WO1998056364A1 - IMMEDIATE RELEASE pH-INDEPENDENT SOLID DOSAGE FORM OF (+)- OR (-)-CISAPRIDE - Google Patents
IMMEDIATE RELEASE pH-INDEPENDENT SOLID DOSAGE FORM OF (+)- OR (-)-CISAPRIDE Download PDFInfo
- Publication number
- WO1998056364A1 WO1998056364A1 PCT/EP1998/003495 EP9803495W WO9856364A1 WO 1998056364 A1 WO1998056364 A1 WO 1998056364A1 EP 9803495 W EP9803495 W EP 9803495W WO 9856364 A1 WO9856364 A1 WO 9856364A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cisapride
- dosage form
- tablet
- tartrate
- solid dosage
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4468—Non condensed piperidines, e.g. piperocaine having a nitrogen directly attached in position 4, e.g. clebopride, fentanyl
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
- A61K9/2018—Sugars, or sugar alcohols, e.g. lactose, mannitol; Derivatives thereof, e.g. polysorbates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/286—Polysaccharides, e.g. gums; Cyclodextrin
- A61K9/2866—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4858—Organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
Definitions
- the present invention concerns solid dosage forms of particular salts of (+)- or
- (-)-cisapride more particularly the (L)-tartrate, (D)-tartrate, sulfate or citrate, which avoid drug-food interaction and which allow co-medication of agents that increase the pH of the stomach.
- the invention further relates to solid oral dosage forms suitable for rapid disintegration and dissolution.
- the present invention also concerns tablets which can be prepared via direct compression.
- the absorption and bioavailability of any particular therapeutic agent can be affected by numerous factors when dosed orally.
- factors include the presence of food in the gastrointestinal (GI) tract because, in general, the gastric residence time of a drug is usually significantly longer in the presence of food than in the fasted state.
- the bioavailability of a drug is affected beyond a certain point due to the presence of food in the GI tract, the drug is said to exhibit a "food effect" or show a drug-food interaction.
- the risk involved with taking drugs exhibiting a food- effect derives from the fact that absorption into the bloodstream may be adversely affected by not taking the drug on the correct point in time so that the patient risks insufficient absorption to remedy the condition for which the drug was administered.
- Cisapride has the following structural formula :
- Cisapride is a racemic mixture of two enantiomers. Cisapride has excellent gastrointestinal motility stimulating properties and is reported to be devoid of antidopaminergic activity. Its utility in a variety of gastro-intestinal disorders has already been reported extensively. It is currently being marketed as a medicine to treat gastro-oesophageal reflux disorders, inter alia oesophagitis, gastroparesis, negative upper digestive discomfort and intestinal pseudo-obstruction.
- Cisapride monohydrate is currently commercially available as tablets, suspension and granules under registered tradenames, such as PREPULSIDTM, PROPULSIDTM, PROPULSINTM, ACENALINTM, ALIMIXTM (this list is not comprehensive).
- Cisapride in its monohydrate form has a pH-dependent solubility and dissolution profile. Hence the bioavailability of cisapride or cisapride monohydrate is pH dependent. Cisapride monohydrate has a low solubility and low dissolution when present in a neutral or basic environment. Therefore, the information leaflet of cisapride monohydrate mentions that the drug should be taken 15 to 30 minutes before meals. The rationale being that the solid dosage form comprising cisapride monohydrate arrives in a more or less empty stomach, where the pH is reasonably low and hence the cisapride can dissolve.
- cisapride monohydrate shows a food effect, which can be expressed as the ratio between the AUC in fed state over AUC in fasted state.
- AUC is the abbreviation of Area Under the Curve, which is an indication of the amount of active ingredient that is present in the blood.
- the ratio for cisapride monohydrate of AUC in fed state over AUC in fasted state is about 1.35 (p > 0.01).
- a drug showing no food effect would have a ratio of 1 (in an ideal case).
- a patient taking cisapride monohydrate has to follow the above described regimen quite strictly in order to create the optimum conditions for high bioavailability of cisapride monohydrate and consequently to maximise the benefit from the drug taken.
- Patients do not always have the necessary discipline to take their medication at the optimum point in time. Consequently, a dosage form that would make the bioavailability independent of a meal (or from any other event for that matter) would mean a serious improvement over the prior art oral dosage forms of cisapride monohydrate and reduce the extent of variability in absorption between patients.
- Cisapride monohydrate is prescribed for infants (children up to 1 year).
- the fact that cisapride monohydrate has to be administered up to 30 minutes before the meal implies that parents often have to wake up the child, administer the cisapride monohydrate and then wait half an hour to feed the child.
- This is very unpractical and it would be desirable to find a form of cisapride which is suitable for administering to infants just before the meal or even after the meal or better still, completely independent of when the meal is taken.
- cisapride is used to treat people having stomach or esophagus problems. Often, these patients receive co-medication to increase the pH of the stomach.
- co-medication are antacids, such as aluminum containing antacids, e.g. Al(OH)3, calcium containing antacids, e.g. CaCO3, or magnesium containing antacids, e.g. Mg(OH)2; ⁇ -antagonists, e.g.
- proton pump inhibitors e.g. omeprazole, lansoprazole, rabeprazole. At the moment the preferred co-medication prescribes proton pump inhibitors.
- WO 94/01112 and WO 94/01111 published on 20 January 1994, disclose very generally methods of using (-)-cisapride respectively ( ⁇ )-cisapride as well as the therapeutically acceptable salts thereof for the treatment of gastro-esophageal reflux disease and other disorders.
- WO 95/34284 published on 21 December 1995, mentions pharmaceutical preparations with a hydrophobic active substance, amongst others cisapride, and an effervescent system.
- EP 670160 published on 6 September 1995, discloses a granular product or tablet containing an effervescent system and an active pharmaceutical substance, and in particular cisapride effervescent tablets are described.
- WO 95/01803 published on January 19, 1995 discloses combinations of H2 antagonists and gastrointestinal motility agents, e.g. cisapride.
- the disadvantage of said prior art combination is that ⁇ -antagonists, but also antacids and especially proton pump inhibitors can cause a considerable raise in the pH in the stomach.
- antacids the pH of the stomach, which is normally between 1 and 1.5, can raise to about 4.5 and with proton pump inhibitors the pH of the stomach can raise to about 6.5.
- cisapride monohydrate does not dissolve quickly enough to give fast appropriate relief.
- WO 96/14070 discloses a matrix-formulation wherein cisapride-(L)-tartrate is embedded in a mixture of viscous polymers. These prior art matrix formulations, however, do not disintegrate and dissolve as rapidly as is required for the solid oral dosage forms of the present invention. On the contrary, these matrix formulations are designed to give a sustained release of cisapride over a much longer period of time.
- the solid dosage forms comprising a salt of (+)- or (-)- cisapride with an acid selected from sulfuric acid, (L)-tartaric acid, (D)-tartaric acid or citric acid, preferably (+)- or (-)-cisapride-(L)-tartrate suitable for rapid dissolution.
- the formulations are preferably suitable for rapid disintegration as well as dissolution.
- the preferred formulations are oral solid dosage forms.
- suitable for rapid dissolution refers to the fact that from the solid dosage forms of the present invention the active ingredient can dissolve for more than 60 % within 1 hour in a pH range from 1 to 7. Said dissolution can be measured according to standard methods described in the European Pharmacoeipea or as set forth in USP test ⁇ 711> in a USP-2 dissolution apparatus. This latter test is described in US Pharmacopeia XXII, pp 1578-1579.
- salts of (+)- or (-)-cisapride have a better dissolution in artificial gastric juice than others : those salts are the (L)-tartrate, (D)-tartrate, the sulfate and the citrate. Moreover, said salts of (+)- or (-)-cisapride show a dissolution profile which is substantially pH independent.
- solid oral dosage forms generally refers to tablets (both swallowable-only and chewable forms) and capsules. Hence, the present composition of salt forms of
- (+)- or (-)-cisapride may be formulated into tablets, caplets, gelcaps or capsules.
- This invention encompasses formulations comprising the salts of (+)- or (-)-cisapride according to the present invention and further comprising a substance which can influence the acidity of the stomach.
- Said substance can be any medication that increases the pH of the stomach (in other words : renders the stomach more basic).
- antacids, ⁇ -antagonists or proton pump inhibitors examples of just medication that increases the pH of the stomach should be mentioned antacids, ⁇ -antagonists or proton pump inhibitors.
- the invention also relates to products containing any of the salt forms of (+)- or (-)-cisapride of the present invention, preferably (+)- or (-)-cisapride-(L)-tartrate, and an antacid or an ⁇ -antagonist or especially a proton pump inhibitor as a combined preparation for simultaneous, separate or sequential use in treating gastrointestinal disorders, especially gastro-esophageal reflux related conditions.
- the formulations of the present invention may optionally include an anti-flatulent, such as simethicone, alpha-D-galactosidase and the like.
- an anti-flatulent such as simethicone, alpha-D-galactosidase and the like.
- Said products comprising combinations of antacids, H 2 antagonists or proton pump inhibitors on the one hand and the (+)- or (-)-cisapride salt forms on the other hand, optionally further combined with an anti flatulent provide the dual action approach to the treatment of gastrointestinal disorders as described in WO 95/01803, i.e. the salt of (+)- or (-)-cisapride as gastrointestinal motility agent offers an enhanced motility while the antacid, the ⁇ -antagonist or the proton pump inhibitor offers a systemic effect of reduced acid production.
- the present invention therefore further provides a method of preventing, treating and relieving heartburn, indigestion, sour stomach, overindulgence, gastro esophageal reflux, constipation, dyspepsia and other gastrointestinal disorders, and gastrointestinal disorders, and optionally flatulence, in mammals, including humans, in need of treatment thereof, comprising administering to such organism : (i) an therapeutically effective amount of an antacid, an H 2 antagonist or a proton pump inhibitor, and (ii) a therapeutically effective amount of a salt form of (+)- or (-)-cisapride of the present invention, and optionally (iii) a therapeutically effective amount of an anti-flatulent, in particular simethicone or alpha-D-galactosidase (ADG).
- ADG alpha-D-galactosidase
- H 2 antagonists such as famotidine, ranitidine and cimetidine are also commercially available under different Tradenames.
- Proton pump inhibitors such as, omeprazole, lansoprazole, rabeprazole and the like are either commercially available or known in the art.
- Simethicone is a well-known and commercially available anti flatulent.
- Alpha-D- galactosidase ADG is a commercially available enzyme preparation used to hydrolyze indigestible sugars found in beans or bean products. The active ingredients, other than the salts of (+)- or (-)-cisapride are therefore readily commercially available.
- the dosages of each of the active ingredients may vary depending upon the severity of the condition and the particular biochemistry and need of the patient.
- the dosages of the active ingredients may also vary depending upon whether the active ingredients are administered in tablet or liquid form or via some other suitable delivery method. A physician or clinician may readily determine suitable dosages.
- the tablets or capsules according to the invention comprise the salt forms of (+)- or (-)-cisapride, preferably (+)- or (-)-cisapride-(L)-tartrate, which are preferably in a microfine or micronized form for some uses.
- Micronized forms of the salt forms of (+)- or (-)-cisapride, especially (+)- or (-)-cisapride-(L)-tartrate may be prepared by micronization techniques known in the art, e.g. by milling in appropriate mills and sieving through appropriate sieves.
- the specific surface area of said micronized material should at least amount to about 10 x 10 3 cm 2 /g (1 x 10 3 m 2 /kg), preferably the specific surface area should amount to more than 12 x 10 3 cm 2 /g (1.2 x 10 3 m 2 /kg), most preferably the specific area should amount to more than 14 x 10 3 cm 2 /g (1.4 x 10 3 m 2 /kg).
- the characteristics of the micronized salt forms of (+)- or (-)-cisapride, especially (+)- or (-)-cisapride-(L)-tartrate, expressed in a different way are as follows. At most 50% of the particles may have a diameter larger than 24 ⁇ m (i.e. 24 x 10 "6 m), hence the dlso has a maximum value of 24 ⁇ m (dl stands for diameter measured via laser diffraction).
- An interesting range of particle size expressed in dlso is from about 10 ⁇ m to about 150 ⁇ m. A more interesting range is from about 20 ⁇ m to 100 ⁇ m.
- the preferred dl 50 is about 24 ⁇ m.
- the preferred d ⁇ o is about 50 ⁇ m.
- the solid oral dosage form when in a unit dose form comprises the equivalent of about 0.1 mg to 100 mg of (+)- or (-)-cisapride in its base form, more particularly are envisaged dosage forms which contain the equivalent of about 5 mg, about 10 mg, and about 20 mg of (+)- or (-)-cisapride in it's base form.
- dosage forms containing about 6.5 mg, about 13 mg and about 26 mg of (+)- or (-)-cisapride-(L)-tartrate are envisaged.
- the excipients of the oral dosage forms of the present invention should be chosen to allow a fast dissolution of the active ingredients.
- Two solid oral dosage forms are preferred, i.e. tablets and capsules.
- excipients Especially with tablets, the choice of excipients is important.
- the excipients should allow a fast dissolution and on the other hand the excipients should allow a convenient industrial production of tablets with an appropriate aspect, an appropriate friability and sufficient hardness.
- Tablets should have an appropriate hardness and friability mainly because said tablets need to be manufactured on an industrial scale at presses with high speed and said tablets have to be packed or filled of in all kinds of containers. If the tablet has an insufficient hardness or is rather friable the tablet that is taken by the patient may be broken or parts of the tablet may have crumbled into powder. As a consequence of this insufficient hardness or friability the patient can no longer be certain that he is taking in the correct amount.
- the minimum required hardness of the tablets should be from about 1.5 daN (deca Newton) as measured by the test as described in the European Pharmacopoeia (3 th
- hardness amongst other properties, of tablets is dependent upon the shape of the tablets.
- Tablets may be circular or oblate or oblong or any other shape that is known in the art.
- the tablets may be scored. It should be noted that also the shape of the tablets may for instance have an influence on the disintegration rate.
- the disintegration time of the present tablets should be less than about 30 minutes, interestingly less than 20 minutes and more interestingly less than about 15 minutes.
- the preferred tablets even have a disintegration time of less than about 3 minutes, even less than about 1.5 minutes.
- the tablets of the present invention comprise tablet disintegrants, such as starch, pregelatinised starch, sodium starch glycolate (Explotab ® ), crosslinked povidone, crosslinked sodium carboxymethylcellulose, clays, microcrystalline cellulose (of the type available under the registered Trademark Avicel®), alginates, gums and others known in the art.
- tablet disintegrants such as starch, pregelatinised starch, sodium starch glycolate (Explotab ® ), crosslinked povidone, crosslinked sodium carboxymethylcellulose, clays, microcrystalline cellulose (of the type available under the registered Trademark Avicel®), alginates, gums and others known in the art.
- Tablets of the present invention preferably comprise as desintegrant crosslinked Carmellose Sodium (Carmellose Sodium is the British Approved Name of sodium carboxymethylcellulose, i.e. the sodium salt of a ether of cellulose, see Martindale , the Extra Pharmacopeia, 29 th edition, page 1433). Said crosslinked Carmellose Sodium is referred to as Croscarmellose Sodium (USP NF, 1995 Edition, page 2238).
- the disintegrant may be present in an amount of about 2 % (w/w) to about 15 % (w/w).
- An interesting range for the disintegrant is from about 3 % (w/w) to about 10 % (w/w). When percentages are used, these percentages are weight per weight (w/w) and represent the ratio (in percent) of the ingredient or the excipient based on the total weight of the tablet (or in the case of coated tablets of the tablet core).
- the "tablet core” is the tablet without the coating.
- internal phase refers to the composition of the granules and the term “external phase” refers to the composition of the compression mixture. It was observed that tablets with disintegrants in the internal and the external phase showed a better disintegration and a better dissolution profile.
- the tablet may further be formulated to include a variety of conventional excipients, depending on the exact formulation, such as binders, flavorings, buffers, diluents, colors, lubricants, sweetening agents, and glidants. Some excipients can serve multiple purposes.
- flavors may be incorporated in the composition, which may be chosen from synthetic flavor oils and flavoring aromatics and/or natural oils, extracts from plant leaves, flowers, fruits and so forth and combinations thereof. These may include cinnamon oil, oil of wintergreen, peppermint oils, bay oil, anise oil, eucalyptus, thyme oil. Also useful as flavors are vanilla, citrus oil, including lemon, orange, grape, lime and grapefruit, and fruit essences, including apple, banana, pear, peach, strawberry, raspberry, cherry, plum, pineapple, apricot and so fort. The amount of flavor may depend on a number of factors including the organoleptic effect desired. Generally the flavor will be present in an amount from about 0.5 % (w/w) to about 3.0 % (w/w), when a flavor is used.
- a variety of materials may be used as fillers or diluents. Examples are spray-dried or anhydrous lactose, sucrose, dextrose, mannitol, sorbitol, starch, cellulose (e.g. micro- crystalline cellulose; Avicel), dihydrated or anhydrous dibasic calcium phosphate, and others known in the art.
- a tablet can comprise one single filler or diluent or a mixture of fillers or diluents. For instance, a mixture of lactose and micro crystalline cellulose can be used. Lactose is used as a pure diluent, while microcrystalline cellulose is a filler that has the property of yielding tablets with an appropriate hardness and it has disintegrant properties because cellulose fibers swell in contact with water.
- lactose is lactose monohydrate DC which corresponds with Pharmatose DCL 11 that is commercially available from DMN International, The Netherlands, said lactose monohydrate DC is spray-dried lactose monohydrate.
- Fillers or diluents may be present in a range from about 50 % (w/w) to about 95 % (w/w) based on the total weight of the tablet or tablet core. Interestingly the amount of fillers or diluents range from about 65 % (w/w) to about 90 % (w/w). Preferably, the amount of fillers or diluents range from about 66 % (w/w) to about 86 % (w/w).
- a spray-dried mixture of lactose monohydrate and microcrystalline cellulose in a ratio of about 75 % by weight of lactose monohydrate and about 25 % by weight of microcrystalline cellulose can be used.
- This mixture is commercially available under the registered tradename MICROCELAC®.
- This spray-dried mixture of lactose monohydrate and microcrystalline cellulose has the advantage that it will promote ordered mixing, which improves the content uniformity of the tablets.
- the solid oral dosage forms contain relatively small amounts of active ingredient in a large amount of filler. In such conditions content uniformity can pose problems, i.e. the tablets prepared in the same batch may not all have the same content of active ingredient due to segregation during manufacturing.
- the spray-dried mixture of lactose monohydrate and microcrystalline cellulose has a porous structure wherein the active ingredient (+)- or (-)-cisapride-(L)- tartrate can be inserted, leading to ordered mixing and consequently a good content uniformity.
- Said MICROCELAC® is present in an amount ranging from about 80 % (w/w) to 95 % (w/w) based on the total weight of the tablet or the tablet core in the case of film coated tablets.
- the MICROCELAC® is present in an amount of about 87 % (w/w).
- Lubricants can also be employed in the manufacture of certain dosage forms, and will usually be employed when producing tablets.
- examples of lubricants are magnesium stearate, stearic acid, sodium stearyl fumarate, magnesium lauryl sulfate, hydrogenated vegetable oil and others known in the art.
- Preferred lubricants are magnesium stearate and sodium stearyl fumarate.
- Lubricants generally are present in an amount ranging from about 0.2 % (w/w) to 7.0 % (w/w) based on the total weight of the tablet or the tablet core in the case of film-coated tablets. Moreover, lubricants are present in amounts ranging from about 0.5 % (w/w) to about 3.0 % (w/w). Preferably, lubricants are present in amounts ranging from about 0.9 % (w/w) to about 1.25 % (w/w). Glidants are normally used in the manufacture of tablets and also capsules. Interesting glidants are calcium silicate, magnesium silicate, colloidal anhydrous silica or talc. Mixtures of glidants may also be used.
- Preferred glidant for the tablet core or the capsule of this invention is colloidal anhydrous silica.
- the type normally used is commercially available under the tradename Aerosil®.
- Glidants are normally present in an amount ranging from about 0.05 % (w/w) to about 1 % (w/w) based on the total weight of the tablet core content. The preferred amount of glidant is about 0.3 %.
- Binders may be acacia, alginic acid, carboxymethylcellulose (sodium), cellulose (microcrystalline), dextrin, ethylcellulose, gelatin, glucose (liquid), guar gum, hydroxypropyl cellulose, hydroxypropyl methylcellulose, methylcellulose, polyethylene oxide, povidone, starch (pregelatinized) or syrup.
- interesting binders are the hydroxypropyl methylcelluloses, especially the low-viscosity hydroxypropyl methylcelluloses.
- Preferred binder is hydroxypropyl methylcellulose 2910 of which a 2 % aqueous solution at 20°C has a viscosity of 15 mPa.s
- Coloring agents and pigments include titanium dioxide and/or dyes approved for use in food and pharmaceuticals.
- a coloring agent is an optional ingredient in the tablet of the present invention, but when used, the coloring agent will be present in an amount up to 3.5 % (w/w) based on the total tablet weight or the tablet core in the case of film-coated tablets.
- the coloring agent is present in the coating of the tablet, where again the coloring agent may be present in an amount ranging from 0.01 % (w/w) to about 10 % (w/w) based upon the total weight of the coating, an interesting range starts from about 0.20 % (w/w) up to about 7.5 % (w/w) based upon the total weight of the coating.
- tablet blends may be dry-granulated or wet-granulated before tabletting.
- (+)- or (-)-cisapride-(L)-tartrate it was possible to prepare tablets using direct compression techniques.
- cisapride monohydrate would be used as an active ingredient the formulation requires a surfactant to obtain the necessary wettability of the cisapride monohydrate.
- a wet-granulation step is required.
- the tablets of the present invention can be prepared by direct compression, i.e. the "usual" wet-granulation step can be omitted. This causes a considerable cost reduction in the production of these tablets.
- Tablets of the present invention may be film-coated to provide ease of swallowing, taste masking and an elegant appearance.
- Many polymeric film-coating materials are known in the art.
- Known film-coating agents are sodium carboxymethylcellulose, cellulose acetate, cellulose acetate phthalate, ethylcellulose, gelatin, pharmaceutical glaze, hydroxypropyl cellulose, hydroxypropyl methylcellulose, hydroxypropyl methyl cellulose phthalate, methacrylic acid copolymer, methylcellulose, polyethylene glycol, polyvinyl acetate phthalate, shellac, sucrose, titanium dioxide, wax, zein.
- a preferred film-coating material is hydroxypropyl methylcellulose (HPMC). HPMC may be obtained commercially.
- Coating agents are normally present in an amount ranging from about 50 % (w/w) to about 95 % (w/w) based upon the total weight of the film coating.
- the interesting range is from about 50 % (w/w) to about 65 % (w/w).
- Anti-adhesives are normally used in the film coating process to avoid sticking effects during film formation and drying.
- the preferred anti-adhesive for this purpose is talc.
- the anti-adhesive and especially talc is present in the film coating in an amount of about 5 % (w/w) to 15 % (w/w) based upon the total weight of the coating.
- plasticizers such as castor oil, diacetylated monoglycerides, dibutyl sebacate, diethyl phthalate, glycerin, polyethylene glycol, propylene glycol, triacetin, triethyl citrate. Also mixtures of plasticizers may be utilized.
- the type of plasticizer depends upon the type of coating agent.
- Preferred plasticizer according to the present invention is propylene glycol.
- Said plasticizer is normally present in an amount ranging from 5 % (w/w) to 30 (w/w) based on total weight of the film coating.
- interesting range of plasticizer is from about 12 % (w/w) to about 16 % (w/w) based on the total weight of the film coating.
- Preferred amount of propylene glycol according to the present invention is about 14 % (w/w).
- An opacifier like titanium dioxide may also be present in an amount ranging from about 10 % (w/w) to about 20 % (w/w) based on the total weight of the coating.
- coloring agents and pigments may be present in the film coating.
- Preferred coloring agents are ferric oxides, which can either be red, yellow, black or blends thereof.
- Said film-coating process may be carried out utilizing spray-coating equipment well- known in the art.
- the coating can be carried out in a perforated pan such as those manufactured under the tradename of Glatt® (for example Glatt Coater 750) AccelaCota® and HiCoater®.
- the tabletting process itself is otherwise standard and readily practiced by forming a tablet from a desired blend or mixture of ingredients into the appropriate shape using a conventional tablet press. Pressures are used ranging from about 0.5 ton/cm 2 (corresponding to about 50 MPa) to about 2.0 ton/cm 2 (corresponding to about 200 MPa). Below the lower limit, the tablets formed will not show appropriate hardness and above the higher limit the tablets may be so hard that they do not dissolve any more. Preferred range is from about 1.1 ton/cm 2 (corresponding to about 110 MPa) to about 1J ton/cm 2 (corresponding to about 170 MPa).
- Capsules according to the present invention comprise, apart form the active ingredient, fillers, glidants, lubricants and disintegrants.
- the same fillers, glidants and lubricants as described above for the tablets may be used in the capsules.
- Preferred filler is lactose.
- Preferred glidants are colloidal silicon dioxide and talc.
- Talc also provides the anti-adherent properties needed to handle the powders.
- Preferred lubricant is magnesium stearate.
- Maize starch can be used as a disintegrant, which is a necessary ingredient for the capsule content in the case the capsule filling equipment uses tamping. In capsule filling equipment using tamping, the capsule content is packed together in several consecutive strokes and at the last stroke the packed capsule content is delivered into the capsule.
- Fillers are present in an amount ranging from about 60 % (w/w) to about 90 % (w/w) based upon the total weight of the capsule content.
- the fillers are present in an amount ranging from about 70 % (w/w) to about 80 % (w/w) based upon the total weight of the capsule content.
- the fillers are present in an amount of about 75 % (w/w).
- Glidants are present in an amount of about 4% (w/w) to 7 % (w/w) based upon the total weight of the content of the capsule.
- the glidants are present in an amount of about 6 % (w/w) based upon the total weight of the content of the capsule.
- the lubricant or lubricants are present in an amount ranging from about 0.5 % (w/w) to about 2.0 % (w/w). Preferably, the lubricant or lubricants are present in an amount of about 1.25 % (w/w) based upon the total weight of the content of the capsule.
- Capsules are normally prepared from gelatin, they may be soft or hard gelatin capsules.
- the capsules are prepared in a conventional way.
- the filler for instance lactose, is milled together with the active ingredient and sieved.
- the resulting mixture is added to a mixture of the remainder of the excipients and mixed in a planetary mixer until a homogenous mixture is obtained.
- This powder is filled off in the capsule using art- known (automatic) capsule filling equipment.
- An advantage of the present solid oral dosage form is that, even when the solid oral dosage form according to the present invention has not yet completely dissolved in the acid environment of the stomach and is passed through to the gut, where the environment is about neutral, i.e. much less acidic, then still the (+)- or (-)-cisapride tartrate is able to dissolve quickly, which is not the case with cisapride monohydrate.
- the pharmaceutical dosage form is to be used as a medicine for treating gastrointestinal disorders, such as, gastroparesis, either idiopathic or associated with diabetic neuropathy, anorexia nervosa, after vagotomy or partial gastrectomy (the symptoms mainly consist of early satiety, anorexia, nausea and vomiting); symptoms of X-ray or endoscopy negative upper digestive discomfort, characterized by early satiety, postprandial fullness, inability to finish a normal sized meal, bloating, excessive belching, anorexia, nausea, vomiting or by ulcer-like complaints (epigastric burning or pain), gastro-esophageal reflux disorders, including the curative and maintenance treatment of oesophagitis; in babies: chronic and excessive regurgitation or vomiting, when positional and dietary measures have failed; intestinal pseudo-obstruction, associated with motility dysfunctions resulting in insufficient propulsive peristaltism and in stasis of gastric and intestinal contents;
- (+)- or (-)-cisapride-(L)-tartrate (+)- or (-)-cisapride-(D)-tartrate, (+)- or (-)-cisapride sulfate, (+)- or (-)-cisapride citrate for the manufacture of an oral dosage for medicament for the treatment of gastro intestinal disorders, which oral dosage form is independent of the interaction of food, is disclosed.
- (+)- or (-)-cisapride-(L)-tartrate, (+)- or (-)-cisapride-(D)- tartrate, (+)- or (-)-cisapride sulfate, (+)- or (-)-cisapride citrate for the manufacture of a medicament for treating gastrointestinal disorders in patients taking medication that increases the pH of the stomach in general or for the manufacture of a medicament for treating gastrointestinal patients taking proton pump inhibitors, ⁇ -inhibitors or antacids in particular is claimed.
- this invention provides a therapeutic package suitable for commercial sale, comprising a container, an oral dosage form of (+)- or (-)-cisapride which does not exhibit an adverse food effect contained therein and associated with said package, written (i.e. printed) matter non-limited as to whether the dosage form can be taken with or without food.
- the written matter is of the type containing information and/or instructions for the physician, pharmacist or patient.
- the written material can be "non-limited as to whether the dosage form can be taken with or without food" by virtue of including no statement regarding whether or not the dosage form can be taken with or without food, i.e. the statement is silent with regard to food effects.
- the written material can be non-limited by containing one or more statements affirmatively informing the user (i.e. the patient, pharmacist, or physician) that the said oral dosage form can be taken by or administered to a patient regardless whether the patient has eaten or otherwise imbibed food (optionally, for example, also stating something like "without regard to the type or quantity of food”).
- the written material cannot contain limiting language with respect to food, e.g., "This dosage form can not be taken with food” or "This dosage form may only be given after the patient has fasted” or the like.
- the container can be in any conventional shape or form as known in the art which is made of a pharmaceutically acceptable material, for example a paper or cardboard box, a glass or plastic bottle or jar, a re-sealable bag, or a blister pack with individual dosages for pressing out of the pack according to a therapeutic schedule.
- a pharmaceutically acceptable material for example a paper or cardboard box, a glass or plastic bottle or jar, a re-sealable bag, or a blister pack with individual dosages for pressing out of the pack according to a therapeutic schedule.
- the container employed can depend on the exact dosage form involved.
- the dosage form of (+)- or (-)-cisapride can be taken in or administered to a patient independently from a meal
- the dosage form can be administered "pro re nat ⁇ ".
- the administration of the dosage form can be symptom driven.
- the patient can take the present dosage form when the patient feels one of the symptoms which are associated with the gastrointestinal disorder he is suffering from. This greatly improves the patient compliance because instead of having to think about taking his medication at the meal, the patient can take in the medication when the symptoms appear.
- Example 1 Tablet A
- the tablet according to the example above comprises : (+)- or (-)-cisapride-(L)-tartrate 1 133..2233 mmgg 7.35 % (w/w)
- microcrystalline cellulose (Avicel®) (38.86 mg, 21.59 % (w/w)), croscarmellose sodium (7.2 mg, (4.00 % (w/w)), colloidal anhydrous silica (0.54 mg,
- (+)- or (-)-cisapride-(L)-tartrate 13.23 mg 7.35 % (w/w) lactose DC 1 16.57 mg 64.76 % (w/w) microcrystalline cellulose 38.86 mg 21.59 % (w/w) croscarmellose sodium 7.2 mg 4.00 % (w/w) colloidal anhydrous silica 0.54 mg 0.3 % (w/w) sodium stearyl fumarate 3.6 mg 2.00 % (w/w)
- Example 3 Tablet C In an analogous manner as described in Examples 1 and 2, tablets with the following composition are prepared :
- (+)- or (-)-cisapride-(L)-tartrate 13.23 mg 7.35 % (w/w) famotidine 10.00 mg 5.56 % (w/w)
- Simethicone or alpha-D-galactosidase may be added to each of the above formulations to provide anti-flatulent relief.
- the quantity of simethicone administered to a patient in need of treatment thereof may vary according to patient need, but may be, for example, the typical known dosage range to treat flatulence (20-40 mg per tablet) or may be increased as necessary.
- the amount of ADG that may be employed in the above formulations ranges from about 675 to about 2250 GalU or may be increased as necessary.
- Example 6 Preparation of film-coated tablets using the wet-granulation step for the tablet cores : Tablet F
- the binder solution prepared as described under 6a) is sprayed unto the powder mixture (process parameter : air flow rate : from about 400 to about 1000 m 3 /h, shaking time : about 7 seconds, shaking time interval : about 35 seconds, diameter of the nozzle : 1.8 mm, position of the nozzle : top, spraying pressure 3 bar, spraying rate : from about 200 to 300 g/min, temperature of the inlet air : from about 45°C to about 60°C, temperature of the outlet air : from about 21°C to about 24°C.) After the spraying homogeneously wetted granules were obtained.
- the drying process starts immediately after the spraying process.
- the drying process is continued until the outlet-air temperature reaches about 38°C.
- process parameters air flow rate : from about 400 to 1000 m 3 /h, shaking time : 7 seconds, shaking time interval 35 seconds, temperature of the inlet air : from about 70°C to about 75°C, temperature of the outlet air : from about 37°C to about 39°C.
- the dried granules prepared as described under 6b) are passed through an oscillating sieving apparatus of the type Frewitt (mesh openings : 1 mm, wire thickness : 0.65 mm) together with 2J72 kg of microcrystalline cellulose, 1.188 kg croscarmellose sodium, 118 g colloidal anhydrous silica and 198 g of magnesium stearate.
- the sieved powder is collected in the bowl of the planetary mixer of the type Collette MP 90 (speed of mixing : mixing arm : 45 rpm and the plateau : 20 rpm) and is mixed during 5 minutes until a homogeneous mixture is obtained.
- the compression mixture prepared as described under 6c) is pressed to tablets using a Killian rotary tablet press.
- Biconvex, white circular tablets with a nominal weight of 180 mg are prepared in this way. These tablets are referred to as tablet core hereinabove.
- mixture B 3.153 kg of purified water, 176 g talc, 264 g titanium dioxide and 33 g yellow ferric oxide are transferred in a stainless steel container of 10 1 and were homogenized for 10 to 15 minutes using a Silverson 2LR homogenizer. This mixture is referred to as mixture B.
- Mixture B is added to mixture A while mixing with a propeller mixer(speed of mixing : from about 200 to about 400 rpm). The total mixture is mixed during 120 minutes to further de-aerate the coating suspension.
- the tablets are filled of in polyethylene bottles or PerlenTM tristar blisters.
- Tablet core of tablet F ingredient amount % (w/w) versus tablet core
- (+)- or (-)-cisapride-(L)-tartrate 13.23 mg 7.35 % (w/w) lactose monohydrate 200 mesh(*l) 107.73 mg 59.85 % (w/w) unmodified maize starch 36.00 mg 20.00 % (w/w)
- HPMC 2910 15 mPa.s(*2) 3.60 mg 2.00 % (w/w) microcrystalline cellulose 12.60 mg 7.00 % (w/w) croscarmellose sodium 5.40 mg 3.00 % (w/w) colloidal anhydrous silica 0.54 mg 0.30 % (w/w) magnesium stearate 0.90 mg 0.50 % (w/w)
- Titanium dioxide (E171) 1.20 mg 16.78 % (w/w)
- (*1) 200 mesh is an indication of the type of lactose monohydrate that is used.
- HPMC means hydroxypropyl methylcellulose
- the number “2910” refers to the type of hydroxypropyl methylcellulose that is used.
- the first two digits, "29”, represent the approximate percentage of methoxylgroups and the third and fourth digit," 10" represents the approximate percentage of hydroxypropyigroups.
- the tablet is coated as described under Example 6.
- the tablet is coated as described under Example 6.
- Example 9 Tablet I : tablet comprising equivalent of 5 mg (+)- or (-)-cisapride base
- Example 10 Tablet J : tablet comprising equivalent of 10 mg (+)- or (-)-cisapride base
- (+)- or (-)-cisapride-(L)-tartrate 13.23 mg 7.35 % lactose monohydrate 200 mesh 107.73 mg 59.85 % unmodified maize starch 36.00 mg 20.00 %
- HPMC 2910 15 mPa.s 3.60 mg 2.00 % microcrystalline cellulose 12.60 mg 7.00 % croscarmellose sodium 5.40 mg 3.00 % colloidal anhydrous silica 0.54 mg 0.30 % magnesium stearate 0.90 mg 0.50 %
- Example 11 Tablet K : tablet comprising equivalent of 20 mg of (+)- or (-)-cisapride base
- Example 12 Capsule A ingredient amount %(w/w) based on total weight of content
- Example 13 Capsule B ingredient amount %(w/w) based on total weight of content
- (+)- or (-)-cisapride-(L)-tartrate 13.23 mg 6.01 % lactose 125 mesh 82.00 mg 37.27 % lactose 200 mesh 81.57 mg 37.08 % maize starch 27.50 mg 12.50 % talc 12.40 mg 5.64 % magnesium stearate 2.75 mg 1.25 % colloidal anhydrous silici a (Aerosil®) 0.55 mg 0.25 %
- the powder is filled in a capsule type number 2
- the capsules as described above are prepared by mixing the ingredients in a planetary mixer and filling the powder in the appropriate capsules.
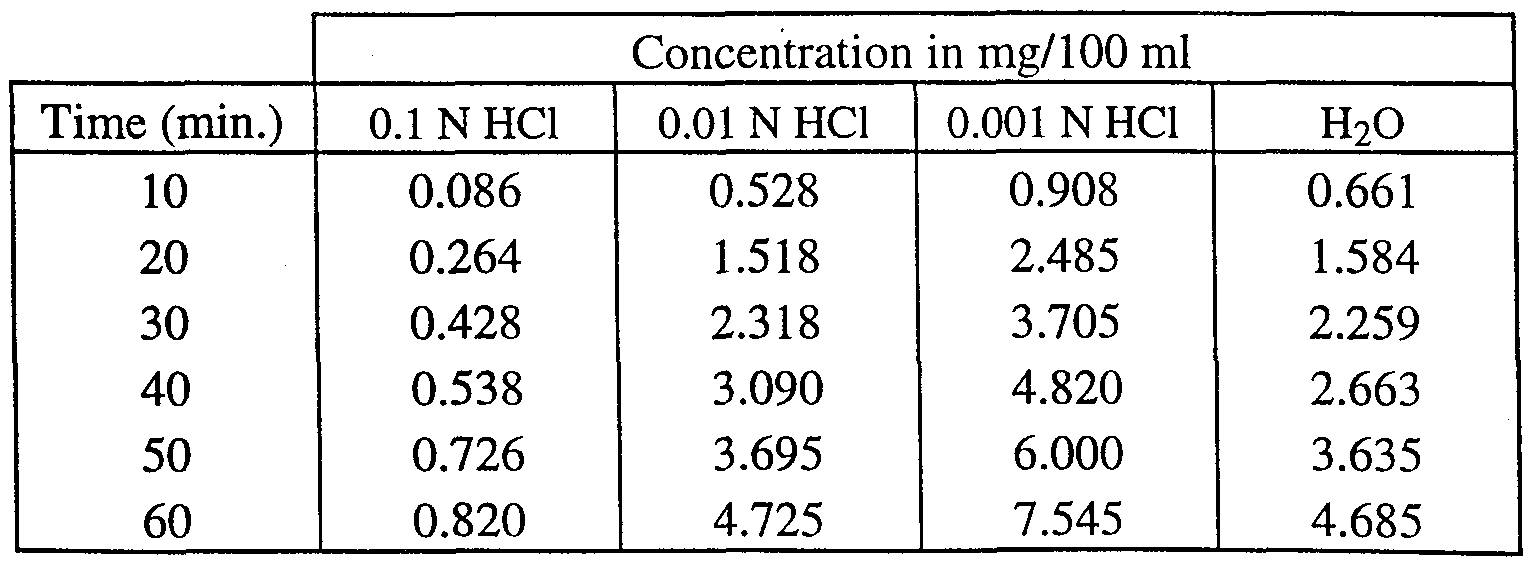
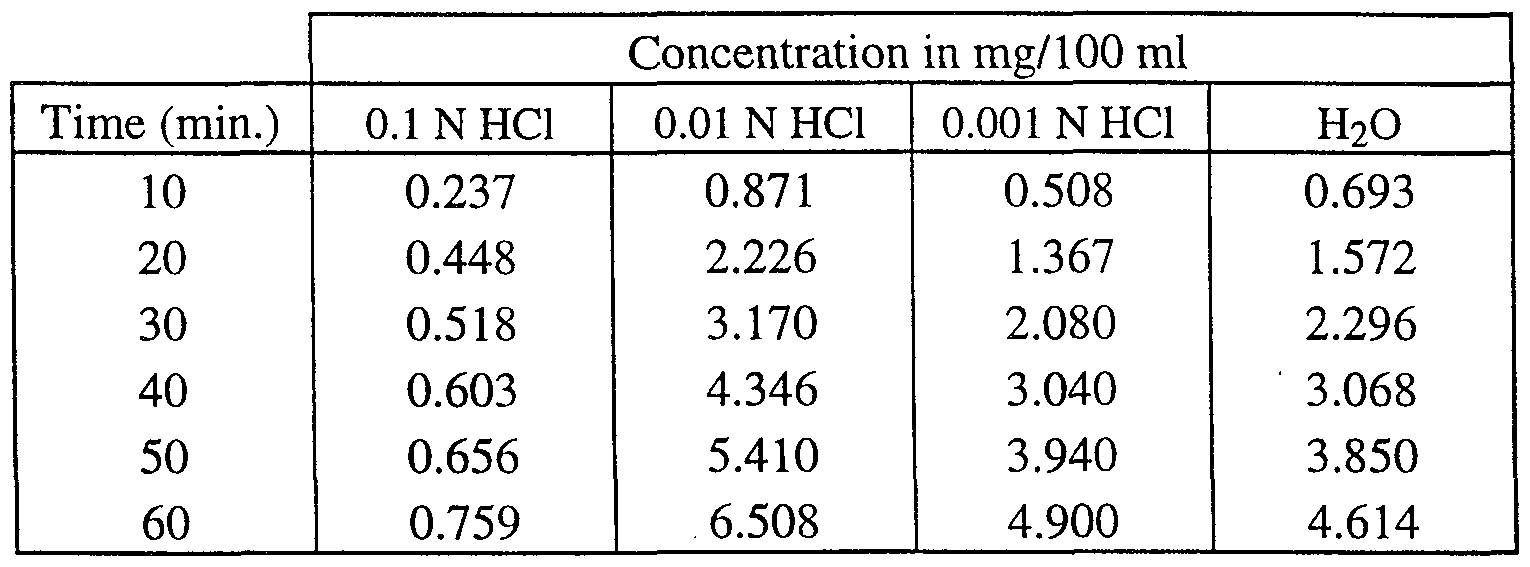
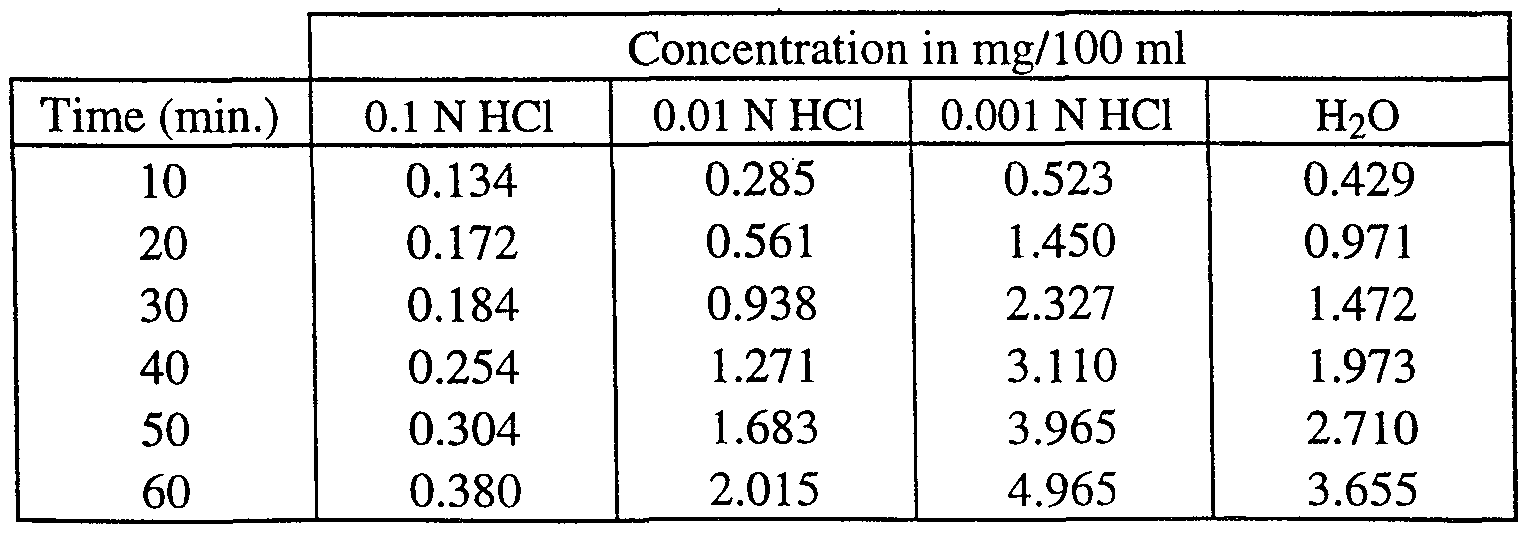
- the tablet was brought in a glass container containing 500 ml of a 0.01 N HCl, 0.001 N HCl or water solution at a temperature of 37°C.
- the stirring was performed by a paddle at a rotational speed of 150 rpm (rotations per minute).
- the concentration of (+)- or (-)-cisapride in solution was measured using its UV absorption at 309 nm.
- Table 2 dissolution data for (+)-cisapride-citrate.
- Table 3 dissolution data for (+ )-cisapride-maleate.
- Table 5 dissolution data for (-)-cisapride-L-tartrate.
- Table 6 dissolution data for (-)-cisapride-citrate.
- Table 7 dissolution data for (-)-cisapride-maleate.
- Table 8 dissolution data for (-)-cisapride-HCl.
- Table 9 dissolution data for cisapride base.
Abstract
Description







Claims
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP50160499A JP2002504107A (en) | 1997-06-11 | 1998-06-04 | Immediate release pH independent solid dosage form of (+)-or (-)-cisapride |
| AU79193/98A AU7919398A (en) | 1997-06-11 | 1998-06-04 | Immediate release ph-independent solid dosage form of (+)- or (-)-cisapride |
| CA002292628A CA2292628A1 (en) | 1997-06-11 | 1998-06-04 | Immediate release ph-independent solid dosage form of (+)- or (-)-cisapride |
| EP98929440A EP0988032A1 (en) | 1997-06-11 | 1998-06-04 | IMMEDIATE RELEASE pH-INDEPENDENT SOLID DOSAGE FORM OF (+)- OR (-)-CISAPRIDE |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP97201768 | 1997-06-11 | ||
| EP97201767 | 1997-06-11 | ||
| EP97201768.5 | 1997-06-11 | ||
| EP97201767.7 | 1997-06-11 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1998056364A1 true WO1998056364A1 (en) | 1998-12-17 |
Family
ID=26146581
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP1998/003495 WO1998056364A1 (en) | 1997-06-11 | 1998-06-04 | IMMEDIATE RELEASE pH-INDEPENDENT SOLID DOSAGE FORM OF (+)- OR (-)-CISAPRIDE |
Country Status (6)
| Country | Link |
|---|---|
| EP (1) | EP0988032A1 (en) |
| JP (1) | JP2002504107A (en) |
| KR (1) | KR20010012104A (en) |
| AU (1) | AU7919398A (en) |
| CA (1) | CA2292628A1 (en) |
| WO (1) | WO1998056364A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004525162A (en) * | 2001-04-10 | 2004-08-19 | サン・ファーマシューティカル・インダストリーズ・リミテッド | Timed pulsed release compositions |
| WO2013049731A1 (en) * | 2011-09-30 | 2013-04-04 | Stomavite Llc | A supplement for ostomy patients |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MXPA06011820A (en) * | 2004-04-16 | 2006-12-15 | Santarus Inc | Combination of proton pump inhibitor, buffering agent, and prokinetic agent. |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994001111A1 (en) * | 1992-07-07 | 1994-01-20 | Sepracor Inc. | Methods of using (+) cisapride for the treatment of gastro-esophageal reflux disease and other disorders |
| WO1994001112A1 (en) * | 1992-07-07 | 1994-01-20 | Sepracor Inc. | Methods of using (-) cisapride for the treatment of gastro-esophageal reflux disease and other disorders |
| WO1995001803A1 (en) * | 1993-07-06 | 1995-01-19 | Merck & Co., Inc. | H2 antagonist-gastrointestinal motility agent combinations |
| EP0670160A1 (en) * | 1994-03-01 | 1995-09-06 | Gerhard Dr. Gergely | Granular product or tablet containing an effervescent system and an active pharmaceutical substance, as well as a method for its preparation |
| WO1995034284A1 (en) * | 1994-06-15 | 1995-12-21 | Gerhard Gergely | Pharmaceutical preparation with a hydrophobic active substance and an effervescent system, and process for preparing said preparation |
| WO1996014070A1 (en) * | 1994-11-02 | 1996-05-17 | Janssen Pharmaceutica N.V. | Cisapride extended release oral compositions |
| EP0803251A1 (en) * | 1996-04-23 | 1997-10-29 | Janssen Pharmaceutica N.V. | Immediate release pH-independent solid dosage form of cisapride |
-
1998
- 1998-06-04 CA CA002292628A patent/CA2292628A1/en not_active Abandoned
- 1998-06-04 AU AU79193/98A patent/AU7919398A/en not_active Abandoned
- 1998-06-04 JP JP50160499A patent/JP2002504107A/en active Pending
- 1998-06-04 EP EP98929440A patent/EP0988032A1/en not_active Withdrawn
- 1998-06-04 WO PCT/EP1998/003495 patent/WO1998056364A1/en not_active Application Discontinuation
- 1998-06-04 KR KR1019997009807A patent/KR20010012104A/en not_active Application Discontinuation
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994001111A1 (en) * | 1992-07-07 | 1994-01-20 | Sepracor Inc. | Methods of using (+) cisapride for the treatment of gastro-esophageal reflux disease and other disorders |
| WO1994001112A1 (en) * | 1992-07-07 | 1994-01-20 | Sepracor Inc. | Methods of using (-) cisapride for the treatment of gastro-esophageal reflux disease and other disorders |
| WO1995001803A1 (en) * | 1993-07-06 | 1995-01-19 | Merck & Co., Inc. | H2 antagonist-gastrointestinal motility agent combinations |
| EP0670160A1 (en) * | 1994-03-01 | 1995-09-06 | Gerhard Dr. Gergely | Granular product or tablet containing an effervescent system and an active pharmaceutical substance, as well as a method for its preparation |
| WO1995034284A1 (en) * | 1994-06-15 | 1995-12-21 | Gerhard Gergely | Pharmaceutical preparation with a hydrophobic active substance and an effervescent system, and process for preparing said preparation |
| WO1996014070A1 (en) * | 1994-11-02 | 1996-05-17 | Janssen Pharmaceutica N.V. | Cisapride extended release oral compositions |
| EP0803251A1 (en) * | 1996-04-23 | 1997-10-29 | Janssen Pharmaceutica N.V. | Immediate release pH-independent solid dosage form of cisapride |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004525162A (en) * | 2001-04-10 | 2004-08-19 | サン・ファーマシューティカル・インダストリーズ・リミテッド | Timed pulsed release compositions |
| WO2013049731A1 (en) * | 2011-09-30 | 2013-04-04 | Stomavite Llc | A supplement for ostomy patients |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20010012104A (en) | 2001-02-15 |
| EP0988032A1 (en) | 2000-03-29 |
| JP2002504107A (en) | 2002-02-05 |
| CA2292628A1 (en) | 1998-12-17 |
| AU7919398A (en) | 1998-12-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6030988A (en) | Immediate release pH-independent solid dosage form of cisapride | |
| JP4573397B2 (en) | Fast disintegrating solid preparation | |
| AU2002320385B2 (en) | Sequential drug delivery systems | |
| JP5002044B2 (en) | Tablet manufacturing method | |
| EP0922464B1 (en) | Quickly disintegrable compression-molded materials and process for producing the same | |
| KR101752014B1 (en) | Orally disintegrating tablet compositions comprising combinations of high and low-dose drugs | |
| US9492541B2 (en) | Phenylepherine containing dosage form | |
| CN107028900A (en) | Rapid dispersion particle, oral disnitegration tablet and method | |
| CN110944641A (en) | Gelatin adhesive composition and methods of making and using the same | |
| JP2009292843A (en) | Solid pharmaceutical preparation | |
| JP2009114113A (en) | Intraorally disintegrable tablet and method for producing the same | |
| WO2019130749A1 (en) | Novel fine particle coating (drug-containing hollow particle and method for manufacturing same) | |
| TWI240638B (en) | A film-coated solid-dosage form pharmaceutical composition | |
| KR100846945B1 (en) | Swallow tablet comprising paracetamol | |
| CN107530335A (en) | New pharmaceutical use | |
| JP5138856B2 (en) | Tablet manufacturing method | |
| EP0803251B1 (en) | Immediate release pH-independent solid dosage form of cisapride | |
| WO1998056364A1 (en) | IMMEDIATE RELEASE pH-INDEPENDENT SOLID DOSAGE FORM OF (+)- OR (-)-CISAPRIDE | |
| US9364451B1 (en) | Alternating sympathomimetic therapy for the treatment of respiratory ailments | |
| JP2021138689A (en) | Tablet, method for producing the same, and pharmaceutical | |
| CN108289849A (en) | The compound formulation of Mosapride and Rabeprazole | |
| MXPA98008710A (en) | Dosage form solid independent of the ph, of immediate release | |
| CN112839636A (en) | Coating method | |
| WO2014027974A1 (en) | Orally disintegrating formulation of paliperidone | |
| Konde | Formulation and Evaluation of Orodispersible Tablets of Lansoprazole |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE GH GM GW HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1998929440 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1019997009807 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09445008 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2292628 Country of ref document: CA Ref country code: CA Ref document number: 2292628 Kind code of ref document: A Format of ref document f/p: F Ref country code: JP Ref document number: 1999 501604 Kind code of ref document: A Format of ref document f/p: F |
|
| WWP | Wipo information: published in national office |
Ref document number: 1998929440 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1019997009807 Country of ref document: KR |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1998929440 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1019997009807 Country of ref document: KR |