WO2000025788A1 - Inhibitors of prenyl-protein transferase - Google Patents
Inhibitors of prenyl-protein transferase Download PDFInfo
- Publication number
- WO2000025788A1 WO2000025788A1 PCT/US1999/024948 US9924948W WO0025788A1 WO 2000025788 A1 WO2000025788 A1 WO 2000025788A1 US 9924948 W US9924948 W US 9924948W WO 0025788 A1 WO0025788 A1 WO 0025788A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- unsubstituted
- aryl
- heterocycle
- alkyl
- Prior art date
Links
- 0 CCC(CC)*(C)[Cn]C(C)CCCCCCC(C)(C)CC1CCCC1 Chemical compound CCC(CC)*(C)[Cn]C(C)CCCCCCC(C)(C)CC1CCCC1 0.000 description 10
- IMRWILPUOVGIMU-UHFFFAOYSA-N Brc1ncccc1 Chemical compound Brc1ncccc1 IMRWILPUOVGIMU-UHFFFAOYSA-N 0.000 description 1
- STYZODGJALKMDC-UHFFFAOYSA-N CC(C)(C)OC(N(C(CCSC)CN1C2(C)C=CC=CC2)C(Cc(cccc2)c2[N+]([O-])=O)C1=O)=O Chemical compound CC(C)(C)OC(N(C(CCSC)CN1C2(C)C=CC=CC2)C(Cc(cccc2)c2[N+]([O-])=O)C1=O)=O STYZODGJALKMDC-UHFFFAOYSA-N 0.000 description 1
- JMZDJUXVJZQLEY-UHFFFAOYSA-N CC(C)(C)OC(N(CC1=O)C(CCSC)CN1c1ccccc1)=O Chemical compound CC(C)(C)OC(N(CC1=O)C(CCSC)CN1c1ccccc1)=O JMZDJUXVJZQLEY-UHFFFAOYSA-N 0.000 description 1
- RAFFKXWNTXTTFO-UHFFFAOYSA-N COC(c(cccn1)c1Br)=O Chemical compound COC(c(cccn1)c1Br)=O RAFFKXWNTXTTFO-UHFFFAOYSA-N 0.000 description 1
- YDXMSJDMYRCVDH-CEBUJLNPSA-N CS(CC[C@@H](CN1c2ccccc2)N(Cc2cnc[n]2Cc2cccc(F)c2)C(Cc2ccccc2[N+]([O-])=O)C1=O)(=O)=O Chemical compound CS(CC[C@@H](CN1c2ccccc2)N(Cc2cnc[n]2Cc2cccc(F)c2)C(Cc2ccccc2[N+]([O-])=O)C1=O)(=O)=O YDXMSJDMYRCVDH-CEBUJLNPSA-N 0.000 description 1
- KFEUOLQKKOVJBP-UHFFFAOYSA-N N#CC1=CCC(C[n]2c(CN(CCN3c4cccc(Cl)c4)C(C4)C3O)cnc2)C=C1Oc1c4cccc1 Chemical compound N#CC1=CCC(C[n]2c(CN(CCN3c4cccc(Cl)c4)C(C4)C3O)cnc2)C=C1Oc1c4cccc1 KFEUOLQKKOVJBP-UHFFFAOYSA-N 0.000 description 1
- GFSGGDUWEIXNGX-UHFFFAOYSA-N O=Cc1cnc[n]1Cc1cc(F)ccc1 Chemical compound O=Cc1cnc[n]1Cc1cc(F)ccc1 GFSGGDUWEIXNGX-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/14—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/22—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains four or more hetero rings
Definitions
- Ras proteins are part of a signalling pathway that links cell surface growth factor receptors to nuclear signals initiating cellular proliferation.
- Biological and biochemical studies of Ras action indicate that Ras functions like a G-regulatory protein.
- Ras In the inactive state, Ras is bound to GDP.
- Ras Upon growth factor receptor activation Ras is induced to exchange GDP for GTP and undergoes a conformational change.
- the GTP-bound form of Ras propagates the growth stimulatory signal until the signal is terminated by the intrinsic GTPase activity of Ras, which returns the protein to its inactive GDP bound form (D.R. Lowy and D.M.
- Mutated ras genes (Ha-r s, Ki4a-r s, Ki4b-r ⁇ s and N-ras) are found in many human cancers, including colorectal carcinoma, exocrine pancreatic carcinoma, and myeloid leukemias. The protein products of these genes are defective in their GTPase activity and constitutively transmit a growth stimulatory signal.
- Ras must be localized to the plasma membrane for both normal and oncogenic functions. At least 3 post-translational modifications are involved with Ras membrane localization, and all 3 modifications occur at the C-terminus of Ras.
- the Ras C-terminus contains a sequence motif termed a "CAAX” or "Cys-Aaa -Aaa -Xaa” box (Cys is cysteine, Aaa is an aliphatic amino acid, the Xaa is any amino acid) (Willumsen et al, Nature 520:583-586 (1984)).
- this motif serves as a signal sequence for the enzymes farnesyl-protein transferase or geranylgeranyl-protein transferase, which catalyze the alkylation of the cysteine residue of the CAAX motif with a C15 or C20 isoprenoid, respectively.
- Ras protein transferases Such enzymes that transfer an isoprenoid moiety to the cysteine sulfur of a protein may be generally termed perenyl-protein transferases.
- the Ras protein is one of several proteins that are known to undergo post-translational farnesylation. Other farnesylated proteins include the Ras-related GTP-binding proteins such as Rho, fungal mating factors, the nuclear lamins, and the gamma subunit of transducin. James, et al., J. Biol. Chem. 269, 14182 (1994) have identified a peroxisome associated protein Pxf which is also farnesylated.
- Farnesyl-protein transferase utilizes farnesyl pyrophosphate to covalently modify the Cys thiol group of the Ras CAAX box with a farnesyl group (Reiss et al, Cell, 62:81-88 (1990); Schaber et al, J. Biol. Chem., 265:14101-14104 (1990); Schafer et al, Science, 545:1133-1139 (1990); Manne et al, Proc. Natl. Acad. Sci USA, 87:7541- 7545 (1990)).
- Inhibition of farnesyl pyrophosphate biosynthesis by inhibiting HMG-CoA reductase blocks Ras membrane localization in cultured cells.
- direct inhibition of farnesyl-protein transferase would be more specific and attended by fewer side effects than would occur with the required dose of a general inhibitor of isoprene biosynthesis.
- FPTase farnesyl-protein transferase
- FPP farnesyl diphosphate
- Ras protein substrates
- the peptide derived inhibitors that have been described are generally cysteine containing molecules that are related to the CAAX motif that is the signal for protein prenylation.
- Such inhibitors may inhibit protein prenylation while serving as alternate substrates for the farnesyl-protein transferase enzyme, or may be purely competitive inhibitors (U.S. Patent 5,141,851, University of Texas; N.E. Kohl et al, Science, 260: 1934- 1937 (1993); Graham, et al., J. Med. Chem., 37, 725
- deletion of the thiol from a CAAX derivative has been shown to dramatically reduce the inhibitory potency of the compound.
- the thiol group potentially places limitations on the therapeutic application of FPTase inhibitors with respect to pharmacokinetics, pharmacodynamics and toxicity. Therefore, a functional replacement for the thiol is desirable.
- farnesyl-protein transferase inhibitors are inhibitors of proliferation of vascular smooth muscle cells and are therefore useful in the prevention and therapy of arteriosclerosis and diabetic disturbance of blood vessels (JP H7-112930).
- an object of this invention to develop peptidomimetic compounds that inhibit a prenyl-protein transferase and thus, the post-translational prenylation of proteins. It is a further object of this invention to develop chemotherapeutic compositions containing the compounds of this invention and methods for producing the compounds of this invention.
- the present invention comprises peptidomimetic piperazine- containing macrocyclic compounds which inhibit a prenyl-protein transferase. Further contained in this invention are chemotherapeutic compositions containing these prenyl-protein transferase inhibitors and methods for their production.
- the compounds of this invention are useful in the inhibition of a prenyl-protein transferase and the prenylation of the oncogene protein Ras.
- the inhibitors of a prenyl- protein transferase are illustrated by the formula A:
- Rla, Rlb ; RIC an( j Rid are independently selected from: a) hydrogen, b) aryl, heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl,
- R b an( j R3a are independently selected from: H; unsubstituted or substituted C1-8 alkyl, unsubstituted or substituted C2-8 alkenyl, unsubstituted or substituted C2-8 alkynyl, unsubstituted or substituted aryl, unsubstituted or substituted heterocycle,
- substituted group is substituted with one or more of:
- R ⁇ a an d R ⁇ a are attached to the same C atom and are combined to form - (CH2)u - wherein one of the carbon atoms is optionally replaced by a moiety selected from: O, S(0) m , -NC(O)-, and -N(COR 10 )- ;
- R ⁇ a nd R ⁇ a are optionally attached to the same carbon atom
- R 4 is selected from: C ⁇ -4 alkyl, C3-6 cycloalkyl. heterocycle, aryl. unsubstituted or substituted with: a) C ⁇ -4 alkoxy, b) aryl or heterocycle, c) halogen. d) HO.
- R5, R6 and R ' are independently selected from: H; Ci-4 alkyl, C3.6 cycloalkyl, heterocycle, aryl, aroyl, heteroaroyl, arylsulfonyl, heteroarylsulfonyl, unsubstituted or substituted with:
- R 6 and R ⁇ may be joined in a ring; and independently, R5 and R ⁇ may be joined in a ring;
- R8 is independently selected from: a) hydrogen, b) unsubstituted or substituted aryl, unsubstituted or substituted heterocycle, C3-C10 cycloalkyl, C2-C6 alkenyl,
- R9 is selected from: a) hydrogen, b) C2-C6 alkenyl, C2-C6 alkynyl, perfluoroalkyl, F, CI, Br,
- RHOC(0)NR 10 - and c) C1-C6 alkyl unsubstituted or substituted by perfluoroalkyl, F, CI, Br, RlOO-, RHS(0) m -, R 10 C(O)NR 10 -, (R 10 )2NC(O)-, R 10 2N-C(NR 1 0)-, CN, Rl0C(O)-, R 10 OC(O)-, N3, -N(R 10 )2, or RllOC(O)NRl0-;
- RlO is independently selected from hydrogen, C -CQ alkyl, benzyl, unsubstituted or substituted aryl and unsubstituted or substituted heterocycle;
- RU is independently selected from C -CQ alkyl unsubstituted or substituted aryl and unsubstituted or substituted heterocycle;
- Al is selected from: a bond, -C(0)-, -C(O)NRl0-, -NRIOC(O)-, O, -N(RlO)-, -S(O) 2 N(Rl0)-, -N(RlO)S(0) 2 -, and S(0) m ;
- A2 is selected from: a bond, -C(0)-, -C(O)NRl0-, -NR 10 C(O)-, O, -N(RlO)-, -S(O)2N(Rl0)., -N(R10)S(0)2-, S(0) m and Gl> G ⁇ and G ⁇ are independently selected from: H2 and 0;
- W is heterocycle
- V is selected from: a) heterocycle, and b) aryl;
- Zl is selected from: unsubstituted or substituted aryl and unsubstituted or substituted heterocycle, wherein the substituted aryl or substituted heterocycle is substituted with one or more of:
- Ci-4 alkyl unsubstituted or substituted with: a) Ci-4 alkoxy, b) NR6R7 c) C3-6 cycloalkyl, d) aryl or heterocycle, e) HO, f) -S(0) m R 4 or g) -C(0)NR 6 R 7 , 2) aryl or heterocycle,
- Z ⁇ is selected from L: a bond, unsubstituted or substituted aryl and unsubstituted or substituted heterocycle, wherein the substituted aryl or substituted heterocycle is substituted with one or more of:
- 3 Z is selected from: 1) a unsubstituted or substituted group selected from aryl, heteroaryl, arylmethyl, heteroarylmethyl, arylsulfonyl, heteroarylsulfonyl, wherein the substituted group is substituted with one or more of the following: a) C ⁇ -4 alkyl, unsubstituted or substituted with: Ci-4 alkoxy, NR R7, C3-6 cycloalkyl, unsubstituted or substituted aryl, heterocycle, HO, -S(0)mR 6a , or -C(0)NR 6 R7, b) aryl or heterocycle, c) halogen, d) OR 6 - e) NR 6 R7, f) CN, g) N0 2 , h) CF 3 ; i) -S(0) m R4 j) -C(0)NR 6 R7, or k) C3-C6 cycloalkyl; or 2) unsub
- n 0, 1, 2, 3 or 4
- p 0, 1, 2, 3 or 4
- q is l or 2
- r is 0 to 5
- s is independently 0, 1, 2 or 3
- u is 4 or 5;
- Rla and R °- are independently selected from: hydrogen and Ci-C ⁇ alkyl
- Rib and Rl° are independently selected from: a) hydrogen, b) aryl, heterocycle, cycloalkyl, Rl°0-, -N(Rl°)2 or C2-C6 alkenyl, and c) unsubstituted or substituted C1-C6 alkyl wherein the substitutent on the substituted C -CQ alkyl is selected from unsubstituted or substituted aryl, heterocycle, cycloalkyl, alkenyl, Rl°0- and -N(RlO)2;
- R2 D and R ⁇ are independently selected from: H and CH3; R2 1S independently selected from H;
- R ⁇ a and R ⁇ a are optionally attached to the same carbon atom
- R 4 is selected from:
- Ci-4 alkyl and C3-6 cycloalkyl unsubstituted or substituted with: a) C 1-4 alkoxy, b) halogen, or c) aryl or heterocycle
- R 6 and R7 are independently selected from: H; C1.4 alkyl, C3-6 cycloalkyl, aryl and heterocycle, unsubstituted or substituted with: a) Ci-4 alkoxy, b) halogen, or c) aryl or heterocycle;
- R8 is independently selected from: a) hydrogen, b) unsubstituted or substituted aryl, unsubstituted or substituted heterocycle, Ci-C ⁇ alkyl, C2-C6 alkenyl, C2-C6 alkynyl, Ci-C ⁇ perfluoroalkyl, F, CI, RlOO-, R1°C(0)NR10-, CN, NO2, (Rl°)2N-C(NRl°)-, Rl°C(0)-, -N(R10) 2 , or
- R9 is selected from: a) hydrogen, b) C2-C6 alkenyl, C2-C6 alkynyl, C ⁇ -C6 perfluoroalkyl,
- RlO is independently selected from hydrogen, C1-C6 alkyl, benzyl, unsubstituted or substituted aryl and unsubstituted or substituted heterocycle;
- RU is independently selected from Ci-C ⁇ alkyl, unsubstituted or substituted aryl and unsubstituted or substituted heterocycle;
- Al is selected from: a bond, -C(O)-, -C(O)NRl0-, -NRlOC(O)-, O, -N(RlO)-, -S(0) 2 N(RlO)-, -N(RlO)S(0) 2 -, and S(0) m ;
- A2 is selected from: a bond, -C(O)-, -C(O)NRl0-, -NR1°C(0)-, O, -N(RlO)-, -S(0)2N(RlO)-, -N(Rl°)S(0)2- ; S(0) m and -C(Rld) 2 -;
- Gl' G ⁇ and G ⁇ are independently selected from: H 2 and O;
- V is selected from: a) heterocycle selected from pyrrolidinyl, imidazolyl, pyridinyl, thiazolyl, pyridonyl, 2-oxopiperidinyl, indolyl, quinolinyl, isoquinolinyl, and thienyl, and b) aryl;
- W is a heterocycle selected from pyrrolidinyl, imidazolyl, pyridinyl, thiazolyl, oxazolyl, pyridonyl, 2-oxopiperidinyl, indolyl, quinolinyl, or isoquinolinyl;
- Zl is selected from: unsubstituted or substituted aryl or unsubstituted or substituted heterocycle, wherein the substituted aryl or substituted heterocycle is independently substituted with one or two of: 1) Ci-4 alkyl, unsubstituted or substituted with: a) C ⁇ -4 alkoxy, b) NR 6 R 7 , c) C3-6 cycloalkyl, d) aryl or heterocycle, e) HO, f) -S(0) m R4 or g) -C(0)NR 6 R7,
- Z ⁇ is selected from .: a bond, unsubstituted or substituted aryl and unsubstituted or substituted heterocycle, wherein the substituted aryl or substituted heterocycle is substituted indep endently with one or two of:
- a unsubstituted or substituted group selected from aryl, heteroaryl, arylmethyl, heteroarylmethyl, arylsulfonyl, heteroarylsulfonyl, wherein the substituted group is substituted with one or more of the following: a) C ⁇ -4 alkyl, unsubstituted or substituted with: C ⁇ -4 alkoxy, NR R7, C3-6 cycloalkyl, unsubstituted or substituted aryl, heterocycle, HO, -S(0) m R 6a , or -C(0)NR 6 R7, b) aryl or heterocycle, c) halogen, d) OR 6 ' e) NR R7, f) CN, g) N0 2 , h) CF3;
- n 0, 1, 2, 3 or 4
- p 0, 1, 2, 3 or 4
- q is l or 2
- r is 0 to 5
- s is independently 0, 1, 2 or 3
- u is 4 or 5; or a pharmaceutically acceptable salt or stereoisomer thereof.
- Rla is selected from: hydrogen or C1-C6 alkyl
- Rib and Rl° are independently selected from: a) hydrogen, b) aryl, heterocycle, cycloalkyl, RlOO-, -N(Rl°)2 or C2-C6 alkenyl, and c) C1-C6 alkyl unsubstituted or substituted by aryl, heterocycle, cycloalkyl, alkenyl, RlOO-, or -N(R 10 )2.
- R ⁇ a is selected from H and CH3; R2a is selected from H;
- Ci-5 alkyl unbranched or branched, unsubstituted or substituted with one or more of: 1) aryl,
- R4 is selected from:
- Ci-4 alkyl and C3..6 cycloalkyl unsubstituted or substituted with: a) C ⁇ -4 alkoxy, b) halogen, or c) aryl or heterocycle;
- R 6 and R are independently selected from: a) hydrogen, b) C ⁇ -C-6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C ⁇ -C-6 perfluoroalkyl, F, CI, RlOO-, Rl0C(O)NRl -, CN, N0 2 , (RlO) 2 N-C(NRlO)-, RlOc(O)-, RlO ⁇ C(O)-, -N(Rl ) 2 , or RllOC(O)NRl0-, and c) C1-C6 alkyl substituted by Ci-C ⁇ perfluoroalkyl, RlOO-, Rl0C(O)NRl0-, (RlO) 2 N-C(NRl )-, RlOC(O)-, Rl°OC(0)-, -N(RlO) 2 , or RllOC(O)NRl0-;
- R8 is independently selected from: a) hydrogen, b) unsubstituted or substituted aryl, C ⁇ -C6 alkyl, C 2 -C6 alkenyl, C2-C6 alkynyl, C ⁇ -C ⁇ perfluoroalkyl, F, CI, Rl°0-, R10C(O)NR10-, CN, NO2, (RlO)2N-C(NRlO)-, R10C(O)-, -N(RlO)2, or RHOC(0)NR10-, and c) C ⁇ -C6 alkyl substituted by: unsubstituted or substituted aryl, C ⁇ -C ⁇ perfluoroalkyl, RlOO-, R10C(0)NR10-, (RlO) 2 N-C(NRlO)-, Rl°C(0)-, -N(RlO) 2 , or RHOC(O)NRl0-;
- R is hydrogen or methyl
- Rl is independently selected from hydrogen, C -CQ alkyl, benzyl and unsubstituted or substituted aryl;
- RU is independently selected from C ⁇ -C6 alkyl and unsubstituted or substituted aryl;
- Al is selected from: a bond, -C(O)- and O;
- Gl' G ⁇ and G ⁇ are independently selected from: H and O;
- V is selected from: a) heterocycle selected from pyrrolidinyl, imidazolyl, pyridinyl, thiazolyl, pyridonyl, 2-oxopiperidinyl, indolyl, quinolinyl, isoquinolinyl, and thienyl, and b) aryl;
- Zl is selected from: unsubstituted or substituted aryl or unsubstituted or substituted heterocycle, wherein the substituted aryl or substituted heterocycle is independently substituted with one or two of:
- a unsubstituted or substituted group selected from aryl, heteroaryl, arylmethyl, heteroarylmethyl, arylsulfonyl, heteroarylsulfonyl, wherein the substituted group is substituted with one or more of the following: a) C ⁇ -4 alkyl, unsubstituted or substituted with:
- n 0, 1, 2, 3 or 4
- p 0, 1, 2, 3 or 4
- r 0 to 5
- s is independently 0, 1, 2 or 3;
- Rla is selected from: hydrogen and C ⁇ -C6 alkyl
- Rib and Rl° are independently selected from: a) hydrogen, b) aryl, heterocycle, cycloalkyl, RlOO-, -N(Rl°)2 or C2-C6 alkenyl, and c) C ⁇ -C ⁇ alkyl unsubstituted or substituted by aryl, heterocycle, cycloalkyl, alkenyl, RlOO-, or -N(Rl°)2;
- R ⁇ a is selected from H and CH3; R ⁇ is selected from H;
- R ⁇ a and R ⁇ a are optionally attached to the same carbon atom;
- R 4 is selected from:
- C ⁇ -4 alkyl and C3-6 cycloalkyl unsubstituted or substituted with: a) C ⁇ -4 alkoxy, b) halogen, or c) aryl or heterocycle;
- R 6 and R7 are independently selected from: a) hydrogen, b) Ci-C ⁇ alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C ⁇ -C 6 perfluoroalkyl, F, CI, Rl°0-, Rl0C(O)NRl0-, CN, N0 2 ,
- R8 is independently selected from: a) hydrogen, b) unsubstituted or substituted aryl, C ⁇ -C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, C ⁇ -C6 perfluoroalkyl, F, CI, Rl O-,
- R9 is hydrogen or methyl
- RlO is independently selected from hydrogen, C ⁇ -C ⁇ alkyl, benzyl and unsubstituted or substituted aryl;
- RU is independently selected from C ⁇ -C6 alkyl and unsubstituted or substituted aryl;
- Al is selected from: a bond, -C(O)- and 0;
- G and G ⁇ are independently selected from: H2 and 0, provided that at least one and only one of Gl and G ⁇ are O;
- Zl is selected from: unsubstituted or substituted aryl or unsubstituted or substituted heterocycle, wherein the substituted aryl or substituted heterocycle is substituted with one or two of: 1) C ⁇ -4 alkyl, unsubstituted or substituted with: a) C ⁇ -4 alkoxy, b) NR 6 R 7 , c) C3-6 cycloalkyl, d) aryl or heterocycle, e) HO, f) -S(0) m R4 ( or g) -C(0)NR 6 R7, 2) aryl or heterocycle,
- a unsubstituted or substituted group selected from aryl, heteroaryl, arylmethyl, heteroarylmethyl, arylsulfonyl, heteroarylsulfonyl, wherein the substituted group is substituted with one or more of the following: a ) C ⁇ -4 alkyl, unsubstituted or substituted with:
- n 0, 1, 2, 3 or 4
- p 0, 1, 2, 3 or 4
- r 0 to 5
- s is independently 0, 1, 2 or 3;
- Rl a is selected from: hydrogen and C ⁇ -C6 alkyl
- Ri and Rl° are independently selected from: a) hydrogen, b) aryl, heterocycle, cycloalkyl, Rl°0-, -N(Rl°) 2 or C -C6 alkenyl, and c) C ⁇ -C(, alkyl unsubstituted or substituted by aryl, heterocycle, cycloalkyl, alkenyl, Rl°0-, or -N(RlO) 2;
- R ⁇ a is selected from H and CH3; R ⁇ a is selected from H; and C ⁇ -5 alkyl, unbranched or branched, unsubstituted or substituted with one or more of: 1) aryl, 2) heterocycle,
- R ⁇ and R ⁇ are optionally attached to the same carbon atom
- R4 is selected from:
- R 6 and R7 are independently selected from: a) hydrogen, b) C ⁇ -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C ⁇ -C 6 perfluoroalkyl, F, CI, Rl°0-, Rl0C(O)NRl0-, CN, NO2, (RlO)2N-C(NRlO)-, RlOC(O)-, RlO ⁇ C(O)-, -N(Rl°)2, or R110C(0)NR10-, and c) C ⁇ -C6 alkyl substituted by C ⁇ -C ⁇ perfluoroalkyl, Rl°0-,
- Rl°C(O)NRl0- (RlO) 2 N-C(NRlO)-, Rl°C(0)-, RlO ⁇ C(O)-, -N(Rl )2, or RllOC(O)NRl0-;
- R8 is independently selected from: a) hydrogen, b) unsubstituted or substituted aryl, C ⁇ -C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, C ⁇ -C6 perfluoroalkyl, F, CI, Rl°0-, Rl0C(O)NRl0-, CN, NO2, (RlO)2N-C(NRlO)-, R10C(O)-, -N(RlO) 2 , or RllOC(O)NRl0-, and c) C ⁇ -C6 alkyl substituted by unsubstituted or substituted aryl, C ⁇ -C6 perfluoroalkyl, RlOO-, R1°C(0)NR10-, (RlO) 2 N-C(NRlO)-, RlOC(O)-, -N(Rl ) 2 , or RH0C(0)NR1°-;
- R9 is hydrogen or methyl
- Rl is independently selected from hydrogen, C ⁇ -C ⁇ alkyl, benzyl and unsubstituted or substituted aryl;
- RU is independently selected from C ⁇ -Cg alkyl and unsubstituted or substituted aryl;
- Al is selected from: a bond, -C(O)- and 0;
- X is a bond
- Zl is selected from: unsubstituted or substituted aryl or unsubstituted or substituted heterocycle, wherein the substituted aryl or substituted heterocycle is substituted with one or two of:
- Ci-4 alkyl unsubstituted or substituted with: a) C ⁇ -4 alkoxy, b) NR 6 R 7 , c) C3-6 cycloalkyl, d) aryl or heterocycle, e) HO, f) -S(0) m R4 or g) -C(0)NR 6 R 7 ,
- a unsubstituted or substituted group selected from aryl, heteroaryl, arylmethyl, heteroarylmethyl, arylsulfonyl, heteroarylsulfonyl, wherein the substituted group is substituted with one or more of the following: a) C ⁇ -4 alkyl, unsubstituted or substituted with:
- n 0, 1, 2, 3 or 4
- p 0, 1, 2, 3 or 4
- r 0 to 5
- s is independently 0, 1, 2 or 3;
- Rla is selected from: hydrogen and Cx-C ⁇ alkyl
- Rib and R ° are independently selected from: a) hydrogen, b) aryl, heterocycle, cycloalkyl, Rl°0-, -N(R 10 )2 or C2-C6 alkenyl, and c) C -CQ alkyl unsubstituted or substituted by aryl, heterocycle, cycloalkyl, alkenyl, RlOO-, or -N(R 10 )2;
- R3a is selected from H and CH3;
- R2 a is selected from H
- R ⁇ and R ⁇ are optionally attached to the same carbon atom
- R4 is selected from:
- R 6 and R7 are independently selected from: a) hydrogen, b) C ⁇ -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C ⁇ -C 6 perfluoroalkyl, F, CI, RlOO-, Rl0C(O)NRl0-, CN, NO2,
- R8 is independently selected from: a) hydrogen, b) unsubstituted or substituted aryl, C ⁇ -C6 alkyl, C -C6 alkenyl, C2-C6 alkynyl, C ⁇ -C6 perfluoroalkyl, F, CI, Rl°0-, Rl°C(O)NRl0-, CN, N0 2 , (R10) 2 N
- R9 is hydrogen or methyl
- RlO is independently selected from hydrogen, C ⁇ -C6 alkyl, benzyl and unsubstituted or substituted aryl;
- RU is independently selected from C ⁇ -CQ alkyl and unsubstituted or substituted aryl;
- Al is selected from: a bond, -C(O)- and O;
- X is a bond
- Zl is selected from: unsubstituted or substituted aryl or unsubstituted or substituted heterocycle, wherein the substituted aryl or substituted heterocycle is substituted with one or two of: 1) C ⁇ -4 alkyl, unsubstituted or substituted with: a) C ⁇ -4 alkoxy, b) NR 6 R 7 , c) C3-6 cycloalkyl, d) aryl or heterocycle, e) HO, f) -S(0) m R 4 , or g) -C(0)NR 6 R7,
- 3 Z is selected from: 1) a unsubstituted or substituted group selected from aryl, heteroaryl, arylmethyl, heteroarylmethyl, arylsulfonyl, heteroarylsulfonyl, wherein the substituted group is substituted with one or more of the following: a) C ⁇ -4 alkyl, unsubstituted or substituted with: C ⁇ -4 alkoxy, NR 6 R 7 , C3-6 cycloalkyl, unsubstituted or substituted aryl, heterocycle, HO, -S(0) m R 6a , or -C(0)NR R7, b) aryl or heterocycle, c) halogen, d) OR 6 - e) NR 6 R7, f) CN, g) N0 ) h) CF3; i) -S(0) m R4, j) -C(0)NR 6 R7, or k) C3-C6 cycloalkyl;
- n 0, 1, 2, 3 or 4
- p 0, 1, 2, 3 or 4
- r 0 to 5
- s is independently 0, 1, 2 or 3;
- the compounds of the present invention may have asymmetric centers and occur as racemates, racemic mixtures, and as individual enantiomers, with all possible isomers, including optical isomers, being included in the present invention.
- any variable e.g. aryl, heterocycle, R , R ⁇ etc.
- its definition on each occurence is independent at every other occurence.
- combinations of substituents/or variables are permissible only if such combinations result in stable compounds.
- alkyl is intended to include both branched and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms; “alkoxy” represents an alkyl group of indicated number of carbon atoms attached through an oxygen bridge.
- Halogen or “halo” as used herein means fluoro, chloro, bromo and iodo.
- aryl is intended to mean any stable monocyclic or bicyclic carbon ring of up to 7 members in each ring, wherein at least one ring is aromatic. Examples of such aryl elements include phenyl, naphthyl, tetrahydronaphthyl, indanyl, biphenyl, phenanthryl, anthryl or acenaphthyl.
- heterocycle or heterocyclic represents a stable 5- to 7-membered monocyclic or stable 8- to 11-membered bicyclic heterocyclic ring which is either saturated or unsaturated, and which consists of carbon atoms and from one to four heteroatoms selected from the group consisting of N, 0, and S, and including any bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring.
- the heterocyclic ring may be attached at any heteroatom or carbon atom which results in the creation of a stable structure.
- heterocycle or heterocyclic includes heteroaryl moieties.
- heterocyclic elements include, but are not limited to, azepinyl, benzimidazolyl, benzisoxazolyl, benzofurazanyl, benzopyranyl, benzothiopyranyl, benzofuryl, benzothiazolyl, benzothienyl, benzoxazolyl, chromanyl, cinnolinyl, dihydrobenzofuryl, dihydrobenzothienyl, dihydrobenzothiopyranyl, dihydrobenzothiopyranyl sulfone, furyl, imidazolidinyl, imidazolinyl, imidazolyl, indolinyl, indolyl, isochromanyl, isoindolinyl, isoquinolinyl, isothiazolidinyl, isothiazolyl, isothiazolidinyl, morpholinyl, naphthyridinyl, oxadiazolyl,
- heteroaryl is intended to mean any stable monocyclic or bicyclic carbon ring of up to 7 members in each ring, wherein at least one ring is aromatic and wherein from one to four carbon atoms are replaced by heteroatoms selected from the group consisting of N, O, and S.
- heterocyclic elements include, but are not limited to, benzimidazolyl, benzisoxazolyl, benzofurazanyl, benzopyranyl, benzothiopyranyl, benzofuryl, benzothiazolyl, benzothienyl, benzoxazolyl, chromanyl, cinnolinyl, dihydrobenzofuryl, dihydrobenzothienyl, dihydrobenzothiopyranyl, dihydrobenzothiopyranyl sulfone, furyl, imidazolyl, indolinyl, indolyl, isochromanyl, isoindolinyl, isoquinolinyl, isothiazolyl, naphthyridinyl, oxadiazolyl, pyridyl, pyrazinyl, pyrazolyl, pyridazinyl, pyrimidinyl, pyrrolyl, quinazolin
- the substituted group intended to mean a substituted C ⁇ -8 alkyl, substituted C 2 -8 alkenyl, substituted C 2 -8 alkynyl, substituted aryl or substituted heterocycle from which the substitutent(s) R ⁇ and R ⁇ are selected.
- substituted C ⁇ _8 alkyl, substituted C3-6 alkenyl, substituted aroyl, substituted aryl, substituted heteroaroyl, substituted arylsulfonyl, substituted heteroarylsulfonyl and substituted heterocycle include moieties containing from 1 to 3 substitutents in addition to the point of attachment to the rest of the compound.
- cyclic moieties When R2 and R ⁇ are combined to form - (CH 2 ) U -, cyclic moieties are formed. Examples of such cyclic moieties include, but are not limited to:
- cyclic moieties may optionally include a heteroatom(s).
- heteroatom-containing cyclic moieties include, but are not limited to:
- Lines drawn into the ring systems from substituents indicate that the indicated bond may be attached to any of the substitutable ring carbon atoms.
- Rla and Ri are independently selected from: hydrogen. -N(Rl°) , R1°C(0)NR10- or unsubstituted or substituted C ⁇ -Cc alkyl wherein the substituent on the substituted C ⁇ -C ⁇ alkyl is selected from unsubstituted or substituted phenyl -N RlO) 2 , R O- and
- Rlc is independently selected from: hydrogen, or unsubstituted or substituted C ⁇ -Cc alkyl wherein the substituent on the substituted C ⁇ -C ⁇ alkyl is selected from unsubstituted or substituted phenyl. -N(R 10 )2, R 10 O- and R10C(O)NR10-.
- R ⁇ a is selected from H,
- substituted group selected from C ⁇ -8 alkyl, C 2 -8 alkenyl and C -8 alkynyl; wherein the substituted group is substituted with one or more of:
- R3a and R3b are independently selected from: hydrogen and C ⁇ -C6 alky
- R4 and R° are hydrogen.
- R and R /a are selected from: hydrogen, unsubstituted or substituted C ⁇ -Cc alkyl. unsubstituted or substituted aryl and unsubstituted or substituted cycloalkyl.
- R a is unsubstituted or substituted C ⁇ -C ⁇ alkyl, unsubstituted or substituted aryl and unsubstituted or substituted cycloalkyl.
- R is hydrogen or methyl. Most preferably, R is hydrogen.
- RlO is selected from H. C ⁇ -Co alkyl and benzyl.
- Al and A ⁇ are independently selected from: a bond, -C(0)NR 10 -. -NRl C(O)-, O. -N(R 10 )-, -S(0)2N(RlO). and
- one of Gl and G ⁇ is O and the other is H2-
- G ⁇ is H .
- V is selected from heteroaryl and aryL More preferably. V is phenyl.
- Zl and Z ⁇ are independently selected from unsubstituted or substituted phenyl, unsubstituted or substituted naphthyl, unsubstituted or substituted pyridyl, unsubstituted or substituted furanyl and unsubstituted or substituted thienyl. More preferably, Zl is selected from unsubstituted or substituted phenyl and unsubstituted or substituted naphthyl. More preferably, Z2 is selected from a bond and unsubstituted or substituted phenyl.
- W is selected from imidazolinyl, imidazolyl, oxazoly], pyrazolyl, pyyrolidinyl, thiazolyl and pyridyl. More preferably, W is selected from imidazolyl and pyridyl.
- n is 0, 1, or 2.
- r is 1 or 2.
- p is 1, 2 or 3.
- s is 0 or 1.
- the moiety is 0 or 1.
- R9 a and R b are independently selected R9. It is intended that the definition of any substituent or variable (e.g., Rla, R , n, etc.) at a particular location in a molecule be independent of its definitions elsewhere in that molecule.
- -N(RlO) 2 represents -NHH, -NHCH3, -NHC2H5, etc. It is understood that substituents and substitution patterns on the compounds of the instant invention can be selected by one of ordinary skill in the art to provide compounds that are chemically stable and that can be readily synthesized by techniques known in the art, as well as those methods set forth below, from readily available starting materials.
- the pharmaceutically acceptable salts of the compounds of this invention include the conventional non-toxic salts of the compounds of this invention as formed, e.g., from non-toxic inorganic or organic acids.
- such conventional non-toxic salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like: and the salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, maleic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicylic, sulfanilic, 2-acetoxy-benzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic, isethionic, trifluoroacetic and the like.
- the pharmaceutically acceptable salts of the compounds of this invention can be synthesized from the compounds of this invention which contain a basic moiety by conventional chemical methods. Generally, the salts are prepared either by ion exchange chromatography or by reacting the free base with stoichiometric amounts or with an excess of the desired salt-forming inorganic or organic acid in a suitable solvent or various combinations of solvents.
- Reactions used to generate the compounds of this invention are prepared by employing reactions as shown in the Schemes 1-16, in addition to other standard manipulations such as ester hydrolysis, cleavage of protecting groups, etc., as may be known in the literature or exemplified in the experimental procedures.
- Substituents R, R a , Rb and R su b as shown in the Schemes, represent the substituents R ⁇ , R3 ( R4, and R° ⁇ and substituents on Zl and Z ⁇ ; however their point of attachment to the ring is illustrative only and is not meant to be limiting.
- Boc-protected amino acids such as I can be coupled to N-arylmethyl acetal amines using a variety of dehydrating agents such as DCC (dicyclohexycarbodiimide) or EDC-HC1 (l-ethyl-3-(3- dimethylaminopropyl)carbodiimide hydrochloride) in a solvent such as methylene chloride , chloroform, dichloroethane, or in dimethylformamide.
- dehydrating agents such as DCC (dicyclohexycarbodiimide) or EDC-HC1 (l-ethyl-3-(3- dimethylaminopropyl)carbodiimide hydrochloride) in a solvent such as methylene chloride , chloroform, dichloroethane, or in dimethylformamide.
- the product II is then deprotected and cyclized with acid, for example hydrogen chloride in chloroform or ethyl acetate, or trifluoroacetic acid in methylene chloride, to give the 5,6-unsaturated piperazinone III.
- acid for example hydrogen chloride in chloroform or ethyl acetate, or trifluoroacetic acid in methylene chloride
- Catalytic hydrogenation of III over palladium on carbon gives the piperazinone IV, which may then be reacted with a suitably substituted benzyloxybenzyl bromide in the presence of a strong base to give intermediate VI.
- the ring carbonyl of intermediate VI may then be reduced with lithium aluminum hydride and the benzyl protecting groups catalytically removed to provide intermediate VII.
- the piperidine nitrogen may be reacted with an activated ester to provide the naphthylamide VIII, which can then be deprotected under acidic conditions to provide intermediate IX.
- the piperidine nitrogen can then be reductively alkylated with a suitably substituted fluorobenzyl- imidazolyl aldehyde X to provide XL
- Cesium carbonate nucleophilic aromatic substitution reaction conditions result in an intramolecular cyclization to yield compound XII of the instant invention.
- This cyclization reaction and other cyclization reactions shown below that are mediated by cesium carbonate depend on the presence of an electronic withdrawing moiety (such as nitro, cyano, and the like) either ortho or para to the fluorine atom.
- Scheme 2 illustrates the synthesis of instant macrocyclic compounds which comprise a piperazinone in the ring.
- the protected piperazinone XIII is alkylated with a naphthylmethyl bromide having a suitably positioned benzyloxy moiety.
- Removal of the Boc protecting group provided intermediate XIV, which may be coupled to a suitably substituted 1-benzylimidazole aldehyde XV to give intermediate XVI.
- Removal of the benzyl protecting group followed by intramolecular cyclization as previously described using the cesium carbonate conditions to provide instant compound XVII.
- Scheme 3 illustrates the preparation of instant compounds which incorporate a piperazinone moiety in the macrocyclic ring wherein the macrocycle is incorporated in the 4- and 5-position of the piperazinone.
- N-protected m-tyrosine XVIII is converted to the corresponding aldehyde XIX.
- Aldehyde XIX is reacted with a suitably substituted amine to provide intermediate XX, which is then treated with bromoacetyl bromide to provide, after base mediated cyclization piperazinone XXI.
- Scheme 5 illustrates incorporation of an indole moiety into the macrocyclic ring.
- the synthesis starts with commercially available 5- hydroxytyrosine XXIX which is converted to the coresponding suitably protected aldehyde XXX.
- the aldehyde then undergoes the reactions described in Scheme 3 hereinabove to provide compound XXXI of the instant invention.
- Scheme 7 illustrates the synthetic strategy that is employed when the R8 substitutent is not an electronic withdrawing moiety either ortho or para to the fluorine atom.
- the intramolecular cyclization can be accomplished via an Ullmann reaction.
- protected imidazolyl- methylacetate is treated with a suitably substituted halobenzylbromide to provide the 1-benzylimidazolyl intermediate XXXII.
- Coupling under standard Ullmann conditions provided compound XXXIV of the instant invention.
- Scheme 8 illustrates the incorporation of a sulfur containing sidechain into the piperazinone ring component of the instant macrocyclic compounds.
- Illustrative examples of the preparation of compounds of the instant invention that incorporate a 2,5-diketopiperazine moiety is shown in Scheme 9.
- Intermediate XXXVI, shown in Scheme 9 may also be used to synthesize a number of other macrocycles that incorporate other heterocyclic "W" moieties, such as illustrated in Scheme 10.
- Scheme 11 illustrates the preparation of macrocyclic compounds of the instant invention that incorporate a 2,3- diketopiperazine moiety.
- Amino acids of the general formula XXXVII which have a sidechain not found in natural amino acids may be prepared by the reactions illustrated in Scheme 12 starting with the readily prepared imine XL VI.
- the compounds of the invention are selective inhibitors of farnesyl-protein transferase.
- a compound is considered a selective inhibitor of farnesyl- protein transferase, for example, when its in vitro farnesyl-protein transferase inhibitory activity, as assessed by the assay described in Example 3, is at least 100 times greater than the in vitro activity of the same compound against geranylgeranyl-protein transferase-type I in the assay described in Example 4.
- a selective compound exhibits at least 1000 times greater activity against one of the enzymatic activities when comparing geranylgeranyl-protein transferase -type I inhibition and farnesyl-protein transferase inhibition.
- the selective inhibitor of farnesyl- protein transferase is further characterized by: b) an IC5 Q (a measure of in vitro inhibitory activity) for inhibition of the prenylation of newly synthesized K-Ras protein more than about 100-fold higher than the IC50 for the inhibition of the farnesylation of hDJ protein.
- IC5 Q a measure of in vitro inhibitory activity
- the selective inhibitor of farnesyl- protein transferase is further characterized by: c) an IC50 (a measurement of in vitro inhibitory activity) for inhibition of K4B-Ras dependent activation of MAP kinases in cells at least 100-fold greater than the IC50 for inhibition of the farnesylation of the protein hDJ in cells.
- the selective inhibitor of farnesyl- protein transferase is further characterized by: d) an IC50 (a measurement of in vitro inhibitory activity) against H-Ras dependent activation of MAP kinases in cells at least 1000 fold lower than the inhibitory activity (IC50) against H-r ⁇ s-CVLL
- the compounds of the invention are dual inhibitors of farnesyl-protein transferase and geranylgeranyl-protein transferase type I. Such a dual inhibitor may be termed a Class II prenyl-protein transferase inhibitor and will exhibit certain characteristics when assessed in in vitro assays, which are dependent on the type of assay employed.
- the dual inhibitor compound has an in vitro inhibitory activity (IC50) that is less than about 12 ⁇ M against K4B-Ras dependent activation of MAP kinases in cells.
- the Class II prenyl-protein transferase inhibitor may also be characterized by: a) an IC5 Q (a measurement of in vitro inhibitory activity) for inhibiting K4B-Ras dependent activation of MAP kinases in cells between 0J and 100 times the IC5 Q for inhibiting the farnesylation of the protein hDJ in cells; and b) an IC50 (a measurement of in vitro inhibitory activity) for inhibiting K4B-Ras dependent activation of MAP kinases in cells greater than 5-fold lower than the inhibitory activity (IC50) against expression of the SEAP protein in cells transfected with the pCMV-
- SEAP plasmid that constitutively expresses the SEAP protein.
- the Class II prenyl-protein transferase inhibitor may also be characterized by: a) an IC50 (a measurement of in vitro inhibitory activity) against H-Ras dependent activation of MAP kinases in cells greater than
- IC50 inhibitory activity against H-ms-CVLL (SEQ.ID.NO.: 1) dependent activation of MAP kinases in cells; and b) an IC50 (a measurement of in vitro inhibitory activity) against H-r ⁇ s-CVLL dependent activation of MAP kinases in cells greater than 5-fold lower than the inhibitory activity (IC50) against expression of the SEAP protein in cells transfected with the pCMV- SEAP plasmid that constitutively expresses the SEAP protein.
- the Class II prenyl-protein transferase inhibitor may also be characterized by: a) an IC50 (a measurement of in vitro inhibitory activity) against
- H-Ras dependent activation of MAP kinases in cells greater than 10-fold lower but less than 2,500 fold lower than the inhibitory activity (IC50) against H-ras-CVLL (SEQ.ID.NO.: 1) dependent activation of MAP kinases in cells; and b) an IC50 (a measurement of in vitro inhibitory activity) against IC50 against H-ras-CVLL (SEQ.ID.NO.: 1) dependent activation of MAP kinases in cells; and b) an IC50 (a measurement of in vitro inhibitory activity) against
- IC50 inhibitory activity
- a method for measuring the activity of the inhibitors of prenyl-protein transferase, as well as the instant combination compositions, utilized in the instant methods against Ras dependent activation of MAP kinases in cells is described in Example 7.
- the instant compounds are useful as pharmaceutical agents for mammals, especially for humans. These compounds may be administered to patients for use in the treatment of cancer.
- Examples of the type of cancer which may be treated with the compounds of this invention include, but are not limited to, colorectal carcinoma, exocrine pancreatic carcinoma, myeloid leukemias and neurological tumors. Such tumors may arise by mutations in the ras genes themselves, mutations in the proteins that can regulate Ras activity (i.e., neurofibromin (NF-1), neu, src, abl, lck, fyn) or by other mechanisms.
- NF-1 neurofibromin
- neu src
- abl abl
- lck lck
- the compounds of the instant invention inhibit farnesyl- protein transferase and the farnesylation of the oncogene protein Ras.
- the instant compounds may also inhibit tumor angiogenesis, thereby affecting the growth of tumors (J. Rak et al. Cancer Research, 55: 4575-4580 (1995)).
- Such anti-angiogenesis properties of the instant compounds may also be useful in the treatment of certain forms of vision deficit related to retinal vascularization.
- the compounds of this invention are also useful for inhibiting other proliferative diseases, both benign and malignant, wherein Ras proteins are aberrantly activated as a result of oncogenic mutation in other genes (i.e., the Ras gene itself is not activated by mutation to an oncogenic form) with said inhibition being accomplished by the administration of an effective amount of the compounds of the invention to a mammal in need of such treatment.
- a component of NF-1 is a benign proliferative disorder.
- the instant compounds may also be useful in the treatment of certain viral infections, in particular in the treatment of hepatitis delta and related viruses (J.S. Glenn et al. Science, 256:1331-1333 (1992).
- the compounds of the instant invention are also useful in the prevention of restenosis after percutaneous transluminal coronary angioplasty by inhibiting neointimal formation (C. Indolfi et al. Nature medicine, 1:541-545(1995).
- the instant compounds may also be useful in the treatment and prevention of polycystic kidney disease (D.L. Schaffner et al. American Journal of Pathology, 142:1051-1060 (1993) and B. Cowley, Jr. et a FASEB Journal, 2.A3160 (1988)).
- the instant compounds may also be useful for the treatment of fungal infections.
- the instant compounds may also be useful as inhibitors of proliferation of vascular smooth muscle cells and therefore useful in the prevention and therapy of arteriosclerosis and diabetic vascular pathologies.
- the compounds of this invention may be administered to mammals, preferably humans, either alone or, preferably, in combination with pharmaceutically acceptable carriers, excipients or diluents, in a pharmaceutical composition, according to standard pharmaceutical practice.
- the compounds can be administered orally or parenterally, including the intravenous, intramuscular, intraperitoneal, subcutaneous, rectal and topical routes of administration.
- compositions containing the active ingredient may be in a form suitable for oral use, for example, as tablets, troches, lozenges, aqueous or oily suspensions, dispersible powders or granules, emulsions, hard or soft capsules, or syrups or elixirs.
- Compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations. Tablets contain the active ingredient in admixture with non-toxic pharmaceutically acceptable excipients which are suitable for the manufacture of tablets.
- excipients may be for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, microcrystalline cellulose, sodium crosscarmellose, corn starch, or alginic acid; binding agents, for example starch, gelatin, polyvinyl-pyrrolidone or acacia, and lubricating agents, for example, magnesium stearate, stearic acid or talc.
- the tablets may be uncoated or they may be coated by known techniques to mask the unpleasant taste of the drug or delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- a water soluble taste masking material such as hydroxypropylmethyl- cellulose or hydroxypropylcellulose, or a time delay material such as ethyl cellulose, cellulose acetate buryrate may be employed.
- Formulations for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water soluble carrier such as polyethyleneglycol or an oil medium, for example peanut oil, liquid paraffin, or olive oil.
- an inert solid diluent for example, calcium carbonate, calcium phosphate or kaolin
- water soluble carrier such as polyethyleneglycol or an oil medium, for example peanut oil, liquid paraffin, or olive oil.
- Aqueous suspensions contain the active material in admixture with excipients suitable for the manufacture of aqueous suspensions.
- excipients are suspending agents, for example sodium carboxymethylcellulose, methylcellulose, hydroxypropylmethyl-cellulose, sodium alginate, polyvinyl-pyrrolidone, gum tragacanth and gum acacia; dispersing or wetting agents may be a naturally-occurring phosphatide, for example lecithin, or condensation products of an alkylene oxide with fatty acids, for example polyoxyethylene stearate, or condensation products of ethylene oxide with long chain aliphatic alcohols, for example heptadecaethylene-oxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol monooleate, or condensation products of ethylene oxide with partial esters derived from fatty acids and hexitol anhydrides, for example polyethylene sorbitan mono
- the aqueous suspensions may also contain one or more preservatives, for example ethyl, or n-propyl p-hydroxybenzoate, one or more coloring agents, one or more flavoring agents, and one or more sweetening agents, such as sucrose, saccharin or aspartame.
- preservatives for example ethyl, or n-propyl p-hydroxybenzoate
- coloring agents for example ethyl, or n-propyl p-hydroxybenzoate
- flavoring agents such as sucrose, saccharin or aspartame.
- sweetening agents such as sucrose, saccharin or aspartame.
- Oily suspensions may be formulated by suspending the active ingredient in a vegetable oil, for example arachis oil, olive oil, sesame oil or coconut oil, or in mineral oil such as liquid paraffin.
- the oily suspensions may contain a thickening agent, for example beeswax, hard paraffin or cetyl alcohol.
- Sweetening agents such as those set forth above, and flavoring agents may be added to provide a palatable oral preparation.
- These compositions may be preserved by the addition of an anti-oxidant such as butylated hydroxyanisol or alpha-tocopherol.
- Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives. Suitable dispersing or wetting agents and suspending agents are exemplified by those already mentioned above. Additional excipients, for example sweetening, flavoring and coloring agents, may also be present. These compositions may be preserved by the addition of an anti-oxidant such as ascorbic acid.
- the pharmaceutical compositions of the invention may also be in the form of an oil-in- water emulsions.
- the oily phase may be a vegetable oil, for example olive oil or arachis oil, or a mineral oil, for example liquid paraffin or mixtures of these.
- Suitable emulsifying agents may be naturally-occurring phosphatides, for example soy bean lecithin, and esters or partial esters derived from fatty acids and hexitol anhydrides, for example sorbitan monooleate, and condensation products of the said partial esters with ethylene oxide, for example polyoxyethylene sorbitan monooleate.
- the emulsions may also contain sweetening, flavouring agents, preservatives and antioxidants.
- Syrups and elixirs may be formulated with sweetening agents, for example glycerol, propylene glycol, sorbitol or sucrose. Such formulations may also contain a demulcent, a preservative, flavoring and coloring agents and antioxidant.
- sweetening agents for example glycerol, propylene glycol, sorbitol or sucrose.
- Such formulations may also contain a demulcent, a preservative, flavoring and coloring agents and antioxidant.
- compositions may be in the form of a sterile injectable aqueous solutions.
- acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution.
- the sterile injectable preparation may also be a sterile injectable oil-in-water microemulsion where the active ingredient is dissolved in the oily phase.
- the active ingredient may be first dissolved in a mixture of soybean oil and lecithin. The oil solution then introduced into a water and glycerol mixture and processed to form a microemulation.
- the injectable solutions or microemulsions may be introduced into a patient's blood-stream by local bolus injection.
- a continuous intravenous delivery device may be utilized.
- An example of such a device is the Deltec CADD-PLUSTM model 5400 intravenous pump.
- the pharmaceutical compositions may be in the form of a sterile injectable aqueous or oleagenous suspension for intramuscular and subcutaneous administration.
- This suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents which have been mentioned above.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally-acceptable diluent or solvent, for example as a solution in 1,3-butane diol.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or diglycerides.
- fatty acids such as oleic acid find use in the preparation of injectables.
- Compounds of Formula A may also be administered in the form of a suppositories for rectal administration of the drug.
- These compositions can be prepared by mixing the drug with a suitable non- irritating excipient which is solid at ordinary temperatures but liquid at the rectal temperature and will therefore melt in the rectum to release the drug.
- suitable non- irritating excipient include cocoa butter, glycerinated gelatin, hydrogenated vegetable oils, mixtures of polyethylene glycols of various molecular weights and fatty acid esters of polyethylene glycol.
- topical use creams, ointments, jellies, solutions or suspensions, etc., containing the compound of Formula A are employed. (For purposes of this application, topical application shall include mouth washes and gargles.)
- the compounds for the present invention can be administered in intranasal form via topical use of suitable intranasal vehicles and delivery devices, or via transdermal routes, using those forms of transdermal skin patches well known to those of ordinary skill in the art.
- the dosage administration will, of course, be continuous rather than intermittent throughout the dosage regimen.
- composition is intended to encompass a product comprising the specified ingredients in the specific amounts, as well as any product which results, directly or indirectly, from combination of the specific ingredients in the specified amounts.
- the daily dosage will normally be determined by the prescribing physician with the dosage generally varying according to the age, weight, sex and response of the individual patient, as well as the severity of the patient's symptoms.
- a suitable amount of compound is administered to a mammal undergoing treatment for cancer.
- Administration occurs in an amount between about 0J mg/kg of body weight to about 60 mg/kg of body weight per day, preferably of between 0.5 mg/kg of body weight to about 40 mg/kg of body weight per day.
- the compounds of the instant invention may also be co-administered with other well known therapeutic agents that are selected for their particular usefulness against the condition that is being treated.
- the compounds of the instant invention may also be co-administered with other well known cancer therapeutic agents that are selected for their particular usefulness against the condition that is being treated. Included in such combinations of therapeutic agents are combinations of the instant farnesyl-protein transferase inhibitors and an antineoplastic agent. It is also understood that such a combination of antineoplastic agent and inhibitor of farnesyl-protein transferase may be used in conjunction with other methods of treating cancer and/or tumors, including radiation therapy and surgery.
- antineoplastic agent examples include, in general, microtubule-stabilizing agents (such as paclitaxel (also known as Taxol®), docetaxel (also known as Taxotere®), epothilone A, epothilone B, desoxvepothilone A, desoxvepothilone B or their derivatives); microtubule- disruptor agents; alkylating agents, anti-metabolites; epidophyllotoxin; an antineoplastic enzyme; a topoisomerase inhibitor; procarbazine; mitoxantrone; platinum coordination complexes; biological response modifiers and growth inhibitors; hormonal/anti-hormonal therapeutic agents and haematopoietic growth factors.
- microtubule-stabilizing agents such as paclitaxel (also known as Taxol®), docetaxel (also known as Taxotere®), epothilone A, epothilone B, desoxvepothilone A, desoxvepot
- Example classes of antineoplastic agents include, for example, the anthracycline family of drugs, the vinca drugs, the mitomycins, the bleomycins, the cytotoxic nucleosides, the taxanes, the epothilones, discodermolide, the pteridine family of drugs, diynenes and the podophyllotoxins.
- Particularly useful members of those classes include, for example, doxorubicin, carminomycin, daunorubicin, aminopterin, methotrexate, methopterin, dichloro-methotrexate, mitomycin C, porfiromycin, 5-fl.uorouracil, 6-mercaptopurine, gemcitabine, cytosine arabinoside, podophyllotoxin or podo-phyllotoxin derivatives such as etoposide, etoposide phosphate or teniposide, melphalan, vinblastine, vincristine, leurosidine, vindesine, leurosine, paclitaxel and the like.
- antineoplastic agents include estramustine, cisplatin, carboplatin, cyclophosphamide, bleomycin, tamoxifen, ifosamide, melphalan, hexamethyl melamine, thiotepa, cytarabin, idatrexate, trimetrexate, dacarbazine, L-asparaginase, camptothecin, CPT-11, topotecan, ara-C, bicalutamide, flutamide, leuprolide, pyridobenzoindole derivatives, interferons and interleukins.
- the preferred class of antineoplastic agents is the taxanes and the preferred antineoplastic agent is paclitaxel.
- Radiation therapy including x-rays or gamma rays which are delivered from either an externally applied beam or by implantation of tiny radioactive sources, may also be used in combination with the instant inhibitor of farnesyl-protein transferase alone to treat cancer. Additionally, compounds of the instant invention may also be useful as radiation sensitizers, as described in WO 97/38697, published on October 23, 1997, and herein incorporated by reference.
- the instant compounds may also be useful in combination with other inhibitors of parts of the signaling pathway that links cell surface growth factor receptors to nuclear signals initiating cellular proliferation.
- the instant compounds may be utilized in combination with farnesyl pyrophosphate competitive inhibitors of the activity of farnesyl-protein transferase or in combination with a compound which has Raf antagonist activity.
- the instant compounds may also be co-administered with compounds that are selective inhibitors of geranylgeranyl protein transferase.
- co-administration with a compound(s) that is a selective inhibitor of geranylgeranyl protein transferase may provide an improved therapeutic effect.
- such administration can be orally or parenterally, including intravenous, intramuscular, intraperitoneal, subcutaneous, rectal and topical routes of administration. It is preferred that such administration be orally. It is more preferred that such administration be orally and simultaneously.
- the protein substrate- competitive inhibitor and farnesyl pyrophosphate-competitive inhibitor are administered sequentially, the administration of each can be by the same method or by different methods.
- the instant compounds may also be useful in combination with an integrin antagonist for the treatment of cancer, as described in U.S. Ser. No. 09/055,487, filed April 6, 1998, which is incorporated herein by reference.
- an integrin antagonist refers to compounds which selectively antagonize, inhibit or counteract binding of a physiological ligand to an integrin(s) that is involved in the regulation of angiogenisis. or in the growth and invasiveness of tumor cells.
- the term refers to compounds which selectively antagonize, inhibit or counteract binding of a physiological ligand to the ⁇ v ⁇ 3 integrin, which selectively antagonize, inhibit or counteract binding of a physiological ligand to the ⁇ v ⁇ integrin, which antagonize, inhibit or counteract binding of a physiological ligand to both the ⁇ v ⁇ 3 integrin and the ⁇ v ⁇ integrin, or which antagonize, inhibit or counteract the activity of the particular integrin(s) expressed on capillary endothelial cells.
- the term also refers to antagonists of the ⁇ l ⁇ l, ⁇ 2 ⁇ l, ⁇ l, ⁇ 6 ⁇ l and ⁇ 6 ⁇ 4 integrins.
- the term also refers to antagonists of any combination of ⁇ v ⁇ 3 integrin, ⁇ v ⁇ integrin, ⁇ l ⁇ l, ⁇ 2 ⁇ l, ⁇ l, ⁇ 6 ⁇ l and ⁇ 6 ⁇ 4 integrins.
- the instant compounds may also be useful with other agents that inhibit angiogenisis and thereby inhibit the growth and invasiveness of tumor cells, including, but not limited to angiostatin and endostatin.
- the instant compounds may also be useful in combination with an inhibitor of 3-hydroxy-3-methylglutaryl-CoA reductase (HMG-CoA reductase) for the treatment of cancer.
- HMG-CoA reductase 3-hydroxy-3-methylglutaryl-CoA reductase
- HMG-CoA reductase inhibitor and “inhibitor of HMG-CoA reductase” have the same meaning when used herein.
- HMG-CoA reductase inhibitors examples include but are not limited to lovastatin (MEVACOR®; see US Patent No. 4,231,938; 4,294,926; 4,319,039), simvastatin (ZOCOR®; see US Patent No. 4,444,784; 4,820,850; 4,916,239), pravastatin (PRAVACHOL®; see US Patent Nos. 4,346,227; 4,537,859; 4,410,629; ⁇ ,030,447 and 5, 180,589), fluvastatin (LESCOL®; see US Patent Nos.
- HMG-CoA reductase inhibitor as used herein includes all pharmaceutically acceptable lactone and open-acid forms (i.e., where the lactone ring is opened to form the free acid) as well as salt and ester forms of compounds which have HMG-CoA reductase inhibitory activity, and therefor the use of such salts, esters, open-acid and lactone forms is included within the scope of this invention.
- An illustration of the lactone portion and its corresponding open-acid form is shown below as structures I and II.
- HMG-CoA reductase inhibitor In HMG-CoA reductase inhibitor's where an open-acid form can exist, salt and ester forms may preferably be formed from the open-acid, and all such forms are included within the meaning of the term "HMG-CoA reductase inhibitor" as used herein.
- the HMG-CoA reductase inhibitor is selected from lovastatin and simvastatin, and most preferably simvastatin.
- the term "pharmaceutically acceptable salts" with respect to the HMG-CoA reductase inhibitor shall mean non-toxic salts of the compounds employed in this invention which are generally prepared by reacting the free acid with a suitable organic or inorganic base, particularly those formed from cations such as sodium, potassium, aluminum, calcium, lithium, magnesium, zinc and tetramethylammonium, as well as those salts formed from amines such as ammonia, ethylenediamine, N-methylglucamine, lysine, arginine, ornithine, choline, N,N'-dibenzylethylenediamine, chloroprocaine, diethanolamine, procaine, N-benzylphenethylamine, l-p-chlorobenzyl-2-pyrrolidine-l'-yl-methylbenzimidazole, diethylamine, piperazine, and tris(hydroxymethyl)aminomethane.
- a suitable organic or inorganic base particularly those formed from c
- salt forms of HMG-CoA reductase inhibitors may include, but are not limited to, acetate, benzenesulfonate, benzoate, bicarbonate, bisulfate, bitartrate, borate, bromide, calcium edetate, camsylate, carbonate, chloride, clavulanate, citrate, dihydrochloride, edetate, edisylate, estolate, esylate, fumarate, gluceptate, gluconate, glutamate, glycollylarsanilate, hexylresorcinate, hydrabamine, hydrobromide, hydrochloride, hydroxynapthoate, iodide, isothionate, lactate, lactobionate, laurate, malate, maleate, mandelate, mesylate, methylsulfate, mucate, napsylate, nitrate, oleate, oxalate, pamao
- Ester derivatives of the described HMG-CoA reductase inhibitor compounds may act as prodrugs which, when absorbed into the bloodstream of a warm-blooded animal, may cleave in such a manner as to release the drug form and permit the drug to afford improved therapeutic efficacy.
- the instant compounds may be useful in combination with agents that are effective in the treatment and prevention of NF-1, restenosis, polycystic kidney disease, infections of hepatitis delta and related viruses and fungal infections. If formulated as a fixed dose, such combination products employ the combinations of this invention within the dosage range described above and the other pharmaceutically active agent(s) within its approved dosage range.
- Combinations of the instant invention may 5 alternatively be used sequentially with known pharmaceutically acceptable agent(s) when a multiple combination formulation is inappropriate.
- the compounds of the instant invention are also useful as a component in an assay to rapidly determine the presence and
- FPTase farnesyl-protein transferase
- composition to be tested may be divided and the two portions contacted with mixtures which comprise a known substrate of FPTase (for example a tetrapeptide having a cysteine at the amine terminus) and farnesyl pyrophosphate and, in one of the mixtures, l ⁇ a compound of the instant invention.
- FPTase for example a tetrapeptide having a cysteine at the amine terminus
- farnesyl pyrophosphate for example a tetrapeptide having a cysteine at the amine terminus
- the chemical content of the assay mixtures may be determined by well known immunological, radiochemical or chromatographic techniques. Because
- a 30 invention may be used in an active site titration assay to determine the quantity of enzyme in the sample.
- a series of samples composed of aliquots of a tissue extract containing an unknown amount of farnesyl- protein transferase, an excess amount of a known substrate of FPTase (for example a tetrapeptide having a cysteine at the amine terminus) and farnesyl pyrophosphate are incubated for an appropriate period of time in the presence of varying concentrations of a compound of the instant invention.
- FPTase for example a tetrapeptide having a cysteine at the amine terminus
- concentration of a sufficiently potent inhibitor i.e., one that has a Ki substantially smaller than the concentration of enzyme in the assay vessel
- concentration of a sufficiently potent inhibitor i.e., one that has a Ki substantially smaller than the concentration of enzyme in the assay vessel
- Step A Preparation of l-triphenylmethyl-4-(hydroxymethyl)- imidazole
- Step E Preparation of l-(4-cyano-3-fluorobenzy ⁇ )-5-(acetoxymethyl)- imidazole hvdrobromide
- Step F Preparation of l-(4-cyano-3-fluorobenzy ⁇ )-5-
- Step G Preparation of l-(4-cyano-3-fluorobenzyl)-5- imidazolecarboxaldehvde
- the amine hydrochloride from Step H (ca. 282 mmol, crude material prepared above) was taken up in 600 mL of THF and ⁇ OO mL of sat. aq. NaHC03 soln., cooled to 0°C, and di-tert-butylpyrocarbonate (61.6 g, 282 mmol) was added. After 30 h, the reaction was poured into EtOAc, 2 ⁇ washed with water and brine, dried (Na2S ⁇ 4), filtered, and concentrated in vacuo to provide the titled carbamate as a brown oil which was used in the next step without further purification.
- Step K Preparation of 4-(ter- butoxycarbonyl)-l-(3-chlorophenyl)-2- piperazinone
- Step N Preparation of 4-(tert-butylcarbonyl)-3-( 2-(benzyloxy)benzyl)- l-(3-chlorophenyl)-2-piperazinone
- a solution of the product from Step K in dry THF was cooled to -78 °C.
- a solution of Sodium bis(trimethylsilyl)amide in THF (1M, 3.2 mL, 3.2 mmol) was added slowly and the reaction stirred for 4 ⁇ minutes.
- a solution of the product from Step M (8 ⁇ 9 mg, 3.2 mmol) in 1 mL of THF was added slowly and the reaction stirred for 2. ⁇ hours.
- reaction was quenched at -78 °C by the addition of H 2 0, warmed to room temperature and diluted with EtOAc. The layers were seperated and the organic portion washed with sat. NaHC0 3 solution and brine, dried (Na2S ⁇ 4), filtered, and concentrated in vacuo to provide of the alkylated piperazinone.
- Step P Preparation of 3-[2-(benzyloxy)benzyl]-l-(3-chlorophenyl)-4- [l-(4-cyano-3-fluorobenzyl)-5-imidazolimidazolylmethyl]-2- piperazinone
- Step Q Preparation of l-(3-chlorophenyl)-4-[l-(4-cyano-3- fluorobenzyl)- ⁇ -imidazolimidazolylmethyl]- of 3-[2- (hydroxy)benzyl]-2-piperazinone
- Step R Preparation of ( ⁇ )18-(3-c-alorophenyl)-16,16a,17, 18, 19,20- hexahydro-17-oxo- ⁇ H-6,10-metheno-22H-benzo- [&]pyrazino[2,l-e]imidazo[4,3-b][l,6,9]oxadiaza- cyclopentadecine-9-carbonitrile dihydrochloride
- cesium carbonate 106 mg, 0.30 mmol
- the reaction was warmed to 50°C under argon for one hour, then heated to 80°C for 1.5 hours, allowed to cool to 50°C and stir for 2 hours more. The reaction was then cooled to room temperature. The solution was poured into EtOAc and washed with water and brine, dried (Na2SO- ⁇ ), filtered, and concentrated in vacuo. The resulting product was purified on four O. ⁇ mm preperative TLC plates run in CHCl 3 :MeOH:NH 4 OH (90:10:1) The compound was converted to the HCI salt by dissolving in a minimal amount of CH2CI2 and treating with
- the titled compound was isolated as a product of Step R of example 1 as a white powder.
- Isoprenyl-protein transferase activity assays are carried out at 30°C unless noted otherwise.
- a typical reaction contains (in a final volume of ⁇ O ⁇ L): [ 3 H]farnesyl diphosphate, Ras protein , ⁇ O mM HEPES, pH 7.5, 5 mM MgCl 2 , 5 mM dithiothreitol, 10 ⁇ M ZnCl 2 , 0.1% polyethyleneglycol (PEG) (15,000-20,000 mw) and isoprenyl-protein transferase.
- PEG polyethyleneglycol
- the FPTase employed in the assay is prepared by recombinant expression as described in Omer, C.A., Krai, A.M., Diehl, R.E., Prendergast, G.C., Powers, S., Allen, CM., Gibbs, J.B. and Kohl, N.E. (1993) Biochemistry 32: ⁇ l67- ⁇ l76. After thermally pre-equilibrating the assay mixture in the absence of enzyme, reactions are initiated by the addition of isoprenyl-protein transferase and stopped at timed intervals (typically ⁇ l ⁇ min) by the addition of 1 M HCI in ethanol (1 mL).
- the quenched reactions are allowed to stand for l ⁇ m (to complete the precipitation process). After adding 2 mL of 100% ethanol, the reactions are vacuum- filtered through Whatman GF/C filters. Filters are washed four times with 2 mL aliquots of 100% ethanol, mixed with scintillation fluid (10 mL) 10 and then counted in a Beckman LS3801 scintillation counter.
- compositions or inhibitors are prepared as concentrated solutions in 100% dimethyl sulfoxide and then diluted 20-fold into the enzyme assay mixture.
- Substrate concentrations for composition or inhibitor IC ⁇ O determinations l ⁇ are as follows: FTase, 6 ⁇ 0 nM Ras-CVLS (SEQ.ID.NO.: 1), 100 nM farnesyl diphosphate.
- Modified-ZVi vitro GGTase inhibition assay 2 ⁇ The modified geranylgeranyl-protein transferase inhibition assay is carried out at room temperature.
- a typical reaction contains (in a final volume of ⁇ O ⁇ L): [ 3 H]geranylgeranyl diphosphate, biotinylated Ras peptide, ⁇ O mM HEPES, pH 7.5, a modulating anion (for example 10 mM glycerophosphate or 5mM ATP), 5 mM MgCl 2 ,
- the GGTase- type I enzyme employed in the assay is prepared as described in U.S. Pat. No. 5,470,832, incorporated by reference.
- the Ras peptide is derived from the K4B-Ras protein and has the following sequence: biotinyl-
- GKKKKKKSKTKCVIM single amino acid code
- Reactions are initiated by the addition of GGTase and stopped at timed intervals (typically l ⁇ min) by the addition of 200 ⁇ L of a 3 mg/mL suspension of streptavidin SPA beads (Scintillation Proximity Assay beads, Amersham) in 0.2 M sodium phosphate, pH 4, containing ⁇ O ⁇ mM EDTA, and 0.5% BSA. The quenched reactions are allowed to stand for 2 hours before analysis on a Packard TopCount scintillation counter.
- compositions or inhibitors are prepared as concentrated solutions in 100% dimethyl sulfoxide and then diluted 25-fold into the enzyme 0 assay mixture.
- IC50 values are determined with Ras peptide near KM concentrations. Enzyme and substrate concentrations for inhibitor IC 50 determinations are as follows: 75 pM GGTase-I, 1.6 ⁇ M Ras peptide, 100 nM geranylgeranyl diphosphate.
- the cell line used in this assay is a v-ras line derived from either Ratl or NIH3T3 cells, which expressed viral Ha-ras p21.
- the 0 assay is performed essentially as described in DeClue, J.E. et al., Cancer Research ⁇ l:712-717, (1991). Cells in 10 cm dishes at ⁇ 0-7 ⁇ % confluency are treated with the test compound or composition (final concentration of solvent, methanol or dimethyl sulfoxide, is 0.1%).
- the cells are labeled in 3 ml methionine-free DMEM supple-mented with ⁇ 10% regular DMEM, 2% fetal bovine serum and 400 mCi[3 S]methionine (1000 Ci/mmol). After an additional 20 hours, the cells are lysed in 1 ml lysis buffer (1% NP40/20 mM HEPES, pH 7.5/5 mM MgCl2/lmM DTT/10 mg/ml aprotinen/2 mg/ml leupeptin/2 mg/ml antipain/0.5 mM PMSF) and the lysates cleared by centrifugation at 100,000 x g for 4 ⁇ min.
- 1 ml lysis buffer 1% NP40/20 mM HEPES, pH 7.5/5 mM MgCl2/lmM DTT/10 mg/ml aprotinen/2 mg/ml leupeptin/2 mg/ml antipain/0.5 mM PMSF
- the immunoprecipitates are washed four times with IP buffer (20 nM HEPES, pH 7.5/1 mM EDTA/1% Triton X- 100.0.5% deoxycholate/0J%/SDS/0J M NaCl) boiled in SDS-PAGE sample buffer and loaded on 13% acrylamide gels. When the dye front reached the bottom, the gel is fixed, soaked in Enlightening, dried and autoradiographed. The intensities of the bands corresponding to farnesylated and nonfarnesylated ras proteins are compared to determine the percent inhibition of farnesyl transfer to protein.
- IP buffer (20 nM HEPES, pH 7.5/1 mM EDTA/1% Triton X- 100.0.5% deoxycholate/0J%/SDS/0J M NaCl
- Rat 1 cells transformed with either v-ras, v-raf, or v-mos are seeded at a density of 1 x 10 4 cells per plate (3 ⁇ mm in diameter) in a 0.3% top agarose layer in medium A (Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum) over a bottom agarose layer (0.6%). Both layers contain 0.1% methanol or an appropriate concentration of the compound or instant composition (dissolved in methanol at 1000 times the final concentration used in the assay).
- the cells are fed twice weekly with 0.5 ml of medium A containing 0.1% methanol or the concentration of the instant compound. Photomicrographs are taken 16 days after the cultures are seeded and comparisons are made.
- SEAP reporter plasmid pDSElOO The SEAP reporter plasmid, pDSElOO was constructed by ligating a restriction fragment containing the SEAP coding sequence into the plasmid pCMV-RE-AKI.
- the SEAP gene is derived from the plasmid pSEAP2-Basic (Clontech, Palo Alto, CA).
- the plasmid pCMV- ⁇ RE-AKI was constructed by Deborah Jones (Merck) and contains ⁇ sequential copies of the 'dyad symmetry response element' cloned upstream of a 'CAT-TATA' sequence derived from the cy tome alo virus immediate early promoter.
- the plasmid also contains a bovine growth hormone poly-A sequence.
- the plasmid, pDSElOO was constructed as follows. A restriction fragment encoding the SEAP coding sequence was cut out of the plasmid pSEAP2-Basic using the restriction enzymes EcoRl and Hpal. The ends of the linear DNA fragments were filled in with the Klenow fragment of E. coli DNA Polymerase I. The 'blunt ended' DNA l ⁇ containing the SEAP gene was isolated by electrophoresing the digest in an agarose gel and cutting out the 1694 base pair fragment. The vector plasmid pCMV- RE-AKI was linearized with the restriction enzyme Bgl-II and the ends filled in with Klenow DNA Polymerase I. The SEAP DNA fragment was blunt end ligated into the pCMV-RE-AKI vector
- the SEAP repotrer plasmid, pDSElOl is also constructed by 30 ligating a restriction fragment containing the SEAP coding sequence into the plasmid pCMV-RE-AKI.
- the SEAP gene is derived from plasmid pGEM7zf(-)/SEAP.
- the plasmid pDSElOl was constructed as follows: A restriction fragment containing part of the SEAP gene coding sequence was cut out of the plasmid pGEM7zf(-)/SEAP using the restriction enzymes Apa I and Kpnl. The ends of the linear DNA fragments were chewed back with the Klenow fragment of E. coli DNA Polymerase I. The "blunt ended" DNA containing the truncated SEAP gene was isolated by 5 electrophoresing the digest in an agarose gel and cutting out the 1910 base pair fragment. This 1910 base pair fragment was ligated into the plasmid pCMV-RE-AKI which had been cut with Bgl-II and filled in with E. coli Klenow fragment DNA polymerase.
- the plasmid pCMV-RE-AKI is derived from plasmid pCMVIE- AKI-DHFR (Whang , Y., Silberklang, M., Morgan, A., Munshi, S., Lenny, A.B., Ellis, R.W., and Kieff, E. (1987) J. Virol., 61, 1796-1807) by removing an EcoRI fragment containing the DHFR and Neomycin markers.
- the plasmid pGEM7zf(-)/SEAP was constructed as follows.
- the SEAP 0 gene was PCRed, in two segments from a human placenta cDNA library (Clontech) using the following oligos.
- Sense strand N-terminal SEAP ⁇ ' GAGAGGGAATTCGGGCCCTTCCTGCAT ⁇ GCTGCTGCTGCTGCTGCTGCTGGGC 3' (SEQ.ID.N0..3)
- Antisense strand N-terminal SEAP ⁇ ' GAGAGAGCTCGAGGTTAACCCGGGT GCGCGGCGTCGGTGGT 3' (SEQ.ID.NO. :4) 0
- Sense strand C-terminal SEAP ⁇ ' GAGAGAGTCTAGAGTTAACCCGTGGTCC CCGCGTTGCTTCCT 3' (SEQ.ID.N0..5)
- Antisense strand C-terminal SEAP ⁇ ' GAAGAGGAAGCTTGGTACCGCCACTG GGCTGTAGGTGGTGGCT 3' (SEQ.ID.N0..6)
- the N-terminal oligos (SEQ.ID.NO.: 4 and SEQ.ID.NO.: ⁇ ) were used to generate a l ⁇ 60 bp N-terminal PCR product that contained EcoRI and Hpal restriction sites at the ends.
- the Antisense N-terminal oligo (SEQ.ID.NO.: 4) introduces an internal translation STOP codon within the SEAP gene along with the Hpal site.
- pGEM7zf(-)/SEAP contains the coding sequence for the SEAP gene from amino acids.
- a constitutively expressing SEAP plasmid pCMV-SEAP 2 ⁇ An expression plasmid constitutively expressing the SEAP protein was created by placing the sequence encoding a truncated SEAP gene downstream of the cy tome galo virus (CMV) IE-1 promoter.
- the expression plasmid also includes the CMV intron A region ⁇ ' to the SEAP gene as well as the 3' untranslated region of the bovine growth hormone 30 gene 3' to the SEAP gene.
- the plasmid pCMVIE-AKI-DHFR (Whang et al, 1987) containing the CMV immediate early promoter was cut with EcoRI generating two fragments. The vector fragment was isolated by agarose electrophoresis and re ligated. The resulting plasmid is named pCMV- AKI.
- the cytomegalovirus intron A nucleotide sequence was inserted downstream of the CMV IE1 promter in pCMV-AKI.
- the intron A sequence was isolated from a genomic clone bank and subcloned into pBR322 to generate plasmid pl6T-286.
- the intron A sequence was mutated at nucleotide 1856 (nucleotide numbering as in Chapman, B.S., Thayer, R.M., Vincent, K.A. and Haigwood, N.L., Nuc.Acids Res. 19, 3979-3986) to remove a Sad restriction site using site directed mutagenesis.
- the mutated intron A sequence was PCRed from the plasmid pl6T-287 using the following oligos.
- Sense strand 5' GGCAGAGCTCGTTTAGTGAACCGTCAG 3' (SEQ.ID.NO.: 7)
- Antisense strand 5' GAGAGATCTCAAGGACGGTGACTGCAG 3' l ⁇ (SEQ.ID.NO.: 8)
- the SEAP gene is cut out of plasmid pGEM7zf(-)/SEAP (described above) using EcoRI and Hindlll. The fragment is filled in with Klenow DNA polymerase and the 1970 base pair fragment isolated from the vector fragment by agarose gel electrophoresis.
- the pCMV-AKI-InA vector is
- pCMV-SEAP contains a modified SEAP sequence downstream of the cytomegalovirus immediately early promoter IE-1 and intron A sequence and upstream of the bovine growth hormone poly-A sequence.
- the plasmid expresses SEAP in a constitutive manner when transfected into mammalian cells.
- a DNA fragment containing viral-H-r ⁇ s can be PCRed from plasmid "H-l” (Ellis R. et al. J. Virol. 36, 408, 1980) or "HB-11 (deposited in the ATCC under Budapest Treaty on August 27, 1997, and designated ATCC 209,218) using the following oligos.
- the sense strand oligo also optimizes the 'Kozak' translation initiation sequence immediately 5' to the ATG start site.
- cysteine 186 would be mutated to a serine by substituting a G residue for a C residue in the C- terminal antisense oligo.
- the PCR primer oligos introduce an Xhol site at the ⁇ ' end and a Xbal site at the 3'end.
- the Xhol-Xbal fragment can be ligated into the mammalian expression plasmid pCI (Promega) cut with Xhol and Xbal. This results in a plasmid in which the recombinant myr- viral-H-ras gene is constitutively transcribed from the CMV promoter of the pCI vector. Cloning of a viral-H-r ⁇ s-CVLL expression plasmid
- a viral-H-r ⁇ s clone with a C-terminal sequence encoding the amino acids CVLL can be cloned from the plasmid "H-l” (Ellis R. et al. J. Virol. 36, 408, 1980) or "HB-11 (deposited in the ATCC under Budapest Treaty on August 27, 1997, and designated ATCC 209,218) by PCR using the following oligos.
- Antisense strand
- the sense strand oligo optimizes the 'Kozak' sequence and adds an Xhol site.
- the antisense strand mutates serine 189 to leucine and adds an Xbal site.
- the PCR fragment can be trimmed with Xhol and Xbal and ligated into the Xhol -Xbal cut vector pCI (Promega). This results in a plasmid in which the mutated viral-H-r ⁇ s-CVLL gene is constitutively transcribed from the CMV promoter of the pCI vector.
- the human c-H-r ⁇ s gene can be PCRed from a human cerebral cortex cDNA library (Clontech) using the following oligonucleotide primers.
- Antisense strand 5'-GAGAGTCGACGCGTCAGGAGAGCACACACTTGC-3' (SEQ.ID.NO.:
- the primers will amplify a c-H-r ⁇ s encoding DNA fragment with the primers contributing an optimized 'Kozak' translation start sequence, an EcoRI site at the N-terminus and
- c-H-ras fragment can be ligated ligated into an EcoRI -Sal I cut mutagenesis vector pAlter-1 (Promega). Mutation of glutamine-61 to a leucine can be accomplished ⁇ using the manufacturer's protocols and the following oligonucleotide:
- the mutated c-H-r ⁇ s-Leu61 can be excised from the pAlter-1 vector, using EcoRI and Sal I, and be directly ligated into the vector pCI
- the human c-N-r ⁇ s gene can be PCRed from a human cerebral cortex cDNA library (Clontech) using the following oligonucleotide primers. 0
- the primers will amplify a c-N-r ⁇ s encoding DNA fragment with the primers contributing an optimized 'Kozak' translation start 0 sequence, an EcoRI site at the N-terminus and a Sal I stite at the C- terminal end.
- the c-N-ras fragment can be ligated into an EcoRI -Sal I cut mutagenesis vector pAlter-1 (Promega). Mutation of glycine-12 to a valine can be accomplished using the manufacturer's protocols and the following ⁇ oligonucleotide:
- the mutated c-N-r ⁇ s- Val- 12 can be excised from the pAlter-1 vector, using EcoRI and Sal I, and be directly ligated into the vector pCI 5 (Promega) which has been digested with EcoRI and Sal I.
- the new recombinant plasmid will constitutively transcribe c-N-r ⁇ s-Val-12 from the CMV promoter of the pCI vector.
- the human c-K-r ⁇ s gene can be PCRed from a human cerebral cortex cDNA library (Clontech) using the following oligonucleotide primers.
- Antisense strand ⁇ '-CTCTGTCGACGTATTTACATAATTACACACTTTGTC-3' 20 (SEQ.ID.NO.: 20)
- the primers will amplify a c-K-r ⁇ s encoding DNA fragment with the primers contributing an optimized 'Kozak' translation start sequence, a Kpnl site at the N-terminus and a Sal I site at the C- 2 ⁇ terminal end.
- the c-K-r ⁇ s fragment can be ligated into a Kpnl -Sal I cut mutagenesis vector pAlter-1 (Promega). Mutation of cysteine-12 to a valine can be accomplished using the manufacturer's protocols and the following oligonucleotide:
- the mutated c-K-r ⁇ s- Val- 12 can be excised from the pAlter-1 3 ⁇ vector, using Kpnl and Sal I, and be directly ligated into the vector pCI (Promega) which has been digested with Kpnl and Sal I.
- the new recombinant plasmid will constitutively transcribe c-K-r ⁇ s-Val-12 from the CMV promoter of the pCI vector.
- the cells are washed with PBS and trypsinized with 1ml of 0.0 ⁇ % trypsin.
- the 1 ml of trypsinized cells is diluted into 10ml of phenol red free DMEM + 0.2% 2 ⁇ charcoal stripped calf serum + IX (Pen Strep, Glutamine and NEAA ).
- Transfected cells are plated in a 96 well microtiter plate (lOO ⁇ l/well) to which drug, diluted in media, has already been added in a volume of lOO ⁇ l. The final volume per well is 200 ⁇ l with each drug concentration repeated in triplicate over a range of half-log steps.
- the heat treated media is assayed for alkaline phosphatase by a luminescence assay using the luminescence reagent CSPD®D (Tropix, Bedford, Mass.).
- a volume of ⁇ O ⁇ l media is combined with 200 ⁇ l of CSPD cocktail and incubated for 60 minutes at room temperature.
- Luminesence is monitored using an ML2200 microplate luminometer (Dynatech). Luminescence reflects the level of activation of the fos reporter construct stimulated by the transiently expressed protein.
- PSN-1 human pancreatic carcinoma
- Subconfluent cells in 100 mm dishes are fed with 3. ⁇ ml of media (methionine-free RPMI supplemented with 2% fetal bovine serum or cysteine-free/methionine-free DMEM supplemented with 0.03 ⁇ ml of 200 mM glutamine (Gibco), 2% fetal bovine serum, respectively) containing the desired concentration of l ⁇ test compound, lovastatin or solvent alone.
- Cells treated with lovastatin ( ⁇ -10 ⁇ M), a compound that blocks Ras processing in cells by inhibiting a rate-limiting step in the isoprenoid biosynthetic pathway serve as a positive control.
- Test compounds or compositions are prepared as lOOOx concentrated solutions in DMSO to yield a final solvent concentration of
- the cells After introducing the label amino acid mixture, the cells are incubated at 37°C for an additional period of time (typically 6 to 24 hours). The media is then removed and the cells are washed once with
- the cells are scraped into 1 ml of cold PBS, collected by centrifugation (10,000 x g for 10 sec at room temperature), and lysed by vortexing in 1 ml of lysis buffer (1% Nonidet P-40, 20 mM HEPES, pH 7.5, 160 mM NaCl, 1 mM EDTA, 0.5% deoxycholate, 0.1% SDS, 1 mM DTT, 10 ⁇ g/ml AEBSF, 10 ⁇ g/ml aprotinin, 2 ⁇ g/ml
- lysis buffer 1% Nonidet P-40, 20 mM HEPES, pH 7.5, 160 mM NaCl, 1 mM EDTA, 0.5% deoxycholate, 0.1% SDS, 1 mM DTT, 10 ⁇ g/ml AEBSF, 10 ⁇ g/ml aprotinin, 2 ⁇ g/ml
- the appropriate volume of lysate is brought to 1 ml with lysis buffer lacking DTT and 8 ⁇ g of the pan Ras monoclonal antibody, Y13-259, added.
- the protein/antibody mixture is incubated on ice at 4°C for 24 hours.
- the immune complex is collected on pansorbin (Calbiochem) coated with rabbit antiserum to rat IgG (Cappel) ⁇ by tumbling at 4°C for 4 ⁇ minutes.
- the pellet is washed 3 times with 1 ml of lysis buffer lacking DTT and protease inhibitors and resuspended in 100 ⁇ l elution buffer (10 mM Tris pH 7.4, 1% SDS).
- the Ras is eluted from the beads by heating at 9 ⁇ °C for ⁇ minutes, after which the beads are pelleted by brief centrifugation (l ⁇ ,000 x g for 30 sec. 0 at room temperature).
- the supernatant is added to 1 ml of Dilution Buffer 0.1% Triton X-100, 5 mM EDTA, 50 mM NaCl, 10 mM Tris pH 7.4) with 2 ⁇ g Kirsten-ras specific monoclonal antibody, c-K-ras Ab-1 (Calbiochem).
- the second protein/antibody mixture is incubated on ice 5 at 4°C for 1-2 hours.
- the immune complex is collected on pansorbin (Calbiochem) coated with rabbit antiserum to rat IgG (Cappel) by tumbling at 4°C for 45 minutes.
- the pellet is washed 3 times with 1 ml of lysis buffer lacking DTT and protease inhibitors and resuspended in Laemmli sample buffer.
- the Ras is eluted from the beads by heating at 0 95°C for 5 minutes, after which the beads are pelleted by brief centrifugation.
- the supernatant is subjected to SDS-PAGE on a 12% acrylamide gel (bis-acrylamide:acrylamide, 1:100), and the Ras visualized by fluorography.
- PSN-1 cells are seeded in 24-well assay plates. For each compound to be tested, the cells are treated with a minimum of seven concentrations in half-log steps. The final solvent (DMSO) concentration is 0.1%. A vehicle-only control is included on each 0 assay plate. The cells are treated for 24 hours at 37°C / 5% C0 2 .
- the growth media is then aspirated and the samples are washed with PBS.
- the cells are lysed with SDS-PAGE sample buffer containing 5% 2-mercaptoethanol and heated to 95°C for ⁇ minutes. After cooling on ice for 10 minutes, a mixture of nucleases is added to reduce viscosity of the samples.
- the plates are incubated on ice for another 10 minutes.
- the samples are loaded onto pre-cast 8% acrylamide gels and ⁇ electrophoresed at l ⁇ mA gel for 3-4 hours.
- the samples are then transferred from the gels to PVDF membranes by Western blotting.
- the membranes are blocked for at least 1 hour in buffer containing 2% nonfat dry milk.
- the membranes are then treated with a monoclonal antibody to HDJ-2 (Neomarkers Cat. # MS-22 ⁇ ), washed, 10 and treated with an alkaline phosphatase-conjugated secondary antibody.
- the membranes are then treated with a fluorescent detection reagent and scanned on a phosphorimager.
- the percent of total signal corresponding to the unprenylated species of HDJ is l ⁇ calculated by densitometry.
- Dose-response curves and IC ⁇ O values are generated using 4-parameter curve fits in SigmaPlot software.
- PSN-1 human pancreatic carcinoma cells are used for analysis of protein processing.
- Subconfluent cells in l ⁇ O mm dishes are fed with 20 ml of media (RPMI supplemented with l ⁇ % fetal bovine serum) containing the desired concentration of test composition, prenyl-
- the cells are incubated at 37°C for 24 hours, the media is then removed and the cells are washed twice with cold PBS.
- the cells are scraped into 2 ml of cold PBS, collected by centrifugation (10,000 x g for 5 min at 4°C) and frozen at -70 °C Cells are lysed by thawing and
- lysis buffer 50 mM HEPES, pH 7.2, ⁇ O mM NaCl, 1% CHAPS, 0.7 ⁇ g/ml aprotinin, 0.7 ⁇ g/ml leupeptin 300 ⁇ g/ml pefabloc, and 0.3 mM EDTA.
- the lysate is then centrifuged at 100,000 x g for 60 min at 4°C and the supernatant saved. The supernatant may be subjected to SDS- PAGE, HPLC analysis, and/or chemical cleavage techniques.
- the lysate is applied to a HiTrap-SP (Pharmacia Biotech) column in buffer A ( ⁇ O mM HEPES pH 7.2) and resolved by gradient in buffer A plus 1 M NaCl. Peak fractions containing Ki4B-Ras are pooled, diluted with an equal volume of water and immunoprecipitated with the pan Ras monoclonal antibody, Yl3-2 ⁇ 9 linked to agarose.
- the protein/antibody mixture is incubated at 4°C for 12 hours.
- the immune complex is washed 3 times with PBS , followed by 3 times with water.
- the Ras is eluted from the beads by either high pH conditions (pH>10) or by heating at 95°C for 5 minutes, after which the beads are pelleted by brief centrifugation.
- the supernatant may be subjected to SDS-PAGE, HPLC analysis, and/or chemical cleavage techniques.
- Rapl For immunoprecipitation of Rapl, samples of lysate supernatant containing equal amounts of protein are utilized. Protein concentration is determined by the bradford method utilizing bovine serum albumin as a standard. The appropriate volume of lysate is brought to 1 ml with lysis buffer lacking DTT and 2 ⁇ g of the Rapl antibody, Rapl/Krevl (121) (Santa Cruz Biotech), is added. The protein/antibody mixture is incubated on ice at 4°C for 1 hour. The immune complex is collected on pansorbin (Calbiochem) by tumbling at 4°C for 45 minutes.
- the pellet is washed 3 times with 1 ml of lysis buffer lacking DTT and protease inhibitors and resuspended in 100 ⁇ l elution buffer (10 mM Tris pH 7.4, 1% SDS).
- the Rapl is eluted from the beads by heating at 9 ⁇ °C for 5 minutes, after which the beads are pelleted by brief centrifugation (l ⁇ ,000 x g for 30 sec. at room temperature).
- the supernatant is added to 1 ml of Dilution Buffer (0.1% Triton X-100, ⁇ mM EDTA, ⁇ O mM NaCl, 10 mM Tris pH 7.4) with 2 ⁇ g Rapl antibody, Rapl/Krevl (121) (Santa Cruz Biotech).
- the second protein/antibody mixture is incubated on ice at 4°C for 1-2 hours.
- the immune complex is collected on pansorbin (Calbiochem) by tumbling at 4°C for 45 minutes.
- the pellet is washed 3 times with 1 ml of lysis buffer lacking DTT and protease inhibitors and resuspended in Laemmli sample buffer.
- Rapl is eluted from the beads by heating at 95°C for ⁇ minutes, after which the beads are pelleted by brief centrifugation. The supernatant is subjected to SDS-PAGE on a 12% acrylamide gel (bis- acrylamide: acrylamide, 1:100), and the Rapl visualized by fluorography.
- PSN-1 cells are passaged every 3-4 days in 10cm plates, splitting near-confluent plates 1:20 and 1:40. The day before the assay is set up, 5x 10 6 cells are plated on 15cm plates to ensure the same stage of confluency in each assay.
- the media for these cells is RPM1 1640 (Gibco), with l ⁇ % fetal bovine serum and lx Pen/Strep antibiotic mix.
- the day of the assay cells are collected from the 15cm plates by trypsinization and diluted to 400,000 cells/ml in media. 0.5ml of these diluted cells are added to each well of 24-well plates, for a final cell number of 200,000 per well. The cells are then grown at 37 C overnight.
- the compounds or compositions to be assayed are diluted in DMSO in 1/2-log dilutions. The range of final concentrations to be assayed is generally 0.1-100 ⁇ M. Four concentrations per compound is typical. The compounds are diluted so that each concentration is lOOOx of the final concentration (i.e., for a lO ⁇ M data point, a lOmM stock of the compound is needed).
- each lOOOx compound stock is diluted into 1ml media to produce a 2X stock of compound.
- a vehicle control solution (2 ⁇ L DMSO to 1ml media), is utilized.
- 0.5 ml of the 2X stocks of compound are added to the cells.
- the media is aspirated from the assay plates.
- Each well is rinsed with 1ml PBS, and the PBS is aspirated.
- 180 ⁇ L SDS- PAGE sample buffer (Novex) containing 5% 2-mercaptoethanol is added to each well.
- the plates are heated to 100°C for ⁇ ⁇ minutes using a heat block containing an adapter for assay plates.
- the plates are placed on ice.
- 20 ⁇ L of an RNAse/ DNase mix is added per well. This mix is lmg/ml DNasel (Worthington
- Each assay plate (usually 3 compounds, each in 4-point titrations, plus controls) requires one l ⁇ -well 14% Novex gel. 2 ⁇ l of each sample is loaded onto the gel. The gel is run at 15mA for about 3.5 hours. It is important to run the gel far enough so that there will l ⁇ be adequate separation between 21kd (Rapl) and 29kd (Rab6).
- the blocking solution is discarded and 20ml fresh blocking solution containing the anti Rap la antibody (Santa Cruz Biochemical SC1482) at 1:1000 (diluted in Western blocking buffer) and the anti
- each of two alkaline phosphatase conjugated antibodies (Alkaline phosphatase conjugated Anti-goat IgG and Alkaline phosphatase conjugated anti-rabbit IgG [Santa Cruz Biochemical]) is then added.
- the membrane is incubated for one hour and washed 3x as above.
- About 2ml per gel of the Amersham ECF detection reagent is placed on an overhead transparency (ECF) and the PVDF membranes are placed face-down onto the detection reagent.
- ECF overhead transparency
- Rapla Minimum Inhibitory Concentration is determined from the lowest concentration of compound or composition that produces a detectable Rapla Western signal.
- the Rapla antibody used recognizes only unprenylated/unprocessed Rapla, so that the precence of a detectable Rapla Western signal is indicative of inhibition of Rapla prenylation.
- mice 10 human Ha-r ⁇ s or Ki-r ⁇ s (10 cells/animal in 1 ml of DMEM salts) are injected subcutaneously into the left flank of 8-12 week old female nude mice (Harlan) on day 0.
- the mice in each oncogene group are randomly assigned to a vehicle, compound or combination treatment group. Animals are dosed subcutaneously starting on day 1 and daily for l ⁇ the duration of the experiment.
- the test combination composition or prenyl-protein transferase inhibitor may be administered by a continuous infusion pump.
- Compound, compound combination or vehicle is delivered in a total volume of 0.1 ml. Tumors are excised and weighed when all of the vehicle-treated animals exhibited lesions of
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Urology & Nephrology (AREA)
- Heart & Thoracic Surgery (AREA)
- Neurology (AREA)
- Vascular Medicine (AREA)
- Virology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Biomedical Technology (AREA)
- Cardiology (AREA)
- Neurosurgery (AREA)
- Ophthalmology & Optometry (AREA)
- Gastroenterology & Hepatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description


































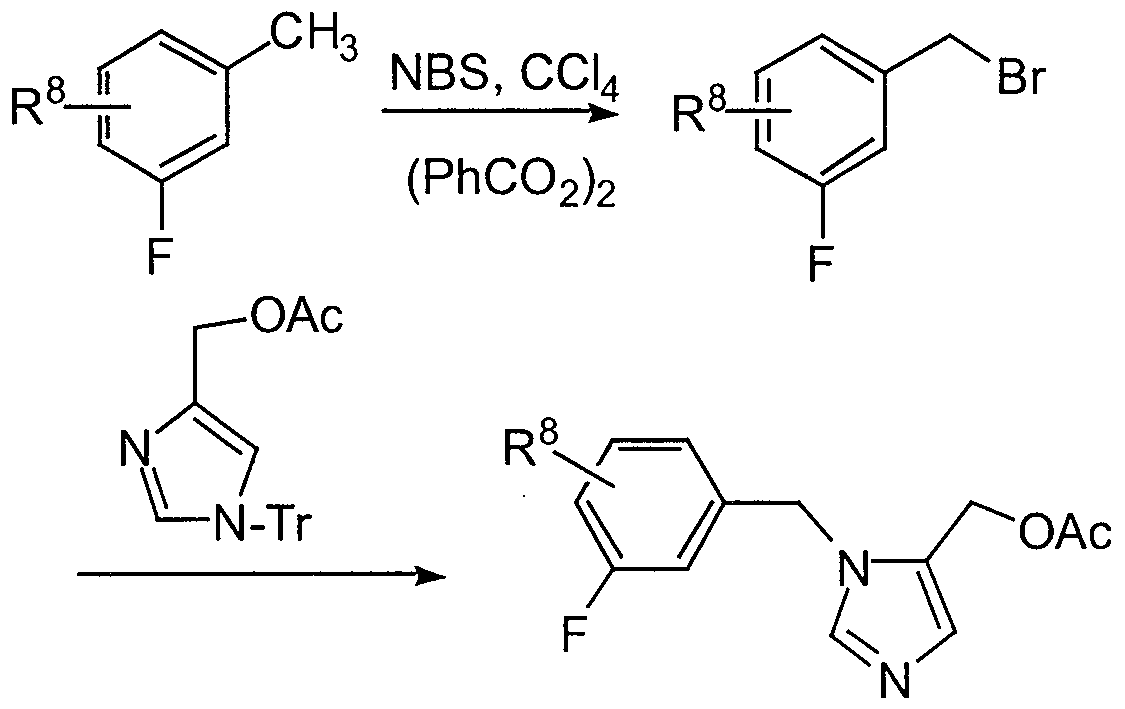



























































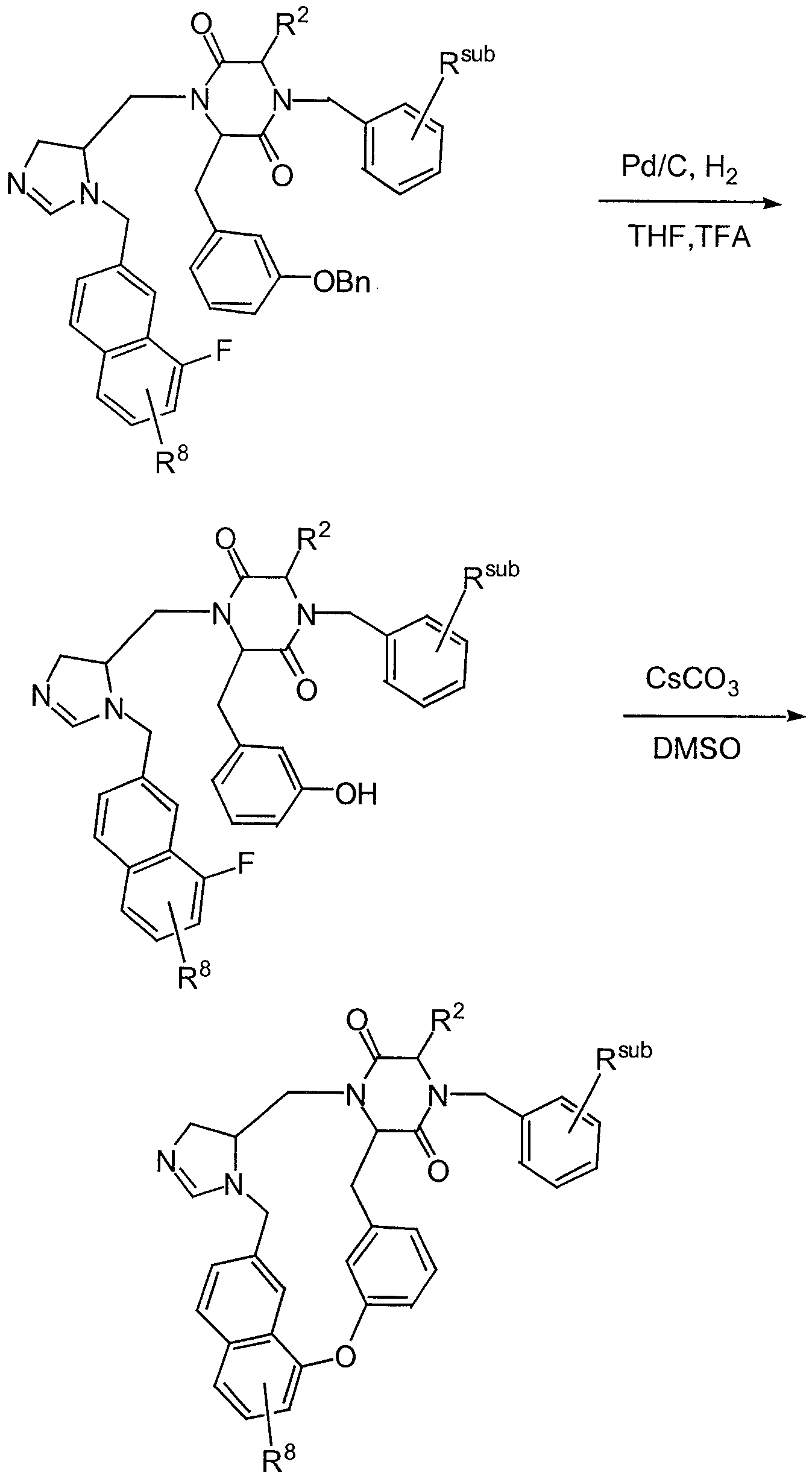














Claims













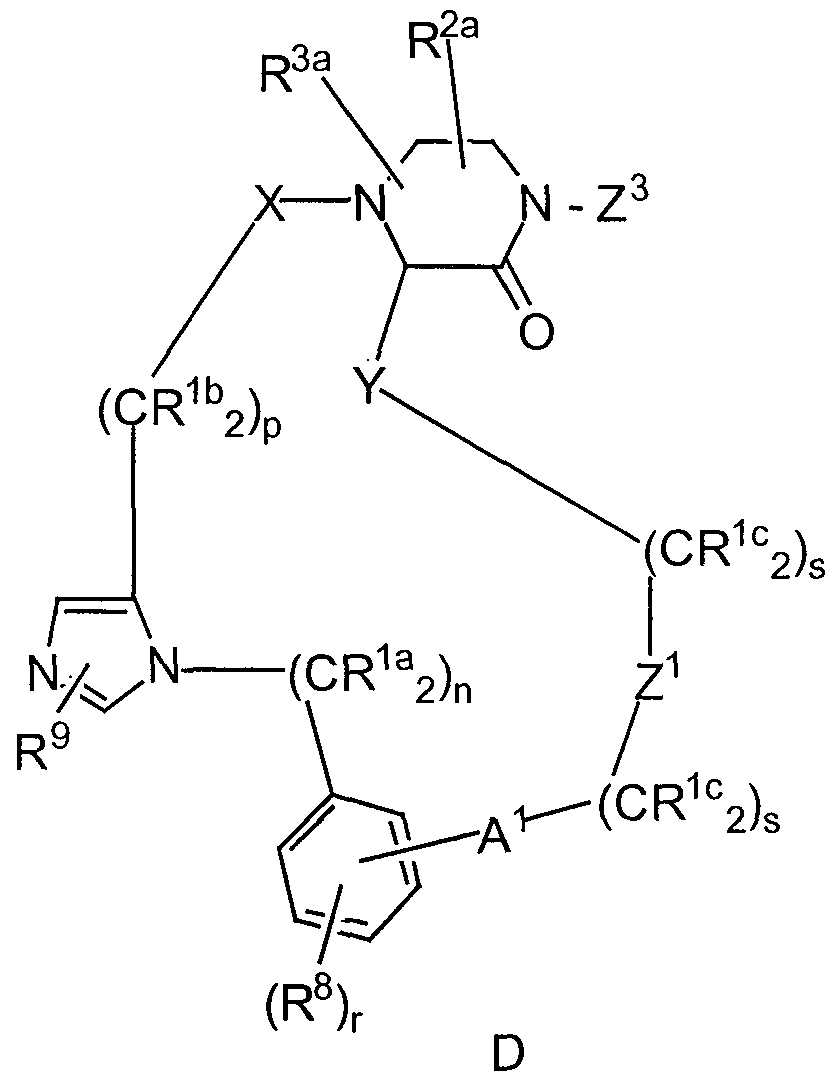




Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP99971320A EP1158984A4 (en) | 1998-10-29 | 1999-10-26 | Inhibitors of prenyl-protein transferase |
| JP2000579229A JP2002528504A (en) | 1998-10-29 | 1999-10-26 | Prenyl protein transferase inhibitors |
| AU12288/00A AU762440B2 (en) | 1998-10-29 | 1999-10-26 | Inhibitors of prenyl-protein transferase |
| CA002348703A CA2348703A1 (en) | 1998-10-29 | 1999-10-26 | Inhibitors of prenyl-protein transferase |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10617798P | 1998-10-29 | 1998-10-29 | |
| US60/106,177 | 1998-10-29 | ||
| GB9900148.9 | 1999-01-05 | ||
| GBGB9900148.9A GB9900148D0 (en) | 1999-01-05 | 1999-01-05 | Inhibitors of prenyl-protein transferase |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2000025788A1 true WO2000025788A1 (en) | 2000-05-11 |
Family
ID=26314955
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1999/024948 WO2000025788A1 (en) | 1998-10-29 | 1999-10-26 | Inhibitors of prenyl-protein transferase |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP1158984A4 (en) |
| JP (1) | JP2002528504A (en) |
| AU (1) | AU762440B2 (en) |
| CA (1) | CA2348703A1 (en) |
| WO (1) | WO2000025788A1 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8580257B2 (en) | 2008-11-03 | 2013-11-12 | Alethia Biotherapeutics Inc. | Antibodies that specifically block the biological activity of kidney associated antigen 1 (KAAG1) |
| US8937163B2 (en) | 2011-03-31 | 2015-01-20 | Alethia Biotherapeutics Inc. | Antibodies against kidney associated antigen 1 and antigen binding fragments thereof |
| US11084872B2 (en) | 2012-01-09 | 2021-08-10 | Adc Therapeutics Sa | Method for treating breast cancer |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8921533B2 (en) | 2011-07-25 | 2014-12-30 | Chromatin Technologies | Glycosylated valproic acid analogs and uses thereof |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5225528A (en) * | 1990-02-27 | 1993-07-06 | Merck & Co., Inc. | Cyclic hexapeptide oxytocin antagonists |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU741725B2 (en) * | 1997-08-27 | 2001-12-06 | Merck & Co., Inc. | Inhibitors of prenyl-protein transferase |
| JP2002519427A (en) * | 1998-07-02 | 2002-07-02 | メルク エンド カムパニー インコーポレーテッド | Prenyl protein transferase inhibitors |
-
1999
- 1999-10-26 WO PCT/US1999/024948 patent/WO2000025788A1/en active IP Right Grant
- 1999-10-26 JP JP2000579229A patent/JP2002528504A/en not_active Withdrawn
- 1999-10-26 EP EP99971320A patent/EP1158984A4/en not_active Withdrawn
- 1999-10-26 AU AU12288/00A patent/AU762440B2/en not_active Ceased
- 1999-10-26 CA CA002348703A patent/CA2348703A1/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5225528A (en) * | 1990-02-27 | 1993-07-06 | Merck & Co., Inc. | Cyclic hexapeptide oxytocin antagonists |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP1158984A4 * |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8580257B2 (en) | 2008-11-03 | 2013-11-12 | Alethia Biotherapeutics Inc. | Antibodies that specifically block the biological activity of kidney associated antigen 1 (KAAG1) |
| US9855291B2 (en) | 2008-11-03 | 2018-01-02 | Adc Therapeutics Sa | Anti-kidney associated antigen 1 (KAAG1) antibodies |
| US8937163B2 (en) | 2011-03-31 | 2015-01-20 | Alethia Biotherapeutics Inc. | Antibodies against kidney associated antigen 1 and antigen binding fragments thereof |
| US9393302B2 (en) | 2011-03-31 | 2016-07-19 | Alethia Biotherapeutics Inc. | Antibodies against kidney associated antigen 1 and antigen binding fragments thereof |
| US9828426B2 (en) | 2011-03-31 | 2017-11-28 | Adc Therapeutics Sa | Antibodies against kidney associated antigen 1 and antigen binding fragments thereof |
| US10597450B2 (en) | 2011-03-31 | 2020-03-24 | Adc Therapeutics Sa | Antibodies against kidney associated antigen 1 and antigen binding fragments thereof |
| US11084872B2 (en) | 2012-01-09 | 2021-08-10 | Adc Therapeutics Sa | Method for treating breast cancer |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2002528504A (en) | 2002-09-03 |
| EP1158984A1 (en) | 2001-12-05 |
| AU1228800A (en) | 2000-05-22 |
| EP1158984A4 (en) | 2003-05-21 |
| AU762440B2 (en) | 2003-06-26 |
| CA2348703A1 (en) | 2000-05-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6562823B1 (en) | Inhibitors of prenyl-protein transferase | |
| US6358985B1 (en) | Inhibitors of prenyl-protein transferase | |
| US20020052363A1 (en) | Inhibitors of prenyl-protein transferase | |
| AU3248600A (en) | Inhibitors of prenyl-protein transferases | |
| EP1014984A1 (en) | Inhibitors of prenyl-protein transferase | |
| US20020052380A1 (en) | Inhibitors of prenyl-protein transferase | |
| US6329376B1 (en) | Inhibitors of prenyl-protein transferase | |
| WO2001060368A1 (en) | Inhibitors of prenyl-protein transferase | |
| US6350755B1 (en) | Inhibitors of prenyl-protein transferase | |
| US20020193283A1 (en) | Inhibitors of prenyl-protein transferase | |
| WO2001045707A1 (en) | Inhibitors of prenyl-protein transferase | |
| US6632818B2 (en) | Inhibitors of prenyl-protein transferase | |
| US6316436B1 (en) | Inhibitors of prenyl-protein transferase | |
| EP1332134A2 (en) | Inhibitors of prenyl-protein transferase | |
| US6335343B1 (en) | Inhibitors of prenyl-protein transferase | |
| AU762440B2 (en) | Inhibitors of prenyl-protein transferase | |
| US6333335B1 (en) | Phenyl-protein transferase inhibitors | |
| US6355643B1 (en) | Inhibitors of prenyl-protein transferase | |
| US6413964B1 (en) | Inhibitors of prenyl-protein transferase | |
| US20040110764A1 (en) | Inhibitors of prenyl-protein transferase | |
| US20020022633A1 (en) | Inhibitors of prenyl-protein transferase | |
| US6534506B2 (en) | Inhibitors of prenyl-protein transferase | |
| WO2001045704A1 (en) | Inhibitors of prenyl-protein transferase | |
| US6380228B1 (en) | Inhibitors of prenyl-protein transferase | |
| WO2001007051A1 (en) | Prenyl-protein transferase inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref country code: AU Ref document number: 2000 12288 Kind code of ref document: A Format of ref document f/p: F |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AL AM AT AU AZ BA BB BG BR BY CA CH CN CR CU CZ DE DK DM EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 12288/00 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1999971320 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 2000 579229 Kind code of ref document: A Format of ref document f/p: F |
|
| ENP | Entry into the national phase |
Ref document number: 2348703 Country of ref document: CA Ref country code: CA Ref document number: 2348703 Kind code of ref document: A Format of ref document f/p: F |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1999971320 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1999971320 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 12288/00 Country of ref document: AU |