a disease or disorder |
| US11667613B2 (en) | 2019-09-26 | 2023-06-06 | Novartis Ag | Antiviral pyrazolopyridinone compounds |
| WO2023111799A1 (en) | 2021-12-13 | 2023-06-22 | Novartis Ag | PYRIDINE-3-CARBOXYLATE COMPOUNDS AS CaV1.2 ACTIVATORS |
| WO2023139534A1 (en) | 2022-01-24 | 2023-07-27 | Novartis Ag | Spirocyclic piperidinyl derivatives as complement factor b inhibitors and uses thereof |
| WO2023187715A1 (en) | 2022-04-01 | 2023-10-05 | Novartis Ag | Complement factor b inhibitors and uses thereof |
| US11806346B2 (en) | 2020-05-13 | 2023-11-07 | Chdi Foundation, Inc. | HTT modulators for treating Huntington's disease |
| WO2024006493A1 (en) | 2022-07-01 | 2024-01-04 | Biogen Ma Inc. | Tyk2 inhibitors |
| WO2024028782A1 (en) | 2022-08-03 | 2024-02-08 | Novartis Ag | Nlrp3 inflammasome inhibitors |
| WO2024079623A1 (en) | 2022-10-12 | 2024-04-18 | Novartis Ag | Tricyclic compounds and their uses |
| WO2024105553A1 (en) | 2022-11-16 | 2024-05-23 | Novartis Ag | Bicyclic heterocycles and their use as wrn inhibitors |
| WO2024105610A1 (en) | 2022-11-18 | 2024-05-23 | Novartis Ag | Pharmaceutical combinations and uses thereof |
| CN118078830A (en) * | 2024-03-13 | 2024-05-28 | 海南海和制药有限公司 | Preparation method of pharmaceutical composition containing fluorouracil |
| WO2024118488A1 (en) | 2022-11-28 | 2024-06-06 | Sumitomo Pharma America, Inc. | 2-phenylmorpholine and 2-phenyl(thio)morpholine compounds and uses thereof |
| US12005134B2 (en) | 2021-06-30 | 2024-06-11 | Abe Pharmaceutical | Composition for stimulating facial hair growth and methods of manufacturing a composition for stimulating facial hair growth |
| WO2024176130A1 (en) | 2023-02-23 | 2024-08-29 | Novartis Ag | Tead- and her2-inhibitor combinations for treating cancer |
| WO2024176131A1 (en) | 2023-02-23 | 2024-08-29 | Novartis Ag | Tead- and kras g12d-inhibitor combinations for treating cancer |
| WO2024211696A1 (en) | 2023-04-07 | 2024-10-10 | Biogen Ma Inc. | 1h-pyrrolo[2,3-b]pyridin-4-yl]-2-oxopyrrolidine-3-carbonitrile derivatives as tyrosine kinase 2 (tyk2) inhibitors for the treatment of inflammatory diseases |
| WO2024218275A1 (en) * | 2023-04-20 | 2024-10-24 | Institut Gustave Roussy | Liquid and solid compositions of imipridone derivatives |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1765379A4 (en) * | 2004-06-17 | 2009-05-27 | Transform Pharmaceuticals Inc | Pharmaceutical co-crystal compositions and related methods of use |
| US7935817B2 (en) | 2008-03-31 | 2011-05-03 | Apotex Pharmachem Inc. | Salt form and cocrystals of adefovir dipivoxil and processes for preparation thereof |
| GB0813709D0 (en) * | 2008-07-26 | 2008-09-03 | Univ Dundee | Method and product |
| JP5558875B2 (en) * | 2009-03-19 | 2014-07-23 | 日本曹達株式会社 | Novel inclusion complex, epoxy resin composition and epoxy resin composition for semiconductor encapsulation |
| CA2876539A1 (en) * | 2012-06-15 | 2013-12-19 | Basf Se | Multicomponent crystals comprising dasatinib and selected co-crystal formers |
| KR101303803B1 (en) | 2012-08-02 | 2013-09-04 | 순천향대학교 산학협력단 | Preparation of co-crystals of various drug substances including cabamazepine-saccharin co-crystals by anti-solvent method |
| KR20180018800A (en) * | 2015-06-19 | 2018-02-21 | 신-낫 프로덕츠 엔터프라이즈 엘엘씨 | Pharmaceutical compositions of the carboplatin based co-crystals and uses thereof |
| CN107847521A (en) * | 2015-06-25 | 2018-03-27 | 新纳特产品公司 | Pharmaceutical co-crystals composition and application thereof |
| JP7348663B2 (en) * | 2018-02-23 | 2023-09-21 | センター フォー インテリジェント リサーチ イン クリスタル エンジニアリング,エセ.エレ. | Co-crystals of ubiquinol and compositions containing them |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001042221A1 (en) * | 1999-12-08 | 2001-06-14 | Pharmacia Corporation | Solid-state form of celecoxib having enhanced bioavailability |
| WO2001042222A1 (en) * | 1999-12-08 | 2001-06-14 | Pharmacia Corporation | Polymorphic crystalline forms of celecoxib |
| WO2001051919A2 (en) * | 2000-01-07 | 2001-07-19 | Transform Pharmaceuticals, Inc. | High-throughput formation, identification, and analysis of diverse solid-forms |
| WO2003033462A2 (en) * | 2001-10-15 | 2003-04-24 | The Regents Of The University Of Michigan | Systems and methods for the generation of crystalline polymorphs |
| WO2003074474A2 (en) * | 2002-03-01 | 2003-09-12 | University Of South Florida | Multiple-component solid phases containing at least one active pharmaceutical ingredient |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB8500862D0 (en) * | 1985-01-14 | 1985-02-20 | Tate & Lyle Plc | Composition |
| ATE550022T1 (en) * | 2003-02-28 | 2012-04-15 | Mcneil Ppc Inc | PHARMACEUTICAL MIXED CRYSTALS OF CELECOXIB-NICOTINAMIDE |
-
2004
- 2004-02-26 CA CA002514733A patent/CA2514733A1/en not_active Abandoned
- 2004-02-26 WO PCT/US2004/006288 patent/WO2004078163A2/en active Application Filing
- 2004-02-26 EP EP04715190A patent/EP1631260A2/en not_active Withdrawn
- 2004-02-26 JP JP2006508979A patent/JP2007524596A/en active Pending
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001042221A1 (en) * | 1999-12-08 | 2001-06-14 | Pharmacia Corporation | Solid-state form of celecoxib having enhanced bioavailability |
| WO2001042222A1 (en) * | 1999-12-08 | 2001-06-14 | Pharmacia Corporation | Polymorphic crystalline forms of celecoxib |
| WO2001051919A2 (en) * | 2000-01-07 | 2001-07-19 | Transform Pharmaceuticals, Inc. | High-throughput formation, identification, and analysis of diverse solid-forms |
| WO2003033462A2 (en) * | 2001-10-15 | 2003-04-24 | The Regents Of The University Of Michigan | Systems and methods for the generation of crystalline polymorphs |
| WO2003074474A2 (en) * | 2002-03-01 | 2003-09-12 | University Of South Florida | Multiple-component solid phases containing at least one active pharmaceutical ingredient |
Non-Patent Citations (1)
| Title |
|---|
| WEISSBUCH I ET AL: "Understanding and control of nucleation, growth, habit, dissolution and structure of two- and three-dimensional crystals using 'tailor-made' auxiliaries" 1995, ACTA CRYSTALLOGRAPHICA. SECTION B, STRUCTURAL SCIENCE, MUNKSGAARD, COPENHAGEN, DK, PAGE(S) 115-148 , XP002960386 ISSN: 0108-7681 the whole document abstract page 145, column 2, paragraph 2 * |
Cited By (559)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7883713B2 (en) | 2001-06-22 | 2011-02-08 | Aicuris Gmbh & Co. Kg | Topical application of thiazolyl amides |
| US7790905B2 (en) | 2002-02-15 | 2010-09-07 | Mcneil-Ppc, Inc. | Pharmaceutical propylene glycol solvate compositions |
| US7927613B2 (en) | 2002-02-15 | 2011-04-19 | University Of South Florida | Pharmaceutical co-crystal compositions |
| US10633344B2 (en) | 2002-03-01 | 2020-04-28 | University Of South Florida | Multiple-component solid phases containing at least one active pharmaceutical ingredient |
| EP1511490A4 (en) * | 2002-05-31 | 2009-03-11 | Transform Pharmaceuticals Inc | Novel conazole crystalline forms and related processes, pharmaceutical compositions and methods |
| EP1511490A2 (en) * | 2002-05-31 | 2005-03-09 | Transform Pharmaceuticals, Inc. | Novel conazole crystalline forms and related processes, pharmaceutical compositions and methods |
| US8183290B2 (en) | 2002-12-30 | 2012-05-22 | Mcneil-Ppc, Inc. | Pharmaceutically acceptable propylene glycol solvate of naproxen |
| JP2007513867A (en) * | 2003-09-04 | 2007-05-31 | セフアロン・インコーポレーテツド | Modafinil composition |
| EP1670753A4 (en) * | 2003-09-04 | 2008-01-02 | Cephalon Inc | Modafinil compositions |
| JP4842819B2 (en) * | 2003-09-04 | 2011-12-21 | セフアロン・インコーポレーテツド | Modafinil composition |
| EP1670753A2 (en) * | 2003-09-04 | 2006-06-21 | Cephalon, Inc. | Modafinil compositions |
| US7507823B2 (en) | 2004-05-06 | 2009-03-24 | Bristol-Myers Squibb Company | Process of making aripiprazole particles |
| US8784887B2 (en) | 2005-03-30 | 2014-07-22 | Aicuris Gmbh & Co. Kg | Pharmaceutical preparation of N-[5-(aminosulfonyl)-4-methyl-1,3-thiazol-2-yl]-N-methyl-2-[4-(2-pyridinyl)phenyl]acetamide |
| WO2006103011A1 (en) * | 2005-03-30 | 2006-10-05 | Aicuris Gmbh & Co. Kg | Pharmaceutical preparation of n -[5-(aminosulfonyl)-4-methyl-1,3-thiazol-2-yl]-n-methyl-2-[4-(2-pyridinyl)phenyl] acetamide |
| AU2006228747B2 (en) * | 2005-03-30 | 2011-11-03 | Aicuris Gmbh & Co. Kg | Pharmaceutical preparation of N -[5-(aminosulfonyl)-4-methyl-1,3-thiazol-2-yl]-N-methyl-2-[4-(2-pyridinyl)phenyl] acetamide |
| WO2006138421A3 (en) * | 2005-06-15 | 2008-07-10 | Elan Pharma Int Ltd | Nanoparticulate azelnidipine formulations |
| WO2006138421A2 (en) * | 2005-06-15 | 2006-12-28 | Elan Pharma International Limited | Nanoparticulate azelnidipine formulations |
| EP1951393A2 (en) * | 2005-10-31 | 2008-08-06 | The Regents of the University of Michigan | Reaction co-crystallization of molecular complexes or co-crystals |
| EP1951393A4 (en) * | 2005-10-31 | 2010-05-19 | Univ Michigan | Reaction co-crystallization of molecular complexes or co-crystals |
| EP2340828B1 (en) | 2005-11-09 | 2020-07-15 | Novartis AG | Pharmaceutical combinations of an angiotensin receptor antagonist and a nep inhibitor |
| US11642329B2 (en) | 2005-11-09 | 2023-05-09 | Novartis Pharmaceuticals Corporation | Amorphous solid form of compounds containing S—N-valeryl-N- {[2′-( 1 H-tetrazole-5-yl)-biphenyl-4-yl]-methyl}-valine and (2R,4S)-5-biphenyl-4-yl-4-(3-carboxy-propionylamino)-2-methyl-pentanoic acid ethyl ester moieties and sodium cations |
| US20160324821A1 (en) * | 2005-11-09 | 2016-11-10 | Lili Feng | Compounds containing s-n-valeryl-n--valine and (2r,4s)-5-biphenyl-4-yl-4-(3-carboxy-propionylamino)-2-methyl-pentanoic acid ethyl ester moieties and cations |
| US11096918B2 (en) | 2005-11-09 | 2021-08-24 | Novartis Pharmaceuticals Corporation | Amorphous solid form of compounds containing S—N-valeryl-N-{[2′-(1H-tetrazole-5-yl)-biphenyl-4-yl]-methyl}-valine and (2R,4S)-5-biphenyl-4-yl-4-(3-carboxy-propionylamino)-2-methyl-pentanoic acid ethyl ester moieties and sodium cations |
| US9388134B2 (en) | 2005-11-09 | 2016-07-12 | Novartis, Ag | Compounds containing S-N-valeryl-N-{[2′-(1H-tetrazole-5-yl)-biphenyl-4-yl]-methyl)-valine and (2R,4S)-5-biphenyl-4-yl-4-(3-carboxy-propionylamino)-2-methyl-pentanoic acid ethyl ester moieties and cations |
| EP1962600A4 (en) * | 2005-12-08 | 2010-09-22 | Ssci Inc | Metronidazole cocrystals and imipramine cocrystals |
| US8163790B2 (en) | 2005-12-08 | 2012-04-24 | New Form Pharmaceuticals, Inc. | Metronidazole cocrystals and imipramine cocrystals |
| EP1962600A2 (en) * | 2005-12-08 | 2008-09-03 | SSCI, Inc. | Metronidazole cocrystals and imipramine cocrystals |
| US9744137B2 (en) | 2006-08-31 | 2017-08-29 | Supernus Pharmaceuticals, Inc. | Topiramate compositions and methods of enhancing its bioavailability |
| WO2008054609A3 (en) * | 2006-10-04 | 2008-06-19 | Univ Michigan | Dissolution and precipitation of cocrystals with ionizable components |
| WO2008054609A2 (en) * | 2006-10-04 | 2008-05-08 | The Regents Of The University Of Michigan | Dissolution and precipitation of cocrystals with ionizable components |
| WO2008063284A2 (en) * | 2006-10-10 | 2008-05-29 | Janssen Pharmaceutica Nv | Novel crystal of (s)-(+)-2-(2-chlorophenyl)-2-hydroxy-ethyl carbamate |
| WO2008063284A3 (en) * | 2006-10-10 | 2008-07-10 | Transform Pharmaceuticals Inc | Novel crystal of (s)-(+)-2-(2-chlorophenyl)-2-hydroxy-ethyl carbamate |
| US8992989B2 (en) | 2006-11-17 | 2015-03-31 | Supernus Pharmaceuticals, Inc. | Sustained-release formulations of topiramate |
| US8298576B2 (en) | 2006-11-17 | 2012-10-30 | Supernus Pharmaceuticals, Inc. | Sustained-release formulations of topiramate |
| US10314790B2 (en) | 2006-11-17 | 2019-06-11 | Supernus Pharmaceuticals, Inc. | Sustained-release formulations of topiramate |
| US8298580B2 (en) | 2006-11-17 | 2012-10-30 | Supernus Pharmaceuticals, Inc. | Sustained-release formulations of topiramate |
| US8889191B2 (en) | 2006-11-17 | 2014-11-18 | Supernus Pharmaceuticals, Inc. | Sustained-release formulations of topiramate |
| US8877248B1 (en) | 2006-11-17 | 2014-11-04 | Supernus Pharmaceuticals, Inc. | Sustained-release formulations of topiramate |
| US8663683B2 (en) | 2006-11-17 | 2014-03-04 | Supernus Pharmaceuticals, Inc. | Sustained-release formulations of topiramate |
| US9622983B2 (en) | 2006-11-17 | 2017-04-18 | Supernus Pharmaceutcals, Inc. | Sustained-release formulations of topiramate |
| US9549940B2 (en) | 2006-11-17 | 2017-01-24 | Supernus Pharmaceuticals, Inc. | Sustained-release formulations of topiramate |
| US9555004B2 (en) | 2006-11-17 | 2017-01-31 | Supernus Pharmaceuticals, Inc. | Sustained-release formulations of topiramate |
| US8859607B2 (en) | 2007-02-09 | 2014-10-14 | Basf Se | Crystalline complexes of agriculturally active organic compounds |
| WO2008108639A1 (en) * | 2007-03-08 | 2008-09-12 | Avantium Holding B.V. | Co-crystalline forms of carbamazepine |
| WO2008153945A3 (en) * | 2007-06-06 | 2009-02-12 | Univ South Florida | Nutraceutical co-crystal compositions |
| US10842797B2 (en) | 2007-06-06 | 2020-11-24 | University Of South Florida | Nutraceutical co-crystal compositions |
| US10376521B2 (en) | 2007-06-06 | 2019-08-13 | University Of South Florida | Nutraceutical co-crystal compositions |
| US20100204204A1 (en) * | 2007-06-06 | 2010-08-12 | University Of South Florida | Nutraceutical co-crystal compositions |
| WO2008153945A2 (en) * | 2007-06-06 | 2008-12-18 | University Of South Florida | Nutraceutical co-crystal compositions |
| US8124603B2 (en) | 2008-01-22 | 2012-02-28 | Thar Pharmaceuticals | In vivo studies of crystalline forms of meloxicam |
| WO2009094155A1 (en) * | 2008-01-22 | 2009-07-30 | Thar Pharmaceuticals | In vivo studies of crystalline forms of meloxicam |
| EP2262488A1 (en) * | 2008-04-07 | 2010-12-22 | Mutual Pharmaceutical Company, Inc. | Colchicine solid complex; methods of making; and methods of use thereof |
| EP2262488A4 (en) * | 2008-04-07 | 2012-07-04 | Mutual Pharmaceutical Co | Colchicine solid complex; methods of making; and methods of use thereof |
| US8519002B2 (en) | 2008-04-07 | 2013-08-27 | Takeda Pharmaceuticals U.S.A., Inc. | Colchicine solid complex; methods of making; and methods of use thereof |
| WO2009140466A2 (en) * | 2008-05-14 | 2009-11-19 | Dr. Reddy's Laboratories Ltd. | Linezolid co-crystals |
| WO2009140466A3 (en) * | 2008-05-14 | 2010-03-04 | Dr. Reddy's Laboratories Ltd. | Linezolid co-crystals |
| US8501802B2 (en) | 2008-05-21 | 2013-08-06 | Laboratorios Del Dr. Esteve, S.A. | Co-crystals of duloxetine and COX-INHIBITORs for the treatment of pain |
| EP2123626A1 (en) * | 2008-05-21 | 2009-11-25 | Laboratorios del Dr. Esteve S.A. | Co-crystals of duloxetine and co-crystal formers for the treatment of pain |
| WO2009141144A1 (en) | 2008-05-21 | 2009-11-26 | Laboratorios Del Dr. Esteve, S.A. | Co-crystals of duloxetine and cox-inhibitors for the treatment of pain |
| US9084774B2 (en) | 2008-05-21 | 2015-07-21 | Laboratorios Del Dr. Esteve, S.A. | Co-crystals of duloxetine and cox-inhibitors for the treatment of pain |
| CN102036946A (en) * | 2008-05-21 | 2011-04-27 | 埃斯蒂文博士实验室股份有限公司 | Co-crystals of duloxetine and Cox-inhibitors for the treatment of pain |
| WO2009152347A3 (en) * | 2008-06-13 | 2010-04-15 | Bionevia Pharmaceuticals Inc. | Crystalline forms of zotepine hydrochloride |
| WO2009152347A2 (en) * | 2008-06-13 | 2009-12-17 | Bionevia Pharmaceuticals Inc. | Crystalline forms of zotepine hydrochloride |
| US8258155B2 (en) | 2008-06-30 | 2012-09-04 | Mutual Pharmaceutical Company, Inc. | Quinine sulfate/bisulfate solid complex; methods of making; and methods of use thereof |
| US8697735B2 (en) | 2008-07-25 | 2014-04-15 | Bionevia Pharmaceuticals, Inc. | Solid forms of epalrestat |
| US9447056B2 (en) | 2008-07-25 | 2016-09-20 | Bionevia Pharmaceuticals, Inc. | Solid forms of epalrestat |
| WO2010017343A3 (en) * | 2008-08-06 | 2010-05-14 | Bionevia Pharmaceuticals, Inc. | Flupirtine hydrochloride maleic acid cocrystal |
| US10464912B2 (en) | 2008-09-06 | 2019-11-05 | Bionevia Pharmaceuticals, Inc. | Choline cocrystal of epalrestat |
| US8906948B2 (en) | 2008-09-06 | 2014-12-09 | Bionevia, LLC | Choline cocrystal of epalrestat |
| US10245276B2 (en) | 2008-10-17 | 2019-04-02 | Laboratorios Del Dr. Esteve, S.A. | Co-crystals of tramadol and coxibs |
| CN102186465B (en) * | 2008-10-17 | 2015-09-16 | 埃斯蒂文博士实验室股份有限公司 | The eutectic of tramadol and NSAID |
| WO2010043412A1 (en) * | 2008-10-17 | 2010-04-22 | Laboratorios Del Dr. Esteve, S.A. | Co-crystals of tramadol and nsaids |
| US10548909B2 (en) | 2008-10-17 | 2020-02-04 | Esteve Pharmaceuticals, S.A. | Co-crystals of tramadol and coxibs |
| RU2599717C2 (en) * | 2008-10-17 | 2016-10-10 | Лабораторьос Дель Др. Эстеве, С.А. | Co-crystalline forms of tramadol and nsaid |
| US11478488B2 (en) | 2008-10-17 | 2022-10-25 | Esteve Pharmaceuticals, S.A. | Co-crystals of tramadol and coxibs |
| EP2199274A1 (en) * | 2008-12-16 | 2010-06-23 | Laboratorios Del. Dr. Esteve, S.A. | Co-crystals of tramadol and paracetamol |
| WO2010069561A1 (en) * | 2008-12-16 | 2010-06-24 | Laboratorios Del Dr. Esteve, S.A. | Cocrystals of tramadol and paracetamol |
| ES2371848A1 (en) * | 2008-12-16 | 2012-01-10 | Laboratorios Del Dr. Esteve, S. A. | Cocrystals of tramadol and paracetamol |
| US8555875B2 (en) | 2008-12-23 | 2013-10-15 | Map Pharmaceuticals, Inc. | Inhalation devices and related methods for administration of sedative hypnotic compounds |
| US9161912B2 (en) | 2008-12-23 | 2015-10-20 | Map Pharmaceuticals, Inc. | Inhalation devices and related methods for administration of sedative hypnotic compounds |
| US8507561B2 (en) | 2009-01-22 | 2013-08-13 | Absorption Pharmaceuticals, LLC | Desensitizing drug product |
| US8563616B2 (en) | 2009-01-22 | 2013-10-22 | Absorption Pharmaceuticals, LLC | Desensitizing drug product |
| US8637577B2 (en) | 2009-01-22 | 2014-01-28 | Absorption Pharmaceuticals, LLC | Desensitizing drug product |
| WO2010090491A3 (en) * | 2009-02-09 | 2011-01-27 | 주식회사 한독약품 | Novel salts of adefovir dipivoxil and preparation method thereof |
| WO2010090491A2 (en) * | 2009-02-09 | 2010-08-12 | 주식회사 한독약품 | Novel salts of adefovir dipivoxil and preparation method thereof |
| WO2010115981A1 (en) | 2009-04-10 | 2010-10-14 | Novartis Ag | 7-azadispiro [3.0.4.1] decane-8-carboxamides as hepatitis c virus inhibitors |
| WO2010116248A1 (en) | 2009-04-10 | 2010-10-14 | Novartis Ag | Organic compounds and their uses |
| WO2010130773A2 (en) | 2009-05-15 | 2010-11-18 | Novartis Ag | Benzoxazolone derivatives as aldosterone symthase inhibitors |
| WO2010130796A1 (en) | 2009-05-15 | 2010-11-18 | Novartis Ag | Aryl pyridine as aldosterone synthase inhibitors |
| EP2432320A4 (en) * | 2009-05-20 | 2013-03-06 | Nutracryst Therapeutics Private Ltd | Pharmaceutical co-crystals of quercetin |
| EP2432320A1 (en) * | 2009-05-20 | 2012-03-28 | Nutracryst Therapeutics Private Limited | Pharmaceutical co-crystals of quercetin |
| EP2594557A1 (en) | 2009-05-28 | 2013-05-22 | Novartis AG | Substituted aminopropionic derivatives as neprilysin inhibitors |
| EP3048100A1 (en) | 2009-05-28 | 2016-07-27 | Novartis AG | Substituted aminobutyric derivatives as neprilysin inhibitors |
| EP2594556A1 (en) | 2009-05-28 | 2013-05-22 | Novartis AG | Substituted aminopropionic derivatives as neprilysin inhibitors |
| WO2010136493A1 (en) | 2009-05-28 | 2010-12-02 | Novartis Ag | Substituted aminopropionic derivatives as neprilysin inhibitors |
| WO2011009852A2 (en) | 2009-07-21 | 2011-01-27 | Novartis Ag | Pharmaceutical compositions |
| WO2011009943A1 (en) | 2009-07-24 | 2011-01-27 | Novartis Ag | Oxazine derivatives and their use as bace inhibitors for the treatment of neurological disorders |
| US8933057B2 (en) | 2009-07-31 | 2015-01-13 | Thar Pharmaceuticals, Inc. | Crystallization method and bioavailability |
| US10323052B2 (en) | 2009-07-31 | 2019-06-18 | Grunenthal Gmbh | Crystallization method and bioavailability |
| US8399023B2 (en) | 2009-07-31 | 2013-03-19 | Thar Pharmaceuticals, Inc. | Crystallization method and bioavailability |
| US10093691B2 (en) | 2009-07-31 | 2018-10-09 | Grunenthal Gmbh | Crystallization method and bioavailability |
| US9334296B2 (en) | 2009-07-31 | 2016-05-10 | Thar Pharmaceuticals, Inc. | Crystallization method and bioavailability |
| US9169279B2 (en) | 2009-07-31 | 2015-10-27 | Thar Pharmaceuticals, Inc. | Crystallization method and bioavailability |
| WO2011015360A1 (en) * | 2009-08-06 | 2011-02-10 | Laboratorios Del Dr. Esteve, S.A. | Pharmaceutical compounds of o-desmethyl-tramadol and cox-inhibitors |
| EP2281558A1 (en) * | 2009-08-06 | 2011-02-09 | Laboratorios Del. Dr. Esteve, S.A. | Pharmaceutical compounds of O-Desmethyl-Tramadol and COX-inhibitors |
| WO2011015652A1 (en) | 2009-08-07 | 2011-02-10 | Novartis Ag | 3-heteroarylmethyl-imidazo[1,2-b]pyridazin-6-yl derivatives as c-met tyrosine kinase modulators |
| WO2011018454A1 (en) | 2009-08-12 | 2011-02-17 | Novartis Ag | Heterocyclic hydrazone compounds and their uses to treat cancer and inflammation |
| WO2011020861A1 (en) | 2009-08-20 | 2011-02-24 | Novartis Ag | Heterocyclic oxime compounds |
| WO2011023677A1 (en) | 2009-08-26 | 2011-03-03 | Novartis Ag | Tetra-substituted heteroaryl compounds and their use as mdm2 and/or mdm4 modulators |
| WO2011026904A1 (en) | 2009-09-04 | 2011-03-10 | Novartis Ag | Pyrazinylpyridines useful for the treatment of proliferative diseases |
| WO2011026917A1 (en) | 2009-09-04 | 2011-03-10 | Novartis Ag | Heteroaryl compounds as kinase inhibitors |
| WO2011026911A1 (en) | 2009-09-04 | 2011-03-10 | Novartis Ag | Bipyridines useful for the treatment of proliferative diseases |
| WO2011029842A1 (en) | 2009-09-10 | 2011-03-17 | Novartis Ag | Sulfonamides as inhibitors of bcl-2 family proteins for the treatment of cancer |
| WO2011029915A1 (en) | 2009-09-10 | 2011-03-17 | Novartis Ag | Ether derivatives of bicyclic heteroaryls |
| EP2325172A1 (en) * | 2009-11-02 | 2011-05-25 | Laboratorios Del. Dr. Esteve, S.A. | Co-crystals of celecoxib and L-proline |
| WO2011054828A1 (en) | 2009-11-04 | 2011-05-12 | Novartis Ag | Heterocyclic sulfonamide derivatives useful as mek inhibitors |
| EP2993169A1 (en) | 2009-11-17 | 2016-03-09 | Novartis AG | Aryl-pyridine derivatives as aldosterone synthase inhibitors |
| WO2011061168A1 (en) | 2009-11-17 | 2011-05-26 | Novartis Ag | Aryl-pyridine derivatives as aldosterone synthase inhibitors |
| WO2011061271A1 (en) | 2009-11-20 | 2011-05-26 | Novartis Ag | Substituted carbamoylmethylamino acetic acid derivatives as novel nep inhibitors |
| EP2735561A1 (en) | 2009-11-20 | 2014-05-28 | Novartis AG | Substituted carbamoylmethylamino acetic acid derivatives as novel NEP inhibitors |
| WO2011064376A1 (en) | 2009-11-30 | 2011-06-03 | Novartis Ag | Imidazole derivatives as aldosterone synthase inhibitors |
| WO2011073316A1 (en) | 2009-12-18 | 2011-06-23 | Novartis Ag | 4-aryl-butane-1,3-diamides |
| WO2011076747A1 (en) | 2009-12-21 | 2011-06-30 | Novartis Ag | Diaza-spiro[5.5]undecanes as orexin receptor antagonists |
| WO2011076744A1 (en) | 2009-12-21 | 2011-06-30 | Novartis Ag | Disubstituted heteroaryl-fused pyridines |
| WO2011076786A1 (en) | 2009-12-22 | 2011-06-30 | Novartis Ag | Substituted isoquinolinones and quinazolinones |
| WO2011080176A1 (en) | 2009-12-31 | 2011-07-07 | Novartis Ag | Pyrazine derivatives and their use in the treatment of neurological disorders |
| WO2011092293A2 (en) | 2010-02-01 | 2011-08-04 | Novartis Ag | Cyclohexyl amide derivatives as crf receptor antagonists |
| WO2011092290A1 (en) | 2010-02-01 | 2011-08-04 | Novartis Ag | Pyrazolo[5,1b]oxazole derivatives as crf-1 receptor antagonists |
| WO2011095450A1 (en) | 2010-02-02 | 2011-08-11 | Novartis Ag | Cyclohexyl amide derivatives as crf receptor antagonists |
| WO2011095452A1 (en) | 2010-02-02 | 2011-08-11 | Novartis Ag | Aryl benzylamine compounds |
| EP2531196A4 (en) * | 2010-02-03 | 2013-07-31 | Laurus Labs Private Ltd | Pterostilbene cocrystals |
| US8399712B2 (en) | 2010-02-03 | 2013-03-19 | Laurus Labs Private Limited | Pterostilbene cocrystals |
| US8513236B2 (en) | 2010-02-03 | 2013-08-20 | Laurus Labs Private Limited | Pterostilbene cocrystals |
| WO2011097372A3 (en) * | 2010-02-03 | 2011-12-29 | Aptuit Laurus Private Limited | Pterostilbene cocrystals |
| US8318807B2 (en) | 2010-02-03 | 2012-11-27 | Laurus Labs Private Limited | Pterostilbene cocrystals |
| EP2531196A2 (en) * | 2010-02-03 | 2012-12-12 | Laurus Labs Private Limited | Pterostilbene cocrystals |
| US8415507B2 (en) | 2010-02-03 | 2013-04-09 | Laurus Labs Private Limited | Pterostilbene cocrystals |
| WO2011104266A1 (en) | 2010-02-25 | 2011-09-01 | Novartis Ag | Dimeric iap inhibitors |
| US11911371B2 (en) | 2010-03-19 | 2024-02-27 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of chronic bronchitis |
| US9365552B2 (en) | 2010-03-19 | 2016-06-14 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of CF |
| USRE46757E1 (en) | 2010-03-19 | 2018-03-20 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of CF |
| WO2011113894A1 (en) | 2010-03-19 | 2011-09-22 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of cf |
| EP2845593A1 (en) | 2010-03-19 | 2015-03-11 | Novartis AG | Pyridine and pyrazine derivative for the treatment of chronic obstructive pulmonary disease |
| US10117858B2 (en) | 2010-03-19 | 2018-11-06 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of CF |
| WO2011131709A1 (en) | 2010-04-21 | 2011-10-27 | Novartis Ag | Furopyridine compounds and uses thereof |
| WO2011158110A3 (en) * | 2010-04-28 | 2012-03-15 | Nuformix Limited | Cilostazol cocrystals and compositions |
| CN102933205A (en) * | 2010-04-28 | 2013-02-13 | 诺弗米克斯有限公司 | Cilostazol cocrystals and compositions |
| US8779146B2 (en) | 2010-04-28 | 2014-07-15 | Nuformix Limited | Cilostazol cocrystals and compositions |
| WO2011144666A1 (en) | 2010-05-20 | 2011-11-24 | Novartis Ag | 2,4-dioxo-1,4-dihydro-2h-quinazolin-3-yl-sulfonamide derivatives |
| WO2011157793A1 (en) | 2010-06-17 | 2011-12-22 | Novartis Ag | Piperidinyl substituted 1,3-dihydro-benzoimidazol-2-ylideneamine derivatives |
| WO2011157787A1 (en) | 2010-06-17 | 2011-12-22 | Novartis Ag | Biphenyl substituted 1,3-dihydro-benzoimidazol-2-ylideneamine derivatives |
| WO2012003189A1 (en) | 2010-06-29 | 2012-01-05 | Irm Llc | Compositions and methods for modulating the wnt signaling pathway |
| WO2012004299A1 (en) | 2010-07-06 | 2012-01-12 | Novartis Ag | Tetrahydro-pyrido-pyrimidine derivatives |
| WO2012006953A1 (en) | 2010-07-13 | 2012-01-19 | Novartis Ag | Oxazine derivatives and their use in the treatment of neurological disorders |
| WO2012007539A1 (en) | 2010-07-14 | 2012-01-19 | Novartis Ag | Ip receptor agonist heterocyclic compounds |
| WO2012010663A1 (en) | 2010-07-22 | 2012-01-26 | Novartis Ag | 2,3,5-trisubstituted thiophene compounds and uses thereof |
| WO2012017020A1 (en) | 2010-08-04 | 2012-02-09 | Novartis Ag | N-((6-amino-pyridin-3-yl)methyl)-heteroaryl-carboxamides as inhibitors of plasma kallikrein |
| WO2012035023A1 (en) | 2010-09-13 | 2012-03-22 | Novartis Ag | Triazine-oxadiazoles |
| WO2012035158A1 (en) | 2010-09-17 | 2012-03-22 | Novartis Ag | Pyrazine derivatives as enac blockers |
| US9540380B2 (en) | 2010-09-20 | 2017-01-10 | Ironwood Pharmaceuticals, Inc. | Imidazotriazinone compounds |
| EP3181566A1 (en) | 2010-09-20 | 2017-06-21 | Ironwood Pharmaceuticals, Inc. | Imidazotriazinone compounds |
| WO2012040230A1 (en) | 2010-09-20 | 2012-03-29 | Envivo Pharmaceuticals, Inc. | Imidazotriazinone compounds |
| WO2012048235A1 (en) | 2010-10-08 | 2012-04-12 | Novartis Ag | Vitamin e formulations of sulfamide ns3 inhibitors |
| WO2012055888A1 (en) | 2010-10-26 | 2012-05-03 | Novartis Ag | Diaza-spiro[5.5]undecanes useful as orexin receptor antagonists |
| WO2012065956A1 (en) | 2010-11-16 | 2012-05-24 | Novartis Ag | Substituted amino bisphenyl pentanoic acid derivatives as nep inhibitors |
| WO2012065953A1 (en) | 2010-11-16 | 2012-05-24 | Novartis Ag | Substituted carbamoylcycloalkyl acetic acid derivatives as nep inhibitors |
| WO2012065958A1 (en) | 2010-11-16 | 2012-05-24 | Novartis Ag | Method of treating contrast-induced nephropathy |
| US10519176B2 (en) | 2010-11-24 | 2019-12-31 | Thar Pharma, Llc | Crystalline forms |
| US9340565B2 (en) | 2010-11-24 | 2016-05-17 | Thar Pharmaceuticals, Inc. | Crystalline forms |
| WO2012080260A1 (en) | 2010-12-13 | 2012-06-21 | Novartis Ag | Dimeric iap inhibitors |
| WO2012080271A1 (en) | 2010-12-13 | 2012-06-21 | Novartis Ag | Dimeric iap inhibitors |
| WO2012087521A1 (en) | 2010-12-20 | 2012-06-28 | Irm Llc | Compositions and methods for modulating farnesoid x receptors |
| WO2012087520A1 (en) | 2010-12-20 | 2012-06-28 | Irm Llc | Compositions and methods for modulating farnesoid x receptors |
| WO2012084873A1 (en) | 2010-12-20 | 2012-06-28 | Novartis Ag | 4- (hetero) aryl - ethynyl - octahydro - indole - 1 - carboxylic acid esters |
| WO2012087519A1 (en) | 2010-12-20 | 2012-06-28 | Irm Llc | Compositions and methods for modulating fxr |
| WO2012101487A1 (en) | 2010-12-22 | 2012-08-02 | Novartis Ag | Di/tri-aza-spiro-c9-c11alkanes |
| US9085555B2 (en) | 2011-01-04 | 2015-07-21 | Novartis Ag | Complement pathway modulators and uses thereof |
| WO2012093101A1 (en) | 2011-01-04 | 2012-07-12 | Novartis Ag | Indole compounds or analogues thereof useful for the treatment of age-related macular degeneration (amd) |
| WO2012095463A1 (en) | 2011-01-12 | 2012-07-19 | Novartis Ag | Oxazine derivatives and their use in the treatment of neurological disorders |
| WO2012095469A1 (en) | 2011-01-13 | 2012-07-19 | Novartis Ag | Novel heterocyclic derivatives and their use in the treatment of neurological disorders |
| WO2012095521A1 (en) | 2011-01-13 | 2012-07-19 | Novartis Ag | Bace-2 inhibitors for the treatment of metabolic disorders |
| WO2012098501A1 (en) * | 2011-01-21 | 2012-07-26 | Ranbaxy Laboratories Limited | Febuxostat co-crystals |
| WO2012101058A1 (en) | 2011-01-24 | 2012-08-02 | Novartis Ag | 4-tolyl-ethynyl-octahydro-indole-1-ester derivatives |
| WO2012101065A2 (en) | 2011-01-28 | 2012-08-02 | Novartis Ag | Pyrimidine biaryl amine compounds and their uses |
| WO2012101064A1 (en) | 2011-01-28 | 2012-08-02 | Novartis Ag | N-acyl pyrimidine biaryl compounds as protein kinase inhibitors |
| WO2012101063A1 (en) | 2011-01-28 | 2012-08-02 | Novartis Ag | N-acyl pyridine biaryl compounds and their uses |
| WO2012101066A1 (en) | 2011-01-28 | 2012-08-02 | Novartis Ag | Pyridine biaryl amine compounds and their uses |
| WO2012104776A1 (en) | 2011-01-31 | 2012-08-09 | Novartis Ag | Novel heterocyclic derivatives |
| WO2012104823A2 (en) | 2011-02-04 | 2012-08-09 | Novartis Ag | Pyridopyrimidinone compounds in the treatment of neurodegenerative diseases |
| WO2012107500A1 (en) | 2011-02-10 | 2012-08-16 | Novartis Ag | [1, 2, 4] triazolo [4, 3 -b] pyridazine compounds as inhibitors of the c-met tyrosine kinase |
| WO2012116217A1 (en) | 2011-02-25 | 2012-08-30 | Irm Llc | Compounds and compositions as trk inhibitors |
| WO2012117224A1 (en) | 2011-03-02 | 2012-09-07 | Sabrepharm Limited | Dipyridinium derivatives |
| WO2012120469A1 (en) | 2011-03-08 | 2012-09-13 | Novartis Ag | Fluorophenyl bicyclic heteroaryl compounds |
| US10130708B2 (en) | 2011-03-24 | 2018-11-20 | University Of South Florida | Lithium cocrystal compositions |
| WO2012129568A3 (en) * | 2011-03-24 | 2012-11-22 | University Of South Florida | Lithium compositions |
| WO2012131633A1 (en) | 2011-04-01 | 2012-10-04 | Novartis Ag | Pyrazolo pyrimidine derivatives |
| WO2012138648A1 (en) | 2011-04-06 | 2012-10-11 | Irm Llc | Compositions and methods for modulating lpa receptors |
| WO2012140597A1 (en) | 2011-04-14 | 2012-10-18 | Novartis Ag | Glycoside derivatives and their uses for the treatment of diabetes |
| WO2012140596A1 (en) | 2011-04-14 | 2012-10-18 | Novartis Ag | Glycoside derivatives and uses thereof |
| EA024422B1 (en) * | 2011-04-28 | 2016-09-30 | Зентива, К.С. | Pharmaceutically acceptable cocrystals of n-[2-(7-methoxy-1-naphtyl)ethyl]acetamide and methods of their preparation |
| WO2012146371A1 (en) * | 2011-04-28 | 2012-11-01 | Zentiva, K.S. | Pharmaceutically acceptable cocrystals of n-[2-(7-methoxy-1-naphtyl)ethyl]acetamide and methods of their preparation |
| CN103501777B (en) * | 2011-04-28 | 2015-08-19 | 赞蒂瓦有限合伙公司 | The pharmaceutically acceptable eutectic of N-[2-(7-methoxy-1-naphthyl) ethyl] acetamide and its preparation method |
| CN103501777A (en) * | 2011-04-28 | 2014-01-08 | 赞蒂瓦有限合伙公司 | Pharmaceutically acceptable cocrystals of n-[2-(7-methoxy-1-naphtyl)ethyl]acetamide and methods of their preparation |
| US8927771B2 (en) | 2011-04-28 | 2015-01-06 | Zentiva K.S. | Pharmaceutically acceptable cocrystals of N-[2-(7-methoxyl-1-naphtyl)ethyl]acetamide and methods of their preparation |
| KR20140008370A (en) * | 2011-04-28 | 2014-01-21 | 젠티바, 케이.에스. | Pharmaceutically acceptable cocrystals of n-[2-(7-methoxy-1-naphtyl)ethyl]acetamide and methods of their preparation |
| WO2012156936A1 (en) | 2011-05-17 | 2012-11-22 | Novartis Ag | Substituted indole derivatives for the treatment of immunological disorders |
| WO2012164473A1 (en) | 2011-05-27 | 2012-12-06 | Novartis Ag | 3-spirocyclic piperidine derivatives as ghrelin receptor agonists |
| WO2012175487A1 (en) | 2011-06-20 | 2012-12-27 | Novartis Ag | Cyclohexyl isoquinolinone compounds |
| WO2012175520A1 (en) | 2011-06-20 | 2012-12-27 | Novartis Ag | Hydroxy substituted isoquinolinone derivatives |
| WO2013008164A2 (en) | 2011-07-08 | 2013-01-17 | Novartis Ag | Method of treating atherosclerosis in high triglyceride subjects |
| WO2013008162A1 (en) | 2011-07-08 | 2013-01-17 | Novartis Ag | Novel trifluoromethyl-oxadiazole derivatives and their use in the treatment of disease |
| WO2013008095A1 (en) | 2011-07-08 | 2013-01-17 | Novartis Ag | Novel pyrrolo pyrimidine derivatives |
| WO2013016197A1 (en) | 2011-07-22 | 2013-01-31 | Novartis Ag | Tetrahydropyrido-pyridine and tetrahydropyrido-pyrimidine compounds and use thereof as c5a receptor modulators |
| WO2013014627A1 (en) | 2011-07-27 | 2013-01-31 | Novartis Ag | Pyrazoline derivatives and their use as selective androgen receptor modulators |
| EP3023423A1 (en) | 2011-08-25 | 2016-05-25 | Novartis AG | 2 -amino-4 - (pyridin- 2 -yl) - 5, 6 -dihydro-4h- 1, 3 -oxazine derivatives and their use as bace-1 and/or bace - 2 inhibitors |
| WO2013027188A1 (en) | 2011-08-25 | 2013-02-28 | Novartis Ag | 2 -amino-4 - (pyridin- 2 -yl) - 5, 6 -dihydro-4h- 1, 3 -oxazine derivatives and their use as bace-1 and/or bace - 2 inhibitors |
| WO2013033167A1 (en) | 2011-09-01 | 2013-03-07 | Irm Llc | Compounds and compositions as c-kit kinase inhibitors |
| WO2013033116A1 (en) | 2011-09-01 | 2013-03-07 | Irm Llc | Compounds and compositions as c-kit kinase inhibitors |
| WO2013030802A1 (en) | 2011-09-01 | 2013-03-07 | Novartis Ag | Bicyclic heterocycle derivatives for the treatment of pulmonary arterial hypertension |
| WO2013033070A1 (en) | 2011-09-01 | 2013-03-07 | Irm Llc | COMPOUNDS AND COMPOSITIONS AS c-KIT KINASE INHIBITORS |
| WO2013033203A1 (en) | 2011-09-01 | 2013-03-07 | Irm Llc | Compounds and compositions as c-kit kinase inhibitors |
| WO2013033620A1 (en) | 2011-09-01 | 2013-03-07 | Irm Llc | Compounds and compositions as pdgfr kinase inhibitors |
| WO2013035047A1 (en) | 2011-09-06 | 2013-03-14 | Novartis Ag | Benzothiazolone compound |
| WO2013038362A1 (en) | 2011-09-15 | 2013-03-21 | Novartis Ag | 6 - substituted 3 - (quinolin- 6 - ylthio) - [1,2,4] triazolo [4, 3 -a] pyradines as tyrosine kinase |
| WO2013038373A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | Pyridine amide derivatives |
| WO2013038381A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | Pyridine/pyrazine amide derivatives |
| WO2013038390A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | N-substituted heterocyclyl carboxamides |
| WO2013038386A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | Heterocyclic compounds for the treatment of cystic fibrosis |
| WO2013038378A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | Pyridine amide derivatives |
| WO2013050987A1 (en) | 2011-10-08 | 2013-04-11 | Novartis Ag | Carbamate/ urea derivatives containing piperidin and piperazin rings as h3 receptor inhibitors |
| WO2013054291A1 (en) | 2011-10-13 | 2013-04-18 | Novartis Ag | Novel oxazine derivatives and their use in the treatment of disease |
| US10251866B2 (en) | 2011-10-19 | 2019-04-09 | Universiteit Gent | Pharmaceutical nanosuspension |
| WO2013057169A1 (en) | 2011-10-19 | 2013-04-25 | Universiteit Gent | Pharmaceutical nanosuspension |
| TWI580442B (en) * | 2011-10-19 | 2017-05-01 | 傑特大學 | Pharmaceutical nanosuspension |
| WO2013057711A1 (en) | 2011-10-21 | 2013-04-25 | Novartis Ag | Quinazoline derivatives as pi3k modulators |
| WO2013061305A1 (en) | 2011-10-28 | 2013-05-02 | Novartis Ag | Novel purine derivatives and their use in the treatment of disease |
| US9532993B2 (en) | 2011-11-25 | 2017-01-03 | Nuformix Limited | Aprepitant L-proline solvates—compositions and cocrystals |
| WO2013080120A1 (en) | 2011-11-28 | 2013-06-06 | Novartis Ag | Novel trifluoromethyl-oxadiazole derivatives and their use in the treatment of disease |
| WO2013080141A1 (en) | 2011-11-29 | 2013-06-06 | Novartis Ag | Pyrazolopyrrolidine compounds |
| WO2013093850A1 (en) | 2011-12-22 | 2013-06-27 | Novartis Ag | Quinoline derivatives |
| WO2013093849A1 (en) | 2011-12-22 | 2013-06-27 | Novartis Ag | Dihydro-benzo-oxazine and dihydro-pyrido-oxazine derivatives |
| WO2013105057A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | Fused pyrroles as ip receptor agonists for the treatment of pulmonary arterial hypertension (pah) and related disorders |
| WO2013105063A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | Fused piperidines as ip receptor agonists for the treatment of pulmonary arterial hypertension (pah) and related disorders |
| WO2013105065A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | Fused piperidines as ip receptor agonists for the treatment of pah and related disorders |
| WO2013105058A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | 7,8- dihydropyrido [3, 4 - b] pyrazines as ip receptor agonists for the treatment of pulmonary arterial hypertension (pah) and related disorders |
| WO2013105061A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | Fused dihydropyrido [2,3 -b] pyrazines as ip receptor agonists for the treatment of pulmonary arterial hypertension (pah) and related disorders |
| EP3272754A1 (en) | 2012-01-26 | 2018-01-24 | Novartis AG | Imidazopyrrolidinone compounds |
| EP3064502A1 (en) | 2012-01-26 | 2016-09-07 | Novartis AG | Imidazopyrrolidinone compounds |
| WO2013111108A1 (en) | 2012-01-27 | 2013-08-01 | Novartis Ag | 5-membered heteroarylcarboxamide derivatives as plasma kallikrein inhibitors |
| WO2013111107A1 (en) | 2012-01-27 | 2013-08-01 | Novartis Ag | Aminopyridine derivatives as plasma kallikrein inhibitors |
| WO2013128421A1 (en) | 2012-03-02 | 2013-09-06 | Novartis Ag | Spirohydantoin compounds and their use as selective androgen receptor modulators |
| US9725453B2 (en) | 2012-03-19 | 2017-08-08 | Ironwood Pharmaceuticals, Inc. | Imidazotriazinone compounds |
| US9969742B2 (en) | 2012-03-19 | 2018-05-15 | Ironwood Pharmaceuticals, Inc. | Imidazotriazinone compounds |
| WO2013163508A1 (en) | 2012-04-27 | 2013-10-31 | Novartis Ag | Tetrahydropyran dgat1 inhibitors |
| WO2013160873A1 (en) | 2012-04-27 | 2013-10-31 | Novartis Ag | Cyclic bridgehead ether dgat1 inhibitors |
| WO2013164790A1 (en) | 2012-05-03 | 2013-11-07 | Novartis Ag | L-malate salt of 2, 7 - diaza - spiro [4.5 ] dec- 7 - yle derivatives and crystalline forms thereof as ghrelin receptor agonists |
| WO2013164802A1 (en) | 2012-05-04 | 2013-11-07 | Novartis Ag | Complement pathway modulators and uses thereof |
| WO2013171690A1 (en) | 2012-05-16 | 2013-11-21 | Novartis Ag | Monocyclic heteroaryl cycloalkyldiamine derivatives |
| WO2013175417A1 (en) | 2012-05-24 | 2013-11-28 | Novartis Ag | Pyrrolopyrrolidinone compounds |
| EP3257848A1 (en) | 2012-06-06 | 2017-12-20 | Novartis AG | Compounds and compositions for modulating egfr activity |
| US9440957B2 (en) | 2012-06-06 | 2016-09-13 | Novartis Ag | Compounds and compositions for modulating EGFR Activity |
| WO2013184766A1 (en) | 2012-06-06 | 2013-12-12 | Irm Llc | Compounds and compositions for modulating egfr activity |
| WO2013184757A1 (en) | 2012-06-06 | 2013-12-12 | Irm Llc | Compounds and compositions for modulating egfr activity |
| WO2013192345A1 (en) | 2012-06-20 | 2013-12-27 | Novartis Ag | Complement pathway modulators and uses thereof |
| WO2013189910A1 (en) * | 2012-06-22 | 2013-12-27 | Basf Se | Multicomponent crystals comprising imatinib mesilate and selected co-crystal formers |
| US9221789B2 (en) | 2012-06-22 | 2015-12-29 | Basf Se | Multicomponent crystals comprising imatinib mesilate and selected co-crystal formers |
| US9388199B2 (en) | 2012-06-28 | 2016-07-12 | Novartis Ag | Pyrrolidine derivatives and their use as complement pathway modulators |
| WO2014002058A2 (en) | 2012-06-28 | 2014-01-03 | Novartis Ag | Complement pathway modulators and uses thereof |
| WO2014002057A1 (en) | 2012-06-28 | 2014-01-03 | Novartis Ag | Pyrrolidine derivatives and their use as complement pathway modulators |
| WO2014002054A1 (en) | 2012-06-28 | 2014-01-03 | Novartis Ag | Pyrrolidine derivatives and their use as complement pathway modulators |
| WO2014002052A1 (en) | 2012-06-28 | 2014-01-03 | Novartis Ag | Pyrrolidine derivatives and their use as complement pathway modulators |
| WO2014002053A1 (en) | 2012-06-28 | 2014-01-03 | Novartis Ag | Pyrrolidine derivatives and their use as complement pathway modulators |
| WO2014002051A2 (en) | 2012-06-28 | 2014-01-03 | Novartis Ag | Complement pathway modulators and uses thereof |
| US9487483B2 (en) | 2012-06-28 | 2016-11-08 | Novartis Ag | Complement pathway modulators and uses thereof |
| US9815819B2 (en) | 2012-06-28 | 2017-11-14 | Novartis Ag | Complement pathway modulators and uses thereof |
| US9464081B2 (en) | 2012-06-28 | 2016-10-11 | Novartis Ag | Pyrrolidine derivatives and their use as complement pathway modulators |
| WO2014009833A2 (en) | 2012-07-12 | 2014-01-16 | Novartis Ag | Complement pathway modulators and uses thereof |
| US9550755B2 (en) | 2012-07-12 | 2017-01-24 | Novartis Ag | Complement pathway modulators and uses thereof |
| WO2014013469A1 (en) | 2012-07-20 | 2014-01-23 | Novartis Ag | Carbamate/urea derivatives |
| EP3564233A1 (en) | 2012-08-13 | 2019-11-06 | Novartis AG | 1,4-disubstituted pyridazine analogs and methods for treating smn-deficiency-related conditions |
| WO2014028459A1 (en) | 2012-08-13 | 2014-02-20 | Novartis Ag | 1,4-disubstituted pyridazine analogs and methods for treating smn-deficiency-related conditions |
| WO2014027300A1 (en) | 2012-08-13 | 2014-02-20 | Novartis Ag | Bicyclic heteroaryl cycloalkyldiamine derivatives as inhibitors of spleen tyrosine kinases (syk) |
| EP3243817A1 (en) | 2012-08-30 | 2017-11-15 | Novartis AG | Salts of benzothiazolone compound as beta-2-adrenoceptor agonist |
| WO2014033654A1 (en) | 2012-08-30 | 2014-03-06 | Novartis Ag | Salts of benzothiazolone compound as beta-2-adrenoceptor agonist |
| WO2014033630A1 (en) | 2012-08-31 | 2014-03-06 | Novartis Ag | Novel aminothiazole carboxamides as kinase inhibitors |
| WO2014033617A1 (en) | 2012-08-31 | 2014-03-06 | Novartis Ag | 2'-ethynyl nucleoside derivatives for treatment of viral infections |
| WO2014033631A1 (en) | 2012-08-31 | 2014-03-06 | Novartis Ag | N-(3-pyridyl) biarylamides as kinase inhibitors |
| WO2014037900A1 (en) | 2012-09-07 | 2014-03-13 | Novartis Ag | Indole carboxamide derivatives and uses thereof |
| WO2014047020A1 (en) | 2012-09-19 | 2014-03-27 | Novartis Ag | Dihydropyrrolidino-pyrimidines as kinase inhibitors |
| WO2014049514A1 (en) | 2012-09-25 | 2014-04-03 | Novartis Ag | Compounds for use in gastric complication |
| WO2014052619A1 (en) | 2012-09-27 | 2014-04-03 | Irm Llc | Piperidine derivatives and compositions as modulators of gpr119 activity |
| WO2014049540A2 (en) | 2012-09-29 | 2014-04-03 | Novartis Ag | Cyclic peptides and use as medicines |
| US10766865B2 (en) | 2012-10-16 | 2020-09-08 | Sumitomo Dainippon Pharma Oncology, Inc. | PKM2 modulators and methods for their use |
| WO2014072911A1 (en) | 2012-11-07 | 2014-05-15 | Novartis Ag | Substituted indole derivatives |
| KR20150082295A (en) * | 2012-11-12 | 2015-07-15 | 노파르티스 아게 | Oxazolidin-2-one-pyrimidine derivatives |
| WO2014072956A1 (en) | 2012-11-12 | 2014-05-15 | Novartis Ag | Oxazolidin-2-one-pyrimidine derivatives |
| EP3199158A1 (en) | 2012-11-12 | 2017-08-02 | Novartis AG | Oxazolidin-2-one-pyrimidine derivatives for use in the treatment of skin fibrosis, scleroderma, hypertrophic scars or keloids |
| KR102153763B1 (en) | 2012-11-12 | 2020-09-09 | 노파르티스 아게 | Oxazolidin-2-one-pyrimidine derivatives |
| WO2014078802A1 (en) | 2012-11-19 | 2014-05-22 | Irm Llc | Compounds and compositions for the treatment of parasitic diseases |
| WO2014078813A1 (en) | 2012-11-19 | 2014-05-22 | Irm Llc | Compounds and compositions for the treatment of parasitic diseases |
| WO2014082935A1 (en) | 2012-11-30 | 2014-06-05 | Novartis Ag | Cyclic nucleoside derivatives and uses thereof |
| WO2014093606A1 (en) | 2012-12-13 | 2014-06-19 | Novartis Ag | Pyridone derivatives and uses thereof in the treatment of tuberculosis |
| WO2014091446A1 (en) | 2012-12-13 | 2014-06-19 | Novartis Ag | Pyrimido [4,5-b]quinoline-4,5 (3h,10h)-diones as nonsense mutation suppressors |
| WO2014099880A1 (en) | 2012-12-19 | 2014-06-26 | Novartis Ag | Aryl-substituted fused bicyclic pyridazine compounds |
| WO2014097147A1 (en) | 2012-12-19 | 2014-06-26 | Novartis Ag | Tricyclic compounds as cftr inhibitors |
| WO2014097151A2 (en) | 2012-12-19 | 2014-06-26 | Novartis Ag | Autotaxin inhibitors |
| WO2014097148A1 (en) | 2012-12-19 | 2014-06-26 | Novartis Ag | Tricyclic compounds for inhibiting the cftr channel |
| WO2014115080A1 (en) | 2013-01-22 | 2014-07-31 | Novartis Ag | Pyrazolo[3,4-d]pyrimidinone compounds as inhibitors of the p53/mdm2 interaction |
| WO2014115077A1 (en) | 2013-01-22 | 2014-07-31 | Novartis Ag | Substituted purinone compounds |
| EP3736271A1 (en) | 2013-01-23 | 2020-11-11 | Novartis AG | Thiadiazole analogs and their use for treating diseases associated with a deficiency of smn motor neurons |
| WO2014116845A1 (en) | 2013-01-23 | 2014-07-31 | Novartis Ag | Thiadiazole analogs thereof and methods for treating smn-deficiency-related-conditions |
| WO2014125413A1 (en) | 2013-02-13 | 2014-08-21 | Novartis Ag | Ip receptor agonist heterocyclic compounds |
| WO2014126979A1 (en) | 2013-02-14 | 2014-08-21 | Novartis Ag | Substituted bisphenyl butanoic phosphonic acid derivatives as nep (neutral endopeptidase) inhibitors |
| WO2014128612A1 (en) | 2013-02-20 | 2014-08-28 | Novartis Ag | Quinazolin-4-one derivatives |
| WO2014132205A1 (en) | 2013-02-28 | 2014-09-04 | Novartis Ag | Formulation comprising benzothiazolone compound |
| US8889190B2 (en) | 2013-03-13 | 2014-11-18 | Upsher-Smith Laboratories, Inc. | Extended-release topiramate capsules |
| US10363224B2 (en) | 2013-03-13 | 2019-07-30 | Upsher-Smith Laboratories, Llc | Extended-release topiramate capsules |
| US8652527B1 (en) | 2013-03-13 | 2014-02-18 | Upsher-Smith Laboratories, Inc | Extended-release topiramate capsules |
| WO2014143638A1 (en) | 2013-03-14 | 2014-09-18 | Novartis Ag | 2-(1h-indol-4-ylmethyl)-3h-imidazo[4,5-b]pyridine-6-carbonitrile derivatives as complement factor b inhibitors useful for the treatment of ophthalmic diseases |
| WO2014151616A1 (en) | 2013-03-14 | 2014-09-25 | Novartis Ag | Biaryl amide compounds as kinase inhibitors |
| EP3299367A1 (en) | 2013-03-14 | 2018-03-28 | Novartis AG | Biaryl amide compounds as kinase inhibitors |
| US10172878B2 (en) | 2013-03-15 | 2019-01-08 | Upsher-Smith Laboratories, Llc | Extended-release topiramate capsules |
| WO2014151784A1 (en) | 2013-03-15 | 2014-09-25 | Irm Llc | Compounds and compositions for the treatment of parasitic diseases |
| US9101545B2 (en) | 2013-03-15 | 2015-08-11 | Upsher-Smith Laboratories, Inc. | Extended-release topiramate capsules |
| US9555005B2 (en) | 2013-03-15 | 2017-01-31 | Upsher-Smith Laboratories, Inc. | Extended-release topiramate capsules |
| WO2014151729A1 (en) | 2013-03-15 | 2014-09-25 | Irm Llc | Compounds and compositions for the treatment of parasitic diseases |
| WO2014160649A1 (en) | 2013-03-29 | 2014-10-02 | Novartis Ag | Hydroxamic acid derivatives as lpxc inhibitors for the treatment of bacterial infections |
| WO2014167528A1 (en) | 2013-04-11 | 2014-10-16 | Novartis Ag | Spiropyrazolopyridine derivatives and uses thereof for the treatment of viral infections |
| WO2014178040A1 (en) | 2013-04-29 | 2014-11-06 | Mapi Pharma Ltd. | Co-crystals of dapagliflozin |
| US9006188B2 (en) | 2013-04-29 | 2015-04-14 | Mapi Pharma Ltd. | Co-crystals of dapagliflozin |
| WO2014184778A1 (en) | 2013-05-17 | 2014-11-20 | Novartis Ag | Pyrimidin-4-yl)oxy)-1h-indole-1-carboxamide derivatives and use thereof |
| WO2014191894A1 (en) | 2013-05-27 | 2014-12-04 | Novartis Ag | Imidazopyrrolidinone derivatives and their use in the treatment of disease |
| EP3412675A1 (en) | 2013-05-27 | 2018-12-12 | Novartis AG | Imidazopyrrolidinone derivatives and their use in the treatment of disease |
| WO2014191896A1 (en) | 2013-05-27 | 2014-12-04 | Novartis Ag | Pyrazolopyrrolidine derivatives and their use in the treatment of disease |
| WO2014191911A1 (en) | 2013-05-28 | 2014-12-04 | Novartis Ag | Pyrazolo-pyrrolidin-4-one derivatives and their use in the treatment of disease |
| WO2014191906A1 (en) | 2013-05-28 | 2014-12-04 | Novartis Ag | Pyrazolo-pyrrolidin-4-one derivatives as bet inhibitors and their use in the treatment of disease |
| EP3299365A1 (en) | 2013-07-15 | 2018-03-28 | Novartis AG | Piperidinyl-indole derivatives complement factor b inhibitors and uses thereof |
| WO2015009616A1 (en) | 2013-07-15 | 2015-01-22 | Novartis Ag | Piperidinyl indole derivatives and their use as complement factor b inhibitors |
| WO2015008230A1 (en) | 2013-07-18 | 2015-01-22 | Novartis Ag | Autotaxin inhibitors comprising a heteroaromatic ring-benzyl-amide-cycle core |
| WO2015008229A1 (en) | 2013-07-18 | 2015-01-22 | Novartis Ag | Autotaxin inhibitors |
| EP4105208A1 (en) | 2013-07-31 | 2022-12-21 | Novartis AG | 1,4-disubstituted pyridazine derivatives and their use for treating smn-deficiency-related conditions |
| WO2015059668A1 (en) | 2013-10-25 | 2015-04-30 | Novartis Ag | Ring-fused bicyclic pyridyl derivatives as fgfr4 inhibitors |
| WO2015066241A1 (en) | 2013-10-30 | 2015-05-07 | Novartis Ag | 2-benzyl-benzimidazole complement factor b inhibitors and uses thereof |
| WO2015066188A1 (en) | 2013-11-01 | 2015-05-07 | Novartis Ag | Aminoheteroaryl benzamides as kinase inhibitors |
| WO2015066413A1 (en) | 2013-11-01 | 2015-05-07 | Novartis Ag | Oxazolidinone hydroxamic acid compounds for the treatment of bacterial infections |
| CN105899519A (en) * | 2013-11-11 | 2016-08-24 | 协同医药发展有限公司 | Metal complexes and methods of treatment |
| WO2015070177A3 (en) * | 2013-11-11 | 2015-10-08 | Collaborative Medicinal Development, Llc | Metal complexes and methods of treatment |
| US11266686B2 (en) | 2013-11-11 | 2022-03-08 | Procypra Therapeutics Llc | Metal complexes and methods of treatment |
| CN105899519B (en) * | 2013-11-11 | 2021-11-09 | 协同医药发展有限公司 | Metal complexes and methods of treatment |
| WO2015075665A1 (en) | 2013-11-21 | 2015-05-28 | Novartis Ag | Pyrrolopyrrolone derivatives and their use as bet inhibitors |
| EP4219478A1 (en) | 2013-11-29 | 2023-08-02 | Novartis AG | Method of preparing amino pyrimidine derivatives |
| EP3689865A1 (en) | 2013-11-29 | 2020-08-05 | Novartis AG | Novel amino pyrimidine derivatives |
| EP3299368A1 (en) | 2013-11-29 | 2018-03-28 | Novartis AG | Novel amino pyrimidine derivatives |
| WO2015079417A1 (en) | 2013-11-29 | 2015-06-04 | Novartis Ag | Novel amino pyrimidine derivatives |
| WO2015095301A2 (en) | 2013-12-17 | 2015-06-25 | Irm Llc | Cytotoxic peptides and conjugates thereof |
| WO2015095477A1 (en) | 2013-12-19 | 2015-06-25 | Irm Llc | [1,2,4]triazolo[1,5-a]pyrimidine derivatives as protozoan proteasome inhibitors for the treatment of parasitic diseases such as leishmaniasis |
| WO2015092740A1 (en) | 2013-12-20 | 2015-06-25 | Novartis Ag | Heteroaryl butanoic acid derivatives as lta4h inhibitors |
| WO2015102929A1 (en) | 2013-12-30 | 2015-07-09 | Novartis Ag | Tricyclic sulfonamide derivatives |
| EP3511328A1 (en) | 2014-03-24 | 2019-07-17 | Novartis AG | Monobactam organic compounds for the treatment of bacterial infections |
| WO2015148379A1 (en) | 2014-03-24 | 2015-10-01 | Novartis Ag | Monobactam organic compounds for the treatment of bacterial infections |
| WO2015161052A1 (en) | 2014-04-17 | 2015-10-22 | Novartis Ag | Polycyclic herg activators |
| WO2015164458A1 (en) | 2014-04-22 | 2015-10-29 | Novartis Ag | Isoxazoline hydroxamic acid derivatives as lpxc inhibitors |
| WO2015162459A1 (en) | 2014-04-24 | 2015-10-29 | Novartis Ag | Amino pyrazine derivatives as phosphatidylinositol 3-kinase inhibitors |
| WO2015162461A1 (en) | 2014-04-24 | 2015-10-29 | Novartis Ag | Pyrazine derivatives as phosphatidylinositol 3-kinase inhibitors |
| WO2015162558A1 (en) | 2014-04-24 | 2015-10-29 | Novartis Ag | Autotaxin inhibitors |
| WO2015162456A1 (en) | 2014-04-24 | 2015-10-29 | Novartis Ag | Amino pyridine derivatives as phosphatidylinositol 3-kinase inhibitors |
| WO2015175487A1 (en) | 2014-05-13 | 2015-11-19 | Novartis Ag | Compounds and compositions for inducing chondrogenesis |
| WO2015173656A1 (en) | 2014-05-14 | 2015-11-19 | Novartis Ag | Carboxamide derivatives |
| WO2015177646A1 (en) | 2014-05-14 | 2015-11-26 | Novartis Ag | Carboxamide derivatives |
| WO2015173659A2 (en) | 2014-05-14 | 2015-11-19 | Novartis Ag | Carboxamide derivatives |
| WO2015175796A1 (en) | 2014-05-14 | 2015-11-19 | Novartis Ag | Carboxamide derivatives |
| WO2015181747A1 (en) | 2014-05-28 | 2015-12-03 | Novartis Ag | Novel pyrazolo pyrimidine derivatives and their use as malt1 inhibitors |
| WO2015186062A1 (en) | 2014-06-03 | 2015-12-10 | Novartis Ag | PYRIMIDO[4,5-b]QUINOLINE-4,5(3H,10H)-DIONE DERIVATIVES |
| WO2015186061A1 (en) | 2014-06-03 | 2015-12-10 | Novartis Ag | Pyridopyrimidinedione derivatives |
| WO2015186063A1 (en) | 2014-06-03 | 2015-12-10 | Novartis Ag | Naphthyridinedione derivatives |
| WO2015189791A1 (en) | 2014-06-13 | 2015-12-17 | Novartis Ag | Auristatin derivatives and conjugates thereof |
| WO2016001876A1 (en) | 2014-07-02 | 2016-01-07 | Novartis Ag | Thiophen-2-yl-pyridin-2-yl-1h-pyrazole-4-carboxylic acid derivatives and the use thereof as soluble guanylate cyclase activators |
| WO2016001878A1 (en) | 2014-07-02 | 2016-01-07 | Novartis Ag | Cyclohexen-1-yl-pyridin-2-yl-1h-pyrazole-4-carboxylic acid derivatives and the use thereof as soluble guanylate cyclase activators |
| WO2016001875A1 (en) | 2014-07-02 | 2016-01-07 | Novartis Ag | Indane and indoline derivatives and the use thereof as soluble guanylate cyclase activators |
| WO2016020836A1 (en) | 2014-08-06 | 2016-02-11 | Novartis Ag | Quinolone derivatives as antibacterials |
| US9573969B2 (en) | 2014-09-12 | 2017-02-21 | Novartis Ag | Compounds and compositions as kinase inhibitors |
| WO2016038581A1 (en) | 2014-09-12 | 2016-03-17 | Novartis Ag | Compounds and compositions as raf kinase inhibitors |
| WO2016038582A1 (en) | 2014-09-12 | 2016-03-17 | Novartis Ag | Compounds and compositions as raf kinase inhibitors |
| WO2016038583A1 (en) | 2014-09-12 | 2016-03-17 | Novartis Ag | Compounds and compositions as kinase inhibitors |
| US9809610B2 (en) | 2014-09-12 | 2017-11-07 | Novartis Ag | Compounds and compositions as kinase inhibitors |
| WO2016049266A1 (en) * | 2014-09-24 | 2016-03-31 | Pain Therapeutics, Inc. | 4:3 naltrexone: 5-methyl-2-furaldehyde cocrystal |
| US9623017B2 (en) | 2014-09-24 | 2017-04-18 | Pain Therapeutics, Inc. | 4:3 naltrexone: 5-methyl-2-furaldehyde cocrystal |
| US10463653B2 (en) | 2014-10-03 | 2019-11-05 | Novartis Ag | Use of ring-fused bicyclic pyridyl derivatives as FGFR4 inhibitors |
| US10507201B2 (en) | 2014-10-03 | 2019-12-17 | Novartis Ag | Use of ring-fused bicyclic pyridyl derivatives as FGFR4 inhibitors |
| WO2016079669A1 (en) | 2014-11-19 | 2016-05-26 | Novartis Ag | Labeled amino pyrimidine derivatives |
| WO2016088082A1 (en) | 2014-12-05 | 2016-06-09 | Novartis Ag | Amidomethyl-biaryl derivatives complement factor d inhibitors and uses thereof |
| WO2016097995A1 (en) | 2014-12-16 | 2016-06-23 | Novartis Ag | Isoxazole hydroxamic acid compounds as lpxc inhibitors |
| WO2016103155A1 (en) | 2014-12-23 | 2016-06-30 | Novartis Ag | Triazolopyrimidine compounds and uses thereof |
| WO2016151499A1 (en) | 2015-03-25 | 2016-09-29 | Novartis Ag | Formylated n-heterocyclic derivatives as fgfr4 inhibitors |
| WO2016164580A1 (en) | 2015-04-07 | 2016-10-13 | Novartis Ag | Combination of chimeric antigen receptor therapy and amino pyrimidine derivatives |
| WO2016174616A1 (en) | 2015-04-30 | 2016-11-03 | Novartis Ag | Fused tricyclic pyrazole derivatives useful for modulating farnesoid x receptors |
| WO2016203432A1 (en) | 2015-06-17 | 2016-12-22 | Novartis Ag | Antibody drug conjugates |
| US11096936B2 (en) | 2015-09-24 | 2021-08-24 | Cassava Sciences, Inc. | Cocrystals of naloxone and naltrexone |
| WO2017053936A1 (en) | 2015-09-24 | 2017-03-30 | Pain Therapeutic, Inc. | Cocrystals of naloxone and naltrexone |
| EP3610883A1 (en) | 2015-10-21 | 2020-02-19 | Hox Therapeutics Limited | Peptides |
| WO2017081641A1 (en) | 2015-11-13 | 2017-05-18 | Novartis Ag | Novel pyrazolo pyrimidine derivatives |
| WO2017089985A1 (en) | 2015-11-26 | 2017-06-01 | Novartis Ag | Diamino pyridine derivatives |
| EP3757097A1 (en) | 2015-11-26 | 2020-12-30 | Novartis AG | Diamino pyridine derivatives |
| CN105541701A (en) * | 2015-12-11 | 2016-05-04 | 吉林大学珠海学院 | Pharmaceutical cocrystal of deferiprone with maleic acid as precursor, and preparation method thereof |
| CN105399665A (en) * | 2015-12-11 | 2016-03-16 | 吉林大学珠海学院 | Deferiprone pharmaceutical cocrystal with p-hydroxybenzoic acid as precursor and preparation method thereof |
| CN105399664A (en) * | 2015-12-11 | 2016-03-16 | 吉林大学珠海学院 | Deferiprone pharmaceutical cocrystal with 2,5-dihydroxybenzoic acid as precursor and preparation method thereof |
| US10688110B2 (en) | 2015-12-16 | 2020-06-23 | Nangenex Nanotechnology Incorporated | Complexes of Celecoxib and its salts and derivatives, process for the preparation thereof and pharmaceutical compositions containing them |
| WO2017103677A1 (en) | 2015-12-16 | 2017-06-22 | Druggability Technologies Ip Holdco Limited | Complexes of celecoxib and its salts and derivatives process for the preparation thereof and pharmaceutical compositions containing them |
| US10307429B2 (en) | 2015-12-16 | 2019-06-04 | Druggability Technologies Ip Holdco Limited | Complexes of celecoxib and its salts and derivatives, process for the preparation thereof and pharmaceutical compositions containing them |
| WO2017103888A1 (en) | 2015-12-18 | 2017-06-22 | Novartis Ag | Indane derivatives and the use thereof as soluble guanylate cyclase activators |
| WO2017103824A1 (en) | 2015-12-18 | 2017-06-22 | Novartis Ag | Tricyclic compounds and compositions as kinase inhibitors |
| WO2017125898A1 (en) | 2016-01-21 | 2017-07-27 | Novartis Ag | Compounds and compositions for the treatment of cryptosporidiosis |
| WO2017140821A1 (en) | 2016-02-19 | 2017-08-24 | Novartis Ag | Tetracyclic pyridone compounds as antivirals |
| WO2017149463A1 (en) | 2016-03-01 | 2017-09-08 | Novartis Ag | Cyano-substituted indole compounds and uses thereof as lsd1 inhibitors |
| US10590079B2 (en) | 2016-03-01 | 2020-03-17 | Novartis Ag | Cyano-substituted indoles as LSD1 inhibitors |
| US10858366B2 (en) | 2016-03-08 | 2020-12-08 | Novartis Ag | Tricyclic compounds useful to treat orthomyxovirus infections |
| US11453677B2 (en) | 2016-03-08 | 2022-09-27 | Novartis Ag | Tricyclic compounds useful to treat orthomyxovirus infections |
| WO2017153919A1 (en) | 2016-03-08 | 2017-09-14 | Novartis Ag | Tricyclic compounds useful to treat orthomyxovirus infections |
| EP4292658A2 (en) | 2016-03-24 | 2023-12-20 | Novartis AG | Alkynyl nucleoside analogs as inhibitors of human rhinovirus |
| WO2017163186A1 (en) | 2016-03-24 | 2017-09-28 | Novartis Ag | Alkynyl nucleoside analogs as inhibitors of human rhinovirus |
| WO2017175185A1 (en) | 2016-04-08 | 2017-10-12 | Novartis Ag | Heteroaryl butanoic acid derivatives as lta4h inhibitors |
| US10195218B2 (en) | 2016-05-31 | 2019-02-05 | Grunenthal Gmbh | Crystallization method and bioavailability |
| WO2017216685A1 (en) | 2016-06-16 | 2017-12-21 | Novartis Ag | Pentacyclic pyridone compounds as antivirals |
| WO2017216686A1 (en) | 2016-06-16 | 2017-12-21 | Novartis Ag | 8,9-fused 2-oxo-6,7-dihydropyrido-isoquinoline compounds as antivirals |
| US11548897B2 (en) | 2016-06-20 | 2023-01-10 | Novartis Ag | Crystalline forms of a triazolopyrimidine compound |
| US11091489B2 (en) | 2016-06-20 | 2021-08-17 | Novartis Ag | Crystalline forms of a triazolopyrimidine compound |
| US10676479B2 (en) | 2016-06-20 | 2020-06-09 | Novartis Ag | Imidazolepyridine compounds and uses thereof |
| WO2017221092A1 (en) | 2016-06-20 | 2017-12-28 | Novartis Ag | Triazolopyridine compounds and uses thereof |
| US10689378B2 (en) | 2016-06-20 | 2020-06-23 | Novartis Ag | Triazolopyridine compounds and uses thereof |
| WO2017221100A1 (en) | 2016-06-20 | 2017-12-28 | Novartis Ag | Imidazopyrimidine compounds useful for the treatment of cancer |
| CN106432136A (en) * | 2016-06-30 | 2017-02-22 | 中国药科大学 | Hydrochlorothiazide and atenolol co-amorphous system and preparation method thereof |
| CN106432136B (en) * | 2016-06-30 | 2018-12-25 | 中国药科大学 | A kind of Hydrochioro and atenolol are total to unformed system and preparation method thereof |
| EP4442317A2 (en) | 2016-07-20 | 2024-10-09 | Novartis AG | Aminopyridine derivatives and their use as selective alk-2 inhibitors |
| EP3971177A1 (en) | 2016-07-20 | 2022-03-23 | Novartis AG | Aminopyridine derivatives and their use as selective alk-2 inhibitors |
| WO2018014829A1 (en) | 2016-07-20 | 2018-01-25 | Novartis Ag | Aminopyridine derivatives and their use as selective alk-2 inhibitors |
| US11912715B2 (en) | 2016-08-29 | 2024-02-27 | Novartis Ag | Fused tricyclic pyridazinone compounds useful to treat orthomyxovirus infections |
| EP3848372A1 (en) | 2016-08-29 | 2021-07-14 | Novartis AG | Fused tricyclic pyridazinone compounds useful to treat orthomyxovirus infections |
| EP4356969A2 (en) | 2016-08-29 | 2024-04-24 | Novartis AG | Fused tricyclic pyridazinone compounds useful to treat orthomyxovirus infections |
| US11098051B2 (en) | 2016-08-29 | 2021-08-24 | Novartis Ag | Fused tricyclic pyridazinone compounds useful to treat orthomyxovirus infections |
| WO2018042303A1 (en) | 2016-08-29 | 2018-03-08 | Novartis Ag | Fused tricyclic pyridazinone compounds useful to treat orthomyxovirus infections |
| WO2018042316A1 (en) | 2016-08-29 | 2018-03-08 | Novartis Ag | N-(pyridin-2-yl)pyridine-sulfonamide derivatives and their use in the treatment of disease |
| WO2018047081A1 (en) | 2016-09-09 | 2018-03-15 | Novartis Ag | Compounds and compositions as inhibitors of endosomal toll-like receptors |
| WO2018047109A1 (en) | 2016-09-09 | 2018-03-15 | Novartis Ag | Polycyclic pyridone compounds as antivirals |
| EP4059934A1 (en) | 2016-09-09 | 2022-09-21 | Novartis AG | Compounds and compositions as inhibitors of endosomal toll-like receptors |
| WO2018055550A1 (en) | 2016-09-23 | 2018-03-29 | Novartis Ag | Indazole compounds for use in tendon and/or ligament injuries |
| WO2018055551A1 (en) | 2016-09-23 | 2018-03-29 | Novartis Ag | Aza-indazole compounds for use in tendon and/or ligament injuries |
| WO2018060926A1 (en) | 2016-09-28 | 2018-04-05 | Novartis Ag | Beta-lactamase inhibitors |
| EP3698796A1 (en) | 2016-09-28 | 2020-08-26 | Novartis AG | Pharmaceutical combination of a tricyclic beta-lactamase inhibitor with specific beta-lactam antibiotics |
| WO2018073753A1 (en) | 2016-10-18 | 2018-04-26 | Novartis Ag | Fused tetracyclic pyridone compounds as antivirals |
| WO2018087677A1 (en) | 2016-11-10 | 2018-05-17 | Novartis Ag | Bmp potentiators |
| WO2018141749A1 (en) | 2017-02-01 | 2018-08-09 | Medivir Ab | Therapeutic applications of malt1 inhibitors |
| WO2018172997A1 (en) | 2017-03-24 | 2018-09-27 | Novartis Ag | Isoxazole carboxamide compounds and uses thereof |
| US10975078B2 (en) | 2017-04-27 | 2021-04-13 | Novartis Ag | Fused indazole pyridone compounds as antivirals |
| EP3998269A1 (en) | 2017-04-27 | 2022-05-18 | Novartis AG | Fused indazole pyridone compounds as antivirals |
| WO2018198079A1 (en) | 2017-04-27 | 2018-11-01 | Novartis Ag | Fused indazole pyridone compounds as antivirals |
| US10301312B2 (en) | 2017-04-27 | 2019-05-28 | Novartis Ag | Fused indazole pyridone compounds as antivirals |
| WO2018203302A1 (en) | 2017-05-05 | 2018-11-08 | Novartis Ag | Tricyclic 2-quinolinones as antibacterials |
| US10604502B2 (en) | 2017-06-19 | 2020-03-31 | Novartis Ag | Substituted 5-cyanoindole compounds and uses thereof |
| WO2018234978A1 (en) | 2017-06-19 | 2018-12-27 | Novartis Ag | Substituted 5-cyanoindole compounds and uses thereof |
| WO2019087163A1 (en) | 2017-11-06 | 2019-05-09 | Novartis Ag | Polycyclic herg activators |
| WO2019087162A1 (en) | 2017-11-06 | 2019-05-09 | Novartis Ag | Polycyclic herg activators |
| WO2019097479A1 (en) | 2017-11-17 | 2019-05-23 | Novartis Ag | Novel dihydroisoxazole compounds and their use for the treatment of hepatitis b |
| WO2019102256A1 (en) | 2017-11-24 | 2019-05-31 | Novartis Ag | Pyridinone derivatives and their use as selective alk-2 inhibitors |
| WO2019123285A1 (en) | 2017-12-20 | 2019-06-27 | Novartis Ag | Fused tricyclic pyrazolo-dihydropyrazinyl-pyridone compounds as antivirals |
| US11234977B2 (en) | 2017-12-20 | 2022-02-01 | Novartis Ag | Fused tricyclic pyrazolo-dihydropyrazinyl-pyridone compounds as antivirals |
| WO2019124507A1 (en) | 2017-12-21 | 2019-06-27 | 杏林製薬株式会社 | Therapeutic agent for nocturnal pollakiuria |
| EP3778557A1 (en) | 2018-02-07 | 2021-02-17 | Novartis AG | Combinations comprising a substituted biphenylbutanoic ester derivative as nep inhibitor and valsartan |
| WO2019154416A1 (en) | 2018-02-07 | 2019-08-15 | Novartis Ag | Substituted bisphenyl butanoic ester derivatives as nep inhibitors |
| WO2019166950A1 (en) | 2018-02-28 | 2019-09-06 | Novartis Ag | 10-(di(phenyl)methyl)-4-hydroxy-8,9,9a,10-tetrahydro-7h-pyrrolo[1 ',2':4,5]pyrazino[1,2-b]pyridazine-3,5-dione derivatives and related compounds as inhibitors of the orthomyxovirus replication for treating influenza |
| WO2019166951A1 (en) | 2018-02-28 | 2019-09-06 | Novartis Ag | Indole-2-carbonyl compounds and their use for the treatment of hepatitis b |
| US12071441B2 (en) | 2018-02-28 | 2024-08-27 | Novartis Ag | 10-(di(phenyl)methyl)-4-hydroxy-8,9,9A,10-tetrahydro-7H-pyrrolo[1′,2′:4,5]pyrazino[1,2-b]pyridazine-3,5-dione derivatives and related compounds as inhibitors of the orthomyxovirus replication for treating influenza |
| US11629149B2 (en) | 2018-02-28 | 2023-04-18 | Novartis Ag | 10-(di(phenyl)methyl)-4-hydroxy-8,9,9A,10-tetrahydro-7H-pyrrolo[1′,2′:4,5]pyrazino[1,2-b]pyridazine-3,5-dione derivatives and related compounds as inhibitors of the orthomyxovirus replication for treating influenza |
| WO2019223632A1 (en) | 2018-05-22 | 2019-11-28 | Js Innomed Holdings Ltd. | Heterocyclic compounds as kinase inhibitors, compositions comprising the heterocyclic compound, and methods of use thereof |
| WO2019244049A1 (en) | 2018-06-19 | 2019-12-26 | Novartis Ag | Cyanotriazole compounds and uses thereof |
| WO2020016594A1 (en) | 2018-07-19 | 2020-01-23 | Benevolentai Bio Limited | Imidazo[1,2-b]pyridazine derivatives as trk inhibitors |
| US11040038B2 (en) | 2018-07-26 | 2021-06-22 | Sumitomo Dainippon Pharma Oncology, Inc. | Methods for treating diseases associated with abnormal ACVR1 expression and ACVR1 inhibitors for use in the same |
| WO2020035779A1 (en) | 2018-08-17 | 2020-02-20 | Novartis Ag | Urea compounds and compositions as smarca2/brm atpase inhibitors |
| EP4219488A1 (en) | 2018-08-17 | 2023-08-02 | Novartis AG | Method for the preparation of urea compounds as smarca2/brm-atpase inhibitors |
| WO2020039209A1 (en) | 2018-08-23 | 2020-02-27 | Benevolentai Bio Limited | Imidazo[1,2-b]pyridazines as trk inhibitors |
| US11072610B2 (en) | 2018-09-12 | 2021-07-27 | Novartis Ag | Antiviral pyridopyrazinedione compounds |
| WO2020053654A1 (en) | 2018-09-12 | 2020-03-19 | Novartis Ag | Antiviral pyridopyrazinedione compounds |
| WO2020058913A1 (en) | 2018-09-21 | 2020-03-26 | Novartis Ag | Isoxazole carboxamide compounds and uses thereof |
| WO2020075046A1 (en) * | 2018-10-09 | 2020-04-16 | Novartis Ag | Solid forms of n-(4-fluoro-3-(6-(3-methylpyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)phenyl)-2,4-dimethyloxazole-5-carboxamide |
| JP2022504259A (en) * | 2018-10-09 | 2022-01-13 | ノバルティス アーゲー | N- (4-fluoro-3- (6- (3-methylpyridine-2-yl)-[1,2,4] triazolo [1,5-A] pyrimidin-2-yl) phenyl) -2,4 -Solid form of dimethyloxazole-5-carboxamide |
| US11891401B2 (en) | 2018-10-09 | 2024-02-06 | Novartis Ag | Solid forms of N-(4-fluoro-3-(6-(3-methylpyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)phenyl)-2,4-dimethyloxazole-5-carboxamide |
| WO2020128768A1 (en) | 2018-12-18 | 2020-06-25 | Novartis Ag | N-(pyridin-2-ylsulfonyl)cyclopropanecarboxamide derivatives and their use in the treatment of a cftr mediated disease |
| WO2020144604A1 (en) | 2019-01-11 | 2020-07-16 | Novartis Ag | Lta4h inhibitors for the treatment of hidradenitis suppurativa |
| EP4442315A2 (en) | 2019-01-11 | 2024-10-09 | Novartis AG | Lta4h inhibitor for the treatment or prevention of hidradenitis suppurativa |
| WO2020150626A1 (en) | 2019-01-18 | 2020-07-23 | Biogen Ma Inc. | Imidazo[1,2-a]pyridinyl derivatives as irak4 inhibitors |
| WO2020154581A1 (en) * | 2019-01-24 | 2020-07-30 | Assia Chemical Industries Ltd | Solid state forms of fedovapagon-salicyclic acid co-crystal |
| US11712433B2 (en) | 2019-03-22 | 2023-08-01 | Sumitomo Pharma Oncology, Inc. | Compositions comprising PKM2 modulators and methods of treatment using the same |
| WO2020198077A1 (en) | 2019-03-22 | 2020-10-01 | Sumitomo Dainippon Pharma Oncology, Inc. | Compositions comprising pkm2 modulators and methods of treatment using the same |
| WO2020250116A1 (en) | 2019-06-10 | 2020-12-17 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of cf, copd, and bronchiectasis |
| WO2020263980A1 (en) | 2019-06-27 | 2020-12-30 | Biogen Ma Inc. | Imidazo[1,2-a]pyridinyl derivatives and their use in the treatment of disease |
| WO2020263967A1 (en) | 2019-06-27 | 2020-12-30 | Biogen Ma Inc. | 2h-indazole derivatives and their use in the treatment of disease |
| WO2021003417A1 (en) | 2019-07-03 | 2021-01-07 | Sumitomo Dainippon Pharma Oncology, Inc. | Tyrosine kinase non-receptor 1 (tnk1) inhibitors and uses thereof |
| US11529350B2 (en) | 2019-07-03 | 2022-12-20 | Sumitomo Pharma Oncology, Inc. | Tyrosine kinase non-receptor 1 (TNK1) inhibitors and uses thereof |
| WO2021038426A1 (en) | 2019-08-28 | 2021-03-04 | Novartis Ag | Substituted 1,3-phenyl heteroaryl derivatives and their use in the treatment of disease |
| WO2021044351A1 (en) | 2019-09-06 | 2021-03-11 | Novartis Ag | Methods of treating liver disease using lta4h inhibitors |
| CN113795484A (en) * | 2019-09-26 | 2021-12-14 | 大峰Ls株式会社 | Eutectic-type efinaconazole and preparation method thereof |
| WO2021060949A1 (en) * | 2019-09-26 | 2021-04-01 | 대봉엘에스 주식회사 | Co-crystalline efinaconazole, and method for producing same |
| US11667613B2 (en) | 2019-09-26 | 2023-06-06 | Novartis Ag | Antiviral pyrazolopyridinone compounds |
| CN113795484B (en) * | 2019-09-26 | 2024-07-09 | 大峰Ls株式会社 | Eutectic type efroconazole and preparation method thereof |
| WO2021124172A1 (en) | 2019-12-18 | 2021-06-24 | Novartis Ag | 3-(5-methoxy-1-oxoisoindolin-2-yl)piperidine-2,6-dione derivatives and uses thereof |
| WO2021148807A1 (en) | 2020-01-22 | 2021-07-29 | Benevolentai Bio Limited | Pharmaceutical compositions and their uses |
| WO2021148805A1 (en) | 2020-01-22 | 2021-07-29 | Benevolentai Bio Limited | Topical pharmaceutical compositions comprising imidazo[1,2-b]pyridazine compounds |
| WO2021191169A1 (en) | 2020-03-23 | 2021-09-30 | Artemiflow GmbH | 1,2,4-trioxane compounds and compositions comprising the same for use in the treatment of covid-19 |
| EP3884938A1 (en) | 2020-03-25 | 2021-09-29 | ArtemiFlow GmbH | 1,2,4-trioxane compounds and compositions comprising the same for use in the treatment of covid-19 |
| WO2021214073A1 (en) | 2020-04-20 | 2021-10-28 | Novartis Ag | Antiviral 1,3-di-oxo-indene compounds |
| WO2021214080A1 (en) | 2020-04-20 | 2021-10-28 | Novartis Ag | Antiviral 1,3-di-oxo-indene compounds |
| WO2021218930A1 (en) * | 2020-04-30 | 2021-11-04 | 江苏恩华药业股份有限公司 | Co-amorphous substance of celecoxib and pregabalin and preparation method therefor |
| CN115551834B (en) * | 2020-04-30 | 2023-11-03 | 苏州恩华生物医药科技有限公司 | Celecoxib and pregabalin co-amorphous substance and preparation method thereof |
| CN113582927A (en) * | 2020-04-30 | 2021-11-02 | 苏州恩华生物医药科技有限公司 | Celecoxib and pregabalin co-amorphous compound and preparation method thereof |
| CN115551834A (en) * | 2020-04-30 | 2022-12-30 | 苏州恩华生物医药科技有限公司 | Celecoxib and pregabalin co-amorphous compound and preparation method thereof |
| CN113582927B (en) * | 2020-04-30 | 2023-07-04 | 苏州恩华生物医药科技有限公司 | Celecoxib and pregabalin co-amorphous substance and preparation method thereof |
| US11806346B2 (en) | 2020-05-13 | 2023-11-07 | Chdi Foundation, Inc. | HTT modulators for treating Huntington's disease |
| WO2021255607A1 (en) | 2020-06-15 | 2021-12-23 | Novartis Ag | Methyl 2-(fluoromethyl)-5-oxo-4-phenyl-4,5,6,7-tetrahydro-1h-cyclopenta[b]pyridine-3-carboxylates and methyl 2-(fluoromethyl)-5-oxo-4-phenyl-1,4,5,7-tetrahydrofuro[3,4-b]pyridine-3-carboxylates as cav1.2 activators |
| WO2021255608A1 (en) | 2020-06-16 | 2021-12-23 | Novartis Ag | Methyl 2-methyl-5-oxo-1,4,5,7-tetradhydrofuro[3,4- b]pyridine-3-carboxylate compounds as cav1.2 activators |
| WO2022058902A1 (en) | 2020-09-17 | 2022-03-24 | Novartis Ag | Compounds and compositions as sppl2a inhibitors |
| CN112552189A (en) * | 2020-11-10 | 2021-03-26 | 中国海洋大学 | Pharmaceutical co-crystal of amantadine hydrochloride and resveratrol and preparation method thereof |
| CN112521292A (en) * | 2020-12-18 | 2021-03-19 | 深圳市萱嘉生物科技有限公司 | Eutectic crystal of betaine and organic acid and preparation method and application thereof |
| WO2022140425A1 (en) | 2020-12-22 | 2022-06-30 | Biogen Ma Inc. | Imidazo[1,2-a]pyridine derivatives as irak4 inhibitors and their use in the treatment of disease |
| WO2022140415A1 (en) | 2020-12-22 | 2022-06-30 | Biogen Ma Inc. | 2h-indazole derivatives as irak4 inhibitors and their use in the treatment of disease |
| WO2022150446A1 (en) | 2021-01-07 | 2022-07-14 | Biogen Ma Inc. | Tyk2 inhibitors |
| WO2022195454A1 (en) | 2021-03-15 | 2022-09-22 | Novartis Ag | Pyrazolopyridine derivatives and uses thereof |
| WO2022195355A1 (en) | 2021-03-15 | 2022-09-22 | Novartis Ag | Benzisoxazole derivatives and uses thereof |
| WO2022201097A1 (en) | 2021-03-26 | 2022-09-29 | Novartis Ag | 1,3-substituted cyclobutyl derivatives and uses thereof |
| WO2022217232A1 (en) | 2021-04-10 | 2022-10-13 | Sunovion Pharmaceuticals Inc. | Chromans and benzofurans as 5-ht1a and taar1 agonists |
| WO2022217248A1 (en) | 2021-04-10 | 2022-10-13 | Sunovion Pharmaceuticals Inc. | Substituted sulfonamide-chroman compounds, and pharmaceutical compositions, and methods of use thereof |
| WO2022219546A1 (en) | 2021-04-16 | 2022-10-20 | Novartis Ag | Heteroaryl aminopropanol derivatives as inhibitors of lta4h |
| WO2022234287A1 (en) | 2021-05-06 | 2022-11-10 | Benevolentai Bio Limited | Imidazopyridazine derivatives useful as trk inhibitors |
| WO2022249060A1 (en) | 2021-05-26 | 2022-12-01 | Novartis Ag | Triazolo-pyrimidine analogues for treating diseases connected to the inhibiton of werner syndrome recq helicase (wrn) |
| WO2022254362A1 (en) | 2021-06-03 | 2022-12-08 | Novartis Ag | 3-(1-oxoisoindolin-2-yl)piperidine-2,6-dione derivatives and medical uses thereof |
| US12005134B2 (en) | 2021-06-30 | 2024-06-11 | Abe Pharmaceutical | Composition for stimulating facial hair growth and methods of manufacturing a composition for stimulating facial hair growth |
| WO2023275796A1 (en) | 2021-07-01 | 2023-01-05 | Novartis Ag | Heterocyclic derivatives as sphingosine-1-phosphate 3 inhibitors |
| WO2023002443A1 (en) | 2021-07-22 | 2023-01-26 | Novartis Ag | Substituted pyridone compounds useful to treat orthomyxovirus infections |
| WO2023049721A1 (en) | 2021-09-23 | 2023-03-30 | Sunovion Pharmaceuticals Inc. | Methods of treating metabolic disorders |
| CN113845438B (en) * | 2021-10-12 | 2023-02-21 | 河北大学 | Acetaminophen-piracetam pharmaceutical co-crystal and preparation method thereof |
| CN113845438A (en) * | 2021-10-12 | 2021-12-28 | 河北大学 | Acetaminophen-piracetam pharmaceutical co-crystal and preparation method thereof |
| WO2023094965A1 (en) | 2021-11-23 | 2023-06-01 | Novartis Ag | Naphthyridinone derivatives for the treatment of a disease or disorder |
| CN114031515A (en) * | 2021-11-29 | 2022-02-11 | 河北大学 | Paracetamol-ibuprofen pharmaceutical co-crystal and preparation method thereof |
| CN114031515B (en) * | 2021-11-29 | 2022-10-21 | 河北大学 | Acetaminophen-ibuprofen pharmaceutical co-crystal and preparation method thereof |
| WO2023111799A1 (en) | 2021-12-13 | 2023-06-22 | Novartis Ag | PYRIDINE-3-CARBOXYLATE COMPOUNDS AS CaV1.2 ACTIVATORS |
| WO2023139534A1 (en) | 2022-01-24 | 2023-07-27 | Novartis Ag | Spirocyclic piperidinyl derivatives as complement factor b inhibitors and uses thereof |
| CN114644669A (en) * | 2022-03-10 | 2022-06-21 | 华润紫竹药业有限公司 | Preparation method and application of progesterone eutectic |
| WO2023187715A1 (en) | 2022-04-01 | 2023-10-05 | Novartis Ag | Complement factor b inhibitors and uses thereof |
| CN114835657A (en) * | 2022-06-10 | 2022-08-02 | 大连工业大学 | Method for regulating and controlling ethenzamide-saccharin eutectic crystal polycrystalline type based on ionic liquid |
| WO2024006493A1 (en) | 2022-07-01 | 2024-01-04 | Biogen Ma Inc. | Tyk2 inhibitors |
| WO2024028782A1 (en) | 2022-08-03 | 2024-02-08 | Novartis Ag | Nlrp3 inflammasome inhibitors |
| WO2024079623A1 (en) | 2022-10-12 | 2024-04-18 | Novartis Ag | Tricyclic compounds and their uses |
| WO2024105553A1 (en) | 2022-11-16 | 2024-05-23 | Novartis Ag | Bicyclic heterocycles and their use as wrn inhibitors |
| WO2024105610A1 (en) | 2022-11-18 | 2024-05-23 | Novartis Ag | Pharmaceutical combinations and uses thereof |
| WO2024118488A1 (en) | 2022-11-28 | 2024-06-06 | Sumitomo Pharma America, Inc. | 2-phenylmorpholine and 2-phenyl(thio)morpholine compounds and uses thereof |
| CN116162053A (en) * | 2023-01-10 | 2023-05-26 | 山东省分析测试中心 | Gliclazide-piperazine eutectic crystal and preparation method thereof |
| WO2024176130A1 (en) | 2023-02-23 | 2024-08-29 | Novartis Ag | Tead- and her2-inhibitor combinations for treating cancer |
| WO2024176131A1 (en) | 2023-02-23 | 2024-08-29 | Novartis Ag | Tead- and kras g12d-inhibitor combinations for treating cancer |
| WO2024211696A1 (en) | 2023-04-07 | 2024-10-10 | Biogen Ma Inc. | 1h-pyrrolo[2,3-b]pyridin-4-yl]-2-oxopyrrolidine-3-carbonitrile derivatives as tyrosine kinase 2 (tyk2) inhibitors for the treatment of inflammatory diseases |
| WO2024218275A1 (en) * | 2023-04-20 | 2024-10-24 | Institut Gustave Roussy | Liquid and solid compositions of imipridone derivatives |
| CN118078830A (en) * | 2024-03-13 | 2024-05-28 | 海南海和制药有限公司 | Preparation method of pharmaceutical composition containing fluorouracil |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2007524596A (en) | 2007-08-30 |
| CA2514733A1 (en) | 2004-09-16 |
| WO2004078163A3 (en) | 2005-01-20 |
| EP1631260A2 (en) | 2006-03-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1631260A2 (en) | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen | |
| JP4923182B2 (en) | Celecoxib and nicotinamide co-crystal and pharmaceutical composition containing the co-crystal | |
| US20070026078A1 (en) | Pharmaceutical co-crystal compositions | |
| US20070059356A1 (en) | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen | |
| US20100311701A1 (en) | Pharmaceutical Co-Crystal Compositions | |
| US7803786B2 (en) | Pharmaceutical co-crystal compositions and related methods of use | |
| EP1755388B1 (en) | Mixed co-crystals and pharmaceutical compositions comprising the same | |
| US8436029B2 (en) | Pharmaceutical forms, and methods of making and using the same | |
| US7566805B2 (en) | Modafinil compositions | |
| CA2534664C (en) | Modafinil compositions | |
| US8809586B2 (en) | Modafinil compositions | |
| US20060287392A1 (en) | Gabapentin compositions | |
| EP2292213A1 (en) | Compositions comprising a polymorphic form of armodafinil | |
| IL199140A (en) | Modafinil compositions | |
| ALMARSSON et al. | Patent 2514733 Summary | |
| ZA200602736B (en) | Modafinil compositions |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2514733 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10546963 Country of ref document: US Ref document number: 2006508979 Country of ref document: JP Ref document number: 2007059356 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004715190 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004715190 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 10546963 Country of ref document: US |