WO2014160649A1 - Hydroxamic acid derivatives as lpxc inhibitors for the treatment of bacterial infections - Google Patents
Hydroxamic acid derivatives as lpxc inhibitors for the treatment of bacterial infections Download PDFInfo
- Publication number
- WO2014160649A1 WO2014160649A1 PCT/US2014/031612 US2014031612W WO2014160649A1 WO 2014160649 A1 WO2014160649 A1 WO 2014160649A1 US 2014031612 W US2014031612 W US 2014031612W WO 2014160649 A1 WO2014160649 A1 WO 2014160649A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- alkoxy
- crc
- optionally substituted
- group
- Prior art date
Links
- 0 *I***C(NO)=O Chemical compound *I***C(NO)=O 0.000 description 20
- DGXMWHJISAVEOJ-UHFFFAOYSA-N CC(N(CC1)CCC1(CCN(C=CC(c1ccccc1)=C1)C1=O)C(NO)=O)=O Chemical compound CC(N(CC1)CCC1(CCN(C=CC(c1ccccc1)=C1)C1=O)C(NO)=O)=O DGXMWHJISAVEOJ-UHFFFAOYSA-N 0.000 description 2
- APXOQKJSQCYXIM-UHFFFAOYSA-N CCC(CC)(CC1)CCS1(=O)=O Chemical compound CCC(CC)(CC1)CCS1(=O)=O APXOQKJSQCYXIM-UHFFFAOYSA-N 0.000 description 2
- JPCXERIJFYEYBE-UHFFFAOYSA-N COc1ccc(-c2ccc(CNC(C3)(CC3O)C(NO)=O)cc2)c(F)c1 Chemical compound COc1ccc(-c2ccc(CNC(C3)(CC3O)C(NO)=O)cc2)c(F)c1 JPCXERIJFYEYBE-UHFFFAOYSA-N 0.000 description 2
- ZRFZKLTVZNNDOF-UHFFFAOYSA-N CC(C)(C)C(C)(CC1)CS1(=O)=O Chemical compound CC(C)(C)C(C)(CC1)CS1(=O)=O ZRFZKLTVZNNDOF-UHFFFAOYSA-N 0.000 description 1
- LCDZQMQYKLQMCC-UHFFFAOYSA-N CC(C)(C)C(CC1)(CS1(=O)=O)C(C)(C)C Chemical compound CC(C)(C)C(CC1)(CS1(=O)=O)C(C)(C)C LCDZQMQYKLQMCC-UHFFFAOYSA-N 0.000 description 1
- LFONTVSSNDSRJG-UHFFFAOYSA-N CC(c(cc1)ccc1-[n]1nccn1)=O Chemical compound CC(c(cc1)ccc1-[n]1nccn1)=O LFONTVSSNDSRJG-UHFFFAOYSA-N 0.000 description 1
- VMYHDBYTGXFZMD-UHFFFAOYSA-N CCC(CC)(CC1)CC1S Chemical compound CCC(CC)(CC1)CC1S VMYHDBYTGXFZMD-UHFFFAOYSA-N 0.000 description 1
- OCXRYDVMQXVJHJ-UHFFFAOYSA-N CCCC(C)(CC1)CC1OC Chemical compound CCCC(C)(CC1)CC1OC OCXRYDVMQXVJHJ-UHFFFAOYSA-N 0.000 description 1
- YCSWTNOOIHXDLP-UHFFFAOYSA-N CCOC(C(C1)(CC1O)NC(OC(C)(C)C)=O)=O Chemical compound CCOC(C(C1)(CC1O)NC(OC(C)(C)C)=O)=O YCSWTNOOIHXDLP-UHFFFAOYSA-N 0.000 description 1
- ZRKSHRHJIFWXJG-UHFFFAOYSA-N CC[O](C)C(C1(CNC1)NCc(cc1)ccc1Br)=O Chemical compound CC[O](C)C(C1(CNC1)NCc(cc1)ccc1Br)=O ZRKSHRHJIFWXJG-UHFFFAOYSA-N 0.000 description 1
- ATEZPJXGOJUIJG-UHFFFAOYSA-N COC(C(CC1)(CCS1(=O)=O)NCc(cc1)ccc1C#Cc1ccccc1)=O Chemical compound COC(C(CC1)(CCS1(=O)=O)NCc(cc1)ccc1C#Cc1ccccc1)=O ATEZPJXGOJUIJG-UHFFFAOYSA-N 0.000 description 1
- JYUODXKBNUQKNV-UHFFFAOYSA-N COC(C(CCCl)(CC1)CCS1(=O)=O)=O Chemical compound COC(C(CCCl)(CC1)CCS1(=O)=O)=O JYUODXKBNUQKNV-UHFFFAOYSA-N 0.000 description 1
- MKFCYQTVSDCXAQ-UHFFFAOYSA-N Cc(c(F)c1)ccc1Cl Chemical compound Cc(c(F)c1)ccc1Cl MKFCYQTVSDCXAQ-UHFFFAOYSA-N 0.000 description 1
- WQOHEWKWIJOAHQ-UHFFFAOYSA-N Cc(ccc(OC)c1)c1F Chemical compound Cc(ccc(OC)c1)c1F WQOHEWKWIJOAHQ-UHFFFAOYSA-N 0.000 description 1
- KTKOKZANTAIMCP-UHFFFAOYSA-N O=S1(CCC2(CCC2)CC1)=O Chemical compound O=S1(CCC2(CCC2)CC1)=O KTKOKZANTAIMCP-UHFFFAOYSA-N 0.000 description 1
- DXXYHEYUHAAQND-UHFFFAOYSA-N OC(C(CC1)(CS1(=O)=O)NCc1n[o]c(-c2ccccc2)c1)=O Chemical compound OC(C(CC1)(CS1(=O)=O)NCc1n[o]c(-c2ccccc2)c1)=O DXXYHEYUHAAQND-UHFFFAOYSA-N 0.000 description 1
- IEKPDBUUDDNSEX-UHFFFAOYSA-N ONC(C(CC1)(CCS1(=O)=O)NCc(cc1)ccc1-c(cc1)ccc1-[n]1nccn1)=O Chemical compound ONC(C(CC1)(CCS1(=O)=O)NCc(cc1)ccc1-c(cc1)ccc1-[n]1nccn1)=O IEKPDBUUDDNSEX-UHFFFAOYSA-N 0.000 description 1
- CJYUPLLILBZFRV-UHFFFAOYSA-N ONC(C(CC1)(CCS1(=O)=O)NCc1n[o]c(-c(cccc2)c2F)c1)=O Chemical compound ONC(C(CC1)(CCS1(=O)=O)NCc1n[o]c(-c(cccc2)c2F)c1)=O CJYUPLLILBZFRV-UHFFFAOYSA-N 0.000 description 1
- OFQKCVKKYABAEG-UHFFFAOYSA-N ONC(C(CC1)(CCS1(=O)=O)NCc1n[o]c(-c2ccccc2)c1)=O Chemical compound ONC(C(CC1)(CCS1(=O)=O)NCc1n[o]c(-c2ccccc2)c1)=O OFQKCVKKYABAEG-UHFFFAOYSA-N 0.000 description 1
- LKUQXDROMCQHBI-UHFFFAOYSA-N ONC(C(CC1)(CS1(=O)=O)NCc1n[o]c(-c2ccccc2)c1)=O Chemical compound ONC(C(CC1)(CS1(=O)=O)NCc1n[o]c(-c2ccccc2)c1)=O LKUQXDROMCQHBI-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D335/00—Heterocyclic compounds containing six-membered rings having one sulfur atom as the only ring hetero atom
- C07D335/02—Heterocyclic compounds containing six-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C259/00—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups
- C07C259/04—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups without replacement of the other oxygen atom of the carboxyl group, e.g. hydroxamic acids
- C07C259/08—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups without replacement of the other oxygen atom of the carboxyl group, e.g. hydroxamic acids having carbon atoms of hydroxamic groups bound to carbon atoms of rings other than six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D205/00—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom
- C07D205/02—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings
- C07D205/04—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/46—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with hetero atoms directly attached to the ring nitrogen atom
- C07D207/48—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/92—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with a hetero atom directly attached to the ring nitrogen atom
- C07D211/96—Sulfur atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D305/00—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms
- C07D305/02—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D305/04—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D305/08—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/04—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D307/18—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D307/24—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D309/08—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/38—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/46—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings substituted on the ring sulfur atom
- C07D333/48—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings substituted on the ring sulfur atom by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/04—Systems containing only non-condensed rings with a four-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
This invention pertains generally to antibacterial organic compounds of Formula I as described herein, and pharmaceutical compositions containing such compounds. In certain aspects, the invention pertains to treating infections caused by Gram-negative bacteria using these compounds and compositions.
Description
HYDROXAMIC ACID DERIVATIVES AS LpxC INHIBITORS FOR THE TREATMENT OF
BACTERIAL INFECTIONS
FIELD OF THE INVENTION
This invention pertains generally to treating bacterial infections. In certain aspects, the invention pertains to treating infections caused by Gram- negative bacteria. More specifically, the invention described herein pertains to treating Gram-negative infections by inhibiting the activity of UDP-3-0-(R-3-hydroxydecanoyl)-N-acetylglucosamine deacetylase (LpxC). The present invention provides small molecule inhibitors of LpxC, pharmaceutical formulations containing such inhibitors, methods of treating patients with such
pharmaceutical formulations, and methods of preparing such pharmaceutical formulations and inhibitors. The inhibitors can be used to treat Gram-negative infections of patients alone and in combination with other antibacterials.
BACKGROUND OF THE INVENTION
Over the past several decades, the frequency of antimicrobial resistance and its association with serious infectious diseases have increased at alarming rates. The increasing prevalence of resistance among nosocomial pathogens is particularly
disconcerting. Of the over 2 million nosocomial infections occuring each year in the United States, 50 to 60% are caused by antimicrobial-resistant strains of bacteria. The high rate of resistance to commonly used antibacterial agents increases the morbidity, mortality, and costs associated with nosocomial infections. In the United States, nosocomial infections are thought to contribute to or cause more than 77,000 deaths per year and cost approximately $5 to $10 billion annually. Among Gram-positive organisms, the most important resistant pathogens are methicillin-(oxacillin-) resistant Staphylococcus aureus, β-lactam-resistant and multidrug-resistant pneumococci, and vancomycin-resistant enterococci. Important causes of Gram-negative resistance include extended-spectrum β-lactamases (ESBLs) in Klebsiella pneumoniae, Escherichia coli, and Proteus mirabilis, high-level third-generation cephalosporin (Amp C) β-lactamase resistance among Enterobacter species and Citrobacter freundii, and multidrug-resistance genes observed in Pseudomonas, Acinetobacter, and Stenotrophomonas.
The problem of antibacterial resistance is compounded by the existence of bacterial strains resistant to multiple antibacterials. For example, Pseudomonas aeruginosa isolates resistant to fluoroquinolones are virtually all resistant to additional antibacterial medicines.
Thus there is a need for new antibacterials, particularly antibacterials with novel mechanisms of action. Most of the antibacterial discovery effort in the pharmaceutical industry is aimed at the development of drugs effective against Gram-positive bacteria.
However, there is also a need for new Gram-negative antibacterials. Gram-negative bacteria are in general more resistant to a large number of antibacterials and chemotherapeutic agents than are gram-positive bacteria.
SUMMARY OF THE INVENTION
The present invention provides novel compounds, pharmaceutical formulations including the compounds, methods of inhibiting UDP-3-0-(R-3-hydroxydecanoyl)-N- acetylglucosamine deacetylase (LpxC), and methods of treating Gram-negative bacterial infections.
In one aspect, the invention provides compounds of Formula I:

A is a divalent radical selected from , and ;
X is -(CH2)nY(CH2)m-;
Y is selected from the group consisting of -C(H,R1)-, -0-, -N(R2)-, and -S(0)2- n is 0 or 1 ;
m is 0 or 1 ;
R is -C6-Ci0aryl, or 4 to 10 membered heteroaryl containing 1 to 3 heteroatoms selected from N, S and O, wherein said aryl and heteroaryl are optionally substituted with a substituent selected from the group consisting of halogen, -OH, -CN, -S(0)2(CrC4)alkyl, d- C4haloalkyl, C3-C7cycloalkyl, CrC4alkoxy, CrC4haloalkoxy, C C alkyl optionally substituted with Ci-C alkoxy, CrC alkoxy optionally substituted with Ci-C alkoxy and a 4 to 7 membered heterocycle containing 1 to 3 heteroatom selected from N, S, and O, wherein said heterocycle is optionally substituted with one or more halogen, C C alkoxy, Cr C4haloalkoxy, CrC4haloalkyl or Ci-C4 alkyl; or
R is -C6-Ci0aryl, or 4 to 10 membered heteroaryl containing 1 to 3 heteroatoms selected from N, S and O, wherein said aryl and heteroaryl are susbstituted by taking the substituents on adjacent atoms of the -C6-Ci0aryl, or 4 to 10 membered heteroaryl and forming a 3 to 7 membered heterocycle, wherein the formed bicycle substituent is further optionally substituted with halogen, Ci-C alkyl, CrC haloalkyl, Ci-C haloalkoxy or C
C4alkoxy;
R1 is selected from the group consisting of -OH, C C4alkoxy and -S(0)2R3;
R2 is selected from the group consisting of hydrogen, C C alkyl, -C(0)R3 and - S(0)2R3;
R3 is selected from the group consisting of C C4alkyl and C3-C6cycloalkyl;
Z is a divalent radical selected from

Z is with the proviso that A is
R5 is selected from the group consisting of hydrogen, halogen, -CN, d-C4alkyl, and C C4haloalkyl;
R6, R6a, R6b or R6c are independently selected from the group consisting of hydrogen, halogen, -CrC4alkyl, and CrC4haloalkyl;
L is a divalent bond, -CH2-, -O- or
In one aspect, the invention provides a method of inhibiting a deacetylase enzyme in Gram-negative bacteria, thereby affecting bacterial growth, comprising administering to a patient in need of such inhibition a compound of formula I.
In another aspect, the invention provides a method of inhibiting LpxC, thereby modulating the virulence of a bacterial infection, comprising administering to a patient in need of such inhibition a compound of formula I.
In another aspect, the invention provides a method for treating a subject with a Gram-negative bacterial infection comprising administering to the subject in need thereof an antibacterially effective amount of a compound of formula I with a pharmaceutically acceptable carrier. In certain embodiments, the subject is a mammal and in some other embodiments, the subject is a human.
In another aspect, the invention provides a method of administering an inhibitory amount of a compound of formula I to fermentative or non-fermentative Gram-negative bacteria. In certain embodiment of the method of administering an inhibitory amount of a compound of formula I to fermentative or non-fermentative Gram-negative bacteria, the Gram-negative bacteria are selected from the group consisting of Pseudomonas aeruginosa and other Pseudomonas species, Stenotrophomonas maltophilia, Burkholderia cepacia and other Burkholderia species, Alcaligenes xylosoxidans, species of Acinetobacter,
Enterobacteriaceae, Haemophilus, Moraxella, Bacteroides, Fransicella, Shigella, Proteus, Vibrio, Salmonella, Bordetella, Helicobactor, Legionella, Citrobactor, Serratia,
Campylobactor, Yersinia and Neisseria.
In another embodiment, the invention provides a method of administering an inhibitory amount of a compound of formula I to Gram-negative bacteria, such as
Enterobacteriaceae which is selected from the group consisting of organisms such as Serratia, Proteus, Klebsiella, Enterobacter, Citrobacter, Salmonella, Providencia,
Morganella, Cedecea, Yersina and Edwardsiella species and Escherichia coli.
Another embodiment of the invention provides a pharmaceutical composition comprising an effective amount of a compound of Formula I with a pharmaceutically acceptable carrier thereof.
Pharmaceutical formulations according to the present invention are provided which include any of the compounds described above and a pharmaceutically acceptable carrier.
Other aspects of the invention are discussed infra.
The present invention provides novel compounds, methods for inhibiting LpxC in Gram-negative bacteria, and novel methods for treating bacterial infections. The compounds provided herein can be formulated into pharmaceutical formulations and medicaments that are useful in the methods of the invention. The invention also provides for the use of the compounds in preparing medicaments and pharmaceutical formulations, for use of the compounds in inhibiting LpxC, and for use of the compounds in treating bacterial infections in a subject.
The following abbreviations and definitions are used throughout this application: "LpxC" is an abbreviation that stands for UDP-3-0-(R-3-hydroxydecan- oyl)-N- acetylglucosamine deacetylase.
This invention is directed to compounds of Formula l-V and subformulae thereof, and intermediates thereto, as well as pharmaceutical compositions containing the compounds for use in treatment of bacterial infections. This invention is also directed to the compounds of the invention or compositions thereof as LpxC inhibitors. The compounds are particularly useful in interfering with the life cycle of Gram-negative bacteria and in treating or preventing a Gram-negative bacterial infection or physiological conditions associated therewith. The present invention is also directed to methods of combination therapy for treating or preventing an Gram-negative bacterial infection in patients using the compounds of the invention or pharmaceutical compositions, or kits thereof in combination with at least one other therapeutic agent.
DETAILED DESCRIPTION OF THE INVENTION
For purposes of interpreting this specification, the following definitions will apply unless specified otherwise and whenever appropriate, terms used in the singular will also include the plural and vice versa.
Definitions
Terms used in the specification have the following meanings:
As used herein, the term "subject" refers to an animal. In certain aspects, the animal is a mammal. A subject also refers to for example, primates (e.g., humans), cows, sheep, goats, horses, dogs, cats, rabbits, rats, mice, fish, birds and the like. In certain
embodiments, the subject is a human.
As used herein, the term "inhibition" or "inhibiting" refers to the reduction or suppression of a given condition, symptom, or disorder, or disease, or a significant decrease in the baseline activity of a biological activity or process.
As used herein, the term "treating" or "treatment" of any disease or disorder refers in one embodiment, to ameliorating the disease or disorder (i.e., slowing or arresting or
reducing the development of the disease or at least one of the clinical symptoms thereof). In another embodiment "treating" or "treatment" refers to alleviating or ameliorating at least one physical parameter including those which may not be discernible by the patient. In yet another embodiment, "treating" or "treatment" refers to modulating the disease or disorder, either physically, (e.g., stabilization of a discernible symptom), physiologically, (e.g., stabilization of a physical parameter), or both. In yet another embodiment, "treating" or "treatment" refers to preventing or delaying the onset or development or progression of the disease or disorder.
As used herein, the term "a," "an," "the" and similar terms used in the context of the present invention (especially in the context of the claims) are to be construed to cover both the singular and plural unless otherwise indicated herein or clearly contradicted by the context.
All methods described herein can be performed in any suitable order unless otherwise indicated herein or otherwise clearly contradicted by context. The use of any and all examples, or exemplary language (e.g. "such as") provided herein is intended merely to better illuminate the invention and does not pose a limitation on the scope of the invention otherwise claimed.
The term "antibacterial agent" refers to agents synthesized or modified in the laboratory that have either bactericidal or bacteriostatic activity. An "active" agent in this context will inhibit the growth of P. aeruginosa and / or other Gram-negative bacteria. The term "inhibiting the growth" indicates that the rate of increase in the numbers of a population of a particular bacterium is reduced. Thus, the term includes situations in which the bacterial population increases but at a reduced rate, as well as situations where the growth of the population is stopped, as well as situations where the numbers of the bacteria in the population are reduced or the population even eliminated. If an enzyme activity assay is used to screen for inhibitors, one can make modifications in bacterial uptake/efflux, solubility, half-life, etc. to compounds in order to correlate enzyme inhibition with growth inhibition.
"Optionally substituted" means the group referred to can be substituted at one or more positions by any one or any combination of the radicals listed thereafter.
"Halo" or "halogen", as used herein, may be fluorine, chlorine, bromine or iodine.
"CrC6-Alkyl", as used herein, denotes straight chain or branched alkyl having 1-8 carbon atoms. If a different number of carbon atoms is specified, such as Ce or C3, then the definition is to be amended accordingly, such as "CrC4-Alkyl" will represent methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl and tert-butyl.
"CrC6-Alkoxy", as used herein, denotes straight chain or branched alkoxy having 1-8 carbon atoms. If a different number of carbon atoms is specified, such as C6 or C3, then the
edefinition is to be amended accordingly, such as "CrC4-Alkoxy" will represent methoxy, ethoxy, propoxy, isopropoxy, butoxy, isobutoxy, sec-butoxy and tert-butoxy.
"CrC4-Haloalkyl", as used herein, denotes straight chain or branched alkyl having 1 - 4 carbon atoms with at least one hydrogen substituted with a halogen. If a different number of carbon atoms is specified, such as C6 or C3, then the definition is to be amended accordingly, such as "Ci-C4-Haloalkyl" will represent methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl and tert-butyl that have at least one hydrogen substituted with halogen, such as where the halogen is fluorine: CF3CF2-, (CF3)2CH-, CH3-CF2-, CF3CF2-, CF3, CF2H-, CF3CF2CHCF3 or CF3CF2CF2CF2-.
"C3-C8-cycloalkyl" as used herein refers to a saturated monocyclic hydrocarbon ring of 3 to 8 carbon atoms. Examples of such groups include cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl. If a different number of carbon atoms is specified, such as C3-C6, then the definition is to be amended accordingly.
"4- to 8-Membered heterocyclyl", "5- to 6- membered heterocyclyl", "3- to 10- membered heterocyclyl", "3- to 14-membered heterocyclyl", "4- to 14-membered
heterocyclyl" and "5- to 14-membered heterocyclyl", refers, respectively, to 4- to 8- membered, 5- to 6-membered, 3- to 10-membered, 3- to 14-membered, 4- to 14-membered and 5- to 14-membered heterocyclic rings containing 1 to 7, 1 to 5 or 1 to 3 heteroatoms selected from the group consisting of nitrogen, oxygen and sulphur, which may be saturated, or partially saturated. The heterocyclic group can be attached at a heteroatom or a carbon atom. The term "heterocyclyl" includes single ring groups, fused ring groups and bridged groups. Examples of such heterocyclyl include, but are not limited to pyrrolidine, piperidine, piperazine, pyrrolidine, pyrrolidinone, morpholine, tetrahydrofuran, tetrahydrothiophene, tetrahydrothiopyran, tetrahydropyran, 1 ,4-dioxane, 1 ,4-oxathiane, 8-aza- bicyclo[3.2.1 ]octane, 3,8-diazabicyclo[3.2.1 ]octane, 3-Oxa-8-aza-bicyclo[3.2.1 ]octane, 8- Oxa-3-aza-bicyclo[3.2.1 ]octane, 2-Oxa-5-aza-bicyclo[2.2.1 ]heptane, 2,5-Diaza- bicyclo[2.2.1 ]heptane, azetidine, ethylenedioxo, oxtane or thiazole.
"Heteroaryl" is a completely unsaturated (aromatic) ring. The term "heteroaryl" refers to a 5-14 membered monocyclic- or bicyclic- or tricyclic-aromatic ring system, having 1 to 8 heteroatoms selected from N, O or S. Typically, the heteroaryl is a 5-10 membered ring system (e.g., 5-7 membered monocycle or an 8-10 membered bicycle) or a 5-7 membered ring system. Typical heteroaryl groups include furan, isotriazole, thiadiazole, oxadiazole, indazole, indazole, indole, quinoline, 2- or 3-thienyl, 2- or 3-furyl, 2- or 3-pyrrolyl, 2-, 4-, or 5- imidazolyl, 3-, 4-, or 5- pyrazolyl, 2-, 4-, or 5-thiazolyl, 3-, 4-, or 5-isothiazolyl, 2-, 4-, or 5- oxazolyl, 3-, 4-, or 5-isoxazolyl, 3- or 5-(1 ,2,4-triazolyl), 4- or 5-(1 ,2, 3-triazolyl), tetrazolyl, triazine, pyrimidine, 2-, 3-, or 4-pyridyl, 3- or 4-pyridazinyl, 3-, 4-, or 5-pyrazinyl, 2-pyrazinyl, and 2-, 4-, or 5-pyrimidinyl.
The term "hydroxy" or "hydroxyl" includes groups with an -OH.
The term "a," "an," "the" and similar terms used in the context of the present invention (especially in the context of the claims) are to be construed to cover both the singular and plural unless otherwise indicated herein or clearly contradicted by the context.
Various embodiments of the invention are described herein. It will be recognized that features specified in each embodiment may be combined with other specified features to provide further embodiments.
In one embodiment, the invention provides compounds of Formula I:

A is a divalent radical selected from and
X is -(CH2)nY(CH2)m-;
Y is selected from the group consisting of -C(H,R1)-, -0-, S, -N(R2)-, and -S(0)2- n is 0 or 1 ; m is 0 or 1 ;
R is C3-6 cycloalkyi, -C6-Ci0aryl, or 4 to 10 membered heteroaryl containing 1 to 3 heteroatoms selected from N, S and O, wherein said cycloalkyi, aryl and heteroaryl are each optionally substituted with up to three substituents selected from the group consisting of halogen, -OH, -CN, -S(0)2(CrC4)alkyl, C C4haloalkyl, C3-C7cycloalkyl, C C4alkoxy, C C4haloalkoxy, C C4alkyl optionally substituted with Ci-C4alkoxy or a 5-6 membered heterocycle containing up to two heteroatoms selected from N, O and S as ring members and optionally substituted with R10, CrC4alkoxy optionally substituted with d-C4alkoxy or Ci_ 3 alkyl or C3-e cycloalkyi where the C1-3 alkyl or C3-e cycloalkyi are each optionally substituted with hydroxy, methoxy, or methyl, and a 4 to 7 membered heterocycle or a 5 to 6 membered heteroaryl wherein the 4 to 7 membered heterocycle or 5 to 6 membered heteroaryl
contains 1 to 3 heteroatoms selected from N, S, and O as ring members and is optionally substituted with one or more halogen, CrC4alkoxy, CrC4haloalkoxy, CrC4haloalkyl or C C4 alkyl; or
R is -C6-Ci0aryl, or 4 to 10 membered heteroaryl containing 1 to 3 heteroatoms selected from N, S and O, wherein said aryl and heteroaryl are susbstituted by taking the substituents on adjacent atoms of the -C6-Ci0aryl, or 4 to 10 membered heteroaryl and forming a 3 to 7 membered heterocycle, wherein the formed bicycle substituent is further optionally substituted with one or two groups selected from halogen, Ci-C alkyl, d- C haloalkyl, C C haloalkoxy and CrC alkoxy;
R1 is selected from the group consisting of -OH, C C alkoxy and -S(0)2R3;
R2 is selected from the group consisting of hydrogen, CrC4alkyl, -C(0)OR3 , -C(0)R3 and -S(0)2R3;
R3 is selected from the group consisting of C C alkyl and C3-C6cycloalkyl;
Z is a divalent radical selected from

; or

with the proviso that A is
R5 is selected from the group consisting of hydrogen, halogen, -CN, d-dalkyl, and CrC4haloalkyl;
R6, R6a, R6b or R6c are independently selected from the group consisting of hydrogen, halogen, -CrC4alkyl, and CrC4haloalkyl;
R10 is selected from halo, C^ alkyl, d-4 haloalkyl, d^ alkoxy, -C(0)R11 and -C(O)-
OR11 ;
R11 is d^ alkyl; and '
L is a divalent bond, -CH2-, -O- or
In certain embodiments of the compounds of Formula I,

A is a divalent radical selected from and
X is -(CH2)nY(CH2)m-;
Y is selected from the group consisting of -C(H,R1)-, -0-, -N(R2)-, and -S(0)2- n is 0 or 1 ; m is 0 or 1 ;
R is -C6-d0aryl, or 4 to 10 membered heteroaryl containing 1 to 3 heteroatoms selected from N, S and O, wherein said cycloalkyi, aryl and heteroaryl are each optionally substituted with substituents selected from the group consisting of halogen, -OH, -CN, - S(0)2(d-d)alkyl, d-dhaloalkyl, d-dcycloalkyl, d-dalkoxy, d-dhaloalkoxy, d-dalkyl optionally substituted with d-dalkoxy, d-dalkoxy optionally substituted with d-dalkoxy,
and a 4 to 7 membered heterocycle wherein the 4 to 7 membered heterocycle contains 1 to 3 heteroatoms selected from N, S, and O as ring members and is optionally substituted with one or more halogen, d-C4alkoxy, CrC4haloalkoxy, CrC4haloalkyl or C C4 alkyl; or
R is -C6-Ci0aryl, or 4 to 10 membered heteroaryl containing 1 to 3 heteroatoms selected from N, S and O, wherein said aryl and heteroaryl are susbstituted by taking the substituents on adjacent atoms of the -C6-Ci0aryl, or 4 to 10 membered heteroaryl and forming a 3 to 7 membered heterocycle, wherein the formed bicycle substituent is further optionally substituted with groups selected from halogen, Ci-C alkyl, Ci-C haloalkyl, Ci- C haloalkoxy and C C alkoxy;
R1 is selected from the group consisting of -OH, C C alkoxy and -S(0)2R3;
R2 is selected from the group consisting of hydrogen, CrC4alkyl, -C(0)R3 and - S(0)2R3;
R3 is selected from the group consisting of C C alkyl and C3-C6cycloalkyl;
Z is a divalent radical selected from

; or

Z is with the proviso that A is
R5 is selected from the group consisting of hydrogen, halogen, -CN, d-C4alkyl, and CrC4haloalkyl;
R6, R6a, R6b or R6c are independently selected from the group consisting of hydrogen, halogen, -CrC4alkyl, and CrC4haloalkyl; and '
L is a divalent bond, -CH2-, -O- or
In certain emodiments of the compounds of Formula I, R is phenyl substituted with one or two substituents selected from the group consisting of halogen, -OH, -CN, -S(0)2(Cr C4)alkyl, CrC4haloalkyl, C3-C7cycloalkyl, C C alkoxy, CrC haloalkoxy, and CrC alkyl optionally substituted with C C alkoxy.
In certain emodiments of the compounds of Formula I, X is -CH2-SO2-CH2-. In some of these embodiments, A is [Z]-CH2-NH-, where [Z] indicates the point where A attaches to Z in Formula I.
In certain emodiments of any of the compounds of Formula I as described above, Z

is or . In these embodiments, R5 is preferably
H, and R6, R6a, R6b and R6c can all be H.
In certain emodiments of any of the compounds of Formula I as described above, L is a bond.
In an embodiment of the invention, the compound or a pharmaceutically acceptable salt represented by formula II:


wherein Q is selected from the group consisting of



and
R2 is selected from the group consisting of hydrogen, C C4alkyl, -C(0)CR3 and
S(0)2R3;
R3 is selected from the group consisting of CrC4alkyl and C3-C6cycloalkyl.
In an embodiment of the invention, the compound or a pharmaceutically acceptable salt represented by formula II:




A is a divalent radical selected from , and ;
R is phenyl optionally substituted with a substituent selected from the group consisting of halogen, -OH, -CN, -S(0)2(CrC4)alkyl, C C4haloalkyl, C3-C7cycloalkyl, C C4alkoxy, Ci-C4haloalkoxy, Ci-C4alkyl optionally substituted with Ci-C4alkoxy, Ci-C4alkoxy optionally substituted with C C alkoxy and a 4 to 7 membered heterocycle containing 1 to 3 heteroatom selected from N, S, and O, wherein said heterocycle is optionally substituted with one or more halogen, Ci-C alkoxy, C C haloalkoxy, Ci-C haloalkyl or C C alkyl; or
R is phenyl susbstituted by taking the substituents on adjacent atoms of the -C6- Ci0aryl, or 4 to 10 membered heteroaryl and forming a 3 to 7 membered heterocycle, wherein the formed bicycle substituent is further optionally substituted with halogen, C C alkyl, CrC haloalkyl, C C haloalkoxy or C C alkoxy;
R2 is selected from the group consisting of hydrogen, C C alkyl, -C(0)CR3 and - S(0)2R3;
R3 is selected from the group consisting of C C alkyl and C3-C6cycloalkyl;
Z is a divalent radical selected from

R5 is selected from the group consisting of hydrogen, halogen, -CN, Ci-C alkyl, and
CrC4haloalkyl;
R6, R6a, R6b or R6c are independently selected from the group consisting of hydrogen, halogen, CrC4alkyl and CrC4haloalkyl;
L is a divalent bond, -CH2-, -O- or
In certain emodiments of the compounds of Formula II described above, R is phenyl substituted with one or two substituents selected from the group consisting of halogen, -OH, -CN, -S(0)2(CrC4)alkyl, C C4haloalkyl, C3-C7cycloalkyl, C C4alkoxy, C C4haloalkoxy, and CrC alkyl optionally substituted with Ci-C alkoxy.

In certain emodiments of these compounds of Formula II, Q is . In some of these embodiments, A is [Z]-CH2-NH-, where [Z] indicates the point where A attaches to Z in Formula II.
In certain emodiments of any of the compounds of Formula II as described above, Z

is or In these embodiments, R5 is preferably
H, and R6, R6a, R6b and R6c can all be H.
In certain emodiments of any of the compounds of Formula II as described above, L is a bond.
In an embodiment of the invention, the compound or a pharmaceutically acceptable salt represented by formula II:


wherein Q is selected from the group consisting of

A is a divalent radical selected from , and
R is phenyl optionally substituted with a substituent selected from the group consisting of halogen, -OH, -CN, -S(0)2(CrC4)alkyl, C C4haloalkyl, C3-C7cycloalkyl, d- C4alkoxy, CrC haloalkoxy, C C alkyl optionally substituted with C C alkoxy, C C alkoxy optionally substituted with C C alkoxy and a 4 to 7 membered heterocycle containing 1 to 3 heteroatom selected from N, S, and O, wherein said heterocycle is optionally substituted with one or more halogen, Ci-C alkoxy, C C haloalkoxy, Ci-C haloalkyl or C C alkyl; or
R is phenyl susbstituted by taking the substituents on adjacent atoms of the -C6-
Ci0aryl, or 4 to 10 membered heteroaryl and forming a 3 to 7 membered heterocycle, wherein the formed bicycle substituent is further optionally substituted with halogen, C C4alkyl, CrC4haloalkyl, CrC4haloalkoxy or CrC4alkoxy;
R2 is selected from the group consisting of hydrogen, C C alkyl and -S(0)2R3;
R3 is selected from the group consisting of C C alkyl and C3-C6cycloalkyl.
Z is a divalent radical selected from

, and ;
R5 is selected from the group consisting of hydrogen, halogen, C C alkyl, and C C haloalkyl;
L is a divalent bond.
In an embodiment of the invention, the compound or a pharmaceutically acceptable salt represented by formula II:

II
wherein Q is selected from the group consisting of




A is
R is phenyl optionally substituted with a substituent selected from the group consisting of halogen, -OH, -CN, -S(0)2(CrC4)alkyl, C C4haloalkyl, C3-C7cycloalkyl, C C4alkoxy, Ci-C4haloalkoxy, Ci-C4alkyl optionally substituted with Ci-C4alkoxy, Ci-C4alkoxy optionally substituted with C C alkoxy, and a 4 to 7 membered heterocycle containing 1 to 3 heteroatom selected from N, S, and O, wherein said heterocycle is optionally substituted with one or more halogen, Ci-C alkoxy, C C haloalkoxy, Ci-C haloalkyl or C C alkyl; or
R is phenyl optionally susbstituted by taking the substituents on adjacent atoms of the phenyl and forming a 3 to 7 membered heterocycle, wherein the formed bicycle substituent is further optionally substituted with halogen, C C alkyl, CrC haloalkyl, C C haloalkoxy or C C alkoxy;
R2 is selected from the group consisting of hydrogen, CrC4alkyl and -S(0)2R3;
R3 is selected from the group consisting of C C alkyl and C3-C6cycloalkyl.

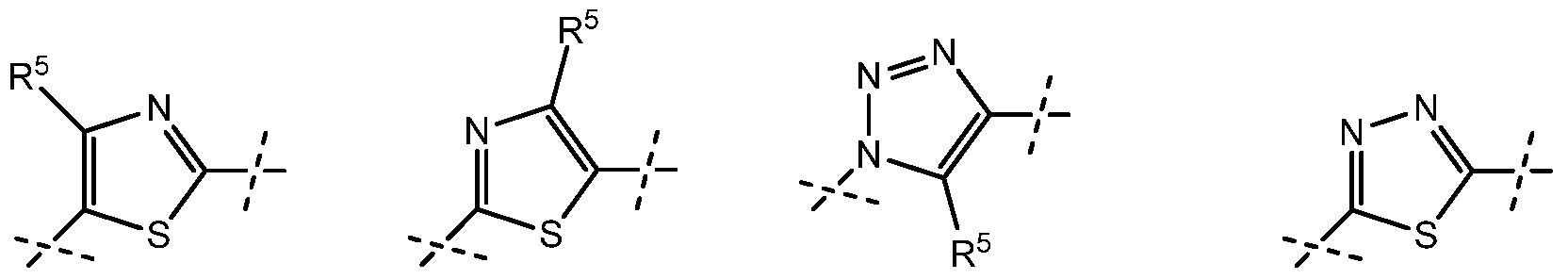
, and
R5 is selected from the group consisting of hydrogen, halogen, CrC4alkyl, and C C4haloalkyl;
L is a divalent bond.
In an embodiment of the invention, the compound or a pharmaceutically acceptable salt represented by formula II:

II wherein Q is selected from the group consisting of


, and

A is ;
R is phenyl optionally substituted with a substituent selected from the group consisting of halogen, -OH, -CN, -S(0)2(CrC4)alkyl, C C4haloalkyl, C3-C7cycloalkyl, C C4alkoxy, Ci-C4haloalkoxy, Ci-C4alkyl optionally substituted with Ci-C4alkoxy, Ci-C4alkoxy optionally substituted with C C alkoxy and a 4 to 7 membered heterocycle containing 1 to 3 heteroatom selected from N, S, and O, wherein said heterocycle is optionally substituted with one or more halogen, Ci-C alkoxy, C C haloalkoxy, Ci-C haloalkyl or C C alkyl;
R2 is selected from the group consisting of hydrogen, C C alkyl and -S(0)2R3;
R3 is selected from the group consisting of C C alkyl and C3-C6cycloalkyl.
Z is a divalent radical selected from

, and
R5 is selected from the group consisting of hydrogen, halogen, -CN, d-C4alkyl, and CrC4haloalkyl;
L is a divalent bond.
In an embodiment of the invention, the compound or a pharmaceutically acceptable salt represented by formula II:



and

R2 is selected from the group consisting of hydrogen, CrC4alkyl and -S(0)2R3; R3 is selected from the group consisting of CrC4alkyl and C3-C6cycloalkyl. Z is a divalent radical selected from


L is a divalent bond or
In an embodiment of the invention, the compound or a pharmaceutically acceptable salt thereof represented by formula III


R is -C6-Ci0aryl, or 4 to 10 membered heteroaryl containing 1 to 3 heteroatoms selected from N, S and O, wherein said aryl and heteroaryl are optionally substituted with a substituent selected from the group consisting of halogen, -OH, -CN, -S(0)2(CrC4)alkyl, Ci- C4haloalkyl, C3-C7cycloalkyl, CrC4alkoxy, CrC4haloalkoxy, C C alkyl optionally substituted with Ci-C alkoxy, CrC alkoxy optionally substituted with Ci-C alkoxy and a 4 to 7 membered heterocycle containing 1 to 3 heteroatom selected from N, S, and O, wherein said heterocycle is optionally substituted with one or more halogen, C C alkoxy, Cr C4haloalkoxy, CrC4haloalkyl or Ci-C4 alkyl; or
R is -C6-Ci0aryl, or 4 to 10 membered heteroaryl containing 1 to 3 heteroatoms selected from N, S and O, wherein said aryl and heteroaryl are susbstituted by taking the substituents on adjacent atoms of the -C6-Ci0aryl, or 4 to 10 membered heteroaryl and forming a 3 to 7 membered heterocycle, wherein the formed bicycle substituent is further optionally substituted with halogen, Ci-C alkyl, CrC haloalkyl, Ci-C haloalkoxy or C C4alkoxy;
R2 is selected from the group consisting of hydrogen, C C alkyl and -S(0)2R3;
R3 is selected from the group consisting of C C alkyl and C3-C6cycloalkyl;
R5 is selected from the group consisting of hydrogen, halogen, -CN, -OH, Ci-C alkyl, CrC haloalkyl, Ci-C haloalkoxy, CrC alkoxy and C3-C7cycloalkyl optionally substituted with halogen or Ci-C alkyl; '
L is a direct bond, -CH2-, -O- or
In certain emodiments of the compounds of Formula III, R is phenyl substituted with one or two substituents selected from the group consisting of halogen, -OH, -CN, -S(0)2(C C )alkyl, Ci-C haloalkyl, C3-C7cycloalkyl, Ci-C alkoxy, Ci-C haloalkoxy, and Ci-C alkyl optionally substituted with C C alkoxy.

In certain emodiments of these compounds of Formula III, Q is
In certain emodiments of any of the compounds of Formula III as described above, L is a bond.
In an embodiment of the invention, the compound or a pharmaceutically acceptable salt thereof according to any proceeding claim, wherein
R is phenyl optionally substituted with a substituent selected from the group consisting of halogen, -OH, -CN, -S(0)2(CrC4)alkyl, C C4haloalkyl, C3-C7cycloalkyl, C C4alkoxy, Ci-C4haloalkoxy, Ci-C4alkyl optionally substituted with Ci-C4alkoxy, Ci-C4alkoxy optionally substituted with C C alkoxy and a 4 to 7 membered heterocycle containing 1 to 3 heteroatom selected from N, S, and O, wherein said heterocycle is optionally substituted with one or more halogen, Ci-C alkoxy, C C haloalkoxy, Ci-C haloalkyl or C C alkyl; or
R is phenyl susbstituted by taking the substituents on adjacent atoms of the -C6- Ci0aryl, or 4 to 10 membered heteroaryl and forming a 3 to 7 membered heterocycle, wherein the formed bicycle substituent is further optionally substituted with halogen, C C alkyl, CrC haloalkyl, C C haloalkoxy or C C alkoxy.
In an embodiment of the invention, the compound or a pharmaceutically acceptable salt thereof according to any proceeding claim, wherein the compound is formula III

III
wherein

Q is selected from the group consisting of

and ;
R is phenyl optionally substituted with a substituent selected from the group consisting of halogen, -OH, -CN, -S(0)2(CrC4)alkyl, C C4haloalkyl, C3-C7cycloalkyl, C C4alkoxy, CrC haloalkoxy, C C alkyl optionally substituted with C C alkoxy, C C alkoxy optionally substituted with C C alkoxy and a 4 to 7 membered heterocycle containing 1 to 3 heteroatom selected from N, S, and O, wherein said heterocycle is optionally substituted with one or more halogen, Ci-C alkoxy, C C haloalkoxy, Ci-C haloalkyl or C C alkyl; or
R is phenyl susbstituted by taking the substituents on adjacent atoms of the -C6- Ci0aryl, or 4 to 10 membered heteroaryl and forming a 3 to 7 membered heterocycle, wherein the formed bicycle substituent is further optionally substituted with halogen, Ci- C alkyl, CrC haloalkyl, C C haloalkoxy or C C alkoxy;
R2 is selected from the group consisting of hydrogen, C C alkyl and -S(0)2R3;
R3 is selected from the group consisting of CrC4alkyl and Cs-Cecycloalkyl; '
L is a direct bond, -CH2-, -O- or
In an embodiment of the invention, the compound or a pharmaceutically acceptable salt thereof wherein the compound is formula III

, and
R is phenyl optionally substituted with a substituent selected from the group consisting of halogen, -OH, -CN, -S(0)2(CrC4)alkyl, C C4haloalkyl, C3-C7cycloalkyl, C C4alkoxy, CrC haloalkoxy, C C alkyl optionally substituted with C C alkoxy, C C alkoxy optionally substituted with Ci-C4alkoxy, and a 4 to 7 membered heterocycle containing 1 to 3 heteroatom selected from N, S, and O, wherein said heterocycle is optionally substituted with one or more halogen, Ci-C alkoxy, C C haloalkoxy, Ci-C haloalkyl or C C alkyl; or
R is phenyl optionally susbstituted by taking the substituents on adjacent atoms of the phenyl and forming a 3 to 7 membered heterocycle, wherein the formed bicycle substituent is further optionally substituted with halogen, C C alkyl, CrC haloalkyl, C C haloalkoxy or C C alkoxyR2 is selected from the group consisting of hydrogen, Ci-C alkyl
and -S(0)2R3;
R3 is selected from the group consisting of CrC4alkyl and C3-C6cycloalkyl;
L is a direct bond or
In an embodiment of the invention, the compound represented by formula IV


, and
R is -C6-Ci0aryl, or 4 to 10 membered heteroaryl containing 1 to 3 heteroatoms selected from N, S and O, wherein said aryl and heteroaryl are optionally substituted with a substituent selected from the group consisting of halogen, -OH, -CN, -S(0)2(CrC4)alkyl, Ci- C4haloalkyl, C3-C7cycloalkyl, CrC4alkoxy, CrC4haloalkoxy, C C alkyl optionally substituted with Ci-C alkoxy, CrC alkoxy optionally substituted with Ci-C alkoxy and a 4 to 7 membered heterocycle containing 1 to 3 heteroatom selected from N, S, and O, wherein said heterocycle is optionally substituted with one or more halogen, C C alkoxy, Cr C4haloalkoxy, CrC4haloalkyl or Ci-C4 alkyl; or
R is -C6-Ci0aryl, or 4 to 10 membered heteroaryl containing 1 to 3 heteroatoms selected from N, S and O, wherein said aryl and heteroaryl are susbstituted by taking the substituents on adjacent atoms of the -C6-Ci0aryl, or 4 to 10 membered heteroaryl and forming a 3 to 7 membered heterocycle, wherein the formed bicycle substituent is further optionally substituted with halogen, Ci-C alkyl, CrC haloalkyl, Ci-C haloalkoxy or C C4alkoxy;
R2 is selected from the group consisting of hydrogen, C C alkyl, -C(0)CR3 and - S(0)2R3;
R3 is selected from the group consisting of C C alkyl and C3-C6cycloalkyl;
R6, R6a, R6b or R6c are independently selected from the group consisting of hydrogen, halogen, -CN, C C alkyl, C3-C7cycloalkyl, C C alkoxy and CrC haloalkyl;
L is a direct bond, -CH2-, -O- or
In an embodiment of the invention, the compound is represented by formula IV

, and
R is phenyl optionally substituted with a substituent selected from the group consisting of halogen, -OH, -CN, -S(0)2(CrC4)alkyl, C C4haloalkyl, C3-C7cycloalkyl, C C4alkoxy, CrC haloalkoxy, C Calkyl optionally substituted with C C alkoxy, C Calkoxy optionally substituted with C Calkoxy, and a 4 to 7 membered heterocycle containing 1 to 3
heteroatom selected from N, S, and O, wherein said heterocycle is optionally substituted with one or more halogen, d-C4alkoxy, CrC4haloalkoxy, CrC4haloalkyl or C C4 alkyl; or
R is phenyl optionally susbstituted by taking the substituents on adjacent atoms of the phenyl and forming a 3 to 7 membered heterocycle, wherein the formed bicycle substituent is further optionally substituted with halogen, C C alkyl, CrC haloalkyl, C C haloalkoxy or C C alkoxyR2 is selected from the group consisting of hydrogen, Ci-C alkyl, -C(0)CR3 and -S(0)2R3;
R3 is selected from the group consisting of CrC4alkyl and Cs-Cecycloalkyl;
R6, R6a, R6b or R6c are independently selected from the group consisting of hydrogen, halogen, C C alkyl, and CrC haloalkyl;
L is a direct bond, or
In certain emodiments of the compounds of Formula IV, R is phenyl substituted with one or two substituents selected from the group consisting of halogen, -OH, -CN, -S(0)2(Cr C )alkyl, Ci-C haloalkyl, C3-C7cycloalkyl, C C alkoxy, CrC haloalkoxy, and CrC alkyl optionally substituted with C C alkoxy.

In certain emodiments of these compounds of Formula III, Q is
In certain emodiments of any of the compounds of Formula IV as described above, L is a bond.
In an embodiment of the invention, the compound is of formula V

V

wherein Q is selected from the group consisting of

R is -C6-Ci0aryl, or 4 to 10 membered heteroaryl containing 1 to 3 heteroatoms selected from N, S and O, wherein said aryl and heteroaryl are optionally substituted with a substituent selected from the group consisting of halogen, -OH, -CN, -S(0)2(CrC4)alkyl, d- C4haloalkyl, C3-C7cycloalkyl, CrC4alkoxy, CrC4haloalkoxy, C C alkyl optionally substituted with Ci-C alkoxy, CrC alkoxy optionally substituted with Ci-C alkoxy and a 4 to 7 membered heterocycle containing 1 to 3 heteroatom selected from N, S, and O, wherein said heterocycle is optionally substituted with one or more halogen, C C alkoxy, Cr C4haloalkoxy, CrC4haloalkyl or Ci-C4 alkyl; or
R is -C6-Ci0aryl, or 4 to 10 membered heteroaryl containing 1 to 3 heteroatoms selected from N, S and O, wherein said aryl and heteroaryl are susbstituted by taking the substituents on adjacent atoms of the -C6-Ci0aryl, or 4 to 10 membered heteroaryl and
forming a 3 to 7 membered heterocycle, wherein the formed bicycle substituent is further optionally substituted with halogen, d-C4alkyl, CrC4haloalkyl, CrC4haloalkoxy or C C4alkoxy;
R2 is selected from the group consisting of hydrogen, CrC4alkyl, -C(0)CR3 and - S(0)2R3;
R3 is selected from the group consisting of CrC4alkyl and C3-C6cycloalkyl.
In an embodiment of the invention, the compound, the compound of formula V

wherein Q is selected from the group consisting of


, and
R is phenyl optionally substituted with halogen, Ci-4alkyl or Ci-C4alkoxy;
R2 is selected from the group consisting of hydrogen, CrC4alkyl, -C(0)CR3 and - S(0)2R3;
R3 is selected from the group consisting of C C4alkyl and Cs-Cecycloalkyl.
In an embodiment of the invention, the compound of formula I to V, wherein L is a direct bond;

R is selected from the group consisting of



and

In an embodiment of the invention, the compound of formula I to V, wherein L is a direct bond;

R is selected from the group consisting of



In an embodiment of the invention, the compound is of formula I to V, wherein L is a direct bond;

R is selected from the group consisting of

, and
In another embodiment of the invention, the compound according to formula I to IV or a pharmaceutically acceptable salt thereof represented by
4-(([1 , 1 '-biphenyl]-4-ylmethyl)amino)-N-hydroxytetrahydro-2H-thiopyran-4- carboxamide 1 , 1 -dioxide;
N-hydroxy-4-(((4'-methoxy-[1 , 1 '-biphenyl]-4-yl)methyl)amino)tetrahydro-2H-thiopyran- 4-carboxamide 1 , 1 -dioxide;
4-(((4'-chloro-2'-fluoro-[1 , 1 '-biphenyl]-4-yl)methyl)amino)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide;
N-hydroxy-4-(((4'-(morpholinomethyl)-[1 , 1 '-biphenyl]-4-yl)methyl)amino)tetrahydro- 2H-thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((4'-(2H-1 ,2,3-triazol-2-yl)-[1 , 1 '-biphenyl]-4-yl)methyl)amino)-N-hydroxytetrahydro- 2H-thiopyran-4-carboxamide 1 , 1 -dioxide;
N-hydroxy-4-(((4'-morpholino-[1 , 1 '-biphenyl]-4-yl)methyl)amino)tetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((4'-cyclopropyl-[1 , 1 '-biphenyl]-4-yl)methyl)amino)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide;
N-hydroxy-4-((4-(naphthalen-2-yl)benzyl)amino)tetrahydro-2H-thiopyran-4- carboxamide 1 ,1 -dioxide;
4-(((2'-fluoro-4'-methoxy-[1 , 1 '-biphenyl]-4-yl)methyl)amino)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1 ,1 -dioxide;
N-hydroxy-4-(((4'-(((1 r,4r)-4-hydroxycyclohexyl)methoxy)-[1 ,1 '-biphenyl]-4- yl)methyl)amino)tetrahydro-2H-thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((2'-fluoro-[1 , 1 '-biphenyl]-4-yl)methyl)amino)-N-hydroxytetrahydro-2H-thiopyran-4- carboxamide 1 ,1 -dioxide;
N-hydroxy-4-((4-(pyridin-4-yl)benzyl)amino)tetrahydro-2H hiopyran-4-carboxamide 1 ,1 -dioxide;
4-(((2-fluoro-[1 , 1 '-biphenyl]-4-yl)methyl)amino)-N-hydroxytetrahydro-2H-thiopyran-4- carboxamide 1 ,1 -dioxide;
4-(((2-fluoro-4'-methoxy-[1 , 1 '-biphenyl]-4-yl)methyl)amino)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1 ,1 -dioxide;
4-(((2,2'-difluoro-[1 , 1 '-biphenyl]-4-yl)methyl)amino)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1 ,1 -dioxide;
4-(((4'-chloro-2,2'-difluoro-[1 , 1 '-biphenyl]-4-yl)methyl)amino)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1 ,1 -dioxide;
4-(((2-fluoro-4'-(2H-1 ,2,3-triazol-2-yl)-[1 , 1 '-biphenyl]-4-yl)methyl)amino)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide;
N-hydroxy-4-(((2-methyl-4'-(2H-1 ,2,3-triazol-2-yl)-[1 , 1 '-biphenyl]-4- yl)methyl)amino)tetrahydro-2H-thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((4'-chloro-2'-fluoro-2-methyl-[1 , 1 '-biphenyl]-4-yl)methyl)amino)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide;
4-(((2'-fluoro-4'-methoxy-2-methyl-[1 , 1 '-biphenyl]-4-yl)methyl)amino)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide;
4-(((4'-cyclopropyl-2-methyl-[1 , 1 '-biphenyl]-4-yl)methyl)amino)-N-hydroxytetrahydro- 2H-thiopyran-4-carboxamide 1 ,1 -dioxide;
4-(((2-chloro-2'-fluoro-4'-methoxy-[1 , 1 '-biphenyl]-4-yl)methyl)amino)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide;
N-hydroxy-4-((4-(phenylethynyl)benzyl)amino)tetrahydro-2H-thiopyran-4- carboxamide 1 ,1 -dioxide;
N-hydroxy-4-(((5-phenylisoxazol-3-yl)methyl)amino)tetrahydro-2H-thiopyran-4- carboxamide 1 ,1 -dioxide;
N-hydroxy-4-(((3-phenylisoxazol-5-yl)methyl)amino)tetrahydro-2H-thiopyran-4- carboxamide 1 ,1 -dioxide;
4-(((5-(4-chloro-2-fluorophenyl)isoxazol-3-yl)methyl)amino)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((5-(4-chloro-2-fluorophenyl)isoxazol-3-yl)methyl)amino)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((5-(2-fluoro-4-methoxyphenyl)isoxazol-3-yl)methyl)amino)-N-hydroxytetrahydro- 2H-thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((5-(4-chloro-2-fluorophenyl)-4-fluoroisoxazol-3-yl)methyl)amino)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((5-(4-(2H-1 ,2,3-triazol-2-yl)phenyl)isoxazol-3-yl)methyl)amino)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((4-chloro-5-(4-chloro-2-fluorophenyl)isoxazol-3-yl)methyl)amino)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 , 1 -dioxide;
N-hydroxy-4-((( 1 -phenyl-1 H-1 ,2,3-triazol-4-yl)methyl)amino)tetrahydro-2H-thiopyran- 4-carboxamide 1 , 1 -dioxide;
4-(2-(4-(4-chloro-2-fluorophenyl)-2-oxopyridin-1 (2H)-yl)ethyl)-N-hydroxytetrahydro- 2H-thiopyran-4-carboxamide 1 , 1 -dioxide;
3-(([1 , 1 '-biphenyl]-4-ylmethyl)amino)-N-hydroxytetrahydrothiophene-3-carboxamide;
N-hydroxy-3-(((5-phenylisoxazol-3-yl)methyl)amino)tetrahydrothiophene-3- carboxamide 1 , 1 -dioxide;
N-hydroxy-3-(((3-phenylisoxazol-5-yl)methyl)amino)tetrahydrothiophene-3- carboxamide 1 , 1 -dioxide;
N-hydroxy-3-((4-(phenylethynyl)benzyl)amino)tetrahydrothiophene-3-carboxamide 1 , 1 -dioxide;
3- (((4'-chloro-2'-fluoro-[1 , 1 '-biphenyl]-4-yl)methyl)amino)-N- hydroxytetrahydrothiophene-3-carboxamide 1 , 1 -dioxide;
1 -acetyl-N-hydroxy-4-(2-(2-oxo-4-phenylpyridin-1 (2H)-yl)ethyl)piperidine-4- carboxamide;
N-hydroxy-1 -(methylsulfonyl)-4-(2-(2-oxo-4-phenylpyridin-1 (2H)-yl)ethyl)piperidine-4- carboxamide;
4- ([1 , 1 '-biphenyl]-4-ylmethyl)-N-hydroxy-1 -(methylsulfonyl)piperidine-4-carboxamide; (1 S,4S)-1 -(([1 , 1 '-biphenyl]-4-ylmethyl)amino)-N,4- dihydroxycyclohexanecarboxamide;
(1 R,4R)-1 -(([1 , 1 '-biphenyl]-4-ylmethyl)amino)-N,4- dihydroxycyclohexanecarboxamide;
(1 R,4R)-1 -(([1 , 1 '-biphenyl]-4-ylmethyl)amino)-N-hydroxy-4- methoxycyclohexanecarboxamide;
3-(((4'-chloro-2'-fluoro-[1 , 1 '-biphenyl]-4-yl)methyl)amino)-N-hydroxytetrahydrofuran-3- carboxamide;
3- (((4'-chloro-2'-fluoro-[1 , 1 '-biphenyl]-4-yl)methyl)amino)-N-hydroxyoxetane-3- carboxamide;
N-hydroxy-4-(2-(2-oxo-4-phenylpyridin-1 (2H)-yl)ethyl)tetrahydro-2H-pyran-4- carboxamide;
4- (2-([1 , 1 '-biphenyl]-4-yl)ethyl)-N-hydroxytetrahydro-2H-pyran-4-carboxamide; and 4-(([1 , 1 '-biphenyl]-4-ylmethyl)amino)-N-hydroxytetrahydro-2H-pyran-4-carboxamide.
Additional embodiments include:
4-(((5-(4-(difluoromethoxy)phenyl)isoxazol-3-yl)methyl)amino)-N-hydroxytetrahydro- 2H-thiopyran-4-carboxamide 1 , 1 -dioxide;
N-hydroxy-4-(((5-(4-methoxyphenyl)isoxazol-3-yl)methyl)amino)tetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((5-(4-fluoro-3-methoxyphenyl)isoxazol-3-yl)methyl)amino)-N-hydroxytetrahydro- 2H-thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((5-(2-fluoro-4-methylphenyl)isoxazol-3-yl)methyl)amino)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((5-(2,3-dichlorophenyl)isoxazol-3-yl)methyl)amino)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((5-(2,4-difluorophenyl)isoxazol-3-yl)methyl)amino)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide;
N-hydroxy-4-(((5-(m-tolyl)isoxazol-3-yl)methyl)amino)tetrahydro-2H-thiopyran-4- carboxamide 1 , 1 -dioxide;
N-hydroxy-4-(((5-(3-methoxyphenyl)isoxazol-3-yl)methyl)amino)tetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((5-(2,2-difluorobenzo[d][1 ,3]dioxol-5-yl)isoxazol-3-yl)methyl)amino)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((5-(3-chloro-5-fluorophenyl)isoxazol-3-yl)methyl)amino)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((5-(3-chlorophenyl)isoxazol-3-yl)methyl)amino)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((5-(4-chloro-2,3-difluorophenyl)isoxazol-3-yl)methyl)amino)-N-hydroxytetrahydro- 2H-thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((5-(2-chloro-4-fluorophenyl)isoxazol-3-yl)methyl)amino)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((5-(2,5-difluorophenyl)isoxazol-3-yl)methyl)amino)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((5-(2-fluorophenyl)isoxazol-3-yl)methyl)amino)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((5-(2-fluoro-4-(trifluoromethoxy)phenyl)isoxazol-3-yl)methyl)amino)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((5-(3-fluoro-4-methoxyphenyl)isoxazol-3-yl)methyl)amino)-N-hydroxytetrahydro- 2H-thiopyran-4-carboxamide 1 , 1 -dioxide;
N-hydroxy-4-(((5-(2,3^-trifluorophenyl)isoxazol-3-yl)methyl)amino)tetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((5-(4-cyanophenyl)isoxazol-3-yl)methyl)amino)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((5-(2,6-difluorophenyl)isoxazol-3-yl)methyl)amino)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide;
N-hydroxy-4-(((5-(5-methylthiophen-2-yl)isoxazol-3-yl)methyl)amino)tetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide;
N-hydroxy-4-(((5-(4-methylthiophen-2-yl)isoxazol-3-yl)methyl)amino)tetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(((5-(4-chloro-2-fluorophenyl)isoxazol-3-yl)methyl)amino)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide;
4-((4-(cyclopropylethynyl)benzyl)amino)-N-hydroxytetrahydro-2H-thiopyran-4- carboxamide 1 , 1 -dioxide;
4-(2-(4-(2-fluoro-4-methoxyphenyl)-2-oxopyridin-1 (2H)-yl)ethyl)-N-hydroxytetrahydro- 2H-thiopyran-4-carboxamide 1 , 1 -dioxide;
N-hydroxy-4-(2-(4-phenyl-1 H-pyrazol-1 -yl)ethyl)tetrahydro-2H-thiopyran-4- carboxamide 1 , 1 -dioxide;
4-(2-(4-(4-chlorophenyl)-2H-1 ,2,3-triazol-2-yl)ethyl)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide;
4-(2-([1 , 1 '-biphenyl]-4-yl)ethyl)-N-hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 , 1 -dioxide;
3-(((5-(4-chloro-2-fluorophenyl)isoxazol-3-yl)methyl)amino)-N- hydroxytetrahydrothiophene-3-carboxamide 1 , 1 -dioxide;
3-(((4'-chloro-2'-fluoro-[1 , 1 '-biphenyl]-4-yl)methyl)amino)-N-hydroxy-1 - (methylsulfonyl)azetidine-3-carboxamide;
1 -(((2'-fluoro-4'-methoxy-[1 , 1 '-biphenyl]-4-yl)methyl)amino)-N,3- dihydroxycyclobutanecarboxamide;
3-(2-(4'-chloro-2'-fluoro-[1 , 1 '-biphenyl]-4-yl)ethyl)-N-hydroxy-1 - (methylsulfonyl)pyrrolidine-3-carboxamide; and
1-(((5-(4-chloro-2-fluorophenyl)isoxazol-3-yl)methyl)amino)-N-hydroxy-3- (methylsulfonyl)cyclobutanecarboxamide.
Each of the compounds in Table 1 is a specific embodiment of the invention.
The compounds as defined in embodiments may be synthesized by the general synthetic routes below, specific examples of which are described in more detail in the Examples.
The invention further includes any variant of the present processes, in which an intermediate product obtainable at any stage thereof is used as starting material and the remaining steps are carried out, or in which the starting materials are formed in situ under the reaction conditions, or in which the reaction components are used in the form of their salts or optically pure material.
Compounds of the present invention and intermediates can also be converted into each other according to methods generally known to those skilled in the art.
Within the scope of this text, only a readily removable group that is not a constituent of the particular desired end product of the compounds of the present invention is designated a "protecting group", unless the context indicates otherwise. The protection of functional groups by such protecting groups, the protecting groups themselves, and their cleavage reactions are described for example in standard reference works, such as J. F. W. McOmie, "Protective Groups in Organic Chemistry", Plenum Press, London and New York 1973, in T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis", Third edition, Wiley, New York 1999, in "The Peptides"; Volume 3 (editors: E. Gross and J.
Meienhofer), Academic Press, London and New York 1981 , in "Methoden der organischen Chemie" (Methods of Organic Chemistry), Houben Weyl, 4th edition, Volume 15/1, Georg Thieme Verlag, Stuttgart 1974, in H.-D. Jakubke and H. Jeschkeit, "Aminosauren, Peptide, Proteine" (Amino acids, Peptides, Proteins), Verlag Chemie, Weinheim, Deerfield Beach, and Basel 1982, and in Jochen Lehmann, "Chemie der Kohlenhydrate: Monosaccharide und Derivate" (Chemistry of Carbohydrates: Monosaccharides and Derivatives), Georg Thieme Verlag, Stuttgart 1974. A characteristic of protecting groups is that they can be removed readily (i.e. without the occurrence of undesired secondary reactions) for example by solvolysis, reduction, photolysis or alternatively under physiological conditions (e.g. by enzymatic cleavage).
Salts of compounds of the present invention having at least one salt-forming group may be prepared in a manner known to those skilled in the art. For example, salts of compounds of the present invention having acid groups may be formed, for example, by
treating the compounds with metal compounds, such as alkali metal salts of suitable organic carboxylic acids, e.g. the sodium salt of 2-ethylhexanoic acid, with organic alkali metal or alkaline earth metal compounds, such as the corresponding hydroxides, carbonates or hydrogen carbonates, such as sodium or potassium hydroxide, carbonate or hydrogen carbonate, with corresponding calcium compounds or with ammonia or a suitable organic amine, stoichiometric amounts or only a small excess of the salt-forming agent preferably being used. Acid addition salts of compounds of the present invention are obtained in customary manner, e.g. by treating the compounds with an acid or a suitable anion exchange reagent. Internal salts of compounds of the present invention containing acid and basic salt-forming groups, e.g. a free carboxy group and a free amino group, may be formed, e.g. by the neutralisation of salts, such as acid addition salts, to the isoelectric point, e.g. with weak bases, or by treatment with ion exchangers.
Salts can be converted into the free compounds in accordance with methods known to those skilled in the art. Metal and ammonium salts can be converted, for example, by treatment with suitable acids, and acid addition salts, for example, by treatment with a suitable basic agent.
Mixtures of isomers obtainable according to the invention can be separated in a manner known to those skilled in the art into the individual isomers; diastereo isomers can be separated, for example, by partitioning between polyphasic solvent mixtures, recrystallisation and/or chromatographic separation, for example over silica gel or by e.g. medium pressure liquid chromatography over a reversed phase column, and racemates can be separated, for example, by the formation of salts with optically pure salt-forming reagents and separation of the mixture of diastereoisomers so obtainable, for example by means of fractional crystallisation, or by chromatography over optically active column materials.
Intermediates and final products can be worked up and/or purified according to standard methods, e.g. using chromatographic methods, distribution methods, (re-) crystallization, and the like.
The following applies in general to all processes mentioned herein before and hereinafter.
All the above-mentioned process steps can be carried out under reaction conditions that are known to those skilled in the art, including those mentioned specifically, in the absence or, customarily, in the presence of solvents or diluents, including, for example, solvents or diluents that are inert towards the reagents used and dissolve them, in the absence or presence of catalysts, condensation or neutralizing agents, for example ion exchangers, such as cation exchangers, e.g. in the H+ form, depending on the nature of the reaction and/or of the reactants at reduced, normal or elevated temperature, for example in a temperature range of from about -100 °C to about 190 °C, including, for example, from
approximately -80 °C to approximately 150 °C, for example at from -80 to -60 °C, at room temperature, at from -20 to 40 °C or at reflux temperature, under atmospheric pressure or in a closed vessel, where appropriate under pressure, and/or in an inert atmosphere, for example under an argon or nitrogen atmosphere.
At all stages of the reactions, mixtures of isomers that are formed can be separated into the individual isomers, for example diastereo isomers or enantiomers, or into any desired mixtures of isomers, for example racemates or mixtures of diastereo isomers, for example analogously to the methods described under "Additional process steps".
The solvents from which those solvents that are suitable for any particular reaction may be selected include those mentioned specifically or, for example, water, esters, such as lower alkyl-lower alkanoates, for example ethyl acetate, ethers, such as aliphatic ethers, for example diethyl ether, or cyclic ethers, for example tetrahydrofuran or dioxane, liquid aromatic hydrocarbons, such as benzene or toluene, alcohols, such as methanol, ethanol or 1- or 2-propanol, nitriles, such as acetonitrile, halogenated hydrocarbons, such as methylene chloride or chloroform, acid amides, such as dimethylformamide or dimethyl acetamide, bases, such as heterocyclic nitrogen bases, for example pyridine or N-methylpyrrolidin-2- one, carboxylic acid anhydrides, such as lower alkanoic acid anhydrides, for example acetic anhydride, cyclic, linear or branched hydrocarbons, such as cyclohexane, hexane or isopentane, methycyclohexane, or mixtures of those solvents, for example aqueous solutions, unless otherwise indicated in the description of the processes. Such solvent mixtures may also be used in working up, for example by chromatography or partitioning.
The compounds of the present invention, including their salts, may also be obtained in the form of hydrates, or their crystals may, for example, include the solvent used for crystallization. Different crystalline forms may be present.
The invention relates also to those forms of the process in which a compound obtainable as an intermediate at any stage of the process is used as starting material and the remaining process steps are carried out, or in which a starting material is formed under the reaction conditions or is used in the form of a derivative, for example in a protected form or in the form of a salt, or a compound obtainable by the process according to the invention is produced under the process conditions and processed further in situ.
All starting materials, building blocks, reagents, acids, bases, dehydrating agents, solvents and catalysts utilized to synthesize the compounds of the present invention are either commercially available or can be produced by organic synthesis methods known to one of ordinary skill in the art (Houben-Weyl 4th Ed. 1952, Methods of Organic Synthesis, Thieme, Volume 21 ).
The term "an optical isomer" or "a stereoisomer" refers to any of the various stereoisomeric configurations which may exist for a given compound of the present invention
and includes geometric isomers. It is understood that a substituent may be attached at a chiral center of a carbon atom. The term "chiral" refers to molecules which have the property of non-superimposability on their mirror image partner, while the term "achiral" refers to molecules which are superimposable on their mirror image partner. Therefore, the invention includes enantiomers, diastereomers or racemates of the compound.
"Enantiomers" are a pair of stereoisomers that are non- superimposable mirror images of each other. A 1 : 1 mixture of a pair of enantiomers is a "racemic" mixture. The term is used to designate a racemic mixture where appropriate. "Diastereoisomers" are stereoisomers that have at least two asymmetric atoms, but which are not mirror-images of each other. The absolute stereochemistry is specified according to the Cahn- Ingold- Prelog R-S system. When a compound is a pure enantiomer the stereochemistry at each chiral carbon may be specified by either R or S. Resolved compounds whose absolute configuration is unknown can be designated (+) or (-) depending on the direction (dextro- or levorotatory) which they rotate plane polarized light at the wavelength of the sodium D line. Certain compounds described herein contain one or more asymmetric centers or axes and may thus give rise to enantiomers, diastereomers, and other stereoisomeric forms that may be defined, in terms of absolute stereochemistry, as (R)- or (S)-.
Depending on the choice of the starting materials and procedures, the compounds can be present in the form of one of the possible isomers or as mixtures thereof, for example as pure optical isomers, or as isomer mixtures, such as racemates and diastereo isomer mixtures, depending on the number of asymmetric carbon atoms. The present invention is meant to include all such possible stereoisomers, including racemic mixtures, diasteriomeric mixtures and optically pure forms. Optically active (R)- and (S)- isomers may be prepared using chiral synthons or chiral reagents, or resolved using conventional techniques. If the compound contains a double bond, the substituent may be E or Z configuration. If the compound contains a disubstituted cycloalkyl, the cycloalkyl substituent may have a cis- or trans-configuration. All tautomeric forms are also intended to be included.
Any resulting mixtures of isomers can be separated on the basis of the
physicochemical differences of the constituents, into the pure or substantially pure geometric or optical isomers, diastereomers, racemates, for example, by chromatography and/or fractional crystallization.
Any resulting racemates of final products or intermediates can be resolved into the optical antipodes by known methods, e.g., by separation of the diastereomeric salts thereof, obtained with an optically active acid or base, and liberating the optically active acidic or basic compound. In particular, a basic moiety may thus be employed to resolve the compounds of the present invention into their optical antipodes, e.g., by fractional crystallization of a salt formed with an optically active acid, e.g., tartaric acid, dibenzoyl
tartaric acid, diacetyl tartaric acid, di-0,0-p-toluoyl tartaric acid, mandelic acid, malic acid or camphor-10-sulfonic acid. Racemic products can also be resolved by chiral
chromatography, e.g., high pressure liquid chromatography (HPLC) using a chiral adsorbent.
Furthermore, the compounds of the present invention, including their salts, can also be obtained in the form of their hydrates, or include other solvents used for their
crystallization. The compounds of the present invention may inherently or by design form solvates with pharmaceutically acceptable solvents (including water); therefore, it is intended that the invention embrace both solvated and unsolvated forms. The term "solvate" refers to a molecular complex of a compound of the present invention (including pharmaceutically acceptable salts thereof) with one or more solvent molecules. Such solvent molecules are those commonly used in the pharmaceutical art, which are known to be innocuous to the recipient, e.g., water, ethanol, and the like. The term "hydrate" refers to the complex where the solvent molecule is water.
The compounds of the present invention, including salts, hydrates and solvates thereof, may inherently or by design form polymorphs.
As used herein, the terms "salt" or "salts" refers to an acid addition or base addition salt of a compound of the present invention. "Salts" include in particular "pharmaceutically acceptable salts". The term "pharmaceutically acceptable salts" refers to salts that retain the biological effectiveness and properties of the compounds of this invention and, which typically are not biologically or otherwise undesirable. In many cases, the compounds of the present invention are capable of forming acid and/or base salts by virtue of the presence of amino and/or carboxyl groups or groups similar thereto.
Pharmaceutically acceptable acid addition salts can be formed with inorganic acids and organic acids, e.g., acetate, aspartate, benzoate, besylate, bromide/hydrobromide, bicarbonate/carbonate, bisulfate/sulfate, camphorsulfonate, chloride/hydrochloride, chlortheophyllonate, citrate, ethandisulfonate, fumarate, gluceptate, gluconate, glucuronate, hippurate, hydroiodide/iodide, isethionate, lactate, lactobionate, laurylsulfate, malate, maleate, malonate, mandelate, mesylate, methylsulphate, naphthoate, napsylate, nicotinate, nitrate, octadecanoate, oleate, oxalate, palmitate, pamoate, phosphate/hydrogen phosphate/dihydrogen phosphate, polygalacturonate, propionate, stearate, succinate, subsalicylate, tartrate, tosylate and trifluoroacetate salts.
Inorganic acids from which salts can be derived include, for example, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like.
Organic acids from which salts can be derived include, for example, acetic acid, propionic acid, glycolic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, mandelic acid, methanesulfonic acid,
ethanesulfonic acid, toluenesulfonic acid, sulfosalicylic acid, and the like. Pharmaceutically acceptable base addition salts can be formed with inorganic and organic bases.
Inorganic bases from which salts can be derived include, for example, ammonium salts and metals from columns I to XII of the periodic table. In certain embodiments, the salts are derived from sodium, potassium, ammonium, calcium, magnesium, iron, silver, zinc, and copper; particularly suitable salts include ammonium, potassium, sodium, calcium and magnesium salts.
Organic bases from which salts can be derived include, for example, primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, basic ion exchange resins, and the like. Certain organic amines include isopropylamine, benzathine, cholinate, diethanolamine, diethylamine, lysine, meglumine, piperazine and tromethamine.
The pharmaceutically acceptable salts of the present invention can be synthesized from a basic or acidic moiety, by conventional chemical methods. Generally, such salts can be prepared by reacting free acid forms of these compounds with a stoichiometric amount of the appropriate base (such as Na, Ca, Mg, or K hydroxide, carbonate, bicarbonate or the like), or by reacting free base forms of these compounds with a stoichiometric amount of the appropriate acid. Such reactions are typically carried out in water or in an organic solvent, or in a mixture of the two. Generally, use of non-aqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile is desirable, where practicable. Lists of additional suitable salts can be found, e.g., in "Remington's Pharmaceutical Sciences", 20th ed., Mack Publishing Company, Easton, Pa., (1985); and in "Handbook of Pharmaceutical Salts:
Properties, Selection, and Use" by Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002).
Any formula given herein is also intended to represent unlabeled forms as well as isotopically labeled forms of the compounds of the present invention. Isotopically labeled compounds have structures depicted by the formulas given herein except that one or more atoms are replaced by an atom having a selected atomic mass or mass number. Examples of isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine, and chlorine, such as 2H, 3H, 11C, 13C, 14C, 15N, 18F 31P, 32P, 35S, 36CI, 125l respectively. The invention includes various isotopically labeled compounds of the present invention, for example those into which radioactive isotopes, such as 3H and 14C, or those into which non-radioactive isotopes, such as 2H and 13C are present. Such isotopically labelled compounds are useful in metabolic studies (with 14C), reaction kinetic studies (with, for example 2H or 3H), detection or imaging techniques, such as positron emission tomography (PET) or single-photon emission computed tomography (SPECT) including drug or substrate tissue distribution assays, or in
radioactive treatment of patients. In particular, an 18F labeled compound of the present invention may be particularly desirable for PET or SPECT studies. Isotopically-labeled compounds of the present invention can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples and Preparations using an appropriate isotopically-labeled reagent in place of the non-labeled reagent previously employed.
Further, substitution with heavier isotopes, particularly deuterium (i.e., 2H or D) may afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements or an improvement in therapeutic index. It is understood that deuterium in this context is regarded as a substituent of a compound of the present invention. The concentration of such a heavier isotope, specifically deuterium, may be defined by the isotopic enrichment factor. The term "isotopic enrichment factor" as used herein means the ratio between the isotopic abundance and the natural abundance of a specified isotope. If a substituent in a compound of this invention is denoted deuterium, such compound has an isotopic enrichment factor for each designated deuterium atom of at least 3500 (52.5% deuterium incorporation at each designated deuterium atom), at least 4000 (60% deuterium incorporation), at least 4500 (67.5% deuterium incorporation), at least 5000 (75% deuterium incorporation), at least 5500 (82.5% deuterium incorporation), at least 6000 (90% deuterium incorporation), at least 6333.3 (95% deuterium incorporation), at least 6466.7 (97% deuterium incorporation), at least 6600 (99% deuterium incorporation), or at least 6633.3 (99.5% deuterium incorporation).
Pharmaceutically acceptable solvates in accordance with the invention include those wherein the solvent of crystallization may be isotopically substituted, e.g. D20, d6-acetone, de-DMSO.
Compounds of the present invention that contain groups capable of acting as donors and/or acceptors for hydrogen bonds may be capable of forming co-crystals with suitable co- crystal formers. These co-crystals may be prepared from compounds of the present invention by known co-crystal forming procedures. Such procedures include grinding, heating, co-subliming, co-melting, or contacting in solution compounds of the present invention with the co-crystal former under crystallization conditions and isolating co-crystals thereby formed. Suitable co-crystal formers include those described in WO 2004/078163. Hence the invention further provides co-crystals comprising a compound of the present invention.
All methods described herein can be performed in any suitable order unless otherwise indicated herein or otherwise clearly contradicted by context. The use of any and all examples, or exemplary language (e.g. "such as") provided herein is intended merely to
better illuminate the invention and does not pose a limitation on the scope of the invention otherwise claimed.
The present invention provides novel compounds, pharmaceutical formulations including the compounds, methods of inhibiting UDP-3-0-(R-3-hydroxydecanoyl)-N- acetylglucosamine deacetylase (LpxC), and methods of treating Gram-negative bacterial infections.
Substitution with heavier isotopes such as deuterium, i.e. 2H, may afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life or reduced dosage requirements, and hence may be preferred in some circumstances. For example, deuterium substitution at non-exchangeable hydrocarbon bonds (e.g., C-H) may retard epimerization and/or metabolic oxidation in vivo.
Isotopically-labeled compounds of the invention, i.e. compounds of formula (I), can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples and Preparations Sections using an appropriate isotopically-labeled reagent in place of the non-labeled reagent previously.
In another aspect, the invention provides a method of inhibiting a deacetylase enzyme in a Gram-negative bacterium, the method comprising the step of contacting the Gram-negative bacteria with a compound of the invention, e.g., a compound of Formula I or salt thereof.
In still another aspect, the invention provides a method for treating a subject with a Gram-negative bacterial infection, the method comprising the step of administering to the subject in need thereof an antibacterially effective amount of a compound of the invention, e.g., a compound of Formula I or salt thereof with a pharmaceutically acceptable carrier.
The compounds of the invention can be used for treating conditions caused by the bacterial production of endotoxin and, in particular, by Gram-negative bacteria and bacteria that use LpxC in the biosynthesis of lipopolysaccharide (LPS) or endotoxin.
The compounds of the invention also are useful in the treatment of patients suffering from or susceptible to pneumonia, sepsis, cystic fibrosis, wound, complicated diabetic foot or complicated urinary track infections and sexually transmitted diseases caused by Gram- negative pathogens. The compounds of the invention also are useful in the conditions that are caused or exacerbated by the bacterial production of lipid A and LPS or endotoxin, such as sepsis, septic shock, systemic inflammation, localized inflammation, chronic obstructive pulmonary disease (COPD) and acute exacerbations of chronic bronchitis (AECB). For these conditions, treatment includes the administration of a compound of the invention, or a combination of compounds of the invention, optionally with a second agent wherein the second agent is a second antibacterial agent or a second non-antibacterial agent.
For sepsis, septic shock, systemic inflammation, localized inflammation, chronic obstructive pulmonary disease (COPD) and acute exacerbations of chronic bronchitis (AECB), preferred second non-antibacterial agents include antiendotoxins including endotoxin receptor-binding antibodies, endotoxin-binding antibodies, antiCD14-binding protein antibodies antilipopolysaccharide-binding protein antibodies and tyrosine kinase inhibitors.
In treatment of serious or chronic respiratory tract infections, the compounds of the present invention may also be used with second non-antibacterial agents administered via inhalation. Preferred non-antibacterial agents used in this treatment include antiinflammatory steroids, non-steroidal anti-inflammatory agents, bronchiodilators, mucolytics, anti-asthma therapeutics and lung fluid surfactants. In particular, the non-antibacterial agent may be selected from a group consisting of albuterol, salbuterol, budesonide,
beclomethasone, dexamethasone, nedocromil, beclomethasone, fluticasone, flunisolide, triamcinolone, ibuprofin, rofecoxib, naproxen, celecoxib, nedocromil, ipratropium, metaproterenol, pirbuterol, salneterol, bronchiodilators, mucolytics, calfactant, beractant, poractant alfa, surfaxin and pulmozyme (also called domase alfa).
The compounds of the invention can be used, alone or in combination with a second antibacterial agent for the treatment of a serious or chronic respiratory tract infection including serious lung and nosocomial infections such as those caused by Enterobacter aerogenes, Enterobacter cloacae, Escherichia coli, Klebsiella pneumoniae, Klebsiella oxytoca, Proteus mirabilis, Serratia marcescens, Stenotrophomonas maltophilia,
Pseudomonas aeruginosa, Burkholderia cepacia, Acinetobacter baumanii, Alcaligenes xylosoxidans, Flavobacterium meningosepticum, Providencia stuartii and Citrobacter freundi, community lung infections such as those caused by Haemophilus influenzae, Legionella species, Moraxella catarrhalis, Enterobacter species, Acinetobacter species, Klebsiella species, and Proteus species, and infections caused by other bacterial species such as Neisseria species, Shigella species, Salmonella species, Helicobacter pylori, Vibrionaceae and Bordetella species as well as the infections is caused by a Brucella species, Francisella tularensis and/or Yersinia Pestis.
A compound of the present invention may also be used in combination with other agents, e.g., an additional antibiotic agent that is or is not of the formula I, for treatment of a bacterial infection in a subject.
By the term "combination", is meant either a fixed combination in one dosage unit form, or a kit of parts for the combined administration where a compound of the present invention and a combination partner may be administered independently at the same time or separately within time intervals that especially allow that the combination partners show a cooperative, e.g., synergistic, effect, or any combination thereof.
When used for treating Gram-negative bacteria, the compounds of the present invention can be used to sensitize Gram-negative bacteria to the effects of a second agent.
An embodiment of the present invention is compounds of the present invention used in combination with a second antibacterial agent, non-limiting examples of antibacterial agents may be selected from the following groups:
(1 ) Macrolides or ketolides such as erythromycin, azithromycin, clarithromycin, and telithromycin;
(2) Beta-lactams including penicillin such as penicillin G, penicillin V, methicillin, oxacillin, cloxacillin, dicloxacillin, nafcillin, ampicillin, amoxicillin, carbenicillin, ticarcillin, mezlocillin, piperacillin, azlocillin, temocillin, cephalosporin such as cepalothin, cephapirin, cephradine, cephaloridine, cefazolin, cefamandole, cefuroxime, cephalexin, cefprozil, cefaclor, loracarbef, cefoxitin, cefinetazole, cefotaxime, ceftizoxime, ceftriaxone,
cefoperazone, ceftazidime, cefixime, cefpodoxime, ceftibuten, cefdinir, cefpirome, cefepime, and carbapenems such as carbapenem, imipenem, meropenem and PZ-601 ;
(3) Monobactams such as aztreonam;
(4) Quinolones such as nalidixic acid, oxolinic acid, norfloxacin, pefloxacin, enoxacin, ofloxacin, levofloxacin, ciprofloxacin, temafloxacin, lomefloxacin, fleroxacin, grepafloxacin, sparfloxacin, trovafloxacin, clinafloxacin, gatifloxacin, moxifloxacin, sitafloxacin,
ganefloxacin, gemifloxacin and pazufloxacin;
(5) Antibacterial sulfonanmides and antibacterial sulphanilamides, including para- aminobenzoic acid, sulfadiazine, sulfisoxazole, sulfamethoxazole and sulfathalidine;
(6) Aminoglycosides such as streptomycin, neomycin, kanamycin, paromycin, gentamicin, tobramycin, amikacin, netilmicin, spectinomycin, sisomicin, dibekalin and isepamicin;
(7) Tetracyclines such as tetracycline, chlortetracycline, demeclocycline, minocycline, oxytetracycline, methacycline, doxycycline, tegacycline;
(8) Rifamycins such as rifampicin (also called rifampin), rifapentine, rifabutin, bezoxazinorifamycin and rifaximin;
(9) Lincosamides such as lincomycin and clindamycin;
(10) Glycopeptides such as vancomycin and teicoplanin;
(1 1 ) Streptogramins such as quinupristin and daflopristin;
(12) Oxazolidinones such as linezolid;
(13) Polymyxin, colistin and colymycin;
(14) Trimethoprim and bacitracin.
(15) Efflux pump inhibitors.
The second antibacterial agent may be administered in combination with the compounds of the present inventions wherein the second antibacterial agent is administered
prior to, simultaneously, or after the compound or compounds of the present invention. When simultaneous administration of a compound of the invention with a second agent is desired and the route of administration is the same, then a compound of the invention may be formulated with a second agent into the same dosage form. An example of a dosage form containing a compound of the invention and a second agent is a tablet or a capsule.
When used for treating serious or chronic respiratory tract infections, the compounds of the invention may be used alone or in combination with a second antibacterial agent administered via inhalation. In the case of inhalation, a preferred second antibacterial agent is selected from a group consisting of tobramycin, gentamicin, aztreonam, ciprofloxacin, polymyxin, colistin, colymycin, azithromycin and clarithromycin.
The language "effective amount" of the compound is that amount necessary or sufficient to treat or prevent a bacterial infection and/or a disease or condition described herein. In an example, an effective amount of the LpxC inhibitor is the amount sufficient to treat bacterial infection in a subject. In another example, an effective amount of the LpxC inhibitor is an amount sufficient to treat a bacterial infection, such as, but not limited to Pseudomonas aeruginosa and the like in a subject. The effective amount can vary depending on such factors as the size and weight of the subject, the type of illness, or the particular compound of the invention. For example, the choice of the compound of the invention can affect what constitutes an "effective amount." One of ordinary skill in the art would be able to study the factors contained herein and make the determination regarding the effective amount of the compounds of the invention without undue experimentation.
The regimen of administration can affect what constitutes an effective amount. The compound of the invention can be administered to the subject either prior to or after the onset of a bacterial infection. Further, several divided dosages, as well as staggered dosages, can be administered daily or sequentially, or the dose can be continuously infused, or can be a bolus injection. Further, the dosages of the compound(s) of the invention can be proportionally increased or decreased as indicated by the exigencies of the therapeutic or prophylactic situation.
Compounds of the invention may be used in the treatment of states, disorders or diseases as described herein, or for the manufacture of pharmaceutical compositions for use in the treatment of these diseases. The invention provides methods of use of compounds of the present invention in the treatment of these diseases or pharmaceutical preparations having compounds of the present invention for the treatment of these diseases.
The language "pharmaceutical composition" includes preparations suitable for administration to mammals, e.g., humans. When the compounds of the present invention are administered as pharmaceuticals to mammals, e.g., humans, they can be given per se or as a pharmaceutical composition containing, for example, 0.1 to 99.5% (more preferably, 0.5
to 90%) of active ingredient in combination with a pharmaceutically acceptable carrier.
The phrase "pharmaceutically acceptable carrier" is art recognized and includes a pharmaceutically acceptable material, composition or vehicle, suitable for administering compounds of the present invention to mammals. The carriers include liquid or solid filler, diluent, excipient, solvent or encapsulating material, involved in carrying or transporting the subject agent from one organ, or portion of the body, to another organ, or portion of the body. Each carrier must be "acceptable" in the sense of being compatible with the other ingredients of the formulation and not injurious to the patient. Some examples of materials which can serve as pharmaceutically acceptable carriers include: sugars, such as lactose, glucose and sucrose; starches, such as corn starch and potato starch; cellulose, and its derivatives, such as sodium carboxymethyl cellulose, ethyl cellulose and cellulose acetate; powdered tragacanth; malt; gelatin; talc; excipients, such as cocoa butter and suppository waxes; oils, such as peanut oil, cottonseed oil, safflower oil, sesame oil, olive oil, corn oil and soybean oil; glycols, such as propylene glycol; polyols, such as glycerin, sorbitol, mannitol and polyethylene glycol; esters, such as ethyl oleate and ethyl laurate; agar; buffering agents, such as magnesium hydroxide and aluminum hydroxide; alginic acid; pyrogen-free water; isotonic saline; Ringer's solution; ethyl alcohol; phosphate buffer solutions; and other non-toxic compatible substances employed in pharmaceutical formulations.
Wetting agents, emulsifiers and lubricants, such as sodium lauryl sulfate and magnesium stearate, as well as coloring agents, release agents, coating agents, sweetening, flavoring and perfuming agents, preservatives and antioxidants can also be present in the compositions.
Examples of pharmaceutically acceptable antioxidants include: water soluble antioxidants, such as ascorbic acid, cysteine hydrochloride, sodium bisulfate, sodium metabisulfite, sodium sulfite and the like; oil-soluble antioxidants, such as ascorbyl palmitate, butylated hydroxyanisole (BHA), butylated hydroxytoluene (BHT), lecithin, propyl gallate, - tocopherol, and the like; and metal chelating agents, such as citric acid, ethylenediamine tetraacetic acid (EDTA), sorbitol, tartaric acid, phosphoric acid, and the like.
Formulations of the present invention include those suitable for oral, nasal, inhalation, topical, transdermal, buccal, sublingual, rectal, vaginal and/or parenteral administration. The formulations may conveniently be presented in unit dosage form and may be prepared by any methods well known in the art of pharmacy. The amount of active ingredient that can be combined with a carrier material to produce a single dosage form will generally be that amount of the compound that produces a therapeutic effect. Generally, out of one hundred per cent, this amount will range from about 1 per cent to about ninety-nine percent of active ingredient, preferably from about 5 per cent to about 70 per cent, most preferably from about 10 per cent to about 30 per cent.
Methods of preparing these formulations or compositions include the step of bringing into association a compound of the present invention with the carrier and, optionally, one or more accessory ingredients. In general, the formulations are prepared by uniformly and intimately bringing into association a compound of the present invention with liquid carriers, or finely divided solid carriers, or both, and then, if necessary, shaping the product.
Formulations of the invention suitable for oral administration may be in the form of capsules, cachets, pills, tablets, lozenges (using a flavored basis, usually sucrose and acacia or tragacanth), powders, granules, or as a solution or a suspension in an aqueous or non-aqueous liquid, or as an oil-in-water or water-in-oil liquid emulsion, or as an elixir or syrup, or as pastilles (using an inert base, such as gelatin and glycerin, or sucrose and acacia) and/or as mouth washes and the like, each containing a predetermined amount of a compound of the present invention as an active ingredient. A compound of the present invention may also be administered as a bolus, electuary or paste.
In solid dosage forms of the invention for oral administration (capsules, tablets, pills, dragees, powders, granules and the like), the active ingredient is mixed with one or more pharmaceutically acceptable carriers, such as sodium citrate or dicalcium phosphate, and/or any of the following: fillers or extenders, such as starches, lactose, sucrose, glucose, mannitol, and/or silicic acid; binders, such as, for example, carboxymethylcellulose, alginates, gelatin, polyvinyl pyrrolidone, sucrose and/or acacia; humectants, such as glycerol; disintegrating agents, such as agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates, and sodium carbonate; solution retarding agents, such as paraffin; absorption accelerators, such as quaternary ammonium compounds; wetting agents, such as, for example, cetyl alcohol and glycerol monostearate; absorbents, such as kaolin and bentonite clay; lubricants, such a talc, calcium stearate, magnesium stearate, solid polyethylene glycols, sodium lauryl sulfate, and mixtures thereof; and coloring agents. In the case of capsules, tablets and pills, the pharmaceutical compositions may also comprise buffering agents. Solid compositions of a similar type may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugars, as well as high molecular weight polyethylene glycols and the like.
A tablet may be made by compression or molding, optionally with one or more accessory ingredients. Compressed tablets may be prepared using binder (for example, gelatin or hydroxypropylmethyl cellulose), lubricant, inert diluent, preservative, disintegrant (for example, sodium starch glycolate or cross-linked sodium carboxymethyl cellulose), surface-active or dispersing agent. Molded tablets may be made by molding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent.
The tablets, and other solid dosage forms of the pharmaceutical compositions of the present invention, such as dragees, capsules, pills and granules, may optionally be scored
or prepared with coatings and shells, such as enteric coatings and other coatings well known in the pharmaceutical-formulating art. They may also be formulated so as to provide slow or controlled release of the active ingredient therein using, for example, hydroxypropylmethyl cellulose in varying proportions to provide the desired release profile, other polymer matrices, liposomes and/or microspheres. They may be sterilized by, for example, filtration through a bacteria-retaining filter, or by incorporating sterilizing agents in the form of sterile solid compositions that can be dissolved in sterile water, or some other sterile injectable medium immediately before use. These compositions may also optionally contain opacifying agents and may be of a composition that they release the active ingredient(s) only, or preferentially, in a certain portion of the gastrointestinal tract, optionally, in a delayed manner. Examples of embedding compositions that can be used include polymeric substances and waxes. The active ingredient can also be in micro-encapsulated form, if appropriate, with one or more of the above-described excipients.
Liquid dosage forms for oral administration of the compounds of the invention include pharmaceutically acceptable emulsions, microemulsions, solutions, suspensions, syrups and elixirs. In addition to the active ingredient, the liquid dosage forms may contain inert diluent commonly used in the art, such as, for example, water or other solvents, solubilizing agents and emulsifiers, such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1 ,3-butylene glycol, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor and sesame oils), glycerol, tetrahydrofuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan, and mixtures thereof.
Besides inert diluents, the oral compositions can also include adjuvants such as wetting agents, emulsifying and suspending agents, sweetening, flavoring, coloring, perfuming and preservative agents.
Suspensions, in addition to the active compounds, may contain suspending agents as, for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar and tragacanth, and mixtures thereof.
Formulations of the pharmaceutical compositions of the invention for rectal or vaginal administration may be presented as a suppository, which may be prepared by mixing one or more compounds of the invention with one or more suitable nonirritating excipients or carriers comprising, for example, cocoa butter, polyethylene glycol, a suppository wax or a salicylate, and which is solid at room temperature, but liquid at body temperature and, therefore, will melt in the rectum or vaginal cavity and release the active compound.
Formulations of the present invention which are suitable for vaginal administration also include pessaries, tampons, creams, gels, pastes, foams or spray formulations containing such carriers as are known in the art to be appropriate.
Dosage forms for the topical or transdermal administration of a compound of this invention include powders, sprays, ointments, pastes, creams, lotions, gels, solutions, patches and inhalants. The active compound may be mixed under sterile conditions with a pharmaceutically acceptable carrier, and with any preservatives, buffers, or propellants that may be required.
The ointments, pastes, creams and gels may contain, in addition to an active compound of this invention, excipients, such as animal and vegetable fats, oils, waxes, paraffins, starch, tragacanth, cellulose derivatives, polyethylene glycols, silicones, bentonites, silicic acid, talc and zinc oxide, or mixtures thereof.
Powders and sprays can contain, in addition to a compound of this invention, excipients such as lactose, talc, silicic acid, aluminum hydroxide, calcium silicates and polyamide powder, or mixtures of these substances. Sprays can additionally contain customary propellants, such as chlorofluorohydrocarbons and volatile unsubstituted hydrocarbons, such as butane and propane.
Transdermal patches have the added advantage of providing controlled delivery of a compound of the present invention to the body. Such dosage forms can be made by dissolving or dispersing the compound in the proper medium. Absorption enhancers can also be used to increase the flux of the compound across the skin. The rate of such flux can be controlled by either providing a rate controlling membrane or dispersing the active compound in a polymer matrix or gel.
Ophthalmic formulations, eye ointments, powders, solutions and the like, are also contemplated as being within the scope of this invention.
Pharmaceutical compositions of this invention suitable for parenteral administration comprise one or more compounds of the invention in combination with one or more pharmaceutically acceptable sterile isotonic aqueous or nonaqueous solutions, dispersions, suspensions or emulsions, or sterile powders which may be reconstituted into sterile injectable solutions or dispersions just prior to use, which may contain antioxidants, buffers, bacteriostats, solutes which render the formulation isotonic with the blood of the intended recipient or suspending or thickening agents.
Examples of suitable aqueous and nonaqueous carriers that may be employed in the pharmaceutical compositions of the invention include water, ethanol, polyols (such as glycerol, propylene glycol, polyethylene glycol, and the like), and suitable mixtures thereof, vegetable oils, such as olive oil, and injectable organic esters, such as ethyl oleate. Proper fluidity can be maintained, for example, by the use of coating materials, such as lecithin, by the maintenance of the required particle size in the case of dispersions, and by the use of surfactants.
These compositions may also contain adjuvants such as preservatives, wetting
agents, emulsifying agents and dispersing agents. Prevention of the action of
microorganisms may be ensured by the inclusion of various antibacterial and antifungal agents, for example, paraben, chlorobutanol, phenol sorbic acid, and the like. It may also be desirable to include isotonic agents, such as sugars, sodium chloride, and the like into the compositions. In addition, prolonged absorption of the injectable pharmaceutical form may be brought about by the inclusion of agents that delay absorption such as aluminum monostearate and gelatin.
In some cases, in order to prolong the effect of a drug, it is desirable to slow the absorption of the drug from subcutaneous or intramuscular injection. This may be accomplished by the use of a liquid suspension of crystalline or amorphous material having poor water solubility. The rate of absorption of the drug then depends upon its rate of dissolution which, in turn, may depend upon crystal size and crystalline form. Alternatively, delayed absorption of a parenterally-administered drug form is accomplished by dissolving or suspending the drug in an oil vehicle.
Injectable depot forms are made by forming microencapsule matrices of the subject compounds in biodegradable polymers such as polylactide-polyglycolide. Depending on the ratio of drug to polymer, and the nature of the particular polymer employed, the rate of drug release can be controlled. Examples of other biodegradable polymers include
poly(orthoesters) and poly(anhydrides). Depot injectable formulations are also prepared by entrapping the drug in liposomes or microemulsions that are compatible with body tissue.
The preparations of the present invention may be given orally, parenterally, topically, or rectally. They are of course given by forms suitable for each administration route. For example, they are administered in tablets or capsule form, by injection, inhalation, eye lotion, ointment, suppository, etc., administration by injection, infusion or inhalation; topical by lotion or ointment; and rectal by suppositories. Oral administration is preferred.
The phrases "parenteral administration" and "administered parenterally" as used herein means modes of administration other than enteral and topical administration, usually by injection, and includes, without limitation, intravenous, intramuscular, intraarterial, intrathecal, intracapsular, intraorbital, intracardiac, intradermal, intraperitoneal, transtracheal, subcutaneous, subcuticular, intraarticular, subcapsular, subarachnoid, intraspinal and intrasternal injection and infusion.
The phrases "systemic administration," "administered systemically," "peripheral administration" and "administered peripherally" as used herein mean the administration of a compound, drug or other material other than directly into the central nervous system, such that it enters the patient's system and, thus, is subject to metabolism and other like processes, for example, subcutaneous administration.
These compounds may be administered to humans and other animals for therapy by
any suitable route of administration, including orally, nasally, as by, for example, a spray, rectally, intravaginally, parenterally, intracisternally and topically, as by powders, ointments or drops, including buccally and sublingually.
Regardless of the route of administration selected, the compounds of the present invention, which may be used in a suitable hydrated form, and/or the pharmaceutical compositions of the present invention, are formulated into pharmaceutically acceptable dosage forms by conventional methods known to those of skill in the art.
Actual dosage levels of the active ingredients in the pharmaceutical compositions of this invention may be varied so as to obtain an amount of the active ingredient which is effective to achieve the desired therapeutic response for a particular patient, composition, and mode of administration, without being toxic to the patient.
The selected dosage level will depend upon a variety of factors including the activity of the particular compound of the present invention employed, or the ester, salt or amide thereof, the route of administration, the time of administration, the rate of excretion of the particular compound being employed, the duration of the treatment, other drugs, compounds and/or materials used in combination with the particular compound employed, the age, sex, weight, condition, general health and prior medical history of the patient being treated, and like factors well known in the medical arts.
A physician or veterinarian having ordinary skill in the art can readily determine and prescribe the effective amount of the pharmaceutical composition required. For example, the physician or veterinarian could start doses of the compounds of the invention employed in the pharmaceutical composition at levels lower than that required in order to achieve the desired therapeutic effect and gradually increase the dosage until the desired effect is achieved.
In general, a suitable daily dose of a compound of the invention will be that amount of the compound that is the lowest dose effective to produce a therapeutic effect. Such an effective dose will generally depend upon the factors described above. Generally, intravenous and subcutaneous doses of the compounds of this invention for a patient, when used for the indicated analgesic effects, will range from about 0.0001 to about 100 mg per kilogram of body weight per day, more preferably from about 0.01 to about 50 mg per kg per day, and still more preferably from about 1.0 to about 100 mg per kg per day. An effective amount is that amount treats a bacterial infection.
If desired, the effective daily dose of the active compound may be administered as two, three, four, five, six or more sub-doses administered separately at appropriate intervals throughout the day, optionally, in unit dosage forms.
While it is possible for a compound of the present invention to be administered alone, it is preferable to administer the compound as a pharmaceutical composition.
The compounds as defined in embodiments may be synthesized by the general synthetic routes below, specific examples of which are described in more detail in the Examples.
General Synthetic Schemes
The compounds of general structure 1-1 can be synthesized by the Scheme 1 shown below. Compound 1 c can be prepared from amine 1 b either through reductive amination with aldehyde 1a or alkylation with appropriate electrophile 1 d. The ester in 1 c can be converted into hydroxamate in 1-1 in standard three step procedure. The ester was hydrolyzed under basic condition to provide carboxylic acid 1 e, which can then be coupled with protected hydroxylamine to give 1f. Finally the protecting group can be cleaved to generate the desired compound of formula 1-1.
Scheme 1


1-1
As is readily apparent to one skilled in the art, the synthetic route in Scheme 1 could be modified and the tail fragment represented by R-L could be coupled at late stage (as shown in Scheme 2). The initial step is to couple amine 1 b and aldehyde 2a by a reductive amination reaction. The product 2b can be hydrolyzed to acid 2c followed by amide coupling to provide protected hyroxamate 2d. The tail fragment can be attached by standard transition metal catalyzed coupling reaction to provide 2e. Finally the protecting group can be cleaved to generate the desired compound of formula IV.
Scheme 2

Step 4
tail fragment coupling

The synthesis of compounds with formula I-2 was illustrated in Scheme 3. As shown in Scheme 1 and 2, ester 3a is a key intermediate, which can be converted to IV by standard three step procedures. The key step in the synthesis of 3a is attaching R-L-Z-CH2CH2 moiety to ester 3c through alkylation. There are a variety of methods to assemble individual [R-L-Z- A] fragments and attach to ester 3c. Such variations are feasible to one of skilled in the art.
Scheme 3

Step 1. hydrolysis
Step 2. couple with NH20-PG
Step 3. deprotection

Synthetic Procedure
Compounds of the present invention are prepared from commonly available compounds using procedures known to those skilled in the art, including any one or more of the following conditions without limitation:
Within the scope of this text, only a readily removable group that is not a constituent of the particular desired end product of the compounds of the present invention is designated a "protecting group," unless the context indicates otherwise. The protection of functional groups by such protecting groups, the protecting groups themselves, and their cleavage reactions are described for example in standard reference works, such as e.g., Science of Synthesis: Houben-Weyl Methods of Molecular Transformation. Georg Thieme Verlag, Stuttgart, Germany. 2005. 41627 pp. (URL: http://www.science-of-synthesis.com (Electronic Version, 48 Volumes)); J. F. W. McOmie, "Protective Groups in Organic
Chemistry", Plenum Press, London and New York 1973, in T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis", Third edition, Wiley, New York 1999, in "The Peptides"; Volume 3 (editors: E. Gross and J. Meienhofer), Academic Press, London and New York 1981 , in "Methoden der organischen Chemie" (Methods of Organic Chemistry), Houben Weyl, 4th edition, Volume 15/1, Georg Thieme Verlag, Stuttgart 1974, in H.-D.
Jakubke and H. Jeschkeit, "Aminosauren, Peptide, Proteine" (Amino acids, Peptides,
Proteins), Verlag Chemie, Weinheim, Deerfield Beach, and Basel 1982, and in Jochen Lehmann, "Chemie der Kohlenhydrate: Monosaccharide und Derivate" (Chemistry of Carbohydrates: Monosaccharides and Derivatives), Georg Thieme Verlag, Stuttgart 1974. A characteristic of protecting groups is that they can be removed readily (i.e. , without the occurrence of undesired secondary reactions) for example by solvolysis, reduction, photolysis or alternatively under physiological conditions (e.g., by enzymatic cleavage).
Salts of compounds of the present invention having at least one salt-forming group may be prepared in a manner known per se. For example, salts of compounds of the present invention having acid groups may be formed, for example, by treating the compounds with metal compounds, such as alkali metal salts of suitable organic carboxylic acids, e.g., the sodium salt of 2-ethyl hexanoic acid, with organic alkali metal or alkaline earth metal compounds, such as the corresponding hydroxides, carbonates or hydrogen carbonates, such as sodium or potassium hydroxide, carbonate or hydrogen carbonate, with corresponding calcium compounds or with ammonia or a suitable organic amine, stoichiometric amounts or only a small excess of the salt-forming agent preferably being used. Acid addition salts of compounds of the present invention are obtained in customary manner, e.g., by treating the compounds with an acid or a suitable anion exchange reagent. Internal salts of compounds of the present invention containing acid and basic salt-forming groups, e.g., a free carboxy group and a free amino group, may be formed, e.g., by the neutralisation of salts, such as acid addition salts, to the isoelectric point, e.g., with weak bases, or by treatment with ion exchangers.
Salts can be converted in customary manner into the free compounds; metal and ammonium salts can be converted, for example, by treatment with suitable acids, and acid addition salts, for example, by treatment with a suitable basic agent.
Mixtures of isomers obtainable according to the invention can be separated in a manner known per se into the individual isomers; diastereoisomers can be separated, for example, by partitioning between polyphasic solvent mixtures, recrystallisation and/or chromatographic separation, for example over silica gel or by, e.g., medium pressure liquid chromatography over a reversed phase column, and racemates can be separated, for example, by the formation of salts with optically pure salt-forming reagents and separation of the mixture of diastereoisomers so obtainable, for example by means of fractional crystallisation, or by chromatography over optically active column materials.
Intermediates and final products can be worked up and/or purified according to standard methods, e.g., using chromatographic methods, distribution methods, (re-) crystallization, and the like.
General process conditions
The following applies in general to all processes mentioned throughout this disclosure.
The process steps to synthesize the compounds of the invention can be carried out under reaction conditions that are known per se, including those mentioned specifically, in the absence or, customarily, in the presence of solvents or diluents, including, for example, solvents or diluents that are inert towards the reagents used and dissolve them, in the absence or presence of catalysts, condensation or neutralizing agents, for example ion exchangers, such as cation exchangers, e.g., in the H+ form, depending on the nature of the reaction and/or of the reactants at reduced, normal or elevated temperature, for example in a temperature range of from about -100 °C to about 190°C, including, for example, from approximately -80°C to approximately 150°C, for example at from -80 to -60°C, at room temperature, at from -20 to 40°C or at reflux temperature, under atmospheric pressure or in a closed vessel, where appropriate under pressure, and/or in an inert atmosphere, for example under an argon or nitrogen atmosphere.
At all stages of the reactions, mixtures of isomers that are formed can be separated into the individual isomers, for example diastereo isomers or enantiomers, or into any desired mixtures of isomers, for example racemates or mixtures of diastereo isomers, for example analogously to the methods described in Science of Synthesis: Houben-Weyl Methods of Molecular Transformation. Georg Thieme Verlag, Stuttgart, Germany. 2005.
The solvents from which those solvents that are suitable for any particular reaction may be selected include those mentioned specifically or, for example, water, esters, such as lower alkyl-lower alkanoates, for example ethyl acetate, ethers, such as aliphatic ethers, for example diethyl ether, or cyclic ethers, for example tetrahydrofurane or dioxane, liquid aromatic hydrocarbons, such as benzene or toluene, alcohols, such as methanol, ethanol or 1- or 2-propanol, nitriles, such as acetonitrile, halogenated hydrocarbons, such as methylene chloride or chloroform, acid amides, such as dimethylformamide or dimethyl acetamide, bases, such as heterocyclic nitrogen bases, for example pyridine or N-methylpyrrolidin-2- one, carboxylic acid anhydrides, such as lower alkanoic acid anhydrides, for example acetic anhydride, cyclic, linear or branched hydrocarbons, such as cyclohexane, hexane or isopentane, or mixtures of those solvents, for example aqueous solutions, unless otherwise indicated in the description of the processes. Such solvent mixtures may also be used in working up, for example by chromatography or partitioning.
The compounds, including their salts, may also be obtained in the form of hydrates, or their crystals may, for example, include the solvent used for crystallization. Different crystalline forms may be present.
The invention relates also to those forms of the process in which a compound obtainable as an intermediate at any stage of the process is used as starting material and
the remaining process steps are carried out, or in which a starting material is formed under the reaction conditions or is used in the form of a derivative, for example in a protected form or in the form of a salt, or a compound obtainable by the process according to the invention is produced under the process conditions and processed further in situ.
The present invention also relates to pro-drugs of a compound of the present invention that are converted in vivo to the compounds of the present invention as described herein. Any reference to a compound of the present invention is therefore to be understood as referring also to the corresponding pro-drugs of the compound of the present invention, as appropriate and expedient.
In accordance with the foregoing the present invention provides in a yet further aspect:
• A pharmaceutical combination comprising a) a first agent which is a compound of the invention, e.g. a compound of formula I or any subformulae thereof, and b) a co- agent, e.g. a second drug agent as defined above.
• A method as defined above comprising co-administration, e.g. concomitantly or in sequence, of a therapeutically effective amount of a compound of the invention, e.g. a compound of formula I or any subformulae thereof, and a co-agent, e.g. a second drug agent as defined above.
The terms "co-administration" or "combined administration" or the like as utilized herein are meant to encompass administration of the selected therapeutic agents to a single patient, and are intended to include treatment regimens in which the agents are not necessarily administered by the same route of administration or at the same time. Fixed combinations are also within the scope of the present invention. The administration of a pharmaceutical combination of the invention results in a beneficial effect, e.g. a synergistic therapeutic effect, compared to a monotherapy applying only one of its pharmaceutically active ingredients.
Each component of a combination according to this invention may be administered separately, together, or in any combination thereof.
The compound of the invention and any additional agent may be formulated in separate dosage forms. Alternatively, to decrease the number of dosage forms
administered to a patient, the compound of the invention and any additional agent may be formulated together in any combination. For example, the compound of the invention inhibitor may be formulated in one dosage form and the additional agent may be formulated together in another dosage form. Any separate dosage forms may be administered at the same time or different times.
Alternatively, a composition of this invention comprises an additional agent as described herein. Each component may be present in individual compositions, combination
compositions, or in a single composition.
Exemplification of the Invention
The invention is further illustrated by the following examples, which should not be construed as further limiting. The assays used throughout the Examples are accepted. Demonstration of efficacy in these assays is predictive of efficacy in subjects.
GENERAL SYNTHESIS METHODS
All starting materials, building blocks, reagents, acids, bases, dehydrating agents, solvents, and catalysts utilized to synthesis the compounds of the present invention are either commercially available or can be produced by organic synthesis methods known to one of ordinary skill in the art (Houben-Weyl 4th Ed. 1952, Methods of Organic Synthesis, Thieme, Volume 21 ). Further, the compounds of the present invention can be produced by organic synthesis methods known to one of ordinary skill in the art as shown in the following examples.
LIST OF ABBREVIATIONS
Ac acetyl
ACN Acetonitrile
AcOEt / EtOAc Ethyl acetate
AcOH acetic acid
aq aqueous
Ar aryl
Bn benzyl
Bu butyl (nBu = n-butyl, tBu = tert-butyl)
CDI Carbonyldiimidazole
CH3CN Acetonitrile
DBU 1 ,8-Diazabicyclo[5.4.0]-undec-7-ene
Boc20 di-tert-butyl dicarbonate
DCE 1 ,2-Dichloroethane
DCM Dichloromethane
DiBAI-H Diisobutylaluminum Hydride
DIPEA N-Ethyldiisopropylamine
DMAP Dimethylaminopyridine
DMF N,N'-Dimethylformamide
DMSO Dimethylsulfoxide
El Electrospray ionisation
Et20 Diethylether
Et3N Triethylamine
Ether Diethylether
EtOAc Ethylacetate
EtOH Ethanol
FC Flash Chromatography
h hour(s)
HATU 0-(7-Azabenzotriazole-1-yl)-N,N,N'N'- tetramethyluronium hexafluorophosphate
HBTU 0-(Benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate
HCI Hydrochloric acid
HMPA Hexamethylphosphoramide
HOBt 1-Hydroxybenzotriazole
HPLC High Performance Liquid Chromatography
H20 Water
L liter(s)
LC-MS Liquid Chromatography Mass Spectrometry
LiHMDS Lithium bis(trimethylsilyl)amide
MgS04 Magnesium Sulfate
Me methyl
Mel lodomethane
MeOH Methanol
mg milligram
min minute(s)
mL milliliter
MS Mass Spectrometry
NaHC03 Sodium Bicarbonate
Na2S04 Sodium Sulfate
NH2OH hydroxylamine
Pd/C palladium on charcoal
Pd(OH)2 palladium hydroxide
PG protecting group
Ph phenyl
Ph3P triphenyl phosphine
Prep Preparative
Rf ratio of fronts
RP reverse phase
Rt Retention time
rt Room temperature
Si02 Silica gel
SOCI2 Thionyl Chloride
TBAF Tetrabutylammonium
TEA Triethylamine
TFA Trifluoroacetic acid
THF Tetrahydrofuran
TLC Thin Layer Chromatography
General Conditions:
Mass spectra were run on LC-MS systems using electrospray ionization. These were WATERS Acquity Single Quard Detector. [M+H]+ refers to mono-isotopic molecular weights.
NMR spectra were run on open access Varian 400 NMR spectrometers. Spectra were measured at 298K and were referenced using the solvent peak.
If not indicated otherwise, the analytical UPLCconditions are as follows:
Method A
Column Phenonemax Kinetix C18 Column; 2.1 mm x 50 mm; 2.6 u core size
Column Temperature 50 °C
Eluents solvent A: water with 0.1 % TFA; solvent B: CH3CN with 0.1 % TFA
Flow Rate 1.2 mL/min
Gradient 2-88% solvent B in 9.5 mins
Synthesis of LpxC inhibitors
1.1.1 Synthesis of compound 1.1.1

Step 1. Synthesis of ethyl 4-(([1,1'-biphenyl]-4-ylmethyl)amino)tetrahydro-2H- thiopyran-4-carboxylate 1 ,1 -dioxide [1.1.1a]

1.1.1a
4-( Bromomethyl)biphenyl (150 mg, 0.607 mmol) and Ethyl 4-aminotetrahydro-2H- thiopyran-4-carboxylate 1 ,1-dioxide (178 mg, 0.728 mmol) were combined in ethanol (5.0 mL) in a microwave vial, and K2C03 (839 mg, 6.07 mmol) was added. The vial was sealed and heated to 60 °C for 24 hours. The mixture was filtered and the filtrate was evaporated. The residue was purified by silica gel column chromatography (EtOAc/heptane) to give product 1.1.1a (31.1 mg, 13 % yield). LCMS (m/z): 388.3 [M+H]+
Step 2. Synthesis of 4-(([1,1'-biphenyl]-4-ylmethyl)amino)tetrahydro-2H- thiopyran-4-carboxylic acid 1,1-dioxide [1.1.1 b]

To a mixture of 1.1.1a (22 mg, 0.057 mmol) in THF (0.4 mL), MeOH (0.1 mL,) and Water (0.1 mL) was added LiOH (7.15 mg, 0.170 mmol) and the resulting mixture was stirred at room temperature overnight. The reaction mixture was acidified by addition of 1 N HCI aqueous solution until pH=2 and then diluted with EtOAc. The organic layer was separated from the water layer and concentrated. Azeotroped 2x with toluene obtaining product
1.1.01 b (20.41 mg, 100 % yield). The crude material was used in the next step with no further purification. LCMS (m/z) 360.2 [M+H]+
Step 3. Synthesis of 4-(([1,1'-biphenyl]-4-ylmethyl)amino)-N-((tetrahydro-2H- pyran-2-yl)oxy)tetrahydro-2H-thiopyran-4-carboxamide 1,1-dioxide [1.1.1c]

To a solution of 1.1.01 b (20 mg, 0.056 mmol) in DCM (1 mL) was added Et3N (0.039 ml, 0.278 mmol), EDC.HCI (16.00 mg, 0.083 mmol), HOBT (15.34 mg, 0.100 mmol) and O- (tetrahydro-2H-pyran-2-yl)hydroxylamine (13.04 mg, 0.1 1 1 mmol). The reaction mixture was stirred at room temperature for 72 hours. The reaction mixture was quenched with water and extracted with EtOAc. The organic layer was washed with water and brine, dried over magnesium sulfate, filtered and concentrated. The residue was purified by silica gel column chromatography to afford product 1.1.1c (13.4 mg, 52.5 % yield). LCMS (m/z) 459.3 [M+H]+
Step 4. Synthesis of 4-(([1,1'-biphenyl]-4-ylmethyl)amino)-N-hydroxytetrahydro- 2H-thiopyran-4-carboxamide 1 ,1-dioxide [1.1.1]

1.1.1
To a solution of 1.1.1c (13.4 mg, 0.029 mmol) in Ethanol (2 mL) was added concentrated HCI aqueous solution (0.009 mL) was added. After stirring at room
temperature for 24 hours, aother 0.1 mL of Cone. HCI added, and the solution was stirred for another 96 hours. The reaction mixture was centrifuged and supernatant was removed. The remaining solid was triturated with Et20, centrifuged again. The solid residue was dried under vacuum to give 1.1.1 (10 mg, 81 % yield) as a white powder. LCMS (m/z) 375.2
[M+H]+ 1H NMR (400 MHz, CD3OD) δ ppm 2.36 - 2.71 (m, 4 H) 2.78 - 2.97 (m, 2 H) 3.25 (m, J=3.13, 1.57 Hz, 2 H) 4.15 (s, 2 H) 7.32 - 7.40 (m, 1 H) 7.42 - 7.49 (m, 2 H) 7.50 - 7.60 (m, 2 H) 7.60 - 7.67 (m, 2 H) 7.71 - 7.76 (m, 2 H)
1.1.2 Synthesis of compound 1.1.2

Step 1. Synthesis of methyl 4-((4-bromobenzyl)amino)tetrahydro-2H-thiopyran- 4-carboxylate 1,1 -dioxide [1.1.2a]

Methyl 4-aminotetrahydro-2H-thiopyran-4-carboxylate 1 ,1-dioxide (1 g, 4.83 mmol) and 4-bromobenzaldehyde (0.893 g, 4.83 mmol) were dissolved in 1 ,2-dichloroethane (48.3 mL) and the resulting solution was stirred overnight at room temperature. To the solution was added acetic acid (0.829 mL, 14.48 mmol) and the reaction mixture was allowed to stir another 24 hours, at which time sodium triacetoxyborohydride (3.07 g, 14.48 mmol) was added and the reaction mixture was stirred for 72 hours. The reaction was quenched with water and extracted with diethyl ether. The organic layer was washed water, saturated NaHC03 aqueous solution and brine, then dried over MgS04 and concentrated to afford product 1.1.2a (1.8 g,100 % yield). The crude material was used in the next step without further purification. LCMS (m/z) 376.5 [M+H]+; 1H NMR (400 MHz, CDCI3) δ ppm 2.19 - 2.29 (m, 2 H) 2.49 - 2.60 (m, 2 H) 2.83 - 2.94 (m, 2 H) 3.41 (td, J=13.38, 3.42 Hz, 2 H) 3.53 (br. s, 2 H) 3.80 (s, 3 H) 7.18 - 7.22 (m, 2 H) 7.44 - 7.50 (m, 2 H)
Step 2. Synthesis of 4-((4-bromobenzyl)amino)tetrahydro-2H-thiopyran-4- carboxylic acid 1,1-dioxide [1.1.2b]

1.1.2b
To a solution of 1.1.2a (3.64 g, 9.67 mmol) in MeOH (55 ml.) and water (9 ml.) was added LiOH H20 (1.2 g, 29.0 mmol) and the resulting solution was stirred at room temperature for 24 hours. The mixture was then concentrated in vacuo to 1/3 of its volume, diluted with water and brought to a pH of 4 with 1.0 M HCI aqueous solution. The white precipitate was collected, washed with diethyl ether, dried on vacuum to afford 1.1.02b (2.83 g, 81 % yield). LCMS (m/z) 364.0 [M+H]+; 1H NMR (400 MHz, DMSO-d6) δ ppm 2.06 - 2.28 (m, 4 H) 2.95 (d, J=12.13 Hz, 2 H) 3.15 - 3.32 (m, 2 H) 3.56 (s, 2 H) 7.30 - 7.35 (m, 2 H) 7.48 - 7.53 (m, 2 H)
Step 3. Synthesis of 4-((4-bromobenzyl)amino)-N-((tetrahydro-2H-pyran-2- yl)oxy)tetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide [1.1.2c]

1.1.2c
EDC HCI (2.097 g, 10.94 mmol) and aza-HOBt (1.914 g, 14.06 mmol) was added to a solution of 1.1.2b (2.83 g, 7.81 mmol), 0-(tetrahydro-2H-pyran-2-yl)hydroxylamine (1.373 g, 1 1.72 mmol) and Et3N (4.12 ml, 29.7 mmol) in DMF (39.1 ml_). After stirring at room temperature for 96 hours, the reaction mixture was quenched with water and saturated aqueous NH4CI solution. The mixture was extracted with diethyl ether and DCM until no more product was present in the aqueous layer. The combined organic layers were dried (MgS04), filtered and concentrated. Purification by silica gel column chromatography (EtOAc/heptane, 50-100%) afforded 1.1.2c (3.1 g, 86 % yield) as a white powder. LCMS (m/z) 463.1 [M+H]+; 1H NMR (400 MHz, CDCI3) δ ppm 1.63 - 2.03 (m, 6 H) 2.14 - 2.36 (m, 2 H) 2.41 - 2.68 (m, 2 H) 3.07 - 3.45 (m, 4 H) 3.48 - 3.73 (m, 3 H) 3.80 - 3.99 (m, 1 H) 4.96 (br. s., 1 H) 7.17 - 7.23 (m, 2 H) 7.42 - 7.52 (m, 2 H) 9.37 (br. s., 1 H)
Step 4. Synthesis of N-hydroxy-4-(((4'-methoxy-[1,1'-biphenyl]-4- yl)methyl)amino)tetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide [1.1.2]

PdCI2(dppf).CH2CI2 (17.70 mg, 0.022 mmol) was added to a degassed solution of 1.1.2c (100 mg, 0.217 mmol), (4-methoxyphenyl) boronic acid (49.4 mg, 0.325 mmol) and Na2C03 (0.325 ml_, 0.650 mmol) in dimethoxy ether (1.1 ml_). The reaction was heated to 110 °C with microwave for 20 minutes, then was placed in a 100 °C sand bath for 1 hour. The reaction mixture was diluted with DCM and neutralized with saturated aqueous NH4CI solution. The aqueous layer was extracted with DCM. The organic was stirred with
Siliabond-DMT palladium-scavenger, then filtered and concentrated. Purification by silica gel column chromatography (EtO Ac/heptane 30-100%) afforded 4-(((4'-methoxy-[1 ,1 '-biphenyl]- 4-yl)methyl)amino)-N-((tetrahydro-2H-pyran-2-yl)oxy)tetrahydro-2H-thiopyran-4-carboxamide 1 ,1-dioxide (15.8 mg, 15 % yield). The white solid was dissolved in HCI in EtOH (1.25 M.1.7 ml_, 2.2 mmol) and stirred for 1 hour. The white precipitate was collected by filtration to afford 1.1.2 (10.6 mg, 10 % yield). LCMS (m/z) 405.2 [M+H]+ 1H NMR (400 MHz, CD3OD) δ ppm 2.31 - 2.69 (m, 4 H) 2.91 (d, J=14.48 Hz, 2 H) 3.16 - 3.24 (m, 2 H) 3.75 - 3.90 (m, 3 H) 4.15 (s, 2 H) 6.95 - 7.07 (m, 2 H) 7.44 - 7.61 (m, 4 H) 7.69 (d, J=8.22 Hz, 2 H)
1.1.3 Synthesis of compound 1.1.3

Compound 1.1.3 was prepared following the procedures described for the synthesis of 1.1.2 using (4-chloro-2-fluorophenyl)boronic acid in Step 4. LCMS (m/z) 427.1 [M+H]+ 1H NMR (400 MHz, CD3OD) ppm 2.53 (d, J=7.43 Hz, 4 H) 2.88 (m, J=13.69 Hz, 4 H) 4.17 (s, 2 H) 7.25 - 7.36 (m, 2 H) 7.45 - 7.54 (m, 1 H) 7.55 - 7.70 (m, 4 H)
1.1.4 Synthesis of compound 1.1.4

Compound 1.1.4 was prepared similarly to the synthesis for 1.1.2 changing the Suzuki coupling conditions in Step 4 as following. Pd(PPh3)4 (50.1 mg, 0.043 mmol) was added to a degassed mixture of 1.1.2c (100 mg, 0.217 mmol), 4-(4-(4,4,5,5-tetramethyl- 1 ,3,2-dioxaborolan-2-yl)benzyl)morpholine (99 mg, 0.325 mmol), Na2C03 (68.9 mg, 0.650 mmol), THF (1.1 mL) and Water (0.36 mL). The reaction mixture was heated to 70 °C for 18 hours. The reaction mixture was diluted with DCM and neutralized with saturated aqueous NH4CI solution. The aqueous layer was extracted with DCM. The combined organic layer was dried over magnesium sulfate, stirred with Siliabond DMT pd. Scavenger, filtered and dried on to silica. Purification by silica gel column chromatography (MeOH/DCM, 1-10%) afforded 4-(((4'-(morpholinomethyl)-[1 , 1 '-biphenyl]-4-yl)methyl)amino)-N-((tetrahydro-2H- pyran-2-yl)oxy)tetrahydro-2H-thiopyran-4-carboxamide 1 ,1-dioxide (23 mg, 19 % yield) 1H NMR (400 MHz, CDCI3) ppm 1.13 - 1.30 (m, 7 H) 1.64 - 1.89 (m, 6 H) 2.17 - 2.40 (m, 2 H) 2.48 (br. s., 7 H) 3.42 (br. s., 2 H) 3.55 (s, 2 H) 3.60 - 3.78 (m, 8 H) 7.39 (d, J=4.30 Hz, 2 H) 7.41 (d, J=3.91 Hz, 2 H) 7.54 (d, J=7.83 Hz, 2 H) 7.58 (d, J=8.22 Hz, 2 H). The product was dissolved in 1.25 M HCI in EtOH (3.5 mL, 4.33 mmol) and the resulting solution was stirred at room temperature for 3 hours. The precipitate was collected by filtration and washed with Et20 to afford product 1.1.4 (13.09 mg, 10 % yield) as a white solid. 1H NMR (400 MHz, CDCI3) δ ppm 3.16 - 3.28 (m, 5 H) 3.35 - 3.46 (m, 4 H) 3.75 (t, J=12.13 Hz, 2 H) 4.06 (d, J=12.13 Hz, 2 H) 4.14 (br. s., 2 H) 4.42 (s, 2 H) 7.64 (d, J=8.22 Hz, 4 H) 7.72 - 7.84 (m, 4 H) LCMS (m/z) 474.2 [M+H]+
1.1.5 Synthesis of compound 1.1.5

StepL Synthesis of (1 E,2E)-1,2-bis(2-phenylhydrazono)ethane. [1.1.5a]

1.1.5a
Phenylhydrazine (1.8 ml_, 18.49 mmol) was dissolved in MeOH (30 ml_). To this solution, a solution of oxalaldehyde (1.0 ml_, 9.25 mmol) in MeOH (10.0 ml_) was added, followed by AcOH (0.032 ml_, 0.555 mmol). The mixture was then stirred at room temperature overnight. The precipitate was collected by filtration, washed with MeOH and dried in the air to afford 1.1.5a (1.37 g, 31.1 % yield) as a pale yellow solid. LCMS (m/z) 239.1 [M+H]+
Step2. Synthesis of 2-phenyl-2H-1,2,3-triazole. [1.1.5b]

1.1.5b
1.1.5a (2.9 g, 12.17 mmol) was suspended in ethylene glycol (50 ml_) in a glass pressure vessel and Copper(ll)Triflate (0.220 g, 0.609 mmol) was added. The mixture was stirred at 180 °C for 2 hours and at room temperature for 24 hours. The reaction mixture was partitioned between water and ethyl acetate. The phases were separated and the aqueous layer was extracted with ethyl acetate. The combined organic layer was washed with water, brine and dried over sodium sulfate. The organic layer was filtered, evaporated onto silica gel and purified by silica gel column chromatography to give product 1.1.5b (1.22 g, 69.1 % yield). LCMS (m/z) 146.2 [M+H]+ 1H NMR (400 MHz, CDCI3) 8.10 (d, J = 7.83 Hz, 2H), 7.82 (s, 2H), 7.50 (t, J = 7.83 Hz, 2H), 7.32 - 7.41 (m, 1 H)
Step3. Synthesis of 2-(4-bromophenyl)-2H-1,2,3-triazole. [1.1.5c]

1.1.5c
1.1.5b (68 mg, 0.468 mmol) was dissolved in concentrated H2S04 (2 mL) and Br2 (0.024 mL, 0.468 mmol) was added, followed by Ag2S04 (175 mg, 0.56 mmol). After stirring at room temperature for 40 minutes, the reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was dried (Na2S04) and concentrated to afford 1.1.5c (40 mg, 38 % yield) as an off white solid. LCMS (m/z) 225.9 [M+H]+ 1H NMR (400 MHz, CDCI3) 7.99 (d, J = 8.61 Hz, 2H), 7.83 (s, 2H), 7.62 (d, J = 8.61 Hz, 2H)
Step 5. 2-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)-2H-1 ,2,3- triazole. [1.1.5d]

A microwave vial was charged with 1.1.5c (1.66 g, 7.41 mmol) and 1 ,4-dioxane (60 mL). KOAc (2.254 g, 22.97 mmol) and bis(pinacolato)diboron (2.258 g, 8.89 mmol) were added and the mixture was purged with N2 for 5 minutes. PdCI2(dppf).CH2CI2 adduct (0.605 g, 0.741 mmol) was then added and the reaction mixture was stirred at 68 °C for 5 hours. The mixture was diluted with EtOAc and stirred with Siliabond DMT overnight, then washed with water, brine, dried filtered and evaporated onto silica gel. Purification by flash column chromatography on silica gel (EtO Ac/Heptane, 0 to 40%) afforded product 1.1.5d (1.38 g, 68.7 % yield) as an off white solid. LCMS (m/z) 272.2 [M+H]+ 1H NMR (400 MHz, CDCI3) δ 8.10 (d, J = 8.61 Hz, 2H), 7.94 (d, J = 8.22 Hz, 2H), 7.81 - 7.86 (m, 2H), 1.34 - 1.42 (m, 12H)
Step 6. Synthesis of 4-(((4,-(2H-1,2,3-triazol-2-yl)-[1,1,-biphenyl]-4- yl)methyl)amino)-N-hydroxytetrahydro-2H-thiopyran-4-carboxamide 1,1-dioxide [1.1.5]

Compound 1.1.5 was prepared following the procedures described for the synthesis 1.1.04 using 1.1.5d in step 4.. LC/MS (m/z) 442.2 [M+H]+. 1H NMR (400 MHz, DMSO-d6) ppm 2.51 - 2.85 (m, 4 H) 2.96 - 3.23 (m, 2 H) 3.29 - 3.63 (m, 2 H) 3.70 - 4.27 (m, 2 H) 7.62 (br. s., 2 H) 7.80 (d, J=7.04 Hz, 2 H) 7.90 (d, J=8.61 Hz, 2 H) 8.02 - 8.27 (m, 4 H)
1.1.6 Synthesis of compound 1.1.6

Compound 1.1.6 was prepared following the procedures described for the synthesis 1.1.4 using 4-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)morpholine in Step 4. LC/MS (m/z) 460.2 [M+H]+. 1H NMR (400 MHz, CD3OD) δ ppm 2.34 (br. s., 2 H) 2.56 (s, 2 H) 3.04 (d, J=1.57 Hz, 2 H) 3.13 (d, J=4.70 Hz, 4 H) 3.23 - 3.30 (m, 2 H) 3.68 - 3.91 (m, 7 H) 6.98 (d, J=8.61 Hz, 2 H) 7.39 (d, J=8.22 Hz, 2 H) 7.44 - 7.49 (m, 2 H) 7.55 (d, J=7.43 Hz, 2
H)
1.1.7 Synthesis of compound 1.1.7

Compound 1.1.7 was prepared following the procedures described for the synthesis 1.1.4 using 2-(4-cyclopropylphenyl)-4, 4, 5, 5-tetramethyl-1 ,3,2-dioxaborolane in step 4.
LC/MS (m/z) 415.2 [M+H]+. 1H NMR (400 MHz, CD3OD) δ ppm 0.58 - 0.67 (m, 2 H) 0.86 - 0.96 (m, 2 H) 1.80 - 1.91 (m, 1 H) 2.28 - 2.44 (m, 4 H) 3.04 (br. s., 2 H) 3.25 - 3.34 (m, 2 H) 3.84 (br. s., 2 H) 7.06 (d, J=8.22 Hz, 2 H) 7.42 (dd, J=8.02, 3.33 Hz, 4 H) 7.56 (d, J=7.83 Hz, 2 H).
1.1.8 Synthesis of compound 1.1.8

Compound 1.1.8 was prepared following the procedures described for the synthesis 1.1.4 using naphthalen-2-ylboronic acid in step 4. LC/MS (m/z) 425.2 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ ppm 3.00 - 3.23 (m, 2 H) 3.28 - 3.54 (m, 2 H) 3.58 - 4.26 (m, 7 H) 7.44 - 7.70 (m, 4 H) 7.80 - 7.97 (m, 3 H) 8.00 (t, J=8.02 Hz, 2 H) 8.24 (s, 1 H)
1.1.9 Synthesis of compound 1.1.9

1.1.10 Synthesis of compound 1.1.10

Compound 1.1.10 was prepared following the procedures described for the synthesis 1.1.04 using 4,4,5,5-tetramethyl-2-(4-(((1 r,4r)-4-((tetrahydro-2H-pyran-2- yl)oxy)cyclohexyl)methoxy)phenyl)-1 ,3,2-dioxaborolane (ref. WO2011/73845 A1 , 201 1 ) in step 4. LC/MS (m/z) 503.2 [M+H]+. 1H NMR (400 MHz, CD3OD) δ ppm 1.02 - 1.30 (m, 4 H) 1.59 - 1.73 (m, 1 H) 1.81 - 1.98 (m, 4 H) 2.28 - 2.39 (m, 2 H) 2.47 - 2.65 (m, 2 H) 3.04 (dt, J=3.28, 1.64 Hz, 2 H) 3.25 (d, J=7.24 Hz, 2 H) 3.38 - 3.50 (m, 1 H) 3.67 - 3.90 (m, 4 H) 6.85 - 6.92 (m, 2 H) 7.39 (d, J=8.27 Hz, 2 H) 7.42 - 7.48 (m, 2 H) 7.53 (d, J=7.97 Hz, 2 H).
1.1.11 Synthesis of compound 1.1.11

Compound 1.1.11 was prepared following the procedures described for the synthesis 1.1.4 using (2-fluorophenyl)boronic acid in step 4. LC/MS (m/z) 393.2 [M+H]+. 1H NMR (400 MHz, <CD3CN>) δ ppm 2.62 (t, J=1 1.93 Hz, 2 H) 2.98 (br. s., 2 H) 3.15 - 3.29 (m, 2 H) 3.44 (d, J=1 1.74 Hz, 2 H) 4.16 (s, 2 H) 7.16 - 7.35 (m, 2 H) 7.39 - 7.48 (m, 1 H) 7.54 (td, J=7.92,
1.76 Hz, 1 H) 7.61 - 7.69 (m, 2 H) 7.74 (d, J=8.22 Hz, 2 H) 9.79 (br. s., 2 H) 1 1.71 (br. s., 1 H)
1.1.12 Synthesis of compound 1.1.12

Compound 1.1.12 was prepared following the procedures described for the synthesis 1.1.4 using pyridin-4-ylboronic acid in step 4. LC/MS (m/z) 276.2 [M+H]+, 1H NMR (400 MHz, <CD3OD>) δ ppm 2.26 - 2.53 (m, 4 H) 3.00 (d, J=12.91 Hz, 2 H) 3.35 - 3.46 (m, 3 H) 3.77 (br. s., 2 H) 7.68 (d, J=8.22 Hz, 2 H) 7.97 (d, J=8.22 Hz, 2 H) 8.31 (d, J=6.26 Hz, 2 H) 8.80 (d, J=6.65 Hz, 2 H).
1.1.13 Synthesis of 4-(((2-f luoro-[1 ,1 '-biphenyl]-4-yl)methyl)amino)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide [1.1.13]

1.1.13
Step 1. Synthesis of Methyl 4-((4-bromo-3-fluorobenzyl)amino)tetrahydro-2H- thiopyran-4-carboxylate 1,1 -dioxide [1.1.13a]

1.1.13a
4-Bromo-3-fluorobenzaldehyde (1.39 g, 6.88 mmol, 1.9 equiv) was mixed with methyl 4-aminotetrahydro-2H-thiopyran-4-carboxylate 1 ,1-dioxide (0.750 g, 3.62 mmol) and DCE (18 mL) was added followed by AcOH (1.24 mL, 21.71 mmol, 6 equiv) and Na(AcO)3BH (2.15 g, 10.13mmol, 2.8 equiv). The mixture was stirred under nitrogen at ambient temperature for 48 hours. The reaction was quenched by slowly pouring it into saturated aqueous NaHC03 solution and then extracted with EtOAc. The combined organic layers were dried over sodium sulfate, filtered and concentrated. The crude material was purified by silica gel column chromatography, (EtOAc/heptane, 0-100%) to afford Methyl 4-((4-bromo-3- fluorobenzyl)amino)tetrahydro-2H-thiopyran-4-carboxylate 1 , 1-dioxide 1.1.13a as a white solid (1.25 g, 88%) MS m/z 394.1 [M+H]+
Step 2. Synthesis of 4-((4-bromo-3-fluorobenzyl)amino)tetrahydro-2H- thiopyran-4-carboxylic acid 1,1-dioxide [1.1.13b]

1.1.13b
Methyl 4-((4-bromo-3-fluorobenzyl)amino)tetrahydro-2H-thiopyran-4-carboxylate 1 ,1- dioxide 1.1.13a (1.25 g, 3.17 mmol) was dissolved in THF/MeOH (16mL, 4/1 ) and 2.0 M LiOH aqueous solution (4.76 mL, 9.51 mmol, 3 equiv) was added. The mixture was stirred at ambient temperature for 17 hours. The volatiles were removed under reduced pressure and 6.0 N HCI aqueous solution was added until the pH was ~1. The white precipitate was collected by filtration to afford Methyl 4-((4-bromo-3-fluorobenzyl)amino)tetrahydro-2H- thiopyran-4-carboxylate 1 ,1-dioxide 1.1.13b as a white solid (1.25g, 100%). MS m/z 380.0
[M+H]+. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.66 (t, J=7.80 Hz, 1 H) 7.43 (dd, J=10.10, 1.59 Hz, 1 H) 7.20 (dd, J=8.24, 1.49 Hz, 1 H) 3.65 (s, 2 H) 3.20 - 3.33 (m, 2 H) 2.97 (d, J=12.96 Hz, 2 H) 2.16 - 2.29 (m, 4 H)
Step 3. Synthesis of 4-((4-bromo-3-fluorobenzyl)amino)-N-((tetrahydro-2H- pyran-2-yl)oxy)tetrahydro-2H-thiopyran-4-carboxamide 1,1-dioxide [1.1.13c]

1.1.13c
Methyl 4-((4-bromo-3-fluorobenzyl)amino)tetrahydro-2H-thiopyran-4-carboxylate 1 ,1- dioxide 1.1.13b (1.20 g, 3.17 mmol), 0-(tetrahydro-2H-pyran-2-yl)hydroxylamine (0.557 g, 4.76 mmol, 1.5 equiv), EDC HCI (0.851 g, 4.44 mmol, 1.4 equiv) and aza-HOBt (0.777g, 5.71 mmol, 1.8 equiv) were dissolved in DMF (16 ml_) and triethylamine (0.795 ml_, 5.71 mmol, 1.8 equiv) was added. The solution was stirred at ambient temperature for 45 hours. The solution was poured into water and extracted with EtOAc. The combined organic layers were washed with brine, dried over sodium sulfate, filtered and concentrated. The crude material was purified by silica gel column chromatography (EtOAc/heptane 0 to 100%) to afford 4-((4-bromo-3-fluorobenzyl)amino)-N-((tetrahydro-2H-pyran-2-yl)oxy)tetrahydro-2H- thiopyran-4-carboxamide 1 ,1-dioxide 1.1.13c as a white solid (1.24g, 82%). MS m/z 497.1 [M+H]+
Step 4. Synthesis of_ 4-(((2-fluoro-[1,1'-biphenyl]-4-yl)methyl)amino)-N- ((tetrahydro-2H-pyran-2-yl)oxy)tetrahydro-2H-thiopyran-4-carboxamide 1,1-dioxide
[1.1.13d]

1.1.13d
Na2C03 aqueous solution (2.0 M, 0.834ml_,1.66 mmol, 4.0 equiv) was added to a mixture of 1.1.13c (0.2 g, 0.417 mmol), phenylboronic acid (63.6 mg, 0.522 mmol, 1.25
equiv), DME (2.0 mL) and EtOH (2.0 mL). The mixture was purged with nitrogen 15 minutes and then Pd(PPh3)4 (14.4mg, 0.013 mmol, 0.03equiv) was added. The mixture was heated at 1 10 °C with microwave for 1 hour. The mixture was diluted with EtOAc, washed with water and brine, dried over sodium sulfate, filtered and concentrated. The residue was purified by silica gel column chromatography, (EtOAc/heptane, 0 to 100%) to afford 4-((4-bromo-3- fluorobenzyl)amino)-N-((tetrahydro-2H-pyran-2-yl)oxy)tetrahydro-2H-thiopyran-4- carboxamide 1 ,1-dioxide 1.1.13d as a yellow solid. (14mg, 7%). MS m/z 477.3 [M+H]+
Step 5. Synthesis of 4-(((2-fluoro-[1,1'-biphenyl]-4-yl)methyl)amino)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide [1.1.13]

1.1.13
4-((4-bromo-3-fluorobenzyl)amino)-N-((tetrahydro-2H-pyran-2-yl)oxy)tetrahydro-2H- thiopyran-4-carboxamide 1 ,1-dioxide 1.1.13c (14 mg, 0.029 mmol) was dissolved in
DCM/MeOH (0.294 mL,4/1 ) and 4.0 M HCI in dioxane (0.0734mL, 0.294 mmol) was added. The reaction was stirred at ambient temperature for 15 minutes. The solvents were removed under reduced pressure. The remaining material was purified by reverse phase HPLC to afford 1.1.13 as a white powder (4mg, 26%). MS m/z 393.1 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.46 - 7.58 (m, 5 H) 7.37 - 7.44 (m, 2 H) 7.32 (d, J=7.83 Hz, 1 H) 3.26 - 3.38 (m, 3 H) 2.95 - 3.05 (m, 2 H) 2.16 - 2.30 (m, 3 H) Note: Benzylic protons were buried under the water peak.
1.1.14 Synthesis of 4-(((2-fluoro-4'-methoxy-[1,1,-biphenyl]-4-yl)methyl)amino)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1,1-dioxide [1.1.14 ]

1.1.14
Compound 1.1.14 was prepared following the procedures described for the synthesis of 1.1.13 using (4-methoxyphenyl)boronic acid in Step 4. (The solvent used in Step 4 was changed to THF). MS m/z 423.1 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ ppm 10.91 (br. s.,
1 H) 7.49 (dd, J=8.75, 1.52 Hz, 3 H) 7.26 - 7.42 (m, 2 H) 6.99 - 7.08 (m, 2 H) 3.78 - 3.83 (m, 5 H) 3.27 - 3.41 (m, 3 H) 3.05 (br. s., 2 H) 2.19 - 2.40 (m, 3 H) Note: Benzylic protons buried under water peak.
1.1.15 Synthesis of 4-(((2-fluoro-4'-methoxy-[1,1,-biphenyl]-4-yl)methyl)am
hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide [1.1.15]

1.1.15
Compound 1.1.15 was prepared following the procedures described for the synthesis of 1.1.13 using (2-fluorophenyl)boronic acid in Step 4. (The solvent used in Step 4 was changed to THF) MS m/z 41 1.0 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.40 - 7.54 (m, 4 H) 7.29 - 7.38 (m, 3 H) 3.34 (t, J=12.13 Hz, 3 H) 3.03 (br. s., 2 H) 2.32 (d, J=1.86 Hz, 3 H) Note: Benzylic protons buried under water peak
1.1.16 Synthesis of 4-(((4'-chloro-2,2'-difluoro-[1,1,-biphenyl]-4-yl)methyl)am
hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide [1.1.16]

1.1.16
Compound 1.1.16 was prepared following the procedures described for the synthesis of 1.1.13 using (2-fluorophenyl)boronic acid in Step 4. (The solvent used in Step 4 was changed to THF). MS m/z 445.0 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ ppm 10.81 (br. s., 1 H) 7.59 (dd, J=9.95, 2.03 Hz, 1 H) 7.41 - 7.53 (m, 4 H) 7.35 (d, J=7.58 Hz, 1 H) 3.33 (br. s., 3 H) 3.02 (br. s., 2 H) 2.18 - 2.33 (m, 3 H) Note: Benzylic protons buried under water peak
1.1.17 Synthesis of 4-(((2-fluoro-4,-(2H-1,2,3-triazol-2-yl)-[1,1,-biphenyl]-4- yl)methyl)amino)-N-hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide
[1.1.17]

1.1.17
Compound 1.1.17 was prepared following the procedures described for the synthesis of 1.1.13 using (2-(4-(4,4, 5, 5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)-2H-1 ,2,3-triazole (prepared as described in Example 1.1.05d) in Step 4 (Step 4 was changed to heating at 80C for 20h in a sand bath using THF as solvent). MS m/z 460.2 [M+H]+. 1H NMR (400 MHz, DMSO-d6) 5 ppm 10.61 - 1 1.07 (m, 1 H) 8.06 - 8.12 (m, 4 H) 7.70 (dd, J=8.73, 1.44 Hz, 2 H) 7.55 (t, J=7.75 Hz, 1 H) 7.27 - 7.42 (m, 2 H) 3.28 (t, J=1 1.22 Hz, 3 H) 2.98 (br. s., 2 H) 2.14 - 2.29 (m, 3 H) Note: Benzylic protons buried under water peak
1.1.18 Synthesis of N-hydroxy^-^-methyl^'^H-l^^-triazol^-y -t r-biphenyl]^- yl)methyl)amino)tetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide [1.1.18]

1.1.18
Compound 1.1.18 was prepared following the procedures described for the synthesis of 1.1.13 using 2-(4-(4,4, 5, 5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)-2H-1 ,2,3-triazole (prepared as described in Example 1.1.05d) in Step 4. (Step 4 was changed to heating at 80C for 20h in a sand bath using THF as solvent). MS m/z 456.3 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.16 (s, 2 H) 8.10 (d, J=8.66 Hz, 2 H) 7.54 (d, J=8.75 Hz, 2 H) 7.25 - 7.42 (m, 3 H) 3.30 - 3.45 (m, 3 H) 2.98 - 3.15 (m, 2 H) 2.31 - 2.41 (m, 3 H) 2.30 (s, 3 H) Note: Benzylic protons buried under water peak
1.1.19 Synthesis of 4-(((2-chloro-2'-fluoro-4'-methoxy-[1,1,-biphenyl]-4- yl)methyl)amino)-N-hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide
[1.1.19]

1.1.19
Compound 1.1.19 was prepared following the procedures described for the synthesis of 1.1.13 using (4-chloro-2-fluorophenyl)boronic acid in Step 4. (Step 4 was changed to heating at 80C for 18h in a sand bath using THF as solvent). MS m/z 441.2 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.55 (dd, J=9.76, 1.98 Hz, 1 H) 7.30 - 7.42 (m, 4 H) 7.19 - 7.28 (m, 1 H) 3.36 (br. s., 3 H) 3.07 (br. s., 2 H) 2.25 - 2.42 (m, 3 H) 2.14 (s, 3 H) Note: Benzylic protons buried under water peak
1.1.20 Synthesis of 4-(((2'-fluoro-4,-methoxy-2-methyl-[1,1'-biphenyl]-4- yl)methyl)amino)-N-hydroxytetrahydro-2H-thiopyran-4-carboxamide 1,1- dioxidedioxide [1.1.20]

1.1.20
Compound 1.1.20 was prepared following the procedures described for the synthesis of 1.1.13 using (2-fluoro-4-methoxyphenyl)boronic acid in Step 4. (Step 4 was changed to heating at 80C for 17h in a sand bath using THF as solvent). MS m/z 437.2 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.30 - 7.41 (m, 3 H) 7.20 (t, J=8.73 Hz, 3 H) 6.84 - 6.96 (m, 3 H) 3.82 (s, 5 H) 3.33 - 3.44 (m, 3 H) 3.03 - 3.14 (m, 2 H) 2.29 - 2.45 (m, 3 H) 2.15 (s, 3 H) Note: Benzylic protons were buried under the water peak
1.1.21 Synthesis of 4-(((4'-cyclopropyl-2-methyl-[1,1,-biphenyl]-4-yl)methyl)amino)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide [1.1.21 ]

1.1.21
Compound 1.1.21 was prepared following the procedures described for the synthesis of 1.1.13 using (4-cyclopropylphenyl)boronic acid in Step 4. (Step 4 was changed to heating at 80C for 17h in a sand bath using THF as solvent). MS m/z 429.3 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.27 - 7.36 (m, 3 H) 7.18 - 7.23 (m, 4 H) 7.12 - 7.16 (m, 3 H) 3.36 (d, J=9.24 Hz, 3 H) 3.08 (br. s., 2 H) 2.30 - 2.45 (m, 3 H) 2.24 (s, 3 H) 1.96 (tt, J=8.38, 5.03 Hz, 1 H) 0.95 - 1.02 (m, 2 H) 0.67 - 0.74 (m, 2 H) Note: Benzylic protons were buried under the water peak
1.1.22 Synthesis of 4 4-(((2-chloro-2'-fluoro-4'-methoxy-[1,1,-biphenyl]-4- yl)methyl)amino)-N-hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide
[1.1.22]

1.1.22
Compound 1.1.22 was prepared following the procedures described for the synthesis of 1.1.13 using (2-fluoro-4-methoxyphenyl)boronic acid in Step 4. (Step 4 was changed to heating at 80C for 17h in a sand bath using THF as solvent). MS m/z 457.1 [M+H]+. 1H NMR (400 MHz, DMSO-d6) ppm 10.99 (br. s., 1 H) 7.66 (s, 1 H) 7.36 - 7.51 (m, 2 H) 7.26 (t, J=8.66 Hz, 1 H) 6.84 - 7.01 (m, 2 H) 3.83 (s, 3 H) 3.36 (t, J=1 1.42 Hz, 3 H) 3.06 (d, J=10.91 Hz, 2 H) 2.32 (dd, J=3.69, 1.83 Hz, 3 H) Note: Benzylic protons were buried under the water peak
1.1.23 Synthesis of compound 1.1.23

Step 1. Synthesis of 4-(phenylethynyl)benzaldehyde [1.1.23a]

1.1.23a
To a degassed solution of ethynylbenzene (0.3 ml_, 2.73 mmol), 4-iodobenzaldehyde (761 mg, 3.28 mmol), and Et3N (0.757 mL, 5.46 mmol) in THF (Volume: 21.6 ml_),
Pd(PPh3)2CI2 (96 mg, 0.137 mmol) and Cul (36.4 mg, 0.191 mmol) were added. After stirring at room temperature overnight, the reaction mixture was concentrated on to silica gel and purified by silica gel column chromatography (EtOAc/heptane, 0-50%). Fractions containing product were collected. Pale yellow crystals crashed out, which were washed with pentane and heptane to afford 4-(phenylethynyl)benzaldehyde [1.1.23a] (419 mg, 74.4 % yield). 1H NMR (400 MHz, CDCI3) δ ppm 7.30 - 7.44 (m, 3 H) 7.45 - 7.59 (m, 2 H) 7.62 - 7.73 (m, 2 H) 7.76 - 7.98 (m, 2 H) 10.02 (s, 1 H)
Step 2. Synthesis of methyl 4-((4-(phenylethynyl)benzyl)amino)tetrahydro-2H- thiopyran-4-carboxylate 1,1 -dioxide [1.1.23b]

Compound 1.1.23b was prepared following the procedures described for the synthesis 1.1. 2a using 1.1.23a in StepL 1H NMR (400 MHz,CDCI3) δ ppm 2.26 (d, J=14.48
Hz, 2 H) 2.46 - 2.65 (m, 2 H) 2.89 (d, J=12.91 Hz, 2 H) 3.34 - 3.52 (m, 2 H) 3.59 (s, 2 H) 3.80 (s, 3 H) 7.28 - 7.40 (m, 5 H) 7.47 - 7.58 (m, 4 H).
Step 3. Synthesis of 4-((4-(phenylethynyl)benzyl)amino)tetrahydro-2H- thiopyran-4-carboxylic acid 1,1 -dioxide [1.1.23c]

Compound 1.1.23c was prepared following the procedures described for the synthesis 1.1.2b in Step 2. LC/MS (m/z) 384.2 [M+H]+.
Step 4. Synthesis of 4-((4-(phenylethynyl)benzyl)amino)-N-((tetrahydro-2H- pyran-2-yl)oxy)tetrahydro-2H-thiopyran-4-carboxamide 1,1 -dioxide [1.1.23d]

Compound 1.1.23d was prepared following the procedures described for the synthesis 1.1.2c in Step 3. LC/MS (m/z) 384.2 [M+H]+.
Step 5. Synthesis of N-hydroxy-4-((4-(phenylethynyl)benzyl)amino)tetrahydro- 2H-thiopyran-4-carboxamide 1,1 -dioxide [1.1.23]

Compound 1.1.23d (148 mg) was dissolved in 4 M HCI in dioxane and the solution was stirred at room temperature of 1 hour, during which time, white precipitated crashed out.
The precipitate was collected by filtration to afford 1.1.23 (71 mg, 41.9 % yield). LC/MS (m/z) 399.1 [M+H]+. 1H NMR (400 MHz, CD3OD) δ ppm 2.53 (br. s., 2 H) 2.88 (d, J=12.13 Hz, 2 H) 3.16 - 3.29 (m, 4 H) 4.14 (s, 2 H) 7.32 - 7.42 (m, 3 H) 7.46 - 7.55 (m, 4 H) 7.57 - 7.68 (m, 2 H).
1.1.24 Synthesis of 4-((4-(cyclopropylethynyl)benzyl)amino)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1,1 -dioxide 1.1.24]

Compound 1.1.24 was prepared following the procedures described for the synthesis 1.1.23. 1H NMR (CD3OD): 7.21-7.63 (m, 4H), 4.10 (br. s., 2H), 3.19 (d, J=1.6 Hz, 4H), 2.88 (br. s., 2H), 2.54 (br. s., 2H), 1.33-1.70 (m, 1 H), 0.84-1.01 (m, 2H), 0.66-0.81 (m, 2H). LCMS m/z 363.3 [M+H]+
1.2.1 Synthesis of A-hydroxy-4-(((5-phenylisoxazol-3-yl)methyl)amino)tetrahydro-2H- thiopyran-4-carboxamide 1,1 -dioxide [1.2.1]

Step 1. Synthesis of methyl 4-(((5-phenylisoxazol-3-yl)methyl)amino)tetrahydro- 2H-thiopyran-4-carboxylate 1,1-dioxide. [1.2.1a]

Step 2. Synthesis of 4-(((5-phenylisoxazol-3-yl)methyl)amino)tetrahydro-2H- thiopyran-4-carboxylic acid 1 ,1-dioxide. [1.2.1 b]

To a solution of methyl 4-(((5-phenylisoxazol-3-yl)methyl)amino)tetrahydro- 2/-/-thiopyran-4-carboxylate 1 ,1-dioxide (192 mg, 0.53 mmol, 1 equiv) in THF/methanol/water (4/1/1 , 2.6 mL) was added LiOH H20 as a solid (66.4 mg, 1.58 mmol, 3.0 equiv). After stirring at room temperature overnight, the reaction mixture was concentrated in vacuo and acidified to pH 1 by addition of 1 N HCI aqueous solution. The white precipitate was filtered and rinsed with ether to afford 4-(((5-phenylisoxazol-3-yl)methyl)amino)tetrahydro-2/-/- thiopyran-4-carboxylic acid 1 ,1-dioxide (144 mg, 78%). MS m/z 351.2 [M+H]+.
Step 3. Synthesis of 4-(((5-phenylisoxazol-3-yl)methyl)amino)-A -((tetrahydro- 2H-pyran-2-yl)oxy)tetrahydro-2H-thiopyran-4-carboxamide 1,1-dioxide. [1.2.1c]

To a solution of 4-(((5-phenylisoxazol-3-yl)methyl)amino)tetrahydro-2/-/-thiopyran-4- carboxylic acid 1 ,1-dioxide (184 mg, 0.53 mmol, 1 equiv) in DMF (2.1 mL) was added O- (tetrahydro-2/-/-pyran-2-yl)hydroxylamine (92 mg, 0.79 mmol, 1.5 equiv) followed by
triethylamine (0.28 mL, 1.99 mmol, 3.8 equiv). After stirring at room temperature for 5 minutes, EDC HCI (140 mg, 0.74 mmol, 1.5 equiv) was added followed by HOAt (129 mg, 0.95 mmol, 1.8 equiv). After stirring at room temperature overnight, the reaction mixture was diluted with saturated solution ammonium chloride solution and dichloromethane. Aqueous layer was extracted with dichloromethane. Combined organic layer was dried with Na2S04, filtered and concentrated in vacuo. The resulting residue was purified by silica gel column chromatorgraphy (acetone/heptane) to afford 4-(((5-phenylisoxazol-3-yl)methyl)amino)-/V- ((tetrahydro-2/-/-pyran-2-yl)oxy)tetrahydro-2/-/-thiopyran-4-carboxamide 1 , 1-dioxide (159 mg, 86%). MS m/z 450.2 [M+H]+.
Step 4. Synthesis of A -hydroxy-4-(((5-phenylisoxazol-3- yl)methyl)amino)tetrahydro-2H-thiopyran-4-carboxamide 1,1-dioxide. [1.2.1]

To a solution of 4-(((5-phenylisoxazol-3-yl)methyl)amino)-A/-((tetrahydro-2/-/-pyran-2- yl)oxy)tetrahydro-2H-thiopyran-4-carboxamide 1 ,1-dioxide (159 mg, 0.35 mmol, 1 equiv) in dichloromethane/methanol (4/1 , 1.8 mL) was added HCI in dioxane (4M, 0.88 mL, 10 equiv). After stirring at room temperature for 1 h, the white precipitate was filtered to afford N- hydroxy-4-(((5-phenylisoxazol-3-yl)methyl)amino)tetrahydro-2/-/-thiopyran-4-carboxamide 1 ,1-dioxide (84 mg, 58%). MS m/z 366.1 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 1 1.01 (br- s, 1 H), 7.86-7.84 (m, 2H), 7.56-7.48 (m, 3H), 7.17 (s, 1 H), 3.82 (s, 2H), 3.38-3.32 (m, 2H), 3.01-2.98 (m, 2H), 2.44-2.31 (m, 4H).
1.2.2 Synthesis of A -hydroxy-4-(((3-phenylisoxazol-5-yl)methyl)amino)tetrahydro-2H- thiopyran-4-carboxamide 1,1-dioxide [1.2.2]

N-hydroxy-4-(((3-phenylisoxazol-5-yl)methyl)amino)tetrahydro^
carboxamide 1 , 1 -dioxide was prepared following the procedures described for the synthesis of 1.2.1 using 3-phenylisoxazole-5-carbaldehyde in step 1 . MS m/z 366.1 [M+H]+. 1 H NMR (400 MHz, DMSO-d6) δ 10.98 (br-s, NH), 7.86 - 7.83 (m, 2H), 7.51 - 7.49 (m, 3H), 7.09 (s, 1 H), 3.92 (s, 2H), 3.37-3.33 (m. , 2H), 3.01 - 2.97 (m, 2H), 2.39-2.25 (m. , 4H).
1.2.3 Synthesis of 4-(((3-(4-chloro-2-fluorophenyl)isoxazol-5-yl)methyl)amino)-A - hydrox -carboxamide 1 ,1 -dioxide [1.2.3]

Step 1. Synthesis of (Z)-4-chloro-2-fluoro-A -hydroxybenzimidoyl chloride.
[1.2.3a]

1.2.3a
To a solution of (E)-4-chloro-2-fluorobenzaldehyde oxime (262 mg, 1 .51 mmol, 1 equiv) in THF (2.9 mL) was added NCS (302 mg, 2.26 mmol, 1 .5 equiv) followed by pyridine ( 12 L, 0.15 mmol, 0.1 equiv). The mixture was stirred at 48 °C for 1 hour. The crude mixture was continued to the next step.
Step 2. Synthesis of (3-(4-chloro-2-fluorophenyl)isoxazol-5-yl)methanol. [1.2.3b]

To the reaction mixture from step 1 was added propargyl alcohol (0.22 mL, 7.55 mmol, 5 equiv) followed by triethylamine (1.05 mL, 7.55 mmol, 5 equiv). After heated at reflux for 1 hour, the reaction mixture was cooled to room temperature and diluted with water and ethyl acetate. Aqueous layer was extracted with ethyl acetate. Combined organic layer was washed with brine, dried with Na2S04, filtered and concentrated in vacuo. The resulting residue was purified by silica gel column chromatography (acetone/heptane) to afford (3-(4- chloro-2-fluorophenyl)isoxazol-5-yl)methanol (222 mg, 65% yield). MS m/z 228 [M+H]+.
Step 3. Synthesis of 3-(4-chloro-2-fluorophenyl)isoxazole-5-carbaldehyde.
[1.2.3c]

To a cooled solution of oxalyl chloride (0.19 mL, 2.2 mmol, 5.0 equiv) in dichloromethane (4.4 mL) at -78 °C was added DMSO (0.47 mL, 6.6 mmol, 15 equiv) dropwise. After stirring at -78 °C for 15 minutes, a solution of (3-(4-chloro-2- fluorophenyl)isoxazol-5-yl)methanol (100 mg, 0.44 mmol, 1 equiv) in dichloromethane (4.4 mL) was added to the reaction mixture followed by triethylamine (0.43 mL, 3.1 mmol, 7.0 equiv). After stirring at room temperature for 15 min, the reaction mixture was diluted with dichloromethane, washed with water, saturated sodium bicarbonate solution, brine, dried with Na2S04, filtered and concentrated in vacuo. The resulting residue was purified by silica gel column chromatography (acetone/heptane) to afford 3-(4-chloro-2- fluorophenyl)isoxazole-5-carbaldehyde (66 mg, 66% yield). MS m/z 226.0 [M+H]+.
Step 4. Synthesis of 4-(((3-chloro-2-fluorophenyl)isoxazol-5-yl)methyl)amino)- A -hydroxytetrahydro-2H-thiopyran-4-carboxamide 1,1 -dioxide. [1.2.3]

4-(((3-chloro-2-fluorophenyl)isoxazol-5-yl)methyl)amino)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1 , 1 -dioxide was prepared following the procedures described for the synthesis of 1.2.1 using 3-(4-chloro-2-fluorophenyl)isoxazole-5-carbaldehyde in step 1 . HRMS m/z 418.0640 [M+H]+. 1H NMR (400 MHz, DMSO-d6) 10.72 (br-s., 1 H), 7.91 (t, J = 8.27 Hz, 1 H), 7.67 (dd, J = 2.05, 10.81 Hz, 1 H), 7.45 (dd, J = 2.20, 8.41 Hz, 1 H), 6.91 (d, J = 2.93 Hz, 1 H), 3.78 (s, 1 H), 3.22 - 3.29 (m, 2H), 2.93-2.96 (m, 2H), 2.14 - 2.31 (m, 4H).
1.2.4 Synthesis of 4-(((5-(4-chloro-2-fluorophenyl)isoxazol-3-yl)methyl)amino)-A - hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide [1.2.4]

Step 1. Synthesis of ethyl 4-(4-chloro-2-fluorophenyl)-2,4-dioxobutanoate.
[1.2.4a]

1.2.4a
To a solution of LHMDS (1 .0 M in THF, 37.7 mL, 37.7 mmol) in ether (45 mL) at -78 °C was added a solution of 1 -(4-chloro-2-fluorophenyl)ethanone (5.0 g, 29 mmol, 1 equiv) in ether (15 mL). After stirring at -78 °C for 30 min, diethyl oxalate was added, and the reaction mixture was stirred at room temperature for 1 hour. The reaction mixture was
quenched with saturated aqueous NH4CI solution. The precipitate was collected by filtration and azeotroped with toluene to afford ethyl 4-(4-chloro-2-fluorophenyl)-2,4-dioxobutanoate which was used in the next step without further purification. MS m/z 273.1 [M+H]+.
Step 2. Synthesis of ethyl 5-(4-chloro-2-fluorophenyl)isoxazole-3-carboxylate.
[1.2.4b]

To a solution of ethyl 4-(4-chloro-2-fluorophenyl)-2,4-dioxobutanoate (7.9 g, 29 mmol, 1 equiv) in ethanol (145 mL) was added NH2OH HCI (6.7 g, 96 mmol, 3.3 equiv) followed by TsOH H20 (5.5 g, 29 mmol, 1 equiv). After refluxing overnight, the reaction mixture was cooled to room temperature and concentrated in vacuo. The resulting residue was partitioned between EtOAc and water. The aqueous layer was extracted with ethyl acetate. The combined organic layers were dried over Na2S04, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (EtOAc/heptane) to afford ethyl 5-(4-chloro-2-fluorophenyl)isoxazole-3-carboxylate (5.9 g, 76% yield). MS m/z 270.0 [M+H]+.
Step 3. Synthesis of (5-(4-chloro-2-fluorophenyl)isoxazol-3-yl)methanol. [1.2.4c]

To a solution of ethyl 5-(4-chloro-2-fluorophenyl)isoxazole-3-carboxylate (5.9 g, 22 mmol, 1 equiv) in THF/MeOH (10/1 , 1 10 mL) at 0 °C was added NaBH4 (2.5 g, 66 mmol, 3.0 equiv). After stirring at room temperature for 1 hour, the reaction mixture was quenched slowly with water, concentrated in vacuo and extracted with ethyl acetate. The combined organic layers were dried with Na2S04, filtered and concentrated in vacuo. The resulting residue was purified by silica gel column chromatography (acetone/heptane) to afford (5-(4- chloro-2-fluorophenyl)isoxazol-3-yl)methanol (4.3 g, 85% yield). MS m/z 228.1 [M+H]+.
Step 4. Synthesis of 5-(4-chloro-2-fluorophenyl)isoxazole-3-carbaldehyde.
[1.2.4d]

To a solution of oxalyl chloride (1 1.6 mL, 132 mmol, 7.0 equiv) in dichloromethane (94 mL) at -78 °C was added DMSO (26.8 mL, 378 mmol, 20 equiv) dropwise. After stirring at -78 °C for 15 min, a solution of (5-(4-chloro-2-fluorophenyl)isoxazol-3-yl)methanol (4.3 g, 18.9 mmol, 1 equiv) in dichloromethane (94 mL) was added to the reaction mixture followed by triethylamine (26 mL, 189 mmol, 10 equiv). After stirring at room temperature for 15 minutes, the reaction mixture was diluted with dichloromethane, washed with water, saturated aqueous NaHC03 solution, brine, dried over Na2S04, filtered, concentrated in vacuo. The resulting residue was azeotroped with toluene to afford 5-(4-chloro-2- fluorophenyl)isoxazole-3-carbaldehyde. The crude material was used in the next step without further purification. MS m/z 226.1 [M+H]+.
Step 5. Synthesis of 4-(((5-(4-chloro-2-fluorophenyl)isoxazol-3- yl)methyl)amino)-A -hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide.
[1.2.4]

4-(((5-(4-chloro-2-fluorophenyl)isoxazol-3-yl)methyl)amino)-/V-hydroxytetrahydro-2/-/- thiopyran-4-carboxamide 1 ,1 -dioxide was prepared following the procedures described for the synthesis of 1.2.1 using 5-(4-chloro-2-fluorophenyl)isoxazole-3-carbaldehyde in step 1. MS m/z (M+1 ) 418.0646 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.80 (br-s., 1 H), 7.90 (t, J = 8.31 Hz, 1 H), 7.68 (dd, J = 1.96, 10.91 Hz, 1 H), 7.44 (dd, J = 1.96, 8.51 Hz, 1 H), 7.03 (d, J = 3.23 Hz, 1 H), 3.69 (br-s., 2H), 3.26-3.20 (m, 2H), 2.90-2.94 (m, 2H), 2.14-2.28 (m, 4H).
1.2.6 Synthesis of 4-(((5-(2-fluoro-4-methoxyphenyl)isoxazol-3-yl)methyl)am hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide [1.2.6]

4-(((5-(2-fluoro-4-methoxyphenyl)isoxazol-3-yl)methyl)amino)-N-hydroxytetrahyd 2/-/-thiopyran-4-carboxamide 1 ,1-dioxide was prepared following the procedures described for the synthesis of 1.2.1 using 1-(2-fluoro-4-methoxyphenyl)ethanone in step 1. MS m/z 414.1 135 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.91 (br-s., 1 H), 7.78 (t, J = 8.78 Hz, 1 H), 7.03 (dd, J = 2.40, 13.16 Hz, 1 H), 6.87 - 6.96 (m, 2H), 3.79 (s, 5H), 3.24 - 3.29 (m, 2H), 2.93-2.96 (m, 2H), 2.23-2.31 (m, 4H).
1.2.8. Synthesis of 4-(((5-(4-chloro-2-fluorophenyl)-4-fluoroisoxazol-3- yl)methyl)amino)-N-hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide [1.2.8]

Step 1. Synthesis of Ethyl 5-(4-chloro-2-fluorophenyl)-4-fluoroisoxazole-3- carboxylate [1.2.8a]

Saturated brine (10 mL) was added the mixture and the mixture was extracted with DCM. The organic layers were combined and concentrated. The residue was purified by silica gel column chromatography (EtO Ac/heptane) to give prodct (127 mg), which contained unreacted starting material. This mixture was continued to the next step. MS m/z 288.0
[M+H]+.
Step 2. Synthesis of (5-(4-chloro-2-fluorophenyl)-4-fluoroisoxazol-3-yl)methanol
[1.2.8b]

Sodium borohydride (50.1 mg, 1.325 mmol) was added to a solution of 1.2.8c (127 mg) in Methanol/THF (2 mL/0.2 mL) at 0 oC and the mixture was stirred at this temperature for 1 hour. Water (5 mL) was added and mixture was extracted with DCM. The orgnaic layers were combined and concentrated. The residue was purified by silica gel column chromatography (EtOAc/heptane) to give product 1.2.8b (36 mg,13.3 % yield over two steps).
Step 3. Synthesis of 4-(((5-(4-chloro-2-fluorophenyl)-4-fluoroisoxazol-3- yl)methyl)amino)-N-hydroxytetrahydro-2H-thiopyran-4-carboxamide 1,1-dioxide [1.2.8]

Compound 1.2.8 was prepared following the procedures described for the synthesis of 1.2.4. HRMS m/z 436.0541 [M+H]+. 1H NMR (400 MHz, CD3OD) 7.77 (t, J=8.02 Hz, 1 H) 7.36 - 7.53 (m, 2 H) 3.87 (s, 2 H) 3.33 - 3.51 (m, 2 H) 2.96 (s, 1 H) 2.99 (s, 1 H) 2.37 - 2.49 (m, 2 H) 2.28 - 2.37 (m, 2 H)
1.2.9 Synthesis of 4-(((5-(4-(2H-1,2,3-triazol-2-yl)phenyl)isoxazol-3-yl)methyl)amino)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide [1.2.9]

Step 1. Synthesis of 1-(4-(2H-1,2,3-triazol-2-yl)phenyl)ethanone

An oven dried round bottom flask was charged with [Pd2(dba)3] (89 mg, 0.097 mmol, 0.75 mol%) and Me4tBuXPhos (1 12 mg, 0.233 mmol, 1.8 mol%), evacuated and backfilled with N2 (three times). Toluene (6.47 ml_) was added and heated to 120 °C for 3 minutes. K3PO4 (5.5 g, 25.9 mmol, 2.0 equiv), 4-chloroacetophenone (2.0 g, 12.94 mmol, 1 equiv), and triazole (1.1 g, 15.52 mmol, 1.2 equiv) were added and heated to 120 °C for 5 hours. The reaction mixture was cooled to room temperature, diluted with ethyl acetate, washed with brine, dried over Na2S04, concentrated in vacuo. The resulting residue was purified by silica gel column chromatography to give 1-(4-(2H-1 ,2,3-triazol-2-yl)phenyl)ethanone (1.4 g, 56 % yield). MS m/z 188.4 [M+H]+.
Step 2. Synthesis of 4-(((5-(4-(2H-1,2,3-triazol-2-yl)phenyl)isoxazol-3- yl)methyl)amino)-N-hydroxytetrahydro-2H-thiopyran-4-carboxamide 1,1 -dioxide [1.2.9]

4-(((5-(4-(2H-1 ,2,3-triazol-2-yl)phenyl)isoxazol-3-yl)methyl)amino)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide was prepared following the procedures described for the synthesis of 1.2.4 using 1.2.9a in step 1. HRMS m/z 433.1296 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.14 (d., 4H), 8.01 (d, J = 8.61 Hz, 2H), 7.19 (br, s, 1 H), 3.73 (br, 2H), 3.29 (br, s, 2H), 2.94 (d, J = 13.69 Hz, 2H), 2.26 (m, 4H).
1.2.10. Synthesis of 4-(((4-chloro-5-(4-chloro-2-fluorophenyl)isoxazol-3- yl)methyl)amino)-N-hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide
[1.2.10]

Step 1. Synthesis of Ethyl 4-chloro-5-(4-chloro-2-fluorophenyl)isoxazole-3- carboxylate [1.2.10a]

N-Cholorosuccinimide (636 mg, 4.76 mmol) was added to a solution of Ethyl 5-(4-chloro-2-fluorophenyl)isoxazole-3-carboxylate (214 mg, 0.794 mmol) in acetic acid (3.9 mL) and the mixture was heated at 95 °C for 17 hours. After cooling to room temperature,
the reaction mixture was quenched by adding water (30 mL) and the mixture was extracted with DCM. The organic layers were combined and concentrated. The residue was purified by silica gel column chromatography (EtOAc/heptane) to afford product 1.2.10a (217 mg, 90% yield). MS m/z 304.0 [M+H]+.
Step 2. Synthesis of 4-(((4-chloro-5-(4-chloro-2-fluorophenyl)isoxazol-3- yl)methyl)amino)-N-hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide
[1.2.10]

Compound 1.2.10 was prepared following the procedures described for the synthesis of 1.2.4. HRMS m/z 452.0240 [M+H]+. 1H NMR (400 MHz, CD3OD) 7.68 (t, J=8.02 Hz, 1 H) 7.24 - 7.47 (m, 2 H) 3.78 (s, 2 H) 3.26 - 3.44 (m, 2 H) 2.89 (s, 1 H) 2.92 (s, 1 H) 2.31 - 2.45 (m, 2 H) 2.15 - 2.31 (m, 2 H)
1.2.11. Synthesis of 4-(((5-(4-(difluoromethoxy)phenyl)isoxazol-3-yl)methyl)amino)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide [1.2.11]

Step 1. Synthesis of tributyl(ethynyl)stannane [1.2.11a]
-= SnBu3
1.2.11a
A flask was charged with tributylchlorostannane (37.8 g, 1 16.1 mmol, 1.0 equiv) and THF (50 ml_). Ethynylmagnesium bromide (15 g, 1 16.1 mmol, 1.0 equiv) was added dropwise at -70 °C and the resulting solution was stirred at -70 °C for 1 hour and at room temperature for 3 hours. The reaction mixture was quenched with saturated aqueous ammonium chloride solution and extracted with EtOAc. The organic layer was washed with brine, dried over sodium sulfate and concentrated. The residue was purified by silica gel column chromatography (100 % Hexane) to afford product 1.2.11a (1 1 g, 30.1 % yield). 1H NMR (400 MHz, CDCI3) δ 4.50 (dq, J = 20.1 , 7.1 Hz, 2H), 1.84 - 1.50 (m, 5H), 1.51 - 1.16 (m, 13H), 1.04 - 0.77 (m, 7H).
ethyl 5-(tributylstannyl)isoxazole-3-carboxylate [1.2.11 b]

1.2.11 b
1.2.11a (4 g, 12.7 mmol, 1.0 equiv) and ethyl (E)-2-chloro-2-(hydroxyimino)acetate (1.92 g, 12.7 mmol, 1.0 equiv) were dissoved in diethyl ether (40 ml_). TEA (6.41 g, 63.5 mmol, 5.0 equiv) was added dropwise and the reaction mixture was stirred at room temperature for 4 hours. The reaction mixture was quenched with water and extracted with EtOAc. The organic layer was washed with brine, dried over sodium sulfate and concentrated. The residue was purified by silica gel column chromatography (10-15 % EtOAc in Hexane) to afford product 1.2.11 b (2.5 g, 45.7 % yield). 1H NMR (400 MHz, CDCI3) δ 6.82 (s, 1 H), 4.46 (q, J = 7.1 Hz, 2H), 1.62 (d, J = 7.0 Hz, 2H), 1.58 - 1.53 (m, 2H), 1.44 (t, J = 7.1 Hz, 3H), 1.34 (ddd, J = 26.0, 16.7, 9.4 Hz, 8H), 1.26 - 1.14 (m, 6H), 0.92 (t, J = 7.3 Hz, 9H).
Step 3. Synthesis of ethyl 5-(4-(difluoromethoxy)phenyl)isoxazole-3- carboxylate [1.2.11c]

1.2.11c
A vial was charged with 1.2.11 b (0.76 g, 1.77 mmol, 1.0 equiv) and 1-bromo-4- uoromethoxy) benzene (0.5 g, 2.13 mmol, 1.2 equiv) were dissolved in 1 ,4-dioxane (10
ml_). Pd(PPh3)2CI2 (0.062 g, 0.085 mmol, 0.05 equiv) was added and the reaction mixture was stirred at 130 °C for 2 hours. The reaction mixture was quenched with water and extracted with EtOAc. The organic layer was washed with brine, dried over sodium sulfate and concentrated. The residue was purified by silica gel column chromatography (0-10 % EtOAc in Hexane) to afford product 1.2.11c (0.6 g, 94 % yield). LCMS (m/z): 284.2 [M+H]. 1H NMR (400 MHz, CDCI3) δ 7.89 - 7.81 (m, 2H), 7.28 - 7.24 (m, 2H), 6.93 (s, 1 H), 6.4 - 6.9 (3s, 1 H), 4.19 (q, J = 7.1 Hz, 2H), 1.31 - 1.29 (t, 3H).
Step 4. Synthesis of 4-(((5-(4-(difluoromethoxy)phenyl)isoxazol-3- yl)methyl)amino)-N-hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide
[1.2.11]

From ester 1.2.11c, hydroxamic acid was prepared following the procedures described for the synthesis of 1.2.4. LCMS (m/z): 432.3 [M+H]. 1 H NMR (400 MHz, DMSO) δ 10.72 (s, 1 H), 8.95 (s, 1 H), 7.94 (d, J = 8.7 Hz, 2H), 7.2-7.56 (3s, 1 H), 7.48 - 7.27 (m, 2H), 7.17 (d, J = 15.3 Hz, 1 H), 3.63 (d, J = 6.8 Hz, 2H), 3.40 (d, J = 7.0 Hz, 2H), 3.27 (d, J = 10.7 Hz, 2H), 2.96 (d, J = 12.7 Hz, 2H), 2.88 (s, 1 H), 2.35 - 2.08 (m, 4H).
Synthesis of N-hydroxy-4-(((5-(4-methoxyphenyl)isoxazol-3- pyran-4-carboxamide 1,1 -dioxide [1.2.12]
Me

Compound 1.2.12 was prepared following the procedures described for the synthesis of 1.2.11. 1H NMR (400 MHz, DMSO) δ 10.72 (s, 1 H), 8.95 (s, 1 H), 7.81 (d, J = 8.8 Hz, 2H), 7.10 (d, J = 8.9 Hz, 2H), 7.00 (s, 1 H), 3.83 (s, 3H), 3.61 (d, J = 6.9 Hz, 2H), 3.27 (d, J = 1 1.6
Hz, 2H), 2.96 (d, J = 12.6 Hz, 2H), 2.85 (s, 1 H), 2.29 - 2.13 (m, 4H). LCMS (m/z): 396.3 [M+H]+.
1.2.13 Synthesis of 4-(((5-(4-fluoro-3-methoxyphenyl)isoxazol-3-yl)methyl)amino)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide [1.2.13]

Compound 1.2.13 was prepared following the procedures described for the synthesis of 1.2.11. 1H NMR (400 MHz, DMSO) δ 10.74 (s, 1 H), 8.96 (s, 1 H), 7.61 (dd, J = 8.2, 2.0 Hz, 1 H), 7.48 (ddd, J = 8.4, 4.5, 2.0 Hz, 1 H), 7.40 (dd, J = 1 1.2, 8.5 Hz, 1 H), 7.17 (s, 1 H), 3.95 (s, 3H), 3.64 (s, 2H), 3.27 (d, J = 1 1.0 Hz, 3H), 2.97 (d, J = 12.9 Hz, 2H), 2.88 (s, 1 H), 2.22 (dd, J = 34.3, 13.2 Hz, 4H). LCMS (m/z): 414.4 [M+H]+.
1.2.14 Synthesis of 4-(((5-(2-fluoro-4-methylphenyl)isoxazol-3-yl)methyl)amino)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1,1 -dioxide [1.2.14]

Compound 1.2.14 was prepared following the procedures described for the synthesis of 1.2.11. 1H NMR (400 MHz, DMSO) δ 10.76 (s, 1 H), 8.94 (s, 1 H), 7.82 (t, J = 8.0 Hz, 1 H), 7.30 (d, J = 12.2 Hz, 1 H), 7.22 (d, J = 8.0 Hz, 1 H), 6.98 (d, J = 3.4 Hz, 1 H), 3.65 (s, 2H), 3.28 (dd, J = 18.5, 7.4 Hz, 2H), 2.97 (d, J = 12.9 Hz, 3H), 2.40 (s, 3H), 2.22 (dd, J = 35.4, 13.5 Hz, 4H). LCMS (m/z): 398.3 [M+H]+.
1.2.15 Synthesis of 4-(((5-(2,3-dichlorophenyl)isoxazol-3-yl)methyl)a hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide [1.2.15]

Compound 1.2.15 was prepared following the procedures described for the synthesis of 1.2.11. 1H NMR (400 MHz, DMSO) δ 10.78 (s, 1 H), 8.96 (s, 1 H), 7.86 (ddd, J = 1 1.1 , 8.0, 1.4 Hz, 2H), 7.57 (t, J = 8.0 Hz, 1 H), 7.27 (s, 1 H), 3.70 (s, 3H), 3.28 (t, J = 1 1.4 Hz, 2H), 2.97 (d, J = 13.1 Hz, 2H), 2.36 - 2.12 (m, 4H). LCMS (m/z): 434.2 [M+H]+.
1.2.16 Synthesis of 4-(((5-(2,4-difluorophenyl)isoxazol-3-yl)methyl)amino)-N- hydrox -4-carboxamide 1 ,1 -dioxide [1.2.16]

Compound 1.2.16 was prepared following the procedures described for the synthesis of 1.2.11. 1H NMR (400 MHz, DMSO) δ 1 1.21 (s, 1 H), 8.02 (td, J = 8.7, 6.5 Hz, 1 H), 7.59 (ddd, J = 11.5, 9.3, 2.4 Hz, 1 H), 7.33 (td, J = 8.7, 2.5 Hz, 1 H), 7.14 (d, J = 25.6 Hz, 1 H), 3.93 (m, 2H), 3.41 (m, 2H), 3.05 (m, 2H), 2.49 - 2.42 (m, 2H), 2.38 (m, 2H). LCMS (m/z): 402.4 [M+H]+.
1.2.17 Synthesis of N-hydroxy-4-(((5-(m-tolyl)isoxazol-3-yl)methyl)amino)tetrahydro- 2H-thio ide [1.2.17]

1.2.17
Compound 1.2.17 was prepared following the procedures described for the synthesis of 1.2.11. 1H NMR (400 MHz, DMSO) δ 7.69 (s, 1H), 7.66 (d, J = 7.5 Hz, 1H), 7.43 (t, J = 7.6 Hz, 1H), 7.33 (d, J = 7.3 Hz, 1H), 7.10 (s, 1H), 3.66 (s, 2H), 3.30 (t, J = 12.1 Hz, 2H), 2.97 (d, J= 13.1 Hz, 2H), 2.39 (s, 3H), 2.32-2.15 (m, 4H). LCMS (m/z): 380.3 [M+H]+.
1.2.18 Synthesis of N-hydroxy-4-(((5-(3-methoxyphenyl)isoxazol-3- yl)met ran-4-carboxamide 1 ,1 -dioxide [1.2.18]

Compound 1.2.18 was prepared following the procedures described for the synthesis of 1.2.11. 1H NMR (400 MHz, DMSO) δ 10.74 (s, 1H), 8.96 (s, 1H), 7.50 - 7.43 (m, 2H), 7.42 - 7.38 (m, 1H), 7.19 (s, 1H), 7.09 (dt, J = 6.7, 2.6 Hz, 1H), 3.85 (s, 3H), 3.64 (s, 2H), 3.28 (d, J= 11.4 Hz, 2H), 2.97 (d, J= 13.3 Hz, 2H), 2.89 (s, 1H), 2.22 (dd, J= 34.5, 13.7 Hz, 4H). LCMS (m/z): 396.3 [M+H]+.
1.2.19 Synthesis of 4-(((5-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)isoxazol-3- yl)methyl)amino)-N-hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide
[1.2.19]

Compound 1.2.19 was prepared following the procedures described for the synthesis of 1.2.11. 1H NMR (400 MHz, DMSO) δ 10.73 (s, 1H), 8.96 (s, 1H), 7.98 (d, J= 11.1 Hz, 1H), 7.77 (d, J= 8.4 Hz, 1H), 7.61 (d, J = 8.4 Hz, 1H), 7.20 (s, 1H), 3.63 (d, J = 6.1 Hz, 2H), 3.28 (t, J= 11.1 Hz, 2H), 2.96 (d, J= 12.9 Hz, 2H), 2.88 (s, 1H), 2.32-2.11 (m, 4H). LCMS (m/z): 446.5 [M+H]+.
1.2.20 Synthesis of 4-(((5-(3-chloro-5-fluorophenyl)isoxazol-3-yl)methyl)amino)-N- hydr rboxamide 1,1 -dioxide [1.2.20]

Compound 1.2.20 was prepared following the procedures described for the synthesis of 1.2.11. 1H NMR (400 MHz, DMSO) δ 10.72 (s, 1 H), 8.97 (s, 1 H), 7.86 (s, 1 H), 7.78 (d, J = 9.1 Hz, 1 H), 7.64 (dd, J = 8.8, 1.9 Hz, 1 H), 7.39 (s, 1 H), 3.64 (d, J = 6.4 Hz, 2H), 3.27 (d, J = 11.4 Hz, 2H), 2.97 (d, J = 12.3 Hz, 2H), 2.89 (s, 1 H), 2.21 (dd, J = 32.6, 13.5 Hz, 4H). LCMS (m/z): 416.3 [M+H]+.
1.2.21 Synthesis of 4-(((5-(3-chlorophenyl)isoxazol-3-yl)methyl)amino)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1,1 -dioxide [1.2.21]
Compound 1.2.21 was prepared following the procedures described for the synthesis of 1.2.11. 1H NMR (400 MHz, DMSO) δ 10.73 (s, 1 H), 8.97 (s, 1 H), 7.97 (d, J = 0.8 Hz, 1 H), 7.91 - 7.81 (m, 1 H), 7.59 (d, J = 5.2 Hz, 2H), 7.31 (s, 1 H), 3.64 (d, J = 5.8 Hz, 2H), 3.28 (d, J = 10.9 Hz, 2H), 2.97 (d, J = 12.9 Hz, 2H), 2.89 (s, 1 H), 2.31 - 2.12 (m, 4H). LCMS (m/z): 400.2 [M+H]+.
1.2.22 Synthesis of 4-(((5-(4-chloro-2,3-difluorophenyl)isoxazol-3-yl)methyl)amino)-N- hydr -carboxamide 1,1 -dioxide [1.2.22]

Compound 1.2.22 was prepared following the procedures described for the synthesis of 1.2.11. 1H NMR (400 MHz, DMSO) δ 7.78 (t, J = 7.8 Hz, 1 H), 7.69 - 7.58 (m, 1 H), 7.17
(s, 1 H), 3.91 (s, 2H), 3.34 (t, J = 1 1.9 Hz, 2H), 3.03 (d, J = 12.7 Hz, 2H), 2.48 - 2.36 (m, 2H), 2.35 - 2.22 (m, 2H). LCMS (m/z): 436.4 [M+H]+.
1.2.23 Synthesis of 4-(((5-(2-chloro-4-fluorophenyl)isoxazol-3-yl)methyl)amino)-N- hydrox -4-carboxamide 1,1 -dioxide [1.2.23]

Compound 1.2.23 was prepared following the procedures described for the synthesis of 1.2.11. 1H NMR (400 MHz, DMSO) δ 10.75 (s, 1 H), 8.95 (s, 1 H), 7.97 (dd, J = 8.8, 6.1 Hz, 1 H), 7.74 (dd, J = 8.9, 2.6 Hz, 1 H), 7.45 (td, J = 8.5, 2.6 Hz, 1 H), 7.19 (s, 1 H), 3.67 (d, J = 5.2 Hz, 2H), 3.27 (dd, J = 18.5, 7.1 Hz, 2H), 2.97 (d, J = 1 1.7 Hz, 3H), 2.31 - 2.10 (m, 4H). LCMS (m/z): 418.3 [M+H]+.
1.2.24 Synthesis of 4-(((5-(2,5-difluorophenyl)isoxazol-3-yl)methyl)amino)-N- hydrox arboxamide 1,1 -dioxide [1.2.24]

Compound 1.2.24 was prepared following the procedures described for the synthesis of 1.2.11. 1H NMR (400 MHz, DMSO) δ 10.76 (s, 1 H), 8.94 (s, 1 H), 7.79 (ddd, J = 8.8, 5.7, 3.2 Hz, 1 H), 7.60 - 7.51 (m, 1 H), 7.47 (dt, J = 12.8, 6.3 Hz, 1 H), 7.14 (d, J = 3.3 Hz, 1 H), 3.66 (d, J = 7.1 Hz, 2H), 3.27 (t, J = 14.3 Hz, 1 H), 2.97 (d, J = 14.1 Hz, 2H), 2.21 (dd, J = 35.9, 13.4 Hz, 3H). LCMS (m/z): 402.2 [M+H]+.
1.2.25 Synthesis of 4-(((5-(2-fluorophenyl)isoxazol-3-yl)methyl)am hydroxytetrahydro-2H-thiopyran-4-carboxamide 1,1 -dioxide [1.2.25]

Compound 1.2.25 was prepared following the procedures described for the synthesis of 1.2.11. 1H NMR (400 MHz, DMSO) δ 10.92 (s, 1 H), 8.93 (s, 1 H), 7.94 (t, J = 6.9 Hz, 1 H), 7.59 (dd, J = 13.7, 5.7 Hz, 1 H), 7.44 (dt, J = 15.1 , 9.3 Hz, 2H), 7.07 (d, J = 3.1 Hz, 1 H), 3.65 (d, J = 6.6 Hz, 2H), 3.26 (t, J = 10.3 Hz, 2H), 3.08 (s, 1 H), 2.97 (d, J = 12.8 Hz, 2H), 2.30 - 2.1 1 (m, 4H). LCMS (m/z): 384.3 [M+H]+.
1.2.26 Synthesis of 4-(((5-(2-fluoro-4-(trifluoromethoxy)phenyl)isoxazol-3- yl)methyl)amino)-N-hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide
[1.2.26]

Compound 1.2.26 was prepared following the procedures described for the synthesis of 1.2.11. 1H NMR (400 MHz, DMSO) δ 10.75 (s, 1 H), 8.93 (s, 1 H), 8.09 (t, J = 8.6 Hz, 1 H), 7.71 (d, J = 12.7 Hz, 1 H), 7.46 (d, J = 8.9 Hz, 1 H), 7.1 1 (d, J = 3.3 Hz, 1 H), 3.67 (d, J = 6.5 Hz, 2H), 3.28 (dd, J = 20.0, 9.1 Hz, 2H), 2.97 (d, J = 12.1 Hz, 3H), 2.22 (dd, J = 36.7, 13.6 Hz, 4H). LCMS (m/z): 468.2 [M+H]+.
1.2.27 Synthesis of 4-(((5-(3-fluoro-4-methoxyphenyl)isoxazol-3-yl)methyl)amino)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1,1 -dioxide [1.2.27]
NHOH
MeO F 1.2.27
Compound 1.2.27 was prepared following the procedures described for the synthesis of 1.2.11. 1H NMR (400 MHz, DMSO) δ 7.72 (dd, J = 12.0, 2.0 Hz, 1 H), 7.66 (d, J = 8.6 Hz, 1 H), 7.32 (t, J = 8.8 Hz, 1 H), 7.01 (s, 1 H), 3.89 (s, 3H), 3.83 (s, 2H), 3.33 (t, J = 1 1.3 Hz, 2H), 3.02 (d, J = 14.5 Hz, 2H), 2.46 - 2.35 (m, 2H), 2.35 - 2.23 (m, 2H). LCMS (m/z): 414.2 [M+H]+.
1.2.28 Synthesis of N-hydroxy-4-(((5-(2,3,4-trifluorophenyl)isoxazol-3- yl)methyl)amino)tetrahydro-2H-thiopyran-4-carboxamide 1,1 -dioxide [1.2.28]

Compound 1.2.28 was prepared following the procedures described for the synthesis of 1.2.11. 1H NMR (400 MHz, DMSO) δ 10.75 (s, 1 H), 8.93 (s, 1 H), 7.81 (d, J = 8.7 Hz, 1 H), 7.55 (dd, J = 16.8, 7.4 Hz, 1 H), 7.13 (d, J = 3.1 Hz, 1 H), 3.67 (d, J = 7.0 Hz, 2H), 3.28 (dd, J = 21.5, 10.7 Hz, 2H), 2.97 (d, J = 13.2 Hz, 3H), 2.31 - 2.10 (m, 3H). LCMS (m/z): 420.3 [M+H]+.
1.2.29 Synthesis of 4-(((5-(4-cyanophenyl)isoxazol-3-yl)methyl)amino)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1,1 -dioxide [1.2.29]

Compound 1.2.29 was prepared following the procedures described for the synthesis of 1.2.11. 1H NMR (400 MHz, DMSO) δ 10.72 (s, 1 H), 8.94 (s, 1 H), 8.06 (q, J = 8.6 Hz, 4H), 7.40 (s, 1 H), 3.66 (d, J = 7.1 Hz, 2H), 3.33 - 3.22 (m, 2H), 3.02 - 2.87 (m, 3H), 2.32 - 2.12 (m, 4H). LCMS (m/z): 390.2 [M+H]+.
1.2.30 Synthesis of 4-(((5-(2,6-difluorophenyl)isoxazol-3-yl)methyl)amino)-N- hydr -carboxamide 1,1 -dioxide [1.2.30]

Compound 1.2.30 was prepared following the procedures described for the synthesis of 1.2.11. 1H NMR (400 MHz, CD3CN) δ 9.76 (s, 1 H), 7.65 - 7.51 (m, 1 H), 7.20 (t, J = 8.8 Hz, 2H), 6.92 (s, 1 H), 3.80 (m, 2H), 3.30 (m, 3H), 3.00 (m, 2H), 2.40 (m, 4H). LCMS (m/z): 402.3 [M+H]+.
1.2.31 Synthesis of N-hydroxy-4-(((5-(5-methylthiophen-2-yl)isoxazol-3- yl)met hiopyran-4-carboxamide 1 ,1 -dioxide [1.2.31 ]

Compound 1.2.31 was prepared following the procedures described for the synthesis of 1.2.11. 1H NMR (400 MHz, CD3CN) δ 7.40 (d, J = 3.6 Hz, 1 H), 6.89 (d, J = 3.7 Hz, 1 H),
6.62 (s, 1 H), 3.68 (s, 2H), 3.38 - 3.26 (m, 2H), 3.02 - 2.93 (m, 2H), 2.56 (d, J = 19.5 Hz, 3H), 2.36 (t, J = 13.2 Hz, 2H), 2.24 (d, J = 15.0 Hz, 2H). LCMS (m/z): 386.3 [M+H]+.
1.2.32 Synthesis of N-hydroxy-4-(((5-(4-methylthiophen-2-yl)isoxazol-3- yl)met opyran-4-carboxamide 1 ,1 -dioxide [1.2.32]
1.2.32
Compound 1.2.32 was prepared following the procedures described for the synthesis of 1.2.11. 1H NMR (400 MHz, CD3CN) δ 7.69 (s, 1 H), 7.18 (s, 1 H), 6.64 (s, 1 H), 3.69 (s, 2H), 3.31 (t, J = 1 1.7 Hz, 2H), 2.97 (d, J = 13.6 Hz, 2H), 2.55 (d, J = 20.5 Hz, 3H), 2.36 (t, J = 12.3 Hz, 2H), 2.24 (d, J = 14.9 Hz, 2H). LCMS (m/z): 386.3 [M+H]+.
1.2.33 Synthesis of 4-(((5-(4-chloro-2-fluorophenyl)isoxazol-3-yl)methyl)amino)-N- hydrox -carboxamide 1,1 -dioxide [1.2.33]

Compound 1.2.33 was prepared following the procedures described for the synthesis of 1.2.11. 1H NMR (400 MHz, DMSO) δ 10.72 (s, 1 H), 8.95 (s, 1 H), 7.99 (dd, J = 10.1 , 1.6 Hz, 1 H), 7.88 - 7.71 (m, 2H), 7.31 (s, 1 H), 3.65 (s, 2H), 3.29 (t, J = 1 1.1 Hz, 2H), 2.97 (d, J = 12.7 Hz, 2H), 2.93 - 2.74 (m, 1 H), 2.32 - 2.10 (m, 4H). LCMS (m/z): 418.3 [M+H]+.
1.3.1. Synthesis of N-hydroxy-4-(((1-phenyl-1 H-1,2,3-triazol-4- yl)methyl)amino)tetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide [1.3.1 ]

Compound 1.3.1 was prepared following the procedures described for the synthesis of 1.2.1 using 1-phenyl-1 H-1 ,2,3-triazole-4-carbaldehyde and methyl 4-aminotetrahydro-2H- thiopyran-4-carboxylate 1 ,1 -dioxide in Step 1. HRMS m/z 366.1230 [M+H]+. 1H NMR (400 MHz, CD3OD) 8.51 (s, 1 H) 7.85 (d, J=7.83 Hz, 2 H) 7.55 - 7.63 (m, 2 H) 7.47 - 7.54 (m, 1 H) 3.96 (br. s., 1 H) 3.48 (s, 2 H) 3.33 - 3.43 (m, 4 H) 3.08 (br. s., 2 H) 2.39 (br. s., 2 H)
1.4.1 Synthesis of compound 1.4.1

Step 1. Synthesis of methyl 4-(2-((tert-butyldimethylsilyl)oxy)ethyl)tetrahydro- 2H-thiopyran-4-carboxylate [1.4.1a]

1.4.1 a
To a solution of methyl tetrahydro-2H-thiopyran-4-carboxylate (817 mg, 5.1 mmol) in THF (10.2 mL) at -78 °C, LDA (3.06 mL, 6.12 mmol) was added dropwise over 15 minutes. The reaction was stirred for 15 minutes at which time a precipitate formed. A solution of (2- bromoethoxy)(tert-butyl)dimethylsilane (1.64 mL, 7.65 mmol) in THF (10.2 mL) was added and the reaction mixture was stirred at -78 °C for 30 minutes at which time the cooling bath was removed and the reaction was allowed to warm to room temperature. The reaction mixture was quenched with saturated aqueous NH4CI solution and extracted with EtOAc.
The organic ayer was washed with brine, dried over magnesium sulfate, filtered and concentrated. The residue was purified by silica gel column chromatography
(EtOAc/heptane, 0-50%) to afford 1.4.1a (967 mg, 59.5 % yield). 1H NMR (400 MHz, CDCI3) δ ppm 0.00 - 0.06 (m, 6 H) 0.87 (s, 9 H) 1.59 - 1.70 (m, 2 H) 1.77 (t, J=7.04 Hz, 2 H) 2.39 (d, J=12.91 Hz, 2 H) 2.52 (d, J=14.48 Hz, 2 H) 2.63 - 2.79 (m, 2 H) 3.60 (t, J=6.85 Hz, 2 H) 3.70 (s, 3 H).
Step 2. Synthesis of 2-oxa-8-thiaspiro[4.5]decan-1-one [1.4.01 b]

1.4.1b
1.4.1a (967 mg, 3.04 mmol) was dissolved in 1.0 M HCI solution in MeOH (9.1 ml_, 9.1 mmol) and the reaction solution was stirred at room temperature for 40 minutes. The reaction mixture was concentrated in vacuo and dried under high vacuum to afford 1.4.1 b (523 mg, 100 % yield) as a white solid. 1H NMR (400 MHz, CDCI3) δ ppm 1.83 (ddd, J=13.69, 7.04, 2.74 Hz, 2 H) 2.07 - 2.21 (m, 4 H) 2.57 - 2.69 (m, 2 H) 2.85 (td, J=6.65, 3.91 Hz, 2 H) 4.29 (t, J=7.04 Hz, 2 H).
Step 3. Synthesis of 4-(2-(4-iodo-2-oxopyridin-1(2H)-yl)ethyl)tetrahydro-2H- thiopyran-4-carboxylic acid [1.4.1c]

A slurry of 1.4.1 b (100 mg, 0.58 mmol), 4-iodopyridin-2(1 H)-one (257 mg, 1.16 mmol), Cs2C03 (378 mg, 1.16 mmol) and DMF (1.16 mL) was heated to 150 °C with miocrowave for 90 minutes. The reaction was quenched water and acidified with 1.0 M HCI aqueous to pH of 4. The aqueous layer was extracted with EtOAc. The organic layer was washed with brine, dried (MgS04), filtered and concentrated to afford a brown oil. The oil was dissolved in DMSO and purified by reverse phase HPLC to afford 1.4.1c (33 mg, 14 % yield) as a white crystalline solid. 1H NMR (400 MHz, CD3OD) ppm 1.61 - 1.74 (m, 2 H) 1.85 - 1.95 (m, 2 H) 2.30 - 2.42 (m, 2 H) 2.55 (d, J=14.48 Hz, 2 H) 2.64 - 2.78 (m, 2 H) 3.87 - 4.00 (m, 2 H) 6.69 (dd, J=7.04, 1.57 Hz, 1 H) 7.02 (d, J=1.17 Hz, 1 H) 7.31 (d, J=7.04 Hz, 1 H).
Step 4. Synthesis of 4-(2-(4-iodo-2-oxopyridin-1(2H)-yl)ethyl)-N-((tetrahydro-2H- pyran-2-yl)oxy)tetrahydro-2H-thiopyran-4-carboxamide [1.4.1d]

1.4.1d
1.4.1c (33 mg, 0.084 mmol) was dissolved in DMF (0.559 ml_). To this solution, Et3N (46.5 μΙ, 0.336 mmol), 0-(tetrahydro-2H-pyran-2-yl)hydroxylamine (14.75 mg, 0.126 mmol), aza-HOBt (20.56 mg, 0.151 mmol) and aza-HOBt (20.56 mg, 0.151 mmol) were added. After stirring at room temperature for 24 hours, the reaction mixture was quenched by addition of saturated aqueous NH4CI solution and extracted with EtOAc. The organic layer was washed with brine, dried over MgS04 and concentrated on to silica gel. Purification by silica gel column chromatography (EtOac/heptane, 10-100%) afforded 1.4.1d (35 mg, 85 % yield). LC/MS (m/z) 408.98 [M+H]+
Step 5. Synthesis of 4-(2-(4-iodo-2-oxopyridin-1(2H)-yl)ethyl)-N-((tetrahydro-2H- pyran-2-yl)oxy)tetrahydro-2H-thiopyran-4-carboxamide 1,1 -dioxide [1.4.1e]

1.4.1e m-CPBA (35.0 mg, 0.142 mmol) was added to a solution of 1.4.1 d (35 mg, 0.07 mmol) in DCM (0.7 ml_) at -10 °C and the reaction was allowed to warm to 0°C for 2 hours. The mixture was quenched with water and extracted with DCM. The organic layer was dried over magnesium sulfate, filtered and concentrated on to silica gel. Purification by silica gel column chromatography (EtOAc/heptane,10-100%) afforded 1.4.1e (25 mg, 67 % yield). LC/MS (m/z) 440.98 [M+H]+.
Step 6. Synthesis of 4-(2-(4-(4-chloro-2-fluorophenyl)-2-oxopyridin-1(2H)- yl)ethyl)-N-hydroxytetrahydro-2H-thiopyran-4-carboxamide 1,1 -dioxide [1.4.1]

A degassed mixture of 1.4.1e (25 mg, 0.048 mmol), (4-chloro-2-fluorophenyl)boronic acid (9.98 mg, 0.057 mmol), K3P04 (25.3 mg, 0.1 19 mmol) and PdCI2(dppf) CH2CI2 adduct (7.79 mg, 0.009 mmol) in THF (0.72 mL) and water (0.24 mL) was stirred at 60 °C for 3 hours. The reaction mixture was partitioned between EtOAc and saturated aqueous NH4CI solution. The organic layer was stirred with Siliabond DMT to remove the Pd residue, dried over magnesium sulfate, filtered and concentrated on to silica. Purification by silica gel column chromatography (EtOAc/heptane 10-100%) afforded 4-(2-(4-(4-chloro-2- fluorophenyl)-2-oxopyridin-1 (2H)-yl)ethyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)tetrahydro-2H- thiopyran-4-carboxamide 1 ,1 -dioxide (1 1.2 mg, 44.6 % yield). This material was then dissolved in 1.0 M HCI in MeOH and the solution was stirred at room temperature. The precipitate was collected by filtration to afford (2.8 mg, 13 % yield) LC/MS (m/z) 443.1
[M+H]+.1 H NMR (400 MHz, CD3OD) ppm 1.97 - 2.06 (m, 2 H) 2.07 - 2.22 (m, 2 H) 2.55 (d, J=14.87 Hz, 2 H) 3.00 - 3.26 (m, 4 H) 3.92 - 4.08 (m, 2 H) 6.60 (d, J=7.04 Hz, 1 H) 6.71 (s, 1 H) 7.27 - 7.40 (m, 2 H) 7.55 (t, J=8.41 Hz, 1 H) 7.68 (d, J=7.04 Hz, 1 H
1.4.2 Synthesis of 4-(2-(4-(2-fluoro-4-methoxyphenyl)-2-oxopyridin-1(2H)-yl)ethyl)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide [1.4.2]

Compound 1.4.2 was synthesized following the procedure described for 1.4.1. 1H NMR (500 MHz, METHANOL-c4) δ ppm 2.03 - 2.1 1 (m, 2 H) 2.17 (t, J=12.30 Hz, 2 H) 2.58 (d, J=14.82 Hz, 2 H) 3.06 - 3.16 (m, 2 H) 3.16 - 3.27 (m, 2 H) 3.89 (s, 3 H) 4.04 - 4.12 (m, 2 H) 6.79 - 6.90 (m, 3 H) 6.92 (dd, J=8.67, 2.05 Hz, 1 H) 7.55 (t, J=8.67 Hz, 1 H) 7.79 (d,
J=6.94 Hz, 1 H). LC/MS (m/z) 439.1 [M+H]+
1.5.1 Synthesis of N-hydroxy-4-(2-(4-phenyl-1 H-pyrazol-1-yl)ethyl)tetrahydro-2H- thiopyran-4-carboxamide 1,1 -dioxide [1.5.1]

StepL Synthesis of methyl 4-(2-chloroethyl)tetrahydro-2H-thiopyran-4- carb

.1a
A solution of methyl tetrahydro-2H-thiopyran-4-carboxylate (1.0 g, 6.24 mmol) in THF (31.2 mL) was cooled at -78 °C. A solution of LDA (3.74 ml, 7.49 mmol, 2.0 M in heptane) was added over 15 min. After 30 mins, 1-bromo-2-chloroethane (1.039 ml, 12.48 mmol) was added over 5 min and the reaction was allowed to stir at -78 °C for 1 hour at which time the cooling bath was removed and the reaction was allowed to warm to room temperature. The reaction was quenched with saturated aqueous ammonium chloride and extracted with EtOAc. The organic layer was dried (MgS04), filtered and concentrated. The material was purified by silica gel column chromatography, EtOAc/heptane 0 to 30%, to afford 1.5.1a (667 mg, 48.0 % yield). 1H NMR (400 MHz, CDCI3) δ ppm 1.56 - 1.71 (m, 2 H) 1.94 - 2.08 (m, 2 H) 2.42 (d, J=12.52 Hz, 2 H) 2.53 (d, J=14.48 Hz, 2 H) 2.60 - 2.77 (m, 2 H) 3.39 - 3.53 (m, 2 H) 3.65 - 3.81 (m, 3 H)
Step 2. Synthesis of methyl 4-(2-chloroethyl)tetrahydro-2H-thiopyran-4- carb [1.5.1 b]

A solution of 1.5.1a (103 mg, 0.462 mmol) in DCM (4.6 ml_) was cooled in an ice water bath. mCPBA (239 mg, 0.971 mmol) was added and the reaction was stirred at 0 °C for 20 mins then at room temperature for 1 hour. The reaction was quenched with water and diluted with DCM. The organic layer was washed with saturated aqueous NaHC03 solution,
water and brine. The organic layer was dried over magnesium sulfate, filtered and concentrated to afford product 1.5.1b (1 18 mg, 100 % yield). 1H NMR (400 MHz, CDCI3) δ ppm 2.04 - 2.19 (m, 4 H) 2.56 (d, J=13.30 Hz, 2 H) 2.92 - 3.14 (m, 4 H) 3.46 (t, J=7.43 Hz, 2 H) 3.80 (s, 3 H)
Step 3. Synthesis of methyl 4-(2-(4-phenyl-1 H-pyrazol-1-yl)ethyl)tetrahydro-2H- thiopy oxide [1.5.1c]

A mixture of 1.5.1 b (1 18 mg, 0.463 mmol), 4-phenyl-1 H-pyrazole (55.7 mg, 0.386 mmol) and Cs2C03 (277 mg, 0.849 mmol) in DMF (Volume: 2.145 ml) was stirred at 45 °C for 24 hours. The reaction mixture was then partitioned between EtOAc and water. The organic layer was washed with brine, dried over Na2S04, filtered and concentrated to afford 1.5.1.C (124.3 mg, 89 % yield) as a white solid. 1H NMR (400 MHz, CDCI3) δ ppm 2.04 - 2.21 (m, 2 H) 2.29 (t, J=7.43 Hz, 2 H) 2.56 (d, J=14.09 Hz, 2 H) 2.91 - 3.15 (m, 6 H) 3.69 -
3.77 (m, 3 H) 4.15 (t, J=7.43 Hz, 2 H) 7.31 - 7.43 (m, 3 H) 7.44 - 7.49 (m, 2 H) 7.59 (s, 1 H)
7.78 (s, 1 H). LC/MS (m/z) 363.6 [M+H]+
Step 4. Synthesis of N-hydroxy-4-(2-(4-phenyl-1 H-pyrazol-1 -yl)ethyl)tetrahydro- 2H-thi -dioxide [1.5.1]

Compound 1.5.1 was prepared from ester 1.5.1c following the procedures described for the synthesis of 1.1.1 step 2-4. 1 H NMR (500 MHz, METHANOL-c 4) δ ppm 2.06 - 2.16 (m, 2 H) 2.16 - 2.23 (m, 2 H) 2.52 (d, J=15.13 Hz, 2 H) 3.02 - 3.1 1 (m, 2 H) 3.12 - 3.23 (m, 2 H) 4.17 - 4.27 (m, 2 H) 7.18 - 7.28 (m, 1 H) 7.37 (t, J=7.88 Hz, 2 H) 7.51 - 7.60 (m, 2 H) 7.89
- 7.95 (m, 1 H) 8.01 - 8.07 (m, 1 H). LC/MS (m/z) 364.2 [M+H]+.
1.5.2 Synthesis of 4-(2-(4-(4-chlorophenyl)-2H-1,2,3-triazol-2-yl)ethyl)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1 ,1 -dioxide [1.5.2]

Step 1. Synthesis of methyl 4-(2-(4-(4-chlorophenyl)-2H-1,2,3-triazol-2- yl)ethyl)tetrahydro-2H-thiopyran-4-carboxylate 1 ,1 -dioxide [1.5.2a]

A mixture of 1.5.1 b (100 mg, 0.393 mmol) and sodium azide (63.8 mg, 0.981 mmol) in DMF (0.491 mL) was stirred at 80 °C for 24 hours. The reaction mixture was cooled to room temperature. Cul (74.8 mg, 0.393 mmol) and 1-chloro-4-ethynylbenzene (53.6 mg, 0.393 mmol) were added and the reaction was then stirred at 80 °C for 72 hours. The reaction was quenched with water and then filtered. The filtrate was extracted with DCM. The combined organic layers were washed with brine, dried over Na2S04, filtered and concentrated. The residue was purified by silica gel column chromatography,
EtOAc/heptane 70% to 100%, to afford product 1.5.2a (55 mg, 35.2 % yield). 1H NMR (CDCI3) δ: 7.77 (d, J=8.5 Hz, 2H), 7.74 (s, 1 H), 7.43 (d, J=8.2 Hz, 2H), 4.42 (t, J=7.7 Hz, 2H), 3.77 (d, J=0.9 Hz, 3H), 3.06-3.16 (m, 2H), 2.97-3.06 (m, 2H), 2.62 (d, J=13.6 Hz, 2H), 2.35 (t, J=7.6 Hz, 2H), 2.14-2.25 (m, 2H). LC/MS (m/z) 398.2 [M+H]+,
Step 2. Synthesis of 4-(2-(4-(4-chlorophenyl)-2H-1,2,3-triazol-2-yl)ethyl)-N- hydroxytetrahydro-2H-thiopyran-4-carboxamide 1,1 -dioxide [1.5.2]

(m/z) 399.1 [M+H]+
1.6.1 Synthesis of 4-(2-([1,1'-biphenyl]-4-yl)ethyl)-N-hydroxytetrahydro-2H-thiopyran-4- carbo

Step 1. Synthesis of methyl 4-(2-([1,1'-biphenyl]-4-yl)-2-oxoethyl)tetrahydro-2H- thiopyran-4-carboxylate [1.6.1a]

A solution of methyl tetrahydro-2H-thiopyran-4-carboxylate (200 mg, 1.248 mmol) in THF (12.5 mL) was cooled in a dry ice acetone bath. LDA (0.749 ml_, 1.498 mmol, 2.0 M in heptane) was added dropwise. The reaction was allowed to stir for 30 min at which time a solution of 1-([1 ,1 '-biphenyl]-4-yl)-2-bromoethanone (515 mg, 1.872 mmol) in THF was added. The reaction was allowed to stir at -78 °C for 1 hour and then warmed to room temperature. The reaction was quenched with saturated aqueous ammonium chloride solution and extracted with Et20. The combined organic layers was washed with brine, dried over MgS04, filtered and concentrated. The residue was purified by silica gel column chromatography, EtOAc/Heptane 0-40% to afford product 1.6.1a (414 mg, 94 % yield). LC/MS (m/z) 355.3 [M+H]+
Step 2. Synthesis of methyl 4-(2-([1,1'-biphenyl]-4-yl)-2-oxoethyl)tetrahydro-2H- thiopyran-4-carboxylate 1,1-dioxide [1.6.1 b]
A solution of 1.6.1a (304 mg, 0.858 mmol) in THF (6.4 mL) and water (2.1 mL) was cooled at 0 °C. Oxone (1 160 mg, 1.887 mmol) was added and the mixture was stirred at 0 °C for 2 hours. The reaction mixture was brought to a neutral pH with 1.0 M aqueous NaOH solution and then extracted with EtOAc. The organic layer was washed with brine, dried over magnesium sulfate, filtered and concentrated. The residue was purified by silica gel column chromatography, EtOAc/heptane 20-80% to afford product 1.6.1 b (141 mg, 42.5 % yield). LC/MS (m/z) 387.2 [M+H]+
Step 3. Synthesis of methyl 4-(2-([1,1'-biphenyl]-4-yl)ethyl)tetrahydro-2H- thiopy e [1.6.1c]

A solution of 1.6.1 b (45 mg, 0.1 16 mmol) in MeOH (5.3 mL) and EtOAc (0.5 mL) was added to a mixture of Pd/C (10% on carbon, 18.59 mg, 0.017 mmol) in MeOH (0.3 mL). The mixture was stirred under 1 atm of hydrogen for 48 hours. The reaction mixture was then filtered through Celite and the filtrate was concentrated. The remaining oil was purified by silica gel column chromatorgaphy, EtOAc/heptane 10-100%, to give product 1.6.1 c (22 mg, 50.7 % yield). 1H NMR (400 MHz, CDCI3) δ ppm 1.37 (d, J=7.04 Hz, 3 H) 2.07 - 2.24 (m, 2 H) 2.48 (dd, J=14.28, 3.33 Hz, 1 H) 2.53 - 2.63 (m, 1 H) 2.89 - 3.01 (m, 3 H) 3.02 - 3.15 (m, 2 H) 3.73 (s, 3 H) 7.15 (d, J=7.83 Hz, 2 H) 7.31 - 7.38 (m, 1 H) 7.44 (t, J=7.63 Hz, 2 H) 7.53 (d, J=7.83 Hz, 2 H) 7.58 (d, J=7.43 Hz, 2 H). LC/MS (m/z) 372.3 [M+H]+
Step 4. Synthesis of 4-(2-([1,1'-biphenyl]-4-yl)ethyl)-N-hydroxytetrahydro-2H- thiopyran-4-carboxamide 1,1 -dioxide [1.6.1]

(m/z) 374.2 [M+H]+
2.1.1 Synthesis of compound 2.1.1

Step 1. Synthesis of methyl 3-(([1,1'-biphenyl]-4-ylmethyl)amino)
tetrahydrothiophene-3-carboxylate [2.1.1a]

A slurry of methyl 3-aminotetrahydrothiophene-3-carboxylate (169 mg, 1.048 mmol), [1 ,1 '-biphenyl]-4-carbaldehyde (159 mg, 0.874 mmol) and acetic acid (0.250 ml, 4.37 mmol) in 1 ,2-dichloroethane (7.9 ml_) was stirred at room temperature overnight and then at 60 °C for 3 hours. The reaction was cooled to room temperature and sodium triacetoxyborohydnde (926 mg, 4.37 mmol) was added. After stirring at room temperature overnight, the reaction mixture was quenched with water and extracted with EtOAc. The organic layer was washed with 1.0 M NaOH aqueous solution and brine, dried over magnesium sulfate filtered and concentrated on to silica gel. Purification by silica gel column chromatography
(EtOAc/heptane, 0 to 50%) afforded 2.1.1a (132 mg, 46 % yield). LC/MS (m/z) 328.2 [M+H]+. 1H NMR (400 MHz, <CDCI3>) δ ppm 2.16 - 2.30 (m, 1 H) 2.42 (dt, J=12.91 , 7.83 Hz, 1 H) 2.84 - 3.00 (m, 2 H) 3.03 - 3.13 (m, 1 H) 3.29 (d, J=10.96 Hz, 1 H) 3.73 (s, 2 H) 3.75 - 3.82 (m, 3 H) 7.30 - 7.76 (m, 9 H)
Step 2. Synthesis of methyl 3-(([1,1'-biphenyl]-4-ylmethyl)amino)
tetrahydrothiophene-3-carboxylic acid [2.1.1 b]

A solution of 2.1.1a (132 mg, 0.403 mmol) and LiOH (2.0 M in water, 2.0 ml_, 4.0 mmol) in MeOH (0.8 ml_) was stirred overnight at room temperature. The reaction mixture was concentrated in vacuo. The remaining salt was dissolved in water and acidified to pH=4 by adding 6 M HCI aqueous solution. The white precipitate was collected and washed with diethyl ether to afford 2.1.1 b (66 mg, 52 % yield) LC/MS (m/z) 314.1 [M+H]+.
Step 3. Synthesis of methyl 3-(([1,1'-biphenyl]-4-ylmethyl)amino)-N- hydroxytetrahydrothiophene-3-carboxamide [2.1.1]

A solution of 2.1.1 b (1 18 mg, 0.38 mmol), 0-(tetrahydro-2H-pyran-2- yl)hydroxylamine (66 mg, 0.563 mmol), EDC HCI (101 mg, 0.526 mmol), aza-HOBt (92 mg, 0.676 mmol) and Et3N (94 μΙ, 0.676 mmol) in DMF (Volume: 3756 μΙ) was stirred for 72 hours. The reaction mixture was diluted with water and brought to a pH of 4 with NH4CI. The aqueous layer was extracted with EtOAc/Et20 (1/1 ). The organic layer was washed with water, brine and dried over MgS04. The organic layer was filtered and concentrated. The remaining material was purified by silica gel column chromatography (EtO Ac/heptane 0 to 50%) to afford 3-(([1 ,1'-biphenyl]-4-ylmethyl)amino)-N-((tetrahydro-2H-pyran-2- yl)oxy)tetrahydrothiophene-3-carboxamide (50 mg, 0.121 mmol, 32.3 % yield). 25 mg of this product was dissolved in a solution of HCI in EtOH (1.25 M, 3.0 ml_, 3.75 mmol) and the reaction solution was allowed to stir at room temperature for 2 hours. Et20 was added and the white precipitate was collected to afford 2.1.1 (6.7 mg, 4.84 % yield).
LC/MS (m/z) 329.1 [M+H]+. 1H NMR (400 MHz, <CD3OD>) δ ppm 2.40 - 2.68 (m, 2 H) 3.09 (t, J=6.85 Hz, 2 H) 3.36 - 3.45 (m, 1 H) 4.19 (br. s., 2 H) 7.30 - 7.40 (m, 1 H) 7.45 (t, J=7.63 Hz, 2 H) 7.57 - 7.68 (m, 4 H) 7.73 (d, J=8.22 Hz, 2 H)
2.2.1 Synthesis of compound 2.2.1

Step 1. Synthesis of methyl 3-((tert- butoxycarbonyl)amino)tetrahydrothiophene-3-carboxylate [2.2.1a]

2.2.1a
Boc20 (3.46 ml, 14.92 mmol) was added to a solution of methyl 3- aminotetrahydrothiophene-3-carboxylate (2.186 g, 13.56 mmol) and Et3N (4.70 ml, 33.9 mmol) in THF (Volume: 67.8 ml) and the reaction was stirred at room temperature for 24 hours. The reaction was quenched with water and extracted with EtOAc. The EtOAc layer was dried (MgS04), filtered and concentrated to afford 2.2.1a (2.7 g, 76 % yield). 1H NMR (400 MHz, CDCI3) δ ppm 1.27 (s, 2 H) 1.43 (s, 9 H) 2.30 - 2.45 (m, 1 H) 2.50 - 2.67 (m, 1 H) 2.83 - 3.02 (m, 3 H) 3.20 - 3.35 (m, 1 H) 3.60 - 3.88 (m, 3 H)
Step 2. Synthesis of methyl 3-((tert- butoxycarbonyl)amino)tetrahydrothiophene-3-carboxylate 1,1 -dioxide [2.2.1 b]

2.2.1b
Oxone (14.0 g, 22.7 mmol) was added was added to a solution 2.2.1a (2.7 g, 10.33 mmol), THF (52 mL) and water (52 mL) at 0 °C. After stirring at 0 °C for 5 hours, the reaction mixture was diluted with water, neutralized by adding 1.0 M NaOH aqueous solution and extracted with EtOAc. The organic layer was dried on to silica gel and purified by silica gel
column chromatography (EtOAc/heptane 25-100%) to afford product 2.2.1 b (1.5 g, 50 % yield) as a white solid.
Step 3. Synthesis of methyl 3-aminotetrahydrothiophene-3-carboxylate 1 ,1- dioxide [2.2.1c]

2.2.1c
A solution of 2.2.1 b (1.4 g, 4.77 mmol) in 4.0 M HCI in dioxane (12.0 mL, 47.7 mmol) was stirred at room temperature for 72 hours. Et20 was added and the precipitate was collected by filtration to afford 2.2.1c as HCI salt (700 mg, 55 % yield). LC/MS (m/z) 194.0 [M+H]+.
Step 4. Synthesis of methyl 3-(((5-phenylisoxazol-3-yl)methyl)amino) tetrahydrothiophene-3-carboxylate 1,1 -dioxide [2.2.1 d]

A solution of 2.2.1c (100 mg, 0.376 mmol), acetic acid (0.064 mL, 1.127 mmol), Et3N (0.156 mL, 1.127 mmol) and 5-phenylisoxazole-3-carbaldehyde (65.1 mg, 0.376 mmol) was stirred at room temperature for 1 hour. Sodium triacetoxyborohydride (239 mg, 1.127 mmol) was added and the mixture was stirred overnight. The reaction was quenched with water, basified to a pH of 8 with 1.0 M aqueous NaOH solution and extracted with EtOAc. The organic layer was washed with water and brine, dried (MgS04), filtered and concentrated on to silica gel. Purification by silica gel column chromatography (EtOAc/heptane 0 to 50% afforded 2.2.1d (75 mg, 57 % yield). LC/MS (m/z) 351.1 [M+H]+.
Step 5. Synthesis of 3-(((5-phenylisoxazol-3-yl)methyl)amino)
tetrahydrothiophene-3-carboxylic acid 1 ,1-dioxide [2.2.1 e]

A solution of 2.2.1d (72 mg, 0.205 mmol) and LiOH (2.0 M in water, 1.5 ml_, 3.0 mmol) in THF (4.1 ml_) was allowed to stir for 24 hours. The reaction mixture was concentrated and acidified to pH=4 with 1.0 N HCI aqueous solution. The precipitate was collected by filtration to afford 2.2.1e (55 mg, 80 % yield). LC/MS (m/z) 337.2 [M+H]+.
Step 6. Synthesis of N-hydroxy-3-(((5-phenylisoxazol-3-yl)methyl)amino) tetrahydrothiophene-3-carboxamide 1,1 -dioxide [2.2.1]

EDC HCI (43.9 mg, 0.229 mmol) and aza-HOBt (40.1 mg, 0.294 mmol) was added to a solution of 2.2.1e (55 mg, 0.164 mmol), 0-(tetrahydro-2H-pyran-2-yl)hydroxylamine (28.7 mg, 0.245 mmol) and Et3N (0.086 ml_, 0.621 mmol) in DMF (1.6 ml_). After stirring at room temperature over night, the reaction mixture was quenched with water, brought to pH=4 by adding saturated aqueous NH4CI solution. The mixture was extracted with Et20 and DCM, until no more product present in the aqueous layer. The combined organic layers were dried (MgS04), filtered and concentrated. The remaining oil was purified by silica gel column chromatography (EtOAc/heptane, 50-100%) to afford 3-(((5-phenylisoxazol-3- yl)methyl)amino)-N-((tetrahydro-2H-pyran-2-yl)oxy) tetrahydro-thiophene-3-carboxamide 1 ,1- dioxide (45 mg, 63.2 % yield). This material was dissolved in 4M HCI in dioxane and stirred for 1 hour. The precipitate was collected by filtration to afford 2.2.1 (32.2 mg, 50% yield). LC/MS (m/z) 352.1 [M+H]+. 1H NMR (400 MHz, CD3OD) δ ppm 2.49 - 2.62 (m, 1 H) 2.74 (dt, J=14.48, 7.24 Hz, 1 H) 3.24 - 3.29 (m, 1 H) 3.40 - 3.47 (m, 1 H) 3.50 (d, J=14.09 Hz, 1 H) 3.85 (d, J=14.48 Hz, 1 H) 4.00 - 4.16 (m, 2 H) 6.91 (s, 1 H) 7.43 - 7.57 (m, 3 H) 7.78 - 7.90 (m, 2 H)
2.2.2 Synthesis of compound 2.2.2

Compound 2.2.2 was prepared following the procedures described for the synthesis of 2.2.1 using 4'-chloro-2'-fluoro-[1 ,1'-biphenyl]-4-carbaldehyde (Ref. Tetrahedron Lett. 2005, 46, 7575-7579) in Step 4. LC/MS (m/z) 413.0 [M+H]+. 1H NMR (400 MHz, CD3OD) δ ppm 2.55 - 2.71 (m, 1 H) 2.90 (br. s., 1 H) 3.32 - 3.40 (m, 1 H) 3.51 (dd, J=13.69, 6.65 Hz, 1 H) 3.66 (s, 2 H) 3.98 (d, J=14.87 Hz, 1 H) 4.10 - 4.22 (m, 2 H) 7.28 - 7.36 (m, 2 H) 7.50 (t, J=8.41 Hz, 1 H) 7.58 - 7.68 (m, 4 H)
2.2.3 Synthesis of compound 2.2.3

Compound 2.2.3 was prepared following the procedures described for the synthesis of 2.2.1 using 3-phenylisoxazole-5-carbaldehyde in Step 4. LC/MS (m/z) 352.1 [M+H]+. 1H NMR (400 MHz, CD3OD) δ ppm 2.31 - 2.51 (m, 2 H) 3.08 - 3.18 (m, 1 H) 3.24 (br. s., 2 H) 3.60 (d, J=13.69 Hz, 1 H) 3.73 - 3.80 (m, 1 H) 3.82 - 3.90 (m, 1 H) 6.75 (s, 1 H) 7.33 - 7.42 (m, 3 H) 7.68 - 7.76 (m, 2 H).
II-2.2.4 Synthesis of compound 2.2.4

Compound 2.2.4 was prepared following the procedures described for the synthesis of 2.2.1 using 4-(phenylethynyl)benzaldehyde in Step 4. LC/MS (m/z) 385.1 [M+H]+. 1 H NMR (400 MHz, CD3OD) δ ppm 2.51 - 2.68 (m, 1 H) 2.85 (br. s. , 1 H) 3.32 - 3.37 (m, 1 H) 3.43 - 3.53 (m, 1 H) 3.59 (d, J=14.48 Hz, 1 H) 3.93 (d, J=14.48 Hz, 1 H) 4.02 - 4.13 (m, 2 H) 7.35 - 7.41 (m, 3 H) 7.48 - 7.54 (m, 4 H) 7.57 - 7.62 (m, 2 H).
2.2.5 Synthesis of 3-(((5-(4-chloro-2-fluorophenyl)isoxazol-3-yl)methyl)amino)-N- hydroxytetrahydrothiophene-3-carboxamide 1 ,1 -dioxide [2.2.5]

Compound 2.2.5 was prepared following the procedures described for the synthesis of 2.2.1 . The racemic material was separated into two enantiomers 2.2.5a and 2.2.5b with chiral HPLC.
2.2.5a: 1 H NMR (400 MHz, DMSO-d6) δ ppm 2.22 - 2.43 (m, 2 H) 3.06 - 3.18 (m, 1 H) 3.23 (t, J=7.24 Hz, 1 H) 3.35 (s, 1 H) 3.52 (d, J=13.69 Hz, 1 H) 3.61 - 3.79 (m, 2 H) 7.02 (d, J=3.52 Hz, 1 H) 7.48 (dd, J=8.61 , 1 .57 Hz, 1 H) 7.72 (dd, J=10.96, 1 .96 Hz, 1 H) 7.93 (t, J=8.41 Hz, 1 H) 9.02 (s, 1 H) 10.82 (br. s. , 1 H). LC/MS (m/z) 404.1 [M+H]+. Rt 5.34 min, chiral HPLC, AD column, Heptane/EtOH 40/60, flow rate 1 .0 mL/min, run time 10 mins.
2.2.5b 1 H NMR (400 MHz, DMSO-d6) δ ppm 2.22 - 2.43 (m, 2 H) 3.06 - 3.18 (m, 1 H) 3.23 (t, J=7.24 Hz, 1 H) 3.35 (s, 1 H) 3.52 (d, J=13.69 Hz, 1 H) 3.61 - 3.79 (m, 2 H) 7.02 (d, J=3.52 Hz, 1 H) 7.48 (dd, J=8.61 , 1 .57 Hz, 1 H) 7.72 (dd, J=10.96, 1 .96 Hz, 1 H) 7.93 (t, J=8.41 Hz, 1 H) 9.02 (s, 1 H) 10.82 (br. s. , 1 H). LC/MS (m/z) 404.1 [M+H]+. Rt 7.67 min, chiral HPLC, AD column, Heptane/EtOH 40/60, flow rate 1 .0 mL/min, run time: 10 mins.
3.1.1 Synthesis of compound 3.1.1

Step 1. Synthesis of 1-tert-butyl 4-ethyl 4-(2-chloroethyl)piperidine-1,4- dicarboxylate [3.1.1a]

3.1.1a
A solution of 1-tert-butyl 4-ethyl piperidine-1 ,4-dicarboxylate (4.68 g, 18.19 mmol) in THF (91 mL) was added to LiHMDS solution in THF (1.0 M, 20.0 ml_, 20.0 mmol) at -78 °C over 50 minutes. 1-bromo-2-chloroethane (3.13 g, 21.8 mmol) was added and the reaction mixture was warmed to room temperature and stirred overnight. The reaction was quenched with saturated aqueous NH4CI solution and extracted with Et20. The organic layer was washed with water and brine, dried over magnesium sulfate, filtered and concentrated. The remaining oil was purified by silica gel column chromatography (EtOAc/heptane 0 to 20%) to afford 3.1.1a (2.0 g, 34.4 % yield) of a colorless oil. 1H NMR (400 MHz, CDCI3) δ ppm 1.29 (t, J=7.04 Hz, 3 H) 1.33 - 1.50 (m, 1 1 H) 1.97 - 2.06 (m, 2 H) 2.12 (d, J=13.30 Hz, 2 H) 2.79 - 2.99 (m, 2 H) 3.41 - 3.51 (m, 2 H) 3.80 - 3.98 (m, 2 H) 4.21 (q, J=7.04 Hz, 2 H).
Step 2. Synthesis of 1-tert-butyl 4-ethyl 4-(2-(4-iodo-2-oxopyridin-1(2H)- yl)ethyl)piperidine-1 ,4-dicarboxylate [3.1.1 b]

3.1.1a (796 mg, 2.489 mmol) was added to a slurry of 4-iodopyridin-2(1 H)-one (500 mg, 2.262 mmol) and Cs2C03 (1622 mg, 4.98 mmol) in DMF (Volume: 1 1.300 mL) and the reaction mixture was heated to 50 °C for 24 hours. The reaction mixture was quenched with water and extracted with Et20. The organic layer was washed with brine, dried over magnesium sulfate filtered and concentrated on to silica gel. Purification by silica gel column chromatography (EtOAc/heptane, 0-30%) afforded product 3.1.1 b (796 mg). 1H NMR (400 MHz, CDCI3) δ ppm 1.31 (t, J=7.24 Hz, 3 H) 1.35 - 1.52 (m, 1 1 H) 1.93 (t, J=8.02 Hz, 2 H) 2.12 (d, J=13.30 Hz, 2 H) 2.84 - 3.10 (m, 2 H) 3.67 - 3.95 (m, 4 H) 4.21 (q, J=7.04 Hz, 2 H) 6.47 (dd, J=7.04, 1.56 Hz, 1 H) 6.87 (d, J=7.04 Hz, 1 H) 7.06 (d, J=1.56 Hz, 1 H).
Step 3. Synthesis of 1-tert-butyl 4-ethyl 4-(2-(2-oxo-4-phenylpyridin-1(2H)- yl)ethyl)piperidine-1,4-dicarboxylate [3.1.1c]

PdCI2(dppf)-CH2CI2Adduct (34.3 mg, 0.042 mmol) was added to a degassed mixture of phenylboronic acid (154 mg, 1.261 mmol), 3.1.1b (424 mg, 0.841 mmol) and potassium phosphate (535 mg, 2.52 mmol) in 2-MeTHF (3.2 mL) and H20 (1.1 mL). The mixture was stirred at 65 °C for 1 hour, after which, the reaction mixture was diluted with water/Et20 and filtered through Celite. The aqueous layer was brought to a pH of 4 with 1.0 M aqueous NaHS04 solution and extracted with Et20. Then combined organic layer was washed with
water and brine, dried over magnesium sulfate, filtered. The filtrate was stirred with
Siliabond-DMT Pd scavenger for 2 hours. The Pd scavenger was then filtered off and the filtrate was concentrated to afford 3.1.1c (331 mg, 87 % yield). 1H NMR (400 MHz, CDCI3) δ ppm 1.32 (t, J=7.24 Hz, 3 H) 1.39 - 1.58 (m, 1 1 H) 2.02 (t, J=7.83 Hz, 2 H) 2.16 (d, J=13.30 Hz, 2 H) 3.00 (br. s., 2 H) 3.78 - 4.07 (m, 4 H) 4.24 (q, J=7.04 Hz, 2 H) 6.45 (d, J=6.26 Hz, 1 H) 6.77 (s, 1 H) 7.32 - 7.51 (m, 4 H) 7.53 - 7.59 (m, 2 H).
Step 4. Synthesis of 1-(tert-butoxycarbonyl)-4-(2-(2-oxo-4-phenylpyridin-1(2H)- yl)ethyl)piperidine-4-carboxylic acid [3.1.1d]

A solution of 3.1.1c (1 10 mg, 0.242 mmol) and LiOH.H20 (102 mg, 2.42 mmol) in MeOH (3.6 mL) and water (1.2 mL) was stirred at room temperature for one week and at 50°C for 48 hours. The reaction mixture was concentrated to 1/3 of its the volume and brought to a pH of 4 by adding 1.0 M HCI aqueous solution. The aqueous layer was extracted with EtOAc/Et20 (1/1 ). The combined organic layers were washed with brine, dried over magnesium sulfate, filtered and concentrated to afford 3.1.1d (66 mg, 64 % yield). 1H NMR (400 MHz, CDCI3) δ ppm 1.37 - 1.48 (m, 1 1 H) 2.05 (s, 1 H) 2.16 (s, 1 H) 2.89 - 3.19 (m, 2 H) 3.69 - 4.1 1 (m, 4 H) 6.48 (dd, J=7.24, 2.15 Hz, 1 H) 6.79 (d, J=1 .96 Hz, 1 H) 7.36 - 7.51 (m, 5 H) 7.37 - 7.37 (m, 1 H) 7.53 - 7.59 (m, 2 H).
Step 5. Synthesis of 4-(2-(2-oxo-4-phenylpyridin-1(2H)-yl)ethyl)piperidine-4- carboxylic acid [3.1.1e]

3.1.1d (66 mg, 0.155 mmol) was dissolved in HCI solution in EtOH (1.25 M, 2.5 mL, 3.1 mmol) and the resulting solution was stirred at room temperature overnight. The reaction mixture was concentrated in vacuo to afford 3.1.1e (56.1 mg, 100 % yield). LC/MS (m/z) 327.23 [M+H]+.
Step 6. Synthesis of 1-acetyl-4-(2-(2-oxo-4-phenylpyridin-1(2H)- yl)ethyl)piperidine-4-carboxylic acid [3.1.1f]

Ac20 (0.016 ml, 0.171 mmol) was added to a solution of 3.1.1e (0.054 g, 0.155 mmol) and Et3N (0.215 ml, 1.550 mmol) in DCM (3.1 mL). After stirring at room temperature for 30 minutes, the reaction mixture was concentrated in vacuo to afford 3.1.1f (quantitative yield). The crude product was continued to the next step without further purification. LC/MS (m/z) 369.2 [M+H]+.
Step 7. Synthesis of 1-acetyl-N-hydroxy-4-(2-(2-oxo-4-phenylpyridin-1(2H)- yl)ethyl)piperidine-4-carboxamide [3.1.1].

A solution of 3.1.1f (57.1 mg, 0.155 mmol), 0-(tetrahydro-2H-pyran-2- yl)hydroxylamine (27.2 mg, 0.233 mmol), EDC HCI (41.6 mg, 0.217 mmol), aza-HOBt (38.0 mg, 0.279 mmol) and Et3N (0.038 mL, 0.279 mmol) in DMF (1.5 mL) was stirred for 72 hours at room temperature. The reaction mixture was diluted with water and brought to a pH of 4 by adding saturated aqueous NH4CI solution. The aqueous layer was extracted with
EtOAc/Et20 (1/1 ). The organic layer was washed with water, brine, dried (MgS04) and concentrated. The remaining material was purified by silica gel column chromatography, (EtOAc/heptane 50-100%) to afford 1-acetyl-4-(2-(2-oxo-4-phenylpyridin-1 (2H)-yl)ethyl)-N- ((tetrahydro-2H-pyran-2-yl)oxy)piperidine-4-carboxamide (32 mg, 44.2 % yield). This material was dissolved in 1.0 M HCI ethanolic solution (1.25 M,. 1.2 mL, 1.75 mmol) and the reaction was stirred for 2 hours. Et20 was added to the reaction mixture was the white precipitate was collected by filtration to afford 3.1.1 (29.1 mg, 48.5 % yield). LC/MS (m/z) 384.2 [M+H]+. 1H NMR (400 MHz, CD3OD) δ ppm 1.47 - 1.68 (m, 2 H) 1.96 - 2.07 (m, 2 H) 2.09 - 2.30 (m, 5 H) 3.03 - 3.13 (m, 1 H) 3.33 - 3.40 (m, 1 H) 3.77 (d, J=13.99 Hz, 1 H) 4.03 - 4.20 (m, 4 H) 7.02 (br. s., 1 H) 7.10 (d, J=6.16 Hz, 1 H) 7.48 - 7.57 (m, 3 H) 7.73 (dd, J=4.23, 1.74 Hz, 2 H) 7.94 (d, J=6.99 Hz, 1 H).
3.1.2 Synthesis of compound 3.1.2


3.1.1c (220 mg, 0.484 mmol) was dissolved in HCI solution (4.0 M in dioxane, 4.84 mL, 19.36 mmol) and the resulting solution was stirred at room temperature for 5 days. The reaction mixture was diluted with water and neutralized by adding aqueous NaOH solution. The mixture was extracted with EtOAc. The organic layer was washed with brine, dried over magnesium sulfate, filtered and concentrated to afford 3.1.2a (150 mg, 87 % yield). LCMS (m/z) 355.2 [M+H]+. 1H NMR (400 MHz, CD3OD) δ ppm 1.31 (t, J=7.24 Hz, 3 H) 1 .49 - 1.65 (m, 2 H) 1.93 - 2.07 (m, 2 H) 2.23 (d, J=13.69 Hz, 2 H) 2.64 - 2.81 (m, 2 H) 3.03 (dt, J=13.1 1 , 3.62 Hz, 2 H) 3.94 - 4.05 (m, 2 H) 4.21 (q, J=7.04 Hz, 2 H) 6.70 (dd, J=7.04, 2.35 Hz, 1 H) 6.74 (d, J= .96 Hz, 1 H) 7.10 - 7.27 (m, 1 H) 7.28 - 7.73 (m, 6 H).
Step 2. Synthesis of ethyl 1-(methylsulfonyl)-4-(2-(2-oxo-4-phenylpyridin-1(2H)- yl)ethyl)piperidine-4-carboxylate [3.1.2b]

Methanesulfonyl chloride (36.3 μΙ, 0.466 mmol) was added to a solution of 3.1.2a (150 mg, 0.423 mmol) and Et3N (0.176 mL, 1.270 mmol) in DCM (4.2 mL) at 0 °C and the reaction mixture was allowed to warm to room temperature and stirred for 6 hours. The reaction mixture was quenched with water and extracted with Et20. The organic layer was washed with water and brine, dried over MgS04, filtered and concentrated on to silica gel. Purification by silica gel column chromatography (EtOAc/heptane 0-50%) afforded 3.1.2b
(152 mg, 83% yield). 1H NMR (400 MHz, CDCI3) δ ppm 1.34 (t, J=7.24 Hz, 3 H) 1.64 - 1.76 (m, 2 H) 1.97 - 2.13 (m, 2 H) 2.31 (d, J=13.69 Hz, 2 H) 2.77 (s, 3 H) 2.84 - 2.99 (m, 2 H) 3.54 - 3.69 (m, 2 H) 3.87 - 3.98 (m, 2 H) 4.25 (q, J=7.17 Hz, 2 H) 6.44 (dd, J=7.04, 1.96 Hz, 1 H) 6.76 (d, J=1.57 Hz, 1 H) 7.39 - 7.50 (m, 3 H) 7.56 (dd, J=7.43, 1.96 Hz, 2 H) 7.75 (d, J=6.65 Hz, 1 H).
Step 3. Synthesis of 1-(methylsulfonyl)-4-(2-(2-oxo-4-phenylpyridin-1(2H)- yl)ethyl)piperidine-4-carboxylic acid [3.1.2c]

A solution of 3.1.2b (152 mg, 0.351 mmol) and LiOH H20 (14.75 mg, 0.351 mmol) in MeOH (2.6 mL) and H20 (0.87 mL) was stirred at room temperature for 4 days, at 40 °C for 24 hours and at 60°C for 3 hours. The reaction mixture was concentrated to 1/3 of its volume then acidified by adding 1.0 M HCI aqueous solution. The precipitate was collected by filtration to afford 3.1.1c (142 mg, 100 % yield). LCMS (m/z) 405.2 [M+H]+.
Step 4. Synthesis of N-hydroxy-1-(methylsulfonyl)-4-(2-(2-oxo-4-phenylpyridin- 1 (2H)-yl)ethyl)piperidine-4-carboxamide [3.1.2]

Compound 3.1.2 was prepared following the procedure described for the synthesis of 3.1.1 using 3.1.2c in Step 7. LC/MS (m/z) 420.2 [M+H]+. 1H NMR (400 MHz, CD3OD) δ ppm 1.70 (ddd, J=13.89, 10.37, 3.91 Hz, 2 H) 1.94 - 2.08 (m, 2 H) 2.26 (d, J=14.09 Hz, 2 H) 2.82 (s, 3 H) 2.92 - 3.04 (m, 2 H) 3.48 - 3.56 (m, 2 H) 4.10 - 4.19 (m, 2 H) 7.03 (d, J=1.57 Hz, 1
H) 7.12 (dd, J=7.04, 1.96 Hz, 1 H) 7.49 - 7.57 (m, 3 H) 7.74 (dd, J=6.85, 2.93 Hz, 2 H) 7.95 (d, J=6.65 Hz, 1 H).
11-3.1.3 Synthesis of compound 3.1.3

3.1.3
Step 1. Synthesis 1-tert-butyl 4-ethyl 4-([1 ,1'-biphenyl]-4-ylmethyl)piperidine- 1,4-dicarboxylate [3.1.3a]

3.1.3a
□HMDS solution in THF (2.9 ml_, 1.0 M, 2.9 mmol) was added to a solution of 1-tert- butyl 4-ethyl piperidine-1 ,4-dicarboxylate (500 mg, 1.943 mmol) in THF (9.7 mL) at -78 °C and the reaction was stirred for 1 hour. To this solution, 4-(bromomethyl)-1 , 1 '-biphenyl (960 mg, 3.89 mmol) was added and the reaction mixture was warmed to room temperature overnight. The reaction was quenched with saturated aqueous NH4CI solution and extracted with EtOAc. The organic layer was washed with water and brine, dried over magnesium sulfate, filtered and concentrated on to silica. Purification by silica gel column
chromatography, (EtOAc/heptane 0-30%) afforded 3.1.3a (762 mg, 93 % yield). LCMS (m/z) 368.25 [M+H]+. 1H NMR (400 MHz, CDCI3) δ ppm 1.20 (t, J=7.24 Hz, 3 H) 1.37 - 1.51 (m, 12 H) 2.13 (d, J=13.30 Hz, 2 H) 2.73 - 2.92 (m, 5 H) 3.83 - 4.05 (m, 2 H) 4.13 (q, J=7.17 Hz, 2 H) 7.12 (d, J=7.83 Hz, 2 H) 7.34 (d, J=7.43 Hz, 1 H) 7.43 (t, J=7.63 Hz, 2 H) 7.49 (d, J=7.83 Hz, 2 H) 7.56 (d, J=7.43 Hz, 2 H).
Step 2. Synthesis of ethyl 4-([1,1'-biphenyl]-4-ylmethyl)piperidine-4-carboxylate
[3.1.3b]

3.1.3b
In a flask charged with 3.1.3a (400 mg, 0.944 mmol), 4.0 M HCI solution in dioxane (4.72 mL, 18.89 mmol) was added and the reaction was stirred at room temperature for 8 hours. The reaction mixture was concentrated in vacuo to afford 3.1.3b (340 mg, 100 % yield). LCMS (m/z) 324.9 [M+H]+. 1H NMR (400 MHz, CD3OD) δ ppm 1.24 (t, J=7.24 Hz, 3 H) 1.72 - 1.86 (m, 2 H) 2.34 (d, J=14.09 Hz, 2 H) 2.87 - 3.05 (m, 4 H) 3.36 (d, J=13.30 Hz, 2 H) 4.19 (q, J=7.30 Hz, 2 H) 7.19 (d, J=8.22 Hz, 2 H) 7.28 - 7.36 (m, 1 H) 7.42 (t, J=7.63 Hz, 2 H) 7.56 (dd, J=10.37, 8.02 Hz, 4 H).
Step 3. Synthesis of ethyl 4-([1,1'-biphenyl]-4-ylmethyl)-1-(methylsulfonyl) piperidine-4-carboxylate [3.1.3c]

3.1.3c
To a solution of 3.1.3b (100 mg, 0.278 mmol) in DCM at 0 °C, methanesulfonyl chloride (0.22 mL, 0.306 mmol) was added and the reaction was stirred at 0 °C for 2 hours. The reaction was quenched with water and extracted with EtOAc. The organic layer was washed with water and brine, dried over magnesium sulfate and concentrated on to silica gel. Purification by silica gel column chromatography (EtO Ac/heptane, 0-50%) afforded
3.1.3c (81 mg, 72.6 % yield). LCMS (m/z) 402.2 [M+H]+. 1 H NMR (400 MHz, CDCI3) δ ppm 1.16 - 1.26 (m, 3 H) 1.67 (td, J=12.91 , 4.30 Hz, 2 H) 2.28 (d, J=12.52 Hz, 2 H) 2.64 - 2.78 (m, 5 H) 2.89 (s, 2 H) 3.71 (d, J=12.13 Hz, 2 H) 4.16 (q, J=7.17 Hz, 2 H) 7.12 (d, J=7.83 Hz, 2 H) 7.30 - 7.37 (m, 1 H) 7.43 (t, J=7.43 Hz, 2 H) 7.50 (d, J=7.83 Hz, 2 H) 7.57 (d, J=7.43 Hz, 2 H).
Step 4. Synthesis of 4-([1,1'-biphenyl]-4-ylmethyl)-1-(methylsulfonyl)piperidine- 4-carboxylic acid [3.1.3d]

3.1.3d
A solution of NaOH aqueous solution (1.0 M in water, 0.24 ml_, 0.24 mmol) was added to a solution of 3.1.3c (81 mg, 0.202 mmol) in THF (1.0 ml_) and the reaction was stirred at room temperature for 1 hour and at 50°C for 24 hours. The reaction mixture was diluted with water, acidified to pH=3 by adding 1.0 M HCI aqueous solution and extracted with Et20. The organic layer was washed with brine, dried over MgS04, filtered and concentrated to afford 3.1.3d (62 mg, 53.5 % yield). LCMS (m/z) 374.2 [M+H]+.
Step 5. Synthesis of 4-([1,1'-biphenyl]-4-ylmethyl)-N-hydroxy-1-(methylsulfonyl) piperidine-4-carboxamide [3.1.3]

3.1.3
Compound 3.1.3 was prepared following the procedure described for the synthesis of 3.1.1 using 3.1.3d in Step 7. LCMS (m/z) 389.2 [M+H]+. 1H NMR (400 MHz, CD3OD) ppm 1.59 - 1.75 (m, 2 H) 2.16 (d, J=14.09 Hz, 2 H) 2.72 - 2.95 (m, 7 H) 3.51 - 3.62 (m, 2 H) 7.21 (d, J=8.22 Hz, 2 H) 7.25 - 7.33 (m, 1 H) 7.41 (t, J=7.63 Hz, 2 H) 7.53 (d, J=7.83 Hz, 2 H) 7.58 (d, J=7.43 Hz, 2 H).
3.2.1 Synthesis of S-i^'-chloro^'-fluoro-n.l'-biphenylJ^-y methy aminoJ-N-hydroxy- 1-(methylsulfonyl)azetidine-3-carboxamide [3.2.1]


A solution of 1-tert-butyl 3-ethyl 3-aminoazetidine-1 ,3-dicarboxylate (1.5 g, 6.14 mmol), 4-bromobenzaldehyde (1.250 g, 6.75 mmol) and acetic acid (1.055 ml, 18.42 mmol) in 1 ,2-Dichloroethane (Volume: 30.7 ml) was stirred at room temperature for 72 hours. Sodium triacetoxyborohydride (3.90 g, 18.42 mmol) was added and the resulting reaction mixture was allowed to stir for another 24 hours. The reaction was quenched with water and extracted with DCM. The organic layer was washed wtih saturated aqueous NaHC03 solution and brine. The organic layer was dried over magnesium sulfate, filtered and concentrated. The residue was was purified by silica gel column chromatography,
EtOAc/heptane 501-100% to afford product 3.2.1a (1.58 g, 62.3 % yield). 1H NMR (400 MHz, CDCI3) δ ppm 1.33 (t, J=7.04 Hz, 3 H) 1.45 (s, 9 H) 3.62 (s, 2 H) 3.85 (d, J=8.61 Hz, 2 H) 4.20 (d, J=9.00 Hz, 2 H) 4.26 (q, J=7.04 Hz, 2 H) 7.19 - 7.25 (m, 2 H) 7.45 (d, J=8.22 Hz, 2 H). LC/MS (m/z) 415.1 [M+H]+
Step 2. Synthesis of ethyl 3-((4-bromobenzyl)amino)azetidine-3-carboxylate
[3.2.1 b]

3.2.1a (1.58 g, 3.82 mmol) was dissolved in a solution of HCI in EtOH (1.0 M, 10 mL) and the resulting solution was stirred at room temperature for 18 hours. Diethyl ether was added to to the solution and after 48 hours a white precipitate crashed out. The solid was collected by filtration to afford amine HCI salt (1.3 g, 88 % yield). 1H NMR (400 MHz, CD3OD) 5 ppm 1.45 (t, J=7.24 Hz, 3 H) 4.19 (br. s., 2 H) 4.44 - 4.60 (m, 4 H) 4.67 (d, J=1 1.74 Hz, 2 H) 7.46 - 7.55 (m, 2 H) 7.63 (d, J=8.61 Hz, 2 H).
Step 3. Synthesis of ethyl 3-((4-bromobenzyl)amino)-1- (methylsulfonyl)azetidine-3-carboxylate [3.2.1c]

A solution of 3.2.1 b (500 mg, 1.295 mmol) and DIEA (0.905 ml_, 5.18 mmol) in DCM (6.5 ml_) was placed in an ice water bath. Methanesulfonyl chloride (0.131 ml_, 1.683 mmol) was slowly added and the resulting mixture was stirred at room temperature over 1 hour. The reaction was quenched with water and extracted with DCM. The DCM layer was washed with water and brine, dried over magnesium sulfate, filtered and concentrated. The remaining oil was purified by silica gel column chromatography, EtOAc/heptane 10-75% to afford product 3.2.1c (373 mg, 73.6 % yield). 1H NMR (500 MHz, CDCI3) δ ppm 1.37 (t, J=7.09 Hz, 3 H) 2.95 (s, 3 H) 3.68 (s, 2 H) 3.92 (d, J=8.51 Hz, 2 H) 4.25 (d, J=8.51 Hz, 2 H) 4.31 (q, J=7.25 Hz, 2 H) 7.24 (d, J=8.20 Hz, 2 H) 7.48 (d, J=8.51 Hz, 2 H). LC/MS (m/z) 393.0 [M+H]+.
Step 3. Synthesis of 3-(((4,-chloro-2'-fluoro-[1 ,1'-biphenyl]-4-yl)methyl)amino)- N-hydroxy-1-(methylsulfonyl)azetidine-3-carboxamide [3.2.1]
Compound 3.2.1 was prepared following the procedure described for the synthesis of 1.1.2. 1H NMR (400 MHz, CD3OD) δ ppm 2.91 (s, 4 H) 3.83 (s, 2 H) 3.96 (d, J=9.39 Hz, 2 H) 4.15 (d, J=9.39 Hz, 2 H) 7.1 1 - 7.29 (m, 2 H) 7.32 - 7.52 (m, 5 H). LC/MS (m/z) 428.1
[M+H]+
4.1.1. Synthesis of compound 4.1.1 and 4.1.2

Step 1. Synthesis of (1s,4s)-methyl 1-(([1,1'-biphenyl]-4-ylmethyl)amino)-4- hydroxycyclohexanecarboxylate [4.1.1a] and (1 r,4r)-methyl 1-(([1,1'-biphenyl]-4- ylmethyl)amino)-4-hydroxycyclohexanecarboxylate [4.1.2a]

To a solution of methyl 1 -amino-4-oxocyclohexanecarboxylate (100 mg, 0.584 mmol, 1.0 equiv) in DCE (2.0 mL) was added para-phenyl benzaldehyde (213 mg, 1 .2 mmol, 2.0 equiv) and HOAc (0.1 mL, 1 .75 mmol, 3.0 equiv). After stirring at room temperature for 10 minutes, sodium triacetoxy borohydride (371 mg, 1 .75 mmol, 3.0 equiv) was added and the resulting solution was stirred at room temperature for 18 hours. The reaction was then quenched by addition of saturated aqueous NaHC03 solution and extracted with EtOAc. The organic layer was washed with water and brine, dried over Na2SC>4 and concentrated. The residue was purified by silica gel column chromatography, (EtOAc/heptane 0 to 60%) to afford product 4.1.1a (50 mg, 25% yield, less polar diastereomer) and 4.1.2a (40 mg, 20% yield, more polar diastereomer). 4.1.1a MS (m/z) 340.6 [M+H]+; 4.1.2a MS (m/z) 340.3
[M+H]+.
Step 2. Synthesis of compound 4.1.1 and 4.1.2

Compounds 4.1.1 and 4.1.2 were synthesized from 4.1.1a and 4.1.2a following the procedures described for the synthesis of 1.2.1 step 2-4.
4.1.1 : MS (m/z) 341 .3 [M+H]+. 1 H NMR (400 MHz, DMSO-c/6) 1 .42 - 1 .74 (m, 4 H) 1 .93 - 2.26 (m, 4 H) 3.62 - 3.76 (m, 1 H) 3.86 - 4.14 (m, 2 H) 5.59 - 5.99 (m, 1 H) 7.38 (d, J=7.43 Hz, 1 H) 7.47 (t, J=7.63 Hz, 2 H) 7.58 - 7.76 (m, 6 H) 7.82 - 7.86 (m, 1 H) 9.06 - 9.46 (m, 2 H) 1 1 .10 - 1 1 .47 (m, 1 H)
4.1 .2: MS (m/z) 341 .3 [M+H]+. 1 H NMR (400 MHz, DMSO-c/6) 1 .07 - 1 .35 (m, 2 H) 1 .54 - 1 .91 (m, 4 H) 2.22 - 2.38 (m, 2 H) 3.39 - 3.51 (m, 1 H) 3.80 - 4.06 (m, 2 H) 7.34 (d, J=7.43 Hz, 1 H) 7.38 - 7.45 (m, 2 H) 7.53 (d, J=7.83 Hz, 2 H) 7.59 - 7.75 (m, 4 H) 9.09 - 9.54 (m, 2 H) 1 1 .13 - 1 1 .42 (m, 1 H)
4.1.3 Synthesis of compound 4.1.3

Step 1. Synthesis of 4.1.3a

4.1.3a
NaBH4 (84 mg, 2.21 1 mmol, 2.0 equiv) was added to a solution of methyl 1 -((tert- butoxycarbonyl)amino)-4-oxocyclohexanecarboxylate (300 mg, 1 .106 mmol) in THF (2 ml_)/MeOH (2 mL) at 0 °C. The mixture was stirred for 30 minutes and then was quenched with 5 mL saturated aqueous NH4CI solution. The aqueous phase was extracted with EtOAc. The organic layer was dried over Na2S04, concentrated. The remaining oil was purified by silica gel column chromatography (EtOAc/heptane 50-100%) to afford product 4.1.3a (309 mg, 100% yield). MS m/z [M+H]+ 274.2
Step 2. Synthesis of 4.1.3b

4.1.3b
Methyl iodide (0.869 mL, 13.90 mmol, 20 equiv) was added to a mixture of 4.1.3a (190 mg, 0.695 mmol, 1.0 equiv), silver oxide (1.289 g, 5.56 mmol, 8.0 equiv) in acetonitrile (9 mL) and the resulting mixture was stirred for 16 hours at room temperature. The mixture was diluted with EtOAc and filtered. The filtrate was concentrated and the residue was purified by silica gel column chromatography (EtOAc/heptane, 30-100%) to afford product 4.1.3b (146 mg, 73% yield). MS m/z [M+H]+ 288.2
Step 3. Synthesis of 4.1.3c

4.1.3c
TFA (0.196 mL, 2.54 mmol, 5.0 equiv) was added to a solution of 4.1.3b (146 mg, 0.508 mmol, 1.0 equiv) in DCM (2 mL) and the resulting solution was stirred at the room temperature for 4 hours. The solution was concentrated and the residue was dissolved in 20 mL DCM, washed with saturated aqueous NaHC03 solution, dried over Na2S04 and concentrated. The material was continued to the next step without further purification. MS m/z [M+H]+ 188.4
Step 4. Synthesis of methyl 1-(([1,1'-biphenyl]-4-ylmethyl)amino)-4- methoxycyclohexanecarboxylate [4.1.3d]

4.1.3d
[1 ,1'-biphenyl]-4-carbaldehyde (238 mg, 1.309 mmol, 2.5 equiv) and AcOH (0.090 mL, 1.570 mmol, 3.0 equiv) were added to a solution of 4.1.3c (98 mg, 0.523 mmol, 1.0 equiv) in dichloroethane (2 mL). After stirring at room temperature for 10 minutes,
NaBH(OAc)3 (333 mg, 1.570 mmol, 3.0 equiv) was added and the solution was stirred at room temperature for 16 hours. To the mixture, EtOAc and saturated aqueous NaHC03 solution were added. The phases were separated and the aqueous layer was extracted with 15 mL EtOAc. The combined organic layers were dried over Na2S04 and concentrated. The
residue was purified by silica gel column chromatography (EtOAc/heptane, 5-30%) to afford product 4.1.3c (123 mg, 66.5% yield). MS m/z [M+H]+ 354.3. 1H NMR (400 MHz,
CHLOROFORM-d) δ ppm 7.50 - 7.62 (m, 4 H) 7.40 - 7.4,9 (m, 4 H) 7.31 - 7.38 (m, 1 H) 3.71 - 3.77 (m, 3 H) 3.61 (s, 2 H) 3.32 - 3.41 (m, 3 H) 3.24 (m, J=3.91 Hz, 1 H) 1.73 - 1.95 (m, 8 H)
Step 5. Synthesis of 1-(([1,1'-biphenyl]-4-ylmethyl)amino)-N-hydroxy-4- methoxycyclohexanecarboxamide [4.1.3]

Compound 4.1.3d was converted to 4.1.3 following the procedure described for the synthesis of 1.2.1 (step2-4). MS m/z 355.3 [M+H]+. 1H NMR (500 MHz, CD3OD) δ ppm 7.74 (m, J=8.20 Hz, 2 H) 7.66 (dd, J=8.20, 0.95 Hz, 2 H) 7.59 (m, J=8.20 Hz, 2 H) 7.46 - 7.50 (m, 2 H) 7.37 - 7.42 (m, 1 H) 4.12 (s, 2 H) 3.52 (br. s., 1 H) 3.38 (s, 3 H) 2.28 (d, J=13.24 Hz, 2 H) 2.12 (br. s., 2 H) 1.91 - 2.03 (m, 2 H) 1.69 - 1.81 (m, 2 H)
5.1.1. Synthesis of 3 S-i^'-chloro^'-fluoro-tl.l'-biphenylJ^-y methy aminoJ-N- hydroxytetrahydrofuran-3-carboxamide [5.1.1]

Compound 5.1.1 was synthesized following the procedures described for the synthesis of 1.2.1 using 4'-chloro-2'-fluoro-[1 ,1 '-biphenyl]-4-carbaldehyde and methyl 3- aminotetrahydrofuran-3-carboxylate in step 1. HRMS m/z 365.1075 [M+H]+. 1H NMR (400 MHz, CD3OD) 7.63 (s, 4 H) 7.50 (t, J=8.41 Hz, 1 H) 7.33 (d, J=2.74 Hz, 1 H) 7.31 (s, 1 H) 3.99 - 4.29 (m, 6 H) 2.37 - 2.60 (m, 2 H).
4.2.1 Synthesis of l-i^'-fluoro^'-methoxy-n.l'-biphenyll^-y methy aminoJ-N^- dihydroxycyclobutanecarboxamide [4.2.1]

Step 1. Synthesis of ethyl 1-((tert-butoxycarbonyl)am
hydroxycyclobutanecarboxylate [4.2.1a]

4.2.1a
Sodium borohydride (0.29 g, 7.7 mmol) was added to a solution of ethyl 1 -((tert- butoxycarbonyl)amino)-3-oxocyclobutanecarboxylate (2.0 g, 7.7 mmol) in THF (26 ml_) at room temerature and the resulting mixture was stirred at room temperature for 2 hours. The reaction was then quenched by saturated aqueous NaHC03 solution. The mixture was extracted with EtOAc. The organic layers were washed with brine, dried over Na2S04 and concentrated. The residue was purified by silica gel column chromatography,
EtOAc/heptane 0-100% to afford product 4.2.1a 1.5 g. The product contained mixtures of diastereomers with 3/1 ratio. LC/MS (m/z) 260.2 [M+H]+.
Step 2. Synthesis of l-^'-fluoro^'-methoxy-II.I'-biphenyl]^- yl)methyl)amino)-N,3-dihydroxycyclobutanecarboxamide [4.2.1]

Compound 4.2.1 was prepared following the procedures described for the synthesis of 4.1.3 Steps 3-5. The product was purified by reverse phase HPLC to give two diastereomers.
4.2.1a: 1H NMR (400 MHz, DMSO-c/6) δ ppm 2.63 - 2.82 (m, 2 H) 3.47 - 3.66 (m, 1 H) 3.97 (br. s., 2 H) 4.51 (t, J=6.65 Hz, 1 H) 6.76 - 7.08 (m, 2 H) 7.37 - 7.67 (m, 5 H) 9.22 - 9.50 (m, 1 H) 9.76 (br. s., 2 H) 1 1.41 (br. s., 1 H). LC/MS (m/z) 361.1 [M+H]+
4.2.1b: 1H NMR (400 MHz, DMSO-c/6) δ ppm 2.16 - 2.38 (m, 2 H) 2.57 - 2.73 (m, 2 H) 3.83 - 3.94 (m, 2 H) 3.99 - 4.33 (m, 1 H) 5.35 - 5.85 (m, 1 H) 6.76 - 7.15 (m, 2 H) 7.48 (s, 6
H) 9.31 - 9.49 (m, 1 H) 9.50 - 9.69 (m, 1 H) 1 1.1 1 - 1 1.33 (m, 1 H). LC/MS (m/z) 361.1 [M+H]+
5.2.1. Synthesis of S-iii^-chloro^'-fluoro-n.l'-biphenyll^-y methy aminoJ-N- hydroxyoxetane-3-carboxamide [5.2.1]

Compound 5.2.1 was synthesized following the procedures described for the synthesis of 1.2.1 using 4'-chloro-2'-fluoro-[1 ,1 '-biphenyl]-4-carbaldehyde and methyl 3- aminooxetane-3-carboxylate in step 1. HRMS m/z 351.0916 [M+H]+. 1H NMR (500 MHz, CD3OD) 7.58 - 7.70 (m, 4 H) 7.53 (t, J=8.51 Hz, 1 H) 7.28 - 7.41 (m, 2 H) 4.93 (br. s., 2 H) 4.82 (d, J=8.20 Hz, 2 H) 4.14 (s, 2 H)
5.3.1. Synthesis of N-hydroxy-4-(2-(2-oxo-4-phenylpyridin-1(2H)-yl)ethyl)tetrahydro- 2H-pyran-4-carboxamide[5.3.1]

Compound 5.3.1 was prepared following the procedures described for the synthesis of 3.1.1. HRMS m/z 343.1654 [M+H]+. 1H NMR (400 MHz, CD3CI) 7.54 - 7.62 (m, 2 H) 7.49 (br. s., 1 H) 7.48 (d, J=1.96 Hz, 2 H) 7.43 (d, J=7.04 Hz, 1 H) 6.93 (s, 1 H) 6.69 (m, 1 H) 4.02 (d, J=8.61 Hz, 2 H) 3.76 (t, J=5.28 Hz, 4 H) 2.17 (d, J=3.13 Hz, 2 H) 2.15 (br. s., 2 H) 1.57 (m, 2 H)
5.3.2 Synthesis of compound 5.3.2
Step 1. Synthesis of ethyl 4-(4-bromophenethyl)tetrahydro-2H-pyran-4- carboxylate [5.3.2a]

5.3.2a
A solution of LDA (2.465 ml, 4.93 mmol) and THF (5.0 mL) was cooled at -78 oC. Ethyl tetrahydro-2H-pyran-4-carboxylate (600 mg, 3.79 mmol) in THF (2.0 mL) was added dropwise to above solution and the solution was stirred at -78 °C for 30 minutes. Then 1- bromo-4-(2-bromoethyl)benzene (0.873 ml, 5.69 mmol) was added. The mixture was stirred at -78 oC for 30 minutes and warmed to room temperature. The reaction was quenched with saturated aqueous NH4CI solution and extracted with EtOAc. The organic layer was washed with water and brine, dried over Na2S04, filtered and concentrated on to silica. Purification by silica gel column chromatography (EtO Ac/heptane, 0 to 70%) to afford 5.3.2a (85.4 mg, 6.6% yield). LCMS (m/z) 343.1 [M+H]+. 1H NMR (400 MHz, CDCI3) δ ppm 1.31 (t, J=7.24 Hz, 3 H) 1.50 - 1.62 (m, 2 H) 1.72 - 1.91 (m, 2 H) 2.15 (d, J=12.13 Hz, 2 H) 2.38 - 2.60 (m, 2H) 3.48 (td, J=1 1.74, 1.96 Hz, 2 H) 3.86 (dt, J=1 1.84, 3.47 Hz, 2 H) 4.21 (q, J=7.17 Hz, 2 H) 7.02 (d, J=8.22 Hz, 2 H) 7.40 (d, J=8.22 Hz, 2 H).
Step 2. Synthesis of ethyl 4-(2-([1,1'-biphenyl]-4-yl)ethyl)tetrahydro-2H-pyran-4- carboxylate [5.3.2b]

Step 3. Synthesis of 4-(2-([1,1'-biphenyl]-4-yl)ethyl)tetrahydro-2H-pyran-4- carboxylic acid [5.3.2c]

LiOH (9.20 mg, 0.384 mmol) was added to a solution of 5.3.2b (13 mg, 0.038 mmol) in THF/MeOH/water (0.2/0.2/0.02 mL) and the solution was stirred at 85 °C for 16 hours. The reaction was concentrated, diluted with water and EtOAc. The aqueous layer was acidified to pH=1 by adding 6.0 N HCI aqueous solution and was extracted with EtOAc. The organic layer was dried over Na2S04 and concentrated to afford 5.3.2c (10 mg, 84 % yield). LCMS (m/z) 293.2 [M-OH]
Step 4. Synthesis 4-(2-([1,1'-biphenyl]-4-yl)ethyl)-N-hydroxytetrahydro-2H- pyran-4-carboxamide [5.3.2]
Oxalyl chloride (0.0028 mL, 0.032 mmol) and DMF (0.249 μΙ, 3.22 μιτιοΙ) was added to a solution of 5.3.2c (10 mg, 0.032 mmol) in DCM (0.4 mL). After stirred at room temperature fpr 1 hour the mixture was concentrated. The remaining material was dissolved in DCM (0.4 mL) and a solution of NH2OH HCI (4.48 mg, 0.064 mmol), Et3N (0.022 mL, 0.161 mmol) in DCM (0.4 mL) was added. The reaction mixture was stirred at room
temperature for 2 h and then was diluted with DCM, washed with water and brine. The organic layer was dried over Na2S04 and concentrated. The residue was purified by reverse phase prep HPLC to give product 5.3.2 (1.5 mg, 10 % yield). LCMS (m/z) 326.1 [M+H]+.
5.3.3 Synthesis of compound 5.3.3

Step 1. Synthesis methyl 4-(([1,1'-biphenyl]-4-ylmethyl)amino)tetrahydro-2H- pyran-4-carboxylate [5.3.3a]

Compound 5.3.3a was prepared following the procedure described for the synthesis of 1.1.1a using methyl 4-aminotetrahydro-2H-pyran-4-carboxylate in Step 1. LCMS (m/z) 326.2 [M+H]+.
Step 2. Synthesis of 4-(([1,1'-biphenyl]-4-ylmethyl)amino)-N-hydroxytetrahydro- 2H-pyran-4-carboxamide [5.3.3]

Hydroxylamine (50% in water, 1.5 mL, 25.6 mmol) and NaOH (1.0 M in water, 0.128 mL, 0.128 mmol) was to a solution of 5.3.3a (83.3 mg, 0.256 mmol) in MeOH (2.5 mL) and the reaction mixture was stirred at room temperature for 72 hours. The mixture was concentrated and the residue was purified by reverse phase HPLC to give product 5.3.3 (12.2 mg, 10.6 % yield). LCMS (m/z) 327.2 [M+H]+. 1H NMR (400 MHz, CD3OD) δ ppm 1.97
(d, J=16.04 Hz, 2 H) 2.46 (d, J=13.30 Hz, 2 H) 3.61 (t, J=10.76 Hz, 2 H) 3.91 - 4.08 (m, 2 H) 4.13 (s, 2H) 7.36 - 7.43 (m, 1 H) 7.47 (t, J=7.43 Hz, 2 H) 7.58 (d, J=8.22 Hz, 2 H) 7.62 - 7.69 (m, 2 H) 7.75 (d, J=7.83 Hz ,2H).
Table 1. UPLC retention time and HRMS


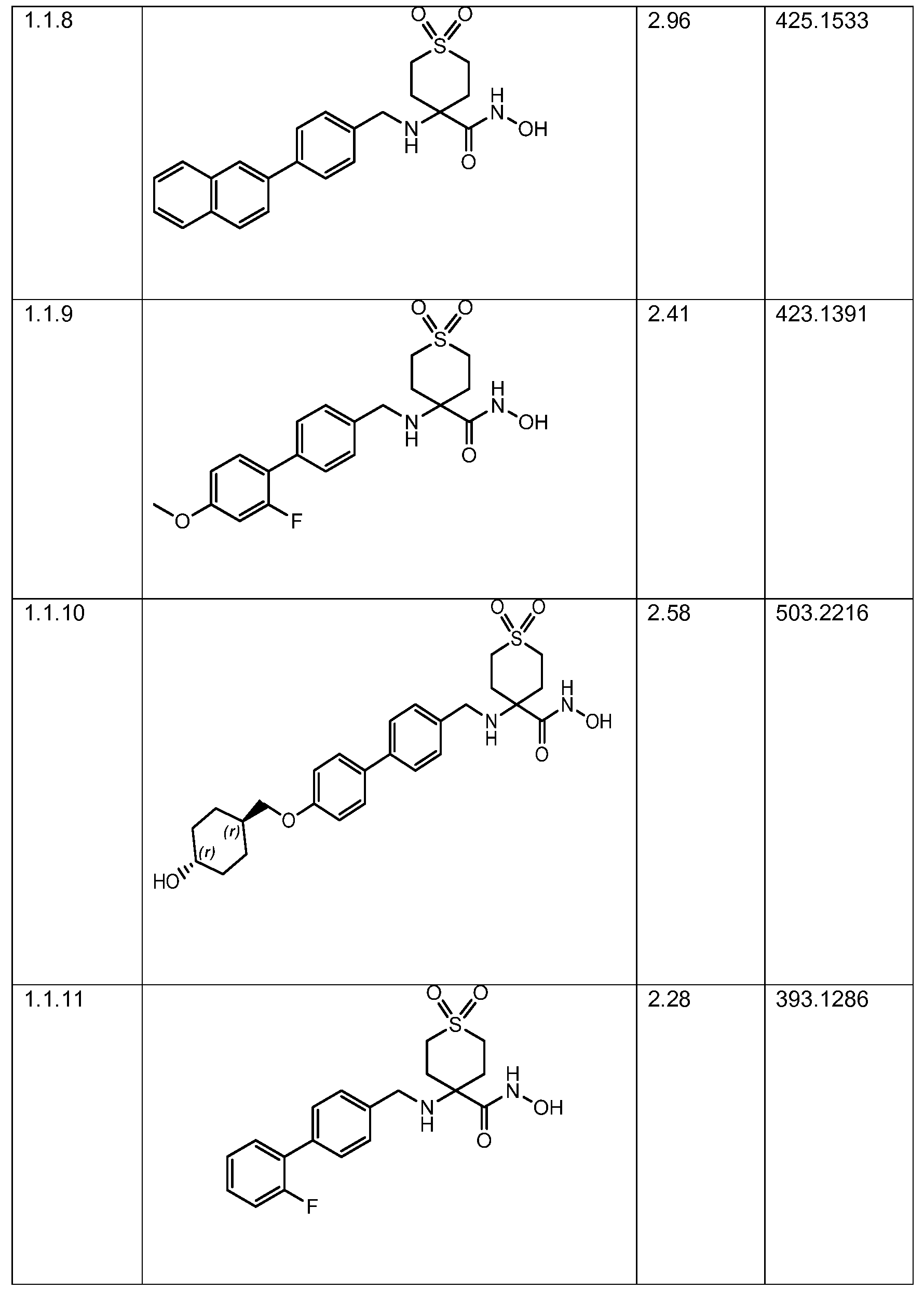
















Pharmaceutical Activity
Example P. aeruginosa LpxC Inhibition Assay
The P. aeruginosa LpxC protein is produced according to the general method of Hyland et al (Journal of Bacteriology 1997 179, 2029-2037: Cloning, expression and purification of UDP-3-O-acyl-GlcNAc deacetylase from Pseudomonas aeruginosa: a metalloamidase of the lipid A biosynthesis pathway). The LC-MS/MS method for quantitation of LpxC product was developed using an Agilent 1200 Capillary HPLC system coupled to an Applied Biosystems MDS Sciex 4000QTRAP mass spectrometer. Both instruments are controlled using the Applied Biosystems MDS Sciex Analyst software. LpxC reaction product (UDP-3-0-(R-3-hydroxyacyl)-glucosamine) was produced by hydrolysis of LpxC substrate catalyzed by P. a. LpxC and purified using reversed phase chromatography on a
Phenomenex Luna C18(2) 4.6 x 50 mm column. An LpxC product calibration curve was generated to evaluate the sensitivity and dynamic range of the LC-MS/MS method. Briefly, compounds are pre-incubated with 1 nM P. aeruginosa LpxC for 30 min. at room
temperature. Reactions are initiated by the addition of 2 μΜ UDP-3-0-(R-3- hydroxydecanoyl)-GlcNAc. Reactions are conducted in a 384-well plate with a total volume
of 50 [it in each well containing 50 mM Sodium phosphate pH 7.5, 0.005% Trition X-100 for 20 min at room temperature. After quenching with 1 .8% HOAc (5 μΙ_ of a 20% HOAc added to each well), reaction mixtures are analyzed using the LC-MS/MS method and peak areas are transformed into product concentration using a LpxC product calibration curve. Total activity (0% inhibition control) is obtained from reactions with no inhibitors and 100% inhibition control is the background using quenched samples before reaction starts. For IC50 determinations, peak areas are converted to percent inhibition in Microsoft Excel. Percent inhibition values are plotted vs. log compound concentration using XLfit. Data is fit to the four-parameter logistic equation using the non-linear regression algorithm in XLfit to return the IC5o and hill slope values.
Bacterial Screens and Cultures
Bacterial isolates were cultivated from -70°C frozen stocks by two consecutive overnight passages at 35°C in ambient air on 5% blood agar (Remel, Lenexa, Kans.).
Quality control and P. aeruginosa ATCC 27853) is from the American Type Culture
Collection (ATCC; Rockville, Md.) and PA01 was received from Dr. K. Poole.
Susceptibility Testing
Minimal Inhibitory Concentrations (MICs) were determined by the broth microdilution method in accordance with Clinical and Laboratories Institute (CLSI) guidelines. In brief, fresh bacterial overnight cultures were resuspended in sterile saline, adjusted to a 0.5 McFarland turbidity standard and then diluted 20010-fold in cation adjusted Mueller-Hinton Broth II (MHB; Remel BBL) to yield a final inoculum of approximately 5x105 colony-forming units (CFU)/mL. Two-fold serial dilutions of compounds were prepared in 100% dimethyl sulfoxide (DMSO) at 100-fold the highest final assay concentration; the resulting dilution series of compounds were diluted 1 : 10 with sterile water. Ten μΙ of the drug dilution series in 10% DMSO was transferred to microtiter wells and 90 μΙ of bacterial suspension was inoculated into the wells. All inoculated microdilution trays were incubated in ambient air at 37 35°C for 20 hours. Following incubation, assay plates were read in a microtiter plate reader at 600 nm and visually inspected to confirm the MIC endpoint well with the OD value. Tthe lowest concentration of the compound that prevented visible growth was recorded as the MIC. Performance of the assay was monitored by testing ciprofloxacin against laboratory quality control strains in accordance with guidelines of the
CLSI. Compounds of Examples 1 -6, 8-19, 21 , 23-26 and 28-53 exhibit an MIC of 64 μg mL against at least one P. aeruginosa strain selected from PA01 and ATCC27853.
The LpxC inhibitory activity for the compounds of Examples is reported in Table A.
Table A: Biological data for the compounds above.

1.2.1 1 0.0003 1
1.2.12 <0.0005 0.75
1.2.13 0.0001 16
1.2.14 0.0003 0.75
1.2.15 0.0008 1
1.2.16 0.0008 2
1.2.17 0.0005 2
1.2.18 0.0004 8
1.2.19 0.002 4
1.2.20 0.0005 8
1.2.21 0.0002 4
1.2.22 0.0003 2
1.2.23 0.0006 2
1.2.24 0.0005 4
1.2.25 0.001 2
1.2.26 0.0002 2
1.2.27 <0.0005 8
1.2.28 <0.0005 4
1.2.29 0.003 16
1.2.30 0.001 8
1.2.31 0.005 2
1.2.32 0.005 8
1.2.33 0.0003 1
1.3.01 0.0051 64 32
1.4.01 0.0006 8 4
1.4.2 0.002 2
1.5.1 0.03 32
1.5.2 0.01 32
1.6.1 0.2 64
2.1.01 0.0019 16 4
2.2.01 0.002 4 2
2.2.02 0.002 16 8
2.2.03 0.001 8 4
2.2.4 0.001 2 1
2.2.5a 0.0005 1
2.2.5b 0.0005 0.5
3.1.01 0.12 > 64 > 64
3.1.02 0.005 32 16
3.1.03 0.054 > 64 > 64
3.2.1 0.0005 1
3.3.1 0.0005 8
4.1.01 < 0.0005 16 8
4.1.02 0.00175 64 16
4.1.03 0.042 64 32
4.2.1 a 0.0005 2
4.2.1 b 0.002 2
4.3.1 a 0.001 4
4.3.1 b 0.01 32
5.1.01 < 0.0005 4 1
5.2.01 < 0.0005 4 1
5.3.01 0.009 32 32
5.3.02 0.002
5.3.03 0.002 32 32
Those skilled in the art will recognize, or be able to ascertain using no more than routine experimentation, many equivalents to the specific embodiments and methods described herein. Such equivalents are intended to be encompassed by the scope of the following claims.
Claims
1. A compound of the formula I:

A is a divalent radical selected from , and ;
X is -(CH2)nY(CH2)m-;
Y is selected from the group consisting of -C(H,R1)-, -0-, S, -N(R2)-, and -S(0)2- n is 0 or 1 ; m is 0 or 1 ;
R is C3-6 cycloalkyi, -C6-Ci0aryl, or 4 to 10 membered heteroaryl containing 1 to 3 heteroatoms selected from N, S and O, wherein said cycloalkyi, aryl and heteroaryl are each optionally substituted with up to three substituents selected from the group consisting of halogen, -OH, -CN , -S(0)2(CrC4)alkyl, C C4haloalkyl, C3-C7cycloalkyl, C C4alkoxy, C C4haloalkoxy, C C alkyl optionally substituted with C C alkoxy or a 5-6 membered heterocycle containing up to two heteroatoms selected from N, O and S as ring members and optionally substituted with R10, C C alkoxy optionally substituted with Ci-C alkoxy or Ci_ 3 alkyl or C3-e cycloalkyi where the C1-3 alkyl or C3-e cycloalkyi are each optionally substituted with hydroxy, methoxy, or methyl, and a 4 to 7 membered heterocycle or a 5 to 6 membered heteroaryl wherein the 4 to 7 membered heterocycle or 5 to 6 membered heteroaryl contains 1 to 3 heteroatoms selected from N, S, and O as ring members and is optionally substituted with one or more halogen, C C alkoxy, CrC haloalkoxy, CrC haloalkyl or Ci-C alkyl; or
R is -C6-Ci0aryl, or 4 to 10 membered heteroaryl containing 1 to 3 heteroatoms selected from N, S and O, wherein said aryl and heteroaryl are susbstituted by taking the substituents on adjacent atoms of the -C6-Ci0aryl, or 4 to 10 membered heteroaryl and forming a 3 to 7 membered heterocycle, wherein the formed bicycle substituent is further optionally substituted with one or two groups selected from halogen, d-C4alkyl, c
C4haloalkyl, CrC4haloalkoxy and CrC4alkoxy;
R1 is selected from the group consisting of -OH, Ci-C4alkoxy and -S(0)2R3;
R2 is selected from the group consisting of hydrogen, C C alkyl, -C(0)OR3 , -C(0)R3 and -S(0)2R3;
R3 is selected from the group consisting of C C alkyl and C3-C6cycloalkyl; Z is a divalent radical selected from

; or

Z is with the proviso that
R5 is selected from the group consisting of hydrogen, halogen, -CN, d-dalkyl, and Ci-C4haloalkyl;
R6, R6a, R6b or R6c are independently selected from the group consisting of hydrogen, halogen, -CrC4alkyl, and C C4haloalkyl;
R10 is selected from halo, d.4 alkyl, d-4 haloalkyl, d_4 alkoxy, -C(0)R11 and -C(O)-
OR11 ;
R11 is d.4 alkyl; and '
L is a divalent bond, -CH2-, -O- or
2. The compound or a pharmaceutically acceptable salt thereof according to claim 1 , represented by formula II:

II

wherein Q is selected from the group consisting of

A is a divalent radical selected from , and
R is -C6-Ci0aryl, or 4 to 10 membered heteroaryl containing 1 to 3 heteroatoms selected from N, S and O, wherein said aryl and heteroaryl are optionally substituted with a substituent selected from the group consisting of halogen, -OH, -CN, -S(0)2(CrC4)alkyl, d- C4haloalkyl, C3-C7cycloalkyl, CrC4alkoxy, CrC4haloalkoxy, C C alkyl optionally substituted with Ci-C alkoxy, CrC alkoxy optionally substituted with Ci-C alkoxy and a 4 to 7 membered heterocycle containing 1 to 3 heteroatom selected from N, S, and O, wherein said heterocycle is optionally substituted with one or more halogen, C C alkoxy, Cr C haloalkoxy, CrC haloalkyl or C C alkyl; or
R is -C6-Ci0aryl, or 4 to 10 membered heteroaryl containing 1 to 3 heteroatoms selected from N, S and O, wherein said aryl and heteroaryl are susbstituted by taking the substituents on adjacent atoms of the -C6-Ci0aryl, or 4 to 10 membered heteroaryl and forming a 3 to 7 membered heterocycle, wherein the formed bicycle substituent is further
optionally substituted with halogen, d-C4alkyl, CrC4haloalkyl, CrC4haloalkoxy or C C4alkoxy;
R1 is selected from the group consisting of -OH, CrC4alkoxy and -S(0)2R3;
R2 is selected from the group consisting of hydrogen, CrC4alkyl, -C(0)CR3 and - S(0)2R3;
R3 is selected from the group consisting of C C alkyl and C3-C6cycloalkyl;
Z is a divalent radical selected from

Z is with the proviso that A is
R5 is selected from the group consisting of hydrogen, halogen, -CN, Ci-C alkyl, and CrC4haloalkyl;
R6, R6a, R6b or R6c are independently selected from the group consisting of hydrogen, halogen, CrC4alkyl, and CrC4haloalkyl; '
L is a divalent bond, -CH2-, -O- or
3. The compound or a pharmaceutically acceptable salt thereof according to claim 1 or 2, represented by formula III


, and
R is -C6-Ci0aryl, or 4 to 10 membered heteroaryl containing 1 to 3 heteroatoms selected from N, S and O, wherein said aryl and heteroaryl are optionally substituted with a substituent selected from the group consisting of halogen, -OH, -CN, -S(0)2(CrC4)alkyl, Ci- C4haloalkyl, C3-C7cycloalkyl, CrC4alkoxy, CrC4haloalkoxy, C C alkyl optionally substituted with Ci-C alkoxy, CrC alkoxy optionally substituted with Ci-C alkoxy and a 4 to 7 membered heterocycle containing 1 to 3 heteroatom selected from N, S, and O, wherein said heterocycle is optionally substituted with one or more halogen, C C alkoxy, Cr C4haloalkoxy, CrC4haloalkyl or Ci-C4 alkyl; or
R is -C6-Ci0aryl, or 4 to 10 membered heteroaryl containing 1 to 3 heteroatoms selected from N, S and O, wherein said aryl and heteroaryl are susbstituted by taking the substituents on adjacent atoms of the -C6-Ci0aryl, or 4 to 10 membered heteroaryl and forming a 3 to 7 membered heterocycle, wherein the formed bicycle substituent is further optionally substituted with halogen, Ci-C alkyl, CrC haloalkyl, Ci-C haloalkoxy or C C4alkoxy;
R2 is selected from the group consisting of hydrogen, C C alkyl, -C(0)CR3 and - S(0)2R3;
R3 is selected from the group consisting of C C alkyl and C3-C6cycloalkyl;
R5 is selected from the group consisting of hydrogen, halogen, -CN, Ci-C alkyl, and CrC haloalkyl; '
L is a direct bond or
4. The compound or a pharmaceutically acceptable salt thereof according to any any one of the proceeding claims, wherein
R is phenyl optionally substituted with a substituent selected from the group
consisting of halogen, -OH, -CN, -S(0)2(CrC4)alkyl, C C4haloalkyl, C3-C7cycloalkyl, C C4alkoxy, CrC haloalkoxy, C C alkyl optionally substituted with C C alkoxy, C C alkoxy optionally substituted with C C alkoxy, and a 4 to 7 membered heterocycle containing 1 to 3 heteroatom selected from N, S, and O, wherein said heterocycle is optionally substituted with one or more halogen, Ci-C alkoxy, C C haloalkoxy, Ci-C haloalkyl or C C alkyl; or R is phenyl optionally susbstituted by taking the substituents on adjacent atoms of the phenyl and forming a 3 to 7 membered heterocycle, wherein the formed bicycle substituent is further optionally substituted with halogen, Ci-C alkyl, CrC haloalkyl, d- C haloalkoxy or C C alkoxy.
5. The compound or a pharmaceutically acceptable salt thereof according to any any one of the proceeding claims, wherein the compound is formula III


and ;
R is phenyl optionally substituted with a substituent selected from the group consisting of halogen, -OH, -CN, -S(0)2(CrC4)alkyl, C C4haloalkyl, C3-C7cycloalkyl, C C4alkoxy, Ci-C4haloalkoxy, Ci-C4alkyl optionally substituted with Ci-C4alkoxy, Ci-C4alkoxy optionally substituted with C C alkoxy, and a 4 to 7 membered heterocycle containing 1 to 3 heteroatom selected from N, S, and O, wherein said heterocycle is optionally substituted with one or more halogen, Ci-C alkoxy, Ci-C haloalkoxy, Ci-C haloalkyl or Ci-C alkyl; or
R is phenyl optionally susbstituted by taking the substituents on adjacent atoms of the phenyl and forming a 3 to 7 membered heterocycle, wherein the formed bicycle substituent is further optionally substituted with halogen, C C alkyl, CrC haloalkyl, C C haloalkoxy or C C alkoxy;
R2 is selected from the group consisting of hydrogen, C C alkyl, C(0)CR3 and - S(0)2R3;
R3 is selected from the group consisting of C C alkyl and C3-C6cycloalkyl; '
L is a direct bond or
6. The compound or a pharmaceutically acceptable salt thereof according to any one of claims 1 to 4, wherein the compound is formula III


Q is selected from the group consisting of
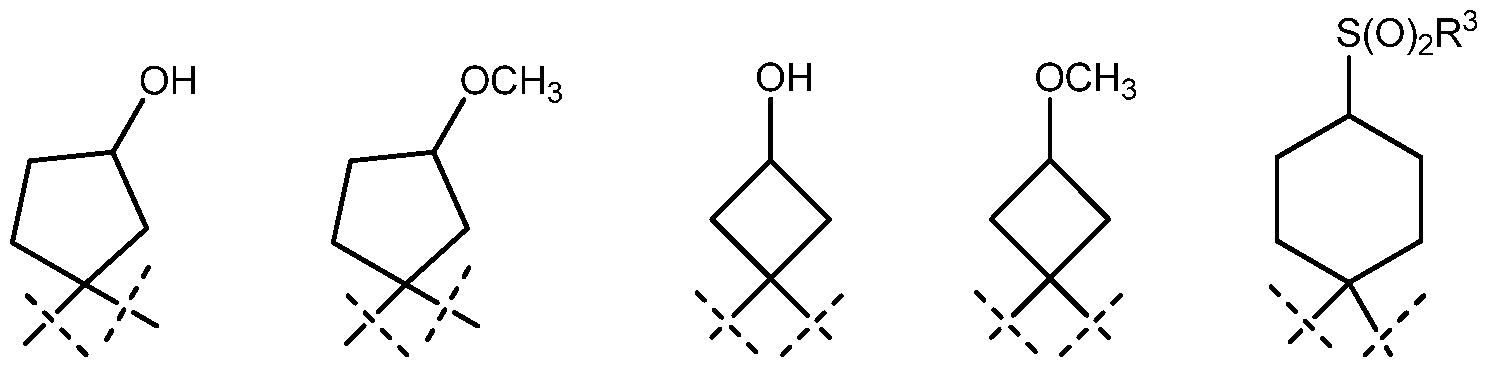

, and
R is phenyl optionally substituted with a substituent selected from the group consisting of halogen, -OH, -CN, -S(0)2(CrC4)alkyl, C C4haloalkyl, C3-C7cycloalkyl, C C4alkoxy, CrC haloalkoxy, C C alkyl optionally substituted with C C alkoxy, C C alkoxy optionally substituted with C C alkoxy, and a 4 to 7 membered heterocycle containing 1 to 3 heteroatom selected from N, S, and O, wherein said heterocycle is optionally substituted with one or more halogen, Ci-C alkoxy, C C haloalkoxy, Ci-C haloalkyl or C C alkyl; or
R is phenyl optionally susbstituted by taking the substituents on adjacent atoms of the phenyl and forming a 3 to 7 membered heterocycle, wherein the formed bicycle substituent is further optionally substituted with halogen, C C alkyl, CrC haloalkyl, C C haloalkoxy or C C alkoxy;
R3 is selected from the group consisting of C C alkyl and C3-C6cycloalkyl;
; '
;' '
L is a direct bond or
7. The compound of claim 1 or 2, represented by formula IV





, and
R is phenyl optionally substituted with a substituent selected from the group consisting of halogen, -OH, -CN, -S(0)2(CrC4)alkyl, C C4haloalkyl, C3-C7cycloalkyl, C C4alkoxy, CrC haloalkoxy, C Calkyl optionally substituted with C C alkoxy, C Calkoxy
optionally substituted with CrC4alkoxy, and a 4 to 7 membered heterocycle containing 1 to 3 heteroatom selected from N, S, and O, wherein said heterocycle is optionally substituted with one or more halogen, d-C4alkoxy, CrC4haloalkoxy, CrC4haloalkyl or C C alkyl; or
R is phenyl optionally susbstituted by taking the substituents on adjacent atoms of the phenyl and forming a 3 to 7 membered heterocycle, wherein the formed bicycle substituent is further optionally substituted with halogen, C C4alkyl, C C4haloalkyl, C C haloalkoxy or C C alkoxy;
R2 is selected from the group consisting of hydrogen, CrC4alkyl, -C(0)CR3 and - S(0)2R3;
R3 is selected from the group consisting of C C alkyl and C3-C6cycloalkyl;
R6, R6a, R6b or R6c are independently selected from the group consisting of hydrogen, halogen, C C alkyl and CrC haloalkyl; '
L is a direct bond, -CH2-, -O- or 7. The compound of claim 1 or 2, represented by formula III

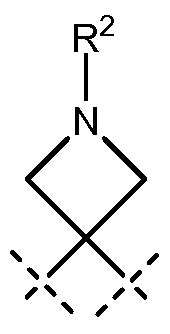
and ;
R is phenyl optionally substituted with a substituent selected from the group consisting of halogen, -OH, -CN, -S(0)2(CrC4)alkyl, C C4haloalkyl, C3-C7cycloalkyl, C C4alkoxy, Ci-C4haloalkoxy, Ci-C4alkyl optionally substituted with Ci-C4alkoxy, Ci-C4alkoxy optionally substituted with C C alkoxy, and a 4 to 7 membered heterocycle containing 1 to 3 heteroatom selected from N, S, and O, wherein said heterocycle is optionally substituted with one or more halogen, Ci-C alkoxy, Ci-C haloalkoxy, Ci-C haloalkyl or Ci-C alkyl; or
R is phenyl optionally susbstituted by taking the substituents on adjacent atoms of the phenyl and forming a 3 to 7 membered heterocycle, wherein the formed bicycle substituent is further optionally substituted with halogen, C C alkyl, CrC haloalkyl, C C haloalkoxy or C C alkoxy;
R2 is selected from the group consisting of hydrogen, C C alkyl, -C(0)CR3 and - S(0)2R3;
R3 is selected from the group consisting of C C alkyl and C3-C6cycloalkyl;
R6, R6a, R6b or R6c are independently selected from the group consisting of hydrogen, halogen, C C alkyl, and CrC haloalkyl; '
L is a direct bond or
8. The compound according to any any one of the proceeding claims, wherein
L is a direct bond;

R is selected from the group consisting of

and
9. A pharmaceutical composition, comprising:
the compound according to claims 1 to 8 and
a pharmaceutically acceptable carrier.
10. A pharmaceutical combination composition, comprising:
the compound according to any one of claims 1 to 8,
an antibacterially effective amount of a second therapeutic agent, and
a pharmaceutically acceptable carrier.
11. The pharmaceutical combination composition according to claim 10, wherein the second therapeutic agent is selected from the group consisting of Ampicillin, Piperacillin, Penicillin G, Ticarcillin, Imipenem, Meropenem, Azithromycin, erythromycin, Aztreonam, Cefepime, Cefotaxime, Ceftriaxone, Cefatazidime, Ciprofloxacin, Levofloxacin, Clindamycin,
Doxycycline, Gentamycin, Amikacin, Tobramycin, Tetracycline, Tegacyclin, Rifampicin, Vancomycin and Polymyxin.
12. A method of inhibiting a deacetylase enzyme in a Gram-negative bacterium, comprising: contacting the Gram-negative bacteria with the compound according to any one of claims 1 to 8.
13. A method for treating a subject with a Gram-negative bacterial infection, comprising: administering to the subject in need thereof an antibacterially effective amount of the compound according to any one of claims 1 to 8 and a pharmaceutically acceptable carrier.
14. The method of claim 13, wherein the Ggram negative bacterial infection is an infection comprising at least one bacterium selected from the group consisting of Pseudomonas, Stenotrophomonas maltophila, Burkholderia, Alcaligenes xylosoxidans, Acinetobacter, Enterobacteriaceae, Haemophilus, Moraxella, Bacteroids, Fransicella, Shigella, Proteus, Vibrio, Salmonella, Bordetella, Helicobactor, Legionella, Citrobactor, Serratia,
Campylobactor, Yersinia and Neisseria.
15. The method of claim 14, wherein the bacterium is a Enterobacteriaceae which is selected from the group consisting of Serratia, Proteus, Klebsiella, Enterobacter, Citrobacter, Salmonella, Providencia, Morganella, Cedecea, Yersinia, and Edwardsiella species and Escherichia coli.
16. A compound according to any one of claims 1 to 8 or a pharmaceutically acceptable salt thereof, for use as a medicament.
17. A compound according to any one of claims 1 to 8 or a pharmaceutically acceptable salt thereof, for use in treatment of a Gram-negative bacterial infection.
18. A compound according to any one of claims 1 to 8 or a pharmaceutically acceptable salt thereof, for use in treatment of a Gram-negative bacterial infection, wherein the bacterial infection is selected from Pseudomonas aeruginosa, Stenotrophomonas maltophila, Burkholderia cepacia, Alcaligenes xylosoxidans, Acinetobacter, Enterobacteriaceae, Haemophilus, and Neisseria species.
19. Use of the compound according to any one of claims 1 to 8, for the preparation of a medicament for the treatment of a Gram-negative bacterial infection in a subject, wherein the bacterial infection is selected from Pseudomonas aeruginosa, Stenotrophomonas maltophila, Burkholderia cepacia, Alcaligenes xylosoxidans, Acinetobacter,
Enterobacteriaceae, Haemophilus, and Neisseria species.
20. The use of claim 19, wherein the bacterial infection is an Enterobacteriaceae selected from the group consisting of Serratia, Proteus, Klebsiella, Enterobacter, Citrobacter, Salmonella, Providencia, Morganella, Cedecea, and Edwardsiella species and Escherichia coli.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361806664P | 2013-03-29 | 2013-03-29 | |
| US61/806,664 | 2013-03-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014160649A1 true WO2014160649A1 (en) | 2014-10-02 |
Family
ID=50771589
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2014/031612 WO2014160649A1 (en) | 2013-03-29 | 2014-03-24 | Hydroxamic acid derivatives as lpxc inhibitors for the treatment of bacterial infections |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2014160649A1 (en) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015066413A1 (en) | 2013-11-01 | 2015-05-07 | Novartis Ag | Oxazolidinone hydroxamic acid compounds for the treatment of bacterial infections |
| US9539305B1 (en) | 2014-03-14 | 2017-01-10 | Fleurir Abx Llc | Pristinamycin compositions, LpxC compositions, their improvements, and combinations thereof |
| US9549916B2 (en) | 2014-12-16 | 2017-01-24 | Novartis Ag | Isoxazole hydroxamic acid compounds as LpxC inhibitors |
| US9637482B2 (en) | 2014-04-22 | 2017-05-02 | Novartis Ag | Isoxazoline hydroxamic acid derivatives as LpxC inhibitors |
| WO2017083434A1 (en) | 2015-11-09 | 2017-05-18 | Forge Therapeutics, Inc. | Pyrone based compounds for treating bacterial infections |
| US10071973B2 (en) | 2016-06-14 | 2018-09-11 | Novartis Ag | Crystalline isoxazole hydroxamic acid compounds |
| US10414735B2 (en) | 2015-11-09 | 2019-09-17 | Forge Therapeutics, Inc. | Substituted hydroxypyrimidinones for treating bacterial infections |
| CN111867600A (en) * | 2018-03-15 | 2020-10-30 | 辉瑞大药厂 | Pyridone and pyrimidone phosphates and borates as antibacterial agents |
| US11021471B2 (en) | 2017-05-10 | 2021-06-01 | Forge Therapeutics, Inc. | Antibacterial compounds |
| CN113166077A (en) * | 2018-09-20 | 2021-07-23 | 福至治疗公司 | Antibacterial compounds |
| WO2023015236A3 (en) * | 2021-08-04 | 2023-03-02 | The Research Foundation For The State University Of New York | Composition and method for treatment of gram negative bacterial infection |
| US11731962B2 (en) | 2020-03-25 | 2023-08-22 | Blacksmith Medicines, Inc. | LpxC inhibitor and methods of making |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004062601A2 (en) * | 2003-01-08 | 2004-07-29 | Chiron Corporation | Antibacterial agents |
| WO2004078163A2 (en) | 2003-02-28 | 2004-09-16 | Transform Pharmaceuticals, Inc. | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
-
2014
- 2014-03-24 WO PCT/US2014/031612 patent/WO2014160649A1/en active Application Filing
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004062601A2 (en) * | 2003-01-08 | 2004-07-29 | Chiron Corporation | Antibacterial agents |
| WO2004078163A2 (en) | 2003-02-28 | 2004-09-16 | Transform Pharmaceuticals, Inc. | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
Non-Patent Citations (17)
| Title |
|---|
| "Methoden der organischen Chemie", vol. 15/1, 1974, GEORG THIEME VERLAG |
| "Methods of Organic Synthesis", vol. 21, 1952, THIEME |
| "Remington's Pharmaceutical Sciences", 1985, MACK PUBLISHING COMPANY |
| "Science of Synthesis: Houben-Weyl Methods of Molecular Transformation", 2005, GEORG THIEME VERLAG |
| "Science of Synthesis: Houben-Weyl Methods of Molecular Transformation", 2005, GEORG THIEME VERLAG, pages: 41627 |
| "The Peptides", vol. 3, 1981, ACADEMIC PRESS |
| H.-D. JAKUBKE; H. JESCHKEIT: "Aminosauren, Peptide, Proteine" |
| H.-D. JAKUBKE; H. JESCHKEIT: "Aminosauren, Peptide, Proteine", 1982, VERLAG CHEMIE |
| HALE MICHAEL R ET AL: "Exploring the UDP pocket of LpxC through amino acid analogs", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, PERGAMON, AMSTERDAM, NL, vol. 23, no. 8, 1 March 2013 (2013-03-01), pages 2362 - 2367, XP028525203, ISSN: 0960-894X, DOI: 10.1016/J.BMCL.2013.02.055 * |
| HYLAND ET AL., JOURNAL OF BACTERIOLOGY, vol. 179, 1997, pages 2029 - 2037 |
| J. F. W. MCOMIE: "Protective Groups in Organic Chemistry", 1973, PLENUM PRESS |
| JOCHEN LEHMANN: "Chemie der Kohlenhydrate: Monosaccharide und Derivate", 1974, GEORG THIEME VERLAG |
| JOCHEN LEHMANN: "Chemistry of Carbohydrates: Monosaccharides and Derivatives", 1974, GEORG THIEME VERLAG, article "Chemie der Kohlenhydrate: Monosaccharide und Derivate" |
| STAHL; WERMUTH: "Handbook of Pharmaceutical Salts: Properties, Selection, and Use", 2002, WILEY-VCH |
| T. W. GREENE; P. G. M. WUTS: "Protective Groups in Organic Synthesis", 1999, WILEY |
| T. W. GREENE; P. G. M. WUTS: "Protective Groups in Organic Synthesis", vol. 3, 1999, ACADEMIC PRESS, article "The Peptides" |
| TETRAHEDRON LETT., vol. 46, 2005, pages 7575 - 7579 |
Cited By (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015066413A1 (en) | 2013-11-01 | 2015-05-07 | Novartis Ag | Oxazolidinone hydroxamic acid compounds for the treatment of bacterial infections |
| US9872884B1 (en) | 2014-03-14 | 2018-01-23 | Fleurir Abx Llc | Pristinamycin compositions, LPXC compositions, their improvements, and combinations thereof |
| US9539305B1 (en) | 2014-03-14 | 2017-01-10 | Fleurir Abx Llc | Pristinamycin compositions, LpxC compositions, their improvements, and combinations thereof |
| US11951148B1 (en) | 2014-03-14 | 2024-04-09 | Fleurir Abx, Llc | Pristinamycin compositions, LpxC compositions, their improvements, and combinations thereof |
| US11141453B1 (en) | 2014-03-14 | 2021-10-12 | Fleurir Abx Llc | Pristinamycin compositions, LpxC compositions, their improvements, and combinations thereof |
| US10307457B1 (en) | 2014-03-14 | 2019-06-04 | Fleurir Abx Llc | Pristinamycin compositions, LpxC compositions, their improvements, and combinations thereof |
| US9718792B2 (en) | 2014-04-22 | 2017-08-01 | Novartis Ag | Isoxazoline hydroxamic acid derivatives as LpxC inhibitors |
| US9637482B2 (en) | 2014-04-22 | 2017-05-02 | Novartis Ag | Isoxazoline hydroxamic acid derivatives as LpxC inhibitors |
| US10029994B2 (en) | 2014-12-16 | 2018-07-24 | Novartis Ag | Isoxazole hydroxamic acid compounds as LpxC inhibitors |
| US9815804B2 (en) | 2014-12-16 | 2017-11-14 | Novartis Ag | Isoxazole hydroxamic acid compounds as LpxC inhibitors |
| US9549916B2 (en) | 2014-12-16 | 2017-01-24 | Novartis Ag | Isoxazole hydroxamic acid compounds as LpxC inhibitors |
| WO2017083434A1 (en) | 2015-11-09 | 2017-05-18 | Forge Therapeutics, Inc. | Pyrone based compounds for treating bacterial infections |
| US10414735B2 (en) | 2015-11-09 | 2019-09-17 | Forge Therapeutics, Inc. | Substituted hydroxypyrimidinones for treating bacterial infections |
| US10611747B2 (en) | 2015-11-09 | 2020-04-07 | Forge Therapeutics, Inc. | Pyrone based compounds for treating bacterial infections |
| US10875832B2 (en) | 2015-11-09 | 2020-12-29 | Forge Therapeutics, Inc. | Substituted pyrimidines for treating bacterial infections |
| US10071973B2 (en) | 2016-06-14 | 2018-09-11 | Novartis Ag | Crystalline isoxazole hydroxamic acid compounds |
| US11021471B2 (en) | 2017-05-10 | 2021-06-01 | Forge Therapeutics, Inc. | Antibacterial compounds |
| CN111867600A (en) * | 2018-03-15 | 2020-10-30 | 辉瑞大药厂 | Pyridone and pyrimidone phosphates and borates as antibacterial agents |
| US11407740B2 (en) | 2018-09-20 | 2022-08-09 | Forge Therapeutics, Inc. | Antibacterial compounds |
| CN113166077A (en) * | 2018-09-20 | 2021-07-23 | 福至治疗公司 | Antibacterial compounds |
| US11731962B2 (en) | 2020-03-25 | 2023-08-22 | Blacksmith Medicines, Inc. | LpxC inhibitor and methods of making |
| WO2023015236A3 (en) * | 2021-08-04 | 2023-03-02 | The Research Foundation For The State University Of New York | Composition and method for treatment of gram negative bacterial infection |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2014160649A1 (en) | Hydroxamic acid derivatives as lpxc inhibitors for the treatment of bacterial infections | |
| AU2017294231B2 (en) | Aromatic acetylene or aromatic ethylene compound, intermediate, preparation method, pharmaceutical composition and use thereof | |
| US9718792B2 (en) | Isoxazoline hydroxamic acid derivatives as LpxC inhibitors | |
| US10029994B2 (en) | Isoxazole hydroxamic acid compounds as LpxC inhibitors | |
| AU2016275764B8 (en) | Efflux-pump inhibitors and therapeutic uses thereof | |
| CN112074507B (en) | Compounds as Antibiotics | |
| WO2015066413A1 (en) | Oxazolidinone hydroxamic acid compounds for the treatment of bacterial infections | |
| US20220226298A1 (en) | Gpr40 agonists | |
| KR102264248B1 (en) | Tetrahydropyridine derivatives and their use as antibacterial agents | |
| CN113677659A (en) | Substituted amide compounds useful as farnesoid X receptor modulators | |
| NZ751511B2 (en) | Tetrahydropyridine derivatives and their use as antibacterial agents |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14725829 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 14725829 Country of ref document: EP Kind code of ref document: A1 |