NOVEL ACTIVATED POLYETHYLENE GLYCOL COMPOUNDS
FIELD OF THE INVENTION
[001] The present invention relates generally to compounds that are useful in modifying biological materials to reduce immunogenicity of the biological material. The biological material may include proteins, liposomes, or cellular compositions that can be modified by attachment of such compounds. In particular, activated polyethylene glycol (PEG) compounds are used to reduce the immunogenicity of such biological materials.
BACKGROUND
[002] Polyethylene glycol (PEG) is used to improve the delivery, safety and efficacy of a variety of therapeutic biological materials by covalent attachment of the PEG to the materials. For example, therapeutic proteins modified by PEG will increase the plasma (circulation) half life of the proteins. The PEG increases the size of the molecule, resulting in decreased clearance by the reticuloendothelial system. In addition, the PEG forms a protective shell around the molecule, protecting it from degradation by proteolytic or protein-modifying enzymes or clearance by an immune response. Due to the variety of biological materials that may be treated by PEG, the optimal conditions for modification needs to be determined for each system. In addition to protein based molecules, PEG can be used to modify liposomes used for drug delivery in order to increase their biological half life or to modify a variety of cellular components, such as red blood cells (RBC), in order to reduce the antigenicity of the cells. It has been demonstrated that the PEG molecule itself, over a very large molecular weight range, has no significant toxicity. PEG is approved for use as an excipient in a variety of pharmaceutical formulations, including injectable, topical, rectal and nasal formulations as well as in foods and cosmetics [Harris et al.,
Clin Pharmacokinet 40(7):539-551 (2001)]. In addition, some PEG modified proteins have been approved for human use, such as PEG-adenosine deaminase and PEG-asparaginase for chemotherapy. It is desirable to be able to readily assess the level of modification of these biological materials by determining the number of PEG molecules bound per molecule of the biological material or per cell in the case of a modified red cell. This allows for the quanitative assessment of reaction conditions, comparison of the properties of different compounds, and quality control of manufacturing methods.
SUMMARY OF THE INVENTION
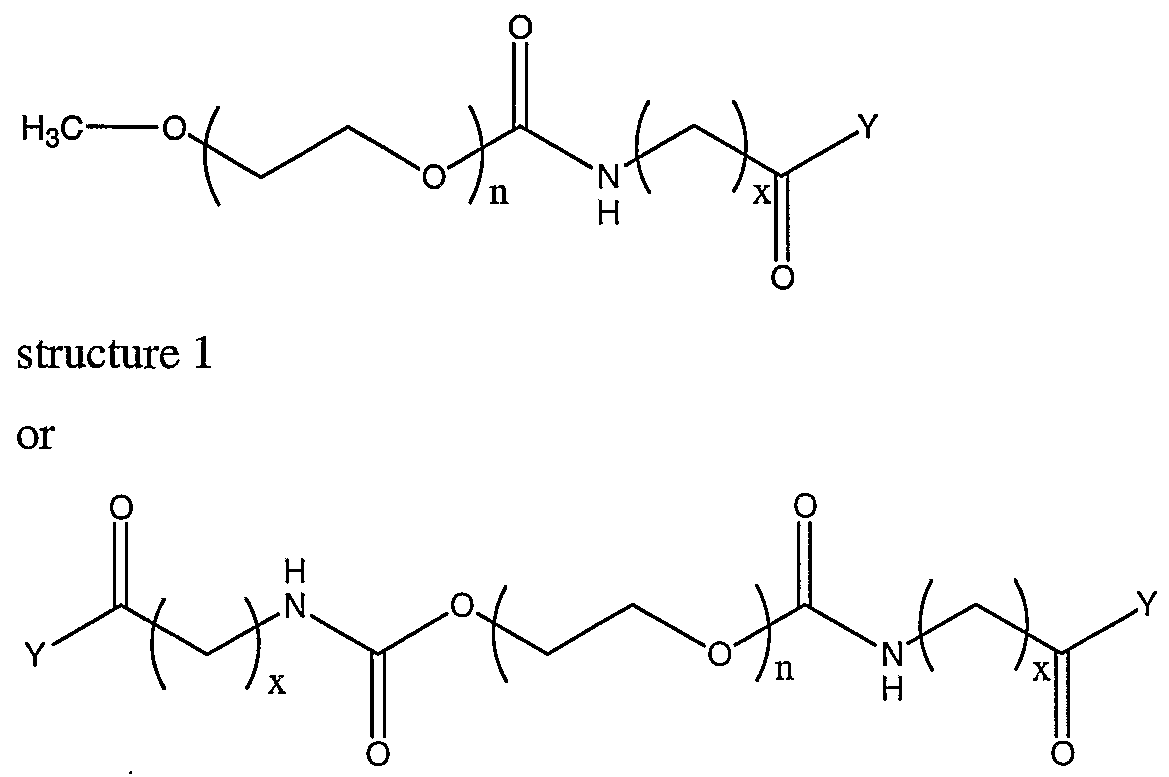
[003] The present invention provides new activated PEG compounds for the modification of materials for in vivo use. The compounds comprise a polyethyleneglycol (PEG) attached to a coupling group, where the region between the mPEG and the coupling group comprises an amino acid, preferably an unnatural amino acid. The amino acid comprises a carbon chain, wherein the length of the carbon chain affects the reactivity of the coupling group. In addition, the coupling group can be modified in order to further effect the kinetics of the reactivity of the coupling group. This allows for better control of the kinetics of the reaction of the activated PEG compounds with biological materials. The kinetics can be optimized for reactivity with the reactive groups of the biological materials, such as primary amino groups. In addition, the amino acid remains attached to the PEG portion when the activated compound is reacted with a biological material. When the amino acid is an unnatural amino acid, the unnatural amino acid can be used to assess the amount of the compound that is bound to a biological material. The compounds are useful for attachment to biological materials such as proteins, liposomes and cellular compositions. The amount of the PEG bound to these materials can be determined quantitatively by doing an amino acid analysis of the modified biological materials. The unnatural amino acid can be determined from the amino acid analysis and can either be calculated based on known amounts of material analyzed, or can be normalized to the natural amino acids in the sample. The compounds of the invention have a general formula of:
structure 2 or
[004] where n is greater than or equal to 3 and x is 1-10, also 2-10, also 3-10, also 4-10, also 5-10, or 6-10. Y is a suitable leaving group, such as N- hydroxysuccinimide, that enhances the reaction with the biological material. The value of x will influence the kinetics of the reaction of the compound with the biological material, where the reaction is slower as x increases. The leaving group can be altered to further influence the kinetics of the reaction. This allows for control of the kinetics of reactivity in order to optimize the modification of the biological materials. When the compounds of structure 2, 3, or 4 are used, the amino acid analysis calculations are based on having two to four unnatural amino acids per mPEG.
DESCRIPTION OF THE DRAWINGS
[005] Fig. 1 is an exemplary plot of the population maximum fluorescence by flow cytometry for the binding of fluorescent antibody to mPEG modified RBC. [006] Fig. 2A is an exemplary plot of a standard curve of fluorescence measurement vs. fluorescently labeled activated PEG (FPEG) concentration spiked into red cell ghosts for quantitation of PEG binding to red cells (1.3 billion ghosts per sample). [007] Fig. 2B is a plot of the FACScan peak (FL1 -Height) vs. the number of PEG molecules bound per red cell.
DESCRIPTION OF THE INVENTION
[008] PEG is a non-toxic, water-soluble polymer that has little or no recognition by the immune system. Because of these properties, PEG molecules are useful in modification of a variety of biological materials. Therapeutic proteins can be modified by covalent binding of PEG molecules to the protein. The modification results in improved safety and efficacy of the therapeutic proteins, in part by decreasing the antigenicity of the proteins and in part by reducing the clearance of the proteins by the reticuloendothelial system. Similar modifications can be made to liposomes for use as drug delivery vehicles. The half-life of the liposomes can be increased, resulting in a longer lasting therapeutic effect of the compounds being delivered by the liposomes. It is also advantageous to mask the antigens of certain cells, such as cells that may elicit an unwanted immune response when transplanted into a subject. This includes, but is not limited to, RBC and endothelial cells of transplant organs. For red cells, it is also possible to improve the rheological properties of the cells by attachment of the PEG . This results in reduced viscosity under low shear rates. Such low viscosity red cells can be used to treat ischemic conditions, such as those resulting from stroke, myocardial infarction, sickle cell anemia, and other conditions relating to vascular occlusion (see US Patent 6,312,685). Additional diseases that can be treated include angina, critical limb ischemia, cerebral vasospasm, and subarrachnoid hemorrhage. The new compounds of the present invention can be used to control the reactivity in aqueous media in order to optimize the efficiency of the PEG modification. In addition, the compounds can be used to quantitatively assess the number of PEG molecules
attached per biological molecule. This can be used as a quality control measure to ensure that an adequate level of modification of the biological material is consistently reproduced. It also allows for the correlation of the amount of modification required to achieve a desired effect, for example, adequate masking of red cell antigens. [009] The compounds of the invention have a general formula of:
[0010] where n is greater than or equal to 3 and x is 1-10, also 2-10, also 3-10, also 4-10, also 5-10, or 6-10, and all salts thereof. In one embodiment, n is about 3-1,000, also about 3-500, also about 10-500, also about 100-500. When the compound is the 3-Arm PEG (structure 3), n is preferably about 3-350, also about 3-250, also about 10-250, also about 10-150. When the compound is the 4-Arm PEG (structure 4), n is preferably about 3-250, also about 3-200, also about 10-200, also about 10-120. The
terminal CO-Y group is a reactive coupling group that is subject to nucleophilic attack on the carbonyl carbon, for example by thiol and amine groups on a biological material, where Y is a suitable leaving group that enhances the reaction with the biological material. As an example where Y is N-hydroxysuccinimide, an amine group of a biological material reacts as follows:
[0011] where B-NH2 represents the amine group of a biological material. Examples of other leaving groups include, but are not limited to, carboxylates, sulfonates, oxycarbonylimidazoles, maleimides, isocyanates, isothiocyanates, substituted triazine, substituted triazole, halogens, chloro substituted phenoxy (e.g. 2,4,5- trichlorophenoxy) and sulfur analogs, fluoro substituted phenoxy (e.g. pentafluorophenoxy) and sulfur analogs (e.g. pentafluorobenzenethiolate, 2,3,5,6- tetrafluorobenzenethiolate and 4-fluorobenzenethiolate), alkoxy (preferably ethoxy), fluoro substituted alkoxy (e.g. 2,2,2-trifluoroethoxy) and sulfur analogs (e.g. alkylthiolates and substituted alkylthiolates such as 2,2,2-trifluoroethanethiolate and ethanethiolate), and chloro substituted alkoxy and their sulfur analogs. A variety of coupling groups for activated PEG compounds are known in the art, a discussion of which can be found in US Provisional Patent Application Serial number 60/338,707 and US Patent Application Serial number 10/310,618, the disclosures of which are hereby incorporated by reference.
[0012] The amino acid group -NH-(CH2)x-CONH- remains bound to the biological material along with the PEG such that upon hydrolysis of the modified material with strong acid, the amino acid NH2-(CH2)x-COOH is released and can be quantitatively measured, for example by HPLC analysis. When the amino acid is an unnatural amino acid, the relative amount of the unnatural amino acid can be compared to the
amounts of natural amino acids from the biological material that is modified to determine the amount of PEG bound per molecule of biological material. While this method has been used on modified proteins, it provides a unique approach to measuring the amount of PEG bound per red cell. In order to evaluate the binding to red cells, they must first be prepared as red cell ghosts so that the only natural amino acids result from those present in the red cell membrane (Example 3). [0013] Exemplary compounds of the invention include N-6- [Methoxypoly(ethyleneglycol)] -øxo-aminocaproate N-hydroxysuccinimide ester (mPEG-6AC-ΝHS), N-5-[Methoxypoly(ethyleneglycol)]-θΛθ-aminovalerate N- hydroxysuccinimide ester (mPEG-5AV-ΝHS), N-4-[Methoxypoly(ethyleneglycol)]- αrø-aminobutyrate N-hydroxysuccinimide ester (mPEG-4AB-ΝHS), N-6- [Memoxypoly(ethyleneglycol)]-øxo-aminocaproate pentafluorophenyl ester (mPEG- 6AC-PFP), N-6-[Methoxypoly(ethyleneglycol)]-θΛθ-aminocaproate 2,2,2- trifluoroethyl ester (mPEG-6AC-TFE), N-6-[Methoxypoly(ethyleneglycol)]-øxo~ aminocaproate ethyl ester (mPEG-6AC-OEt), N-6-[Methoxypoly(ethyleneglycol)]- øxø-aminocaproate pentafluorobenzenethio ester (mPEG-6AC-PFT), N-6- [Methoxypoly(ethyleneglycol)]-oxo-aminocaproate 2,3,5, 6-tetrafluorobenzene thio ester (mPEG-6AC-TFT), N-6-[Methoxypoly(ethyleneglycol)]-oxo-aminocaproate 4- fluorobenzenethio ester (mPEG-6AC-4FTP), N-6-[Methoxypoly(ethyleneglycol)]- αxø-aminocaproate 2,2,2-trifluoroethylthio ester (mPEG-6AC-TFET), and N-6- [Methoxypoly(ethyleneglycol)]-oxo-aminocaproate ethylthio ester (mPEG-6AC-SEt). The corresponding compounds with the reactive coupling group at each end of the PEG or the 3-Arm and 4-Arm compounds with the reactive coupling group at each end of the PEG are also envisioned. The structures of linear compounds with one reactive coupling group are as follows:
[0014] The efficacy of these compounds for use in masking red cell antigens can be assessed, for example, by looking at the modification density on the red cells, evaluating the masking of binding of fluorescent anti-type antibody to the modified red cells by FACScan™ (Becton, Dickinson and Co., NJ), assaying for agglutination with antibodies to red cell antigens, and assessing in vivo function by various methods as given in the following examples. In order to estimate the reactivity of these compounds, the kinetics of hydrolysis can be determined by quantitative measurement of the release of the leaving group as a function of time under appropriate conditions. The hydrolysis rate can be used to estimate appropriate conditions under which the compounds can be reacted with biological materials in order to optimize the reactivity with such materials. It is possible that certain reactive nucleophilic sites on a biological material are not as accessible to the compounds as others such that slower reacting compounds may provide more complete binding of the compound to the biological material. The slower reacting
compounds are able to react with such sites before the coupling group undergoes hydrolysis. It may also be advantageous to use a combination of activated PEG compounds, wherein one compound has relatively fast kinetics and another has relatively slow kinetics. This may provide a more efficient method of modifying a biological material, wherein the more reactive activated PEG compounds will bind to the more accessible nucleophilic sites on the biological material and the less reactive activated PEG will bind to the less accessible nucleophilic sites. Another possibility is that nucleophilic sites within a biological material will have slightly different pKa values dependent on their local environment. As such, a combination of slow and fast reacting compounds may provide reactivity with certain sites at one pH and other sites at another pH. The fast reacting compounds can be used to bind those sites that are reactive at a first pH and the pH can then be adjusted for reaction of the slow reacting compound with any remaining sites.
[0015] The invention further comprises the use of the compounds of the invention for modification of biological materials, such as proteins, liposomes and cellular compositions, such as red blood cells. The biological materials are modified by incubation with the compounds of the invention under appropriate conditions. In one embodiment, the biological material is modified by incubation in a suitable buffer with the compounds of the invention. In a preferred embodiment, the biological material is washed with a suitable buffer and then reacted in the buffer with the compounds of the invention. Modification of proteins and lipids using similar compounds are described in the art, for example Harris et al., Clin Pharmacokinet 40(7): 539-551 (2001). The modification of red cells is discussed in detail below and some exemplary methods are described in the following examples. In a preferred embodiment, the red cells are washed with a suitable buffer, e.g. a buffer with a pH in the range of about 8-10, prior to reacting with the compounds of the invention. The compounds are reacted at a suitable concentration, depending on the size of the compound, in order to adequately mask the red cell antigens. The compounds can be evaluated to detennine their effect on the function of the modified red cells. Such function can be assessed by looking at a variety of in vitro parameters as discussed in the examples. In addition, the invention comprises the use of biological materials modified by the compounds of the present invention, such as transfusion of modified cells, therapeutic use of modified proteins, and delivery of therapeutic compounds using modified liposomes.
[0016] The methods of reacting compounds of the invention with red cells can be optimized to provide adequate masking of the antigens and adequate function of the resulting red cell composition for use in vivo. The methods can be optimized with respect to the buffers used during processing of the RBC. A thorough discussion of the buffering of the solution for optimal reaction of an activated antigen masking compound with the red cells can be found in US Patent Application Serial Number 10/310,618, the contents of which are hereby incorporated by reference. In one embodiment, a red cell composition is mixed with a suitable reaction solution and activated antigen masking compound. This mixture is incubated so that the activated antigen masking compound covalently binds the surface of the red cells. The incubation is done at a temperature ranging from about 4-40 °C, preferably about 20- 25 °C, for at least 30 minutes, typically 30-240 minutes, preferably approximately 60- 120 minutes. In one embodiment, it is preferable to wash the red cells with a suitable buffer prior to reacting with the activated antigen masking compounds. In one embodiment, the pre reaction wash solution and reaction solution comprise buffers that provide adequate buffering of the system to optimize the reaction of the activated antigen masking compound with the red cell surface. In one embodiment, the buffering is adequately provided by the pre reaction wash solution such that the reaction solution need not be buffered. The buffering solutions of the invention will preferably have a pH in the range of approximately 8-10, preferably approximately 9 and comprise a buffer at a concentration of approximately 50-350 niM, preferably about 100-200 mM. Buffers for use in the present invention include, but are not limited to, [(2-Hydroxy-l,l-bis[hydroxymethyl]ethyl)amino]-l-propanesulfonic acid (TAPS, pKa 8.40), 2-Amino-2-methyl-l,3-propanediol (AMPD, pKa 8.80), N-tris- (Hydroxymethyl)methyl-4-aminobutanesulfonic acid (TABS, pKa 8.90), 3-([l,l- Dimethyl-2-hydroxyethyl]amino)-2-hydroxypropanesulfonic acid (AMPSO, pKa 9.00), N-(2-Hydroxyethyl)piperazine-N'-(2-ethanesulfonic acid) (HEPES, pKa 7.48), 3-(Cyclohexylamino)-2-hydroxy-l-propanesulfonic acid (CAPSO, pKa 9.60), 2-(N- Cyclohexylamino)ethanesulfonic acid (CHES, pKa 9.30), phosphate buffers, and Triethanolamine (pKa 7.8).
[0017] In one embodiment, the buffers discussed above are used as a wash buffer to prepare the red cells for reaction. When the red cells are adequately washed with these buffers, the reaction solution need not be buffered. For example, the reaction solution could be unbuffered blood bank saline, or an unbuffered dextrose solution
that is isotonic or hypertonic. In one embodiment, the reaction solution is an isotonic or hypertonic dextrose solution that lacks chloride ions. In another embodiment, when the red cells are washed with the above-mentioned buffers, the reaction solution can be a buffer with a pH range of about 6-10, also about 7-9. In one embodiment, the red cells are washed with a solution comprising a buffer having a concentration of about 50-350 mM, preferably about 100-200 mM, and a pH of about 8-10. In one embodiment, the washed red cells are then reacted in a reaction solution comprising blood bank saline. In another embodiment, the washed red cells are reacted in a reaction solution comprising unbuffered dextrose at a concentration that is effectively isotonic or hypertonic, such as about 125-300 mM, preferably about 125-200 mM. In a preferred embodiment, the unbuffered dextrose solution lacks chloride ions. In another embodiment, the pH is maintained by use of a resin. Appropriate buffering conditions for both reaction of activated antigen masking compound and for long term storage may be achieved by addition of a resin material to alter the buffering capacity of the red cell solution. A resin is defined as any solid material that can achieve the change of pH without being dissolved in the solution and encompasses man-made or naturally occurring materials, such as solid minerals. Such a resin could reduce or eliminate any washing requirements in order to achieve a suitable pH for the reaction of the compounds with the red cells. A thorough discussion of such methods can be found in US Provisional Patent Application Serial Number 60/338,707 and US Patent Application Serial Number 10/310,618. [0018] After reaction with the activated antigen masking compound, the cells can be washed with a buffer that provides suitable conditions for storage of the modified red cells, such as a buffer that restores the pH to physiological value (i.e. approximately pH 7). The post reaction wash will preferably have a pH of about 7-7.5, preferably about 7 and comprise a buffer at a concentration of approximately 50-350 mM, preferably about 100-200 mM. In addition to the buffering provided by these solutions, the solutions used in the methods of the present invention are optimized to reduce the amount of hemolysis of the red cells during the processing. Additives such as dextrose and L-carnitine may be included in these solutions in order to minimize hemolysis. In another embodiment, these solutions lack chloride ions. Any or all of the solutions used in the processing of the red cells comprise dextrose at a concentration of about 50-300 mM, preferably about 75-200 mM, more preferably approximately 100 mM or L-carnitine at a concentration of about 2-100 mM,
preferably about 2-10 mM, more preferably about 5 mM. In some embodiments, any or all of the solutions lack chloride ions. In some embodiments, any or all of the solutions used will comprise both dextrose and L-carnitine. In some embodiments, the solutions comprising both dextrose and L-carnitine will also lack chloride ions. In a preferred embodiment, all solutions used in the processing of the red cells will comprise both dextrose and L-carnitine. In a further embodiment, all solutions used in the processing will lack chloride ions.
[0019] The methods for reaction of the antigen masking compounds with red cells can also be improved by reacting at higher hematocrit. Reaction of red cells with a 5 kDa mPEG-SPA-NHS at the same extracellular concentration results in the same level of antigen masking of the red cells at 40, 60 or 80% hematocrit as determined by FACScan analysis of fluorescent labeled antibody binding to the cells. As hematocrit increases, a lesser amount of the mPEG is used with a higher number of red cells. Since level of antigen masking does not change, it is far more cost effective to react at the higher hematocrit. Therefore, it is preferred for this invention to carry out the reaction at a hematocrit of greater than about 50%, also greater than about 60%, or at approximately 60-95%. A detailed discussion of the reaction of antigen masking compounds with red cells at high hematocrit can be found in US Provisional Patent Application Serial Number 06/431,215, the contents of which are hereby incorporated by reference.
[0020] The present invention includes compositions comprising the compounds of the invention and a biological material. In one embodiment, the invention includes a composition comprising the compounds of the invention and a biological material selected from the group consisting of proteins, liposomes, and cells. In one embodiment, the composition comprises the compounds of the invention and red blood cells. In one embodiment, the composition comprises the compounds of the invention, a biological material, and a suitable buffer. The invention includes a biological material that has been reacted with the compounds of the invention. In one embodiment, the invention includes a composition comprising red blood cell that have been modified by covalent attachment of antigen masking compounds using the activated antigen masking compounds of the invention.
EXPERIMENTAL
EXAMPLE 1 Synthesis of mPEG-6AC-NHS, mPEG-5AV-NHS and mPEG-4AB-NHS esters.
[0021] N-6-[Methoxypoly(ethyleneglycol)]-oxo-aminocaproic acid 20 kDa (mPEG- 6AC):
[0022] 6-Aminocaproic acid (840 mg, 6.40 mmol) and NaHC03 (538 mg, 6.40 mmol) were dissolved in a mixture of H20 (160 mL) and ethanol (50 mL). Methoxypoly(ethyleneglycol) succinimidyl carbonate (n = 453, 64.2 g, 3.20 mmol, prepared from the Methoxypoly(ethyleneglycol)-OH (Shearwater Polymers, Huntsville, AL) by known methods) was added and the mixture was stirred at room temperature for 3 hours. HC1 (I N) was added until the solution reached pH 5. The resulting solution was extracted with CH2C1 (3 x 150 mL) and the combined organic extracts were dried (Na2S0 ), filtered and concentrated under vacuum to give mPEG- 6AC (60.11 g, 2.99 mmol, 93%) as a white solid.
[0023] 1H NMR (200 MHz, DMSO-J6): δH 7.14-7.29 (m, 1 H), 4.07 (t, / = 4.44, 2 H), 3.60-3.30 (m, 1810 H), 3.25 (s, 3 H), 2.89-2.99 (m, 2 H), 2.20 (t, J = 7.32, 2 H), 1.10-1.60 (m, 6 H). IR; nujol, cm"1,1721, 1643, 1099. N-6-[Methoxypoly(ethyleneglycol)]-oxo-aminocaproate N-hydroxysuccinimide ester
20 kDa (mPEG-6AC-ΝHS):
[0024] N-6-[methoxypoly(ethyleneglycol)]-oxo-aminocaproic acid (n = 453, 55.16 g, 2.74 mmol) was dissolved in CH2C12 (200 mL) and cooled in an ice bath under an atmosphere of argon. N-Hydroxysuccinimide (631 mg, 5.48 mmol) and
dicyclohexylcarbodiimide (DOC, 1.13 g, 5.48 mmol, in CH2C12 (5 mL)) were added and the resulting mixture was stirred at room temperature for 17 hours. The reaction mixture was filtered to remove the crystals which had formed and the filtrate was concentrated under vacuum. The resulting white solid was washed with ether (600 mL), filtered and covered with isopropanol (800 mL). The suspension as warmed to 50 °C until the solid completely dissolved after which the solution was cooled in an ice bath for 2 hours. The suspension was filtered, and the solid redissolved in CH2C12 (160 mL). Ether (1600 mL) was added and the resulting white precipitate was collected by filtration and dried under vacuum to give mPEG-6AC-NHS (53.2 g, 2.63 mmol, 96%) as a white solid.
[0025] 1H NMR (200 MHz, DMSO-- 6): δH 7.09-7.29 (m, 1 H), 3.98-4.09 (m, 1 H), 3.11-3.93 (m, 1810 H), 3.25 (s, 3 H), 2.91-3.04 (m, 2 H), 2.82 (s, 4 H), 2.65 (t, J = 7.59, 2 H), 1.51-1.67 (m, 2 H), 1.26-1.49 (m, 4 H). IR; nujol, cm"1, 1813, 1783, 1740, 1713, 1148, 1109, 1061.
[0026] N-5-[Methoxypoly(ethylenegιycol)]-oxø-amino valeric acid 5 kDa (mPEG- 5AV):
[0027] 5-Aminovaleric acid (91 mg, 0.778 mmol) and NaHC03 (65 mg, 0.778 mmol) were dissolved in a mixture of H20 (14 mL) and ethanol (6 mL). Methoxypoly(ethyleneglycol) succinimidyl carbonate (n = 114, 2.0 g, 0.39 mmol) was added and the mixture was strrred at room temperature for 2 hours. HC1 (I N) was added until the solution reached pH 3. The resulting solution was extracted with CH2C12 (3 x 50 mL) and the combined organic extracts were dried (Na2S0 ), filtered and concentrated under vacuum to give mPEG-5-AV acid (1.66 g, 83%) as a white solid.
[0028] 1H NMR (200 MHz, DMSO-J
6): δ
H 7.14-7.29 (m, 1 H), 4.05 (m, 2 H), 3.55 (s, 454 H), 3.25 (s, 3 H), 2.95 (q, J = 5.94 Hz, 2 H), 2.20 (t, J = 7.32, 2 H), 1.25-1.6 (m, 4H). IR; nujol,
1103, 1099.
[0029] N-5-[Methoxypoly(ethyleneglycol)]-oxø-ammovalerate N- hydroxysuccinimide ester(mPEG-5 AV-ΝHS) :
[0030] N-5-[methoxypoly(ethyleneglycol)]-oxo-aminovaleric acid (n = 114, 1.66 g, 0.323 mmol) was dissolved in CH C12 (8.5 mL) and cooled in an ice bath under an atmosphere of argon. N-Hydroxysuccinimide (74 mg, 0.65 mmol) was added, and DCC (133 mg, 0.65 mmol, in CH C12 (0.5 mL) was added. The resulting mixture was stirred at room temperature for 17 hours. The reaction mixture was filtered to remove the crystals which had formed and the filtrate was concentrated under vacuum. The resulting white solid was recrystallized with isopropanol (10 mL). The solid was redissolved in CH2C1 (3 mL). Ether (50 mL) was added and the resulting white precipitate was collected by filtration and dried under vacuum to give mPEG- 5AV-ΝHS (1.23 g, 0.24 mmol, 72%) as a white solid.
[0031] 1H NMR (200 MHz, DMSO-J6): δH 7.09-7.29 (m, 1 H), 3.98-4.09 (m, 1 H), 3.55 (s, 454 H), 3.25 (s, 3 H), 3.0 (q, J = 5.78, 2H), 2.83 (s, 4 H), 2.70 (t, J = 6.31 Hz, 2 H), 1.42-1.80 (m, 4 H). IR; nujol, cm"1, 1813, 1783, 1740, 1713, 1148, 1109, 1061. [0032] N-4-[Methoxypoly(ethyleneglycol)]-c»xo-aminobutyric acid 5 kDa (mPEG- 4AB):
[0033] 4-Aminobutyric acid (80 mg, 0.778 mmol) and NaHC03 (65 mg, 0.778 mmol) were dissolved in a mixture of H20 (14 mL) and ethanol (6 mL).
Methoxypoly(ethyleneglycol) succinimidyl carbonate (n = 114, 2.0 g, 0.39 mmol) was added and the mixture was stirred at room temperature for 2 hours. HC1 (I N) was added until the solution reached pH 3. The resulting solution was extracted with
CH
2C1
2 (3 x 50 mL) and the combined organic extracts were dried (Na
2S0 ), filtered and concentrated under vacuum to give mPEG-4-AB acid (1.77 g, 88%) as a white solid.
[0034] 1H NMR (200 MHz, DMSO--f
6): δ
H 7.14-7.29 (m, 1 H), 4.05 (t, / = 4.44, 2 H), 3.55 (s, 454 H), 3.25 (s, 3 H), 3.0 (q, J = 6.31 Hz, 2 H), 2.22 (t, / = 7.4, 2 H), 1.62 (pent, J = 7.4 Hz, 2H). IR; nujol,
1103, 1099. [0035] N-4-[Methoxypoly(ethyleneglycol)]-αxσ-aminobutyrate N- hydroxysuccinimide ester 5 kDa (mPEG-4AB-ΝHS):
[0036] N-4-[methoxypoly(ethyleneglycol)]-oxo-aminobutyric acid (n = 114, 1.77 g, 0.345 mmol) was dissolved in CH2C12 (8.2 mL) and cooled in an ice bath under an atmosphere of argon. N-Hydroxysuccinimide (79 mg, 0.69 mmol) was added, and DCC (142 mg, 0.69 mmol, in CH2C12 (0.5 mL) was added. The resulting mixture was stirred at room temperature for 17 hours. The reaction mixture was filtered to remove the crystals which had formed and the filtrate was concentrated under vacuum. The resulting white solid was recrystallized with isopropanol (15 mL). The solid was redissolved in CH2CI2 (5 mL). Ether (100 mL) was added and the resulting white precipitate was collected by filtration and dried under vacuum to give mPEG- 4AB-ΝHS (1.35 g, 0.26 mmol, 75%) as a white solid.
[0037] 1H NMR (200 MHz, DMSO- ): δH 7.09-7.29 (m, 1 H), 3.98-4.09 (m, 1 H), 3.55 (s, 454 H), 3.25 (s, 3 H), 2.98 (q, J = 5.85, 2H), 2.83 (s, 4 H), 2.68 (t, J = 7.59, 2 H), 1.52-1.72 (m, 2 H). IR; nujol, cm"1, 1813, 1783, 1740, 1713, 1148, 1109, 1061. [0038] The above reactions can be done starting with PEG in place of the mPEG to make the succinimidyl carbonates, so that the succinimidyl carbonate will be at both ends of the compound (di-succinimidyl carbonate PEG), and subsequently the oxo- amino acid N-hydroxysuccinimide ester will be at both ends of the compound (di- 6AC-ΝHS-PEG, see Example 3).
EXAMPLE 2 Synthesis of various mPEG-6AC esters and thio esters.
[0039] The following procedures can be followed to make a variety of active mPEG-6AC derivatives. As per Example 1, the mPEG-6AC could also be prepared as di-6AC-PEG by making the di-succinimidyl carbonate PEG from PEG instead of
mPEG. The resulting compounds would have the αxø-6-aminocaproic acid esters and thiols at both ends. Procedure A:
[0040] N-6-[methoxypoly(ethyleneglycol)]-oxo-aminocaproic acid (e.g. 1 g, 49.7 μmol) was dissolved in CH2C12 (10 mL). l-[3-(dimethylamino)propyl]-3- ethylcarbodimide (EDCI, 1.5 eq., Aldrich), and 4-(dimethylamino)pyridine (DMAP, 0.1 eq., Aldrich) were mixed with the alcohol (10 eq.) or thiol (10 eq.), and stirred at room temperature under an atmosphere of nitrogen until Thin Layer Chromatography (TLC, reverse phase, C8, CH3OHH20 4/1, v/v) indicated complete conversion to a less polar product. Ether (100 mL) was added and the resulting suspension was cooled to 0 °C for 1 hour. The suspension was filtered and the white solid recrystallized from isopropanol (20 mL). The solid was redissolved in CH2C12 (10 mL) and ether (100 mL) was added. The resulting solution was cooled to 0 °C for 1 hour, after which the white precipitate was collected by filtration and dried under vacuum. Procedure B:
[0041] N-6-[methoxypoly(ethyleneglycol)]-oxo-aminocaproic acid (e.g. 1 g, 49.7 μmol) was dissolved in CH3CΝ (10 mL). Benzotriazole-1-yl-oxy-tris-pyrrolidino- phosphonium hexafluorophosphate (PyBOP, 1 eq., Calbiochem, San Diego, CA), and 1-hydroxybenzotriazole (HOBT, 1 eq., Aldrich), were mixed with triethylamine (Et3N, 2 eq., Aldrich) and the alcohol (5 eq.) or thiol (5 eq.), and stirred at room temperature under an atmosphere of nitrogen until TLC (reverse phase, C8, CH3OH/H204/1, v/v) indicated complete conversion to a less polar product. Ether (100 mL) was added and the resulting suspension was cooled to 0 °C for 1 hour. The suspension was filtered and the white solid recrystallized from isopropanol (20 mL). The solid was redissolved in CH2C12 (10 mL) and ether (100 mL) was added. The resulting solution was cooled to 0 °C for 1 hour, after which the white precipitate was collected by filtration and dried under vacuum. Procedure C:
[0042] N-6-[methoxypoly(ethyleneglycol)]-oxo-aminocaproic acid (1 g, 49.7 μmol) was dissolved in CH3CΝ (10 mL). 2-(lH-Benzotriazole-l-yl)-l, 1,3,3- tetramethyluronium hexafluorophosphate (HBTU, 1 eq., Calbiochem, San Diego, CA) and Et3N (2 eq.) were mixed with the alcohol (5 eq.) or thiol (5 eq.), and stirred at room temperature under an atmosphere of nitrogen until TLC (reverse phase, C8,
CH3OH H204/1, v/v) indicated complete conversion to a less polar product. Ether (100 mL) was added and the resulting suspension was cooled to 0 °C for 1 hour. The suspension was filtered and the white solid recrystallized from isopropanol (20 mL). The solid was redissolved in CH2C12 (10 mL) and ether (100 mL) was added. The resulting solution was cooled to 0 °C for 1 hour, after which the white precipitate was collected by filtration and dried under vacuum.
[0043] N-6-[Methoxypoly(ethyleneglycol)]-oxo-aminocaproate pentafluorophenyl ester 20 kDa (mPEG-6AC-PFP):
[0044] Prepared using procedure C (n = 453, 1 g, 49.7 μmol). Obtained 953 mg, 47.0 μmol, 87% as a white solid.
[0045] 1H NMR (200 MHz, DMSO--4): δH 7.17-7.30 (m, 1 H), 3.99-4.10 (m, 2 H), 3.11-3.93 (m, 1810 H), 3.25 (s, 3 H), 2.89-3.04 (m, 2 H), 2.80 (m, 2 H), 1.57-1.79 (m, 2 H), 1.26-1.51 (m, 4 H). IR; nujol, cm"1, 1788, 1719, 1674, 1105, 1061. [0046] N-6-[Methoxypoly(ethyleneglycol)]-oxo-aminocaproate 2,2,2-trifluoroethyl ester 20 kDa (mPEG-6AC-TFE):
[0047] Prepared using procedure A (n = 453, 1 g, 49.7 μmol). Obtained 957 mg,
47.4 μmol, 82% as a white solid.
[0048] 1H NMR (200 MHz, DMSO-J6): δH 7.19-7.31 (m, 1 H), 4.65-4.84 (m, 2 H),
3.97-4.10 (m, 2 H), 3.18-3.95 (m, 1813 H), 2.87-3.06 (m, 2 H), 2.37-2.48 (m, 2 H),
1.14-1.62 (m, 6 H). IR; nujol, cm"1, 1756, 1717, 1641, 1148, 1109, 1057.
[0049] N-6-[Methoxypoly(ethyleneglycol)]-øxø-aminocaproate ethyl ester 20 kDa
(mPEG-6AC-OEt):
mPEG-6AC mPEG-6AC-OEt
[0050] Prepared using procedure A (n = 453, 1 g, 49.7 μmol). Obtained 940 mg,
46.7 μmol, 79% as a white solid.
[0051] 1H NMR (200 MHz, DMSO-J6): δH 7.05-7.26 (m, 1 H), 4.01-4.19 (m, 2 H),
3.11-4.01 (m, 1813 H), 2.92-3.04 (m, 2 H), 2.20-2.36 (m, 2 H), 1.10-1.65 (m, 9 H),.
IR; nujol, cm"1, 1720, 1141, 1111, 1060.
[0052] N-6- [Methoxypoly(ethyleneglycol)] -øxoaminocaproate pentafluorobenzenethio ester 20 kDa (mPEG-6AC-PFT):
[0053] Prepared using procedure C (n = 453, 1 g, 49.7 μmol). Obtained 0.84 g, 41.4 μmol, 89% as a white solid. !H NMR (200 MHz, DMSO-J6): δH 7.13-7.25 (m, 1 H), 3.99-4.12 (m, 2 H), 3.08-3.95 (m, 1813 H), 2.79-3.07 (m, 4 H), 1.19-1.71 (m, 6 H). IR; nujol, cm"1, 1719, 1640, 1145, 1111, 1061.
[0054] N-6-[Methoxypoly(ethyleneglycol)]-oxo-aminocaproate 2,3,5,6- tetrafluorobenzene thio ester 20 kDa (mPEG-6AC-TFT):
[0055] Prepared using procedure C (n = 453, 1 g, 49.7 μmol). Obtained 1.10 g, 54.2 μmol, 98% as a white solid. 1H NMR (200 MHz, DMSO-J6): δH 8.06-8.31 (m, 1 H), 7.13-7.25 (m, 1 H), 3.99-4.11 (m, 2 H), 3.08-3.95 (m, 1810 H), 3.26 (s, 3 H), 2.77- 3.07 (m, 4 H), 1.53-1.75 (m, 2 H), 1.22-1.53 (m, 4 H). J-R; nujol, cm"1, 1720, 1633, 1148, 1110, 1061.
[0056] N-6- [Methoxypoly(ethyleneglycol)] - xo-aminocaproate 4-fluorobenzenethio ester 20 kDa (mPEG-6AC-4FTP):
mPEG-6AC mPEG-6AC-4FTP
[0057] Prepared using procedure B (n = 453, 1 g, 49.7 μmol). Obtained 0.94 g, 54.2 μmol, 88% as a white solid. tø NMR (200 MHz, DMSO-J
6): δ
H 7.25-7.59 (m, 3 H), 3.97-4.11 (m, 2 H), 3.07-3.96 (m, 1813 H), 2.90-3.03 (m, 2 H), 2.64-2.77 (m, 2 H), 1.53-1.76 (m, 2 H), 1.25-1.49 (m, 4 H). IR; nujol, cm
"1, 1710, 16.76, 1633, 1147, 1108, 1061.
[0058] N-6-[Methoxypoly(ethyleneglycol)]-oxo-aminocaρroate 2,2,2- trifluoroethylthio ester 20 kDa (mPEG-6AC-TFET):
[0059] Prepared using procedure B (n = 453, 1 g, 49.7 μmol). Obtained 1.04 g, 51.4 μmol, 86 % as a white solid. JH NMR (200 MHz, DMSO-J6): δH 7.07-7.25 (m, 1 H), 3.99-4.10 (m, 2 H), 3.09-3.94 (m, 1813 H), 2.89-3.00 (m, 2 H), 2.62-2.77 (m, 2 H), 1.51-1.74 (m, 2 H), 1.19-1.48 (m, 4 H). IR; nujol, cm"1, 1722, 1142, 1108, 1062. [0060] N-6-[Methoxypoly(ethyleneglycol)]-oxo-aminocaproate ethylthio ester 20 kDa (mPEG-6AC-SEt):
Ethanethiol
[0061] Prepared using procedure A (n = 453, 1 g, 49.7 μmol). Obtained 0.86 g, 42.6 μmol, 85% as a white solid. XH ΝMR (200 MHz, DMSO-J6): δH 7.03-7.30 (m, 1 H), 3.98-4.11 (m, 2 H), 3.04-3.97 (m, 1813 H), 2.78-3.03 (m, 4 H), 1.06-1.78 (m, 9 H). IR; nujol, cm"1, 1785, 1718, 1646, 1148, 1109, 1061.
EXAMPLE 3 Synthesis of di-6AC-ΝHS-PEG
[0062] The following procedure can be used to make a variety of sizes of activated branched PEG compounds based on the size of the starting material.
[0063] Poly(ethyleneglycol)- ,CD-di-(succinimidyl carbonate) (SC-PEG-SC)
[0064] Poly(ethyleneglycol) (n=453, 20 kDa, 20 g, 1.00 mmol, Fluka) was dissolved in a mixture of CH2C12 (30 mL) and CH3CN (30 mL) under an atmosphere of argon by stirring at room temperature for 5 minutes. Pyridine (2.02 mL, 25 mmol) and DSC (2.05 g, 8 mmol) were added and the reaction mixture was stirred under argon for 20 hours to give a light brown colored solution. The solution was filtered and the filtrate concentrated under vacuum. Ether (200 mL) was added to the residue followed by stirring to give an off white solid which was isolated by filtration. The solid was re-dissolved in ethyl acetate (200 mL) at 50 °C to give a cloudy brown solution which was subsequently filtered. The clear filtrate was triturated with ether (200 mL) and cooled in an ice bath for 2 hours. The resulting white solid was isolated by filtration to give SC-PEG-SC (19.29 g, 0.952 mmol, 95%). [0065] lR NMR (200 MHz, DMSO-J6): δH 4.42-4.50 (m, 4 H), 3.30-3.78 (m, 1808 H), 2.81 (s, 8 H). IR; nujol, cm"1, 1811, 1788, 1742, 1716, 1641, 1101.
[0066] Poly(ethyleneglycol)-α,ω-di-oxo-aminocaproic acid (6AC-PEG-6AC).
[0067] 6-Aminocaproic acid (468 mg, 3.57 mmol) and NaHC03 (300 mg, 3.57 mmol) were dissolved in a mixture of H20 (45 mL) and ethanol (15 mL). Poly(ethyleneglycol)-α,ω-di-(succinimidyl carbonate) (18.1 g, 0.893 mmol) was added and the mixture was stirred at room temperature for 2.5 hours. HC1 (1 N) was added until the solution reached pH 4. The resulting solution was extracted with CH2C12 (3 x 50 mL) and the combined organic extracts were dried (Na2S04), filtered and concentrated under vacuum to give poly(ethyleneglycol)-α,ω-di-oxo-
aminocaproic acid (17.78 g, 0.876 mmol, 98%) as a white solid.
1H NMR (200 MHz, DMSO-J6): δH 7.17-7.25 (m, 2 H), 4.05-4.10 (m, 4 H), 3.40-
3.57 (m, 1808 H), 2.95 (m, 4 H), 2.20 (t, / = 7.3, 4 H), 1.10-1.55 (m, 12 H). IR; nujol, cm"1, 1719, 1648, 1099
[0068] Poly(ethyleneglycol)-α,ω-di-oxo-aminocaproate N-hydroxysuccinimide ester
(ΝHS-6AC-PEG-6AC-ΝHS).
[0069] Poly(ethyleneglycol)-α,ω-di-oxo-aminocaproic acid (17.01 g, 0.838 mmol) was dissolved in CH C12 (70 mL) and cooled in an ice bath under an atmosphere of argon. N-Hydroxysuccinimide (386 mg, 3.35 mmol) and DCC (691mg, 3.35 mmol, in CH2C12 (5 mL)) were added and the resulting mixture was stirred at room temperature for 18 hours. The reaction mixture was filtered to remove the white solid which had formed and the filtrate was concentrated under vacuum. The resulting white solid was washed with ether (200 mL), filtered and covered with isopropanol (250 mL). The suspension was warmed to 50 °C until the solid completely dissolved after which the solution was cooled in an ice bath for 2 hours. The suspension was filtered, and the solid re-dissolved in CH2C12 (50 mL). Ether (500 mL) was added and the resulting white precipitate was collected by filtration and dried under vacuum to give ΝHS-6AC-PEG-6AC-ΝHS (16.48 g, 0.804 mmol, 96%) as a white solid.
[0070] 1H NMR (200 MHz, DMSO-J6): δH 7.15-7.24 (m, 2 H), 4.00-4.09 (m, 4 H), 3.30-3.58 (m, 1808 H), 2.90-2.98 (m, 4 H), 2.82 (s, 8 H), 2.65 (t, 4 H), 1.51-1.70 (m, 4 H), 1.30-1.49 (m, 8 H). IR; nujol, cm"1, 1812, 1783, 1740, 1713, 1641, 1148, 1107.
EXAMPLE 4 Synthesis of 4-Arm-PEG-6AC-NHS
[0071] The following procedure can be used to make a variety of sizes of activated branched PEG compounds based on the size of the starting material. Further, the corresponding 3 arm compound can be made by starting with the 3 arm PEG (available from Shearwater).
[0072] Pentaerythritoxy poly(ethylene glycol) Succinimidyl Carbonate (4-Arm-PEG- SC)
[0073] Disuccinimidyl carbonate (DSC, 1.54 g, 6 mmol, Aldrich) was added with stirring to pentaerythritol poly(ethylene glycol) (n=114, 20 kDa, 10 g, 0.5 mmol (2 mmol OH groups), Shearwater) in dichloromethane (20 mL) and acetonitrile (40 mL). Pyridine (1 mL) was added. The mixture was stirred at ambient temperature for 4 hours. The reaction solution was filtered, and concentrated in vacuo to a minimal volume. The residue was dissolved in ethyl acetate (90 mL) with heating. Ether (90 mL) was added to this solution, and the resulting solution was cooled with an ice bath. A white precipitate was collected under reduced pressure, and dried under vacuum at room temperature overnight, 10.2 g.
1H NMR (200 MHz, DMSO-J6): δH 4.5 (m, 8H), 3.60-3.30 (m, 1824 H), 2.82 (s, 16H). IR; nujol, cm"1, 1811, 1789, 1742, 1717, 1633, 1099.
[0074] Pentaerythritoxy poly(ethylene glycol) oxo-aminocaproic acid 20 kD (4-Arm- PEG-6AC):
[0075] 6-Aminocaproic acid (500 mg, 3.91 mmol, Aldrich) and NaHC03 (330 mg, 3.91 mmol, EM Science) were dissolved in H20 (60 mL). Pantaerythritoxy poly(ethylene glycol) succinimidyl carbonate (10 g, 0.49 mmol (1.95 mmol NHS ester groups)) from above was added and the mixture was stirred at room temperature for 2 hours. H20 (200 mL) was added to the reaction solution. HCl (1 N) was added until the solution reached pH 5. The resulting solution was extracted with CH2C12 (3
x 50 mL) and the combined organic extracts were dried (Na2S0 ), filtered and concentrated under vacuum to give Pentaerythritoxy poly(ethylene glycol) oxo- aminocaproic acid (9.47 g, 0.46 mmol, 95%) as a white solid. 1H NMR (200 MHz, DMSO-J6): δH 4.07 (t, / = 4.44, 8 H), 3.60-3.30 (m, 1832 H), 2.20 (t, /= 7.32, 8 H), 1.10-1.60 (m, 24 H). IR; nujol, cm"1,1721, 1643, 1099.
[0076] Succinimidyl Pentaerythritoxy polyethylene glycol) oxo-aminocaproate (4- Arm-PEG-6AC NHS):
[0077] Pentaerythritoxy poly(ethylene glycol) oxo-aminocaproic acid (9 g, 0.5 mmol (2 mmol carboxylic acid groups), from previous preparation) was dissolved in CH2C12 (50 mL) and cooled in an ice bath under an atmosphere of argon. N- Hydroxysuccinimide (402 mg, 3 mmol, Aldrich) and dicyclohexylcarbodiimide (DCC) (0.721 g, 3 mmol, Aldrich), in CH2C12 (5 mL)) were added and the resulting mixture was stirred at room temperature for 17 hours. The reaction mixture was filtered to remove the crystals which had formed and the filtrate was concentrated under vacuum. The resulting white solid was washed with ether (100 mL), recrystallized in IPA (105 mL), and the solid redissolved in CH2C12 (25 mL). Ether (150 mL) was added and the resulting white precipitate was collected by filtration and dried under vacuum to give Succinimidyl Pentaerythritoxy polyethylene glycol) oxo-aminocaproate (8.72 g, 0.42 mmol, 95%) as a white solid. 1H ΝMR (200 MHz, DMSO÷J6): δH 7.09-7.29 (m, 4 H), 4.01-4.1 (m, 8 H), 3.55 (s, 1824 H), 2.91-3.04 (m, 8 H), 2.82 (s, 16 H), 2.65 (t, / = 7.59, 8 H), 1.51-1.67 (m, 8 H), 1.26-1.49 (m, 16 H). IR; nujol, cm"1, 1813, 1783, 1740, 1713, 1148, 1109, 1061.
EXAMPLE 5 Analysis of hydrolysis and leaving group content of compounds.
[0078] The purity of the compounds were estimated by measuring the amount of the leaving group (e.g. ΝHS) that is released upon hydrolysis of a known amount of the compound. For ΝHS, the mPEG-ΝHS derivative was dissolved in water to a final
concentration of 1 mM. A 300 μl aliquot of this was added to 2.7 mL of 0.1 M NH OH and mixed for 2 minutes. The UV absorbance was measured at 260 nm and the NHS was calculated based on a standard curve for NHS concentration. For thioester compounds, the thiol content was measured by use of EUman's assay to measure free thiol released. The mPEG-thioester was dissolved in IN NaOH to a final concentration of 1 mM. A 10 μl aliquot of this was added to a mixture of 890 μl of sodium phosphate (300 mM pH 9) and 100 μl EUman's solution. EUman's solution was 10 mg 5,5'-dithiobis(2-nitrobenzoic acid) (Sigma) in 100 mL of 1% sodium citrate. The UV absorbance was measured at 412 nm and the thiol content was determined from a standard curve prepared using known amounts of the free thiol.
[0079] In addition, the relative reactivity of these compounds was assessed by measuring the kinetics of hydrolysis. For mPEG-NHS derivatives, the activated mPEG was dissolved in 5 mM HCl to a final concentration of 4.67 mM. A 300 μl aliquot of this was added to 2.7 mL of 0.1 M phosphate buffer pH 8. The UV absorbance at 290 nm was measured every 5 minutes for 2.5 hours and the t 2 is determined as the time to hydrolyze half of the starting material. For mPEG-PFP derivatives, the PFP release was determined by measuring the change in absorbance at 265 nm. The mPEG-PFP was prepared in HCl as per the NHS but the dilution of 300 μL was into 2.7 mL of 0.1 M CHES pH 9. The absorbance at 265 nm was monitored every 5 minutes overnight and the tι/2 was determined. Results for some of the compounds are given in Table 1. Also included are data for mPEG-SβA-NHS, where the unnatural amino acid group is β alanine, wherein x=2 and Y=N- hydroxysuccinimide in the formula given above for compounds. Comparison of the mPEG-SβA-NHS to the 4AB, 5AV, and 6AC compounds demonstrates the change in reactivity as a function of x. For thioesters that do not have sufficient absorbance, the EUman's assay discussed above could be used to monitor the release of thiol with time.
Table 1 NHS or thiol content and tυ2 for various compounds.
* These concentrations are presented as percent of expected values based on having one NHS or thiol per PEG molecule (4 for the 4-Arm compound).
EXAMPLE 6 Measurement of modification density for PEG Modified RBC
[0080] Leukofiltered RBC (approximately 60% hematocrit) containing a suitable additive solution such as Eiythrosol are centrifuged to an 80-95% hematocrit (red cell concentrate), washed twice with buffer (e.g. pH 9.0 CHES, 150 mM CEDES, 50 mM NaCl) and subsequently diluted to a hematocrit of 40% into a CHES buffer solution containing mPEG-6AC-NHS (or other activated PEG) at an appropriate concentration. In addition, the activated PEG is a mixture of the activated mPEG plus FITC labeled activated PEG (FPEG) with the same coupling group, which bears a fluorescent label on the end opposite the coupling group. Alternatively, the activated mPEG is modified with other detectable labels, such as a radioactive isotope. A 50:50 mixture of mPEG-6AC-NHS to FPEG-6AC-NHS is typically used for this experiment. The reaction is allowed to proceed for 2 hours at room temperature (RT) and the cells are subsequently washed to remove the reaction side products and any fluorescent label that is not attached to the red cells. RBC concentrate (200 μ ) is subsequently used to make ghost membranes through controlled lysis with chilled hypotonic lysis buffer (1600 L, 7.5 mM sodium phosphate, lmM NaEDTA, pH 7.5). The resulting ghosts are isolated through centrifugation (14000 x g; 2 min) and washed a total of 4 times with chilled lysis
buffer and then suspended in a 250/xL volume of the same buffer. SDS is added to the suspension to a final concentration of 1% SDS in order to achieve complete dissolution of the membranes. The resulting solution is further diluted 5 fold in lysis buffer and then analyzed for fluorescent label content (λexc=490nm, λemπι=525nm). The amount of fluorescent label is quantitated versus a standard curve prepared by adding specific amounts of FPEG in the dissolved ghost membranes in a lysis buffer containing SDS, prepared as per the reacted samples above. The fluorescence reading is plotted against the known concentration of FPEG added to the ghost membrane preparation and this curve is used to calculate the FPEG concentration corresponding to a fluorescence reading in the ghosts that have been reacted with the activated FPEG. Based on this calculated concentration of FPEG on the ghosts (or the concentration of another suitable label) and the number of cells used to prepare the ghosts, the amount of FPEG per cell (membrane modification density) can be calculated for a given experiment (Figure 2A). From the FPEG:mPEG ratio, the number of total PEG molecules per cell can be calculated. An aliquot of the red cells that are modified with the FPEG can also be analyzed by FACScan (counting a set number of red cells). The calculated PEG molecules per cell can be plotted against the FACScan peak value (FL1 -Height) and this curve can be used on new samples to calculate the amount of binding directly from the FACScan reading of the modified red cells (Figure 2B).
[0081] As a an example of the calculations involved, ghosts were prepared at a level of 1.3 x 109 cells and dosed with known concentrations of FPEG and the fluorescence measured to generate the line from Figure 2A. Ghosts were prepared from 5 kDa mPEG-SPA-NHS / FPEG-SPA-NHS modified red cells (reacted as described above, only using pH 8.0 HEPES, 150 mM HEPES, 50 mM NaCl, 75 mM glucose) and the modification density was measured at each concentration of PEG used in the reaction. The 13.2 mM PEG sample gave a fluorescence reading of 1495, which from Figure 2A was calculated to be 9.8 μM. As the actual samples were only 50% FPEG, the total PEG for this sample was 19.6 μM. Based on the known cell number in the sample (1 mL volume), the total number of moles of PEG per cell was calculated as 19.6 μM x 10"6 (mole/μmole) x 10"3 (L/mL). The moles of PEG per cell was then 1.96 x 10"8 moles / 1.3 x 109 cells. Using Avogadros number, the total amount of PEG per cell was calculated to be 9 x 106.
[0082] An additional method of use of the FPEG approach for the measurement of RBC PEG modification is achieved through the use of flow cytometry analysis of the pegylated RBC using a FACScan device. The RBC are directly analyzed for fluorescence intensity through a commercial device. The number of PEG molecules attached to the RBC surface is proportional to the percent of active FPEG in the active PEG. The FACScan fluorescent signal intensity is proportional to the PEG content. A standard curve of PEG modification done either through the method above or by comparison to beads containing known amounts of fluorescent molecules on them can be used to quantify fluorescent label amounts. Beads used in a FACScan device are commercially available and can be prepared to custom specifications (Bangs Laboratories, Fishers, IN).
[0083] An alternative method for the quantitation of the PEG molecules is the use of radioactively labeled activated mPEG (labeled with covalently attached 3H, 14C or other appropriate radioactive atom). The RBC are washed after the end of the PEG modification procedure and then the washed RBC are lysed, decolorized and the radioactivity content is measured through liquid scintillation. The extent of PEG modification is calculated using the specific activity of the radiolabeled activated mPEG.
[0084] Another method involves the use of compounds of the present invention containing an unnatural amino acid in the coupling group, such as mPEG-6AC-NHS (6-amino caproic acid is an unnatural amino acid). The reaction of this with red cells will deposit a number of the unnatural amino acids on the surface of the red cells that corresponds to the number of mPEG molecules on the surface of the red cells. The RBC are lysed after PEG modification along with control RBC that are unmodified. Ghosts are prepared from both populations for a known number of cells, the samples are treated to release free amino acids, and the amino acid content is measured for both preparations through the use of an HPLC assay (Sartore et al., Applied Biochemistry and Biotechnology 31:213, 1991). For example, with mPEG-6AC- NHS, the 6AC content will be compared to the number of natural amino acids. Since the control sample will give you the number of natural amino acids per red cell, the ratio between 6AC and the natural amino acids can be used to quantify the amount of 6AC per red cell, which gives the amount of mPEG per red cell. Alternatively, the number of 6AC can be determined for a known number of red cells and the mPEG per red cell can be calculated directly. The methods above using FPEG could be
used to compare the FPEG results to the amino acid analysis results. The modification density for an antigen masking compound of a certain size can be determined as a function of the concentration used and correlated with agglutination assays or antibody binding assays to estimate the level of modification density necessary to get adequate coverage of the red cell antigens.
EXAMPLE 7 Measurement of modification density for RBC modified with mPEG-6AC-NHS.
[0085] Leukofiltered RBC (approximately 60% hematocrit) containing a suitable additive solution such as Erythrosol ™ (Erythrosol consists of 94 mL of Part A (25.0 mM sodium citrate, 16.0 mM disodium phosphate, 4.4 mM monosodium phosphate, 1.5 mM adenine, 39.9 mM mannitol), and 20 mL of Part B (8% dextrose)) are centrifuged at 4 °C at 4100 x g for 6 min and the plasma is removed to provide a red cell concentrate. These are washed with a buffer comprising 150 mM CHES pH 9, 100 mM dextrose, 5 mM L-carnitine. A sample of 5 kDa mPEG-6AC-NHS is dissolved in the same buffer and added to the washed red cell concentrate at a hematocrit of approximately 40% (22 mM 6 AC mPEG in the extracellular volume). This is gently mixed and incubated at room temperature for approximately 1 hour. Following incubation, the cells are washed with 150 mM Na2HP04 pH 7, 100 mM dextrose, 5 mM L-carnitine to remove reaction side products and unreacted mPEG- 6AC-NHS. The modified RBC concentrate (200 μL) is subsequently used to make ghost membranes through controlled lysis with chilled hypotonic lysis buffer (1600 iL) (7.5 mM sodium phosphate, lmM NaEDTA, pH 7.5). A sample of unmodified RBC concentrate with a know red cell count is also treated to make ghost membranes. The resulting ghosts are isolated through centrifugation (14000 x g; 2 min) and washed a total of 4 times with chilled lysis buffer and then taken up in a 250μL volume of the same buffer. SDS is added to the suspension to a final concentration of 1% SDS in order to achieve complete dissolution of the membranes. The modified and unmodified red cell ghosts are diluted into concentrated HCl and hydrolyzed under vacuum at 110 °C for 24 hours and the resulting samples are analyzed by HPLC [Sartore et al, Applied Biochemistry and Biotechnology, 31:213, 1991]. The amino acid content per red cell is evaluated for the unmodified control. For the modified sample, the 6 amino caproic acid (6 AC) content is compared to the
content of natural amino acids. The ratio between 6 amino caproic acid and the natural amino acids can be used to quantify the amount of 6AC per red cell, which gives the amount of mPEG per red cell.
EXAMPLE 8 Determination of agglutination reaction of RBC of the present invention.
[0016] The process of antigen masking of red cells is carried out under appropriate conditions on CPDA-1 collected RBC using PEG derivatives of the present invention. Agglutination reactions of the treated RBC are assayed by standard techniques as described in Walker et al., AABB Technical Manual, 10th Ed., pp. 528-537 (1990). The agglutination reaction is assessed on serially diluted samples. The dilution level at which agglutination no longer is observed is recorded for treated RBC compared to untreated RBC. This assay is carried out using type A RBC and anti-A antibody or anitserum or type B RBC and anti-B antibody or antiserum. The processing of the RBC with respect to antigen masking can be optimized in part based on this assay.
[0017] Similar assays can be done using Rh positive RBC and anti-D antiserum. In this assay, the agglutination will be scored as described in the AABB technical manual. The treated RBC will be compared to an untreated control sample to assess ability of the process to mask the D antigen.
[0018] Similarly, a non-immunogenic red cell composition prepared using compounds of the present invention can be assayed using an A B/D Monoclonal Grouping Card™ kit (Micro Typing Systems, Pompano Beach, FL). The desired red cell sample at a hematocrit of approximately 40 % is diluted to approximately 4 % with MTS Diluent 2 Plus (typically, 50 μl of red cells are diluted with 0.5 mL of diluent). A 10-12.5 μl aliquot of the red cell sample is added to an Anti-A/B/D microtube. Typically, six microtubes are prepared as a Gel Card and centrifuged using the MTS centrifuge. The Gel Card is then observed and scored for agglutination. Agglutination is graded as 0, 1+, 2+, 3+, or 4+. This range has 0 indicating no reaction with the red cells, all cells pelleting at the bottom of the microtube and 4+ indicating complete agglutination with a layer of cells at the top of the gel). There may be cases where a mixed field results, i.e. there are some cells at both the top and bottom of the gel.
EXAMPLE 9 Assessment in vitro of RBC function after processing of RBC.
[0089] The intracellular adenosine-5'-triphosphate (ATP), intracellular 2,3- diphosphoglyceric acid (2,3-DPG), extracellular potassium, intracellular and extracellular pH and hemolysis levels are readily assessed following processing of the RBC with compounds of the present invention. The results are compared to untreated control samples to assess whether the treated RBC are suitable for their intended use, such as transfusion. Intracellular ATP and 2,3-DPG are measured using a Sigma ATP Kit or 2,3-DPG kit respectively (Sigma, St. Louis, Mo.). The ATP kit was used following Sigma procedure No. 366-UV hereby incorporated by reference. Extracellular potassium levels can be measured using a Ciba Corning 614 K+ Na+ Analyzer (Ciba Corning Diagnostics Corp., Medfield, Ma.). The extracellular pH can be measured by centrifuging the cells at 4 °C for 15 minutes at 12,000 x g and removing the supernatant. The supernatant pH is measured on a standard pH meter at room temperature (e.g. Beckman, Epoxy Calomel electrode). For the intracellular pH, the remaining pellet in a centrifuge tube is capped and stored at approximately -80 oC for at least 2 hours, then lysed by adding deionized water. The lysed sample is well mixed and the pH of the solution is measured either at room temperature using a standard pH meter or at 37 °C using a Ciba-Corning model 238 blood gas analyzer.
EXAMPLE 10 Evaluation of the oxygen affinity of the processed RBC.
[0090] Following the processing of RBC with the compounds of the present invention, oxygen affinity of the RBC samples is measured with a Hemox analyzer. The Hemox analyzer is pre-equilibrated at 37 °C. Fifty μL of the RBC sample is mixed with 3.97 mL Hemox buffer solution (TCS Scientific Corp., New Hope, PA), containing 20 μL of 20% Bovine Serum Albumin (TCS Scientific Corp.) and 10 μL anti-foaming reagent (TCS Scientific Corp.) before transferring into the Hemox Analyzer cuvette. After the diluted sample is drawn into the cuvette, the temperature of the mixture is equilibrated with stirring for 8 minutes at 37 °C. Subsequently, the
diluted sample is fully oxygenated by exposure to air for 8 minutes. The instrument is calibrated for the partial pressure reading and the degree of hemoglobin saturation for each sample. The log ratio of the solution absorption at 560 to the absorption at 570 nm is recorded on the Y-axis while the partial pressure of oxygen (pθ2) obtained from a Clark electrode is recorded on the X-axis. The X-axis is calibrated by assigning values of 0 and the maximum calculated pθ2 for the day to readings obtained from 100% nitrogen and 100% air. The Y-axis is calibrated by assigning values of 0 and 1 to readings obtained from hemoglobin equilibrated under nitrogen or oxygen, respectively. For each sample an oxygen affinity curve is obtained by lowering the pθ2 through the introduction of nitrogen to the space above the liquid sample and measuring the percent of oxygen saturation of hemoglobin. The numerical data is converted to a graph of the oxygen affinity curve through the use of the computer program Kaleidagraph 3.0.5 (Synergy Software, Reading, PA) and the P50 is determined from the half point of the curve. Measurements can be made on treated samples and compared to measurements of untreated control samples.
EXAMPLE 11 Evaluation of the osmotic fragility of the processed RBC.
[0091] The osmotic fragility of samples is measured for RBC processed with compounds of the present invention and compared to untreated control samples. Reagent is prepared at 0.1, 0.2, 0.3, 0.35, 0.4, 0.45, 0.5, 0.55, 0.6, 0.65, 0.75, and 0.9 % PBS (1.0% PBS is 9g NaCl, 1.365g Na2HP04, and 0.186g NaH2P04 to a final volume of 1 liter in water). A 10 μL aliquot of RBC sample is added to 1.0 mL of each of these solutions, mixed gently and incubated at room temperature for 30 minutes. After incubation, the sample is mixed gently and centrifuged for 2 minutes at 2,000 x g. A spectrophotometer is zeroed with water and the absorption of the supernatant of the sample is measured at 540 nm. The % lysis is calculated using the following formula, in which the 0.9% PBS sample is considered background lysis and the 0.1% PBS sample is considered to be 100% lysis. % lysis = (A540 - 0.9% A540) ÷ (0.1% A540% - 0.9% A540) x 100
[0092] The % lysis is plotted as a function of the %PBS and the plots are compared for treated RBC and untreated control RBC.
EXAMPLE 12 Flow Cytometry analysis of RBC to assess levels of PEG modification.
[0093] A unit of ABO-typed whole blood (Sacramento Blood Center, CA) is leukofiltered according to standard blood banking methods. The RBC are washed with a buffer comprising 150 mM CHES at a pH of 9.0 to eliminate plasma proteins and adjust the pH of the extracellular domain to the desired value for the reaction. A solution of a compound of the present invention is prepared in the CHES buffer and an aliquot of the RBC suspension is added to this solution resulting in a final concentration of the compound of 22 mM (for a 5 kDa compound) in the extracellular volume at a hematocrit of 40%. The solution is mixed by gentle vortexing and inversion and incubated for 1 hour at room temperature. Following this room temperature incubation, the solution is washed three times with blood bank saline (BBS, 154 mM NaCl, Baxter Healthcare) to remove any excess compound and any other reaction side products. Following this wash, Adsol™, comprising 154 mM NaCl, 2.0 mM adenine, 41.2 mM mannitol, and 111.0 mM dextrose (Baxter Healthcare, IL), is added to a final hematocrit of 40%. The resulting RBC suspension is stored at 4°C.
[0094] The modified cells are analyzed for their ability to bind fluorescent labeled antibody with a flow cytometry method using a FACScan™ (Becton, Dickinson and Co., NJ). An aliquot of cells is centrifuged and the supernatant removed. A 50 μL portion of RBC (approximately lxlO6 cells) is incubated at room temperature for 1 hour with 5μL of an appropriate stock antibody solution (i.e. antibody would bind non compound modified RBC, e.g. anti-A FITC conjugate BRIC-145, anti-B FITC conjugate BGRL1, or anti-D FJTC conjugate BRAD-3 depending on the blood type, International Blood Group Reference Laboratory, UK). The cells are subsequently washed with BBS to remove the excess of the antibody and are analyzed by flow cytometry for bound fluorescent antibodies. The level of bound fluorescent antibodies is compared to either non compound modified cells (positive control) or cells which are not incubated with FITC antibody (negative control). The relative degree of PEG modification is estimated based on the ratio of the population maximum fluorescence (test article - negative control) / (positive control - negative control). This is represented as A2/A1 in Figure 1. This calculation can be reported as a percent binding of antibody relative to a positive control.
EXAMPLE 13 Antigen masking of red cells reacted with mPEG-6AC, 5AV or 4AB-NHS and mPEG-6AC-PFP.
[0095] A unit of A+ whole blood (Sacramento Blood Center, CA) was leukofiltered according to standard blood banking methods. The RBC were centrifuged at 4 °C at 4100 x g for 6 minutes and the plasma was removed to give a red cell concentrate. The red cell concentrate was washed with an equal volume of reaction buffer, centrifuged as above, the supernatant removed and the wash repeated. The washed red cell concentrate was reacted with an activated mPEG by dissolving the mPEG in reaction buffer and adding it to the red cell concentrate to a hematocrit of approximately 40% and the desired mPEG concentration in the extracellular volume. The reaction buffer was either 150 mM CHES pH 9 with 100 mM dextrose and 5 mM L-carnitine (CHES-GC) or 150 mM CHES pH 9 with 50 mM NaCl (CHES-Na). The reaction mixture was incubated for 2 hours at room temperature and an equal volume of wash buffer was added, the samples centrifuged as above, the supernatant removed and the wash repeated. The wash buffer was either 150 mM Na2HP04 pH 7, 50 mM NaCl (PBS), or 150 mM Na2HP04 pH 7, 100 mM dextrose, 5 mM L- carnitine (PB-GC). The final red cell solution is stored in Erythrosol and assayed for anti-A antibody binding as per Example 12. The results are found in Table 2. All compounds showed effective masking of the red cell antigens. Table 2 Modification of red cells with various activated mPEGs as measured by anti-type antibody binding.
EXAMPLE 14
Antigen masking of red cells reacted with 4-Arm-PEG-6AC-NHS.
[0096] A unit of A+ whole blood was prepared as red cell concentrate per Example 13. The red cell concentrate was washed with an equal volume of CHES GC pH 9.0 (150 mM CHES, 100 mM glucose, 5 mM L-carnitine), incubated for 5 minutes at room temperature, then centrifuged at 4100 x g for 6 minutes at room temperature and the supernatant removed. This wash was repeated, resulting in a washed red cell concentrate of approximately 80% hematocrit. An aliquot of this was put into separate tubes labeled control and 1-4 (see Table 3). Samples of PEG were weighed into additional tubes similarly labeled (see Table 3). A 0.5 mL of CHES GC was added to each PEG sample. These were sonicated and vortexed to dissolve the PEGs and then added to the appropriate tube containing 0.5 mL of washed red cell concentrate according to Table 3. The samples were mixed with gentle vortexing and incubated static for 1 hour at room temperature. Following incubation, 0.5 mL of PBGC pH 7.0 (150 mM phosphate, 100 mM glucose, 5 mM L-carnitine) was added to each sample with mixing. After 5 minutes at room temperature, the samples were centrifuged at 8,000 x g for 2 minutes at room temperature and the supernatant discarded. This wash was repeated and a 50-100 mL aliquot of the concentrate was removed for FACScan analysis. For all samples, the inhibition of anti-A or anti-D antibody binding was assessed by FACScan per Example 12, and the agglutination assay with gel cards per Example 8. For the gel card assay, the 5 mM mPEG-6AC- NHS control and the 2.5 mM 4-Arm-PEG-6AC-NHS samples showed no agglutination. The 1.25 mM 4-Arm-PEG-6AC-NHS sample showed no agglutination with anti-A and a mixed field for anti-D. The 0.5 mM 4-Arm-PEG- 6AC-NHS showed complete agglutination for both anti-A and anti-D. The FACScan results are indicated in Table 3. The results indicate some efficacy for the 4-Arm-PEG-6AC-NHS, but this was not as effective as the single arm mPEG-6AC- NHS.
Table 3 Modification of red cells with 4-Arm-PEG-6AC-NHS as measured by anti-type antibody binding.
* concentration of PEG based on extracellular volume.
EXAMPLE 15 Reaction of a full RBC unit with 20 kDa mPEG-6AC-NHS.
[0097] A unit of A+ whole blood was leukofiltered according to standard blood banking methods. The RBC, contained in a blood bag, were centrifuged at 4 °C for 6 minutes at 4100 x g and the plasma was removed. The red cell concentrate (approximately 80% hematocrit) was washed with approximately an equal volume of 150 mM CHES pH 9, 100 mM dextrose, 5 mM L-carnitine (approximately 200 mL red cell concentrate and 200 mL buffer) and centrifuged as above, and the supernatant was removed. The wash procedure was repeated. A 10.7 g sample of 20 kDa mPEG-6AC-NHS was dissolved in 67 mL of 150 mM CHES pH 9, 100 mM dextrose, 5 mM L-carnitine and added to the approximately 200 mL washed red cell concentrate to give approximately 5 mM mPEG-6AC-NHS in the extracellular volume at a hematocrit of approximately 60%. The reaction mixture was mixed by grasping each end of the blood bag and using a figure 8 motion approximately 30 times and incubated at room temperature for approximately 1 hour. Following incubation 200 mL of 150 mM Na2HP0 pH 7, 100 mM dextrose, 5 mM L-carnitine was added to the approximately 267 mL RBC sample. This was mixed using the figure 8 technique and centrifuged at 4 °C for 6 minutes at 4100 x g. The supernatant was removed and the red cell concentrate was washed again with 200 mL of the phosphate buffer. The wash buffer was removed and the final red cell concentrate was suspended in Erythrosol (Erythrosol is added as 94 mL part A and 20 mL part B(8% dextrose)), and stored at 4 °C. The amount of anti-A antibody binding was assessed as per Example 12, agglutination tested as per Example 8 initially and after
21 and 42 days storage at 4 °C. The anti-A antibody binding was 0% at all points and the gel cards showed no reaction as well. The hemolysis, ATP levels, potassium levels, glutathione levels (GSH), and both intracellular and extracellular pH, were measured initially and after storage at 4 °C for 2, 7, 14, 21, 28, 35, and 42 days. These were compared to control samples, either red cells stored by standard methods (4 °C control prepared directly with Erythrosol) or red cells that are processed as above without the mPEG-6AC-NHS (wash control) and red cells that are processed with 20 kDa mPEG-OH. The in vitro function results are found in Tables 4A-H. The results show that a full unit can be modified to adequately mask antigens and provide suitable values for hemolysis, potassium, glutathione, ATP, and both intracellular and extracellular pH.
[0098] Table 4A Day 0 in vitro measurements of red cell function for a full unit modified with 20 kDa mPEG-6AC-NHS.
Table 4B Day 2 in vitro measurements of red cell function for a full unit modified with 20 kDa mPEG-6AC-NHS.
Table 4C Day 7 in vitro measurements of red cell function for a full unit modified with 20 kDa mPEG-6AC-NHS.
Table 4D Day 14 in vitro measurements of red cell function for a full unit modified with 20 kDa mPEG-6AC-NHS.
Table 4E Day 21 in vitro measurements of red cell function for a full unit modified with 20 kDa mPEG-6AC-NHS.
- data not measured.
Table 4F Day 28 in vitro measurements of red cell function for a full unit modified with 20 kDa mPEG-6AC-NHS.
- data not measured.
Table 4G Day 35 in vitro measurements of red cell function for a full unit modified with 20 kDa mPEG-6AC-NHS.
Table 4H Day 42 in vitro measurements of red cell function for a full unit modified with 20 kDa mPEG-6AC-NHS.
EXAMPLE 16 The effect of hematocrit on the extent of PEG modification of RBC.
[0099] A unit of A+ whole blood is leukofiltered according to standard blood banking methods. A 20 mL sample is centrifuged at 4 °C at 3800 rpm (4100 x g) for 6 minutes and the plasma is removed. The red cell concentrate (RCC) is washed with an equal volume of 150 mM CHES pH 9, 100 mM dextrose, 5 mM L-carnitine and centrifuged as above, the supernatant is removed, and the wash repeated. Samples of 5 kDa mPEG-6AC-NHS, or 5 kDa mPEGOH as a non reacting control, were prepared in the 150 mM CHES pH 9, 100 mM dextrose, 5 mM L-carnitine and added to 0.5 mL of the washed red cell concentrate. The amounts of the mPEG samples and volumes added were adjusted to give the approximate values indicated in Table 1. The samples were gently mixed and incubated at room temperature for approximately 1 hour. Following incubation, an equal volume of 150 mM Na2HP0 pH 7, 100 mM dextrose, 5 mM L-carnitine was added to each sample, the sample was gently mixed and centrifuged at 4 °C for 6 minutes at 4100 x g. The supernatant was removed and the red cell concentrate was washed again with an equal volume of the pH 7 buffer. After centrifuging the final wash, the final red cell pellet was suspended in an approximately equal volume of Erythrosol and stored at 4 °C. Each sample was assayed using the FACScan assay described in Example 12, using an anti-A FITC conjugate. The level of hemolysis was also measured for each sample. This experiment was done with two different red cell units. The results are given in Table 5. The results indicate that for a given extracellular concentration of mPEG-
6AC-NHS, the extent of modification of the red cells is the same from 40% to greater than 70% hematocrit. Similarly, for a given mass of mPEG-6AC-NHS, a higher extent of modification can be achieved with a higher hematocrit. The amount of activated mPEG used per red cell can be greatly reduced to give the same level of antigen masking by going to higher hematocrit.
Table 5. Extent of mPEG modification of red cells as a function of reaction hematocrit.
EXAMPLE 17 The effect of hematocrit on the extent of PEG modification of RBC with 5 kDa or 20 kD MPEG-6AC-NHS.
[00100] An experiment was done similarly to Example 16, the difference being that instead of using the same amount of RCC per reaction, the total reaction volume was constant at 1 mL. It was observed with a 40% hematocrit reaction that the hemolysis decreases going from a 0.5 mL to 1.0 mL reaction volume, with further decrease as the volume was increased up to 5 mL. This may have contributed to the high hemolysis levels of the higher hematocrit samples in Example 16. In addition, a 20 kDa mPEG-6AC-NHS was also used, and the controls were done without addition of any mPEG. Table 6 indicates the amounts and volumes of materials used in this example, with the anti-A antibody binding and hemolysis results. Contrary to Example 16, the hemolysis results were improved with higher hematocrit, possibly due to the sample volume increase. While the 20 kDa sample did not show quite as
much masking of anti-A antibody binding at the highest hematocrit, the experiment confirms the observation that the reaction is more efficient at higher hematocrit. Table 6. Extent of mPEG modification of red cells as a function of reaction hematocrit using equal reaction volumes, 5 kDa vs. 20 kDa mPEG-
6AC-NHS.
EXAMPLE 18 The effect of hematocrit up to 85% on the extent of PEG modification of RBCs with
20 kDa MPEG-6AC-NHS.
[00101] A unit of B+ whole blood was leukofiltered according to standard blood banking methods. A 120 mL sample was centrifuged at 4 °C at 4100 x g for 6 minutes at room temperature and the plasma was removed. The red cell concentrate (RCC) was washed with an equal volume of 150 mM CHES pH 9, 100 mM dextrose, 5 mM L-carnitine (CHES GC), incubated for 5 minutes at room temperature, and centrifuged as above, the supernatant was removed, and the wash repeated. The hematocrit of this was measured to be 79%. A second RCC stock was prepared by taking a 20 mL aliquot of the first RCC. This was then centrifuged in multiple eppendorf tubes (2 mL per each of ten tubes) and centrifuged 14,000 x g for 5
minutes, and the supernatant removed. This resulted in a hematocrit of 90%. Samples of 20 kDa mPEG-6AC-NHS were prepared in the CHES GC and added to 0.5 ml of the appropriate red cell concentrate according to Table 7. The CHES GC was added to the mPEG-6AC-NHS and sonicated with mixing to dissolve completely (approximately 1 minute). The dissolved samples were then added to the appropriate volume of RCC (Table 7A), mixed with vortexing, and incubated for 1 hour at room temperature (mPEG-6AC-NHS at 5 mM in extracellular volume). Following incubation, samples were centrifuged for 6 minutes at room temperature at 4100 x g. The supernatant was removed and the red cell concentrate was washed with 150 mM Na2HP04 pH 7, 100 mM dextrose, 5 mM L-carnatine buffer (PBGC), using a volume equal to the PEG reaction volume (Table 7A). This was centrifuged and the wash repeated. After centrifuging the final wash, the final red cell pellet was suspended in Erythrosol to a hematocrit of approximately 40% and stored at 4 °C. Each sample was assayed using the FACScan assay described in Example 12, using an anti-B FITC conjugate. The percent of anti-B antibody binding is indicated in Table 7A. The gel card analysis showed no agglutination with any of the modified red cell samples. This study was repeated with an A+ unit of blood, with the details and results in Table 7B. These examples demonstrate reasonable reaction efficiency up to a hematocrit of 85%.
Table 7A Reaction conditions and results for reaction of red cells with 5 mM 20 kDa mPEG-6AC-NHS at various hematocrit.
Table 7B Reaction conditions and results for reaction of red cells with 5 mM 20 kDa mPEG-6AC-NHS at various hematocrit.























































