USRE46830E1 - Method for solid phase peptide synthesis - Google Patents
Method for solid phase peptide synthesis Download PDFInfo
- Publication number
- USRE46830E1 USRE46830E1 US14/659,770 US200514659770A USRE46830E US RE46830 E1 USRE46830 E1 US RE46830E1 US 200514659770 A US200514659770 A US 200514659770A US RE46830 E USRE46830 E US RE46830E
- Authority
- US
- United States
- Prior art keywords
- glu
- gly
- peptide
- pro
- phe
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active, expires
Links
- 0 *C(C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1Cl.[1*]C Chemical compound *C(C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1Cl.[1*]C 0.000 description 22
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/81—Protease inhibitors
- C07K14/815—Protease inhibitors from leeches, e.g. hirudin, eglin
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/04—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length on carriers
- C07K1/042—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length on carriers characterised by the nature of the carrier
Definitions
- the present invention relates to an improved method of solid phase peptide synthesis of the anticoagulant peptide bivalirudin, a so-called ‘hirulog’. It further relates to the respective peptide-solid phase conjugate products comprising the still protected peptide bound to the resin.
- Thrombin inhibitors are considered as promising antithrombotics: Proteolytic processing by thrombin is pivotal in the control of blood clotting. Hirudin, a potent clinical thrombin peptide inhibitor from the blood-sucking leech Hirudo medicinalis, consists of 65 amino acids.
- Hirulogs Shorter peptide analogs of the peptide segment amino acid positions 45-65 of Hirudin, the so-called Hirulogs, have proven effective in treatment of thrombosis, a life-threatening condition.
- Acidolytic cleavage from the Wang resin is applied under strongly acidic conditions and is known to inevitably incur undesirable alkylation of Trp residues as a side reaction, despite the use of scavenging reagents during acidolysis (Giraud et al., 1999, J. Peptide Science 5:457-461). In particular C-terminal Trp is prone to such side reaction (Atherton et al., 1988, Tetrahedron 44:843-857). Alkylation is caused by aromatic carbenium ions generated from the Wang resin linkers phenoxy moiety. —Whilst the Hirulogs do not contain Trp residues, they do comprise in the C-proximal position a Tyr residue. We found and report here for the first time that this Tyr residue is equally prone to erratic alkylation upon cleavage from Wang resin, negatively affecting product purity.
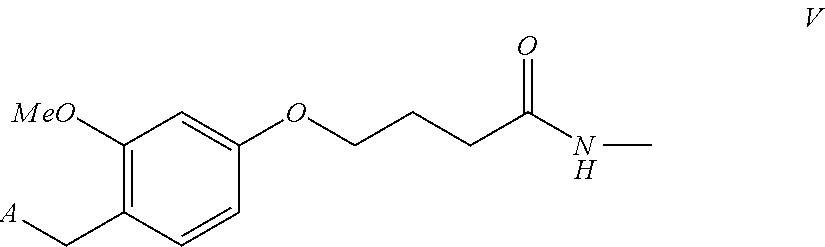
- a method for detaching and deprotecting a peptide-solid phase conjugate to yield finally a peptide, preferably a peptide of the formula D-Phe-Pro-Arg-Pro-Gly-Gly-Gly-Gly-Asn-Gly-Asp-Phe-Glu-Glu-Tyr-Leu (SEQ ID NO: 8).
- Said peptide-solid phase conjugate is comprising a 2-chloro-trityl handle of formula I
- Hirulog-8 (described in EP-489 070). It is a 20mer bivalent derivative of hirudin (a 65mer), a naturally occurring potent thrombin inhibitor. It is made up from functionally important, linked structural motifs from Hirudin: The active site binding motif D-Phe-Pro-Arg-Pro (SEQ ID NO: 12) and the carboxy-terminal sequence Asn 9 to Leu 20 from Hirudin, bridged by a tetraglycine spacer (SEQ ID NO: 1).
- -D-Phe- means D-phenylalanine, as opposed to the naturally occurring L-enantiomer of a given amino acid, in this case Phe.
- radical A in formula I may be any of the following:
- A P-X1-Tyr(R9)-X2- (SEQ ID NO: 13) wherein X1 is a peptidyl moiety, optionally comprising protection groups on individual amino acid side chains, of 0 to 200, preferably 1 to 100, most preferably 2 to 50 amino acids, and wherein X2 is a single, optionally side chain protected, amino acid residue linked to the solid phase via —O— or —NH—, wherein preferably X2 is not Trp, Cys or Arg, and wherein P is either H (i.e.
- the protection group is an orthogonal protection group or is one removable under strongly acidic condition as defined below, more preferably the protection group is selected from the group consisting of Boc, Fmoc, Dde, Nps, Alloc, Z.
- A P-X1-Tyr(R9)-Leu-O (SEQ ID NO: 14) or is P-X1-Tyr(R9)-X2 (SEQ ID NO: 13)
- A P-X1-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-Leu-O (SEQ ID NO: 2) or is P-X1-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-X2 (SEQ ID NO: 3)
- A P-X1-[Gly] 0-3 -Asn(R3)-Gly-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-Leu-O (SEQ ID NO: 4) or is P-X1-[Gly] 0-3 -Asn(R3)-Gly-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-X2 (SEQ ID NO: 5)
- A P-X1-Arg(R2)-Pro-Gly-Gly-Gly-Asn(R3)-Gly-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-Leu-O— (SEQ ID NO: 6) or is P-X1-Arg(R2)-Pro-Gly-Gly-Gly-Asn(R3)-Gly-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-X2 (SEQ ID NO: 7)
- the peptide-solid phase conjugate of the present invention can be synthesized by routine solid phase methods well-known in the art, and well described and referenced in Bodanszky et al., Principles of Peptide Synthesis, 2 nd ed., Springer Verlag Berlin Heidelberg 1989). Necessarily, due to the acid-lability of the solid phase attachment, such synthetic strategy employs Fmoc chemistry for carrying out the coupling reactions during solid-phase synthesis. Only the last, terminating D-Phe residue may either be Boc- or Fmoc protected. Such Fmoc protection may be eliminated still on-resin, by standard treatment with e.g.
- the terminating D-Phe residue is Boc-protected or is protected with another protection group that can be easily removed in strongly acid condition, for avoiding the need of a separate Fmoc deprotection step.
- this includes e.g. Z-(benzyloxy-carbonyl-) protection group, which may be cleaved, inter alia, by strongly acidic conditions as defined in the present context, though hydrogenolytic or HF promoted cleavage is known to be more efficient.
- a separate Fmoc deprotection step on the terminating D-Phe residue exposing early on the N-terminus (terminating in free a-amino group, which may be equally denoted as H-D-Phe- . . . or NH 2 -D-Phe . . . in formula I) and rendering the now free N-terminal D-Phe prone to racemisation e.g.
- Said one-step detachment or cleavage along with global deprotection may be carried out in a solvent mixture such as aqueous TFA and DCM, for instance.
- the present invention it is possible either to cleave the protected peptide of formula I from the resin concomittant with or, in initial step, to cleave the protected peptide of formula I from the resin preceding the deprotection or global deprotection of amino acid side chains and, preferably, the N-terminal protection group.
- it is sequentially subjected firstly to weakly acidic condition for cleavage from resin and secondly to strongly acidid condition for cleavage of all remaining protection groups (global deprotection).
- the CTC resin allows of effecting resin cleavage of the still protected peptide and tyrosine under very mild acidolytic reaction conditions, e.g. in 0.5% trifluoro acetic acid (TFA) in dichloromethane (DCM), a condition at which most side chain and N-terminal protection groups will normally not be affected and hence alkylation is prevented by segregation of the different deprotection events in time.
- TFA trifluoro acetic acid
- DCM dichloromethane
- a strongly acidic condition as being opposed to a weakly acidic condition means applying at least 50% (v/v) trifluoro acetic acid (TFA) in the solvent.
- a protection group requiring strongly acid condition for removal is a protection group that can be removed, at the very least, by 80% TFA. Accordingly, protection groups that require even stronger acids such as HF do not come under the afore mentioned definition in the context of the present invention.
- a weakly acidic condition is defined by having 0.01% (v/v) to ⁇ 50% TFA, preferably having 0.1% to 30% TFA.
- the peptidyl moiety of the present invention notably shows an unexpected absence of undesirable alkylation of the juxtaproximal tyrosine and it is entirely devoid of diketopiperazine side reaction, another possible side reaction that happens upon cleavage from resin and is known to be particular sensitive to the nature of the last two C-terminal amino acids.
- a tyrosine at position 2 of the peptide chain next to the CTC resin handle is just at the optimum distance and spacing as to show some stabilising, hydrophobic stacking of the aromatic phenyl moieties, avoiding e.g. the cyclic arrangement that is the prelude to diketopiperazine formation.
- Loading of the CTC resin commonly takes place by nucleophilic substitution of the diphenyl-2′-chlorophenyl-chlormethan derivative (hence CTC, short for chloro-trityl-chloride) and is known to be effective.
- CTC diphenyl-2′-chlorophenyl-chlormethan derivative
- preloaded Fmoc-amino-acid-CTC resins are commercially available.
- Protection groups and their chemistry are further well-known and well-referenced in the art (see Bodanszky, supra). It is needless to say that of course different protection groups R2 to R9 are suited for protection of individual amino acid side chains, different chemical moieties requiring different protection groups. Examples are e.g. histidine being conventionally protectable with trityl or Boc, lysine being protectable with Boc or allyloxycarbonyl, aspartate being protectable as tert.butylester or allylester. Threonine, serine and tyrosine are usually protected as tert-butyl ether. The protection of arginine will be further discussed below. Different modes of deprotection may be applied, e.g.
- allylic protection groups are laborously removed by Pd-catalyzed reductive acyl-transfer reaction.
- Z (benzyloxycarbonyl) groups are less expediently employed since requiring hydrogenolysis for efficient removal.
- the protection groups R2 to R9 are acid-labile, ‘labile’ meaning a cleavage rate of at least 20% of said respective protection group when incubated in DCM solution for up to 5 hours under either weakly or strongly acidic conditions. More preferably according to the present invention, the protection groups R2 to R9 are removed and are only removable under strongly acidic condition as defined above only, that is by way of acidolysis under strongly acidic condition.
- R1 is an insoluble, normally polymeric solid phase, e.g. a crosslinked polystyrene/1% divinylbenzol co-polymer.
- solid phase R1 will of course display further, multiple 2-chloro-trityl-handle moities functionalized with peptide radical A beyond the one shown explicitedly in formula I.
- the polymeric solid phase will have a minimum particle size in order to give a true suspension of easily filtratable or pelletable particles of sufficient size, rather than colloidal behaviour.
- polystyrene base polymer Apart from polystyrene base polymer either directly derivatized with a CTC handle or linker (such as Bayer's 4-carboxytrityl liner, Bayer et al, 13 th American Peptide Symposium, Hodges et al., Ed., ESCOM, Leiden, 1994, page 156) or wherein individual benzene moieties of the base polymer have been derivatized to form part of the 2-Chloro-trityl function, further other base polymers such as pure or mixed PEG resins (e.g. Tentagel) or optionally hybrid or grafted resins, wherein e.g.
- a CTC handle or linker such as Bayer's 4-carboxytrityl liner, Bayer et al, 13 th American Peptide Symposium, Hodges et al., Ed., ESCOM, Leiden, 1994, page 156) or wherein individual benzene moieties of the
- a 2-CTC linker (such as the Bayer linker) has been grafted onto a polystyrene base polymer via a PEG spacer moiety instead of directly reacting the linker with the polystyrene base polymer.
- Including PEG into a resin provides a more amphilic resin and hence better handling e.g in DCM/TFA mixtures for one-step detachment and deprotection, though loading capacity may then become an issue.
- PEG resins which are strictly insoluble of course.
- the solid phase has a mesh size of less than 700 mesh (mesh size as defined by the US Bureau of Standards, retrievable e.g. in Römpps Chemie-Lexikon, 7. Auflage, 1973, Franck'sche Verlags Stuttgart, W. Keller & Co. Stuttgart/Germany).
- the 2-chloro-trityl-functionalized solid phase of the present invention has a mesh size of from 50 to 600 mesh (as defined by US Bureau of Standards), more preferably of from 60 to 400 mesh, most preferably of from 100 to 300 mesh.
- the Tyrosin of the present invention may be protected by different protection groups, e.g. tert.butyl ether or Z- or more preferably 2-Bromo-Z esters. It is equally possible to use tritylalkohol protection groups such as 2-chloro-trityl or 4-methoxy or 4,4′ methoxy-trityl groups.
- R9 is a trityl or a tert.butyl protection group. More preferably, R9 is a tertiary butyl (tBu) protection group, meaning the tyrosyl side chain is modified to a tertiary-butyl ether. The tBu group is only efficiently removed under strongly acidic condition.
- the arginine protection group R2 is selected from the group consisting of pentamethyldihydrobenzofuranyl-(Pbf), adamantyloxy-carbonyl and isobornyl-oxy-carbonyl, pentamethylenchromanesulfonyl (Pmc), 4-methoxy-2,3,6-trimethylbenzenesulfonyl (Mtr) and its 4-tert.butyl-2,3,5,6-tetramethyl homologue (Tart) or Boc, which are only cleaved under strongly acidic conditions as defined above.
- R2 is Pbf, Pmc, Mtr, most preferably, it is Pbf; upon global deprotection of side chains under strongly acidic conditions, in usually aqueous medium, bystander-alkylation of deprotected tyrosine is not observed with Pmc, Mtr and esp. Pbf. Pbf's cleavage rate is the highest ever.
- Carboxy-protection groups for Glu, Asp are well known, e.g. Mpe, O-1-Adamantyl, O-benzyl and even simply alkyl esters may be used, though less commonly used.
- Mpe Mpe
- O-1-Adamantyl O-benzyl
- alkyl esters may be used, though less commonly used.
- typically and preferably tert.butyl groups are used, independently, for protection groups R4, R5, R6, R7, R8.
- Protection group R3 may be of paramount importance because of occurring in above sequence Gly-Asp in Hirulog-8, which dipeptide sequence is particularly prone to aspartimide formation as a side reaction. Aspartimide formation may occur in the protected peptide over each subsequent cycle of coupling during linear synthesis to a minor extent (0.1-0.5%), having cumulative effect in the end. Whilst again protection with a trityl protection group or 2-chloro and 4-methyl or 4-methoxy derivatives thereof, is preferred, likewise adamantyl protection group may be used. Most preferably, a trityl protection group is employed.
- Na-alkyl protected dipeptide modules may be used for coupling during linear synthesis; such dipeptides have secondary structure disrupting effect, easing yield and purity of synthesis.
- Fmoc-Gly-(N-Hmb)Gly-OH and Fmoc-Gly-(N-Dmb)Gly-OH are commercially available from EMD Biosciences (Novabiochem). It is to be understood that such N-alkyl groups are not considered protection groups in the sense of the present invention, hence their use or presence is optional and not excluded by the structure of formula I.
- the two step sequential scheme of first conducting an acidolysis under weakly acidic conditions for cleaving the protected peptide from the CTC-resin and secondly removing the remaining protection groups under strongly acidic conditions is applied.
- a method is devised of detaching and deprotecting the peptide-solid-phase conjugate of formula I as defined above to give a peptide of formula D-Phe-Pro-Arg-Pro-Gly-Gly-Gly-Gly-Asn-Gly-Asp-Phe-Glu-Glu-Ile-Pro-Glu-Glu-Glu-Tyr-Leu (SEQ ID NO: 15), characterized in that in a first step, the protected peptide is cleaved from the 2-chloro trityl handle by treatment under weakly acidic condition, preferably with 0.1 to 10% TFA in an polar, aprotic solvent, and that in a second step, the protection groups are removed under strongly acidic condition as defined above.
- the first step is conducted in a polar, aprotic solvent that is dichloromethane.
- a polar, aprotic solvent that is dichloromethane.
- This is the best solvent to carry out such reaction, in contrast to other solvents such as NMP (N-methylpyrrolidone).
- NMP N-methylpyrrolidone
- Such scavenger intercept reactive alkyl-carbenium ions intermediates that are generated upon removal of the protection groups (which may already happen to a minor extent during cleavage reaction in the first step).
- scavenger is e.g. thioanisol (which also has second, acidolysis-promoting effect—such secondary role and substitutes for aniosol are discussed in Bodanszky M. et al., Int. J. Peptide Protein Res. 23:287).
- Other examples of scavengers having no such acidolysis effect are phenol and/or trialkylsilanes are used (Stierandova et al., Int. J. Peptide Protein Res. 43, 1994, 31-38).
- the reaction is directly quenched by admixing with pyridine and subsequently recovering the product of step 1 by admixing with water. This way, the product is most simply and efficiently recovered.
- peptide-solid phase conjugate of formula I is claimed but with the sole difference that the -Arg(R2)-Pro- which is the thrombin cleavage site, is not a standard peptide bond but a chemically modified, pseudoscissile or ‘psi’ bond (the replacement of an amide bond is indicated by the atoms designated in an extra bracket preceded by the akronym ‘psi’, see. Rudinger et al., Drug Design Vol. II, Ed. Ariens, E., Academic Press, New York, p. 319 (1971). More preferably, such psi replacement is -Arg[psiCH 2 NH]Pro- (Kline, T.
- A may be any of the above defined embodiments for A, optionally comprising individual amino acid side chain protection groups and wherein R2 to R9 are defined as above where present, wherein and wherein W is a, preferably insoluble, solid phase or solid phase composite which allows of cleaving the peptide moiety under weakly acidic conditions and which is comprising a resin handle or linker of a.
- R′′′ is the solid phase and wherein R′′1, R′′2, R′′3 are, independently, hydrogen, 4- or 4′-(C 1 -C 4 alkyl) or 4- or 4′-(C 1 -C 4 alkoxy), and may be the same or different with the proviso that only one of R′′1, R′′2 may be hydrogen, and wherein R′′2 may optionally be 2-Cl with the proviso that then R′′1 is H, and wherein more and most preferably, the handle or linker of formula II is selected from the group consisting of 2-chloro-trityl, 4-methoxy-trityl, 4,4′-dimethoxytrityl, 4-methyltrityl, b. or of the formula III
- the resin handle is of formula VI, the above definitions for radicals R′′′, R′′1 and R′′2 applying,
- resin or resin handle is of formula VII, the above definitions for radicals R′′′, R′′1 and R′′2 applying,
- R′′1, R′′2 are independently hydrogen, methyl or methoxy with the provisio that only one of R′′1, R′′2 may be hydrogen, and that, where A including a residue X2 is linked via —N— to said handle or linker of formula VII, independently are methyl or methoxy, preferably are methoxy.
- A, also where comprising X2, is bound to the handle via a —O— function, R′′1 is hydrogen and R′′2 is methyl or methoxy and preferably A is a resin or resin handle. Most preferably, R′′2 is methyl.
- the resin or resin handle composite entity may in principle be any resin employed for synthesis, such as for example a polystyrene-divinylbenzene resin as used by Merrifield along with hydroxybenzyl-phenyl integral linker moieties or by Wang with hydroxy-benzyl-p-benzyloxy moieties, such as for example moieties to which e.g. more acid-labile linkers may be further grafted, or alternatively the latter linkers may be integrally or directly linked to the resin.
- a solid phase resin for use in synthesis necessarily comprises at least an integral linker or handle which is part of the solid phase core material; such linker or handle may be considered as an immobilized protection group (Guillier et al., Chem. Rev.
- Examples are e.g. Sieber resin, related xanthenyl type PAL-handle resins, Rink amide resin, Rink acid resin, more complex PEG-grafted polystyrene resins such as tentagel-based Novasyn TG (Novabiochem, Merck Biosciences, Germany) which are available with different grafted handles such as 2′-chloro-trityl, or resins that are constituted by grafting functional handles onto matrix material such as silica gels.
- the resin is a trityl resin or resin handle
- such resin is a 4-methoxy or 4,4′-dimethoxy-trityl resin.
- Resins as used in the present invention are of standard mesh size, which is about 50-500 mesh, more preferably 100 to 400 mesh.
- a resin or solid-phase R′′′ as shown in formula IV is to be construed as to comprise a crosslinked, polymeric matrix material which may be bound to the handle moiety specified in formulas IV to VII by way of any kind of chemically inert alkyl, alkyloxy, aryloxy or alkylester spacer or linker which is to be considered an integral part of R′′′.
- the chemical nature of the resin material and in particular the chemical nature of the handle group may well influence synthetic efficiency of coupling and especially lactamisation reactions in a yet poorly understood fashion.
- the resin or resin handle is of formula IV as set forth in the claims in detail, more preferably of formula VI and most preferably of formula VII as set forth in the claims in detail.
- examples of such resins or resin handles are (4-methoxyphenyl)-methyl- and (4-methylphenyl)-methyl-polystyrene (Atkinson et al., 2000, J. Org. Chem. 65, 5048), resins in O- or N-linkage to the peptide moiety and their PEG-resin derivatives, respectively. Further examples are e.g.
- acid-labile refers to essentially quantitative cleavage in 2-10% TFA in dichloromethane at ambient temperature for at least an hour.
- acid-labile refers to essentially quantitative cleavage in 2-10% TFA in dichloromethane at ambient temperature for at least an hour.
- resins having the diphenyl-methyl structural core motif allow for more efficient coupling reaction during linear synthesis and lactamisation; notably, such resins also allow a lower reaction temperature of 15-25° C. as compared to the standard 40° C. required for efficient coupling on e.g. tritylresins.
- Fmoc deprotection was carried out with 3-4 cycles of 20% piperidine in NMP at 30° C., with suitable rinsing with NMP in between.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Life Sciences & Earth Sciences (AREA)
- Molecular Biology (AREA)
- Gastroenterology & Hepatology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Analytical Chemistry (AREA)
- Peptides Or Proteins (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
A novel method for synthesizing a Hirulog peptide is devised.
Description
This application is the US national phase of international application PCT/EP2005/011226 filed 19 Oct. 2005, which designated the U.S. and claims benefit of EP 04024812.2 filed 19 Oct. 2004, the entire contents of each of which are hereby incorporated by reference.
The instant application contains a Sequence Listing which has been submitted electronically in ASCII format and is hereby incorporated by reference in its entirety. Said ASCII copy, created on Apr. 22, 2015, is named LZAS-69-RI_SL.txt and is 22,633 bytes in size.
The present invention relates to an improved method of solid phase peptide synthesis of the anticoagulant peptide bivalirudin, a so-called ‘hirulog’. It further relates to the respective peptide-solid phase conjugate products comprising the still protected peptide bound to the resin.
Thrombin inhibitors are considered as promising antithrombotics: Proteolytic processing by thrombin is pivotal in the control of blood clotting. Hirudin, a potent clinical thrombin peptide inhibitor from the blood-sucking leech Hirudo medicinalis, consists of 65 amino acids.
Shorter peptide analogs of the peptide segment amino acid positions 45-65 of Hirudin, the so-called Hirulogs, have proven effective in treatment of thrombosis, a life-threatening condition.
Okayama et al. (1996, Chem. Pharm. Bull. 44:1344-1350) and Steinmetzer et al. (1999, Eur. J. Biochem. 265:598-605) devise solid phase synthesis of different hirulogs on Wang resin, that is using ester bonding of the C-terminal Fmoc amino acid to a resin that is esterified to a p-benzyloxy-benzyl alcohol radical. The Wang resin requires cleavage of the peptide from resin with concentrated trifluoroacetic acid, for which the resin cleavage amounts to concomittant global deprotection of peptide.
Acidolytic cleavage from the Wang resin is applied under strongly acidic conditions and is known to inevitably incur undesirable alkylation of Trp residues as a side reaction, despite the use of scavenging reagents during acidolysis (Giraud et al., 1999, J. Peptide Science 5:457-461). In particular C-terminal Trp is prone to such side reaction (Atherton et al., 1988, Tetrahedron 44:843-857). Alkylation is caused by aromatic carbenium ions generated from the Wang resin linkers phenoxy moiety. —Whilst the Hirulogs do not contain Trp residues, they do comprise in the C-proximal position a Tyr residue. We found and report here for the first time that this Tyr residue is equally prone to erratic alkylation upon cleavage from Wang resin, negatively affecting product purity.
It is the object of the present invention to devise another or improved method of synthesizing the respective Hirulog peptides that lacks the disadvantages of the prior art.
This object is solved by the peptide-resin conjugates and respective method of synthesis devised by the present invention.
According to the present invention, a method is devised for detaching and deprotecting a peptide-solid phase conjugate to yield finally a peptide, preferably a peptide of the formula D-Phe-Pro-Arg-Pro-Gly-Gly-Gly-Gly-Asn-Gly-Asp-Phe-Glu-Glu-Tyr-Leu (SEQ ID NO: 8). Said peptide-solid phase conjugate is comprising a 2-chloro-trityl handle of formula I

wherein A=Boc-D-Phe-Pro-Arg(R2)-Pro-Gly-Gly-Gly-Gly-Asn(R3)-Gly-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-Leu-O— (SEQ ID NO: 9) or A=Fmoc-D-Phe-Pro-Arg(R2)-Pro-Gly-Gly-Gly-Gly-Asn(R3)-Gly-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-Leu-O— (SEQ ID NO: 10) or A=NH2-D-Phe-Pro-Arg(R2)-Pro-Gly-Gly-Gly-Gly-Asn(R3)-Gly-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-Leu-O— (SEQ ID NO: 11) and wherein R2, R3, R4, R5, R7, R8, R9 are amino side chain protection groups and wherein R1 is an insoluble solid phase.
The above peptide sequence is that of Hirulog-8 (described in EP-489 070). It is a 20mer bivalent derivative of hirudin (a 65mer), a naturally occurring potent thrombin inhibitor. It is made up from functionally important, linked structural motifs from Hirudin: The active site binding motif D-Phe-Pro-Arg-Pro (SEQ ID NO: 12) and the carboxy-terminal sequence Asn9 to Leu20 from Hirudin, bridged by a tetraglycine spacer (SEQ ID NO: 1). For sake of definition, herein ‘-D-Phe-’ means D-phenylalanine, as opposed to the naturally occurring L-enantiomer of a given amino acid, in this case Phe.
Optionally, in a further object of the present invention, radical A in formula I may be any of the following:
1. A=P-X1-Tyr(R9)-X2- (SEQ ID NO: 13) wherein X1 is a peptidyl moiety, optionally comprising protection groups on individual amino acid side chains, of 0 to 200, preferably 1 to 100, most preferably 2 to 50 amino acids, and wherein X2 is a single, optionally side chain protected, amino acid residue linked to the solid phase via —O— or —NH—, wherein preferably X2 is not Trp, Cys or Arg, and wherein P is either H (i.e. gives α-NH2) or a protection group, preferably the protection group is an orthogonal protection group or is one removable under strongly acidic condition as defined below, more preferably the protection group is selected from the group consisting of Boc, Fmoc, Dde, Nps, Alloc, Z.
2. A=P-X1-Tyr(R9)-Leu-O (SEQ ID NO: 14) or is P-X1-Tyr(R9)-X2 (SEQ ID NO: 13)
3. A=P-X1-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-Leu-O (SEQ ID NO: 2) or is P-X1-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-X2 (SEQ ID NO: 3)
4. A=P-X1-[Gly]0-3-Asn(R3)-Gly-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-Leu-O (SEQ ID NO: 4) or is P-X1-[Gly]0-3-Asn(R3)-Gly-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-X2 (SEQ ID NO: 5)
5. A=P-X1-Arg(R2)-Pro-Gly-Gly-Gly-Asn(R3)-Gly-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-Leu-O— (SEQ ID NO: 6) or is P-X1-Arg(R2)-Pro-Gly-Gly-Gly-Asn(R3)-Gly-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-X2 (SEQ ID NO: 7)
The definitions for P, X1, X2 consistingly apply to all these possible embodiments for A and the resulting peptide-solid phase conjugates.
We found and report here for the first time that said Tyr residue is equally prone to erratic alkylation upon cleavage from Wang resin, negatively affecting product purity. In case of the Hirulog, such modification appears to be promoted by a proximity effect similar to the observations made for Trp by Atherton et al.; however, alkylation of Tyr e.g. in case of Arginine deprotection has been never reported as a general issue, quite in contrast to Trp (Atherton et al., 1989, Solid phase synthesis: A practical approach, IRL press, Oxford). Further, Atherton's observations pertained to C-terminal Trp only, whereas the Tyr residue in the Hirulog peptide, synthesized in the C to N-terminal direction, is only the juxtaproximal, that is the second last residue next to the C-terminus of the growing peptide chain. In hindsight, without wanting to be bound by theory, this may be explained by that phenoxy moieties are more reactive than average arylic compounds in electrophilic substitution. Indeed phenols are used as scavenging agents in acidolytic cleavage from resin (D. S. King et al., 1990, Int. J. Peptid Protein Res., 36, 255). Still then, said side-reaction has not yet been described or suggested by the skilled person, only terminal Trp's having been believed up to now to be vulnerable in this regard. Consequently Wang resin has been widely employed in the prior art, up to recent, for Hirulog synthesis.
The peptide-solid phase conjugate of the present invention can be synthesized by routine solid phase methods well-known in the art, and well described and referenced in Bodanszky et al., Principles of Peptide Synthesis, 2nd ed., Springer Verlag Berlin Heidelberg 1989). Necessarily, due to the acid-lability of the solid phase attachment, such synthetic strategy employs Fmoc chemistry for carrying out the coupling reactions during solid-phase synthesis. Only the last, terminating D-Phe residue may either be Boc- or Fmoc protected. Such Fmoc protection may be eliminated still on-resin, by standard treatment with e.g. 20% piperidine or other Fmoc deprotecting base reagent to yield the peptide-resin conjugate of the present invention but with a free N-terminal amino group. However, such early Fmoc deprotection exposing early on the N-terminus renders would render said free N-terminal D-Phe residue much more prone to undergo racemisation when subjected to detachment from the resin by acidolysis or in particular global deprotection along with detachment under strongly acidic condition. Hence more preferably, the terminating D-Phe residue is Boc-protected or is protected with another protection group that can be easily removed in strongly acid condition, for avoiding the need of a separate Fmoc deprotection step. For the sake of clarity, this includes e.g. Z-(benzyloxy-carbonyl-) protection group, which may be cleaved, inter alia, by strongly acidic conditions as defined in the present context, though hydrogenolytic or HF promoted cleavage is known to be more efficient. Again, a separate Fmoc deprotection step on the terminating D-Phe residue, exposing early on the N-terminus (terminating in free a-amino group, which may be equally denoted as H-D-Phe- . . . or NH2-D-Phe . . . in formula I) and rendering the now free N-terminal D-Phe prone to racemisation e.g. when subjected to global deprotection along with detachment from the resin by acidolysis, is not as good an option though it is another feasible embodiment of the present invention. Said one-step detachment or cleavage along with global deprotection may be carried out in a solvent mixture such as aqueous TFA and DCM, for instance.
In general, according to the present invention, it is possible either to cleave the protected peptide of formula I from the resin concomittant with or, in initial step, to cleave the protected peptide of formula I from the resin preceding the deprotection or global deprotection of amino acid side chains and, preferably, the N-terminal protection group. In the latter embodiment, it is sequentially subjected firstly to weakly acidic condition for cleavage from resin and secondly to strongly acidid condition for cleavage of all remaining protection groups (global deprotection).
Anyway in both conditions, especially the 2-chloro-trityl-resin (CTC resin for short), and e.g. commercially available, closely similar 4-methoxy- or 4-methyl-trityl-resin or to equal or lesser extent the other resins claimed by the present invention, is well suited for avoiding unwanted modification of the juxtaproximal tyrosine residue upon cleavage and/or deprotection. It prevents undesirable alkylation of a juxtaproximal tyrosine, that is a tyrosine that is second last on the C-terminal side, when the tyrosine is concomittantly deprotected upon cleavage from resin. By virtue of the halogeno substituent, optionally the CTC resin allows of effecting resin cleavage of the still protected peptide and tyrosine under very mild acidolytic reaction conditions, e.g. in 0.5% trifluoro acetic acid (TFA) in dichloromethane (DCM), a condition at which most side chain and N-terminal protection groups will normally not be affected and hence alkylation is prevented by segregation of the different deprotection events in time. —In the following, embodiments referred to with regard to CTC resin in particular, as the most preferred embodiment for the solid phase or resin, tacidly refer to the other resins described and claimed in the present invention.
By definition, according to the present invention, a strongly acidic condition as being opposed to a weakly acidic condition means applying at least 50% (v/v) trifluoro acetic acid (TFA) in the solvent. Further, conversely, a protection group requiring strongly acid condition for removal is a protection group that can be removed, at the very least, by 80% TFA. Accordingly, protection groups that require even stronger acids such as HF do not come under the afore mentioned definition in the context of the present invention. A weakly acidic condition is defined by having 0.01% (v/v) to <50% TFA, preferably having 0.1% to 30% TFA.
Either mode, the peptidyl moiety of the present invention notably shows an unexpected absence of undesirable alkylation of the juxtaproximal tyrosine and it is entirely devoid of diketopiperazine side reaction, another possible side reaction that happens upon cleavage from resin and is known to be particular sensitive to the nature of the last two C-terminal amino acids. Without wanting to be limited by theory, it is speculated that a tyrosine at position 2 of the peptide chain next to the CTC resin handle is just at the optimum distance and spacing as to show some stabilising, hydrophobic stacking of the aromatic phenyl moieties, avoiding e.g. the cyclic arrangement that is the prelude to diketopiperazine formation.
Loading of the CTC resin commonly takes place by nucleophilic substitution of the diphenyl-2′-chlorophenyl-chlormethan derivative (hence CTC, short for chloro-trityl-chloride) and is known to be effective. As an option, preloaded Fmoc-amino-acid-CTC resins are commercially available.
Protection groups and their chemistry are further well-known and well-referenced in the art (see Bodanszky, supra). It is needless to say that of course different protection groups R2 to R9 are suited for protection of individual amino acid side chains, different chemical moieties requiring different protection groups. Examples are e.g. histidine being conventionally protectable with trityl or Boc, lysine being protectable with Boc or allyloxycarbonyl, aspartate being protectable as tert.butylester or allylester. Threonine, serine and tyrosine are usually protected as tert-butyl ether. The protection of arginine will be further discussed below. Different modes of deprotection may be applied, e.g. allylic protection groups are laborously removed by Pd-catalyzed reductive acyl-transfer reaction. Z (benzyloxycarbonyl) groups are less expediently employed since requiring hydrogenolysis for efficient removal. Preferably, the protection groups R2 to R9 are acid-labile, ‘labile’ meaning a cleavage rate of at least 20% of said respective protection group when incubated in DCM solution for up to 5 hours under either weakly or strongly acidic conditions. More preferably according to the present invention, the protection groups R2 to R9 are removed and are only removable under strongly acidic condition as defined above only, that is by way of acidolysis under strongly acidic condition.
R1 is an insoluble, normally polymeric solid phase, e.g. a crosslinked polystyrene/1% divinylbenzol co-polymer. Typically, but not strictly required for working the present invention, such solid phase R1 will of course display further, multiple 2-chloro-trityl-handle moities functionalized with peptide radical A beyond the one shown explicitedly in formula I. More importantly, for being useful in solid-phase synthesis as devised first by Merrifiled, the polymeric solid phase will have a minimum particle size in order to give a true suspension of easily filtratable or pelletable particles of sufficient size, rather than colloidal behaviour. Apart from polystyrene base polymer either directly derivatized with a CTC handle or linker (such as Bayer's 4-carboxytrityl liner, Bayer et al, 13th American Peptide Symposium, Hodges et al., Ed., ESCOM, Leiden, 1994, page 156) or wherein individual benzene moieties of the base polymer have been derivatized to form part of the 2-Chloro-trityl function, further other base polymers such as pure or mixed PEG resins (e.g. Tentagel) or optionally hybrid or grafted resins, wherein e.g. a 2-CTC linker (such as the Bayer linker) has been grafted onto a polystyrene base polymer via a PEG spacer moiety instead of directly reacting the linker with the polystyrene base polymer. Including PEG into a resin provides a more amphilic resin and hence better handling e.g in DCM/TFA mixtures for one-step detachment and deprotection, though loading capacity may then become an issue. —It is to be noted that there are PEG resins which are strictly insoluble of course. However, a technique described by Bayer, et al., Nature 1972, vol. 237, page 512f, described a PEG polymer-borne technique mimicking solid phase separation principle whilst strictly working in solution, the peptide-resin conjugate still being soluble and providing homogenous one phase system. In its preferred meaning in the present context, such resin behaviour is included by the present definition of ‘insoluble’ since essentially allowing of quick and simple, size-based separation by micro- or ultrafiltration techniques at the microscopic level. In a more preferred meaning, ‘insoluble’ refers to, in a given solvent system for peptide synthesis, two phase system, one phase being a truly solid, suspended phase.
Preferably, the solid phase has a mesh size of less than 700 mesh (mesh size as defined by the US Bureau of Standards, retrievable e.g. in Römpps Chemie-Lexikon, 7. Auflage, 1973, Franck'sche Verlagshandlung, W. Keller & Co. Stuttgart/Germany).
Preferably, the 2-chloro-trityl-functionalized solid phase of the present invention has a mesh size of from 50 to 600 mesh (as defined by US Bureau of Standards), more preferably of from 60 to 400 mesh, most preferably of from 100 to 300 mesh.
The Tyrosin of the present invention may be protected by different protection groups, e.g. tert.butyl ether or Z- or more preferably 2-Bromo-Z esters. It is equally possible to use tritylalkohol protection groups such as 2-chloro-trityl or 4-methoxy or 4,4′ methoxy-trityl groups. Preferably, R9 is a trityl or a tert.butyl protection group. More preferably, R9 is a tertiary butyl (tBu) protection group, meaning the tyrosyl side chain is modified to a tertiary-butyl ether. The tBu group is only efficiently removed under strongly acidic condition.
Preferably, alone and in particular in combination with the further preferred embodiments, the arginine protection group R2 is selected from the group consisting of pentamethyldihydrobenzofuranyl-(Pbf), adamantyloxy-carbonyl and isobornyl-oxy-carbonyl, pentamethylenchromanesulfonyl (Pmc), 4-methoxy-2,3,6-trimethylbenzenesulfonyl (Mtr) and its 4-tert.butyl-2,3,5,6-tetramethyl homologue (Tart) or Boc, which are only cleaved under strongly acidic conditions as defined above. More preferably, R2 is Pbf, Pmc, Mtr, most preferably, it is Pbf; upon global deprotection of side chains under strongly acidic conditions, in usually aqueous medium, bystander-alkylation of deprotected tyrosine is not observed with Pmc, Mtr and esp. Pbf. Pbf's cleavage rate is the highest ever.
Carboxy-protection groups for Glu, Asp are well known, e.g. Mpe, O-1-Adamantyl, O-benzyl and even simply alkyl esters may be used, though less commonly used. For sake of ease, typically and preferably tert.butyl groups are used, independently, for protection groups R4, R5, R6, R7, R8.
Protection group R3 may be of paramount importance because of occurring in above sequence Gly-Asp in Hirulog-8, which dipeptide sequence is particularly prone to aspartimide formation as a side reaction. Aspartimide formation may occur in the protected peptide over each subsequent cycle of coupling during linear synthesis to a minor extent (0.1-0.5%), having cumulative effect in the end. Whilst again protection with a trityl protection group or 2-chloro and 4-methyl or 4-methoxy derivatives thereof, is preferred, likewise adamantyl protection group may be used. Most preferably, a trityl protection group is employed.
It is also to be noted that instead of coupling both side chain and Na protected amino acids, Na-alkyl protected dipeptide modules may be used for coupling during linear synthesis; such dipeptides have secondary structure disrupting effect, easing yield and purity of synthesis. E.g. Fmoc-Gly-(N-Hmb)Gly-OH and Fmoc-Gly-(N-Dmb)Gly-OH are commercially available from EMD Biosciences (Novabiochem). It is to be understood that such N-alkyl groups are not considered protection groups in the sense of the present invention, hence their use or presence is optional and not excluded by the structure of formula I.
In a preferred method of detaching and deprotecting the peptide-conjugate of formula I as essentially set forth in the respective claims, the two step sequential scheme of first conducting an acidolysis under weakly acidic conditions for cleaving the protected peptide from the CTC-resin and secondly removing the remaining protection groups under strongly acidic conditions, is applied.
The reason for this is that a one-step global deprotection of the peptide-solid phase conjugate of formula I suffered from opposing solvent requirements of the fully deprotected product and the hydrophobic, conjugated educt, the need for compromise negatively affecting both product purity and yield. The sequential, stepwise approach eliminates such intrinsic drawbacks, allows of better controlling different reactions and hence allows of optimal yield. According to the present invention, it further enjoys the surprising effect of fully suppressing diketopiperazine formation as a side reaction.
Accordingly, a method is devised of detaching and deprotecting the peptide-solid-phase conjugate of formula I as defined above to give a peptide of formula D-Phe-Pro-Arg-Pro-Gly-Gly-Gly-Gly-Asn-Gly-Asp-Phe-Glu-Glu-Ile-Pro-Glu-Glu-Tyr-Leu (SEQ ID NO: 15), characterized in that in a first step, the protected peptide is cleaved from the 2-chloro trityl handle by treatment under weakly acidic condition, preferably with 0.1 to 10% TFA in an polar, aprotic solvent, and that in a second step, the protection groups are removed under strongly acidic condition as defined above.
Preferably, the first step is conducted in a polar, aprotic solvent that is dichloromethane. This is the best solvent to carry out such reaction, in contrast to other solvents such as NMP (N-methylpyrrolidone). It is possible, but not mandatory, to further include a scavenging reagent in the solvent, especially in the solvent system for the second deprotection step, that are present in an amount of 0.1 to 10% (w/w) to the reaction broth for preventing unwanted alkylation of the tyrosine's aromatic core again. Such scavenger intercept reactive alkyl-carbenium ions intermediates that are generated upon removal of the protection groups (which may already happen to a minor extent during cleavage reaction in the first step).
Examples of scavenger is e.g. thioanisol (which also has second, acidolysis-promoting effect—such secondary role and substitutes for aniosol are discussed in Bodanszky M. et al., Int. J. Peptide Protein Res. 23:287). Other examples of scavengers having no such acidolysis effect are phenol and/or trialkylsilanes are used (Stierandova et al., Int. J. Peptide Protein Res. 43, 1994, 31-38).
Preferably, after the first step of cleavage or detachment from resin, the reaction is directly quenched by admixing with pyridine and subsequently recovering the product of step 1 by admixing with water. This way, the product is most simply and efficiently recovered.
In a further embodiment of the present invention, essentially the peptide-solid phase conjugate of formula I is claimed but with the sole difference that the -Arg(R2)-Pro- which is the thrombin cleavage site, is not a standard peptide bond but a chemically modified, pseudoscissile or ‘psi’ bond (the replacement of an amide bond is indicated by the atoms designated in an extra bracket preceded by the akronym ‘psi’, see. Rudinger et al., Drug Design Vol. II, Ed. Ariens, E., Academic Press, New York, p. 319 (1971). More preferably, such psi replacement is -Arg[psiCH2NH]Pro- (Kline, T. et al., 1991, Hirulog peptides with scissile bond replacements resistant to thrombin cleavage, Biochem. Biophys. Res. Commun. 177, 1049-1055). Most easily, such psi bond is e.g. introduced during solid-phase synthesis by normal coupling of the growing, conjugated peptide with the premade, Fmoc-protected psi-dipeptide right away.
It is a further object of the present invention, to extend the above described embodiments and methods to peptide-solid phase conjugates comprising a resin moiety other than the above said CTC resin which, still then, similarly allows of cleaving the peptide moiety from the resin under weakly or mildly acidic conditions as defined above. 2-CTC and related trityl and 4-methoxy- and 4-methyl-trityl resins as defined below are still then considered the best embodiment of the present invention, in accordance with the above said.
As a further object, a peptide-resin conjugate of the formula A-W
is devised wherein A may be any of the above defined embodiments for A, optionally comprising individual amino acid side chain protection groups and wherein R2 to R9 are defined as above where present, wherein and wherein W is a, preferably insoluble, solid phase or solid phase composite which allows of cleaving the peptide moiety under weakly acidic conditions and which is comprising a resin handle or linker of
a. the formula II
a. the formula II

with the proviso that then A where including a residue X2 is always linked via —O— to said handle or linker,
and wherein R′″ is the solid phase and wherein R″1, R″2, R″3 are, independently, hydrogen, 4- or 4′-(C1-C4 alkyl) or 4- or 4′-(C1-C4 alkoxy), and may be the same or different with the proviso that only one of R″1, R″2 may be hydrogen, and wherein R″2 may optionally be 2-Cl with the proviso that then R″1 is H, and wherein more and most preferably, the handle or linker of formula II is selected from the group consisting of 2-chloro-trityl, 4-methoxy-trityl, 4,4′-dimethoxytrityl, 4-methyltrityl,
b. or of the formula III

(which may derived from an amino- or hydroxy functionalized resin by acylation with Bayer's 4-carboxytrityl linker, see E. Bayer, supra) with the proviso that then A, also where including a residue X2, is linked via —O— to said handle or linker, R′″ being defined as above,
c. or of the formula IV

-
- wherein R′″ is a solid phase or polymeric resin, and R″1, R″2, R″3 are, independently, hydrogen, C1-C4 alkyl or C1-C4 alkoxy, and may be the same or different with the provisio that only one of R″1, R″2 may be hydrogen, and wherein L is A (L=A) or wherein L is of formula V

In a further preferred embodiment, the resin handle is of formula VI, the above definitions for radicals R′″, R″1 and R″2 applying,

Again even more preferred is that the resin or resin handle is of formula VII, the above definitions for radicals R′″, R″1 and R″2 applying,

In a further even more preferred embodiment, it is preferred that, where A, optionally including a residue X2, is linked via —O— to said handle or linker of formula VII, R″1, R″2 are independently hydrogen, methyl or methoxy with the provisio that only one of R″1, R″2 may be hydrogen, and that, where A including a residue X2 is linked via —N— to said handle or linker of formula VII, independently are methyl or methoxy, preferably are methoxy. Even more preferably then, A, also where comprising X2, is bound to the handle via a —O— function, R″1 is hydrogen and R″2 is methyl or methoxy and preferably A is a resin or resin handle. Most preferably, R″2 is methyl.
The resin or resin handle composite entity may in principle be any resin employed for synthesis, such as for example a polystyrene-divinylbenzene resin as used by Merrifield along with hydroxybenzyl-phenyl integral linker moieties or by Wang with hydroxy-benzyl-p-benzyloxy moieties, such as for example moieties to which e.g. more acid-labile linkers may be further grafted, or alternatively the latter linkers may be integrally or directly linked to the resin. In principle, a solid phase resin for use in synthesis necessarily comprises at least an integral linker or handle which is part of the solid phase core material; such linker or handle may be considered as an immobilized protection group (Guillier et al., Chem. Rev. 100, 2091-2157, 2000). Examples are e.g. Sieber resin, related xanthenyl type PAL-handle resins, Rink amide resin, Rink acid resin, more complex PEG-grafted polystyrene resins such as tentagel-based Novasyn TG (Novabiochem, Merck Biosciences, Germany) which are available with different grafted handles such as 2′-chloro-trityl, or resins that are constituted by grafting functional handles onto matrix material such as silica gels. Preferably, where the resin is a trityl resin or resin handle, such resin is a 4-methoxy or 4,4′-dimethoxy-trityl resin. Resins as used in the present invention are of standard mesh size, which is about 50-500 mesh, more preferably 100 to 400 mesh. A resin or solid-phase R′″ as shown in formula IV is to be construed as to comprise a crosslinked, polymeric matrix material which may be bound to the handle moiety specified in formulas IV to VII by way of any kind of chemically inert alkyl, alkyloxy, aryloxy or alkylester spacer or linker which is to be considered an integral part of R′″. However, it should be noted that apart from impacting the conditions of cleavage from the resin, the chemical nature of the resin material and in particular the chemical nature of the handle group may well influence synthetic efficiency of coupling and especially lactamisation reactions in a yet poorly understood fashion. The yields of mature peptide at the on-resin stage may differ depending on the type of resin or resin handle employed. For this reason, in an preferred embodiment according to the present invention the resin or resin handle is of formula IV as set forth in the claims in detail, more preferably of formula VI and most preferably of formula VII as set forth in the claims in detail. Examples of such resins or resin handles are (4-methoxyphenyl)-methyl- and (4-methylphenyl)-methyl-polystyrene (Atkinson et al., 2000, J. Org. Chem. 65, 5048), resins in O- or N-linkage to the peptide moiety and their PEG-resin derivatives, respectively. Further examples are e.g. acid-labile HMPB-MBHA o HMPB-BHA resin (Sieber et al., 1987, Tetrahedron Lett. 28, 6147), acid-labile Rink amide resin or Rink acid resin (Rink et al., 1987, Tetrahedron Lett. 28,3787). The term ‘acid-labile’ refers to essentially quantitative cleavage in 2-10% TFA in dichloromethane at ambient temperature for at least an hour. Surprisingly, using such preferred resins having the diphenyl-methyl structural core motif allow for more efficient coupling reaction during linear synthesis and lactamisation; notably, such resins also allow a lower reaction temperature of 15-25° C. as compared to the standard 40° C. required for efficient coupling on e.g. tritylresins.
All reagents were sourced from EMD Biosciences (Madison, Wis./U.S.A.; Novabiochem-brand). Polystyrene-based 2-C1Trt (CTC) resin (Cbl Patras, Greece), preloaded with Fmoc-Leu-OH, was of 100-200 mesh as regards the base polymer and of 60-200 mesh as regards the preloaded, final CTC resin product. Loading density was about 0.60 mmol/g Individual amino acids were sourced as either Fmoc amino acids or, in case of D-Phe, as readily Boc-protected Boc-D-Phe. Couplings were carried out with TCTU in dichloromethane/N-methylpyrrolidone (NMP), in the presence of Hünig-Base (disopropyl-ethyl-amine, DIEA). Usually, 1.5 eq. of the Fmoc or Boc protected amino acid were used, except for coupling of Fmoc-Arg(Pbf), where 2.5 eq. were used. Similarly, the standard coupling reaction time of 60 min. (at 30° C.) was extended to 90 min. in case of Fmoc-Arg (Pbf). In process control of coupling efficiency was effected by means of the Kaiser test or Chloranil tests.
Fmoc deprotection was carried out with 3-4 cycles of 20% piperidine in NMP at 30° C., with suitable rinsing with NMP in between.
Cleavage from 48.3 g resin (about 100 ml swollen resin) as generated in experiment 1 above was achieved with 3 cycles of 15 min. each at 15° C., 2% (w/w) TFA, 1% (w/w) triethylsilane (TES) in dichloromethane. The reaction was stirred by nitrogen bubbling; the colour of the reaction changed from cycle to cycle from yellow/orange to brownish. After each cycle, cleavage reaction was directly quenched by pouring the whole reaction broth into dilute pyridin (pyridine/ethanol 1:9 (v/v)). Resin was then removed by filtration with a frit and subjected to the next cycle. All filtrates were pooled, concentrated to an orange semi-liquid under vacuo (RotaVap), washed with DCM, resuspended in 400 ml double distilled water, stirred at room temperature, filtrated, washed with water and dried. Yield was 28.8 g of a slightly yellow powder of analytical quality (˜90% pure). Product was analyzed by HPLC and LC-MS.
Global deprotection was carried out in DCM diluted with cleavage cockatail (‘CC’), DCM: ‘CC’=1:10 (v/v). ‘CC’ was made up of TFA/thioanisole/phenol/water/TES in the mixing ratio (% w/w): 89:2.5:2.5:5.0:1.0.1 g of dry product from experiment 2 was dissolved in 10 ml DCM diluted as said above with ‘CC’ and stirred for 5 hours at room temperature. The product was then recovered by addition of 50 ml methyl-tertbutyl-ether (MTBE, Fluka Chemie, Buchs/Switzerland), cooling the reaction down to 0° C. in a water bath for 30 min. under stirring and filtrating off the salt precipitate that has formed in the whiletime. The filter cake is rinsed with MTBE several times which is then dried at room temperature, yielding 0.8 g of a crude product of about 55% purity as determined by HPLC. The total yield jointly over steps 2 and 3 is about 55%.
Using HPLC LC-MS analytics, it could be shown that upon cleavage from resin and global deprotection at strongly acidic conditions, 1-10% of the peptide product proved alkylated in case of Wang resin, whereas no such modification could be observed upon cleavage from CTC resin. MS analysis allowed of mapping that modification to the tyrosyl residue. Synthetic procedure as described above.
Claims (42)
1. A peptide-resin conjugate A-W, wherein A=P-X1-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-X2 (SEQ ID NO:3), wherein X1 is a peptidyl moiety of 0 to 200 amino acids, X1 optionally comprising protection groups on individual amino acid side chains, wherein R9 is an amino side chain protection group and wherein X2 is a single amino acid residue linked to the solid phase via —O— and optionally being side chain or C-terminally protected, and wherein P is H or is a protection group selected from the group consisting of Boc, Fmoc, Dde, Nps, Alloc, Z, and R4, R5, R6, R7 and R8 are amino acid side chain protection groups, and
wherein W is a solid phase composite comprising a resin handle or linker
a) of the formula II

with the proviso that then when A, where including includes a residue X2, A is always linked via —O— to said handle or linker,
and wherein R′″ is a solid phase, and wherein R″1, and R″2, R″3 are, independently, H, 4-(C1-C4 alkyl) or 4′-(C1-C4 alkyl) or 4-(C1-C4 alkoxy) or 4′-(C1-C4 alkoxy), and may be are the same or different with the proviso that only one of R″1, R″2 may can be H, and wherein R″2 may optionally be 2-Cl with the proviso that then when R″1 is H,
b) or of the formula III

with the proviso that then when A, where including includes a residue X2, A is linked via —O— to said handle or linker, and R′″ being defined as above is a solid phase,
c) or of the formula IV

wherein R′″ is defined as above and R″1, R″2, R″3 are, independently, H, C1-C4 alkyl or C1-C4 alkoxy, and may be are the same or different with the provisio proviso that only one of R″1, R″2 may can be H, and
wherein L is A(L=A) or wherein L is of formula V

and wherein W allows of cleaving the peptide moiety under weakly acidic conditions of 0.1% to 30% trifluoroacetic acid.
2. The peptide-resin conjugate of claim 1 , characterized in that wherein W is of formula II as defined or is of formula VI,

the above definitions for radicals R′″, R″1 and R″ 2 applying.
3. The peptide-resin conjugate of claim 2 , characterized in that wherein W is of formula VII,

the above definitions for radicals R″1 and R″2 applying and wherein R″1, R″ 2 are, independently, H, methyl or methoxy with the provisio proviso that only one of R″1, R″2 may can be H, and that, where A including a residue X2 is linked via —N— to said handle or linker of formula VII, independently are methyl or methoxy, preferably are methoxy.
4. The peptide-resin conjugate of claim 1 , wherein the handle or linker of formula II is selected from the group consisting of 2-chloro-trityl, 4-methoxy-trityl, 4,4′-dimethoxytrityl and 4-methyltrityl.
5. The peptide-resin conjugate according to claim 1 , characterized in that wherein X2 is not Trp, Cys or Arg.
6. The peptide-resin conjugate according to claim 1 , characterized in that wherein X1 comprises 0 to 50 amino acid residues.
7. The peptide-resin conjugate according to claim 1 , characterized in that wherein A=P-X1-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-Leu-O (SEQ ID NO:2).
8. The peptide-resin conjugate of claim 1 , characterized in that wherein R9 is tertiary-butyl.
9. The peptide-resin conjugate according to claim 1 , wherein A=Boc-D-Phe-Pro-Arg(R2)-Pro-Gly-Gly-Gly-Gly-Asn(R3)-Gly-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-Leu-O— or A=Fmoc-D-Phe-Pro-Arg(R2)-Pro-Gly-Gly-Gly-Gly-Asn(R3)-Gly-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-Leu-O— or A=NH2-D-Phe-Pro-Arg(R2)-Pro-Gly-Gly-Gly-Gly-Asn(R3)-Gly-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-Leu-O— and wherein R2, R3, R4, R5, R6, R7, R8, R9 are amino side chain protection groups and wherein R1 is an insoluble solid phase.
10. The peptide-resin conjugate according to claim 1 , characterized in that wherein the solid phase is polymeric and has a mesh size of less than 700 (US Bureau of Standards).
11. The peptide-resin conjugate according to claim 9 , characterized in that wherein R2 is pentamethyldihydrobenzofuranyl, adamantyloxy-carbonyl or isobornyloxycarbonyl, R9 is tert-butyl or a derivative thereof and that R3 to R8 are acid-labile protection groups.
12. The peptide-resin conjugate according to claim 9 , characterized in that wherein R2 is Pbf and that R4 to R9 are acid-labile protection groups that require at least 50% trifluoroacetic acid for removal.
13. The peptide-resin conjugate according to claim 12 , characterized in that wherein R3 is trityl- and that R4, R5, R6, R7 and R8 are tertiary-butyl.
14. The peptide-resin conjugate according to claim 13 , characterized in that wherein R9 is tertiary-butyl.
15. The peptide-resin conjugate according to claim 1 , characterized in that wherein the -Arg(R2)-Pro- which is the thrombin cleavage site, is -Arg[psiCH2NH]Pro-.
16. A Hirulog peptide synthesized using the peptide-resin conjugate according to claim 1.
17. The Hirulog peptide of claim 16, wherein the Hirulog peptide comprises bivalirudin.
18. A process of using a peptide resin conjugate A-W for the synthesis of Bivalirudin, the process comprising
cleaving a protected peptide from the peptide resin conjugate A-W,
wherein
A=Boc-D-Phe-Pro-Arg(R2)-Pro-Gly-Gly-Gly-Gly-Asn(R3)-Gly-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-Leu-O— or
A=Fmoc-D-Phe-Pro-Arg(R2)-Pro-Gly-Gly-Gly-Gly-Asn(R3)-Gly-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-Leu-O— or
A=NH2-D-Phe-Pro-Arg(R2)-Pro-Gly-Gly-Gly-Gly-Asn(R3)-Gly-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-Leu-O—
wherein R2, R3, R4, R5, R6, R7, R8, R9 are amino side chain protection groups, and
wherein W is a solid phase composite comprising a resin handle or linker
a) of the formula II

with the proviso that when A includes a residue X2, A is linked via —O— to said handle or linker,
wherein R′″ is a solid phase, and wherein R″1 and R″2 are independently, H, 4-(C1-C4 alkyl) or 4′-(C1-C4 alkyl) or 4-(C1-C4 alkoxy) or 4′-(C1-C4 alkoxy), and are the same or different with the proviso that only one of R″1, R″2 can be H, and wherein R″2 may optionally be 2-Cl when R″1 is H,
b) or of the formula III

with the proviso that when A includes a residue X2, A is linked via —O— to said handle or linker and R′″ is a solid phase,
c) or of the formula IV

wherein R′″ is defined as above and R″1, R″2, R″3 are, independently, H, C1-C4 alkyl or C1-C4 alkoxy, and are the same or different with the proviso that only one of R″1, R″2 can be H, and
wherein L is A(L=A) or wherein L is of formula V
and wherein W allows of cleaving the peptide moiety under weakly acidic conditions of 0.1% to 30% trifluoroacetic acid.
19. The process according to claim 18, further comprising deprotecting the protected peptide to provide a deprotected peptide.
20. The process according to claim 19, wherein the deprotecting is conducted concomitant with cleaving.
21. The process according to claim 19, wherein the deprotecting is conducted after cleaving.
22. The process according to claim 19, wherein the deprotecting is conducted with a composition comprising a strong acid.
23. The process according to claim 19, wherein the deprotecting is conducted with a composition comprising trifluoroacetic acid.
24. The process according to claim 22, wherein the composition further comprises a scavenger.
25. The process according to claim 24, wherein the scavenger comprises thioanisole, phenol, trialkylsilane, or a combination thereof.
26. The process according to claim 18, wherein the cleaving is conducted with a composition comprising a weak acid.
27. The process according to claim 18, wherein the cleaving is conducted with a composition comprising trifluoroacetic acid.
28. The process according to claim 19, further comprising precipitating the deprotected peptide.
29. The process according to claim 19, further comprising contacting the deprotected peptide with methyl-tertbutyl-ether.
30. The process according to claim 18, wherein W is of formula II.
31. The process according to claim 30, wherein formula II is selected from the group consisting of 2-chloro-trityl, 4-methoxy-trityl, 4,4′-dimethoxytrityl and 4-methyltrityl.
32. The process according to claim 18, wherein R9 is tertiary-butyl.
33. The process according to claim 18, wherein R2 is selected from the group consisting of pentamethyldihydrobenzofuranyl, adamantyloxy-carbonyl, isobornyl-oxy-carbonyl, pentamethylenchromanesulfonyl, 4-methoxy-2,3,6-trimethylbenzenesulfonyl and its 4-tert.butyl-2,3,5,6-tetramethyl homologue or Boc.
34. The process according to claim 18, wherein R3 is trityl- and that R4, R5, R6, R7 and R8 are tertiary-butyl.
35. A process of using a peptide resin conjugate A-W for the synthesis of a Hirulog peptide, the process comprising
cleaving a protected peptide from the peptide resin conjugate A-W, and
deprotecting the protected peptide,
wherein A=P-X1-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-X2 (SEQ ID NO:3), wherein X1 is a peptidyl moiety of 0 to 200 amino acids, X1 optionally comprising protection groups on individual amino acid side chains, wherein R9 is an amino side chain protection group and wherein X2 is a single amino acid residue linked to the solid phase via —O— and optionally being side chain or C-terminally protected, and wherein P is H or is a protection group selected from the group consisting of Boc, Fmoc, Dde, Nps, Alloc, Z, and R4, R5, R6, R7 and R8 are amino acid side chain protection groups, and
wherein W is a solid phase composite comprising a resin handle or linker
a) of the formula II

with the proviso that when A includes a residue X2, A is linked via —O— to said handle or linker,
wherein R′″ is a solid phase, and wherein R″1 and R″2 are independently, H, 4-(C1-C4 alkyl) or 4′-(C1-C4 alkyl) or 4-(C1-C4 alkoxy) or 4′-(C1-C4 alkoxy), and are the same or different with the proviso that only one of R″1, R″2 can be H, and wherein R″2 may optionally be 2-Cl when R″1 is H,
b) or of the formula III

with the proviso that when A includes a residue X2, A is linked via —O— to said handle or linker and R′″ is a solid phase,
c) or of the formula IV

wherein R′″ is defined as above and R″1, R″2, R″3 are, independently, H, C1-C4 alkyl or C1-C4 alkoxy, and are the same or different with the proviso that only one of R″1, R″2 can be H, and
wherein L is A(L=A) or wherein L is of formula V

and wherein W allows of cleaving the peptide moiety under weakly acidic conditions of 0.1% to 30% trifluoroacetic acid.
36. The process according to claim 35, wherein the deprotecting is conducted concomitant with cleaving.
37. The process according to claim 35, wherein the deprotecting is conducted after the cleaving.
38. The process according to claim 35, wherein W is of formula II and is selected from the group consisting of 2-chloro-trityl, 4-methoxy-trityl, 4,4′-dimethoxytrityl and 4-methyltrityl.
39. The process according to claim 35, wherein X2 is not Trp, Cys or Arg.
40. The process according to claim 35 wherein A=P-X1-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-Leu-O (SEQ ID NO:2).
41. The process according to claim 35, wherein A=Boc-D-Phe-Pro-Arg(R2)-Pro-Gly-Gly-Gly-Gly-Asn(R3)-Gly-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-Leu-O— or A=Fmoc-D-Phe-Pro-Arg(R2)-Pro-Gly-Gly-Gly-Gly-Asn(R3)-Gly-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-Leu-O— or A=NH2-D-Phe-Pro-Arg(R2)-Pro-Gly-Gly-Gly-Gly-Asn(R3)-Gly-Asp(R4)-Phe-Glu(R5)-Glu(R6)-Ile-Pro-Glu(R7)-Glu(R8)-Tyr(R9)-Leu-O— and wherein R2, R3, R4, R5, R6, R7, R8, R9 are amino side chain protection groups.
42. The process according to claim 35, wherein the Hirulog peptide is Bivalirudin.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/659,770 USRE46830E1 (en) | 2004-10-19 | 2005-10-19 | Method for solid phase peptide synthesis |
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP04024812 | 2004-10-19 | ||
| EP04024812 | 2004-10-19 | ||
| PCT/EP2005/011226 WO2006045503A1 (en) | 2004-10-19 | 2005-10-19 | Method for solid phase peptide synthesis |
| US11/665,541 US7939629B2 (en) | 2004-10-19 | 2005-10-19 | Method for solid phase peptide synthesis |
| US14/659,770 USRE46830E1 (en) | 2004-10-19 | 2005-10-19 | Method for solid phase peptide synthesis |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| USRE46830E1 true USRE46830E1 (en) | 2018-05-08 |
Family
ID=35427609
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/659,770 Active 2027-07-25 USRE46830E1 (en) | 2004-10-19 | 2005-10-19 | Method for solid phase peptide synthesis |
| US11/665,541 Ceased US7939629B2 (en) | 2004-10-19 | 2005-10-19 | Method for solid phase peptide synthesis |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/665,541 Ceased US7939629B2 (en) | 2004-10-19 | 2005-10-19 | Method for solid phase peptide synthesis |
Country Status (11)
| Country | Link |
|---|---|
| US (2) | USRE46830E1 (en) |
| EP (1) | EP1737889B1 (en) |
| JP (1) | JP4903709B2 (en) |
| CN (2) | CN102225966B (en) |
| AT (1) | ATE480561T1 (en) |
| DE (1) | DE602005023429D1 (en) |
| DK (1) | DK1737889T3 (en) |
| ES (1) | ES2352204T3 (en) |
| IL (1) | IL182698A (en) |
| PT (1) | PT1737889E (en) |
| WO (1) | WO2006045503A1 (en) |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MX2008003552A (en) * | 2005-09-14 | 2008-11-12 | Novetide Ltd | Process for production of bivalirudin. |
| US20080287650A1 (en) * | 2007-03-01 | 2008-11-20 | Avi Tovi | High purity peptides |
| US7598343B1 (en) | 2008-07-27 | 2009-10-06 | The Medicines Company | Pharmaceutical formulations of bivalirudin and processes of making the same |
| US7582727B1 (en) | 2008-07-27 | 2009-09-01 | The Medicinces Company | Pharmaceutical formulations of bivalirudin and processes of making the same |
| CA2736126C (en) * | 2008-09-03 | 2016-11-29 | Scinopharm Taiwan Ltd. | Process for making bivalirudin |
| DK2382232T3 (en) * | 2008-12-29 | 2013-12-16 | Lonza Braine Sa | Process for the preparation of bivalirudin |
| CN101475631B (en) * | 2009-01-08 | 2011-08-17 | 苏州中科天马肽工程中心有限公司 | Liquid phase synthesizing method for bivalirudin |
| US20110160431A1 (en) | 2009-04-06 | 2011-06-30 | Novetide, Ltd. | Production of peptides containing poly-gly sequences using fmoc chemistry |
| CN101555274B (en) * | 2009-05-15 | 2013-08-21 | 海南双成药业股份有限公司 | Preparation method of polypeptide solid-phase synthesis bivalirudin crude product |
| WO2013042129A1 (en) | 2011-09-23 | 2013-03-28 | Natco Pharma Limited | Improved process for preparation of bivalirudin |
| US9388212B2 (en) | 2013-02-21 | 2016-07-12 | Chemical & Biopharmaceutical Laboratories Of Patras S.A. | Solid phase peptide synthesis via side chain attachment |
| JP6382488B2 (en) * | 2013-02-21 | 2018-08-29 | ケミカル アンド バイオファーマシューティカル ラボラトリーズ オブ パトラ エス.エー. | Solid phase peptide synthesis via side chain bonds |
| GB201310921D0 (en) | 2013-06-19 | 2013-07-31 | Chemical & Biopharmaceutical Lab Of Patras S A | Peptide-resin conjugate and use thereof |
| CN107001409A (en) * | 2014-09-26 | 2017-08-01 | 株式会社钟化 | The manufacture method of hydrophobic peptide |
| CN108383905A (en) * | 2016-12-30 | 2018-08-10 | 江苏金斯瑞生物科技有限公司 | A kind of preparation method of bivalirudin |
| CN112111001B (en) * | 2019-06-19 | 2021-10-29 | 翰宇药业(武汉)有限公司 | Method for synthesizing thymosin T alpha-1 |
| JP2020138975A (en) * | 2020-05-18 | 2020-09-03 | ケミカル アンド バイオファーマシューティカル ラボラトリーズ オブ パトラ エス.エー. | Solid-phase peptide synthesis via side chain bond |
Citations (151)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4093610A (en) | 1977-01-31 | 1978-06-06 | American Home Products Corporation | Process for producing triglycyl-lysine vasopressin and intermediates therefor |
| US4108846A (en) | 1977-02-01 | 1978-08-22 | Hoffmann-La Roche Inc. | Solid phase synthesis with base N alpha-protecting group cleavage |
| US4169141A (en) | 1978-01-30 | 1979-09-25 | Shering Corporation | 1-Peptidyl derivatives of di-O-aminoglycosyl-1,3-diaminocyclitol antibacterial agents |
| WO1991002750A1 (en) | 1989-08-18 | 1991-03-07 | Biogen, Inc. | Novel inhibitors of thrombin |
| US5166394A (en) | 1990-05-23 | 1992-11-24 | Hoechst Aktiengesellschaft | Coupling reagent for peptide synthesis |
| WO1993005065A1 (en) | 1991-08-30 | 1993-03-18 | Porton Developments Limited | Preparation of peptides by a solid-phase synthesis and intermediates therefor |
| US5242810A (en) | 1990-12-07 | 1993-09-07 | Biogen, Inc. | Bifunctional inhibitors of thrombin and platelet activation |
| CA2120302A1 (en) | 1993-04-01 | 1994-10-02 | Alfred Jonczyk | Linear adhesion inhibitors |
| US5362858A (en) | 1992-02-20 | 1994-11-08 | Transgene S.A. | Polyethylene glycol-hirudin conjugates, process for preparing them and their use for the treatment of thromboses |
| US5371184A (en) | 1992-02-05 | 1994-12-06 | Mallinckrodt Medical, Inc. | Radiolabelled peptide compounds |
| US5393873A (en) | 1991-02-07 | 1995-02-28 | Basf Aktiengesellschaft | Peptides with anticoagulant activity |
| US5425936A (en) | 1989-08-18 | 1995-06-20 | Biogen, Inc. | Inhibitors of thrombin |
| US5443827A (en) | 1993-05-03 | 1995-08-22 | President And Fellows Of Harvard College | Fibrin-targeted inhibitors of thrombin |
| US5449761A (en) | 1993-09-28 | 1995-09-12 | Cytogen Corporation | Metal-binding targeted polypeptide constructs |
| US5455181A (en) | 1991-11-26 | 1995-10-03 | Basf Aktiengesellschaft | Thrombin-inhibitory proteins from terrestrial leeches |
| US5516656A (en) | 1990-11-08 | 1996-05-14 | Nippon Mining Company, Limited | Production of a new hirudin analog and anticoagulant pharmaceutical composition containing the same |
| US5541161A (en) | 1990-02-13 | 1996-07-30 | Merrell Pharmaceuticals Inc. | Stabilized sulfonate, sulfate, phosphonate and phosphate derivatives of hirudin |
| US5574012A (en) | 1990-07-24 | 1996-11-12 | Merrell Pharmaceuticals Inc. | Analogs of hirudin having anti-platelet activity |
| US5602231A (en) | 1991-06-14 | 1997-02-11 | Zeneca Limited | Process for making peptides |
| US5624822A (en) | 1989-12-22 | 1997-04-29 | Basf Aktiengesellschaft | Hirudin fusion proteins and preparation of hirudin |
| US5656600A (en) | 1993-03-25 | 1997-08-12 | Corvas International, Inc. | α-ketoamide derivatives as inhibitors of thrombosis |
| US5659041A (en) | 1993-07-19 | 1997-08-19 | Resolution Pharmaceuticals, Inc. | Hydrazino-type radionuclide chelators having an N3 S configuration |
| US5663141A (en) | 1989-12-01 | 1997-09-02 | Basf Aktiengesellschaft | Hirudin/polyalkylene glycol conjugates and hirudin muteins |
| US5662885A (en) | 1994-07-22 | 1997-09-02 | Resolution Pharmaceuticals Inc. | Peptide derived radionuclide chelators |
| US5674838A (en) | 1994-02-10 | 1997-10-07 | Hoechst Aktiengesellschaft | Hirudin derivatives and a process for their preparation |
| US5681721A (en) | 1993-07-15 | 1997-10-28 | Gruenenthal Gmbh | Bifunctional urokinase variants with improved fibrinolytic characteristics and thrombin inhibiting effect |
| US5681925A (en) | 1993-06-11 | 1997-10-28 | Merrell Pharmaceuticals Inc. | Trifunctional antithrombin and antiplatelet peptides |
| US5681926A (en) | 1993-06-16 | 1997-10-28 | Merck & Co., Inc. | Thrombin receptor binding peptides |
| US5686564A (en) | 1992-04-25 | 1997-11-11 | Novartis Corporation | Peptide derivatives corresponding to the carboxy terminal sequence of hirudin |
| US5698104A (en) | 1994-09-07 | 1997-12-16 | Dong Kook Pharmaceutical Co., Ltd. | Purification process for hirudin using affinity chromatography |
| US5719128A (en) | 1991-02-04 | 1998-02-17 | Akzo Nobel N.V. | Factor IIa inhibitors |
| US5723576A (en) | 1993-04-16 | 1998-03-03 | Development Biotechnological Processes S.N.C. | Thrombin inhibitors, the preparation thereof and the use thereof for therapeutical, prophylactic and diagnostic applications |
| US5747453A (en) | 1995-06-06 | 1998-05-05 | Alza Corporation | Method for increasing the electrotransport flux of polypeptides |
| US5759542A (en) | 1994-08-05 | 1998-06-02 | New England Deaconess Hospital Corporation | Compositions and methods for the delivery of drugs by platelets for the treatment of cardiovascular and other diseases |
| US5767078A (en) | 1995-06-07 | 1998-06-16 | Johnson; Dana L. | Agonist peptide dimers |
| US5767235A (en) | 1991-03-05 | 1998-06-16 | Nippon Mining Company Limited | Anticoagulant hirudin variants and methods for their production |
| US5786330A (en) | 1993-10-07 | 1998-07-28 | Adir Et Compagnie | Peptide compounds which are therapeutically active in the cascade of blood coagulation, process for preparing them and pharmaceutical compositions containing them |
| US5789540A (en) | 1987-01-23 | 1998-08-04 | Merrell Pharmaceuticals Inc. | Anticoagulant peptides |
| US5817758A (en) | 1995-06-07 | 1998-10-06 | Terrapin Technologies, Inc. | P-nitrobenzyl side-chain protection for solid-phase synthesis |
| WO1998050563A1 (en) | 1997-05-01 | 1998-11-12 | Ppl Therapeutics (Scotland) Ltd. | Methods of production of an amidated peptide through the use of a fusion protein |
| US5837808A (en) | 1991-08-20 | 1998-11-17 | Baxter International Inc. | Analogs of hirudin |
| US5886146A (en) | 1992-02-14 | 1999-03-23 | Corvas International, Inc. | Inhibitors of thrombosis |
| US5910481A (en) | 1995-11-13 | 1999-06-08 | Immuno Ag | Hybrid proteins with modified activity |
| US5968476A (en) | 1992-05-21 | 1999-10-19 | Diatide, Inc. | Technetium-99m labeled peptides for thrombus imaging |
| US5972648A (en) | 1993-09-28 | 1999-10-26 | Japan Energy Corporation | Hirudin analogs, methods of manufacture thereof and anticoagulant compositions having these as active ingredients |
| US5976841A (en) | 1994-11-17 | 1999-11-02 | Gruenenthal Gmbh | Proteins having fibrinolytic and coagulation--inhibiting properties |
| US6005071A (en) | 1987-01-23 | 1999-12-21 | Merrell Pharmaceuticals Inc. | Anticoagulant peptides |
| US6060451A (en) | 1990-06-15 | 2000-05-09 | The National Research Council Of Canada | Thrombin inhibitors based on the amino acid sequence of hirudin |
| US6127337A (en) | 1993-10-25 | 2000-10-03 | National Research Council Of Canada | Bivalent thrombin inhibitors |
| US6133011A (en) | 1994-11-30 | 2000-10-17 | Gruenenthal Gmbh | Chimeric proteins having fibrinolytic and thrombin-inhibiting properties |
| US6143719A (en) | 1995-06-09 | 2000-11-07 | The Regents Of The University Of Michigan | Bradykinin analogs as selective thrombin inhibitors |
| US6156540A (en) | 1993-12-22 | 2000-12-05 | Human Genome Sciences, Inc. | Thrombin inhibitor |
| US6239101B1 (en) | 1989-07-05 | 2001-05-29 | Oklahoma Medical Research Foundation | Thrombin binding polypeptides |
| US6265204B1 (en) | 1997-01-17 | 2001-07-24 | Genencor International, Inc. | DNA sequences, vectors, and fusion polypeptides for secretion of polypeptides in filamentous fungi |
| US6281331B1 (en) | 1998-03-23 | 2001-08-28 | Trimeris, Inc. | Methods and compositions for peptide synthesis |
| US6333189B1 (en) | 1996-06-06 | 2001-12-25 | Alza Corporation | Method of making an electrotransport device |
| US20020045589A1 (en) | 2000-03-30 | 2002-04-18 | Gary Shen | Hirulog-like peptide and gene therapy |
| US6432921B2 (en) | 1995-11-03 | 2002-08-13 | Akzo Nobel N.V. | Thrombin inhibitors |
| US6506761B1 (en) | 2000-04-14 | 2003-01-14 | Corvas International, Inc. | Substituted hydrazinyl heteroaromatic inhibitors of thrombin |
| US6514730B1 (en) | 1991-03-21 | 2003-02-04 | Consortium für elektrochemische Industrie GmbH | Secretion of hirudin derivatives |
| US6544750B1 (en) | 1999-08-17 | 2003-04-08 | Thromgen, Inc. | Peptide analogs as selective inhibitors of thrombin activation of protease activated receptor 1 |
| EP1314745A1 (en) | 2001-11-27 | 2003-05-28 | Beadtech Inc. | Process for preparing trityl group containing polystyrene resins |
| US6579678B1 (en) | 1996-12-18 | 2003-06-17 | Maxygen, Inc. | Methods and compositions for polypeptide engineering |
| CA2496739A1 (en) | 2002-08-26 | 2004-03-04 | A & Pep Inc. | Method for synthesizing peptides |
| US6703364B2 (en) | 2001-07-23 | 2004-03-09 | Cleveland State University | Thrombin generation inhibitors |
| US6706512B2 (en) | 2001-06-08 | 2004-03-16 | Emory University | Antithrombotic thrombin variants |
| US6710031B2 (en) | 2000-10-04 | 2004-03-23 | Ajinomoto Co., Inc. | Protein having antithrombotic activity and method for producing the same |
| US6825168B2 (en) | 1997-03-13 | 2004-11-30 | Auburn University | Antithrombin protein and DNA sequences from black fly |
| US6875893B2 (en) | 2002-05-23 | 2005-04-05 | Cephalon, Inc. | Preparations of a sulfinyl acetamide |
| US20050090651A1 (en) | 2001-09-27 | 2005-04-28 | Adprotech Limited Cesterford Research Park, Little Chesterford Saffr | Polymeric compounds |
| US6897289B1 (en) | 1999-05-20 | 2005-05-24 | Lipotec, S.A. | Peptide synthesis procedure in solid phase |
| US20050165217A1 (en) | 2003-12-31 | 2005-07-28 | Guinn Martin R. | Process and systems for peptide synthesis |
| US6927279B2 (en) | 1998-08-20 | 2005-08-09 | Auburn University | Antithrombin nucleotides and proteins from horn fly |
| US20060063699A1 (en) | 1998-03-09 | 2006-03-23 | Larsen Bjarne D | Pharmacologically active peptide conjugates having a reduced tendency towards enzymatic hydrolysis |
| US7060484B1 (en) | 1993-11-12 | 2006-06-13 | Gilead Sciences, Inc. | Polypeptides and coagulation therapy |
| US7074765B2 (en) | 2003-05-01 | 2006-07-11 | The Regents Of The University Of Michigan | Synthetic peptide analogs of Arg-Pro-Pro-Gly-Phe as selective inhibitors of thrombin and thrombin activation of protease activated receptors 1 and 4 |
| US7081447B2 (en) | 2001-09-10 | 2006-07-25 | Novel Science International Gmbh | Organic compounds with biological activity as thrombin inhibitors and use thereof |
| US20060172319A1 (en) | 2004-07-12 | 2006-08-03 | Applera Corporation | Mass tags for quantitative analyses |
| US7135534B2 (en) | 2003-07-24 | 2006-11-14 | Rajiv Gandhi Centre For Biotechnology | Polymer support for solid phase peptide synthesis and process for preparation thereof |
| US7138489B2 (en) | 2002-04-11 | 2006-11-21 | Daiichi Asubio Pharma Co., Ltd. | Method for producing a modified peptide |
| US7144902B1 (en) | 1999-04-09 | 2006-12-05 | Abbott Gmbh & Co. Kg | Prodrugs of thrombin inhibitors |
| US20060276626A1 (en) | 2005-05-03 | 2006-12-07 | Avi Tovi | Methods for the production of peptide derivatives |
| US7176282B1 (en) | 1996-09-09 | 2007-02-13 | Zealand Pharma A/S | Solid-phase peptide synthesis and agent for use in such synthesis |
| US20070042946A1 (en) | 2003-02-27 | 2007-02-22 | Feng Ni | Peptide inhibitors of thrombin as potent anticoagulants |
| US20070055048A1 (en) | 2003-07-04 | 2007-03-08 | Vincent Cool | Process for supported phase synthesis |
| US20070093423A1 (en) | 2005-09-14 | 2007-04-26 | Avi Tovi | Process for production of Bivalirudin |
| WO2007067979A2 (en) | 2005-12-09 | 2007-06-14 | Bracco International B.V. | Targeting vector-phospholipid conjugates |
| US20080025966A1 (en) | 2004-01-30 | 2008-01-31 | Currie Mark G | Methods And Compositions For The Treatment Of Gastrointestinal disorders |
| US20080051558A1 (en) | 2006-03-10 | 2008-02-28 | Yiming Zhou | Method of preparing bivalirudin |
| US7348404B2 (en) | 1996-09-09 | 2008-03-25 | Zealand Pharma A/S | Solid-phase peptide synthesis and agent for use in such synthesis |
| US7351771B2 (en) | 2002-12-20 | 2008-04-01 | Hoffmann-La Roche Inc. | Process for regenerating 2-chlorotrityl resins |
| US7407982B2 (en) | 2001-01-23 | 2008-08-05 | Haemosys Gmbh | Oligo or polyalkylene glycol-coupled thrombin inhibitors |
| US7423021B2 (en) | 2003-01-15 | 2008-09-09 | Nsci Novel Science International Gmbh | Peptidic thrombin inhibitors |
| WO2008109079A2 (en) | 2007-03-01 | 2008-09-12 | Novetide, Ltd. | High purity peptides |
| US7425533B2 (en) | 2003-06-26 | 2008-09-16 | Merck Patent Gmbh | Modified hirudin proteins and T-cell epitopes in hirudin |
| US7427260B2 (en) | 2001-03-30 | 2008-09-23 | Mayo Foundation For Medical Education And Research | Efficient methods for solid phase synthesis using trityl chloride resins |
| US20080253992A1 (en) | 2006-10-03 | 2008-10-16 | Neose Technologies, Inc. | Methods for the purification of polypeptide conjugates |
| WO2008155658A2 (en) | 2007-06-18 | 2008-12-24 | Institute Of Zoology Of The Slovak Academy Of Sciences | Thrombin inhibitor |
| CN101372512A (en) | 2007-08-23 | 2009-02-25 | 中国人民解放军军事医学科学院生物工程研究所 | Anticoagulated blood polypeptides and uses thereof |
| US20090054319A1 (en) | 2005-03-17 | 2009-02-26 | Microbia, Inc. | Methods and Compositions for the Treatment of Hypertension and Gastrointestinal Disorders |
| US20090062511A1 (en) | 2007-09-05 | 2009-03-05 | Raghavendracharyulu Venkata Palle | Process for the preparation of bivalirudin and its pharmaceutical compositions |
| US20090131636A1 (en) | 2002-03-01 | 2009-05-21 | Bracco International B.V. | Targeting vector-phospholipid conjugates |
| CN101475631A (en) | 2009-01-08 | 2009-07-08 | 苏州中科天马肽工程中心有限公司 | Liquid phase synthesizing method for bivalirudin |
| US7582727B1 (en) | 2008-07-27 | 2009-09-01 | The Medicinces Company | Pharmaceutical formulations of bivalirudin and processes of making the same |
| US7598343B1 (en) | 2008-07-27 | 2009-10-06 | The Medicines Company | Pharmaceutical formulations of bivalirudin and processes of making the same |
| US20090269422A1 (en) | 2004-04-12 | 2009-10-29 | Cheng-Der Tony Yu | Methods for controlling angiogenesis and cell proliferation |
| US7645858B2 (en) | 2003-08-02 | 2010-01-12 | Almac Sciences (Scotland) Limited | Method of peptide synthesis of peptides containing a proline residue or hydroxyproline residue at or adjacent to the C-terminal end of the peptide |
| US20100056755A1 (en) | 2008-09-03 | 2010-03-04 | Scinopharm Taiwan Ltd. | Process for Making Bivalirudin |
| US7691968B2 (en) | 2002-05-03 | 2010-04-06 | Avecia Biologics Limited | Process for the synthesis of peptides amides by side-chain attachment to a solid phase |
| US7713928B1 (en) | 2009-08-20 | 2010-05-11 | The Medicines Company | Ready-to-use bivalirudin compositions |
| WO2010054503A1 (en) | 2008-11-17 | 2010-05-20 | 中国人民解放军军事医学科学院生物工程研究所 | Anticoagulant polypeptides and uses thereof |
| US20100160604A1 (en) | 2006-11-21 | 2010-06-24 | Ipsen Manufacturing Ireland Limited | Boc and fmoc solid phase peptide synthesis |
| US20100168443A1 (en) | 2006-11-02 | 2010-07-01 | University Of Virginia Patent Foundation | Ethoid-Containing Compounds, Methods for Preparing Ethoid-Containing Compounds, and Methods of Use |
| US20100184952A1 (en) | 2007-07-25 | 2010-07-22 | Ajinomoto Co., Inc | Method for selective removal of dibenzofulvene derivative |
| WO2010117725A2 (en) | 2009-04-06 | 2010-10-14 | Novetide, Ltd. | Production of peptides containing poly-gly sequences using fmoc chemistry |
| US7824677B2 (en) | 1997-03-10 | 2010-11-02 | Genentech, Inc. | Method for using antibodies for inhibiting blood coagulation |
| US7829659B2 (en) | 2006-05-02 | 2010-11-09 | Allozyne, Inc. | Methods of modifying polypeptides comprising non-natural amino acids |
| US20100292436A1 (en) | 2009-05-15 | 2010-11-18 | Shanghai Ambiopharm, Inc. | Method for producing bivalirudin |
| US7879792B2 (en) | 2005-06-02 | 2011-02-01 | The Regents Of The University Of Michigan | Synthetic peptide inhibitors of thrombin and thrombin activation of protease activated receptors 1 and 4 |
| WO2011071799A2 (en) | 2009-12-11 | 2011-06-16 | Dr. Reddy's Laboratories Ltd. | Purification of bivalirudin |
| US7985733B1 (en) | 2010-01-06 | 2011-07-26 | The Medicines Company | Buffer-based method for preparing bivalirudin drug product |
| US20110190475A1 (en) | 2008-08-06 | 2011-08-04 | Ajinomoto Co., Inc. | Processes for removal of dibenzofulvene |
| US8008434B2 (en) | 2004-05-28 | 2011-08-30 | Deutsches Krebsforschungszentrum Stiftung Des Offentlichen Rechts | Preparation of solid phase bound peptides or PNAs |
| US8022181B2 (en) | 2006-05-03 | 2011-09-20 | Mallinckrodt Llc | Composition and method for the release of protected peptides from a resin |
| US20110251372A1 (en) | 2008-12-29 | 2011-10-13 | Geoffroy Sommen | Process for the production of bivalirudin |
| US8063018B2 (en) | 2004-06-23 | 2011-11-22 | National Research Council Of Canada | Bivalent thrombin binding molecules comprising linkers |
| US20110288235A1 (en) | 2008-09-03 | 2011-11-24 | Scinopharm Taiwan Ltd. | Process for the Preparation of Pramlintide |
| CN102260323A (en) | 2011-05-30 | 2011-11-30 | 杭州诺泰制药技术有限公司 | Method for preparing bivalirudin by combining solid phase with liquid phase and detection method |
| US20110319594A1 (en) | 2010-06-28 | 2011-12-29 | Shanghai Ambiopharm, Inc. | Method for producing bivalirudin |
| US8101379B2 (en) | 2006-12-15 | 2012-01-24 | Institute of Radiation Medicine Academy of Military Medical Science | Preparation of low bleeding anticoagulant fusion protein and its use |
| CN102336813A (en) | 2011-07-01 | 2012-02-01 | 上海苏豪逸明制药有限公司 | Preparation method for synthesizing proteidin with solid phase polypeptide |
| US20120041173A1 (en) | 2010-08-16 | 2012-02-16 | Cem Corporation | Water soluble solid phase peptide synthesis |
| US8153761B2 (en) | 2003-06-23 | 2012-04-10 | Cem Corporation | Microwave-assisted peptide synthesis |
| US20120135931A1 (en) | 2009-05-05 | 2012-05-31 | Natural Environment Research Council | Method of modifying serine protease inhibitors |
| US20120149868A1 (en) | 2008-05-15 | 2012-06-14 | Novo Nordisk A/S | Purification of peptides prepared by solid phase synthesis |
| US8206967B2 (en) | 2007-07-06 | 2012-06-26 | Medimmune Limited | Method for production of recombinant human thrombin |
| KR20120069288A (en) | 2010-12-20 | 2012-06-28 | 주식회사 씨트리 | Purification method of bivalirudin using soluble sugar alcohol |
| CN102532274A (en) | 2012-02-13 | 2012-07-04 | 成都圣诺生物制药有限公司 | Method for preparing bivalirudin |
| CN102641506A (en) | 2012-04-11 | 2012-08-22 | 承德医学院中药研究所 | Bivalirudin-polyethylene glycol compound |
| US20120232014A1 (en) | 2009-09-04 | 2012-09-13 | Imperial Innovations Limited | Antithrombotic compounds |
| CN102702325A (en) | 2012-06-19 | 2012-10-03 | 深圳翰宇药业股份有限公司 | Preparation method of anticoagulant polypeptide |
| CN102731624A (en) | 2012-06-14 | 2012-10-17 | 无锡市凯利药业有限公司 | Method for synthesis of bivalirudin in solid-phase fragment approach |
| US8314208B2 (en) | 2006-02-10 | 2012-11-20 | Cem Corporation | Microwave enhanced N-fmoc deprotection in peptide synthesis |
| WO2012165546A1 (en) | 2011-05-31 | 2012-12-06 | 味の素株式会社 | Method for producing peptide |
| WO2012174816A1 (en) | 2011-06-23 | 2012-12-27 | 成都圣诺科技发展有限公司 | Bivalirudin preparation method |
| US8361965B2 (en) | 2007-02-16 | 2013-01-29 | Lunan Pharmaceutical Group Corporation | Recombinant chimeric protein of neutrophil inhibitory factor and hirugen, and pharmaceutical composition thereof |
| US20130034547A1 (en) | 2011-08-02 | 2013-02-07 | Kelly Jeffery W | Reliable stabilization of n-linked polypeptide native states with enhanced aromatic sequons located in polypeptide tight turns |
| CN102924575A (en) | 2012-10-31 | 2013-02-13 | 深圳翰宇药业股份有限公司 | Preparation method of bivalirudin |
| US8404724B2 (en) | 2006-12-08 | 2013-03-26 | Millennium Pharmaceuticals, Inc. | Unit dose formulations and methods of treating thrombosis with an oral factor Xa inhibitor |
| WO2013042129A1 (en) | 2011-09-23 | 2013-03-28 | Natco Pharma Limited | Improved process for preparation of bivalirudin |
| US8415454B2 (en) | 2006-07-21 | 2013-04-09 | Solvay (Société Anonyme) | Process for the manufacture of peptides |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK0570493T3 (en) * | 1991-02-08 | 2000-06-26 | Diatide Inc | Technetium-99m-labeled polypeptides for imaging. |
| DE4140381A1 (en) * | 1991-12-07 | 1993-06-09 | Hoechst Ag, 6230 Frankfurt, De | NEW SYNTHETIC ISOHIRUDINE WITH IMPROVED STABILITY |
| CN1088836A (en) * | 1992-12-30 | 1994-07-06 | 中国科学院生物物理研究所 | Lepirudin 023 ludon and complex thereof are used for preparation prevention and treatment thrombotic disease medicine |
-
2005
- 2005-10-19 WO PCT/EP2005/011226 patent/WO2006045503A1/en active Application Filing
- 2005-10-19 US US14/659,770 patent/USRE46830E1/en active Active
- 2005-10-19 EP EP05796944A patent/EP1737889B1/en active Active
- 2005-10-19 DE DE602005023429T patent/DE602005023429D1/en active Active
- 2005-10-19 PT PT05796944T patent/PT1737889E/en unknown
- 2005-10-19 JP JP2007537195A patent/JP4903709B2/en active Active
- 2005-10-19 DK DK05796944.6T patent/DK1737889T3/en active
- 2005-10-19 CN CN2011101120914A patent/CN102225966B/en active Active
- 2005-10-19 AT AT05796944T patent/ATE480561T1/en not_active IP Right Cessation
- 2005-10-19 ES ES05796944T patent/ES2352204T3/en active Active
- 2005-10-19 US US11/665,541 patent/US7939629B2/en not_active Ceased
- 2005-10-19 CN CN2005800435731A patent/CN101094867B/en active Active
-
2007
- 2007-04-19 IL IL182698A patent/IL182698A/en active IP Right Grant
Patent Citations (200)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4093610A (en) | 1977-01-31 | 1978-06-06 | American Home Products Corporation | Process for producing triglycyl-lysine vasopressin and intermediates therefor |
| US4108846A (en) | 1977-02-01 | 1978-08-22 | Hoffmann-La Roche Inc. | Solid phase synthesis with base N alpha-protecting group cleavage |
| US4169141A (en) | 1978-01-30 | 1979-09-25 | Shering Corporation | 1-Peptidyl derivatives of di-O-aminoglycosyl-1,3-diaminocyclitol antibacterial agents |
| US5789540A (en) | 1987-01-23 | 1998-08-04 | Merrell Pharmaceuticals Inc. | Anticoagulant peptides |
| US6005071A (en) | 1987-01-23 | 1999-12-21 | Merrell Pharmaceuticals Inc. | Anticoagulant peptides |
| US6239101B1 (en) | 1989-07-05 | 2001-05-29 | Oklahoma Medical Research Foundation | Thrombin binding polypeptides |
| US5691311A (en) | 1989-08-18 | 1997-11-25 | Biogen, Inc. | Methods for coating invasive devices with inhibitors of thrombin |
| US5196404A (en) | 1989-08-18 | 1993-03-23 | Biogen, Inc. | Inhibitors of thrombin |
| WO1991002750A1 (en) | 1989-08-18 | 1991-03-07 | Biogen, Inc. | Novel inhibitors of thrombin |
| US5425936A (en) | 1989-08-18 | 1995-06-20 | Biogen, Inc. | Inhibitors of thrombin |
| US5433940A (en) | 1989-08-18 | 1995-07-18 | Biogen, Inc. | Inhibitors of thrombin |
| US5196404B1 (en) | 1989-08-18 | 1996-09-10 | Biogen Inc | Inhibitors of thrombin |
| US5663141A (en) | 1989-12-01 | 1997-09-02 | Basf Aktiengesellschaft | Hirudin/polyalkylene glycol conjugates and hirudin muteins |
| US5624822A (en) | 1989-12-22 | 1997-04-29 | Basf Aktiengesellschaft | Hirudin fusion proteins and preparation of hirudin |
| US5541161A (en) | 1990-02-13 | 1996-07-30 | Merrell Pharmaceuticals Inc. | Stabilized sulfonate, sulfate, phosphonate and phosphate derivatives of hirudin |
| US5166394A (en) | 1990-05-23 | 1992-11-24 | Hoechst Aktiengesellschaft | Coupling reagent for peptide synthesis |
| US6060451A (en) | 1990-06-15 | 2000-05-09 | The National Research Council Of Canada | Thrombin inhibitors based on the amino acid sequence of hirudin |
| US5574012A (en) | 1990-07-24 | 1996-11-12 | Merrell Pharmaceuticals Inc. | Analogs of hirudin having anti-platelet activity |
| US5516656A (en) | 1990-11-08 | 1996-05-14 | Nippon Mining Company, Limited | Production of a new hirudin analog and anticoagulant pharmaceutical composition containing the same |
| US5242810A (en) | 1990-12-07 | 1993-09-07 | Biogen, Inc. | Bifunctional inhibitors of thrombin and platelet activation |
| US5719128A (en) | 1991-02-04 | 1998-02-17 | Akzo Nobel N.V. | Factor IIa inhibitors |
| US5393873A (en) | 1991-02-07 | 1995-02-28 | Basf Aktiengesellschaft | Peptides with anticoagulant activity |
| US5767235A (en) | 1991-03-05 | 1998-06-16 | Nippon Mining Company Limited | Anticoagulant hirudin variants and methods for their production |
| US5880258A (en) | 1991-03-05 | 1999-03-09 | Japan Energy Corporation | Anticoagulant hirudin variants and methods for their production |
| US6514730B1 (en) | 1991-03-21 | 2003-02-04 | Consortium für elektrochemische Industrie GmbH | Secretion of hirudin derivatives |
| US5602231A (en) | 1991-06-14 | 1997-02-11 | Zeneca Limited | Process for making peptides |
| US6028170A (en) | 1991-08-20 | 2000-02-22 | Baxter International Inc. | Analogs of hirudin |
| US5837808A (en) | 1991-08-20 | 1998-11-17 | Baxter International Inc. | Analogs of hirudin |
| WO1993005065A1 (en) | 1991-08-30 | 1993-03-18 | Porton Developments Limited | Preparation of peptides by a solid-phase synthesis and intermediates therefor |
| US5455181A (en) | 1991-11-26 | 1995-10-03 | Basf Aktiengesellschaft | Thrombin-inhibitory proteins from terrestrial leeches |
| US5371184A (en) | 1992-02-05 | 1994-12-06 | Mallinckrodt Medical, Inc. | Radiolabelled peptide compounds |
| US5886146A (en) | 1992-02-14 | 1999-03-23 | Corvas International, Inc. | Inhibitors of thrombosis |
| US5362858A (en) | 1992-02-20 | 1994-11-08 | Transgene S.A. | Polyethylene glycol-hirudin conjugates, process for preparing them and their use for the treatment of thromboses |
| US5686564A (en) | 1992-04-25 | 1997-11-11 | Novartis Corporation | Peptide derivatives corresponding to the carboxy terminal sequence of hirudin |
| US5968476A (en) | 1992-05-21 | 1999-10-19 | Diatide, Inc. | Technetium-99m labeled peptides for thrombus imaging |
| US5670479A (en) | 1993-03-25 | 1997-09-23 | Corvas International, Inc. | α-ketoamide derivatives as inhibitors of thrombosis |
| US5656600A (en) | 1993-03-25 | 1997-08-12 | Corvas International, Inc. | α-ketoamide derivatives as inhibitors of thrombosis |
| CA2120302A1 (en) | 1993-04-01 | 1994-10-02 | Alfred Jonczyk | Linear adhesion inhibitors |
| US5723576A (en) | 1993-04-16 | 1998-03-03 | Development Biotechnological Processes S.N.C. | Thrombin inhibitors, the preparation thereof and the use thereof for therapeutical, prophylactic and diagnostic applications |
| US5443827A (en) | 1993-05-03 | 1995-08-22 | President And Fellows Of Harvard College | Fibrin-targeted inhibitors of thrombin |
| US5681925A (en) | 1993-06-11 | 1997-10-28 | Merrell Pharmaceuticals Inc. | Trifunctional antithrombin and antiplatelet peptides |
| US5681926A (en) | 1993-06-16 | 1997-10-28 | Merck & Co., Inc. | Thrombin receptor binding peptides |
| US5681721A (en) | 1993-07-15 | 1997-10-28 | Gruenenthal Gmbh | Bifunctional urokinase variants with improved fibrinolytic characteristics and thrombin inhibiting effect |
| US5659041A (en) | 1993-07-19 | 1997-08-19 | Resolution Pharmaceuticals, Inc. | Hydrazino-type radionuclide chelators having an N3 S configuration |
| US5449761A (en) | 1993-09-28 | 1995-09-12 | Cytogen Corporation | Metal-binding targeted polypeptide constructs |
| US5578288A (en) | 1993-09-28 | 1996-11-26 | Cytogen Corporation | Metal-binding targeted polypeptide constructs |
| US5972648A (en) | 1993-09-28 | 1999-10-26 | Japan Energy Corporation | Hirudin analogs, methods of manufacture thereof and anticoagulant compositions having these as active ingredients |
| US5593656A (en) | 1993-09-28 | 1997-01-14 | Cytogen Corporation | Metal-binding targeted polypeptide constructs |
| US5609847A (en) | 1993-09-28 | 1997-03-11 | Cytogen Corporation | Treatment methods using metal-binding targeted polypeptide constructs |
| US5786330A (en) | 1993-10-07 | 1998-07-28 | Adir Et Compagnie | Peptide compounds which are therapeutically active in the cascade of blood coagulation, process for preparing them and pharmaceutical compositions containing them |
| US6127337A (en) | 1993-10-25 | 2000-10-03 | National Research Council Of Canada | Bivalent thrombin inhibitors |
| US7060484B1 (en) | 1993-11-12 | 2006-06-13 | Gilead Sciences, Inc. | Polypeptides and coagulation therapy |
| US6156540A (en) | 1993-12-22 | 2000-12-05 | Human Genome Sciences, Inc. | Thrombin inhibitor |
| US5674838A (en) | 1994-02-10 | 1997-10-07 | Hoechst Aktiengesellschaft | Hirudin derivatives and a process for their preparation |
| US5976495A (en) | 1994-07-22 | 1999-11-02 | Resolution Pharmaceuticals, Inc. | Peptide derived radionuclide chelators |
| US5780006A (en) | 1994-07-22 | 1998-07-14 | Resolution Pharmaceuticals Inc. | Peptide derived radionuclide chelators |
| US5662885A (en) | 1994-07-22 | 1997-09-02 | Resolution Pharmaceuticals Inc. | Peptide derived radionuclide chelators |
| US5759542A (en) | 1994-08-05 | 1998-06-02 | New England Deaconess Hospital Corporation | Compositions and methods for the delivery of drugs by platelets for the treatment of cardiovascular and other diseases |
| US5698104A (en) | 1994-09-07 | 1997-12-16 | Dong Kook Pharmaceutical Co., Ltd. | Purification process for hirudin using affinity chromatography |
| US5976841A (en) | 1994-11-17 | 1999-11-02 | Gruenenthal Gmbh | Proteins having fibrinolytic and coagulation--inhibiting properties |
| US6133011A (en) | 1994-11-30 | 2000-10-17 | Gruenenthal Gmbh | Chimeric proteins having fibrinolytic and thrombin-inhibiting properties |
| US5747453A (en) | 1995-06-06 | 1998-05-05 | Alza Corporation | Method for increasing the electrotransport flux of polypeptides |
| US6313092B1 (en) | 1995-06-06 | 2001-11-06 | Alza Corporation | Method for increasing the electrotransport flux of polypeptides |
| US5767078A (en) | 1995-06-07 | 1998-06-16 | Johnson; Dana L. | Agonist peptide dimers |
| US5817758A (en) | 1995-06-07 | 1998-10-06 | Terrapin Technologies, Inc. | P-nitrobenzyl side-chain protection for solid-phase synthesis |
| US6143719A (en) | 1995-06-09 | 2000-11-07 | The Regents Of The University Of Michigan | Bradykinin analogs as selective thrombin inhibitors |
| US6432921B2 (en) | 1995-11-03 | 2002-08-13 | Akzo Nobel N.V. | Thrombin inhibitors |
| US6051418A (en) | 1995-11-13 | 2000-04-18 | Immuno Ag | Hybrid proteins with modified activity |
| US5910481A (en) | 1995-11-13 | 1999-06-08 | Immuno Ag | Hybrid proteins with modified activity |
| US6333189B1 (en) | 1996-06-06 | 2001-12-25 | Alza Corporation | Method of making an electrotransport device |
| US7348404B2 (en) | 1996-09-09 | 2008-03-25 | Zealand Pharma A/S | Solid-phase peptide synthesis and agent for use in such synthesis |
| US7176282B1 (en) | 1996-09-09 | 2007-02-13 | Zealand Pharma A/S | Solid-phase peptide synthesis and agent for use in such synthesis |
| US6579678B1 (en) | 1996-12-18 | 2003-06-17 | Maxygen, Inc. | Methods and compositions for polypeptide engineering |
| US6586182B1 (en) | 1996-12-18 | 2003-07-01 | Maxygen, Inc. | Methods and compositions for polypeptide engineering |
| US6265204B1 (en) | 1997-01-17 | 2001-07-24 | Genencor International, Inc. | DNA sequences, vectors, and fusion polypeptides for secretion of polypeptides in filamentous fungi |
| US6590078B2 (en) | 1997-01-17 | 2003-07-08 | Genencor International, Inc. | DNA sequences, vectors, and fusion polypeptides for secretion of polypeptides in filamentous fungi |
| US7824677B2 (en) | 1997-03-10 | 2010-11-02 | Genentech, Inc. | Method for using antibodies for inhibiting blood coagulation |
| US6825168B2 (en) | 1997-03-13 | 2004-11-30 | Auburn University | Antithrombin protein and DNA sequences from black fly |
| WO1998050563A1 (en) | 1997-05-01 | 1998-11-12 | Ppl Therapeutics (Scotland) Ltd. | Methods of production of an amidated peptide through the use of a fusion protein |
| US20110312878A1 (en) | 1998-03-09 | 2011-12-22 | Zealand Pharma A/S | Pharmacologically active peptide conjugates having a reduced tendency towards enzymatic hydrolysis |
| US7414107B2 (en) | 1998-03-09 | 2008-08-19 | Zealand Pharma A/S | Pharmacologically active peptide conjugates having a reduced tendency towards enzymatic hydrolysis |
| US20080139785A1 (en) | 1998-03-09 | 2008-06-12 | Bjarne Due Larsen | Pharmacologically active peptide conjugates having a reduced tendency towards enzymatic hydrolysis |
| US20080227954A1 (en) | 1998-03-09 | 2008-09-18 | Larsen Bjarne D | Pharmacologically active peptide conjugates having a reduced tendency towards enzymatic hydrolysis |
| US20080015152A1 (en) | 1998-03-09 | 2008-01-17 | Larsen Bjarne D | Pharmacologically active peptide conjugates having a reduced tendency towards enzymatic hydrolysis |
| US20080234467A1 (en) | 1998-03-09 | 2008-09-25 | Bjarne Due Larsen | Pharmacologically active peptide conjugates having a reduced tendency towards enzymatic hydrolysis |
| US20070293418A1 (en) | 1998-03-09 | 2007-12-20 | Larsen Bjarne D | Pharmacologically active peptide conjugates having a reduced tendency towards enzymatic hydrolysis |
| US7935786B2 (en) | 1998-03-09 | 2011-05-03 | Zealand Pharma A/S | Pharmacologically active peptide conjugates having a reduced tendency towards enzymatic hydrolysis |
| US20060063699A1 (en) | 1998-03-09 | 2006-03-23 | Larsen Bjarne D | Pharmacologically active peptide conjugates having a reduced tendency towards enzymatic hydrolysis |
| US6281331B1 (en) | 1998-03-23 | 2001-08-28 | Trimeris, Inc. | Methods and compositions for peptide synthesis |
| US6927279B2 (en) | 1998-08-20 | 2005-08-09 | Auburn University | Antithrombin nucleotides and proteins from horn fly |
| US7144902B1 (en) | 1999-04-09 | 2006-12-05 | Abbott Gmbh & Co. Kg | Prodrugs of thrombin inhibitors |
| US6897289B1 (en) | 1999-05-20 | 2005-05-24 | Lipotec, S.A. | Peptide synthesis procedure in solid phase |
| US6544750B1 (en) | 1999-08-17 | 2003-04-08 | Thromgen, Inc. | Peptide analogs as selective inhibitors of thrombin activation of protease activated receptor 1 |
| US20020045589A1 (en) | 2000-03-30 | 2002-04-18 | Gary Shen | Hirulog-like peptide and gene therapy |
| US20040229806A1 (en) | 2000-03-30 | 2004-11-18 | Gary Shen | Hirulog-like peptide and gene therapy |
| US7582602B2 (en) | 2000-03-30 | 2009-09-01 | Shen Garry X | Hirulog-like peptide and gene therapy |
| US6506761B1 (en) | 2000-04-14 | 2003-01-14 | Corvas International, Inc. | Substituted hydrazinyl heteroaromatic inhibitors of thrombin |
| US7084113B2 (en) | 2000-10-04 | 2006-08-01 | Ajinomoto Co., Inc. | Protein having antithrombotic activity and method for producing the same |
| US6710031B2 (en) | 2000-10-04 | 2004-03-23 | Ajinomoto Co., Inc. | Protein having antithrombotic activity and method for producing the same |
| US7407982B2 (en) | 2001-01-23 | 2008-08-05 | Haemosys Gmbh | Oligo or polyalkylene glycol-coupled thrombin inhibitors |
| US7427260B2 (en) | 2001-03-30 | 2008-09-23 | Mayo Foundation For Medical Education And Research | Efficient methods for solid phase synthesis using trityl chloride resins |
| US6706512B2 (en) | 2001-06-08 | 2004-03-16 | Emory University | Antithrombotic thrombin variants |
| US6703364B2 (en) | 2001-07-23 | 2004-03-09 | Cleveland State University | Thrombin generation inhibitors |
| US7074892B2 (en) | 2001-07-23 | 2006-07-11 | Cleveland State University | Thrombin generation inhibitors |
| US7081447B2 (en) | 2001-09-10 | 2006-07-25 | Novel Science International Gmbh | Organic compounds with biological activity as thrombin inhibitors and use thereof |
| US20050090651A1 (en) | 2001-09-27 | 2005-04-28 | Adprotech Limited Cesterford Research Park, Little Chesterford Saffr | Polymeric compounds |
| EP1314745A1 (en) | 2001-11-27 | 2003-05-28 | Beadtech Inc. | Process for preparing trityl group containing polystyrene resins |
| US20090131636A1 (en) | 2002-03-01 | 2009-05-21 | Bracco International B.V. | Targeting vector-phospholipid conjugates |
| US7138489B2 (en) | 2002-04-11 | 2006-11-21 | Daiichi Asubio Pharma Co., Ltd. | Method for producing a modified peptide |
| US7691968B2 (en) | 2002-05-03 | 2010-04-06 | Avecia Biologics Limited | Process for the synthesis of peptides amides by side-chain attachment to a solid phase |
| US6875893B2 (en) | 2002-05-23 | 2005-04-05 | Cephalon, Inc. | Preparations of a sulfinyl acetamide |
| CA2496739A1 (en) | 2002-08-26 | 2004-03-04 | A & Pep Inc. | Method for synthesizing peptides |
| US7351771B2 (en) | 2002-12-20 | 2008-04-01 | Hoffmann-La Roche Inc. | Process for regenerating 2-chlorotrityl resins |
| US7423021B2 (en) | 2003-01-15 | 2008-09-09 | Nsci Novel Science International Gmbh | Peptidic thrombin inhibitors |
| US20070042946A1 (en) | 2003-02-27 | 2007-02-22 | Feng Ni | Peptide inhibitors of thrombin as potent anticoagulants |
| US20090137779A1 (en) | 2003-02-27 | 2009-05-28 | Feng Ni | Peptide inhibitors of thrombin as potent anticoagulants |
| US7456152B2 (en) | 2003-02-27 | 2008-11-25 | National Research Council Of Canada | Peptide inhibitors of thrombin as potent anticoagulants |
| US7074765B2 (en) | 2003-05-01 | 2006-07-11 | The Regents Of The University Of Michigan | Synthetic peptide analogs of Arg-Pro-Pro-Gly-Phe as selective inhibitors of thrombin and thrombin activation of protease activated receptors 1 and 4 |
| US8153761B2 (en) | 2003-06-23 | 2012-04-10 | Cem Corporation | Microwave-assisted peptide synthesis |
| US7425533B2 (en) | 2003-06-26 | 2008-09-16 | Merck Patent Gmbh | Modified hirudin proteins and T-cell epitopes in hirudin |
| US20070055048A1 (en) | 2003-07-04 | 2007-03-08 | Vincent Cool | Process for supported phase synthesis |
| US7135534B2 (en) | 2003-07-24 | 2006-11-14 | Rajiv Gandhi Centre For Biotechnology | Polymer support for solid phase peptide synthesis and process for preparation thereof |
| US7645858B2 (en) | 2003-08-02 | 2010-01-12 | Almac Sciences (Scotland) Limited | Method of peptide synthesis of peptides containing a proline residue or hydroxyproline residue at or adjacent to the C-terminal end of the peptide |
| US7439222B2 (en) | 2003-12-31 | 2008-10-21 | Roche Palo Alto Llc | Process and systems for peptide synthesis |
| US20050165217A1 (en) | 2003-12-31 | 2005-07-28 | Guinn Martin R. | Process and systems for peptide synthesis |
| EP1701969B1 (en) | 2003-12-31 | 2007-10-24 | F.Hoffmann-La Roche Ag | Process for peptide synthesis using a reduced amount of deprotection agent |
| US20080025966A1 (en) | 2004-01-30 | 2008-01-31 | Currie Mark G | Methods And Compositions For The Treatment Of Gastrointestinal disorders |
| US20110224150A1 (en) | 2004-04-12 | 2011-09-15 | Canyon Pharmaceuticals, Inc. | Methods for effecting regression of tumor mass and size in a metastasized tumor |
| US20090269422A1 (en) | 2004-04-12 | 2009-10-29 | Cheng-Der Tony Yu | Methods for controlling angiogenesis and cell proliferation |
| US7795205B2 (en) | 2004-04-12 | 2010-09-14 | Canyon Pharmaceuticals, Inc. | Methods for effecting regression of tumor mass and size in a metastasized pancreatic tumor |
| US8008434B2 (en) | 2004-05-28 | 2011-08-30 | Deutsches Krebsforschungszentrum Stiftung Des Offentlichen Rechts | Preparation of solid phase bound peptides or PNAs |
| US8063018B2 (en) | 2004-06-23 | 2011-11-22 | National Research Council Of Canada | Bivalent thrombin binding molecules comprising linkers |
| US20060172319A1 (en) | 2004-07-12 | 2006-08-03 | Applera Corporation | Mass tags for quantitative analyses |
| US20090054319A1 (en) | 2005-03-17 | 2009-02-26 | Microbia, Inc. | Methods and Compositions for the Treatment of Hypertension and Gastrointestinal Disorders |
| US20060276626A1 (en) | 2005-05-03 | 2006-12-07 | Avi Tovi | Methods for the production of peptide derivatives |
| US7879792B2 (en) | 2005-06-02 | 2011-02-01 | The Regents Of The University Of Michigan | Synthetic peptide inhibitors of thrombin and thrombin activation of protease activated receptors 1 and 4 |
| US20070093423A1 (en) | 2005-09-14 | 2007-04-26 | Avi Tovi | Process for production of Bivalirudin |
| US20130196916A1 (en) | 2005-09-14 | 2013-08-01 | Teva Pharmaceuticals Usa, Inc. | Process for production of bivalirudin |
| US20100029916A1 (en) | 2005-09-14 | 2010-02-04 | Avi Tovi | Process for production of bivalirudin |
| US20130196917A1 (en) | 2005-09-14 | 2013-08-01 | Teva Pharmaceuticals Usa, Inc. | Process for production of bivalirudin |
| US20130196919A1 (en) | 2005-09-14 | 2013-08-01 | Teva Pharmaceuticals Usa, Inc. | Process for production of bivalirudin |
| US20100273982A1 (en) | 2005-09-14 | 2010-10-28 | Avi Tovi | Process for production of bivalirudin |
| WO2007067979A2 (en) | 2005-12-09 | 2007-06-14 | Bracco International B.V. | Targeting vector-phospholipid conjugates |
| US8314208B2 (en) | 2006-02-10 | 2012-11-20 | Cem Corporation | Microwave enhanced N-fmoc deprotection in peptide synthesis |
| US20080051558A1 (en) | 2006-03-10 | 2008-02-28 | Yiming Zhou | Method of preparing bivalirudin |
| US7829659B2 (en) | 2006-05-02 | 2010-11-09 | Allozyne, Inc. | Methods of modifying polypeptides comprising non-natural amino acids |
| US8022181B2 (en) | 2006-05-03 | 2011-09-20 | Mallinckrodt Llc | Composition and method for the release of protected peptides from a resin |
| US8415454B2 (en) | 2006-07-21 | 2013-04-09 | Solvay (Société Anonyme) | Process for the manufacture of peptides |
| US20080253992A1 (en) | 2006-10-03 | 2008-10-16 | Neose Technologies, Inc. | Methods for the purification of polypeptide conjugates |
| US20100168443A1 (en) | 2006-11-02 | 2010-07-01 | University Of Virginia Patent Foundation | Ethoid-Containing Compounds, Methods for Preparing Ethoid-Containing Compounds, and Methods of Use |
| US20100160604A1 (en) | 2006-11-21 | 2010-06-24 | Ipsen Manufacturing Ireland Limited | Boc and fmoc solid phase peptide synthesis |
| US8383770B2 (en) | 2006-11-21 | 2013-02-26 | Ipsen Manufacturing Ireland Limited | Boc and Fmoc solid phase peptide synthesis |
| US8404724B2 (en) | 2006-12-08 | 2013-03-26 | Millennium Pharmaceuticals, Inc. | Unit dose formulations and methods of treating thrombosis with an oral factor Xa inhibitor |
| US8101379B2 (en) | 2006-12-15 | 2012-01-24 | Institute of Radiation Medicine Academy of Military Medical Science | Preparation of low bleeding anticoagulant fusion protein and its use |
| US8361965B2 (en) | 2007-02-16 | 2013-01-29 | Lunan Pharmaceutical Group Corporation | Recombinant chimeric protein of neutrophil inhibitory factor and hirugen, and pharmaceutical composition thereof |
| WO2008109079A2 (en) | 2007-03-01 | 2008-09-12 | Novetide, Ltd. | High purity peptides |
| US20080287650A1 (en) | 2007-03-01 | 2008-11-20 | Avi Tovi | High purity peptides |
| WO2008155658A2 (en) | 2007-06-18 | 2008-12-24 | Institute Of Zoology Of The Slovak Academy Of Sciences | Thrombin inhibitor |
| US8206967B2 (en) | 2007-07-06 | 2012-06-26 | Medimmune Limited | Method for production of recombinant human thrombin |
| US20100184952A1 (en) | 2007-07-25 | 2010-07-22 | Ajinomoto Co., Inc | Method for selective removal of dibenzofulvene derivative |
| CN101372512A (en) | 2007-08-23 | 2009-02-25 | 中国人民解放军军事医学科学院生物工程研究所 | Anticoagulated blood polypeptides and uses thereof |
| US20090062511A1 (en) | 2007-09-05 | 2009-03-05 | Raghavendracharyulu Venkata Palle | Process for the preparation of bivalirudin and its pharmaceutical compositions |
| US20120149868A1 (en) | 2008-05-15 | 2012-06-14 | Novo Nordisk A/S | Purification of peptides prepared by solid phase synthesis |
| US7582727B1 (en) | 2008-07-27 | 2009-09-01 | The Medicinces Company | Pharmaceutical formulations of bivalirudin and processes of making the same |
| US7598343B1 (en) | 2008-07-27 | 2009-10-06 | The Medicines Company | Pharmaceutical formulations of bivalirudin and processes of making the same |
| US20110190475A1 (en) | 2008-08-06 | 2011-08-04 | Ajinomoto Co., Inc. | Processes for removal of dibenzofulvene |
| US20100056755A1 (en) | 2008-09-03 | 2010-03-04 | Scinopharm Taiwan Ltd. | Process for Making Bivalirudin |
| US20110288235A1 (en) | 2008-09-03 | 2011-11-24 | Scinopharm Taiwan Ltd. | Process for the Preparation of Pramlintide |
| WO2010028122A1 (en) | 2008-09-03 | 2010-03-11 | Scinopharm Taiwan Ltd. | Process for making bivalirudin |
| US20100081788A1 (en) | 2008-09-03 | 2010-04-01 | Scinopharm Taiwan Ltd. | Process for the Preparation of Pramlintide |
| US8252896B2 (en) | 2008-09-03 | 2012-08-28 | ScnioPharm Taiwan, Ltd. | Process for making bivalirudin |
| WO2010054503A1 (en) | 2008-11-17 | 2010-05-20 | 中国人民解放军军事医学科学院生物工程研究所 | Anticoagulant polypeptides and uses thereof |
| US20110251372A1 (en) | 2008-12-29 | 2011-10-13 | Geoffroy Sommen | Process for the production of bivalirudin |
| CN101475631A (en) | 2009-01-08 | 2009-07-08 | 苏州中科天马肽工程中心有限公司 | Liquid phase synthesizing method for bivalirudin |
| US20110160431A1 (en) | 2009-04-06 | 2011-06-30 | Novetide, Ltd. | Production of peptides containing poly-gly sequences using fmoc chemistry |
| WO2010117725A2 (en) | 2009-04-06 | 2010-10-14 | Novetide, Ltd. | Production of peptides containing poly-gly sequences using fmoc chemistry |
| US20120135931A1 (en) | 2009-05-05 | 2012-05-31 | Natural Environment Research Council | Method of modifying serine protease inhibitors |
| US20100292436A1 (en) | 2009-05-15 | 2010-11-18 | Shanghai Ambiopharm, Inc. | Method for producing bivalirudin |
| US7803762B1 (en) | 2009-08-20 | 2010-09-28 | The Medicines Company | Ready-to-use bivalirudin compositions |
| US20110046063A1 (en) | 2009-08-20 | 2011-02-24 | Nagesh Palepu | Ready-to-use bivalirudin compositions |
| US7713928B1 (en) | 2009-08-20 | 2010-05-11 | The Medicines Company | Ready-to-use bivalirudin compositions |
| US20120232014A1 (en) | 2009-09-04 | 2012-09-13 | Imperial Innovations Limited | Antithrombotic compounds |
| WO2011071799A2 (en) | 2009-12-11 | 2011-06-16 | Dr. Reddy's Laboratories Ltd. | Purification of bivalirudin |
| US7985733B1 (en) | 2010-01-06 | 2011-07-26 | The Medicines Company | Buffer-based method for preparing bivalirudin drug product |
| US20110319594A1 (en) | 2010-06-28 | 2011-12-29 | Shanghai Ambiopharm, Inc. | Method for producing bivalirudin |
| US20120041173A1 (en) | 2010-08-16 | 2012-02-16 | Cem Corporation | Water soluble solid phase peptide synthesis |
| KR20120069288A (en) | 2010-12-20 | 2012-06-28 | 주식회사 씨트리 | Purification method of bivalirudin using soluble sugar alcohol |
| CN102260323A (en) | 2011-05-30 | 2011-11-30 | 杭州诺泰制药技术有限公司 | Method for preparing bivalirudin by combining solid phase with liquid phase and detection method |
| US20140088291A1 (en) | 2011-05-31 | 2014-03-27 | Ajinomoto Co., Inc. | Method for producing peptide |
| WO2012165546A1 (en) | 2011-05-31 | 2012-12-06 | 味の素株式会社 | Method for producing peptide |
| WO2012174816A1 (en) | 2011-06-23 | 2012-12-27 | 成都圣诺科技发展有限公司 | Bivalirudin preparation method |
| US20140187745A1 (en) | 2011-06-23 | 2014-07-03 | Chengdu Shengnuo Tech Co., Ltd. | Method for preparing bivalirudin |
| CN102336813A (en) | 2011-07-01 | 2012-02-01 | 上海苏豪逸明制药有限公司 | Preparation method for synthesizing proteidin with solid phase polypeptide |
| US20130034547A1 (en) | 2011-08-02 | 2013-02-07 | Kelly Jeffery W | Reliable stabilization of n-linked polypeptide native states with enhanced aromatic sequons located in polypeptide tight turns |
| WO2013042129A1 (en) | 2011-09-23 | 2013-03-28 | Natco Pharma Limited | Improved process for preparation of bivalirudin |
| CN102532274A (en) | 2012-02-13 | 2012-07-04 | 成都圣诺生物制药有限公司 | Method for preparing bivalirudin |
| CN102641506A (en) | 2012-04-11 | 2012-08-22 | 承德医学院中药研究所 | Bivalirudin-polyethylene glycol compound |
| CN102731624A (en) | 2012-06-14 | 2012-10-17 | 无锡市凯利药业有限公司 | Method for synthesis of bivalirudin in solid-phase fragment approach |
| CN102702325A (en) | 2012-06-19 | 2012-10-03 | 深圳翰宇药业股份有限公司 | Preparation method of anticoagulant polypeptide |
| CN102924575A (en) | 2012-10-31 | 2013-02-13 | 深圳翰宇药业股份有限公司 | Preparation method of bivalirudin |
Non-Patent Citations (17)
| Title |
|---|
| Albericio et al., Convergent Solid-Phase Peptide Synthesis, Methods in Enzymology, vol. 289, 1997: pp. 313-336. |
| Burgess & Lim: "Resin type can have important effects on solid phase asymmetric alkylation reactions" Chem. Commun., vol. 1997, 1997, pp. 785-786. |
| EMEA Article on Angiox, 2005, pp. 1-32. |
| Freund, E. et al "Solid-phase synthesis of a putative heptapeptide intermediate in vancomycin biosynthesis" Chem. Commun., 1999, 2509-2510. |
| Giraud et al.: "A side-reaction in the SPPS of trp-containing peptides," J. Peptide Sci., vol. 5, 1999, pp. 457-461. |
| Goulas et al., Convergent solid-phase synthesis of hirudin, Journal of Peptide Science, vol. 12, No. 2. 2006, pp. 116-123. |
| Guillier et al.: "Linkers and cleavage strategies in solid-phase organic synthesis and combinatorial chemistry" Chem. Rev., vol. 100, 2000, pp. 2091-2157. |
| International Preliminary Report on Patentability for PCT/EP2004/014599 completed Apr. 24, 2006. |
| International Search Report for PCT/EP2004/014599 dated Jul. 15, 2005. |
| International Search Report for PCT/EP2005/011226 dated Dec. 16, 2005. |
| Lloyd-Williams et al., Solid-Phase Synthesis of Protected Peptide Segments, Solid-Phase Synthesis—A Practical Guide, 2000, pp. 378-381. |
| Luo, Juan, Bivalirudin, a direct inhibitor drug against thrombin, Zhongguo Yaoxue Zazhi (Beijing, China), vol. 37, No. 10, 2002, pp. 789-790. |
| Maraganore et al., Design and characterization of hirulogs: a novel class of bivalent peptide inhibitors of thrombin, Biochemistry, vol. 29, No. 30, 1990, pp. 7095-7101. |
| Okayama et al., Anticoagulant peptides; synthesis, stability and antithrombin activity of hirudin C-terminal-related peptides and their disulfated analog, Chemical and Pharmaceutical Bulletin (Tokyo), vol. 44, No. 7, 1996, pp. 1344-1350, XP001207786. |
| OKAYAMA T, ET AL.: "ANTICOAGULANT PEPTIDES; SYNTHESIS, STABILITY AND ANTITHROMBIN ACTIVITY OF HIRUDIN C-TERMINAL-RELATED PEPTIDES AND THEIR DISULFATED ANALOG", CHEMICAL AND PHARMACEUTICAL BULLETIN, PHARMACEUTICAL SOCIETY OF JAPAN, JP, vol. 44, no. 07, 1 January 1996 (1996-01-01), JP, pages 1344 - 1350, XP001207786, ISSN: 0009-2363 |
| Scatena, Roberto, Bivalirudin, Biogen, Current Opinion in Cardiovascular, Pulmonary & Renal Investigational Drugs, 2(2): 189-194, 2000. * |
| Wellings et al., Standard Fmoc Protocols, Methods in Enzymology, vol. 289, pp. 44-67. |
Also Published As
| Publication number | Publication date |
|---|---|
| CN101094867B (en) | 2011-08-24 |
| JP2008517018A (en) | 2008-05-22 |
| ATE480561T1 (en) | 2010-09-15 |
| PT1737889E (en) | 2010-12-13 |
| WO2006045503A1 (en) | 2006-05-04 |
| JP4903709B2 (en) | 2012-03-28 |
| DE602005023429D1 (en) | 2010-10-21 |
| US20080287648A1 (en) | 2008-11-20 |
| DK1737889T3 (en) | 2011-01-03 |
| IL182698A (en) | 2013-11-28 |
| CN102225966B (en) | 2012-12-26 |
| IL182698A0 (en) | 2007-09-20 |
| CN102225966A (en) | 2011-10-26 |
| US7939629B2 (en) | 2011-05-10 |
| ES2352204T3 (en) | 2011-02-16 |
| EP1737889B1 (en) | 2010-09-08 |
| CN101094867A (en) | 2007-12-26 |
| EP1737889A1 (en) | 2007-01-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| USRE46830E1 (en) | Method for solid phase peptide synthesis | |
| CN107406480B (en) | Peptide synthesis method | |
| JP4405594B2 (en) | Improved solid phase peptide synthesis and reagents for use in such synthesis | |
| KR102397271B1 (en) | Method for preparing amg 416 | |
| DK175042B1 (en) | Synthetic resin, process for its preparation and method for producing peptides and peptide amides using the synthetic resin | |
| US20110160431A1 (en) | Production of peptides containing poly-gly sequences using fmoc chemistry | |
| WO2015100876A1 (en) | Method for preparing liraglutide | |
| US20100249370A1 (en) | Process for the production of pramlintide | |
| US20100081788A1 (en) | Process for the Preparation of Pramlintide | |
| US20240116980A1 (en) | Compound or salt thereof and preparation method and application of same | |
| JP5328345B2 (en) | Method for producing peptide thioester compound | |
| CN114401981A (en) | Process for producing glucagon | |
| US20220033440A1 (en) | An improved process for the preparation of plecanatide | |
| Thurieau et al. | New N. alpha.-Guanidinobenzoyl Derivatives of Hirudin-54-65 Containing Stabilized Carboxyl or Phosphoryl Groups on the Side Chain of Phenylalanine-63 | |
| JP4878031B2 (en) | Solid phase peptide synthesis | |
| US5516642A (en) | Polypeptides derived from major histocompatibility complex Class I antigen | |
| WO2003093301A2 (en) | Process for the synthesis of peptides | |
| CN107556363B (en) | Peptide or salt thereof and process for producing the same | |
| JPH1067796A (en) | Synthesis of cyclic peptide | |
| WO2024079043A1 (en) | Method of manufacturing a peptide with a lysine derivative | |
| US20230242581A1 (en) | Synthesis of a guanylate cyclase agonist by fragments based approach | |
| Slaninova et al. | Analogs of arginine vasopressin modified in position 3 with (R)-a-hydroxymethylphenylalanine |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| MAFP | Maintenance fee payment |
Free format text: PAYMENT OF MAINTENANCE FEE, 8TH YEAR, LARGE ENTITY (ORIGINAL EVENT CODE: M1552); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY Year of fee payment: 8 |
|
| MAFP | Maintenance fee payment |
Free format text: PAYMENT OF MAINTENANCE FEE, 12TH YEAR, LARGE ENTITY (ORIGINAL EVENT CODE: M1553); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY Year of fee payment: 12 |