WO2002088101A2 - Inhibitors of bace - Google Patents
Inhibitors of bace Download PDFInfo
- Publication number
- WO2002088101A2 WO2002088101A2 PCT/US2002/013741 US0213741W WO02088101A2 WO 2002088101 A2 WO2002088101 A2 WO 2002088101A2 US 0213741 W US0213741 W US 0213741W WO 02088101 A2 WO02088101 A2 WO 02088101A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- trifluoromethyl
- ppm
- phenyl
- carboxylic acid
- phenoxymethyl
- Prior art date
Links
- HQGBLJKANVZPHV-UHFFFAOYSA-N C1CC2=CC2C1 Chemical compound C1CC2=CC2C1 HQGBLJKANVZPHV-UHFFFAOYSA-N 0.000 description 1
- 0 CCC(C)(CC*1C)C1=O Chemical compound CCC(C)(CC*1C)C1=O 0.000 description 1
- GLXOHWLGZMRLRM-UHFFFAOYSA-N CN(CCC1)CCC1O Chemical compound CN(CCC1)CCC1O GLXOHWLGZMRLRM-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/75—Amino or imino radicals, acylated by carboxylic or carbonic acids, or by sulfur or nitrogen analogues thereof, e.g. carbamates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/68—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member
- C07D211/70—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/68—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member
- C07D211/72—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D211/74—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/80—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members
- C07D211/82—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/22—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the nitrogen-containing ring
- C07D217/26—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/48—Two nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/04—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D243/00—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms
- C07D243/06—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms having the nitrogen atoms in positions 1 and 4
- C07D243/08—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms having the nitrogen atoms in positions 1 and 4 not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/12—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly or doubly bound nitrogen atoms
- C07D295/135—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly or doubly bound nitrogen atoms with the ring nitrogen atoms and the substituent nitrogen atoms separated by carbocyclic rings or by carbon chains interrupted by carbocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/10—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Definitions
- the present invention relates to inhibitors of aspartic proteinases, particularly, BACE.
- the present invention also relates to compositions thereof and methods therewith for inhibiting BACE activity in a mammal, and for treating Alzheimer's Disease and other BACE-mediated diseases.
- BACE-1 Aspartic proteinases are found in a variety of pathways in different eukaryotic organisms, including mammals, viral, fungal and parasitic organisms.
- BACE-1 (hereinafter “BACE”), as discussed below, has been implicated in the pathogenesis of Alzheimer's
- BACE-2 an aspartic proteinase with high homology to BACE, is a glycosylated transmembrane protein with potentially similar disease implications as BACE.
- Renin a well-known aspartic proteinase, is part of a critical signaling pathway that creates balance in blood pressure. See, e.g., Tamura K. et al . , "Recent Advances in the Study of Renin and Angiotensinogen genes : from molecules to the whole body," Hypertens . Res . , 18(1) pp. 7-18 (1995) . Renin has been implicated in hypertension and other cardiovascular conditions. Napsin-A and Napsin-B are closely related aspartic proteinases.
- Napsin-A is expressed in lung and kidney tissue and has been implicated in lung adenocarcinoma .
- Chuman, Y. et al . "Napsin A, a member of the aspartic protease family, is abundantly expressed in normal lung and kidney tissue and also expressed in lung adenocarcinomas, " FEBS Lett . , 462(1-2): pp. 129-34 (1999).
- Cathepsin-D a lysosomal aspartic proteinase, is expressed in all tissues and is implicated in protein catabolism, antigen processing, degenerative diseases and breast cancer progression. See, e.g., Erickson, J. W. , et al . , "Structure of human Cathepsin D: comparison of inhibitor binding and subdomain displacement with other aspartic proteinases, " Adv. Exp . Med. Biol . , 362, pp. 181-192 (1995).
- Cathepsin-E a non-lysosomal aspartic proteinase
- Cathepsin-E may play a role in proteolytic degradation of antigen, which is a major regulatory step in the activation of a T- lymphocyte response.
- Bennet, K. et al. "Antigen processing for presentation by Class II major histocompatibility complex requires cleavage by cathepsin E, " Eur . J. Immunol . , 22 (6) , pp 1519-24 (1992) .
- Pepsinogen-A and Pepsinogen-C both aspartic proteinase secreted in the stomach, are involved in the digestion of proteins in the stomach. Richter, C. et al .
- AD Alzheimer's AD experience progressive loss of memory and other cognitive functions that compromise their ability to work, interact socially, and care for themselves. These impairments are associated with widespread damage to several classes of neurons and different neurotransmitter systems in the brain. The symptoms and pathology of AD are progressive. People with AD eventually become dependent on others for all aspects of their care.
- AD Alzheimer's Disease
- Drugs that augment cholinergic neurotransmission by inhibiting the enzyme acetylcholinesterase have been approved for use in humans. These drugs have been shown to improve cognitive function modestly but not to slow underlying disease progression.
- the pathological hallmarks of AD are senile plaques (SPs) and neurofibrillary tangles (NFTs) .
- SSPs senile plaques
- NFTs neurofibrillary tangles
- Senile plaques comprise extracellular aggregates of A ⁇ protein, dystrophic neurites, activated microglia, and reactive astrocytes.
- a ⁇ is 40-42-residue endoproteolytic fragment of the amyloid precursor protein ("APP”) .
- APP amyloid precursor protein
- AD has not been established, but a growing body of data indicates that A ⁇ plays a central role in disease pathogenesis .
- a ⁇ is produced in vivo following proteolytic cleavage of the membrane-anchored APP at the ⁇ site by ⁇ - secretase, followed by cleavage at the ⁇ site by ⁇ - secretase.
- the ⁇ site lies on the lumenal side of the membrane.
- the ⁇ site lies in the transmembrane domain and is more variable, ⁇ Cleavage at residue 711 yields A ⁇ - 40 .
- ⁇ Cleavage at residue 713 yields A ⁇ ! _ 42 • Cleavage at the ⁇ site is the rate-limiting step in production of A ⁇ in vivo . Tang et al .
- BACE ⁇ secretase
- Asp 2 ⁇ amyloid converting enzyme
- memapsin 2 this enzyme is an aspartic proteinase.
- BACE is expressed as a 501 amino acid pro-polypeptide containing an N-terminal signal sequence and pro region that is cleaved post-translationally.
- BACE also contains a C- terminal trans-membrane domain and exists in cells as a membrane-bound protein.
- peptidyl inhibitors of BACE are not readily suitable for therapy because, typically, they do not cross the blood-brain barrier. Thus, there is a need for peptidyl inhibitors of BACE that readily cross the blood- brain barrier. There are no reported non-peptidyl inhibitors of BACE.
- non- peptidyl BACE inhibitors and compositions thereof there is a need for non- peptidyl BACE inhibitors and compositions thereof.
- inhibitors of other aspartic proteinases and methods for designing such inhibitors of aspartic proteinases There is also a need for compounds and compositions useful in treating BACE-mediated diseases.
- methods for treating diseases such as Alzheimer's Disease and related neurological disorders.
- HB-1 is a first hydrogen-bonding moiety capable of forming up to four hydrogen bonds with the carboxylate oxygen atoms of Asp-228 and Asp-32 of BACE.
- HPB-2 is a second hydrophobic moiety capable of associating with substantially all residues in the Flap binding pocket of BACE;
- HPB-3 is a third hydrophobic moiety capable of associating with substantially all residues in the P2' binding pocket of BACE;
- HPB-4 is a fourth hydrophobic moiety capable of inducing favorable interactions with the phenyl ring of at least two of Tyr-71, Phe-108 and Trp-76 of BACE.
- V is a 3-4 membered acyclic group or a 5-7 membered, fully or partially saturated cyclic group; wherein V comprises a first moiety selected from NH, CH-OH, or a CH-NH 2 , and a second moiety selected from carbon, CH, or N; wherein said first moiety and said second moiety in V are non-adjacent; and
- V is attached to R through said second moiety; wherein V is optionally substituted with R 10 ; R is a suitable linker; p is 0 or 1; R 10 is P1-R1-P2-R2- ;
- T is a five to eleven membered monocyclic or bicyclic, aromatic or non-aromatic ring having zero to three heteroatoms independently selected from O, S, N or NH, wherein T has at least one R 10 substituent and up to three more substituents selected from R 10 or J;
- J is halogen, -R' , -OR', -N0 2 , -CN, -CF 3 , -OCF 3 , oxo, 1,2-methylenedioxy, -N(R') 2 , -SR' , -S(0)R', -S(0)N(R') 2/ -S0 2 R', -C(0)R', -C0 2 R' , -C(0)N(R') 2 , -N(R )C(0)R / , -N(R' ) C(O) OR' , -
- R' is independently selected from hydrogen, aliphatic, heterocyclyl , heterocycly-alkyl, aryl, aralkyl, heteroaryl, or heteroaralkyl ; wherein R' is optionally substituted with up to
- R 11 is hydrogen, (C-i-Cg) -alkyl, (C 2 -C 6 ) -alkenyl or alkynyl, or (C 3 ⁇ C 6 ) cycloalkyl;
- PI and P2 each are independently: - absent ; or
- RI and R2 each are independently:
- W is five to eleven membered monocyclic or bicyclic, aromatic or non-aromatic ring having zero to three heteroatoms independently selected from 0, S, N, or NH, wherein W has up to 3 J substituents.
- compositions comprising inhibitors of BACE.
- Figure 1 depicts the interaction between binding sites/subsites of BACE and four features of the inhibitors of the present invention, namely: first hydrogen bonding moiety ("HB-1"), second hydrophobic moiety (“HPB-2”), third hydrophobic moiety (“HPB-3”) and a fourth hydrophobic moiety (“HPB-4").
- HB-1 first hydrogen bonding moiety
- HPB-2 second hydrophobic moiety
- HPB-3 third hydrophobic moiety
- HPB-4" fourth hydrophobic moiety
- Figure 2 depicts the interaction between binding sites/subsites of BACE and five features of the inhibitors of the present invention, namely: HB-1, first hydrophobic moiety ("HPB-1"), HPB-2, HPB-3 and HPB-4.
- Figure 3 depicts the interaction between binding sites/subsites of BACE and six features of the inhibitors of the present invention, namely: HB-1, HPB-1, HPB-2, HPB-3, HPB-4 and a second hydrogen-bonding moiety (“HB- 2”) .
- Figure 4 depicts the interaction between binding sites/subsites of BACE and six features of the inhibitors of the present invention, namely: HB-1, HPB-1, HPB-2, HPB-3, HPB-4 and a third hydrogen bonding moiety ("HB- 3") .
- Figure 5 depicts the interaction between binding sites/subsites of BACE and seven features of the inhibitors of the present invention, namely: HB-1, HB-2, HB-3, HPB-1, HPB-2, HPB-3 and HPB-4.
- P2 binding pocket refers to the substrate binding site on the BACE molecule defined by at least Thr-231, Thr-232, Asn-233, Arg-235 and Ser-325.
- P2' binding pocket refers to the substrate binding site on the BACE molecule defined by at least Asn-37, Ala-39, Val-69, Trp-76, Ile-118 and Arg- 128.
- flap binding pocket refers to the pocket defined by at least Trp-76, Phe-108, Phel09, Trp-115 and Ile-102.
- the flap In the absence of an inhibitor, the flap can be in the closed conformation. However, in the presence of an inhibitor, the flap shifts into a more open conformation to make room for the part of the inhibitor that interacts with the above residues in the flap binding pocket.
- hydrophobic refers to a non-polar moiety that tends not to dissolve in water and is fat-soluble.
- Hydrophobic moieties include, but are not limited to, hydrocarbons, such as alkanes, alkenes, alkynes, cycloalkanes, ethers, cycloalkenes, cycloalkynes and aromatic compounds, such as aryls, certain saturated and unsaturated heterocycles and moieties that are substantially similar to the side chains of hydrophobic natural and unnatural ⁇ -amino acids, including valine, leucine, isoleucine, methionine, phenylanine, ⁇ -amino isobutyric acid, alloisoleucine, tyrosine, and tryptophan.
- association refers to a condition of proximity between an inhibitor or portions thereof to the BACE molecule or portions thereof wherein the juxtaposition is energetically favored by electrostatic or van der Waals interactions.
- hydrogen bond refers to a favorable interaction that occurs whenever a suitable donor atom, X, bearing a proton, H, and a suitable acceptor atom, Y, have a separation of ⁇ 3.5A and where the angle X-H - - - Y is greater than 90 degrees.
- a single proton on a donor atom X may form a plurality of suitable acceptor atoms, Y.
- the proton on a -NH- group may form a separate hydrogen bond with each of the two oxygen atoms in a carboxylate anion.
- Suitable donor and acceptor atoms are well understood in medicinal chemistry (G.C. Pimentel and A.L. McClellan, The Hydrogen Bond, Freeman, San Francisco, 1960; R. Taylor and
- hydrogen bonding moiety refers to a chemical structure containing one or more suitable hydrogen bond donor moieties or hydrogen bond acceptor moieties.
- hydrophilicity donor moiety refers to a chemical structure containing a suitable hydrogen bond donor atom bearing one or more protons .
- donor atoms having one proton are -OH, -SH and -NH- .
- donor atoms having more than one proton are -NH 2 , [-NH 3 ] + and [-NH 2 -] + .
- hydrogen bonding acceptor moiety refers to a chemical structure containing a suitable hydrogen bond acceptor atoms .
- acceptor atoms include fluorine, oxygen, sulfur and nitrogen.
- stacking interaction refers to the favorable attractive interactions between two aromatic ring systems, wherein the two rings are juxtaposed such that they are oriented either parallel, perpendicular or at an intermediate angle to each other.
- salt bridge refers to the non-covalent attractive interaction between a positively charged moiety (P) and a negatively charged moiety (N) when the distance between the centers of mass of P and N is between 2 and 6 Angstroms .
- atoms which may contain a formal charge and atoms immediately adjacent to these are included.
- a salt bridge may be formed between the positively charged guanidinium side chain of an arginine residue and the negatively charged carboxylate side chain of a glutamate residue. Salt bridges are well known in medicinal chemistry (L. Stryer, Biochemistry, Freeman, San Francisco, (1975) ; K.A. Dill, "Dominant Forces in Protein Folding", Biochemistry, 29, No. 31, pp. 7133- 7155, (1990) ) .
- center of mass refers to a point in three-dimensional space that represents a weighted average position of the masses that make up an object.
- backbone chain and “backbone” refer to the portion of a polypeptide which comprises the repeating unit -CO-CH-NH- .
- minimized geometry refers to the systematic altering of the atomic geometry of a molecule or molecular complex so that any further minor perturbation of the atomic geometry would cause the total energy of the system as measured by a molecular mechanics force-field to increase. Minimization and molecular mechanics force-fields are well understood in computational chemistry [U. Burkert and N.L. Allinger, Molecular Mechanics, ACS Monograph 177, American Chemical Society, Washington, D.C. 1982 pages 59-78] .
- strain energy is used in this application to refer to the difference between the free conformation energy of a compound and the bound conformation energy of that compound when bound to BACE.
- the strain energy can be determined by the following steps: Determine the bound conformational energy, determine and then subtract from this the un-bound conformational energy. This is the free conformation energy.
- a more comprehensive definition of strain energy can be found in Bostrom, J. , Norrby, P.-O.; Liljefors, T., "Conformational Energy Penalties of Protein-Bound Ligands", J. Comput . Aided Mol . Design, 1998, 383.
- the strain energy for binding of a potential inhibitor to BACE is the difference between the free conformation energy and the bound conformation energy.
- the strain energy of an inhibitor of the present invention is less than about 10 kcal/mol.
- an optionally substituted group may have a substituent at each substitutable atom of the group (including more than one substituent on a single atom) , and the identity of each substituent is independent of the others.
- aliphatic or "aliphatic group” as used herein means :
- bicyclic C 8 ⁇ C 12 hydrocarbon that is completely saturated or that contains one or more units of unsaturation, but which is not aromatic (also referred to herein as "carbocycle” ) , that has a single point of attachment to the rest of the molecule wherein any individual ring in said bicyclic ring system has three to seven members.
- suitable aliphatic groups include, but are not limited to, linear or branched or alkyl, alkenyl, alkynyl groups, carbocyclic groups and hybrids thereof, such as (cycloalkyl) alkyl, (cycloalkenyl) alkyl or (cycloalkyl) alkenyl .
- / up to 2 carbons may be independently replaced by 0, S, N, or NH.
- alkyl used alone or as part of a larger moiety include both straight and branched chains, wherein up to 2 carbons may be independently replaced by 0, S, N, or NH. Unless prefixed with a specific chain length, alkyl, alkenyl and alkynyl contain one to twelve carbon atoms and at least two carbon atoms and one double bond in the case of alkenyl and at least two carbon atoms and one triple bond, in the case of alkynyl.
- halo and halogen used alone or as part of a larger moiety means F, Cl, Br, or I.
- heteroatom includes oxygen and any oxidized form of nitrogen and sulfur, and the quaternized form of any basic nitrogen.
- aryl used alone or as part of a larger moiety as in “aralkyl”, “aralkoxy”, or “aryloxyalkyl” , refers to monocyclic, bicyclic and tricyclic ring systems having a total of five to fourteen ring members, wherein at least one ring in the system is aromatic and wherein each ring in the system contains three to seven ring members.
- aryl may be used interchangeably with the term “aryl ring”.
- heterocycle means non-aromatic, monocyclic, bicyclic or tricyclic ring systems having five to fourteen ring members in which one or more ring members is a heteroatom, wherein each ring in the system contains three to seven ring members .
- heteroaryl used alone or as part of a larger moiety as in “heteroaralkyl” or “heteroarylalkoxy” , refers to monocyclic, bicyclic and tricyclic ring systems having a total of five to fourteen ring members, wherein at least one ring in the system is aromatic, at least one ring in the system contains one or more heteroatoms, and wherein each ring in the system contains three to seven ring members.
- heteroaryl may be used interchangeably with the term “heteroaryl ring” or the term “heteroaromatic” .
- the BACE inhibitors of this invention may contain one or more "asymmetric" carbon atoms and thus may occur as racemates and racemic mixtures, single enantiomers, diastereomeric mixtures and individual diastereomers . All such isomeric forms of these compounds are expressly included in the present invention.
- Each stereogenic carbon may be of the R or S configuration.
- specific compounds and scaffolds exemplified in this application may be depicted in a particular stereochemical configuration, compounds and scaffolds having either the opposite stereochemistry at any given chiral center or mixtures thereof are also envisioned. Combinations of substituents and variables envisioned by this invention are only those that result in the formation of stable compounds.
- chemically stable arrangement refers to a compound structure that possesses stability sufficient to allow manufacture and administration to a mammal by methods known in the art. Typically, such compounds are stable at a temperature of 40°C or less, in the absence of moisture or other chemically reactive conditions, for at least a week.
- abbreviations are used herein to represent the features present within the BACE inhibitors of the present invention: HB-1 - a first hydrogen bonding moiety capable of forming up to four hydrogen bonds with the carboxylate oxygen atoms of Asp-228 and Asp-32 of BACE.
- HB-2 - a second hydrogen-bonding moiety capable of forming a hydrogen bond with the carbonyl oxygen atom of Gly-34 of BACE.
- HB-3 - a third hydrogen-bonding moiety capable of forming a hydrogen bond with the carbonyl oxygen of Gly- 230 of BACE.
- HPB-1 - a first hydrophobic moiety capable of associating with substantially all residues in the P2 binding pocket of BACE .
- HPB-2 - a second hydrophobic moiety capable of associating with substantially all residues in the Flap binding pocket of BACE .
- HPB-3 - a third hydrophobic moiety capable of associating with substantially all residues in the P2 ' binding pocket of BACE .
- HPB-4 - a fourth hydrophobic moiety capable of inducing favorable interactions with the phenyl ring of at least two of Tyr-71, Phe-108 and Trp-76 of BACE.
- the present invention provides inhibitors of BACE having the following features: (a) HB-1; (b) HPB-4; and at least one of the following (c) and (d) :
- the inhibitor contains features (a) , (b) and (c) .
- the inhibitor contains features (a) , (b) and (d) .
- the present invention provides a BACE inhibitor having the following features :
- the present invention provides a BACE inhibitor having the following features:
- the inhibitor contains features (a) , (b) , (c) , and (d) .
- the inhibitor contains features (a) , (b) , (c) and (e) .
- the BACE inhibitor of the present invention further comprises a HB-2 feature. This embodiment is illustrated in Fig. 3.
- the BACE inhibitor of the present invention further comprises a HB-3 feature. This embodiment is illustrated in Fig. 4.
- the BACE inhibitor of the present invention comprises both, HB-2 and HB-3 features. This embodiment is illustrated in Fig. 5.
- each of the HB-1, HB-2 and HB-3 is independently less than about 3.5 A in length.
- each of HB-1, HB-2 and HB-3 is independently less about 3.0 A.
- HB-1 of the BACE inhibitor of the present invention is replaced with a electropositive moiety comprising one or more positively charged atoms, wherein said electropositive moiety forms a salt bridge with the carboxylate oxygen atoms of Asp- 228 and Asp-32.
- the HPB-1 moiety is capable of associating with the P2 binding pocket of BACE such that the distance between the center of mass of the HPB-1 moiety and the C- ⁇ atom of substantially all of Thr-231, Thr-232, Asn-233, Arg-235 and Gln-73 is between about 4.0 A to about 12 A. More preferably, the HPB-1 moiety is capable of associating with the P2 binding pocket of BACE such that the distance between the center of mass of the hydrophobic moiety and the C- ⁇ atom of substantially all of Thr-231, Thr-232, Asn-233, Arg-235 and Gln-73 is between about 5.0 A to about 10 A.
- the HPB-1 moiety is capable of associating with the P2 binding pocket of BACE such that the distance between the center of mass of HPB-1 and the C- ⁇ atom of substantially all of Thr-231, Thr-232, Asn- 233, Arg-235 and Gln-73 is as follows:
- the HPB-2 moiety is capable of associating with the Flap binding pocket such that the distance between the center of mass of the HPB-2 moiety and the C- ⁇ atom of substantially all of Trp-76, Phe-108, Phe-109, Trp-115 and Ile-102 is between about 3.0 A to about 8.5 A.
- the distance between the center of mass of the HPB-2 moiety and the C- ⁇ atom of substantially all of Trp-76, Phe-108, Phe-109, Trp-115 and Ile-102 is between about 3.5 A to about 8.0 A.
- the distance between the center of mass of the HPB-2 moiety and the C- ⁇ atom of substantially all of Trp-76, Phe-108, Phe-109, Trp-115 and Ile-102 is as follows: Trp-76 - about 8 A; Phe-108 - about 3.5 A; Phe-109 - about 6 A; Trp-115 - about 8 A; and Ile-102 - about 6 A.
- the HPB-3 moiety binds to the P2 ' pocket such that the distance between the center of mass of the HPB-3 moiety and the C- ⁇ atom of substantially all of Asn-37, Ala-39, Val-69, Trp-76, Ile-118 and Arg-128 is between 3.5 A to 8 A.
- the distance between the center of mass of the HPB-3 moiety and the C- ⁇ atom of substantially all of Asn-37, Ala-39, Val-69, Trp-76, Ile- 118 and Arg-128 is between 4 A to 7.5 A.
- the distance between the center of mass of the HPB-3 moiety and the C- ⁇ atom of substantially all of Asn-37, Ala-39, Val-69, Trp-76, Ile- 118 and Arg-128 is as follows:
- HPB-4 is an aromatic stacking moiety that interacts favorably with the phenyl ring of at least two of Tyr-71, Phe-108 and Trp-76.
- the HPB-4 moiety interacts with at least two of Tyr-71, Phe-108 and Trp-76 such that the distance between the center of mass of the HPB-4 moiety and the C- ⁇ atom of at least two of Tyr-71, Phe-108 and Trp-76 is between 5.5 A and 8.5 A.
- the HPB-4 moiety interacts with at least two of Tyr-71, Phe-108 and Trp-76 such that the distance between the center of mass of the HPB-4 moiety and the C- ⁇ atom of at least two of Tyr-71, Phe-108 and Trp-76 is between 6.0 A and 8.0 A.
- the HPB-4 moiety interacts with at least two of Tyr-71, Phe-108 and Trp-76 such that the distance between the center of mass of the HPB-4 moiety and the C- ⁇ atom of at least two each of Tyr-71, Phe-108 and Trp-76 is as follows: Tyr-71 - about 6.0 A;
- the HPB-4 moiety interacts with Tyr-71 and Phe-108.
- HPB-4 moiety interacts with Try-71.
- the distance between the HB-1 moiety and other moieties in the inhibitor, when present, is in the range as set forth below in Table 1:
- the BACE inhibitor is characterized by a neutral or favorable enthalpic contribution from the sum of all electrostatic interactions between the inhibitor and BACE when the inhibitor is bound thereto.
- the BACE inhibitor is characterized by an ability to cross the blood-brain barrier.
- an ability to cross the blood-brain barrier One of skill in the art will be well aware of methods for determining whether an inhibitor has such ability. See, e.g., Murcko et al . , "Designing Libraries with CNS activity," J. Med . Chem . , 42(24), pp. 4942-51 (1999).
- the present invention provides an enzyme-inhibitor complex, wherein said enzyme is BACE and said inhibitor is as described above.
- the present invention provides a method of inhibiting BACE activity in a mammal, comprising the step of administering to said mammal a BACE inhibitor selected from any one of the above embodiments .
- each of the above aspartic proteases has a corresponding hydrogen bonding interactions (HB-1, HB-2 and HB-3), a P2 binding pocket, a P2' binding pocket, a flap-binding pocket and amino acid resides corresponding to Tyr-71, Phe-108 and Trp-76 that have favorable interactions with HPB-4 in BACE.
- Trp-78 of BACE and Trp-40 of Cathepsin-D occupy structurally equivalent positions although their main chains are far apart.
- Table 2 illustrates the substantial similarity in the enzyme-inhibitor interactions between BACE and Cathepsin-D.
- the hydrogen bonding residues and the hydrophobic residues present in the BACE binding sites are substantially present in the analogous residues in the corresponding Cathepsin-D binding sites.
- the moieties present in the BACE inhibitors of the present invention, and the interactions that they engender, are also present in Cathepsin-D inhibitors. Consequently, one of skill in the art will readily recognize that the binding features that render the inhibitors of the present invention effective against BACE also render them effective against Cathepsin-D. Therefore, the inhibitors of BACE, described above are also useful as inhibitors of other aspartic proteinases in general, and those listed above, in particular.
- the present invention provides inhibitors of aspartic proteinases.
- the present invention provides inhibitors of BACE-2, Renin, Napsin-A, Napsin-B, Cathepsin-D, Cathepsin-E, Pepsinogen- A and Pepsinogen-C.
- the present invention provides inhibitors of aspartic proteinases other than renin.
- the present invention provides enzyme-inhibitor complexes, wherein said enzyme is an aspartic proteinase and said inhibitor is as described above.
- said aspartic proteinase in said enzyme- inhibitor complex is BACE-2, BACE, Renin, Napsin-A, Napsin-B, Cathepsin-D, Cathepsin-E, Pepsinogen-A or Pepsinogen-C.
- said aspartic proteinase in said enzyme-inhibitor complex is other than renin.
- the present invention provides methods for designing a specific compound as an inhibitor of aspartic proteinases. Such a method is described below for BACE. But, one of skill in the art will readily appreciate that because aspartic proteinases share substantially similar inhibitor-enzyme binding interactions, the methods described below may readily, without undue experimentation, be extended to other aspartic proteinases.
- GRID (Goodford, P.J. A Computational Procedure for Determining Energetically Favorable Binding Sites on Biologically Important Macromolecules . J. Med. Chem. , 28, pp. 849-857 (1985)). GRID is available from Oxford University, Oxford, UK.
- MCSS is available from Molecular Simulations, Inc., San Diego, CA, a division of Pharmacopiea, Princeton, NJ.
- AUTODOCK (Goodsell, D.S.; Olsen, A.J. Automated Docking of Substrates to Proteins by Simulated Annealing. PROTEINS: Structure, Function and Genetics, 8, pp. 195- 202 (1990)). AUTODOCK is available from the Scripps Research Institute, La Jolla, CA.
- DOCK (Kuntz, I.D.; Blaney, J.M. ; Oatley, S.J.; Langridge, R. ; Ferrin, T.E. A Geometric Approach to Macromolecule-Ligand Interactions. J. Mol . Biol. , 161, pp. 269-288 (1982)). DOCK is available from the University of California, San Francisco, CA.
- suitable binding moieties Once suitable binding moieties have been selected, they can be assembled into a single inhibitor. This assembly may be accomplished by connecting the various moieties to a central scaffold through suitable linkers. The assembly process may, for example, be done by visual inspection followed by manual model building, again using software such as QUANTA or SYBYL. A number of other programs may also be used to help select ways to connect the various moieties. These include:
- CAVEAT Bartlett, P. . ; Shea, G.T. ; Telfer, S.J.; Waterman, S. CAVEAT: A Program to Facilitate the Structure-Derived Design of Biologically Active
- 3D Database systems such as MACCS-3D (MDL Information Systems, San Leandro, CA) . This area has been recently reviewed by Martin (Martin, Y.C. 3D Database Searching in Drug Design. J. Med . Che . , 35, pp. 2145-2154 (1992)).
- the inhibitors of this invention may be constructed "de novo" using either an empty active site or optionally including some portions of a known inhibitor (Walters, W. P., M. T. Stahl, et al . (1998). “Virtual Screening - An Overview.” Drug Disovery Today 3: 160-178) .
- Such methods are well known in the art. They include, for example: 1.
- LUDI (Bohm, H.J. The Computer Program LUDI : A New Method for the De Novo Design of Enzyme Inhibitors. J. Comp. Aid. Molec. Design., 6, 61-78 (1992)). LUDI is available from Biosym Technologies, Princeton, NJ.
- LEGEND (Nishibata, Y. , Itai, A., Tetrahedron, 47, 8985 (1991)). LEGEND is available from Molecular Simulations, Princeton, NJ.
- LeapFrog available from Tripos associates, St. Louis, MO
- the skilled artisan can advantageously avoid time consuming and expensive experimentation to determine enzymatic inhibition activity of particular compounds.
- the method also is useful to facilitate rational design of BACE inhibitors and therapeutic and prophylactic agents against BACE mediated diseases. Accordingly, the present invention envisions such inhibitors and uses.
- a variety of conventional techniques may be used to carry out each of the above evaluations as well as the evaluations necessary in screening a candidate compound for BACE inhibiting activity.
- these techniques involve determining the location and binding proximity of a given moiety, the occupied space of a bound inhibitor, the deformation energy of binding of a given compound and electrostatic interaction energies.
- Examples of conventional techniques useful in the above evaluations include: quantum mechanics, molecular mechanics, molecular dynamics, Monte Carlo sampling, systematic searches and distance geometry methods (G.R. Marshall, Ann. Rev. Pharmacol. Toxicol., 27, p. 193 (1987) ) . Specific computer software has been developed for use in carrying out these methods .
- Examples of programs designed for such uses include: Gaussian 92, revision E.2 (M.J. Frisch, Gaussian, Inc., Pittsburgh, PA ⁇ 1993); AMBER, version 4.0 (P.A. Kollman, University of California at San Francisco, ⁇ 1993) ; QUANTA/CHARMM and Insight II/Discover [Molecular Simulations, Inc., San Diego, CA, a division of Pharmacopiea, Inc., Princeton, NJ ⁇ 1992] . These programs may be implemented, for instance, using a Silicon Graphics Octane workstation or IBM RISC/6000 workstation model 550. Other hardware systems and software packages will be known and of evident applicability to those skilled in the art.
- BACE inhibitors of this invention may also use different scaffolds or core structures, but all of these cores will allow the necessary moieties to be placed in the active site such that the specific interactions necessary for binding may be obtained.
- These compounds are best defined in terms of their ability to match the pharmacophore, i.e., their structural identity relative to the shape and properties of the active site of BACE. Distances between the different moieties of the pharmacophore may be readily determined using any modeling software and other suitable chemical structure software.
- specialized, commercially available pharmacophore modeling software enables one to determine pharmacophore models from a variety of structural information and data.
- This software may also be used to search a database of three- dimensional structures in order to identify compounds that meet the above specific pharmacophore requirements.
- Examples of this software include: 1. DISCO (Martin, Y.C., Bures, M.G., Danaher, E.A., DeLazzer, J. , Lico, A., Pavlik, P.A., J. Comput . Aided Mol . Design, 1993, 7, 83). DISCO is available from Tripos Associates, St. Louis, MO.
- APEX-3D which is part of the Insight molecular modeling program, distributed by Molecular Simulations, Inc., San Diego, CA.
- CATALYST is distributed by Molecular Simulations, Inc., San Diego, CA.
- a method known in the art utilizes scaffolds from known drugs in the market. These "drug-like" scaffolds may provide the requisite cores useful in tailoring the requisite moieties to match the pharmacophore such that their interactions with the active site of BACE is optimal. See, e.g., WO 98/57155, and Fesjo, J. , et al . , "The SHAPES Strategy: an NMR-based approach for lead generation in drug discovery," Chemistry & Biology, 6: 755-769 (1999) .
- the BACE inhibitor of the present invention has the following formula (I) :
- X N-, -N(R)-, -NH-, -NH 2 or -CHOH; wherein R is H, (Cx-Cg) alkyl, (C 2 -C 6 ) alkenyl or alkynyl ;
- A-X-B moiety is optionally fused with a non-aromatic or aromatic carbocyclic or heterocyclic ring; and wherein the A-X-B moiety contains up to 3 substituents having the formula -(L) n ⁇ M, wherein: n is 0 or 1;
- L is a suitable linker, optionally containing a hydrogen bonding moiety
- M is independently selected from HB-1, HB-2, HPB-1, HPB-2, HPB-3 or HPB-4. According to a preferred embodiment, M is an aromatic stacking moiety such as a carbocyclic aromatic or heterocyclic aromatic moiety.
- suitable linker R when present, has the formula:
- T 1 and T 2 are independently selected from C ⁇ -C 6 alkyl, C 2 -C 6 alkenyl or alkynyl, wherein any carbon in T 1 and T 2 may be replaced by a heteroatom group in a chemically stable arrangement selected from -0-, -S-, -NH-, -NR'-, -C(0)-, -S(0)- and -S(0) 2 -;
- R' is H or aliphatic
- L 1 is -CH(OH)-, -CH(OR)-, -CH(NRR)-, -CO-, -0-, -NR'-, -SO-, -S02-, -NR'S02-, -CONR'-, -NR'-CO-, -0-C0-, -CO-0-, -O-CO-NR'-, -NR'-CO-O-, or -NR' -CO-NR' - .
- suitable linker R is -CH 2 -, -0-, -S-, -SO-, -S0 2 -, -NR'-, -C(0)0-, -OC(O)-, -C(0)NR'-, - NR'-C(O)-, -0-C(0)-0-, -O-C(O) -NR 1 -, -NR ' -C (O) -NR' - , - NR'-C(0)-0-, -SO-NR', -NR'-SO-, -NR'-S0 2 -, -S0 2 -NR'-, - CHOR'-, -CHNR'-, or -C(O)-.
- Preferred embodiments of formula (I) include the following:
- M is an aromatic carbocyclic or aromatic heterocyclic moiety; and the ring attached to TI is optionally substituted with up to 2 substituents.
- T 1 is C ⁇ -C 6 alkyl (i.e., m is 1) ;
- L 1 is 0, NH or S;
- T 2 is absent (i.e., m is zero);
- M is a phenyl ring optionally substituted with up to 4 substituents selected from (C1-C6) alkyl, (C2-C6) alkenyl, -OMe or halogen.
- substituents selected from (C1-C6) alkyl, (C2-C6) alkenyl, -OMe or halogen.
- T 1 is (C1-C6) alkyl (i.e., m is 1); more preferably T 1 is methyl;
- R is (C1-C6) alkyl; L 1 is CHOH; T 2 is (C1-C6) alkyl (i.e., m is 1); more preferably T 2 is methyl; and
- M is a phenyl ring optionally substituted with up to 4 substituents selected from (C1-C6) alkyl, (C2-C6) alkenyl, -OMe or halogen.
- substituents selected from (C1-C6) alkyl, (C2-C6) alkenyl, -OMe or halogen.
- the present invention provides a method of inhibiting BACE activity in a mammal, comprising the step of administering to said mammal a compound of formula IA:
- V is a 3-4 membered acyclic group or a 5-7 membered, fully or partially saturated cyclic group; wherein V comprises a first moiety selected from NH, CH-OH, or a CH-NH 2 , and a second moiety selected from carbon, CH, or N; wherein said first moiety and said second moiety in V are non-adjacent; and
- V is attached to R through said second moiety; wherein V is optionally substituted with R 10 ; R is a suitable linker; p is 0 or 1; R 10 is P1-R1-P2-R2-W;
- T is a five to eleven membered monocyclic or bicyclic, aromatic or non-aromatic ring having zero to three heteroatoms independently selected from O, S, N or NH, wherein T has at least one R 10 substituent and up to three more substituents selected from R 10 or J;
- J is halogen, -R' , -OR', -N0 2 , -CN, -CF 3 , -OCF 3 , oxo, 1,2-methylenedioxy, -N(R') 2 , -SR' , -S(0)R', -S(0)N(R') 2 , -S0 2 R' , -C(0)R', -C0 2 R' , -C(0)N(R') 2 , -N(R')C(0)R' # -N (R' ) C (O) OR' , -
- R' is independently selected from hydrogen, aliphatic, heterocyclyl, heterocycly-alkyl, aryl, aralkyl, heteroaryl, or heteroaralkyl ; wherein R' is optionally substituted with up to
- R 11 is hydrogen, (C ⁇ -C 6 ) -alkyl, (C 2 -C 6 ) -alkenyl or alkynyl, or (C 3 -C 6 ) cycloalkyl;
- PI and P2 each are independently: - absent ; or
- RI and R2 each are independently:
- W is five to eleven membered monocyclic or bicyclic, aromatic or non-aromatic ring having zero to three heteroatoms independently selected from O, S, N, or NH, wherein W has up to 3 J substituents.
- p is 0. According to another embodiment of the present invention, p is 1. According to one embodiment, suitable linker R, when present, has the formula:
- T 1 and T 2 are independently selected from C ⁇ - 6 alkyl
- C 2 _ 6 alkenyl or alkynyl wherein any carbon in T 1 and T 2 may be replaced by a heteroatom group in a chemically stable arrangement selected from -0-, -S-, -NH-, -NR'-, -C(0)-, -S(O)- and -S(0) 2 - ;
- R' is independently selected from hydrogen, aliphatic, cycloalkyl, cycloalkyl-alkyl, heterocyclyl, aryl, aralkyl, heteroaryl, or heteroaralkyl ; wherein R' is optionally substituted with up to 3 substituents selected independently from -R 11 ,
- R 11 is hydrogen, (C ⁇ -C 6 ) -alkyl, (C 2 -C 6 ) -alkenyl or alkynyl, or (C 3 -C 6 ) cycloalkyl; and L 1 is selected from -CH(OR')-, -CH(NR'R')-,
- R is -CH 2 -, -0-, -S-, -SO-, -S0 2 ⁇ , -NR'-, -C(0)0-, -OC(O)-, -C(0)NR'-, -NR'C(O)-, -O- ,
- R 10 is P1-R1-P2-R2-W, wherein one of PI and P2 is absent and the other of PI and P2 is aliphatic, and/or one of RI and R2 is absent and the other of RI and R2 is R.
- W is a five to seven membered monocyclic, aromatic or non-aromatic ring having zero to three heteroatoms independently selected from O, S, N, or NH, wherein W has up to 3 substituents independently selected from J.
- W is a five to six membered monocyclic, aromatic ring having one to three heteroatoms independently selected from 0, S, N, or NH, wherein W has up to 3 substituents independently selected from J.
- Preferred five or six membered aromatic rings having one to three heteroatoms include 2-furanyl, 3-furanyl, 3-furazanyl, N-imidazolyl , 2-imidazolyl, 4-imidazolyl, 5-imidazolyl, 3-isoxazolyl, 4-isoxazolyl, 5-isoxazolyl, 2-oxadiazolyl , 5-oxadiazolyl, 2-oxazolyl, 4-oxazolyl, 5-oxazolyl, 1-pyrrolyl, 2- pyrrolyl, 3-pyrrolyl, 1-pyrazolyl, 2-pyrazolyl, 3- pyrazolyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 2-pyrimidyl, 4-pyrimid
- W is a five to six membered monocyclic, non-aromatic ring having one to three heteroatoms independently selected from 0, S, N, or NH, wherein W has up to 3 substituents independently selected from J.
- Preferred five or six membered non-aromatic rings having one to three heteroatoms include 2-tetrahydrofuranyl, 3- tetrahydrofuranyl , 2-tetrahydropyranyl, 3- tetrahydropyranyl , 4-tetrahydropyranyl, [1, 3] -dioxalanyl, [1, 3] -dithiolanyl, [1, 3] -dioxanyl, 2- tetrahydrothiophenyl , 3-tetrahydrothiophenyl, 2- morpholinyl, 3-morpholinyl, 4-morpholinyl, 2- thiomorpholinyl , 3-thiomorpholinyl, 4-thiomorpholinyl, 1- pyrrolidin
- W is a five to seven membered monocyclic, aromatic or non- aromatic ring having zero heteroatoms independently selected from 0, S, N, or NH, wherein W has up to 3 substituents independently selected from J. More preferably, W is cyclopentyl, cyclohexyl, or phenyl, wherein W has up to 3 substituents independently selected from J. Most preferably, W is phenyl, wherein W has up to 3 substituents independently selected from J.
- W is an eight to eleven membered bicyclic ring, wherein either or both rings may be aromatic or non-aromatic, and either or both rings may have zero to three heteroatoms independently selected from 0, S, N, or NH, wherein W has up to 3 substituents independently selected from J.
- Preferred aromatic or non-aromatic bicyclic rings having one to three heteroatoms include naphthyl, decalinyl, tetrahydro- naphthyl, 3-lH-benzimidazol-2-one, (1-substituted) -2-oxo- benzimidazol-3-yl, 1-phthalimidinyl, benzoxanyl, benzopyrrolidinyl , benzopiperidinyl, benzoxolanyl, benzothiolanyl, benzothianyl, indolinyl, chromanyl, phenanthridinyl , tetrahydroquinolinyl, carbazolyl, benzimidazolyl, benzothienyl, benzofuranyl , indolyl , quinolinyl, benzotriazolyl, benzothiazolyl , benzooxazolyl, benzimidazolyl, isoquinolinyl, indo
- R 10 is independently selected from substituents present in compounds in any of Table 1 through Table 5, infra .
- V in compounds of formula IA is a 3-4 membered acyclic group, wherein V comprises a first moiety selected from NH, CH-OH, or a CH-NH 2 , and a second moiety selected from carbon, CH, or N; wherein said first moiety and said second moiety in V are non-adjacent; and
- V in compounds of formula IA is 5-7 membered cyclic group, wherein V comprises a first moiety selected from NH, CH-OH, or a CH-NH 2 , and a second moiety selected from carbon, CH, or N; wherein said first moiety and said second moiety in V are non- djacent; and
- V is attached to R through said second moiety; wherein V is optionally substituted with R 10 .
- V in compounds of formula IA is a 5 membered cyclic group, wherein V comprises a first moiety selected from NH, CH-OH, or a CH-NH 2 , and a second moiety selected from carbon, CH, or N; wherein said first moiety and said second moiety in V are non-adjacent; and
- V is attached to R through said second moiety; wherein V is optionally substituted with R 10 .
- V in compounds of formula IA is selected from IA-1 through IA-9 shown below:
- V in compounds of formula IA is a 6-7 membered cyclic group, wherein V comprises a first moiety selected from NH, CH-OH, or a CH-NH 2 , and a second moiety selected from carbon, CH, or N; wherein said first moiety and said second moiety in V are non-adjacent; and
- V is attached to R through said second moiety; wherein V is optionally substituted with R 10 .
- V in compounds of formula IA is selected from formula IB-1 to formula IB-6 shown below:
- V in compounds of formula IA is selected from IB-1 or IB-5. Most preferably, V is IB-5
- the present invention provides a method of inhibiting BACE activity in a mammal, comprising the step of administering to said mammal a compound of formula IAB:
- V is selected from IA1 , IBl , IB2 , IB4 , IB5 , or IB6 ;
- T is a five to eleven membered monocyclic or bicyclic , aromatic or non-aromatic ring having zero to three heteroatoms independently selected from O, S , N or NH, wherein T has at least one R 10 substituent and up to three more substituents selected from R 10 or J;
- T and V share a ring atom
- J is halogen, -R' , -OR', -N0 2 , -CN, -CF 3 , -OCF 3 , oxo, 1,2-methylenedioxy, -N(R') 2 , -SR' , -S(0)R',
- R' is independently selected from hydrogen, aliphatic, heterocyclyl, heterocycly-alkyl, aryl, aralkyl, heteroaryl, or heteroaralkyl ; wherein R' is optionally substituted with up to 3 substituents selected independently from -R 11 , -OR 11 , -N0 2 , -CN, -CF 3 , -OCF 3 , oxo, 1,2- methylenedioxy, -N(R 1X ) 2 , -SR 11 , -S(0)R 1
- R 11 is hydrogen, (C ! -C 6 ) -alkyl, (C 2 -C 6 ) -alkenyl or alkynyl, or (C 3 -C 6 ) cycloalkyl ;
- R 10 is P1-R1-P2-R2-W; PI and P2 each are independently:
- RI and R2 each are independently:
- R is a suitable linker
- W is five to eleven membered monocyclic or bicyclic, aromatic or non-aromatic ring having zero to three heteroatoms independently selected from O, S, N, or NH, wherein W has up to 3 J substituents.
- the compound of formula IA is selected from formula ICa or formula ICb:
- R is a suitable linker; p is zero or one;
- R 12 is absent or R' 10
- R ,1 i 0 U is P1-R1-P2-R2-W;
- T is a five to eleven membered monocyclic or bicyclic, aromatic or non-aromatic ring having zero to three heteroatoms independently selected from O, S, N or NH, wherein T has at least one R 10 substituent and up to three more substituents selected from R 10 or J;
- J is halogen, -R' , -OR', -N0 2 , -CN, -CF 3 , -OCF 3 , oxo, 1,2-methylenedioxy, -N(R') 2 , -SR' , -SOR' , -S0 2 R' , -C(0)R', -C(0)0R' or -C(0)N(R' ) 2 , wherein R' is independently selected from hydrogen, aliphatic, heterocyclyl, heterocyclyl-alkyl, aryl, aralkyl, heteroaryl, or heteroaralkyl ;
- PI and P2 each are independently:
- RI and R2 each are independently:
- W is five to eleven membered monocyclic or bicyclic, aromatic or non-aromatic ring having zero to three heteroatoms independently selected from O, S, N, or NH, wherein W has up to 3 substituents independently selected from J.
- V is ICa-1.
- Naphthalene-1-carboxylic acid (4-piperazin-l-yl- 3' -trifluoromethyl-biphenyl-3 -yl) -amide
- Naphthalene-1-carboxylic acid (4-piperazin-l-yl*
- the present invention provides a method of inhibiting BACE activity in a mammal, comprising the step of administering to said mammal a compound of formula ID:
- A is a five or six membered aryl ring having zero to two heteroatoms independently selected from nitrogen, oxygen or sulfur, wherein:
- A has at least one R 10 substituent and up to three more substituents selected from R 10 or J; k is 0 or 1; n is 0-2;
- J is halogen, -R' , -OR', -N0 2 , -CN, -CF 3/ -0CF 3/ oxo, 1,2-methylenedioxy, -N(R') 2 , -SR' , -S(0)R', -S(0)N(R') 2 , -S0 2 R' , -C(0)R', -C0 2 R' ,
- R' is independently selected from hydrogen, aliphatic, heterocyclyl , heterocycly-alkyl, aryl, aralkyl, heteroaryl, or heteroaralkyl ; wherein R' is optionally substituted with up to 3 substituents selected independently from -R 11 , -OR 11 , -N0 2 , -CN, -CF 3 , -OCF 3 , oxo, 1,2- methylenedioxy, -N(R ⁇ : ⁇ ) 2 , -SR 11 , -S(0)R 11 , - S(0)N(R 1:L )2, -SO2R 11 , -C(0)R 11 , -CO 2 R 11 ,
- R 11 is hydrogen, (C ⁇ -C 6 ) -alkyl, (C 2 -C 6 ) -alkenyl or alkynyl, or (C 3 -C 6 ) cycloalkyl ;
- R 10 is P1-R1-P2-R2-W; PI and P2 each are independently: - absent; or
- RI and R2 each are independently:
- R is a suitable linker
- W is a five to eleven membered monocyclic or bicyclic, aromatic or non-aromatic ring having zero to three heteroatoms independently selected from 0, S, N, or NH, wherein W has up to 3 substituents independently selected from J.
- R 10 and R in compounds of formula ID are as recited above for R 10 and R in compounds of formula IA.
- the present invention provides a method of inhibiting BACE activity in a mammal, comprising the step of administering to said mammal a compound of formula IE: H
- Wi is -NH-, -CH 2 -NH-, -C(0)-NH-, or -C(0)-0-;
- W 2 is P1-R1-P2-R2-W;
- PI and P2 each are independently:
- RI and R2 each are independently:
- W is five to eleven membered monocyclic or bicyclic, aromatic or non-aromatic ring having zero to three heteroatoms independently selected from O, S, N, or NH, wherein W has up to 3 substituents independently selected from J;
- R is -CH 2 -, -O-, -S-, -SO-, -S0 2 -, -NR'-, -C(0)0-, -OC(O)-, -C(0)NR'-, -NR'C(O)-, -O- , -0C(0)NR'-, -NR'C(0)NR'-, -NR'C(0)0-, -SO-NR', -NR'SO-, -NR'S0 2 -, -S0 2 NR'-, -CHOR'-, -CHNR'-, or -C(O)-;
- J is halogen, -R' , -OR', -N0 2 , -CN, -CF 3/ -OCF 3 , oxo, 1,2-methylenedioxy, -N(R') 2 , -SR", -S(0)R', -S(0)N(R') 2 , -S0 2 R', -C(0)R', -C0 2 R' or -C(0)N(R') 2 , wherein R' is independently selected from hydrogen, aliphatic, heterocyclyl, heterocycly- alkyl, aryl, aralkyl, heteroaryl, or heteroaralkyl ; wherein R' is optionally substituted with up to 3 substituents selected independently from -R 11 , -OR 11 , -N0 2 , -CN, -CF 3 , -OCF 3 , oxo, 1,2- methylenedioxy, -N(R 1:L ) 2 , -S
- T is a five to eleven membered monocyclic or bicyclic, aromatic or non-aromatic ring having zero to three heteroatoms independently selected from 0, N or NH, wherein T has at least one R 10 substituent and up to three more substituents selected from R 10 or J;
- i in compounds of formula IE is -NH-, -CH 2 -NH- or -C (O) -NH- .
- R, p, and T in compounds of formula IE are as recited for R, P, and T in compounds of formula IA.
- p is 0 and T is selected from phenyl or naphthyl, wherein T has at least one R 10 substituent and up to three more substituents selected from R 10 or J.
- T has three R 10 substituents. More preferably, T has two R 10 substituents.
- Vi is selected from:
- Vi is optionally substituted with R 10 ;
- W 3 is hydrogen or
- W6 is selected from -0-, -S-, or -NH- ; j is 0 to 3; W 4 is hydrogen or a 5-11 membered monocyclic or bicyclic aromatic ring having 0-3 heteroatoms independently selected from O, S, N, or NH, wherein W 4 has up to 3 J substituents;
- W 5 is hydrogen or R 10 ; provided that at least two or W 3 , W 4 , and W 5 are simultaneously non-hydrogen;
- R 10 is P1-R1-P2-R2-W
- J is halogen, -R' , -OR', -N0 2 , -CN, -CF 3 , -OCF 3 , oxo, 1,2-methylenedioxy, -N(R') 2 / -SR' , -S(0)R',
- R' is independently selected from hydrogen, aliphatic, heterocyclyl, heterocycly-alkyl, aryl, aralkyl, heteroaryl, or heteroaralkyl ; wherein R' is optionally substituted with up to 3 substituents selected independently from -R 11 , -OR 11 , -N0 2 , -CN, -CF 3 , -OCF 3 , oxo, 1,2- methylenedioxy, -N(R 13 -) 2 , -SR 11 , -S(0)R
- R 11 is hydrogen, (C ⁇ -C 6 ) -alkyl, (C 2 -C 6 ) -alkenyl or alkynyl, or (C 3 -C 6 ) cycloalkyl ;
- PI and P2 each are independently:
- RI and R2 each are independently: - absent; or
- R is a suitable linker; and W is five to eleven membered monocyclic or bicyclic, aromatic or non-aromatic ring having zero to three heteroatoms independently selected from 0, S, N, or NH, wherein W has up to 3 J substituents.
- j is selected from 1, 2 or 3.
- W 3 is 2- trifluoromethyl-phenoxymethyl .
- V x is unsubstituted 3 , 4-didehydropiperidyl .
- V ⁇ is unsubstituted piperazyl .
- W or W 4 is independently phenyl or a five to seven membered monocyclic, aromatic ring having 1-3 heteroatoms independently selected from O, S, N, or NH, wherein W or W 4 has up to 3 substituents independently selected from J.
- W or W 4 is selected from 2-furanyl, 3-furanyl, 3-furazanyl, N- imidazolyl, 2-imidazolyl, 4-imidazolyl, 5-imidazolyl , 3- isoxazolyl, 4-isoxazolyl, 5-isoxazolyl, 2-oxadiazolyl, 5- oxadiazolyl, 2-oxazolyl, 4-oxazolyl, 5-oxazolyl, 1- pyrrolyl, 2-pyrrolyl, 3-pyrrolyl, 1-pyrazolyl, 2- pyrazolyl, 3-pyrazolyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 2-pyrimidyl, 4-pyrimidyl, 5-pyrimidyl, 3-pyridazinyl, 2- thiazolyl, 4-thiazolyl, 5-thiazolyl, 5-tetrazolyl, 2- triazolyl, 5-triazolyl, 2-thien
- W or W 4 is an eight to eleven membered bicyclic ring, wherein either or both rings is aromatic, and either or both rings has zero to three heteroatoms independently selected from 0, S, N, or NH, wherein W or W 4 has up to 3 substituents independently selected from J.
- W or W 4 is selected from naphthyl, 3-lH-benzimidazol-2-one, (1- substituted) -2-oxo-benzimidazol-3-yl, 1-phthalimidinyl, benzoxanyl, benzopyrrolidinyl, benzopiperidinyl, benzoxolanyl, benzothiolanyl, benzothianyl , indolinyl, chromanyl, phenanthridinyl, tetrahydroquinolinyl, carbazolyl, benzimidazolyl, benzothienyl, benzofuranyl, indolyl, quinolinyl, benzotriazolyl , benzothiazolyl , benzooxazolyl, benzimidazolyl, isoquinolinyl, indolyl, isoindolyl, acridinyl, benzoisoxazolyl, tetrahydr
- W 4 is phenyl or 5-hydroxyphenyl .
- W 5 is Pl-Rl-W or R1-P2-W.
- each of PI and P2 is independently (C1-C6) -alkyl, and RI is R.
- R is selected from -CH 2 -, -0-, -S-, -SO-, -S0 2 -, -NR'-, -C(0)0-, -0C(0)-, -C(0)NR'-, -NR'C(O)-, -0- , -OC(0)NR'-, - NR'C(0)0-, -NR'C(0)NR'-, -NR'C(0)0-, -SO-NR' , -NR'SO-, -NR'S0 2 -, -S0 2 NR'-, -CHOR'-, -CHNR'-, or -C(O)-.
- each of PI and P2 is methylene
- - RI is -0-, -NH-C(O)-, -C(0)-NH-, or -NH- ;
- J is independently selected from halogen, -R' , -OR', -N0 2 , -CN, -CF 3 , -0CF 3 , oxo, 1,2-methylenedioxy, -N(R') 2 , -SR' , -S(0)R', -S(0)N(R') 2 , -S0 2 R' , -C(0)R', -C0 2 R' or
- R' is independently selected from hydrogen or (C1-C6) -alkyl .
- a stable compound or chemically feasible compound is one in which the chemical structure is not substantially altered when kept at a temperature of 40°C or less, in the absence of moisture or other chemically reactive conditions, for at least a week.
- the BACE inhibitors of this invention may contain one or more "asymmetric" carbon atoms and thus may occur as racemates and racemic mixtures, single enantiomers, diastereomeric mixtures and individual diastereomers . All such isomeric forms of these compounds are expressly included in the present invention.
- Each stereogenic carbon may be of the R or S configuration.
- structures depicted herein are also meant to include compounds that differ only in the presence of one or more isotopically enriched atoms.
- compounds having the present structures except for the replacement of a hydrogen by a deuterium or tritium, or the replacement of a carbon by a 13 C- or 14 C-enriched carbon are within the scope of this invention.
- Reagents (a) Cs 2 C0 3 , N-BOC piperazine, DMF, 55°; (b) NiCl 2 , NaBH 4 , CH 2 C1 2 , CH 3 OH, 0°C; (c) ArC(0)Cl, (i- Pr) 2 N(Et), room temperature; (d) R 10 -B(OH) 2 , PdCl 2 (dppf), K 3 P0 4 , DME, 70°C; (e) TFA, CHC1 2 , room temperature.
- Scheme I above shows a general route for the preparation of compounds of formula IA.
- Substituent R 10 was then introduced using a boronic acid under palladium catalysis followed by trifluoroacetic acid mediated cleavage of the BOC protecting group to give compounds of formula IA.
- Reagents (a) R 10 -NH 2 , EDC, HOBt, Et 3 N, CH 2 C1 2 , room temperature; (b) 10% Pd/C, CH 3 OH, H 2 (l atm); (c) ArC(0)Cl, pyridine, CH 2 C1 2 , room temperature; (d) R -. l 1 u 0-OH, K 2 C0 3 , acetone, 50°C; (e) IN HCl in Et 2 0, CH 3 OH, 50°C.
- Scheme II above shows another general route for the preparation of compounds of formula IA.
- Commercially available acid 5a was converted to amide intermediate 6a.
- Hydrogenolysis of the Cbz protecting group followed by acylation provided intermediate 8a.
- Displacement of the benzyl chloride in 8a with R 10 phenol followed by ethereal HCl mediated removal of the BOC protecting group afforded compounds of formula IA.
- Reagents (a) NaOMe, EtOH, reflux; (b) BzlBr, CHC1 3 , CH 3 OH, 65 °C; (c) NaBH 4 , CH 3 OH, H 2 0; (d) BOC 2 0, CH 3 OH, EtOAc, 10%Pd/C H 2 (1 atm) ; (e) R 10 -Br, K 2 C0 3 , acetone, 55°C (f) IN HCl in Et 2 0, CH 3 OH, 50°C.
- Scheme III above shows another general route for the preparation of compounds of formula IA.
- Commercially available amidino pyridine 9a was cyclo-condensed with commercially available ethyl ester 10a to provide pyrimidine intermediate 11a.
- Alkylation and subsequent reduction provided 12a.
- Reduction and benzyl deprotection with in situ reprotection with BOC anhydride afforded intermediate 13a.
- Alkylation with a suitable R 10 benzyl halide followed by ethereal HCl mediated removal of the BOC protecting group afforded compounds of formula IA.
- Reagents (a) N-bromosuccinimide, benzoyl peroxide, CC1 4 , 100°C; (b) R 10 OH, K 2 C0 3 , acetone, 70°C; (c) PdCl 2 (dppf), K 3 P0 4 , DME, 70 °C; (d) TFA, CH 2 Cl 2/ room temperature.
- Scheme IV above shows another general route for the preparation of compounds of formula IA.
- Commercially available dibromoxylene 14a was converted to tetrabromide 15a and further displaced with R 10 phenols to give intermediate dibromide 16a.
- a Suzuki type coupling with cyclic boronates 17a and 18a yielded intermediate 19a.
- Boronate 17a was prepared according to the method reported in Tetrahedron Letters, 41(19), 3705-3708 (2000) .
- Final tri luoroacetic acid mediated cleavage of the BOC protecting group gave compounds of formula IA.
- Reagents (a) BOC 2 0, Et 3 N, CH 3 OH, room temperature; (b) 2N NaOH, EtOH, 50 °C; (c) R 10 -NH 2 , EDC, HOBt, Et 3 N, CH 2 C1 2 , room temperature (d) TFA, CH 2 C1 2 , room temperature.
- Scheme V above shows a general route for the preparation of compounds of formula IB.
- Commercially available azepine ester 20a was N-protected followed by ester hydrolysis to give intermediate acid 22a.
- Coupling with a suitable R 10 -amine followed by trifluoroacetic acid mediated deprotection provided compounds of formula IB.
- Reagents (a) BOC 2 0, THF, 0°C; (b) 4-fluoro-3-nitro-4 ' - R 10 -phenyl, CH 3 CN, K 2 C0 3 , reflux; (c) 10%Pd/C, EtOH, H2 (1 atm) (d) NaH, DMF, 0°C, R 10 -Br, then 50°C; (e) trifluoroacetic acid, CH 2 C1 2 , room temperature.
- Scheme VI above shows another general route for the preparation of compounds of formula IA.
- Commercially available diamine 24a was N-protected then used to displace a commercially available aryl flouride to give intermediate 26a.
- Palladium mediated nitro reduction gave intermediate 27a which was then alkylated with a suitable R 10 bromide to afford intermediate 28a.
- N-BOC deprotection with trifluoroacetic acid gave compounds of formula IA.
- Reagents (a) glyoxylic acid, H 2 0, room temperature, then 6M HCl, 80 °C, then K 2 C0 3 , 120 °C; (b) BOC 2 0, Et 3 N, DMF; (c) R 10 -Br, K 2 C0 3 , (n-Bu) 4 NI, CH 3 CN, reflux. (d) NaH, DMF,
- Scheme VII above shows a general route for the preparation of compounds of formula IB.
- Commercially available 5-hydroxytryptamine 29a was converted to intermediate carboline 30a. Further N-protection with Boc anhydride gives compound 31a. ⁇ therification with a suitable R 10 -bromide, followed by N alkylation with another R 10 -bromide and final N-Boc removal with trifluoroacetic acid gave compounds of formula lb.
- Reagents (a) BOC 2 0, DMF, ⁇ t 3 N room temperature; (b) R 10 - Br, NaH, (n-Bu) NI, DMF, 50°C. (c) trifluoroacetic acid, CH 2 C1 2 , room temperature .
- Scheme VIII above shows a general route for the preparation of compounds of formula IA.
- Commercially available pyrazole 33a was N-protected with Boc anhydride to provide intermediate 34a.
- Pyrazole alkylation followed by deprotection of the N-Boc group with trifluoroacetic acid provided compounds of formula IA.
- Reagents (a) CH 3 OH, H 2 S0 4 , reflux; (b) NBS, benzoyl peroxide, benzene, reflux; (c) R 10 -OH, K 2 C0 3 , acetone, 50°C; (d) Ar-Br, Pd(dppf)Cl2, K 2 C0 3 , KOt-Bu, DMF, 80°C; (e) 1M DIBAL-hexanes, THF, -78 °C (f) MsCl , CH 2 C1 2 , pyridine, Et 3 N, 0°C (g) R 10 -OH, K 2 C0 3 , acetone, 60°C (h) IN HCl in Et 2 0 CH 3 OH, 50°C.
- the present invention provides a composition for inhibit BACE activity in a mammal, comprising compounds of formula IA, formula IB, formula ICa, formula ICb, formula ID or formula IE or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier, adjuvant, or vehicle.
- the amount of compound in the compositions of this invention is such that it is effective to detectably inhibit an aspartic proteinase, particularly BACE in a biological sample or in a patient.
- the composition of this invention is formulated for administration to a patient in need of such composition.
- the composition of this invention is formulated for oral administration to a patient.
- the pharmaceutical composition of the present invention is comprised of a compound of formula IA, formula IB, formula ICa, formula ICb, formula ID, or formula IE, a pharmaceutically acceptable carrier, and a neurotrophic factor.
- neurotrophic factor refers to compounds which are capable of stimulating growth or proliferation of nervous tissue.
- Numerous neurotrophic factors have been identified in the art and any of those factors may be utilized in the compositions of this invention.
- These neurotrophic factors include, but are not limited to, nerve growth factor (NGF) , insulin-like growth factor (IGF-1) and its active ⁇ truncated derivatives such as gIGF-1 and Des (1-3) IGF-I, acidic and basic fibroblast growth factor (aFGF and bFGF, respectively) , platelet-derived growth factors (PDGF) , brain-derived neurotrophic factor (BDNF) , ciliary neurotrophic factors (CNTF) , glial cell line-derived neurotrophic factor (GDNF) , neurotrophin-3 (NT-3)and neurotrophin 4/5 (NT-4/5) .
- the most preferred neurotrophic factor in the compositions of this invention is NGF.
- patient means an animal, preferably a mammal, and most preferably a human.
- compositions of this invention refers to a non-toxic carrier, adjuvant, or vehicle that does not destroy the pharmacological activity of the compound with which it is formulated.
- Pharmaceutically acceptable carriers, adjuvants or vehicles that may be used in the compositions of this invention include, but are not limited to, ion exchangers, alumina, aluminum stearate, lecithin, serum proteins, such as human serum albumin, buffer substances such as phosphates, glycine, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, cellulose-based substances, polyethylene glycol, sodium carboxymethylcellulose, polyacrylates, waxes, polyethylene-polyoxypropylene-block
- detectably inhibit means a measurable change in BACE activity between a sample comprising said composition and a BACE proteinase and an equivalent sample comprising BACE proteinase in the absence of said composition.
- a “pharmaceutically acceptable salt” means any non- toxic salt, ester, salt of an ester or other derivative of a compound of this invention that, upon administration to a recipient, is capable of providing, either directly or indirectly, a compound of this invention or an inhibitorily active metabolite or residue thereof.
- Pharmaceutically acceptable salts of the compounds of this invention include those derived from pharmaceutically acceptable inorganic and organic acids and bases.
- suitable acid salts include acetate, adipate, alginate, aspartate, benzoate, benzenesulfonate, bisulfate, butyrate, citrate, camphorate, camphorsulfonate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptanoate, glycerophosphate, glycolate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide , hydroiodide, 2-hydroxyethanesulfonate, lactate, maleate, malonate, methanesulfonate, 2- naphthalenesulfonate, nicotinate, nitrate, oxalate, palmoate,
- Salts derived from appropriate bases include alkali metal (e.g., sodium and potassium), alkaline earth metal (e.g., magnesium), ammonium and N + (C ⁇ - 4 alkyl) 4 salts.
- alkali metal e.g., sodium and potassium
- alkaline earth metal e.g., magnesium
- ammonium and N + (C ⁇ - 4 alkyl) 4 salts e.g., sodium and potassium
- alkali metal e.g., sodium and potassium
- alkaline earth metal e.g., magnesium
- ammonium and N + (C ⁇ - 4 alkyl) 4 salts e.g., sodium and potassium
- compositions of the present invention may be administered orally, parenterally, by inhalation spray, topically, rectally, nasally, buccally, vaginally or via an implanted reservoir.
- parenteral as used herein includes subcutaneous, intravenous, intramuscular, intra-articular, intra-synovial, intrasternal, intrathecal, intrahepatic, intralesional and intracranial injection or infusion techniques.
- the compositions are administered orally, intraperitoneally or intravenously.
- Sterile injectable forms of the compositions of this invention may be aqueous or oleaginous suspension. These suspensions may be formulated according to techniques known in the art using suitable dispersing or wetting agents and suspending agents.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally-acceptable diluent or solvent, for example as a solution in 1, 3-butanediol .
- a non-toxic parenterally-acceptable diluent or solvent for example as a solution in 1, 3-butanediol .
- acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or di-glycerides .
- Fatty acids such as oleic acid and its glyceride derivatives are useful in the preparation of injectables, as are natural pharmaceutically-acceptable oils, such as olive oil or castor oil, especially in their polyoxyethylated versions.
- These oil solutions or suspensions may also contain a long-chain alcohol diluent or dispersant, such as carboxymethyl cellulose or similar dispersing agents that are commonly used in the formulation of pharmaceutically acceptable dosage forms including emulsions and suspensions.
- Other commonly used surfactants such as Tweens, Spans and other emulsifying agents or bioavailability enhancers which are commonly used in the manufacture of pharmaceutically acceptable solid, liquid, or other dosage forms may also be used for the purposes of formulation.
- compositions of this invention may be orally administered in any orally acceptable dosage form including, but not limited to, capsules, tablets, aqueous suspensions or solutions.
- carriers commonly used include lactose and corn starch.
- Lubricating agents such as magnesium stearate, are also typically added.
- useful diluents include lactose and dried cornstarch.
- aqueous suspensions are required for oral use, the active ingredient is combined with emulsifying and suspending agents. If desired, certain sweetening, flavoring or coloring agents may also be added.
- compositions of this invention may be administered in the form of suppositories for rectal administration.
- suppositories for rectal administration.
- suppositories can be prepared by mixing the agent with a suitable non- irritating excipient that is solid at room temperature but liquid at rectal temperature and therefore will melt in the rectum to release the drug.
- suitable non- irritating excipient include cocoa butter, beeswax and polyethylene glycols.
- compositions of this invention may also be administered topically, especially when the target of treatment includes areas or organs readily accessible by topical application, including diseases of the eye, the skin, or the lower intestinal tract. Suitable topical formulations are readily prepared for each of these areas or organs .
- Topical application for the lower intestinal tract can be effected in a rectal suppository formulation (see above) or in a suitable enema formulation. Topically- transdermal patches may also be used.
- the pharmaceutically acceptable compositions may be formulated in a suitable ointment containing the active component suspended or dissolved in one or more carriers.
- Carriers for topical administration of the compounds of this invention include, but are not limited to, mineral oil, liquid petrolatum, white petrolatum, propylene glycol, polyoxyethylene, polyoxypropylene compound, emulsifying wax and water.
- the pharmaceutically acceptable compositions can be formulated in a suitable lotion or cream containing the active components suspended or dissolved in one or more pharmaceutically acceptable carriers.
- Suitable carriers include, but are not limited to, mineral oil, sorbitan monostearate, polysorbate 60, cetyl esters wax, cetearyl alcohol, 2-octyldodecanol, benzyl alcohol and water.
- the pharmaceutically acceptable compositions may be formulated as micronized suspensions in isotonic, pH adjusted sterile saline, or, preferably, as solutions in isotonic, pH adjusted sterile saline, either with or without a preservative such as benzylalkonium chloride.
- the pharmaceutically acceptable compositions may be formulated in an ointment such as petrolatum.
- compositions of this invention may also be administered by nasal aerosol or inhalation.
- Such compositions are prepared according to techniques well-known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other conventional solubilizing or dispersing agents.
- compositions of this invention are formulated for oral administration.
- compositions should be formulated so that a dosage of between 0.01 - 100 mg/kg body weight/day of the inhibitor can be administered to a patient receiving these compositions.
- a specific dosage and treatment regimen for any particular patient will depend upon a variety of factors, including the activity of the specific compound employed, the age, body weight, general health, sex, diet, time of administration, rate of excretion, drug combination, and the judgment of the treating physician and the severity of the particular disease being treated.
- the amount of a compound of the present invention in the composition will also depend upon the particular compound in the composition.
- compositions of this invention may also be present in the compositions of this invention.
- the amount of additional therapeutic agent present in the compositions of this invention will be no more than the amount that would normally be administered in a composition comprising that therapeutic agent as the only active agent.
- the amount of additional therapeutic agent in the presently disclosed compositions will range from about 50% to 100% of the amount normally present in a composition comprising that agent as the only therapeutically active agent.
- the invention relates to a method of inhibiting BACE activity in a biological sample comprising the step of contacting said biological sample with a compound of this invention, or composition comprising said compound.
- the invention relates to a method of inhibiting BACE proteinase activity in a biological sample comprising the step of contacting said biological sample with a compound of formula IA, formula IB, formula ICa, formula ICb, formula ID or formula IE.
- biological sample includes, without limitation, cell cultures or extracts thereof; biopsied material obtained from a mammal or extracts thereof; and blood, saliva, urine, feces, semen, tears, or other body fluids or extracts thereof.
- Inhibition of BACE activity in a biological sample is useful for a variety of purposes which are known to one of skill in the art. Examples of such purposes include, but are not limited to, blood transfusion, organ-transplantation, biological specimen storage, and biological assays .
- the invention provides a method for treating or lessening the severity of a BACE-mediated disease or condition in a patient comprising the step of administering to said patient a composition according to the present invention.
- BACE-mediated disease means any disease or other deleterious condition or disease in which BACE is known to play a role.
- a disease or condition includes Alzheimer's Disease, MCI ("mild cognitive impairment"), Down's syndrome, hereditary cerebral hemorrhage, cerebral amyloid angiopathy, dementia, including dementia of mixed vascular and degenerative origin, dementia associated with Parkinson's disease, progressive supranuclear palsy or cortical basal degeneration.
- the methods of this invention that utilize compositions that do not contain an additional therapeutic agent comprise the additional step of separately administering to said patient an additional therapeutic agent.
- additional therapeutic agents When these additional therapeutic agents are administered separately they may be administered to the patient prior to, sequentially with or following administration of the compositions of this invention.
- the compounds of this invention or pharmaceutical compositions thereof may also be incorporated into compositions for coating an implantable medical device, such as prostheses, artificial valves, vascular grafts, stents and catheters.
- an implantable medical device such as prostheses, artificial valves, vascular grafts, stents and catheters.
- Vascular stents for example, have been used to overcome restenosis (re-narrowing of the vessel wall after injury) .
- patients using stents or other implantable devices risk clot formation or platelet activation. These unwanted effects may be prevented or mitigated by pre-coating the device with a pharmaceutically acceptable composition comprising a kinase inhibitor.
- Suitable coatings and the general preparation of coated implantable devices are described in US Patents 6,099,562; 5,886,026; and 5,304,121.
- the coatings are typically biocompatible polymeric materials such as a hydrogel polymer, polymethyldisiloxane, polycaprolactone, polyethylene glycol, polylactic acid, ethylene vinyl acetate, and mixtures thereof.
- the coatings may optionally be further covered by a suitable topcoat of fluorosilicone, polysaccarides, polyethylene glycol, phospholipids or combinations thereof to impart controlled release characteristics in the composition.
- Implantable devices coated with a compound of this invention are another embodiment of the present invention.
- Method B The piperidine N-boc cleavage by HCl: The starting material (10 to 30 mg) was dissolved or suspended in methanol (3 ml) and a solution of 1 N HCl- ether (3 ml) was added. After stirring at 50°C for 40 min, the reaction was evaporated to dryness to give the HCl salt of the substituted piperidines.
- HPLC HPLC
- a HP series 1100 system was used, with a 3.0 X 150 mm YMC ODS-AQ 5.5 ⁇ 120A column, the solvents system were run according to the following order:
- reaction mixture was diluted with ethyl acetate, filtered, and the filtrate concentrated to an oil which was purified by silica chromatography (15% ethyl acetate/hexanes eluent) to give the t-boc protected product MS MH+ 576.0.
- This material was dissolved in 1 mL methylene chloride and 1 mL TFA was added and the reaction mixture let stand for 1 hr.
- naphthalene-1-carboxylic acid (2 ' , 5 ' -dichloro-4-piperazin-l-yl-biphenyl-3-yl) -amide as a TFA salt, 30 mg, 0.051 mmol, 51 % yield.
- naphthalene-1-carboxylic acid (3 ' , 4' -dichloro-5- piperazin-l-yl-2-tri luoromethyl -biphenyl -4-yl) -amide as the TFA salt, 0.025g, 0.05 mmol 79 %.
- reaction mixture was stirred for 2 hours, concentrated to an oil, applied to a column with methylene chloride and eluted with 20 to 30 % ethyl acetate/hexanes to give 4- ⁇ 5-benzyloxy-2- [ (naphthalene-1- carbonyl) -amino] -phenyl ⁇ -piperazine-1-carboxylic acid tert-butyl ester as a white foam, 0.67 g, 1.2 mmol, 96 % yield.
- reaction mixture was concentrated, applied to silica with methylene chloride and eluted with 20 % ethyl acetate/hexane to give the t-Boc protected product (ms MH+ 576) .
- This material was dissolved in 1 mL methylene chloride and 1 mL TFA was added and the reaction mixture let stand for 1 hr. The solvent was then removed and the residue purified by reverse-phase HPLC.
Abstract
Description









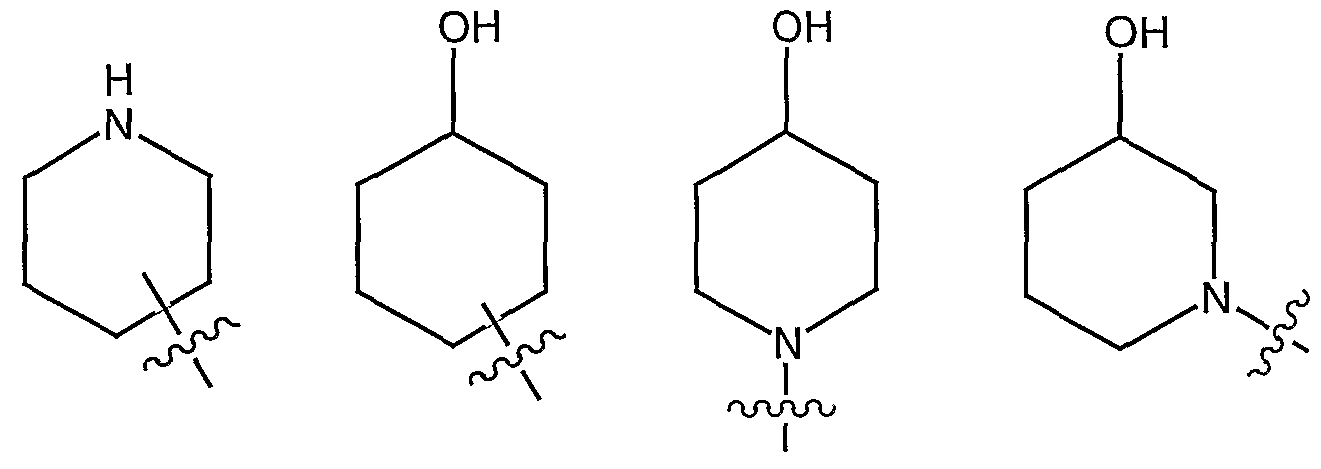







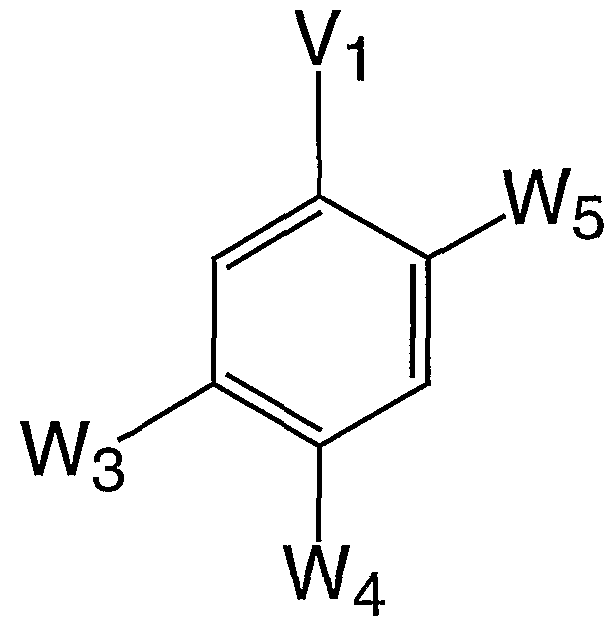




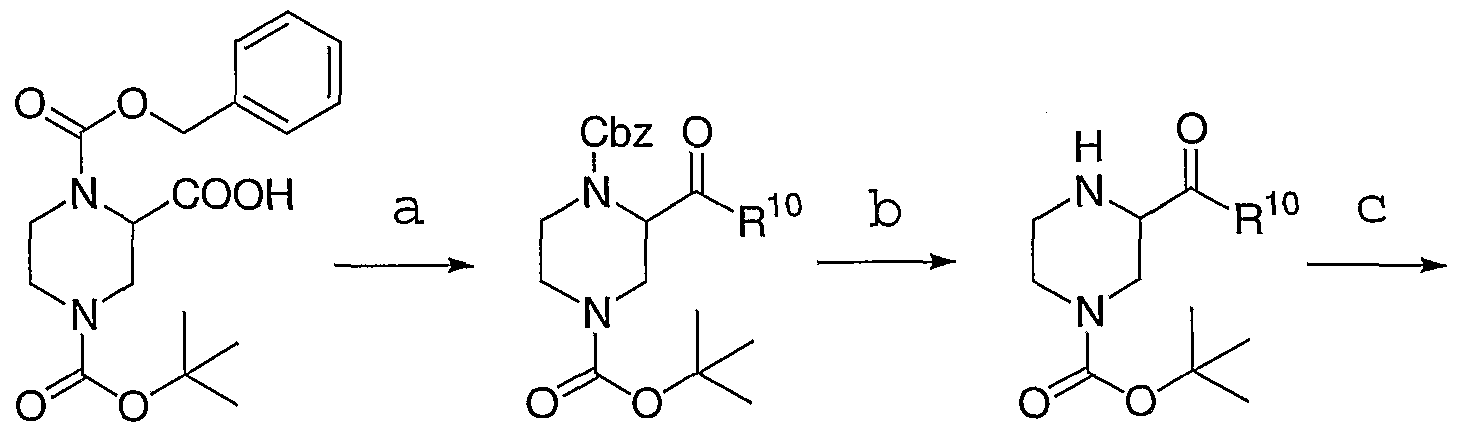












Claims














Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002585403A JP2004534017A (en) | 2001-04-27 | 2002-04-29 | BACE inhibitors |
| AU2002256418A AU2002256418A1 (en) | 2001-04-27 | 2002-04-29 | Inhibitors of bace |
| EP02725881A EP1389194A2 (en) | 2001-04-27 | 2002-04-29 | Inhibitors of bace |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US28716901P | 2001-04-27 | 2001-04-27 | |
| US60/287,169 | 2001-04-27 | ||
| US30104901P | 2001-06-26 | 2001-06-26 | |
| US60/301,049 | 2001-06-26 | ||
| US34226301P | 2001-12-18 | 2001-12-18 | |
| US60/342,263 | 2001-12-18 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2002088101A2 true WO2002088101A2 (en) | 2002-11-07 |
| WO2002088101A3 WO2002088101A3 (en) | 2003-01-03 |
Family
ID=27403680
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2002/013741 WO2002088101A2 (en) | 2001-04-27 | 2002-04-29 | Inhibitors of bace |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20030095958A1 (en) |
| EP (1) | EP1389194A2 (en) |
| JP (1) | JP2004534017A (en) |
| AU (1) | AU2002256418A1 (en) |
| WO (1) | WO2002088101A2 (en) |
Cited By (85)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003043987A2 (en) * | 2001-11-19 | 2003-05-30 | Elan Pharmaceuticals, Inc. | (4-phenyl) piperidin-3-yl-phenylcarboxylate derivatives and related compounds as beta-secretase inhibitors for the treatment of alzheimer's disease |
| WO2004089903A1 (en) * | 2003-04-10 | 2004-10-21 | Warner-Lambert Company Llc | Piperidine derivatives as renin inhibitors for the treatment of hypertension |
| JP2005060401A (en) * | 2003-08-13 | 2005-03-10 | Syrrx Inc | Dipeptidyl peptidase inhibitor |
| US6947847B2 (en) | 2002-03-08 | 2005-09-20 | Wisconsin Alumni Research Foundation | Method to design therapeutically important compounds |
| WO2006038684A1 (en) | 2004-10-01 | 2006-04-13 | Takeda Pharmaceutical Company Limited | Method of screening transmembrane enzyme inhibitory substance |
| WO2006005741A3 (en) * | 2004-07-09 | 2006-07-06 | Speedel Experimenta Ag | Piperdine derivatives as renin inhibitors |
| WO2006069788A1 (en) * | 2004-12-30 | 2006-07-06 | Novartis Ag | Organic compounds |
| WO2006124865A2 (en) * | 2005-05-19 | 2006-11-23 | Vertex Pharmaceuticals Incorporated | Biaryls derivatives useful as modulators of ion channels |
| WO2006129237A2 (en) * | 2005-05-27 | 2006-12-07 | Actelion Pharmaceuticals Ltd | Novel piperidine carboxylic acid amide derivatives |
| JP2007509038A (en) * | 2003-10-03 | 2007-04-12 | ノバルティス アクチエンゲゼルシャフト | Certain substituted spirocyclic lactams and their use as pharmaceuticals |
| WO2007060028A1 (en) * | 2004-12-31 | 2007-05-31 | Gpc Biotech Ag | Napthyridine compounds as rock inhibitors |
| JPWO2005068427A1 (en) * | 2004-01-14 | 2007-09-06 | 武田薬品工業株式会社 | Carboxamide derivatives and uses thereof |
| US7504420B2 (en) | 2005-04-08 | 2009-03-17 | Comentis, Inc. | Compounds which inhibit beta-secretase activity and methods of use |
| US7592348B2 (en) | 2003-12-15 | 2009-09-22 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US7598250B2 (en) | 2003-08-08 | 2009-10-06 | Schering Corporation | Cyclic amine BACE-1 inhibitors having a heterocyclic substituent |
| US7652003B2 (en) | 2004-07-28 | 2010-01-26 | Schering-Plough Corporation | Macrocyclic β-secretase inhibitors |
| US7662816B2 (en) | 2003-08-08 | 2010-02-16 | Schering Corporation | Cyclic amine BACE-1 inhibitors having a benzamide substituent |
| WO2010032856A1 (en) | 2008-09-19 | 2010-03-25 | 武田薬品工業株式会社 | Nitrogen-containing heterocyclic compound and use of same |
| US7696204B2 (en) | 2005-10-11 | 2010-04-13 | Ludwig Institute For Cancer Research | Pharmaceutical compounds |
| US7700603B2 (en) | 2003-12-15 | 2010-04-20 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US7754727B2 (en) | 2003-11-26 | 2010-07-13 | Novartis Ag | 4-phenylpiperidine derivatives as renin inhibitors |
| US7763609B2 (en) | 2003-12-15 | 2010-07-27 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US7803802B2 (en) | 2004-07-22 | 2010-09-28 | Schering Corporation | Substituted amide beta secretase inhibitors |
| US7880008B2 (en) | 2005-05-31 | 2011-02-01 | Vertex Pharmaceuticals Incorporated | Heterocycles useful as modulators of ion channels |
| EP2303859A2 (en) * | 2008-06-20 | 2011-04-06 | Metabolex Inc. | Aryl gpr119 agonists and uses thereof |
| US8093254B2 (en) | 2006-12-12 | 2012-01-10 | Schering Corporation | Aspartyl protease inhibitors |
| US8129411B2 (en) | 2005-12-30 | 2012-03-06 | Novartis Ag | Organic compounds |
| US8138340B2 (en) | 2004-08-25 | 2012-03-20 | Actelion Pharmaceuticals Ltd. | Bicyclononene derivatives |
| US8163773B2 (en) | 2005-07-11 | 2012-04-24 | Novartis Ag | Organic compounds |
| US8188117B2 (en) | 2005-07-26 | 2012-05-29 | Sanofi-Aventis | Piperidinyl-substituted isoquinolone derivatives |
| US8278294B2 (en) | 2006-12-27 | 2012-10-02 | Sanofi | Substituted isoquinoline and isoquinolinone derivatives as inhibitors of Rho-kinase |
| US8383650B2 (en) | 2007-06-25 | 2013-02-26 | Novartis Ag | Organic compounds |
| US8399482B2 (en) | 2008-06-24 | 2013-03-19 | Sanofi | 6-substituted isoquinolines and isoquinolinones |
| US8420640B2 (en) | 2008-08-29 | 2013-04-16 | Treventis Corporation | Methods of treating amyloid disease using analogs of 1-(4-nitrophenyl) piperazine |
| US8501736B2 (en) | 2005-06-28 | 2013-08-06 | Sanofi | Isoquinoline derivatives |
| US8524737B2 (en) | 2008-06-24 | 2013-09-03 | Sanofi | Bi- and polycyclic substituted isoquinoline and isoquinolinone derivatives |
| US8541449B2 (en) | 2008-06-24 | 2013-09-24 | Sanofi | Substituted isoquinolines and isoquinolinones as Rho kinase inhibitors |
| US8541408B2 (en) | 2007-04-24 | 2013-09-24 | Shionogi & Co., Ltd. | Aminodihydrothiazine derivatives substituted with a cyclic group |
| US8609691B2 (en) | 2005-07-26 | 2013-12-17 | Sanofi | Cyclohexylamin isoquinolone derivatives |
| US8618299B2 (en) | 2009-07-01 | 2013-12-31 | Albany Molecular Research, Inc. | Azinone-substituted azapolycycle MCH-1 antagonists, methods of making, and use thereof |
| US8629158B2 (en) | 2009-07-01 | 2014-01-14 | Albany Molecular Research, Inc. | Azabicycloalkane-indole and azabicycloalkane-pyrrolo-pyridine MCH-1 antagonists, methods of making, and use thereof |
| US8637501B2 (en) | 2009-07-01 | 2014-01-28 | Albany Molecular Research, Inc. | Azinone-substituted azepino[b]indole and pyrido-pyrrolo-azepine MCH-1 antagonists, methods of making, and use thereof |
| US8653067B2 (en) | 2007-04-24 | 2014-02-18 | Shionogi & Co., Ltd. | Pharmaceutical composition for treating Alzheimer's disease |
| US8697700B2 (en) | 2010-12-21 | 2014-04-15 | Albany Molecular Research, Inc. | Piperazinone-substituted tetrahydro-carboline MCH-1 antagonists, methods of making, and uses thereof |
| US8710077B2 (en) | 2006-12-27 | 2014-04-29 | Sanofi | Cycloalkylamine substituted isoquinoline and isoquinolinone derivatives |
| US8710228B2 (en) | 2006-12-27 | 2014-04-29 | Sanofi | Cycloalkylamine substituted isoquinoline derivatives |
| US8716308B2 (en) | 2008-01-11 | 2014-05-06 | Albany Molecular Research, Inc. | (1-azinone)-substituted pyridoindoles |
| US8742116B2 (en) | 2006-12-27 | 2014-06-03 | Sanofi | Cycloalkylamine substituted isoquinolone derivatives |
| US8748614B2 (en) | 2006-12-27 | 2014-06-10 | Sanofi | Substituted isoquinoline and isoquinolinone derivatives |
| US8772492B2 (en) | 2006-12-27 | 2014-07-08 | Sanofi | Substituted isoquinoline and isoquinolinone derivatives |
| US8846675B2 (en) | 2007-07-19 | 2014-09-30 | Cymabay Therapeutics, Inc. | N-linked heterocyclic receptor agonists for the treatment of diabetes and metabolic disorders |
| US8921350B2 (en) | 2006-12-28 | 2014-12-30 | Cymabay Therapeutics, Inc. | Heterocyclic receptor agonists for the treatment of diabetes and metabolic disorders |
| US8993765B2 (en) | 2010-12-21 | 2015-03-31 | Albany Molecular Research, Inc. | Tetrahydro-azacarboline MCH-1 antagonists, methods of making, and uses thereof |
| US9018214B2 (en) | 2011-04-21 | 2015-04-28 | Astex Therapeutics Limited | Bicyclic heterocycle compounds and their uses in therapy |
| US9029358B2 (en) | 2005-10-25 | 2015-05-12 | Shionogi & Co., Ltd. | Aminodihydrothiazine derivatives |
| US9073925B2 (en) | 2009-07-01 | 2015-07-07 | Albany Molecular Research, Inc. | Azinone-substituted azabicycloalkane-indole and azabicycloalkane-pyrrolo-pyridine MCH-1 antagonists, methods of making, and use thereof |
| US9150567B2 (en) | 2009-10-01 | 2015-10-06 | Cymabay Therapeutics, Inc. | Substituted tetrazol-1-yl-phenoxymethyl-thiazol-2-yl-piperidinyl-pyrimidine salts |
| US9241924B2 (en) | 2010-06-23 | 2016-01-26 | Cymabay Therapeutics, Inc. | Compositions of 5-ethyl-2-{4-[4-(4-tetrazol-1-yl-phenoxymethyl)-thiazol-2-yl]-piperidin-1-yl}-pyrimidine |
| EP2934514A4 (en) * | 2012-12-21 | 2016-06-08 | Univ Leland Stanford Junior | Transthyretin stabilizers and their use for inhibiting transthyretin amyloidosis and protein-protein interactions |
| US9546156B2 (en) | 2012-11-13 | 2017-01-17 | Array Biopharma Inc. | N-bicyclic aryl,N'-pyrazolyl urea, thiourea, guanidine cyanoguanidine compounds as TrkA kinase inhibitors |
| US9562055B2 (en) | 2011-05-13 | 2017-02-07 | Array Biopharma Inc. | Pyrrolidinyl urea, pyrrolidinyl thiourea and pyrrolidinyl guanidine compounds as TrkA kinase inhibitors |
| US9617283B2 (en) | 2012-10-19 | 2017-04-11 | Astex Therapeutics Limited | Bicyclic heterocycle compounds and their uses in therapy |
| US9617248B2 (en) | 2012-10-19 | 2017-04-11 | Astex Therapeutics Limited | Bicyclic heterocycle compounds and their uses in therapy |
| EP2456759B1 (en) * | 2009-07-24 | 2017-04-19 | Bayer CropScience AG | Pesticidal carboxamides |
| US9650371B2 (en) | 2008-06-13 | 2017-05-16 | Shionogi & Co., Ltd. | Sulfur-containing heterocyclic derivative having beta secretase inhibitory activity |
| US9656974B2 (en) | 2009-12-11 | 2017-05-23 | Shionogi & Co., Ltd. | Oxazine derivatives |
| US9663512B2 (en) | 2012-10-19 | 2017-05-30 | Astex Therapeutics Limited | Bicyclic heterocycle compounds and their uses in therapy |
| WO2017147701A1 (en) * | 2016-03-01 | 2017-09-08 | Ontario Institute For Cancer Research (Oicr) | Inhibitors of wdr5 protein-protein binding |
| US9758513B2 (en) | 2012-10-24 | 2017-09-12 | Shionogi & Co., Ltd. | Dihydrooxazine or oxazepine derivatives having BACE1 inhibitory activity |
| US9783538B2 (en) | 2013-12-20 | 2017-10-10 | Astex Therapeutics Limited | Bicyclic heterocycle compounds and their uses in therapy |
| US9790210B2 (en) | 2012-11-13 | 2017-10-17 | Array Biopharma Inc. | N-(monocyclic aryl),N'-pyrazolyl-urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US9790178B2 (en) | 2012-11-13 | 2017-10-17 | Array Biopharma Inc. | Pyrrolidinyl urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US9809578B2 (en) | 2012-11-13 | 2017-11-07 | Array Biopharma Inc. | Pyrazolyl urea, thiourea, guanidine and cyanoguanidine compounds as trkA kinase inhibitors |
| US9822118B2 (en) | 2012-11-13 | 2017-11-21 | Array Biopharma Inc. | Bicyclic heteroaryl urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US9828360B2 (en) | 2012-11-13 | 2017-11-28 | Array Biopharma Inc. | Pyrrolidinyl urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US9833420B2 (en) | 2003-02-27 | 2017-12-05 | JoAnne McLaurin | Methods of preventing, treating, and diagnosing disorders of protein aggregation |
| US9896435B2 (en) | 2012-11-13 | 2018-02-20 | Array Biopharma Inc. | N-pyrrolidinyl,N′-pyrazolyl-urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US9969694B2 (en) | 2012-11-13 | 2018-05-15 | Array Biopharma Inc. | N-(arylalkyl)-N′-pyrazolyl-urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US9981959B2 (en) | 2012-11-13 | 2018-05-29 | Array Biopharma Inc. | Thiazolyl and oxazolyl urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US9980973B2 (en) | 2012-10-19 | 2018-05-29 | Astex Therapeutics Limited | Bicyclic heterocycle compounds and their uses in therapy |
| US10292983B2 (en) | 2016-08-03 | 2019-05-21 | Cymabay Therapeutics, Inc. | Oxymethylene aryl compounds for treating inflammatory gastrointestinal diseases or gastrointestinal conditions |
| US10351575B2 (en) | 2012-11-13 | 2019-07-16 | Array Biopharma Inc. | Bicyclic urea, thiourea, guanidine and cyanoguanidine compounds useful for the treatment of pain |
| US10835533B2 (en) | 2014-05-15 | 2020-11-17 | Array Biopharma Inc. | 1 -((3S,4R)-4-(3-fluorophenyl)-1-(2-methoxyethyl)pyrrolidin-3-yl)-3-(4-methyl-3-(2-methylpyrimidin-5-yl)-1-phenyl-1H-pyrazol-5-yl)urea as a TrkA kinase inhibitor |
| US11319299B2 (en) | 2016-03-01 | 2022-05-03 | Propellon Therapeutics Inc. | Substituted carboxamides as inhibitors of WDR5 protein-protein binding |
| USRE49582E1 (en) | 2009-06-29 | 2023-07-18 | Agios Pharmaceuticals, Inc. | Therapeutic compounds and compositions |
Families Citing this family (58)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7806980B2 (en) * | 2000-12-23 | 2010-10-05 | Elan Pharmaceuticals, Inc. | Method for crystallizing human beta secretase in complex with an inhibitor |
| US7217556B1 (en) | 2000-12-23 | 2007-05-15 | Pfizer Inc | Crystallization and structure determination of glycosylated human beta secretase, an enzyme implicated in Alzheimer's disease |
| US7601528B1 (en) | 2000-12-23 | 2009-10-13 | Elan Pharmaceuticals, Inc. | Crystallization and structure determination of glycosylated human beta secretase, an enzyme implicated in alzheimer's disease |
| US7384773B1 (en) * | 2001-05-10 | 2008-06-10 | Pfizer Inc | Crystal of HIV protease-cleaved human beta secretase and method for crystallization thereof |
| US7524668B1 (en) * | 2001-05-10 | 2009-04-28 | Elan Pharmaceuticals, Inc. | Crystal of human beta secretase having monoclinic space group symmetry C2 and methods for crystallization thereof |
| US7442537B1 (en) | 2002-05-10 | 2008-10-28 | Elan Pharmaceuticals, Inc. | Crystals of unliganded beta secretase and/or beta secretase-like proteins and the use thereof |
| US7829694B2 (en) | 2002-11-26 | 2010-11-09 | Medtronic, Inc. | Treatment of neurodegenerative disease through intracranial delivery of siRNA |
| US7618948B2 (en) | 2002-11-26 | 2009-11-17 | Medtronic, Inc. | Devices, systems and methods for improving and/or cognitive function through brain delivery of siRNA |
| US7605249B2 (en) | 2002-11-26 | 2009-10-20 | Medtronic, Inc. | Treatment of neurodegenerative disease through intracranial delivery of siRNA |
| US7732591B2 (en) * | 2003-11-25 | 2010-06-08 | Medtronic, Inc. | Compositions, devices and methods for treatment of huntington's disease through intracranial delivery of sirna |
| US7994149B2 (en) | 2003-02-03 | 2011-08-09 | Medtronic, Inc. | Method for treatment of Huntington's disease through intracranial delivery of sirna |
| US20050208090A1 (en) * | 2004-03-18 | 2005-09-22 | Medtronic, Inc. | Methods and systems for treatment of neurological diseases of the central nervous system |
| EP1797052A1 (en) * | 2004-09-17 | 2007-06-20 | Comentis, Inc. | Bicyclic compounds which inhibit beta-secretase activity and methods of use thereof |
| KR20070097590A (en) * | 2005-01-25 | 2007-10-04 | 에픽스 델라웨어, 인코포레이티드 | Substituted arylamine compounds and their use as 5-ht6 modulators |
| MX2007011234A (en) * | 2005-03-14 | 2007-11-12 | Transtech Pharma Inc | Benzazole derivatives, compositions, and methods of use as b-secretase inhibitors. |
| US20060253068A1 (en) * | 2005-04-20 | 2006-11-09 | Van Bilsen Paul | Use of biocompatible in-situ matrices for delivery of therapeutic cells to the heart |
| US7902352B2 (en) * | 2005-05-06 | 2011-03-08 | Medtronic, Inc. | Isolated nucleic acid duplex for reducing huntington gene expression |
| EP1885854B1 (en) * | 2005-05-06 | 2012-10-17 | Medtronic, Inc. | Methods and sequences to suppress primate huntington gene expression |
| US9133517B2 (en) | 2005-06-28 | 2015-09-15 | Medtronics, Inc. | Methods and sequences to preferentially suppress expression of mutated huntingtin |
| US20080280843A1 (en) * | 2006-05-24 | 2008-11-13 | Van Bilsen Paul | Methods and kits for linking polymorphic sequences to expanded repeat mutations |
| EP1747779A1 (en) * | 2005-07-28 | 2007-01-31 | Laboratorios Del Dr. Esteve, S.A. | Tetrahydro-b-carbolin-sulfonamide derivatives as 5-HT6 ligands |
| US9273356B2 (en) | 2006-05-24 | 2016-03-01 | Medtronic, Inc. | Methods and kits for linking polymorphic sequences to expanded repeat mutations |
| US20080039415A1 (en) * | 2006-08-11 | 2008-02-14 | Gregory Robert Stewart | Retrograde transport of sirna and therapeutic uses to treat neurologic disorders |
| US8324367B2 (en) | 2006-11-03 | 2012-12-04 | Medtronic, Inc. | Compositions and methods for making therapies delivered by viral vectors reversible for safety and allele-specificity |
| US9375440B2 (en) * | 2006-11-03 | 2016-06-28 | Medtronic, Inc. | Compositions and methods for making therapies delivered by viral vectors reversible for safety and allele-specificity |
| US7819842B2 (en) | 2006-11-21 | 2010-10-26 | Medtronic, Inc. | Chronically implantable guide tube for repeated intermittent delivery of materials or fluids to targeted tissue sites |
| US7988668B2 (en) * | 2006-11-21 | 2011-08-02 | Medtronic, Inc. | Microsyringe for pre-packaged delivery of pharmaceuticals |
| US20080171906A1 (en) * | 2007-01-16 | 2008-07-17 | Everaerts Frank J L | Tissue performance via hydrolysis and cross-linking |
| KR20100051668A (en) * | 2007-07-26 | 2010-05-17 | 코멘티스, 인코포레이티드 | Isophthalamide derivatives inhibiting beta-secretase activity |
| US20120121515A1 (en) | 2009-03-13 | 2012-05-17 | Lenny Dang | Methods and compositions for cell-proliferation-related disorders |
| AU2010234526B2 (en) | 2009-04-06 | 2016-07-21 | Agios Pharmaceuticals, Inc. | Pyruvate kinase M2 modulators, therapeutic compositions and related methods of use |
| EP2427441B1 (en) | 2009-05-04 | 2016-12-14 | Agios Pharmaceuticals, Inc. | Pkm2 activators for use in the treatment of cancer |
| EP2448581B1 (en) | 2009-06-29 | 2016-12-07 | Agios Pharmaceuticals, Inc. | Therapeutic compositions and related methods of use |
| EP2491145B1 (en) | 2009-10-21 | 2016-03-09 | Agios Pharmaceuticals, Inc. | Methods and compositions for cell-proliferation-related disorders |
| EP2515908A2 (en) * | 2009-12-22 | 2012-10-31 | Merck Sharp & Dohme Corp. | 1,4-substituted piperazine derivatives and methods of use thereof |
| US8927721B2 (en) | 2010-10-29 | 2015-01-06 | Shionogi & Co., Ltd. | Naphthyridine derivative |
| US9018219B2 (en) | 2010-10-29 | 2015-04-28 | Shionogi & Co., Ltd. | Fused aminodihydropyrimidine derivative |
| EP2651898B1 (en) | 2010-12-17 | 2015-12-09 | Agios Pharmaceuticals, Inc. | Novel n-(4-(azetidine-1-carbonyl)phenyl)-(hetero-)arylsulfonamide derivatives as pyruvate kinase m2 (pmk2) modulators |
| EP2655350B1 (en) | 2010-12-21 | 2016-03-09 | Agios Pharmaceuticals, Inc. | Bicyclic pkm2 activators |
| TWI549947B (en) | 2010-12-29 | 2016-09-21 | 阿吉歐斯製藥公司 | Therapeutic compounds and compositions |
| JPWO2012147763A1 (en) | 2011-04-26 | 2014-07-28 | 塩野義製薬株式会社 | Oxazine derivatives and BACE1 inhibitors containing the same |
| ME03074B (en) | 2011-05-03 | 2019-01-20 | Agios Pharmaceuticals Inc | Pyruvate kinase activators for use in therapy |
| CN103608016A (en) | 2011-05-03 | 2014-02-26 | 安吉奥斯医药品有限公司 | Pyruvate kinase activators for use in therapy |
| CN102827073A (en) | 2011-06-17 | 2012-12-19 | 安吉奥斯医药品有限公司 | Therapeutically active compositions and application methods thereof |
| CN102827170A (en) | 2011-06-17 | 2012-12-19 | 安吉奥斯医药品有限公司 | Active treatment compositions and use method thereof |
| US9474779B2 (en) | 2012-01-19 | 2016-10-25 | Agios Pharmaceuticals, Inc. | Therapeutically active compositions and their methods of use |
| WO2013183718A1 (en) * | 2012-06-06 | 2013-12-12 | 国立大学法人京都大学 | Screening method, protein instability and/or stability inducers, and protein activity assesment |
| EP2906212A4 (en) | 2012-10-15 | 2016-06-08 | Agios Pharmaceuticals Inc | Therapeutic compounds and compositions |
| CA2917671A1 (en) | 2013-07-11 | 2015-01-15 | Agios Pharmaceuticals, Inc. | 2,4-or 4,6-diaminopyrimidine compounds as idh2 mutants inhibitors for the treatment of cancer |
| US9579324B2 (en) | 2013-07-11 | 2017-02-28 | Agios Pharmaceuticals, Inc | Therapeutically active compounds and their methods of use |
| WO2015003355A2 (en) | 2013-07-11 | 2015-01-15 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| WO2015003360A2 (en) | 2013-07-11 | 2015-01-15 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US20150031627A1 (en) | 2013-07-25 | 2015-01-29 | Agios Pharmaceuticals, Inc | Therapeutically active compounds and their methods of use |
| AU2015229214B2 (en) | 2014-03-14 | 2019-07-11 | Les Laboratoires Servier | Pharmaceutical compositions of therapeutically active compounds |
| EP4344703A1 (en) | 2015-06-11 | 2024-04-03 | Agios Pharmaceuticals, Inc. | Methods of using pyruvate kinase activators |
| EP3362065B1 (en) | 2015-10-15 | 2024-04-03 | Les Laboratoires Servier | Combination therapy comprising ivosidenib, cytarabine and daunorubicin or idarubicin for treating acute myelogenous leukemia |
| PT3362066T (en) | 2015-10-15 | 2021-11-16 | Celgene Corp | Combination therapy for treating malignancies |
| US10980788B2 (en) | 2018-06-08 | 2021-04-20 | Agios Pharmaceuticals, Inc. | Therapy for treating malignancies |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997038989A1 (en) * | 1996-04-18 | 1997-10-23 | Neurogen Corporation | N-aminoalkyl-2-anthraquinonecarboxamides; new dopamine receptor subtype specific ligands |
| WO1998027081A1 (en) * | 1996-12-19 | 1998-06-25 | Smithkline Beecham Plc | Sulphonamide derivatives, process for their preparation, and their use as medicaments |
| WO1999033793A2 (en) * | 1997-12-24 | 1999-07-08 | Vertex Pharmaceuticals Incorporated | Prodrugs of aspartyl protease inhibitors |
| WO2001052845A1 (en) * | 2000-01-18 | 2001-07-26 | Vertex Pharmaceuticals Incorpoated | Gyrase inhibitors and uses thereof |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6737038B1 (en) * | 1998-11-12 | 2004-05-18 | Bristol-Myers Squibb Company | Use of small molecule radioligands to discover inhibitors of amyloid-beta peptide production and for diagnostic imaging |
| EP1233021A3 (en) * | 2001-02-20 | 2002-11-20 | Pfizer Products Inc. | An inhibitor of Beta amyloid cleavage enzyme |
-
2002
- 2002-04-29 AU AU2002256418A patent/AU2002256418A1/en not_active Abandoned
- 2002-04-29 JP JP2002585403A patent/JP2004534017A/en active Pending
- 2002-04-29 EP EP02725881A patent/EP1389194A2/en not_active Withdrawn
- 2002-04-29 WO PCT/US2002/013741 patent/WO2002088101A2/en not_active Application Discontinuation
- 2002-04-29 US US10/136,576 patent/US20030095958A1/en not_active Abandoned
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997038989A1 (en) * | 1996-04-18 | 1997-10-23 | Neurogen Corporation | N-aminoalkyl-2-anthraquinonecarboxamides; new dopamine receptor subtype specific ligands |
| WO1998027081A1 (en) * | 1996-12-19 | 1998-06-25 | Smithkline Beecham Plc | Sulphonamide derivatives, process for their preparation, and their use as medicaments |
| WO1999033793A2 (en) * | 1997-12-24 | 1999-07-08 | Vertex Pharmaceuticals Incorporated | Prodrugs of aspartyl protease inhibitors |
| WO2001052845A1 (en) * | 2000-01-18 | 2001-07-26 | Vertex Pharmaceuticals Incorpoated | Gyrase inhibitors and uses thereof |
Cited By (132)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7338965B2 (en) | 2001-11-19 | 2008-03-04 | Pharmacia & Upjohn Company | 3,4-disubstituted, 3,5-disubstituted and 3,4,5-substituted piperidines |
| WO2003043987A3 (en) * | 2001-11-19 | 2003-07-10 | Elan Pharm Inc | (4-phenyl) piperidin-3-yl-phenylcarboxylate derivatives and related compounds as beta-secretase inhibitors for the treatment of alzheimer's disease |
| WO2003043987A2 (en) * | 2001-11-19 | 2003-05-30 | Elan Pharmaceuticals, Inc. | (4-phenyl) piperidin-3-yl-phenylcarboxylate derivatives and related compounds as beta-secretase inhibitors for the treatment of alzheimer's disease |
| US6947847B2 (en) | 2002-03-08 | 2005-09-20 | Wisconsin Alumni Research Foundation | Method to design therapeutically important compounds |
| US9833420B2 (en) | 2003-02-27 | 2017-12-05 | JoAnne McLaurin | Methods of preventing, treating, and diagnosing disorders of protein aggregation |
| WO2004089903A1 (en) * | 2003-04-10 | 2004-10-21 | Warner-Lambert Company Llc | Piperidine derivatives as renin inhibitors for the treatment of hypertension |
| US9062007B2 (en) | 2003-08-08 | 2015-06-23 | Merck Sharp & Dohme Corp. | Cyclic amide BACE-1 inhibitors having a benzamide substituent |
| US8278334B2 (en) | 2003-08-08 | 2012-10-02 | Merck, Sharp & Dohme Corp. | Cyclic amine BACE-1 inhibitors having a benzamide substituent |
| US7910590B2 (en) | 2003-08-08 | 2011-03-22 | Schering Corporation | Cyclic amine bace-1 inhibitors having a heterocyclic substituent |
| US7662816B2 (en) | 2003-08-08 | 2010-02-16 | Schering Corporation | Cyclic amine BACE-1 inhibitors having a benzamide substituent |
| US7598250B2 (en) | 2003-08-08 | 2009-10-06 | Schering Corporation | Cyclic amine BACE-1 inhibitors having a heterocyclic substituent |
| JP2005060401A (en) * | 2003-08-13 | 2005-03-10 | Syrrx Inc | Dipeptidyl peptidase inhibitor |
| JP2007509038A (en) * | 2003-10-03 | 2007-04-12 | ノバルティス アクチエンゲゼルシャフト | Certain substituted spirocyclic lactams and their use as pharmaceuticals |
| US8362040B2 (en) | 2003-11-26 | 2013-01-29 | Novartis Ag | 4-phenylpiperidine derivatives as renin inhibitors |
| US7754727B2 (en) | 2003-11-26 | 2010-07-13 | Novartis Ag | 4-phenylpiperidine derivatives as renin inhibitors |
| US7592348B2 (en) | 2003-12-15 | 2009-09-22 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US8242112B2 (en) | 2003-12-15 | 2012-08-14 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US7763609B2 (en) | 2003-12-15 | 2010-07-27 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US8178513B2 (en) | 2003-12-15 | 2012-05-15 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US8937093B2 (en) | 2003-12-15 | 2015-01-20 | Merck Sharp & Dohme Corp. | Heterocyclic aspartyl protease inhibitors |
| US7973067B2 (en) | 2003-12-15 | 2011-07-05 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US7700603B2 (en) | 2003-12-15 | 2010-04-20 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US9416108B2 (en) | 2003-12-15 | 2016-08-16 | Merck Sharp & Dohme Corp. | Heterocyclic aspartyl protease inhibitors |
| JPWO2005068427A1 (en) * | 2004-01-14 | 2007-09-06 | 武田薬品工業株式会社 | Carboxamide derivatives and uses thereof |
| WO2006005741A3 (en) * | 2004-07-09 | 2006-07-06 | Speedel Experimenta Ag | Piperdine derivatives as renin inhibitors |
| US8367712B2 (en) | 2004-07-22 | 2013-02-05 | Merck, Sharp & Dohme, Corp. | Substituted amide beta secretase inhibitors |
| US7803802B2 (en) | 2004-07-22 | 2010-09-28 | Schering Corporation | Substituted amide beta secretase inhibitors |
| US7652003B2 (en) | 2004-07-28 | 2010-01-26 | Schering-Plough Corporation | Macrocyclic β-secretase inhibitors |
| US8012953B2 (en) | 2004-07-28 | 2011-09-06 | Schering Corporation | Macrocyclic beta-secretase inhibitors |
| US8138340B2 (en) | 2004-08-25 | 2012-03-20 | Actelion Pharmaceuticals Ltd. | Bicyclononene derivatives |
| WO2006038684A1 (en) | 2004-10-01 | 2006-04-13 | Takeda Pharmaceutical Company Limited | Method of screening transmembrane enzyme inhibitory substance |
| US8178559B2 (en) | 2004-12-30 | 2012-05-15 | Novartis Ag | Organic compounds |
| US8084450B2 (en) | 2004-12-30 | 2011-12-27 | Novartis Ag | Organic compounds |
| WO2006069788A1 (en) * | 2004-12-30 | 2006-07-06 | Novartis Ag | Organic compounds |
| WO2007060028A1 (en) * | 2004-12-31 | 2007-05-31 | Gpc Biotech Ag | Napthyridine compounds as rock inhibitors |
| US7504420B2 (en) | 2005-04-08 | 2009-03-17 | Comentis, Inc. | Compounds which inhibit beta-secretase activity and methods of use |
| WO2006124865A3 (en) * | 2005-05-19 | 2007-01-11 | Vertex Pharma | Biaryls derivatives useful as modulators of ion channels |
| US7705002B2 (en) | 2005-05-19 | 2010-04-27 | Vertex Pharmaceuticals Incorporated | Biaryls useful as modulators of ion channels |
| WO2006124865A2 (en) * | 2005-05-19 | 2006-11-23 | Vertex Pharmaceuticals Incorporated | Biaryls derivatives useful as modulators of ion channels |
| WO2006129237A2 (en) * | 2005-05-27 | 2006-12-07 | Actelion Pharmaceuticals Ltd | Novel piperidine carboxylic acid amide derivatives |
| US7799805B2 (en) | 2005-05-27 | 2010-09-21 | Actelion Pharmaceuticals Ltd. | Piperidine carboxylic acid amide derivatives |
| WO2006129237A3 (en) * | 2005-05-27 | 2007-03-22 | Actelion Pharmaceuticals Ltd | Novel piperidine carboxylic acid amide derivatives |
| KR100963455B1 (en) | 2005-05-27 | 2010-06-18 | 액테리온 파마슈티칼 리미티드 | Novel piperidine carboxylic acid amide derivatives |
| US7880008B2 (en) | 2005-05-31 | 2011-02-01 | Vertex Pharmaceuticals Incorporated | Heterocycles useful as modulators of ion channels |
| US8329702B2 (en) | 2005-05-31 | 2012-12-11 | Vertex Pharmaceuticals Incorporated | Heterocycles useful as modulators of ion channels |
| US8501736B2 (en) | 2005-06-28 | 2013-08-06 | Sanofi | Isoquinoline derivatives |
| US8722671B2 (en) | 2005-06-28 | 2014-05-13 | Sanofi | Isoquinoline derivatives |
| US8163773B2 (en) | 2005-07-11 | 2012-04-24 | Novartis Ag | Organic compounds |
| US8188117B2 (en) | 2005-07-26 | 2012-05-29 | Sanofi-Aventis | Piperidinyl-substituted isoquinolone derivatives |
| US8609691B2 (en) | 2005-07-26 | 2013-12-17 | Sanofi | Cyclohexylamin isoquinolone derivatives |
| US8796458B2 (en) | 2005-07-26 | 2014-08-05 | Sanofi | Cyclohexylamine isoquinolone derivatives |
| US7696204B2 (en) | 2005-10-11 | 2010-04-13 | Ludwig Institute For Cancer Research | Pharmaceutical compounds |
| US9029358B2 (en) | 2005-10-25 | 2015-05-12 | Shionogi & Co., Ltd. | Aminodihydrothiazine derivatives |
| US8129411B2 (en) | 2005-12-30 | 2012-03-06 | Novartis Ag | Organic compounds |
| US8093254B2 (en) | 2006-12-12 | 2012-01-10 | Schering Corporation | Aspartyl protease inhibitors |
| US8710077B2 (en) | 2006-12-27 | 2014-04-29 | Sanofi | Cycloalkylamine substituted isoquinoline and isoquinolinone derivatives |
| US8461144B2 (en) | 2006-12-27 | 2013-06-11 | Sanofi | Substituted isoquinoline and isoquinolinone derivatives |
| US8710228B2 (en) | 2006-12-27 | 2014-04-29 | Sanofi | Cycloalkylamine substituted isoquinoline derivatives |
| US8742116B2 (en) | 2006-12-27 | 2014-06-03 | Sanofi | Cycloalkylamine substituted isoquinolone derivatives |
| US8772492B2 (en) | 2006-12-27 | 2014-07-08 | Sanofi | Substituted isoquinoline and isoquinolinone derivatives |
| US8748614B2 (en) | 2006-12-27 | 2014-06-10 | Sanofi | Substituted isoquinoline and isoquinolinone derivatives |
| US8278294B2 (en) | 2006-12-27 | 2012-10-02 | Sanofi | Substituted isoquinoline and isoquinolinone derivatives as inhibitors of Rho-kinase |
| US9925189B2 (en) | 2006-12-28 | 2018-03-27 | Cymabay Therapeutics, Inc. | Heterocyclic receptor agonists for the treatment of diabetes and metabolic disorders |
| US8921350B2 (en) | 2006-12-28 | 2014-12-30 | Cymabay Therapeutics, Inc. | Heterocyclic receptor agonists for the treatment of diabetes and metabolic disorders |
| US9737537B2 (en) | 2006-12-28 | 2017-08-22 | Cymabay Therapeutics, Inc. | Heterocyclic receptor agonists for the treatment of diabetes and metabolic disorders |
| US8975258B2 (en) | 2006-12-28 | 2015-03-10 | Cymabay Therapeutics, Inc. | Heterocyclic receptor agonists for the treatment of diabetes and metabolic disorders |
| US8653067B2 (en) | 2007-04-24 | 2014-02-18 | Shionogi & Co., Ltd. | Pharmaceutical composition for treating Alzheimer's disease |
| US8541408B2 (en) | 2007-04-24 | 2013-09-24 | Shionogi & Co., Ltd. | Aminodihydrothiazine derivatives substituted with a cyclic group |
| US8383650B2 (en) | 2007-06-25 | 2013-02-26 | Novartis Ag | Organic compounds |
| US8497286B2 (en) | 2007-06-25 | 2013-07-30 | Novartis Ag | Organic compounds |
| US8846675B2 (en) | 2007-07-19 | 2014-09-30 | Cymabay Therapeutics, Inc. | N-linked heterocyclic receptor agonists for the treatment of diabetes and metabolic disorders |
| US9650378B2 (en) | 2008-01-11 | 2017-05-16 | Albany Molecular Research, Inc. | (1-azinone)-substituted pyridoindoles |
| US8716308B2 (en) | 2008-01-11 | 2014-05-06 | Albany Molecular Research, Inc. | (1-azinone)-substituted pyridoindoles |
| US9296743B2 (en) | 2008-01-11 | 2016-03-29 | Albany Molecular Research, Inc. | (1-azinone)-substituted pyridoindoles |
| US9650371B2 (en) | 2008-06-13 | 2017-05-16 | Shionogi & Co., Ltd. | Sulfur-containing heterocyclic derivative having beta secretase inhibitory activity |
| US20110294836A1 (en) * | 2008-06-20 | 2011-12-01 | Metabolex, Inc. | Aryl gpr119 agonists and uses thereof |
| EP2303859A2 (en) * | 2008-06-20 | 2011-04-06 | Metabolex Inc. | Aryl gpr119 agonists and uses thereof |
| EP2303859A4 (en) * | 2008-06-20 | 2012-08-22 | Metabolex Inc | Aryl gpr119 agonists and uses thereof |
| US8541449B2 (en) | 2008-06-24 | 2013-09-24 | Sanofi | Substituted isoquinolines and isoquinolinones as Rho kinase inhibitors |
| US8524737B2 (en) | 2008-06-24 | 2013-09-03 | Sanofi | Bi- and polycyclic substituted isoquinoline and isoquinolinone derivatives |
| US8399482B2 (en) | 2008-06-24 | 2013-03-19 | Sanofi | 6-substituted isoquinolines and isoquinolinones |
| US8420640B2 (en) | 2008-08-29 | 2013-04-16 | Treventis Corporation | Methods of treating amyloid disease using analogs of 1-(4-nitrophenyl) piperazine |
| USRE49686E1 (en) | 2008-09-19 | 2023-10-10 | Takeda Pharmaceutical Company Limited | Nitrogen-containing heterocyclic compound and use of same |
| US8592454B2 (en) | 2008-09-19 | 2013-11-26 | Takeda Pharmaceutical Company Limited | Nitrogen-containing heterocyclic compound and use of same |
| WO2010032856A1 (en) | 2008-09-19 | 2010-03-25 | 武田薬品工業株式会社 | Nitrogen-containing heterocyclic compound and use of same |
| USRE48334E1 (en) | 2008-09-19 | 2020-12-01 | Takeda Pharmaceutical Company Limited | Nitrogen-containing heterocyclic compound and use of same |
| USRE49582E1 (en) | 2009-06-29 | 2023-07-18 | Agios Pharmaceuticals, Inc. | Therapeutic compounds and compositions |
| US9073925B2 (en) | 2009-07-01 | 2015-07-07 | Albany Molecular Research, Inc. | Azinone-substituted azabicycloalkane-indole and azabicycloalkane-pyrrolo-pyridine MCH-1 antagonists, methods of making, and use thereof |
| US8618299B2 (en) | 2009-07-01 | 2013-12-31 | Albany Molecular Research, Inc. | Azinone-substituted azapolycycle MCH-1 antagonists, methods of making, and use thereof |
| US8629158B2 (en) | 2009-07-01 | 2014-01-14 | Albany Molecular Research, Inc. | Azabicycloalkane-indole and azabicycloalkane-pyrrolo-pyridine MCH-1 antagonists, methods of making, and use thereof |
| US8637501B2 (en) | 2009-07-01 | 2014-01-28 | Albany Molecular Research, Inc. | Azinone-substituted azepino[b]indole and pyrido-pyrrolo-azepine MCH-1 antagonists, methods of making, and use thereof |
| EP2456759B1 (en) * | 2009-07-24 | 2017-04-19 | Bayer CropScience AG | Pesticidal carboxamides |
| US9150567B2 (en) | 2009-10-01 | 2015-10-06 | Cymabay Therapeutics, Inc. | Substituted tetrazol-1-yl-phenoxymethyl-thiazol-2-yl-piperidinyl-pyrimidine salts |
| US9656974B2 (en) | 2009-12-11 | 2017-05-23 | Shionogi & Co., Ltd. | Oxazine derivatives |
| US9241924B2 (en) | 2010-06-23 | 2016-01-26 | Cymabay Therapeutics, Inc. | Compositions of 5-ethyl-2-{4-[4-(4-tetrazol-1-yl-phenoxymethyl)-thiazol-2-yl]-piperidin-1-yl}-pyrimidine |
| US8993765B2 (en) | 2010-12-21 | 2015-03-31 | Albany Molecular Research, Inc. | Tetrahydro-azacarboline MCH-1 antagonists, methods of making, and uses thereof |
| US8697700B2 (en) | 2010-12-21 | 2014-04-15 | Albany Molecular Research, Inc. | Piperazinone-substituted tetrahydro-carboline MCH-1 antagonists, methods of making, and uses thereof |
| US9676768B2 (en) | 2011-04-21 | 2017-06-13 | Astex Therapeutics Limited | Bicyclic heterocycle compounds and their uses in therapy |
| US11866428B2 (en) | 2011-04-21 | 2024-01-09 | Astex Therapeutics Limited | Bicyclic heterocycle compounds and their uses in therapy |
| US9018214B2 (en) | 2011-04-21 | 2015-04-28 | Astex Therapeutics Limited | Bicyclic heterocycle compounds and their uses in therapy |
| US10323022B2 (en) | 2011-05-13 | 2019-06-18 | Array Biopharma Inc. | Pyrrolidinyl urea, pyrrolidinyl thiourea and pyrrolidinyl guanidine compounds as TrkA kinase inhibitors |
| US9562055B2 (en) | 2011-05-13 | 2017-02-07 | Array Biopharma Inc. | Pyrrolidinyl urea, pyrrolidinyl thiourea and pyrrolidinyl guanidine compounds as TrkA kinase inhibitors |
| US9878997B2 (en) | 2011-05-13 | 2018-01-30 | Array Biopharma Inc. | Pyrrolidinyl urea, pyrrolidinyl thiourea and pyrrolidinyl guanidine compounds as TrkA kinase inhibitors |
| US9617283B2 (en) | 2012-10-19 | 2017-04-11 | Astex Therapeutics Limited | Bicyclic heterocycle compounds and their uses in therapy |
| US9617248B2 (en) | 2012-10-19 | 2017-04-11 | Astex Therapeutics Limited | Bicyclic heterocycle compounds and their uses in therapy |
| US9980973B2 (en) | 2012-10-19 | 2018-05-29 | Astex Therapeutics Limited | Bicyclic heterocycle compounds and their uses in therapy |
| US9663512B2 (en) | 2012-10-19 | 2017-05-30 | Astex Therapeutics Limited | Bicyclic heterocycle compounds and their uses in therapy |
| US9758513B2 (en) | 2012-10-24 | 2017-09-12 | Shionogi & Co., Ltd. | Dihydrooxazine or oxazepine derivatives having BACE1 inhibitory activity |
| US9822118B2 (en) | 2012-11-13 | 2017-11-21 | Array Biopharma Inc. | Bicyclic heteroaryl urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US10889589B2 (en) | 2012-11-13 | 2021-01-12 | Array Biopharma Inc. | Bicyclic urea, thiourea, guanidine and cyanoguanidine compounds useful for the treatment of pain |
| US9896435B2 (en) | 2012-11-13 | 2018-02-20 | Array Biopharma Inc. | N-pyrrolidinyl,N′-pyrazolyl-urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US9809578B2 (en) | 2012-11-13 | 2017-11-07 | Array Biopharma Inc. | Pyrazolyl urea, thiourea, guanidine and cyanoguanidine compounds as trkA kinase inhibitors |
| US9969694B2 (en) | 2012-11-13 | 2018-05-15 | Array Biopharma Inc. | N-(arylalkyl)-N′-pyrazolyl-urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US9981959B2 (en) | 2012-11-13 | 2018-05-29 | Array Biopharma Inc. | Thiazolyl and oxazolyl urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US9790178B2 (en) | 2012-11-13 | 2017-10-17 | Array Biopharma Inc. | Pyrrolidinyl urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US9828360B2 (en) | 2012-11-13 | 2017-11-28 | Array Biopharma Inc. | Pyrrolidinyl urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US9790210B2 (en) | 2012-11-13 | 2017-10-17 | Array Biopharma Inc. | N-(monocyclic aryl),N'-pyrazolyl-urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| US10351575B2 (en) | 2012-11-13 | 2019-07-16 | Array Biopharma Inc. | Bicyclic urea, thiourea, guanidine and cyanoguanidine compounds useful for the treatment of pain |
| US10851080B2 (en) | 2012-11-13 | 2020-12-01 | Array Biopharma Inc. | Methods of treatment using pyrrolidinyl urea, thiourea, guanidine and cyanoguanidine compounds |
| US9546156B2 (en) | 2012-11-13 | 2017-01-17 | Array Biopharma Inc. | N-bicyclic aryl,N'-pyrazolyl urea, thiourea, guanidine cyanoguanidine compounds as TrkA kinase inhibitors |
| US10842777B2 (en) | 2012-12-21 | 2020-11-24 | The Board Of Trustees Of The Leland Stanford Junior University | Compounds and compositions that bind and stabilize transthyretin and their use for inhibiting transthyretin amyloidosis and protein-protein interactions |
| US10398681B2 (en) | 2012-12-21 | 2019-09-03 | The Board Of Trustees Of The Leland Stanford Junior University | Compounds and compositions that bind and stabilize transthyretin and their use for inhibiting transthyretin amyloidosis and protein-protein interactions |
| EP2934514A4 (en) * | 2012-12-21 | 2016-06-08 | Univ Leland Stanford Junior | Transthyretin stabilizers and their use for inhibiting transthyretin amyloidosis and protein-protein interactions |
| US10618895B2 (en) | 2013-12-20 | 2020-04-14 | Astex Therapeutics Limited | Bicyclic heterocycle compounds and their uses in therapy |
| US11225476B2 (en) | 2013-12-20 | 2022-01-18 | Astex Therapeutics Limited | Bicyclic heterocycle compounds and their uses in therapy |
| US11643410B2 (en) | 2013-12-20 | 2023-05-09 | Astex Therapeutics Limited | Bicyclic heterocycle compounds and their uses in therapy |
| US9783538B2 (en) | 2013-12-20 | 2017-10-10 | Astex Therapeutics Limited | Bicyclic heterocycle compounds and their uses in therapy |
| US10835533B2 (en) | 2014-05-15 | 2020-11-17 | Array Biopharma Inc. | 1 -((3S,4R)-4-(3-fluorophenyl)-1-(2-methoxyethyl)pyrrolidin-3-yl)-3-(4-methyl-3-(2-methylpyrimidin-5-yl)-1-phenyl-1H-pyrazol-5-yl)urea as a TrkA kinase inhibitor |
| US11174250B2 (en) | 2016-03-01 | 2021-11-16 | Propellon Therapeutics Inc. | Substituted carboxamides as inhibitors of WDR5 protein-protein binding |
| US11319299B2 (en) | 2016-03-01 | 2022-05-03 | Propellon Therapeutics Inc. | Substituted carboxamides as inhibitors of WDR5 protein-protein binding |
| WO2017147701A1 (en) * | 2016-03-01 | 2017-09-08 | Ontario Institute For Cancer Research (Oicr) | Inhibitors of wdr5 protein-protein binding |
| US10292983B2 (en) | 2016-08-03 | 2019-05-21 | Cymabay Therapeutics, Inc. | Oxymethylene aryl compounds for treating inflammatory gastrointestinal diseases or gastrointestinal conditions |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2002088101A3 (en) | 2003-01-03 |
| EP1389194A2 (en) | 2004-02-18 |
| JP2004534017A (en) | 2004-11-11 |
| AU2002256418A1 (en) | 2002-11-11 |
| US20030095958A1 (en) | 2003-05-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1389194A2 (en) | Inhibitors of bace | |
| AU2017258187B2 (en) | Isoquinolin-3-yl carboxamides and preparation and use thereof | |
| EP3630755B1 (en) | 5-methyl-1,3,4-oxadiazol-2-yl compounds | |
| EP2968297B1 (en) | Multisubstituted aromatic compounds as serine protease inhibitors | |
| JP5739527B2 (en) | N-cyclic-3- (cyclic carbonylaminomethyl) benzamide derivatives as Rho kinase inhibitors | |
| JP2021020939A (en) | Substituted pyrazole compounds as serine protease inhibitors | |
| EP2864318B1 (en) | 2-aminopyrazine derivatives as csf-1r kinase inhibitors | |
| US20020119968A1 (en) | Aminoheterocyclic derivatives as antithrombotic or anticoagulant agents | |
| KR20010073000A (en) | Arylpiperazines and their use as metalloproteinase inhibiting agents(mmp) | |
| CZ381397A3 (en) | ARTYLSULFONYLAMINOBENZENE DERIVATIVES AND THEIR USE AS Xa FACTOR INHIBITORS | |
| WO2000044726A1 (en) | 2h-phthalazin-1-one derivatives and drugs comprising these derivatives as the active ingredient | |
| JP2014525443A (en) | Kynurenin-3-monooxygenase inhibitors, pharmaceutical compositions, and methods of use thereof | |
| EA022076B1 (en) | Hexafluoroisopropyl carbamate derivatives, their preparation and their therapeutic application | |
| CA2643859A1 (en) | Soluble epoxide hydrolase inhibitors and methods of using same | |
| JPWO2003089410A1 (en) | Phenylalanine derivative | |
| JP2004002368A (en) | Pharmaceutical composition | |
| PL217872B1 (en) | Alkyl ether derivative or a salt thereof, which is a 1-{3-[2-(1-benzothiophen-5-yl)ethoxy] propyl}-3-azetydynol or a salt thereof | |
| CN102325754A (en) | Cyclic compound having hetero atom | |
| US9951025B2 (en) | Halogenopyrazoles as inhibitors of thrombin | |
| CA3153437A1 (en) | Selective dihydropyrrolopyrimidine jak2 inhibitors | |
| JP4250423B2 (en) | Aryl-substituted alicyclic compound and pharmaceutical composition containing the same | |
| TW200418808A (en) | Novel pyrimidine-4,6-dicarboxylic acid diamides for selectively inhibiting collagenases | |
| JPWO2003011812A1 (en) | Novel amine derivative having human-β-tryptase inhibitory activity and medicament containing the same | |
| WO2007055183A1 (en) | Benzene derivative or salt thereof | |
| CA3181351A1 (en) | Nampt modulators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SD SE SG SI SK SL TJ TM TN TR TT TZ UA UG US UZ VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SD SE SG SI SK SL TJ TM TN TR TT TZ UA UG US UZ VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2002585403 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002725881 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2002725881 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2002725881 Country of ref document: EP |