US8604148B2 - Functionalization of vinyl terminated polymers by ring opening cross metathesis - Google Patents
Functionalization of vinyl terminated polymers by ring opening cross metathesis Download PDFInfo
- Publication number
- US8604148B2 US8604148B2 US13/306,263 US201113306263A US8604148B2 US 8604148 B2 US8604148 B2 US 8604148B2 US 201113306263 A US201113306263 A US 201113306263A US 8604148 B2 US8604148 B2 US 8604148B2
- Authority
- US
- United States
- Prior art keywords
- mol
- polymer
- propylene
- oligomer
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related, expires
Links
- 229920000642 polymer Polymers 0.000 title claims abstract description 140
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 title claims abstract description 124
- 229920002554 vinyl polymer Polymers 0.000 title claims abstract description 95
- 238000005686 cross metathesis reaction Methods 0.000 title description 13
- 238000007142 ring opening reaction Methods 0.000 title description 7
- 238000007306 functionalization reaction Methods 0.000 title description 4
- -1 cyclic olefin Chemical class 0.000 claims abstract description 102
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 claims abstract description 100
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 claims description 88
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 claims description 86
- 125000001183 hydrocarbyl group Chemical group 0.000 claims description 85
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 79
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 claims description 73
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 claims description 70
- 239000005977 Ethylene Substances 0.000 claims description 70
- 239000001257 hydrogen Substances 0.000 claims description 61
- 229910052739 hydrogen Inorganic materials 0.000 claims description 61
- 150000001336 alkenes Chemical class 0.000 claims description 54
- 238000005160 1H NMR spectroscopy Methods 0.000 claims description 48
- 229920001577 copolymer Polymers 0.000 claims description 44
- 125000004432 carbon atom Chemical group C* 0.000 claims description 39
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 claims description 36
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 35
- LPIQUOYDBNQMRZ-UHFFFAOYSA-N cyclopentene Chemical group C1CC=CC1 LPIQUOYDBNQMRZ-UHFFFAOYSA-N 0.000 claims description 32
- 239000000203 mixture Substances 0.000 claims description 29
- 125000000746 allylic group Chemical group 0.000 claims description 27
- 229910052799 carbon Inorganic materials 0.000 claims description 25
- 229920000098 polyolefin Polymers 0.000 claims description 21
- SJYNFBVQFBRSIB-UHFFFAOYSA-N norbornadiene Chemical group C1=CC2C=CC1C2 SJYNFBVQFBRSIB-UHFFFAOYSA-N 0.000 claims description 20
- JFNLZVQOOSMTJK-KNVOCYPGSA-N norbornene Chemical group C1[C@@H]2CC[C@H]1C=C2 JFNLZVQOOSMTJK-KNVOCYPGSA-N 0.000 claims description 20
- HECLRDQVFMWTQS-RGOKHQFPSA-N 1755-01-7 Chemical group C1[C@H]2[C@@H]3CC=C[C@@H]3[C@@H]1C=C2 HECLRDQVFMWTQS-RGOKHQFPSA-N 0.000 claims description 19
- IAQRGUVFOMOMEM-UHFFFAOYSA-N butene Natural products CC=CC IAQRGUVFOMOMEM-UHFFFAOYSA-N 0.000 claims description 19
- DCTOHCCUXLBQMS-UHFFFAOYSA-N 1-undecene Chemical group CCCCCCCCCC=C DCTOHCCUXLBQMS-UHFFFAOYSA-N 0.000 claims description 18
- ZGEGCLOFRBLKSE-UHFFFAOYSA-N methylene hexane Chemical group CCCCCC=C ZGEGCLOFRBLKSE-UHFFFAOYSA-N 0.000 claims description 18
- 239000000178 monomer Substances 0.000 claims description 18
- 125000005842 heteroatom Chemical group 0.000 claims description 17
- VXNZUUAINFGPBY-UHFFFAOYSA-N 1-Butene Chemical group CCC=C VXNZUUAINFGPBY-UHFFFAOYSA-N 0.000 claims description 16
- SNRUBQQJIBEYMU-UHFFFAOYSA-N Dodecane Chemical group CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 claims description 16
- RGSFGYAAUTVSQA-UHFFFAOYSA-N pentamethylene Chemical group C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 claims description 16
- URYYVOIYTNXXBN-UPHRSURJSA-N cyclooctene Chemical group C1CCC\C=C/CC1 URYYVOIYTNXXBN-UPHRSURJSA-N 0.000 claims description 15
- 239000004913 cyclooctene Chemical group 0.000 claims description 15
- 229910052757 nitrogen Inorganic materials 0.000 claims description 14
- 239000000314 lubricant Substances 0.000 claims description 13
- RRKODOZNUZCUBN-CCAGOZQPSA-N (1z,3z)-cycloocta-1,3-diene Chemical group C1CC\C=C/C=C\C1 RRKODOZNUZCUBN-CCAGOZQPSA-N 0.000 claims description 12
- LIKMAJRDDDTEIG-UHFFFAOYSA-N 1-hexene Chemical group CCCCC=C LIKMAJRDDDTEIG-UHFFFAOYSA-N 0.000 claims description 12
- YCNYCBYHUAGZIZ-UHFFFAOYSA-N 7-oxabicyclo[2.2.1]hept-2-ene Chemical group O1C2CCC1C=C2 YCNYCBYHUAGZIZ-UHFFFAOYSA-N 0.000 claims description 12
- YKCNBNDWSATCJL-UHFFFAOYSA-N 7-oxabicyclo[2.2.1]hepta-2,5-diene Chemical group C1=CC2C=CC1O2 YKCNBNDWSATCJL-UHFFFAOYSA-N 0.000 claims description 12
- HYPABJGVBDSCIT-UPHRSURJSA-N cyclododecene Chemical group C1CCCCC\C=C/CCCC1 HYPABJGVBDSCIT-UPHRSURJSA-N 0.000 claims description 12
- ZXIJMRYMVAMXQP-UHFFFAOYSA-N cycloheptene Chemical group C1CCC=CCC1 ZXIJMRYMVAMXQP-UHFFFAOYSA-N 0.000 claims description 12
- KWKAKUADMBZCLK-UHFFFAOYSA-N 1-octene Chemical group CCCCCCC=C KWKAKUADMBZCLK-UHFFFAOYSA-N 0.000 claims description 11
- 229910052782 aluminium Inorganic materials 0.000 claims description 10
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 claims description 10
- CRSBERNSMYQZNG-UHFFFAOYSA-N 1 -dodecene Chemical group CCCCCCCCCCC=C CRSBERNSMYQZNG-UHFFFAOYSA-N 0.000 claims description 9
- 229940069096 dodecene Drugs 0.000 claims description 9
- AFFLGGQVNFXPEV-UHFFFAOYSA-N n-decene Chemical group CCCCCCCCC=C AFFLGGQVNFXPEV-UHFFFAOYSA-N 0.000 claims description 9
- YWAKXRMUMFPDSH-UHFFFAOYSA-N pentene Chemical group CCCC=C YWAKXRMUMFPDSH-UHFFFAOYSA-N 0.000 claims description 9
- 150000001993 dienes Chemical class 0.000 claims description 6
- 229920000089 Cyclic olefin copolymer Polymers 0.000 claims description 4
- 239000000853 adhesive Substances 0.000 claims description 3
- 230000001070 adhesive effect Effects 0.000 claims description 3
- 239000002816 fuel additive Substances 0.000 claims description 3
- 239000004034 viscosity adjusting agent Substances 0.000 claims description 3
- 125000000383 tetramethylene group Chemical group [H]C([H])([*:1])C([H])([H])C([H])([H])C([H])([H])[*:2] 0.000 claims description 2
- 238000000034 method Methods 0.000 abstract description 68
- 230000008569 process Effects 0.000 abstract description 39
- 238000004519 manufacturing process Methods 0.000 abstract description 3
- 239000003054 catalyst Substances 0.000 description 91
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 54
- 125000004122 cyclic group Chemical group 0.000 description 41
- 239000003446 ligand Substances 0.000 description 38
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 36
- 239000000047 product Substances 0.000 description 33
- 238000005649 metathesis reaction Methods 0.000 description 32
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 31
- 238000006243 chemical reaction Methods 0.000 description 29
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 29
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 28
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 28
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 27
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 26
- 229910052707 ruthenium Inorganic materials 0.000 description 26
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 26
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 25
- 0 [1*]/C=C\C1CC(C=C)c([3*])c1[2*] Chemical compound [1*]/C=C\C1CC(C=C)c([3*])c1[2*] 0.000 description 24
- 150000004820 halides Chemical class 0.000 description 24
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 24
- 125000003118 aryl group Chemical group 0.000 description 23
- 125000000217 alkyl group Chemical group 0.000 description 22
- 229910052751 metal Inorganic materials 0.000 description 22
- 239000002184 metal Substances 0.000 description 22
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 21
- 150000001875 compounds Chemical class 0.000 description 21
- 239000000460 chlorine Substances 0.000 description 20
- 229910052762 osmium Inorganic materials 0.000 description 18
- 238000006116 polymerization reaction Methods 0.000 description 17
- 239000002904 solvent Substances 0.000 description 17
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 16
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 15
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 15
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 15
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 15
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical group [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 description 14
- 125000000129 anionic group Chemical group 0.000 description 14
- 229910052736 halogen Inorganic materials 0.000 description 14
- 150000002367 halogens Chemical class 0.000 description 14
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 14
- 150000004703 alkoxides Chemical class 0.000 description 13
- 150000004696 coordination complex Chemical class 0.000 description 13
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 13
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 13
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 12
- 238000005984 hydrogenation reaction Methods 0.000 description 12
- 230000007935 neutral effect Effects 0.000 description 12
- 239000000377 silicon dioxide Substances 0.000 description 12
- YAYGSLOSTXKUBW-UHFFFAOYSA-N ruthenium(2+) Chemical compound [Ru+2] YAYGSLOSTXKUBW-UHFFFAOYSA-N 0.000 description 11
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 10
- 229910052801 chlorine Inorganic materials 0.000 description 10
- 125000002573 ethenylidene group Chemical group [*]=C=C([H])[H] 0.000 description 10
- 229910052760 oxygen Inorganic materials 0.000 description 10
- ADLVDYMTBOSDFE-UHFFFAOYSA-N 5-chloro-6-nitroisoindole-1,3-dione Chemical compound C1=C(Cl)C([N+](=O)[O-])=CC2=C1C(=O)NC2=O ADLVDYMTBOSDFE-UHFFFAOYSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 150000008052 alkyl sulfonates Chemical class 0.000 description 9
- 238000006471 dimerization reaction Methods 0.000 description 9
- 150000003003 phosphines Chemical class 0.000 description 9
- 239000000243 solution Substances 0.000 description 9
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 8
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 8
- 150000001412 amines Chemical class 0.000 description 8
- AFABGHUZZDYHJO-UHFFFAOYSA-N dimethyl butane Natural products CCCC(C)C AFABGHUZZDYHJO-UHFFFAOYSA-N 0.000 description 8
- PXHVJJICTQNCMI-UHFFFAOYSA-N nickel Substances [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 8
- 239000000376 reactant Substances 0.000 description 8
- 239000000126 substance Substances 0.000 description 8
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 7
- 150000008064 anhydrides Chemical class 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- 150000002466 imines Chemical class 0.000 description 7
- HZVOZRGWRWCICA-UHFFFAOYSA-N methanediyl Chemical compound [CH2] HZVOZRGWRWCICA-UHFFFAOYSA-N 0.000 description 7
- 239000003921 oil Substances 0.000 description 7
- SYQBFIAQOQZEGI-UHFFFAOYSA-N osmium atom Chemical group [Os] SYQBFIAQOQZEGI-UHFFFAOYSA-N 0.000 description 7
- 239000007858 starting material Substances 0.000 description 7
- 238000003786 synthesis reaction Methods 0.000 description 7
- WLPUWLXVBWGYMZ-UHFFFAOYSA-N tricyclohexylphosphine Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1 WLPUWLXVBWGYMZ-UHFFFAOYSA-N 0.000 description 7
- KNDQHSIWLOJIGP-UMRXKNAASA-N (3ar,4s,7r,7as)-rel-3a,4,7,7a-tetrahydro-4,7-methanoisobenzofuran-1,3-dione Chemical compound O=C1OC(=O)[C@@H]2[C@H]1[C@]1([H])C=C[C@@]2([H])C1 KNDQHSIWLOJIGP-UMRXKNAASA-N 0.000 description 6
- RELMFMZEBKVZJC-UHFFFAOYSA-N 1,2,3-trichlorobenzene Chemical compound ClC1=CC=CC(Cl)=C1Cl RELMFMZEBKVZJC-UHFFFAOYSA-N 0.000 description 6
- TVFPFYXSBWXCBJ-UHFFFAOYSA-N CC(C)=C(C)(C)(C)C Chemical compound CC(C)=C(C)(C)(C)C TVFPFYXSBWXCBJ-UHFFFAOYSA-N 0.000 description 6
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 6
- 239000000654 additive Substances 0.000 description 6
- 229940045714 alkyl sulfonate alkylating agent Drugs 0.000 description 6
- 229910052794 bromium Inorganic materials 0.000 description 6
- 239000007795 chemical reaction product Substances 0.000 description 6
- 239000003085 diluting agent Substances 0.000 description 6
- 239000000539 dimer Substances 0.000 description 6
- 125000005678 ethenylene group Chemical group [H]C([*:1])=C([H])[*:2] 0.000 description 6
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 6
- GYNNXHKOJHMOHS-UHFFFAOYSA-N methyl-cycloheptane Natural products CC1CCCCCC1 GYNNXHKOJHMOHS-UHFFFAOYSA-N 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- 235000019198 oils Nutrition 0.000 description 6
- 239000002002 slurry Substances 0.000 description 6
- 239000007787 solid Substances 0.000 description 6
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 description 5
- LSMWOQFDLBIYPM-UHFFFAOYSA-N 1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydro-2h-imidazol-1-ium-2-ide Chemical group CC1=CC(C)=CC(C)=C1N1[C-]=[N+](C=2C(=CC(C)=CC=2C)C)CC1 LSMWOQFDLBIYPM-UHFFFAOYSA-N 0.000 description 5
- HIXDQWDOVZUNNA-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-hydroxy-7-methoxychromen-4-one Chemical compound C=1C(OC)=CC(O)=C(C(C=2)=O)C=1OC=2C1=CC=C(OC)C(OC)=C1 HIXDQWDOVZUNNA-UHFFFAOYSA-N 0.000 description 5
- GVNVAWHJIKLAGL-UHFFFAOYSA-N 2-(cyclohexen-1-yl)cyclohexan-1-one Chemical compound O=C1CCCCC1C1=CCCCC1 GVNVAWHJIKLAGL-UHFFFAOYSA-N 0.000 description 5
- 101150065749 Churc1 gene Proteins 0.000 description 5
- 102100038239 Protein Churchill Human genes 0.000 description 5
- 238000005865 alkene metathesis reaction Methods 0.000 description 5
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 5
- 125000000753 cycloalkyl group Chemical group 0.000 description 5
- ZSWFCLXCOIISFI-UHFFFAOYSA-N cyclopentadiene Chemical compound C1C=CC=C1 ZSWFCLXCOIISFI-UHFFFAOYSA-N 0.000 description 5
- 235000019441 ethanol Nutrition 0.000 description 5
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 5
- 125000000524 functional group Chemical group 0.000 description 5
- QWTDNUCVQCZILF-UHFFFAOYSA-N isopentane Chemical compound CCC(C)C QWTDNUCVQCZILF-UHFFFAOYSA-N 0.000 description 5
- 125000000654 isopropylidene group Chemical group C(C)(C)=* 0.000 description 5
- 229910052759 nickel Inorganic materials 0.000 description 5
- 238000001228 spectrum Methods 0.000 description 5
- 125000001424 substituent group Chemical group 0.000 description 5
- 229910052717 sulfur Inorganic materials 0.000 description 5
- QVLAWKAXOMEXPM-DICFDUPASA-N 1,1,1,2-tetrachloro-2,2-dideuterioethane Chemical compound [2H]C([2H])(Cl)C(Cl)(Cl)Cl QVLAWKAXOMEXPM-DICFDUPASA-N 0.000 description 4
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 4
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 4
- 125000006374 C2-C10 alkenyl group Chemical group 0.000 description 4
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 4
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 4
- MCMNRKCIXSYSNV-UHFFFAOYSA-N Zirconium dioxide Chemical compound O=[Zr]=O MCMNRKCIXSYSNV-UHFFFAOYSA-N 0.000 description 4
- 125000000304 alkynyl group Chemical group 0.000 description 4
- DMEGYFMYUHOHGS-UHFFFAOYSA-N cycloheptane Chemical compound C1CCCCCC1 DMEGYFMYUHOHGS-UHFFFAOYSA-N 0.000 description 4
- 229930195733 hydrocarbon Natural products 0.000 description 4
- NNPPMTNAJDCUHE-UHFFFAOYSA-N isobutane Chemical compound CC(C)C NNPPMTNAJDCUHE-UHFFFAOYSA-N 0.000 description 4
- UAEPNZWRGJTJPN-UHFFFAOYSA-N methylcyclohexane Chemical compound CC1CCCCC1 UAEPNZWRGJTJPN-UHFFFAOYSA-N 0.000 description 4
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 4
- 238000007152 ring opening metathesis polymerisation reaction Methods 0.000 description 4
- 239000000523 sample Substances 0.000 description 4
- 229920006395 saturated elastomer Polymers 0.000 description 4
- 150000003573 thiols Chemical class 0.000 description 4
- UCPDHOTYYDHPEN-UPHRSURJSA-N (4z)-cyclooct-4-en-1-ol Chemical compound OC1CCC\C=C/CC1 UCPDHOTYYDHPEN-UPHRSURJSA-N 0.000 description 3
- OJOWICOBYCXEKR-KRXBUXKQSA-N (5e)-5-ethylidenebicyclo[2.2.1]hept-2-ene Chemical compound C1C2C(=C/C)/CC1C=C2 OJOWICOBYCXEKR-KRXBUXKQSA-N 0.000 description 3
- QVLAWKAXOMEXPM-UHFFFAOYSA-N 1,1,1,2-tetrachloroethane Chemical class ClCC(Cl)(Cl)Cl QVLAWKAXOMEXPM-UHFFFAOYSA-N 0.000 description 3
- PBKONEOXTCPAFI-UHFFFAOYSA-N 1,2,4-trichlorobenzene Chemical compound ClC1=CC=C(Cl)C(Cl)=C1 PBKONEOXTCPAFI-UHFFFAOYSA-N 0.000 description 3
- VYXHVRARDIDEHS-UHFFFAOYSA-N 1,5-cyclooctadiene Chemical compound C1CC=CCCC=C1 VYXHVRARDIDEHS-UHFFFAOYSA-N 0.000 description 3
- 239000004912 1,5-cyclooctadiene Substances 0.000 description 3
- CXOZQHPXKPDQGT-UHFFFAOYSA-N 3-Methylcyclopentene Chemical compound CC1CCC=C1 CXOZQHPXKPDQGT-UHFFFAOYSA-N 0.000 description 3
- INYHZQLKOKTDAI-UHFFFAOYSA-N 5-ethenylbicyclo[2.2.1]hept-2-ene Chemical compound C1C2C(C=C)CC1C=C2 INYHZQLKOKTDAI-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- QUUZPTHNMBTPKJ-WSELPHFCSA-N C=CC1CC(/C=C\CC([Cm])CC([Cm])CC(C)C)C2C(=O)OC(=O)C12 Chemical compound C=CC1CC(/C=C\CC([Cm])CC([Cm])CC(C)C)C2C(=O)OC(=O)C12 QUUZPTHNMBTPKJ-WSELPHFCSA-N 0.000 description 3
- CROQCBPSWGGZDQ-UHFFFAOYSA-N CCC([Cm])CC([Cm])CC(C)C Chemical compound CCC([Cm])CC([Cm])CC(C)C CROQCBPSWGGZDQ-UHFFFAOYSA-N 0.000 description 3
- DPBMNGOWCCVNRQ-UHFFFAOYSA-N CCC([Cm])CC([Cm])CC[Cm] Chemical compound CCC([Cm])CC([Cm])CC[Cm] DPBMNGOWCCVNRQ-UHFFFAOYSA-N 0.000 description 3
- 238000005481 NMR spectroscopy Methods 0.000 description 3
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical group O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 3
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 3
- QWGXPNSOYZOCHH-IHWYPQMZSA-N [(4z)-cyclooct-4-en-1-yl] acetate Chemical compound CC(=O)OC1CCC\C=C/CC1 QWGXPNSOYZOCHH-IHWYPQMZSA-N 0.000 description 3
- OXLURKCRXVAJQS-UHFFFAOYSA-L [1,3-bis(2,4,6-trimethylphenyl)imidazolidin-2-ylidene]-dichloro-[[5-(dimethylsulfamoyl)-2-propan-2-yloxyphenyl]methylidene]ruthenium Chemical compound CC(C)OC1=CC=C(S(=O)(=O)N(C)C)C=C1C=[Ru](Cl)(Cl)=C1N(C=2C(=CC(C)=CC=2C)C)CCN1C1=C(C)C=C(C)C=C1C OXLURKCRXVAJQS-UHFFFAOYSA-L 0.000 description 3
- 150000001299 aldehydes Chemical class 0.000 description 3
- 229910000074 antimony hydride Inorganic materials 0.000 description 3
- RBFQJDQYXXHULB-UHFFFAOYSA-N arsane Chemical compound [AsH3] RBFQJDQYXXHULB-UHFFFAOYSA-N 0.000 description 3
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 3
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 230000010354 integration Effects 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 125000001434 methanylylidene group Chemical group [H]C#[*] 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 239000003607 modifier Substances 0.000 description 3
- 125000001400 nonyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- CHWKGJOTGCSFNF-UHFFFAOYSA-N norbornene anhydride Chemical compound C1CC2C3C(=O)OC(=O)C3=C1C2 CHWKGJOTGCSFNF-UHFFFAOYSA-N 0.000 description 3
- 125000004043 oxo group Chemical group O=* 0.000 description 3
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 3
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 3
- 125000005538 phosphinite group Chemical group 0.000 description 3
- XRBCRPZXSCBRTK-UHFFFAOYSA-N phosphonous acid Chemical compound OPO XRBCRPZXSCBRTK-UHFFFAOYSA-N 0.000 description 3
- OJMIONKXNSYLSR-UHFFFAOYSA-N phosphorous acid Chemical compound OP(O)O OJMIONKXNSYLSR-UHFFFAOYSA-N 0.000 description 3
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Substances [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 3
- 229920000573 polyethylene Polymers 0.000 description 3
- 229920001155 polypropylene Polymers 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 3
- 150000003254 radicals Chemical class 0.000 description 3
- OUULRIDHGPHMNQ-UHFFFAOYSA-N stibane Chemical compound [SbH3] OUULRIDHGPHMNQ-UHFFFAOYSA-N 0.000 description 3
- 150000003462 sulfoxides Chemical class 0.000 description 3
- 229920001897 terpolymer Polymers 0.000 description 3
- 150000007970 thio esters Chemical class 0.000 description 3
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 3
- 125000000027 (C1-C10) alkoxy group Chemical group 0.000 description 2
- LCOFYVWULBZOTA-UHFFFAOYSA-L 1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazole;dichloro(3-methylbut-2-enylidene)ruthenium;tricyclohexylphosphane Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1.CC(C)=CC=[Ru](Cl)(Cl)=C1N(C=2C(=CC(C)=CC=2C)C)CCN1C1=C(C)C=C(C)C=C1C LCOFYVWULBZOTA-UHFFFAOYSA-L 0.000 description 2
- ZRPFJAPZDXQHSM-UHFFFAOYSA-L 1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazole;dichloro-[(2-propan-2-yloxyphenyl)methylidene]ruthenium Chemical compound CC(C)OC1=CC=CC=C1C=[Ru](Cl)(Cl)=C1N(C=2C(=CC(C)=CC=2C)C)CCN1C1=C(C)C=C(C)C=C1C ZRPFJAPZDXQHSM-UHFFFAOYSA-L 0.000 description 2
- RUKVGXGTVPPWDD-UHFFFAOYSA-N 1,3-bis(2,4,6-trimethylphenyl)imidazolidine Chemical group CC1=CC(C)=CC(C)=C1N1CN(C=2C(=CC(C)=CC=2C)C)CC1 RUKVGXGTVPPWDD-UHFFFAOYSA-N 0.000 description 2
- LSIXBBPOJBJQHN-UHFFFAOYSA-N 2,3-Dimethylbicyclo[2.2.1]hept-2-ene Chemical compound C1CC2C(C)=C(C)C1C2 LSIXBBPOJBJQHN-UHFFFAOYSA-N 0.000 description 2
- XUKLTPZEKXTPBT-UHFFFAOYSA-N 3-oxatricyclo[5.2.1.01,5]dec-5-ene-2,4-dione Chemical compound C1CC2C=C3C(=O)OC(=O)C13C2 XUKLTPZEKXTPBT-UHFFFAOYSA-N 0.000 description 2
- UQRONKZLYKUEMO-UHFFFAOYSA-N 4-methyl-1-(2,4,6-trimethylphenyl)pent-4-en-2-one Chemical group CC(=C)CC(=O)Cc1c(C)cc(C)cc1C UQRONKZLYKUEMO-UHFFFAOYSA-N 0.000 description 2
- 238000012935 Averaging Methods 0.000 description 2
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 2
- KAKZBPTYRLMSJV-UHFFFAOYSA-N Butadiene Chemical compound C=CC=C KAKZBPTYRLMSJV-UHFFFAOYSA-N 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 2
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 2
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 2
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 2
- 239000004793 Polystyrene Substances 0.000 description 2
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- 239000012190 activator Substances 0.000 description 2
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 125000003302 alkenyloxy group Chemical group 0.000 description 2
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 2
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 2
- 125000004414 alkyl thio group Chemical group 0.000 description 2
- 125000005133 alkynyloxy group Chemical group 0.000 description 2
- 229910000323 aluminium silicate Inorganic materials 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 125000004104 aryloxy group Chemical group 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- YDOACSUCKSODBO-UHFFFAOYSA-L benzylidene(dichloro)ruthenium;1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazole;3-bromopyridine Chemical compound BrC1=CC=CN=C1.BrC1=CC=CN=C1.CC1=CC(C)=CC(C)=C1N(CCN1C=2C(=CC(C)=CC=2C)C)C1=[Ru](Cl)(Cl)=CC1=CC=CC=C1 YDOACSUCKSODBO-UHFFFAOYSA-L 0.000 description 2
- PNPBGYBHLCEVMK-UHFFFAOYSA-L benzylidene(dichloro)ruthenium;tricyclohexylphosphane Chemical compound Cl[Ru](Cl)=CC1=CC=CC=C1.C1CCCCC1P(C1CCCCC1)C1CCCCC1.C1CCCCC1P(C1CCCCC1)C1CCCCC1 PNPBGYBHLCEVMK-UHFFFAOYSA-L 0.000 description 2
- FCDPQMAOJARMTG-UHFFFAOYSA-M benzylidene-[1,3-bis(2,4,6-trimethylphenyl)imidazolidin-2-ylidene]-dichlororuthenium;tricyclohexylphosphanium Chemical compound C1CCCCC1[PH+](C1CCCCC1)C1CCCCC1.CC1=CC(C)=CC(C)=C1N(CCN1C=2C(=CC(C)=CC=2C)C)C1=[Ru](Cl)(Cl)=CC1=CC=CC=C1 FCDPQMAOJARMTG-UHFFFAOYSA-M 0.000 description 2
- 150000001649 bromium compounds Chemical class 0.000 description 2
- 239000001273 butane Substances 0.000 description 2
- 150000001718 carbodiimides Chemical class 0.000 description 2
- 150000001244 carboxylic acid anhydrides Chemical class 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical group ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 2
- 125000000068 chlorophenyl group Chemical group 0.000 description 2
- 239000004927 clay Substances 0.000 description 2
- 229910052570 clay Inorganic materials 0.000 description 2
- 238000010924 continuous production Methods 0.000 description 2
- 150000001923 cyclic compounds Chemical class 0.000 description 2
- 150000001925 cycloalkenes Chemical class 0.000 description 2
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cyclohexene Chemical compound C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 description 2
- 239000003599 detergent Substances 0.000 description 2
- 125000003963 dichloro group Chemical group Cl* 0.000 description 2
- UZAFZWUWHHFWOK-UHFFFAOYSA-L dichloro-(3-phenylinden-1-ylidene)ruthenium;tricyclohexylphosphane Chemical compound C12=CC=CC=C2C(=[Ru](Cl)Cl)C=C1C1=CC=CC=C1.C1CCCCC1P(C1CCCCC1)C1CCCCC1.C1CCCCC1P(C1CCCCC1)C1CCCCC1 UZAFZWUWHHFWOK-UHFFFAOYSA-L 0.000 description 2
- KMKCJXPECJFQPQ-UHFFFAOYSA-L dichloro-[(2-propan-2-yloxyphenyl)methylidene]ruthenium;tricyclohexylphosphane Chemical compound CC(C)OC1=CC=CC=C1C=[Ru](Cl)Cl.C1CCCCC1P(C1CCCCC1)C1CCCCC1 KMKCJXPECJFQPQ-UHFFFAOYSA-L 0.000 description 2
- DHCWLIOIJZJFJE-UHFFFAOYSA-L dichlororuthenium Chemical compound Cl[Ru]Cl DHCWLIOIJZJFJE-UHFFFAOYSA-L 0.000 description 2
- 229910001873 dinitrogen Inorganic materials 0.000 description 2
- 239000002270 dispersing agent Substances 0.000 description 2
- 230000032050 esterification Effects 0.000 description 2
- 238000005886 esterification reaction Methods 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 125000001153 fluoro group Chemical group F* 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 238000004817 gas chromatography Methods 0.000 description 2
- 238000005227 gel permeation chromatography Methods 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 229920001519 homopolymer Polymers 0.000 description 2
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical compound [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- JCYWCSGERIELPG-UHFFFAOYSA-N imes Chemical group CC1=CC(C)=CC(C)=C1N1C=CN(C=2C(=CC(C)=CC=2C)C)[C]1 JCYWCSGERIELPG-UHFFFAOYSA-N 0.000 description 2
- 125000001841 imino group Chemical group [H]N=* 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 239000001282 iso-butane Substances 0.000 description 2
- 239000012948 isocyanate Substances 0.000 description 2
- 150000002513 isocyanates Chemical class 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 239000011976 maleic acid Substances 0.000 description 2
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 2
- 229910021645 metal ion Inorganic materials 0.000 description 2
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 2
- 239000010705 motor oil Substances 0.000 description 2
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 2
- LJDZFAPLPVPTBD-UHFFFAOYSA-N nitroformic acid Chemical compound OC(=O)[N+]([O-])=O LJDZFAPLPVPTBD-UHFFFAOYSA-N 0.000 description 2
- 230000000269 nucleophilic effect Effects 0.000 description 2
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 2
- 125000004430 oxygen atom Chemical group O* 0.000 description 2
- 125000001037 p-tolyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 150000002989 phenols Chemical class 0.000 description 2
- 125000003356 phenylsulfanyl group Chemical group [*]SC1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- 229910052698 phosphorus Inorganic materials 0.000 description 2
- 229910052697 platinum Inorganic materials 0.000 description 2
- 125000003367 polycyclic group Chemical group 0.000 description 2
- 229920002223 polystyrene Polymers 0.000 description 2
- 239000012429 reaction media Substances 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 230000003595 spectral effect Effects 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 125000003107 substituted aryl group Chemical group 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 125000004434 sulfur atom Chemical group 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 235000010215 titanium dioxide Nutrition 0.000 description 2
- 125000005425 toluyl group Chemical group 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 150000004670 unsaturated fatty acids Chemical class 0.000 description 2
- 235000021122 unsaturated fatty acids Nutrition 0.000 description 2
- 239000003039 volatile agent Substances 0.000 description 2
- 239000008096 xylene Substances 0.000 description 2
- YUHZIUAREWNXJT-UHFFFAOYSA-N (2-fluoropyridin-3-yl)boronic acid Chemical class OB(O)C1=CC=CN=C1F YUHZIUAREWNXJT-UHFFFAOYSA-N 0.000 description 1
- GGQQNYXPYWCUHG-RMTFUQJTSA-N (3e,6e)-deca-3,6-diene Chemical compound CCC\C=C\C\C=C\CC GGQQNYXPYWCUHG-RMTFUQJTSA-N 0.000 description 1
- QTYUSOHYEPOHLV-FNORWQNLSA-N 1,3-Octadiene Chemical compound CCCC\C=C\C=C QTYUSOHYEPOHLV-FNORWQNLSA-N 0.000 description 1
- 239000004322 Butylated hydroxytoluene Substances 0.000 description 1
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 1
- 125000006539 C12 alkyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- YHQXBTXEYZIYOV-UHFFFAOYSA-N C=CC(C)C Chemical compound C=CC(C)C YHQXBTXEYZIYOV-UHFFFAOYSA-N 0.000 description 1
- BZHMBWZPUJHVEE-UHFFFAOYSA-N CC(C)CC(C)C Chemical compound CC(C)CC(C)C BZHMBWZPUJHVEE-UHFFFAOYSA-N 0.000 description 1
- MBUYJORXAWWKJS-UHFFFAOYSA-N CC1=C(C)[C]1.CN1[C]C(C)(C)CC1.CN1[C]N(C)C=C1.CN1[C]SC=C1.CP1[C]P(C)CC1 Chemical compound CC1=C(C)[C]1.CN1[C]C(C)(C)CC1.CN1[C]N(C)C=C1.CN1[C]SC=C1.CP1[C]P(C)CC1 MBUYJORXAWWKJS-UHFFFAOYSA-N 0.000 description 1
- WBEOMFTVCDCHGR-UHFFFAOYSA-N CC1=CC(C)=C(N2CC(C)(C)CC2(C)C)C(C)=C1.CCC1=CC=CC(CC)=C1N1CC(C)(C)CC1(C)C Chemical compound CC1=CC(C)=C(N2CC(C)(C)CC2(C)C)C(C)=C1.CCC1=CC=CC(CC)=C1N1CC(C)(C)CC1(C)C WBEOMFTVCDCHGR-UHFFFAOYSA-N 0.000 description 1
- AEKLIWYAKFVADW-SCYJYYCWSA-N CCCC(C1C)[C@@H]2C=CC=CC2C1NC1=C(C)C(CCC)c2c1cccc2 Chemical compound CCCC(C1C)[C@@H]2C=CC=CC2C1NC1=C(C)C(CCC)c2c1cccc2 AEKLIWYAKFVADW-SCYJYYCWSA-N 0.000 description 1
- DCJYHMWWEQQQAH-UHFFFAOYSA-N CN1C=CN(C)C1.CN1CCN(C)C1 Chemical compound CN1C=CN(C)C1.CN1CCN(C)C1 DCJYHMWWEQQQAH-UHFFFAOYSA-N 0.000 description 1
- QKYWADPCTHTJHQ-UHFFFAOYSA-N CNCC(C)C Chemical compound CNCC(C)C QKYWADPCTHTJHQ-UHFFFAOYSA-N 0.000 description 1
- HKQRKLJWAQVSBC-UHFFFAOYSA-N CNCNC Chemical compound CNCNC HKQRKLJWAQVSBC-UHFFFAOYSA-N 0.000 description 1
- 229910020314 ClBr Inorganic materials 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- KNDQHSIWLOJIGP-UHFFFAOYSA-N O=C(C1C2C3C=CC1C3)OC2=O Chemical compound O=C(C1C2C3C=CC1C3)OC2=O KNDQHSIWLOJIGP-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- ACIAHEMYLLBZOI-ZZXKWVIFSA-N Unsaturated alcohol Chemical compound CC\C(CO)=C/C ACIAHEMYLLBZOI-ZZXKWVIFSA-N 0.000 description 1
- 229920002522 Wood fibre Polymers 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 238000010535 acyclic diene metathesis reaction Methods 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 150000001335 aliphatic alkanes Chemical group 0.000 description 1
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 125000001118 alkylidene group Chemical group 0.000 description 1
- 150000001414 amino alcohols Chemical class 0.000 description 1
- 239000007866 anti-wear additive Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 150000001491 aromatic compounds Chemical class 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 150000005840 aryl radicals Chemical group 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 229940095259 butylated hydroxytoluene Drugs 0.000 description 1
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 150000001728 carbonyl compounds Chemical class 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 239000012018 catalyst precursor Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 238000005352 clarification Methods 0.000 description 1
- WHDPTDWLEKQKKX-UHFFFAOYSA-N cobalt molybdenum Chemical compound [Co].[Co].[Mo] WHDPTDWLEKQKKX-UHFFFAOYSA-N 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 238000007334 copolymerization reaction Methods 0.000 description 1
- 238000007405 data analysis Methods 0.000 description 1
- 230000009849 deactivation Effects 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 238000005906 dihydroxylation reaction Methods 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 238000006735 epoxidation reaction Methods 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 150000002191 fatty alcohols Chemical class 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 239000003063 flame retardant Substances 0.000 description 1
- 238000010528 free radical solution polymerization reaction Methods 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000003365 glass fiber Substances 0.000 description 1
- 239000004519 grease Substances 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- AHAREKHAZNPPMI-UHFFFAOYSA-N hexa-1,3-diene Chemical compound CCC=CC=C AHAREKHAZNPPMI-UHFFFAOYSA-N 0.000 description 1
- 230000036571 hydration Effects 0.000 description 1
- 238000006703 hydration reaction Methods 0.000 description 1
- 238000005913 hydroamination reaction Methods 0.000 description 1
- 238000006197 hydroboration reaction Methods 0.000 description 1
- 238000007037 hydroformylation reaction Methods 0.000 description 1
- 238000006459 hydrosilylation reaction Methods 0.000 description 1
- 238000005470 impregnation Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 239000002198 insoluble material Substances 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- JEIPFZHSYJVQDO-UHFFFAOYSA-N iron(III) oxide Inorganic materials O=[Fe]O[Fe]=O JEIPFZHSYJVQDO-UHFFFAOYSA-N 0.000 description 1
- 125000005647 linker group Chemical group 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 238000005461 lubrication Methods 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- 239000000395 magnesium oxide Substances 0.000 description 1
- CPLXHLVBOLITMK-UHFFFAOYSA-N magnesium oxide Inorganic materials [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 1
- AXZKOIWUVFPNLO-UHFFFAOYSA-N magnesium;oxygen(2-) Chemical compound [O-2].[Mg+2] AXZKOIWUVFPNLO-UHFFFAOYSA-N 0.000 description 1
- 238000010128 melt processing Methods 0.000 description 1
- AUHZEENZYGFFBQ-UHFFFAOYSA-N mesitylene Substances CC1=CC(C)=CC(C)=C1 AUHZEENZYGFFBQ-UHFFFAOYSA-N 0.000 description 1
- 125000001827 mesitylenyl group Chemical group [H]C1=C(C(*)=C(C([H])=C1C([H])([H])[H])C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 150000002736 metal compounds Chemical class 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- NFWSQSCIDYBUOU-UHFFFAOYSA-N methylcyclopentadiene Chemical compound CC1=CC=CC1 NFWSQSCIDYBUOU-UHFFFAOYSA-N 0.000 description 1
- SNVLJLYUUXKWOJ-UHFFFAOYSA-N methylidenecarbene Chemical compound C=[C] SNVLJLYUUXKWOJ-UHFFFAOYSA-N 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 229910003455 mixed metal oxide Inorganic materials 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 229910052750 molybdenum Inorganic materials 0.000 description 1
- 150000005673 monoalkenes Chemical class 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 239000012434 nucleophilic reagent Substances 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 150000002978 peroxides Chemical class 0.000 description 1
- 125000004437 phosphorous atom Chemical group 0.000 description 1
- 239000002952 polymeric resin Substances 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 239000012041 precatalyst Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 238000011045 prefiltration Methods 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- 238000013139 quantization Methods 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 229910052703 rhodium Inorganic materials 0.000 description 1
- 238000006798 ring closing metathesis reaction Methods 0.000 description 1
- 125000006413 ring segment Chemical group 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 150000004671 saturated fatty acids Chemical class 0.000 description 1
- 229910052711 selenium Inorganic materials 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 239000011949 solid catalyst Substances 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 239000003351 stiffener Substances 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 229920003002 synthetic resin Polymers 0.000 description 1
- 229910052714 tellurium Inorganic materials 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 238000012956 testing procedure Methods 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 229910052723 transition metal Inorganic materials 0.000 description 1
- 150000003623 transition metal compounds Chemical class 0.000 description 1
- 150000003624 transition metals Chemical class 0.000 description 1
- 150000008648 triflates Chemical class 0.000 description 1
- MCULRUJILOGHCJ-UHFFFAOYSA-N triisobutylaluminium Chemical compound CC(C)C[Al](CC(C)C)CC(C)C MCULRUJILOGHCJ-UHFFFAOYSA-N 0.000 description 1
- LFXVBWRMVZPLFK-UHFFFAOYSA-N trioctylalumane Chemical compound CCCCCCCC[Al](CCCCCCCC)CCCCCCCC LFXVBWRMVZPLFK-UHFFFAOYSA-N 0.000 description 1
- 229910052721 tungsten Inorganic materials 0.000 description 1
- 230000007306 turnover Effects 0.000 description 1
- 125000002948 undecyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000007740 vapor deposition Methods 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 239000002025 wood fiber Substances 0.000 description 1
- 150000003738 xylenes Chemical class 0.000 description 1
- 239000010457 zeolite Substances 0.000 description 1
- 239000004711 α-olefin Substances 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F222/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a carboxyl radical and containing at least one other carboxyl radical in the molecule; Salts, anhydrides, esters, amides, imides, or nitriles thereof
- C08F222/04—Anhydrides, e.g. cyclic anhydrides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F293/00—Macromolecular compounds obtained by polymerisation on to a macromolecule having groups capable of inducing the formation of new polymer chains bound exclusively at one or both ends of the starting macromolecule
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G61/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G61/02—Macromolecular compounds containing only carbon atoms in the main chain of the macromolecule, e.g. polyxylylenes
- C08G61/04—Macromolecular compounds containing only carbon atoms in the main chain of the macromolecule, e.g. polyxylylenes only aliphatic carbon atoms
- C08G61/06—Macromolecular compounds containing only carbon atoms in the main chain of the macromolecule, e.g. polyxylylenes only aliphatic carbon atoms prepared by ring-opening of carbocyclic compounds
- C08G61/08—Macromolecular compounds containing only carbon atoms in the main chain of the macromolecule, e.g. polyxylylenes only aliphatic carbon atoms prepared by ring-opening of carbocyclic compounds of carbocyclic compounds containing one or more carbon-to-carbon double bonds in the ring
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G61/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G61/12—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule
- C08G61/122—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides
- C08G61/123—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides derived from five-membered heterocyclic compounds
- C08G61/124—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides derived from five-membered heterocyclic compounds with a five-membered ring containing one nitrogen atom in the ring
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G61/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G61/12—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule
- C08G61/122—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides
- C08G61/123—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides derived from five-membered heterocyclic compounds
- C08G61/125—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides derived from five-membered heterocyclic compounds with a five-membered ring containing one oxygen atom in the ring
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G61/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G61/12—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule
- C08G61/122—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides
- C08G61/123—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides derived from five-membered heterocyclic compounds
- C08G61/126—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides derived from five-membered heterocyclic compounds with a five-membered ring containing one sulfur atom in the ring
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/30—Monomer units or repeat units incorporating structural elements in the main chain
- C08G2261/33—Monomer units or repeat units incorporating structural elements in the main chain incorporating non-aromatic structural elements in the main chain
- C08G2261/332—Monomer units or repeat units incorporating structural elements in the main chain incorporating non-aromatic structural elements in the main chain containing only carbon atoms
- C08G2261/3324—Monomer units or repeat units incorporating structural elements in the main chain incorporating non-aromatic structural elements in the main chain containing only carbon atoms derived from norbornene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/30—Monomer units or repeat units incorporating structural elements in the main chain
- C08G2261/33—Monomer units or repeat units incorporating structural elements in the main chain incorporating non-aromatic structural elements in the main chain
- C08G2261/334—Monomer units or repeat units incorporating structural elements in the main chain incorporating non-aromatic structural elements in the main chain containing heteroatoms
- C08G2261/3342—Monomer units or repeat units incorporating structural elements in the main chain incorporating non-aromatic structural elements in the main chain containing heteroatoms derived from cycloolefins containing heteroatoms
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/40—Polymerisation processes
- C08G2261/41—Organometallic coupling reactions
- C08G2261/418—Ring opening metathesis polymerisation [ROMP]
Definitions
- This invention is directed toward functionalization of polymers, particularly vinyl-terminated polymers.
- polymers may be functionalized to improve toughness, enhance the acceptance of flame retardants, mineral stiffeners, glass or wood fibers, or other desired ingredients.
- Polymers may also be modified to help them combine more usefully or deliver higher value when recycled. Modifications can improve wetting, aid mix dispersion, filler adhesion, melt processing, surface-to-surface attraction, and other performance features.
- Such polymers are of interest for use in a broad range of applications as lubricants, compatibilizers, tie-layer modifiers, surfactants, and surface modifiers, among other things.
- Metathesis is generally thought of as the interchange of radicals between two compounds during a chemical reaction.
- metathesis reactions such as ring opening metathesis, acyclic diene metathesis, ring closing metathesis, and cross metathesis.
- R. T. Mathers and G. W. Coates, Chem. Commun., 2004, pp. 422-423 disclose examples of using cross-metathesis to functionalize polyolefins containing pendant vinyl groups to form polar-functionalized products with a graft-type structure.
- ROCM ring-open cross metathesis
- U.S. 2008/0064891 discloses ROCM reaction of cyclic olefins with seed oils and the like comprising contacting: (a) at least one olefinic substrate selected from (i) an unsaturated fatty acid, (ii) an unsaturated fatty alcohol, (iii) an esterification product of an unsaturated fatty acid with an alcohol, and (iv) an esterification product of a saturated fatty acid with an unsaturated alcohol; with (b) at least one cyclic olefin as a cross-metathesis partner; in the presence of (c) a ruthenium alkylidene olefin metathesis catalyst; and (d) under conditions effective to allow ring insertion cross-metathesis whereby the cyclic olefin is simultaneously opened and inserted into the olefinic substrate.
- olefinic substrate selected from (i) an unsaturated fatty acid, (ii) an unsaturated fatty alcohol, (iii) an
- WO 98/40373 discloses ROCM on solid supports to isolate the olefin immobilized on the resin, preventing unwanted olefin polymerization. Additional references of interest include: U.S. Pat. Nos. 4,988,764; 6,225,432; EP 1 693 357; U.S. Ser. No. 12/487,739; and U.S. Ser. No. 12/143,663.
- This invention relates to a polymer represented by the formula (A):
- R 1 is a hydrocarbyl group having greater than 25 carbon atoms
- R 2 and R 3 are the same or different and each is hydrogen or a hydrocarbyl group having from 1 to 40 carbon atoms, or R 2 and R 3 are joined to form a five-membered or six-membered ring, or substituted analogs thereof
- X is C, N, or O
- n is an integer from 1 to 10,000
- the dotted line indicates an optional double bond
- the polymer comprises one or more vinyl terminated macromonomer derived units.
- This invention also relates to a polymer represented by the formula (A):
- R 2 and R 3 are the same or different and each is hydrogen or a hydrocarbyl group having from 1 to 40 carbon atoms, or R 2 and R 3 are joined to form a five-membered or six-membered ring, or substituted analogs thereof;
- X is C, N, or O;
- n is an integer from 1 to 10,000; and the dotted line indicates an optional double bond;
- R 1 is (i) a polymer comprising of one or more C 4 to C 40 higher olefin derived units, where the higher olefin polymer comprises substantially no propylene derived units; and or (ii) a copolymer comprising (a) from about 20 mol % to about 99.9 mol % of at least one C 5 to C 40 higher olefin and (b) from about 0.1 mol % to about 80 mol % of propylene; and or (iii) a copolymer comprising (a) from about 80 mol % to about 99.
- This invention also relates to a process to produce the polymers described above.
- This invention also relates to a composition comprising the polymers described above.
- FIG. 1 is a representation of some of the possible outcomes of ring opening cross-metathesis.
- Polyolefin means an oligomer or polymer of two or more olefin mer units and specifically includes oligomers and polymers as defined below.
- An “olefin,” alternatively referred to as “alkene,” is a linear, branched, or cyclic compound of carbon and hydrogen having at least one double bond.
- a “mono-olefin” has one double bond, for example, an alpha, omega, pendant, or internal double bond.
- a “polymer” has two or more of the same or different mer units.
- a “homopolymer” is a polymer having mer units that are the same.
- a “copolymer” is a polymer having two or more mer units that are different from each other.
- a “terpolymer” is a polymer having three mer units that are different from each other.
- the term “different” as used to refer to mer units indicates that the mer units differ from each other by at least one atom or are different isomerically. Accordingly, the definition of copolymer, as used herein, includes terpolymers and the like.
- An oligomer is typically a polymer having a low molecular weight (such as an Mn of less than 25,000 g/mol, preferably less than 2,500 g/mol) or a low number of mer units (such as 75 mer units or less, typically 50 mer units or less, even 20 mer units or less, even 10 mer units or less).
- a low molecular weight such as an Mn of less than 25,000 g/mol, preferably less than 2,500 g/mol
- a low number of mer units such as 75 mer units or less, typically 50 mer units or less, even 20 mer units or less, even 10 mer units or less.
- a polymer or copolymer is referred to as comprising an olefin, including, but not limited to ethylene, propylene, and butene
- the olefin present in such polymer or copolymer is the polymerized form of the olefin.
- a copolymer is said to have an “ethylene” content of 35 wt % to 55 wt %, it is understood that the mer unit in the copolymer is derived from ethylene in the polymerization reaction and said derived units are present at 35 wt % to 55 wt %, based upon the weight of the copolymer.
- an ethylene polymer or oligomer contains at least 50 mol % of ethylene
- a propylene polymer or oligomer contains at least 50 mol % of propylene
- a butene polymer or oligomer contains at least 50 mol % of butene, and so on.
- Mn is number average molecular weight
- Mw is weight average molecular weight
- Mz is z average molecular weight.
- Molecular weight distribution is defined to be Mw divided by Mn. Unless otherwise noted, all molecular weight units (e.g., Mw, Mn, Mz) are g/mol.
- substituted means that a hydrogen group has been replaced with a hydrocarbyl group, a heteroatom or a heteroatom containing group.
- methyl cyclopentadiene is a cyclopentadiene (Cp) group substituted with a methyl group
- ethyl alcohol is an ethyl group substituted with an —OH group.
- hydrocarbyl radical is defined to be C 1 to C 20 radicals, that may be linear, branched, or cyclic (aromatic or non-aromatic); and include substituted hydrocarbyl radicals as defined below.
- Substituted hydrocarbyl radicals are radicals in which at least one hydrogen atom has been substituted with a heteroatom or heteroatom containing group, preferably with at least one functional group such as halogen (Cl, Br, I, F), NR* 2 , OR*, SeR*, TeR*, PR* 2 , AsR* 2 , SbR* 2 , SR*, BR* 2 , SiR* 3 , GeR* 3 , SnR* 3 , PbR* 3 , and the like or where at least one heteroatom has been inserted within the hydrocarbyl radical, such as halogen (Cl, Br, I, F), O, S, Se, Te, NR*, PR*, AsR*, SbR*, BR*, SiR* 2 , GeR* 2 , SnR* 2 , PbR* 2 , and the like, where R* is, independently, hydrogen or a hydrocarbyl.
- halogen Cl, Br, I
- a “substituted alkyl” is an alkyl radical made of carbon and hydrogen where at least one hydrogen is replaced by a heteroatom, a heteroatom containing group, or a cyclic substituted or unsubstituted hydrocarbyl group having 1 to 30 carbon atoms.
- a “substituted aryl” group is an aryl radical made of carbon and hydrogen where at least one hydrogen is replaced by a heteroatom, a heteroatom containing group, or a linear, branched, or cyclic substituted or unsubstituted hydrocarbyl group having 1 to 30 carbon atoms.
- a “heteroatom containing ring” is a cyclic ring where one or more ring vertices are occupied by a heteroatom (N, O, P, S).
- tetrahydrofuran is a heteroatom containing ring, having an oxygen atom as part of the ring backbone.
- anionic ligand is a negatively charged ligand which donates one or more pairs of electrons to a metal ion.
- neutral donor ligand is a neutrally charged ligand which donates one or more pairs of electrons to a metal ion.
- catalysts are described as comprising neutral stable forms of the components, it is well understood by one of ordinary skill in the art, that the ionic form of the component is the form that reacts with the monomers to produce polymers.
- the transition metal compound used for catalysis may be described as a catalyst precursor, a pre-catalyst compound, a catalyst, or a catalyst compound, and these terms are used interchangeably.
- a “reactor” is any container(s) in which a chemical reaction occurs.
- Mol % means mole percent
- wt % means weight percent
- vol % means volume percent
- This invention relates to a new class of functionalized polymers and processes to produce them.
- These polymers are end-functionalized with a cyclic functional group and possess the ability to be further functionalized through a terminal vinyl group.
- This ability to add further functionality post-polymerization affords appreciable synthetic flexibility that may be of tremendous commercial utility. For instance, bulk polymer properties such as viscosity may be tailored by utilizing this synthetic handle to increase the size and viscosity of the polymer.
- functionalized vinyl-terminated polymers also referred to as functionalized vinyl terminated macromonomers
- This invention relates to a polymer represented by the formula (A):
- R 1 is a hydrocarbyl group having greater than 25 carbon atoms, preferably greater than 30 carbon atoms, greater than 40 carbon atoms, or greater than 50 carbon atoms; preferably from 31 to 100,000 carbons, from 40 to 75,000 carbons, from 50 to 60,000 carbons; preferably R 1 is represented by the formula (B):
- Cm is a C 4 to C 40 olefin derived unit (preferably Cm is one or more derived units of butene, pentene, hexene, heptene, octene, nonene, decene, undecene, dodecene, norbornene, norbornadiene, dicyclopentadiene, cyclopentene, cycloheptene, cyclooctene, cyclooctadiene, cyclododecene, 7-oxanorbornene, 7-oxanorbornadiene, substituted derivatives thereof, and isomers thereof); m is an integer from 3 to 39 (preferably from 3 to 29, preferably from 4 to 19, preferably from 4 to 11); and p is an integer greater than 1 (preferably from 1 to 10,000, from 1 to 5,000, from 1 to 2,500, from 1 to 1,000, from 1 to 500, or from 1 to 50); preferably R 1 is represented by the formula (C
- Cm is a C 3 to C 40 olefin derived unit (preferably Cm is one or more derived units of propylene, butene, pentene, hexene, heptene, octene, nonene, decene, undecene, dodecene, norbornene, norbornadiene, dicyclopentadiene, cyclopentene, cycloheptene, cyclooctene, cyclooctadiene, cyclododecene, 7-oxanorbornene, 7-oxanorbornadiene, substituted derivatives thereof, and isomers thereof); m is an integer from 2 to 39 (preferably from 2 to 29, preferably from 3 to 19, preferably from 4 to 11); and p is an integer greater than 1 (preferably from 1 to 10,000, from 1 to 5,000, from 1 to 2,500, from 1 to 1,000, from 1 to 500, or from 1 to 50); R 2 and R 3 are the
- R 1 is preferably not an ethylene polymer. In any embodiment described herein, R 1 is preferably not a propylene polymer. In any embodiment described herein, R 1 is preferably not a butene polymer.
- R 1 is derived from vinyl terminated polymer (i) described above. In a preferred embodiment of the invention R 1 is derived from vinyl terminated polymer (ii) described above. In a preferred embodiment of the invention R 1 is derived from vinyl terminated polymer (iii) described above.
- V is a C X to C y olefin derived unit” (where x and y are the integers described above) means that Cm as depicted in the formulae has one less carbon than the olefin it was derived from, i.e., one carbon of the originating olefin is incorporated into the polymer backbone.
- the polymer is represented by the formula (D):
- X is C, N, or O (preferably C or O, preferably C); n is an integer from 1 to 10,000 (preferably from 1 to 5,000, from 1 to 2,500, from 1 to 1,000, from 1 to 500, or from 1 to 50); Cm is a C 3 to C 40 olefin derived unit (preferably Cm is one or more derived units of propylene, butene, pentene, hexene, heptene, octene, nonene, decene, undecene, dodecene, norbornene, norbornadiene, dicyclopentadiene, cyclopentene, cycloheptene, cyclooctene, cyclooctadiene, cyclododecene, 7-oxanorbornene, 7-oxanorbornadiene, substituted derivatives thereof, and isomers thereof); m is an integer from 2 to 39 (preferably from 2 to 29, preferably from 3 to 19,
- the vinyl terminated macromonomer derived units are derived from (i), (ii) or (iii) above.
- this invention relates to a polymer represented by the formula (A):
- R 1 is (i) a polymer, preferably having an Mn of at least 200 g/mol (measured by 1 H NMR), comprising of one or more C 4 to C 40 higher olefin derived units (preferably C 5 to C 40 higher olefin derived units), where the higher olefin polymer comprises substantially no propylene derived units; and or (ii) a copolymer, preferably having an Mn of at least 300 g/mol (measured by 1 H NMR), comprising (a) from about 20 mol % to about 99.9 mol % of at least one C 5 to C 40 higher olefin and (b) from about 0.1 mol % to about 80 mol % of propylene; and or (iii) a copolymer, preferably having an Mn of at least 300 g/mol (measured by 1 H NMR), comprising (a) from about
- the polymer is hydrogenated.
- the polymers produced herein may be hydrogenated by contacting the polymer with hydrogen and a hydrogenation catalyst. This hydrogenation step is often used to reduce the bromine number (preferably below 2.0, preferably below 1.8). Bromine number is determined by ASTM D 1159. In a preferred embodiment, the bromine number of the hydrogenated polymer decreases by at least 50% (preferably at least 75%) as compared to the starting polymer.
- the hydrogenation catalyst is selected from the group consisting of supported Group 7, 8, 9, and 10 metals, preferably the hydrogenation catalyst selected from the group consisting of one or more of Ni, Pd, Pt, Co, Rh, Fe, Ru, Os, Cr, Mo, and W, supported on silica, alumina, clay, titania, zirconia, or mixed metal oxide supports.
- a preferred hydrogenation catalyst is nickel supported on kieselguhr, or platinum or palladium supported on alumina, or cobalt-molybdenum supported on alumina Usually, a high nickel content catalyst, such as 60% Ni on Kieselguhr catalyst, is used, or a supported catalyst with high amount of Co—Mo loading. Alternately, the hydrogenation catalyst is nickel supported on Kieselguhr, silica, alumina, clay, or silica-alumina.
- the polymer is contacted with hydrogen (preferably at a hydrogen pressure of from 25 psi to 2500 psi (0.17 MPa to 17.24 MPa), preferably from 100 psi to 2000 psi (0.69 MPa to 13.79 MPa)), and a hydrogenation catalyst at a temperature from 25° C. to 350° C., preferably 100° C. to 300° C., and/or a time period from 5 minutes to 100 hours, preferably from 5 minutes to 24 hours.
- hydrogen preferably at a hydrogen pressure of from 25 psi to 2500 psi (0.17 MPa to 17.24 MPa), preferably from 100 psi to 2000 psi (0.69 MPa to 13.79 MPa
- a hydrogenation catalyst at a temperature from 25° C. to 350° C., preferably 100° C. to 300° C., and/or a time period from 5 minutes to 100 hours, preferably from 5 minutes to 24 hours.
- the hydrogenation process can be accomplished in a slurry reactor in a batch operation or in a continuous stirred tank reactor (CSTR), where the catalyst, hydrogen, and the polymer are continuously added to the reactor to allow for certain residence time, usually 5 minutes to 10 hours to allow complete hydrogenation of the unsaturated olefins.
- the amount of catalyst added is usually very small, for example, 0.001 wt % to 20 wt % of the polymer feed or preferably 0.01 wt % to 10 wt %, to compensate for the catalyst deactivation.
- the catalyst and hydrogenated polymer are continuously withdrawn from the reactor.
- the product mixture may then be filtered, centrifuged, or settled to remove the solid hydrogenation catalyst.
- the catalyst can be regenerated and reused.
- the hydrogenation process can also be accomplished by a fixed bed process, in which the solid catalyst is packed inside a tubular reactor and heated to reactor temperature.
- compositions comprising the polymers produced herein are also disclosed.
- the composition is a lubricant or lubricant base stock, an adhesive, a viscosity modifier, or a fuel additive.
- a novel lubricant comprises the polymers produced in this invention, alone or together with one or more other base stocks, including Group I to Group V base stocks with kinematic viscosity (ASTM D445) range from 1.5 cSt to 100 cSt at 100° C. to formulate suitable viscosity grades.
- additives of one or more of: thickeners, viscosity index improvers, antioxidants, anti-wear additives, detergent/dispersant/inhibitor packages, and/or anti-rust additives may be added.
- the polymers produced herein are combined with one or more of dispersants, detergents, friction modifiers, traction improving additives, demulsifiers, defoamants, chromophores (dyes), and/or haze inhibitors.
- dispersants detergents, friction modifiers, traction improving additives, demulsifiers, defoamants, chromophores (dyes), and/or haze inhibitors.
- These fully formulated lubricants can be used in automotive crank case oil (engine oil), industrial oil, grease, or gas turbine engine oil.
- additives used in finished lubricant formulations are examples of additives used in finished lubricant formulations. Additional information on additives used in product formulation can be found in “Lubricants and Lubrications”, Ed. By T. Mang and W. Dresel, by Wiley-VCH GmbH, Weinheim 2001.
- the polymers prepared herein may be further functionalized by reacting a heteroatom containing group (preferably amines, aldehydes, alcohols, acids, succinic acid, maleic acid, and/or maleic anhydride) with the polymer, with or without a catalyst.
- a heteroatom containing group preferably amines, aldehydes, alcohols, acids, succinic acid, maleic acid, and/or maleic anhydride
- a heteroatom containing group preferably amines, aldehydes, alcohols, acids, succinic acid, maleic acid, and/or maleic anhydride
- a heteroatom containing group preferably amines, aldehydes, alcohols, acids, succinic acid, maleic acid, and/or maleic anhydride
- Examples include catalytic hydrosilylation, hydroformylation, hydroboration, epoxidation, hydration, dihydroxylation, hydroamination, or maleation with or without activators such as free radical generators (e.g
- the functionalized polymers can be used in lubricant, oil additivation, and many other applications.
- the vinyl end group of the polymers of the present invention is synthetically facile thereby allowing for functionalization of the resultant polymer.
- Some examples of functionalized polymers include those that are functionalized with maleic acid or maleic anhydride groups.
- the functionalized polymers can in turn be derivatized with a derivatizing compound, such as described in U.S. Pat. No. 6,022,929; A. Toyota, T. Tsutsui, and N. Kashiwa, Polymer Bulletin 48, 213-219, 2002; and J. Am. Chem. Soc., 1990, 112, 7433-7434.
- the derivatizing compound can react with the functional groups of the functionalized capped polymer by any means known in the art, such as nucleophilic substitution, Mannich Base condensation, and the like.
- the derivatizing compound can be polar and/or contain reactive derivative groups.
- Preferred derivatizing compounds are selected from hydroxy containing compounds, amines, metal salts, anhydride containing compounds, and acetyl halide containing compounds.
- the derivatizing compounds can comprise at least one nucleophilic group and preferably at least two nucleophilic groups.
- An exemplary derivatized polymer may be made by contacting a functionalized polymer, for example, one substituted with a carboxylic acid/anhydride or ester, with a nucleophilic reagent, for example, amines, alcohols (including polyols), amino alcohols, reactive metal compounds and the like.
- a nucleophilic reagent for example, amines, alcohols (including polyols), amino alcohols, reactive metal compounds and the like.
- the functionalized polymers of the present invention may be produced in any manner known to one of skill in the art. These functionalized polymers are advantageously produced from vinyl terminated macromonomers, such as those described in U.S. Ser. No. 13/072,288; U.S. Ser. No. 13/072,249; and U.S. Ser. No. 12/143,663. More particularly, in embodiments of the present invention, processes for producing a polymer comprise contacting at least (A) one cyclic olefin with (B) at least one vinyl terminated macromonomer in the presence of (C) a metathesis catalyst, under suitable polymerization conditions.
- the product comprises a ROCM product of a cyclic olefin and a vinyl terminated macromonomer.
- this invention also relates to a process to produce a polymer represented by the formula (A):
- R 1 is a hydrocarbyl group having greater than 25 carbon atoms
- R 2 and R 3 are the same or different and each is hydrogen or a hydrocarbyl group having from 1 to 40 carbon atoms, or R 2 and R 3 are joined to form a five-membered or six-membered ring, or substituted analogs thereof
- X is C, N, or O
- n is an integer from 1 to 10,000
- the dotted line indicates an optional double bond, wherein the polymer comprises one or more vinyl terminated macromonomer derived units; the process comprising contacting: at least one cyclic olefin (preferably norbornene, norbornadiene, dicyclopentadiene, cyclopentene, cycloheptene, cyclooctene, cyclooctadiene, cyclododecene, 7-oxanorbornene, 7-oxanorbornadiene, and substituted derivatives therefrom; more preferably cyclo
- M is a Group 8 metal, preferably Ru or Os, preferably Ru;
- X and X 1 are, independently, any anionic ligand, preferably a halogen (preferably chlorine), an alkoxide or a triflate, or X and X 1 may be joined to form a dianionic group and may form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
- L and L 1 are, independently, a neutral two electron donor, preferably a phosphine or a N-heterocyclic carbene, L and L 1 may be joined to form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
- L and X may be joined to form a multidentate monoanionic group and may form a single ring of up to 30 non-hydrogen atoms or a multinu
- M is Os or Ru, preferably Ru;
- X, X 1 , L, and L 1 are as described above for Formula E; and
- R 9 and R 10 may be different or the same and may be hydrogen, substituted or unsubstituted alkyl, or substituted or unsubstituted aryl; and/or (iii) a metathesis catalyst represented by the formula (G):
- M* is a Group 8 metal, preferably Ru or Os, preferably Ru;
- X* and X 1 * are, independently, any anionic ligand, preferably a halogen (preferably chlorine), an alkoxide or an alkyl sulfonate, or X* and X 1 * may be joined to form a dianionic group and may form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
- L* is N—R**, 0, P—R**, or S, preferably N—R** or O (R** is a C 1 to C 30 hydrocarbyl or substituted hydrocarbyl, preferably methyl, ethyl, propyl or butyl);
- R* is hydrogen or a C 1 to C 30 hydrocarbyl or substituted hydrocarbyl, preferably methyl;
- M′′ is a Group 8 metal (preferably M is ruthenium or osmium, preferably ruthenium); each X is independently an anionic ligand (preferably selected from the group consisting of halides, alkoxides, aryloxides, and alkyl sulfonates, preferably a halide, preferably chloride); R′′ 1 and R′′ 2 are independently selected from the group consisting of hydrogen, a C 1 to C 30 hydrocarbyl, and a C 1 to C 30 substituted hydrocarbyl (preferably R′′ 1 and R′′ 2 are independently selected from the group consisting of methyl, ethyl, propyl, isopropyl, butyl, tert-butyl, sec-butyl, pentyl, cyclopentyl, hexyl, cyclohexyl, heptyl, octyl, cyclooctyl, and substituted analogs and isomers thereof, preferably selected from the group 8
- M′′ is a Group 8 metal (preferably M is ruthenium or osmium, preferably ruthenium); each X is independently an anionic ligand (preferably selected from the group consisting of halides, alkoxides, aryloxides, and alkyl sulfonates, preferably a halide, preferably chloride); R′′ 1 and R′′ 2 are independently selected from the group consisting of hydrogen, a C 1 to C 30 hydrocarbyl, and a C 1 to C 30 substituted hydrocarbyl (preferably R′′ 1 and R′′ 2 are independently selected from the group consisting of methyl, ethyl, propyl, isopropyl, butyl, tert-butyl, sec-butyl, pentyl, cyclopentyl, hexyl, cyclohexyl, heptyl, octyl, cyclooctyl, and substituted analogs and isomers thereof, preferably selected from the group 8
- the catalyst is represented by formula F. In a preferred embodiment, the catalyst is represented by formula G. In a preferred embodiment, the catalyst is represented by formula H. In a preferred embodiment, the catalyst is represented by formula I.
- the ROCM involves a tandem sequence in which a cyclic olefin is opened and a vinyl terminated macromonomer is crossed onto the newly formed termini. After the initial ring opening of the cyclic olefin, the metal-bound intermediate has two options: reaction with another cyclic olefin or reaction with the vinyl terminated macromonomer. It will be appreciated that a ROCM reaction between a cyclic olefin and a vinyl terminated macromonomer reactant can result in several different types of reaction products, depending, in large part, on the relative rates of the ring-opening metathesis reaction and the cross-metathesis reaction between the vinyl terminated macromonomer and the cyclic olefin, as shown in FIG.
- R is a hydrocarbyl group derived from the vinyl terminated macromonomer and having greater than 25 carbon atoms (preferably greater than 30 carbon atoms, greater than 40 carbon atoms, or greater than 50 carbon atoms).
- a cyclic olefin will undergo a ring opening reaction in the presence of the catalyst at a rate constant k RO
- the vinyl terminated macromonomer will undergo a cross-metathesis reaction with the ring opened cyclic olefin at a rate constant k CM .
- k CM is greater than or equal to k RO
- the ROCM product is predominantly a monomer, dimer, and/or oligomer. More specifically, when k CM is approximately equal to k RO , the ROCM product is predominantly a dimer, while when k RO is greater than k CM , the ROCM product is predominantly higher Mw.
- Oligomers are of particular interest because their internal olefin moieties may be further functionalized by metathesis or other transformations. It should be appreciated that k RO will be higher for moderately and highly strained cyclic olefins such as norbornadiene, but lower for low-strain olefins such as cyclopentene and cyclohexene.
- the reactants are typically combined in a reaction vessel at a temperature of 20° C. to 200° C. (preferably 50° C. to 160° C., preferably 60° C. to 140° C.) and a pressure of 0 MPa to 1000 MPa (preferably 0.5 MPa to 500 MPa, preferably 1 MPa to 250 MPa) for a residence time of 0.5 seconds to 10 hours (preferably 1 second to 5 hours, preferably 1 minute to 1 hour).
- the molecular weight of the polymer products may be controlled by, inter alia, choice of catalyst, ratio of vinyl terminated macromonomer to cyclic olefin, and/or possibly temperature.
- the olefin pressure is typically greater than 5 psig (34.5 kPa); preferably, greater than 10 psig (68.9 kPa); and more preferably, greater than 45 psig (310 kPa).
- the aforementioned pressure ranges may also be suitably employed as the total pressure of olefin and diluent.
- the aforementioned pressure ranges may be suitably employed for the inert gas pressure.
- the quantity of metathesis catalyst that is employed in the process of this invention is any quantity that provides for an operable metathesis reaction.
- the ratio of moles of monomers (e.g., cyclic olefins and vinyl terminated macromonomer) to moles of metathesis catalyst is typically greater than 10:1, preferably greater than 100:1, preferably greater than 1,000:1, preferably greater than 10,000:1, preferably greater than 25,000:1, preferably greater than 50,000:1, preferably greater than 100,000:1).
- 0.00001 moles to 1.0 moles, preferably 0.0001 moles to 0.05 moles, preferably 0.0005 moles to 0.01 moles of catalyst are charged to the reactor per mole of vinyl terminated macromonomer charged.
- 0.00001 moles to 1.0 moles, preferably 0.0001 moles to 0.05 moles, preferably 0.0005 moles to 0.01 moles of catalyst are charged to the reactor per mole of cyclic olefin charged.
- the ratio of vinyl terminated macromonomer to cyclic olefin monomer is preferably 0.01:1 to 1000:1, preferably 1:1 to 100:1 depending on the final polymer sought. It has been noted that in the instant invention, the ratio of vinyl terminated macromonomer to cyclic olefin monomer has an effect on molecular weight.
- the process is typically a solution process, although it may be a bulk or high pressure process. Homogeneous processes are preferred. (A homogeneous process is defined to be a process where at least 90 wt % of the product is soluble in the reaction media.) A bulk homogeneous process is particularly preferred. (A bulk process is defined to be a process where reactant concentration in all feeds to the reactor is 70 vol % or more.) Alternately, no solvent or diluent is present or added in the reaction medium, (except for the small amounts used as the carrier for the catalyst or other additives, or amounts typically found with the reactants, e.g., propane in propylene).
- Suitable diluents/solvents for the process include non-coordinating, inert liquids.
- Examples include straight and branched-chain hydrocarbons, such as isobutane, butane, pentane, isopentane, hexanes, isohexane, heptane, octane, dodecane, and mixtures thereof; cyclic and alicyclic hydrocarbons, such as cyclohexane, cycloheptane, methylcyclohexane, methylcycloheptane, and mixtures thereof such as can be found commercially (IsoparTM); perhalogenated hydrocarbons, such as perfluorinated C 4-10 alkanes, chlorobenzene, and aromatic and alkyl substituted aromatic compounds, such as benzene, toluene, mesitylene, and xylene.
- straight and branched-chain hydrocarbons such as isobutane, butane,
- aliphatic hydrocarbon solvents are preferred, such as isobutane, butane, pentane, isopentane, hexanes, isohexane, heptane, octane, dodecane, and mixtures thereof; cyclic and alicyclic hydrocarbons, such as cyclohexane, cycloheptane, methylcyclohexane, methylcycloheptane, and mixtures thereof.
- the solvent is not aromatic.
- aromatics are present in the solvent at less than 1 wt %, preferably at 0.5 wt %, preferably at 0 wt % based upon the weight of the solvents.
- suitable diluents/solvents also include aromatic hydrocarbons, such as toluene or xylenes, and chlorinated solvents, such as dichloromethane.
- the feed for the process comprises 60 vol % solvent or less, based on the total volume of the feed, preferably 40 vol % or less, preferably 20 vol % or less.
- the process is a slurry process.
- slurry process or “slurry polymerization process” means a polymerization process where a supported catalyst is employed and monomers are polymerized on the supported catalyst particles. At least 95 wt % of polymer products derived from the supported catalyst is in granular form as solid particles (not dissolved in the diluent).
- the process may be batch, semi-batch, or continuous.
- continuous means a system that operates without interruption or cessation.
- a continuous process to produce a polymer would be one where the reactants are continually introduced into one or more reactors and polymer product is continually withdrawn.
- Useful reaction vessels include reactors (including continuous stirred tank reactors, batch reactors, reactive extruders, pipes, or pumps).
- the productivity of the process is at least 200 g of polymer (preferably polymer represented by formula (A)) per mmol of catalyst per hour, preferably at least 5000 g/mmol/hour, preferably at least 10,000 g/mmol/hr, preferably at least 300,000 g/mmol/hr.
- This invention further relates to a process, preferably an in-line process, preferably a continuous process, to produce polymer represented by formula (A), comprising introducing cyclic olefin, vinyl terminated macromonomer, and alkene metathesis catalyst into a reactor, obtaining a reactor effluent containing polymer, optionally removing (such as flashing off) solvent, unused monomer and/or other volatiles, obtaining polymer then hydrogenating or functionalizing the polymer.
- a process preferably an in-line process, preferably a continuous process, to produce polymer represented by formula (A), comprising introducing cyclic olefin, vinyl terminated macromonomer, and alkene metathesis catalyst into a reactor, obtaining a reactor effluent containing polymer, optionally removing (such as flashing off) solvent, unused monomer and/or other volatiles, obtaining polymer then hydrogenating or functionalizing the polymer.
- reaction zone also referred to as a “polymerization zone” is defined as an area where activated catalysts and monomers are contacted and a polymerization reaction takes place.
- each reactor is considered as a separate polymerization zone.
- each polymerization stage is considered as a separate polymerization zone.
- the process produces the metathesis product (polymers having the formula A disclosed herein) in good yield (greater than 50%).
- the process produces little or no dimerization.
- the extent of dimerization is determined by the comparison of the degree of polymerization (ROCM) as obtained from methods A and B, described in the Examples.
- the degree of ROCM obtained from Method A differs from that obtained from Method B by less than 25%, preferably less than 20%, preferably less than 15%, and most preferably less than 10%.
- the cyclic olefin may be a single cyclic olefin, or a combination of cyclic olefins, that is a mixture of two or more different cyclic olefins.
- the cyclic olefins may be strained or unstrained, monocyclic, or polycyclic; and may optionally include heteroatoms and/or one or more functional groups.
- dicyclopentadiene, norbornene, norbornadiene, ethylidene norbornene, and vinyl norbornene are polycyclic.
- Suitable cyclic olefins include, but are not limited to norbornene, norbornadiene, dicyclopentadiene, cyclopentene, cycloheptene, cyclooctene, cyclooctadiene, cyclododecene, 7-oxanorbornene, 7-oxanorbornadiene, and substituted derivatives therefrom.
- substituents include, but are not limited to, hydroxyl, thiol, ketone, aldehyde, ester, ether, amine, imine, amide, nitro, carboxylic acid, disulfide, carbonate, isocyanate, carbodiimide, carboalkoxy, and halogen.
- Preferred cyclic olefins include cyclooctene, 1,5-cyclooctadiene, 1-hydroxy-4-cyclooctene, 1-acetoxy-4-cyclooctene, 5-methylcyclopentene, cyclopentene, dicyclopentadiene, norbornene, norbornadiene, and their respective homologs and derivatives, preferably norbornene, norbornadiene, cis-5-norbornene-endo-2,3-dicarboxylic anhydride, dimethyl norbornene carboxylate, norbornene-exo-2,3-carboxylic anhydride, and dicyclopentadiene.
- the cyclic olefin is derived from substituted or unsubstituted cyclopentadiene, such as dicyclopentadiene, norbornene, norbornadiene, cis-5-norbornene-endo-2,3-dicarboxylic anhydride, dimethyl norbornene carboxylate, norbornene-exo-2,3-carboxylic anhydride, ethylidene norbornene, vinyl norbornene, and the like.
- substituted or unsubstituted cyclopentadiene such as dicyclopentadiene, norbornene, norbornadiene, cis-5-norbornene-endo-2,3-dicarboxylic anhydride, dimethyl norbornene carboxylate, norbornene-exo-2,3-carboxylic anhydride, ethylidene norbornene, vinyl norbornene, and the like.
- a “vinyl terminated macromonomer,” as used herein, refers to one or more of:
- a copolymer having an Mn of 300 g/mol or more (measured by 1 H NMR) comprises (a) from about 80 mol % to
- the vinyl terminated macromonomer has an Mn of at least 200 g/mol, (preferably 200 g/mol to 100,000 g/mol, preferably 200 g/mol to 75,000 g/mol, preferably 200 g/mol to 60,000 g/mol, preferably 300 g/mol to 60,000 g/mol, or preferably 750 g/mol to 30,000 g/mol) (measured by 1 H NMR), and comprise one or more (preferably two or more, three or more, four or more, and the like) C 4 to C 40 (preferably C 4 to C 30 , C 4 to C 20 , or C 4 to C 12 , preferably butene, pentene, hexene, heptene, octene, nonene, decene, undecene, dodecene, norbornene, norbornadiene, dicyclopentadiene, cyclopentene, cycloheptene, cyclooctene
- the vinyl terminated macromonomers may also comprise ethylene derived units, preferably at least 5 mol % ethylene (preferably at least 15 mol % ethylene, preferably at least 25 mol % ethylene, preferably at least 35 mol % ethylene, preferably at least 45 mol % ethylene, preferably at least 60 mol % ethylene, preferably at least 75 mol % ethylene, or preferably at least 90 mol % ethylene).
- ethylene derived units preferably at least 5 mol % ethylene (preferably at least 15 mol % ethylene, preferably at least 25 mol % ethylene, preferably at least 35 mol % ethylene, preferably at least 45 mol % ethylene, preferably at least 60 mol % ethylene, preferably at least 75 mol % ethylene, or preferably at least 90 mol % ethylene).
- the vinyl terminated macromonomers described herein have an Mn (measured by 1 H NMR) of greater than 200 g/mol (preferably 300 g/mol to 60,000 g/mol, 400 g/mol to 50,000 g/mol, 500 g/mol to 35,000 g/mol, 300 g/mol to 15,000 g/mol, 400 g/mol to 12,000 g/mol, or 750 g/mol to 10,000 g/mol), and comprise:
- the vinyl terminated macromonomer has an Mn of 300 g/mol or more (measured by 1 H NMR, preferably 300 g/mol to 60,000 g/mol, 400 g/mol to 50,000 g/mol, 500 g/mol to 35,000 g/mol, 300 g/mol to 15,000 g/mol, 400 g/mol to 12,000 g/mol, or 750 g/mol to 10,000 g/mol), and comprises:
- allyl chain ends preferably at least 50% allyl chain ends, at least 60% allyl chain ends, at least 70% allyl chain ends, or at least 80% allyl chain ends, at least 90% allyl chain ends, at least 95% allyl chain ends
- an isobutyl chain end to allyl chain end ratio of less than 0.70:1, less than 0.65:1, less than 0.60:1, less than 0.50:1, or less than 0.25:1, and in further embodiments, an allyl chain end to vinylidene group ratio of more than 2:1, more than 2.5:1, more than 3:1, more than 5:1, or more than 10:1.
- Such macromonomers are also further described in U.S.
- the vinyl terminated macromonomer is a propylene co-oligomer having an Mn of 300 g/mol to 30,000 g/mol as measured by 1 H NMR (preferably 400 g/mol to 20,000 g/mol, preferably 500 g/mol to 15,000 g/mol, preferably 600 g/mol to 12,000 g/mol, preferably 800 g/mol to 10,000 g/mol, preferably 900 g/mol to 8,000 g/mol, preferably 900 g/mol to 7,000 g/mol), comprising 10 mol % to 90 mol % propylene (preferably 15 mol % to 85 mol %, preferably 20 mol % to 80 mol %, preferably 30 mol % to 75 mol %, preferably 50 mol % to 90 mol %) and 10 mol % to 90 mol % (preferably 85 mol % to 15 mol %, preferably 20 mol % to 80 mol %, preferably 85 mol % to 15
- the vinyl terminated macromonomer is a propylene oligomer, comprising more than 90 mol % propylene (preferably 95 mol % to 99 mol %, preferably 98 mol % to 9 mol %) and less than 10 mol % ethylene (preferably 1 mol % to 4 mol %, preferably 1 mol % to 2 mol %), wherein the oligomer has: at least 93% allyl chain ends (preferably at least 95%, preferably at least 97%, preferably at least 98%); a number average molecular weight (Mn) of about 400 g/mol to about 30,000 g/mol, as measured by 1 H NMR (preferably 500 g/mol to 20,000 g/mol, preferably 600 g/mol to 15,000 g/mol, preferably 700 g/mol to 10,000 g/mol, preferably 800 g/mol to 9,000 g/mol, preferably 900 g/mol to 8,000 g/mol,
- the vinyl terminated macromonomer is a propylene oligomer, comprising: at least 50 mol % (preferably 60 mol % to 90 mol %, preferably 70 mol % to 90 mol %) propylene and from 10 mol % to 50 mol % (preferably 10 mol % to 40 mol %, preferably 10 mol % to 30 mol %) ethylene, wherein the oligomer has: at least 90% allyl chain ends (preferably at least 91%, preferably at least 93%, preferably at least 95%, preferably at least 98%); an Mn of about 150 g/mol to about 20,000 g/mol, as measured by 1 H NMR (preferably 200 g/mol to 15,000 g/mol, preferably 250 g/mol to 15,000 g/mol, preferably 300 g/mol to 10,000 g/mol, preferably 400 g/mol to 9,500 g/mol, preferably 500 g/mol to 9,000 g/mol
- the vinyl terminated macromonomer is a propylene oligomer, comprising: at least 50 mol % (preferably at least 60 mol %, preferably 70 mol % to 99.5 mol %, preferably 80 mol % to 99 mol %, preferably 90 mol % to 98.5 mol %) propylene, from 0.1 mol % to 45 mol % (preferably at least 35 mol %, preferably 0.5 mol % to 30 mol %, preferably 1 mol % to 20 mol %, preferably 1.5 mol % to 10 mol %) ethylene, and from 0.1 mol % to 5 mol % (preferably 0.5 mol % to 3 mol %, preferably 0.5 mol % to 1 mol %) C 4 to C 12 olefin (such as butene, hexene or octene, preferably butene), wherein the oligomer has: at least 90% ally
- the vinyl terminated macromonomer is a propylene oligomer, comprising: at least 50 mol % (preferably at least 60 mol %, preferably 70 mol % to 99.5 mol %, preferably 80 mol % to 99 mol %, preferably 90 mol % to 98.5 mol %) propylene, from 0.1 mol % to 45 mol % (preferably at least 35 mol %, preferably 0.5 mol % to 30 mol %, preferably 1 mol % to 20 mol %, preferably 1.5 mol % to 10 mol %) ethylene, and from 0.1 mol % to 5 mol % (preferably 0.5 mol % to 3 mol %, preferably 0.5 mol % to 1 mol %) diene (such as C 4 to C 12 alpha-omega dienes (such as butadiene, hexadiene, octadiene), norbornene, e
- diene such as C
- the vinyl terminated macromonomer is a propylene homo-oligomer, comprising propylene and less than 0.5 wt % comonomer, preferably 0 wt % comonomer, wherein the oligomer has:
- At least 93% allyl chain ends (preferably at least 95%, preferably at least 96%, preferably at least 97%, preferably at least 98%, preferably at least 99%);
- a number average molecular weight (Mn) of about 500 g/mol to about 20,000 g/mol, as measured by 1 H NMR preferably 500 g/mol to 15,000 g/mol, preferably 700 g/mol to 10,000 g/mol, preferably 800 g/mol to 8,000 g/mol, preferably 900 g/mol to 7,000 g/mol, preferably 1,000 g/mol to 6,000 g/mol, preferably 1,000 g/mol to 5,000 g/mol
- Mn number average molecular weight
- the vinyl terminated macromonomers may be homopolymers, copolymers, terpolymers, and so on.
- Vinyl terminated macromonomers generally have a saturated chain end (or terminus) and/or an unsaturated chain end, or terminus
- the unsaturated chain end of the vinyl terminated macromonomer comprises an “allyl chain end” or a “3-alkyl” chain end.
- An allyl chain end is represented by CH 2 CH—CH 2- , as shown in the formula:
- M represents the polymer chain.
- “Allylic vinyl group,” “allyl chain end,” “vinyl chain end,” “vinyl termination,” “allylic vinyl group,” and “vinyl terminated” are used interchangeably in the following description.
- the number of allyl chain ends, vinylidene chain ends, vinylene chain ends, and other unsaturated chain ends is determined using 1 H NMR at 120° C. using deuterated tetrachloroethane as the solvent on an at least 250 MHz NMR spectrometer, and in selected cases, confirmed by 13 C NMR.
- a 3-alkyl chain end (where the alkyl is a C 1 to C 38 alkyl), also referred to as a “3-alkyl vinyl end group” or a “3-alkyl vinyl termination”, is represented by the formula:
- R b is a C 1 to C 38 alkyl group, preferably a C 1 to C 20 alkyl group, such as methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, dodecyl, and the like.
- the amount of 3-alkyl chain ends is determined using 13 C NMR as set out below.
- 13 C NMR data is collected at 120° C. at a frequency of at least 100 MHz, using a Bruker 400 MHz NMR spectrometer.
- a 90 degree pulse, an acquisition time adjusted to give a digital resolution between 0.1 and 0.12 Hz, at least a 10 second pulse acquisition delay time with continuous broadband proton decoupling using swept square wave modulation without gating is employed during the entire acquisition period.
- the spectra is acquired with time averaging to provide a signal to noise level adequate to measure the signals of interest.
- Samples are dissolved in tetrachloroethane-d 2 at concentrations between 10 wt % to 15 wt % prior to being inserted into the spectrometer magnet.
- the “allyl chain end to vinylidene chain end ratio” is defined to be the ratio of the percentage of allyl chain ends to the percentage of vinylidene chain ends.
- the “allyl chain end to vinylene chain end ratio” is defined to be the ratio of the percentage of allyl chain ends to the percentage of vinylene chain ends.
- Vinyl terminated macromonomers typically also have a saturated chain end.
- the saturated chain end may be a C 4 or greater (or “higher olefin”) chain end, as shown in the formula below:
- n is an integer selected from 4 to 40. This is especially true when there is substantially no ethylene or propylene in the polymerization.
- the polymer chain may initiate growth in an ethylene monomer, thereby generating a saturated chain end which is an ethyl chain end.
- the polymer chain may initiate growth in a propylene monomer, thereby generating an isobutyl chain end.
- An “isobutyl chain end” is defined to be an end or terminus of a polymer, represented as shown in the formula below:
- Mn ( 1 H NMR) is determined according to the following NMR method.
- 1 H NMR data is collected at either room temperature or 120° C. (for purposes of the claims, 120° C. shall be used) in a 5 mm probe using a Varian spectrometer with a 1 H frequency of 250 MHz, 400 MHz, or 500 MHz (for the purpose of the claims, a proton frequency of 400 mHz is used).
- Data is recorded using a maximum pulse width of 45° C., 8 seconds between pulses and signal averaging 120 transients.
- Spectral signals are integrated and the number of unsaturation types per 1000 carbons are calculated by multiplying the different groups by 1000 and dividing the result by the total number of carbons.
- Mn is calculated by dividing the total number of unsaturated species into 14,000, and has units of g/mol.
- the chemical shift regions for the olefin types are defined to be between the following spectral regions.
- Unsaturation Type Region (ppm) Number of hydrogens per structure Vinyl 4.95-5.10 2 Vinylidene (VYD) 4.70-4.84 2 Vinylene 5.31-5.55 2 Trisubstituted 5.11-5.30 1
- Mn may also be determined using a GPC-DRI method, as described below.
- Mn is determined by 1 H NMR.
- Mn, Mw, and Mz may be measured by using a Gel Permeation Chromatography (GPC) method using a High Temperature Size Exclusion Chromatograph (SEC, either from Waters Corporation or Polymer Laboratories), equipped with a differential refractive index detector (DRI).
- GPC Gel Permeation Chromatography
- SEC High Temperature Size Exclusion Chromatograph
- DRI differential refractive index detector
- the nominal flow rate is 0.5 cm 3 /min and the nominal injection volume is 300 ⁇ L.
- the various transfer lines, columns and differential refractometer (the DRI detector) are contained in an oven maintained at 135° C.
- Solvent for the SEC experiment is prepared by dissolving 6 grams of butylated hydroxy toluene as an antioxidant in 4 liters of Aldrich reagent grade 1,2,4 trichlorobenzene (TCB). The TCB mixture is then filtered through a 0.7 ⁇ m glass pre-filter and subsequently through a 0.1 ⁇ m Teflon filter. The TCB is then degassed with an online degasser before entering the SEC.
- Polymer solutions are prepared by placing dry polymer in a glass container, adding the desired amount of TCB, then heating the mixture at 160° C. with continuous agitation for about 2 hours. All quantities are measured gravimetrically.
- the TCB densities used to express the polymer concentration in mass/volume units are 1.463 g/mL at room temperature and 1.324 g/mL at 135° C.
- the injection concentration is from 1.0 to 2.0 mg/mL, with lower concentrations being used for higher molecular weight samples.
- the DRI detector and the injector Prior to running each sample the DRI detector and the injector are purged. Flow rate in the apparatus is then increased to 0.5 mL/minute, and the DRI is allowed to stabilize for 8 to 9 hours before injecting the first sample.
- K DRI is a constant determined by calibrating the DRI
- (dn/dc) is the refractive index increment for the system.
- (dn/dc) 0.104 for propylene polymers and ethylene polymers, and 0.1 otherwise.
- a suitable metathesis catalyst is a compound that catalyzes the reaction between a cyclic olefin and a vinyl terminated macromonomer to produce a polymer represented by the formula (A).
- the alkene metathesis catalyst is represented by the Formula (E):
- M is a Group 8 metal, preferably Ru or Os, preferably Ru;
- X and X 1 are, independently, any anionic ligand, preferably a halogen (preferably chlorine), an alkoxide or a triflate, or X and X 1 may be joined to form a dianionic group and may form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
- L and L 1 are, independently, a neutral two electron donor, preferably a phosphine or a N-heterocyclic carbene, L and L 1 may be joined to form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
- L and X may be joined to form a multidentate monoanionic group and may form a single ring of up to 30 non-hydrogen atoms or a multinu
- Preferred alkoxides include those where the alkyl group is a phenol, substituted phenol (where the phenol may be substituted with up to 1, 2, 3, 4, or 5 C 1 to C 12 hydrocarbyl groups) or a C 1 to C 10 hydrocarbyl, preferably a C 1 to C 10 alkyl group, preferably methyl, ethyl, propyl, butyl, or phenyl.
- Preferred phosphines are represented by the formula: PR 3 ′ R 4 ′ R 5 ′, where R 3 ′ is a secondary alkyl or cycloalkyl (preferably a C 3 to C 12 secondary alkyl or cycloalkyl), and R 4 ′ and R 5 ′ are aryl, C 1 to C 10 primary alkyl, secondary alkyl, or cycloalkyl. R 4 ′ and R 5 ′ may be the same or different.
- Preferred phosphines include P(cyclohexyl) 3 , P(cyclopentyl) 3 , and/or P (isopropyl) 3 .
- Preferred triflates are represented by the Formula (J):
- R A is hydrogen or a C 1 to C 30 hydrocarbyl group, preferably a C 1 to C 12 alkyl group, preferably methyl, ethyl, propyl, butyl, or phenyl.
- N-heterocyclic carbenes are represented by the Formula (II) or the Formula (III):
- each R B is independently a hydrocarbyl group or substituted hydrocarbyl group having 1 to 40 carbon atoms, preferably methyl, ethyl, propyl, butyl (including isobutyl and n-butyl), pentyl, cyclopentyl, hexyl, cyclohexyl, octyl, cyclooctyl, nonyl, decyl, cyclodecyl, dodecyl, cyclododecyl, mesityl, adamantyl, phenyl, benzyl, toluoyl, chlorophenyl, phenol, substituted phenol, or CH 2 C(CH 3 ) 3 ; and each R C is hydrogen, a halogen, or a C 1 to C 12 hydrocarbyl group, preferably hydrogen, bromine, chlorine, methyl, ethyl, propyl, butyl, or phenyl.
- one of the N groups bound to the carbene in formula (II) or (III) is replaced with an S, O, or P atom, preferably an S atom.
- N-heterocyclic carbenes include the compounds described in Hermann, W. A. Chem. Eur. J., 1996, 2, pp. 772 and 1627; Enders, D. et al. Angew. Chem. Int. Ed., 1995, 34, pg. 1021; Alder R. W., Angew. Chem. Int. Ed., 1996, 35, pg. 1121; and Bertrand, G. et al., Chem. Rev., 2000, 100, pg. 39.
- the metathesis catalyst is one or more of tricyclohexylphosphine[1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene][3-phenyl-1H-inden-1-ylidene]ruthenium(II)dichloride, tricyclohexylphosphine[3-phenyl-1H-inden-1-ylidene][1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydro-imidazol-2-ylidene]ruthenium(II)dichloride, tricyclohexylphosphine[1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene][(phenylthio)methylene]ruthenium(II)dichloride, bis(tricyclohexylphosphine)-3-phenyl-1H-inden-1-ylidene
- the catalyst is 1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene[2-(i-propoxy)-5-(N,N-dimethylaminosulfonyl)phenyl]methyleneruthenium(II)dichloride and/or tricyclohexylphosphine[3-phenyl-1H-inden-1-ylidene][1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene]ruthenium(II)dichloride.
- the metathesis catalyst is represented by Formula (E) above, where: M is Os or Ru; R 5 is hydrogen; X and X 1 may be different or the same and are any anionic ligand; L and L 1 may be different or the same and are any neutral electron donor; and R 4 may be hydrogen, substituted or unsubstituted alkyl, or substituted or unsubstituted aryl.
- R 4 is preferably hydrogen, C 1 to C 20 alkyl, or aryl.
- the C 1 to C 20 alkyl may optionally be substituted with one or more aryl, halide, hydroxy, C 1 to C 20 alkoxy, or C 2 to C 20 alkoxycarbonyl groups.
- the aryl may optionally be substituted with one or more C 1 to C 20 alkyl, aryl, hydroxyl, C 1 to C 5 alkoxy, amino, nitro, or halide groups.
- L and L 1 are preferably phosphines of the formula PR 3 ′ R 4 ′ R 5 ′, where R 3 ′ is a secondary alkyl or cycloalkyl, and R 4 ′ and R 5 ′ are aryl, C 1 to C 10 primary alkyl, secondary alkyl, or cycloalkyl.
- R 4 ′ and R 5 ′ may be the same or different.
- L and L 1 are preferably the same and are —P(cyclohexyl) 3 , —P(cyclopentyl) 3 , or —P(isopropyl) 3 .
- X and X 1 are most preferably the same and are chlorine.
- the metathesis catalyst is a ruthenium and/or osmium carbene compound represented by the Formula (F):
- R 9 and R 10 may be different or the same and may be hydrogen, substituted or unsubstituted alkyl, or substituted or unsubstituted aryl.
- the R 9 and R 10 groups may optionally include one or more of the following functional groups: alcohol, thiol, ketone, aldehyde, ester, ether, amine, imine, amide, nitro, carboxylic acid, disulfide, carbonate, isocyanate, carbodiimide, carboalkoxy, and halogen groups.
- Such compounds and their synthesis are described in, inter alia, U.S. Pat. No. 6,111,121.
- the metathesis catalyst useful herein may be any of the catalysts described in U.S. Pat. Nos. 6,111,121; 5,312,940; 5,342,909; 7,329,758; 5,831,108; 5,969,170; 6,759,537; 6,921,735; and U.S. Patent Publication No. 2005-0261451 A1, including, but not limited to,
- the metathesis catalyst is represented by the formula (G):
- M* is a Group 8 metal, preferably Ru or Os, preferably Ru;
- X* and X 1 * are, independently, any anionic ligand, preferably a halogen (preferably chlorine), an alkoxide or an alkyl sulfonate, or X* and X 1 * may be joined to form a dianionic group and may form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
- L* is N—R**, O, P—R**, or S, preferably N—R** or O (R** is a C 1 to C 30 hydrocarbyl or substituted hydrocarbyl, preferably methyl, ethyl, propyl or butyl);
- R* is hydrogen or a C 1 to C 30 hydrocarbyl or substituted hydrocarbyl, preferably methyl;
- any two adjacent R groups may form a fused ring having from 5 to 8 non-hydrogen atoms.
- the non-hydrogen atoms are C and/or O.
- the adjacent R groups form fused rings of 5 to 6 ring atoms, preferably 5 to 6 carbon atoms.
- adjacent is meant any two R groups located next to each other, for example R 3 * and R 4 * can form a ring and/or R 11 * and R 12 * can form a ring.
- the metathesis catalyst compound comprises one or more of:
- the invention relates to a metathesis catalyst comprising: a Group 8 metal complex represented by the formula (H):
- M′′ is a Group 8 metal (preferably M is ruthenium or osmium, preferably ruthenium); each X′′ is independently an anionic ligand (preferably selected from the group consisting of halides, alkoxides, aryloxides, and alkyl sulfonates, preferably a halide, preferably chloride); R′′ 1 and R′′ 2 are independently selected from the group consisting of hydrogen, a C 1 to C 30 hydrocarbyl, and a C 1 to C 30 substituted hydrocarbyl (preferably R′′ 1 and R′′ 2 are independently selected from the group consisting of methyl, ethyl, propyl, isopropyl, butyl, tert-butyl, sec-butyl, pentyl, cyclopentyl, hexyl, cyclohexyl, heptyl, octyl, cyclooctyl, and substituted analogs and isomers thereof, preferably selected from the
- a “cyclic carbene” may be defined as a cyclic compound with a neutral dicoordinate carbon center featuring a lone pair of electrons. Such cyclic carbenes may be represented by the formula (IV) below:
- n is a linking group comprising from one to four ring vertices selected from the group consisting of C, Si, N, P, O, and S, with available valences optionally occupied by H, oxo, hydrocarbyl, or substituted hydrocarbyl groups; preferably, n comprises two ring vertices of carbon with available valences occupied by H, oxo, hydrocarbyl or substituted hydrocarbyl groups; preferably n is C 2 H 2 , C 2 H 4 , or substituted versions thereof; each E is independently selected from the group comprising C, N, S, O, and P, with available valences optionally occupied by Lx, Ly, Lz, and Lz′; preferably, at least one E is a C; preferably, one E is a C and the other E is a N; preferably, both E's are C; and Lx, Ly, Lz, and Lz′ are independently selected from the group comprising hydrogen, hydrocarbyl groups, and substitute
- Useful substituents include C 1-10 alkyl, C 2-10 alkenyl, C 2-10 alkynyl, aryl, C 1-10 alkoxy, C 2-10 alkenyloxy, C 2-10 alkynyloxy, aryloxy, C 2-10 alkoxycarbonyl, C 1-10 alkylthio, C 1-10 alkylsulfonyl, fluoro, chloro, bromo, iodo, oxo, amino, imine, nitrogen heterocycle, hydroxy, thiol, thiono, phosphorous, and carbene groups.
- Lx, Ly, and Lz are as defined above.
- at least two of Lx, Ly, Lz, and Lz′ may be joined to form a 3- to 12-membered spirocyclic ring, with available valences optionally occupied by H, oxo, halogens, hydrocarbyl or substituted hydrocarbyl groups.
- Useful substituents include C 1-10 alkyl, C 2-10 alkenyl, C 2-10 alkynyl, aryl, C 1-10 alkoxy, C 2-10 alkenyloxy, C 2-10 alkynyloxy, aryloxy, C 2-10 alkoxycarbonyl, C 1-10 alkylthio, C 1-10 alkylsulfonyl, fluoro, chloro, bromo, iodo, oxo, amino, imine, nitrogen heterocycle, hydroxy, thiol, thiono, phosphorous, and carbene groups.
- NHCs N-heterocyclic carbenes
- E is N and the available valences on the N are occupied by Lx and Ly.
- NHCs may be represented by the formula:
- n, Lx, and Ly are as described above in Formula (IV).
- Some particularly useful NHCs include:
- NHCs include the compounds described in Hermann, W. A. Chem. Eur. J. 1996, 2, 772 and 1627; Enders, D. et al., Angew. Chem. Int. Ed. 1995, 34, 1021; Alder R. W., Angew. Chem. Int. Ed. 1996, 35, 1121; U.S. Ser. No. 61/314,388; and Bertrand, G. et al., Chem. Rev. 2000, 100, 39.
- CAACs cyclic alkyl amino carbines
- CAACs are cyclic carbenes of the types described in Formula II above, where one E is N and the other E is C, and the available valences on the N and C are occupied by Lx, Ly, and Lz.
- CAACs may be represented by the formula:
- n, Lx, Ly, and Lz are as described above in Formula (IV).
- CAACs include:
- CAACs include the compounds described in U.S. Pat. No. 7,312,331; U.S. Ser. No. 61/259,514; and Bertrand et al, Angew. Chem. Int. Ed. 2005, 44, 7236-7239.
- carbenes useful in embodiments of the present invention include thiazolyldenes, P-heterocyclic carbenes (PHCs), and cyclopropenylidenes.
- M′′ is a Group 8 metal (preferably M is ruthenium or osmium, preferably ruthenium); each X′′ is independently an anionic ligand (preferably selected from the group consisting of halides, alkoxides, aryloxides, and alkyl sulfonates, preferably a halide, preferably chloride); R′′ 1 and R′′ 2 are independently selected from the group consisting of hydrogen, a C 1 to C 30 hydrocarbyl, and a C 1 to C 30 substituted hydrocarbyl (preferably R′′ 1 and R′′ 2 are independently selected from the group consisting of methyl, ethyl, propyl, isopropyl, butyl, tert-butyl, sec-butyl, pentyl, cyclopentyl, hexyl, cyclohexyl, heptyl, octyl, cyclooctyl, and substituted analogs and isomers thereof, preferably selected from the
- At least one phosphine ligand is a secondary phosphine ligand.
- R′′ 3 and R′′ 4 or R′′ 5 and R′′ 6 are selected from the group consisting of C 1 to C 12 hydrocarbyl groups, substituted C 1 to C 12 hydrocarbyl groups, and halides.
- At least one donor ligand is a primary phosphine ligand.
- one of R′′ 3 and R′′ 4 or one of R′′ 5 and R′′ 6 is selected from the group consisting of C 1 to C 12 hydrocarbyl groups, substituted C 1 to C 12 hydrocarbyl groups, and halides.
- both donor ligands are primary phosphine ligands and one of R′′ 3 and R′′ 4 and one of R′′ 5 and R′′ 6 is selected from the group consisting of C 1 to C 12 hydrocarbyl groups, substituted C 1 to C 12 hydrocarbyl groups, and halides.
- R′′ 3 and R′′ 4 form a ring.
- L′′ is a phosphine having a formula PHR′′ 5 R′′ 6
- R′′ 5 and R′′ 6 form a ring.
- R′′ 3 and R′′ 4 form a ring and R′′ 5 and R′′ 6 form a ring.
- R′′ 3 and at least one of R′′ 5 and R′′ 6 may form a ring, thereby forming a chelating phosphine ligand.
- R′′ 4 and at least one of R′′ 5 and R′′ 6 may form a ring, thereby forming a chelating phosphine ligand.
- the Group 8 metal complex is selected from:
- the catalyst employed in the process of this invention may be bound to or deposited onto a solid support.
- the Group 8 metal complex may be bound to or deposited onto a solid support, which may simplify catalyst recovery.
- the support may increase catalyst strength and attrition resistance.
- Suitable catalyst supports include, without limitation, silicas; aluminas; silica-aluminas; aluminosilicates, including zeolites and other crystalline porous aluminosilicates; as well as titanias; zirconia; magnesium oxide; carbon; and cross-linked polymeric resins, such as functionalized cross-linked polystyrenes, e.g., chloromethyl-functionalized cross-linked polystyrenes; preferably silica or alumina
- the Group 8 metal complex may be deposited onto the support by any method known to those skilled in the art, including, for example, impregnation, ion-exchange, deposition-precipitation, and vapor deposition.
- a component of the catalyst such as the Group 8 metal complex
- the catalyst may be immobilized by one or more covalent bonds with one or more of substituents of a ligand of the Group 8 metal complex.
- the Group 8 metal complex may be deposited onto a silica support. Further, the Group 8 metal complex may be preloaded onto the solid support before forming the catalyst of the present invention. Alternatively, the supported catalyst may be generated in situ.
- the catalyst compound may be loaded onto the catalyst support in any amount, provided that the metathesis process of this invention proceeds to the metathesis products.
- the catalyst compound is loaded onto the support in an amount based on the weight of the transition metal, preferably the Group 8 metal, preferably ruthenium or osmium, relative to the total weight of the catalysts plus support.
- the catalyst compound may be loaded onto the support in an amount greater than about 0.01 wt % of the Group 8 metal, based upon the weight of the catalysts plus support and preferably, greater than about 0.05 wt % of the Group 8 metal.
- the catalyst compound is loaded onto the support in an amount that is less than about 20 wt % of the Group 8 metal, and preferably less than about 10 wt % of the Group 8 metal.
- this invention relates to:
- R 1 is a hydrocarbyl group having greater than 25 carbon atoms (preferably greater than 30 carbon atoms, greater than 40 carbon atoms, or greater than 50 carbon atoms; preferably from 31 carbon atoms to 100,000 carbon atoms, from 40 carbon atoms to 75,000 carbon atoms, from 50 carbon atoms to 60,000 carbon atoms; preferably R 1 is represented by the formula (B):
- Cm is a C 4 to C 40 olefin derived unit (preferably Cm is one or more units derived from butene, pentene, hexene, heptene, octene, nonene, decene, undecene, dodecene, norbornene, norbornadiene, dicyclopentadiene, cyclopentene, cycloheptene, cyclooctene, cyclooctadiene, cyclododecene, 7-oxanorbornene, 7-oxanorbornadiene, substituted derivatives thereof, and isomers thereof); m is an integer from 3 to 39 (preferably from 3 to 29, preferably from 4 to 19, preferably from 5 to 11); and p is an integer greater than 1 (preferably from 1 to 10,000, from 1 to 5,000, from 1 to 2,500, from 1 to 1,000, from 1 to 500, or from 1 to 50); preferably R 1 is represented by the formula (C
- Cm is a C 3 to C 40 olefin derived unit (preferably Cm is one or more derived units of propylene, butene, pentene, hexene, heptene, octene, nonene, decene, undecene, dodecene, norbornene, norbornadiene, dicyclopentadiene, cyclopentene, cycloheptene, cyclooctene, cyclooctadiene, cyclododecene, 7-oxanorbornene, 7-oxanorbornadiene, substituted derivatives thereof, and isomers thereof); m is an integer from 2 to 39 (preferably from 2 to 29, preferably from 3 to 19, preferably from 4 to 11); and p is an integer greater than 1 (preferably from 1 to 10,000, from 1 to 5,000, from 1 to 2,500, from 1 to 1,000, from 1 to 500, or from 1 to 50); R 2 and R 3 are the
- n is an integer from 1 to 10,000 (preferably from 1 to 5,000, from 1 to 2,500, from 1 to 1,000, from 1 to 500, or from 1 to 50);
- Cm is a C 3 to C 40 olefin derived unit (preferably Cm is one or more derived units of propylene, butene, pentene, hexene, heptene, octene, nonene, decene, undecene, dodecene, norbornene, norbornadiene, dicyclopentadiene, cyclopentene, cycloheptene, cyclooctene, cyclooctadiene, cyclododecene, 7-oxanorbornene, 7-oxanorbornadiene, substituted derivatives thereof, and isomers thereof); m is an integer from 2 to 39 (preferably from 2 to 29, preferably from 3 to 19, preferably from 4 to 11); and p is an integer greater than 1 (preferably
- M is a Group 8 metal, preferably Ru or Os, preferably Ru;
- X and X 1 are, independently, any anionic ligand, preferably a halogen (preferably chlorine), an alkoxide or a triflate, or X and X 1 may be joined to form a dianionic group and may form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
- L and L 1 are, independently, a neutral two electron donor, preferably a phosphine or a N-heterocyclic carbene, L and L 1 may be joined to form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
- L and X may be joined to form a multidentate monoanionic group and may form a single ring of up to 30 non-hydrogen atoms or a multinu
- M is Os or Ru, preferably Ru;
- X, X 1 , L, and L 1 are as described above for Formula E; and
- R 9 and R 10 may be different or the same and may be hydrogen, substituted or unsubstituted alkyl, or substituted or unsubstituted aryl; and/or (iii) a metathesis catalyst represented by the formula (G):
- M* is a Group 8 metal, preferably Ru or Os, preferably Ru;
- X* and X 1 * are, independently, any anionic ligand, preferably a halogen (preferably chlorine), an alkoxide or an alkyl sulfonate, or X* and X 1 * may be joined to form a dianionic group and may form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
- L* is N—R**, O, P—R**, or S, preferably N—R** or O (R** is a C 1 to C 30 hydrocarbyl or substituted hydrocarbyl, preferably methyl, ethyl, propyl or butyl);
- R* is hydrogen or a C 1 to C 30 hydrocarbyl or substituted hydrocarbyl, preferably methyl;
- M′′ is a Group 8 metal (preferably M is ruthenium or osmium, preferably ruthenium); each X′′ is independently an anionic ligand (preferably selected from the group consisting of halides, alkoxides, aryloxides, and alkyl sulfonates, preferably a halide, preferably chloride); R′′ 1 and R′′ 2 are independently selected from the group consisting of hydrogen, a C 1 to C 30 hydrocarbyl, and a C 1 to C 30 substituted hydrocarbyl (preferably R′′ 1 and R′′ 2 are independently selected from the group consisting of methyl, ethyl, propyl, isopropyl, butyl, tert-butyl, sec-butyl, pentyl, cyclopentyl, hexyl, cyclohexyl, heptyl, octyl, cyclooctyl, and substituted analogs and isomers thereof, preferably selected from the
- M′′ is a Group 8 metal (preferably M is ruthenium or osmium, preferably ruthenium); each X′′ is independently an anionic ligand (preferably selected from the group consisting of halides, alkoxides, aryloxides, and alkyl sulfonates, preferably a halide, preferably chloride); R′′ 1 and R′′ 2 are independently selected from the group consisting of hydrogen, a C 1 to C 30 hydrocarbyl, and a C 1 to C 30 substituted hydrocarbyl (preferably R′′ 1 and R′′ 2 are independently selected from the group consisting of methyl, ethyl, propyl, isopropyl, butyl, tert-butyl, sec-butyl, pentyl, cyclopentyl, hexyl, cyclohexyl, heptyl, octyl, cyclooctyl, and substituted analogs and isomers thereof, preferably selected from the
- the molar ratio of products was calculated based on the ratios of the 1 H-NMR integration of CH 2 olefin in the functionalized polymer (5.1 ppm-5.3 ppm) and the polymer starting material (4.9 ppm-5.0 ppm), plus the CH ⁇ CH olefin of the norbornene anhydride starting material (6.3 ppm).
- the degree of ROCM, or number of norbornene anhydride units on the end of the functionalized copolymer was calculated by the ratio of the 1 H-NMR integration of total internal olefin (5.3 ppm-5.7 ppm) to the CH 2 olefin in the functionalized polymer (5.1 ppm-5.3 ppm):
- the degree of ROCM can be calculated by the ratio of the 1 H-NMR integration of product ring methine protons (2.75 ppm-3.5 ppm) to the CH 2 olefin in the functionalized polymer (5.1 ppm-5.3 ppm).
- This measure of catalyst activity is determined from the molar ratio of VTM to catalyst multiplied by the VTM conversion and the degree of ROCM:
- a 2 L autoclave was filled with 600 mls isohexanes, 0.5 ml 1.0 M triisobutyl aluminum (in hexanes), 200 mls propylene, 100 mls hexene and the contents heated to 70° C.
- a catalyst solution was made by reacting 10.7 mg of Metallocene E (structure below; synthesis of Metallocene E is disclosed in U.S. Ser. No. 13/072,288), with 24.1 mg of dimethylanilinium tetrakis(perfluoronaphthyl)borate in 10 ml toluene. This solution (3.3 mls) was injected into the autoclave under high N 2 pressure. The reaction proceeded for 30 minutes.
- the autoclave was then cooled to room temperature, depressurized and the contents transferred to a beaker. Volatiles were removed and the product dried in vacuo at 70° C. for 4 hrs. Yield was 126 g.
- the copolymer had 97% vinyls, 3% vinylidenes, and Mn of 1600 g/mol by 1 H NMR.
- Metallocene E as the catalyst, and dimethylaniliniumtetrakis(perfluoronaphthyl)borate as the activator.
- Metallocene E was premixed with dimethylaniliniumtetrakis(perfluoronaphthyl)borate in a 1:1 ratio and fed into the reactor at a rate of 3.3 ⁇ 10 ⁇ 7 moles/minute.
- Propylene (C 3 ) was fed into the reactor at a rate of 15 g/minute, isohexane at a feed rate of 59.4 g/minute, butene (C 4- ) at a feed rated of 8.0 g/minute, and tri-n-octyl aluminum at a feed rate of 5.2 ⁇ 10 ⁇ 6 moles/minute.
- Catalyst activity 18,400 kgP/molcat.
- the copolymer had 92.4% vinyls, 4.4% vinylidenes, and Mn of 1726 g/mol by 1 H NMR.
- the vinyl terminated hexene propylene copolymer (1.0 g, described above) was placed in a 20 ml scintillation vial with 5 mls of tetrachloroethane-d 2 .
- Cis-5-norbornene-endo-2,3-dicarboxylic anhydride (0.10 g, Sigma Aldrich, St. Louis, Mo.) was added to the mixture.
- the solubility of the carboxylic anhydride was noted visually as minimal
- a 3.0 mg amount of ((t-Bu) 2 PH) 2 Cl 2 Ru ⁇ CHCH ⁇ C(CH 3 ) 2 was added to the reaction mixture and the slurry was heated to 50° C. for three hours.
- the vinyl terminated hexene propylene copolymer (1.4 g, as described above) was placed in a 20 ml scintillation vial with 5 mls of tetrachloroethane-d 2 .
- Dimethyl norbornene dicarboxylate (0.14 g, Sigma Aldrich) was added to the mixture.
- a 3.0 mg amount of ((t-Bu) 2 PH) 2 Cl 2 Ru ⁇ CHCH ⁇ C(CH 3 ) 2 was added to the reaction mixture and the slurry was heated to 50° C. for three hours.
- the reaction was further diluted into 10 mls of dichloromethane, filtered through 1.0 grams of silica and dried under vacuum.
- the product oil was characterized using 1 H-NMR: 400 MHz (C 2 D 2 Cl 4 ): ⁇ 5.9 (m, 0.45H), 5.3-5.7 (m, 1.05H), 5.1-5.3 (m, 0.86H), 5.0 (m, 0.15), 3.35-3.55 (m, 0.98H), 3.2-3.35 (m, 0.21H), 2.9-3.1 (m, 0.77H), 0.5-2.5 (m, 160H).
- the polymer product was analyzed by 1 H-NMR: 400 MHz (C 2 D 2 Cl 4 ): ⁇ 5.9 (m, 0.08H), 5.3-5.7 (m, 0.89H), 5.1-5.3 (m, 0.17H) 5.0 (m, 0.08), 3.1-3.4 (m, 0.91H), 2.75-3.0 (m, 0.53H), 0.5-2.5 (m, 97.2H).
- the catalyst ((t-Bu) 2 PH) 2 Cl 2 Ru ⁇ CHCH ⁇ C(CH 3 ) 2 , was added in one portion and stirred at 50° C. for 2 hours.
- the reaction was quenched using 1 g silica, and the CHCl 3 was removed under vacuum at 45° C. overnight.
- the translucent brown polymer was treated with approximately 150 mL of pentane.
- the mixture was heated to 40° C. and stirred for several hours to homogenize the mixture.
- the mixture was then cooled for 2 hours to ⁇ 25° C. This mixture was then filtered using a plug of silica, followed by further filtration through 1 micron syringe filters.
- the solvent was removed under a stream of nitrogen gas.
- the polymer was further dried by sparging nitrogen gas directly into the polymer while heating it to 80° C.
- the dried polymer was analyzed by 1 H-NMR: 400 MHz (C 2 D 2 Cl 4 ): ⁇ 5.9 (m, 0.32H), 5.3-5.7 (m, 1.35H), 5.1-5.3 (m, 0.57H), 2.75-3.5 (m, 2.51H), 0.5-2.5 (m, 93.9H).
- compositions, an element or a group of elements are preceded with the transitional phrase “comprising,” it is understood that we also contemplate the same composition or group of elements with transitional phrases “consisting essentially of,” “consisting of,” “selected from the group of consisting of,” or “is” preceding the recitation of the composition, element, or elements and vice versa.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
Abstract
Description

where R1 is a hydrocarbyl group having greater than 25 carbon atoms;
R2 and R3 are the same or different and each is hydrogen or a hydrocarbyl group having from 1 to 40 carbon atoms, or R2 and R3 are joined to form a five-membered or six-membered ring, or substituted analogs thereof;
X is C, N, or O;
n is an integer from 1 to 10,000;
the dotted line indicates an optional double bond; and
wherein the polymer comprises one or more vinyl terminated macromonomer derived units.

where
R2 and R3 are the same or different and each is hydrogen or a hydrocarbyl group having from 1 to 40 carbon atoms, or R2 and R3 are joined to form a five-membered or six-membered ring, or substituted analogs thereof;
X is C, N, or O;
n is an integer from 1 to 10,000; and
the dotted line indicates an optional double bond;
where R1 is
(i) a polymer comprising of one or more C4 to C40 higher olefin derived units, where the higher olefin polymer comprises substantially no propylene derived units; and or
(ii) a copolymer comprising (a) from about 20 mol % to about 99.9 mol % of at least one C5 to C40 higher olefin and (b) from about 0.1 mol % to about 80 mol % of propylene; and or
(iii) a copolymer comprising (a) from about 80 mol % to about 99.9 mol % of at least one C4 olefin and (b) from about 0.1 mol % to about 20 mol % of propylene.

where R1 is a hydrocarbyl group having greater than 25 carbon atoms, preferably greater than 30 carbon atoms, greater than 40 carbon atoms, or greater than 50 carbon atoms; preferably from 31 to 100,000 carbons, from 40 to 75,000 carbons, from 50 to 60,000 carbons; preferably R1 is represented by the formula (B):

wherein Cm is a C4 to C40 olefin derived unit (preferably Cm is one or more derived units of butene, pentene, hexene, heptene, octene, nonene, decene, undecene, dodecene, norbornene, norbornadiene, dicyclopentadiene, cyclopentene, cycloheptene, cyclooctene, cyclooctadiene, cyclododecene, 7-oxanorbornene, 7-oxanorbornadiene, substituted derivatives thereof, and isomers thereof); m is an integer from 3 to 39 (preferably from 3 to 29, preferably from 4 to 19, preferably from 4 to 11); and p is an integer greater than 1 (preferably from 1 to 10,000, from 1 to 5,000, from 1 to 2,500, from 1 to 1,000, from 1 to 500, or from 1 to 50); preferably R1 is represented by the formula (C):

wherein Cm is a C3 to C40 olefin derived unit (preferably Cm is one or more derived units of propylene, butene, pentene, hexene, heptene, octene, nonene, decene, undecene, dodecene, norbornene, norbornadiene, dicyclopentadiene, cyclopentene, cycloheptene, cyclooctene, cyclooctadiene, cyclododecene, 7-oxanorbornene, 7-oxanorbornadiene, substituted derivatives thereof, and isomers thereof); m is an integer from 2 to 39 (preferably from 2 to 29, preferably from 3 to 19, preferably from 4 to 11); and p is an integer greater than 1 (preferably from 1 to 10,000, from 1 to 5,000, from 1 to 2,500, from 1 to 1,000, from 1 to 500, or from 1 to 50);
R2 and R3 are the same or different and each is hydrogen or a hydrocarbyl group having from 1 to 40 carbon atoms (preferably from 1 to 30, from 2 to 15, and from 2 to 12), or R2 and R3 are joined to form a five-membered or six-membered ring, or substituted analogs thereof (preferably R2 and R3 are joined to form a heteroatom containing ring; preferably R2 and R3 are joined to form a dicarboxylic anhydride);
X is C, N, or O (preferably X is C or O, preferably X is C);
n is an integer from 1 to 10,000 (preferably from 1 to 5,000, from 1 to 2,500, from 1 to 1,000, from 1 to 500, or from 1 to 50); and
the dotted line indicates an optional double bond,
wherein the polymer, preferably R1, comprises one or more vinyl terminated macromonomer derived units (preferably the vinyl terminated macromonomer is one or more of:
(i) a vinyl terminated polymer having an Mn of at least 200 g/mol (measured by 1H NMR) comprising of one or more C4 to C40 higher olefin derived units, where the higher olefin polymer comprises substantially no propylene derived units; and wherein the higher olefin polymer has at least 5% allyl chain ends;
(ii) a copolymer having an Mn of 300 g/mol or more (measured by 1H NMR) comprising (a) from about 20 mol % to about 99.9 mol % of at least one C5 to C40 higher olefin, and (b) from about 0.1 mol % to about 80 mol % of propylene, wherein the higher olefin copolymer has at least 40% allyl chain ends;
(iii) a copolymer having an Mn of 300 g/mol or more (measured by 1H NMR), and comprises
(a) from about 80 mol % to about 99.9 mol % of at least one C4 olefin; and (b) from about 0.1 mol % to about 20 mol % of propylene; and wherein the vinyl terminated macromonomer has at least 40% allyl chain ends relative to total unsaturation;
(iv) a co-oligomer having an Mn of 300 to 30,000 g/mol (measured by 1H NMR) comprising 10 mol % to 90 mol % propylene and 10 mol % to 90 mol % of ethylene, wherein the oligomer has at least X % allyl chain ends (relative to total unsaturations), where: 1) X=(−0.94*(mol % ethylene incorporated)+100), when 10 mol % to 60 mol % ethylene is present in the co-oligomer; 2) X=45, when greater than 60 mol % and less than 70 mol % ethylene is present in the co-oligomer; and 3) X=(1.83*(mol % ethylene incorporated)-83), when 70 mol % to 90 mol % ethylene is present in the co-oligomer;
(v) a propylene oligomer, comprising more than 90 mol % propylene and less than 10 mol % ethylene wherein the oligomer has: at least 93% allyl chain ends, a number average molecular weight (Mn) of about 500 g/mol to about 20,000 g/mol, an isobutyl chain end to allylic vinyl group ratio of 0.8:1 to 1.35:1.0, and less than 100 ppm aluminum;
(vi) a propylene oligomer, comprising: at least 50 mol % propylene and from 10 mol % to 50 mol % ethylene, wherein the oligomer has: at least 90% allyl chain ends, an Mn of about 150 g/mol to about 10,000 g/mol, and an isobutyl chain end to allylic vinyl group ratio of 0.8:1 to 1.2:1.0, wherein monomers having four or more carbon atoms are present at from 0 mol % to 3 mol %;
(vii) a propylene oligomer, comprising: at least 50 mol % propylene, from 0.1 mol % to 45 mol % ethylene, and from 0.1 mol % to 5 mol % C4 to C12 olefin, wherein the oligomer has: at least 90% allyl chain ends, an Mn of about 150 g/mol to about 10,000 g/mol, and an isobutyl chain end to allylic vinyl group ratio of 0.8:1 to 1.35:1.0;
(viii) a propylene oligomer, comprising: at least 50 mol % propylene, from 0.1 mol % to 45 mol % ethylene, and from 0.1 mol % to 5 mol % diene, wherein the oligomer has: at least 90% allyl chain ends, an Mn of about 150 g/mol to about 10,000 g/mol, and an isobutyl chain end to allylic vinyl group ratio of 0.7:1 to 1.35:1.0; and
(ix) a homo-oligomer, comprising propylene, wherein the oligomer has: at least 93% allyl chain ends, an Mn of about 500 g/mol to about 20,000 g/mol, an isobutyl chain end to allylic vinyl group ratio of 0.8:1 to 1.2:1.0, and less than 1400 ppm aluminum).

wherein X is C, N, or O (preferably C or O, preferably C); n is an integer from 1 to 10,000 (preferably from 1 to 5,000, from 1 to 2,500, from 1 to 1,000, from 1 to 500, or from 1 to 50); Cm is a C3 to C40 olefin derived unit (preferably Cm is one or more derived units of propylene, butene, pentene, hexene, heptene, octene, nonene, decene, undecene, dodecene, norbornene, norbornadiene, dicyclopentadiene, cyclopentene, cycloheptene, cyclooctene, cyclooctadiene, cyclododecene, 7-oxanorbornene, 7-oxanorbornadiene, substituted derivatives thereof, and isomers thereof); m is an integer from 2 to 39 (preferably from 2 to 29, preferably from 3 to 19, preferably from 4 to 11); and p is an integer greater than 1 (preferably from 1 to 10,000, from 1 to 5,000, from 1 to 2,500, from 1 to 1,000, from 1 to 500, or from 1 to 50).

where
R2 and R3; X, n, and the dotted line are as defined above and R1 is
(i) a polymer, preferably having an Mn of at least 200 g/mol (measured by 1H NMR), comprising of one or more C4 to C40 higher olefin derived units (preferably C5 to C40 higher olefin derived units), where the higher olefin polymer comprises substantially no propylene derived units; and or
(ii) a copolymer, preferably having an Mn of at least 300 g/mol (measured by 1H NMR), comprising (a) from about 20 mol % to about 99.9 mol % of at least one C5 to C40 higher olefin and (b) from about 0.1 mol % to about 80 mol % of propylene; and or
(iii) a copolymer, preferably having an Mn of at least 300 g/mol (measured by 1H NMR), comprising (a) from about 80 mol % to about 99.9 mol % of at least one C4 olefin and (b) from about 0.1 mol % to about 20 mol % of propylene.

where R1 is a hydrocarbyl group having greater than 25 carbon atoms;
R2 and R3 are the same or different and each is hydrogen or a hydrocarbyl group having from 1 to 40 carbon atoms, or R2 and R3 are joined to form a five-membered or six-membered ring, or substituted analogs thereof;
X is C, N, or O;
n is an integer from 1 to 10,000; and
the dotted line indicates an optional double bond,
wherein the polymer comprises one or more vinyl terminated macromonomer derived units; the process comprising contacting:
at least one cyclic olefin (preferably norbornene, norbornadiene, dicyclopentadiene, cyclopentene, cycloheptene, cyclooctene, cyclooctadiene, cyclododecene, 7-oxanorbornene, 7-oxanorbornadiene, and substituted derivatives therefrom; more preferably cyclooctene, 1,5-cyclooctadiene, 1-hydroxy-4-cyclooctene, 1-acetoxy-4-cyclooctene, 5-methylcyclopentene, cyclopentene, dicyclopentadiene, norbornene, norbornadiene, cis-5-norbornene-endo-2,3-dicarboxylic anhydride, dimethyl norbornene carboxylate, norbornene-exo-2,3-carboxylic anhydride, and their respective homologs and derivatives) with at least one vinyl terminated macromonomer in the presence of one or more metathesis catalysts (preferably one or more of:
(i) an alkene metathesis catalyst represented by the Formula (E):

where
M is a Group 8 metal, preferably Ru or Os, preferably Ru;
X and X1 are, independently, any anionic ligand, preferably a halogen (preferably chlorine), an alkoxide or a triflate, or X and X1 may be joined to form a dianionic group and may form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
L and L1 are, independently, a neutral two electron donor, preferably a phosphine or a N-heterocyclic carbene, L and L1 may be joined to form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
L and X may be joined to form a multidentate monoanionic group and may form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
L1 and X1 may be joined to form a multidentate monoanionic group and may form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
R4 and R5 are, independently, hydrogen or C1 to C30 substituted or unsubstituted hydrocarbyl (preferably a C1 to C30 substituted or unsubstituted alkyl or a substituted or unsubstituted C4 to C30 aryl);
R5 and L1 or X1 may be joined to form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms; and
R4 and L or X may be joined to form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms; and/or
(ii) a metathesis catalyst represented by the Formula (F):

where M is Os or Ru, preferably Ru;
X, X1, L, and L1 are as described above for Formula E; and
R9 and R10 may be different or the same and may be hydrogen, substituted or unsubstituted alkyl, or substituted or unsubstituted aryl; and/or
(iii) a metathesis catalyst represented by the formula (G):

where
M* is a Group 8 metal, preferably Ru or Os, preferably Ru;
X* and X1* are, independently, any anionic ligand, preferably a halogen (preferably chlorine), an alkoxide or an alkyl sulfonate, or X* and X1* may be joined to form a dianionic group and may form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
L* is N—R**, 0, P—R**, or S, preferably N—R** or O (R** is a C1 to C30 hydrocarbyl or substituted hydrocarbyl, preferably methyl, ethyl, propyl or butyl);
R* is hydrogen or a C1 to C30 hydrocarbyl or substituted hydrocarbyl, preferably methyl;
R1*, R2*, R3*, R4*, R5*, R6*, R7*, and R8* are, independently, hydrogen or a C1 to C30 hydrocarbyl or substituted hydrocarbyl, preferably methyl, ethyl, propyl or butyl, preferably R1*, R2*, R3*, and R4* are methyl;
each R9* and R13* are, independently, hydrogen or a C1 to C30 hydrocarbyl or substituted hydrocarbyl, preferably a C2 to C6 hydrocarbyl, preferably ethyl;
R10*, R11*, R12* are, independently hydrogen or a C1 to C30 hydrocarbyl or substituted hydrocarbyl, preferably hydrogen or methyl;
each G, is, independently, hydrogen, halogen or C1 to C30 substituted or unsubstituted hydrocarbyl (preferably a C1 to C30 substituted or unsubstituted alkyl or a substituted or unsubstituted C4 to C30 aryl); and
where any two adjacent R groups may form a single ring of up to 8 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms; and/or
(iv) a Group 8 metal complex represented by the formula (H):

wherein
M″ is a Group 8 metal (preferably M is ruthenium or osmium, preferably ruthenium);
each X is independently an anionic ligand (preferably selected from the group consisting of halides, alkoxides, aryloxides, and alkyl sulfonates, preferably a halide, preferably chloride);
R″1 and R″2 are independently selected from the group consisting of hydrogen, a C1 to C30 hydrocarbyl, and a C1 to C30 substituted hydrocarbyl (preferably R″1 and R″2 are independently selected from the group consisting of methyl, ethyl, propyl, isopropyl, butyl, tert-butyl, sec-butyl, pentyl, cyclopentyl, hexyl, cyclohexyl, heptyl, octyl, cyclooctyl, and substituted analogs and isomers thereof, preferably selected from the group consisting of tert-butyl, sec-butyl, cyclohexyl, and cyclooctyl);
R″3 and R″4 are independently selected from the group consisting of hydrogen, C1 to C12 hydrocarbyl groups, substituted C1 to C12 hydrocarbyl groups, and halides (preferably R″3 and R″4 are independently selected from the group consisting of methyl, ethyl, propyl, isopropyl, butyl, tert-butyl, sec-butyl, pentyl, cyclopentyl, hexyl, cyclohexyl, heptyl, octyl, cyclooctyl, and substituted analogs and isomers thereof, preferably selected from the group consisting of tert-butyl, sec-butyl, cyclohexyl, and cyclooctyl); and
L″ is a neutral donor ligand, preferably L″ is selected from the group consisting of a phosphine, a sulfonated phosphine, a phosphite, a phosphinite, a phosphonite, an arsine, a stibine, an ether, an amine, an imine, a sulfoxide, a carboxyl, a nitrosyl, a pyridine, a thioester, a cyclic carbene, and substituted analogs thereof; preferably a phosphine, a sulfonated phosphine, an N-heterocyclic carbene, a cyclic alkyl amino carbene, and substituted analogs thereof (preferably L″ is selected from a phosphine, an N-heterocyclic carbene, a cyclic alkyl amino carbene, and substituted analogs thereof); and/or
(v) a Group 8 metal complex represented by the formula (I):

wherein
M″ is a Group 8 metal (preferably M is ruthenium or osmium, preferably ruthenium);
each X is independently an anionic ligand (preferably selected from the group consisting of halides, alkoxides, aryloxides, and alkyl sulfonates, preferably a halide, preferably chloride);
R″1 and R″2 are independently selected from the group consisting of hydrogen, a C1 to C30 hydrocarbyl, and a C1 to C30 substituted hydrocarbyl (preferably R″1 and R″2 are independently selected from the group consisting of methyl, ethyl, propyl, isopropyl, butyl, tert-butyl, sec-butyl, pentyl, cyclopentyl, hexyl, cyclohexyl, heptyl, octyl, cyclooctyl, and substituted analogs and isomers thereof, preferably selected from the group consisting of tert-butyl, sec-butyl, cyclohexyl, and cyclooctyl);
R″3, R″4, R″5, and R″6 are independently selected from the group consisting of hydrogen, C1 to C12 hydrocarbyl groups, substituted C1 to C12 hydrocarbyl groups, and halides (preferably R″3, R″4, R″5, and R″6 are independently selected from the group consisting of methyl, ethyl, propyl, isopropyl, butyl, tert-butyl, sec-butyl, pentyl, cyclopentyl, hexyl, cyclohexyl, heptyl, octyl, cyclooctyl, and substituted analogs and isomers thereof, preferably selected from the group consisting of tert-butyl, sec-butyl, cyclohexyl, and cyclooctyl); and at a temperature of 20° C. to 200° C. (preferably 50° C. to 160° C., preferably 60° C. to 140° C.) and a pressure of 0 MPa to 1000 MPa (preferably 0.5 MPa to 500 MPa, preferably 1 MPa to 250 MPa) for a residence time of 0.5 seconds to 10 hours (preferably 1 second to 5 hours, preferably 1 minute to 1 hour).
(ii) a copolymer having an Mn of 300 g/mol or more (measured by 1H NMR) comprising (a) from about 20 mol % to about 99.9 mol % of at least one C5 to C40 higher olefin, and (b) from about 0.1 mol % to about 80 mol % of propylene, wherein the higher olefin copolymer has at least 40% allyl chain ends;
(iii) a copolymer having an Mn of 300 g/mol or more (measured by 1H NMR), and comprises (a) from about 80 mol % to about 99.9 mol % of at least one C4 olefin, (b) from about 0.1 mol % to about 20 mol % of propylene; and wherein the vinyl terminated macromonomer has at least 40% allyl chain ends relative to total unsaturation;
(iv) a co-oligomer having an Mn of 300 g/mol to 30,000 g/mol (measured by 1H NMR) comprising 10 mol % to 90 mol % propylene and 10 mol % to 90 mol % of ethylene, wherein the oligomer has at least X % allyl chain ends (relative to total unsaturations), where: 1) X=(−0.94*(mol % ethylene incorporated)+100), when 10 mol % to 60 mol % ethylene is present in the co-oligomer; 2) X=45, when greater than 60 mol % and less than 70 mol % ethylene is present in the co-oligomer; and 3) X=(1.83*(mol % ethylene incorporated)-83), when 70 mol % to 90 mol % ethylene is present in the co-oligomer;
(v) a propylene oligomer, comprising more than 90 mol % propylene and less than 10 mol % ethylene wherein the oligomer has: at least 93% allyl chain ends, a number average molecular weight (Mn) of about 500 g/mol to about 20,000 g/mol, an isobutyl chain end to allylic vinyl group ratio of 0.8:1 to 1.35:1.0, and less than 100 ppm aluminum;
(vi) a propylene oligomer, comprising: at least 50 mol % propylene and from 10 mol % to 50 mol % ethylene, wherein the oligomer has: at least 90% allyl chain ends, an Mn of about 150 g/mol to about 10,000 g/mol, and an isobutyl chain end to allylic vinyl group ratio of 0.8:1 to 1.2:1.0, wherein monomers having four or more carbon atoms are present at from 0 mol % to 3 mol %;
(vii) a propylene oligomer, comprising: at least 50 mol % propylene, from 0.1 mol % to 45 mol % ethylene, and from 0.1 mol % to 5 mol % C4 to C12 olefin, wherein the oligomer has: at least 90% allyl chain ends, an Mn of about 150 g/mol to about 10,000 g/mol, and an isobutyl chain end to allylic vinyl group ratio of 0.8:1 to 1.35:1.0;
(viii) a propylene oligomer, comprising: at least 50 mol % propylene, from 0.1 mol % to 45 mol % ethylene, and from 0.1 mol % to 5 mol % diene, wherein the oligomer has: at least 90% allyl chain ends, an Mn of about 150 g/mol to about 10,000 g/mol, and an isobutyl chain end to allylic vinyl group ratio of 0.7:1 to 1.35:1.0; and
(ix) a homo-oligomer, comprising propylene, wherein the oligomer has: at least 93% allyl chain ends, an Mn of about 500 g/mol to about 20,000 g/mol, an isobutyl chain end to allylic vinyl group ratio of 0.8:1 to 1.2:1.0, and less than 1400 ppm aluminum.
(b) from about 0.1 mol % to 80 mol % (preferably from about 5 mol % to 70 mol %, from about 10 mol % to about 65 mol %, from about 15 mol % to about 55 mol %, from about 25 mol % to about 50 mol %, or from about 30 mol % to about 80 mol %) of propylene;
wherein the vinyl terminated macromonomer has at least 40% allyl chain ends (preferably at least 50% allyl chain ends, at least 60% allyl chain ends, at least 70% allyl chain ends, or at least 80% allyl chain ends, at least 90% allyl chain ends, at least 95% allyl chain ends) relative to total unsaturation; and, optionally, an isobutyl chain end to allyl chain end ratio of less than 0.70:1, less than 0.65:1, less than 0.60:1, less than 0.50:1, or less than 0.25:1; and further optionally, an allyl chain end to vinylidene chain end ratio of greater than 2:1 (preferably greater than 2.5:1, greater than 3:1, greater than 5:1, or greater than 10:1); and even further optionally, an allyl chain end to vinylene ratio is greater than 1:1 (preferably greater than 2:1 or greater than 5:1). Such macromonomers are further described in U.S. Ser. No. 13/072,249, hereby incorporated by reference.
iii) an isobutyl chain end to allylic vinyl group ratio of 0.8:1 to 1.3:1.0; and
iv) less than 1400 ppm aluminum, (preferably less than 1200 ppm, preferably less than 1000 ppm, preferably less than 500 ppm, preferably less than 100 ppm). Such macromonomers are also further described in U.S. Ser. No. 12/143,663.
where M represents the polymer chain. “Allylic vinyl group,” “allyl chain end,” “vinyl chain end,” “vinyl termination,” “allylic vinyl group,” and “vinyl terminated” are used interchangeably in the following description. The number of allyl chain ends, vinylidene chain ends, vinylene chain ends, and other unsaturated chain ends is determined using 1H NMR at 120° C. using deuterated tetrachloroethane as the solvent on an at least 250 MHz NMR spectrometer, and in selected cases, confirmed by 13C NMR. Resconi has reported proton and carbon assignments (neat perdeuterated tetrachloroethane used for proton spectra, while a 50:50 mixture of normal and perdeuterated tetrachloroethane was used for carbon spectra; all spectra were recorded at 100° C. on a Bruker spectrometer operating at 500 MHz for proton and 125 MHz for carbon) for vinyl terminated oligomers in J. American Chemical Soc., 114, 1992, pp. 1025-1032 that are useful herein. Allyl chain ends are reported as a molar percentage of the total number of moles of unsaturated groups (that is, the sum of allyl chain ends, vinylidene chain ends, vinylene chain ends, and the like).

where “••••” represents the polyolefin chain and Rb is a C1 to C38 alkyl group, preferably a C1 to C20 alkyl group, such as methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, dodecyl, and the like. The amount of 3-alkyl chain ends is determined using 13C NMR as set out below.
| Chain End | 13C NMR Chemical Shift | ||
| P~i-Bu | 23-5 to 25.5 and 25.8 to 26.3 ppm | ||
| E~i-Bu | 39.5 to 40.2 ppm | ||
| P~Vinyl | 41.5 to 43 ppm | ||
| E~Vinyl | 33.9 to 34.4 ppm | ||
where M represents the polymer chain and n is an integer selected from 4 to 40. This is especially true when there is substantially no ethylene or propylene in the polymerization. In an ethylene/(C4 or greater monomer) copolymerization, the polymer chain may initiate growth in an ethylene monomer, thereby generating a saturated chain end which is an ethyl chain end. In polymerizations where propylene is present, the polymer chain may initiate growth in a propylene monomer, thereby generating an isobutyl chain end. An “isobutyl chain end” is defined to be an end or terminus of a polymer, represented as shown in the formula below:

where M represents the polymer chain. Isobutyl chain ends are determined according to the procedure set out in WO 2009/155471.
| Unsaturation Type | Region (ppm) | Number of hydrogens per structure |
| Vinyl | 4.95-5.10 | 2 |
| Vinylidene (VYD) | 4.70-4.84 | 2 |
| Vinylene | 5.31-5.55 | 2 |
| Trisubstituted | 5.11-5.30 | 1 |
c=K DRI I DRI/(dn/dc)
where KDRI is a constant determined by calibrating the DRI, and (dn/dc) is the refractive index increment for the system. The refractive index, n=1.500 for TCB at 135° C. and λ=690 nm. For purposes of this invention and the claims thereto (dn/dc)=0.104 for propylene polymers and ethylene polymers, and 0.1 otherwise. Units of parameters used throughout this description of the SEC method are: concentration is expressed in g/cm3, molecular weight is expressed in g/mol, and intrinsic viscosity is expressed in dL/g.
C. Metathesis Catalysts

where
M is a Group 8 metal, preferably Ru or Os, preferably Ru;
X and X1 are, independently, any anionic ligand, preferably a halogen (preferably chlorine), an alkoxide or a triflate, or X and X1 may be joined to form a dianionic group and may form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
L and L1 are, independently, a neutral two electron donor, preferably a phosphine or a N-heterocyclic carbene, L and L1 may be joined to form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
L and X may be joined to form a multidentate monoanionic group and may form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
L1 and X1 may be joined to form a multidentate monoanionic group and may form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
R4 and R5 are, independently, hydrogen or C1 to C30 substituted or unsubstituted hydrocarbyl (preferably a C1 to C30 substituted or unsubstituted alkyl or a substituted or unsubstituted C4 to C30 aryl);
R5 and L1 or X1 may be joined to form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms; and
R4 and L or X may be joined to form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms.

where RA is hydrogen or a C1 to C30 hydrocarbyl group, preferably a C1 to C12 alkyl group, preferably methyl, ethyl, propyl, butyl, or phenyl.

where
each RB is independently a hydrocarbyl group or substituted hydrocarbyl group having 1 to 40 carbon atoms, preferably methyl, ethyl, propyl, butyl (including isobutyl and n-butyl), pentyl, cyclopentyl, hexyl, cyclohexyl, octyl, cyclooctyl, nonyl, decyl, cyclodecyl, dodecyl, cyclododecyl, mesityl, adamantyl, phenyl, benzyl, toluoyl, chlorophenyl, phenol, substituted phenol, or CH2C(CH3)3; and
each RC is hydrogen, a halogen, or a C1 to C12 hydrocarbyl group, preferably hydrogen, bromine, chlorine, methyl, ethyl, propyl, butyl, or phenyl.

where M is Os or Ru, preferably Ru; X, X1, L, and L1 are as described above for Formula (E); and R9 and R10 may be different or the same and may be hydrogen, substituted or unsubstituted alkyl, or substituted or unsubstituted aryl. The R9 and R10 groups may optionally include one or more of the following functional groups: alcohol, thiol, ketone, aldehyde, ester, ether, amine, imine, amide, nitro, carboxylic acid, disulfide, carbonate, isocyanate, carbodiimide, carboalkoxy, and halogen groups. Such compounds and their synthesis are described in, inter alia, U.S. Pat. No. 6,111,121.
- benzylidene-bis(tricyclohexylphosphine)dichlororuthenium,
- benzylidene[1,3-bis(2,4,6-trimethylphenyl)-2 imidazolidinylidene]dichloro(tricyclohexyl phosphine) ruthenium,
- dichloro(o-isopropoxyphenylmethylene)(tricyclohexylphosphine)ruthenium(II),
- (1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene)dichloro(o-isopropoxyphenylmethylene)ruthenium,
- 1,3-bis(2-methylphenyl)-2-imidazolidinylidene]dichloro(2-isopropoxyphenylmethylene) ruthenium(II),
- [1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene]dichloro[3-(2-pyridinyl)propylidene]ruthenium(II),
- [1,3-bis(2-methylphenyl)-2-imidazolidinylidene]dichloro(phenylmethylene) (tricyclohexylphosphine)ruthenium(II),
- [1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene]dichloro(3-methyl-2-butenylidene) (tricyclohexylphosphine)ruthenium(II), and
- [1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene]dichloro(benzylidene)bis(3-bromopyridine)ruthenium(II).

where
M* is a Group 8 metal, preferably Ru or Os, preferably Ru;
X* and X1* are, independently, any anionic ligand, preferably a halogen (preferably chlorine), an alkoxide or an alkyl sulfonate, or X* and X1* may be joined to form a dianionic group and may form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
L* is N—R**, O, P—R**, or S, preferably N—R** or O (R** is a C1 to C30 hydrocarbyl or substituted hydrocarbyl, preferably methyl, ethyl, propyl or butyl);
R* is hydrogen or a C1 to C30 hydrocarbyl or substituted hydrocarbyl, preferably methyl;
R1*, R2*, R3*, R4*, R5*, R6*, R7*, and R8* are, independently, hydrogen or a C1 to C30 hydrocarbyl or substituted hydrocarbyl, preferably methyl, ethyl, propyl or butyl, preferably R1*, R2*, R3*, and R4* are methyl;
each R9* and R13* are, independently, hydrogen or a C1 to C30 hydrocarbyl or substituted hydrocarbyl, preferably a C2 to C6 hydrocarbyl, preferably ethyl;
R10*, R11*, R12* are, independently hydrogen or a C1 to C30 hydrocarbyl or substituted hydrocarbyl, preferably hydrogen or methyl;
each G, is, independently, hydrogen, halogen or C1 to C30 substituted or unsubstituted hydrocarbyl (preferably a C1 to C30 substituted or unsubstituted alkyl or a substituted or unsubstituted C4 to C30 aryl); and
where any two adjacent R groups may form a single ring of up to 8 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms.
- 2-(2,6-diethylphenyl)-3,5,5,5-tetramethylpyrrolidine[2-(i-propoxy)-5-(N,N-dimethylamino sulfonyl)phenyl]methylene ruthenium dichloride;
- 2-(mesityl)-3,3,5,5-tetramethylpyrrolidine[2-(i-propoxy)-5-(N,N-dimethylaminosulfonyl)phenyl]methylene ruthenium dichloride;
- 2-(2-isopropyl)-3,3,5,5-tetramethylpyrrolidine[2-(i-propoxy)-5-(N,N-dimethylaminosulfonyl)phenyl]methylene ruthenium dichloride;
- 2-(2,6-diethyl-4-fluorophenyl)-3,3,5,5-tetramethylpyrrolidine[2-(i-propoxy)-5-(N,N-dimethylaminosulfonyl)phenyl]methylene ruthenium dichloride; and mixtures thereof.

wherein
M″ is a Group 8 metal (preferably M is ruthenium or osmium, preferably ruthenium);
each X″ is independently an anionic ligand (preferably selected from the group consisting of halides, alkoxides, aryloxides, and alkyl sulfonates, preferably a halide, preferably chloride);
R″1 and R″2 are independently selected from the group consisting of hydrogen, a C1 to C30 hydrocarbyl, and a C1 to C30 substituted hydrocarbyl (preferably R″1 and R″2 are independently selected from the group consisting of methyl, ethyl, propyl, isopropyl, butyl, tert-butyl, sec-butyl, pentyl, cyclopentyl, hexyl, cyclohexyl, heptyl, octyl, cyclooctyl, and substituted analogs and isomers thereof, preferably selected from the group consisting of tert-butyl, sec-butyl, cyclohexyl, and cyclooctyl);
R″3 and R″4 are independently selected from the group consisting of hydrogen, C1 to C12 hydrocarbyl groups, substituted C1 to C12 hydrocarbyl groups, and halides (preferably R″3 and R″4 are independently selected from the group consisting of methyl, ethyl, propyl, isopropyl, butyl, tert-butyl, sec-butyl, pentyl, cyclopentyl, hexyl, cyclohexyl, heptyl, octyl, cyclooctyl, and substituted analogs and isomers thereof, preferably selected from the group consisting of tert-butyl, sec-butyl, cyclohexyl, and cyclooctyl); and
L″ is a neutral donor ligand, preferably L″ is selected from the group consisting of a phosphine, a sulfonated phosphine, a phosphite, a phosphinite, a phosphonite, an arsine, a stibine, an ether, an amine, an imine, a sulfoxide, a carboxyl, a nitrosyl, a pyridine, a thioester, a cyclic carbene, and substituted analogs thereof; preferably a phosphine, a sulfonated phosphine, an N-heterocyclic carbene, a cyclic alkyl amino carbene, and substituted analogs thereof (preferably L″ is selected from a phosphine, an N-heterocyclic carbene, a cyclic alkyl amino carbene, and substituted analogs thereof).

where
n is a linking group comprising from one to four ring vertices selected from the group consisting of C, Si, N, P, O, and S, with available valences optionally occupied by H, oxo, hydrocarbyl, or substituted hydrocarbyl groups; preferably, n comprises two ring vertices of carbon with available valences occupied by H, oxo, hydrocarbyl or substituted hydrocarbyl groups; preferably n is C2H2, C2H4, or substituted versions thereof;
each E is independently selected from the group comprising C, N, S, O, and P, with available valences optionally occupied by Lx, Ly, Lz, and Lz′; preferably, at least one E is a C; preferably, one E is a C and the other E is a N; preferably, both E's are C; and
Lx, Ly, Lz, and Lz′ are independently selected from the group comprising hydrogen, hydrocarbyl groups, and substituted hydrocarbyl groups; preferably, Lx, Ly, Lz, and Lz′ are independently selected from the group comprising a hydrocarbyl group and substituted hydrocarbyl group having 1 to 40 carbon atoms; preferably, Lx, Ly, Lz, and Lz′ are independently selected from the group comprising C1-10 alkyl, substituted C1-10 alkyl, C2-10 alkenyl, substituted C2-10 alkenyl, C2-10 alkynyl, substituted C2-10 alkynyl, aryl, and substituted aryl; preferably, Lx, Ly, Lz, and Lz′ are independently selected from the group comprising methyl, ethyl, propyl, butyl (including isobutyl and n-butyl), pentyl, cyclopentyl, hexyl, cyclohexyl, octyl, cyclooctyl, nonyl, decyl, cyclodecyl, dodecyl, cyclododecyl, mesityl, adamantyl, phenyl, benzyl, toluoyl, chlorophenyl, 2,6-diethylphenyl, 2,6-diisopropylphenyl, 2-isopropylphenyl, 2-ethyl-6-methylphenyl, 3,5-ditertbutylphenyl, 2-tertbutylphenyl, and 2,3,4,5,6-pentamethylphenyl. Useful substituents include C1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, aryl, C1-10 alkoxy, C2-10 alkenyloxy, C2-10 alkynyloxy, aryloxy, C2-10 alkoxycarbonyl, C1-10 alkylthio, C1-10 alkylsulfonyl, fluoro, chloro, bromo, iodo, oxo, amino, imine, nitrogen heterocycle, hydroxy, thiol, thiono, phosphorous, and carbene groups.

where Lx, Ly, and Lz are as defined above. In some embodiments, at least two of Lx, Ly, Lz, and Lz′ may be joined to form a 3- to 12-membered spirocyclic ring, with available valences optionally occupied by H, oxo, halogens, hydrocarbyl or substituted hydrocarbyl groups. Useful substituents include C1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, aryl, C1-10 alkoxy, C2-10 alkenyloxy, C2-10 alkynyloxy, aryloxy, C2-10 alkoxycarbonyl, C1-10 alkylthio, C1-10 alkylsulfonyl, fluoro, chloro, bromo, iodo, oxo, amino, imine, nitrogen heterocycle, hydroxy, thiol, thiono, phosphorous, and carbene groups.

where
n, Lx, and Ly are as described above in Formula (IV).
where Lx and Ly are as described above. Other useful NHCs include the compounds described in Hermann, W. A. Chem. Eur. J. 1996, 2, 772 and 1627; Enders, D. et al., Angew. Chem. Int. Ed. 1995, 34, 1021; Alder R. W., Angew. Chem. Int. Ed. 1996, 35, 1121; U.S. Ser. No. 61/314,388; and Bertrand, G. et al., Chem. Rev. 2000, 100, 39.

where
n, Lx, Ly, and Lz are as described above in Formula (IV).

Other useful CAACs include the compounds described in U.S. Pat. No. 7,312,331; U.S. Ser. No. 61/259,514; and Bertrand et al, Angew. Chem. Int. Ed. 2005, 44, 7236-7239.

wherein
M″ is a Group 8 metal (preferably M is ruthenium or osmium, preferably ruthenium);
each X″ is independently an anionic ligand (preferably selected from the group consisting of halides, alkoxides, aryloxides, and alkyl sulfonates, preferably a halide, preferably chloride);
R″1 and R″2 are independently selected from the group consisting of hydrogen, a C1 to C30 hydrocarbyl, and a C1 to C30 substituted hydrocarbyl (preferably R″1 and R″2 are independently selected from the group consisting of methyl, ethyl, propyl, isopropyl, butyl, tert-butyl, sec-butyl, pentyl, cyclopentyl, hexyl, cyclohexyl, heptyl, octyl, cyclooctyl, and substituted analogs and isomers thereof, preferably selected from the group consisting of tert-butyl, sec-butyl, cyclohexyl, and cyclooctyl); and
R″3, R″4, R″5, and R″6 are independently selected from the group consisting of hydrogen, C1 to C12 hydrocarbyl groups, substituted C1 to C12 hydrocarbyl groups, and halides (preferably R″3, R″4, R″5, and R″6 are independently selected from the group consisting of methyl, ethyl, propyl, isopropyl, butyl, tert-butyl, sec-butyl, pentyl, cyclopentyl, hexyl, cyclohexyl, heptyl, octyl, cyclooctyl, and substituted analogs and isomers thereof, preferably selected from the group consisting of tert-butyl, sec-butyl, cyclohexyl, and cyclooctyl).
- [(HP(tert-butyl)2)2Ru(C5H8)Cl2],
- [(H2P(tert-butyl))2Ru(C5H8)Cl2],
- [(HP(cyclohexyl)2)2Ru(C5H8)Cl2],
- [(H2P(cyclohexyl))2Ru(C5H8)Cl2],
- [(HP(cyclopentyl)2)2Ru(C5H8)Cl2],
- [(H2P(cyclopentyl))2Ru(C5H8)Cl2],
- [(HP(n-butyl)2)2Ru(C5H8)Cl2],
- [(H2P(n-butyl))2Ru(C5H8)Cl2],
- [(HP(sec-butyl)2)2Ru(C5H8)Cl2],
- [(H2P(sec-butyl))2Ru(C5H8)Cl2], and
fluoride and bromide derivatives thereof (preferably, wherein the Cl2 in the above list is replaced with F2, Br2, ClF, ClBr or FBr).
- 1. A polymer represented by the formula (A):

where R1 is a hydrocarbyl group having greater than 25 carbon atoms (preferably greater than 30 carbon atoms, greater than 40 carbon atoms, or greater than 50 carbon atoms; preferably from 31 carbon atoms to 100,000 carbon atoms, from 40 carbon atoms to 75,000 carbon atoms, from 50 carbon atoms to 60,000 carbon atoms; preferably R1 is represented by the formula (B):

wherein Cm is a C4 to C40 olefin derived unit (preferably Cm is one or more units derived from butene, pentene, hexene, heptene, octene, nonene, decene, undecene, dodecene, norbornene, norbornadiene, dicyclopentadiene, cyclopentene, cycloheptene, cyclooctene, cyclooctadiene, cyclododecene, 7-oxanorbornene, 7-oxanorbornadiene, substituted derivatives thereof, and isomers thereof); m is an integer from 3 to 39 (preferably from 3 to 29, preferably from 4 to 19, preferably from 5 to 11); and p is an integer greater than 1 (preferably from 1 to 10,000, from 1 to 5,000, from 1 to 2,500, from 1 to 1,000, from 1 to 500, or from 1 to 50); preferably R1 is represented by the formula (C):

wherein Cm is a C3 to C40 olefin derived unit (preferably Cm is one or more derived units of propylene, butene, pentene, hexene, heptene, octene, nonene, decene, undecene, dodecene, norbornene, norbornadiene, dicyclopentadiene, cyclopentene, cycloheptene, cyclooctene, cyclooctadiene, cyclododecene, 7-oxanorbornene, 7-oxanorbornadiene, substituted derivatives thereof, and isomers thereof); m is an integer from 2 to 39 (preferably from 2 to 29, preferably from 3 to 19, preferably from 4 to 11); and p is an integer greater than 1 (preferably from 1 to 10,000, from 1 to 5,000, from 1 to 2,500, from 1 to 1,000, from 1 to 500, or from 1 to 50);
R2 and R3 are the same or different and each is hydrogen or a hydrocarbyl group having from 1 to 40 carbon atoms (preferably from 1 to 30, from 2 to 15, and from 2 to 12), or R2 and R3 are joined to form a five-membered or six-membered ring, or substituted analogs thereof (preferably R2 and R3 are joined to form a heteroatom containing ring; preferably R2 and R3 are joined to form a dicarboxylic anhydride);
X is C, N, or O (preferably X is C or O, preferably X is C);
n is an integer from 1 to 10,000 (preferably from 1 to 5,000, from 1 to 2,500, from 1 to 1,000, from 1 to 500, or from 1 to 50); and
the dotted line indicates an optional double bond;
wherein the polymer comprises one or more vinyl terminated macromonomer derived units (preferably the vinyl terminated macromonomer is one or more of:
(i) a vinyl terminated polymer having an Mn of at least 200 g/mol (measured by 1H NMR) comprising of one or more C4 to C40 higher olefin derived units, where the higher olefin polymer comprises substantially no propylene derived units; and wherein the higher olefin polymer has at least 5% allyl chain ends;
(ii) a copolymer having an Mn of 300 g/mol or more (measured by 1H NMR) comprising (a) from about 20 mol % to about 99.9 mol % of at least one C5 to C40 higher olefin, and (b) from about 0.1 mol % to about 80 mol % of propylene, wherein the higher olefin copolymer has at least 40% allyl chain ends;
(iii) a copolymer having an Mn of 300 g/mol or more (measured by 1H NMR) and comprises (a) from about 80 mol % to about 99.9 mol % of at least one C4 olefin and (b) from about 0.1 mol % to about 20 mol % of propylene, wherein the vinyl terminated macromonomer has at least 40% allyl chain ends relative to total unsaturation;
(iv) a co-oligomer having an Mn of 300 g/mol to 30,000 g/mol (measured by 1H NMR) comprising 10 mol % to 90 mol % propylene and 10 mol % to 90 mol % of ethylene, wherein the oligomer has at least X % allyl chain ends (relative to total unsaturations), where: 1) X=(−0.94*(mol % ethylene incorporated)+100), when 10 mol % to 60 mol % ethylene is present in the co-oligomer, 2) X=45, when greater than 60 mol % and less than 70 mol % ethylene is present in the co-oligomer, and 3) X=(1.83*(mol % ethylene incorporated)−83), when 70 mol % to 90 mol % ethylene is present in the co-oligomer;
(v) a propylene oligomer, comprising more than 90 mol % propylene and less than 10 mol % ethylene wherein the oligomer has: at least 93% allyl chain ends, a number average molecular weight (Mn) of about 500 g/mol to about 20,000 g/mol, an isobutyl chain end to allylic vinyl group ratio of 0.8:1 to 1.35:1.0, and less than 100 ppm aluminum;
(vi) a propylene oligomer, comprising: at least 50 mol % propylene and from 10 mol % to 50 mol % ethylene, wherein the oligomer has: at least 90% allyl chain ends, an Mn of about 150 g/mol to about 10,000 g/mol, and an isobutyl chain end to allylic vinyl group ratio of 0.8:1 to 1.2:1.0, wherein monomers having four or more carbon atoms are present at from 0 mol % to 3 mol %;
(vii) a propylene oligomer, comprising: at least 50 mol % propylene, from 0.1 mol % to 45 mol % ethylene, and from 0.1 to 5 mol % C4 to C12 olefin, wherein the oligomer has: at least 90% allyl chain ends, an Mn of about 150 to about 10,000 g/mol, and an isobutyl chain end to allylic vinyl group ratio of 0.8:1 to 1.35:1.0;
(viii) a propylene oligomer, comprising: at least 50 mol % propylene, from 0.1 to 45 mol % ethylene, and from 0.1 mol % to 5 mol % diene, wherein the oligomer has: at least 90% allyl chain ends, an Mn of about 150 g/mol to about 10,000 g/mol, and an isobutyl chain end to allylic vinyl group ratio of 0.7:1 to 1.35:1.0; and
(ix) a homo-oligomer, comprising propylene, wherein the oligomer has: at least 93% allyl chain ends, an Mn of about 500 g/mol to about 20,000 g/mol, an isobutyl chain end to allylic vinyl group ratio of 0.8:1 to 1.2:1.0, and less than 1400 ppm aluminum).
2. The polymer of paragraph 1, wherein the polymer is represented by the formula (D):

wherein n is an integer from 1 to 10,000 (preferably from 1 to 5,000, from 1 to 2,500, from 1 to 1,000, from 1 to 500, or from 1 to 50); Cm is a C3 to C40 olefin derived unit (preferably Cm is one or more derived units of propylene, butene, pentene, hexene, heptene, octene, nonene, decene, undecene, dodecene, norbornene, norbornadiene, dicyclopentadiene, cyclopentene, cycloheptene, cyclooctene, cyclooctadiene, cyclododecene, 7-oxanorbornene, 7-oxanorbornadiene, substituted derivatives thereof, and isomers thereof); m is an integer from 2 to 39 (preferably from 2 to 29, preferably from 3 to 19, preferably from 4 to 11); and p is an integer greater than 1 (preferably from 1 to 10,000, from 1 to 5,000, from 1 to 2,500, from 1 to 1,000, from 1 to 500, or from 1 to 50).
3. The polymer of paragraphs 1 and 2, wherein the polymer is hydrogenated.
4. The use of the polymer of paragraphs 1 to 3 as a lubricant or lubricant base stock, an adhesive, a viscosity modifier, or a fuel additive.
5. A process to produce the polymer of paragraphs 1 to 3, the process comprising contacting at least one cyclic olefin (preferably norbornene, norbornadiene, dicyclopentadiene, cyclopentene, cycloheptene, cyclooctene, cyclooctadiene, cyclododecene, 7-oxanorbornene, 7-oxanorbornadiene, and substituted derivatives therefrom; more preferably cyclooctene, 1,5-cyclooctadiene, 1-hydroxy-4-cyclooctene, 1-acetoxy-4-cyclooctene, 5-methylcyclopentene, cyclopentene, dicyclopentadiene, norbornene, norbornadiene, cis-5-norbornene-endo-2,3-dicarboxylic anhydride, dimethyl norbornene carboxylate, norbornene-exo-2,3-carboxylic anhydride, and their respective homologs and derivatives) with at least one vinyl terminated macromonomer in the presence of one or more metathesis catalysts (preferably one or more of:
(i) an alkene metathesis catalyst represented by the Formula (E):

where:
M is a Group 8 metal, preferably Ru or Os, preferably Ru;
X and X1 are, independently, any anionic ligand, preferably a halogen (preferably chlorine), an alkoxide or a triflate, or X and X1 may be joined to form a dianionic group and may form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
L and L1 are, independently, a neutral two electron donor, preferably a phosphine or a N-heterocyclic carbene, L and L1 may be joined to form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
L and X may be joined to form a multidentate monoanionic group and may form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
L1 and X1 may be joined to form a multidentate monoanionic group and may form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
R4 and R5 are, independently, hydrogen or C1 to C30 substituted or unsubstituted hydrocarbyl (preferably a C1 to C30 substituted or unsubstituted alkyl or a substituted or unsubstituted C4 to C30 aryl);
R5 and L1 or X1 may be joined to form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms; and
R4 and L or X may be joined to form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms; and/or
(ii) a metathesis catalyst represented by the Formula (F):

where M is Os or Ru, preferably Ru;
X, X1, L, and L1 are as described above for Formula E; and
R9 and R10 may be different or the same and may be hydrogen, substituted or unsubstituted alkyl, or substituted or unsubstituted aryl; and/or
(iii) a metathesis catalyst represented by the formula (G):

where
M* is a Group 8 metal, preferably Ru or Os, preferably Ru;
X* and X1* are, independently, any anionic ligand, preferably a halogen (preferably chlorine), an alkoxide or an alkyl sulfonate, or X* and X1* may be joined to form a dianionic group and may form a single ring of up to 30 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms;
L* is N—R**, O, P—R**, or S, preferably N—R** or O (R** is a C1 to C30 hydrocarbyl or substituted hydrocarbyl, preferably methyl, ethyl, propyl or butyl);
R* is hydrogen or a C1 to C30 hydrocarbyl or substituted hydrocarbyl, preferably methyl;
R1*, R2*, R3*, R4*, R5*, R6*, R7*, and R8* are, independently, hydrogen or a C1 to C30 hydrocarbyl or substituted hydrocarbyl, preferably methyl, ethyl, propyl or butyl, preferably R1*, R2*, R3*, and R4* are methyl;
each R9* and R13* are, independently, hydrogen or a C1 to C30 hydrocarbyl or substituted hydrocarbyl, preferably a C2 to C6 hydrocarbyl, preferably ethyl;
R10*, R11*, R12* are, independently hydrogen or a C1 to C30 hydrocarbyl or substituted hydrocarbyl, preferably hydrogen or methyl;
each G, is, independently, hydrogen, halogen or C1 to C30 substituted or unsubstituted hydrocarbyl (preferably a C1 to C30 substituted or unsubstituted alkyl or a substituted or unsubstituted C4 to C30 aryl); and
where any two adjacent R groups may form a single ring of up to 8 non-hydrogen atoms or a multinuclear ring system of up to 30 non-hydrogen atoms; and/or
(iv) a Group 8 metal complex represented by the formula (H):

wherein
M″ is a Group 8 metal (preferably M is ruthenium or osmium, preferably ruthenium);
each X″ is independently an anionic ligand (preferably selected from the group consisting of halides, alkoxides, aryloxides, and alkyl sulfonates, preferably a halide, preferably chloride);
R″1 and R″2 are independently selected from the group consisting of hydrogen, a C1 to C30 hydrocarbyl, and a C1 to C30 substituted hydrocarbyl (preferably R″1 and R″2 are independently selected from the group consisting of methyl, ethyl, propyl, isopropyl, butyl, tert-butyl, sec-butyl, pentyl, cyclopentyl, hexyl, cyclohexyl, heptyl, octyl, cyclooctyl, and substituted analogs and isomers thereof, preferably selected from the group consisting of tert-butyl, sec-butyl, cyclohexyl, and cyclooctyl);
R″3 and R″4 are independently selected from the group consisting of hydrogen, C1 to C12 hydrocarbyl groups, substituted C1 to C12 hydrocarbyl groups, and halides (preferably R″3 and R″4 are independently selected from the group consisting of methyl, ethyl, propyl, isopropyl, butyl, tert-butyl, sec-butyl, pentyl, cyclopentyl, hexyl, cyclohexyl, heptyl, octyl, cyclooctyl, and substituted analogs and isomers thereof, preferably selected from the group consisting of tert-butyl, sec-butyl, cyclohexyl, and cyclooctyl); and
L″ is a neutral donor ligand, preferably L″ is selected from the group consisting of a phosphine, a sulfonated phosphine, a phosphite, a phosphinite, a phosphonite, an arsine, a stibine, an ether, an amine, an imine, a sulfoxide, a carboxyl, a nitrosyl, a pyridine, a thioester, a cyclic carbene, and substituted analogs thereof; preferably a phosphine, a sulfonated phosphine, an N-heterocyclic carbene, a cyclic alkyl amino carbene, and substituted analogs thereof (preferably L″ is selected from a phosphine, an N-heterocyclic carbene, a cyclic alkyl amino carbene, and substituted analogs thereof); and/or
(v) a Group 8 metal complex represented by the formula (I):

wherein
M″ is a Group 8 metal (preferably M is ruthenium or osmium, preferably ruthenium);
each X″ is independently an anionic ligand (preferably selected from the group consisting of halides, alkoxides, aryloxides, and alkyl sulfonates, preferably a halide, preferably chloride);
R″1 and R″2 are independently selected from the group consisting of hydrogen, a C1 to C30 hydrocarbyl, and a C1 to C30 substituted hydrocarbyl (preferably R″1 and R″2 are independently selected from the group consisting of methyl, ethyl, propyl, isopropyl, butyl, tert-butyl, sec-butyl, pentyl, cyclopentyl, hexyl, cyclohexyl, heptyl, octyl, cyclooctyl, and substituted analogs and isomers thereof, preferably selected from the group consisting of tert-butyl, sec-butyl, cyclohexyl, and cyclooctyl); and
R″3, R″4, R″5, and R″6 are independently selected from the group consisting of hydrogen, C1 to C12 hydrocarbyl groups, substituted C1 to C12 hydrocarbyl groups, and halides (preferably R″3, R″4, R″5, and R″6 are independently selected from the group consisting of methyl, ethyl, propyl, isopropyl, butyl, tert-butyl, sec-butyl, pentyl, cyclopentyl, hexyl, cyclohexyl, heptyl, octyl, cyclooctyl, and substituted analogs and isomers thereof, preferably selected from the group consisting of tert-butyl, sec-butyl, cyclohexyl, and cyclooctyl);
at a temperature of 20° C. to 200° C. (preferably 50° C. to 160° C., preferably 60° C. to 140° C.) and a pressure of 0 MPa to 1000 MPa (preferably 0.5 MPa to 500 MPa, preferably 1 MPa to 250 MPa) for a residence time of 0.5 seconds to 10 hours (preferably 1 second to 5 hours, preferably 1 minute to 1 hour).
6. The process of paragraph 5, wherein the metathesis catalyst is one or more of:
- tricyclohexylphosphine[1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene][3-phenyl-1H-inden-1-ylidene]ruthenium(II)dichloride,
- tricyclohexylphosphine[3-phenyl-1H-inden-1-ylidene][1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydro-imidazol-2-ylidene]ruthenium(II)dichloride,
- tricyclohexylphosphine[1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene][(phenylthio)methylene]ruthenium(II)dichloride,
- bis(tricyclohexylphosphine)-3-phenyl-1H-inden-1-ylideneruthenium(II)dichloride,
- 1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene[2-(i-propoxy)-5-(N,N-dimethylaminosulfonyl)phenyl]methyleneruthenium(II)dichloride,
- [1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene]-[2-[[(4-methylphenyl)imino]methyl]-4-nitrophenolyl]-[3-phenyl-1H-inden-1-ylidene]ruthenium(II) chloride,
- benzylidene-bis(tricyclohexylphosphine)dichlororuthenium,
- benzylidene[1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene]dichloro(tricyclohexylphosphine)ruthenium,
- dichloro(o-isopropoxyphenylmethylene)(tricyclohexylphosphine)ruthenium(II),
- (1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene)dichloro(o-isopropoxyphenylmethylene)ruthenium,
- 1,3-bis(2-methylphenyl)-2-imidazolidinylidene]dichloro(2-isopropoxyphenylmethylene) ruthenium(II),
- [1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene]dichloro[3-(2-pyridinyl)propylidene]ruthenium(II),
- [1,3-bis(2-methylphenyl)-2-imidazolidinylidene]dichloro(phenylmethylene) (tricyclohexylphosphine)ruthenium(II),
- [1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene]dichloro(3-methyl-2-butenylidene) (tricyclohexylphosphine)ruthenium(II),
- [1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene]dichloro(benzylidene)bis(3-bromopyridine)ruthenium(II),
- 2-(2,6-diethylphenyl)-3,5,5,5-tetramethylpyrrolidine[2-(i-propoxy)-5-(N,N-dimethylaminosulfonyl)phenyl]methylene ruthenium dichloride,
- 2-(mesityl)-3,3,5,5-tetramethylpyrrolidine[2-(i-propoxy)-5-(N,N-dimethylaminosulfonyl)phenyl]methylene ruthenium dichloride,
- 2-(2-isopropyl)-3,3,5,5-tetramethylpyrrolidine[2-(i-propoxy)-5-(N,N-dimethylaminosulfonyl)phenyl]methylene ruthenium dichloride,
- 2-(2,6-diethyl-4-fluorophenyl)-3,3,5,5-tetramethylpyrrolidine[2-(i-propoxy)-5-(N,N-dimethylaminosulfonyl)phenyl]methylene ruthenium dichloride,
- [(HP(tert-butyl)2)2Ru(C5H8)Cl2],
- [(H2P(tert-butyl))2Ru(C5H8)Cl2],
- [(HP(cyclohexyl)2)2Ru(C5H8)Cl2],
- [(H2P(cyclohexyl))2Ru(C5H8)Cl2],
- [(HP(cyclopentyl)2)2Ru(C5H8)Cl2],
- [(H2P(cyclopentyl))2Ru(C5H8)Cl2],
- [(HP(n-butyl)2)2Ru(C5H8)Cl2],
- [(H2P(n-butyl))2Ru(C5H8)Cl2],
- [(HP(sec-butyl)2)2Ru(C5H8)Cl2],
- [(H2P(sec-butyl))2Ru(C5H8)Cl2], and
fluoride and bromide derivatives thereof
7. The process of paragraphs 5 and 6, wherein substantially no dimerization occurs.
Method B
Comparison of Methods A and B
General

Vinyl Terminated Butene Propylene Copolymer

An oven dried 100 mL round bottom flask was charged with the vinyl terminated hexene-propylene copolymer (VT hexene propylene copolymer) as described in Example 1 (15.2 g, ˜9 mmol), cis-5-norbornene-endo-2,3-dicarboxylic anhydride (2.1 g, 12.8 mmol) and CHCl3 (15 mL). The suspension was then stirred and heated to 50° C. and the ((t-Bu)2PH)2Cl2Ru═CHCH═C(CH3)2 was added in one portion. After 5 hours a large portion of the insoluble material had dissolved, indicating progression of the ROCM reaction. The reaction was allowed to stir overnight at room temperature. The reaction was observed to still be a suspension on the following morning. The reaction was then diluted with methylene chloride and filtered through a plug of silica.


Claims (13)

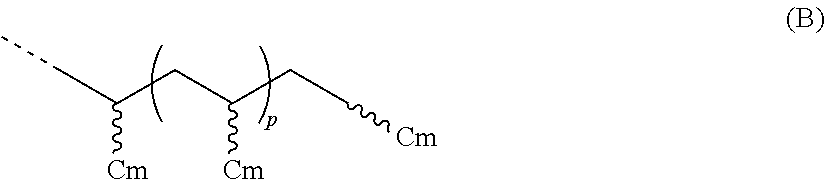



Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US13/306,263 US8604148B2 (en) | 2011-11-29 | 2011-11-29 | Functionalization of vinyl terminated polymers by ring opening cross metathesis |
| EP12854390.7A EP2785763A4 (en) | 2011-11-29 | 2012-10-08 | Functionalization of vinyl terminated polymers by ring opening cross metathesis |
| CN201280056901.1A CN103987755B (en) | 2011-11-29 | 2012-10-08 | Functionalization by the metathetic vinyl terminated polymer of open loop crossover |
| PCT/US2012/059191 WO2013081726A1 (en) | 2011-11-29 | 2012-10-08 | Functionalization of vinyl terminated polymers by ring opening cross metathesis |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US13/306,263 US8604148B2 (en) | 2011-11-29 | 2011-11-29 | Functionalization of vinyl terminated polymers by ring opening cross metathesis |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| US20130137829A1 US20130137829A1 (en) | 2013-05-30 |
| US8604148B2 true US8604148B2 (en) | 2013-12-10 |
Family
ID=48467433
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/306,263 Expired - Fee Related US8604148B2 (en) | 2011-11-29 | 2011-11-29 | Functionalization of vinyl terminated polymers by ring opening cross metathesis |
Country Status (1)
| Country | Link |
|---|---|
| US (1) | US8604148B2 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021126691A1 (en) | 2019-12-16 | 2021-06-24 | Exxonmobil Chemical Patents. Inc. | Tire tread compounds |
| WO2021178235A1 (en) | 2020-03-03 | 2021-09-10 | Exxonmobil Chemical Patents Inc. | Rubber compounds for heavy-duty truck and bus tire treads and methods relating thereto |
| WO2021242636A1 (en) | 2020-05-29 | 2021-12-02 | Exxonmobil Chemical Patents Inc. | Processes for producing cyclic olefins from polymers and re-polymerization thereof |
| US11912861B2 (en) | 2020-10-29 | 2024-02-27 | ExxonMobil Engineering & Technology Co. | Rubber composition for lighter weight tires and improved wet traction |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102294024B1 (en) | 2013-09-27 | 2021-08-27 | 바스프 에스이 | Polyolefin compositions for building materials |
| US9382354B2 (en) * | 2013-11-22 | 2016-07-05 | Exxonmobil Chemical Patents Inc. | Polyesters containing polyolefin arms |
| WO2015076958A1 (en) * | 2013-11-22 | 2015-05-28 | Exxonmobil Chemical Patents Inc. | Novel polyesters containing polyolefin arms |
| US10683388B2 (en) | 2017-08-15 | 2020-06-16 | University Of Florida Research Foundation, Incorporated | High temperature metathesis chemistry |
| CN110170077B (en) * | 2019-03-26 | 2021-09-28 | 南京理工大学 | Polyion type biological lubricant and preparation method thereof |
| WO2022008946A1 (en) * | 2020-07-08 | 2022-01-13 | Centre National De La Recherche Scientifique | Optically pure enantiomers of ruthenium complexes and uses thereof |
Citations (91)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3022305A (en) | 1957-06-17 | 1962-02-20 | Du Pont | Process for preparing pyridazine derivatives and novel fluorine-containing pyridazine derivatives |
| US3235484A (en) | 1962-03-27 | 1966-02-15 | Lubrizol Corp | Cracking processes |
| GB1310847A (en) | 1971-03-12 | 1973-03-21 | Lubrizol Corp | Fuel compositions |
| US4069023A (en) | 1975-05-01 | 1978-01-17 | Exxon Research & Engineering Co. | Carboxylate esters of 1-aza-3,7-dioxabicyclo[3.3.0] oct-5-yl methyl alcohols, their preparation and use as additives for oleaginous compositions |
| US4110377A (en) | 1976-10-28 | 1978-08-29 | Anic S.P.A. | Process for the alkylation of secondary aliphatic amines in the presence of an amide of a transition metal |
| US4197398A (en) | 1974-12-04 | 1980-04-08 | Exxon Research & Engineering Co. | Process for neutralizing and deashing polypropylene |
| US4619756A (en) | 1985-04-11 | 1986-10-28 | Exxon Chemical Patents Inc. | Method to inhibit deposit formation |
| JPH0264115A (en) | 1988-08-30 | 1990-03-05 | Nippon Unicar Co Ltd | Polydialkylosiloxane-olefin block copolymer, production thereof, composition containing the same copolymer and molded article made from the composition |
| US4973414A (en) | 1987-06-02 | 1990-11-27 | Bayer Aktiengesellschaft | Polyethers, process for their preparation and lubricants containing these polyethers |
| US4988764A (en) | 1987-09-29 | 1991-01-29 | Toyota Jidosha Kabushiki Kaisha | Impact-resistant polypropylene resin compositions |
| US5026948A (en) | 1989-02-21 | 1991-06-25 | Mobil Oil Corporation | Disproportionation of alpha-olefin dimer to liquid lubricant basestock |
| US5211834A (en) | 1992-01-31 | 1993-05-18 | Betz Laboratories, Inc. | Method for controlling fouling deposit formation in a liquid hydrocarbonaceous medium using boronated derivatives of polyalkenylsuccinimides |
| US5229022A (en) | 1988-08-01 | 1993-07-20 | Exxon Chemical Patents Inc. | Ethylene alpha-olefin polymer substituted mono- and dicarboxylic acid dispersant additives (PT-920) |
| US5252677A (en) | 1990-11-20 | 1993-10-12 | Mitsubishi Petrochemical Company Limited | Functionalized olefin polymers |
| US5266186A (en) | 1989-10-12 | 1993-11-30 | Nalco Chemical Company | Inhibiting fouling employing a dispersant |
| JPH05320260A (en) | 1992-05-20 | 1993-12-03 | Idemitsu Kosan Co Ltd | Vinyl-terminated ethylene-propylene copolymer and method for producing the same |
| US5382634A (en) | 1991-03-15 | 1995-01-17 | Tonen Corporation | Terminal-modifed polyolefins |
| US5439607A (en) | 1993-12-30 | 1995-08-08 | Exxon Chemical Patents Inc. | Multifunctional viscosity index improver-dispersant antioxidant |
| WO1995027717A1 (en) | 1994-04-06 | 1995-10-19 | Montell Technology Company Bv | Metallocene compounds and use thereof in catalysts for the polymerization of olefins |
| EP0767182A2 (en) | 1995-10-03 | 1997-04-09 | Ethyl Corporation | Copolymers of atactic propene polymers, dispersants derived therefrom and lubricating oils and fuel compositions containing the dispersants |
| US5621047A (en) | 1993-05-27 | 1997-04-15 | Amoco Corporation | Process for preparing linear monofunctional and telechelic difunctional polymers and compositions obtained thereby |
| EP0802216A1 (en) | 1996-04-15 | 1997-10-22 | DOW CORNING ASIA, Ltd. | Copolymer of polypropylene and organopolysiloxane and method for preparation thereof |
| WO1997047665A1 (en) | 1996-06-13 | 1997-12-18 | University Of Waterloo | Hydrosilylation of polypropylene |
| US5741946A (en) | 1994-10-27 | 1998-04-21 | Mobil Oil Corporation | Polyether lubricants and method for their production |
| US5750815A (en) | 1992-04-03 | 1998-05-12 | California Institute Of Technology | Olefin metathesis coupling using ruthenium and osmium carbene complexes |
| US5756428A (en) | 1986-10-16 | 1998-05-26 | Exxon Chemical Patents Inc. | High functionality low molecular weight oil soluble dispersant additives useful in oleaginous composition |
| WO1998040373A1 (en) | 1997-03-14 | 1998-09-17 | Sepracor, Inc. | Ring opening metathesis of alkenes |
| WO1999005182A1 (en) | 1997-07-22 | 1999-02-04 | Bp Chemicals Limited | Polymerisation catalysts |
| WO1999046270A1 (en) | 1998-03-11 | 1999-09-16 | The Dow Chemical Company | Integrated process for preparation of diene complexes |
| EP0958309A1 (en) | 1997-02-07 | 1999-11-24 | Exxon Chemical Patents Inc. | Preparation of vinyl-containing macromers |
| WO2000000576A1 (en) | 1998-06-30 | 2000-01-06 | Chevron Chemical Company Llc | Ashless lubricating oil formulations for natural gas engines |
| US6017859A (en) | 1996-06-17 | 2000-01-25 | Exxon Chemical Patents Inc | Polymers derived from olefins useful as lubricant and fuel oil additives, processes for preparation of such polymers and additives and use thereof |
| JP2000038420A (en) | 1998-07-24 | 2000-02-08 | Idemitsu Petrochem Co Ltd | Propylene copolymer and its production |
| US6117962A (en) | 1997-12-10 | 2000-09-12 | Exxon Chemical Patents Inc. | Vinyl-containing stereospecific polypropylene macromers |
| WO2000055218A1 (en) | 1999-03-18 | 2000-09-21 | California Institute Of Technology | Novel aba triblock and diblock copolymers and methods of preparing the same |
| US6143686A (en) | 1994-08-03 | 2000-11-07 | Exxon Chemical Patents, Inc. | Supported ionic catalyst compositions |
| US6197910B1 (en) | 1997-12-10 | 2001-03-06 | Exxon Chemical Patents, Inc. | Propylene polymers incorporating macromers |
| US6225432B1 (en) | 1998-08-26 | 2001-05-01 | Exxon Chemical Patents Inc. | Branched polypropylene compositions |
| US6268518B1 (en) | 1997-03-29 | 2001-07-31 | Montell Technology Company B.V. | Metallocene compounds and their use in catalysts for the polymerization of olefins |
| US6448350B1 (en) | 1998-04-21 | 2002-09-10 | Basell Technology Company Bv | Process for the preparation of copolymers of ethylene with alpha-olefins |
| US20020137978A1 (en) | 2000-06-23 | 2002-09-26 | Grubbs Robert H. | Synthesis of functionalized and unfunctionalized olefins via cross and ring-closing Metathesis |
| WO2002079127A1 (en) | 2001-03-30 | 2002-10-10 | California Institute Of Technology | Selective ring-opening cross-metathesis of cycloolefins |
| US6476167B2 (en) | 2000-12-14 | 2002-11-05 | Bayer Corporation | End-functionalized polyolefin prepared via ring opening metathesis polymerization in the presence of a novel chain transfer agent, and a process for the preparation of the end-functionalized polyolefin via ring opening metathesis polyermization |
| US6590048B1 (en) | 1997-12-03 | 2003-07-08 | Studiengesellschaft Kohle Mbh | Highly active cationic ruthenium and osmium complexes for olefin metathesis reactions |
| US20030161752A1 (en) | 2002-01-25 | 2003-08-28 | Sydney Luk | Powder metallurgy lubricant compositions and methods for using the same |
| EP1361232A2 (en) | 1997-12-10 | 2003-11-12 | ExxonMobil Chemical Patents Inc. | Polypropylene polymers incorporating macromers |
| US6703457B2 (en) | 2000-05-24 | 2004-03-09 | Basell Polyolefine Gmbh | Propylene polymers and process for the preparation thereof |
| WO2004031250A1 (en) | 2002-10-02 | 2004-04-15 | Dow Global Technologies Inc. | Liquid and gel-like low molecular weight ethylene polymers |
| US6750307B2 (en) | 1997-02-07 | 2004-06-15 | Exxon Mobil Chemical Patents Inc. | Propylene polymers incorporating polyethylene macromers |
| US20040127649A1 (en) | 2002-10-24 | 2004-07-01 | Palanisamy Arjunan | Branched crystalline polypropylene |
| US20040214953A1 (en) | 2003-04-28 | 2004-10-28 | Tosoh Corporation | Polyethylene composition and process for producing same |
| US20040249046A1 (en) | 2002-10-15 | 2004-12-09 | Ramin Abhari | Polyolefin adhesive compositions and articles made therefrom |
| US20050054793A1 (en) | 2003-09-09 | 2005-03-10 | Reinking Mark K. | Hydrosilane additives for increased polyolefin molecular weight |
| US6897261B1 (en) | 1999-07-26 | 2005-05-24 | Idemitsu Kosan Co., Ltd. | Branched olefinic macromonomer, olefin graft copolymer, and olefin resin composition |
| JP2005139284A (en) | 2003-11-06 | 2005-06-02 | National Institute Of Advanced Industrial & Technology | Method for producing functionalized polypropylene |
| US20050159299A1 (en) | 2004-01-16 | 2005-07-21 | George Rodriguez | Hydrophobization and silica for supported catalyst |
| WO2005092935A1 (en) | 2004-02-24 | 2005-10-06 | Stichting Dutch Polymer Institute | Catalyst system comprising magnesium halide |
| US20050261440A1 (en) | 2004-05-20 | 2005-11-24 | Dickakian Ghazi B | Dispersant material for mitigating crude oil fouling of process equipment and method for using same |
| JP2005336092A (en) | 2004-05-26 | 2005-12-08 | Mitsubishi Chemicals Corp | Novel transition metal compound and method for producing propylene polymer using the transition metal compound |
| US20060052553A1 (en) | 2002-12-04 | 2006-03-09 | Basell Polyolefine Gmbh | Process for preparing 1-butene polymers |
| EP1693357A1 (en) | 2005-02-22 | 2006-08-23 | Miltitz Aromatics GmbH | Process and apparatus for carying out a ring opening cross-metathesis reaction between cyclic and acyclic olefins |
| US7126031B2 (en) | 2002-04-24 | 2006-10-24 | Symyx Technologies, Inc. | Bridged bi-aromatic ligands, catalysts, processes for polymerizing and polymers therefrom |
| WO2006127483A1 (en) | 2005-05-20 | 2006-11-30 | Bridgestone Corporation | Method for preparing low molecular weight polymers |
| US20060270814A1 (en) | 2003-08-27 | 2006-11-30 | Haruyuki Makio | Polyolefin functional at one end |
| JP2007169340A (en) | 2005-12-19 | 2007-07-05 | Tosoh Corp | Olefin polymerization catalyst and process for producing olefin polymer |
| US7247385B1 (en) | 1998-08-14 | 2007-07-24 | University Of Waterloo | Melt phase hydrosilylation of polypropylene |
| CN101011062A (en) | 2007-02-06 | 2007-08-08 | 张家港市骏博化工有限公司 | Organosilicon pesticide booster and preparing method thereof |
| JP2007246433A (en) | 2006-03-15 | 2007-09-27 | Tosoh Corp | Organic transition metal compound, catalyst for olefin polymerization using the same, and method for producing polyolefin |
| US7276567B2 (en) | 2004-04-16 | 2007-10-02 | Exxonmobil Chemical Patents Inc. | Heterocyclic substituted metallocene compounds for olefin polymerization |
| EP1849757A1 (en) | 2005-02-18 | 2007-10-31 | Idemitsu Kosan Co., Ltd. | Method for producing unsaturated hydrocarbon compound |
| EP1862491A1 (en) | 2005-03-22 | 2007-12-05 | Zeon Corporation | Thermoplastic resin, method for producing same and molding material |
| US20070293640A1 (en) | 2002-10-15 | 2007-12-20 | Peijun Jiang | Multiple catalyst system for olefin polymerization and polymers produced therefrom |
| JP2008050278A (en) | 2006-08-22 | 2008-03-06 | Tosoh Corp | Transition metal compound, catalyst for olefin polymerization, and method for producing polyolefin |
| US20080064891A1 (en) | 2006-07-12 | 2008-03-13 | Lee Choon W | Ring opening cross-metathesis reaction of cyclic olefins with seed oils and the like |
| US20080228017A1 (en) | 2005-06-06 | 2008-09-18 | Dow Global Technologies Inc. | Metathesis Process For Preparing An Alpha, Omega-Functionalized Olefin |
| US20080234451A1 (en) | 2004-12-23 | 2008-09-25 | Imperial Chemical Industries Plc | Olefin Metathesis Polymerisation |
| US20090247441A1 (en) | 2008-03-31 | 2009-10-01 | Exxonmobil Research And Engineering Company | High viscosity index pao with polyurea thickeners in grease compositions |
| US20090318647A1 (en) | 2008-06-20 | 2009-12-24 | Hagadorn John R | Olefin Functionalization By Metathesis Reaction |
| US20090318646A1 (en) | 2008-06-20 | 2009-12-24 | Patrick Brant | Functionalized High Vinyl Terminated Propylene Based Oligomers |
| US20090318644A1 (en) | 2008-06-20 | 2009-12-24 | Patrick Brant | High Vinyl Terminated Propylene Based Oligomers |
| US20090318640A1 (en) | 2008-06-20 | 2009-12-24 | Patrick Brant | Polymacromonomer And Process For Production Thereof |
| JP2010037555A (en) | 2008-07-11 | 2010-02-18 | Mitsui Chemicals Inc | Method for producing silylated polyolefin and additive containing the silylated polyolefin |
| US20100069573A1 (en) | 2006-08-25 | 2010-03-18 | Dow Global Technologies Inc. | Production of meta-block copolymers by polymer segment interchange |
| US20100152388A1 (en) | 2005-06-22 | 2010-06-17 | Peijun Jiang | In-Reactor Polymer Blends |
| US20100152387A1 (en) | 2007-05-23 | 2010-06-17 | Basf Se | Isotactic polystyrene having reactive groups |
| US7820607B2 (en) | 2004-09-10 | 2010-10-26 | Mitsui Chemicals, Inc. | Viscosity modifier for lubricating oils, additive composition for lubricating oils, and lubricating oil compositions |
| JP2011026448A (en) | 2009-07-24 | 2011-02-10 | Mitsui Chemicals Inc | Composition having mold releasability |
| US7943716B2 (en) | 2004-03-12 | 2011-05-17 | Basell Polyolefine Gmbh | Process for polymerizing 1-hexene or higher alpha-olefins |
| US7960487B2 (en) | 2006-02-22 | 2011-06-14 | Chevron Phillips Chemical Company Lp | Dual metallocene catalysts for polymerization of bimodal polymers |
| JP2012051859A (en) | 2010-09-03 | 2012-03-15 | Mitsui Chemicals Inc | Method for producing diol compound |
| JP2012052062A (en) | 2010-09-03 | 2012-03-15 | Mitsui Chemicals Inc | Method for producing epoxy compound |
-
2011
- 2011-11-29 US US13/306,263 patent/US8604148B2/en not_active Expired - Fee Related
Patent Citations (100)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3022305A (en) | 1957-06-17 | 1962-02-20 | Du Pont | Process for preparing pyridazine derivatives and novel fluorine-containing pyridazine derivatives |
| US3235484A (en) | 1962-03-27 | 1966-02-15 | Lubrizol Corp | Cracking processes |
| GB1310847A (en) | 1971-03-12 | 1973-03-21 | Lubrizol Corp | Fuel compositions |
| US4197398A (en) | 1974-12-04 | 1980-04-08 | Exxon Research & Engineering Co. | Process for neutralizing and deashing polypropylene |
| US4069023A (en) | 1975-05-01 | 1978-01-17 | Exxon Research & Engineering Co. | Carboxylate esters of 1-aza-3,7-dioxabicyclo[3.3.0] oct-5-yl methyl alcohols, their preparation and use as additives for oleaginous compositions |
| US4110377A (en) | 1976-10-28 | 1978-08-29 | Anic S.P.A. | Process for the alkylation of secondary aliphatic amines in the presence of an amide of a transition metal |
| US4619756A (en) | 1985-04-11 | 1986-10-28 | Exxon Chemical Patents Inc. | Method to inhibit deposit formation |
| US5756428A (en) | 1986-10-16 | 1998-05-26 | Exxon Chemical Patents Inc. | High functionality low molecular weight oil soluble dispersant additives useful in oleaginous composition |
| US4973414A (en) | 1987-06-02 | 1990-11-27 | Bayer Aktiengesellschaft | Polyethers, process for their preparation and lubricants containing these polyethers |
| US4988764A (en) | 1987-09-29 | 1991-01-29 | Toyota Jidosha Kabushiki Kaisha | Impact-resistant polypropylene resin compositions |
| US5229022A (en) | 1988-08-01 | 1993-07-20 | Exxon Chemical Patents Inc. | Ethylene alpha-olefin polymer substituted mono- and dicarboxylic acid dispersant additives (PT-920) |
| JPH0264115A (en) | 1988-08-30 | 1990-03-05 | Nippon Unicar Co Ltd | Polydialkylosiloxane-olefin block copolymer, production thereof, composition containing the same copolymer and molded article made from the composition |
| US5026948A (en) | 1989-02-21 | 1991-06-25 | Mobil Oil Corporation | Disproportionation of alpha-olefin dimer to liquid lubricant basestock |
| US5266186A (en) | 1989-10-12 | 1993-11-30 | Nalco Chemical Company | Inhibiting fouling employing a dispersant |
| US5252677A (en) | 1990-11-20 | 1993-10-12 | Mitsubishi Petrochemical Company Limited | Functionalized olefin polymers |
| US5382634A (en) | 1991-03-15 | 1995-01-17 | Tonen Corporation | Terminal-modifed polyolefins |
| US5211834A (en) | 1992-01-31 | 1993-05-18 | Betz Laboratories, Inc. | Method for controlling fouling deposit formation in a liquid hydrocarbonaceous medium using boronated derivatives of polyalkenylsuccinimides |
| US5750815A (en) | 1992-04-03 | 1998-05-12 | California Institute Of Technology | Olefin metathesis coupling using ruthenium and osmium carbene complexes |
| JPH05320260A (en) | 1992-05-20 | 1993-12-03 | Idemitsu Kosan Co Ltd | Vinyl-terminated ethylene-propylene copolymer and method for producing the same |
| US5621047A (en) | 1993-05-27 | 1997-04-15 | Amoco Corporation | Process for preparing linear monofunctional and telechelic difunctional polymers and compositions obtained thereby |
| US5439607A (en) | 1993-12-30 | 1995-08-08 | Exxon Chemical Patents Inc. | Multifunctional viscosity index improver-dispersant antioxidant |
| WO1995027717A1 (en) | 1994-04-06 | 1995-10-19 | Montell Technology Company Bv | Metallocene compounds and use thereof in catalysts for the polymerization of olefins |
| US6143686A (en) | 1994-08-03 | 2000-11-07 | Exxon Chemical Patents, Inc. | Supported ionic catalyst compositions |
| US5741946A (en) | 1994-10-27 | 1998-04-21 | Mobil Oil Corporation | Polyether lubricants and method for their production |
| EP0767182A2 (en) | 1995-10-03 | 1997-04-09 | Ethyl Corporation | Copolymers of atactic propene polymers, dispersants derived therefrom and lubricating oils and fuel compositions containing the dispersants |
| US5744541A (en) | 1996-04-15 | 1998-04-28 | Dow Corning Asia, Ltd. | Copolymer of polypropylene and organopolysiloxane and method for preparation thereof |
| EP0802216A1 (en) | 1996-04-15 | 1997-10-22 | DOW CORNING ASIA, Ltd. | Copolymer of polypropylene and organopolysiloxane and method for preparation thereof |
| WO1997047665A1 (en) | 1996-06-13 | 1997-12-18 | University Of Waterloo | Hydrosilylation of polypropylene |
| US6114445A (en) | 1996-06-13 | 2000-09-05 | University Of Waterloo | Hydrosilylation of polypropylene |
| US6017859A (en) | 1996-06-17 | 2000-01-25 | Exxon Chemical Patents Inc | Polymers derived from olefins useful as lubricant and fuel oil additives, processes for preparation of such polymers and additives and use thereof |
| EP0958309A1 (en) | 1997-02-07 | 1999-11-24 | Exxon Chemical Patents Inc. | Preparation of vinyl-containing macromers |
| US6444773B1 (en) | 1997-02-07 | 2002-09-03 | Exxonmobile Chemical Patents Inc. | Preparation of vinyl-containing macromers |
| US6750307B2 (en) | 1997-02-07 | 2004-06-15 | Exxon Mobil Chemical Patents Inc. | Propylene polymers incorporating polyethylene macromers |
| WO1998040373A1 (en) | 1997-03-14 | 1998-09-17 | Sepracor, Inc. | Ring opening metathesis of alkenes |
| US6268518B1 (en) | 1997-03-29 | 2001-07-31 | Montell Technology Company B.V. | Metallocene compounds and their use in catalysts for the polymerization of olefins |
| WO1999005182A1 (en) | 1997-07-22 | 1999-02-04 | Bp Chemicals Limited | Polymerisation catalysts |
| US6590048B1 (en) | 1997-12-03 | 2003-07-08 | Studiengesellschaft Kohle Mbh | Highly active cationic ruthenium and osmium complexes for olefin metathesis reactions |
| EP1361232A2 (en) | 1997-12-10 | 2003-11-12 | ExxonMobil Chemical Patents Inc. | Polypropylene polymers incorporating macromers |
| US6197910B1 (en) | 1997-12-10 | 2001-03-06 | Exxon Chemical Patents, Inc. | Propylene polymers incorporating macromers |
| US6117962A (en) | 1997-12-10 | 2000-09-12 | Exxon Chemical Patents Inc. | Vinyl-containing stereospecific polypropylene macromers |
| WO1999046270A1 (en) | 1998-03-11 | 1999-09-16 | The Dow Chemical Company | Integrated process for preparation of diene complexes |
| US6448350B1 (en) | 1998-04-21 | 2002-09-10 | Basell Technology Company Bv | Process for the preparation of copolymers of ethylene with alpha-olefins |
| WO2000000576A1 (en) | 1998-06-30 | 2000-01-06 | Chevron Chemical Company Llc | Ashless lubricating oil formulations for natural gas engines |
| JP2000038420A (en) | 1998-07-24 | 2000-02-08 | Idemitsu Petrochem Co Ltd | Propylene copolymer and its production |
| US7247385B1 (en) | 1998-08-14 | 2007-07-24 | University Of Waterloo | Melt phase hydrosilylation of polypropylene |
| US6225432B1 (en) | 1998-08-26 | 2001-05-01 | Exxon Chemical Patents Inc. | Branched polypropylene compositions |
| WO2000055218A1 (en) | 1999-03-18 | 2000-09-21 | California Institute Of Technology | Novel aba triblock and diblock copolymers and methods of preparing the same |
| US6410666B1 (en) | 1999-03-18 | 2002-06-25 | California Institute Of Technology | ABA triblock and diblock copolymers and methods of preparing the same |
| US6897261B1 (en) | 1999-07-26 | 2005-05-24 | Idemitsu Kosan Co., Ltd. | Branched olefinic macromonomer, olefin graft copolymer, and olefin resin composition |
| US6703457B2 (en) | 2000-05-24 | 2004-03-09 | Basell Polyolefine Gmbh | Propylene polymers and process for the preparation thereof |
| US20020137978A1 (en) | 2000-06-23 | 2002-09-26 | Grubbs Robert H. | Synthesis of functionalized and unfunctionalized olefins via cross and ring-closing Metathesis |
| US6476167B2 (en) | 2000-12-14 | 2002-11-05 | Bayer Corporation | End-functionalized polyolefin prepared via ring opening metathesis polymerization in the presence of a novel chain transfer agent, and a process for the preparation of the end-functionalized polyolefin via ring opening metathesis polyermization |
| WO2002079127A1 (en) | 2001-03-30 | 2002-10-10 | California Institute Of Technology | Selective ring-opening cross-metathesis of cycloolefins |
| US6803429B2 (en) | 2001-03-30 | 2004-10-12 | California Institute Of Technology | Selective ring-opening cross-metathesis of cycloolefins |
| US20030161752A1 (en) | 2002-01-25 | 2003-08-28 | Sydney Luk | Powder metallurgy lubricant compositions and methods for using the same |
| US7126031B2 (en) | 2002-04-24 | 2006-10-24 | Symyx Technologies, Inc. | Bridged bi-aromatic ligands, catalysts, processes for polymerizing and polymers therefrom |
| WO2004031250A1 (en) | 2002-10-02 | 2004-04-15 | Dow Global Technologies Inc. | Liquid and gel-like low molecular weight ethylene polymers |
| US20040249046A1 (en) | 2002-10-15 | 2004-12-09 | Ramin Abhari | Polyolefin adhesive compositions and articles made therefrom |
| US20070293640A1 (en) | 2002-10-15 | 2007-12-20 | Peijun Jiang | Multiple catalyst system for olefin polymerization and polymers produced therefrom |
| US20040127649A1 (en) | 2002-10-24 | 2004-07-01 | Palanisamy Arjunan | Branched crystalline polypropylene |
| US20060052553A1 (en) | 2002-12-04 | 2006-03-09 | Basell Polyolefine Gmbh | Process for preparing 1-butene polymers |
| US7589160B2 (en) | 2002-12-04 | 2009-09-15 | Basell Polyolefine Gmbh | Process for preparing 1-butene polymers |
| US20040214953A1 (en) | 2003-04-28 | 2004-10-28 | Tosoh Corporation | Polyethylene composition and process for producing same |
| US20060270814A1 (en) | 2003-08-27 | 2006-11-30 | Haruyuki Makio | Polyolefin functional at one end |
| US6939930B2 (en) | 2003-09-09 | 2005-09-06 | Equistar Chemicals, Lp | Hydrosilane additives for increased polyolefin molecular weight |
| US20050054793A1 (en) | 2003-09-09 | 2005-03-10 | Reinking Mark K. | Hydrosilane additives for increased polyolefin molecular weight |
| JP2005139284A (en) | 2003-11-06 | 2005-06-02 | National Institute Of Advanced Industrial & Technology | Method for producing functionalized polypropylene |
| US20050159299A1 (en) | 2004-01-16 | 2005-07-21 | George Rodriguez | Hydrophobization and silica for supported catalyst |
| WO2005092935A1 (en) | 2004-02-24 | 2005-10-06 | Stichting Dutch Polymer Institute | Catalyst system comprising magnesium halide |
| US7943716B2 (en) | 2004-03-12 | 2011-05-17 | Basell Polyolefine Gmbh | Process for polymerizing 1-hexene or higher alpha-olefins |
| US7276567B2 (en) | 2004-04-16 | 2007-10-02 | Exxonmobil Chemical Patents Inc. | Heterocyclic substituted metallocene compounds for olefin polymerization |
| US20050261440A1 (en) | 2004-05-20 | 2005-11-24 | Dickakian Ghazi B | Dispersant material for mitigating crude oil fouling of process equipment and method for using same |
| JP2005336092A (en) | 2004-05-26 | 2005-12-08 | Mitsubishi Chemicals Corp | Novel transition metal compound and method for producing propylene polymer using the transition metal compound |
| US7820607B2 (en) | 2004-09-10 | 2010-10-26 | Mitsui Chemicals, Inc. | Viscosity modifier for lubricating oils, additive composition for lubricating oils, and lubricating oil compositions |
| US20080234451A1 (en) | 2004-12-23 | 2008-09-25 | Imperial Chemical Industries Plc | Olefin Metathesis Polymerisation |
| EP1849757A1 (en) | 2005-02-18 | 2007-10-31 | Idemitsu Kosan Co., Ltd. | Method for producing unsaturated hydrocarbon compound |
| EP1693357A1 (en) | 2005-02-22 | 2006-08-23 | Miltitz Aromatics GmbH | Process and apparatus for carying out a ring opening cross-metathesis reaction between cyclic and acyclic olefins |
| US20090221750A1 (en) * | 2005-03-22 | 2009-09-03 | Zeon Corporation | Thermoplastic Resin, Method for Producing Same and Molding Material |
| EP1862491A1 (en) | 2005-03-22 | 2007-12-05 | Zeon Corporation | Thermoplastic resin, method for producing same and molding material |
| WO2006127483A1 (en) | 2005-05-20 | 2006-11-30 | Bridgestone Corporation | Method for preparing low molecular weight polymers |
| US8058351B2 (en) | 2005-05-20 | 2011-11-15 | Bridgestone Corporation | Method for preparing low molecular weight polymers |
| US20080228017A1 (en) | 2005-06-06 | 2008-09-18 | Dow Global Technologies Inc. | Metathesis Process For Preparing An Alpha, Omega-Functionalized Olefin |
| US20100152388A1 (en) | 2005-06-22 | 2010-06-17 | Peijun Jiang | In-Reactor Polymer Blends |
| JP2007169340A (en) | 2005-12-19 | 2007-07-05 | Tosoh Corp | Olefin polymerization catalyst and process for producing olefin polymer |
| US7960487B2 (en) | 2006-02-22 | 2011-06-14 | Chevron Phillips Chemical Company Lp | Dual metallocene catalysts for polymerization of bimodal polymers |
| JP2007246433A (en) | 2006-03-15 | 2007-09-27 | Tosoh Corp | Organic transition metal compound, catalyst for olefin polymerization using the same, and method for producing polyolefin |
| US20080064891A1 (en) | 2006-07-12 | 2008-03-13 | Lee Choon W | Ring opening cross-metathesis reaction of cyclic olefins with seed oils and the like |
| JP2008050278A (en) | 2006-08-22 | 2008-03-06 | Tosoh Corp | Transition metal compound, catalyst for olefin polymerization, and method for producing polyolefin |
| US20100069573A1 (en) | 2006-08-25 | 2010-03-18 | Dow Global Technologies Inc. | Production of meta-block copolymers by polymer segment interchange |
| CN101011062A (en) | 2007-02-06 | 2007-08-08 | 张家港市骏博化工有限公司 | Organosilicon pesticide booster and preparing method thereof |
| US20100152387A1 (en) | 2007-05-23 | 2010-06-17 | Basf Se | Isotactic polystyrene having reactive groups |
| US20090247441A1 (en) | 2008-03-31 | 2009-10-01 | Exxonmobil Research And Engineering Company | High viscosity index pao with polyurea thickeners in grease compositions |
| US20090318640A1 (en) | 2008-06-20 | 2009-12-24 | Patrick Brant | Polymacromonomer And Process For Production Thereof |
| US20090318644A1 (en) | 2008-06-20 | 2009-12-24 | Patrick Brant | High Vinyl Terminated Propylene Based Oligomers |
| US20090318646A1 (en) | 2008-06-20 | 2009-12-24 | Patrick Brant | Functionalized High Vinyl Terminated Propylene Based Oligomers |
| US20090318647A1 (en) | 2008-06-20 | 2009-12-24 | Hagadorn John R | Olefin Functionalization By Metathesis Reaction |
| JP2010037555A (en) | 2008-07-11 | 2010-02-18 | Mitsui Chemicals Inc | Method for producing silylated polyolefin and additive containing the silylated polyolefin |
| JP2011026448A (en) | 2009-07-24 | 2011-02-10 | Mitsui Chemicals Inc | Composition having mold releasability |
| JP2012051859A (en) | 2010-09-03 | 2012-03-15 | Mitsui Chemicals Inc | Method for producing diol compound |
| JP2012052062A (en) | 2010-09-03 | 2012-03-15 | Mitsui Chemicals Inc | Method for producing epoxy compound |
Non-Patent Citations (37)
| Title |
|---|
| Amin et al., "Versatile Pathways for In Situ Polyolefin Functionalization with Heteroatoms: Catalytic Chain Transfer", Angew. Chem. Int. Ed., 2008, vol. 47, pp. 2006-2025. |
| Balboni et al., C2-Symmetric Zirconocenes for High Molecular Weight Amorphous Poly(propylene), Macromolecular Chemistry and Physics, 2001, vol. 202, No. 10, pp. 2010-2028. |
| Britovsek et al., Iron and Cobalt Ethylene Polymerization Catalysts Bearing 2, 6-Bis(Imino)Pyridyl Ligands: Synthesis, Structures, and Polymerization Studies, Journal of the American Chemical Society, 1999, vol. 121, No. 38, pp. 8728-8740. |
| Britovsek et al., Novel Olefin Polymerization Catalysts Based on Iron and Cobalt, Chemical Communications, 1998, No. 7, pp. 849-850. |
| Brzezinska et al., Synthesis of ABA Triblock Copolymers via Acyclic Diene Metathesis Polymerization and Living Polymerization of alpha-Amino Acid-N-Carboxyanhydrides, Macromolecules, 2001, vol. 34, pp. 4348-4354. |
| Brzezinska et al., Synthesis of ABA Triblock Copolymers via Acyclic Diene Metathesis Polymerization and Living Polymerization of α-Amino Acid-N-Carboxyanhydrides, Macromolecules, 2001, vol. 34, pp. 4348-4354. |
| Bujadoux et al., Use of bridged and non-bridged metallocene catalysts in high pressure/high temperature ethylene/alpha-olefin copolymerization, Metallocene Polymers, 1995, pp. 377-402. |
| Bujadoux et al., Use of bridged and non-bridged metallocene catalysts in high pressure/high temperature ethylene/α-olefin copolymerization, Metallocene Polymers, 1995, pp. 377-402. |
| Chen et al., Reactive & Functional Polymers, 2008, vol. 68, No. 9, pp. 1307-1313. |
| Chung, "Synthesis of Functional Polyolefin Copolymers with Graft and Block Structures", Prog. Polym. Sci., 2002, vol. 27, pp. 39-85. |
| Corey et al., "Reactions of Hydrosilanes and Olefins in the Presence of Cp2 MCl2 /nBuLi", Organometallics, 1992, vol. 11, pp. 672-683. |
| Cossy et al., "Cross-Metathesis reaction. Generation of Highly Functionalized Olefins from Unsaturated Alcohols", Journal of Organometallic Chemistry, 2001, vol. 634, Issue 2, pp. 216-221. |
| Hansell et al., Additive-Free Clicking for Polymer Functionalization and Coupling by Tetrazine-Norbornene Chemistry, Journal of the American Chemical Society, 2011, vol. 133, No. 35, pp. 13828-13831. |
| Herzon et al., "Direct, Catalytic Hydroaminoalkylation of Unactivated Olefins with N-Alkyl Arylamines", JACS, 2007, vol. 129, pp. 6690-6691. |
| Herzon et al., "Hydroaminoalkylation of Unactivated Olefins with Dialkylamines", JACS, 2007, vol. 130, pp. 14940-14941. |
| Katayama, et al. "Vinylideneruthenium Complexes in Catalysis", Coordination Chemistry Reviews, 2004, vol. 248, pp. 1703-1715. |
| Kesti et al., "Group 4 Metallocene Olefin Hydrosilyation Catalysts", Organometallics, 1992, vol. 11, pp. 1095-1103. |
| Kolodka et al., "Copolymerization of Propylene with Poly(Ethylene-Co-Propylene) Macromonomer and Branch Chain-Length Dependence of Rheological Properties", Macromolecules, 2002, vol. 35, pp. 10062-10070. |
| Koo et al., "Silicon-Modified Ziegler-Natta Polymerization. Catalytic Approaches to Silyl-Capped and Silyl-Linked Polyolefins Using "Single-Site" Cationic Ziegler-Natta Catalysts", Journal of American Chemical Society, 1999, vol. 121, pp. 8791-8802. |
| Liu et al., Kinetics of Initiation, Propagation, and Termination for the [rac-(C2H4(1-indenyl)2)ZrMe]{MeB(C6F5)3-Catalyzed Polymerization of 1-Hexene, Journal of the American Chemical Society, 2001, vol. 123, pp. 11193-11207. |
| Lopez et al., "Synthesis of Well-Defined Polymer Architectures by, Successive Catalytic Olefin Polymerization and Living/Controlled Polymerization Reactions ", Prog. Polym. Sci., 2007, vol. 32, pp. 419-454. |
| Markel, et al., "Metallocene-Based Branch-Block Thermoplastic Elastomers", Macromolecules, 2000, vol. 33, pp. 8541-8548. |
| Mathers et al., "Cross Metathesis Functionalization of Polyolefins", Chem Commun, 2004, pp. 422-423. |
| Nagai et al., Novel Well-defined Funcationalized Polyolefins and Polyolefin-polar Polymer Block Copolymers: Formations and Their Features, Poly Preprints, 2008, vol. 49, No. 2, 776-777. |
| Ornelas et al., Cross Olefin Metathesis for the Selective Functionalization, Ferrocenylation, and Solubilization in Water of Olefin-Terminated Dendrimers, Polymers, and Gold Nanoparticles and for a Divergent Dendrimer Constructions, Journal of American Chemical Soc., 2008, vol. 130, No. 4, pp. 1495-1506. |
| Passaglia et al., "Grafting of Diethyl Maleate and Maleic Anhydride Onto Styrene-b-(Ethylene-co-1-Butene)-b-Styrene Triblock Copolymer (SEBS)", Polymer, 2000, vol. 41, pp. 4389-4400. |
| Quirk et al., "Anionic Synthesis of Secondary Amine Functionalized Polymers by Reaction of Polymeric Organolithiums with N-Benzylidenemethylamine" , Macromolecular Chemistry and Physics, 2002, vol. 203, pp. 1178-1187. |
| Resconi et al., Chain Transfer Reactions in Propylene Polymerization with Zirconocene Catalysts, Topics in Catalysis, 1999, vol. 7, No. 1-4, pp. 145-163. |
| Rodriguez et al., Poly(4-vinylpyridazine). First Synthesis, Characterization and Properties, Polymeric Materials Science and Engineering, Proceedings of the ACS Division of Polymeric Materials Science and Engineering, 1990, vol. 63, pp. 376-382 (Abstract). |
| Rose et al., "Poly(ethylene-co-propylene macromonomer)s: Synthesis and Evidence for Starlike Conformaitons in Dilute Solution", Macromolecules, 2008, vol. 41, pp. 559-567. |
| Rybak et al., "Acyclic Diene Metathesis with a Monomer with a Monomer from Renewable Resources: Control of Molecular Weight and One-Step Preparation of Block Copolymers", ChemSusChem, 2008, vol. 1, pp. 542-547. |
| Shiono et al., Copolymerization of poly(propylene) macromonomer with ethylene by (tertbutanamide)dimethyl(tetramethyl-qscyclopentadienyl) silane titanium dichloride/methylaluminoxane catalyst, Macromol. Chem. Phys., 1997, vol. 198, pp. 3229-3237. |
| Weng et al., Long Chain Branched Isotactic Polypropylene, Macromolecules, 2002, vol. 35, pp. 3838-3843. |
| Weng et al., Synthesis of Long-Chain Branched Propylene Polymers via Macromonomer Incorporation, Macromol. Rapid Commun., 2001, vol. 22, No. 18, pp. 1488-1492. |
| Weng et al., Synthesis of Vinyl-Terminated Isotactic Poly(propylene), Macromol. Rapid Commun., 2000, 21, No. 16, pp. 1103-1107. |
| Wu et al., Synthesis of Polynorbornene-poly(ethylene-co-propylene) Diblock Copolymers, Polymeric Materials Science and Engineering, 1998, vol. 78, pp. 158-159. |
| Xu et al., Ethylene Copolymerization with 1-Octene Using a 2-Methylbenz[e] indenyl-Based ansa-Monocyclopentadienylamido Complex and Methylaluminoxane Catalyst, Macromolecules, 1998, vol. 31, pp. 4724-4729. |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021126691A1 (en) | 2019-12-16 | 2021-06-24 | Exxonmobil Chemical Patents. Inc. | Tire tread compounds |
| WO2021178235A1 (en) | 2020-03-03 | 2021-09-10 | Exxonmobil Chemical Patents Inc. | Rubber compounds for heavy-duty truck and bus tire treads and methods relating thereto |
| WO2021242636A1 (en) | 2020-05-29 | 2021-12-02 | Exxonmobil Chemical Patents Inc. | Processes for producing cyclic olefins from polymers and re-polymerization thereof |
| US11912861B2 (en) | 2020-10-29 | 2024-02-27 | ExxonMobil Engineering & Technology Co. | Rubber composition for lighter weight tires and improved wet traction |
Also Published As
| Publication number | Publication date |
|---|---|
| US20130137829A1 (en) | 2013-05-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8604148B2 (en) | Functionalization of vinyl terminated polymers by ring opening cross metathesis | |
| US9181360B2 (en) | Polymers prepared by ring opening / cross metathesis | |
| US8669330B2 (en) | Olefin triblock polymers via ring-opening metathesis polymerization | |
| JP5427887B2 (en) | Functionalization of olefins by metathesis reaction | |
| US8283419B2 (en) | Olefin functionalization by metathesis reaction | |
| US8940839B2 (en) | Diblock copolymers prepared by cross metathesis | |
| US8785562B2 (en) | Amphiphilic block polymers prepared by alkene metathesis | |
| WO2013025284A1 (en) | Polymers prepared by ring opening/cross metathesis | |
| US9169334B2 (en) | Preparation of bottlebrush polymers via ring-opening metathesis polymerization | |
| EP2897992B1 (en) | Functionalized resins obtained via olefin metathesis | |
| WO2013081726A1 (en) | Functionalization of vinyl terminated polymers by ring opening cross metathesis | |
| CN103443136B (en) | By Amphipathilic block polymer prepared by olefin metathesis | |
| US9382354B2 (en) | Polyesters containing polyolefin arms | |
| EP2688954A2 (en) | Olefin triblock polymers via ring-opening metathesis polymerization | |
| EP2688921B1 (en) | Diblock copolymers prepared by cross metathesis | |
| CN103459435B (en) | Diblock copolymers prepared by cross-metathesis | |
| WO2014120433A1 (en) | Preparation of bottlebrush polymers via ring-opening metathesis polymerization | |
| WO2015076958A1 (en) | Novel polyesters containing polyolefin arms |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: EXXONMOBIL CHEMICAL PATENTS INC., TEXAS Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:HOLTCAMP, MATTHEW W.;CROWTHER, DONNA J.;HUFF, CAOL P.;AND OTHERS;SIGNING DATES FROM 20111215 TO 20120110;REEL/FRAME:027549/0250 |
|
| STCF | Information on status: patent grant |
Free format text: PATENTED CASE |
|
| FPAY | Fee payment |
Year of fee payment: 4 |
|
| FEPP | Fee payment procedure |
Free format text: MAINTENANCE FEE REMINDER MAILED (ORIGINAL EVENT CODE: REM.); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY |
|
| LAPS | Lapse for failure to pay maintenance fees |
Free format text: PATENT EXPIRED FOR FAILURE TO PAY MAINTENANCE FEES (ORIGINAL EVENT CODE: EXP.); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY |
|
| STCH | Information on status: patent discontinuation |
Free format text: PATENT EXPIRED DUE TO NONPAYMENT OF MAINTENANCE FEES UNDER 37 CFR 1.362 |
|
| FP | Lapsed due to failure to pay maintenance fee |
Effective date: 20211210 |